B515935k.dvi W 36 00b4951e4e9ff263e1000000
User Manual: W-36
Open the PDF directly: View PDF ![]() .
.
Page Count: 9

PAPER www.rsc.org/dalton | Dalton Transactions
Inorganic crown: the host–guest chemistry of a high nuclearity ‘Celtic-ring’
isopolyoxotungstate [H12W36O120]12−†
De-Liang Long, Oliver Br¨
ucher, Carsten Streb and Leroy Cronin*
Received 9th November 2005, Accepted 2nd February 2006
First published as an Advance Article on the web 3rd May 2006
DOI: 10.1039/b515935k
A range of complexes based on the high-nuclearity {W36}isopolyoxotungstate cluster, [H12W36O120]12−,
with a triangular topology has been isolated by using the organic cation, protonated triethanolamine. In
analogy to an 18-crown-6 crown ether with six oxygen donors on a ring, the cluster can form alkali and
alkaline earth metal complexes {M⊂W36}(M =K+,Rb
+,Cs
+,NH
4+,Sr
2+ and Ba2+,1–6, respectively).
Compounds 1–6 were characterized by single-crystal X-ray diffraction, elemental analysis, IR
spectroscopy. Comparisons between the structures of 1–6 and 18-crown-6 as well as the symmetry
directing influence of the organo-cations in the isolation of the overall cluster architecture are discussed.
Introduction
The self-assembly of clusters based on molybdenum, tungsten or
vanadium oxo anions gives rise to an almost unrivalled range
of polyoxometalate cluster anions. The versatile nature of these
clusters originates from the ability to polymerize metal-based poly-
hedra to form a range of clusters from low to high nuclearities.1
In particular, the ability for molybdenum- and tungsten-based
systems to form very large clusters has been demonstrated by a
number of nano-sized cluster systems with over 100 tungsten2
and molybdenum3,4 atoms in a single cluster molecule. Therefore
it is not surprising that polyoxometalates have been subjected to
a vast number of studies due to their attractive electronic and
molecular properties that give rise to a variety of applications
e.g. in catalysis,5,6 magnetism,7,8 redox chemistry,9,10 medicine,11,12
and materials science.13–15 Despite this great range of cluster types
and properties, the ability to assemble large cluster systems from
smaller known building blocks in a pre-determined way is a great
challenge, as such routes could be a direct way to systematically
control the overall cluster architecture and properties. This is
because the understanding and manipulation of the self assembly
processes that underpin the formation of POM clusters has to be an
attractive route to enable the design of designer clusters and multi-
functional materials, which take advantage of the unique physical
properties associated with this extraordinary class of molecules.1
The true enormity of this challenge can be further clarified
when one realizes the main route to synthesize POM clusters
(large or small) often employs ‘one-pot’ reactions.1–4 Therefore,
the manipulation of some of the many reaction parameters often
represents a straight-forward, but rather tedious, route to new,
self-assembled POM architectures. To overcome such problems,
the design of larger architectures (based on clusters) using POM
building blocks as synthons could provide a step change in the
design and assembly of such systems. This is because the ability
to assemble large cluster systems from smaller known building
WestCHEM, Department of Chemistry, The University of Glasgow, Glasgow,
UK G12 8QQ. E-mail: L.Cronin@chem.gla.ac.uk
† Based on the presentation given at Dalton Discussion No. 9, 19–21st
April 2006, Hulme Hall, Manchester, UK.
blocks could be a direct way to systematically control the overall
cluster architecture and properties while retaining the geometries
of the building blocks. Thus, such building blocks of well-defined
shape and connectivity might form the basis for work towards
the growth of nanoscopic clusters of predetermined structure and
function.16 However, the major problem with this approach lies in
establishing routes to produce reactive building blocks present in
solution in significant concentrations that can be reliably utilized
in the formation of larger architectures, without re-organizing or
isomerization to other fragments. Access to such building blocks
has been the major limitation in stepwise growth of W- and Mo-
based POM clusters. Such limitations may be circumvented by
adopting an approach that kinetically stabilizes the building block
in solution, thereby effectively preventing its reorganization to
other structure types.17–21
One possible route to this goal is to use bulky organic cations
to isolate a new structure type by virtue of the cations used
to ‘encapsulate’ the new building blocks, thereby limiting their
reorganization to simpler structural types.17–21 By trapping clusters
during the self-assembly process it may be possible to restrain
the cluster from reorganizing into other well-known structure
types, see Fig. 1. Also, in many cases, synthetic strategies to
W- and Mo-based cluster systems are guided by the fact that
structures of polyoxomolybdate clusters are frequently derived
from highly stable, low-nuclearity structural archetypes such as
the Oh-symmetric Lindqvist22 anion [Mo6O19]2– and the various
Fig. 1 A schematic showing the ‘encapsulation’ of the [H2Mo16 O52 ]10−
cluster units during the cluster assembly process (shown schematically by
the central view within the dotted circle) in the presence of the bulky
organo-cation HMTAH (protonated hexamethylenetetramine).
2852 |Dalton Trans., 2006, 2852–2860 This journal is ©The Royal Society of Chemistry 2006

isomers of the Keggin structure,23 [M12O36 (XO4)]n–, or the Dawson
structure,24 [M18 O46(XO4)2]n–(X =S, P, As, Si etc.)(M=W, M o ) . 25
Previously, by using protonated hexamethylenetetramine
(HMTAH+) as counter ions, we were able to stabilize and isolate
a highly charged polyoxomolybdate anion, [H2MoV4MoVI16O52 ]10–,
{Mo16}, which represents a new structural type.17,18 The {Mo16 }
anion displays an unusual flat shape and can itself be formally
decomposed into a highly condensed {Mo12}building block which
incorporates two pairs of MoVcentres and to which two edge-
sharing {Mo2}groups are attached via corners. As we previously
reported,17,18 the {Mo16}is stable in the solid state as the salt
(C6H13N4)10[H2Mo16 O52 ]·34H2O since the highly negative cluster
anion is virtually completely wrapped by the organic HMTAH+
cations. In an extension to this approach we also recently isolated
a family of sulfite-based Dawson-type mixed-valence polyoxo-
molybdates [Mo18O54 (SO3)2]n–, using the same type of synthetic
approach. Furthermore these [Mo18O54 (SO3)2]n–clusters possess
unusual electronic properties and display S ···S interactions
between the lone pairs of the two sulfite anions inside the cluster.19
Thus, the use of bulky organic cations in the formation of Mo-
based POMs appears to restrict aggregation to the more highly
symmetrical cluster types, allowing a fundamentally more diverse
set of clusters and cluster-based building blocks to be isolated,
that display unprecedented structural17,18 or physical19 features.
This observation was further explored by us when we used
bulky organo-cations to rationally isolate and connect {Mo8Ag2}
building blocks to larger polymeric architectures.21
In previous work we showed that this simple but very efficient
strategy can successfully be extended to polyoxotungstate chem-
istry, where we were able to isolate a {W36 }-based cluster. This
has the formula {(H2O)4K⊂[H12 W36O120 ]}11−and represents the
largest isopolyoxotungstate so far discovered.26 Interestingly, the
centre of this “Celtic-ring” cluster has a metal oxo framework that
resembles the 18-crown-6 ether and also shows main features of
crown ethers, for instance the ability to bind different metal ions in
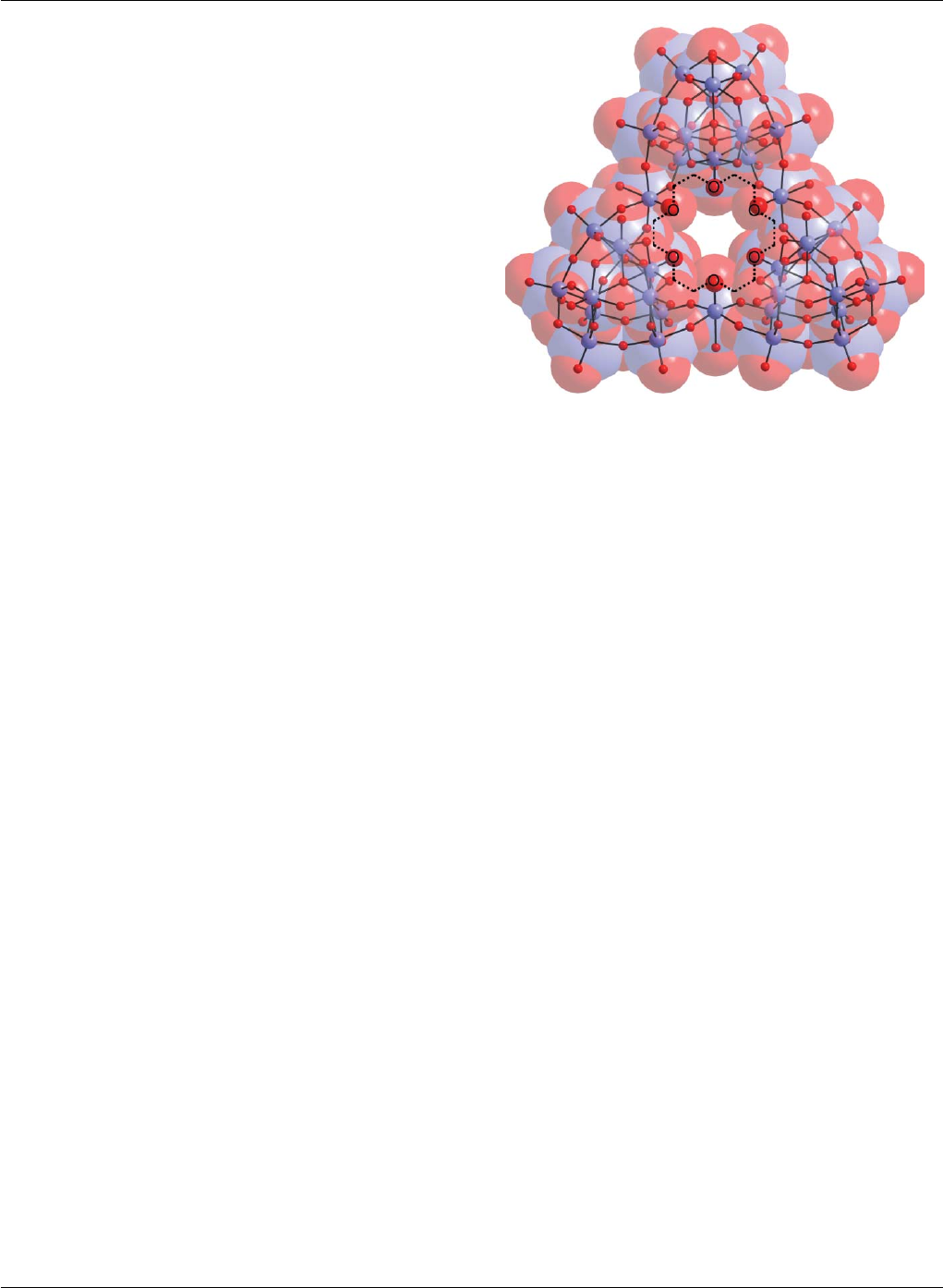
the central cavity of the cluster, see Fig. 2. The {K⊂W36 }11−anion
is approximately C3v-symmetric with a Celtic ring-like shape, and
comprises three {W11}cluster subunits linked together by three
{W1}bridges (shown as the cyan coloured polyhedra in Fig. 2).
The {W11}cluster consists of a ring of six basal W positions,
an additional W position in the centre of this ring, and four
apical W positions in a butterfly configuration. Every W centre
has a distorted WO6octahedral coordination geometry with one
terminal W=O(W–O∼1.70 A
˚) extending away from the cluster.
Within the {W11}moieties, two protons form hydrogen bonds
between the four central l3/4-oxo ligands. The three bridging
{W1}groups also display a distorted WO6octahedral coordination
geometry, each sharing four bridging oxo ligands in the equatorial
plane with the {W11}clusters; one W=Oterm (W–O ∼1.70 A
˚)
points to the central potassium ion and one water ligand (W–OH2
∼2.20 A
˚) points outside the cluster. The potassium ion has a rather
distorted coordination geometry and is coordinated by 10 oxygen
atoms; four of these 10 are water ligands (K–O distances are in
the range 2.75(2) and 3.03(2) A
˚) the other six are Oterm ligands
coming from the WO6moieties. The potassium ion is displaced
slightly above the equatorial plane, formed by the six Oterm ligands,
with three water molecules coordinating from above (RHS inset on
Fig. 2), and one below the equatorial plane (LHS inset on Fig. 2).
Overall the coordination geometry of the potassium ion can be
Fig. 2 Molecular structure of {(H2O)4K⊂[H12W36O120 ]}11– .Themiddle
view shows a ball-and-stick representation of the W (blue) and O
(red) framework. The central ten-coordinate K+ion is represented by
a purple sphere. The K+ion is coordinated to the terminal oxygen
positions of a {W6O6}moiety (shown by the dotted lines) and each of
these metal-centered polyhedra are shown in blue or cyan. Below, all
the WO-based units interacting with the potassium ion are shown by
polyhedral representations ({W1}linkers in cyan, and units from the {W11 }
fragments in blue). This view also reveals the “cavity” in which the K+
ion resides. The top right inset shows the top view of the coordination
around the potassium ion showing the trigonal face formed by the three
coordinating water ligands on the front of the cluster. The top left inset
shows the view from the bottom of the cluster with the single water ligand
coordinated to the potassium ion.
described as a distorted polyhedron with 10 vertices, 16 faces and
24 edges and can be considered to be a kind of distorted dipyramid.
It is interesting to see that the six WO6moieties, which form the
cluster cavity in which the potassium ion is ligated, map extremely
well onto the structure of the crown ether 18-crown-6. Using this
observation as inspiration, we have extended our investigations to
see how far this analogy can be taken. The present work extends
this initial observation and focuses on the investigation of the host–
guest chemistry of this new {W36}cluster by extending its potas-
sium ion complex to other alkali and alkali-earth metal ions.26
Results and discussion
Synthesis of {W36}derivatives with alkali and alkali-earth metal
cations
The {W36}cluster was initially isolated from an aqueous solu-
tion containing NaWO4·2H2O and triethanolamine, which was
adjusted to a pH of 2.0 with hydrochloric acid followed by the
addition of Na2S2O4as reducing agent.26 As revealed by X-ray
crystallographic studies, the product has an approximate formula
of (TEAH)9Na2{(H2O)4K⊂[H12W36O120 ]}·17H2O(1). Although
there was no potassium added to the reaction mixture, the
X-ray crystallographic data clearly supports the unambiguous
assignment of a K+ion coordinated to the central cavity of
the cluster. Therefore the small amount of potassium needed to
produce the cluster compound 1was assumed to originate from
This journal is ©The Royal Society of Chemistry 2006 Dalton Trans., 2006, 2852–2860 | 2853
the glassware used. This assumption was confirmed by atomic
absorption analysis on the postulated {K⊂W36 }system, which
indeed revealed the presence of potassium at the required concen-
tration. The slow sequestration of potassium from the glassware
was also supported by the observation that it took over four weeks
for the product to form in the initial synthetic system, whereas the
addition of potassium directly to the reaction mixture reduces this
time to only a few days. Subsequent studies also demonstrated
that no product was obtained when plastic flasks were used for the
original reaction system. Furthermore the experiment confirmed
that the addition of Na2S2O4is not necessary for the formation of
the cluster 1, although the addition of Na2S2O4can form a more
soluble reduced polyoxotungstate at the starting point, and this
serves to prevent precipitation of insoluble polymers. However we
have now discovered that warming of the solution can also prevent
precipitation of the intermediates whilst the pH is lowered to the
crystallization pH for compound 1.
Comparisons between the {W36}cluster anions, crown ether
chemistry and other polyoxometalates
Today the design of highly sophisticated macrocycles based on
polyethers, the crown ethers, is continuing to accelerate since their
first discovery by Pederson in the mid-1960s.27,28 This is because
they are an extremely important class of molecules capable of
recognition and binding of cations. They are able to form selective
and stable complexes with various cations, most notably metal
ions, and their selectivity as complexing agents results from the
definite size of the crown cavity, which only accepts cations of
comparable ionic radii. The well known 18-crown-6 ether for
instance, has an estimated cavity radius of 2.6–3.2 A
˚and is most
suitable for complexing K+,NH
4+and Rb+, but is also known
to bind to other metal cations.28 As such, the crown ethers find
ubiquitous application from ionophores in cation sensing, to phase
transfer catalysis in chemical synthesis, to chiral separators in
chromatography to name but a few.29–31
During the last two decades chemists realized that the size selec-
tivity, so effectively applied by crown ethers in the differentiation
and complexation of cations, could also be utilized to recognize,
design, and apply inorganic ring-type compounds that resemble
organic crown ethers for binding cations. Notable examples are
reported by Pecoraro et al. such as [Mn11 (salicylhydroximate)]42−,32
[C45H50N10 O10Cu5Sm]33 and [CuII (picha)]5(picha =picoline hy-
droxamic acid),34 which are often referred to as metallacrowns.
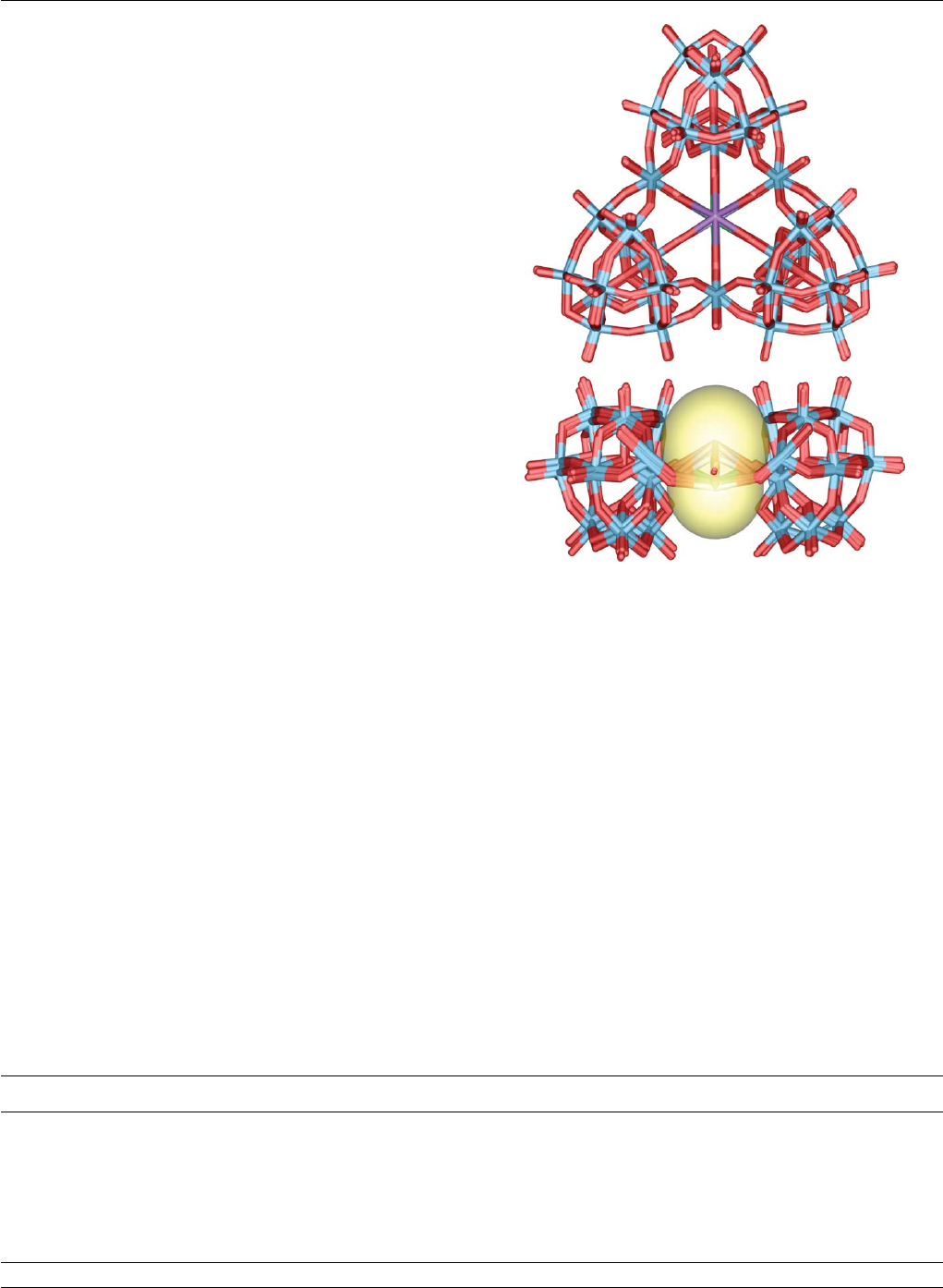
This comparison may also be made with the {W36}cluster, which
in analogy to 18-crown-6, has a very similar cavity radius of ca.
2.8 A
˚, see Fig. 3.
Polyoxometalates can also encapsulate metal ions such as
potassium in a {W65}cluster,35 and a {Mo80V22},36 but these
do not involve a cavity that can be compared to classical
crown ethers. However one novel compound, the Keplerate ball,
reported by M ¨
uller et al.,37 has a spherical framework with a high
charge and accessible inner chamber. This cluster, of the form
{MoVI72 MoV60 L30}n−(n=42 when L =acetate, n=72 when L =
sulfate) has been investigated as a type of ‘inorganic-cell’ whereby
the cluster behaves as a semi-permeable inorganic membrane open
for H2O and small cations.37 This is because the cavities, or pores
shown in the Keplerate have the form {Mo9O9}and provide a
structural motif rather similar to that of the classical crown ethers.
Fig. 3 Comparative illustration of the {W36 }cluster framework and the
18-crown-6 structure (show to scale). W and O atoms shown as blue and
red spheres with the crown ether superimposed on O atoms forming the
‘cavity’ of the cluster (these are larger for clarity). The ball and stick
structure is also superimposed on the space-filling CPK representation of
the {W36}to show the central cavity.
Complexes of {W36}with Rb+,Cs
+,NH
4
+,Sr
2+ and Ba2+
As already discussed, the {W36 }-based cluster type has a similar
oxygen coordination environment as in the 18-crown-6 ether
and also shows the ability to capture potassium ions from
aqueous solution, even though the oxygen atoms that comprise
the ‘crown-like’ cavity are terminal oxo ligands rather than the
bridging oxo ligands found in both organic-based crown ethers
and the metallacrowns.32–34 Extension of this work was relatively
straightforward in so far as our experiments were conducted in
plastic vessels (to avoid sequestration of potassium from glassware;
the potassium originates from the KOH/alcohol base bath used
to clean the glassware),26 and the new metal cations added to
the solution of the cluster at the correct pH. Interestingly, in the
presence of only Na+,the{W36}cluster could not be isolated, even
if the concentration of Na+, was elevated in the reaction mixture.
Therefore experiments, with Rb+,Cs
+,NH
4+,Sr
2+ and Ba2+
were carried out as a function of pH, concentration of tungstate,
triethanolamine and total ionic strength. By varying these pa-
rameters and adjusting an aqueous solution of sodium tungstate
and triethanolamine hydrochloride to a certain pH with HCl
solution, followed by a short period of heating, the corresponding
chlorides of these cations were added to the reaction mixture
which was then stored for crystallization. In addition to the use
of plastic vessels, special attention was focussed on preventing
possible contamination of the reactions with potassium sources
(e.g. from the reagents, pH meter etc). As a further internal control,
samples of the reaction mixtures, without added metal salt were
kept as control samples and were never observed to lead to the
formation of the {W36}cluster. This indicated that the potassium-
free approach was efficient and the results of these investigations
are shown below (Table 1).
During the reactions, the {W36 }cluster emerged as colourless
needles of {W36}-based clusters 1–6 which were obtained for all
2854 |Dalton Trans., 2006, 2852–2860 This journal is ©The Royal Society of Chemistry 2006
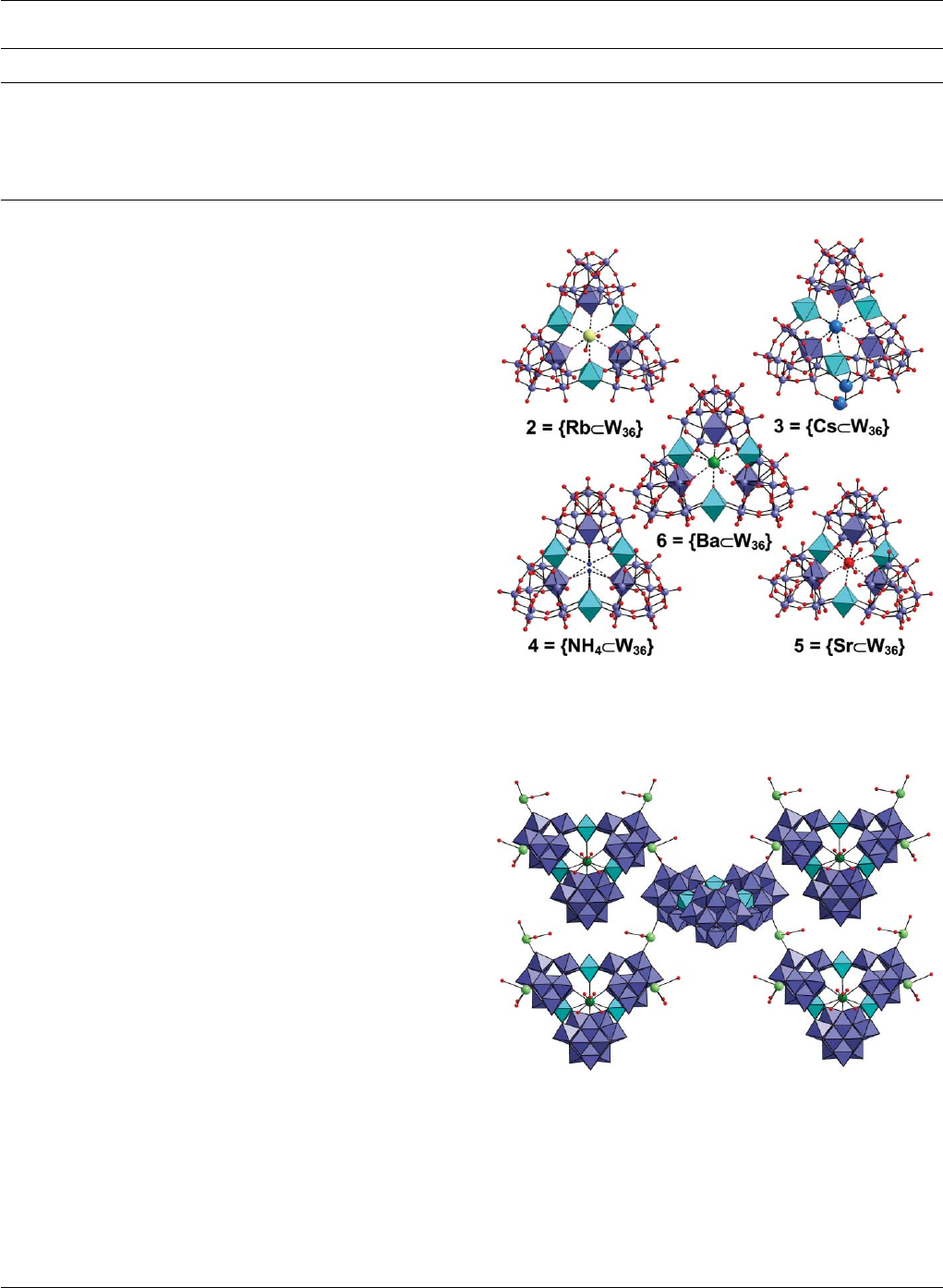
Table 1 Results from the formation of {W36}derivatives with K+,Rb
+,Cs
+,NH
4+,Sr
2+ and Ba2+ chloride salts
Metal salt pH W/M+ratio Yield (%) Product formula
KCl 2.2 15 : 1 49 (TEAH)9Na2{(H2O)4K⊂[H12W36 O120 ]}·17H2O(1)
RbCl 2.2 15 : 1 39 (TEAH)9Na2{(H2O)4Rb⊂[H12W36 O120 ]}·17H2O(2)
CsCl 2.2 15 : 1 32 (TEAH)9Na2{(H2O)4Cs⊂[H12 W36 O120 ]}·15H2O(3)
NH4Cl 1.8 10 : 1 38 (TEAH)9Na2{(NH4)⊂[H12W36 O120 ]}·19H2O(4)
SrCl2·6H2O 2.2 15 : 1 52 (TEA)(TEAH)8Na2{(H2O)4Sr⊂[H12W36 O120 ]}·17H2O(5)
BaCl2·2H2O 1.3 30 : 1 48 (TEA)(TEAH)8Na{(H2O)4Ba1.5⊂[H12 W36 O120]}·17H2O(6)
of the applied cations. For K+,Rb
+,Cs
+,NH
4+,Sr
2+, the reaction
condition with a pH of 2.2 seems to be optimal for the formation of
the {W36}cluster, whereas for Ba2+ no crystals could be obtained
(but material quickly precipitates from solution) at this pH value
directly. Therefore the Ba2+ compound was produced as follows:
the barium chloride was added to the tungstate solution at pH 1.3
and then the pH of the solution was brought up to 2.2 for
crystallization. It would appear that a lower pH is needed for
the {W36Ba}-unit formation and then again a significant higher
pH to allow crystallization. Further, although the ionic radii of the
potassium- and the ammonium ion are almost the same, crystals
from the {W36 NH4}cluster 4were only obtained in lower yields
and took a longer time to crystallize out.
Structures of compounds 2–6
The structures of all cluster derivatives 1–6 were determined by
single-crystal X-ray diffraction. Except that 1was previously
reported,26 crystal structures of 2–6 are included in this paper.
All six compounds are essentially isomorphous and crystallize
in the same orthorhombic system with space group Pnma. The
asymmetric unit contains only half a {W36}cluster with a
crystallographic mirror plane passing through the cluster and
the central coordinated metal ions (K+to Ba2+). Even though
the TEAH+, sodium cations and solvent water molecules are
highly disordered, the main structures, i.e. the cluster anion
[H12W36O120 ]12−together with the coordinated metal ions K+to
Ba2+, are well defined. Compounds 2{W36Rb}and 5{W36Sr}
incorporate Rb+and Sr2+ in the centre of the {W36}cavity at full
occupancy as determined by crystallographic analysis, similar to
the K+found in 1, see Fig. 4. However, the NH4+ion in 4is
disordered over two positions nearly half up the O6plane of the
{W36}cluster and half down, see Fig. 4. In compound 3{W36Cs},
the Cs site at the centre of the {W36 }cavity is not fully occupied
and additional electron density that can be assigned as partially
occupied Cs ion positions was found elsewhere between two {W11 }
cluster subunits, see Fig. 4.
These positions can only be assigned to partially occupied
Cs centres according to the bond distances to neighbouring
oxygen atoms. Interestingly, the {W36Ba}complex, compound 6,
shows some difference in the crystal structure to the other metal
cation derivatives, see Fig. 4. As revealed by single-crystal X-ray
diffraction and chemical analysis, this compound contains one
and a half Ba2+ cations per cluster {W36 }unit. The extra half Ba2+
cation was found to sit between {W36}frameworks and link them
together to form 3-D polymers in the solid state, see Fig. 5. This
observation also helps explain the poor solubility of compound
6in water. While compound 3{W36Cs}is the most soluble
derivative, compound 6is only about half as soluble. The decreased
Fig. 4 Representations of the crystal structures of the {W36 }framework
complexed with Rb+(2), Cs+(3), NH4+(4), Sr2+ (5)andBa
2+ (6). Colour
scheme same as for Fig. 2 except the central complexed ions (Rb+:pale
green, Cs+:blue,NH
4+: small blue spheres, Sr+: red, Ba2+: green).
Fig. 5 Representation of the network formed in compound 6.The{W36 }
is shown in polyhedral view ({W11}-units-blue) with the linking {W1}units
in cyan. The Ba2+ inthecavityareshownindarkgreenandthose
connecting the clusters light green. The coordinating water ligands are
shown as red spheres.
solubility of the latter can be explained with the stated linking
of the cluster frameworks by the additional Ba2+ ions present in
the structure. Interestingly, the terminal oxo ligands of the {W36 }
This journal is ©The Royal Society of Chemistry 2006 Dalton Trans., 2006, 2852–2860 | 2855
cluster that are connected to the polymeric network by the Ba2+
were previously postulated (on the basis of DFT calculations26 )
as to be one of the possible sites (high nucleophilicity) for linkage
with electrophiles.
Given the {W36 }cluster has a triangular shape (this is
unique among the known family of isopolyoxometalates, even
though there are a few examples of triangular heteroPOMs,
[P5Co9W27O119H17 ]16−,38 [(a-SiW11MnO38OH)3]15−.39 ), with the cav-
ity centre resting on the approximate C3-symmetry axis (Fig. 1),
it is possible to examine the displacement of the complexed metal
ions from the equatorial plane formed by the six terminal oxo-
ligands. The degree of displacement from the equatorial plane
above or below should reflect the increasing ionic radii of the
cations as they fit less well into the cavity. These displacements are
pronounced in the metal ion derivatives 1–3 and 4,5,whichcanbe
seen in distances between the corresponding central ions and the
six surrounding oxygen atoms from the W=O ligands of the {W36}
crown. These distances are summarized in Table 2 and visualized
in Fig. 6.
As seen in the table, the found average metal–oxygen distances
in the {W36}host–guest complexes 1–6 are very close to those
shown in the corresponding 18-crown-6 complexes. The metal
cations in the synthesized compounds show different distances
to the equatorial plane of the cluster framework which reflect the
different sizes of their ionic radii, see Fig. 6.
Although there are some great similarities between the {W36}
system and 18-crown-6, there are some features that are not
common to both systems. Firstly the six-coordinated oxygen atoms
on {W36}are not planar but in a manner with three oxo ligands
up and three down alternatively forming two parallel planes
distant at ∼0.5 A
˚, while those on 18-crown-6 can adopt a planar
conformation. Further, the {W=O}6donor groups of the {W36 }
crown are considerably more rigid unlike 18-crown-6, which is
much more flexible. This means that 18-crown-6 can deform to
form metal complexes with small ions like Na+,Ca
2+, lanthanide
ions and d-transition metal ions. This is because 18-crown-6, along
with other similar crown ethers is able to distort and wrap itself
around these smaller metal cations in an attempt to maximize the
electrostatic interactions. This increases the strain of the ligand,
which makes these complexes less stable than the ones with metal
cations of optimal spatial fit. However, the {W36}framework, due
to its high rigidity, simply cannot change conformation in such
a way to bind these metal cations. This is partly confirmed by
the observation that the diameter of the central cavity present in
the family of {W36}clusters presented here is very well defined
and shows no significant differences in structures 1–6. In addition,
Fig. 6 An overlay of the six structures of 1–6. The top view shows that
the framework of {W36}is basically unchanged as a function of the cation
complexed. The bottom view shows the maximum extent of the distortions
found for all the complexes characterized; the largest distortions out of
the plane upwards was found in compound 3(Cs-surface in yellow) and
downwards was found in compound 2(Rb-surface in yellow). The front
{W11}unit is omitted in the bottom side view for clarity.
other studies also confirmed that small ions cannot be complexed
by the cavity present in the {W36}cluster. This is because we
have attempted to synthesize the {W36}cluster in the presence
of Ca2+, lanthanide ions, and first row transition metal ions
without success. Although experiments with CaCl2also yielded
in colourless crystals, the presence of the Ca2+ ion in the cluster
cavity could not be confirmed and chemical analysis showed only
a low concentration of Ca2+ to be present in the system. In the
case of lanthanide ions, the electrophilic nature of these cations
appeared to complex rapidly with other tungstate-based species
yielding polymeric compound precipitates rapidly upon reaction.
Table 2 Comparison of acquired metal-oxygen distances and displacements of metal ions to the cavity centre in the {W36 }cluster and the corresponding
figures for the 18-crown-6 ether (esds are all within 0.02 A
˚)
Mn+Ionic radius/A
˚Av. dM–O
a/A
˚({W36}cluster) Av. dM–O/A
˚(18-crown-6) dM-c
b/A
˚({W36}cluster) dM-c /A
˚(18-crown-6)
K+1.38 2.80 2.80 0.70 0
Rb+1.52 2.87 2.95 0.84 0.93
Cs+1.67 3.16 3.18 1.61 1.47
Sr2+ 1.18 2.70 2.73 0.53 0
Ba2+ 1.35 2.82 2.82 0.73 0
aAv. dM–O is the average distance from Mn+to the six oxo ligands on the ring. bdM–c is the distance from Mn+to the cavity centre, which is defined as the
intersection of the main molecular axis and the equatorial plane defined by the six oxo ligands.
2856 |Dalton Trans., 2006, 2852–2860 This journal is ©The Royal Society of Chemistry 2006
pH and symmetry transferring effects on cluster formation
In our work we have been investigating the formation of new
POM cluster types using a number of control parameters, in
particular pH, and cation types, and have identified a very
strong dependence upon both parameters. Indeed, there have
been a number of reports that discuss the syntheses of a range
of isopolyoxotungstates where the structural types are precisely
controlled by the pH.40–46 However there is no system reported
previously concerning the uses of water-soluble organic cations to
help isolation of new isopolyoxotungstates.
By using TEAH+cations, we have also isolated a number of
heteroPOM Keggin ions and [H2W12O40]6−,aswellasthe{W36 }
(see in ref. 26) and a new isopolyoxotungstate [H4W19O62]6−.47
The pH effects on cluster formation in the presence of TEAH+
cations are summarized in Scheme 1. At pH around 4, the reaction
always gives rise to the production of Keggin anion [H2W12 O40]6−
as main product, however at a lower pH of ca. 2.0, the Keggin ion,
[H2W12O40 ]6−, can also be produced as a by-product as well as the
main [H12W36O120 ]12−product. Of relevance to the work reported
here, is that all the {W36}-based complexes, 1–6 ([H12 W36 O120]12−),
can only be produced at pH around 2.0 and the yields can be
increased if the solution is warmed to dissolve the intermediate
tungstate precipitates before the addition of the metal ions. By
going to even lower pH values, and by refluxing for longer time, the
system produces another new isopolyoxotungstate [H4W19O62]6−,47
which has a Dawson-like structure of three-fold symmetry but with
a{WO6}as central template rather than two hetero-tetrahedral
{XO4}templates.
Scheme 1 pH effects in the synthesis of new POM clusters with TEA.
One extremely interesting observation we have made in all the
work done utilizing the TEAH+cations, is that clusters with
three-fold symmetry are exclusively produced from the reaction
systems. These observations may be entirely circumstantial, but we
have isolated over four distinct structure types using the TEAH+
cations, and this has led us to propose that the TEAH+cation
is able to transfer its symmetry onto the clusters in solution,
see Scheme 2. Indeed, supramolecular interactions between the
TEAH+cation and the cluster building blocks via the three
hydroxyl groups on the TEAH+cation, can form H-bonds to inter-
mediate components of the final product and possibly even guide
their assembly to the overall cluster architecture. The first example
we discovered was in the synthesis of reduced molybdosulfite
cluster [Mo18O54(SO3)2]6−, which was produced by the reaction of
sodium molybdate with Na2S2O4reducing agent in the presence
of TEA at a pH of ca. 4.19 The cluster anion [Mo18 O54(SO3)2]6−has
a Dawson-like structure with main symmetry of a three-fold axis,
which is therefore postulated to be derived from the templating
influence of the three-fold symmetric TEAH+cation. Although
appealing, we treated this idea with a degree of scepticism but
Scheme 2 The proposed symmetry transfer process, from the
organo-cation to the cluster architecture via supramolecular interactions
in solution, which allow the isolation of the new cluster type {Mo18 }19 and
{W36}26 with the same symmetry as the cation.
this hypothesis is strengthened further by the observation that
similar reaction conditions (e.g., pH value, concentrations, ionic
strength, and temperature), but with different large cations, e.g.
protonated hexamethylenetetramine, results in the formation of
low symmetrical-type POM clusters of [H2Mo16O52]10−.17,18 In
the tungsten system, the observations of the cluster formation
in (TEAH)6[H2W12O40]atpH4,
26 and (TEAH)6[H4W19O62]47 at
pH 0.8 also verify the symmetry-transferring hypothesis as these
two clusters also demonstrate three-fold symmetry. Finally, the
{W36}clusters shown in this paper all comprise three {W11}cluster
subunits linked together by three {W1}bridges to form the overall
triangular shaped cluster. However, the three {W11 }subunits
are analogous to the framework geometry of the [H4W11O38 ]6−
cluster, which was isolated by Lehmann et al. in the 1980s
by using K+as counter cations at pH 1.3.45 This [H4W11O38 ]6−
cluster has a spherical shape with approximate mirror symmetry.
Therefore, the isolation of [H4W11O38]6−as K+salt suggests
that in solution, the {W11 }cluster is the precursor for the
{W36}clusters and the isolation of {W36}only become possible
when three-fold symmetric TEAH+cations are present; indeed
attempts to assemble the {W36}cluster in the presence of other
cations, but under the same reaction conditions, has not been
possible.
Conclusions
A range of new complexes of the triangular {W36}cluster,
[H12W36O120 ]12−that incorporate cations within the central cavity
complexed by six W=O pendant donor groups, have been
characterized. These complexes demonstrate the {W36}can act
like a type of inorganic “crown ether” with similar preferences to
18-crown-6, but with much greater rigidity, and therefore, potential
to distinguish between different cations. The synthetic routes to
the {W36}cluster, along with other clusters we have previously
characterized, using the TEAH+cation imply a possible route
to ‘imprinting’ or transferring symmetry from the organo-cation
to the cluster architecture. In future work we will attempt to
This journal is ©The Royal Society of Chemistry 2006 Dalton Trans., 2006, 2852–2860 | 2857

exploit the rigid nature of the {W36}and attempt to determine
selectivity for a range of metal ions, as well as attempt to build
new architectures by complexation of rigid organo-amines with
primary amino groups that can be bound in the cavity of the
{W36}cluster. We will also attempt to extend our observation of
apparent symmetry transfer between the organo-cations and the
cluster type to gain access to new polyoxometalates structure types
and symmetries.47
Experimental
General procedures
All reagents and chemicals were purchased from commercial
sources and used without further purification. Infrared spectra
were recorded as KBr disc using a Perkin-Elmer paragon 1000
PC or Nicolet Magna 550 series II FTIR spectrometer. Elemental
analyses were carried with dried samples. It should be noted that
there are differences between the analytical values determined and
theoretical values calculated from crystallographic formula. This
is because these compounds contain a large amount of solvent
of crystallization and drying occurs very quickly. However these
problems were minor and did not hinder the overall formula
determination for the main cluster {W36}and the guest metal
ions encapsulated.
Synthetic procedures
Synthesis of (TEAH)9Na2{(H2O)4K⊂[H12W36 O120 ]}·17H2O(1).
Na2WO4·2H2O (2.00 g, 6.06 mmol) and triethanolamine-
hydrochloride (2.5 g, 13.47 mmol) were dissolved in H2O(50ml)
in a plastic flask and the solution was adjusted to a pH of 2.2 with
2.0 ml HCl (4 M). After heating the reaction mixture to 80 ◦Cin
a water bath for 30 min, 0.030 g (0.40 mmol) KCl were added to
the hot solution and the mixture was cooled down without further
stirring. The sample was stored undisturbed for crystallization,
which yielded colourless needles of 1in 2–3 days. The crystals
were collected on a filter paper and dried. Yield 0.869 g (49%
based on W). IR (KBr disk): m/cm−1: 3447, 1622, 1449, 1401,
1320, 1256, 1205, 1093, 1065, 950, 895, 780, 654, 419; UV/VIS
(H2O): kmax =276 nm (br sh, 0.34); elemental analysis: calc. for
C54H198KN9Na2O168 W36: C 6.26, H 1.93, K 0.38, N 1.22, Na 0.44,
W 63.9% found: C 6.69, H 2.00, K 0.38, N 1.39, Na 0.45, W 61.7%.
Synthesis of (TEAH)9Na{(H2O)4Rb⊂[H12W36 O120 ]}·17H2O(2).
The same procedure as preparing 1, with RbCl (0.048 g, 0.40
mmol) replacing KCl, yielded colourless needles of 2in 2–3 days.
Yield 0.695 g (39% based on W). IR (KBr disk): m/cm−1: 3443,
1620, 1449, 1404, 1321, 1255, 1203, 1093, 1065, 950, 894, 778,
656, 421; UV/VIS (H2O): kmax =276 nm (br sh, 0.34); elemental
analysis: calc. for C54 H198N9Na2O168 RbW36 : C 6.23, H 1.92, N 1.21,
Na 0.44, Rb 0.82, W 63.6%; found: C 6.96 H 1.64 N 1.35, Na 0.44,
Rb 0.81, W 64.7%.
Synthesis of (TEAH)9Na2{(H2O)4Cs⊂[H12W36 O120 ]}·15H2O
(3). The same procedure as preparing 1, with CsCl (0.067 g, 0.40
mmol) replacing KCl, yielded colourless needles of 3in 3–4 days.
Yield 0.689 g (39% based on W). IR (KBr disk): m/cm−1: 3450,
1620, 1449, 1404, 1320, 1255, 1203, 1093, 1065, 950, 894, 781,
652, 431; UV/VIS (H2O): kmax =276 nm (br sh, 0.34); elemental
analysis: calc. for C54 H194CsN9Na2O166 W36 : C 6.22, H 1.88, N 1.21,
Na 0.44, Cs 1.28, W 63.5; found: C 6.93 H 1.58 N 1.34, Na 0.55,
Cs 0.81, W 64.1%
Synthesis of (TEAH)9Na2{(NH4)⊂[H12W36O120 ]}·19H2O(4).
Na2WO4·2H2O (2.00 g, 6.06 mmol) and triethanolamine hy-
drochloride (2.5 g, 13.47 mmol) were dissolved in H2O(50ml)
in a plastic flask and the solution was adjusted to a pH of 1.8 with
2.5 ml HCl (4 M). After heating the reaction mixture to 80 ◦Cin
a water bath for 30 min, 0.027 g (0.50 mmol) NH4Cl dissolved in
5mlH
2O were dropped to the stirred hot solution and the mixture
was cooled down without further stirring. The sample was stored
undisturbed for crystallization, which yielded colourless needles of
4in 4–5 days. The crystals were washed with cold water, collected
by filtration and dried. Yield was 0.662 g (38% based on W).
IR (KBr disk): m/cm−1: 3442, 1623, 1485, 1456, 1404, 1321, 1255,
1197, 1093, 950, 897, 781, 666, 414; UV/VIS (H2O): kmax =276 nm
(br sh, 0.34); elemental analysis: calc. for C54 H198N10 Na2O166 W36:
C 6.29, H 1.94, N 1.36, Na 0.45, W 64.2; found: C 6.92 H 1.66 N
1.37, Na 0.46, W 66.5%.
Synthesis of (TEA)(TEAH)8Na2{(H2O)4Sr⊂[H12W36 O120 ]}·
17H2O(5). The same procedure as preparing 1,withSrCl
2·6H2O
(0.107 g, 0.40 mmol) replacing KCl, yielded colourless needles of
6in 2–3 days. Yield 0.93 g (53% based on W). IR (KBr disk):
m/cm−1: 3433, 1620, 1449, 1398, 1320, 1257, 1204, 1093, 1066, 953,
898, 779, 660, 416; UV/VIS (H2O): kmax =276 nm (br sh, 0.34);
elemental analysis: calc. for C54 H197N9Na2O168SrW36 : C 6.23, H
1.91, N 1.21, Na 0.44, Sr 0.84, W 63.6; found: C 6.76 H 1.58 N
1.29, Na 0.48, Sr 0.81, W 64.0%.
Synthesis of (TEA)(TEAH)8Na{(H2O)4Ba1.5⊂[H12 W36 O120]}·
17H2O(6). Na2WO4·2H2O (1.00 g, 3.03 mmol) and tri-
ethanolamine hydrochloride (1.25 g, 6.73 mmol) were dissolved
in H2O (35 ml) and the solution was adjusted to pH 1.3 with
ca. 3 ml HCl (4 M). After heating the reaction mixture to 80 ◦C
in a water bath for 30 min, BaCl2·2H2O (0.025 g, 0.10 mmol)
dissolved in H2O (5 ml) were added to the hot solution. The
pH was then immediately adjusted to 2.2 with a few drops of
aqueous triethanolamine solution (3M) and the mixture was
cooled down without further stirring. After a little amount of white
precipitate was filtered off, the sample was stored undisturbed for
crystallization, which yielded colourless needles of 7in 2–3 days.
The crystals were washed with cold water, collected by filtration
and dried. Yield was 0.422 g (48% based on W). IR (KBr disk):
m/cm−1: 3434, 1618, 1450, 1386, 1320, 1257, 1203, 1093, 1067, 956,
895, 778, 647, 436; UV/VIS (H2O): kmax =276 nm (br sh, 0.34);
elemental analysis: calc. for C54 H198Ba1.5 N9O168 Na1W36: C 6.17, H
1.89, N 1.20, Na 0.22, Ba 1.96; W 62.8 found: C 6.29 H 1.74 N
1.20, Na 0.45, Ba 2.1, W 60.1%.
NMR measurements
Compounds 1–6 show basically the same signals in 1HNMR
spectra. These comprise two triplets for the methylene protons of
the surrounding TEAH+cations at 3.4–3.6 and 3.8–4.0 ppm, small
singlets between 5.2–5.6 ppm, which belong to the six protons
forming hydrogen bonds inside the W11 moieties and various small
signals in the H2O area, which can not be clearly assigned to the
different types of water molecules present in the cluster framework.
2858 |Dalton Trans., 2006, 2852–2860 This journal is ©The Royal Society of Chemistry 2006

Table 3 Crystallographic data collection, intensity measurements and structure refinement parameters for 2–6
23456
Chemical formula C54H198N9Na2O168 RbW36 C54H198 CsN9Na2O166 W36 C54H198 N10 Na2O166W36 C54 H197 N9Na2O168SrW36 C54 H197 Ba1.5N9NaO168 W36
M/g mol−110412.26 10423.67 10308.8 10413.41 10508.81
Symmetry Orthorhombic Orthorhombic Orthorhombic Orthorhombic Orthorhombic
Space group Pnma Pnma Pnma Pnma Pnma
a/A
˚27.3088(3) 26.8369(10) 27.0818(4) 27.5918(3) 27.3739(4)
b/A
˚35.1380(4) 34.8568(13) 35.1386(4) 34.9442(3) 35.1269(6)
c/A
˚21.2632(3) 21.0337(8) 21.2120(3) 20.7727(2) 20.9520(4)
V/A
˚320403.7(4) 19675.9(13) 20185.7(5) 20028.5(3) 20146.6(6)
Z44444
Dc/g cm−33.39 3.52 3.39 3.45 3.47
l/mm−120.55 21.25 20.53 20.96 20.86
F(000) 18608 18600 18424 18608 18748
Crystal size/mm 0.30 ×0.14 ×0.14 0.30 ×0.12 ×0.10 0.30 ×0.09 ×0.08 0.25 ×0.22 ×0.16 0.30 ×0.06 ×0.06
No. data measured 111232 79977 92755 96316 84809
No. unique data 15357 15583 15979 15960 17021
No. observed data 10618 11664 9561 11392 11213
No. variables 1121 1112 1170 1182 1097
R1 0.0460 0.0386 0.0474 0.0370 0.0652
R2 (all data) 0.1146 0.1013 0.1001 0.0838 0.1780
Goodness of fit, S1.039 1.062 1.009 1.039 1.056
Maximum shift/error 0.002 0.002 0.003 0.002 0.005
The exact chemical shift values for the methylene groups of the
TEAH+cations depend on the included metal ion and differ
slightly.
Crystallographic structure determinations
Details of data collection procedures and structure refinements
aregiveninTable3.Singlecrystalsofsuitablesizewereattached
to glass fibres using Fomblin YR-1800 oil, and mounted. Some
samples suffered solvent loss, and were glued to the glass fibre
under solvent and transferred as rapidly as possible to the
cold stream of the Oxford Instruments Cryostream. All data
were collected on a Nonius KappaCCD or Bruker Apex II
CCD diffractometer, equipped with graphite monochromated X-
radiation (k=0.71073 A
˚), running under the Collect software.
The structures were solved by SHELXS-97.48,49 Most of the non-
hydrogen atoms were refined anisotropically. In some structures
the hydrogen atoms on the TEAH+cations were refined using a
riding model using the SHELXL HFIX instructions. In all other
cases hydrogen atoms on the TEAH+cations were not present
due to disorders on the TEAH+cations themselves. No attempts
were made to add hydrogen atoms to water molecules as they
are quite disordered and have partial occupancy in the structures.
Refinement was with SHELXL-97 using full-matrix least squares
on F2and all the unique data. All samples showed the presence
of disordered solvent molecules. All calculations were carried out
using the WinGX package50 of crystallographic programs.
CCDC reference numbers 289116–289120.
For crystallographic data in CIF or other electronic format see
DOI: 10.1039/b515935k
Acknowledgements
This work was supported by the Leverhulme Trust (London), The
Royal Society, The University of Glasgow and the EPSRC. The
EPSRC provided funds for the X-ray diffractometer. We would
like to acknowledge Prof. A. Slavin (the University of St Andrews)
for help with the preliminary analysis of compounds 5and 6.
References
1 L. Cronin, High Nuclearity Clusters: Iso and Heteropolyoxoanions
and Relatives, in Comprehensive Coordination Chemistry II,vol.7,ed.
J. A. McCleverty and T. B. Meyer, Elsevier, Amsterdam, 2004, pp. 1–56;
M. T. Pope and A. M ¨
uller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34;
C. L. Hill, Chem. Rev., 1998, 98,1.
2 K. Wassermann, M. H. Dickman and M. T. Pope, Angew. Chem., Int.
Ed. Engl., 1997, 36, 1445.
3 L. Cronin, C. Beugholt, E. Krickemeyer, M. Schmidtmann, H. B ¨
ogge,
P. K ¨
ogerler, T. K. K. Luong and A. M¨
uller, Angew. Chem., Int. Ed.,
2002, 41, 2805.
4A.M
¨
uller, E. Beckmann, H. B¨
ogge, M. Schmidtmann and A. Dress,
Angew. Chem., Int. Ed., 2002, 41, 1162.
5 I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171.
6 J. T. Rhule, W. A. Neiwert, K. I. Hardcastle, B. T. Do and C. L. Hill,
J. Am. Chem. Soc., 2001, 123, 12101.
7A. M
¨
uller, M. Luban, C. Schr¨
oder, R. Modler, P. K ¨
ogerler, M.
Axenovich, J. Schnack, P. Canfield, S. Bud’ko and N. Harrison,
ChemPhysChem, 2001, 2, 517.
8A.M
¨
uller, P. K ¨
ogerler and A. W. M. Dress, Coord. Chem. Rev., 2001,
222, 193.
9D.M.Way,A.M.BondandA.G.Wedd,Inorg. Chem., 1997, 36,
2826.
10 P. J. S. Richardt, J. M. White, P. A. Tregloan, A. M. Bond and A. G.
Wedd, Can. J. Chem., 2001, 79, 613.
11 K. F. Aguey-Zinsou, P. V. Bernhardt, U. Kappler and A. G. McEwan,
J. Am. Chem. Soc., 2003, 125, 530.
12 D.A.Judd,J.H.Nettles,N.Nevins,J.P.Snyder,D.C.Liotta,J.Tang,
J. Ermolieff, R. F. Schinazi and C. L. Hill, J. Am. Chem. Soc., 2001,
123, 886.
13 H. D. Zeng, G. R. Newkome and C. L. Hill, Angew. Chem., Int. Ed.,
2000, 39, 1772.
14 F. Ogliaro, S. P. de Visser, S. Cohen, P. K. Sharma and S. Shaik, J. Am.
Chem. Soc., 2002, 124, 2806.
15D.-L.Long,H.Abbas,P.K
¨
ogerler and L. Cronin, Angew. Chem., Int.
Ed., 2005, 44, 3415.
16 P. K¨
ogerler P and L. Cronin, Angew. Chem., Int. Ed., 2005, 44,
844.
17 D.-L.Long,P.K
¨
ogerler, L. J. Farrugia and L. Cronin, Angew. Chem.,
Int. Ed., 2003, 42, 4180.
18 D.-L. Long, P. Kogerler, L. J. Farrugia and L. Cronin, Dalton Trans.,
2005, 1372.
19D.-L.Long,P.K
¨
ogerler and L. Cronin, Angew. Chem., Int. Ed., 2004,
43, 1817.
20 D.-L.Long,P.K¨
ogerler, D. Orr, G. Seeber, L. J. Farrugia and L. Cronin,
J. Cluster Sci., 2003, 14, 313.
21 H. Abbas, A. L. Pickering, D.-L. Long, P. K ¨
ogerler and L. Cronin,
Chem.Eur.J., 2005, 11, 1071.
This journal is ©The Royal Society of Chemistry 2006 Dalton Trans., 2006, 2852–2860 | 2859

22 D. Hagrman, P. J. Hagrman and J. Zubieta, Angew. Chem., Int. Ed.,
1999, 38, 3165; S. S. Kuduva, N. Avarvari and M. Fourmigue, J. Chem.
Soc., Dalton Trans., 2002, 3686.
23 P. Mialane, A. Dolbecq, L. Lisnard, A. Mallard, J. Marrot and F.
Secheresse, Angew. Chem., Int. Ed., 2002, 41, 2398.
24 T. Hori, O. Tamada and S. Himeno, J. Chem. Soc., Dalton Trans., 1989,
1491; S. Juraja, T. Vu, P. J. S. Richardt, A. M. Bond, T. J. Cardwell,
J. D. Cashion, G. D. Fallon, G. Lazarev, B. Moubaraki, K. S. Murray
and A. G. Wedd, Inorg. Chem., 2002, 41, 1072.
25 M. T. Pope, in Comprehensive Coordination Chemistry, ed. G. Wilkin-
son, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, 1987,
vol. 3.
26D.-L.Long,H.Abbas,P.K
¨
ogerler and L. Cronin, J. Am. Chem. Soc.,
2004, 126, 13880.
27 C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017.
28 C. J. Pedersen, Fed. Proc., 1965, 27, 1305.
29 C. L. Liotta and H. P. Harris, J. Am. Chem. Soc., 1974, 97, 224.
30 A. P. d. Silva, H. Q. Gunaratne, T. Gunnlaugsson, A. J. Huxley,
C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97,
1515.
31 M. H. Hyun and S. C. Han, J. Biochem. Biophys. Methods, 2002, 54,
235.
32 M. S. Lah and V. L. Pecoraro, J. Am. Chem. Soc., 1989, 111, 7258.
33 A. D. Cutland-Van Noord, J. W. Kampf and V. L. Pecoraro, Angew.
Chem., Int. Ed., 2002, 41, 4667.
34 A. J. Stemmler, J. W. Kampf, M. L. Kirk, B. H. Atasi and V. L. Pecoraro,
Inorg. Chem., 1999, 38, 2807.
35 U. Kortz, M. G. Savelieff, B. S. Bassil and M. H. Dickman, Angew.
Chem., Int. Ed., 2002, 41, 3384.
36 A. M¨
ulle r, B. Bot ar, H. B ¨
ogge, P. K¨
ogerler and A. Berkle, Chem.
Commun., 2002, 2944.
37 A. M ¨
uller, D. Rehder, E. T. K. Haupt, A. Merca, H. B ¨
ogge, M.
Schmidtmann and G. Heinze-Br¨
uckner, Angew. Chem., Int. Ed., 2004,
43, 4466.
38 T. J. R. Weakley, J. Chem. Soc., Chem. Commun., 1984, 1406.
39 U. Kortz and S. Matta, Inorg. Chem., 2001, 40, 815.
40 J. Fuchs, R. Palm and H. Hartl, Angew. Chem., Int. Ed. Engl., 1996, 35,
2651.
41 R. Bhattacharyya, S. Biswas, J. Armstrong and E. M. Holt, Inorg.
Chem., 1989, 28, 4297.
42 H. Hartl, R. Palm and J. Fuchs, Angew. Chem., Int. Ed. Engl., 1993, 32,
1492.
43 J. Fuchs and E. P. Flindt, Z. Naturforsch., Teil B, 1979, 34, 412.
44 Y. Sasaki, T. Yamase, Y. Ohashi and Y. Sasada, Bull. Chem. Soc. Jpn.,
1987, 60, 4285.
45 T. Lehmann and J. Z. Fuchs, Z. Naturforsch., TeilB, 1988, 43, 89.
46 I. Br¨
udgam, J. Fuchs, H. Hartl and R. Palm, Angew. Chem., Int. Ed.,
1998, 37, 2668, and references therein.
47 D. L. Long, P. K¨
ogerler, A. D. C. Parenty, J. Fielden and L. Cronin,
Angew. Chem., Int. Ed., 2006, submitted.
48 G. M. Sheldrick, Acta Crystallogr., Sect. A, 1998, 46, 467.
49 G. M. Sheldrick, SHELXL-97. Program for Crystal Structure Analysis,
University of G¨
ottingen, Germany, 1997.
50 L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837.
2860 |Dalton Trans., 2006, 2852–2860 This journal is ©The Royal Society of Chemistry 2006
