Content 532010.p65 TAN 14 NF P 19022010224341 PJSIR 53 2010 Enc
User Manual: TAN 14 NF P
Open the PDF directly: View PDF ![]() .
.
Page Count: 59

Pakistan Journal of Scientific and Industrial Research
Vol. 53, No. 1 Contents January - February 2010
Physical Sciences
Effect of Thermal Shocking and Quenching on the Degradation Behaviour of a
Thin PZT Disc
Riffat Asim Pasha and Muhammad Zubair Khan 1
Comparison of Ion Chromatography with Ion Selective Electrodes for the
Determination of Inorganic Anions in Drinking Water Samples
Muhammad Hakim, Farhat Waqar, Saida Jan, Bashir Mohammad, Wasim Yawar and
Shah Alam Khan 6
Physical and Chemical Evaluation of Oils of Two Varieties of Carthamus tinctorius
Grown in Pakistan
Razia Sultana, Rubina Saleem and Ambrat 14
Analysis of Caffeine and Heavy Metal Contents in Branded and Unbranded Tea
Available in Pakistan
Asma Inayat, Shahid Rehman Khan, Muhammad Nawaz Chowdhry and Amran Waheed 20
Measurement of Atmospheric Concentrations of CO, SO2, NO and NOx in Urban Areas
of Karachi City, Pakistan
Durdana Rais Hashmi, Farooq Ahmad Khan, Akhtar Shareef, Farhan Aziz Abbasi,
Ghulam Hussain Sheikh and Alia Bano Munshi 25
Seasonal and Year Wise Variations of Water Quality Parameters in the
Dhanmondi Lake, Dhaka, Bangladesh
Shamshad Begum Qureshi 30
Biological Sciences
Salt Tolerance Evaluation of Rice (Oryza sativa L.) Genotypes Based on Physiological
Characters Contributing to Salinity Resistance
Jalal-ud-Din, Samiullah Khan and Ali Raza Gurmani 37
Parasitic Contamination in the Table Vegetables Planted in Shiraz Plain, Iran
Meraj Madadi 42
Microbiological Quality of Drinking Water and Beverages in Karachi, Pakistan
Anila Siddiqui, Korish Hasnain Sahir and Seema Ismat Khan 46
Short Communication
Feeding Inter-Relationship of Caranx hippos (Linneaus), Chrysichthys nigrodigitatus
(Lacepede), Ethmalosa fimbriata (Bowdich) and Mugil cephalus (Linneaus) in
Lagos Lagoon, Nigeria
Adebiyi Adenike Fatimat 50

Effect of Thermal Shocking and Quenching on the
Degradation Behaviour of a Thin PZT Disc
Riffat Asim Pasha* and Muhammad Zubair Khan
Department of Mechanical and Aeronautical Engineering, University of Engineering and Technology, Taxila, Pakistan
(received October 22, 2008; revised November 16, 2009; accepted November 21, 2009)
Introduction
Piezoelectric materials are used in various electromechanical
applications where they are influenced by various cyclic
loadings. Thermal cycling or thermal fatigue in most electro-
nics materials may cause degradation in their internal charac-
teristics. Thermal fatigue test methods include quench
method and repeated heating method for thermal shocks
which have been earlier discussed (Lamon and Pherson, 1991;
Lamon, 1981). Influence of temperature on the electromecha-
nical and fatigue behaviour of piezoelectric ceramics has
been studied by Wang et al. (1998). Temperature gradient is
developed due to sudden change in temperature in the
ceramic materials and therefore, thermal stress is generated.
Effect of thermal shocks has been studied by developing
newly designed equipment. There are various popular thermal
shock methods available in ascending and descending orders.
Some of them popular for ascending thermal shocks, include
hot jet gas method, high power radiation, melt immersion test,
ribbon test method and high power laser heating method.
Similarly, various test methods for descending thermal shocks
are quenching in water, fluidized bed or a cold air jet impinging
on hot discs; quenching in contact with huge brass rods and
indentation method have been mentioned by Panda et al.
(2002). Earlier, thermal shocks in a plate of finite thickness had
been attempted. Thermal shock and thermal fatigue of ferro-
electric thin films were investigated by Zheng et al. (2005). In
all of the above methods, the water quenching method is
mostly used for thermal shock tests in which samples are
heated to a particular temperature and then quenched in
water bath. Fatigue studies show that material degradation
of PZT ceramics are strongly influenced by temperature.
Lead zirconate titanate ceramics show decrease in the dielec-
tric constant and the resonance frequency when subjected
to thermal shocks. Importance of temperature stability for
dielectric constants and resonance frequencies have been
discussed by Lee and Kim (2005). Earlier thermal shock resis-
tance of the materials was evaluated by water quenching.
Degradation of various properties of the piezoelectric devices
in the presence of water and AC voltage was investigated by
Xiang et al. (2007). They concluded that water is an important
cause of degradation of piezoelectric (PZT) ceramics.
However, limited work has been published on the effect of
thermal shocking, quenching and on the degradation beha-
viour of thin piezoelectric ceramic discs. In this study, the
degradation phenomenon of thin PZT ceramic disc have
been investigated when exposed to repeated heating and
quenching cycles below its curie temperature.
Materials and Methods
Lead zirconate titanate piezoelectric discs, nickel electroded
on major faces, 0.191 mm thick and 12.7 mm in diameter, were
used for the experimentation. The thin piezoelectric ceramic
discs were heated at the heating rate of 9 °C/sec up to 100 °C,
and 150 °C, using a thermal chamber and then quenched in
deionized water at a temperature of about 20 °C. For all
thermal cycling and quenching experiments, 2 PZT test
samples were used and subjected to identical conditions.
The temperature of the PZT samples was recorded using a
spring loaded thermocouple and data acquisition system
attached directly to the samples. In order to observe degrada-
tion phenomenon of the PZT ceramic, the capacitance, dissi-
pation factor and impedance were measured at a frequency of
1 kHz at the start and after every five heating and quenching
Pak. J. Sci. Ind. Res. 2010 53 (1) 1-5
Abstract. Thin lead zirconate titanate discs were subjected to thirty five thermal shocks from two different temperatures
in deionized water and their relative dielectric constant, coupling factor and impedance values were measured with a view
to investigating the behaviour of thin piezoelectric (PZT) discs at frequency of maximum and minimum impedance.
Noticeable differences were observed in the electrical properties of the material, probably due to the change in dipole
lengths and their orientations during thermal shocking. The results can be useful in modeling and designing of smart
components for predicting their behaviour during such expected shocking conditions prior to fabrication.
Keywords: piezoelectric material, thermal shock, deionized water, dielectric constant, impedance, PZT
*Author for correspondence; E-mail: asimpasha@uettaxila.edu.pk
Physical Sciences
1

cycles. Data was collected for a total of thirty-five thermal
shocks and their relative frequencies of maximum and
minimum impedance were observed between 100 kHz and
200 kHz. The capacitance and impedance at these frequencies
were recorded using impedance analyzer and dielectric test
fixture (model 1645 B). The fixture was attached to an LCR
meter and impedance analyzer 4294 A which uses a 4 pair
terminal measurement configuration. The values of capa-
citance measured with impedance analyzer were used to
calculate dielectric constant (K3
T and effective coupling
factor (Keff) by using the following equations from IEEE
Standard 177 (IEEE Standard, 1976) and Moulson and
Herbert (2005):
ta × Cp
K3
T = ————
A ×
∈
0
Effective and transverse coupling factors (Keff) were deter-
mined by using the following relationships:
Keff = SQRT (fn2 - fm2) / fn2
K31 = SQRT (Ψ/(1+Ψ),
where Ψ = π/2 (fn/fm) × tan | π/2 × (fn - fm) / fm |
Abbreviations used are as follows:
fm = frequency of maximum impedance [Hz]
fn = frequency of minimum impedance [Hz]
Cp = equivalent parallel capacitance [F]
ta = average thickness of testing material [m]
A = area of guarded electrode [m2]
K3
T = dielectric constant
Keff = effective coupling factor
K31 = coupling factor with transverse excitation
∈0 = permittivity at free space (8.854 × 10-12)
Ψ = phase angle
Results and Discussion
The changes in dielectric constant and coupling factor were
measured as a function of frequency of maximum and mini-
mum impedance (fm and fn, respectively). Increase in the value
of the capacitance of the as-received PZT ceramic was
observed to be 5.8 × 104 pF which gradually decreased with
increasing thermal cycling (100 °C - 20 °C) to 1.72 × 104 pF. A
corresponding change in the fm was observed with a value of
160 kHz for the PZT sample at the start and then the value
decreased to 116.5 kHz. This represented a 28% decrease in
the fm after the ceramic was thermal cycled. A similar change
was observed for the fn which decreased from 165.5 kHz for
the as-received to 153.3 kHz after 35 thermal cycles. For the
thermal cycling (150 °C - 20 °C) change in fm was observed
from 160.2 kHz to 141 kHz and from 165.175 kHz to 157.5 kHz
in fn. Change in dielectric constant and coupling factor for
thirty five shocks in deionized water has been tabulated in
Table 1.
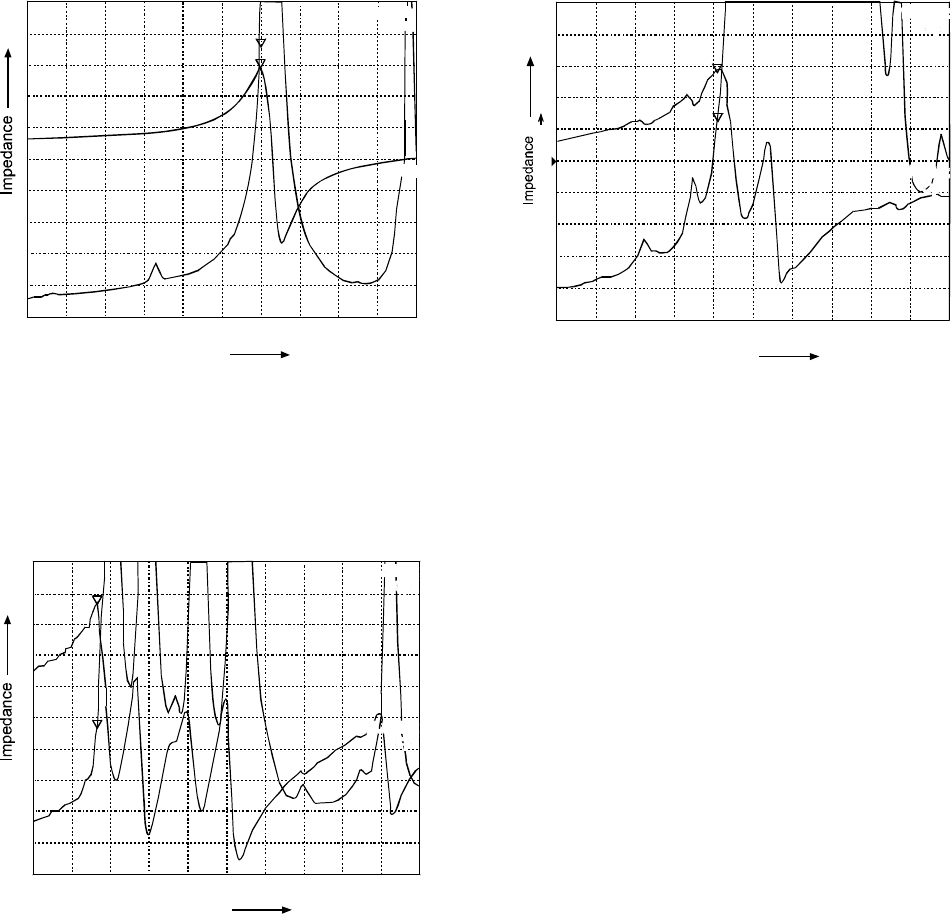
Figure 1 indicates the value of capacitance directly measured
by impedance analyzer for the unshocked discs at frequency
of maximum impedance. Discs were shocked in deionized
water from 100 °C to 20 °C for thirty five shocks when their
capacitance value decreased from 58.041 nF to 17.237 nF
(Fig. 1 and 2). Interestingly, PZT discs shocked from 150 °C
to 20 °C showed a less decrease in capacitance value after
having thirty five shocks. In this case capacitance value at
frequency of maximum impedance decreased to 24.189 nF
(Fig. 3).
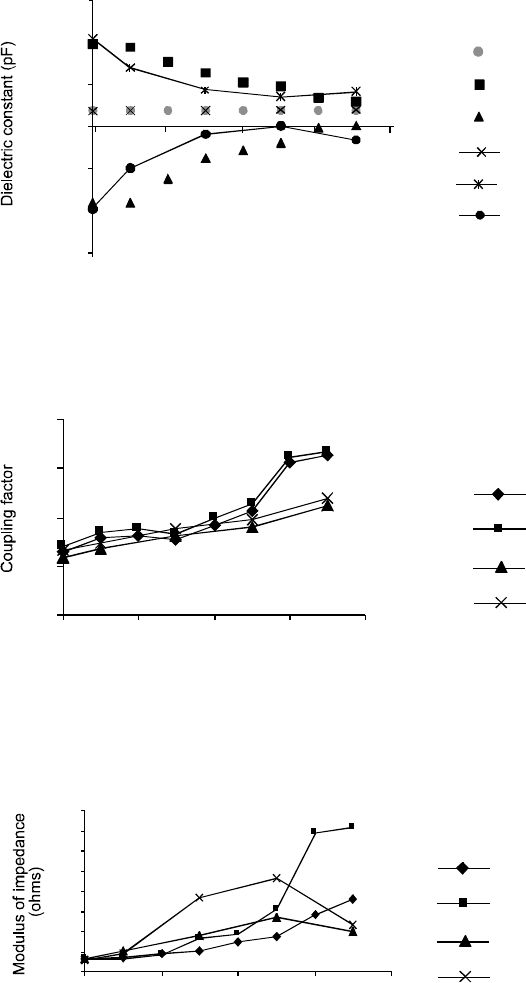
A comparison of the graphical output for dielectric constant
for the PZT samples before thermal cycling and then after
exposing the ceramic to thirty five heating and quenching
shocks is shown in Fig. 4. Dielectric constant remains inde-
pendent when measured at 1kHz. The dielectric constant is an
Table 1. Change in dielectric constant and coupling factor for two different thermal shocking conditions
Shocking from 100 °C to 20 °C Shocking from 150 °C to 20 °C
K3
TK3
TK3
TK3
T
Shocks # at 1kHz at fmKeff K31 at 1kHz at fmKeff K31
0 1853 9888 0.255 0.279 1869 10462 0.23 0.26
5 1891 9481 0.313 0.34 1915 8532 0.27 0.29
10 1911 7726 0.324 0.352 1923 7005 0.3 0.32
15 1930 6392 0.306 0.333 1932 4366 0.32 0.35
20 1918 5291 0.364 0.393 1954 3926 0.34 0.37
25 1922 4790 0.427 0.457 1976 3504 0.36 0.38
30 1928 3450 0.621 0.644 1976 3912 0.41 0.41
35 1928 2935 0.651 0.671 1976 4121 0.44 0.47
2Riffat Asim Pasha and Muhammad Zubair Khan
intrinsic property of the ceramic material and the results
show that this value decreased with increasing thermal
cycles at fm and vice versa. The relative difference in the
frequencies of the maximum and minimum impedance values
depends on the material coupling factor and the resonator
geometry (i.e., dimensions of the ceramic PZT sample). For
this reason, quantities known as the effective coupling
factor (Keff) and the transverse excitation factor (K31) were
calculated and compared as a function of the number of
thermal shocks (Fig. 5). It was found that both the values of
K31 and Keff increased with increasing thermal cycles to
which the PZT ceramic was exposed.
The change in modulus of impedance for two different
shocking conditions were evaluated (Fig. 6). It can be seen
that the modulus of impedance for both fm and fn, when
shocked from 100 °C to 20 °C, increased whereas for the other
conditions, it started decreasing after twenty five shocks.
Another interesting result is that when impedance at fm and
fn increased, the difference became larger at later shocks.
Decrease in dielectric constant in thermal shocking is the
expected normal behaviour. Various coupling factors were
close to each other but a noticeable change was observed
due to shocking and quenching effect. All changes may have
occurred due to change in dipole moments and their expected
random orientations. This reorientation may change the
length of dipoles, due to which specimen undergoes a change
in its piezoelectric properties.
The change in fm and fn causes the change of mechanical
quality factor. This response of the material can be utilized in
designing of oscillators. It is observed that the difference in
these two stated frequencies (fm and fn) is small as compared
to their impedance peaks during thermal shocking. The
3
Fig. 1. Value of capacitance for un-shocked disc w.r.t.
frequency (100 kHz-200 kHz); capacitance in nF at
frequency of maximum impedance.
A: Cp
B: D SCALE
SCALE 20
100 nF/div REF
mU/div REF 0 F
500 mU 58.0411 nF
854.265 mU
160 kHz
Cp l
START 100 kHz OSC 500 mVolt STOP 200 kHz
0
0
Frequency
160 kHz
Cp l
Thermal Shocking and Quenching Effect on PZT Disc
Fig. 2. Value of capacitance after thirty five shocks w.r.t.
frequency (100 kHz-200 kHz); capacitance in nF at
frequency of maximum impedance when shocked
from 100 °C - 20 °C.
0
0
START 100 kHz OSC 500 mVolt STOP 200 kHz
Frequency
A: Cp
B: D SCALE
SCALE 2
100 nF/div REF
mU/div REF 10 F
500 mU 17.2376 nF
472.24 mU
Cp l
116.5 kHz
Cp l
116.5 kHz
Fig. 3. Value of capacitance after thirty five shocked w.r.t.
frequency (100 kHz-200 kHz); capacitance in nF at
frequency of maximum impedance when shocked
from 150 °C - 20 °C.
0
24.1897 nF
624.817 mU
START 100 kHz OSC 500 mVolt STOP 200 kHz
Frequency
A: Cp
B: D SCALE
SCALE 5
100 nF/div REF
mU/div REF 10 nF
500 mU
EX1 Cp l
141 kHz
Cp l
141 kHz
0
results suggest that the PZT ceramics suffer a noticeable
change in polarization when exposed to repeated heating
and quenching cycles, well below the curie temperature
(350 °C) for the PZT ceramic. It is thought that significant
depolarization of the PZT ceramic occurs due to the disorien-
tation of the ferroelectric domains and this reorientation is
affecting the critical piezoelectric properties by thermal
shocking and quenching. The behaviour is normal but the
number of peaks increases due to expected change in length
and reorientation of dipoles. Development of dipole moments
4Riffat Asim Pasha and Muhammad Zubair Khan
Fig. 5. Change in coupling factor (K31, Keff) against number of shocks from 100 °C - 20 °C and from 150 °C - 20 °C in deionized
water.
0.2
0.4
0.6
0.8
Number of shocks
Shocked from
K
eff
,
K
31
,
K
eff
,
K
31
,
C
O
C
O
C
O
C
O
100 C 20
O
-
100 C 20
O
-
150 C 20
O
-
150 C 20
O
-
00 10203040
Fig. 4. Change in dielectric constant against number of shocks, at frequency 1kHz and frequencies of maximum and minimimum
impedance.
Fig. 6. Change in modulus of impedance (|Z|) against number of shocks from 100 °C - 20 °C and from 150 °C - 20 °C in deionized
water.
0
20
40
60
80
100
120
140
160
0 10203040
Number of c
y
cles
Shocked from
at f
m
100 C 20
O
-
at f
m
150 C 20
O
-
at f
n
100 C 20
O
-
at f
n
150 C 20
O
-
C
O
C
O
C
O
C
O
010 20 30 40
Number of shocks
-15000
-10000
-5000
0
5000
10000
15000 Shocked from
C at 1kHz
C at f
m
C at f
n
C at f
m
C at f
n
O
O
O
O
O
100 C 20
O
-
100 C 20
O
-
100 C 20
O
-
150 C 20
O
-
150 C 20
O
-
C at 1kHz
O
150 C 20
O
-

Comparison of Ion Chromatography with Ion Selective Electrodes for
the Determination of Inorganic Anions in Drinking Water Samples
Muhammad Hakima*, Farhat Waqara, Saida Jana, Bashir Mohammada, Wasim Yawara
and Shah Alam Khanb
aCentral Analytical Facility Division, PINSTECH, P. O. Nilore, Islamabad, Pakistan
bPCSIR Laboratories Complex, Jamrud Road, Peshawar - 25120, Pakistan
(received July 8, 2009; revised December 15, 2009; accepted December 19, 2009)
Introduction
Due to increase in population, urbanization and continued
industrial growth, per capita water availability in Pakistan
has decreased from 5000 m3/annum in 1951 to 1100 m3/annum
in 2007 (WWF, 2007). The increasing gap between water
demand and supply has led to severe water shortage in
almost all sectors and has adversely affected the quality of
drinking water; consequently, water pollution has become
a serious problem in the country and most of the reported
health problems are directly or indirectly related to water
(PCRWR, 2008).
There are various sources of contaminants in drinking water
which, when exceeding certain levels, are harmful to man.
These contaminants are microorganisms, inorganic and
organic chemicals and certain radioisotopes. Inorganic
anions may affect the quality of water. Fluoride, chloride
and nitrate have considerable importance in the quality of
drinking water. Specially, the excess of nitrate and fluoride
in drinking water has intense effects on human health
(Meenakshi and Maheshwari, 2006; Fraser and Chilvers, 1981).
Excess nitrate in drinking water could cause serious illness
in infants below the age of six months. Fluoride might be the
reason for different bone diseases and tenderness of bones
in children (US EPA, 2009).
Fluoride, chloride and nitrate in groundwater and surface
water originate from natural sources, sewage, industrial
effluents, different food additives and as a result of leaching
or runoff from agricultural land (WHO, 2004).
Various analytical methods have been proposed for the deter-
mination of fluoride in aqueous solutions, such as colorimet-
ric, conductometric, complexometric and potentiometric
methods (APHA, 1985). Some methods are rapid, sensitive,
precise and relatively free of interferences. Traditional methods
used for determination of fluoride, chloride and nitrate anions
are based on colorimetric method, which due to interference
by various ions, require special treatment of the samples like
distillation or reduction and special analytical skills. Ion
chromatography is becoming more popular for the analysis
of water samples and is also recognized by the US Environ-
mental Protection Agency (US EPA, 2009) as a method of choice
for the determination of anions in water samples (Bosch et al.,
1995; Cheam, 1992; Pereira, 1992; Frankenberger et al., 1990).
Potentiometry has been widely used for quite some time due
to its simplicity and prompt results. However, the selectivity
is rather limited, especially if chemically similar ions are
present in the sample. Recent developments in separation
techniques have led to an improvement especially in the
determination of fluoride in terms of selectivity and sensi-
tivity (Weiss et al., 1995; Vasconcelos et al., 1994). Results of
determination of bromide, chloride, fluoride, nitrate and
sulphate using ion chromatography (IC) had been compared
with those obtained by colorimetry for rainfall, cloud water
and stream waters. According to that, there was no significant
difference in chloride and nitrate measurements between the
Pak. J. Sci. Ind. Res. 2010 53 (1) 6-13
Abstract. Fluoride, chloride and nitrate anions were determined in drinking water samples using techniques of ion selective
electrodes (ISE) and non-suppressed/suppressed ion chromatography (IC). Detection limit, percentage recovery and run
time were evaluated for the two methods. Detection limits for ISE [0.02, 0.20 and 1.7 ppm (μg/mL) for fluoride, chloride
and nitrate, respectively], were better than those for non suppressed IC (2.0, 1.0 and 2.0 ppm for fluoride, chloride and
nitrate, respectively). Suppressed IC was used to measure fluoride. Statistical analysis of the data revealed no evidence of
systematic difference between ISE and non suppressed IC for chloride and nitrate. Fluoride concentrations in all water
samples were lower, while chloride and nitrate concentrations in some samples were higher than the maximum contaminant
levels established by the United States Environmental Protection Agency.
Keywords: drinking water, nitrate, chloride, fluoride, ion selective electrode, ion chromatography
*Author for correspondence; E-mail: hakimsiwag@hotmail.com
6
two methods. For fluoride, the IC method gave lower values
than the colorimetry, especially for the stream waters. Since,
the colorimetric method determines total fluorine, differences
in the values might be expected, for example fluoride forms
complexes with the available aluminium, especially in the
stream water (Neal et al., 2007). Statistical analysis of
fluoride concentrations in rain water samples as obtained
by capillary electrophoresis (CE), IC and ISE indicated that
there were no systematic differences between CE and ISE,
but the fluoride concentrations obtained by IC were signi-
ficantly higher. The observed differences are most likely
due to presence of aluminium cations (Van den Hoop et al.,
1996). A fully validated dual ion chromatographic method,
complying with ISO 17025, has been developed at the
chemical laboratory of the Athens Water Supply and
Sewerage Company (EYDAP SA) for the concurrent deter-
mination of ten ions (F-, Cl-, NO3-, Br-, PO43-, SO42-, Na+, K+,
Ca2+ and Mg2+) in surface, ground and potable water samples
(Miskaki et al., 2007).
The aim of the present study was to optimize a simple, selec-
tive and efficient method for simultaneous determination of
chloride, fluoride and nitrate ions in drinking water samples
collected from various sources by using ion chromatography
and ion selective electrodes.
Materials and Methods
Reagents. High purity distilled deionized water was used
throughout the work. Anion standards solutions were
prepared using sodium salt of fluoride (Merck, Germany),
chloride (Merck, Germany) and nitrate (BDH Chemical,
England). Other chemicals were analytically pure reagents
from RDH Chemicals, Germany.
Ion selective electrode. Cole-Parmer ion selective electrode
chloride model EW-27502-13 (USA), Cole-Parmer combination
ion selective electrode fluoride model EW-27504-14 (USA) and
Cole-Parmer combination ion selective electrode nitrate model
EW-27504-22 (USA) were used. The response was in mV given
by OAKTON pH/mV/Ion Meter (pH 2100 series USA). Glacial
acetic acid and sodium chloride were used as low level total
ionic strength adjuster buffer (TISAB-2) for low level fluoride
measurement by ion selective electrodes. Sodium nitrate and
ammonium sulphate were used as ionic strength adjuster for
chloride and nitrate, respectively.
By serial dilution, 10 ppm fluoride standard was prepared by
diluting 1000 ppm standard solution. 50 mL low level TISAB-2
was added to 50 mL of the above standard solution. In a 150 mL
beaker, 50 mL of distilled water and 50 mL low level TISAB-2
were added. The volume of real water samples and TISAB-2
were same as that of the standard. This solution was stirred
at constant rate. The electrode tip was dipped in solution
while the meter was in mV mode. Increments of 10 ppm
standard solution were made after 90 second intervals to
get 0.01, 0.02, 0.04, 0.06, 0.10, 0.29, 0.48 and 1.10 ppm concen-
trations. For nitrate and chloride, 1, 10, 50, 100, 500 ppm
standard solutions were used and 2 mL of ISA (ion strength
adjuster) was added to 100 mL of standard solution. Same
amount of ISA was added to 100 mL of water samples. Rest of
the procedure was same. Calibration curve was obtained by
plotting a graph between electrode potential and concentra-
tions from which the unknown concentrations of F-, Cl- and
NO3
- in water samples were calculated.
Ion chromatography. Ion chromatograph consisted of
Kanauer HPLC quaternary pump Model K-1001 (Germany)
with maximum operating pressure of 400 bars and flow range
of 0.001-9.999 ml/min. HAMILTON PRP-X-100 polymer base
reverse phase No. R-79439 (USA) anion exchange column
PRP X-100 (150 mm × 4.1 mm) having 10 μm particle size with
comparative guard column was used. A comparative guard
column was also used. Alltech model 650 conductivity
detector (USA) was used as detector. Alltech model 640
suppressor (USA) and Metrosep A supp 3 (Metrohm,
Switzerland) anion exchange column (250 mm × 4.6 mm)
having particle size 9 μm, packed with polystyrene/divinyl-
benzene copolymer were used with comparative guard
column in suppressed ion chromatography. The volume of
sample loop used for injection was 20 μL. 4 mM solution of
p-hydroxy benzoic acid was used as mobile phase for non
suppressed ion chromatography while 1.8 mM Na2CO3/
1.7 mM NaHCO3 solution was used as mobile phase for
suppressed ion chromatography.
Optimal mobile phase and its flow rate were used for sepa-
ration of F-, Cl- and NO3
-, using the standard solutions.
Standard solutions of varying concentrations of fluoride,
chloride and nitrate were prepared from standard stock
solutions. These solutions were injected into ion chromato-
graph. Peak areas and heights of all these solutions were
measured and calibration curves for fluoride, chloride and
nitrate were obtained. All water samples were filtered through
0.45 μm pore diameter membrane syringe filters and injected.
The concentration of these anions in samples was determined
using these calibration curves.
ICP-OES instrument. The ICP-OES instrument used in the
present work is ARL 3580 model, made by Applied Research
Laboratories, Switzerland. The instrument is equipped with
a monochromator, a polychromator and a spark excitation
source besides ICP source. Both the monochromator and the
7
Inorganic Ion Detection by ISE and IC
polychromator are 1 meter focal length Paschen-Runge spec-
trometers having 1080 groves/mm concave grating mounted
in Rowland circles. The operating conditions of the ICP-OES
used are given in Table 1. Metal ions were investigated in
some water samples and their emission wavelengths were as
follows: Al (309.271 nm), Ca (393.366 nm), Fe (261.187 nm),
K (766.490 nm), Mg (279.553 nm), Mn (257.610 nm), Na
(588.995 nm), Ni (221.647 nm), Pb (220.353 nm), Si (251.611 nm),
Sr (407.771 nm) and Zn (213.856 nm).
injected at 1.0 mL/min flow rate at various pH of mobile phase.
The retention was decreased by increasing the pH of the
mobile phase. Variation of pH of the mobile phase led to shift
in the dissociation equilibrium and thus to change the
retention time. The peak height and peak area decreased, by
increasing the pH of mobile phase. This might be due to the
decrease in retention time. There was a shift in the base line
showing incomplete separation of anions. So optimum pH
was determined which was based on good resolution. The
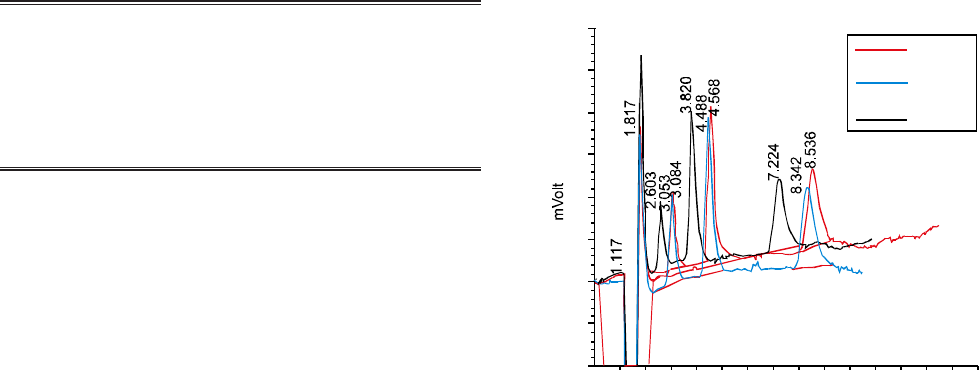
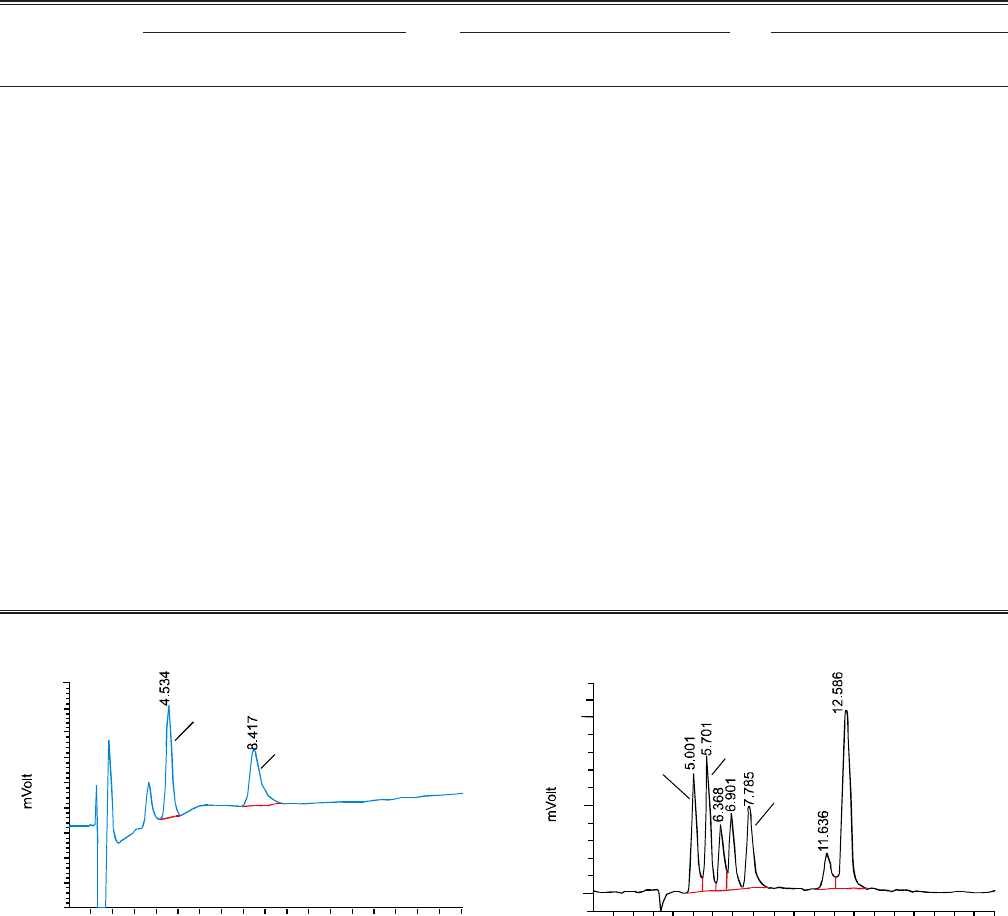
clearer picture is given in Fig. 1. Consequently optimal pH
necessary for complete and in-time separation was 8.5.
Table 1. Operating conditions of ICP-OES
Generator frequency 27MHz
Incident power 1.25 kW
Out gas flow 12 L/min.
Intermediate gas flow 0.8 L/min
Carrier gas flow 1 L/min
Observation height 16 mm above the coil
Sample uptake 1-3 mL/min
Samples. Surface water and groundwater are the main water
sources available to the residents of Islamabad. Drinking
water samples were collected from water sources in the month
of October 2008, from different sectors and nearby villages of
Islamabad. The water was allowed to flow from the source for
about 1-2 min in order to stabilize different parameters i.e.,
conductivity and pH. The collected samples were stored in
pre-cleaned, sterilized polyethylene bottles of one litre
capacity. The samples were cooled to 4 °C in clean and dust
free environment.
Statistical analysis. A paired t-test was performed to check
the validity of two methods (Miller and Miller, 1997). The
formula of paired t-test is:
_
xd √_
n
tcal = _____
Sd
where:
Sd = standard deviation
_
xd = mean of group one minus group two
n = the number of values
If tcal is less than ttab at a specific confidence limit then there is
no significant difference between the two methods.
Results and Discussion
Optimization of mobile phase for non-suppressed IC. In
order to obtain optimal separation, pH and flow rate of mobile
phase was optimized. Standards containing 10 ppm, 20 ppm
and 40 ppm of fluoride, chloride and nitrate, respectively, were
8Muhammad Hakim et al.
To see the effect of flow rate on retention time, a single
standard containing 10 ppm, 20 ppm and 40 ppm of fluoride,
chloride and nitrate, respectively, was injected by using
mobile phase of optimal pH 8.5. The effect of flow rate was
studied in the range of 0.8-1.2 mL/min. The results are shown
in Fig. 2. By increasing the flow rate, the retention time
decreased. The peak height and area also decreased with
retention time. This was due to faster separation of anions
resulting in incomplete separation of ions. The optimum
flow rate was 1 mL/min.
Performance characteristics. Performance characteristics in
terms of detection limit, percent recovery and total run time of
the analytical response were calculated from reproducibility
experiments which are shown in Table 2. The detection limits
for ISE were estimated based on three times standard devia-
tion of response plus mean response from determination of
Fig. 1. Chromatogram of F- 10 ppm, Cl- 20 ppm and NO3
-
40 ppm at different pH at flow rate of 1.0 mL/min by
non-suppressed IC.
60
50
40
30
20
10
0
-10
-20
pH 8.3
pH 8.5
pH 8.7
510 15
Time (min)
six blank samples. The detection limit is thus the correspon-
ding concentration of the response from calibration curve
of each anion (Skoog et al., 2005). The detection limit for IC is
three times signal-to-noise ratio. Hence the detection limits
were found by using the standard whose response was three
times signal to noise. In order to evaluate the accuracy of
method, percentage recovery was calculated by adding
known amount of fluoride, chloride and nitrate to drinking
water samples according to the following equation:
spiked conc. _ actual conc.
Recovery (%) = ( ______________________ ) × 100
conc. of standard added
The total run time includes sample introduction, purging/
washing time and run time, whereas, the time needed for
pretreatment of the sample and to calculate the correspon-
ding concentration were not taken into account.
Analysis of water samples. Measurement of pH. pH of all the
collected samples was measured which was in the range of
6.85-8.65 (Table 3). pH of most samples was in good agree-
ment with US EPA which is 6.5-8.5 except that of sample no.11
which was slightly higher. This sample was from Malal
stream in periphery of the village of Islamabad. People living
nearby this stream wash their clothes in the stream so pH may
be higher due to mixing of soapy water.
Determination of anions. Determination of concentrations
of fluoride, chloride and nitrate was carried out using ISE and
suppressed/non-suppressed ion chromatography. The results
are given in Table 3.
For the analysis of water samples, optimized non-suppressed
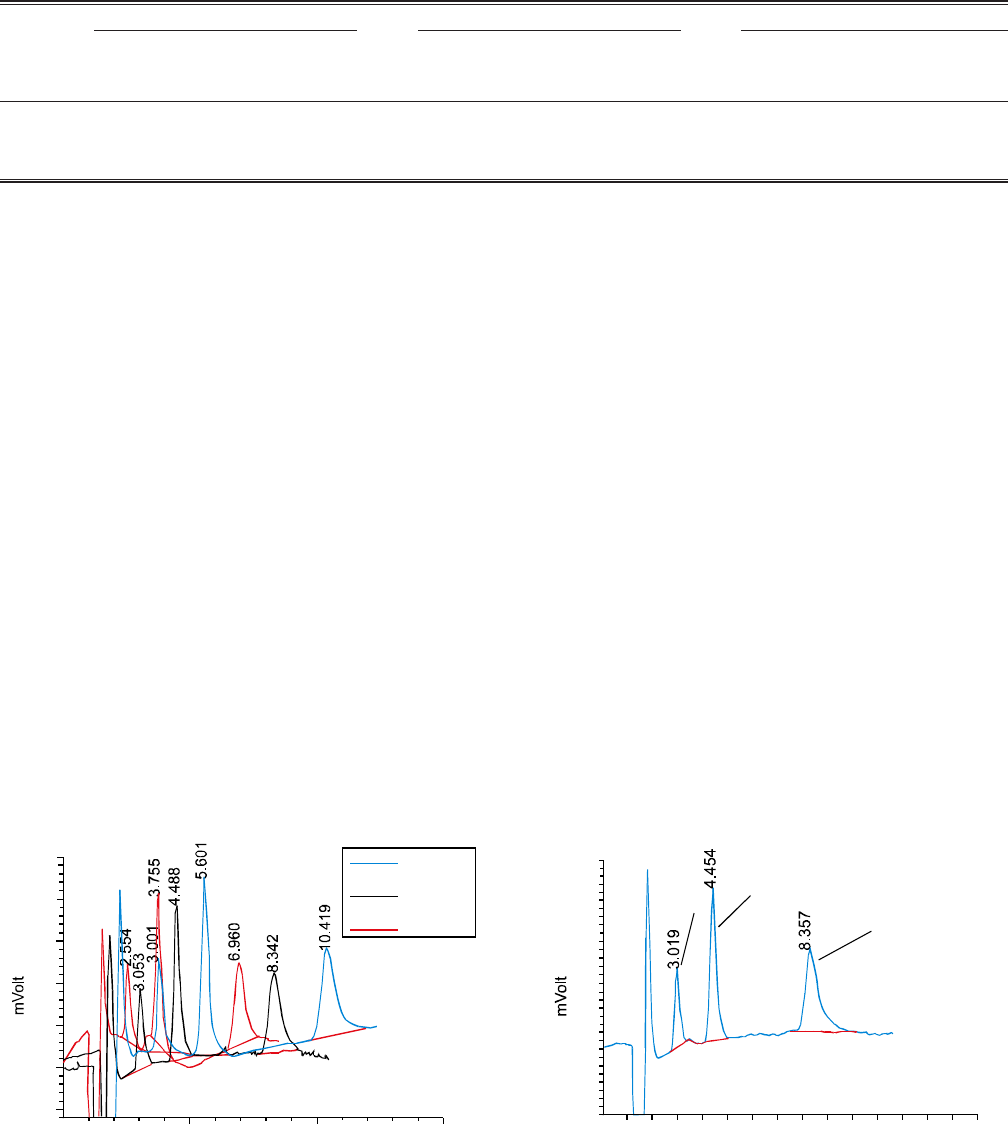
IC conditions were used. The chromatograms obtained by
injecting the samples were compared to standard chromato-
gram; peaks of these chromatograms were quite sharp and
resolution was also very good. Some of the chromatograms of
a standard and a sample are shown in Fig. 3 and 4, respectively.
Samples were analyzed by non suppressed/suppressed
IC as described above. Results obtained for chloride and
nitrate concentration by non suppressed IC are shown in
Table 3.
Table 2. Performance characteristics of the applied techniques
Ion selective electrode Non-suppressed IC Suppressed IC
Anions Detection Run Recovery Detection Run Recovery Detection Run Recovery
limit time (%) limit time (%) limit time (%)
(ppm) (min) (ppm) (min) (ppm) (min)
Fluoride 0.02 3 98.6 2 20 102.7 0.05 25 99.3
Chloride 0.2 3 101.5 1 20 103.2 0.05 25 102.6
Nitrate 1.7 3 109.4 2 20 98.5 0.1 25 106.5
9
Inorganic Ion Detection by ISE and IC
Fig. 2. Chromatogram of F- 10 ppm, Cl- 20 ppm and NO3
-
40 ppm at different flow rates at pH 8.5 by non-
suppressed IC.
50
40
30
20
10
0
-10
510 15
Time (min)
0.8 ml/min
1.0 ml/min
1.2 ml/min
Fig. 3. Chromatogram of F- 10 ppm, Cl- 20 ppm and NO3
-
40 ppm by non-suppressed IC at flow rate of
1.0 mL/min.
50
40
30
20
10
0
-10
Fluoride Chloride
Nitrate
510 15
Time (min)
10 Muhammad Hakim et al.
Fluoride was not measured by non-suppressed ion chroma-
tography due to its low concentration in the samples. So,
suppressed IC was used. Seven anion standards were injected
at the flow rate of 1 mL/min. The chromatogram is shown in
Fig. 5. Quantitative determination of fluoride in the water
samples was made by comparison of peak areas in the
chromatograms of the samples and that of the standard;
chromatogram of a sample is shown in Fig. 6. The results for
Table 3. Results obtained by ion selective electrodes/pH electrode and non-suppressed ion chromatography (suppressed ion
chromatography for fluoride only)
Sample p H Ion selective electrodes/pH electrode Non-suppressed ion chromatography Suppressed ion chromatography
Fluoride Chloride Nitrate Fluoride Nitrate Chloride Fluoride
(ppm) (ppm) (ppm) (ppm) (ppm) (ppm) (ppm)
1 7.45 0.34 92.11 90.72 nd 80.61 90.10 0.31
2 7.45 0.77 114.10 287.69 nd 222.70 118.25 0.73
3 7.30 0.15 53.93 42.31 nd 38.20 50.23 0.15
4 7.20 0.43 67.10 42.73 nd 39.10 65.53 0.41
5 7.30 0.64 128.09 226.11 nd 205.23 122.50 0.60
6 6.85 0.33 1062.07 1321.69 nd 1080.60 855.50 0.41
7 7.50 0.13 39.79 19.05 nd 15.50 37.90 0.10
8 7.35 0.79 96.14 70.59 nd 63.54 95.69 0.81
9 7.30 0.96 67.96 39.24 nd 35.42 65.21 0.92
10 7.35 0.59 30.13 38.27 nd 23.45 20.28 0.62
11 8.65 0.38 29.11 6.01 nd 5.24 22.50 0.36
12 7.05 0.23 13.02 16.39 nd 11.34 9.52 0.22
13 7.85 0.19 13.53 15.90 nd 10.59 10.15 0.20
14 8.00 0.23 12.79 19.15 nd 14.69 9.24 0.24
15 7.80 0.89 19.97 22.48 nd 16.22 19.12 0.90
16 7.25 1.08 55.81 31.94 nd 26.45 18.20 1.12
17 7.30 0.25 10.92 23.75 nd 18.65 9.10 0.24
18 7.55 0.21 8.79 28.03 nd 23.57 6.24 0.26
19 8.10 0.20 9.63 16.88 nd 13.82 8.13 0.22
20 7.55 0.07 2.92 6.82 nd 4.56 2.25 0.05
21 8.00 0.08 3.20 6.91 nd 4.89 3.10 0.06
22 7.65 0.08 251.34 379.91 nd 251.32 240.53 0.09
23 7.05 0.25 80.24 2.88 nd 2.13 78.20 0.27
24 7.15 0.08 12.34 23.52 nd 21.52 10.88 0.07
nd = not detected.
fluoride analysis in water samples by suppressed IC are
given in Table 3.
The concentration of fluoride in all the water samples was
within the limits established by USEPA (4.0 ppm). The
chloride level was also within the permissible range i.e.,
Fig. 5. Chromatogram of F- 3 ppm, Cl- 3 ppm, NO2
-
4 ppm, Br- 4 ppm, NO3
- 4 ppm, PO4
3- 8 ppm and
SO4
2- 8 ppm at flow rate of 1.0 mL/min by supp-
ressed IC.
510 15 20
Chloride
Nitrate
Fluoride
Time (min)
100
50
0
Fig. 4. Chromatogram of sample # 2 by non-suppressed
IC.
60
50
40
30
20
10
0
-10
-20
-30
Chloride
Nitrate
Time (min)
510 15
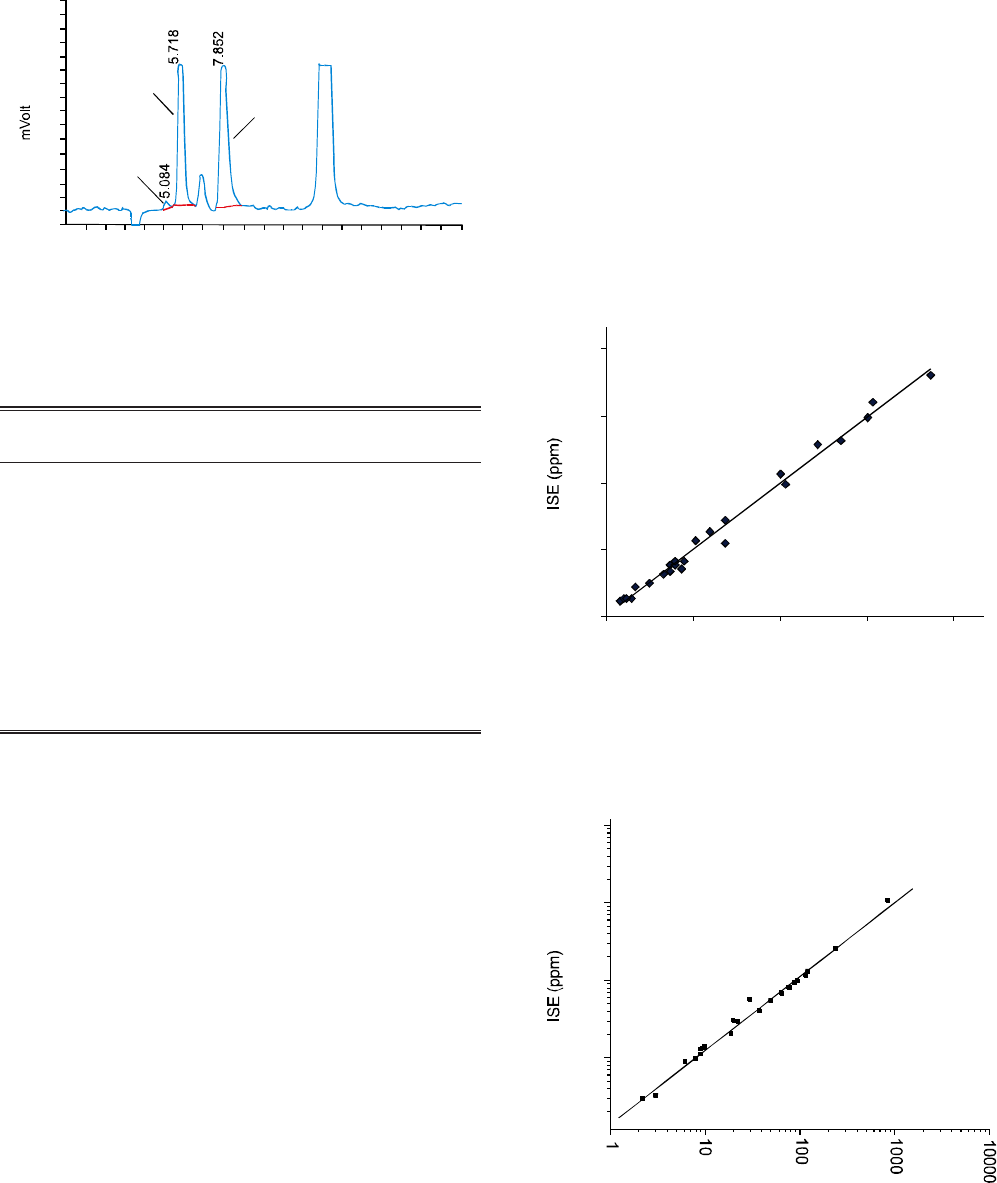
250 ppm in all the samples except sample no. 6. In most of the
samples, nitrate level was higher than US EPA standard for
safe drinking water i.e. 10 ppm. Sample # 6 has the maximum
level of chloride and nitrate exceeding 1000 ppm. This is the
water obtained from house pump installed by boring in Nilore
colony situated in the surrounding area of Islamabad. The
underground water in these areas is in the narrow channels
rather than in large reservoirs. So the water may be in contact
with some rocks containing salts of nitrates and chlorides.
Thus, metal ion analysis especially of samples 6, 7 and 17
were performed using Inductive Coupled Plasma Optical
Emission Spectroscopy by conditions given in the experimen-
tal section. The results are given in the Table 4. It is clear from
the results that sample 6 contained a high concentration of
sodium, magnesium and calcium ions. So most probably the
nitrate and chloride of these cations may exist.
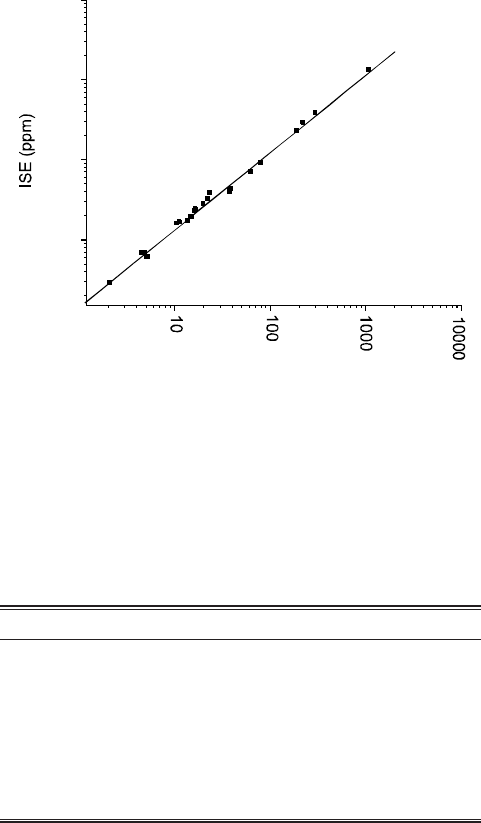
The results obtained by the two methods are compared in
Fig. 7-9. The correlation coefficient in each case shows good
y = 0.9905x + 0.0033
r = 0.9951
0
0.3
0.6
0.9
1.2
0 0.3 0.6 0.9 1.2
IC (ppm)
Fig. 7. Comparison of the results for the determination of
fluoride in drinking water samples (n = 23) using IC
(suppressed) and ISE.
Table 4. Concentration of cations by ICP-OES
Cations Sample 6 Sample 7 Sample 16
(ppm) (ppm) (ppm)
Al nd nd nd
Ca 176.39 32.77 30.64
Fe nd nd nd
K 5.50 8.94 1.70
Mg 328.42 17.79 37.41
M n nd nd nd
Na 481.06 61.51 185.66
Ni nd nd nd
Pb nd nd nd
Si 6.30 5.58 4.86
Sr 7.40 0.22 1.20
Zn nd nd nd
nd = not detected.
11
Inorganic Ion Detection by ISE and IC
linearity in the results of the two methods from low concentra-
tion to the higher concentration.
A paired t-test was also performed to check the validity of
two methods. According to this test, if tcal is less than ttab
at a specific confidence limit then there is no significant
difference between the two methods. The results are given
in Table 5. The results of statistical analysis, according to
student’s t-test, shows that there is no significant difference
between the results obtained with non-suppressed ion
chromatography and ion selective electrodes for chloride
and nitrate determination in water samples.
Fig. 6. Chromatogram of sample # 10 by suppressed IC at
flow rate of 1.0 mL/min.
105
100
50
0
Chloride
Nitrate
Fluoride
510 15 20
Time (min)
Fig. 8. Comparison of the results for the determination of
chloride in drinking water samples (n = 23) using IC
(non-suppressed) and ISE.
10
100
1000
10000
r = 0.9941
y = 0.137+0.955 log(x)
IC (ppm)
Conclusion
Both ion chromatography and ion selective electrode were
employed for the determination of three anions (fluoride,
chloride and nitrate) in drinking water samples. ISE is a
preferred technique due to shorter analysis time and less
operational cost of the equipment. Ion chromatography is
sophisticated and reliable for simultaneous determination of
anions in routine water analysis. This technique can be used
for comparison and validation of methods.
Acknowledgement
Authors gratefully acknowledge the efforts of Central
Analytical Facility Division, PINSTECH, Islamabad in the
analysis of the water samples.
References
APHA, 1985. Standard Methods for the Examination of
Water and Wastewater, 16th edition, American Public
Health Association, Washington DC., USA.
Bosch, N.B., Mata, M.G., Penuela, M.J., Galan, T.R., Ruiz, B.L.
1995. Determination of nitrite levels in refrigerated and
frozen spinach by ion chromatography. Journal of
Chromatography A, 706: 221-228.
Cheam, V. 1992. Comparison of ion chromatographic
methods for the determination of organic and inorganic
acids in precipitation samples. Analyst, 117: 1137-
1144.
Frankenberger Jr., W.T., Mehra, H.C., Gjerde, D.T. 1990.
Environmental application of ion chromatography.
Journal of Chromatography A, 504: 211-245.
Fraser, P., Chilvers, C. 1981. Health aspects of nitrate in
drinking water. Science of The Total Environment, 18:
103-116.
Meenakshi, Maheshwari, R.C. 2006. Fluoride in drinking
water and its removal. Journal of Hazardous Materials,
137: 456-463.
Miller, J.C., Miller, J.N. 1997. Statistics for Analytical
Chemistry, 3rd edition, Ellis Horwood Limited, UK.
Miskaki, P., Lytras, E., Kousouris, L., Tzoumerkas, P. 2007.
Data quality in water analysis: validation of ion chro-
matographic method for the determination of routine
ions in potable water. Desalination, 213: 182-188.
Neal, M., Neal, C., Wickham, H., Harman, S. 2007. Determina-
tion of bromide, chloride, fluoride, nitrate and sulphate
by ion chromatography: Comparison of methodologies
for rainfall, cloud water and river waters at the Plynlimon
catchments of mid-Wales. Hydrology and Earth System
Sciences, 11: 294-300.
PCRWR, 2008. Water Quality Report. National Water Quality
Monitoring Programme. Pakistan Council of Research in
Water Resources, Ministry of Science & Technology,
Islamabad, Pakistan.
Pereira, C.F. 1992. Application of ion chromatography to
determination of inorganic anions in foodstuffs. Journal
of Chromatography A, 624: 451-470.
Skoog, D.A., Holler, F.J., Nieman, T.A. 2005. Principles of
Instrumental Analysis, 5th edition, Thomson Asia Pvt.,
Ltd., Singapore.
US EPA, 2009. Drinking Water Contaminants, United States
Environmental Protection Agency, Office of Ground
Water and Drinking Water, Washington DC., USA.
Van den Hoop, M.A.G.T., Cleven, R.F.M.J., Van Staden, J.J.,
12 Muhammad Hakim et al.
Table 5. Statistical analysis of the correlation between
chloride and nitrate concentrations by suppressed/non-
suppressed IC and ISE
Anions Intercept Slope r2tcal ttab
Fluoride 0.0033 0.9905 0.99022 - 0.70200 2.074
±0.0005 ±0.01201
Chloride 0.16042 0.9637 0.99398 -1.46362 2.074
±0.02570 ±0.01636
Nitrate 0.15521 0.9682 0.99386 -2.06386 2.074
±0.02604 ±0.0166
Confidence limits = ±95; two tail; n=23.
Fig. 9. Comparison of the results for the determination of
nitrate in drinking water samples (n = 23) using IC
(non-suppressed) and ISE.
10
100
1000
10000
r = 0.9969
y = 0.0.155+0.968 log(x)
IC (ppm)
13
Neele, J. 1996. Analysis of flouride in rain water: Com-
parison of capillary electrophoresis with ion chromato-
graphy and ion selective electrode potentiometry.
Journal of Chromatography A, 739: 241-248.
Vasconcelos, M.T.S.D., Gomes, C.A.R., Machado, A.A.S.C.
1994. Ion chromatographic determination of fluoride in
welding fumes with elimination of high contents of iron
by solid-phase extraction. Journal of Chromatography
A, 685: 53-60.
Weiss, J., Reinhard, S., Pohl, C., Saini, C., Narayaran, L. 1995.
Stationary phase for the determination of fluoride and
other inorganic anions. Journal of Chromatography A,
706: 81-92.
WHO, 2004. Guidelines for Drinking-Water Quality Recom-
mendations, vol. 1, 3rd edition, World Health Organiza-
tion, Geneva, Swizerland.
WWF, 2007. Pakistan Water at Risk. Special Report, WWF-
Pakistan, Ferozepur Road, Lahore-54600, Pakistan.
Inorganic Ion Detection by ISE and IC

Pak. J. Sci. Ind. Res. 2010 53 (1) 14-19
*Author for correspondence; E-mail: razias@hotmail.com
Physical and Chemical Evaluation of Oils of Two Varieties of
Carthamus tinctorius Grown in Pakistan
Razia Sultana*, Rubina Saleem and Ambrat
Applied Chemistry Research Center, PCSIR Laboratories Complex,
Shahrah-e-Dr. Salimuzzaman Siddiqui, Karachi-75280, Pakistan
(received August 25, 2009; revised December 8, 2009; accepted December 12, 2009)
Abstract. On evaluation of oils of two spineless varieties of Carthamus tinctorius, Thori-78 and Pawari-95 growing in
Sindh, Pakistan, the quality of the oil was found to be similar, only the oil content differed. The hexane-extracted oil
content of Thori-78 and Pawari-95 was 28.33±1.15 and 33.07±1.12, respectively. The oils contained 90.97% and
89.55% unsaturated fatty acids and 8.44% and 9.69%, saturated fatty acids, respectively. Linoleic acid was 75.42±0.59%
and 76.40±1.0% and oleic acid was 15.55±0.30% and 13.15±0.49% by weight, respectively, and were the predominant
fatty acids present in the oil.
Keywords: safflower oil, Thori-78, Pawari-95, linoleic acid, oleic acid
Introduction
Safflower (Carthamus tinctorius) is an annual herb belong-
ing to the family Compositae. It is widely distributed through-
out the world such as in Pakistan, India, Bangladesh, Afgha-
nistan, Middle East, Thailand, China, Japan, Ethiopia, Sudan,
Tanzania, Keneya, Tunisia, Europe, Argintina, USA, Canada
and Australia (Knights et al., 2001). C. tinctorius flowers, seeds
and oil have wide range of medicinal uses in different coun-
tries. Flowers are used for the preparation of dyes and drugs
which are used for treating a number of disorders such as for
dilation of arteries, reduction of hypertension, increasing blood
flow, decreasing blood cholesterol, in treatment of rheumotoid
arthritis, menstrual problems, skin diseases, urinary problems
and jaundice etc. (Kaffka et al., 2001; Sastri, 1950). Seed
decoction is used as laxative in Pakistan. The oil is used in
Iran to treat liver and heart ailments and in charred state, used
in India in treatment of sores and rheumatism. In Northern
America, it is cultivated for using as bird feed, animal meal
and for industrial applications (Oyen et al., 2007; Mündel
et al., 2004; Oplinger et al., 1992).
Safflower is used as a substitute for saffron; its flowers are
commonly mixed with rice, pickles and other foods to give
an attractive colour (Sastri, 1950). America, India and
Africa are the main producers of safflower oil. Its seeds are
edible and are eaten after roasting. The seed oil content varies
from 24 to 36%, depending on the variety of safflower, soil
texture, climate and other conditions (Pritchard, 1991; Swern,
1964a). There are two types of safflower oil: high oleic (high
in mono-unsaturated fatty acids) and high linoleic (high in
polyunsaturated fatty acids). Gas chromatography has been
an indispensable analytical technique ever since its first use
in the fatty acid determination of plant seed oil (Echard
et al., 2007; Peris-Vicente et al., 2006; Seppänen-Laakso
et al., 2002). High performance liquid chromatography
(HPLC) with ultraviolet and fluorescence detectors are the
alternative methods for separation of volatile short chain and
long chain fatty acids (Peris-Vicente et al., 2005; 2004; Chen
and Chuang, 2002).
Safflower oil can be used in cosmetics, foods, nutritional
supplements, personal care products, soaps and shampoos.
Cold press oil is golden yellow and is used for culinary
purposes. The oil obtained by dry hot distillation is dark and
sticky and is used only for greasing ropes and leather goods
which are exposed to water. Developed countries have
created the most significant market for safflower oil for use as
salad oil and cooking oil and in making margarine; being non-
allergenic, it is considered to be one of the healthiest oils for
human consumption because it has a high ratio of polyunsatu-
rated/saturated fatty acids.
Safflower was introduced as oilseed crop in Pakistan in 1960.
It is mainly cultivated in Sindh and Baluchistan provinces.
Being a drought-tolerant crop, it is recommended for planting
in rainfed areas. In Sindh it is cultivated after the rice crop on
residual moisture. Due to the increasing interest in the
safflower oil for edible purposes based on its high content of
linoleic acid, our studies are mainly focused on the content
and physical and chemical evaluation of the oils of two spine-
less varieties of safflower, Thori-78 and Pawari-95, grown in
14
Sindh, Pakistan. Proximate analysis of oils were carried out
for the content, glycerides composition and physical and
chemical parameters such as free fatty acid, acid value, per-
oxide value, iodine value, refractive index, saponification
value, unsaponifiable matter, specific gravity and colour; the
fatty acid composition of both the varieties of oils were inves-
tigated as methyl esters by gas chromatography.
Materials and Methods
Plant material. Two varieties of Carthamus tictorius seeds,
Thori-78 and Pawari-95, were collected from Tandojam,
Sindh, Pakistan. The fruit is an achene (dry, one seeded with a
thin hull) and resembles sunflower seed but is smaller in size
and creamish in colour. It is irregularly pear-shaped, smooth
and shiny up to 10 mm long.
Reagents. Solvents and chemicals such as n-hexane (95%),
n-heptane (99%), ethanol (95%), carbon tetrachloride (95.5%),
chloroform (99.5%), methanol (98.8%), sulphuric acid
(95.98%), hydrochloric acid (37%), acetic acid (100%),
glacial acetic acid (99.5%); sodium hydroxide (98%), potas-
sium hydroxide (98%), sodium thiosulphate pentahydrate
(R.G), oxalic acid (extra pure), potassium dichromate (extra
pure), potassium iodide (extra pure), iodine monochloride
(R.G) and anhydrous sodium sulphate were purchased from
E. Merck (Damstadt, Germany) and Labscan (Bankok,
Thialand). Standards of fatty acid methyl esters were purchased
from Supelco (Bellefonte, PA, USA) and Sigma Aldrich Co.
(St. Louis, MO, USA).
Apparatus. The apparatus used included gas chromatograph
with flame ionization detector, model Clarus 500, from Perkin
Elmer Instruments LLC, (Shelton, CT, USA), capillary
column Rtx-2330 (60 × 0.25 mm × 0.20 ìm, film thickness)
from Supelco (Bellefonte, PA, USA), Lovibond model E
tintometer (Salisbury, UK), Abbé refractometer model 2W
(Shijiazhuang, China), Gallen Kamp air oven (West Mildlands,
UK) and vacuum oven (Melrose Park, IL, USA).
Oil extraction. Safflower, Thori-78 and Pawari-95, seeds
(500 g each) were crushed and finely ground to flour and then
subjected to extraction with n-hexane (0.5 litre) in a one litre
Soxhlet extractor for 8 h (AOCS, 2004). The fat was recov-
ered using a rotary evaporator. The extracted fat was placed
in an oven at 60 °C for 1 h, transferred to a capped reagent
bottle and stored at 4 °C until required.
Quantitative separation of tri-, di- and mono-acylglycerols
of oil. The lipid class composition, comprising of TAGs,
DAGs, and MAGs mixture in C. tinctorius seed oil was
determined by solid-liquid adsorption chromatography
(SLAC), using silica gel as the adsorbent and eluted with
different solvent systems by following the AOCS method
(AOCS, 2004) with little modification. The effectiveness of
separation was verified by thin layer chromatography, using
solvent system (petroleum-ether and acetone: 9:1).
Fatty acid composition. Methyl esters of fatty acids were
prepared according to standard IUPAC method 2.301 (IUPAC,
1987). The chemical composition of fatty acid methyl esters
was accomplished with a Perkin Elmer gas chromatograph
model Clarus 500 fitted with a polar capillary column Rtx-
2330 (60×0.25 mm×0.20 μm, film thickness) and a flame
ionization detector. Nitrogen was used as carrier gas at a flow
rate of 3 mL/min. Other conditions were as follows: initial
oven temperature, 70 °C was maintained for 5 min then ramped
at 10 °C/min to 180 °C, followed by 3 °C/min to final tem-
perature of 220 °C, where it was held for 15 mins; injector
temperature and detector temperature was 270 °C. A sample
volume of 0.3 μL was injected (splitless). Fatty acid methyl
esters were identified by comparing their relative and abso-
lute retention times to those of authentic standards of fatty
acid methyl esters purchased from Supelco Sigma-Aldrich Co.
Quantification was done by a built-in data-handling
programme, provided by the manufacturer of the gas chro-
matograph. Analyses were performed in triplicate.
Physical and chemical analysis of the extracted oils. The
following tests for refractive index, specific gravity, colour,
free fatty acid, acid value, peroxide value, iodine value,
saponification value and unsaponifiable matter of the extracted
oils were performed by the standard methods of AOCS (2004).
Colour of the oils was determined by a Lovi bond tintometer
(Tintometer Ltd., Salisbury, UK) using a one inch cell.
Results and Discussion
Hexane extracted oil content of the two varieties of
C. tinctorius, Thori-78 and Pawari-95, seeds was found to be
28.33±1.15 and 33.07±1.12%, respectively; the high percent-
age of oil gives these varieties distinct potential for the oil
industry, because the average oil content of the seeds exceeds
those of conventional oil seeds i.e., cotton (15.0-24.0%),
canola (17-21%), soyabean (17-21%), olive (20-25%) which
are grown in the USA, Brazil and Asia (Pritchard, 1991) but
oil content is slightly lower than that of sunflower (25-35%).
Physical and chemical parameters of the oils are depicted in
Table 1. At room temperature, both varieties of seed oil were
present in a liquid state. The refractive index and specific
gravity of Thori-78 and Pawari-95 oils were determined at
40 °C, which were concordant with the reported value and
comparable with other vegetable oils (Rossell, 1991a; Swern,
1964a; 1964b). The values determined for free fatty acids as
15
Carthamus tinctorius Oil Quality

OA and acid values are comparable with the reported values
of the crude oil (Dhellot et al., 2006; El-Adawy and Taha,
2001). Very low value of free fatty acid and acid value in the
present analysis is an indication of the good quality of crude
oil. Peroxide values (Table 1), indicating the presence of hy-
droperoxides in oils, were high, thus showing low resistance
to oxidation (Onyeike and Acheru et al., 2002); thus oils could
be used after slight refining. The analyzed crude oils were
high in colour index 2.13R + 45.4Y + 0.71N (Thori-78) and
2.48R + 46.16Y + 0.81N (Pawari-95). Intense colour of
vegetable oils depend mainly on the presence of various
colouring pigments of plants such as carotenoids, chlorophyll
etc., which are effectively removed during refining and bleach-
ing steps of oil processing. Vegetable oils with minimum
values of colour index are good for edible purpose.
Iodine values were comparatively high due to the presence of
high content of unsaturated fatty acids and are comparable
with the values of poppy, soybean and sunflower oils (Rossell,
1991a). High iodine value shows that both the varieties of
seed oils have good qualities of oils, required for edible and
drying purposes (Eromosele et al., 1994). Values of saponifi-
cation and unsaponifiable matter of Thori-78 and Pawari-95
oils are concordant with the values of sunflower, poppy seed
and soybean oils (Rossell, 1991a; Swern, 1964a; 1964b), in-
dicating them to be good source of industrial oil which can be
used in the manufacture of soap and liquid soap.
Table 2 shows glyceride composition of oils of both the
varieties of C. tinctorius. The content of triacylglycerides is
over 83%. Comparison of free fatty acid value, acid value,
saponification value, unsaponifiable matter, iodine value, re-
fractive index, specific gravity and colour of the studied oils
with those of the known edible oils reveal that the quality of
both the varieties of oil have great potential for edible usage.
Table 2: Glyceride composition of C. tinctorius seed oils
(wt. %)
C. tinctorius Monoglyceride Diglyceride Triglyceride
variety
Thori-78 5.70±0.55 7.62±0.87 85.61±0.61
Pawari-95 5.77±0.50 8.72±0.53 83.93±0.88
Values are means ± SD, analyzed in triplicate.
Table 1. Proximate and physicochemical characteristics of
C. tinctorius oils
Parameters Thori-78 Pawari-95
Oil content 28.33±1.15 33.07±1.12
(27.11-29.88) (31.5-34.0)
Free fatty acid 0.52±0.012 0.53±0.004
(% as OA) (0.51-0.54) (0.53-0.54)
Acid value (mg/kg) 1.12±0.15 1.06±0.02
(0.99-1.34) (1.04-1.09)
Peroxide value 17.2±0.25 20.75±0.38
(Meq/kg) (16.94-17.54) (20.2-21.1)
Iodine value 134.82±0.68 136.16±0.96
(g of I/100 g of oil) (133.99-135.67) (134.89-137.21)
Saponification value 187.56±2.34 188.96±2.18
(mg of KOH/ g of oil) (184.89-190.60) (186.5-191.8)
Unsaponifiable 0.41±0.06 0.57±0.05
matter (%) (0.32-0.46) (0.50-0.62)
Refractive index 1.4734±0.0005 1.4679±0.0007
at 40 oC (1.4730-1.4742) (1.4672-1.4689)
Specific gravity 0.9064±0.002 0.9240±0.0004
at 40 oC (0.9031-0.9085) (0.9234-0.9245)
Colour (Red unit) 2.13±0.04 2.48±0.08
(2.1-2.2) (2.4-2.6)
(Yellow unit) 45.4±0.29 46.16±0.62
(45.0-45.7) (45.5-47.0)
(Blue unit) 0 0
(Neutral unit) 0.71±0.06 0.81±0.01
(0.65-0.80) (0.79-0.83)
Fatty acid composition of the oils of the two varieties was
determined using gas chromatography (Fig. 1 and 2 and
Table 3). The principal fatty acid components in Thori-78 and
Pawari-95 were palmitic (C16:0), stearic (C18:0), oleic (C18:1) and
linoleic (C18:2) acids. Linoleic acid is predominantly present
in both the varieties as compared to other varieties, U.S.-10,
S.-208 and V. F.-stp (53-1) grown in Pakistan (Table 3) (Raie,
2008). Fatty acid composition was more or less similar to that
of sunflower, soybean, corn, and cotton seed oils and safflower
oil originating from different geographic regions (Cosge
et al., 2007; Rossell, 1991b). These results suggest that these
varieties of C. tinctorius can serve as potential dietary sources
of mono unsaturated fatty acid (MUFA) and poly unsaturated
fatty acid (PUFA).
Present studies revealed that seed oils of C. tinctorius variet-
ies, Thori-78 and Pawari-95, indigenous to Pakistan have very
good potential for edible and industrial usage and also for use
in developing nutritionally balanced formulations blended with
other high stearic or high oleic oils. These Oils are used in the
food and pharmaceutical industries to produce cooking oils,
16 Razia Sultana et al.
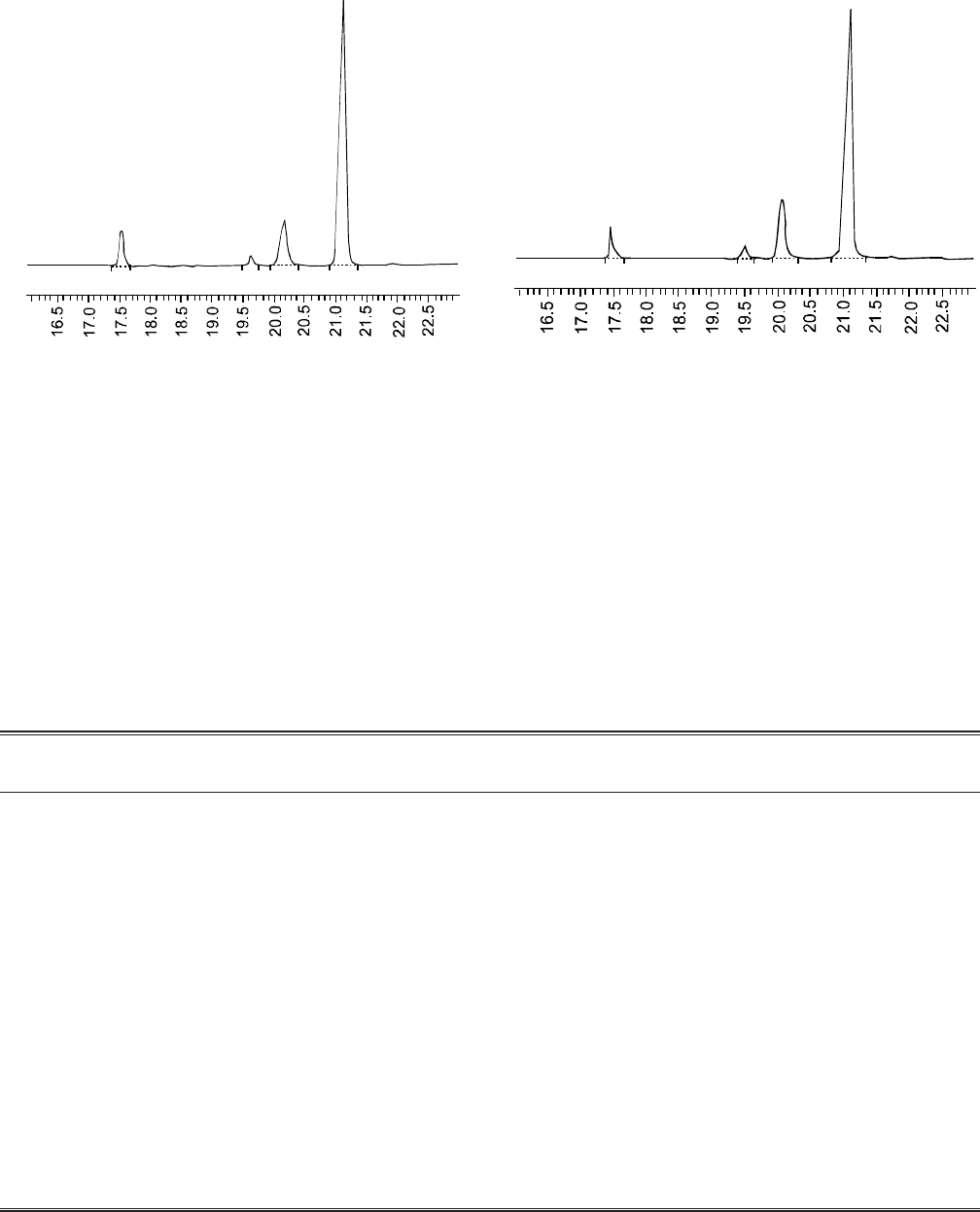
Table 3. Fatty acid composition of high linoleic C. tinctorius varieties grown in Pakistan (wt. %)
Fatty acids Thori-78 Pawari-95 US-10 S.-208 V.F.-stp
(53-1)
Myristic Acid - - 3.1 0.9 2.8
(C14:0)
Palmitic Acid 6.45±0.57 6.92±0.37
(C16:0) (5.66-7.02) (6.41-7.28) 10.2 9.4 12.0
Stearic Acid 1.99±0.09 2.77±0.49
(C18:0) (1.89-2.12) (2.24-3.42) 5.5 2.3 3.6
Oleic Acid 15.55±0.30 13.15±0.49
(C18:1) (15.31-15.98) (12.67-13.81) 14.4 14.0 15.7
Linoleic Acid 75.42±0.59 76.40±1.0
(C18:2) (74.65-76.11) (75.01-77.32) 66.8 73.4 65.9
Others 0.59±0.04 0.76±0.06
(0.54-0.64) (0.68-0.83) - - -
Total saturated
fatty acids 8.44 9.69 18.8 12.6 18.4
Total unsaturated
fatty acids 90.97 89.55 81.2 87.4 81.6
Values are mean ± SD, analyzed in triplicate.
Fig. 1: Gas chromatogram of fatty acids of safflower
(Thori-78) seed oil; major components are
labelled.
Fig. 2: Gas chromatogram of fatty acids of safflower
(Pawari-95) seed oil; major components are
labelled.
food supplements and skin care products. The good drying
property and high content of linoleic acid and absence of
linolenic acid and wax and low content of free fatty acid, colour
and unsaponifiable compounds make them suitable for use in
the production of high quality paints, alkyd resins, coatings,
varnishes and linoleum. They can also be used in the produc-
tion of biodiesel.
Pakistan imports huge amount of palm oil and soybean from
foreign countries to fulfil the increasing demand of oil in
the country. Moreover, Pakistan has suitable atmosphere for
cultivating all the conventional and non-conventional oilseed
crops. Cultivation of safflower varieties at larger scale could
fulfil the requirements of the country and save enormous
amount of foreign exchange spent otherwise.
C16:0
C18:0
C18:1
C18:2
C16:0 C18:0
C18:1
C18:2
17
Carthamus tinctorius Oil Quality
Retention time (min) Retention time (min)
Conclusion
These studies were focussed on the yield and physical and
chemical evaluation of seed oils of C. tinctorius varieties,
Thori-78 and Pawari-95, cultivated in the region of Sindh,
Pakistan. It was revealed that oils of both the varieties have
very good potential for developing nutritionally blended
formulations balanced with other high saturated fatty oils, as
well as for different industrial usage due to the presence of
high percentage of polyunsaturated fatty acids. These can also
be used in the production of biodiesel and thus in different
industries in Pakistan.
References
AOCS, 2004. Official Methods and Recommended Practices
of American Oil Chemists Society, 5th edition, American
Oil Chemists Society, Champaign, Illinoise, USA.
Chen, S.H., Chuang, Y.J. 2002. Analysis of fatty acids by
column liquid chromatography. Analytica Chimica Acta,
465: 145-155.
Cosge, B., Gûrbûz, B., Kiralan, M. 2007. Oil content and
fatty acid composition of some safflower (Carthamus
tinctorius L.) varieties sown in spring and winter. Inter-
national Journal of Natural and Engineering Sciences,
1: 11-15.
Dhellot, J.R., Matouba, E., Maloumbi, M.G., Nzikou, J.M.,
Safou Ngoma, D.G., Linder, M., Desobry, S., Parmentier,
M. 2006. Extraction, chemical composition and nutri-
tional characterization of vegetable oils: Case of
Amaranthus hybridus (var1 and 2) of Congo Brazzaville.
African Journal of Biotechnology, 5: 1095-1101.
Echard, J.P., Benoit, C., Peris-Vicente, J., Malecki, V., Gimeno-
Adelantado, J.V., Vaiedelich, S. 2007. Gas chromatogra-
phy/ mass spectrometry characterization of historical
varnishes of ancient Italian lutes and violin. Analytica
Chimica Acta, 584: 172-180.
El-Adawy, T.A., Taha, K.M. 2001.Characteristics and
composition of different seed oils and flours. Food
Chemistry, 74: 47-54.
Eromosele, I.C., Eromosele C.O., Akintoye, A.O., Komolafe,
T.O. 1994. Characterization of oils and chemical analy-
ses of the seeds of wild plants. Plant Foods for Human
Nutrition, 46: 361-365.
IUPAC, 1987. Standard Methods for the Analysis of Oils,
Fats and Derivativs, C. Paquot and A. Hautfenne (eds.),
96 pp., 7th revised and enlarged edition, Blackwell Scien-
tific Publications, London, UK.
Kaffka, S.R., Kearny, T.E., Knowles, P.D., Miller, M.D. 2001.
Safflower production in California. [http://agric.ucdavis.
edu/agronomy/crops/oilseed/safflower.html].
Knights, S.E., Wachsman, N.G. and Norton, R.M. 2001.
Safflower (Carthamus tinctorius L.) Production and re-
search in southern Australia. (Abstract: Fifth International
Safflower Conference).[http://www.sidney.ars. usda. gov/
state/saffcon/abstracts/ProductionManagement/
knights.html].
Mündel, H.H., Blackshaw, R.E., Byers, J.R., Huang, H.C.,
Johnson, D.L., Keon, R., Kubik, J., Mckenzie, R., Otto,
B., Roth, B., Standford, K. 2004. Safflower production
on the Canadian prairies: revisited in 2004. In: Agricul-
ture and Agri-Food Canada, pp. 4-5, Graphcom Printers
Ltd., Lethbridge, Alberta, Canada. [http://Safflower.wsu.
edu/SafflowerProduction_Canada.pdf]or//res2.agr.ca/
lethbridge/safflo/part 1- e.htm.].
Onyeike, E.N., Acheru, G.N. 2002. Chemical composition of
selected Nigerian oil seeds and physicochemical proper-
ties of the oil extracts. Food Chemistry, 77: 431-437.
Oplinger, E.S., Oelke, E.A., Kaminski, A. R., Putnam, D. T.,
Teynor, T.M., Doll, D.J., Kelling, K.A., Durgan, B. R.,
Noetzel, D.M. 1991. Safflower. In: Alternative Field
Crops Manual.[http://www. hort.purdue.edu/NEW-
CROP/AFCM/Safflower.html].
Oyen, L.P.A., Umali, B.E. 2007. Carthamus tinctorius L. In:
PROTA 14: Vegetable Oils/ Oléagineux, H.A.M. van der
Vossen, and G.S. Mkamilo (eds.), PROTA, Wageningen,
Netherlands.[http://database.prota.org/PROTAhtml/
Carthamus%20tinctorius_En.html]
Peris-Vicente, J., Gimeno Adelantado, J.V., Doménech-Carbó,
M.T., Mateo-Castro, R., Bosch-Reig, F. 2006. Charac-
terization of waxes used in pictorial art works according
to their relative amount of fatty acids and hydrocarbons
by gas chromatography. J. Chromatography A, 1101: 254-
260.
Peris-Vicente, J., Gimeno Adelantado, J.V., Doménech-Carbó,
M.T., Mateo-Castro, R., Bosch-Reig, F. 2005. Identifi-
cation of lipid binders in old oil paintings by separation
of 4-bromomethyl-7-methoxycoumarin derivatives of
fatty acids by liquid chromatography with fluorescence
detection. Journal of Chromatography A, 1076: 44-50.
Peris-Vicente, J. Gimeno Adelantado, J.V., Doménech-Carbó,
M.T., Mateo-Castro, R., Bosch-Reig, F. 2004. Identifi-
cation of drying oil used in pictorial works of art by
liquid chromatography of the 2-nitro phenyl hydrazides
derivatives of fatty acids. Talanta, 64: 326-333.
Pritchard, J.L.R. 1991. Analysis and properties of oilseeds.
In: Analysis of Oilseeds, Fats and Fatty Foods, J.B.
Rossell and J.L.R. Pritchard (eds.), pp. 82-93, Elsevier
Applied Science, New York, USA.
Raie, M.Y. 2008. Nutritional aspects. In: Oils, Fats and Waxes,
pp. 123, National Book Foundation, Combine Printers,
18 Razia Sultana et al.
Lahore, Pakistan.
Rossell, J.B. 1991a. Vegetable Oils and Fats. In: Analysis of
Oilseeds, Fats and Fatty Foods, J.B. Rossell, and J.L.R.
Pritchard (eds.), pp. 266-268, Elsevier Applied Science,
New York, USA.
Rossell, J.B. 1991b. Vegetable oils and fats. In: Analysis of
Oilseeds, Fats and Fatty Foods, J.B. Rossell and J.L.R.
Pritchard (eds.), pp. 290-296, Elsevier Applied Science,
New York, USA.
Sastri, B.N. (ed.) 1950. The Wealth of India, A Dictionary of
Indian Raw Materials and Industrial Product. vol. II,
pp. 84-88, Publication and Information Directorate, CSIR
New Dehli, India.
Seppänen-Laakso, T., Laakso, I., Hiltunen, R. 2002. Analysis
of fatty acids by gas chromatography and its relevance to
research on health and nutrition. Analytica Chimica Acta,
465: 39-62.
Swern, D. 1964a. Structure and composition of fats and
oils. In: Bailey’s Industrial Oil and Fat Products,
D. Swern (ed.), pp. 211-212, John Wiley & Sons, New
York, USA.
Swern, D. 1964b. Structure and composition of fats and oils.
In: Bailey’s Industrial Oil and Fat Products, D. Swern
(ed.), pp. 207, John Wiley & Sons, New York, USA.
19
Carthamus tinctorius Oil Quality

Analysis of Caffeine and Heavy Metal Contents in Branded and
Unbranded Tea Available in Pakistan
Asma Inayata*, Shahid Rehman Khana, Muhammad Nawaz Chowdhryb and Amran Waheedb
aApplied Chemistry Research Centre, PCSIR Laboratories Complex Lahore - 54600, Pakistan
bCollege of Earth and Environmental Science, University of Punbjab, Lahore, Pakistan
(received November 10, 2008; revised December 2, 2009; accepted December 10, 2009)
Introduction
Tea (Camellia sinensis) is one of the most popular beverages
all over the world. According to an estimate, 2.5 million metric
tonnes of dried tea is manufactured annually, 75% of which is
processed as black tea and consumed in different countries.
In UK, on an average, one litre of tea is consumed per person
per day (Al-Oud, 2003). Different brands of tea are manufac-
tured to meet the increasing demands of consumers world-
wide. Positive and negative effects of tea on the health have
been investigated by many researchers, recently (Yao et al.,
2006a).
Caffeine ascribes quality characteristics to tea, such as
briskness, taste etc., and has been considered an important
quality parameter in the evaluation of tea quality (Yao et al.,
2006b). Caffeine is a pharmacologically active substance and,
depending on the dose, can be a mild nervous system
stimulant. Caffeine does not accumulate in the body and is
normally excreted within several hours of consumption
(Mumin et al., 2006; Obanda et al., 1999). Human body
requires both metallic and non-metallic elements for healthy
growth and development within certain permissible limits.
The optimum concentration needed for this purpose varies
widely from one element to another, from infant to childhood
to adult and from male to female (Atta, 1995). Determination
of these elements in beverages, water, food, plant and soil is
thus of utmost important. Tremendous research has been
rendered on finding tolerance limits for daily intake of nearly
all essential elements needed for healthy growth and sound
physiological changes in human body. There is a fairly
narrow gap between the essential and the toxic levels of
metals and essential trace elements that can otherwise
accumulate in bone, hair and soft tissues such as liver,
kidney, brain or lungs (Tautkus et al., 2004).
Materials and Methods
Caffeine. Preparation of tea solution. Two grams of tea were
added to boiling water, (200 mL) in a 250 mL conical flask placed
on a hot plate at 90 °C while stirring for 10 min by a magnetic
bar. Then the tea solution was filtered through cotton wool
and the residue was washed thrice with distilled water
(10 mL). The tea solution was cooled to room temperature and
washings were diluted to 250 mL with distilled water. The
sample was analyzed in duplicate.
Measurement. To 10 mL of tea solution, 5 mL HCl (0.9 mL of
36% HCl was diluted to 1000 mL with distilled water) and 1 mL
lead acetate solution (100 g of lead acetate was dissolved in
small quantity of water and diluted to 200 mL with distilled
water) were added and diluted upto 100 mL with distilled
water. The solution was then filtered through Whatman filter
paper # 42. Filtrate 25 mL and 0.3 mL sulphuric acid (167 mL of
98% H2SO4 was diluted to 1000 mL with distilled water) were
placed in a volumetric flask and diluted to 50 mL with distilled
water. The solution was filtered using the same type of filter
paper. Absorbance of the filtrate was measured using spec-
trophotometer (Spectronic Unicam) at 274 nm. Readings were
taken in duplicate.
Standard curve. Caffeine stock solution (20 mg caffeine/10 mL,
w/v distilled water) was diluted to 200 mL with distilled water.
Next, 0, 10, 20, 30, 40 and 50 mL of the diluted caffeine solution
were separately mixed, each with 4 mL HCl in a volumetric
flask and diluted to 100 mL with distilled water. Thereafter, the
Pak. J. Sci. Ind. Res. 2010 53 (1) 20-24
Abstract. In the investigation of caffeine and heavy metal contents in four branded and six unbranded tea samples
collected from local markets of Lahore, Faisalabad and Peshawar, the amount of caffeine and heavy metals in all the
branded tea samples were in agreement with the international standards. In unbranded tea samples, though the amount
of caffeine was within standard limits but two of the samples collected from Peshawar had high concentrations of lead
being, 13.69 and 15.78 mg/kg, consumption of which can lead to serious problems.
Keywords: tea, caffeine, heavy metals
*Author for correspondence; E-mail: asimainayat@hotmail.com
20

measuring steps were repeated as described above. Readings
of the absorption of the standard solution against its concen-
tration were used to prepare the standard curve. Following
formula was used to calculate the caffeine contents:
Caffeine (%) = (E/1000) × Vo × (100 × V1) × (50/25) / W
or
= 0.2 EVo /V1 /W
where:
E is caffeine (mg) from the standard curve against the read-
ing of the spectrophotometer
E/1000 is used to convert ‘mg’ into ‘g’,
Vo is the total volume of tea solution (250 mL),
V1 is the volume used for the measurement (10 mL),
100/V1 indicates 10 mL tea solution that were diluted to
1000 mL,
50/25 shows another dilution from 25 mL filtrate made to
50 mL in the measurement and
W is the dry weight of the tea sample.
Heavy metals. Double distilled water and analytical grade
reagents were used in all the experiments. Standard solution
(10 ppm) of heavy metals i.e., Zn, Cu, Cd, Co, Pb, Mn and Ni
were prepared by taking 1 mL of each heavy metal stock
solution (1000 ppm) in a 100 mL volumetric flask and diluted
upto the mark with double distilled water. Heavy metal
working standard solutions i.e., 0.5, 1.0, 1.5 and 2.0 ppm were
prepared by diluting 5, 10, 15 and 20 mL, respectively, standard
solution (10 ppm) of each metal made upto 100 mL with double
distilled water.
The calibration curve used for the determination of heavy
metals in tea samples by flame atomic absorption spectro-
metry (FAAS) was established using the working standard
solutions. For the determination of metals in tea, 3 g of the
oven dried (105 °C) samples were preheated for 30 min at
250 °C and burned for one hour at 800-850 °C. The resulting
ash was wetted with double distilled water and mixed with 10
mL of diluted HCl (1:1). The mixture was mildly boiled, cooled
to room temperature, filtered (if necessary), transferred to
100 mL volumetric flask and diluted with double distilled
water.
A Varian (AA 240) flame atomic absorption spectrophoto-
meter equipped with hollow cathode lamps was used for the
analysis. The instrumental setup was adjusted according to
the manufacturer’s instructions.
Results and Discussion
The spectrophotometric method is the most common
method used for the determination of caffeine. Flame atomic
absorption spectrometry (FAAS) is one of the techniques
most extensively used for determining various elements with
a significant precision and accuracy. This analytical technique
is remarkable for its selectivity, speed and fairly low operation
cost. However, in some cases it is rather difficult to determine
traces of heavy metals in environmental samples due to unsuf-
ficient sensitivity or matrix interferences. Thus, a pre-concen-
tration or/and separation step is necessary.
Caffeine. Caffeine content in different tea samples ranged from
3.41 to 3.74% with a mean of 3.56% of the dry mass (Table 1).
Earlier studies showed that caffeine content in black tea was
affected by clone of plant, season and stage of plucking of
leaves (Obanda and Owuor, 1995). Caffeine content can vary
in tea leaves from 24% to 40% depending on the maturity i.e.,
the young leaves may contain more caffeine than the older tea
leaves (Owuor and Orchard, 1992; Owuor et al., 1987). The
results (Table 1) of the current study showed that the mean
caffeine content in tea varieties marketed in Pakistan was
Table 1. Concentrations of caffeine and heavy metals in branded and unbranded tea samples
Tea sample Caffeine Heavy metals (mg/kg)
(code) (%) Cu Cd Mn Co Ni Zn Pb
Branded (S 1) 3.41 1.20 nd 2.75 0.69 4.58 nd 4.64
Branded (S 2) 3.45 1.05 nd 40.50 0.60 4.58 nd 3.28
Branded (S 3) 3.42 0.75 nd 0.25 0.65 4.59 0.25 4.75
Branded (S 4) 3.46 0.75 nd 39.75 0.61 4.58 1.00 4.28
Unbranded (LHR 1) (S 5) 3.64 0.50 0.25 19.50 0.10 9.71 nd 4.64
Unbranded (LHR 2) (S 6) 3.72 1.0 0.28 15.70 1.29 9.27 0.76 4.59
Unbranded (FSD 1) (S 7) 3.53 0.50 0.25 16.10 1.20 9.98 0.84 4.20
Unbranded (FSD 2) (S 8) 3.49 0.75 0.50 25.75 1.14 8.66 0.25 3.75
Unbranded (PWR 1) (S 9) 3.74 1.45 0.34 25.50 1.02 10.01 nd 13.69
Unbranded (PWR 2) (S 10) 3.73 0.75 0.25 16.50 0.89 9.69 0.75 15.78
nd = not detected.
21
Caffeine and Heavy Metals in Tea
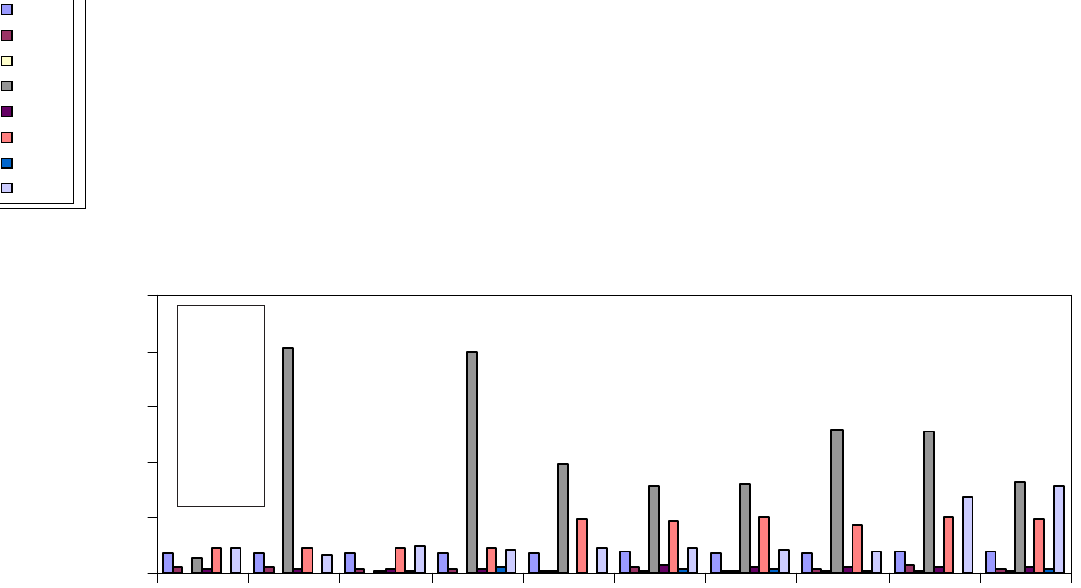
similar to the values reported in literature, 3.32 to 31.81%, and
also that the tea varieties originating from different sources,
with differences in clones, seasons and maturity, differ in
caffein content (Gulati et al., 1999).
Heavy metals. Results of the study show that different tea
samples contain the elements Cu, Cd, Mn, Co, Ni, Zn and Pb
in various proportions (Table 1 and Fig. 1). Variations in
elemental contents from sample to sample can be attributed to
the differences in botanical profile as well as the mineral com-
position of the soil in which plants are cultivated. Other
factors responsible for variation in elemental contents may be
pre-ferential absorbability of the plant, fertilizers, irrigation
water, climatic conditions and, most of all, differences in the
methods of processing and packing of tea leaves. Deposition
of various metals from the vehicular emission and other
sources on the open tea leaves could contribute to differ-
ences in analytical results.
Lead. Lead was found in variable amounts in all tea samples
(Table 1). For example high concentration of lead i.e., 13.69
and 15.78 mg/kg were found in two tea samples (S-9 and 10,
respectively), while in other samples the amount of Pb varied
from 3.28 to 4.75 mg/kg which is lower than the prescribed
limit of WHO (1998) for Pb content in herbs i.e. 10 mg/kg. It is
believed that 95% of Pb present in plants is due to foliar
uptake from the surroundings. As majority of the tea varieties
are imported from other countries, so source plants with
different origins have different air, water and soil environ-
ment. The high concentration of Pb in plants growing above
the ground is due to air borne Pb and the surrounding
polluted environment.
Cadmium. Cd is another toxic metal not required by any
living being. In the four branded tea samples (1, 2, 3 and 4)
Cd was not detected while in other samples its value ranged
from 0.20 to 0.50 mg/kg (Table 1). It may have been introduced
during packaging or the tea plants might have absorbed this
pollutant from the surrounding air and the soil. Thus con-
sumption of such tea for long period of time is dangerous due
to Cd contamination. Cd damages nerve cells; it inhibits the
release of acetylcholine and activates cholinesterase enzyme,
resulting in hyper activity of the nervous system. By altering
calcium and phosphorus metabolism, toxic level of Cd can
contribute to arthritis, osteoporosis and neuromuscular
diseases. In cardio-vascular system, Cd replaces Zn in the
arteries making them brittle and inflexible. Cd accumulates
in the kidneys and once accumulated, it stays there and is not
removed through excretion, resulting in high blood pressure
and kidney diseases.
Copper. In all the tea samples, high Cu concentration was
found. In sample-9, concentration of Cu was 1.45 mg/kg
(Table 1), while in all other samples, its concentration varied
from 0.50 to 1.20 mg/kg. Although Cu is an essential enzymatic
element for normal plant growth and development but at
excessive levels, it can be toxic. Phytotoxicity can occur if its
concentration in plants is higher than 20 mg/kg. Copper
accumulation in body can result in a tendency for hyper-
activity in autistic children. An excess of Cu can cause
stuttering, insomnia and hypertension as well as oily skin,
loss of skin tone (due to blocking of vitamin C absorption)
and dark pigmentation of the skin, usually around the face. Cu
can also make nails brittle and thin and may contribute to hair
loss especially in women.
22 Asma Inayat et al.
Fig. 1. Concentration of caffeine and heavy metals in different tea samples.
Caffine
Cu
Cd
Mn
Co
Ni
Zn
Pb
S-1 S-2 S-3 S-4 S-5 S-6 S-7 S-8 S-9 S-10
Sample number
50
40
30
20
10
0
Conc. (mg/kg)
1
2
3
4
5
6
7
8
123456 78 123456 78 123456 78 123456 78 123456 78 123456 78 123456 78 123456 78 123456 78 123456 78
Caffeine

Zinc. Zn is an important metal required by both plants and
animals. The highest concentrations of Zn (1.0 mg/kg) was
found in one branded sample (sample-4), while in other
samples, its amount varied from 0.25 to 0.84 mg/kg (Table 1).
In few tea samples, its concentration was very low. Zn is very
important for plant and human life. In the blood about 85%
of Zn combines with proteins for the transport of the latter
after its absorption and its turnover is rapid in the pancreas.
Deficiency of Zn causes diabetic hyposmia, hypogensia or
coma.
Nickel. In all the studied unbranded tea samples, higher
amount of Ni was recorded: 10.01 mg/kg in sample-9, followed
by 8.66-9.98 mg/kg in five samples (samples 5, 6, 7, 8 and 10),
while equal amounts of Ni (4.58 mg/kg) was found in all the
branded samples. Ni plays an important role in the production
of insulin in the pancreas. Its deficiency results in disorder of
the liver. However, higher concentration, of Ni have adverse
effects on health. For example Ni tends to accumulate in
kidneys causing kidney damage. Being a common ingredient
in fashion jewellery, Ni can cause allergic reactions in some
wearers; eczema and even asthma attacks may develop. A
steady exposure ot Ni can cause cancer of lungs and nasal
sinus.
Cobalt. As shown in Table 1, the highest concentration of Co
was found in one sample (sample 6, 1.29 mg/kg), followed
by 1.20 mg/kg in sample-7. Other tea samples had a con-
centration of 0.60 mg/kg to 1.14 mg/kg. Thus as a whole,
considerable variations were recorded in all the tea samples
which may be due to the differences in the air, soil and water
environment of the source plants as well as local activities
in their nearby surroundings. Human body needs Co in trace
amounts and it is toxic in higher concentrations. Co in the
form of vitamin B12 is in its physiologically active form.
Manganese. Mn was the most abundant metal found in tea
samples in variable amounts (Table 1). For example high Mn
concentration was found in two branded samples No.2 and 4,
being 40.50 and 39.75 mg/kg, respectively, and 25.75 and 25.50
mg/kg in two unbranded samples, samples 8 and 9, respec-
tively. The least amount of Mn was found in one branded
sample (sample 3) being 0.25 mg/kg. The presence of Mn in
tea samples may be due to its use as a colouring material for
tea leaves.
Daily intake of heavy metals. Due to lack of information about
the maximum allowable levels of heavy metals in tea leaves
the discussion will be extended to the acceptable daily intake
that can be taken into account through foods and drinking
water. For intance, the expected calculated intake of Mn in the
present study was 121.38 mg/day for tea (Table 2). This value
is much higher than the intake through food (0.008) and at the
same time nearly equal to 3.8 mg/day through drinking water
according to the standards of US Environmental Protection
Agency (MAFF, 1997). The calculated overall mean intake of
Ni through tea was 49.39 mg/day. This value is in agreement
with the human requirements (50 mg/day). However, this value
is higher than the average intake in the UK (0.13 mg/day)
recorded for the total diet studied (MAFF, 1999). According
to MAFF (1998) tea beverage has considerable amount of Ni
that could significantly contribute to daily intake of metals.
Calculated intake of Cu through tea was 5.22 μg daily or
0.005 mg daily. This value was much lower than that of 1.2 mg
daily set by UK and WHO (1998).
23
Table 2. Daily intake of different heavy metals
Heavy metal Total (ppm) Intake (mg/day)
Cu 0.87 5.22
Cd 0.18 1.12
Mn 20.23 121.38
Co 0.91 5.46
Ni 7.56 49.39
Zn 0.38 2.31
Pb 6.36 38.16
The mentioned values were calculated based on the assump-
tion that the average consumption of tea beverage for a single
person is three cups a day with one packet of 2 g in a cup, i.e.,
6 g of tea particles/day.
The results show (Table 2) that only small part of heavy metal
content through tea leaves may be introduced into beverage
preparation. All branded and unbranded tea samples were
found to contain heavey metal contents within safe levels
except for only two unbranded samples collected from
Peshawar, which had high concentrations of Pb.
References
Al-Oud, S.S. 2003. Heavy metal contents in tea and herb
leaves. Pakistan Journal of Biological Sciences, 6:
208-212.
Atta, M.B. 1995. Aluminium content in dust black tea leaves
and its beverages. Menofiya. Journal of Agricultural
Research, 20: 137-150.
Gulati, A., Ravindranath, S.D. 1999. Seasonal variations in
quality of kangra tea Amellia sinensis (L.) O kuntze) in
Himachal Pradesh. Journal of the Science of Food and
Agriculture, 71: 231-236.
Caffeine and Heavy Metals in Tea
MAFF, 1999. Total diet study: Aluminium, arsenic, cadmium,
chromium, copper, lead, mercury, nickel, selenium, tin
and zinc. Food Surveillance Information Sheet No. 191.
Ministry of Agriculture Fisheries and Food (1997), UK.
MAFF 1998. Lead, Arsenic and other metals in food. Food
Surveillance Paper No.52. London, UK.
MAFF 1997. Total diet study : metals and other elements.
Food Surveillance Information Sheet No. 131, Ministry
of Agriculture Fisheries and Food, UK.
Mumin, M.A., Akhter, K.F., Abedin, M.Z., Hossain, M.Z. 2006.
Determination and characterization of caffeine in tea,
coffee and soft drinks by solid phase extraction and
high performance liquid chromatography (SPE-HPLC).
Malaysian Journal of Chemistry, 8: 45-51.
Obanda, M., Owuor P.O., Taylor, S.J. 1999. Flavanol compo-
sition and caffeine content of green leaf as quality
potential indicators of Kenyan black teas. Journal of
the Science of Food and Agriculture, 74: 209-215.
Obanda, M., Owuor, P.O. 1995. Changes in black tea quality
chemical parameter due to storage, duration and packa-
ging method. Tea, 16: 34-40.
Owuor, P.O., Orchard, J.E. 1992. Effects of storage time in a
two stage withering process on the quality of seedling
black tea. Food Chemistry, 45: 45-49.
Owuor, P.O., Obanda, A.M., Tsushida, T., Horita, H., Murai, T.
1987. Geographical variation of theaflavin, thearubigins
and caffeine in Kenyan clonal black teas. Food Chemistry,
26: 223-230.
Tautkus, S., Kazlauskas, R., Kareiva, A. 2004. Determination
of copper in tea leaves by flame atomic absorption
spectrometry. Chemistry, 15: 49-52.
WHO, 1998. Guidelines for Drinking Water Quality, 2nd
edition, Addendum to volume 2: Health Criteria and
other Supporting Information, pp. 46-61, World Health
Organization, Geneva, Switzerland.
Yao, L.H., Jiang, Y.M., Caffin, N., Arcy, B.D., Datta, N., Liu, X.,
Singanusong, R. Xu, Y. 2006a. Phenol compounds in tea
from Australian supermarkets. Food Chemistry, 96: 614-620.
Yao, L., Liu, X., Jiang, Y., Caffin, N., Arcy, B.D., Singanusong,
R., Datta, N., Xu, Y. 2006b. Compositional analysis of
teas from Australian supermarkets. Food Chemistry,
94: 115-122.
24 Asma Inayat et al.

Measurement of Atmospheric Concentrations of CO, SO2, NO and NOx
in Urban Areas of Karachi City, Pakistan
Durdana Rais Hashmi*, Farooq Ahmad Khan, Akhtar Shareef, Farhan Aziz Abbasi,
Ghulam Hussain Sheikh and Alia Bano Munshi
Centre for Environmental Studies, PCSIR Laboratories Complex, Sharah-e-Dr. Salimuzzaman Siddiqui,
Karachi-75280, Pakistan
(received May 28, 2009; revised December 8, 2009; accepted December 12, 2009)
*Author for correspondence; E-mail: drhpak@yahoo.com
Abstract. In the assessment of variation trends in ambient air quality at five selected regions of Karachi city, four air
pollutants namely carbon monoxide, sulphur dioxide, nitrogen oxide and nitrogen dioxide were monitored, along with
metrological parameters, for eight consecutive days. The results suggested that all the pollutants were mainly due to
the emissions from motor vehicles and industries, owing to the absence of regulatory laws/standards about ambient air
quality in Pakistan. The results have been discussed with reference to recommendations of the World Health Organiza-
tion for the same.
Keywords: air pollution, industrial emission, vehicular emission, atmosphere
Introduction
Karachi is the largest metropolitan city of Pakistan having an
estimated population of above 10 million. Total amount and
complexity of toxic pollutants in the environment of Karachi are
increasing day by day with the rapid increase of population
and proportional increase of industries, vehicular traffic and open
air garbage burning. Rate of atmospheric pollution is 40 percent
higher in Karachi than the other cities of Pakistan (Qureshi, 1997).
Typical major ambient air pollutants in the urban environ-
ment include CO, SO2, NO, NOx, HC and PM10. CO is
formed during combustion of carbon containing compounds.
It is a toxic gas and its prolonged exposure, even at very low
levels, may adversely affect central nervous system. When
inhaled, it reacts with the haemoglobin of the blood stream
to form carboxy-haemoglobin. CO attaches to haemoglobin
roughly about 210 times more than the oxygen (All
Refer.com, 2005). SO2 is also generated by the combustion
of high sulphur fuels. SO2 is toxic to human body especially
for persons having previous history of respiratory diseases,
such as emphysema; besides, it also causes pneumonia.
Nitrogen oxides are generated at high temperatures during
combustion. Their ultimate effect on human beings is still
not clearly understood, but they act as irritants to breathing
and create discomfort to eyes and also destroy the celia in
the respiratory system.
Present study was carried out in various industrial, residential/
commercial and down-town regions of Karachi city to gene-
rate base-line data on these localities by air pollution moni-
toring analyzers, to identify major sources of air pollution and
suggest their remedial measures. The data so generated may
assist in the formulation of the country air quality standards.
Information about the industries was obtained from different
civic agencies and the Department of Industries.
Materials and Methods
The subtropical city of Karachi is located in a semiarid zone.
It is the largest industrial and commercial centre in Pakistan
and declared as one of the twenty mega cities of the world
(Mage et al., 1996). Growing urban population, industrializa-
tion and traffic congestion are the main causes of air pollution
in Karachi city. In order to assess the load of air pollutants in
the environments of the city, monitoring of different air
pollutants was carried out at five different locations (as shown
in location map) of the city, categorized as follows:
1- Region A: the site with urban background, moderately
populated, having low vehicular traffic density. It is
one km distant from the main super highway. The area
around the sampling site is sparsely populated.
2- Region B: a commercial site, densely populated, having
high vehicular traffic density. This site is the busiest
intersection of Karachi, surrounded by multistoried
commercial as well as residential buildings. The popula-
tion around this site mostly belongs to high income group.
3- Region C: an industrial area in district South of Karachi,
with nearly 2000 different types of industries, approxi-
mately 60 percent comprising of textile mills, while
Pak. J. Sci. Ind. Res. 2010 53(1) 25-29
25

other industries are related to pharmaceuticals, chemi-
cals, detergents, iron and steel, sulphur refining, vegetable
oils, beverages and food products.
4- Region D: an industrial area in district East of Karachi
with approximately 2000 various types of industries
including tanneries (more than 100 units), pharmaceuti-
cal, textile and chemical units and refineries etc.
5- Region E: also an industrial area in district East of
Karachi with 300 industrial units of different catego-
ries including textile, food, chemical, pharmaceutical
and engineering units.
Measurement of major ambient air pollution components
such as CO, SO2, NO and NO2 was carried out in summer
season, for eight consecutive days at each of the five
stations. Average variations of the pollutants were
recorded for 11 hours at hourly intervals, at the selected
regions (A-E).
Air quality measurements were performed, using air ana-
lyzer, designed and fabricated by Environmental SA,
France. Average values of CO, SO2, NO, NO2 and NOx
concentrations for 15 min were used for determining daily
average hourly concentrations. The daily hourly average
concentration values were further averaged for determin-
ing values for 8 days and for time weighted average (TWA)
values for 1 h, 8 h and 24 h for each region.
UV fluorescent SO2 Analyzer Model AF21 M consisted of
zinc ray UV lamp with stabilized power supply, continuous
energy monitor and compensation for measurement at
constant energy level and integrated carbon kicker for
continuous removal of interfering hydrocarbons.
The chemiluminescent NO-NOx Analyzer Model AC 31M
was of two channel type coupled with serial R232 output
signal processing and continuous zero control by the micro-
processor. The air sampled by a pump placed at the circuit
end, is carried, on the one hand via a converter oven towards
the NOx chamber and on the other hand, directly into NO
chamber. The radiation emitted in the NOx chamber is
proportional to NO+NO2 (reduced to NO).
Concentration of carbon monoxide was measured by Snift
CO Analyzer (Model 50). The meter was kept at about 1.2 m
26 Durdana Rais Hashmi et al.
Location map: sample collection points
above the ground level and readings were taken at intervals
of 15 minutes.
Results and Discussion
Hourly average variations of the pollutants, recorded at five
selected regions A, B, C, D and E are graphically presented in
Fig. 1 to 5. Table 1 gives the time weighted average (TWA)
values for 1, 8, and 24 h, along with permissible ambient air
quality limits, recommended by WHO.
Maximum average concentration of CO was found to be 4,
21, 11, 9, and 6 ppm in regions A, B, C, D and E, respectively
(Fig. 1). The main source of CO at regions A and B may be
motor vehicles plying on nearby main super highway and
University Road, where traffic density is high, whereas, at
regions C, D and E, the combustion of fuels in nearby indus-
tries and power generation plants.
Maximum hourly average concentrations of SO2 were 5.7,
349.1, 74.5, 42.2 and 21.2 ppb at the regions A, B, C, D, and
E, respectively, the highest concentration of SO2 being
349.1, recorded at region B (Fig. 2). The main cause of this
high concentration of SO2 at this region may be very high
traffic density due to narrow and congested roads, surrounded
by high rising buildings. At region A, the main source of
27
Inorganic Ion Concentrations in Urban Karachi Atmosphere
SO2 is considered to be the vehicular traffic, whereas, at
regions C, D, and E, the combustion of fuels in the nearby
industries.
The highest average concentrations of NO were found to be
17.2, 231.3, 193.5, 157.4 and 132.5 ppb (Fig. 3), and those of
NO2, 9.8, 127.3, 122.2, 103.2 and 79.2 ppb (Fig. 4), whereas
the highest average concentrations of NOx were found
to be 27.0, 358.6, 315.7, 260.6 and 211.7 ppb (Fig. 5) in the
selected regions A, B, C, D and E, respectively.
Table 1. Concentration of CO, SO2 and NO2 evaluated for 1, 8 and 24 h (TWA) and permissible limits of WHO.
Pollutants Time weighted average (TWA) values Averaging
Region A Region B Region C Region D Region E WHO Unit time
CO 3 12.25 6.7 3.7 3.5 30 mg/m31 h
2.1 15.75 8.8 4.1 3.8 10 8 h
SO24 159.1 42.1 30.6 15.9 350 μg/m31 h
100-150 24 h
NO25.6 79.4 47.7 35.9 32.9 400 μg/m31 h
150 24 h
Fig. 2. Hourly average SO2 concentrations in regions A-E.
Fig. 3. Hourly average NO concentrations in regions A-E.
Fig. 1. Hourly average CO concentrations in regions A-E.
25
20
15
10
5
08:00 9:00 10:00 11:00 12:00 1:00 2:00 3:00 4:00 5:00 6:00 7:00
A
B
C
D
E
3
13
7
6
4
4
21
9
8
5
2
10
5
4
3
2
11
6
5
3
4
13
9
6
4
3
15
10
7
6
2
19
7
5
3
4
11
5
3
2
2
13
7
5
3
3
17
9
7
4
3
19
11
6
5
4
21
10
9
6
Local time in hr
1000
100
10
18:00 9:00 10:00 11:00 12:00 1:00 2:00 3:00 4:00 5:00 6:00 7:00
A
B
C
D
E
3.4
175.2
29.1
41.3
10.2
5.7
349.1
58.9
15.2
21.1
3.9
123.6
31.1
15.7
6.7
2.9
142.1
19.3
13.9
6.7
5.1
179.6
29.6
39.6
10.2
2.1
202.4
34.2
24.1
13.4
2.9
221.3
36.7
21.5
11.9
4.3
140.6
23.6
16.3
9.2
2.1
197.2
41.9
20.1
8.1
1.7
213.7
67.3
42.2
17.6
3.1
250.9
74.5
34.4
21.2
2.8
249.1
61.3
27.1
19.3
Local time in hr
8:00 9:00 10:00 11:00 12:00 1:00 2:00 3:00 4:00 5:00 6:00 7:00
A
B
C
D
E
13.5
167.5
182.2
131.4
105.6
10.1
152.9
193.5
112.2
91.3
9.6
135.3
112.3
98.6
80.5
9
117.6
100.4
80.5
61.4
8.6
96.2
79.1
61.3
46.7
8.1
84.8
68.8
53.5
41.6
7.5
80.5
63.7
51.2
40.2
6.7
95.1
60.4
47.8
38.1
7.9
112.7
81.3
64.5
51.1
9.2
130.5
112.3
97.2
82.6
12.3
119.3
134.4
123.3
107.9
17.2
231.3
186.5
157.4
132.5
Local time in hr
250
200
150
50
0
28 Durdana Rais Hashmi et al.
Fig. 5. Hourly average NOx concentrations in regions A-E.
In region B, the pollution generation is mainly due to vehicu-
lar traffic. This site is the busiest intersection on M.A. Jinnah
Road having high traffic density and traffic jams with high-
rising buildings on both sides of the road creating tunnel
effect. On the contrary, in regions C,D, and E, the
pollution generation may be due to the emissions from
nearby industries, power generation plants and boilers of the
industries.
Sulphur dioxide originates mostly from the combustion of
trace amounts of inorganic and organic sulphur, contained in
the fuel. The estimated background concentration of SO2 is
0.2 ppb and calculated atmospheric residence time is 4 days
(Bhatia, 2005). Short term high level of SO2 may enhance
respiratory diseases, lung function disturbance and even
mortality in adults and children (Nautiyal et al., 2007). The
maximum average concentrations of SO2 at regions A and B
were found to be 5.7 and 349.1 ppb, respectively, during 7.00
to 9.00 a.m. At these stations, the high concentration of SO2
may be due to the fuel combustion by vehicular traffic. At
regions C, D and E the highest values of SO2 were 74.5, 42.2
and 21.2 ppb, respectively, at 6:00 p.m. The main source of
SO2 at station C may be power plants and boilers of the indus-
tries and at stations D and E, a large oil refinery, all located in
SW directionof the areas. Relatively high concentration of
SO2 obtained at regions C, D, and E, during specified period
may be due to emissions from the nearby industrial units.
Nitrogen oxides are of great concern, being precursors in
ozone production in the presence of sun light. NO and NO2
are emitted together from combustion sources and exist in
equilibrium in the atmosphere; together, they are usually
referred to as NOx. The diurnal pattern of NO and NOx
has correlation with solar energy. A distinct photochemical
relation between NO, NOx and solar energy has been estab-
lished and as the solar energy increases during the day time,
the level of NO, NOx decreases. The reaction of photochemi-
cal oxidants has a time scale of one to a few minutes (Clark,
1988). At region C, the highest concentrations of NO and
NOx were found to be 193.5 ppb and 306.1 ppb, respectively,
between 7.00 to 9.00 a.m. which may originate from the com-
bustion of industries and power plants, about 45-60 meters
away. The reaction is therefore, even more rapid here, having
a time scale of only few seconds. The chemical reaction
between the two mixing species was not completed due to
time lag and shows high concentration of NO and NOx
during the day time. However, at regions A, B, D and E, the
highest values of hourly average concentration of NO and
NOx were found before the sunrise which started decreasing
as the ultra-violet radiations from the sun increased, and again
increasing with the decrease of ultra-violet radiations from
the sun. The main contributor of NO and NO2 at regions A
and B were the emissions from the vehicular traffic due to
Carbon monoxide is not easily detected by olfactory senses.
High concentration of this pollutant in central parts of the
cities due to traffic jams may creat serious problems. (ALA,
2000; WDNR, 2000). Exposure to carbon monoxide may lead
to head ache, tiredness, dizziness, nausea, vomiting and
drowsiness and in very acute situations, to unconsciousness
and even death (Malakootian and Yaghmaeian, 2004). Expo-
sure to elevated carbon monoxide level is associated
with impairment of visual perception, work capacity, manual
dexterity, learning ability and performance of complex tasks
(Aziz and Qureshi, 2003).
In region A, the highest hourly average concentration of CO
was recorded to be 4 ppm, from 7:00 to 9:00 a.m. and from
5:00 to 7:00 p.m. In the morning, the movement of traffic is
down town and is reverse in the evening. Variations in the
concentrations of carbon monoxide show that the concentra-
tion gradually increases till 9:00 a.m. and then comes down at
1:00 p.m. and again increases around 6:00 p.m. which are the
rush hours. In region A, the air pollution was generated by
vehicular traffic as the air currents were coming from main
Super Highway that has quite high traffic density.
Fig. 4. Hourly average NO2 concentrations in regions A-E.
8:00 9:00 10:00 11:00 12:00 1:00 2:00 3:00 4:00 5:00 6:00 7:00
A
B
C
D
E
5.2
96.4
103.5
71.2
57.5
4.1
90.1
122.2
64.2
50.3
3.3
81.2
63.3
98.6
80.5
3.1
69.1
52.5
40.3
31.4
2.7
61.6
41.6
30.5
23.7
2.1
54.5
36.8
27.6
20.8
1.8
52.1
32.1
23.2
20.1
3.1
51.5
30.6
27.1
17.2
4.2
67.7
41.4
36.7
24.6
6.4
91.3
58.2
47.6
32.7
7.9
116.5
101.2
84.7
59.9
9.8
127.3
119.6
103.2
79.2
Local time in hr
140
120
100
80
60
40
20
0
8:00 9:00 10:00 11:00 12:00 1:00 2:00 3:00 4:00 5:00 6:00 7:00
A
B
C
D
E
18.7
263.9
285.7
202.6
163.1
14.2
243
315.7
176.4
141.6
12.9
186.7
152.9
120.8
122.7
12.1
186.7
152.9
120.8
92.8
11.3
157.8
120.7
91.8
70.4
10.2
139.3
105.6
81.1
62.4
9.3
132.6
95.8
74.4
60.3
9.8
146.6
91
74.9
55.3
12.1
180.4
122.7
101.2
75.7
15.6
221.8
170.5
144.8
115.3
20.2
307.8
235.6
208
167.8
27
358.6
306.1
103.2
79.2
Local time in hr
400
350
300
250
200
150
100
50
0
combustion of fuel, whereas, at regions D and E, the combus-
tion gases emitted by the industries.
A comparison of the time weighted average values of all
the measured pollutants at the selected stations, with those of
the WHO recommended air quality guidelines shows that
the concentrations of ambient air pollutants found at these
stations are well within the WHO limits.
Air pollution has become a world wide public health prob-
lem, particularly in large towns and cities of the developing
countries where people are commonly exposed daily to very
high levels of pollution for 3-7 hours for the last many years
(Engel et al., 1998). Effect of air pollution on human health
varies according to the intensity, duration of exposure and
health status of exposed population. Air pollution increases
the risk of chronic obstructive pulmonary diseases and acute
respiratory infections in childhood, lung and chest cancer,
tuberculosis, prenatal outcomes including low birth weight
and eye diseases. The worst affected age group had been
between 50-60 years, followed by the lower age group of
45-55 years (Maddission, 1997).
Conclusion
The baseline data generated for major ambient air pollutants
at different urban sites of Karachi show that the concentration
of ambient air pollutants such as CO, SO2, NO and NOx, are
all within WHO threshold limits. The values recorded
indicate that all the pollutants are emitted by the industries
and motor vehicles. It is expected that the generated data
will play a part in laying the foundation for developing appro-
priate ambient air quality standards for Pakistan and their
implementation.
References
AllRefer.com 2005. Hemoglobin derivatives. Available at:
http://health.allrefer.com/health/hemoglobinderatives-
info.html.Accessed April 6, 2005.
ALA, 2000. Carbon Monoxide and the Environment,
American Lung Association,USA, available at: http;//
www.lunguas.org/air/carbon.
Aziz, J.A., Qureshi, T.A. 2003. Measurement of ambient
particulate matter and carbon monoxide in Peshawar.
Science Technology and Development, 22: 1-4.
Bhatia, S.C. 2005. Environmental Pollution and Control in
Chemical Process Industry, Delhi, pp. 104, published by
Khanna Publishers, Nai Sarak, India.
Clark, .P.A. 1988. Mixing models for simulation of plume
interaction with ambient air. Atmospheric Environment,
22: 1097-1106.
Engel, P., Hortodo, E., Ruel, M. 1998. Smoke Exposure of women
and young children in highland Guatemala, predictions
recall Accuracy. Human Organization, 54: 408-417.
Mage, D., Ozolins, G., Peterson, P., Webster, A., Orthofer, R.,
Vandeweerd, V., Gwynne, M. 1996. Urban air pollution
in megacities of the world. Atmospheric Environment,
30: 681-686.
Malakootian, Yaghmaeian, K. 2004. Investigation of carbon
monoxide in heavy traffic intersections of Municipal dis-
tricts. International Journal of Environmental Science
and Technology, 1: 227-231.
Maddission, D.A. 1997. Meta Analysis of Air Pollution Epi-
demiological Studies, London Centre for Social and Eco-
nomic Research on the Global Environment, University
College London, UK.
Nautiyal, J., Garg, M.I., Kumar, M.S., Khan, A.A., Thakur,
J.S., Kumar, R. 2007. Air pollution and cardiovascular
health in Mandi Gobindgarh, Punjab, India, -A pilot study.
International Journal of Environmental Research
and Public Health, 4: 268-282. (doi10.3390/)
Qureshi, O.R. 1997. Atmospheric pollution in Karachi 40
percent higher than other cities. Frontier Post, June 07,
Karachi.
WDNR, 2000. Carbon monoxide sources and health
effects. Wisconsin Department of Natural Resources,
USA, (http://www.dnr.state.wi.us).
29
Inorganic Ion Concentrations in Urban Karachi Atmosphere

Seasonal and Year Wise Variations of Water Quality Parameters in the
Dhanmondi Lake, Dhaka, Bangladesh
Shamshad Begum Qureshi
Chemistry Division, Atomic Energy Centre, Dhaka-1000, Bangladesh
(received April 18, 2008; revised December 11, 2009; accepted December 18, 2009)
Introduction
Over the past 25 years, the quality of water bodies around
Dhaka city has deteriorated a lot due to unplanned
discharge of untreated effluents from factories and sewage.
Dhanmondi Lake is one of the biggest lake and a great recrea-
tion place for the people of Dhaka city. But, this Lake is being
contaminated due to the increase in human activities
during the last few years. Lot of construction work had also
been done during 1980 to 2006 along the valley of this Lake,
which had directly influenced the water quality of the Lake.
In addition, frequent floods during recent years have also
contributed in polluting the lake water. The number of tourists
has also increased in recent years, directly affecting the
quality of water. Fishing activities around the lake are another
source of contamination. Thus, a constant and systematic
monitoring is essential to study long term pollution in the
Lake environment especially when it is impacted by the
increasing tourist population which disturbs normal
activities in the area. Some short-term research work had been
carried out in the past on water quality parameters of the river,
and lake water in our laboratory (Quraishi et al., 2006; Azim
2005; Chowdhury et al., 2005; Hossain, 2005; Hadi et al., 1996;
1991; Maroof et al., 1985). But long-term monitoring is neces-
sary to evaluate the pollution sources and to get a clear trend
of pollution. Therefore, a long-term monitoring program was
initiated in 2002 spread over a period of 5 years during 2002-
2007, for a wide range of water quality parameters. The main
objectives of this work were (i) to establish background levels
for Mn, Fe, Zn, Cr, Ni, Co, Cu, Cd, and Pb in the lake water of
Dhaka City and (ii) to examine the seasonal and year-wise
variability of trace metals in lake water on seasonal basis.
Trace toxic metals and physicochemical parameters e.g., Pb,
Cd, Cr, Ni, Co, Cu, Fe, Mn, Zn, EC, pH, Cl, F, SO4, PO4, CN, NO3
concentrations were monitored three times at three different
locations over a period of five years between March 2002 and
September 2007; data for the year 2003 is not available.
Materials and Methods
Reagents. All chemicals were of analytical reagent grade.
HNO3, HCl and H2SO4 were of analar grade from BDH
Laboratories. Certified reference material was obtained from
the National Institute of Standards and Technology, USA.
Commercially available 1000 mg/L (ICP grade) single element
standard solutions (Merck or SPEX Certiprep, Metuchen, NJ,
USA) were used in preparation of the working standards. Stan-
dard solutions were freshly prepared from 1000 ppm stock by
dilution with deionized water (DI ).
Sample collection, preparation and analysis. Water samples
were collected from three locations thrice in a year between
March 2002 and September 2007. Sampling was done in March
(pre-monsoon), July (monsoon) and September (post-mon-
soon). The locations of the three sampling sites are shown in
Fig. 1.
Each sample was divided into two portions, one for the
analysis of metals ions and another for that of anion. pH of
the portion for the analysis of metals was adjusted below 1 by
addition of nitric acid to prevent adsorption to the bottle and
the portion of anions was filtered, using Whatman filter #41 to
remove suspended matter and stored at 4 °C. Water sample
(250 mL) was quantitatively transferred to 250 mL beaker and
then heated on a hot plate with 2 mL of HNO3 until the total
volume was reduced to approximately 5 mL. The concentrate
Abstract. The quality of the surface water through 16 physicochemical variables was monitored at three sites of
Dharmondi Lake of Dhaka, Bangladesh, over 5 years during 2002-2007. The concentration of heavy metals (Pb, Cd, Cr,
Co, Ni, Cu) was below detection limits with few exceptions. No clear seasonal variation trend for Fe, Mn, Zn, PO43-, SO42,
Cl- and F- was observed which differed from year to year. Slight increasing tendency in case of sulphate, phosphate,
chloride concentrations and electrical conductivity was observed but it was not clear in other parameters. The levels of
all parameters were found well below the standards for drinking water.
Keywords: lake water, seasonal variability, pollution trend, water quality, heavy metals
Pak. J. Sci. Ind. Res. 2010 53(1) 30-36
E-mail: mumu3222@yahoo.com
30
obtained was transferred to a 10 mL volumetric flask and made
up to volume.
Electrical Conductivity (EC) and pH were measured using
Jenway Conductivity Meter, model No. 4070 and WTW
Multiline P4 Universal Meter, respectively. Concentration of
anions (Cl-, F-, NO3-) were determined by ion selective elec-
trode (ISE). For fluoride determination, 1:1 (sample: TISAB)
low level TISAB was used whereas for chloride and nitrate,
2% of 5M NaNO3 and 2% of 2M (NH4)2SO4 were used, respec-
tively, as ionic strength adjuster (ISA) according to the users
manuals (Quraishi et al., 2006; Chowdhury et al., 2005). The
concentrations of chloride, fluoride and nitrate in samples
were measured by the ISE method based on direct calibration.
50 mL of each of the calibration standard solutions (0.01, 0.10,
1.0, 10.0, 100 ppm) were taken in a 100 mL beaker and the
required amount of ISA buffer was added to it. The electrode
potentials (mV) of the standards were measured using the
digital ion-selective electrode meter (Orion Ionalyzer/model
470 A). After measurement, a calibration curve was drawn by
plotting electrode potentials of the standards against their
respective concentrations. Then the target electrode was
connected to the meter for determination of target anion in the
real samples similarly treated as the standards. From the cali-
bration curve constructed by the instrument as mentioned
above, the slope was found to be -57±3 mV/decad. The com-
puter code in the instrument provided the concentration of
anion in the sample directly by carrying out the calculations
based on the calibration factor (slope: -57±3 mV/decad) and
the electrode potential value of the sample. SO42- and PO43-
concentration were measured by Shimadzu 1201 uv-visible
spectrophotometer. After digestion, CRM and samples were
analyzed for metal content using a three point calibration by
atomic absorption spectrophotometer (Perkin Elmer 3110
and 560, USA). Standard solutions were prepared from single
element standards in acid media matching with sample
solution. Calibration curve was constructed for all elements
using at least three different concentrations. Very good R
square value of 0.9995 was obtained for all the elements.
Quality assurance. Quality assurance measures included the
calculation of method detection limit, recovery and analysis
of standard reference materials. To determine the DL, a low
concentration standard solution was analyzed several times
and the standard deviation (δ) was calculated for the data.
The detection limits of the method were calculated using 10 δ,
recommended by IUPAC, including preconcentration factors
for the elements Pb, Cd, Cr, Ni, Co, Cu, Fe, Mn, Zn, as 10.0,
4.46, 5.07, 5.30, 1.50, 3.38, 3.0, 4.0 and 12.0 μg/L, respectively.
The accuracy of the method was checked by recovery assays
of known amounts of analyte added to the samples. The
recovery values obtained for spiked samples were 95-102 % in
case of all elements. Procedural blanks were used throughout
the sample preparation and analysis to evaluate contamina-
tion from reagents, containers etc. and the procedures were
validated by triplicate analysis of water samples, reference
material and blanks.
Results and Discussion
The baseline-monitoring programme was initiated in 2001 to
monitor heavy metals in different lakes in Dhaka city by the
Table 1. Analytical results for analysis of NIST SRM 1643d
and 1640
Elements Method NIST certified Measured Trueness
values for SRM values (%)
1643d, (μg/L) (μg/L)
Fe AAS 91.2 ± 3.9 91.5 ± 0.94 100
Mn AAS 37.66 ± 0.83 35.55 ± 0.36 94.4
Zn AAS 72.48 ± 0.65 69 ± 1.86 95.2
Cu AAS 20.5 ± 3.8 21.77 ± 0.22 106
Cd AAS 6.47 ± 0.37 7 ± 0.07 108
Cr AAS 18.53 ± 0.20 18.75 ± 0.19 101
Ni AAS 58.1 ± 2.7 54.54 ± 0.56 93.9
NIST SRM 1640, Trace elements in natural water (μg/L)
Cu AAS 85.2 ± 1.2 80.28 ± 0.81 94.2
Mn AAS 121.5 ± 1.1 129.12 ± 1.31 106
Fig. 1. Map of Dhanmondi lake
31
Seasonal Quality Variations of Dhanmondi Lake Water
E
NS
W
DP3
DP1
DP2
Map of D............. lake
32
0
0.2
0.4
0.6
0.8
1
2002 2004 2005 2006 2007
Year
PO4 (mg/l)
Mar
July
Sept
0
10
20
30
40
50
60
2002 2004 2005 2006 2007
Year
Cl (mg/L)
Mar
July
Sept
0
10
20
30
40
50
2002 2004 2005 2006 2007
Year
SO4(mg/l)
Mar
July
Sept
0
0.1
0.2
0.3
0.4
2002 2004 2005 2006 2007
Year
F (mg/l)
Mar
July
Sept
0
20
40
60
80
2002 2004 2005 2006 2007
Year
Mn (ug/l)
Mar
July
Sept
0
0.2
0.4
0.6
0.8
1
1.2
2002 2004 2005 2006 2007
Year
Fe (mg/l)
Mar
July
Sept
0
20
40
60
80
100
2002 2004 2005 2006 2007
Year
Zn (ug/l)
Mar
July
Sept
0
100
200
300
400
500
2002 2004 2005 2006 2007
Year
EC ( uS/cm2)
Mar
July
Sept
Fig. 2. Seasonal variations of different parameters of the Lake water during five years (2002-2007).
PO4 (mg/L)
Cl (mg/L)
Fe (mg/L)
Zn (μg/L)
SO4 (mg/L)
F (mg/L)
Mn (μg/L)
EC (μS/cm2)
Year
Year
Year
Year
Year Year
Year
Year
Shamshad Begum Qureshi
33
Seasonal Quality Variations of Dhanmondi Lake Water
0
0.2
0.4
0.6
0.8
1
2002 2004 2005 2006 2007
Year
Annual Arithmeti
c
Mean (mg/l
PO4
30
32
34
36
38
40
2002 2004 2005 2006 2007
Year
Annual Arithmeti
c
Mean (mg/l
Cl
0
10
20
30
40
50
60
2002 2004 2005 2006 2007
Year
Annual Arithmeti
c
Mean (ug/l
Mn
0
0.2
0.4
0.6
0.8
1
2002 2004 2005 2006 2007
Year
Annual Arithmetic
Mean (mg/l
Fe
0
10
20
30
40
2002 2004 2005 2006 2007
Year
Annual Arithmetic
Mean (mg/l
SO4
0
0.05
0.1
0.15
0.2
0.25
0.3
2002 2004 2005 2006 2007
Year
Annual Arithmetic Mean
(mg/l)
F
Fig. 3. Year wise variations of different parameters of Lake water during five years (2002, 2004-2007).
0
0.2
0.4
0.6
0.8
1
2002 2004 2005 2006 2007
Year
Annual Arithmetic
Mean (mg/l
Fe
0
10
20
30
40
50
2002 2004 2005 2006 2007
Year
Annual Arithmetic Mean
(ug/l
Zn
Annual arithmetic
mean (mg/L)
Annual arithmetic
mean (mg/L)
Annual arithmetic
mean (mg/L)
Annual arithmetic
mean (mg/L)
Annual arithmetic
mean (mg/L)
Annual arithmetic
mean (μS/cm2)
Annual arithmetic
mean (μg/L)
Year
Year
Year
Year
Year
Annual arithmetic
mean (μg/L)
2002 2004 2005 2006 2007
2002 2004 2005 2006 2007
Year
2002 2004 2005 2006 2007
Year
40
38
36
34
32
30
0.3
0.25
0.2
0.15
0.1
0.05
0
40
30
20
10
0
1
0.8
0.6
0.4
0.2
0
Year
PO4
SO4
Cl
1
0.8
0.6
0.4
0.2
0
1
0.8
0.6
0.4
0.2
02002 2004 2005 2006 20072002 2004 2005 2006 2007
EC
34
0
10
20
30
40
50
Sept
2002
July
2004
March
2005
Sept
2005
Sept
2006
Sept
2007
So4
DP 1
DP 2
DP 3
0
10
20
30
40
50
60
70
Sept
2002
July
2004
March
2005
Sept
2005
Sept
2006
Sept
2007
Cl
DP 1
DP 2
DP 3
0
20
40
60
80
100
120
Sep t
2002
July
2004
March
2005
Sep t
2005
Sep t
2006
Sep t
2007
Mn
DP 1
DP 2
DP 3
0
0.5
1
1.5
2
Sept
2002
July
2004
March
2005
Sept
2005
Sept
2006
Sept
2007
Fe
DP 1
DP 2
DP 3
0
0.2
0.4
0.6
0.8
1
1.2
1.4
Mar
2002
Sept
2002
July
2004
March
2005
Sept
2005
Sept
2006
Sept
2007
po4
DP 1
DP 2
DP 3
0
0.1
0.2
0.3
0.4
Sept
2002
July
2004
Mar ch
2005
Sept
2005
Sept
2006
Sept
2007
F
DP 1
DP 2
DP 3
0
20
40
60
80
100
120
Sep t
2002
July
2004
March
2005
Sep t
2005
Sep t
2006
Sept
2007
Zn
DP 1
DP 2
DP 3
0
100
200
300
400
500
Sept
2002
July
2004
March
2005
Sep t
2005
Sept
2006
Sept
2007
EC
DP1
DP2
DP3
Fig. 4. Variations in different parameters of water between the sampling locations of the Lake (DP1, DP2 and DP3).
SO
4
PO4
0.4
0.3
0.2
0.1
0
F
Sept July March Sept Sept Sept
2002 2004 2005 2005 2006 2007
Sept July March Sept Sept Sept
2002 2004 2005 2005 2006 2007
Sept July March Sept Sept Sept
2002 2004 2005 2005 2006 2007
Sept July March Sept Sept Sept
2002 2004 2005 2005 2006 2007
Sept July March Sept Sept Sept
2002 2004 2005 2005 2006 2007
Sept July March Sept Sept Sept
2002 2004 2005 2005 2006 2007
Fe
Zn
500
400
300
200
100
0
Shamshad Begum Qureshi
Cl
Mn
EC
Chemistry Division of Atomic Energy Centre. Analytical
results for analysis of NIST SRM 1643d and 1640 are summa-
rized in Table 1. Good agreement was found between the
measured and the certified values. The data of heavy metals
and various physical parameter of water for 2002 and 2004-
2007 (five years) are presented in Fig. 2-4. It was observed
that during the study period (2002 and 2004-2007), the levels
of various parameters including toxic metals were much lower
than the drinking water standard of Bangladesh. Annual mean
concentrations of phosphate in 2006 and 2007 were 0.45 and
0.81 mg/L, respectively, (Fig. 3) (MEF,1997) which were much
lower than the standard set as 6 mg/L. The mean fluoride
concentrations (0.20 mg/L) observed during the study period
was also much lower than the standard, which is set at 1.0 mg/L.
Mean sulphate concentration in Dhanmondi lake water
was 17.5 mg/L which was much lower than the drinking water
standard (400 mg/L). Concentrations of Pb, Cd, Cr, Co, Ni,
known as toxic metals, were found to be below the method of
detection limits. The levels of Fe, Mn and Zn, which are known
to be the essential elements, were found to be in the range of
60-1000, 11.6-60 and 3.2-89 μg/L, respectively, the levels of
Mn and Zn were much lower than the Bangladesh drinking
water standards (Mn: 100 μg/L and Zn: 5000 μg/L) but were
found much higher as compared to those of other lakes (Nojiri
et al., 1985).
Seasonal variations. Seasonal variations of phosphate,
sulphate, chloride, fluoride, electrical conductivity, iron,
manganese and zinc in the lake water are shown in Fig. 2
which were found to be inconsistent. Fluoride level in
September 2002, was markedly high as compared to those
in March and July. In the year 2004, a systematic decrease
of fluoride was noticed during March to September. In case
of phosphate, concentration was higher in March (pre-
monsoon) than in the July (rainy season) in the year 2002.
But in the year 2005, just reverse trend was observed and
in the year 2006, the highest concentration was found in
March and the lowest in September. Therefore, it could be
concluded that seasonal variations of phosphate, sulphate,
chloride, fluoride, electrical conductivity, iron, manganese
and zinc were not consistent (Fig. 2). This inconsistency
indicated that there is no particular or permanent source of
contaminants but sudden contamination events appear
which cannot be related to the local pollution events or
other local anthropogenic origins but rather to the influ-
ence of tourism-related activities (Topalian et al., 1999).
Contamination by metals can be related to the metallic
objects used in fishing activities, metallic containers and/
or packing materials of food which are directly thrown
into the lake by the tourists and thus contamination can
increase with the increase in the number of tourists (Conde
and Garcia - Montelongo, 2004).
Year wise variations. Time course changes in the concentra-
tions of phosphate, sulphate, chloride, fluoride, electrical con-
ductivity, iron, manganese and zinc in the lake water are shown
in Fig. 3. Phosphate concentration decreased till 2005 and
increased again in 2006 and 2007. Cl- level increased from
2002 to 2004, decreased in 2005 and again increased in 2006
and fell in 2007. Sulphate concentration significantly decreased
from 2002 to 2005 and again remarkably increased in 2006 and
2007. Fluoride concentration fell from 2002 to 2004 and
remained almost constant until 2007. Zinc concentration
increased from 2002 to 2005 and then sharply decreased in
2006 followed by 2007. On the other hand, iron concentration
showed a zigzag pattern as shown in Fig. 3. In case of
manganese, concentration increased till 2006 and then
suddenly decreased in 2007. An increasing tendency was
observed for EC from the year 2004 to 2007.
Variations between locations. The distribution of different
parameters was not uniform in surface water of Dhanmondi
Lake. Significant variations in iron, manganese, zinc and
phosphate concentrations were observed with regard to
sampling stations throughout the study period (Fig. 4).
Therefore, it was very difficult to find out a particular sam-
pling station having the highest or the lowest contamination
point in a particular season during the monitoring period. It
also indicates that contamination sources are not fixed and
these differences can be related to tourist activities. It can be
also seen in this figure that no significant variations
between the sampling stations were observed in case of
sulphate, chloride, fluoride and electrical conductivity.
Conclusion
This study was focused on the evaluation of water quality of
Lake and consequently on the determination of the pollution
level of this aquatic environment. Seasonal variations did not
show any clear pattern and were difficult to explain. A possible
explanation might be made based on the fact that the lake is
acceptor of both regular and non-regular pollution pulses. An
increasing tendency was observed in case of electrical conduc-
tivity, phosphate, chloride and sulphate, whereas the level was
unchanged in case of fluoride during the last four years of the
study. Concentration of zinc, iron and manganese decreased in
2007 as compared to the previous year.
References
Azim, A. 2005. Study on Trace Elements and other Micronutri-
ents in the Water Samples from the Rivers Buriganga and
35
Seasonal Quality Variations of Dhanmondi Lake Water
Turga. M.Sc Thesis, 127 pp. University of Dhaka,
Bangladesh.
Chowdhury, M.N., Akhter, S., Khan, M.M.K., Quraishi, S.B.
2005. Studies on water quality parameters in flood
water. Journal of Bangladesh Academy of Sciences, 29:
195-200.
Conde, J.E., Garcia-Montelongo, F.J. 2004. Heavy metals in
the coastal environment of Las Galletas, Tenerife, Canary
Islands, Spain. Bulletin on Environmental Contamina-
tion and Toxicology, 73: 77-84.
Hadi, D.A., Tarafdar, S.A., Akhter, S. 1996. The level of
aluminium in drinking water supplies and river water.
Bangladesh Journal of Scientific and Industrial
Research, 31: 41-46.
Hadi, D.A., Tarafdar, S.A., Mia, Y. 1991. The study of nitrate,
nitrite and phosphate in some river water around Dhaka
City. Nuclear Science and Applications, 3: 69-75.
Hossain, Z. 2005. Study on Quality of Lake Water in Dhaka
City: Monitoring of Seasonal Variation of Water Quality
Parameters for Evaluation of Water Pollution. M.Sc.
Thesis, pp. 151, Gogonath College, Dhaka, Bangladesh.
Maroof, F.B.A., Hadi, D.A., Khan, A.H. 1985. Cadmium level
of surface water of the Dhaka City. Nuclear Science and
Applications, 16&17, Series B: 145-146.
MEF, 1997. Drinking water quality criteria. In: A Compilation
of Environmental Laws (Bangladesh Gazette Additional
28), M. E. Huq (ed.), pp. 52-54, Department of Environ-
ment, The Ministry of Environment and Forests,
Bangladesh.
Nojiri, Y., Kawai, T., Otsuki, A., Fuwa, K. 1985. Simultaneous
multi element determination of trace metals in lake waters
by ICP emission spectrometry with preconcentration and
their background levels in Japan. Water Research, 19:
503-509.
Quraishi, S.B., Chowdhury, M.N., Khan, M.M.K, Akhter, S. 2006.
Impact of flood on some water quality parameters of lake
waters in Dhaka City area. Nuclear Science and Applica-
tions, 15: 82-85.
Topalian, M.L, Rovedatti, M. G., Castane, P.M., Salibian, A.
1999. Pollution in a low land river system. A case study:
The Reconquista River ( Buenos Aires, Argentina).
Water, Air and Soil Pollution, 114: 287-302.
36 Shamshad Begum Qureshi

Introduction
Salinization of agricultural soils is one of the major abiotic
stresses reducing crop productivity worldwide. Over 6 % of
the global land area and over 20% of the irrigated land are
currently affected by salinity (Munns, 2005). As irrigated
system supplies roughly one-third of the world food supply,
therefore, addressing the problem of salinity is of great
concern especially with an increasing global population. Rice
is one of the most important food crops, but the yield of the
grain is very susceptible to salinity (Akbar et al., 1985). In
Pakistan out of 6.8 million hectares of salt-affected land, over
1.5 million hectares are under rice cultivation (Khan, 1998).
Thus, selection of rice cultivar having salt tolerant potential
that would grow over a range of soil salinity is a prerequisite
for generating income for rice farmers having sizable salt-
affected lands.
Salinity resistance in rice is a complex character and many
factors contribute to such resistance as occurs in species.
Physiological studies suggest that in rice, restriction of
sodium entry, higher potassium uptake, plant vigour, tissue
tolerance to absorbed ions and water-use efficiency are the
main factors contributing towards salinity resistance (Yeo
et al., 1990). Reduction in shoot growth under salinity limits
the volume of tissue for the uptake of newly arriving salt
and once it starts, the situation worsens. Accumulation of Na+
and Cl- in the leaves has been found to reduce photosynthetic
activity, with ultra-structural and metabolic damage (Flowers
et al., 1985). Salinity tolerance in rice can be enhanced by
reducing the influx of excessive amounts of sodium chloride
in the transpiration stream. The salt concentration in the shoot
can be reduced by lowering sodium transport to the shoot and/
or increasing plant vigour. Normally the more vigorous plants
under non-saline conditions show greater resistance to salts
(Yeo and Flowers, 1986). Some traditional cultivars and
landraces are more tolerant to various abiotic stresses than
elite cultivars. These cultivars are good source of tolerant traits;
however, they generally have poor agronomic traits.
Maximizing the salt tolerance of crop species mainly depends
on two factors: availability of genetic variation to tolerance
and exploitation of the genetic variation by screening and
selection of plants with superior performance under the
applied stress (Yamaguchi and Blumwald, 2005; Shannon
et al., 1994). Sensitivity of rice to salinity varies with the stage
of growth. Generally, it is very sensitive to salinity stress at
Salt Tolerance Evaluation of Rice (Oryza sativa L.) Genotypes Based on
Physiological Characters Contributing to Salinity Resistance
Jalal-ud-Din*, Samiullah Khan and Ali Raza Gurmani
Plant Physiology Programme, Crop Sciences Institute, National Agricultural Research Centre,
Islamabad, Pakistan
(received May 22, 2009; revised December 9, 2009; accepted December 20, 2009)
Abstract. Seven newly developed rice cultivars i.e., KS-133, DR-83, DR-64, BR-601, Gomal, JP-5 and Gomal-6,
were evaluated for salinity tolerance in a glasshouse along with three varieties of known salinity tolerance i.e., KS-282
(tolerant), IR-6 (medium tolerant) and Basmati-385 (susceptible). Based on the survival percentage at 50 mol/m3
sodium chloride salinity imposed at seedling stage, rice cultivars KS-133, Gomal, and DR-83 showed high survival
comparable to that of salinity tolerant cultivars like KS-282, and were thus placed in tolerance range. Survival percent-
age of JP-5, Gomal-6 and DR-64 remained in medium tolerance range (35 to 38%) as that of IR-6. The rice cultivar
BR-601 showed only 13% survival and was found to be as sensitive towards salinity as Basmati-385. The results of
rice survival in saline medium showed good uniformity and the check varieties showed results corresponding to
those found elsewhere. Sodium (Na+) and potassium (K+) concentrations in the third leaf showed variations among
different rice cultivars under salinity. There was an inverse correlation between varietal leaf Na+ vs survival percentage
(r = -0.808) and Na+ vs leaf chlorophyll (r = -0.857). The correlation between K+ and final survival percentage was
direct (r = 0.744) and also leaf chlorophyll vs survival (r = 0.952). The shoot fresh and dry weights were greater in the
rice genotypes having higher final survival percentage under saline conditions. Therefore, in addition to final survival
percentage, the higher shoot fresh and dry weight under salinity could be also used as criterion for evaluation of
salinity tolerance of rice.
Keywords: salinity, rice, chlorophyll, salinity tolerance
*Author for correspondence; E-mail: jalaludin10@yahoo.com.uk
Pak. J. Sci. Ind. Res. 2010 53 (1) 37-41
37
Biological Sciences
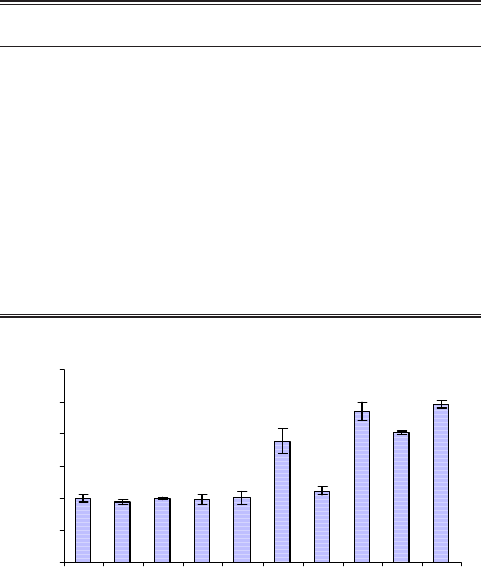
young seedling stage and less at reproductive stage (Lutts
et al., 1995). Hence, for selection a specific growth stage that
is more sensitive to salinity stress should be targeted. Rice is
considered to be generally salt sensitive; there is genetic
variation for salt tolerance at critical stages in the cultivated
gene pool (Moradi et al., 2003; Yeo and Flowers, 1983). There-
fore, selection of highly salt tolerant rice cultivars could be
expected to provide useful material for breeding and for
experimental comparison with unselected lines, in order to
examine possible mechanism of salt tolerance. In the present
study, we have reported the screening of some newly deve-
loped rice cultivars for overall performance (survival) and
other physiological characters, such as leaf chlorophyll,
Na+ and K+ concentrations and biomass accumulation under
salt stress.
Materials and Methods
Plant material and growth conditions. The experiment was
conducted in a glasshouse with temperature controlled between
25 ºC to 35±3 ºC. Seven newly developed rice cultivars, along
with three varieties of known salinity tolerance were used in
the study. The three rice varieties of known salinity tolerance
used as check were; KS-282 (salt resistant), IR-6 (moderate
resistant) and Basmati-385 (salinity sensitive). Sterilized rice
seeds were germinated and seedlings were grown in a sand
culture. Seven days old seedlings were transplanted into black
boxes filled with 5 L rice culture solution (Yoshida et al., 1976).
The solution was renewed once a week. Each cultivar was
replicated thrice in separate boxes having 30 plants per repli-
cate. At day 11, the solution was salinized with NaCl at a
concentration of 50 mol/m3 (total electrolyte concentration
resulting in electrical conductivity of 6 ds/m). The concentra-
tion of 50 mol/m3 NaCl is an established and useful working
level for eliciting a wide range of varietal response (Yeo
et al., 1990).
Leaf sodium, potassium and chlorophyll concentrations.
Six days after salinization, third leaves of 10 plants of each
cultivar was analyzed for sodium (Na+), potassium (K+)
and chlorophyll concentration as described by Din (1997).
Chlorophyll contents were extracted in 80% ethanol and
calculated according to Arnon (1949).
Plant growth and survival tests. After salinization for
24 days, 30 plants (10 from each replicate) of each cultivar
were harvested and the shoot fresh and dry weights were
recorded. The number of dead plants was recorded every
day after the first plant had died and continued till more than
50% plants of the sensitive check variety, Basmati-385, were
dead; a plant was considered dead when it was totally bleached.
The final survival was calculated by subtracting the number
of dead plants from the total number of plants and expressed
as percentage.
Results and Discussion
Survival rate was used as an indicator of genotypic
performance to salinity. Based on the survival in saline
medium, the cultivars were divided into three tolerance ranks.
Survival of KS-133, Gomal and DR-83 was as high as that of
KS-282 and therefore, these three varieties were ranked as
tolerant. Survival percentage of JP-5, Gomal-6 and DR-64
was 35-36% as that of IR-6, and so these were ranked as
medium tolerant, while, BR-601 showed only 13% survival
as that of the sensitive, Basmati-385 (Table 1).
The concentrations of Na+, measured 5 days after salinization,
showed differences in various rice cultivars (Fig. 1). There
was an inverse correlation between leaf Na+ and survival
Table 1. Ranking of salinity tolerance of rice cultivars based
on the survival under NaCl salinity (50 mM)
Salinity rank Rice genotypes Survival (%)
Resistant KS-133 66
Resistant Gomal 72
Resistant DR-83 56
Resistant KS-282 63
Medium JP-5 36
Medium Gomal-6 36
Medium IR-6 35
Medium DR-64 29
Sensitive Bas-385 13
Sensitive BR-601 13
Fig. 1. Mean Na+ concentration (mg/g dry weight) of
third leaf of rice cultivars under NaCl (50 mol/
m3) salinity. Leaves were sampled 5 days after
salinization. Each bar represents standard error
of the mean.
0.0
0.5
1.0
1.5
2.0
2.5
3.0
KS-282
KS-133
DR-83
Gomal
JP-5
Gomal-6
IR-6
DR-64
Bas-385
BR-601
Leaf Na+ (mg/g d.wt.)
Rice cultivars
38 Jalal-ud-Din et al.
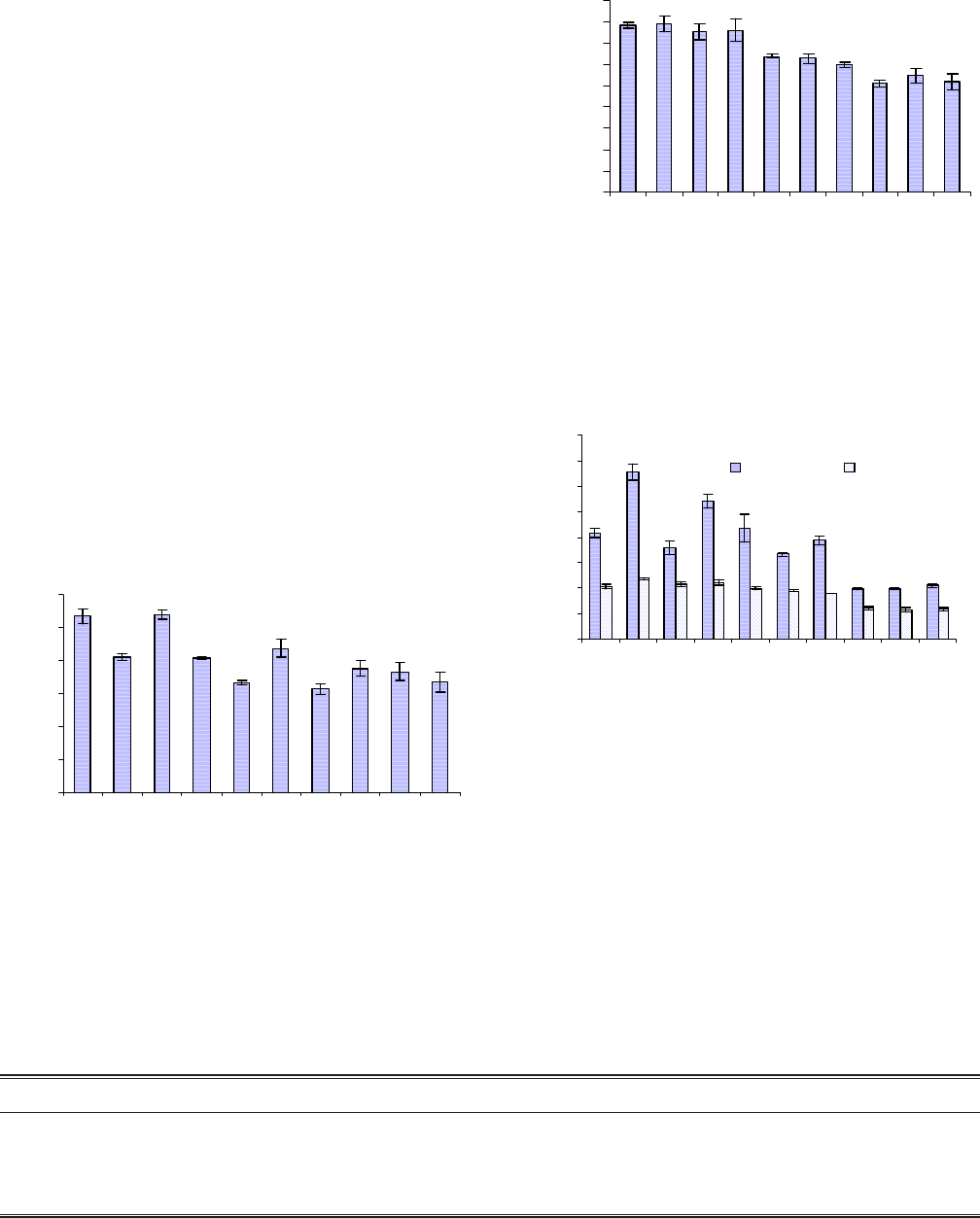
Fig. 2. Mean K+ concentration (mg/g dry weight) of third
leaf of rice cultivars under NaCl (50 mol/m3)
salinity. Leaves were sampled 5 days after
salinization. Each bar represents standard error
of the mean.
0.0
0.5
1.0
1.5
2.0
2.5
3.0
KS-282
KS-133
DR-83
Gomal
JP-5
Gomal-6
IR-6
DR-64
Bas-385
BR-601
Leaf K+ (mg/g d.wt.)
Rice cultivars
(r = -0.808). Leaf K+ showed good correlation with the salinity
tolerance of rice cultivars (Fig. 2) where correlation with
survival under salinity was (r=0.744, Table 2). Total
chlorophyll contents measured in the third leaf after
salinization was greater in the tolerant as compared to the
sensitive (Fig. 3). The shoot fresh and dry weight showed
good correlation with the final survival under NaCl salinity
(Table 2). KS-133, Gomal and DR-83 had greater shoot fresh
and dry weights and DR-64 had the least (Fig. 4).
In this study survival was used as a quantification of genotypic
performance to salinity. This assessment criterion is used in
the field for evaluation of salinity damage during mass
screening of rice cultivars (IRRI, 1996). Based on the survival
in saline medium, the cultivars were divided into three
tolerance classes (Table 1). Generally, survival or visual assess-
ment of salt damage is the criterion for overall measurement
of plant performance. The characteristics would normally be
chosen on their better correlation with overall performance.
Yeo et al. (1990) concluded that survival under salinity strongly
correlates with the salinity resistance of rice. In the present
research study, the survival experiment was repeated three
times and the results showed good uniformity; the check
Fig. 4. Mean fresh and dry weights (g/plant) of shoots
of rice cultivars, when grown for 24 days in NaCl
(50 mol/m3) salinity. Each bar represents standard
error of the mean.
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
KS-282
KS-133
DR-83
Gomal
JP-5
Gomal-6
IR-6
DR-64
Bas-385
BR-601
SFW SDW
Rice cultivars
Weight (g/plant)
varieties, KS-282 and Basmati-385, showed corresponding
salinity tolerance as found earlier (Khan and Abdullah, 2003)
under field conditions.
In monocots, generally salinity tolerance is associated with
the ability of plant to exclude Na+ from shoot tissues (Tester
39
Fig. 3. Chlorophyll concentration (mg/g dry weight) of
third leaf of rice cultivars under NaCl salinity
(50 mol/m3). Leaves were sampled 5 days after
salinization. Each bar represents standard error
of the mean.
0
1
2
3
4
5
6
7
8
9
KS-282
KS-133
DR-83
Gomal
JP-5
Gomal-6
IR-6
DR-64
Bas-385
BR-601
Rice cultivars
Chlorophyll (mg/g d.wt.)
Table 2. Correlation coefficient (r) for relationship between overall performance (survival) and individual traits of the
available data
Shoot Na+Chlorophyll SFW SDW K+
Survival 0.808 ( - )* 0.952 (+ )* 0.762 (+ ) 0.886 (+ ) 0.744 (+ )
(n = 30) (n = 30) (n = 30) (n = 30) (n = 30)
Chlorophyll 0.857 ( - )
(n = 30)
* = signs + and – represent the positive and negative correlation, respectively.
Salt Tolerance in Rice Genotypes
Fresh weight Dry weight
and Davenpor, 2003). In the present study, there were
differences in the leaf Na+ concentration among the rice
cultivars; those having higher leaf sodium had poor survival
rate (Table 1). Potassium accumulation also showed good
correlation with the salinity tolerance of rice cultivars
(r = 0.744). In rice, genotypic variations in Na+ and K+ uptake
have already been reported; low concentration of Na+ and
higher K+ were correlated with the salinity tolerance under
salinity stress (Babu et al., 2007; Kader et al., 2006; Walia
et al., 2005). The chlorophyll contents measured in the third
leaf after salinization was greater in the tolerant as compared
to the sensitive varieties (Fig. 3). There was inverse
correlation between leaf chlorophyll and Na+ concentration
(r = -0.857). Yeo and Flowers (1983) established inverse
relationship between leaf chlorophyll and Na+ concentration.
The shoot fresh and dry weights were the greatest in the
tolerant genotypes followed by the medium tolerant varieties
and the least in sensitive rice cultivars (Fig. 4) and showed
high correlation with the final survival under saline conditions
(Table 2). Sankar et al. (2006) recorded the highest total
biomass and vigour index in salt tolerant cultivars. The
greater plant vigour (shoot fresh and dry weights) may
provide dilution of salt concentrations with growth and
tolerance within the tissues; these are the traits that may be
expected to be helpful in achieving greater salinity tolerance
(Yeo and Flowers, 1986). Richards (1983) concluded that
vigour is essential for plant survival and productivity under
saline environment. Akbar et al. (1985) identified more
vigorous accessions, which were non-dwarf land races, as
the most salt resistant in mass screening trials.
Conclusion
The data demonstrate that rice cultivars with low sodium and
high potassium uptake could lead to higher survival of rice
under saline conditions. The additional aspects of less
chlorophyll damage under such conditions are the potential
of resistant cultivars. Chlorophyll content could be used as an
index of salt tolerance for selection of rice tolerance against
salinity stress. It also demonstrates that amount of biomass of
rice seedlings under NaCl salinity could be used as a criterion
in ranking for salinity tolerance.
References
Akbar, M., Khush, G.S., Hellerislamber, D. 1985. Genetics
of salt tolerance in rice. In: Rice Genetics, Proceedings
of the International Rice Genetics Symposium IRRI, pp.
399-409, Manila, Phillipines.
Arnon, D.I. 1949. Copper enzymes in isolated chloroplasts:
polyphenol-oxidase in Beta vulgaris. Plant Physiology,
24: 1-15.
Babu, S., Sheeba, A., Yogameenakshi, P., Anbumalarmathi,
J., Rangasamy, P. 2007. Effect of salt stress in the selection
of salt tolerant hybrids in rice (Oryza sativa L.) under in
vitro and in vivo condition. Asian Journal of Plant
Sciences, 6: 137-142.
Din, J. 1997. Effect of Hormonal Pretreatments on the
Response of Rice (Oryza sativa L.) and Wheat (Triticum
aestivum L.) to Salinity. D. Phil. Thesis, pp. 25-26, Univer-
sity of Sussex, UK.
Flowers, T.J., Duque, E., Hajibagheri, M.A., McGonigle, T.P.,
Yeo, A.R. 1985. The effect of salinity on leaf ultra
structure and net photosynthesis of two varieties of rice:
Further evidence for cellular component of salt-resistance.
New Phytologist, 100: 37-43.
IRRI, 1996. Standard Evaluation System for Rice, The
International Rice Research Institute, Los Banos, Manila,
Philippines.
Kader, M.A., Seidel, T., Golldack, D., Lindberg, S. 2006.
Expressions of OsHKT1, OsHKT2, and OsVHA are
differentially regulated under NaCl stress in salt-sensitive
and salt-tolerant rice (Oryza sativa L.) cultivars. Journal
of Experimental Botany, 57: 4257-4268.
Khan, G.S. 1998. Soil Salinity/Sodicity Status in Pakistan. pp.
1-59, Soil Survey of Pakistan, Lahore, Pakistan.
Khan, M.A., Abdullah, Z. 2003. Salinity-sodicity induced
changes in the reproductive physiology of rice (Oryza
sativa L.) under dense soil conditions. Environmental and
Experimental Botany, 49: 145-157.
Lutts, S., Kinet, J.M., Bouharmont, J. 1995. Changes in plant
response to NaCl during development of rice (Oryza
sativa L.) varieties differing in salinity resistance. Journal
of Experimental Botany, 46: 1843-1852.
Moradi, F., Ismail, A.M., Gregorio, G., Egdane, J. 2003.
Salinity tolerance of rice during reproductive development
and association with tolerance at seedling stage. Indian
Journal of Plant Physiology, 8: 105-116.
Munns, R. 2005. Genes and salt tolerance: bringing them
together. New Phytologist, 167: 645-663.
Richards, R.A. 1983. Should selection for yield in saline
regions be made on saline or non-saline soils? Euphytica,
32: 431-438.
Sankar, P.D., Subbaraman, N., Narayanan, S.L. 2006. Ranking
of salt tolerant rice lines based on germination and
seedling growth under salt stress conditions. Research
on Crops, 7: 798-803.
Shannon, M.C., Grieve, C.M., Francois, L.E. 1994. Whole
plant response to salinity. In: Plant-Environment
Interaction, R.E. Wilkinson (ed.), pp. 199-244, Marcel
Dekker Inc., New York, USA.
40 Jalal-ud-Din et al.
Tester, M., Davenpor, R. 2003. Na+ tolerance and Na+ transport
in higher plants. Annals of Botany, 91: 503-527.
Walia, H., Wilson, C., Condamine, P., Liu, X., Ismail, A.M., Zeng,
L., Wanamaker, S.I., Mandal, J., Xu, J., Cui, X., Close, T.J.
2005. Comparative transcriptional profiling of two
contrasting rice genotypes under salinity stress during the
vegetative growth stage. Plant Physiology, 139: 822-835.
Yamaguchi, T., Blumwald, E. 2005. Developing salt-tolerant
crop plants: challenges and opportunities. Trends in Plant
Sciences, 10: 615-620.
Yeo, A.R., Yeo, M.E., Flowers, S.A., Flowers, T.J. 1990.
Screening of rice (Oryza sativa L.) genotypes for
physiological characters contributing to salinity resistance,
and their relationship to overall performance. Theoretical
and Applied Genetics, 79: 377-384.
Yeo, A.R., Flowers, T.J. 1986. Salinity resistance in rice (Oryza
sativa L.) and a pyramidal approach to breeding varieties
for saline soils. Australian Journal of Plant Physiology,
13: 161-167.
Yeo, A.R., Flowers, T.J. 1983. Varietal differences in the
toxicity of sodium ions in rice leaves. Physiologia
Plantarum, 59: 189-195.
Yoshida, S., Forno, D.A., Cock, J.H., Gomez, K.A. 1976.
Laboratory Manual for Physical Studies of Rice,
3rd edition, International Rice Research Institute, Los
Banos, Laguna, Philippines.
41
Salt Tolerance in Rice Genotypes

Parasitic Contamination in the Table Vegetables Planted
in Shiraz Plain, Iran
Meraj Madadi
Scientific Board of Islamic Azad University, Firoozabad, Iran
(received June 10, 2008; revised June 17, 2009; accepted September 2, 2009)
Introduction
Table vegetables, particularly those irrigated with raw
sewage, play an effective role in the transfer of parasites
specially the soil parasites and thus in spreading contagious
and parasitic diseases to the consumers (Shariatpanahi, 2001;
Fereydoun, 1987). The vegetables irrigated with Firoozabad-
Tehran creek in 1989 and 1990 and table vegetables used
in Yasooj in 1996, were found to be contaminated with
parasitic worm eggs (Sarkari, 1997; Vosooghi, 1990). In south
Louisiana in USA, where city and industrial sewage contami-
nated the agricultural lands, vegetables such as spinach,
parsley, onion, asparagus, spearmint, tomato, pea, carrot and
cabbage were found to be contaminated with heavy metals,
the latter being 1.60% more than the permissible limit of the
American National Health Society. Moreover 28.20% of the
farms were contaminated with the parasitic eggs found in
sewage (Ramelow et al., 1992).
In Japan, the researchers of Agriculture College, Tokyo
University, in 1962 conducted necessary tests for finding the
contamination of leek, parsley, sweet basil, spearmint and
green pepper growing in the fields which were irrigated with
city and industrial raw sewage and found these vegetables,
heavily contaminated with parasites whereas the content of
heavy metals was 2.13% more than the standards (Chino
et al., 1991).
The researches conducted in Iran show that the vegetables
may transfer eggs of worms such as Ascaris, Trichocephalus,
Hymenolepis nana, Taenia, Fasciolia hepatica, larvae of
worms such as Trichostrongylus and hookworms, unicellular
creatures such as Antamoeba histolytica, Giardia lamblia,
Toxoplasma gondii which cause amoebiasis, giardiasis,
(lambliasis) and toxoplasmosis and other diseases (Fereydoun,
1987).
Though irrigation method should be defined in relation
to the amount of water used and the type of plants being
irrigated, as each plant needs a different rate of water depen-
ding on the environment and geographical conditions such
as temperature, raining rate, latitude etc., it was observed that
these factors are ignored in Shiraz plain irrigation; here the
irrigation system is deep water which is traditional and uses
about 4,000-12,000 cubic meter water per hectare, yearly.
This system causes problems relating to drainage of water,
environmental contamination, agricultural damages and soil
errosion etc. (Rastegar, 1992).
The present study was undertaken to find the extent of
contamination of vegetables irrigated directly by Shiraz
Roodkhaneh Khoshk River and Soltanabad River and by
nearby lying well water. Taking into consideration the
diseases, such as diarrhoea, resulting from the above
mentioned practice, it is intended to propose safe means of
growing vegetables and prevent regional contamination of
vegetables with parasites and their transfer to the consumers.
Materials and Methods
City sewage is discharged into Shiraz Roodkhaneh Khoshk
River passing through the city centre. Some districts of the
city do not have proper sewage disposal means and the
factories beside the river also dump their waste products
Pak. J. Sci. Ind. Res. 2010 53 (1) 42-45
Abstract. Contamination with parasites of the vegetables grown in Shiraz plains and irrigated by urban and industrial
sewage-laden Shiraz Rookdhaneh Khoshk River and seasonal Soltanabad River was studied. It was found that 31.5% of the
farms irrigated by the river water directly, 30.9% of the farms irrigated by water of nearby located shallow wells and 33.7%
of the farms using water from the wells at a distance of one kilometer from the River were contaminated by Ascaris ova.
32.20% vegetables of farms irrigated by the wells located near Soltanabad river were contaminated with insects and larvae
and 24.5% with Ascaris worm. After Ascaris ova, the larvae of different insects, Strongyloides parasite, Sterocoralis and
Trichostrongylus were the contaminants most present.
Keywords: parasites, irrigation, vegetables, Shiraz rivers, Ascaris
E-mail: navid_madadi@yahoo.com
42

into it. Due to disposal of garbage into the river, some agri-
cultural irrigation places have high BOD (the index indicating
sewage contamination) related to city sewage (WHO, 1989).
This study covers table vegetables planted in the farms
around Shiraz where the vegetables are consumed either raw
or cooked. The areas studied included Gheisar Aboonasr,
Mehraghan, Eghbal Abad, Torkan, Nasirabad, Kooshkak,
Mahfiroozan, Dasht Khezr, Noortaban, Khaljooy and
Sharifabad villages, mountainous region and Kaftarak
village, all of them located beside Roodkhaneh Khoshk River.
Parameters taken into consideration were area of the land
under cultivation, type and amount of harvest, fertilizer and
disinfectants, herbicides, fungicides and means of irrigation
including deep and semi-deep wells and river.
The land for cultivation of table vegetables, domestic animals
provender, wheat, barley and potato was about 1,372 hectares
of which 128 hectares was under cultivation of the provender
including alfalfa, 168 hectares that of barley, wheat and maize
and 100 hectares were used for planting of potato. The land
used for cultivation of vegetables was 276 hectares of which
90 hectares were irrigated with river and 186 hectares irrigated
mostly by shallow wells.
Samples (140) of different vegetables cultivated in Kaftarak
and Soltanabad were selected on the basis of the factors
such as means of irrigation including shallow wells, wells
within one km radius of the river and the river itself; one type
of vegetable from each farm was sampled. 45 samples were
taken from farms irrigated with river and 95 samples, from farms
irrigated with deep, semi-deep and shallow wells.
The following vegetables, 250-500 g of each, were sampled:
a) Sweet basil, leek, spearmint, cress, purslane, parsley
lettuce, tarragon, common dill and spinach (eaten raw).
b) Radish, tomato, cucumber, eggplant, green pepper,
carrot, onion, cabbage and squash (eaten cooked except
carrot)
c) Green bean, pea and okra (eaten cooked).
The samples were collected with gloved hands and put into
nylex bags and carried to the related laboratories (Gholami
and Mohammadi, 1998). Domestic animals provender was
sampled in the same way.
Results and Discussion
Samples of 58 shallow, semi deep and deep wells were
subjected to bacteriological tests. Results indicated that 98%
of the wells were contaminated with coliform, 100% of which
were soil coliform. Physical and chemical properties of water
did not conform to the standards, except that of 12 deep wells
lying at a distance of 1,500 m from the river.
Findings show that the parasitic contamination of table
vegetables depends on location of the place/farm to be irri-
gated, source of irrigation and type of the parasites (Table 1).
Most of the farms around Roodkhaneh Khoshk river were
contaminated with “Ascaris” and larvae of different insects.
31.50% farms irrigated directly by river (Fig. 1), 31% farms
irrigated by the wells located around the Roodkhaneh
Khoshk river (Fig. 2) and 33.70% by the wells within one
kilometer radius of the river were contaminated with ascaris
eggs; mostly the fertilizer used was human faeces (Table 1);
bacteriological tests of shallow regional well water indicated
faecal coliform contamination, 3 to 45 MPN/100 mL, and total
coliform, 70 to 800 MPN/100 mL, whereas, total coliforms in
deep well water were 5 to 45 MPN/100 mL. Physical and
chemical specifications of the water used for irrigation of
Shiraz plains and Roodkhaneh Khoshk farms were not within
the standard limits necessary for agriculture with BOD more
Table 1: Parasitic contamination in the vegetables according to the irrigating source and the type of parasite
Parasite Roodkhaneh Wells near Well within radius Wells near
Khoshk water Roodkhaneh Khoshk of 1 km from the river Soltanabad river
No. of % No. of % No. of % No. of %
parasites parasites parasites parasites
Trichostrongylus 35 15.5 19 16.8 13 16.3 12 19.6
Strongyloides stercoralis 32 14.2 13 11.5 19 23.7 10 16.4
Ascaris lumbricoides 71 31.5 35 31 27 33.7 15 24.6
Trichocephalus 4 1.78 0 0.0 0 0.0 0 0.0
Oxyure 19 8.5 0 0.0 2 2.6 4 6.6
Different insect
larvae and eggs 64 28.5 46 40.7 19 23.7 20 32.8
Total 225 100.0 113 100.0 80 100.0 61 100.0
43
Parasitic Contamination of Vegetables in Iran
than 100 mg/L. Also the level of some heavy metals was not
within the range applicable for agricultural use. Water EC
(electrical conductivity) of some well water was high, but all
the wells had coliform contamination.
In transfer of parasites, three factors are involved: source of
infection, means of transfer and a sensible host, which
means the process depends on the way of distribution of the
parasite in a defined place at a defined time. The means by
which the parasite reaches from the source to the host are
clear (Neva and Brown, 2001). Some parasites reach the host
through direct contact whereas others have a more complica-
ted life cycle and need to pass through some growth stages
such as free life or need an intermediate host to become infec-
tive. The transfer is accomplished through direct and indirect
contact, such as by means of food, water, soil and vertebrates
and arthropodes. Studies show that rate of contamination
with some worms in different parts of Iran is high; in Iran,
around 32 human parasites were found, some of which were
very common and frequent such as Ascaris (Mahvi, 1996).
The vegetables, contaminated with parasite eggs, transfer
them to the consumers (Monzavi, 1985). Raw sewage, human
faeces and contaminated water, used as fertilizer and for
irrigating farms, are dangerous sources of contamination for
the consumers of the final products because traditional
and deep water systems are still used here as the sources of
potable water.
Fig. 4. Parasitic pollution (%) of different vegetables irrigated
via wells close to Khoshk river.
Pollution (%)
12
10
8
6
4
2
0
87
5
8
6
8
545
9
7
910
68
44
40
35
30
25
20
15
10
5
0
21
13
25
17
10 14
97912
35
18
726
10 10
Fig. 3. Parasitic pollution (%) of different vegetables irrigated
via water of Khoshk river.
Pollution (%)
Tests showed that all the table vegetables cultivated in Shiraz
plains and irrigated with Roodkhaneh Khoshk river, and the
wells close to the river were contaminated with one or several
faecal parasites. The range of pollution in vegetables irrigated
directly by the river was (with the exception of 2% in cabbage)
from 6% (in gourd) to 35% (in green beans) (Fig. 3) and in
those irrigated by close-by lying wells was 4% (in gourd
cabbage and onion) to 10% (in green beans) (Fig. 4). Thus it
is clear that most of the contamination relates to the means
of irrigation and use of man faeces (US EPA, 1981).
44 Meraj Madadi
Fig. 1. Pollution (%) of vegetables irrigated via water of Khoshk
river with different parasites.
Other insects,
28.45%
Oxyure,
14.23%
Trichocephalus,
1.78% Ascaris,
31.55%, 30%
Stercoralis,
14.23%
TricostrongyIus,
15.56%
Fig. 2. Pollution (%) of vegetables irrigated via wells close to
Khoshk river with different parasites.
Ascaris,
31% Stercoralis,
11.50%
Other insects,
40.70%
TricostrongyIus,
16.82%
The land measuring 276 hectares is used for cultivation of
table vegetables and is irrigated with Roodkhaneh Khoshk
river; these raw vegetables are distributed in Shiraz and other
adjoining cities. Also the wells located within a radius of one
km of the river, when tested bacteriologically, whenever less
deep and farther from the river, were more contaminated; it
shows that the wells were influenced by the river. The distant
well water had parasitic eggs found in human faeces (used
in the farm) and the differences were as follows:
a) The parasitic contamination was more in the vegetables
of farms irrigated with the river water directly.
b) Contamination of the vegetables with parasite eggs was
less in the farms irrigated by hand and shallow wells
farther from the river than the farms irrigated directly by
the river water .
c) Contamination in the farms, irrigated by deep wells more
than one km distant from the river, was less than the
above-mentioned two cases.
So, it is clear that the river water contaminated by sewage
played main role in contaminating the consumable raw
vegetables with parasite eggs.
It was found that green beans, sweet basil and tarragon were
more contaminated with the eggs and larvae of Ascaris and
Trichostrongylus than with the other insects. Also the farms
irrigated by the shallow wells and the wells, within a radius of
50 m of the river, had vegetables contaminated with parasite
eggs similar to the farms irrigated by the river water.
It was also found that the farms irrigated by the wells around
the seasonal river, Soltanabad, had eggs of strongylosis,
sterocoralis, ascaris and trichostrongylosis, more than other
parasite eggs.
The contamination of vegetables with parasite eggs of farms
irrigated by Roodkhaneh Khoshk river was found to be more
frequent as compared to those irrigated by the wells beside
the seasonal river, Soltanabad.
Conclusion
The results show that the vegetables cultivated in Shiraz
plain, irrigated/fertilized by sewage-containing water, play an
important role in transferring important parasites such as
ascaris, trichostrongylus, trichocephalus, S. stercoralis and
oxyure. Thus sewage and human fertilizers have been the
cause of contamination of farm vegetables irrigated by such
means. Irrigation of agricultural farms with such sources of
contamination is harmful from hygienic point of view of the
consumers of crops, grown in such farms.
It is therefore, proposed that the standards and regulations
relating to the irrigation water used specially for raw
vegetable crops should be observed. It is recommended that
unrefined as well as refined sewage may not be used for
irrigating vegetable crops and for fertilization; instead animal
fertilizer, whose conditions of maintenance have been
observed, be used.
References
Chino, M., Moriyama, K., Saito, H., Mori, T. 1991. The amount
of heavy metals derived from domestic sources in Japan.
Water, Air and Soil Pollution, 57: 829-837.
Fereydoun, A. 1987. Medical Helminthology, vol. 1 & 2, 3rd
edition, Danesh Pajouh Publications, Tehran, Iran.
Gholami, M., Mohammadi, H. 1998. Water and Wastewater
Microbiology, Hayan Publications, Tehran, Iran.
Mahvi, A.H. 1996. Sewage Filtration in Torrid Zone, Jahad
Daneshgahi Publications Organization, Tehran, Iran.
Monzavi, M.T.I. 1985. Water Supply, 4th edition, Tehran
University Publications, Tehran, Iran.
Neva, F.A., Brown, H.W. 2001. Basic Clinical Parasitology
(Translated by A. Athari), Pezhman Publications, Tehran,
Iran.
Ramelow, G.J., Biven, S.L., Zhang, Y., Beck, J.N., Young, J.C.,
Callahan, J.D., Marcon, M.F. 1992. The identification
of point sources of heavy metals in an industrially
impacted water way by periphyton and surface sediment
monitoring. Water, Air and Soil Pollution, 65: 175-190.
Rastegar, M. 1992. Dry Farming, 1st edition, Behramand
Publications, Tehran, Iran.
Sarkari, B. 1997. Examining parasitic contamination of the
vegetables used in Yasuj city. Journal of Yasuj Univer-
sity of Medical Sciences, 3: 15-18.
Shariatpanahi, M. 2001. Principles of Quality and Treatment
of Water and Wastewater, 6th edition, Tehran University
Publications, Tehran, Iran.
US EPA, 1981. Process Design Manual for Land Treatment of
Municipal Wastewater, US Environmental Protection
Agency, Rept. 625 11-77-008, US. Govt. Printing Office,
Washington DC., USA.
Vosooghi, M. A. 1990. Examining the Worm Parasitic Eggs
Contamination of the Vegetables Irrigated by Firoozabad
River, MS. Thesis, Tehran Medical Sciences University,
Tehran, Iran.
WHO, 1989. Health Guidelines for the Use of Wastewater
in Agriculture and Aquaculture, WHO Technical
Report Series 778, World Health Organization, Geneva,
Switzerland.
45
Parasitic Contamination of Vegetables in Iran

Microbiological Quality of Drinking Water and Beverages
in Karachi, Pakistan
Anila Siddiqui, Korish Hasnain Sahir and Seema Ismat Khan*
PCSIR Laboratories Complex, Shahrah-e-Dr. Salimuzzaman Siddiqui, Karachi-75280, Pakistan
(received February 20, 2009; revised October 21, 2009; accepted October 28, 2009)
Introduction
Raw water itself does not contain large number of micro-
organisms. Drinking water contains assimilable organic
compounds that allow a certain degree of bacterial growth
(Exner et al., 2005). Improperly installed hand pumps permit
infiltration of contaminated surface water, whereas unclean
storage devices and other factors contribute to disease cycle,
malnutrition and high mortality. Infectious diseases caused
by pathogenic bacteria are the most common and wide
spread health risk associated with drinking water. Some of
the pathogens, which are transmitted through contaminated
drinking water, lead to severe and sometimes life threatening
diseases particularly in children. The potential of drinking
water to transport microbial pathogens to large number of
population, causing subsequent illness, is well documented
in different countries.
The most common and widespread risk associated with drink-
ing water is its contamination by human or animal excreta. The
potential consequences of microbial contamination necessi-
tates that its control be of paramount importance. Pathogens
in drinking water, presenting serious risk of diseases, include
Salmonella sp., pathogenic Escherichia coli, Pseudomonas,
Vibrio cholerae etc. Faecal specific bacteria such as coliforms,
faecal coliforms and E. coli are the parameters of importance
in monitoring faecal pollution.
The survival and growth of microorganisms in processing
environments of foods, such as beverages, sherbets, ice
creams, ice-lollies, etc. may lead to contamination of the
finished products, resulting in reduction of microbiological
safety and quality (Kohnen et al., 2005). However, several
studies have demonstrated that the total counts and number
of pathogens may get reduced due to the acidity and the
effect of CO2 during storage. (Mugochi et al., 1999; Simango
and Rukure, 1992; Sheth et al., 1988; Zschaler, 1979; King and
Nagel, 1975; 1967).
In Pakistan people lack access to adequate supply of safe
water for daily use. The basic sanitary facilities are very poor
and are not available for half of the population. Sources of
microbial contamination of water and beverages include raw
materials, processing equipment or utensils, human activities,
sanitation practices, workers or handlers, waste materials,
animal and insect pests and microbial growth niches. Chemical
composition of foods and beverages and the environmental
factors, such as water activity, pH, temperature, etc., determine
the types of organisms that can grow there.
The present study was undertaken keeping in view the
hazards of polluted or contaminated drinking water and the
effect of its use in beverages and ice lollies etc. The survey
provides base line data for authorities to set the guidelines
for microbiological quality of water and beverages within
the country.
Materials and Methods
Sample collection. A total of 780 water and 1220 beverage
samples were collected from 490 different schools/educational
institutes of Karachi, located in various areas including those
inhabited by low income, middle class and upper middle class
population, as well as rich and wealthy people. Both the
government and the private institutions were included in the
Pak. J. Sci. Ind. Res. 2010 53 (1) 46-49
Abstract. Microbiological assay of 780 water samples and 1220 beverage samples (412 branded and 808 unbranded),
collected from 490 different schools, both government (98 schools) and private (392 schools), situated in different areas
of the city of Karachi, was conducted for bacterial heterotrophic plate count, total coliforms, faecal coliforms, E. coli,
faecal streptococci, Pseudomonas and Salmonella species. The counts ranged from 0 to 2.5 × 105 cfu/mL and from 0 to
106 cfu/mL in water and beverage samples, respectively. About 36% of water samples and 48% of unbranded beverage
samples were contaminated with the indicator and the pathogenic bacteria; all the branded beverage samples were found
fit for human consumption from microbiological viewpoint.
Keywords: drinking water, beverages, microbiological quality
*Author for correspondence; E-mail: seema_ismat@hotmail.com
46
study, out of these 98 were government schools whereas 392
were private institutions. Insulated ice chest with ice packs
was used for collection and transportation of samples. The
collected samples were labelled with date and laboratory
code. Other necessary information about the samples, like
area or location of school, collection point etc. was recorded
on prescribed forms. Samples were collected in sterilized
screw-capped glass bottles.
Beverage samples were categorized as branded and un-
branded. The unbranded beverages consisted of gola-gandas
(local name for an item made by crushed ice and additives),
sherbets (drinks) and carbonated and other drinks sold on
vending carts (thailas). All the water and beverage samples
were tested microbiologically for their heterotrophic plate
counts, total coliforms, fecal coliforms, E. coli 0157:H7, faecal
streptococci, Pseudomonas and Salmonella species.
Heterotrophic plate count (HPC), coliforms and faecal coliforms
were tested according to the Standard Methods for the Exami-
nation of Water and Wastewater (APHA, 1998; ISO 9308-1:
2000; 9308-2: 1990; 6222: 1988a; 8199: 1988b). E. coli 0157:H7
was tested by serological kit method (Pro-Lab Diagnostics)
according to Thompson et al. (1990) and faecal streptococci
were determined using ISO method 7899-1 (1984). Other
parameters were tested according to on-line Bacteriological
Analytical Manual of US FDA (2006; 2002; 2001). Salmonella
was confirmed using antisera (Pro-Lab Diagnostics).
Results and Discussion
Figures 1 and 2 present total heterotrophic plate count (HPC)
in water and beverage samples, respectively. The heterotrophic
plate count in water and beverage samples ranged from 0 to
2.5 × 105 cfu/ml and from 0 to <106 cfu/ml, respectively. HPC
may be used to assess the general bacterial content as well as
the efficiency of water treatment. The HPC standards for
drinking water vary a lot from country to country. According
to WHO (1999) guidelines for drinking water quality, the limit
for HPC is 100 cfu/mL which has also been adopted by
Pakistan Standards and Quality Control Authority (PSQCA;
2004).
However, WHO guidelines for drinking water quality are
intended to be used as a basis for the development of national
standards in the context of local or national environmental,
socio-economical and cultural conditions (WHO, 1999). The
HPC standard for drinking water in Sri Lanka is 10,000 cfu/mL
(SLS, 2001; 1995), which is a pretty relaxed standard, compared
to the stringent standard of WHO. According to Canadian
standard, HPC is not considered as a parameter of drinking
water quality. A review study conducted by Allen et al. (2004)
reveals that there is no evidence to support health-based
regulations of HPC concentrations.
The national standards should be influenced by national
priorities and economic factors. Stringent standards could
limit the availability of water supplies, which is a significant
consideration in regions of water shortage. However, public
health must never be endangered. For this very reason, it is
suggested that the WHO standard for HPC must not be
adopted as the criterion for rejection of line water samples for
drinking purpose and the acceptance limit must be adopted
according to Sri Lankan standard or the Canadian guidelines.
The presence of indicator organisms and/or pathogens only
must be considered as the criterion for declaring a sample
unfit for human consumption. However, we do support
adoption of WHO guidelines for bottled water according to
which the samples must not contain any count at all as the
manufacturers/bottlers are claiming it to be pure and charging
extra money for that.
Figure 2 presents total bacterial counts in beverage samples.
According to the PSQCA (2002) standard PS: 1654-2002 R,
the freshly prepared carbonated drinks may contain <100
bacteria per mL of the sample whereas the count must
47
Microbiological Quality of Drinking Water
Fig. 1. Heterotrophic plate counts of water samples.
48
343
207 177
5
0
50
100
150
200
250
300
350
<10 <100 <1000 <10000 <100000
HPC (cfu/mL)
Fig. 2. Heterotrophic plate counts of beverage samples.
272
0
121
16 19
89
0 0
318
0
164
0
50
100
150
200
250
300
350
<10 <100 <1000 <10000 <100000 <1000000
HPC (cfu/mL)
Branded
Unbranded 221
decrease to <30 within three days of storage. The counts in
the branded samples were mostly within the limit of PS
standard with only 19 of the 412 tested samples having counts
exceeding 100 mL.
Coliform organisms are recognized as suitable microbial
indicators of drinking water quality. Coliforms include
heterogeneous lactose fermenting bacteria found in faeces
and environment. Detection of coliforms suggests inade-
quate treatment, post-treatment contamination or excessive
nutrients. Although coliforms may not always be related to
the presence of faecal contamination or pathogens, they are
useful for monitoring the microbial quality of public water
supplies. Presence of coliforms in the absence of faecal
coliforms suggests the use of secondary indicators like
faecal streptococci for confirmation of faecal contamination.
Figures 3 and 4 present the occurrence of organisms of public
health significance in water and beverage samples, respec-
tively. The microbiological analyses revealed that a total of
36% water samples and 48% of unbranded beverages were
unfit for human consumption due to the presence of organ-
isms of public health significance i.e. indicator organisms
and/or pathogenic bacteria. The branded samples, on the
other hand were all free from organisms of public health
significance.
In the present study, a total of 269 out of 780 water samples
(34.5%; Fig. 3) and 549 out of 808 unbranded beverages
samples (45%, Fig. 4) were found contaminated with coliform
bacteria (Noble et al., 2004, 2003; Reasoner and Galdreich,
1985).
Thermotolerant (faecal) coliform group comprises of
Escherichia, Klebsiella, Enterobacter and Citrobacter,
which are able to ferment lactose at 44-45 °C. Out of these
organisms, only E. coli is specifically of faecal origin; other
thermotolerant coliforms may originate from organically
enriched waters such as industrial effluents or from decaying
plant materials and soils.
Out of 780 water samples, 185 (23.5%) and 156 (20%) (Fig. 3)
and out of 808 unbranded beverages, 549 (45%) and 463 (38%)
(Fig. 4) samples were found contaminated with faecal coliforms
and E. coli, respectively.
The presence of Pseudomonas aeruginosa in potable water
also indicates serious deterioration in bacteriological quality
and is often associated with complaints about taste, odour
and turbidity linked to low rates of flow in distribution system
and a rise in water temperature.
Conclusion
The present study has shown that the microbiological quality
of water and locally made beverages vended in schools is not
satisfactory and approximately 36% water samples and 48%
unbranded beverage samples were found to be unfit for
human consumption due to the presence of organisms of
public health significance i.e. indicator organisms and/or
pathogenic bacteria.
Water sources must be protected from contamination by the
human and animal excreta and other wastes to protect the
community from the risks of outbreaks of intestinal and other
infectious diseases. Moreover, effective treatment and
regular monitoring must be carried out in order to protect the
water reservoirs. Furthermore, strict sanitary and hygienic
regulations must be imposed on the local manufacturers of
beverages; the sanitary conditions of the vending carts and
the beverages being sold through them must also be effec-
tively monitored.
48 Seema Ismat Khan et al.
Fig. 3. Incidence of organisms of public health significance
in water samples.
269
185
156
14
76
0
0
50
100
150
200
250
300
Fig. 4. Incidence of organisms of public health significance
in beverage samples.
0
549
0
549
0
463
00012 00
0
100
200
300
400
500
600
Branded
Unbranded
References
Allen, M.J., Edberg, S.C., Reasoner, D.J. 2004. Heterotrophic
plate count bacteria - what is their significance in drink-
ing water. Intternational Journal of Food and Micro-
biology, 92: 265-274.
APHA, 1998. Microbiological examination. In: Standard
Methods for the Examination of Water and Wastewater,
20th edition, American Public Health Association,
Washington DC., USA.
Exner, M., Vacata, V., Gebel, J. 2005. Public health aspects of
the role of HPC - An introduction. In: Heterotrophic Plate
Counts and Drinking Water Safety, J. Bartram Cotruvo,
M. Exner and A. Glasmacher (eds.), AITBS Publishers,
New Delhi, India.
ISO 9308-1:2000(E). Detection and enumeration of
Escherichia coli and coliform bacteria. Part 1. Membrane
Filtration Method.
ISO 9308-2:1990(E). Water quality - detection and enumera-
tion of coliform organisms, thermotolerant coliform
organisms, and presumptive Escherichia coli. Part 2:
Multiple Tube (Most Probable Number) Method.
ISO 6222:1988a. Enumeration of viable microorganisms.
Colony count by inoculation in or on nutrient culture
medium.
ISO 8199:1988b. General guide to enumeration of microorgan-
isms by inoculation in or on culture medium.
ISO 7899-1:1984. Detection and enumeration of faecal strepto-
cocci-Part 1: Method by enrichment in a liquid medium.
King, Jr., A.D., Nagel, Ch. W. 1975. lnfluence of carbon dioxide
upon the metabolism of Pseudomonas aeruginosa.
Journal of Food Science, 40: 362-366.
King, Jr., A.D., Nagel, Ch. W. 1967. Growth inhibition of a
Pseudomonas by carbon dioxide. Journal of Food
Science, 32: 575-579.
Kohnen, W., Teske-Keiser, S., Meyer, H.G., Loos, A.H., Pietsch,
M., Jansen, B. 2005. Microbiological quality of carbona-
ted drinking water produced with in-home carbonation
systems. International Journal of Hygiene and Envi-
ronmental Health, 208: 415-423.
Mugochi, T., Parawira, W., Mpofu, A., Simango, C., Zvauya,
R. 1999. Survival of some species of Salmonella and
Shigella in mukumbi, a traditional Zimbabwean wine.
International Journal of Food Science and Nutrition,
50: 451-455.
Noble, R.T., Leecaster, M.K., McGee, C.D., Weisberg, S.B.,
Ritter, K. 2004. Comparison of bacterial indicator analysis
methods in stormwater-affected coastal waters. Water
Research, 38: 1183-1188.
Noble, R.T., Moore, D.F., Leecaster, M.K., McGee, C.D.,
Weisberg, S.B. 2003. Comparison of total coliform, fecal
coliform and enterococcus bacterial indicator response
for ocean recreational water quality testing. Water
Research, 37: 1637-1643.
PSQCA, 2004. Pakistan Standard Specification for Drinking
Water. PS: 4639-2004 (R), Pakistan Standards & Quality
Control Authority, Standards Development Centre.
PSQCA, 2002. Pakistan Standard Specification for Carbo-
nated Beverages. PS: 1654-2002 (R), Pakistan Standards
and Quality Control Authority, Standards Development
Centre.
Reasoner, D.J., Geldreich, E.E. 1985. A new medium for the
enumeration and subculture of bacteria from potable
water. Applied Environmental Microbiology, 49: 1-7.
SLS, 2001. Sri Lankan Standards 894, specification for pack-
aged bottled drinking water.
SLS, 1995. Sri Lankan Standards 1038. Specification for bottled
natural mineral water.
Sheth, N.K., Wisniewski, T.R., Franson, T.R. 1988. Survival of
enteric pathogens in common beverages: an in vitro study.
American Journal of Gastroenterology, 83: 658-660.
Simango, C., Rukure, G. 1992. Survival of bacterial enteric
pathogens in traditional fermented foods. Journal of
Applied Bacteriology, 73: 37-40.
Thompson, J.S., Hodge, D.S., Borczyk, A.A. 1990. Rapid
biochemical test to identify verocytotoxin-positive strains
of Escherichia coli serotype O157. Journal of Clinical
Microbiology, 28: 2165-2168.
US FDA, 2006. Bacteriological Analytical Manual, An
Online publication of Center for Food Safety & Applied
Nutrition of USFDA. Chapter 3 (2001), Chapter 4 (2002),
Chapter 5 (2006).
WHO, 1999. Guidelines for Drinking-Water Quality. Health
Criteria and Other Supporting Information, vol. 2, 2nd
edition, AITBS Publishers, World Health Organization,
Geneva, Switzerland.
Zschaler, R. 1979. Influence of carbon dioxide on the micro-
biology of beverages. Antonie van Leeuwenhoek 45:
158-160.
49
Microbiological Quality of Drinking Water

Study of food and feeding habits of fish requires continuous
research, since successful fishery management, aquaculture and
capture fishery programmes are based on it (Oso et al., 2006).
Caranx hippos, Chrysichthys nigrodigitatus, Ethmalosa
fimbriata and Mugil cephalus are some of the fish species readily
available in the Lagos Lagoon, Nigeria in West Africa, and make
up an important part of artisanal fisheries. Several studies of
the food and feeding habits of these four fish species have been
made, some of which include the work of Oronsaye and
Nakpodia, (2005) and Blay (1995). However, information on
the feeding inter-relationship of these species is lacking. In
this paper, a report on the feeding inter-relationship among
the four fish species is presented.
Forty specimens of the above-mentioned four fish species were
caught in the Lagos lagoon each month during February to
May 2001. Body weights and lengths of fish were measured
and the stomach contents were studied. The organisms found
were identified to the species level and analyzed by numeri-
cal and frequency of occurrence method.
Analysis of the food of C. hippos revealed diatoms and algae
to form the major food items. Other food items were molluscs
(Aloidis trigona, bivalve shell and Tympanotonus fuscatus),
crustaceans (Calanus finmarchicus, shrimp and shrimp parts),
fish (eggs, bones, scales and flesh) and detritus and other
unidentifiable matter (Fig. 1).
Diatoms formed the major food items of C. nigrodigitatus.
Other food items were molluscs (Aloidis trigona, bivalve shell
and Tympanotonus fuscatus), crustaceans (shrimp parts,
Feeding Inter-Relationship of Caranx hippos (Linneaus),
Chrysichthys nigrodigitatus (Lacepede), Ethmalosa fimbriata (Bowdich)
and Mugil cephalus (Linneaus) in Lagos Lagoon, Nigeria
Adebiyi Adenike Fatimat
Department of Marine Sciences, Faculty of Science, Univeristy of Lagos, Lagos, Nigeria
(received July 17, 2009; revised November 23, 2009; accepted December 5, 2009)
E-mail: Adebiyi_fatima@yahoo.com
Abstract. Study of the feeding inter-relationship of Caranx hippos, Chrysichthys nigrodigitatus, Ethmalosa fimbriata
and Mugil cephalus in the Lagos Lagoon, Nigeria, revealed that algae and diatoms formed the main food items of the
four fish species; other food items were crustaceans, molluscs and detritus. Utilization of nearly identical food items
suggested inter-specific competition for food.
Keywords: feeding habits, Caranx hippos, Chrysichthys nigrodigitatus, Ethmalosa fimbriata, Mugil cephalus, Nigeria
Calanus finmarchicus, cladocera and crab appendages),
fish (fins, scales, eggs and bones), algae, plant material and
unidentifiable matter (Fig. 2).
Diatoms were the major food items of E. fimbriata as well.
Other food items were crustaceans (Calanus finmarchicus,
shrimp parts, isopods and cladocera), fish (bones, scales
and eggs), algae, plant materials and unidentifiable matter
(Fig. 3).
Major food items in the gut of M. cephalus were diatoms.
Other food items were crustaceans (Calanus finmarchicus and
shrimp parts), fish (scales and bones), algae, plant materials
and unidentifiable matter (Fig. 4).
Analysis of the food items in the gut of four fish species
revealed that C. hippos did not feed on fish in February and
April, molluscs in April and did not feed on plant material at
all. C. nigrodigitatus fed on plant material in Februaury and
fish in May. E. fimbriata did not feed on fish in February and
April, on plant material in April and did not feed on molluscs
at all throughout the months studied. M. cephalus did not feed
on fish in April and May and did not feed on molluscs at all.
The study reveals the food and feeding habits of the four fish
species. M. cephalus is a plankton feeder, feeding mainly on
algae and diatoms (Ramirez-Luna et al., 2008). In this study
the important food item of M. cephalus comprised of diatoms,
while other food items were algae, crustaceans, plant material
and detritus. The food items of C. nigrodigitatus included plant
materials, molluscs, crustaceans, fish and detritus. Dada and
Araoye (2008) also discovered similar food items in the
stomach of C. nigrodigitatus. Ajah et al. (2006) reported
Pak. J. Sci. Ind. Res. 2010 53 (1) 50-51 Short Communication
50
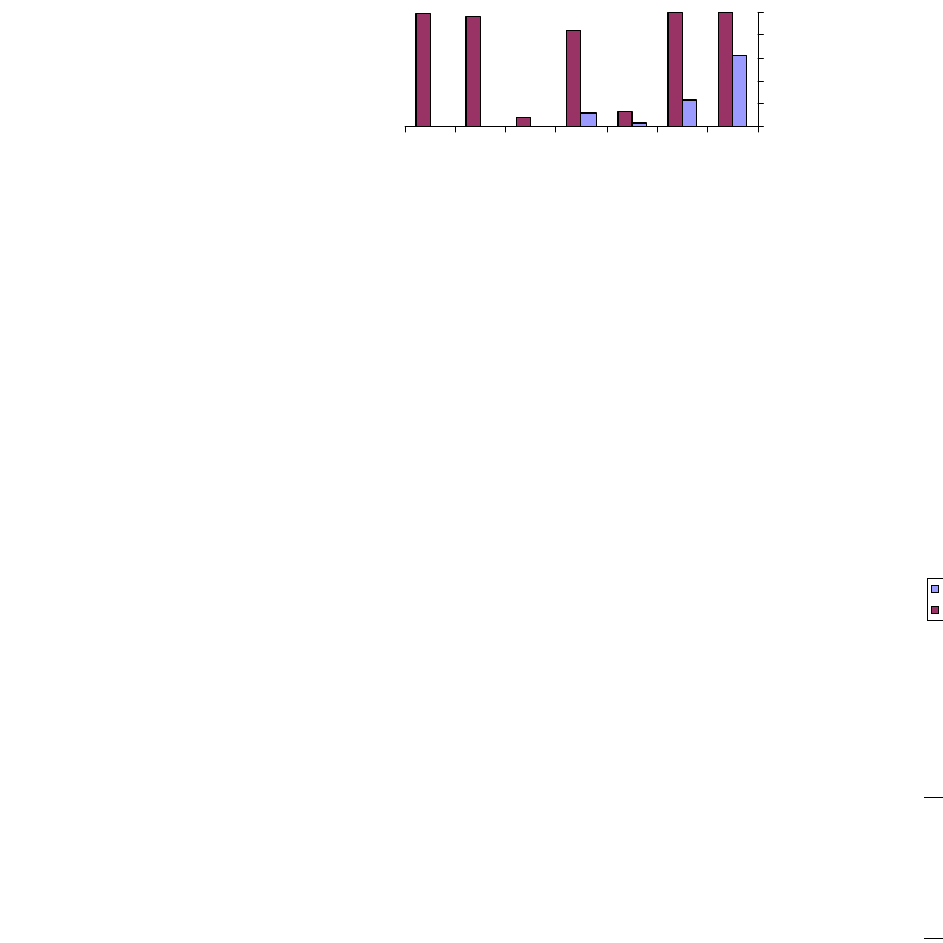
as another important food. It can, therefore, be concluded that
there was an inter-relationship in the food and feeding habits
of the four fish species. This inter-relationship would lead to
a high competition for food in the Lagos lagoon.
Acknowledgement
The author is grateful to the Department of Marine Sciences,
University of Lagos, for providing the facilities used for this
study and to Prof. K. Kusemiju for academic assistance.
References
Ajah, P.O., Georgewill, M.N., Ajah, M.O. 2006. The food and
feeding habits of five freshwater and brackish-water fish
species in Nigeria. African Journal of Aquatic Science,
31: 313-318.
Blay, J. (Jr.) 1995. Food and feeding habits of four species of
juvenile mullet (Mugilidae) in a tidal lagoon in Ghana.
Journal of Fish Biology, 46: 134-141.
Dada, J.O., Araoye, P.A. 2008. Some aspects of the biology
of Chrysichthys nigrodigitatus (Pisces: Siluroidae) in Asa
Lake, Ilorin, Nigeria. Nigerian Journal of Fisheries, 5:
73-84.
Oronsaye, C.G., Nakpodia, F.A. 2005. A comparative study
of the food and feeding habits of Chrysichthys
nigrodigitatus and Brycinus nurse in a tropical River.
Pakistan Journal of Scientific and Industrial Research,
48: 118-121.
Oso, J.A., Ayodele I.A., Fagbuaro, O. 2006. Food and feeding
habits of Oreochromis niloticus (L) and Sarotherodon
galilaeus (L) in a tropical reservoir. World Journal of
Zoology, 1: 118-121.
Ramirez-Luna, V., Navia, A.F., Rubio, E.A. 2008. Food habits
and feeding ecology of an estuarine fish assemblage of
Northern Pacific Coast of Ecuador. Pan-American
Journal of Aquatic Sciences, 3: 361-372.
the food items of C. nigrodigitatus to include gastropods,
nematodes, diatoms and crustaceans. However, in this study,
nematodes were not recorded.
From this study, it appears that there is likely to be inter-spe-
cific competition for food among the four fish species, due to
the fact that they all seem to have the same important food
item, i.e. diatoms, in common and thus competition for dia-
toms was high. C. hippos was an exception which had algae
Short Communication: Feeding Inter-relationship of Fish Species in Lagos Lagoon 51
0
20
40
60
80
100
Fig. 1. Food items of Caranx hippos.
Food items
Percentage (%)
Diatiom
Algae
Molluscs
Crustaceans
Fish
Detritus
Unidentified matter
2
4
6
8
1
0
d
Fig. 2. Food items of Chrysichthys nigrodigitatus.
Food items
Percentage (%)
Diatiom
Algae
Mollusc
Crustaceans
Fish
Plant material
Detritus
Unidentified matter
Fi
g
2
4
6
8
10
Fig. 3. Food items of Ethmalosa fimbriata.
Food items
Percentage (%)
Diatiom
Algae
Crustaceans
Fish
Plant material
Detnitus
Unidentified matter
Fig. 4. Food items of Mugil cephalus.
Fig
0
20
40
60
80
100
d
Food items
Percentage (%)
Diatiom
Algae
Crustaceans
Fish
Plant material
Detritus
Unidentified matter
Production and Characterization of Chitosan from
Shrimp (Penaeus semisulcatus ) Shell Waste of UAE
Fazilatun Nessa*, Saeed Ahmed Khan and Farah Mohammad Anas Al Khatib
Dubai Pharmacy College, Al Muhaisanah 1, Al Mizhar, P.O.Box. 19099, Dubai - United Arab Emirates
(received June 11, 2009; revised December 1, 2009; accepted December 16, 2009)
Abstract. Chitosan was prepared from shrimp (Penaeus semisulcatus) shell waste by a chemical process involving
demineralization, deproteinization and deacetylation; conversion of chitin to chitosan (deacetylation) was achieved by
treatment with concentrated sodium hydroxide solution (55%) at room temperature (25 ºC). The present study was
undertaken to evaluate the influence of deacetylation process during chitosan production on the physicochemical and
functional properties of shrimp shell chitosan. Four experimental chitosan samples were prepared with deacetylation
for 40 h, for 50 h, with and without stirring as well as for 60 h and were subjected to physicochemical and functional
characteristic analysis. Change in duration of deacetylation process yielded some differences in each characteristic;
deacetylation for 40 h led to lower viscosity, solubility, water/fat binding capacity and degree of deacetylation and for
60 h resulted in increase in solubility but decrease in viscosity. Stirring during deacetylation process led to lower
viscosity, higher degree of deacetylation and higher fat binding capacity of the product. In contrast non-stirred sample
produced product with lower degree of deacetylation and higher viscosity. It was concluded that duration of
deacetylaylation process should be monitored constantly for optimal chitosan production depending on its intended
usages in food, pharmaceutical and biomedical industries.
Keywords: shrimp shell waste, deacetylation, chitosan, chitin
Introduction
Chitosan is a fiber-like substance derived from chitin, a
homopolymer of ß-(1→4)-linked N-acetyl-D-glucosamine.
Chitin is widely distributed in marine invertebrates, insects,
fungi, and yeast (Subasingle, 1995; Austin et al., 1981); how-
ever, it is not present in higher plants and higher animals.
Generally, the shells of selected crustaceans consist of 30-40%
protein, 30-50% calcium carbonate and calciumc phosphate
and 20-30% chitin (Acosta et al., 1993; Knorr, 1984). Chitin is
widely available from a variety of sources among which, the
principal source is shellfish waste such as that of shrimps, crabs
and crawfish (Rinaudo, 2006; Allan and Hadwiger, 1979). It
also exists naturally in a few species of fungi (Franco et al.,
2004; Andrade et al., 2000; Chung et al., 1994). Chitin and
chitosan have similar chemical structures (Fig. 1). Chitin is made
up of a linear chain of acetylglucosamine groups while chitosan
is obtained by removing enough acetyl groups (CH3-CO) from
the molecule so that it becomes soluble in most diluted acids.
This process is called deacetylation. The actual difference
between chitin and chitosan is the acetyl content of the poly-
mer. Chitosan having a free amino group is the most useful
derivative of chitin (No and Meyers, 1992).
Chitosan is a non toxic, biodegradable polymer of high
molecular weight (Zhang and Neau, 2001; Tomihata and Ikada,
1997). Over the last several years, chitinous polymers, espe-
cially chitosan, have received increased attention as one of
the promising renewable polymeric materials for their exten-
sive applications in the pharmaceutical and biomedical indus-
tries for enzyme immobilization and purification, in chemical
plants for wastewater treatment and in food industries for use
in food formulations as binding, gelling, thickening and stabi-
lizing agent (Prashanth and Tharanathan, 2007; Franco et al.,
2004; Knorr, 1984).
Traditional isolation of chitosan from crustacean shell waste
consists of four basic steps: demineralization, deproteinization,
decolourization and deacetylation (Galed et al., 2008; No and
Meyers, 1995). Several procedures have been developed and
proposed by many researchers over the years for preparation
of chitosan from different crustacean shell wastes (Galed
Pak. J. Sci. Ind. Res. 2010 53 (1) 52-58
*Author for correspondence; E-mail: nessa1995@yahoo.com
Fig. 1. Structure of chitin and chitosan.
52
Technology
HO
HO
HO
HO
HO
HO
NHCOCH
3
NHCOCH
3
NH
2
NH
2
NH
2
NHCOCH
3
CH OH
2
CH OH
2
CH OH
2
CH OH
2
CH OH
2
CH OH
2
O
OO
OO
O
O
O
O
O
O
O
Chitin
Chitosan n
n
et al., 2008; No and Meyers, 1995; No et al., 1989). Some of
these formed the basis of chemical processes for industrial
production of chitosan. But most of the reported processes
were carried out with 45% concentrated sodium hydroxide
solution at 100 °C or higher temperature with autoclaving
(Galed et al., 2008; Prashanth and Tharanathan, 2007; Domard
and Rinaudo, 1983; Horton and Lineback, 1965). Therefore,
the specific objectives of this work were to develop an
optimum shrimp shell chitosan production process at room
temperature (25 °C) with increased alkali strength without
decolourization step and to study the influence of deacetylation
process on the physicochemical and functional properties of
shrimp shell chitosan.
Materials and Methods
Shrimp shell chitosan production. Penaeus semisulcatus,
Metapenaeus mastersii and Penaeus latisulcatus are the
shrimp species found in UAE waters of which Penaeus
semisulcatus is the most common and commercially impor-
tant species. Undersized shrimp shell waste of Penaeus
semisulcatus was obtained from a commercial shrimp shell
processor of Dubai, UAE. Upon receipt, shells (head, body
and tail) were washed under running warm tap water to
remove soluble organics, adherent proteins and other impuri-
ties. The shells were then dried in the oven (Mammert,
Germany) at 70 °C for a period of 24 h or longer until
completely dried shells were obtained. The moisture content
of dried shell was 0.48%. To obtain a uniform size product,
the dried shell was ground through a centrifugal grinding mill
and sifted with 20-mesh (0.841 mm) and 40-mesh (0.425 mm)
sieves. Dried ground shell powder was placed in opaque
plastic bottles and stored at room temperature until used. The
production of chitosan from shrimp shell waste was carried
out with a modified method of No et al. (1989). The dried
shrimp shell powder (5 kg) was demineralized with 8-10%
hydrochloric acid at ambient temperature with a solid to
solvent ratio of 1:15 (w/v), in an acid resistant vessel with
stirrer for 20-22 h until deminerali-zation was completed. The
demineralized shells were deproteinized with 8-10% sodium
hydroxide solution for 20-22 h at 65 °C with constant stirring
or without stirring at a solid to solvent ratio of 1:10 (w/v).
Samples were then washed with tap water and dried under
vacuum for 2-3 h until the powder was crispy. Removal of
acetyl groups from chitin was achieved by using concentrated
sodium hydroxide solution (55%) with a solid to solvent ratio
of 1:10 (w/v). Samples of four experimental shrimp shell
chitosans were prepared. The chemical reactions were car-
ried out at room temperature (25 °C). Duration of deacetylation
process was 40 h for sample C40, 50 h for C50S (with magnetic
stirring), 50 h for C50WS (without stirring) and 60 h for C60.
The resulting chitosans were washed to neutrality in running
tap water, rinsed with distilled water, filtered and dried at
60 °C for 24 h in the oven. The obtained shrimp shell chitosan
was white to off white in colour and it was not necessary to
decolourize or bleach it.
Physicochemical and functional properties. Measurement
of nitrogen. Nitrogen of the crawfish chitosan was determined
using a microprocessor-based, software-controlled instrument
Model-TruSpec CN (Model # FP-428 Leco Corporation,
USA). There were three phases during an analysis cycle, i.e.,
purging, burning and analysis. The encapsulated sample was
purged of any atmospheric gases that had entered during
sample loading. During the burning phase, the sample was
dropped into a hot furnace (850 °C) and flushed with pure
oxygen for a very rapid combustion. Finally, in the analysis
phase, the remaining combustion product (nitrogen) was mea-
sured by the thermal conductivity cell. The final result was
displayed as percent nitrogen.
Ash. Ash of the crawfish chitosan was calculated according
to the standard method # 923.03 (AOAC, 1990). 2.0 g of
chitosan (triplicate) were placed into previously ignited,
cooled, and tarred crucible. The samples were heated in a
muffle furnace preheated to 600 °C for 6 h. The crucibles
were allowed to cool in the furnace to less than 200 ºC
and then placed in desiccator with a vented top. Crucibles
were cooled, weighed and ash content was recorded.
Degree of deacetylation. Chitosan samples prepared in the
form of KBr discs were studied for the degree of deacetylation
(DD) (Kassai, 2008; Khan et al., 2002). The prepared chitosan
KBr discs were kept in desiccators for 12 h and then placed in
sealed plates before scanning. The DD of chitosan was estab-
lished using a FTIR (Fourier Transform Infrared Spectros-
copy) instrument (Model # M2000, Midac Corp. USA) with
frequency of 4000-4/cm. The degree of deacetylation (DD)
of the chitosan was calculated using the baseline reported by
Khan et al. (2002). The computation equation for the baseline
is given below:
DD = 100 – [(A1655 / A3450) × 100 / 1.33]
where A1655 and A3450 are the absorbance at 1655 cm-1 of
the amide-I band as a measure of the N-acetyl group con-
tent and at 3450 cm-1 of the hydroxyl band as an internal
standard to correct for disc thickness. The factor ‘1.33’
denotes the value of the ratio of A1655/A3450 for fully
N-acetylated chitosan.
Viscosity. Viscosity of chitosan was determined with a
Brookfield viscometer (Model DV-II + Brookfield Engineering
53
Production of Chitosan from Shrimp Shell Waste

Laboratories Inc., Stonghton, MA.). Chitosan solution was
prepared in 1% acetic acid at 1% concentration on dry
basis. Measurement was made in duplicate using a No. 27
spindle at 50 rpm on solutions at 25 °C with values reported
in centipoise (cP) unit.
Solubility. Crawfish chitosan sample (0.1 g in triplicate) was
placed in a centrifuge tube (known weight) then dissolved with
10 ml of 1% acetic acid for 30 min. The solution was then
centrifuged at 10,000 rpm for 10 min. The supernatant was
decanted. The undissolved particles were washed in
distilled water (25 ml) then centrifuged at 10,000 rpm. The
supernatant was removed and undissolved pellets were dried
at 60 °C for 24 h. Finally, the particles were weighed and the
percentage solubility was determined.
Water binding capacity (WBC). WBC of chitosan was
measured using a modified method of Knorr (1982). Initially
a centrifuge tube containing 0.5 g of sample was weighed,
10 ml of water was added and mixing was carried out on a
vortex mixer for one min to disperse the sample. The con-
tents were left at ambient temperature for 30 min with inter-
mittent shaking for 5 s every 10 min and then centrifuged
(Model # Z383K, HERMLE-National Labnet Company,
USA) at 3,500 rpm (6,000 × g) for 25 min. After the super-
natant was decanted, the tube was weighed again. WBC was
calculated as follows:
WBC (%) = [water bound (g)/ initial sample weight (g)]× 100.
All experiments were carried out in triplicate.
Fat binding capacity (FBC). FBC of chitosan was measured
using a modified method of Knorr (1982). Initially a centri-
fuge tube containing 0.5 g of sample was weighed, 10 ml of
oil (three types of oil were used namely soybean, corn and
sunflower oils) were added and mixing was carried out on a
vortex mixer for 1 min to disperse the sample. The contents
were left at ambient temperature for 30 min with shaking
for 5 s every10 min and then centrifuged at 3,500 rpm
(6,000 × g) for 25 min. After the supernatant was decanted,
the tube was weighed again. FBC was calculated as follows:
FBC (%) = [fat bound (g)/ initial sample weight (g)] × 100.
Experiments were performed in triplicate.
Statistical analysis. All experiments were carried out in
triplicate, Average values (means) and standard deviations
were reported. Mean separations were analyzed using the
ANOVA and Tukey’s student range tests at á = 0.05.
Results and Discussion
Yield. Yield was calculated as the dry weight of chitin
obtained from 5 kg of dried shrimp shell powder. The yield of
chitin was 20% and that of chitosan ranged from 16-19%.
The highest yields were obtained from sample C40 (19%),
followed by C50WS (18%), C60 (17%), and C50S (16%). Results
are shown in Table 1. Brzeski (1982) reported about 14% yield
of chitosan from krill and 18.6% from prawn waste (Alimuniar
and Zainuddin, 1992). The yield of chitosan obtained (15-
18%) is lower than that (approximately 23%) of chitin
reported in the literature (No and Meyers, 1989). This may be
due to loss of sample mass/weight during deacetylation pro-
cess as we used here 55% concentrated sodium hydroxide
solution, whereas in other methods 45% sodium hydroxide
solution was used. The moisture content of the shrimp shell
chitosan, determined by the gravimetric method (Black, 1965),
was in the range of 0.3% to 0.4% (Table 1).
Table 1. Proximate analysis of shrimp shell and commercial
chitosans (dry weight basis)
Sample Yield Moisture Nitrogen Ash
(%) (%) (%) (%)
C40 19 0.4 (0.25)a*8.33 (0.02)a*0.29 (0.07)a*
C50S 16 0.3 (0.20)a8.19 (0.01)a0.3 (0.99)a
C50WS 18 0.4 (0.25)a8.11 (0.05)a0.3 (0.23)a
C60 17 0.4 (0.22)a7.91 (0.05)a0.3 (0.98)a
Sigma 91** 2.5 (0.11)b8.23 (0.09)a1.5 (0.25)a
* = numbers in parentheses are standard deviations; means with
different letters in each column are significantly different (P < 0.05);
** Sigma 91 is a commercial crab chitosan.
Nitrogen content. Nitrogen content of the shrimp shell
chitosan samples varied between 7.91% and 8.33% on a dry
basis, showing no significant differences (P >0.05) in nitro-
gen content, but the values were slightly higher than that
(7.06% to 7.97%) reported by No and Meyers (1995), for
chitosan from crab and shrimp shell on a dry basis. This is
probably due to the presence of protein residues as mentioned
by Rutherford and Austin (1978). Protein is bound by cova-
lent bonds forming stable complex with chitin and chitosan.
Thus, it is very difficult to achieve 100% deproteinization.
Even with complete deprotinization, nitrogen was still present
since chitosan has the amino (-NH2) group.
Ash. Table 1 shows the ash content of shrimp shell chitosan
in the range of 0.29-0.3%. Ash measurement is an indicator of
the effectiveness of the demineralization step for removal of
calcium carbonate. Elimination of demineralization step
results in products having 31-36% ash (Bough et al., 1978).
Some residual ash of chitosans may affect their solubility,
consequently contributing to lower viscosity, or can affect other
more important characteristics of the final product. A high
54 Fazilatun Nessa et al.

quality grade of chitosan should have less than 1% of ash
content (No and Meyers, 1995). An ash content of less than
1% from crab chitosans has been reported by No and Meyers
(1995). The results presented in Table 1 indicate that the
resultant chitosan sample was completely demineralized and
contained less than 1% ash.
Degree of deacetylation. The degree of deacetylation
(DD) of the studied shrimp shell chitosan samples ranged
from 55% to 76% (Table 2). According to No and Meyers
(1995), DD of chitosan ranges from 56% to 99% with an
average of 80%. Sample C50S (76%) had the highest DD,
followed by C50WS, C60 and C40 (75%, 74%, and 55%,
respectively).
As in the Table 2, C40 had a very low solubility and viscosity
which may be due to the lower DD value. Therefore,
comparison among samples C50S, C50WS and C60, sample C50S
gave lower viscosity (136.6 cP) and higher DD (76%) value
which are very important characteristics of chitosan. The
medical and pharmaceutical applications of chitosan as
antitumor, hemostatic, hypocholesterolemic, antimicrobial
and antioxidant depends mostly upon DD and solubility (Jian
et al., 2008; Muzzarelli and Muzzarelli, 2005). However, we
expected that samples C50S, C50WS and C60 would have higher
DD with higher solubility but the values obtained were lower
than the expected ones. According to Kassai (2008) and Khan
et al. (2002) , the IR spectroscopic method is commonly used
for the estimation of chitosan DD values for its advantages: it
is relatively fast and does not require dissolution of the chitosan
sample in an aqueous solvent. DD values are not only highly
dependent on the source and method of purification (No et
al., 1989) but also on the type of analytical methods employed,
sample preparation and type of instrument used; other condi-
tions may also influence the analysis of DD (Kassai 2008;
Khan et al., 2002).
Viscosity. The viscosity of chitosan solutions, reported
in the literature, generally ranges from 60 to 780 cP
(Alimuniar and Zainuddin, 1992). This range of viscosity
was also observed by Cho et al. (1998) for five commer-
cially available chitosans. The results of viscosity, solubility
and degree of deacetylation of our shrimp shell chitosans
are shown in Table 2.
Bough et al. (1978) stated that viscosity of chitosans varied
considerably from 60 to 5,110 cP depending on the species.
Our shrimp shell samples had viscosity ranging from 90.7 to
170.2 cP. C40 had the lowest viscosity (90.7 cP) comparable
to that of other samples as of lower solubility may be due
to incomplete deacetylation of the sample. Whereas C50WS had
a very high viscosity (170.2 cP) (Table 2). Some factors
affect viscosity during the production of chitosan such as the
degree of deacetylation, molecular weight, concentration, ionic
strength, pH and temperature, etc. Moorjani et al. (1975)
reported that viscosity of chitosan decreased with increasing
time of demineralization. The viscosity of chitosan in acetic
acid tends to increase with decreasing pH but decrease with
decreasing pH in HCl. Intrinsic viscosity of chitosan is a
function of the degree of ionization as well as ion strength
(Bough et al., 1978). Deproteinization with 3% NaOH and
elimination of the demineralization step in chitin preparation,
decreased the viscosities of the final chitosan samples (Bough
et al., 1978). Moorjani et al. (1975) stated that it is not
desirable to bleach the material at any stage since bleaching
considerably reduces the viscosity of the final chitosan prod-
uct. Our product, prepared without bleaching step, gave lower
viscosity which was desirable for preservation of foods against
microbial deterioration, formation of biodegradable films and
medical applications (Liu et al., 2008; Zeng et al., 2008).
Solubility. Three shrimp shell chitosan samples demonstrated
excellent solubility ranging from 98.01 to 99% with no
significant difference (Table 2), except sample C40, which
showed comparatively lower solubility (60.3%); it may be due
to lower degree of deacetylation. Brine and Austin (1981)
noted that lower solubility values suggested incomplete
removal of protein and acetyl group. Since solubility of
chitosan depends on the removal of acetyl group from chitin
therefore lower DD value and the presence of protein
contaminants remaining in the sample during the analysis
process could adversely interfere with the results.
Water binding capacity (WBC). Water binding capacity of
shrimp shell and commercial chitosans are shown in Table 3.
WBC differed among crawfish chitosan samples, ranging from
299.6 % to 745.4%. There were no significant differences in
Table 2. Viscosity, solubility and degree of deacetylation of
shrimp shell and commercial chitosans
Sample Viscosity Solubility Degree of
(cP) (%) deacetylation
(%)
C40 90.7 (5.07)*a 60.3 (0.61)a55
C50S 136.6 (2.09)b98.2 (0.66)b76
C50WS 170.2 (3.66)c98.01 (0.45)b75
C60 154.29 (2.69)d99.00 (0.56)b74
Sigma 91** 380.15 (3.44)e89.88 (0.42)c74
* = numbers in parentheses indicate standard deviation; means with
different letters in each column are significantly different (P < 0.05);
** Sigma 91 is a commercial crab chitosan.
55
Production of Chitosan from Shrimp Shell Waste

WBC between C50S, C50WS and C60. These values were in agree-
ment, except C40, with those reported by Cho et al. (1998)
where WBC for chitosans ranged from 458% to 805% for
five commercial chitosans from shrimp and crab shell. Sample
C40 had a lower WBC (299.6 %) than that of other samples; it
may be due to lower DA value.
Fat binding capacity (FBC). Fat binding capacity (FBC) of
four shrimp shell chitosans was measured using three types of
oils including soybean, corn, and sunflower oil. The results
are shown in Table 3. FBC differed among chitosan products,
ranging from 245.5% to 599.2%. Among our crawfish chitosan
samples, C50S showed the highest FBC values: 587.3% with
soybean oil, 599.2% with corn oil and 586.8% with sunflower
oil, although C50S had low viscosity (136.6 cP); C50S, C50WS
and C60 showed no significant difference in FBC.
The sample C40 showed the lowest FBC (245.5%-258.7%) as
it was not properly deacetylated; it seems higher deacetylation
facilitates oil binding capacity of chitosan. Several workers
suggested that the DD of chitosan is an important factor which
influences fat binding capacity of chitosan (Shahidi et al.,
2002). They suggested that increased DD causes increased
electrostatic force between chitosan and fatty and bile acid
and increased FBC. Moorjani et al. (1975) advocated that
changing the sequence of steps, when demineralization is
conducted prior to deproteinization and finally deacetylation,
results in an increase in FBC than when deproteinization is
conducted prior to demineralization and finally deacetylation.
Amongst the three types of oil used, soyabean oil generally
demonstrated more FBC with shrimp shell chitosan samples,
whereas sunflower oil showed the least FBC. Regardless of
the type of vegetable oils, the four prepared shrimp shell
chitosan samples showed desirable FBC ranging from 566.9%
(with sunflower) to 599.2% (with corn) which is in agreement
with those (314 to 535% with an average of 417%) reported
by No et al. (2000). Sample C40 showed lower value than the
reported value. It seemed degree of deacetylation influenced
the fat binding capacity of chitosan.
Conclusion
Throughout the literature on chitosan, the main emphasis is
on its quality and physicochemical properties which vary
widely with the crustacean species and the preparation
methods. Most of the reported preparation methods used high
temperature with 45% concentrated alkali and sometimes used
autoclave. Based on the reported practice this research study
attempted to present a process for the production of shrimp
shell chitosan at room temperature (25°C) with increased
alkali strength (55%); it could help to develop small industry
without wasting energy. This study also demonstrated that the
duration of deacetylation process affects the quality of the
products. In view of the foregoing, it is our recommendation
that for the purpose of achieving uniformity and proper
product quality control for particular usage of chitosan, the
relationship between the process protocols/conditions and the
resulting specific characteristics of chitosan products must be
monitored constantly and properly.
References
Acosta, N., Jimenez, C., Borau, V., Heras, A. 1993. Extrac-
tion and characterization of chitin from crustaceans.
Biomass and Bioenergy, 5: 145-153.
Alimuniar, A., Zainuddin, R. 1992. An economical technique
for producing chitosan. In: Advances in Chitin and
Chitosan, C.J. Brine, P.A. Sanford and J.P. Zikakis (eds.),
pp.627-632. Elsevier Applied Science, Essex, UK.
Allan, R., Hadwiger, L.A. 1979. The fungicidal effect of
chitosan on fungi of varying cell wall composition.
Experimental Mycology, 3: 285-287.
Andrade, V.S., Neto, B.B., Souza, W., Campos-Takaki, G.M.
2000. A factorial design analysis of chitin production by
Cunninghamella elegans. Canadian Journal of Micro-
biology, 46: 1042-1045.
AOAC. 1990. Official Methods of Analysis of Association of
Official Analytical Chemists, 15th edition, AOAC
Washington, DC., USA.
Table 3. Water binding capacity and fat binding capacity of shrimp shell and commercial chitosans
Sample WBC (%) Fat binding capacity (%)
Soybean oil Corn oil Sunflower oil
C40 299.6 (9.97)*a258.7(8.9)a245.5 (4.8)a255.7 (5.3)a
C50S 738.8 (5.6)b587.3 (5.3)b599.2 (8.5)b586.8 (9.9)b
C50WS 745.4 (4.9)b571.5 (7.9)b583.6 (7.3)b579.4 (5.6)b
C60 732.2 (4.04)b575.8 (6.5)b577.5 (6.7)b566.9 (7.6)b
Sigma 91** 538.5 (4.99)c379.7 (5.9)c444.3 (5.3)c398.6 (6.6)c
* = numbers in parentheses indicate standard deviation; means with different letters in each column are significantly different (P < 0.05);
** = Sigma 91 is a commercial crab chitosan.
56 Fazilatun Nessa et al.
Austin, P.R., Brine, C.J., Castle, J.E., Zikakis, J.P. 1981. Chitin:
New facets of research. Science, 212: 749-753.
Black, C.A. 1965. Methods of Soil Analysis: Part I. Physical
and Mineralogical Properties, 2nd edition, American
Society of Agronomy, Madison, Wisconsin, USA.
Bough, W.A., Salter, W.L., Wu, A.C.M., Perkins, B.E. 1978.
Influence of manufacturing variables on the characteris-
tics and effectiveness of chitosan products. 1. Chemical
composition, viscosity, and molecular weight distribution
of chitosan products. Biotechnology and Bioengineering,
20: 1931-1942.
Brine, C.J., Austin, P.R. 1981. Chitin variability with species
and method of preparation. Comparative Biochemistry
and Physiology, 69B: 283-286.
Brzeski, M.M. 1982. Concept of chitin/chitosan isolation from
Antarctic krill (Euphausia superba) shells on a technique
scale. In: Proceedings of the Second International Con-
ference on Chitin and Chitosan, S. Hirano and S. Tokura
(eds.), The Japan Society of Chitin and Chitosan, Sapporo,
Japan.
Cho, Y.I., No, H.K., Meyers, S.P. 1998. Physicochemical
Characteristics and Functional Properties of Various
Commercial Chitin and Chitosan Products. Journal of
Agriculture and Food Chemistry, 46: 3839-3843.
Chung, L.Y., Schmidt, R.J., Hamlyn, P.F., Sagar, B.F.,
Andrews, A.M., Turner, T.D. 1994. Biocompatibility of
potential wound management products: fungal mycelia
as a source of chitin/chitosan and their effect on the
proliferation of human F1000 fibroblasts in culture.
Journal of Biomedical Materials Research, 28: 463-469.
Domard, A., Rinaudo, M. 1983. Preparation and character-
ization of fully deacetylated chitosan. International Jour-
nal of Biological Macromolecules, 5: 49-52.
Franco, L.O., Maia, R.C.C., Porto, A.L.F., Messias, A.S.,
Fukushima, K., Campos-Takaki, G.M. 2004. Heavy metal
biosorption by chitin and chitosan isolated from
Cunninghamella elegans (IFM 46109). Brazilian
Journal of Microbiology, 35: 243-247.
Galed, G., Diaz, E., Heras, A. 2008. Conditions of N-
deacetylation on chitosan production from alpha chitin.
Natural Product Communications, 3: 543-550.
Horton, D., Lineback, D.R. 1965. N-deacetylation, chitosan
from chitin. In: Methods in Carbohydrate Chemistry,
R.L.Whistler and M.L. Wolfson (eds.), pp.403, New York,
USA.
Jian, Y., Feng, T., Zheng, W., Qing, W., Yan-Jun, Z., Shi-Qian,
C. 2008. Effect of chitosan molecular weight and
deacetylation degree on hemostasis. Journal of Biomedical
Materials Research, 84B: 131-137.
Kassai, M.A. 2008. Review of several reported procedures to
determine the degree of N-acetylation for chitin and
chitosan using infrared spectroscopy. Carbohydrate
Polymers, 71: 497-508.
Khan, T.A., Peh, K.K., Ch’ng, H.S. 2002. Reporting degree
of deacetylation values of chitosan: The influence of
analytical methods. Journal of Pharmacy and Pharma-
ceutical Sciences, 5: 205-212.
Knorr, D. 1984. Use of chitinous polymers in food−A
challenge for food research and development. Food
Technology, 38: 85-97.
Knorr, D. 1982. Functional properties of chitin and chitosan.
Journal of Food Science, 47: 593-595.
Liu, J., Zhang, J., Xia, W. 2008. Hypocholesterolaemic
effects of different chitosan samples in vitro and in vivo.
Food Chemistry, 107: 419-25.
Moorjani, M.N., Achutha, V., Khasim, D.I. 1975. Parameters
affecting the viscosity of chitosan from prawn waste.
Journal of Food Science and Technology, 12: 187-189.
Muzzarelli, R.A.A., Muzzarelli, C. 2005. Chitosan Chemis-
try: Relevance to the biomedical sciences. Advances in
Polymer Science, 186: 151-209.
No, H.K., Lee, K.S., Meyers, S.P. 2000. Correlation between
physicochemical characteristics and binding capacities
of chitosan products. Journal of Food Science, 65: 1134-
1137.
No, H.K., Meyers, S.P. 1995. Preparation and characteriza-
tion of chitin and chitosan - A Review. Journal of Aquatic
Food Product Technology, 4: 27-52.
No, H.K., Meyers, S.P. 1992. Utilization of crawfish process-
ing wastes as carotenoids, chitin and chitosan sources.
Journal of Korean Society of Food and Nutrition, 21:
319-326.
No, H.K., Meyers, S.P. Lee, K.S. 1989. Isolation and charac-
terization of chitin from crawfish shell waste. Journal of
Agriculture and Food Chemistry, 37: 575-579.
No, H.K., Meyers, S.P. 1989. Crawfish chitosan as a coagu-
lant in recovery of organic compounds from seafood
processing streams. Journal of Agriculture and Food
Chemistry, 37: 580-583.
Prashanth, R., Tharanathan, R. 2007. Chitin/chitosan: Modi-
fications and their unlimited application potential - an
overview. Trends in Food Science and Technology, 18:
117-131.
Rinaudo, M. 2006. Chitin and chitosan: Properties and appli-
cations. Progress in Polymer Science, 31: 603-632.
Rutherford, F.A., Austin, P.R. 1978. Marine chitin properties
and solvents. In: Proceedings of the First International
Conference on Chitin/Chitosan, R.A.A. Muzzarrelli and
P.R. Austin (eds.), pp.182-192, MIT Sea Grant Pro-
gramme, Cambridge, MA, USA.
57
Production of Chitosan from Shrimp Shell Waste
Shahidi, F., Kamil J., Jeon, Y.J., Kim, S.K. 2002. Antioxidant
role of chitosan in a cooked cod (Cadus morhua) model
system. Journal of Food Lipids, 9: 57-64.
Subasingle, S. 1995. The development of crustacean and
mollusc industries for chitin and chitosan resources. In:
Chitin and Chitosan, M.B. Zakaria, W.M. Wan Muda
and M.P. Abdullah (eds.), pp. 27-34, Penerbit Universiti
Kebangsaan, Malaysia.
Tomihata, K., Ikada, Y. 1997. In vitro and in vivo degradation
of films of chitins and its deacetylated derivatives.
Biomaterials, 18: 567-575.
Zeng, L., Qin, C., Wang, W., Chi, W., Li, W. 2008. Absorption
and distribution of chitosan in mice after oral
administration. Carbohydrate Polymers, 71: 435-440.
Zhang, H., Neau, S.H. 2001. In vitro degradation of chitosan
by a commercial enzyme preparation: effect of molecular
weight and degree of deacetylation. Biomaterials, 22:
1653-1658.
58 Fazilatun Nessa et al.
