29015
User Manual: 29015
Open the PDF directly: View PDF ![]() .
.
Page Count: 40

3,300+
OPEN ACCESS BOOKS
107,000+
INTERNATIONAL
AUTHORS AND EDITORS 113+ MILLION
DOWNLOADS
BOOKS
DELIVERED TO
151 COUNTRIES
AUTHORS AMONG
TOP 1%
MOST CITED SCIENTIST
12.2%
AUTHORS AND EDITORS
FROM TOP 500 UNIVERSITIES
Selection of our books indexed in the
Book Citation Index in Web of Science™
Core Collection (BKCI)
Chapter from the book
Tande m Mass Spectrometry - Applications and Principles
Downloaded from: http://www.intechopen.com/books /tandem-mass-spectrometry-
applications-and-principles
PUBLISHED BY
World's largest Science,
Technology & Medicine
Open Access book publisher
Interested in publishing with InTechOpen?
Contact us at book.department@intechopen.com

21
HPLC-MS/MS
of Highly Polar Compounds
Luigi Silvestro1, Isabela Tarcomnicu2 and Simona Rizea Savu1
13S-Pharmacological Consultation & Research GmbH, Harpstedt,
2Pharma Serv Int’l SRL, Bucharest,
1Germany
2Romania
1. Introduction
Complicate high-performance liquid chromatographic (HPLC) interfaces like the moving
belt, direct liquid interface, thermospray (Arpino, 1985) fast atom bombardment (FAB)
(Caprioli et al., 1986; Garcia & Barceló, 1993; Dell et al., 1988; Larry et al., 1989) characterized
the early days of HPLC used in tandem with mass-spectrometry (MS); all of them, apart
FAB, were more effective with moderately polar compounds (Strege et al., 1999). After years
of difficulties and struggles the introduction of atmospheric pressure ionization by
electrospray (ESI) or chemical ionization marked a clear breakthrough: an effective, robust
and user friendly HPLC interface was finally available.
As a matter of fact, considering the physicochemical principles of the source, the polar
compounds were the first compounds successfully ionized (Griffits, 2008).
Despite the huge potential of ESI for polar and highly polar compounds the applications for
the latter proved to be critical due to the fact that effective HPLC separations often require
non volatile buffers and/or high water content that are not suitable in MS detection.
Along the years, the development of HPLC columns, specific derivatization methods, new
chromatographic techniques and sample preparation approaches have increased the
number of applications with highly polar compounds. Three classes of not so widely
studied molecules have been selected in this study in order to illustrate the application of
HPLC-MS/MS to highly polar compounds; updates as well a review of past data will be
presented.
The groups of compounds taken in consideration are: glycosaminoglycans, bisphosphonates
and amynoglycoside antibiotics; the first-ones are both endogenous and exogenous while
the last 2 classes are of typical pharmaceutical interest.
Obviously other more well–known molecules like peptides and polynucleotides are also
included in this class of highly polar compounds but HPLC-MS/MS methods in these
particular fields are widely described and probably represent the most common areas of
application. It is also important to keep in mind that this chapter doesn’t intend to be an
exhaustive presentation of the published analytical methods for heparins, bisphosphonates
or aminoglycosides, but these compounds are used as models to emphasize the potential of
HPLC-MS/MS in the analysis of highly polar molecules.
www.intechopen.com
Tandem Mass Spectrometry – Applications and Principles
494
2. Glycosaminoglycans
2.1 Chemical structure and main features of glycosaminoglycans
Glycosaminoglycans (GAGs) are a family of highly sulphated, complex, polydispersed
linear polysaccharides that display a variety of important biological roles. They contain
amino-sugars with alternating disaccharide units, which comprise an acidic monosaccharide
such as glucuronic or iduronic acid and a basic monosaccharide such as N-
acetylglucosamine or N-acetyl-galactosamine; both monosaccharides can be sulphated. In
natural biological systems GAGs are mainly linked to proteins to form proteoglycans, with
great interest for medical science being structural proteins or receptor as well adhesion
molecules (just to mention a few examples) and can be categorized, based on the GAGs
structure, into three main structural groups: heparin/heparan sulphate group,
dermatan/chondroitin sulphate group and hyaluronan (Mao et al., 2002; Tiayu Peng, 2002).
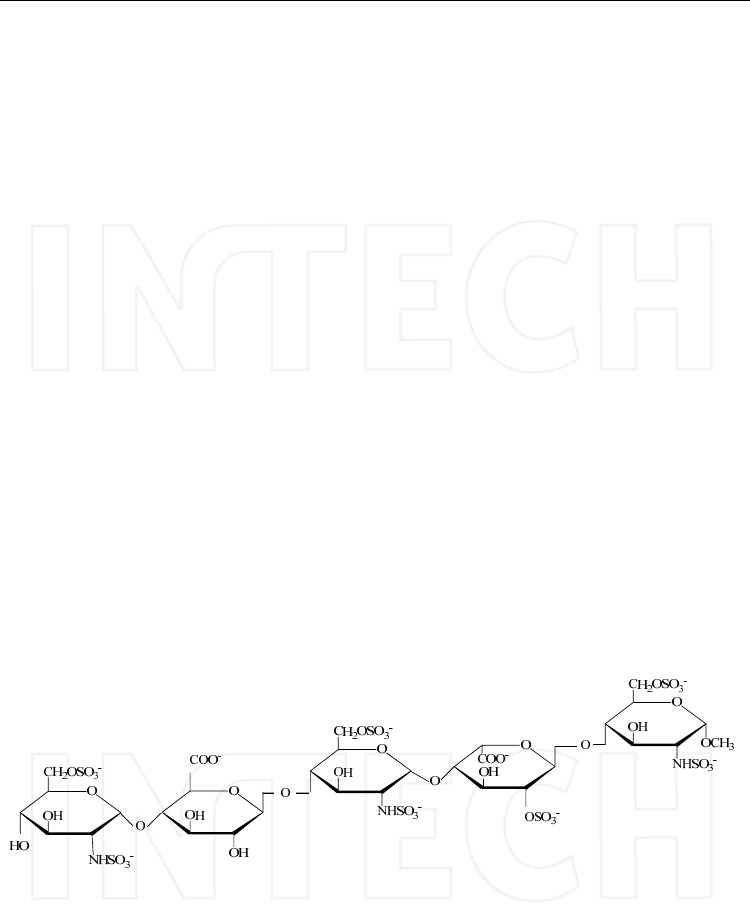
Heparin and heparan sulphate occur as proteoglycans in which the glycosaminoglycan chains
have different lengths and are composed of disaccharide units consisting of L-iduronic or D-
glucuronic acids (1-4) linked to glucosamine units that can be N-acetylated or N-sulphated
(Figure 1).
Heparin and heparan sulphate have closely related structures and consist of similar
disaccharide composition except that heparin has a greater content of iduronic acid and is
more highly sulphated per polysaccharide chain than is heparan sulphate. The differences
between the chemical structure of the sequences of heparin and heparan sulphate explain
the biological function related to them. As an example, the specific interaction of heparin
with the protein antithrombin III, determining its anticoagulant activity, is mediated by a
characteristic pentasaccharide that is not normally present in heparan sulphate and it
accounts for just a few percent of the heparin mass (Saad & Leary, 2003).
Fig. 1. Main sequence structure of heparin/heparan sulphate.
The dermatan/chondroitin sulphate are the main components of connective tissues and the
chemical structure comprises alternating uronic acid and N-acetyl-galactosamine residues;
depending on their composition 3 main types of chondroitin sulphates have been identified
(Gunay & Linhardt, 1999). The structures of these types of chondroitin sulphate are
presented in Figure 2.
www.intechopen.com
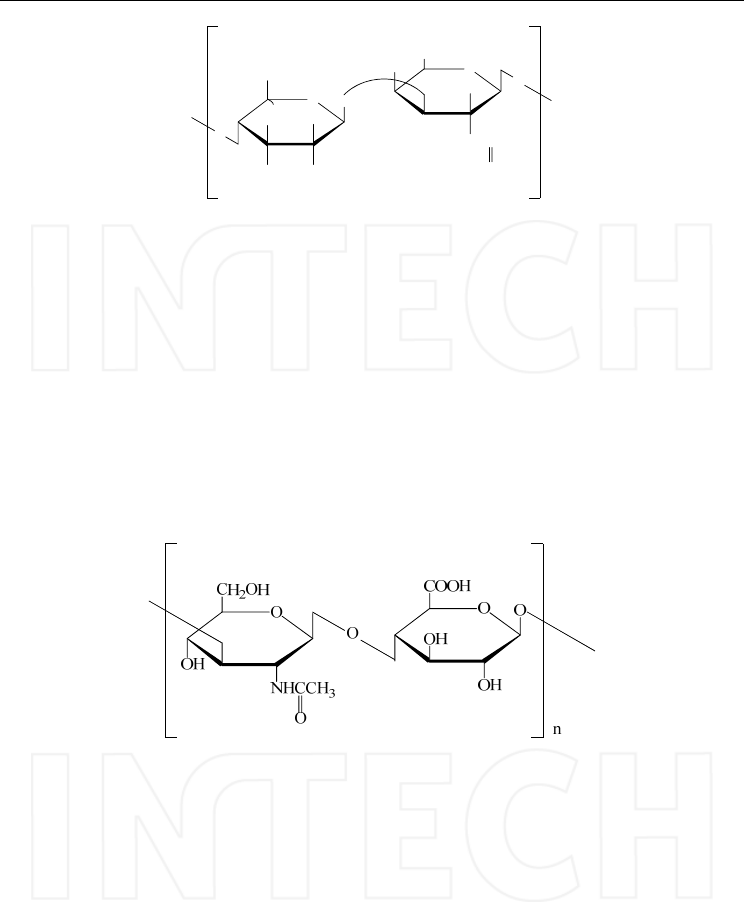
HPLC-MS/MS of Highly Polar Compounds
495
OO
O
H2COR2
COOH
OH
OH
R1O
NHCHCH3
O
O
O
n
R
Chondroitin Sulphate Type A: R=H, R1=SO3H, R2=H, R3=COOH
Chondroitin Sulphate Type B: R=COOH, R1= SO3H, R2=H, R3=H
Chondroitin Sulphate Type C: R=H, R1=H, R2=SO3H, R3=COOH
Fig. 2. Structure of chondroitin sulphate.
Chondroitin sulphate A and chondroitin sulphate C contain D-glucuronic acid, N-
acetylgalactosamine and sulphate residues in equimolar quantities but differ in the position
of the sulphate ester group (Saito et al., 1968). Dermatan sulphate formerly called
chondroitin sulphate B is similar to chondroitin sulphate A but instead of D-glucuronic acid
it consists mainly of L-iduronic acid.
Hyaluronic acid has alternating repeating units of the structure, (1→3) -N-acetyl-D-
glucosamine (1→4) -D-glucuronic acid, but tipically it is not sulphated and the molecules
are larger than the other glycosaminoglycans, often several million Da, with special
rheologic characteristic (Mao et al., 2002).
Fig. 3. Structure of the repeating units of Hyaluronic acid.
2.2 Special problems of these molecules and potential approach to solve them
GAGs are extremely difficult to analyze because of their polydispersity and
microheterogeneity of chemical structure combined with high polarity. The first two aspects
(polydispersity and microheterogeneity) push to get an optimal separation of all species
presented in a sample in order to may accurately characterize each component. It is in fact
important to consider that these molecules are closely related polymers with common
disaccharides/oligosaccharides and if not well separated it is also impossible to get detailed
structure information.
In the meantime the high polarity of GAGs make them difficult compounds for optimal
chromatographic separation and an adequate separation of all components present in a
GAGs sample is virtually impossible.
In such conditions two approaches are feasible:
www.intechopen.com

Tandem Mass Spectrometry – Applications and Principles
496
1. Application of analytical techniques able to gather structure data also of unfractionated
or partially fractionated complex mixtures
2. Structure specific degradation of the GAGs in order to reduce such complex samples
with almost infinite variant of similar polymers to more discrete mixtures of oligomers
corresponding to their “building blocks” that can be properly analyzed and quantified
obtaining quite detailed information of the polymers themselves intended as a mean
structure or a fingerprint.
Regarding the first approach, only NMR permits to get in-depth chemical structure
information of almost unfractionated materials but the instrumental sensitivity is a major
drawback when studying limited biological samples and in any case the information
obtained are practically a mean of the individual structures of the components present in the
sample. Despite the fact that hyphenation of NMR and HPLC (as separation method) is
feasible, due to sensitivity problems its application on such complex samples is more
theoretical then practical so far.
Before to close this parenthesis on NMR, outside the scope of this chapter, it is evident that
NMR spectroscopy is also a very effective technique for the characterization of the
oligosaccharides (Yung, 2011). It gives valuable information on monosaccharide
composition, glycosidic linkage, uronic acid type and sulfation patterns (Bo et al., 2011), but
it requires always relatively large amount of material and is not suitable for the analysis of
very small samples, like most biological samples. By coupling capillary isotachophoresis
with on-line microcoil NMR detection (CITP-NMR) the sensitivity for the characterization
of heparin – derived oligosaccharides has been improved (Korir & Larive, 2007).
Considering the limitations related to this first analytical approach, based on intact
molecules, the focus in the next paragraphs will be on presenting methods to analyze GAGs
following depolimerization; clearly that in rare cases of small oligosaccharide (i.e. from
synthesis) or highly purified fractions such step can be avoided.
2.3 Overview on methods of GAGs depolimerization
As previously pointed out most often the structure characterization of GAGs involves
degradation steps to obtain disaccharides and/or a range of oligosaccharide fragments
allowing an efficient disaccharide/oligosaccharide mapping (Bo et al., 2011). These
structural analyses of GAGs may involve complete or partial chemical degradation, as well
as enzymatic degradation permitting a quite selective cut of the polysaccharide chains
(Ruiz-Calero et al., 2003; Gatti et al., 2010). Highly specific enzymes have been isolated and
they are now commercially available making their application viable (Johnson, 1982; Jandik
et al., 1996; Cohen & Linhardt, 1990). In figure 4 the main used enzymes are shown.
An important aspect of these enzymes is the high specificity for the substrate, fact that if it is
welcomed in biochemical studies can be critical sometimes when analyzing semisynthetic
derivatives.
Chemical degradation is a very interesting alternative to enzymatic degradation giving also
quite specific structure information. The degradation with nitrous acid and the periodic
oxidation followed by Smith degradation are two classical tools for GAGs analysis.
In several cases depolimerization methods are followed by derivatization procedures
generally to enhance the analytical detection when using conventional HPLC or CE
detectors.
www.intechopen.com
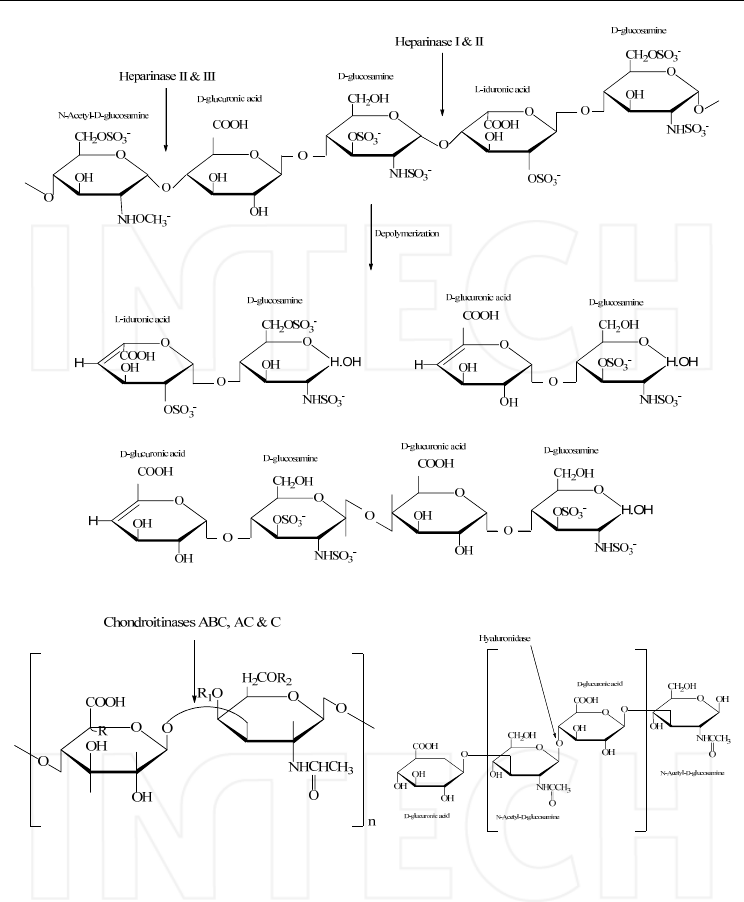
HPLC-MS/MS of Highly Polar Compounds
497
(A) Heparin and heparan sulphate
(B) Chondroitin sulphate (C) Hyaluronic acid
Fig. 4. The enzymatic cleavage of GAGs; A) heparin, heparan sulphate and the specific
enzymes: Heparinase I, Heparinase II and Heparinase III; B) Chondroitin sulphate and the
specific enzymes: Chondroitinase ABC for Chondroitin sulphate A, B and C, Chondroitinase
AC for Chondroitin sulphate A and C and Chondroitinase C for Chondroitin sulphate A
and C; C) Hyaluronic acid and the specific enzyme: Hyaluronidase
2.4 Overview on HPLC analysis of GAGs
For the analysis of intact GAGs, a variety of chromatographic techniques have been
developed mainly to separate different group of GAGS and/or to characterize the molecular
www.intechopen.com

Tandem Mass Spectrometry – Applications and Principles
498
weight distribution. Concerning this last aspect gel permeation chromatography (both as
HPLC or low pressure chromatography) is the preferred approach.
Ion exchange chromatography and reversed phase ion-pair chromatography are the most
employed techniques to separate the main classes of intact GAGs, which present differences
in polarity, both as low pressure chromatography (Patel et al., 2009; Jin et al., 2009) or high
performance liquid chromatography (Shao et al., 2004; Trehy et al., 2009). More recently also
capillary electrophoresis (CE) (Malsch & Harenberg, 1996; Malsch et al., 1996) have been
used successfully. The main interest is, however, on methods for the separation of
depolymerisation fragments of GAG, obtained by different digestion methods (chemical or
enzymatic), by HPLC (Du & Eddington, 2002; Plaas et al., 1996; Toyoda et al., 2000) or CE
(Malavaki et al., 2008; Ruiz-Calero, et al., 2003) followed by detection with conventional
spectral system (UV or fluorescence in case of adequate derivatization), as in the previous
papers, but in the last years often by mass spectrometry (Oguma et al., 2007; Yang et al.,
2011; Barosso et al., 2005; Silvestro et al., 1992; Da Col et al., 1993; Hemstrom & Irgum,
2006; Silvestro et al., 1996).
The peculiar aspects of separation in connection with MS detection will be detailed in the
following paragraph.
2.5 Overview on mass-spectrometric analysis of GAGs
Methodologies using fast atom bombardment (FAB) were first employed; however a direct
coupling to HPLC separation was not achieved, as in the case of peptides, due to limitations
in mobile phase composition (Dell et al., 1988; Mallis et al., 1989). The introduction of soft
ionization methods (ESI), permitting an easy interfacing to HPLC has first provided an on-
line separation and structure elucidation of complex oligosaccharide mixtures (Henriksen et
al., 2006; Barosso et al., 2005; Thanawiroon et al., 2004). As a complement, matrix-assisted
laser desorption ionization (MALDI) has proved to be a valuable tool for the analysis of
protein/peptide-heparin/HS oligosaccharide complexes (Ori et al., 2009; Venkataram et al.,
1999).
The first important point to consider is the kind of oligosaccharides and disaccharides to be
analyzed in particular if they are sulphated or not sulphated.
2.5.1 Sulphated oligosaccharides
As discussed previously (Chapter 2.4), sulphated oligosaccharides and disaccharides are
separated, when using conventional detectors (UV, fluorescence, electrochemical),
employing strong ion exchange columns with gradients of molarity of salts from strong
acids (sulphates, phosphates, chloride etc.). It is evident that such mobile phases, being non-
volatile, cannot be used in case of MS interfacing and alternative solutions have been
developed.
An option is the application of weak anion exchange columns permitting the elution of
sulphated derivatives also with gradients of ammonium acetate, compatible with an ESI
source; nonetheless it cannot be excluded that highly sulphated large oligomers are not
eluted from this kind of columns.
Another possibility, probably the most widely used, is the separation on a reversed phase
column in presence of an ion-pairing reagent. In an early paper by our group (Da Col et al.,
1993) tetrapropylammonium (TPA) was selected as ion pair reagent for this kind of
separation; smaller quaternary ammonium derivatives (tetramethylammonium and
tetraethylammonium) didn’t improve at all the chromatographic separation while larger
www.intechopen.com
HPLC-MS/MS of Highly Polar Compounds
499
derivative (tetrabutylammonium and higher) showed critical problems of volatility in the
ionization source without offering other advantages. It is important to note that the choice of
the stationary phase can be quite critical: we observed in fact that in case of reversed phase
columns with longer aliphatic chain (C8 or longer) the sulphated oligosaccharides couldn’t
be eluted being probably retained by an HILIC-like retention mechanism. Best conditions
were obtained on C4 – C6 columns with gradients of acetonitrile water (from 100% to 50%
water) in presence of 3.3 mM tetrapropylammonium at pH 4.5 with formic acid. To prepare
the mobile phase most common tetrapropylammonium salts like the sulphate (common in
conventional HPLC) were avoided due to volatility problems and preparations were made
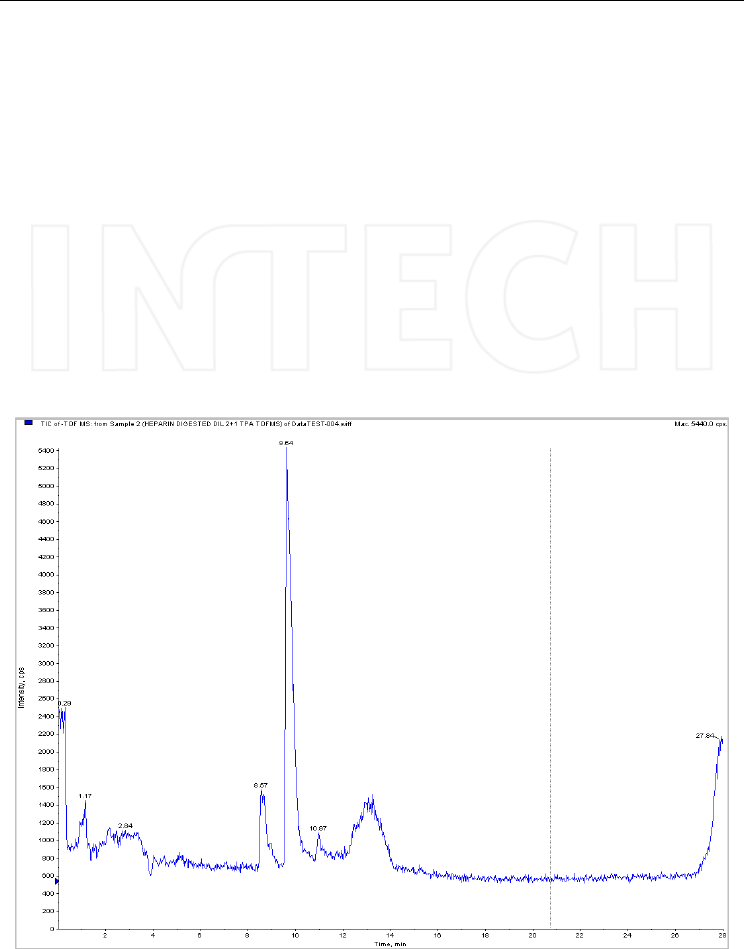
starting from tetrapropylammonium hydroxide. An example of TIC chromatograms
recorded on a digest of heparin is presented in Figure 5.
More recently, several groups have successfully introduced the use of tertiary aliphatic
amines (tributylammonium acetate in acetonitrile as ion pair reagents) claiming that a better
volatility can be obtained (Thanawiroon et al., 2004). It must be observed that short chain
amines like triethylamine, widely used in HPLC, are not able to form ion-pair with these
sulphated oligosaccharides.
Fig. 5. Total ion chromatogram obtained in TOF full scan after injecting a sample of heparin
digested with heparinase II. Column: Kromasil C4 (100x2.1mm, 3μm); mobile phase: (A)
TPA 3.3 mM in water, pH 4 and (B) TPA 3.3 mM in acetonitrile/water (90/10, v/v), pH 4.
Mass spectrometer: QTOF Qstar XL, operated in negative ESI mode.
www.intechopen.com
Tandem Mass Spectrometry – Applications and Principles
500
The addition of an ion pair reagent to sulphated oligosaccharides brings to the formation of
complexes stable enough to be ionized as such. Complex pseudomolecular ions with one or
more molecules of the ion-pair reagent, sometimes multiple charged, are predominant in
spectra of chromatographic separations of such di- and oligo-saccharides; 2 examples of
similar spectra are presented in Figure 6 and Figure 7.
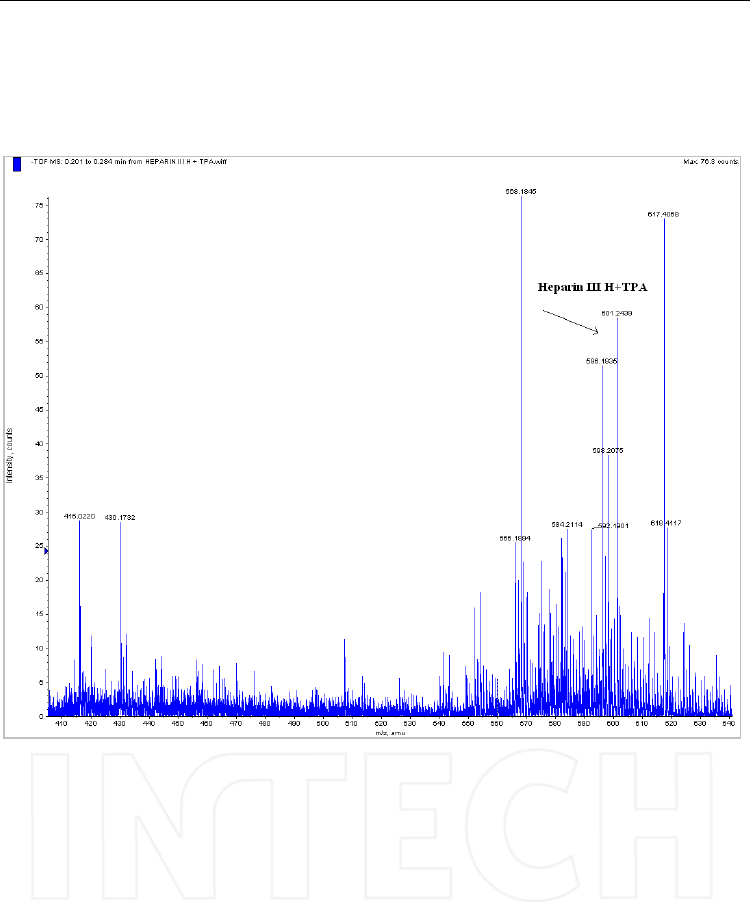
Fig. 6. The molecular ion of the adduct formed by the disaccharide Heparin III H and TPA
[M+TPA-H]- = 601.2439. Spectrum recorded by direct infusion with the syringe of a solution
diluted at 1 μg/mL. Mass spectrometer: QTOF Qstar XL, operated in negative ESI mode.
It is evident that the use of high resolution instruments is a valuable tool to clarify the
charge state of these ions while to clarify the stoichiometry of ion-pair
reagent/oligosaccharide ratio the utilization of a labelled ion-pairing reagent is very
interesting (Silvestro et al., 1996).
In an effort to obtain more easily interpretable spectra an attractive approach is also the use
model MMPC of the ion suppressor (i.e. Dionex…) to remove the tetrapropylammonium
from the chromatographic mobile phase. The experiments carried out have really showed an
efficient removal of tetrapropylammonium (an example is shown in Figure 8). Anyway, the
oligosaccharides as free acidic compounds are much more prone to non-sequence related in
source fragmentation during ionization (mainly desulphation), making sometimes very
difficult to understand the oligosaccharide real structure.
www.intechopen.com
HPLC-MS/MS of Highly Polar Compounds
501
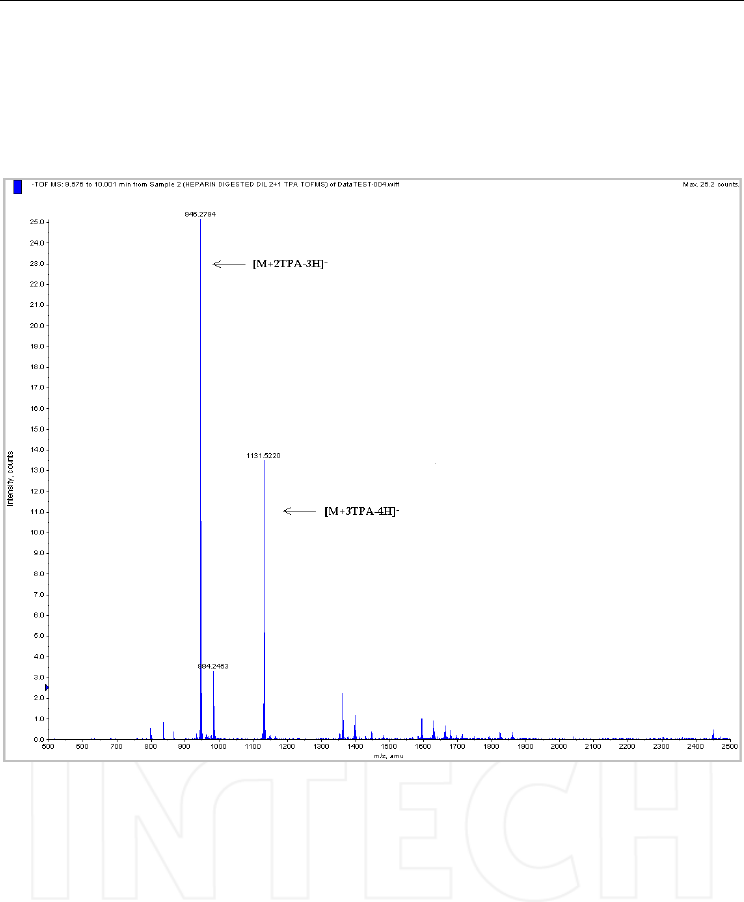
Fig. 7. Extracted spectrum obtained after injecting a sample of heparin digested with
heparinase II. Column: Kromasil C4 (100x2.1mm, 3μm); mobile phase: (A) TPA 3.3 mM in
water, pH 4 and (B) TPA 3.3 mM in acetonitrile/water (90/10, v/v), pH 4. Mass
spectrometer: QTOF Qstar XL, operated in negative ESI mode. M: trisulphated disaccharide
obtained from heparin by enzymatic digestion.
The MS/MS properties of these di- and oligo-saccharides are also noteworthy: in case of
parent ions (single or multiple charged) the desulphation fragments are generally the most
abundant being easily formed and generally very few weak signals coming from
fragmentation of the sugar back bone can be observed. The situation is similar in case of
TPA complexes with the tendency to an even less favourable fragmentation.
www.intechopen.com
Tandem Mass Spectrometry – Applications and Principles
502
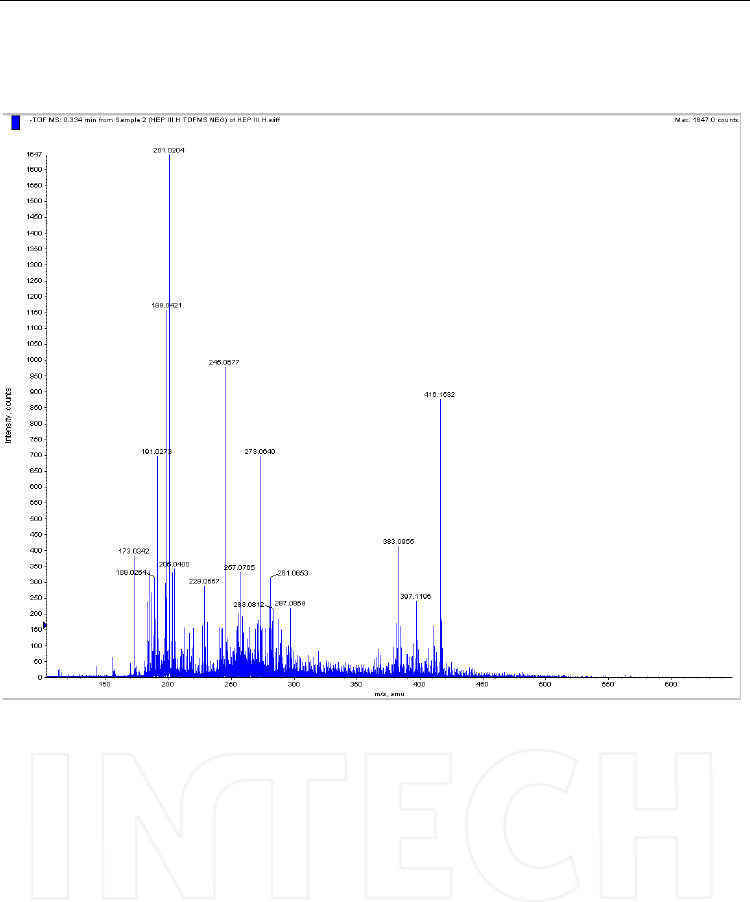
Fig. 8. The molecular ion of the oligomer Heparin III H after ion suppression; [M-H]- =
416.1532 can be easily seen. Spectrum recorded by direct infusion with the syringe of a
solution diluted at 1 μg/mL. Mass spectrometer: QTOF Qstar XL, operated in negative ESI
mode.
2.5.2 Non sulphated oligosaccharides
The analytical separations of these molecules are less challenging than those of the more
charged sulphated products. In this case the use of ion exchange columns eluted with
gradients of volatile buffers (i.e. ammonium acetate) at high ionic strength is effective and
permits a good separation of oligomers with different chain length. The application of ion-
pair chromatography is practically not needed and in general the formation of ion-pair
complexes having only carboxylic groups is not effective.
Deprotonated molecular ions, sometimes multiple charged, are typically observed, as shown
in Figure 9.
www.intechopen.com
HPLC-MS/MS of Highly Polar Compounds
503
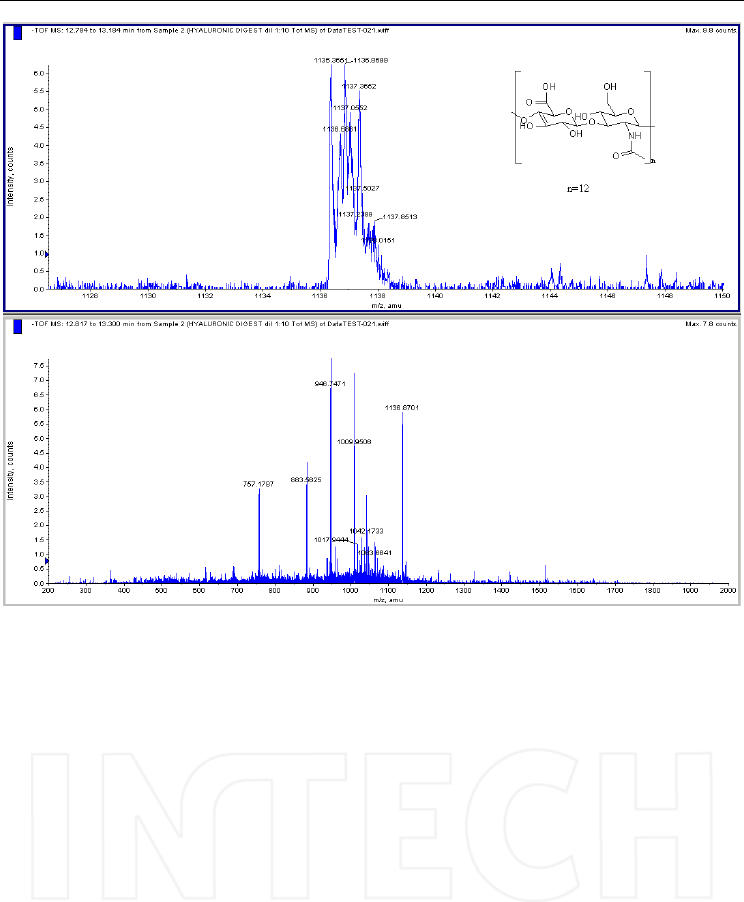
Fig. 9. Mass spectrum obtained in TOF full scan mode after injecting a sample of a
hyaluronic acid digested with hyaluronidase. Column: Supelcosil LC-SAX 1 (250x4.6mm,
5μm); mobile phase: (A) water/methanol (1/1, v/v) (B) ammonium acetate 0.5 M in
water/methanol (1/1, v/v). Mass spectrometer: QTOF Qstar XL, operated in negative ESI
mode. The main ions in the spectrum are multiple charged; they all correspond to
dissacahride oligomers resulted from hyaluronic acid degradation. A detail can be seen in
the spectra above for [M-2H]2-=1136.8, a doubly charged ion corresponding to a
dodecasaccharide.
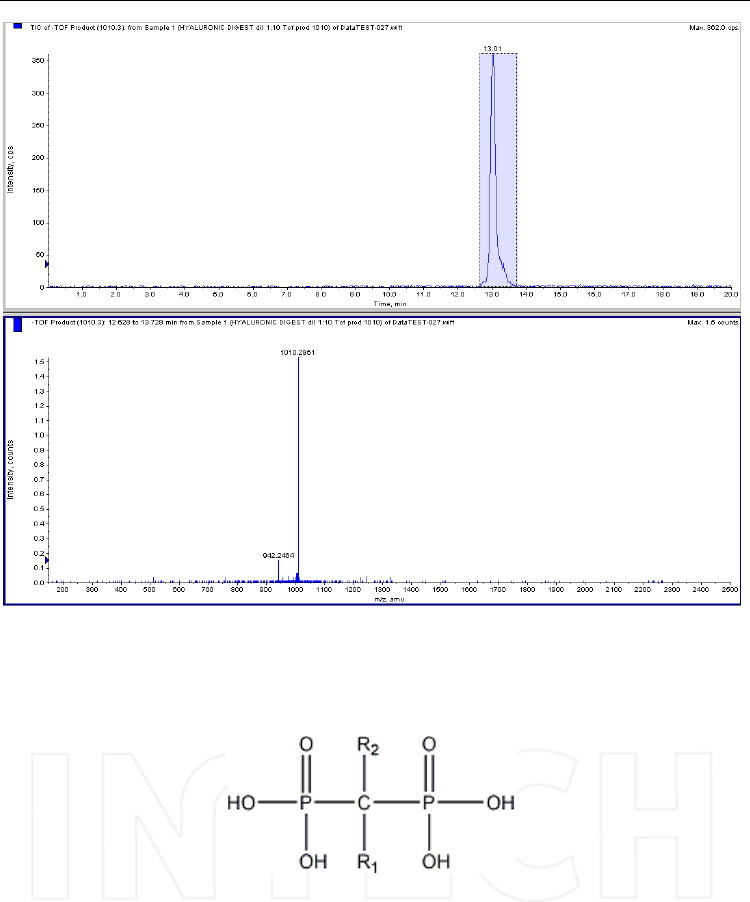
Daughter spectra can be obtained as it is shown in Figure 10. However, it can be seen, these
kinds of molecules are hard to break and only scarce structural information can be gathered.
3. Bisphosphonates analysis
3.1 Chemical structure and main features of bisphosphonates
Bisphosphonates, polar structures with two phosphonate groups covalently bonded to
carbon (called P-C-P bridge, Fig. 11) are a class of drugs used in the treatment of
osteoporosis, osteolytic metastasis, Paget’s disease (osteitis deformans), and other
disorders involving bone fragility. They have also non-therapeutic usage, based on their
chelating properties (as water softeners, in agriculture, in paper, detergent and cosmetics
industries).
www.intechopen.com
Tandem Mass Spectrometry – Applications and Principles
504
Fig. 10. Product ion spectrum of one of the oligomers presented in Figure 9 - [M-2H]2- =
1010. Column: Supelcosil LC-SAX 1 (250x4.6mm, 5μm); mobile phase: (A) water/methanol
(1/1, v/v) (B) ammonium acetate 0.5 M in water/methanol (1/1, v/v). Mass spectrometer:
QTOF Qstar XL, operated in negative ESI mode; collision energy: -30V.
Fig. 11. General structure of bisphosphonates.
The long-chain substituent (R2 in figure) determines the chemical properties and mechanism
of action, while the short-chain substituent (R1) influences mainly the pharmacokinetics of
the bisphophonate drug (Fleish 2004). Obviously, bisphosphonates are extremely
hydrophilic and their structures similar to endogenous phosphorylated compounds. They
can be grouped in two classes: nitrogen (N) – containing (pamidronate, neridronate,
olpadronate, alendronate, ibandronate, risedronate, zoledronate) and non-N-containing
(etidronate, clodronate, tiludronate) bisphosphonates.
www.intechopen.com

HPLC-MS/MS of Highly Polar Compounds
505
3.2 Special problems of these molecules and potential approach to solve them:
derivatization or not
Analysis of bishposphonates from biological matrices is very challenging, due to the
aforementioned features of these molecules: high polarity and similarity with endogenous
phosphates. Finding appropriate conditions for their extraction from plasma or urine is very
difficult; chromatographic separation raise typical problems of hydrophilic compounds and
last but not least, bisphosphonate molecules often lack chromophores or fluorescent groups
needed for sensitive UV or fluorescence detection in liquid chromatography. As an
approach to facilitate either the isolation from biological samples, or the chromatographic
separation, or the detection, or all of them, derivatization is widely used in the analysis of
bisphosphonates. Mass spectrometric detection was used more recently to develop sensitive
quantification methods, but also most of the times in combination with derivatization
procedures. In gas-chromatography (GC), derivatization is needed to increase the volatility
and thermal stability of the molecules. The N-containing bisphosphonate undergo reaction
of the amino- group, and for all compounds the derivatization of the hydroxyls from the
phosphonate group can give good results.
3.3 HPLC analysis of bisphosphonates
Various analytical procedures for the determination of bisphosphonates from biological
samples or pharmaceutical formulations have been presented so far; review papers were
previously published by Sparidans and den Hartigh (1999), Zacharis and Tzanavaras (2007).
One of the first methods, developed for dichloromethylene bisphposphonates, was
described by Chester et al (1981). The separation is based on ion-exchange chromatography,
without derivatization, using an AG1 X8 resin column and a mobile phase containing
hydrochloric acid and a flame-photometric detector. The coprecipitation with calcium
phosphate was used to extract the target compounds from urine. This method has a limit of
detection of 2µmol/l.
Separation procedures without derivatization are performed on ion-exchange columns like
Waters IC-Pak HR (Tsai et al, 1992) or Dionex OmniPac PAX-100; the mobile phases contain
mineral acids (mostly diluted nitric acid without or with small percentage of organic
modifier) and the detection is realized through flame-photometry (Chester et al, 1981),
conductivity (Tsai et al, 1992) or inductive coupled plasma mass spectrometry (ICP-MS)
(Kovacevic et al, 2004).
Mineral acids are replaced with organic acids (especially formic acid) in researches that use
ion-chromatography in tandem with mass-spectrometry (Ip et al, 1994). An alternative to
ion-exchange chromatography is ion-pairing reversed-phase chromatography. Xie et al
(2006) used ion-pair chromatography with evaporative light-scattering detection to
determine ibandronate in pharmaceutical formulations. The C8 Inertsil column was eluted
in isocratic conditions with a mobile phase consisted of ammonium acetate containing
amylamine/acetonitrile/methanol. Tetrabutyl ammonium hydrogen sulphate (Rao et al,
2005), tetrabutyl-ammonium phosphate (Aluoch et al, 2005), 1-octyltrimethylammonium
phosphate (Vallano et al, 2003) are among the ion-pair reagents employed in separations
without derivatization.
As mentioned above, a pre- or post-column derivatization step is often needed to introduce
in the molecule a chromophore group, in order to increase the sensitivity of the detection by
UV or fluorescence. The pre-column derivatization, if possible directly in biological samples,
is also very important for the isolation of bisphosphonates, introducing in the hydrophilic
www.intechopen.com

Tandem Mass Spectrometry – Applications and Principles
506
molecule a large non-polar group that facilitates their subsequent extraction in organic
solvents.
Most of the published methods involve therefore a derivatization approach, either of the
amino, or of the hydroxyl groups, or of both of them in some cases.
Fluorescamine was one of the most used derivatization reagents of the amino group (Flesh
et al, 1991, Wong et al, 2004). Aminoalkylphosphonic acids were isolated from urine or
plasma by coprecipitation with calcium phosphate; then the precipitate was dissolved in
Na2EDTA pH10 and reacted with fluorescamine. The reaction products were
chromatographed on an octadecyl stationary phase (Nucleosil C18) and detected by
fluorescence.
9-Fluorenylmethyl-chloroformate was also widely applied in the determination of
alendronate from different matrices like pharmaceutical preparations, plasma or urine. The
derivative was chromatographed on C18 columns, with mobile phases consisting of
acetonitrile, methanol and pyrophosphate buffer, and detection in UV or fluorescence (de
Marco et al, 1989, Ptacek et al, 2002, Yun et al, 2006, Apostolou et al, 2007). As an example,
Ptacek et al (2002) obtained with this derivatization a limit of quantification as low as 5
ng/ml from urine, with reproducible results that could be used for bioavailability studies.
The reaction was conducted at alkaline pH after bisphosphonates isolation from biological
sample by coprecipitation with calcium phosphate and the calcium ions removed on ion-
exchange resins; the chromatography was carried out on a C18 stationary phase, in gradient
conditions, with fluorescence detection.
The derivatization of the amino group with isobutyl chloroformate was proposed by
Sakyiama et al (1996) prior to GC separation. A fused-silica capillary column HP-1 (5m x
0.53 mm i.d., 2.65μm film thickness) was used and flame photometric detection. The same
reaction was later used by our group directly on biological samples (urine); this reduced the
polarity of the molecule and allowed the extraction with organic solvent (Tarcomnicu et al,
2007).
An interesting reaction described by Kline et al (1992) uses 2,3 naphtalene-
dicarboxaldehyde, in presence of cyanide ions as nucleophiles. The substituted
cyanobenzo[f]indole obtained was measured by fluorescence. Alendronate was determined
from urine samples using this technique. The same authors studied also the use of acetyl-
penicillamine as nucleophile for improved sensitivity; electrochemical detection was
preferred over fluorescence.
Introducing a chromophore in order to get sensitive UV or fluorescence measurements is
also possible post-column, after the separation by ion-exchange chromatography. Daley-
Yates et al (1989) described such a derivatization with ammonium persulphate and
molybdenum ascorbate; a phosphomolibdate detected in visible (820 nm) was obtained.
Nevertheless, in the LC methods above presented, although the separation is performed on
octadecyl stationary phase, the mobile phases consisted of phosphate, pyrophosphate,
citrate buffers in combination with various percentages of organic modifiers like methanol
or acetonitrile. These inorganic buffers, as well as mineral acids, are not suitable for LC-MS.
3.4 Overview on mass spectrometric analysis of biphosphonates
When coupling gas chromatography with mass spectrometry, derivatization is needed in
order to obtain volatile compounds. In this case the hydroxyls of the phosphonate group are
silylated or alkylated. Sakiyama et al (2005) described a method for alendronate analysis in
urine that employs double derivatization: first the amino group was reacted directly in the
www.intechopen.com

HPLC-MS/MS of Highly Polar Compounds
507
biological matrix with isobutyl chloroformate and extracted with organic solvent, then
diazomethane was used to methylate the phosphonate groups. A fused silica capillary
column containing cross-linked OV-1 stationary phase (Quadrex 12mx25mm id, 0.25μm film
thikness) eluted with helium at 1 ml/min was employed for chromatographic separation.
The limit of detection was 20 ng/ml.
The studies conducted for (aminoalkan)phosphonic acids, widely used either as pesticides
or as pharmaceuticals, are good starting points also for bisphoshonate analysis. A large
variety of derivatization approaches is described in the literature.
Silylation with N-methyl N-(tert-butyldimethylsilyl)trifluoroacetamide (MTBSTFA) was
proposed by Moye and Deyrup (1984) for GC/MS analysis of the herbicide glyphosate and
its metabolite, aminomethylphosphonic acid. In order to obtain high yield of the reaction,
coating of the glassware by exposing it to a diluted solution of phosphoric acid in ethanol
was necessary; however authors reported problems at low ppb levels.
Also in GC/MS pentafluorobenzyl bromide (Palit et al, 2004) and trimethyl orthoacetate
(Kudzin et al, 2003) were used. The two techniques were applied in the determination of
alkyl and aryl bisphosphonic drugs and in trace analysis of organophosphorous pesticides
or nerve agents and their metabolites from biological or environmental samples. Palit et al
(2004) studied the derivatization of methyl phosphonic and pinacolyl methyl phosphonic
acid, degradation products of organophosphorous compounds, with pentafluorobenzyl
bromide (PFBBr) at alkaline pH, in different conditions. They further compared the stability
of the pentafluorobenzyl phosphonic esters with that of the sylylated derivatives, the later
more widely used in GC/MS, but more sensitive to humidity.
The same reaction was tested by our group on two drugs from the bisphosphophonate class,
and the obtained compounds were separated by LC-MS/MS (unpublished data). A phenyl
stationary phase (Supelcosil LC-DP 15cmx3mm, 5μm) was used, eluted in gradient with a
mobile phase containing 50mM ammonium acetate and acetonitrile. High resolution mass
spectrometric detection on a quadrupole-time of flight (QTOF) instrument model Qstar
Pulsar i (AB Sciex), equipped with an electrospray ionisation source (ESI) operated in
negative ions mode was employed. Di- and triesters resulted mainly from the derivatization
of clodronate, as shown in figure 12; further optimisation is needed to increase the yield of
one of the derivatives in order to use this approach for quantitative purposes.
Zhu et al (2006) reported a methodology for the quantification of risedronate and
alendronate by LC-MS/MS in biological samples after derivatization of the phosphonate
groups with diazomethane. As a general approach, the urine or serum samples were loaded
onto Bond-Elut SAX solid-phase extraction (SPE) columns that were washed with water
prior to on-column derivatization with diazomethane. The methylated compounds were
eluted with methanol and separated on an ion-exchange Zorbax 300-SCX column, with
ammonium formate pH 2.5 and acetonitrile (75/25, v/v) as mobile phase. Triple quadrupole
mass-spectrometers model API 4000 or 5000 were used, operated in ESI positive mode. Low
limits of quantification (LLOQs) as of 0.2 ng/ml for risedronate and 1 ng/ml for alendronate
in urine were achieved using this technique.
A general drawback of the methods using diazomethane is the toxic and explosive nature of
the reagent; special care needs to be taken to ensure safety of all operations.
Alternative alkylations of aminoalkyl phosphonic acids could be performed with
fluorinated alcohols/perfluorinated anhydrides mixtures (Deyrup et al, 1985) or trimethyl
orthoacetate (Royer et al, 2000, Kudzin et al (2003).
www.intechopen.com
Tandem Mass Spectrometry – Applications and Principles
508
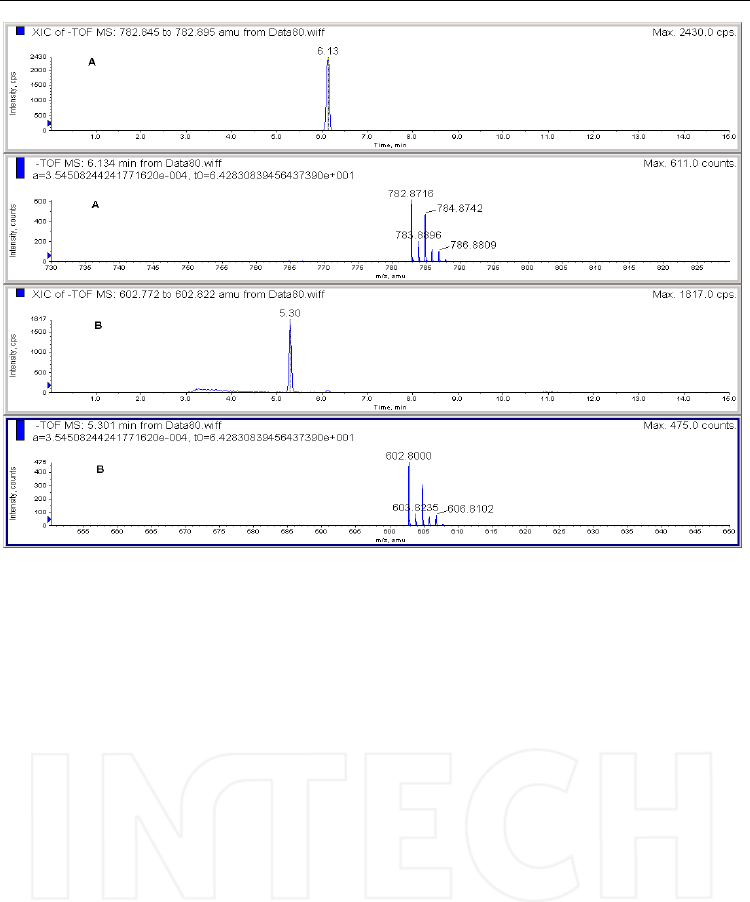
Fig. 12. Extracted chromatogram of the negative molecular ions corresponding to the triester
- [MH]- =782.8716 (A) and diester - [MH]- =602.800 (B) obtained in the reaction of clodronate
and PFBBr. Column: Supelcosil LC-DP 15cmx3mm, 5μm, mobile phase: ammonium acetate
and acetonitrile, in gradient. Mass spectrometer: QTOF Qstar Pulsar i, operated in negative
ESI mode.
Deyrup et al (1985) analysed glyphosate (N-phosphonomethylglycine) and its major
metabolite, aminomethylphosphonic acid, from water samples by GC with
flamephotometric, electron capture or mass spectrometric detection, after derivatization
with trifluoroethanol (TFE) and trifluoroacetic anhydride (TFAA). With this approach the
amino and hydroxyl groups are derivatized in the same step. No special glass coating is
needed, as in the case of MTBSTFA, and no particular safety issues are raised, as in the case
of diazomethane derivatization. An LLOQ of 0.5 μg/L in deionized water was obtained
with electron capture detection.
Trimethylorthoacetate (TMOA) was preferred by Royer et al (2000) for the derivatization of
another herbicide, gluphosinate (DL homoalanine-4-yl (methyl)phosphonic acid), and two
of its metabolites, 3-methylphosphonico-propionic and 2-methylphosphonico-acetic acids,
prior to analysis by GC-MS/MS with chemical ionization. This method was applied for the
quantification of gluphosinate and its metabolites in water samples, over a range of 10-150
μg/L.
Kudzin et al (2003) combined anhydride and TMOA approaches for the derivatization of a
series of biologically active aminoalcanphosphonic acids. Briefly, the neat standards or dried
water extracts were dissolved in 0.1 ml of trifluoroacetic acid/trifluoroacetic anhydride
www.intechopen.com

HPLC-MS/MS of Highly Polar Compounds
509
mixture (1/1, v/v), incubated for 90 min at 40 ºC, then 0.4 ml TMOA were added, followed
by another incubation step at 100 ºC for 120 min. Both amino and hydroxyl groups were
derivatized and the reaction mixtures were analized after preconcentration under vacuum,
by 31P NMR or GC-MS. The derivatization products were stable for several weeks at
ambient temperature and the quantifiable levels by GC-FID (flame ionization detection)
were in the low μmol/L range.
More recently our group studied the application of TMOA derivatization in the analysis of
bisphosphonate drugs. The hydroxyl groups reacted with TMOA in acidic conditions
independently from the presence of amino groups and all tested drugs (alendronate,
pamidronate, etidronate, clodronate, risedronate, ibandronate) were successfully
derivatized. Triethylorthoacetate (TEOA), trimethylorthoformiate (TMOF),
trimethylorthobutirate (TMOB) were also tested as alkylation reagents and found effective
(unpublished data). All reagents do not require special safety measures which is an
important advantage.
Studying these reactions, the expected alkylation products were found, but also other
compounds with higher molecular mass were obtained in the case of bisphosphonates
containing a hydroxyl group at the carbon in the P-C-P bridge. This hydroxyl is also
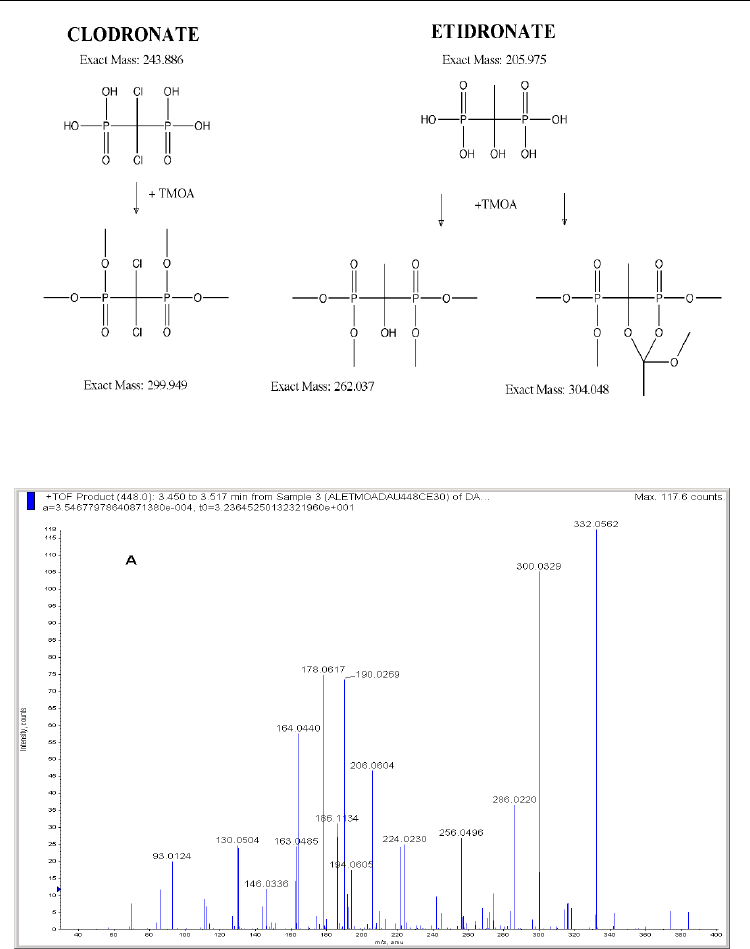
involved in the reaction probably resulting in cyclic structures. In the case of clodronate, the
carbon in the bridge is substituted with two chlorine atoms and only the tetraalkylated
structure is formed, as it can be seen in Figure 13. When the reagent was TMOF, an increase
of 28 mass units was observed; for TMOA and TEOA the shift was 42, and for TMOB 70 Da.
Based on these results, we have further developed a double derivatization method for the
quantification of alendronate in urine by LC-MS/MS (Tarcomnicu et al, 2007). First, the amino
group was reacted with isobutyl chloroformate (IBCF), to facilitate the extraction of the polar
drug from biological matrix with organic solvent. The dried extract was redissolved in acetic
acid, and incubated with TMOA for 1 h at 100 ºC. The methylation of the phosphonate groups
significantly reduced the polarity of the molecule permitting the separation on a reversed-
phase column (Supelco Discovery HSC18) with a gradient of mobile phase containing formic
acid 0.1% in water/formic acid 0.1% in acetonitrile, which was suitable for LC/MS and
enhanced the ionization in positive mode. The clyclic compounds, as described above, were
obtained with highest yield for alendronate and pamidronate; a product ion spectrum of the
alendronate derivative is presented in Figure 14.
The method vas fully validated according to FDA guidelines over a range of concentrations
of 6.667 – 4860 ng/mL alendronate in urine, and it was applied to a bioequivalence study.
With the help of a switching valve 2 columns were run in parallel, resulting in an analysis
time of 5 min/sample, that was very short compared with HPLC-fluorescence or with GC
methods.
Trimethylsilyl diazomethane (TMSD) was another derivatization reagent successfully used
in LC-MS/MS, in order to analyze ibandronate from plasma samples for pharmacokinetic
applications (Tarcomnicu et al, 2009). A high-throughput method with an LLOQ of 200
pg/mL was optimized and fully validated according to FDA guidelines. Owing to its
structure (N-substituted aminobisphosphonate), ibandronate was isolated from plasma
extracts with the same technique previously proposed for alendronate (Tarcomnicu et al,
2007). To reduce the polarity of the molecule and make it suitable for reversed-phase LC, in
the next step we decided to methylate the phosphonic groups. First, TMOA was tried as
derivatization reagent, but in the case of plasma extracts this was found not as effective as
for urine extracts. TMSD proved to be very reactive also with plasma extracts, as already
www.intechopen.com
Tandem Mass Spectrometry – Applications and Principles
510
Fig. 13. Clodronate and etidronate derivatization with TMOA; for the latter both hypotheses
(methylation and methylation plus cyclization) are shown.
Fig. 14. Product ion spectrum of the alendronate derivative with IBCF and TMOA ([M+H]+=
448), during chromatographic separation. Column: Supelco Discovery HSC18, 10cmx2.1mm,
5μm, mobile phase: (A) formic acid 0.1% in water and (B) formic acid 0.1% in acetonitrile;
gradient conditions. Mass spectrometer: QTOF Qstar Pulsar i, operated in positive ESI
mode.
www.intechopen.com
HPLC-MS/MS of Highly Polar Compounds
511
studied by another group (Ranz et al 2008) and compared to diazomethane it is a stable and
safe reagent. The alkylation reaction was carried out for 30 min at 70 ºC and the fully
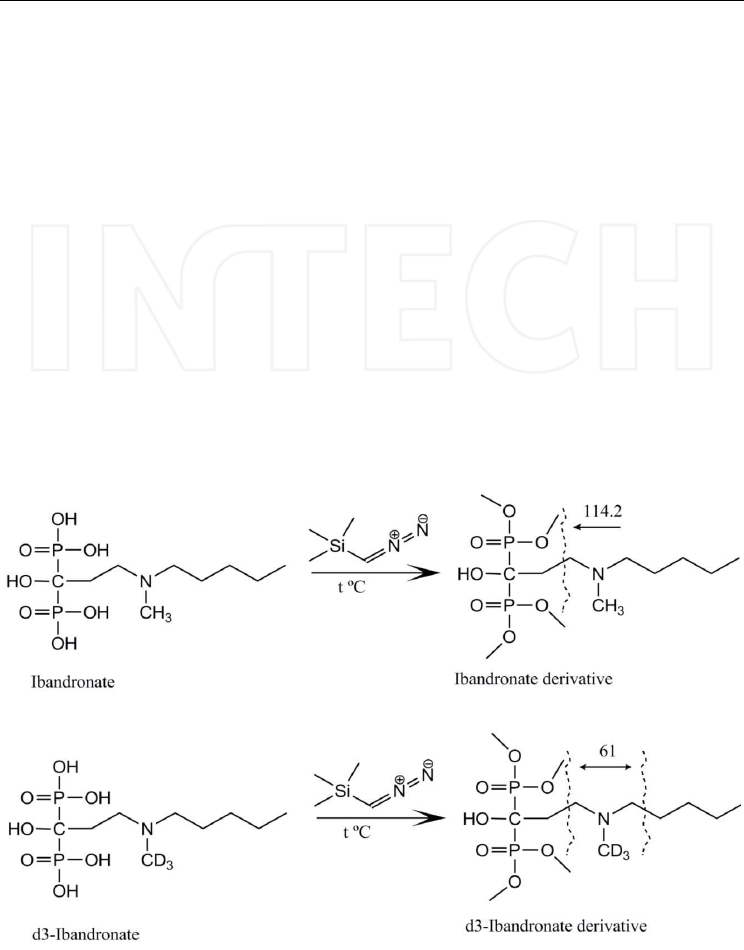
methylated compound was obtained. The derivatization scheme is presented in Figure 15,
the product ion spectrum of the ibandronate derivative in Figure 16, and Figure 17 shows
selected chromatograms obtained on a blank, calibrator 1 (LLOQ) and calibrator 8 extracted
from plasma. The separation was performed on a Discovery HSC18 column (10cm×2.1mm
i.d., 5μm particle size) and a mobile phase consisting of formic acid 0.1% in water and
formic acid 0.1% in acetonitrile, with a composition gradient. An alternative satisfactory
approach using alkaline mobile phase (ammonium hydroxide 0.05% in water/ammonium
hydroxide 0.05% in acetonitrile) on a Purospher Star RP-18e (3cm×2.1mm, 2.7μm) was also
tested; good peak intensities, similar to the ones with acidic mobile phase, were achieved.
Elution at acidic pH was preferred for this study due to better background, but the basic
mobile phase can be also considered.
Fig. 15. Derivatization scheme of ibandronate and its internal standard, d3-ibandronate.
www.intechopen.com
Tandem Mass Spectrometry – Applications and Principles
512
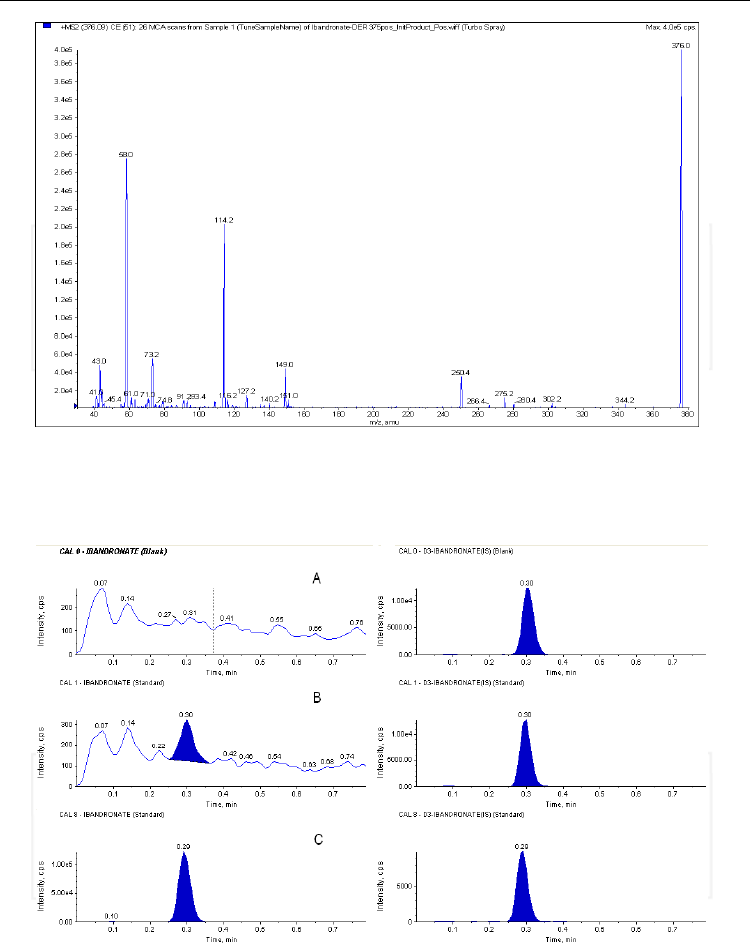
Fig. 16. Product ion spectrum of the ibandronate derivative with TMSD - [M+H]+ = 376
obtained with direct infusion via syringe of a 1μg/mL solution water/methanol (1/1, v/v).
Mass spectrometer: quadrupole-linear ion trap API4000QTrap, operated in positive ESI
mode; collision energy: 25V.
Fig. 17. HPLC traces of the MRM transitions selected for ibandronate (left) and d3-
ibandronate (right) recorded on a blank plasma sample (A), plasma spiked with ibandronate
for CAL1—0.2 ng/ml (B) and CAL8—175.0 ng/ml (C) concentrations. Note: The retention
time shown in the figure is relative, as the analyses were performed in overlapping mode;
1.5 min must be added to get the total retention time.
www.intechopen.com

HPLC-MS/MS of Highly Polar Compounds
513
3.5 Remarks
Considering existing data for the analysis of bisphosphonates, especially with MS detection,
the derivatization approach has given so far the best results. Nevertheless, the new
developments in column manufacturing could open possibilities for the analysis of
underivatized bisphosphonates. A promising direction is represented by HILIC (hydrophilic
interaction liquid chromatography) which is gaining interest in the analysis of polar
compounds, with applications in various fields (A.J. Alpert, 2011, Van Nuijs et al, 2011).
HILIC is a version of normal phase liquid chromatography; the typical mobile phase
consists of acetonitrile with a small percentage of water, while the stationary phases are
polar surfaces (silica, diol, amino, amide, zwitterionic bonded phases). The retention of
polar compounds is not based on liquid-solid partition like in reversed phase separations,
but on a liquid-liquid partition mechanism between the water-deficient bulk eluent and the
water-enriched layer immobilized on the HILIC stationary phases (Alpert et al, 1990).
Therefore polar analytes early eluting in reversed-phase are strongly retained in HILIC
conditions. HILIC is particularly useful in the analysis of aminoglycoside antibiotics (more
details will be provided in the following chapter), other various pharmaceuticals (e.g.
metformin, salicylic and acetyl salicylic acid, tetracyclines), nucleosides, organic phosphates,
peptides and proteins, sugars, warfare agents or drugs of abuse (cocaine, benzoylecgonine)
etc. The use of mobile phases with high percentage of organic solvent in HILIC separation is
beneficial for MS detection, because of enhanced ionization which results in an increased
sensitivity. Methanol, ethanol, 2-propanol, tetrahydrofuran can be used for some
applications. The most common buffers used to control pH and ionic strength are
ammonium acetate or ammonium formate, at low molarities, also favouring the ionization
in MS; only rarely high molarities or non-volatile buffers are required. Another advantage is
the simpler sample preparation, being no need for derivatization. The functional groups on
the phase surface influence the selectivity of the separation, thus the capacity to differentiate
similar compounds. Other interactions which could be involved in HILIC are electrostatic
interaction, hydrogen-bonding, dipole–dipole interaction, molecular shape selectivity, and
even hydrophobic interaction.
The retention mechanisms have been studied by several groups in order to characterize and
classify HILIC stationary phases (Dinh et al, 2011, Kawachi et al, 2011).
4. Analysis of aminoglycoside antibiotics
4.1 Chemical structure and main features
Aminoglycosides are antimicrobial agents used in the treatment of both animals and
humans against aerobic gram-negative bacteria. These molecules are natural products or
semisynthetic derivatives of compounds produced by different varieties of actinomycetes
isolated from soil (Higgins and Kastner, 1967); the class include very important antibiotics
as gentamicin, tobramycin, amikacin, netilmicin, kanamycin, streptomycin and neomycin.
The first drug of the class, Streptomycin, was isolated from a strain of Streptomyces griseus by
Waksman and coworkers in 1944 and it is an important agent for the treatment of tuberculosis.
Gentamicin and netilmicin are derived from species of the actinomycete Micromonospora.
Tobramycin is produced by S.tenebrarius (Chambers, 2006). Amikacin, a derivative of
kanamycin, and netilmicin, a derivative of sisomicin, are semisynthetic products.
The antimicrobial activity of aminoglycosides is based on their ability to selectively inhibit
protein synthesis, in bacteria; the most important pathogens treated with aminoglycosides
are pseudomonas, enterococci, coliforms, and salmonellae.
www.intechopen.com
Tandem Mass Spectrometry – Applications and Principles
514
The molecule of aminoglycosides presents two or more amino sugars linked by a glycosidic
bridge to a hexose nucleus (aminocyclitol) that is either streptidine (found in streptomycin)
or 2-deoxystreptamine (in all other available aminoglycosides).
The aminoglycosides are divided into the following subclasses (Chambers, 2006):
a. The neomycin family, which includes neomycin B and paromomycin, presents three
amino sugars attached to the central 2-deoxystreptamine.
Neomycin B
b. The kanamycin family (kanamycin A - B, amikacin and tobramycin) presents two amino
sugars attached to the central 2-deoxystreptamine moiety; one of these being a 3-
aminohexose.
Kanamycin A
c. The gentamicin family, including gentamicin, sisomicin and netilmicin, contains two
amino sugars attached to the central 2-deoxystreptamine, one of these being
garosamine.
www.intechopen.com
HPLC-MS/MS of Highly Polar Compounds
515
Gentamicin C
d. Streptomycin contains streptidine that is not in the central position like in the other
aminoglycosides.
Streptomycin
www.intechopen.com

Tandem Mass Spectrometry – Applications and Principles
516
4.2 Chromatography of aminoglycosides
Aminoglycoside antibiotics are very polar compounds, highly water soluble, practically
insoluble in hydrophobic organic solvents, lacking chromophores for UV absorption or
fluorescent groups. They have been determined by bioassays, GC–MS and HPLC, in this
case often following derivatization (to improve detection and/or separation), adding ion-
pair agents in the mobile phase or by ion-exchange techniques. More recently, HILIC was
also proposed as worthwhile approach and there is an increasing number of applications; in
literature brief reviews about the application of these methods for the separation of
aminoglycoside antibiotics can be found (Tawa et al, 1998, Isoherranen and Sobak, 1999,
Stead, 2000).
Because of the nonvolatile and very polar nature of aminoglycosides the gas
chromatographic analysis invariably involves derivatization of the amino and hydroxyl
groups of these molecules. trimethylsilylimidazole (TMSI) and heptafluoro-
butyrylimidazole (HFBI) have been used as derivatizing agents for the analysis of
gentamicin, tobramycin, netilmicin, amikacin and paromomycin in serum by Mayhew et al
(1978). Trimethylsilyldiethylamine (TMSDEA) was also tested as derivatization reagent for
both amino and hydroxyl groups but with poor results as repeatability and linearity
(Margosis and Tsuji, 1978).
Owing to their hydrophilic properties, aminoglycosides are not adequately separated also
by reversed-phase liquid chromatography, and therefore derivatization with non polar
agents or mobile phase containing counter-ions reagents, forming ion pairs, have been
widely employed in HPLC.
As derivatization reagents o-phthalaldehyde (OPA), 1-fluoro-2,4-dinitrobenzene (FDNB)
and dansyl chloride are the most commonly used for the analysis of aminoglycosides
(Isoherranen and Sobak, 1999). OPA reacts in the presence of mercaptan or other strong
reducing agents with primary amines to form fluorescent derivatives. The reagent is stable
in different buffer solutions and the reaction has a fast rate at room temperature allowing
the derivatization to be performed either pre- or post- column. FDNB easily reacts with both
primary and secondary amines, in basic conditions, producing UV absorbing derivatives
with maximum absorption wavelength at 365 nm.
The separation columns used for the derivatized aminoglycoside generally contain C8 or
C18 silica as stationary phase; the mobile phase consist of acidic buffers with methanol
and/or acetonitrile.
Gentamicins C1, C1a and C2 were determined as their 2,4-dinitrophenyl derivatives in
plasma and urine using a C18 RP column and as mobile phase 680ml/l acetonitrile-320ml/l
Tris buffer (8.3mmol/l titrated to pH 7.0) (Isoherranen and Sobak, 2000). A HPLC method
for the analysis of tobramycin in urine samples used pre-column derivatization of
tobramycin with fluorescein isothyociante followed by fluorescence detection; the
chromatographic separation was carried out on a Phenomenex Luna C18 column and the
limit of quantitation of the method in urine was 250 ng/mL (Mashat et al, 2008).
Another derivatization agent used for the RP-HPLC analysis of aminoglycoside antibiotics
was phenylisocyanate in the presence of triethylamine. Phenylisocianate groups easily react
with amino groups of aminoglycosides like kanamicin, neomycin and gentamicin (Kim et al,
2001 and 2003); the complex structures were confirmed by ESI-MS.
Clarot et al (2005) described a method for the determination of neomycin sulphate,
framycetin sulphate and other related compounds by evaporative light scattering detection
(ELSD) thus avoiding the need of derivatization for detection. The chromatographic
separation was performed on a Polaris C18 column using a mobile phase with 170 mM
www.intechopen.com

HPLC-MS/MS of Highly Polar Compounds
517
trifluoroacetic acid; in this case trifluoroacetate ions act like counter-ions forming ion pairs
with aminoglycosides. The ELSD elution of the complexes with antibiotics was confirmed
by MS.
Ion-pair chromatography is probably the most popular approach for aminoglycosides
analysis; when using standard detectors mainly alkyl sulphonates (pentane, heptane and
hexane sulphonates) in acetate or phosphate buffers are selected as counter-ions. For HPLC-
MS applications the volatile fluorinated carboxylic acids are preferred. Pre-column and post
column derivatization with o-phthalaldehyde are often used for the detection of these
molecules also when separated by ion-pair HPLC. As an example, Aubin et al separated
nine aminoglycoside antibiotics (streptomycin, dihydrostreptomycin, neomycin, sisomicin
gentamicin C1, C1a, C2, C2a, netilmicin) on an Atlantis C18 column eluted with 0.02M
potassium dihydrogen phosphate (adjusted to pH 3.0 with phosphoric acid) containing 35
g/l sodium sulfate, 500mg/l sodium octanesulfonate and 15ml/l tetrahydrofuran; the
detection was achieved with a pulsed electrochemical detector. Tobramycin and colistin
sulphate were simultaneous determined from pharmaceutical formulations using a Zorbax
SBC18 column eluted with acetonitrile / water / trifluoroacetic acid and evaporative light
scattering detection (Clarot et al, 2009).
Pulsed electrochemical detection was applied also for the analysis of underivatized
tobramycin (Shruti et al, 2010) and kanamycin by ion-pairing with octansulfonate in
phosphate buffer (Manyanga et al, 2010). A system for the determination of gentamicin by
ion chromatography using pulsed amperometric detector after isocratic elution on a
polystyrene column was developed by Metrohm in 2005.
4.3 Analysis of aminoglycosides by mass spectrometry
The concentrations usually monitored in plasma or serum of patients treated with
aminoglycoside antibiotics are in the low ng/mL range. Due to their non negligible toxicity
in biological systems and to side-effects like inducing resistance in bacteria or alteration of
the normal microbial flora (thus influencing various biological systems), antibiotics are also
carefully monitored in environmental or food and feed samples. Maximum residue limits
(MRLs) have been set in most countries and they are in the low ppb ranges, too. Therefore,
mass spectrometry, being a very sensitive and selective analytical technique is extremely
important in the analysis of these antibiotics. With respect to aminoglycosides, the GC/MS
methods are based on complex and lengthy derivatizations as above mentioned; Preu et al,
1998 adapted the method using trimehtylsilylimidazole and heptafluorobutyrylimidazole
for capillary GC/MS.
LC-MS/MS methods have been preferred recently owing to their high sensitivity and ease
of use compared to GC/MS. Two non-derivatization approaches have been developed in
parallel, reversed phase ion-pair chromatography and HILIC.
Aminoglycoside antibiotics are among the best candidates to be analyzed with HILIC and
such methods were applied for difficult matrices like plasma, kidney or meat. Oertel et al
(2004) described an automated method for the simultaneous quantification of amikacin,
gentamicin, kanamycin, neomycin, paromomycin, and tobramycin in human serum by
HILIC-MS/MS. Separation was carried out on a zwitterionic SeQuant ZIC-HILIC column,
100mm × 2.1mm with a Phenomenex SecurityGuard C18, 4mm × 2mm i.d. The mobile phase
was composed of acetonitrile, 2mM ammonium acetate and formic acid, in gradient
conditions, and data acquisition was performed with a Quattro Micro triple quadrupole
www.intechopen.com

Tandem Mass Spectrometry – Applications and Principles
518
mass spectrometer equipped with ESI source, operated in positive ions mode. Low limits of
quantification (LLOQ) of 100ng/mL were obtained using 500μL serum extracted by solid
phase extraction (SPE).
Ishii et al (2008) reported a quantification method for seven aminoglycosides from swine
and bovine meat and kidney that also employed a SeQuant ZIC–HILIC column (100 mm ×
2.1 mm, 5 μm) eluted with a mobile phase composed of (A) ammonium acetate 150 mM +
1% formic acid in water and (B) AcN, at a flow rate of 0.3 mL/min. MS detection was
performed in positive mode. LOQs in swine bovine kidney were 25 ng/g for gentamicin, 50
ng/g for spectinomycin, dihydrostreptomycin, kanamycin and apramycin, and 100 ng/g for
streptomycin and neomycin, well below the existing FAO MRLs.
Beside HILIC the most common separation technique for aminoglycoside antibiotics is by
ion-pair chromatography, performed mostly on octyl (C8) or octadecyl (C18) columns,
eluted with mobile phases containing trifluoroacetic (TFA), pentafluoropropionic (PFPA),
heptafluorobutiric (HFBA) or nonafluoropentanoic (NFPA) acids as ion-pair reagents. Good
peak shape and high sensitivities are obtained, thus compensating the negative aspect that
the ion-pair reagents are serious contaminants for the MS interface.
Shen et al (2008) studied the separation of amikacin, streptomycin, spectinomicyn and
gentamicin by ion-pair and HILIC. The ion-pair chromatography was performed on an
Agilent Zorbax SB-C8 (30 X 2.0 mm, 5 μm column), eluted with a mobile phase consisting of
(A) 10 mM NFPA with 10 mM ammonium hydroxide in water and (B) 5 mM NFPA acid
with 5 mM ammonium hydroxide in a 10:90 mixture of water:acetonitrile, with a gradient
from 20% to 90% (B). HILIC was carried out on a SeQuant Zic-HILIC (50 X 2.0 mm, 5 μm)
column, with a mobile phase composed of (A) 5 or 25 mM ammonium formate in water
with formic acid (pH =~2.5) and (B) acetonitrile with 1% (v:v) formic acid; the gradient
started from 90% B and was decreased to 10%B. A simple protein precipitation with cold
acetonitrile was used for clean-up of the different samples (mouse, rat or Guinea pig
plasma) and triple quadrupole (API 4000 or 5000) mass spectrometers were used for
detection. An LLOQ of 20ng/mL for all four antibiotics was achieved with the ion pair
method, while the HILIC method was highly sensitive only for spectinomycin, needing
further optimization for the other analytes.
Amikacin, neomycin and gentamicin in plasma or tissue samples were also analyzed by
Zimmer et al (2008) using a similar approach. They tested HILIC on a Varian Inertsil 5 Si
(50X2.0mm column) with a mobile phase composed of acetonitrile /ammonium acetate 100
mM in water, pH 3, and ion pair separation on a Discovery HS-C18 (2.1x50mm, 3μm)
column eluted with 0.1% HFBA in water / 0.1% HFBA in acetonitrile (all methods were
carried out using a composition gradient). LLOQs of 5 ng/mL were obtained using ion-pair
LC, which also in this case has proven to be more sensitive than HILIC.
Granja et al (2009) described a method for the determination of streptomycin residues in
honey that employs ion-pairing with HFBA in water/acetonitrile (85:15, v/v) as a mobile
phase (isocratic conditions), on a C18 column (Gemini 5 micron C18, 50x2mm). Also
Hammel et al (2009) presented a multi-screening approach for 42 antibiotic residues in
honey and 3 aminoglycosides where among them. HPLC analyses were run on a Zorbax SB-
C18 reverse phase column (2.1×50 mm, 1.8μm). The mobile phases were constituted with
solvent A:water containing 1mM NFPA mixed with 0.5% formic acid (v/v) and solvent B:
acetonitrile/methanol (50/50, v/v) containing 0.5% formic acid (v/v).
The following paragraphs present the practical experiments of our group on aminoglycoside
antibiotics by LC-MS/MS (unpublished data). The first method was developed for residues
of streptomycin in honey, by ion-exchange chromatography, on a Grom-Sil 300WCX
www.intechopen.com
HPLC-MS/MS of Highly Polar Compounds
519
(100x4.6mm, 7μm) eluted in gradient with (A) methanol/water (60/40, v/v) and (B)
ammonium acetate 500mM in methanol/water (60/40, v/v); the initial mobile phase was
100% A. The honey sample was simply diluted 1/1 with water and injected; detection was
achieved with an API 4000 triple quadrupole mass-spectrometer operated in positive ESI
ionization. The calibration curve in honey was built over a range of 10 - 250 ng/mL and
gentamicin was used as internal standard. In order to improve the sensitivity and
robustness of the determination, later we switched to ion-pair chromatography on a
pentafluorophenylpropyl column (Discovery HSF5 100x2.1 mm, 5μm), using a gradient of
PFPA 0.2% in water/PFPA 0.2% in acetonitrile as mobile phase. An LLOQ of 3.3 ng/mL was
obtained with the same instrument set-up and same sample preparation; gentamicin was
replaced as internal standard by cimetidine. Figure 18 shows two examples of
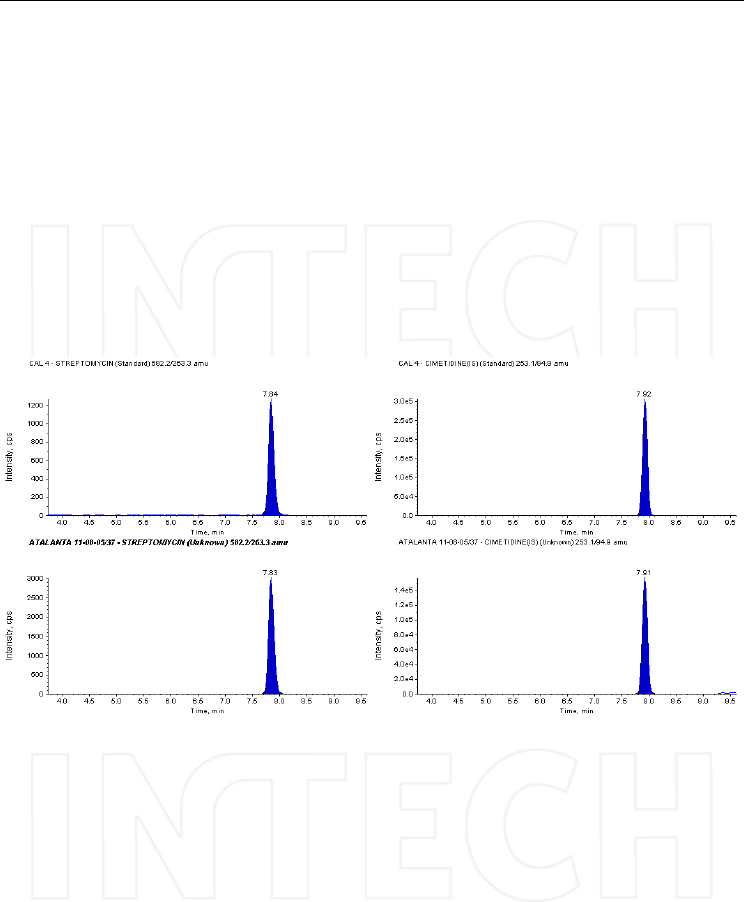
chromatograms, recorded on honey samples.
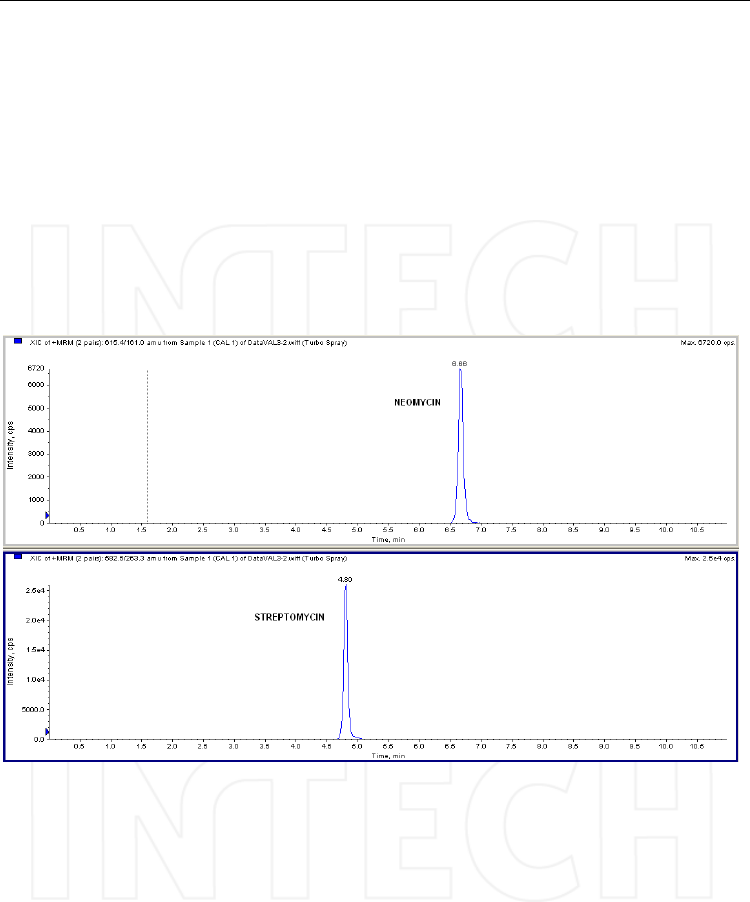
Fig. 18. HPLC traces of the MRM transitions selected for streptomycin and its internal
standard cimetidine, recorded on a spiked honey sample – concentration 90 ng/mL (top),
and on an unknown sample (bottom chromatograms) that was obviously contaminated with
streptomycin. Column: Discovery HSF5 (10cmx2.1mm, 5μm), mobile phase: A) 0.2% PFPA
in water and B) 0.2% PFPA in acetonitrile; flow: 0.3 ml/min; injection volume: 50μL. Mass
spectrometer: Triple quadrupole API4000, operated in ESI positive mode.
The same method was later adapted for the analysis of neomycin in ophthalmic solutions,
and streptomycin was used as internal standard. The samples were just diluted 1:1000 with
the initial mobile phase (water/acetonitrile (95/5, v/v) + 0.2 % pentafluoropropionic acid)
and injected in the LC-MS system; being the concentrations quite high (1-5 μg/mL) no
further optimization was needed. An example of chromatogram is presented in Figure 19.
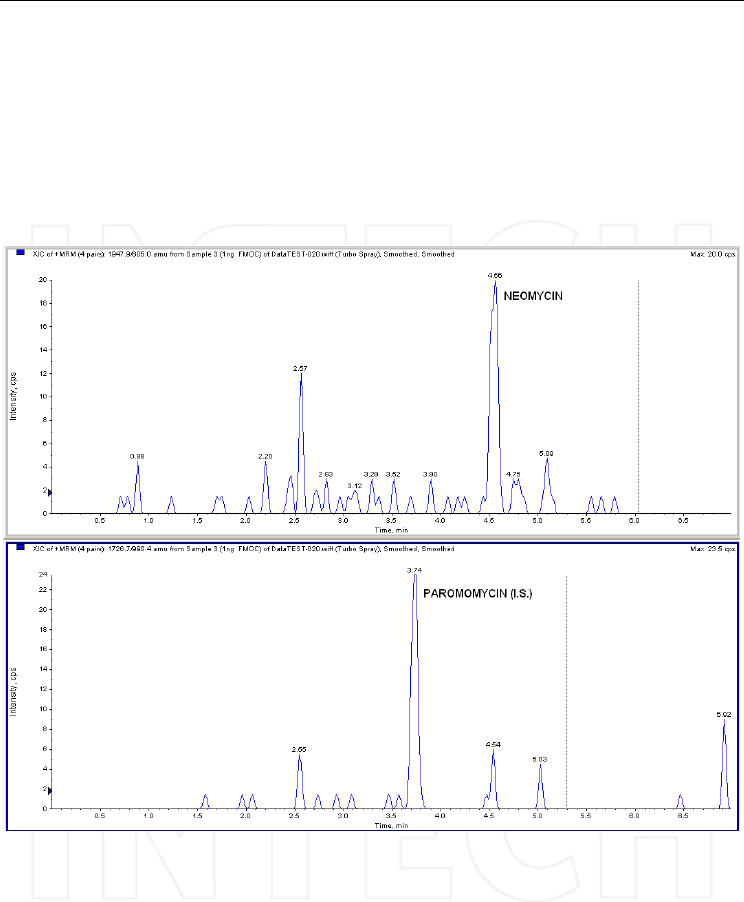
Another approach in aminoglycoside analysis by HPLC remains pre-column
derivatization, aiming to reduce polarity and make the compounds suitable for reversed-
www.intechopen.com
Tandem Mass Spectrometry – Applications and Principles
520
phase separations. Based on a previous research of Lauser and Bergner-Lang (1995), we
tested in our laboratory the derivatization of neomycin with FMOC. The reaction needs to
be optimized, because partial and fully substituted derivatives (all six amino groups
reacted with FMOC) are formed and in order to obtain a better sensitivity the equilibrium
should be ideally directed towards one product. We performed the chromatographic
separation on a C8 column eluted with water, acetonitrile and formic acid and
preliminary tests on plasma extracts have given promising results. The chromatographic
traces corresponding to the fully substituted neomycin and paromomycin derivatives (the
selected internal standard) on plasma samples are shown in Figure 20, while the product
ion spectrum of neomycin derivative in Figure 21.
Fig. 19. Chromatographic traces of neomycin and its internal standard streptomycin,
recorded on spiked sample (concentration 1 μg/ml). Column: Discovery HSF5
(10cmx2.1mm, 5μm), mobile phase: A) 0.2% PFPA in water and B) 0.2% PFPA in acetonitrile;
flow: 0.3 ml/min; injection volume: 10μL. Mass spectrometer: quadrupole-linear ion trap
API4000 QTrap, operated in ESI positive mode.
Last by not least, we have studied HILIC for the determination of vancomycin in plasma,
using neomycin as internal standard. The column employed was an Ascentis Express HILIC
(100x2.1mm, 2.7μm), the separation being carried out in isocratic conditions with
acetonitrile/water (90/10, v/v) containing 0.1% formic acid. A good sensitivity in the range
of few ng/mL, adequate for therapeutic drug monitoring, was obtained.
www.intechopen.com
HPLC-MS/MS of Highly Polar Compounds
521
Fig. 20. The MRM transitions of neomycin and paromomycin (internal standard) derivatives
with FMOC, obtained after injection of a derivatized plasma extract (spiked concentration
1ng/mL). Column: Ascentis Express C8 (100x2.1mm, 2.7 μm); mobile phase: A)
water/acetonitrile (90/10, v/v) and B) acetonitrile, with composition gradient; injection
volume: 50μL. Mass spectrometer: triple quadrupole API 4000 operated in ESI positive
mode.
www.intechopen.com
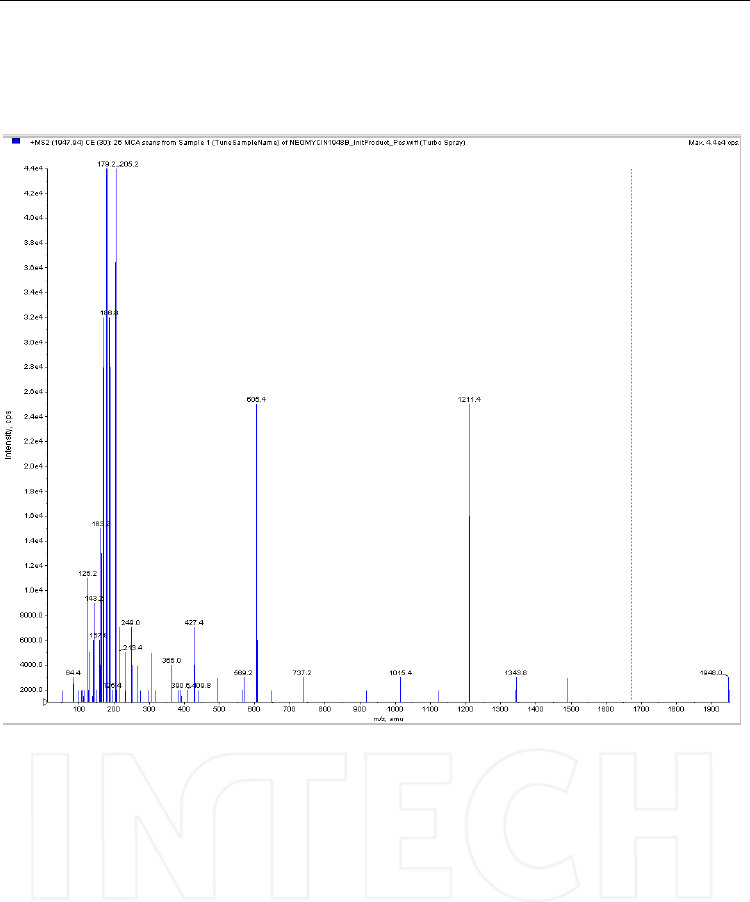
Tandem Mass Spectrometry – Applications and Principles
522
Fig. 21. Product ion spectrum of the neomycin derivative with FMOC - [M+H]+ = 1948
obtained with direct infusion via syringe of a 1μg/mL solution in water/methanol (1/1,
v/v). Mass spectrometer: triple quadrupole API4000, operated in positive ESI mode;
collision energy: 30V.
5. Conclusions
The data presented above clearly show that, despite the fact that highly polar compounds
present optimal characteristics to be analyzed by MS, they are difficult molecules due to
critical separation problems generated by the need to employ mobile phases compatible
with the ionization source and process. It is therefore essential, when developing a new
method for such compounds, to first consider the available options to get an optimal
separation compatible with the MS interfacing.
www.intechopen.com

HPLC-MS/MS of Highly Polar Compounds
523
Between the various options HILIC is gaining a lot of interest as a separation for highly
polar compounds; the development of several columns dedicated to this technique are
widening a lot its application and the growing mass of publications in the field is a prove.
HILIC employs high percentage of organic solvent in the mobile phase usually leading to
enhanced ionization and lesser contamination of the ion source.
Ion exchange remains an interesting alternative especially when considering weak
exchanger stationary phases; in comparison to HILIC however the technical development is
moving much slower. Mixed mode columns (ion exchange + reversed phase) are an
interesting opportunity to get improved separation methods.
The use of ion-pair reagents in connection with reversed-phase columns is another
opportunity but it presents a few pitfalls: restrictions on ionization polarity (in general ion-
pair suppress ionization in the same polarity, i.e. quaternary ammonium salt and positive
ionization), often complex spectra, reduced analytical sensitivity and finally a tendency to
contaminate the ionization source and quadrupoles. It is however interesting, see the GAGs
examples, that ion-pair reagents can, in some cases, positively help the ionization reducing
source fragmentation.
Globally the previously indicated strategies are preferable whenever possible. As a final
option derivatization has to be considered, without meaning that it is the last desirable
approach. It has to be considered especially in case of amphoteric compounds bearing both
positive and negative charges; such compounds are generally difficult to extract and the
ionization is not optimal; aminobisphosphonates are a good example in this context. The
development of derivatization methods can be time consuming but often very solid
methods and a quite good analytical sensitivity can be obtained. It is interesting to see a
continuous development for obtaining optimal derivatization reagents for LC-MS detection.
GC derivatization agents are indeed aiming to get non-polar and volatile compounds,
aspects not always desirable in LC-MS, while fluorescence and UV detection reagents can be
chemically very complex without favoring the ionization process.
The analytical approach described can be usefully applied as well to other classes of highly
polar compounds (for examples polynucleotides, phosphorylated carbohydrates, several
pharmaceuticals).
6. References
Alpert, A.J. (1990). Hydrophilic-interaction chromatography for the separation of peptides,
nucleic acids and other polar compounds. Journal of Chromatography 499: 177–196
Alpert, A.J. (2011). HILIC at 21: Reflections and perspective. Journal of Chromatography A
1218, 5879
Aluoch, A., Tatini, R., Parsons, D.M. & Sadik, O. (2005). Stability Indicating Ion-Pair HPLC
Methods for the Determination of Risedronate in A Commercial Formulation,
Journal of Liquid Chromatography Related Technologies, 27, 2799–2813
Apostolou, C., Dotsikas, Y., Kousoulos, C., Tsatsou, G., Colocouri, F., Soumelas, G.S. &
Loukas, Y. (2007). Application of a Semi-Automated 96-well Format Solid-Phase
Extraction, Column-Switching, Fluorescence Detection Protocol for the
Determination of Alendronate in Human Urine Samples Obtained from a
Bioequivalence Study, Journal of Pharmaceutical and Biomedical Analysis, 43 (3), 1151
Arpino, P. J. (1985). Ten years of liquid chromatography-mass spectrometry, Journal of
Chromatography A 323 (1), 3-11
www.intechopen.com

Tandem Mass Spectrometry – Applications and Principles
524
Aubin, A., Analysis of aminoglycoside antibiotics with Waters 2465 electrochemical
detector, Waters Application Note
Barosso, B., Didraga, M. & Bischoff, R. (2005). Analysis of proteoglycans derived sulphated
disaccharides by liquid chromatography/mass spectrometry, Journal of
Chromatography A, 1080(1), 43-48
Caprioli, R.M., Fan, T. & Cottrell,J.S. (1986). A continuous flow sample probe for fast atom
bombardment mass spectrometry, Analytical Chemistry, 58, 2949-2954
Chambers, H. F., (2006). Aminoglycosides, in Goodman & Gilman’s The Pharmacological Basis
of Therapeutics, 11e, pag.1155, Brunton, L.L., Lazo, J.S. & Parker., K.L. (Ed) McGraw Hill
Companies Inc.
Chester, T.L., Lewis, E.C., Benedict, J.J., Sunberg, R.J. & Tettenhorst, W.C. (1981).
Determination of (dichloromethylene) Diphosphonate in Physiological Fluids by
Ion-exchange Chromatography with Phosphorous-selective Detection, Journal of
Chromatography, 225, 17
Chopra, S., Vanderheyden, G., Hoogmartens, J., Schepdael, A. & Adams E. (2010).
Comparative study on the performance of different detectors for the LC analysis of
tobramycin, Journal of Pharmaceutical and Biomedical Analysis, 53(2), 151-157
Clarot, I., Storme-Paris, I., Chaminade, P., Estevenon, O., Nicolas, A. & Rieutord, A. (2009).
Simultaneous quantitation of tobramycin and colistin sulphate by HPLC with
evaporative light scattering detection, Journal of Pharmaceutical and Biomedical
Analysis, 50(1), 2009, 64-67
Clarot, I., Regazzeti, A., Auzeil, N., Laadani, F., Citton, M., Netter, P. & Nicolas, A. (2005).
Analysis of neomycin sulphate and framycetin sulphate by HPLC using
evaporative light scattering detection , Journal of Chromatography A, 1087(1-2), 236-
244.
Cohen, D.M. & Linhardt, R.J. (1990). Randomness in the heparin polymer: Computer
simulations of alternative action patterns of heparin lyase, Biopolymers 30(7-8), 733-
741
Da Col, R., Silvestro, L., Naggi, A., Torri, G., Baiocchi, C., Moltrasio, D., Cedro, A. & Viano, I.
(1993). Characterization of the chemical structure of sulphated glycosaminoglycans
after enzymatic digestion: Application of liquid chromatography—mass
spectrometry with an atmospheric pressure interface, Journal of Chromatography,
647, 289-300
Daley-Yates, P.T., Gifford, L.A. & Hoggarth, C.R. (1989). Assay of 1-hydroxy-3-
aminopropylidene-1,1-bisphosphonate and related bisphosphonates in human
urine and plasma by high-performance ion chromatography, Journal of
Chromatography, 490 (2), 329
Dell, A., Rogers, M.E. & Thomas–Oates, J.E. (1988). Fast-atom-bombardment mass-
spectrometric strategies for sequencing sulphated oligosaccharides, Carbohydrate
Researches, 179, 7
Deyrup, C.L., Chang, S.M., Weintraub, R. & Moye, H.A. (1985). Simultaneous esterification
and acylation of pesticides for analysis by gas chromatography. 1. Derivatization of
glyphosate and (aminomethyl)phosphonic acid with fluorinated alcohols-
perfluorinate anhydrides, Journal of Agricultural and Food Chemistry, 33, 944-947
Dinh, N.P, Jonsson, T, Irgum, K. (2011). Probing the interaction mode in hydrophilic
interaction chromatography. Journal of Chromatography A 1218, 5880-5891
www.intechopen.com

HPLC-MS/MS of Highly Polar Compounds
525
Du,J. & Eddington, N. (2002). Determination of the Chondroitin Sulphate Disaccharides in
Dog and Horse Plasma by HPLC Using Chondroitinase Digestion, Precolumn
Derivatization, and Fluorescence Detection, Analytical Biochemistry, 306, 252-258
Flesch, G., Tominaga, N. & Degen ,P.(1991). Improved Determination of the Bisphosphonate
Pamidronate Disodium in Plasma and Urine by Pre-Column Derivatization with
Fluorescamine, High-Performance Liquid Chromatography and Fluorescence
Detection, Journal of Chromatography, 568, 261
Fleisch, H. (2004). Development of biphosphonates, Breast Cancer Research, 4 (1), 30
Food and Drug Administration, Guidance for Industry / Bioanalytical method validation, May
2001
Garcia, J. F. & Barceló, D. (1993). An overview of LC–MS interfacing systems with selected
applications, Journal of High, Resolution Chromatography, 16(11), 633–641
Gatti, R., Andreatta, P, Gioia, M.G. & Boscchetti, S. (2010). A Simple and validated LC
method for the Simultaneous Analysis of Glucosamine and Chondroitin Sulphate
Equivalent in Dietary products, Journal of Liquid Chromatography and Related
Technologies, 33(19), 1760-1775
Granja, RH., Nino, AM, Zucchetti RA, Nino RE., Patel, R., Salerno, AG. (2009).
Determination of streptomycin residues in honey by liquid chromatography-
tandem mass spectrometry. Analitica Chimica Acta 637, (1-2), 64-47.
Gunay, N.S. & Linhardt, R.J. (1999). Heparinoids: Structure, Biological Activity and
Therapeutic Applications, Planta Medica, 65, 301-306
Hammel, Y-A., Mohamed, R., Gremaud, E., LeBreton, M-H., Guy, P. (2008). Multi-screening
approach to monitor and quantify 42 antibiotic residues in honey by liquid
chromatography–tandem mass spectrometry. Journal of Chromatography A, 1177, 58-
76
Hemstrom, P. & Irgum, K., (2006). Hydrophilic interaction chromatography, Journal of
Separation Sciences, 29(12),1784-1821
Henriksen, J., Roepstorff , P. & Ringborg, L.H. (2006). Ion-pairing reversed-phased
chromatography/mass spectrometry of heparin, Carbohydrate Researches, 341(3),
382-387
Higgins, C.E. & Kastner, R.E. (1967). Nebramycin, a new broad-spectrum antibiotic complex.
II. Description of Streptomyces tenebrarius, Antimicrobial Agents Chemotherapy, 7,
324-31.
Ip, D.P., Qin, X.Z., Sakuma, T. & Tsai, E.W. (1994). Pharmaceutical Application of Liquid
Chromatography-Mass spectrometry .2. Ion Chromatography Ion-spray Mass-
Spectrometric Characterization of Alendronate, Journal of Chromatography A, 686,
205
Ishii, R., Horie, M., Chan, W., MacNeil, J. (2008). Multi-residue quantitation of
aminoglycoside antibiotics in kidney and meat by liquid chromatography with
tandem mass spectrometry. Food Addit. Contam. A 25 (2008) 1509.
Isoherranen, N. & Soback, S. (1999). Chromatographic methods for analysis of
aminoglycoside antibiotics, Journal of Association of Analytical Chemists International,
82 (5), 1017-1045
Isoherranen, N. & Soback, S. (2000). Determination of Gentamicins C1, C1a and C2 in
plasma and urine by HPLC, Clinical Chemistry 46(6) 837-842
Jandik, K.A., Kruep, D., Cartier, M. & Linhardt, R.J. (1996). Accelerated stability studies of
heparin, Journal of Pharmaceutical Sciences, 85(1), 45-51
www.intechopen.com

Tandem Mass Spectrometry – Applications and Principles
526
Jin, P., Ma,J., Wu,X., Zou,D., Sun,C. & Hu,X. (2009). Simultaneous determination of
chondroitin sulphate sodium, allantoin and pyridoxine hydrochloride in
pharmaceutical eye drops by an ion-pair high-performance liquid chromatography,
Journal Pharmaceutical and Biomedical Analysis 50(3), 293-297
Johnson, E.A. (1982). Characterisation and separation of sulphated glycosaminoglycuronans,
Pharmacological Research Communications, 14(4), 289-320
Kawachi, Y., Tohru Ikegami, T., Hirotaka Takubo, H, Ikegami, Y., Miyamoto, M., Tanaka, N.
(2011). Chromatographic characterization of hydrophilic interaction liquid
chromatography stationary phases: Hydrophilicity, charge effects, structural
selectivity, and separation efficiency. Journal of Chromatography A 1218, 5903-5919
Kim, B.H., Lee, S.C., Lee, H.J. & Ok, J.H. (2003). Reversed-phase liquid chromatographic
method for the analysis of aminoglycoside antibiotics using pre-column
derivatisation with phenylisocyanate, Biomedical Chromatography 17(6), 396-403
Kim, B.H., Lee, S.C., Lee, H.J. & Ok, J.H. (2001). Development of liquid chromatographic
method for the analysis of kanamycin residues in varicella vaccine using
phenylisocyanate as a derivatization agent, Biomedical Chromatography, 752(1), 173-7
Kline, W.F. & Matuszewski, B.K. (1992). Improved determination of the bisphosphonate
alendronate in human plasma and urine by automated precolumn derivatization
and high-performance liquid chromatography with fluorescence and
electrochemical detection, Journal of Chromatography, 583, 183
Korir, A.K. & Larive, C.K. (2007). On-line NMR detection of microgram quantities of
heparin-derived oligosaccharides and their structure elucidation by microcoil
NMR, Analytical and Bioanalytical Chemistry, 388(8), 1707-1716
Kovacevic, M., Gartner, A. & Novic, M.(2004). Determination of Bisphosphonates by Ion
Chromatography–Inductively Coupled Plasma Mass Spectrometry, Journal of
chromatography A, 1039, 77
Kudzin, Z.H., Gralak, D.K., Andrijewski, G., Drabowicz J. & Luczak, J.(2003). Simultaneous
analysis of biologically active aminoalkanephosphonic acids, Journal of
Chromatography A, 998, 183
Lauser, G., Bergner-Lang, B. (1995). Flurimetrische Bestimmung von Aminoglykosid
Antibiotika nach Vörsaulenderivatisierung im Autosampler mit 9-Fluorenylmethyl
Chloroformiat und anschliessender HPLC-Trennung. Deutsche Lebensmittel
Rundschau 91 (12), 390-396
Malavaki, C. J., Asimakopoulou, A.P, Lamari, F.N., Teocharis, A.D.,.Tzanakakis, G.N. &
Karamanos, N.K. (2008). Capillary electrophoresis for the quality control of
chondroitin sulphates in raw materials and formulations, Analytical Biochemistry,
374(1), 213-220
Mallis, L.M., Wang, H.M., Loganathan, D. & Linhardt, R.J. (1989). Sequence analysis of
highly sulphated, heparin-derived oligosaccharides using fast atom bombardment
mass spectrometry, Analytical Chemistry, 61, 1453
Malsch, R. & Harenberg, J. (1996) Purity of glycosaminoglycan-related compounds using
capillary electrophoresis, Electrophoresis, 17, 401-405
Malsch, R., Harenberg, J., Piazolo, L., Huhle, G. & Heene, D.L. (1996). Chromatographic and
electrophoretic applications for the analysis of heparin and dermatan sulphate,
Journal of Chromatography B, 685, 223-231
www.intechopen.com

HPLC-MS/MS of Highly Polar Compounds
527
Manyanga, V., Dhulipalla, R. L., Hoogmartens, J. & Adams, E. (2010). Improved LC
method with pulsed electrochemical detection for the analysis of kanamycin,
Journal of Chromatography A, 1217(24), 3748-3753.
Mao, W., Thanawiroon, C. & Linhardt, R.J. (2002). Capillary electrophoresis for the Analysis
of Glycosaminoglycan-Derived Oligosaccharides, Biomedical Chromatography, 16, 77-
94
Mayhew, J.W. & Gorbach, S. L. (1978). Gas-liquid chromatographic method for the assay of
aminoglycoside antibiotics in serum, Journal of Chromatography, 151, 133-146
de Marco, J.D., Biffar, S.E., Reed, D.G. & Brooks, M.A. (1989). The Determination of 4-amino-
1-hydroxybutane-1,1-diphosphonic Acid Monosodium Salt Trihydrate in
Pharmaceutical Dosage Forms by High-Performance Liquid Chromatography,
Journal of Pharmaceutical and Biomedical Analusis, 7(12),1719–1727.
Margosis, M. &Tsuji, K. (1973), Journal of Pharmaceutical Sciences, 62, 1873
Mashat, M., Chrystyn, H., Clark, B.J. & Assi, K.H. (2008). Development and validation of
HPLC method for the determination of tobramycin in urine samples post-
inhalation using pre-column derivatization with fluorescein isothiocyanate, Journal
of Chromatography B, 869(1-2), 59-66
Metrohm. Gentamicin determination by ion chromatography. Laboratorytalk editorial team,
(2005). http://www.laboratorytalk.com/news/mea/mea456.html
Moye, HA, Deyrup, CL, (1984). Simultaneous esterification and acylation of pesticides for
analysis by gas chromatography. 1. Derivatization of glyphosate and
(aminomethyl)phosphonic acid with fluorinated alcohols-perfluorinated
anhydrides, Journal of Agricultural and Food Chemistry, 32, 192
Oertel, R,. Neumeister, V., Kirch, W. (2004). Hydrophilic interaction chromatography
combined with mass spectrometry to determine six aminoglycosides in serum.
Journal of Chromatography A, 1058, 197-201
Oguma, T., Tomatsu, S., Montano, A. M. & Okazaki, O. (2007). Analytical method for the
determination of disaccharides derived from keratan, heparan, and dermatan
sulphates in human serum and plasma by high-performance liquid
chromatography/turbo ionspray ionization tandem mass spectrometry, Analytical
Biochemistry 368(1), 79-86
Ori, A., Free, P., Courty, J., Wilkinson & M.C., Fernig, D.G. (2009). Identification of Heparin-
binding Sites in Proteins by Selective Labeling, Molecular & Cellular Proteomics 8(10),
2256-2265
Palit, M., Gupta, A., Jain, R. & Raza, S.K. (2004). Determination of pentafluorobenzyl
derivatives of phosphonic and phosphonothioic acids by gas-chromatography-
mass spectrometry, Journal of Chromatography A, 1043, 275
Patel, R.P., Narkowicz, C. & Jacobson, G.A. (2009) Effective reversed-phase ion pair high-
performance liquid chromatography method for the separation and
characterization of intact low-molecular-weight heparins, Analytical Biochemistry
367(1), 113-121
Plaas, A.H., Hascall, V.C. & Midura, R.J. (1996). Ion exchange HPLC microanalysis of
chondroitin sulphate: quantitative derivatization of chondroitin lyase digestion
products with 2-aminopyridine, Glycobiology, 6(8), 823-9
Ptacek, P., Klima, J. & Macek, J. (2002). Determination of Alendronate in Human Urine as 9-
fluorenylmethyl Derivative by High Performance Liquid Chromatography, Journal
of Chromatography B, 767, 111
www.intechopen.com

Tandem Mass Spectrometry – Applications and Principles
528
Rao, B.M., Srinivasu, M.K., Rani, Ch.P., Kumar, S.S., Kumar, P.R., Chandrasekhar, K.B. &
Veerender, M., (2005). A Validated Stability Indicating Ion-Pair RP-LC Method for
Zoledronic Acid, Journal of Pharmaceutical and Biomedical Analysis, 39(3-4), 781–790
Ranz, A., Korpecka, J. & Lankmayr, E. (2008) Optimized derivatization of acidic herbicides
with trimethylsilyldiazomethane for GC analysis, Journal of Separation Science 31 (4),
746
Royer, A., Beguin, S., Sochor, H., Communal, P., (2000). Determination of gluphosinate
ammonium ant its metabolite (AE F064619 and AE F061517) residues in water by
gas-chormatography with tandem mass spectrometry after ion exchange clean-up
and derivatization, Journal of Agricultural and Food Chemistry, 48, 5184-5189
Ruiz-Calero, V., Puignou, L. & Galceran, M.T., Determination of glycosaminoglycan
monosaccharides by capillary electrophoresis using laser-induced fluorescence
detection, Journal of Chromatography B, 791(1-2), 193-202
Yang, B., Solakyidirim, K., Chang, Y. & Linhardt, R.J. (2011). Hyphenated techniques for the
analysis of heparin and heparan sulphate, Analytical and Bioanalytical Chemistry,
399, 541-557
Yun, M.H. & Kwon, K.L. (2006). High performance Liquid Chromatography for
Determining Alendronate Sodium in Human Plasma by Detecting Fluorescence:
Application to a Pharmacokinetic Study, Journal of Pharmaceutical and Biomedical
Analysis, 40, 168–172Saad, O.M & Leary, J. A. (2003). Composition Analysis and
Quantification of Heparin and Heparan Sulphate by Electrospray Ionization Ion
Trap Mass Spectrometry, Analytical Chemistry, 75(13), 2985-2995
Saito, H., Yamagata, T. & Suzuki, S. (1968). Enzymatic Methods for the Determination of
Small Quantities of Isomeric Chondroitin Sulphates, Jouranl of Biological Chemistry,
243, 1536-1542
Shao,Y., Alluri,R., Mummert,M., Koetter,U. & Lech, S. (2004). A stability-indicating HPLC
method for the determination of glucosamine in pharmaceutical formulations,
Journal of Pharmaceutical and Biomedical Analysis, 35(3), 625-631
Silvestro, L., Rizea Savu, S., van Veelen, P.A. & Jacobs, P.L. (1996). Application of Mass
Spectrometry to the Analysis of Natural and Synthetic sulphated Oligosaccharides,
in Nonanticoagulant Actions of Glycosaminoglycans, Harenberg, J. & Casu, B. (Ed),
Plenum Press, New York
Silvestro, L., Viano, I., Naggi, A., Torri, G., Col, R.Da. & Baiocchi, C. (1992). High-
performance liquid chromatographic-mass spectrometric analysis of
oligosaccharides from enzymatic digestion of glycosaminoglycans: Application to
human samples, Journal of Chromatography, 591, 225-232
Shen, A. Q., Morgan, L., Barroso, M.L., Zhang X., & Nguyen, T. (June 2008). Method
development of LC-MS/MS analysis of aminoglycoside drugs: challenges and
solutions, Presented at the 2008 ASMS Conference, Denver, CO, June 2008,
Available from: http://tandemlabs.com/documents/ASMS08-Angela-Web.pdf
Sakiyama, N., Kataoka, H. & Makita, M. (1996). Gas chromatographic analysis of 3-amino-1-
hydroxypropilidende-1,1-bisphosphonate and related bisphosphonate as their N-
isobutoxycarbonyl methyl ester derivatives, Journal of Chromatography A, 724, 279
Sakiyama, N., Kataoka, H.& Makita, M. (1995). Selective and sensitive determination of
pamidronate in human plasma and urine by gas-chromatography with flame
photometric detection, Biomedical Chromatography 9 (5), 243
www.intechopen.com

HPLC-MS/MS of Highly Polar Compounds
529
Sparidans, R.W. & den Hartigh, J. (1999). Chromatographic Analysis of Biphosphonates,
Pharmaceutical World & Science, 21(1), 1–10
Stead, D.A. (2000). Current methodologies for the analysis of aminoglycosides, Journal of
Chromatography B, 747(1-2), 69-93
Strege, M. A., (1999). High-performance liquid chromatographic–electrospray ionization
mass spectrometric analyses for the integration of natural products with modern
high-throughput screening, Journal of Chromatography B, 725 (1-2), 67-78
Tarcomnicu, I., Silvestro, L., Rizea Savu, S., Gherase, A. & Dulea, C. (2007). Development
and application of an HPLC-MS/MS method to determine alendronate in human
urine, Journal of Chromatography A, 1160, 21-33
Tarcomnicu, I., Gheorghe, MC., Silvestro, L., Rizea Savu, S., Boaru, I., Tudoroniu, A. (2009).
High-throughput HPLC-MS/MS method to determine ibandronate in
humanplasma for pharmacokinetic applications. Journal of Chromatography B, 877,
3159-3168
Tawa, R., Matsunaga, H. & Fujimoto, T. (1998). HPLC analysis of aminoglycoside
antibiotics, Journal of Chromatography A, 812(1-2), 141-150
Thanawiroon, C., Rice, K. G., Toida, T. & Linhardt, R. J. (2004). Liquid
Chromatography/Mass Spectrometry Sequencing Approach for Highly Sulphated
Heparin-derived Oligosaccharides, Journal of Biological Chemistry, 279(4), 2608-2615
The Merck Index, (2006) Fourtheenth Edition, Published by Merck Research Laboratories,
Merck and Co. Inc., Whitehouse Station NJ, USA
Tiayu Peng, (2002). Analysis of Saccharides by Capillary Electrodriven Separation Methods,
Ph.D Thesis, Department of organic Chemistry, Faculty of Science, University of Ghent,
Belgium
Toyoda, H., Kinoshita-Toyoda, A., Fox, B., Selleck, S.B. (2000). Structural Analysis of
Glycosaminoglycans inDrosophila and Caenorhabditis elegans and Demonstration
That tout-velu, a Drosophila Gene Related to EXT Tumor Suppressors, Affects
Heparan Sulphate in Vivo, Journal Biological Chemistry , 275(4), 2269-2275
Trehy, M.L.,.Reepmeyer, J.C., Kolinski, R.E., Westenberger, B.J. & Buhse, L.F. (2009).
Analysis of heparin sodium by SAX/HPLC for contaminants and impurities,
Journal of Pharmaceutical and Biomedical Analysis 49(3), 670-673
Tsai, E. W., Ip, D.P. & Brooks, M.A. (1992). Determination of Alendronate in Pharmaceutical
Dosage Formulations by Ion Chromatography with Conductivity Detection, Journal
of Chromatography A, 556, 217
Vallano, P.T., Shugarts, S.B., Kline, W.F., Woolf E.J. & Matuszewski, B.K. (2003).
Determination of Risedronate in Human Urine by Column-Switching Ion-Pair
High-Performance Liquid Chromatography with Ultraviolet Detection Journal of
Chromatography B. 794, 23–33
Van Nuijs A., Tarcomnicu I., Covaci A. (2011) Application of hydrophilic interaction
chromatography for the analysis of polar contaminants in food and environmental
samples. Journal of Chromatography A, 1218, 5964-5974
Venkataram, G., Shriver, Z., Raman, R., & Sasisekharan, R. (1999) Sequencing Complex
Polysaccharides, Science, 286, 537-542
Xie, Z., Jiang, Y. & D. Zhang (2006). Simple Analysis of Four Bisphosphonates
Simultaneously by Reverse Phase Liquid Chromatography Using n-amylamine as
Volatile Ion-Pairing Agent, Journal of Chromatography A, 1004(1-2), 173
www.intechopen.com

Tandem Mass Spectrometry – Applications and Principles
530
Zhu, L.S. , Lapko, V.N., Lee, J.W., Basir, Y.J., Kafonek, C., Olsen, R. & Briscoe, C. (2006). A
general approach for the quantitative analysis of bisphosphonates in human serum
and urine by high-performance liquid chromatography/tandem mass
spectrometry, Rapid Communications. Mass Spectrometry 20, 2426–3421
Zimmer, J. S.D., Needham, S. R., McKinnell, J., Cass, B. & Karr, D. (June 2008). Development
of a Bioanalytical LC/MS/MS Assay for the Quantitative Analysis of Amikacin,
Neomycin and Gentamicin in Plasma and Tissue Samples. Presented at the 2008
ASMS Conference, Denver, CO, June 2008. Available from:
http://www.alturasanalytics.com/home/pdf/ASMS2008AlturasAnalyticsPosterR
eprint.pdf
Wong, J.A., Renton, K.W., Crocker, J.F., O’Regan, R.A. & Acott, P.D. (2004). Determination
of Pamidronate in Human Whole Blood and Urine by Reversed-Phase HPLC with
Fluorescence Detection, Biomedical Chromatography, 18, 98
www.intechopen.com

Tandem Mass Spectrometry - Applications and Principles
Edited by Dr Jeevan Prasain
ISBN 978-953-51-0141-3
Hard cover, 794 pages
Publisher InTech
Published online 29, February, 2012
Published in print edition February, 2012
InTech Europe
University Campus STeP Ri
Slavka Krautzeka 83/A
51000 Rijeka, Croatia
Phone: +385 (51) 770 447
Fax: +385 (51) 686 166
www.intechopen.com
InTech China
Unit 405, Office Block, Hotel Equatorial Shanghai
No.65, Yan An Road (West), Shanghai, 200040, China
Phone: +86-21-62489820
Fax: +86-21-62489821
Tandem Mass Spectrometry - Applications and Principles presents comprehensive coverage of theory,
instrumentation and major applications of tandem mass spectrometry. The areas covered range from the
analysis of drug metabolites, proteins and complex lipids to clinical diagnosis. This book serves multiple groups
of audiences; professional (academic and industry), graduate students and general readers interested in the
use of modern mass spectrometry in solving critical questions of chemical and biological sciences.
How to reference
In order to correctly reference this scholarly work, feel free to copy and paste the following:
Luigi Silvestro, Isabela Tarcomnicu and Simona Rizea Savu (2012). HPLC-MS/MS of Highly Polar Compounds,
Tandem Mass Spectrometry - Applications and Principles, Dr Jeevan Prasain (Ed.), ISBN: 978-953-51-0141-3,
InTech, Available from: http://www.intechopen.com/books/tandem-mass-spectrometry-applications-and-
principles/hplc-ms-ms-of-highly-polar-compounds
