AABB Technical Manual 15th Ed. 2005
AABB%20Technical%20Manual%2015TH
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 931 [warning: Documents this large are best viewed by clicking the View PDF Link!]

Technical Manual
15th Edition
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.
Other related publications available from the AABB:
Technical Manual and Standards for Blood Banks and
TransfusionServicesonCD-ROM
Transfusion Therapy: Clinical Principles and Practice, 2nd Edition
Edited by Paul D. Mintz, MD
Transfusion Medicine Self-Assessment and Review
By Pam S. Helekar, MD; Douglas P. Blackall, MD; Jeffrey L. Winters, MD;
and Darrell J. Triulzi, MD
Blood Transfusion Therapy: A Physician’s Handbook, 8th Edition
Edited by Jerry Gottschall, MD
Practical Guide to Transfusion Medicine
By Marian Petrides, MD, and Gary Stack, MD, PhD
Transfusion Medicine Interactive: A Case Study Approach CD-ROM
By Marian Petrides, MD; Roby Rogers, MD; and Nora Ratcliffe, MD
To purchase books, please call our sales department at (866)222-2498 (within the United
States) or (301)215-6499 (outside the United States); fax orders to (301)907-6895 or email
orders to sales@aabb.org. View the AABB Publications Catalog and order books on the
AABB Web site at www.aabb.org. For other book services, including chapter reprints and
large quantity sales, ask for the Senior Sales Associate.
Copyright © 2005 by the AABB. All rights reserved.

Mention of specific products or equipment by contributors to this AABB publication
does not represent an endorsement of such products by the AABB nor does it necessar-
ily indicate a preference for those products over other similar competitive products. Any
forms and/or procedures in this book are examples. AABB does not imply or guarantee
that the materials meet federal, state, or other applicable requirements. It is incumbent
on the reader who intends to use any information, forms, policies, or procedures con-
tained in this publication to evaluate such materials for use in light of particular circum-
stances associated with his or her institution.
Efforts are made to have publications of the AABB consistent in regard to acceptable
practices. However, for several reasons, they may not be. First, as new developments in
the practice of blood banking occur, changes may be recommended to the Standards for
Blood Banks and Transfusion Services. It is not possible, however, to revise each publica-
tion at the time such a change is adopted. Thus, it is essential that the most recent edi-
tion of the Standards be consulted as a reference in regard to current acceptable prac-
tices. Second, the views expressed in this publication represent the opinions of authors.
The publication of this book does not constitute an endorsement by the AABB of any
view expressed herein, and the AABB expressly disclaims any liability arising from any
inaccuracy or misstatement.
Copyright © 2005 by AABB. All rights reserved. No part of this book may be reproduced
or transmitted in any form or by any means, electronic or mechanical, including photo-
copying, recording, or by any information storage and retrieval system, without permis-
sion in writing from the Publisher.
AABB ISBN No. 1-56395-196-7
8101 Glenbrook Road Printed in the United States
Bethesda, Maryland 20814-2749
Cataloging-in-Publication Data
Technical manual / editor, Mark E. Brecher. —15th ed.
p.;cm.
Including bibliographic references and index.
ISBN 1-56395-196-7
1. Blood Banks—Handbooks, manuals, etc. I. Brecher, Mark E. II. AABB.
[DNLM: 1. Blood Banks—laboratory manuals. 2. Blood Transfusion—
laboratory manuals. WH 25 T2548 2005]
RM172.T43 2005
615’.39—dc23
DNLM/DLC
Copyright © 2005 by the AABB. All rights reserved.
Technical Manual
Program Unit
Chair and Editor
Mark E. Brecher, MD
Associate Editors
Regina M. Leger, MSQA, MT(ASCP)SBB, CQMgr(ASQ)
Jeanne V. Linden, MD, MPH
Susan D. Roseff, MD
Members/Authors
Martha Rae Combs, MT(ASCP)SBB
Gregory Denomme, PhD, FCSMLS(D)
Brenda J. Grossman, MD, MPH
N. Rebecca Haley, MD, MT(ASCP)SBB
Teresa Harris, MT(ASCP)SBB, CQIA(ASQ)
Betsy W. Jett, MT(ASCP), CQA(ASQ)CQMgr
Regina M. Leger, MSQA, MT(ASCP)SBB, CQMgr(ASQ)
Jeanne V. Linden, MD, MPH
Janice G. McFarland, MD
James T. Perkins, MD
Susan D. Roseff, MD
Joseph Sweeney, MD
Darrell J. Triulzi, MD
Liaisons
Gilliam B. Conley, MA, MT(ASCP)SBB
MichaelC.Libby,MSc,MT(ASCP)SBB
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.
Acknowledgments
The Technical Manual Program Unit extends special thanks to those volunteers who
provided peer review and made other contributions:
James P. AuBuchon, MD
Lucia M. Berte, MA,
MT(ASCP)SBB, DLM,
CQA(ASQ)CQMgr
Arthur Bracey, MD
Linda Braddy,
MT(ASCP)SBB
Donald R. Branch,
MT(ASCP)SBB, PhD
Ritchard Cable, MD
Sally Caglioti,
MT(ASCP)SBB
Loni Calhoun,
MT(ASCP)SBB
Tony S. Casina,
MT(ASCP)SBB
Geoff Daniels, PhD,
MRcPath
Robertson Davenport, MD
Richard J. Davey, MD
Walter Dzik, MD
Ted Eastlund, MD
Anne F. Eder, MD, PhD
Ronald O. Gilcher, MD,
FACP
Lawrence T. Goodnough,
MD
Linda Hahn,
MT(ASCP)SBB, MPM
Heather Hume, MD
Mark A. Janzen, PhD
SusanT.Johnson,MSTM,
MT(ASCP)SBB
W. John Judd, FIBMS,
MIBiol
Michael H. Kanter, MD
Louis M. Katz, MD
Debra Kessler, RN, MS
Thomas Kickler, MD
Karen E. King, MD
Joanne Kosanke,
MT(ASCP)SBB
Thomas A. Lane, MD
Alan H. Lazarus, PhD
German F. Leparc, MD
Douglas M. Lublin, MD,
PhD
Dawn Michelle,
MT(ASCP)SBB
Kenneth Moise, Jr., MD
S. Breanndan Moore, MD
Tania Motschman, MS,
MT(ASCP)SBB,
CQA(ASQ)
Marilyn K. Moulds,
MT(ASCP)SBB
Nancy C. Mullis,
MT(ASCP)SBB
Scott Murphy, MD
Patricia Pisciotto, MD
Mark A. Popovsky, MD
Marion E. Reid, PhD,
FIBMS
Jennifer F. Rhamy, MBA,
MA, MT(ASCP), SBB, HP
Scott D. Rowley, MD
ArellS.Shapiro,MD
R. Sue Shirey, MS,
MT(ASCP)SBB
Bruce Spiess, MD, FAHA
JerryE.Squires,MD,PhD
Marilyn J. Telen, MD
Susan Veneman,
MT(ASCP)SBB
Phyllis S. Walker, MS,
MT(ASCP)SBB
Dan A. Waxman, MD
Robert Weinstein, MD
Connie M. Westhoff, PhD,
MT(ASCP)SBB
Members of AABB com-
mittees who reviewed
manuscriptsaspartof
committee resource
charges
The staff of the Armed
Services Blood Program
Office
The staff of the US Food
and Drug Administra-
tion, Center for
Biologics Evaluation
and Research
The staff of the Transplan-
tation and Transfusion
Service, McClendon
Clinical Laboratories,
UNC Hospitals
Special thanks are due to Laurie Munk, Janet McGrath, Nina Hutchinson, Jay Penning-
ton, Frank McNeirney, Kay Gregory, MT(ASCP)SBB, and Allene Carr-Greer,
MT(ASCP)SBB of the AABB National Office for providing support to the Program Unit
during preparation of this edition.
Copyright © 2005 by the AABB. All rights reserved.
Introduction
The 15th edition of the AABB Tech-
nical Manual is the first in the
second half century of this publica-
tion. The original Technical Manual (then
called Technical Methods and Procedures)
was published in 1953 and the 14th edi-
tion marked the 50th anniversary of this
publication.
Over the years, this text has grown and
matured, until today it is a major textbook
used by students (medical technology and
residents) and practicing health-care pro-
fessionals (technologists, nurses, and phy-
sicians) around the world. Selected editions
or excerpts have been translated into
French, Hungarian, Italian, Japanese, Span-
ish, Polish, and Russian. It is one of only
two AABB publications that are referenced
by name in the AABB Standards for Blood
Banks and Transfusion Services (the other
being the Circular of Information for the
Use of Human Blood Components). All
branches of the US Armed Services have
adopted the AABB Technical Manual as
their respective official manuals for blood
banking and transfusion medicine activi-
ties.
The Technical Manual serves a diverse
readership and is used as a technical refer-
ence, a source for developing policies and
procedures, and an educational tool. The
Technical Manual is often the first reference
consulted in many laboratories; thus, it is
intended to provide the background infor-
mation to allow both students and experi-
enced individuals to rapidly familiarize
themselves with the rationale and scientific
basis of the AABB standards and current
standards of practice. As in previous edi-
tions, the authors and editors have tried to
provide both breadth and depth, including
substantial theoretical and clinical material
as well as technical details. Due to space
limitations, the Technical Manual cannot
provide all of the advanced information on
any specific topic. However, it is hoped that
sufficient information is provided to answer
the majority of queries for which individu-
als consult the text, or at a minimum, to di-
rect someone toward additional pertinent
references.
Readers should be aware that, unlike
most textbooks in the field, this book is
subjected to extensive peer review (by ex-
perts in specific subject areas, AABB com-
mittees, and regulatory bodies such as the
Food and Drug Administration). As such,
this text is relatively unique, and represents
ix
Copyright © 2005 by the AABB. All rights reserved.
a major effort on the part of the AABB to
provide an authoritative and balanced
reference source.
As in previous recent editions, the con-
tent is necessarily limited in order to retain
thesizeoftheTechnical Manual to that of a
textbook that can be easily handled. Never-
theless, readers will find extensive new and
updated information, including expanded
coverage of quality approaches, apheresis
indications, cellular nomenclature, molec-
ular diagnostics, hematopoietic progenitor
cell processing, and transfusion-transmitted
diseases.
Techniques and policies outlined in the
Technical Manual are, to the best of the
Technical Manual Program Unit's ability, in
conformance with AABB Standards.They
are not to be considered the only permissi-
ble way in which requirements of Stan-
dards can be met. Other methods, not in-
cluded, may give equally acceptable results.
If discrepancy occurs between techniques
or suggestions in the Technical Manual and
the requirements of Standards,authority
resides in Standards. Despite the best ef-
forts of both the Program Unit and the ex-
tensive number of outside reviewers, errors
mayremaininthetext.Aswithprevious
editions, the Program Unit welcomes sug-
gestions, criticisms, or questions about the
current edition.
I would like to thank the members of the
Technical Manual Program Unit for their
dedication and long hours of work that
went into updating this edition. I would
also like to thank all the AABB committees,
the expert reviewers, and the readers who
have offered numerous helpful suggestions
that helped to make this edition possible. I
would particularly like to thank my three
associate editors—Gina Leger, Jeanne Lin-
den, and Sue Roseff—who have provided
countless invaluable hours in the prepara-
tion of this edition. Finally I would like to
thank Laurie Munk, AABB Publications Di-
rector, whose tireless efforts on behalf of
the Technical Manual never cease to amaze
me, and who has made the publication of
this book a pleasure.
This edition is my third and final Techni-
cal Manual. I served as associate editor for
the 13th edition and chief editor for the
14th and 15th editions. It has been an
honor to help shepherd these editions to
fruition and it is my hope that the AABB
Technical Manual will continue to be one
of the AABB's premier publications for de-
cades to come.
Mark E. Brecher, MD
Chief Editor
Chapel Hill, NC
x AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.
Contents Contents
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ix
Quality Issues
1. Quality Systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
Quality Control, Quality Assurance, and Quality Management . . . . . . . . . . . . . 2
Quality Concepts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
Practical Application of Quality Principles . . . . . . . . . . . . . . . . . . . . . . . . . 6
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
Appendix 1-1. Glossary of Commonly Used Quality Terms . . . . . . . . . . . . . . . 30
Appendix 1-2. Code of Federal Regulations Quality-Related References . . . . . . . 32
Appendix 1-3. Statistical Tables for Binomial Distribution Used to
DetermineAdequateSampleSizeandLevelofConfidencefor
Validation of Pass/Fail Data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
Appendix 1-4. Assessment Examples: Blood Utilization. . . . . . . . . . . . . . . . . 36
2. Facilities and Safety. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
Facilities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
Safety Program. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
Fire Prevention . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
Electrical Safety . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
Biosafety . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
Chemical Safety . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
Radiation Safety . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
Shipping Hazardous Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
Waste Management. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
Disaster Planning . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
Suggested Reading . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .70
Appendix 2-1. Safety Regulations and Recommendations Applicable to
Health-Care Settings. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
Appendix 2-2. General Guidelines for Safe Work Practices, Personal Protective
Equipment, and Engineering Controls . . . . . . . . . . . . . . . . . . . . . . . . . 73
Appendix 2-3. Biosafety Level 2 Precautions . . . . . . . . . . . . . . . . . . . . . . . 77
Appendix 2-4. Sample Hazardous Chemical Data Sheet. . . . . . . . . . . . . . . . . 78
Appendix 2-5. Sample List of Hazardous Chemicals in the Blood Bank . . . . . . . . 80
Appendix 2-6. Specific Chemical Categories and How to Work Safely with
These Chemicals . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
Appendix 2-7. Incidental Spill Response . . . . . . . . . . . . . . . . . . . . . . . . . . 84
Appendix 2-8. Managing Hazardous Chemical Spills . . . . . . . . . . . . . . . . . . 87
xi
Copyright © 2005 by the AABB. All rights reserved.
3. Blood Utilization Management. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
Minimum and Ideal Inventory Levels . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
Determining Inventory Levels. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
Factors that Affect Outdating . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
Improving Transfusion Service Blood Ordering Practices . . . . . . . . . . . . . . . . 91
Special Product Concerns . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
Blood Donation and Collection
4. Allogeneic Donor Selection and Blood Collection . . . . . . . . . . . . . . . . . . . 97
Blood Donation Process . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
Collection of Blood . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
Appendix 4-1. Full-Length Donor History Questionnaire . . . . . . . . . . . . . . . 110
Appendix 4-2. Medication Deferral List. . . . . . . . . . . . . . . . . . . . . . . . . . 113
Appendix 4-3. Blood Donor Education Materials . . . . . . . . . . . . . . . . . . . . 114
Appendix 4-4. Some Drugs Commonly Accepted in Blood Donors . . . . . . . . . 115
5. Autologous Blood Donation and Transfusion . . . . . . . . . . . . . . . . . . . . . 117
Preoperative Autologous Blood Collection . . . . . . . . . . . . . . . . . . . . . . . . 118
Acute Normovolemic Hemodilution . . . . . . . . . . . . . . . . . . . . . . . . . . . 126
Intraoperative Blood Collection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 130
Postoperative Blood Collection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 133
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 135
6. Apheresis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 139
Separation Techniques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 139
Component Collection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 140
Therapeutic Apheresis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 144
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
7. Blood Component Testing and Labeling . . . . . . . . . . . . . . . . . . . . . . . . 163
Testing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 163
Labeling, Records, and Quarantine . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 174
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 174
8. Collection, Preparation, Storage, and Distribution of Components from
Whole Blood Donations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 175
Blood Component Descriptions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 175
Collection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 178
Prestorage Processing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 179
Storage. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184
xii AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Inspection, Shipping, Disposition, and Issue . . . . . . . . . . . . . . . . . . . . . . 194
Blood Component Quality Control . . . . . . . . . . . . . . . . . . . . . . . . . . . . 197
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 199
Appendix 8-1. Component Quality Control . . . . . . . . . . . . . . . . . . . . . . . 202
Immunologic and Genetic Principles
9. Molecular Biology in Transfusion Medicine . . . . . . . . . . . . . . . . . . . . . . 203
From DNA to mRNA to Protein . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
Genetic Mechanisms that Create Polymorphism . . . . . . . . . . . . . . . . . . . . 207
Genetic Variability . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 208
Molecular Techniques. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 209
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 220
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 221
Appendix 9-1. Molecular Techniques in Transfusion Medicine . . . . . . . . . . . . 222
10. Blood Group Genetics. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223
Basic Principles. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223
Genetics and Heredity. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 225
Patterns of Inheritance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 232
Population Genetics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 236
Blood Group Nomenclature . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 238
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 239
Appendix 10-1. Glossary of Terms in Blood Group Genetics . . . . . . . . . . . . . 241
11. Immunology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 243
Immune Response. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 243
Organs of the Immune System. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 249
Cells of the Immune System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 249
Soluble Components of the Immune Response . . . . . . . . . . . . . . . . . . . . . 256
Immunology Relating to Transfusion Medicine . . . . . . . . . . . . . . . . . . . . . 263
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 267
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 268
Appendix 11-1. Definitions of Some Essential Terms in Immunology . . . . . . . . 269
12. Red Cell Antigen-Antibody Reactions and Their Detection . . . . . . . . . . . . 271
Factors Affecting Red Cell Agglutination . . . . . . . . . . . . . . . . . . . . . . . . . 272
Enhancement of Antibody Detection . . . . . . . . . . . . . . . . . . . . . . . . . . . 276
The Antiglobulin Test . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 277
Other Methods to Detect Antigen-Antibody Reactions. . . . . . . . . . . . . . . . . 283
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 286
Contents xiii
Copyright © 2005 by the AABB. All rights reserved.
Blood Groups
13. ABO, H, and Lewis Blood Groups and Structurally Related Antigens . . . . . . 289
The ABO System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 289
The H System. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 303
The Lewis System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 304
The I/i Antigens and Antibodies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 306
The P Blood Group and Related Antigens . . . . . . . . . . . . . . . . . . . . . . . . 308
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 311
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 312
14. The Rh System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 315
The D Antigen and Its Historical Context. . . . . . . . . . . . . . . . . . . . . . . . . 315
Genetic and Biochemical Considerations . . . . . . . . . . . . . . . . . . . . . . . . 316
Rh Terminology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 318
Serologic Testing for Rh Antigen Expression . . . . . . . . . . . . . . . . . . . . . . . 319
Weak D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 322
Other Rh Antigens . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 324
Rhnull Syndrome and Other Deletion Types . . . . . . . . . . . . . . . . . . . . . . . . 325
Rh Antibodies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 327
Rh Typing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 328
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 332
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 333
15. Other Blood Groups . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 335
Distribution of Antigens . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 335
MNS System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 337
Kell System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 340
Duffy System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 343
Kidd System. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 345
Other Blood Group Systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 346
Blood Group Collections . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 355
High-Incidence Red Cell Antigens Not Assigned to a Blood Group
System or Collection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 356
Low-Incidence Red Cell Antigens Not Assigned to a Blood Group
System or Collection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 357
Antibodies to Low-Incidence Antigens . . . . . . . . . . . . . . . . . . . . . . . . . . 358
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 358
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 360
16. Platelet and Granulocyte Antigens and Antibodies . . . . . . . . . . . . . . . . 361
Platelet Antigens . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 361
Granulocyte Antigens . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 377
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 380
xiv AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
17. The HLA System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 385
Genetics of the Major Histocompatibility Complex . . . . . . . . . . . . . . . . . . 386
Biochemistry, Tissue Distribution, and Structure . . . . . . . . . . . . . . . . . . . . 390
Nomenclature . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 391
Biologic Function . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 393
Detection of HLA Antigens and Alleles . . . . . . . . . . . . . . . . . . . . . . . . . . 394
The HLA System and Transfusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 397
HLA Testing and Transplantation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 400
Parentage and Other Forensic Testing . . . . . . . . . . . . . . . . . . . . . . . . . . 402
HLA and Disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 402
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 404
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 405
Serologic Principles and Transfusion Medicine
18. Pretransfusion Testing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 407
Transfusion Requests . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 407
Blood Sample. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 409
Serologic Testing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 410
Crossmatching Tests. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 413
Interpretation of Antibody Screening and Crossmatch Results . . . . . . . . . . . . 415
Labeling and Release of Crossmatched Blood at the Time of Issue. . . . . . . . . . 416
Selection of Units . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 418
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 420
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 420
19. Initial Detection and Identification of Alloantibodies to Red Cell Antigens . . . . 423
Significance of Alloantibodies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 423
General Procedures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 424
Basic Antibody Identification Techniques . . . . . . . . . . . . . . . . . . . . . . . . 427
Complex Antibody Problems. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 431
Selecting Blood for Transfusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 439
Selected Serologic Procedures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 443
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 450
20. The Positive Direct Antiglobulin Test and Immune-Mediated Red
Cell Destruction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 453
The Direct Antiglobulin Test . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 454
Immune-Mediated Hemolysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 458
Serologic Problems with Autoantibodies . . . . . . . . . . . . . . . . . . . . . . . . . 469
Drug-Induced Immune Hemolytic Anemia . . . . . . . . . . . . . . . . . . . . . . . 472
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 477
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 479
Contents xv
Copyright © 2005 by the AABB. All rights reserved.
Appendix 20-1. An Example of an Algorithm for Investigating a Positive DAT
(Excluding Investigation of HDFN) . . . . . . . . . . . . . . . . . . . . . . . . . . 480
Appendix 20-2. Some Drugs Associated with Immune Hemolysis and/or
Positive DATs Due to Drug-Induced Antibodies . . . . . . . . . . . . . . . . . . . 481
Clinical Considerations in Transfusion Practice
21. Blood Transfusion Practice . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 483
Red Blood Cell Transfusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 483
Platelet Transfusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 488
Granulocyte Transfusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 492
Special Cellular Blood Components. . . . . . . . . . . . . . . . . . . . . . . . . . . . 492
Replacement of Coagulation Factors . . . . . . . . . . . . . . . . . . . . . . . . . . . 493
Cryoprecipitated AHF Transfusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . 500
Special Transfusion Situations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 508
Pharmacologic Alternatives to Transfusion . . . . . . . . . . . . . . . . . . . . . . . 512
Oversight of Transfusion Practice . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 514
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 515
22. Administration of Blood and Components . . . . . . . . . . . . . . . . . . . . . . 521
Pre-Issue Events . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 521
Blood Issue and Transportation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 524
Pre-Administration Events . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 525
Administration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 527
Post-Administration Events . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 531
Quality Assurance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 532
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 532
23. Perinatal Issues in Transfusion Practice . . . . . . . . . . . . . . . . . . . . . . . . 535
Hemolytic Disease of the Fetus and Newborn . . . . . . . . . . . . . . . . . . . . . 535
Neonatal Immune Thrombocytopenia . . . . . . . . . . . . . . . . . . . . . . . . . . 551
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 554
24. Neonatal and Pediatric Transfusion Practice . . . . . . . . . . . . . . . . . . . . . 557
Fetal and Neonatal Erythropoiesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . 557
Unique Aspects of Neonatal Physiology . . . . . . . . . . . . . . . . . . . . . . . . . 558
Cytomegalovirus Infection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 562
Red Cell Transfusions in Infants Less than 4 Months of Age . . . . . . . . . . . . . . 562
Transfusion of Other Components . . . . . . . . . . . . . . . . . . . . . . . . . . . . 568
Neonatal Polycythemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 572
Extracorporeal Membrane Oxygenation . . . . . . . . . . . . . . . . . . . . . . . . . 572
Leukocyte Reduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 573
Transfusion Practices in Older Infants and Children . . . . . . . . . . . . . . . . . . 574
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 577
xvi AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
25. Cell Therapy and Cellular Product Transplantation . . . . . . . . . . . . . . . . 581
Diseases Treated with Hematopoietic Cell Transplantation . . . . . . . . . . . . . . 583
Sources of Hematopoietic Progenitor Cells . . . . . . . . . . . . . . . . . . . . . . . 583
Donor Eligibility . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 589
Collection of Products. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 591
Processing of Hematopoietic Progenitor Cells. . . . . . . . . . . . . . . . . . . . . . 596
Freezing and Storage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 604
Transportation and Shipping. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 606
Thawing and Infusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 607
Evaluation and Quality Control of Hematopoietic Products. . . . . . . . . . . . . . 607
Regulations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 608
Standards . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 609
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 609
26. Tissue and Organ Transplantation . . . . . . . . . . . . . . . . . . . . . . . . . . . 617
Transplant-Transmitted Diseases and Preventive Measures. . . . . . . . . . . . . . 617
Bone Banking. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 623
Skin Banking . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 625
Heart Valves. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 625
Records of Stored Tissue Allografts . . . . . . . . . . . . . . . . . . . . . . . . . . . . 626
FDA Regulation of Tissue . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 626
The Importance of ABO Compatibility . . . . . . . . . . . . . . . . . . . . . . . . . . 627
The Role of Transfusion in Kidney Transplants . . . . . . . . . . . . . . . . . . . . . 627
Liver Transplants. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 627
Other Organ Transplants . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 629
Transfusion Service Support for Organ Transplantation . . . . . . . . . . . . . . . . 629
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 630
27. Noninfectious Complications of Blood Transfusion . . . . . . . . . . . . . . . . 633
Manifestations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 633
Acute Transfusion Reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 639
Evaluation of a Suspected Acute Transfusion Reaction. . . . . . . . . . . . . . . . . 652
Delayed Consequences of Transfusion . . . . . . . . . . . . . . . . . . . . . . . . . . 656
Records of Transfusion Complications . . . . . . . . . . . . . . . . . . . . . . . . . . 660
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 661
28. Transfusion-Transmitted Diseases . . . . . . . . . . . . . . . . . . . . . . . . . . . 667
Hepatitis. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 667
Human Immunodeficiency Viruses. . . . . . . . . . . . . . . . . . . . . . . . . . . . 675
Human T-Cell Lymphotropic Viruses . . . . . . . . . . . . . . . . . . . . . . . . . . . 682
West Nile Virus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 683
Herpesviruses and Parvovirus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 686
Transmissible Spongiform Encephalopathies . . . . . . . . . . . . . . . . . . . . . . 689
Bacterial Contamination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 690
Syphilis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 695
Tick-Borne Infections . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 695
Contents xvii
Copyright © 2005 by the AABB. All rights reserved.
Other Nonviral Infectious Complications of Blood Transfusion . . . . . . . . . . . 697
Reducing the Risk of Infectious Disease Transmission . . . . . . . . . . . . . . . . . 699
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 703
Suggested Reading. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 711
Methods
Methods Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 713
1. General Laboratory Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 715
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 715
Method 1.1. Transportation and Shipment of Dangerous Goods . . . . . . . . . . . 716
Method 1.2. Treatment of Incompletely Clotted Specimens . . . . . . . . . . . . . . . 722
Method 1.3. Solution Preparation—Instructions . . . . . . . . . . . . . . . . . . . . 723
Method 1.4. Serum Dilution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 725
Method 1.5. Dilution of % Solutions. . . . . . . . . . . . . . . . . . . . . . . . . . . . 726
Method 1.6. Preparation of a 3% Red Cell Suspension . . . . . . . . . . . . . . . . . 727
Method 1.7. Preparation and Use of Phosphate Buffer . . . . . . . . . . . . . . . . . 728
Method 1.8. Reading and Grading Tube Agglutination . . . . . . . . . . . . . . . . . 728
2. Red Cell Typing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 731
Method 2.1. Slide Test for Determination of ABO Type of Red Cells . . . . . . . . . 731
Method 2.2. Tube Tests for Determination of ABO Group of Red Cells and
Serum . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 732
Method 2.3. Microplate Test for Determination of ABO Group of Red Cells
and Serum . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 733
Method 2.4. Confirmation of Weak A or B Subgroup by Adsorption and Elution. . 735
Method 2.5. Saliva Testing for A, B, H, Lea,andLe
b. . . . . . . . . . . . . . . . . . . 736
Method 2.6. Slide Test for Determination of Rh Type . . . . . . . . . . . . . . . . . 739
Method 2.7. Tube Test for Determination of Rh Type . . . . . . . . . . . . . . . . . 740
Method 2.8. Microplate Test for Determination of Rh Type . . . . . . . . . . . . . . 741
Method 2.9. Test for Weak D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 741
Method 2.10. Preparation and Use of Lectins . . . . . . . . . . . . . . . . . . . . . . 743
Method 2.11. Use of Sulfhydryl Reagents to Disperse Autoagglutination . . . . . . 744
Method 2.12. Gentle Heat Elution for Testing Red Cells with a Positive DAT . . . . 745
Method 2.13. Dissociation of IgG by Chloroquine for Red Cell Antigen
Testing of Red Cells with a Positive DAT . . . . . . . . . . . . . . . . . . . . . . . . 746
Method 2.14. Acid Glycine/EDTA Method to Remove Antibodies from
Red Cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 747
Method 2.15. Separation of Transfused from Autologous Red Cells by
Simple Centrifugation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 748
Method 2.16. Separation of Transfused Red Cells from Autologous
Red Cells in Patients with Hemoglobin S Disease . . . . . . . . . . . . . . . . . . 749
xviii AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
3. Antibody Detection, Antibody Identification, and Serologic Compatibility
Testing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 751
Method 3.1. Immediate-Spin Compatibility Testing to Demonstrate
ABO Incompatibility . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 751
Method 3.2. Indirect Antiglobulin Test (IAT) for the Detection of Antibodies
to Red Cell Antigens . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 752
Method 3.3. Prewarming Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . 754
Method 3.4. Saline Replacement to Demonstrate Alloantibody in the
Presence of Rouleaux . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 755
Method 3.5. Enzyme Techniques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 756
Method 3.6. Direct Antiglobulin Test (DAT) . . . . . . . . . . . . . . . . . . . . . . . 760
Method 3.7. Antibody Titration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 761
Method 3.8. Use of Sulfhydryl Reagents to Distinguish IgM from IgG
Antibodies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 764
Method 3.9. Plasma Inhibition to Distinguish Anti-Ch and -Rg from
Other Antibodies with HTLA Characteristics . . . . . . . . . . . . . . . . . . . . . 765
Method 3.10. Dithiothreitol (DTT) Treatment of Red Cells . . . . . . . . . . . . . . 766
Method 3.11. Urine Neutralization of Anti-Sda. . . . . . . . . . . . . . . . . . . . . . 767
Method 3.12. Adsorption Procedure . . . . . . . . . . . . . . . . . . . . . . . . . . . 768
Method 3.13. Using the American Rare Donor Program . . . . . . . . . . . . . . . . 769
4. Investigation of a Positive Direct Antiglobulin Test . . . . . . . . . . . . . . . . . 771
Elution Techniques
Method 4.1. Cold-Acid Elution. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 772
Method 4.2. Glycine-HCl/EDTA Elution . . . . . . . . . . . . . . . . . . . . . . . . . 772
Method 4.3. Heat Elution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 773
Method 4.4. Lui Freeze-Thaw Elution. . . . . . . . . . . . . . . . . . . . . . . . . . . 774
Method 4.5. Methylene Chloride Elution. . . . . . . . . . . . . . . . . . . . . . . . . 775
Immune Hemolytic Anemia Serum/Plasma Methods
Method 4.6. Cold Autoadsorption . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 775
Method 4.7. Determining the Specificity of Cold-Reactive Autoagglutinins. . . . . 776
Method 4.8. Cold Agglutinin Titer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 778
Method 4.9. Autologous Adsorption of Warm-Reactive Autoantibodies . . . . . . . 779
Method 4.10. Differential Warm Adsorption Using Enzyme- or ZZAP-Treated
Allogeneic Red Cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 781
Method 4.11. One-Cell Sample Enzyme or ZZAP Allogeneic Adsorption . . . . . . 782
Method 4.12. Polyethylene Glycol Adsorption. . . . . . . . . . . . . . . . . . . . . . 783
Method 4.13. The Donath-Landsteiner Test . . . . . . . . . . . . . . . . . . . . . . . 784
Method 4.14. Detection of Antibodies to Penicillin or Cephalosporins by
Testing Drug-Treated Red Cells. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 786
Method 4.15. Demonstration of Immune-Complex Formation Involving Drugs. . 788
Method 4.16. Ex-Vivo Demonstration of Drug/Anti-Drug Complexes . . . . . . . . 789
Contents xix
Copyright © 2005 by the AABB. All rights reserved.
5. Hemolytic Disease of the Fetus and Newborn . . . . . . . . . . . . . . . . . . . . 793
Method 5.1. Indicator Cell Rosette Test for Fetomaternal Hemorrhage . . . . . . . 793
Method 5.2. Acid-Elution Stain (Modified Kleihauer-Betke). . . . . . . . . . . . . . 794
Method 5.3. Antibody Titration Studies to Assist in Early Detection of
Hemolytic Disease of the Fetus and Newborn . . . . . . . . . . . . . . . . . . . . 796
6. Blood Collection, Storage, and Component Preparation. . . . . . . . . . . . . . 799
Method 6.1. Copper Sulfate Method for Screening Donors for Anemia . . . . . . . 799
Method 6.2. Arm Preparation for Blood Collection . . . . . . . . . . . . . . . . . . . 800
Method 6.3. Phlebotomy and Collection of Samples for Processing and
Compatibility Tests . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 801
Method 6.4. Preparation of Red Blood Cells . . . . . . . . . . . . . . . . . . . . . . . 804
Method 6.5. Preparation of Prestorage Red Blood Cells Leukocytes
Reduced . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 805
Method 6.6. Rejuvenation of Red Blood Cells . . . . . . . . . . . . . . . . . . . . . . 806
Method 6.7. Red Cell Cryopreservation Using High-Concentration
Glycerol—Meryman Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 807
Method 6.8. Red Cell Cryopreservation Using High-Concentration
Glycerol—Valeri Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 810
Method 6.9. Checking the Adequacy of Deglycerolization of Red Blood Cells . . . 812
Method 6.10. Preparation of Fresh Frozen Plasma from Whole Blood . . . . . . . . 813
Method 6.11. Preparation of Cryoprecipitated AHF from Whole Blood . . . . . . . 814
Method 6.12. Thawing and Pooling Cryoprecipitated AHF . . . . . . . . . . . . . . 815
Method 6.13. Preparation of Platelets from Whole Blood . . . . . . . . . . . . . . . 815
Method 6.14. Preparation of Prestorage Platelets Leukocytes Reduced
from Whole Blood . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 817
Method 6.15. Removing Plasma from Platelet Concentrates (Volume
Reduction) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 817
7. Quality Control . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 819
Method 7.1. Quality Control for Copper Sulfate Solution . . . . . . . . . . . . . . . 819
Method 7.2. Standardization and Calibration of Thermometers . . . . . . . . . . . 821
Method 7.3. Testing Blood Storage Equipment Alarms . . . . . . . . . . . . . . . . 823
Method 7.4. Functional Calibration of Centrifuges for Platelet Separation . . . . . 826
Method 7.5. Functional Calibration of a Serologic Centrifuge . . . . . . . . . . . . 828
Method 7.6. Performance Testing of Automatic Cell Washers . . . . . . . . . . . . 830
Method 7.7. Monitoring Cell Counts of Apheresis Components . . . . . . . . . . . 832
Method 7.8. Manual Method for Counting Residual White Cells in
Leukocyte-Reduced Blood and Components . . . . . . . . . . . . . . . . . . . . 832
Appendices
Appendix 1. Normal Values in Adults . . . . . . . . . . . . . . . . . . . . . . . . . . . 835
Appendix 2. Selected Normal Values in Children . . . . . . . . . . . . . . . . . . . . 836
Appendix 3. Typical Normal Values in Tests of Hemostasis and Coagulation
(Adults). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 837
xx AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Appendix 4. Coagulation Factor Values in Platelet Concentrates . . . . . . . . . . . 838
Appendix 5. Approximate Normal Values for Red Cell, Plasma, and
Blood Volumes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 839
Appendix 6. Blood Group Antigens Assigned to Systems. . . . . . . . . . . . . . . . 840
Appendix 7. Examples of Gene, Antigen, and Phenotype Terms . . . . . . . . . . . 844
Appendix 8. Examples of Correct and Incorrect Terminology . . . . . . . . . . . . . 844
Appendix 9. Distribution of ABO/Rh Phenotypes by Race or Ethnicity . . . . . . . 845
Appendix 10. Suggested Quality Control Performance Intervals . . . . . . . . . . . 846
Appendix 11. Directory of Organizations . . . . . . . . . . . . . . . . . . . . . . . . . 848
Appendix 12. Resources for Safety Information . . . . . . . . . . . . . . . . . . . . . 850
Index. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 853
Contents xxi
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 1: Quality Systems
Chapter 1
Quality Systems
APRIMARYGOALOFbloodcenters
and transfusion services is to pro-
mote high standards of quality in
all aspects of production, patient care, and
service. This commitment to quality is re-
flected in standards of practice set forth by
the AABB.1(p1) A quality system includes the
organizational structure, responsibilities,
policies, processes, procedures, and re-
sources established by the executive man-
agement to achieve quality.1(p1) A glossary of
quality terms used in this chapter is in-
cluded in Appendix 1-1.
The establishment of a formal quality as-
surance program is required by regulation
under the Centers for Medicare and Medi-
caid Services (CMS)2Clinical Laboratory
Improvement Amendments (CLIA) and the
Food and Drug Administration (FDA)3-5 cur-
rent good manufacturing practice (cGMP).
The FDA regulations in 21 CFR 211.22 re-
quire an independent quality control or
quality assurance unit that has responsibil-
ity for the overall quality of the finished
product and authority to control the pro-
cesses that may affect this product.4(See
Code of Federal Regulations quality-related
citations in Appendix 1-2.) Professional and
accrediting organizations, such as the
AABB,1Joint Commission on Accreditation
of Healthcare Organizations (JCAHO),6Col-
lege of American Pathologists (CAP),7and
the Clinical and Laboratory Standards Insti-
tute (formerly NCCLS),8have also estab-
lished requirements and guidelines to ad-
dress quality issues. The International
Organization for Standardization (ISO)
quality management standards (ISO 9001)
are generic to any industry and describe
the key elements of a quality system.9In
addition, the Health Care Criteria for Per-
formance Excellence10 published by the
Baldrige National Quality Program provide
an excellent framework for implementing
quality on an organizational level. The
AABB defines the minimum elements that
1
1
Copyright © 2005 by the AABB. All rights reserved.
must be addressed in a blood bank or
transfusion service quality system in its
Quality System Essentials (QSEs).11 The
AABB QSEs were developed to be compati-
ble with ISO 9001 standards and the FDA
Guideline for Quality Assurance in Blood
Establishments.5Table 1-1 shows a compar-
ison of the AABB QSEs and ISO 9001:2000
requirements.
Quality Control, Quality
Assurance, and Quality
Management
The purpose of quality control (QC) is to
provide feedback to operational staff
about the state of a process that is in pro-
gress. It tells staff whether to continue
(everything is acceptable), or whether to
stop until a problem has been resolved
(something is found to be out of control).
Product QC is performed to determine
whether the product or service meets
specifications. Historically, blood banks
and transfusion services have employed
many QC measures as standard practice
in their operations. Examples include re-
agent QC, clerical checks, visual inspec-
tions, and measurements such as temper-
ature readings on refrigerators and volume
or cell counts performed on finished
blood components.
Quality assurance activities are not tied
to the actual performance of a process.
They include retrospective review and anal-
ysis of operational performance data to de-
termine if the overall process is in a state of
control and to detect shifts or trends that
require attention. Quality assurance pro-
vides information to process managers re-
garding levels of performance that can be
used in setting priorities for process im-
provement. Examples in blood banking in-
clude record reviews, monitoring of quality
indicators, and internal assessments.
Quality management considers interre-
lated processes in the context of the organi-
zation and its relations with customers and
suppliers. It addresses the leadership role of
executive management in creating a com-
mitment to quality throughout the organi-
zation, the understanding of suppliers and
customers as partners in quality, the man-
agement of human and other resources,
and quality planning. The quality systems
approach described in this chapter encom-
passes all of these activities. It ensures the
application of quality principles through-
out the organization and reflects the chang-
ing focus of quality efforts from detection
to prevention.
Quality Concepts
Juran’s Quality Trilogy
Juran’s Quality Trilogy is one example of a
quality management approach. This model
centers around three fundamental pro-
cesses for the management of quality in
any organization: planning, control, and
improvement.12(p2.5)
The planning process for a new product
or service includes activities to identify re-
quirements, to develop product and pro-
cess specifications to meet those require-
ments, and to design the process. During
the planning phase, the facility must per-
form the following steps:
1. Establish quality goals for the pro-
ject.
2. Identify the customers.
3. Determine customer needs and ex-
pectations.
4. Develop product and service specifi-
cations to meet customer, opera-
tional, regulatory, and accreditation
requirements.
2 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 1: Quality Systems 3
Table 1-1. Comparison of the AABB Quality System Essentials and the ISO 9001
Categories*
AABB Quality System Essentials ISO 9001:2000
Organization 4.1 General requirements
5.1 Management commitment
5.2 Customer focus
5.3 Quality policy
5.4 Planning
5.5 Responsibility, authority, and communication
5.6 Management review
Resources 6.1 Provision of resources
6.2 Human resources
Equipment 6.3 Infrastructure
7.6 Control of monitoring and measuring devices
Supplier and Customer Issues 7.2 Customer-related processes
7.4 Purchasing
Process Control 7.1 Planning of product realization
7.3 Design and development
7.5 Production and service provision
Documents and Records 4.2 Documentation requirements
Deviations, Nonconformances, and
Complications
8.3 Control of nonconforming product
Assessments: Internal and External 8.2 Monitoring and measuring
8.4 Analysis of data
Process Improvement 8.1 General
8.4 Analysis of data
8.5 Improvement
Facilities and Safety 6.3 Infrastructure
6.4 Work environment
*This table represents only one way of comparing the two systems.
Copyright © 2005 by the AABB. All rights reserved.

5. Develop operational processes for
production and delivery, including
written procedures and resources re-
quirements.
6. Develop process controls and vali-
date the process in the operational
setting.
The results of the planning process are
referred to as design output.9
Once implemented, the control process
provides a feedback loop for operations
that includes the following:
1. Evaluation of actual performance.
2. Comparison of performance to goals.
3. Action to correct any discrepancy
between the two.
It addresses control of inputs, produc-
tion, and delivery of products and services
to meet specifications. Process controls
should put operational staff in a state of
self-control such that they can recognize
when things are going wrong, and either
make appropriate adjustments to ensure
the quality of the product or stop the pro-
cess. An important goal in quality manage-
ment is to establish a set of controls that
ensure process and product quality but that
are not excessive. Controls that do not add
valueshouldbeeliminatedinordertocon
-
serve limited resources and to allow staff to
focus attention on those controls that are
critical to the operation. Statistical tools,
such as process capability measurement
and control charts, allow the facility to eval-
uate process performance during the plan-
ning stage and in operations. These tools
help determine whether a process is stable
(ie, in statistical control) and whether it is
capable of meeting product and service
specifications.12(p22.19)
Quality improvement is intended to at-
tain higher levels of performance, either by
creating new or better features that add
value, or by removing existing deficiencies
in the process, product, or service. Oppor-
tunities to improve may be related to defi-
ciencies in the initial planning process; un-
foreseen factors that are discovered upon
implementation; shifts in customer needs;
or changes in starting materials, environ-
mental factors, and other variables that af-
fect the process. Improvements must be
based on data-driven analysis; an ongoing
program of measurement and assessment
is fundamental to this process.
Process Approach
In its most generic form, a process in-
cludes all of the resources and activities
that transform an input into an output.
An understanding of how to manage and
control processes in the blood bank or
transfusion service is based on the simple
equation:
INPUT PROCESS OUTPUT
For example, a key process for donor cen-
ters is donor selection. The “input” in-
cludes 1) the individual who presents for
donation and 2) all of the resources re-
quired for the donor health screening.
Through a series of activities including
verification of eligibility (based on results
of prior donations, mini-physical, and
health history questionnaire), an individ-
ual is deemed an “eligible donor.” The
“output” is either an eligible donor who
can continue to the next process (blood
collection) or an ineligible donor who is
deferred. When the selection process re-
sults in a deferred donor, the resources
(inputs) associated with that process are
wasted and contribute to the cost of qual-
ity. One way that donor centers attempt to
minimize this cost is to educate potential
donors before the health screening so that
those who are not eligible do not enter the
selection process.
4 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Strategies for managing a process should
consider all of its components, including its
interrelated activities, inputs, outputs, and
resources. Supplier qualification, formal
agreements, supply verification, and inven-
tory control are strategies for ensuring that
the inputs to a process meet specifications.
Personnel training and competency assess-
ment, equipment maintenance and con-
trol, management of documents and re-
cords, and implementation of appropriate
in-process controls provide assurance that
the process will operate as intended. End-
product testing and inspection, customer
feedback, and outcome measurement pro-
vide information to help evaluate the qual-
ity of the product and to improve the process
as a whole. These output measurements and
quality indicators are used to evaluate the
effectiveness of the process and process
controls.
In order to manage a system of pro-
cesses effectively, the facility must under-
stand how its processes interact and any
cause-and-effect relationships between
them. In the donor selection example, the
consequences of accepting a donor who is
not eligible reach into almost every other
process in the facility. One example would
be a donor with a history of high-risk be-
havior that is not identified during the se-
lection process. The donated product may
test positive for one of the viral marker as-
says, triggering follow-up testing, look-back
investigations, and donor deferral and noti-
fication procedures. Components must be
quarantined and their discard documented.
Staff involved in collecting and processing
theproductareatriskofexposuretoinfec
-
tious agents. Part of quality planning is to
identify those relationships so that quick
and appropriate corrective action can be
taken if process controls fail. It is important
to remember that operational processes in-
clude not only product manufacture or ser-
vice creation, but also the delivery of a
product or service. Delivery generally in-
volves interaction with the customer. The
quality of this transaction is critical to
customer satisfaction and should not be
overlooked in the design and ongoing
assessment of the quality system.
Service vs Production
Quality principles apply equally to a
broad spectrum of activities, from those
involved in processing and production, to
those involving the interactions between
individuals in the delivery of a service.
However, different strategies may be ap-
propriate when there are differing expec-
tations related to customer satisfaction.
Although the emphasis in a production
process is to minimize variation in order
to create a product that consistently meets
specifications, service processes require a
certain degree of flexibility to address cus-
tomer needs and circumstances at the
time of the transaction. In production,
personnel need to know how to maintain
uniformity in the day-to-day operation. In
service, personnel need to be able to
adapt the service in a way that meets cus-
tomer expectations but does not compro-
mise quality. To do this, personnel must
have sufficient knowledge and under-
standing of interrelated processes to use
independent judgment appropriately, or
they must have ready access to higher
level decision-makers. When designing
quality systems for production processes,
it is useful to think of the process as the
driver, with people providing the over-
sight and support needed to keep it run-
ning smoothly and effectively. In service,
people are the focus; the underlying pro-
cess provides a foundation that enables
staff to deliver safe and effective services
that meet the needs of the customers in
almost any situation.
Chapter 1: Quality Systems 5
Copyright © 2005 by the AABB. All rights reserved.
Quality Management as an Evolving
Science
It is important to remember that quality
management is an evolving science. The
principles and tools in use today will
change as research provides new knowl-
edge of organizational behavior, as tech-
nology provides new solutions, and as the
field of transfusion medicine presents
new challenges. Periodic assessments of the
quality management systems will help
identify practices that are no longer effec-
tive or that could be improved through
the use of new technology or new tools.
Practical Application of
Quality Principles
The remainder of this chapter discusses
the elements of a quality system and prac-
tical application of quality principles to the
blood bank and transfusion service envi-
ronment. These basic elements include:
■Organizational management
■Human resources
■Customer and supplier relations
■Equipment management
■Process management
■Documents and records
■Deviations and nonconforming prod-
ucts and services
■Monitoring and assessment
■Process improvement
■Work environment
Organizational Management
The facility should be organized in a man-
ner that promotes effective implementa-
tion and management of its quality system.
The structure of the organization must be
documented and the responsibilities for
the provision of blood, components, pro-
ducts, and services must be clearly de-
fined. These should include a description
of the relationships and avenues of com-
munication between organizational units
and those responsible for key quality
functions. Each facility may define its
structure in any format that suits its oper-
ations. Organizational trees or charts that
show the structure and relationships are
helpful.
The facility must define in writing the
authority and responsibilities of manage-
ment to establish and maintain the quality
system. These include oversight of opera-
tions and regulatory and accreditation
compliance as well as periodic review and
assessment of quality system effectiveness.
Executive management support for quality
system goals, objectives, and policies is
critical to the success of the program. Man-
agement must participate in the review and
approval of quality and technical policies,
processes, and procedures.
The individual designated to oversee the
facility’s quality functions must report di-
rectly to management. This person has the
responsibility to coordinate, monitor, and
facilitate quality system activities and has
the authority to recommend and initiate
corrective action when appropriate.5The
designated individual need not perform all
of the quality functions personally. Ideally,
this person should be independent of the
operational functions of the donor center
or transfusion service. In small facilities,
however, this may not always be possible.
Depending on the size and scope of the or-
ganization, the designated oversight person
mayworkinadepartment(eg,transfusion
service), may have responsibilities covering
several areas (eg, laboratory-wide), may
have a staff of workers (eg, quality unit), or
may be part of an organization-wide unit
(eg, hospital quality management). Individ-
uals with dual quality and operational re-
sponsibilities should not provide quality
oversight for operational work they have
performed (21 CFR 211.194).
6 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Quality oversight functions may include
the following5:
■Review and approval of standard op-
erating procedures (SOPs) and train-
ing plans.
■Review and approval of validation
plans and results.
■Review and approval of document
control and record-keeping systems.
■Audit of operational functions.
■Development of criteria for evaluat-
ing systems.
■Review and approval of suppliers.
■Review and approval of product
specifications, ie, requirements to be
met by the products used in the man-
ufacturing, distribution, or transfusion
of blood and components.
■Review of reports of adverse reactions,
deviations in the manufacturing pro-
cess, nonconforming products and ser-
vices, and customer complaints.
■Participation in decisions to deter-
mine blood and component suitabil-
ity for use, distribution, or recall.
■Review and approval of corrective
action plans.
■Surveillance of problems (eg, error
reports, inspection deficiencies, cus-
tomer complaints) and the effective-
ness of corrective actions implemented
to solve these problems.
■Use of information resources to
identify trends and potential prob-
lems before a situation worsens and
products or patients are affected.
■Preparation of periodic (as specified
by the organization) reports of qual-
ity issues, trends, findings, and cor-
rective and preventive actions.
Quality oversight functions may be
shared among existing staff, departments,
and facilities, or, in some instances, may be
contracted to an outside firm. The goal is to
provide as much of an independent evalua-
tion of the facility’s quality activities as pos-
sible. Policies, processes, and procedures
must exist to define the roles and responsi-
bilities of all individuals in the development
and maintenance of these quality goals.
Quality system policies and processes
should be applicable across the entire facil-
ity. A blood bank or transfusion service
need not develop its own quality policies if
it is part of a larger entity whose quality
management system addresses all of the
minimum requirements. The quality sys-
tem must address all matters related to
compliance with federal, state, and local
regulations and accreditation standards ap-
plicable to the organization.
Human Resources
This element of the quality system is aim-
ed at management of personnel, includ-
ing selection, orientation, training, com-
petency assessment, and staffing.
Selection
Each blood bank, transfusion service, or
donor center must have a process to pro-
vide adequate numbers of qualified per-
sonnel to perform, verify, and manage all
activities within the facility.1(p3),3 Qualifica-
tion requirements are determined based
on job responsibilities. The selection pro-
cess should consider the applicant’s qual-
ifications for a particular position as
determined by education, training, exper-
ience, certifications, and/or licensure. For
laboratory testing staff, the standards for
personnel qualifications must be compat-
ible with the regulatory requirements es-
tablished under CLIA.2Job descriptions are
required for all personnel involved in pro-
cesses and procedures that affect the
quality of blood, components, tissues,
and services. Effective job descriptions
clearly define the qualifications, responsi-
bilities, and reporting relationships of the
position.
Chapter 1: Quality Systems 7
Copyright © 2005 by the AABB. All rights reserved.
Orientation, Training, and Competency
Assessment
Once hired, employees must be oriented
to their position and to the organization’s
policies and procedures. The orientation
program should include facility-specific
requirements and an introduction to poli-
cies that address issues such as safety,
quality, computers, security, and confi-
dentiality. The job-related portion of the
orientation program covers the opera-
tional issues specific to the work area.
Training must be provided for each proce-
dure for which employees have responsi-
bility. The ultimate result of the orienta-
tion and training program is to deem new
employees competent to work independ-
ently in performing the duties and re-
sponsibilities defined in their job descrip-
tions. Time frames should be established
to accomplish this goal. Before the intro-
duction of a new test or service, existing
personnel must be trained to perform
their newly assigned duties and must be
deemed competent. During orientation
and training, the employee should be
given the opportunity to ask questions
and seek additional help or clarification.
All aspects of the training must be docu-
mented and the facility trainer or desig-
nated facility management representative
and the employee should mutually agree
upon the determination of competence.
FDA cGMP training is required for staff
involved in the manufacture of blood and
blood components.4It should provide staff
with an understanding of the regulatory ba-
sis for the facility’s policies and procedures
as well as train them in facility-specific ap-
plication of the cGMP requirements as de-
scribedintheirownwrittenoperatingpro
-
cedures. This training must be provided at
periodic intervals to ensure that staff re-
main familiar with regulatory require-
ments.
To ensure that skills are maintained, the
facility must have regularly scheduled com-
petence evaluations of all staff whose activ-
ities affect the quality of blood, compo-
nents, tissues, or services.2,6 Depending
upon the nature of the job duties, such as-
sessments may include: written evalua-
tions; direct observation of activities; review
of work records or reports, computer re-
cords, and QC records; testing of unknown
samples; and evaluation of the employee’s
problem-solving skills.5
A formal competency plan that includes
a schedule of assessments, defined minimum
acceptable performance, and remedial
measures is one way to ensure appropriate
and consistent competence assessments.
Assessments need not be targeted at each
individual test or procedure performed by
the employee; instead, they can be grouped
together to assess like techniques or meth-
ods. Written tests can be used effectively to
evaluate problem-solving skills and to rap-
idly cover many topics by asking one or
more questions for each area to be as-
sessed. For testing personnel, CMS requires
that employees who perform testing be as-
sessed semiannually during the first year
and annually thereafter.2
The quality oversight personnel should
assist in the development, review, and ap-
proval of training programs, including the
criteria for retraining.5Quality oversight
personnel also monitor the effectiveness of
thetrainingprogramandcompetenceeval
-
uations and make recommendations for
changes as needed. In addition, JCAHO re-
quires the analysis of aggregate compe-
tency assessment data for the purpose of
identifying staff learning needs.6
Staffing
Management should have a staffing plan
that describes the number and qualifica-
tions of personnel needed to perform the
8 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
functions of the facility safely and effec-
tively. JCAHO requires that hospitals eval-
uate staffing effectiveness by looking at
human resource indicators (eg, overtime,
staff injuries, staff satisfaction) in con-
junction with operational performance
indicators (eg, adverse events, patient’s
complaints).6The results of this evalua-
tion should feed into the facility’s human
resource planning process along with
projections based on new or changing
operational needs.
Customer and Supplier Relations
Materials, supplies, and services used as
inputs to a process are considered “criti-
cal” if they affect the quality of products
and services being produced. Examples of
critical supplies are blood components,
blood bags, test kits, and reagents. Exam-
ples of critical services are infectious
disease testing, blood component irradia-
tion, transportation, equipment calibration,
and preventive maintenance services. The
suppliers of these materials and services
may be internal (eg, other departments
within the same organization) or external
(outside vendors). Supplies and services used
in the collection, testing, processing, pre-
servation, storage, distribution, transport,
and administration of blood, components,
and tissue that have the potential to affect
quality should be qualified before use and
obtained from suppliers who can meet the
facility’s requirements.1(pp8,9) The quality
system must include a process to evaluate
the suppliers’ abilities to meet these re-
quirements. Three important elements
are supplier qualification; agreements;
and receipt, inspection, and testing of in-
coming supplies.
Supplier Qualification
Critical supplies and services must be
qualified on the basis of defined require-
ments. Similarly, the supplier should be
qualified to ensure a reliable source of
materials. The facility should clearly de-
fine requirements or expectations for the
suppliers and share this information with
staff and the supplier. The ability of sup-
pliers to consistently meet specifications
for a supply or service should be evalu-
ated along with performance relative to
availability, delivery, and support. Exam-
ples of factors that could be considered to
qualify suppliers are:
■Licensure, certification, or accredita-
tion.
■Supply or product requirements.
■Review of supplier-relevant quality
documents.
■Results of audits or inspections.
■Review of quality summary reports.
■Review of customer complaints.
■Review of experience with supplier.
■Cost of materials or services.
■Delivery arrangements.
■Financial security, market position,
and customer satisfaction.
■Support after the sale.
A list of approved suppliers should be
maintained, including both primary suppli-
ers and suitable alternatives for contingency
planning. Critical supplies and services
should be purchased only from those sup-
pliers who have been qualified. Once quali-
fied, periodic evaluation of the supplier’s
performance helps to ensure its continued
ability to meet requirements. Tracking the
supplier’s ability to meet expectations gives
the facility valuable information about the
stability of the supplier’s processes and its
commitment to quality. Documented fail-
ures of supplies or suppliers to meet de-
fined requirements should result in imme-
diate action by the facility. These actions
include notifying the supplier, quality over-
sight personnel, and management with
contracting authority, if applicable. Sup-
plies may need to be replaced or quaran-
Chapter 1: Quality Systems 9
Copyright © 2005 by the AABB. All rights reserved.
tined until all quality issues have been re-
solved.
Agreements
Contracts and agreements define expec-
tations and reflect concurrence of the par-
ties involved.1(p8) Periodic review of agree-
ments ensures that expectations of all
partiescontinuetobemet.Changesmust
be mutually agreed upon and incorpo-
rated as needed.
Blood banks and transfusion services
should maintain written contracts or agree-
ments with outside suppliers of critical ma-
terials and services such as blood compo-
nents, irradiation, compatibility testing, or
infectious disease marker testing. The out-
side supplier may be another department
within the same facility that is managed in-
dependently, or it may be another facility
(eg, contract manufacturer). The contract-
ing facility assumes responsibility for the
manufacture of the product; ensuring the
safety, purity, and potency of the product;
and ensuring that the contract manufac-
turer complies with all applicable product
standards and regulations. Both the con-
tracting facility and the contractor are
legally responsible for the work performed
by the contractor.
It is important for the blood bank or
transfusion service to participate in the
evaluation and selection of suppliers. They
should review contracts and agreements to
ensure that all aspects of critical materials
and services are addressed. Examples of is-
sues that could be addressed in an agree-
ment or a contract include: responsibility
for a product or blood sample during ship-
ment; the responsibility of the supplier to
promptly notify the facility when changes
that could affect the safety of blood, com-
ponents, or patients have been made to the
materials or services; and the responsibility
of the supplier to notify the facility when
informationthataproductmaynotbe
considered safe is discovered, such as
during look-back procedures.
Receipt, Inspection, and Testing of
Incoming Supplies
Before acceptance and use, critical mate-
rials, such as reagents and blood compo-
nents, must be inspected and tested (if
necessary) to ensure that they meet speci-
fications for their intended use.1(pp8,9),4 It is
essential that supplies used in the collec-
tion, processing, preservation, testing,
storage, distribution, transport, and ad-
ministration of blood and components
also meet FDA requirements.
The facility must define acceptance cri-
teria for critical supplies (21 CFR 210.3) and
develop procedures to control materials
that do not meet specifications to prevent
their inadvertent use. Corrective action
may include returning the material to the
vendor or destroying it. Receipt and inspec-
tion records provide the facility with a
means to trace materials that have been
used in a particular process and also pro-
vide information for ongoing supplier
qualification.
Equipment Management
Equipmentthatmustoperatewithinde
-
fined specifications to ensure the quality
of blood, components, tissues, and ser-
vices is referred to as “critical” equipment
in the quality system.1(p4) Critical equip-
ment may include instruments, measur-
ing devices, and computer systems (hard-
ware and software). Activities designed to
ensure that equipment performs as in-
tended include qualification, calibration,
maintenance, and monitoring. Calibration,
functional and safety checks, and preven-
tive maintenance must be scheduled and
performed according to the manufac-
turer’s recommendations and regulatory
10 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
requirements of the FDA3and CMS.2Writ-
ten procedures for the use and control of
equipment must comply with the manu-
facturer’s recommendations unless an al-
ternative method has been validated by
the facility and approved by the appropri-
ate regulatory and accrediting agencies.
When selecting new equipment, it is im-
portant to consider not only the perfor-
mance of equipment as it will be used in
the facility, but also any supplier issues re-
garding ongoing service and support. There
should be a written plan for installation,
operational, and performance qualifica-
tion. After installation, there must be docu-
mentation of any problems and the fol-
low-up actions taken. Recalibration and
requalification may be necessary if repairs
are made that affect the critical operating
functions of the equipment. Recalibration
and requalification should also be consid-
ered when existing equipment is relocated.
The facility must develop a mechanism
to uniquely identify and track all critical
equipment, including equipment software
versions, if applicable. The unique identi-
fier may be the manufacturer’s serial num-
ber or a facility’s unique identification
number. Maintaining a list of all critical
equipment helps in the control function of
scheduling and performing functional and
safety checks, calibrations, preventive
maintenance, and repair. The equipment
listing can be used to ensure that all appro-
priate actions have been performed and re-
corded. Evaluation and analysis of equip-
ment calibration, maintenance, and repair
data will assist the facility in assessing the
suitability of the equipment. They will also
allow for better control in managing defec-
tive equipment and in identifying equip-
ment that may need replacement. When
equipment is found to be operating outside
acceptable parameters, the potential effects
on the quality of products or test results
must be evaluated and documented.
Process Management
Written, approved policies, processes, and
procedures must exist for all critical func-
tions performed in the facility and must
be carried out under controlled condi-
tions. Each facility should have a system-
atic approach for identifying, planning, and
implementing policies, processes, and pro-
cedures that affect the quality of blood,
components, tissues, and services. These
documents must be reviewed by manage-
ment personnel with direct authority over
the process and by quality oversight per-
sonnel before implementation. Changes
must be documented, validated, reviewed,
and approved. Additional information on
policies, processes, and procedures can
be found in the Documents and Records
section.
Once a process has been implemented,
the facility must have a mechanism to en-
sure that procedures are performed as
defined and that critical equipment, re-
agents, and supplies are used in confor-
mance with manufacturers’ written instruc-
tions and facility requirements. Table 1-2
lists elements that constitute sound process
control. A facility using reagents, supplies,
or critical equipment in a manner that is
different from the manufacturer’s direc-
tions must have validated such use and
may be required to request FDA approval to
operate at variance to 21 CFR 606.65(e) if
the activity is covered under regulations for
blood and blood components (21 CFR
640.120). If a facility believes that changes
to the manufacturer’s directions would be
appropriate, it should encourage the man-
ufacturer to make such changes in the la-
beling (ie, package insert or user manual).
Process Validation
Validation is used to demonstrate that a
process is capable of achieving planned
results.9It is critical to validate processes
Chapter 1: Quality Systems 11
Copyright © 2005 by the AABB. All rights reserved.
in situations where it is not feasible to
measure or inspect each finished product
or service in order to fully verify confor-
mance with specifications. However, even
when effective end-product testing can be
achieved, it is advisable to validate impor-
tant processes to generate information that
can be used to optimize performance.
Prospective validation is used for new or
revised processes. Retrospective validation
may be used for processes that are already
in operation but were not adequately vali-
dated before implementation. Concurrent
validation is used when required data
cannot be obtained without performance
of a “live” process. If concurrent valida-
tion is used, data are reviewed at prede-
fined intervals before final approval for full
implementation occurs. Modifications to
a validated process may warrant revalida-
tion, depending on the nature and extent
of the change. It is up to the facility to de-
termine the need for revalidation based
on its understanding of how the proposed
changes may affect the process.
Validation Plan
Validation must be planned if it is to be
effective. Development of a validation
plan is best accomplished after obtaining
an adequate understanding of the system,
or framework, within which the process
will occur. Many facilities develop a tem-
plate for the written validation plan to
ensure that all aspects are adequately ad-
dressed. Although no single format for a
validation plan is required, the following
elements are common to most:
■System description
■Purpose/objectives
■Risk assessment
■Responsibilities
■Validation procedures
■Acceptance criteria
■Approval signatures
■Supporting documentation
The validation plan must be reviewed
and approved by quality oversight person-
nel. Staff responsible for carrying out the
validation activities must be trained in the
process before the plan is implemented. The
12 AABB Technical Manual
Table 1-2. Elements of a Sound Process Control System
■Systematic approach to developing policies, processes, or procedures and controlling changes.
■Validation of policies, processes, and procedures.
■Development and use of standard operating procedures.
■Equipment qualification processes.
■Staff training and competence assessment.
■Acceptance testing for new or revised computer software involved in blood bank procedures.
■Establishment of quality control, calibration, and preventive maintenance schedules.
■Monitoring of quality control, calibration, preventive maintenance, and repairs.
■Monitoring and control of production processes.
■Processes to determine that supplier qualifications and product specifications are maintained.
■Participation in proficiency testing appropriate for each testing system in place.
■Processes to control nonconforming materials, blood, components, and products.
Copyright © 2005 by the AABB. All rights reserved.
results and conclusions of these activities
may be appended to the approved valida-
tion plan or recorded in a separate docu-
ment. This documentation typically contains
the following elements:
■Expected and observed results
■Interpretation of results as accept-
able or unacceptable
■Corrective action and resolution of
unexpected results
■Conclusions and limitations
■Approval signatures
■Supporting documentation
■Implementation time line
When a validation process does not pro-
duce the expected outcome, its data and
corrective actions must be documented as
well. The responsible quality oversight per-
sonnel should have final review and ap-
proval of the validation plan, results, and
corrective actions and determine whether
new or modified processes and equipment
may be implemented, or implemented with
specified limitations.
Equipment Validation
Validation of new equipment used in a
process should include installation quali-
fication, operational qualification, and
performance qualification.13
■Installation qualification demonstrates
that the instrument is properly in-
stalled in environmental conditions
that meet the manufacturer’s specifi-
cations.
■Operational qualification demon-
strates that the installed equipment
operates as intended. It focuses on
the capability of the equipment to
operate within the established limits
and specifications supplied by the
manufacturer.
■Performance qualification demon-
strates that the equipment performs
as expected for its intended use in
the processes established by the
facility and that the output meets
specifications. It evaluates the ade-
quacy of equipment for use in a spe-
cific process that employs the
facility’s own personnel, procedures,
and supplies in a normal working
environment.
Computer System Validation
The FDA considers computerized systems
to include: “hardware, software, periph-
eral devices, personnel, and documenta-
tion.”14 End-user validation of computer
systems and the interfaces between
systems should be conducted in the envi-
ronment where it will be used. Testing
performed by the vendor or supplier of
computer software is not a substitute for
computer validation at the facility. End-
user acceptance testing may repeat some
of the validation performed by the devel-
oper, such as load or stress testing and
verification of security, safety, and control
features, in order to evaluate performance
under actual operating conditions. In ad-
dition, the end user must evaluate the
ability of personnel to use the computer
system as intended within the context of
actual work processes. Staff must be able
to successfully navigate the hardware and
software interface and respond appropri-
ately to messages, warnings, and other
functions. Depending upon the nature of
the computer functionality, changes to the
computer system may result in changes to
how a process is performed. If this occurs,
process revalidation must also be per-
formed. As with process validation, qual-
ity oversight personnel should review and
approve validation plans, results, and cor-
rective actions and determine whether
implementation may proceed with or
without limitations. Facilities that de-
velop their own software should refer to
Chapter 1: Quality Systems 13
Copyright © 2005 by the AABB. All rights reserved.
FDA guidance regarding general princi-
ples of software validation for additional
information.15
Quality Control
QC testing is performed to ensure the
proper functioning of materials, equip-
ment, and methods during operations.
QC performance expectations and ac-
ceptable ranges must be defined and
readily available to staff so that they will
recognize unacceptable results and trends
and respond appropriately. The frequency
for QC testing is determined by the facil-
ity in accordance with the applicable
CMS, FDA, AABB, state, and manufac-
turer’s requirements. QC results must be
documented concurrently with perfor-
mance.3Records of QC testing must include
identification of personnel, identification
of reagent (including lot number, expira-
tion dates, etc), identification of equip-
ment, testing date and time (when appli-
cable), results, interpretation, and reviews.
Unacceptable QC results must be investi-
gated and corrective action implemented,
if indicated, before repeating the QC pro-
cedure or continuing the operational pro-
cess. Specific examples of suggested qual-
ity control intervals for blood banks and
transfusion services are included in Ap-
pendix 10 at the end of the book, and in-
formation regarding methods of quality
control are found in the methods section
devoted to QC.
Documents and Records
Documentation provides a framework for
understanding and communication through-
out the organization. Documents describe
the way that processes are intended to
work, how they interact, where they must
be controlled, what their requirements
are, and how to implement them. Records
provide evidence that the process was
performedasintendedandinformation
needed to assess the quality of products
and services. Together, documents and
records are used by quality oversight per-
sonnel to evaluate the effectiveness of a fa-
cility’s policies, processes, and procedures.
An example of quality system documenta-
tion is provided in ISO 9001 and includes
the following items9:
1. The quality policy and objectives.
2. A description of the interactions be-
tween processes.
3. Documented procedures for the control
of documents, control of records,
control of a nonconforming product,
corrective action, preventive action,
and internal quality audits.
4. Records related to the quality sys-
tem, operational performance, and
product/service conformance.
5. All other documents needed by the
organization to ensure the effective
planning, operation, and control of
its processes.
Written policies, process descriptions, pro-
cedures, work instructions, labels, forms,
and records are all part of the facility’s doc-
umentation system. They may be pa-
per-based or electronic. Documents pro-
vide a description or instructions of what is
supposed to happen; records provide evi-
dence of what did happen. A document
management system provides assurance
that documents are comprehensive, cur-
rent, and available, and that records are
accurate and complete. A well-structured
document management system links poli-
cies, process descriptions, procedures, forms,
and records together in an organized and
workable system.
Documents
Documents should be developed in a for-
mat that conveys information clearly and
provides staff with instructions and forms.
14 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
The Clinical and Laboratory Standards In-
stitute offers guidance regarding general
levels of documentation8as well as de-
tailed instructions on how to write proce-
dures.16 General types of documentation
are described below.
Policies. Policies communicate the high-
est level goals, objectives, and intent of the
organization. The rest of the organization’s
documentation will interpret and provide
instruction regarding implementation of
these policies.
Processes. Process documents describe
a sequence of actions and identify respon-
sibilities, decision points, requirements,
and acceptance criteria. Table 1-3 lists ex-
amples of process documents that might be
in place to support a quality system. Pro-
cessdiagramsorflowchartsareoftenused
for this level of documentation. It is helpful
to show process control points on the dia-
gram as well as flow of information and hand-
offs between departments or work groups.
Procedures and Work Instructions.
These documents provide step-by-step di-
rections on how to perform job tasks and
procedures. Procedures and work instruc-
tions should include enough detail to per-
form the task correctly, but not so much as
to make them difficult to read. The use of
standardized formats will help staff know
where to find specific elements and facili-
tates implementation and control.1(p66) Proce-
dures may also be incorporated by refer-
ence, such as those from a manufacturer’s
manual. Relevant procedures must be
available to staff in each area where the cor-
responding job tasks are performed.1(p66),3
Forms. Forms provide a template for
capturing data either on paper or electroni-
cally. These documents specify the data re-
quirements called for in SOPs and pro-
cesses. Forms should be carefully designed
for ease of use, to minimize the likelihood
of errors, and to facilitate retrieval of infor-
mation. They should include instructions
for use when it is not immediately evident
what information should be recorded or
how to record it. For quantitative data, the
form should indicate units of measure.
Computer data entry and review screens
are a type of form. Forms must be designed
to effectively capture outcomes and sup-
port process traceability.
Labels. Blood component labels are a
critical material subject to the requirements
of a document management system. Many
facilities maintain a master set of labels that
can be used as reference to verify that only
current approved stock is in use. New label
stock must be verified as accurate before it
is put into inventory; comparison against a
master label provides a mechanism for ac-
complishing this. Change control procedures
must be established for the use of on-demand
label printers to prevent nonconforming
modification of label format or content.
Each facility must have a defined process
for developing and maintaining docu-
ments. It should include: basic elements re-
quired for document formats; procedures
for review and approval of new or revised
documents; a method for keeping docu-
ments current; control of document distri-
bution; and a process for archiving, pro-
tecting, and retrieving obsolete documents.
Training must be provided to staff responsi-
ble for the content of new or revised docu-
ments. Document management systems in-
clude established processes to:
1. Verify the adequacy of the document
before approval and issue.
2. Periodically review, modify, and re-
approve as needed to keep documents
current.
3. Identify changes and revision status.
4. Ensure that documents are legible,
identifiable, and readily available.
5. Prevent unintended use of outdated
or obsolete documents.
6. Protect documents from unintended
damage or destruction.
Chapter 1: Quality Systems 15
Copyright © 2005 by the AABB. All rights reserved.

16 AABB Technical Manual
Table 1-3. Examples of Quality System Process Documents
Organization ■Management review process
Resources ■Personnel hiring process
■Training process
■Competence assessment process
Equipment ■Equipment management process
■Installation qualification process
Supplier and Customer Issues ■Supplier qualification process
■Contract review process
■Process for qualification of critical materials
■Ordering and inventory control of critical materials
■Receipt, inspection, and testing of incoming critical
materials
Process Control ■Change control process
■Validation process
■Process for acceptance testing of computer software
■Process for handling proficiency testing
■Process for handling, storage, distribution, and transport
of blood components
Documents and Records ■Process for creation and approval of documents
■Document management process
■Records management process
Deviations, Nonconformances, and
Complications
■Event management process
■Process for handling customer complaints
■Process for notification of external sources
Assessments ■Internal audit process
■Process for quality monitoring
■Process for handling external assessments
Process Improvement ■Corrective and preventive action processes
Facilities and Safety ■Process for handling disasters
■Employee safety management process
Copyright © 2005 by the AABB. All rights reserved.
External documents that are incorpo-
rated by reference become part of the doc-
ument management system and must be
identified and controlled. The facility must
have a mechanism to detect changes to ex-
ternal documents in its system, such as a
manufacturer’s package inserts or user
manuals, so that corresponding changes to
procedures and forms can be made. When
new or revised policies, process descrip-
tions, procedures, or forms are added to or
replaced in the facility’s manual, the docu-
mentsmustbemarkedwiththeeffective
date. One copy of retired documents must
be retained as defined by existing and ap-
plicable standards and regulations.
A master list of all current policies, pro-
cess descriptions, procedures, forms, and
labels is useful for maintaining document
control. It should include the document ti-
tle, the individual or work group responsible
for maintaining it, the revision date and
number (if one is assigned), and the area
where it is used. It should also identify the
number and location of controlled copies in
circulation. Copies of documents that will be
used in the workplace should be identified
and controlled to ensure that none are
overlooked when changes are implemented.
Records
Records provide evidence that critical
steps in a procedure have been performed
appropriately and that products and ser-
vices conform to specified requirements.
Review of records is an important tool to
help evaluate the effectiveness of the qua-
lity system. Records must be created con-
currently with the performance of each
significant step and clearly indicate the
identity of individuals who performed
each step and when it occurred.3The
quality system must include a process for
managing records that addresses the fol-
lowing items:
■Creation and identification of records
■Protection from accidental or unau-
thorized modification or destruction
■Verification of completeness, accu-
racy, and legibility
■Storage and retrieval
■Creation of copies or backups
■Retention periods
■Confidentiality
Record-keeping systems must allow for
ready retrieval within time frames estab-
lished by the facility and must permit trace-
ability of blood components as required by
federal regulations.3Specific requirements
for records to be maintained by blood
banks and transfusion services are included
in the AABB Standards for Blood Banks and
Transfusion Services1(pp69-80) and in 21 CFR
606.160.
When forms are used for capturing data
or recording steps or test results, the forms
become records. Data must be recorded in
aformatthatisclearandconsistent.The
facility must define a process and time
frames for the record review to ensure ac-
curacy, completeness, and appropriate fol-
low-up. It must determine how reports and
records are to be archived and define their
retention period. When copies of records
are retained, the facility must verify that the
copy contains complete, legible, and acces-
sible content of the original record before
the original is destroyed.
If records are maintained electronically,
adequate backup must exist in case of sys-
tem failure. Electronic records must be
readable for the entire length of their reten-
tion period. Obsolete computer software,
necessary to reconstruct or trace records,
must be archived appropriately. If the
equipment or software used to access ar-
chived data cannot be maintained, the re-
cords should be converted to another for-
mat or copied to another medium to
permit continued access. Converted data
must be verified against the original to en-
Chapter 1: Quality Systems 17
Copyright © 2005 by the AABB. All rights reserved.
sure completeness and accuracy. Electronic
media such as magnetic tapes, optical
disks, and online computer data storage are
widely used for archiving documents. Re-
cords kept in this manner must meet FDA
requirements for electronic record-keep
-
ing.17 Microfilm or microfiche may be used
to archive written records. The medium se-
lected should be appropriate for the reten-
tion requirements.
Privacy of patient and donor informa-
tion must be addressed in the quality
system with established policies and proce-
dures to maintain the security and confi-
dentiality of records. Computer systems must
be designed with security features to pre-
vent unauthorized access and use. This sys-
tem may include levels of security defined
by job responsibility and administered by
the use of security codes and passwords.
Each facility should have a policy for al-
tering or correcting records. A common
practice is to indicate the date, the change,
the identity of the person making the
change, and evidence of review by a re-
sponsible person. The original recording
must not be obliterated in written records;
it may be crossed out with a single line, but
it should remain legible. Electronic records
must permit tracking of both original and
corrected data and include the date and
user identification of the person making
the change. There should be a process for
controlling changes.1(p10) Amethodforrefer
-
encing changes to records, linked to the
original records, and a system for reviewing
changes for completeness and accuracy are
essential. Audit trails for changed data in
computerized systems are required by the
FDA.17
Thefollowingareissuesthatmightbe
considered when planning record storage:
■Storage of records in a manner that
protectsthemfromdamageandfrom
accidental or unauthorized destruc-
tion or modification.
■Degree of accessibility of records in
proportion to frequency of their use.
■Method and location of record stor-
age related to the volume of records
and the amount of available storage
space.
■Availability of properly functioning
equipment, computer hardware, and
software to view archived records.
■Documentation that microfiched re-
cords legitimately replace original
documents that may be stored else-
where or destroyed.
■Retention of original color-coded re-
cords when only black-and-white re-
productions are available.
Considerations for electronic records in-
clude:
■A method of verifying the accuracy
of data entry.
■Prevention of unintended deletion of
data or access by unauthorized per-
sons.
■Adequate protection against inad-
vertent data loss (eg, when a storage
device is full).
■Validated safeguards to ensure that a
record can be edited by only one
person at a time.
■Security and access of confidential
data.
Abackupdiskortapeshouldbemain
-
tained in the event of unexpected loss of
information from the storage medium.
Backup or archived computer records and
databases must be stored off-site.1(p68) The
storage facility should be secure and main-
tain appropriate conditions, in accordance
with the manufacturer’s recommendations
and instructions. An archival copy of the
computer operating system and applica-
tions software should be stored in the same
manner.
The facility should develop and maintain
alternative systems to ensure information
access if computerized data are not avail-
18 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
able. The backup and recovery procedures
forcomputerdowntimemustbedefined,
with validation documentation to show
that the backup system works properly. The
associated processes must be checked peri-
odically to ensure that the backup system
remains effective. Special consideration
should be given to staff competence and
readiness to use the backup system.
To link relevant personnel to recorded
data, the facility must maintain a record of
names, inclusive dates of employment, sig-
natures, and identifying initials or identifi-
cation codes of personnel authorized to
create, sign, initial, or review reports and
records. Magnetically coded employee
badges and other computer-related identi-
fying methods are generally accepted in lieu
of written signatures provided they meet elec-
tronic record-keeping requirements.
Deviations and Nonconforming Products
or Services
The quality system must include a process
for detecting, investigating, and responding
to events that result in deviations from ac-
cepted policies, processes, and procedures
or in failures to meet requirements, as de-
fined by the donor center or transfusion
service, AABB standards, or applicable re-
gulations.1(p81),3 This includes the discovery
of nonconforming products and services
as well as adverse reactions to blood do-
nation and transfusion.1(pp81-85),2 The facility
should define how to:
■Document and classify occurrences.
■Determine the effect, if any, on the
quality of products or services.
■Evaluate the impact on interrelated
activities.
■Implement corrective action, includ-
ing notification and recall, as appro-
priate.
■Analyze the event to understand root
causes.
■Implement preventive actions as ap-
propriate on the basis of root-cause
analysis.
■Report to external agencies, when
required.
Facility personnel should be trained to
recognize and report such occurrences. De-
pending upon the severity of the event and
risk to patients, donors, and products, as
well as the likelihood of recurrence, investi-
gation into contributing factors and under-
lying cause(s) may be warranted. The
cGMP regulations require an investigation
and documentation of the results if a spe-
cific event could adversely affect patient
safety or the safety, purity, potency, or effi-
cacy of blood or components.2,3 Tools and
approaches for performing root-cause
analysis and implementing corrective ac-
tion are discussed in the section addressing
process improvement. A summary of the
event, investigation, and any follow-up
must be documented. Table 1-4 outlines
suggested components of an internal event
report.
Fatalities related to blood collection or
transfusion must be reported as soon as
possible to the FDA Center for Biologics
Evaluation and Research (CBER) [21 CFR
606.170(b)]. Instructions for reporting to
CBER are available in published guidance20
and at http://www.fda.gov/cber/transfu-
sion.htm. A written follow-up report is sub-
mitted within 7 days of the fatality and
should include a description of any new pro-
cedures implemented to avoid recurrence.
AABB Association Bulletin #04-06 provides
additional information, including a form to
be used for reporting donor fatalities.21
Regardless of their licensure and regis-
tration status with the FDA, all donor cen-
ters, blood banks, and transfusion services
must promptly report biologic product de-
viations (previously known as errors and
Chapter 1: Quality Systems 19
Copyright © 2005 by the AABB. All rights reserved.

20 AABB Technical Manual
Table 1-4. Components of an Internal Event Report18,19
WHO ■Identity of reporting individual(s)
■Identity of individuals involved (by job title) in committing, compounding,
discovering, investigating, and initiating any immediate action
■Patient or donor identification
■Reviewer(s) of report
WHAT ■Brief description of event
■Effects on and outcome to patient, donor, or blood component
■Name of component and unit identification number
■Manufacturer, lot number, and expiration date of applicable reagents and
supplies
■Immediate action taken
WHEN ■Date of report
■Date and time of event occurrence
■Date and time of discovery
■Collection and shipping dates of blood component(s)
WHERE ■Physical location of event
■Where in process detected
■Where in process initiated
WHY/HOW ■Explanation of how event occurred
■Contributing factors
■Root cause(s)
FOLLOW-UP ■External reports or notifications (eg, FDA*, manufacturer, or patient’s
physician)
■Corrective actions
■Implementation dates
■Effectiveness of actions taken
*The following are
some
examples identified by the FDA as reportable events if components or products
are released for
distribution
:
–Arm preparation not performed or done incorrectly
–Units from donors who are (or should have been) either temporarily or permanently deferred because of their medical
history or a history of repeatedly reactive viral marker tests
–Shipment of unit with repeatedly reactive viral markers
–ABO/Rh or infectious disease testing not done in accordance with the manufacturer’s package insert
–Units from donors for whom test results were improperly interpreted because of testing errors related to the improper
use of equipment
–Units released before completion of all tests (except as emergency release)
–Sample used for compatibility testing that contains the incorrect identification
–Testing error that results in the release of an incorrect unit
–Incorrectly labeled blood components (eg, ABO, expiration date)
–Incorrect crossmatch label or tag
–Storage of biological products at the incorrect temperature
–Microbial contamination of blood components when the contamination is attributed to an error in manufacturing
Copyright © 2005 by the AABB. All rights reserved.
accidents) and information relevant to
these events to the FDA3,22 using Form
FDA-3486 when the event:
■Is associated with manufacturing (ie,
testing, processing, packing, labeling,
storing, holding, or distributing).
■Represents a deviation from current
good manufacturing practice, appli-
cable regulations or standards, or
established specifications, or is un-
expected or unforeseen.
■May affect the safety, purity, or po-
tency of the product.
■Occurs while the facility had control
of or was responsible for the product.
■Involves a product that has left the
control of the facility (ie, distributed).
There must also be a mechanism to re-
port medical device adverse events to the
FDA.23 The JCAHO encourages reporting of
sentinel events, including hemolytic trans-
fusion reactions involving the administra-
tion of blood or components having major
blood group incompatibilities.6
Each facility should track reported events
and look for trends. The use of classifica-
tion schemes may facilitate trend analysis
and typically involves one or more of the
following categories: the nature of the
event, the process (or procedure) in which
the event occurred, event severity, and
causes. If several events within a relatively
short period involve a particular process or
procedure, that process or procedure should
be further investigated. The most useful
schemes involve use of multiple categories
for each event, which allow data to be
sorted in a variety of ways so that patterns
canemerge(seeexampleinTable1-5).
Such sorting can result in identification of
situations that require closer monitoring or
of problems needing corrective action. The
extent of monitoring and length of time to
monitor processes will depend on the fre-
quency of the occurrence and the critical
aspects of the occurrences. Reporting and
monitoring of events are essential problem
identification methods for process im-
provement activities in a quality manage-
ment system.
Occasionally, the blood bank or transfu-
sion service may need to deviate from ap-
proved procedures in order to meet the
unique medical needs of a particular pa-
tient. When this situation arises, a medi-
cally indicated exception is planned and
approved in advance by the facility’s medi-
cal director. The rationale and nature of the
planned exception must be documented.
Careful consideration should be given to
maintaining a controlled process and to
verifying the safety and quality of the re-
sulting product or service. Any additional
risk to the patient must be disclosed.
Chapter 1: Quality Systems 21
Table 1-5. Example of Event Classification
Event: A unit of Red Blood Cells from a directed donor was issued to an incorrect patient.
■Classification of event
Type of event – patient
Procedure involved – issuing products
Process involved – blood administration
Product involved – Red Blood Cells
Other factors – directed donor
■Investigation revealed
Proximate cause – two patients with similar names had crossmatched blood available
Root cause – inadequate procedure for verification of patient identification during issue
Copyright © 2005 by the AABB. All rights reserved.
Monitoring and Assessment
The quality system should describe how
the facility monitors and evaluates its
processes. The AABB Standards1(p93) de-
fines assessment as a systematic, inde-
pendent examination that is performed at
defined intervals and at sufficient fre-
quency to determine whether actual ac-
tivities comply with planned activities,
are implemented effectively, and achieve
objectives. Evaluations typically include
comparison of actual results to expected
results. Depending on the focus, this can
include evaluation of process outputs (eg,
results), the activities that make up a pro-
cessaswellasitsoutputs,oragroupof
related processes and outputs (ie, the
system). Types of assessments include ex-
ternal assessments, internal assessments,
quality assessments, peer review, and self-
assessments.
Internal Assessments
Internal assessments may include evalua-
tion of quality indicator data, targeted au-
dits of a single process, or system audits
that are broader in scope and cover a set
of interrelated processes. These assess-
ments should be planned and scheduled.
The details of who performs the assess-
ments and how they are performed should
be addressed. Assessments should cover
the quality system and major operating
systems found in the blood bank, transfu-
sion service, or donor center.
In addition, there must be a process for
responding to the issues raised as a result of
the assessment, including review processes
andtimeframes.Theresultsshouldbe
documented and submitted to manage-
ment personnel with authority over the
process assessed as well as to executive
management. Management should develop
corrective and preventive action plans with
input from operational staff and quality
oversight personnel for any deficiencies
noted in the assessment. Quality oversight
personnel should track progress toward im-
plementation of corrective and preventive
actions and monitor them for effectiveness.
In order to make the best use of these as-
sessments, there must be a process to track,
trend, and analyze the problems identified
so that opportunities for improvement can
be recognized.1(pp86,87) Early detection of trends
makes it possible to develop preventive ac-
tions before patient safety or blood compo-
nents are adversely affected. Evaluation
summaries provide information useful in
correcting individual or group performance
problems and ensuring adequacy of test
methods and equipment. In addition to re-
view of assessment results, executive man-
agement must review any associated cor-
rective or preventive action.
Quality Indicators
Quality indicators are specific perfor-
mance measurements designed to moni-
tor one or more processes during a defined
time and are useful for evaluating service
demands, production, adequacy of per-
sonnel, inventory control, and process
stability. These indicators can be pro-
cess-based or outcome-based. Process-
based indicators measure the degree to
which a process can be consistently per-
formed. An example of a process-based
indicator is measurement of turnaround
time from blood component ordering un-
til transfusion. Outcome-based indicators
are often used to measure what does or
does not happen after a process is or is
not performed. Counting incorrect test
result reports is an example of such an in-
dicator. For each indicator, thresholds are
set that represent warning limits and/or
action limits. These thresholds can be de-
termined from regulatory or accreditation
22 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
requirements, benchmarking, or inter-
nally derived data.
Tools frequently used for displaying
quality indicator data are run charts and
control charts. In a run chart, time is plot-
ted on the x-axis and values on the y-axis.
In control charts, the mean of the data and
upper and lower control limits, which have
been calculated from the data, are added to
thechart.Singlepointsoutsidetheupper
and lower control limits result from special
causes. Statistical rules for interpreting con-
secutive points outside 1 standard devia-
tion (SD), 2 SD, and 3 SD should be used to
recognize a process that is out of control;
therootcauseshouldbedeterminedand
corrective action should be initiated if indi-
cated.
Blood Utilization Assessment
The activities of blood usage review com-
mittees in the transfusion setting are an
example of internal assessment. Guide-
lines are available from the AABB for both
adult and pediatric utilization review.24-26
Peer review of transfusion practices, re-
quired by the AABB, is also required by
the JCAHO6for hospital accreditation, by
the CMS2for hospitals to qualify for
Medicare reimbursement, and by some
states for Medicaid reimbursement.
Transfusion audits provide a review of
policies and practices to ensure safe and
appropriate transfusions and are based on
measurable, predetermined performance
criteria. Transfusion services should inves-
tigate an adequate sampling of cases (eg,
5% of the number of cases occurring within
a defined time frame or 30 cases, whichever
is larger). Audits assess the facility’s perfor-
mance and effectiveness in:
■Blood ordering practices for all cate-
gories of blood and components.
■Minimizing wastage of blood com-
ponents.
■Distribution, handling, use, and ad-
ministration of blood components.
■Evaluating all confirmed transfusion
reactions.
■Meeting patients’ transfusion needs.
■Informing patients and physicians
in a timely and confidential manner
of possible infectious disease trans-
mission.
One method of assessing the blood ad-
ministration process is to observe a prede-
termined number of transfusions by follow-
ing the unit of blood as it is issued for
transfusion and as it is transfused.25
Assessments of transfusion safety policy
and practice may include a review of trans-
fusion reactions and transfusion-transmit-
ted diseases. The review committee may
monitor policies and practices for notifying
recipients of recalled products (look-back
notification) and donors of abnormal test
results. Other assessments important in
transfusion practice include the review of
policies for informed consent, indication
for transfusion, release of directed donor
units, and outpatient or home transfusion.
Additional assessments should include,
where appropriate: therapeutic apheresis,
use of cell-saver devices, procurement and
storage of hematopoietic progenitor cells,
perioperative autologous blood collection,
procurement and storage of tissue, and
evaluation of evolving technologies and
products. Appendix 1-4 lists blood utiliza-
tion assessment examples.
External Assessments
External assessments include inspections,
surveys, audits, and assessments per-
formed by those not affiliated with the or-
ganization, such as the FDA, AABB, CAP,
or JCAHO. Participation in an external
assessment program provides an inde-
pendent, objective view of the facility’s
performance. External assessors often bring
Chapter 1: Quality Systems 23
Copyright © 2005 by the AABB. All rights reserved.
broad-based experience and knowledge
of best practices that can be shared. In the
preparation phase of scheduled assess-
ments, there is typically some data gath-
ering and information to submit to the or-
ganization performing the assessment.
Coordinated scheduling and planning will
help ensure that adequate time is allotted
foreachareatobecoveredandthatade
-
quatestaffareavailabletoanswerques
-
tions and assist in the assessment activi-
ties. During the assessment phase, it is
important to know who is responsible for
the assessors or inspectors during the
time they are in the facility. Clear descrip-
tions of what information can be given to
these individuals, and in what form, will
help the facility through the assessment
or inspection process. After the assess-
ment, identified issues must be addressed.
Usually a written response is submitted.
Proficiency Testing for Laboratories
Proficiency testing (PT) is one means for
determining that test systems (including
methods, supplies, and equipment) are
performing as expected. As a condition
for certification, the CMS requires labora-
tories to participate successfully in an ap-
proved PT program for each specialty and
analyte that they routinely test. When no
approved PT program exists for a particu-
lar analyte, the laboratory must have an-
other means to verify the accuracy of the
test procedure at least twice annually.2
Proficiency testing must be performed us-
ing routine work processes and conditions
if it is to provide meaningful information.
Handling and testing of PT samples should
be the same as those for patient or donor
specimens. Supervisory review of the sum-
mary evaluation report must be documented
along with investigation and corrective
action for results that are unacceptable.
Quality oversight personnel should moni-
tor the proficiency testing program and
verify that test systems are maintained in
a state of control and that appropriate cor-
rective action is taken when indicated.
Process Improvement
Continuous improvement is a fundamen-
tal goal in any quality management sys-
tem. In transfusion medicine, this goal is
tied to patient safety goals and expecta-
tions for the highest quality health care.
The importance of identifying, investigat-
ing, correcting, and preventing problems
cannot be overstated. The process of de-
veloping corrective and preventive action
plans includes identification of problems
and their causes, and identification and
evaluation of solutions to prevent future
problems. It must include a mechanism
for data collection and analysis, as well as
follow-up to evaluate the effectiveness of
the actions taken. Statistical tools and
their applications may be found in publi-
cations from the AABB and the American
Society for Quality.27,28 The JCAHO stan-
dards for performance improvement are
outlined in Table 1-6.6
Corrective action is defined as the action
taken to eliminate the causes of an existing
nonconformance or other undesirable situ-
ation in order to prevent recurrence.1(p94)
Preventive action is defined as the action
taken to eliminate the causes of a potential
nonconformance or other undesirable situ-
ation in order to prevent occurrence.1(p97)
Corrective action can be thought of as a re-
active approach to reported problems that
includes a preventive component, whereas
preventive action can be thought of as a
proactive approach resulting from the anal-
ysis of data and information. In contrast,
remedial action is defined as the action
taken to alleviate the symptoms of existing
nonconformances or any other undesirable
24 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

situation.27,28 Remedial action addresses
only the visible indicator of a problem, not
the actual cause (see comparisons in Table
1-7). Effective corrective and preventive ac-
tions cannot be implemented until the un-
derlying cause is determined and the pro-
cess is evaluated in relationship to other
processes. Pending such evaluation, it may
be desirable to implement interim remedial
action.
Identification of Problems and Their Causes
Sources of information for process im-
provement activities include the follow-
ing: blood product and other deviations;
nonconforming products and services;
customer complaints; QC records; profi-
ciency testing; internal audits; quality in-
dicators; and external assessments. Active
monitoring programs may be set up to
help identify problem areas. These pro-
grams should be representative of the
facility processes, consistent with organi-
zational goals, and reflect customer needs.
Preparation of an annual facility quality
report, in which data from all these sources
are collated and analyzed, can be a valu-
able tool to identify issues for performance
improvement.
Once identified, problems must be ana-
lyzed to determine their scope, potential ef-
fects on the quality and operational sys-
tems, relative frequency, and the extent of
variation. This analysis is important to
avoid tampering with processes that are
showing normal variation or problems with
little impact.
Chapter 1: Quality Systems 25
Table 1-6. Applicable JCAHO Performance Improvement Standards6
■Data are collected to measure the performance of potentially high-risk processes, including
blood utilization.
■Performance data are systematically aggregated and analyzed to determine current performance
levels, patterns, and trends over time.
■Undesirable patterns and trends in performance are evaluated. All confirmed transfusion
reactions are analyzed.
■There is a defined process for identification and management of serious adverse events.
Root-cause analysis and corrective action are documented.
■Information from data analysis is used to improve performance and patient safety and minimize
the risk of serious adverse events.
■The facility defines and implements a program to proactively identify opportunities for
improvement. Preventive actions are implemented and monitored for effectiveness.
Table 1-7. Comparison of Remedial, Corrective, and Preventive Action29
Action Problem Approach Outcome
Remedial Existent Reactive Alleviates symptoms
Corrective Existent Reactive Prevents recurrence
Preventive Nonexistent Proactive Prevents occurrence
Copyright © 2005 by the AABB. All rights reserved.
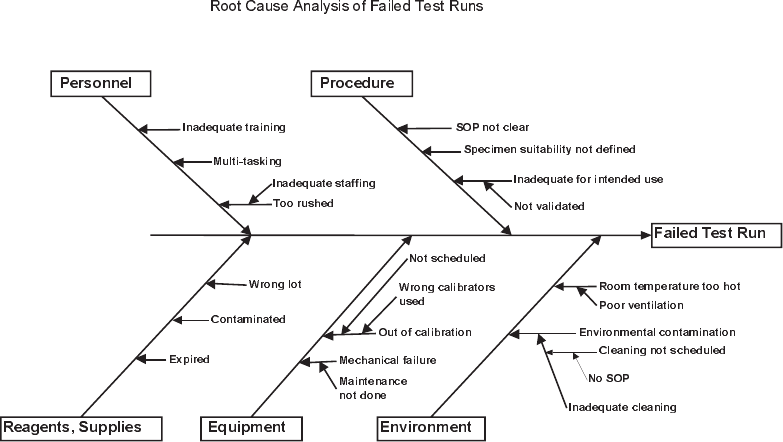
Identifying underlying causes for an un-
desirable condition or problem can be ac-
complished by an individual or a group.
Themorecomplextheproblemandthe
more involved the process, the greater the
need to enlist a team of individuals and to
formalize the analysis. The three most com-
monly used tools for identifying underlying
causes in an objective manner are process
flowcharting, use of the “repetitive why,”
and the cause-and-effect diagram. A pro-
cess flowchart gives a detailed picture of the
multiple activities and important decision
points within the process. By examining
this picture, problem-prone areas may be
identified. The “repetitive why” is used to
work backward through the process. One
repeatedly asks the question “why did this
happen?” until: 1) no new information can
be gleaned, 2) the causal path cannot be
followed because of missing information,
or 3) further investigation is impractical,
impossible, or outside the boundaries of
the organization. Use of the “repetitive
why” prevents the mistake of interpreting
an effect as a cause.
The cause-and-effect diagram, also known
as the Ishikawa or fish-bone diagram, em-
ploys a specialized form of brainstorming
that breaks down problems into “bite-size”
pieces. An example of a cause-and-effect
diagramisshowninFig1-1.Itisamethod
designed to focus ideas around the compo-
nent parts of a process, as well as give a pic-
torial representation of the ideas that are
generated and their interactions. When us-
ing the cause-and-effect diagram, one looks
at equipment, materials, methods, environ-
ment, and human factors. These tools iden-
tify both active and latent failures. Active
failures are those that have an immediate
adverse effect. Latent failures are those
more global actions and decisions with po-
tential for damage that may lie dormant
26 AABB Technical Manual
Figure 1-1. Example of a cause-and-effect diagram (SOP = standard operating procedure).
Copyright © 2005 by the AABB. All rights reserved.
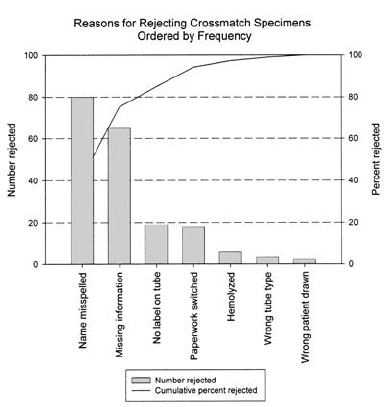
and become evident only when triggered
bythepresenceoflocalizedfactors.Thekey
to successfully determining root cause is
not to stop too soon or get caught in the
trap of placing blame on an individual.
Most problems, particularly those that
are complex, have several root causes. A
method that can be of use when this occurs
is the Pareto analysis. A chart of causes, laid
out in order of decreasing frequency, is pre-
pared. Those that occur most frequently are
considered the “vital few”; the rest are con-
sidered the “trivial many.” This method of-
fers direction about where to dedicate re-
sources for maximal impact. An example of
aParetochartisshowninFig1-2.
Identification and Evaluation of Solutions
Potential solutions to problems are iden-
tified during the creative phase of process
improvement. Brainstorming and process
flowcharting can be particularly helpful in
this phase. Possible solutions should be
evaluated relative to organizational con-
straints and narrowed down to those most
reasonable. Individuals who perform the
process are usually the most knowledge-
able about what will work. They should be
included when possible solutions are be-
ing considered. Individuals with knowl-
edge of the interrelationships of processes
and the more “global” view of the organi-
zation should also be included. Solutions
may fail if representatives with these per-
spectives are not involved.
Potential solutions should be tested be-
fore full implementation, with a clear plan
relative to methods, objectives, timelines,
decision points, and algorithms for all pos-
sible results of the trial. Large-scale solu-
tions can be tried on a limited basis and
expanded if successful; smaller scale solu-
tions can be implemented pending an ef-
fectiveness evaluation. Nonetheless, data
should be collected to evaluate the effec-
tiveness of the proposed change. Data can
be collected by the methods used initially
to identify the problems or by methods
specially designed for the trial. Once solu-
tions have been successfully tested, full im-
plementation can occur. Following imple-
mentation, data should be collected, on at
least a periodic basis, to ensure adequate
control of the process.
Work Environment
The facility must provide a safe workplace
with adequate environmental controls
and emergency procedures for the safety
of the employees, donors, patients, and all
other inhabitants or visitors.1(p88) Proce-
dures must be in place to address:
■General safety
■Disaster preparedness
■Biological safety (blood-borne patho-
gens)
■Chemical safety
■Fire safety
■Radiation safety, if applicable
Chapter 1: Quality Systems 27
Figure 1-2. Example of a Pareto chart.
Copyright © 2005 by the AABB. All rights reserved.
■Discard of blood, components, and
tissue
Current good manufacturing practice
regulations require quality planning and
control of the physical work environment,
including:
■Adequate space and ventilation
■Sanitation and trash disposal
■Equipment for controlling air quality
and pressure, humidity, and temper-
ature
■Water systems
■Toilet and hand-washing facilities
An evaluation of the infrastructure and
its limitations before implementation of
procedures or equipment will help to en-
sure maximum efficiency and safety. A
more thorough discussion of facilities and
safety can be found in Chapter 2.
References
1. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
2. Code of federal regulations. Title 42 CFR Part
493. Washington, DC: US Government Print-
ing Office, 2004 (revised annually).
3. Code of federal regulations. Title 21 CFR Parts
606, 610, 630, and 640. Washington, DC: US
Government Printing Office, 2004 (revised
annually).
4. Code of federal regulations. Title 21 CFR Parts
210 and 211. Washington, DC: US Govern-
ment Printing Office, 2004 (revised annually).
5. Food and Drug Administration. Guideline for
quality assurance in blood establishments.
Docket #91N-0450. (July 11, 1995) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 1995.
6. Hospital accreditation standards. Oakbrook
Terrace, IL: Joint Commission Resources,
Inc., 2004.
7. College of American Pathologists Laboratory
Accreditation Program checklists. Chicago,
IL: College of American Pathologists, 2003.
8. A quality system model for health care;
NCCLS approved guideline (HS1-A). Wayne,
PA: National Committee for Clinical Labora-
tory Standards, 2002.
9. ANSI/ISO/ASQ Q9000-2000 series—quality
management standards. Milwaukee, WI: ASQ
Quality Press, 2000.
10. Baldrige National Quality Program. Health
care criteria for performance excellence.
Gaithersburg, MD: National Institute of Stan-
dards and Technology, 2004 (revised annu-
ally).
11. Quality program implementation. Associa-
tion Bulletin 97-4. Bethesda, MD: AABB, 1997.
12. Juran JM, Godfrey AB. Juran’s quality hand-
book. 5th ed. New York: McGraw-Hill, 1999.
13. Food and Drug Administration. Guidance on
general principles of process validation. (May
1, 1987) Rockville, MD: CBER Office of Com-
munication, Training, and Manufacturers
Assistance, 1987.
14. Food and Drug Administration. Glossary of
computerized system and software develop-
ment terminology. (August 1995) Rockville,
MD: Division of Field Investigations, Office of
Regional Operations, Office of Regulatory
Affairs, 1995.
15. Food and Drug Administration. Guidance for
industry: General principles of software vali-
dation: Final guidance for industry and FDA
staff. (January 11, 2002) Rockville, MD: CBER
Office of Communication, Training, and
Manufacturers Assistance, 2002.
16. Clinical laboratory technical procedure man-
uals. NCCLS approved guideline. 4th ed.
(GP2-A4). Wayne, PA: National Committee for
Clinical Laboratory Standards, 2002.
17. Code of federal regulations. Title 21 CFR Part
11. Washington, DC: US Government Printing
Office, 2004 (revised annually).
18. Motschman TL, Santrach PJ, Moore SB. Er-
ror/incident management and its practical
application. In: Duckett JB, Woods LL,
Santrach PJ, eds. Quality in action. Bethesda,
MD: AABB, 1996:37-67.
19. Food and Drug Administration. Biological
products: Reporting of biological product
deviations in manufacturing. Docket No.
97N-0242. (November 7, 2000) Fed Regist
2000;65:66621-35.
20. Food and Drug Administration. Guidance for
industry: Notifying FDA of fatalities related to
blood collection or transfusion. (September
22, 2003) Rockville, MD: CBER Office of Com-
munication, Training, and Manufacturers
Assistance, 2003.
21. Reporting donor fatalities. Association Bulle-
tin #04-06. Bethesda, MD: AABB, 2004.
22. Food and Drug Administration. Draft guid-
ance for industry: Biological product devia-
tion reporting for blood and plasma estab-
lishments. (August 10, 2001) Rockville, MD:
28 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
CBER Office of Communication, Training,
and Manufacturers Assistance, 2001.
23. Code of federal regulations. Title 21 CFR Part
803. Washington, DC: US Government Print-
ing Office, 2004 (revised annually).
24. Shulman IA, Lohr K, Derdiarian AK, et al.
Monitoring transfusionist practices: A strat-
egy for improving transfusion safety. Transfu-
sion 1994;34:11-15.
25. Becker J, Blackall D, Evans C, et al for the Sci-
entific Section Coordinating Committee.
Guidelines for blood utilization review.
Bethesda, MD: AABB, 2001.
26. Strauss RG, Blanchette VS, Hume H. National
acceptability of American Association of
Blood Banks Hemotherapy Committee guide-
lines for auditing pediatric transfusion prac-
tices. Transfusion 1993;33:168-71.
27. Anderson TD. Tools for statistical process
control. In: Ziebell LW, Kavemeier K, eds.
Quality control: A component of process con-
trol in blood banking and transfusion medi-
cine. Bethesda, MD: AABB Press, 1999:13-48.
28. Russell JP, Regel T. After the quality audit. 2nd
ed. Milwaukee, WI: ASQ Quality Press, 2000.
29. Motschman T. Corrective versus preventive
action. AABB News 1999;21(8):5,33.
Chapter 1: Quality Systems 29
Copyright © 2005 by the AABB. All rights reserved.

30 AABB Technical Manual
Appendix 1-1. Glossary of Commonly Used Quality Terms
Calibration Comparison of measurements performed by an instrument to those
made by a more accurate instrument or standard for the purpose of de-
tecting, reporting, and eliminating errors in measurement.
Change control Established procedures for planning, documenting, communicating, and
executing changes to infrastructure, processes, products, or services.
This includes the submission, analysis, decision making, approval, im-
plementation, and postimplementation review of the change. Formal
change control provides a measure of stability and safety and avoids ar-
bitrary changes that might affect quality.
Control chart A graphic tool used to determine whether the distribution of data values
generated by a process is stable over time. A control chart plots a sta-
tistic vs time and helps to determine whether a process is in control or
out of control according to defined criteria, eg, a shift from a central line
or a trend toward upper or lower acceptance limits.
Design output Documents, records, and evidence in any other format used to verify
that design goals have been met. Design output should identify charac-
teristics of a product or service that are crucial to safety and function
and to meeting regulatory requirements. It should contain or make ref-
erence to acceptance criteria. Examples of design output include stan-
dard operating procedures, specifications for supplies, reagents and
equipment, identification of quality control requirements, and the re-
sults of verification and validation activities.
End-product test and
inspection
Verification through observation, examination, and/or testing that the
finished product or service conforms to specified requirements.
Process capability Ability of a controlled process to produce a service or product that ful-
fills requirements. Also, a statistical measure of the inherent process
variability for a given characteristic relative to design specifications. The
most widely accepted formula for process capability is six sigma.
Process control Activities intended to minimize variation within a process in order to
produce a predictable output that meets specifications.
Qualification Demonstration that an entity is capable of fulfilling specified require-
ments. Verification of attributes that must be met or complied with in
order for a person or thing to be considered fit to perform a particular
function. For example, equipment may be qualified for an intended use
by verifying performance characteristics such as linearity, sensitivity, or
ease of use. An employee may be qualified based on technical, aca-
demic, and practical knowledge and skills developed through training,
education, and on-the-job performance.
Quality assurance Activities involving quality planning, control, assessment, reporting,
and improvement necessary to ensure that a product or service meets
defined quality standards and requirements.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 1: Quality Systems 31
Quality control Operational techniques and activities used to monitor and eliminate
causes of unsatisfactory performance at any stage of a process.
Quality indicators Measurable aspects of processes or outcomes that provide an indica-
tion of the condition or direction of performance over time. Used to
monitor progress toward stated quality goals and objectives.
Quality management The organizational structure, processes, and procedures necessary to
ensure that the overall intentions and direction of an organization’s
quality program are met and that the quality of the product or service is
ensured. Quality management includes strategic planning, allocation of
resources, and other systematic activities such as quality planning, im-
plementation, and evaluation.
Requirement A stated or obligatory need or expectation that can be measured or ob-
served and is necessary to ensure quality, safety, effectiveness, or cus-
tomer satisfaction. Requirements can include things that the system or
product must do, characteristics it must have, and levels of perfor-
mance it must attain.
Specification Description of a set of requirements to be satisfied by a product, mate-
rial, or process indicating, if appropriate, the procedures to be used to
determine whether the requirements are satisfied. Specifications are of-
ten in the form of written descriptions, drawings, professional stan-
dards, and other descriptive references.
Validation Demonstration through objective evidence that the requirements for a
particular application or intended use have been met. Validation pro-
vides assurance that new or changed processes and procedures are ca-
pable of consistently meeting specified requirements before implemen-
tation.
Verification Confirmation, by examination of objective evidence, that specified re-
quirements have been met.
Appendix 1-1. Glossary of Commonly Used Quality Terms (cont’d)
Copyright © 2005 by the AABB. All rights reserved.

32 AABB Technical Manual
Appendix 1-2. Code of Federal Regulations Quality-Related References
21 CFR Citation Topic
606.20 Personnel
606.40 Facilities
606.60 Equipment quality control
606.65 Supplies, reagents
606.100 Standard operating procedures
606.140 Laboratory controls
606.160 Records
606.170 Adverse reactions
606.171 Biological product deviations
211.22 Quality control/quality assurance unit responsibilities
211.25 Personnel qualifications
211.28 Personnel responsibilities
211.160 Laboratory controls
211.192 Production record review
211.194 Laboratory records and reviews
Copyright © 2005 by the AABB. All rights reserved.
Chapter 1: Quality Systems 33
Appendix 1-3. Statistical Tables for Binomial Distribution* Used to Determine
Adequate Sample Size and Level of Confidence for Validation of Pass/Fail Data
Confidence Levels (%) for Percent Conforming
Requirement for
% Conforming
Requirement for
% Conforming
Sample
Size
No. of
Failures
90% 95%
Sample
Size
No. of
Failures
90% 95%
% Confidence % Confidence
10 0 65.1 – 50 0 99.5 92.3
1 26.4 – 1 96.6 72.1
20 0 87.8 64.2 2 88.8 45.9
1 60.8 26.4 3 75.0 –
2 32.3 – 4 56.9 –
30 0 95.8 78.5 5 38.4 –
1 81.6 44.6 60 0 99.8 95.4
2 58.9 – 1 98.6 80.8
3 35.3 – 2 94.7 58.3
40 0 98.5 87.1 3 86.3 35.3
1 91.9 60.1 4 72.9 –
2 77.7 32.3 5 56.3 –
3 57.7 – 6 39.4 –
4 37.1 –
This table answers the question,
“How confident am I that [90 or 95]% of all products manufac-
tured will meet specifications if I have tested __ number of samples and found __ number to
be nonconforming (failures)?”
*Data from Reliability Analysis Center, http://rac.alionscience.com/Toolbox/.
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

34 AABB Technical Manual
Minimum Sample Size for Percent Conforming
Requirement for Percent Conforming
90% 95% 99%
Confidence Level Confidence Level Confidence Level
No. of
Failures
90% 95% 99% 90% 95% 99% 90% 95% 99%
Sample Size Sample Size Sample Size
0 22 29 45 46 59 90 230 299 459
1 39 47 65 77 94 130 388 467 662
2 53 63 83 106 125 166 526 625 838
3 66 76 98 133 180 198 664 773 1002
4 78 90 113 159 208 228 789 913 1157
5 91 103 128 184 235 258 926 1049 1307
6 104 116 142 209 260 288 1051 1186 1453
7 116 129 158 234 286 317 1175 1312
8 128 143 170 258 310 344 1297 1441
9 140 154 184 282 336 370 1418
10 152 167 197 306 361 397
This table answers the question,
“How many samples do I need to test with ___ number of fail-
ures if I want to have [90, 95, or 99] % confidence that [90, 95, or 99]% of all products will
meet specifications?”
*Data from Reliability Analysis Center, http://rac.alionscience.com/Toolbox/.
Appendix 1-3. Statistical Tables for Binomial Distribution* Used to Determine
Adequate Sample Size and Level of Confidence for Validation of Pass/Fail Data
(cont'd)
Copyright © 2005 by the AABB. All rights reserved.

Chapter 1: Quality Systems 35
Example of Minimum Sample Size and Number of Failures Allowed to Meet AABB Requirements
for Product Validation and Quality Control
Product Requirement1
Sample
Size2
Number of
Failures
%
Confidence
Level
Platelets Pheresis At least 90% of units sampled
contain ≥3×1011 platelets
and have a pH ≥6.2 at the
end of allowable storage.
10
10
0
1
65
26
Platelets Pheresis,
Leukocytes
Reduced
At a minimum, 95% of units
sampled shall contain a re-
sidual leukocyte count
<5 ×106.
20
20
0
1
64
26
Red Blood Cells
Pheresis
At least 95% of units sampled
shall have >50 g of hemo-
globin (or 150 mL red cell
volume) per unit.
20
20
0
1
64
26
Granulocytes
Pheresis
Prepared by a method known
to yield a minimum of
1.0 ×1010 granulocytes in
at least 75% of the units
tested.
4
4
0
1
68
26
1. From Silva MA, ed. Standards for blood banks and transfusion services. 23rd ed. Bethesda, MD: AABB,
2005. Although the AABB
Standards
does not require a specific confidence level, the facility may use
this as a way to assess the degree of certainty that each product manufactured will meet
specifications.
2. Period of time used to define a population for sampling is determined by the facility. (NOTE: The longer
the period, the more difficult it may be to identify causes of failure, and the more products already in
distribution that may be involved in a recall.)
*Data from Reliability Analysis Center, http://rac.alionscience.com/Toolbox/.
Appendix 1-3. Statistical Tables for Binomial Distribution* Used to Determine
Adequate Sample Size and Level of Confidence for Validation of Pass/Fail Data
(cont'd)
Copyright © 2005 by the AABB. All rights reserved.

36 AABB Technical Manual
Appendix 1-4. Assessment Examples: Blood Utilization1,2
A blood usage review committee should consider the following areas of practice and develop specific
measurements for monitoring blood transfusion processes. Some measurements provide data for
several processes.
Ordering of Appropriate Blood Components
1. Preanalytical errors Errors in specimen collection, verbal orders, transfusion orders.
2. Units transfused Figures for each type of blood component and special preparation.
Use of autologous and directed donor collections. Analyze by clin-
ical service or by prescriber.
3. Patients transfused Total number of patients receiving each of the components or
products listed in item 2.
4. Units transfused
per patient transfused
Average number of units of each component or product given to
patients receiving that component. May be useful to analyze by di-
agnosis or surgical/medical procedure.
5. Special components
prepared and trans-
fused
Number and relative percent of leukocyte-reduced, irradiated,
cytomegalovirus-negative units; aliquots prepared and transfused;
outpatient and home transfusions.
6. Units returned unused Number and percent of units issued and later returned unused.
Analyze by ward, by clinical service, or by prescriber.
7. Crossmatch-to-trans-
fusion (C:T) ratio
Number of units crossmatched divided by the number of units
transfused. Analysis could be by institutional total, by emergency
vs routine requests, or by clinical service, surgical procedure, or
prescriber, as needed.
8. Transfusion guidelines Verification that guidelines are current, appropriate for the patient
population being treated, and readily available to physicians.
Distributing, Handling, and Dispensing Blood Components
1. Turnaround time Interval between the time a transfusion request is received and
time the unit is available for transfusion and/or is transported to
the patient’s bedside. May analyze by emergency, routine, or oper-
ative requests.
2. Emergency requests Number and percent may be analyzed by clinical service,
prescriber, diagnosis, or time (week, day, shift).
3. Uncrossmatched units Number and percent of units issued uncrossmatched or with ab-
breviated pretransfusion testing. May analyze by clinical service,
or prescriber.
4. Age distribution
of units
Age of units in inventory and crossmatched by ABO and Rh type,
age of units when received from the supplier, age at the time of
transfusion, age when returned to the supplier.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 1: Quality Systems 37
5. Surgical cancellations
due to unavailability of
blood
Number and percent of cases delayed due to the unavailability of
blood; number of hours or days of delay, analyzed by surgical
procedure and by cause (eg, antibody problem in an individual
patient, general shortage, or shortage of a particular ABO or Rh
type).
6. Significant type
switches due to
unavailability of blood
Number of Rh-negative patients given Rh-positive red cells or
platelets; transfusions with ABO-incompatible plasma.
7. Outdate rate Number of units outdated (expired unused) divided by the num-
ber of units received; should be monitored for all blood compo-
nents and derivatives. Analysis by ABO and Rh type may prove in-
formative.
8. Wastage rates Number of units wasted due to breakage, improper preparation,
improper handling or storage; units prepared for a patient but not
used; number of units that failed to meet inspection requirements.
9. Adequacy of service
from the blood
supplier
Number of orders placed that could be filled as requested; aver-
age time between the order and receipt of emergency delivery;
number of orders associated with an error such as improper unit
received or units improperly shipped.
10. Compatibility testing
requirements
Adequacy, currency, and appropriateness of policies and proce-
dures.
11. Quality control policies
and procedures
Percent of records of temperature, equipment, component prepa-
ration, or testing that are incomplete or have deviations.
Administration of Blood and Components
1. Blood issue/delivery
errors
Number of wrong units issued; number of units delivered to
wrong patient-care area or improperly transported.
2. Blood administration
policies and
procedures
Adherence to facility-specific requirements when monitoring pa-
tients for signs and symptoms of adverse reactions. Availability of
copies of current policies and procedures and of current
Circular
of Information for the Use of Human Blood and Blood Compo-
nents
.
3. Blood administration
audits
Summary of on-site performance reviews, to include number of
deviations by category (eg, identification of patient and donor
unit, documentation and completeness of medical record). May
include audit for documentation of transfusion order and indica-
tion for transfusion or informed consent.
4. Transfusion
equipment
Review of quality control documentation for equipment, including
blood warmers, infusion pumps, special filters or administration
sets; documentation in the medical record that devices were used;
number of situations where their use was inappropriate.
Appendix 1-4. Assessment Examples: Blood Utilization1,2 (cont'd)
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

38 AABB Technical Manual
5. Special transfusion
situations
Review of compliance with policies for out-of-hospital transfu-
sions and perioperative and postoperative collection of autolo-
gous blood.
Monitoring Transfusion Results
1. Compliance with trans-
fusion guidelines
Number and percent of inappropriate transfusions, as determined
by the blood usage review committee; analysis of reasons for in-
appropriate transfusion.
2. Transfusion reactions Number and percent of reported transfusion reactions; turn-
around time for complete investigation; documentation of transfu-
sion service and committee review; documentation in the medical
record.
3. Transfusion-trans-
mitted disease
Number of cases by infectious agent; turnaround time of investi-
gation; completeness of review and recording.
4. Look-back investi-
gations
Number of cases by infectious agent; turnaround time of investi-
gation; completeness of case-finding, notification, review, and re-
cording.
5. Review of policies
and procedures
Adequacy, currency, and appropriateness of policies and proce-
dures for detection and reporting of adverse effects of transfu-
sion.
Management Data
1. Workload and produc-
tivity
Evaluation of activities and efficiency of the laboratory; may be
analyzed by day of week and by shift. Hours worked per unit
transfused or patient transfused may be more valuable as an effi-
ciency measure than data obtained from traditional productivity
calculations.
2. Event reports Number of events dealing with laboratory processes (eg, labeling,
preparation, testing, issue); procedural events in blood adminis-
tration; errors, accidents, and recalls by blood supplier(s).
3. Staff training and
competency
Documentation of training and continuing competency of labora-
tory and nursing staff to perform transfusion-related procedures
and policies.
1. Comprehensive accreditation manual for hospitals: The official handbook. Oakbrook Terrace, IL: Joint
Commission Resources, Inc., 2002.
2. Becker J, Blackall D, Evans C, et al for the Scientific Section Coordinating Committee. Guidelines for
blood utilization review. Bethesda, MD: AABB, 2001.
Appendix 1-4. Assessment Examples: Blood Utilization1,2 (cont'd)
Copyright © 2005 by the AABB. All rights reserved.

Chapter 2: Facilities and Safety
Chapter 2
Facilities and Safety
FACILITY DESIGN AND mainte-
nance are critical to ensure that op-
erational needs are met and that
the work environment is safe for both
staff and visitors. The layout of the physi-
cal space; management of utilities such as
water and air ventilation; flow of person-
nel, materials, and waste; and ergonomic
factors should all be considered in the fa-
cility management plan.
In addition to providing adequate facili-
ties, the organization must develop and
implement a safety program that defines
policies and procedures for safe work
practices. This includes hazard communi-
cation, use of protective equipment, train-
ing, and competency assessment in accor-
dance with regulations for emergency and
disaster preparedness, chemical hygiene,
blood-borne pathogens, and radiation
safety when applicable. All employees are
responsible for protecting their own safety
and the safety of others by adhering to poli-
cies set forth in the facility safety program.
The AABB requires accredited blood
banks and transfusion services to plan, im-
plement, and maintain a program to mini-
mize risks to the health and safety of do-
nors, patients, volunteers, and employees
from biological, chemical, and radiological
hazards.1(p88) Other professional and accred-
iting organizations have similar or more de-
tailed safety program requirements, includ-
ing the College of American Pathologists
(CAP), the Clinical and Laboratory Stan-
dards Institute (formerly NCCLS), and the
Joint Commission on Accreditation of
Healthcare Organizations (JCAHO).2-5
Several federal agencies have issued reg-
ulations and recommendations to protect
the safety of workers and the public. Those
relevant to health-care settings are listed in
Appendix 2-1. The contents of these regula-
tions and guidelines are discussed in more
39
2
Copyright © 2005 by the AABB. All rights reserved.
detail in each section of this chapter. Blood
banks and transfusion services should con-
sult with state and local agencies as well to
identify any additional safety requirements.
Trade and professional organizations also
provide safety recommendations that are
relevant to blood banks and transfusion
services. These organizations are also listed
in Appendix 2-1.
Facilities
Facility Design and Workflow
Proper design and maintenance of facili-
ties and organization of work can reduce
or eliminate many potential hazards. De-
sign, maintenance, and organization also
affect efficiency, productivity, error rates,
employee and customer satisfaction, and
the quality of products and services. State
and local building codes should be con-
sulted in the design planning stages for
architectural safety regarding space, fur-
nishings, and storage.
During the design phase for a new space,
the location and flow of personnel, materi-
als, and equipment should be considered
in the context of the processes to be per-
formed. Adequate space must be allotted
for personnel movement, location of sup-
plies and large equipment, and private or
“distraction-free” zones for certain manu-
facturing tasks (eg, donor interviewing,
record review, and blood component label-
ing). The facility must be able to accommo-
date designated “clean” and “dirty” spaces
andprovideforcontrolledmovementof
materials and waste in and out of these ar-
eas so as to avoid contamination. Chemical
fume hoods and biological safety cabinets
should be located away from drafts and
high-traffic areas. The number and location
of eyewashes and emergency showers must
also be considered. Water sources for re-
agent preparation must be considered. Staff
handling hazardous materials must have
ready access to hand-washing sinks. For
certain pieces of heavy equipment, such as
irradiators, load-bearing capacity must be
taken into account.
Laboratories must be designed with ade-
quate illumination, electrical power, and
conveniently located outlets. Emergency
backup power sources, such as uninter-
ruptible power supplies and backup gener-
ators, should be considered to ensure that
loss of blood products does not occur dur-
ing power failures. The National Electrical
Code6is routinely used as a national guide-
line for the design of essential electrical dis-
tribution systems, with modifications ap-
proved by the local building authority having
jurisdiction.
Appropriate systems for heating, ventila-
tion, and air conditioning must be used.
Environmental monitoring systems should
be considered for laboratories that require
positive or negative air pressure differen-
tials to be maintained, or where air filtra-
tion systems are used to control particle
levels. The nationally accepted specifica-
tions for ventilation are published by the
American Society of Heating, Refrigerating,
and Air-Conditioning Engineers, Inc.7
Housekeeping
The workplace should be kept clean and
free of clutter. Work surfaces and equip-
ment should be regularly cleaned and dis-
infected. Items that may accumulate dust
anddebrisshouldnotbestoredabove
clean supplies or work surfaces. Exits and
fire safety equipment must not be block-
ed or obstructed in any way. Receptacles
and disposal guidelines for nonhazardous
solid waste, biohazardous, chemical, and
radiation waste should be clearly delin-
eated. Housekeeping responsibilities,
methods, and schedules should be de-
fined for every work area. Written proce-
40 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
dures, initial training, continuing educa-
tion of personnel, and ongoing monitoring
of housekeeping effectiveness are essen-
tial to safe operations.
Restricted Areas
Hazardous areas should be clearly and
uniformly identified with warning signs in
accordance with federal Occupational
Safety and Health Administration (OSHA)
and Nuclear Regulatory Commission (NRC)
standards so that personnel entering or
working around them are aware of exist-
ing biological, chemical, or radiation dan-
gers.8-11 Staff not normally assigned to
these areas should receive adequate train-
ing to avoid endangering themselves. Risk
areas can be stratified. For example, “high-
risk” areas might include chemical fume
hoods, biological safety cabinets, and stor-
age areas for volatile chemicals or radio-
isotopes. Technical work areas could be
considered “moderate risk” and restricted
to laboratory personnel. Administrative
and clerical areas could be considered
“low risk” and not restricted.
Whenever possible, functions not requir-
ing special precautions should be separated
from those performed in restricted areas.
Every effort should be made to prevent the
contamination of designated “clean” areas
and common equipment. Work area tele-
phones can be equipped with speakers to
eliminate the need to pick up the receiver.
Computer keyboards and telephones can
be covered with plastic. They should be
cleaned on a regular basis and when visibly
soiled. Employees should remove their per-
sonalprotectivebarrierssuchasglovesand
laboratory coats and wash their hands with
soap and water when leaving a “contami-
nated” area.
Concerns for safety dictate that there be
no casual visitors in areas where laboratory
hazards may be encountered.12 Children,
especially, should not be allowed in areas
where they could be exposed to hazards
andshouldbecloselysupervisedinthose
areas where their presence is permitted. Fa-
cilities should consider establishing specific
safety guidelines for visitors with business
in restricted areas and documenting that this
information was received and understood.
Mobile Sites
Mobile blood collection operations can
present special problems. An individual
trained in safety principles should make an
advance visit to the collection site to ensure
that hazards are minimized. All mobile per-
sonnel should be trained to recognize un-
safe conditions and understand infection
control policies and procedures, but re-
sponsibility for site safety should be as-
signed to a senior-level employee.
Hand-washing access is essential at all
collection sites. Carpeted or difficult-to-
clean surfaces may be protected with an
absorbent overlay with waterproof backing
to protect from possible blood spills. Porta-
ble screens and ropes are helpful in direct-
ing traffic flow to maintain safe work areas.
Food service areas should be physically
separated from areas for blood collection
and storage. Blood-contaminated waste must
be either returned to a central location for
disposal or packaged and decontaminated
using thermal (autoclave, incinerator) or
chemical disinfectant in accordance with
local regulations for medical wastes. Train-
ed staff must perform this decontamination
with particular attention paid to cleanup of
mobile sites after blood collection.
Ergonomics
Consideration in physical design should
be given to ergonomics and accommoda-
tions for individuals covered under the
Americans with Disabilities Act (42 U.S.C.
Chapter 2: Facilities and Safety 41
Copyright © 2005 by the AABB. All rights reserved.
§ 12101-12213, 1990). Several factors may
contribute to employee fatigue, musculo-
skeletal disorder syndromes, or injury, in-
cluding the following13:
■Awkward postures—positions that
place stress on the body such as
reaching overhead, twisting, bend-
ing, kneeling, or squatting.
■Repetition—performing the same
motions continuously or frequently.
■Force—the amount of physical effort
used to perform work.
■Pressure points—pressing the body
against hard or sharp surfaces.
■Vibration—continuous or high-intensity
hand-arm or whole-body vibration.
■Other factors—extreme high or low
temperatures; lighting too dark or
too bright.
Both the total time per work shift and
the length of uninterrupted periods of work
can be significant in contributing to prob-
lems. Actions to correct problems associ-
ated with ergonomics may include:
■Engineering improvements to reduce
or eliminate the underlying cause,
such as making changes to equip-
ment, workstations, or materials.
■Administrative improvements, such
as providing variety in tasks; adjust-
ing work schedules and work pace;
providing recovery or relaxation time;
modifying work practices; ensuring
regular housekeeping and maintenance
of work spaces, tools, and equipment;
and encouraging exercise.
■Provision of safety gear such as gloves,
knee and elbow pads, footwear, and
other items that employees wear to
protect themselves against injury.
Safety Program
An effective safety program starts with a
well-thought-out safety plan. This plan
identifies the applicable regulatory re-
quirements and describes how they will
be met. In general, institutions are re-
quired to:
■Provideaworkplacefreeofrecog
-
nized hazards.
■Evaluate all procedures for potential
exposure risks.
■Evaluate each employment position
for potential exposure risks.
■Identify hazardous areas or materi-
als with appropriate labels and signs.
■Educate staff, document training,
and monitor compliance.
■Apply Standard Precautions (includ-
ing Universal and Blood and Body
Fluid Precautions) to the handling of
blood, body fluids, and tissues.
■Dispose of hazardous waste appro-
priately.
■Report incidents and accidents and
provide treatment and follow-up.
■Provide ongoing review of safety pol-
icies, procedures, operations, and
equipment.
■Develop facility policies for disaster
preparedness and response.
Safety programs should consider the
needs of all persons affected by the work
environment. Most obvious is the safety of
technical staff, but potential risks for blood
donors, ancillary personnel, volunteers, vis-
itors, housekeeping staff, and maintenance
and repair workers must also be evaluated.
Appropriate provisions must be applied if
these individuals cannot be excluded from
risk areas.
Laboratories should appoint a safety of-
ficer who can provide general guidance and
expertise.3This individual might develop
the safety program, oversee orientation and
training, perform safety audits, survey work
sites, recommend changes, and serve on or
direct the activities of safety committees. It
is recommended that facilities using haz-
ardous chemicals and radioactive materials
42 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
appoint a chemical hygiene officer and ra-
diation safety officer to oversee chemical
and radiation protection programs.8,11 A
general safety officer with sufficient exper-
tise may fill these roles, or separate officers
maybeappointedandprogramoversight
given to a safety committee.
There are five basic elements that must
be addressed for each type of hazard cov-
ered in the safety program:
1. Training.
2. Hazard identification and communi-
cation.
3. Engineering controls and personal
protective equipment.
4. Safe work practices, including waste
disposal.
5. Emergency response plan.
In addition, management controls should
be implemented to ensure that these ele-
ments are in place and effective. Manage-
ment is responsible for:
1. Developing and communicating the
written plan.
2. Ensuring implementation and pro-
viding adequate resources.
3. Providing access to employee health
services related to prevention strate-
gies and treatment of exposures.
4. Monitoring compliance and effec-
tiveness.
5. Evaluating and improving the safety
plan.
Basic Elements of a Safety Program
Training
Employees must understand the hazards
in their workplace and the appropriate
precautions to take in order to manage
them safely. The mandate for employee
training programs is based on good gen-
eral practice as well as OSHA require-
ments.9-11 All persons must be trained to
protect themselves appropriately before
beginning work with hazardous materials.
Supervisors or their designees are res-
ponsible for documenting the employee’s
understanding of and ability to apply safety
precautions before independent work is
permitted. Safety training must precede
even temporary work assignments if signifi-
cant potential for exposure exists. Staff who
do not demonstrate the requisite under-
standing and skills must undergo retrain-
ing. These requirements apply not only to
laboratory staff, but also to housekeeping
and other personnel who may come into
contact with hazardous substances or
waste.Table2-1liststopicstocoverin
work safety training programs.
Hazard Identification and Communication
Employees must know when they are
working with hazardous substances and
must know where they are located in the
workplace. Employers are required to pro-
vide information about workplace haz-
ards to their employees to help reduce the
risk of occupational illnesses and injuries.
This is done by means of signage, labels
on containers, written information, and
training programs.
Engineering Controls and Personal
Protective Equipment
Whenever possible, the physical work-
space should be designed to eliminate the
potential for exposure. When this is not
possible, protective gear must be pro-
vided to protect the employee. Engineer-
ing controls are physical plant controls or
equipment such as sprinkler systems,
chemical fume hoods, and needleless sys-
tems that isolate or remove the hazard from
the workplace. Personal protective equip-
ment (PPE) is specialized clothing or
equipment worn by an employee for pro-
tection against a hazard, such as gloves,
Chapter 2: Facilities and Safety 43
Copyright © 2005 by the AABB. All rights reserved.

masks, and laboratory coats. General
guidance on the use of engineering con-
trols and PPE is included in Appendix 2-2.
Safe Work Practices
Employees must be trained to know how
to work with hazardous materials in a way
that protects themselves, their co-workers,
and the environment. Safe work practices
are defined as tasks performed in a man-
ner that reduces the likelihood of expo-
sure to workplace hazards. General rec-
ommendations for safe work practices are
included in Appendix 2-2.
Emergency Response Plan
When engineering and work practice con-
trols fail, employees must know how to re-
spond. The purpose of advance planning
is to control the hazardous situation as
quickly and safely as possible. Regular
testing of the emergency response plan
will identify areas for improvement and
will also build confidence in staff to re-
spond effectively in a real situation. OSHA
requires a written plan for facilities with
more than 10 employees. Verbal commu-
nication of the plan is acceptable for 10 or
fewer employees.14
44 AABB Technical Manual
Table 2-1. Topics to Cover in a Work Safety Training Program
Work safety training programs should ensure that all personnel:
■Have access to a copy of pertinent regulatory texts and an explanation of the contents.
■Understand the employer’s exposure control plan and how to obtain a copy of the written plan.
■Understand how hepatitis and human immunodeficiency virus (HIV) are transmitted and how
often; be familiar with the symptoms and consequences of hepatitis B virus (HBV), hepatitis C
virus (HCV), and HIV infection.
■Know that they are offered vaccination against HBV.
■Recognize tasks that pose infectious risks and distinguish them from other duties.
■Know what protective clothing and equipment are appropriate for the procedures they will
perform and how to use them.
■Know and understand the limitations of protective clothing and equipment (eg, different types of
gloves are recommended based on the permeability of the hazardous material to be used).
Employers and staff should be forewarned against a false sense of security.
■Know where protective clothing and equipment are kept.
■Are familiar with and understand all requirements for work practices specified in standard
operating procedures (SOPs) for the tasks they perform, including the meaning of signs and
labels.
■Know how to remove, handle, decontaminate, and dispose of contaminated material.
■Know the appropriate actions to take and the personnel to contact if exposed to blood or other
biological, chemical, or radiological hazards.
■Know the corrective actions to take in the event of spills or personal exposure to fluids, tissues,
and contaminated sharps, the appropriate reporting procedures, and the medical monitoring
recommended when parenteral exposure may have occurred.
■Know their right for access to medical treatment and medical records.
Copyright © 2005 by the AABB. All rights reserved.
Management Controls
Supervisory personnel must monitor safe-
ty practices in their areas of responsibility.
Continuing attention to safety issues should
be addressed in routine staff meetings
and training sessions. Periodic audits per-
formed by a safety professional help in-
crease safety awareness. Management
should seek staff input into the design and
improvement of the facility’s safety plan.
The safety program, with its policies,
guidelines, and supporting references to
regulatory documents, should be detailed
in a safety manual and made available to all
personnel at risk. This manual, along with
operational procedures manuals, should be
reviewed at least annually and updated as
technology evolves and new information
becomes available. Work sites and safety
equipment also should be inspected regu-
larly to ensure compliance and response
readiness. Checklists are helpful in docu-
menting these audits and assessing safety
preparedness. Checklist items and essential
elements for safety and environmental
management audits can be obtained from
other sources2,3,15 or can be developed inter-
nally.
Employee Health Services
Hepatitis Prophylaxis
All employees routinely exposed to blood
must be offered hepatitis B virus (HBV)
vaccine if they do not already have HBV-
protective antibodies (ie, anti-HBs). OSHA
requires that the vaccine be offered at no
cost to the employee, and if the employee
refuses the vaccine, that the refusal be
documented.10
Monitoring Programs
The employer must provide a system for
monitoring exposure to certain substances
as defined in the OSHA standard if there
is reason to believe that exposure levels
routinely exceed the action level.16
Medical First Aid and Follow-up
When requested by a worker who has sus-
tained known or suspected blood expo-
sure, monitoring for HBV, hepatitis C virus
(HCV), and human immunodeficiency vi-
rus (HIV) antibodies should be provided
with appropriate counseling. Informed
consent is required for this voluntary test-
ing; rejection of offered testing must be
documented. The usual schedule would
includeimmediatetestsontheworker
and on the source of the potentially infec-
tious material, with follow-up of the work-
er at intervals after exposure.9,10 All aspects
of accident follow-up should be appropri-
ately documented.
The Centers for Disease Control and Pre-
vention (CDC) has published recommen-
dations for both pre- and postexposure
prophylaxis if the contaminating material is
HBV-positive or if this information is un-
known.17 Hepatitis B Immune Globulin is
usually given concurrently with hepatitis B
vaccine in cases of penetrating injuries.
When administered in accordance with the
manufacturer’s directions, both products
are very safe and carry no documented risk
for infection with HBV, HCV, or HIV. Post-
exposure prophylaxis for HIV is continu-
ally evolving; policies are generally based on
Public Health Service recommendations17
and current standards of practice.
Reporting Accidents and Injuries
When an injury occurs, as much relevant
information as possible should be docu-
mented (see Table 2-2). In addition, the
supervisor should complete any accident
reports and investigation forms required
by the institution’s insurer and worker’s
compensation agencies. Medical records
for individual employees should be pre-
Chapter 2: Facilities and Safety 45
Copyright © 2005 by the AABB. All rights reserved.

served for the duration of employment
plus 30 years, with few exceptions.17
OSHA requires health service employers
with 11 or more workers to maintain re-
cords of occupational injuries and illnesses
that require care that exceeds the capabili-
ties of a person trained in first aid.18 Records
of first aid provided by a nonphysician for
minor injuries such as cuts or burns do not
have to be retained. Initial documentation
must be completed within 6 days of the in-
cident.Alllogs,summaries,andsupple
-
mental records must be preserved for at
least 5 years beyond the calendar year of
occurrence. Employers must report fatali-
ties and injuries resulting in the hospital-
ization of three or more employees to
OSHA within 8 hours of the accident.18
Latex Allergies
With the increased use of gloves, there has
been a rise in the number of health-care
workers with latex allergies. Adverse reac-
tions associated with latex and/or powdered
gloves include contact dermatitis, allergic
dermatitis, urticaria, and anaphylaxis.
Medical devices that contain latex must
bear a caution label. The National Insti-
tute for Occupational Safety and Health
offers the following recommendations19:
■Make nonlatex gloves available as an
alternative to latex. Encourage use of
nonlatex gloves for activities and
work environments where there is
minimal risk of exposure to infec-
tious materials.
■If latex gloves are used, provide re-
duced protein, powder-free gloves.
(Note: This is not a requirement, but
a recommendation to reduce expo-
sure.)
■Use good housekeeping practices to
remove latex-containing dust from
the workplace.
46 AABB Technical Manual
Table 2-2. Information to Be Included in Injury Reports
■Name and address of the injured person.
■Time of the injury (hour, day, month, year).
■Specified place where the injury occurred.
■Details of the injured person’s activities at the time of injury.
■Nature of the injury (eg, bruise, laceration, burn, etc).
■Part of the body injured (eg, head, arm, leg, etc).
■Nature of the known or potential agent, in cases of exposure to pathologic organisms or other
hazardous materials.
■Nature of medical attention or first aid applied in the workplace.
■Date the injured person stopped work.
■Date the injured person returned to work.
■Estimated cost of damage to property or to equipment.
■Injured person’s statement of the events leading to the injury.
■Statements from witnesses.
■Cause of the injury.
■Corrective action taken or recommendations for corrective action.
Copyright © 2005 by the AABB. All rights reserved.
■Use work practices that reduce the
chance of reaction, such as hand wash-
ing and avoiding oil-based hand lo-
tions.
■Provide workers with education pro-
grams and training materials about
latex allergy.
■Periodically screen high-risk workers
for latex allergy symptoms.
■Evaluate current prevention strategies.
■If symptoms of latex allergy develop,
avoid direct contact with latex and
consult a physician about allergy
precautions.
Fire Prevention
Fire prevention relies on a combination of
facility design based on the National Fire
Protection Association (NFPA) Life Safety
Code,20 defined processes to maintain fire
protection systems in good working order,
and fire safe work practices. The Life
Safety Code includes both active and pas-
sive fire protection systems (eg, alarms,
smoke detectors, sprinklers, egress lights
and corridors, and fire-rated barriers).
Training
Fire safety training is recommended at the
start of employment and at least annually
thereafter. Training should emphasize
prevention and an employee’s awareness
of the work environment, including how
to recognize and report unsafe condi-
tions, how to report fires, the locations of
the nearest alarm and fire containment
equipment and their use, and evacuation
policies and routes.
All staff are required to participate in fire
drills at least annually by the CAP and the
JCAHO.2,4 In areas where patients are
housed or treated, the JCAHO requires
quarterly drills on each shift. Staff partici-
pation and understanding should be docu-
mented.
Hazard Identification and Communication
Emergency exits must be clearly marked
with an “EXIT” sign. Additional signage
must be posted along the route of egress
to show the direction of travel if it is not
immediately apparent. All flammable ma-
terials should be labeled with appropriate
hazard warnings and flammable storage
cabinets should be clearly marked.
Engineering Controls and Personal
Protective Equipment
Laboratories storing large volumes of
flammable chemicals are usually built
with 2-hour fire separation walls, or with
1-hour separation if there is an automatic
fire extinguishing system.3Permanent exit
routes must be designed to provide free
and unobstructed egress from all parts of
the facility to an area of safety. Secondary
exits may be required for areas larger than
1000 square feet; consult local safety au-
thority having jurisdiction such as the
local fire marshal and the NFPA. Fire de-
tection and alarm systems should be pro-
vided in accordance with federal, state,
and local regulations.
Safe Work Practices
All fire equipment should be inspected on
a regular basis to ensure good working or-
der. Fire extinguishers should be made
readily available and staff should be
trained to use them properly. Nothing
should be stored along emergency exit
routes that would obstruct evacuation ef-
forts. Exit doors cannot be locked from the
inside. Housekeeping and inventory man-
agement plans should be designed to
control the accumulations of flammable
and combustible materials stored in the
Chapter 2: Facilities and Safety 47
Copyright © 2005 by the AABB. All rights reserved.
facility. In areas where sprinkler systems
are installed, all items should be stored at
least 18 inches below the sprinkler head.
Local fire codes may require greater clear-
ance.
Emergency Response Plan
The fire emergency response plan should
encompass both facility-wide and area-
specific situations. It should describe re-
porting and alarm systems; location and
use of emergency equipment; roles and
responsibilities for staff during the re-
sponse; “defend in place” strategies; and
conditions for evacuation, evacuation pro-
cedures, and routes of egress.4,14 When a
fire occurs, the general sequence for im-
mediateresponseshouldbeto1)rescue
anyone in immediate danger, 2) activate
the fire alarm system and alert others in
thearea,3)confinethefirebyclosingdoors
and shutting off fans or other oxygen
sources if possible, and 4) extinguish the
fire with a portable extinguisher if the fire
is small, or evacuate if it is too large to
manage.
Electrical Safety
Electrical hazards, including fire and
shock, may arise from use of faulty elec-
trical equipment, damaged receptacles,
connectors or cords, or unsafe work prac-
tices. Proper use of electrical equipment,
periodic inspection and maintenance,
and hazard recognition training are es-
sential to help prevent accidents that may
result in electric shock or electrocution.
The severity of shock depends on the path
that the electrical current takes through
the body, the amount of current flowing
through the body, and the length of time
that it is flowing through the body. Even
low-voltage exposures can lead to serious
injury.21
Training
Safety training should be designed to make
employees aware of electrical hazards as-
sociated with receptacles and connectors
and help them recognize potential prob-
lems such as broken receptacles and con-
nectors, improper electrical connections,
damaged cords, and inadequate ground-
ing.
Hazard Identification and Communication
The safety plan should address the proper
use of receptacles and connectors. Equip-
ment that does not meet safety standards
should be marked to prevent accidental use.
Engineering Controls and Personal
Protective Equipment
OSHA requires that electrical systems and
equipment be constructed and installed
in a way that minimizes the potential for
workplace hazards. When purchasing
equipment, the facility should verify that
it bears the mark of an OSHA-approved
independent testing laboratory such as
Underwriters Laboratories (UL).22 Ade-
quate working space should be provided
around equipment to allow easy access
for safe operation and maintenance.
Ground-fault circuit interrupters should
be installed in damp or wet areas.
Safe Work Practices
Electrical safety practices are focused
around 1) proper use of electrical equip-
ment and 2) proper maintenance and re-
pair. Staff should not plug or unplug
equipment from an electrical source when
their hands are wet. Overloading circuits
with too many devices may cause the cur-
rent to heat the wires to a very high tem-
perature and generate a fire. Damaged re-
ceptacles and faulty electrical equipment
must be tagged and removed from service
48 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
until they have been repaired and check-
ed for safety. Flexible cords should be se-
curedtopreventtrippingandshouldbe
protected from damage from heavy or
sharp objects. Slack in flexible cords
should be kept to prevent tension on elec-
trical terminals and cords should be
checked for cut, broken, or cracked insu-
lation. Extension cords should not be
used in lieu of permanent wiring.
Emergency Response Plan
When it is not possible to decrease the
power or disconnect equipment, the power
supply should be shut off from the circuit
breaker. If it is not possible to interrupt
the power supply, a nonconductive mate-
rial such as dry wood should be used to
pry a victim from the source of current.21
Victims must not be touched directly.
Emergency first aid for victims of electri-
cal shock must be sought. Water-based
fire extinguishers are not to be used on
electrical fires.
Biosafety
The blood bank or transfusion service must
define and enforce measures to minimize
theriskofexposuretobiohazardousma
-
terials in the workplace. Requirements
published by OSHA (Bloodborne Pathogen
Standard)10 and recommendations pub-
lished by the US Department of Health and
Human Services9,12 provide the basis for
an effective biosafety plan.
Bloodborne Pathogen Standard
The OSHA Bloodborne Pathogen Stan-
dard is intended to protect employees in
all occupations where there is a risk of ex-
posure to blood and other potentially in-
fectious materials. It requires that the fa-
cility develop an Exposure Control Plan
and describes appropriate engineering
controls, personal protective equipment,
and work practice controls to minimize
the risk of exposure. It also requires em-
ployers to provide hepatitis B vaccination
for staff with occupational exposure, to
provide medical follow-up in case of acci-
dental exposure, and to keep records re-
lated to accidents and exposures.
Standard Precautions
Standard Precautions represent the most
current recommendations by CDC to re-
duce the risk of transmission of blood-
borne and other pathogens in hospitals.
Published in 1996 in the Guidelines for
Isolation Precautions in Hospitals,9they
build on earlier recommendations, in-
cluding Body Substance Isolation (1987),
Universal Precautions (1986), and Blood
and Body Fluid Precautions (1983). The
Bloodborne Pathogen Standard refers to
the use of Universal Precautions; however,
OSHA recognizes the more recent guide-
lines from the CDC and, in Directive CPL
2-2.69, allows hospitals to use acceptable
alternatives, including Standard Precau-
tions,aslongasallotherrequirementsin
the standard are met.23
Standard Precautions apply to all patient
care activities regardless of diagnosis where
there is a risk of exposure to 1) blood; 2) all
body fluids, secretions, and excretions, ex-
cept sweat; 3) nonintact skin; and 4) mucous
membranes.
Biosafety Levels
Recommendations for biosafety in labo-
ratories are based on the potential haz-
ards for specific infectious agents and the
activities performed.12 They include guid-
ance on both engineering controls and
safe work practices. The four biosafety
levels are designated in ascending order,
with increasing protection for personnel,
the environment, and the community.
Chapter 2: Facilities and Safety 49
Copyright © 2005 by the AABB. All rights reserved.
Biosafety Level 1 (BSL-1) involves work
with agents of no known or of minimal po-
tential hazard to laboratory personnel and
the environment. Activities are usually con-
ducted on open surfaces and no contain-
ment equipment is needed.
Biosafety Level 2 (BSL-2) work involves
agents of moderate potential hazard to per-
sonnel and the environment, usually from
contact-associated exposure. Most blood
bank laboratory activities are considered
BSL-2. Precautions described in this section
will focus on BSL-2 requirements. Labora-
tories should consult the CDC or National
Institutes of Health (NIH) guidelines for
precautions appropriate for higher levels of
containment.
Biosafety Level 3 (BSL-3) includes work
with indigenous or exotic agents that may
cause serious or potentially lethal disease
as a result of exposure to aerosols (eg, My-
cobacterium tuberculosis) or by other routes
that would result in grave consequences to
the infected host (eg, HIV). Recommenda-
tions for work at BSL-3 are designed to con-
tain biohazardous aerosols and minimize
the risk of surface contamination.
Biosafety Level 4 (BSL-4) applies to work
with dangerous or exotic agents that pose
high individual risk of life-threatening dis-
ease from aerosols (eg, agents of hemor-
rhagic fevers, filoviruses). BSL-4 is not
applicable to routine blood-bank-related
activities.
Training
OSHA requires annual training for all em-
ployees whose tasks carry risk of infec-
tious exposure.10,23 Training programs
must be tailored to the target group, both
in level and content. General background
knowledge of biohazards, understanding
of control procedures, or work experience
cannot meet the requirement for specific
training, although assessment of such
knowledge is a first step in planning pro-
gram content. Workplace volunteers require
at least as much safety training as paid
staff performing similar functions.
Hazard Identification and Communication
The facility’s Exposure Control Plan com-
municatestheriskspresentinthework
-
place and describes controls to minimize
exposure. BSL-2 through BSL-4 facilities
must have a biohazard sign posted at the
entrance when infectious agents are in
use. It serves to notify personnel and
visitors about the agents used, a point of
contact for the area, and any special pro-
tective equipment or work practices re-
quired.
Biohazard warning labels must be placed
on containers of regulated waste; refrigera-
tors and freezers containing blood or other
potentially infectious material; and other
containers used to store, transport, or ship
blood or other potentially infectious mate-
rials. Blood components that are labeled to
identify their contents and have been re-
leased for transfusion or other clinical use
are exempted.
Engineering Controls and Personal
Protective Equipment
OSHA requires that hazards be controlled
by engineering or work practices when-
ever possible. Engineering controls for
BSL-2 laboratories include limited access
to the laboratory when work is in progress
and biological safety cabinets or other
containment equipment for work that may
involve infectious aerosols or splashes.
Hand-washing sinks and eyewash stations
must be available. The work space should
be designed so that it can be easily cleaned
and bench-tops should be impervious to
water and resistant to chemicals and sol-
vents.
50 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Biological safety cabinets (BSCs) are pri-
mary containment devices for handling
moderate- and high-risk organisms. There
are three types—Class I, II, and III—with
Class III providing the highest protection to
the worker. A comparison of the features
and applications for the three classes of
cabinets is provided in Table 2-3.24 BSCs are
not required for Standard Precautions, but
centrifugation of open blood samples or
manipulation of units known to be positive
for HBsAg or HIV are examples of blood
bank procedures for which a BSC could be
useful. The effectiveness of the BSC is a
function of directional airflow inward and
downward, through a high-efficiency filter.
Efficacy is reduced by anything that dis-
rupts the airflow pattern, eg, arms moving
rapidly in and out of the BSC, rapid move-
ments behind the employee using the BSC,
downdrafts from ventilation systems, or
open laboratory doors. Care should be
taken not to block the front intake and rear
exhaust grills. Performance should be certi-
fied annually.25
Injuries from contaminated needles and
other sharps continued to be a major con-
cern in health-care settings even after the
Bloodborne Pathogens Standard went into
effect. In 2001, OSHA revised the standard
to include reference to engineered sharps
injury protections and needleless systems.26
It requires that employers implement ap-
propriate new control technologies and safer
medical devices in their Exposure Control
Plan and that they solicit input from their
employees to identify, evaluate, and select
engineering and work practice controls. Ex-
amples of safer devices are needleless sys-
tems and self-sheathing needles in which
the sheath is an integral part of the device.
Decontamination
Reusable equipment and work surfaces
that may be contaminated with blood re-
quire daily cleaning and decontamination.
Obvious spills on equipment or work sur-
faces should be cleaned up immediately;
routine wipe-downs with disinfectant
should occur at the end of each shift or on
a regular basis that provides equivalent
safety. Equipment that is exposed to blood
or other potentially infectious material
must be decontaminated before servicing
or shipping. When decontamination of all
or a portion of the equipment is not feasi-
ble, a biohazard label stating which por-
tions remain contaminated should be at-
tached before servicing or shipping.
Choice of Disinfectants
The Environmental Protection Agency
(EPA) maintains a list of chemical prod-
ucts that have been shown to be effective
antimicrobial disinfectants.27 (See http://
www.epa.gov/oppad001/chemreginex.htm
for a current list.) The Association for Pro-
fessionals in Infection Control and Epide-
miology also publishes a guideline to as-
sist health-care professionals in their
decisions involving judicious selection
and proper use of specific disinfectants.28
For facilities covered under the Blood-
borne Pathogens Rule, OSHA allows the
use of EPA-registered tuberculocidal dis-
infectants, EPA-registered disinfectants
that are effective against both HIV and
HBV, and/or a diluted bleach solution to
decontaminate work surfaces.23
Before selecting a product, workers
should consider several factors. Among
them are the type of material or surface to
be treated, the hazardous properties of the
chemical such as corrosiveness, and the
level of disinfection required. After select-
ing a product, procedures need to be writ-
ten to ensure effective and consistent
cleaning and treatment of work surfaces.
Some factors to consider for effective de-
Chapter 2: Facilities and Safety 51
Copyright © 2005 by the AABB. All rights reserved.

52 AABB Technical Manual
Table 2-3. Comparison of Class I, II, and III Biological Safety Cabinets*
Main Features Intended Use Common Applications
Class I Unfiltered room air is drawn into the cabi-
net. Inward airflow protects personnel
from exposure to materials inside the
cabinet. Exhaust is HEPA filtered to pro-
tect the environment. Maintains airflow at
a minimum velocity of 75 linear feet per
minute (lfpm) across the front opening
(face velocity).
Personal and environ-
mental protection
To enclose equipment (eg, centrifuges) or
procedures that may generate aerosols
Class II (General-
applies to all
types of Class II
cabinets)
Uses laminar flow (air moving at a constant
velocity in one direction along parallel
lines). Room air is drawn into the front
grille. HEPA filtered air is forced down-
ward in a laminar flow to minimize
cross-contamination of materials in the
cabinet. Exhaust is HEPA filtered.
Personal, environmen-
tal, and product pro-
tection
Work with microorganisms assigned to
biosafety levels 1, 2, or 3
Handling of products where prevention of
contamination is critical, such as cell cul-
ture propagation or manipulation of
blood components in an open system
Class II, A 75% of air is recirculated after passing
through a HEPA filter. Face velocity = 75
lfpm.
See Class II, general See Class II, general
Class II, B1 70% of air exits through the rear grille, is
HEPA filtered, and then discharged from
the building. The other 30% is drawn into
the front grille, HEPA filtered, and
recirculated. Face velocity = 100 lfpm.
See Class II, general Allows for safe manipulation of small quan-
tities of hazardous chemicals and
biologics
Copyright © 2005 by the AABB. All rights reserved.

Chapter 2: Facilities and Safety 53
Class II, B2 100% of air is exhausted; none is
recirculated. A supply blower draws air
from the room or outside and passes it
through a HEPA filter to provide the
downward laminar flow. Face velocity =
100 lfpm.
See Class II, general Provides both chemical and biological con-
tainment. More expensive to operate be-
cause of the volume of conditioned room
air being exhausted.
Class II, B3 Similar in design to Type A, but the system
is ducted and includes a negative pres-
sure system to keep any possible con-
tamination within the cabinet. Face veloc-
ity = 100 lfpm.
See Class II, general Allows for safe manipulation of small quan-
tities of hazardous chemicals and
biologics
Class III Cabinet is airtight. Materials are handled
with rubber gloves attached to the front
of the cabinet. Supply air is HEPA fil-
tered. Exhaust air is double HEPA filtered
or may have one filter and an air incinera-
tor. Materials are brought in and out of
the cabinet either through a dunk tank or
a double-door pass-through box that can
be decontaminated. Cabinet is kept under
negative pressure.
Maximum protection to
personnel and envi-
ronment.
Work with biosafety level 4 microorganisms
*Data from the US Department of Health and Human Services.24
Copyright © 2005 by the AABB. All rights reserved.
contamination include the contact time,
thetypeofmicroorganisms,thepresenceof
organic matter, and the concentration of
the chemical agent. Workers should review
the basic information on decontamination
and follow the manufacturer’s instructions.
Storage
Hazardous materials must be segregated
and areas for different types of storage
must be clearly demarcated. Blood must
be protected from unnecessary exposure
to other materials and vice versa. If trans-
fusion products cannot be stored in a sep-
arate refrigerator from reagents, specimens,
and unrelated materials, areas within the
refrigerator must be clearly labeled, and
extra care must be taken to reduce the
likelihood of spills and other accidents.
Storage areas must be kept clean and or-
derly; food or drink is never allowed where
biohazardous materials are stored.
Personal Protective Equipment
Where hazards cannot be eliminated, OSHA
requires employers to provide appropriate
PPE and clothing, and to clean, launder,
or dispose of PPE at no cost to their em-
ployees.10 Standard PPE and clothing in-
clude uniforms, laboratory coats, gloves,
face shields, masks, and safety goggles. In-
dications and guidelines for their use are
discussed in Appendix 2-2.
Safe Work Practices
Safe work practices appropriate for Stan-
dard Precautions include the following:
■Wash hands after touching blood,
body fluids, secretions, excretions, and
contaminated items, whether or not
gloves are worn.
■Wear gloves when touching blood,
body fluids, secretions, excretions, and
contaminated items, and change them
between tasks.
■Wear a mask and eye protection or a
face shield during activities that are
likely to generate splashes or sprays
of blood, body fluids, secretions, and
excretions.
■Wear a gown during activities that
are likely to generate splashes or
sprays of blood, body fluids, secre-
tions, or excretions.
■Handle soiled patient-care equipment
in a manner that prevents expo-
sures; ensure that reusable equip-
ment is not used for another patient
until it has been cleaned and repro-
cessed appropriately; and ensure
that single-use items are discarded
properly.
■Ensure that adequate procedures are
defined and followed for the routine
care, cleaning, and disinfection of
environmental surfaces and equip-
ment.
■Handle soiled linen in a manner that
prevents exposures.
■Handle needles, scalpels, and other
sharp instruments or devices in a
manner that minimizes the risk of
exposure.
■Use mouthpieces, resuscitation bags,
or other ventilation devices as an al-
ternative to mouth-to-mouth resus-
citation methods.
■Place in a private room those pa-
tients who are at risk of contaminat-
ing the environment or who are not
able to maintain appropriate hy-
giene (eg, tuberculosis).
Laboratory Biosafety Precautions
Several factors need to be considered when
assessing the risk of blood exposures among
laboratory personnel. Some factors in-
clude the number of specimens processed,
personnel behaviors, laboratory tech-
niques, and type of equipment.29 The lab-
54 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
oratory director may wish to institute
BSL-3 practices for procedures that are
considered to be higher risk than BSL-2.
When there is doubt whether an activity is
BSL-2 or BSL-3, the safety precautions for
BSL-3 should be followed. BSL-2 precau-
tions that are applicable to the laboratory
setting are summarized in Appendix 2-3.
Considerations for the Donor Room
The Bloodborne Pathogen Standard ac-
knowledges a difference between hospital
patients and healthy donors, in whom the
prevalence of infectious disease markers
is significantly lower. The employer in a
volunteer blood donation facility may de-
termine that routine use of gloves is not
required for phlebotomy as long as10:
■The policy is periodically reevaluated.
■Glovesaremadeavailabletothose
whowanttousethem,anduseisnot
discouraged.
■Gloves are required when an em-
ployee has cuts, scratches, or breaks
in skin; when there is a likelihood
that contamination will occur; while
drawing autologous units; while per-
forming therapeutic procedures;
and during training in phlebotomy.
Procedures used in the donation of blood
should be assessed for risks of biohazard-
ous exposures and risks inherent in work-
ing with a donor or patient. Some proce-
dures are more likely to cause injury than
others, such as using lancets for finger
puncture, handling capillary tubes, crush-
ing vials for arm cleaning, handling any un-
sheathed needle, cleaning scissors, and giv-
ing cardiopulmonary resuscitation.
In some instances, it may be necessary
to collect blood from donors known to pose
a high risk of infectivity (eg, collection of
autologous blood or Source Plasma for the
production of other products such as vac-
cines). The Food and Drug Administration
(FDA) provides guidance for collecting
blood from such “high-risk” donors.30 The
most recent regulations and guidelines
should be consulted for changes or addi-
tions.
Emergency Response Plan
Blood Spills
Every facility handling blood should be
prepared for spills in advance. Table 2-4
lists steps to be taken when a spill occurs.
Cleanup is easier when preparation in-
cludes the following elements:
■Design work areas so that cleanup is
relatively simple.
■Prepareaspillkitorcartthatcon
-
tains all necessary supplies and equip-
ment with instructions for their use.
Place it near areas where spills are
anticipated.
■Assign responsibility for kit/cart main-
tenance, spill handling, record-keep-
ing, and review of significant inci-
dents.
■Train personnel in cleanup proce-
dures and reporting of significant in-
cidents.
Biohazardous Waste
Medical waste is defined as any waste
(solid, semisolid, or liquid) generated in
the diagnosis, treatment, or immunization
of human beings or animals in related re-
search, production, or testing of biologics.
Infectious waste includes disposable equip-
ment, articles, or substances that may
harbor or transmit pathogenic organisms
or their toxins. In general, infectious
waste should either be incinerated or de-
contaminated before disposal in a sani-
tary landfill. Blood and components,
suctioned fluids, excretions, and secre-
tions may be carefully poured down a
drain connected to a sanitary sewer if
state law allows. Sanitary sewers may also
Chapter 2: Facilities and Safety 55
Copyright © 2005 by the AABB. All rights reserved.

be used to dispose of other potentially in-
fectious wastes that can be ground and
flushed into the sewer. State and local
health departments should be consulted
about laws and regulations on disposal of
biological waste into the sewer.
Laboratories should clearly define what
will be considered hazardous waste. For ex-
ample, in the blood bank items contami-
nated with liquid or semiliquid blood are
biohazardous. Items contaminated with
dried blood are considered hazardous if
there is potential for the dried material to
flake off during handling. Contaminated
sharps are always considered hazardous
because of the risk for percutaneous injury.
However,itemssuchasusedgloves,swabs,
plastic pipettes with excess liquid removed,
or gauze contaminated with small droplets
of blood may be considered nonhazardous
if the material is dried and will not be
released into the environment during han-
dling.
Guidelines for Biohazardous Waste Dis-
posal. Employees must be trained before
handling or disposing of biohazardous
waste, even if it is packaged. The following
disposal guidelines are recommended31:
■Identify biohazardous waste consis-
tently; red seamless plastic bags (at
least 2 mil thick) or containers carry-
ing the biohazard symbol are recom-
mended.
■Place bags in a protective container
with closure upward to avoid break-
age and leakage during storage or
transport.
■When transported over public roads,
the waste must be prepared and
shipped according to US Department
of Transportation regulations.
■Discard sharps (eg, needles, broken
glass, glass slides, wafers from sterile
connecting devices) in rigid, punc-
ture-proof, leakproof containers.
56 AABB Technical Manual
Table 2-4. Blood Spill Cleanup
■Contain the spill if possible.
■Evacuate the area for 30 minutes if an aerosol has been created.
■Post warnings to keep the area clear.
■Remove clothing if it is contaminated.
■If the spill occurs in the centrifuge, turn the power off immediately and leave the cover closed
for 30 minutes. The use of overwraps helps prevent aerosolization and helps contain the spill.
■Wear appropriate protective clothing and gloves. If sharp objects are involved, gloves must be
puncture-resistant, and a broom or other instrument should be used during cleanup to avoid
injury.
■Use absorbent material to mop up most of the liquid contents.
■Clean the spill area with detergent.
■Flood the area with disinfectant and use it as described in the manufacturer’s instructions. Allow
adequate contact time with the disinfectant.
■Wipe up residual disinfectant if necessary.
■Dispose of all materials safely in accordance with biohazard guidelines. All blood-contaminated
items must be autoclaved or incinerated.
Copyright © 2005 by the AABB. All rights reserved.
■Put liquids only in leakproof, un-
breakable containers.
■Do not compact waste materials.
Storage areas for infectious material
must be secured to reduce accident risk.
Infectious waste must never be placed in
the public trash collection system. Most fa-
cilities hire private carriers to decontami-
nate and dispose of infectious or hazardous
waste. Contracts with these companies
should include disclosure of the risks of
handling the waste by the facility, and an
acknowledgment by the carrier that all fed-
eral, state, and local laws for biohazardous
(medical) waste transport, treatment, and
disposal are known and followed.
Treating Infectious or Medical Waste.
Facilities that incinerate hazardous waste
must comply with EPA standards of perfor-
mance for new stationary sources and
emission guidelines for existing sources.32
In this regulation, a hospital/medical/in-
fectious waste incinerator (HMIWI) is any
device that combusts any amount of hospi-
tal waste or medical/infectious waste.
Decontamination of biohazardous waste
by autoclaving is another common method
for decontamination/inactivation of blood
samples and blood components. The fol-
lowing elements are considered in deter-
mining processing time for autoclaving:
■Size of load being autoclaved.
■Type of packaging of item(s) being
autoclaved.
■Density of items being autoclaved.
■Number of items in single autoclave
load.
■Placement of items in the autoclave,
to allow for steam penetration.
It is useful to place a biological indicator
in the center of loads that vary in size and
contents to evaluate optimal steam pene-
tration times. The EPA provides detailed in-
formation about choosing and operating
such equipment.31
Longer treatment times are needed for
sterilization, but decontamination requires
a minimum of 1 hour. A general rule is to
process 1 hour for every 10 pounds of waste
being processed. Usually, decontaminated
laboratory wastes can be disposed of as
nonhazardous solid wastes. Staff should
check with the local solid waste authority to
ensure that the facility is in compliance
with the regulations for their area. Waste
containing broken glass or other sharp
items should be disposed of in a method
consistent with policies for the disposal of
other sharp or potentially dangerous mate-
rials.
Chemical Safety
One of the most effective preventive mea-
sures a facility can take to reduce hazard-
ous chemical exposure is to evaluate the
use of alternative nonhazardous chemi-
cals whenever possible. A review of order-
ing practices of hazardous chemicals can
result in the purchase of smaller quanti-
ties of hazardous chemicals, thus reduc-
ing the risk of storing excess chemicals
and later dealing with the disposal of
these chemicals.
OSHA requires that facilities using haz-
ardous chemicals develop a written Chemi-
cal Hygiene Plan (CHP) and that the plan
be accessible to all employees. The CHP
should outline procedures, equipment,
personal protective equipment, and work
practices that are capable of protecting em-
ployees from hazardous chemicals used in
the facility.11,16 This plan must also provide
assurance that equipment and protective
devices are functioning properly and that
criteria to determine implementation and
maintenance of all aspects of the plan are
in control. Employees must be informed of
all chemical hazards in the workplace and
be trained to recognize chemical hazards,
Chapter 2: Facilities and Safety 57
Copyright © 2005 by the AABB. All rights reserved.
to protect themselves when working with
these chemicals, and where to find infor-
mation on particular hazardous chemicals.
Appendix 2-4 provides an example of a haz-
ardous chemical data safety sheet that may
be used in the CHP. Safety audits and an-
nual reviews of the CHP are important con-
trol steps to help ensure that safety prac-
tices comply with the policies set forth in
the CHP and that the CHP is up to date.
Establishing a clear definition of what
constitutes hazardous chemicals is some-
times difficult. Generally, hazardous chemi-
cals are those that pose a significant health
risk if an employee is exposed to them or
pose a significant physical risk, such as fire
or explosion, if handled or stored improp-
erly. Categories of health and physical
hazards are listed in Tables 2-5 and 2-6. Ap-
pendix 2-5 lists examples of hazardous che-
micals that may be found in the blood
bank.
The facility should identify a qualified
chemical hygiene officer to be responsible
for determining guidelines for hazardous
materials.16 The chemical hygiene officer is
also accountable for monitoring and docu-
menting accidents and initiating process
change as needed.
Training
Initial training is required for all employ-
ees who may be exposed to hazardous
chemicals—before they begin work in an
area where hazards exist. If an individual
has received prior training, it may not be
necessary to retrain them, depending on
the employer’s evaluation of the new em-
ployee’s level of knowledge. New employee
training is likely to be necessary regarding
such specifics as the location of the rele-
vant Material Safety Data Sheets (MSDS),
details of chemical labeling, the personal
protective equipment to be used, and
site-specific emergency procedures.
Training must be provided whenever a
new physical or health hazard is introduced
into the workplace, but not for each new
chemical that falls within a particular haz-
ard class.11 Forexample,ifanewsolventis
brought into the workplace and it has haz-
58 AABB Technical Manual
Table 2-5. Categories of Health Hazards
Hazard Definition
Carcinogens Cancer-producing substances
Irritants Agents causing irritations (edema, burning, etc) to skin or mu-
cous membranes upon contact
Corrosives Agents causing destruction of human tissue at the site of con-
tact
Toxic or highly toxic agents Substances causing serious biologic effects following inhala-
tion, ingestion, or skin contact with relatively small amounts
Reproductive toxins Chemicals that affect reproductive capabilities, including chro-
mosomal damages and effects on fetuses
Other toxins Hepatotoxins, nephrotoxins, neurotoxins, agents that act on the
hematopoietic systems, and agents that damage the lungs,
skin, eyes, or mucous membranes
Copyright © 2005 by the AABB. All rights reserved.

ards similar to existing chemicals for which
training has already been conducted, then
the employer need only make employees
aware of the new solvent’s hazard category
(eg, corrosive, irritant). However, if the
newly introduced solvent is a suspected
carcinogen and carcinogenic hazard train-
ing has not been provided before, then new
training must be conducted for employees
with potential exposure. Retraining is ad-
visable as often as necessary to ensure that
employees understand the hazards, partic-
ularly the chronic and specific target-organ
health hazards, linked to the materials with
which they work.
Hazard Identification and Communication
Hazard Communication
Employers must prepare a comprehensive
hazard communication program for all
areas using hazardous chemicals to com-
plement the CHP and to “ensure that the
hazards of all chemicals produced or im-
ported are evaluated, and that information
concerning their hazards is transmitted to
employers and employees.”11 The pro-
gram should include labeling hazardous
chemicals, when and how to post warning
labels for chemicals, managing MSDS re-
ports for hazardous chemicals in the facil-
ities, and employee training.
Safety materials made available to em-
ployees should include:
■The facility’s written CHP.
■The facility’s written program for
hazard communication.
■Identification of work areas where
hazardous chemicals are located.
■Required list of hazardous chemicals
and their MSDS. (It is the responsi-
bility of the facility to determine
which chemicals may present a haz-
ard to employees. This determina-
tion should be based on the quantity
of chemical used; the physical prop-
erties, potency, and toxicity of the
chemical; the manner in which the
chemical is used; and the means
available to control the release of, or
exposure to, the chemical.)
Hazardous Chemical Labeling and Signs
The Hazard Communication Standard re-
quires manufacturers of chemicals and
hazardous materials to provide the user
with basic information about the hazards
of these materials through product label-
ing and Material Safety Data Sheets.11 Em-
Chapter 2: Facilities and Safety 59
Table 2-6. Categories of Physical Hazards
Hazard Definition
Combustible or flammable
chemicals
Chemicals that can burn (includes combustible and flammable
liquids, solids, aerosols, and gases)
Compressed gases A gas or mixture of gases in a container under pressure
Explosives Unstable or reactive chemicals that undergo violent chemical
change at normal temperatures and pressure
Unstable (reactive) chemicals Chemicals that could be self-reactive under conditions of
shocks, pressure, or temperature
Water-reactive chemicals Chemicals that react with water to release a gas that is either
flammable or presents a health hazard
Copyright © 2005 by the AABB. All rights reserved.
ployers are required to provide the follow-
ing to employees who are expected to work
with these hazardous materials: informa-
tion about the hazards of the materials,
how to read the labeling, how to interpret
symbols and signs on the labels, and how
to read and use the MSDS. Table 2-7 lists
theelementstobeincludedinanMSDS.
At a minimum, hazardous chemical con-
tainer labels must include the name of the
chemical, the name and address of the
manufacturer, hazard warnings, labels,
signs, placards, and other forms of warning
to provide visual reminders of specific haz-
ards. The label may refer to the MSDS for
additional information. Labels applied by
the manufacturer must remain on contain-
ers. The user may add storage requirements
and dates of receipt, opening, and expira-
tion. If chemicals are aliquotted into sec-
ondary containers, the secondary container
must be labeled with the name of the che-
mical and appropriate hazard warnings.
Additional information such as precaution-
ary measures, concentration if applicable,
and date of preparation are helpful but not
mandatory. It is a safe practice to label all
containers with the content, even water.
Transfer containers used for temporary
storage need not be labeled if the person
performing the transfer retains control and
intends them for immediate use. Informa-
tion regarding acceptable standards for
hazard communication labeling is provided
by the NFPA33 and the National Paint and
Coatings Association.34
Signs meeting OSHA requirements must
be posted in areas where hazardous chemi-
cals are used. Decisions on where to post
warning signs are based on the manufac-
turer’s recommendations on the chemical
hazards, the quantity of the chemical in the
room or laboratory, and the potency and
toxicity of the chemical.
Material Safety Data Sheets
The MSDS identifies the physical and
chemical properties of a hazardous chem-
ical (eg, flash point, vapor pressure), its
physical and health hazards (eg, potential
for fire, explosion, signs and symptoms of
exposure), and precautions for safe han-
dling and use. Specific instructions in an
individual MSDS take precedence over
60 AABB Technical Manual
Table 2-7. Required Elements of a Material Safety Data Sheet
■Identity of product as it appears on label
■Chemical and common name(s) of all hazardous ingredients
■Physical/chemical characteristics
■Fire and explosion hazard data
■Reactivity data
■Health hazard data, including primary route(s) of entry and exposure limits
■Precautions for safe handling and use
■Exposure control measures
■Emergency and first aid procedures
■Manufacturer information, MSDS revision date
Copyright © 2005 by the AABB. All rights reserved.
generic information in the Hazardous Ma-
terials (HAZMAT) program.
Employers must maintain copies of the
required MSDS in the workplace for each
hazardous chemical and must ensure that
theyarereadilyaccessibleduringeach
work shift to employees when they are in
their work areas. When household con-
sumer products are used in the workplace
in the same manner that a consumer would
use them, ie, where the duration and fre-
quency of use (and therefore exposure) are
not greater than those the typical consumer
would experience, OSHA does not require
that an MSDS be provided to purchasers.
However, if exposure to such products ex-
ceeds that normally found in consumer ap-
plications, then employees have a right to
know about the properties of such hazard-
ous chemicals. OSHA does not require or
encourage employers to maintain an MSDS
for nonhazardous chemicals.
Engineering Controls and Personal
Protective Equipment
Guidelines for laboratory areas in which
hazardous chemicals are used or stored
must be established. Physical facilities,
especially ventilation, must be adequate
for the nature and volume of work con-
ducted. Chemicals must be stored accord-
ing to chemical compatibility (eg, corrosives,
flammables, oxidizers, etc) and in mini-
mal volumes. Bulk chemicals should be
kept outside work areas. NFPA standards
and others provide guidelines for proper
storage.3,33,35
Chemical fume hoods are recommended
for use with organic solvents, volatile liq-
uids, and dry chemicals with a significant
inhalation hazard.3Although constructed
with safety glass, most fume hood sashes
are not designed as safety shields. Hoods
should be positioned in an area where
there is minimal foot traffic to avoid dis-
rupting the airflow and compromising the
containment field.
Personal protective equipment that may
be provided depending on the hazardous
chemicals used includes chemical resistant
gloves and aprons, shatterproof safety gog-
gles, and respirators.
Emergency showers should be available
to areas where caustic, corrosive, toxic,
flammable, or combustible chemicals are
used.3,36 Thereshouldbeunobstructedac
-
cess, within 10 seconds, from the areas
where hazardous chemicals are used. Safety
showers should be periodically flushed and
tested for function, and associated floor
drains should be checked to ensure that
drain traps remain filled with water.
Safe Work Practices
Hazardous material should not be stored
or transported in open containers. Con-
tainers and their lids or seals should be
designed to prevent spills or leakage in all
reasonably anticipated conditions. Con-
tainers should be able to safely store the
maximum anticipated volume and should
be easy to clean. Surfaces should be kept
clean and dry at all times. When working
with a chemical fume hood, all materials
should be kept at a distance of at least six
inches behind the face opening; the verti-
cal sliding sash should be positioned at
the height specified on the certification
sticker. The airfoil, baffles and rear venti-
lation slot must not be blocked. Appendix
2-6 lists specific chemicals and sugges-
tions on how to work with them safely.
Emergency Response
The time to prepare for a chemical spill is
before a spill occurs. A comprehensive
employee training program should pro-
vide the employee with all tools necessary
to act responsibly at the time of a chemi-
Chapter 2: Facilities and Safety 61
Copyright © 2005 by the AABB. All rights reserved.
cal spill. The employee should know re-
sponse procedures, be able to assess the
severity of a chemical spill, know or be
able to quickly look up the basic physical
characteristics of the chemicals, and know
where to find emergency response phone
numbers. The employee should be able
to: assess, stop, and confine the spill; ei-
ther clean up the spill or call for a spill
clean-up team; and follow up on the re-
port of the spill. The employee must know
when to ask for assistance, when to iso-
late the area, and where to find cleanup
materials.
Chemical spills in the workplace can be
categorized as follows37:
■Incidental releases are spills that are
limited in quantity and toxicity and
pose no significant safety or health
hazard to the employee. They may
be safely cleaned up by the employ-
ees familiar with the hazards of the
chemical involved in the spill. Waste
from the cleanup may be classified
as hazardous and must be disposed
of in the proper fashion. Appendix
2-7 describes appropriate responses
to incidental spills.
■Releases that may be incidental or
may require an emergency response
are spills that may pose an exposure
risk to the employees depending
upon the circumstances. Consider-
ations such as the hazardous sub-
stance properties, the circumstances
of release, and mitigating factors
play a role in determining the ap-
propriate response. The facility’s
emergency response plan should
provideguidanceinhowtodeter
-
mine whether the spill is incidental
or requires an emergency response.
■Emergency response releases are
spills that pose a threat to health and
safety regardless of the circum-
stances surrounding their release.
The spill may require evacuation of
theimmediatearea.Theresponsetyp
-
ically comes from outside the imme-
diate release area by personnel trained
as emergency responders. These
spills include but are not limited to:
immediate danger to life or health,
serious threat of fire or explosion,
and high levels of toxic substances.
Appendix 2-8 addresses the manage-
ment of hazardous chemical spills. Spill
cleanup kits or carts tailored to the specific
hazards present should be available in each
area. These may contain the following: rub-
ber gloves and aprons, shoe covers, goggles,
suitable aspirators, general absorbents,
neutralizingagents,broom,dustpan,ap
-
propriate trash bags or cans for waste dis-
posal, and cleanup directions. Chemical
absorbents such as clay absorbents or spill
blankets can be used for cleaning up a
number of chemicals and thus may be eas-
ier for the employee to use in spill situa-
tions.
With any spill of a hazardous chemical,
but especially with a carcinogenic agent, it
is essential to refer to the MSDS and to con-
tact a designated supervisor or designee
trained to handle these spills and hazard-
ous waste disposal.3Facility environmental
health and safety personnel also can offer
assistance. The employer must assess the
extent of the employee’s exposure. After an
exposure, the employee must be given an
opportunity for medical consultation to
determine the need for a medical examina-
tion.
Another source of a workplace hazard is
the unexpected release of hazardous vapors
into the environment. OSHA has set limits
for exposure to hazardous vapors from
toxic and hazardous substances.38 The po-
tential risk associated with the chemical is
determined by the manufacturer and listed
on the MSDS. See Table 2-8 for a listing of
the limits of exposure.
62 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chemical Waste Disposal
Most laboratory chemical waste is consid-
ered hazardous and is regulated by the
EPA through the Resource Conservation
and Recovery Act (42 U.S.C. § 6901 et seq,
1976). This regulation specifies that haz-
ardous waste can only be legally disposed
of at an EPA-approved disposal facility.
Disposal of chemical waste into a sanitary
sewer is regulated by the Clean Water Act
(33 U.S.C. § 1251 et seq, 1977), and most
states have strict regulations concerning
disposal of chemicals in the water system.
Federal and applicable state regulations
should be consulted when setting up and
reviewing facility waste disposal policies.
Radiation Safety
Radiation can be defined as energy in the
form of waves or particles emitted and
propagated through space or a material
medium. Gamma rays are electromag-
netic radiation, whereas alpha and beta
emitters are examples of particulate radi-
ation. The presence of radiation in the
blood bank, either from radioisotopes
used in laboratory testing or from self-
contained blood irradiators, requires ad-
ditional precautions and training.3,39
Radiation Measurement Units
The measurement unit quantifying the
amount of energy absorbed per unit mass
of tissue is the Gray (Gy) or rad (radiation
absorbed dose); 1 Gy equals 100 rads.
Dose equivalency measurements are
more useful than simple energy measure-
ments because they take into account the
effectiveness of the different types of radia-
tiontocausebiologiceffects.Theabilityof
radiation to cause damage is assigned a
number called a quality factor (QF). For ex-
ample, exposure to a given amount of alpha
particles (QF = 20) is far more damaging
than exposure to an equivalent amount of
gamma rays (QF = 1). The common unit of
measurement for dose equivalency is the
rem (rad equivalent man). To obtain dose
from a particular type of radiation in rem,
multiply the number of rad by the quality
factor (rad ×QF = rem). Because the quality
factor for gamma rays, x-rays, and most
betaparticlesis1,thedoseinradisequal
to the dose in rem for these types of radia-
tion.
Biologic Effects of Radiation
Anyharmtotissuebeginswiththeab
-
sorption of radiation energy and subse-
quent disruption of chemical bonds. Mol-
Chapter 2: Facilities and Safety 63
Table 2-8. Regulatory Limits for Exposure to Toxic and Hazardous Vapors32
Limit Definition
Permissible exposure
limit
The maximum concentration of vapors in parts per million (ppm) that
an employee may be exposed to in an 8-hour day/40-hour work week.
Short-term exposure
limit
The maximum allowable concentration of vapors that an employee
may be exposed to in a 15-minute period, with a maximum of four ex-
posures per day allowed with at least 1 hour between each.
Ceiling limit The maximum concentration of vapors that may not be exceeded in-
stantaneously at any time.
Copyright © 2005 by the AABB. All rights reserved.
ecules and atoms become ionized and/or
excited by absorbing this energy. The “di-
rect action” path leads to radiolysis or for-
mation of free radicals that, in turn, alter
the structure and function of molecules in
the cell.
Molecular alterations can cause cellular
or chromosomal changes, depending upon
the amount and type of radiation energy
absorbed. Cellular changes can manifest as
a visible somatic effect, eg, erythema.
Changes at the chromosome level may
manifest as leukemia or other cancers, or
possibly as germ cell defects that are trans-
mitted to future generations.
The type of radiation, the part of the
body exposed, the total absorbed dose, and
thedoserateinfluencebiologicdamage.
The total absorbed dose is the cumulative
amount of radiation absorbed in the tissue.
Thegreaterthedose,thegreaterthepoten-
tial for biologic damage. Exposure can be
acute or chronic. The low levels of ionizing
radiation likely to occur in blood banks
should not pose any detrimental risk.40-43
Regulations
TheNRCcontrolsuseofradioactivema
-
terials by establishing licensure require-
ments. States and municipalities may also
have requirements for inspection and/or
licensure. The type of license for using ra-
dioisotopes or irradiators will depend on
the scope and magnitude of the use of ra-
dioactivity. Facilities should contact the
NRC and appropriate state agencies for li-
cense requirements and application as
soon as such activities are proposed.
NRC-licensed establishments must have
a qualified radiation safety officer who is re-
sponsible for establishing personnel pro-
tection requirements and for proper dis-
posal and handling of radioactive materials.
Specific radiation safety policies and proce-
dures should address dose limits, employee
training, warning signs and labels, shipping
and handling guidelines, radiation moni-
toring, and exposure management. Emer-
gency procedures must be clearly defined
and readily available to staff.
Exposure Limits
The NRC sets standards for protection
against radiation hazards arising from li-
censed activities, including dose limits.8
These limits, or maximum permissible
dose equivalents, are a measure of the ra-
diation risk over time and serve as stan-
dards for exposure. The occupational total
effective-dose-equivalent limit is 5 rem/
year. The shallow dose equivalent (skin) is
50 rem/year, the extremity dose equiva-
lent limit is 50 rem/year, and the eye dose
equivalent limit is 15 rem/year.8,41 Dose
limits to an embryo/fetus must not ex-
ceed 0.5 rem during the pregnancy.8,41,44
Employers are expected not only to main-
tain radiation exposure below allowable
limits, but also to keep exposure levels as
far below these limits as can reasonably be
achieved.
Radiation Monitoring
Monitoring is essential for early detection
and prevention of problems due to radia-
tion exposure. It is used to evaluate the
environment, work practices, and proce-
dures, and to comply with regulations and
NRC licensing requirements. Monitoring is
accomplished with the use of dosimeters,
bioassay, survey meters, and wipe tests.3
Dosimeters, such as film or thermo-
luminescent badges and/or rings, measure
personnel radiation doses. The need for do-
simeters depends on the amount and type
of radioactive materials in use; the facility
radiation safety officer will determine indi-
vidual dosimeter needs. Film badges must
64 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
be changed at least quarterly and in some
instances monthly, protected from high
temperature and humidity, and stored at
work away from sources of radiation.
Bioassay, such as thyroid and whole
body counting or urinalysis, may be used to
determine if there is radioactivity inside the
body and, if so, how much. If necessary,
bioassays are usually performed quarterly
and after an incident where accidental in-
take may have occurred.
Survey meters are sensitive to low levels
of gamma or particulate radiation and pro-
vide a quantitative assessment of radiation
hazard. They can be used to monitor stor-
age areas for radioactive materials or wastes,
testing areas during or after completion of a
procedure, and packages or containers of
radioactive materials. Survey meters must
be calibrated annually by an authorized
NRC licensee. Selection of appropriate me-
ters should be discussed with the radiation
safety officer.
In areas where radioactive materials are
handled, work surfaces, equipment, and
floors that may be contaminated should be
checked regularly with a wipe test. In the
wipe test, a moistened absorbent material
(the wipe) is passed over the surface and
then counted for radiation. Kits are avail-
able for this purpose. In most clinical labo-
ratories, exposure levels of radiation are
well below the limits set by federal and
state regulations.
Training
Personnel who handle radioactive materi-
als or work with blood irradiators must re-
ceive radiation safety training before be-
ginning work. This training should cover
an explanation of the presence and po-
tential hazards of radioactive materials
found in the employee’s specific work area,
general health protection issues, emer-
gency procedures, and radiation warning
signs and labels in use. Instruction in the
following is also suggested:
■NRC regulations and license condi-
tions.
■The importance of observing license
conditions and regulations and re-
porting violations or conditions of
unnecessary exposure.
■Precautions to minimize exposure.
■Interpretation of results of monitor-
ing devices.
■Requirements for pregnant workers.
■Employees’ rights.
■Documentation and record-keeping
requirements.
The need for refresher training is deter-
mined by the license agreement between
the NRC and the facility.
Engineering Controls and Personal
Protective Equipment
Although self-contained blood irradiators
present little risk to laboratory staff and
film badges are not required for routine
operation, blood establishments with ir-
radiation programs must be licensed by
the NRC.41
The manufacturer of the blood irradiator
usually accepts responsibility for radiation
safety requirements during transportation,
installation, and validation of the unit as
part of the purchase contract. The radiation
safety officer can help oversee the installa-
tion and validation processes and confirm
that appropriate training, monitoring sys-
tems, procedures, and maintenance proto-
cols are in place before use and reflect the
manufacturer’s recommendations. Sus-
pected malfunctions must be reported im-
mediately to defined facility authorities so
that appropriate actions can be initiated.
Blood irradiators should be located in
secure areas with limited access so that
only trained individuals have access. Fire
protection for the unit must also be consid-
Chapter 2: Facilities and Safety 65
Copyright © 2005 by the AABB. All rights reserved.
ered. Automatic fire detection and control
systems should be readily available in the
immediate area. Blood components that
have been irradiated are not radioactive
and pose no threat to staff or the general
public.
Safe Work Practices
Each laboratory should establish policies
and procedures for the safe use of radio-
active materials. They should include
requirements for following general labo-
ratory safety principles, appropriate stor-
age of radioactive solutions, and proper
disposal of radioactive wastes. Radiation
safety can be improved with the follow-
ing:
■Minimize time of exposure by work-
ing as efficiently as possible.
■Maximize distance from the source
of the radiation by staying as far
from the source as possible.
■Maximize shielding (eg, by using a
self-shielded irradiator or by wear-
ing lead aprons when working with
certain radioactive materials). These
requirements are usually stipulated
in the license conditions.
■Use good housekeeping practices to
minimize spread of radioactivity to
uncontrolled areas.
Emergency Response Plan
Radioactive contamination is the dis-
persal of radioactive material into or onto
areas where it is not intended; for exam-
ple, the floor, work areas, equipment,
personnel clothing, or skin. The NRC reg-
ulations state that gamma or beta radio-
active contamination cannot exceed 2200
dpm/100 cm2in the posted (restricted)
area or 220 dpm/100 cm2in an unre-
stricted area such as corridors; for alpha
emitters, these values are 220 dpm/100
cm2and 22 dpm/100 cm2, respectively.45
If a spill occurs, contaminated skin sur-
faces must be washed several times and the
radiation safety officer must be notified im-
mediately for further guidance. Others
must not be allowed to enter the area until
emergency response personnel arrive.
Waste Management
Policies for the disposal of radioactive
waste, whether liquid or solid, should be
established, with input from the radiation
safety officer and the disposal contractor,
if an approved company is used.
Liquid radioactive waste may be col-
lected into large sturdy bottles labeled with
an appropriate radiation waste tag. The
rules for separation by chemical compati-
bility apply. Bottles must be carefully stored
to protect against spillage or breakage. Dry
or solid waste may be sealed in a plastic
bag and tagged as radiation waste. The iso-
tope, activity of the isotope, and date that
the activity was measured should be placed
on the bag. Radiation waste must never be
discharged into the drain system without
prior approval of the radiation safety offi-
cer.
Shipping Hazardous
Materials
Local surface transport of blood specimens,
components, and biohazardous materials
from one facility (or part thereof) to an-
other may be made by a local approved
courier service. The safe transport of these
materialsrequiresthattheybepackaged
in such a way that the possibility of leak-
age or other release from the package un-
der normal conditions of transport does
not occur. See Method 1.1 for detailed
shipping instructions for diagnostic spec-
imens and infectious substances.
66 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Waste Management
Those responsible for safety must be con-
cerned with protecting the environment,
as well as staff. Every effort should be
made to establish facility-wide programs
to reduce solid wastes, including nonhaz-
ardous and especially hazardous wastes
(ie, biohazardous, chemical, and radia-
tion wastes). A hazardous waste reduction
program instituted at the point of use of
the material achieves several goals. It re-
duces the institutional risk for occupa-
tional exposures to hazardous agents and
“cradle to grave” liability for disposal as
well as enhances compliance with envi-
ronmental requirements to reduce pollu-
tion generated from daily operations of
the laboratory.31,46,47 These requirements
necessitate that a facility minimize pollu-
tion of the environment by the “three R’s”
(reduce, reuse, and recycle). Seeking suit-
able alternatives to the use of materials
that create hazardous waste and separat-
ing hazardous waste from nonhazardous
waste can reduce the volume of hazard-
ous waste and decrease costs for its dis-
posal.
A goal of waste management should be
to reduce to a minimum the volume of
hazardous material. Noninfectious waste
should always be separated from infectious
waste. Changes in techniques or materials,
which reduce the volume of infectious
wasteorrenderitlesshazardous,shouldbe
carefully considered and employees should
be encouraged to identify safer alternatives
wherever possible.
Facilities should check with state and lo-
cal health and environmental authorities
for current requirements for storage and
disposal of a particular multihazardous
wastebeforecreatingthatwaste.Ifthe
multihazardous waste cannot be avoided,
the volume generated should be mini-
mized. In some states, copper sulfate con-
taminated with blood is considered a multi-
hazardous waste. The disposal of this waste
poses several problems with transportation
from draw sites to a central facility to dis-
posal of the final containers. State and local
health departments must be involved in the
review of transportation and disposal prac-
tices where this is an issue, and procedures
must be developed in accordance with
their regulations as well as those of the US
Department of Transportation.
Disaster Planning
Blood banks and transfusion services should
establish action plans for uncommon but
potential dangers (eg, floods; hurricanes;
tornadoes; earthquakes; fires; explosions;
biological, chemical, or radiation emer-
gencies; structural collapse; hostage situ-
ations; bomb threats and other acts of ter-
rorism; or other events in which mass
casualties might occur). These events re-
quire a plan to ensure the safety of pa-
tients, visitors, workers, and the blood
supply. Such disasters may involve the fa-
cility alone, the surrounding area, or both,
and can be categorized by severity level:
minor impact on normal operations;
moderate to substantial reduction in op-
erations; or severe, prolonged loss of op-
erations. The JCAHO requires a plan to
address four phases of activities: mitiga-
tion, preparedness, response, and recov-
ery.4Policies and procedures may address:
■Notification procedures.
■Ongoing communication (ie, com-
mand center).
■Evacuation or relocation.
■Isolation or containment.
■Personal safety and protection.
■Provision of additional staffing.
Typically, in a disaster situation, the first
person who becomes aware of the disaster
takes immediate action and notifies others,
Chapter 2: Facilities and Safety 67
Copyright © 2005 by the AABB. All rights reserved.
either through an alarm activation system
(eg, fire alarm) or by notifying an individual
in authority, who then implements the ini-
tial response steps and contacts the facil-
ity’s disaster coordinator. Emergency tele-
phone numbers should be prominently
posted. Employees should be trained in the
facility’s disaster response policies. Because
the likelihood of being involved in an actual
disaster is minimal, drills should be con-
ducted to ensure appropriate responses
and prepare staff to act quickly. Every disas-
ter is a unique occurrence. Modifications
must be made as necessary; flexibility is
important. Once the disaster is under con-
trol and recovery is under way, actions
should be evaluated and modifications
made to the disaster plan as needed. How-
ever, the single most effective protection a
facility has against unexpected danger is the
awareness that safety-minded employees
have for their surroundings.
References
1. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005:88.
2. Laboratory Accreditation Program Labora-
tory general checklist. Chicago, IL: College of
American Pathologists Commission on Labo-
ratory Accreditation, 2002.
3. Clinical laboratory safety; approved guide-
line. NCCLS document GP17-A. National
Committee for Clinical Laboratory Standards.
Wayne, PA: NCCLS, 1996.
4. 2003 Hospital accreditation standards. Oak-
brook Terrace, IL: Joint Commission on Ac-
creditation of Healthcare Organizations, 2003.
5. 2002-2003 Standards for pathology and labo-
ratory services. Oakbrook Terrace, IL: Joint
Commission on Accreditation of Healthcare
Organizations, 2002.
6. NFPA 70 - National electrical code. Quincy, MA:
National Fire Protection Association, 2002.
7. ANSI/ASHRAE Standard 62-1999. Ventilation
for acceptable indoor air quality. Atlanta, GA:
American Society of Heating, Refrigerating,
and Air-Conditioning Engineers, Inc., 1999.
8. Code of federal regulations. Standards for
protection against radiation. Title 10 CFR
Part 20. Washington, DC: US Government
Printing Office, 2004 (revised annually).
9. Garner JS, for the CDC Hospital Infection Con-
trol Practices Advisory Committee. Guide-
lines for isolation precautions in hospitals.
Infect Control Hosp Epidemiol 1996;17:53-
80.
10. Code of federal regulations. Occupational ex-
posure to bloodborne pathogens, final rule.
Title 29 CFR Part 1910.1030. Washington, DC:
US Government Printing Office, 2004 (revised
annually).
11. Code of federal regulations. Hazard communi-
cation standard. Title 29 CFR Part 1910.1200.
Washington, DC: US Government Printing
Office, 2004 (revised annually).
12. Richmond JY, McKinney RW, eds. Biosafety in
microbiological and biomedical laboratories.
4th ed. Washington, DC: US Government
Printing Office, 1999.
13. Bernard B, ed. Musculoskeletal disorders and
workplace factors: A critical review of epide-
miologic evidence for work-related musculo-
skeletal disorders of the neck, upper extrem-
ity, and low back. NIOSH Publication 97-141.
Cincinnati, OH: US Department of Health
and Human Services, Public Health Service,
Centers for Disease Control and Prevention,
National Institute for Occupational Safety
and Health, 1997.
14. Code of federal regulations. Emergency ac-
tion plans. Title 29 CFR Part 1910.38. Wash-
ington, DC: US Government Printing Office,
2004 (revised annually).
15. Wagner KD, ed. Environmental management
in healthcare facilities. Philadelphia: WB
Saunders, 1998.
16. Code of federal regulations. Occupational ex-
posure to hazardous chemicals in laborato-
ries. Title 29 CFR Part 1910.1450. Washington,
DC: US Government Printing Office, 2004 (re-
vised annually).
17. Centers for Disease Control. Public Health
Service guidelines for the management of oc-
cupational exposures to HBV, HCV, and HIV
and recommendations for post-exposure
prophylaxis. MMWR Morb Mortal Wkly Rep
2001;50:1-52.
18. Code of federal regulations. Access to em-
ployee exposure and medical records. Title 29
CFR Part 1910.1020. Washington, DC: US
Government Printing Office, 2004 (revised
annually).
19. National Institute for Occupational Safety
and Health. NIOSH Alert: Preventing allergic
reactions to natural rubber latex in the work-
place. (June 1997) NIOSH Publication No. 97-
135. Washington, DC: National Institute for
68 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Occupational Safety and Health, 1997. [Avail-
able at http://www.cdc.gov/niosh/latexalt.html.]
20. NFPA 101: Code for safety to life from fire in
buildings and structures. Quincy, MA: Na-
tional Fire Protection Association, 2000.
21. Fowler TW, Miles KK. Electrical safety: Safety
and health for electrical trades student man-
ual. (January 2002) NIOSH Publication No.
2002-123. Washington, DC: National Institute
for Occupational Safety and Health, 2002.
22. Occupational Safety and Health Administra-
tion. OSHA technical manual: TED 1-0.15A.
Washington, DC: US Department of Labor, 1999.
23. Occupational Safety and Health Administra-
tion. Enforcement procedures for the occu-
pational exposure to bloodborne pathogens,
Directive CPL 2-2.69. Washington, DC: US
Department of Labor, 2001.
24. US Department of Health and Human Ser-
vices, CDC, and NIH. Primary containment
for biohazards: Selection, installation and use
of biological safety cabinets. (September,
1995) Bethesda, MD: National Institutes of
Health, 1995. [Available at http://www.niehs.
nih.gov/odhsb/biosafe/bsc/bsc.htm.]
25. Richmond JY. Safe practices and procedures
for working with human specimens in bio-
medical research laboratories. J Clin Im-
munoassay 1988;11:115-9.
26. Code of federal regulations. Occupational ex-
posure to bloodborne pathogens; needlestick
and other sharps injuries; final rule. Title 29 CFR
Part 1910.1030. Fed Regist 2001;66:5317-25.
27. Environmental Protection Agency. Registered
hospital disinfectants and sterilants (TS767C).
Washington, DC: Antimicrobial Program
Branch, 1992.
28. Rutala WA. APIC guideline for selection and
use of disinfectants. Am J Infect Control 1996;
24:313-42.
29. Evans MR, Henderson DK, Bennett JE. Poten-
tial for laboratory exposures to biohazardous
agents found in blood. Am J Public Health
1990;80:423-7.
30. Food and Drug Administration. Memoran-
dum: Guideline for collection of blood prod-
ucts from donors with positive tests for infec-
tious disease markers (“high risk” donors).
(October 26, 1989) Rockville, MD: CBER Of-
fice of Communication, Training, and Manu-
facturers Assistance, 1989.
31. Environmental Protection Agency. EPA guide
for infectious waste management. EPA/530-
SW-86-014. NTIS #PB86-199130. Washington,
DC: National Technical Information Service,
1986.
32. Code of federal regulations. Standards of per-
formance for new stationary sources and
emission guidelines for existing sources: Hos-
pital/medical/infectious waste incinerators.
Title 40 CFR Part 60. Washington, DC: US
Government Printing Office, 2004 (revised
annually).
33. NFPA 704—standard for the identification of
the hazards of materials for emergency re-
sponse. Quincy, MA: National Fire Protection
Association, 2001.
34. HMIS implementation manual. 3rd ed. Neenah,
WI: JJ Keller and Associates, Inc., 2001.
35. Lisella FS, Thomasston SW. Chemical safety
in the microbiology laboratory. In: Fleming
DO, Richardson JH, Tulis JJ, Vesley D, eds.
Laboratory safety, principles and practices.
2nd ed. Washington, DC: American Society
for Microbiology Press, 1995:247-54.
36. American National Standards Institute.
American national standards for emergency
eyewash and shower equipment. ANSI
Z358.1-1998. New York, NY: ANSI, 1998.
37. Occupational Safety and Health Administra-
tion. Inspection procedures for the hazard-
ous waste operations and emergency re-
sponse standard, 29 CFR 1910.120 and
1926.65, paragraph (q): Emergency response
to hazardous substance releases. OSHA direc-
tive CPL 2-2.59A. Washington, DC: US Gov-
ernment Printing Office, 1998.
38. Code of federal regulations. Air contaminants:
Toxic and hazardous substances. Title 29 CFR
Part 1910.1000. Washington, DC: US Govern-
ment Printing Office, 2004 (revised annually).
39. Cook SS. Selection and installation of self-
contained irradiators. In: Butch S, Tiehen A,
eds. Blood irradiation: A user’s guide. Bethesda,
MD: AABB Press, 1996:19-40.
40. Beir V. Health effects of exposure to low levels
of ionizing radiation. Washington, DC: Na-
tional Academy Press, 1990:1-8.
41. Regulatory Guide 8.29: Instruction concern-
ing risks from occupational radiation expo-
sure. Washington, DC: Nuclear Regulatory
Commission, 1996.
42. NCRP Report No. 115: Risk estimates for radi-
ation protection: Recommendations of the
National Council on Radiation Protection
and Measurements. Bethesda, MD: National
Council on Radiation Protection and Mea-
surements, 1993.
43. NCRP Report No. 105: Radiation protection
for medical and allied health personnel: Rec-
ommendations of the National Council on
Radiation Protection and Measurements.
Bethesda, MD: National Council on Radia-
tion Protection and Measurements, 1989.
44. NRC Regulatory Guide 8.13: Instruction con-
cerning prenatal radiation exposure. Washing-
ton, DC: Nuclear Regulatory Commission, 1999.
Chapter 2: Facilities and Safety 69
Copyright © 2005 by the AABB. All rights reserved.
45. NRC Regulatory Guide 8.23: Radiation surveys
at medical institutions. Washington, DC: Nu-
clear Regulatory Commission, 1981.
46. United States Code. Pollution Prevention Act.
42 U.S.C. § 13101 and 13102 et seq. Washing-
ton, DC: US Government Printing Office, 1990.
47. Clinical laboratory waste management. Ap-
proved Standard Doc GP5-A. Wayne, PA: Na-
tional Committee for Clinical Laboratory
Standards, 1993.
Suggested Reading
CDC Office of Biosafety. Radiation safety manual.
Atlanta, GA: Centers for Disease Control, 1992.
Disaster operations handbook: Coordinating the
nation’s blood supply during disasters and biolog-
ical events. Bethesda, MD: AABB, 2003.
Disaster plan development procedure manual. In:
Developing a disaster plan. Bethesda, MD: AABB,
1998.
Handbook of compressed gases. 3rd ed. Compressed
Gas Association. New York: Chapman and Hall, 1990.
Heinsohn PA, Jacobs RR, Concoby BA, eds. Biosafety
reference manual. 2nd ed. Fairfax, VA: American
Industrial Hygiene Association Biosafety Commit-
tee, 1995.
Liberman DF, ed. Biohazards management hand-
book. 2nd ed. New York: Marcel Dekker, Inc, 1995.
NIH guide to waste disposal. Bethesda, MD: Na-
tional Institutes of Health, 2003. [Available at http://
www.nih.gov/od/ors/ds/wasteguide.]
Prudent practices for handling hazardous chemi-
cals in laboratories. Washington, DC: National
Academy Press, 1981.
Risk management and safety procedure manual.
In: Developing a disaster plan. Bethesda, MD:
AABB, 1998.
Vesley D, Lauer JL. Decontamination, sterilization,
disinfection and antisepsis. In: Fleming DO, Rich-
ardson JH, Tulis JJ, Vesley D, eds. Laboratory
safety, principles and practices. 2nd ed. Washing-
ton, DC: American Society for Microbiology Press,
1995:219-37.
70 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 2: Facilities and Safety 71
Appendix 2-1. Safety Regulations and Recommendations Applicable to Health-Care
Settings
Agency/Organization Reference Title
Federal Regulations and Recommendations
Nuclear Regulatory Com-
mission (NRC)
10 CFR 20 Standards for Protection Against
Radiation
Guide 8.29 Instruction Concerning Risks
from Occupational Radiation
Exposure
Department of Labor, Occu-
pational Safety and
Health Administration
(OSHA)
29 CFR 1910.1030 Occupational Exposure to
Bloodborne Pathogens
29 CFR 1910.1096 Ionizing Radiation
29 CFR 1910.1200 Hazard Communication Standard
29 CFR 1910.1450 Occupational Exposure to Haz-
ardous Chemicals in Labora-
tories
Department of Transporta-
tion (DOT)
49 CFR 171-180 Hazardous Materials Regulations
Environmental Protection
Agency (EPA)
EPA Guide for Infectious Waste
Management
Centers for Disease Control
and Prevention (CDC)
Guideline for Isolation Precau-
tions in Hospitals
Food and Drug Administra-
tion (FDA)
21 CFR 606.40, 606.60,
and 606.65
Current Good Manufacturing
Practice for Blood and Blood
Components
Guideline for Collection of Blood
Products from Donors with
Positive Tests for Infectious
Disease Markers
21 CFR 801.437 User Labeling for Devices that
Contain Natural Rubber
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

72 AABB Technical Manual
Agency/Organization Reference Title
Trade and Professional Organizations
National Fire Protection
Association (NFPA)
NFPA 70 National Electrical Code
NFPA 70E Electrical Safety Requirements
for Employee Workplaces
NFPA 101 Code for Safety to Life from Fire
in Buildings and Structures
NFPA 704 Standard for the Identification of
the Hazards of Materials for
Emergency Response
National Paint and Coatings
Association
Hazardous Materials Identifica-
tion System (HMIS) Imple-
mentation Manual
International Air Traffic
Association (IATA)
Dangerous Goods Regulations
Appendix 2-1. Safety Regulations and Recommendations Applicable to Health-Care
Settings (cont'd)
Copyright © 2005 by the AABB. All rights reserved.

Chapter 2: Facilities and Safety 73
Appendix 2-2. General Guidelines for Safe Work Practices, Personal Protective
Equipment, and Engineering Controls
Uniforms and Laboratory Coats
Closed laboratory coats or full aprons over long-sleeved uniforms or gowns should be worn
when personnel are exposed to blood, corrosive chemicals, or carcinogens. The material of re-
quired coverings should be appropriate for the type and amount of hazard exposure. Plastic dis-
posable aprons may be worn over cotton coats when there is a high probability of large spills or
splashing of blood and body fluids; nitrile rubber aprons may be preferred when pouring caustic
chemicals.
Protective coverings should be removed before leaving the work area and should be discarded or
stored away from heat sources and clean clothing. Contaminated clothing should be removed
promptly, placed in a suitable container, and laundered or discarded as potentially infectious.
Home laundering of garments worn in Biosafety Level 2 areas (see below) is not permitted be-
cause unpredictable methods of transportation and handling can spread contamination, and
home laundering techniques may not be effective.1
Gloves
Gloves or equivalent barriers should be used whenever tasks are likely to involve exposure to
hazardous materials. Latex or vinyl gloves are adequate for handling most blood specimens and
chemicals (see latex allergy issues below).
Types of Gloves
Glove type varies with the task:
■Sterile gloves: for procedures involving contact with normally sterile areas of the body.
■Examination gloves: for procedures involving contact with mucous membranes, unless otherwise
indicated, and for other patient care or diagnostic procedures that do not require the use of
sterile gloves.
■Rubber utility gloves: for housekeeping chores involving potential blood contact, for instrument
cleaning and decontamination procedures, for handling concentrated acids and organic solvents.
Utility gloves may be decontaminated and reused but should be discarded if they show signs of
deterioration (peeling, cracks, discoloration) or if they develop punctures or tears.
■Insulated gloves: for handling hot or frozen material.
Indications for Use
The following guidelines should be used to determine when gloves are necessary1:
■For donor phlebotomy when the health-care worker has cuts, scratches, or other breaks in his or
her skin.
■For phlebotomy of autologous donors or patients (eg, therapeutic apheresis procedures,
intraoperative red cell collection).
■For persons who are receiving training in phlebotomy.
■When handling “open” blood containers or specimens.
■When collecting or handling blood or specimens from patients or from donors known to be
infected with a blood-borne pathogen.
■When examining mucous membranes or open skin lesions.
■When handling corrosive chemicals and radioactive materials.
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

74 AABB Technical Manual
■When cleaning up spills or handling waste materials.
■When likelihood of exposure cannot be assessed because of lack of experience with a procedure
or situation.
The Occupational Safety and Health Administration (OSHA) does not require routine use of gloves
by phlebotomists working with healthy prescreened donors or the changing of unsoiled gloves
between donors if gloves are worn.1,2 Experience has shown that the phlebotomy process is low
risk because donors have low rates of infectious disease markers. Also, exposure to blood is rare
during routine phlebotomy, and other alternatives can be utilized to provide barrier protection,
such as using a folded gauze pad to control any blood flow when the needle is removed from the
donor’s arm.
The employer whose policies and procedures do not require routine gloving should periodically
reevaluate the potential need for gloves. Employees should never be discouraged from using
gloves, and gloves should always be available.
Guidelines on Use
Guidelines for the safe use of gloves include the following3,4:
■Securely bandage or cover open skin lesions on hands and arms before putting on gloves.
■Change gloves immediately if they are torn, punctured, or contaminated; after handling high-risk
samples; or after performing a physical examination, eg, on an apheresis donor.
■Remove gloves by keeping their outside surfaces in contact only with outside and by turning the
glove inside out while taking it off.
■Use gloves only where needed and avoid touching clean surfaces such as telephones, door
knobs, or computer terminals with gloves.
■Change gloves between patient contacts. Unsoiled gloves need not be changed between donors.
■Wash hands with soap or other suitable disinfectant after removing gloves.
■Do not wash or disinfect surgical or examination gloves for reuse. Washing with surfactants may
cause “wicking” (ie, the enhanced penetration of liquids through undetected holes in the glove).
Disinfecting agents may cause deterioration of gloves.
■Use only water-based hand lotions with gloves, if needed; oil-based products cause minute
cracks in latex.
Face Shields, Masks, and Safety Goggles
Where there is a risk of blood or chemical splashes, the eyes and the mucous membranes of the
mouth and nose should be protected.5Permanent shields, fixed as a part of equipment or bench
design, are preferred, eg, splash barriers attached to tubing sealers or centrifuge cabinets. All
barriers should be cleaned and disinfected on a regular schedule.
Safety glasses alone provide impact protection from projectiles but do not adequately protect
eyes from biohazardous or chemical splashes. Full-face shields or masks and safety goggles are
recommended when permanent shields cannot be used. Many designs are commercially avail-
able; eliciting staff input on comfort and selection can improve compliance on use.
Appendix 2-2. General Guidelines for Safe Work Practices, Personal Protective
Equipment, and Engineering Controls (cont'd)
Copyright © 2005 by the AABB. All rights reserved.

Chapter 2: Facilities and Safety 75
Masks should be worn whenever there is danger from inhalation. Simple, disposable dust masks
are adequate for handling dry chemicals, but respirators with organic vapor filters are preferred
for areas where noxious fumes are produced, eg, for cleaning up spills of noxious materials. Res-
pirators should be fitted to their specific wearers and checked annually.
Hand Washing
Frequent effective hand washing is the first line of defense in infection control. Blood-borne
pathogens generally do not penetrate intact skin, so immediate removal reduces the likelihood of
transfer to a mucous membrane or broken skin area or of transmission to others. Thorough
washing of hands (and arms) also reduces the risks from exposure to hazardous chemicals and
radioactive materials.
Hands should always be washed before leaving a restricted work area, before using a biosafety
cabinet, between medical examinations, immediately after becoming soiled with blood or hazard-
ous materials, after removing gloves, and after using the toilet. Washing hands thoroughly before
touching contact lenses or applying cosmetics is essential.
OSHA allows the use of waterless antiseptic solutions for hand washing as an interim method.2
These solutions are useful for mobile donor collections or in areas where water is not readily
available for cleanup purposes. If such methods are used, however, hands must be washed with
soap and running water as soon as feasible thereafter. Because there is no listing or registration
of acceptable hand wipe products similar to the one the Environmental Protection Agency main-
tains for surface disinfectants, consumers should request data from the manufacturer to support
advertising claims.
Eye Washes
Laboratory areas that contain hazardous chemicals must be equipped with eye wash stations.3,6
Unobstructed access, within 10 seconds from the location of chemical use, must be provided for
these stations. Eye washes must operate so that both of the user’s hands are free to hold open
the eyes. Procedures and indications for use must be posted and routine function checks must be
performed. Testing eye wash fountains weekly helps ensure proper function and flushes out the
stagnant water. Portable eye wash systems are allowed only if they can deliver flushing fluid to
the eyes at a rate of at least 1.5 liters per minute for 15 minutes. They should be monitored rou-
tinely to ensure the purity of their contents.
Employees should be trained in the proper use of eye wash devices, although prevention, through
consistent and appropriate use of safety glasses or shields, is preferred. If a splash occurs, the
employee should be directed to keep the eyelids open and use the eye wash according to proce-
dures, or go to the nearest sink and direct a steady, tepid stream of water into the eyes. Solutions
other than water should be used only upon a physician’s direction.
After adequate flushing (many facilities recommend 15 minutes), follow-up medical care should
be sought, especially if pain or redness develops. Whether eye washing is effective in preventing
infection has not been demonstrated but it is considered desirable when accidents occur.
Appendix 2-2. General Guidelines for Safe Work Practices, Personal Protective
Equipment, and Engineering Controls (cont'd)
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

76 AABB Technical Manual
1. Code of federal regulations. Occupational exposure to bloodborne pathogens, final rule. Title 29 CFR
Part 1910.1030. Fed Regist 1991;56(235):64175-82.
2. Occupational Safety and Health Administration. Enforcement procedures for the occupational
exposure to bloodborne pathogens. OSHA Instruction CPL2-2.44D. Washington, DC: US Government
Printing Office, 1999.
3. Clinical and Laboratory Standards Institute. Clinical laboratory safety; approved guideline. NCCLS
document GP17-A. Wayne, PA: CLSI, 1996.
4. Food and Drug Administration. Medical glove powder report. (September 1997) Rockville, MD: Center
for Devices and Radiological Health, 1997. [Available at http://www.fda.gov/cdrh/glvpwd.html.]
5. Inspection checklist: General laboratory. Chicago, IL: College of American Pathologists, 2001.
6. American National Standards Institute. American national standards for emergency eyewash and
shower equipment. ANSI Z358.1-1998. New York: ANSI, 1998.
Appendix 2-2. General Guidelines for Safe Work Practices, Personal Protective
Equipment, and Engineering Controls (cont'd)
Copyright © 2005 by the AABB. All rights reserved.

Chapter 2: Facilities and Safety 77
Appendix 2-3. Biosafety Level 2 Precautions
Biosafety Level 2 precautions as applied in the blood establishment setting include at least the
following1,2:
■High-risk activities are appropriately segregated from lower risk activities, and the boundaries
are clearly defined.
■Bench tops are easily cleaned and are decontaminated daily with a hospital disinfectant
approved by the Environmental Protection Agency.
■Laboratory rooms have closable doors and sinks. An air system with no recirculation is
preferred, but not required.
■Workers are required to perform procedures that create aerosols (eg, opening evacuated tubes,
centrifuging, mixing, or sonicating) in a biologic safety cabinet or equivalent, or to wear masks
and goggles in addition to gloves and gowns during such procedures. (Note: Open tubes of
blood should not be centrifuged. If whole units of blood or plasma are centrifuged,
overwrapping is recommended to contain leaks.)
■Gowns and gloves are used routinely and in accordance with general safety guidelines. Face
shields or their equivalent are used where there is a risk from splashing.
■Mouth pipetting is prohibited.
■No eating, drinking, smoking, application of cosmetics, or manipulation of contact lenses occurs
in the work area. All food and drink are stored outside the restricted area, and laboratory
glassware is never used for food or drink. Personnel are instructed to avoid touching their face,
ears, mouth, eyes, or nose with their hands or other objects, such as pencils and telephones.
■Needles and syringes are used and disposed of in a safe manner. Needles are never bent,
broken, sheared, replaced in sheath, or detached from syringe before being placed in
puncture-proof, leakproof containers for controlled disposal. Procedures are designed to
minimize exposure to sharp objects.
■All blood specimens are placed in well-constructed containers with secure lids to prevent
leaking during transport. Blood is packaged for shipment in accordance with regulatory agency
requirements for etiologic agents or clinical specimens, as appropriate.
■Infectious waste is not compacted and is decontaminated before its disposal in leakproof
containers. Proper packaging includes double, seamless, tear-resistant, orange or red bags
enclosed in protective cartons. Both the carton and the bag inside display the biohazard symbol.
Throughout delivery to an incinerator or autoclave, waste is handled only by suitably trained
persons. If a waste management contractor is used, the agreement should clearly define
respective responsibilities of the staff and the contractor.
■Equipment to be repaired or submitted for preventive maintenance, if potentially contaminated
with blood, must be decontaminated before its release to a repair technician.
■Accidental exposure to suspected or actual hazardous material is reported to the laboratory
director or responsible person immediately.
1. Clinical and Laboratory Standards Institute. Clinical laboratory safety; approved guideline. NCCLS
document GP17-A. Wayne, PA: CLSI, 1996.
2. Fleming DO. Laboratory biosafety practices. In: Fleming DO, Richardson JH, Tulis JJ, Vesley D, eds.
Laboratory safety, principles and practices. 2nd ed. Washington, DC: American Society for
Microbiology Press, 1995:203-18.
Copyright © 2005 by the AABB. All rights reserved.

78 AABB Technical Manual
Appendix 2-4. Sample Hazardous Chemical Data Sheet
The following information should be a part of the procedures for use of hazardous chemicals.
Facility Identification : ________________________________________________________
Laboratory Name : ________________________________________________________
Room Number : ________________________________________________________
Name of Chemical : ________________________________________________________
Synonyms : ________________________________________________________
Chemical Abstract No.
(Case #) : ________________________________________________________
Common Name : ________________________________________________________
Primary Hazard Carcinogen: _______________________________________________
Reproductive toxin: _________________________________________
High acute toxicity: _________________________________________
Other health hazard: ________________________________________
Safety hazard: _____________________________________________
MSDS or other reference available: ____________________________
Is prior approval required for use of the chemical; if so,
by whom? ________________________________________________
General and Special Precautions:
Signs Required (Warning signs indicating presence of hazardous chemicals/operations):
____________________________________________________________________________
Storage (Secondary containment, temperature-sensitive, incompatibilities, water-reactive,
etc): ________________________________________________________________________
____________________________________________________________________________
Special Controls and Location (Fume hood, glove box, etc):____________________________
____________________________________________________________________________
Copyright © 2005 by the AABB. All rights reserved.

Chapter 2: Facilities and Safety 79
Special Equipment and Location (Vacuum line filter, liquid or other traps, special shielding):
____________________________________________________________________________
Personal Protective Equipment (Glove type, eye protection, special clothing, etc): __________
____________________________________________________________________________
Emergency Procedures:
Spill or release: _______________________________________________________________
Fire:_________________________________________________________________________
Decontamination procedures: ____________________________________________________
Disposal procedures: ___________________________________________________________
____________________________________________________________________________
____________________________________________________________________________
Appendix 2-4. Sample Hazardous Chemical Data Sheet (cont'd)
Copyright © 2005 by the AABB. All rights reserved.

80 AABB Technical Manual
Appendix 2-5. Sample List of Hazardous Chemicals in the Blood Bank
Chemical Hazard
Ammonium chloride Irritant
Bromelin Irritant, sensitizer
Calcium chloride Irritant
Carbon dioxide, frozen (dry ice) Corrosive
Carbonyl iron powder Oxidizer
Chloroform Toxic, suspected carcinogen
Chloroquine Irritant, corrosive
Chromium-111 chloride hexahydrate Toxic, irritant, sensitizer
Citric acid Irritant
Copper sulfate (cupric sulfate) Toxic, irritant
Dichloromethane Toxic, irritant
Digitonin Toxic
Dimethyl sulfoxide (DMSO) Irritant
Dry ice (carbon dioxide, frozen) Corrosive
Ethidium bromide Carcinogen, irritant
Ethylenediaminetetraacetic acid (EDTA) Irritant
Ethyl ether Highly flammable and explosive, toxic, irritant
Ficin (powder) Irritant, sensitizer
Formaldehyde solution (34.9%) Suspected carcinogen, combustible, toxic
Glycerol Irritant
Hydrochloric acid Highly toxic, corrosive
Imidazole Irritant
Isopropyl (rubbing) alcohol Flammable, irritant
Liquid nitrogen Corrosive
Lyphogel Corrosive
2-Mercaptoethanol Toxic, stench
Mercury Toxic
Mineral oil Irritant, carcinogen, combustible
Papain Irritant, sensitizer
Polybrene Toxic
Potassium hydroxide Corrosive, toxic
Saponin Irritant
Sodium azide Toxic, irritant, explosive when heated
Sodium ethylmercurithiosalicylate (thimerosal) Highly toxic, irritant
Sodium hydrosulfite Toxic, irritant
Sodium hydroxide Corrosive, toxic
Sodium hypochlorite (bleach) Corrosive
Copyright © 2005 by the AABB. All rights reserved.

Chapter 2: Facilities and Safety 81
Chemical Hazard
Sodium phosphate Irritant, hygroscopic
Sulfosalicylic acid Toxic, corrosive
Trichloroacetic acid (TCA) Corrosive, toxic
Trypsin Irritant, sensitizer
Xylene Highly flammable, toxic, irritant
Appendix 2-5. Sample List of Hazardous Chemicals in the Blood Bank (cont'd)
Copyright © 2005 by the AABB. All rights reserved.

82 AABB Technical Manual
Appendix 2-6. Specific Chemical Categories and How to Work Safely with These
Chemicals
Chemical
Category Hazard Precautions Special Treatment
Acids, alkalis,
and corrosive
compounds
Irritation
Severe burns
Tissue damage
During transport, protect
large containers with
plastic or rubber bucket
carriers
During pouring, wear eye
protection and chemical-
resistant-rated gloves
and gowns as recom-
mended
Always ADD ACID TO
WATER, never water to
acid
When working with large
jugs, have one hand on
the neck and the other at
the base, and position
them away from the face
Store concentrated acids in
acid safety cabinets
Limit volumes of concen-
trated acids to 1 liter
Post cautions for materials
in the area
Report changes in appear-
ance (perchloric acid
may be explosive if it be-
comes yellowish or
brown) to chemical
safety officer
Acrylamide Neurotoxic
Carcinogenic
Adsorbed
through the
skin
Wear chemically rated
gloves
Wash hands immediately
after exposure
Store in a chemical cabinet
Compressed
gases
Explosive Label as to contents
Leave valve safety covers on
until use
Open valves slowly for use
Label empty tanks
Transport using hand trucks
or dollies
Place cylinders in a stand or
secure them to prevent
falling
Store in well-ventilated sep-
arate rooms
Oxygen should not be
stored close to combusti-
ble gas or solvents
Check connections for leaks
with soapy water
Liquid nitrogen Freeze injury
Severe burns to
skin or eyes
Use heavy insulated gloves
and goggles when work-
ingwithliquidnitrogen
The tanks should be se-
curely supported to avoid
being tipped over
The final container of liquid
nitrogen (freezing unit)
must be securely sup-
ported to avoid tipping
over
Copyright © 2005 by the AABB. All rights reserved.

Chapter 2: Facilities and Safety 83
Chemical
Category Hazard Precautions Special Treatment
Flammable
solvents
Classified accord-
ingtoflash
point—see
MSDS
Classified accord-
ing to volatility
Use extreme caution when
handling
Post NO SMOKING signs in
working area
Have a fire extinguisher and
solvent cleanup kit in the
room
Pour volatile solvents under
suitable hood
Use eye protection when
pouring and chemical-re-
sistant neoprene gloves
No flame or other source of
possible ignition should
be in or near areas where
flammable solvents are
being poured
Label as FLAMMABLE
Make every attempt to re-
place hazardous materi-
als with less hazardous
materials
Store containers larger than
1galloninaflammable
solvent storage room or
in a fire safety cabinet
Ground metal containers by
connecting the can to a
water pipe or ground
connection; if recipient
container is also metal, it
should be electrically
connected to the delivery
container while pouring
Appendix 2-6. Specific Chemical Categories and How to Work Safely with These
Chemicals (cont'd)
Copyright © 2005 by the AABB. All rights reserved.

84 AABB Technical Manual
Appendix 2-7. Incidental Spill Response*
Chemicals Hazards PPE Control Materials
Acids
Acetic
Hydrochloric
Nitric
Perchloric
Sulfuric
Photographic
chemicals
(acid)
Severe irritant if inhaled
Contact causes burns to skin
and eyes
Corrosive
Fire or contact with metal may
produce irritating or poison-
ous gas
Nitric, perchloric, and sulfuric
acids are water-reactive oxi-
dizers
Acid-resistant gloves
Apron and coveralls
Goggles and face shield
Acid-resistant foot
covers
Acid neutralizers/
absorbent
Absorbent boom
Leakproof containers
Absorbent pillow
Mat (cover drain)
Shovel or paddle
Bases and caustics
Potassium hydroxide
Sodium hydroxide
Photographic chemicals
(basic)
Corrosive
Fire may produce irritating or
poisonous gas
Gloves; impervious apron or
coveralls
Goggles or face shield; impervi-
ous foot covers
Base control/neutralizer
Absorbent pillow
Absorbent boom
Drain mat
Leakproof container
Shovel/paddle
Chlorine
Bleach
Sodium hypochlorite
Inhalation can cause respiratory
irritation
Liquid contact can produce irri-
tation of the eyes or skin
Toxicity due to alkalinity, possi-
ble chlorine gas generation,
and oxidant properties
Gloves (double set 4H
undergloves and butyl or
nitrile overgloves);
impervious apron
or coveralls
Goggles or face shield
Impervious foot covers (neo-
prene boots for emergency
response releases)
Self-contained breathing appara-
tus (emergency response
releases)
Chlorine control powder
Absorbent pillow
Absorbent
Absorbent boom
Drain mat
Vapor barrier
Leakproof container
Shovel or paddle
Copyright © 2005 by the AABB. All rights reserved.
Chapter 2: Facilities and Safety 85
Cryogenic gases
Carbon dioxide
Nitrous oxide
Liquid nitrogen
Contact with liquid
nitrogen can produce
frostbite
Asphyxiation (displaces oxygen)
Anesthetic effects
(nitrous oxide)
Full face shield or goggles; neo-
prene boots; gloves (insu-
lated to protect from the cold)
Hand truck (to transport cylinder
outdoors if necessary)
Soap solution (to check for
leaks)
Putty (to stop minor pipe and
line leaks)
Flammable gases
Acetylene
Oxygen gases
Butane
Propane
Simple asphyxiate
(displaces air)
Anesthetic potential
Extreme fire and explosion
hazard
Release can create an oxy-
gen-deficient
atmosphere
Face shield and goggles; neo-
prene boots; double set of
gloves; coveralls with hood
and feet
Hand truck (to transport cylinder
outdoors if needed)
Soap solution (to check for
leaks)
Flammable liquids
Acetone
Xylene
Methyl alcohol toluene
Ethyl alcohol
Other alcohols
Vapors harmful if inhaled
(central nervous system
depressants)
Harmful via skin absorption
Extreme flammability
Liquid evaporates to form flam-
mable vapors
Gloves (double 4H undergloves
and butyl or nitrile
overgloves); impervious
apron or coveralls; goggles or
face shield; impervious foot
covers
Absorbent
Absorbent boom
Absorbent pillow
Shovel or paddle (nonmetal,
nonsparking)
Drain mat
Leakproof containers
Formaldehyde and
glutaraldehyde
4% formaldehyde
37% formaldehyde
10% formalin
2% glutaraldehyde
Harmful if inhaled or absorbed
through skin;
Irritation to skin, eyes, and
respiratory tract
Formaldelyde is a suspected
human carcinogen
Keep away from heat, sparks,
and flame (37% formalde-
hyde)
Gloves (double set 4H
undergloves and butyl or
nitrile overgloves); impervi-
ous apron or coveralls; gog-
gles; impervious foot covers
Aldehyde neutralizer/absorbent
Absorbent boom
Absorbent pillow
Shovel or pallet (nonsparking)
Drain mat
Leakproof container
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

86 AABB Technical Manual
Chemicals Hazards PPE Control Materials
Mercury
Cantor tubes
Thermometers
Barometers
Sphygmomanometers
Mercuric chloride
Mercury and mercury vapors are
rapidly absorbed in respira-
tory tract, GI tract, skin
Short-term exposure may cause
erosion of respiratory/GI
tracts, nausea, vomiting,
bloody diarrhea, shock, head-
ache, metallic taste
Inhalation of high concentrations
can cause pneumonitis, chest
pain, dyspnea, coughing
stomatitis, gingivitis, and sali-
vation
Avoid evaporation of mercury
from tiny globules by quick
and thorough cleaning
Gloves (double set 4H
underglove and butyl or nitrile
overglove); impervious apron
or coveralls; goggles; imper-
vious foot covers
Mercury vacuum or spill kit
Scoop
Aspirator
Hazardous waste containers
Mercury indicator powder
Absorbent
Spatula
Disposable towels
Sponge with amalgam
Vapor suppressor
*This list of physical and health hazards is not intended as a substitute for the specific MSDS information. In the case of a spill or if any questions arise, always refer to the chem-
ical-specific MSDS for more complete information. GI = gastrointestinal; MSDS = material safety data sheet; PPE = personal protective equipment.
Appendix 2-7. Incidental Spill Response* (cont'd)
Copyright © 2005 by the AABB. All rights reserved.

Chapter 2: Facilities and Safety 87
Appendix 2-8. Managing Hazardous Chemical Spills
Actions Instructions for Hazardous Liquids, Gases, and Mercury
De-energize Liquids: For 37% formaldehyde, de-energize and remove all sources of ig-
nition within 10 feet of spilled hazardous material. For flammable liq-
uids, remove all sources of ignition.
Gases: Remove all sources of heat and ignition within 50 feet for flamma-
ble gases.
Remove all sources of heat and ignition for nitrous oxide release.
Isolate, evacuate,
and secure the
area
Isolate the spill area and evacuate everyone from the area surrounding the
spill except those responsible for cleaning up the spill. (For mercury,
evacuate within 10 feet for small spills, 20 feet for large spills.) Secure
area.
Have the appropriate
PPE
See Appendix 2-2 for recommended PPE.
Contain the spill Liquids or mercury: Stop the source of spill if possible.
Gases: Assess the scene; consider the circumstances of the release (quan-
tity, location, ventilation). If circumstances indicate it is an emergency
response release, make appropriate notifications; if release is deter-
mined to be incidental, contact supplier for assistance.
Confine the spill Liquids: Confine spill to initial spill area using appropriate control equip-
ment and material. For flammable liquids, dike off all drains.
Gases: Follow supplier’s suggestions or request outside assistance.
Mercury: Use appropriate materials to confine the spill (see Appendix 2-2).
Expel mercury from aspirator bulb into leakproof container, if applicable.
Neutralize the spill Liquids: Apply appropriate control materials to neutralize the chemical—
see Appendix 2-2.
Mercury: Use mercury spill kit if needed.
Spill area cleanup Liquids: Scoop up solidified material, booms, pillows, and any other mate-
rials. Put used materials into a leakproof container. Label container with
name of hazardous material. Wipe up residual material. Wipe spill area
surface three times with detergent solution. Rinse areas with clean wa-
ter. Collect supplies used (goggles, shovels, etc) and remove gross
contamination; place into separate container for equipment to be
washed and decontaminated.
Gases: Follow supplier’s suggestions or request outside assistance.
Mercury: Vacuum spill using a mercury vacuum or scoop up mercury
paste after neutralization and collect it in designated container. Use
sponge and detergent to wipe and clean spill surface three times to re-
move absorbent. Collect all contaminated disposal equipment and put
into hazardous waste container. Collect supplies and remove gross con-
tamination; place them into a separate container for equipment that will
be thoroughly washed and decontaminated.
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

88 AABB Technical Manual
Actions Instructions for Hazardous Liquids, Gases, and Mercury
Disposal Liquids: For material that was neutralized, dispose of it as solid waste. Fol-
low facility’s procedures for disposal. For flammable liquids, check with
facility safety officer for appropriate waste determination.
Gases: The manufacturer or supplier will instruct facility on disposal if ap-
plicable.
Mercury: Label with appropriate hazardous waste label and DOT diamond
label.
Report Follow appropriate spill documentation and reporting procedures. Investi-
gate the spill; perform root cause analysis if needed. Act on opportuni-
ties for improving safety.
DOT = Department of Transportation; PPE = personal protective equipment.
Appendix 2-8. Managing Hazardous Chemical Spills (cont'd)
Copyright © 2005 by the AABB. All rights reserved.

Chapter 3: Blood Utilization Management
Chapter 3
Blood Utilization
Management
THE GOAL OF BLOOD utilization
management is to ensure effective
use of limited blood resources. It
includes the policies and practices related
to inventory management and blood us-
age review. Although regional blood cen-
ters and transfusion services approach
utilization management from different
perspectives, they share the common goal
of providing appropriate, high-quality
blood products with minimum waste. This
chapter reviews the elements of utiliza-
tion management, emphasizing the trans-
fusion service.
Minimum and Ideal
Inventory Levels
Transfusion services should establish both
minimum and ideal inventory levels. In-
ventory levels should be evaluated period-
ically and adjusted if needed. Important
indicators of performance include, but are
not limited to, outdate rates, the frequency
of emergency blood shipments, and delays
in scheduling elective surgery. Inventory
levels should also be reevaluated when-
ever a significant change is planned or
observed. Examples of significant change
may include adding more beds; perform-
ing new surgical procedures; or changing
practices in oncology, transplantation, neo-
natology, or cardiac surgery.
Determining Inventory
Levels
The ideal inventory level provides ade-
quate supplies of blood for routine and
emergency situations and minimizes out-
dating. Forecasting is an attempt to pre-
dict future blood product use from data
collected about past usage. The optimal
number of units to keep in inventory can
be estimated using mathematical formu-
las, computer simulations, or empirical
89
3
Copyright © 2005 by the AABB. All rights reserved.
calculations. Three less complicated meth-
ods of estimating minimum inventory are
described below. When the minimum in-
ventory level has been calculated, a buffer
margin for emergencies should be added
to obtain an ideal inventory level.
Average Weekly Use Estimate
This method gives an estimate of the av-
erage weekly blood usage of each ABO
group and Rh type.
1. Collect weekly blood and product
usage data over a 26-week period.
2. Record usage by ABO group and Rh
type for each week.
3. Disregard the single highest usage
for each type to correct for unusual
week-to-week variation (eg, a large
volume used for an emergency).
4. Total the number of units of each
ABO group and Rh type, omitting
the highest week in each column.
5. Divide each total by 25 (total num-
ber of weeks minus the highest week).
This gives an estimate of the average
weekly blood usage of each ABO
group and Rh type.
Average Daily Use Estimate
Facilities that transfuse on a daily basis
may calculate daily blood usage by the
following method.
1. Determine the total use over several
months.
2. Divide the total use by the number
of days in the period covered.
3. Determine the percentage of each of
the blood types used during one or
more representative months.
4. Multiply the average blood use per
day by the percentage of blood use
by type.
5. Determine the minimum inventory
level by multiplying the daily use by
the number of days of blood supply
required to be on hand (this may be 3,
5, or 7 days depending on the blood
supplier’s delivery schedule).
A transfusion service may find the aver-
age daily use calculation more helpful
when blood shipments are made once or
more per day.
Moving Average Method
Themovingaveragemethodcanbeuse
-
ful in facilities with any level of activity.
1. Determine the preferred recording
period (such as day or week).
2. Add the number of units used in each
period to obtain the total use.
3. Divide the total number of units by
the number of recording periods.
4. Delete old data as new data are added.
This method tends to minimize variation
from one period to another.
Factors that Affect Outdating
Benchmark data on component outdating
have been published by the National Blood
Data Resource Center (NBDRC).1The data
from this report showed that approxi-
mately 2,190,000 total components out-
dated in 2001, a 6.6% decrease from 1999.
Whole-blood-derived platelet concen-
trates accounted for more than half of the
outdated components (49%), or 1,074,000
units. Outdated allogeneic RBCs (directed
and nondirected) accounted for 26.8% of
all outdated components, or 588,000 units.
The outdate rate for each component is
shown in Table 3-1. Outdating continues to
be a problem, particularly for autologous
units and platelets. The outdate rate is af-
fected by many factors other than inven-
tory level (eg, the size of the hospital, the
extent of services provided, the shelf life
of the products, the shipping distance
and frequency, and ordering policies).
90 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

In addition to establishing both mini-
mum and ideal inventory levels, maximum
inventory levels may assist staff in deter-
mining when to arrange for return or trans-
fer of in-date products to avoid outdating.
Both transfusion services and donor cen-
ters should establish record-keeping sys-
tems that allow personnel to determine the
number of units ordered and the number of
units received or shipped. The responsibil-
ity for ordering may be centralized, and or-
ders should be based on established poli-
cies for minimal and maximal levels.
Standing orders can simplify inventory
planning for both transfusion services and
blood centers. Blood centers may send a
predetermined number of units on a regu-
lar schedule or may keep the transfusion
service inventory at established levels by
replacing all units reported as transfused.
Optimal inventory management requires
distribution and transfusion of the oldest
blood first and this requires clearly written
policies on blood storage and blood selec-
tion. Technologists generally find it easier
to select, crossmatch, and issue the oldest
units first when inventories are arranged by
expiration date.
Policies on blood selection must be flexi-
ble, to allow use of fresher blood when indi-
cated (eg, for infants). Generally, however,
oldest units are crossmatched for patients
most likely to need transfusion.
Improving Transfusion
Service Blood Ordering
Practices
The shelf life decreases each time a unit is
held or crossmatched for a patient who
does not use it. When physicians order more
blood than needed, it is unavailable for
other patients, which may increase the
outdate rate. Providing testing guidelines,
such as type and screen (T/S) policies and
Chapter 3: Blood Utilization Management 91
Table 3-1. Blood Component Units Processed, Transfused, and Outdated in United
States in 20011
Component
Units
Processed
Units
Transfused
Units
Outdated
Percent
Outdated
RBCs (allogeneic,
nondirected)
14,259,000 13,361,000 576,000 4.0
RBCs (autologous) 619,000 359,000 263,000 42.5
RBCs (directed) 169,000 95,000 12,000 7.1
Platelets
(whole-blood-
derived)
4,164,000 2,614,000 1,074,000 25.8
Platelets Pheresis 1,456,000 1,264,000 160,000 11.0
FFP 4,437,000 3,926,000 77,000 1.7
Cryoprecipitated AHF 1,068,000 898,000 28,000 2.6
FFP = Fresh Frozen Plasma; RBCs = Red Blood Cells.
Copyright © 2005 by the AABB. All rights reserved.

maximum surgical blood order schedules
(MSBOS),2as well as monitoring cross-
match-to-transfusion (C:T) ratios may be
helpful. A C:T ratio greater than 2.0 usu-
ally indicates excessive crossmatch re-
quests. In some situations, it may be use-
ful to determine C:T ratios by service to
identify areas with the highest ratio.
Some institutions define those proce-
dures that normally do not use blood in a
“type and screen” guideline. Both the T/S
guidelineandtheMSBOSusedataabout
past surgical blood use to recommend a
T/S order or a maximum number of units
that should be ordered initially for common
elective surgical procedures. With the
MSBOS, physicians may order the number
of units believed to be appropriate for the
patient since the MSBOS is intended to be a
guideline for appropriate patient care.
Some institutions have modified the MSBOS
concept into a “standard” blood order (SBO)
system for surgical procedures.3
Ordering guidelines such as those in Ta-
ble 3-2 are derived by reviewing a facility’s
blood use over a suitable period. Conclu-
sions can then be drawn about the likeli-
hood of transfusion and probable blood
use for each surgical procedure. A T/S order
is a recommended SBO for procedures that
require on average less than 0.5 unit of
blood per patient per procedure. An SBO
often represents the average number of
units transfused for each procedure, where-
as the MSBOS often defines the number of
units needed to meet the needs of 80% to
90% of patients undergoing a specific surgi-
cal procedure.3
An institution’s guidelines must reflect
local patterns of surgical practice and pa-
tient population. These may be compared
to published guidelines to ensure that local
practice does not markedly deviate from
generally accepted standards of care.
(Transfusion audits are discussed in Chap-
ter1.)OncetheT/S,MSBOS,SBO,orother
92 AABB Technical Manual
Table 3-2. Example of a Maximum
Surgical Blood Order Schedule
Procedure Units*
General Surgery
Breast biopsy T/S
Colon resection 2
Exploratory laparotomy T/S
Gastrectomy 2
Laryngectomy 2
Mastectomy, radical T/S
Pancreatectomy 4
Splenectomy 2
Thyroidectomy T/S
Cardiac-Thoracic
Aneurism resection 6
Redo coronary artery bypass graft 4
Primary coronary artery bypass graft 2
Lobectomy T/S
Lung biopsy T/S
Vascular
Aortic bypass with graft 4
Endarterectomy T/S
Femoral-popliteal bypass with graft 2
Orthopedics
Arthroscopy T/S
Laminectomy T/S
Spinal fusion 3
Total hip replacement 3
Total knee replacement T/S
OB-GYN
Abdomino-perineal repair T/S
Cesarean section T/S
Dilation and curettage T/S
Hysterectomy, abdominal/
laparoscopic
T/S
Hysterectomy, radical 2
Urology
Bladder, transurethral resection T/S
Nephrectomy, radical 3
Radical prostatectomy, perineal 2
Prostatectomy, transurethral T/S
Renal transplant 2
*Numbers may vary with institutional practice.
Copyright © 2005 by the AABB. All rights reserved.
schedule is accepted, inventory levels often
can be reduced. Ordering guidelines should
be periodically reviewed to keep pace with
changing methods and practices. A change
in the C:T ratio might signal a significant
modification in clinical practice.
The T/S, MSBOS, or SBO systems are in-
tended for typical circumstances. Surgeons
or anesthesiologists may individualize spe-
cific requests and override the system to ac-
commodate special needs. The transfusion
service must give special consideration to
patients with a positive antibody screen.
The antibody should be identified and, if it
is potentially clinically significant, an ap-
propriate number of antigen-negative units
should be identified (eg, two, if the original
order was a type and screen).
Facilities using the immediate spin or
computer crossmatch can provide addi-
tional crossmatched units more rapidly if
required. This capability can allow such fa-
cilities to adjust their T/S, MSBOS, and SBO
schedules accordingly. More procedures
can be safely handled as type and screens
and fewer crossmatched units may be nec-
essary.
Routine vs Emergency Orders
Transfusion services should establish pro-
cedures that define ideal stocking levels for
each blood type and critical levels at which
emergency orders are indicated. Transfu-
sion service staff should have institutional
policies identifying the following:
■Who monitors inventory levels?
■Who is responsible for placing orders?
■When and how are orders to be
placed (by telephone or facsimile)?
■How are orders documented?
The addresses and telephone numbers of
approved blood suppliers and any needed
courier or cab services should be immedi-
ately available. Transfusion services need to
establish guidelines for handling blood
shortages and unexpected emergencies.
Equally important is specifying the actions
to take if transfusion requests cannot im-
mediately be met.
Transfusion services should develop pol-
icies defining the following:
■When ABO-compatible units may be
given instead of ABO-identical units.
■When Rh-positive units may be given
to Rh-negative recipients.
■When units crossmatched for a sur-
gical procedure may be released be-
fore the standard interval.
■If units may be crossmatched for more
than one patient at a time.
■What resources are available for trans-
fer of inventory.
■Mechanisms to notify physicians of
critical blood shortages.
■When cancellation of elective proce-
dures should be considered.
■Methods to notify staff and patients
of surgery cancellations.
Inventory Counts and Inspection
On-hand units may be counted once or
several times a day to determine ordering
needs; computerized facilities may prefer
to take inventory electronically. Individ-
ual units must be visually inspected for
signs of contamination or atypical ap-
pearance before issue or shipping. Units
that do not meet inspection criteria must
be quarantined for further evaluation.
An organizational format for storage
should be established and followed. Unpro-
cessed or incompletely processed units,
autologous units, and unsuitable units
must be clearly segregated (quarantined)
from routine stock.4(p15) Most institutions or-
ganize their blood inventory by status
(quarantined, retype unconfirmed, retype
confirmed, available, crossmatched, etc), by
product, by ABO group and Rh type, and,
within these categories, by expiration date.
Chapter 3: Blood Utilization Management 93
Copyright © 2005 by the AABB. All rights reserved.
Attention to detail in placing blood into
storage is necessary because a placement
error could be critical if a quarantined unit
is issued or a group O Rh-positive unit, in-
correctly placed among group O Rh-nega-
tive units, is issued without careful check-
ing in an emergency situation.
Special Product Concerns
Platelets
Few articles address the management of
platelet inventory. Optimal levels are diffi-
cult to determine because demand is epi-
sodicandtheshelflifeisshort.Often,the
effective shelf life is 3 days because, of the
allowable 5 days after phlebotomy, day 1
may be taken for testing and day 2 for
shipment. Planning is further complicated
by requests for special products, such as
leukocyte-reduced, crossmatched, HLA-
matched, or cytomegalovirus (CMV)-sero-
negative platelets.
Platelet inventory management requires
good communication and cooperation
among patient care providers, transfusion
services, and blood centers. Information
about patients’ diagnoses and expected
transfusion schedules helps the blood cen-
ter plan how many platelets to prepare and
which donors to recruit for plateletpheresis.
Transfusion services with low platelet
use usually order platelets only when they
receive a specific request. If the transfusion
service staff follows daily platelet counts
and special transfusion requirements of
known platelet users, they can often antici-
pate needs and place orders in advance.
Transfusion services with high use may find
it helpful to maintain platelets in inventory.
Transfusion services should define selec-
tion and transfusion guidelines for ABO
group, Rh type, irradiation, CMV serologic
testing, and leukocyte reduction of platelet
products. The formulas described in the
beginning of the chapter may be used to
estimate ideal inventory ranges.
Platelet usage often increases the day af-
ter a holiday because elective procedures
and oncology transfusions will have been
postponed. Planning ahead to stock plate-
lets helps meet postholiday demand.
Frozen Plasma Products
Because plasma components can be stored
frozen up to 1 year, these inventories are
easier to manage. Optimal inventory lev-
els are determined by assessing statistics
on patient populations and usage pat-
terns. Production goals and schedules can
then be established. Most centers find it
best to maintain consistent production
levels throughout the year, to achieve
evenly distributed expiration dates.
Some facilities prefer to freeze plasma
from group AB and A donors because these
units will be ABO-compatible with most
potential recipients. Plasma can be col-
lected by apheresis to increase general
stock and provide for special needs.
Cryoprecipitated AHF is a labor-inten-
sive product to prepare and supplies can-
not easily be increased to meet large acute
needs. It is prudent to maintain inventories
at close-to-maximum levels.
Autologous and Directed Units
If autologous and directed donor units
constitute an increasing fraction of inven-
tory, their management becomes a signif-
icant and controversial issue both for the
intended recipients and for institutions.
An extended discussion of autologous
blood collection and transfusion methods
can be found in Chapter 5.
Autologous and directed units should be
stored in separate designated areas in the
blood refrigerator. Such units are often ar-
ranged alphabetically by the intended re-
94 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
cipient’s last name. Available units must be
clearly identified and monitored to ensure
issue in the proper sequence. Autologous
blood should always be used first, followed
by directed donor blood, and, finally, allo-
geneic units from general stock. Policies
about the reservation period for directed
donor units and possible release to other
recipients should be established at both the
transfusion service and donor center and
should be made known to laboratory staff,
to potential recipients, and to their physi-
cians.
Special Inventories
Donor centers and transfusion services
are faced with requests for specialty prod-
ucts such as CMV-reduced-risk units,
HLA-matched platelets, antigen-matched
red cells, or irradiated components. The
appropriate use of these products is dis-
cussed in other chapters. Depending on
how and when they were prepared, these
products may have shortened expiration
dates.
If demand and inventory levels are very
high, a transfusion service may need to
keep separate inventories of these products
to make them easier to locate and monitor.
These special units can be rotated into gen-
eral stock as they near their outdate be-
cause they can be given safely to others.
References
1. Comprehensive report on blood collection
and transfusion in the United States in 2001.
Bethesda, MD: National Blood Data Resource
Center, 2003.
2. Friedman BA, Oberman HA, Chadwick AR, et
al. The maximum surgical blood order sched-
ule and surgical blood use in the United States.
Transfusion 1976;16:380-7.
3. Devine P, Linden JV, Hoffstadter L, et al. Blood
donor-, apheresis-, and transfusion-related
activities: Results of the 1991 American Asso-
ciation of Blood Banks Institutional Member-
ship Questionnaire. Transfusion 1993;33:779-
82.
4. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005:15.
Chapter 3: Blood Utilization Management 95
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 4: Allogeneic Donor Selection and Blood Collection
Chapter 4
Allogeneic Donor Selection
and Blood Collection
BLOOD CENTERS AND transfusion
services depend on volunteer do-
nors to provide the blood neces-
sary to meet the needs of the patients
they serve. To attract volunteer donors
and encourage their continued participa-
tion, it is essential that conditions sur-
rounding blood donation be as pleasant,
safe, and convenient as possible. To pro-
tect donors and recipients, donors are
questioned about their medical history
and are given a miniphysical examination
to help blood center staff determine
whether they are eligible donors. The
phlebotomy is conducted carefully to
minimize any potential donor reactions
or bacterial contamination of the unit.
Blood Donation Process
The donor area should be attractive, ac-
cessible, and open at hours convenient
for donors. It must be well lighted, com-
fortably ventilated, orderly, and clean.
Personnel should be friendly, understand-
ing, professional, and well trained. The area
mustprovideadequatespaceforprivate
and accurate examinations of individuals
to determine their eligibility as blood do-
nors, and for the withdrawal of blood
from donors with minimum risk of con-
tamination or exposure to activities and
equipment unrelated to blood collection.
Registration
The information obtained from the donor
during registration must fully identify the
donor and link the donor to existing re-
cords.1(p13) Some facilities require photo-
graphic identification. Current informa-
tion must be obtained and recorded for
each donation. Selected portions of dona-
tion records must be kept indefinitely and
must make it possible to notify the donor
of any information that needs to be con-
veyed.1(p69) The following information should
be included:
97
4
Copyright © 2005 by the AABB. All rights reserved.
1. Date and time of donation.
2. Name: Last, first (and middle initial
if available).
3. Address: Residence and/or business.
4. Telephone: Residence and/or busi-
ness.
5. Gender.
6. Age or date of birth. Blood donors must
be at least 17 years of age or the age
stipulated by applicable state law.
7. A record of reasons for previous de-
ferrals, if any. Persons who have been
placed on a deferral or surveillance
list must be identified before any
unitdrawnfromthemismadeavail
-
able for release. Ideally, a donor de-
ferral registry should be available to
identify ineligible donors before
blood is drawn. If such a registry is
notavailable,theremustbeaproce-
dure to review prior donation re-
cords and/or deferral registries be-
fore releasing the components from
quarantine.2
The following information may also be
useful:
1. Additional identification such as so-
cial security or driver’s license num-
beroranyothernameusedbythe
donor on a previous donation. The
Social Security Act specifically al-
lows the use of the social security
number for this purpose. These data
are required for information retrieval
in some computerized systems. Iden-
tification of other names used by a
donor is particularly important to
ensure that the appropriate donor
file is accessed or that a deferral sta-
tusisaccurate.
2. Name of patient or group to be ac-
knowledged.
3. Race. Although not required, this in-
formation can be particularly useful
when blood of a specific phenotype
is needed for patients who have un-
expected antibodies. Care should be
taken to be sure that minority popu-
lations understand the medical im-
portance and scientific applications
of this information.3,4
4. Unique characteristics of the donor.
Certain information about the donor
may enable the blood bank to make
optimal use of the donation. For ex-
ample, blood from donors who are
seronegative for cytomegalovirus
(CMV), or who are group O, Rh neg-
ative, is often designated for neona-
tal patients. The blood center may
specify that blood from these indi-
viduals be drawn routinely into col-
lection bags suitable for pediatric
transfusion. Individuals known to
have clinically significant antibodies
may be identified so that their blood
can be processed into components
that contain only minimal amounts
of plasma.
5. A record of special communications
to a donor, special drawing of blood
samples for studies, etc.
6. If the donation is directed to a spe-
cific patient, information about when
and where the intended recipient will
be hospitalized should be obtained.
An order from the intended recipi-
ent’s physician should be provided
to the blood center staff. The in-
tended recipient’s date of birth, so-
cial security number, or other identi-
fiers may be required by the transfusion
service. If the donor is a blood relative
of the intended recipient, this infor-
mation must be noted so that cellular
components can be irradiated.1(p43)
Information Provided to the Prospective
Donor
All donors must be given educational ma-
terials informing them of significant risk
98 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
of the procedure, the clinical signs and
symptoms associated with human immu-
nodeficiency virus (HIV) infection and
AIDS, high-risk activities for transmission,
and the importance of refraining from do-
nating blood if they have engaged in these
activities or experienced associated signs
or symptoms. Before donating, the pro-
spectivedonorsmustdocumentthatthey
have read the material and have been
given the opportunity to ask questions
about the information. This information
must include a list of activities defined by
the Food and Drug Administration (FDA)
that increase the risk of exposure to HIV. A
description of HIV-associated clinical signs
and symptoms, including the following,
must be provided5:
1. Unexplained weight loss.
2. Night sweats.
3. Blue or purple spots on or under the
skin or on mucous membranes.
4. Swollen lymph nodes lasting more
than 1 month.
5. White spots or unusual sores in the
mouth.
6. Temperature greater than 100.5 F for
more than 10 days.
7. Persistent cough and shortness of
breath.
8. Persistent diarrhea.
The donor should be provided with
information about the tests to be done on
his or her blood, the existence of registries
of ineligible donors, and regulations or lo-
cal standard operating procedures (SOPs)
that require notification to government
agencies of the donor’s infectious disease
status. The requirement to report positive
test results may differ from state to state;
they may include HIV, syphilis, and hepati-
tis testing. Prospective donors must also be
informed if there are routine circumstances
in which some tests for disease markers are
not to be performed.1(p15) The donor should
be told that he or she will be notified when
abnormal test results are recorded and the
donor has been placed on a deferral list.
When applicable, the donor must also be
informed that his or her blood is to be
tested with an investigational test such as a
nucleic acid amplification test. The possi-
bility that testing may fail to identify infec-
tive individuals in an early seronegative
stage of infection should be included as
well.6The same educational material can
be used to warn the prospective donor of
possible reactions and provide suggestions
for postphlebotomy care.
This information should be presented in
a way that the donor will understand.5Pro-
visionsshouldbemadeforthehearing-or
vision-impaired, and interpreters should be
available for donors not fluent in English.
The use of interpreters known to the donor
should be discouraged. If such a practice is
necessary, a signed confidentiality state-
ment should be obtained. In some loca-
tions, it may be helpful to have brochures
in more than one language. It is also helpful
to provide more detailed information for
first-time donors. Information about alter-
native sites or other mechanisms to obtain
HIV tests should be available to all prospec-
tive donors.
Donor Selection
The donor screening process is one of the
most important steps in protecting the
safety of the blood supply. The process is
intended to identify elements of the med-
ical history and behavior or events that
put a person at risk for transmissible dis-
ease or at personal medical risk. It is,
therefore, imperative that proper guide-
lines and procedures be followed to make
the donor screening process effective.
A qualified physician must determine
the eligibility of donors. The responsibility
may be delegated to a designee working
Chapter 4: Allogeneic Donor Selection and Blood Collection 99
Copyright © 2005 by the AABB. All rights reserved.
under the physician’s direction after appro-
priate training.7Donor selection criteria are
established through regulations, recom-
mendations, and standards of practice.
When a donor’s condition is not covered or
addressed by any of these, a qualified phy-
sician should determine the eligibility.
Donor selection is based on a medical
history and a limited physical examination
done on the day of donation to determine
if giving blood will harm the donor or if
transfusion of the unit will harm a recipi-
ent.1(p17) The medical history questions (in-
cluding questions pertaining to risk behav-
ior associated with HIV infections) may be
asked by a qualified interviewer or donors
may complete their own record, which
must then be reviewed with the donor and
initialed by a trained knowledgeable staff
member of the donor service according to
local SOPs, state, and FDA approval.5,8 Some
donor centers have instituted an FDA-ap-
proved computer-generated questionnaire.
The interview and physical examination
must be performed in a manner that en-
sures adequate auditory and visual privacy,
allays apprehensions, and provides time for
any necessary discussion or explanation.
Details explaining a donor’s answers that
require further investigation should be doc-
umented by the staff on the donor form.
Resultsofobservationsmadewhenaphys
-
ical examination is given and when tests
are performed must be recorded concur-
rently.
Donors must understand the informa-
tion that is presented to them in order to
make an informed decision to donate their
blood. Effective communication is vital for
conveying important information and elimi-
nating ineligible donors from the donor
pool. Of equal importance is the training of
donor center staff. Screening can be effec-
tive only if the staff members are proficient
in their jobs and understand thoroughly the
technical information required to perform
the job. Good interpersonal and public re-
lations skills are essential for job
competency. Because donor center staff are
in constant contact with donors, knowl-
edgeable personnel and effective commu-
nication contribute to positive public
perception and to the success of donor
screening programs.
Medical History
While the medical history is obtained,
some very specific questions are neces-
sary to ensure that, to the greatest extent
possible, it is safe for the donor to donate
and for the blood to be transfused. The in-
terviewer should review and evaluate all
responses to determine eligibility for do-
nation and document the decision. To be
sure that all the appropriate questions are
asked and that donors are given a consis-
tent message, use of the most recent FDA-
approved AABB donor history question-
naire is recommended. The most recent
FDA-approved version is found on the AABB
Web site. (See Appendices 4-1 through 4-3,
whichwerecurrentatthetimeofthiswrit-
ing.)
One area of the medical history—medi-
cations and drugs taken by the donor—of-
ten requires further investigation. New pre-
scription drugs and over-the-counter
medications enter the marketplace daily,
and donors may report use of a drug not
specifically noted in the facility’s SOP man-
ual. Although there is consensus on those
drugs that are always or never a cause for
deferral, many drugs fall into a category
over which disagreement exists. In these
cases, the reason for taking the drug (rather
than the drug itself) is usually the cause for
deferral. Appendix 4-4 lists drugs that many
blood banks do consider acceptable with-
out approval from a donor center physician.
Prospective donors who have taken iso-
tretinoin (Accutane) or finasteride (Proscar
100 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

or Propecia) within the 30 days preceding
donation; dutasteride (Avodart) within the
6 months preceding donation; acitretin
(Soriatane) within the 3 years preceding do-
nation; or etretinate (Tegison) at any time
must be deferred.1(p62) The Armed Services
Blood Program Office makes its drug defer-
ral list available to the public.9
Deferring or rejecting potential donors
often leaves those persons with negative
feelings about themselves as well as the
blood donation process. Donors who are
deferred must be given a full explanation of
the reason and be informed whether or
when they can return to donate. It may be
prudent to document this notification.
Confidential Unit Exclusion
Donorsmaybegiventheopportunityto
indicate confidentially whether their blood
is or is not suitable for transfusion to oth-
ers. This should be done by a mechanism
that allows the donor to avoid face-to-
face admission of risk behaviors.
The donor must be given instructions to
the effect that he or she may call the blood
bank after the donation and ask that the
unit collected not be used. A mechanism
should exist to allow retrieval of the unit
without obtaining the donor’s identity (eg,
use of Whole Blood number).
If the donor indicates that blood col-
lected should not be used for transfusion,
he or she should be informed that the
blood will be subjected to testing and that
there will be notification of any positive
results. Counseling or referral must be pro-
vided for positive HIV test results, or if any
other medically significant test results have
been detected.
Physical Examination
The following variables must be evaluated
for each donor. The donor center physi-
cian must approve exceptions to routinely
acceptable findings. For special donor
categories, the medical director may pro-
vide policies and procedures to guide de-
cisions. Other donors may require indi-
vidual evaluation.
1. General appearance: If the donor
looks ill, or is excessively nervous, it
is best to defer the donation.
2. Weight: No more than 10.5 mL of
whole blood per kilogram of body
weight shall be collected at a dona-
tion.1(p61) This amount shall include
samples for testing. If it is necessary
to draw a smaller amount than ap-
propriate for a standard collection
container, then the amount of anti-
coagulant in the container must be
adjusted appropriately. The formula
inTable4-1maybeusedtodeter
-
mine the amount of anticoagulant to
remove. The volume of blood drawn
must be measured carefully and ac-
curately.
3. Temperature: The donor’s tempera-
ture must not exceed 37.5 C (99.5 F)
if measured orally, or its equivalent if
measured by another method. Lower
Chapter 4: Allogeneic Donor Selection and Blood Collection 101
Table 4-1. Calculations for Drawing Donors Weighing Less than 50 kg (110 lb)
A. Volume to draw* = (Donor’s weight in kg/50) ×450 mL
B. Amount of anticoagulant†needed = (A/100) ×14
C. Amount of anticoagulant to remove from collection bag = 63 mL – B
*Approximately 12% of total blood volume.
†CPD or CPDA-1 solutions for which the desired anticoagulant:blood ratio is 1.4:10.
Copyright © 2005 by the AABB. All rights reserved.

than normal temperatures are usu-
ally of no significance in healthy in-
dividuals; however, they should be
repeated for confirmation.
4. Pulse: The pulse rate should be counted
for at least 15 seconds. It should ex-
hibit no pathologic irregularity, and
the frequency should be between 50
and 100 beats per minute. If a pro-
spective donor is a known athlete
with high exercise tolerance, a pulse
rate below 50 may be noted and
should be acceptable. A donor cen-
ter physician should evaluate marked
abnormalities of pulse and recom-
mend acceptance, deferral, or refer-
ral for additional evaluation.
5. Blood pressure: The blood pressure
should be no higher than 180 mm Hg
systolic and 100 mm Hg diastolic. Pro-
spective donors whose blood pres-
sure is above these values should not
be drawn without individual evalua-
tion by a qualified physician.
6. Hemoglobin or packed cell volume
(hematocrit): Before donation, the
hemoglobin or hematocrit must be
determined from a sample of blood
obtained at the time of donation.
Although this screening test is in-
tended to prevent collection of blood
from a donor with anemia, it does
not ensure that the donor has an ad-
equatestoreofiron.Table4-2gives
the lower limits of hemoglobin for
accepting allogeneic donors. Indi-
viduals with unusually high hemo-
globin or hematocrit levels may need
to be evaluated by a physician be-
cause the elevated levels may reflect
pulmonary, hematologic, or other
abnormalities. Methods to evaluate
hemoglobin concentration include
1) specific gravity determined by cop-
per sulfate (see Method 6.1), 2) spec-
trophotometric measurement of he-
moglobin or determination of the
hematocrit, or 3) alternate accepted
methods to rule out erroneous re-
sults that may lead to rejection of a
donor. Earlobe puncture is not an
acceptable source for a blood sam-
ple.1(p61),10
7. Skin lesions: The skin at the site of
venipuncture must be free of lesions.
Both arms must be examined for
signs of repeated parenteral entry,
especially multiple needle puncture
marks and/or sclerotic veins as seen
with drug use. Such evidence is rea-
son for indefinite exclusion of a pro-
spective donor. Mild skin disorders
or the rash of poison ivy should not
be cause for deferral unless unusu-
ally extensive and/or present in the
antecubital area. Individuals with
boils, purulent wounds, or severe
skin infections anywhere on the
102 AABB Technical Manual
Table 4-2. Minimum Levels of Hemoglobin, Hematocrit, and Red Cell Density for
Accepting an Allogeneic Blood Donor
Donor Test Method Minimal Acceptable Value1
Hemoglobin
Hematocrit
Copper sulfate
12.5 g/dL
38%
1.053 sp gr
Copyright © 2005 by the AABB. All rights reserved.
body should be deferred, as should
anyone with purplish-red or hemor-
rhagic nodules or indurated plaques
suggestive of Kaposi’s sarcoma.
The record of the physical examination
and the medical history must identify and
contain the examiner’s initials or signature.
Any reasons for deferral must be recorded
and explained to the donor. A mechanism
must exist to notify the donor of clinically
significant abnormal findings in the physi-
cal examination, medical history, or post-
donation laboratory testing.11 Abnormalities
found before donation may be explained
verbally by qualified personnel. Test results
obtained after donation that preclude fur-
ther donation may be reported in person,
by telephone, or by letter. Donors should be
asked to report any illness developing with-
in a few days after donation and, especially,
to report a positive HIV test or the occur-
rence of hepatitis or AIDS that develops
within 12 months after donation.
Donor Consent
Written consent that allows donor center
personnel to collect and use blood from
the prospective donor is required.1(p15) The
consent form is part of the donor record
and must be completed before donation.
Theproceduremustbeexplainedin
terms that donors can understand, and
theremustbeanopportunityforthepro
-
spective donor to ask questions. The
signed donor record or consent form
should also indicate that the donor has
read and understood the information
about infectious diseases transmissible by
transfusion and has given accurate and
truthful answers to the medical history
questions. Wording equivalent in mean-
ing to the following is suggested:
“I have read and understand the infor-
mation provided to me regarding the spread
of the AIDS virus (HIV) by blood and
plasma. If I am potentially at risk for
spreading the virus known to cause AIDS, I
agree not to donate blood or plasma for
transfusion to another person or for further
manufacture. I understand that my blood
will be tested for HIV and other disease
markers; however, there may be unforeseen
circumstances when infectious disease test-
ing may not be performed. If this testing in-
dicates that I should no longer donate blood
or plasma because of the risk of transmitting
an infectious disease, my name will be en-
tered on a list of permanently deferred do-
nors. I understand that I will be notified of a
positive laboratory test result(s). If, instead,
the results of the testing are not clearly nega-
tive or positive, my blood will not be used
and my name may be placed on a deferral
list.”
If units are occasionally used for reasons
other than transfusion, such as research,
then the informed consent should address
such occasions.
Special Donor Categories
Exceptions to the usual eligibility require-
ments may be made for special donor cat-
egories:
1. Autologous donors: The indications
for collection and variations from
usual donor procedures are discus-
sed in Chapter 5.
2. Hemapheresis: Special requirements
and recommendations for cytaphere-
sisdonorsorfordonorsinaplasma
-
pheresis program are detailed in
Chapter 6.
3. Recipient-specific “designated” dona-
tions: Under certain circumstances,
it may be important to use blood or
components from a specific donor
for a specific patient. Examples in-
clude the patient with an antibody
to a high-incidence antigen or a
combination of antibodies that makes
Chapter 4: Allogeneic Donor Selection and Blood Collection 103
Copyright © 2005 by the AABB. All rights reserved.
it difficult to find compatible blood;
the infant with neonatal alloimmune
thrombocytopenia whose mother
can provide platelets; the patient
awaiting a kidney transplant from a
living donor; or the multitransfused
patient whose family members can
provide components.
The repeated use of a single donor
to supply components needed for a
single patient is allowed, provided it
is requested by the patient’s physi-
cian and approved by the donor cen-
ter physician. The donor must meet
all the usual requirements for dona-
tion, except that the frequency of
donation can be as often as every 3
days, as long as the predonation he-
moglobin level meets or exceeds the
minimum value for routine allo-
geneic blood donation.
The blood must be processed ac-
cording to AABB Standards for Blood
Banks and Transfusion Services.1(pp24-32)
Special tags identifying the donor
unit number and the intended recip-
ient must be affixed to the blood or
component bag, and all such units
must be segregated from the normal
inventory. A protocol for handling
such units must be included in the
SOP manual.
4. Directed donors: The public’s con-
cern about the safety of transfusion
has generated demands from poten-
tial recipients to choose the donors
to be used for their transfusions.
Several states have laws establishing
this as an acceptable procedure that
must be offered by a donor service
in nonemergency situations, if re-
quested by a potential blood recipi-
ent or ordered by a physician. Despite
logistic and philosophic problems
associated with these “directed” do-
nations, most blood centers and
hospitals provide this service. The
selection and testing of directed do-
nors should be the same as for other
allogeneic donors, although special
exemptions to the 56-day (or 112
days for double red cell donation)
waiting period between donations
may be made. Federal regulations
state that a person may serve as a
source of Whole Blood more than
once in 8 weeks only if at the time of
donation the donor is examined and
certified by a physician to be in good
health.12 To avoid misunderstand-
ings, it is important to establish SOPs
that define the interval required be-
tween collection of the blood and its
availability to the recipient; the policy
about determining ABO group before
collection; and the policy for releas-
ing units for use by other patients.
Collection of Blood
Blood is to be collected only by trained
personnel working under the direction of
a qualified licensed physician. Blood col-
lection must be by aseptic methods, using
a sterile closed system. If more than one
skin puncture is needed, a new container
and donor set must be used for each addi-
tional venipuncture unless the SOP allows
the use of an FDA-approved device to at-
tach a new needle while preserving steril-
ity. The phlebotomist must sign or initial
the donor record, even if the phlebotomy
did not result in the collection of a full
unit.
Blood Containers
Blood must be collected into an FDA-ap-
proved container that is pyrogen-free and
sterile and contains sufficient anticoagu-
lant for the quantity of blood to be col-
104 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
lected. The container label must state the
type and amount of anticoagulant and the
approximate amount of blood collected.
Blood bags may be supplied in packages
containing more than one bag. The manu-
facturer’s directions should be followed for
the length of time unused bags may be
stored in packages that have been opened.
Identification
Identification is essential in each step
from donor registration to final disposi-
tion of each component. A numeric or al-
phanumeric system must be used that
identifies, and relates to, the source do-
nor, the donor record, the specimens used
for testing, the collection container, and
all components prepared from the unit.
Extreme caution is necessary to avoid any
mix-up or duplication of numbers. All re-
cords and labels should be checked be-
fore use for printing errors. If duplicate
numbers are found, they must be re-
moved and may be investigated to ascer-
tain the reason for the duplication (eg,
supplier error, etc). A record must be kept
of all voided numbers.
Before beginning the collection, the
phlebotomist should:
1. Identify the donor record (at least by
name) with the donor and ask the
donor to state or spell his or her
name.
2. Attach numbered labels to the donor
record and ensure that it matches
the blood collection container, at-
tached satellite bags, and tubes for
donor blood samples. Attaching the
numbers at the donor chair, rather
than during the examination proce-
dures, helps reduce the likelihood of
identification errors.
3. Be sure that the processing tubes are
correctly numbered and that they
accompany the container during the
collection of blood. Tubes may be at-
tached in any convenient manner to
theprimarybagorintegraltubing.
4. Recheck all numbers.
Preparation of the Venipuncture Site
Blood should be drawn from a large firm
vein in an area (usually the antecubital
space) that is free of skin lesions. Both
arms must be inspected for evidence of
drug use, skin disease, or scarring. A tour-
niquet or a blood pressure cuff inflated to
40 to 60 mm Hg makes the veins more
prominent. Having the donor open and
close the hand a few times is also helpful.
Once the vein is selected, the pressure de-
vice should be released before the skin
site is prepared.
There is no way to make the veni-
puncture site completely aseptic, but surgi-
cal cleanliness can be achieved to provide
the best assurance of an uncontaminated
unit. Several acceptable procedures exist
(see Method 6.2). After the skin has been
prepared, it must not be touched again to
repalpate the vein. The entire site prepara-
tion must be repeated if the cleansed skin is
touched.
Phlebotomy and Collection of Samples
A technique for drawing a donor unit and
collecting samples for testing appears in
Method 6.3. The unit should be collected
from a single venipuncture after the pres-
sure device has again been inflated. Dur-
ing collection, the blood should be mixed
with the anticoagulant. The amount of
blood collected should be monitored care-
fully so that the total, including samples,
does not exceed 10.5 mL per kilogram of
donor weight per donation.1(p61) When the
appropriate amount has been collected,
segments and specimen tubes must be
filled. The needle and any blood-contam-
inatedwastemustbedisposedofsafelyin
Chapter 4: Allogeneic Donor Selection and Blood Collection 105
Copyright © 2005 by the AABB. All rights reserved.
accordance with universal precaution
guidelines. The needle must not be re-
capped unless a safety recapping device is
used. Disposal of the needle must be in a
puncture-proof container. After collec-
tion, there must be verification that the
identifiers on the unit, the donor history,
and the tubes are the same. Gloves must
be available for use during phlebotomy
and must be worn by phlebotomists when
collecting autologous blood and when in-
dividuals are in training.
Care of the Donor After Phlebotomy
After removing the needle from the vein,
the phlebotomist should:
1. Apply firm pressure with sterile gauze
over the point of entry of the needle
into vein. (The donor may be in-
structed to continue application of
pressure for several minutes.) Check
arm and apply a bandage only after
all bleeding stops.
2. Have donor remain reclining on the
bedorinthedonorchairforafew
minutes under close observation by
staff.
3. Allow the donor to sit up under ob-
servation until his or her condition
appears satisfactory. The donor should
be observed in the upright position
before release to the observation/re-
freshment area. Staff should monitor
donors in this area. The period of ob-
servation and provision of refreshment
should be specified in the SOP manual.
4. Give the donor instructions about
postphlebotomy care. The medical
directormaywishtoincludesome
or all of the following recommenda-
tions or instructions:
a. Eat and drink something before
leaving the donor site.
b. Do not leave until released by a
staff member.
c. Drink more fluids than usual in
the next 4 hours.
d. Avoid consuming alcohol until
something has been eaten.
e. Do not smoke for 30 minutes.
f. If there is bleeding from the
phlebotomy site, raise arm and
apply pressure to the site.
g. If fainting or dizziness occurs,
either lie down or sit with the
head between the knees.
h. If any symptoms persist, either
telephone or return to the do-
norcenterorseeadoctor.
i. Resume all normal activities if
asymptomatic. Donors who work
in certain occupations (eg, con-
struction workers, operators of
machinery) or persons working
at heights should be cautioned
that dizziness or faintness may
occur if they return to work im-
mediately after giving blood.
j. Remove bandage after a few
hours.
k. Maintain high fluid intake for
several days to restore blood
volume.
5. Thank the donor for an important
contribution and encourage repeat
donation after the proper interval.
All personnel on duty throughout
the donor area, volunteer or paid,
should be friendly and qualified to
observe for signs of a reaction such
as lack of concentration, pallor, rapid
breathing, or excessive perspiration.
Donor room personnel should be
trained and competent to interpret
instructions, answer questions, and
accept responsibility for releasing
the donor in good condition.
6. Note on the donor record any ad-
verse reactions that occurred. If the
donor leaves the area before being
released, note this on the record.
106 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Adverse Donor Reactions
Most donors tolerate giving blood very
well, but adverse reactions occur occa-
sionally. Personnel must be trained to rec-
ognize adverse reactions and to provide
initial treatment.
Donor room personnel should be train-
ed in cardiopulmonary resuscitation (CPR).
Special equipment to handle emergency
situations must be available.
Syncope (fainting or vasovagal syn-
drome)maybecausedbythesightof
blood, by watching others give blood, or by
individual or group excitement; it may also
happen for unexplained reasons. Whether
caused by psychologic factors or by neuro-
physiologic response to blood donation, the
symptoms may include weakness, sweat-
ing, dizziness, pallor, loss of consciousness,
convulsions, and involuntary passage of fe-
ces or urine. On occasion, the skin feels
cold and blood pressure falls. Sometimes,
the systolic blood pressure levels fall as low
as 50 mm Hg or cannot be heard with the
stethoscope. The pulse rate often slows sig-
nificantly. This can be useful in distinguish-
ing between vasovagal attack and cardio-
genic or hypovolemic shock, in which cases
the pulse rate rises. This distinction, al-
though characteristic, is far from abso-
lute.
Rapid breathing or hyperventilation may
cause the anxious or excited donor to lose
excessive amounts of carbon dioxide. This
may cause generalized sensations of suffo-
cation or anxiety, or localized problems
such as tingling or twitching.
The donor center physician must pro-
vide written instructions for handling do-
nor reactions, including a procedure for ob-
taining emergency medical help. Sample
instructions might be as follows:
1. General.
a. Remove the tourniquet and with-
draw the needle from the arm if
signs of a reaction occur during
the phlebotomy.
b. If possible, remove any donor
who experiences an adverse re-
action to an area where he or
she can be attended in privacy.
c. Apply the measures suggested
below and, if they do not lead
to rapid recovery, call the blood
bank physician or the physician
designated for such purposes.
2. Fainting.
a. Apply cold compresses to the
donor’s forehead or the back of
the neck.
b. Place the donor on his or her
back, with their legs raised above
the level of the head.
c. Loosen tight clothing.
d. Be sure the donor has an ade-
quate airway.
e. Monitor blood pressure, pulse,
and respiration periodically un-
til the donor recovers.
Note: Some donors who experi-
ence prolonged hypotension may re-
spondtoaninfusionofnormalsa-
line. The decision to initiate such
therapy should be made by the do-
nor center physician either on a case-
by-case basis or in a policy stated in
the facility’s SOP manual.
3. Nausea and vomiting.
a. Make the donor as comfortable
as possible.
b. Instruct the donor who is nau-
seated to breathe slowly and
deeply.
c. Apply cold compresses to the
donor’s forehead and/or back
of neck.
d. Turn the donor’s head to the side.
e. Provide a suitable receptacle if
the donor vomits and have clean-
sing tissues or a damp towel
ready. Be sure the donor’s head
Chapter 4: Allogeneic Donor Selection and Blood Collection 107
Copyright © 2005 by the AABB. All rights reserved.
is turned to the side because of
the danger of aspiration.
f. After vomiting has ended, give
the donor some water to rinse
out his or her mouth.
4. Twitching or muscular spasms. Ex-
tremely nervous donors may hyper-
ventilate, causing faint muscular
twitching or tetanic spasm of their
hands or face. Donor room person-
nel should watch closely for these
symptoms during and immediately
after the phlebotomy.
a. Divert the donor’s attention by
engaging in conversation, to
interrupt the hyperventilation
pattern.
b. Have the donor cough if he or
she is symptomatic. Do not give
oxygen.
5. Hematoma during or after phlebotomy.
a. Remove the tourniquet and the
needle from the donor’s arm.
b. Place three or four sterile gauze
squares over the venipuncture
site and apply firm digital pres-
sure for 7 to 10 minutes, with
the donor’s arm held above the
heart level. An alternative is to
apply a tight bandage, which
should be removed after 7 to 10
minutes to allow inspection.
c. Apply ice to the area for 5 min-
utes, if desired.
d. Should an arterial puncture be
suspected, immediately with-
draw needle and apply firm
pressure for 10 minutes. Apply
pressure dressing afterwards.
Check for the presence of a ra-
dial pulse. If pulse is not palpa-
ble or is weak, call a donor cen-
ter physician.
6. Convulsions.
a. Call for help immediately. Pre-
vent the donor from injuring him-
self or herself. During severe
seizures, some people exhibit
great muscular power and are
difficult to restrain. If possible,
hold the donor on the chair or
bed; if not possible, place the
donor on the floor. Try to pre-
vent injury to the donor and to
yourself.
b. Be sure the donor has an ade-
quate airway. A padded device
should separate the jaws after
convulsion has ceased.
c. Notify the donor center physi-
cian.
7. Serious cardiac difficulties.
a. Call for medical aid and/or an
emergency care unit immediately.
b. If the donor is in cardiac arrest,
begin CPR immediately and
continue it until help arrives.
The nature and treatment of all reactions
should be recorded on the donor’s record
or a special incident report form. This should
include a notation of whether the donor
should be accepted for future donations.
The medical director should decide what
emergency supplies and drugs should be in
the donor area. The distance to the nearest
emergency room or emergency care unit
heavily influences decisions about neces-
sary supplies and drugs. Most donor cen-
ters maintain some or all of the following:
1. Emesis basin or equivalent.
2. Towels.
3. Oropharyngeal airway, plastic or hard
rubber.
4. Oxygen and mask.
5. Emergency drugs: Drugs are seldom
required to treat a donor’s reaction.
If the donor center physician wishes
to have any drugs available, the kind
andamounttobekeptonhandmust
be specified in writing. In addition,
the medical director must provide
written policies stating when and by
108 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
whom any of the above medical sup-
plies or drugs may be used.
References
1. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
2. Code of federal regulations. Title 21 CFR
606.160(e). Washington, DC: US Government
Printing Office, 2004 (revised annually).
3. Beattie KM, Shafer AW. Broadening the base
of a rare donor program by targeting minority
populations. Transfusion 1986;26:401-4.
4. Vichinsky EP, Earles A, Johnson RA, et al. Allo-
immunization in sickle cell anemia and
transfusion of racially unmatched blood. N
Engl J Med 1990;322:1617-21.
5. Food and Drug Administration. Memoran-
dum: Revised recommendations for the pre-
vention of human immunodeficiency virus
(HIV) transmission by blood and blood prod-
ucts. (April 23, 1992) Rockville, MD: CBER Of-
fice of Communication, Training, and Manu-
facturers Assistance, 1992.
6. Centers for Disease Control. Update: Univer-
sal precautions for prevention of transmis-
sion of human immunodeficiency virus, hep-
atitis B virus, and other bloodborne pathogens
in health-care settings. JAMA 1988;260:528-31.
7. Code of federal regulations. Title 21 CFR
640.4(a). Washington, DC: US Government
Printing Office, 2004 (revised annually).
8. Food and Drug Administration. Guidance for
Industry: Streamlining the donor interview
process: Recommendations for self-adminis-
tered questionnaires. (July 3, 2003) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 2003.
9. Armed Services Blood Program Office. Drugs
and medications. [Available at http://www.
tricare.osd.mil/asbpo/library/policies/
downloads/medication_list.doc.]
10. Newman B. Blood donor suitability and
allogeneic whole blood donation. Transfus
Med Rev 2001;15:234-44.
11. Food and Drug Administration. General re-
quirements for blood, blood components, and
blood derivatives; donor notification. Title 21
CFR 630.6. Fed Regist 2001;66:31165-77.
12. Code of federal regulations. Title 21 CFR
640.3(f). Washington, DC: US Government
Printing Office, 2004 (revised annually).
Suggested Reading
Code of federal regulations. Title 21 CFR 640.3.
Washington, DC: US Government Printing Office,
2004 (revised annually). [History of viral hepatitis
before the 11th birthday.]
Food and Drug Administration. Memorandum:
Revised recommendations for the prevention of
human immunodeficiency virus (HIV) transmis-
sion by blood and blood products. (April 23, 1992)
Rockville, MD: CBER Office of Communication,
Training, and Manufacturers Assistance, 1992.
Food and Drug Administration. Guidance for in-
dustry: Revised preventive measures to reduce the
possible risk of transmission of Creutzfeldt-Jakob
disease (CJD) and new variant Creutzfeldt-Jakob
disease (nvCJD) by blood and blood products.
(January 9, 2002) Rockville, MD: CBER Office of
Communication, Training, and Manufacturers
Assistance, 2002.
Infectious disease testing for blood transfusions.
NIH Consensus Statement 13:1, January 1995.
Bethesda, MD: National Institutes of Health, 1995.
Kasprisin C, Laird-Fryer B, eds. Blood donor col-
lection practices. Bethesda, MD: AABB, 1993.
Linder J, ed. Practical solutions to practical prob-
lems in transfusion medicine and tissue banking.
[Supplement 1 to Am J Clin Pathol 1997;107(4).]
Chicago, IL: American Society of Clinical Patholo-
gists, 1997.
Schmuñis GA. Trypanosoma cruzi,theetiologic
agent of Chagas’ disease: Status in the blood sup-
ply in endemic and nonendemic countries. Trans-
fusion 1991;31:547-57.
Smith KJ, Simon TL. Recruitment and evaluation
of blood and plasma donors. In: Rossi EC, Simon
TL, Moss GS, eds. Principles of transfusion medi-
cine. 2nd ed. Baltimore, MD: Williams and Wilkins,
1995:871-9.
Tan L, Williams MA, Khan MK, et al. Risk of trans-
mission of bovine spongiform encephalopathy to
humans in the United States. JAMA 1999;281:2330-8.
Chapter 4: Allogeneic Donor Selection and Blood Collection 109
Copyright © 2005 by the AABB. All rights reserved.

110 AABB Technical Manual
Appendix 4-1. Full-Length Donor History Questionnaire*
Copyright © 2005 by the AABB. All rights reserved.

Chapter 4: Allogeneic Donor Selection and Blood Collection 111
Appendix 4-1. Full-Length Donor History Questionnaire (cont’d)*
Copyright © 2005 by the AABB. All rights reserved.

112 AABB Technical Manual
Appendix 4-1. Full-Length Donor History Questionnaire (cont’d)*
*Downloaded from http://www.aabb.org on April 19, 2005. Check web site for updates.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 4: Allogeneic Donor Selection and Blood Collection 113
Appendix 4-2. Medication Deferral List*
*Downloaded from http://www.aabb.org on April 19, 2005. Check web site for updates.
Copyright © 2005 by the AABB. All rights reserved.

114 AABB Technical Manual
Appendix 4-3. Blood Donor Education Materials*
*Downloaded from http://www.aabb.org on April 19, 2005. Check web site for updates.
Copyright © 2005 by the AABB. All rights reserved.
Chapter 4: Allogeneic Donor Selection and Blood Collection 115
Appendix 4-4. Some Drugs Commonly Accepted in Blood Donors
In many blood centers, blood donation may be allowed by individuals who have taken the follow-
ing drugs:
■Tetracyclines and other antibiotics taken to treat acne.
■Topical steroid preparations for skin lesions not at the venipuncture site.
■Blood pressure medications, taken chronically and successfully so that pressure is at or below
allowable limits. The prospective donor taking antihypertensive drugs should be free from side
effects, especially episodes of postural hypotension, and should be free of any cardiovascular
symptoms.
■Over-the-counter bronchodilators and decongestants.
■Oral hypoglycemic agents in well-controlled diabetics without any vascular complications of the
disease.
■Tranquilizers, under most conditions. A physician should evaluate the donor to distinguish
between tranquilizers and antipsychotic medications.
■Hypnotics used at bedtime.
■Marijuana (unless currently under the influence), oral contraceptives, mild analgesics, vitamins,
replacement hormones, or weight reduction pills.
Note: Acceptance of donors must always be with the approval of the blood bank’s medical director.
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 5: Autologous Blood Donation and Transfusion
Chapter 5
Autologous Blood Donation
and Transfusion
AUTOLOGOUS BLOOD TRANSFU-
sion is an alternative therapy for
many patients anticipating trans-
fusion. Different categories of autologous
transfusion are:
1. Preoperative collection (blood is drawn
and stored before anticipated need).
2. Perioperative collection and admin-
istration.
a. Acute normovolemic hemodi-
lution (blood is collected at the
start of surgery and then in-
fused during or at the end of
the procedure).
b. Intraoperative collection (shed
blood is recovered from the
surgical field or circulatory de-
vice and then infused).
c. Postoperative collection (blood
is collected from drainage de-
vices and reinfused to the pa-
tient).
Each type of autologous transfusion
practice offers potential benefits and risks
depending on the type of surgery, condition
of the patient, and technology available.
Each facility must analyze its own transfu-
sion practices, transfusion practices of
other similarly situated institutions, and its
own capabilities to determine the appropri-
ate services to be offered.
However, it is generally accepted that,
when feasible, the patient should have the
option to use his or her own blood. The US
Supreme Court has ruled that asymptom-
atic infection with HIV is a disability pro-
tected under the Americans with Disabili-
ties Act.1Therefore, if institutions offer
autologous services to any patient, they
should consider offering such services to
HIV-positive patients.2Patients who are
likely to require transfusion therapy and
whoalsomeetthedonationcriteriashould
be told about the options for autologous
transfusion therapies. Patients considering
autologous transfusion therapy should be
informed about the risks and benefits of both
the autologous donation and the auto-
117
A
5
Copyright © 2005 by the AABB. All rights reserved.
logous transfusion process. Specific issues
unique to the use of autologous transfusion
in the anticipated surgical procedure should
be identified, including the possibility of
administrative error. In addition, patients
need information about any special fees for
autologous services, the level of infectious
disease testing that will be performed, and
the possibility that additional, allogeneic,
units may be used.
Preoperative Autologous
Blood Collection
Frequently cited advantages and disad-
vantages of preoperative autologous blood
donation (PAD) are summarized in Table
5-1. Candidates for preoperative collec-
tion are stable patients scheduled for sur-
gical procedures in which blood transfu-
sion is likely. For procedures that are
unlikely to require transfusion (ie, a maxi-
mal surgical blood ordering schedule
does not suggest that crossmatched blood
be available), the use of preoperative blood
collection is not recommended.
In selected patient subgroups, preopera-
tive collection of autologous blood can sig-
nificantly reduce exposure to allogeneic
blood. PAD collections should be considered
for patients likely to receive transfusion,
such as patients undergoing major ortho-
pedic procedures, vascular surgery, and
cardiac or thoracic surgery.3,4 The most
common surgical procedures for which
autologous blood is donated are total joint
replacements.4Autologous blood should not
be collected for procedures that seldom
(less than 10% of cases) require transfusion,
such as cholecystectomy, herniorrhaphy,
vaginal hysterectomy, and uncomplicated
obstetric delivery.5
Special Patient Categories
In special circumstances, preoperative
autologous blood collection can be per-
formed for patients who would not ordi-
narily be considered for allogeneic dona-
tion. The availability of medical support is
important in assessing patient eligibility.
With suitable volume modification, pa-
rental cooperation, and attention to prep-
aration and reassurance, pediatric patients
118 AABB Technical Manual
Table 5-1. Autologous Blood Donation
Advantages Disadvantages
1. Prevents transfusion-transmitted disease. 1. Does not eliminate risk of bacterial contam-
ination.
2. Prevents red cell alloimmunization. 2. Does not eliminate risk of ABO incompati-
bility error.
3. Supplements the blood supply. 3. Is more costly than allogeneic blood.
4. Provides compatible blood for patients with
alloantibodies.
4. Results in wastage of blood that is not
transfused.
5. Prevents some adverse transfusion reac-
tions.
5. Increased incidence of adverse reactions to
autologous donation.
6. Provides reassurance to patients con-
cerned about blood risks.
6. Subjects patients to perioperative anemia
and increased likelihood of transfusion.
Copyright © 2005 by the AABB. All rights reserved.

can participate in preoperative collection
programs.6The successful use of autolo-
gous blood in a patient with sickle cell
disease has been reported,7and it may be
particularly useful for a sickle cell patient
with multiple alloantibodies; however, the
patient may derive greater benefit from
allogeneic transfusions that provide he-
moglobin A. Red cells containing hemo-
globin S require special handling during
the cryopreservation process.8
Patients with significant cardiac disease
are considered poor risks for autologous
blood donation. Despite reports of safety in
small numbers of patients who underwent
autologous blood donation,9the risks that
are associated with autologous blood dona-
tion10 in these patients are probably greater
than the current estimated risks of allo-
geneic transfusion.11,12 Table 5-2 summa-
rizes the contraindications to a patient’s
participation in an autologous blood dona-
tion program.13
The collection of autologous blood from
women during routine pregnancy is unwar-
ranted,14 because blood is so seldom needed.
Many centers give serious consideration to
autologous collection for women with allo-
antibodies to multiple or high-incidence
antigens, placenta previa, or other conditions
placing them at high risk for ante- or intra-
partum hemorrhage.5Apolicyshouldbe
developed for situations in which maternal
red cells are considered for transfusion to
the infant.
Voluntary Standards
AABB Standards for Blood Banks and Trans-
fusion Services offers uniform standards to
be followed in determining patient eligi-
bility; collecting, testing, and labeling the
unit; and pretransfusion testing.15(pp18,39,51)
These AABB standards apply to preopera-
tive autologous blood collection. Stan-
dards for Perioperative Autologous Blood
Collection and Administration have been
established to enhance the quality and
safety of perioperative autologous trans-
fusion activities (intra- and postoperative
blood recovery, perioperative autologous
component production, and intraopera-
tive acute normovolemic hemodilution).16
Compliance Considerations
Food and Drug Administration (FDA) re-
quirements have evolved over time. The
FDA first issued guidance for autologous
blood and blood components in March of
1989.17 This guidance was clarified in a
second memorandum issued in February
of 1990.18 Much of the information in pre-
vious guidance has been superseded by
regulations. The FDA included requirements
regarding autologous blood in regulations
issued June 11, 2001.19,20
Testing
The FDA requires tests for evidence of in-
fection resulting from the following com-
Chapter 5: Autologous Blood Donation and Transfusion 119
Table 5-2. Contraindications to
Participation in Autologous Blood
Donation Programs
1. Evidence of infection and risk of
bacteremia.
2. Scheduled surgery to correct aortic steno-
sis.
3. Unstable angina.
4. Uncontrolled seizure disorder.
5. Myocardial infarction or cerebrovascular
accident within 6 months of donation.
6. Patients with significant cardiac or pulmo-
nary disease who have not yet been cleared
for surgery by their treating physician.
7. High-grade left main coronary artery dis-
ease.
8. Cyanotic heart disease.
9. Uncontrolled hypertension.
Copyright © 2005 by the AABB. All rights reserved.
municable diseases: HIV-1, HIV-2, HBV,
HCV, HTLV-I, and HTLV-II (21 CFR 610.40)
and a serologic test for syphilis [21 CFR
610.40(a)(1) and 610.40(I)]. Such tests in-
clude nucleic acid tests for HCV and HIV.21
Testing of autologous donations is not
required unless the donations are to be
used for allogeneic transfusion [21 CFR
610.40(d)(1)]. Autologous donations that
are to be shipped to another facility that
does allow autologous units to be used for
allogeneic transfusion must be tested [21
CFR 610.40(d)(2)]. For autologous dona-
tions shipped to another establishment
that does not allow autologous donations
to be used for allogeneic transfusion, the
first donation in each 30-day period must
be tested [21 CFR 610.40(d)(3)].15(p34)
Autologous donations found to be reac-
tive by a required screening test must be re-
tested whenever a supplemental (additional,
more specific) test has been approved by
the FDA. At a minimum, the first reactive
donation in each 30-day period must be
tested unless a record exists for a positive
supplemental test result for that donor [21
CFR 610.40(e)(1,2)]. Both the AABB Stan-
dards15(p34) and the FDA requirements [21
CFR 630.6(d)] state that the patient and the
patient’s physicians must be notified of any
medically significant abnormalities.
Donor Deferral
If an autologous donor has a reactive
screening test for a communicable dis-
ease agent or a reactive screening test for
syphilis (21 CFR 610.41), the donor must
be deferred from making future allogeneic
donations. Within 8 weeks, the patient
and referring physician must be notified
of the reason for deferral, including the
results of supplemental testing and, where
appropriate, the types of donation that the
autologous donor should not make in the
future (21 CFR 630.6).
Special Labeling Considerations
Each autologous unit must be labeled
“Autologous Donor.” Another special label,
“BIOHAZARD,” is required for any unit
that is reactive in the current collection or
reactive in the last 30 days. Autologous
units that are untested must be labeled
“DONOR UNTESTED.” If the autologous
unit tested negative within the last 30
days,itmustbelabeled“DONORTESTED
WITHIN THE LAST 30 DAYS” [21 CFR
610.40(d)(4)].
Shipping
Blood or components (including reactive
donations) intended for autologous use
may be shipped provided that the units
have been tested as required and are la-
beled appropriately [21 CFR 610.40(d)]. If
distributed on a common carrier not un-
der the direct control of the collection fa-
cility, the transportation of the product
must meet provisions for shipping an in-
fectious agent.22
Establishing a Preoperative Autologous
Blood Collection Program
Each blood center or hospital that decides
to conduct an autologous blood collection
program must establish its own policies,
processes, and procedures. Guidelines ex-
ist for establishing a new program, moni-
toring utilization, or improving an exist-
ing one.13,23,24
Physician Responsibility
A successful autologous program requires
cooperation and communication among
all the physicians involved. Responsibility
for the health and safety of the patient
during the collection process rests with the
medical director of the collecting facility;
during the transfusion, responsibility rests
120 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
with the patient’s physician and the medi-
cal director of the transfusion service. The
patient’s physician initiates the request
for autologous services, which must be
approved by the transfusion service phy-
sician. There should be a transfusion
medicine physician available to help as-
sess patients whose medical history sug-
gests a risk for complications if a donor
reaction occurs during blood collection.
Supplemental Iron
The patient should be advised about tak-
ing supplemental iron. Ideally, supple-
mental iron is prescribed by the requesting
physician before the first blood collection,
in time to allow maximum iron intake.
Iron-restricted erythropoiesis is one of the
limiting factors in collecting multiple
units of blood over a short interval. Oral
iron is commonly provided but may be
insufficient to maintain iron stores.25 The
dose and administration schedule should
be adjusted to minimize gastrointestinal
side effects.
Collection
The collection of autologous blood has
many elements in common with collec-
tion from regular volunteer donors, but
numerous special considerations exist.
Requests for autologous blood collection
aremadeinwritingbythepatient’sphysi
-
cian; a request form (which may be a sim-
ple prescription or a form designed for
the purpose) is kept by the collecting fa-
cility. The request should include the
patient’s name, a unique identification
number, the number of units and kind of
component requested, the date of sched-
uled surgery, the nature of the surgical
procedure, and the physician’s signature.
It is important to establish guidelines for
the appropriate number of units to be col-
lected. A sufficient number of units should
be drawn, whenever possible, so that the
patient can minimize exposure to allo-
geneic blood. However, excessive collection
and/or collection close to the date of sur-
gery increases the patient’s likelihood of re-
quiring transfusion. A hospital’s surgical
blood order schedule can provide estimates
of transfusion levels for specific procedures.
Two-unit collections via an automated red
cell apheresis system may be an option. The
collection of units for liquid blood storage
should be scheduled as far in advance of
surgery as possible, in order to allow comp-
ensatory erythropoiesis to minimize anemia.
A schedule for blood collections should
be established with the patient. A weekly
schedule is often used. Table 5-3 details the
value of beginning autologous blood dona-
tion early in the known preoperative inter-
val, in order to allow optimal compensatory
erythropoiesis (shown here as equivalent
RBC units).26 Ordinarily, the last collection
should occur no sooner than 72 hours be-
fore the scheduled surgery and preferably
longer, to allow time for adequate volume
repletion. Programs should notify the re-
questing physician of the total number of
units donated when the requested number
of units cannot be collected. Each program
should establish a policy regarding re-
scheduling of surgery beyond the expira-
tion date of autologous units and whether
discarding or freezing the unit are options.
Donor Screening
Because of the special circumstances re-
garding autologous blood transfusion,
rigid criteria for donor selection are not
required. In situations where require-
ments for allogeneic donor selection or
collection are not applied, alternative re-
quirements must be established by the
medical director and recorded in the pro-
cedures manual. The hemoglobin con-
centration of the donor should be no less
Chapter 5: Autologous Blood Donation and Transfusion 121
Copyright © 2005 by the AABB. All rights reserved.

than 11.0 g/dL and the hematocrit, if sub-
stituted, should be no less than 33%. Indi-
vidual deviations from the alternate re-
quirements must be approved by the blood
bank medical director, usually in consul-
tation with the donor-patient’s physician.
Medical Interview
The medical interview should be struc-
tured to meet the special needs of autolo-
gous donors. For example, more attention
should be given to questions about medi-
cations, associated medical illnesses, and
cardiovascular risk factors.10 Questions
should elicit any possibility of intermittent
bacteremia. Because crossover is not rou-
tinely permitted, a substantially shortened
set of interview questions can be used for
autologous donations; for example, ques-
tions related to donor risks for transfusion-
transmitted diseases are not necessary.
Volume Collected
For autologous donors weighing >50 kg,
the 450-mL collection bag is usually used
instead of the 500-mL bag, in case the do-
nor cannot give a full unit. If a low-volume
(300-405 mL) unit is collected, the red cells
are suitable for storage and subsequent
autologous transfusion. The plasma from
low-volume units cannot be transfused
because of the abnormal anticoagulant/
plasma ratio. Under-collected units (<300
mL) may be suitable for autologous use
with approval of the medical director. For
patients weighing <50 kg, there should be
a proportional reduction in the volume of
blood collected. Regardless of donor weight,
the volume collected should not exceed
10.5 mL/kg of the donor’s estimated body
weight, including the samples for testing.
Serologic Testing
The collecting facility must determine
ABO and Rh type on all units. Transfusing
facilities must retest ABO and Rh type on
units drawn at other facilities, unless the
collecting facility tests segments from the
unit according to AABB Standards.15(p37)
Testing for ABO and Rh type must be
performed on a properly labeled blood
sample from the patient. An antibody
screen should be performed to provide for
the possible need for allogeneic blood.
Labeling
Units should be clearly labeled with the
patient’s name and an identifying num-
ber, the expiration date of the unit, and, if
available, the name of the facility where
the patient is to be transfused. The unit
should be clearly marked “For Autologous
Use Only” if intended for autologous use
only. If components have been prepared,
122 AABB Technical Manual
Table 5-3. Timing and Red Cell Regeneration During Preoperative Autologous
Donation26
Time from Donation to
Surgery (days)
No. of
Patients
Mean RBC Units
Regenerated
5% CI
of Mean
6-13 39 0.52 0.25-0.79
14-20 127 0.54 0.40-0.68
21-27 128 0.75 0.61-0.90
28-34 48 1.16 0.96-1.36
35-41 30 1.93 1.64-2.2
Copyright © 2005 by the AABB. All rights reserved.
the container of each component must be
similarly labeled. A biohazard label must
be applied when indicated by FDA require-
ments (see Compliance Considerations).
Labeling requirements for autologous units
are detailed in the AABB Standards,15(p51)
which parallels the FDA regulations.
Storage
Collection should be scheduled to allow
for the longest possible shelf life for col-
lected units. This increases flexibility for
the patient and the collecting facility and
allows time for the patient to rebuild red
cell mass during the interval between
blood collection and surgery. Liquid stor-
age is feasible for up to 6 weeks. Some
programs store autologous units as Whole
Blood for 35 days rather than as RBCs;
Whole Blood is simpler to store, and the
risk of volume overload subsequent to
transfusion is low. The collection of autol-
ogous units more than 6 weeks before
scheduled surgery has been described,
but requires that the red cells be frozen.
Although this provides more time for the
donor to recover lost red cell mass, freez-
ing and thawing add to the cost of the
program, reduce the volume of red cells
through processing losses, and compli-
cate blood availability during the peri-
operative period.
Transfusion of Autologous Units
Autologous transfusion programs should
have a system to ensure that if autologous
blood is available, it be issued and used
before allogeneic components are given.
A special “autologous” label may be used
with numbering to ensure that the oldest
units are issued first. Anesthesiologists,
surgeons, and physicians should be edu-
cated about the importance of selecting
autologous components before allogeneic
units are given, and a policy should be in
placeregardingtheissueofautologous,
allogeneic, and/or directed units to the
operating room.
Records
AABB standards for the proper issue and
return of unused autologous units are the
same as for allogeneic units.15(pp45,71) Re-
cords must be maintained that identify
the unit and all components made from
it, from collection and processing through
their eventual disposition.
Adverse Reactions
The investigation of suspected adverse
transfusion events should be the same for
autologous and allogeneic units. Autolo-
gous transfusions have a lower risk of in-
fectious and immune complications but
carry a similar risk of bacterial contami-
nation, volume overload, and misadminis-
tration compared with volunteer allogeneic
units. For these reasons, autologous blood
should not be transfused without a clear
indication for transfusion.
Continuous Quality Improvement
Several quality improvement issues have
been identified for PAD practices.5The
most important indicator for autologous
blood practice is how effectively it reduces
allogeneic transfusions to participating
patients. The “wastage” rate of autologous
units for surgical procedures can also be
monitored. However, even for procedures
such as joint replacement or radical pros-
tatectomy, a well-designed program may
result in 50% of collected units being un-
used (Fig 5-1).5,27 Nevertheless, as much as
25% of autologous blood is collected for
procedures that seldom require transfu-
sion, such as vaginal hysterectomies and
normal vaginal deliveries. Up to 90% of
units collected for these procedures are
wasted.14 The additional costs associated
Chapter 5: Autologous Blood Donation and Transfusion 123
Copyright © 2005 by the AABB. All rights reserved.
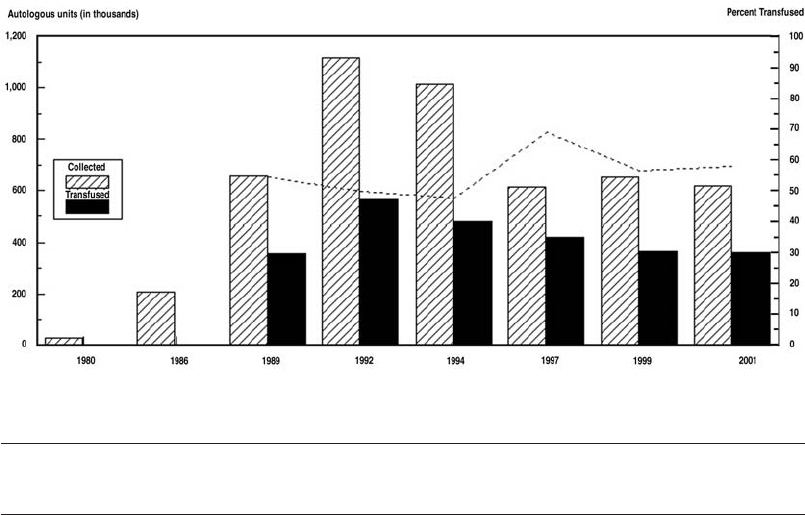
with the collection of autologous units,
along with advances in the safety of allo-
geneic blood, have altered the cost-effec-
tiveness of PAD in many situations.28 Such
cost-effectiveness analyses do not consider
an immunomodulatory effect of avoiding
allogeneic leukocytes, which remains
controversial.29,30
Criteria can be established to monitor
the appropriateness of autologous transfu-
sions.Thesecriteriamaybethesameas,or
different from, those established for allo-
geneic units.24 As with allogeneic blood,
transfusion of preoperatively donated auto-
logous blood carries the same risks associ-
ated with administrativeerrororbacterial
contamination. Autologous programs should
be monitored for unavailability of autolo-
gous blood when needed, the transfusion
of allogeneic blood before autologous
blood, and identification errors.
Evolving Issues in Preoperative
Autologous Services
Selection of Patients
Attempts to stratify patients into groups
at high and low risk for needing transfu-
sion based on the baseline level of hemo-
globin and on the type of procedure show
some promise. In a Canadian study using
a point score system, 80% of patients un-
dergoing orthopedic procedures were
identified to be at low risk (<10%) for
transfusion, so autologous blood procure-
ment for these patients would not be rec-
ommended.31 However, one problem with
algorithms that consider the estimated
blood loss and preoperative hematocrit is that
blood losses are difficult to measure32,33 or
predict because specific surgical proce-
dures performed even by the same sur-
124 AABB Technical Manual
Figure 5-1. Autologous RBC collection and transfusion data from 1980 to 2001 in the United States il-
lustrate the rise and fall of interest in PAD. The dashed line in the chart indicates the percent (right
axis) of collected PAD units transfused. (Modified with permission from Brecher and Goodnough.27)
Percent of Total RBCs that Were PAD RBCs
1980 1986 1989 1992 1994 1997 1999 2001
Collected 0.3 1.5 4.8 8.5 7.8 4.9 4.7 4.0
Transfused <1.0 3.1 5.0 4.3 3.7 3.0 2.6
Copyright © 2005 by the AABB. All rights reserved.
geon can be accompanied by a wide range
of blood losses.
The Role of Aggressive Phlebotomy and the
Use of Erythropoietin
The efficacy of PAD is dependent on the
degree to which the patient’s erythro-
poiesis increases the production of red
cells.25,34-36 The endogenous erythropoietin
response and compensatory erythropoie-
sis are suboptimal under “standard” con-
ditions of one blood unit donated weekly.
Weekly PAD is accompanied by an 11%
(with no oral iron supplementation) to 19%
(with oral iron supplementation) expan-
sion in red cell volume over a 3-week pe-
riod, which is not sufficient to prevent in-
creasing anemia in patients undergoing
PAD.34,35 If the erythropoietic response to
autologous blood phlebotomy is not able
to maintain the patient’s hematocrit level
during the donation interval, the dona-
tion of autologous blood may be harm-
ful.37 This outcome was confirmed in a
study of patients undergoing hysterecto-
mies,38 in which it was shown that PAD
resulted in perioperative anemia and an
increased likelihood of any blood transfu-
sion. A published mathematical model37
illustrates the relationship between antic-
ipated surgical blood losses, the level of
hematocrit that the physician may want
to maintain perioperatively, and the need
for autologous blood donation for indivi-
dual patients (Fig 5-2). Models such as
this may be helpful in designing autolo-
gous procurement programs or monitor-
ing their value through quality assurance.
In contrast to autologous blood donation
under “standard” conditions, studies of “ag-
gressive” autologous blood phlebotomy
(twice weekly for 3 weeks, beginning 25 to
35 days before surgery) have demonstrated
that endogenous erythropoietin levels do
increase, along with enhanced erythropoie-
sis representing 19% to 26% red cell volume
expansion.39-41 Exogenous (pharmacologic)
erythropoietin therapy to further stimulate
erythropoiesis (up to 50% red cell volume
expansion39-41) during autologous phlebot-
omy has been approved in Canada and Ja-
panbutnotintheUnitedStates.
42
Transfusion Trigger
Disagreement exists about the proper he-
moglobin/hematocrit level (“transfusion
trigger”) at which autologous blood should
be given.23 Autologous blood transfusion
is not without risks to the recipient; these
include misidentification of patients or
units, bacterial contamination of stored
units, and volume overload. The case can
be made that autologous and allogeneic
blood transfusion triggers should be simi-
lar because the additional mortality risks
of allogeneic blood now approach the risks
of mortality from administrative errors as-
sociated with both autologous and allo-
geneic blood.43 Data from a well-designed
clinical trial indicate that even critical care
patients can tolerate substantial anemia
(to hemoglobin ranges of 7 to 9 g/dL) with
no apparent benefit from more aggressive
transfusion therapy.44
Cost-Effectiveness
Although autologous blood collections have
become popular, the costs associated with
their collection are usually higher than
those associated with the collection of
allogeneic blood. The continued need for
autologous blood programs has been ques-
tioned because of the reduced risk of
allogeneic blood transfusions and pressure
to reduce health-care costs.27 Table 5-4 lists
suggestions for improving the efficiency
of hospital-based autologous blood pro-
grams without sacrificing safety.45
Chapter 5: Autologous Blood Donation and Transfusion 125
Copyright © 2005 by the AABB. All rights reserved.
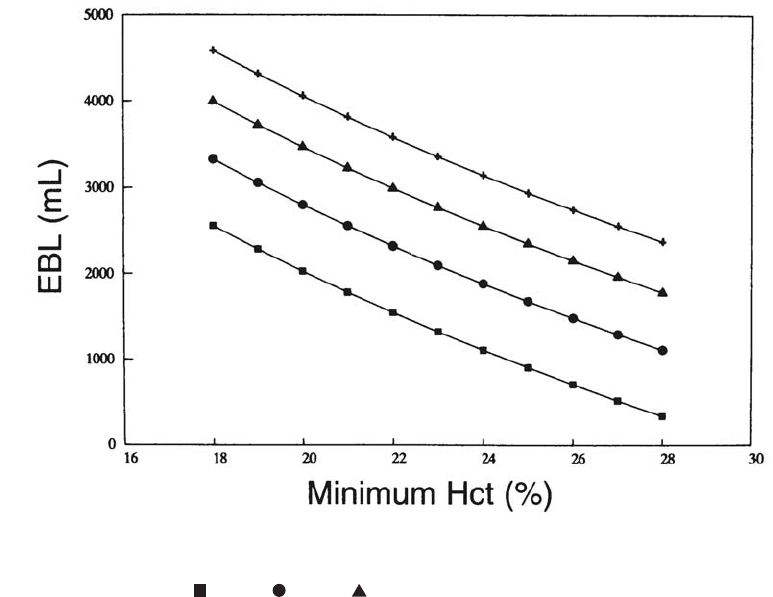
Preoperative Collection of Components
Some workers believe that preoperative or
intraoperative collection of platelet-rich
plasma during cardiopulmonary bypass
surgery may improve hemostasis and de-
crease allogeneic exposures, but others
have found no benefit.46 Preoperative col-
lection of autologous platelets, especially
in cardiac surgery, is often impractical be-
cause patients may be taking antiplatelet
drugs; surgery is often scheduled on an
emergency basis; and relatively low num-
bers of platelets are harvested.
Recently, autologous platelet collection
using commercial point-of-care collection
systems has been advocated to produce a
platelet gel for topical use.47 Platelet gel is
created by adding calcium chloride and
thrombin to a platelet concentrate. The
platelet gel serves as a rich source of pla-
telet-derived growth factors, which have
been reported in small studies to enhance
tissue repair and wound healing.48,49 Larger
randomized clinical trials are needed to es-
tablish the clinical efficacy of this product.
Platelet gel is not FDA-approved yet.
Acute Normovolemic
Hemodilution
Acute normovolemic hemodilution (ANH)
is the removal of whole blood from a pa-
tient, with concurrent restoration of the
circulating blood volume with an acellular
fluid shortly before an anticipated signifi-
cant surgical blood loss. To minimize the
manual labor associated with hemodilu-
tion, the blood should be collected in
standard blood bags containing anticoag-
126 AABB Technical Manual
Figure 5-2. Relationship of estimated blood loss (EBL) and minimum (nadir) hematocrit during hospi-
talization at various initial hematocrit levels (30%,35%,40%,45%) in a surgical patient with a whole
blood volume of 5000 mL. = 30%; = 35%; = 40%; + = 45%.(Reprinted with permission from Co-
hen and Brecher.37)
Copyright © 2005 by the AABB. All rights reserved.
ulant on a tilt-rocker with automatic cut-
off via volume sensors. Then, the blood is
stored at room temperature and reinfused
during surgery after major blood loss has
ceased, or sooner if indicated. Simultaneous
infusions of crystalloid (3 mL crystalloid
for each 1 mL of blood withdrawn) and
colloid (dextrans, starches, gelatin, albu-
min, 1 mL for each 1 mL of blood with-
drawn) have been recommended.50
Subsequent intraoperative fluid manage-
ment is based on the usual surgical require-
ments. Blood units are reinfused in the
reverse order of collection. The first unit
collected, and therefore the last unit trans-
fused, has the highest hematocrit and con-
centration of coagulation factors and plate-
lets. Although this technique has been
primarily developed and used in Europe,
increasing interest in the United States has
led to data that show promise as an alterna-
tive method of autologous blood procure-
ment.51 Augmented hemodilution (replace-
ment of ANH collected or surgical blood
lost in part by oxygen therapeutics) has the
advantage of not being limited by anemia.
Its use is restricted to the investigational
setting until these solutions are approved
by the FDA.
Physiologic Considerations
Conserved Red Cell Mass
The chief benefit of ANH is the reduction
of red cell losses when whole blood is shed
perioperatively at lower hematocrit levels
after ANH has been completed.52 Mathe-
matical modeling has suggested that se-
vere ANH to preoperative hematocrit lev-
els of less than 20%, accompanied by
substantial blood losses, would be re-
quired before the red cell volume “saved”
by ANH would become clinically impor-
tant.53 However, the equivalent of one blood
unit54 can be “saved” by ANH,55 which ap-
proaches the red cell volume expansion
generated by PAD under standard condi-
tions (Table 5-3).
Improved Oxygenation
Withdrawal of whole blood and replace-
ment with crystalloid or colloid solution
decrease arterial oxygen content, but com-
pensatory hemodynamic mechanisms and
the existence of surplus oxygen-delivery
capacity make ANH safe. A sudden de-
crease in red cell concentration lowers
blood viscosity, thereby decreasing pe-
ripheral resistance and increasing cardiac
Chapter 5: Autologous Blood Donation and Transfusion 127
Table 5-4. Suggestions for Making Autologous Blood Transfusion Protocols
Cost-Effective
1. Use standardized indications for preoperative autologous blood collection and transfusion.
2. Streamline the autologous blood donor interview.
3. Discontinue serologic tests for infectious disease markers following autologous blood
collections.
4. Simplify the donation process for uncomplicated patients.
5. Limit the use of frozen blood.
6. Store autologous whole blood, rather than components.
7. Use standardized indications and appropriate technology for intraoperative and postoperative
autologous blood recovery.
8. Share intraoperative blood recovery resources among institutions.
9. Cautiously adopt new research applications for autologous blood techniques.
Copyright © 2005 by the AABB. All rights reserved.
output. If cardiac output can effectively
compensate, oxygen delivery to the tis-
sues at a hematocrit of 25% to 30% is as
good as, but no better than, oxygen deliv-
ery at a hematocrit of 30% to 35%.56
Preservation of Hemostasis
Because blood collected by ANH is stored
at room temperature and is usually re-
turned to the patient within 8 hours of
collection, there is little deterioration of
platelets or coagulation factors. The
hemostatic value of blood collected by
ANH is of questionable benefit for ortho-
pedicorurologicsurgerybecauseplasma
and platelets are rarely indicated in this
setting. Its value in protecting plasma and
platelets from the acquired coagulopathy
of extracorporeal circulation in cardiac
surgery is better established.46,57
Clinical Studies
Prospective randomized studies in radical
prostatectomy,58 knee replacement,59 and
hip replacement60 suggest that ANH can
be considered equivalent to PAD as a
method of autologous blood procure-
ment. Additional, selected clinical trials of
ANH are summarized in Table 5-5.61-67 Re-
views68,69 and commentaries70 on the mer-
its of ANH have been published. However,
a recently published meta-analysis of 42
clinical trials of ANH found only a modest
benefit with unproven safety.71 When ANH
and reinfusion are accomplished in the
operating room by on-site personnel, the
procurement and administration costs
are minimized. Blood obtained during
ANH does not require the commitment of
the patient’s time, transportation, costs,
and loss of work time that can be associ-
ated with PAD. The wastage of PAD units
(approximately 50% of units collected)
also is eliminated with ANH. Additionally,
autologous blood units procured by ANH
require no inventory or testing costs. Be-
cause the blood never leaves the patient’s
room, ANH minimizes the possibility of
an administrative or a clerical error that
could lead to an ABO-incompatible blood
transfusion and death, as well as bacterial
contamination associated with prolonged
storage at 4 C.
Practical Considerations
The following considerations are impor-
tant in establishing an ANH program:
1. Decisions about ANH should be
based on the surgical procedure and
on the patient’s preoperative blood
volume and hematocrit, target hemo-
dilution hematocrit, and other phys-
iologic variables.
2. The institution’s policy and proce-
dures and the mechanisms for edu-
cating staff should be established
and periodically reviewed.
3. Thereshouldbecarefulmonitoring
of the patient’s circulating volume
and perfusion status during the pro-
cedure.
4. Blood must be collected in an asep-
tic manner, ordinarily into standard
blood collection bags with citrate
anticoagulant.
5. Units must be properly labeled and
stored. The label must contain, at a
minimum, the patient’s full name,
medical record number, date, and
time of collection, and the statement
“For Autologous Use Only.” Room
temperature storage should not ex-
ceed 8 hours. Units maintained at
room temperature should be re-
infused in the reverse order of col-
lection to provide the maximum
number of functional platelets and
coagulation factors in the last units
infused. If more time elapses be-
tween collection and transfusion,
128 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 5: Autologous Blood Donation and Transfusion 129
Table 5-5. Selected Clinical Trials of Acute Normovolemic Hemodilution (ANH)
Estimated Blood Loss (mL)
Postoperative
Hematocrit (%)
Allogeneic RBC-Containing
Units or Liters Transfused
Surgery Control ANH p Value Control ANH p Value Control ANH p Value Reference
Vascular 2250 2458 NS NR 33.0 NR 6.0 2.6 <0.01 61
Liver resection 1479 1284 NS 37.9 33.8 <0.01 3.8 0.4 <0.001 62
Hip arthroplasty 1800 2000 NS 38.4 32.4 NS (2.1) (0.9) NR 63
Spinal fusion 5490 1700 <0.005 NR 28.7 NR 8.6 <1.0 <0.001 64
Colectomy NR NR NR 37.0 35.0 NR 2.4 0 NR 65
Prostate 1246 1106 NS 35.5 31.8 <0.001 0.16 0 NS 66
Prostate 1717 1710 NS 29.5 27.9 <0.5 0.30 0.13 NS 67
Modified from Brecher and Rosenfeld.53 NR = not reported; NS = not significant.
Copyright © 2005 by the AABB. All rights reserved.

the blood should be stored in a
monitored refrigerator. ANH blood
collected from an open system (eg,
from a central venous line or an ar-
terial catheter) may be stored for up
to 8 hours at room temperature or 24
hours in a monitored refrigerator.
Policies, procedures, and guidelines
must be developed for ANH by an
experienced group of anesthesiolo-
gists in conjunction with the operat-
ing room’s nursing staff and the hos-
pital’s blood bank and transfusion
services.68 These will include indica-
tions for ANH, monitoring require-
ments, endpoints for blood with-
drawal and transfusion, types and
amounts of replacement fluids (ie,
colloid/crystalloid ratios), and full
adherence to AABB guidelines. Some
practical considerations are listed in
Table 5-6. Suggested criteria for pa-
tient selection are listed in Table 5-7.
Intraoperative Blood
Collection
The term intraoperative blood collection
or recovery describes the technique of
collecting and reinfusing blood lost by a
patient during surgery. The oxygen-trans-
port properties of recovered red cells are
equivalent to stored allogeneic red cells.
The survival of recovered blood cells ap-
pears to be at least comparable to that of
130 AABB Technical Manual
Table 5-6. Practical Considerations for Acute Normovolemic Hemodilution (ANH)
■There must be a physician responsible for the perioperative blood recovery program.
Responsibilities shall include compliance with AABB standards,16 the establishment of written
policies and procedures, and periodic review of those policies and procedures.
■The blood bank or transfusion service should participate in the development of policies and
procedures related to the perioperative blood recovery program.
■Blood collected perioperatively shall not be transfused to other patients.
■Methods for perioperative blood collection and reinfusion shall be safe and aseptic and ensure
accurate identification of all blood and components collected. The equipment used shall be
pyrogen-free, shall include a filter capable of retaining particles potentially harmful to the
recipient, and must preclude air embolism. If the blood is warmed before infusion, warming
protocols apply.
■A complete written protocol of all perioperative collection procedures should be maintained,
including selection of anticoagulants and solutions used for processing, labeling of collected
blood or components, and procedures for the prevention and treatment of adverse reactions.
■All facilities regularly collecting blood by perioperative procedures should establish a program of
quality control and quality assurance. Written procedures should include criteria for acceptable
performance. Records of results should be reviewed and retained. Quality control measurements
should address the safety and quality of the blood or components collected for the recipient.
■Units collected for ANH shall be stored under one of the following conditions before the start of
transfusion:
—At room temperature, for up to 8 hours
—At 1 to 6 C for up to 24 hours, provided that storage at 1 to 6 C is begun within 8 hours of
initiating the collection.
Copyright © 2005 by the AABB. All rights reserved.
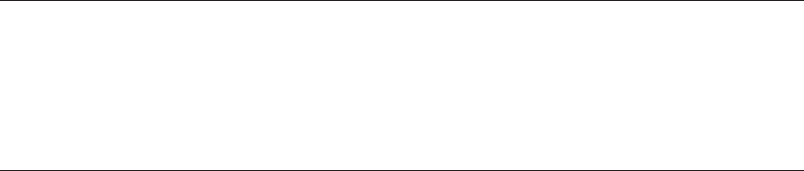
transfused allogeneic red cells.72 Intra-
operative collection is contraindicated
when certain procoagulant materials (eg,
topical collagen) are applied to the surgi-
cal field because systemic activation of
coagulation may result. Microaggregate
filters (40 microns) are used most often
because recovered blood may contain tis-
sue debris, small blood clots, or bone
fragments.
Cell washing devices can provide the
equivalent of 12 units of banked blood per
hour to a massively bleeding patient.72 Data
regarding adverse events of reinfusion of
recovered blood have been published.73 Air
embolusisapotentiallyseriousproblem.
Three fatalities from air embolus were re-
ported over a 5-year interval to the New
York State Department of Public Health, for
an overall fatality risk of one in 30,000.43
Hemolysis of recovered blood can occur
during suctioning from the surface instead
of from deep pools of shed blood. For this
reason, manufacturers’ guidelines recom-
mend a maximum vacuum setting of no
more than 150 torr. One study found that
vacuumsettingsashighas300torrcould
be used when necessary, without causing
excessive hemolysis.74 The clinical impor-
tance of free hemoglobin in the concentra-
tions usually seen has not been established,
although excessive free hemoglobin may
indicate inadequate washing. Positive bac-
terial cultures from recovered blood are
sometimes observed; however, clinical in-
fection is rare.75
Most programs use machines that col-
lect shed blood, wash it, and concentrate
the red cells. This process typically results
in 225-mL units of saline-suspended red
cells with a hematocrit of 50% to 60%. Pa-
tients exhibit a level of plasma-free hemo-
globin that is usually higher than after
allogeneic transfusion. Sodium and chlo-
ride concentrations are the same as in the
saline wash solution, and potassium con-
centration is low. The infusate contains
minimal coagulation factors and platelets.
Clinical Studies
As with PAD and ANH, collection and re-
covery of intraoperative autologous blood
should undergo scrutiny with regard to
both safety and efficacy. Controlled stud-
ies in cardiothoracic surgery have re-
ported conflicting results when transfu-
sion requirements and clinical outcome
were followed.75,76 Although the collection
of a minimum of one blood unit equiva-
lent is possible for less expensive (with
unwashed blood) methods, it is generally
agreed that at least two blood unit equiva-
lents need to be recovered using a cell-re-
covery instrument (with washed blood) in
order to achieve cost-effectiveness.77 The
value of intraoperative blood collection
has been best defined for vascular surger-
ies with large blood losses, such as aortic
aneurysm repair and liver transplanta-
tion.78 However, a prospective randomized
Chapter 5: Autologous Blood Donation and Transfusion 131
Table 5-7. Criteria for Selection of Patients for Acute Normovolemic Hemodilution
1. Likelihood of transfusion exceeds 10% (ie, blood requested for crossmatch according to
maximum surgical blood order schedule).
2. Preoperative hemoglobin level of at least 12 g/dL.
3. Absence of clinically significant coronary, pulmonary, renal, or liver disease.
4. Absence of severe hypertension.
5. Absence of infection and risk of bacteremia.
Copyright © 2005 by the AABB. All rights reserved.
trial79 of intraoperative recovery and re-
infusion in patients undergoing aortic an-
eurysm repair showed no benefit in the
reduction of allogeneic blood exposure. A
mathematical model of cell recovery sug-
gests that when it is combined with normo-
volemic anemia, the need for allogeneic
transfusion can be avoided—even with
large blood loss, eg, 5 to 10 liters.80 The
value of this technology may rest on cost
savings and blood inventory consider-
ations in patients with substantial blood
losses.
Medical Controversies
Collection devices that neither concentrate
nor wash shed blood before reinfusion in-
crease the risk of adverse effects. Shed
blood has undergone varying degrees of
coagulation/fibrinolysis and hemolysis,
and infusion of large volumes of washed
or unwashed blood has been described in
association with disseminated intravascular
coagulation.81 Factors that affect the degree
of coagulation and clot lysis include:
1. Whether the patient had received
systemic anticoagulation.
2. The amount and type of anticoagu-
lant used.
3. The extent of contact between blood
and serosal surfaces.
4. The extent of contact between blood
and artificial surfaces.
5. Thedegreeofturbulenceduringcol
-
lection.
In general, blood collected at low flow rates
or during slow bleeding from patients who
are not systemically anticoagulated will
have undergone coagulation and fibrinoly-
sis and will not contribute to hemostasis
upon reinfusion.
The high suction pressure and surface
skimming during aspiration and the turbu-
lence or mechanical compression that oc-
curs in roller pumps and plastic tubing
make some degree of hemolysis inevitable.
High concentrations of free hemoglobin
may be nephrotoxic to patients with im-
paired renal function. Many programs limit
the quantity of recovered blood that may be
reinfused without processing.
Practical Considerations
Collection and recovery services require
the coordinated efforts of surgeons, anes-
thesiologists, transfusion medicine spe-
cialists, and specific personnel trained in
the use of special equipment. Equipment
options may include:
1. Devices that collect recovered blood
for direct reinfusion.
2. Devices that collect recovered blood,
which is then concentrated and wash-
ed in a separate cell washer.
3. High-speed machines that automat-
ically concentrate and wash recov-
ered red cells.
Some hospitals develop their own pro-
grams, whereas others contract with outside
services. Each hospital’s needs should dic-
tate whether blood collection and recovery
are used and how they are achieved.
Processing Before Reinfusion
Several devices automatically process re-
covered blood before reinfusion. Vacuum
suction and simultaneous anticoagula-
tion are used for collection. To minimize
hemolysis, the vacuum level should ordi-
narily not exceed 150 torr, although higher
levels of suction may occasionally be
needed during periods of rapid bleeding.
Either citrate (ACD) or heparin may be
used as an anticoagulant. Blood is held in
a reservoir until centrifuged and washed
withavolumeofsalinethatvariesbe
-
tween 500 and 1500 mL. If not infused im-
mediately, the unit must be labeled with
the patient’s name and identification
132 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
number, the date and time collection was
initiated, and the statement “For Autolo-
gous Use Only.”
An alternative approach is to collect
blood in a canister system designed for di-
rect reinfusion and then concentrate and
wash the recovered red cells in a blood
bank cell washer. Intraoperatively collected
and recovered blood must be handled in
the transfusion service laboratory like any
other autologous unit. The unit should be
reinfused through a filter.
Direct Reinfusion
Systems are available that collect recovered
blood and return it directly. These sys-
tems generally consist of a suction cathe-
ter attached to a disposable collection bag
or rigid plastic canister, to which antico-
agulant (citrate or heparin) may have
been added. Blood is suctioned into the
holding canister before being reinfused
through a microaggregate filter. Low-vac-
uum suction and minimal hemolysis are
preferred in nonwashed systems.
Requirements and Recommendations
The AABB requires a process that includes
patient and storage bag identification and
time collected with expiration date.16(p10)
Units collected intraoperatively should be
labeled with the patient’s first and last
name, hospital identification number, the
date and time of collection and expira-
tion, and the statement “For Autologous
Use Only.”16(p10)
Conditions for storage and expiration of
autologous components collected in the
operating room are listed in Table 5-8.16(p14) If
the blood leaves the patient for washing or
storage in a remote location, there must be
appropriate procedures to ensure proper
labeling of the blood according to AABB
standards.16(pp9,10)
Hospitals with collection and recovery
programs should establish written policies
and procedures that are regularly reviewed
by a physician who has been assigned re-
sponsibility for the program. Transfusion
medicine specialists should play an active
role in design, implementation, and opera-
tion of the program. Written policies must
be in place for the proper collection, label-
ing, and storage of intraoperative autolo-
gous blood. Equipment and techniques for
collection and infusion must ensure that
the blood is aseptic. Quality management
should include evaluation of the appropri-
ate use of blood collection and recovery
services and adequate training of person-
nel. Written protocols, procedure logs, ma-
chine maintenance, procedures for han-
dling adverse events, and documentation
are recommended.24
Postoperative Blood
Collection
Postoperative blood collection denotes
the recovery of blood from surgical drains
followed by reinfusion, with or without
processing. In some programs, postopera-
tive shed blood is collected into sterile
canisters and reinfused, without process-
ing, through a microaggregate filter. Re-
covered blood is dilute, partially hemolyzed
and defibrinated, and may contain high
concentrations of cytokines. For these
reasons, most programs set an upper limit
on the volume (eg, 1400 mL) of unprocessed
blood that can be reinfused. If transfusion
of blood has not begun within 6 hours of
initiating the collection, the blood must be
discarded. Hospitals should establish
written policies, procedures, labeling re-
quirements, quality assurance, and review
consistent with AABB standards.16(p2)
Chapter 5: Autologous Blood Donation and Transfusion 133
Copyright © 2005 by the AABB. All rights reserved.

Clinical Studies
The evolution of cardiac surgery has been
accompaniedbyabroadexperiencein
postoperative conservation of blood. Post-
operative autologous blood transfusion is
practiced widely, but not uniformly. Pro-
spective and controlled trials have dis-
agreed over the efficacy of postoperative
blood recovery in cardiac surgery pat-
ients; at least three such studies have
demonstrated lack of efficacy,82-84 but at
least two studies have shown benefit.85,86
The disparity of the results in these stud-
ies may be explained, in part, by differences
in transfusion practices. Modification of
physician transfusion practices may have
been an uncredited intervention in these
blood conservation studies.
In the postoperative orthopedic surgical
setting, several reports have similarly des-
cribed the successful recovery and reinfu-
sion of washed87 and unwashed88,89 wound
drainage from patients undergoing arthro-
plasty. Red cells recovered in this setting
appear to have normal survival in the circu-
lation.90 The volume of reinfused drainage
blood has been reported to be as much as
3000 mL and averages more than 1100 mL
in patients undergoing cementless knee re-
134 AABB Technical Manual
Table 5-8. Handling, Storage, and Expiration of Intraoperative Blood Collections
Collection
Type
Storage
Temperature Expiration
Special
Conditions
Acute normovolemic
hemodilution
Room tempera-
ture
8 hours from start
of collection
None
1-6 C 24 hours from start
of collection
Storage at 1-6 C shall
begin within 8 hours
of start of collection
Intraoperative blood
recovered with
Room tempera-
ture
4 hours from end of
collection
None
processing 1-6 C 24 hours from start
of collection
Storage at 1-6 C shall
begin within 4 hours
of start of collection
Intraoperative blood
recovered without
processing
Room tempera-
ture or 1-6 C
4 hours from end of
collection
None
Shed blood under post-
operative or post-
traumatic conditions
with or without pro-
cessing
N/A 6 hours from start
of collection
None
Non-red-cell component
preparation
Room tempera-
ture
Shall be used before
leaving the oper-
ating room
None
Copyright © 2005 by the AABB. All rights reserved.
placement.89 Because the red cell content of
the fluid collected is low (hematocrit levels
of 20%), the volume of red cells reinfused is
often small.91 A prospective randomized
study of postoperative recovery and reinfu-
sion in patients undergoing total knee or
hip replacement found no differences in
perioperative hemoglobin levels or allo-
geneic blood transfusions between patients
who did or did not have joint drainage de-
vices.92 The safety of reinfused unwashed
orthopedic wound drainage has been con-
troversial. Theoretical concerns have been
expressed regarding infusion of potentially
harmful materials in recovered blood, in-
cluding free hemoglobin, red cell stroma,
marrow fat, toxic irritants, tissue or metha-
crylate debris, fibrin degradation products,
activated coagulation factors, and comple-
ment. Although two small studies have
reported complications,93,94 several larger
studies have reported no serious adverse
effects when drainage was passed through
a standard 40-micron blood filter.88,89,95
Patient Selection
The potential for decreasing exposure to
allogeneic blood among orthopedic patients
undergoing postoperative blood collection
(whether washed or unwashed) is greatest
for cementless bilateral total knee replace-
ment, revision hip or knee replacement,
and long segment spinal fusion. As in the
case of intraoperative recovery, blood loss
must be sufficient to warrant the addi-
tional cost of processing technology.96,97 As
in the selection of patients who can bene-
fit from PAD and ANH, prospective identi-
fication of patients who can benefit from
intra- and postoperative autologous blood
recovery is possible if anticipated surgical
blood losses and the perioperative “trans-
fusion trigger” are taken into account (Fig
5-2).
References
1. Pub. Law No. 101-336, 104 Stat. 327 (1990).
Codified at 42 USC §12101-213.
2. The ADA, HIV, and autologous blood dona-
tion. Association Bulletin 98-5. Bethesda, MD:
AABB, 1998.
3. Goodnough LT. Preoperative autologous do-
nation. In: Spence RK, ed. Problems in gen-
eral surgery. Philadelphia: Lippincott Wil-
liams & Wilkins, 2000;17:25-31.
4. Bierbaum BE, Callaghan JJ, Galante JO,
Rubash HE. An analysis of blood manage-
ment in patients having total hip or knee
arthroplasty. J Bone J Surg 1999;81A:2-10.
5. Renner SW, Howanitz PJ, Bachner P. Preoper-
ative autologous blood donation in 612 hos-
pitals. Arch Pathol Lab Med 1992;116:613-9.
6. Silvergleid AJ. Safety and effectiveness of pre-
deposit autologous transfusions in preteen
and adolescent children. JAMA 1987;257:
3403-4.
7. ChaplinH,MischeauxJR,InksterMD,
Sherman LA. Frozen storage of 11 units of
sickle cell red cells for autologous transfusion
of a single patient. Transfusion 1986;26:341-5.
8. Meryman HT, Hornblower M. Freezing and
deglycerolizing sickle trait red blood cells.
Transfusion 1976;16:627-32.
9. Mann M, Sacks HJ, Goldfinger D. Safety of
autologous blood donation prior to elective
surgery for a variety of potentially high risk
patients. Transfusion 1983;23:229-32.
10. Popovsky MA, Whitaker B, Arnold NL. Severe
outcomes of allogeneic and autologous blood
donation: Frequency and characterization.
Transfusion 1995;35:734-7.
11. GoodnoughLT,BrecherME,KanterMH,
AuBuchon JP. Transfusion medicine. First of
two parts. Blood transfusion. N Engl J Med
1999;340:438-47.
12. DoddRY,NotariEP,StramerSL.Currentprev
-
alence and incidence of infectious disease
markers and estimated window-period risk in
the American Red Cross blood donor popula-
tions. Transfusion 2002;42:975-9.
13. Thomas MJG, Gillon J, Desmond MJ. Preop-
erative autologous blood donation. Transfu-
sion 1996;36:633-9.
14. Sayers MH. Controversies in transfusion med-
icine. Autologous blood donation in preg-
nancy: Con. Transfusion 1990;30:172-4.
15. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
16. Santrach P, ed. Standards for perioperative
autologous blood collection and administra-
tion. 1st ed. Bethesda, MD: AABB, 2001.
Chapter 5: Autologous Blood Donation and Transfusion 135
Copyright © 2005 by the AABB. All rights reserved.
17. Food and Drug Administration. Memorandum:
Guidance for autologous blood and blood
components. (March 15, 1989) Rockville, MD:
CBER Office of Communication, Training,
and Manufacturers Assistance, 1989.
18. Food and Drug Administration. Memorandum:
Autologous blood collection and processing
procedures. (February 12, 1990) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 1990.
19. Food and Drug Administration. General re-
quirements for blood, blood components,
and blood derivatives; donor notification. 21
CFR 630.6. Fed Regist 2001;66(112):31165-77.
20. Food and Drug Administration. Require-
ments for testing human blood donors for ev-
idence of infection due to communicable dis-
ease agents. 21 CFR 610.40. Fed Regist 2001;
66(112):31146-65.
21. Food and Drug Administration. Guidance for
industry: Use of nucleic acid tests on pooled
and individual samples from donors of whole
blood and blood components (including
Source Plasma and Source Leukocytes) to ad-
equately and appropriately reduce the risk of
transmission of HIV-1 and HCV. (October 21,
2004) Rockville, MD: CBER Office of Commu-
nication, Training, and Manufacturers Assis-
tance, 2004.
22. Code of federal regulations. Title 42 CFR Part
72.3. Washington, DC: US Government Print-
ing Office, 2004 (revised annually).
23. National Heart, Lung, and Blood Institute
Autologous Transfusion Symposium Working
Group. Autologous transfusion: Current trends
and research issues. Transfusion 1995;35:525-
31.
24. Becker J, Blackall D, Evans C, et al for the Sci-
entific Section Coordinating Committee.
Guidelines for blood utilization review.
Bethesda, MD: AABB, 2001:20-4.
25. Goodnough LT, Skikne B, Brugnara C. Eryth-
ropoietin, iron, and erythropoiesis. Blood
2000;96:823-33.
26. Toy P, Ahn D, Bacchetti P. When should the
first of two autologous donations be made?
(abstract) Transfusion 1994;34(Suppl):14S.
27. Brecher ME, Goodnough LT. The rise and fall
of preoperative autologous blood donation
(editorial). Transfusion 2001;41:1459-62.
28. Etchason J, Petz L, Keeler E, et al. The cost-ef-
fectiveness of preoperative autologous blood
donations. N Engl J Med 1995;332:719-24.
29. Vamvakas EC. Meta-analysis of randomized
controlled trials comparing the risk of post-
operative infection between recipients of
allogeneic and autologous blood. Vox Sang
2002;83:339-46.
30. Vanderlinde E, Heal JM, Blumberg N. Auto-
logous transfusion. Br Med J 2002;324:772-5.
31. Larocque BJ, Gilbert K, Brien WF. Prospective
validation of a point score system for predict-
ing blood transfusion following hip or knee
replacement. Transfusion 1998;38:932-7.
32. Brecher MA, Monk TG, Goodnough LT. A
standardized method for calculating blood
loss. Transfusion 1997;37:1070-4.
33. Rosencher N. Orthopedic surgery transfusion
hemoglobin overview (OSTHEO) study.
Transfusion 2003;43:459-69.
34. Kasper SM, Gerlich W, Buzello W. Preopera-
tiveredcellproductioninpatientsundergo
-
ing weekly autologous blood donation.
Transfusion 1997;37:1058-62.
35. Kasper SM, Lazansky H, Stark C, et al. Effi-
cacy of oral iron supplementation is not en-
hanced by additional intravenous iron during
autologous blood donation. Transfusion
1998;38:764-70.
36. WeisbachV,SkodaP,RippelR,etal.Oralor
intravenous iron as an adjunct to autologous
blood donation in elective surgery: A ran-
domized, controlled study. Transfusion 1999;
39:465-72.
37. Cohen JA, Brecher ME. Preoperative autolo-
gous blood donation: Benefit or detriment? A
mathematical analysis. Transfusion 1995;35:640-4.
38. Kanter MH, Van Maanen D, Anders KH, et al.
Preoperative autologous blood donation be-
fore elective hysterectomy. JAMA 1996;276:
798-801.
39. GoodnoughLT,RudnickS,PriceTH,etal.In
-
creased preoperative collection of autologous
blood with recombinant human erythropoie-
tin therapy. N Engl J Med 1989;321:1163-8.
40. Goodnough LT, Price TH, Rudnick S, Soegiarso
RW. Preoperative red blood cell production in
patients undergoing aggressive autologous
blood phlebotomy with and without erythro-
poietin therapy. Transfusion 1992;32:441-5.
41. Goodnough LT, Price TH, Friedman KD, et al.
A phase III trial of recombinant human eryth-
ropoietin therapy in non-anemic orthopedic
patients subjected to aggressive autologous
blood phlebotomy: Dose, response, toxicity,
efficacy. Transfusion 1994;34:66-71.
42. Goodnough LT, Monk TG, Andriole GL. Eryth-
ropoietin therapy. N Engl J Med 1997;336:
933-8.
43. LindenJV,WagnerK,VoytovichAE,Sheehan
J.TransfusionerrorsinNewYorkState:An
analysis of 10 years’ experience. Transfusion
2000;40:1207-13.
44. Hébert PC, Wells G, Blajchman MA, et al. A
multicenter, randomized, controlled clinical
trial of transfusion requirements in critical
care. N Engl J Med 1999;340:409-17.
136 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
45. Kruskall MS, Yomtovian R, Dzik WH, et al. On
improving the cost effectiveness of autolo-
gous blood transfusion practices. Transfusion
1994;34:259-64.
46. Triulzi DJ, Gilmor GD, Ness PM, et al. Efficacy
of autologous fresh whole blood or platelet-
rich plasma in adult cardiac surgery. Transfu-
sion 1995;35:627-34.
47. Kevy SV, Jacobson MS. Comparison of meth-
ods for point of care preparation of autolo-
gous platelet gel. J Extra Corpor Technol
2004;36:28-35.
48. Crovetti G, Martinelli G, Issi M, et al. Platelet
gel for healing cutaneous chronic wounds.
Transfus Apheresis Sci 2004;30:145-51.
49. Mazzucco L, Medici D, Serra M, et al. The use
of autologous platelet gel to treat difficult to
heal wounds: A pilot study. Transfusion 2004;
44:1013-8.
50. Goodnough LT, Brecher ME, Monk TG. Acute
normovolemic hemodilution in surgery. He-
matology 1992;2:413-20.
51. GoodnoughLT,BrecherME,KanterMH,
AuBuchon JP. Transfusion medicine. Second
of two parts. Blood conservation. N Engl J
Med 1999;340:525-33.
52. Messmer K, Kreimeier M, Intagliett A. Present
state of intentional hemodilution. Eur Surg
Res 1986;18:254-63.
53. Brecher ME, Rosenfeld M. Mathematical and
computer modeling of acute normovolemic
hemodilution. Transfusion 1994;34:176-9.
54. Goodnough LT, Bravo J, Hsueh Y, et al. Red
blood cell volume in autologous and homolo-
gous units: Implications for risk/benefit
assessment for autologous blood “crossover”
and directed blood transfusion. Transfusion
1989;29:821-2.
55. GoodnoughLT,GrishaberJE,MonkTG,
Catalona WJ. Acute normovolemic hemodilu-
tion in patients undergoing radical supra-
pubic prostatectomy: A case study analysis.
Anesth Analg 1994;78:932-7.
56. Weiskopf RB. Mathematical analysis of isovo-
lemic hemodilution indicates that it can de-
crease the need for allogeneic blood trans-
fusion. Transfusion 1995;35:37-41.
57. Petry AF, Jost T, Sievers H. Reduction of ho-
mologous blood requirements by blood pool-
ing at the onset of cardiopulmonary bypass. J
Thorac Cardiovasc Surg 1994;1097:1210-14.
58. Monk TG, Goodnough LT, Brecher ME, et al. A
prospective, randomized trial of three blood
conservation strategies for radical prostatec-
tomy. Anesthesiology 1999;91:24-33.
59. Goodnough LT, Merkel K, Monk TG, Despotis
GJ. A randomized trial of acute normovo-
lemic hemodilution compared to preopera-
tive autologous blood donation in total knee
arthroplasty. Vox Sang 1999;77:11-16.
60. Goodnough LT, Despotis GJ, Merkel K, Monk
TG. A randomized trial of acute normo-
volemic hemodilution compared to preoper-
ative autologous blood donation in total hip
arthroplasty. Transfusion 2000;40:1054-7.
61. Davies MJ, Cronin KD, Domanique C. Hae-
modilution for major vascular surgery using
3.5% polygeline (Haemaccel). Anaesth Inten-
sive Care 1982;10:265-70.
62. Sejourne P, Poirier A, Meakins JL, et al. Effects
of haemodilution on transfusion requirements
in liver resection. Lancet 1989;ii:1380-2.
63. Rosenberg B, Wulff K. Regional lung function
following hip arthroplasty and preoperative
normovolemic hemodilution. Acta Anaesthe-
siol Scand 1979;23:242-7.
64. Kafer ER, Isley MR, Hansen T, et al. Auto-
mated acute normovolemic hemodilution re-
duces blood transfusion requirements for
spinal fusion (abstract). Anesth Analg 1986;
65(Suppl):S76.
65. Rose D, Coustoftides T. Intraoperative normo-
volemic hemodilution. J Surg Res 1981;31:375-81.
66. Ness PM, Bourke DL, Walsh PC. A random-
ized trial of perioperative hemodilution ver-
sus transfusion of preoperatively deposited
autologous blood in elective surgery. Transfu-
sion 1991;31:226-30.
67. Monk TG, Goodnough LT, Birkmeyer JD, et al.
Acute normovolemic hemodilution is a cost-
effective alternative to preoperative autolo-
gous blood donation by patients undergoing
radical retropubic prostatectomy. Transfu-
sion 1995;35:559-65.
68. Shander A. Acute normovolemic hemodilu-
tion. In: Spence RK, ed. Problems in general
surgery. Philadelphia: Lippincott Williams &
Wilkins, 1999;17:32-40.
69. Monk TG, Goodnough LT, Brecher ME, et al.
Acute normovolemic hemodilution can re-
place preoperative autologous blood dona-
tion as a standard of care for autologous
blood procurement in radical prostatectomy.
Anesth Analg 1997;85:953-8.
70. Goodnough LT, Monk TG, Brecher ME. Acute
normovolemic hemodilution should replace
preoperative autologous blood donation be-
fore elective surgery. Transfusion 1998;38:
473-7.
71. Segal JB, Blasco-Colmenares E, Norris EJ,
Guallar E. Preoperative acute normovolemic
hemodilution: A meta-analysis. Transfusion
2004;44:632-44.
72. Williamson KR, Taswell HF. Intraoperative
blood salvage: A review. Transfusion 1991;31:
662-75.
Chapter 5: Autologous Blood Donation and Transfusion 137
Copyright © 2005 by the AABB. All rights reserved.
73. Domen RE. Adverse reactions associated with
autologous blood transfusion: Evaluation
and incidence at a large academic hospital.
Transfusion 1998;38:296-300.
74. Gregoretti S. Suction-induced hemolysis at
various vacuum pressures: Implications for
intraoperative blood salvage. Transfusion
1996;36:57-60.
75. Bell K, Stott K, Sinclair CJ, et al. A controlled
trial of intra-operative autologous transfu-
sion in cardiothoracic surgery measuring ef-
fect on transfusion requirements and clinical
outcome. Transfus Med 1992;2:295-300.
76. TempeDK,BanjerjeeA,VirmaniS,etal.
Comparison of the effects of a cell saver and
low dose aprotinin on blood loss and homol-
ogous blood use in patients undergoing valve
surgery. J Cardiothorac Vasc Anesth 2001;15:
326-30.
77. Bovill DF, Moulton CW, Jackson WS, et al. The
efficacy of intraoperative autologous transfu-
sion in major orthopaedic surgery: A regres-
sion analysis. Orthopedics 1986;9:1403-7.
78. Goodnough LT, Monk TG, Sicard G, et al.
Intraoperative salvage in patients undergoing
elective abdominal aortic aneurysm repair.
An analysis of costs and benefits. J Vasc Surg
1996;24:213-8.
79. Claggett GP, Valentine RJ, Jackson MR, et al. A
randomized trial of intraoperative transfu-
sion during aortic surgery. J Vasc Surg 1999;
29:22-31.
80. Waters JH, Karafa MT. A mathematical model
of cell salvage efficiency. Anesth Analg 2002;
95:1312-7.
81. de Haan J, Boonstra P, Monnink S, et al. Re-
transfusion of suctioned blood during cardio-
pulmonary bypass impairs hemostasis. Ann
Thorac Surg 1995;59:901-7.
82. WardHB,SmithRA,CandisKP,etal.Apro
-
spective, randomized trial of autotransfusion
after routine cardiac surgery. Ann Thorac
Surg 1993;56:137-41.
83. Thurer RL, Lytle BW, Cosgrove DM, Loop FD.
Autotransfusion following cardiac opera-
tions: A randomized, prospective study. Ann
Thorac Surg 1979;27:500-6.
84. Roberts SP, Early GL, Brown B, et al. Auto-
transfusion of unwashed mediastinal shed
blood fails to decrease banked blood require-
ments in patients undergoing aorta coronary
bypass surgery. Am J Surg 1991;162:477-80.
85. SchaffHV,HauerJM,BellWR,etal.Auto
-
transfusion of shed mediastinal blood after
cardiac surgery. A prospective study. J Thorac
Cardiovasc Surg 1978;75:632-41.
86. EngJ,KayPH,MurdayAJ,etal.Post-opera
-
tive autologous transfusion in cardiac sur-
gery. A prospective, randomized study. Eur J
Cardiothorac Surg 1990;4:595-600.
87. Semkiw LB, Schurman OJ, Goodman SB,
Woolson ST. Postoperative blood salvage us-
ing the cell saver after total joint arthroplasty.
J Bone Joint Surg (Am) 1989;71A:823-7.
88. Faris PM, Ritter MA, Keating EM, Valeri CR.
Unwashed filtered shed blood collected after
knee and hip arthroplasties. J Bone Joint Surg
(Am) 1991;73A:1169-77.
89. MartinJW,WhitesideLA,MillianoMT,Reedy
ME. Postoperative blood retrieval and trans-
fusion in cementless total knee arthroplasty. J
Arthroplasty 1992;7:205-10.
90. Umlas J, Jacobson MS, Kevy SV. Survival and
half-life of red cells salvaged after hip and knee
replacement surgery. Transfusion 1993;33:591-3.
91. Umlas J, Foster RR, Dalal SA, et al. Red cell
loss following orthopedic surgery: The case
against postoperative blood salvage. Transfu-
sion 1994;34:402-6.
92. Ritter MA, Keating EM, Faris PM. Closed
wound drainage in total hip or knee replace-
ment: A prospective, randomized study. J
Bone J Surg 1994;76:35-8.
93. Clements DH, Sculco TP, Burke SW, et al.
Salvage and reinfusion of postoperative
sanguineous wound drainage. J Bone Joint
Surg (Am)1992;74A:646-51.
94. Woda R, Tetzlaff JE. Upper airway oedema
following autologous blood transfusion from
a wound drainage system. Can J Anesth 1992;
39:290-2.
95. Blevins FT, Shaw B, Valeri RC, et al. Re-
infusion of shed blood after orthopedic pro-
cedures in children and adolescents. J Bone
Joint Surg (Am) 1993;75A:363-71.
96. Goodnough LT, Verbrugge D, Marcus RE. The
relationship between hematocrit, blood lost,
and blood transfused in total knee replace-
ment: Implications for postoperative blood
salvage and reinfusion. Am J Knee Surg 1995;
8:83-7.
97. Jackson BR, Umlas J, AuBuchon JP. The cost-
effectiveness of postoperative recovery of
RBCs in preventing transfusion-associated
virus transmission after joint arthroplasty.
Transfusion 2000;40:1063-6.
138 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 6: Apheresis
Chapter 6
Apheresis
APHERESIS, FROM THE Greek
pheresis meaning “to take away,”
involves the selective removal of
blood constituents from blood donors or
patients.
The AABB provides standards1for volun-
tary compliance for apheresis activities.
The Food and Drug Administration (FDA)
has established specific requirements that
aresetforthintheCode of Federal Regula-
tions2for apheresis activities. The American
Society for Apheresis (ASFA)3has published
additional guidelines and recommendations.
In addition, hemapheresis practitioner (HP)
and apheresis technician (AT) certifications
are available through the American Society
of Clinical Pathology Board of Registry. All
personnel involved with apheresis activities
should be familiar with these sources and
should have documentation that they are
qualified by training and experience to per-
form apheresis.
Separation Techniques
Automated blood processing devices are
used for both component preparation and
therapeutic applications of apheresis.
Manual apheresis, in which whole blood
is collected in multiple bags and centri-
fuged offline, requires great care to ensure
that the bags are labeled correctly and are
returned to the correct donor. With the
currently available automated technology,
this process is seldom used.
Separation by Centrifugation
In most apheresis instruments, centrifu-
gal force separates blood into components
on the basis of differences in density. A
measured amount of anticoagulant solu-
tion is added to the whole blood as it is
drawn from the donor or patient. The blood
is pumped into a rotating bowl, chamber,
or tubular rotor in which layering of com-
139
6
Copyright © 2005 by the AABB. All rights reserved.
ponents occurs on the basis of their den-
sities. The desired fraction is diverted and
the remaining elements are returned to the
donor (or patient) by intermittent or con-
tinuous flow.
All systems require prepackaged dispos-
able sets of sterile bags, tubing, and centrif-
ugal devices unique to the instrument. Each
system has a mechanism to allow the sepa-
ration device to rotate without twisting the
attached tubing. In the intermittent flow
method, the centrifuge container is alternately
filled and emptied. Most instruments in use
today employ a method that involves the
continuous flow of blood through a separa-
tion chamber. Depending on the procedure
and device used, the apheresis procedure
time varies from 30 minutes to several hours.
Each manufacturer supplies detailed in-
formation and operational protocols. Each
facility must have, in a manual readily
available to nursing and technical person-
nel, detailed descriptions of each type of
procedure performed, specific for each type
of blood processor.4
Separation by Adsorption
Selective removal of a pathologic material
has theoretical advantages over the re-
moval of all plasma constituents. Centrif-
ugal devices can be adapted to protocols
that selectively remove specific soluble
plasma constituents by exploiting the
principles of affinity chromatography.5Se-
lective removal of low-density lipoproteins
(LDLs) in patients with familial hypercho-
lesterolemia has been accomplished using
both immunoaffinity (anti-LDL) and che-
mical affinity (eg, dextran sulfate) columns.6
Adsorbents such as staphylococcal protein
A (SPA), monoclonal antibodies, blood
group substances, DNA-collodion, and
polymers with aggregated IgG attached
can extract antibodies, protein antigens,
and immune complexes. Returning the
depleted plasma along with the cellular
components reduces or eliminates the
need for replacement fluids. Immuno-
adsorption can be performed online, or
the plasma can be separated from the cel-
lular components, passed through an off-
line column, and then reinfused.
Component Collection
Whenever components intended for trans-
fusion are collected by apheresis, the do-
nor must give informed consent. Although
apheresis collection and preparation pro-
cesses are different from those used for
whole-blood-derived components, stor-
age conditions, transportation require-
ments, and some quality control steps are
thesame.SeeChapter8formoredetailed
information. The facility must maintain
written protocols for all procedures used
and must keep records for each procedure
as required by AABB Standards for Blood
Banks and Transfusion Services.1(p2)
Platelets Pheresis
Plateletpheresis is used to obtain platelets
from random volunteer donors, from pat-
ients’ family members, or from donors with
matched HLA or platelet antigen pheno-
types. Because large numbers of platelets
can be obtained from a single individual,
collection by apheresis reduces the num-
ber of donor exposures for patients. AABB
Standards requires the component to
contain at least 3 ×1011 platelets in 90% of
sampled units.1(p31) When a high yield is
obtained, the original apheresis unit may
be divided into multiple units, each of which
must meet minimum standards inde-
pendently. Some instruments are pro-
grammed to calculate the yield from the
donor’s hematocrit, platelet count, height,
and weight. For alloimmunized patients
who are refractory to random allogeneic
140 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
platelets (see Chapters 16 and 21), platelets
from an apheresis donor selected on the
basis of a compatible platelet crossmatch
or matched for HLA antigens may be the
only way to achieve a satisfactory post-
transfusion platelet increment. Within the
United States, the use of apheresis plate-
lets has been steadily increasing over the
last 25 years. Currently, it is estimated that
77% of therapeutic platelet doses are
transfused as apheresis platelets.7
Donor Selection and Monitoring
Plateletpheresis donors may donate more
frequently than whole blood donors but
must meet all other donor criteria. The in-
terval between donations should be at
least 2 days, and donors should not un-
dergo plateletpheresis more than twice
in a week or more than 24 times in a
year.1(pp19,20) If the donor donates a unit of
Whole Blood or if it becomes impossible
to return the donor’s red cells during pla-
teletpheresis, at least 8 weeks should elapse
before a subsequent plateletpheresis pro-
cedure, unless the extracorporeal red cell
volume is less than 100 mL.1(p20) Platelets
may be collected from donors who do not
meet these requirements if the compo-
nent is expected to be of particular value
to a specific intended recipient, and if a
physician certifies in writing that the do-
nor’s health will not be compromised (eg,
an HLA-matched donor). Donors who
have taken aspirin-containing medica-
tions within 36 hours of donation are usu-
ally deferred because the platelets ob-
tained by apheresis are often the single
source of platelets given to a patient.
Vasovagal and hypovolemic reactions are
rare in apheresis donors, but paresthesias
and other reactions to the citrate antico-
agulant are common (see Complications,
later in this chapter). Serious reactions
occur less often among apheresis donors
than among whole blood donors.
Plateletpheresis donors should meet
usual donor requirements, including he-
moglobin or hematocrit level. A platelet
count is not required before the first
apheresis collection or if 4 weeks or more
have elapsed since the last procedure. If the
donation interval is less than 4 weeks, the
donor’s platelet count should be above
150,000/µL before subsequent platelet-
pheresis occurs. AABB Standards permits
documentation of the platelet count from a
sample collected immediately before the
procedure or from a sample obtained either
before or after the previous procedure.1(p21)
Exceptions to these laboratory criteria should
be approved in writing by the apheresis phy-
sician. The FDA specifies that the total vol-
ume of plasma collected should be no more
than 500 mL (or 600 mL for donors weighing
more than 175 pounds).8The platelet count
of each unit should be kept on record but
need not be written on the product label.8
Some plateletpheresis programs collect
plasma for use as Fresh Frozen Plasma
(FFP) in a separate bag during platelet col-
lection. Apheresis can also be used to col-
lect plasma for FFP without platelets, ie,
plasmapheresis. The FDA has provided
guidance with regard to the volume of
plasma that is allowed to be collected using
automated devices.9A total serum or plasma
protein determination and a quantitative
determination of IgG and IgM (or a serum
protein electrophoresis) must be deter-
mined at 4-month intervals for donors un-
dergoing large-volume plasma collection, if
the total annual volume of plasma collected
exceeds 12 liters (14.4 L for donors weigh-
ing more than 175 pounds) or if the donor
is a frequent (more often than every 4
weeks) plasma donor.10 AABB Standards re-
quires that the donor’s intravascular volume
deficit must be less than 10.5 mL per kilo-
gram of body weight at all times.1(p24)
Chapter 6: Apheresis 141
Copyright © 2005 by the AABB. All rights reserved.
Laboratory Testing
Tests for ABO group and Rh type, unex-
pected alloantibodies, and markers for
transfusion-transmitted diseases must be
performed by the collecting facility in the
same manner as for other blood compo-
nents. Each unit must be tested unless the
donor is undergoing repeated procedures
to support a single patient, in which case
testing for disease markers need be re-
peated only at 30-day intervals.1(p34)
If red cells are visible in a product, the
hematocrit should be determined. FDA
guidelines require that if the component
contains more than 2 mL of red cells, a
sample of donor blood for compatibility
testing be attached to the container.8In
some instances, it may be desirable for the
donorplasmatobeABO-compatiblewith
the recipient’s red cells—for example, if the
recipient is a child or an ABO-mismatched
allogeneic progenitor cell transplant recipi-
ent. In order to be considered leukocyte-
reduced, apheresis platelets must contain
less than 5 ×106leukocytes and must meet
the specifications of the apheresis device
manufacturer. Chapter 8 describes addi-
tional quality control measures that apply
to all platelet components.
Records
Complete records (see Chapter l) must be
kept for each procedure. All adverse reac-
tions should be documented along with
the results of their investigation and fol-
low-up. Records of all laboratory findings
and collection data must be periodically
reviewed by a knowledgeable physician
and found to be within acceptable limits.
FDA guidelines require review at least
once every 4 months.8Facilities must have
policies and procedures in place to ensure
that donor red cell loss during each pro-
cedure does not exceed acceptable limits.
Plasma
Apheresis can be used to collect plasma as
FFP or for Source Plasma for subsequent
manufacturing. FDA requirements for
plasma collection are different from those
for whole blood or plateletpheresis; per-
sonnel who perform serial plasmapheresis
must be familiar with both AABB stan-
dards and FDA requirements. If plasma is
intended for transfusion, testing require-
ments are the same as those for red cell
components. Plasma collected for manu-
facture of plasma derivatives is subject to
different requirements for infectious dis-
ease testing.
A distinction is made between “occa-
sional plasmapheresis,” in which the donor
undergoes plasmapheresis no more often
than once in 4 weeks, and “serial plasma-
pheresis,” in which donation is more fre-
quent than every 4 weeks. For donors in an
occasional plasmapheresis program, donor
selection and monitoring are the same as
for whole blood donation. For serial plasma-
pheresis using either automated instru-
ments or manual techniques, the following
principles apply:
1. Donors must provide informed con-
sent. They must be observed closely
during the procedure and emergency
medicalcaremustbeavailable.
2. Red cell losses related to the proce-
dure, including samples collected
for testing, must not exceed 25 mL
perweek,sothatnomorethan200
mL of red cells are removed in 8
weeks. If the donor’s red cells cannot
be returned during an apheresis pro-
cedure, hemapheresis or whole blood
donation should be deferred for 8
weeks.
3. In manual plasma collection systems,
there must be a mechanism to en-
sure safe reinfusion of the autologous
red cells. Before the blood container
142 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
is separated from the donor for pro-
cessing, there should be two sepa-
rate, independent means of identifi-
cation, so that both the donor and
the phlebotomist can ascertain that
the contents are those of the donor.
Often, the donor’s signature is one
identifier, along with a unique iden-
tification number.
4. In manual procedures for donors
weighing 50 to 80 kg (110-176 lb), no
more than 500 mL of whole blood
should be removed at one time, or
1000 mL during the session or within
a 48-hour period. The limits for do-
nors who weigh more than 80 kg are
600 mL and 1200 mL, respectively.
For automated procedures, the allow-
able volume has been determined
for each instrument by the FDA.9
5. At least 48 hours should elapse be-
tween successive procedures; ordi-
narily, donors should not undergo
more than two procedures within a
7-day period. Exceptions are permis-
sible when plasma is expected to
have special therapeutic value for a
single recipient.
6. Atthetimeofinitialplasmapheresis
and at 4-month intervals thereafter
for donors undergoing plasmaphere-
sis more often than once every 4
weeks, serum or plasma must be
tested for total protein and serum
protein electrophoresis or quantita-
tive immunoglobulins. Results must
be within normal limits.2
7. A qualified, licensed physician, know-
ledgeable in all aspects of hema-
pheresis, must be responsible for the
program.
Red Cells
Both AABB standards and FDA-approved
protocols address the removal of two allo-
geneic or autologous Red Blood Cell units
every 16 weeks by an automated aphere-
sis method. Saline infusion is used to min-
imizevolumedepletion,andtheproce
-
dure is limited to persons who are larger
and have higher hematocrits than current
minimum standards for whole blood do-
nors (for males: weight 130 lb, height 5′1″;
for females: weight 150 lb, height 5′5″;
hematocrit 40% for both genders).11
Granulocytes
The indications for granulocyte transfu-
sion are controversial (see Chapter 21). A
meta-analysis of randomized controlled
trials of granulocyte transfusion indicates
that effectiveness depends on an adequate
dose (>1 ×1010 granulocytes/day) and
crossmatch compatibility (no recipient
antibodies to granulocyte antigens).12
There is renewed interest in granulocyte
transfusion therapy for adults because
much larger cell doses can be delivered
when cells are collected from donors who
receive colony-stimulating factors.13 Some
success with granulocyte transfusions has
been observed in the treatment of septic
infants,14 possibly because the usual dose
is relatively larger in these tiny recipients
and because HLA alloimmunization is ab-
sent.
Drugs Administered for Leukapheresis
A daily dose of at least 1 ×1010 granulo-
cytes is necessary to achieve a therapeutic
effect.15 Collection of this number of cells
requires administration of drugs or other
adjuvants to the donor. The donor’s con-
sent should include specific permission
for any drugs or sedimenting agents to be
used.
Hydroxyethyl Starch. A common sedi-
menting agent, hydroxyethyl starch (HES),
causes red cells to aggregate and thereby
sediment more completely. Sedimenting
Chapter 6: Apheresis 143
Copyright © 2005 by the AABB. All rights reserved.
agents enhance granulocyte harvest and re-
sult in minimal red cell content. Because
HES can be detected in donors for as long
as a year after infusion, AABB Standards re-
quires facilities performing granulocyte col-
lections to have a process to control the
maximal cumulative dose of any sedi-
menting agent administered to the donor
within a given interval.1(p24) Because HES is a
colloid, it acts as a volume expander, and
donors who have received HES may experi-
ence headaches or peripheral edema because
of expanded circulatory volume.
Corticosteroids. Corticosteroids can
double the number of circulating granulo-
cytes by mobilizing them from the marginal
pool. A protocol using 60 mg of oral predni-
sone as a single or divided dose before do-
nation gives superior granulocyte harvests
with minimal systemic steroid activity.16 Al-
ternatively, 8 mg of oral dexamethasone may
be used. Before administration of cortico-
steroids, donors should be questioned about
any history or symptoms of hypertension,
diabetes, cataracts,17 and peptic ulcer.
Growth Factors. Recombinant hemato-
poietic growth factors—specifically, granulo-
cyte colony-stimulating factor (G-CSF)—
can effectively increase granulocyte yields.
Hematopoietic growth factors alone can re-
sult in collection of up to 4 to 8 ×1010 granu-
locytes per apheresis procedure.13 Typical
doses of G-CSF employed are 5 to 10 µg/kg
given 8 to 12 hours before collection.18 Pre-
liminary evidence suggests that in-vivo re-
covery and survival of these granulocytes
are excellent and that growth factors are
well tolerated by donors.13
Laboratory Testing
Testing for ABO and Rh, alloantibodies,
and infectious disease markers on a sam-
pledrawnatthetimeofphlebotomyare
required. Red cell content in granulocyte
concentrates is inevitable; the red cells
should be ABO-compatible with the re-
cipient’s plasma and, if more than 2 mL
are present, the component should be
crossmatched. Ideally, D-negative recipi-
ents should receive granulocyte concentrates
from D-negative donors. Leukocyte (HLA)
matching is recommended in alloim-
munized patients.
Storage and Infusion
Because granulocyte function deteriorates
during storage, concentrates should be
transfused as soon as possible after prep-
aration. AABB Standards prescribes a
storage temperature of 20 to 24 C, for no
longer than 24 hours.1(p57) Agitation during
storage is probably undesirable. Irradiation
is required before administration to im-
munodeficient recipients and will proba-
bly be indicated for nearly all recipients
because of their primary disease. Infusion
through a microaggregate or leukocyte re-
duction filter is contraindicated.
Hematopoietic Progenitor Cells
Cytapheresis for collection of hematopoietic
progenitor cells is useful for obtaining
progenitor cells for marrow reconstitution
in patients with cancer, leukemia in re-
mission, and various lymphomas (see
Chapter 25). Cytapheresis procedures can
also be used to collect donor lymphocytes
forinfusionasanimmunetherapyinthese
patients (see Chapter 25). The AABB has
published Standards for Cellular Therapy
Product Services.19 Additional requirements
are reviewed in Chapter 25.
Therapeutic Apheresis
Therapeutic apheresis has been used to
treat many different diseases. Cells, plasma,
or plasma constituents may be removed
from the circulation and replaced by nor-
mal plasma, crystalloid, or colloid solu-
144 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
tions of starch or albumin. The term
“therapeutic apheresis” is used for the
general procedure and the term “thera-
peutic plasma exchange” (TPE) is used for
procedures in which the goal is the re-
moval of plasma, regardless of the solu-
tion used as replacement.
The theoretical basis for therapeutic
apheresisistoreducethepatient’sloadofa
pathologic substance to levels that will al-
low clinical improvement. In some condi-
tions, replacement with normal plasma is
intended to supply an essential substance
that is absent. In the absence of the need to
replace plasma constituents, colloidal solu-
tions and/or saline should be used as re-
placement fluids. Other possible outcomes
of therapeutic apheresis include alteration
of the antigen-to-antibody ratio, modifica-
tion of mediators of inflammation or im-
munity, and clearance of immune com-
plexes. Some perceived benefit may result
from a placebo effect. Despite difficulties in
documentation, there is general agreement
that therapeutic apheresis is effective treat-
ment for the conditions listed in Table 6-1
as Category I or Category II.3,20-22
General Considerations
Appropriate use of therapeutic apheresis
requires considerable medical knowledge
and judgment. The patient should be
evaluated for treatment by his or her per-
sonal physician and by the apheresis phy-
sician. Close consultation between these
physicians is important, especially if the
patient is small or elderly, has poor vascu-
lar access or cardiovascular instability, or
has a condition for which apheresis is of
uncertain benefit. The apheresis physi-
cian should make the final determination
about appropriateness of the procedure
and eligibility of the patient (see Table
6-1).3,20-22 When therapeutic apheresis is
anticipated, those involved with the pa-
tient’s care should establish a treatment
plan and the goal of therapy. The end-
point may be an agreed-upon objective
outcome or a predetermined duration for
the therapy, whichever is achieved first. It
is helpful to document these mutually ac-
ceptable goals in the patient’s medical re-
cord. The nature of the procedure, its ex-
pected benefits, its possible risks, and the
available alternatives should be explained
to the patient by a knowledgeable individ-
ual, and the patient’s consent should be
documented. The procedure should be
performed only in a setting where there is
ready access to care for untoward reac-
tions, including equipment, medications,
and personnel trained in managing seri-
ous reactions.
Vascular Access
For most adults needing a limited number
of procedures, the antecubital veins are
suitable for removal and return of blood.
For critically ill adults and for children,
indwelling central or peripheral venous
catheters are typically used. Especially ef-
fective are rigid-wall, large-bore, double-
lumen catheters placed in the subclavian,
femoral, or internal jugular vein. Cathe-
ters of the type used for temporary hemo-
dialysis allow both removal and return of
blood at high flow rates. Central catheters
can be maintained for weeks if multiple
procedures are necessary. Tunnel catheters
canbeusedwhenlong-termapheresisis
anticipated.
Removal of Pathologic Substances
During TPE, plasma that contains the
pathologic substance is removed and a re-
placement fluid is infused. The efficiency
with which material is removed can be
estimated by calculating the patient’s
plasma volume and using Fig 6-1. This es-
timate depends on the following assump-
Chapter 6: Apheresis 145
Copyright © 2005 by the AABB. All rights reserved.

146 AABB Technical Manual
Table 6-1. Indication Categories for Therapeutic Apheresis20
Disease Procedure
Indication
Category
Renal and metabolic diseases
Antiglomerular basement membrane antibody
disease
Plasma exchange I
Rapidly progressive glomerulonephritis Plasma exchange II
Hemolytic uremic syndrome Plasma exchange III
Renal transplantation
Rejection Plasma exchange IV
Sensitization Plasma exchange III
Recurrent focal glomerulosclerosis Plasma exchange III
Heart transplant rejection Plasma exchange III
Photopheresis III
Acute hepatic failure Plasma exchange III
Familial hypercholesterolemia Selective adsorption I
Plasma exchange II
Overdose or poisoning Plasma exchange III
Phytanic acid storage disease Plasma exchange I
Autoimmmune and rheumatic diseases
Cryoglobulinemia Plasma exchange II
Idiopathic thrombocytopenic purpura Immunoadsorption II
Raynaud's phenomenon Plasma exchange III
Vasculitis Plasma exchange III
Autoimmune hemolytic anemia Plasma exchange III
Rheumatoid arthritis Immunoadsorption II
Lymphoplasmapheresis II
Plasma exchange IV
Scleroderma or progressive systemic
sclerosis
Plasma exchange III
Systemic lupus erythematosus Plasma exchange III
Lupus nephritis Plasma exchange IV
Psoriasis Plasma exchange IV
Hematolic diseases
ABO-mismatched marrow transplant RBC removal (marrow) I
Plasma exchange
(recipient)
II
Copyright © 2005 by the AABB. All rights reserved.

Chapter 6: Apheresis 147
Disease Procedure
Indication
Category
Erythrocytosis or polycythemia vera Phlebotomy I
Erythrocytapheresis II
Leukocytosis and thrombocytosis Cytapheresis I
Thrombotic thrombocytopenia purpura Plasma exchange I
Posttransfusion purpura Plasma exchange I
Sickle cell diseases RBC exchange I
Myeloma, paraproteins, or hyperviscosity Plasma exchange II
Myeloma or acute renal failure Plasma exchange II
Coagulation factor inhibitors Plasma exchange II
Aplastic anemia or pure RBC aplasia Plasma exchange III
Cutaneous T-cell lymphoma Photopheresis I
Leukapheresis III
Hemolytic disease of the fetus and newborn Plasma exchange III
Platelet alloimmunization and refractoriness Plasma exchange III
Immunoadsorption III
Malaria or babesiosis RBC exchange III
AIDS Plasma exchange IV
Neurologic disorders
Chronic inflammatory demyelinating
polyradiculoneuropathy
Plasma exchange I
Acute inflammatory demyelinating
polyradiculoneuropathy
Plasma exchange I
Lambert-Eaton myasthenia syndrome Plasma exchange II
Multiple sclerosis
Relapsing Plasma exchange III
Progressive Plasma exchange III
Lymphocytapheresis III
Myasthenia gravis Plasma exchange I
Acute central nervous system inflammatory
demyelinating disease
Plasma exchange II
Paraneoplastic neurologic syndromes Plasma exchange III
Immunoadsorption III
Table 6-1. Indication Categories for Therapeutic Apheresis20 (cont'd)
(cont'd)
Copyright © 2005 by the AABB. All rights reserved.

tions: 1) the patient’s blood volume does
not change; 2) mixing occurs immedi-
ately; and 3) there is relatively little pro-
duction or mobilization of the pathologic
material to be removed during the proce-
dure. As seen in Fig 6-1, removal is great-
est early in the procedure and diminishes
progressively during the exchange. Ex-
change is usually limited to 1 or 1.5
plasma volumes, or approximately 40 to
60 mL plasma exchanged per kg of body
weight in patients with normal hemato-
crit and average body size. This maxi-
mizes the efficacy per procedure but may
make it necessary to repeat the process.
Rarely are two or more plasma volumes
exchanged in one procedure. Although
larger volume exchange causes greater ini-
tial diminution of the pathologic sub-
stance, overall it is less efficient and re-
quires considerably more time. Larger
volume exchanges can increase the risk of
coagulopathy, citrate toxicity, or electro-
lyte imbalance, depending on the re-
placement fluid.
The rates at which a pathologic sub-
stance is synthesized and distributed be-
tween intravascular and extravascular com-
148 AABB Technical Manual
Disease Procedure
Indication
Category
Demyelinating polyneuropathy with IgG and
IgA
Plasma exchange I
Immunoadsorption III
Sydenham's chorea Plasma exchange II
Polyneuropathy with IgM (with or without
Waldenstrom's)
Plasma exchange
Immunoadsorption
II
III
Cryoglobulinemia with polyneuropathy Plasma exchange II
Multiple myeloma with polyneuropathy Plasma exchange III
POEMS syndrome Plasma exchange III
Systemic (AL) amyloidosis Plasma exchange IV
Polymyositis or dermatomyositis Plasma exchange III
Leukapheresis IV
Inclusion-body myositis Plasma exchange III
Leukapheresis IV
Rasmussen's encephalitis Plasma exchange III
Stiff-person syndrome Plasma exchange III
PANDAS Plasma exchange II
Amyotrophic lateral sclerosis Plasma exchange IV
POEMS = polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin lesions; PANDAS = pediat-
ric autoimmune neuropsychiatric disorders; Category I = standard acceptable therapy; Category II = sufficient evidence
to suggest efficacy usually as adjunctive therapy; Category III = inconclusive evidence of efficacy or uncertain risk/bene-
fit ratio; Category IV = lack of efficacy in controlled trials.
Table 6-1. Indication Categories for Therapeutic Apheresis20 (cont'd)
Copyright © 2005 by the AABB. All rights reserved.
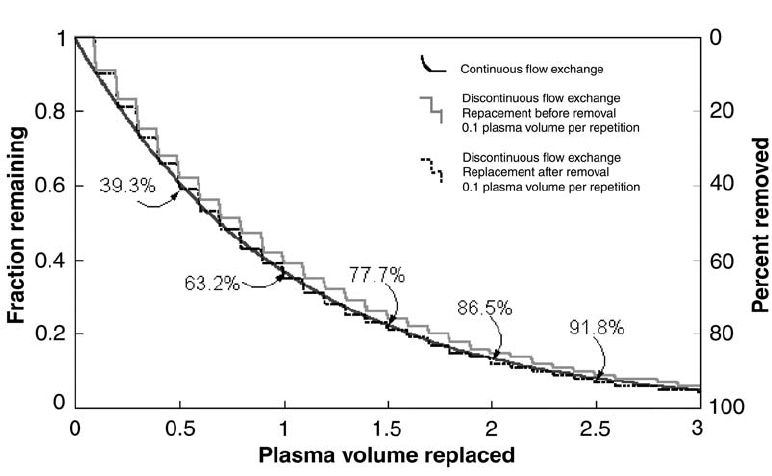
partments affect the outcome of TPE. For
example, the abnormal IgM of Walden-
strom’s macroglobulinemia is synthesized
slowly and remains almost entirely (about
75%) intravascular, making apheresis par-
ticularly effective in removing it.23 In addi-
tion, a relatively small change in intra-
vascular protein concentration may result
in a large change in blood viscosity. In con-
trast, efforts to prevent hydrops fetalis with
intensive TPE to lower the mother’s level of
IgG anti-D have been less successful. This is
due in part to the fact that about 55% of IgG
is in the extravascular fluid. In addition,
rapid reduction of IgG may cause antibody
synthesis to increase rapidly and “rebound”
over pretreatment levels.24 Rebound synthe-
sis may also complicate TPE treatment of
autoimmune diseases. Immuno-suppressive
agents such as cyclophosphamide, azathio-
prine, or prednisone may be administered
to blunt the autoantibody rebound response
to apheresis.
Plasma removed during TPE should be
handledcarefullyanddisposedofproperly.
Such plasma cannot be used for subse-
quent manufacture of transfusable plasma
derivatives.
Removal of Normal Plasma Constituents
When the quantity of plasma removed
during TPE exceeds 1.5 times the plasma
volume, different rates of removal and re-
constitution are observed for different
constituents.25 In the case of fibrinogen,
the third component of complement (C3),
and immune complexes, 75% to 85% of the
original substance is removed after a 1.5
plasma-volume procedure. Pretreatment
levels are restored in 3 to 4 days. The con-
centrations of electrolytes, uric acid, Fac-
tor VIII, and other proteins are less af-
fected by a plasma exchange. A 10% or
more decrease in platelet count generally
occurs, with 2 to 4 days needed for a re-
Chapter 6: Apheresis 149
Figure 6-1. The relationship between the volume of plasma exchange and the patient’s original plasma
remaining.
Copyright © 2005 by the AABB. All rights reserved.
turn to pretreatment values.26 Coagulation
factors other than fibrinogen generally re-
turn to pretreatment values within 24
hours. Immunoglobulin removal occurs at
about the expected rate of 65% per plasma
volume, but recovery patterns vary for dif-
ferent immunoglobulin classes, depend-
ing on intravascular distribution and rates
of synthesis. (Table 11-3 describes immu-
noglobulin characteristics.) Plasma IgG
levels return to approximately 60% of the
pretreatment value within 48 hours be-
cause of reequilibration with protein in the
extravascular space. These issues are im-
portant in planning the frequency of ther-
apeutic procedures. Weekly apheresis per-
mits more complete recovery of normal
plasma constituents; daily procedures can
be expected to deplete many normal, as
well as abnormal, constituents. Addition-
ally, intensive apheresis reduces the con-
centration of potentially diagnostic plasma
constituents, so blood for testing should be
drawn before TPE.
Replacement Fluids
Available replacement solutions include:
crystalloids, albumin solutions, plasma
(FFP, cryosupernatant plasma, or Plasma
Frozen within 24 Hours of Collection), and
HES.27 Table 6-2 presents advantages and
disadvantages of each. A combination is
often used, the relative proportions being
determined by the physician on the basis
of the patient’s disease and physical con-
dition, the planned frequency of proce-
dures, and cost. Acute treatment of
immediately life-threatening conditions
usually requires a series of daily plasma
exchange procedures, often producing a
significant reduction of coagulation factors.
Monitoring the platelet count, prothrom-
bin time, activated partial thromboplastin
time, and fibrinogen level helps deter-
mine the need for supplemental platelets,
plasma, or cryoprecipitate. Because plasma
contains citrate, its use may increase the
risk of citrate toxicity.
Complications
With careful patient selection and atten-
tion to technical details, most therapeutic
apheresis procedures are completed with-
out complications. Adverse effects of ther-
apeutic apheresis were reported in only
4% of patients in one large study.28 How-
ever, therapeutic apheresis is often required
for patients who are critically ill and at risk
for a variety of complications.
Vascular Access. Patients requiring ther-
apeutic apheresis have often been subjected
to multiple venipunctures and achieving
peripheral vascular access may be difficult.
Frequently, special venous access, such as
placement of an indwelling double-lumen
apheresis/dialysis catheter, is required. Ve-
nous access devices may cause further
vascular damage, sometimes resulting in
thrombosis. Infrequently, they may result in
severe complications such as pneumo-
thorax or perforation of the heart or great
vessels.28 Other complications include
arterial puncture, deep hematomas, and
arteriovenous fistula formation. Bacterial
colonization often complicates long-term
placement and may lead to catheter-associ-
ated sepsis, especially in patients who are
receiving steroids or other immunosup-
pressants. Inadvertent disconnection of
catheters may produce hemorrhage or air
embolism.
Alteration of Pharmacodynamics. TPE
can lower blood levels of drugs, especially
those that bind to albumin. Apheresis re-
duces plasma levels of antibiotics and anti-
convulsants, but few clinical data exist to
suggest adverse patient outcomes due to
apheresis-associated lowering of drug lev-
els. Nevertheless, the pharmacokinetics of
all drugs being given to a patient should be
150 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

considered before starting apheresis and
dosage schedules adjusted if necessary. It is
prudent to withhold the administration of
drugs scheduled to be given during or up to
an hour before apheresis until after the pro-
cedure has finished. Removal of plasma
cholinesterase may complicate the admin-
istration of paralyzing agents such as
succinylcholine in the immediate post-
exchange period.
Hypocalcemia. Most patients and donors
with normal parathyroid and liver function
maintain calcium homeostasis during aphere-
sis. However, symptoms of hypocalcemia
related to citrate toxicity are the most com-
mon adverse effect reported in 3.0% of
therapeutic apheresis procedures in one
large study.28 Symptoms of reduced plasma
levels of ionized calcium (perioral pare-
sthesias, tingling, a feeling of vibrations) re-
flect the rate at which citrate anticoagulant
is returned, ionized and bound calcium are
removed, and ionized calcium is bound to
“calcium-stripped” albumin replacement.
Hyperventilation, hypothermia, hypo-
magnesemia, and the use of plasma as a re-
placement solution exacerbate citrate tox-
icity. Hypocalcemia can usually be controlled
by reducing the proportion of citrate or
slowing the reinfusion rate. If untreated,
symptoms may progress to muscle twitch-
ing, chills, pressure in the chest, nausea,
vomiting, and hypotension. Low ionized
calcium concentrations can induce severe
cardiac arrhythmias. Asking the patient to
report any vibrations or tingling sensations
can help determine the appropriate rein-
fusion rate. Extra precautions must be
taken in patients who are unable to com-
municate or who may metabolize citrate
poorly (eg, those with liver failure). Hypo-
calcemic toxicity can usually be managed
Chapter 6: Apheresis 151
Table 6-2. Comparison of Replacement Fluids
Replacement
Solution Advantages Disadvantages
Crystalloids Low cost
Hypoallergenic
No viral risk
2-3 volumes required
Hypo-oncotic
No coagulation factors
No immunoglobulins
Albumin Iso-oncotic
No contaminating
“inflammatory mediators”
No viral risk
High cost
No coagulation factors
No immunoglobulins
Hydroxyethyl starch Moderate cost
Iso-oncotic
No contaminating “inflammatory
mediators”
No coagulation factors
Long-term residual levels of HES
Contraindicated with renal failure
Possible coagulopathy
Plasma Maintains normal levels of:
immunoglobulins
complement
antithrombin
other proteins
Viral transmission risk
Citrate load
ABO incompatibility risk
Allergic reactions
Sensitization
Copyright © 2005 by the AABB. All rights reserved.
by administering oral calcium carbonate or
intravenous calcium.22,23,29
Circulatory Effects. Hypovolemia and
subsequent hypotension may occur during
apheresis, especially when the volume of
extracorporeal blood exceeds 15% of the to-
tal blood volume. Hypotension tends to oc-
cur in ill children, the elderly, neurology pa-
tients, anemic patients, and those treated
with intermittent-flow devices that have
large extracorporeal volumes. Continuous-
flow devices typically do not require large
extracorporeal volumes but can produce
hypovolemia if return flow is inadvertently
diverted to a waste collection bag, either
through operator oversight or mechanical
or software failures. Hypovolemia may also
be secondary to inadequate volume or pro-
tein replacement. During all procedures, it
is essential to maintain careful and contin-
uous records of the volumes removed and
returned. The use of antihypertensive med-
ications, especially angiotensin-converting
enzyme (ACE) inhibitors combined with al-
bumin replacement, may also contribute to
hypotensive reactions (see Chapter 27). Pa-
tients taking agents that inhibit ACE have
experienced severe hypotensive episodes
when treated with SPA columns and with
other immunosorbents.30 Patients should not
receive these medications for 72 hours be-
fore undergoing immunoabsorption treat-
ment. Because infusion of cold fluids
through a central venous catheter may in-
duce arrhythmias, some apheresis programs
use blood warmers for selected patients.
Infections. Plasma is the only commonly
used replacement solution with the risk of
transmitting infectious viruses. Bacterial
colonization and infection related to re-
peated apheresis usually arise from within
the vascular catheter. Intensive apheresis
regimens decrease levels of immunoglobu-
lins and the opsonic components of com-
plement. In addition, immunosuppressive
drugs used to prevent rebound antibody
production may further compromise de-
fense mechanisms.
Mechanical Hemolysis and Equipment
Failures. Collapsed or kinked tubing, mal-
functioning pinch valves, or improper
threading of tubing may damage donor or
patient red cells in the extracorporeal cir-
cuit. Machine-related hemolysis was ob-
served in 0.07% of over 195,000 apheresis
procedures performed in the United King-
dom.31 Similar rates of hemolysis, 0.06% and
0.01%, were reported in response to uniform
questionnaires regarding therapeutic and
donor apheresis procedures, respectively.32,33
Hemolysis can also occur with incom-
patible replacement fluids such as D5W
(eg, D5W used to dilute 25% albumin) or
ABO-discrepant plasma. The operator
should carefully observe plasma collection
lines for pink discoloration suggestive of
hemolysis. Other types of equipment fail-
ure, such as problems with the rotating
seal, leaks in the plastic, and roller pump
failure, are rare.34
Allergic Reactions and Respiratory Dis-
tress. Respiratory difficulty during or im-
mediately following apheresis can have
many causes: pulmonary edema, massive
pulmonary embolism, air embolism, ob-
struction of the pulmonary microvascula-
ture, anaphylactic reactions, and transfusion-
related acute lung injury.35 Hemothorax or
hemopericardium due to vascular erosion
by a central venous catheter is typically un-
suspected yet may be fatal.36,37 Pulmonary
edema that results from volume overload or
cardiac failure is usually associated with
dyspnea, an increase in the diastolic blood
pressure, and characteristic chest X-ray
findings. Acute pulmonary edema can also
arise from damage to alveolar capillary
membranes secondary to an immune reac-
tion or to vasoactive substances in FFP or
colloid solutions prepared from human
plasma. The use of FFP as a replacement
fluid has been associated with complement
152 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
activation and with allergic reactions that
produce urticaria, swelling of oral mucosa,
and bronchospasm; these usually respond
to antihistamines and corticosteroids. Hypo-
tension and flushing associated with the
rapid infusion of albumin in patients taking
ACE inhibitors are discussed in Chapter 27.
Predominantly ocular (periorbital edema,
conjunctival swelling, and tearing) reac-
tions have occurred in donors sensitized to
the ethylene oxide gas used to sterilize dis-
posable plastic apheresis kits.38
Fatalities During Apheresis. Despite the
fact that patients undergoing therapeutic
apheresis are often critically ill, fatalities
during apheresis are comparatively rare.
Estimates of fatality rates range from 3 in
10,00039 to 1 in 50040 procedures. Most deaths
were due to cardiac arrhythmias or arrest
during or shortly after the procedure or to
acute pulmonary edema or adult respira-
tory distress syndrome occurring during a
procedure. Rare fatalities resulted from
anaphylaxis, vascular perforation, hepatitis,
sepsis, thrombosis, and hemorrhage.
Indications for Therapeutic Apheresis
Although therapeutic apheresis has been
used in the treatment of many diseases,
most published studies are case reports or
small uncontrolled series, often providing
insufficient evidence of efficacy. Publica-
tion bias tends to favor positive results,
and physicians should avoid subjecting
patients to the risks and high costs of
apheresis procedures based on marginal
clinical studies. Controlled, randomized,
blinded studies of therapeutic apheresis
are difficult to conduct, especially be-
cause using sham treatments as a control
is expensive and carries some risk. How-
ever, the complicated apheresis instru-
ments and associated attention from
nursing and medical personnel may cre-
ate or amplify a placebo effect and bias
the evaluation of clinical improvement.
For many of the diseases being treated,
the etiology, pathogenesis, and natural his-
tory are incompletely understood, and re-
ductions in such measured variables as
complement components, rheumatoid fac-
tor, or immune complexes cannot be corre-
lated reliably with changes in disease activ-
ity. An example is the use of the erythrocyte
sedimentation rate (ESR) as an index of dis-
ease activity in rheumatoid arthritis. The
ESR invariably decreases during intensive
TPE, but this reflects removal of fibrinogen
and not necessarily a decrease in disease
activity. For the same reasons, the optimal
volume and frequency of exchange are of-
ten not established. For severe imminently
life-threatening disease, when albumin/sa-
line is the replacement fluid, TPE is initially
performed daily. After a few days, the
fibrinogen or platelet count may be low
enough to significantly increase the risk of
bleeding. Clinical judgment must then be
exercised to decide whether to proceed
with TPE using clotting factor/platelet
transfusions or to withhold TPE until these
parameters normalize. The conditions dis-
cussed below are established indications
for therapeutic apheresis.3,20,21,41,42
Hematologic Conditions
Serum Hyperviscosity Syndrome. Serum
hyperviscosity resulting from multiple
myeloma or Waldenstrom’s macroglobu-
linemia can cause congestive heart failure;
reduced blood flow to the cerebral, car-
diac, or pulmonary circulation; or symp-
toms of headache, vertigo, somnolence, or
obtundation. Paraproteins may interfere
with hemostasis, leading to hemorrhagic
symptoms.
Thepresenceofhyperviscositycorrelates
only in very general terms with the concen-
Chapter 6: Apheresis 153
Copyright © 2005 by the AABB. All rights reserved.
tration of paraprotein. Measurement of se-
rum viscosity relative to water is a simple
procedure that provides more objective in-
formation. For some pathologic proteins,
serum viscosity is highly temperature de-
pendent, so serum viscosity should be
measured at physiologically relevant tem-
peratures. Normal serum viscosity ranges
from 1.4 to 1.8 relative to water. Because
most patients are not symptomatic until
their relative serum viscosity is more than
4.0 or 5.0, patients with mild elevations may
not require treatment. For symptomatic
hyperviscosity, a single apheresis procedure
is usually highly effective.23
Hyperleukocytosis. Leukapheresisisof
-
ten used to treat the dramatically elevated
white cell count that can occur in acute leu-
kemia. Several different thresholds have
been used: fractional volume of leukocytes
(leukocrit) above 10%; total circulating leu-
kocytes above 100,000/µL; and circulating
blasts above 50,000/µL.43 However, the use
of a single laboratory value as an indication
for treatment is an oversimplification. Such
factors as erythrocyte concentration, leuke-
mic cell type, rate at which the count is ris-
ing, potential obstructions to cerebral or
pulmonary blood flow, and the patient’s co-
agulation status and general condition must
be considered. Most leukemic patients with
extreme leukocytosis have significant ane-
mia. Reduced red cell mass reduces blood
viscosity, so unless there is an acute need to
increase oxygen-carrying capacity, red cells
should not be transfused until the hypervis-
cosity crisis has been resolved.43
In some patients with acute blast crisis
or in unusual types of leukemia, both the
hematocrit and leukocrit are elevated. If
there is evidence of cerebral or pulmonary
symptoms, rapid reduction of leukocyte
concentration should be considered, al-
though the efficacy of such leukocyte re-
duction is unproved. More commonly,
however, the white cell count rises over
weeks or longer, and leukocyte reduction
can be effected with chemotherapy, with or
without leukapheresis. Leukapheresis is
sometimes used to reduce the white cell
count to <100,000/µLbeforethestartof
chemotherapy, to reduce the likelihood of
tumor lysis syndrome. However, there have
been no controlled clinical trials to sub-
stantiate this approach, and it must be rec-
ognized that more malignant cells are
present outside the circulation than within
the bloodstream.
Thrombocythemia. Therapeutic plate-
letpheresis is usually undertaken for symp-
tomatic patients with platelet counts above
1,000,000/µL. The measured count, by itself,
should not determine whether platelet
reduction is indicated. In patients with
evidence of thrombosis secondary to
thrombocythemia, plateletpheresis can be
beneficial. The rationale for platelet reduc-
tion in bleeding patients with thrombocy-
themia is less clear. There are no accepted
indications for prophylactic plateletphere-
sis in asymptomatic patients, although the
risk of placental infarction and fetal death
may justify the procedure in a pregnant
woman with severe thrombocythemia.43
Thrombotic Thrombocytopenic Purpura/
Hemolytic-Uremic Syndrome (TTP/HUS).
The conditions described as TTP/HUS are
multisystem disorders, in which platelet/fi-
brin thrombi occlude the microcirculation.
They are characterized by varying degrees
of thrombocytopenia, microangiopathic
hemolytic anemia, renal dysfunction, neu-
rologic abnormalities, and fever. Patients
presenting with fulminant TTP usually have
platelet counts below 50,000/µLandlactic
dehydrogenase (LDH) levels above 1000
IU/mL, resulting from systemic ischemia
and hemolysis.44 The peripheral blood
smear characteristically shows increased
numbers of schistocytes. Evidence for dis-
seminated intravascular coagulation is gen-
erally absent.
154 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
TTP usually develops without obvious
cause, although episodes may occur after
infections, pregnancy, or use of some com-
mon drugs such as ticlopidine, or clopido-
grel. Recent reports suggest that it is caused
by a transient antibody to a protease
(ADAMTS13) that normally cleaves large
von Willebrand factor (vWF) multimers.
The unusually large vWF multimers avidly
aggregate circulating platelets, triggering
the syndrome.45,46 Increasingly, cases of re-
current or relapsing TTP are being recognized.
HUS is a similar condition that occurs
morecommonlyinchildrenthanadults.
HUS may follow diarrheal infections with
verotoxin-secreting strains of Escherichia
coli (strain 0157:H7) or Shigella.Compared
with patients who have classic TTP, those
withHUShavemorerenaldysfunctionand
less prominent neurologic and hematologic
findings. Most patients with HUS do not
have antibody to the vWF protease and
have normal concentrations of vWF prote-
ase. TTP/HUS can occur after treatment with
certain cytotoxic drugs, including mito-
mycin C. A microangiopathic hemolysis
similar to TTP/HUS can occur in organ or
stem cell transplant recipients receiving
cyclosporine and tacrolimus.47,48 Transplant-
associated microangiopathic hemolysis ap-
pearstobelessresponsivetotherapyand
probably represents a different disease pro-
cess.48
TPE with plasma or Plasma Cryopreci-
pitate Reduced replacement has become
the treatment of choice for TTP/HUS.49,50
Protease levels have been shown to be sta-
ble for at least 2 weeks in citrated plasma
stored at 37 C.51 TPE is now thought to re-
move both antibody to the protease and
vWF and to replace deficient protease.
Other largely unproved treatments include
prednisone, antiplatelet agents, splenec-
tomy, vincristine, rituximab, and intrave-
nous immunoglobulin. Because platelet
transfusions have anecdotally been associ-
ated with disease exacerbation and death,
they are usually contraindicated (except in
the presence of life-threatening hemorrhage).
TPE is typically performed daily for l to 2
weeks, but the intensity and duration of
treatment should be guided by the individ-
ual patient’s course. Occasionally, pro-
longed courses of treatment are required.
Therapeutic plasma exchange has impres-
sively improved the survival rate in TTP,
from being almost universally fatal before
1964 to 80% survival in a recent series.49
Signs of response to therapy include a ris-
ing platelet count and reduction of LDH
between procedures. As patients recover to
near normal LDH and platelet count (100-
150,000/µL), TPE is discontinued. Some
programs switch from intensive TPE to in-
termittent plasma exchange or simple
plasma infusion, but the efficacy of this ap-
proach has not been established.52 Despite
the success of TPE, TTP/HUS remains a
serious condition. Treatment failures con-
tinue to occur and to cause major organ
damage or death.
Complications of Sickle Cell Disease.
Several complications of sickle cell disease
are syndromes that can be treated by red
cell exchange. These conditions include
stroke or impending stroke, acute chest
syndrome, and multiorgan failure. Either
manual or automated techniques can be
used for red cell exchange, but automated
techniques are faster and better controlled.
The goal is to replace red cells containing
hemoglobin S with a sufficient number of
red cells containing hemoglobin A so that
the overall proportion of hemoglobin A in
the blood is 60% to 80%. Some centers pro-
vide partially phenotypically matched red
cells (eg, C, E, and K1) to avoid alloimmuni-
zing long-term transfusion recipients to
these antigens. At the end of the procedure,
the patient’s hematocrit should be no higher
than 30% to 35% to avoid increased blood
viscosity. Chronic erythrocytapheresis can be
Chapter 6: Apheresis 155
Copyright © 2005 by the AABB. All rights reserved.
used to manage iron overload in patients
requiring long-term transfusion therapy.53
Cryoglobulinemia
Significant elevations of cryoglobulins may
cause cold-induced vascular occlusion,
abnormalities of coagulation, renal insuf-
ficiency, or peripheral nerve damage. Re-
moval of cryoglobulins by apheresis can
be used to treat acute symptomatic epi-
sodes, but definitive therapy depends on
identifying and treating the underlying
causative condition.
Neurologic Conditions
Myasthenia Gravis. Myasthenia gravis re-
sults from autoantibody-mediated block-
ade of the acetylcholine receptor located
on the postsynaptic motor endplate of
muscles. Standard treatment includes ste-
roids and acetylcholinesterase inhibitors.
TPEisusedasadjunctivetreatmentfor
patients experiencing exacerbations not
controlled by medications and for pa-
tients being prepared for thymectomy. A
typical treatment protocol is five or six
TPE procedures over l to 2 weeks. Concur-
rent immunosuppression to prevent anti-
body rebound is recommended. Chronic
TPE has been used with some success in a
small number of patients.
Acute Guillain-Barré Syndrome.
Guillain-Barré syndrome is an acute auto-
immune demyelinating polyneuropathy
that can produce dramatic paralysis in oth-
erwise healthy individuals. The cause is un-
known; many cases appear to follow benign
viral infections or Campylobacter jejuni in-
fection. Most patients recover spontane-
ously,butasmanyasoneinsixmaybe
-
come unable to walk or may develop
respiratory failure requiring ventilatory
support. Early treatment is beneficial for
patients with rapidly progressive disease.
The response to therapy is inferior in pa-
tients who remain untreated for several
weeks. Recent controlled studies suggest
that intravenous immunoglobulin gives re-
sults equivalent to five TPE procedures over
a 2-week period.54 A cost-effectiveness anal-
ysis has suggested that TPE is less costly
than a course of intravenous immune glob-
ulin.55 Multicenter trials have suggested that
TPE, if initiated early, can decrease the pe-
riod of minimal sensorimotor function.56
Patients whose illness is not acute in onset,
is not characteristic of Guillain-Barré syn-
drome, or in whom nerve conduction stud-
ies show complete axonal block may have a
poorer prognosis and less response to
apheresis therapy.
Chronic Inflammatory Demyelinating
Polyneuropathy. Chronic inflammatory
demyelinating polyneuropathy (CIDP), of-
ten seen in HIV patients, is a group of disor-
ders with slow onset and progressive or
intermittent course, characterized by ele-
vated spinal fluid protein, marked slowing
of nerve conduction velocity, and segmen-
tal demyelination of peripheral nerves. Var-
ious sensorimotor abnormalities result.
Polyneuropathy may also occur in the
POEMS syndrome, characterized by poly-
neuropathy, organomegaly, endocrino-
pathy, MGUS (monoclonal gammopathy of
unknown significance), and skin changes.
Corticosteroids are the first-line treatment
for CIDP. TPE and intravenous immuno-
globulin have equivalent efficacy in patients
unresponsive to corticosteroids.57
Polyneuropathy Associated with Mono-
clonal Gammopathy of Undetermined Sig-
nificance. When polyneuropathy is associ-
ated with monoclonal paraproteins of un-
certain significance, TPE has been shown to
be effective for all variants.58,59
Renal Diseases
Rapidly progressive glomerulonephritis
(RPGN) associated with antibodies to
156 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
basement membranes of glomeruli and al-
veoli, which may result in pulmonary
hemorrhage (Goodpasture’s disease), usu-
ally responds to TPE as an adjunct to im-
munosuppressive drugs.41 TPE accelerates
the disappearance of antibodies to base-
ment membranes and improves renal
function. TPE is particularly effective in
halting pulmonary hemorrhage in these
patients, even if renal function does not
completely normalize following treat-
ment. Therapeutic apheresis has been
used in treating the vasculitis associated
with RPGN and the presence of anti-
neutrophil cytoplasmic antibody (ANCA-
positive RPGN).60,61 TPE is most effective
inthemoreseverecases.
41
Myeloma light chains may be toxic to re-
nal tubular epithelium and cause renal fail-
ure in up to 10% of cases. TPE is useful as
adjunctive therapy in some myeloma pa-
tients with cast nephropathy but is not as-
sociated with improved survival.41,62
Other Conditions
TPE has been used as adjunctive treatment
for a variety of multisystem diseases. A
combination of steroid, cytotoxic agents,
and TPE has been used for severely ill pa-
tients with polyarteritis nodosa,63 al-
though most rheumatologic conditions
respond poorly to TPE. Clinical trials us-
ing standard TPE have not shown benefit
in the treatment of systemic lupus erythe-
matosus, polymyositis, dermatomyositis, or
scleroderma.21 Immunoadsorption col-
umns are of benefit in patients with rheu-
matoid arthritis refractory to medical
management.64
Homozygous Type II Familial Hyper-
cholesterolemia. Homozygous hypercho-
lesterolemia, a rare disorder of the receptor
for low-density lipoproteins, results in se-
vere premature atherosclerosis and early
death from coronary artery disease. Pro-
longed reduction in circulating lipids can
be achieved with repeated TPE, often with
selective adsorption or filtration tech-
niques.43 Heterozygous hypercholesterol-
emia results from several gene defects in
the LDL receptor. Some patients with het-
erozygous hypercholesterolemia also de-
velop high levels of cholesterol and are at
increased risk for developing premature
atherosclerotic heart disease.
Two apheresis systems for selective LDL
removal in patients with homozygous or
heterozygous hypercholesterolemia have
been cleared by the FDA. The Liposorber
LA-150(KanekaPharmaAmerica,New
York, NY) is based on a dextran sulfate ad-
sorption system. The H.E.L.P. LDL system
(B. Braun, Melsugen, Germany) is a hepa-
rin-induced LDL cholesterol precipitation
system. The LDL apheresis procedure se-
lectively removes apolipoprotein-B-con-
taining cholesterols such as LDL and very
LDL, sparing high-density lipoprotein
(HDL) cholesterol. This provides an advan-
tage over standard apheresis, which re-
moves all plasma proteins, including the
“protective” HDL. The procedure acutely
lowers levels of targeted cholesterols by
60% to 70%. Treatment is usually per-
formed every 1 to 2 weeks, and cholesterol-
lowering drugs are generally employed si-
multaneously. Several studies have shown
that use of LDL apheresis can achieve sig-
nificant lowering of lipids in nearly all pa-
tients with severe hypercholesterolemia.65,66
Promising results have also been reported
from a system capable of direct adsorption
of LDL and Lp(a) (DALI) from whole
blood.67
Refsum’s Disease (Phytanic Acid Dis-
ease). Refsum’s disease is a rare inborn er-
ror of metabolism resulting in toxic levels of
phytanic acid, causing neurologic, cardiac,
skeletal, and skin abnormalities.21 TPE is
useful in conjunction with a phytanic-
acid-deficient diet and should be started as
Chapter 6: Apheresis 157
Copyright © 2005 by the AABB. All rights reserved.
soon as possible, before permanent dam-
age occurs.
Staphylococcal Protein A
Immunoadsorption
SPA immunoadsorption is approved by the
FDA for treatment of acute and chronic
immune thrombocytopenic purpura. The
device is also FDA-approved to treat
adults with rheumatoid arthritis unre-
sponsive to disease-modifying antirheu-
matic drugs.64 Although not FDA-ap-
proved, this technique has been used,
with limited success, to treat other auto-
immune thrombocytopenias.68 Many of
these protocols are still experimental and
randomized trials have not been con-
ducted. Anecdotal cases of TTP/HUS re-
fractory to TPE that have responded to SPA
immunoadsorption have also been re-
ported.69
Photopheresis
Photopheresis is a technique that involves
the treatment of patients with psoralens,
the separation of lymphocytes by aphere-
sis, and treatment of the cells with ultravi-
olet radiation. This renders the lympho-
cytes and other nucleated cells incapable
of division. The treated cells are then re-
infused. This procedure, also known as
extracorporeal photochemotherapy, has
been approved by the FDA for the treat-
ment of cutaneous T-cell lymphoma and
is considered the first line of treatment for
the erythrodermic phase of this disease.70
Clinical trials are under way to determine
the efficacy of photopheresis in the fol-
lowing conditions: cellular-mediated re-
jection of heart and lung allografts, and
acute and chronic graft-vs-host disease
following allogeneic marrow transplant.
This technology is not available in most
apheresis centers.
References
1. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
2. Code of federal regulations. Title 21 CFR Part
640. Washington, DC: US Government Print-
ing Office, 2004 (revised annually).
3. McLeod BC. Introduction to the third special
issue: Clinical applications of therapeutic
apheresis. J Clin Apheresis 2000;15(½):2-3.
4. Burgstaler EA. Current instrumentation for
apheresis. In: McLeod BC, Price TH, Weinstein
R, eds. Apheresis: Principles and practice.
2nd ed. Bethesda, MD: AABB Press, 2003:95-
130.
5. Vamvakas EC, Pineda AA. Selective extraction
of plasma constituents. In: McLeod BC, Price
TH, Weinstein R, eds. Apheresis: Principles and
practice.2nded.Bethesda,MD:AABBPress,
2003:437-76.
6. BergerGM,FirthJC,JacobsP,etal.Threedif
-
ferent schedules of low-density lipoprotein
apheresis compared with plasmapheresis in
patients with homozygous familial hypercho-
lesterolemia. Am J Med 1990;88:94-100.
7. Silva MA, Brecher ME. Summary of the AABB
interorganizational taskforce on bacterial
contamination of platelets, fall 2004 impact
survey. Transfusion 2005 (in press).
8. Food and Drug Administration. Memoran-
dum: Revised guideline for the collection of
Platelets, Pheresis. (October 7, 1988) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 1988.
9. Food and Drug Administration. Memoran-
dum: Volume limits for automated collection
of source plasma. (November 4, 1992) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 1992.
10. Food and Drug Administration. Memoran-
dum: Requirements for infrequent plasma-
pheresis donors. (March 10, 1995) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 1995.
11. Food and Drug Administration. Guidance for
industry: Recommendations for collecting
red blood cells by automated apheresis meth-
ods. (January 30, 2001; technical correction
February 13, 2001) Rockville, MD: CBER Of-
fice of Communication, Training, and Manu-
facturers Assistance, 2001.
12. Vamvakas EC, Pineda AA. Determinants of the
efficacy of prophylactic granulocyte transfu-
sions: A meta-analysis. J Clin Apheresis 1997;
12:74-81.
13. StroncekDF,YauYY,OblitasJ,LeitmanSF.
Administration of G-CSF plus dexametha-
sone produces greater granulocyte concen-
158 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
trate yields while causing no more donor tox-
icity than G-CSF alone. Transfusion 2001;41:
1037- 44.
14. Strauss RG. Neutrophil collection and trans-
fusions.In:SimonTL,DzikWH,SnyderEL,et
al, eds. Rossi’s principles of transfusion medi-
cine.3rded.Baltimore,MD:LippincottWil
-
liams and Wilkins, 2002:258-67.
15. McCullough J. Granulocyte transfusion. In:
PetzLD,SwisherSN,KleinmanS,etal,eds.
Clinical practice of transfusion medicine. 2nd
ed. New York: Churchill-Livingstone, 1996:
413-32.
16. Barnes A, DeRoos A. Increased granulocyte
yields obtained with an oral three-dose pred-
nisone premedication schedule (abstract).
Am J Clin Pathol 1982;78:267.
17. Strauss RG, Ghodsi Z. Cataracts in neutrophil
donors stimulated with adrenal cortico-
steroids. Transfusion 2001;41:1464-8.
18. Stroncek DF, Matthews CL, Follman D,
Leitman SF. Kinetics of G-CSF induced
granulocyte mobilization in healthy subjects:
Effects of route of administration and addi-
tion of dexamethasone. Transfusion 2002;42:
597-602.
19. Szczepiorkowski ZM, ed. Standards for cellu-
lar therapy product services. 1st ed. Bethesda,
MD: AABB, 2004.
20. Smith JW, Weinstein R, Hillyer KL for the
AABB Hemapheresis Committee. Therapeutic
apheresis: A summary of current indications
categories endorsed by the AABB and the
American Society for Apheresis. Transfusion
2003;43:820-2.
21. Strauss RG, Ciavarella D, Gilcher RO, et al. An
overview of current management. J Clin
Apheresis 1993;8:189-272.
22. Jones HG, Bandarenko N. Management of the
therapeutic apheresis patient. In: McLeod
BC,PriceTH,WeinsteinR,eds.Apheresis:
Principles and practice. 2nd ed. Bethesda,
MD: AABB Press, 2003:253-82.
23. McLeod BC. Therapeutic plasma exchange.
In:SimonTL,DzikWH,SnyderEL,etal,eds.
Rossi’s principles of transfusion medicine.
3rd ed. Baltimore, MD: Lippincott Williams
and Wilkins, 2002:662-82.
24. Williams WJ, Katz VL, Bowes WA. Plasma-
pheresis during pregnancy. Obstet Gynecol
1990;76:451-7.
25. OrlinJB,BerkmanEM.Partialplasmare
-
placement: Removal and recovery of normal
plasma constituents. Blood 1980;56:1055-9.
26. Weinstein R. Basic principles of therapeutic
blood exchange. In: McLeod BC, Price TH,
Weinstein R, eds. Apheresis: Principles and
practice.2nded.Bethesda,MD:AABBPress,
2003:295-320.
27. Brecher ME, Owen HG, Bandarenko N. Alter-
natives to albumin: Starch replacement for
plasma exchange. J Clin Apheresis 1997;12:
146-53.
28. KiprovDD,GoldenP,RoheR,etal.Adverse
reactions associated with mobile therapeutic
apheresis; analysis of 17,940 procedures. J
Clin Apheresis 2001;16:130-3.
29. Weinstein R. Prevention of citrate reactions
during therapeutic plasma exchange by con-
stant infusion of calcium gluconate with the
return fluid. J Clin Apheresis 1996;11:204-10.
30. Olbricht CJ, Schaumann D, Fischer D. Ana-
phylactoid reactions, LDL apheresis with
dextran sulphate, and ACE inhibitors. Lancet
1993;341:60-1.
31. Robinson A. Untoward reactions and inci-
dents in machine donor apheresis. Transfus
Today 1990;7:7-8.
32. McLeodBC,SniecinskiI,CiavarellaD,etal.
Frequency of immediate adverse effects asso-
ciated with therapeutic apheresis. Transfu-
sion 1999;39:282-8.
33. McLeodBC,PriceTH,OwenH,etal.Frequency
of immediate adverse effects associated with
apheresis donation. Transfusion 1998;38:
938-43.
34. Westphal RG. Complications of hemapheresis.
In: Westphal RG, Kasprisin DO, eds. Current
status of hemapheresis: Indications, technol-
ogy and complications. Arlington, VA: AABB,
1987:87-104.
35. Askari S, Nollet K, Debol SM, et al. Transfu-
sion-related acute lung injury during plasma
exchange: Suspecting the unsuspected. J Clin
Apheresis 2002;17:93-6.
36. DuntleyP,SieverJ,KorwesML,etal.Vascular
erosion by central venous catheters. Clinical
features and outcome. Chest 1992;101:1633-
8.
37. Quillen K, Magarace L, Flanagan J, Berkman
EM. Vascular erosion caused by a double-lu-
men central venous catheter during thera-
peutic plasma exchange. Transfusion 1995;
35:510-2.
38. Leitman SF, Boltansky H, Alter HJ, et al. Aller-
gic reactions in healthy plateletpheresis do-
nors caused by sensitization to ethylene ox-
ide gas. N Engl J Med 1986;315:1192-6.
39. Gilcher RO. Apheresis: Principles and tech-
nology of hemapheresis. In: Simon TL, Dzik
WH,SnyderEL,etal,eds.Rossi’sprinciplesof
transfusion medicine. 3rd ed. Baltimore, MD:
Lippincott Williams and Wilkins, 2002:648-
58.
40. Schmitt E, Kundt G, Klinkmann H. Three
years with a national apheresis registry. J Clin
Apheresis 1992;7:58-72.
Chapter 6: Apheresis 159
Copyright © 2005 by the AABB. All rights reserved.
41. Madore F, Lazarus JM, Brady HR. Therapeutic
plasma exchange in renal diseases. J Am Soc
Nephrol 1996;7:367-86.
42. McLeod BC. Apheresis principles and prac-
tice. 2nd ed. Bethesda, MD: AABB Press, 2003.
43. Klein HG. Principles of apheresis. In: Ander-
son KC, Ness PM, eds. Scientific basis of
transfusion medicine. 2nd ed. Philadelphia:
WB Saunders, 2000:553-68.
44. Cohen JA, Brecher ME, Bandarenko N. Cellu-
lar source of serum lactate dehydrogenase el-
evation in patients with thrombotic thrombo-
cytopenic purpura. J Clin Apheresis 1998;13:
16-9.
45. Furlan M, Robles R, Galbusera M, et al. von
Willebrand factor-cleaving protease in throm-
botic thrombocytopenic purpura and the
hemolytic-uremic syndrome. N Engl J Med
1998;339:1578-84.
46. Tsai H-M, Chun-Yet Lian E. Antibodies to von
Willebrand factor-cleaving protease in acute
thrombotic thrombocytopenic purpura. N Engl
J Med 1998;339:1585-94.
47. McLeod BD. Thrombotic microangiopathies
in bone marrow and organ transplant pa-
tients. J Clin Apheresis 2002;17:118-23.
48. GeorgeJN,LiX,McMinnJR,TerrellDR,etal.
Thrombotic thrombocytopenic purpura-
hemolytic uremic syndrome following allo-
geneic HPC transplantation: A diagnostic di-
lemma. Transfusion 2004;44:294-304.
49. George JN, Rizui MA. Thrombocytopenia. In:
Beutler E, Lichtman MA, Coller BS, et al, eds.
Williams’ hematology. 6th ed. New York:
McGraw-Hill, 2001:1495-539.
50. Rock G, Shumak KH, Sutton DM, et al. Cryo-
supernatant as replacement fluid for plasma
exchange in thrombotic thrombocytopenic
purpura. Br J Haematol 1996;94:383-6.
51. Gerritsen HE, Robles R, Lammle B, Furlan M.
Partial amino acid sequence of purified von
Willebrand factor-cleaving protease. Blood 2001;
98;1654-61.
52. Bandarenko N, Brecher ME, and members of
the US TTP Apheresis Study Group. United
States Thrombotic Thrombocytopenic Pur-
pura Apheresis Study Group (US TTP ASG):
Multicenter survey and retrospective analysis
of current efficacy of therapeutic plasma ex-
change. J Clin Apheresis 1998;13:133-41.
53. Adans DM, Schultz WH, Ware RE, Kinney TR.
Erythrocytapheresis can reduce iron overload
and prevent the need for chelation therapy in
chronically transfused pediatric patients. J
Pediatr Hematol Oncol 1996;18:46-50.
54. Plasma Exchange/Sandoglobulin Guillain-
Barré Syndrome Trial Group. Randomized
trial of plasma exchange, intravenous immu-
noglobulin, and combined treatments in
Guillain-Barré syndrome. Lancet 1997;349:
225-30.
55. Nagpal S, Benstead T, Shumak K, et al. Treat-
ment of Guillain-Barré syndrome: A cost-ef-
fectiveness analysis. J Clin Apheresis 1999;14:
107-13.
56. Guillain-Barré Syndrome Study Group.
Plasmapheresis and acute Guillain-Barré syn-
drome. Neurology 1985;35:1096-104.
57. vanDoorn PA, Vermeulen M, Brand A. Intra-
venous immunoglobulin treatment in pa-
tients with chronic inflammatory demye-
linating polyneuropathy. Arch Neurol 1991;
48:217-20.
58. DyckPJ,LowPA,WindebankAJ,etal.Plasma
exchange in polyneuropathy associated with
monoclonal gammopathy of undetermined
significance. N Engl J Med 1991;325:1482-6.
59. Simovic D, Gorson KC, Popper AH. Compari-
son of IgM-MGUS and IgG-MGUS polyneuro-
pathy. Acta Neurol Scand 1998;97:194-200.
60. Frasca GM, Zoumparidis NG, Borgnino LC, et
al. Plasma exchange treatment in rapidly pro-
gressive glomerulonephritis associated with
anti-neutrophil cytoplasmic autoantibodies.
Int J Artif Organs 1992;3:181-4.
61. Pusey CD, Rees AJ, Evans JJ, et al. A random-
ized controlled trial of plasma exchange in
rapidly progressive glomerulonephritis with-
out anti-GBM antibodies. Kidney Int 1991;40:
757-63.
62. Johnson WJ, Kyle RA, Pineda AA, et al. Treat-
ment of renal failure associated with multiple
myeloma. Arch Intern Med 1990;150:863-9.
63. Guillevin L, Lhote F, Leon A, et al. Treatment
of polyarteritis nodosa related to hepatitis B
virus with short-term steroid therapy associ-
ated with antiviral agents and plasma ex-
changes: A prospective trial in 33 patients J
Rheumatol 1993;20:289-98.
64. Felson DT, LaValley MP, Baldassare AR, et al.
The Prosorba column for treatment of refrac-
tory rheumatoid arthritis: A randomized,
double-blind, sham-controlled trial. Arthritis
Rheum 1999;42:2153-9.
65. Kroon AA, Aengevaeren WRM, van der Werf T,
et al. LDL-apheresis atherosclerosis regres-
sion study (LAARS): Effect of aggressive ver-
sus conventional lipid lowering treatment on
coronary atherosclerosis. Circulation 1996;
93:1826-35.
66. Thompson GR, Maher VMG, Matthews S, et
al. Familial hypercholesterolemia regression
study: A Randomized trial of low-density-li-
poprotein apheresis. Lancet 1995;345:811-16.
67. BoschT,LennertzA,SchenzleD,etal.Direct
adsorption of low density lipoprotein and li-
poprotein (a) from whole blood: Results of
the first clinical long-term multicenter study
160 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
using DALI apheresis. J Clin Apheresis 2002;
17:161-9.
68. Handelsman H. Office of health technology
assessment report, No. 7. Protein A columns
for the treatment of patients with idiopathic
thrombocytopenic purpura and other indica-
tions.Rockville,MD:DHHS,PHS,Agencyfor
Health Care Policy and Research, 1991:1-8.
69. Gaddis TG, Guthrie TH, Drew MJ, et al. Treat-
ment of refractory thrombotic thrombocyto-
penic purpura with protein A immuno-
adsorption. Am J Hematol 1997;55:55-8.
70. Lim HW, Edelson RL. Photopheresis for treat-
ment of cutaneous T-cell lymphoma. Hema-
tol Oncol Clin North Am 1995;9:1117-26.
Chapter 6: Apheresis 161
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 7: Blood Component Testing and Labeling
Chapter 7
Blood Component Testing
and Labeling
EACH DONOR UNIT must be tested
and properly labeled before its re-
lease for transfusion. Although the
scope and characteristics of donor tests
changed with the release of new tests and
the advent of new regulatory require-
ments, the intent of donor testing remains
constant: to enhance the safety of the
blood supply. This chapter presents the
general principles that apply to testing
and labeling donor blood, and it provides
a description of the specific tests that are
required or done voluntarily at most
blood banks on each donation. Discus-
sion of the infectious complications of
blood transfusion is found in Chapter 28.
Other aspects of component preparation
are covered in Chapter 8.
Testing
General Requirements
Each laboratory needs to develop standard
operating procedures for the performance
of blood component testing strictly in
compliance with current instructions pro-
vided by the test manufacturers. Testing
must be performed in a planned, orderly
manner under a quality plan and a writ-
ten set of procedures that instruct the
staff how to perform testing, under what
circumstances additional testing needs to
be done, and what to do if things go
wrong. The facilities and equipment must
be adequate for the activity being con-
ducted. Access to the area must be lim-
ited. The environment must be controlled
so that temperature specifications for the
tests will be met, and the test will not be
adversely affected by the environment.
The test materials and equipment in use
must be those previously approved and
validated by the facility. If a facility uses
reagents or equipment from several dif-
ferent manufacturers, the facility is respon-
sible for documentation that validation of
the equipment or reagent combination
for each test in use has occurred and that
staff have been trained on the most cur-
163
7
Copyright © 2005 by the AABB. All rights reserved.
rent applicable instructions. For tests re-
quired by the Food and Drug Administra-
tion (FDA)1and/or AABB Standards for
Blood Banks and Transfusion Services, all
reagents used must meet or exceed the re-
quirements of the FDA.2(p9) If the manufac-
turer of a licensed test supplies controls,
they must be used for that test. However,
if these controls are used for calibration,
different controls must be used to verify
test performance. These controls may need
to be purchased separately. The manufac-
turer defines acceptable sample (speci-
men) requirements and considerations
that usually include the presence and na-
ture of anticoagulant, the age of suitable
samples, and permissible storage inter-
vals and conditions. Tests must be per-
formed on a properly identified sample
from the current donation. Testing must
be completed for each blood donation be-
fore release. Each test result must be re-
corded concurrently with its observation;
interpretation is to be recorded only when
testing has been completed. Testing re-
sults must be recorded and records main-
tained so that any results can be traced for
a specific unit and/or component.
The facility should have a policy for noti-
fying donors of positive infectious disease
test results. Test results are confidential and
must not be released to anyone (other than
the donor) without the donor’s written con-
sent. At the time of donation, the donor
must be told if the policy is to release posi-
tive test results to state or local public
health agencies, and the donor must agree
to those conditions before phlebotomy. Do-
nors must sign a consent form before they
donate blood acknowledging that the facil-
ity maintains a registry of donors who gave
disqualifying donor histories or have posi-
tive infectious disease results. The donor
must also be informed if the sample will
undergo research testing, including investi-
gational new drug (IND) protocols. The
most problematic notifications are those in
which the donor has a false-positive test re-
sult. For some analytes [eg, antibodies to
hepatitis C virus (anti-HCV) or human im-
munodeficiency virus, types ½ (anti-
HIV-1/2)], confirmatory or supplemental
testing is routinely performed for donor
counseling purposes and possible donor
reentry, if applicable. In the case of a minor,
state and local laws apply.3
Required Tests
ABO group and Rh type must be determined
at each donation. A sample from each do-
nation intended for allogeneic use must
be tested for the following1,2(p33):
■Syphilis
■Hepatitis B surface antigen (HbsAg)
■HIV nucleic acid (individual or com-
bined HIV/HCV assay)
■HCV nucleic acid (individual or
combined HIV/HCV assay)
■Anti-HIV-1
■Anti-HIV-2
■Antibodies to hepatitis B core anti-
gen (anti-HBc)
■Anti-HCV
■Antibodies to human T-cell lympho-
tropic viruses, types I/II (anti-HTLV-
I/II)
A combination test for anti-HIV-1/2 may
be used. A test for alanine aminotransferase
(ALT) is not required by the FDA or the
AABB. Recommendations for labeling units
associated with an elevated ALT have been
released by the FDA.4
Equipment Requirements
All equipment used for testing must be
properly calibrated and validated upon
installation, after repairs, and periodically.
There must be a schedule for planned
maintenance. All calibration, mainte-
nance, and repair activities must be docu-
mented for each instrument. Software
164 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
used to control the instrument or to inter-
face with the institution’s computer sys-
tem must also be properly validated.5
Records Requirements
Records must show each production step
associated with each blood component
from its source to its final disposition.6,7
Recordsmustbekeptinamannerthat
protects the identity and personal infor-
mation of the donor from discovery by
anyone other than the facility doing the
donor recruitment, qualification, and
blood collection, with the exception of
government agencies that require certain
test-positive results to be reported by law
for public health purposes. Testing records
on donor units must be kept in a manner
that makes it possible to investigate ad-
verse consequences to a recipient. In ad-
dition, donor testing records must be suit-
able for look-back to previously donated
components when a donor’s blood gives
positive results on a new or improved in-
fectious disease test.
Previous records of a donor’s ABO and D
typing results must be reviewed and com-
pared with the ABO and D test findings on
the current donation. This is a very valuable
quality check on both the sample identity
correctness and the operation of the labo-
ratory. If a discrepancy is found between
any current or historic test required, the
unit must not be released until there is un-
equivocal resolution of the discrepancy.2(p39)
ABO and D Testing
Every unit of blood intended for transfu-
sion must be tested for ABO and D.2(p32),8
TheABOgroupmustbedeterminedby
testing donor red cells with reagent anti-A
and anti-B, and donor serum or plasma
with A1andBredcells.TheRhtypemust
be determined by testing donor red cells
with anti-D serum. Red cells that are
nonreactive with anti-D in direct aggluti-
nation tests must be tested by a method
designed to detect weak D. Red cells that
react with anti-D either by direct aggluti-
nation or by the weak D test must be
labeled Rh positive. Red cells that are
nonreactive with anti-D by direct aggluti-
nation and the weak D test must be la-
beled Rh negative. Some of the automated
techniques have sufficient D sensitivity to
obviate the need for a weak D test. These
instruments add reagents, incubate re-
agent and sample appropriately, read the
reaction, and provide a result ready for in-
terpretation. In addition, the automated
devices incorporate positive sample iden-
tification with the use of barcode readers
and use anticoagulated blood so that only
one tube is needed for both red cell and
plasma sampling. See Chapter 13 and
Chapter 14 for a more complete discus-
sion of the principles of ABO and D test-
ing.
Antibody Screening
Blood from donors with a history of trans-
fusion or pregnancy must be tested for
unexpected antibodies. Because it is usu-
ally impractical to segregate blood that
should be tested from units that need not
be tested, most blood centers test all do-
nor units for unexpected red cell antibod-
ies. Donor serum or plasma may be tested
against individual or pooled reagent red
cells of known phenotypes. Methods must
be those that demonstrate clinically sig-
nificant red cell antibodies. See Methods
Section 3 for antibody detection techni-
ques and Chapter 19 for a discussion of
antibody detection.
Serologic Test for Syphilis
Serologic testing for syphilis (STS) has been
carriedoutondonorsamplesforover50
years. Although experimental studies in
Chapter 7: Blood Component Testing and Labeling 165
Copyright © 2005 by the AABB. All rights reserved.
the 1980s showed that survival of spiro-
chetes at 4 C is dependent on the concen-
tration, it is not known how long the spiro-
chete (Treponema pallidum)survivesat
refrigerated temperatures in a naturally
infected blood component.9The last re-
ported transfusion transmitted case of
syphilis was reported in fresh blood com-
ponents in 1969.10
The majority of screening tests for syphi-
lis in US blood collection centers are micro-
hemagglutinin or cardiolipin-based tests
that are typically automated. Donor units
testing positive for syphilis (STS) may not
be used for allogeneic transfusion. Results
can be confirmed before a donor is noti-
fied. Volunteer donors are much more
likely to have a false-positive test result
than a true-positive one.
Viral Marker Testing
Two screening methods are widely used
to detect viral antigens and/or antibodies.
The first is the enzyme-linked immuno-
sorbent assay (EIA). The EIA tests for the
viral antigen HBsAg employ a solid sup-
port (eg, a bead or microplate) coated
with an unlabeled antiserum against the
antigen. The indicator material is the
same or another antibody, labeled with an
enzyme whose presence can be detected
by a color change in the substrate. If the
specimen contains antigen, it will bind to
the solid-phase antibody and will, in turn,
be bound by the enzyme-labeled indica-
tor antibody. To screen for viral antibod-
ies, ie, anti-HIV-1, anti-HIV-2, anti-HBc,
anti-HCV, or anti-HTLV-I/II, the solid
phase (a bead or microtiter well) is coated
with antigens prepared from the appro-
priate viral recombinant proteins or syn-
thetic peptides. The second technology
for virus detection is based on nucleic
acid amplification and detection of viral
nuclear material.11 The RNA viruses being
tested for routinely are HIV and HCV. Two
experimental tests for West Nile virus
(WNV) are undergoing study nation
-
wide.12,13 Nucleic acid testing (NAT) for
HBsAg is undergoing clinical trials in
some centers.
In the capture approach frequently used
in assays, serum or plasma is incubated
with fixed antigen. If present, antibody
binds firmly to the solid phase and remains
fixed after excess fluid is washed away. An
enzyme-conjugated preparation of antigen
or antiglobulin is added; if fixed antibody is
present, it binds the labeled antigen or
antiglobulin, and the antigen-antibody-an-
tigen (or antiglobulin) complex can be
quantified by measuring enzyme activity.
One assay for anti-HBc uses an indirect
capture method (competitive assay), in
whichanenzyme-antibodyconjugateis
added to the solid-phase antigen along
with the unknown specimen. Any antibody
present in the unknown specimen will
compete with the enzyme-conjugated anti-
body and significantly reduce the level of
enzyme fixed, compared with results seen
when nonreactive material is present. Anti-
gens used in the viral antibody screening
tests may be made synthetically by recom-
binant technology or extracted from viral
particles.
NAT is a powerful but expensive technol-
ogy that reduces the exposure window for
HIV and HCV by detecting very low num-
bers of viral copies after they appear in the
bloodstream. Primers for HCV and HIV vi-
ruses are placed in microplate wells, either
separately or in combination according to
the specific test design. In the use of pooled
sera,16to24donorsamplesaremixedand
tested. If viral RNA matching the fragments
already in the well is present in the donor
samples, heat-cycling nucleic acid amplifi-
cation using a heat-cycling technique will
cause the viral fragments to multiply and
be easily detectable. The microplate with
166 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
the aliquot of pooled sera is placed in the
well with the viral primers and substrate so
that if the primers and viral material in the
donor samples are the same, the primer
and viral particles will increase geometri-
cally with each cycle. Viral presence can
then be detected reliably. When a pool is
found to be positive, all the individual sam-
ples making up the pool are tested sepa-
rately for the individual viruses HIV and
HCVtofindthepositivedonorsample.The
second major advantage of this test is the
relative lack of false-positive results as long
as sample and laboratory cross-contamina-
tion are controlled.12
For most of the assays, samples giving
nonreactive results on the initial screening
test, as defined by the manufacturer’s pack-
age insert, are considered negative and
need not be tested further. Samples that are
reactive on the initial screening test must
be repeated in duplicate. Reactivity in one
or both of the repeated tests constitutes a
positive result and is considered repeatedly
reactive. All components must be discarded
in this case. If both the duplicate repeat
tests are nonreactive, the test is interpreted
as having a negative result.
Before a donor is designated as antigen-
or antibody-positive, a status that may have
significant clinical and social consequences
and cause permanent exclusion from blood
donation, it is important to determine
whether the screening result is a true- or
false-positive result.
Invalidation of Test Results
In the course of viral marker testing, it
may be necessary to invalidate test results
if the test performance did not meet the
requirements of the manufacturer’s pack-
age insert (eg, faulty equipment, im-
proper procedure, compromised reagents),
or if the control results do not meet the
acceptance criteria defined in the pack-
age insert. All results, both the reactive
and the nonreactive, obtained in the run
must be declared invalid; all specimens
involved must be tested in a new run,
which becomes the initial test of record.13
However, if the batch controls are ac-
ceptable and no error is recognized in test
performance, the reactive and nonreactive
results from the initial run remain as the
initial test of record for the specimens in-
volved. Specimens with reactive results
must be retested in duplicate, as required
by the manufacturer’s instructions.
Before a test run is invalidated, the prob-
lems observed should be reviewed by a su-
pervisor or equivalent, causes should be
analyzed, and corrective action should be
taken, if applicable. A record of departure
from normal standard operating proce-
dures should be prepared, with a complete
description of the reason for invalidation
and the nature of corrective actions.13
Use of External Controls
Other considerations may need to be ad-
dressed before the invalidation of test re-
sults when external controls are used.13
Internal controls are the validation mate-
rials provided with the licensed assay kit;
they are used to demonstrate that the test
performs as expected. External controls
generally consist of at least one positive
control and one negative control. If the
negative control from the kit is used to
calculate the assay cutoff, it cannot be
used as an assay control reagent for test-
ing. An external negative control should
be used in its place (see Table 7-1). Exter-
nal controls are frequently used to dem-
onstrate the ability of the test to identify
weakly reactive specimens. External con-
trols are surrogate specimens, either pur-
chased commercially or developed by the
institution, that are not a component of
the test kit; they are used for surveillance
Chapter 7: Blood Component Testing and Labeling 167
Copyright © 2005 by the AABB. All rights reserved.

of test performance. External controls are
tested in the same manner as donor sam-
ples to augment blood safety efforts and
to alert the testing facility to the possibil-
ity of an increasing risk of error.
Before being entered into routine use,
external controls must be qualified, lot by
lot, because each control lot may vary with
the test kit. One way to qualify an external
control is:
1. Run the external control for 2 days,
four replicates per day, using two to
three different test kit lots. The per-
formance of the external controls
must meet specified requirements
before use.
2. If the external controls do meet the
specifications, the acceptable sam-
ple-to-cutoff ratio for the external
control (eg, within three standard
deviations of the mean) must be
determined.
Additional qualification testing must be
performed whenever a new lot of test kits
or external controls is introduced.
When a change of test kit lot occurs, rep-
licates (eg, 20 replicates) of the external
control should be run with the current kit
lot and the new lot. The new sample-to-
cutoff ratio and limits should be deter-
mined.
Users should verify special requirements
for external control handling in pertinent
state, federal, and international regulations,
as applicable.
A facility may invalidate nonreactive test
results on the basis of external controls, but
if the assay was performed in accordance
with manufacturer’s specifications and the
internal controls performed as expected,
external controls cannot be used to invali-
date reactive test results. The use of external
controls may be more stringent than, but
must be consistent with, the package in-
sert’s criteria for rejection of test results.
Observation of donor population data,
such as an unexpectedly increased reactive
rate within a test run, may cause non-
reactive results to be considered invalid.
The next assay, performed on a single
aliquot from affected specimens, becomes
the initial test of record for those samples
nonreactive in the invalidated run. How-
ever, reactive results obtained in a run with
an unexpectedly increased reactive rate
may not be invalidated unless the entire
run fails to meet the performance criteria
specified in the package insert. Such reac-
tive results remain the initial test of record.
The samples must be tested in duplicate as
the repeat test.
External controls may also be used to in-
validate a duplicate repeat test run when an
assay run is valid by test kit acceptance cri-
teria and both the repeated duplicate tests
are nonreactive. The duplicate samples
168 AABB Technical Manual
Table 7-1. Use of External Controls
Test Kit Reagents
Used to Calculate
Assay Cutoff? External Controls Required?
Negative control only Yes Yes–negative control
No No
Positive control only Yes Yes–positive control
No No
Both positive and negative
controls
Yes Yes–positive and negative
controls
Copyright © 2005 by the AABB. All rights reserved.
may be repeated in duplicate; the second
duplicate test becomes the test of record. If
either of the original duplicate repeat tests
is reactive, the donor(s) must be classified
as repeatedly reactive and no further repeat
screening tests should be performed.
When samples are reactive on the initial
screening test, allogeneic donor units must
be quarantined until the results of dupli-
cate repeat testing are available. Compo-
nents associated with repeatedly reactive
test results must not be used for allogeneic
transfusion. Supplemental or confirmatory
testing may be performed on samples that
are repeatedly reactive to obtain additional
information for donor counseling and pos-
sible reentry, depending on the viral marker
and availability of approved assays.
Supplemental Tests: Neutralization
In confirmatory antigen neutralization
tests (eg, HBsAg), the reactive specimen is
incubated with human serum known to
contain antibody specific for the antigen
in question. If incubation causes the posi-
tive reaction to disappear or to diminish
by at least 50% (or the percentage speci-
fied in the package insert) and all controls
behave as expected, the presence of anti-
gen is confirmed and the original result is
considered a true positive. If incubation
with a known antibody does not affect
subsequent reactivity, the original reactiv-
ity is considered a false-positive result.
Known positive and negative control sam-
ples must be tested in parallel with donor
or patient samples. Parallel incubations
must be performed with a preparation
known to contain antibody specific for
the antigen in question and with a prepa-
rationknowntobefreeofbothantigen
and antibody. If the positive and negative
control values do not fall within limits
stated in the package insert, the test must
be repeated.
Supplemental Test for EIA-Reactive
Anti-HIV-1/2, -HTLV I/II, and -HCV Tests
Western blot is the method most fre-
quently used for the confirmation of re-
peatedly reactive anti-HIV EIA tests. The
technique separates antigenic viral mate-
rial into bands according to molecular
weight. The material is transferred to
nitrocellulose membranes. Antibody in
the test serum reacts with the individual
bands, depending on the specificity.
Most persons infected with HIV, whether
asymptomatic or exhibiting AIDS, show
multiple bands, representing antibodies
to virtually all of the various gene prod-
ucts. A fully reactive test serum should re-
act with the p17, p24, and p55 gag pro-
teins; the p31, p51, and p66 pol proteins;
and the gp41, gp120, and gp160 env
glycoproteins.Western blot results in
EIA-reactive blood donor samples are
classified as positive, negative, or indeter-
minate. Positive results are those with re-
activity to at least two of the following HIV
proteins: p24, core protein; gp41, trans-
membrane protein; and gp120/160, exter-
nal protein and external precursor pro-
tein. Indeterminate results are those with
other patterns of reactivity.
An immunofluorescence assay (IFA) is
used in some blood centers as an alterna-
tive to Western blot testing. Cells infected
with virus are fixed on a slide. The sample is
incubated with the fixed cells. Antibody in
the sample will bind to the antigen sites on
the viral particles. The reaction mixture is
incubated with fluorescent-labeled anti-
human IgG. Following incubation and
washing, binding of the labeled antihuman
IgG is read using a fluorescence micro-
scope, with subsequent interpretation of
the fluorescence pattern.
Although there are FDA-approved West-
ern blot confirmatory tests for anti-HIV, the
Western blot test using recombinant DNA
Chapter 7: Blood Component Testing and Labeling 169
Copyright © 2005 by the AABB. All rights reserved.
and viral lysate antigens for anti-HTLV-I/II
has not been approved by the FDA. An ap-
propriate supplemental test to confirm a
reactive anti-HTLV test result is to repeat
the test using another manufacturer’s EIA
test. If that test is repeatedly reactive, the
result is considered confirmed. If the test is
negative, the anti-HTLV test result is
considered a false-positive result.
The FDA has approved a recombinant
immunoblot assay (RIBA) system to further
differentiate anti-HCV EIA repeatedly reac-
tive samples. The RIBA system is based on
the fusion of HCV antigens to human
superoxide dismutase in the screening test
and to a recombinant superoxide dismu-
tase in the confirmatory test to detect non-
specific reactions. A positive result requires
reactivity to two HCV antigens and no reac-
tivity to superoxide dismutase. Reactivity to
only one HCV antigen or to one HCV anti-
gen and superoxide dismutase is classified
as an indeterminate reaction. Results are
usually presented as positive, negative, or
indeterminate. As with all procedures, it is
essential to follow the manufacturer’s in-
structions for classification of test results.
The use of nucleic acid amplification test-
ing is discussed in Chapter 28.
Cytomegalovirus Testing
Optional tests may be performed on units
intended for recipients with special needs.
For example, cytomegalovirus (CMV)
testing is a commonly performed optional
test. CMV can persist in the tissues and
leukocytes of asymptomatic individuals
for years after initial infection. Blood from
persons lacking antibodies to the virus
has reduced risk of transmitting infection
compared with untested (nonleukocyte-
reduced) units. Only a small minority of
donor units with positive test results for
anti-CMV will transmit infection. However,
there is presently no way to distinguish
infective antibody-positive units from
noninfective units containing anti-CMV.
Routine testing for anti-CMV is not re-
quired by AABB Standards,2(p43) but, if it is
performed, the usual quality assurance
considerations apply. The most common
CMV antibody detection methods in use
are EIA and latex agglutination. Other
methods, such as indirect hemagglutina-
tion, complement fixation, and immuno-
fluorescence, are also available.
Labeling, Records, and
Quarantine
Labeling
Labeling is a process that includes a final
review of records of donor history, collec-
tion, testing, blood component modifica-
tion, quality control functions, and any
additional information obtained after do-
nation. This also includes a review of la-
bels attached to the components and checks
to ensure that all labels meet regulatory
requirements and are an accurate reflec-
tion of the contents of the blood or blood
components.1,8
All labeling of blood components must
be performed in compliance with AABB
Standards2(p12) and FDA regulations. Blood
centers and transfusion services must en-
sure that labeling is specific and controlled.
Before the labeling process begins, there
should be a mechanism or procedure in
place that ensures the use of appropriate
labels. This process should include assur-
ance of acceptable label composition, in-
spection on receipt, secure storage and dis-
tribution of labels, archiving of superseded
labels, and availability of a master set of la-
bels in use. In addition, procedures should
address generation of labels, changes in la-
bels, and modification of labels to reflect la-
170 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
bel control of altered or new components.
Labels should be checked for the proper
product code for the component being la-
beled, which is based on collection method,
anticoagulant, and modifications to the
component during processing. All aspects
of labeling (the bag label as well as the Cir-
cular of Information for the Use of Human
Blood and Blood Components,14 including
the label size, type size, wording, spacing,
and the base label adhesive) are strictly
controlled.
ISBT 128
The ISBT 128 labeling system is an inter-
nationally defined system based on bar-
code symbology called Code 128. It stan-
dardizes the labeling of blood so that
bar-coded labels can be read by blood
centers and transfusion services around
the world. The system allows for each
number assigned to a unit of blood (blood
identification number) to be unique. The
unique number will allow tracking of a
unit of blood from donor to recipient, re-
gardless of where the unit was drawn or
transfused. As outlined in the United States
Industry Consensus Standard for the Uni-
form Labeling of Blood and Blood Compo-
nents Using ISBT 128,15 the information
appearing on the label, the location of the
label, and the exact wording on the label
are standardized.
ISBT 128 differs from its predecessor,
which used CODABAR symbology, by in-
cluding more specific information on the
label. One advantage of the standardized
system is that additional information on
the label allows for better definition of
product codes. Other changes include an
expanded donation identification number
to include the collection facility’s identifica-
tion; bar-coded manufacturer’s lot number,
bag type, etc; bar-coded expiration date;
and special testing barcode. A benefit will
be that the use of standardized computer-
generated barcode labels (with better dif-
ferentiation between components, prepara-
tion methods, and expiration dates) en-
hances efficiency, accuracy, and ultimately
safety of labeled components. For example,
the ISBT 128 label shows conspicuously
that an autologous, biohazard unit is not a
standard unit by making the blood type a
different, smaller size and filling the space
usually reserved for blood type with a bio-
hazard symbol. Adherence to the guidelines
ensures compliance with AABB standards2(p12)
and FDA regulations. The United States
blood banking community has recently
been prompted by a general directive from
the Secretary of Health and Human Ser-
vices and a subsequent proposal from the
FDA for uniform acceptance of this more
comprehensive labeling system. Until the
new international guidelines are imple-
mented, the 1985 FDA Uniform Labeling
Guideline remains in effect. More informa-
tion on ISBT 128 is available from the Inter-
national Council on Commonality in Blood
Banking Automation at www.iccbba.com.
Label Requirements
The following pieces of information are
required2(p12),16,17 in clear readable letters on
a label firmly attached to the container of
all blood and component units:
■Thepropernameofthecomponent,
in a prominent position.
■A unique numeric or alphanumeric
identification that relates the origi-
nal unit to the donor and each com-
ponent to the original unit.
■For components, the name, address,
and FDA license number or registra-
tion number (whichever is appropri-
ate) of the facility that collected the
blood and/or the component. The la-
belmustincludethenameandloca
-
tion of all facilities performing any
Chapter 7: Blood Component Testing and Labeling 171
Copyright © 2005 by the AABB. All rights reserved.
part of component manufacturing.
This includes facilities that wash, ir-
radiate, and reduce leukocytes by fil-
tration. If a process is performed
under contract, and the process is
performed under processes con-
trolled by the contracting facility,
only that facility’s name is required
in this case. There should not be
more than two alphanumeric identi-
fiers on the unit.
■The expiration dates, including the
date and year; if the shelf life is 72
hoursorless,thehourofexpiration
must be stated.
■The amount of blood collected.
■The kind and quantity of anticoagu-
lant (not required for frozen, degly-
cerolized, rejuvenated, or washed red
cells).
■For all blood and blood components,
including pooled components, the
approximate volume of the compo-
nent must appear on the container.
■Recommended storage temperature.
■ABO group and Rh type.
■Interpretation of unexpected red cell
antibody tests for plasma-containing
components when positive (not re-
quired for cryoprecipitate or frozen,
deglycerolized, rejuvenated, or washed
RBCs).
■Results of unusual tests or procedures
performed when necessary for safe
and effective use. Routine tests per-
formed to ensure the safety of the
unit need not be on the label if they
are listed in the Circular of Informa-
tion for the Use of Human Blood and
Blood Components.14
■Reference to the Circular of Informa-
tion for the Use of Human Blood and
Blood Components,14 which must be
available for distribution, and con-
tains information about actions, in-
dications, contraindications, dosage,
administration, side effects, and
hazards.
■Essential instructions or precautions
for use, including the warning that
the component may transmit infec-
tious agents, and the statements: “Rx
only” and “Properly Identify In-
tended Recipient.”
■The appropriate donor classification
statement—“autologous donor,” “paid
donor,” or “volunteer donor”—in
type no less prominent than that
used for the proper name of the
component.
■Any additives, sedimenting agents,
or cryoprotective agents that might
still be present in the component.
Special Labeling
■Cellular blood components issued as
“Leukocytes Reduced” must be la-
beled as such.
■Thenameandfinalvolumeofthe
component and a unique identifier
for a pool must appear on all pooled
components.
■The number of units in a pool and
their ABO group and Rh type must
be on the label or an attached tie tag.
■Identification numbers of the indi-
vidual units in a pool should not be
on the label but must be in the re-
cords of the facility preparing the
pool.
■Cellular blood components issued as
“CMV negative” must be labeled as
such.
■Irradiated blood components must
carry the appropriate irradiated label.
Records
Current good manufacturing practice reg-
ulations, as defined by Title 21 CFR Parts
200 and 600,6,7,16,18 state that master pro-
duction and control records must be a
172 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
part of the labeling process. These records
must be described in the facility’s proce-
dures. Before labeling, these records must
be reviewed for accuracy and complete-
ness. Appropriate signatures and dates
(either electronic or manual) must docu-
ment the review process.
Control Records
Control records include but may not be
limited to:
■Donation process: that all questions
are answered on the donor card,
consent is signed, all prequalifying
tests are acceptable (eg, hemoglo-
bin, blood pressure), and a final re-
view is documented by qualified
supervisory personnel.
■Infectious disease testing: if per-
formed at the collecting facility, that
tests have acceptable quality control
and performance; that daily equip-
ment maintenance was performed
and was acceptable; and that final
results are reviewed to identify the
date and person performing the
review.
■Donor deferral registry: That the list
of deferred donors has been checked
to ensure that the donor is eligible.
■Component preparation: that all
blood and blood components were
processed and/or modified under
controlled conditions of tempera-
ture and other physical require-
ments of each component.
■Transfer of records: if testing is per-
formed at an outside facility, that all
records of that facility are up to date
and that the appropriate licensure is
indicated. Records, either electronic
or manual, must transfer data ap-
propriately. All electronically trans-
ferred test records must be transmitted
by a previously validated system.
Transfer of those results must be
performed by a system that properly
identifies test results to all appropri-
ate blood and blood components.
■Quarantine: that any nonconforming
unit is appropriately isolated.
Production Records
Master production records must be trace-
able back to:
■Dates of all processing or modifica-
tion.
■Identification of the person and equip-
ment used in the process steps.
■Identification of batches and in-pro-
cess materials used.
■Weights and measures used in the
course of processing.
■In-laboratory control results (tem-
peratures, refrigerator, etc).
■Inspection of labeling area before
and after use.
■Results of component yield when
applicable.
■Labeling control.
■Secondary bag and containers used
in processing.
■Any sampling performed.
■Identification of person performing
and checking each step.
■Any investigation made on noncon-
forming components.
■Results of examinations of all review
processes.
Quarantine
Before final labeling, there must be a pro-
cess to remove nonconforming blood and
blood components from the labeling pro-
cess until further investigation has oc-
curred. This process must be validated to
capture and isolate all blood and blood
components that do not conform to re-
quirements in any of the critical areas of
collecting, testing, and processing. This
Chapter 7: Blood Component Testing and Labeling 173
Copyright © 2005 by the AABB. All rights reserved.
process must also capture verbal (eg, tele-
phone calls) information submitted to the
collection facility after the collection pro-
cess. All nonconforming units must re-
main in quarantine until they are investi-
gated and all issues are resolved. The units
may then be discarded, labeled as non-
conforming units (eg, autologous units),
or labeled appropriately for transfusion if
the investigation resolved the problems. If
the nonconformance cannot be resolved
and the units are from an allogeneic do-
nation, they must be discarded.
References
1. Code of federal regulations. Title 21 CFR
610.40. Washington, DC: US Government
Printing Office, 2004 (revised annually).
2. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
3. Dodd RY, Stramer SL. Indeterminate results
in blood donor testing: What you don’t know
can hurt you. Transfus Med Rev 2000;14:151-9.
4. Food and Drug Administration. Memoran-
dum: Recommendations for labeling and use
of units of whole blood, blood components,
source plasma, recovered plasma or source
leukocytes obtained from donors with ele-
vated levels of alanine aminotransferase
(ALT). (August 8, 1995) Rockville, MD: CBER
Office of Communication, Training, and
Manufacturers Assistance, 1995.
5. Code of federal regulations. Title 21 CFR
606.60. Washington, DC: US Government
Printing Office, 2004 (revised annually).
6. Code of federal regulations. Title 21 CFR
606.160. Washington, DC: US Government
Printing Office, 2004 (revised annually).
7. Code of federal regulations. Title 21 CFR
606.165. Washington, DC: US Government
Printing Office, 2004 (revised annually).
8. Code of federal regulations. Title 21 CFR
640.5. Washington, DC: US Government
Printing Office, 2004 (revised annually).
9. van der Sluis JJ, ten Kate FJ, Vuzevski VD, et
al. Transfusion syphilis, survival of Tre-
ponema pallidum in donor blood. II. Dose
dependence of experimentally determined
survival times. Vox Sang 1985;49:390-9.
10. ChambersRW,FoleyHT,SchmidtPJ.Trans
-
mission of syphilis by fresh blood compo-
nents. Transfusion 1969;9:32-4.
11. Busch MP, Stramer SL, Kleinman SH. Evolving
applications of nucleic acid amplification as-
says for prevention of virus transmission by
blood components and derivatives. In:
Garratty G, ed. Applications of molecular bi-
ology to blood transfusion medicine. Bethesda,
MD: AABB, 1997:123-76.
12. Vargo K, Smith K, Knott C, et al. Clinical spec-
ificity and sensitivity of a blood screening as-
say for detection of HIV-1 and HCV RNA.
Transfusion 2002;42:876-85.
13. Food and Drug Administration. Guidance for
industry. Revised recommendations regard-
ing invalidation of test results of licensed and
510(k)-cleared blood-borne pathogen assays
used to test donors. (July 11, 2001) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 2001.
14. AABB, American Red Cross, and America’s
Blood Centers. Circular of information for the
use of human blood and blood components.
Bethesda, MD: AABB, 2002.
15. Food and Drug Administration. Guidance for
Industry: United States industry consensus
standard for the uniform labeling of blood
and blood components using ISBT 128, Ver-
sion 1.2.0. (November 28, 1999) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 1999.
16. Code of federal regulations. Title 21 CFR
606.210, and 606.211. Washington, DC: US
Government Printing Office, 2004 (revised
annually).
17. Food and Drug Administration. Guidelines
for the uniform labeling of blood and blood
components. (August 1985) Rockville, MD:
CBER Office of Communication, Training and
Manufacturers Assistance, 1985.
18. Code of federal regulations. Title 21 CFR Part
210, 211 and 606. Washington, DC: US Gov-
ernment Printing Office, 2004 (revised annu-
ally).
Suggested Reading
AABB, American Red Cross, and America’s Blood
Centers. Circular of information for the use of hu-
man blood and blood components. Bethesda, MD:
AABB, 2002.
174 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 8: Components from Whole Blood Donations
Chapter 8
Collection, Preparation,
Storage, and Distribution of
Components from Whole
Blood Donations
DONOR CENTERS AND transfusion
services share a common goal in
blood component production: to
provide a safe and efficacious component
that benefits the intended recipient. To this
end and in keeping with Food and Drug
Administration (FDA) current good man-
ufacturing practice regulations, all pro-
cesses involved in the collection, testing,
preparation, storage, and transport of blood
and components are monitored for qual-
ity, including procedures, personnel, re-
agents, equipment, and the contents of the
components themselves. Processes should
ensure the potency and purity of the final
product, minimize microbial contamina-
tion and proliferation, and prevent or de-
lay the detrimental physical and chemical
changes that occur when blood is stored.
Blood Component
Descriptions
Readers should refer to Chapters 21, 23, and
24 and the current Circular of Information
for the Use of Human Blood and Blood
Components1for more detailed indica-
tions and contraindications for transfusion.
Whole Blood
Fresh Whole Blood contains all blood ele-
ments plus the anticoagulant-preservative
in the collecting bag. It is used commonly
as a source for component production.
After 24-hour storage, it essentially be-
comes red cells suspended in a protein
solution equivalent to liquid plasma, with
a minimum hematocrit of approximately
33%.
175
8
Copyright © 2005 by the AABB. All rights reserved.

Red Blood Cells
Red Blood Cells (RBCs) are units of Whole
Blood with most of the plasma removed
(see Method 6.4). If prepared from whole
blood collected into citrate-phosphate-
dextrose (CPD), citrate-phosphate-dextrose-
dextrose (CP2D), or citrate-phosphate-
dextrose-adenine (CPDA-1), the final
hematocrit must be ≤80%. Additive red
cell preservative systems consist of a pri-
mary collection bag containing an antico-
agulant-preservative with at least two sat-
ellite bags integrally attached; one is
empty and one contains an additive solu-
tion (AS). AS contains sodium chloride,
dextrose, adenine, and other substances that
supportredcellsurvivalandfunctionup
to 42 days2(seeTable8-1).Thevolumeof
the AS in a 450-mL collection set is 100
mL and the volume in 500-mL sets is 110
mL. AS is added to the red cells remaining
intheprimarybagaftermostofthe
plasma has been removed. This allows
blood centers to use or recover a maxi-
mum amount of plasma, yet still prepare
a red cell component with a final hemato-
crit between 55% and 65%, a level that fa-
cilitates excellent flow rates and allows
easy administration.
RBCs can be prepared at any time during
their shelf life, but AS must be added within
the time frame specified by the manufac-
turer, generally within the first 72 hours of
storage. Shelf life at 1 to 6 C storage de-
pends on the anticoagulant-preservative
used and the method of preparation.
Platelets
Platelet concentrates (Platelets) are prepared
from units of whole blood that have not
been allowed to cool below 20 C. Plate-
let-rich plasma (PRP) is separated within
4 hours after completion of the phlebot-
omy or within the time frame specified in
the directions for the use of the blood col-
lecting, processing, and storage system—
typically8hours.
3The platelets are concen-
trated by an additional centrifugation step
and the removal of most of the superna-
tant plasma. A procedure for preparation
of platelets from single units of whole
blood appears in Method 6.13. The final
component should contain resuspended
platelets in an amount of plasma ade-
quate to maintain an acceptable pH; gen-
erally, 40 to 70 mL is used. Although not
approved in the United States, platelet
concentrates are commonly manufac-
176 AABB Technical Manual
Table 8-1. Content of Additive Solutions (mg/100 mL)
AS-1
(Adsol)
AS-3
(Nutricel)
AS-5
(Optisol)
Dextrose 2200 1100 900
Adenine 27 30 30
Monobasic sodium phosphate 0 276 0
Mannitol 750 0 525
Sodium chloride 900 410 877
Sodium citrate 0 588 0
Citric acid 0 42 0
Copyright © 2005 by the AABB. All rights reserved.
tured in Europe using buffy coat as an in-
termediate product. In this schema, the
initial centrifugation is a “hard-spin” in
which the platelets are concentrated in the
buffy coat. The supernatant platelet-poor
plasma and the red cells can be expressed
using a top-and-bottom device. The buffy
coats can then be centrifuged in a “soft-
spin” to remove the white cells or, more
commonly, pooled before storage (not
currently allowed by the FDA), then soft-
spun as a pooled concentrate, with ex-
pression of the PRP.4-6
Plasma
Plasma in a unit of Whole Blood can be
separated at any time during storage, up
to 5 days after the expiration date of the
Whole Blood. When stored frozen at –18 C
or colder, this component is known as
Plasma and can be used up to 5 years af-
ter the date of collection. If not frozen, it
is called Liquid Plasma, which is stored at
1 to 6 C and transfused up to 5 days after
the expiration date of the Whole Blood
from which it was prepared.
Fresh Frozen Plasma (FFP) is plasma
prepared from whole blood, either from the
primary centrifugation of whole blood into
red cells and plasma or from a secondary
centrifugation of PRP. The plasma must be
frozen within 8 hours of collection.3See
Methods 6.10 and 6.13.
Blood centers often convert plasma and
liquid plasma to an unlicensed component,
“Recovered Plasma (plasma for manufac-
ture),” which is usually shipped to a frac-
tionator and processed into derivatives
such as albumin and/or immune globulins.
To ship recovered plasma, the collecting fa-
cility must have a “short supply agreement”
with the manufacturer.7Because recovered
plasma has no expiration date, records for
this component must be retained indefi-
nitely.
See further discussion of additional
plasma products later in the chapter.
Cryoprecipitated AHF
Cryoprecipitated antihemophilic factor
(AHF) is the cold-insoluble portion of
plasma that precipitates when FFP is
thawed between 1 to 6 C. It is essentially a
concentrate of high-molecular-weight
glycoproteins also known as CRYO. This
component is prepared from a single
Whole Blood unit collected into CPDA-1,
CPD, or CP2D and suspended in less than
15 mL of plasma. It contains ≥80 IU Factor
VIII (AHF), >150 mg of fibrinogen, and
most of the Factor XIII originally present
inthefreshplasma.CRYOcontainsboth
the procoagulant activity (Factor VIII) and
the von Willebrand factor of the Factor
VIII von Willebrand complex.
Once separated, CRYO is refrozen within
1 hour of preparation and stored at –18 C or
colder for up to 1 year after the date of phle-
botomy. See Method 6.11 for a preparation
procedure.
Plasma Cryoprecipitate Reduced
If cryoprecipitate has been removed from
plasma, this must be stated on the label.
When stored at –18 C or colder, this com-
ponent has a 12-month expiration date
fromthedateofcollection.
8(p59) This com-
ponent is used primarily in the treatment
of thrombotic thrombocytopenic purpura.9
Granulocytes
Granulocytes are usually collected by aphere-
sis techniques; however, buffy coats har-
vested from fresh Whole Blood units can
provide a source (<1 ×109)ofgranulocytes
in urgent neonatal situations. Their effec-
tiveness is controversial, however.10 Granu-
locytes should be transfused as soon as
possible after collection but may be
stored at 20 to 24 C without agitation for
Chapter 8: Components from Whole Blood Donations 177
Copyright © 2005 by the AABB. All rights reserved.

up to 24 hours. Arrangements for pre-
collection testing are often useful. This
product is becoming obsolete.
Collection
Blood component quality begins with a
healthy donor and a clean venipuncture
site to minimize bacterial contamination.
To prevent activation of the coagulation
system during collections, blood should
be collected rapidly and with minimal
trauma to tissues. Although the target col-
lection time is usually 4 to 10 minutes,
one study has shown platelets and fresh
frozen plasma to be satisfactory after col-
lection times of up to 15 minutes.11 The fa-
cility’s written procedures should be fol-
lowed regarding these collection times.
There should be frequent, gentle mixing
of the blood with the anticoagulant. If pre-
storage filtration is not intended after col-
lection, the tubing to the donor arm may be
stripped into the primary collection bag, al-
lowed to fill, and segmented, so that it will
represent the contents of the donor bag for
compatibility testing. Blood is then cooled
toward1to6Cunlessitistobeusedfor
room temperature component production,
in which case it should be cooled toward,
but not below, 20 C. Chapter 4 discusses
blood collection in detail.
Anticoagulants and Preservatives
Whole blood is collected into a bag that
contains an FDA-approved anticoagulant-
preservative solution designed to prevent
clotting and to maintain cell viability and
function during storage. Table 8-2 com-
pares some common solutions. Citrate
prevents coagulation by chelating cal-
cium, thereby inhibiting the several cal-
cium-dependent steps of the coagulation
cascade.2
The FDA approves 21-day storage at 1 to
6 C for red cells from whole blood collected
in CPD and CP2D and 35-day storage for
red cells collected in CPDA-1.12 Most blood
centers now collect up to 500 mL ± 50 mL
(450-550 mL) whole blood in bags specifi-
cally designed for this larger volume. Blood
bags intended for a collection volume of
450 mL ± 45 mL of whole blood (ie, 405-495
mL) contain 63 mL of anticoagulant-pre-
178 AABB Technical Manual
Table 8-2. Anticoagulant-Preservative Solutions (mg in 63 mL) for 450 mL
Collections
CPD CP2D CPDA-1
Ratio (mL solution to blood) 1.4:10 1.4:10 1.4:10
FDA-approved shelf life (days) 21 21 35
Content
Sodium citrate 1660 1660 1660
Citric acid 206 206 206
Dextrose 1610 3220 2010
Monobasic sodium phosphate 140 140 140
Adenine 0 0 17.3
With 500 mL collections, the volume is 70 mL and the content 10% to 11% higher.
Copyright © 2005 by the AABB. All rights reserved.
servative.Thevolumeofanticoagulant-pre
-
servative in 500 mL ±50 mL bags is 70 mL.
The allowable range of whole blood col-
lected is dependent upon the collection bag
selected and can vary with manufacturer,
but the total amount collected including
testing samples must not exceed 10.5
mL/kg donor weight per donation.
If only 300 to 404 mL of blood is col-
lected into a blood bag designed for a
450-mL8(p28) collection, the red cells may be
used for transfusion provided the unit is la-
beled “Red Blood Cells Low Volume.” How-
ever, other components should not be pre-
pared from these low-volume units. If a
whole blood collection of less than 300 mL
is planned, the volume of anticoagulant-
preservative solution in that bag should be
reduced proportionately (see Chapter 4 for
calculations), to ensure that the correct
amount of anticoagulant is used (ratio of
anticoagulant: whole blood).
Transportation from a Collection Site
Whole blood should be transported from
the collection site to the component pre-
paration laboratory as soon as possible.
Units should be cooled toward 1 to 6 C
unless platelets are to be harvested, in
which case, units should be cooled toward,
but not below, 20 C. The time between
collection and the separation of compo-
nents must not exceed the time frame
specified in the directions for use of the
blood collection,3processing, and storage
system.
Prestorage Processing
Differential Centrifugation
To simplify the separation of whole blood
into its component parts, blood is collected
into primary bags to which up to three
satellite bags are attached.5Set design is
based on intended use: RBCs, platelets,
FFP, Cryoprecipitated AHF, or neonatal
aliquots. Refer to Methods Section 6 for
specific component preparation proce-
dures. Whole blood must be separated
andpreparedcomponentsplacedintotheir
required storage temperatures within the
anticoagulant-preservative manufacturer’s
recommended times of collection.3Re-
cords of component preparation should
identify each individual performing a sig-
nificant step in processing.
Because red cells, platelets, and plasma
have different specific gravities, they are
separated using differential centrifugation.13
Rotor size, speed, and duration of spin are
critical variables in centrifugation. Method
7.4 describes how to calibrate a centrifuge
for platelet separation, but each centrifuge
must be calibrated for optimal speeds and
spin times for each combination of compo-
nents prepared in like fashion and for each
different type of collection bag. Times in-
clude the time of acceleration and “at
speed,” not deceleration time. Once the op-
erating variables are identified for compo-
nent production, timer accuracy, rpm, and
temperature (if appropriate), centrifuges
should be monitored periodically to verify
equipment performance.
Another practical way to assess centrifu-
gation is to monitor quality control data on
components prepared in each centrifuge. If
component quality does not meet defined
standards, eg, if platelet concentrate yields
areinconsistent,theentireprocessshould
be evaluated. Factors affecting the quality
assessment are the calibration of the centri-
fuge, the initial platelet count on the whole
blood donations, storage time, conditions
between blood collection and platelet prep-
aration, sampling technique, and counting
methods.
Large centrifuges rotate at high speeds,
exerting gravitational forces of thousands of
pounds on blood bags. Weaknesses in these
Chapter 8: Components from Whole Blood Donations 179
Copyright © 2005 by the AABB. All rights reserved.
blood bags or the seals between tubing seg-
ments can cause rupture and leakage. The
addition of filters for blood sets presents
different challenges for cup insertion.
Blood bags may be overwrapped with plas-
tic bags to contain any leaks. Bags should
be positioned so that a broad surface faces
the outside wall of the centrifuge to reduce
thecentrifugalforceonbloodbagseams.
Contents in opposing cups of the centri-
fuge must be equal in weight to improve
centrifuge efficiency and prevent damage to
the rotor. Dry balancing materials are pref-
erable to liquid material. Weighted rubber
discs and large rubber bands are excellent
and come in several thicknesses to provide
flexibility in balancing. Swinging centrifuge
cups provide better separation between cells
and plasma than fixed-angle cups.
Testing and Labeling of Donor Units
Chapter 7 contains detailed information
on testing and labeling blood components.
Filtration
During inline filtration of whole blood, the
anticoagulated whole blood is filtered by
gravity through an inline leukocyte reduc-
tion filter contained in the collection sys-
tem. The filtered whole blood may be
manufactured into leukocyte-reduced
RBCs. Whole blood leukocyte reduction
filters retain the platelets to a variable de-
gree, so platelet concentrates cannot be
routinely prepared. However, newly de-
signed platelet sparing filters are under
investigation. This filtration should occur
within the time specified by the filter
manufacturer.
A leukocyte reduction filter can be at-
tached to a unit of Whole Blood or RBCs
using a sterile connection device. Ideally,
such filtration should occur as early as pos-
sible after collection but must conform to
the manufacturer’s recommendations for
the specific filter in use. This method may
be preferable if special units are to be se-
lected for leukocyte reduction.
During inline filtration of red cells, plate-
lets and/or plasma are first removed from
the whole blood donation and the additive
solution is transferred to the red cells. The
red cells are then filtered through an inline
filter into a secondary storage container.
This filtration step should occur as early in
the shelf life as possible and within the al-
lowed time frame for the specific filter in
use.
Leukocyte-reduced platelets may be pre-
pared from PRP using inline leukocyte re-
duction filtration.14 FFP manufactured using
this intermediate step typically will have a
leukocyte content of <5 ×106.
Freezing
Acellular Components
When stored at –18 C or colder, FFP con-
tains maximum levels of labile and non-
labile clotting factors (about 1 IU per mL)
and has a shelf life of 12 months from the
date of collection. FFP frozen and main-
tainedat–65Cmaybestoredupto7years.
See Method 6.10 for preparation details.
Plasma can be rapidly frozen by placing the
bag 1) in a dry ice-ethanol or dry ice-anti-
freeze bath, 2) between layers of dry ice, 3)
in a blast freezer, or 4) in a mechanical
freezer maintained at –65 C or colder.
Plasma frozen in a liquid bath should be
overwrapped with a plastic bag to protect
the container from chemical alteration.
When a mechanical freezer is used, care
must be taken to avoid slowing the freezing
process by introducing too many units at
one time.
It is prudent practice to use a method to
facilitate detection of inadvertent thawing
of plasma during storage, such as:
1. Pressing a tube into the bag during
freezing to leave an indentation that
180 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
disappears if the unit thaws. Remove
tube(s).
2. Freezing the plasma bag in a flat,
horizontal position but storing it up-
right. Air bubbles trapped along the
bag’s uppermost broad surface dur-
ing freezing will move to the top if
the unit is thawed in a vertical posi-
tion.
3. Placing a rubber band around the
liquid plasma bag and removing it
after freezing to create an indenta-
tion that disappears with thawing.
Plasma separated and frozen at –18 C
between 8 and 24 hours (eg, plasma that
does not meet the stricter time require-
ments of FFP) may be labeled as “Plasma
Frozen Within 24 Hours after Phlebotomy.”
It contains all the stable proteins found in
FFP (see Table 21-3). FFP thawing guide-
lines apply.
Cellular Components
Frozen storage can significantly extend the
shelf life of red cell components. Unfortu-
nately, the process can also cause cell dam-
age and add considerable expense.
Effects of Freezing and Thawing. When
unprotected cells are frozen, damage may
result from cellular dehydration and from
mechanical trauma caused by intracellular
ice crystals. At rates of freezing slower than
10 C/minute, extracellular water freezes be-
fore intracellular water, producing an osmotic
gradient that causes water to diffuse from
inside the cell to outside the cell. This leads
to intracellular dehydration. Controlling the
freezing rate, however, is not sufficient by
itself to prevent cellular damage, so cryo-
protective agents must be used.
Cryoprotective agents are classified as
penetrating and nonpenetrating. Penetrat-
ing agents such as glycerol are small mole-
cules that freely cross the cell membrane
into the cytoplasm. The intracellular cryo-
protectant provides an osmotic force that
prevents water from migrating outward as
extracellular ice is formed. A high concen-
tration of cryoprotectant prevents forma-
tion of ice crystals and consequent mem-
brane damage.15 Glycerol, a trihydric alcohol,
is a colorless, sweet-tasting, syrup-like fluid
that is miscible with water. Pharmacologi-
cally, glycerol is relatively inert.
Nonpenetrating cryoprotective agents
(eg, hydroxyethyl starch) are large mole-
cules that do not enter the cell. The mole-
cules protect the cells by a process called
“vitrification” because they form a non-
crystalline “glassy” shell around the cell.
This prevents loss of water and dehydration
injury and alters the temperature at which
the solution undergoes transition from
liquid to solid.
Freezing of RBCs. Frozen preservation of
RBCs with glycerol is primarily used for
storing units with rare blood types and
autologous units. Frozen cells can be effec-
tively stockpiled for military mobilization
or civilian disasters, but the high cost and
the 24-hour shelf life after deglycerolization
make them less useful for routine inventory
management. Recently, an effectively closed
system was approved with a 2-week post-
thaw shelf life when the blood is collected
in CPDA-1, frozen within 6 days, and stored
at –80 C.
Two concentrations of glycerol have been
used to cryopreserve red cells, as shown in
Table 8-3. This chapter and Methods 6.7
and 6.8 discuss only the high-concentration
glycerol technique used by most blood
banks. Modifications have been developed
for glycerolizing, freezing, storing, thawing,
and deglycerolizing red cells and are dis-
cussed elsewhere.16 Several instruments are
available that partially automate glyceroli-
zation and deglycerolization of red cells.
The manufacturer of each instrument pro-
videsdetailedinstructionsforitsuse.
Chapter 8: Components from Whole Blood Donations 181
Copyright © 2005 by the AABB. All rights reserved.

Blood intended for freezing can be col-
lected into CPD, CP2D, or CPDA-1 and stored
as Whole Blood or RBCs (including AS-
RBCs). Ordinarily, red cells are glycerolized
andfrozenwithin6daysofcollectionorre
-
juvenated and frozen up to 3 days after they
expire, but RBCs preserved in AS have been
frozen up to 42 days after collection, with
adequate recovery.17,18
Some glycerolization procedures require
removalofmostoftheplasmaoradditive
from the RBCs; others do not. The concen-
tration of glycerol used for freezing is hyper-
tonic to blood. Its rapid introduction can
cause osmotic damage to red cells, which
manifests as hemolysis only after thawing.
Therefore, when initiating the cryopreser-
vation process, glycerol should be intro-
duced slowly to allow equilibration within
the red cells.
The US Department of Defense has
adopted a method for high-concentration
glycerolization that uses an 800-mL pri-
mary collection container suitable for
freezing (see Method 6.8). Because the cyto-
protective agent for freezing is transferred
directly into the primary collection con-
tainers, there is less chance of contamina-
tion and/or identification error. In addition,
the amount of extracellular glycerol is
smaller and it is more efficient to store and
ship units prepared by this method.
Storage Bags. Storage bag composition can
affect the freezing process; less hemolysis
mayoccurinpolyolefinthaninsomepoly
-
vinyl chloride (PVC) bags. Contact between
red cells and the PVC bag surface may cause
an injury that slightly increases hemolysis
upon deglycerolization. In addition, polyo-
lefin bags are less brittle at –80 C and less
likely to break during shipment and han-
dling than PVC bags.
Freezing Process. Red cells frozen within
6 days of collection with a final glycerol
182 AABB Technical Manual
Table 8-3. Comparison of Two Methods of Red Blood Cell Cryopreservation
Consideration
High-Concentration
Glycerol
Low-Concentration
Glycerol
Final glycerol concentration (wt/vol) Approx. 40% Approx. 20%
Initial freezing temperature –80 C –196 C
Freezing rate Slow Rapid
Freezing rate controlled No Yes
Type of freezer Mechanical Liquid nitrogen
Storage temperature (maximum) –65 C –120 C
Change in storage temperature Can be thawed and
refrozen
Critical
Type of storage Polyvinyl chloride;
polyolefin
Polyolefin
Shipping Dry ice Liquid nitrogen
Special deglycerolizing equipment
required
Yes No
Deglycerolizing time 20-40 minutes 30 minutes
Hematocrit 55-70% 50-70%
Copyright © 2005 by the AABB. All rights reserved.
concentration of 40% (wt/vol) must be
stored at –65 C or colder. Red cells are usu-
ally placed in canisters to protect the plastic
bagduringfreezing,storage,andthawing.
Although up to 18 hours at room tem-
perature may elapse between glycerolizing
and freezing without increased postthaw
hemolysis, an interval not exceeding 4
hours is recommended.16 With current 40%
(wt/vol) glycerol methods, controlled rate
freezing is unnecessary; freezing is accom-
plished by placing the RBC container into a
–80 C freezer.
Refreezing Deglycerolized RBCs. It may
occasionally be desirable to refreeze thawed
RBC units that have not been used as ex-
pected or have been unintentionally thawed.
Units that were deglycerolized, stored 20
hours at refrigerator temperature, and then
reglycerolized and refrozen showed no loss
of adenosine triphosphate (ATP), 2,3-
diphosphoglycerate (2,3-DPG), or in-vivo
survival,19 and RBCs subjected three times
to glycerolizing, freezing, and thawing ex-
hibited a 27% loss of total hemoglobin.20
AABB Standards for Blood Banks and Trans-
fusion Services does not address refreezing
thawed units because this should not be
considered a routine practice. If thawed
units are refrozen, the records should docu-
mentthevaluablenatureofsuchunitsand
the reasons for refreezing them.
Freezing of Platelets. Perhaps because of
their greater complexity, platelets appear to
sustain greater injury during cryopreserva-
tion than red cells, although several proto-
cols have successfully used dimethyl sul-
foxide as a cryoprotectant.21,22 Because
postthaw platelet recovery and function are
significantly reduced when compared with
those of liquid-stored platelets, the clinical
use of cryopreserved platelets is not wide-
spread. The primary use of this procedure
is to freeze autologous platelets for future
use. Platelet cryopreservation is essentially
a research technique.
Irradiation
Cellular blood components may be irradi-
ated before storage to prevent transfusion-
associated graft-vs-host disease (GVHD).
This does not shorten the shelf life of pla-
telets or granulocytes, but red cells expire
28 days after irradiation or at the end of
the storage period, whichever comes first.
Pooling
Sterile connection devices are used to at-
tach additional bags and compatible tub-
ing to a blood bag without breaking the
sterile integrity of the system. The shelf
life of components thus prepared is the
same as those prepared in a closed system
except for Pooled Platelets, which expire 4
hours after pooling. All sterile connection
device welds must be inspected for com-
pleteness, integrity, leakage, and air bub-
bles; procedures must address the action
to take if the weld is not satisfactory. Re-
cord-keeping should include documenta-
tion of the products welded, weld quality
control, and lot numbers of disposables.23
Cryoprecipitated AHF
Units of CRYO may be pooled before la-
beling, freezing, and storage. If pooled
promptly after preparation using aseptic
technique and refrozen immediately, the
resulting component is labeled “Cryopre-
cipitated AHF Pooled,” with the number
of units pooled stated on the label. The
volume of saline, if added to facilitate
pooling, must also appear on the label.
The statement may appear in the Circular
of Information for the Use of Human Blood
and Blood Components1instead of on the
container label.
The facility preparing the pool must
maintain a record of each individual donor
traceable to the unique identifier used for
the pooled component.8(p26)
Chapter 8: Components from Whole Blood Donations 183
Copyright © 2005 by the AABB. All rights reserved.
If an open-system pool or component is
to be stored frozen, it should be placed in
the freezer within 6 hours after the seal has
been broken. The AABB and the FDA re-
quire transfusion within 4 hours for pooled
thawed components stored at 20 to 24
C.8(p58)
Platelets
Prestorage pooling of PRP whole-blood-
derived platelets is possible using a sterile
connection device and a storage con-
tainer suitable for storage of a high-yield
platelet concentrate. Platelet concentrates
can be leukocyte-reduced using inline fil-
tration14 or they can be pooled into a
pooling container, then subsequently leu-
kocyte-reduced by filtration and stored in
a storage container. Although current FDA
requirements limit the dating of these pools
to 4 hours, studies of such prestorage leu-
kocyte-reduced pooled platelet concen-
trates show good preservation of platelet
function without any evidence of a mixed
lymphocyte reaction, with up to 7 days of
storage.24 However, the AABB and FDA re-
quire transfusion of pooled platelets within
4 hours of pooling.
As indicated earlier, buffy-coat-derived
platelets are commonly pooled before stor-
age with filtration of the PRP after centri-
fugation.4-6
Storage
Refrigerated Storage
Blood must be stored only in refrigerators
that, by design and capacity, maintain the
required blood storage temperatures of 1
to 6 C throughout their interior space.
There must be a system to monitor tem-
peratures continuously and record them
at least every 4 hours, and an alarm sys-
tem with an audible signal that activates
before blood reaches unacceptable storage
temperatures.
Interiors should be clean, adequately
lighted, and well organized. Clearly desig-
nated and segregated areas are needed for:
1) unprocessed blood; 2) labeled blood
suitable for allogeneic transfusion; 3) re-
jected, outdated, or quarantined blood; 4)
autologous blood; and 5) biohazardous
autologous blood. Refrigerators used for
the storage of blood and blood components
may also be used for blood derivatives, tis-
sues, patient and donor specimens, and
blood bank reagents.
Refrigerators for blood storage outside
the blood bank, as may be found in surgical
suites or emergency rooms, must meet
these same standards. Temperature records
are required at all times when blood is pres-
ent. It is usually most practical to make
blood bank personnel responsible for mon-
itoring these refrigerators.
Frozen Storage
The FDA licenses Red Blood Cells Frozen
for storage up to 10 years when prepared
with high glycerol (40% wt/vol) methods.
Units stored for up to 21 years have been
transfused successfully. A facility’s medi-
cal director may wish to extend the stor-
age period; however, storage beyond 10
years requires exceptional circumstances.
The distinctive nature of such units and
the reason for retaining them past the
10-year storage period should be docu-
mented. As units are put into long-term
storage, many consider it prudent to freeze
samples of serum or plasma for subse-
quent testing should new donor screening
testsbeintroducedinthefuture.Thetype
of any specimen saved, date of collection,
date of freezing, and specimen location, if
necessary, should be included in records
of frozen blood.
184 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Not all such specimens may meet the
sample requirements of new tests. If stored
samples are not available or inappropriate
for testing, blood centers may attempt to
call the donor back for subsequent testing.
Frozen rare RBCs that have not been tested
for all required disease markers should be
transfused only after weighing the risks and
benefits to the patient. The label should in-
dicate that the unit has not been com-
pletely tested and should identify the miss-
ing test(s) results.
Bloodfreezershavethesametempera
-
ture monitoring and alarm requirements as
blood refrigerators and must be kept clean
and well organized. Freezers designated for
plasma storage must maintain tempera-
tures colder than –18 C (many function at
–30 C or colder); RBC freezers must main-
tain temperatures colder than –65 C (many
maintain temperatures colder than –80 C).
Self-defrosting freezers must maintain ac-
ceptable temperatures throughout their
defrost cycle.
Freezer alarm sensors should be accessi-
ble and located near the door, although
older units may have sensors located be-
tween the inner and outer freezer walls
where they are neither apparent nor acces-
sible. In such cases, the location of the sen-
sor can be obtained from the manufacturer
and a permanent mark placed on the wall
at that location. Clinical engineers may be
able to relocate the sensor thermocouple
foreasieruse.
Liquid nitrogen tanks used for blood
storage also have alarm system require-
ments. The level of liquid nitrogen should
be measured and the sensor placed some-
where above the minimum height needed.
Room Temperature Storage
Platelets require gentle, continuous agita-
tion during storage because stationary
platelets display increased lactate pro-
duction and a decrease in pH. Elliptical,
circular, and flat-bed agitators are avail-
able for tabletop or chamber use. Ellipti-
cal rotators are not recommended for use
with storage bags made of polyolefin
without plasticizer (PL-732).25
Other components that require 20 to 24
C temperatures, eg, cryoprecipitate, can be
stored on a tabletop in any room with an
appropriate ambient temperature, pro-
vided the temperature is recorded every 4
hours during storage. Because room tem-
peratures fluctuate, “environmental” or
“platelet chambers” have been developed to
provide consistent, controlled room temper-
atures. These chambers are equipped with
circulating fans, temperature recorders, and
alarm systems.
Liquid Storage
Red Blood Cells
Biochemical changes occur when red cells
are stored at 1 to 6 C; these changes, some
of which are reversible, contribute to the
“storage lesion” of red cells and to a re-
duction in viability and levels of 2,3-DPG
affecting oxygen delivery to tissues.26 The
most striking biochemical changes that
affect stored red cells are listed in Table
8-4, but some of these changes rarely
have clinical significance, even in mas-
sively transfused recipients. Hemoglobin
becomes fully saturated with oxygen in
the lungs but characteristically releases
only some of its oxygen at the lower oxy-
gen pressure (pO2) of normal tissues. The
relationship between pO2and oxygen satu-
ration of hemoglobin is shown by the oxy-
gendissociationcurve(seeFig8-1).Re
-
lease of oxygen from hemoglobin at a
given pO2is affected by ambient pH,
intracellular red cell levels of 2,3-DPG, and
other variables. High levels of 2,3-DPG in
the red cells cause greater oxygen release
at a given pO2, which occurs as an adap-
Chapter 8: Components from Whole Blood Donations 185
Copyright © 2005 by the AABB. All rights reserved.

186 AABB Technical Manual
Table 8-4. Biochemical Changes in Stored Non-Leukocyte-Reduced Red Blood Cells
CPD CPDA-1 AS-1 AS-3 AS-5
Variable Whole Blood
Whole
Blood
Red Blood
Cells
Whole
Blood
Red Blood
Cells
Red Blood
Cells
Red Blood
Cells
Red Blood
Cells
Days of Storage 0 21 0 0 35 35 42 42 42
% Viable cells
(24 hours
posttransfusion)
100 80 100 100 79 71 76 (64-85) 84 80
pH (measure at 37 C) 7.20 6.84 7.60 7.55 6.98 6.71 6.6 6.5 6.5
ATP (% of initial
value)
100 86 100 100 56 (± 16) 45 (± 12) 60 59 68.5
2,3-DPG (% of initial
value)
100 44 100 100 <10 <10 <5 <10 <5
Plasma K+ (mmol/L) 3.9 21 4.20 5.10 27.30 78.50* 50 46 45.6
Plasma hemoglobin 17 191 82 78 461 658.0* N/A 386 N/A
% Hemolysis N/A N/A N/A N/A N/A N/A 0.5 0.9 0.6
*Values for plasma hemoglobin and potassium concentrations may appear somewhat high in 35-day stored RBC units; the total plasma in these units is only about 70 mL.
Copyright © 2005 by the AABB. All rights reserved.
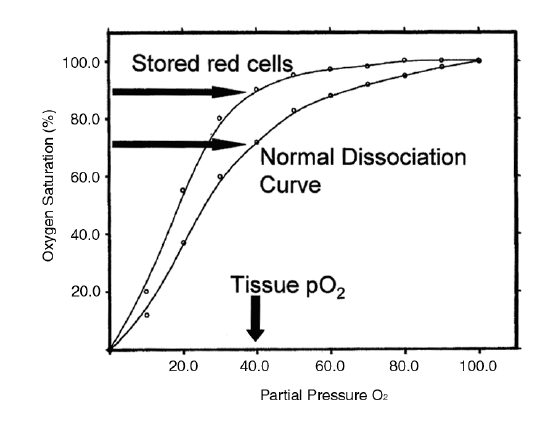
tive change in anemia; lower red cell lev-
els of 2,3-DPG increase the affinity of he-
moglobin for oxygen, causing less oxygen
release at the same pO2. In red cells stored
in CPDA-1 or in current additive systems,
2,3-DPG levels fall at a linear rate to zero
after approximately 2 weeks of storage.
This is caused by a decrease in intra-
cellular pH caused by lactic acid, which
increases the activity of a diphosphatase.26
This causes the dissociation curve to shift
to the left, resulting in less oxygen release
(Fig 8-1).
Upon entering the recipient’s circulation,
stored red cells regenerate ATP and 2,3-DPG,
resuming normal energy metabolism and
hemoglobin function as they circulate in
the recipient. It takes approximately 12
hours for severely depleted red cells to re-
generate half their 2,3-DPG levels, and
about 24 hours for complete restoration of
2,3-DPG and normal hemoglobin func-
tion.26,27
Red cells lose potassium and gain so-
dium during the first 2 to 3 weeks of storage
at 1 to 6 C because sodium/potassium
adenosine triphosphatase, which pumps
sodium out of red cells and replaces it with
potassium, has a very high temperature co-
efficient and functions poorly in the cold.
Supernatant levels of potassium in a unit of
CPDA-1 RBCs have been reported to in-
crease from 5.1 mmol/L on the day of col-
lection to 23 mmol/L on day 7 and 75
mmol/L on day 35. Intracellular levels of
potassium will be replenished after transfu-
sion.
Supernatant potassium levels in red cell
components seem high when compared
with levels in units of Whole Blood of
equivalent age. However, the smaller
supernatant fluid volumes must be consid-
ered when determining total potassium
load. Blood stored at 1 to 6 C for more than
24 hours has few functional platelets, but
levels of coagulation Factors II, VII, IX, X,
Chapter 8: Components from Whole Blood Donations 187
Figure 8-1. Oxygen dissociation of hemoglobin under normal circumstances and in Red Blood Cells
(RBCs) stored in excess of 14 days. At tissue pO2(40 mm Hg), 25% to 30% of the oxygen is normally re-
leased. In stored RBCs, this will decrease to 10% to 15%.
Copyright © 2005 by the AABB. All rights reserved.
and fibrinogen are well maintained. Labile
factors (Factors V and VIII) decrease with
time and are not considered sufficient to
correct specific deficiencies in bleeding pa-
tients, although levels of 30% for Factor V
and 15% to 20% for Factor VIII have been
reported in Whole Blood stored for 21 days,
and platelets stored at room temperature
have been shown to have Factor V levels of
47% (see Chapter 21) and Factor VIII levels
of 68% after 72 hours.13 Forbetterpreserva
-
tion of Factors V and VIII and platelets,
whole blood is separated into its compo-
nent parts and the plasma is stored as FFP.
Platelets
Platelets stored in the liquid state at 20 to
24 C are suspended in either autologous
anticoagulated plasma (United States and
Europe) or in platelet additive solutions
(Europe). Under these conditions, the
current shelf life of the platelets in most
countriesis5days(Table8-5).Thistime
limitation is partly related to concerns
about storage-related deterioration in
product potency28 and partly to the poten-
tial for bacteria to grow rapidly in this
temperature range.29-30 With regard to po-
tency, liquid-stored platelets undergo
in-vitro changes, which are related to the
duration of storage and are collectively
known as the platelet storage lesion.31,32
This is characterized by a change in pla-
telet shape from discoid to spherical; the
generation of lactic acid from glycolysis,
with an associated decrease in pH; the re-
lease of cytoplasmic and granule con-
tents; a decrease in various in-vitro mea-
sures of platelet function, particularly
osmotic challenge; shape changes in-
duced by adenosine diphosphate; and re-
duction in in-vivo recovery and survival.
The in-vitro measures are useful measures
of qualitative potency, but controversy
still exists regarding their utility as practi-
cal surrogates for predicting in-vivo via-
bility and function.33 Attempts to date to
define the biochemical nature of the
platelet storage lesion have not been con-
clusive. These observed changes may rep-
resent a normal aging process, which is
attenuated by the lower temperature of
storage (20-24 C), rather than the in-vivo
temperature of 37 C. However, a role for
mitochondrial injury as a contributing
cause of these changes is plausible. Rest-
ing platelets derive substantial energy
from β-oxidation of fatty acids.34 Alter-
ation in mitochondrial integrity would re-
sult in a reduction in carbon flux through
the tricarboxylic acid cycle and require
energy metabolism through glycolysis,
with increased lactate production. Such a
reduction would compromise the genera-
tion of efficient ATP and result in a de-
crease in the metabolic pool of ATP and,
therefore, the energy charge of the pla-
telet.35 This decrease in the energy charge
wouldbeexpectedtoaffectmembrane
integrity, resulting in a leakage of cyto-
plasmic contents, a diminished response
to physiologic stimuli, and an inability to
repair oxidized membrane lipids, with
subsequent distortions in platelet mor-
phology.36
Shelf Life
The maximum allowable storage time for
a blood component held under acceptable
temperatures and conditions is called its
“shelf life.” For red cells, the criteria for
determining shelf life for an approved an-
ticoagulant-preservative require that at
least 75% of the original red cells (from a
normal allogeneic donor) be in the recipi-
ent’s circulation 24 hours after transfusion.
For other components, their shelf life is
based on functional considerations. Stor-
age times are listed in Table 8-5.
188 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 8: Components from Whole Blood Donations 189
Table 8-5. Expiration Dates for Selected Blood Components8(pp53-60)
Category Expiration
Whole Blood ACD/CPD/CP2D – 21 days
CPDA-1 – 35 days
Whole Blood Irradiated Original outdate (see outdates above per anticoagulant) or
28 days form date of irradiation, whichever is sooner
Red Blood Cells (RBCs) ACD/CPD/CP2D – 21 days
CPDA-1 – 35 days
Open system – 24 hours
Additive solutions – 42 days
RBCs Washed 24 hours
RBCs Leukocytes Reduced ACD/CPD/CP2D – 21 days
CPDA-1 – 35 days
Open system – 24 hours
Additive solutions – 42 days
RBCs Rejuvenated 24 hours
RBCs Rejuvenated Washed 24 hours
RBCs Irradiated Original outdate above or 28 days from date of irradiation,
whichever is sooner
RBCs Frozen 40% Glycerol 10 years
RBCs Frozen 20% Glycerol 10 years
RBCs Deglycerolized 24 hours – or 14 days depending on method
RBCs Open System 24 hours
RBCs Open System – Frozen 10 years, 24 hours after thaw
RBCs Closed System – Frozen 10 years, 2 weeks after thaw as approved by the FDA
RBCs Frozen – Liquid Nitrogen 10 years
Platelets 24 hours to 5 days, depending on collection system*
Platelets Pooled or in Open
System
4 hours, unless otherwise specified
Platelets Leukocytes Reduced Open system – 4 hours
Closed system – no change from original expiration date*
Platelets Irradiated Open system – 4 hours
Closed system – no change from original expiration date
Granulocytes 24 hours
FFP 12 months (–18 C)
7 years (–65 C), as approved by the FDA
(cont'd)
Copyright © 2005 by the AABB. All rights reserved.

Poststorage Processing
Additional discussion of some of the fol-
lowing topics can be found in Chapters 21
and 22.
Filtration for Leukocyte Reduction
Only special leukocyte reduction filters
reliably provide the ≥99.9% (log 3) re-
moval needed to meet the 5 ×106specifi-
cation.37 Red cell leukocyte reduction fil-
ters contain multiple layers of synthetic
nonwoven fibers that retain white cells and
platelets, allowing red cells to flow through.
Leukocyte reduction filters are commer-
cially available in a number of set config-
urations to facilitate filtration during the
separation process at the bedside or in the
laboratory before issue.38 Intact leukocyte
removal efficiency is best when per-
formed soon after collection; therefore, pre-
storage leukocyte reduction is preferred.39
There were concerns that early removal
of leukocytes would allow bacteria, present
at the time of collection, to proliferate.
However, studies suggest that early removal
(within24hours)inthecaseofRBCsmay
reduce the likelihood of significant bacte-
rial contamination.40 Bedside filtration, par-
ticularly of platelets, may not be as effective
in preventing reactions in multitransfused
patients and is less desirable for this reason
than prestorage leukocyte reduction.41 Bed-
side filtration has also caused hypotensive
reactions.42 Cytokines and other substances
that accumulate during storage (particu-
larly in platelet components) may account
for some failures of bedside filtration to
prevent febrile reactions43 (see Chapter 27).
Furthermore, quality control is difficult to
attain at the bedside.44
190 AABB Technical Manual
Category Expiration
FFP Thawed 24 hours
FFP Thawed – Open System 24 hours
Plasma Frozen within 24 hours
after Phlebotomy
12 months
Plasma Frozen within 24 hours
after Phlebotomy Thawed
24 hours
Thawed Plasma <5 days if whole blood derived
24 hours if apheresis
Plasma Liquid 5 days after expiration of RBCs
Plasma Cryoprecipitate
Reduced Frozen
12 months
Plasma Cryoprecipitate
Reduced Frozen
24 hours to 5 days
Cryoprecipitated AHF 12 months
Cryoprecipitated AHF Thawed 4 hours if open system or pooled, 6 hours if single unit
*Maximum time without agitation is 24 hours.
Table 8-5. Expiration Dates for Selected Blood Components8(pp53-60) (cont'd)
Copyright © 2005 by the AABB. All rights reserved.
Thawing
Thawing FFP
FFP is thawed either at temperatures be-
tween 30 and 37 C or in an FDA-cleared
device.8(p59) It is then known as “FFP
Thawed” and should be transfused imme-
diately or stored between 1 and 6 C for no
more than 24 hours. The expiration date
and time must be indicated on the label.
FFP thawed in a waterbath should be
protected so that entry ports are not con-
taminated with water. This can be accom-
plished by wrapping the container in a
plastic overwrap, or by positioning the con-
tainer upright with entry ports above the
water level. Microwave devices should be
shown not to exceed temperature limits
and not to damage the plasma proteins,
and there should be a warning device to in-
dicate if the temperature rises unaccept-
ably.Aswithanydevice,thereshouldbea
procedure for the quality control of indi-
cated functions.
When whole-blood-derived FFP pre-
pared in a closed system is thawed but not
transfused within 24 hours, the label must
be modified. This product should be la-
beled “Thawed Plasma” and can be stored
at 1 to 6 C and transfused up to 5 days after
thawing. It is similar to FFP except for a re-
duction in both Factor V and Factor VIII,
particularly Factor VIII.
Thawing CRYO
CRYO is thawed at temperatures between
30 to 37 C for no more than 15 minutes
[CFR 606.122(n) (4)]. Bags should not re-
main at 30 to 37 C once thawed, so that
degradation of Factor VIII is minimized.
As with FFP, entry ports should be pro-
tected from water contamination if the
unit is thawed in a waterbath. Single-unit
thawed CRYO must be transfused imme-
diately or can be stored at room tempera-
ture (20 to 24 C) for no more than 6 hours.
All pooled CRYO, whether prepared in an
open or a closed system, must be trans-
fused within 6 hours after thawing, or 4
hours after pooling, whichever comes
first. CRYO may be pooled into one bag
after thawing to simplify transfusion to a
patient requiring multiple units. The
pooled product is assigned a unique pool
number, but records must document the
individual units included. See Method 6.12
for guidelines on how to thaw and pool
CRYO for transfusion.
Thawing and Deglycerolizing RBCs
The protective canister and enclosed frozen
cells may be placed directly in a 37 C dry
warmer or can be overwrapped and im-
mersed in a 37 C waterbath. Units frozen
in the primary collection bag system
should be thawed at 42 C.16 The thawing
process takes at least 20 to 25 minutes and
should not exceed 40 minutes. Gentle agi-
tation may be used to speed thawing.
Thawed cells contain a high concentra-
tion of glycerol that must be reduced grad-
ually to avoid in-vivo or in-vitro hemolysis.
Deglycerolization is achieved by washing
the red cells with solutions of decreasing
osmolarity. In one procedure (see Method
6.9), glycerolized cells are diluted with 150
mL of 12% saline, then washed with 1 L of
1.6% saline, followed by 1 L of 0.9% saline
with 0.2% dextrose. The progressive de-
crease in osmolarity of the washing solu-
tions causes osmotic swelling of the cells,
so each solution must be added slowly, with
adequate time allowed for mixing and os-
motic equilibration. Any of the commer-
cially available instruments for batch or
continuous-flow washing can be used to
deglycerolize red cells frozen in a high con-
centration of glycerol. Because there are
many potentially important variations in
deglycerolization protocols for each instru-
Chapter 8: Components from Whole Blood Donations 191
Copyright © 2005 by the AABB. All rights reserved.
ment, personnel in each facility should not
only follow the manufacturer’s instructions,
but also validate the local process. The pro-
cess selected must ensure adequate re-
moval of cryoprotectant agents, minimal
free hemoglobin, and recovery of greater
than 80% of the original red cell volume af-
ter the deglycerolization process.8(p27)
When deglycerolization is complete, the
integrally connected tubing should be filled
with an aliquot of red cells and sealed in
such a manner that it can be detached for
subsequent compatibility testing. The label
must identify both the collecting facility
and the facility that prepares the degly-
cerolized unit if it is different from the col-
lection facility.
When glycerolized frozen red cells from
persons with sickle cell trait are suspended
in hypertonic wash solutions during de-
glycerolization and centrifuged, they form a
jelly-like mass and hemolyze.16 Modified
wash procedures using only 0.9% saline
with 0.2% dextrose after the addition of
12% saline can eliminate this problem.45 In
some cryopreservation programs, dona-
tions are screened for the presence of he-
moglobin S before being frozen.
When glycerolization or deglyceroliza-
tion involves entering the blood bag, the
system is considered “open” and the result-
ing suspension of deglycerolized cells can
be stored for only 24 hours at 1 to 6 C. A
method for glycerolization and degly-
cerolization in an effectively closed system
allows for the resulting suspension of
deglycerolized red cells to be stored for 2
weeks at 1 to 6 C. This method allows more
effective inventory management of the
deglycerolized RBC units.
When deglycerolized RBCs are stored at
1 to 6 C for periods up to 14 days, the major
changes observed are increased concentra-
tions of potassium and hemoglobin in the
supernatant fluid. Red cells that have un-
dergone gamma irradiation and subse-
quent storage at 1 to 6 C tolerate freezing
with no more detectable damage than
unirradiated cells.46,47
Irradiation
Blood components that contain viable
lymphocytes (including red cell, platelet,
and granulocyte components) should be
irradiated to prevent proliferation of trans-
fused T lymphocytes in recipients at risk
of acquiring, or from donors at risk of caus-
ing, GVHD. The AABB and FDA recom-
mend a minimum 25 Gy dose of gamma
radiation to the central portion of the
container, with no less than 15 Gy deliv-
ered to any part of the bag.8(p26)
Irradiation is accomplished using ce-
sium-137 or cobalt-60, in self-contained
blood irradiators or hospital radiation ther-
apy machines. More recently, an x-ray de-
vice has been developed that is capable of
adequate dose delivery. Measurement of
dose distribution; verification of exposure
time, proper mechanical function, and
turntable rotation; and adjustment of expo-
sure time as the radioactive source decays
should be addressed in the facility’s proce-
dures.48 Records must be maintained, and
all steps, supplies, and equipment used in
the irradiation process must be documented.
To confirm the irradiation of individual
units, radiochromic film labels (available
commercially) may be affixed to bags be-
fore irradiation. When exposed to an ade-
quate amount of radiation, the film portion
of the label darkens, indicating that the
component has been exposed to an ade-
quate radiation dosage. Because irradiation
damages red cells and reduces the overall
viability (24-hour recovery),49 red cell com-
ponents that have been irradiated expire on
their originally assigned outdate or 28 days
from the date of irradiation, whichever co-
mes first. Platelets sustain minimal damage
from irradiation, so their expiration date
192 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
does not change.50 Irradiated blood is es-
sential for patients at risk from transfu-
sion-associated GVHD, including fetuses
receiving intrauterine transfusion, select
immunocompetent or immunocompro-
mised recipients, recipients who are under-
going hematopoietic transplantation, recip-
ients of platelets selected for HLA or platelet
compatibility, and recipients of donor units
from blood relatives.
Washing
Washing a unit of RBCs with 1 to 2 L of
sterile normal saline removes about 99%
of plasma proteins, electrolytes, and anti-
bodies. Automated and manual washing
methods remove some of the leukocytes
in the RBCs, but not enough to prevent
alloimmunization. Up to 20% of the red
cell mass may be lost depending on the
protocol used. Washed red cells must be
used within 24 hours because preparation
is usually accomplished in an open sys-
tem, and removal of the anticoagulant-
preservative solution compromises long-
term preservation of cell viability and
function.
Platelets can be washed with normal sa-
line or saline-buffered with ACD-A or citrate,
using manual or automated methods. The
procedures may result in a reduction in
radiolabeled recovery (about 33% less), but
not in survival of the washed platelets51;
white cell content is not significantly
changed. Washed platelets must be used
within 4 hours of preparation.
Pooling
When a patient requires multiple units of
Platelets, pooling them into a single bag
simplifies issue and transfusion. This prod-
uct should be labeled “Platelets Pooled.”
If Platelets contain a significant number
of red cells and ABO groups are mixed,
plasma antibodies should be compatible
with any visible red cells present in the
pool. Only one unique number is affixed
to the final component, but records must
reflect the pooling process and all units
included in the pool. This pooled product
has an expiration time of 4 hours.
Single CRYO units may be pooled after
thawing and labeled appropriately. The
AABB and FDA require transfusion of CRYO
within 4 hours of pooling and subsequent
storageat20to24C.
Volume Reduction
Platelets may be volume-reduced in order
to decrease the total volume of the com-
ponent transfused or partially remove
supernatant substances, such as ABO allo-
antibodies. The former need may arise in
patients with small intravascular volumes
or those with fluid overload (eg, resulting
from renal or cardiac failure). The latter
need may be better addressed by washing
(see below). If sterility is broken, the expi-
ration of the product becomes 4 hours. If
sterility is not broken (eg, a sterile con-
nection device is used23), removal of the
supernatant still reduces glucose avail-
ability and buffering capacity, and the
subsequent storage of the platelets is in a
suboptimal environment. Transfusion as
soon as possible is generally advocated.
Aliquoting
Blood components may be aliquoted in
smaller volumes into other containers in
order to meet the needs of very-low-vol-
ume transfusion recipients or to provide a
component to meet the needs of patients
with fluid overload. The composition of
red cell and plasma components is not al-
tered by aliquoting, unlike volume reduc-
tion. Therefore, the expiration date is not
altered if a sterile connection device is
used to perform the aliquoting.23 The shelf
life and viability of platelets, however, are
Chapter 8: Components from Whole Blood Donations 193
Copyright © 2005 by the AABB. All rights reserved.
dependent on the storage bag, plasma
volume, and storage environment. Re-
moving aliquots of platelets from the
“mother” bag changes the storage envi-
ronment of the platelets remaining in the
“mother” bag. If the altered storage envi-
ronment does not meet the storage bag
manufacturer’s requirements, the expira-
tion period of the remaining component
should also be modified.
Rejuvenation
It is possible to restore levels of 2,3-DPG
and ATP in red cells stored in CPD or
CPDA-1 solutions by adding an FDA-li-
censed solution containing pyruvate,
inosine, phosphate, and adenine (see
Method 6.6). RBCs may be rejuvenated af-
ter 1 to 6 C storage up to 3 days after expi-
ration; then, they may be glycerolized and
frozeninthesamemannerasfreshred
cells. If rejuvenated RBC units are to be
transfused within 24 hours, they may be
stored at 1 to 6 C; however, they must be
washed before use to remove the inosine,
which might be toxic to the recipient. The
blood label and component records must
indicate the use of rejuvenating solutions.
Inspection, Shipping,
Disposition, and Issue
Inspection
Stored blood components are inspected
immediately before issue for transfusion
or shipment to other facilities.8(pp14,15) These
inspections must be documented; records
should include the date, donor number,
description of any abnormal units, the ac-
tion taken, and the identity of personnel
involved. Visual inspections cannot al-
ways detect contamination or other dele-
terious conditions; nonetheless, blood
components that look abnormal must not
be shipped or transfused.
Deleterious conditions should be sus-
pected if52: 1) segments appear much
lighter in color than what is in the bag (for
AS-RBCs),2)theredcellmasslookspurple,
3) a zone of hemolysis is observed just
above the cell mass, 4) clots are visible, 5)
blood or plasma is observed in the ports or
at sealing sites in the tubing, or (6) the
plasma or supernatant fluid is murky, pur-
ple, brown, or red. A green hue from
light-induced changes in bilirubin pig-
ments need not cause the unit to be re-
jected. Mild lipemia, characterized by a
milky appearance, does not render a dona-
tion unsuitable provided that all infectious
disease testing can be performed. Grossly
lipemic specimens are unsuitable.
A component that is questioned for any
reason should be quarantined until a re-
sponsible person determines its disposi-
tion. Evaluation might include inverting the
unit gently a few times to mix the cells with
the supernatant fluid because considerable
undetected hemolysis, clots, or other alter-
ations may be present in the undisturbed
red cells. If, after resuspension, resettling,
and careful examination, the blood no lon-
ger appears abnormal, it may be returned
to inventory. Appropriate records should be
maintained documenting the actions
taken, when, and by whom. Units of FFP
and CRYO should be inspected when they
are removed from frozen storage for evi-
dence of thawing and refreezing and for ev-
idence of cracks in the tubing or plastic
bag. Unusual turbidity in thawed compo-
nents may be cause for discard.
All platelet components should be in-
spected before release and issue. Units with
macroscopically visible platelet aggregates
should not be used for transfusion. Some
facilities assess the “swirling” appearance
of platelets by holding platelet bags up to a
light source and gently tapping them. This
194 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
swirl phenomenon correlates well with pH
values associated with adequate platelet
in-vivo viability.53 Some platelet compo-
nents have been noted to contain small
amounts of particulate matter. These com-
ponents are suitable for use.
Bacterial contamination of transfusion
components is rare because of the use of
aseptic technique and screening for bacte-
ria in platelets, the availability of closed sys-
tems for collection and preparation, and
careful control of storage conditions. Steril-
ity testing of blood components plays a role
in validating initial production processes. If
a transfusion component has an abnormal
appearance, or if an adverse clinical reac-
tion appears to be related to contaminated
donor blood, culturing should be performed
and a Gram’s stain should be evaluated.
Microbiologists can best advise blood cen-
ter staff on sample requirements and appro-
priate test methods for detecting potential
blood contaminants, including cryophilic mi-
croorganisms. Making cultures directly from
the contents of the bag and from the recipi-
ent can provide useful diagnostic information.
Any facility that collected a reportedly
contaminated component should be noti-
fied so that donor bacteremia, potentially
inadequate donor arm preparation, or im-
proper handling or pooling technique can
be investigated. The donor’s health should
be reviewed, and other components prepared
from that collection should be withdrawn.
Shipping
Shipment to areas outside the facility re-
quires additional packaging. Transport
containers or coolers and packaging pro-
cedures must be validated before use to
verify that they are able to maintain blood
components at required temperatures for
the intended time and conditions. Con-
tainersmustalsobeabletowithstand
leakage, pressure, and other conditions
incidental to routine handling. Refer to
Chapter 2 for more information on ship-
ping regulations and guidelines.
Simple exposure to temperatures outside
the acceptable range does not necessarily
render blood unsuitable for transfusion. Ex-
ceptions may be made under unusual cir-
cumstances such as for autologous units or
cells of a rare phenotype, but the records
must document the reasons for preserving
the unit, the evaluation of its continued
suitability for transfusion, and the identity
of the person responsible for the decision.
Other factors to consider when assessing
component acceptability after transport in-
clude the length of time in shipment, mode
of transportation, magnitude of variance
above or below the acceptable range, pres-
ence of residual ice in the shipping box, ap-
pearance of the unit(s), age of the unit(s),
and likelihood of additional storage before
transfusion. The shipping facility should be
notified when a receiving facility observes
that acceptable transport temperatures
have been exceeded.
Whole Blood and RBCs
Liquid Whole Blood and RBCs shipped
from the collection facility to another fa-
cility must be transported in a manner
that ensures a temperature of 1 to 10 C.
The upper limit of 10 C can be reached in
30 minutes if a unit of blood taken from 5
C storage is left at an ambient tempera-
ture of 25 C. Smaller units, as are com-
monly used for pediatric patients, can
warm even more quickly.
Wet ice, securely bagged to prevent leak-
age, is the coolant of choice to maintain re-
quired temperatures during transport and
shipping. An appropriate volume is placed
on top of the units within the cardboard
box or insulated container. Clinical coolant
packs or specially designed containers may
also be used to maintain acceptable trans-
Chapter 8: Components from Whole Blood Donations 195
Copyright © 2005 by the AABB. All rights reserved.
port temperatures. Super-cooled ice (eg,
large blocks of ice stored at –18 C) in con-
tact with Whole Blood or RBCs may result
in temperatures below 1 C, with resultant
hemolysis of the red cells.
Platelets and Granulocytes
Every reasonable effort must be made to
ensure that platelets and granulocytes are
maintained at 20 to 24 C during ship-
ment. A well-insulated container without
added ice, or with a commercial coolant
designed to keep the temperature at 20 to
24 C, is recommended. Fiber-filled enve-
lopes or newspaper are excellent insula-
tors. For very long distances or travel
times in excess of 24 hours, double-insu-
lated containers may be needed.
Frozen Components
Frozen components must be packaged for
transport in a manner designed to keep
them frozen. This may be achieved by us-
ing a suitable quantity of dry ice in well-
insulated containers or in standard ship-
ping cartons lined with insulation mate-
rial, such as plastic air bubble packaging
or dry packaging fragments. The dry ice,
obtained as sheets, may be layered at the
bottom of the container, between each
layer of frozen components, and on top.
Shipping facilities should determine op-
timal conditions for shipping frozen com-
ponents, which depend on the temperature
requirements of the component, the dis-
tance to be shipped, the shipping container
used, and ambient temperatures to be en-
countered. Procedures and shipping con-
tainers should be validated and periodically
monitored. The receiving facility should al-
ways observe the shipment temperature
and report unacceptable findings to the
shipping facility.
Red cells cryopreserved with high-con-
centration glycerol (40% wt/vol) tolerate
fluctuations in temperatures between –85 C
and –20 C with no significant change in
in-vitro recovery or 24-hour posttrans-
fusion survival, so transport with dry ice is
acceptable. Blood shipments containing
dry ice as a coolant are considered “danger-
ous goods,” and special packaging and la-
beling requirements apply (see Chapter 2).
Shipping boxes containing dry ice must not
be completely sealed so that the carbon di-
oxide gas released as the dry ice sublimes
can escape without risk of explosion.
Disposition
Both blood collection sites and transfu-
sion services must maintain records on all
blood components handled so that units
can be tracked from collection to final
disposition. Units of blood that cannot be
released for transfusion should be re-
turned to the provider or discarded as
biohazardous material. The nature of the
problem disqualifying the unit should be
investigated and the results reported to
the blood supplier. Findings may indicate
a need to improve phlebotomy techni-
ques, donor screening methods, or the
handling of units during processing, stor-
age, or transport.
Disposal procedures must conform to the
local public health code for biohazardous
waste. Autoclaving or incineration is rec-
ommended. If disposal is carried out off-
site, a contract with the waste disposal firm
must be available and should specify that
appropriate Environmental Protection Agency,
state, and local regulations are followed
(see Chapter 2 for the disposal of biohazar-
dous waste).
Issuing from a Transfusion Service
Blood transported short distances within
a facility, eg, to the patient care area for
transfusion, requires no special packaging
other than that dictated by perceived safety
196 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
concerns and institutional preferences.
However, blood should never unnecessar-
ily be allowed to reach temperatures out-
side its accepted range. Specific transport
guidelines may be warranted if transport
time is prolonged.
Units that have left the control of the
transfusion service or donor center and
then have been returned must not be reis-
sued for transfusion unless the following
conditions have been met:
1. The container closure has not been
penetrated, entered, or modified in
any manner.
2. Red cell components have been main-
tained continuously between 1 and
10 C, preferably 1 to 6 C. Blood cen-
ters and transfusion services usually
donotreissueRBCunitsthathave
remained out of a monitored refrig-
erator or a validated cooler for lon-
ger than 30 minutes because, beyond
that time, the temperature of the
component may have risen above 10 C.
3. At least one sealed segment of inte-
gral donor tubing remains attached
to the red cell component, if the
blood has left the premises of the is-
suing facility.
4. Records indicate that the blood has
been reissued and has been inspected
before reissue.
Blood Component Quality
Control
Ensuring safe and efficacious blood com-
ponents requires applying the principles
of quality assurance to all aspects of com-
ponent collection, preparation, testing,
storage, and transport. All procedures and
equipment in use must be validated be-
fore their implementation and periodi-
cally monitored thereafter. Staff must be
appropriately trained and their compe-
tency evaluated. The contents of final
products should be periodically assessed
to make sure that they meet expectations.
How much quality control is performed is
best determined by the institution, with
input from the compliance officer and con-
sideration of AABB and FDA requirements.
See Appendix 8-1.
Quality Control of Equipment
Continuous Temperature Monitoring
Systems
Most blood refrigerators, freezers, and
chambers have built-in temperature mon-
itoring sensors connected to recording
charts or digital readout systems for easy
surveillance. Digital recording devices
measure the differences in potential gen-
erated by a thermocouple; this difference
is converted to temperature. Because warm
air rises, temperature-recording sensors
should be placed on a high shelf and im-
mersed in a volume of liquid not greater
than the volume of the smallest compo-
nent stored. Either a glass container or a
plastic blood bag may be used. Recording
charts and monitoring systems must be in-
spected daily to ensure proper function.
When recording charts or tapes are
changed, they should be dated inclusively
(ie, start and stop dates), and labeled to
identify the facility, the specific refrigerator
or freezer, and the person changing the
charts. Any departure from normal temper-
ature should be explained in writing on the
chart beside the tracing or on another doc-
ument and should include how the stored
components were managed. A chart with a
perfect circle tracing may indicate that the
recorder is not functioning properly or is
not sensitive enough to record the expected
variations in temperature that occur in any
actively used refrigerator.
Blood banks with many refrigerators and
freezers may find it easier to use a central
Chapter 8: Components from Whole Blood Donations 197
Copyright © 2005 by the AABB. All rights reserved.
alarm monitoring system that monitors all
equipment continuously and simultane-
ously and prepares a hard copy tape of
temperatures at least once every 4 hours.
These systems have an audible alarm that
sounds as soon as any connected equip-
ment reaches its predetermined tempera-
ture alarm point and indicates the equip-
ment in question. Blood storage equipment
so monitored does not require a separate
independent recording chart.
Thermometers
Visual thermometers in blood storage equip-
ment provide ongoing verification of tem-
perature accuracy. One should be immersed
in the container with the continuous mon-
itoring sensor. The temperature of the
thermometer should be compared peri-
odically with the temperature on the re-
cording chart. If the two do not agree
within 2 C, both should be checked
against a thermometer certified by the
National Institute of Standards and Tech-
nology (NIST) and suitable corrective ac-
tion should be taken (see Method 7.2). (A
2 C variation between calibrated ther-
mometers allows for the variation that
may occur between thermometers cali-
brated against the NIST thermometers.)
Thermometers also help verify that the
temperature is appropriately maintained
throughout the storage space. Large refrig-
erators or freezers may require several ther-
mometers to assess temperature fluctua-
tions. In addition to the one immersed with
the continuous monitoring sensor (usually
located on a high shelf), at least one other
in a similar container is placed on the low-
est shelf on which blood is stored. The tem-
perature in both areas must be within the
required range at all times.
Either liquid-in-glass (analog) thermom-
eters or electronic and thermocouple (digi-
tal) devices can be used for assessing stor-
age temperatures, as long as their accuracy
is calibrated against a NIST-certified ther-
mometer or a thermometer with a NIST-
traceable calibration certificate (see Method
7.2). Of equal importance is that they be
used as intended, according to the manu-
facturer’s recommendations.
Alarm Systems
To ensure that alarm signals will activate
at a temperature that allows personnel to
take proper action before blood reaches
undesirable temperatures, both tempera-
ture of activation and power source are
tested periodically. The electrical source
for the alarm system must be separate
from that of the refrigerator or freezer; ei-
ther a continuously rechargeable battery
or an independent electrical circuit served
by an emergency generator is acceptable.
Method 7.3.1 provides a detailed proce-
dure to test the temperatures of activation
for refrigerator alarms. Suggestions for
freezer alarms are in Method 7.3.2 Ther-
mocouple devices that function at freezer
temperatures are especially useful for de-
termining the temperature of activation
with accuracy when sensors are accessible.
When they are not, approximate activation
temperatures can be determined by check-
ing a freezer’s thermometer and recording
chart when the alarm sounds after it is shut
down for periodic cleaning or mainte-
nance. It can also be assessed by placing a
water bottle filled with cold tap water
against the inner freezer wall where the
sensor is located.
When the alarm goes off, usually in a
short time, the recording chart can be
checked immediately for the temperature
of activation. There must be written in-
structions for personnel to follow when the
alarm sounds. These instructions should
include steps to determine the immediate
cause of the temperature change and ways
198 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
to handle temporary malfunctions, as well
as steps to take in the event of prolonged
failure. It is important to list the names of
keypeopletobenotifiedandwhatsteps
should be taken to ensure that proper stor-
age temperature is maintained for all blood,
components, and reagents.
Quality Control of Red Blood Cells
RBCs prepared without additive solutions
must have a hematocrit ≤80%. This
should be established during process vali-
dation and periodically confirmed by
quality control procedures. RBCs Leuko-
cytes Reduced should contain <5 ×106re-
sidual leukocytes and retain 85% of the
original red cells.54 Quality control should
demonstrate that at least 95% of units
sampled meet this specification. Units
tested must meet the leukocyte reduction
specification.6,8(p28) (See Appendix 8-1.)
Quality Control of Platelets
The quality of every method of platelet
preparation must be assessed periodi-
cally.Datamustshowthatatleast90%of
components tested contain an acceptable
number of platelets (5.5 ×1010 for whole-
blood-derived platelets) and have a plasma
pH of 6.2 or higher at the end of the allow-
able storage period.8(p36)
Prestorage leukocyte-reduced Platelets
derived from filtration of platelet-rich
plasma must contain less than 8.3 ×105re-
sidual leukocytes per unit to be labeled as
leukocyte reduced. Validation and quality
control should demonstrate that at least
95% of units sampled meet this require-
ment.8(p31)
Quality Control of Cryoprecipitated AHF
AABB Standards for Blood Banks and
Transfusion Services requires that all
tested individual units of CRYO contain a
minimum of 80 IU of Factor VIII and 150
mg of fibrinogen.8(p30) Each pool must have
aFactorVIIIcontentofatleast80mg
times the number of donor units in the
pool; for fibrinogen, the content should be
150 mg times the number of donor units.8(p30)
(See Appendix 8-1.)
References
1. AABB, American National Red Cross, and
America’s Blood Centers. Circular of informa-
tion for the use of human blood and blood
components. Bethesda, MD: AABB, 2002.
2. Buetler E. Liquid preservation of red blood
cells.In:SimonTL,DzikWH,SnyderEL,etal,
eds. Rossi’s principles of transfusion medi-
cine.3rded.Philadelphia:LippincottWil
-
liams & Wilkins, 2002:50-61.
3. Code of federal regulations. Title 21 CFR
640.24(b) and 640.34(b). Washington, DC: US
Government Printing Office, 2004 (revised
annually).
4. Pietersz RN, van der Meer PF, Steneker I, et al.
Preparation of leukodepleted platelet con-
centrates from pooled buffy-coats: Prestorage
filtration with Autostop BC. Vox Sang 1999;76:
231-6.
5. Boomgaard MN, Joustra-Dijkhuis AM,
Gouwerok CW, et al. In vitro evaluation of
platelets, prepared from pooled buffy-coats,
stored for 8 days after filtration. Transfusion
1994;34:311-6.
6. van der Meer PF, Pietersz RN, Tiekstra MJ, et
al. WBC-reduced platelet concentrates from
pooled buffy-coats in additive solution: An
evaluation of in vitro and in vivo measures.
Transfusion 2001;41:917-22.
7. Code of federal regulations. Title 21 CFR
601.22. Washington, DC: US Government
Printing Office, 2004 (revised annually).
8. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
9. Owens MR, Sweeney JD, Tahhan RH, et al. In-
fluenceoftypeofexchangefluidonsurvival
in therapeutic apheresis for thrombotic
thrombocytopenic purpura. J Clin Apheresis
1995;10:178-82.
10. RosenthalJ,MitchellSC.Neonatalmyelo
-
poiesis and immunomodulation of host de-
fenses.In:PetzLD,SwisherSN,KleinmanS,
etal,eds.Clinicalpracticeoftransfusion
medicine.3rded.NewYork:ChurchillLiving
-
stone, 1996:685-703.
Chapter 8: Components from Whole Blood Donations 199
Copyright © 2005 by the AABB. All rights reserved.
11. Huh YO, Lichtiger B, Giacco GG, et al. Effect
of donation time on platelet concentrates and
fresh-frozen plasma. Vox Sang 1989;56:21-4.
12. Code of federal regulations. Title 21 CFR
610.53 (c). Washington, DC: US Government
Printing Office, 2004 (revised annually).
13. Calhoun L. Blood product preparation and
administration. In: Petz LD, Swisher SN,
Kleinman S, et al, eds. Clinical practice of
transfusion medicine. 3rd ed. New York:
Churchill Livingstone, 1996:305-33.
14. Sweeney JD, Holme S, Heaton WAL, Nelson E.
White cell-reduced platelet concentrates pre-
pared by in-line filtration of platelet-rich
plasma. Transfusion 1995;35:131-6.
15. ValeriCR.Frozenpreservationofredblood
cells.In:SimonTL,DzikWH,SnyderEL,etal,
eds. Rossi’s principles of transfusion medi-
cine.3rded.Philadelphia:LippincottWil
-
liams & Wilkins, 2002:62-8.
16. Meryman HT, Hornblower M. A method for
freezing and washing RBCs using a high glyc-
erol concentration. Transfusion 1972;12:145-
56.
17. Lovric VA, Klarkowski DB. Donor blood
frozen and stored between –20 C and –25 C
with 35-day liquid post-thaw shelf-life. Lan-
cet 1989;i:71-3.
18. Rathbun EJ, Nelson EJ, Davey RJ. Post-
transfusion survival of red cells frozen for 8
weeks after 42-day liquid storage in AS-3.
Transfusion 1989;29:213-7.
19. Kahn RA, Auster MJ, Miller WV. The effect of
refreezing previously frozen deglycerolized
red blood cells. Transfusion 1978;18:204-5.
20. Myhre BA, Nakasako YUY, Schott R. Studies
on 4 C stored frozen reconstituted red blood
cells. III. Changes occurring in units which
have been repeatedly frozen and thawed.
Transfusion 1978;18:199-203.
21. Angelini A, Dragani A, Berardi A, et al. Evalua-
tion of four different methods for platelet
freezing:Invitroandinvivostudies.VoxSang
1992;62:146-51.
22. BorziniP,AssaliG,RivaMR,etal.Plateletcryo
-
preservation using dimethylsulfoxide/poly-
ethylene glycol/sugar mixture as cyto-
preserving solution. Vox Sang 1993;64:248-9.
23. Food and Drug Administration. Memoran-
dum: Use of an FDA cleared or approved ster-
ile connection device (STCD) in blood bank
practice. (July 29, 1994) Rockville, MD: CBER
Office of Communication, Training, and
Manufacturers Assistance, 1994.
24. Sweeney JD, Kouttab NM, Holme SH, et al.
Prestorage pooled whole blood derived
leukoreduced platelets stored for seven days
preserve acceptable quality and do not show
evidence of a mixed lymphocyte reaction.
Transfusion 2004;44:1212-9.
25. Moroff G, Holme S. Concepts about current
conditions for the preparation and storage of
platelets. Transfus Med Rev 1991;5:48-59.
26. Högman CF, Meryman HT. Storage parame-
ters affecting red blood cell survival and
functions after transfusion. Transfus Med Rev
1999;13:275-6.
27. Heaton A, Keegan T, Holme S. In vivo regener-
ation of red cell 2,3-diphosphoglycerate fol-
lowing transfusion of DPG-depleted AS-1,
AS-3 and CPDA-1 red cells. Br J Haematol
1989;71:131-6.
28. Sweeney JD, Holme S, Heaton WAL. Quality of
platelet concentrates. In: Van Oss CJ, ed.
Transfusion immunology and medicine. New
York: Marcel Dekker, 1995:353-70.
29. Heal JM, Singl S, Sardisco E, et al. Bacterial
proliferation in platelet concentrates. Trans-
fusion 1996;26:388-9.
30. Punsalang A, Heal JM, Murphy PJ. Growth of
gram-positive and gram-negative bacteria in
platelet concentrates. Transfusion 1989;29:
596-9.
31. Murphy S. Platelet storage for transfusion.
Semin Hematol 1985;22:165-77.
32. Murphy S, Rebulla P, Bertolini F, et al. In vitro
assessment of the quality of stored platelet
concentrates. The BEST (Biomedical Excel-
lence of Safer Transfusion) Task Force of the
International Society of Blood Transfusion.
Transfus Med Rev 1994;8:29-36.
33. Rinder HM, Smith BR. In vitro evaluation of
stored platelets: Is there hope for predicting
post-transfusion platelet survival and func-
tion? Transfusion 2003;43:2-6.
34. Murphy S. The oxidation of exogenously
added organic anions by platelets facilitates
maintenance of pH during their storage for
transfusion at 22 C. Blood 1995;85:1929-35.
35. Holme S, Heaton WA, Courtright M. Platelet
storage lesion in second-generation contain-
ers: correlation with platelet ATP levels. Vox
Sang 1987;53:214-20.
36. Holme S, Sawyer S, Heaton A, Sweeney JD.
Studies on platelets exposed or stored at tem-
peratures below 20 C or above 24 C. Transfu-
sion 1997;37:5-11.
37. Dzik S. Leukodepletion blood filters: Filter
design and mechanisms of leukocyte re-
moval. Transfus Med Rev 1993;7:65-77.
38. Leukocyte reduction. Association Bulletin
99-7. Bethesda, MD: AABB, 1999.
39. Heaton A. Timing of leukodepletion of blood
products. Semin Hematol 1991;28:1-2.
40. Buchholz DH, AuBuchon JP, Snyder EL, et al.
Effects of white cell reduction on the resis-
200 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
tance of blood components to bacterial mul-
tiplication. Transfusion 1994;34:852-7.
41. Sweeney JD, Kouttab N, Penn LC, et al. A
comparison of prestorage leukoreduced
whole blood derived platelets with bedside
filtered whole blood derived platelets in
autologous stem cell transplant. Transfusion
2000;40:794-800.
42. CyrM,HumeH,SweeneyJD,etal.Anomaly
of the des-Arg9-bradykinin metabolism asso-
ciated with severe hypotensive reaction dur-
ing blood transfusions: A preliminary report.
Transfusion 1999;39:1084-8.
43. HeddleNM,KlamaL,SingerJ,etal.Therole
of the plasma from platelet concentrates in
transfusion reactions. N Engl J Med 1994;331:
625-8.
44. Sweeney JD. Quality assurance and standards
for red cells and platelets. Vox Sang 1998;74:
201-5.
45. Meryman HT, Hornblower M. Freezing and
deglycerolizing sickle-trait red blood cells.
Transfusion 1976;16:627-32.
46. SudaBA,LeitmanSF,DaveyRJ.Characteris-
tics of red cells irradiated and subsequently
frozen for long-term storage. Transfusion
1993;33:389-92.
47. MiragliaCC,AndersonG,MintzPD.Effectof
freezing on the in vivo recovery of irradiated
cells. Transfusion 1994;34:775-8.
48. Food and Drug Administration. Guidance for
industry: Gamma irradiation of blood and
blood components: A pilot program for li-
censing. (March 15, 2000) Rockville, MD:
CBER Office of Communication, Training,
and Manufacturers Assistance, 2000.
49. Moroff G, Holme S, AuBuchon JP, et al. Viabil-
ity and in vitro properties of gamma irradi-
ated AS-1 red blood cells. Transfusion 1999;
39:128-34.
50. Sweeney JD, Holme S, Moroff G. Storage of
apheresis platelets after gamma radiation.
Transfusion 1994;34:779-83.
51. Pineda AA, Zylstra VW, Clare DE, et al. Viabil-
ity and functional integrity of washed plate-
lets. Transfusion 1989;29:524-7.
52. Kim DM, Brecher ME, Bland LA, et al. Visual
identification of bacterially contaminated red
cells. Transfusion 1992;32:221-5.
53. Bertoloni F, Murphy S. A multicenter inspec-
tion of the swirling phenomenon in platelet
concentrates prepared in routine practice.
Transfusion 1996;36:128-32.
54. Dumont LJ, Dzik WH, Rebulla P, Brandwein
H, and the Members of the BEST Working
Party of the ISBT. Practical guidelines for pro-
cess validation and process control of white
cell-reduced blood components: Report of
theBiomedicalExcellenceforSaferTransfu-
sion (BEST) Working Party of the Interna-
tional Society of Blood Transfusion (ISBT).
Transfusion 1996;36:11-20.
Chapter 8: Components from Whole Blood Donations 201
Copyright © 2005 by the AABB. All rights reserved.

202 AABB Technical Manual
Appendix 8-1. Component Quality Control
Component
Specifications
and Standards*
AABB
Standards
†
Red Blood Cells Hematocrit ≤80% (in all) 5.7.5.1
Red Blood Cells
Leukocytes
Reduced
Retain 85% of original red cells, 95% of tested
units <5 ×106leukocytes in the final con-
tainer
5.7.5.6
Cryoprecipitated AHF Factor VIII: ≥80 IU/bag (100%)
Fibrinogen ≥150 mg/bag (100%)
5.7.5.14
Platelets ≥5.5 ×1010 platelets per unit and pH ≥6.2 in
90% of units tested
5.7.5.16
Platelets Leukocytes
Reduced
≥5.5 ×1010 platelets in 75% of units tested,
≥6.2 pH in 90% of units tested, and
<8.3 ×105leukocytes in 95% of units tested
5.7.5.17
Platelets Pheresis ≥3.0 ×1011 platelets in final container of compo-
nents tested; and pH ≥6.2 in 90% of units
tested
5.7.5.19
Platelets Pheresis
Leukocytes
Reduced
<5.0 ×106leukocytes in 95% of components
tested
and
≥3.0 ×1011 platelets in the final
container and pH ≥6.2 in 90% tested units
5.7.5.20
Granulocytes
Pheresis
≥1.0 ×1010 granulocytes in at least 75% of com-
ponents tested
5.7.5.21
Irradiated compo-
nents
25 Gy delivered to the central portion of the con-
tainer; minimum of 15 Gy at any point in the
component
5.7.4.2
*The
specification
is the threshold value; the
standard
is the percentage of tested units meeting or exceeding this thresh-
old. The manufacturing procedures used should be validated as capable of meeting these standards before implementa-
tion and routine QC. The number of units tested during routine QC should be such as to have a high level of assurance
that conformance with these standards is being achieved.
†Silva MA, ed. Standards for blood banks and transfusion services. 23rd ed. Bethesda, MD: AABB, 2005.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 9: Molecular Biology in Transfusion Medicine
Chapter 9
Molecular Biology in
Transfusion Medicine
PROTEINS ARE MACROMOLECULES
composed of amino acids, the se-
quences of which are determined
by genes. Lipids and carbohydrates are
not encoded directly by genes; genetic de-
termination of their assembly and func-
tional structures results from the action of
different protein enzymes. Blood group
antigens can be considered gene prod-
ucts, either directly, as polymorphisms of
membrane-associated proteins, or indi-
rectly, as carbohydrate configurations cat-
alyzed by glycosyltransferases.
From DNA to mRNA to
Protein
Structure of DNA
A gene consists of a specific sequence of
nucleotides located at a specific position
(locus) along a chromosome. Each chro-
mosome consists of long molecules or
strands of deoxyribonucleic acid (DNA).
DNA is composed of the sugar deoxyribose,
a phosphate group, the purine bases ade-
nine (A) and guanine (G), and the pyrimi-
dine bases thymine (T) and cytosine (C).
The combination of a sugar, a phosphate
group, and a base is called a nucleotide. A
double strand of DNA consists of two com-
plementary (nonidentical) single strands
held together by hydrogen bonds between
specific base pairings of A-T and G-C. The
two strands form a double helix configu-
ration with the sugar-phosphate back-
bone on the outside and the paired bases
ontheinside(seeFig9-1).DNAsynthesis
is catalyzed by DNA polymerase, which
adds a deoxyribonucleotide to the 3′end
oftheexistingchain.The3′and 5′nota-
tion refers to the carbon position of the
deoxyribose linkage to the phosphate
group. Phosphate groups bridge the sugar
groups between the fifth carbon atom of
one deoxyribose molecule and the third
carbon atom of the adjacent deoxyribose
203
9
Copyright © 2005 by the AABB. All rights reserved.
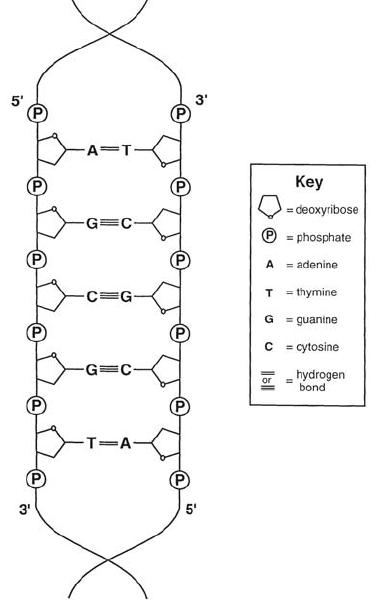
molecule and thus create the backbone of
the DNA strand. DNA polymerase-depend-
ent synthesis always occurs in the direc-
tion of 5′to 3′.
DNA Transcription
Linear sequences of nucleotides along the
DNA strands constitute the genes. Genes
occupy a constant location (locus) in the
DNA of a specific chromosome; the loci of
most known genes have been identified due
to the human genome project. For protein
synthesis to occur (see Fig 9-2), the infor-
mation encoded in the DNA sequence
must be copied into RNA (transcription)
and transported to the cytoplasmic organ-
elles called ribosomes where protein as-
sembly takes place (translation). Tran-
scription is done by copying one strand of
the DNA into a primary ribonucleic acid
transcript, which is then modified into
messenger ribonucleic acid (mRNA). The
mRNA represents a single stranded linear
sequence of nucleotides that differs from
DNA in the sugar present in its backbone
(ribose instead of deoxyribose) and the re-
placement of thymine by uracil (U), which
also pairs with adenine. DNA transcrip-
tion is catalyzed by the enzyme RNA poly-
merase. RNA polymerase binds tightly to a
specific DNA sequence called the pro-
moter, which contains the site at which
RNA synthesis begins (see Fig 9-3). Pro-
teins called transcription factors are re-
quired for RNA polymerase to bind to
DNA and for transcription to occur. Regu-
lation of transcription can lead to in-
creased, decreased, or absent expression
of a gene. For instance, a single base-pair
mutation in the transcription factor bind-
ing site of the Duffy gene promoter im-
pairs the promoter activity and is respon-
sible for the Fy(a–b–) phenotype.1
After binding to the promoter, RNA poly-
merase opens up the double helix of a local
region of DNA, exposing the nucleotides on
each strand. The nucleotides of one ex-
posed DNA strand act as a template for com-
plementary base pairing; RNA is synthe-
sized by the addition of ribonucleotides to
theelongatingchain.AsRNApolymerase
movesalongthetemplatestrandofDNA,
the double helix is opened before it and
closes behind it like a zipper. The process
continues, usually 0.5 to 2 kb downstream
of the poly-A signal, whereupon the en-
zyme halts synthesis and releases both the
DNAtemplateandthenewRNAchain.
mRNA Processing
Shortly after the initiation of transcrip-
tion, the newly formed chain is capped at
204 AABB Technical Manual
Figure 9-1. Schematic representation of the base
pairing of double-stranded DNA.
Copyright © 2005 by the AABB. All rights reserved.
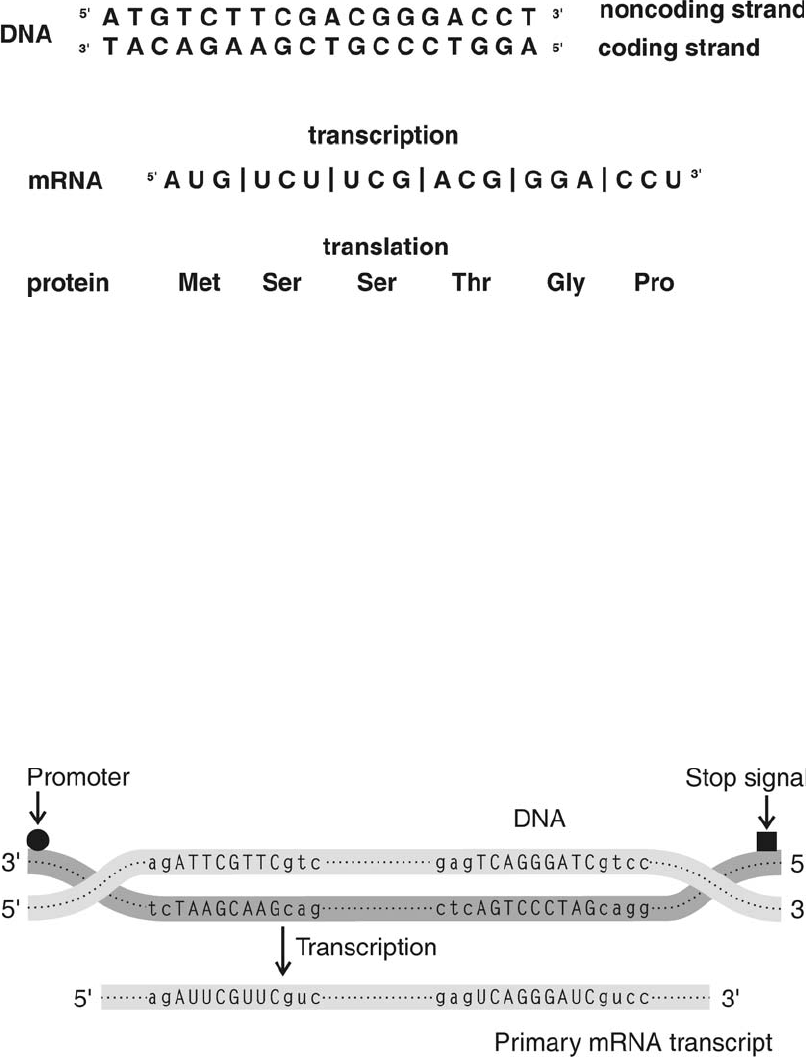
its 5′end by the addition of a methylated
G nucleotide. The 5′cap is important for
initiating protein synthesis and possibly
for protecting the mRNA molecule from
degradation during its transport to the cy-
toplasm. At the 3′end of the mRNA, a
multiprotein cleavage-polyadenylation
complex carries out a two-step process
that cleaves the new RNA at a specific se-
quence and then attaches 100 to 200 cop-
ies of adenylic acid, called the poly-A tail.
The poly-A tail functions in the export of
mature mRNA from the cell nucleus to the
cytoplasm, in the stabilization of the mRNA,
and as a ribosomal recognition signal re-
quired for efficient translation.
In eukaryotic cells, the nucleotide se-
quence of a gene often contains certain re-
gions that are represented in the mRNA and
other regions that are not represented. The
Chapter 9: Molecular Biology in Transfusion Medicine 205
Figure 9-2. Model of a nucleotide sequence. The sequence is fictitious.
↓
↓
Figure 9-3. The promoter sequence (•) contains the starting site for RNA synthesis. RNA polymerase
binds to the promoter and opens up a local region of the DNA sequence. One strand of DNA (the lower
one in this figure) acts as a template for complementary base pairing. The RNA polymerase copies the
DNA in a 5′to 3′direction until it encounters a stop signal (■). Lowercase letters represent nucleo-
tides in introns; uppercase letters represent coding bases in exons. The sequence is fictitious.
Copyright © 2005 by the AABB. All rights reserved.
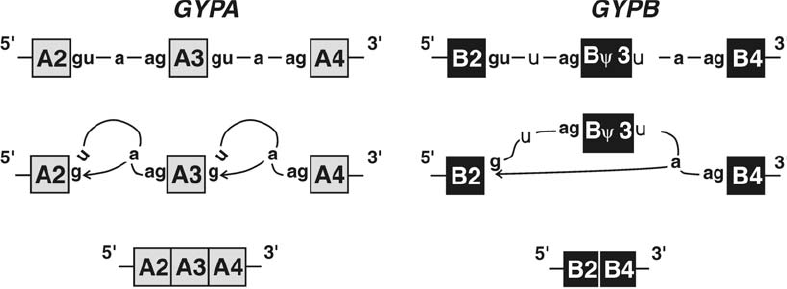
regions of the gene that are represented in
mRNA are called exons, which specify the
protein-coding sequences and the se-
quences of the untranslated 5′and 3′re-
gions. The regions that are not represented
in the mRNA are called intervening se-
quences or introns. In the initial transcrip-
tion of DNA to RNA, the introns and exons
are copied in their entirety, and the result-
ing product is known as the primary RNA
transcript or pre-mRNA. Processing occurs
while pre-mRNA is still in the nucleus, and
the introns are cut out by a process known
as RNA splicing (see Fig 9-4).
RNAsplicingdependsonthepresenceof
certain highly conserved sequences con-
sisting of GU at the 5′splice site (donor site)
and AG at the 3′splice site (acceptor site).
Additionally, an adenosine residue within a
specific sequence in the intron participates
in a complex reaction along with a very
large ribonucleoprotein complex called the
spliceosome. The reaction results in cleav-
age and joining of the 5′and 3′splice sites,
with release of the intervening sequence as
a lariat. Substitution of any of these highly
conserved sequences can result in inaccu-
rate RNA splicing (see Fig 9-4). Splicing of
pre-RNA is highly regulated during differ-
entiation and is tissue-specific. For example,
acetylcholinesterase, which bears the Cart-
wright blood group antigen, is spliced in a
manner to produce a glycosylphosphati-
dylinositol-linked protein in red cells, but it
is a transmembrane protein in nerve cells.2
Alternative splicing may also occur in a sin-
gletissuetypeandmayresultinthepro
-
ductionofmorethanoneproteinfromthe
same gene; an example of this type is the
production of glycophorins C and D from a
single glycophorin C (GPC) gene.3
Translation of mRNA
The bases within a linear mRNA sequence
are read (or translated) in groups of three,
called codons. Each three-base combina-
tion codes for one amino acid. There are
only 20 amino acids commonly used for
protein synthesis, and there are 64 (4 ×4×
4) possible codons. Most amino acids can
be specified by each of several different
codons, a circumstance known as “degen-
eracy” or “redundancy” in the genetic
code. For example, lysine can be specified
206 AABB Technical Manual
Figure 9-4. Substitution of two nucleotides (boldface) in the intron sequences flanking exon 3 of
GYPB
prevents normal splicing. Instead, all nucleotides between the 5′donor site of the second intron and
the 3′acceptor site of the third intron are excised. Because exon 3 is not translated, it is called a
pseudoexon (Ψ).
Copyright © 2005 by the AABB. All rights reserved.
by either AAA or AAG. Methionine has
only the single codon AUG. Three codons
(UAA, UGA, and UAG) function as stop sig-
nals; when the translation process en-
counters one of them, peptide synthesis
stops.
For the translation of codons into amino
acids, cytoplasmic mRNA requires the as-
sistance of transfer RNA (tRNA) molecules.
The tRNA molecules interact with the
mRNA through specific base pairings and
bring with them the amino acid specified
by the mRNA codon. Thus, the amino acids
are linked in amino to carboxyl peptide
bonds forming a growing polypeptide
chain. Proteins are synthesized such that
the initial amino acid has an unlinked
amine (NH2)groupandendswithatermi-
nal carboxylic acid (COOH) group. Protein
synthesis occurs on ribosomes, which are
large complexes of RNA and protein mole-
cules; ribosomes bound to the rough endo-
plasmic reticulum are the site of synthesis
of membrane and secretory proteins, where-
asfreeribosomesarethesiteofsynthesisof
cytosolicproteins.Theribosomebindsto
the tRNA and to the mRNA, starting at its 5′
end (amino terminal). Therefore, protein
synthesis occurs from the amino-terminal
end toward the carboxyl-terminal end. The
protein is produced sequentially until a stop
codon is reached, which terminates the
translation process and releases the newly
synthesized protein.
Many of the steps in the pathway of RNA
synthesis to protein production are closely
regulated at different levels to control gene
expression. Control steps include: initiation
of transcription, proofreading of the tran-
scription process, addition of the poly-A tail,
transportation of mRNA to the cytosol, ini-
tiation of translation, and elongation of the
polypeptide chain. Moreover, tissue-specific
and differentiation or stage-specific tran-
scriptionfactors,aswellashormonere
-
sponse elements and regulatory elements
at the 5´ and 3´ ends of mRNA, can affect
gene expression.
Genetic Mechanisms that
Create Polymorphism
Despite the redundancy inherent in de-
generacy of the genetic code, molecular
events such as substitution, insertion, or
deletion of a nucleotide may have far-
reaching effects on the protein encoded.
Some of the blood group polymorphisms
observed at the phenotypic level can be
traced to small changes at the nucleotide
level. The sequence in Fig 9-2, which is
not meant to represent a known sequence,
can be used to illustrate the effects of
minute changes at the nucleotide level,
discussed below.
Nucleotide Substitution
Nucleotide substitutions in the genomic
DNA can have profound effects on the re-
sultant protein. Many blood group anti-
gens are the result of single nucleotide
changes at the DNA level. The nucleotide
changes are transcribed into the RNA,
which alters the sequence of the codon. In
some instances, the codon change results
in the incorporation of a different amino
acid. For example, a change in the DNA
sequencefromaTtoaCatagivenposi
-
tion would result in an mRNA change
from U to G. Any one of three possible
outcomes can follow the substitution of a
single nucleotide:
1. Silent mutation. For example, the
substitution of an A with a C in the
third position of the DNA coding
strand for serine (UCU) in Fig 9-2 also
codes for serine (UCG). Thus, there
wouldbenoeffectontheprotein
because the codon would still be
translated as serine.
Chapter 9: Molecular Biology in Transfusion Medicine 207
Copyright © 2005 by the AABB. All rights reserved.
2. Missense response. The substitution
of a G with an A in the second posi-
tion of the DNA coding strand for the
second serine changes the product
of the codon from serine (UCG) to
leucine (UUG). Many blood group
polymorphisms reflect a single amino
acid change in the underlying mole-
cule. For example, the K2 antigen has
threonine, but K1 has methionine, as
the amino acid at position 193. This
results from a single C to T substitu-
tion in exon 6 of the Kell (KEL)gene.
4
3. Nonsense response. The substitu-
tion of a G with a T in the second po-
sition of the DNA coding strand for
serine (UCG) results in the creation of
the codon (UAG), which is one of the
three stop codons. No protein syn-
thesis will occur beyond this point,
resulting in a shortened or truncated
protein. Depending upon where this
nonsense substitution occurs, the
synthesized protein may be rapidly
degraded or may retain some func-
tion in its abbreviated form. The
Cromer blood group antigens reside
on the decay-accelerating factor
(DAF) membrane protein. In the null
(Inab) phenotype, a G to A nucleo-
tide substitution of the DAF gene
creates a stop codon at position 53,
andtheredcellhasnoDAFexpres
-
sion.5
A nucleotide substitution outside an
exon sequence may alter splicing and lead
to the production of altered proteins, as
seen with the altered expression of glyco-
phorin B (GPB)6or in the failure to produce
a normal amount of protein, as in the
Dr(a–) phenotype of Cromer.7In GYPB,the
gene that encodes GPB, substitutions of
two conserved nucleotides required for
RNA splicing and present in the
glycophorin A gene result in the excision of
exon 3 as well as introns 2 and 3 of GYPB
(see Fig 9-4). Because exon 3 is a coding
exon in GYPA but is noncoding in GYPB,it
is called a pseudoexon in GYPB.
Nucleotide Insertion and Deletion
Insertion of an entirely new nucleotide re-
sults in a frameshift, described as +1, be-
cause a nucleotide is being added. Nucle-
otide deletion causes a –1 frameshift. A
peptide may be drastically altered by the
insertion or deletion of a single nucleo-
tide. For example, the insertion of an A af-
ter the second nucleotide of the noncoding
DNA strand of Fig 9-2 would change the
readingframeofthemRNAtoUAUCAG
AAG CUG CCC UGG and represent the
polypeptide isoleucine-valine-phenylala-
nine-aspartic acid-glycine-threonine.
Genetic Variability
Gene conversion and crossing over may
occur between homologous genes located
on two copies of the same chromosome
that are misaligned during meiosis. Exam-
ples of homologous genes encoding blood
group antigens are the RHD and RHCE
genes of the Rh blood group system and
GYPA and GYPB, which encode the anti-
gens of the MNS blood group system.
Single Crossover
A single crossover is the mutual exchange
of nucleotides between two homologous
genes. If crossover occurs in a region
where paired homologous chromosomes
are misaligned, two hybrid genes are
formed in reciprocal arrangement (see Fig
9-5). The novel amino acid sequences en-
coded by the nucleotides at the junction
of the hybrid gene may result in epitopes
recognized by antibodies in human se-
rum and are known to occur in at least
three blood group systems: Rh, MNS, and
Gerbich.
208 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
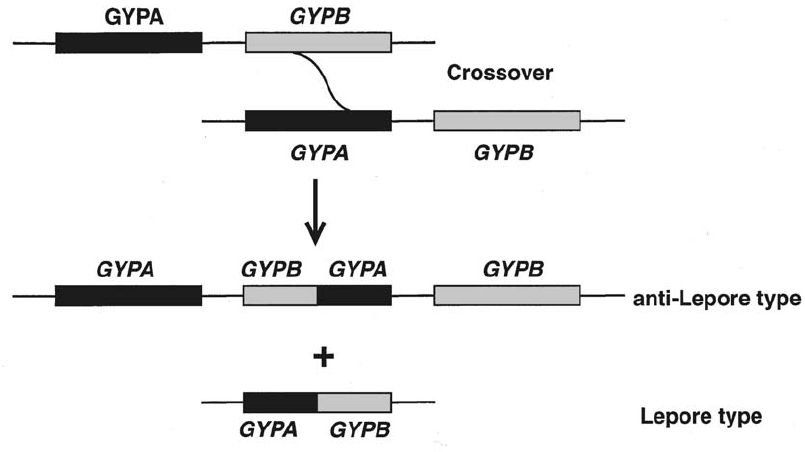
Gene Conversion
The process of gene conversion is thought
to consist of crossover, a general DNA re-
combination process, and DNA repair
duringmeiosis.Theresultisthatnucleo
-
tides from one homologous gene are in-
serted into another gene without recipro-
cal exchange. At the site of chromosome
crossover during meiosis, a heteroduplex
joint can form; this is a staggered joint be-
tween nucleotide sequences on two par-
ticipating DNA strands (see Fig 9-6). A
second type of gene conversion occurs
in meiosis when the DNA polymerase
switches templates and copies informa-
tion from a homologous sequence. This
event is usually the result of mismatch
repair; nucleotides removed from one
strand are replaced by repair synthesis us-
ing the homologous strand as a template.
Molecular Techniques
The development of modern molecular
techniques has greatly expanded our
knowledge of all biologic systems. These
same techniques are also applicable to the
diagnosis of disease, the practice of foren-
sic science, the generation of recombi-
nant proteins, and the production of func-
tional genes for gene therapy. Many of
these processes begin with DNA typing
and analysis, techniques that are reviewed
below.
Isolation of Nucleic Acids
The first step in most molecular biology
techniques is the isolation and purifica-
tion of nucleic acid, either DNA or RNA.
For applications of interest to the blood
banking community, the desired nucleic
Chapter 9: Molecular Biology in Transfusion Medicine 209
Figure 9-5. Single crossover: exchange of nucleotides between misaligned homologous genes. The
products are reciprocal.
Copyright © 2005 by the AABB. All rights reserved.
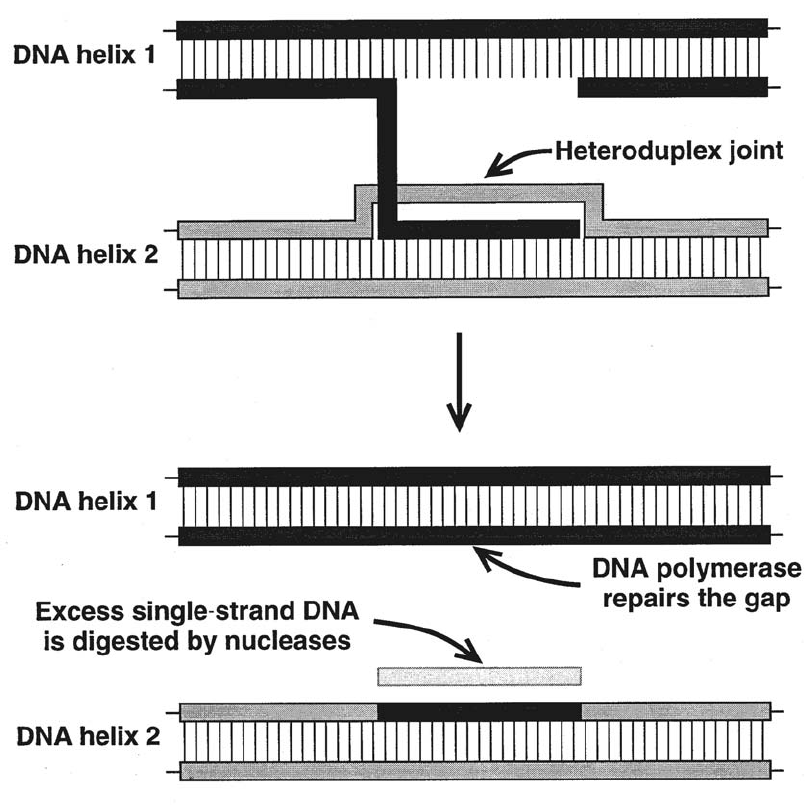
acid is typically human genomic DNA and
mRNA. Genomic DNA is present in all nu-
cleated cells and can be isolated from pe-
ripheral blood white cells or from buccal
tissue obtained by a simple cheek swab.
Both nucleated cells and reticulocytes are
cell-specific sources of mRNA.
Manufacturers offer kits for the isolation
of human genomic DNA from whole blood,
cells, and tissues. These kits vary in the
quantity and quality of the DNA isolated, in
rough proportion to the cost and ease of
use of the kit. High-quality DNA is of high
molecular weight and is relatively free of
contamination by protein or RNA. DNA pu-
rity is assessed by the ratio of its optical
density (OD) at 260 nm to that at 280 nm,
with the OD 260/280 ratio for pure DNA be-
210 AABB Technical Manual
Figure 9-6. Gene conversion: a heteroduplex joint forms between homologous sequences on two genes.
DNA polymerase repairs the double strands. Any excess single-stranded DNA is degraded by nu-
cleases, producing a hybrid gene on one chromosome but not on the other.
Copyright © 2005 by the AABB. All rights reserved.
ing 1.8. Low ratios (<1.6) indicate that the
DNA is contaminated with protein or mate-
rials used in the isolation procedure, and
high ratios (>2.0) indicate that the DNA is
contaminated with RNA. If the DNA is pure
and of sufficient concentration, it can be
quantitated by measurement of the OD at
260 nm. If the DNA is impure or in low con-
centration, it is best quantitated by electro-
phoresis in agarose gel along with DNA
standards of known concentration, fol-
lowed by visualization of the DNA with
ethidium bromide staining.
For certain molecular biology techniques
such as polymerase chain reaction (PCR),
the quantity and quality of the genomic
DNA used as starting material are not cru-
cial, and good results can be obtained with
even nanogram quantities of DNA that has
been degraded into small fragments. For
other molecular biology techniques, such
as cloning, larger quantities of high-molec-
ular-weight DNA are required.
One nucleated cell contains about 6 pg
of genomic DNA. Based on an average
white cell count of 5000/µL, each milliliter
of peripheral blood contains about 30 µgof
DNA. Commercial DNA isolation kits typi-
cally yield in excess of 15 µgofDNAper
milliliter of whole blood processed.
Polymerase Chain Reaction
The introduction of the PCR technique has
revolutionized the field of molecular ge-
netics.8This technique permits specific
DNA sequences to be multiplied rapidly
and precisely in vitro. PCR can amplify, to
a billionfold, a single copy of the DNA se-
quence under study, provided a part of the
nucleotide sequence is known. The inves-
tigator must know at least some of the
gene sequence in order to synthesize DNA
oligonucleotides for use as primers. Two
primers are required: a forward primer (5′)
and a reverse primer (3′). These are de-
signed so that one is complementary to
each strand of DNA, and, together, they
flank the region of interest. Primers can
be designed that add restriction sites to
the PCR product to facilitate its subse-
quent cloning or labeled to facilitate its
detection. Labels may incorporate radio-
activity, or, more frequently, a nonradio-
active tag, such as biotin or a fluorescent
dye. The PCR reaction is catalyzed by one
of several heat-stable DNA polymerases
isolated from bacterial species that are na-
tive to hot springs or to thermal vents on
the ocean floor. The thermostability of
these enzymes allows them to withstand
repeated cycles of heating and cooling.
Reaction Procedure
Theamplificationtechniqueissimpleand
requires very little DNA (typically <100 ng
ofgenomicDNA).TheDNAunderstudy
is mixed together with a reaction buffer,
excess nucleotides, the primers, and poly-
merase (see Fig 9-7). The reaction cocktail
is placed in a thermocycler programmed
to produce a series of heating and cooling
cycles that result in exponential amplifi-
cation of the DNA. The target DNA is ini-
tially denatured by heating the mixture,
which separates the double-stranded DNA
into single strands. Subsequent cooling in
the presence of excess quantities of sin-
gle-stranded forward and reverse primers
allows them to bind or anneal with com-
plementary sequences on the single-
stranded template DNA. The specific cool-
ing temperature is calculated to be appro-
priate for the primers being used. The re-
action mixture is then heated to the
optimal temperature for the thermo-sta-
ble DNA polymerase, which generates a
new strand of DNA on the single-strand
template, using the nucleotides as build-
Chapter 9: Molecular Biology in Transfusion Medicine 211
Copyright © 2005 by the AABB. All rights reserved.
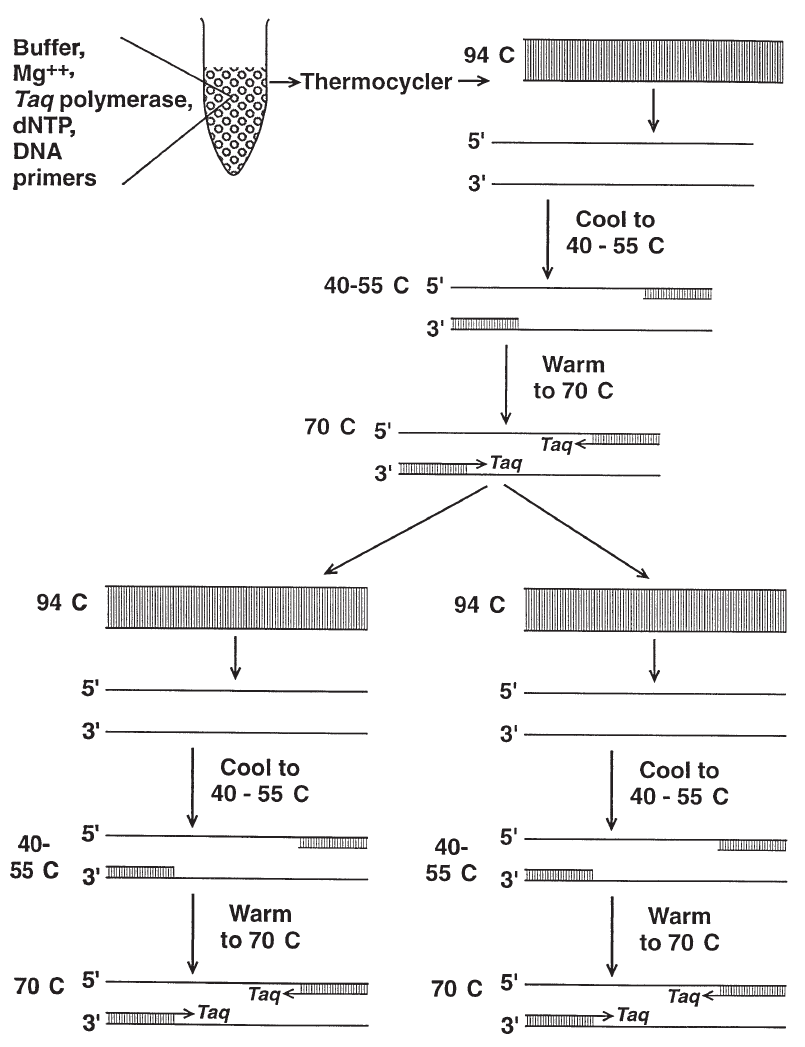
212 AABB Technical Manual
Figure 9-7. The polymerase chain reaction results in the exponential amplification of short DNA se-
quences such that the target sequence is amplified over a billionfold after 20 cycles. (
Taq
polymerase
is used here as an example of a thermostable DNA polymerase.)
Copyright © 2005 by the AABB. All rights reserved.
ing blocks for elongation. The next cycle
denatures newly formed double strands,
and the single-stranded DNA copies serve as
templates for subsequent synthesis. The
number of DNA copies doubles with each
cycle, such that after 20 cycles, there is a
billionfold amplification of the target DNA.
The amplified DNA may be analyzed by
agarose gel electrophoresis in the presence
of ethidium bromide, which binds to DNA
and is visible under ultraviolet light. The
DNA will be present as a single discrete
band equivalent in length to the distance
between the 5′ends of the primers. The
amplified DNA can also be differentiated by
size using capillary electrophoresis. Alter-
natively, the DNA sample can be blotted
onto a membrane and hybridized to a la-
beled, allele-specific probe. This is known
as a dot blot and is particularly useful when
multiple samples are being analyzed for the
same polymorphism.
Variations of the PCR have been devel-
oped to meet specific needs. For instance,
“long-distance” PCR, which has a mixture
of thermostable polymerases, can amplify
much larger targets (up to 40 kilobases in
length) than those typically amplified by
conventional PCR (up to 2 kilobases in
length). It is even possible to perform PCR
in situ, in tissue and cells. A related tech-
nology, the ligase chain reaction (LCR), uses
a thermostable DNA ligase instead of a
thermostable DNA polymerase. Rather
than amplification of a DNA target segment
located between two flanking primers using
a DNA polymerase as occurs in PCR, in the
LCR direct primer ligation occurs with no
amplification of an intervening DNA seg-
ment. Other PCR variations include multi-
plex PCR, in which multiple independent
segments of DNA are co-amplified in the
same reaction, and kinetic PCR, in which
the amplification product is measured in
real time in order to quantitate the amount
of starting nucleic acid.
Applications of PCR
Amplification of minute quantities of DNA
to detectable levels may significantly af-
fect the practice of transfusion medicine.
In screening donor blood for infectious
agents, PCR has become the procedure of
choice and thus eliminate our reliance on
seroconversion, which occurs well after
exposure to viruses or other pathogens
(see Chapter 28). Blood centers have been
using nucleic acid amplification testing
(NAT) to identify the presence of HIV and
HCV RNA in donor samples; detection of
WestNilevirusRNAhasbeenperformed
as an investigational test. Detection of vi-
ral RNA involves three steps: extraction,
amplification, and detection. Extraction
may occur following centrifugation steps
to concentrate the virus and remove con-
taminants. Otherwise, RNA extraction can
occur first followed by capture of viral
RNA onto a molecule that is immobilized
on a solid phase or onto magnetic parti-
cles in solution (this procedure is referred
to as target capture). Once immobilized,
the impurities may be removed by a series
of wash steps. Before amplification, viral
RNA must be converted to DNA; this is ac-
complished by the enzyme, reverse tran-
scriptase (RT). Amplification of the DNA
then can occur through multiple interme-
diates. In the case of PCR, the amplified
product is DNA (and is synthesized using
thermostable Taq DNA polymerase),
whereas, in the case of transcription-me-
diated amplification, the amplified prod-
uct is RNA (and is synthesized using T7
RNA polymerase) (see Fig 9-8). Detection
of the amplified product can occur by
capture of the amplified DNA on nitro-
cellulose or by enzyme immunoassay or
chemiluminescence. Currently, NAT for
blood donor screening has been imple-
mented for HIV and hepatitis C; NAT for
hepatitis B is under development. NAT for
Chapter 9: Molecular Biology in Transfusion Medicine 213
Copyright © 2005 by the AABB. All rights reserved.
other agents (eg, human T-cell lympho-
tropic virus, hepatitis A virus, parvovirus
B19) is not widely available.
PCR is being used for prenatal determi-
nation of many inheritable disorders, such
as sickle cell disease, in evaluating hemolytic
disease of the fetus and newborn, to type
fetal amniocytes,9to quantitate residual
white cells in filtered blood, and for tracing
donor leukocytes in transfusion recipients
(chimerism). Long-distance PCR is used for
cloning, sequencing, and chromosome
mapping, and reverse transcriptase (RT)-
PCR is used for studying gene expression
and cDNA cloning. LCR has special appli-
cability in transfusion medicine because of
its powerful ability to detect genetic vari-
ants. In the field of transplantation, PCR us-
ing sequence-specific oligonucleotide
probes or sequence-specific primers is
used to determine HLA types (see Chapter
17).
Restriction Endonucleases
The discovery of bacterial restriction endo-
nucleases provided the key technique for
DNA analysis. These enzymes, found in
different strains of bacteria, protect a bac-
teria cell from viral infection by degrading
viral DNA after it enters the cytoplasm.
Each restriction endonuclease recognizes
only a single specific nucleotide sequence,
typically consisting of four to six nucleo-
tides. These enzymes cleave the DNA
strand wherever the recognized sequence
occurs, generating a number of DNA frag-
ments whose length depends upon the
number and location of cleavage sites in
the original strand. Many endonucleases
have been purified from different species
of bacteria; the name of each enzyme re-
flects its host bacterium, eg, Eco RI is iso-
lated from Escherichia coli, Hind III is
from Hemophilus influenzae,andHpaIis
from Hemophilus parainfluenzae.
Restriction Fragment Length Polymorphism
Analysis
The unique properties of restriction endo-
nucleases make analysis of restriction
fragment length polymorphism (RFLP)
suitable for the detection of a DNA poly-
morphism. The changes in nucleotide se-
quence described above (substitution, in-
sertion, deletion) can alter the relative
locations of restriction nuclease cutting
sites and thus alter the length of DNA
fragments produced. RFLPs are detected
using Southern blotting and probe hy-
bridization (see Fig 9-9).
The isolated DNA is cleaved into frag-
ments by digestion with one or more re-
striction endonucleases. The DNA frag-
ments are separated by electrophoresis
through agarose gel and then transferred
onto a nylon membrane or nitrocellulose
paper. Once fixed to a nylon membrane or
nitrocellulose paper, the DNA fragments
are examined by application of a probe,
which is a small fragment of DNA whose
nucleotide sequence is complementary to
the DNA sequence under study. A probe
may be an artificially manufactured oligo-
nucleotide or may derive from cloned com-
plementary DNA. The probe is labeled with
a radioisotope or another indicator that
permits visualization of the targeted DNA
restriction fragments and is then allowed
to hybridize with the Southern blot. Un-
bound excess probe is washed off, and hy-
bridized DNA is visualized as one or more
bands of specific size, dictated by the spe-
cific nucleotide sequence. If several indi-
viduals are analyzed for polymorphism,
several different banding patterns may be
observed.
RFLP analysis has been used in gene
mapping and analysis, linkage analysis,
characterization of HLA genes in transplan-
tation, paternity testing, and forensic sci-
ence.
214 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
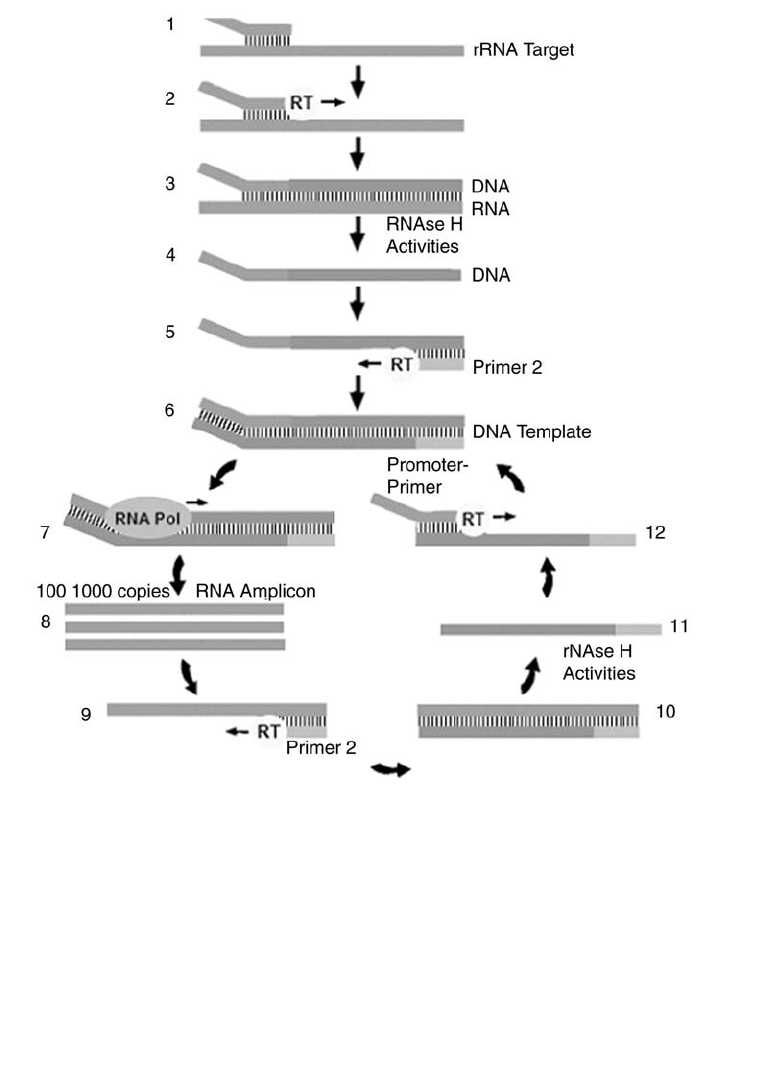
Chapter 9: Molecular Biology in Transfusion Medicine 215
Figure 9-8. Transcription-mediated amplification cycle.
Step 1: Promoter-primer binds to rRNA target.
Step 2: Reverse transcriptase (RT) creates DNA copy of rRNA target.
Step 3: RNA:DNA duplex.
Step 4: RNAse H activities of RT degrade the rRNA.
Step 5: Primer 2 binds to the DNA and RT creates a new DNA copy.
Step 6: Double-stranded DNA template with a promoter sequence.
Step 7: RNA polymerase (RNA Pol) initiates transcription of RNA from DNA template.
Step 8: 100-1000 copies of RNA amplicon are produced.
Step 9: Primer 2 binds to each RNA amplicon and RT creates a DNA copy.
Step 10: RNA:DNA duplex.
Step 11: RNAse H activities of RT degrade the rRNA.
Step 12: Promoter-primer binds to the newly synthesized DNA. RT creates a double-stranded DNA and the autocatalytic cycle
repeats, resulting in a billion-fold amplification.
-
Copyright © 2005 by the AABB. All rights reserved.
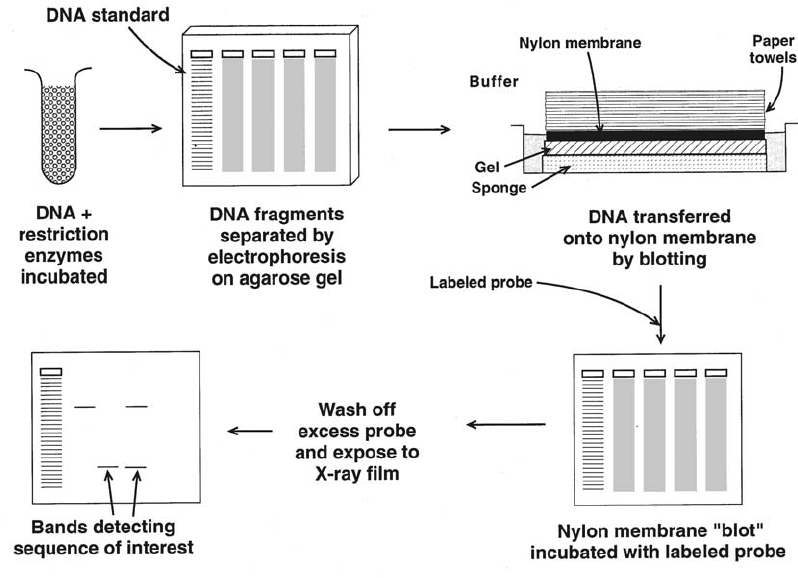
DNA Profiling
Regions of DNA that show great allelic vari-
ability (“minisatellites” and “microsatel-
lites”) can be studied by the application of
RFLP mapping and/or PCR analysis (a pro-
cess sometimes called DNA profiling, DNA
typing, or DNA fingerprinting). Minisatel-
lites or variable number of tandem repeat
(VNTR) loci consist of tandem repeats of a
medium-sized (6-100 base-pair) sequence,
whereas microsatellites or short tandem
repeat (STR) loci consist of tandem re-
peats of a short (typically four base pair)
sequence. These regions are almost al-
ways found in the noncoding regions of
DNA. Variability stems from differences in
the number of repeat units contained
within the fragments. There is so much
variation between individuals that the
chances are very low that the same num-
bers of repeats will be shared by two indi-
viduals, even if related. The VNTR and/or
STR patterns observed at four to eight dif-
ferent loci may be unique for an individ-
ual and thus constitute a profile or “fin-
gerprint” that identifies his or her DNA.
When DNA profiling was developed,
testing was performed by RFLP analysis.
DNA profiling is now increasingly done by
amplification of selected, informative
VNTR and/or STR loci using locus-specific
oligonucleotide primers, followed by mea-
surementofthesizeofthePCRproducts
produced. PCR products can be separated
by size by electrophoresis through poly-
acrylamide gel and detected by silver stain-
ing, or, if the PCR products incorporate a
216 AABB Technical Manual
Figure 9-9. Southern blotting: a technique for the detection of polymorphism by gel-transfer and hy-
bridization with known probes.
Copyright © 2005 by the AABB. All rights reserved.
fluorescent tag, by fluorescent detection
systems including those designed for auto-
mated DNA sequencing. Comparison of the
VNTR and STR PCR products with a stan-
dard-size ladder distinguishes the alleles
present in the sample.
DNA profiling is a technique that is ex-
tremely powerful for identifying the source
of human DNA; therefore, it has applica-
tions in forensic and paternity testing, as well
as in the documentation of chimerism, which
is of special importance in monitoring
allogeneic hematopoietic transplantation.
DNA Cloning
PCR may also be used for analysis of mRNA,
which is an especially useful source of ge-
netic material because only the exons of
the gene are present. By a modification of
PCR called RT-PCR, the single-stranded
mRNA is converted to double-stranded
DNA.TheenzymeRTisusedtogenerate
a single strand of DNA, which serves as a
template for a second strand generated by
DNA polymerase. The product is comple-
mentary DNA (cDNA) and it is the DNA
molecule of choice for cloning and se-
quencing.
In gene cloning, the DNA containing the
gene of interest is inserted into a vector,
which is a self-replicating genetic element
such as a virus (eg, the bacteriophage lambda
gt11) or a plasmid. Plasmids are small cir-
cular molecules of double-stranded DNA
that occur naturally in bacteria and typi-
cally confer antibiotic resistance. After the
gene has been inserted into the DNA of the
vector, the recombinant DNA can be intro-
duced into a bacterial host where it under-
goes replication. Because many vectors
carry genes for antibiotic resistance, this
characteristic can be exploited by growing
the host bacteria in the presence of the ap-
propriate antibiotic; only bacteria that have
successfully incorporated the recombinant
vector will survive to form colonies or
clones. Each individual vector potentially
contains a different cDNA sequence. The
sum of bacterial clones harboring recombi-
nant vectors is called a DNA library. Librar-
ies can be obtained from many commercial
sources or can be produced by the individ-
ual investigator. The library can be probed
through a technique similar to Southern
blotting, with an oligonucleotide probe
basedonpartofaknownsequence.Posi
-
tive clones can be selected and a pure cul-
ture grown in large quantities. Once puri-
fied, the cloned DNA can be recovered for
use as a probe or for detailed molecular
characterization.
The ability to insert genes into the
genomes of virtually any organism, includ-
ing bacteria, plants, invertebrates (such as
insects), and vertebrates (such as mam-
mals), permits not only gene characteriza-
tion but also genetic engineering, including
the production of recombinant proteins (see
below) and gene therapy. Although still in
the developmental stages, gene therapy
promises to have a role in the management
of disorders as diverse as inherited genetic
diseases, human immunodeficiency virus,
and cancer, and in the development of
novel vaccines.10,11
DNA Sequencing
A major worldwide scientific effort called
the Human Genome Project has obtained
thecompletenucleotidesequenceofthe
human genome as well as the genomes of
several other key organisms. The initiative
also improved DNA sequencing technol-
ogy. Realization of both goals has had a
positive impact on transfusion practice.
The identification of all human genes12,13
provides a complete blueprint of the pro-
teins that are relevant in transfusion med-
icine. In turn, this information has helped
Chapter 9: Molecular Biology in Transfusion Medicine 217
Copyright © 2005 by the AABB. All rights reserved.
in the development of recombinant pro-
teins for use as transfusion components and
in-vitro test reagents. It also plays a role in
clarifying the disorders that afflict trans-
fusion recipients.
Advances in DNA sequencing have taken
thefieldalongwayfromthecumbersome
manual techniques common in research
laboratories until recently.14 Automated
DNA sequencers using laser detection of
fluorescently labeled sequencing products
detect all four nucleotide bases in a single
lane on polyacrylamide gel and can be op-
timized for specialty applications such as
heterozygote detection and sizing of PCR
fragments. Automated DNA sequencers us-
ing capillary electrophoresis are especially
useful for the rapid sequencing of short
DNA templates. DNA sequence can also be
obtained using mass spectrometry. Auto-
mated sequencers will become increasingly
common in clinical laboratories as this
technology evolves. If it can be made
cost-effective for routine use, then DNA se-
quencing could become a routine genotyp-
ing method.
DNA Microarrays
The complexity of the human genome re-
quires that the differential expression of
multiple genes be analyzed at once to un-
derstand normal biologic processes as well
as changes in diseases. A powerful tech-
nique to accomplish this goal is DNA
microarrays or gene chips.15 In this method,
tens of thousands of separate DNA mole-
cules are spotted or synthesized on a small
area of a solid support, often a glass slide.
The DNA can be generated by PCR or oligo-
nucleotide synthesis. This microarray is
then probed, in a process analogous to
Southern blotting, using cDNA created
from the total mRNA expressed at a given
time by a cell or tissue. The result is a pic-
ture of the gene expression profile of the
tissue for all of the genes on the micro-
array. Also, microarrays can be used for
comparative genomics and genotyping, an
application for blood groups.
Recombinant Proteins
The technology to make recombinant
proteins includes in-vitro systems in bac-
teria, yeast, insect cells, and mammalian
cells, as well as in-vivo systems involving
transgenic plants and animals.16 First, a
source of DNA corresponding in nucleic
acid sequence to the desired protein is
prepared, typically by cloning the cDNA
and ligating it into a suitable expression
vector. Then, the expression vector con-
taining the DNA of interest is transfected
into the host cell, and the DNA of interest
is transcribed under the control of the
vector promoter. Next, the resulting mRNA
is translated into protein by the host cell.
Posttranslational modifications such as
the addition of carbohydrates to the new
protein will be carried out by the host cell.
If specific posttranslational modifications
required for the new protein’s function can-
not be carried out by the host cell, then it
may be necessary to endow the host cell
with additional capabilities; for example, by
cotransfection with the cDNA for a specific
enzyme. In some cases, posttranslational
modification may not be crucial for a re-
combinant protein to be effective; for
instance, granulocyte colony-stimulating
factor (G-CSF) is produced in a nonglyco-
sylated form in E. coli (filgrastim) and in a
glycosylated form in yeast (lenograstim).
Recombinant proteins are finding multi-
ple uses in transfusion medicine, as thera-
peutic agents and vaccine components, in
component preparation, in virus diagnosis,
and in serologic testing. Recombinant hu-
man erythropoietin,17 G-CSF and GM-CSF,18
interferon-alpha, interleukin-2, interleu-
kin-11, and Factors VIII,19 IX,20 VII, and VIIa21
218 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
are all available and finding clinical accep-
tance. For instance, recombinant erythro-
poietin can be used to increase red cell pro-
duction in anemic patients before surgery,
reducing the need for allogeneic blood.22 It
can also be used to increase the amount of
autologous red cells that can be withdrawn
before surgery from non-anemic patients.23
Moreover, it has revolutionized the man-
agement of renal transplant candidates
whose kidneys are too impaired to produce
endogenous erythropoietin.
Recombinant human thrombopoietin
may be of value in augmenting platelet
yields from apheresis donors but is unlikely
to significantly reduce the need for platelet
transfusions when given to thrombo-
cytopenic patients.24 In the coagulation
arena, recombinant proteins such as pro-
tein C, antithrombin, and hirudin are ap-
provedbytheFoodandDrugAdministra-
tion for use, and tissue factor pathway
inhibitor, Factor XIII, and von Willebrand
factor appear promising. Recombinant
myeloid growth factors such as G-CSF are
used to enhance yields of progenitor cells
during apheresis and to support patients
following chemotherapy and hemato-
poietic transplantation.25
Recombinant proteins can be used as
transfusion components.26 Recombinant
human hemoglobin has been produced in
a number of in-vitro expression systems
and in vivo in transgenic swine and may be
useful as a noninfectious blood substitute.27
Recombinant human serum albumin has
been produced in yeast. Alphagalactosi-
dase, an enzyme that is capable of convert-
ing group B cells into group O cells, has
been produced in a recombinant form that
can modify group B units for transfusion to
group A and O recipients.28 These recombi-
nant proteins and other products under de-
velopment will undoubtedly affect the vari-
ety of transfusion components that will
become available in the future.
Recombinant proteins corresponding to
proteins from clinically relevant viruses,
bacteria, and parasites, some of which may
be transmitted by blood transfusion, may
be used as vaccine components29 and as
antigens in test kits for the detection of an-
tibodies. Cells transfected with appropriate
vectors can be induced to express recombi-
nant proteins on the membrane surface
and, as such, may become useful as geneti-
cally engineered reagent cells for in-vitro
testing.
Gene Therapy
Gene therapy refers to the introduction of
nonself genetic material into cells to treat
or prevent disease. At present, gene ther-
apy is restricted to somatic cells because
of ethical concerns about the transfer of
genes to germ-line cells. More than 3500
patients have been administered gene
therapy in clinical trials,30 but results over-
all have been disappointing largely due to
problems with delivery (transfection) of
the genetic material (transgene) into cells
and the limited lifespan of the success-
fully transfected cells.
There are different types of vectors (gene-
delivery vehicles) that deliver genes to cells,
including viral vectors (retrovirus, adeno-
virus, adeno-associated virus, vaccinia, her-
pes simplex), naked DNA, and modified DNA.
Genetic material can also be transferred to
cells by physical means such as electro-
poration (use of an electric field). As meth-
ods for gene delivery improve, it is antici-
pated that the benefits of gene therapy will
become more apparent.
Gene therapy can be used to replace a
defective gene, leading to increased pro-
duction of a specific protein as in the re-
placement of Factor VIII or IX for patients
with hemophilia,31 or it can be used to
downregulate (reduce) expression of an un-
desirable gene. The latter can be accom-
Chapter 9: Molecular Biology in Transfusion Medicine 219
Copyright © 2005 by the AABB. All rights reserved.
plished by the introduction of DNA se-
quences (antisense oligonucleotides)
corresponding to the antisense strand of
the mRNA, which then interferes with
translation of the mRNA into protein.
Protein and RNA Targeted Inactivation
More recently, novel pharmacologic agents
have been developed that interfere with
specific molecules produced by cancer
cells or infectious particles. One such ex-
ample is imatinib mesylate (Gleevec,
Novartis Pharmaceuticals, East Hanover,
NJ), which binds specifically to the Bcr-
Abl protein and blocks its tyrosine kinase
activity in malignant white cells. It specif-
ically blocks the binding site for adeno-
sine triphosphate on the kinase, thus
inhibiting its ability to phosphorylate
intracellular proteins.32 The inactivation
prevents Bcr-Abl-induced malignant cell
proliferation and anti-apoptosis. Normal
kinase signaling pathways are largely un-
affected.
Another promising therapy is RNA inter-
ference, which has its historical roots as a
research tool used to characterize the func-
tion of known genes. RNA interference is
based on an antiviral mechanism in which
dsRNA is delivered to a cell and is subse-
quently processed into small (21-25 bp) in-
terfering RNA (siRNA) molecules. The siRNA
molecules silence the expression of a target
gene in a sequence-specific manner. More
important, RNA interference has potential
as a therapeutic strategy to silence cancer-re-
lated genes or infectious diseases like viral
hepatitis.33
References
1. Tournamille C, Colin Y, Cartron JP, Le Van
Kim C. Disruption of a GATA motif in the
Duffy gene promoter abolishes erythroid
gene expression in Duffy-negative individu-
als. Nat Genet 1995;10:224-8.
2. Li Y, Camp S, Taylor P. Tissue-specific expres-
sion and alternative mRNA processing of the
mammalian acetylcholinesterase gene. J Biol
Chem 1993;268:5790-7.
3. Le Van Kim C, Mitjavila MT, Clerget M, et al.
An ubiquitous isoform of glycophorin C is
produced by alternative splicing. Nucleic Ac-
ids Res 1990;18:3076.
4. LeeS,WuX,ReidM,etal.Molecularbasisof
the Kell (K1) phenotype. Blood 1995;85:912-
16.
5. Lublin DM, Mallinson G, Poole J, et al. Molec-
ular basis of reduced or absent expression of
decay-accelerating factor in Cromer blood
group phenotypes. Blood 1994;84:1276-82.
6. Kudo S, Fukuda M. Structural organization of
glycophorin A and B genes: Glycophorin B
gene evolved by homologous recombination
at Alu repeat sequences. Proc Natl Acad Sci
U S A 1989;86:4619-23.
7. Lubin DM, Thompson ES, Green AM, et al.
Dr(a–) polymorphism of decay accelerating
factor. Biochemical, functional, and molecu-
lar characterization and production of allele-
specific transfection. J Clin Invest 1991;87:
1945-52.
8. Erlich HA. Principles and applications of the
polymerase chain reaction. Rev Immuno-
genet 1999;1:127-34.
9. BennettPR,LeVanKimC,ColinY,etal.Pre-
natal determination of fetal RhD type by DNA
amplification. N Engl J Med 1993;329:607-10.
10. Friedmann T. Overcoming the obstacles to
gene therapy. Sci Am 1997;276:80-5.
11. Hillyer CD, Klein HG. Immunotherapy and
gene transfer in the treatment of the oncol-
ogy patient: Role of transfusion medicine.
Transfus Med Rev 1996;10:1-14.
12. International Human Genome Sequencing
Consortium. Initial sequencing and analysis
of the human genome. Nature 2001;409:860-
921.
13. VenterJC,AdamsMD,MyersEW,etal.The
sequence of the human genome. Science
2001;291:1304-51.
14. Griffin HG, Griffin AM. DNA sequencing. Re-
cent innovations and future trends. Appl Bio-
chem Biotechnol 1993;38:147-59.
15. Duggan DJ, Bittner M, Chen Y, et al. Expres-
sion profiling using cDNA microarrays. Nat
Genet 1999;21:S10-14.
16. Lubon H, Paleyanda RK, Velander WH,
Drohan WN. Blood proteins from transgenic
animal bioreactors. Transfus Med Rev 1996;
10:131-43.
17. CazzolaM,MercurialiF,BrugnaraC.Useof
recombinant human erythropoietin outside
the setting of uremia. Blood 1997;89:4248-67.
220 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
18. Ganser A, Karthaus M. Clinical use of hema-
topoietic growth factors. Curr Opin Oncol 1996;
8:265-9.
19. VanAken WG. The potential impact of recom-
binant factor VIII on hemophilia care and the
demand for blood and blood products. Trans-
fus Med Rev 1997;11:6-14.
20. White GC II, Beebe A, Nielsen B. Recombi-
nant factor IX. Thromb Haemost 1997;78:
261-5.
21. Lusher JM. Recombinant factor VIIa (Novo-
Seven) in the treatment of internal bleeding
in patients with factor VIII and IX inhibitors.
Haemostasis 1996;26(Suppl 1):124-30.
22. Braga M, Gianotti L, Gentilini O, et al. Ery-
thropoietic response induced by recombi-
nanthumanerythropoietininanemiccancer
patients candidate to major abdominal sur-
gery. Hepatogastroenterology 1997;44:685-90.
23. Cazenave JP, Irrmann C, Waller C, et al. Epo-
etin alfa facilitates presurgical autologous
blood donation in non-anaemic patients
scheduled for orthopaedic or cardiovascular
surgery. Eur J Anaesthesiol 1997;14:432-42.
24. Kuter DJ. What ever happened to thrombo-
poietin? Transfusion 2002;42:279-83.
25. Ketley NJ, Newland AC. Haemopoietic growth
factors. Postgrad Med J 1997;73:215-21.
26. Growe GH. Recombinant blood components:
Clinical administration today and tomorrow.
World J Surg 1996;20:1194-9.
27. Kumar R. Recombinant hemoglobins as
blood substitutes: A biotechnology perspec-
tive. Proc Soc Exp Biol Med 1995;208:150-8.
28. Lenny LL, Hurst R, Zhu A, et al. Multiple-unit
and second transfusions of red cells enzymat-
ically converted from group B to group O: Re-
port on the end of phase 1 trials. Transfusion
1995;35:899-902.
29. Ellis RW. The new generation of recombinant
viral subunit vaccines. Curr Opin Biotechnol
1996;7:646-52.
30. Mountain A. Gene therapy: The first decade.
Trends Biotechnol 2000;18:119-28.
31. Kaufman RJ. Advances toward gene therapy
for hemophilia at the millennium. Hum Gene
Therapy 1999;10:2091-107.
32. Druker BJ, Tamura S, Buchdunger E, et al. Ef-
fects of a selective inhibitor of the Abl tyro-
sine kinase on the growth of Bcr-Abl positive
cells. Nat Med 1996;2:561-6.
33. Radhakrishnan SK, Layden TJ, Gartel AL. RNA
interference as a new strategy against viral
hepatitis. Virology 2004;323:173-81.
34. Farese AM, Schiffer CA, MacVittie TJ. The im-
pact of thrombopoietin and related mpl-lig-
ands on transfusion medicine. Transfus Med
Rev 1997;11:243-55.
35. Barbara JA, Garson JA. Polymerase chain re-
action and transfusion microbiology. Vox
Sang 1993;64:73-81.
36. Power EG. RAPD typing in microbiology—a
technical review. J Hosp Infect 1996;34:247-
65.
37. LarsenSA,SteinerBM,RudolphAH,WeissJB.
DNA probes and PCR for diagnosis of para-
sitic infections. Clin Microbiol Rev 1995;8:1-
21.
38. WeissJB.DNAprobesandPCRfordiagnosis
of parasitic infections. Clin Microbiol Rev
1995;8:113-30.
39. Majolino I, Cavallaro AM, Scime R. Peripheral
blood stem cells for allogeneic transplanta-
tion. Bone Marrow Transplant 1996;18(Suppl
2):171-4.
40. Gretch DR. Diagnostic tests for hepatitis C.
Hepatology 1997;26:43S-7S.
41. PenaSD,PradoVF,EpplenJT.DNAdiagnosis
of human genetic individuality. J Mol Med
1995;73:555-64.
42. van Belkum A. DNA fingerprinting of medi-
cally important microorganisms by use of PCR.
Clin Microbiol Rev 1994;7:174-84.
43. Siegel DL. Research and clinical applications
of antibody phage display in transfusion
medicine. Transfus Med Rev 2001;15:35-52.
44. Hyland CA, Wolter LC, Saul A. Identification
and analysis of Rh genes: Application of PCR
and RFLP typing tests. Transfus Med Rev
1995;9:289-301.
Suggested Reading
Denomme G, Lomas-Francis C, Storry RJ, Reid ME.
Approaches to molecular blood group genotyping
and their applications. In: Stowell C, Dzid W, eds.
Emerging technologies in transfusion medicine.
Bethesda, MD: AABB Press, 2003:95-129.
Garratty G, ed. Applications of molecular biology
to blood transfusion medicine. Bethesda, MD:
AABB, 1997.
Lewin B. Genes VII. Oxford: Oxford University Press,
2000.
Sheffield WP. Concepts and techniques in molecu-
lar biology—an overview. Transfus Med Rev 1997;
11:209-23.
Chapter 9: Molecular Biology in Transfusion Medicine 221
Copyright © 2005 by the AABB. All rights reserved.

222 AABB Technical Manual
Appendix 9-1. Molecular Techniques in Transfusion Medicine
Technique Applications Examples References
PCR Infectious disease
testing
Viruses, bacteria,
parasites
35
36
37, 38
Polymorphism detec-
tion
HLA, blood group
antigens
1, 39
Prenatal detection Rh 9
Recombinant proteins/
DNA cloning
Therapy Erythropoietin,
thrombopoietin,
G-CSF
17, 18, 34
Apheresis G-CSF 39
Infectious disease
testing
Viral testing 40
Recombinant
components
Coagulation factors 19
Hemoglobin 27
Component processing Alpha-galactosidase 28
Vaccine production Hepatitis, malaria, HIV 27-29
DNA profiling Human identification
Bacterial identification
Chimerism, forensic
science
Bacterial typing
41
42
DNA sequencing Polymorphism detec-
tion, heterozygote
detection
HLA 14
Phage display/
repertoire cloning
Monoclonal antibody
production
Anti-D 43
RFLP Polymorphism
detection
HLA, blood group
antigens
44
PCR = polymerase chain reaction; G-CSF = granulocyte colony-stimulating factor; HIV = human immunodefi-
ciency virus; RFLP = restriction fragment length polymorphism.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 10: Blood Group Genetics
Chapter 10
Blood Group Genetics
LANDSTEINER’S DISCOVERY OF
the ABO blood group system dem-
onstrated that human blood ex-
pressed inheritable polymorphic structures.
Shortly after the discovery of the ABO sys-
tem, red cells proved to be an easy and ac-
cessible means to test for blood group
polymorphisms in individuals of any age.
As more blood group antigens were de-
scribed, blood group phenotyping pro-
vided a wealth of information about the
polymorphic structures expressed on pro-
teins, glycoproteins and glycolipids on
red cells, and the genetic basis for their
inheritance.
Basic Principles
Inheritance of transmissible characteris-
tics or “traits,” including blood group an-
tigens, forms the basis of the science of
genetics. The genetic material that deter-
mines each trait is found in the nucleus
of a cell. This nuclear material is called
chromatin and is primarily made up of DNA
(see Chapter 9). When a cell divides, the
chromatin loses its homogenous appear-
ance and forms a number of rod-shaped
organelles called chromosomes. Encoded
in the chromatin or chromosomal DNA
are units of genetic information called
genes. The genes are arranged in a spe-
cific order along a chromosome with the
precise gene location known as the locus.
Chromosomes
The number of chromosomes and chro-
mosome morphology are specific for each
species. Human somatic cells have 46
chromosomes that exist as 23 pairs (one-
half of each pair inherited from each par-
ent). Twenty-two of the pairs are alike in
both males and females and are called
autosomes; the sex chromosomes, XX in
females and XY in males, are the remain-
ing pair.
223
10
Copyright © 2005 by the AABB. All rights reserved.
Each chromosome consists of two arms
joined at a primary constriction, the centro-
mere. The two arms are usually of different
lengths: the short, or petite, arm is termed
“p,” and the long arm is termed “q.” The
arms of individual chromosomes are indi-
cated by the chromosome number followed
by a p or q (ie, Xp is the short arm of the X
chromosome; 12q is the long arm of chro-
mosome 12). When banded and stained,
each chromosome displays a unique pat-
tern of bands, which are numbered from
the centromere outward (see Fig 10-1).
Chromosomes are identified by the loca-
tion of the centromere and their banding
patterns. The locations of individual genes
along the chromosome may be physically
“mapped” to specific band locations.
Lyonization
In the somatic cells of females, only one X
chromosome is active. Inactivation of one
of the X chromosomes is a random pro-
cess that occurs within days of fertiliza-
tion. Once an X chromosome has become
inactivated, all of that cell’s clonal descen-
dants have the same inactive X. Hence, in-
activation is randomly determined but
once the decision is made, the choice is
permanent. This process is termed lyoni-
zation.
Mitosis and Meiosis
Cells must replicate their chromosomes
as they divide so that each daughter cell
receives the full complement of genetic
information. Cell division is of two kinds:
mitosis and meiosis. Mitosis is the pro-
cess whereby the body grows or replaces
dead or injured somatic cells. This process
consists of five stages: prophase, pro-
metaphase, metaphase, anaphase, and
telophase. The end result after cytokinesis
is two complete daughter cells, each with
a nucleus containing all the genetic infor-
mation of the original parent cell (Fig
10-2).
Meiosis occurs only in primordial cells
destined to mature into haploid gametes.
Diploid cells that undergo meiosis give rise
to haploid gametes (sperm and egg cells with
only 23 chromosomes). Hence, somatic
cells divide by mitosis, giving rise to diploid
cells that have a 2N chromosome comple-
ment. Gametes formed following meiosis
are haploid, with a 1N chromosome com-
plement. It is during meiosis that genetic
diversity occurs. One type of diversity is the
224 AABB Technical Manual
Figure 10-1. Diagram of Giemsa-stained normal
human chromosome 7. With increased resolution
(left to right), finer degrees of banding are evi-
dent. Bands are numbered outward from the
centromere (c), which divides the chromosome
intopandqarms.
1
Copyright © 2005 by the AABB. All rights reserved.
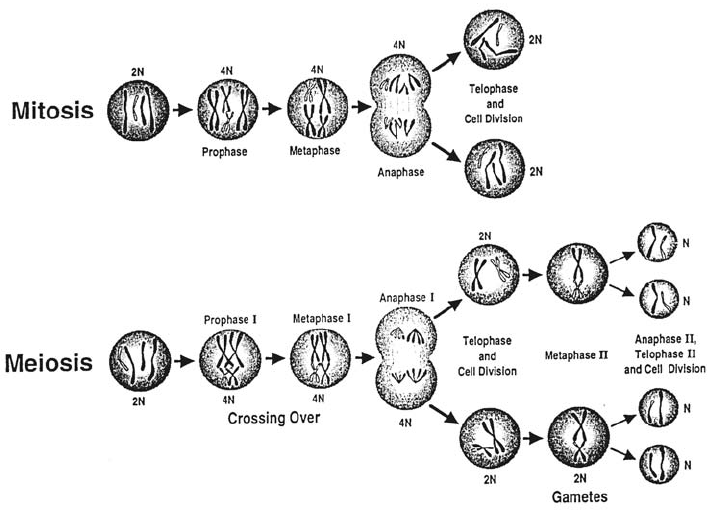
independent assortment of maternal and
paternal homologous chromosomes that
occurs during division I of meiosis. By the
end of meiosis, gametes are formed with an
assortment of maternal and paternal chro-
mosomes. The other type of diversity oc-
curs when homologous pairs of chromo-
somes line up during the first prophase.
The homologous chromosomes genetically
recombine in a process called chromo-
somal crossing over (see below). Therefore,
not only are the chromosomes shuffled
during meiosis, but also portions of chro-
mosomes are recombined to shuffle the
genes of an individual chromosome.
Genetics and Heredity
Alleles
Alternative forms of genes, any one of
which may occupy a single locus on ho-
mologous chromosomes, are called al-
leles. The ISBT terminology distinguishes
between the alleles for blood group anti-
gens (ie, the genetic polymorphisms) and
the antigens that they encode. For exam-
ple, the major antigens of the ABO system
are A, B, and O, yet the alleles are A1,B1,
and O1. In the Kell system, two alleles, K
and k, determine the K and k antigens, re-
spectively. Individuals who have identical
alleles at a given locus on both chromo-
somes are homozygous for the allele (eg,
A1/A1or K/K or k/k). In the heterozygous
condition, the alleles present at the par-
ticular locus on each chromosome are
nonidentical (eg, A1/O1or A1/B1or K/k). Ta-
ble 10-1 summarizes genotypes and chro-
mosomal locations for the 29 blood group
antigen systems.
Individuals who are homozygous for an
allele in some blood group systems may
have more antigen expressed on their red
cells than persons who are heterozygous for
that allele. For example, red cells from a
Chapter 10: Blood Group Genetics 225
Figure 10-2. The two types of cell division are mitosis and meiosis.
Copyright © 2005 by the AABB. All rights reserved.

226 AABB Technical Manual
Table 10-1. Chromosomal Locations of Human Blood Group System Genes*
System ISBT No.
ISBT
Symbol
Gene(s)
Designation
(ISGN) Location
ABO 001 ABO
ABO
9q34.2
MNS 002 MNS
GYPA, GYPB, GYPE
4q28.2-q31.1
P 003 P1
P1
22q11.2-qter
Rh 004 RH
RHD, RHCE
1p36.13-p34.3
Lutheran 005 LU
LU
19q13.2
Kell 006 KEL
KEL
7q33
Lewis 007 LE
FUT3
19p13.3
Duffy 008 FY
DARC
1q22-q23
Kidd 009 JK
SLC14A1
18q11-q12
Diego 010 DI
SLC4A1
17q21-q22
Yt 011 YT
ACHE
7q22.1
Xg 012 XG
XG
Xp22.32
Scianna 013 SC
SC
1p34
Dombrock 014 DO
DO
12p12.3
Colton 015 CO
AQP1
7p14
Landsteiner-Wiener 016 LW
LW
19p13.3
Chido/Rodgers 017 CH/RG
C4A, C4B
6p21.3
H 018 H
FUT1
19q13.3
Kx 019 XK
XK
Xp21.1
Gerbich 020 GE
GYPC
2q14-q21
Cromer 021 CROM
DAF
1q32
Knops 022 KN
CR1
1q32
Indian 023 IN
CD44
11p13
OK 024 OK
CD147
19p13.3
RAPH 025 MER2
MER2
11p15.5
JMH 026 JMH SEMA7A 15q22.3-q23
I 027 I CGNT2 6p24
Globoside 028 P B3GALT3 3q25
GIL 029 GIL AQP3 9q13
*Modified from Zelinski1; Garratty et al2; and Denomme et al.3
Copyright © 2005 by the AABB. All rights reserved.
person whose phenotype is Jk(a+b–) have a
“double dose” of the Jkaallele and, as a re-
sult, express more Jkaantigenonthered
cell surface than an individual whose phe-
notype is Jk(a+b+) (a single dose of the Jka
allele). The difference in amount of antigen
expressed on the red cell membrane be-
tween a homozygous and a heterozygous
phenotype can often be detected serologi-
cally and is termed the dosage effect. For
example, some anti-Jkasera may give the
following pattern of reactivity:
Phenotype of RBC Donor
Antibody Jk(a+b–) Jk(a+b+)
Anti-Jka3+ 2+
Dosage effect is not seen with all blood
group antigens or even with all antibodies
of a given specificity. Antibodies that typi-
callydemonstratedosageincludethosein
the Rh, MNS, Kidd, and Duffy blood group
systems.
Alleles arise by genetic changes at the
DNA level and may result in a different ex-
pressed phenotype. Some of the changes
found among blood group alleles may re-
sult from:
■Missense mutations (a single nucle-
otide substitution leading to the
coding of a different amino acid)
■Nonsense mutations (a single nucle-
otide substitution leading to the
coding of a stop codon)
■Mutations in motifs involved in tran-
scription
■Mutations leading to alternate RNA
splicing
■Deletion of a gene, exon, or nucleo-
tide(s)
■Insertionofanexonornucleotide(s)
■Alternate transcription initiation site
■Chromosome translocation
■Gene conversion or recombination
■Crossing over
Mutations may result in the creation of
new polymorphisms associated with the
altered gene. Figure 10-3 illustrates how
mutations in the genes that code for the
MNS blood group antigens have resulted
in the creation of various low-incidence
MNS system antigens.
Allele (Gene) Frequencies
The frequency of an allele (or its gene fre-
quency) is the proportion that it contrib-
utes to the total pool of alleles at that lo-
cuswithinagivenpopulationatagiven
time. This frequency can be calculated
from phenotype frequencies observed
within a population. The sum of allele fre-
quencies at a given locus must equal 1.
The Hardy-Weinberg Law
The Hardy-Weinberg law is based on the
assumption that genotypes are distrib-
uted in proportion to the frequencies of
individual alleles in a population and will
remain constant from generation to gen-
eration if the processes of mutation, mi-
gration, etc do not occur. For example, the
Kidd blood group system is basically a
two-allele system (Jkaand Jkb; the silent Jk
allele is extremely rare) that can be used
to illustrate the calculation of gene fre-
quencies. This calculation uses the Hardy-
Weinberg equation for a two-allele sys-
tem. If p is the frequency of the Jkaallele
and q is the frequency of the Jkballele,
then the frequencies of the three combi-
nations of alleles can be represented by
the equation p2+2pq+q
2= 1 where:
p = frequency of Jkaallele
q = frequency of Jkballele
p2=frequencyofJka/Jkagenotype
2pq = frequency of Jka/Jkbgenotype
q2=frequencyofJkb/Jkbgenotype
Chapter 10: Blood Group Genetics 227
Copyright © 2005 by the AABB. All rights reserved.
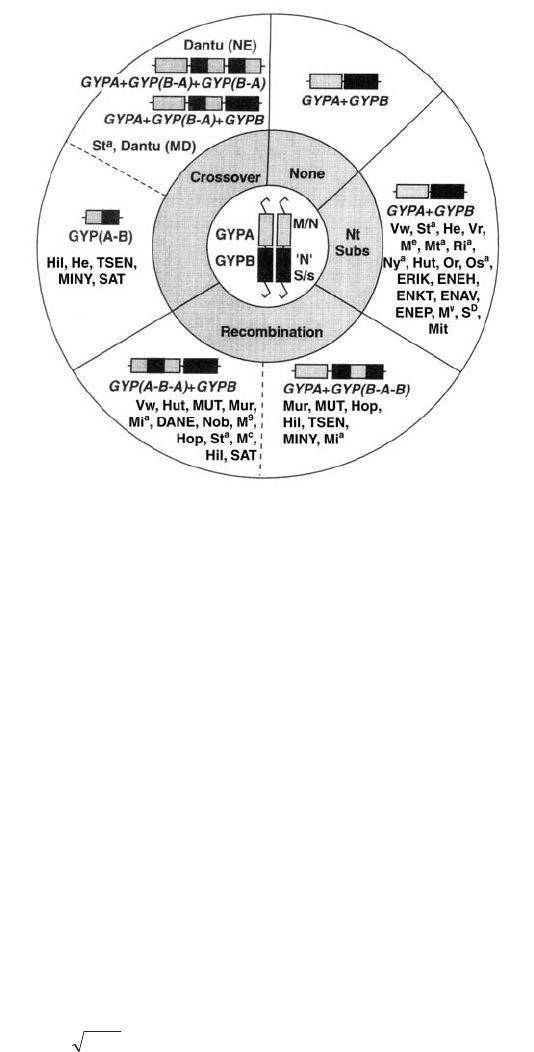
Using the observation that 77% of indi-
viduals within a population express Jkaanti-
gen on their red cells, then:
p2+2pq = frequency of
persons who
are Jk(a+) and
carry the Jkaallele
=0.77
q2=1–(p
2+2pq)= frequencyof
persons who
are Jk(a–)
(homozygous for
the Jkballele)
q2= 1 – 0.77 = 0.23
q=
0.23
q = 0.48 (allele
frequency of Jkb)
Because the sum of frequencies of both
alleles must equal 1.00,
p+q = 1
p=1–q
p = 1 – 0.48
p = 0.52 (allele frequency of Jka)
Once the allele frequencies have been
calculated, the number of Jk(b+) individu-
als (both homozygous and heterozygous)
can be calculated as:
2pq + q2= frequency of Jk(b+)
=2(0.52×0.48) + (0.48)2
=0.73
If both anti-Jkaand anti-Jkbsera are avail-
able, allele frequencies can be determined
more easily by direct counting. As shown in
Table 10-2, the random sample of 100 peo-
ple tested for Jkaand Jkbantigens possess a
total of 200 alleles at the Jk locus (each per-
son inherits two alleles, one from each par-
ent). There are two Jkaalleles inherited by
228 AABB Technical Manual
Figure 10-3. How crossover, recombination, and nucleotide substitution (nt subs) result in variations
of genes producing glycophorin A and B. The changes are associated with the presence of various
low-incidence MNS system antigens. (Modified from Reid.4)
Copyright © 2005 by the AABB. All rights reserved.

each of the 28 individuals who phenotype
as Jk(a+b–), for a total of 56 alleles. There
are 49 Jkaalleles in the individuals who are
Jk(a+b+), for a total of 105 alleles or a gene
frequency of 0.52 (105 ÷ 200). The fre-
quency of Jkbis 95 ÷ 200 = 0.48.
The Hardy-Weinberg law is generally
used to calculate allele and genotype fre-
quencies in a population when the fre-
quency of one genetic trait (eg, antigen
phenotype) is known. However, it relies on
certain assumptions: no mutation; no mi-
gration (in or out) of the population; lack of
selective advantage/disadvantage of a par-
ticular trait; and a large enough population
so that chance alone cannot alter an allele
frequency. If all of these conditions are
present, the gene pool is in equilibrium and
allele frequencies will not change from one
generation to the next. If these assumptions
do not apply, changes in allele frequencies
may occur over a few generations and can
explain many of the differences in allele
frequencies between populations.
Segregation
The term segregation refers to the con-
cept that the two members of a single
gene pair (alleles) are never found in the
same gamete but always segregate and
pass to different gametes. In blood group
genetics, this can be illustrated by the in-
heritance of the ABO alleles (Fig 10-4). In
this example, the members of the paren-
tal generation (P1) are homozygous for an
Aallele and an Oallele. All members of
the first filial generation (F1) will be het-
erozygous (A/O) but will still express the
blood group A antigen (Ois a silent al-
lele). If an F1individual mates with an A/O
genotypic individual, the resulting prog-
eny [termed the second filial generation:
(F2)] will blood group as A (either hetero-
zygous or homozygous) or group O. If the
F1individual mated with a heterozygous
group B person (B/O), the offspring could
have the blood group A, B, AB, or O.
Independent Assortment
Mendel’s law of independent assortment
states that genes determining various
traits are inherited independently from
each other. For example, if one parent is
group A (homozygous for A)andK+k+,
and the other parent is group B (homozy-
gous for B) and K–k+ (homozygous for k),
all the F1children would be group AB; half
would be K+k+ and half K–k+ (Fig 10-5). A
second filial generation could manifest
any of the following phenotypes: group A,
Chapter 10: Blood Group Genetics 229
Table 10-2. Gene Frequencies in the Kidd Blood Group System Calculated Using
Direct Counting Method*
Phenotype
No. of
Individuals
No. of
Kidd Genes
Gene Frequencies (%)
Jk
a
Jk
Jk(a+b–) 28 56 56 0
Jk(a+b+) 49 98 49 49
Jk(a–b+) 23 46 0 46
Totals 100 200 105 95
Gene Frequency 0.525 0.475
*Assumes absence of silent
Jk
allele.
Copyright © 2005 by the AABB. All rights reserved.
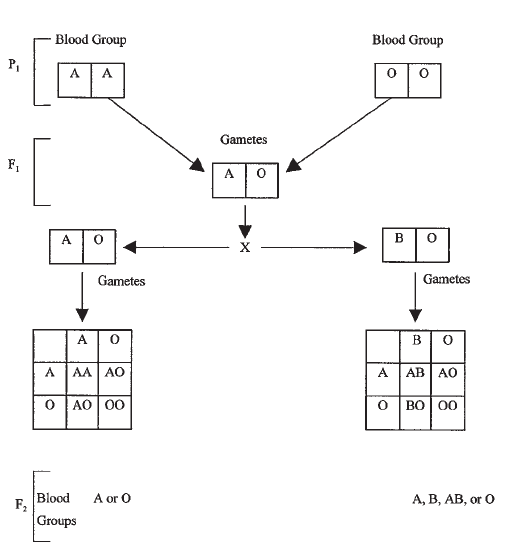
K+k+; group AB, K+k+; group B, K+k+;
group A, K–k+; group AB, K–k+; group B,
K–k+. The proportions would be
1:2:1:1:2:1.
Independent assortment applies if the
genes are on different chromosomes or on
distant portions of the same chromosome.
One exception to this rule is that closely
linked genes on the same chromosome do
not sort independently but often remain to-
gether from one generation to another. This
observation is termed linkage.
Linkage
Genetic linkage is defined as the tendency
for alleles close together on the same
chromosome to be transmitted together.
During mitosis, each pair of homologous
chromosomes undergoes a series of re-
combinations. The resultant reciprocal
exchange of segments between the chro-
matids is termed crossing over (Fig 10-6).
Genes close together on a chromosome
tend to be transmitted together during
these recombinations and their alleles,
therefore, do not segregate independently.
Sometimes, the linkage is very tight so that
recombination rarely occurs. The strength
of linkage can be used as a unit of mea-
surement to estimate the distance between
different loci. This type of analysis can
help in identifying, mapping, and diag-
nosing the genes responsible for certain
inherited diseases.
The demonstration of linkage between
the gene controlling ABH secretion (Se)and
the expression of Lutheran blood group an-
tigens (Lua,Lu
b) was the first recognized ex-
ample of autosomal linkage in humans.5
Analysis of this relationship also provided
the first evidence in humans of recombina-
tion due to crossing-over and helped dem-
230 AABB Technical Manual
Figure 10-4. Mendel’s law of independent segregation demonstrated by the inheritance of ABO genes.
Copyright © 2005 by the AABB. All rights reserved.
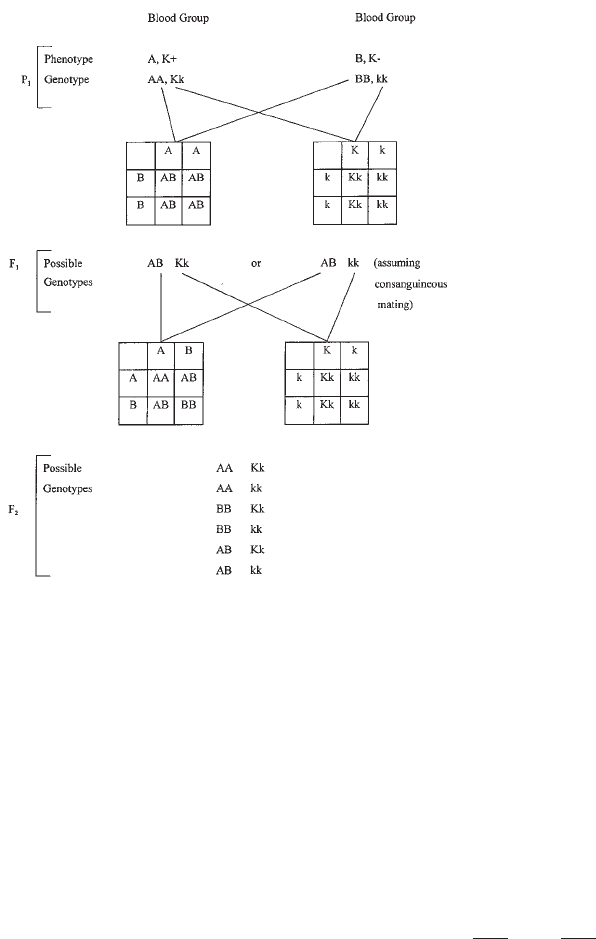
onstrate that crossing-over occurs more
ofteninfemalesthaninmales.
Linkage Disequilibrium
When two loci are closely linked, alleles at
those loci tend to be inherited together
and are said to constitute a haplotype.
Again, the close linkage between the loci
controlling expression of M and N and of
S and s is an example of linkage disequi-
librium. The approximate frequencies of
each of the four alleles are:
M=0.53 S=0.33
N=0.47 s=0.67
If the alleles of the M, N, S, and s anti-
gens segregated independently, the ex-
pected frequency of each haplotype would
be the product of the frequencies of the in-
dividual alleles. However, the frequencies
observed are not those expected:
Expected Observed
Frequency Frequency
MS =0.53×0.33 = 0.17 0.24
Ms =0.53×0.67 = 0.36 0.28
NS =0.47×0.33 = 0.16 0.08
Ns =0.47×0.67 = 0.31 0.40
Total 1.00 1.00
This is an example of linkage disequilib-
rium: the tendency of specific combina-
tions of alleles at two or more linked loci
to be inherited together more frequently
than would be expected by chance.
Another commonly cited example of
linkage disequilibrium occurs in the HLA
system (see Chapter 17). The combination
Chapter 10: Blood Group Genetics 231
Figure 10-5. Mendel’s law of independent assortment demonstrated by the inheritance of ABO and Kell
genes.
Copyright © 2005 by the AABB. All rights reserved.
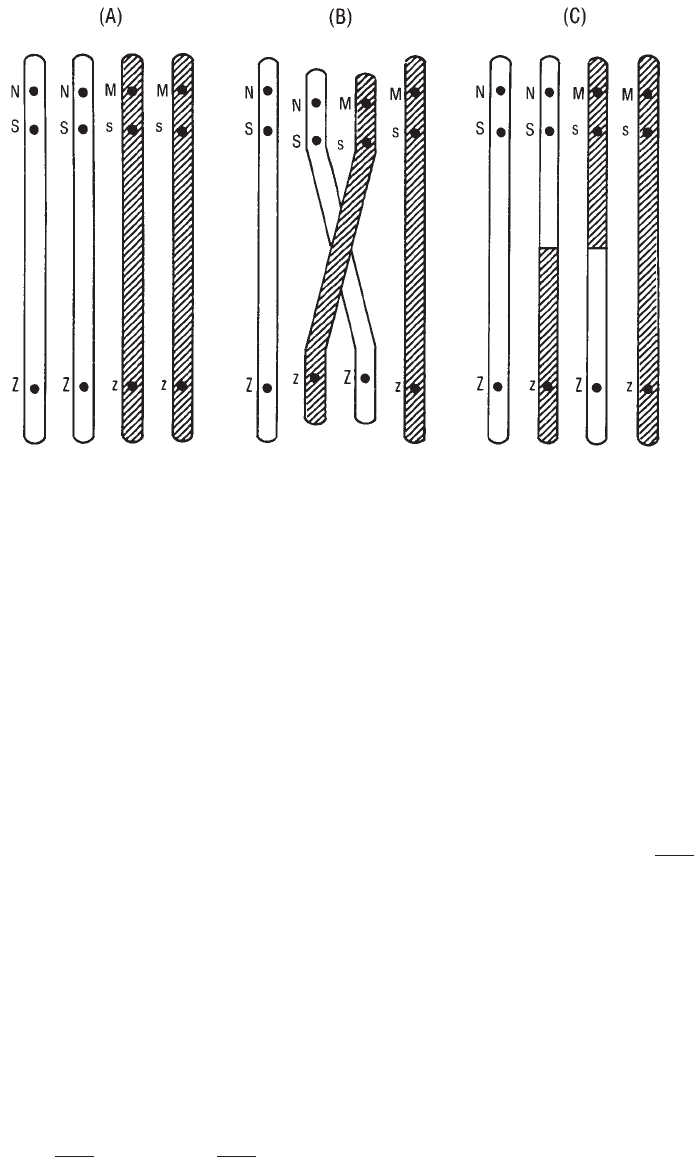
of HLA-A1 with HLA-B8 occurs in some
populations approximately five times more
frequently than would be expected based
on the frequencies of the individual alleles,
an example of positive linkage disequilib-
rium. Linkage disequilibrium may be posi-
tive or negative, and it may indicate a selec-
tive advantage of one haplotype over another.
Over many generations, the alleles of even
closely linked loci may reach equilibrium
and associate according to their individual
frequencies in the population.
When there is linkage equilibrium, the
alleles at two loci associate with frequen-
cies that reflect their individual frequencies.
For example, if alleles in the population
have the following frequencies:
Y0.53 Z0.30
y0.47 z0.70
Total 1.00 1.00
then the frequencies of the combination
should be the product of the frequency of
each allele:
YZ 0.53 ×0.3 = 0.16
Yz 0.53 ×0.7 = 0.37
yZ 0.47 ×0.3 = 0.14
yz 0.47 ×0.7 = 0.33
Total 1.00
In such a case, the alleles are in linkage
equilibrium because they are inherited
independently.
Patterns of Inheritance
Dominant and Recessive Traits
Traits are the observed expression of genes.
A trait that is observable when the deter-
mining allele is present is called domi-
232 AABB Technical Manual
Figure 10-6. Very closely linked loci are rarely affected by crossing over so that alleles of those loci
are inherited together (N and S, M and s in the example shown). Loci on the same chromosome that
are not closely linked (the
Ss
locus and the
Zz
locus shown) can demonstrate crossing over. Crossing
over is one kind of recombination. It occurs between homologous chromatids during meiosis, resulting
in segregation of alleles on the same chromosome.
Copyright © 2005 by the AABB. All rights reserved.
nant; when different alleles on homologous
chromosomes each produce an observable
trait, the term co-dominant is used. A re-
cessive trait is observable only when the
allele is not paired with a dominant allele
(two recessive alleles are present). De-
scribing traits as dominant and recessive
depends on the method used to detect gene
products. Observable traits are called phe-
notypes. Thus, blood group antigen typing
using antisera identifies a phenotype. In
some cases, genotypes may be inferred
from the phenotype, especially when
family studies are performed, but geno-
types are not usually determined directly
by typing red cells.
Autosomal Dominant Trait
An autosomal dominant trait shows a
characteristic pattern of inheritance. The
trait appears whenever an individual pos-
sesses the allele. Figure 10-7(A) presents a
pedigree showing the pattern of auto-
somal dominant inheritance. Typically,
each person with the trait has at least one
parent with the trait, continuing back-
ward through generations.
Autosomal Recessive Trait
People who exhibit a recessive trait are
homozygous for the encoding allele. Their
parents may or may not express the trait.
However, parents who lack the trait must
be carriers, ie, heterozygotes for an allele
whose presence is not phenotypically
apparent.
If the frequency of the variant allele is
low,therecessivetraitwillberareandgen
-
erally will occur only in members of one
generation, not in preceding or successive
generations unless consanguineous mating
occurs. Blood relatives are more likely to
carry the same rare allele than unrelated
persons from a random population. When
offspring are homozygous for a rare allele
(frequency: <1:10,000) and display the trait,
the parents are often blood relatives [Fig
10-7(B)]. Recessive traits may remain unex-
pressed for many generations, so that the
appearance of a rare recessive trait does not
necessarily imply consanguinity, although
family ethnicity and geographic origin may
be informative. A higher frequency for a re-
cessive allele indicates the less likelihood of
consanguinity. Traits inherited in either
autosomal dominant or autosomal reces-
sive fashion typically occur with equal fre-
quency in males and females.
Sex-Linked Dominant or Co-dominant
Trait
A male always receives his single X chro-
mosome from his mother. The predomi-
nant feature of X-linked inheritance, of
either dominant or recessive traits, is ab-
sence of male-to-male (father-to-son)
transmission of the trait. Because a male
passes his X chromosome to all his dau-
ghters, all daughters of a man expressing
adominant X-linked trait also possess the
allele and the trait. If a woman expresses a
dominant trait, but is heterozygous, each
child, male or female, has a 50% chance of
inheriting that allele and thus the trait
[Fig 10-7(C)]. If the mother possesses the
determining allele on both X chromo-
somes, all her children will express the
trait. X-linked dominant traits tend to ap-
pear in each generation of a kindred, but
without male-to-male transmission. A
sex-linked dominant trait of interest in
blood group genetics is the Xg blood
group system.
Sex-Linked Recessive Trait
Hemophilia A provides a classic example
of X-linked recessive inheritance [Fig
10-7(D)]. Males inherit the trait from car-
rier mothers or, very rarely, from a mother
who is homozygous for the allele and
Chapter 10: Blood Group Genetics 233
Copyright © 2005 by the AABB. All rights reserved.
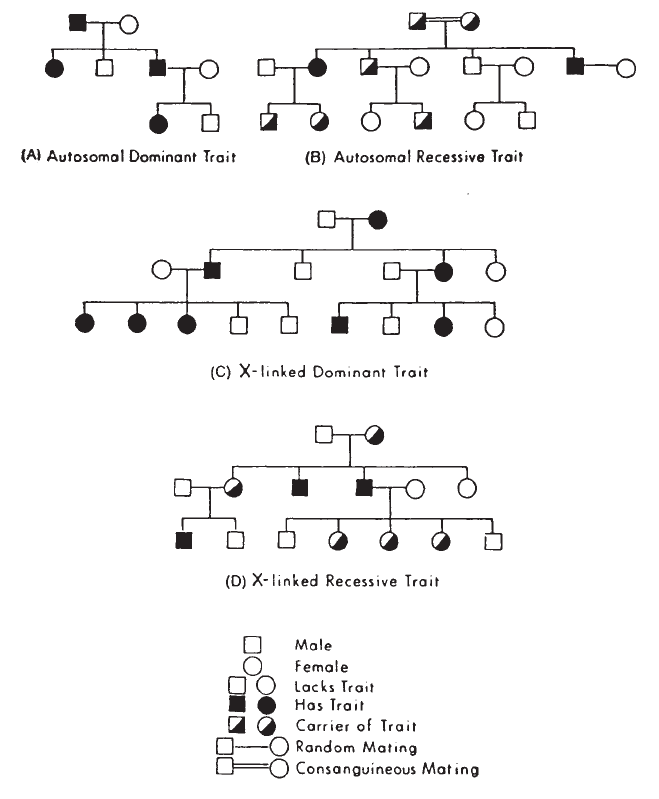
therefore expresses the trait. In the mat-
ing of a normal male and a carrier female,
one half of the male offspring are affected
and one half of the females are carriers.
Among the children of an affected male
and a female who lacks the determining
allele, all sons are normal and all daugh-
ters are carriers.
If the recessive X-linked allele is rare, the
trait will be exhibited almost exclusively in
males. If the X-linked allele occurs more
frequently in the population, affected fe-
males will be seen because the likelihood
increases that an affected male will mate
with a carrier female and produce daugh-
ters, half of whom will be homozygous for
the abnormal allele.
Blood Group Co-dominant Traits
Blood group antigens, as a rule, are ex-
pressed as co-dominant traits: heterozy-
gotes express the products of both alleles.
If an individual’s red cells type as both K+
and k+, the K/k genotype may be inferred.
234 AABB Technical Manual
Figure 10-7. Four pedigrees showing different patterns of inheritance.
Copyright © 2005 by the AABB. All rights reserved.
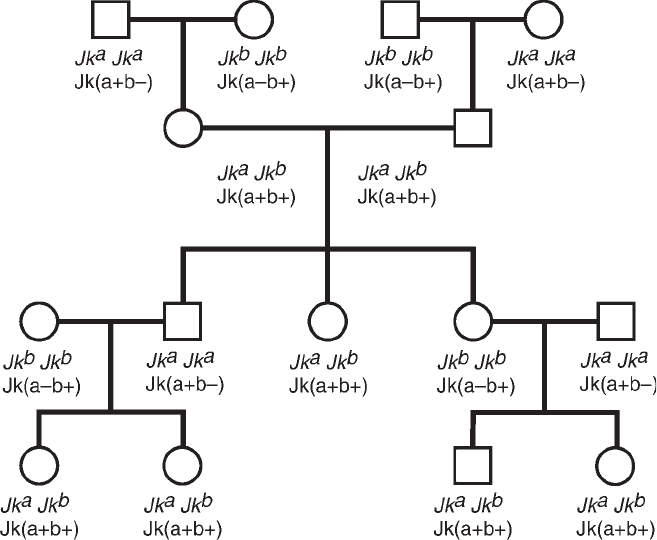
Figure 10-8 shows the inheritance patterns
of the two active alleles of the Kidd blood
group system (Jkaand Jkb)andtheco-
dominant phenotypic expression of the
two respective antigens Jkaand Jkb.
In the ABO system, the situation is more
complex. The genes of the ABO system do
not code for membrane proteins but con-
trol production of enzymes termed glyco-
syltransferases. These enzymes add specific
sugars to a precursor structure on the red
cell membrane, resulting in specific antigen
expression. In an A1/A2heterozygote, the
phenotype is A1; the presence of the A2al-
lele cannot be inferred. Although the A1al-
lele appears dominant to that of the A2al-
lele by simple cell typing, techniques that
identify the specific transferases reveal that
an A1/A2heterozygote does generate the
products of both alleles, ie, both A1and A2
transferases. Similarly, in an A2/O person, A2
is dominant to O.TheOallele codes for a
specific protein, but this protein (trans-
ferase) is nonfunctional. The presence of
ABO genes can be demonstrated by molec-
ular techniques (see Chapter 13).
Chromosomal Assignment
The loci of all major blood group genes
have been mapped to one or another of
the 22 pairs of autosomes, as shown in Ta-
ble 10-1. The Xg and XK loci are the only
bloodgroupgenesmappedtotheXchro
-
mosome.
Interaction among alleles or the prod-
ucts of different genes may modify the ex-
pression of a trait. The terms “suppressor”
and “modifier” are used to describe genes
that affect the expression of other genes;
Chapter 10: Blood Group Genetics 235
Figure 10-8. Inheritance and co-dominant expression of Kidd blood group antigens.
Copyright © 2005 by the AABB. All rights reserved.
however, the mechanism of these postu-
lated gene interactions is not always fully
understood. Some observations in blood
group serology have been explained by
gene interaction: weakening of the D anti-
gen expression when the Callele is present
in cis (on the same chromosome) or in
trans (on the paired chromosome),6and the
suppression of Lutheran antigen expression
by the dominant modifier gene, In(Lu).7
When products of two different genes
are important in the sequential develop-
ment of a biochemical end product, the
gene interaction is called epistasis. Failure
to express A or B antigens if H substance
has not first been produced (absence of the
Hgene) is an example of epistasis. A muta-
tion database of gene loci encoding com-
mon and rare blood group antigens has
been established (Blood Group Antigen
Mutation Database) and is available on the
Internet (see http://www.bioc.aecom.yu.
edu/bgmut/index.htm).
Population Genetics
Some understanding of population genet-
ics is essential for parentage testing and
helpful in such clinical situations as pre-
dicting the likelihood of finding blood
compatible with a serum that contains
multiple antibodies. Calculations use
published phenotype frequencies.
Phenotype Frequencies
The frequencies of blood group pheno-
types are obtained by testing many ran-
domly selected people of the same race or
ethnic group and observing the propor-
tion of positive and negative reactions
with a specific blood group antibody. In a
blood group system, the sum of pheno-
type frequencies should equal 100%. For
example, in a Caucasian population, 77% of
randomly selected individuals are Jk(a+).
The frequency of Jk(a–) individuals should
be 23%. If blood is needed for a patient
with anti-Jka, 23% or approximately one in
four ABO-compatible units of blood should
be compatible.
Calculations for Combined Phenotypes
If a patient has multiple blood group anti-
bodies, it may be useful to estimate the
number of units that will have to be tested
in order to find units of blood negative for
all the antigens. For example, if a patient
has anti-c, anti-K, and anti-Jka,howmany
ABO-compatible units of blood would
have to be tested to find 4 units of the ap-
propriate phenotype?
Phenotype Frequency (%)
c– 20
K– 91
Jk(a–) 23
To calculate the frequency of the com-
bined phenotype, the individual frequen-
cies are multiplied because the phenotypes
are independent of one another. Thus, the
proportion of persons who are c– is 20%. Of
the 20% of c– individuals, 91% are K–;
hence, 18% (0.20 ×0.91 = 0.18) are c– and
K–. Of this 18% of c–K– individuals, 23%
will be Jk(a–); therefore, only 4% of individ-
uals will have c–K–Jk(a–) blood (0.2 ×
0.91 ×0.23 = 0.04). Therefore, of 100 units
tested, 4 compatible units should be found.
Calculations such as this influence deci-
sions about asking for assistance from the
local blood supplier or reference laboratory
when trying to find compatible blood for an
alloimmunized patient.
Parentage Testing
Blood group antigens, many of which are
expressed as co-dominant traits with sim-
ple Mendelian modes of inheritance, are
useful in parentage analyses. If one as-
236 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
sumes maternity and that test results are
accurate, paternity can be excluded in
either of two ways:
1. Direct exclusion of paternity is es-
tablished when a genetic marker is
present in the child but is absent
from the mother and the alleged fa-
ther. Example:
Blood Group Phenotype
Child Mother Alleged Father
BO O
The child has inherited a Bgene, which
could not be inherited from either the
mother or the alleged father, assuming that
neither the mother nor the alleged father is
oftherareO
hphenotype. Based on the phe-
notypes of mother and child, the Bgene
must have been inherited from the biologic
father and is called a paternal obligatory gene.
2. Exclusion is indirect when the child
lacks a genetic marker that the alleged
father (given his observed pheno-
type)musttransmittohisoffspring.
Example:
Blood Group Phenotype
Child Mother Alleged Father
Jk(a+b–) Jk(a+b–) Jk(a–b+)
In this case, the alleged father is presum-
ably homozygous for Jkbandshouldhave
transmitted Jkbto the child.
Direct exclusion is more convincing than
indirect exclusion when trying to establish
parentage. Apparent indirect exclusion can
sometimes result from the presence of a si-
lent allele. In the example above, the al-
leged father could have one silent allele
(Jk), which was transmitted to the child.
Thechild’sgenotypecouldbeJkaJk instead
of the far more common JkaJka. Interpreta-
tion of phenotypic data must take into ac-
count all biologic and analytic factors
known to influence results.
When the alleged father cannot be ex-
cluded from paternity, it is possible to cal-
culate the probability of paternity. The
probability that the alleged father transmit-
ted the paternal obligatory genes is com-
pared with the probability that any other
randomly selected man from the same eth-
nic/racial population could have transmit-
ted the genes. The result is expressed as a
likelihood ratio (paternity index) or as a
percentage (posterior probability of pater-
nity given some prior probability). Methods
for parentage analysis often include the
study of many genetic systems other than
red cell blood groups [ie, HLA and short
tandem repeat (STR) systems]. Many par-
entage testing laboratories employ the STR
method of DNA analysis (see Chapter 9) as
a means of evaluating cases of disputed
parentage. The AABB has developed stan-
dards for laboratories that perform parent-
age studies.8
Chimerism
A chimera is one whose cells are derived
from more than one distinct zygotic line.
Although rare, this may occur when an
anastomosis occurs within the vascular
tissues of twin embryos, or when two fer-
tilized zygotes fuse to form one individ-
ual. This condition, although not heredi-
tary, leads to dual (multiple) phenotypic
populations of cells within one individual.
Blood types of such rare individuals may
demonstrate a mixed-field appearance,
with distinct populations of cells of the
person’s true genetic type, as well as cells
of the implanted type. Chimeras also
demonstrate immune tolerance: a geneti-
cally group O person with implanted A
cells does not produce anti-A. More com-
monly, chimeras are artificial and arise
from the transfer of actively dividing cells,
eg, through hematopoietic transplanta-
tion (see Chapter 25).
Chapter 10: Blood Group Genetics 237
Copyright © 2005 by the AABB. All rights reserved.
238 AABB Technical Manual
Blood Group Nomenclature
The terminology and notations for blood
group systems embody many inconsis-
tencies because blood group serologists
failed to follow conventions of classic
Mendelian genetics. Listed below are a
few examples of the confusion engen-
dered by many decades of uncoordinated
scientific publications.
1. An allele that determines a dominant
trait often is signified by a capital
letter; one that determines a reces-
sive trait is denoted by both lower-
case letters. The Aand Bco-domi-
nant genes of the ABO system are
signified by a capital letter. The O
gene is also given a capital but does
not present as a dominant trait.
Without prior knowledge, it would
be impossible for one to recognize
that these notations represent allelic
products in a blood group system.
2. Some co-dominant traits have been
designated with capital letters and
allelic relationships with lowercase
letters; for example, Kand kof the
Kell blood group system and Cand c
of the Rh system.
3. Some co-dominant traits have iden-
tical base symbols but different su-
perscript symbols, such as Fyaand
Fyb(Duffy system) and Luaand Lub
(Lutheran system).
4. In some allelic pairs, the lower fre-
quency antigen is expressed with an
“a” superscript ( Wrahas a lower fre-
quency than Wrb). In other allelic
pairs, the “a” superscript denotes the
higher incidence antigen (Coahas a
higher frequency than Cob).
5. Some authors have denoted the ab-
sence of a serologic specificity with a
base symbol devoid of superscripts,
and others use a lowercase version
ofthebasesymbol.IntheLutheran
system, the assumed amorphic gene
is called Lu,notlu,whereasthe
amorph in the Lewis system is le.
6. Numeric terminology was intro-
duced for some blood group sys-
tems, resulting in mixtures of letters
and numbers for antigen designa-
tions, eg, K, Kpa, and K11.
Colloquial use of these terminologies,
even in some published articles and texts,
has compounded their improper use. Early
model computers or printers also did not
easily accept certain terminologies (eg, su-
perscripts, subscripts, unusual fonts).
In recent years, concerted attempts have
been made to establish rational, uniform
criteria for the notations used to designate
phenotype, genotype, and locus informa-
tion for blood group systems. Issitt and
Crookston9and Garratty et al2presented
guidelines for the nomenclature and termi-
nology of blood groups. The International
Society of Blood Transfusion (ISBT) Work-
ing Party on Terminology for Red Cell Sur-
face Antigens has provided a standardized
system for classifying blood group antigens
(see Appendix 6). Similar international
committees have established principles for
assigning nomenclature of the hemoglo-
bins, immunoglobulin allotypes, histo-
compatibility antigens, clusters of differen-
tiation, STR sequences, and other serum
protein and red cell enzyme systems. Al-
though many of the older terminologies
must be retained to avoid even further
confusion, common conventions now exist
for correct usage.
The ISBT terminology for red cell anti-
gens was devised as a numeric nomencla-
ture suitable for computerization. A six-
digit designation indicates each blood
group specificity. The first three numbers
identify the blood group system and the
last three numbers identify the individual
specificity. This numeric terminology is de-
signed mainly for computer databases and
Copyright © 2005 by the AABB. All rights reserved.
is not necessarily intended to supplant more
common usage.
For ISBT classification, each defined
blood group system must be genetically
distinct. Assignment of antigens to a spe-
cific blood group system is dependent on
genetic, serologic, and/or biochemical rela-
tionships. Gene cloning has made the task
of assignment more definitive and has al-
lowed some designations previously un-
proved by traditional family studies (ie, the
expansion of the Diego system to include a
number of low-incidence antigens).
Some antigens, however, have not yet
been proven to be part of a recognized sys-
tem. Collections (termed the 200 series) are
apparently related sets of antigens for which
definitive genetic information is lacking.
Other isolated antigens of high (901 series)
or low (700 series) incidence are listed to-
gether until genetic information becomes
available. In recent years, the number of
antigens in these three series has dramati-
cally declined as further genetic and bio-
chemical data allow reassignment.
Correct Terminology
The following are accepted conventions
for expressing red cell antigen pheno-
types and genotypes.2
1. Genes encoding the expression of
blood group antigens are written in
italics (or underlined if italics are not
available). If the antigen name in-
cludes a subscript (A1), generally the
encoding gene is expressed with a
superscript (A1).
2. Antigen names designated by a su-
perscript or a number (eg, Fya,Fy:1)
arewritteninnormal(Roman)script.
Numeric designations are written on
thesamelineastheletters.Super
-
script letters are lowercase. (Some
exceptions occur, based on historic
usage: hrS,hr
B.)
3. When antigen phenotypes are ex-
pressed using single letter designa-
tions, results are usually written as +
or –, set on the same line as the let-
ter(s) of the antigen: K+ k–.
4. To express phenotypes of antigens
designated with a superscript letter,
that letter is placed in parentheses
onthesamelineasthesymbolde
-
fining the antigen: Fy(a+) and Fy(a–).
5. For antigens designated by num-
bers, the symbol defining the system
is notated in capital letters followed
by a colon, followed by the number
representing the antigen tested. Plus
signs do not appear when test re-
sults are positive (K:1), but a minus
sign is placed before negative test re-
sults: K:1, K:–1. If tests for several an-
tigens in one blood group have been
done, the phenotype is designated
by the letter(s) of the locus or blood
group system followed by a colon,
followed by antigen numbers sepa-
rated by commas: K:–1,2,–3,4. Only
antigens tested are listed; if an anti-
body defining a specific antigen was
not tested, the number of the anti-
gen is not listed: K:–1,–3,4.
Although numeric terminology has been
devised for various systems and antigens, it
shouldnotbeassumedthatitmustreplace
conventional terminology. The use of con-
ventional antigen names is also acceptable.
In some systems, notably Rh, multiple ter-
minologies exist and not all antigens within
the system have names in each type.
References
1. Zelinski T. Chromosomal localization of hu-
man blood group genes. In: Silberstein LE, ed.
Molecular and functional aspects of blood
group antigens. Bethesda, MD: AABB, 1995:
41-73.
2. Garratty G, Dzik W, Issitt PD, et al. Terminol-
ogy for blood group antigens and genes—his-
Chapter 10: Blood Group Genetics 239
Copyright © 2005 by the AABB. All rights reserved.
torical origins and guidelines in the new mil-
lennium. Transfusion 2000;40:477-89.
3. Denomme G, Lomas-Francis C, Storry JR, Reid
ME. Approaches to blood group molecular
genotyping and its applications. In: Stowell C,
Dzik W, eds. Emerging diagnostic and thera-
peutic technologies in transfusion medicine.
Bethesda, MD: AABB Press, 2003: 95-129.
4. Reid ME. Molecular basis for blood groups and
functions of carrier proteins. In: Silberstein
LE, ed. Molecular and functional aspects of
blood group antigens. Bethesda, MD: AABB,
1995:75-125.
5. Mohr J. A search for linkage between the Lu-
theran blood group and other hereditary
characters. Acta Path Microbiol Scand 1951;
28:207-10.
6. Araszkiewicz P, Szymanski IO. Quantitative
studies on the Rh-antigen D effect of the C
gene. Transfusion 1987;27:257-61.
7. CrawfordNM,GreenwaitTJ,SasakiT.The
phenotype Lu(a–b–) together with unconven-
tional Kidd groups in one family. Transfusion
1961;1:228-32.
8. Gjertson D, ed. Standards for parentage test-
ing laboratories. 6th ed. Bethesda, MD: AABB,
2004.
9. Issitt PD, Crookston MC. Blood group termi-
nology: Current conventions. Transfusion 1984;
24:2-7.
240 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Appendix 10-1. Glossary of Terms in Blood Group Genetics
Allelic: Pairs of genes located at the same site on chromosome pairs.
Centromere: A constricted region of a chromosome that connects the chroma-
tids during cell division.
Chromatid: One of the two potential chromosomes formed by DNA replication
of each chromosome before mitosis and meiosis. They are joined
together at the centromere.
Chromatin: The deeply staining genetic material present in the nucleus of a cell
that is not dividing.
Chromosome: A linear thread made of DNA in the nucleus of the cell.
Co-dominant: A gene that expresses a trait regardless of whether or not an alter-
native allele at the same locus is also expressed on the other paren-
tal chromosome.
Crossing over: The process of breaking single maternal and paternal DNA double
helices in each of two chromatids and rejoining them to each other
in a reciprocal fashion, which results in the exchange of parts of ho-
mologous chromosomes.
Dominant: A gene that expresses a trait that does not allow the expression of a
trait encoded by an alternative allele at the same locus on the other
parental chromosome.
Gene: The basic unit of heredity, made of DNA. Each gene occupies a spe-
cific location on a chromosome.
Locus: The site of a gene on a chromosome.
Lyonization: TheinactivationofoneofthefemaleXchromosomesduring
embryogenesis. This inactivated chromosome forms the Barr body
in the cell nucleus.
Meiosis: A process of two successive cell divisions producing cells, egg, or
sperm that contain half the number of chromosomes found in so-
matic cells.
Mitosis: Division of somatic cells resulting in daughter cells containing the
same number of chromosomes as the parent cell.
Recessive: A gene that in the presence of its dominant allele does not express
itself. A recessive trait is apparent only if both alleles are recessive.
Sex-linked: A gene contained within the X or Y chromosome.
Somatic cell: Nonreproductive cells or tissues.
X-linked: A gene on the X chromosome for which there is no corresponding
gene on the Y chromosome.
Chapter 10: Blood Group Genetics 241
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 11: Immunology
Chapter 11
Immunology
THE IMMUNE RESPONSE is a
highly evolved innate and adap-
tive system that is fundamental for
survival. It has a sophisticated ability to
distinguish self from nonself and provides
a memory bank that allows the body to
rapidly respond to recurring foreign or-
ganisms. A healthy immune response can
recognize foreign material or pathogens
that invade the body and can initiate a se-
ries of events to eliminate these pathogens
with minimal or no prolonged morbidity
to the host.
The numerous components of the im-
mune system work in delicate balance to
ensure a state of health. This may include
destruction of abnormal/malignant cells,
removal of harmful bacteria or viruses,
and/or an inflammatory response to pro-
mote healing. If the immune system be-
comes hyperreactive, the body may attack
itsowntissueororgans(autoimmunedis
-
ease), or allergies may develop. If hyporeac-
tive, the host may be susceptible to a wide
variety of infectious agents or proliferation
of malignant cells. The ultimate goal of im-
mune activity is to maintain this delicate
balance.
Immune Response
The immune response can be classified
into two categories: the innate response
and the adaptive (acquired) response. In-
nate responses are indiscriminate: the
same mechanisms can be deployed against
invasive organisms or harmful stimuli. In
contrast, the adaptive response recognizes
specific features of the harmful stimuli
and provides a customized response based
on previous experiences (Fig 11-1). The
adaptive response is a late evolutionary
development, found only in vertebrates.
Innate immunity, on the other hand, uses
universal properties and processes such as
epithelial barriers, proteolytic enzymes,
243
11
Copyright © 2005 by the AABB. All rights reserved.
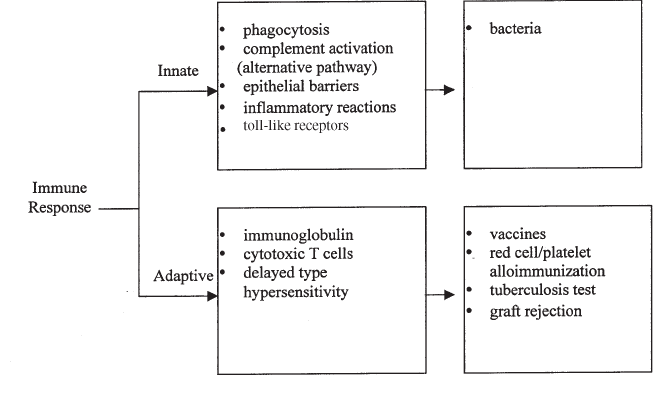
cellular phagocytosis, and inflammatory
reactions. It is important to note that in-
nate and adaptive immunity are comple-
mentary—not mutually exclusive—immune
responses. Regardless of the classification,
the immune response reflects the complex
interaction of cells, tissues, organs, and
soluble factors. Appendix 11-1 describes
some frequently used immunology terms.
Immunoglobulin Superfamily
Molecules that belong to the immuno-
globulin superfamily (IgSF) of receptors
play a critical role in the recognition of
foreign antigens. Antigen recognition is
accomplished through receptors that are
found in a soluble form and on the sur-
face of lymphoid cells. Figure 11-2 illus-
trates the similar structure of some of
these immunology receptors. The overall
structure of these receptors is very simi-
lar, and it has been proposed that they
arose from a common ancestral gene. The
basic structure is similar to the domains
of the immunoglobulin molecule; hence,
these receptors have been grouped into a
single superfamily of molecules. Examples
of these receptors include: immunoglobu-
lins, T-cell receptor (TCR), major histo-
compatibility complex (MHC) Class I and
Class II molecules, and receptors for growth
factors and cytokines. There are several
hundred members of the IgSF (ie, immu-
noglobulin receptors, integrins, etc).2
Major Histocompatibility Complex
The MHC is a large cluster of genes. In
humans, the MHC includes the genes of
the HLA system and genes that encode
other proteins such as tumor necrosis fac-
tor (TNF), some complement compo-
nents, and some heat shock proteins.3The
genes that encode MHC molecules are lo-
cated on the short arm of chromosome 6.
There are three classes of these molecules.
MHC Class I Molecules
MHC Class I molecules are found on al-
most all cells in the body. The molecules
are defined by three major Class I genes
244 AABB Technical Manual
Figure 11-1. Examples of the factors used in innate and adaptive immunity and examples of the two
types of immunity.
Copyright © 2005 by the AABB. All rights reserved.
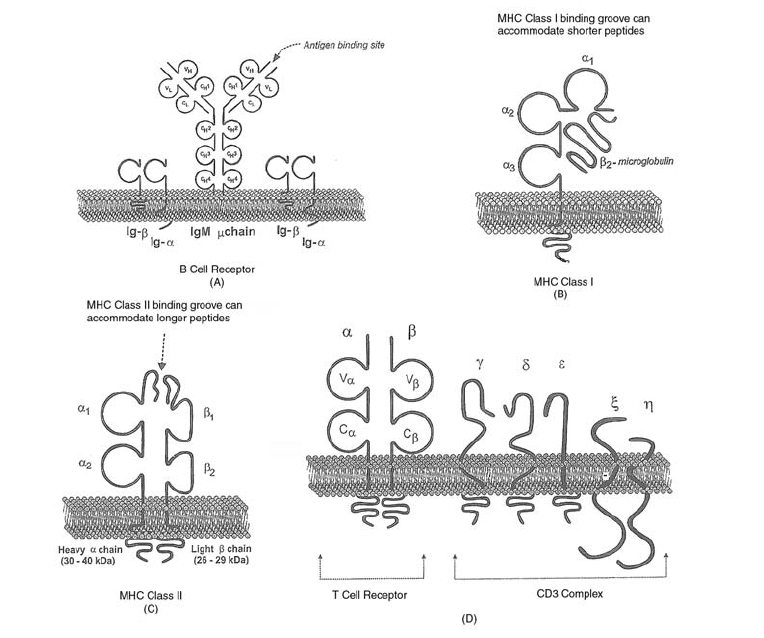
designated HLA-A, -B, and -C. Hence,
each individual will express six distinct
Class I HLA molecules: two HLA-A, two
HLA-B, and two HLA-C. Each locus has
many alleles; more than 50 have been de-
fined for HLA-A; more than 75 for HLA-B;
and more than 30 for HLA-C.
The structure of a Class I HLA molecule
is illustrated in Fig 11-2. Each molecule
contains a heavy chain (45 kDa) and a
smaller (12 kDa) peptide chain called
β2-microglobulin. The heavy chain has a cy-
toplasmic tail, a transmembrane region,
and three extracellular immunoglobu-
lin-like domains. β2-microglobulin is non-
covalently associated with the heavy chain
and is not a transmembrane protein. This
protein is required for Class I MHC expres-
sion and function on the cell surface. The
antigen-binding groove of the Class I mole-
cule is formed by the α1and α2domains of
the heavy chain. The structure of the anti-
gen-binding groove consists of a platform
made up of eight parallel βstrands that is
supported by two αhelices.4The peptides
displayed in the antigen-binding groove are
8to12aminoacidslongandrepresenthy
-
drolyzed proteins that have been synthe-
sized within the antigen-presenting cell;
hence, they are referred to as endogenous
antigens. The endogenous source of pro-
teins indicates that the genes encoding the
protein also must reside in the cell. These
proteins could be the product of host genes
including tumorigenic genes or genes from
viruses or intracellular bacteria.
Chapter 11: Immunology 245
Figure 11-2. Structures of some receptors found on various cells in the immune system.1
Copyright © 2005 by the AABB. All rights reserved.
MHC Class II Molecules
TherearethreemajorClassIIgeneloci:
HLA-DR, -DQ, and -DP. As with the Class I
molecules, there are many different al-
leles that could occupy each locus. Each
individual expresses four DR molecules, four
DQ molecules, and two DP molecules, for
a total of 10 forms of Class II HLA mole-
cules.
ClassIImoleculesareheterodimeric
structures consisting of a heavy αchain (30
to 34 kDa) and a light βchain (26 to 29
kDa). Each chain has a cytoplasmic tail, a
transmembrane region, and an extracellu-
lar portion. There are two immunoglobu-
lin-like domains on each chain (α1and α2
and β1and β2). The α2and β2domains for
each gene have a constant structure; the α1
and β1domains are diverse. The antigen-
bindinggrooveislocatedwithintheα1,β1
domains and is similar in structure to the
ClassImolecules.However,thegrooveis
larger, accommodating peptides that are 12
to 20 amino acids in length. Class II mole-
cules are expressed on monocytes, macro-
phages, dendritic cells, and B lymphocytes.
The antigenic peptides displayed in the
groove of Class II HLA molecules come
from proteins that have been phagocytosed
or endocytosed by antigen-presenting cells.
These proteins are termed exogenous anti-
gens and include most bacteria, parasites,
and viral particles released from other
cells.5
MHC Class III Molecules
There are approximately 20 genes in the
Class III region of the MHC. These genes
code for proteins of the complement sys-
tem and proinflammatory molecules such
as TNF.
Cluster of Differentiation (CD) Molecules
The CD designation is a nomenclature
system used to describe numerous mole-
cules expressed on cells and components
of the blood and lymphoid organs. Over
200 CD markers have been described to
date.3Some of the major CD markers on
cells of the immune system are summa-
rized in Table 11-1.
Cell Adhesion Molecules
For normal immune function to occur,
leukocytes must be able to attach to extra-
cellular matrices and each other. Three
families of adhesion molecules facilitate
this attachment process: selectins, inte-
grins, and IgSF adhesion molecules. The
members of the selectin family (L-selec-
tin, E-selectin, and P-selectin) are found
on leukocytes and participate in the pro-
cess of leukocyte rolling along the vascu-
lar endothelium. The integrins (VAL,
LFA-1,andMAC-I)andtheIgSFadhesion
molecules (ICAMs, VCAMs, LFA-2, and
LFA-3) are required to stop leukocyte roll-
ing and mediate leukocyte aggregation
and transendothelial migration.6Most ad-
hesion molecules have CD designations
(eg, LFA-2 is CD2 and LFA-3 is CD58).7
Signal Transduction
Signal transduction is the process of send-
ing signals between or within cells that
results in the initiation or inhibition of
gene transcription. Cell surface activation
signals associated with the immune re-
sponse are initiated by extracellular inter-
action of various ligands and receptors.
Many of these ligands and receptors have
CD designations. Any defect or deficiency
in the signal transduction process can have
significant consequences on the normal
functioning of the immune system. For
example, severe combined immunodefi-
ciency disease can result from a defi-
ciency of Jak3, which impairs signal
transduction of a specific cytokine recep-
tor subunit.7
246 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 11: Immunology 247
Table 11-1. Some Major CD Antigens on Cells of the Immune System
CD
Designation
Cell
Population
Other Cells
with Antigen Comments
CD1 Cortical thymocytes Some APCs, some B cells Strength of expression is inverse to
expression of TCR/CD3
CD2 Pan-T marker, present on early
thymocytes
NK cells This is a sheep-cell rosette receptor;
activation and adhesion function
(LFA-2)
CD3 T lymphocytes —— Functions as a signal transduction
complex
CD4 Developing and mature thymocytes
and on 2/3 of peripheral T cells
T helper cells and some macrophages Adhesion molecule that mediates MHC
restriction; signal transmission; HIV
receptor
CD5 Pan-T marker, from late cortical stage B cells of chronic lymphocytic leuke-
mia; possibly long-lived
autoreactive B cells
Function unknown; possibly involved
in costimulatory effects of
cell-to-cell adhesion
CD8 Developing and mature thymocytes
and cytotoxic T lymphocytes
None Adhesion molecule that mediates MHC
restriction; signal transmission
CD14 Monocytes —— LPS receptor
CD16 Macrophages, neutrophils, NK cells —— FcγRIII (low-affinity Fc receptor for
IgG)
CD19 B lymphocytes —— Signaling (also called B4)
CD20 B lymphocytes —— Signaling (also called B1)
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

248 AABB Technical Manual
Table 11-1. Some Major CD Antigens on Cells of the Immune System (cont’d)
CD
Designation
Cell
Population
Other Cells
with Antigen Comments
CD21 Mature B cells Possibly macrophages This is a receptor for C3d (CR2); also
receptor for EBV
CD25 Activated T and B cells Macrophages; virally transformed cells High-affinity IL-2 receptor, earlier
called TaC
CD34 Stem cells Hematopoietic cells; endothelial cells Called “stem cell antigen”; used in the
laboratory to isolate hematopoietic
precursor cells; physiologic func-
tion unknown
CD35 Mature and activated B cells Red cells, macrophages, granulocytes,
dendritic cells
This is a receptor for C3b (CR4)
CD45 Immature and mature B and T cells All cells of hematopoietic origin except
red cells
Also called leukocyte common antigen
(LCA); different leukocytes have dif-
ferent isoforms
CD56 NK cells NKT cells NK cell adhesion and lineage marker
for NK cells; innate immunity
CD71 Early thymocytes; activated T and B
cells
Activated hematopoietic cells; prolifer-
ating cells of other somatic lines;
reticulocytes
This is the transferrin receptor
CD = clusters of differentiation; APCs = antigen-presenting cells; NK = natural killer; TCR = T-cell receptor; MHC = major histocompatibility complex; HIV = human immunodefi-
ciency virus; IL = interleukin; NKT = natural killer T.
Copyright © 2005 by the AABB. All rights reserved.
Organs of the Immune
System
Numerous organs are involved in the im-
mune system. The central organs include
the marrow, liver, and thymus. Peripheral
organs are the lymph nodes and spleen.
The gastrointestinal tract-associated lym-
phoid tissue and bronchus-associated
lymphoid tissue also play an important
role involving both central and peripheral
functions.
Cells of the Immune System
The primary cells involved in the adaptive
immune system are lymphocytes—B cells,
T cells, and antigen-presenting cells.
Other cells such as macrophages are in-
volved in the induction of the immune re-
sponse, and both macrophages and poly-
morphonuclear leukocytes participate in
various inflammation responses associated
with the immune response. All of these cells
are of hematopoietic origin (ie, marrow-
derived) and are formed from a single pro-
genitor called a pluripotent hematopoietic
stem cell. As illustrated in Fig 11-3, the
pluripotent stem cell can give rise to two
lineages: myeloid and lymphoid. Granulo-
cytes (neutrophils, basophils, and eosino-
phils), platelets, mast cells, red cells, and
macrophages arise from myeloid progeni-
tors. T and B lymphocytes are formed from
lymphoid progenitors. Dendritic cells and
natural killer (NK) cells are also derived
from the pluripotent stem cell; however,
their precise origin is unknown.
Lymphocytes
The two major lineages of lymphocytes
are B cells and T cells. B cells are derived
from the marrow and T cells are derived
from T-cell precursors produced in the
marrow, which migrate to the thymus. B
cells are the precursors of the cells that
make antibody (plasma cells). T cells con-
sist of subpopulations that either help in
antibody formation (helper T cells), kill
target cells (cytotoxic T cells), induce in-
flammation (delayed hypersensitivity T
cells), or inhibit the immune response
(regulatory T cells).
B Lymphocytes
Each B cell can recognize one (or a lim-
ited set) of antigen epitopes through re-
ceptors on the cell surface. These recep-
tors are similar to IgM and IgD molecules,
which are produced and transported to
themembranesurface.Whentheimmu
-
noglobulin receptor binds to a specific
antigen, the cell is stimulated to divide
and differentiate into a plasma cell. The
plasma cell can secrete a soluble form of
the immunoglobulin “receptor” known as
antibody. The antibody secreted is specific
for the same antigen that interacted with
the cell-bound immunoglobulin receptor.
The B-cell receptor (BCR) has one addi-
tional heavy chain domain (CH4) compared
to secreted immunoglobulin. This domain
is required to anchor the receptor to the cell
membrane. Several accessory molecules on
the surface of B cells are closely associated
with the BCR and are important for signal
transduction. Some of these accessory mol-
ecules include Igα,Igβ,MHCClassIImole
-
cules, complement receptors, and some CD
markers (see Table 11-2). All of these com-
ponents are part of the BCR antigen com-
plex (some of these structures are illustra-
ted in Fig 11-2).
ThechallengeforthebodyistohaveB
lymphocytes that can recognize each of the
thousands of different foreign antigens en-
countered over a lifetime. The immune sys-
tem has a unique approach to ensure this
diversity.Thehumangenomeisknownto
contain approximately 40,000 genes. The B
Chapter 11: Immunology 249
Copyright © 2005 by the AABB. All rights reserved.

cellsinthebodyprobablymakemorethan
100 million different antibody proteins that
are expressed as immunoglobulin recep-
tors. Because the human body does not
have enough genes to code for the millions
of different foreign proteins that the im-
mune system must have the capability to
recognize, a process called gene rearrange-
ment is used to create the required diver-
sity.9
Several genes code for the heavy chain
and light chain that make up the immuno-
globulin receptor on B cells. The loci for
genes that code for the heavy chain are on
chromosome 14 and the loci that code for
the light chains are on chromosome 2 (κ
light chain) or chromosome 22 (λlight
chain). Three gene loci contribute to the di-
versity of the immunoglobulin receptor: V
(variable), D (diversity), and J (joining). The
250 AABB Technical Manual
Table 11-2. Receptors/Markers Present on Macrophages, Monocytes, and B
Lymphocytes
Marker Function
Macrophages and Monocytes
Complement receptors
CR1 (C3b receptors, CD35) Binds to cells coated with C3b
CR3 (C3bi receptor, CDIIb) Adhesion and activation
Cytokine receptors (IL-1, IL-4, IFNγ) Receptors that bind cytokines signaling
and migration-inhibition factor activation and other cell functions
Fc receptors
FcγRI (CD64) High affinity for IgG
FcγRII (CD32) Medium affinity for IgG
FcγRIII (CD16) Low affinity for IgG
FcγRII (CD23) Low-affinity receptor for the Fc of IgE
Leukocyte function antigen Adhesion and activation
(LFA1 or CD11a)
Mannose/fucose receptors Binds sugars on microorganisms
P150,95 (cD11c) Adhesion and activation
B Lymphocytes
CD5 Cell marker that identifies a subset of B cells
predisposed to autoantibody production
CD19, 20, and 22 Primary cell markers used to distinguish
B cells
CD72-78 Other cell markers that identify B cells
Complement receptors Play a role in cell activation and “homing”
C3b (CR1, CD35); C3d (CR2, CD21) of cells
IgαTransport and assemble IgM monomers in
the cell membrane
IgβAccessory molecules that interact with the
transmembrane segments of IgM
MHC Class II (DP, DQ, DR) Present on antigen-presenting cells and is
critical for initiating T-cell-dependent
immune responses
Copyright © 2005 by the AABB. All rights reserved.
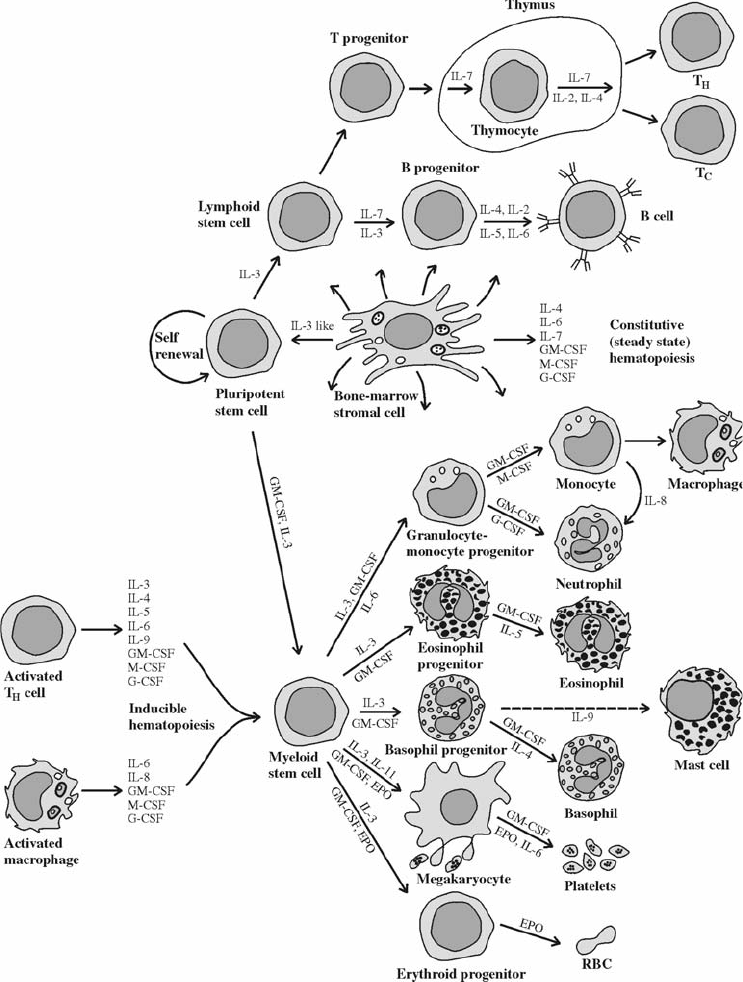
Chapter 11: Immunology 251
Figure 11-3. The pluripotent stem cell, in the upper middle part of the diagram, gives rise to the lym-
phoid stem cell and to the myeloid stem cell, from which all other lines of blood cells derive.
Cytokines from marrow stromal cells influence the replication and differentiation of stem and later
cells. Cytokines from activated members of the highly differentiated T-cell and macrophage lines exert
major effects at all stages of myeloid and lymphoid development. (Used with permission from Goldsby
et al.8)
Copyright © 2005 by the AABB. All rights reserved.
fourth locus codes for the C (constant) region
of the immunoglobulin receptor and de-
fines the expression of isotypes. The C region
does not affect antigen binding but codes
for the biologic functions associated with
the immunoglobulin such as complement
activation and binding to specific receptors.
Isotype switching (changing from IgM or
IgD to one of the other isotypes) is a T-cell-
dependent process that occurs at the DNA
level. The process of isotype switching al-
lows the specificity of antibody molecule to
be maintained regardless of the heavy chain
isotype.9,10 Table 11-3 summarizes the bio-
logic properties of the different immuno-
globulin isotypes.
At the pre-B-cell stage of development,
one heavy chain gene is randomly selected
from each of the four segments (VH,D
H,J
H,
and CH). There are over 104possible combi-
nations because of the large number of
possible genes at each segment. The light
chain gene has only three segments (VL,J
L,
and CL), and one gene is selected from each
of these segments in a process similar to
the heavy chain gene selection. This pro-
cess results in over 1000 possible light chain
combinations. When the heavy chain and
light chains are combined, approximately
10 million different combinations could be
formed, each one representing an immu-
noglobulin receptor with unique antigen
specificity. In addition to gene rearrange-
ment, several other processes contribute to
this diversity. These processes include so-
matic mutations that occur at the time of
B-cell activation, combinatorial shuffling
that occurs when the heavy and light
chains are assembled, and the addition of
random DNA bases to the end of the genes
during the joining process. The combina-
tion of all of these processes ensures that B
cells have immunoglobulin receptors spe-
cific for any foreign antigen that could be
encountered (approximately 1011 antigen
specificities).9-11
Because the formation of the B-cell im-
munoglobulin receptor occurs through
random rearrangement, some of the recep-
tors produced will react to the body’s own
cells. To prevent autoimmune disease, the
body must eliminate or downregulate these
B cells. This is accomplished through a pro-
cess of negative selection.12 When a B cell is
formed, it encounters large quantities of self
antigen. If a B cell binds strongly to self an-
tigen, the immunoglobulin receptor sends
a signal that activates enzymes within the
cell to cleave nuclear DNA. This causes the
cell to die, a process termed apoptosis (pro-
grammed cell death). Only 25% of the B cells
that mature in the marrow reach the circu-
lation. The majority of B cells undergo
apoptosis. This process results in B cells
that have low affinity to self antigen but still
bind to foreign antigens that enter the
body. When mature B cells enter the pe-
ripheral blood circulation, they bind for-
eign antigens that are specific for their im-
munoglobulin receptors. When specific
antigen binds to the immunoglobulin re-
ceptor, a signal occurs causing the recep-
tor/antigen complex to be internalized. In-
side the cell, the antigen is degraded into
small peptides that bind to MHC Class II
molecules within the cell. This MHC-pep-
tide complex is transported to the outer
membrane of the cell where it can interact
with the TCR. This interaction signals the
cell to produce various cytokines, causing
the B cell to proliferate into a memory cell
or a plasma cell. The antibodies produced
by a plasma cell are always of the same im-
munoglobulin class. However, each time
B-cell proliferation occurs, somatic muta-
tions result in slight differences in the bind-
ing affinity of the immunoglobulin recep-
tor. Because immunoglobulin receptors
with the highest binding affinity will be the
ones most likely to encounter antigen, this
process preferentially results in prolifera-
tion of B cells with the highest affinity for
252 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

antigen. This preferential selection is termed
a focused immune response.5,13,14
T Lymphocytes
There are two major types of T cells: cyto-
toxic T cells and helper T cells. These two
cell types can be differentiated by the pre-
senceofspecificCDmarkersonthecell.
Cytotoxic T cells are positive for the sur-
face marker CD8 and negative for CD4
and make up approximately one-third of
the circulating T cells in the peripheral
blood. Helper T cells are CD4 positive and
CD8 negative and represent approximately
Chapter 11: Immunology 253
Table 11-3. Characteristics and Biologic Properties of Human Immunoglobulins
Class IgG IgA IgM IgD IgE
Structure
H-chain isotype
Number of subclasses
L-chain, types
Molecular weight (daltons)
γ
4
κ,λ
150,000
α
2
κ,λ
180,000-
500,000
µ
1
κ,λ
900,000
δ
?
κ,λ
180,000
ε
?
κ,λ
200,000
Exists as polymer No Yes Yes No No
Electrophoretic mobility γγbetween γ
and β
between γ
and β
fast γ
Sedimentation constant
(in Svedberg units)
6-7S 7-15S 19S 7S 8S
Gmallotypes(Hchain) +0000
Km allotypes (Kappa L chain:
formerly Inv)
+++??
Amallotypes 0+000
Serum concentration (mg/dL) 1000-1500 200-350 85-205 3 0.01-0.07
Total immunoglobulin (%) 80 15 5 <0.1 <0.1
Synthetic rate (mg/kg/day) 33 24 6-7 <0.4 <0.02
Serum half-life (days) 23 6 5 2-8 1-5
Distribution (% of total
in intravascular space)
45 42 76 75 51
Present in epithelial secretions No Yes No No No
Antibody activity Yes Yes Yes Probably
no
Yes
Serologic characteristics Usually
nonagglu-
tinating
Usually
nonagglu-
tinating
Usually
agglu-
tinating
??
Fixes complement Yes No Yes No No
Crosses placenta Yes No No No No
Copyright © 2005 by the AABB. All rights reserved.
two-thirds of the circulating T cells. Helper
T cells recognize antigen presented by
Class II HLA molecules, whereas cytotoxic
T cells recognize antigen in the context of
Class I HLA molecules. Approximately 5%
of peripheral blood T cells are negative for
both CD4 and CD8.
T cells go through a process of positive
and negative selection in the thymus dur-
ing T-cell ontogeny. If a T cell is able to rec-
ognize self MHC antigens, it survives (posi-
tive selection) and migrates to the medulla
of the thymus. In the medulla, the T cells
undergo a process (negative selection) that
deletes T cells with high affinity for self
MHC antigens. T cells that fail to recognize
self MHC antigens undergo apoptosis. The
primary goal is to select T cells that recog-
nize self MHC molecules that have foreign
peptides in their groove. The process is so
exquisite that approximately 10% of T cells
have the ability to react with foreign MHC
complexes, which forms the major basis of
transplantation rejection. In the end, only
5% of the cells in the thymus survive both
positive and negative selection and become
mature T cells.5,13,15
The receptor on the T cell that is respon-
sible for MHC/peptide recognition is the TCR
(see Fig 11-2). There are two major types of
TCRs: those that express αand βchains or γ
and δchains as part of the TCR complex.
Approximately 90% of all T cells bear α,β
chains. Each transmembrane chain has two
domains (one variable, the other constant).
These chains are produced through a pro-
cess of gene rearrangement in a manner
similar to MHC and immunoglobulin re-
ceptors. The number of possible TCR αand
βspecificities is estimated at 1015.TheTCRs
bearing γand δare expressed on 5% to 15%
of T cells, predominantly by those T cells in
the mucosal endothelium. These T cells ap-
pear to play an important role in protecting
the mucosal surfaces of the body from for-
eign bacteria.
The TCR is noncovalently associated with
theCD3complex,whichismadeupofthree
pairs of dimers. This CD3 complex is re-
sponsible for signal transduction once pep-
tide is recognized by the TCR.16
Recognition by Cytotoxic T Cells.
Cytotoxic T cells recognize peptides associ-
ated with Class I MHC molecules. As dis-
cussed previously, these peptides may be
derived from self proteins or proteins from
intracellular viruses or microbes. The
TCR-2 receptor on the cytotoxic T cell rec-
ognizes the peptide-MHC Class I molecule
in combination with a co-receptor (CD8) on
the T cell. These interactions signal the cell
to produce proteins (eg, perforin) that dis-
rupt the integrity of the target cell mem-
brane, resulting in cell death. During this
process, cytokines [TNF-αand interferon-γ
(IFNγ)] are also produced. These cytokines
prevent replication of virus that may be shed
from the cell during cell death; hence, the
infection is stopped through these processes.
Although the process is an extremely effec-
tive mechanism for killing cells infected with
virus, the process can be harmful to the host.
The cytokines produced to prevent viral rep-
lication can cause adverse effects including
the damage or destruction of healthy host
tissue. Liver damage associated with hepa-
titis B infection is an example of morbidity
caused by the cytotoxic T-cell response.5,11
Stimulation of B Cells. B-cell activation
can occur through activation by T cells or by
a mechanism independent of T-cell interaction.
These two mechanisms are described below.
T-Cell-Dependent Stimulation. Helper
T-cell receptors recognize foreign peptides
in the antigen-binding groove of Class II
MHC molecules in combination with CD4.
This TCR-CD4 interaction with MHC Class
II upregulates the expression of CD80/86
on the surface of antigen-specific B cells.
CD80/86 reacts with the ligand CD28 on
the T-cell surface, causing upregulation of
the CD40 ligand (CD40L), which engages
254 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
with CD40 on B cells. This cascade of sig-
nals is important for cytokine production,
which results in the isotype switch response
by the B cell. The production of interleukin
(IL)-4 causes B cells to switch from IgM to
IgG4 and IgE. The production of transform-
ing growth factor β(TGF-β)andIL-10
causes the B cell to switch to IgA1 and IgA2.
If there is an absence or impaired function
of the ligand interaction isotype, switching
can be affected. For example, if the CD40L-
CD40 interaction is impaired, isotype
switching will not occur and only IgM anti-
body is produced. This clinical situation is
termed hyper-IgM immune deficiency.7
T-Cell-Independent Stimulation. B cells
can be activated to produce antibodies
by polysaccharides, lipopolysaccharides,
and polymeric proteins independent of
T-cell interaction. The B cells react directly
with these molecules, producing a rapid
immune response to pathogens. However,
there are disadvantages to this mechanism:
the process is ineffective for the production
of memory B cells; antibody affinity matu-
ration is poor; and isotype switching is not
induced. The T-cell-independent process
can also cause antibody production by B
cells whose immunoglobulin is specific for
antigens other than those found on the
pathogen; hence, both protective antibodies
and autoantibodies may be produced. When
autoantibodies are produced through this
mechanism, transient clinical symptoms of
autoimmune disease may occur.7
NK Cells
NK cells do not express T-cell receptors or
B-cell receptors and represent approxi-
mately 10% of the lymphocyte popula-
tion. These cells have the ability to kill
some cells infected with viruses and some
tumor cells. NK cells do this by a mecha-
nism termed antibody-dependent cellular
cytotoxicity or ADCC. ADCC occurs when
viral proteins are expressed on the surface
of an infected cell, usually as a result of
viral budding. The proteins are recog-
nized as foreign by the immune system
and antibodies are produced, which later
bind to the viral proteins expressed on the
infected cell. NK cells recognize the pres-
ence of antibody bound on the surface of
the cells via their Fcγreceptors. The NK
cells produce perforins, which cause lysis
of the virus-infected cells by a mechanism
that is not entirely understood.5
Phagocytic Cells
Phagocytes include cells such as monocytes
and polymorphonuclear granulocytes
(eosinophils, basophils, and neutrophils).
Some monocytes migrate into tissues
(liver, lungs, spleen, kidney, lymph nodes,
and brain) and become tissue macrophages.
The polymorphonuclear granulocytes are
rapidly produced and live only for a short
time (several days). The neutrophil is the
most abundant granulocyte. These cells
respond to chemotactic agents such as
complement fragments and cytokines,
causing them to migrate to the site of in-
flammation. Eosinophils represent only
2% to 15% of the white cells and play an
important role in regulating the inflam-
matory response by releasing an antihis-
tamine. These cells may also play a role in
phagocytosing and killing microorgan-
isms. Basophils make up less than 0.2% of
the total leukocyte pool. These cells also
respond to chemotactic factors and are
involved in the allergic response. Histori-
cally, this network of phagocytic cells was
called the reticuloendothelial system but is
now termed the mononuclear phagocytic
system.
The ability of cells to phagocytose is ac-
complished through the presence of recep-
tors on the cell membrane. There are many
types of receptors including: mannose-fucose
Chapter 11: Immunology 255
Copyright © 2005 by the AABB. All rights reserved.
receptors that bind to sugars on the surface
of microorganisms, Fc receptors that bind
to IgG, and complement receptors. A sum-
mary of some membrane receptors found
on macrophages and monocytes is found
in Table 11-2. The internalization and pro-
cessing of particulate matter occur through
the production of enzymes (peroxidase and
acid hydrolases).5Under optimal cytokine
conditions, macrophages and monocytes
can become formal antigen-presenting
cells; therefore, their ability to ingest and
process foreign molecules is an essential
part of the adaptive immune response.
Soluble Components of the
Immune Response
Therearethreemajorsolublecomponents
of the immune response: immunoglobu-
lins, complement, and cytokines.
Immunoglobulins
Immunoglobulins are the proteins that can
be cell bound and serve as antigen recep-
tors on B cells (see section on B lympho-
cytes) or can be secreted in a soluble form
as antibodies. The molecular development
of the immunoglobulin molecule is dis-
cussed in the section on B lymphocytes.
The structures of the different types of
immunoglobulin are discussed below.
Each monomeric immunoglobulin mol-
ecule consists of two identical heavy chains
and two identical light chains. The heavy
chains consist of approximately 450 amino
acids with a molecular weight of approxi-
mately 50 to 77 kDa. There are five heavy
chain classes termed isotypes. They include
mu (µ), gamma (γ), alpha (α), delta (δ), and
epsilon (ε). The light chains are smaller (ap-
proximately 210 amino acids; molecular
weight 25 kDa) and can be either kappa (κ)
or lambda (λ). The heavy and light chains
of the immunoglobulin molecule are held
together by disulfide bonds. The polypep-
tide chains (both heavy and light) are
looped, forming globular structures called
the immunoglobulin domain. On each
chain, there is a variable domain in which
the amino acid sequence is diverse, giving
the immunoglobulin its specificity. The epi-
topes expressed in this region are termed
idiotypes. The remaining domains on both
the heavy and light chains are called con-
stant domains and have similar amino acid
sequences, depending on the isotype. The
hinge area of the molecule (between CH1
and CH2, as shown in Fig 11-4) gives the
molecule flexibility, allowing the two anti-
gen-binding components to operate inde-
pendently. The biologic functions of the
molecule are associated with the constant
domains on the heavy chain. These func-
tions include: placental transfer, macro-
phage binding, and complement activa-
tion.4
Interchain Bonds
Each light chain is joined to one heavy chain
by a disulfide bond. One or more disulfide
bonds link the two heavy chains at a point
between CH1andC
H2, in an area of con-
siderable flexibility called the hinge re-
gion. It is these interchain disulfide bonds
that are the target of reducing agents used
to produce “chemically modified” anti-D
reagent.
Fab and Fc Fragments
Polypeptides can be cleaved at predictable
sites by proteolytic enzymes. Much infor-
mation about immunoglobulin structure
and function is derived from the study of
cleavage fragments generated by papain
digestion of Ig molecules. Papain cleaves
theheavychainatapointjustabovethe
hinge, creating three separate fragments.
Two are identical, each consisting of one
256 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
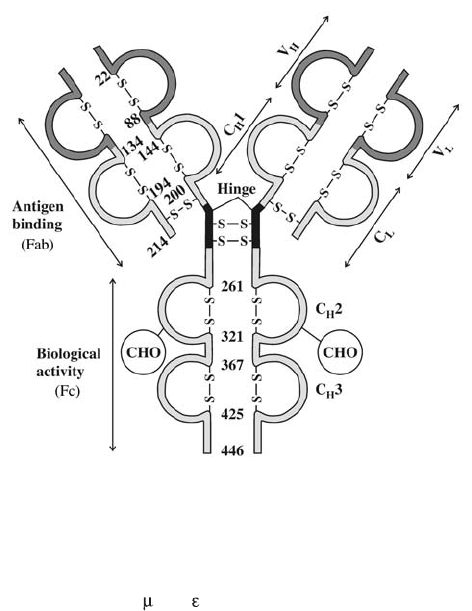
light chain linked to the N-terminal half
of one heavy chain; the other fragment
consists of the C-terminal halves of the
heavy chains, still joined to one another
by the hinge-region disulfide bonds. The
two identical N-terminal fragments,
which retain the specificity of the anti-
body, are called Fab fragments. The joined
C-terminal halves of the heavy chains
constitute a nonantibody protein frag-
ment capable of crystallization, called the
Fc fragment.
Immunoglobulin Polymers
Disulfide bonds may also join some Ig
monomeric units to one another to form
larger polymeric molecules; only IgM and
IgAcanformpolymers.IgG,IgE,andIgD
exist only in the monomeric form; there
are no polymeric forms of these Ig classes.
The IgM synthesized by unstimulated B
cellsandexpressedonthemembraneas
the immunoglobulin receptor is expres-
sed in the monomeric form. As men-
tioned previously, the µheavy chain has
four constant domains in the membrane
form of IgM. The fourth domain allows
the IgM monomer to bind to the cell
membrane as the immunoglobulin recep-
tor. Following clonal expansion and differ-
entiation to a plasma cell, the activated
cell produces µchains with one less con-
stant domain. While still in the plasma-
cell cytoplasm, five IgM monomers are
joined by the formation of disulfide bonds
between the CH3 domains and CH4do
-
Chapter 11: Immunology 257
Figure 11-4. The basic four-chain immunoglobulin unit. Idiotypic specificity resides in the variable do-
mains of heavy and light chains (VHand VL). Antigen-binding capacity depends on intact linkage be-
tween one light chain (VLand CL) and the amino-terminal half of one heavy chain (VHand CH1), the Fab
fragments of the molecule. Disulfide bonds in the hinge region join carboxy-terminal halves of both
heavy chains (CH1 and CH3, plus CH4for and heavy chains), to form the Fc fragment. (Used with per-
mission from Goldsby et al.8)
Copyright © 2005 by the AABB. All rights reserved.
mains.5The result is a pentamer that is
secreted to the exterior of the cell and
constitutes the form in which IgM accu-
mulates in body fluids.
Secreted IgA exists in both monomeric
and polymeric forms. Monomeric forms
predominate in the bloodstream, but dimers
and trimers that are secreted by B cells in
mucosal surfaces and exocrine tissue are
the biologically active form.
Other Chains
Pentameric IgM and the dimers and tri-
mers of IgA contain a 15-kDa polypeptide
called the J chain. Before the polymer
leaves the plasma-cell cytoplasm, this
chain attaches to the terminal constant
domain of two adjacent monomers. No
matter how many monomers constitute
the polymer, there will be only one J chain.
Its function is not fully understood.
The polymeric Ig molecules present in
epithelial secretions also exhibit a subunit
called the secretory component. The secre-
tory component appears to protect the bio-
logically important surface antibodies from
proteolysis in the enzyme-rich secretions of
respiratory and alimentary tracts.
Individual Immunoglobulin Classes
IgM. IgM is the first Ig class produced by
the maturing B cell. It is the first to appear
in the serum of maturing infants and the
first to become detectable in a primary
immune response. Secreted pentameric
IgM normally constitutes 5% to 10% of the
immunoglobulin in normal serum. Very
few of these large molecules diffuse into
interstitial fluid.
Although the five monomers comprise
10 antigen-combining sites, only five sites
are readily available to combine with most
antigens; hence, IgM is described as penta-
valent.Becauseoftheirlargesizeand
multivalency, IgM molecules readily bind to
antigens on particulate surfaces, notably
those on red cells or microorganisms. IgM
antibodies can crosslink cells expressing a
specific antigen, forming a clump of cells
(agglutination). Although extremely useful
as a laboratory endpoint, agglutination
probably plays a relatively minor role in
biologic events.
The most important biologic effect of IgM
is its ability to activate the complement
cascade, which enhances inflammatory
and phagocytic defense mechanisms and
may produce lysis of antigen-bearing cells.
IgG. Immunoglobulin G exists only as a
monomer and accounts for 75% to 80% of
the immunoglobulins present in serum. It
is equally distributed in the intravascular
and extravascular compartments. In vivo,
cells or particles coated with IgG undergo
markedly enhanced interactions with cells
that have receptors for the Fc portion of γ
chains, especially neutrophils and macro-
phages.
IgG molecules can be classified into four
subclasses: IgG1, IgG2, IgG3, and IgG4.
Structurally, these subclasses differ primar-
ily in the characteristics of the hinge region
and the number of inter-heavy-chain
disulfide bonds. Biologically, they have sig-
nificantly different properties. IgG3 has the
greatest ability to activate complement, fol-
lowed by IgG1 and, to a much lesser extent,
IgG2. IgG4 is incapable of complement ac-
tivation. IgG1, IgG3, and IgG4 readily cross
the placenta. IgG1, IgG2, and IgG4 have a
half-life of 23 days, significantly longer than
that of other circulating immunoglobulins;
however, the half-life of IgG3 is only slightly
longer than IgA and IgM. IgG1 and IgG3
readily interact with the Fc receptors on
phagocytic cells, but IgG4 and IgG2 have
low affinity for these receptors with the ex-
ception that IgG2 has an affinity similar to
IgG1 and IgG3 for an allotype of Fcγrecep-
tor IIa.
258 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
IgA. Although there is a large body con-
tent of IgA (10% to 15% of serum immuno-
globulin concentration), relatively little is
found in the blood. Most of the IgA exists in
mucosal secretions. Secretory IgA protects
the underlying epithelium from bacterial
and viral penetration. Polymeric IgA is
thought to combine with environmental
antigens, forming complexes that are elimi-
nated as surface secretions are excreted.
This process may be important in control-
ling the development of hypersensitivity.
The heavy chain of IgA has no comple-
ment-binding site; hence, IgA cannot acti-
vate complement through the classical
pathway. IgA can activate complement
through the alternative pathway (see be-
low).
IgE. The concentration of serum IgE is
measured in nanograms, compared with
milligram levels for other immunoglobu-
lins. Even when patients with severe aller-
gies have markedly elevated serum concen-
trations of IgE, the absolute level is minimal
compared with other immunoglobulins.
Most IgE is present as monomers tightly
bound to the membrane of basophilic
granulocytes or mast cells. IgE is responsible
for immediate hypersensitivity events, such
as allergic asthma, hay fever, and systemic
anaphylactic reactions. Although IgE ap-
pears to be involved in reactions to proto-
zoal parasites, no specific protective mecha-
nisms have been identified.
IgD. Serum contains only trace amounts
of IgD. Most IgD exists as membrane im-
munoglobulin on unstimulated B cells. The
function of IgD is unknown, but it may be
important for lymphocytic differentiation,
which is triggered by antigen binding, and
in the induction of immune tolerance.
Complement
Complement is the term applied to a sys-
temof25to30serumandmembrane
proteins that act in a cascading manner—
similar to the coagulation, fibrinolytic,
and kinin systems—to produce numerous
biologic effects. The participating pro-
teins remain inactive until an event initi-
ates the process, following which the
product of one reaction becomes the cata-
lyst for the next step (see Fig 11-5). Each
evolving enzyme or complex can act on
multiple substrate molecules, creating the
potential for tremendous amplification of
an initially modest or localized event.
Complement has three major roles: pro-
motion of acute inflammatory events; alter-
ation of surfaces so that phagocytosis is en-
hanced; and the modification of cell mem-
branes, which leads to cell lysis. These ac-
tions cause destruction of bacteria, protect
against viral infection, eliminate protein
complexes, and enhance development of
immune events. However, activation of
complement can also initiate inflammatory
and immune processes that may harm the
host and mediate destruction of host cells,
especially those in the blood.
Different mechanisms17 exist for activat-
ing the complement cascade: the classical
pathway, which is initiated by interaction
between an antibody and its antigen, and
the alternative pathway, which is usually not
antibody mediated.
The Classical Pathway
For the classical pathway of complement
activation to occur, an immunoglobulin
must react with target antigen. Combina-
tion with antigen alters the configuration
of the Fc portion of the immunoglobulin,
rendering accessible an area in one of the
heavy-chain constant domains that inter-
acts with the C1 component of comple-
ment. C1 can combine only with Ig mole-
cules having an appropriate heavy-chain
configuration. This configuration exists in
Chapter 11: Immunology 259
Copyright © 2005 by the AABB. All rights reserved.
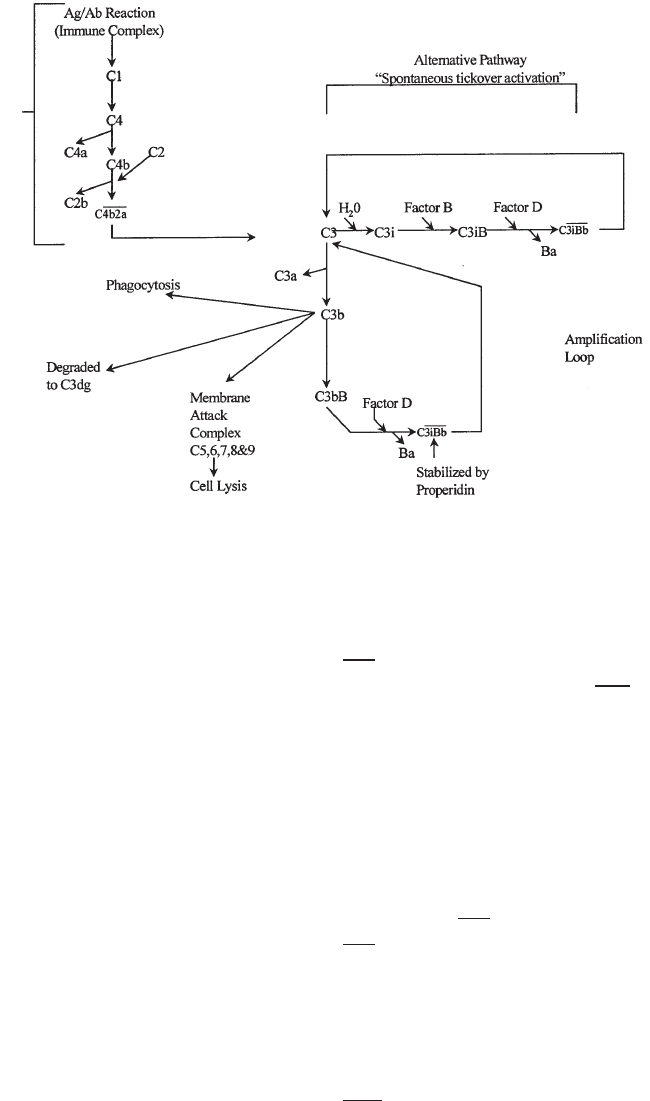
the µheavy chain and in the γchains in
IgGsubclasses1,2,and3.
The C1 component of complement atta-
ches to activation sites on the Fc portion of
two or more Ig monomers. The pentameric
IgM molecule provides an abundance of
closely configured Fc monomers; hence, a
single IgM molecule can initiate the com-
plement cascade. For IgG antibodies to ac-
tivate the sequence, two separate molecules
must attach to antigen sites close together.3
Hence, complement activation by IgG anti-
bodies depends not only on the concentra-
tion and avidity of the antibody, but also on
the topography of the antigen.
Circulating C1 is a macromolecule con-
sisting of three distinct proteins (C1q, C1r,
and C1s). When an antigen-antibody reac-
tion occurs, two or more chains of C1q at-
tach to the CH2 domain of IgG or the CH3
domain of IgM. This causes a conforma-
tional change, resulting in activation of two
C1r molecules that then cleave two C1s
molecules into activated C1s (a strong
serine esterase). The C1r, C1s, and C1q
complex is stabilized by Ca2+ ions. In the ab-
sence of Ca2+, the complex dissociates.
Thus, chelating agents often used in the
laboratory, such as citrate and oxalate anti-
coagulants, prevent the stabilization of the
C1 complex and subsequent activation of
complement proteins.
Activated C1s works on two substrates.
C1s cleaves C4 into two fragments: a large
fragment (C4b) and a smaller fragment
(C4a) that has modest anaphylatoxic activ-
ity. Most of the C4b generated is inacti-
vated; however, some C4b binds to the cell
surface and acts as a binding site for C2.
C1s also cleaves the bound C2, releasing a
fragment called C2b. The C2a fragment that
remainsboundtoC4formsanactivated
260 AABB Technical Manual
Figure 11-5. Diagram illustrating the activation of complement by the classical and alternative path-
ways. (Used with permission from Heddle.1)
Classical Pathway
Copyright © 2005 by the AABB. All rights reserved.
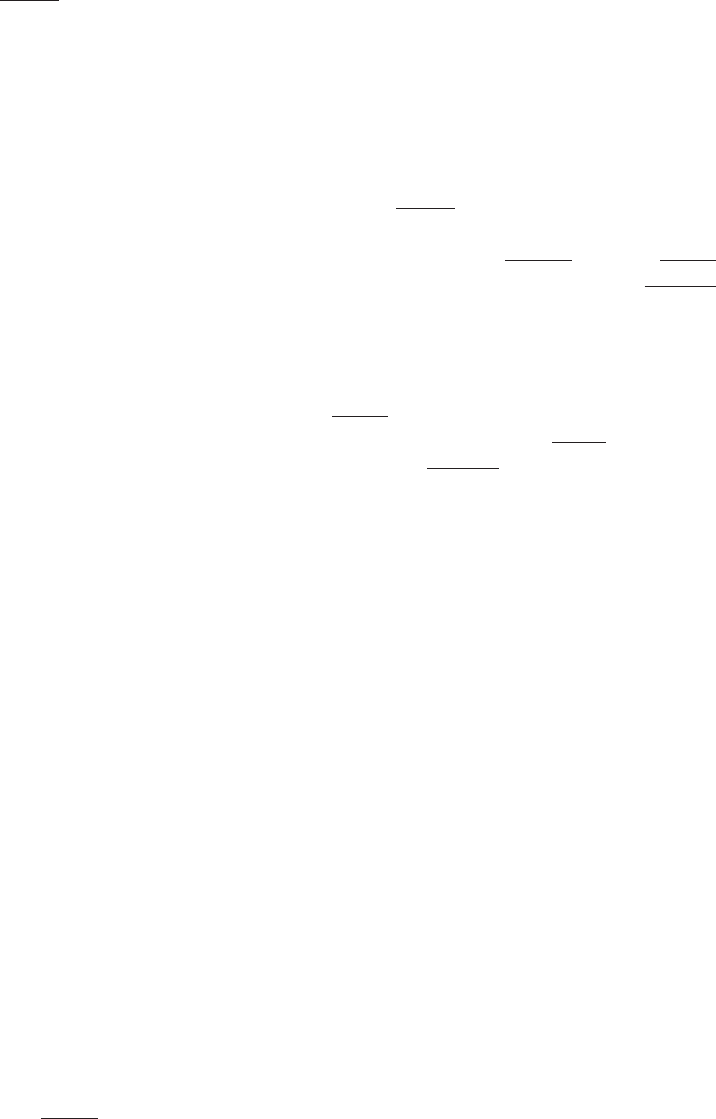
complex C4b2a,alsoknownasC3con
-
vertase. Each C3 convertase can cleave more
than 200 native C3 molecules, splitting the
molecule into two fragments. A small frag-
ment (C3a) that has anaphylatoxic activity
is released into the plasma; the larger frag-
ment (C3b) attaches to proteins and sugars
on the cell surface.
The classical pathway and the alternative
pathway are both means of generating a C3
convertase. Once C3 has been cleaved, the
same events occur in the two pathways.
The Alternative Pathway
The alternative pathway allows comple-
ment activation in the absence of an anti-
gen/antibody interaction. This pathway is
a first-line antimicrobial defense for ver-
tebrates and a mechanism whereby pre-
vertebrates can enhance their inflamma-
tory effectiveness. The alternative pathway
is a surface-active phenomenon that can
be triggered by such initiators as dialysis
membranes; the cell wall of many bacte-
ria, yeasts, and viruses; protein com-
plexes, including those containing anti-
bodies that do not bind complement;
anionicpolymerssuchasdextran;and
some tumor cells. The alternative path-
way of complement activation can also
occur spontaneously in the plasma at a
slow but steady rate (tickover activation)
(see Fig 11-5).
Four proteins participate in the alterna-
tive pathway: factor B, factor D, properdin
(factor P), and C3. Fluid-phase C3 under-
goes continuous but low-level spontaneous
cleavage, resulting in C3i that is rapidly in-
activated by fluid-phase control proteins. If
C3i encounters factor B, a complex called
C3iB is formed and additional interactions
can occur. Factor D acts on bound factor B,
generating a C3iBb complex capable of
cleaving C3 into C3a and C3b. Most of the
C3b generated in the fluid phase is inacti-
vated; however, if C3b binds to a foreign
surface such as a bacteria cell wall, the acti-
vation of the complement cascade can be
accelerated.
When C3b binds to a cell surface, factor
B is bound to give C3bB. Factor D can also
react with this cell-bound substrate, releas-
ing the small Ba fragment leaving cell-
bound C3bBb. This complex will dissociate
unless it is stabilized by properdin, result-
ing in the complex C3bBbP
.LiketheC4b2a
complex of the classical pathway, C3bBbP
iscapableofconvertingmoreC3intoC3b.
In summary, both the classical and alterna-
tive pathways result in C3b generation. The
C3 convertase of the classical pathway is
C4b2a, whereas the C3 convertases of the
alternative pathway are C3iBb in the fluid
phase and C3bBbP when cell bound.
The Membrane Attack Complex
The final phase of activation is called the
membrane attack complex and can occur
once C3b has been cleaved through either
the classical or alternative pathway. C3
convertase cleaves C5 into two fragments:
a small peptide (C5a) having potent ana-
phylatoxin activity and a larger fragment
(C5b). The C5b fragment binds C6, C7,
C8, and up to 14 monomers of C9, result-
inginalyticholeinthemembrane.Small
amounts of lysis can occur when C8 is
bound; however, the binding of C9 facili-
tates cell lysis.
The binding of C3b to a cell membrane
is the pivotal stage of the pathway. The
cell-bound C3b can proceed to activate the
membrane attack complex (C5-C9) and
cause cell lysis; alternatively, inhibitors may
stop the activation sequence, leaving the
cell coated with C3b. Factor I is an inhibitor
that can cleave cell-bound C3b, leaving
iC3b on the membrane. These two sub-
components of C3 (C3b, iC3b) can facilitate
phagocytosis by acting as opsonins. How-
Chapter 11: Immunology 261
Copyright © 2005 by the AABB. All rights reserved.
ever, C3b can be further cleaved, leaving a
small fragment called C3dg. It is the C3dg
molecule that is detected by the anti- C3d
component in anticomplement reagents
used for the direct antiglobulin test.
Complement Receptors
Some phagocytic cells have receptors that
can bind to C3 on the cell. Four different
complement receptors have been identi-
fied on phagocytic cells: CR1, CR2, CR3,
and CR4.18
CR1. CR1 is found on a variety of cells.
On red cells and platelets, the CR1 receptor
plays an important role in clearing immune
complexes. On phagocytic cells and B lym-
phocytes, it is an opsonic receptor that is
involved in lymphocyte activation. CR1 also
plays a regulatory role in complement acti-
vation by assisting factor I in cleaving C3b
into iC3b and C3dg.
CR2. CR2 is found on B cells, some epi-
thelial cells, and follicular dendritic cells. It
plays an important role in mediating B-cell
activation. It is also the receptor for inter-
feron αand the Epstein-Barr virus.
CR3. CR3 is found on cells of the myeloid
lineage. CR3 mediates phagocytosis of parti-
cles coated with iC3b and is also an impor-
tant adhesion molecule capable of binding
to certain types of bacteria and yeast.
CR4. CR4 is found on both lymphoid
and myeloid cells. Its function is not well
characterized, but it appears to have op-
sonic activity for iC3b, and it plays a role in
adhesion.
Regulation of Complement Activation
There is a need to control or regulate the
enzyme and activation factors of the com-
plement cascade. The regulatory actions
of these control proteins prevent damage
to host tissue. These control systems
(Table 11-4) act by several different mecha-
nisms: direct inhibition of serine proteases,
decay and destruction of convertases, and
control of membrane attack complexes.19
Physiologic Effects of Complement
Activation
Opsonization. Neutrophils and macro-
phages phagocytose any particle that pro-
trudes from the surface of a cell or micro-
organism with no regard to the nature of
the material. Phagocytosis is more intense
if the particle adheres firmly to the mem-
brane of the phagocytic cell. To achieve
adherence, phagocytic cells have various
receptor molecules such as the Fc recep-
tors for certain immunoglobulins and
receptors for C3b. The enhancement of
phagocytosis resulting from antibody or
complement coating of cells is called
opsonization.
Anaphylatoxins Promote Inflammation.
Complement fragments C3a and C5a are
anaphylatoxins and they play an important
role in acute inflammation. These ana-
phylatoxins bind to receptors on mast cells
and basophils, causing them to release his-
tamine and other biologic response modifi-
ers that can be associated with anaphylaxis.
More frequently, anaphylatoxins affect vas-
cular permeability, membrane adhesion
properties, and smooth-muscle contraction
that constitute a large part of the acute in-
flammatory response. C5a and C3a also
cause neutrophils and macrophages to mi-
grate to the site of complement activation.
Cytokines
Throughout this chapter, inference has
been made to a number of soluble media-
tors termed cytokines. Cytokines are a
diverse group of intracellular signaling
peptides and glycoproteins that have mo-
lecular weights ranging from 6,000 to
60,000 daltons. Each cytokine is secreted
by particular cell types in response to dif-
262 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

ferent stimuli and has been implicated in
a wide variety of important regulatory bi-
ologic functions such as inflammation,
tissue repair, cell activation, cell growth,
fibrosis, and morphogenesis. One cytokine
can have several different functions, de-
pending on the type of cell to which it
binds. Several hundred different cytokines
have been described. Some of the cyto-
kines involved in immune functions are
summarized in Table 11-5.1,5 Cytokines
have been implicated in a variety of clini-
cally important factors of transfusion
medicine (hemolytic and febrile transfu-
sion reactions, immunomodulation, stem
cell collection, etc) (see Chapter 27).
Immunology Relating to
Transfusion Medicine
Several examples of how the immune sys-
tem relates to situations encountered in
transfusion medicine are described below,
including red cell alloimmunization, pla-
telet alloimmunization, immune-medi-
ated red cell destruction, and reagent an-
tibody production.
Chapter 11: Immunology 263
Table 11-4. Summary of the Inhibitors of Complement Activation
Complement Control Proteins Function
Inhibition of Serine Protease
C1 inhibitor
Decay/Destruction of Convertases
A serine protease inhibitor that binds and acti-
vates Clr and Cls
Factor I plus C4 binding protein (C4-bp)
C4-bp
Catabolizes C4 in the fluid phase
Causes dissociation of C2a from C4b2a
Decay accelerating factor
(DAF, CD55)
Inhibits the binding of C2 to C4b
Causes dissociation of C2 from C4b
Complement receptor 1 (CR1, CD35) Accelerates the dissociation of C3bBb
Membrane cofactor protein (MCP) Prompts catabolism of C4b by factor I and is a
cofactor with factor I to cleave C3b
Factor H Causes dissociation of Bb from C3I and C3b and
is a cofactor to factor I for catabolism of C3i
and C3b
Factor I Cleaves and degrades C3b using one of three
cofactors: factor H (plasma), CR1, or MCP
(membrane-bound)
Membrane Attack Complex (MAC)
S protein (Vitronectin) Binds to the C5b67 complex preventing insertion
into the lipid bilayer
C8-binding protein (CD59) This cell-bound protein binds to C8 preventing
C9 from inserting into the membrane
MAC-inhibiting protein (MIP) Inhibitor of C9, also known as homologous
restriction factor (HRF)
Copyright © 2005 by the AABB. All rights reserved.

264 AABB Technical Manual
Table 11-5. Summary of Some Cytokines Involved in the Immune System, Their
Site of Production, and Function1
Cytokine Produced by Primary Functions
Interleukins
IL-1 Many cells (eg, APCs,
endothelial cells, B cells,
fibroblasts)
T-cell activation, neutrophil
activation, stimulates
marrow, pyrogenic, acute-
phase protein synthesis
IL-2 Activated THcells T-cell growth, chemotaxis,
macrophage activation
IL-4 Activated THcells B-cell activation, B-cell
differentiation, T-cell
growth, TH2 differentiation
IL-6 Many cells (eg, T cells,
APCs, B cells, fibroblasts,
and endothelial cells)
B-cell differentiation,
pyrogenic, acute-phase
protein synthesis
IL-8 Many cells (eg, macrophages
and endothelial cells)
Inflammation, cell migration/
chemotaxis
IL-10 Activated TH2 cells Suppression of TH1 cells,
inhibits antigen
presentation, inhibits
cytokine production (IL-1, IL-6,
TNFα, and IFN)
Others
TNF Macrophages and
lymphocytes
Neutrophil activation,
pyrogenic, acute-phase
protein synthesis
Interferon γ(IFNγ) T cells Phagocyte activation
Transforming growth
factor-β(TGF-β)
Various cells Stimulates connective tissue growth
and collagen formation, inhibi-
tory function
Colony-stimulating
factors (GM-CSF,
M-CSF, G-CSF)
Various cells Growth and activation of phagocytic
cells
APCs = antigen-presenting cells; G-CSF = granulocyte colony-stimulating factor; GM-CSF = granulocyte macrophage–
colony-stimulating factor; M-CSF = macrophage colony-stimulating factor; TNF = tumor necrosis factor.
Copyright © 2005 by the AABB. All rights reserved.
Red Cell Alloimmunization
The mechanism of red cell alloimmuniza-
tion is not well understood. When allo-
geneic red cells are transfused, some of
the red cells fragment as they age or when
they pass through the spleen, thus releas-
ing membrane-bound red cell proteins
into the bloodstream as the fragments de-
grade. Both formal antigen-presenting
cells and B lymphocytes can present anti-
gen either as a primary immune response
(formal antigen presentation by dendritic
cells) or in the secondary immune re-
sponse by B cells.
HLA Alloimmunization to Platelets
Leukocyte reduction of red cells and pla-
telets to a threshold of 5 × 106per product
has been shown to reduce HLA alloim-
munization, possibly by the following
mechanism. Donor leukocytes express
both Class I and Class II MHC antigens.
Some of the donor Class II MHC antigens
will contain peptides that originated from
the MHC Class I antigens on the donor’s
cells. Most of the transfusion recipient’s T
cells recognize self-MHC-carrying foreign
peptides. However, as mentioned previ-
ously, a small percentage of T cells (<10%)
areabletorecognizeforeign-MHC-carry
-
ing foreign peptides. When platelets are
transfused, the recipient’s T helper cells
recognize the foreign MHC Class II com-
plex on the donor leukocytes. If a signal
occurs, the T cells activate recipient B
cells, which have also bound donor MHC
Class I antigen fragments to their Ig re-
ceptor, resulting in cell proliferation and
MHC Class I (HLA) antibody production
(Fig 11-6). In this case, the donor’s leuko-
cytes serve as the antigen-presenting
cells. This major mechanism is termed
“direct allorecognition” because the recip-
ient’s T cells are directly stimulated by do-
nor antigen-presenting cells.20 Alterna-
tively, the process of antigen presentation
and immune recognition of foreign HLA
peptides can occur by the recipient’s own
immune system. This alternative form of
allorecognition is termed “indirect” and is
the classical alloimmune response seen for
most foreign antigens. Leukocyte reduc-
tionappearstobeeffectiveinreducing
HLA alloimmunization because antigen
presentation by donor leukocytes is re-
duced and the amount of HLA Class I an-
tigens transfused is greatly reduced.
Immune-Mediated Red Cell Destruction
The activation of complement and/or the
presence of IgG on the red cell surface can
trigger red cell destruction by predomi-
nantly two mechanisms: intravascular
hemolysis and extravascular red cell de-
struction (Fig 11-7). Intravascular hemolysis
occurs when complement is activated,
resulting in activation of the membrane
attack complex. As the integrity of the
membrane is disrupted, hemoglobin is re-
leased into the plasma. Free hemoglobin
binds to haptoglobin and is excreted in
the urine. The heme portion of hemoglo-
bin binds to albumin, forming methemal-
bumin, which can be visually detected in
plasma by a brownish green discolor-
ation. At times, the inhibitors of comple-
ment stop the cascade, leaving C3b on the
red cells. This complement fragment has
chemotactic activity for phagocytic cells
and these cells have receptors for C3b.
C3b-coated red cells adhere to phagocytic
cells but are relatively ineffective at trig-
gering phagocytosis. Enzymes can cleave
the cell-bound C3b, leaving a small frag-
ment (C3dg) present on the cells. C3dg-
sensitized red cells survive normally be-
cause phagocytic cells do not have recep-
tors for C3dg. If IgG is present on the red
cells, binding to Fcγreceptors will occur,
resulting in phagocytosis. If both IgG and
Chapter 11: Immunology 265
Copyright © 2005 by the AABB. All rights reserved.
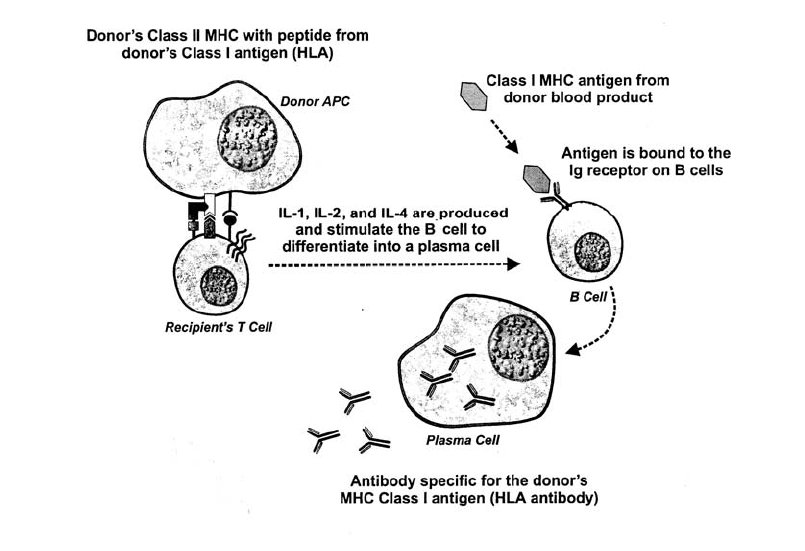
C3b are present on the red cells, clearance
of the cells may be enhanced, probably
because of the chemotactic function of
C3b and its adherence capability.
Reagent Antibodies
Heterogeneous antibodies are not opti-
mal as reagents for use in serologic testing
because they can vary in concentration,
serologic properties, and epitope recogni-
tion and can contain other antibodies of
unwanted specificity. The ideal serum for
serologic testing is a concentrated sus-
pension of highly specific, well-character-
ized, uniformly reactive, immunoglobulin
molecules. Until the 1970s, the only way
to obtain reagents was to immunize ani-
mals or humans with purified antigens and
then perform time-consuming and some-
times unpredictable separation techni-
ques in an attempt to purify the resulting
sera. Monoclonal antibody production
provided an alternative to human and an-
imal sources of these proteins. Using this
approach, a single B-cell clone is propa-
gated in cell culture and the supernatant
fluid from the culture contains antibody
of a single specificity. However, there are
problems with this approach. Normal B
cells reproduce themselves only a limited
number of times; hence, the cultured cell
lines survive only a short time.
In 1976, Köhler and Milstein21 provided a
solution to this problem. Plasma cells of
normal antibody-producing capacity were
fused to neoplastic plasma cells of infinite
reproductive capacity (ie, myeloma cells).
Techniques had previously been developed
that cause cell membranes to merge, allow-
ing the cytoplasm and the nucleus of two
266 AABB Technical Manual
Figure 11-6. Diagram illustrating the major mechanism of HLA alloimmunization due to leukocytes
present in platelet transfusion. (Used with permission from Heddle.1)
Copyright © 2005 by the AABB. All rights reserved.
different kinds of cells to fuse into a single
cell. These plasma cell/myeloma cell hy-
brids can be maintained in cell culture for
prolonged periods, producing large quanti-
ties of the selected antibody.
The exquisite specificity of monoclonal
antibodies is both an advantage and a dis-
advantage for reagent use. An antibody that
gives strong and specific reactions with one
epitope of a multivalent antigen molecule
may fail to react with cells whose antigen
expression lacks that particular configura-
tion. Thus, reagent preparations typically
used in the laboratory are blends of several
different monoclonal products, thereby in-
creasing the range of variant phenotypes
that the antiserum can identify. Single or
blended monoclonal preparations often re-
act more strongly than immune-serum
preparations when tested against cells with
weakly expressed antigens.
Phage technology is under investigation
as an approach for producing genetically
engineered antibodies for a variety of thera-
peutic treatments,22 as well as for use as
typing reagents.23
References
1. Heddle NM. Overview of immunology. In:
ReidME,NanceSJ,eds.Redcelltransfusion.
A practical guide. Totowa, NJ: Humana Press,
1998:13-37.
2. Barclay AN. Membrane proteins with immu-
noglobulin-like domains—a master super-
family of interaction molecules. Semin
Immunol 2003;15:215-3.
Chapter 11: Immunology 267
*Ineffective mediator of phagocytosis.
†Due to the chemotactic ability of C3b, extravascular destruction may be enhanced when both IgG and C3b are present on the
red cells.
Figure 11-7. Summary of intravascular and extravascular red cell destruction.
Copyright © 2005 by the AABB. All rights reserved.
3. MarshSG,AlbertED,BodmerWF,etal.No
-
menclature for factors of the HLA system, 2002.
Hum Immunol 2002;63:1213-68.
4. Santos-Aguado J, Barbosa JA, Biro A, Stro-
minger JL. Molecular characterization of se-
rologic recognition sites in the human HLA-
A2 molecule. J Immunol 1988;141:2811-18.
5. RoittI,BrostoffJ,MaleD.Immunology.5th
ed. St. Louis: Mosby, 1998.
6. Ono SJ, Nakamura T, Miyazaki D, et al.
Chemokines: Roles in leukocyte develop-
ment, trafficking, and effector function. J Al-
lergy Clin Immunol 2003;111:1185-99.
7. Huston DP. The biology of the immune sys-
tem. JAMA 1997;278:1804-14.
8. Goldsby RA, Kindt TJ, Osborne BA. Kuby im-
munology. 4th ed. New York: WH Freeman
and Company, 2000.
9. Tonegawa S. Somatic generation of antibody
diversity. Nature 1983;302:575-81.
10. Alt FW, Backwell TK, Yancopoulos GD. Devel-
opment of the primary antibody repertoire.
Science 1987;238:1079-87.
11. Janeway CA Jr. How the immune system rec-
ognizes invaders. Sci Am 1993;269(3):73-9.
12. Russell DM, Dembic Z, Morahan G, et al. Pe-
ripheral deletion of self-reactive B cells. Na-
ture 1991;354:308-11.
13. Weissman IL, Cooper MD. How the immune
system develops. Sci Am 1993;269(3):64-71.
14. Paul WE. Infectious diseases and the immune
system. When bacteria, viruses and other
pathogens infect the body, they hide in differ-
ent places. Sci Am 1993;269(3):90-7.
15. MarrackP,LoD,BrinsterR,etal.Theeffects
of thymus environment on T cell develop-
ment and tolerance. Cell 1988;53:627-34.
16. Clevers H, Alarcon B, Willeman T, Terhorst C.
The T cell receptor/CD3 complex: A dynamic
protein ensemble. Annu Rev Immunol 1988;
6:629-62.
17. Sakamoto M, Fujisawa Y, Nishioka K. Physio-
logic role of the complement system in host
defense, disease, and malnutrition. Nutrition
1998;14:391-8.
18. RoittI.Essentialimmunology.8thed.Oxford:
Blackwell Scientific Publications, 1994.
19. Devine DV. The regulation of complement on
cell surfaces. Transfus Med Rev 1991;5:123-31.
20. Semple JW, Freedman J. Recipient antigen-
processing pathways of allogeneic platelet
antigens: Essential mediators of immunity.
Transfusion 2002;42:958-61.
21. Köhler G, Milstein C. Derivation of specific
antibody-producing tissue culture and tumor
lines by cell fusion. Eur J Immunol 1976;6:
511-19.
22. MarksC,MarksJD.Phagelibraries—anew
route to clinically useful antibodies. N Engl J
Med 1996;335:730-3.
23. Siegel DL. Phage display tools for automated
blood typing (abstract). Transfusion 2004;
44(Suppl):2A.
Suggested Reading
AbbasAK,LichtmanAH,PoberJS,eds.Cellular
and molecular immunology. 4th ed. Philadelphia:
WB Saunders, 2000.
Anderson KC, Ness PM, eds. Scientific basis of
transfusion medicine. 2nd ed. Philadelphia: WB
Saunders, 1999.
Barclay AN, Brown MH, Law SKA, et al. The leuko-
cyte antigen factsbook. 2nd ed. San Diego, CA: Ac-
ademic Press, 1997.
Carroll MC. The role of complement and comple-
ment receptors in induction and regulation of im-
munity. Annu Rev Immunol 1998;16:545-68.
Janeway CA. Immunobiology: The immune system
in health and disease. 5th ed. New York: Garland
Publishing, 2001.
Marchalonis JJ, Schluter SF, Bernstein RM, et al.
Phylogenetic emergence and molecular evolution
of the immunoglobulin family. Adv Immunol 1998;
70:417-506.
Muller D. The molecular biology of autoimmunity.
Immunol Allerg Clin North Am 1996;16:659-82.
PaulWE.Fundamentalimmunology.5thed.Phila
-
delphia: Lippincott Williams & Wilkins, 2003.
Paul W, Raghavan M, Bjorkman PJ. Fc receptors
and their interactions with immunoglobulins.
Annu Rev Cell Dev Biol 1996;12:181-220.
Stites DP, Terr AI, Parslow TG, eds. Medical immuno-
logy. 10th ed. Stamford, CT: Appleton & Lange, 2001.
Vamvakas EC, Blajchman MA, eds. Immunomodu-
latory effects of blood transfusion. Bethesda, MD:
AABB Press, 1999.
268 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Appendix 11-1. Definitions of Some Essential Terms in Immunology
Adhesion molecule:
Any of the many membrane
molecules expressed on white cells and en-
dothelial cells that allow cells to come into
close apposition with each other.
Allotypic:
Variations in the amino acid structure
of heavy and light chains unrelated to anti-
body specificity. Present in some but
not all
members of the species.
Antibody:
Immunoglobulin secreted by the
plasma-cell progeny of B lymphocytes after
stimulation by a specific immunogen. Immu-
noglobulin molecules on the surface of un-
stimulated lymphocytes serve as antigen
receptors.
Antigen:
Any material capable of specific combi-
nation with antibody or with cell-surface re-
ceptors of T lymphocytes. Often used as a
synonym for “immunogen,” although some
antigens that react with products of the im-
mune response are not capable of eliciting
an immune response.
Antigen-presenting cell:
A cell capable of incor-
porating antigenic epitopes into MHC Class II
molecules and displaying the epitope-MHC
complex on its membrane.
Clone:
A population of genetically identical cells
derived from successive divisions of a single
progenitor cell.
Cytokine:
A low-molecular-weight protein, se-
creted from an activated cell, that affects the
function or activity of other cells.
Epitope:
The small portion of an immunogen,
usually5to15aminoacidsor3to5glyco-
sides, that combines specifically with the an-
tigen receptor of a T or B lymphocyte.
Idiotype:
The molecular configuration unique to
the variable portion of an antigen-receptor
molecule, reflecting the DNA rearrangement
occurring in earliest lymphocyte differentia-
tion and conferring upon the cell its specific-
ity of antigen recognition.
Immune system:
A collective term for all the
cells and tissues involved in immune activity.
It includes, in addition to lymphocytes and
cells of monocyte/macrophage lineage, the
thymus, lymph nodes, spleen, marrow, por-
tions of the liver, and the mucosa-associated
lymphoid tissue.
Immunogen:
A material capable of provoking an
immune response when introduced into an
immunocompetent host to whom it is for-
eign.
Ligand:
A molecule, either free in a fluid milieu
or present on a membrane, whose three-di-
mensional configuration allows it to form a
tightly fitting complex with a cell-surface
molecule (its receptor) of complementary
shape.
MHC Class I molecules:
Heterodimeric membrane
proteins determined by genes in the MHC,
consisting of a highly polymorphic αchain
linked noncovalently with the nonpolymorphic
β2-microglobulin chain; these molecules
present antigen to CD8+ T cells and are the
site of HLA antigens of the HLA-A, HLA-B,
and HLA-C series.
MHC Class II molecules:
Heterodimeric mem-
brane proteins determined by genes in the
MHC, consisting of two transmembrane
polypeptide chains; these molecules present
antigen to CD4+ T cells and exhibit the
HLA-DP, HLA-DQ, and HLA-DR series of an-
tigens.
Phagocytosis:
The process whereby macro-
phages and granulocytes ingest particulate
material present in the surrounding milieu
and subject it to intracellular alteration.
Receptor:
A cell-membrane protein molecule
whose three-dimensional configuration al-
lows it to form a tightly fitting complex with
another molecule (called its ligand) of com-
plementary shape.
Chapter 11: Immunology 269
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 12: Red Cell Antigen-Antibody Reactions and Their Detection
Chapter 12
Red Cell Antigen-Antibody
Reactions and Their
Detection
DEMONSTRATION OF RED cell an-
tigen-antibody reactions is key to
immunohematology. The combi-
nation of antibody with antigen may pro-
duce a variety of observable results. In
blood group serology, the most com-
monly observed reactions are agglutina-
tion, hemolysis, and precipitation.
Agglutination is the antibody-mediated
clumping of particles that express antigen
on their surface. Agglutination of red cells
occurs because antibody molecules bind to
antigenic determinants on multiple adja-
cent red cells, linking them together to
form a visible aggregate. Agglutination is
the endpoint for most tests involving red
cells and blood group antibodies and is the
primary reaction type discussed in this
chapter. In some tests, antibody directly
bridges the gap between adjacent cells; in
others, antibody molecules attach to, but
do not agglutinate, the red cells, and an ad-
ditional step is needed to induce visible ag-
glutination or to otherwise measure the
reaction.
Hemolysis is the rupture of red cells with
release of intracellular hemoglobin. In-vitro
antibody-mediated hemolysis depends on
activity of the membrane attack unit of
complement. Hemolysis does not occur if
the antigen and antibody interact in serum
that lacks complement, or in plasma in
which the anticoagulant has chelated the
cations (calcium and magnesium) neces-
sary for complement activation. In tests for
antibodies to red cell antigens, hemolysis is
a positive result because it demonstrates
the union of antibody with antigen that ac-
tivates the complement cascade. (The ac-
tions of complement are described in
Chapter 11.) Pink or red supernatant fluid
in a test system of serum and red cells is an
important observation that may indicate
that antigen-antibody binding has taken
place. Some antibodies that are lytic in vitro
(eg, anti-Vel, anti-Lea,andanti-Jk
a)may
271
12
Copyright © 2005 by the AABB. All rights reserved.
cause intravascular hemolysis in a transfu-
sion recipient.
Precipitation is the formation of an in-
soluble, usually visible, complex when solu-
ble antibody reacts with soluble antigen.
Such complexes are seen in test tubes as a
sediment or ring and in agar gels as a white
line. Precipitation is the endpoint of proce-
duressuchasimmunodiffusionandim
-
munoelectrophoresis.
Precipitation may not occur, even when
soluble antigen and its specific antibody
are present. Precipitation of the antigen-an-
tibody complex requires that antigen and
antibody be present in optimal propor-
tions. If antibody is present in excess, too
few antigen sites exist to crosslink the mol-
ecules and a lattice structure is not formed.
Antigen-antibody complexes form but do
not accumulate sufficiently to form a visi-
ble lattice. This phenomenon is called a
prozone.
The combination of soluble antigen with
soluble antibody may also result in a full or
partial neutralization of the antibody. Al-
though a visible precipitate is often not
produced, such inhibition can be useful in
antibody identification procedures by se-
lectively eliminating specific antibodies.
Factors Affecting Red Cell
Agglutination
Agglutination is a reversible chemical re-
action and is thought to occur in two
stages: 1) sensitization, the attachment of
antibody to antigen on the red cell mem-
brane; and 2) formation of bridges be-
tween the sensitized red cells to form the
lattice that constitutes agglutination. Vari-
ous factors affect these two stages and
can be manipulated to enhance (or de-
crease) agglutination. The effects of en-
hancement techniques on the two stages
cannot always be clearly differentiated.1
Stage One: Sensitization
Initially, the antigen and antibody must
come together and interact in a suitable
spatial relationship. The chance of associ-
ation between antibody and antigen can
be enhanced in a number of ways, such as
agitation or centrifugation, or by varying
the relative concentration of antibody and
antigen.AsshowninFig12-1,antibody
and antigen must complement each other
with both a structural (steric) and a chem-
ical fit.
For sensitization to occur, a noncovalent
chemical bond must form between antigen
and antibody. The forces holding antigens
and antibodies together are generally weak
(compared with covalent bonds that hold
molecules together) and are active only
over a very short distance. The antigen-an-
tibody combination is reversible, and ran-
dom bonds are constantly made and bro-
ken until a state of equilibrium is attained.
272 AABB Technical Manual
Figure 12-1. Antigen-antibody “goodness of fit.”
For maximum complementarity, both structural
fit and complementary distribution of chemical
groups must be achieved. (A) Good structural fit
with complementary chemical attraction.
(B) Chemical groups are complementary, but
structural fit is poor. (C) Good structural fit, but
chemical groupings are not attractive and may
repel each other. (Reprinted with permission
from Moore.2)
Copyright © 2005 by the AABB. All rights reserved.

Chemical Bonding
Various types of chemical bonds are re-
sponsible for the binding of antibody to
antigen, including hydrogen bonds, hy-
drophobic bonds, electrostatic or ionic
bonds, and van der Waals forces. These
types of chemical bonds are relevant to
immunohematology because different types
of bonds have different thermodynamic
characteristics; they are either exothermic
or endothermic. Thermodynamic charac-
teristics, in turn, may affect the serologic
phenomena observed in the test system.
For example, carbohydrate antigens tend
to form exothermic hydrogen bonds with
the antibody-combining site, so the bond
is stronger at lower temperatures. In con-
trast, hydrophilic bonds formed with pro-
tein antigens are endothermic, so these
bonds are enhanced at higher reaction
temperatures.
Equilibrium (Affinity) Constant of the
Antibody
The equilibrium constant or affinity con-
stant (Ko) of a reaction is determined by
the relative rates of association and disso-
ciation (see Fig 12-2). For each antigen-
antibody reaction, the Kovaries. The Kore-
flects the degree to which antibody and
antigen associate and bind to one another
(“goodness of fit”) and the speed of the re-
action. The higher the Kovalue, the better
the association or “fit.” When the Kois
large, the reaction occurs more readily
and is more difficult to dissociate; such
antibodies may have a greater clinical im-
portance. When the Kois small, a higher
ratioofantibodytoantigenmaybere
-
quired for detection.
The degree of antigen-antibody “fit” is
influenced by the type of bonds predomi-
nating. Hydrophobic bonds are usually as-
sociated with higher Kothan hydrogen
bonds. The Kois also affected by physical
conditions such as the temperature at
which the reaction occurs, the pH and ionic
strength of the suspending medium, and
the relative antigen-to-antibody concentra-
tions. In laboratory tests that use agglutina-
tion as an endpoint, altering the physical
conditions of the system can increase or
decrease the test’s sensitivity.
Temperature
Most blood group antibodies react within
restricted temperature ranges. Typically,
these antibodies fall into two broad cate-
gories: those optimally reactive at “cold”
temperatures (eg, 4 to 25 C) and those op-
timally reactive at “warm” temperatures
(eg, 30 to 37 C). Antibodies that react in
vitro only at temperatures below 30 C
rarely cause destruction of transfused an-
Chapter 12: Red Cell Antigen-Antibody Reactions and Their Detection 273
All chemical reactions are reversible. Anti-
gen (Ag)-antibody (Ab) reactions may be
expressed as
Ag Ab AgAb+→
←
The reaction proceeds until a state of
equilibrium is reached. This is controlled
by the rate constants of association (ka)
and dissociation (kd).
Ag Ab AgAb
k
k
d
a
+→
←
By the law of mass action, the speed of
the reaction is proportional to the concen-
trations of the reactants and their prod-
uct. The equilibrium constant (Ko)isa
function of these intrinsic association
constants for the antibody being tested.
[AbAg]
[Ab][Ag]
k
kK
a
d
o
==
Figure 12-2. The law of mass action and the equi-
librium constant.
Copyright © 2005 by the AABB. All rights reserved.
tigen-positive red cells and are generally
considered clinically insignificant. Many
of these “cold-reactive” antibodies have been
found to be IgM, whereas their “warm-re-
active” counterparts have often been found
to be IgG. This has led to the mistaken
conclusion by many that antibody class
determines the temperature of bonding
(and clinical significance). Instead, how-
ever, the temperature of optimal antigen-
antibody reactivity has more to do with
thetypeofreactionandthechemicalna
-
ture of the antigen than with the antibody
class. Carbohydrate antigens are more
commonly associated with “cold-reactive”
antibodies and protein antigens with
“warm-reactive” antibodies.
pH
Changes in pH can affect electrostatic
bonds. For most clinically significant
blood group antibodies, optimal pH has
not been determined but is assumed to
approximate the physiologic pH range.
Occasional antibodies, notably some ex-
amples of anti-M, react best at a lowered
pH. For most routine testing, a pH around
7.0 should be used. Stored saline often has
a pH of 5.0 to 6.0. Buffered saline is an al-
ternativeandmaybeparticularlyhelpful
in solid-phase testing.3
Incubation Time
The time needed to reach equilibrium dif-
fers for different blood group antibodies.
Significant variables include temperature
requirements, immunoglobulin class, and
specific interactions between antigen
configuration and the Fab site of the anti-
body. The addition of enhancement agents
to the system can decrease the incubation
time needed to reach equilibrium.
For saline systems in which antiglobulin
serum is used to demonstrate antibody at-
tachment, 30 to 60 minutes of incubation at
37 C is adequate to detect most clinically
significant antibodies. For some weakly re-
active antibodies, association may not
reach equilibrium at 30 minutes and ex-
tending the incubation time may increase
sensitivity of the test. Prolonging the incu-
bation time beyond 60 minutes has few dis-
advantages except for the delay before re-
sults are available.
Incubation time at 37 C can usually be
reduced to 10 to 15 minutes if a low-ionic-
strength saline (LISS) solution is used (in-
cluding LISS additive solutions). The use of
water-solublepolymerssuchaspolyethyl
-
ene glycol (PEG) can also reduce the neces-
sary incubation time, although for different
reasons. (See section on Enhancement of
Antibody Detection and Method Section 3.)
Ionic Strength
In normal saline, Na+and Cl–ions cluster
around and partially neutralize opposite
charges on antigen and antibody mole-
cules. This hinders the association of anti-
body with antigen. By lowering the ionic
strength of the reaction medium, how-
ever, this shielding effect can be weak-
ened and electrostatic attractions enhanced.
Reducing the salt concentration of the se-
rum-cell system increases the rate at
which antibody and antigen come into
proximity and may increase the amount
of antibody bound. Extending the incuba-
tion time in LISS systems may result in a
loss of sensitivity.4(See section on LISS and
LISS Additives.)
Antigen-Antibody Proportions
An excess of antigen to antibody should
result in increased antibody uptake. For
inhibition or adsorption tests, such an ex-
cess of antigen is desirable. For most red
cell tests, however, antigen excess reduces
the number of antibody molecules bound
274 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
per red cell, limiting their ability to agglu-
tinate. Antibody excess is, therefore, de-
sirable in most routine test systems. A
commonly used ratio in red cell serology
is 2 drops of serum to 1 drop of a 2% to
5% red cell suspension. If the antibody is
weakly reactive, increasing the quantity of
antibody present can increase the test’s
sensitivity. Very rarely, significant anti-
body excess may inhibit direct agglutina-
tion, producing a prozone phenomenon
comparable to what occurs with precipi-
tation reactions. Usually, however, in-
creasing antibody concentration en-
hances the sensitivity of agglutination
tests. Reducing the concentration of red
cells from 5% to 2% or 3% doubles the se-
rum-to-cell ratio, as does adding 4 drops
of serum to the standard cell suspension.
Sometimes, it is useful to increase the vol-
ume of serum to 10 or even 20 drops, par-
ticularly during an investigation of a
hemolytic transfusion reaction in which
routine testing reveals no antibody. Alter-
ations in the volume of serum or plasma
significantly affect the ionic strength of
test systems in which LISS has reduced
the dielectric constant, so procedures
must be modified so that the appropriate
ratio of serum to LISS is maintained.
Chapters 18 and 19 give more details
about antibody detection and pretrans-
fusion testing.
Stage Two: Agglutination
Once antibody molecules attach to anti-
gens on the red cell surface, the sensitized
cells must be linked into a lattice. This al-
lows visualization of the reaction. The size
and physical properties of the antibody
molecules, the concentration of antigen
sites on each cell, and the distance be-
tween cells all have an effect on the devel-
opment of agglutinates.
The bridges formed between antibodies
interlinked to antigen sites on adjacent red
cells usually result from chance collision of
the sensitized cells. Under isotonic condi-
tions, red cells cannot approach each other
closer than a distance of 50 to 100 Å.1IgG
molecules characteristically fail to bridge
this distance between red cells and cause
sensitization without lattice formation. For
larger, multivalent IgM molecules, however,
direct agglutination occurs easily. The loca-
tion and density of antigen sites on the cells
may also allow some IgG antibodies to
cause direct agglutination; A, B, M, and N
antigens, for example, are on the outer
edges of red cell glycoproteins and have rel-
atively high densities, allowing IgG antibod-
ies to crosslink.
Red cells suspended in saline have a net
negative charge at their surface and there-
fore repel one another. Negatively charged
molecules on the red cell membrane cause
mutual repulsion of red cells. This repul-
sion may be decreased by various labora-
tory manipulations and by inherent or al-
tered red cell membrane characteristics.
Various strategies are used to overcome
this repulsion and to enhance agglutina-
tion. Centrifugation physically forces the
cells closer together. The indirect anti-
globulin test (IAT) uses antiglobulin serum
to crosslink the reaction. Other methods in-
clude reducing the negative charge of sur-
face molecules (eg, proteolytic enzymes),
reducing the hydration layer around the cell
(eg, albumin), and introducing positively
charged macromolecules (eg, Polybrene®)
that aggregate the cells.1
Inhibition of Agglutination
In agglutination inhibition tests, the pres-
ence of either antigen or antibody is de-
tected by its ability to inhibit agglutina-
tion in a system with known reactants
(see Chapter 19). For example, the saliva
Chapter 12: Red Cell Antigen-Antibody Reactions and Their Detection 275
Copyright © 2005 by the AABB. All rights reserved.
from a secretor contains soluble blood
group antigens that combine with anti-A,
-B, or -H. The indicator system is a stan-
dardized dilution of antibody that aggluti-
nates the corresponding cells to a known
degree. If the saliva contains blood group
substance, incubating saliva with anti-
body will wholly or partially abolish ag-
glutination of cells added to the incu-
bated mixture. The absence of expected
agglutination indicates the presence of
soluble antigen in the material under test.
Agglutination of the indicator cells is a
negative result.
Enhancement of Antibody
Detection
Albumin Additives
Although used routinely for many years as
an enhancement medium, albumin itself
probably does little to promote antibody
uptake (Stage 1). Much of the enhance-
ment effect attributed to albumin may be
due to its attributes as a low-ionic-strength
buffer. Albumin may influence agglutina-
tion by reducing the repulsion between
cells, thus predisposing antibody-coated
cells to agglutinate. Bovine serum albu-
min is available as solutions of 22% or
30% concentration.
Enzymes
The proteolytic enzymes used most often
in immunohematology laboratories are
bromelin, ficin, papain, and trypsin. While
enhancing agglutination by some anti-
bodies, the enzymes destroy certain red
cellantigens,notablyM,N,S,Fy
a,andFy
b.
Proteolytic enzymes reduce the red cell
surfacechargebycleavingpolypeptides
containing negatively charged sialic acid
molecules from polysaccharide chains.
Sialic acid is a major contributor to the net
negative charge at the red cell surface. Any
mechanism that reduces the net charge
should enhance red cell agglutination, and
red cells pretreated with proteolytic en-
zymes often show enhanced agglutination
by IgG molecules.
Polyethylene Glycol
PEG is a water-soluble linear polymer
used as an additive to potentiate anti-
gen-antibody reactions.5It has been sug-
gested that PEG promotes antibody up-
take through steric exclusion of water
molecules in the diluent, such that anti-
gens and antibody molecules come into
closer proximity, resulting in increased
cell-antibody collisions and subsequent
antibody binding. Multiple studies have
shown that PEG increases the detection of
potentially clinically significant antibod-
ies and decreases the detection of clini-
cally insignificant antibodies.6
Anti-IgG is usually the antihuman globu-
lin (AHG) reagent of choice with PEG test-
ing, to avoid false-positive reactions with
some polyspecific AHG reagents. Commer-
cially available PEG reagents may be pre-
pared in a LISS solution. PEG can be used
intestswitheluates,aswellaswithserum
or plasma. PEG can enhance warm-reactive
autoantibodies and thus may be advanta-
geous in detecting weak serum autoanti-
bodies for diagnostic purposes. On the
other hand, this enhancement may be dis-
advantageous when trying to detect allo-
antibodies in the presence of autoanti-
bodies. In such cases, testing the serum by
LISS or a saline IAT may allow for the detec-
tion or exclusion of alloantibodies without
interference from autoantibodies.
Centrifugation of PEG with test serum
and red cells before washing should be
avoided because the nonspecific aggregates
276 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
generated by PEG may not disperse. After
incubation with PEG, test cells should be
washed immediately in saline for the
antiglobulin test.
Precipitation of serum proteins when
PEG is added has been reported; this prob-
lem appears to be related to elevated serum
globulin levels.6This problem becomes ap-
parent when the IgG-coated red cells are
nonreactive. At least four washes of the red
cells at the antiglobulin phase, with agita-
tion, will ensure that the red cells are fully
resuspended and will serve to prevent this
problem from occurring.
LISS and LISS Additives
LISS (approximately 0.03 M) greatly in-
creases the speed of antibody sensitiza-
tion of red cells, compared with normal
saline (approximately 0.17 M). To prevent
lysis of red cells at such a low ionic strength,
a nonionic substance such as glycine is
incorporated in the LISS.
Most laboratories use a LISS additive re-
agent, rather than LISS itself. These com-
mercially available LISS additives may con-
tain albumin in addition to ionic salts and
buffers. LISS solutions increase the rate of
antibody association (Stage 1) when vol-
umeproportionsarecorrect.(SeeMethods
3.2.2 and 3.2.3.) Increasing the volume of
serum used in a test will increase the ionic
strength of the test; hence, any alteration in
prescribed volumes of serum used requires
adjustment of the LISS volume or omission
of LISS. For this reason, the use of LISS for
routine titration studies and for some other
testsisproblematic.WhenLISSisusedas
an additive reagent, the manufacturer’s in-
structions must be followed.
The Antiglobulin Test
In 1945, Coombs, Mourant, and Race7de-
scribed procedures for detecting attach-
ment of antibodies that did not produce
agglutination. This test uses antibody to
human globulins and is known as the
antiglobulin test. It was first used to dem-
onstrate antibody in serum, but later the
same principle was used to demonstrate
in-vivo coating of red cells with antibody
or complement components. As used in
immunohematology, antiglobulin testing
generates visible agglutination of sensi-
tized red cells. The direct antiglobulin test
(DAT) is used to demonstrate in-vivo sen-
sitization of red cells. An IAT is used to
demonstrate in-vitro reactions between
red cells and antibodies that sensitize, but
do not agglutinate, cells that express the
corresponding antigen.
Principles of the Antiglobulin Test
All antibody molecules are globulins. Ani-
mals injected with human globulins pro-
duce antibody to the foreign protein. Af-
ter the animal serum is adsorbed to remove
unwanted agglutinins, it will react specifi-
cally with human globulins and can be
called AHG serum. AHG sera with varying
specificities can be produced, notably,
anti-IgG and antibodies to several com-
plement components. Hybridoma tech-
niques are used for the manufacture of
most AHG. These techniques are more
fully described in Chapter 11.
Anti-IgG combines mainly with the Fc
portion of the sensitizing antibody mole-
cules, not with any epitopes native to the
red cell (see Fig 12-3). The two Fab sites of
the AHG molecule form a bridge between
adjacent antibody-coated cells to produce
visible agglutination. Cells that have no
globulin attached will not be agglutinated.
The strength of the observed agglutination
is usually proportional to the amount of
bound globulin.
AHG will react with human antibodies
and complement molecules that are bound
Chapter 12: Red Cell Antigen-Antibody Reactions and Their Detection 277
Copyright © 2005 by the AABB. All rights reserved.
to red cells or are present, free, in serum.
Unbound globulins may react with AHG,
causing false-negative antiglobulin tests.
Unlesstheredcellsarewashedfreeofun-
bound proteins before addition of AHG se-
rum, the unbound globulins may neutralize
AHG and cause a false-negative result.
Direct Antiglobulin Testing
The DAT is used to demonstrate in-vivo
coating of red cells with antibodies or
complement, in particular IgG and C3d.
Washed red cells from a patient or donor
are tested directly with AHG reagents (see
Method 3.6). The DAT is used in investi-
gating autoimmune hemolytic anemia
(AIHA), drug-induced hemolysis, hemolytic
disease of the fetus and newborn, and
alloimmune reactions to recently trans-
fused red cells.
Indirect Antiglobulin Testing
In indirect antiglobulin procedures, se-
rum (or plasma) is incubated with red
cells, which are then washed to remove
unbound globulins. Agglutination that oc-
curs when AHG is added indicates that
antibody has bound to a specific antigen
present on the red cells. The specificity of
the antibody may be known and the anti-
gen unknown, as in blood group pheno-
typing with an AHG-reactive reagent such
as anti-Fya. The presence or specificity of
antibody may be unknown, as in antibody
detection and identification tests. Or, in
other applications, such as the crossmatch,
the serum and cells are unknown. This
procedure is used to determine whether
any sort of antigen-antibody interaction
has occurred.
Methods have been developed that obvi-
ate the need to wash coated red cells before
adding antiglobulin reagent. Column ag-
glutination technology, described later in
this chapter, is an example. It uses a micro-
column filled with mixtures of either glass
beads or gel, buffer, and sometimes re-
agents and can be used for direct or indi-
rect antiglobulin procedures. Density barri-
ers allow separation of test serum (or plasma)
from red cells, making a saline washing
phase unnecessary.
Antiglobulin Reagents
Monospecific antibodies to human globu-
lins can be prepared by injecting animals
with purified IgG, IgA, IgM, C3, or C4.
Such sera require adsorption to remove
unwanted (eg, heterophile) antibodies
from the monospecific AHG reagent.
These animal-made antisera are poly-
clonal in nature. Monospecific mono-
clonal reagents can also effectively be
prepared from hybridomas (see Chapter
11). Monospecific animal or hybridoma-
derived antibodies can be combined into
reagent preparations containing any de-
sired combination of specificities, or dif-
278 AABB Technical Manual
Figure 12-3. The antiglobulin reaction. Anti-
human IgG molecules are shown reacting with
the Fc portion of human IgG coating adjacent red
cells (eg, anti-D coating D-positive red cells).
Copyright © 2005 by the AABB. All rights reserved.

ferent clones all recognizing the same an-
tigen specificity can be combined into a
single reagent. Thus, reagents may be
polyclonal, monoclonal, blends of mono-
clonal, or blends of monoclonal and poly-
clonal antibodies.
The Food and Drug Administration
(FDA) has established definitions for a vari-
ety of AHG reagents,8as shown in Table 12-1.
Antisera specific for other immunoglobulins
(IgA, IgM) or subclasses (IgG1, IgG3, etc) ex-
ist but are rarely standardized for routine
test tube methods and must be used with
rigorous controls.
Polyspecific AHG
Polyspecific AHG reagents are used for
DATs and, in some laboratories, for rou-
tine compatibility tests and antibody de-
tection. These reagents contain antibody
to human IgG and to the C3d component
of human complement. Other comple-
ment antibodies may be present, includ-
ing anti-C3b, -C4b, and -C4d. Currently
available, commercially prepared, poly-
specific antiglobulin sera contain little, if
any, activity against IgA and IgM heavy
chains. However, some reagents may re-
act with IgA or IgM molecules because the
polyspecific mixture may react with lambda
and kappa light chains, which are present
in immunoglobulins of all classes.
Because most clinically significant anti-
bodies are IgG, the most important func-
tion of polyspecific AHG, in most procedures,
is detection of IgG. The anticomplement
Chapter 12: Red Cell Antigen-Antibody Reactions and Their Detection 279
Table 12-1. Antihuman Globulin Reagents
Antibody Designation on
Container Label Definition*
(1) Anti-IgG, -C3d;
Polyspecific
Contains anti-IgG and anti-C3d (may contain other
anticomplement and anti-immunoglobulin antibodies).
(2) Anti-IgG Contains anti-IgG with no anticomplement activity (not neces-
sarily gamma chain specific).
(3) Anti-IgG; heavy
chains
Contains only antibodies reactive against human gamma
chains.
(4) Anti-C3b Contains only C3b antibodies with no anti-immunoglobulin ac-
tivity. Note: The antibody produced in response to immuni-
zation is usually directed against the antigenic determinant,
which is located in the C3c subunit; some persons have
called this antibody “anti-C3c.” In product labeling, this an-
tibody should be designated anti-C3b.
(5) Anti-C3d Contains only C3d antibodies with no anti-immunoglobulin
activity.
(6) Anti-C4b Contains only C4b antibodies with no anti-immunoglobulin
activity.
(7) Anti-C4d Contains only C4d antibodies with no anti-immunoglobulin
activity.
*As defined by the FDA.8
Copyright © 2005 by the AABB. All rights reserved.
component has limited usefulness in cross-
matching and in antibody detection be-
cause antibodies detectable only by their
ability to bind complement are quite rare.
Anti-C3d activity is important, however, for
the DAT, especially in the investigation of
AIHA. In some patients with AIHA, C3d
may be the only globulin detectable on
their red cells.9
Monospecific AHG Reagents
Licensed monospecific AHG reagents in
common use are anti-IgG and anti-C3b,
-C3d. The FDA has established labeling
requirements for other anticomplement
reagents, including anti-C3b, anti-C4b,
and anti-C4d, but these products are not
generally available. If the DAT with a
polyspecific reagent reveals globulins on
red cells, monospecific AHG reagents are
used to characterize the coating proteins.
Anti-IgG
Reagents labeled “anti-IgG” contain no
anticomplement activity. The major com-
ponent of anti-IgG is antibody to human
gamma heavy chains, but unless labeled
as “heavy-chain-specific,” these reagents
may exhibit some reactivity with light
chains, which are common to all immu-
noglobulin classes. An anti-IgG reagent
not designated “heavy-chain-specific”
must be considered theoretically capable
of reacting with light chains of IgA or IgM.
A positive DAT with such an anti-IgG does
not definitively prove the presence of IgG,
although it is quite rare to have an in-vivo
coating with IgA or IgM in the absence of
IgG. Many workers prefer anti-IgG over
polyspecific AHG in antibody detection
and compatibility tests because anti-IgG
AHG does not react with complement
bound to red cells by cold-reactive anti-
bodies that are not clinically significant.
Anti-C3b, -C3d
Anti-C3b, -C3d reagents prepared by ani-
mal immunization contain no activity
against human immunoglobulins and are
used in situations described for anti-C3d.
This type of anti-C3d characteristically re-
acts with C3b and possibly other epitopes
present on C3-coated red cells. Murine
monoclonal anti-C3b, -C3d reagent is a
blend of hybridoma-derived antibodies.
Role of Complement in Antiglobulin
Reactions
Complement components may attach to
red cells in vivo or in vitro by one of two
mechanisms:
1. Complement-binding antibody spe-
cific for a red cell antigen may cause
attachment of complement to the cell
surface as a consequence of comple-
ment activation by the antigen-anti-
body complex.
2. Immune complexes, not specific for
red cell antigens, may be present in
plasma and may activate comple-
ment components that adsorb onto
red cells in a nonspecific manner.
Attachment of complement to the
membrane of cells not involved in
the specific antigen-antibody reac-
tion is often described as “innocent
bystander” complement coating.
Red cells coated with elements of the
complement cascade may or may not un-
dergo hemolysis. If the cascade does not go
to completion, the presence of bound early
components of the cascade can be detected
by anticomplement reagents. The compo-
nent most readily detected is C3 because
several hundred C3 molecules may be
280 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
bound to the red cell by the attachment of
only a few antibody molecules. C4 coating
also can be detected, but C3 coating has
more clinical significance.
Complement as the Only Coating Globulin
Complement alone, without detectable im-
munoglobulin, may be present on washed
red cells in certain situations.
1. IgM antibodies reacting in vitro oc-
casionally attach to red cell antigens
without agglutinating the cells, as is
seen with IgM antibodies to Lewis
antigens. IgM coating is difficult to
demonstrate in AHG tests, partly be-
cause IgM molecules tend to disso-
ciateduringthewashingprocess
and partly because polyspecific AHG
contains little if any anti-IgM activ-
ity. IgM antibodies may activate
complement, and the IgM reactivity
can be demonstrated by identifying
the several hundred C3 molecules
bound to the cell membrane near
the site of antibody attachment.
2. About 10% to 20% of patients with
warm AIHA have red cells with a pos-
itive DAT due to C3 coating alone.10
No IgG, IgA, or IgM coating is de-
monstrable with routine procedures,
although some specimens may be
coated with IgG at levels below the
detection threshold for the DAT.
3. In cold agglutinin syndrome, the
cold-reactive autoantibody can react
with red cell antigens at tempera-
tures up to 32 C.11 Red cells passing
through vessels in the skin at this
temperature become coated with
autoantibody, which activates com-
plement. If the cells escape hemoly-
sis, they return to the central circula-
tion, where the temperature is 37 C,
and the autoantibody dissociates
from the cells, leaving complement
components firmly bound to the red
cell membrane. The component
usually detected by AHG reagents is
C3d.
4. Immune complexes that form in the
plasma and bind weakly and non-
specifically to red cells may cause
complement coating. The activated
complement remains on the red cell
surfaceaftertheimmunecomplexes
dissociate.C3remainsastheonly
detectable surface globulin.
IgG-Coated Cells
The addition of IgG-coated cells to nega-
tive antiglobulin tests (used to detect IgG)
is required for antibody detection and
crossmatching procedures.12(p33) These
sensitized red cells should react with the
antiglobulin sera, verifying that the AHG
reagent was functional. Reactivity with
IgG-sensitized cells demonstrates that, in-
deed, AHG was added and it had not been
neutralized. Tests need to be repeated if
the IgG-coated cells are not reactive.
Testing with IgG-sensitized cells does
not detect all potential failures of the
antiglobulin test.13-15 Partial neutralization of
the AHG may not be detected at all, partic-
ularly if the control cells are heavily coated
with IgG. Errors in the original test, such as
omission of test serum, improper centri-
fuge speed, or inappropriate concentra-
tions of test red cells, may yield negative
test results but positive results with control
cells. Oversensitized control cells may ag-
glutinate when centrifuged.
Complement-coated cells can also be
prepared and are commercially available.
They can be used in some cases to control
tests with complement-specific reagents.
Some sources of error in antiglobulin
tests are listed in Tables 12-2 and 12-3.
Chapter 12: Red Cell Antigen-Antibody Reactions and Their Detection 281
Copyright © 2005 by the AABB. All rights reserved.

282 AABB Technical Manual
Table 12-2. Sources of Error in Antiglobulin Testing—False-Negative Results
Neutralization of Antihuman Globulin (AHG) Reagent
■Failure to wash cells adequately to remove all serum/plasma. Fill tube at least ¾ full of saline for
each wash. Check dispense volume of automated washers.
■If increased serum volumes are used, routine wash may be inadequate. Wash additional times or
remove serum before washing.
■Contamination of AHG by extraneous protein. Do not use finger or hand to cover tube.
Contaminated droppers or wrong reagent dropper can neutralize entire bottle of AHG.
■High concentration of IgG paraproteins in test serum; protein may remain even after multiple
washes.13
Interruption in Testing
■Bound IgG may dissociate from red cells and either leave too little IgG to detect or may
neutralize AHG reagent.
■Agglutination of IgG-coated cells will weaken. Centrifuge and read immediately.
Improper Reagent Storage
■AHG reagent may lose reactivity if frozen. Reagent may become bacterially contaminated.
■Excess heat or repeated freezing/thawing may cause loss of reactivity of test serum.
■Reagent red cells may lose antigen strength on storage. Other subtle cell changes may cause
loss of reactivity.
Improper Procedures
■Overcentrifugation may pack cells so tightly that agitation required to resuspend cells breaks up
agglutinates. Undercentrifugation may not be optimal for agglutination.
■Failure to add test serum, enhancement medium, or AHG may cause negative test.
■Too heavy a red cell concentration may mask weak agglutination. Too light suspension may be
difficult to read.
■Improper/insufficient serum:cell ratios.
Complement
■Rare antibodies, notably some anti-Jka,-Jk
b, may only be detected when polyspecific AHG is
used and active complement is present.
Saline
■Low pH of saline solution can decrease sensitivity.3Optimal saline wash solution for most
antibodies is pH 7.0 to 7.2.
■Some antibodies may require saline to be at specific temperature to retain antibody on the cell.
Use37Cor4Csaline.
Copyright © 2005 by the AABB. All rights reserved.

Other Methods to Detect
Antigen-Antibody Reactions
The following methods represent alterna-
tives to traditional tube testing and use of
antiglobulin serum. Some methods do
not allow detection of both IgM and IgG
antibodies and may not provide informa-
tion on the phase and temperature of re-
activity of antibodies that is obtained
when traditional tube tests are used.
Solid-Phase Red Cell Adherence Tests
Solid-phase microplate techniques use
immobilized antigen or antibody. In a di-
rect test, antibody is fixed to a microplate
well and red cells are added. If the cells
express the corresponding antigen, they
will adhere across the sides of the well; if
no antigen-antibody reaction occurs, the
red cells pellet to the bottom of the well
when centrifuged.16 An indirect test uses
red cells of known antigenic composition
Chapter 12: Red Cell Antigen-Antibody Reactions and Their Detection 283
Table 12-3. Sources of Error in Antiglobulin Testing—False-Positive Results
Cells Agglutinated Before Washing
■If potent agglutinins are present, agglutinates may not disperse during washing. Observe cells
before the addition of antihuman globulin (AHG) or use control tube substituting saline for AHG;
reactivity before the addition of AHG or in saline control invalidates AHG reading.
Particles or Contaminants
■Dust or dirt in glassware may cause clumping (not agglutination) of red cells. Fibrin or
precipitates in test serum may produce cell clumps that mimic agglutination.
Improper Procedures
■Overcentrifugation may pack cells so tightly that they do not easily disperse and appear positive.
■Centrifugation of test with polyethylene glycol or positively charged polymers before washing
may create clumps that do not disperse.
Cells That Have Positive Direct Antiglobulin Test (DAT)
■Cells that are positive by DAT will be positive in any indirect antiglobulin test. Procedures for
removing IgG from DAT-positive cells are given in Methods 2.13 and 2.14.
Complement
■Complement components, primarily C4, may bind to cells from clots or from CPDA-1 donor
segments during storage at 4 C and occasionally at higher temperatures. For DATs, use red cells
anticoagulated with EDTA, ACD, or CPD.
■Samples collected in tubes containing silicone gel may have spurious complement attachment.14
■Complement may attach to cells in specimens collected from infusion lines used to administer
dextrose-containing solutions. Strongest reactions are seen when large-bore needles are used or
when sample volume is less than 0.5 mL.15
Copyright © 2005 by the AABB. All rights reserved.
bound to a well. The test sample is added
to the red-cell-coated wells and allowed
to react with the cells, after which the
plates are washed free of unbound pro-
teins. The indicator for attached antibody
is a suspension of anti-IgG-coated red
cells. The reaction is positive if the indica-
tor cells adhere across the sides of the
well. If they pellet to the bottom when
centrifuged, it demonstrates that no anti-
gen-antibody reaction has occurred (see
Fig 12-4).17 In the indirect test, isolated
membrane components (eg, specific pro-
teins), rather than intact cells, can be af-
fixed to the microwell. Solid-phase at-
tachment of antigen or antibody is an
integral part of other tests, such as the
monoclonal antibody-specific immobili-
zation of erythrocyte antigens (MAIEA)
assay, discussed below. Solid-phase sys-
tems have also been devised for use in de-
tecting platelet antibodies and in tests for
syphilis, cytomegalovirus, and hepatitis B
surface antigen.17-20
Column Agglutination Technology
Various methods have been devised in
which red cells are filtered through a col-
umn containing a medium that separates
selected red cell populations. As commer-
cially prepared, the systems usually em-
ploy a card or strip of microtubes, rather
than conventional test tubes. They allow
simultaneous performance of several tests.
Usually, a space or chamber at the top of
each column is used for red cells alone or
to incubate red cells and serum or plasma.
As the cells pass through the column
(usually during centrifugation), the column
medium separates agglutinated from
unagglutinated red cells, based on aggre-
gate size.17 Alternatively, in some tests,
specific antisera or proteins can be in-
cluded in the column medium itself; cells
bearing a specific antigen are selectively
captured as they pass through the me-
dium. When the column contains anti-
globulin serum or a protein that specifi-
cally binds immunoglobulin, the selected
284 AABB Technical Manual
Figure 12-4. An indirect solid-phase test. A monolayer of red cells is affixed to a microwell (1). Test
serum is added. If antibody (2) is present, it binds to antigens on the affixed red cells (3). Indicator red
cells coated with IgG and anti-IgG (4) are added. The anti-IgG portion binds to any antibody attached to
the fixed red cells (5). In a positive test, the indicator red cells are effaced across the microwell. In a
negative test, the indicator cells do not bind but pellet to the center of the well when centrifuged. Weak
reactions give intermediate results.
Copyright © 2005 by the AABB. All rights reserved.
cells will be those sensitized with immu-
noglobulin. By using a centrifuge time
and speed that allow red cells to enter the
column but leave serum or plasma above,
theneedforsalinewashingforanti
-
globulin tests can be eliminated.21
Typically in column tests, negative test
cells pellet to the bottom of the column. In
positive tests, the cells are captured at the
top, or in the body, of the column. An ad-
vantage of most such systems is the stabil-
ity of the final reaction phase, which can be
read by several individuals or, in some
cases, documented by photocopying. Col-
umn tests generally have sensitivity similar
to LISS antiglobulin methods, but such
tests have reportedly performed less well in
detecting weak antibodies, especially those
in the ABO system.22
In 1986, Lapierre et al developed a pro-
cess leading to a technology that uses a col-
umn of gel particles.23 As commercially pre-
pared, the gel test uses six microtubes
instead of test tubes, contained in what is
called a card or strip. The gel particles func-
tion as filters that trap red cell agglutinates
when the cards are centrifuged. Gels con-
taining antiglobulin serum are used to cap-
ture sensitized, but unagglutinated, cells.
Gels with various antisera can be used for
phenotyping cells (see Fig 12-5).
In another column agglutination tech-
nology, a column of glass microbeads in a
diluent is used instead of gel. As with the
gel test, the beads may either entrap agglu-
tinated cells or antisera, such as anti-IgG,
can be added to the diluent.24
Automated Testing Platforms
Several automated devices have been de-
veloped for the detection of antigen-anti-
body reactions. All steps in the testing
process, from sample aliquotting to re-
porting results, are performed by the sys-
tem. These test systems permit the per-
formance of multiple tests, using solid-
phase, gel-column, and/or microtiter
plate technology. The systems are con-
trolled by a computer program and posi-
tive sample identity can be ensured by
barcode technology. The test system’s
computer may be integrated into a labo-
ratory’s information system for results re-
porting. Automated test systems may be
particularly useful in institutions per-
forming large volumes of patient or donor
testing.
Immunofluorescence
Immunofluorescence testing allows iden-
tification and localization of antigens
inside or on the surface of cells. A fluoro-
chrome such as fluorescein or phyco-
erythrin can be attached to an antibody
molecule, without altering its specificity
or its ability to bind antigen. Attachment
of fluorescein-labeled antibody to cellular
antigen makes the antibody-coated cells
appear brightly visible yellow-green or
red (depending on the fluorochrome).
Immunofluorescent antibodies can be
used in direct or indirect procedures, with
the fluorescence analogous to agglutination
as an endpoint. In a direct test, the fluores-
cein-labeled antibody is specific for a single
antigen of interest. In an indirect test,
Chapter 12: Red Cell Antigen-Antibody Reactions and Their Detection 285
Figure 12-5. Gel test.
Copyright © 2005 by the AABB. All rights reserved.
fluorescein-labeled antiglobulin serum is
added to cells that have been incubated
with an unlabeled antibody of known spec-
ificity. Immunofluorescent techniques were
initially used to detect antigens in or on
lymphocytes or in tissue sections. More re-
cently, immunofluorescent antibodies have
been used in flow cytometry. Among their
many applications, they have been used to
quantify fetomaternal hemorrhage, to iden-
tify transfused cells and follow their survival
in recipients, to measure low levels of
cell-bound IgG, and to distinguish homozy-
gous from heterozygous expression of
blood group antigens.25
Enzyme-Linked Immunosorbent Assay
Enzyme-linked immunosorbent assays
(ELISAs) are used to measure either anti-
gen or antibody. Enzymes such as alkaline
phosphatase can be bound to antibody
molecules without destroying either the
antibody specificity or the enzyme activ-
ity. ELISAs have been used to detect and
measure cell-bound IgG and to demon-
strate fetomaternal hemorrhage. When
red cells are examined, the test often is
called an enzyme-linked antiglobulin test
(ELAT).
Monoclonal Antibody-Specific
Immobilization of Erythrocyte Antigens
Assay
IntheMAIEAassay,redcellsareincu
-
bated with two antibodies. One contains
human alloantibody to a blood group an-
tigen and the other is a nonhuman (usu-
ally mouse monoclonal) antibody that re-
acts with a different portion of the same
membrane protein. The red cells are lysed
and the membrane solubilized, then
added to a microwell coated with goat
antimouse antibody. This antibody then
captures the mouse antibody attached to
the membrane protein (with the human
antibody also attached). Conjugated anti-
human antibody is added, which reacts
with the bound human antibody and
gives an ELISA-readable reaction. Thus
far, this method has been used primarily
to isolate specific membrane structures
for blood group antigen studies.26,27
References
1. van Oss CJ. Immunological and physio-
chemical nature of antigen-antibody interac-
tions. In: Garratty G, ed. Immunobiology of
transfusion medicine. New York: Marcel
Dekker, Inc., 1994:327-64.
2. Moore BPL. Antibody uptake: The first stage
of the hemagglutination reaction. In: Bell CA,
ed. A seminar on antigen-antibody reactions
revisited. Arlington, VA: AABB, 1982:47-66.
3. RolihS,ThomasR,FisherE,TalbotJ.Anti-
body detection errors due to acidic or un-
buffered saline. Immunohematology 1993;
9:15-18.
4. Jørgensen J, Nielsen M, Nielsen CB, Nørmark
J. The influence of ionic strength, albumin
and incubation time on the sensitivity of the
indirect Coombs’ test. Vox Sang 1980;36:186-
91.
5. Nance SJ, Garratty G. Polyethylene glycol: A
new potentiator of red blood cell antigen-an-
tibody reactions. Am J Clin Pathol 1987;87:
633-5.
6. Issitt PD, Anstee DJ. Applied blood group se-
rology. 4th ed. Durham, NC: Montgomery
Scientific Publications, 1998:47-8.
7. Coombs RRA, Mourant AE, Race RR. A new
test for the detection of weak and “incom-
plete” Rh agglutinins. Br J Exp Pathol 1945;
26:255-66.
8. Code of federal regulations. Title 21 CFR
660.55. Washington, DC: US Government
Printing Office, 2004 (revised annually).
9. Packman CH. Acquired hemolytic anemia
due to warm-reacting autoantibodies. In:
Beutler E, Lichtman MA, Coller BS, et al, eds.
Williams’ hematology. 6th ed. New York:
McGraw Hill, 2001:639-48.
10. Sokol RJ, Hewitt S, Stamps BK. Autoimmune
haemolysis: An 18-year study of 865 cases re-
ferred to a regional transfusion centre. Br
Med J 1981;282:2023-7.
11. Packman CH. Cryopathic hemolytic syn-
dromes. In: Beutler E, Lichtman MA, Coller
286 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
BS, et al, eds. Williams’ hematology. 6th ed.
New York: McGraw-Hill, 2001:649-55.
12. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
13. Ylagen ES, Curtis BR, Wildgen ME, et al. In-
validation of antiglobulin tests by a high ther-
mal amplitude cryoglobulin. Transfusion
1990;30:154-7.
14. Geisland JR, Milam JD. Spuriously positive di-
rect antiglobulin tests caused by silicone gel.
Transfusion 1980;20:711-13.
15. Grindon AJ, Wilson MJ. False-positive DAT
caused by variables in sample procurement.
Transfusion 1981;21:313-14.
16. Rolih SD, Eisinger RW, Moheng JC, et al. Solid
phase adherence assays: Alternatives to con-
ventional blood bank tests. Lab Med 1985;16:
766-70.
17. Walker P. New technologies in transfusion
medicine. Lab Med 1997;28:258-62.
18. Plapp FV, Sinor LT, Rachel JM, et al. A solid
phase antibody screen. Am J Clin Pathol 1984;
82:719-21.
19. Rachel JM, Sinor LT, Beck ML, Plapp FV. A
solid-phase antiglobulin test. Transfusion
1985;25:24-6.
20. Sinor L. Advances in solid-phase red cell ad-
herence methods and transfusion serology.
Transfus Med Rev 1992;6:26-31.
21. Malyska H, Weiland D. The gel test. Lab Med
1994;25:81-5.
22. Phillips P, Voak D, Knowles S, et al. An expla-
nation and the clinical significance of the
failure of microcolumn tests to detect weak
ABO and other antibodies. Transfus Med
1997;7:47-53.
23. LapierreY,RigalD,AdamJ,etal.Thegeltest:
A new way to detect red cell antigen-antibody
reactions. Transfusion 1990;30:109-13.
24. ReisKJ,ChachowskiR,CupidoA,etal.Col
-
umn agglutination technology: The antiglo-
bulin test. Transfusion 1993;33:639-43.
25. Garratty G, Arndt P. Applications of flow cyto-
fluorometry to transfusion science. Transfu-
sion 1994;35:157-78.
26. Petty AC. Monoclonal antibody-specific im-
mobilisation of erythrocyte antigens (MAIEA).
A new technique to selectively determine an-
tigenic sites on red cell membranes. J Immunol
Methods 1993;161:91-5.
27. PettyAC,GreenCA,DanielsGL.Themono
-
clonal antibody-specific antigens assay
(MAIEA) in the investigation of human red-
cell antigens and their associated membrane
proteins. Transfus Med 1997;7:179-88.
Chapter 12: Red Cell Antigen-Antibody Reactions and Their Detection 287
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens
Chapter 13
ABO, H, and Lewis Blood
Groups and Structurally
Related Antigens
THE ABO, AS well as the H, Lewis, I,
and P, blood group antigens reside
on structurally related carbohydrate
molecules. The antigens arise from the
action of specific glycosyltransferases that
add individual sugars sequentially to sites
on short chains of sugars (oligosaccha-
rides) on common precursor substances. In-
teractions of the ABO, Hh, Sese, and Lele
gene products affect the expression of the
ABO, H, and Lewis antigens. Refer to Ap-
pendix 6 for ISBT numbers and nomencla-
ture for the blood groups.
The ABO System
The ABO system was discovered when Karl
Landsteiner recorded the agglutination of
human red cells by the sera of other indi-
viduals in 19001and, in the following year,
detailed the patterns of reactivity as three
types,nowcalledgroupsA,B,andO.
2,3 He
found that serum from group A individu-
als agglutinated the red cells from group B
individuals, and, conversely, the serum from
group B individuals agglutinated group A
red cells. A and B were thus the first red
cell antigens to be discovered. Red cells
that were not agglutinated by the serum
of either the group A or group B individu-
als were later called group O; the serum
from group O individuals agglutinated the
red cells from both group A and group B
individuals. Von Decastello and Sturli in
1902 discovered the fourth group, AB.4
TheimportanceofLandsteiner’sdiscov
-
ery is the recognition that antibodies to A
and B antigens are present when the cor-
responding antigen is missing. Routine ABO
typing procedures developed from these and
later studies.5
The ABO antigens and antibodies remain
the most significant for transfusion practice.
It is the only blood group system in which
the reciprocal antibodies (see Table 13-1)
289
13
Copyright © 2005 by the AABB. All rights reserved.

are consistently and predictably present in
the sera of most people who have had no
exposure to human red cells. Due to these
antibodies, transfusion of ABO-incompati-
ble blood may cause severe intravascular
hemolysis as well as the other manifesta-
tions of an acute hemolytic transfusion re-
action (see Chapter 27). Testing to detect ABO
incompatibility between a recipient and the
donor is the foundation on which all pre-
transfusion testing is based.
Genetics and Biochemistry
The genes for all of the carbohydrate anti-
gens discussed in this chapter encode spe-
cific glycosyltransferases, enzymes that
transfer specific sugars to the appropriate
carbohydrate chain acceptor; thus, the an-
tigens are indirect products of the genes.
Genes at three separate loci (H,Se, and
ABO) control the occurrence and the loca-
tion of the A and B antigens. The Hand Se
(secretor) loci, officially named FUT1 and
FUT2, respectively, are on chromosome
19 and are closely linked. Each locus has
two recognized alleles, one of which has
no demonstrable product and is consid-
ered an amorph. The active allele at the H
locus, H, produces a transferase that acts
at the cellular level to form the H antigen
on red cells. The amorph, h,isveryrare.
The active allele at the Se locus, Se,pro-
duces a transferase that also acts to form H
antigen, but primarily in secretions such
as saliva.6Eighty percent of individuals are
secretors. The amorphic allele is se.The
enzymes produced by Hand Se alleles are
both fucosyltransferases, but they have
slightly different activity. H antigen on red
cells and in secretions is the substrate for
the formation of A and B antigens.
There are three common alleles at the
ABO locus on chromosome 9, A,B,andO.7
The Aand Balleles encode glycosyltrans-
ferases that produce the A and B antigens
respectively; the Oallele does not encode a
functional enzyme.8The red cells of group
O individuals lack A and B antigens but carry
an abundant amount of H antigen, the un-
converted precursor substance on which A
and B antigens are built.
The carbohydrate chains (oligosaccha-
rides) that carry ABH antigens can be at-
tached to either protein (glycoprotein),
sphingolipid (glycosphingolipid), or lipid
(glycolipid) carrier molecules. Glycopro-
teins and glycosphingolipids carrying A or
B antigens are integral parts of the mem-
290 AABB Technical Manual
Table 13-1. Routine ABO Typing
Reaction of Cells
Tested with
Reaction of Serum
Tested Against
Interpre-
tation
Incidence (%) in
US Population
Anti-A Anti-B A1Cells B Cells O Cells
ABO
Group Whites Blacks
0
+
0
+
0
0
+
+
+
0
+
0
+
+
0
0
0
0
0
0
0
A
B
AB
45
40
11
4
49
27
20
4
+ = agglutination; 0 = no agglutination.
Copyright © 2005 by the AABB. All rights reserved.
branes of red cells, epithelial cells, and
endothelial cells (Fig 13-1) and are also
present in soluble form in plasma. Glyco-
proteins secreted in body fluids such as sa-
liva contain molecules that may, if the per-
son possesses an Se allele, carry A, B, and H
antigens. A and B antigens that are unat-
tached to carrier protein or lipid molecules
are also found in milk and urine as free oli-
gosaccharides.
The transferases encoded by the A,B,H,
and Se alleles add a specific sugar to a pre-
cursor carbohydrate chain. The sugar that
is added is referred to as immunodominant
because when it is removed from the struc-
ture, the specific blood group activity is
lost. The sugars can be added only in a se-
quential manner. H structure is made first,
then sugars for A and B antigens are added
to H. The Hand Se alleles encode a fuco-
syltransferase that adds fucose (Fuc) to the
precursor chain; thus, fucose is the im-
munodominant sugar for H (see Fig 13-2).
The Aallele encodes N-acetyl-galactosa-
minyltransferase that adds N-acetyl-D-
galactosamine (or GalNAc) to H to make A
antigen on red cells. The Ballele encodes
galactosyltransferase that adds D-galactose
(or Gal) to H to make B antigen. Group AB
individuals have alleles that make transfer-
ases to add both GalNAc and Gal to the pre-
cursor H antigen. Attachment of the A or B
immunodominant sugars diminishes the se-
rologic detection of H antigen so that the ex-
pressions of A or B antigen and of H antigen
are inversely proportional. Rare individuals
who lack both Hand Se alleles (genotype hh
and sese) have no H and, therefore, no A or B
antigens on their red cells or in their secre-
tions (see Ohphenotype below). However, H,
A, and B antigens are found in the secretions
of some hh individuals who appear, through
family studies, to possess at least one Se allele
(see para-Bombay phenotype below).
The oligosaccharides to which the A or B
immunodominant sugars are attached may
exist as simple repeats of a few sugar mole-
cules linked in linear fashion, or as part of
more complex structures, with many sugar
residues linked in branching chains. Differ-
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 291
Figure 13-1. Schematic representation of the red cell membrane showing antigen-bearing glycosyla-
tion of proteins and lipids. GPI = glycophosphatidylinositol. (Courtesy of ME Reid, New York Blood
Center.)
Copyright © 2005 by the AABB. All rights reserved.
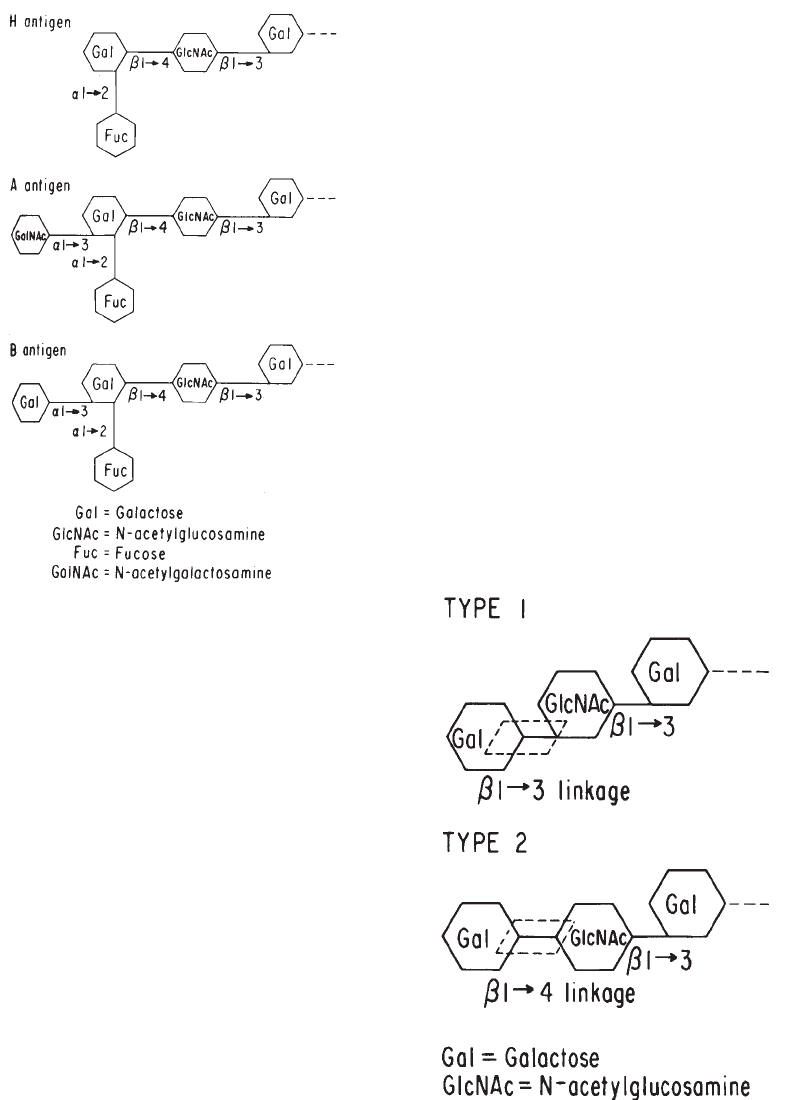
ences between infants and adults in cellular
A, B, and H activity may be related to the
number of branched structures present on
cellular membranes at different ages. The
red cells of infants are thought to carry pre-
dominantly linear carbohydrate chains,
which have only one terminus to which the
H (and subsequent A and/or B) sugars can
be added. In contrast, the red cells of adults
carry a high proportion of branched carbo-
hydrate chains, providing additional sites for
conversion to H and then to A and B anti-
gens.
A, B, and H antigens are constructed on
carbohydrate chains that are characterized
by different linkages and composition of the
terminal disaccharide; there are at least six
of these types of disaccharide linkages.9
Type 1 chains and Type 2 chains differ in
the linkage of the terminal Gal to GlcNAc
disaccharide(seeFig13-3).Type1A,B,and
H structures are present in secretions, plasma,
andendodermallyderivedtissues.Theyare
not synthesized by red cells but are incor-
porated into the red cell membrane from
the plasma. Type 2 chains are the predomi-
nant ABH-carrying oligosaccharides on red
cells and are also found in secretions. Type
3 chains (repetitive form) are found on red
cells from group A individuals.10 They are
synthesized by the addition of Gal to the
terminal GalNAc Type 2 A chains, thus
formingType3H;Type3Hchainsaresub
-
sequently converted to Type 3 A by the
addition of GalNAc through the action of
A1-transferase, but not by A2-transferase
(see Fig 13-4).
292 AABB Technical Manual
Figure 13-2. Gal added to the subterminal Gal
confers B activity; GalNAc added to the subtermi-
nal Gal confers A activity to the sugar. Unless the
fucose moiety that determines H activity is at-
tached to the number 2 carbon, galactose does
not accept either sugar on the number 3 carbon.
Figure 13-3. Type 1 and 2 oligosaccharide chains
differ only in the linkage between the GlcNAc and
the terminal Gal.
Copyright © 2005 by the AABB. All rights reserved.
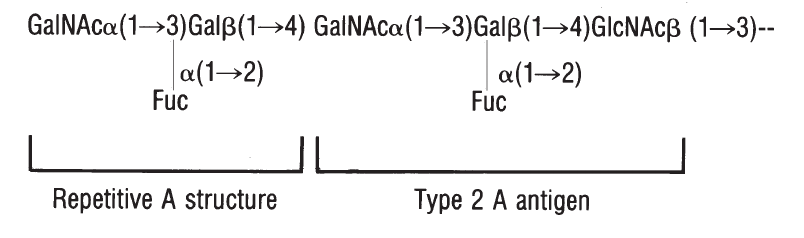
Active alleles Hand Se,oftheFUT1 and
FUT2 genes, encode fucosyltransferases with
a high degree of homology. The enzyme
produced by Hacts primarily on Type 2
chains, which are prevalent on the red cell
membranes. The enzyme produced by Se
prefers, but is not limited to, Type 1 chains
and acts primarily in the secretory glands.
ABO Genes at the Molecular Level
Yamamoto et al11 have shown that Aand B
alleles differ from one another by seven
nucleotide differences, four of which re-
sulted in amino acid substitutions at posi-
tions 176, 235, 266, and 268 in the protein
sequence of the A and B transferases. Re-
cent crystallography of A- and B-transfer-
ases demonstrated the role of these criti-
cal amino acids in substrate recognition.12
The initial Oallele examined had a single
nucleotide deletion that resulted in a frame
shift and premature stop codon resulting in
the predicted translation of a truncated (ie,
inactive) protein. Subsequently, other Oal-
leles have been identified as well as muta-
tions of Aand Balleles that result in weak-
ened expression of A and B antigens (reviewed
elsewhere).13,14 The A2allele encodes a pro-
tein with an additional 21 amino acids.
Antigens
Agglutination tests are used to detect A and
B antigens on red cells. Reagent antibod-
ies frequently produce weaker reactions
with red cells from newborns than with
red cells from adults. Although A and B
antigens can be detected on the red cells
of 5- to 6-week-old embryos, A and B anti-
gens are not fully developed at birth, pre-
sumably because the branching carbohy-
drate structures develop gradually. By 2 to
4 years of age, A and B antigen expression
is fully developed and remains fairly con-
stant throughout life.
Subgroups
ABO subgroups are phenotypes that differ
in the amount of antigen carried on red
cells and, for secretors, soluble antigen
present in the saliva. Subgroups of A are
more commonly encountered than sub-
groups of B. The two principal subgroups
of A are A1and A2. Red cells from A1and A2
persons both react strongly with reagent
anti-A in direct agglutination tests. The
serologic distinction between Aland A2
cells can be determined by testing with
anti-A1lectin (see anti-A1below). There is
both a qualitative and quantitative differ-
ence between A1and A2.15 The A1-trans-
ferase is more efficient at converting H
substance into A antigen and is capable of
makingtherepetitiveType3Astructures.
There are about 10.5 ×105Aantigensites
on adult A1red cells, and about 2.21 ×105
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 293
Figure 13-4. Type 3 A antigen structure.
Copyright © 2005 by the AABB. All rights reserved.
A antigen sites on adult A2red cells.9Ap-
proximately 80% of group A or group AB
individuals have red cells that are aggluti-
nated by anti-A1and thus are classified as
A1or A1B. The remaining 20%, whose red
cells are strongly agglutinated by anti-A but
not by anti-A1, are called A2or A2B. Rou-
tine testing with anti-A1is unnecessary for
donors or recipients.
Subgroups weaker than A2occur infre-
quently and, in general, are characterized
by decreasing numbers of A antigen sites
on the red cells and a reciprocal increase in
H antigen activity. Subgroups are most of-
ten recognized when there is a discrepancy
between the red cell (forward) and serum
(reverse) grouping. Generally, classification
of weak A subgroups (A3,A
x,A
m,A
el)isbased
on the:
1. Degree of red cell agglutination by
anti-A and anti-A1.
2. Degree of red cell agglutination by hu-
man and some monoclonal anti-A,B.
3. Degree of red cell agglutination by
anti-H (Ulex europaeus).
4. Presence or absence of anti-A1in the
serum.
5. Presence of A and H substances in
the saliva of secretors.
6. Adsorption/elution studies.
7. Family (pedigree) studies.
Identification of the various A subgroups
is not routinely done. The serologic classifi-
cation of A (and B) subgroups was devel-
oped using human polyclonal anti-A,
anti-B, and anti-A,B reagents. These re-
agents have been replaced by murine
monoclonal reagents, and the reactivity is
dependent upon which clone(s) is selected
by the manufacturer. There are, however,
some characteristics that should be noted.
A3red cells give a characteristic mixed-field
pattern when tested with anti-A from group
BorOdonors.A
xred cells are characteristi-
cally not agglutinated by human anti-A
from group B persons but are agglutinated
by anti-A,B from group O persons. Axred
cells may react with some monoclonal anti-A
reagents, depending on which monoclonal
antibody is selected for the reagent. Ael red
cells are not agglutinated by anti-A or
anti-A,B of any origin, and the presence of
A antigen is demonstrable only by adsorp-
tion and elution studies. Subgroups of B are
even less common than subgroups of A.
Molecular studies have confirmed that A
and B subgroups are heterogeneous, and the
serologic classification does not consistently
correlate with genomic analysis; multiple
alleles yield the same weakened phenotype,
and, in some instances, more than one
phenotypehasthesameallele.
16
Antibodies to A and B
Ordinarily, individuals possess antibodies
directed toward the A or B antigen absent
from their own red cells (see Table 13-1).
This predictable complementary relation-
ship permits ABO testing of sera as well as
of red cells (see Methods 2.2 and 2.3). One
hypothesis for the development of these
antibodies is based on the fact that the
configurations that confer A and B anti-
genic determinants also exist in other bio-
logic entities, notably bacteria cell walls.
Bacteria are widespread in the environ-
ment, and their presence in intestinal
flora, dust, food, and other widely distrib-
uted agents ensures a constant exposure
of all persons to A-like and B-like antigens.
Immunocompetent persons react to the
environmental antigens by producing an-
tibodies to those that are absent from
their own systems. Thus, anti-A is pro-
duced by group O and group B persons
and anti-B is produced by group O and
group A persons. Group AB people, hav-
ing both antigens, make neither antibody.
This “environmental” explanation for the
emergence of anti-A and anti-B remains a
hypothesis that has not been proven.
294 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Time of Appearance
Anti-A and anti-B produced by an infant
can generally be detected in serum after 3
to 6 months of life. Most of the anti-A and
anti-B present in cord blood are of ma-
ternal origin, acquired by the placental
transfer of maternal IgG; occasionally, in-
fants can be found who produce these
antibodies at the time of birth.17(p124) Thus,
anti-A and anti-B detected in the sera of
newborns or infants younger than 3 to 6
months cannot be considered valid. Anti-
body production increases, reaching the
adult level at 5 to 10 years of age, and de-
clines later in life. Elderly people usually
have lower anti-A and anti-B levels than
young adults.
Reactivity of Anti-A and Anti-B
IgM is the predominant immunoglobulin
class of anti-A produced by group B indi-
viduals and anti-B produced by group A
individuals, although small quantities of
IgG antibody are also present. IgG is the
dominant class of anti-A and anti-B of group
Oserum.
17(p124) Because IgG readily crosses
the placenta and IgM does not, group A or
B infants of group O mothers are at higher
risk for ABO hemolytic disease of the fetus
and newborn (HDFN) than the infants of
group A or B mothers; but severe HDFN
can also occur in infants of group A and
group B mothers.
Both IgM and IgG anti-A and anti-B pref-
erentially agglutinate red cells at room tem-
perature (20-24 C) or below and efficiently
activate complement at 37 C. The comple-
ment-mediated lytic capability of these an-
tibodies becomes apparent if serum testing
includes an incubation phase at 37 C. Sera
from some people will cause hemolysis of
ABO-incompatible red cells at tempera-
turesbelow37C.HemolysisduetoABO
antibodies should be suspected when the
supernatant fluid of the serum test is pink
to red or when the cell button is absent or
reduced in size. Hemolysis must be inter-
preted as a positive result. Because the
hemolysis is complement-mediated, it will
not occur if plasma is used for testing, or if
reagent red cells are suspended in solutions
that contain EDTA or other agents that pre-
vent complement activation.
Reactivity of Anti-A,B (Group O Serum)
Serum from a group O individual contains
an antibody designated as anti-A,B because
it reacts with both A and B red cells, and
the anti-A and anti-B cannot be separated
by differential adsorption. In other words,
after adsorption of group O serum, an elu-
ate prepared from the group A or group B
adsorbing cells reacts with both A and B
test cells. Saliva from a secretor of either A
or B substance inhibits the activity of
anti-A,B against A or B red cells, respec-
tively.
Anti-A
1
Anti-A1occurs as an alloantibody in the
serumof1%to2%ofA
2individuals and
25% of A2B individuals.15 Sometimes,
Anti-A1can also be found in the sera of in-
dividuals with other weak subgroups of A.
Anti-A1can cause discrepancies in ABO
testing and incompatibility in cross-
matches with A1or A1B red cells. Anti-A1
usually reacts better or only at tempera-
tures well below 37 C and is considered
clinically insignificant unless there is re-
activity at 37 C. When reactive at 37 C, only
A2or O red cells should be used for trans-
fusion.
In simple adsorption studies, the anti-A
of group B serum appears to contain sepa-
rable anti-A and anti-A1.NativegroupBse
-
rum agglutinates A1and A2red cells; after
adsorption with A2red cells, group B serum
reacts only with A1red cells. If further tests
are performed, however, the differences in
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 295
Copyright © 2005 by the AABB. All rights reserved.
A antigen expression between A1and A2red
cells appear to be quantitative rather than
qualitative.9
A reliable anti-A1reagent from the lectin
of Dolichos biflorus is commercially avail-
able or may be prepared (see Method 2.10).
The raw plant extract will react with both A1
and A2red cells, but an appropriately diluted
reagent preparation will not agglutinate A2
cells and thus constitutes an anti-A1.
Routine Testing for ABO
Routine testing for determining the ABO
group consists of testing the red cells with
anti-A and anti-B (cell or forward type)
and testing the serum or plasma with A1
and B red cells (serum or reverse type).
Both red cell and serum testing are re-
quired for routine ABO tests on donors and
patients because each serves as a check
on the other.18(pp32,37) The two exceptions to
performing both cell and serum testing
are confirmation testing of the ABO type of
donor units that have already been la-
beled and testing blood of infants less
than 4 months of age; in both of these in-
stances, only ABO testing of red cells is re-
quired.
Anti-A and anti-B typing reagents agglu-
tinate most antigen-positive red cells on di-
rect contact, even without centrifugation.
Anti-A and anti-B in the sera of some pa-
tients and donors are too weak to aggluti-
nate red cells without centrifugation or pro-
longed incubation. Serum tests should be
performed by a method that will adequa-
tely detect the antibodies—eg, tube, micro-
plate, or column agglutination techniques.
Procedures for ABO typing by slide, tube,
and microplate tests are described in Meth-
ods 2.1, 2.2, and 2.3.
Additional reagents, such as anti-A,B for
red cell tests and A2and O red cells for serum
tests, are not necessary for routine testing
but are helpful in resolving typing discrep-
ancies (see below). The use of anti-A,B may
not have the same benefit in detecting weak
subgroups when testing with monoclonal
reagents (depending on the clones used) as
when human polyclonal reagents were in
use. Many monoclonal ABO typing re-
agents have been formulated to detect
some of the weaker subgroups. Manufac-
turers’ inserts should be consulted for spe-
cific reagent characteristics. Special techni-
ques to detect weak subgroups are not
routinely necessary because a typing dis-
crepancy (eg, the absence of expected se-
rum antibodies) usually distinguishes these
specimens from group O specimens.
The A2red cells are intended to facilitate
the recognition of anti-A1. Because most group
A specimens do not contain anti-A1,routine
use of this reagent is not necessary.
Discrepancies Between Red Cell and
Serum Tests
Table 13-1 shows the results and interpre-
tations of routine red cell and serum tests
for ABO. A discrepancy exists when the
results of red cell tests do not agree with
serum tests. When a discrepancy is en-
countered, the discrepant results must be
recorded, but interpretation of the ABO
group must be delayed until the discrep-
ancy has been resolved. If the specimen is
from a donor unit, the unit must not be
released for transfusion until the discrep-
ancy has been resolved. When the blood
is from a potential recipient, it may be
necessary to administer group O red cells
oftheappropriateRhtypebeforethein
-
vestigation has been completed. It is im-
portant to obtain sufficient pretransfusion
blood samples from the patient to com-
plete any additional studies that may be
required.
296 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Red cell and serum test results may be
discrepant because of intrinsic problems
with red cells or serum, or technical errors.
Discrepancies may be signaled either be-
cause negative results are obtained when
positive results are expected, or positive re-
sults are found when tests should have been
negative (see Table 13-2).
Specimen-Related Problems in Testing Red
Cells
ABO testing of red cells may give unex-
pected results for many reasons.
1. Red cells from individuals with vari-
ant Aor Balleles may carry poorly
expressed antigens. Antigen expres-
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 297
Table 13-2. Possible Causes of ABO Typing Discrepancies
Category Causes
Red cell weak/
missing reactivity
ABO subgroup
Leukemia/malignancy
Transfusion
Intrauterine fetal transfusion
Transplantation
Excessive soluble blood group substance
Extra red cell
reactivity
Autoagglutinins/excess protein coating red cells
Unwashed red cells: plasma proteins
Unwashed red cells: antibody in patient’s serum to reagent
constituent
Transplantation
Acquired B antigen
B(A) phenomenon
Out-of-group transfusion
Mixed-field red cell
reactivity
Recent transfusion
Transplantation
Fetomaternal hemorrhage
Twin or dispermic (tetragametic) chimerism
Serum weak/missing
reactivity
Age related (<4-6 months old, elderly)
ABO subgroup
Hypogammaglobulinemia
Transplantation
Serum extra reactivity Cold autoantibody
Cold alloantibody
Serum antibody to reagent constituent
Excess serum protein
Transfusion of plasma components
Transplantation
Infusion of intravenous immune globulin
Copyright © 2005 by the AABB. All rights reserved.
sion may also be weakened on the red
cells of some persons with leukemia
or other malignancies.
2. A patient who has received red cell
transfusions or a marrow transplant
may have circulating red cells of more
than one ABO group and constitute
a transfusion or transplantation chi-
mera (see Mixed-Field Agglutination
below).
3. Exceptionally high concentrations of
A or B blood group substances in the
serum can combine with and neutra-
lize reagent antibodies to produce an
unexpected negative reaction against
serum- or plasma-suspended red cells.
4. A patient with potent autoagglutinins
may have red cells so heavily coated
with antibody that the red cells ag-
glutinate spontaneously in the pres-
ence of diluent, independent of the
specificity of the reagent antibody.
5. Abnormal concentrations of serum
proteins or the presence in serum of
infused macromolecular solutions may
cause the nonspecific aggregation of
serum-suspended red cells that sim-
ulates agglutination.
6. Serum- or plasma-suspended red cells
may give false-positive results with
monoclonal reagents if the serum or
plasma contains a pH-dependent
autoantibody.19,20 Serum- or plasma-
suspended red cells may also give
discrepant results due to an antibody
to a reagent dye/constituent21 or to
proteins causing rouleaux.
7. Red cells of some group B individu-
als are agglutinated by a licensed
anti-A reagent that contains a partic-
ular murine monoclonal antibody,
MHO4. These group B individuals had
excessively high levels of Ballele-
specified galactosyltransferase, and
the designation B(A) was given to
this blood group phenotype.22
8. Red cells of individuals with the
acquired B phenotype typically ag-
glutinate strongly with anti-A and
weakly with anti-B, and the serum
contains strong anti-B. The acquired
B phenotype arises when microbial
deacetylating enzymes modify the A
antigen by altering the A-determin-
ing sugar (N-acetylgalactosamine)
so that it resembles the B-determin-
ing galactose. The acquired B phe-
nomenon is found most often in in-
dividuals with the A1phenotype.
9. Inherited or acquired abnormalities
of the red cell membrane can lead to
what is called a polyagglutinable state.
The abnormal red cells can be unex-
pectedly agglutinated by human re-
agent anti-A, anti-B, or both because
human reagents will contain anti-
bodies to the so-called cryptantigens
that are exposed in polyagglutinable
states. In general, monoclonal anti-A
and anti-B reagents will not detect
polyagglutination.
Specimen-Related Problems in Testing
Serum or Plasma
ABO serum/plasma tests are also subject
to false results.
1. Small fibrin clots that may be mis-
taken for agglutinates may be seen
in ABO tests with plasma or incom-
pletely clotted serum.
2. Negative or weak results are seen in
serum tests from infants under 4 to 6
months of age. Serum from newborns
is not usually tested because anti-
bodies present are generally passively
transferred from the mother.
3. Unexpected absence of ABO aggluti-
ninsmaybeduetothepresenceof
an A or B variant.
4. Patients who are immunodeficient
due to disease or therapy may have
298 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
such depressed immunoglobulin levels
that there is little or no ABO aggluti-
nin activity. Samples from elderly
patients whose antibody levels have
declined with age or from patients
whose antibodies have been greatly
diluted by plasma exchange proce-
dures may also have unexpectedly weak
agglutinins.
5. If the patient has received a marrow
transplant of a dissimilar ABO group,
serum antibodies will not agree with
red cell antigens. For example, a group
A individual who receives group O
marrow may have circulating group
O red cells and produce only anti-B in
the serum. Refer to Chapter 25 for more
information on the effects of ABO-
mismatched transplants.
6. Cold allo- or autoantibodies that re-
act at room temperature can react
with one or both reverse grouping
cells. For example, if the patient has
a room-temperature-reactive anti-M,
it may cause an unexpected reaction
with M-positive A1and/or B reagent
red cells.
7. Antibodies to constituents of the dil-
uents used to preserve reagent A1and
B red cells can agglutinate the cells
independent of ABO antigens and
antibodies.
8. Abnormal concentrations of proteins,
altered serum protein ratios, or the
presence of high-molecular-weight
plasma expanders can cause non-
specific red cell aggregation or roule-
aux that is difficult to distinguish from
true agglutination. Rouleaux forma-
tion is easily recognized on micro-
scopic examination if the red cells
assume what has been described as
a “stack of coins” formation.
9. Recent transfusion with plasma com-
ponents containing ABO agglutinins
may cause unexpected reactions.
10. Recent infusion of intravenous im-
mune globulin that may contain ABO
isohemagglutinins can cause unex-
pected reactions.
Mixed-Field Agglutination
Occasional samples are encountered that
contain two distinct, separable populations
of red cells. Usually, this reflects the re-
cent transfusion of group O red cells to a
non-group-O recipient or receipt of a
marrow transplant of an ABO group dif-
ferent from the patient’s own. Red cell
mixtures also occur in a condition called
blood group chimerism, resulting either
from intrauterine exchange of erythro-
poietic tissue by fraternal twins or from
mosaicism arising through dispermy. In all
such circumstances, ABO red cell tests may
give a mixed-field pattern of agglutina-
tion. Mixed-field reactions due to transfu-
sion last only for the life of the transfused
red cells. After hematopoietic transplanta-
tion, the mixed-field reaction usually dis-
appears when the patient’s own red cells
are no longer produced. Persistent mixed-
cell populations do occur in some marrow
recipients. Mixed-field reactions that arise
through blood group chimerism may per-
sist throughout the life of the individual.
For more information regarding the trans-
fusion and evaluation of hematopoietic
transplant patients, refer to Chapters 21
and 25.
Technical Errors
Technical errors leading to ABO discrep-
ancies include:
1. Specimen mix-up.
2. Red cell suspensions are too heavy
or too light.
3. Failure to add reagents.
4. Missed observation of hemolysis.
5. Failure to follow manufacturer’s in-
structions.
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 299
Copyright © 2005 by the AABB. All rights reserved.
6. Under- or overcentrifugation of tests.
7. Incorrect interpretation or recording
of test results.
Resolving ABO Discrepancies
The first step in resolving an apparent se-
rologic problem should be to repeat the
tests on the same sample. If initial tests
were performed on red cells suspended in
serum or plasma, the testing should be re-
peated after washing the red cells several
times with saline. Washing the red cells
can eliminate many problems for red cell
typing that are associated with plasma
proteins. If the discrepancy persists, the
following initial steps can be incorporated
into the investigation.
1. Obtain the patient’s diagnosis, his-
torical blood group, and history of
previous transfusions, transplanta-
tion, and medications.
2. Review the results of the antibody
detection test against group O red cells
and autologous red cells to detect
possible interference from allo- or
autoantibodies.
3. Obtain a new blood specimen and test
the new sample if a discrepancy due
to a contaminated specimen is sus-
pected.
In addition to a discrepancy between the
red cell and serum tests, an ABO discrep-
ancy may also exist when the observed re-
activity is not in agreement with a previous
type on record. The first step in resolving
this type of discrepancy is to obtain a new
blood specimen.
Resolving Discrepancies Due to Absence of
Expected Antigens
The cause of a discrepancy can some-
times be inferred from the strength of the
reactions obtained in red cell or serum
tests. For example, serum that strongly
agglutinates group B red cells but not A1
cells probably comes from a group A per-
son, even though the red cells are not
agglutinated by anti-A or anti-B. The fol-
lowing procedures can be used to en-
hance the detection of weakly expressed
antigens.
1. Incubate washed red cells with anti-A,
anti-B, and anti-A,B for 15 minutes
at room temperature to increase the
association of antibody with antigen.
Incubating the test system for 15 to
30 minutes at 4 C may further en-
hance antibody attachment. An inert
(eg, 6% albumin) or autologous con-
trol for room temperature and 4 C
tests is recommended. The manufac-
turer’s directions for any reagent
should be consulted for possible
comments or limitations.
2. Treat the patient’s red cells with a
proteolyticenzymesuchasficin,
papain, or bromelin. Enzyme treat-
ment increases the antigen-antibody
reaction with anti-A or anti-B. In
some instances, reactions between
reagent antibody and red cells ex-
pressing antigens will become de-
tectable at room temperature within
30 minutes if enzyme-treated red
cells are employed. Enzyme-treated
group O red cells and an autologous
control must be tested in parallel as
a control for the specificity of the
ABO reaction. The manufacturer’s
directions should be consulted for
possible comments or limitations.
3. Incubate an aliquot of red cells at
room temperature or at 4 C with
anti-A or anti-B (as appropriate) to
adsorb antibody to the correspond-
ing red cell antigen for subsequent
elution (see Method 2.4). Group A or
B (as appropriate) and O red cells
should be subjected to parallel ad-
sorption and elution with any re-
agent to serve as positive and nega-
300 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
tive controls. Anti-A1lectin should
not be used for adsorption/elution
studies because in a more concen-
tratedform,suchasaneluate,itmay
react nonspecifically with red cells.
TesttheeluateagainstgroupA
1,B,
and O cells. Unexpected reactivity in
control eluates invalidates the re-
sults obtained with the patient’s red
cells. This indicates that the adsorp-
tion/elution procedure was not per-
formed correctly, another antibody is
present (ie, in a polyclonal reagent),
or the specificity of a monoclonal re-
agent is not distinct enough for the
reagent to be used by this method.
4. TestthesalivaforthepresenceofH
and A or B substances (see Method
2.5). Saliva tests help resolve ABO
discrepancies only if the person is a
secretor. This may be surmised from
the Lewis phenotype but may not be
known until after the saliva testing is
complete. See the discussion on Lewis
antigens.
Resolving Discrepancies Due to Absence of
Expected Antibodies
1. Incubate the serum with A1and B red
cells for 15 to 30 minutes at room
temperature. If there is still no reac-
tion, incubate at 4 C for 15 to 30
minutes. It is recommended to in-
clude an autocontrol and group O
red cells for room temperature and 4
C testing to control for reactivity of
common cold autoagglutinins.
2. Treat the A1and B reagent cells with
a proteolytic enzyme such as ficin,
papain, or bromelin. Enzyme-treated
group O and autologous red cells
must be tested in parallel as a con-
trol for reactivity. The manufacturer’s
directions should be consulted for
possible comments or limitations.
Resolving Discrepancies Due to Unexpected
Red Cell Reactions with Anti-A and Anti-B
Red cell ABO tests sometimes give unex-
pected positive reactions. For example,
reagent anti-A may weakly agglutinate red
cells from a sample in which the serum
gives reactions expected of a normal
group B or O sample. The following para-
graphs describe some events that can
cause unexpected reactions in ABO typing
tests and the steps that can be taken to
identify them.
B(A) Phenotype. When cells react weakly
with monoclonal anti-A and strongly with
anti-B, and the serum reacts with A1red
cells, but not B cells, the B(A) phenotype
should be suspected. Verification that the
anti-A reagent contains the discriminating
MHO4 clone confirms the suspicion. B(A)
red cells can show varying reactivity with
anti-A; the majority of examples react
weakly, and the agglutinates are fragile and
easily dispersed, although some examples
have reacted as strongly as 2+.22 Sera from
these individuals agglutinate both A1and A2
cells. Except for newborns and immuno-
compromised patients, serum testing should
distinguish this phenomenon from the AB
phenotype in which a subgroup of A is ac-
companied by anti-A1. Testing with an
anti-A without the MHO4 clone should re-
solve the discrepancy. The recipient can be
considered a group B.
Acquired B Phenotype. Red cells aggluti-
nated strongly by anti-A and weakly by
anti-B and a serum containing strong
anti-B suggest the acquired B state. The ac-
quired B phenomenon is found most often
in individuals with the A1phenotype; a few
examples of A2with acquired B have been
found. Most red cells with acquired B anti-
gens react weakly with anti-B, but occa-
sional examples are agglutinated quite
strongly. Behavior with monoclonal anti-B
reagents varies with the particular clone
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 301
Copyright © 2005 by the AABB. All rights reserved.
used. Acquired B antigens had been ob-
served with increased frequency in tests with
certain FDA-licensed monoclonal anti-B
blood grouping reagents containing the ES-4
clone,23 but the manufacturers have lowered
the pH of the anti-B reagent so the fre-
quency of the detection of acquired B is
similar to polyclonal anti-B or have discon-
tinued the use of that clone. To confirm that
group A red cells carry the acquired B struc-
ture:
1. Check the patient’s diagnosis. Ac-
quired B antigens are usually associ-
ated with tissue conditions that al-
low colonic bacteria to enter the
circulation, but acquired B antigens
have been found on the red cells of
apparently normal blood donors.23
2. Test the patient’s serum against auto-
logous red cells or known acquired B
cells. The individual’s anti-B will not
agglutinate his or her own red cells
or red cells known to be acquired B.
3. Test the red cells with monoclonal
anti-B reagents for which the manu-
facturer’s instructions give a detailed
description. Unlike most human poly-
clonal antibodies, some monoclonal
antibodies do not react with the ac-
quired B phenotype; this information
may be included in the manufac-
turer’s directions.
4. Test the red cells with human anti-B
serum that has been acidified to pH
6.0. Acidified human anti-B no lon-
ger reacts with the acquired B anti-
gen.
Antibody-Coated Red Cells. Red cells
from infants with HDFN or from adults suf-
fering from autoimmune or alloimmune
conditions may be so heavily coated with
IgG antibody molecules that they aggluti-
nate spontaneously in the presence of re-
agent diluents containing high protein con-
centrations. Usually, this is at the 18% to
22% range found in some anti-D reagents,
but, sometimes, the sensitized red cells also
agglutinate in ABO reagents with protein
concentrations of 6% to 12%. Methods 2.12
or 2.14 may be used to remove much of this
antibody from the red cells so that the cells
can be tested reliably with anti-A and anti-B.
Red cells from a specimen containing cold-
reactive IgM autoagglutinins may autoag-
glutinate in saline tests. Incubating the cell
suspension briefly at 37 C and then wash-
ing the cells several times with saline
warmed to 37 C can usually remove the an-
tibodies. If the IgM-related agglutination is
not dispersed by this technique, the red cells
can be treated with the sulfhydryl com-
pound dithiothreitol (DTT) (see Method 2.11).
Resolving Discrepancies Due to Unexpected
Serum Reactions
The following paragraphs describe some
events that can cause unexpected or erro-
neous serum test results and the steps that
can be taken to resolve them.
1. Reactivity of the A1reagent cells
when anti-A is strongly reactive with
the red cells suggests the presence of
anti-A1in the serum of an A2or A2B
individual. To demonstrate this as
the cause of the discrepancy:
a. Test the red cells with anti-A1
lectin to differentiate group A1
from A2red cells.
b. Test the serum against several
examples of each of the follow-
ing: A1,A
2,andOredcells.Only
if the antibody agglutinates all
A1red cells and none of the A2
or O red cell samples can it be
called anti-A1.
2. Strongly reactive cold autoaggluti-
nins, such as anti-I, anti-IH, anti-IA,
and anti-IB, can agglutinate red cells
of adults, including autologous cells
and reagent red cells, at room tem-
perature (20-24 C). With few excep-
302 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
tions, agglutination caused by the cold
autoagglutinin is weaker than that
caused by anti-A and anti-B. The fol-
lowing steps can be performed when
such reactivity interferes to the point
that the interpretation of serum tests
is difficult.
a. Warm the serum and reagent red
cells to 37 C before mixing and
testing. Incubate at 37 C for 1
hour and perform a “settled”
reading (ie, observe for aggluti-
nation without centrifugation).
Rare weakly reactive examples
of IgM anti-A or anti-B may not
be detected by this method.
b. Remove the cold autoagglutinin
from the serum using a cold
autoadsorption method as de-
scribedinMethod4.6.Thead-
sorbed serum can then be tested
against A1and B reagent red cells.
3. Unexpected alloantibodies that react
at room temperature, such as anti-P1
or anti-M, may agglutinate the red
cells used in serum tests if the cells
carry the corresponding antigen. One
or more of the reagent cells used in
the antibody detection test may also
be agglutinated if the serum and cell
mixture was centrifuged for either a
room temperature or 37 C reading; a
rare serum may react with an anti-
genoflowincidenceontheserum
testing cells that is not present on cells
used for antibody detection. Steps to
determine the correct ABO type of sera
containing cold-reactive alloantibodies
include:
a. Identify the room temperature
alloantibody, as described in
Chapter 19, and test the re-
agent A1and B cells to determine
which, if either, carries the cor-
responding antigen. Obtain A1
and B red cells that lack the an-
tigen and use them for serum
testing.
b. Raisethetemperatureto30to
37 C before mixing the serum
and cells, incubate for 1 hour, and
perform a “settled” reading (ie,
without centrifugation). If the
thermal amplitude of the allo-
antibody is below the tempera-
ture at which anti-A and anti-B
react, this may resolve the dis-
crepancy.
c. If the antibody detection test is
negative, test the serum against
several examples of A1and B
red cells. The serum may con-
tain an antibody directed against
an antigen of low incidence,
which will be absent from most
randomly selected A1and B red
cells.
4. Sera from patients with abnormal
concentrations of serum proteins, or
with altered serum protein ratios, or
who have received plasma expand-
ers of high molecular weight can ag-
gregate reagent red cells and mimic
agglutination. Some of these sam-
ples cause aggregation of the type
described as rouleaux. More often,
the aggregates appear as irregularly
shaped clumps that closely resemble
antibody-mediated agglutinates. The
results of serum tests can often be
corrected by diluting the serum 1:3
in saline to abolish its aggregating
properties or by using a saline re-
placement technique (see Method
3.4).
The H System
On group O red cells, there is no A or B
antigen, and the membrane expresses
abundant H. Because H is a precursor of A
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 303
Copyright © 2005 by the AABB. All rights reserved.
and B antigens, A and B persons have less
H substance than O persons. The amount
of H antigen detected on red cells with the
anti-H lectin Ulex europaeus is, in order of
diminishing quantity, O>A2>B>A2B>A1>A1B.
Occasionally, group A1,A
1B, or (less com-
monly) B individuals have so little uncon-
verted H antigen on their red cells that they
produce anti-H. This form of anti-H is gen-
erally weak, reacts at room temperature or
below, and is not considered clinically sig-
nificant. Individuals of the rare Ohpheno-
type (see below), whose red cells lack H,
have a potent and clinically significant
alloanti-H in their serum (in addition to
anti-A and anti-B).
OhPhenotype
The term Ohor Bombay phenotype has
been used for the very rare individuals
whose red cells and secretions lack H, A,
and B antigens and whose plasma con-
tains potent anti-H, anti-A, and anti-B.6
This phenotype was first discovered in
Bombay, India. The phenotype initially
mimics normal group O but becomes ap-
parentwhenserumfromtheO
hindivid-
ual is tested against group O red cells, and
strong immediate-spin agglutination
and/or hemolysis occurs. The anti-H of
an Ohperson reacts over a thermal range
of 4 to 37 C with all red cells except those
of other Ohpeople. Ohpersons must be
transfused only with Ohblood because
their non-red-cell-stimulated antibodies
rapidly destroy cells with A, B, or H anti-
gens. If other examples of Ohred cells are
available, further confirmation can be ob-
tained by demonstrating compatibility of
the serum with Ohred cells. At the geno-
typic level, the Ohphenotype arises from
the inheritance of hh at the Hlocus and
sese at the Se locus. Because the Se allele is
necessary for the formation of Leb,O
hred
cells will be Le(a+b–) or Le(a–b–).
Para-Bombay Phenotype
The para-Bombay phenotype designation,
Ah,B
h,andAB
h, is classically used for indi-
viduals who are H-deficient secretors, ie,
those who have an inactive H-transferase
but have active Se-transferase. The red
cells lack serologically detectable H anti-
gen but carry small amounts of A and/or
B antigen (sometimes detectable only by
adsorption/elution tests), depending on
the individual’s alleles at the ABO locus.
Tests with anti-A or anti-B reagents may
or may not give weak reactions, but the
cells are nonreactive with anti-H lectin or
with anti-H serum from Ohpersons. Indi-
viduals with the para-Bombay phenotype
have a functional Se allele and thus will
express A, B, and H antigens in their
plasma and secretions. The sera of Ahand
Bhpeople contain anti-H and/or anti-IH
in addition to the expected anti-A or
anti-B.
H-deficient secretors may also be group
O. These individuals will have traces of H
antigen, but no A or B antigen, on their red
cells and only have H in their secretions.
In 1994, Kelly et al reported the molecu-
lar bases for the Bombay and para-Bombay
phenotypes.24 Many mutations at the Hlo-
cus have subsequently been associated
with H-deficiency.
The Lewis System
The Lewis system antigens, Leaand Leb,
result from the action of a glycosyltrans-
ferase encoded by the Le allele that, like
theA,B,andHglycosyltransferases,adds
a sugar to a precursor chain. Leais pro-
duced when Le is inherited with sese and
Lebis produced when Le is inherited with
at least one Se allele. When the silent or
amorphic allele le is inherited, regardless
of the secretor allele inherited, no Leaor
Lebis produced. Thus, Leaand Lebare not
304 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

antithetical antigens produced by alleles;
rather, they result from the interaction of
independently inherited alleles.
The Lewis antigens are not intrinsic to
red cells but are expressed on glycosphing-
olipid Type 1 chains adsorbed from plasma
onto red cell membranes. Plasma lipids ex-
change freely with red cell lipids.
Gene Interaction and the Antigens
The synthesis of the Lewis antigens is de-
pendent upon the interaction of two dif-
ferent fucosyltransferases: one from the
Se locusandonefromtheLewislocus.
Both enzymes act upon the same precur-
sorType1substratechains.Thefuco
-
syltransferase encoded by the Le allele at-
taches fucose in α(1→4) linkage to the
subterminal GlcNAc; in the absence of the
transferase from the Se allele, this config-
uration has Leaactivity. This product can-
not be further glycosylated. Leboccurs
when the Type 1 precursor is converted to
Type 1 H by the fucosyltransferase from
the secretor allele, and subsequently acted
upon by the fucosyltransferase from the
Le allele. This Lebconfiguration has two
fucose moieties.9Thus, Lebreflects the
presence of both the Le and Se alleles. Le
without Se results in Leaactivity only; Se
with the amorphic allele le will result in
no secretion of Leaor Lebandtheredcells
will have the Le(a–b–) phenotype.
Table 13-3 shows phenotypes of the Lewis
system and their frequencies in the popula-
tion. Red cells that type as Le(a+b+) are rare
in people of European and African origin
but are relatively common in persons of
Asian origin, due to a fucosyltransferase en-
coded by a variant secretor allele that com-
petes less efficiently with the Le fuco-
syltransferase.9
Lewis Antibodies
Lewis antibodies occur almost exclusively
in the sera of Le(a–b–) individuals, usually
without known red cell stimulus. Those
individuals whose red cell phenotype is
Le(a–b+) do not make anti-Leabecause
small amounts of unconverted Leaare
present in their saliva and plasma. It is
most unusual to find anti-Lebin the sera
of Le(a+b–) individuals, but anti-Lebmay
exist along with anti-Leain the sera of
Le(a–b–) individuals. Lewis antibodies are
often found in the sera of pregnant women
who transiently demonstrate a Le(a–b–)
phenotype. The Lewis antibodies, however,
are almost always IgM and do not cross
the placenta. Because of this and because
Lewis antigens are poorly developed at
birth, the antibodies are not associated
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 305
Table 13-3. Phenotypes in the Lewis System and Their Incidence
Reactions with Anti- Adult Phenotype Incidence %
LeaLebPhenotype Whites Blacks
+ 0 Le(a+b−)22 23
0+Le(a−b+) 72 55
00Le(a−b−)6 22
+ + Le(a+b+) Rare Rare
+ = agglutination; 0 = no agglutination.
Copyright © 2005 by the AABB. All rights reserved.
with HDFN. Lewis antibodies may bind
complement, and fresh serum that con-
tains anti-Lea(or infrequently anti-Leb)
may hemolyze incompatible red cells in
vitro. Hemolysis is more often seen with
enzyme-treated red cells than with un-
treated red cells.
Most Lewis antibodies agglutinate sa-
line-suspended red cells of the appropriate
phenotype. The resulting agglutinates are
often fragile and are easily dispersed if red
cell buttons are not resuspended gently af-
ter centrifugation. Agglutination sometimes
is seen after incubation at 37 C, but rarely
of the strength seen in tests incubated at
room temperature. Some examples of
anti-Lea, and less commonly anti-Leb,canbe
detected in the antiglobulin phase of test-
ing. Sometimes this reflects complement
bound by the antibody if a polyspecific re-
agent (ie, containing anticomplement) is
used. In other cases, antiglobulin reactivity
results from an IgG component of the anti-
body.
Sera with anti-Lebactivity can be divided
into two categories. The more common type
reacts best with Le(b+) red cells of group O
and A2; these antibodies have been called
anti-LebH. Antibodies that react equally well
with the Lebantigen on red cells of all ABO
phenotypes are called anti-LebL.
Transfusion Practice
Lewis antigens readily adsorb to and elute
from red cell membranes. Transfused red
cells shed their Lewis antigens and assume
the Lewis phenotype of the recipient
within a few days of entering the circula-
tion. Lewis antibodies in a recipient’s se-
rum are readily neutralized by Lewis
blood group substance in donor plasma.
For these reasons, it is exceedingly rare for
Lewisantibodiestocausehemolysisof
transfused Le(a+) or Le(b+) red cells. It is
not considered necessary to type donor
blood for the presence of Lewis antigens
before transfusion or when crossmatch-
ing for recipients with Lewis antibodies;
red cells that are compatible in tests at 37
C can be expected to survive normally in
vivo.25,26
Lewis Antigens in Children
Red cells from newborn infants usually do
not react with either human anti-Leaor
anti-Leband are considered to be Le(a–b–).
Some can be shown to carry small amounts
of Leawhen tested with potent mono-
clonal anti-Leareagents. Among children,
the incidence of Le(a+) red cells is high
and that of Le(b+) red cells low, reflecting
a greater production of the Le allele-spe-
cific transferase in infants; the Se allele-
specific transferase is produced in lower
levels. The phenotype Le(a+b+) may be
transiently observed in children as the Se
allele transferase levels increase toward
adult levels. Reliable Lewis typing of young
children may not be possible because test
reactions may not reflect the correct phe-
notype until approximately 2 to 3 years of
age.25
The I/i Antigens and
Antibodies
Cold agglutinins with I specificity are fre-
quently encountered in sera of normal in-
dividuals. The antibody is usually not
clinically significant and reacts with all
red cells except cord cells and the rare i
adult phenotype. I and i antigens, however,
are not antithetical but are expressed in a
reciprocal relationship. At birth, infant red
cells are rich in i; I is almost undetectable.
Thus, for practical purposes, cord cells are
considered to be I–, i+. During the first 2
years of life, I antigen gradually increases
at the expense of i. The red cells of most
306 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
adults are strongly reactive with anti-I
and react weakly or not at all with anti-i.
Rare adults have red cells that carry high
levels of i and only trace amounts of I;
these red cells have the i adult phenotype.
ThereisnotrueI–ori–phenotype.
The I and i antigens on red cells are in-
ternal structures carried on the same glyco-
proteins and glycosphingolipids that carry
H, A, or B antigens and in secretions on the
same glycoproteins that carry H, A, B, Lea,
and Lebantigens. The I and i antigens are
located closer to the membrane than the
terminal sugars that determine the ABH
antigens. The i structure is a linear chain of
at least two repeating units of N-acetyl-
galactosamine [Galβ(1→4)GlcNAcβ(1→3)].
On the red cells of adults, many of these
linear chains are modified by the addition
of branched structures consisting of GlcNAc
in a β(1→6) linkage to a galactose residue
internal to the repeating sequence. The
branching configuration confers I specific-
ity.27,28 Different examples of anti-I appear to
recognize different portions of the branch-
ed oligosaccharide chain. As branching oc-
cursandasthesugarsforH,A,andBanti-
gens are added, access of anti-i and anti-I
may be restricted.25
The I antigen, together with the i anti-
gen, used to comprise the Ii Blood Group
Collection in the ISBT nomenclature. Re-
cent cloning of the gene that encodes the
transferase responsible for converting i ac-
tive straight chains into I active branched
chains and identification of several muta-
tions responsible for the rare i adult pheno-
type have caused the I antigen to be as-
signed to the new I Blood Group System;
the i antigen remains in the Ii collection.29
Antibodies to I/i
Anti-I is a common, benign autoantibody
found in the serum of many normal healthy
individuals that behaves as a cold aggluti-
nin at 4 C with a titer of <64. Agglutination
with adult red cells and weaker or no ag-
glutination with cord cells is the classic
reactivity. Some stronger examples agglu-
tinate cells at room temperature; others
may react only with the strongest I+ red
cells and give inconsistent reactions. Incu-
bating tests in the cold enhances anti-I re-
activity and helps to confirm its identity;
albumin and testing enzyme-treated red
cells also enhance anti-I reactivity.
Autoanti-I assumes pathologic signifi-
cance in cold agglutinin syndrome (CAS),
in which it behaves as a complement-bind-
ing antibody with a high titer and high ther-
mal amplitude. The specificity of the auto-
antibody in CAS may not be apparent when
the undiluted sample is tested (I adult and
cord cells may react to the same strength
even at room temperature); titration studies
and/or thermal amplitude studies may be
necessary to define the specificity (see
Chapter 20). Anti-I is often made by pa-
tients with pneumonia due to Mycoplasma
pneumoniae. These patients may experience
transient hemolytic episodes due to the an-
tibody.
Autoanti-i is less often implicated in
symptomatic disease than anti-I. On rare
occasions, anti-i may be seen as a relatively
weak cold autoagglutinin reacting preferen-
tially at 4 C. Anti-i reacts strongest with
cord and i adult red cells, and weakest with
I adult red cells. Patients with infectious
mononucleosis often have transient but
potent anti-i.
Table 13-4 illustrates the serologic be-
havior of anti-I and anti-i at 4 C and 22 C.
Reaction strengths should be considered
relative; clear-cut differences in reactivity
between the two are seen only with weaker
examples of the antibodies. Titration stud-
ies may be needed to differentiate strong
examples of the antibodies.
Serum containing anti-I or anti-i is some-
times reactive at the antiglobulin phase of
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 307
Copyright © 2005 by the AABB. All rights reserved.

testing when polyspecific antihuman glob-
ulin is used. Such reactions rarely indicate
antibody activity at 37 C. Rather, comple-
ment components are bound when serum
and cells interact at lower temperatures;
during the 37 C incubation, the antibody
dissociates but the complement remains
bound to the red cells. Thus, the anti-
globulin phase reactivity is usually due to
the anticomplement in a polyspecific anti-
globulin reagent. Usually, avoiding room
temperature testing and using anti-IgG in-
stead of a polyspecific antihuman globulin
help to eliminate detection of cold auto-
antibodies. Cold autoadsorption to remove
the autoantibody from the serum may be
necessary for stronger examples; cold auto-
adsorbed serum or plasma can also be used
in ABO typing (see Method 4.6). The pre-
warmingtechniquecanalsobeusedonce
the reactivity has been confirmed as cold
autoantibody (see Method 3.3).
Complex Reactivity
Some antibodies appear to recognize I de-
terminants with attached H, A, or B im-
munodominant sugars. Anti-IH occurs
quite commonly in the serum of A1indi-
viduals; it reacts stronger with red cells
that have high levels of H as well as I (ie,
group O and A2red cells) and weaker, if at
all, with group A1cells of adults or cord
cells of any group. Anti-IH should be sus-
pected when serum from a group A pa-
tient causes direct agglutination of all
cells used for antibody detection but is
compatible with all or most group A do-
nor blood. Other examples of complex re-
activity include anti-IA, -IP1,-IBH,-ILe
bH,
and -iH.
The P Blood Group and
Related Antigens
The P blood group has traditionally con-
sisted of the P, P1,andP
kantigens, and
later Luke (LKE). However, the biochemis-
try and molecular genetics, although not
yet completely understood, make it clear
that at least two biosynthetic pathways
and genes at different loci are involved in
the development and expression of these
antigens. Thus, in ISBT nomenclature, the
P antigen is assigned to the new globoside
(GLOB) blood group system, the P1anti-
gen is assigned to the P blood group system,
and Pkand LKE remain in the globoside
collection of antigens.29 For simplicity,
these antigens are often referred to as the
P blood group.
308 AABB Technical Manual
Table 13-4. Comparative Serologic Behavior of the I/i Blood Group Antibodies with
Saline Red Cell Suspensions
Temperature Cell Type Anti-I Anti-i
4 C I adult 4+ 0-1+
icord 0-2+ 3+
i adult 0-1+ 4+
22 C I adult 2+ 0
i cord 0 2-3+
i adult 0 3+
Copyright © 2005 by the AABB. All rights reserved.
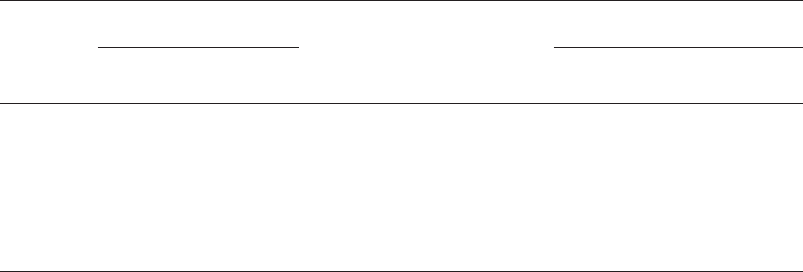
The first antigen of the P blood group
was discovered by Landsteiner and Levine
in 1927, in a series of animal experiments
that led also to the discovery of M and N.
Originally called P, the name of the antigen
was later changed to P1.ThedesignationP
has since been reassigned to an antigen
present on almost all human red cells. The
Pkantigen is also present on almost all hu-
man red cells, but it is not readily detected
unless P is absent, eg, in the rare P1
kor P2
k
phenotypes. The null phenotype, p, is very
rare. LKE antigen is present on almost all
red cells except those of the rare phenotypes
porP
kand in about 2% of P+ red cells.
Common and Rare Phenotypes
There are two common phenotypes asso-
ciated with the P blood group, P1and P2,
and three rare phenotypes, p, P1
k,andP
2
k,
asshowninTable13-5.TheP
1phenotype
describes those red cells that react with
anti-P1and anti-P; red cells that do not re-
act with anti-P1, but do react with anti-P,
are of the P2phenotype. When red cells
are tested only with anti-P1and not with
anti-P, the phenotype should be written as
P1+or P1–.
Biochemistry and Genetics
ThePbloodgroupantigens,liketheABH
antigens, are sequentially synthesized by
the addition of sugars to precursor chains.
The different oligosaccharide determi-
nants of the P blood group antigens are
showninFig13-5.Alltheantigensareex
-
clusively expressed on glycolipids on hu-
man red cells, not on glycoproteins.30 The
precursor of P1can also be glycosylated to
Type 2H chains, which carry ABH antigens.
There are two distinct pathways for the
synthesis of the P blood group antigens as
showninFig13-6.Thecommonprecursor
is lactosylceramide, also known as cera-
mide dihexose or CDH. One pathway re-
sults in the formation of paragloboside and
P1. Paragloboside is also the Type 2 precur-
sor for ABH antigens. The other pathway
results in the formation of the globoside se-
ries of antigens: Pk,P,andLKE.
The genes encoding the glycosyltrans-
ferases that are responsible for synthesizing
Pkfrom lactosylceramide and for converting
Pkto P were cloned in 2000. Several muta-
tions that result in the p and Pkphenotypes
have been identified.31-33 The genetic rela-
tionship between P1,PandP
kis still not un-
derstood. Red cells of the p phenotype are
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 309
Table 13-5. Phenotypes of the P Blood Group and Related Antigens
Reactions with Anti- Phenotype Incidence (%)
P1PP
kP1+P+PkPhenotype Whites Blacks
++ 0 + P
179 94
0+ 0 + P
221 6
00* 00p
+0 + + P
1kAll extremely rare
00 + + P
2k
*Usually negative, occasionally weakly positive.
Copyright © 2005 by the AABB. All rights reserved.
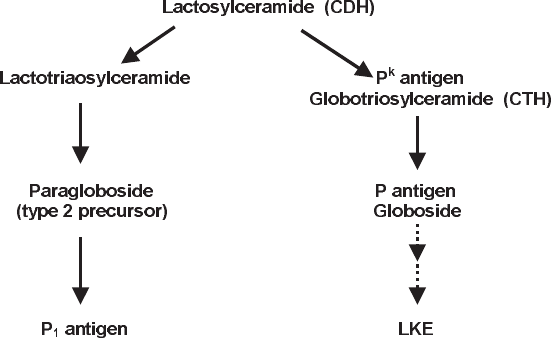
P1– in addition to being P– and Pk–; the P1–
status of p red cells cannot be explained.
The P1gene is located on chromosome 22
and the Pgene is located on chromosome 3.
Anti-P1
The sera of P1– individuals commonly
contain anti-P1. If sufficiently sensitive
techniques are applied, it is likely that
anti-P1would be detected in the serum of
virtually every person with P1– red cells.25
The antibody reacts optimally at 4 C but
may occasionally be detected at 37 C.
Anti-P1is nearly always IgM and has not been
reported to cause HDFN. Only rarely has
it been reported to cause hemolysis in
vivo.17(p139),34
The strength of the P1antigen varies widely
among different red cell samples, and anti-
gen strength has been reported to diminish
when red cells are stored. These character-
istics sometimes create difficulties in iden-
tifying antibody specificity in serum with a
positive antibody screen. An antibody that
reacts weakly in room temperature testing
can often be shown to have anti-P1specific-
ity by incubation at lower temperatures or
by the use of enzyme-treated red cells.
Hydatid cyst fluid or P1substance derived
from pigeon eggs inhibits the activity of
anti-P1. Inhibition may be a useful aid to
310 AABB Technical Manual
Figure 13-6. Biosynthesis of P blood group antigens.
Lactosylceramide (CDH) Galβ(1→4)Glc-Cer
PkGlobotriosylceramide (CTH) Galα(1→4)Galβ(1→4)Glc-Cer
P Globoside GalNAcβ(1→3)Galα(1→4)Galβ(1→4)Glc-Cer
LKE Sialosylgalactosylgloboside NeuAcα(2→3)Galβ(1→3)GalNAcβ(1→3)Galα(1→4)Galβ(1→4)Glc-Cer
Paragloboside Galβ(1→4)GlcnNAcβ(1→3)Galβ(1→4)Glc-Cer
P1Galactosylparagloboside Galα(1→4)Galβ(1→4)GlcnNAcβ(1→3)Galβ(1→4)Glc-Cer
Figure 13-5. Some biochemical structures of P blood group antigens.
Copyright © 2005 by the AABB. All rights reserved.
antibody identification, especially if anti-P1
is present in a serum with antibodies of
other specificities.
Rare Antibodies
Alloanti-P, found as a naturally occurring
potent hemolytic antibody in the sera of
P1
kand P2
kindividuals, reacts with all red
cells except those of the rare p and Pkphe-
notypes. Anti-P can be IgM or a mixture of
IgM and IgG. Anti-PP1Pk, formerly called
anti-Tja, is produced by individuals of the
p phenotype without red cell stimulation
and reacts with all red cells except those
of the rare p phenotype. Anti-PP1Pkcan be
separated into its components (anti-P,
anti-P1,andanti-P
k) through adsorptions.
These components can be IgM and/or
IgG, react over a broad thermal range, and
can efficiently bind complement, which
make them potent hemolysins. Anti-
PP1Pkhas caused hemolytic transfusion
reactions and, occasionally, HDFN.17(p139)
There is an association between both
anti-P and anti-PP1Pkand spontaneous
abortions occurring early in pregnancy 35,36
Autoanti-P associated with paroxysmal
cold hemoglobinuria is a cold-reactive IgG
autoantibody that is described as a biphasic
hemolysin.17(pp220-1) The antibody typically
does not react in routine test systems, but is
demonstrable only by the Donath-Land-
steiner test (see Chapter 20).
P Antigens as Receptors for Pathogens
The P blood group antigens are receptors
for several pathogens. P, P1,P
k,andLKE
are receptors for uropathogenic Esche-
richia coli;P
kand P1are receptors for tox-
ins from enterohemorrhagic E. coli37;and
the meningitis-causing bacterium Strep-
tococcus suis binds to Pkantigen.38 The P
antigen (globoside) has also been shown
to serve as a receptor for erythrovirus (par-
vovirus) B19, which causes erythema in-
fectiosum (Fifth disease). Individuals of p
phenotype who lack globoside are natu-
rally resistant to infection with this patho-
gen.39
References
1. Landsteiner K. Zur Kenntnis der antifer-
mentativen, lytischen und agglutinierenden
Wirkungen des Blutserums und der Lymph.
Zbl Balk 1900;27:367.
2. Landsteiner K. Uber Agglutinationserschers-
chein ungen normalen menschlichen Blutes.
Wien Klin Wochenschr 1901;14:1132-4.
3. Garratty G, Dzik W, Issitt PD, et al. Terminol-
ogy for blood group antigens and genes—his-
torical origins and guidelines in the new mil-
lennium. Transfusion 2000;40:477-89.
4. Von Decastello A, Sturli A. Yber due usiaglutinie
im Serumgesunder und lronker Menschen.
Munchen Med Wochenschr 1902;26:1090-5.
5. Watkins WM. The ABO blood group system:
Historical background. Transfus Med 2001;
11:243-65.
6. Oriol R, Candelier JJ, Mollicone R. Molecular
genetics of H. Vox Sang 2000;78(Suppl 2):105-
8.
7. Yamamoto F. Molecular genetics of ABO. Vox
Sang 2000;78(Suppl 2):91-103.
8. Yamamoto F. Molecular genetics of the ABO
histo-blood group system. Vox Sang 1995;69:
1-7.
9. DanielsG.Humanbloodgroups.2nded.Ox
-
ford: Blackwell Science, 2002.
10. Clausen H, Levery SB, Nudelman E, et al. Re-
petitive A epitope (type 3 chain A) defined by
group A1-specific monoclonal antibody
TH-1: Chemical basis of qualitative A1and A2
distinction. Proc Natl Acad Sci U S A 1985;82:
1199-203.
11. Yamamoto F, Clausen H, White T, et al. Mo-
lecular genetic basis of the histo-blood group
ABO system. Nature 1990;345:229-33.
12. Patenaude SI, Seto NOL, Borisova SN, et al.
The structural basis for specificity in human
ABO(H) blood group biosynthesis (letter).
Nat Struct Biol 2002;9:685-90.
13. Lee AH, Reid ME. ABO blood group system: A
review of molecular aspects. Immunohema-
tology 2000;16:1-6.
14. Olsson ML, Chester MA. Polymorphism and
recombination events at the ABO locus: A
major challenge for genomic ABO blood
grouping strategies. Transfus Med 2001;11:
295-313.
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 311
Copyright © 2005 by the AABB. All rights reserved.
15. Reid ME, Lomas-Francis C. The blood group
antigen factsbook. 2nd ed. San Diego, CA: Ac-
ademic Press, 2004.
16. Olsson ML, Irshaid NM, Hosseini-Maaf B, et
al. Genomic analysis of clinical samples with
serologic ABO blood grouping discrepancies:
Identification of 15 novel A and B subgroup
alleles. Blood 2001;98:1585-93.
17. Mollison PL, Engelfriet CP, Contreras M.
Blood transfusion in clinical medicine. 10th
ed. Oxford: Blackwell Scientific Publications,
1997.
18. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
19. SpruellP,ChenJ,CullenK.ABOdiscrepancies
in the presence of pH-dependent autoagglu-
tinins (abstract). Transfusion 1994;34(Suppl):
22S.
20. KennedyMS,WaheedA,MooreJ.ABOdis
-
crepancy with monoclonal ABO reagents
caused by pH-dependent autoantibody. Im-
munohematology 1995;11:71-3.
21. Garratty G. In vitro reactions with red blood
cells that are not due to blood group antibod-
ies: A review. Immunohematology 1998;14:1-
11.
22. Beck ML, Yates AD, Hardman J, Kowalski MA.
Identification of a subset of group B donors
reactive with monoclonal anti-A reagent. Am
J Clin Pathol 1989;92:625-9.
23. Beck ML, Kowalski MA, Kirkegaard JR, Korth
JL. Unexpected activity with monoclonal anti-B
reagents (letter). Immunohematology 1992;8:
22.
24. KellyRJ,ErnstLK,LarsenRD,etal.Molecular
basis for H blood group deficiency in Bombay
(Oh) and para-Bombay individuals. Proc Natl
Acad Sci U S A 1994;91:5843-7.
25. Issitt PD, Anstee DJ. Applied blood group se-
rology. 4th ed. Durham, NC: Montgomery
Scientific Publications, 1998.
26. Waheed A, Kennedy MS, Gerhan S, Senhauser
DA. Success in transfusion with cross-
match-compatible blood. Am J Clin Pathol
1981;76:294-8.
27. Yu L-C, Twu Y-C, Chang C-Y, Lin M. Molecu-
lar basis of the adult I phenotype and the
gene responsible for the expression of the hu-
man blood group I antigen. Blood 2001;98:
3840-5.
28. YuL-C,TwuY-C,ChouM-L,etal.Themolec
-
ular genetics of the human Ilocus and mo-
lecular background explain the partial asso-
ciation of the adult I phenotype with congenital
cataracts. Blood 2003;101:2081-8.
29. Daniels GL, Cartron JP, Fletcher A, et al. Inter-
national Society of Blood Transfusion Com-
mittee on terminology for red cell surface an-
tigens: Vancouver report. Vox Sang 2003;84:
244-7.
30. Yang Z, Bergstrom J, Karlsson KA. Glycopro-
teins with Galα4Gal are absent from human
erythrocyte membranes, indicating that
glycolipids are the sole carriers of blood group
P activities. J Biol Chem 1994;269:14620-4.
31. Hellberg A, Poole J, Olsson ML. Molecular ba-
sis of the globoside-deficient Pkblood group
phenotype. J Biol Chem 2002;277;29455-9.
32. Furukawa K, Iwamura K, Uchikawa M, et al.
Molecular basis for the p phenotype. J Biol
Chem 2000;275:37752-6.
33. Koda Y, Soejima M, Sato H, et al. Three-base
deletion and one-base insertion of the α(1,4)
galactosyltransferase gene responsible for the
p phenotype. Transfusion 2002;42:48-51.
34. Arndt PA, Garratty G, Marfoe RA, Zeger GD.
An acute hemolytic transfusion reaction
caused by an anti-P1that reacted at 37 C.
Transfusion 1998;38:373-7.
35. Shirey RS, Ness PM, Kickler TS, et al. The as-
sociation of anti-P and early abortion. Trans-
fusion 1987;27:189-91.
36. Cantin G, Lyonnais J. Anti-PP1Pkand early
abortion. Transfusion 1983;23:350-1.
37. Spitalnik PF, Spitalnik SL. The P blood group
system: Biochemical, serological, and clinical
aspects. Transfus Med Rev 1995;9:110-22.
38. Haataja S, Tikkanen K, Liukkonen J, et al.
Characterization of a novel bacterial adhe-
sion specificity of Streptococcus suis recogniz-
ing blood group P receptor oligosaccharides.
J Biol Chem 1993;268:4311-17.
39. Brown KE, Hibbs JR, Gallinella G, et al. Resis-
tance to parvovirus B19 infection due to lack
of virus receptor (erythrocyte P antigen). N
Engl J Med 1994;330:1192-6.
Suggested Reading
Chester MA, Olsson ML. The ABO blood group
gene: A locus of considerable genetic diversity.
Transfus Med Rev 2001;15:177-200.
Daniels G. Human blood groups. 2nd ed. Oxford:
Blackwell Science Publications, 2002.
Hanfland P, Kordowicz M, Peter-Katalinic J, et al.
Immunochemistry of the Lewis blood-group sys-
tem: Isolation and structure of Lewis-c active and
related glycosphingolipids from the plasma of
blood-group O Le(a–b–) nonsecretors. Arch
Biochem Biophys 1986;246:655-72.
Issitt PD, Anstee DJ. Applied blood group serology.
4th ed. Durham, NC: Montgomery Scientific Pub-
lications, 1998.
312 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Judd WJ. Methods in immunohematology. 2nd ed.
Durham, NC: Montgomery Scientific Publications,
1994.
Lee AH, Reid ME. ABO blood group system: A re-
view of molecular aspects. Immunohematology
2000;16:1-6.
Morgan WTJ, Watkins WM. Unraveling the bio-
chemical basis of blood group ABO and Lewis an-
tigenic specificity. Glycoconj J 2000;17:501-30.
Olsson ML, Chester MA. Polymorphism and re-
combination events at the ABO locus: A major
challenge for genomic ABO blood grouping strate-
gies. Transfus Med 2001;11:295-313.
Palcic MM, Seto NOL, Hindsgual O. Natural and
recombinant A and B gene encoded glycosyltrans-
ferases. Transfus Med 2001;11:315-23.
Rydberg L. ABO-incompatibility in solid organ
transplant. Transfus Med 2001;11:325-42.
Watkins WM. The ABO blood group system: His-
torical background. Transfus Med 2001;11:243-65.
Yamamoto F. Cloning and regulation of the ABO
genes. Transfus Med 2001;11:281-94.
Chapter 13: ABO, H, and Lewis Blood Groups and Structurally Related Antigens 313
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 14: The Rh System
Chapter 14
The Rh System
THIS CHAPTER USES the DCE no-
menclature—a modification of the
nomenclature originally proposed
by Fisher and Race,1which has been able
to accommodate our present understand-
ing of the genetics and biochemistry of this
complex system. The Rh-Hr terminology
of Wiener is presented only in its histori-
cal context, as molecular genetic evidence
does not support Wiener’s one-locus theory.
The D Antigen and Its
Historical Context
Discovery of D
The terms “Rh positive” and “Rh negative”
refer to the presence or absence of the red
cell antigen D. The first human example
of the antibody against the antigen later
called D was reported in 1939 by Levine
and Stetson2; the antibody was found in
the serum of a woman whose fetus had
hemolytic disease of the fetus and new-
born (HDFN) and who experienced a
hemolytic reaction after transfusion of
her husband’s blood. In 1940, Landsteiner
and Wiener3described an antibody ob-
tained by immunizing guinea pigs and
rabbits with the red cells of Rhesus mon-
keys; it agglutinated the red cells of ap-
proximately 85% of humans tested, and
they called the corresponding determi-
nant the Rh factor. In the same year, Le-
vine and Katzin4found similar antibodies
in the sera of several recently delivered
women, and at least one of these sera
gave reactions that paralleled those of the
animal anti-Rhesus sera. Also in 1940,
Wiener and Peters5observed antibodies of
the same specificity in the sera of persons
whose red cells lacked the determinant
and who had received ABO-compatible
transfusions in the past. Later evidence
established that the antigen detected by
315
14
Copyright © 2005 by the AABB. All rights reserved.
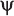
animal anti-Rhesus and human anti-D
were not identical, but, by that time, the
Rh blood group system had already re-
ceived its name. Soon after anti-D was
discovered, family studies showed that
the D antigen is genetically determined;
transmission of the trait follows an auto-
somal dominant pattern.
Clinical Significance
After the A and B antigens, D is the most
important red cell antigen in transfusion
practice. In contrast to A and B, however,
persons whose red cells lack the D anti-
gen do not regularly have anti-D. Forma-
tion of anti-D results from exposure,
through transfusion or pregnancy, to red
cells possessing the D antigen. The D an-
tigen has greater immunogenicity than
other red cell antigens; it is estimated that
30% to 85%6,7 of D– persons who receive a
D+ transfusion will develop anti-D. There-
fore, in most countries, the blood of all re-
cipients and all donors is routinely tested
for D to ensure that D– recipients are
identified and given D– blood.
Other Rh Antigens
By the mid-1940s, four additional anti-
gens—C, E, c, and e—had been recog-
nized as belonging to what is now called
the Rh system. Subsequent discoveries
have brought the number of Rh-related
antigens to 49 (Table 14-1), many of
which exhibit both qualitative and quan-
titative variations. The reader should be
aware that these other antigens exist (see
the suggested reading list), but, in most
transfusion medicine settings, the five
principal antigens (D, C, E, c, e) and their
corresponding antibodies account for the
vast majority of clinical issues involving
the Rh system.
Although Rh antigens are fully expressed
at birth with antigen detection as early as 8
weeks’ gestation,10 they are present on red
cells only and are not detectable on plate-
lets, lymphocytes, monocytes, neutrophils,
or other tissues.11,12
Genetic and Biochemical
Considerations
Attempts to explain the genetic control of
Rh antigen expression were fraught with
controversy. Wiener13 proposed a single
locus with multiple alleles determining
surface molecules that embody numerous
antigens. Fisher and Race14 inferred from
the existence of antithetical antigens the
existence of reciprocal alleles at three in-
dividual but closely linked loci. Tippett’s
prediction15 that two closely linked struc-
tural loci on chromosome 1 determine
the production of Rh antigens has been
shown to be correct.
RH
Genes
Two highly homologous genes on the
short arm of chromosome 1 encode the
nonglycosylated polypeptides that ex-
press the Rh antigens (Fig 14-1).16,17 One
gene, designated RHD, determines the
presence of a membrane-spanning pro-
tein that confers D activity on the red cell.
In Caucasian D– persons, the RHD gene is
deleted; the D– phenotype in some other
populations (persons of African descent,
Japanese, and Chinese) is associated with
an inactive, mutated, or partial RHD
gene.18 The inactive RHD gene or pseudo-
gene (RHD ) responsible for the D– phe-
notype in some Africans has been de-
scribed.19
The RHCE gene determines the C, c, E,
and e antigens; its alleles are RHCe,RHCE,
RHcE,andRHce.20 Evidence derived from
316 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

transfection studies21 indicates that both
C/c and E/e reside on a single polypeptide
product.
Biochemical and Structural Observations
The predicted products of both RHD and
RHCE are proteins of 417 amino acids
that, modeling studies suggest, traverse
the red cell membrane 12 times and dis-
play only short exterior loops of amino
acids (Fig 14-1). The polypeptides are fatty
acylated and, unlike most blood-group-
associated proteins, carry no carbohy-
drate residues.
Chapter 14: The Rh System 317
Table 14-1. Antigens of the Rh Blood Group System and Their Incidence
Numeric
Designation
Antigen
Name
Incidence (%)*
Numeric
Designation
Antigen
Name
Incidence (%)*
White Black Overall White Black Overall
Rh1 D 85 92 Rh32 Rh32 <0.01 1
Rh2 C 68 27 Rh33 Har <0.01
Rh3 E 29 22 Rh34 Bastiaan >99.9
Rh4 c 80 96 Rh35 Rh35 <0.01
Rh5 e 98 Rh36 Bea<0.1
Rh6 f 65 92 Rh37 Evans <0.01
Rh7 Ce 68 27 Rh39 C-like >99.9
Rh8 Cw2 1 Rh40 Tar <0.01
Rh9 Cx<0.01 Rh41 Ce-like 70
Rh10 V 1 30 Rh42 Ces<0.1 2
Rh11 Ew<0.01 Rh43 Crawford <0.01
Rh12 G 84 92 Rh44 Nou >99.9
Rh17 Hr0>99.9 Rh45 Riv <0.01
Rh18 Hr >99.9 Rh46 Rh46 >99.9
Rh19 hrs98 Rh47 Dav >99.9
Rh20 VS <0.01 32 Rh48 JAL <0.01
Rh21 CG68 Rh49 STEM <0.01 6
Rh22 CE <1 Rh50 FPTT <0.01
Rh23 Dw<0.01 Rh51 MAR >99.9
Rh26 80 96 Rh52 BARC <0.01
Rh27 cE 28 22 Rh53 JAHK <0.01
Rh28 <0.01 Rh54 DAK <0.01
Rh29 total Rh >99.9 Rh55 LOCR <0.01
RH30 Goa0 <0.01 Rh56 CENR <0.01
Rh31 hrB98
*Incidence in White and Black populations where appropriate.8,9
Copyright © 2005 by the AABB. All rights reserved.
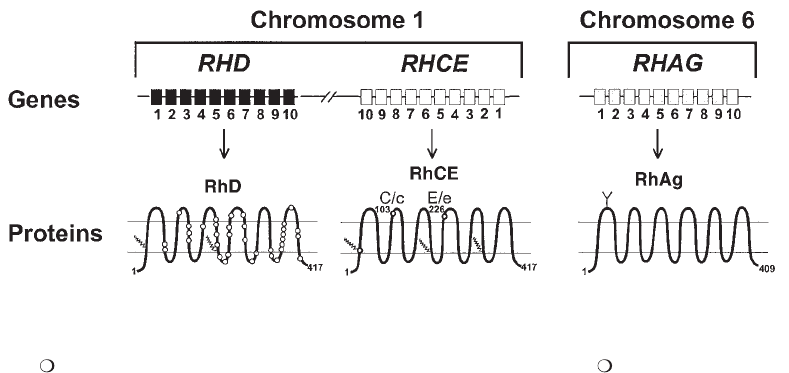
Considerable homology exists between
the products of RHD and RHCE;theprod-
ucts of the different alleles of RHCE are
even more similar.18 C and c differ from one
another in only four amino acids, at posi-
tions 16, 60, 68, and 103, of which only the
difference between serine and proline at
103appearstobecritical.Thepresenceof
proline or alanine at position 226 distin-
guishes E from e. The D polypeptide, by
contrast, possesses 32 to 35 amino acids
that will be perceived as foreign by D–
individuals.
Within the red cell membrane, the Rh
polypeptides form a complex with the Rh-
associated glycoprotein (RhAG), which has
37% sequence homology with the Rh poly-
peptides but is encoded by the RHAG gene
on chromosome 6 (Fig 14-1).22
The study of Rhnull red cells, which lack all
Rh antigens, reveals that this complex (Rh
proteins and RhAG) is essential for expres-
sion of other membrane proteins. Rhnull cells
lack LW antigens, are negative for Fy5 of the
Duffy system, and have weakened expres-
sion of the antigens carried on glycophorin
B(S,s,andU).
23 Although Rh/RhAG pro-
teins play a structural role in the red cell
membrane as evidenced by red cell mor-
phology changes in Rhnull syndrome (see
later in this chapter), their function remains
unknown. There is evidence, however, that
the RhAG protein plays a role in ammo-
nium transport.24,25
Rh Terminology
Three systems of nomenclature were de-
veloped to convey genetic and serologic
information about the Rh system before
the recent advances in our understanding
of the genetics.
System Terminology
The Rh-Hr terminology derives from
Wiener,13 who believed the RH gene prod-
uct to be a single entity he called an
agglutinogen. An agglutinogen was char-
acterized by numerous individual speci-
ficities, called factors, that were identified
by specific antibodies. This theory was in-
correct, but for the designation of pheno-
type, particularly in conversation, many
serologists use a shorthand system based
on Wiener’s Rh-Hr notation. The pheno-
type notations convey haplotypes with the
single letters R and r. R is used for haplo-
318 AABB Technical Manual
Figure 14-1. Schematic representation of
RHD, RHCE,
and
RHAG
genes and RhD, RhCE, and RhAG pro-
teins. on RhD represents amino acid differences between RhD and RhCE. on RhCE indicates the
critical amino acids involved in C/c and E/e antigen expression.
Copyright © 2005 by the AABB. All rights reserved.

types that produce D, r for haplotypes that
do not produce D. Subscripts or, occa-
sionally, superscripts indicate the combi-
nations of other antigens present. For ex-
ample, R1indicates DCe haplotype; R2
indicates DcE;rindicatesdce;R
0indicates
Dce; and so on (Table 14-2).
CDE terminology was introduced by
Fisher and Race,1who postulated three sets
of closely linked genes (Cand c,Dand d,
and Eand e). Both gene and gene product
have the same letter designation, with ital-
icsusedforthenameofthegene.Amodi
-
fied CDE terminology is now commonly
used to communicate research and sero-
logic findings. Rosenfield and coworkers26
proposed a system of nomenclature based
on serologic observations. Symbols were
not intended to convey genetic informa-
tion, merely to facilitate communication of
phenotypic data. Each antigen is given a
number, generally in the order of its discov-
ery or its assignment to the Rh system. Ta-
ble 14-1 lists the Rh system antigens by
number designation, name, and incidence.
Determining Phenotype
In clinical practice, five blood typing re-
agents are readily available: anti-D, -C, -E,
-c, and -e. Routine pretransfusion studies
include only tests for D. Other reagents
are used principally in the resolution of
antibody problems or in family studies.
The assortment of antigens detected on a
person’s red cells constitutes that person’s
Rh phenotype. Table 14-3 shows reaction
patterns of cells tested with antibodies to
thefiveantigensandtheprobableRh
phenotype in modified Wiener terminol-
ogy.
Serologic Testing for Rh
Antigen Expression
Expression of D
D– persons either lack RHD, which en-
codes for the D antigen, or have a non-
functional RHD gene. Most D– persons
are homozygous for RHce, the gene en-
coding c and e; less often they may have
RHCe or RHcE, which encode C and e or c
and E, respectively. The RHCE gene,
whichproducesbothCandE,isquite
rare in D– or D+ individuals.
The D genotype of a D+ person cannot
be determined serologically; dosage studies
are not effective in showing whether an in-
dividual is homozygous or heterozygous for
RHD. Using serologic tests, RHD genotype
can be assigned only by inference from the
antigens associated with the presence of D.
Molecular techniques, however, allow the
determination of D genotype.19,27,28
Interaction between genes results in so-
called “position effect.” If the interaction is
between genes or the product of genes on
the same chromosome, it is called a cis ef-
fect. If a gene or its product interacts with
one on the opposite chromosome, it is
called a trans effect. Examples of both ef-
fects were first reported in 1950 by Lawler
and Race,29 who noted as a cis effect that the
E antigen produced by DcE is quantitatively
Chapter 14: The Rh System 319
Table 14-2. The Principal
RH
Gene
Complexes and the Antigens Encoded
Haplotype
Genes
Present
Antigens
Present
Pheno-
type
R
1
RHD,RHCe
D,C,e R1
R
2
RHD,RHcE
D,c,E R2
R
o
RHD,RHce
D,c,e Ro
R
z
RHD,RHCE
D,C,E Rz
r
′
RHCe
C,e r′
r
″
RHcE
c,E r″
r RHce
c,e r
r
y
RHCE
C,E r
y
Copyright © 2005 by the AABB. All rights reserved.

weaker than E produced by cE.Theynoted
as trans effects that both C and E are
weaker when they result from the genotype
DCe/DcE than when the genotypes are
DCe/ce or DcE/ce, respectively.
Expression of C, c, E, e
To determine whether a person has genes
that encode C, c, E, and e, the red cells are
tested with antibody to each of these anti-
gens. If the red cells express both C and c
or both E and e, it can be assumed that
the corresponding genes are present in the
individual. If the red cells carry only C or
c, or only E or e, the person is assumed to
be homozygous for the particular allele.
Ethnic Origin
Ethnic origin influences deductions about
genotype because the incidence of Rh
genes differs from one geographic group
to another. For example, a White person
with the phenotype Dce would probably
be Dce/ce,but,inaBlackperson,thege
-
notype could as likely be either Dce/Dce or
Dce/ce. Table 14-4 shows the incidence of
D, C, E, c, and e antigens in White and
Black populations.
Gene Frequency
The phenotype DCcEe (line 3 of Table
14-3) can arise from any of several geno-
types. In any population, the most proba-
ble genotype is DCe/DcE.Boththese
haplotypes encode D; a person with this
phenotype will very likely be homozygous
for the RHD gene, although heterozygous
for the RHCE gene (Ce/cE). Some less
likely genotypes could result if the person
is heterozygous at the Dlocus (for exam-
320 AABB Technical Manual
Table 14-3. Determination of Likely Rh Phenotypes from the Results of Tests with
the Five Principal Rh Blood Typing Reagents
Reagent
Antigens
Present
Probable
Phenotype
Anti-D Anti-C Anti-E Anti-c Anti-e
++0++D,C,c,eR
1r
++00+D,C,eR
1R1
+++++D,C,c,E,eR
1R2
+00++D,c,eR
0R0/R0r
+0+++D,c,E,eR
2r
+0++0D,c,ER
2R2
+++0+D,C,E,eR
1Rz
++++0D,C,c,ER
2Rz
+++00D,C,ER
zRz
000++c,err
0+0++C,c,er′r
00+++c,E,er″r
0++++C,c,E,er′r″
Copyright © 2005 by the AABB. All rights reserved.

ple, DCe/cE,DcE/Ce,orDCE/ce), but these
are uncommon in all populations. Table
14-4 gives the incidence of the more com-
mon genotypes in D+ persons. The figures
given are for Whites and Blacks. The ab-
sence of the RHD gene is uncommon in
other ethnic groups.
Determining Genotype
Identifying antigens does not always al-
low confident deduction of genotype. Pre-
sumptions regarding the most probable ge-
notype rest on the incidence of antigenic
combinations determined from population
studies in different ethnic groups. Infer-
ences about genotype are useful in popu-
lation studies, paternity tests, and in pre-
dicting the Rh genes transmitted by the
husband/partner of a woman with Rh an-
tibodies (Table 14-4).
Molecular techniques are now available
that can determine Rh genotype. Determi-
Chapter 14: The Rh System 321
Table 14-4. Incidence of the More Common Genotypes in D+ Persons*
Likelihood of Zygosity for D (%)
Antigens
Present
Genotype Incidence (%) Homo- Hetero- Homo- Hetero-
DCE
Mod.
Rh-hr Whites Blacks Whites Blacks
D,C,c,e
DCe/ce R1r
31.1 8.8
DCe/Dce R1R0
3.4 15.0 10 90 59 41
Dce/Ce R0r
0.2 1.8
D,C,e
DCe/DCe R1R1
17.6 2.9 91 9 81 19
DCe/Ce R1r
1.7 0.7
D,c,E,e
DcE/ce R2r
10.4 5.7 10 90 63 37
DcE/Dce R2R0
1.1 9.7
D,c,E
DcE/DcE R2R2
2.0 1.3 87 13 99 1
DcE/cE R2r
0.3 <<0.1
D,C,c,E,e
DCe/DcE R1R2
11.8 3.7
DCe/cE R1r
0.8 <0.1 89 11 90 10
DcE/Ce R2r
0.6 0.4
D,c,e
Dce/ce R0r
3.0 22.9 6 94 46 54
Dce/Dce R0R0
0.2 19.4
*For the rare phenotypes and genotypes not shown in this table, consult the Suggested Readings listed at the end of this
chapter.
Copyright © 2005 by the AABB. All rights reserved.
nations of genotype with polymerase chain
reaction methods can be made using DNA
harvested from white cells or amnio-
cytes19,27,28 or from noncellular fetal DNA in
maternal plasma.30 Rarely, DNA genotype
results will disagree with serologic findings.
Weak D
Most D+ red cells show clear-cut macro-
scopic agglutination after centrifugation
with reagent anti-D and can be readily
classified as D+. Red cells that are not im-
mediately or directly agglutinated cannot
as easily be classified. For some D+ red
cells, demonstration of the D antigen re-
quires incubation with the anti-D reagent
or addition of antihuman globulin (AHG)
serum after incubation with anti-D [indi-
rect antiglobulin test (IAT)]. These cells are
considered D+, even if an additional step
in testing is required.
In the past, red cells that required addi-
tional steps for the demonstration of D
were classified as Du.ThetermD
uis no lon-
ger considered appropriate; red cells that
carry weak forms of D are classified as D+
and should be described as “weak D.” Im-
provement of polyclonal reagents and the
more widespread use of monoclonal anti-D
reagents have resulted in the routine detec-
tion of some D+ red cells that would have
been considered weak D when tested with
less sensitive polyclonal reagents. Addition-
ally, monoclonal anti-D may react by direct
agglutination with epitopes of D that had
previously required more sensitive test
methods or, occasionally, may fail to react
with some epitopes of the D antigen. Con-
versely, some monoclonal anti-D may react
by direct testing with rare D epitopes that
were not detected with polyclonal reagents
(eg, DHAR and Crawford). It is important to
realize that anti-D reagents differ among
manufacturers and to know the character-
istics of the product being used.
Quantitative Weak D
In the majority of cases, this form of the
weak D phenotype is due to an RHD gene
encoding an altered RhD protein associ-
ated with reduced D antigen expression on
the red cell membrane. The transmemb-
rane or cytoplasmic location of the amino
acid changes in the altered D protein does
notresultinthelossofDepitopes;thus,
the production of alloanti-D as in the par-
tial D phenotype (see partial D) would not
be expected.31,32 Weak D expression is fairly
common in Blacks, often occurring as part
of a Dce haplotype. Genes for weak D ex-
pression are less common in Whites and
may be seen as part of an unusual DCe or
DcE haplotype.
Red cell samples with a quantitative
weak D antigen either fail to react or react
very weakly in direct agglutination tests with
most anti-D reagents. However, the cells will
reactstronglybyanIAT.
Red cells from some persons of the ge-
notype Dce/Ce have weakened expression
of D, a suppressive effect exerted by RHC in
the trans position to RHD on the opposite
chromosome. Similar depression of D can
be seen with other RHD haplotypes accom-
panied by RHCe in trans position. Many of
the weak D phenotypes due to position ef-
fect that were reported in the early litera-
ture appear as normal D.
Partial D
The concept that the D antigen consists of
multiple constituents arose from observa-
tions that some people with D+ red cells
produced alloanti-D that was nonreactive
with their own cells. Most D+ persons who
produce alloanti-D have red cells that re-
act strongly when tested with anti-D. But
some, especially those of the DVI pheno-
322 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
type, give weaker reactions than normal
D+ red cells or react only in the AHG test.
Red cells lacking components of the D an-
tigen have been referred to in the past as
“D mosaic” or “D variant.” Current termi-
nology more appropriately describes these
red cells as “partial D.”
Categorization of partial D phenotypes
was performed by cross-testing red cells
with alloanti-D produced by D+ persons.
The four categories initially described by
Wiener have been expanded considerably
over the years. Tests of many monoclonal
anti-D reagents with red cells of various D
categories suggest that the D antigen com-
prises numerous epitopes. Partial D pheno-
types can be defined in terms of their D
epitopes. Tippett et al33 established at least
10 epitopes but point out that the D anti-
gen is not large enough to accommodate
more than eight distinct epitopes and there
must be considerable overlap between them.
A current model describes 30 epitopes,34
thus demonstrating the dynamic nature of
the D-epitope model as it is revised due to
variation in reagents and techniques. Dogma
regarding the existence of many discrete
epitopes is giving way to a model that is more
topographic in nature, with the fit of anti-
body to antigen described as a “footprint.”35
Molecular studies have elucidated the
genetic mechanisms behind many of the
partial D phenotypes and have shown that
the phenotypes arise as the result of ex-
change between the RHCE gene and the RHD
gene or from single-point mutations.9,18
Significance of Weak/Partial D in Blood
Donors
AABB Standards for Blood Banks and
Transfusion Services36(p32) requires donor
blood specimens to be tested for weak ex-
pression of D and to be labeled as D+ if
the test is positive. Transfusion of blood
with weak expression of the D antigen to
D– recipients is not recommended due to
the fact that some weak or partial D red
cells could elicit an immune response to
D.37 Weak forms of the D antigen, how-
ever, seem to be less immunogenic than
normal D+ blood; transfusion of a total of
68 units of blood with weak D to 45 D– re-
cipients failed to stimulate production of
a single example of anti-D.38
Hemolytic transfusion reactions and HDFN
due to weak D red cells were reported in
the early literature, but it is probable that,
with currently available reagents, the re-
sponsible cells would have been considered
D+.39
Significance of Weak/Partial D in
Recipients
The transfusion recipient whose red cells
test as weak D is sometimes a topic of de-
bate. Most of these patients can almost al-
ways receive D+ blood without risk of im-
munization, but if the weak D expression
reflects the absence of one or more D epi-
topes, the possibility exists that transfu-
sion of D+ blood could elicit the produc-
tion of alloanti-D. This is especially true in
persons of the DVI phenotype. The same
possibility exists, however, for persons
whose partial D red cells react strongly
with anti-D reagents. AABB Standards for
Blood Banks and Transfusion Services36(p48)
only requires recipients’ specimens to be
tested with anti-D by direct agglutination.
The test for weak D is not required. Cur-
rently available, licensed anti-D reagents
are sufficiently potent that most patients
with weak D are found to be D+. The few
patients classified as D–, whose D+ status
is only detectable by an IAT, can receive
D– blood without problems. Some
serologists consider this practice wasteful
of D– blood and prefer to test potential re-
cipients for weak D, then issue D+ blood
when indicated.
Chapter 14: The Rh System 323
Copyright © 2005 by the AABB. All rights reserved.
If D+ blood is given to recipients of the
weak D phenotype, it is important to safe-
guard against careless or incorrect interpre-
tation of tests. D– recipients erroneously
classified as D+, possibly due to a positive di-
rect antiglobulin test (DAT) causing a false-
positive test for weak D, run the risk of im-
munization to D if given D+ blood. Individu-
als whose weakly expressed D antigen is de-
tectable only by an IAT will be classified as
D– recipients if an IAT is not performed.
However, if they donate blood subsequently,
they will be classified as D+ at the time of
blood donation. Personnel in blood centers
and transfusion services should be prepared
to answer questions from puzzled donors or
their physicians. This can present special
problems in autologous donations, when the
D– patient’s own blood is labeled as D+. In
this case, confirmation of the patient’s D sta-
tus by the IAT resolves the apparent discrep-
ancy between recipient and donor types.
Other Rh Antigens
Numbers up to 56 have been assigned to
Rh red cell antigens (see Table 14-1); some
of the numbers are now obsolete because
antigens have been rescinded or reas-
signed. Of the currently included 49, most
beyond D, C, c, Cw,E,andeandtheircor
-
responding antibodies are encountered
much less frequently in routine blood
transfusion therapy.
Cis
Product Antigens
Themembranecomponentsthatexhibit
Rh activity have numerous possible anti-
genic subdivisions. Each gene or gene
complex determines a series of interre-
lated surface structures, of which some
portions are more likely than others to
elicit an immune response. The poly-
peptides determined by the genes in the
haplotype DCe express determinants ad-
ditional to those defined as D, C, and e.
These include Ce (rhi), a cis product that
almost always accompanies C and e when
they are encoded by the same haplotype.
The Ce antigen is absent from red cells on
which the C and e were encoded by differ-
ent haplotypes, for example, in a person
of the genotype DCE/ce. Similar cis prod-
uct antigens exist for c and e determined
by the same haplotype (the antigen called
ce or f), for c and E (cE), and for C and E
(CE).
Although antibodies directed at cis prod-
uct antigens are encountered infrequently,
it would not be correct to consider them
rare. Such antibodies may be present but
unnoticed in serum containing antibodies
of the more obvious Rh specificities; only
adsorption with red cells of selected pheno-
types would demonstrate their presence.
Anti-f (ce) may be present, for example, as a
component of some anti-c and anti-e sera,
but its presence would have little practical
significance. The additional antibody
should not confuse the reaction patterns
given by anti-c and anti-e because all red
cells that react with anti-f will express both
c and e. A person with the genotype
DCe/DcE who makes anti-f may receive c–
or e– red cells because cells of either phe-
notype would also be f–.
Anti-Ce is frequently the true specificity
of the apparent anti-C that a DcE/DcE per-
son produces after immunization with C+
blood. This knowledge can be helpful in es-
tablishing an individual’s Rh genotype. If
anti-Ce is the predominant specificity in a
reagent anti-C, the individual whose weak
C antigen resulted from a DCE haplotype
may be mistyped unless test methods and
control red cells are chosen carefully.40
The G Antigen and Cross-Reactions
The G antigen results from serine at posi-
tion 103 of the Rh polypeptides and is en-
324 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
coded by either RHD or RHCE.41 As a re-
sult, the G antigen is almost invariably
present on red cells possessing either C or
D. Antibodies against G appear superfi-
cially to be anti-C+D, but the anti-G activ-
ity cannot be separated into anti-C and
anti-D. The fact that G appears to exist as
an entity common to C and D explains the
fact that D– persons immunized by C–D+
red cells sometimes appear to have made
anti-C as well as anti-D, and why D– per-
sons who are exposed to C+D– red cells
develop antibodies appearing to contain
an anti-D component. Differentiation of
anti-D, -C, and -G is not necessary in the
pretransfusion setting because virtually
all D–C– red cells are G–. In obstetric pa-
tients, however, some serologists believe
it is essential to distinguish the antibody
specificities to determine the need for Rh
immune globulin prophylaxis.42 Differen-
tial adsorption and elution studies to dis-
tinguish anti-D, -C, and -G are outlined by
Issitt and Anstee.43
Rare red cells have been described that
possess G but lack D and C. The rGpheno-
type is found mostly in Blacks; generally,
the G antigen is weakly expressed and is as-
sociated with the presence of the VS anti-
gen. The rGphenotype has been described
in Whites but is not the same as rGin
Blacks. Red cells also exist that express par-
tial D but lack G entirely, for example, per-
sons of the DIIIb phenotype.39
Variant Antigens
Although red cells from most people give
straightforward reactions with reagent
anti-D, anti-C, anti-E, anti-c, and anti-e
sera, some cells give atypical reactions
and other seemingly normal red cells
stimulate the production of antibodies
that do not react with red cells of com-
mon Rh phenotypes. It has been conve-
nient to consider C and c, and E and e as
antithetical antigens at specific surface
sites. Antigens that behave as if they have
an antithetical relationship to C/c or E/e
have been found, mainly in Whites. The
most common is Cw,buttherelationship
is phenotypic only because Cwand Cxare
antithetical to the high-incidence antigen,
MAR. Variant forms of the e antigen have
been identified, for example, hrSor hrBan-
tigens (Rh19 and Rh31, respectively). Per-
sons who are e+ and hrS–and/orhr
B–are
found more frequently in Black popula-
tions.9The absence of hrBis associated in
most cases with the presence of the VS
antigen.44
Rhnull Syndrome and Other
Deletion Types
Rhnull
The literature reports at least 43 persons
in 14 families whose red cells appear to
have no Rh antigens; others are known
but have not been reported. The pheno-
type, described as Rhnull, is produced by at
least two different genetic mechanisms.
Inthemorecommonregulatortypeof
Rhnull, mutations occur in the RHAG gene
that result in the complete absence of the
core Rh complex (Rh polypeptides and
RhAG) that is necessary for the expression
of Rh antigens.18 Such persons transmit
normal RHD and RHCE genes to their off-
spring.
The other form of Rhnull, the amorph
type, has a normal RHAG gene; however,
there is a mutation in each RHCE gene to-
gether with the common deletion of RHD
(as in D– individuals).18 Theamorphtypeof
Rhnull is considerably rarer than the regula-
tor type. Parents and offspring of this type
of Rhnull are obligate heterozygotes for the
amorph.
Chapter 14: The Rh System 325
Copyright © 2005 by the AABB. All rights reserved.
Red Cell Abnormalities
Whatever the genetic origin, red cells lack-
ing Rh/RhAG proteins have membrane
abnormalities and a compensated hemoly-
tic anemia. The severity of hemolysis and
resulting anemia varies among affected
persons, but stomatocytosis, shortened
red cell survival, absence of LW and Fy5
antigens, and variably altered activity of S,
s, and U have been consistent features.
Serologic Observations
AfewRh
null individuals were recognized
because their sera contained Rh antibod-
ies. Some came to light, however, when
routine Rh phenotyping of their red cells
revealed the absence of any Rh antigens.
In three cases, the discovery resulted from
deliberate testing for Rh antigens in pa-
tients with morphologically abnormal red
cells and hemolytic anemia. Immunized
Rhnull people have produced antibodies
varying in specificity from apparently
straightforward anti-e or anti-C to several
examples that reacted with all red cells
tested except those from other Rhnull peo-
ple. This antibody, considered to be “anti-
total Rh,” has been given the numerical
designation anti-Rh29.
Rhmod
The Rhmod phenotype represents less com-
plete suppression of Rh antigen expres-
sion. Unlike Rhnull red cells, those classi-
fied as Rhmod do not completely lack Rh
and LW antigens. Rhmod red cells show
much reduced and sometimes varied ac-
tivity, depending on the Rh system genes
the individual possesses and on the po-
tency and specificity of the antisera used
in testing. Sometimes, the Rh antigens
have sufficiently weakened expression
that only adsorption-elution techniques
will demonstrate their presence. As in
Rhnull, hemolytic anemia is a feature of the
Rhmod condition. It may be appropriate to
think of the two abnormalities as being
essentially similar, differing only in de-
gree. As in the regulator type of Rhnull,a
mutation in RHAG was shown to result in
the Rhmod phenotype.18
Deleted Phenotypes
Rare RH haplotypes encode D antigen but
fail to encode some or all of the CE anti-
gens. Some examples of deleted phenotypes
includeD––,D••,DC
w–, and Dc–. These
rare phenotypes have been shown to arise
from replacement of large portions of RHCE
with RHD.18 Red cells that lack C/c and/or
E/e antigens may show exceptionally
strong D activity, an observation by which
such red cells have sometimes been rec-
ognized during routine testing with anti-D.
TheD––phenotypemaybeidentifiedin
the course of studies to investigate an un-
expected antibody. Such persons may have
alloantibody of complex specificity be-
cause the person’s red cells lack all the
epitopes expressed on the RhCE poly-
peptide. Antibody with Rh17 (Hro) speci-
ficity is often made by persons of this rare
phenotype, although some sera have been
reported to contain apparently separable
specificities, such as anti-e.
TheD••phenotypeissimilartoD––,ex
-
cept that the D antigen is not elevated to
the same degree. D•• red cells may be ag-
glutinated weakly by some examples of sera
from immunized Rh-deletion persons if the
serum sample contains anti-Rh47 in addi-
tion to anti-Rh17.39 Rh47 is a high-inci-
dence antigen encoded by the RHCE gene
and is absent from other deleted pheno-
types (eg, D– –, DCw–,Dc–).Anotherdistin
-
guishing characteristic of D•• red cells is
that they possess the low-incidence antigen
Rh37 (Evans).
326 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Rh Antibodies
Most Rh antibodies result from exposure
to human red cells through pregnancy or
transfusion. Occasionally, Rh antibodies
(eg, anti-E, anti-Cw) are naturally-occur-
ring. D is the most potent immunogen,
followed by c and E. Although a few exam-
ples of Rh antibodies behave as saline ag-
glutinins, most react best in high-protein,
antiglobulin, or enzyme test systems. Even
sera containing potent saline-reactive
anti-D are usually reactive at higher dilu-
tions in antiglobulin testing. Some work-
ers find enzyme techniques especially use-
ful for detecting weak or developing Rh
antibodies.
Antibody usually persists for many years.
If serum antibody levels fall below detect-
able thresholds, subsequent exposure to the
antigen characteristically produces a rapid
secondary immune response. With exceed-
ingly rare exceptions, Rh antibodies do not
bind complement, at least to the extent rec-
ognizable by techniques currently used. As
a result, primarily extravascular hemolysis,
instead of intravascular hemolysis, occurs
in transfusion reactions involving Rh anti-
bodies.
Dosage Effect
Anti-D seldom shows any difference in re-
activity between red cells from individu-
als homozygous or heterozygous for RHD,
but D expression seems to vary somewhat
with the accompanying alleles of the ge-
notype. For example, red cells from a
DcE/DcE individual carry more D antigen
sites than red cells from a DCe/DCe per-
son and may show higher titration scores
with anti-D. Dosage effects can some-
times be demonstrated with some anti-
bodies directed at the E, c, and e antigens
and, occasionally, at the C antigen.
Anti-D in D+ Individuals
Alloanti-D may be produced in D+ indi-
viduals with partial D phenotype (see Par-
tial D), although not all persons who are
D+ and produce what appears to be anti-D
should be assumed to have epitope-defi-
cient red cells. Weakly reactive anti-LWab
or anti-LWamay react with D+ cells but
not with D– cells. A D+ person whose an-
tibody is a weakly reactive anti-LWamay
be indistinguishable on initial serologic
testing from an individual with a partial D
antigen who has made anti-D to missing
epitopes. (See the section on the LW sys-
tem in Chapter 15.) Anti-LW can be differ-
entiated from anti-D by testing the anti-
body with 0.02M DTT-treated red cells
(Method 3.10); the LW antigen is de-
stroyed by sulfhydryl reagents, whereas D
is unaffected.
Concomitant Antibodies
Some Rh antibodies tend to occur in con-
cert. For example, the DCe/DCe person
manifesting immune anti-E almost cer-
tainly has been exposed to c as well as E.
Anti-c may be present in addition to
anti-E, although substantially weaker and
possibly undetectable at the time the
anti-E is found, and transfusion of seem-
ingly compatible E–c+ blood may elicit an
immediate or delayed reaction. Generally,
it is not a sound practice to select donor
blood that is negative for antigens absent
from the recipient’s red cells when anti-
body has not been detected, but some
practitioners feel that the DCe/DCe recipi-
ent with anti-E is a case that merits avoid-
ance of c+ blood as well.45 Anti-E less con-
sistently accompanies anti-c because the
patient can easily have been exposed to c
without being exposed to E. There is little
clinical value in pursuing anti-E in serum
known to contain anti-c because the vast
Chapter 14: The Rh System 327
Copyright © 2005 by the AABB. All rights reserved.
majority of c– donor blood will be nega-
tive for the E antigen.
Rh Typing
Routine Rh typing for donors and patients
involves only the D antigen. Tests for the
other Rh antigens are performed when
identifying unexpected Rh antibodies, ob-
taining compatible blood for a patient
with an Rh antibody, investigating parent-
age or other family studies, selecting a
panel of phenotyped cells for antibody
identification, or evaluating whether a
person is likely to be homozygous or het-
erozygous for RHD.
In finding compatible blood for a recipi-
ent with a comparatively weak Rh antibody,
tests with potent blood typing reagents
more reliably confirm the absence of anti-
gen than mere demonstration of a compat-
ible crossmatch. Determination of the pa-
tient’s Rh phenotype may help confirm the
antibody specificity and indicate which
other Rh antibodies could also be present.
Routine Testing for D
Until recently, high-protein anti-D re-
agents of human polyclonal origin that
were suitable for slide, tube, or microplate
tests were used for most routine testing.
More recently, monoclonal anti-D re-
agents have become widely available.
Tests may employ red cells suspended in
saline, serum, or plasma, but test condi-
tions should be confirmed by reading the
manufacturer’s directions before use. Pro-
cedures for microplate tests are similar
to those for tube tests, but very light sus-
pensions of red cells are used.
Slidetestsproduceoptimalresultsonly
when a high concentration of red cells and
protein are combined at a temperature of
37 C. A disadvantage of the slide test is
evaporation of the reaction mixture, which
can cause the red cells to aggregate and be
misinterpreted as agglutination. There are
also greater biohazardous risks associated
with increased potential for spillage of the
specimen during manipulation. Represen-
tative procedures for tube, slide, and micro-
plate tests are given in Methods 2.6, 2.7,
and 2.8.
Techniques to demonstrate weak D are
required by AABB Standards only for donor
blood or for testing blood from neonates
born to Rh-negative women to determine
Rh immune globulin candidacy.36(pp32,48)
When there is an indication to test for weak
D, an IAT should be performed (Method
2.9). A reliable test for weak D expression
cannot be performed on a slide.
High-Protein Reagents
Some anti-D reagents designated for use
in slide, rapid tube, or microplate tests
contain high concentrations of protein
(20-24%) and other macromolecular addi-
tives. Such reagents are nearly always pre-
pared from pools of human sera and give
rapid reliable results when used in accor-
dance with manufacturers’ directions.
High-protein levels and macromolecular
additives may cause false-positive reac-
tions. A false-positive result could cause a
D– patient to receive D+ blood and be-
come immunized. An appropriate control
tested according to the manufacturer’s di-
rections must be performed.
Control for High-Protein Reagents
Manufacturers offer their individual dilu-
ent formulations for use as control re-
agents. The nature and concentration of
additives differ significantly among re-
agents from different manufacturers and
may not produce the same pattern of
false-positive reactions. If red cells exhibit
aggregation in the control test, the results
of the anti-D test cannot be considered
328 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
valid. In most cases the presence or ab-
sence of D can be determined with other
reagents, as detailed later in this chapter.
Misleading Results with High-Protein
Reagents
False Positives. The following circum-
stances can produce false-positive red cell
typing results.
1. Cellular aggregation resulting from
immunoglobulin coating of the pa-
tient’s red cells or serum factors that
induce rouleaux will give positive re-
sultsinboththetestandthecontrol
tubes. Serum factors can be elimi-
nated by thoroughly washing the red
cells (with warm saline if cold agglu-
tinins are present or suspected) and
retesting. If the cells in the control
test remain unagglutinated and the
anti-D test gives a positive result, the
red cells are D+. If agglutination still
occurs in the control tube, the most
likely explanation is immunoglobu-
lin coating of the red cells, which
should then be tested with low-pro-
tein reagents.
2. Red cell aggregation, simulating ag-
glutination, may occur if red cells and
anti-D are incubated too long and
excessive evaporation occurs during
the slide test. It is important to fol-
low the manufacturer’s recommen-
dations to interpret the test within
the recommended period.
False Negatives. The following circum-
stances can produce false-negative red
cell typing results.
1. Too heavy a red cell suspension in
thetubetestortooweakasuspen
-
sion in the slide test may weaken ag-
glutination.
2. Saline-suspended red cells must not
be used for slide testing.
3. Red cells possessing weakly expres-
sed D antigen may not react well
within the 2-minute limit of the slide
test or upon immediate centrifuga-
tion in the tube test.
Low-Protein Reagents
The low-protein Rh reagents in current
use are formulated predominantly with
monoclonal antibodies. Immunoglobu-
lin-coated red cells can usually be suc-
cessfully typed with low-protein Rh re-
agents that contain saline-agglutinating
antibodies.
Monoclonal Source Anti-D
Monoclonal anti-D reagents are made
predominantly from human IgM antibod-
ies, which require no potentiators and ag-
glutinate most D+ red cells from adults
and infants in a saline system. Monoclonal
anti-D reagents usually promote reactions
stronger than those with polyclonal IgG
reagents, but they may fail to agglutinate
red cells of some partial D categories.
Adding small amounts of IgG anti-D to
the monoclonal IgM antibodies provides
a reagent that will react with weak or par-
tial D red cells in antiglobulin tests. Cer-
tain rare D+ red cells (DHAR, DVa, DVc,
Rh43+)mayreactatimmediatespinwith
some of these blended reagents, but, with
other reagents, these same cells may be
nonreactive in an IAT or reactive only in
an IAT.
Licensed monoclonal/polyclonal or
monoclonal/monoclonal blends can be
used by all routine typing methods and are
as satisfactory as high-protein reagents in
an IAT for weak D. False-negative findings
can result, however, if tests using mono-
clonal reagents are incubated in excess of a
manufacturer’s product directions. These re-
agents, prepared in a low-protein medium,
can be used to test red cells with a positive
Chapter 14: The Rh System 329
Copyright © 2005 by the AABB. All rights reserved.
DAT, provided those tests are not subjected
to an IAT.
Control for Low-Protein Reagents
Most monoclonal blended reagents have
a total protein concentration approximat-
ing that of human serum. False-positive
reactions due to spontaneous aggregation
of immunoglobulin-coated red cells occur
nomoreoftenwiththiskindofreagent
than with other saline-reactive reagents.
False-positive reactions may occur in any
saline-reactive test system if the serum
contains cold autoagglutinins or a protein
imbalance causing rouleaux and the red
cells are tested unwashed. It is seldom
necessary to perform a separate control
test. Absence of spontaneous aggregation
can usually be demonstrated by observ-
ing absence of agglutination by anti-A
and/or anti-B in the cell tests for ABO. For
red cell specimens that show agglutina-
tion in all tubes (ie, give the reactions of
group AB, D+), a control should be per-
formed as described by the reagent man-
ufacturer; this is not required when do-
nors’ cells are tested. In most cases, a
suitable control is a suspension of the pa-
tient’s red cells with autologous serum or
with 6% to 8% bovine albumin, although
exceptions have been noted.46 If the test is
one of several performed concurrently
and in a similar manner, any negative re-
sult serves as an adequate control. For ex-
ample, a separate control tube would be
required only for a red cell specimen that
gives positive reactions with all the Rh
reagents (ie, is typed as D+C+E+c+e+).
Testing for D in Hemolytic Disease of the
Fetus and Newborn
Because red cells from an infant suffering
from HDFN are coated with immunoglob-
ulin, a low protein reagent is usually nec-
essary for Rh testing. Occasionally, the in-
fant’s red cells may be so heavily coated
with antibody that all antigen sites are oc-
cupied, leaving none available to react
with a low protein antibody of appropri-
ate specificity. This “blocking” phenome-
non should be suspected if the infant’s
cells have a strongly positive DAT and are
not agglutinated by a low protein reagent
of the same specificity as the maternal an-
tibody.
Anti-D is the specificity responsible for
nearly all cases of blocking by maternal an-
tibody. It is usually possible to obtain cor-
rect typing results with a low protein anti-D
after 45 C elution of the maternal antibody
from the cord blood red cells. (See Method
2.12.) Elution liberates enough antigen sites
to permit red cell typing, but it must be per-
formed cautiously because overexposure to
heat may denature or destroy Rh antigens.
Tests for Antigens Other than D
Reagents are readily available to test for
the other principal Rh antigens: C, E, c,
and e. These are formulated as either low-
protein (usually monoclonal or mono-
clonal/polyclonal blends) or high-protein
reagents. High-protein reagents of any
specificity have the same problems with
false-positive results as high-protein anti-D
and require a comparable control test
performed concurrently and under the
same conditions. Observation of a nega-
tive result in the control test for anti-D
may not properly control the tests for
other Rh antigens because results with
anti-D are usually obtained after immedi-
ate centrifugation; tests for the other Rh
antigens are generally incubated at 37 C
before centrifugation.
Rh reagents may give weak or negative
reactions with red cells possessing variant
antigens. This is especially likely to happen
if anti-e is used to test the red cells from
Blacks, among whom variants of e are rela-
330 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
tively common.9It is impossible to obtain
anti-e reagents that react strongly and con-
sistently with the various qualitative and
quantitative variants of e. Variable reactivity
with anti-C reagents may occur if the DCE
or CE haplotypes are responsible for the ex-
pression of C on red cells. Variant E and c
antigens have been reported but are con-
siderably less common.
Whatever reagents are used, the manu-
facturer’s directions must be carefully fol-
lowed. The IAT must not be used unless the
manufacturer’s instructions state explicitly
that the reagent is suitable for this use. The
pools of human source sera (nonmono-
clonal) used to prepare reagents for the
other Rh antigens may contain anti-
globulin-reactive, “contaminating” speci-
ficities. Positive and negative controls
should be tested; red cells selected for the
positive control should have a single dose
of the antigen or be known to show weak
reactivity with the reagent.
Additional Considerations in Rh Testing
The following limitations are common to
all Rh typing procedures, including those
performed with high-protein reagents.
False-Positive Reactions
The following circumstances can produce
false-positive red cell typing results.
1. The wrong reagent was inadvertently
used.
2. An unsuspected antibody of another
specificity was present in the human
source reagent. Antibodies for anti-
gens having an incidence of less than
1% in the population may occasion-
ally be present and cause false-posi-
tive reactions, even when the manu-
facturer’s directions are followed.
For crucial determinations, many
workers routinely perform replicate
tests using reagents from different
sources. Replicate testing is not an
absolute safeguard, however, because
reagents from different manufactur-
ers may not be derived from different
sources.
3. Polyagglutinable red cells may be ag-
glutinated by any reagent containing
human serum. Although antibodies
that agglutinate these surface-altered
red cells are present in most adult
human sera, polyagglutinins in re-
agents very rarely cause problems.
Aging, dilution, and various steps in
the manufacturing process tend to
eliminate these predominantly IgM
antibodies.
4. Autoagglutinins and abnormal pro-
teins in the patient’s serum may cause
false-positive reactions when un-
washed red cells are tested.
5. Reagent vials may become contami-
nated with bacteria, with foreign
substances, or with reagent from an-
other vial. This can be prevented by
the use of careful technique and the
periodic inspection of the vials’ con-
tents. However, bacterial contamina-
tion may not cause recognizable tur-
bidity because the refractive index of
bacteria is similar to that of high-
protein reagents.
False-Negative Reactions
The following circumstances can produce
false-negative red cell typing results.
1. The wrong reagent was inadvertently
used.
2. The reagent was not added to the tube.
It is good practice to add serum to
all the tubes before adding the red
cells and any enhancement medium.
3. A specific reagent failed to react with
a variant form of the antigen.
4. A reagent that contains antibody di-
rected predominantly at a cis-prod-
Chapter 14: The Rh System 331
Copyright © 2005 by the AABB. All rights reserved.
uct Rh antigen failed to give a reli-
ably detectable reaction with red
cells carrying the individual antigens
as separate gene products. This oc-
cursmostoftenwithanti-Csera.
5. The manufacturer’s directions were
not followed.
6. The red cell button was shaken so
roughly during resuspension that
small agglutinates were dispersed.
7. Contamination, improper storage,
or outdating cause antibody activity
to deteriorate. Chemically modified
IgG antibody appears to be particu-
larly susceptible to destruction by
proteolytic enzymes produced by cer-
tain bacteria.
References
1. Race RR. The Rh genotypes and Fisher’s the-
ory. Blood 1948; special issue 2:27-42.
2. Levine P, Stetson RE. An unusual case of
intragroup agglutination. JAMA 1939;113:
126-7.
3. Landsteiner K, Wiener AS. An agglutinable
factor in human blood recognized by im-
mune sera for rhesus blood. Proc Soc Exp
Biol NY 1940;43:223.
4. Levine P, Katzin EM. Isoimmunization in
pregnancy and the variety of isoagglutinins
observed. Proc Soc Exp Biol NY 1940;43:
343-6.
5. Wiener AS, Peters HR. Hemolytic reactions
following transfusions of blood of the homol-
ogous group, with three cases in which the
same agglutinogen was responsible. Ann In-
tern Prn Med 1940;13:2306-22.
6. FrohnC,DümbgenL,BrandJ-M,etal.Proba
-
bility of anti-D development in D– patients
receiving D+ RBCs. Transfusion 2003;43:
893-8.
7. Mollison PL, Engelfriet CP, Contreras M.
Blood transfusion in clinical medicine. 10th
ed. Oxford: Blackwell Scientific Publishers,
1997.
8. Daniels GL, Fletcher A, Garratty G, et al.
Blood group terminology 2004: From the In-
ternational Society of Blood Transfusion
committee on terminology for red cell sur-
face antigens. Vox Sang 2004;87:304-16.
9. Reid ME, Lomas-Francis C. The blood group
antigen factsbook. 2nd ed. London: Academic
Press, 2004.
10. Gemke RJBJ, Kanhai HHH, Overbeeke MAM,
et al. ABO and Rhesus phenotyping of fetal
erythrocytes in the first trimester of preg-
nancy. Br J Haematol 1986;64:689-97.
11. Dunstan RA, Simpson MB, Rosse WF. Erythro-
cyte antigens on human platelets. Absence of
Rh, Duffy, Kell, Kidd, and Lutheran antigens.
Transfusion 1984;24:243-6.
12. Dunstan RA. Status of major red cell blood
group antigens on neutrophils, lymphocytes
and monocytes. Br J Haematol 1986;62:301-9.
13. Wiener AS. Genetic theory of the Rh blood
types. Proc Soc Exp Biol Med 1943;54:316-19.
14. Fisher RA, Race RR. Rh gene frequencies in
Britain. Nature 1946;157:48-9.
15. Tippett P. A speculative model for the Rh
blood groups. Ann Hum Genet 1986;50:241-7.
16. ColinY,Chérif-ZaharB,LeVanKimC,etal.
Genetic basis of RhD-positive and RhD-nega-
tive blood group polymorphisms as deter-
mined by southern analysis. Blood 1991;78:
2747-52.
17. ArceMA,ThomsonES,WagnerS,etal.Mo-
lecular cloning of RhD cDNA derived from a
gene present in RhD-positive, but not RhD-
negative individuals. Blood 1993;82:651-5.
18. Huang CH, Liu PZ, Cheng JG. Molecular biol-
ogy and genetics of the Rh blood group sys-
tem. Semin Hematol 2000;37:150-65.
19. SingletonBK,GreenCA,AventND,etal.The
presence of an RHD pseudogene containing a
37 base pair duplication and a nonsense mu-
tation in Africans with the Rh D-negative
blood group phenotype. Blood 2000;95:12-8.
20. Mouro I, Colin Y, Chérif-Zahar B, et al. Molec-
ular genetic basis of the human Rhesus blood
group system. Nat Genet 1993;5:62-5.
21. SmytheJS,AventND,JudsonPA,etal.Expres
-
sion of RHD and RHCE gene products using
retroviral transduction of K562 cells estab-
lishes the molecular basis of Rh blood group
antigens. Blood 1996;87:2968-73.
22. Ridgwell K, Spurr NK, Laguda B, et al. Isola-
tion of cDNA clones for a 50 kDa glycoprotein
of the human erythrocyte membrane associ-
ated with Rh (rhesus) blood-group antigen
expression. Biochem J 1992;287:223-8.
23. Tippett P. Regulator genes affecting red cell
antigens. Transfus Med Rev 1990;4:56-68.
24. HemkerMB,GoedelC,vanZwietenR,etal.
The Rh complex exports ammonium from
human red blood cells. Br J Haematol 2003;
122:333-40.
25. Westhoff CM, Seigel D, Burd C, Foskett JK.
Mechanism of genetic complementation of
ammonium transport in yeast by human
332 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
erythrocyte Rh-associated glycoprotein. J
Biol Chem 2003;279:17443-8.
26. Rosenfield RE, Allen FH Jr, Swisher SN,
Kochwa S. A review of Rh serology and pre-
sentation of a new terminology. Transfusion
1962;2:287-312.
27. Wagner FF, Flegel WA. RHD gene deletion oc-
curred in the Rhesus box. Blood 2000;95:
3662-8.
28. Chiu RW, Murphy MF, Fidler C, et al. Determi-
nation of RhD zygosity: Comparison of a
double amplification refractory mutation
system approach and a multiplex real-time
quantitative PCR approach. Clin Chem 2003;
47:667-72.
29. Lawler SD, Race RR. Quantitative aspects of
Rh antigens. Proceedings of the International
Society of Hematology 1950:168-70.
30. Lo YMD, Hjelm NM, Fidler C, et al. Prenatal
diagnosis of fetal RhD status by molecular
analysis of maternal plasma. N Engl J Med
1998;339:1734-8.
31. Wagner F, Gassner C, Müller T, et al. Molecu-
lar basis of weak D phenotypes. Blood 1999;
93:385-93.
32. Wagner FF, Frohmajer A, Ladewig B, et al.
Weak D alleles express distinct phenotypes.
Blood 2000;95:2699-708.
33. Tippett P, Lomas-Francis C, Wallace M. The
Rh antigen D: Partial D antigens and associ-
ated low incidence antigens. Vox Sang 1996;
70:123-31.
34. Scott ML. Section 1A: Rh serology. Coordina-
tor’s report. 4th International Workshop on
Monoclonal Antibodies Against Human Red
Cell Surface Antigens, Paris. Transfus Clin Biol
2002;9:23-9.
35. Chang TY, Siegel DL. Genetic and immuno-
logical properties of phage-displayed human
anti-Rh(D) antibodies: Implications for Rh(D)
epitope topology. Blood 1998;91:3066-78.
36. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
37. Flegel WA, Khull SR, Wagner FF. Primary
anti-D immunization by weak D type 2 RBCs.
Transfusion 2000;40:428-33.
38. Schmidt PJ, Morrison EG, Shohl J. The antige-
nicity of the RhoDublood factor. Blood 1962;
20:196-202.
39. DanielsG.Humanbloodgroups.2nded.Ox
-
ford: Blackwell Scientific Publications, 2002.
40. Van Loghem JJ. Production of Rh agglutinins
anti-C and anti-E by artificial immunization
of volunteer donors. Br Med J 1947;ii:958-9.
41. FaasBHW,BeckersEAM,SimsekS,etal.In
-
volvement of Ser103 of the Rh polypeptides
in G epitope formation. Transfusion 1996;36:
506-11.
42. ShireyRS,MirabellaDC,LumadueJA.Differ
-
entiation of anti-D, -C, and -G: Clinical rele-
vance in alloimmunized pregnancies. Trans-
fusion 1997;37:493-6.
43. Issitt PD, Anstee DJ. Applied blood group se-
rology. 4th ed. Durham, NC: Montgomery
Scientific Press, 1998:350-3.
44. ReidME,StorryJR,IssittPD,etal.Rhhaplo
-
typesthatmakeebutnothr
Busually make
VS. Vox Sang 1997;72:41-4.
45. Shirey RS, Edwards RE, Ness PM. The risk of
alloimmunization to c (Rh4) in R1R1patients
who present with anti-E. Transfusion 1994;34:
756-8.
46. Rodberg K, Tsuneta R, Garratty G. Discrepant
Rh phenotyping results when testing IgG-
sensitized rbcs with monoclonal Rh reagents
(abstract). Transfusion 1995;35(Suppl):67S.
Suggested Reading
Agre P, Cartron JP. Molecular biology of the Rh an-
tigens. Blood 1991;78:551-3.
Avent ND, Reid ME. The Rh blood group system: A
review. Blood 2000;95:375-87.
Cartron JP. Defining the Rh blood group antigens.
Blood Rev 1994;8:199-212.
Daniels G. Human blood groups. 2nd ed. Oxford:
Blackwell Scientific Publications, 2002.
IssittPD.Aninvitedreview:TheRhantigene,its
variants, and some closely related serological ob-
servations. Immunohematology 1991;7:29-36.
Issitt PD. The Rh blood group. In: Garratty G, ed.
Immunology of transfusion medicine. New York:
Marcel Dekker, 1994:111-47.
Issitt PD. The Rh blood group system 1988: Eight
new antigens in nine years and some observations
on the biochemistry and genetics of the system.
Transfus Med Rev 1989;3:1-12.
Issitt PD, Anstee DJ. Applied blood group serology.
4th ed. Durham, NC: Montgomery Scientific Press,
1998.
Lomas-Francis C, Reid ME. The Rh blood group
system: The first 60 years of discovery. Immuno-
hematology 2000;16:7-17.
Race RR, Sanger R. Blood groups in man. 6th ed.
Oxford: Blackwell Scientific Publications, 1968.
Reid ME, Ellisor SS, Frank BA. Another potential
source of error in Rh-Hr typing. Transfusion 1975;
15:485-8.
Chapter 14: The Rh System 333
Copyright © 2005 by the AABB. All rights reserved.
Reid ME, Lomas-Francis C. The blood group anti-
gen factsbook. 2nd ed. London: Academic Press,
2004.
Sonneborn H-H, Voak D, eds. A review of 50 years
of the Rh blood group system. Biotest Bulletin 1997;
5(4):389-528.
Vengelen-Tyler V, Pierce S, eds. Blood group sys-
tems: Rh. Arlington, VA: American Association of
Blood Banks, 1987.
White WD, Issitt CH, McGuire D. Evaluation of the
use of albumin controls in Rh typing. Transfusion
1974;14:67-71.
334 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 15: Other Blood Groups
Chapter 15
Other Blood Groups
THERE ARE MANY antigens on red
cells in addition to the ones men-
tioned in previous chapters. These
antigens are grouped into blood group
systems, collections, and a series of inde-
pendent antigens, composed mostly of
antigens of low or high incidence. A blood
group system is a group of one or more
antigens governed by a single gene locus
or by a complex of two or more closely
linked homologous genes that have been
shown to be phenotypically and geneti-
cally related to each other and genetically
distinct from other blood group systems.
A collection is a group of antigens shown
to have a phenotypic, biochemical, or ge-
netic relationship to each other; however,
there is insufficient information or data that
shows them to be a distinct blood group
system genetically independent from other
blood group systems. Table 10-1 lists the
blood group systems, as defined by the
International Society of Blood Transfusion
(ISBT) working party on blood group
terminology, and their gene location.1-3
Additional information on ISBT terminol-
ogy for all the antigens mentioned in this
chapter can be found in Appendix 6. Table
15-1 shows the serologic behavior and
characteristics of the major blood group
antibodies derived from human sources.
The major systems will be discussed first
in the chapter, followed by the other blood
group systems, collections, and independ-
ent high-incidence and low-incidence an-
tigens. In each grouping, the order will
reflect the ISBT number order.
Distribution of Antigens
Antigens present in almost all persons are
known as high-incidence antigens, where-
as antigens found in very few persons are
termed very low-incidence antigens. The
frequency of these high- or low-incidence
antigens may also differ by ethnic group.
335
15
Copyright © 2005 by the AABB. All rights reserved.

336 AABB Technical Manual
Table 15-1. Serologic Behavior of the Principal Antibodies of Different Blood Group
Systems
Antibody
In-Vitro
Hemolysis
Saline Albumin Papain/Ficin
Associated
with
4C 22C 37C AHG 37C AHG HDFN HTR
Anti-M 0 Most Some Few Few 0 0 Few Few
Anti-N 0 Most Few Occ. Occ. 0 0 Rare No
Anti-S 0 Few Some Some Most See text Yes Yes
Anti-s 0 No Few Few Most See text Yes Yes
Anti-U 0 No Occ. Some Most Most Most Yes Yes
Anti-Lua0 Some Most Few Few Few Few No No
Anti-Lub0 Few Few Few Most Few Few Mild Yes
Anti-K 0 Few Some Most Some Most Yes Yes
Anti-k 0 Few Few Most Some Most Yes Yes
Anti-Kpa0 Some Some Most Some Most Yes Yes
Anti-Kpb0 Few Few Most Some Most Yes Yes
Anti-Jsa0 Few Few Most Few Most Yes Yes
Anti-Jsb0 0 0 Most Few Most Yes Yes
Anti-LeaSome Most Most Some Some Some Most No Rare
Anti-LebSome Most Most Some Some Some Most No No
Anti-Fya0 Rare Rare Most 0 0 Yes Yes
Anti-Fyb0 Rare Rare Most 0 0 Mild Yes
Anti-JkaSome Few Few Most Some Most Mild Yes
Anti-JkbSome Few Few Most Some Most Mild Yes
Anti-Xga0 Few Few Most 0 0 No report
Anti-Dia0 Some Some Most Some Some Yes Yes
Anti-Dib0 Most Some Some Yes Yes
Anti-Yta000Most0SomeNoYes
Anti-Ytb0 All No report
Anti-Doa0 0 0 Some Some Most No Yes
Anti-Dob0 All All All No Yes
Anti-Coa0 0 0 Some Some Most Yes Yes
Anti-Cob0 0 0 Some Some Most Mild Yes
Anti-Sc1 0 All No No
Anti-Sc2 0 Some Some Most Most Most No No
AHG = Antihuman globulin; HDFN = Hemolytic disease of the fetus and newborn; HTR = Hemolytic transfusion reaction;
Occ. = Occasionally. The reactivity shown in the table is based on the tube methods in common use. If tests are carried
out by more sensitive test procedures (such as in capillary tubes, in microtiter plates, or by the albumin layering
method), direct agglutination (before the antiglobulin phase) may be observed more often with some antibodies. Blank
spaces indicate a lack of sufficient data for generalization about antibody behavior.
Copyright © 2005 by the AABB. All rights reserved.

Antigens that occur as codominant traits,
such as Jkaand Jkb, may have a variable in-
cidence and may differ in ethnic groups.
For an illustration, the Duffy glycoprotein
is known to be a receptor for the parasite
Plasmodium vivax, one of the causative
agents of malaria. In West Africa, where
malaria is endemic, the Fy(a–b–) red cell
phenotype, very rare in Whites, occurs with
an incidence of greater than 80%.
Each of the known antigens described
in this chapter was initially identified
through the detection of its specific anti-
body in a serum. Tables listing phenotype
frequencies among Whites and Blacks in the
US population are given throughout this
chapter. Frequencies among other groups
in the population are not given because
data are scanty and wide differences be-
tween groups of diverse Asian, South Ameri-
can, or Native American origins make gen-
eralizations about phenotypes inappropriate.
MNS System
M, N, S, s, and U Antigens
The MNS system is a complex system of
over 40 antigens carried on two glycophorin
molecules or hybrid molecules of the two
proteins. The M, N, S, s, and U antigens
are the most important antigens of the MNS
system with regard to transfusion medi-
cine. They have also been important to
our understanding of biochemistry and
genetics. The M and N antigens are lo-
cated on glycophorin A (GPA). The S, s,
and U antigens are located on glycophorin
B (GPB). Table 15-2 shows the frequencies
of the common phenotypes of the MNS
system. There is considerable linkage dis-
equilibrium between M,N and S,s due to
the gene location on the chromosome.
For example, the gene complex producing
N with s is much more common than that
producing N with S. The MNSs genes
GYPA and GYPB are in very close proxim-
ity on chromosome 4.4See the section be-
low on Genes Encoding Glycophorins and
Chapters 9 and 10 for more information
about gene interactions.
Red cells that lack S and s may be nega-
tive for a high-incidence antigen called U;
persons who lack U may make anti-U when
exposed to U+ red cells.
Low-Incidence Antigens of the MNS System
The MNS system includes several low-inci-
dence antigens. Recent biochemical data
Chapter 15: Other Blood Groups 337
Table 15-2. Phenotypes and Frequencies in the MNS System
Reactions with Anti- Phenotype Frequency (%)
M N S s U Phenotype Whites Blacks
+ 0 M+N– 28 26
+ + M+N+ 50 44
0 + M–N+ 22 30
+ 0 + S+s–U+ 11 3
+ + + S+s+U+ 44 28
0 + + S–s+U+ 45 69
0 0 0 S–s–U– 0 Less than 1
0 0 (+) S–s–U+w 0 Rare*
*May not be detected by some antisera and are listed as U–.
Copyright © 2005 by the AABB. All rights reserved.
attribute the reactivity of various low-in-
cidence determinants to one or more amino
acid substitutions, variation in the extent
or type of glycosylation, or the existence
of a hybrid sialoglycoprotein (SGP).
Genes Encoding Glycophorins
The genes encoding the MNS system anti-
gens are located on chromosome 4 at
4q28-q31. The gene that encodes GPA is
called GYPA and the gene that encodes
GPB is GYPB. The similarities in amino
acid sequences of GPA and GPB suggest
that both genes derive from a common
ancestral gene. GYPA and GYPB consist of
seven and five exons, respectively. The
genes share >95% identity. Although the
genes are highly homologous, GYPB re-
sults in a shorter protein because a point
mutation at the 5′splicing site of the third
intron prevents transcription of exon 3,
called pseudo exon 3. Following the ho-
mologous sequences, GYPA and GYPB dif-
fer significantly in the 3′end sequences.
Hybrid Molecules
Pronounced SGP modifications occur in
hybrid molecules that may arise from un-
equal crossing over or gene conversion
between GYPA and GYPB.HybridSGPs
may carry the amino-terminal portion of
GPA and the carboxy-terminal portion of
GPB, or vice versa. Other hybrids appear
as a GPB molecule with a GPA insert or a
GPA molecule with a GPB insert. The low-
incidence antigens Hil (MNS20), Sta
(MNS15), Dantu (MNS25), and Mur
(MNS10), among others, are associated with
hybrid SGPs. Some variants are found in
specificethnicgroups.Forexample,the
Dantu antigen occurs predominantly in
Blacks, although the antigen is of low inci-
dence.
Many of the MNS low-incidence anti-
gens were categorized into a subsystem
called the Miltenberger system, based on
reactivity with selected sera. As more anti-
gens have been identified and knowledge of
the genetic events that give rise to these
novel antigens has increased, it is clear that
the Miltenberger subsystem is outdated.
These antigens, such as Mia, Vw, Hil, etc,
should be considered glycophorin variants.
Biochemistry of the MNS System
Antigens of the MNS system are carried
on GPA and GPB, which are single-pass trans-
membrane glycoproteins. The carboxy (C)
terminal of each glycophorin extends into
the cytoplasm of the red cell, and a hydro-
phobic segment is embedded within the
lipid bilayer. An amino (N) terminal seg-
ment extends into the extracellular envi-
ronment. The molecules are sensitive to
cleavage at varying positions by certain
proteases (see Fig 15-1).
There are approximately 1,000,000 cop-
ies of GPA per red cell. M and N blood
group antigen activity resides on the extra-
cellular segment, a sequence of 72 amino
acids with carbohydrate side chains at-
tached within the first 50 residues of the
amino terminal. When GPA carries M anti-
gen activity (GPAM), the first amino acid res-
idue is serine and the fifth is glycine. When
it carries N antigen activity (GPAN), leucine
and glutamic acid replace serine and gly-
cine at positions one and five, respectively
(see Fig 15-1).
Red cells that lack most or all of GPA are
described as En(a–). These rare En(a–) indi-
viduals may produce antibodies (collec-
tively called anti-Ena) that react with vari-
ous portions of the extracellular part of the
glycoprotein. Some En(a–) persons may
produce an antibody against an antigen
called Wrbthat is part of the Diego blood
group system. Wrbarises from an interac-
tion between GPA and the anion exchange
molecule, AE-1 (also known as band 3).5
338 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
GPB is a smaller protein than GPA, and
there are fewer (approximately 200,000)
copies per red cell. GPB carries S, s, and U
antigens. GPB that expresses S activity has
methionine at position 29; GPB with s ac-
tivity has threonine at that position (see Fig
15-1). The N-terminal 26 amino acids of
GPB are identical to the sequence of GPAN,
which accounts for the presence of an N
antigen (known as ‘N’) on all red cells of
normal MNS types. Red cells that lack GPB
altogether lack not only S, s, and U activity
but also ‘N’. Immunized individuals of the
rare M+N–S–s–U– phenotype can produce
a potent anti-N (anti-U/GPB) that reacts
with all red cells of normal MNS types,
whether N-positive or N-negative, and
should be considered clinically significant.
Some S–s– red cells also have a variant GPB.
The Effect of Proteolytic Enzymes on MNS
Antigens
Proteolytic enzymes, such as ficin or papain,
cleave red cell membrane SGPs at well-
defined sites. Reactivity with anti-M and
anti-N is abolished by commonly used
enzyme techniques. The effect of different
enzymes on the expression of MNS sys-
tem antigens reflects the point at which
the particular enzyme cleaves the anti-
gen-bearing SGP and the position of the
antigen relative to the cleavage site (see
Fig 15-1). Sensitivity of the antigens to
Chapter 15: Other Blood Groups 339
Figure 15-1. Schematic diagram of glycophorin A and glycophorin B. The amino acid sequences that
determine M, N, S, and s are given. indicates an
O
-linked oligosaccharide side chain, indicates
an
N
-linked polysaccharide side chain. Approximate locations of protease cleavage sites are indi-
cated. (Courtesy New York Blood Center.)
Copyright © 2005 by the AABB. All rights reserved.
proteases may help in the identification
of antibodies to M and N antigens, but the
effects of proteases on tests for the S and s
antigens are more variable. In addition,
the S antigen is sensitive to trace amounts
of chlorine bleach.6
MNS System Antibodies
The antibodies most commonly encoun-
teredaredirectedattheM,N,S,andsan
-
tigens.
Anti-M
Anti-M is detected frequently as a saline
agglutinin if testing is done at room tem-
perature. Anti-M is often found in the sera
of persons who have had no exposure to
human red cells. Although M antibodies
are generally thought to be predominantly
IgM, many examples that are partly or
wholly IgG are frequently found. However,
these antibodies are rarely clinically sig-
nificant. Some examples of anti-M cause
stronger agglutination if the pH of the test
system is reduced to 6.5 and when testing
red cell samples possessing a double-dose
expression of the M antigen. Examples that
react at 37 C or at the antiglobulin phase
of testing should be considered potentially
significant. Compatibility testing per-
formed by a strictly prewarmed method
(see Method 3.3) should eliminate the re-
activity of most examples of anti-M. In a
few exceptional cases, anti-M detectable
at the antiglobulin phase has caused
hemolytic disease of the fetus and newborn
(HDFN) or hemolysis of transfused cells.
Anti-N
Anti-N is comparatively rare. Examples are
usually IgM and typically appear as weakly
reactive cold agglutinins. Some powerful and
potentially significant IgG examples have
been observed in a few persons of the rare
phenotypes M+N–S–s–U– and M+N–S–s–U+w
because these people lack or possess an
altered form of GPB.
Antibodies to S, s, and U
Unlike anti-M and anti-N, antibodies to S,
s, and U usually occur following red cell
immunization. All are capable of causing
hemolytic transfusion reactions (HTRs)
and HDFN. Although a few saline-reactive
examples have been reported, antibodies
to S, s, and U are usually detected in the
antiglobulin phase of testing. Most, but
not all, investigators7have found that
papain or ficin destroys the reactivity of
S+ red cells with anti-S. Depending on the
enzyme solution used, the reactivity of
anti-s with s+ cells can be variable.8(p477)
Most examples of anti-U react equally
with untreated and enzyme-treated red
cells, but there have been examples of
broadly reactive anti-U, which detect an
enzyme-sensitive determinant.
Anti-U is rare but should be considered
when serum from a previously transfused
or pregnant Black person contains anti-
body to a high-incidence antigen.
Antibodies to Low-Incidence Antigens
There are many examples of antibodies to
low-incidence antigens, such as anti-Mg
or anti-VW, in the MNS blood group sys-
tem. Many of these antibodies occur as a
saline agglutinin in sera from persons who
have no known exposure to human red
cells. The rarity of these antigens makes it
unlikely that the antibodies will be de-
tected if present.
Kell System
Kell System Antigens
Kell system antigens are expressed on the
red cell membrane in low density and are
weakened or destroyed by treatment with
340 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

reducing agents and with acid. The anti-
gens are carried on one protein and en-
coded by a single gene. For an in-depth
review, see Lee, Russo, and Redman.9
K and k
The K antigen was first identified in 1946
because of an antibody that caused HDFN.
ThealleleresponsiblefortheKantigenis
present in 9% of Whites and approxi-
mately 2% of Blacks. The existence of the
expected allele for k was confirmed when
an antithetical relationship was estab-
lished between K and the antigen de-
tected by anti-k. Anti-k reacts with the red
cells of over 99% of all individuals.
Other Kell Blood Group Antigens
Other antithetical antigens of the Kell sys-
tem include Kpa,Kp
b,andKp
c;Js
aand Jsb;
K11 and K17; and K14 and K24. Not all
theoretically possible genotype combina-
tions have been recognized in the Kell
system. For example, Kpaand Jsahave
never been found to be produced by the
same chromosome. Kpais an antigen
found predominantly in Whites, and Jsais
found predominantly in Blacks. The
haplotype producing K and Kpahas also
not been found. Table 15-3 shows some
phenotypes of the Kell system. The table
also includes Ko,anullphenotypein
which the red cells lack all of the antigens
of the Kell system. Several high-incidence
antigens were assigned to the Kell system
because the identifying antibodies were
found to be nonreactive with Kored cells.
For simplicity, various Kell antigens of
high and low incidence have not been
included in the table.
Phenotypes with Depressed Kell Antigens
Kmod is an umbrella term used to describe
phenotypes characterized by weak ex-
pression of Kell system antigens. Adsorp-
tion/elution tests are often necessary for
their detection. The Kmod phenotype is
thought to arise through several different
point mutations of the KEL gene. Red cells
of persons with some Gerbich negative
Chapter 15: Other Blood Groups 341
Table 15-3. Some Phenotypes and Frequencies in the Kell System
Reactions with Anti- Frequency (%)
KkKp
aKpbJsaJsbPhenotype Whites Blacks
+ 0 K+k– 0.2 Rare
++ K+k+ 8.8 2
0 + K–k+ 91.0 98
+ 0 Kp(a+b–) Rare 0
+ + Kp(a+b+) 2.3 Rare
0 + Kp(a–b+) 97.7 100
+ 0 Js(a+b–) 0.0 1
+ + Js(a+b+) Rare 19
0 + Js(a–b+) 100.0 80
000000 K
0Exceedingly rare
Copyright © 2005 by the AABB. All rights reserved.
phenotypes also exhibit depressed Kell
phenotypes (see the Gerbich System).
Persons of the Ge:–2,–3 and Ge:–2,–3,–4
(Leach) phenotypes have depression of at
least some Kell system antigens.
ThepresenceoftheKp
aallele weakens
the expression of other Kell antigens when
in cis position. For example, the k antigen
of Kp(a+) red cells reacts more weakly than
expected and, when tested with weaker ex-
amples of anti-k, may be interpreted as k–.
Biochemistry of the Kell System
The Kell system antigens are carried on a
93-kD single-pass red cell membrane pro-
tein. Kell system antigens are easily inac-
tivated by treating red cells with sulfhy-
dryl reagents such as 2-mercaptoethanol
(2-ME), dithiothreitol (DTT), or 2-amino-
ethylisothiouronium bromide (AET).
Such treatment is useful in preparing red
cells that artificially lack Kell system anti-
gens to aid in the identification of Kell-re-
lated antibodies. Treatment with sulfhydryl
reagents may impair the reactivity of
other antigens (LWa,Do
a,Do
b,Yt
a, and oth-
ers). Thus, although treatment with these
reagents may be used in antibody prob-
lem solving, specificity must be proven by
other means. As expected, Kell system an-
tigens are also destroyed by ZZAP, a mix-
ture of DTT and enzyme (see Methods
3.10, 4.6, and 4.9). This susceptibility to
sulfhydryl reagents suggests that disulfide
bonds are essential to maintain activity of
the Kell system antigens. This hypothesis
has been supported by the biochemical
characterization of Kell proteins deduced
from cloned DNA10; they exhibit a number
of cysteine residues in the extracellular
region. Cysteine readily forms disulfide
bonds, which contribute to the folding of
a protein. Antigens that reflect protein
conformation will be susceptible to any
agent that interferes with its tertiary struc-
ture. Kell antigens are also destroyed by
treatment with EDTA-glycine acid.
The function of the Kell protein is un-
known,butithasstructuralsimilaritiestoa
family of zinc-binding neutral endopep-
tidases. It has most similarity with the
common acute lymphoblastic leukemia
antigen (CALLA or CD10), a neutral endo-
peptidase on leukocytes.8(pp647-648)
Kx Antigen, the McLeod Phenotype, and
Their Relationship to the Kell System
Although the Kell system locus is on the
long arm of chromosome 7 and the Kx lo-
cus (XK) is on the Xp21 region of the X
chromosome, evidence suggests that the
Kell and Kx proteins form a covalently
linked complex on normal red cells.8On
red cells that carry normal expressions of
Kell antigens, Kx appears to be poorly ex-
pressed. It is believed that this finding
represents steric interference by the Kell
glycoprotein in the approach of anti-Kx to
its antigen. Red cells of Koindividuals re-
act strongly with anti-Kx. Similarly, the re-
moval or denaturation of Kell glycopro-
teins with AET or DTT renders the cells
strongly reactive with anti-Kx.
It is believed that the presence of the
glycoprotein on which Kx is carried is es-
sential for the antigens of the Kell system to
attach to or be expressed normally on red
cells. Therefore, a lack of Kx is associated
with poor expression of Kell system antigens.
Red cells that lack Kx exhibit not only
markedly depressed expression of Kell sys-
tem antigens but also shortened survival,
reduced deformability, decreased perme-
ability to water, and acanthocytic morphol-
ogy. This combination or group of red cell
abnormalities is called the McLeod pheno-
type, after the first person in whom these
observations were made. Persons with McLeod
red cells also have a poorly defined abnor-
mality of the neuromuscular system, char-
342 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
acterized by persistently elevated serum
levels of the enzyme creatine phospho-
kinase and, in older people, disordered
muscular function. The McLeod phenotype
arises through deletion and mutations of
the XK locus of chromosome X.
In a few instances, the McLeod pheno-
type has been found in patients with chronic
granulomatous disease (CGD), in which
granulocytes exhibit normal phagocytosis
of microorganisms but an inability to kill
ingested pathogens. The McLeod pheno-
type associated with CGD appears to result
from deletion of a part of the X chromo-
some that includes the XK locusaswellas
the gene responsible for X-linked CGD.
Kell System Antibodies
Anti-K and Anti-k
Because the K antigen is strongly immuno-
genic, anti-K is frequently found in sera
from transfused patients. Rare examples
of anti-K have appeared as a saline agglu-
tinin in sera from subjects never exposed
to human red cells. Most examples are of
immune origin and are reactive in anti-
globulin testing; some bind complement.
Some workers have observed that exam-
ples of anti-K react less well in tests that in-
corporate low-ionic-strength-saline (LISS)
solutions (notably the Polybrene test) than
in saline tests or tests that include albumin.
Others, however, have not shown differ-
ences in antibody reactivity, testing many
examples of anti-K in low ionic systems.
Anti-KhascausedHTRsonnumerousoc
-
casions, both immediate and delayed.
Anti-K can cause severe HDFN and fetal
anemia may be caused by the immune de-
struction of K+ erythroid progenitor cells by
macrophages in the fetal liver.11
Becauseover90%ofdonorsareK–,itis
not difficult to find compatible blood for
patients with anti-K. Anti-k has clinical and
serologic characteristics similar to anti-K
but occurs much less frequently. Only about
one person in 500 lacks the k antigen and
finding compatible blood is correspond-
ingly more difficult.
Other Kell System Antibodies
Anti-Kpa,anti-Kp
b,anti-Js
a,andanti-Js
bare
allmuchlesscommonthananti-Kbut
show similar serologic characteristics and
are considered clinically significant. Any of
them may occur following transfusion or
fetomaternal immunization. Antibody fre-
quency is influenced by the immunogeni-
city of the particular antigen and by the
distribution of the relevant negative phe-
notypes among transfusion recipients and
positive phenotypes among donors. In
Black patients frequently transfused with
phenotypically matched blood, usually
from other Black donors, anti-Jsais rela-
tively common. This is due to the approx-
imate 20% incidence of the Jsaantigen in
the Black population (see Table 15-3). Ordi-
narily, however, these antibodies are rare.
Assistance from a rare donor file is usually
needed to find compatible blood for patients
immunized to the high-incidence antigens
Kpband Jsb. Anti-Ku is the antibody char-
acteristically seen in immunized Koper-
sons. It has been reported to cause a fatal
HTR,12 and it appears to be directed at a
singledeterminantbecauseithasnotbeen
separable into other Kell specificities.
However, antibodies to other Kell system
antigens may be present in serum contain-
inganti-Ku.SomepeopleoftheK
mod pheno-
type have made a Ku-like antibody.
Duffy System
Duffy System Antigens
The antigens Fyaand Fybare encoded by a
pair of codominant alleles at the Duffy
Chapter 15: Other Blood Groups 343
Copyright © 2005 by the AABB. All rights reserved.

(FY) locus on chromosome 1. Anti-Fyaand
anti-Fybdefine the four phenotypes observed
in this blood group system, namely:
Fy(a+b–), Fy(a+b+), Fy(a–b+), and Fy(a–b–)
(see Table 15-4). In Whites, the first three
phenotypes are common and Fy(a–b–) in-
dividuals are extremely rare. However, the
incidence of the Fy(a–b–) phenotype among
Blacks is 68%.
The Duffy gene encodes a glycoprotein
that is expressed in other tissues, including
thebrain,kidney,spleen,heart,andlung.
The Fy(a–b–) individual can be the result of
the FyFy genotype or null phenotype. How-
ever, in many Black Fy(a–b–) individuals,
the transcription in the marrow is pre-
vented and Duffy protein is absent from the
red cells. These individuals have an allele
that is the same in the structural region as
the Fybgene that prevents the transcrip-
tion.8(p439) However, the Duffy protein is ex-
pressed normally in nonerythroid cells of
these persons.13 Other Fy(a–b–) individuals
either appear to have a total absence or
markedly altered Duffy glycoprotein.8(pp457-458)
This affects other cell lines and tissues, not
only the red cells. Those individuals who
have absent or altered glycoprotein can
make anti-Fy3, which will react with cells
that are Fy(a+) and/or Fy(b+).
A rare inherited form of weak Fybcalled
Fyxhas been described and is probably due
to a point mutation. The Fyxantigen may go
undetected unless potent anti-Fybis used in
testing. The Fy5 antigen appears to be de-
fined by an interaction of the Duffy and Rh
gene products because it is not expressed
on Rhnull red cells. The Fy6 antigen has been
described only by murine monoclonal anti-
bodies and is not present on red cells that
are Fy(a–b–) and Fy:–3,–5.8(p448)
Biochemistry of the Duffy System
In red cells, the Duffy gene encodes a multi-
pass membrane glycoprotein. The antigens
Fya,Fy
b, and Fy6 are located on the N-ter-
minal of the Duffy glycoprotein and are
sensitive to denaturation by proteases such
as ficin, papain, and α-chymotrypsin, un-
like Fy3 or Fy5. Fy3 has been located on
the last external loop of the Duffy glyco-
protein. It is unaffected by protease treat-
ment (reviewed in Pierce and Mac-
pherson).14 The glycoprotein is the recep-
tor for the malarial parasite Plasmodium
vivax, and persons whose red cells lack
Fyaand Fybare resistant to that form of
the disease. In sub-Saharan Africa, nota-
bly West Africa, the resistance to P. v i v a x
malaria conferred by the Fy(a–b–) pheno-
type may have favored its natural selec-
tion, and most individuals are Fy(a–b–).
The Duffy gene has been cloned13 and
the Duffy glycoprotein has been identified
as an erythrocyte receptor for a number of
344 AABB Technical Manual
Table 15-4. Phenotypes and Frequencies in the Duffy System
Reactions with Anti- Adult Phenotype Frequency (%)
FyaFybPhenotype Whites Blacks
+ 0 Fy(a+b–) 17 9
+ + Fy(a+b+) 49 1
0 + Fy(a–b+) 34 22
0 0 Fy(a–b–) Very rare 68
Copyright © 2005 by the AABB. All rights reserved.
chemokines, notably interleukin-8.15 Be-
cause chemokines are biologically active
molecules, it has been postulated that Duffy
acts as a sponge for excess chemokines,
without ill effect on the red cells.
Duffy System Antibodies
Anti-Fyais quite common and may cause
HDFN and HTRs. Anti-Fybis rare and gen-
erally is weakly reactive. Anti-Fybcan cause
rare mild HDFN and has been responsible
for mostly mild HTRs. Both antibodies are
usually IgG and react best by antiglobulin
testing. The glycoprotein that expresses
the antigens is cleaved by most proteases
used in serologic tests, so anti-Fyaand
anti-Fybare usually nonreactive in en-
zyme test procedures.
Weak examples of anti-Fyaor anti-Fyb
may react only with red cells that have a
double dose of the antigen. In Whites, red
cells that express only one of the two anti-
gens are assumed to come from persons
homozygous for the gene and to carry a
double dose of the antigen. In Blacks, such
cells may express the antigen only in single
dose and may not give the expected strong
reaction with antibodies that show dosage.
For example, the patient typing Fy(a+b–)
may be FyaFy.
Anti-Fy3 was first described in the serum
of a White person of the Fy(a–b–) pheno-
type and is directed at the high-incidence
antigen Fy3. The only cells with which it is
nonreactive are Fy(a–b–). Unlike Fyaand
Fyb, the Fy3 antigen is unaffected by prote-
ase treatment, and anti-Fy3 reacts well with
enzyme-treated cells positive for either Fya
or Fyb. Anti-Fy3 is rare but is sometimes
made by Black Fy(a–b–) patients lacking
Fy3 who have been immunized by multiple
transfusions.
Two other rare antibodies have been de-
scribed, both reactive with papain-treated
red cells. One example of anti-Fy4 has been
reported. It reacted with red cells of the
Fy(a–b–) phenotype and with some Fy(a+b–)
and Fy(a–b+) red cells from Blacks but not
with Fy(a+b+) red cells, suggesting reactiv-
ity with a putative product of the Fy gene.
However, different reference laboratories
obtained equivocal results and evidence for
the existence of the Fy4 antigen is weak.
Anti-Fy5 is similar to anti-Fy3, except
that it fails to react with Rhnull red cells that
express Fy3 and is nonreactive with cells
from Fy(a–b–) Blacks. It may react with the
red cells from Fy(a–b–) Whites. This pro-
vided a previously unrecognized distinction
between the Fy(a–b–) phenotype so com-
mon in Blacks and the one that occurs, but
very rarely, in Whites.8(p447)
Anti-Fy6 is a murine monoclonal anti-
body that describes a high-incidence anti-
gen in the same region as Fyaand Fyb.The
antibody reacts with all Fy(a+) and/or
Fy(b+) red cells, is nonreactive with Fy(a–b–)
red cells, but, unlike anti-Fy3, is nonreac-
tive with enzyme-treated red cells.
Kidd System
Jkaand JkbAntigens
The Jkaand Jkbantigens are located on the
urea transporter, encoded by the HUT 11
gene on chromosome 18. Jk(a–b–) red cells,
which lack the JK protein, are more resis-
tant to lysis by 2M urea.16 Red cells of nor-
malJkphenotypeswellandlyserapidlyin
asolutionof2Murea.
The four phenotypes identified in the Kidd
system are shown in Table 15-5. The
Jk(a–b–) phenotype is extremely rare, ex-
cept in some populations of Pacific Island
origin. Two mechanisms have been shown
to produce the Jk(a–b–) phenotype.17 One is
the homozygous presence of the silent Jk
allele. The other is the action of a dominant
inhibitor gene called In(Jk). The dominant
suppression of Kidd antigens is similar to
Chapter 15: Other Blood Groups 345
Copyright © 2005 by the AABB. All rights reserved.

the In(Lu) suppression of the Lutheran sys-
tem.
Kidd System Antibodies
Anti-Jk
a
and Anti-Jk
b
Anti-Jkawas first recognized in 1951 in the
serum of a woman who had given birth
to a child with HDFN. Two years later,
anti-Jkbwas found in the serum of a pa-
tient who had suffered a transfusion reac-
tion. Both antibodies react best in anti-
globulin testing, but saline reactivity is
sometimes observed in freshly drawn
specimens or when antibodies are newly
forming. Both anti-Jkaand anti-Jkbare of-
ten weakly reactive, perhaps because,
sometimes, they are detected more readily
through the complement they bind to red
cells. Some examples may become unde-
tectable on storage. Other examples may
react preferentially with red cells from
homozygotes.
Some workers report no difficulties in
detecting anti-Jkaand anti-Jkbin low ionic
tests that incorporate anti-IgG. Others find
that an antiglobulin reagent containing an
anticomplement component may be im-
portant for the reliable detection of these
inconsistently reactive antibodies. Stronger
reactions may be obtained with the use of
polyethylene glycol (PEG) or enzyme-
treated red cells in antiglobulin testing.
Kidd system antibodies occasionally
causeHDFN,butitisusuallymild.These
antibodies are notorious, however, for their
involvement in severe HTRs, especially de-
layed hemolytic transfusion reactions
(DHTRs). DHTRs occur when antibody de-
velops so rapidly in an anamnestic re-
sponse to antigens on transfused red cells
that it destroys the still-circulating red cells.
In many cases, retesting the patient’s pre-
transfusion serum confirms that the anti-
body was undetectable in the original tests.
Anti-Jk3
Sera from some rare Jk(a–b–) persons
have been found to contain an antibody
that reacts with all Jk(a+) and Jk(b+) red
cells but not with Jk(a–b–) red cells. Al-
though a minor anti-Jkaor anti-Jkbcom-
ponent is sometimes separable, most of
the reactivity has been directed at an anti-
gen called Jk3, which is present on both
Jk(a+) and Jk(b+) red cells.
Other Blood Group Systems
So far, this chapter has been devoted to
blood group systems of red cell antigens
of which the principal antibodies may be
seen fairly frequently in the routine blood
typing laboratory. The other blood group
systems listed in Table 10-1 will be re-
346 AABB Technical Manual
Table 15-5. Phenotypes and Frequencies in the Kidd System
Reactions with Anti- Phenotype Frequency (%)
JkaJkbPhenotype Whites Blacks
+ 0 Jk(a+b–) 28 57
+ + Jk(a+b+) 49 34
0 + Jk(a–b+) 23 9
0 0 Jk(a–b–) Exceedingly rare
Copyright © 2005 by the AABB. All rights reserved.

viewed here briefly; the interested reader
should refer to other texts and reviews for
greater detail.
Lutheran System
Lu
a
and Lu
b
Antigens
The phenotypes of the Lutheran system,
as defined by anti-Luaand anti-Lub,are
showninTable15-6.TheLu(a–b–)pheno
-
type is very rare and may arise from one
of three distinct genetic circumstances
(reviewed in Pierce and Macpherson14). In
the first, a presumably amorphic Lu-
theran gene (Lu) is inherited from both
parents. In the second and most com-
mon, the negative phenotype is inherited
as a dominant trait attributed to the inde-
pendently segregating inhibitor gene,
In(Lu), which prevents the normal expres-
sion of Lutheran and certain other blood
group antigens (notably P1,I,AnWj,In
a,
and Inb). The third Lu(a–b–) phenotype is
due to an X-borne suppressor, recessive in
its effect.
Other Lutheran Blood Group Antigens
A series of high-incidence antigens (Lu4,
Lu5, Lu6, Lu7, Lu8, Lu11, Lu12, Lu13,
Lu16, Lu17, and Lu20) has been assigned
to the Lutheran system because the corre-
sponding antibodies do not react with
Lu(a–b–) red cells of any of the three ge-
netic backgrounds. Two low-incidence
antigens, Lu9 and Lu14, have gained ad-
mission to the Lutheran system because
of their apparent antithetical relationship
to the high-incidence antigens Lu6 and
Lu8, respectively.
Aua(Lu18), an antigen of relatively high
incidence [90% of all populations are
Au(a+)] and its antithetical partner, Aub
(Lu19), present in 50% of Whites and 68%
of Blacks, have been shown to belong to the
Lutheran system.18,19
Biochemistry of the Lutheran System
Lutheran antigens are carried on a glyco-
protein bearing both N-linked and O-linked
oligosaccharides. This protein exists in
two forms and has been shown to have a
role in cell adhesion. The antigens are de-
stroyed by trypsin, α-chymotrypsin, and
sulfhydryl-reducing agents.20 These re-
sults and results of immunoblotting ex-
periments suggest the existence of inter-
chain or intrachain disulfide bonds. Tests
performed with monoclonal anti-Lubsug-
gest that the number of Lubantigen sites
per red cell is low, approximately 600-
1600 per Lu(a+b+) red cell and 1400-3800
per Lu(a–b+) red cell.21
The Lu and Se (secretor) loci were shown
to be linked in 1951, the first recorded ex-
ample of autosomal linkage in humans.
The two loci have been assigned to chro-
mosome19.ThegeneencodingtheLu
glycoproteins has been cloned.22
Lutheran System Antibodies
Thefirstexampleofanti-Lu
a(-Lu1) was
found in 1945 in a serum that contained
several other antibodies. Anti-Luaand
anti-Lubare not often encountered. They
Chapter 15: Other Blood Groups 347
Table 15-6. Phenotypes and Frequencies
in the Lutheran System in Whites*
Reactions with
Anti- Phenotype
Frequency
(%)
LuaLubPhenotype
+ 0 Lu(a+b–) 0.15
+ + Lu(a+b+) 7.5
0 + Lu(a–b+) 92.35
0 0 Lu(a–b–) Very rare
*Insufficient data exist for the reliable calculation of fre-
quencies in Blacks.
Copyright © 2005 by the AABB. All rights reserved.
are most often produced in response to
pregnancy or transfusion but have oc-
curred in the absence of obvious red cell
stimulation. Lutheran antigens are poorly
developed at birth. It is not surprising that
anti-Luahas not been reported to cause
HDFN; neither has it been associated with
HTRs. Anti-Lubhas been reported to shor-
ten the survival of transfused red cells but
causes no, or at most very mild, HDFN.
Most examples of anti-Luaand some anti-
Lubwill agglutinate saline-suspended red
cells possessing the relevant antigen, char-
acteristically producing a mixed-field ap-
pearance with small to moderately sized,
loosely agglutinated clumps of red cells
interspersed among many unagglutinated
red cells.
Diego System
Diego System Antigens
The Diego system consists of two inde-
pendent pairs of antithetical antigens,
called Dia/Diband Wra/Wrb. The system
also contains a large number of low-inci-
dence antigens as seen in Appendix 6.
The antigens are located on AE-1 (band
3), which is encoded by a gene on chro-
mosome 17. The Diaand Dibantigens are
useful as anthropologic markers because
the Diaantigen is almost entirely confined
to populations of Asian origin and Native
NorthandSouthAmericans,inwhichthe
incidence of Diacanbeashighas54%.
8(p583)
The Wraand Wrbantigens are located on
AE-1 in close association with GPA. Wrbex-
pression is dependent upon the presence of
GPA (see MNS Blood Group System).
Diego System Antibodies
Anti-Diamay cause HDFN or destruction
of transfused Di(a+) red cells. Anti-Dibis
rare but clinically significant. Anti-Wrais
fairly common and can occur without red
cell alloimmunization. It is a rare cause of
HTR or HDFN. Anti-Wrbis a rarely en-
countered antibody that may be formed
by rare Wr(a+b–) and some En(a–) indi-
viduals. Anti-Wrbmay recognize an en-
zyme-resistant or enzyme-sensitive anti-
gen.23 It should be considered to have
potential to destroy Wr(b+) red cells.8(p589)
Cartwright System
Cartwright Antigens
The Yt (Cartwright) blood group system
consists of two antigens, Ytaand Ytb(see
Table 15-7). A gene on chromosome 7 en-
codes the antigens. The Yt antigens are lo-
cated on red cell acetylcholinesterase
(AChE),24 an enzyme important in neural
transmission, but the function of which is
unknown on red cells. Enzymes have a
variable effect on the Ytaantigen but 0.2M
DTT appears to destroy the Ytaantigen ex-
pression.
Cartwright Antibodies
Some examples of anti-Ytaare benign. A
few cases of anti-Ytahave shown acceler-
ated destruction of transfused Yt(a+) red
cells. Prediction of the clinical outcome by
the monocyte monolayer assay has proved
successful.25,26 Anti-Ytais not known to cause
HDFN. Anti-Ytbis rare and has not been
implicated in HTR or HDFN.
Xg System
Xg
a
Antigen
In 1962, an antibody was discovered that
identified an antigen more common among
womenthanamongmen.Thiswouldbe
expected of an X-borne characteristic be-
cause females inherit an X chromosome
from each parent, whereas males inherit
X only from their mother. The antigen was
named Xgain recognition of its X-borne
manner of inheritance. Table 15-8 gives
the phenotype frequencies among White
348 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

males and females. Enzymes, such as pa-
pain and ficin, denature the antigen. The
gene encoding Xgahas been cloned.27,28
Xg
a
Antibody
Anti-Xgais an uncommon antibody that
usually reacts only in antiglobulin testing.
Anti-Xgahas not been implicated in HDFN
or HTRs. Anti-Xgamay be useful for trac-
ing the transmission of genetic traits asso-
ciated with the X chromosome, although
linkage with the Xg locus has been dem-
onstrated for few traits to date.
Scianna System
Scianna Antigens
Five antigens—Sc1, Sc2, Sc3, Rd, and
STAR—are recognized as belonging to the
Scianna blood group system. Scianna an-
tigens are expressed by the red cell adhe-
sion protein ERMAP.3Sc1 is a high-inci-
Chapter 15: Other Blood Groups 349
Table 15-7. Phenotype Frequencies in Other Blood Group Systems with
Co-Dominant Antithetical Antigens
System Reactions with Anti- Phenotype
Phenotype
Frequency in
Whites (%)*
Yt YtaYtb
+ 0 Yt(a+b–) 91.9
+ + Yt(a+b+) 7.9
0 + Yt(a–b+) 0.2
Dombrock DoaDob
+ 0 Do(a+b–) 17.2
+ + Do(a+b+) 49.5
0 + Do(a–b+) 33.3
Colton CoaCob
+ 0 Co(a+b–) 89.3
+ + Co(a+b+) 10.4
0 + Co(a–b+) 0.3
0 0 Co(a–b–) Very rare
Scianna Sc1 Sc2
+ 0 Sc:1,–2 99.7
+ + Sc:1,2 0.3
0 + Sc:–1,2 Very rare
0 0 Sc:–1,–2 Very rare
Indian InaInb
+ 0 In(a+b–) Very rare
+ + In(a+b+) <1
0 + In(a–b+) >99
Diego DiaDib
+ 0 Di(a+b–) Rare
+ + Di(a+b+) Rare
0 + Di(a–b+) >99.9
*There are insufficient data for the reliable calculation of frequencies in Blacks.
Copyright © 2005 by the AABB. All rights reserved.

dence antigen, whereas Sc2 occurs very
infrequently. Sc1 and Sc2 behave as prod-
ucts of allelic genes (see Table 15-7). Sc3
is thought to be present on the red cells of
any individual who inherits a functional
Sc1or Sc2gene, analogous to Fy3. Rd and
STAR are low-incidence antigens recently
assigned to the Scianna system. The anti-
gens are resistant to enzymes routinely
used in blood group serology. The gene
encoding the Scianna antigens is located
on chromosome 1.
Scianna Antibodies
The antibodies are rare. Anti-Sc1 has not
been reported to cause HTR or HDFN.
Anti-Sc2 has caused positive DATs in the
neonate but no clinical HDFN.
Dombrock Blood Group System
Dombrock Antigens
Initially, this blood group system consisted
of Doaand Dob, with the phenotype fre-
quency as shown in Table 15-7. The dis-
covery that red cells negative for the high-
incidence antigen Gyawere Do(a–b–)29 has
led to the recent expansion of the Dombrock
blood group system. Three high-incidence
antigens are Gya,Hy,andJo
a.Theinterre
-
lationship of the phenotypes is shown in
Table 15-9.30 Gy(a–) red cells represent the
null phenotype. The Gy(a+w), Hy–, and
Jo(a–) phenotypes have been found exclu-
sively in Blacks. The Dombrock antigens
are located on a glycoprotein of 46 to 58
kD, the function of which is unknown.
Dombrock Antibodies
Anti-Doaand anti-Dobare uncommon an-
tibodies and are usually found in sera
containing multiple red cell antibodies.
This can make the detection and identifi-
cation of anti-Doaand anti-Dobdifficult.
Anti-Doahas caused HDFN and HTR.31
HDFN due to anti-Dobhas not been re-
ported, but examples have caused HTR.
Antibodies to Gya,Hy,andJo
amay cause
shortened survival of transfused anti-
gen-positive red cells or mild HDFN. Re-
activity of Dombrock antibodies may be
enhanced by papain or ficin treatment of
350 AABB Technical Manual
Table 15-8. Frequencies of the Xg(a+)
and Xg(a–) Phenotypes in White Males
and Females
Phenotype Frequency (%)
Phenotype Males Females
Xg(a+) 65.6 88.7
Xg(a–) 34.4 11.3
Frequencies are based on the combined results of testing
nearly 7000 random blood samples from populations of
Northern European origin. There are insufficient data for
reliable calculation of frequencies in Blacks.
Table 15-9. Relationship of the Dombrock Blood Group Antigens
Phenotype DoaDobGyaHy Joa
Normal Do(a+b–) +0+++
Normal Do(a+b+) +++++
Normal Do(a–b+) 0++++
Gy(a–) 00000
Hy– 0 (+) (+) 0 (+)
Jo(a–) (+) (+) + (+) 0
(+) = weak antigen expression.
Copyright © 2005 by the AABB. All rights reserved.
red cells but is weakened or destroyed by
sulfhydryl reagents.
Colton System
Colton Antigens
The Colton system consists of Coa,ahigh-
incidence antigen; Cob, a low-incidence
antigen; and Co3, an antigen (like Fy3)
considered to be the product of either the
Coaor Cobgene. The antigens are products
of a gene on chromosome 7. The pheno-
type frequencies in Whites are shown in
Table 15-7. The Colton antigens have
been located on membrane protein CHIP
28 (Aquaporin), which functions as the
red cell water transporter.32
Colton Antibodies
Anti-Coahas been implicated in HTRs and
HDFN.8Anti-Cobhas caused an HTR and
mild HDFN. Enzyme treatment of red
cells enhances reactions with the Colton
antibodies.
LW System
LW Antigens
Table 15-10 shows the LW phenotypes and
their frequencies. The antigens are dena-
tured by sulfhydryl reagents, such as DTT,
and by pronase but are unaffected by
papain or ficin. There are reported cases
of both inherited and acquired LW(a–b–)
individuals.
Association with Rh
LWais more strongly expressed on D+
than D– red cells. However, the gene en-
coding the LW antigens is independent of
the genes encoding the Rh proteins. Ge-
netic independence was originally estab-
lished through the study of informative
families in which LW has been shown to
segregate independently of the RH genes.
The LW gene has been assigned to chro-
mosome 19 and has been cloned. The
glycoprotein it encodes has homology
with cell adhesion molecules.33
Red cells from persons of the Rhnull phe-
notype are LW(a–b–). It appears that the LW
glycoprotein requires an interaction with
Rh proteins for expression, although the
basis of this interaction is not clear. (For a
review of the evolution of the LW system,
see Storry.34)
LW Antibodies
Anti-LWahas not been reported to cause
HTRs or HDFN and both D+ and D–
LW(a+) red cells have been successfully
transfused into patients whose sera con-
tained anti-LWa. Reduced expression of
LW antigens can occur in pregnancy and
some hematologic diseases. LW antibod-
ies may occur as an autoantibody or as an
apparent alloantibody in the serum of
such individuals. Anti-LWab has been re-
ported in LW(a–b–) individuals as well as
in patients with suppressed LW antigens.19
Chido/Rodgers System
Chido/Rodgers Antigens
The Chido (Ch) and Rodgers (Rg) antigens
are high-incidence antigens present on
the complement component C4. The an-
Chapter 15: Other Blood Groups 351
Table 15-10. Phenotypes and
Frequencies in the LW System in
Whites
Reactions with
Anti-
Phenotype
Phenotype
Frequency
(%)
LWaLWb
+ 0 LW(a+b–) >99%
+ + LW(a+b+) <1%
0 + LW(a–b+) Very rare
0 0 LW(a–b–) Very rare
Copyright © 2005 by the AABB. All rights reserved.

tigens are not intrinsic to the red cell. In
antigen-positive individuals, the antigens
are adsorbed onto red cells from the
plasma through an attachment mecha-
nism that remains unclear.35 Ch has been
subdivided into six antigens and Rg into
two antigens. A ninth antigen, WH, re-
quires the interaction of Rg1 and Ch6 for
expression. C4 is encoded by two linked
genes, C4A and C4B, on chromosome 6.
Existing sera are poorly classified, but
the phenotype frequency may be consid-
ered as in Table 15-11. The antigens are de-
stroyed by papain/ficin treatment but unaf-
fected by DTT/AET treatment.
Chido/Rodgers Antibodies
Antibodies to Ch and Rg are generally be-
nign but may be a great nuisance in sero-
logic investigations. Rapid identification
is possible using red cells coated with C4
or by inhibition with pooled plasma from
antigen-positive individuals. (See Method
3.9.) The antibodies are nonreactive with
enzyme-treated red cells.
Gerbich System
Gerbich Antigens
The Gerbich system includes eight anti-
gens, of which three (Ge2, Ge3, and Ge4)
are of high incidence and five (Wb, Lsa,
Ana,Dh
a,andGEIS)areoflowincidence.
Several phenotypes that lack one or more
of the high-incidence antigens are shown
in Table 15-12; all are rare. Red cells with
the Gerbich or Leach phenotype have a
weakened expression of some Kell system
antigens.Ge2,Ge4,Wb,Ls
a,An
a,Dh
a,and
GEIS are destroyed by papain and ficin,
but Ge3 resists protease treatment.
352 AABB Technical Manual
Table 15-11. Some Blood Group Antigens with Phenotypic Relationships
Approximate Frequency (%)
Antigens Phenotypes Whites Blacks
Chido (Ch) and Rodgers (Rg) Ch+,Rg+ 95.0
Ch–,Rg+ 2.0
Ch+,Rg– 3.0
Ch–,Rg– Very rare
Cost (Csa)* and York (Yka) Cs(a+),Yk(a+) 82.5 95.6
Cs(a+),Yk(a–) 13.5 3.2
Cs(a–),Yk(a+) 2.1 0.6
Cs(a–),Yk(a–) 1.9 0.6
Knops-Helgeson (Kna) and McCoy (McCa) Kn(a+),McC(a+) 97.0 95.0
Kn(a+),McC(a–) 2.0 4.0
Kn(a–),McC(a+) 1.0 1.0
Kn(a–),McC(a–) Rare Rare
*Although Csais not part of the Knops blood group system, there is a phenotypic association between Ykaand Csa.
Copyright © 2005 by the AABB. All rights reserved.

The antigens of the Gerbich blood group
system are carried on glycophorin C (GPC)
and glycophorin D (GPD). GPC carries Ge3
and Ge4, whereas GPD carries Ge2 and
Ge3. Anais carried on an altered form of
GPD. DhaandWbarelocatedonaltered
forms of GPC. Lsaisfoundonanaltered
form of GPC and GPD.36 The proteins are
the product of a single gene, GYPC,on
chromosome 2. GPC is approximately four
times more abundant than GPD. The
mechanism whereby these two proteins are
derived from a single gene involves an al-
ternative initiation site in the gene. GPC
and GPD interact directly with protein
band 4.1 in the membrane skeleton. It is
clear that the interaction is important in
maintaining cell shape because deficien-
cies of either band 4.1 or GPC/D cause
elliptocytosis.36
Gerbich Antibodies
The antibodies that Ge-negative individu-
als may produce are shown in Table 15-12;
they may be immune or occur without red
cell stimulation. Anti-Ge is usually IgG
but may have an IgM component. The
clinical significance of the antibodies is
variable. Antibodies to the Gerbich anti-
gensmaybeararecauseofHDFN.
Cromer System
Cromer Antigens
A total of 13 antigens have been assigned
to the Cromer blood group system: 10 high-
incidence antigens and three low-inci-
dence antigens (see Table 15-13). Tcais
antithetical to the low-incidence antigen
Tcbin Blacks and to Tccin Whites. WESbis
the high-incidence antigen antithetical to
WESa.Cr
a,Dr
a,Es
a,UMC,GUTI,SERF,and
ZENA are not associated with low-inci-
dence antigens. IFC is absent only in the
null phenotype (Inab).
The antigens are located on the comple-
ment regulatory protein called decay-accel-
erating factor (DAF). The protein is en-
coded by DAF, one gene of the regulators of
complement activation (RCA) complex, on
chromosome 1. The antigens are on leuko-
cytes, platelets, and trophoblasts of the pla-
centa as well as in soluble form in the se-
rum/plasma and urine.37 The antigens are
not affected by ficin or papain. DTT or AET
may weaken the antigens but do not com-
pletely destroy them.37
Cromer Antibodies
Antibodies to antigens of the Cromer sys-
tem are immune-mediated and extremely
uncommon. Most examples of anti-Cra,
-WESb,and-Tc
ahave been found in the
sera of Black individuals. Anti-GUTI was
found in a Canadian of Chilean ancestry.37
The clinical significance of the antibodies
is variable, and some examples cause de-
creased survival of transfused red cells.
The antibodies will not cause HDFN be-
cause the placenta tissue is a rich source
of DAF, which is thought to adsorb the
maternal antibodies.37
Chapter 15: Other Blood Groups 353
Table 15-12. Ge– Phenotypes
Phenotype Antibody Produced
Ge: –2,3,4 (Yus type) Anti-Ge2
Ge:–2,–3,4 (Gerbich type) Anti-Ge2 or -Ge3
Ge:–2,–3,–4 (Leach type) Anti-Ge2, -Ge3, or -Ge4
Copyright © 2005 by the AABB. All rights reserved.

Knops System
Knops Antigens
Most of the eight Knops system antigens
(Kna,Kn
b,McC
a,McC
b,Sl
a,Yk
a, Vil, and Sl3)
have been located on the C3b/C4b recep-
tor (CR1), the primary complement re-
ceptor on red cells. A gene on chromo-
some 1 encodes CR1. Kna,McC
a,Sl
a,Yk
a,
and Sl3 are high-incidence antigens. Knb,
McCb, and Vil are low-incidence antigens.
Some variation in frequency is observed
between the red cells of Whites and Blacks
(see Table 15-11).
The Knops system antigens are not de-
stroyed by ficin or papain but may be weak-
ened or destroyed by DTT or AET.
Knops Antibodies
The antibodies commonly show variable
weak reactivity in the antiglobulin phase
of testing but may continue to react even
at high dilutions. Moulds et al38 have shown
that the variable reactivity of anti-CR1-re-
lated sera is a direct reflection of the
number of CR1 sites that exhibit both size
and expression polymorphisms and vary
widely among individuals. The antibodies
are of no clinical significance.
Indian System
Inaand Inbare located on CD44, a protein
of wide tissue distribution with the char-
acteristics of a cell adhesion molecule. Ina
is a low-incidence antigen, and Inbis of
high incidence (see Table 15-7). Inbshows
reduced expression on Lu(a–b–) red cells
of the In(Lu) type but is normally ex-
pressed on Lu(a–b–) red cells from per-
sons homozygous for the amorph or pos-
sessing the X-borne suppressor gene. The
antigens are destroyed by papain and ficin
as well as by reducing agents such as 0.2
M DTT. There are few data on the clinical
significance of the corresponding antibod-
ies.
Other Blood Group Systems
Ok System
The Ok system consists of a single high-
incidence antigen, Oka.Thefewrare
Ok(a–) individuals to date have been Jap-
anese. Proteases do not seem to weaken
expression of Okain routine agglutination
tests. Anti-Okareacts optimally by an indi-
rect antiglobulin test and appears to be
clinically significant in transfusion ther-
apy, causing rapid destruction of Ok(a+)
red cells.
Raph System
The Raph system consists of a single anti-
gen, MER2. Anti-MER2 has been reported
in three Israeli Jews. All three were on re-
nal dialysis, raising the possibility that an-
tibody production may be associated with
kidney disease. The MER2 antigen has
354 AABB Technical Manual
Table 15-13. Antigens of High and Low
Incidence in the Cromer Blood Group
System
Antigen Incidence (%)
Cra>99
Tca>99
Tcb<1
Tcc<1
Dra>99
Esa>99
IFC >99
WESa<1
WESb>99
UMC >99
GUTI >99
SERF >99
ZENA >99
Copyright © 2005 by the AABB. All rights reserved.
been detected on the red cells of 92% of
those tested. The MER2 antigen is sensi-
tive to DTT but is not affected by treat-
ment with ficin, papain, or chloroquine.
The antibodies have been IgG and some
have bound complement. To date, there
has been no information on whether these
antibodies are capable of causing HTR or
HDFN.
John Milton Hagen System
The John Milton Hagen (JMH) antigen is
carried on a GPI-linked CD108 glycopro-
tein. JMH antigen decreases over time. The
JMH– phenotype can be transient. The
JMH– phenotype can be acquired or in-
herited. The antigen is destroyed or al-
tered by ficin or papain treatment and by
AET or DTT treatment.
Antibodies that react with JMH show
variation in reactivity and are usually weak.
Autoanti-JMH can often be found in older
people, along with an acquired absent or
weak JMH antigen expression. The anti-
body is not routinely considered capable of
HTR or HDFN.
GIL System
There is one antigen of high frequency, GIL,
in this system. GIL is located on aquaporin
3 (AQP3), which is a glycerol transporter.
The antibody has not been reported to
cause HDFN or HTR.
Blood Group Collections
In addition to the blood group systems,
there are collections of antigens that ex-
hibit shared characteristics but do not as
yet meet the criteria for blood group sys-
tem status defined by the ISBT. They in-
clude Cost (ISBT 205), Er (ISBT 208), and
Vel (ISBT 211).
Cost
Csaand Csbare all that remain of this col-
lection after the Knops antigens were
identified on CR1. Csaoccurs in a fre-
quency greater that 98% in most popula-
tions, whereas Csbappears in about 34%
of the population.19 There is, however, an
unexplained connection between the Yka
and Csaantigens, such that red cells nega-
tive for one antigen are often weak or neg-
ativefortheother.(SeeTable15-11.)The
antigens are not destroyed by ficin, papain,
or DTT. Anti-Csabehaves similarly to anti-
bodies produced to the Knops system an-
tigens and is not considered clinically sig-
nificant.
Er
The Er collection consists of two antigens,
which give rise to four phenotypes: Er(a+b–),
Er(a+b+), Er(a–b+), and Er(a–b–). Erais a
high-incidence antigen present on the red
cells of >99% of all individuals, but Erbhas
a prevalence of less than 1%. The presence
of a silent third allele, Er, is thought to ac-
countfortheEr(a–b–)phenotype,asde-
monstrated by family studies. The antigens
are not destroyed by ficin, papain, or DTT
but are destroyed by EDTA-glycine acid.
Vel
The Vel collection was recently created to
include two serologically related antigens
of high incidence, Vel and ABTI.
Vel is a high-incidence antigen that is
unaffected by protease and sulfhydryl treat-
ment. It is well developed at birth, but anti-
gen expression is variable.8Despite its oc-
currence after known immunizing stimuli,
anti-Vel is most commonly IgM. It has been
reported to range from causing only a posi-
tive DAT in the neonate to causing severe
HDFN.19 It has been implicated in HTRs.
Anti-Vel binds complement, and in-vitro
hemolysis of incompatible red cells is often
Chapter 15: Other Blood Groups 355
Copyright © 2005 by the AABB. All rights reserved.
seen when testing freshly drawn serum
containing this antibody. Reactivity of
anti-Vel is usually enhanced by enzyme
treatment of red cells expressing the anti-
gen.
High-Incidence Red Cell
Antigens Not Assigned to a
Blood Group System or
Collection
Table 15-14 lists the antigens of high inci-
dence that are independent of a blood
group system or collection. Persons who
make alloantibody to a specific blood
group antigen necessarily have red cells
lacking that antigen. For this reason, anti-
bodies directed at high-incidence antigens
are rarely encountered. The antibodies
corresponding to these antigens usually
react best by antiglobulin testing.
Lan Antigen
Lan is a high-incidence antigen that is re-
sistant to enzyme-treatment and to 0.2 M
DTT treatment. A weak form of the Lan
antigen has been reported.19 Anti-Lan is
characteristically IgG, may bind comple-
ment, and may cause HTRs. Cases of
HDFN due to anti-Lan have been mild
even though the Lan antigen is present on
cord red cells.19
AtaAntigen
Atais a high-incidence antigen that is re-
sistant to enzyme treatment and to 0.2 M
DTT treatment. The At(a–) phenotype has
been found only in Black individuals.19
Anti-Atais characteristically IgG. The anti-
body appears to cause only moderate
HTRs and no clinical HDFN.19
JraAntigen
Jrais a high-incidence antigen that is re-
sistant to enzyme treatment and to 0.2 M
DTT treatment. The Jr(a–) phenotype is more
commonly found in Japanese individuals
but has been found in other populations
as well.8(pp805-806) Anti-Jrahas been shown to
cause reduced red cell survival.8(pp805-806)
Other examples of anti-Jrahave shown lit-
tle or no clinical significance in HDFN or
HTR.8(p806),39
AnWj Antigen
AnWj is a high-incidence antigen that is
resistant to enzyme treatment but weak-
ened by 0.2 M DTT treatment.19 The anti-
geniscarriedonCD44,whichalsocarries
the Indian blood group system antigens.
The AnWj antigen is weakened on the red
cells from individuals with the In(Lu)
gene (see section on Lutheran System).
Some patients with Hodgkin’s disease
may experience a long-term suppression
of the AnWj antigen.8(pp783-784) The AnWj an-
tigen is the receptor for Haemophilus
influenzae.8(pp784-785) Anti-AnWj has been
356 AABB Technical Manual
Table 15-14. Some Antigens of High
Incidence Not Assigned to a Blood
Group System or Collection
Name Symbol
August Ata
Langereis Lan
Sid Sda
Duclos
Jra
Emm
AnWj
PEL
MAM
Copyright © 2005 by the AABB. All rights reserved.
implicated in severe HTRs but not in HDFN
because the antigen is not present on cord
red cells.
SdaAntigen
Sdais an antigen of fairly high incidence,
widely distributed in mammalian tissues
and body fluids. The antigen is variably
expressed on the red cells of Sd(a+) indi-
viduals. Sdaexpression may diminish dur-
ing pregnancy and the Sd(a–) phenotype
is observed in 30% to 75% of pregnant
women. The antigen is not present on
cord cells. The strongest expression of Sda
has been observed on polyagglutinable
red cells of the Cad phenotype. The fre-
quency of Sd(a–) blood is considered to
be around 9%, but weakly positive reac-
tions are often difficult to distinguish from
negative ones.
Anti-Sdacan be reactive by antiglobulin
testing. Microscopic examination of posi-
tive reactions generally shows mixed-field
agglutination, with relatively small, tightly
agglutinated clumps of red cells present
against a background of free red cells.
These agglutinates are refractile and may
have a shiny appearance. Because the ma-
jority of the examples of anti-Sdaare IgM,
thewideuseofanti-IgGmeansthatmany
examples of anti-Sdaare no longer de-
tected. Anti-Sdais not considered to be clin-
ically significant. However, there have been
reported cases of HTRs with red cells with
strong Sdaexpression.8(pp816-817)
The immunodominant sugar of Sdais
N-acetylgalactosamine (GalNAc), also the
immunodominant sugar of the A blood
group antigen and of the Tamm-Horsfall
glycoprotein, found in human and guinea
pig urine. Anti-Sdaactivity can be inhibited
by incubation with urine from guinea pigs
or from Sd(a+) humans. See Method 3.11
for the performance of a urine neutraliza-
tion of anti-Sda.
Low-Incidence Red Cell
Antigens Not Assigned to a
Blood Group System or
Collection
Many independent low-incidence red cell
antigens have been recognized in addition
to a growing number that have been as-
signed to the MNS, Rh, and Diego systems.
Table 15-15 lists those that have been
studied and shown to be inherited in a
dominant manner. Antibodies specific for
these low-incidence antigens react with
so few random blood samples that they
virtually never cause difficulties in select-
ing blood for transfusion but may be im-
plicated in some rare cases of HDFN. The
antibodies are of interest to the serologist,
however, because of the unexpectedly
high incidence with which they occur,
often without an identifiable antigenic
stimulus.
Chapter 15: Other Blood Groups 357
Table 15-15. Antigens of Low Incidence
Not Assigned to a Blood Group System
or Collection
Batty (By) Livesay (Lia)
Biles (Bi) Milne
Box (Bxa)Oldeide(Ol
a)
Christiansen (Chra) Peters (Pta)
HJK Rasmussen (RASM)
HOFM Reid (Rea)
JFV REIT
JONES SARA
Jensen (Jea) Torkildsen (Toa)
Katagiri (Kg)
The antigens occur with a frequency of 1 in 500 or less.
Copyright © 2005 by the AABB. All rights reserved.
Antibodies to Low-Incidence
Antigens
Antibodies to low-incidence antigens have
sometimes been implicated in transfusion
reactions and HDFN. These antibodies
are usually encountered by chance, when
the red cells used for antibody detection
or selected for crossmatching happen to
carry the corresponding antigen.
Antibodies to low-incidence antigens may
also be present as unsuspected contaminants
in blood typing reagents prepared from hu-
man serum and may cause false-positive
test results if the red cells tested carry the
antigen. Testing with reagents from differ-
ent manufacturers may not eliminate this
error because it is not uncommon for a sin-
gle individual with an uncommon antibody
to provide the serum used for reagent prep-
aration by different manufacturers.
Some antibodies to low-incidence anti-
gens react as saline agglutinins. They can
also occur as IgG antibodies reactive only
by antiglobulin testing, even if there has
been no exposure to red cell immunization.
It is common for several low-incidence an-
tibodies to occur together in a single serum;
multiple specificities are especially likely in
sera from patients with autoimmune con-
ditions.
Bg (Bennett-Goodspeed) Antigens
Antibodies directed at certain leukocyte
antigens sometimes cause confusing re-
actions in serologic tests with red cells. At
least three separate specificities have been
given names as Bg antigens: Bgacorresponds
to HLA-B7; Bgbcorresponds to HLA-B17;
and Bgccorresponds to HLA-A28. A fourth
antibody in some antileukocyte sera re-
acts with red cells of persons who express
HLA-A10.Theso-calledBgantigensare
expressed to variable degrees on red cells,
with the result that reactions of differing
strengths are observed when a single se-
rum containing “anti-Bg” is tested with
different Bg+ red cells. Reactivity is most
commonly observed in antiglobulin test-
ing, but highly potent anti-Bg sera may di-
rectly agglutinate red cells with an unusu-
ally strong expression of the Bg antigens.
Confident and precise classification of
reactivity is made difficult by the weak ex-
pression of these antigens on some red cells
and by multiple specificities among differ-
ent examples of the Bg antibodies. These
antibodies may also occur as unsuspected
contaminants in human source blood typ-
ing sera, where they may cause false-posi-
tive reactions with cells having unusually
strong expression of the corresponding Bg
antigen. Bg-related antigens are denatured
by chloroquine diphosphate or a solution
of glycine-HCl/EDTA.
References
1. Garratty G, Dzik W, Issitt PD, et al. Terminol-
ogy for blood group antigens and genes—his-
torical origins and guidelines in the new mil-
lennium. Transfusion 2000;40:477-89.
2. Daniels GL, Anstee DJ, Cartron JP, et al. Ter-
minology for red cell surface antigens. ISBT
Working Party Oslo Report. International So-
ciety of Blood Transfusion. Vox Sang 1999;77:
52-7.
3. Daniels GL, Cartron JP, Fletcher A, et al. Inter-
national Society of Blood Transfusion Com-
mittee on terminology for red cell surface an-
tigens: Vancouver Report. Vox Sang 2003;84:
244-7.
4. Race RR, Sanger R. Blood groups in man. 6th
ed. Oxford: Blackwell Scientific Publications,
1975.
5. Bruce LJ, Ring SM, Anstee DJ, et al. Changes
in the blood group Wright antigens are asso-
ciated with a mutation at amino acid 658 in
human erythrocyte band 3: A site of interac-
tion between band 3 and glycophorin A un-
der certain conditions. Blood 1995;85:299-
306.
6. Rygiel SA, Issitt CH, Fruitstone MJ. Destruc-
tion of the S antigen by Clorox (abstract).
Transfusion 1983;23:410.
7. Case J. The behavior of anti-S antibodies with
ficin-treated human red cells. In: Abstracts of
358 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
volunteer papers. 30th Annual Meeting of the
American Association of Blood Banks. Wash-
ington, DC: American Association of Blood
Banks, 1977:36.
8. Issitt PD, Anstee DJ. Applied blood group se-
rology. 4th ed. Durham, NC: Montgomery
Scientific, 1998.
9. LeeS,RussoD,RedmanCM.TheKellblood
group system: Kell and XK membrane pro-
teins. Semin Hematol 2000;37:113-21.
10. Zelinski T, Coghlan G, Myal Y, et al. Genetic
linkage between the Kell blood group system
and prolactin-inducible protein loci: Provi-
sional assignment of KEL to chromosome 7.
Ann Hum Genet 1991;55:137-40.
11. DanielsG,HadleyA,GreenCA.Causesoffe
-
tal anemia in hemolytic disease due to anti-K
(letter). Transfusion 2003;43:115-16.
12. Lin M, Wang CL, Chen FS, et al. Fatal hemoly-
tic transfusion reaction due to anti-Ku in a
Knull patient. Immunohematol 2003;19:19-
21.
13. ChaudhuriA,PolyakovaJ,ZbrezeznaV,etal.
Cloning of glycoprotein D cDNA which en-
codes the major subunit of the Duffy blood
group system and the receptor for the Plasmo-
dium vivax malaria parasite. Proc Natl Acad
Sci U S A 1993;90:10793-7.
14. Pierce SP, Macpherson CR, eds. Blood group
systems: Duffy, Kidd and Lutheran. Arlington,
VA: American Association of Blood Banks,
1988.
15. Horuk R, Chitnis C, Darbonne W, et al. A re-
ceptor for the malarial parasite Plasmodium
vivax: The erythrocyte chemokine receptor.
Science 1993;261:1182-4.
16. Heaton DC, McLoughlin K. Jk(a–b–) red blood
cells resist urea lysis. Transfusion 1982;22:70-1.
17. Mougey R. The Kidd blood group system. In:
Pierce SR, Macpherson CR, eds. Blood group
systems: Duffy, Kidd and Lutheran. Arlington,
VA: American Association of Blood Banks,
1988:53-71.
18. Zelinski K, Kaita H, Coghlan G, Philipps S. As-
signment of the Auberger red cell antigen
polymorphism to the Lutheran blood group
system: Genetic justification. Vox Sang 1991;
61:275-6.
19. Reid ME, Lomas-Francis C. The blood group
antigen factsbook. 2nd ed. San Diego, CA: Ac-
ademic Press, 2004.
20. Daniels G. Effect of enzymes on and chemical
modifications of high-frequency red cell anti-
gens. Immunohematology 1992;8:53-7.
21. Merry AH, Gardner B, Parsons SF, Anstee DJ.
Estimation of the number of binding sites for
a murine monoclonal anti-Lubon human
erythrocytes. Vox Sang 1987;53:57-60.
22. Crew VK, Green C, Daniels G. Molecular
bases of the antigens of the Lutheren blood
group system. Transfusion 2003;43:1729-37.
23. Storry JR, Reid ME, Chiofolo JT, et al. A new
Wr(a+b–) Proband with anti-Wrbrecognizing
a ficin sensitive antigen (abstract). Transfu-
sion 2001;41(Suppl):23S.
24. Spring FA. Characterization of blood-group-
active erythrocyte membrane glycoproteins
with human antisera. Transfus Med 1993;3:
167-78.
25. Eckrich RJ, Mallory DM. Correlation of mono-
cyte monolayer assays and posttransfusion
survival of Yt(a+) red cells in patients with
anti-Yta(abstract). Transfusion 1993;33
(Suppl):18S.
26. Garratty G, Arndt P, Nance S. The potential
clinical significance of blood group alloanti-
bodies to high frequency antigens (abstract).
Blood 1997;90(Suppl):473a.
27. Ellis NA, Ye T-Z, Patton S, et al. Cloning of
PBDX,aMIC2-related gene that spans the
pseudoautosomal boundary on chromosome
Xp. Nat Genet 1994;6:394-9.
28. Ellis NA, Tippett P, Petty A, et al. PBDX is the
XG blood group gene. Nat Genet 1994;8:285-
90.
29. Banks JA, Parker N, Poole J. Evidence to show
that Dombrock antigens reside on the Gya/
Hy glycoprotein. Transfus Med 1992;(Suppl)
1:68.
30. Scofield TL, Miller JP, Storry JR, et al. Evi-
dence that Hy– RBCs express weak Joaanti-
gen. Transfusion 2004;44:170-2.
31. JuddWJ,SteinerEA.Multiplehemolytic
transfusion reactions caused by anti-Doa.
Transfusion 1991;31:477-8.
32. Smith BL, Preston GM, Spring F, et al. Human
red cell aquaporin CHIP. J Clin Invest 1994;94:
1043-9.
33. Bailly P, Hermand P, Callebaut I, et al. The LW
blood group glycoprotein is homologous to
intercellular adhesion molecules. Proc Natl
Acad Sci U S A 1994;91:5306-10.
34. Storry JR. Review: The LW blood group sys-
tem. Immunohematology 1994;8:87-93.
35. Moulds JM, Laird-Fryer B, eds. Blood groups:
Chido/Rodgers, Knops/McCoy/York and Cro-
mer. Bethesda, MD: American Association of
Blood Banks, 1992.
36. Reid ME, Spring FA. Molecular basis of glyco-
phorin C variants and their associated blood
group antigens. Transfus Med 1994;4:139-46.
37. Storry JR, Reid ME, The Cromer blood group
system: A review. Immunohematology 2002;
18:95-101.
38. Moulds JM, Moulds JJ, Brown M, Atkinson JP.
Antiglobulin testing for CR1-related (Knops/
McCoy/Swain-Langley/York) blood group an-
Chapter 15: Other Blood Groups 359
Copyright © 2005 by the AABB. All rights reserved.
tigens: Negative and weak reactions are
caused by variable expression of CR1. Vox Sang
1992;62:230-5.
39. Kwon M, Ammeus M, Blackall D. A Japanese
patient with a Jraantibody: Apparent lack of
clinical significance despite multiple incom-
patible transfusions (abstract). Transfusion
2001;41(Suppl):58S.
Suggested Reading
Akane A, Mizukami H, Shiono H. Classification of
the standard alleles of the MN blood group sys-
tem. Vox Sang 2000;79:183-7.
Liu M, Jiang D, Liu S, et al. Frequencies of the ma-
jor alleles of Diego, Dombrock, Yt, and Ok blood
group systems in the Chinese, Han, Hui, and Ti-
betan nationalities. Immunohematology 2003;19:
22-5.
Lögdberg L, Reid M, Miller J. Cloning and genetic
characterization of blood group carrier molecules
and antigens. Transfus Med Rev 2002;16:1-10.
Pogo AO, Chaudhuri A. The Duffy protein: A ma-
larial and chemokine receptor. Semin Hematol
2000;37:122-9.
Reid ME. The Dombrock blood group system: A re-
view. Transfusion 2003;43:107-14.
Reid ME, Rios M, Yazdanbakhsh K. Applications of
molecular biology techniques to transfusion med-
icine. Semin Hematol 2000;37:166-76.
Reid ME, Storry JR. Low-incidence MNS antigens
associated with single amino acid changes and their
susceptibility to enzyme treatment. Immunohema-
tology 2001;17:76-81.
360 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 16: Platelet and Granulocyte Antigens and Antibodies
Chapter 16
Platelet and Granulocyte
Antigens and Antibodies
ANTIBODIES REACTIVE WITH an-
tigens expressed on platelets and
leukocytes are assuming increas-
ing importance. Some blood group anti-
gens are shared by red cells, white cells,
and platelets; others are specific to cer-
tain cell types. This chapter discusses an-
tibodies directed at antigens expressed on
platelets and neutrophils, with an empha-
sis on those specific for these cells. HLA
antibodies and antigens are covered more
fully in Chapter 17.
Platelet Antigens
Antigens Shared with Other Tissues
Platelets express a variety of antigenic
markers on their surface. Some of these
antigens are shared with other cell types,
as in the case of ABH antigens and HLA
antigens, which are shared with virtually
all nucleated cells in the body. Others are
observed to be essentially platelet spe-
cific.
ABH Antigens
The ABH antigens expressed on platelets
are a combination of structures intrinsic
totheplasmamembraneandthosead
-
sorbed from the plasma. The amount of
ABH antigen present on platelets is quite
variable from individual to individual,
with from 5% to 10% of non-group-O in-
dividuals expressing extremely elevated
amounts of A or B substance on their
platelets. These people appear to have a
“high-expresser” form of glycosyltrans-
ferase in their sera. Although platelets are
often transfused without regard to ABO
compatibility, in some cases, ABO anti-
bodies (particularly IgG antibodies of
high titer in group O recipients) may react
with platelets carrying large amounts of A
or B antigen.1High-expresser platelets are
particularly vulnerable to this type of im-
361
16
Copyright © 2005 by the AABB. All rights reserved.
mune destruction. ABO antibodies in re-
cipients may also cause reduced survival
of ABO-incompatible platelets from nor-
mal-expresser phenotype donors, causing
occasional patients to exhibit refractori-
ness to platelet transfusions on this basis.
Other red cell antigens—including Lea,
Leb,Ii,andP
2as well as the Cromer antigens
associated with decay accelerating fac-
tor3—are also found on platelets, but there
is no evidence that antibodies to these anti-
gens significantly reduce platelet survival in
vivo.
HLA Antigens
HLA antigens are found on the surfaces of
both platelets and white cells (see Chap-
ter 17). In fact, platelets are the major
source of Class I HLA antigens in whole
blood.2Recent evidence indicates that
most Class I HLA molecules on platelets
are integral membrane proteins, and
smaller amounts may be absorbed from
surrounding plasma.3
HLA alloantibodies do not occur natu-
rally, arising only after sensitization by
pregnancy or blood transfusion. Studies of
HLA alloimmunization in patients trans-
fused with platelets document the develop-
ment of antibodies within 3 to 4 weeks after
primary exposure and as early as 4 days af-
ter secondary exposure in patients previ-
ously transfused or pregnant.4The likeli-
hood of HLA alloimmunization by
transfusion in patients not previously sensi-
tized is variable,4,5 and the risk of HLA
alloimmunization appears to be related to
the underlying disease as well as to the
immunosuppressive effects of treatment
regimens. Platelets carry Class I HLA anti-
gens but lack Class II antigens, which are
necessary for primary sensitization. There-
fore, exposure to leukocytes expressing
HLA antigens during transfusion is the
principal cause of primary HLA alloim-
munization.
Platelet Transfusion Refractoriness
A less-than-expected increase in platelet
count occurs in about 20% to 70% of
multitransfused thrombocytopenic pa-
tients,6and patients treated for malignant
hematopoietic disorders are particularly
likely to become refractory to platelet
transfusions. A widely accepted definition
of refractoriness was used in a random-
ized controlled clinical trial of platelet
transfusion therapy, sponsored by the Na-
tional Institutes of Health (NIH). In this
study, two consecutive 1-hour posttrans-
fusion platelet corrected count increments
(CCI) of less than 5000 platelets ×m2body
surface area/µL indicated refractoriness.7
Others have used less stringent criteria
(eg, three platelet transfusions over a
2-week period that yield inadequate post-
transfusion platelet counts).8,9 Response is
often determined by calculating either a
CCI or a posttransfusion platelet recovery
(PPR) between 10 and 60 minutes after
transfusion (see Table 16-1). Responses of
7500 platelets ×m2body surface area/µL
or 20% can be considered acceptable from
the CCI or the PPR calculation, respec-
tively.
Alloimmune platelet refractoriness is
most often the result of HLA sensitization
and can be diagnosed by demonstration of
significant levels of HLA antibodies. Patient
serum is tested against a panel of lympho-
cytes (or synthetic beads bearing Class I an-
tigens) that represent most of the Class I
HLA specificities in the population. A
panel-reactive antibody (PRA) score of 20%
or higher is evidence that HLA sensitization
may be contributing to the platelet refrac-
toriness (see Chapter 17).
Although platelet alloimmunization is
one cause of refractoriness, there are multi-
362 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

ple, nonimmune reasons why transfused
platelets may not yield the expected in-
crease in platelet count [eg, sepsis, dissemi-
nated intravascular coagulation (DIC), or
the administration of certain drugs]. Some
of the most commonly cited nonimmune
causes of platelet refractoriness are listed in
Table 16-2. A study of patients undergoing
marrow transplant suggested that pa-
tient-related variables such as total body ir-
radiation, advanced disease status, and liver
dysfunction are important predictors of
poor platelet count increments as well.10,11
Even when possible immune causes of re-
fractoriness are identified, nonimmune fac-
tors are often simultaneously present.
Several strategies may be considered
when selecting platelets for patients with
immune-mediated refractoriness. When
antibodies to HLA antigens are demon-
strated, a widely used approach is to supply
apheresis platelets matched to the patient’s
HLA type.12 A disadvantage is that a pool of
several thousand HLA-typed potential
apheresis donors is necessary to find suffi-
cient HLA-compatible matches.13 Moreover,
donor selection on the basis of HLA type
can lead to the exclusion of donors with
HLA types different from that of the recipi-
ent but potentially effective if the recipient
is alloimmunized to other antigenic deter-
minants.14 For patients who are likely to re-
quiremultipleplatelettransfusions,HLA
typing should be performed in advance of a
planned course of treatment.
It is important to understand the degree
of match that may be provided (see Table
16-3). Platelets received following a request
for “HLA-matched” platelets are typically
the closest match obtainable within the
constraints of time and donor availability.
In one study,15 43% of platelets provided as
Chapter 16: Platelet and Granulocyte Antigens and Antibodies 363
Table 16-1. Determination of Response to Transfused Platelets
Calculation of Corrected Count Increment (CCI)
CCI = Body Surface Area (m2)×Platelet Count Increment ×1011
No. of Platelets Transfused
EXAMPLE: If 4 ×1011 platelets are transfused to a patient whose body surface area is 1.8 m2and
the increase in posttransfusion platelet count is 25,000/µL, then:
CCI 1.8 m 25,000 / L 10
410 11,250 plat
211
11
=××
×=
µelets m / L
2
×µ
Calculation of Posttransfusion Platelet Recovery (PPR)
PPR(%) Estimated Total Blood Volume* Platelet
=×Count Increment
No. of Platelets Transfused
*Total blood volume can be estimated in adult patients as 75 mL/kg
EXAMPLE: If 4 ×1011 platelets are transfused to a 70-kg patient and the increase in
posttransfusion platelet count is 25,000/µL, then:
PPR 70 kg 70 mL / kg 25,000 plts / L 10
41
3
=×× ×
×
µ
0 platelets 30.6%
11 =
Copyright © 2005 by the AABB. All rights reserved.

HLA-matched were relatively poor grade B
or C matches. The most successful re-
sponses occur with the subset of grade A
and B1U or B2U HLA matches, but mis-
matches for some antigens (B44, 45) that
are poorly expressed on platelets can be
useful. According to AABB Standards for
Blood Banks and Transfusion Services,16(p43)
HLA-matched platelets should be irradi-
ated to prevent transfusion-associated
graft-vs-host disease.
A second approach to provide effective
platelets is to use a pretransfusion platelet
crossmatching assay. This approach can be
used to predict and, therefore, avoid subse-
quent platelet transfusion failures.17 The
solid-phase red cell adherence test (SPRCA)
is the most widely used method for platelet
crossmatching, and test results are reason-
ably predictive of posttransfusion platelet
counts.18-20 Compared with HLA matching,
crossmatching can prove both more conve-
nient and economically advantageous. It
avoids exclusion of HLA-mismatched but
compatible donors and has the added ad-
vantage of selecting platelets when the an-
tibody (-ies) involved is (are) directed at a
platelet-specific antigen. Platelet cross-
matching, however, will not always be suc-
cessful, particularly when patients are
highly alloimmunized (PRA >50%). In these
instances, finding sufficient compatible
units may be problematic, and selection of
HLA-matched platelets may be more prac-
tical. Although the incidence of platelet-
specific antibodies causing patients to be
refractory to most or all attempted platelet
transfusions is very small, this possibility
should be investigated when most of the at-
tempted crossmatches are positive. If pla-
telet-specific antibodies are present, donors
of known platelet antigen phenotype or
364 AABB Technical Manual
Table 16-2. Some Nonimmune Causes
of Platelet Refractoriness
Massive bleeding
Fever
Sepsis
Splenomegaly (splenic sequestration)
Disseminated intravascular coagulation
Allogeneic transplantation
Poor storage of platelets before transfusion
Effects of drugs (may include immune
mechanisms)
Intravenous amphotericin B
Thrombotic thrombocytopenic purpura
Table 16-3. Degree of Matching for HLA-Matched Platelets
Match
Grade Description
Examples of Donor
Phenotypes for a Recipient
Who Is A1,3;B8,27
A 4-antigen match A1,3;B8,27
B1U 1 antigen unknown or blank A1,-;B8,27
B1X 1 cross-reactive group A1,3;B8,7
B2UX 1 antigen blank and 1 cross-reactive A1,-;B8,7
C 1 mismatched antigen present A1,3;B8,35
D 2 or more mismatched antigens present A1,32;B8,35
R Random A2,28;B7,35
Copyright © 2005 by the AABB. All rights reserved.
family members, who are more likely to
share the patient’s phenotype, should be
tested.
An alternative approach to supplying
HLA-compatible transfusions is to deter-
mine the specificity of the patient’s HLA an-
tibodies and select donors whose platelets
lack the antigens with which the antibodies
react. This is termed the antibody specific-
ity prediction (ASP) method.8One study
compared the effectiveness of transfused
platelets selected by the ASP method with
those selected on the basis of HLA match-
ing, platelet crossmatching, or on a random
basis.8Platelets selected by the ASP method
were equally effective as those selected by
HLA matching or by crossmatching, and
superior to randomly selected platelets. In
addition, from a file of HLA-typed donors,
many more potential donors were identi-
fied by the ASP method than were available
using traditional HLA matching criteria,
making the acquisition of compatible
platelets for alloimmunized refractory pa-
tients much more feasible.
A further refinement of HLA matching
was proposed by Duquesnoy.21 Acomputer-
ized algorithm—HLA Matchmaker, available
at http://tpis.upmc.edu/tpis/HLAMatchmaker—
is employed for evaluation of the molecular
similarities and differences between HLA
Class I epitopes. First developed to aid in
locating compatible organs for alloim-
munized prospective renal transplant pa-
tients, the strategy is based on the concept
that immunogenic epitopes are repre-
sented by amino acid triplets on exposed
partsofproteinsequencesoftheClassI
alloantigens that are accessible to allo-
antibodies. Using this scheme, many Class I
HLA antigens classified as mismatches to a
patient’s HLA type have no incompatible
exposed amino acid triplets and, therefore,
would not be expected to elicit an antibody
response. The pool of potentially compati-
ble HLA-selected donors is thereby greatly
expanded. Although this strategy may
prove useful in selecting platelet donors for
refractory patients, it has not yet been eval-
uated in a clinical trial for this purpose and
remains to be validated for this indication.
Prevention of Platelet Alloimmunization
Once refractoriness resulting from plate-
let alloimmunization is established, it is
very difficult, if not impossible, to reverse.
Therefore, in addition to developing meth-
ods of selecting compatible platelet do-
nors for the refractory patient, several
strategies have been evaluated to prevent
alloimmunization to platelets from occur-
ring in the first place. They include reduc-
tion in the number of leukocytes in the
platelet products and ultraviolet B (UVB)
irradiation. The report of the Trial to Re-
duce Alloimmunization to Platelets (TRAP)
Study Group7indicated that use of either
leukocyte-filtered or UVB-irradiated blood
components reduced the incidence of HLA
antibody generation from 45% to between
17% and 21%. The incidence of platelet
refractoriness was reduced from 16% to
between 7% and 10%. Although a rela-
tionship had been reported between allo-
immunization and the number of donor
exposures in one report,22 a second study23
found no relationship between the num-
ber of donor exposures and the rate or se-
verity of alloimmunization. The TRAP
study found that leukocyte reduction, not
the number of donor exposures, was sig-
nificant in modifying the rate of allo-
immunization. Thus, leukocyte-reduced,
pooled, whole-blood-derived platelets
appear to be clinically equivalent to
apheresis platelets, at least in terms of re-
ducing primary alloimmunization.24
Antibodies to HLA antigens may be de-
tected by lymphocytotoxicity tests or by
many of the platelet antibody tests dis-
cussed below. Lymphocytotoxicity tests de-
Chapter 16: Platelet and Granulocyte Antigens and Antibodies 365
Copyright © 2005 by the AABB. All rights reserved.
tect complement-binding antibodies capa-
ble of killing lymphocytes. One strategy for
managing patients who are receiving multi-
ple platelet transfusions and who have de-
veloped clinical refractoriness is to test for
thepresenceofHLAantibodiesusinga
lymphocytotoxicity antibody screen against
a panel of lymphocytes representing most
of the Class I HLA antigens present in the
donor population. Reactivity to greater
than 20% of the cells in the panel (PRA
>20%) indicates that HLA sensitization may
be at least a contributing cause of the
platelet refractoriness. Newer, more sensi-
tive HLA antibody detection techniques
such as the Flow PRA25 (One Lambda,
Canoga Park, CA) have been adapted to a
PRA result format, and some HLA testing
laboratories use these methods instead of
the traditional lymphocytotoxicity test.
The laboratory detection of lymphocyto-
toxic antibodies does not necessarily indi-
cate that the patient will experience re-
duced survival of transfused platelets.
Moreover, HLA antibodies may disappear
from the patient’s plasma despite contin-
ued exposure through transfusions.26 A
fuller discussion of HLA antibody and anti-
gen testing can be found in Chapter 17.
Platelet-Specific Alloantigens
To date, 22 platelet-specific alloantigens
have been characterized as to their local-
ization to platelet surface glycoprotein
structures, quantification of their density
on the platelet surface, and determination
of DNA polymorphisms in genes encod-
ing for them (see Table 16-4).27 Several
others have been described serologically,
but genetic polymorphisms underlying
them have not yet been determined.28 The
term “platelet specific,” is a misnomer for
some of these markers because they may
be found on other types of cells as well
(especially endothelial cells). However,
their chief clinical importance remains
linked to their presence on platelets. Of
the dozens of recognized platelet mem-
brane glycoproteins, at least five [GPIa, Ib
(alpha and beta), IIb, IIIa, and CD109] are
polymorphic and have been demon-
strated to be alloimmunogenic.28 In addi-
tion, rare individuals who lack a sixth
membrane glycoprotein, GPIV (CD36),
may become sensitized to this antigen.29
Approximately 3% to 5% of individuals of
Asian or African ethnicity lack GPIV on
their platelets30 and can become immu-
nized by transfusion or pregnancy.31 Al-
though antibodies to these various mem-
brane glycoproteins may be associated, in
rare instances, with refractoriness to pla-
telet transfusions, alloantibodies to plate-
let-specific antigens are more often asso-
ciated with the alloimmune syndromes
posttransfusion purpura (PTP) and neo-
natal alloimmune thrombocytopenia
(NAIT).
Several antigen systems on platelets are
now recognized32 (Table 16-4).27,33 The no-
menclature adopted by the International
Society of Blood Transfusion classifies the
systems numerically according to the date
of publication and alphabetically to reflect
their frequency in the population.34 As with
red cells, different terminologies for platelet
antigens often coexist. The first recognized
antigen,35 Zwa, is now designated HPA-1a of
the HPA-1 system. The HPA-1a antigen is
often better known as PlA1.HPA-1aispres
-
ent on the platelets of about 98% of persons
of European ethnicity and anti-HPA-1a
(anti-PlA1) is the most frequently encoun-
tered clinically significant platelet-specific
antibody in this population. Its antithetical
antigen, HPA-1b (PlA2), occurs in 27% of this
population.
The HPA-1a and HPA-1b alleles reside on
the platelet membrane glycoprotein GPIIIa.
Patients with Glanzmann’s thrombasthenia
Type I, a disorder of platelet function, lack
366 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 16: Platelet and Granulocyte Antigens and Antibodies 367
Table 16-4. Alloantigenic Polymorphisms of Platelet Glycoproteins that Have Been Implicated in Alloimmune Syndromes*
HPA System
Name
Antigens (Familiar
Names)
Phenotypic
Frequencies GP Location
Amino Acid
Substitution
Alloimmune
Syndromes
POLYMORPHISMS OF GLYCOPROTEIN IIIa
HPA-1 HPA-1a, (PlA1,Zw
a)98% IIIa Leu↔Pro33 NAIT, PTP
HPA-1b, (PlA2,Zw
b)27% IIIa
HPA-4 HPA-4a (Pena,Yuk
b)†99.9% IIIa Arg↔Gln143 NAIT, PTP
HPA-4b (Penb,Yuk
a)<1% IIIa
HPA-6 HPA-6bw (Caa,Tu
a)<1% IIIa Arg↔Gln489 NAIT
HPA-7 HPA-7bw (Mo) <1% IIIa Pro↔Ala407 NAIT
HPA-8 HPA-8bw (Sra) <1% IIIa Arg↔Cys636 NAIT
HPA-10 HPA-10bw (Laa) <1% IIIa Arg↔Gln62 NAIT
HPA-11 HPA-11bw (Groa) <1% IIIa Arg↔His633 NAIT
HPA-14 HPA-14bw (Oea) <1% IIIa Lys611 Deleted NAIT
HPA-16 HPA-16bw (Duva) <1% IIIa Ile↔Thr140 NAIT
POLYMORPHISMS OF GLYCOPROTEIN IIb
HPA-3 HPA-3a (Baka,Lek
a) 85% IIb IIe↔Ser843 NAIT, PTP
HPA-3b (Bakb, Lekb)63% IIb
HPA-9 HPA-9bw (Maxa) 0.6% IIb Val↔Met837 NAIT
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

368 AABB Technical Manual
HPA System
Name
Antigens (Familiar
Names)
Phenotypic
Frequencies GP Location
Amino Acid
Substitution
Alloimmune
Syndromes
POLYMORPHISMS OF GLYCOPROTEIN Ia
HPA-5 HPA-5a (Brb,Zav
b) 99% Ia glu↔Lys505 NAIT, PTP
HPA-5b (Bra,Zav
a) 20% Ia
HPA-13 HPA-13bw (Sita) <0.2% Ia Thr↔Met799 NAIT
POLYMORPHISMS OF GLYCOPROTEIN Ib
HPA-2 HPA-2a (Kob,Sib
b) 99% Ib alpha Thr↔Met145 NAIT
HPA-2b (Koa,Sib
a) 15% Ib alpha
HPA-12 HPA-12bw (Iya)0.3%IbbetaGly↔Glu15 NAIT
OTHER PROBABLE PLATELET ALLOANTIGEN SPECIFICITIES
HPA-15 HPA-15a (Govb) 80% CD109 Tyr↔Ser703 NAIT, PTP
HPA-15b (Gova) 60% CD109
*Modified from Kroll.27 GP = glyprotein; NAIT = neonatal alloimmune thrombocytopenia; PTP = posttransfusion purpurpa.
Table 16-4. Alloantigenic Polymorphisms of Platelet Glycoproteins that Have Been Implicated in Alloimmune Syndromes*
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.
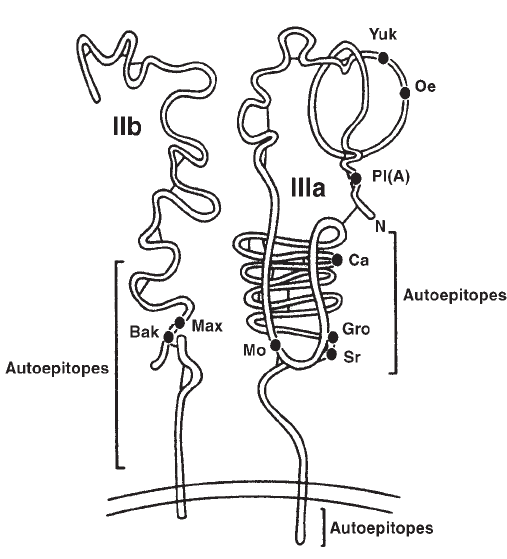
this glycoprotein and, therefore, do not ex-
press HPA-1 antigens. The HPA-1 polymor-
phism arises by the substitution of a single
base pair (leucine in HPA-1a and proline in
HPA-1b)ataminoacidposition33ofthe
protein’s DNA coding sequence. GPIIIa is
also the carrier of HPA-4, -6, -7, -8, -10, -11,
-14, and -16 antigens. Alleles in each of
these systems also arise as a result of single
amino acid substitutions at different posi-
tions. The HPA-2 antigen system is situated
on GPIb alpha; the HPA-3 system on GPIIb;
and the HPA-5 system on GPIa.36,37
On the platelet membrane, most of the
glycoproteins that carry these “platelet-spe-
cific” antigens are present as heterodimeric
compounds, ie, each consists of two differ-
ent glycoprotein molecules (see Fig 16-138).
Therefore, platelet glycoprotein names are
often paired (eg, Ia/IIa, IIb/IIIa, or Ib/IX),
referring to the alpha and beta chains in
each complex. GPIb/IX is a leucine-rich
membrane glycoprotein that serves as a re-
ceptor for von Willebrand factor on plate-
lets. The Ia/IIa and IIb/IIIa complexes are
members of a broadly distributed family of
adhesion molecules called integrins. Inte-
grins are essential for platelet adhesion and
aggregation because the molecules serve as
receptors for ligands such as fibrinogen
(IIb/IIIa), von Willebrand factor (Ib/IX), and
collagen (Ia/IIa). When present on other
cells, the glycoprotein pairings may differ.
For example, on platelets, GPIIIa is nor-
mally paired with GPIIb. On endothelial
cells,fibroblasts,andsmoothmuscle,how
-
ever, GPIIIa is paired with a different
glycoprotein. Thus, these cells share the
Chapter 16: Platelet and Granulocyte Antigens and Antibodies 369
Figure 16-1. Schematic diagram of platelet glycoprotein complex IIb/IIIa. Dots and letters (Yuk, Oe,
Pla, Ca, Gro, Sr, Mo, Bak, Max) designate positions and names of recognized allotypic epitopes. The
molecular regions where autoepitopes have been recognized are indicated by brackets.38
Copyright © 2005 by the AABB. All rights reserved.
HPA alloantigens found on the GPIIIa mol-
ecule, but not those found on the GPIIb
molecule.
CD109 is an exception to the hetero-
dimeric rule, occurring as a monomeric
structure on the platelet membrane. This
GPI-linked protein is found on activated T
cells, cultured endothelial cells, several tu-
mor cell lines, as well as platelets.39 Two
alloantigens designated HPA-15wb (Gova)
and HPA-15wa (Govb) have been localized
to platelet CD109.39 Unlike most platelet-
specific alloantigens, both alleles are highly
expressed [0.53% (Govb) and 0.47% (Gova)]
in persons of European ethnicity (Table
16-4). Sensitization to Gov alloantigens has
been associated with platelet refractoriness,
NAIT, and PTP, albeit usually together with
other alloantibodies to platelet antigens.40
Clinical Importance of Platelet-Specific
Antigens and Antibodies
Neonatal Alloimmune Thrombocytopenia
Neonatal alloimmune thrombocytopenia
(variously abbreviated NAIT, NATP, etc) is
described in Chapter 23.
Posttransfusion Purpura
Posttransfusion purpura (PTP) is charac-
terized by the development of dramatic,
sudden, and self-limiting thrombocytopenia
5 to 10 days after a blood transfusion in a
patient with a history of sensitization by
pregnancy or transfusion. Coincident with
the thrombocytopenia is the development
of a potent platelet-specific alloantibody
in the patient’s serum, usually anti-HPA-
1a. Other specificities have been implicated,
almost always associated with antigens
on GPIIb/IIIa.37,41 PTP differs from transfu-
sion reactions caused by red cell antibod-
ies because the patient’s own antigen-
negative (usually HPA-1a-negative) plate-
lets as well as any transfused antigen-pos-
itive platelets (which may be accompa-
nied by clinically severe reactions) are de-
stroyed. Transfusion of antigen-negative
platelets may be of value during the acute
phaseofPTP;however,suchplatelets
have a reduced in-vivo survival.42 Plasma-
pheresis—once the treatment of
choice—has largely been supplanted by
the use of intravenous immune globulin
(IGIV). The mechanism by which these
treatments are efficacious is unknown.
Platelet antibody assays usually reveal a
serum antibody with HPA-1a specificity.
Typing of the patient’s platelets after re-
covery will document a HPA-1a-negative
phenotype or analogous typing for other
platelet-specific antigen systems. Follow-
ing recovery, future transfusions should
be provided using washed antigen-nega-
tive RBC units if possible. Washed RBC
units may offer some protection against
recurrence, although at least one case of
PTPcausedbyantibodytoHPA-5bwas
precipitated by transfusion of a washed
RBC unit.43
Testing for Platelet-Specific Antigens and
Antibodies
Clinically useful platelet antibody assays
emerged later than serologic assays to di-
agnose immunologic disorders involving
redcells.Thisismainlybecauseitisdiffi
-
cult to separate platelets from whole blood
specimens and to distinguish antibody-
dependent endpoints from nonspecific
changes that occur in platelets under as-
say conditions. Three types of platelet an-
tibody detection methods have been de-
veloped44 (Table 16-5). The earliest were
Phase I assays that involved mixing pa-
tient serum with normal platelets and
used platelet function-dependent end-
points such as alpha granule release, ag-
gregation, or agglutination. Phase II tests
measured either surface or total platelet-
associated immunoglobulin on patient
370 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

platelets or on normal platelets after sen-
sitization with patient serum. Phase III
solid-phase assays were developed in
which the binding of antibodies to iso-
lated platelet surface glycoproteins is de-
tected. The test methods are examples of
Phase I, II, and III assays; variations of each
test method are also used. Lymphocyto-
toxicity tests are discussed in Chapter 17.
Mixed Passive Hemagglutination Assay
(MPHA). A Phase II assay used for the de-
tection of platelet-specific antibodies as
well as for platelet crossmatching is the
MPHA. Shibata et al were the first to use
this method to detect and identify clinically
significant platelet alloantibodies.45 Amodi
-
fication of MPHA, the SPRCA, is widely
used.18 In this assay, intact platelets are im-
mobilized in the round-bottom wells of a
microtiter tray and are sensitized with anti-
body to be detected. After washing, detec-
tor red cells previously coated with an anti-
body specific for human immunoglobulin
are added. After incubation from several
hours to overnight, the tray is subjected to a
slow centrifugation and examined visually.
If antibody is bound to the immobilized
platelets, the indicator red cells fail to form
a compact button in the center of the well
because they are evenly distributed like a
Chapter 16: Platelet and Granulocyte Antigens and Antibodies 371
Table 16-5. Platelet Antibody Assays
Phase I Assays
Platelet aggregation
Inhibition of platelet aggregation
Inhibition of clot retraction
Inhibition of platelet migration
Complement fixation
Platelet factor 3 release
51Chromium release
14C-Serotonin release
Phase II Assays
Detection of platelet surface-associated immunoglobulin
■Platelet suspension immunofluorescence test (PSIFT)
■Flow cytometry
■Radioimmunoassay (125I staphylococcal Protein A,125I antihuman immunoglobulin, polyclonal or
monoclonal)
■Antiglobulin consumption (two-stage assay)
■Solid-phase red cell adherence
Detection of total platelet-associated immunoglobulin
■Nephelometry
■Electroimmunoassay
■Radial immunodiffusion
Phase III Assays
Monoclonal antibody immobilization of platelet antigens (MAIPA)
Antigen capture ELISA (ACE)
Modified antigen capture ELISA (MACE)
Immunobead assay
Immunoblotting
Copyright © 2005 by the AABB. All rights reserved.
“carpet” over the antibody-coated platelets.
In a negative reaction, a red cell button forms
in the center of the well. A limitation of the
MPHA assay is that it fails to distinguish
platelet-specific from non-platelet-specific
antibodies. A modification of the MPHA as-
say, the Capture-P (Imucor Gamma, Norcross,
GA), is available as a commercial kit and is
most often marketed for platelet cross-
matching.18 SPRCA testing may be modified
by treatment of target platelets with chloro-
quine or acid,46,47 which disrupts the Class I
HLA heavy chain-peptide-β2-microglobulin
trimolecular complex. This modifies anti-
genic epitopes, reducing the binding of
specific antibodies directed against HLA on
platelets. However, strong HLA antibodies
may still bind, giving the impression that
the antibody is directed to non-HLA anti-
gens.
Flow Cytometry. Another example of a
Phase II assay is platelet antibody detection
using immunofluorescence. Originally a
slide-based method,48 thetechniquenow
uses flow cytometry to detect platelet-reac-
tive antibody in patient sera that binds to
intact platelets.49 In the assay, washed plate-
lets are sensitized with patient or control
serum for up to 60 minutes, usually at room
temperature. The platelets are then washed
repeatedly to remove nonspecific immuno-
globulins, and platelet-bound antibodies
are detected with a fluorescent-labeled
(usually fluorescein isothiocyanate, FITC)
polyclonal or monoclonal antibody specific
for human immunoglobulin. The platelets
are analyzed in the flow cytometer and re-
sults can be expressed as a ratio of the
mean or peak channel fluorescence of nor-
mal platelets sensitized with patient serum
over that of normal platelets incubated in
normal serum. In order to prevent nonspe-
cific binding of the immunoglobulin probe
via Fc receptors on the target platelets, the
probe antibodies are enzyme treated to re-
move the Fc end of the molecule. There-
fore, binding of the labeled probe can be
assumed to be via its F(ab’)2or antigen-spe-
cific end. A second fluorescent label [eg,
phycoerythrin (PE)] can be attached to an
antihuman IgM probe to detect IgM plate-
let antibodies. Because FITC and PE fluo-
resce with peak light intensities at different
wavelengths when exposed to the mono-
chromatic argon laser in the flow cytometer
(520-nm green light and 580-nm reddish-
orange light, respectively), cells labeled
with FITC can be distinguished from those
labeled with PE. Both anti-IgG and anti-
IgM labeled with different fluorochromes
can be added to the same tube with washed
sensitized platelets for the simultaneous
detection of antiplatelet IgG and IgM.
Flow cytometry has proven to be a very
sensitive method for detection of alloanti-
bodies. The assay is capable of detecting
very small numbers of antibody molecules
bound to platelets as is the case with allo-
antibodies specific for antigens of the
HPA-5 (Br) system having only 1000 to 2000
sites per platelet. Moreover, some alloanti-
bodies that are specific for labile epitopes
that are unreliably detected in Phase III as-
says can be detected on intact platelets
using flow cytometry.
Because the target platelets used in the
assay are intact, flow cytometry does not
differentiate between platelet-specific (ie,
platelet glycoprotein directed) and non-
platelet-specific antibodies. Examples of
the latter are HLA and ABO antibodies. This
is an advantage when the method is used to
detect antibodies that will affect the success
of a platelet transfusion, and, for this rea-
son, flow cytometry has been advocated as
a platelet crossmatching method. However,
when used to investigate cases of suspected
NAIT or PTP, the method has a potential
drawback—the more relevant platelet-spe-
cific antibodies characteristic in these dis-
eases can be obscured by non-platelet-spe-
cific reactivity.
372 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Monoclonal Antibody-Specific Immobi-
lization of Platelet Antigen (MAIPA). An ex-
ample of a Phase III assay is the MAIPA,50-52
perhaps the most widely used assay to de-
tect platelet-specific antibodies. The assay
requires the use of murine monoclonal an-
tibodies (MoAbs) that recognize the target
antigens of interest but do not compete
with the human antibody being detected.
In the assay, target platelets are simulta-
neously sensitized with patient serum and
a murine MoAb recognizing the desired tar-
get molecule on the platelet surface. After
the initial sensitization step, platelets are
washed and solubilized in a nonionic de-
tergent. After centrifugation to remove
cytoskeletal fragments, an aliquot of the
supernatant lysate is added to wells of a
microtiter tray containing immobilized
goat antibody specific for mouse IgG. The
MoAb is thereby captured and the platelet
surface glycoprotein with its bound human
antibody is immobilized. After a wash step,
the human antibody is detected with an
enzyme-labeled goat antihuman immuno-
globulin probe.
There are several other versions of Phase
III assays in use today, including the anti-
gen capture enzyme-linked immunosorbent
assay (ACE), the modified antigen capture
ELISA (MACE),53,54 and the commercially
available GTI PAKPLUS55 (GTI, Waukesha,
WI). Each relies on MoAbs to immobilize
only the glycoproteins of interest, thereby
reducing or eliminating interfering reactions
due to non-platelet-specific antibodies, es-
peciallyanti-HLA,which,ifpresent,isde
-
tected only in wells containing pools of im-
mobilized HLA Class I antigens.
Platelet Typing Using Molecular Meth-
ods. Molecular typing by polymerase chain
reaction (PCR) is available for many platelet
antigens. Because immunophenotyping is
limited by the shortage of characterized
typing antisera and by low platelet counts,
several DNA-based HPA typing techniques,
such as restriction fragment length poly-
morphism (RFLP) analysis and sequence-
specific oligonucleotide hybridization, have
been developed.56,57 All of these techniques
are reliable, but they are also laborious and
time-consuming. For this reason, PCR
genotyping with sequence-specific primers
(SSP) appears to be much more practical to
use.58,59 In a recent workshop, SSP-PCR was
the most common and reliable method of
determining platelet antigens,60 making it
feasible for genotyping HPAs independent
of the patient’s platelet count and of rare
typing sera.61
Autoimmune Platelet Disorders
Idiopathic (Autoimmune)
Thrombocytopenic Purpura
Autoantibodies directed against platelet
antigens may result in thrombocytopenia.
Chronic idiopathic thrombocytopenic
purpura (ITP), most often a disease in
adults, is characterized by an insidious
onset and moderate thrombocytopenia
that may exist for months to years before
diagnosis. Females are twice as likely to
be affected as males. Spontaneous remis-
sions are rare, and treatment is usually
required to raise the platelet count. First-
line therapy consists of steroids, high-
dose IGIV or Rh immunoglobulin (RhIG),
followed by splenectomy in nonresponders.
Many other therapies have been used in
patients who fail to respond to splenec-
tomy. Results have varied. Chronic auto-
immune thrombocytopenia may be idio-
pathic or associated with other diseases
(eg, HIV infection, malignancy, other au-
toimmune conditions). Acute ITP is mainly
a childhood disease characterized by abrupt
onset of severe thrombocytopenia and
bleeding symptomatology, often after a
viral infection. The majority of cases re-
solve spontaneously over a 2- to 6-month
Chapter 16: Platelet and Granulocyte Antigens and Antibodies 373
Copyright © 2005 by the AABB. All rights reserved.
period. If treatment is required, IGIV or
RhIG infusions are usually effective in
raising the platelet count. Steroids are
used less often because of serious side ef-
fects in children. Splenectomy, if used, is
reserved for those children whose disease
is severe and lasts longer than 6 months,
similar to chronic ITP in adults.
Testing for Platelet Autoantibodies
Numerous Phase I, II, and III platelet anti-
body assays have been developed to de-
tect relevant autoantibodies in ITP pa-
tients. Although many tests have been
demonstrated to be quite sensitive, par-
ticularly in detecting total or cell surface
platelet-associated immunoglobulins (Phase
II assays),62 none has been sufficiently
specific to be particularly useful in either
the diagnosis or management of ITP. The
American Society of Hematology’s prac-
tice guidelines for ITP state that serologic
testing is unnecessary, assuming the clini-
cal findings are compatible with the diag-
nosis.63 However, platelet antibody tests
may be helpful in the evaluation of patients
suspected of having ITP when other, non-
immune causes may be present.
The goal of serologic testing in ITP is to
detect autoantibody bound to the patient’s
own platelets with or without demonstra-
tion of similar reactivity in the patient’s
plasma. Most of the newer assays offered
for evaluation of patients suspected of hav-
ing ITP are Phase III assays, designed to de-
tect immunoglobulin binding to platelet-
specific epitopes found on platelet glyco-
protein complexes GPIIb/IIIa, GPIa/IIa,
and/or GPIb/IX.
These solid-phase GP-specific assays ap-
pear to have improved specificity in distin-
guishing ITP from nonimmune thrombo-
cytopenia when compared to Phase II assays,
but this is often balanced by a decrease in
sensitivity.64 Moreover, all of these methods
have limited usefulness in patients who
have very low platelet counts that prevent
adequate numbers of platelets to be collected
for use in the tests.
One commercially available Phase III
test, the GTI PAKAUTO,65 uses eluates pre-
pared from washed patient platelets. The
eluates are tested against a panel of MoAb-
immobilized platelet GP complexes, and
antibody binding is detected using an en-
zyme-linked antihuman immunoglobulin
probe. In the indirect phase of the assay,
patient plasma is tested against the same
glycoprotein panel. In general, plasma anti-
bodies are detected less often than anti-
bodies in the eluates. ITP patients may
have antibodies that are reactive with one
or several GP targets. To date, there is no
correlation between the specificity of auto-
antibodies in ITP and disease severity.
Drug-Induced Immune Platelet Disorders
Thrombocytopenia associated with specific
drugs is not uncommon. Drugs often im-
plicated include quinidine/quinine, sulfa
drugs, heparin, and colloidal gold. Both
drug-dependent and drug-independent
antibodies may be produced. Drug-inde-
pendent antibodies, although stimulated
by drugs, do not require the continued
presence of the drug to react with plate-
lets and are serologically indistinguish-
able from other platelet autoantibodies.
(Unlike typical autoimmune thrombocyto-
penia, these antibodies are transient ex-
cept when caused by therapy with gold,
which is excreted very slowly.) Drug-de-
pendent antibodies result when a drug
combines with platelets in such a way as to
create neoantigens to which antibodies are
formed. The drug must be present for the
antibody to react. These antibodies can
cause a thrombocytopenia of sudden and
rapid onset, usually resolving when the
drug is discontinued.
374 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Testing for Drug-Dependent Platelet-
Reactive Antibodies
Serology. Virtually any platelet serology
test that is used to detect platelet-bound
immunoglobulin can be modified for use
in the detection of drug-dependent pla-
telet-reactive antibodies. In performing
drug-dependent antibody testing, it is es-
sential to establish the proper positive
and negative controls for the assay. Each
serum or plasma sample suspected of con-
taining drug-dependent antibody must be
tested against normal target platelets in
thepresenceandabsenceofdrug.More
-
over, at least one normal serum should be
tested with and without drug to control
for any possible drug-related platelet ef-
fect that does not require specific anti-
body. Finally, a positive control serum
known to be reactive with the drug being
assayedshouldbetestedwithandwith-
out drug to complete the evaluation. A
positive result must show that the serum
is positive against normal target platelets
inthepresenceofdrugandnotwithout
drug, and that the drug did not non-
specifically cause a positive result in the
target platelet. Likewise, the positive con-
trol must be positive with the drug and
negative without it.
Flow Cytometry. The flow cytometry test
can be readily adapted to detect both IgG
and IgM drug-dependent platelet antibod-
ies.53,66 In this modification, fluorescence of
normal platelets sensitized with the pa-
tient’s serum in the presence of drug can be
compared with that of the patient’s sample
without drug or to a normal serum with
drug to determine relative intensity of la-
beling. Flow cytometry has proven to have
superior sensitivity to other assays for de-
tection of quinine-quinidine- and sulfona-
mide-dependent platelet-reactive antibod-
ies.53 Table 16-6 shows other agents for
which drug-dependent platelet-reactive an-
tibodies have been detected and confirmed
by flow cytometry in a large platelet immu-
nology reference laboratory.
Flow cytometry has its limitations, as do
other antibody detection methods, in de-
tecting drug-dependent antibodies. For
many drugs, the optimal concentration to
demonstrate in-vitro binding of antibody
has not been determined. Probably the
most extensively studied drugs in this re-
gard are quinine or quinidine.67 Another
cause of poor sensitivity is the weak bind-
ing of drug to platelets, leading to rapidly
declining numbers of drug molecules on
the platelet surface once drug is removed
from the environment of the platelet. It is
therefore important to maintain a critical
concentration of drug in all washing buffers
before addition of probe at the end of an
assay.66 Yet another potential reason for in-
sensitivity is that a patient may not be sen-
sitized to the native drug but, rather, to a
metabolite of the drug. Antibodies depend-
ent on metabolites of acetaminophen and
sulfamethoxazole have been reported.68,69
A number of other assays have been
adopted for the detection of drug-depend-
ent platelet antibodies. Among these are
the SPRCA.70,71 Phase I assays have also been
modified for detection of heparin-depend-
ent platelet antibodies (see below). In some
cases, determination of the specific glyco-
protein to which antibody is directed by
Phase III assays may provide useful clinical
information. For example, drug-dependent
antibody to GPIb/IX was associated with a
more acute, but reversible, quinine-in-
duced thrombocytopenia, whereas anti-
body to GPIIb/IIIa was associated with a more
prolonged course.72
Heparin-Induced Thrombocytopenia
Two types of heparin-induced thrombo-
cytopenia (HIT) have been recognized.
Type I, of nonimmune origin, presents with
Chapter 16: Platelet and Granulocyte Antigens and Antibodies 375
Copyright © 2005 by the AABB. All rights reserved.
mild transient thrombocytopenia within
minutes to several days after heparin ex-
posure but generally resolves despite on-
going heparin therapy and is not clinically
important. In contrast, immune, or Type
II, HIT may lead to life- and limb-threat-
ening thrombotic complications and requires
careful evaluation and management of af-
flicted patients.
The exact incidence of immune HIT is
unknown,butitmaydevelopinupto3%of
patients treated with unfractionated hepa-
rin. Low-molecular-weight heparin is less
likely to be associated with either antibody
production or thrombocytopenia.73 Bovine
heparin appears somewhat more likely to
causeHITthanporcineheparin.
74 Areduc
-
tion in baseline platelet count by at least
50% occurs generally within 5 to 14 days af-
ter primary exposure and sooner after sec-
ondary exposure to the drug. The platelet
count is often less than 100,000/µLand
usually recovers within 5 to 7 days upon
discontinuation of heparin.
About 30% of patients with HIT, or ap-
proximately 0.9% of patients who receive
heparin, develop thrombosis, which can
occur in the arterial, venous, or both sys-
tems.75,76 Patients may develop cardiovascu-
lar problems, myocardial infarction, limb
ischemia, deep venous thrombosis, or
ischemia of other organs. The thrombotic
complications may force limb amputation
or may prove fatal.
Thrombosis or an unexplained decrease
in platelet count while on heparin therapy
should raise concern about HIT. Heparin,
including heparin flushes and heparin-
coated catheters, should be discontinued,
and the patient should be evaluated for
laboratory evidence of HIT and signs of
thrombosis. In mild-to-moderate thrombo-
cytopenia, monitoring of platelet counts
and observation may be sufficient, but be-
cause of the high risk of thrombosis, treat-
ment with alternative anticoagulants is
generally recommended. Warfarin should
be avoided in the early treatment of HIT
because it does not prevent thrombosis in
this setting and may provoke limb-threat-
ening venous gangrene by reducing levels
of naturally occurring anticoagulants faster
than it reduces activated coagulation fac-
tors. However, warfarin can and should be
used after the patient is anticoagulated
with alternative drugs. Suitable anticoagu-
lants approved by the Food and Drug Ad-
ministration (FDA) include hirudin (a natu-
ral thrombin inhibitor) and argatroban.
Selected patients may also benefit from
376 AABB Technical Manual
Table 16-6. Drugs Confirmed to Elicit Drug-Dependent Platelet-Reactive Antibodies
In Vitro Using Flow Cytometry Testing
abciximab
acetaminophen
carbamazepine
ceftazidime
ceftizoxime
ceftriaxone
ciprofloxacin
eptifibatide
esomeprazole
fentanyl
fexofenadine
heparin
ibuprofen
levofloxacin
loracarbef
naproxen
orbofiban
oxaliplatin
phenytoin
propoxyphene
quinidine
quinine
ranitidine
rifampin
sulfamethoxazole
sulfisoxazole
suramin
tirofiban
trimethoprim
vancomycin
xemilofiban
Copyright © 2005 by the AABB. All rights reserved.
thrombolectomy or thrombolytic therapy.
Platelet transfusion should be avoided,
given that bleeding is a rare complication in
HIT, and administration of platelets may
precipitate thrombosis.77-79
Heparin forms a complex with platelet
factor 4 (PF4), a tetrameric protein released
from platelet alpha granules. Antibodies
(IgG, IgM, and some IgA) form against vari-
ous epitopes on this complex and attach to
platelet FcγIIa receptors, whereby platelets
become activated. The antibody may also
bind to the complexes at other sites, nota-
bly on endothelial cells. Thus, HIT might
involve activation and damage not only of
platelets but also of endothelium, causing
increased susceptibility to thrombosis. This
new understanding of the mechanism of
heparin antibodies is exploited by ELISA
tests in which microwells are coated with
the complexes rather than with the plate-
lets themselves.80
Testing for Heparin-Dependent Antibodies
The PF4 ELISA is an example of a Phase
III assay for HIT. Target complexes of PF4
and heparin or heparin-like molecules are
immobilized on a solid phase. To perform
the test, patient serum is added to premade
complexes of PF4 and heparin or hepa-
rin-like molecules (eg, polyvinyl sulfate,
PVS) alone and in the presence of high-
dose (100 U/mL) heparin. Heparin-de-
pendent antibody binds to the complexes
and is detected via enzyme-conjugated
antihuman immunoglobulin. An optical
density value above 0.4 in the PF4-PVS
well that is inhibited by high-dose hepa-
rin confirms the presence of a heparin-
dependent antibody in the patient’s sam-
ple. Although IgG antibodies are the most
clinically relevant antibodies causing this
syndrome,81 occasional patients with HIT
appear to have only non-IgG (IgM or IgA)
antibodies.82 The PF4 ELISA detects but
does not differentiate IgG, IgM, and IgA
antibodies that bind to the PF4-heparin
complex.
The 14C-serotonin release assay (SRA) is
an example of a Phase I assay for detection
of heparin-dependent antibodies.83 Normal,
fresh target platelets are incubated with
14C-serotonin that is taken up into the
dense granules of the platelets. Then, target
platelets are exposed to patient serum in
thepresenceoflowandhighconcentra
-
tions of heparin. Release of at least 20% of
the radioactive label at the low dose of hep-
arin and inhibition of this release at the
high dose confirms the presence of hepa-
rin-dependent antibodies. Other functional
tests used to detect heparin-dependent an-
tibodies include the heparin-induced
platelet aggregation test and the heparin-
induced platelet activation test.
ThePF4ELISAandtheSRAareboth
more sensitive and specific than the plate-
let aggregation test for the detection of hep-
arin-dependent platelet antibodies in pa-
tients for whom there is clinical suspicion
of HIT.83-85 However, in asymptomatic pa-
tients receiving heparin or in those who
have not yet received the drug, neither test
is sufficiently predictive of HIT to warrant
its use in screening.
Granulocyte Antigens
Analogous to platelet alloantigens, neu-
trophil alloantigens are implicated in
clinical syndromes including neonatal
alloimmune neutropenia (NAN), transfu-
sion-related acute lung injury (TRALI),
immune neutropenia after marrow trans-
plantation, refractoriness to granulocyte
transfusion, and chronic benign autoim-
mune neutropenia of infancy. A neutrophil
equivalent of PTP has not been described.
To date, seven neutrophil alloantigens have
been described, including localization to
Chapter 16: Platelet and Granulocyte Antigens and Antibodies 377
Copyright © 2005 by the AABB. All rights reserved.

neutrophil surface glycoprotein structures
and, in some cases, determination of DNA
polymorphisms in genes encoding for them
(see Table 16-7).
The first granulocyte-specific antigen,
NA1 (HNA-1a), was described in 1966 by
Lalezari and Bernard.86 HNA-1a and its anti-
thetical antigen, HNA-1b, are present on
FcγRIIIb. Antibodies to HNA-1a and -1b
have been implicated in TRALI, NAN, and
autoimmune neutropenia of infancy. About
0.1% of individuals of European ethnicity
have neutrophils with no detectable
FcγRIIIb (NAnull). The FcγRIII protein also
carries the neutrophil alloantigen SH
(HNA-1c).87 NB1 (HNA-2a) is found on an-
other granulocyte surface glycoprotein,
CD177, the function of which is still unde-
termined. HNA-2a has been reported to
have an allele, NB2, but the product of this
gene cannot be reliably identified with
alloantisera, and no MoAb specific for NB2
has been identified; therefore, the existence
of a second allele to HNA-2a (NB1)isun
-
proven.88 HNA-2a has been associated with
TRALI and NAN. The DNA sequence of the
NB1(HNA-2a) gene has been determined as
have the molecular polymorphisms associ-
ated with HNA-1a and HNA-1b and HNA-1c
on the gene for FcγRIII. Therefore, genotyp-
ing for these specificities can be performed
on genomic DNA using PCR-SSP.89 Reduced
expression of granulocyte antigens occurs in
paroxysmal nocturnal hemoglobinuria,
chronic myelogenous leukemia, and in pre-
mature infants.
Additional antigens on granulocytes are
shared with other cells and are not granulo-
cyte-specific. These include 5b (HNA-3a),
MARTa(HNA-4a), and ONDa(HNA-5a). The
HNA-3a is located on a 70- to 95-Kd protein
that has not yet been cloned. HNA-3a is also
expressed on the surface of lymphocytes.
The antibodies directed at this antigen are
378 AABB Technical Manual
Table 16-7. Neutrophil Alloantigens
Antigen Frequency (%)
Antigen
System Allele
HNA
Designation White Black
Glycoprotein
Location
Neutrophil-specific
NA
NA1
HNA-1a 46 46 FcγRIIIb (CD16)
NA
NA2
HNA-1b 88 84 FcγRIIIb (CD16)
SH
SH
HNA-1c 5 22 FcγRIIIb (CD16)
NB
NB1
HNA-2a 97 CD177
Neutrophil-nonspecific
5
5b
HNA-3a 97 70-95 kD GP
MART
MART
a
HNA-4a 99 CD11b
OND
OND
a
HNA-5a 99 CD11a
Copyright © 2005 by the AABB. All rights reserved.
usually agglutinins; they occasionally occur
in women after pregnancy and may be as-
sociated with febrile transfusion reactions.
Potent anti-HNA-3a agglutinins in trans-
fused plasma have been responsible for fa-
tal TRALI.90-92 MARTaand ONDa,bothhigh-
incidence antigens, are also present on
monocytes and lymphocytes. MARTahas
been localized to the alpha M chain
(CD11b) of the C3bi receptor (CR3) and re-
sults from a single nucleic acid substitu-
tion. MARTahas been recently reported to
cause NAN.93 ONDais expressed on the al-
pha L integrin unit, leukocyte function an-
tigen-1 (CD11a) and also results from a sin-
gle nucleotide substitution. This marker,
found in a chronically transfused aplastic
anemia patient, has not been reported to
be associated with clinical disease.
Clinical Syndromes in Which
Granulocyte-Specific Alloantigens Are
Implicated
Nonhemolytic, febrile transfusion reac-
tions are often associated with granulo-
cyte antibodies. Although such reactions
are more often caused by antibodies to
Class I HLA antibodies directed at Class I
epitopes present on granulocytes, granu-
locyte-specific antibodies have been asso-
ciated with clinical syndromes similar to
those seen with antibodies to red cell and
platelet antigens.
Neonatal Alloimmune Neutropenia
NAN is caused by maternal antibodies
against alloantigens of fetal neutrophils;
the most frequent specificities seen are
against NA1, NA2, and NB1 antigens (see
Table 16-7). NAN most often occurs in
women of the neutrophil alloantigen phe-
notypes NA1/NA1 and NA2/NA2; it may
also occur in women of the rare NAnull
phenotype who lack the FcγRIII protein.
The neutropenia in all these cases can oc-
casionally be life-threatening because of
increased susceptibility to infection. Man-
agement with antibiotics, IGIV, granulo-
cyte colony-stimulating growth factor,
and/or plasma exchange may be helpful.
TRALI
TRALI is an acute, often life-threatening
reaction characterized by respiratory dis-
tress, hypo- or hypertension, and non-
cardiogenic pulmonary edema that oc-
curs within 6 hours of a transfusion of a
plasma-containing blood component.
TRALI has been reported to be induced by
neutrophil antibodies, although more re-
cent reports are more likely to implicate
antibodies to both Class I and II HLA anti-
gens.94 In TRALI, the causative antibodies
are most often found in the plasma of the
blood donor (see Chapters 17 and 27). Re-
cent reports have postulated that another
etiology for TRALI is possible. This theory
suggests that two events must coincide
for TRALI to occur: 1) the patient must
have a predisposing clinical condition
that releases cytokines or other factors
that prime neutrophils, causing adher-
ence to endothelium, and 2) the patient
must receive a transfusion of biologically
active lipids (which also stimulate neutro-
phils) from stored blood components.95 In
one study, neutrophil antibodies were de-
tected in only three of 10 incidents of
TRALI, and none of the donors had HLA
antibodies.
Autoimmune neutropenia, usually oc-
curring in adults, may be idiopathic or may
occur secondary to such diseases as rheu-
matoid arthritis, systemic lupus erythe-
matosus, or bacterial infections. In autoim-
mune neutropenia of infancy, usually
occurring in children between the ages of 6
months to 2 years, the autoantibody has a
neutrophil antigen specificity (usually
HNA-1a and -1b) in about half the cases.
Chapter 16: Platelet and Granulocyte Antigens and Antibodies 379
Copyright © 2005 by the AABB. All rights reserved.
The condition is usually self-limiting (with
recovery usually in 7-24 months) and the
condition is relatively benign and manage-
able with antibiotics.96 Drug-dependent an-
tibodies can also cause neutropenia.
Testing for Granulocyte Autoantibodies
Tests for granulocyte antibodies are not
widely performed, although the implica-
tion of neutrophil antibodies as a cause of
TRALI has increased the demand for this
laboratory resource. Agglutination tests
performed in tube, capillary, or micro-
plate formats use heat-inactivated serum
inthepresenceofEDTAandrequirefresh
granulocytes. Immunofluorescence tests,
read with either a fluorescence micro-
scope or a flow cytometer, are also used
and are capable of detecting granulocyte-
bound antiglobulin. A combination of
agglutination and immunofluorescence
testsisbeneficial.
97 Other methods include
chemiluminescence and a MoAb-specific
immobilization of granulocyte antigens
(MAIGA) assay, similar to the MAIPA as-
say. An advantage of the MAIGA assay is
its ability to differentiate readily between
HLA and granulocyte-specific antibodies.
References
1. Ogasawara K, Ueki J, Takenaka M, Furihata K.
Study on the expression of ABO antigens on
platelets. Blood 1993;82:993-9.
2. Bialek J, Bodmer W, Bodmer J, Payne R. Dis-
tribution and quantity of leukocyte antigens
in the formed elements of blood. Transfusion
1966;6:193-204.
3. Feuerstein N, Mono DS, Cooper HL. Phorbo-
lester effect in platelets, lymphocytes, and
leukemic cells (HL-60) is associated with en-
hanced phosphorylation of Class I HLA anti-
gens. Biochem Biophys Res Commun 1985;
126:206-13.
4. Howard JA, Perkins HA. The natural history of
alloimmunization to platelets. Transfusion
1978;18:496-503.
5. Godeau B, Fromont P, Seror T, et al. Platelet
alloimmunization after multiple transfu-
sions: A prospective study of 50 patients. Br J
Haematol 1992;81:395-400.
6. Dzik WH. Leukoreduced blood components:
Laboratory and clinical aspects. In: Simon
TL, Dzik WH, Snyder EL, et al, eds. Rossi’s
principles of transfusion medicine. 3rd ed.
Philadelphia, PA: Lippincott Williams and
Wilkins, 2002:270-87.
7. The Trial to Reduce Alloimmunization to
Platelets Study Group. Leukocyte reduction
and ultraviolet B irradiation of platelets to
prevent alloimmunization and refractoriness
to platelet transfusions. N Engl J Med 1997;
337:1861-9.
8. Petz LD, Garratty G, Calhoun L, et al. Select-
ing donors of platelets for refractory patients
on the basis of HLA antibody specificity.
Transfusion 2000;40:1446-56.
9. Koerner TAW, Vo TL, Wacker KE, Strauss RG.
The predictive value of three definitions of
platelet transfusion refractoriness (abstract).
Transfusion 1988;28:33S.
10. Ishida A, Handa M, Wakui M, et al. Clinical
factors influencing post-transfusion platelet
increment in patients undergoing hema-
topoietic progenitor cell transplantation—a
prospective analysis. Transfusion 1998;38:
839-47.
11. Bishop J, McGrath K, Wolf M, et al. Clinical
factors influencing the efficacy of pooled
platelet transfusions. Blood 1988;71:383-7.
12. Moroff G, Garratty G, Heal JM, et al. Selection
of platelets for refractory patients by HLA
matching and prospective crossmatching.
Transfusion 1992;32:633-40.
13. Bolgiano DC, Larson EB, Slichter SJ. A model
to determine required pool size for HLA-
typed community donor apheresis programs.
Transfusion 1989;29:306-10.
14. Duquesnoy RJ, Filip DJ, Rodey G, et al. Suc-
cessful transfusion of platelets “mismatched”
for HLA antigens to alloimmunized thrombo-
cytopenic patients. Am J Hematol 1997;2:
219-26.
15. DahlkeMB,WeissKL.Platelettransfusions
from donors mismatched from cross-reactive
HLA antigens. Transfusion 1984;24:299-302.
16. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
17. RachelJM,SummersTC,SinorLT,etal.Use
of a solid phase red blood cell adherence
method for pretransfusion platelet compati-
bility testing. Am J Clin Pathol 1988;90:63-8.
18. Rachel JM, Sinor LT, Tawfik OW, et al. A solid-
phase red cell adherence test for platelet
crossmatching. Med Lab Sci 1985;42:194-5.
19. O’Connell BA, Schiffer CA. Donor selection
for alloimmunized patients by platelet cross-
380 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
matching of random-donor platelet concen-
trates. Transfusion 1990;20:314-7.
20. FriedbergRC,DonnellySF,MintzPD.Inde
-
pendent roles for platelet crossmatching and
HLA in the selection of platelets for allo-
immunized patients. Transfusion 1994;34:
215-20.
21. Duquesnoy RJ. HLA Matchmaker: A molecu-
larly based algorithm for histocompatibility
determination. I. Description of the Algo-
rithm. Hum Immunol 2002;63:339-52.
22. Gmür J, von Felten A, Osterwaider B, et al.
Delayed alloimmunization using random sin-
gle donor platelet transfusions: A prospective
study of thrombocytopenic patients with
acute leukemia. Blood 1983;62:473-9.
23. Dutcher JP, Schiffer CA, Aisner J, Wiernik PH.
Alloimmunization following platelet transfu-
sion: The absence of a dose-response rela-
tionship. Blood 1981;57:395-8.
24. Kruskall MS. The perils of platelet transfusions
(editorial). N Engl J Med 1997;337:1914-5.
25. Gebel HM, Bray RA. Sensitization and sensi-
tivity: Defining the unsensitized patient.
Transplantation 2000;69:1370-4.
26. MurphyMF,MetcalfeP,OrdJ,etal.Disap-
pearance of HLA and platelet-specific anti-
bodies in acute leukemia patients allo-
immunized by multiple transfusions. Br J
Haematol 1987;67:255-60.
27. Kroll H. Human platelet alloantigens (HPA).
[Available at http://www.uniklinikum-gies-
sen.de/immunologie/hkroll/HPAs.htm (accessed
January 2005).]
28. Newman PJ, Valetin N. Human platelet
alloantigens: Recent findings, new perspec-
tives. Thromb Haemost 1995;84:234-9
29. Greenwalt DE, Lipsky RH, Ockenhouse CF, et
al. Membrane glycoprotein CD36: A review of
its roles in adherence, signal transduction, and
transfusion medicine. Blood 1992;80:1105-15.
30. CurtisBR,AsterRH.IncidenceoftheNak(a)-
negative platelet phenotype in African Ameri-
cans is similar to that of Asians. Transfusion
1996;36:331-4.
31. Curtis GR, Ali S, Glazier AM, et al. Isoimmuni-
zation against CD36 (glycoprotein IV): De-
scription of four cases of neonatal isoimmune
thrombocytopenia and brief review of the lit-
erature. Transfusion 2002;42:1173-9.
32. Curtis BR, McFarland JG, Gottschall JL. Pla-
telet immunology and alloimmunization. In:
Simon TL, Dzik WH, Snyder EL, et al, eds.
Rossi’s principles of transfusion medicine.
3rd ed. Philadelphia, PA: Lippincott Williams
and Wilkins, 2002:203-18.
33. McFarland J. Nomenclature update: HLA,
HPA and RBCs. Platelet-specific antigen no-
menclature. In: The compendium: A selec-
tion of short topic presentations. Bethesda,
MD: AABB, 1997.
34. Metcalfe P, Watkins NA, Ouwehand WH, et al.
Nomenclature of human platelet antigens.
Vox Sang 2003;85:240-5.
35. van Loghem JJ, Dorfmeijer H, van der Hart M.
Serological and genetical studies on a platelet
antigen (Zw). Vox Sang 1959;4:161-9.
36. Aster RH. Platelet-specific alloantigen sys-
tems: History, clinical significance and mo-
lecular biology. In: Nance ST, ed. Allo-
immunity: 1993 and beyond. Bethesda, MD:
AABB, 1993:83-116.
37. vondemBorneAEGKr,SimsekS,vanderSchoot
S, et al. Platelet and neutrophil alloantigens:
Their nature and role in immune-mediated
cytopenias. In: Garratty G, ed. Immunobiology
of transfusion medicine. New York: Marcel
Dekker, 1994:149-71.
38. Müeller-Eckhardt C. Platelet autoimmunity.
In: Silberstein LE, ed. Autoimmune disorders
of blood. Bethesda, MD: AABB, 1996:115-50.
3 9 . S m i t h J W, H a y w a r d C P, H o r s e w o o d P, e t a l .
Characterization and localization of the Gova/b
alloantigens to the glycosylphosphatidylino-
sitol-anchored protein CDw109 on human
platelets. Blood 1995;86; 2807-14.
40. Berry JE, Murphy CM, Smith GA, et al. Detec-
tion of Gov system antibodies by MAIPA re-
veals an immunogenicity similar to the
HPA-5 alloantigens. Br J Haematol 2000;110:
735-42.
41. McFarland JG. Posttransfusion purpura. In:
Popovsky MA, ed. Transfusion reactions. 2nd
ed. Bethesda, MD: AABB Press, 2001:187-212.
42. Brecher ME, Moore SB, Letendre L. Post-
transfusion purpura: The therapeutic value of
PlA1-negative platelets. Transfusion 1990;30:
433-5.
43. ChristieD,PulkrabekS,PutnamJ,etal.Post
-
transfusion purpura due to an alloantibody
reactive with glycoprotein Ia/IIa (anti-HPA-
5b). Blood 1991;77:2785-9.
44. Sinha RK, Kelton JG. Current controversies
concerning the measurement of platelet as-
sociated IgG. Transfus Med Rev 1990;2:121-35.
45. ShibataY,JujiT,NishizawaY,etal.Detection
of platelet antibodies by a newly developed
mixed passive agglutination with platelets.
Vox Sang 1981;41:25-31.
46. Neumuller J, Tohidast-Akrad M, Fisher M,
Mayr WR. Influence of chloroquine or acid
treatment of human platelets on the antige-
nicity of HLA and the “thrombocyte-specific”
glycoproteins Ia/IIa, IIb, and IIb/IIIa. Vox
Sang 1993;65:223-31.
47. Gouttefangeas C, Diehl M, Keilholz W, et al.
Thrombocyte HLA molecules retain non-
renewable endogenous peptides of mega-
Chapter 16: Platelet and Granulocyte Antigens and Antibodies 381
Copyright © 2005 by the AABB. All rights reserved.
karyocyte lineage and do not stimulate direct
allocytotoxicity in vitro. Blood 2000;95:3168-
75.
48. von dem Borne AEGKr, Verheugt FWA,
Oosterhof F, et al. A simple immunofluores-
cence test for the detection of platelet anti-
bodies. Br J Haematol 1978;39:195-207.
49. Garratty G, Arndt P. Applications of flow
cytofluorometry to transfusion science.
Transfusion 1995;35:157-78.
50. Kiefel V, Santoso S, Weisheit M, Müeller-
Eckhardt C. Monoclonal antibody-specific
immobilization of platelet antigens (MAIPA):
A new tool for the identification of platelet-
reactive antibodies. Blood 1987;70:1722-6.
51. Kiefel V. The MAIPA assay and its applica-
tions in immunohematology. Transfus Med
1992;2:181-8.
52. Morel-KoppMC,DavietL,McGregorJ,etal.
Drawbacks of the MAIPA technique in char-
acterizing human anti-platelet antibodies.
Blood Coag Fibrinol 1996;7:144-6.
53. Visentin GP, Wolfmeyer K, Newman PJ, Aster
RH. Detection of drug-dependent, platelet-
reactive antibodies by antigen-capture ELISA
and flow cytometry. Transfusion 1990;30:694-
700.
54. Menitove JE, Pereira J, Hoffman R, et al. Cy-
clic thrombocytopenia of apparent autoim-
mune etiology. Blood 1989;73:1561-9.
55. GTI PAKPLUSTM platelet antibody screening
kit (package insert). Waukesha, WI: GTI, 1996.
56. McFarland JG, Aster RH, Bussel JB, et al. Pre-
natal diagnosis of neonatal alloimmune
thrombocytopenia using allele-specific oligo-
nucleotide probes. Blood 1991;78:2276-82.
57. Simsek S, Faber NM, Bleeker PM, et al. Deter-
mination of human platelet antigen frequen-
cies in the Dutch population by immuno-
phenotyping and DNA (allele-specific
restriction enzyme) analysis. Blood 1993;82:
835-40.
58. Skogen B, Bellissimo D, Hessner M, et al.
Rapid determination of platelet alloantigen
genotypes by polymerase chain reaction us-
ing allele-specific primers. Transfusion 1994;
34:955-60.
59. Kluter H, Fehlau K, Panzer S, et al. Rapid typ-
ing for human platelet antigen systems-1, -2,
-3 and -5 by PCR amplification with sequence-
specific primers. Vox Sang 1996;71: 121-5.
60. Panzer S. Report on the Tenth International
Platelet Genotyping and Serology Workshop
on behalf of the International Society of
Blood Transfusion. Vox Sang 2001;80:72-8.
61. Meyer O, Hildebrandt M, Schulz B, et al. Si-
multaneous genotyping of human platelet
antigens (HPA) 1 through 6 using new se-
quence-specific primers for HPA-5. Transfu-
sion 1999;39:1256-8.
62. George JN. Platelet immunoglobulin G: Its
significance for the evaluation of thrombo-
cytopenia and for understanding the origin of
α-granule proteins. Blood 1990;76:859-70.
63. George JN, Woolf SH, Raskob GE, et al. Idio-
pathic thrombocytopenic purpura: A practice
guideline developed by explicit methods for
the American Society of Hematology. Blood
1996;88:3-40.
64. Warner MN, Moore JC, Warkentin TE, et al. A
prospective study of protein-specific assays
used to investigate idiopathic thrombocyto-
penic purpura. Br J Haematol 1999;104:442-7.
65. GTI PAKAUTOTM ELISA screening test for auto-
antibodies to platelet glycoproteins IIb/IIIa,
Ib/IX and Ia/IIa (package insert). Waukesha,
WI: GTI, 2004.
66. CurtisBR,McFarlandJG,WuGG,etal.Anti
-
bodies in sulfonamide-induced immune
thrombocytopenia recognize calcium-de-
pendent epitopes on the glycoprotein IIb/IIIa
complex. Blood 1994;84:176-83.
67. Christie DJ, Aster RH. Drug-antibody-platelet
interaction in quinine- and quinidine-in-
duced thrombocytopenia. J Clin Immunol
1982;70:989-98.
68. Eisner EV, Shahidi NT. Immune thrombo-
cytopenia due to a drug metabolite. N Engl J
Med 1972;87:376-81.
69. Kiefel V, Santoso S, Schmidt S, et al. Metabo-
lite-specific IgG and drug-specific antibodies
IgG, IgM in two cases of trimethoprim-
sulfamethoxazole-induced immune throm-
bocytopenia. Transfusion 1987;27:262-5.
70. Sinor LT, Stone DL, Plapp FV, et al. Detection
of heparin-IgG immune complexes on plate-
lets by solid phase red cell adherence assays.
(Immucorrespondence) Norcross, GA: Immucor,
1990.
71. Leach MF, Cooper LK, AuBuchon JP. Detec-
tion of drug-dependent, platelet-reactive an-
tibodies by solid-phase red cell adherence as-
says. Br J Haematol 1997;97:755-61.
72. Nieminen U, Kekomäki R. Quinidine-induced
thrombocytopenia purpura: Clinical presen-
tation in relation to drug-dependent and
drug-independent platelet antibodies. Br J
Haematol 1992;80:77-82.
73. Warkentin TE, Levine M, Hirsh J, et al. Hepa-
rin-induced thrombocytopenia in patients
treated with low molecular weight heparin or
unfractionated heparin. N Engl J Med 1995;
332:1330-5.
74. Warkentin TE. Pork or beef? Ann Thorac Surg
2003;75:15-6.
382 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
75. Nand S, Wong W, Yuen B, et al. Heparin-in-
duced thrombocytopenia with thrombosis.
Am J Hematol 1997;56:12-6.
76. Warkentin TE, Kelton JG. A 14-year study of
heparin-induced thrombocytopenia. Am J
Med 1996;101:502-7.
77. Warkentin TE, Chong BA, Greinacher A. Hep-
arin-induced thrombocytopenia: Towards
consensus. Thromb Haemost 1998;79:1-7.
78. CancioLC,CohenDJ.Heparin-induced
thrombocytopenia thrombosis. J Am Coll
Surg 1998;186:76-91.
79. Gupta AK, Kovacs MJ, Sauder DN. Heparin-
induced thrombocytopenia. Ann Pharmacother
1998;32:55-9.
80. GTI-HATTM for the detection of heparin-as-
sociated antibodies (package insert). Waukesha,
WI: GTI, 1997.
81. Suh JS, Malik MI, Aster RH, Visentin GP. Char-
acterization of the humoral immune re-
sponse in heparin induced thrombocyto-
penia. Am J Hematol 1997;54:196-201.
82. Amiral J, Wolf M, Fischer A, et al. Pathogenic-
ity of IgA and/or IgM antibodies to hepa-
rin-PF4 complexes in patients with heparin
induced thrombocytopenia. Br J Haematol
1996;92:954-9.
83. Sheridan D, Carter C, Kelton JG. A diagnostic
test for heparin-induced thrombocytopenia.
Blood 1986;67:27-30.
84. Pouplard C, Amiral J, Borg JY, et al. Decision
analysis for use of platelet aggregation test,
carbon 14-serotonin release assay, and hepa-
rin platelet factor 4 enzyme-linked immuno-
sorbent assay for diagnosis of heparin-in-
duced thrombocytopenia. Am J Clin Pathol
1999;111:700-6.
85. Lindhoff-Last E, Gerdsen F, Ackermann H, et
al. Determination of heparin platelet factor 4
IgG antibodies improves diagnosis of hepa-
rin-induced thrombocytopenia. Br J Haema-
tol 2001;113:886-90.
86. Lalezari P, Bernard JE. An isologous antigen-
antibody reaction with human neutrophils
related to neonatal neutropenia. J Clin Invest
1966;45:1741-50.
87. Steffensen R, Gulen T, Varming K, Jersild C.
FcγRIIIb polymorphism: Evidence that NA1/
NA2 and SH are located in two closely linked
loci and that the SH allele is linked to the NA1
allele in the Danish population. Transfusion
1999;39:593-8.
88. Stroncek D. Neutrophil alloantigens. Transfus
Med Rev 2002;16:67-75.
89. Hessner MJ, Curtis BR, Endean DJ, Aster RH.
Determination of neutrophil antigen gene
frequencies in five ethnic groups by polymer-
ase chain reaction with sequence-specific
primers. Transfusion 1996;36:895-9.
90. Nordhagen R, Conradi M, Promtord SM. Pul-
monary reaction associated with transfusion
of plasma containing anti-5b. Vox Sang 1986;
51:102-8.
91. DavorenA,CurtisRBR,ShulmanIA,etal.
TRALI due to granulocyte-agglutinating hu-
man neutrophil antigen-3a (5b) alloanti-
bodies in donor plasma: A report of 2 fatali-
ties. Transfusion 2003;43:641-5.
92. Kopko PM, Marshall CS, MacKenzie MR, et al.
Transfusion-related acute lung injury: Report
of a clinical look-back investigation. JAMA
2002;287:1968-71.
93. FungUL,WillettJE,PitcherLA,etal.Con-
firming an alloimmune neonatal neutropenia
due to anti-HLA-4a (MART) by DNA charac-
terization. Presented at the 7th European
symposium on platelet, granulocyte and red
cell immunobiology, Lago Maggiore, Italy,
April 11-14, 2002.
94. Flesch BK, Neppert J. Transfusion-related
acute lung injury caused by human leukocyte
antigen Class II antibody. Br J Haematol
2002;116:673-6.
95. Silliman R, Paterson AJ, Dickey WO, et al. The
association of biologically active lipids with
the development of transfusion-related acute
lung injury: A retrospective study. Transfu-
sion 1997;37:719-26.
96. Bux J, Behrens G, Jaeger G, et al. Diagnosis
and clinical course of autoimmune neutro-
penia in infancy: Analysis of 240 cases. Blood
1998;91:81-6.
97. Bux J, Chapman J. Report on the second in-
ternational granulocyte serology workshop.
Transfusion 1997;37:977-83.
Chapter 16: Platelet and Granulocyte Antigens and Antibodies 383
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 17: The HLA System
Chapter 17
The HLA System
THE HLA SYSTEM includes a com-
plex array of genes and their pro-
tein products. HLA antigens con-
tribute to the recognition of self and
nonself, to the immune responses to anti-
genic stimuli, and to the coordination of
cellular and humoral immunity. The HLA
genes, which are located in the major
histocompatibility complex (MHC) on the
short arm of chromosome 6, code for
glycoprotein molecules found on cell sur-
face membranes. Class I molecules are
found on the surface of platelets and of all
nucleated cells of the body. Mature red
cells usually lack HLA antigens demon-
strable by conventional methods, but nu-
cleated immature erythroid cells express
them. MHC Class II antigens are restricted
to a few cell types; the most important are
B lymphocytes, macrophages, and den-
dritic cells. Other terms that have been
applied to antigens of the HLA system are:
major histocompatibility locus antigens,
transplantation antigens, and tissue anti-
gens.
The HLA antigen molecules play a key
role in antigen presentation. Immunologic
recognition of differences in HLA antigens
is probably the first step in the rejection of
transplanted tissue. The HLA system is sec-
ond in importance only to the ABO anti-
gens in influencing the long-term survival
of transplanted solid organs and is of para-
mount significance in hematopoietic pro-
genitor cell (HPC) transplantation. HLA an-
tigens and antibodies are also important in
such complications of transfusion therapy
as platelet refractoriness, febrile non-
hemolytic transfusion reactions (FNHTRs),
transfusion-related acute lung injury (TRALI),
and posttransplant and posttransfusion
graft-vs-host disease (GVHD).
Studies correlating HLA polymorphisms
with susceptibility and disease resistance
began soon after serologic techniques for
HLA Class I typing were developed. Histori-
cally, HLA antigen typing has been of value
in parentage testing and in forensic investi-
gations. Molecular analysis of the HLA re-
gion permits selection of more closely
385
17
Copyright © 2005 by the AABB. All rights reserved.
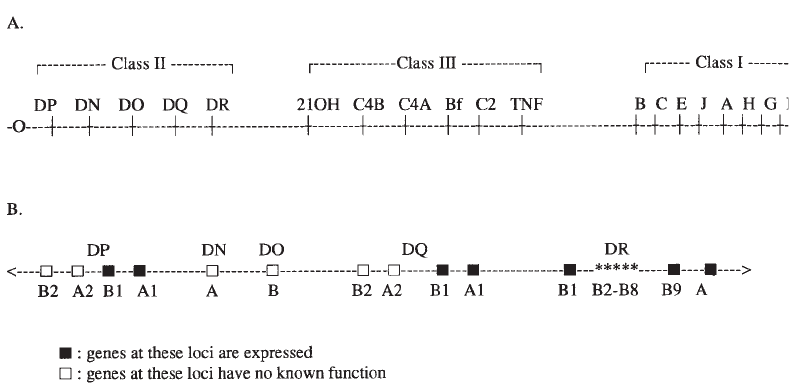
matched donors for HPC transplantation,
for investigation of disease associations,
and for anthropologic population studies.
Becauseofthepolymorphicnatureofthe
HLA genes, a complex nomenclature has
been developed to refer to the unique allele
sequences based on the relationship of the
allele to the serologic specificity of the cor-
responding antigen.1
Genetics of the Major
Histocompatibility Complex
Class I and II HLA antigens are cell sur-
face glycoproteins that are products of
closely linked genes mapped to the p21.3
band on the short arm of chromosome 6
(Fig 17-1). This genomic region is called
the MHC and is usually inherited en bloc
as a haplotype. Each of the several loci
has multiple alleles with codominant ex-
pression of the products from each chro-
mosome. The HLA system is the most
polymorphic system of genes described in
humans.
The HLA-A,HLA-B,andHLA-C genes
encode the corresponding Class I antigens
A, B, and C. The HLA-DR,HLA-DQ,and
HLA-DP gene cluster codes for the synthe-
sis of correspondingly named Class II anti-
gens. Located between the Class I and Class
II genes is a group of non-HLA genes that
code for molecules that include the com-
plement proteins C2, Bf, C4A, C4B; a ste-
roid enzyme (21-hydroxylase); and a
cytokine (tumor necrosis factor). This re-
gion is referred to as MHC Class III.
Organization of HLA Genetic Regions
The HLA Class I region contains, in addi-
tion to the classical genes HLA-A,HLA-B,
and HLA-C, other gene loci designated
HLA-E, HLA-F, HLA-G, HFE, HLA-J, HLA-K,
HLA-L, MICA,andMICAB. These latter
genes encode the nonclassical or Class Ib
HLA proteins, characterized by limited
polymorphism and low levels of expres-
386 AABB Technical Manual
Figure 17-1. (A) The major histocompatibility complex located on the short arm of chromosome 6. The
centromere is to the left. The key Class I, II, and III genetic loci are shown. The Class III region con-
tains complement system genes (C2, Bf, C4A, C4B), the 21-hydroxylase gene (21OH), and the gene for
tumor necrosis factor (TNF). (B) Greater detail of the Class II region.
Copyright © 2005 by the AABB. All rights reserved.
sion.2Some Class I genes express non-
functional proteins or are not able to ex-
press a protein. Genes unable to express a
functional protein product are termed
pseudogenes and presumably represent
an evolutionary dead end. HLA-E regu-
lates natural killer cells. HLA-G is ex-
pressed by the trophoblast and may be in-
volved in the development of maternal
immune tolerance of the fetus. Hereditary
hemochromatosis (HH), an iron overload
disorder with a 10% carrier frequency in
Northern Europeans, is associated with
two missense mutations in a Class I-like
gene.3ThegeneconferringHHwasini
-
tially named HLA-H; however, the HLA-H
designation had already been assigned to
an HLA Class I pseudogene by the World
Health Organization (WHO) Nomencla-
ture Committee.4ThegeneconferringHH
is now called HFE. Class I molecules are
also located outside the MHC, such as
CD1, which can present nonprotein anti-
gens (such as lipids) to T cells.
The genomic organization of the MHC
Class II region (HLA-D region) is more
complex. An MHC Class II molecule con-
sists of a noncovalent complex of two struc-
turally similar chains, the α-chain and the
β-chain. Both of these chains are encoded
within the MHC. The polymorphism of
HLA Class II molecules results from differ-
ences in both the α-chain and the β-chain;
this depends on the Class II isoform. For
example, with HLA-DR, the α-chain is
monomorphic, but the β-chain is very poly-
morphic. Multiple loci code for either alpha
or beta chains of the Class II MHC proteins.
Different haplotypes have different num-
bers of Class II genes and pseudogenes.
The proteins coded by the DRA gene and
the DRB1 gene result in HLA-DR1 through
HLA-DR18. The products of the Agene and
the B3 gene (if present) express HLA-DR52;
those of the Agene and the B4 gene (if pres-
ent) express HLA-DR53; and those of the A
gene and B5 gene (if present) express
HLA-DR51. The HLA-DQ1 through DQ9
antigens are expressed on the glycoproteins
coded by the DQA1 and DQB1 genes in the
DQ gene cluster. Many of the other genes of
the DQ cluster are probably pseudogenes. A
similar organization is found in the HLA-
DP gene cluster.
The MHC Class III region contains four
complement genes, whose alleles are gen-
erally inherited together as a unit, termed a
complotype.Therearemorethan10differ
-
ent complotypes inherited in humans. Two
of the Class III genes, C4A and C4B,code
for variants of the C4 molecule. These vari-
ants have distinct protein structure and
function; the C4A molecule (if present) car-
ries the Rodgers antigen and the C4B mole-
cule (if present) carries the Chido antigen,
both of which are adsorbed onto the red
cells of individuals who possess the gene.
Patterns of Inheritance
Although the organization of the MHC is
complicated, its inheritance follows the
established principles of genetics. Every
person has two different copies of chro-
mosome 6 and, thus, possesses two HLA
haplotypes, one from each parent. An in-
dividual’s haplotype is typically determined
by typing multiple family members from
different generations and observing which
alleles are inherited together. The ex-
pressed gene products constitute the phe-
notype, which can be determined for an
individual by typing for the HLA antigens.
Because the HLA genes are autosomal
and codominant, the phenotype repre-
sents the combined expression of both
haplotypes. Figure 17-2 illustrates inheri-
tance of haplotypes.
Finding HLA-Identical Siblings
Each child inherits one copy of chromo-
some 6 from each parent; hence, one MHC
Chapter 17: The HLA System 387
Copyright © 2005 by the AABB. All rights reserved.
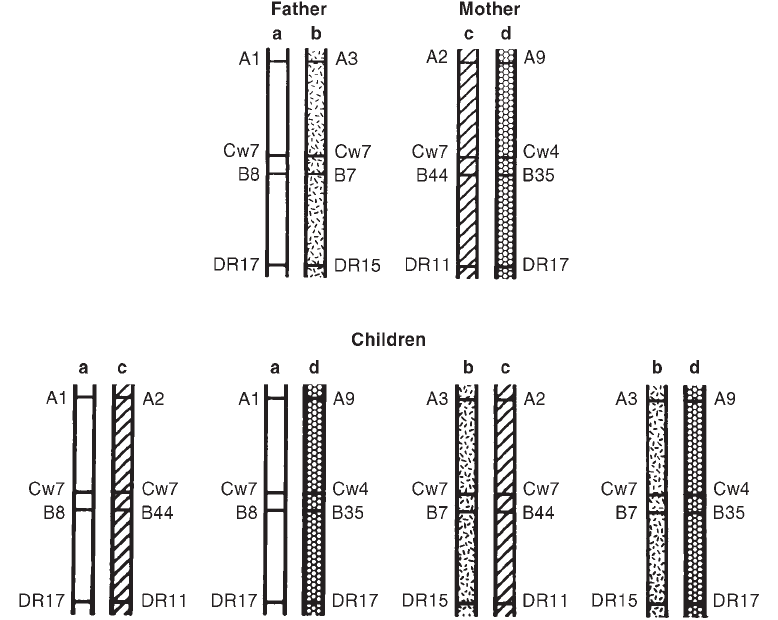
haplotype is inherited from each parent.
Because each parent has two different
copies of chromosome 6, four different
combinations of haplotypes are possible
in the offspring (assuming no recombina-
tion). This inheritance pattern is impor-
tant in predicting whether family members
will be compatible donors for transplan-
tation. The chance that two siblings will
be HLA-identical is 25%. The chance that
any one patient with “n” siblings will have
at least one HLA-identical sibling is 1-(3/4)n.
Having two siblings provides a 44% chance
and three siblings a 58% chance that one
sibling will be HLA-identical. No matter
how many siblings are available for typing
(aside from identical twins), the probabil-
ity will never be 100% for finding an HLA-
identical sibling.
Absence of Antigens
Usually, both copies of the genes within
the MHC are expressed as antigens; how-
388 AABB Technical Manual
Figure 17-2. The linked genes on each chromosome constitute a haplotype. To identify which haplo-
types a person possesses, one must know the antigens present and also the inheritance pattern in the
specific kindred. The observed typing results of the father in this family are interpreted into the follow-
ing phenotype: A1,3;B7,8;Cw7,-;DR15,17. The observed results plus the family study reveal the
haplotypes of the father to be: a = A1,Cw7,B8,DR17 andb=A3,Cw7,B7,DR15.Offspringofasingle
mating pair must have one of only four possible combinations of haplotypes, assuming there has been
no crossing-over.
Copyright © 2005 by the AABB. All rights reserved.
ever, in certain individuals, only one anti-
gen can be identified. This may occur if
the individual is homozygous for the al-
lele, or if appropriate antisera are not
available to type the individual’s antigen
(referred to as a blank allele). Very rarely,
the absence of an antigen can result from
a null allele. A null allele is characterized
by substitutions within the coding region
ofthegenethatpreventtheexpressionof
a functional protein at the cell surface. Such
inactivation of a gene may be caused by
nucleotide substitutions, deletions, or in-
sertions, which lead to a premature cessa-
tion in the antigen’s synthesis. When re-
ferring to phenotypes, a blank is often
written as “x” (for A locus), “y” (for B lo-
cus), or “–” (for any locus) (eg, A1,x;B7,40
or A1,–;B7,40). Family studies must be per-
formed to determine the correct genotype.
Crossing-Over
The genes of the HLA region occasionally
demonstrate chromosome crossover, in
which segments containing linked genetic
material are exchanged between the two
chromosomes during meiosis or gameto-
genesis (see Fig 10-6). These recombinants
are then transmitted as new haplotypes to
the offspring. Crossover frequency is in
part related to the physical distance be-
tween genes. For example, the HLA-A,
HLA-B,andHLA-DR loci are close to-
gether, with 0.8% crossover between the A
and Bloci and 0.5% between the Band
DR loci. In family studies and in parent-
age testing, the possibility of recombina-
tion must be considered.
Linkage Disequilibrium
The MHC system is so polymorphic that,
theoretically, the number of possible uni-
que HLA phenotypes is greater than the
global human population. Moreover, new
HLA alleles are constantly being discov-
ered and characterized. As of 2004, there
were 309 HLA-A, 563 HLA-B, and 368
DRB1 alleles. In reality, many HLA haplo-
types are overrepresented compared with
what would be expected if the distribution
of HLA genes were random. The phenom-
enon of linkage disequilibrium accounts
for the discrepancy between expected and
observed HLA haplotype frequencies.
Expected frequencies for HLA haplo-
types are derived by multiplication of the
frequencies of each allele. For example, in
individuals of European ancestry, the over-
all frequency of the gene coding for HLA-A1
is 0.15 and that for HLA-B8 is 0.10; there-
fore, 1.5% (0.15 ×0.10) of all HLA haplo-
types in this population would be expected
to contain genes coding for both HLA-A1
and HLA-B8 if they were randomly distrib-
uted. The actual frequency of the A1 and B8
combination, however, is 7% to 8% in this
population. Certain allelic combinations
occur with increased frequency in different
racial groups and constitute common haplo-
types in those populations. These are called
ancestral haplotypes because they appear
to be inherited from a single common an-
cestor. The most common ancestral haplo-
type in Northern Europeans, the A1, B8,
DR3, DQ2 haplotype, includes both Class I
and Class II regions. It is unclear whether
ancestral haplotypes represent relatively
young haplotypes that have not had suffi-
cient time to undergo recombination, or
whether they are old haplotypes that are re-
sistant to recombination because of selec-
tion. Linkage disequilibrium in the HLA
system is important in studies of parent-
age because haplotype frequencies in the
relevant population make the transmis-
sion of certain gene combinations more
likely than others. Linkage disequilibrium
also affects the likelihood of finding suit-
able unrelated donors for HLA-matched
platelet transfusions and for HPC trans-
plantation.
Chapter 17: The HLA System 389
Copyright © 2005 by the AABB. All rights reserved.
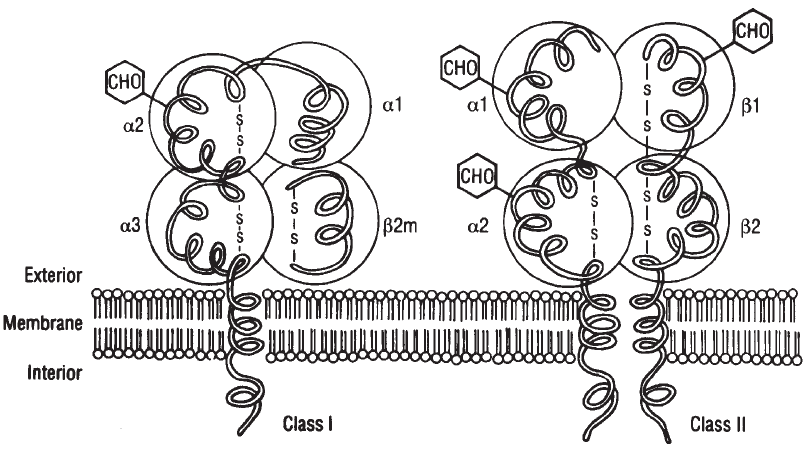
Biochemistry, Tissue
Distribution, and Structure
The HLA antigens are cell surface glyco-
proteins. HLA Class I molecules contain
one copy of two polypeptides: a heavy
chain, which is attached to the mem-
brane, and a light chain, which is called
β2-microglobulin. The HLA Class II mole-
cules are composed of one copy each of
an α-chain and a β-chain, both of which
are attached to the cell surface. HLA anti-
gens are divided into Class I and Class II
based on their function, tissue distribu-
tion, and biochemistry.
Characteristics of Class I and Class II
Antigens
Class I antigens (HLA-A, -B, and -C) have
a molecular weight around 57,000 daltons
and consist of two chains: a glycoprotein
heavy chain (45,000 daltons) encoded on
the short arm of chromosome 6 and, as a
light chain, the β2-microglobulin molecule
(12,000 daltons) encoded by a gene on
chromosome 15. The heavy chain pene-
trates the cell membrane. β2-microglobu-
lin is not attached to the cell membrane;
it associates with the heavy chain via the
latter’s nonvariable (α3) domain but is not
covalently bound to it (see Fig 17-3). The
external portion of the heavy chain con-
sists of three amino acid domains (α1, α2,
and α3), of which the outermost α1and
α2 domains contain the polymorphic re-
gions conferring the HLA antigen speci-
ficity.
Class I molecules are found on platelets
and on most nucleated cells in the body,
with some exceptions such as neurons, cor-
390 AABB Technical Manual
Figure 17-3. Stylized diagram of Class I and Class II MHC molecules showing αand βpolypeptide
chains, their structural domains, and attached carbohydrate units.
Copyright © 2005 by the AABB. All rights reserved.
neal epithelium, trophoblast, and germinal
cells. Only vestigial amounts remain on
mature red cells, with certain allotypes
better expressed than others. These Class I
polymorphisms were independently recog-
nized as red cell alloantigens by serologists
and were designated as Bennett-Good-
speed (Bg) antigens. The specificities called
Bga,Bg
b,andBg
care identified as HLA-B7,
HLA-B17, and HLA-A28, respectively. Plate-
lets express primarily HLA-A and HLA-B
antigens. HLA-C antigens are present at
very low levels and Class II antigens are
generally not expressed at all on platelets.
ClassIIantigens(HLA-DR,-DQ,and
-DP) have a molecular weight of approxi-
mately 63,000 daltons and consist of two
structurally similar glycoprotein chains (α
and β), both of which traverse the mem-
brane (see Fig 17-3). The extramembranous
portion of each chain has two amino acid
domains, of which the outermost domain
contains the variable regions of the Class II
alleles. The expression of Class II antigens
is more restricted than that of Class I. Class
II antigens are expressed constitutively on
B lymphocytes, monocytes, and cells de-
rived from monocytes such as macro-
phages and dendritic cells, intestinal epi-
thelium, and early hematopoietic cells.
There is also constitutive expression of
Class II antigens on some endothelial cells,
especially those lining the microvasculature.
However, in general, endothelium, particu-
larly that of larger blood vessels, is negative
for Class II antigen expression, although its
presence can be induced (for instance, by
interferon-gamma during immune activa-
tion). T lymphocytes are negative for Class
II antigen expression but become positive
when activated. Class II antigens are ex-
pressed abnormally in autoimmune dis-
ease and on some tumor cells.
Soluble HLA Class I and Class II antigens
shed from cells are found in blood and
body fluids and may play a role in modulat-
ing immune reactivity.5Levels of soluble
HLA increase with infection [including hu-
man immunodeficiency virus (HIV)], in-
flammatory disease, and transplant rejec-
tion, and decline with progression of
malignancy. Levels of soluble HLA in blood
components are proportionate to the num-
ber of residual donor leukocytes and to the
length of storage.6Soluble HLA in blood
components may be involved in the im-
munomodulatory effect of blood transfu-
sion.
Configuration
A representative three-dimensional struc-
ture of these molecules can be obtained
by X-ray crystallographic analysis of puri-
fied HLA antigens. The outer domains,
which contain the regions of amino acid
variability and the antigenic epitopes of
the molecules, form a structure known as
the “peptide-binding groove.” Alleles de-
fined by polymorphisms in the HLA gene
sequences have unique amino acid se-
quences and, therefore, form unique
binding grooves, each able to bind differ-
ent classes of peptides. The peptide-bind-
inggrooveiscriticalforthefunctionalas
-
pects of HLA molecules (see section on
Biologic Function).
Nomenclature
An international committee sponsored by
the World Health Organization estab-
lishes the nomenclature of the HLA sys-
tem. It is updated regularly to incorporate
new HLA alleles. HLA antigens are desig-
nated by a number following the letter
that denotes the HLA series (eg, HLA-A1
or HLA-B8). Previously, antigenic speci-
ficities that were not fully confirmed car-
ried the prefix “w” (eg, HLA-Aw33). When
identification of the antigen became de-
finitive,theWHONomenclatureCommit
-
Chapter 17: The HLA System 391
Copyright © 2005 by the AABB. All rights reserved.
tee dropped the “w” from the designation.
The Committee meets regularly to update
nomenclature by recognizing new speci-
ficities or genetic loci. The “w” prefix is no
longer applied in this manner and is now
used only for the following: 1) Bw4 and
Bw6, to distinguish these “public” antigens
from other B locus alleles; 2) all C locus
specificities, to avoid confusion with
members of the complement system; and
3) Dw and DP specificities that were de-
fined by mixed lymphocyte reactions and
primed lymphocyte typing. The numeric
designations for the HLA-A and HLA-B
specificities were assigned based on the
order of their discovery.
Splits
Refinement of serologic methods permit-
ted antigens previously believed to repre-
sent a single specificity to be “split” into
specificities that were serologically (and,
later, genetically) distinct. The designa-
tion for an individual antigen that is a
split of an earlier recognized antigen of-
ten includes the number of the parent an-
tigen in parentheses, eg, HLA-B44 (12).
Shared Determinants
As the specificity of HLA typing sera im-
proved, it was found that some epitopes,
the part of the antigen that binds anti-
body, were common to several antigens.
Antibodies to these epitopes identified
antigens that constituted a group, of which
the individual members could be identi-
fied by antisera with more restricted ac-
tivity.
Cross-Reactive Groups
In addition to “splits,” HLA antigens and
antigen groups may have other epitopes
in common. Antibodies that react with
these shared determinants often cause
cross-reactions in serologic testing. The
collective term for a group of HLA anti-
gens that exhibit such cross-reactivity is
cross-reactive group (CREG).
“Public” Antigens
In addition to splits and CREGs, HLA pro-
teins have reactivity that is common to
many different HLA specificities. Called
“public” antigens, these common amino
acid sequences appear to represent the
less variable portion of the HLA molecule.
Two well-characterized “public” antigens,
HLA-Bw4 and HLA-Bw6, are found in the
HLA-B series. The Bw4 antigen is also
found on some A locus molecules. “Pub-
lic” antigens are clinically important be-
cause patients exposed to foreign HLA
antigens via pregnancy, transfusion, or
transplantation can make antibodies to
them. A single antibody, when directed
against a “public” antigen, can resemble
the sum of multiple discrete alloanti-
bodies.
Nomenclature for HLA Alleles
As nucleotide sequencing is used to in-
vestigate the HLA system, increasing
numbers of HLA alleles are being identi-
fied, many of which share a common se-
rologic phenotype. The minimum re-
quirement for designation of a new allele
is the sequence of exons two and three for
HLA Class I and exon two for HLA Class II
(DRB1). These exons encode the variable
amino acids that confer HLA antigen spe-
cificity. A uniform nomenclature has been
adopted that takes into account the locus,
the major serologic specificity, and the al-
lele determined by molecular typing tech-
niques. For example, isoelectric focusing,
amino acid sequencing, and nucleotide
sequencing have identified several unique
variants of HLA-DR4. The first HLA-DR4
variant is designated DRB1*0401,indicat
-
392 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

ing the locus (DR), the protein (β1chain),
an asterisk to represent that an allele
name follows, the major serologic speci-
ficity (04 for HLA-DR4), and the allele
number (variant 01). For Class I alleles, the
name of the locus, for example HLA-B, is
followed by an asterisk and then a num-
ber of digits. The first two digits corre-
spond to the serologic specificity of the
antigen. The third and fourth digits are
used to list the subtypes, numbers being
assigned in the order in which the DNA
sequences have been determined. There-
fore, B*2704 represents the HLA-B locus,
with a serologic specificity of B27, and
was the fourth allele described in this
family (see Table 17-1). Finally, the no-
menclature can accommodate alleles
with silent mutations, ie, those that have
different DNA sequences but identical
amino acid sequences and null alleles.
Biologic Function
The essential function of the HLA system
is self/nonself discrimination. Discrimina-
tion of self from nonself is accomplished
by the interaction of T lymphocytes with
peptide antigens. T lymphocytes interact
with peptide antigen only when the T-cell
receptor (TCR) for antigen engages both
an HLA molecule and the antigenic pep-
tide contained with its peptide-binding
groove. This limitation is referred to as
“MHC restriction.”7
In the thymus, T lymphocytes whose
TCRs bind to a self HLA molecule are se-
lected (positive selection), with the excep-
tion of those whose TCRs also bind to a
peptide derived from a self antigen, in
which case they are deleted (negative selec-
tion). Some self-reactive T cells escape neg-
ative selection, however. If not functionally
inactivated, for instance, by the mechanism
of anergy, these self-reactive T cells may be-
come involved in an autoimmune process.
(See Chapter 11.)
Role of Class I
ClassImoleculesaresynthesized,and
peptide antigens are inserted into the
peptide-binding groove, in the endo-
plasmic reticulum. Peptide antigens that
fit into the Class I peptide-binding groove
are typically eight or nine amino acids in
lengthandarederivedfromproteinsthat
Chapter 17: The HLA System 393
Table 17-1. HLA Nomenclature
Genetic
Locus
Antigenic
Specificity Allele
Number of
Identified
Alleles
Polypeptide
Location
HLA-A
HLA-B
HLA-C
DRA
DRB1
DQA1
DQB1
DPA1
DPB1
A1 to A80
B7 to B81
Cw1toCw10
DR1 to DR18
DQ1 to DQ9
DPw1 to DPw6
A*0101 to *8001
B*0702 to *8301
Bw*0102 to *1802
DRA*0101 to *0102
DRB1*0101 to *1608
DQA1*0101 to *0601
DQB1*0501 to *0402
DPA1*0103 to *0401
DPB1*0101 to *8901
207
412
100
2
271
20
45
19
93
a
a
a
a
β1
a
β1
a1
β1
Copyright © 2005 by the AABB. All rights reserved.
are made by the cell (endogenous pro-
teins). These endogenous proteins, which
may be normal self proteins, altered self
proteins such as those found in cancer
cells, or viral proteins such as those found
in virus-infected cells, are degraded in the
cytosol by a large multifunctional prote-
ase (LMP) and transported to the endo-
plasmic reticulum by a transporter associ-
ated with antigen processing (TAP). The
LMP and TAP genes are both localized to
the MHC.
ClassImoleculesaretransportedtothe
cell surface where they are available to in-
teract with CD8-positive T lymphocytes. If
the TCR of a T lymphocyte can bind the an-
tigenic peptide in the context of the specific
Class I molecule displaying it, then this
binding activates the cytotoxic properties of
the T cell, which will then attack the cell,
characteristically eliciting an inflammatory
response. The presentation of antigen by
ClassImoleculesisespeciallyimportantin
a host’s defense against viral pathogens and
against malignant transformation. Tumor
cells that do not express Class I escape this
immune surveillance.
Role of Class II
Class II molecules, like Class I molecules,
are synthesized in the endoplasmic retic-
ulum, but peptide antigens are not in-
serted into the peptide-binding groove
here. Instead, an invariant chain (Ii) is in-
serted. The Class II-invariant chain mole-
cule is transported to an endosome where
the invariant chain is removed by a spe-
cialized Class II molecule called DM
(whose locus is also localized to the MHC).
A Class II antigenic peptide is then in-
serted into the peptide-binding groove.
Peptide antigens that fit into the Class II
peptide-binding groove are typically 12 to
25 amino acids in length and are derived
from proteins that are taken up by the cell
by endocytosis (exogenous proteins). Ex-
ogenous proteins, which may be normal
self proteins or proteins derived from
pathogens such as bacteria, are degraded
to peptides by enzymes in the endosomal
pathway. Class II molecules are then
transported to the cell surface where they
are available to interact with CD4-positive
T lymphocytes, which secrete immuno-
stimulatory cytokines in response. This
mechanism is especially important for the
production of antibodies.
Detection of HLA Antigens
and Alleles
Methods for the detection of HLA antigens
and alleles fall into three groups: molecu-
lar (DNA-based), serologic, and cellular
assays. Detailed procedures of commonly
used assays are provided in the current
edition of the American Society for Histo-
compatibility and Immunogenetics Labo-
ratory Manual. Depending on the clinical
situation, a particular HLA antigen detec-
tion or typing method may be preferable
(Table 17-2).
DNA-Based Assays
DNA-based typing has several advantages
over serologic and cellular assays: high
sensitivity and specificity; small sample
volumes; decreased turnaround time, for
some methods, as short as a few hours;
and absence of the need for cell surface
antigen expression or cell viability. Al-
though serologic methods can readily dis-
tinguish only about 21 serologic specifi-
cities, high-resolution DNA-based methods
can detect up to 368 alleles.
Polymerase Chain Reaction Testing
Polymerase chain reaction (PCR) technol-
ogy allows amplification of large quanti-
394 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

ties of a particular target segment of geno-
mic DNA. Low- to intermediate-resolution
typing detects the HLA serologic equiva-
lents with great accuracy; eg, it distin-
guishes DR15 from DR16, whereas high-
resolution typing distinguishes individual
alleles, eg, DRB1*0101 from DRB1*0102.
Several PCR-based methods have been
developed, of which two general approaches
are described below.
Oligonucleotide Probes. The first tech-
nique uses sequence-specific oligonucleo-
tide probes (SSOPs) and is known as PCR-
SSO, PCR-SSOP, or allele-specific oligo-
nucleotide (ASO) hybridization.8APCR
product amplified from genomic DNA is
applied to a membrane or filter to which
the labeled SSOPs are hybridized. These
short DNA probes will hybridize with the
complementary sequences and identify
groups of alleles or individual alleles. Ad-
vantages are that all Class II loci can be
typed and highly specific information ob-
tained. Disadvantages include potential
difficulty in interpretation of results and the
need to use multiple filters and perform
multiple amplifications and subsequent
hybridizations. A variation of this tech-
nique, the reverse line or dot blot, elimi-
nates the need for multiple filters and hy-
bridizations by incorporation of a label
(such as biotin) into the PCR product dur-
ing its amplification from genomic DNA.
The PCR product is then hybridized to a
membrane containing all the relevant
SSOPs and its pattern of hybridization with
the SSOPs revealed by detection of the in-
corporated label. In the past few years, a
new method for establishing HLA geno-
types that features arrays of oligonucleotide
probes on a solid phase has begun to ap-
pear in HLA typing laboratories. The
microbead array assay is an SSOP method
for HLA-A, -B, and -DR antigen level typing.
Chapter 17: The HLA System 395
Table 17-2. HLA Typing Methods and Appropriate Applications
Method Clinical Application Resolution
SSP (PCR) Solid organ, related and unrelated HPC
transplantation
Serologic to allele level,
higher resolution with
large number of primers
DNA sequencing Unrelated HPC transplantation, resolu-
tion of typing problems with other
methods, characterization of new
alleles
Allele level
Forward SSOP
hybridization
Solid organ and HPC transplantation
(can accommodate high-volume
testing)
Serologic to allele level
Reverse SSOP
hybridization
Solid organ, related and unrelated HPC
transplantation
Serologic, higher resolution
with larger number of
probes
Microlympho-
cytotoxicity
Solid organ transplantation, evaluation
of platelet refractoriness, HLA typing
(Class I only) of platelet recipients
and platelet donors
Serologic specificity
Copyright © 2005 by the AABB. All rights reserved.
This technique also has the ability for low-
to-intermediate resolution DNA-based tis-
sue typing, with a reduction in sample pro-
cessing time.
Sequence-Specific Primers. A second
major technique uses sequence-specific
primer pairs (SSPs) that target and amplify
a particular DNA sequence.9This method
requires the performance of multiple PCR
reactions in which each reaction is specific
for a particular allele or group of alleles. Di-
rect visualization of the amplified alleles is
seen after agarose gel electrophoresis. Be-
cause SSPs have such specific targets, pres-
ence of the amplified material indicates
presence of the corresponding allele(s). The
pattern of positive and negative PCR ampli-
fications is examined to determine the HLA
alleles present. Primer pair sets are avail-
able that can determine the full HLA-A, -B,
-C,-DR,-DQ,and-DPtype.
Sequence-Based Typing
High-resolution nucleic acid sequencing
of HLA alleles generates allele-level se-
quences that are used to characterize new
allele(s). With the ever-increasing avail-
ability and ease of use of automated se-
quencers, sequence-based typing has be-
come a routine HLA typing method in
some HLA laboratories.
Serologic Assays
Lymphocytotoxicity
The microlymphocytotoxicity test can be
used to detect HLA-A, -B, -C, -DR, and
-DQ antigens. Lymphocytes are used for
testing because they are readily obtained
from anticoagulated peripheral blood
and, unlike granulocytes, give reproduc-
ible results. Lymphocytes obtained from
lymph nodes or spleen may also be used.
HLA typing sera are obtained primarily
from multiparous women. Some mouse
monoclonal antisera are also available.
HLA sera of known specificities are
placed in wells of a microdroplet test plate.
A suspension of lymphocytes is added to
each well. Rabbit complement is then
added and, if sufficient antibody has bound
to the lymphocyte membranes, the com-
plement cascade will be activated through
the membrane attack complex, leading to
lymphocytotoxicity. Damage to the cell
membrane can be detected by the addition
of dye: cells that have no attached antibody,
no activated complement, and no damage
to the membrane keep the vital dyes from
penetrating; cells with damaged mem-
branes allow the dye to enter. The cells are
examined for dye exclusion or uptake un-
der phase contrast microscopy. If a fluores-
cent microscope is available, fluorescent vi-
tal dyes can also be used.
Because HLA-DR and HLA-DQ antigens
are expressed on B cells and not on resting
T cells, typing for these antigens usually re-
quires that the initial lymphocyte prepara-
tion be manipulated before testing to yield
an enriched B-cell population. This is typi-
cally accomplished by the use of magnetic
beads, to which monoclonal antibodies to
B cells have been bound.
The interpretation of serologic reactions
requires skill and experience. Control wells
of known reactivity and careful quality con-
trol of reagents are required, especially for
the activity of the complement used to in-
duce lymphocytotoxicity. In addition, anti-
gen assignments can be made only on the
basis of results obtained with multiple anti-
sera because few reagent antisera have suf-
ficient monospecific reliability to be used
alone. The extreme polymorphism of the
HLA system, the variation in antigen fre-
quencies among different racial groups, the
reliance on biologic antisera and living tar-
get cells, and the complexities introduced
by splits, CREGs, and “public” antigens all
contribute to difficulties in accurate sero-
logic HLA typing.
396 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Antibodies in Patients
Microlymphocytotoxicity testing can be
used to test serum specimens against se-
lected target cells. This is routinely done
in HLA crossmatching, which consists of
testing serum from a potential recipient
against unfractionated lymphocytes (or
fractionated T and B lymphocytes) from a
potential donor. A variation of the micro-
lymphocytotoxicity test, which uses an anti-
globulin reagent, is one of the methods
used to increase sensitivity. Flow cyto-
metryisalsousedasanindependent
method to increase the sensitivity of the
crossmatch.
Testing the patient’s serum against a
panelof30to60ormoredifferenttarget
cells can assess the extent of HLA allo-
immunization. The percent of the panel
cells to which the recipient has formed
cytotoxicantibodiesisreferredtoasthe
panel reactive antibody (PRA) level. Deter-
mination of PRA can be useful in the inves-
tigation of FNHTRs, in the workup of
platelet refractoriness, and in following pa-
tients who are awaiting cadaver solid organ
transplants. This “HLA antibody screen” not
only detects the presence of HLA antibod-
ies but also may allow their specificity to be
determined. The presence of HLA antibod-
ies can also be demonstrated by using an
enzyme-linked immunosorbent assay with
solid-phase HLA antigens or by flow cyto-
metric analysis using antigen-coated beads.
Cellular Assays
Historically, the mixed lymphocyte cul-
ture (MLC) (also called mixed leukocyte
culture, mixed lymphocyte reaction, or
MLR) was used to detect genetic differ-
ences in the Class II region. In the MLR,
lymphocytes from different individuals
are cultured together and have the oppor-
tunity to recognize foreign HLA-D region
antigens and to respond by proliferating.
The HLA System and
Transfusion
HLA system antigens and antibodies play
important roles in a number of transfu-
sion-related events. They include allo-
immunization and platelet refractoriness,
FNHTR, TRALI, and posttransfusion GVHD.
HLA antigens are highly immunogenic. In
response to pregnancy, transfusion, or
transplantation, immunologically normal
individuals are more likely to form anti-
bodies to HLA antigens than to any other
antigen system.
Platelet Refractoriness
The incidence of HLA alloimmunization
and platelet refractoriness among pa-
tients receiving repeated transfusions of
cellular components is 20% to 71%.10 The
refractory state exists when transfusion of
suitably preserved platelets fails to in-
crease the recipient’s platelet count. Pla-
telet refractoriness may be due to clinical
factors such as sepsis, high fever, dissemi-
nated intravascular coagulopathy, medi-
cations, hypersplenism, complement-me-
diated destruction, or a combination of
these, or it may have an immune basis.
(See Chapter 16 for more information
about platelets.)
Antibody Development
Antibodies against HLA antigens usually
cause immune-mediated platelet refrac-
toriness, but antibodies to platelet-spe-
cific or ABH antigens may also be in-
volved. HLA alloimmunization can follow
pregnancy, transfusion, or organ trans-
plantation because the foreign antigens
are the donor MHC antigens themselves.
A common example of this is the develop-
ment of HLA antibodies directed against
Class I antigens that occurs with transfu-
sion of platelets, which express only Class
Chapter 17: The HLA System 397
Copyright © 2005 by the AABB. All rights reserved.
I antigens. The presence, in the trans-
fused component, of leukocytes bearing
Class I and II antigens elicits alloimmuni-
zation. The likelihood of immunization
can be lessened with leukocyte-reduced
blood components, or by treatment with
ultraviolet light, which alters the co-
stimulatory molecules or impairs anti-
gen-presenting cell activity. The threshold
level of leukocytes required to provoke a
primary HLA alloimmune response is un-
clear and probably varies among different
recipients. Some studies have suggested
that 5 ×106leukocytes per transfusion
may represent an immunizing dose. In
patients who have been previously sensi-
tized by pregnancy or transfusion, expo-
sure to even lower numbers of allogeneic
cells is likely to provoke an anamnestic
antibody response.
Finding Compatible Donors
The HLA antibody response of transfused
individuals may be directed against indi-
vidual specificities present on donor cells
or against “public” alloantigens. Precise
characterization may be difficult. An over-
all assessment of the degree of HLA allo-
immunization can be obtained by mea-
suring the PRA of the recipient’s serum.
Platelet-refractory patients with a high
PRA are broadly alloimmunized and may
be difficult to support with platelet trans-
fusions. HLA-matched platelets, obtained
by plateletpheresis, benefit some, but not
all, of these refractory patients. Because
donors with a four-antigen match for an
immunized recipient are hard to find, strat-
egies for obtaining HLA-matched platelets
vary. Selection of partially mismatched
donors, based on serologic cross-reactive
groups, has been emphasized, but such
donors may fail to provide an adequate
transfusionresponseinvivo.Analterna
-
tive approach to the selection of donors is
based on matching for “public” speci-
ficities rather than cross-reactive private
antigens. Obtaining an adequate number
of readily available HLA-typed donors can
prove difficult; it has been estimated that
a pool of 1000 to 3000 or more donors
would be needed to provide the transfu-
sion requirements of most HLA-allo-
immunized patients.11 Useofsingleanti
-
gen beads to identify HLA antibody
specificities precisely can allow a better
selection of donors who have acceptable
mismatched antigens.12
In the past, it was recommended that
patients who were at risk of becoming
alloimmunized and refractory be serologi-
cally Class I HLA-typed early in the course
of their illness, when enough lymphocytes
were present in the peripheral blood to ob-
tain a reliable HLA type. Intensive chemo-
therapy makes it very difficult to obtain
enough cells for such typing. More recently,
with the advent of molecular typing tech-
niques, a patient’s HLA alleles can be deter-
mined using genomic DNA isolated from
very small numbers of white cells or even
nonblood tissue (eg, buccal swabs).
HLA-alloimmunized patients often re-
spond to crossmatch-compatible platelets
selected using patient serum and samples
of apheresis platelets in a platelet antibody
assay. Crossmatching techniques may as-
sess compatibility for both HLA and plate-
let-specific antibodies.13 These histocom-
patible platelet components are further
discussed in Chapter 21.
Febrile Nonhemolytic Transfusion
Reactions
HLA antibodies, as well as granulocyte
and platelet-specific antibodies, have
been implicated in the pathogenesis of
FNHTRs. The recipient’s antibodies, re-
acting with transfused antigens, elicit the
release of cytokines (eg, interleukin-1) ca-
398 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
pableofcausingfever.Serologicinvesti
-
gation, if undertaken, may require multi-
ple techniques and target cells from a
number of different donors (see Chapter
27).
Transfusion-Related Acute Lung Injury
In TRALI, a transfusion reaction that is
being recognized with increasing fre-
quency, acute noncardiogenic pulmonary
edema develops in response to transfusion.
Pathogenesis appears to reflect the pres-
ence of HLA antibodies in donor blood,
which react with and fix complement to
granulocytes of the recipient, leading to
severe capillary leakage and pulmonary
edema. Rarely, HLA antibodies of the re-
cipient react with transfused leukocytes
from the donor (see Chapter 27). Cases of
TRALI have been reported that appear to
be caused by donor antibodies against
Class II antigens in recipients. Because
ClassIIantigensarenotexpressedon
neutrophils, an alternate explanation for
activation of neutrophils in these instances
is required. One hypothesis is that Class II
antigens on the recipient’s pulmonary
macrophages are targeted by these com-
plement-activating antibodies. Subse-
quent release of cytokines and chemokines
results in the recruitment and activation
of neutrophils in the lungs.14
Chimerism and Posttransfusion
Graft-vs-Host Disease
Chimerism refers to the presence of do-
nor cells in the recipient. Persistent chi-
merism after blood transfusion may lead
to the development of GVHD in the recip-
ient. The development of posttransfusion
GVHD depends on several factors: the de-
gree to which the recipient is immuno-
compromised; the number and viability
of lymphocytes in the transfused compo-
nent; and the degree of HLA similarity be-
tween donor and recipient. The observa-
tion of posttransfusion GVHD with the use
of fresh blood components from blood
relatives has highlighted the role of the
HLA system in GVHD.
Figure 17-4 illustrates the conditions for
increased risk of GVHD. The parents have
one HLA haplotype in common. Each child,
therefore, has a one in four chance of inher-
iting the same haplotype from each parent,
and Child #1 is homozygous for the shared
parental HLA haplotype. Transfusion of
blood from this person to an unrelated re-
cipient who did not have this haplotype
would have no untoward consequences. If,
however, Child #1 were a directed donor for
the relatives heterozygous for that haplo-
type (both parents and Child #3), the recipi-
ent would not recognize any foreign anti-
gens on the transfused lymphocytes and
would not eliminate them. The donor cells,
however, would recognize the recipient’s
foreign HLA antigens, would become acti-
vated, proliferate, and attack the host. To
avoid this situation, it is recommended that
all cellular components known to be from
blood relatives be irradiated before transfu-
sion. Other specially chosen donor units,
such as HLA-matched platelets, may also
present an increased risk of posttransfusion
GVHD. Rarely, transfusion-associated GVHD
has occurred after the transfusion of blood
from an unrelated donor.15
Chimerism is also proposed to be re-
sponsible for the maintenance of tolerance
in some organ transplant recipients16 and
for the maintenance of HLA sensitization.17
It has been postulated that scleroderma is a
form of GVHD resulting from chimeric cells
derived from fetal cells transferred across
the placenta during pregnancy.18
Hemolytic Transfusion Reactions
HLA incompatibility has rarely been im-
plicated as a cause of shortened red cell
Chapter 17: The HLA System 399
Copyright © 2005 by the AABB. All rights reserved.
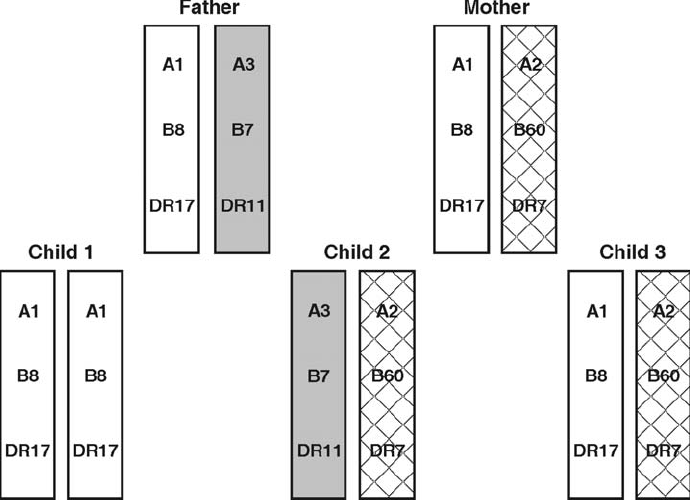
survival in patients with antibodies to HLA
antigens such as Bga(B7), Bgb(B17), and
Bgc(A28) that are expressed, although
weakly, on red cells (see Chapter 15). Such
an incompatibility may not be detected
with conventional pretransfusion testing.
HLA Testing and
Transplantation
HLA testing is an integral part of organ
transplantation. The extent of testing dif-
fers for different types of transplants. Or-
gan transplantation is discussed in great-
er detail in Chapter 26.
Hematopoietic Progenitor Cell Transplants
It has long been recognized that disparity
within the HLA system represents an im-
portant barrier to successful HPC trans-
plantation.19 HLA similarity and compati-
bility between the donor and the recipient
are required for engraftment and to pre-
vent GVHD, but some degree of rejection
or GVHD remain common problems for
recipients of allogeneic HPCs, despite
immunosuppressive conditioning.
Candidate donors and recipients are
typed for their HLA-A, -B, -C, -DR and -DQ
alleles. The goal is to match, as closely as
possible, the alleles of the prospective do-
nor and recipient at the HLA-A,-B,and
-DRB1 loci, with the optimal match being
an allele-level match.20 Some transplant
400 AABB Technical Manual
Figure 17-4. HLA haplotypes in a family at risk for transfusion-associated GVHD. In contrast to the fam-
ily shown in Fig 17-2, each parent shares a common HLA haplotype, HLA-A1,B8,DR17. Child 1 is ho-
mozygous for the haplotype shared by the parents and by child 3. The lymphocytes of child 1 are capa-
ble of producing posttransfusion GVHD if transfused to either parent or to child 3.
Copyright © 2005 by the AABB. All rights reserved.
programs additionally match for HLA-C
and -DQ alleles. Molecular HLA typing is
performed on samples from both the donor
and recipient for optimal assessment of
Class I and II region compatibility. Al-
though HLA-identical sibling donors re-
main the best choice for HPC transplanta-
tion, there is increasing use of unrelated
donors identified by searching the file of 5
million HPC donors listed in the National
Marrow Donor Program’s registry of volun-
teer donors. The use of umbilical cord
blood stem cells and hematopoietic stem
cell grafts that have undergone T-cell deple-
tion may allow greater donor-recipient mis-
matches.21,22
Kidney and Pancreas Transplants
ABO compatibility is the most important
factor determining the immediate survival
of kidney transplants. Because ABH anti-
gens are expressed in varying amounts on
all cells of the body, transplanted ABO-in-
compatible tissue comes into continuous
contact with the recipient’s ABO antibod-
ies. Of particular importance is the ex-
pression of ABH antigens on vascular en-
dothelial cells because the vascular supply
in the transplant is a common site for re-
jection.
Both the recipient and the donor are or-
dinarilytestedforABO,HLA-A,-B,and-DR
antigens. HLA-C and -DQ testing is also
usually performed. Before surgery, a major
crossmatch of recipient serum against do-
nor lymphocytes is required. ASHI Stan-
dards for Histocompatibility Testing23 re-
quire that the crossmatch be performed
using a method more sensitive than routine
microlymphocytotoxicity testing, such as
prolonged incubation, washing, augmenta-
tion with antihuman globulin reagents, or
flow cytometry. Flow cytometry is the most
sensitive method and is especially useful
because it can best predict early acute re-
jection and delayed graft function, both of
which are strong predictors of chronic re-
jection and long-term allograft survival.24 In
patients undergoing cadaveric kidney re-
transplantation, the 7-year graft survival
rate using the T-cell flow crossmatch to se-
lect the donor kidney was comparable to
that of patients undergoing primary cada-
veric transplantation (68% vs 72%) and was
significantly better than that of regraft pa-
tients for whom only the antiglobulin
lymphocytotoxicity crossmatch was used
(45%).25 Because HLA antibody responses
are dynamic, the serum used for the cross-
match is often obtained within 48 hours of
surgery and is retained in the frozen state
for any required subsequent testing. An in-
compatible crossmatch with unfractionated
or T lymphocytes is a contraindication to
kidney transplantation. The significance of
a positive B-cell crossmatch is unclear.
Serumfromapatientawaitingcadaver-
donor kidney transplant surgery is tested at
regular intervals for the degree of alloim-
munization by determining the PRA. In ad-
dition, many laboratories identify the
specificities of HLA alloantibodies formed.
If an antibody with a defined HLA specific-
ity is identified in a recipient, it is a com-
mon practice to avoid the corresponding
antigen when allocating a deceased donor
allograft. The serum samples used for peri-
odic PRA testing are usually frozen. The
samples with the highest PRA are often
used, in addition to the preoperative sam-
ple, for pretransplant crossmatching. The
necessity of a prospective crossmatch for
recipients with no evidence of HLA sensiti-
zation has been questioned. Prompt trans-
plantation with reduced cold ischemia time
for the renal allograft may provide greater
benefit to the patient than prospective
crossmatching, provided 1) a very sensitive
method for antibody detection, such as
flow cytometry, has been used26,27 and2)itis
absolutely certain that the patient has had
Chapter 17: The HLA System 401
Copyright © 2005 by the AABB. All rights reserved.
no additional sensitizing event (ie, immuni-
zation or transfusions within 2 weeks be-
fore or any time since that serum was
screened).
The approach to kidney transplants us-
ing living donors is different. In the past,
when several prospective living donors
were being considered, MLC testing be-
tween the recipient and the donors was
sometimes performed, but it is rarely per-
formed today. HLA matching of recipients
with kidney donors (both living and cada-
veric donor) contributes to long-term allo-
graft survival by decreasing the likelihood
of chronic rejection. In 1996, the projected
20-year allograft survival rates were 57% for
two-haplotype-matched sibling donors,
30% for one-haplotype-matched parental
donors, and 18% for cadaver donors.28 For
cadaver donors with no mismatches with
the recipient for HLA-A, -B, and -DR, the
projected 20-year allograft survival was
40%. Surprisingly, projected survival for
allografts from living, unrelated donors is
similar to those from parental donors.29 Re-
cently, the 1-year survival rates for grafts
from living and cadaver renal donors were
93.9% and 87.7% respectively, and the half-
lives of living-donor and cadaver-donor re-
nal allografts were 21.6 and 13.8 years, re-
spectively.30
Other Solid Organ Transplants
For liver, heart, lung, and heart/lung trans-
plants, ABO compatibility remains the
primary immunologic system for donor
selection, and determining pretransplant
ABO compatibility between donor and re-
cipient is mandatory. HLA-A, -B, and -DR
testing of potential recipients is required,
and the transplant crossmatch must be
available before transplantation when the
recipient has demonstrated presensitiza-
tion, except for emergency situations.
Levels of HLA compatibility do correlate
with graft survival after heart transplanta-
tion, but prospective HLA matching has
been difficult to implement.31
Parentage and Other
Forensic Testing
HLA typing (particularly DNA-based HLA
typing) has proven useful in forensic test-
ing. In parentage testing, HLA typing
alone can exclude about 90% of falsely ac-
cused males. With the addition of red cell
antigen typing, the exclusion rate rises to
95% and exceeds 99% when typing for red
cell enzymes and serum proteins is in-
cluded. Haplotype frequencies, rather
than gene frequencies, are used in these
calculations because linkage disequilib-
rium is so common in the HLA system. It
is important, however, to keep in mind
the racial differences that exist in HLA
haplotype frequencies; recombination
events must also be considered.
Other useful DNA-based assays for fo-
rensic testing are detection of alleles with
variable numbers of tandem repeats and al-
leles with variation in the number of short
tandem repeats, which assess other poly-
morphic, non-HLA genetic regions. DNA-
based assays allow identification of individ-
uals on the basis of extremely small sam-
ples of fluid or tissue, such as hairs, epithe-
lial cells, or semen.
HLA and Disease
For some conditions, especially those be-
lieved to have an autoimmune etiology,
an association exists between HLA phe-
notype and occurrence of clinical disease
(see Table 17-3).32-34 HLA-associated dis-
eases have several features in common.
They are known or suspected to be inher-
ited, display a clinical course with acute
402 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

exacerbations and remissions, usually have
characteristics of autoimmune disorders,
and the exact cause is unknown. Evidence
has been accumulating that implicates
the HLA molecules themselves in disease
susceptibility. For instance, resistance to
cerebral malaria results from a strong
cytotoxic T-cell response to particular ma-
larial peptides that are restricted by (fit
into the peptide-binding grooves of) two
specific HLA molecules.35 Another mecha-
nism that could lead to the association of
HLA phenotype and disease is the pres-
ence of a Class I or II heterodimer en-
coded by a specific allele that preferen-
tially presents autoantigens to the T-cell
receptor.
The ancestral haplotype A1, B8, DR3,
DQ2 discussed previously (under Linkage
Disequilibrium) is associated with suscepti-
bility to Type 1 diabetes, lupus, celiac dis-
ease, common variable immunodeficiency
and IgA deficiency, myasthenia gravis, and
also with an accelerated course of HIV in-
fection, likely due to the presence of multi-
ple genes.36 However, HLA typing has only
limited value in assessing risk for most dis-
eases because the association is incom-
plete, often giving false-negative and
false-positive results. The association of
HLA-B27 and ankylosing spondylitis in
those of European ancestry is instructive.
The test is highly sensitive; more than 90%
of such patients with ankylosing spondylitis
possess the HLA-B27 antigen. On the other
hand, specificity is low; only 20% of individ-
uals with the B27 antigen will develop an-
kylosing spondylitis. A second condition,
narcolepsy, is strongly associated with the
HLA allele DQB1*0602.37 As with the case of
HLA-B27 and ankylosing spondylitis, over
90% of individuals with narcolepsy are pos-
itive for HLA-DQB1*0602, but only a minor-
ity of individuals with this marker develop
the disease.
The degree of association between a
given HLA type and a disease is often de-
scribed in terms of relative risk (RR), which
is a measure of how much more frequently
a disease occurs in individuals with a spe-
cific HLA type when compared to individu-
als not having that HLA type. Calculation of
RR is usually based on the cross-product
ratio of a 2 ×2 contingency table. However,
because the HLA system is so highly poly-
morphic, there is an increased possibility of
Chapter 17: The HLA System 403
Table 17-3. HLA-Associated Diseases
Disease HLA RR 32-37
Celiac disease DQ2 >250
Ankylosing spondylitis B27 >150
Narcolepsy DQ6 >38
Subacute thyroiditis B35 14
Type I diabetes DQ8 14
Multiple sclerosis DR15, DQ6 12
Rheumatoid arthritis DR4 9
Juvenile rheumatoid arthritis DR8 8
Grave’s disease DR17 4
RR = relative risk.
Copyright © 2005 by the AABB. All rights reserved.
finding an association between an HLA an-
tigen and a disease by chance alone. There-
fore, calculation of RR for HLA disease as-
sociations is more complex and is typically
done by Haldane’s modification of Woolf’s
formula.38-39 The RR values for some dis-
eases associated with HLA are shown in Ta-
ble 17-3.
References
1. Schreuder GMTh, Hurley CK, Marsh SGE, et
al. The HLA dictionary 2001: A summary of
HLA-A, -B, -C, -DRB1/3/4/5, -DQB1 alleles and
their association with serologically defined
HLA-A, -B, -C, -DR, and -DQ antigens. Hum
Immunol 2001;62:826-49.
2. Braud VM, Allan DSJ, McMichael AJ. Func-
tions of nonclassical MHC and non-MHC-en-
coded class I molecules. Curr Opin Immunol
1999;11:100-8.
3. Feder JN, Gnirke A, Thomas W, et al. A novel
MHC class I-like gene is mutated in patients
with hereditary haemochromatosis. Nat Genet
1996;13:399-408.
4. BodmerJG,ParhamP,AlbertED,MarshSG.
Putting a hold on “HLA-H.” Nat Genet 1997;
15:234-5.
5. McDonald JC, Adamashvili I. Soluble HLA: A
review of the literature. Hum Immunol 1998;
59:387-403.
6. Ghio M, Contini P, Mazzei C, et al. Soluble
HLA class 1, HLA class II, and Fas ligand in
blood components: A possible key to explain
the immunomodulatory effects of allogeneic
blood transfusion. Blood 1999;93:1770-7.
7. Zinkernagel RM, Doherty PC. The discovery
of MHC restriction. Immunol Today 1997;18:
14-7.
8. CaoK,ChopekM,Fernandez-VinaMA.High
and intermediate resolution DNA typing sys-
tems for class I HLA-A,-B,-C genes by hy-
bridization with sequence-specific oligo-
nucleotide probes (SSOP). Rev Immunogenet
1999;1:177-208.
9. Welsh K, Bunce M. Molecular typing for the
MHC with PCR-SSP. Rev Immunogenet 1999;
1:157-76.
10. Dzik WH. Leukoreduced blood components:
Laboratory and clinical aspects. In: Simon
TL, Dzik WH, Snyder EL, et al, eds. Rossi’s
principles of transfusion medicine. 3rd ed.
Baltimore, MD: Lippincott Williams and
Wilkins, 2002:270-87.
11. Bolgiano DC, Larson EB, Slichter SJ. A model
to determine required pool size for HLA-
typed community donor apheresis programs.
Transfusion 1989;29:306-10.
12. PeiR,LeeJH,ShihNJ,etal.Singlehuman
leukocyte antigen flow cytometry beads for
accurate identification of human leukocyte
antigen antibody specificities. Transplanta-
tion 2003;75:43-9.
13. Friedberg RC. Independent roles for platelet
crossmatching and HLA in the selection of
platelets for alloimmunized patients. Trans-
fusion 1994;34:215-20.
14. Kopko PM, Popovsky MA, MacKenzie MR, et
al. HLA class II antibodies in transfusion-re-
lated acute lung injury. Transfusion 2001;41:
1244-8.
15. Gorman TE, Julius CJ, Barth RF, et al. Transfu-
sion-associated graft-vs-host disease. A fatal
case caused by blood from an unrelated HLA
homozygous donor. Am J Clin Pathol 2000;
113:732-7.
16. Starzl TE, Demetris AJ, Murase N, et al.
Chimerism after organ transplantation. Curr
Opin Nephrol Hypertens 1997;6:292-8.
17. SivasaiKSR,JendrisakM,DuffyBF,etal.
Chimerism in peripheral blood of sensitized
patients waiting for renal transplantation.
Transplantation 2000;69:538-44.
18. Artlett CM, Smith JB, Jimenez SA. Identifica-
tion of fetal DNA and cells in skin lesions
from women with system sclerosis. N Engl J
Med 1998;338:1186-91.
19. Thomas ED. Bone marrow transplantation: A
review. Semin Hematol 1999;36:95-103.
20. Mickelson EM, Petersdorf E, Anasetti PM, et
al. HLA matching in hematopoietic cell trans-
plantation. Hum Immunol 2000;61:92-100.
21. KurtzbergJ,LaughlinM,GrahamML,etal.
Placental blood as a source of hematopoietic
stem cells for transplantation into unrelated
recipients. N Engl J Med 1996;335:157.
22. Aversa F, Tabilio A, Velardi A. Treatment of
high-risk acute leukemia with T cell depleted
stem cells from related donors with one fully
mismatched HLA haplotype. N Engl J Med
1998;339:1186-93.
23. Standards for histocompatibility testing. Mt.
Laurel, NJ: American Society for Histo-
compatibility and Immunogenetics, 1998.
24. Utzig MJ, Blumke M, Wolff-Vorbeck G, et al.
Flow cytometry cross-match: A method for
predicting graft rejection. Transplantation
1997;63:551-4.
25. Bryan CF, Baier KA, Nelson PW, et al. Long-
term graft survival is improved in cadaveric
renal retransplantation by flow cytometric
crossmatching. Transplantation 2000;66:
1827-32.
404 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
26. Taylor CJ, Smith SI, Morgan CH, et al. Selec-
tive omission of the donor crossmatch before
renal transplantation: Efficacy, safety, and ef-
fects of cold storage time. Transplantation
2000;69:719-23.
27. Gebel HM, Bray RA. Sensitization and sensi-
tivity: Defining the unsensitized patient.
Transplantation 2000;69:1370-4.
28. Tera sa ki PI, Cho Y, Takemot o S, et al. Twent y-
year follow-up on the effect of HLA matching
on kidney transplant survival and prediction
of future twenty-year survival. Transplant
Proc 1996;28:1144-5.
29. Terasaki PI, Cecka JM, Gjertson DW, Takemoto
S. High survival rates of kidney transplants
from spousal and living unrelated donors. N
Engl J Med 1995;333:333-6.
30. HariharanS,JohnsonCP,BresnahanBA,etal.
Improved graft survival after renal transplan-
tation in the United States, 1988 to 1996. N
Engl J Med 2000;342:605-12.
31. Ketheesan N, Tay GK, Witt CS, et al. The sig-
nificance of HLA matching in cardiac trans-
plantation. J Heart Lung Transplant 1999;18:
226-30.
32. Thorsby E. Invited anniversary review: HLA
associated diseases. Hum Immunol 1997;53:
1-11.
33. Pile KS. HLA and disease associations. Pa-
thology 1999;31:202-12.
34. Howell WM, Jones DB. The role of human
leukocyte antigen genes in the development
of malignant disease. J Clin Pathol Mol Pathol
1995;48:M302-6.
35. Hill AV. The immunogenetics of resistance to
malaria. Proc Assoc Am Physicians 1999;111:
272-7.
36. PriceP,WittC,AllcockR,etal.Thegenetic
basis for the association of the 8.1 ancestral
haplotype (A1, B8, DR3) with multiple im-
munopathological diseases. Immunol Rev
1999;167:257-74.
37. PelinZ,GuilleminaultC,RischN,etal.HLA-
DQB1*0602 homozygosity increases relative
risk for narcolepsy but not for disease sever-
ity in two ethnic groups. US Modafinil in
Narcolepsy Multicenter Study Group. Tissue
Antigens 1998;51:96-100.
38. Haldane JBS. The estimation and significance
of the logarithm of a ratio of frequencies. Ann
Hum Genet 1955;20:309-11.
39. Woolf B. On estimating the relation between
blood groups and disease. Ann Hum Genet
1955;19:251-3.
Suggested Reading
ASHI Clinical Affairs Committee. Guidelines for
clinical histocompatibility practice. Mt. Laurel,
NJ: American Society for Histocompatibility and
Immunogenetics, 1999.
PhelanDL,MickelsonEM,NoreenHS,etal.ASHI
laboratory manual. 4th ed. Mt. Laurel, NJ: Ameri-
can Society for Histocompatibility and Immuno-
genetics, 2001.
Standards for histocompatibility testing. Mt. Lau-
rel, NJ: American Society for Histocompatibility
and Immunogenetics, 1998.
Chapter 17: The HLA System 405
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 18: Pretransfusion Testing
Chapter 18
Pretransfusion Testing
THE PURPOSE OF pretransfusion
testing is to select blood compo-
nents that will not cause harm to
the recipient and will have acceptable
survival when transfused. If performed
properly, pretransfusion tests will confirm
ABO compatibility between the compo-
nent and the recipient and detect most
clinically significant unexpected antibod-
ies.
The AABB Standards for Blood Banks
and Transfusion Services1requires that the
following procedures be performed before
blood components are issued for transfu-
sion:
■Positive identification of the recipi-
ent and the recipient’s blood sample.
■ABO group and Rh typing of the re-
cipient’s blood.
■Red cell antibody detection tests for
clinically significant antibodies us-
ing the recipient’s serum or plasma.
■Comparison of current findings on
the recipient’s sample with records
of previous results.
■Confirmation of the ABO group of
red cell components.
■Confirmation of the Rh type of Rh-
negative red cell components.
■Selection of components of ABO
group and Rh type appropriate for
the recipient.
■Performanceofaserologicorcom
-
puter crossmatch.
■Labeling of products with the recipi-
ent’s identifying information.
Transfusion Requests
Requests for transfusion may be submit-
ted electronically or on paper and must
contain sufficient information for positive
recipient identification. Standards1(p36) re-
quires two independent identifiers to
identify the patient. These identifiers
could be the patient’s first and last names,
an identification number unique to that
individual, a birth date, or other identify-
ing system. Other information necessary
407
18
Copyright © 2005 by the AABB. All rights reserved.
to process the request includes identifica-
tion of the component needed, the quan-
tity, any special requests such as irradia-
tion, gender and age of the recipient (42
CFR 493.1241), and the name of the re-
sponsible physician. The diagnosis and
the recipient’s history of transfusion and
pregnancy may be helpful in problem-
solving. Each facility should have a writ-
ten policy defining request acceptance
criteria. Blood requests that lack the re-
quired information, are inaccurate, or are
illegible should not be accepted.1(p36) Tele-
phoned requests are acceptable in urgent
situations but should be documented, for
example, in a telephone log; a subsequent
request as a written authorization is to be
made within 30 days.2
Patient Identification
Collection of a properly labeled blood
sample from the intended recipient is
critical to safe blood transfusion. Most
hemolytic transfusion reactions result
from errors in sample or patient identifi-
cation.3,4 Thepersondrawingtheblood
sample must identify the intended recipi-
ent in a positive manner. Each facility
must develop and implement policies and
procedures for patient identification and
specimen collection.
Most hospitals identify patients with an
identification wristband. Ideally, this wrist-
band is placed on the patient before speci-
mencollectionandremainsonthepatient
until discharge. The same identifying infor-
mation on the specimen tube submitted for
testing will be used to label blood compo-
nents and this information will be com-
pared against the patient’s wristband at the
time of transfusion. Some hospitals use an
internally generated or commercially avail-
able identification band with a substitute or
additional “blood bank number” as the
unique patient identifier. Commercial sys-
tems vary in design: color-coded numbers
on wristbands, tubes, and units; a wristband
with an embosser for label printing; and a sys-
tem for barcoding that provides positive sam-
ple and patient identification.
Hospital policies generally require phle-
botomists to collect blood specimens only
from patients who have an attached patient
identification wristband. However, in some
circumstances, it may not be possible for
the patient to wear an identification wrist-
band, and an alternative means of positive
patient identification may be needed. The
use of wristbands is difficult when the pa-
tient has total body burns, when the patient
is an extremely premature infant, or when
the wristband is inaccessible during sur-
gery. Some facilities allow identifying infor-
mation to be placed on a patient’s ankle or
forehead. Intraoperative patient identifica-
tion procedures may allow the use of an al-
ternative identification process in lieu of an
inaccessible wristband. It is important to
remind clinical personnel that transfusions
should not be administered to a patient
who lacks positive identification.
When the patient’s identity is unknown,
an emergency identification method may
be used. This patient identification must be
attached to the patient and affixed or repro-
duced on blood samples. This identifica-
tion must be cross-referenced with the pa-
tient’s name and hospital identification
number or code when they become known.
When hospitals allow the use of confiden-
tial or alias names, the facility must have
policies and procedures that govern their
use.
Outpatients may be identified with the
use of a patient wristband for the purpose
of blood sample collection. Alternative
methods of positive patient identification
include a driver’s license or other photo-
graphic identification. Whenever possible,
the patient should be asked to state his or
her name and to provide confirmation of
408 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
birth date and address. If a discrepancy is
noted, the sample must not be collected
until the patient’s identity has been clari-
fied.
The identification of specimens used for
preadmission testing must meet the same
requirements as those used for inpatient
transfusion—there must be no doubt about
the identity of the specimen and the pa-
tient. With the advent of patient admission
on the same day as surgery, hospitals have
devised several mechanisms to identify pa-
tients when specimens have been collected
several days or weeks before surgery. One
option is to require that the patient wear an
identification wristband. Other facilities use
a unique number on specimens and on a
patient identification form that the patient
must provide on the day of surgery in order
for the preadmission specimen to be valid
for transfusion.5An alternative procedure is
to place on the patient’s medical record the
wristbandusedduringspecimencollec-
tion. This wristband would be attached to
the patient upon arrival on the day of sur-
gery after proper identification of the pa-
tient.6
Regardless of the system used, it must be
well known to all those who collect blood
specimens and be followed routinely.
Ideally, patient identification procedures
should be used as a matter of course to
identify patients for all treatment methods,
not solely transfusions.
Sample Labeling
Before leaving the patient, the phleboto-
mist must label the blood sample tubes
with two independent patient identifiers
and the date of collection. Either hand-
written or imprinted labels may be used
as long as the information on the label is
identical to that on the wristband and
request. There must be a mechanism to
identify the phlebotomist1(p37); this certifi-
cation may be placed on the label of the
tube, placed on the requisition, or docu-
mented in a computer system.
Confirming Sample Identity in the
Laboratory
When a sample is received in the labora-
tory, a trained member of the staff must
confirm that the information on the label
and on the transfusion request is identi-
cal. If there is any doubt about the iden-
tity of the patient, a new sample must be
obtained.1(p37) It is unacceptable for any-
one to correct identifying information on
an incorrectly labeled sample. Each labo-
ratory should establish policies and pro-
cedures that define identifying information
and describe how to document receipt of
mislabeled specimens.
Blood Sample
Pretransfusion testing may be performed
on either serum or plasma. Plasma may be
the preferred specimen for some methods
such as tests using gel technology. Incom-
pletely clotted blood samples may cause
small fibrin clots that trap red cells into
aggregates that could resemble aggluti-
nates. Plasma collected from patients with
high levels of fibrinogen or patients with
dysproteinemia may demonstrate rouleaux.
Rouleaux formation can be mistaken for
agglutination. Clotting may be incomplete
in specimens not intended to be anti-
coagulated, such as patients who have been
treated with heparin. Adding thrombin or
protamine sulphate to the sample usually
corrects the problem.
It is permissible to collect blood from an
infusion line. To avoid interference from re-
sidual intravenous fluid, the tubing should
be flushed with saline, and 5 mL or a vol-
ume of blood approximately twice the fluid
Chapter 18: Pretransfusion Testing 409
Copyright © 2005 by the AABB. All rights reserved.
volume in the line should be withdrawn
and discarded before sample collection.7
Appearance of Sample
The appearance of the serum or plasma
may create difficulties in detecting antibody-
induced hemolysis. Whenever possible, a
hemolyzed sample should be replaced with
a new specimen. Test results observed with
lipemic serum can be difficult to evaluate.
On occasion, it may be necessary to use
hemoglobin-tinged or lipemic serum or
plasma. If hemolyzed samples are used, it
should be noted in the patient testing re-
cords to differentiate hemolysis as a result
of an antigen-antibody reaction. Each in-
stitution should have a procedure describ-
ing the indications for using hemolyzed
and lipemic specimens.
Age of Sample
Blood samples intended for use in cross-
matching should be collected no more
than 3 days before the intended transfu-
sion unless the patient has not been preg-
nant or transfused within the preceding 3
months. If the patient’s transfusion or
pregnancy history is uncertain or unavail-
able, compatibility tests must be per-
formed on blood samples collected within
3 days of RBC transfusions.1(p38) This is to
ensure that the sample used for testing
reflects the recipient’s current immuno-
logic status because recent transfusion or
pregnancy may stimulate production of
unexpected antibodies. Because it is not
possible to predict whether or when such
antibodies will appear, a 3-day limit has
been selected as an arbitrary interval ex-
pected to be both practical and safe. It is
short enough to reflect acute changes in
immunologic status but long enough to
allow the results of preadmission testing
completed on Friday (day 0) to be used
for surgical cases performed on Monday
(day 3). The 3-day requirement applies
only to patients who have been transfused
or pregnant within the last 3 months, but
many laboratories prefer to standardize
their operations by setting a 3-day limit
on all specimens used for pretransfusion
testing.
Each institution should have a policy that
defines the length of time samples may be
used. Testing of stored specimens should
be based on the specimen storage limita-
tions in the reagent manufacturer’s infor-
mation circulars. Lack of appropriate stor-
age space may also limit the length of time
specimens are stored.
Retaining and Storing Blood Samples
The recipient’s blood specimen and a
sample of the donor’s red cells must be
stored at refrigerator temperature for at
least 7 days after each transfusion.1(p37) Do-
nor red cells may be from the remainder
of the segment used in the crossmatch or
a segment removed before issuing the
blood. If the opened crossmatch segment
is saved, it should be placed in a tube la-
beled with the unit number and sealed or
stoppered. Keeping the patient’s and do-
nor’s samples allows repeat or additional
testing if the patient experiences adverse
effects.
Serologic Testing
The patient’s ABO group and Rh type must
be determined in order to transfuse ABO-
and Rh-compatible components. Stan-
dards1(pp37,38) requires that the red cells of
the intended recipient be typed for ABO
and Rh and the serum/plasma be tested
for expected and unexpected antibodies
before components containing red cells
are issued for transfusion. There should
be written procedures for exceptions dur-
ing emergencies. When only plasma and
410 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

platelets or Cryoprecipitated AHF are be-
ing infused, historical testing information
in the patient’s record may be used. Refer
to Table 18-1 for selection of the ABO types
for red cell and plasma transfusion when
ABO-identical products are not available.
ABO Grouping and Rh Typing of the
Recipient
To determine the ABO group of the recipi-
ent, red cells must be tested with anti-A
and anti-B, and the serum or plasma with
A1and B red cells. The techniques used
and interpretation of the results are de-
scribed in Chapter 13. Any discrepant re-
sults should be resolved before blood is
given. If transfusion is necessary before
resolution, the patient should receive group
O red cells.
The patient’s red cells must be tested
with anti-D, with suitable observations or
controls to avoid a false-positive interpreta-
tion. Chapter 14 contains a more extensive
discussion of Rh typing reagents, appropri-
ate control techniques, and weak D types. If
problems in D typing arise, the patient
should be given Rh-negative blood until the
problem has been resolved. Testing a recipi-
ent’s red cells for weak D is not necessary
because giving Rh-negative cells causes no
harm to recipients with the weak D pheno-
type. Omitting the test for weak D prevents
misinterpretations arising from the presence
of a positive direct antiglobulin test (DAT).
However, some transfusion services test pa-
tient pretransfusion specimens for weak D
in order to identify patients who could be
given Rh-positive blood components, thus
reserving the Rh-negative components for
patients who are D–. Routine testing for
other Rh antigens is not required.
Detecting Unexpected Antibodies to Red
Cell Antigens
Before deciding upon routine procedures
for antibody detection, the blood bank di-
rector must approve which antibodies are
considered potentially clinically signifi-
cant. In general, an antibody is consid-
ered potentially clinically significant if an-
tibodies of that specificity have been
associated with hemolytic disease of the
fetus and newborn, a hemolytic transfusion
reaction, or notably decreased survival of
transfused red cells. Antibodies reactive at
37 C and/or in the antiglobulin test are
more likely to be clinically significant
than cold-reactive antibodies.8
Numerous serologic techniques have
been developed that are suitable for detec-
Chapter 18: Pretransfusion Testing 411
Table 18-1. Selection of Components When ABO-Identical Donors Are Not Available
ABO Requirements
Whole Blood Must be identical to that of the recipient.
Red Blood Cells Must be compatible with the recipient’s plasma.
Granulocytes Pheresis Must be compatible with the recipient’s plasma.
Fresh Frozen Plasma* Must be compatible with the recipient’s red cells.
Platelets Pheresis All ABO groups are acceptable; components compatible
with the recipient’s red cells are preferred.
Cryoprecipitated AHF All ABO groups are acceptable.
*Also see Table 21-5.
Copyright © 2005 by the AABB. All rights reserved.
tion of blood group antibodies (see Chapter
12 and Chapter 19). Goals in providing
compatible blood for a recipient are to:
■Detect as many clinically significant
antibodies as possible.
■Detect as few clinically insignificant
antibodies as possible.
■Complete the procedure in a timely
manner.
Standards1(p38) requires that tests for unex-
pected antibodies use unpooled reagent
red cells in a method that detects clinically
significant antibodies and includes an anti-
globulin test preceded by incubation at 37 C.
Each negative antiglobulin test must be
followed by a control system of IgG-sensi-
tized cells (check cells). If alternative proce-
dures are used, there must be documenta-
tion of equivalent sensitivity, and the
manufacturer’s specified controls must be
used.
The method chosen should have suffi-
cient sensitivity to detect very low levels of
antibody in a recipient’s serum. Transfusion
of antigen-incompatible red cells to a recip-
ient with a weakly reactive antibody may
result in rapid anamnestic production of
antibody, with subsequent red cell destruc-
tion. The same antibody detection proce-
dure may be used for all categories of spec-
imens, including pretransfusion and prenatal
testsonpatientsandscreeningofdonor
blood. Once a procedure has been adopted,
the method must be described in the facil-
ity’s standard operating procedures manual.
Reading and Interpreting Reactions
In serologic testing, the hemolysis or ag-
glutination that constitutes the visible end-
point of a red cell antigen-antibody inter-
action must be observed accurately and
consistently. The strength of agglutination
or degree of hemolysis observed with each
cell sample should be recorded immedi-
ately after reading. All personnel in a lab-
oratory should use the same interpreta-
tions and notations and be consistent in
grading (eg, 0-4+) reactions. Some labora-
tories prefer to use a numeric scoring (eg,
0-12) system to indicate reaction strength.
Refer to Method 1.8 for grading and scor-
ing. An optical aid such as a concave mir-
ror enhances visualization in reading tube
tests. Microscopic observation is not rou-
tinely recommended in manufacturer’s in-
serts for enhancement media. A micro-
scope can be useful in distinguishing
rouleaux from true agglutination. Micro-
scopic reading may also allow for the
detection of specific patterns of aggluti-
nation that are characteristic of some an-
tibodies. For example, anti-Sdatypically
produces small refractile agglutinates in a
sea of free red cells, giving the appearance
of a mixed-field appearance. If gel or solid
phase is used, the manufacturer’s direc-
tions must be followed for reading and in-
terpreting positive and negative reactions.
Autologous Control
An autologous control or DAT is not re-
quired as a part of pretransfusion testing.
It is of limited value, even for patients
who have recently been transfused, and
should be used only when antibody iden-
tification is required.
Practical Considerations
Antibody detection tests may be performed
in advance of, or together with, a cross-
match between the patient’s serum/plasma
and donor red cells. Performing antibody
detection tests before crossmatching per-
mits early recognition and identification
of clinically significant antibodies. This
allows for the selection of the appropriate
crossmatch procedure and acquisition of
units with special antigen blood types (eg,
e– units).
412 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Comparison with Previous Records
Results of ABO and Rh tests on a current
specimen must be compared with previ-
ous transfusion service records if there has
been prior testing during the past 12
months, and the comparison must be doc-
umented.1(p39) Errors in identification and/
or testing may be detected when discrep-
ancies are found between previous and
current ABO and Rh results.
Records are also to be reviewed for the
presence of clinically significant red cell an-
tibodies, for difficulties in testing, for the
occurrence of significant adverse reactions,
and for special transfusion requirements.1(p39)
Clinically significant red cell alloantibodies
may become undetectable in a recipient’s
serum over time. Between 30% and 35% of
antibodies become undetectable within 1
year and nearly 50% become undetectable
after10ormoreyears.
9
Crossmatching Tests
Unless there is an urgent need for blood, a
crossmatch must be performed for red
cell transfusion. The crossmatch shall use
procedures to demonstrate ABO incom-
patibility and clinically significant anti-
bodies to red cell antigens. Blood lacking
the relevant antigens is to be selected for
transfusion when a patient has clinically
significant antibody identified currently
or historically, even though the antibody
is presently nonreactive.1(pp39,40) The cross-
match shall include the antiglobulin test.
When no clinically significant antibodies
are detected in current antibody screen-
ing tests and there is no record of previ-
ous detection of such antibodies,1(p40) then
only a method to detect ABO incompati-
bility, such as an immediate-spin or com-
puter crossmatch, is required. It is very
rare for the antiglobulin phase of the
crossmatch to detect a clinically significant
unexpected antibody if the patient’s anti-
body detection test is negative.10,11
The potential benefits of omitting a rou-
tine antiglobulin crossmatch include de-
creased turnaround time, decreased work-
load, reduced reagent costs, and more effective
use of blood inventory. Omitting the anti-
globulin phase of the crossmatch for patients
who meet the criteria must be in approved
written standard operating procedures. The
methods used for serologic crossmatching
may be the same as those used for red cell
antibody detection or identification or they
may be different. For example, gel methods
may be used for both antibody detection
and identification, but the crossmatch may
be performed using a tube test.
Repeat Testing of Donor Blood
A serologic test to confirm the ABO group
of all RBC units and the Rh type of RBC
units labeled as Rh-negative must be per-
formed before transfusion. Confirmatory
testing for weak D is not required. This
confirmatory testing is to be performed
on a sample obtained from an attached
segment. Any discrepancies are to be re-
solved before the unit is issued for trans-
fusion.1(p37)
Suggested Procedures for Routine
Crossmatching
Red cells used for crossmatching must be
obtained from a segment of tubing origi-
nally attached to the blood container. For
routine tube testing, the cells may be
washed and resuspended to 2% to 5% in
saline. Washing the donor’s red cells re-
moves small fibrin clots and some cold
agglutinins that may interfere with inter-
pretation of results. Because the ratio of
serum to cells markedly affects the sensi-
tivity of agglutination tests, it is important
to stay within the 2% to 5% cell suspen-
sion range or as specified by the manufac-
Chapter 18: Pretransfusion Testing 413
Copyright © 2005 by the AABB. All rights reserved.
turer’s instructions. For example, if too many
red cells are present, weak antibodies may
be missed because too few antibody mol-
ecules bind to each cell. Many laborato-
rians find that a 2% to 3% concentration
yields the best results. For tests using col-
umn (gel) or solid-phase microplate sys-
tems, follow the manufacturer’s directions.
The simplest serologic crossmatch method
is the immediate-spin saline technique, in
which serum is mixed with saline-sus-
pended red cells at room temperature and
the tube is centrifuged immediately. The
immediate-spin crossmatch method is de-
signed to detect ABO incompatibilities be-
tween donor red cells and recipient serum.
It can be used as the sole crossmatch
method only if the patient has no present
or previous clinically significant antibodies.
Because the testing is performed at room
temperature, antibodies such as anti-M, -N,
and -P1may be detected that were not ob-
served if antibody detection tests omitted
room-temperature testing. A sample imme-
diate-spin crossmatch technique is de-
scribed in Method 3.1. An antiglobulin
crossmatch procedure that meets the re-
quirements of Standards for all routine situ-
ations is described in Method 3.2.
Type and Screen (Antibody Detection Test)
Type and screen is a policy in which the
patient’s blood sample is tested for ABO,
Rh, and unexpected antibodies, then stored
in the transfusion service for future cross-
match if a unit is needed for transfusion.
Crossmatched blood is not labeled and
reserved for patients undergoing surgical
procedures that rarely require transfu-
sion. The blood bank must have enough
donor blood available to meet unexpected
needs of patients undergoing operations
under a type and screen policy. If transfu-
sion becomes necessary, ABO- and Rh-
compatible blood can be safely released
after an immediate-spin or computer
crossmatch, if the antibody screen is neg-
ative and there is no history of clinically
significant antibodies. However, if the anti-
body screen is positive, the antibody(ies)
must be identified and antigen-negative
units for the clinically significant antibod-
ies identified must be available for use if
needed.
Routine Surgical Blood Orders
Blood ordering levels for common elective
procedures can be developed from previ-
ous records of blood use. Because surgical
requirements vary among institutions,
routine blood orders should be based on
local transfusion utilization patterns. The
surgeons, anesthesiologists, and the med-
ical director of the blood bank should
agree on the number of units required for
each procedure. See Chapter 3 for a more
detailed discussion of blood ordering pro-
tocols. Routine blood order schedules are
successful only when there is cooperation
and confidence among the professionals
involved in setting and using the guidelines.
Once a surgical blood ordering schedule
has been established, the transfusion ser-
vice routinely crossmatches the predeter-
mined number of units for each patient un-
dergoing the designated procedures. Routine
orders may need to be modified for pa-
tients with anemia, bleeding disorders, or
other conditions in which increased blood
use is anticipated. As with other circum-
stances that require rapid availability of
blood, the transfusion service staff must be
prepared to provide additional blood if the
need arises.
Computer Crossmatch
When no clinically significant antibodies
have been detected by antibody screening
414 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
and history review, it is permissible to omit
the antiglobulin phase of the crossmatch
and perform only a procedure to detect
ABO incompatibility. Computerized match-
ing of blood can be used to fulfill the re-
quirement, provided that the following
conditions have been met1(pp40,41):
■The computer system has been vali-
dated, on site, to ensure that only
ABO-compatible whole blood or red
cells have been selected for transfu-
sion.
■Two determinations of the recipi-
ent’s ABO group as specified in Stan-
dards are made, one on a current
specimen and a second one by one
of the following methods: by retest-
ing the same sample, by testing a
second current sample, or by com-
parisonwithpreviousrecords.
■The computer system contains the
unit number, component name,
ABOgroup,andRhtypeofthecom-
ponent; the confirmed donor unit
ABO group; two unique recipient
identifiers; and the recipient’s ABO
group, Rh type, and antibody screen
results.
■A method exists to verify correct en-
try of data before the release of
blood components.
■The system contains logic to alert
the user to discrepancies between
the donor ABO group and Rh type
on the unit label and the interpreta-
tion of the blood group confirmatory
test, and to ABO incompatibility be-
tween recipient and donor unit.
Butch et al12,13 and Safwenberg et al14 have
described in detail a model computer cross-
match system. Potential advantages of a
computer crossmatch include decreased
workload, reduced sample volume required
for testing, reduced exposure of personnel
to blood specimens, and better use of blood
inventory.
Compatibility Testing for Neonates Less
than 4 Months of Age
Requirements for compatibility testing for
neonates less than 4 months of age are
discussed in Chapter 24. An initial pre-
transfusionspecimenmustbeobtained
from the infant to determine ABO and Rh
type. For ABO typing, testing the cells
with anti-A and anti-B is the only test re-
quired. Serum or plasma from either the
infant or the mother may be used to de-
tect unexpected red cell antibodies and
for crossmatching. The infant’s serum
need not be tested for ABO antibodies un-
less a non-group-O infant is to receive
non-group-O cells that are incompatible
with passively acquired anti-A or anti-B.
This antibody most often comes from the
mother but can be present from group O
RBCs or plasma from incompatible plate-
lets. Test methods in this case are to in-
clude an antiglobulin phase using either
donororreagentA
1or B red cells. If no
clinically significant unexpected antibod-
ies are present, it is unnecessary to cross-
match donor red cells for the initial or
subsequent transfusions. Repeat testing
may be omitted for infants less than 4
months of age during any one hospital
admission as long as they are receiving
only group O cells.1(p42)
Interpretation of Antibody
Screening and Crossmatch
Results
Most samples tested have a negative anti-
body screen and are crossmatch-compati-
ble with units selected. A negative anti-
body screen, however, does not guarantee
that the serum does not have clinically
significant red cell antibodies, only that it
contains no antibodies that react with the
screening cells by the techniques employed.
Chapter 18: Pretransfusion Testing 415
Copyright © 2005 by the AABB. All rights reserved.
Furthermore, a compatible crossmatch
does not guarantee normal red cell sur-
vival.
Table18-2reviewsthepossiblecausesof
positive pretransfusion tests. Most of what
is known about red cell antigen and anti-
body reactions comes from work per-
formed in tube testing. This information is
not necessarily applicable to antibody de-
tection tests using other technologies such
as solid-phase microtiter plates or column
technologies. Spurious or unexpected reac-
tions in these other technologies may have
the same or other causes than those seen in
the tube tests. Depending on the antigen/
antibody reaction strength and the testing
conditions, not all of the scenarios will
result in positive tests.
The cause of the serologic problems
should be identified before transfusion.
Chapter 19 reviews techniques for problem
resolution. If the patient is found to have
clinically significant antibodies, units is-
sued for transfusion should be nonreactive
for such antigens when tested with licensed
reagents (see Selection of Units, Other
Blood Groups). When the antibody identi-
fied is considered clinically insignificant
(eg, anti-A1,-M,-N,-P
1,-Le
a,and/or-Le
b)
and is not reactive at 37 C,15 random units
may be selected for crossmatch.
Labeling and Release of
Crossmatched Blood at the
Time of Issue
Standards1(p44) requires that the following
activities take place at the time of issue:
■A tag or label indicating the recipi-
ent’s two independent identifiers,
donor unit number, and compatibil-
ity test interpretation, if performed,
must be attached securely to the
blood container.
■A final check of records maintained
in the blood bank for each unit of
blood or component must include:
1. Two independent identifiers,
one of which is usually the pa-
tient’s name.
2. The recipient’s ABO group and
Rh type.
3. The donor unit or pool identifi-
cation number.
4. Donor’s ABO and, if required,
Rh type.
5. The interpretation of the cross-
match tests (if performed).
6. Thedateandtimeofissue.
7. Special transfusion requirements
(eg, cytomegalovirus-reduced-
risk, irradiated, or antigen-neg-
ative).
■There must be a process to confirm
that the identifying information, the
request, the records, and the blood
or component are in agreement and
that discrepancies have been re-
solved before issue.
Additional records that may be of use in-
clude those that identify the person issuing
the blood and the person to whom the
blood was issued or the destination of the
unit.
After the transfusion, a record of the
transfusion shall be made a part of the pa-
tient’s medical record. This information
may be part of a computer record or a pa-
per form. Records must contain the identi-
fication of the person(s) performing the test
and, if blood is issued before the resolution
of compatibility problems, the final status
of the serologic findings.
There should be a system to ensure that
the proper blood component is issued for
theintendedpatient.Beforeissuingaunit
of blood or component, personnel must
ensure that the product is acceptable for
use, the unit is checked to make sure it
does not have an abnormal color or ap-
416 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 18: Pretransfusion Testing 417
Table 18-2. Causes of Positive Pretransfusion Tests*
Negative Antibody Screen, Incompatible Immediate-Spin Crossmatch
■Donor red cells are ABO-incompatible, caused by error in selecting donor unit, patient
specimen, or labeling the donor unit.
■Donor red cells are ABO-incompatible, caused by failure to detect weak expressions of antigens.
■Donor red cells are polyagglutinable.
■Anti-A1in the serum of an A2or A2B individual.
■Other alloantibodies are reactive at room temperature (eg, anti-M).
■Rouleaux formation.
■Cold autoantibodies (anti-I), especially if immediate spin is not tested on antibody detection
screen.
Negative Antibody Screen, Incompatible Antiglobulin Crossmatch
■Donor red cells have a positive direct antiglobulin test.
■Antibody reacts only with red cells having strong expression of a particular antigen either
because of dosage effect (eg, Rh, Kidd, Duffy, and MN antigens) or because of intrinsic variation
in antigen strength (eg, P1).
■Antibody reacts with a low-incidence antigen present on the donor red cells.
■Passively transferred antibody is present—significant levels of circulating anti-A or -B may
be present after infusion of ABO-incompatible platelets to a recipient.
Positive Antibody Screen, Compatible Crossmatches
■Auto-anti-IH (-H).
■Anti-LebH.
■Antibodies are dependent on reagent cell diluent.
■Antibodies demonstrating dosage effect and red cells of the unit are from heterozygotes
(ie, express a single dose of antigen).
Positive Antibody Screen, Incompatible Crossmatches, Negative Auto Control
■Alloantibody(ies).
■Unexpected interactions with reagent red cells.
Positive Antibody Screen, Incompatible Crossmatches, Positive Auto Control, Negative
Direct Antiglobulin Test
■Antibody to ingredient in enhancement media.
Positive Antibody Screen, Incompatible Crossmatches, Positive Auto Control
■Alloantibody is present and patient is experiencing either a delayed serologic or hemolytic
transfusion reaction.
■Passively transferred alloantibody from a derivative reactive with the recipient’s cells (eg,
intravenous immune globulin).
■Cold-reactive autoantibody.
■Warm-reactive autoantibody.
■Rouleaux formation.
■Reagent-related problems.
*Causes are dependent upon serologic methods used.
Copyright © 2005 by the AABB. All rights reserved.
pearance, the container is not leaking, and
the product is not outdated.
Final identification of the recipient and
the blood container rests with the trans-
fusionist, who must identify the patient and
donor unit and certify that identifying in-
formation on forms, tags, and labels is in
agreement (see Chapter 22).
Selection of Units
ABO Compatibility
Whenever possible, patients should re-
ceive ABO-identical blood; however, it
may be necessary to make alternative se-
lections. If the component to be trans-
fused contains 2 mL or more of red cells,
the donor’s red cells must be ABO-com-
patible with the recipient’s plasma.1(p40) Be-
cause plasma-containing components
can affect the recipient’s red cells, the
ABO antibodies in transfused plasma
should be compatible with the recipient’s
red cells when feasible. Requirements for
components and acceptable alternative
choices are summarized in Table 18-1.
Rh Type
Rh-positive blood components should
routinely be selected for D+ recipients.
Rh-negative units will be compatible but
should be reserved for D– recipients. D–
patients should receive red-cell-contain-
ing components that are Rh-negative to
avoid immunization to the D antigen. Oc-
casionally, ABO-compatible Rh-negative
components may not be available for D–
recipients. In this situation, the blood
bank physician and the patient’s physi-
cian should weigh alternative courses of
action. Depending on the childbearing
potential of the patient and the volume of
red cells transfused, it may be desirable to
administer Rh Immune Globulin to a D–
patient given Rh-positive blood.16
Other Blood Groups
Antigens other than ABO and D are not
routinely considered in the selection of
units of blood. However, if the recipient
has a clinically significant unexpected an-
tibody, antigen-negative blood should be
selected for crossmatching. If the anti-
body is weakly reactive or no longer de-
monstrable, a licensed reagent should be
used to confirm that the donor units are
antigen negative. If there is an adequate
quantity of the patient’s serum, or if an-
other patient’s serum with the same anti-
body specificity is available, and that anti-
body reacts well with antigen-positive red
cells, that serum may be used to screen
for antigen-negative units. Those units
found to be antigen negative must be
confirmed with a licensed reagent, when
available. When licensed reagents are not
available (eg, anti-Lan or anti-Yta), expired
reagents or stored serum specimens from
patients or donors can be used, provided
that controls tested on the day of use are
acceptable (see the Food and Drug Ad-
ministration Compliance Program Guid-
ance Manual, Chapter 42, Blood and Blood
Products). When crossmatch-compatible
units cannot be found, the medical direc-
torshouldbeinvolvedinthedecisionto
transfuse the patient. (See Chapter 19 and
Chapter 20 for additional information on
issuing crossmatch-incompatible units.)
Antigen-negative units are not usually
provided for the patient who has antibod-
ies that are not clinically significant.
Sometimes, problems associated with
crossmatching units for patients with
these antibodies may be avoided by alter-
ing the serologic technique used for the
crossmatch.
418 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Blood Administered in Urgent Situations
When blood is urgently needed, the pa-
tient’s physician must weigh the risk of
transfusing uncrossmatched or partially
crossmatched blood against the risk of
delaying transfusion until compatibility
testing is complete. Ideally, a transfusion
service physician should provide consul-
tation. The risk that the transfused unit
might be incompatible may be judged to
be less than the risk of depriving the pa-
tient of oxygen-carrying capacity of that
transfusion. See Chapter 21.
Required Procedures
When blood is released before pretrans-
fusion testing is complete, the records
must contain a signed statement of the
requesting physician indicating that the
clinical situation was sufficiently urgent
to require release of blood.1(p46) Such a
statement does not absolve blood bank
personnel from their responsibility to is-
sue properly labeled donor blood that is
ABO-compatible with the patient. When
urgent release is requested, blood bank
personnel should:
1. Issue uncrossmatched blood, which
should be:
a. Group O Red Blood Cells if the
patient’s ABO group is unknown.
It is preferable to give Rh-nega-
tive blood if the recipient’s Rh
type is unknown, especially if the
patient is female with the po-
tential to bear children.
b. ABOandRhcompatible,ifthere
has been time to test a current
specimen. Previous records must
not be used, nor should infor-
mation be taken from other re-
cordssuchascards,identifica
-
tion tags, or driver’s license.
2. Indicate in a conspicuous fashion on
the attached tag or label that com-
patibility testing was not complete
atthetimeofissue.
3. Begin compatibility tests and com-
plete them promptly (for massive
transfusion, see below). If incompat-
ibility is detected at any stage of test-
ing, the patient’s physician and the
transfusion service physician should
be notified immediately.
Massive Transfusion
Massive transfusion is defined as infusion,
within a 24-hour period, of a volume of
blood approximating the recipient’s total
blood volume. Exchange transfusion of an
infant is considered a massive transfusion.
Following massive transfusion, the pre-
transfusion sample no longer represents
the blood currently in the patient’s circula-
tion and its use for crossmatching has lim-
ited benefit. It is only important to confirm
ABO compatibility of units administered
subsequently. The blood bank director may
implement a more limited pretransfusion
testing protocol to be used in these situa-
tions. This protocol should be in writing to
ensure consistent application by all labora-
tory personnel.
Blood Administered After
Non-Group-Specific Transfusion
Transfusion services sometimes release
units for transfusion during emergencies
before they receive a sample for blood
typing. When it arrives, the sample is a
pretransfusion specimen. In most cases,
thesampleistestedandunitsofthatABO
group are issued for transfusion without
concern for anti-A and/or anti-B remain-
ing from the initial emergency-release
units. Because most donor units are RBCs
with comparatively little supernatant
plasma, or RBCs (Additive Solution Added)
with even less residual plasma, the risks
involved in following this practice are
Chapter 18: Pretransfusion Testing 419
Copyright © 2005 by the AABB. All rights reserved.
minimal. For example, the patient may
exhibit a transient positive DAT.
In some cases, when large volumes of
red cells are transfused, or when young
children or infants receive transfusions,
passively acquired ABO antibodies may be
detected,17 and it may be appropriate to
demonstrate compatibility of red cells of
the patient’s original ABO group with a
freshly drawn serum specimen. If the cross-
match is incompatible because of ABO an-
tibodies, transfusion with red cells of the al-
ternative group should be continued.
If the change in blood type involves only
the Rh system, return to type-specific blood
is simple because antibodies are unlikely to
be present in the plasma of either the recip-
ient or the donor. If a patient has received
blood of an Rh type other than his or her
own before a specimen has been collected
for testing, it may be difficult to determine
the correct Rh type. If there is any question
about the recipient’s D type, Rh-negative
blood should be transfused if possible. The
use of Rh Immune Globulin prophylaxis
should be considered when Rh-positive
components are transfused to Rh-negative
patients. See Chapter 21.
References
1. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
2. Code of federal regulations. Title 42 CFR
493.1241. Washington, DC: US Government
Printing Office, 2004 (revised annually).
3. Sazama K. Reports of 355 transfusion associ-
ated deaths: 1976 through 1985. Transfusion
1990;30:583-90.
4. LindenJV,WagnerK,VoytovichAE,Sheehan
J.TransfusionerrorsinNewYorkState:An
analysis of 10 years’ experience. Transfusion
2000;40:1207-13.
5. Butch SH, Stoe M, Judd WJ. Solving the same-
day admission identification problem (ab-
stract). Transfusion 1994;34(Suppl):93S.
6. AuBuchon JP. Blood transfusion options: Im-
proving outcomes and reducing costs. Arch
Pathol Lab Med 1997;121:40-7.
7. Procedures for the collection of diagnostic
blood specimens by venipuncture. 3rd ed.
NCCLS document H3-A2, approved standard.
Villanova, PA: National Committee for Clini-
cal Laboratory Standards, 1991.
8. Issitt PD, Anstee DJ. Applied blood group se-
rology. Durham, NC: Montgomery Scientific
Publications, 1998:873-905.
9. Ramsey G, Smietana SJ. Long term follow-up
testing of red cell alloantibodies. Transfusion
1994;34:122-4.
10. Oberman HA. The present and future cross-
match. Transfusion 1992;32:794-5.
11. Meyer EA, Shulman IA. The sensitivity and
specificity of the immediate-spin crossmatch.
Transfusion 1989;29:99-102.
12. Butch SH, Judd WJ, Steiner EA, et al. Elec-
tronic verification of donor-recipient com-
patibility: The computer crossmatch. Trans-
fusion 1994;34:105-9.
13. Butch SH, Judd WJ. Requirements for the
computer crossmatch (letter). Transfusion 1994;
34:187.
14. SafwenbergJ,HögmanCF,CassemarB.Com-
puterized delivery control a useful and safe
complement to the type and screen compati-
bility testing. Vox Sang 1997;72:162-8.
15. Shulman IA, Petz LD. Red cell compatibility
testing: Clinical significance and laboratory
methods. In: Petz LD, Swisher SN, Kleinman
S, eds. Clinical practice of transfusion medi-
cine.3rded.NewYork:ChurchillLivingstone,
1996:199-244.
16. Pollack W, Ascari WQ, Crispen JF, et al. Stud-
ies on Rh prophylaxis II: Rh immune prophy-
laxis after transfusion with Rh-positive blood.
Transfusion 1971;11:340-4.
17. Garratty G. Problems associated with pas-
sively transfused blood group alloantibodies.
Am J Clin Pathol 1998;109:169-77.
Suggested Reading
Beck ML, Tilzer LL. Red cell compatibility testing:
A perspective for the future. Transfus Med Rev 1996;
10:118-30.
Brecher ME. Collected questions and answers. 7th
ed. Bethesda, MD: AABB, 2001:49-56.
Butch SH for the Scientific Section Coordinating
Committee. Guidelines for implementing an elec-
tronic crossmatch. Bethesda, MD: AABB, 2003.
420 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Butch SH, Oberman HA. The computer or elec-
tronic crossmatch. Transfus Med Rev 1997;11:256-
64.
Frohn C, Dumbgen L, Brand JM, et al. Probability
of anti-D development in D– patients receiving D+
RBCs. Transfusion 2003;43:893-8.
Garratty G. How concerned should we be about
missing antibodies to low-incidence antigens?
(editorial) Transfusion 2003;43:844-7.
Issitt PD. From kill to overkill: 100 years of (per-
haps too much) progress. Immunohematology 2000;
16:18-25.
Lumadue JA, Biyd JS, Ness PM. Adherence to a strict
specimen-labeling policy decreases the incidence
of erroneous blood grouping of blood bank speci-
mens. Transfusion 1997;37:1169-72.
Padget BJ, Hannon JL. Variations in pretransfusion
practices. Immunohematology 2003;19:1-6.
Rossmann SN for the Scientific Section Coordinat-
ing Committee. Guidelines for the labeling of spec-
imens for compatibility testing. Bethesda, MD:
AABB, 2002.
Schonewille H, van Zijl AM, Wijermans PW. Im-
portance of antibodies against low-incidence RBC
antigens in complete and abbreviated cross-
matching. Transfusion 2003;43:939-44.
Chapter 18: Pretransfusion Testing 421
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens
Chapter 19
Initial Detection and
Identification of
Alloantibodies to Red Cell
Antigens
RED CELL ALLOANTIBODIES other
than naturally occurring anti-A or
-B are called unexpected red cell
alloantibodies. Depending upon the group
of patients or donors studied and the sen-
sitivity of the test methods used, alloanti-
bodies can be found in 0.3% to 38% of the
population.1,2 Alloantibodies react only with
allogeneic red cells, whereas red cell auto-
antibodies react with the red cells of the
antibody producer. Immunization to red
cell antigens may result from pregnancy,
transfusion, transplantation or from in-
jections with immunogenic material. In
some instances, no specific immunizing
event can be identified. These naturally
occurring antibodies are a result of expo-
sure to environmental, bacterial, or viral
antigens that are similar to blood group
antigens. Also, antibodies detected in se-
rologic tests can be passively acquired.
These antibodies may be acquired from
injected immunoglobulins, donor plasma,
or passenger lymphocytes in transplanted
organs or hematopoietic progenitor cells.
Significance of
Alloantibodies
Alloantibodies to red cell antigens may be
initially detected in any test that uses se-
rum or plasma (eg, ABO test, antibody de-
tection test, crossmatch) or in an eluate
prepared from red cells coated with allo-
antibody. Once an antibody is detected, its
specificity should be determined and its
clinical significance assessed.
A clinically significant red cell antibody,
although difficult to define, could be char-
acterized as an antibody that shortens the
survival of transfused red cells or has been
associated with hemolytic disease of the fe-
tus and newborn (HDFN). The degree of
clinical significance varies with antibodies
ofthesamespecificity.Someantibodies
423
19
Copyright © 2005 by the AABB. All rights reserved.
cause destruction of incompatible red cells
within hours or even minutes, others de-
crease the survival by only a few days, and
some cause no discernible red cell destruc-
tion. Antibodies of some specificities are
known to cause HDFN, whereas others may
cause a positive direct antiglobulin test
(DAT) in the fetus without clinical evidence
of HDFN.
Reported experience with other exam-
ples of antibody with the same specificity
can be used in assessing clinical signifi-
cance. Table 15-1 summarizes the expected
reactivity and clinical significance of com-
monly encountered alloantibodies. Daniels
et al3have published a review of these and
other specificities. For some antibodies, few
or no data exist, and decisions must be
based on the premise that clinically signifi-
cant antibodies are usually those active at
37 C and/or by an indirect antiglobulin test
(IAT). It is not true, however, that all anti-
bodies active in vitro at 37 C and/or by an
IAT are clinically significant.
Antibodies encountered in pretransfu-
sion testing should be identified to assess
the need to select antigen-negative red cell
components for transfusion. Patients with
clinically significant antibodies should,
whenever practical, receive red cells that
have been tested and found to lack the cor-
responding antigen. In prenatal testing, the
specificity and immunoglobulin class of an
antibody influence the likelihood of HDFN.
The results of antibody identification tests
on donor blood may be used to character-
ize the units for labeling before transfusion
and to procure blood typing reagents or
teaching samples.
General Procedures
The techniques employed for antibody de-
tection and antibody identification are
similar. Antibody identification methods
can be more focused and based on the re-
activity patterns seen in the antibody de-
tection test.
Each facility should establish which
techniques for antibody detection and
identification will be employed routinely.
Sometimes, it is valuable to develop flow-
charts to clearly guide the technologist
through the process of selecting additional
techniques to identify antibody specifici-
ties. This approach is helpful in expediting
the identification process and minimizing
unnecessary testing.
Specimen Requirements
Either serum or plasma may be used for
antibody detection and identification.
Plasma is not suitable for detecting com-
plement-activating antibodies. A 5- to
10-mL aliquot of whole blood usually
contains enough serum or plasma for
identifying simple antibody specificities;
more may be required for complex studies.
When autologous red cells are studied,
the use of a sample anticoagulated with
EDTA avoids problems associated with
the in-vitro uptake of complement com-
ponents by red cells, which may occur in
clotted samples.
Medical History
It is useful to know a patient’s clinical di-
agnosis, history of transfusions or preg-
nancies, and recent drug therapy when
performing an antibody identification.
For example, in patients who have had re-
cent red cell transfusions, the circulating
blood may contain sufficient donor cells
to make red cell phenotyping studies dif-
ficult to interpret. Special procedures to
separate the autologous red cells for typ-
ing may be required (see Method 2.15).
Other special procedures may be required
for patients with autoantibodies.
424 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Reagents
Antibody Detection Red Cells
Group O red cells suitable for antibody
screening are commercially available and
are offered as sets of either two or three
vials of single-donor red cells. Pooled an-
tibody detection cells (usually from two
donors) can only be used in testing serum
samples from donors.
Thedecisiontousetwoorthreecellsin
an antibody detection test should be based
on circumstances in each individual labo-
ratory. The reagent red cells are selected to
express the antigens associated with most
commonly encountered antibodies. Re-
agent cells licensed by the Food and Drug
Administration (FDA) for this purpose must
express the following antigens: D, C, E, c, e,
M, N, S, s, P1,Le
a,Le
b,K,k,Fy
a,Fy
b,Jk
a,and
Jkb.4Some weakly reactive antibodies react
only with red cells from donors who are ho-
mozygous for the genes controlling the ex-
pression of these antigens, a serologic phe-
nomenon called dosage. Antibodies in the
Rh,Duffy,MNS,andKiddsystemsmost
commonly demonstrate dosage. Reagent
red cells should be refrigerated when not in
use and should not be used for antibody
detection beyond their expiration date.
Antibody Identification Panels
Identification of an antibody to red cell an-
tigen(s) requires testing the serum against
a panel of selected red cell samples with
known antigen composition for the major
blood groups. Usually, they are obtained
from commercial suppliers, but institutions
may assemble their own by using red cells
from local sources. Panel cells are (except
in special circumstances) group O, allow-
ing serum of any ABO group to be tested.
Each cell of the panel is from a different
individual. The cells are selected so that,
taking all the cells into account, a distinc-
tive pattern of positive and negative reac-
tions exists for each of many antigens. To
be functional, a reagent red cell panel must
make it possible to identify with confidence
those clinically significant alloantibodies
that are most frequently encountered, such
as anti-D, -E, -K, and -Fya. The phenotypes
of the reagent red cells should be distrib-
uted such that single specificities of the
common alloantibodies can be clearly
identified and most others excluded. Ide-
ally, the pattern of reactivity for most exam-
ples of single alloantibodies will not overlap
with any other; eg, all of the K+ samples
should not be the only ones that are also
E+. It may also be valuable to include red
cell samples with a double dose of the anti-
gen in question for antibodies that fre-
quently show dosage. To lessen the possi-
bility that chance alone has caused an
apparently definitive pattern, there must be
a sufficient number of red cell samples that
lack, and sufficient red cell samples that ex-
press, most of the antigens listed in Table
19-1.
Commercially prepared panels are gen-
erally issued every 2 to 4 weeks. Each panel
contains different red cell samples with dif-
ferent antigen patterns, so it is essential to
use the phenotype listing sheet that comes
with the panel in use. Commercial cells
usually come as a 2% to 5% suspension in a
preservative medium that can be used di-
rectly from the vial. Washing is generally
unnecessary unless the media in which the
reagent cells are suspended are suspected
of interfering with alloantibody identifica-
tion.
Panel cells should not be used beyond
the expiration date; however, this is not always
practical. Most serologists use in-date reagent
cells for initial antibody identification pan-
els and, if necessary, use expired reagent
cells for exclusion or confirmation of speci-
ficity. Each laboratory must establish and
validate a policy for the use of expired re-
agent cells.5
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 425
Copyright © 2005 by the AABB. All rights reserved.

426 AABB Technical Manual
Table 19-1. A Reagent Red Cell Panel for Alloantibody Identification
Sample
#
Rh
Phenotype
Rh Kell Duffy Kidd P Lewis MNS
CC
wcDEe K Fy
aFybJkaJkbP1LeaLebMNS s
1r′r +0+00+ 0 +0 ++ + 0+ ++0+
2R
1w++0+0+ + ++ 0+ + +0 ++++
3R
1+00+0+ 0 ++ ++ 0 0+ +0+0
4R
200+++0 0 0+ 0+ + +0 0+0+
5r″r 00+0++ 0 ++ 0+ 0 0+ +++0
6 r 00+00+ 0 0+ +0 + 00 ++0+
7 r 00+00+ + 0+ +0 + 0+ +0+0
8 r 00+00+ 0 +0 0+ + +0 0+0+
9 r 00+00+ 0 0+ +0 0 0+ 0++0
10 R000++0+ 0 00 ++ + 00 ++++
+ Denotes presence of antigen; 0 denotes absence of antigen.
Copyright © 2005 by the AABB. All rights reserved.
Antiglobulin Reagents
To detect clinically significant antibodies,
most antibody identification tests include
an antiglobulin phase. Either polyspecific
or IgG-specific antiglobulin reagents may
be used. Polyspecific reagents may detect,
or detect more readily, antibodies that bind
complement. This may be of value in the
detection of certain Kidd antibodies.6Al-
though this may be advantageous in some
instances, many serologists prefer to use
IgG-specific reagents to avoid unwanted
reactivity resulting from in-vitro comple-
ment binding by cold-reactive antibodies.
Enhancement Media
Although the test system may consist solely
of serum and red cells (reagent red cells as
provided by the manufacturer or saline-
suspended red cells), most serologists use
some type of enhancement medium. Many
different media are available, including
low-ionic-strength saline (LISS), polyeth-
ylene glycol (PEG), and 22% to 30% albu-
min. For the initial antibody identifica-
tion panel, most laboratories use the
same enhancement method used in their
routine antibody detection tests. Addi-
tional enhancement techniques may be
employed for more complex studies. En-
hancement techniques are discussed later
in this chapter.
Autologous Control (Autocontrol)
It may be helpful to know how a serum
under investigation reacts with autologous
red cells. This helps determine whether
alloantibody, autoantibody, or both are
present. Serum that reacts only with the
reagent red cells usually contains only
alloantibody, whereas reactivity with both
reagent and autologous red cells suggests
the presence of autoantibody or autoanti-
body plus alloantibody. However, a pa-
tient with alloantibodies to antigens ex-
pressed on recently transfused red cells may
have circulating donor red cells coated
with alloantibodies, resulting in a positive
autocontrol. Because this result may be
misinterpreted as being due to autoanti-
body, a detailed history of recent transfu-
sions should be obtained for all patients
with a positive DAT or positive auto-
control.
The autologous control, in which serum
and autologous cells undergo the same test
conditions as serum and reagent cells, is
not the same as a DAT. Incubation and the
presence of enhancement reagents may
cause reactivity in the autologous control
that is only an in-vitro phenomenon. If the
autocontrol is positive in the antiglobulin
phase, a DAT should be performed. If the
DAT is positive, elution studies should be
considered if the patient has been recently
transfused, if there is evidence of immune
hemolysis, or if the results of serum studies
prove inconclusive. A reactive DAT may
also indicate the presence of autoantibody.
If autoantibody is detected in the serum,
adsorption studies may be necessary to
detect coexisting alloantibodies.
Basic Antibody Identification
Techniques
For initial panels, it is common to use the
same methods and test phases used in the
antibody detection test or crossmatch.
Some serologists may choose to include
an immediate centrifugation reading and/
or a room temperature incubation and
reading without adding an enhancement
medium. This may enhance the detection
of certain antibodies (anti-M, -N, -P1,-I,
-Lea,or-Le
b) and may help to explain re-
actions detected at other phases. Many in-
stitutions omit these steps to avoid finding
antibodies that react only at lower tem-
peratures and have little or no clinical sig-
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 427
Copyright © 2005 by the AABB. All rights reserved.
nificance. Test observation after 37 C in-
cubation may detect some antibodies (eg,
potent anti-D, -K, or -E) that can cause di-
rect agglutination of red cells. Other anti-
bodies (eg, anti-Lea,-Jk
a) may be detected
by their lysis of antigen-positive red cells
during the 37 C incubation. Some serolo-
gists believe that because clinically signif-
icant antibodies will be detected with the
IAT, the reading after 37 C can be safely
omitted. This omission will lessen the de-
tection of unwanted positive reactions re-
sulting from clinically insignificant cold-
reactive auto- and alloantibodies.7,8
Thephenotypeofthereactiveantibody
detection cells will provide clues to the
specificity or help exclude specificities. This
information is useful for selecting cells that
would be most informative in additional
testing.
If the patient has previously identified
antibodies, this may affect panel selection.
For example, if the patient is known to have
anti-e,itwillnotbehelpfultotestthese-
rum against a panel of 10 red cell samples,
nine of which are e+. Testing a panel of se-
lected e– red cell samples will better reveal
any newly formed antibodies.
Sometimes, the patient’s phenotype in-
fluences the selection of reagent cells. For
example, if the patient is D– and the serum
is reactive with D+ cells in the screening
test, an abbreviated panel or select cell panel
of D– red cell samples may be tested. This
can both confirm the presence of anti-D
and demonstrate the presence or absence
of additional antibodies, while minimizing
the amount of testing required.9
Interpreting Results
Antibody screening results are interpreted
as positive or negative based on the pres-
ence or absence of reactivity (eg, aggluti-
nation). Interpretation of panel results can
be a more complex process combining
technical knowledge and intuitive skills.
Panel results generally will include both
positive and negative results at different
phases of testing, each of which should be
explained by the final conclusion. Deter-
mination of the patient’s red cell pheno-
type and the probability of antibody spec-
ificity can also play roles in the final
interpretation.
Positives and Negatives
Both positive and negative reactions are
important in antibody identification. Pos-
itive reactions indicate the phase and
strength of reactivity (see Method 1.8 for
grading agglutination), which can suggest
certain specificities. Positive reactions also
can be compared to the antigen patterns
expressed by the panel cells to help assign
specificity. Single alloantibodies usually
yield definite positive and negative reac-
tions that create a clear-cut pattern with
antigen-positive and -negative reagent
red cell samples. For example, if a serum
reacts only with cells 4 and 5 of the re-
agent red cell panel shown in Table 19-1,
anti-E is very likely present. Both reactive
samples express E and all nonreactive
samples lack E.
Negative reactions are important in anti-
body identification because they allow ten-
tative exclusion of antibodies to antigens
expressed on the nonreactive cells. Exclu-
sion of antibodies is an important step in
the interpretation process and must be per-
formed to ensure proper identification of
all the antibodies present.
Exclusion or “Crossing Out”
A widely used first approach to the inter-
pretation of panel results is to exclude
specificities based on nonreactivity with
the serum tested. Such a system is some-
times referred to as a “cross-out” or “rule-
428 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
out” method. Once results have been re-
corded on the worksheet, the antigen pro-
file of the first nonreactive cell is exam-
ined. If an antigen is present on the cell
and the serum did not react with the cell,
the presence of the corresponding anti-
body may be, at least tentatively, excluded.
Many technologists will cross out that an-
tigen from the listing on the panel sheet
to facilitate the process. After all antigens
present on that cell have been crossed off,
interpretation proceeds with the other non-
reactive cells and additional specificities
are excluded. In most cases, this process
will leave a group of antibodies that still
have not been excluded.
Next, the cells reactive with the serum
are evaluated. The pattern of reactivity for
each nonexcluded specificity is compared
to the pattern of reactivity obtained with
the test serum. If there is a pattern that
matches exactly, that is most likely the
specificity of the antibody in the serum.
However, if there are remaining speci-
ficities that have not been excluded, addi-
tional testing may be needed to eliminate
remaining possibilities and to confirm the
specificity identified. This requires testing
the serum against cells selected for specific
antigenic characteristics. For example, this
approach could be employed if the pattern
of positive reactions exactly fits anti-Jka,but
anti-K and anti-S have not been excluded.
Then serum should be tested against se-
lected cells, ideally with the following phe-
notypes: Jk(a–), K–, S+; Jk(a–), K+, S–; and
Jk(a+), K–, S–. The reaction pattern with
these cells should both confirm the pres-
ence of anti-Jkaand include or exclude
anti-K and anti-S.
Although the exclusion (cross-out) ap-
proach often identifies simple antibody
specificities, it should be considered only a
provisional step, particularly if the cross-out
was completed based on the nonreactivity
of cells with weaker (eg, heterozygous) ex-
pression of an antigen (see Variations in
Antigen Expression).
Probability
To ensure that an observed pattern is not
theresultofchancealone,conclusivean
-
tibody identification requires serum to be
tested against sufficient reagent red cell
samples that lack, and that express, the
antigen that corresponds to the apparent
specificity of the antibody.
A standard approach (based on Fisher’s
exact method10) has been to require, for
each specificity identified, three antigen-
positive cells that react and three anti-
gen-negative cells that fail to react. This
standard is not always possible, but it works
well in practice, especially if cells with
strong antigen expression are available. A
somewhat more liberal approach is derived
from calculations by Harris and Hochman,11
whereby minimum requirements for a
probability (p) value of 0.05 are met by hav-
ing two positive and three negative cells, or
one positive and seven negative cells (or
the reciprocal of either combination). Com-
parative p values are shown in Table 19-2.
The use of two positive and two negative
cells is also an acceptable approach for an-
tibody confirmation.12 Additional details on
calculating probability may be found in the
suggested readings by Race and Sanger,
Menitove, and Kanter. The possibility of
false-negative results with antigen-positive
cells must be considered as well as unex-
pected positives, ie, false-positive results
due either to the presence of an additional
antibody specificity or an error in the pre-
sumptive antibody identification.
Phenotype of Autologous Red Cells
Once an antibody has been tentatively
identified in a serum, it is often helpful to
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 429
Copyright © 2005 by the AABB. All rights reserved.

demonstrate the presence or absence of
the corresponding antigen on the autolo-
gous red cells. For example, if serum from
an untransfused individual appears to
contain anti-Fyabut the autologous red
cells have a negative DAT and type as
Fy(a+), the data are clearly in conflict and
further testing is indicated.
Determination of the patient’s pheno-
type can be difficult if the patient has been
transfused recently, generally within 3
months. If a pretransfusion specimen is
available, these red cells should be used to
determine the phenotype. Alternatively, the
patient’s own red cells can be separated
from the transfused red cells and then
typed (see Methods 2.15 and 2.16). The use
of potent blood typing reagents, appropri-
ate controls, and observation for mixed-
field reactions often allow an unseparated
specimen to be phenotyped. Phenotyping
resultsonposttransfusionsamplescanbe
misleading, however, and should be inter-
preted with caution.13 If there is little uncer-
tainty about antibody identification, exten-
sive efforts to separate and type the patient’s
own red cells are not necessary. Compatible
antiglobulin crossmatch,14(p40) of antigen-
negative donor units provides additional
confirmation of antibody specificity. Defini-
tive testing can be performed on the pa-
tient’s red cells after a sufficient period
without red cell transfusion. In a chroni-
cally transfused patient, definitive testing
can be performed after an interval during
which only antigen-negative blood has
been given. Any antigen-positive red cells
detected after prolonged transfusion of an-
tigen-negative blood would presumably be
the patient’s own.
430 AABB Technical Manual
Table 19-2. Probability Values
No. Tested No. Positive No. Negative p (Fisher10) p (Harris and Hochman11)
5 3 2 0.100 0.035
6 4 2 0.067 0.022
6 3 3 0.050 0.016
7 5 2 0.048 0.015
7 4 3 0.029 0.008
8 7 1 0.125 0.049
8 6 2 0.036 0.011
8 5 3 0.018 0.005
8 4 4 0.014 0.004
9 8 1 0.111 0.043
9 7 2 0.028 0.008
9 6 3 0.012 0.003
10 9 1 0.100 0.039
10 8 2 0.022 0.007
10 7 3 0.008 0.002
10 6 4 0.005 0.001
10 5 5 0.004 0.001
Copyright © 2005 by the AABB. All rights reserved.
Complex Antibody Problems
Not all antibody identifications are sim-
ple. The exclusion procedure does not al-
ways lead directly to an answer and addi-
tional approaches may be required. Figure
19-1 shows some approaches to identify-
ing antibodies in a variety of situations
when the autocontrol is negative. Addi-
tional approaches may be needed if the
autocontrol is positive; they are discussed
later in this chapter.
Variations in Antigen Expression
For a variety of reasons, antibodies do not
always react with all cells positive for the
corresponding antigen. Basic interpreta-
tion by exclusion, as described previously,
may result in a given specificity being ex-
cluded because the sample is nonreactive
with an antigen-positive red cell sample,
despite the presence of the antibody.
Technical error, weak antibody reactivity,
and variant or weak antigenic expression
are all possible causes. Therefore, when-
ever possible, antibody specificities should
be excluded only on the basis of cells
known to bear a strong expression of the
antigen. Enhancement techniques often help
resolve problems associated with varia-
tions in antigen expression (see Methods
3.2.2, 3.2.3, 3.2.4, 3.5.5, and 3.5.6).
Zygosity
Reaction strength of some antibodies may
vary from one red cell sample to another
due to a phenomenon known as dosage,
in which antibodies react preferentially
with red cells from persons homozygous
for the gene that determines the antigen
(ie, possessing a “double dose” of the anti-
gen). Red cells from individuals heterozy-
gous for the gene may express less antigen
and may react weakly or be nonreactive.
Alloantibodies vary in their tendency to
recognize dosage. Many antibodies to an-
tigensintheRh,Duffy,MNS,andKidd
systems have this trait.
Variation in Adults and Infants
Some antigens (eg, I, P1,Le
a,andSd
a)are
expressed to varying degrees on red cells
from different adult donors. This varia-
tion is unrelated to zygosity; however, the
antigenic differences can be demon-
strated serologically. Certain antibodies
(eg, anti-I, -Lea) demonstrate weaker reac-
tivity with cord red cells than with red
cells from adults (see Table 19-3).
Changes with Storage
Blood group antibodies may give weaker
reactions with stored red cells than with
fresh red cells. Some antigens (eg, Fya,Fy
b,
M, P1,Kn
a/McCa,Bg)
17 deteriorate during
storage more rapidly than others and the
rate varies among red cells from different
donors. Because red cells from donors are
often fresher than commercial reagent cells,
some antibodies give stronger reactions
with suspensions of donor cells than with
reagent cells. Frozen storage of red cells
may result in antigen deterioration that
can cause misleading antibody identifica-
tion results.
The pH or other characteristics of stor-
age media can affect the rate of antigen de-
terioration.17,18 For example, Fyaand Fyban-
tigens may be weakened when the cells are
stored in a suspending medium of low pH
and low ionic strength. Alternatively, cer-
tain antibodies may demonstrate stronger
or weaker reactions with red cells from dif-
ferent manufacturers using different sus-
pending media. The age and nature of the
specimen must also be considered when
typing red cells. Antigens on cells from clot-
ted samples tend to deteriorate faster than
antigens on cells collected in citrate antico-
agulants such as ACD or CPD. Red cells in
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 431
Copyright © 2005 by the AABB. All rights reserved.
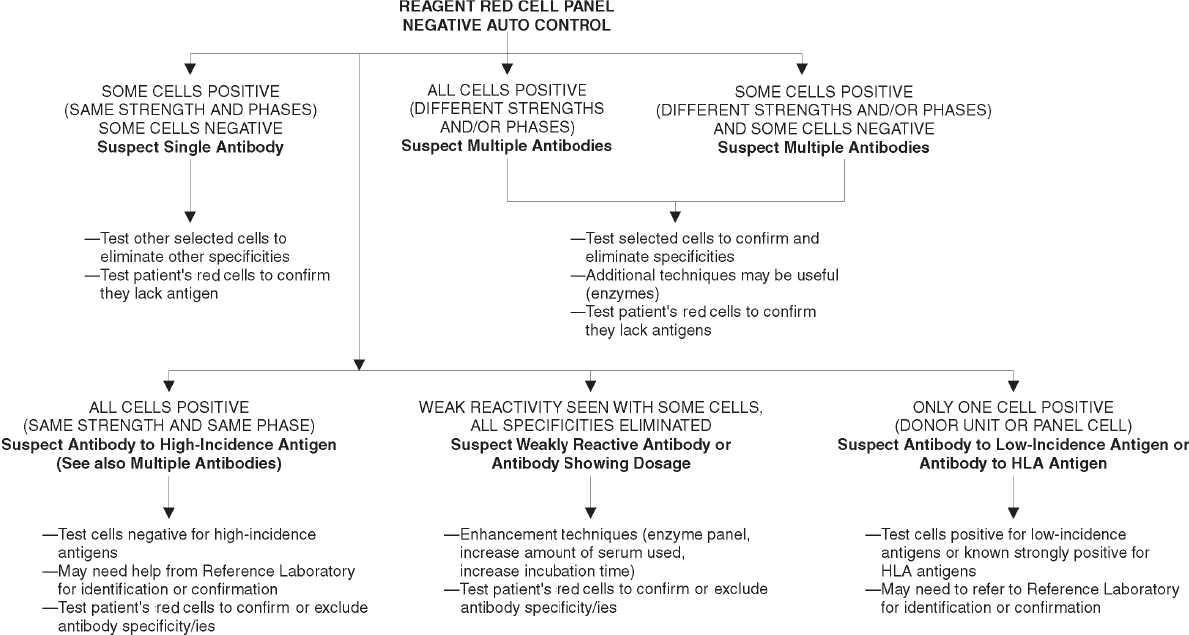

donor units collected into these anticoagu-
lants generally retain their antigens through-
out the standard shelf life of the blood com-
ponent. EDTA samples up to 14 days old
are suitable for antigen typing; however, the
manufacturer’s instructions should be con-
sulted when using commercial typing re-
agents.19
No Discernible Specificity
Factors other than variation in antigen ex-
pression may contribute to difficulty in
interpreting results of antibody identifica-
tion tests. If the reactivity obtained with
the serum is very weak and/or if the cross-
out process has excluded all likely speci-
ficities, alternative approaches to inter-
pretation should be used.
Antigens Present in Common
Instead of excluding antibodies to anti-
gens on nonreactive cells, one can ob-
serve what antigens are common to the
reactive cells. For example, if the cells re-
acting at room temperature are all P1+, yet
not all the P1+ cells react, the antibody
could be an anti-P1that does not react
with cells having a weaker expression of
the antigen. (Sometimes, such cells are
marked on the panel sheet as “+w.”) With
this in mind, one could use a method to
enhance anti-P1, such as testing at colder
temperatures.
If all the reactive cells are Jk(b+), but not
all the Jk(b+) cells react, the reactive ones
might all be Jk(a–b+), with a double-dose
expression of the antigen. Enhancement
techniques, such as enzymes, LISS, or PEG,
may then help demonstrate reactivity with
all the remaining Jk(b+) cells. Typing the
patient’s cells to confirm they lack the cor-
responding antigen can also be very help-
ful.
Inherent Variability
Nebulous reaction patterns that do not
appear to fit any particular specificity are
characteristic of antibodies, such as anti-
Bga, that react with HLA antigens on red
cells. These antigens vary markedly in
their expression on red cells from differ-
ent individuals. Rarely, a pattern of clear-
cut reactive and nonreactive tests that
cannot be interpreted is due to the incor-
rect typing of reagent red cells. If the cell
is from a commercial source, the manu-
facturer should be notified immediately
of the discrepancy.
Unlisted Antigens
Sometimes a serum sample reacts with an
antigen not routinely listed on the antigen
profile supplied by the reagent manufac-
turer; Ytbis one example. Even though se-
rum studies yield clear-cut reactive and
nonreactive tests, anti-Ytbmay not be sus-
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 433
Table 19-3. Antigen Expression on Cord Red Blood Cells*
Expression Antigens
Negative Lea,Le
b,Sd
a,Ch,Rg,AnWj
Weak I, H, P1,Lu
a,Lu
b,Yt
a,Vel,Bg,McC
a,Yk
a,S1
a,Cs
a,Hy,Gy,Jo
a,Do
a,Do
b, Fy3
Strong i, LWa,LW
b
*Modified from Reid.16
Copyright © 2005 by the AABB. All rights reserved.
pected. In such circumstances, it is useful
to ask the manufacturer for additional
phenotype information. If the appropri-
ate blood typing reagent is available, reac-
tive and nonreactive red cell samples, as
well as the autologous red cells, can be
tested. These problems often have to be
referred to an immunohematology refer-
ence laboratory.
ABO Type of Red Cells Tested
A serum sample may react with many or
all of the group O reagent red cell sam-
ples, but not with red cells of the same
ABO phenotype as the autologous red
cells. This occurs most frequently with
anti-H,-IH,or-Le
bH.GroupOandA
2red
cells have large amounts of H antigen; A1
and A1B red cells express very little H (see
Chapter 13). Sera containing anti-H or -IH
react strongly with group O reagent red
cell samples, but autologous A1or A1Bred
cells or donor cells used for crossmatch-
ing may be weakly reactive or nonreac-
tive. Anti-LebH reacts strongly with group
O, Le(b+) red cells, but reacts weakly or
not at all with Le(b+) red cells from A1or
A1B individuals. Such antibodies should
be suspected when the antibody screen,
which uses group O red cells, is strongly
reactive, but serologically compatible A1
or A1B donor samples can be found with-
out difficulty.
Multiple Antibodies
When a serum contains two or more allo-
antibodies, it may be difficult to interpret
the results of testing performed on a sin-
gle panel of reagent red cells. The pres-
ence of multiple antibodies may be sug-
gested by a variety of test results.
1. The observed pattern of reactive and
nonreactive tests does not fit that of a
single antibody. When the exclusion
approach fails to indicate a specific
pattern, it is helpful to see if the pat-
tern matches any two combined
specificities. For example, if the reac-
tive cells (see Table 19-1) are num-
bers 2, 4, 5, and 7, none of the speci-
ficities remaining after crossing-out
exactly fits that pattern, but if both K
and E are considered together, a pat-
tern is discerned. Cells 2 and 7 react
because of anti-K, cells 4 and 5 be-
cause of anti-E. If the typing pat-
terns for no two specificities fit the
reaction pattern, the possibility of
more than two antibodies must be
considered. The more antibodies a
serum contains, the more complex
the identification and exclusion of
specificities will be, but the basic
process remains the same.
2. Reactivity is present at different test
phases.
When reactivity occurs at several
phases, each phase should be evalu-
ated separately. The pattern seen at
room temperature may indicate a
different specificity from the pattern
of antiglobulin results. It is also
helpful to look at variability in the
strength of reactions seen at each
phase of testing. Table 15-2 provides
information on the characteristic re-
activity phase of several antibodies.
3. Unexpected reactions are obtained when
attempts are made to confirm the spec-
ificity of a suspected single antibody.
If a serum suspected of containing
anti-e reacts with additional samples
that are e–, another antibody may be
present or the suspected antibody
may not be anti-e. Testing a panel of
selected e– red cell samples may
help indicate an additional specific-
ity.
4. No discernible pattern emerges.
When uniform or variable reaction
strengths are observed, and dosage or
434 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
other variation in antigen strength does
not provide an explanation, addi-
tional approaches and methods of
testing are indicated. Some helpful
steps include:
a. If strong positive results were
obtained, use the exclusion
method with nonreactive cells
to eliminate some specificities
from initial consideration.
b. If weak or questionable positive
results were obtained, test the se-
rum against cells carrying a strong
expression of antigens corre-
sponding to any suspected spe-
cificities and combine this with
methods to enhance reactivity.
c. If the patient has not been re-
cently transfused, type the pa-
tient’s red cells and eliminate
from consideration specificities
that correspond to antigens on
the autologous cells.
d. Use methods to inactivate cer-
tain antigens on the red cells,
eg, enzyme treatment to render
cells negative for antigens such
as Fya,Fy
b,andS.
e. Use adsorption/elution methods
to separate antibodies.
f. Enhance antibody reactivity by
using a more sensitive method
(eg, PEG). These and other meth-
ods that may be helpful are dis-
cussed below.
Antibodies to High-Incidence Antigens
If all reagent red cell samples are reactive,
but the autocontrol is nonreactive, an
alloantibody to a high-incidence antigen
should be considered, especially if the
strength and test phase of reactions are
uniform for all cells tested. Antibodies to
high-incidence antigens can be identified
by testing red cells of selected rare pheno-
types and by testing the patient’s autologous
red cells with sera known to contain anti-
bodies to high-incidence antigens. Knowing
theraceorethnicoriginoftheantibody
producer can help in selecting additional
tests to be performed. Cells that are null
for all antigens in a system (eg, Rhnull or Ko)
or modified red cells (eg, dithiothreitol-
treated cells, see Method 3.10) can help
limit possible specificities to a particular
blood group.
If cells negative for particular high-inci-
dence antigens are not available, cells posi-
tive for lower-incidence alleles can some-
times be helpful. Weaker reactivity with
Co(a+b+) cells when compared with com-
mon Co(a+b–) cells, for instance, might
suggest anti-Coa. Antibodies to high-inci-
dence antigens may be accompanied by
other antibodies to common antigens,
which can make identification much more
difficult. Because the availability of cells
negative for high-incidence antigens is lim-
ited, it may be necessary to refer specimens
suspected of containing antibodies to high-
incidence antigens to an immunohemato-
logy reference laboratory.
Serologic Clues
Knowledge of the serologic characteristics
of particular antibodies to high-incidence
antigens can help in identification.
1. Reactivity in tests at room tempera-
ture suggests anti-H, -I, -P1,-P,-PP
1Pk
(-Tja), -LW (some), -Ge (some), -Sda,
or -Vel.
2. Lysis of reagent red cells when test-
ing with fresh serum is characteristic
of anti-Vel, -P, - PP1Pk, and -Jk3. It is
also seen with some examples of
anti-H and -I.
3. Reduced or absent reactivity in en-
zyme tests occurs with anti-Ch, -Rg,
-Inb, -JMH, or -Ge2 and is seen with
some examples of anti-Yta.
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 435
Copyright © 2005 by the AABB. All rights reserved.
4. Weak nebulous reactions in the anti-
globulin phase are often associated
with anti-Kna,-McC
a,-Yk
a,and-Cs
a.
Complement-binding autoantibodies,
such as anti-I or anti-IH, give similar
results when polyspecific antiglobu-
lin reagents are used.
5. Antibodies such as anti-U, -McCa,
-Sla,-Js
b,-Hy,-Jo
a,-Tc
a,-Cr
a,and-At
a
should be considered if the serum is
from a Black individual because the
antigen-negative phenotypes occur
almost exclusively in Blacks. Individ-
uals with anti-Kpbare almost always
White. Anti-Dibis usually found among
Asian, South American Indians, and
Native American populations.16(pp526,527)
Interpreting a Positive DAT
When a patient produces antibody di-
rected to a high-incidence antigen after
transfusion, the posttransfusion red cells
may have a positive DAT, and both serum
and eluate may react with all cells tested.
Because this pattern of reactivity is identi-
cal to that produced by many warm-reac-
tive autoantibodies that may also appear
after transfusion, these two scenarios can
be very difficult to differentiate. A post-
transfusion alloantibody to a high-inci-
dence antigen would be expected to pro-
duce a DAT of mixed-field appearance (ie,
some cells agglutinated among many
unagglutinated cells) because only the
transfused red cells would be coated with
antibody. In practice, however, weak sen-
sitization and mixed-field sensitization
can be difficult to differentiate. If a pre-
transfusion red cell sample is not available,
it may be helpful to use cell separation
procedures to isolate autologous cells for
testing. Performing a DAT on autologous
cells and/or testing the posttransfusion
serum with DAT-negative autologous cells
may help to distinguish autoantibody
from alloantibody. Chapter 15 discusses
additional serologic characteristics of an-
tibodies reacting with high-incidence red
cell antigens.
Antibodies to Low-Incidence Antigens
Reactions between a serum sample and a
single donor or reagent red cell sample
maybecausedbyanantibodytoalow-
incidence antigen, such as anti-Wra.Ifred
cells known to carry low-incidence anti-
gens are available, the serum can be
tested against them, or the one reactive
red cell sample can be tested with known
examples of antibodies to low-incidence
antigens. A single serum often contains
multiple antibodies to low-incidence an-
tigens; therefore, the expertise and re-
sources of an immunohematology refer-
ence laboratory may be required to confirm
the suspected specificities.
Serologic Strategies
If an antibody to a low-incidence antigen
is suspected, transfusion should not be
delayed while identification studies are
undertaken.Ifanantibodyintheserum
of a pregnant woman is thought to be di-
rected against a low-incidence antigen,
testing the father’s red cells can predict
the possibility of incompatibility with the
fetus, and identifying the antibody is un-
necessary. If a newborn has a positive
DAT, testing of the mother’s serum or an
eluate from the infant’s cells against the
father’s red cells (assuming they are ABO-
compatible) can implicate an antibody to
a low-incidence antigen as the probable
cause; identifying the antibody is usually
of little importance.
Some reference laboratories do not attempt
to identify antibodies to low-incidence an-
tigens because they are often only of aca-
demic interest. Identification may be made
436 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
when time permits and suitable reagents
are available.
Unexpected Positive Results
When a serum reacts with a panel cell
designated as positive for a low-incidence
antigen, further testing to exclude the an-
tibody is usually unnecessary. For every
antigen of low incidence represented on a
panel, there are many more that are not
represented and are also not excluded by
routine testing. Reactivity against low-in-
cidence antigens is not uncommon; al-
though the antigens are rare, antibodies
against some of the low-incidence anti-
gens are much less rare. Presumably, the
testing is being performed because the
serum contains some other antibody and
reactivity with the cell expressing the
low-incidence antigen is a coincidental
finding. This may complicate interpreta-
tion of the panel results but rarely re-
quires confirmation of antibody specific-
ity or typing of donor blood to ensure the
absence of the antigen. If typing is desired,
a negative crossmatch with the patient’s
serum is sufficient demonstration that the
antigen is absent. Many antibodies to
low-incidence antigens are reactive only
at temperatures below 37 C and are of
doubtful clinical significance.
When the serum reacts only with red
cells from a single donor unit or reagent
cell, the other possibilities to consider are
that the reactive donor red cells are ABO-
incompatible, have a positive DAT, or are
polyagglutinable.
Antibodies to Reagent Components and
Other Anomalous Serologic Reactions
Antibodies to a variety of drugs and addi-
tives can cause positive results in antibody
detection and identification tests. The
mechanisms are probably similar to those
discussed in Chapter 20.
Most of these anomalous reactions are
in-vitro phenomena and have no clinical
significance in transfusion therapy other
than causing laboratory problems that de-
lay needed transfusions. They rarely cause
erroneous interpretations of ABO typing
that could endanger the patient. For a more
detailed discussion, see the suggested read-
ing by Garratty.
Ingredients in the Preservative Solution
Antibodies that react with an ingredient
in the solution used to preserve reagent
red cells (eg, chloramphenicol, neomycin,
tetracycline, hydrocortisone, EDTA, sodium
caprylate, or various sugars) may aggluti-
nate cells suspended in that solution. Re-
activity may occur with cells from several
commercial sources or may be limited to
cells from a single manufacturer. The
autologous control is often nonreactive,
unless the suspension of autologous red
cells is prepared with the manufacturer’s
red cell diluent or a similar preservative.
Such reactions can often be circumvented
by washing the reagent cells with saline
beforetesting.Theroleofthepreservative
can often be confirmed by adding the me-
dium to the autologous control and con-
verting a nonreactive test to a positive test.
In some cases, however, washing the re-
agent cells does not circumvent reactivity
and the resolution may be more complex.
Ingredients in Enhancement Media
Antibodies reactive with ingredients in
other reagents, such as commercially pre-
pared LISS additives or albumin, can cause
agglutination in tests using reagent, donor,
and/or autologous red cells. Ingredients that
have been implicated include parabens (in
some LISS additives), sodium caprylate (in
some albumins), and thimerosal (in some
LISS/saline preparations). Antibody to in-
gredients in enhancement media may be
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 437
Copyright © 2005 by the AABB. All rights reserved.
suspected if the autologous control is
positive but the DAT is negative. Omitting
the enhancement medium will usually
circumvent this reactivity.
In some cases, antibodies dependent upon
reagent ingredients will show blood group
specificity, eg, paraben-dependent anti-Jka,
caprylate-dependent anti-c. The auto-
control may be reactive if the patient’s own
red cells carry the antigen, but the DAT
should be negative.
Problems with Red Cells
The age of the red cells can cause anoma-
lous serologic reactions. Antibodies exist
that react only with stored red cells; they
can cause agglutination of reagent red cells
by all techniques and enhanced reactivity
in tests with enzyme-treated red cells. Such
reactivity is not affected by washing the
red cells, and the autocontrol is usually
nonreactive. No reactivity will be seen in
tests on freshly collected red cells, ie, from
freshly drawn donor or autologous blood
samples.
The Patient with a Positive Autocontrol
No Recent Transfusions
Reactivity of serum with the patient’s own
cells may indicate the presence of auto-
antibody (see Chapter 20). If this reactiv-
ity occurs at room temperature or below,
the cause is often anti-I or another cold
autoagglutinin. Reactivity of the auto-
control in the antiglobulin phase usually
signifies a positive DAT and the possibility
of autoantibody. If, in addition, the serum
reacts with all cells tested, autoadsorption
or other special procedures may be nec-
essary to determine whether autoantibody
in the serum is masking any significant
alloantibodies. If the serum is not reactive
or shows only weak reactivity, an eluate may
demonstratemorepotentautoantibody.
A negative DAT but a positive auto-
control by an IAT is unusual and may indi-
cate antibody to a reagent constituent
causing in-vitro reactivity with all cells, in-
cluding the patient’s own. It may also indi-
cate the presence of warm autoantibodies
or cold autoagglutinins such as anti-I, -IH,
or -Pr reacting by IAT when enhancement
media are used.
Cold Autoantibodies. Potent cold auto-
agglutinins that react with all cells, includ-
ing the patient’s own, can create special
problems, especially when reactivity per-
sists at temperatures above room tempera-
ture. Cold autoagglutinins may be benign
or pathologic. (See Chapter 20 for a more
detailed discussion.)
There are different approaches to testing
a serum with a potent cold agglutinin. One
approach is to determine if the thermal
amplitude is high enough (usually 30 C or
above) that the antibody has clinical signifi-
cance. For identification purposes and de-
termination of thermal amplitude, in-vitro
autoadsorption of the serum must be avoided
by keeping the freshly collected blood
warm (37 C) until the serum is separated.
For purposes of detecting potentially clini-
cally significant antibodies, methods that
circumvent the cold autoantibody are com-
monly used.
Procedures for the detection of alloanti-
bodies in the presence of cold-reactive auto-
antibodies are discussed in Chapter 20 and
include:
1. Prewarmed techniques, in which red
cells and serum to be tested, and sa-
line used for washing, are incubated
at 37 C before they are combined (see
Method 3.3).
2. The use of anti-IgG rather than poly-
specific antiglobulin serum.
3. Cold autoadsorption, to remove auto-
antibodies but not alloantibodies.
438 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
4. Adsorption with rabbit red cells.
Dealing with Warm Autoantibodies. Pa-
tients with warm-reactive autoantibody
present in their sera create a special prob-
lem because the antibody reacts with virtu-
ally all cells tested. If such patients are to be
transfused, it is important to detect any
clinically significant alloantibodies that the
autoantibody may mask. Techniques are
discussed in Chapter 20 and Methods 4.9,
4.10, 4.11, and 4.12.
Reactivity of most warm-reactive auto-
antibodies is greatly enhanced by such
methodsasPEGandenzymes,andtolesser
extent by LISS and albumin. It may be ad-
vantageous to perform antibody detection
tests without the enhancement media usu-
ally employed. If tests are nonreactive, the
same procedure can be used for compati-
bility tests, without the need for adsorptions.
Recent Transfusions
If the autocontrol is positive in the anti-
globulin phase, there may be antibody-
coated cells in the patient’s circulation,
causing a positive DAT, which may show
mixed-field reactivity. Elution may be help-
ful, especially when tests on serum are
inconclusive. For example, a recently trans-
fused patient may have a positive auto-
control and serum that reacts weakly with
most but not all Fy(a+) red cells. It may be
possible to confirm anti-Fyaspecificity by
elution, which concentrates into a small
fluid volume the immunoglobulin mole-
cules present in small numbers on the red
cells in the whole blood sample. It is rare
for transfused cells to make the autocontrol
positive at other test phases, but it can oc-
cur, especially with a newly developing or
cold-reactive alloantibody.
IfthepositiveDATdoesnothavea
mixed-field appearance and, especially, if
the serum is reactive with all cells tested,
the possibility of autoantibody should be
considered. Detection of masked alloanti-
bodies may require allogeneic adsorptions.
Accurate phenotyping of red cells may
be difficult if the DAT is reactive in any pa-
tient, whether or not there has been recent
transfusion. A positive DAT will cause the
cells to be reactive in any test requiring the
addition of antiglobulin serum and with
some reagent antibodies (notably those in
the Rh system) in a high protein medium.
With rare exception, most monoclonal re-
agents not tested by an IAT can give valid
phenotyping results despite a positive DAT.20
Immunohematology Reference
Laboratories
When antibody problems cannot be re-
solved or when rare blood is needed, im-
munohematology reference laboratories
can provide consultation and assistance
through their access to the American Rare
Donor Program (see Method 3.13).
Selecting Blood for
Transfusion
Once an antibody has been identified, it is
important to decide its clinical signifi-
cance. Antibodies reactive at 37 C and/or
by IAT are potentially clinically significant
and those reactive at room temperature
and below are not; however, there are
many exceptions. For example, anti-Ch,
anti-Rg, and many of the Knops and Cost
antibodies have little or no clinical effect
despite reactivity by an IAT. Anti-Vel, -P,
and -PP1Pk(-Tja)mayreactonlyatcold
temperatures yet may cause red cell des-
truction in vivo. Comparison with docu-
mented cases in the literature and con-
sultation with immunohematology refer-
ence laboratories should provide guidance
about previous examples of similar speci-
ficities.
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 439
Copyright © 2005 by the AABB. All rights reserved.
Phenotyping Donor Units
Whenever possible, red cell units selected
for transfusion to a patient with a poten-
tially clinically significant antibody should
be tested and found to be negative for the
appropriate antigen. Even if the antibody
is no longer detectable, the red cells of all
subsequent transfusions to that patient
should lack the antigen, to prevent a sec-
ondary immune response. The transfu-
sion service must maintain records of all
patients in whom significant antibodies
have been previously identified.14(pp38,72) An
antiglobulin crossmatch procedure is re-
quired if the serum contains, or has previ-
ously contained, a significant antibody.
A potent example of the antibody should
be used to identify antigen-negative blood.
Often, this is a commercial antiserum, but
to save expensive or rare reagents, units can
first be tested with the patient’s serum. The
absence of antigen, in nonreactive units, can
then be confirmed with the commercial re-
agent. If the antibody is of unusual specific-
ity or one for which commercial reagents
are not available, a stored sample from the
sensitized patient can be used to select
units for transfusion at a later time, espe-
cially if the patient’s later specimens lose
reactivity. If a patient’s serum is to serve as a
typing reagent, it should be well character-
ized and retain its reactivity after storage,
and appropriate negative and weak-posi-
tive controls should be used at the time of
testing. The FDA has established the follow-
ing criteria for licensing some reagents21:
1. Anti-K, anti-k, anti-Jka,anti-Fy
a,and
anti-Cw: dilution of 1:8 to give at least
1+ reaction.
2. Anti-S, anti-s, anti-P1,anti-M,anti-I,
anti-c (saline), anti-e (saline), and
anti-A1: dilution of 1:4 to give at least
1+ reaction.
3. Most other specificities: undiluted,
must give at least a 2+ reaction.
Reagents prepared in-house from sera
that meet these dilution criteria can be
used.
Source of Antibodies
When selecting units for patients with clini-
cally significant antibodies, some serolo-
gists recommend typing the chosen units
with antibodies from two different sources,
but others consider it unnecessary, espe-
cially when potent reagents are available.
Different lots of antibody from the same
manufacturer and even reagents from dif-
ferent manufacturers may not have been
prepared from different source material
because manufacturers often share the
same resources.
Labeling Units
If a donor unit from a blood establishment
is to be labeled with the results of special
antigen typing, use of licensed (commer-
cial) reagents is preferred. If no licensed
reagent is available, the unit may be la-
beled with appropriate wording (eg,
“TestedandfoundtobenegativeforXX
antigen using unlicensed typing rea-
gents”).22 Except for results of ABO and D
typing, there is no requirement that re-
sults of antigen typing appear on the label
of donor units. The establishment may
use a tie tag attached to the unit for the
additional labeling.
When to Test
For certain antibody specificities, typing
of donor units may not be necessary and
the patient’s serum can be used to select
serologically compatible red cells. This is
especially true for antibodies that charac-
teristically react below 37 C (eg, anti-M,
-N, -P1,-Le
a,-Le
b,-A
1) and do not ordi-
narily exhibit an anamnestic response to
the transfusion of antigen-positive red
cells.
440 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
It is rarely necessary to provide anti-
gen-negative donor units as a prophylactic
measure for patients whose cells lack an
antigen but who do not have detectable an-
tibody. However, special consideration is
sometimes given to certain Rh antibodies.
When a patient of the R1R1phenotype has
anti-E detected in the serum, some workers
suggest that donor blood be negative for
both the E and c antigens,23 based on the
assumption that the stimulus to produce
the anti-E may also have stimulated an
anti-c or anti-cE that remains undetected
by routine tests. For an R2R2patient with
demonstrated anti-C, the use of C–, e– do-
nor blood may be considered. When an
antibody has not been specifically demon-
strated, but cannot conclusively be ex-
cluded, it may be appropriate to transfuse
blood that lacks the antigen.
Tests to Predict Clinical Significance
Certain laboratory procedures have been
used to predict the significance of partic-
ular antibodies. The monocyte monolayer
assay, which quantifies rosetting and/or
phagocytosis of antibody-sensitized red
cells, can be used to predict the in-vivo
clinical significance of some antibodies.
The test for antibody-dependent cellular
cytotoxicity (ADCC), which measures lysis
of antibody-coated cells, and the chemi-
luminescence assay, which measures the
respiratory release of oxygen radicals after
phagocytosis of antibody-coated cells,
have been helpful in predicting in-vivo
antibody reactivity, particularly for sever-
ity of HDFN. For cold-reactive antibodies,
in-vitro thermal amplitude studies can
predict the likelihood of in-vivo problems.
In-vivotestsmayalsobeusedtoevalu
-
ate significance of a given antibody. The
most common technique is infusion of
radiolabeled, antigen-positive red cells,
usually tagged with 51Cr. It is possible to
measure survival of 1 mL or less of infused
cells. Flow cytometry can also be used to
measure the survival of infused cells, but a
larger aliquot of red cells (about 10 mL) is
generally required. Small aliquots of incom-
patible cells may have a faster rate of de-
structionthananentireunitofredcells.
When Blood of Rare Type Is Needed
Blood of a rare type includes not only units
negative for high-incidence antigens but
also blood negative for a combination of
common antigens. When a patient has
multiple antibodies, it can be helpful to
determine the frequency of compatible
donors. To calculate this, the frequency of
random donors negative for one antigen
must be multiplied by the frequency of
donors negative for each of the other anti-
gens. For example, if a serum contains
anti-c, -Fya,and-S,and,amongrandom
donors, 18% are c–, 34% are Fy(a–), and
45% are S–, the frequency of compatible
units would be: 0.18 ×0.34 ×0.45 = 0.028.
If the patient is group O, then, because
45% of random donors are group O, 1.3%
(0.028 ×0.45) of random donors would be
compatible with the patient’s serum. If
any of these three antibodies occurred
singly, finding compatible blood would
not be too difficult. Clearly, when all three
antibodies are present, a large number of
random donors would be necessary to
provide even one unit. The preceding cal-
culation uses frequencies in populations
of European ethnicity. If the donor popu-
lation is predominantly of a different ori-
gin, frequencies for that group, if avail-
able, should be used.
When units of rare (<1 in 5000) or un-
common (<1 in 1000) type are needed, the
American Rare Donor Program can be very
helpful. This program, which can be ac-
cessed only by personnel of an accredited
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 441
Copyright © 2005 by the AABB. All rights reserved.
immunohematology reference laboratory,
can identify blood suppliers known either
to have units available (usually frozen red
cells) or to have eligible donors who may be
asked to donate (see Method 3.13).
Family members offer another potential
source of rare blood donors. Siblings are of-
ten the best source of serologically compat-
ible blood for patients with multiple anti-
bodies or antibodies to high-incidence
antigens. The absence of high-incidence
antigens usually reflects inheritance of the
same rare blood group gene from each par-
ent, and offspring of the same parents are
farmorelikelytohavethesametworare
genes than someone in the general popula-
tion. In most cases, blood from the patient’s
parents or children (and some siblings) will
carry only a single dose of the relevant anti-
gen; if transfusion is essential, and there is
no alternative to giving incompatible blood,
these heterozygous donors would be con-
sidered preferable to random donors. Occa-
sionally, blood from a parent or child also
lacks the high-incidence antigen.
In HDFN or other alloantibody-associ-
ated problems in infants, the mother, if ABO
compatible, is often the logical donor. If the
mother’s red cells are transfused, it is helpful
to retain the plasma for use as a rare reagent.
If the clinical situation allows, autologous
transfusion should be considered for patients
for whom compatible blood is difficult to
find. For some patients with multiple anti-
bodies for whom autologous transfusion is
not an option, it may be necessary to deter-
mine whether any of the antibodies is likely
to be significantly less destructive than the
others and, in a critical situation, give blood
incompatible for that particular antigen.
Frequency of Antibody Testing
Once an antibody has been identified in a
patient’s serum, how frequently should
antibody detection and identification tests
be performed? A primary antibody re-
sponse will produce detectable antibody
as early as 7 to 10 days but typically over a
period of 2 weeks to several months. A
secondary immune response produces
detectable antibody in a shorter time, as
early as 2 to 7 days and usually within 20
days. Shulman24 found that, in a small
number of patients, “new” antibodies could
be detected within 1 to 2 days after trans-
fusion. AABB Standards for Blood Banks
and Transfusion Services14(p38) requires that,
for a patient who has been pregnant or re-
ceived red cells within the preceding 3
months, antibody detection and compati-
bility tests must be performed on a speci-
men obtained within 3 days of the next
scheduled transfusion. The transfusion
service may consider testing a fresher
specimen if clinical evidence suggests
failure of recently transfused red cells to
survive as expected.
If a patient has previously identified clin-
ically significant antibodies, antigen-nega-
tive red cells must be selected for all future
transfusions, even if the antibodies are no
longer detectable. In addition, an antiglo-
bulin crossmatch must be performed using
antigen-negative red cells.
It is rarely necessary to repeat identifica-
tion of known antibodies. AABB Standards
states that in patients with previously iden-
tified antibodies, methods of testing shall
be those that identify additional clinically
significant antibodies.14(p38) Each laboratory
should define and validate methods for the
detection of additional antibodies in these
patients. Depending on the specificity of
the known antibody, repeated testing of the
patient’s serum against routine antibody
detection cells is often not informative. It is
more useful to test against cells negative for
the antigen(s) to which the patient has anti-
body and positive for other major antigens.
This allows detection of most additional
antibodies that might develop. Usually, ap-
442 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
propriate cells can be selected from avail-
able red cell panels. Selection of test cells
may be simplified if the patient’s cells are
known to express a given antigen. The se-
lected cells need not be positive for that an-
tigen because the corresponding antibody
would not be anticipated.
Selected Serologic
Procedures
Many techniques and methods may be
useful in antibody identification. Some of
the methods given here are used routinely
by many laboratories; others are alterna-
tives that may apply only in special cir-
cumstances. It is important to remember
that no single method is optimal for de-
tecting all antibodies in all samples. Any
laboratory performing antibody detection
or identification should have standard pro-
cedures for routine testing and have access
to at least some alternative approaches.
Additional procedures are available in a
variety of references (see Suggested Read-
ing).
Enhancement Techniques
When a pattern of weak reactions fails to
indicate specificity, or when the presence
of an antibody is suspected but cannot be
demonstrated, use of the following proce-
dures may be helpful. An autologous con-
trol should be included with each test
performed.
LISS and PEG
The rationale for these procedures and
some technical details are discussed in
Chapter 12 and Method 3.2. Each may be
used to enhance reactivity and reduce in-
cubation time. LISS methods include the
use of low-ionic-strength saline for resus-
pension of test cells and, more commonly,
the use of commercially available low-
ionic-strength additive media. The use of a
LISS additive requires no preparatory
stages, but care should be taken to adhere
closely to the manufacturer’s product
insert to ensure that the appropriate pro-
portion of serum to LISS is achieved.
Commercially prepared LISS additives
may include other enhancement compo-
nents besides low-ionic-strength saline.
Commercially prepared PEG additives are
also available and may contain additional
enhancing agents. Because LISS and PEG
enhance autoantibody activity, their use
may create problems with certain sam-
ples.25,26
Enzyme Techniques
Treatment of red cells with proteolytic en-
zymes enhances their reactivity with anti-
bodies in the Rh, P, I, Kidd, Lewis, and
some other blood group systems and si-
multaneously destroys or weakens reac-
tivity with other antibodies, most notably
those in the Duffy and MNS systems (see
Table 19-4). The clinical significance of
antibodies that react only with enzyme
techniques is questionable. The literature
indicates that “enzyme-only” antibodies
may have no clinical significance.28 Proce-
dures for the preparation and use of pro-
teolytic enzyme solutions are given in
Methods 3.5 through 3.5.6.
Temperature Reduction
Some alloantibodies (eg, anti-M, -N, -P1,
-Lea,-Le
b,-A
1) that react at room tempera-
ture react better at lower temperatures;
specificity may be apparent only below 22
C. An autocontrol is especially important
for tests at cold temperatures because many
sera also contain anti-I or other cold-re-
active autoantibodies.
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 443
Copyright © 2005 by the AABB. All rights reserved.
Increased Serum-to-Cell Ratio
Increasing the volume of serum incubated
with a standard volume of red cells may
enhance the reactivity of antibodies pres-
ent in low concentration. One acceptable
procedure is to mix 5 to 10 volumes of se-
rum with one volume of a 2% to 5% saline
suspension of red cells and incubate for
60 minutes at 37 C; periodic mixing dur-
ing incubation promotes contact between
red cells and antibody molecules. It is
helpful to remove the serum before wash-
ing the red cells for the antiglobulin test
because the standard three or four washes
may be insufficient to remove all the un-
bound immunoglobulin present in the
additional volume. Additional washes are
not recommended because bound anti-
body molecules may dissociate. Increas-
ing the serum-to-red cell ratio is not
appropriate for tests using a low-ionic-
strength medium or requiring specific
proportions of serum and additive.
Increased Incubation Time
For most antibodies, a 15-minute incuba-
tion period is insufficient to achieve equi-
librium and the observed reactions may
be weak, particularly in saline or albumin
media. Extending incubation to 30 to 60
minutes may improve reactivity and help
clarify the observed pattern of reactions.
Extended incubation may have a nega-
tive effect when LISS or PEG are used. If in-
cubation exceeds the recommended times
for these methods, antibody reactivity may
belost.Caremustbetakentouseallre
-
agents according to the manufacturer’s di-
rections.
Alteration of pH
Decreasing the pH of the reaction system
to 6.5 enhances the reactivity of certain
antibodies, notably some examples of
anti-M.29 If anti-M is suspected because
the only cells agglutinated are M+N–,
modifying the serum to a pH of 6.5 may
reveal a definitive pattern of anti-M reac-
tivity. The addition of one volume of 0.1 N
HCl to nine volumes of serum brings the
pH to approximately 6.5. The acidified se-
rum should be tested against known M–
cells as a control for nonspecific aggluti-
nation. Similarly, some examples of anti-P
may benefit from a lower pH.30
Low pH, however, significantly decreases
reactivity of some antibodies.31 If unbuffered
saline used for cell suspensions and for
washing has a pH much below 6.0, anti-
bodies in the Rh, Duffy, Kidd, and MNS sys-
tems may lose reactivity. Use of phos-
phate-buffered saline (see Method 1.7) can
control pH and enhance detection of anti-
bodies poorly reactive at a lower pH.32
Techniques to Isolate, Remove, or
Depress Antibody Reactivity
It is sometimes useful to decrease or elim-
inate the reactivity of an antibody. This
can be done by inhibiting the antibody with
specific substances, by physically remov-
ing immunoglobulin molecules, or by re-
moving (or weakening) corresponding anti-
gens from the red cells. Such methods can
help confirm suspected specificities and pro-
mote identification of additional antibodies.
Inhibition Tests
Soluble forms of some blood group anti-
gens exist in such body fluids as saliva,
urine, or plasma, or can be prepared from
other sources. These substances can be used
to inhibit reactivity of the corresponding
antibody. If, for example, a suspected
anti-P1does not give a definitive aggluti-
nation pattern, loss of reactivity after ad-
dition of soluble P1substance strongly
suggeststhatthisisthespecificity.Apar
-
allel dilution control with saline is essen-
tial.
444 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Inhibitioncanalsobeusedtoneutralize
antibodies that mask the concomitant pres-
ence of nonneutralizable antibodies. The
following soluble blood group substances
can be used in antibody identification tests:
1. Lewis substances.Le
aand/or Lebsub-
stances are present in the saliva of
persons who possess the Le gene. Lea
substance is present in the saliva of
Le(a+b–) individuals, and Le(a–b+)
persons have both Leaand Lebsub-
stances in their saliva (see Method
2.5). Commercially prepared Lewis
substance is also available.
2. P1substance.SolubleP
1substance is
present in hydatid cyst fluid and can
be prepared from pigeon egg whites.
P1substance is available commercially.
3. Sdasubstance. Soluble Sdablood group
substance is present in various body
fluids; the most abundant source is
urine.33 To confirm anti-Sdaspecific-
ity in a serum sample, urine from a
known Sd(a+) individual (or a pool
of urine specimens) can be used to
inhibit reactivity. Urine known to lack
Sdasubstance, or saline, should be used
as a negative control (see Method 3.11).
4. Chido and Rodgers substances. Ch
and Rg antigens are epitopes of the
fourth component of human com-
plement (C4).34,35 Anti-Ch and -Rg re-
act by an IAT with the trace amounts
of C4 present on normal red cells. If
red cells are coated in vitro with ex-
cess C4,36 these antibodies may cause
direct agglutination. A useful test to
identify anti-Ch and -Rg is by the in-
hibition of the antibodies with plasma
from Ch+, Rg+ individuals (see Method
3.9).
5. Blood group sugars. Sugars that cor-
respond to the immunodominant
configurations of A, B, H, and some
other red cell structures can be used
to inhibit antibodies. Inhibiting anti-A
or -B may allow a serum to be tested
against non-group-O cells.
Inactivation of Blood Group Antigens
Certain blood group antigens can be de-
stroyed or weakened by suitable treatment
of the cells (see Table 19-4). Modified cells
canbeusefulbothinconfirmingthepres-
ence of suspected antibodies and in de-
tecting additional antibodies. This can be
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 445
Table 19-4. Alteration of Antigens by Various Agents*
Agent Antigens Usually Denatured or Altered†
Proteolytic enzymes‡M, N, S, Fya,Fy
b,Yt
a,Ch,Rg,Pr,Tn,Mg,Mi
a/Vw, Cla,Je
a,Ny
a,
JMH, some Ge, Inb
DTT Yta,JMH,Kn
a,McC
a,Yk
a,LW
a,LW
b, all Kell, Lutheran, Dombrock,
and Cromer blood group antigens
ZZAP (a combination of
DTT and proteolytic
enzymes)
Alteration of all the antigens listed above
*Modified from Wilkinson.27
†Some antigens listed may be weakened rather than completely denatured. Appropriate controls should be used with
modified cells.
‡Different proteolytic enzymes may have different effects on certain antigens.
Copyright © 2005 by the AABB. All rights reserved.
especially helpful if the antigen is one of
high incidence and antigen-negative cells
are rare.
Proteolytic enzymes are commonly used
to alter red cell antigens. Ficin, papain,
trypsin, and bromelin, the enzymes most
frequently used, remove antigens such as
M, N, Fya,Fy
b,Xg
a,JMH,Ch,andRg(seeTa
-
ble 19-4). Depending on the specific enzyme
and method used, other antigens may be
altered or destroyed. Antigens inactivated
by one proteolytic enzyme will not neces-
sarily be inactivated by other enzymes.
Sulfhydryl reagents such as 2-amino-
ethylisothiouronium bromide (AET) or
dithiothreitol (DTT) (see Method 3.10) can
be used to weaken or destroy antigens in the
Kell system and some other anti-gens.37-39
ZZAP reagent, which contains proteolytic
enzyme and DTT,40 denatures antigens sen-
sitive to DTT (eg, all Kell system antigens)
in addition to enzyme-sensitive antigens
(see Method 4.9). Glycine-HCl/EDTA treat-
ment of red cells also destroys Bg and Kell
system antigens. However, with the excep-
tion of Eraantigen,41 other antigens outside
the Kell system that are often destroyed by
sulfhydryl reagents remain intact (see Meth-
ods 2.14 and 4.2). Chloroquine diphosphate
can be used to weaken the expression of
Class I HLA antigens (Bg antigens) on red
cells.42 Chloroquine treatment also weakens
some other antigens, including Rh antigens
(see Method 2.13).
Adsorption
Antibody can be removed from a serum
sample by adsorption to red cells carrying
the corresponding antigen. After the anti-
body attaches to the membrane-bound
antigens and the serum and cells are sep-
arated, the specific antibody remains at-
tached to the red cells. It may be possible
to harvest the bound antibody by elution.
Adsorption techniques are useful in such
situations as:
1. Separating multiple antibodies pres-
ent in a single serum.
2. Removing autoantibody activity to
permit detection of coexisting allo-
antibodies.
3. Removing unwanted antibody (often
anti-A and/or anti-B) from serum that
contains an antibody suitable for re-
agent use.
4. Confirming the presence of specific
antigens on red cells through their
ability to remove antibody of corre-
sponding specificity from previously
characterized serum.
5. Confirming the specificity of an anti-
body by showing that it can be ad-
sorbed only to red cells of a particu-
lar blood group phenotype.
Adsorption serves different purposes in
different situations; there is no single pro-
cedure that is satisfactory for all purposes.
A basic procedure for an antibody adsorp-
tion can be found in Method 3.12. The
usual serum-to-cell ratio is one volume of
serum to an equal volume of washed red
cells. To enhance antibody uptake, the pro-
portion of antigen can be increased by us-
ing a larger volume of cells. The incubation
temperature should be that at which the
antibody is optimally reactive. Pretreating
red cells with a proteolytic enzyme may en-
hance antibody uptake and reduce the
number of adsorptions required for com-
plete removal of antibody. Because some
antigens are destroyed by proteases, anti-
bodies directed against these antigens will
not be removed by enzyme-treated red
cells.
In separating mixtures of antibodies, the
selection of red cells of the appropriate
phenotype is extremely important and de-
pends on the object of the separation. If
none of the antibodies in the serum has
been identified, weakly reactive cells may
446 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
be used, on the assumption that they are
reactive with only a single antibody. The
phenotype of the person producing the an-
tibody gives a clue to what specificities
might be present, and cells intended to
separate those particular antibodies can be
chosen. If one or more antibodies have
been identified, cells lacking those antigens
are usually chosen so that only one anti-
body is removed. Adsorption requires a
substantial volume of red cells. Vials of re-
agent red cells usually will not suffice, and
blood samples from staff members or donor
units are the most convenient sources.
Elution
Elution frees antibody molecules from
sensitized red cells. Bound antibody may
be released by changing the thermody-
namics of antigen-antibody reactions, by
neutralizing or reversing forces of attrac-
tion that hold antigen-antibody complexes
together, or by disturbing the structure of
the antigen-antibody binding site. The
usual objective is to recover bound anti-
body in a usable form.
Various elution methods have been de-
scribed. Selected procedures are given in
Methods 4.1 through 4.5. No single method
is best in all situations. Use of heat or
freeze-thaw elution is usually restricted to
the investigation of HDFN due to ABO in-
compatibility because these elution proce-
dures rarely work well for antibodies out-
side the ABO system. Acid or organic solvent
methods are used for elution of warm-reac-
tive auto- and alloantibodies.
Technical factors that influence the suc-
cess of elution procedures include:
1. Incorrect technique. Such factors as
incomplete removal of organic solvents
or failure to correct the tonicity or
pH of an eluate may cause the red
cells used in testing the eluate to
hemolyze or to appear “sticky.” The
presence of stromal debris may in-
terfere with the reading of tests. Careful
technique and strict adherence to
protocols should eliminate such prob-
lems.
2. Incomplete washing. The sensitized
red cells must be thoroughly washed
before elution to prevent contami-
nation of the eluate with residual se-
rum antibody. If it is known that the
serum does not contain antibody,
saline washing may not be necessary.
Six washes with saline are usually
adequate, but more may be needed
if the serum contains a high-titer an-
tibody. To determine the efficacy of
the washing process, supernatant
fluid from the final wash phase should
be tested for antibody activity and
should be nonreactive.
3. Binding of proteins to glass surfaces.
Iftheeluateispreparedinthesame
test tube that was used during the
sensitization phase (eg, in an ad-
sorption/elution process), antibody
nonspecifically bound to the test
tube surface may dissociate during
the elution. Similar binding can also
occur from a whole blood sample if
the patient has a positive DAT and
free antibody in the serum. To avoid
such contamination, the washed red
cells should be transferred into a clean
test tube before the elution procedure
is begun.
4. Dissociation of antibody before elu-
tion. IgM antibodies, such as anti-A
or -M, may spontaneously dissociate
from the cells during the wash phase.
To minimize this loss of bound anti-
body, cold (4 C) saline can be used
for washing. Although this is not a
concern with most IgG antibodies,
some low-affinity IgG antibodies can
also be lost during the wash phase. If
such antibodies are suspected, wash-
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 447
Copyright © 2005 by the AABB. All rights reserved.
ing with cold LISS instead of normal
saline may help maintain antibody
association.
5. Instability of eluates. Dilute protein
solutions, such as those obtained by
elution into saline, are unstable. Elu-
ates should be tested as soon after
preparation as possible. Alternatively,
bovine albumin may be added to a
final concentration of 6% w/v and the
preparation stored frozen. Eluates can
also be prepared directly into anti-
body-free plasma, 6% albumin, or a
similar protein medium instead of
into saline.
Elution techniques are useful for:
1. Investigation of a positive DAT (see
Chapter 20).
2. Concentration and purification of
antibodies, detection of weakly ex-
pressed antigens, and identification
of multiple antibody specificities.
Such studies are used in conjunction
with an appropriate adsorption tech-
nique, as described above and in
Method 2.4.
3. Preparation of antibody-free red cells
for use in phenotyping or autologous
adsorption studies. Procedures used
to remove cold- and warm-reactive
autoantibodies from red cells are
discussed in Method 4.6 and Method
4.9, and a discussion of autologous
adsorption of warm-reactive auto-
antibodies appears in Chapter 20.
Combined Adsorption-Elution
Combined adsorption-elution tests can be
used to separate mixed antibodies from a
single serum, to detect weakly expressed
antigens on red cells, or to help identify
weakly reactive antibodies. The process
consists of first incubating serum with se-
lected cells, then eluting antibody from
the adsorbing red cells. Both the eluate
and treated serum can be used for further
testing. Unmodified red cells are generally
used for adsorption and subsequent elu-
tion; elution from enzyme- or ZZAP-
treated cells may create technical prob-
lems.
Use of Sulfhydryl Reagents
Sulfhydryl reagents, such as DTT and
2-mercaptoethanol (2-ME), cleave the
disulfide bonds that join the monomeric
subunits of the IgM pentamer. Intact 19S
IgM molecules are cleaved into 7S subunits,
which have altered serologic reactivity.43
The interchain bonds of 7S Ig monomers
are relatively resistant to such cleavage
(see Chapter 11 for the structure of immu-
noglobulin molecules). Sulfhydryl re-
agents are used to diminish or destroy
IgM antibody reactivity. DTT also destroys
certain red cell antigens. The applications
of DTT and 2-ME in immunohematology
include:
1. Determining the immunoglobulin
class of an antibody (see Method 3.8).
2. Identifying specificities in a mixture
of IgM and IgG antibodies, particu-
larly when an agglutinating IgM an-
tibody masks the presence of IgG
antibodies.
3. Determining the relative amounts of
IgG and IgM components of a given
specificity (eg, anti-A or -B).
4. Dissociating red cell agglutinates
caused by IgM antibodies (eg, the
spontaneous agglutination of red
cells caused by potent autoantibodies)
(see Method 2.11).
5. Dissociating IgG antibodies from red
cells using a mixture of DTT and a
proteolytic enzyme (ZZAP reagent)
(see Method 4.9).
6. Converting nonagglutinating IgG an-
tibodies into direct agglutinins.44
Commercially prepared, chemically
448 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
modified, blood typing reagents for
use in rapid saline tube, slide, or
microplate tests have been manu-
factured in this manner (see Chapter
12).
7. Destroying selected red cell antigens
(eg, those of the Kell, Dombrock,
Cartwright, and LW systems) for use
in antibody investigations (see Method
3.10).
Titration
The titer of an antibody is usually deter-
mined by testing serial twofold dilutions
of the serum against selected red cell
samples. Results are expressed as the re-
ciprocal of the highest serum dilution that
shows macroscopic agglutination. Titra-
tion values can provide information about
the relative amount of antibody present in
a serum, or the relative strength of anti-
gen expression on red cells.
Titration studies are useful in the follow-
ing situations:
1. Prenatal studies. When the antibody
is of a specificity known to cause HDFN
or its clinical significance is un-
known, the results of titration stud-
ies may contribute to the decision
about performing invasive proce-
dures, eg, amniocentesis (see Chap-
ter 23 and Method 5.3).
2. Antibody identification.Someanti
-
bodies that agglutinate virtually all
reagent red cell samples may pro-
duce an indication of specificity by
demonstrating reactivity of different
strength with different samples in ti-
tration studies. For example, potent
autoanti-I may react in the undi-
luted state with both adult and cord
red cells, but titration may reveal re-
activity at a higher dilution with
adult I+ red cells than with cord red
cells.
Most weakly reactive antibodies
lose reactivity when diluted even
modestly, but some antibodies that
give weak reactions when undiluted
continue to react at dilutions as high
as 1 in 2048. Such antibodies include
anti-Ch, -Rg, -Csa,-Yk
a,-Kn
a,-McC
a,
-JMH, and other specificities. When
weak reactions are observed in indi-
rect antiglobulin tests, titration may
be used to indicate specificity within
this group. Not all antibodies of the
specificities mentioned demonstrate
such “high titer, low avidity” charac-
teristics. Thus, although demonstra-
tion of these serologic characteristics
may help point to certain specifici-
ties, failure to do so does not elimi-
nate those possibilities. Antibodies
of other specificities may sometimes
react at high titers. Details of titra-
tion are given in Method 3.7 and
Method 3.9.
3. Separating multiple antibodies. Titra-
tion results may suggest that one an-
tibody reacts at higher dilutions than
another. This information can allow
the serum to be diluted before test-
ing against a cell panel, effectively
removing one antibody and allowing
identification of the other.
Other Methods
Methods other than traditional tube tech-
niques may be used for antibody identifi-
cation. Some are especially useful for
identifying individual antibody speci-
ficities, for dealing with small volumes of
test reagents, for batch testing, or for use
with automated systems. Such methods
include testing in capillary tubes, micro-
plates, or by solid phase; enzyme-linked
immunosorbent assays; and column ag-
glutination (eg, gel techniques). Other
methods useful in laboratories with spe-
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 449
Copyright © 2005 by the AABB. All rights reserved.
cialized equipment include radioimmuno-
assay, immunofluorescence (including flow
cytometric procedures), immunoblotting,
and immunoelectrode biosensoring. Some
of these methods are discussed in Chap-
ter 12.
References
1. Giblett ER. Blood group alloantibodies: An
assessment of some laboratory practices.
Transfusion 1977;17:299-308.
2. WalkerRH,LinDT,HatrickMB.Alloimmuni
-
zation following blood transfusion. Arch
Pathol Lab Med 1989;113:254-61.
3. Daniels G, Poole J, deSilva M, et al. The clini-
cal significance of blood group antibodies.
Tranfus Med 2002;12:287-95.
4. Code of federal regulations. Title 21 CFR Part
660.33. Washington, DC: US Government
Printing Office, 2004 (revised annually).
5. Standards Source – 4.3.2.1. (January 2004)
Bethesda, MD: AABB, 2004.
6. Howard JE, Winn LC, Gottlieb CE, et al. Clini-
cal significance of anti-complement compo-
nent of antiglobulin antisera. Transfusion
1982;22:269-72.
7. Judd WJ, Fullen DR, Steiner EA, et al. Revisit-
ing the issue: Can the reading for serologic re-
activity following 37 C incubation be omitted?
Transfusion 1999;39:295-9.
8. Issitt PD. Antibody screening: Elimination of
another piece of the test (editorial). Transfu-
sion 1999;39:229-30.
9. Shulman IA, Calderon C, Nelson JM, Nakayama
R. The routine use of Rh-negative reagent red
cells for the identification of anti-D and the
detection of non-D red cell antibodies. Trans-
fusion 1994;34:666-70.
10. Fisher RA. Statistical methods and scientific
inference. 2nd ed. Edinburgh, Scotland: Oli-
ver and Boyd, 1959.
11. Harris RE, Hochman HG. Revised p values in
testing blood group antibodies. Transfusion
1986;26:494-9.
12. Kanter MH, Poole G, Garratty G. Misinterpre-
tation and misapplication of p values in anti-
body identification: The lack of value of a p
value. Transfusion 1997;37:816-22.
13. ReidME,OyenR,StorryJ,etal.Interpreta
-
tion of RBC typing in multi-transfused pa-
tients can be unreliable (abstract). Transfu-
sion 2000;40 (Suppl):123.
14. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
15. Brendel WL. Resolving antibody problems.
In:PierceSR,WilsonJK,eds.Approachesto
serological problems in the hospital transfu-
sion service. Arlington, VA: AABB, 1985:51-72.
16. Reid ME, Lomas-Francis C. The blood group
antigenfactsbook.2nded.NewYork:Academic
Press, 2004.
17. Issitt PD, Anstee DJ. Applied blood group se-
rology. 4th ed. Durham, NC: Montgomery
Scientific Publications, 1998.
18. Malyska H, Kleeman JE, Masouredis SP, et al.
Effects on blood group antigens from storage
at low ionic strength in the presence of
neomycin. Vox Sang 1983;44:375-84.
19. Westhoff CM, Sipherd BD, Toalson LD. Red cell
antigen stability in K3EDTA. Immunohema-
tology 1993;9:109-11.
20. Rodberg K, Tsuneta R, Garratty G. Discrepant
Rh phenotyping results when testing IgG-
sensitized rbcs with monoclonal Rh reagents
(abstract). Transfusion 1995;35(Suppl):67.
21. Code of federal regulations. Title 21 CFR Part
660.25. Washington, DC: US Government
Printing Office, 2004 (revised annually).
22. Food and Drug Administration. Compliance
program guidance manual. Chapter 42; in-
spection of licensed and unlicensed blood
banks, brokers, reference laboratories, and
contractors—program 7342.001. Attachment
C—Product testing system, blood grouping
and typing (ABO and Rh), and compatibility
testing. Rockville, MD: CBER Office of Com-
munication, Training, and Manufacturers As-
sistance, 2003. [Available at http://www.fda.
gov/cber/cpg.htm.]
23. Shirey RS, Edwards RE, Ness PM. The risk of
alloimmunization to c (Rh4) in R1R1patients
who present with anti-E. Transfusion 1994;34:
756-8.
24. ShulmanIA.Controversiesinredbloodcell
compatibility testing. In: Nance SJ, ed. Immune
destruction of red blood cells. Arlington, VA:
AABB, 1989:171-99.
25. ReisnerR,ButlerG,BundyK,MooreSB.
Comparison of the polyethylene glycol anti-
globulin test and the use of enzymes in anti-
body detection. Transfusion 1996;36:487-9.
26. IssittPD,CombsMR,BumgarnerDJ,etal.
Studies of antibodies in the sera of patients
who have made red cell autoantibodies.
Transfusion 1996;36:481-6.
27. Wilkinson SL. Serological approaches to
transfusion of patients with allo- or auto-
antibodies. In: Nance SJ, ed. Immune de-
struction of red blood cells. Arlington, VA:
AABB, 1989:227-61.
28. Issitt PD, Combs MR, Bredehoeft SJ, et al.
Lack of clinical significance of “enzyme-only”
450 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
red cell alloantibodies. Transfusion 1993;33:
284-93.
29. Beattie KM, Zuelzer WW. The frequency and
properties of pH-dependent anti-M. Transfu-
sion 1965;5:322-6.
30. Judd WJ. A pH dependent autoagglutinin with
anti-P specificity. Transfusion 1975;15:373-6.
31. Bruce C, Watt AH, Hare V, et al. A serious
source of error in antiglobulin testing. Trans-
fusion 1986;26:177-81.
32. Rolih S, Thomas R, Fisher F, Talbot J. Antibody
detection errors due to acidic or unbuffered
saline. Immunohematology 1993;9:15-8.
33. Morton J, Pickles MM, Terry AM. The Sda
blood group antigen in tissues and body flu-
ids. Vox Sang 1970;19:472-82.
34. O’Neil GJ, Yang SY, Tegoli J, et al. Chido and
Rodgers blood groups are distinct antigenic
components of human complement, C4. Na-
ture 1978;273:668-70.
35. Tilley CA, Romans DG, Crookston MC. Local-
ization of Chido and Rodgers to the C4d frag-
ment of human C4 (abstract). Transfusion
1978;18:622.
36. Judd WJ, Kreamer K, Moulds JJ. The rapid
identification of Chido and Rodgers antibod-
ies using C4d-coated red blood cells. Transfu-
sion 1981;21:189-92.
37. AdvaniH,ZamorJ,JuddWJ,etal.Inactiva-
tion of Kell blood group antigens by 2-amino-
ethylisothiouronium bromide. Br J Haematol
1982;51:107-15.
38. Branch DR, Muensch HA, Sy Siok Hian AL,
Petz LD. Disulfide bonds are a requirement
for Kell and Cartwright (Yta) blood group an-
tigen integrity. Br J Haematol 1983;54:573-8.
39. Moulds J, Moulds MM. Inactivation of Kell
blood group antigens by 2-amino-ethylisothio-
uronium bromide. Transfusion 1983;23:274-5.
40. Branch DR, Petz LD. A new reagent (ZZAP)
having multiple applications in immuno-
hematology. Am J Clin Pathol 1982;78:161-7.
41. LiewYW,UchikawaM.LossofEr
aantigen in
very low pH buffers. Transfusion 1987;27:442-3.
42. Swanson JL, Sastamoinen R. Chloroquine
stripping of HLA A,B antigens from red cells
(letter). Transfusion 1985;25:439-40.
43. FreedmanJ,MastersCA,NewlandsM,etal.
Optimal conditions for use of sulphydryl
compounds in dissociating RBC antibodies.
Vox Sang 1976;30:231-9.
44. Romans DG, Tilley CA, Crookston MC, et al.
Conversion of incomplete antibodies to di-
rect agglutinins by mild reduction. Evidence
for segmental flexibility within the Fc frag-
ment of immunoglobulin G. Proc Natl Acad
Sci U S A 1977;74:2531-5.
Suggested Reading
Boorman KE, Dodd BE, Lincoln PJ. Blood group
serology.6thed.Edinburgh,Scotland:Churchill
Livingstone, 1988.
Crookston MC. Soluble antigens and leukocyte re-
lated antibodies. Part A. Blood group antigens in
plasma: An aid in the identification of antibodies.
In: Dawson RD, ed. Transfusion with “crossmatch
incompatible” blood. Washington, DC: AABB, 1975:
20-5.
Daniels G. Human blood groups. 2nd ed. Oxford,
England: Blackwell Scientific Publications, 2002.
Engelfriet CP, Overbeeke MAM, Dooren MC, et al.
Bioassays to determine the clinical significance of
red cell antibodies based on Fc receptor-induced
destruction of red cells sensitized by IgG. Transfu-
sion 1994;14:617-26.
Garratty G. In-vitro reactions with red blood cells
that are not due to blood group antibodies: A re-
view. Immunohematology 1998;14(1):1-11.
Issitt PD, Anstee DJ. Applied blood group serology.
4th ed. Durham, NC: Montgomery Scientific Pub-
lications, 1998.
JohnsonST,RudmannSV,WilsonSM,eds.Sero-
logic problem-solving strategies: A systematic ap-
proach. Bethesda, MD: AABB, 1996.
Judd WJ. Elution of antibody from red cells. In:
Bell CA, ed. A seminar on antigen-antibody reac-
tions revisited. Washington, DC: AABB, 1982:175-
221.
Judd WJ. Methods in immunohematology. 2nd ed.
Durham, NC: Montgomery Scientific Publications,
1994.
Kanter MH. Statistical analysis. In: Busch MP, Brecher
ME, eds. Research design and analysis. Bethesda,
MD: AABB, 1998:63-104.
Mallory D, ed. Immunohematology methods and
procedures. Rockville, MD: American Red Cross,
1993.
Marsh WL, Reid ME, Kuriyan M, et al. A handbook
of clinical and laboratory practices in the transfu-
sion of red blood cells. Moneta, VA: Moneta Medi-
cal Press, 1993.
Menitove JE. The Hardy-Weinberg principle: Se-
lection of compatible blood based on mathematic
principles. In: Fridey JL, Kasprisin CA, Chambers
LA,RudmannSV,eds.Numbersforbloodbankers.
Bethesda, MD: AABB, 1995:1-11.
Chapter 19: Initial Detection and Identification of Alloantibodies to Red Cell Antigens 451
Copyright © 2005 by the AABB. All rights reserved.
Mollison PL, Engelfriet CP, Contreras M. Blood
transfusion in clinical medicine. 10th ed. London:
Blackwell Scientific Publications, 1997.
Race RR, Sanger R. Blood groups in man. 6th ed.
Oxford, England: Blackwell Scientific Publications,
1975.
Reid ME, Lomas-Francis C. The blood group anti-
genfactsbook.2nded.NewYork:AcademicPress,
2004.
Rolih S. A review: Antibodies with high-titer, low-
avidity characteristics. Immunohematology 1990;
6:59-67.
Telen MJ. New and evolving techniques for anti-
body and antigen identification. In: Nance ST, ed.
Alloimmunity: 1993 and beyond. Bethesda, MD:
AABB, 1993:117-39.
452 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction
Chapter 20
The Positive Direct
Antiglobulin Test and
Immune-Mediated Red
Cell Destruction
THE DIRECT ANTIGLOBULIN test
(DAT) is generally used to determine
if red cells have been coated in vivo
with immunoglobulin, complement, or both.
A positive DAT, with or without shortened
red cell survival, may result from:
1. Autoantibodies to intrinsic red cell
antigens.
2. Alloantibodies in a recipient’s circu-
lation, reacting with antigens on re-
cently transfused donor red cells.
3. Alloantibodies in donor plasma,
plasma derivatives, or blood frac-
tions that react with antigens on the
red cells of a transfusion recipient.
4. Alloantibodies in maternal circula-
tion that cross the placenta and coat
fetal red cells.
5. Antibodies directed against certain
drugs that bind to red cell mem-
branes (eg, penicillin).
6. Nonspecifically adsorbed proteins,
including immunoglobulins, associ-
ated with hypergammaglobulinemia
or recipients of high-dose intravenous
gammaglobulin,1,2 or modification of
the red cell membrane by certain
drugs, eg, some cephalosporins.
7. Red-cell-bound complement. This may
be due to complement activation by
alloantibodies, autoantibodies, drugs,
or bacterial infection.
8. Antibodies produced by passenger
lymphocytes in transplanted organs
or hematopoietic components.3
A positive DAT does not necessarily
mean that a person’s red cells have short-
ened survival. Small amounts of both IgG
and complement appear to be present on
all red cells. A range of 5 to 90 IgG mole-
cules/red cell4and5to40C3dmolecules/
red cell5appears to be normal on the red
cells of healthy individuals.
The DAT can detect a level of 100 to 500
molecules of IgG/red cell and 400 to 1100
molecules of C3d/red cell, depending on
the reagent and technique used. Positive
DATs without clinical manifestations of im-
453
20
Copyright © 2005 by the AABB. All rights reserved.
mune-mediated red cell destruction are re-
ported in 1 in 1000 up to 1 in 14,000 blood
donors and 1% to 15% of hospital patients.4
Most blood donors with positive DATs
appear to be perfectly healthy, and most
patients with positive DATs have no obvi-
ous signs of hemolytic anemia, although
some may show slight evidence of in-
creased red cell destruction.4,6(p222) Elevated
levels of IgG or complement have been
noted on the red cells of patients with sickle
cell disease, β-thalassemia, renal disease,
multiple myeloma, autoimmune disorders,
AIDS, and other diseases with elevated se-
rum globulin or blood urea nitrogen (BUN)
levels with no clear correlation between a
positive DAT and anemia.1,2,7 Interpretation
of positive DATs should include the pa-
tient’s history, clinical data, and the results
of other laboratory tests.
The Direct Antiglobulin Test
The principles of the DAT are discussed in
Chapter 12. Although any red cells may be
tested, EDTA-anticoagulated blood sam-
ples are preferred to prevent in-vitro fixa-
tion of complement. If red cells from a
clotted blood sample have a positive DAT
due to complement, the results should be
confirmed on cells from a freshly col-
lected or EDTA-anticoagulated specimen
if those results are to be used for diagnos-
tic purposes.
Most DATs are initially performed with
a polyspecific antihuman globulin (AHG)
reagent capable of detecting both IgG and
C3d (see Method 3.6). If positive, tests with
specific anti-IgG and anticomplement re-
agents may be appropriate. Occasionally,
polyspecific AHG reagents react with cell-
bound proteins other than IgG or C3d (eg,
IgM, IgA, or other complement compo-
nents); specific reagents to distinguish
these proteins are not readily available in
most laboratories. If cord blood samples
aretobetested,itisappropriatetouse
anti-IgG only because hemolytic disease of
the fetus and newborn (HDFN) results from
the fetal red cells becoming sensitized with
maternally derived IgG antibody and com-
plement activation rarely occurs.3
The Pretransfusion DAT and the
Autologous Control
Neither the AABB, in the Standards for
Blood Banks and Transfusion Services,8nor
any other accrediting agency requires a DAT
or an autologous control (autocontrol) as
part of pretransfusion testing. Studies
have shown that eliminating the DAT/
autocontrol portion of routine pretransfu-
sion testing carries minimal risk.9
Evaluation of a Positive DAT
Extent of Testing
Clinical considerations should dictate the
extent to which a positive DAT is evalu-
ated. Dialogue with the attending physi-
cian is important. Interpretation of the
significance of serologic findings requires
knowledge of the patient’s diagnosis; re-
cent drug, pregnancy, and transfusion
history; and information on the presence
of acquired or unexplained hemolytic
anemia. The results of serologic tests alone
are not diagnostic; their significance must
be assessed in conjunction with clinical
information and such laboratory data as
hematocrit, bilirubin, haptoglobin, and
reticulocyte count. When investigating a
transfusion reaction, performance of the
DAT on postreaction specimens is part of
the initial transfusion reaction investiga-
tion. The DAT may be positive if sensi-
tized red cells have not been destroyed or
negative if hemolysis and rapid clearance
have occurred. Positive DAT results should
be further evaluated. The patient’s history
454 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
is important in interpreting a posttransfu-
sion reaction positive DAT (see Chapter 27).
Answers to the following questions may
help decide what investigations are appro-
priate:
1. Is there any evidence of in-vivo red
cell destruction? Reticulocytosis,
spherocytes observed on the periph-
eral blood film, hemoglobinemia,
hemoglobinuria, decreased serum
haptoglobin, and elevated levels of
serum unconjugated bilirubin or
lactate dehydrogenase (LDH), espe-
cially LDH1, may be associated with
increased red cell destruction. If an
anemic patient with a positive DAT
does show evidence of hemolysis,
testingtoevaluateapossibleim-
mune etiology is appropriate. IF
THEREISNOEVIDENCEOFIN-
CREASED RED CELL DESTRUC-
TION,NOFURTHERSTUDIESARE
NECESSARY, unless the patient needs
transfusion and the serum contains
incompletely identified unexpected
antibodies to red cell antigens.
2. Has the patient been recently trans-
fused? Many workers routinely at-
tempt to determine the cause of a
positive DAT when the patient has
received transfusions within the pre-
vious 3 months because the first in-
dication of a developing immune re-
sponse may be the attachment of
antibody to recently transfused red
cells. Antibody may appear as early
as7to10days(buttypically2weeks
to several months) after transfusion
in primary immunization and as
earlyas2to7days(butusuallywith
-
in 20 days) in a secondary response;
these alloantibodies could shorten the
survival of red cells already trans-
fused or given subsequently.
Studies have shown that the posi-
tive DAT and reactive eluates can
persist for more than 300 days fol-
lowing a transfusion reaction, which
is far longer than the transfused cells
would be expected to survive, sug-
gesting that autologous as well as
transfused red cells are sensitized
following a transfusion reaction.10,11 A
mixed-field appearance in the post-
transfusion DAT may or may not be
observed.
3. Is the patient receiving any drugs, such
as cephalosporins, procainamide, intra-
venous penicillin, or -methyldopa?
Cephalosporins are associated with
positive DATs; the second- and third-
generation cephalosporins can be as-
sociated with immune red cell destruc-
tion.12 In one study, 21% of patients
receiving procainamide developed a
positive DAT (three of whom had evi-
dence of hemolytic anemia).13 Ahighin-
cidence (39%) of positive DATs has been
reported in patients taking Unasyn.14
Although not commonly seen in recent
years, approximately 3% of patients re-
ceiving intravenous penicillin, at very
high doses, and 15% to 20% of patients
receiving α-methyldopa will develop a
positive DAT. However, fewer than 1%
of those patients who develop a posi-
tive DAT have hemolytic anemia. Posi-
tive DATs associated with other drugs
are rare. If a positive DAT is found in a
patient receiving such drugs, the at-
tending physician should be alerted so
that appropriate surveillance for red
cell destruction can be maintained. If
redcellsurvivalisnotshortened,no
further studies are necessary.
4. Has the patient received marrow, pe-
ripheral blood stem cells, or an organ
transplant? Passenger lymphocytes of
donor origin produce antibodies di-
rected against ABO or other antigens
on the recipient’s cells, causing a pos-
itive DAT.3
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 455
Copyright © 2005 by the AABB. All rights reserved.
5. Is the patient receiving IGIV or intra-
venous RhIG? Immune Globulin, In-
travenous (IGIV) may contain ABO
antibodies, anti-D, or, sometimes, other
antibodies. Intravenous Rh Immune
Globulin (RhIG) causes Rh-positive
patients to develop a positive DAT.15
6. Is the patient septic? Complement
activation can occur in septic patients,
leading to intravascular hemolysis.
This is most often seen in cases of
polyagglutination resulting from or-
ganisms that produce neuraminidase.
Serologic Studies
Three investigative approaches are help-
ful in the evaluation of a positive DAT.
1. Test the DAT-positive red cells with
anti-IgG and anti-C3d reagents to
characterize the types of proteins
coating the red cells.
2. Test the serum/plasma to detect and
identify clinically significant anti-
bodies to red cell antigens.
3. Test an eluate (see Methods 4.1 through
4.5) prepared from the coated red
cells with a panel of reagent red cells
to define whether the coating protein
has red cell antibody activity. When
the only coating protein is comple-
ment, eluates are frequently nonreac-
tive. However, an eluate from the pa-
tient’s red cells coated only with
complement should be tested if there
is clinical evidence of antibody-me-
diated hemolysis. The eluate prepa-
ration can concentrate small amounts
of IgG that may not be detectable on
direct testing using routine methods.
Results of these tests combined with
the patient’s history and clinical data
should assist in classification of the
problems involved.
See Appendix 20-1 for an example of an
algorithm for investigating a positive DAT
(excluding investigation of HDFN).
Elution
Elution frees antibody from sensitized red
cells and recovers antibody in a usable form.
Details of eluate preparation are given in
Chapter 19 and in Methods 4.1 through
4.5. Commercial elution kits are also
available. Table 20-1 lists the advantages
and disadvantages of several common
elution methods; no single elution method
is ideal in all situations. Although many
elution methods damage or destroy the
red cells, certain techniques (see Methods
2.11, 2.12, 2.13, and 2.14) remove anti-
body but leave the cells sufficiently intact
to allow testing for various antigens or for
use in adsorption procedures. Some anti-
gens may be altered by elution, however,
and appropriate controls are essential.
In cases of HDFN or hemolytic transfu-
sion reactions, specific antibody (or anti-
bodies) is usually detected in the eluate,
which may or may not be detectable in the
serum.Inthecaseoftransfusionreactions,
newly developed antibodies initially detect-
able only in the eluate are usually detect-
able in the serum after about 14 to 21 days.
Eluate preparation from the patient’s red
cells often concentrates antibody activity
and may facilitate identification of weakly
reactive serum antibodies.
When the eluate reacts with all the cells
tested, autoantibody is the most likely ex-
planation, especially if the patient has not
been recently transfused. WHEN NO UN-
EXPECTED ANTIBODIES ARE PRESENT IN
THE SERUM, AND IF THE PATIENT HAS
NOT BEEN RECENTLY TRANSFUSED, NO
FURTHER SEROLOGIC TESTING OF AN
AUTOANTIBODY IS NECESSARY.
A nonreactive eluate prepared from IgG-
coated red cells may have several causes.
456 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Onecausemaybethattheeluatewasnot
tested against cells positive for the corre-
sponding antigen, notably group A or
group B cells, or antigens of low incidence,
which are absent from most reagent cell
panels. If a non-group-O patient has re-
ceived plasma containing anti-A or anti-B
(as in transfusion of group O platelets), and
the recipient appears to have immune
hemolysis, the eluate can be tested against
A and/or B cells. If the expected ABO anti-
bodies are not detected, other causes of the
positive DAT should be sought. It may be
appropriate to test the eluate against red
cells from recently transfused donor units,
which could have caused immunization to
a rare antigen, or, in HDFN, against cells
from the father, from whom the infant may
have inherited a rare gene. Pursuing the
cause of a nonreactive eluate for patients
with no evidence of hemolysis is usually
not indicated. Toy et al1showed that 79% of
hospital patients with a positive DAT have a
nonreactive eluate. It is suggested that at
least one contributing factor to these posi-
tive DAT results is nonspecific uptake of
proteins on the red cells, which occurs in
patients with elevated gamma globulin lev-
els.1,2
Reactivity of eluates can be enhanced by
testing them against enzyme-treated cells
or by the use of enhancement techniques
such as polyethylene glycol (PEG). Anti-
body reactivity can be increased by the use
of a concentrated eluate, either by alter-
ation of the fluid-to-cell ratio or by use of
commercial concentration devices. Wash-
ing the red cells with low-ionic-strength sa-
line (LISS) or cold wash solutions may pre-
vent the loss of antibody while the cells are
being prepared for elution.
Certain elution methods give poor re-
sults with certain antibodies. When eluates
are nonreactive yet clinical signs of red cell
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 457
Table 20-1. Antibody Elution Techniques
Method Advantages Disadvantages
Heat (56 C) Good for ABO-HDFN; quick and
easy
Poor recovery of other blood
group allo- and
autoantibodies
Freeze-thaw Good for ABO-HDFN; quick
method; requires small vol-
ume of red cells
Poor recovery of other blood
group allo- and
autoantibodies
Cold acid Quick and easy; adequate for
most warm auto- and
alloantibodies; commercial
kits available
Possible false-positive elution
(see Leger et al16)
Digitonin acid Nonhazardous; good recovery of
most antibodies
Time-consuming washing of
stroma
Dichloromethane/
Methylene chloride
Noncarcinogenic, nonflamma-
ble; good for IgG auto- and
alloantibodies
Vapors harmful
Compiled from Judd17 andSouthetal.
18
Copyright © 2005 by the AABB. All rights reserved.
destruction are present, elution by a differ-
ent method may be helpful. If both serum
and eluate are nonreactive at all test phases
and if the patient has received high-dose
intravenous penicillin or other drug ther-
apy, testing to demonstrate drug-related
antibodies should be considered.
Immune-Mediated
Hemolysis
Immune-mediated hemolysis (immune
hemolysis) is the shortening of red cell
survival by the product(s) of an immune
response. If marrow compensation is ade-
quate, the reduced red cell survival may
not result in anemia. Immune hemolysis
is only one cause of hemolytic anemia,
and many causes of hemolysis are unre-
lated to immune reactions. The serologic
investigations carried out in the blood bank
do not determine whether a patient has a
“hemolytic” anemia. The diagnosis of
hemolytic anemia rests on clinical find-
ings and such laboratory data as hemo-
globin or hematocrit values; reticulocyte
count; red cell morphology; bilirubin,
haptoglobin, and LDH levels; and, some-
times, red cell survival studies. The sero-
logic findings help determine whether the
hemolysis has an immune basis and, if so,
what type of immune hemolytic anemia is
present. This is important because the
treatment for each type is different.
The terms hemolysis and hemolytic are
frequently used to indicate both intravas-
cular and extravascular red cell destruction;
however, this may be misleading. In-vivo
lysis of cells and release of free hemoglobin
within the intravascular compartment (ie,
resulting in hemoglobinemia and hemoglo-
binuria) is uncommon and, often, dramatic.
Extravascular hemolysis, which is more
common, is characterized by an increase in
serum bilirubin, but not by hemoglobinemia
and hemoglobinuria. As a description of
in-vitro antibody reactivity, hemolysis or
lysis of the red cells with release of free he-
moglobin to the surrounding media is both
obvious and rare.
Immune hemolytic anemias can be clas-
sified in various ways. One classification
system is shown in Table 20-2. Autoim-
mune hemolytic anemias (AIHAs) are sub-
divided into five major types: warm anti-
body AIHA (WAIHA), cold agglutinin
syndrome (CAS), mixed-type AIHA, parox-
ysmal cold hemoglobinuria (PCH), and
DAT-negative AIHA. Not all cases fit neatly
into these categories. Drugs (discussed in a
later section of this chapter) may also in-
duce immune hemolysis. The prevalence of
each type can vary depending on the pa-
tient population studied. Table 20-3 shows
the serologic characteristics of the autoim-
muneanddrug-inducedhemolyticane-
mias.
DATs performed with IgG- and C3-spe-
cific AHG reagents as well as the serum and
eluate studies described earlier can be used
to help classify AIHAs. Three additional pro-
cedures may be useful: a cold agglutinin ti-
ter and thermal amplitude studies (Meth-
ods 4.7 and 4.8) and the Donath-Landsteiner
test for PCH (Method 4.13).
The binding of antibody to red cells does
not, in itself, damage the cells. It is the phe-
nomena that the bound antibody-antigen
complex promotes that may eventually
damage cells. These include complement
binding, adherence to Fc receptors on
macrophages leading to phagocytosis, and
cytotoxic lysis. The IgG subclass of bound
antibody may be significant. IgG1 is the
subclass most commonly found, some-
times alone but often in combination with
other subclasses. The other IgG subclasses
occur more often in combination with
other subclasses than alone. In general,
IgG3 antibodies have the most destructive
458 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

effects, followed by IgG1. IgG2 antibodies
are associated with less destruction and
IgG4 with little to no destruction.
The number of antibody molecules per
red cell also plays a role. The number of an-
tibody molecules on the red cells of appar-
ently healthy blood donors with positive
DATs (<200 molecules/red cell) is far less
than that usually seen in patients with
AIHA.4,5 Some patients with apparent im-
mune hemolysis may have negative DATs.
Warm Antibody Autoimmune Hemolytic
Anemia
The most common type of AIHA is associ-
ated with warm-reactive (37 C) antibod-
ies. Typical serologic findings are described
below.
DAT
When IgG-specific and complement-spe-
cificAHGreagentsareused,threepat
-
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 459
Table 20-2. Classification of Immune Hemolytic Anemias
Autoimmune Hemolytic Anemia (AIHA)
1. Warm autoimmune hemolytic anemia
a. primary (idiopathic)
b. secondary [to such conditions as lymphoma, systemic lupus erythematosus (SLE),
carcinoma, or to drug therapy]
2. Cold agglutinin syndrome
a. primary (idiopathic)
b. secondary (to such conditions as lymphoma, mycoplasma pneumonia, infectious
mononucleosis)
3. Mixed-type AIHA
a. primary (idiopathic)
b. secondary (to such conditions as SLE, lymphoma)
4. Paroxysmal cold hemoglobinuria
a. primary (idiopathic)
b. secondary (to such conditions as syphilis, viral infections)
5. DAT-negative AIHA
a. primary (idiopathic)
b. secondary (to such conditions as lymphoma, SLE)
Drug-Induced Hemolytic Anemia
Alloimmune Hemolytic Anemia
1. Hemolytic disease of the fetus and newborn
2. Hemolytic transfusion reaction
Copyright © 2005 by the AABB. All rights reserved.

460 AABB Technical Manual
Table 20-3. Serologic Findings in Immune Hemolytic Anemias
WAIHA CAS Mixed-Type AIHA PCH Drug-Induced
Percent of cases 48%19 to 70%316%3to 32%19 7%19 to 8%20 Rare in adults; 32% in
children21
12%3to 18%19
DAT IgG:
20%3to 66%19
IgG + C3:
24%19 to 63%3
C3:
7%19 to 14%3
C3 only:
91%19 to 98%3
IgG + C3:
71%19 to 100%3
C3:
13%3
C3 only:
94%19 to 100%3
IgG:
94%19
IgG + C3:
6%19
Immuno-
globulin type
IgG (sometimes IgA or
IgM, rarely alone)
IgM IgG, IgM IgG IgG
Eluate IgG antibody Nonreactive IgG antibody Nonreactive IgG antibody
Serum 57% react by saline-IAT;
13% hemolyze
enzyme-treated RBCs
at 37 C; 90%
agglutinate enzyme-
treated RBCs at 37 C;
30% agglutinate
untreated RBCs at
20 C; rarely
agglutinate untreated
RBCs at 37 C3
IgM agglutinating
antibody; titer usually
>1000 at 4 C; usually
react at 30 C in
albumin; monoclonal
antibody in chronic
disease3
IgG IAT-reactive antibody
plus IgM
agglutinating
antibody, usually
react at 30-37 C in
saline; high titer at
4 C (classic CAS) or
low titer (<64) at
4C
3,19,20,22
IgG biphasic hemolysin
(Donath-Landsteiner
antibody)3
IgG antibody similar to
WAIHA3
Specificity Rh specificity; other
specificities have
been reported*
Usually anti-I but can be
anti-i; rarely anti-Pr23
Usually specificity is
unclear19,20,22
Can be anti-I, -i, or other
cold agglutinin
specificities
Anti-P (nonreactive with
pandP
kRBCs)
Specificity often Rh
related23
*See text.
Copyright © 2005 by the AABB. All rights reserved.
terns of reactivity may be found: coating
with IgG alone, with complement alone,
or with both. In approximately 1% of cases,
the DAT will be positive with a poly-
specific AHG reagent but negative with
IgG- and complement-specific AHG re-
agents. Some of these may be due to at-
tachment of IgM or IgA alone if reactivity
with these immunoglobulins has not been
excluded by the manufacturer.3,19
Serum
Autoantibody in the serum typically is IgG
and reacts by indirect antiglobulin testing
against all cells tested.3If the autoanti-
body has been adsorbed by the patient’s
red cells in vivo, the serum may contain
verylittlefreeantibody.Theserumwill
contain antibody after all the specific an-
tigen sites on the red cells have been oc-
cupied and no more antibody can be
bound in vivo. In such cases, the DAT is
usually strongly positive. Approximately
50% of patients with WAIHA have serum
antibodies that react with untreated sa-
line-suspended red cells. When testing
with PEG, enzyme-treated red cells, or
solid-phase methods, over 90% of these
sera can be shown to contain autoantibody.
Approximately one-third of patients with
WAIHA have cold-reactive autoagglutinins
demonstrable in tests at 20 C, but cold ag-
glutinin titers at 4 C are normal. The pres-
ence of this cold agglutinin does not
mean the patient has CAS in addition to
WAIHA.3
Eluate
The presence of the IgG autoantibody on
the red cells may be confirmed by elution
at least upon initial diagnosis and/or at
pretransfusion testing. (See Methods 4.1,
4.2, and 4.5.) Typically, the eluate reacts
with virtually all cells tested, with reactiv-
ity enhanced in tests against enzyme-
treated cells or when PEG is used. The
eluate will usually have no serologic activ-
ity if the only protein coating the red cells
is complement components. Occasion-
ally, antibody not detected by the DAT will
be detected in the eluate, possibly due to
the concentrating effect of eluate prepa-
ration.
Specificity of Autoantibody
The specificity of autoantibodies associ-
ated with WAIHA is complex. In routine
tests, all cells tested are usually reactive.
Some autoantibodies that have weaker or
negative reactivity with cells of rare Rh
phenotypes, such as D– – or Rhnull, appear
to have broad specificity in the Rh system.
Apparent specificity for simple Rh antigens
(D,C,E,c,e)isoccasionallyseen,either
as the sole autoantibody or as a predomi-
nant portion, based on stronger reactivity
with cells of certain phenotypes. Such re-
activity is often termed a “relative” speci-
ficity. Such relative specificity in a serum
may be mistaken for alloantibody, but cells
negative for the apparent target antigen
can adsorb and remove the “mimicking”
specificity.23,24
Unusual Specificities. Apart from Rh
specificity, warm autoantibodies with many
other specificities have been reported, eg,
specificities in the LW, Kell, Kidd, Duffy, and
Diego systems.24 Dilution and selective ad-
sorption of eluates may uncover specificity
or relative specificity of autoantibodies. Pa-
tients with autoantibodies of Kell, Rh, LW,
Ge, Sc, Lu, and Lan specificities may have
depressed expression of the respective anti-
gen and the DAT may be negative or very
weakly positive.24
Practical Significance. Tests against red
cells of rare phenotype and by special tech-
niques have limited clinical application. In
rare instances of WAIHA involving IgM ag-
glutinins, determining autoantibody speci-
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 461
Copyright © 2005 by the AABB. All rights reserved.
ficity may help differentiate such cases
from typical CAS.25 It is rarely, if ever, neces-
sary to ascertain autoantibody specificity in
order to select antigen-negative blood for
transfusion. If apparent specificity is di-
rected to a high-incidence antigen (eg,
anti-U), or when the autoantibody reacts
with all red cells except those of a rare Rh
phenotype (eg, D– –, Rhnull), compatible do-
nor blood is unlikely to be available and
there is little point in determining specific-
ity. Such blood, if available, should be
reserved for alloimmunized patients of that
uncommon phenotype.
Transfusion-Stimulated Autoantibodies.
Transfusion itself may lead to the produc-
tion of autoantibodies that may persist and
cause positive DATs for some time after
transfusion yet not cause obvious red cell
destruction. Such cell-bound autoantibodies
sometimes display blood group specificity
(eg, E, K, Jka). The positive DAT may persist
long after transfused red cells should have
disappeared from the circulation, appar-
ently adsorbed to the patient’s own anti-
gen-negative red cells.11
Transfusion of Patients with Warm-Reactive
Autoantibodies
Inherent Risks. Patients with warm-reac-
tive autoantibodies range from those with
no apparent decreased red cell survival to
those with life-threatening anemia. Pa-
tients with little or no evidence of signifi-
cant in-vivo red cell destruction tolerate
transfusion quite well.
When autoantibody is active in serum, it
may be difficult to exclude the presence of
alloantibodies, which increases the risk of
an adverse reaction. Transfusion may stim-
ulate alloantibody production, complicat-
ing subsequent transfusions. Transfusion
may intensify the autoantibody, inducing
or increasing hemolysis and making sero-
logic testing more difficult. Transfusion
may depress compensatory erythropoiesis.
Destruction of transfused cells may increase
hemoglobinemia and hemoglobinuria. In pa-
tients with active hemolysis, transfused red
cells may be destroyed more rapidly than the
patient’s own red cells. In rare cases, this may
promote hypercoagulability and dissemi-
nated intravascular coagulation (DIC). Trans-
fusion reactions, if they occur, may be diffi-
cult to investigate.
TransfusioninWAIHA.Transfusion is
especially problematic for patients with
rapid in-vivo hemolysis, who may present
with a very low hemoglobin level and hypo-
tension. Reticulocytopenia may accompany
a rapidly falling hematocrit, and the patient
may exhibit coronary insufficiency, conges-
tive heart failure, cardiac decompensation,
or neurologic impairment. Under these cir-
cumstances, transfusion is usually required
as a lifesaving measure. The transfused
cells may support oxygen-carrying capacity
until the acute hemolysis diminishes or
other therapies can effect a more lasting
benefit. These patients represent a signifi-
cant challenge because serologic testing
may be complex while clinical needs are
acute.
Transfusion should not be withheld
solely because of serologic incompatibility.
The volume transfused should usually be
the smallest amount required to maintain
adequate oxygen delivery, not necessarily
toreachanarbitraryhemoglobinlevel.Vol
-
umes of about 100 mL may be appropriate.3
The patient should be carefully monitored
throughout the transfusion.
TransfusioninChronicWAIHA.Most
patients with WAIHA have a chronic stable
anemia, often at relatively low hemoglobin
levels. Those with hemoglobin levels above
8 g/dL rarely require transfusion, and many
patients with levels of 5 g/dL (or even
lower) can be managed with bed rest and
no transfusions. Transfusion will be re-
quired if the anemia progresses or is ac-
462 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
companied by such symptoms as severe
angina, cardiac decompensation, respira-
tory distress, and cerebral ischemia.
Most patients with chronic anemia due
to WAIHA tolerate transfusion without
overt reactions, even though the transfused
cells may not survive any better than their
own. Because transfusion may lead to cir-
culatory overload or to increased red cell de-
struction, the decision to transfuse should
be carefully considered. A few patients
without acute hemolysis have had severe
hemolytic reactions after transfusion. This
may be due to the sudden availability of
large volumes of donor cells and the expo-
nential curve of decay, by which the num-
ber of cells hemolyzed is proportional to
the number of cells present.3,6(pp230,369)
Selecting Blood. If the decision is made
to transfuse, selection of appropriate donor
blood is essential. It is important to deter-
mine the patient’s ABO and Rh type and, if
time permits, to detect potentially clinically
significant alloantibodies. Adsorption and
other special techniques described later in
this chapter can greatly reduce the risk of
undetected alloantibodies but may be
time-consuming. If clinically significant
alloantibodies are present, the transfused
cells should lack the corresponding anti-
gen(s).
If the autoantibody has apparent and
relatively clear-cut specificity for a single
antigen (eg, anti-e) and there is active on-
going hemolysis, blood lacking that antigen
may be selected. There is evidence that, in
some patients, such red cells survive better
than the patient’s own red cells.6(p230),20 In the
absence of hemolysis, autoantibody speci-
ficity is not important, although donor units
negative for the antigen may be chosen be-
cause this is a simple way to circumvent the
autoantibody and detect potential alloanti-
bodies. If the autoantibody shows broader
reactivity, reacting with all cells but show-
ing some relative specificity (eg, it reacts
preferentially with e+ red cells), the use of
blood lacking the corresponding antigen is
debatable. It may be undesirable to expose
the patient to Rh antigens absent from
autologous cells, especially D and espe-
cially in females who may bear children
later, merely to improve serologic compati-
bility testing with the autoantibody (eg,
when a D– patient has autoanti-e).
In many cases of WAIHA, no autoanti-
body specificity is apparent. The patient’s
serum reacts with all red cell samples to the
same degree or reacts with red cells from
different donors to varying degrees for rea-
sons seemingly unrelated to Rh phenotypes.
Even if specificity is identified, the exotic
cells used for such identification are not
available for transfusion. The most impor-
tant consideration in such cases is to ex-
clude the presence of clinically important
alloantibodies before selecting either pheno-
typically similar or dissimilar, crossmatch-
incompatible red cells for transfusion. In
extremely rare cases in which there is se-
vere and progressive anemia, it may be es-
sential to transfuse blood that does not re-
act with the patient’s autoantibody.
Frequency of Testing. Although AABB
Standards8(p38) requires that a sample be
tested every 3 days, some serologists con-
tend that, in these difficult cases, the con-
tinued collection and testing (to include
antibody investigation) of patient samples
are unnecessary.26 Others disagree with that
opinion. In studies of patients with WAIHA,
there was 12% to 40% alloimmunization,
with many alloantibodies developing after
recent transfusions.27,28 These two papers
offer methods to assist in the detection of
alloantibodies in the presence of autoanti-
bodies. For patients with previously identi-
fied clinically significant antibodies, Stan-
dards8(p38) requires that methods of testing
shall be those that identify additional clini-
cally significant antibodies. It is the exclu-
sion of newly formed alloantibodies that is
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 463
Copyright © 2005 by the AABB. All rights reserved.
of concern. Autoantibodies that react with
all reagent red cells, even weakly, are capa-
ble of masking alloantibody reactivity; the
serologic reactivity is not necessarily addi-
tive.29 Due to the presence of autoantibodies,
all crossmatches will be incompatible. This
is unlike the case of clinically significant
alloantibodies, where a compatible cross-
match with antigen-negative red cells can
be obtained. Monitoring for evidence of red
cell destruction due to alloantibodies is dif-
ficult in patients who already have AIHA;
the patient’s own red cells and transfused
red cells will have shortened survival. In pa-
tients who have autoantibodies without
hemolytic anemia, transfused red cells
should have normal survival.
An alternative transfusion management
protocol proposed by one group uses pro-
phylactic antigen-matched units for pa-
tients with warm autoantibodies where fea-
sible, in combination with streamlined
adsorption procedures.30 Such a protocol
depends on the ability to maintain an
adequate inventory of antigen-negative
units.31
IgM Warm AIHA
AIHA associated with IgM agglutinins that
react at 37 C is unusual but is character-
ized by severe hemolysis.25,32-34 The prog-
nosisforthesepatientsispoor.
DAT
The patient’s red cells are typically spon-
taneously agglutinated, requiring disrup-
tion of the IgM agglutinin by dithiothrei-
tol in order to obtain accurate DAT (and
ABO/Rh) results. Complement is usually
detected on the red cells. In one series,
IgG was detected in 17% of cases and IgM
in 28%. By a more sensitive flow cyto-
metric method, red cell-bound IgM was
detected on 82% of patients’ red cells not
reacting with anti-IgM by tube DAT.33
Serum
Warm IgM autoagglutinins are typically
weak and sometimes are enhanced in the
presence of albumin or when the serum is
acidified.33 Occasionally, optimal reactiv-
ity is between 20 C and 30 C, rather than
37 C. These antibodies have low or negli-
gible antibody titers; a 4 C titer of <64 eas-
ily differentiates this IgM warm antibody
from those seen in CAS.
Eluate
IgM agglutinins are often detected in an
eluate when inspected at the agglutinin
phase before proceeding to the antiglo-
bulin test.
Cold Agglutinin Syndrome
Cold agglutinin syndrome (also called cold
hemagglutinin disease, CHD) is the hemoly-
tic anemia most commonly associated with
cold-reactive autoantibodies and accounts
for approximately 16% to 32% of all cases
of immune hemolysis.3,19 (See Table 20-3.)
It occurs as an acute or chronic condition.
The acute form is often secondary to
lymphoproliferative disorders (eg, lym-
phoma) or Mycoplasma pneumoniae in-
fection. The chronic form is often seen in
elderly patients, sometimes associated
with lymphoma, chronic lymphocytic
leukemia, or Waldenstrom’s macroglo-
bulinemia. Acrocyanosis and hemoglo-
binuria may occur in cold weather. CAS is
often characterized by rapid agglutina-
tion, at room temperature, of red cells in
an EDTA specimen. Clumping of red cells
may be obvious in such a sample, some-
times so strong that the cells appear to be
clotted. Problems with ABO and Rh typing
and other tests are not uncommon. Main-
taining the EDTA specimen at 37 C and
washing the red cells with 37 C saline is
usually necessary to disperse the cold
464 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
autoagglutinin before performing ABO and
Rh typing and the DAT.
DAT
Complement is the only protein detected
on the red cells in almost all cases. If other
proteins are detected, a negative control
for the DAT, eg, 6% to 10% albumin,
should be tested to ensure that the cold
autoagglutinin is not causing a false-posi-
tive test.
The cold-reactive autoagglutinin is usu-
ally IgM, which binds to red cells in the
comparatively low temperature of the pe-
ripheral circulation and causes comple-
ment components (C3 and C4 in particular)
to attach to the red cells. As the red cells cir-
culate to warmer areas, the IgM dissociates,
but the complement remains. Red-cell-
bound C3b can react with the CR1 or CR3
receptors of macrophages in the reticulo-
endothelial system. More of the red cell de-
struction occurs in the liver. Regulatory
proteins convert the bound C3 and C4 to
C3dg and C4d, and it is the anti-C3d com-
ponent of polyspecific AHG reagents that
accounts for the positive DAT. The presence
of C3dg alone does not shorten red cell sur-
vival because macrophages have no C3dg
or C3d receptors.
Serum
IgM cold-reactive autoagglutinins associ-
ated with immune hemolysis usually re-
act ≥30 C and have a titer ≥1000 when
testedat4C;theyrarelyreactwithsa
-
line-suspended red cells above 32 C. If
30% bovine albumin is included in the re-
action medium, 100% and 70% of clini-
cally significant examples will react at 30
C or 37 C, respectively.35 Occasionally,
pathologic cold agglutinins will have a
lower titer (ie, <1000), but they will have a
high thermal amplitude (ie, reactive at 30
C with or without the addition of albu-
min). Hemolytic activity against untreated
red cells can be demonstrated sometimes
at 20 to 25 C, and, except in rare cases
with Pr specificity, enzyme-treated red
cells are hemolyzed in the presence of ad-
equate complement.
Determination of the true thermal am-
plitude or titer of the cold autoagglutinin
requires that the specimen be collected and
maintained strictly at 37 C until the serum
and cells are separated, to avoid in-vitro
autoadsorption. Alternatively, plasma can
be used from an EDTA-anticoagulated
specimen that has been warmed for 10 to
15 minutes at 37 C (with repeated mixing)
and then separated from the cells, ideally at
37 C. This should release autoadsorbed an-
tibody back into the plasma.
In chronic CAS, the IgM autoagglutinin
is usually a monoclonal protein with kappa
light chains. In the acute form induced by
Mycoplasma or viral infections, the anti-
body is polyclonal IgM with normal kappa
and lambda light-chain distribution. Rare
examples of IgA and IgG cold-reactive
autoagglutinins have also been described.
Eluate
Elution is seldom necessary in obvious
cases of CAS. If the red cells have been
collected properly and washed at 37 C,
there will be no immunoglobulin on the
cells and no reactivity will be found in the
eluate.
Specificity of Autoantibody
The autoantibody specificity in CAS is
usually of academic interest only. CAS is
most often associated with antibodies with
I specificity.3,23 Less commonly, i specific-
ity is found, usually associated with infec-
tious mononucleosis.3On rare occasions,
cold-reactive autoagglutinins with Pr or
other specificities are seen3,23 (see Method
4.7). Dilution of the serum may be neces-
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 465
Copyright © 2005 by the AABB. All rights reserved.
sary to demonstrate specificity of very
high-titer antibodies.
Autoantibody specificity is not diagnos-
tic for CAS. Autoanti-I may be seen in
healthysubjectsaswellaspatientswith
CAS. The nonpathologic forms of auto-
anti-I, however, rarely react to titers above
64 at 4 C, and are usually nonreactive with
I– (i cord and i adult) red cells at room tem-
perature. In contrast, the autoanti-I of CAS
may react quite strongly with I– red cells in
tests at room temperature, and equal or
even stronger reactions are observed with
I+ red cells. Autoanti-i reacts in the oppo-
site manner, demonstrating stronger reac-
tions with I– red cells than with red cells
that are I+. Procedures to determine the
titers and specificities of cold-reactive auto-
antibodies are given in Method 4.6 and
Method 4.7.
Pretransfusion Testing. Antibody detec-
tion tests should be performed in ways that
minimize cold-reactive autoantibody activ-
ity yet still permit detection of clinically sig-
nificant alloantibodies. The use of albumin
and other potentiators may increase the re-
activity of the autoantibodies. To avoid the
detection of bound complement, most
serologists use an IgG-specific reagent,
rather than a polyspecific AHG serum. Ad-
ditionally, a prewarming technique may be
used (see Method 3.3).
Adsorption Procedures. When cold-re-
active autoantibody reactivity continues to
interfere with antibody detection tests (eg,
when performed strictly at 37 C), cold
autoadsorption studies (see Method 4.6)
can be helpful. One or two cold auto-
adsorptions should remove enough auto-
antibody to make it possible to detect
alloantibodies at 37 C that were otherwise
masked by the cold-reactive autoantibody;
many cold autoadsorptions would be re-
quired to remove enough of the cold-reac-
tive autoantibody for room temperature
testing. If the patient has been recently
transfused, rabbit red cells may be used to
remove autoanti-I and -IH from sera36;clini
-
cally significant alloantibodies, notably
anti-B,-D,-E,Vel,andothers,havebeenre
-
moved by this method.37,38 A preparation of
rabbit red cell stroma is commercially avail-
able. Alternatively, allogeneic adsorption
studiesat4Ccanbeperformedasfor
WAIHA (see below).
Mixed-Type AIHA
Although about one-third of patients with
WAIHA have nonpathologic IgM antibod-
ies that react to high titer at low tempera-
ture, another group of patients with
WAIHA have cold agglutinins that react at
or above 30 C. This latter group is referred
to as “mixed-type” AIHA and can be sub-
divided: patients with high titer, high
thermal amplitude IgM cold antibodies
(therareWAIHAplusclassicCAS)andpa-
tients with normal titer (<64 at 4 C), high
thermal amplitude cold antibodies.19,20,22,39
Patients with mixed-type AIHA often
present with hemolysis and complex se-
rum reactivity present in all phases of
testing. Typical serologic findings are de-
scribed below.
DAT
When the patient has WAIHA plus classic
CAS, both IgG and C3 are usually detect-
able on the patient’s red cells. When the
cold agglutinin has a normal titer, but
high thermal amplitude (greater than or
equalto30C),IgGand/orC3maybede
-
tectable on the red cells.3
Serum
Both warm-reactive IgG autoantibodies
and cold-reactive, agglutinating IgM auto-
antibodies are present in the serum. These
usually result in reactivity at all phases of
testing, with virtually all cells tested. The
IgM agglutinating autoantibody(ies) re-
466 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
acts at 30 C or above. If adsorption studies
are done to detect alloantibodies, it may
be necessary to perform adsorptions at both
warmandcoldtemperatures.
Eluate
A suitably prepared eluate will contain a
warm-reactive IgG autoantibody.
Specificity of Autoantibodies
The unusual cold-reactive IgM agglutinat-
ing autoantibody can have specificities
typical of CAS (ie, I or i) but often has no
apparent specificity.19,20,22 The warm-reac-
tive IgG autoantibody often appears sero-
logically indistinguishable from specifi-
cities encountered in typical WAIHA.
Transfusion in Mixed-Type AIHA
If blood transfusions are necessary, the
considerations in the selection of blood
for transfusion are identical to those de-
scribed for patients with acute hemolysis
due to WAIHA (see above).
Paroxysmal Cold Hemoglobinuria
The rarest form of DAT-positive AIHA is
PCH. In the past, it was characteristically
associated with syphilis, but this associa-
tion is now unusual. More commonly, PCH
presents as an acute transient condition
secondary to viral infections, particularly
in young children. In such cases, the
biphasic hemolysin (see below) may only
be transiently detectable. PCH can also
occur as an idiopathic chronic disease in
older people. One large study found that
none of 531 adults having well-defined
immune hemolytic anemias had Donath-
Landsteiner hemolysins, whereas 22 of 68
(32%) children were shown to have Donath-
Landsteiner hemolysins.21
DAT
PCH is caused by an IgG complement-fix-
ing antibody, but, as with IgM cold-reac-
tive autoagglutinins, it reacts with red
cells in colder areas of the body (usually
the extremities), causes C3 to bind irre-
versibly to red cells, and then the anti-
body dissociates from the red cells as the
blood circulates to warmer parts of the
body. Red cells washed in a routine man-
nerfortheDATareusuallycoatedonly
with complement components, but IgG
may be detectable on cells that have been
washed with cold saline and tested with
cold anti-IgG reagent.3Keeping the system
nearer its optimal binding temperature
allows the cold-reactive IgG autoantibody
to remain attached to its antigen.
Serum
The IgG autoantibody in PCH is classically
described as a biphasic hemolysin because
binding to red cells occurs at low temper-
atures but hemolysis does not occur until
the coated red cells are warmed to 37 C.
This is the basis of the diagnostic test for
the disease, the Donath-Landsteiner test
(see Method 4.13). The autoantibody may
agglutinate normal red cells at 4 C but
rarely to titers greater than 64. Because the
antibody rarely reacts above 4 C, the se-
rum is usually compatible with random
donor cells by routine crossmatch proce-
dures and pretransfusion antibody detec-
tion tests are usually nonreactive.
Eluate
Because complement components are
usually the only globulins present on cir-
culating red cells, eluates prepared from
red cells of patients with PCH are almost
always nonreactive.
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 467
Copyright © 2005 by the AABB. All rights reserved.
Specificity of Autoantibody
The autoantibody of PCH has most fre-
quently been shown to have P specificity,
reacting with all red cells by the Donath-
Landsteiner test (including the patient’s
own red cells) except those of the very
rare p or Pkphenotypes. Exceptional ex-
amples with other specificities have been
described.6(p221),23
Transfusion in PCH
Transfusion is rarely necessary for adult
patients with PCH, unless their hemolysis
is severe. In children, especially under age
6, the thermal amplitude of the antibody
tends to be much wider than in adults
and hemolysis more brisk, so transfusion
may be required as a lifesaving measure.
Althoughthereissomeevidencethatp
red cells survive better than P+ (P1+orP
1–)
red cells,6(p221) theprevalenceofpbloodis
approximately 1 in 200,000 and the urgent
need for transfusion usually precludes at-
tempts to obtain this rare blood. Transfu-
sion of random donor blood should not
be withheld from PCH patients whose
need is urgent. Red cells negative for the P
antigen should be considered only for
those patients who do not respond ade-
quately to random donor blood.3
DAT-Negative AIHA
Clinical evidence of hemolytic anemia is
present in some patients whose DAT is non-
reactive. Frequently, autoantibody cannot
be detected in either eluate or serum.
There may be several reasons the DAT is
negative. The autoantibody may be IgA or
IgM.3,40 Antibodies with low binding affin-
ity may dissociate from the red cells dur-
ing saline washing of the cells for the DAT.
Washing with ice cold (eg, 4 C) LISS or sa-
line may help retain antibody on the cells;
a control (eg, 6-10% albumin) is necessary
to confirm that cold autoagglutinins are
not causing the positive results.3,40 There
may be too few antibody molecules on
the cell for detection by routine methods
but enough to be demonstrable by meth-
odssuchasflowcytometry,enzyme-
linked antiglobulin tests, solid phase,
PEG, direct Polybrene, column agglutina-
tion, or concentrated eluate.
Nonroutine Reagents
The causative antibody may be IgM or IgA
not detected by routine AHG reagents.
Anti-IgG, anti-C3d, and the combined
anti-C3b, -C3d reagents are the only li-
censed products available in the United
States for use with human red cells. AHG
reagents that react with IgA, IgM, or C4
are available commercially but have been
prepared for use with endpoints other
than agglutination. These must be used
cautiously and their hemagglutinating re-
activity carefully standardized by the
user.3Quality control must be rigorous
because agglutination with AHG reagents
is more sensitive than precipitation; a se-
rum that appears to be monospecific by
precipitation tests may react with several
different proteins when used in agglutina-
tion tests.
Antigen Depression
Patients with autoantibodies of Kell, Rh,
LW, Ge, Sc, Lu, and Lan specificities may
have depressed red cell expression of the
respective antigens. When this occurs, an-
tibody may be detected in the serum and
eluate, but the DAT may be negative or very
weakly positive. This may provide in-vivo
protection of autologous cells. Donor cells
of common specific antigen type may be
destroyed, but cells lacking the corre-
sponding antigen (usually high-incidence)
may survive well. When the autoantibody
subsides, autologous cells again express
normal amounts of antigen.6(p228),24
468 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Serologic Problems with
Autoantibodies
In pretransfusion tests on patients with auto-
antibodies, the following problems may
arise:
1. Cold-reactive autoantibodies can cause
autoagglutination, resulting in erro-
neous determinations of ABO and
Rh type.
2. Red cells strongly coated with globu-
lins may undergo spontaneous ag-
glutination with high-protein, anti-
Rh, blood-typing reagents, and occa-
sionally even with low-protein re-
agents.41
3. Thepresenceoffreeautoantibodyin
theserummaymakeantibodyde-
tection and crossmatching tests dif-
ficult to interpret. If time permits, the
presence or absence of unexpected,
clinically important alloantibody(ies)
should be determined (see Methods
4.9 through 4.12) before blood is trans-
fused.
Although resolving these serologic prob-
lems is important, delaying transfusion in
the hope of finding serologically compati-
ble blood may cause greater danger to the
patient in some cases. Only clinical judg-
ment can resolve this dilemma; therefore,
dialogue with the patient’s physician is im-
portant.
Resolution of ABO Problems
There are several approaches to the reso-
lution of ABO typing problems associated
with cold-reactive autoagglutinins. Often,
it is only necessary to maintain the blood
sample at 37 C immediately after collec-
tion and to wash the red cells with warm
(37 C) saline before testing. It is helpful to
perform a parallel control test, using 6%
to 10% bovine albumin in saline, to deter-
mine if autoagglutination persists. If the
control test is nonreactive, the results ob-
tained with anti-A and anti-B are usually
valid. If autoagglutination still occurs, it
may be necessary to treat the red cells
with sulfhydryl reagents.
Because cold-reactive autoagglutinins
are almost always IgM and sulfhydryl re-
agents denature IgM molecules, reagents
such as 2-mercaptoethanol (2-ME) or
dithiothreitol (DTT) can be used to abolish
autoagglutination (see Method 2.11). Treat-
ing the red cells with ZZAP reagent as in the
preparation for adsorptions can also be used
(see Method 4.10). Appropriate controls are
essentialforalltests.
When the serum agglutinates group O
reagent red cells, the results of serum tests
may be unreliable. Repeating the tests us-
ing prewarmed serum and group A, B, and
O red cells at 37 C will often resolve any dis-
crepancy, but weak anti-A and/or -B in
some patients’ sera may not react at 37 C.
Alternatively, adsorbed serum (either auto-
adsorbed or adsorbed with allogeneic
group O red cells) can be used. Because
rabbit red cells express a B-like antigen,
sera adsorbed with rabbit red cells or
stroma may not contain anti-B, and sera
adsorbed in this manner should not be
used for ABO serum tests.
Resolution of Rh Problems
Autoagglutination of red cells by cold- or
spontaneous agglutination of red cells by
warm-reactive autoantibodies may also
cause discrepant Rh typing. The same
procedures described for the resolution of
ABO problems, with the exception of us-
ing ZZAP-treated red cells, may be useful.
Also, IgG antibody can be dissociated from
the cells by treatment with chloroquine
diphosphate (Method 2.13), or by glycine-
HCl/EDTA (Method 2.14), methods that
leave red cells intact for subsequent typ-
ing. IgM-coated cells can be treated with
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 469
Copyright © 2005 by the AABB. All rights reserved.
sulfhydryl reagents (such as 2-ME or DTT,
Method 2.11) to circumvent autoagglu-
tination and spontaneous agglutination.
Detection of Alloantibodies in the
Presence of Warm-Reactive
Autoantibodies
If the patient who has warm-reactive
autoantibodies in the serum needs trans-
fusion, it is important to evaluate the
possible simultaneous presence of allo-
antibodies to red cell antigens. Some
alloantibodies may make their presence
known by reacting more strongly or at dif-
ferent phases than the autoantibody, but
quite often studies may not suggest the
existence of masked alloantibodies.29 It is
helpful to know which of the common red
cell antigens are lacking on the patient’s
red cells, to predict which clinically signif-
icant alloantibodies the patient may have
produced or may produce. Antigens ab-
sent from autologous cells could well be
the target of present or future alloanti-
bodies. When the red cells are coated with
IgG, antiglobulin-reactive reagents can-
not be used to test IgG-coated cells unless
the IgG is first removed (see Methods 2.13
and 2.14). Low-protein antisera (eg, mono-
clonal reagents) that do not require an
antiglobulin test may be helpful in typing
the DAT-positive red cells. Cell separation
procedures (see Methods 2.15 and 2.16)
may be necessary if the patient has been
transfused recently.
Methods to detect alloantibodies in the
presence of warm-reactive autoantibodies
attempt to remove, reduce, or circumvent
the autoantibody. Antibody detection meth-
ods that use PEG, enzymes, column aggluti-
nation, or solid-phase red cell adherence
generally enhance autoantibodies. Testing
LISS- or saline-suspended red cells may
avoid autoantibodies but allow detection of
most significant alloantibodies. Other pro-
cedures involve adsorption, the principles
of which are discussed in Chapter 19. Two
widely used approaches are discussed be-
low.
Autologous Adsorption
In a patient who has not been recently
transfused, autologous adsorption (see
Method 4.9) is the best way to detect allo-
antibodies in the presence of warm-reac-
tive autoantibodies. The adsorbed serum
canbeusedintheroutineantibodyde
-
tection procedure.
Autoadsorption generally requires some
initial preparation of the patient’s red cells.
At 37 C, in-vivo adsorption will have oc-
curred and all antigen sites on the patient’s
own red cells may be blocked. It may be
necessary, therefore, to remove autoanti-
body from the red cells to make sites avail-
able for adsorption. A gentle heat elution at
56 C for 5 minutes can dissociate some of
the bound IgG. This can be followed by
treatment of the autologous red cells with
proteolytic enzymes to increase their ca-
pacity to adsorb autoantibody. Treatment
of the red cells with ZZAP, a mixture of
papain or ficin and DTT (see Method 4.9)
accomplishes both of these actions in one
step; the sulfhydryl component makes the
IgG molecules more susceptible to the pro-
tease and dissociates the antibody mole-
cules from the cell. Multiple sequential
autoadsorptions with new aliquots of red
cells may be necessary if the serum con-
tains high levels of autoantibody. Once the
autoantibody has been removed, the ad-
sorbed serum is examined for alloantibody
activity.
If the patient is to be transfused, it can
be advantageous to collect and save addi-
tional aliquots of pretransfusion cells, to be
used for later adsorptions.
Autologous adsorption is not recom-
mended for patients who have been re-
470 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
cently transfused, because they may have
an admixture of transfused red cells that
might adsorb alloantibody. Red cells nor-
mally live for about 110 to 120 days. In pa-
tients with AIHA, autologous and trans-
fused red cells can be expected to have
shortened survival. However, determining
how long transfused red cells remain in cir-
culation in patients who need repeated
transfusions is not feasible. It has been
demonstrated that very small amounts
(<10%) of antigen-positive red cells are ca-
pable of removing alloantibody reactivity in
in-vitro studies42; therefore, it is recom-
mended to wait for 3 months after transfu-
sion before autologous adsorptions are per-
formed.
Allogeneic Adsorption
The use of allogeneic red cells for adsorp-
tion may be helpful when the patient has
been recently transfused or when insuffi-
cient autologous red cells are available.
The goal is to remove autoantibody and
leave the alloantibody in the adsorbed se-
rum. The adsorbing cells must not have
the antigens against which the alloanti-
bodies react. Because alloantibody speci-
ficity is unknown, red cells of different
phenotypes will usually be used to adsorb
several aliquots of the patient’s serum.
Given the number of potential alloanti-
bodies, the task of selecting cells may ap-
pear formidable. However, the selected
cells need only demonstrate those few allo-
antibodies of clinical significance likely to
be present. These include the common Rh
antigens (D, C, E, c, and e), K, Fyaand Fyb,
Jkaand Jkb, and S and s. Cell selection is
made easier by the fact that some antigens
can be destroyed by appropriate treatment
(eg, with enzymes) before use in adsorption
procedures. Antibodies to high-incidence
antigens cannot be excluded by allogeneic
adsorptions because the adsorbing cells
will almost invariably express the antigen
and adsorb the alloantibody along with
autoantibody.
Patient’s Phenotype Unknown. When
thepatient’sphenotypeisnotknown,
group O red cell samples of three different
Rh phenotypes (R1R1,R
2R2, and rr) should
be selected (see Method 4.10). One should
lack Jkaand another Jkb.Iftreatedwith
ZZAP, these cells would also lack all anti-
gens of the Kell system and enzyme-sensi-
tive antigens (see Table 19-3). If ZZAP is not
available, cells treated only with proteolytic
enzyme can be used, but at least one of the
adsorbing cells must be K– because Kell
system antigens will not be destroyed. Un-
treated cells may be used, but antibody
maybemoredifficulttoremoveandthe
adsorbing cells must, at a minimum, in-
clude at least one negative for the S, s, Fya,
Fyb, and K antigens in addition to the Rh
andKiddrequirementsabove.
Each aliquot may need to be adsorbed
two or three times. The fully adsorbed
aliquots are tested against reagent red cells
knowneithertolackortocarrycommon
antigensoftheRh,MNS,Kidd,Kell,and
Duffy blood group systems. If an adsorbed
aliquot is reactive, that aliquot (or an addi-
tional specimen similarly adsorbed) should
be tested to identify the antibody. Adsorb-
ing several aliquots with different red cell
samples provides a battery of potentially
informative specimens. For example, if the
aliquot adsorbed with Jk(a–) red cells sub-
sequently reacts only with Jk(a+) red cells,
thepresenceofalloanti-Jk
acan confidently
be inferred.
Patient’s Phenotype Known. If the pa-
tient’s Rh and Kidd phenotypes are known
or can be determined, adsorption can be
performed with a single sample of allo-
geneic ZZAP-treated red cells of the same
Rh and Kidd phenotypes as the patient (see
Method 4.11).
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 471
Copyright © 2005 by the AABB. All rights reserved.
Problems Encountered. Occasionally
autoantibody will not be removed by three
sequential adsorptions. Further adsorptions
canbedone,butmultipleadsorptionshave
the potential to dilute the serum. If the ad-
sorbing cells do not appear to remove the
antibody, the autoantibody may have an
unusual specificity that does not react with
the cells used for adsorption. For example,
autoantibodies with Kell, LW, or EnaFS
specificity would not be removed by ZZAP-
treated cells (see Table 19-3 for a list of anti-
gens altered by various agents).
Autoantibodies Mimicking Alloantibodies
Sometimes, autoantibodies have patterns
of reactivity that are easily mistaken for
alloantibody. For example, the serum of a
D– patient may have apparent anti-C and
-e reactivity. The anti-C reactivity may re-
flect warm-reactive autoantibody even if
the patient’s cells lack C. The autoantibody
nature of the reactivity can be demon-
strated by autologous and allogeneic ad-
sorption studies. In this case, the appar-
ent alloanti-C would be adsorbed by C–
red cells, both autologous and allogeneic.
This is quite unlike the behavior of a true
alloanti-C, which would be adsorbed only
by C+ red cells. In one study,43 the serum
prepared from an initial autoadsorption
would often retain autoantibodies that
mimicked alloantibodies in addition to
the true alloantibody(ies) present. Serum
prepared from an initial alloadsorption
most often contains only alloantibodies.
The differences in the auto- or alloanti-
body nature of specificities detected in
the autoadsorbed serum as compared to
the alloadsorbed serum reflect an ineffi-
ciency of autologous adsorption. This is
primarily due to limited volumes of auto-
logous cells available for removing all
autoantibody reactivity from the serum.43
Detection of Alloantibodies in the
Presence of Cold-Reactive Autoantibodies
Cold-reactive autoagglutinins rarely mask
clinically significant alloantibodies if se-
rum tests are conducted at 37 C and if
IgG-specific reagents are used for the
antiglobulin phase. In rare instances, it
may be necessary to perform autoadsorp-
tion at 4 C (see Method 4.6). Achieving the
complete removal of potent cold-reactive
autoagglutinins is very time-consuming
and is usually unnecessary. Removal of
sufficient cold autoagglutinins may be fa-
cilitated by treating the patient’s cells with
enzymes or ZZAP before adsorption.
Drug-Induced Immune
Hemolytic Anemia
Drugs sometimes induce the formation of
antibodies, either against the drug itself
or against intrinsic red cell antigens, that
may result in a positive DAT, immune red
cell destruction, or both. Some of the an-
tibodies produced appear to be depend-
ent on the presence of the drug (ie, drug
dependent) for their detection or destruc-
tive capability, whereas others do not (ie,
drug independent). In some instances, a
reactive DAT may result from nonim-
munologic effects of the drugs. Drugs that
have been reported to cause hemolytic
anemia and/or a positive DAT are listed in
Appendix 20-2.
Theories of the Immune Response and
Drug-Dependent Antibodies
Numerous theories have been suggested
to explain how drugs induce immune re-
sponses and what relation such responses
may have to the positive DAT and immune-
mediated cell destruction observed in some
patients. For many years, drug-associated
472 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
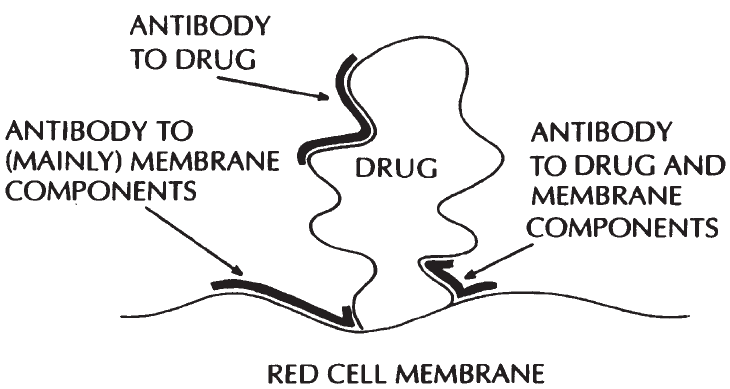
positive DATs were classified into four
mechanisms: drug adsorption (penicil-
lin-type), immune complex formation,
autoantibody production, and nonspe-
cific adsorption. Such classification has
been useful, but many aspects lacked de-
finitive proof. In addition, some drugs
created immune problems involving as-
pects of more than one mechanism. More
recent theories, still unproven, tend to-
ward a more comprehensive approach.44-48
Most drugs are probably capable of
binding loosely, or firmly, to circulating
cells, which can lead to an immune re-
sponse. Figure 20-1 illustrates this concept.
Antibodies can be formed to the drug itself
ortothedrugplusmembranecomponents.
When an antibody is formed to the drug
plus membrane components, the antibody
may recognize primarily the drug or primar-
ily the membrane. One or all three of these
antibody populations may be present.
Serologic and Clinical Classification
Drug-induced antibodies can be classi-
fied into three groups according to their
clinical and serologic characteristics.3In
one group, the drug binds firmly to the
cell membrane and antibody is appar-
ently largely directed against the drug it-
self. This was called the drug adsorption
mechanism. Antibodies to penicillin are
the best described of this group.
The second group of drug-dependent
antibodies reacts with drugs that do not
bind well to the cell membrane (eg, quini-
dine, ceftriaxone). The reactive mechanism
of these antibodies was previously thought
to be due to drug/antidrug immune com-
plex formation, but the theory has never
been proven.48 Antibodies in this group may
cause acute intravascular hemolysis and
may be difficult to demonstrate serologi-
cally. Testing for this type of drug antibody
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 473
Figure 20-1. Proposed unifying theory of drug-induced antibody reactions (based on a cartoon by
Habibi as cited by Garratty23). The thicker darker lines represent antigen-binding sites on the F(ab) re-
gion of the drug-induced antibody. Drugs (haptens) bind loosely, or firmly, to cell membranes and anti-
bodies may be made to: a) the drug [producing in-vitro reactions typical of a drug adsorption (penicil-
lin-type) reaction]; b) membrane components, or mainly membrane components (producing in-vitro
reactions typical of autoantibody); or c) part-drug, part-membrane components (producing an in-vitro
reaction typical of the so-called immune complex mechanism).23(p55)
Copyright © 2005 by the AABB. All rights reserved.
is still referred to as the “immune complex”
method.
Antibodies of the third group (eg, methyl-
dopa, procainamide, and fludarabine) have
serologic reactivity independent of the drug,
despite the fact that it was the drug that
originally induced the immune response.
Serologically, they behave as autoantibodies.
Drug-Dependent Antibodies Reactive with
Drug-Treated Red Cells: Penicillin-Type
Antibodies
The clinical and laboratory features of drug-
induced immune hemolytic anemia oper-
ating through this mechanism are:
1. The DAT is strongly positive due to
IgG coating. Complement coating may
also be present.
2. Antibody eluted from the patient’s
red cells reacts with drug-treated red
cells but not with untreated red cells.
3. The serum contains a high-titer IgG
antibody (especially when the target
is penicillin or cefotetan) reactive with
the drug-treated red cells but not with
the untreated red cells, unless the
patient also has alloantibodies to red
cell antigens.
4. For penicillin, the hemolysis-induc-
ing dose is millions of units daily for
a week or more; for other drugs, eg,
cefotetan, a single 1 to 2 g dose has been
implicated in immune hemolysis.
5. Hemolysis develops gradually but
may be life-threatening if the etiol-
ogy is unrecognized and drug ad-
ministration is continued.
6. Discontinuation of the drug is usu-
ally followed by increased cell sur-
vival, although hemolysis of decreas-
ing severity may persist for several
weeks.
Approximately 3% of patients receiving
large doses of penicillin intravenously (ie,
millions of units per day) will develop a
positive DAT; only occasionally will these
patients develop hemolytic anemia.6(p231) A
possible mechanism for the positive DAT is
given in Fig 20-2. The penicillin becomes
covalently linked to the red cells in vivo. If
the patient has antibodies to penicillin, they
bind to the penicillin bound to the red cells.
The result is that the penicillin-coated red
cells become coated with IgG. If cell destruc-
tion occurs, it takes place extravascularly,
probably in the same way that red cells
coated with IgG alloantibodies are de-
stroyed. Intravascular hemolysis is rare.
Many cephalosporins, which are related
to penicillins, behave in a similar manner.
The cephalosporins are generally classified
by “generations,” based on their effective-
ness against gram-negative organisms (see
Table 20-4). Approximately 4% of patients
receiving first- or second-generation cephalos-
porins develop a positive DAT.48 Dramati-
cally reduced red cell survival has been
associated with second- and third-genera-
tion cephalosporins.12,47,49-53 The prevalence
and severity of cephalosporin-induced im-
mune red cell destruction appear to be in-
creasing.3
Drug-Dependent Antibodies Reacting by the
“Immune Complex” Mechanism
Many drugs have been reported as caus-
ing hemolytic anemia by this mechanism.
Some of the second- and third-generation
cephalosporins react by this mechanism;
anti-ceftriaxone has been detected only
by the immune complex method.49 The
following observations are characteristic:
1. Complement may be the only globu-
lin easily detected on the red cells,
but IgG may be present.
2. The serum antibody can be either
IgMorIgG,orIgMwithIgG.
3. A drug (or metabolite) must be pres-
ent in vitro for demonstration of the
474 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
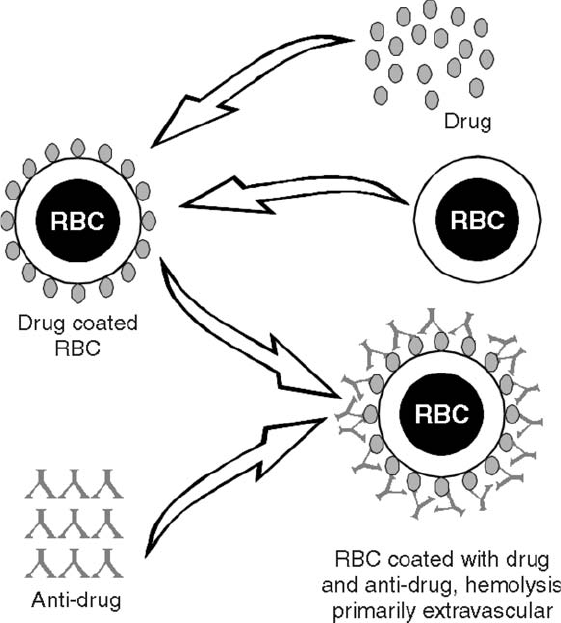
antibody in the patient’s serum. An-
tibodies may cause hemolysis, ag-
glutination, and/or sensitization of
red cells in the presence of the drug.
4. The patient need only take a small
amount of the drug.
5. Acute intravascular hemolysis with
hemoglobinemia and hemoglobinuria
is the usual presentation. Renal fail-
ure is quite common.
6. Once antibody has been formed, se-
vere hemolytic episodes may recur
after exposure to very small quanti-
ties of the drug.
Drug-Independent Antibodies: Autoantibody
Production
Some drugs induce autoantibodies that
appear serologically indistinguishable
from those of WAIHA. Red cells are coated
with IgG, and the eluate as well as the se-
rum react with virtually all cells tested in
the absence of the drug. Blood group spe-
cificity has been demonstrated at times,
similar to that seen in AIHA. The antibody
has no in-vitro activity with the drug, di-
rectly or indirectly.
The best studied of such cases are those
induced by α-methyldopa. A closely related
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 475
Figure 20-2. The drug-adsorption mechanism. The drug binds tightly to the red cell membrane pro-
teins. If a patient develops a potent drug antibody, it will react with the cell-bound drug. Such red cells
will yield a positive result in the DAT using anti-IgG reagents. Complement is usually not activated and
lysis is primarily extravascular in nature. Penicillin-G is the prototype drug.
Copyright © 2005 by the AABB. All rights reserved.

drug, L-dopa, has been implicated, as have
several drugs unrelated to α-methyldopa,
including procainamide, nonsteroidal
anti-inflammatory drugs (eg, mefenamic
acid), second- and third-generation cep-
halosporins, and fludarabine. In some
cases, drug-dependent antibodies are also
present.
Proof that a drug causes autoantibody
production is difficult to obtain. Sufficient
evidence would include: demonstration
that autoantibody production began after
drug administration; resolution of the im-
mune process after withdrawal of the drug;
and recurrence of hemolytic anemia or
autoantibodies if the drug is readmini-
stered. The last requirement is crucial and
the most difficult to demonstrate.
Nonimmunologic Protein Adsorption
The positive DAT associated with some
drugs is due to a mechanism independent
of antibody production. Hemolytic ane-
mia associated with this mechanism oc-
curs rarely.
Cephalosporins (primarily cephalothin)
are the drugs with which this was originally
associated. Red cells coated with cephalo-
thin (Keflin) and incubated with normal
plasma will adsorb albumin, IgA, IgG, IgM,
and C3 in a nonimmunologic manner. If
this occurs, a positive indirect antiglobulin
test will be seen with AHG reagents.
Other drugs that may cause nonimmuno-
logic adsorption of proteins and a positive
DAT include diglycoaldehyde, suramin,
cisplatin, clavulanate (in Timentin and
Augmentin), sulbactam in Unasyn,54 and
tazobactam (in Tazocin and Zosyn).55,56
Laboratory Investigation of Drug-Induced
Antibodies
The drug-related problems most com-
monly encountered in the blood bank are
those associated with a positive DAT. Typ-
476 AABB Technical Manual
Table 20-4. Some Cephalosporins
Generic Name Trade Name*
First Generation
cefadroxil Duricef
cefazolin Ancef, Kefzol,
Zolicef
cephalexin Keflex
cephalothin Keflin
cephapirin Cefadyl
cephradine Anspor
Second Generation
cefaclor Ceclor
cefamandole Mandol
cefmetazole Zefazone
cefonicid Monocid
cefotetan Cefotan
cefoxitin Mefoxin
cefuroxime Zinacef, Kefurox,
Ceftin
cefuroxime axetil Ceftin
Third Generation
cefixime Suprax
cefoperazone Cefobid
cefotaxime Claforan
ceftazidime Fortaz, Ceptaz,
Pentacef, Tazicef,
Tazidime
ceftizoxime Ceftizox
ceftriaxone Rocephin
Fourth Generation
cefepime Maxipime
*Several forms are marketed under other trade names.
This list is intended to be informative, not inclusive.
Copyright © 2005 by the AABB. All rights reserved.
ical DAT results are shown in Table 20-3.
Recent red cell transfusions and/or dra-
matic hemolysis may result in a weak DAT
by the time hemolysis is suspected.
The patient’s serum should be tested for
unexpected antibodies by routine proce-
dures. If the serum does not react with un-
treated red cells, the tests should be re-
peated against ABO-compatible red cells in
the presence of the drug(s) suspected of
causing the problem. Techniques are given
in Method 4.14 and Method 4.15.
If the drug has already been reported as
causing hemolytic anemia, testing methods
maybeavailableinthecasereports.Ifsuch
information is not available, an initial
screening test can be performed with a so-
lution of the drug at a concentration of ap-
proximately 1 mg/mL in phosphate-buf-
fered saline at a pH optimal for solubility of
the drug.
If these tests are not informative, at-
tempts can be made to coat normal red cells
with the drug, and the patient’s serum and
an eluate from the patient’s red cells can be
tested against the drug-coated red cells.
This is the method of choice when penicil-
lin or cephalosporins are thought to be im-
plicated. Results definitive for a penicil-
lin-induced positive DAT are reactivity of
the eluate against penicillin-coated red
cells and absence of reactivity between the
eluate and uncoated red cells.
The immune response may be due to a
metabolite of a drug rather than the drug it-
self. If the clinical picture is consistent with
immune-mediated hemolysis and the above
tests are noninformative, it may be helpful
to test drug metabolites (see Method 4.16).
Normal sera commonly agglutinate and/
or sensitize cephalosporin-treated red cells
due to the nonspecific uptake of protein
discussed above. This problem can be over-
come by testing a 1 in 20 dilution of the pa-
tient’s serum and a normal serum control
against the cephalosporin-treated red cells.
During the testing of cefotetan-treated red
cells, a 1 in 100 dilution of the patient’s se-
rum should be tested because it has been
shown that some normal sera appear to
contain “naturally occurring” antibodies to
cefotetan, some of which still react weakly at
a1in20dilution.
57,58 Cefotetan antibodies
associated with drug-induced immune
hemolytic anemia have very high anti-
globulin titers (4000 to 256,000).49
Two other observations have been made
regarding the testing of cefotetan antibod-
ies: 1) the last wash from the eluate prepa-
ration may react with cefotetan-treated red
cells (possibly due to the high-titer anti-
bodies and/or the antibody affinity), and 2)
drug-independent antibodies may be detected
in the serum and eluate and hemolysis may
be inadvertently attributed to idiopathic
WAIHA.49
References
1. Toy PT, Chin CA, Reid ME, Burns MA. Factors
associated with positive direct antiglobulin
tests in pretransfusion patients: A case con-
trol study. Vox Sang 1985;49:215-20.
2. Heddle NM, Kelton JG, Turchyn KL, Ali MAM.
Hypergammaglobulinemia can be associated
with a positive direct antiglobulin test, a non-
reactive eluate, and no evidence of hemolysis.
Transfusion 1988;28:29-33.
3. Petz LD, Garratty G. Immune hemolytic
anemias. 2nd ed. Philadelphia: Churchill
Livingstone, 2004.
4. Garratty G. The significance of IgG on the red
cell surface. Transfus Med Rev 1987;1:47-57.
5. Freedman J. The significance of complement
on the red cell surface. Transfus Med Rev
1987;1:58-70.
6. Mollison PL, Engelfriet CP, Contreras M.
Blood transfusion in clinical medicine. 10th
ed. Oxford, England: Blackwell Scientific Pub-
lications, 1997.
7. Clark JA, Tanley PC, Wallas CH. Evaluation of
patients with positive direct antiglobulin
tests and nonreactive eluates discovered dur-
ing pretransfusion testing. Immunohematology
1992;8:9-12.
8. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 477
Copyright © 2005 by the AABB. All rights reserved.
9. Judd WJ, Barnes BA, Steiner EA, et al. The
evaluation of a positive direct antiglobulin
test (autocontrol) in pretransfusion testing
revisited. Transfusion 1986;26:220-4.
10. Salama A, Mueller-Eckhardt C. Delayed
hemolytic transfusion reactions. Evidence for
complement activation involving allogeneic
and autologous red cells. Transfusion 1984;
24:188-93.
11. Ness PM, Shirey RS, Thoman SK, Buck SA.
The differentiation of delayed serologic and
delayed hemolytic transfusion reactions: In-
cidence, long-term serologic findings, and
clinical significance. Transfusion 1990;30:
688-93.
12. Garratty G. Immune cytopenia associated with
antibiotics. Transfus Med Rev 1993;7:255-67.
13. Kleinman S, Nelson R, Smith L, Goldfinger D.
Positive direct antiglobulin tests and immune
hemolytic anemia in patients receiving pro-
cainamide. N Engl J Med 1984;311:809-12.
14. Lutz P, Dzik W. Very high incidence of a posi-
tive direct antiglobulin test (+DAT) in pa-
tients receiving Unasyn®(abstract). Transfu-
sion 1992;32(Suppl):23S.
15. Garratty G. Problems associated with pas-
sively transfused blood group alloantibodies.
Am J Clin Pathol 1998;109:769-77.
16. Leger RM, Arndt PA, Ciesielski DJ, Garratty G.
False-positive eluate reactivity due to the
low-ionic wash solution used with commer-
cial acid-elution kits. Transfusion 1998;38:
565-72.
17. Judd WJ. Elution—dissociation of antibody
from red blood cells: Theoretical and practi-
cal considerations. Transfus Med Rev 1999;
13:297-310.
18. South SF, Rea AE, Tregellas WM. An evalua-
tion of 11 red cell elution procedures. Trans-
fusion 1986;26:167-70.
19. Sokol RJ, Hewitt S, Stamps BK. Autoimmune
haemolysis: An 18 year study of 865 cases re-
ferred to a regional transfusion centre. Br Med
J 1981;282:2023-7.
20. Shulman IA, Branch DR, Nelson JM, et al. Au-
toimmune hemolytic anemia with both cold
and warm autoantibodies. JAMA 1985;253:
1746-8.
21. Göttsche B, Salama A, Mueller-Eckhardt C.
Donath-Landsteiner autoimmune hemolytic
anemia in children. A study of 22 cases. Vox
Sang 1990;58:281-6.
22. Sokol RJ, Hewitt S, Stamps BK. Autoimmune
haemolysis. Mixed warm and cold antibody
type. Acta Haematol 1983;69:266-74.
23. Garratty G. Target antigens for red-cell-
bound autoantibodies. In: Nance SJ, ed. Clin-
ical and basic science aspects of immuno-
hematology. Arlington, VA: AABB, 1991:33-72.
24. Garratty G. Specificity of autoantibodies re-
acting optimally at 37 C. Immunohematology
1999;15:24-40.
25. FreedmanJ,WrightJ,LimFC,GarveyMB.
Hemolytic warm IgM autoagglutinins in au-
toimmune hemolytic anemia. Transfusion
1987;27:464-7.
26. Judd WJ. Investigation and management of
immune hemolysis: Autoantibodies and drugs.
In: Wallace ME, Levitt JS, eds. Current appli-
cations and interpretations of the direct anti-
globulin test. Arlington, VA: AABB, 1988:47-
103.
27. Leger RM, Garratty G. Evaluation of methods
for detecting alloantibodies underlying warm
autoantibodies. Transfusion 1999;39:11-16.
28. Branch DR, Petz LD. Detecting alloantibodies
in patients with autoantibodies (editorial).
Transfusion 1999;39:6-10.
29. Church AT, Nance SJ, Kavitsky DM. Predicting
the presence of a new alloantibody underly-
ing a warm autoantibody (abstract). Transfu-
sion 2000;40(Suppl):121S.
30. Shirey RS, Boyd JS, Parwani AV, et al. Prophy-
lactic antigen-matched donor blood for pa-
tients with warm autoantibodies: An algorithm
for transfusion management. Transfusion
2002;42:1436-41.
31. Garratty G, Petz LD. Approaches to selecting
blood for transfusion to patients with auto-
immune hemolytic anemia (editorial). Trans-
fusion 2002;42:1390-2.
32. Garratty G, Arndt P, Domen R, et al. Severe
autoimmune hemolytic anemia associated
with IgM warm autoantibodies directed
against determinants on or associated with
glycophorin A. Vox Sang 1997;72:124-30.
33. Garratty G, Arndt P, Leger R. Serological find-
ings in autoimmune hemolytic anemia asso-
ciated with IgM warm autoantibodies (ab-
stract). Blood 2001;98(Suppl 1):61a.
34. Nowak-Wegrzyn A, King KE, Shirey RS, et al.
Fatal warm autoimmune hemolytic anemia
resulting from IgM autoagglutinins in an in-
fant with severe combined immunodefi-
ciency. J Pediatr Hematol Oncol 2001;23:250-
2.
35. Garratty G, Petz LD, Hoops JK. The correla-
tion of cold agglutinin titrations in saline and
albumin with haemolytic anemia. Br J Haematol
1975;35:587-95.
36. Marks MR, Reid ME, Ellisor SS. Adsorption of
unwanted cold autoagglutinins by formalde-
hyde-treated rabbit erythrocytes (abstract).
Transfusion 1980;20:629.
37. Dzik W, Yang R, Blank J. Rabbit erythrocyte
stroma treatment of serum interferes with
recognition of delayed hemolytic transfusion
reactions (letter). Transfusion 1986;26:303-4.
478 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
38. Mechanic SA, Maurer JL, Igoe MJ, et al. Anti-
Vel reactivity diminished by adsorption with
rabbit RBC stroma. Transfusion 2002;42:
1180-3.
39. Garratty G, Arndt PA, Leger RM. Serological
findings in autoimmune hemolytic anemia
(AIHA) associated with both warm and cold
autoantibodies (abstract). Blood 2003;102
(Suppl 1):563a.
40. Garratty G. Autoimmune hemolytic anemia.
In: Garratty G, ed. Immunobiology of transfu-
sionmedicine.NewYork:MarcelDekker,
1994:493-521.
41. Garratty G, Postoway N, Nance SJ, Brunt DJ.
Spontaneous agglutination of red cells with a
positive direct antiglobulin test in various
media. Transfusion 1984;24:214-7.
42. Laine EP, Leger RM, Arndt PA, et al. In vitro
studies of the impact of transfusion on the
detection of alloantibodies after autoadsorp-
tion. Transfusion 2000;40:1384-7.
43. IssittPD,CombsMR,BumgarnerDJ,etal.
Studies of antibodies in the sera of patients
who have made red cell autoantibodies.
Transfusion 1996;36:481-6.
44. Salama A, Mueller-Eckhardt C. Immune-me-
diated blood cell dyscrasias related to drugs.
Semin Hematol 1992;29:54-63.
45. Petz LD, Mueller-Eckhardt C. Drug-induced
immune hemolytic anemia. Transfusion
1992;32:202-4.
46. Shulman NR, Reid DM. Mechanisms of drug-
induced immunologically mediated cyto-
penias. Transfus Med Rev 1993;7:215-29.
47. Christie DJ. Specificity of drug-induced im-
mune cytopenias. Transfus Med Rev 1993;7:
230-41.
48. Garratty G. Drug-induced immune hemolytic
anemia. In: Garratty G, ed. Immunobiology of
transfusion medicine. New York: Marcel
Dekker, 1994:523-51.
49. Arndt PA, Leger RM, Garratty G. Serology of
antibodies to second- and third-generation
cephalosporins associated with immune
hemolytic anemia and/or positive direct
antiglobulin tests. Transfusion 1999;39:1239-
46.
50. Gallagher NI, Schergen AK, Sokol-Anderson
ML, et al. Severe immune-mediated hemolytic
anemia secondary to treatment with cefote-
tan. Transfusion 1992;32:266-8.
51. Garratty G, Nance S, Lloyd M, Domen R. Fatal
immune hemolytic anemia due to cefotetan.
Transfusion 1992;32:269-71.
52. Stroncek D, Procter JL, Johnson J. Drug-in-
duced hemolysis: Cefotetan-dependent
hemolytic anemia mimicking an acute
intravascular immune transfusion reaction.
Am J Hematol 2000;64:67-70.
53. Viraraghavan R, Chakravarty AG, Soreth J.
Cefotetan-induced haemolytic anaemia. A re-
view of 85 cases. Adv Drug React Toxicol Rev
2002;21:101-7.
54. Garratty G, Arndt PA. Positive direct antiglo-
bulin tests and haemolytic anemia following
therapy with beta-lactamase inhibitor con-
taining drugs may be associated with non-
immunologic adsorption of protein onto red
blood cells. Br J Haematol 1998;100:777-83.
55. Broadberry RE, Farren TW, Kohler JA, et al.
Haemolytic anaemia associated with Tazobac-
tam (abstract). Vox Sang 2002;83(Suppl 2):
227.
56. Arndt PA, Leger RM, Garratty G. Positive di-
rect antiglobulin tests and haemolytic anae-
mia following therapy with the beta-lacta-
mase inhibitor, tazobactam, may also be
associated with non-immunologic adsorp-
tion of protein onto red blood cells (letter).
Vox Sang 2003;85: 53.
57. Arndt P, Garratty G. Is severe immune hemoly-
tic anemia, following a single dose of cefo-
tetan, associated with the presence of “natu-
rally-occurring” anti-cefotetan? (abstract)
Transfusion 2001;41(Suppl):24S.
58. Arndt PA. Practical aspects of investigating
drug-induced immune hemolytic anemia
due to cefotetan or ceftriaxone—a case study
approach. Immunohematology 2002;18:27-
32.
Suggested Reading
Dacie J. Historical review. The immune haemolytic
anaemias: A century of exciting progress in under-
standing. Br J Haematol 2001;114:770-85.
Engelfriet CP, Overbeeke MAM, von dem Borne
AEGKr. Autoimmune hemolytic anemia. Semin
Hematol 1992;29:3-12.
Garratty G. Novel mechanisms for immune destruc-
tion of circulating autologous cells. In: Silberstein
LE, ed. Autoimmune disorders of blood. Bethesda,
MD: AABB, 1996:79-114.
Garratty G. Autoantibodies induced by blood
transfusion (editorial). Transfusion 2004:445-9.
Mack P, Freedman J. Autoimmune hemolytic ane-
mia: A history. Transfus Med Rev 2000;14:223-33.
Petz LD, Garratty G. Immune hemolytic anemias.
2nd ed. Philadelphia: Churchill Livingstone, 2004.
Petz LD. A physician’s guide to transfusion in auto-
immune haemolytic anaemia. Br J Haematol 2004;
124:712-16.
Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 479
Copyright © 2005 by the AABB. All rights reserved.
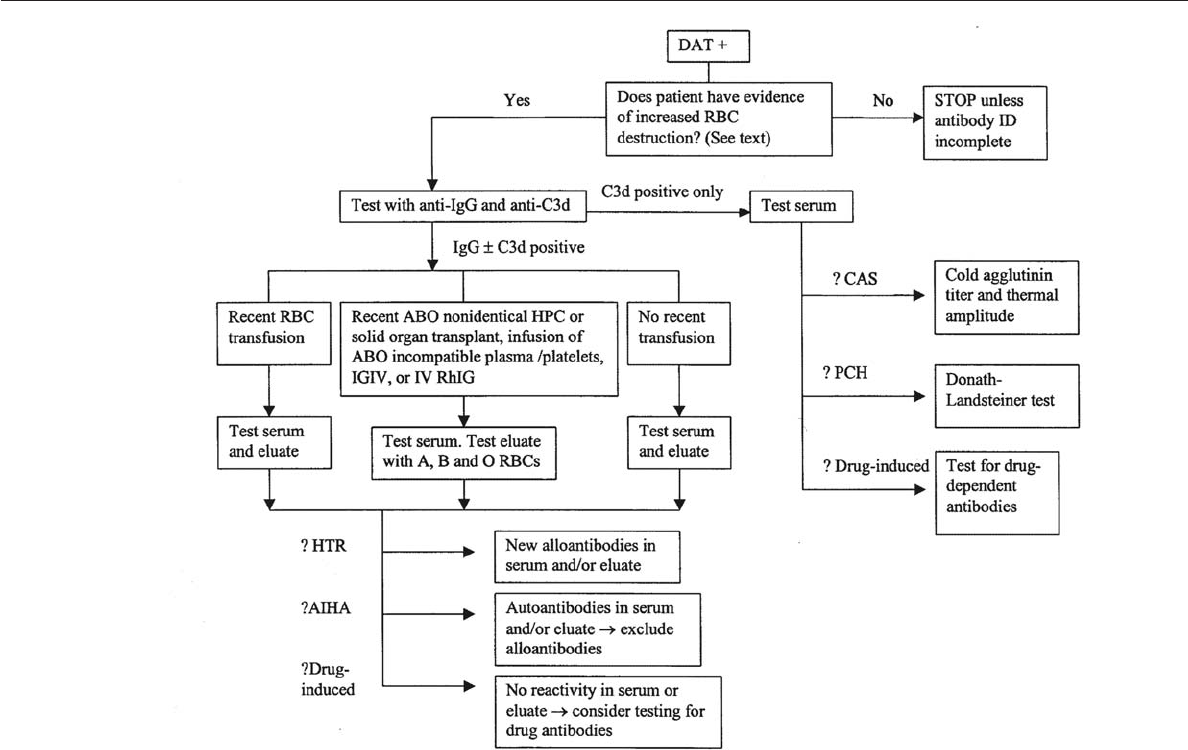
480 AABB Technical Manual
Appendix 20-1. An Example of an Algorithm for Investigating a Positive DAT (Excluding Investigation of HDFN)
Copyright © 2005 by the AABB. All rights reserved.

Chapter 20: The Positive Direct Antiglobulin Test and Immune-Mediated Red Cell Destruction 481
Appendix 20-2. Some Drugs Associated with Immune Hemolysis and/or Positive
DATs Due to Drug-Induced Antibodies
Drug Therapeutic Category Possible Mechanism
Acetaminophen Analgesic, antipyretic DD-IC
Aminopyrine Analgesic, antipyretic DD-IC
Amphotericin B Antifungal, antibiotic DD-IC
Ampicillin Antibacterial DD-IC
Antazoline Antihistamine DD-IC
Apazone (azapropazone) Anti-inflammatory, analgesic DI, DD-DA
Buthiazide (butizide) Diuretic, antihypertensive DD-IC
Carbenicillin Antibacterial DD-DA
Carbimazole Thyroid inhibitor DD-IC
Carboplatin Antineoplastic DD-DA, DD-IC
Carbromal Sedative; hypnotic DD-DA
Catergen Diarrheal astringent, treatment of hepatic disease DI
Cephalosporins Antibacterials
First generation NIA, DD-DA
Second generation DD-IC, DD-DA, DI
Third generation DD-IC, DD-DA, DI
Chaparral DI
Chlorpropamide Antidiabetic DD-IC
Chlorpromazine Antipsychotic DI, DD-IC
Cisplatin Antineoplastic NIA
Cladribine
(chlorodeoxyadenosine)
Antineoplastic DI
Clavulanate potassium β-lactamase inhibitor/antibacterial NIA
Cyanidanol DI, DD-DA, DD-IC
Cyclofenil Gonad-stimulating principle DI
Cyclosporine Immunosuppressive DI
Diclofenac Anti-inflammatory DI, DD-IC
Diethylstilbestrol Estrogen DD-IC
Diglycoaldehyde Antineoplastic NIA
Dipyrone Analgesic, antipyretic DD-IC, DD-DA
Elliptinium acetate Antineoplastic DD-IC
Erythromycin Antibacterial DD-DA
Etodolac Anti-inflammatory, analgesic IC
Fenfluramine Anorexic ?
Fenoprofen Anti-inflammatory, analgesic DI, DD-IC
Fludarabine Antineoplastic DI
Fluorescein Injectable dye DD-DA, DD-IC
Fluorouracil Antineoplastic DD-IC
Glafenine Analgesic DI, DD-IC
Hydralazine Antihypertensive DD-IC
Hydrochlorothiazide Diuretic DD-IC
Ibuprofen Anti-inflammatory DI
Insulin Antidiabetic DD-DA?, DD-IC
Interferon Antineoplastic, antiviral DI
Isoniazid Antibacterial, tuberculostatic DD-DA?, DD-IC
Levodopa Antiparkinsonian, anticholinergic DI
Mefenamic acid Anti-inflammatory DI
Mefloquine Antimalarial DD-IC (cont’d)
Copyright © 2005 by the AABB. All rights reserved.

482 AABB Technical Manual
Appendix 20-2. Some Drugs Associated with Immune Hemolysis and/or Positive
DATs Due to Drug-Induced Antibodies (cont’d)
Drug Therapeutic Category Possible Mechanism
Melphalan Antineoplastic DD-IC
6-Mercaptopurine Antineoplastic DD-DA
Methadone Narcotic analgesic ?
Methicillin Antibacterial DD-DA
Methotrexate Antineoplastic, antimetabolite DD-IC
Methyldopa Antihypertensive DI
Moxalactam (latamoxef) Antibacterial DD-IC, DI
Nafcillin Antibacterial DD-DA
Nomifensine Antidepressant DI, DD-IC
p-Aminosalicylic acid Antitubercular DD-IC
Penicillin G Antibacterial DD-DA
Phenacetin Analgesic, antipyretic DI, DD-IC
Piperacillin Antibacterial DD-DA, DD-IC
Podophyllotoxin Antineoplastic, cathartic ?
Probenecid Uricosuric DD-IC
Procainamide Cardiac depressant, antiarrhythmic DI
Propyphenazone Analgesic, antipyretic, anti-inflammatory DD-IC
Pyramidon Analgesic, antipyretic DD-IC
Quinidine Cardiac depressant, antiarrhythmic DD-DA, DD-IC
Quinine Antimalarial DD-IC
Ranitidine Antagonist (to histamine H2receptors) ?
Rifampin (rifampicin) Antibacterial, antitubercular DD-IC
Sodium pentothal Anesthetic DD-IC
Stibophen Antischistosomal DD-IC
Streptomycin Antibacterial, tuberculostatic DI, DD-DA, DD-IC
Sulbactam sodium β-lactamase inhibitor/antibacterial NIA
Sulfonamides Antibiotics DD-IC
Sulfonylurea derivatives Antidiabetic DD-IC
Sulindac Anti-inflammatory DD-DA, DI
Suprofen Anti-inflammatory, analgesic DD-IC, DI
Suramin Antitrypanosomal, antifilarial NIA
Temafloxacin Antibacterial DD-IC
Teniposide Antineoplastic DI, DD-IC
Tetracycline Antibacterial, antirickettsial, antiamebic DD-DA?, DD-IC
Thiopental Anesthetic DD-IC
Tolbutamide Antidiabetic DD-DA
Tolmetin Anti-inflammatory DI, DD-IC
Triamterene Diuretic DD-IC
Trimellitic anhydride Used in preparation of dyes, resins, etc ?
Zomepirac Analgesic, anti-inflammatory DD-DA, DD-IC, DI
Mechanisms listed are based on descriptions in the literature.3,44-48
DAT = Direct antiglobulin test.
DD-DA = Drug-dependent. Drug adsorbed onto red cells; antibody reacts with drug on cells.
DD-IC = Drug-dependent. “Immune complex mechanism.” Requires drug, serum, and red cells for serologic
demonstration. For most of these drugs, there are only single or very few case reports.
DI = Drug-independent. Associated with autoantibodies similar to those in AIHA. Drug not required for in-vitro
demonstration. Mechanisms of autoantibody production may vary.
NIA = Nonimmunologic adsorption of proteins.
? = Mechanism unclear or unknown.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 21: Blood Transfusion Practice
Chapter 21
Blood Transfusion Practice
THE DECISION TO transfuse, like
any other therapeutic decision,
should be based on the risks, ben-
efits, and alternatives of treatment. Unfor-
tunately, data regarding the indications
for transfusion are frequently not available
and recipients run the risk of both over-
transfusion and undertransfusion. Trans-
fusions based solely on laboratory test
“triggers,” in particular, are problematic.
Consensus statements on the use of blood
components such as those produced by
the National Institutes of Health (NIH)
help guide therapy but cannot substitute
for clinical judgment.
Red Blood Cell Transfusion
Physiologic Principles
The primary indication for transfusion of
Red Blood Cells (RBCs) is to restore or main-
tain oxygen-carrying capacity to meet tis-
sue demands; transfusion to replace red
cells destined for destruction in hemoly-
tic disease of the newborn is discussed in
Chapter 24. Because demand for oxygen
varies greatly among different individuals
in different clinical circumstances, mea-
surement of only the hematocrit or the
hemoglobin concentration (“the hemo-
globin”) cannot accurately assess the need
for transfusion.1-3
Normal Oxygen Supply and Demand
Tissues at rest have a baseline demand for
oxygen, particularly the heart, kidneys,
brain, liver, and gastrointestinal tract;
consumption by muscle is very low at
rest. The oxygen content of blood (mL
O2/mL blood) is determined by the hemo-
globin, the binding coefficient of hemo-
globin for oxygen, the oxygen saturation
of hemoglobin, and a small quantity of
483
21
Copyright © 2005 by the AABB. All rights reserved.
oxygen dissolved in the plasma. This is
described as:
O2content = (Hb ×1.39 ×%sat)
+(pO
2×0.003)
Tissue oxygen consumption is calculated
as the difference between oxygen delivery
in the arterial blood and oxygen return by
the venous blood:
O2consumption = Cardiac output ×
Hb ×1.39 ×(%satarterial –%sat
venous)/100
which is expressed as
(mL O2/minute) =
L/minute×g/L×mLO
2/g
The oxygen saturation of arterial and ve-
nous hemoglobin varies with the partial
pressure of oxygen. Under normal circum-
stances the pO2falls from 100 mm Hg in the
arteries to 40 mm Hg in the veins as the tis-
sues extract oxygen, and hemoglobin satu-
ration falls from near 100% in the arteries to
approximately 75% in the veins; thus, the
oxygen extraction ratio is 0.25. That is, the
hemoglobin “gives up” only 25% of its oxy-
gen. When tissue demand for oxygen in-
creases or the supply of oxygen decreases,
the tissues extract a greater fraction of oxy-
gen from the plasma and from hemoglobin;
this results in a lower venous pO2and de-
creased oxygen saturation of the venous
blood. Studies in primates suggest that a
critical point of limited oxygen delivery is
reached when the oxygen extraction ratio
approaches twice normal or 0.50.2
Under normal resting conditions, the
body has a large reserve of oxygen supply
relative to demand. In the average adult,
approximately 1000 mL/minute is available
to the tissues and only 250 mL/minute is
consumed as follows:
O2supply (Calculation assumes a hemo-
globin of 140 g/L and a pO2of 100)
= Cardiac output × O2contentarterial
= 5 L/minute × [(140 × 1.39 × 100%)
+ (100 × 0.003)]
= 5 L/minute × 200 mL O2/L
= 1000 mL O2/minute
O2consumption = Cardiac output × (O2
contentarterial –O
2contentvenous)
= 5 L/minute × (200 mL O2/L –
150 mL O2/L)
=5L/minute×50mLO
2/L =
250 mL O2/minute
Compensation for Anemia
The above equations demonstrate that any
decrease in oxygen content due to anemia
can be compensated for by an increase in
cardiac output.1,2 This occurs because of
increased cardiac work and also because
anemia decreases blood viscosity, and
thus peripheral vascular resistance. The
increase in oxygen supply provided by in-
creased cardiac output is augmented by
increased oxygen extraction. Oxygen ex-
traction is augmented acutely by a de-
crease in tissue oxygen tension, acting at
the steep portion of the hemoglobin-oxy-
gendissociationcurve
1(see Fig 8-1), and
by acidosis, which promotes oxygen dis-
sociation from hemoglobin. Over time, an
increase in red cell 2,3-diphosphogly-
cerate (2,3-DPG) concentration also has a
significant positive effect on oxygen un-
loading.
Measuring the Adequacy of Oxygen Supply
As shown above, multiple factors deter-
mine oxygen delivery to the tissues, ex-
cept at the lower extremes. Therefore,
measurements in addition to the hemo-
globin must be used to guide most trans-
fusion decisions. The adequacy of the
oxygen supply depends on the partial
484 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
pressure of inspired oxygen, gas exchange
in the lungs, the patient’s cardiac perfor-
mance, hemoglobin, oxygen-hemoglobin
affinity, and current oxygen demand; all
but the oxygen-hemoglobin affinity are
subject to substantial variation. For pa-
tients in an intensive care unit or in the
operating room, direct measurement of
the cardiac output and mixed venous oxy-
gentension,inassociationwithhemoglo
-
bin level, may serve as more physiologic
guides for transfusion decisions than he-
moglobin alone. Nonetheless, most such
decisions will continue to be made based
on hemoglobin and standard clinical as-
sessment.
Determinants of cardiac performance
include the patient’s intravascular volume,
the anemia-related reduction in peripheral
vascular resistance, the presence of coro-
nary artery disease or other forms of heart
disease, and the patient’s age. Tissue oxy-
gen debt results when oxygen demand ex-
ceeds supply; tissues convert to anaerobic
metabolism and produce increased quanti-
ties of lactic acid. Metabolic acidosis, in
turn, impairs cardiac performance, further
decreasing perfusion and tissue oxygen de-
livery, leading to greater tissue hypoxia in a
vicious cycle.
Treating Inadequate Oxygen Supply
RBC transfusion is the most direct means
of raising the hemoglobin concentration.
Other ways to improve oxygen supply rel-
ative to demand include increasing tissue
perfusion (maximizing cardiac perfor-
mance), increasing oxygen saturation
with supplemental oxygen or mechanical
ventilation, and decreasing tissue oxygen
demands with bed rest, antipyretics, and
avoidance of hypertension.
For example, a patient with a hemoglo-
bin of 6 g/dL and a cardiac index of 5 L/
minute/m2has decreased oxygen delivery.
Assuming that oxygen extraction remains
constant, oxygen delivery could be normal-
ized by an increase in the hemoglobin con-
centration to 9 g/dL, or by an increase in
the cardiac index to 7 L/minute/m2.A
smaller increase in hemoglobin would
blunt the required increase in cardiac in-
dex.
Red-Cell-Containing Components
Whole Blood
Whole Blood provides oxygen-carrying
capacity, stable coagulation factors (the
concentrations of Factors V and VIII de-
crease during storage), and blood volume
expansion. Thus, it is useful for patients
with concomitant red cell and volume
deficits, such as actively bleeding pa-
tients, and will help support coagulation
in appropriate clinical settings such as
liver transplantation.4In fact, Whole Blood
is rarely available for allogeneic transfu-
sion; RBCs and asanguinous solutions
have become the standard for most cases
of active bleeding in trauma and surgery,
with supplementation of hemostatic ele-
ments as needed. The major use of Whole
Blood in the United States today is for
autologous transfusion (see Chapter 5).
Red Blood Cells
Red cell components are indicated for the
treatment of patients who require an in-
crease in oxygen-carrying capacity and
red cell mass.5The transfusion of red cells
increases oxygen-carrying capacity with
less expansion of blood volume per unit
than Whole Blood. This is important for
patients who are at risk for circulatory
overload (eg, neonates or patients with
congestive heart failure). In a typical adult
in the absence of bleeding, hemolysis, or
major fluid shifts, one RBC unit is ex-
pected to raise the hemoglobin concen-
Chapter 21: Blood Transfusion Practice 485
Copyright © 2005 by the AABB. All rights reserved.

trationbyapproximately1g/dL,orthe
hematocrit by 3%.
Selection of Whole Blood and Red Cell
Components
Whole Blood must be ABO identical. RBC
units need not be ABO identical but must
be compatible with ABO antibodies in the
recipient’s plasma. Rh-negative recipients
should receive Rh-negative Whole Blood
or red cell components. In cases of
trauma or massive transfusion, it may be
necessary to use Rh-positive components,
as discussed below. See Table 21-1 for
blood group selection in red cell transfu-
sion. Selection of RBC units for patients
with blood group alloantibodies is dis-
cussed in Chapter 19.
Indications for Transfusion
Several organizations have published guide-
lines for RBC transfusion.3,6,7 Reviews of
indications for transfusion are also avail-
able.1,8,9
Blood Loss and Perioperative Transfusion
For actively bleeding patients, the goal of
initial treatment should be to prevent the
development of hypovolemic shock by
stoppingthebleedingandrestoringintra
-
vascular volume. Efforts to restore volume
by the infusion of crystalloid or colloid so-
lutions take precedence over restoration
of oxygen-carrying capacity and should
be started immediately. In situations of
acute bleeding, guidelines suggest trans-
fusing patients who have lost 30% to 40%
of their blood volume in conjunction with
other measures to correct and maintain
total blood volume.3,6 Patients with car-
diac or other disease may require replace-
ment sooner. Healthy resting adults have
been demonstrated to tolerate acute iso-
volemic hemodilution to hemoglobin
concentrations as low as 5 g/dL without
demonstrating evidence of inadequate
oxygenation.10 In patients who refuse
transfusion on religious grounds and un-
dergo surgery, mortality increases pro-
gressively below postoperative hemoglo-
bin levels of 5 or 6 g/dL, particularly for
those with cardiovascular disease.11
Perioperative transfusion accounts for
55% to 65% of red cell component use.12
Randomized trials have demonstrated the
safety of a “transfusion trigger” of 8 g/dL of
hemoglobin in patients undergoing cardio-
vascular surgery,13,14 orthopedic surgery,15
and acute gastrointestinal bleeding.16 Even
before most of this evidence was available,
an NIH consensus conference on periopera-
tive red cell transfusion7emphasized that a
hemoglobin of 10 g/dL was inappropriate
as a guideline or transfusion trigger in the
perioperative setting and suggested a he-
486 AABB Technical Manual
Table 21-1. Suggested ABO Group Selection Order for Transfusion of RBCs
Recipient
ABO Group
Component ABO Group
1st Choice 2nd Choice 3rd Choice 4th Choice
AB AB A B O
AAO
BBO
OO
Copyright © 2005 by the AABB. All rights reserved.
moglobin of 7 g/dL as a level at which
transfusion was frequently required in oth-
erwise healthy individuals with acute ane-
mia. Factors to be considered in making an
individual transfusion decision included
“the duration of the anemia, the intravas-
cular volume, the extent of the operation,
the probability of massive blood loss, and
thepresenceofcoexistingconditionssuch
as impaired pulmonary function, inade-
quate cardiac output, myocardial ischemia,
or cerebrovascular or peripheral circulatory
disease.”7The guidelines also emphasized
that transfusion does not improve wound
healing, which depends on pO2rather than
total oxygen content of the blood.
A guideline by an American Society of
Anesthesiologists task force3cited a hemo-
globin below 6 g/dL as “almost always” in-
dicating transfusion, a hemoglobin above
10 g/dL as rarely indicating transfusion,
and the range in between as the realm of
clinical judgment. A task force of the Col-
lege of American Pathologists reached a
similar conclusion and proposed several
objective measures that might indicate red
cell transfusion for hemoglobin levels in the
range of 6 to 10 g/dL, including tachycardia
or hypotension in the face of normovole-
mia, a mixed venous pO2of <25 torr, an ox-
ygen extraction ratio >50%, or a total oxy-
gen consumption of <50% of baseline.6
In the past, there was some concern that
transfusions of a single RBC unit were likely
to represent unnecessary intervention. How-
ever, if transfusion of a single unit will
achieve the desired clinical outcome, then
only one unit should be transfused. Trans-
fusing additional units in this setting will
increase the risk of transfusion without any
additional benefit.
Anemia
Among medical patients, those with car-
diovascular and malignant diseases ac-
count for a large proportion of those re-
ceiving RBC units.13 In a prospective ran-
domized trial of red cell transfusion, ane-
mic but euvolemic patients in the
intensive care unit (ICU) were assigned to
either “restrictive” or “liberal” transfusion
regimens that maintained the hemoglo-
bin between 7 and 9 g/dL or between 10
and 12 g/dL, respectively.17 The mortality
rate during hospitalization (but not at 30
days) was significantly lower in the re-
strictive-strategy group (22.3% vs 28.1%, p
= 0.05). No difference in mortality rate
was seen among all patients with clini-
cally significant cardiac disease. One third
of the restrictive group avoided transfu-
sion, and total red cell use was half that of
the liberal group. These authors con-
cluded that a restrictive strategy of red
cell transfusion is at least as effective as,
and possibly superior to, a liberal transfu-
sion strategy in critically ill patients, with
the possible exception of the subset of pa-
tients with acute myocardial infarction
(MI) and unstable angina. Reanalysis of
the patients in this study with cardiovas-
cular disease showed a trend, albeit not
statistically significant, toward increased
mortality in the restrictive group among
patients with MI and unstable angina.18
A retrospective study of a large number
of elderly (>65 years old) patients hospital-
ized with acute MI divided into groups ac-
cording to admission hematocrit compared
30-day mortality rates in patients who re-
ceived transfusion and those who did not.19
Transfusion appeared beneficial in patients
with a hematocrit <30%. This result per-
sisted when data were adjusted for multiple
clinical and institutional factors.
Patients with chronic anemia tolerate a
low hemoglobin better than those with
acute anemia because of cardiovascular
compensation and increased oxygen ex-
traction. Moreover, patients at bed rest
who are not febrile, who do not have con-
Chapter 21: Blood Transfusion Practice 487
Copyright © 2005 by the AABB. All rights reserved.
gestive heart failure, and who are not
hypermetabolic have low oxygen require-
ments and may tolerate anemia remark-
ably well. However, the high oxygen needs
of cardiac muscle may precipitate angina
in patients with cardiac disease and ane-
mia. A hemoglobin concentration of 8 g/
dL adequately meets the oxygen needs of
most patients with stable cardiovascular
disease.
Although it is desirable to prevent un-
necessary transfusions, anemic patients
who are symptomatic should receive ap-
propriate treatment. Anemia may cause
symptoms of generalized weakness, head-
ache, dizziness, disorientation, breathless-
ness, palpitations, or chest pain, and signs
include pallor (not cyanosis) and tachycar-
dia. Elwood and coworkers20 could not cor-
relate symptoms with the hemoglobin level
in patients with chronic iron deficiency
anemia and a hemoglobin as low as 8 g/dL.
A study of the use of erythropoietin demon-
strated improvement in symptoms as he-
moglobin is raised to 10 g/dL, but no
change above that level.21 Patients with
chronic hypoproliferative anemia who are
known to be transfusion dependent should
be maintained at a level that prevents
symptoms by establishing a transfusion
schedule and then adjusting it as needed.
Platelet Transfusion
Physiologic Principles
Hemostasis occurs in four phases: the
vascular phase, the formation of a platelet
plug, the development of fibrin clot on
the platelet plug, and the ultimate lysis of
the clot. Platelets are essential to the for-
mation of the primary hemostatic plug
and provide the surface upon which fibrin
forms. Deficiencies in platelet number
and/or function can have unpredictable
effects that range from clinically insignifi-
cant prolongation of the bleeding time to
major life-threatening hemorrhage. Pla-
telet plug formation results from the com-
bined processes of adhesion, activation and
release, aggregation, and procoagulant
activity.22 Platelet adhesion to damaged
endothelium is mediated largely by the
von Willebrand factor (vWF), which binds
to the surface glycoprotein (GP) receptor
GPIb-IX-V complex. The process of acti-
vation and release causes a dramatic
change in platelet shape, with extension
of pseudopod-like structures, a change in
the binding properties of membrane acti-
vation proteins, secretion of internal gran-
ule contents, and activation of several
metabolic pathways. These changes have
many effects, including the recruitment of
additional platelets, which aggregate with
the help of fibrinogen or vWF binding to
platelet surface glycoproteins. Finally, the
platelet membrane procoagulant activity
localizes and directs formation of fibrin.
Assessing Platelet Function
Decreased platelet numbers may result
from decreased production, increased de-
struction, or splenic sequestration. Plate-
let function may be adversely affected by
such factors as drugs, liver or kidney dis-
ease, sepsis, fibrin(ogen) degradation
products, cardiopulmonary bypass, and
primary marrow disorders. Platelet hemo-
stasis is assessed by the medical history,
physical examination, and laboratory
tests including platelet count and bleed-
ing time or in-vitro platelet function as-
says (eg, PFA100). Patients with inadequate
platelet number or function may demon-
strate petechiae, easy bruising, mucous
membrane bleeding, nose and gum bleed-
ing, and hematuria. Preprocedure platelet
counts have predicted bleeding in some
studies23,24 but not in others.25-27
488 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Bleeding time measures both the vascu-
lar phase and the platelet phase of hemo-
stasis. Although the bleeding time may be a
useful diagnostic test in the evaluation of
patients with known or suspected abnor-
malities of platelet function, it is a poor pre-
dictor of surgical bleeding28 and is not a re-
liable indicator of the need for platelet
transfusion therapy.29 The in-vitro measure-
ment of platelet function is useful but, like
the bleeding time, is a poor predictor of
bleeding.30
Platelet Life Span and Kinetics
Platelets normally circulate with a life span
of 10.5 days,31 and platelets that have been
properly collected and stored have a near
normal residual mean life span of 4 to 5
days when reinfused into the original do-
nor. Conditions that shorten platelet life
span include splenomegaly, sepsis, drugs,
disseminated intravascular coagulation
(DIC), auto- and alloantibodies, endothe-
lial cell activation, and platelet activation
(eg, cardiopulmonary bypass or intra-aor-
tic balloon pumps). Because a relatively
constant number of platelets (7,000-
10,000/µL/day) are consumed by routine
plugging of minor endothelial defects, the
fraction of the circulating platelet pool re-
quired for maintenance functions increases
as the total number of platelets declines.
Therefore, the life span of native and
transfused platelets decreases with pro-
gressive thrombocytopenia.31 The re-
sponse to platelet transfusion is best as-
sessed by observing whether bleeding
stops and by measuring the posttransfu-
sion platelet increment. The posttransfu-
sion increment is generally measured
between 10 minutes and 1 hour after
completion of the transfusion and is ex-
pressed as a corrected count increment
(CCI) or percent recovery, as outlined in
Chapter 16. Two consecutive poor re-
sponses suggest platelet refractoriness.
Platelet Components
Platelets
A single unit of Platelets prepared from an
individual unit of Whole Blood may be
adequate for transfusion to neonates or
infants, but, for adults, 4 to 6 units are or-
dinarily pooled for transfusion to achieve
a dose greater than 3.0 × 1011 platelets.
This should increase the platelet count by
30,000 to 60,000/µL.
Platelets Pheresis
Units of platelets prepared by apheresis
technology (“single-donor platelets”) have
a platelet content similar to that of pooled
platelets from four to six donors and, de-
pending on the equipment used, may
have a reduced leukocyte content. The
fact that a single such unit can provide an
entire transfusion facilitates provision of
compatible platelets to recipients who are
refractory because of alloimmunization.
Transfusion of recipients who are refrac-
tory to platelet transfusions is discussed
in Chapter 16.
Selection of Platelets
ABO Matching
Because ABO antigens are present on the
platelet surface, recovery of group A pla-
telets transfused into group O patients is
somewhat decreased,32 but this effect is
not usually clinically significant (see
Chapter 16). Transfusion of ABO-incom-
patible plasma present in platelet compo-
nents may also result in a blunted post-
transfusion platelet count increment.33
Hemolysis occurs rarely in this setting but
it frequently causes a positive direct
antiglobulin test (DAT), which may in-
crease costs and charges to the patient for
Chapter 21: Blood Transfusion Practice 489
Copyright © 2005 by the AABB. All rights reserved.
serologic investigation. Moreover, a retro-
spective analysis suggested that survival
after marrow transplantation was signifi-
cantly reduced in patients who received
substantial amounts of ABO-incompati-
ble plasma from platelet transfusion,34
and it has been suggested that infusion of
soluble A and B antigen in platelet or
plasma components may have a similar
adverse effect mediated by immune com-
plex function.35 Therefore, it may be pru-
dent to use ABO-matched platelets, par-
ticularly for patients requiring repeated
transfusions. However, urgently needed
transfusions should not be delayed in or-
der to obtain them.
For infants, it is desirable to avoid ad-
ministration of plasma that is incompatible
with the infant’s red cells; if platelets con-
taining compatible plasma are not avail-
able, the plasma can be reduced (see
Method 6.15). This is rarely necessary in
adults or older children, although signifi-
cant hemolysis has been reported after
transfusion of group O Platelets Pheresis
with high-titer anti-A or B.36 If transfused
ABO antibodies are detectable in the recipi-
ent, it may become necessary to use group
ORBCunits.
Matching for Rh
The D antigen is not detectable on plate-
lets, and posttransfusion survival of plate-
lets from Rh-positive donors is normal in
recipients with anti-D. However, platelet
components contain small numbers of
red cells so Rh-negative individuals may
become alloimmunized by platelet com-
ponents from Rh-positive donors. For im-
munocompetent normal Rh-negative fe-
males of childbearing potential, it is
especially desirable to avoid administra-
tion of platelets from Rh-positive donors;
however, if this is unavoidable, Rh Im-
mune Globulin (RhIG) should be admin-
istered. If hematoma formation is an is-
sue, an intravenous form of RhIG is avail-
able. A full dose of RhIG, which is consid-
ered immunoprophylactic for up to 15 mL
of Rh-positive red cells, should protect
against the red cells in a minimum of 30
unitsofRh-positivePlateletsor7unitsof
Rh-positive Platelets Pheresis.
Therapeutic Platelet Transfusion
Significant bleeding due to thrombo-
cytopenia or abnormal platelet function is
an indication for “therapeutic” platelet
transfusion.37 The decision to transfuse
platelets depends on the cause of bleed-
ing, the patient’s clinical condition, and
the number and function of the circulat-
ing platelets.3,38-40 Platelet transfusions are
most likely to be of benefit when thrombo-
cytopenia is the primary hemostatic de-
fect. The goal is to maintain counts
>50,000/µL. Other blood components
may also be required in patients with
multiple defects. Bleeding due to the de-
fects in platelet function that follow
cardiopulmonary bypass surgery or to the
ingestion of aspirin-containing com-
pounds, glycoprotein IIb/IIIa antagonists
(eg, abciximab), and P2 inhibitors (eg,
clopidigrel and ticlopidine) often re-
sponds to platelet transfusion. Other de-
fects such as those found in uremia or von
Willebrand disease respond less well be-
cause the transfused platelets tend to ac-
quire the same defect.
Prophylactic Platelet Transfusion
Indications for prophylactic platelet
transfusion are more controversial than
for therapeutic rationales. A threshold of
20,000/µL or less for patients with chemo-
therapy-induced thrombocytopenia has
been used by many physicians, but pro-
spective randomized trials have shown
that a threshold of 10,000/µLinstablepa
-
490 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
tients is equally safe and results in signifi-
cant decreases in platelet usage.41-43 A
higher transfusion trigger is often used for
patients with fever, evidence of rapid con-
sumption, high white cell counts, coagu-
lation defects, and intracranial lesions.38
In contrast, many stable thrombocyto-
penic patients can tolerate platelet counts
as low as 5000/µL.41
Despite the widespread use of prophy-
lactic platelet transfusions, few studies have
documented their clinical benefit. One
study comparing patients given prophylac-
tic transfusions with patients transfused
only for clinically significant bleeding dem-
onstrated a significant decrease in bleed-
ing, but the number of patients in the study
was too small to show a difference between
the groups in overall survival or in deaths
due to bleeding.44 Of interest, the prophy-
lactic group received twice as many platelet
transfusions, and there was a suggestion
that refractoriness developed more often in
this group. This observation raises the ca-
veat that prophylactic platelet transfusion
may be most relevant to patients in whom
thrombocytopenia is expected to be a tem-
porary condition.38 If thrombocytopenia or
platelet dysfunction will be prolonged, the
development of refractoriness may limit the
response to platelets, particularly if im-
mune function is normal as it is in patients
with aplastic anemia or congenital thrombo-
cytopathies.
Patients with severe preoperative thrombo-
cytopenia are generally assumed to benefit
from prophylactic platelet transfusion, but
this has not been demonstrated in experi-
mental studies. The threshold for such pro-
phylaxis is typically set at platelet counts
between 50,000 and 100,000/µL.3Prophy-
lactic transfusion of platelets has been in-
vestigated in circumstances in which
thrombocytopenia is expected to develop
intraoperatively, either because of dilution45
or cardiopulmonary bypass46;inbothcases,
prophylaxis was ineffective. Although it en-
dorsed the logic of prophylactic platelet
transfusion for thrombocytopenic patients
undergoing surgery, the NIH consensus
panel38 suggested that such transfusions
were most appropriate for patients in
whom hemorrhage could not be observed
or in whom it occurred at a site where it
could be critical in small amounts (eg, in
the central nervous system). Published
guidelines3,39 suggest a platelet transfusion
trigger of 50,000/µL for most major surgery,
with counts “near” 100,000/µL possibly re-
quired for patients undergoing neurosur-
gery or ophthalmic procedures.39 Prophy-
lactic platelet transfusion may also be
useful for patients who are having surgery
and who have a platelet function defect,39
including that due to treatment with
abciximab.47
Refractoriness to Platelet Transfusion
Platelet refractoriness, defined as a poor
increment following a dose of platelets, can
result from either immune or nonimmune
mechanisms and is discussed in detail in
Chapter 16 and other reviews.48,49 The anti-
bodies that cause immune refractoriness
may have either allo- or autoreactivity, with
alloantibodies most commonly directed
against Class I HLA antigens. Autoantibodies
occur in immune thrombocytopenic pur-
pura (ITP) (see Chapter 16). Nonimmune
causes of the refractory state include infec-
tion, splenomegaly, drugs (particularly
amphotericin B), and accelerated platelet
consumption (see Table 16-3).
Contraindications to Platelet Transfusion
There are several conditions for which
platelet transfusions may be requested
but are contraindicated. Relative contra-
indications include conditions in which
the likelihood of benefit is remote; trans-
fusion in this setting merely wastes a valu-
Chapter 21: Blood Transfusion Practice 491
Copyright © 2005 by the AABB. All rights reserved.
able component. Examples include pro-
phylactic platelet transfusions in stable
patients with ITP50 or platelet refractoriness.
For ITP patients undergoing splenectomy,
transfusion of platelets should be delayed
until the vascular pedicle is clamped.
Platelet transfusion should be avoided for
patients with thrombotic thrombocyto-
penic purpura (TTP) or active heparin-in-
duced thrombocytopenia except in life- or
organ-threatening hemorrhage. These
conditions are associated with platelet
thrombi, and major thrombotic compli-
cations may follow platelet transfusions.51
Granulocyte Transfusion
The use of granulocyte transfusions for
adult recipients is rare. New antibiotics,
adverse effects attributable to granulocyte
transfusions, the advent of recombinant
growth factors, and difficulty demonstrat-
ing efficacy have contributed to this de-
cline. Nevertheless, in selected patients,
transfused granulocytes may produce
clinical benefits,52 particularly with the
larger granulocyte doses available from
donors treated with granulocyte colony-
stimulating factor.53 Attention to HLA
compatibility is also required for allo-
immunized recipients.54 The preparation,
storage, and pretransfusion testing of
granulocytes are discussed in Chapter 6,
andtheiruseinneonatesisdiscussedin
Chapter 24.
Indications and Contraindications
The goals of granulocyte transfusion should
be clearly defined before a course of ther-
apy is initiated. In general, the patient
should meet the following minimum con-
ditions:
1. Neutropenia (granulocyte count less
than 500/µL).
2. Fever for 24 to 48 hours, positive
bacterial or fungal blood cultures, or
progressive parenchymal infection
unresponsive to appropriate antibi-
otic therapy.
3. Myeloid hypoplasia.
4. A reasonable chance for recovery of
marrow function.
Patients with documented granulocyte
dysfunction, such as those with chronic
granulomatous disease, may also be candi-
dates to receive granulocyte transfusions
during life-threatening episodes of infec-
tion or while awaiting hematopoietic pro-
genitor cell transplantation.
Other Considerations
Granulocyte components contain signifi-
cant amounts of red cells, which must be
crossmatch compatible and Rh specific,
particularly for females with childbearing
potential. Granulocytes should be irradi-
ated to avoid the risk of graft-vs-host dis-
ease (GVHD). If cytomegalovirus (CMV)
transmission is an issue, its risk can be re-
duced by the use of a CMV-seronegative
donor (leukocyte reduction filters are
contraindicated). For alloimmunized re-
cipients, donors should be matched by HLA
typing or leukocyte crossmatching.52,54
Special Cellular Blood
Components
Leukocyte Reduction
The approximate leukocyte content of com-
mon blood components is summarized in
Table 21-2.55-59 Leukocyte reduction has
been used for some time for select groups
of patients. Current federal guidelines57,58
and AABB Standards for Blood Banks and
Transfusion Services59(pp25,28,29,31,32) define a
leukocyte-reduced component as one
with <5 ×106residual donor leukocytes
492 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

per final product (this includes RBCs;
Platelets Pheresis; and pooled Platelets).
AABB Standards59(p31) requires <8.3 ×105
leukocytes in Platelets Leukocytes Re-
duced, which are prepared from a single
unit of Whole Blood, to achieve the re-
quirement for a pool of 6 platelet units.
Draft guidance from the Food and Drug
Administration (FDA) recommends qual-
ity control to indicate with 95% confi-
dence that more than 95% of blood units
meet these criteria. By comparison, Euro-
pean guidelines define leukocyte-reduced
components as those with <1 ×106resid-
ual leukocytes per unit and require that
thereshouldbenomorethana10%fail
-
urerateintheprocess.
Published data demonstrate that leuko-
cyte reduction reduces the risk of:
1. Febrile nonhemolytic reactions (see
Chapter 27).
2. CMV transmission.60
3. HLA alloimmunization that may lead
to patients becoming refractory to
platelet transfusions.61
Controversial and unproven indications
for leukocyte reduction include:
1. Reduction of immunomodulation that
may lead to an increased risk of can-
cer recurrence or bacterial infections.62
2. Reductionintheriskofpriondisease.
3. ReductionintheriskofYersinia entero-
colitica contamination of RBC units.63
Many studies have investigated the pos-
sibility that leukocyte reduction can reduce
the incidence of clinical outcomes due to
transfusion-related immunomodulation,
but the results are contradictory.62 One such
proposal was that savings related to re-
duced immunomodulation could offset the
costs of leukocyte reduction, but this was
not demonstrable in a large prospective
randomized trial.64 Nonetheless, several
countries have converted to a leukocyte-re-
duced blood supply, and this subject re-
mains controversial.65
Irradiation
Irradiation of a cellular blood component
is the only accepted method to prevent
GVHD. GVHD has been reported after
transfusion of leukocyte-reduced compo-
nents.66 For more details on GVHD and in-
dications for irradiation, see Chapter 27.
Replacement of Coagulation
Factors
Physiologic Principles
Coagulation results from a complex but
ordered enzyme cascade occurring on the
surface of platelets and cells that express
tissue factor (see Fig 21-1). The coagula-
tion cascade is typically divided into the
intrinsic and the extrinsic pathways, the
Chapter 21: Blood Transfusion Practice 493
Table 21-2. Approximate Leukocyte
Content of Blood Components (per
Unit)55-59
Whole Blood 109
RBCs 108
RBCs Washed 107
RBCs Deglycerolized 106-107
RBCs Leukocytes Reduced
(by filtration)*
<5×10
6
Platelets Pheresis 106-108
Platelets 107
Platelets Pheresis
Leukocytes Reduced*
<5×10
6
Platelets Leukocytes Reduced <8.3 × 105
Platelets Pooled
Leukocytes Reduced
<5×10
6
FFP Thawed <0.6 × 106-
1.5×10
7
*Leukocyte reduction with third-generation leukocyte ad-
sorption filter.
Copyright © 2005 by the AABB. All rights reserved.
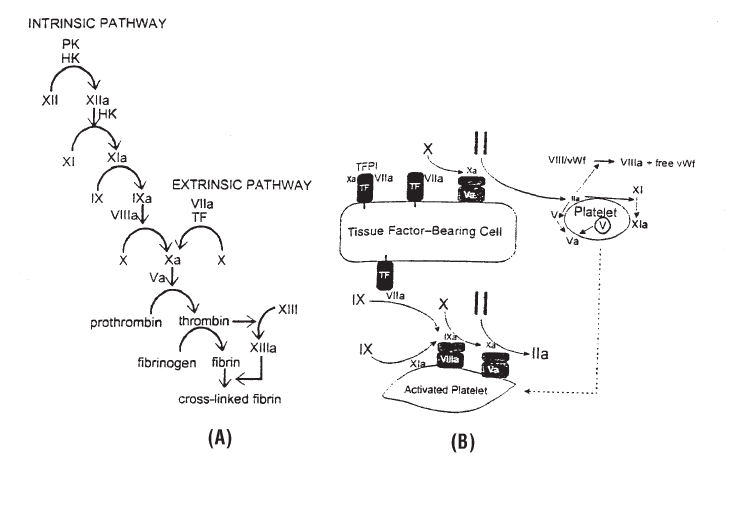
in-vitro activity of which can be measured
by the activated partial thromboplastin
time (aPTT) and prothrombin time (PT),
respectively, but, in vivo, the cascades are
interdependent.67 The central procoagu-
lant enzyme is thrombin, which is acti-
vated by both pathways.
Minimal levels of coagulation factors (see
Table 21-3) are required for normal forma-
tion of fibrin and hemostasis, so normal
plasma contains coagulation factors in ex-
cess, a reserve that usually allows patients
to tolerate replacement of one or more
blood volumes of red cells and crystalloid
without needing Fresh Frozen Plasma (FFP).
Patients with liver disease have less physio-
logic reserve and are more susceptible to
dilutional coagulopathy.
Monitoring Hemostasis
The PT, aPTT, and measurement of
fibrinogen level are commonly used to
monitor coagulation. Results should be
interpreted with four considerations in
mind: 1) mild prolongations of the PT or
aPTT occur before the residual factor con-
centration falls below the level normally
needed for hemostasis; 2) conversely, the
PT and aPTT are relatively insensitive to
low fibrinogen levels; 3) significant defi-
ciencies of coagulation factors (or the
presence of coagulation factor inhibitors)
cause clearly prolonged values for the PT
or aPTT; and 4) an infusion of FFP that in-
creases the concentration of factors by 20%
will have a far greater impact on a greatly
494 AABB Technical Manual
Figure 21-1. (A) The classic cascade model of coagulation reactions was based on in-vitro experimen-
tal data in cell-free systems. The term extrinsic reflects the fact that tissue factor does not circulate in
plasma. (B) More recent evidence emphasizes that the coagulation reactions occur on the surfaces of
tissue factor-bearing cells at the site of injury and on the surface of platelets that are subsequently re-
cruited. HK = high-molecular-weight kininogen, PK = prekallikrein, TF = tissue factor, TFPI = tissue
factor pathway inhibitor. (Adapted with permission from Roberts et al.67)
Roberts HR, Monroe DM III, Hoffman M. Molecular biology and biochemistry of the coagulation factors and pathways of
hemostasis. In: Beutler E, Lichtman MA, Coller BS, et al. Williams’ hematology. 6th ed. New York: McGraw-Hill, 2001:1409-34.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 21: Blood Transfusion Practice 495
Table 21-3. Coagulation Factors
Factor Name
In-vivo
Half-life
In-vitro, 4 C
Half-life
%ofNormal
Needed for
Hemostasis
% In-vivo
Recovery Initial Therapeutic Dose
I Fibrinogen 3-6 days Years 12-50 50-70 1 bag cryoprecipitate/7 kg body weight
II Prothrombin 2-5 days >21 days 10-25 50 10-20 units/kg body weight
V Labile factor, Proaccelerin 4.5-36 hours 10-14 days 10-30 ~80 10-20 mL plasma/kg body weight
VII Stable factor, Proconvertin 2-5 hours >21 days >10 100 10-20 units/kg body weight
VIII Antihemophilic factor 8-12 hours 7 days 30-40 60-70 See Table 21-6
IX Plasma thromboplastin
component, Christmas factor
18-24 hours >21 days 15-40 20 See Table 21-6
X Stuart-Prower factor 20-42 hours >21 days 10-40 50-95 10-20 units/kg body weight
XI Plasma thromboplastin
antecedent (PTA)
40-80 hours >28 days 20-30 90 10-20 mL/kg body weight
XIII Fibrin stabilizing factor 12 days >21 days <5 50-100 500 mL plasma every 3 weeks
AT Antithrombin 60-90 hours >42 days 80-120 50-100 40-50 IU/kg body weight
Notes:
1. All dosings are provided as a general guideline for initial therapy; the exact loading dose and maintenance intervals should be individualized for each patient.
2. One unit of coagulation factor is present in each mL of Fresh Frozen Plasma.
3. DDAVP is the treatment of choice for patients with hemophilia A who are responders.
4. Composite data from the following references:
a. Beutler E, Lichtman MA, Coller BS, Kipps TL, eds. Williams’ hematology. 5th ed. New York: McGraw-Hill, 1995:1413-58, 1657.
b. Mollison PL, Engelfriet CP, Contreras M. Blood transfusion in clinical medicine. 10th ed. Oxford: Blackwell Scientific Publications, 1997:459-88.
c. Huestis DW, Bove JR, Case J, eds. Practical blood transfusion. 4th ed. Boston, MA: Little Brown and Co, 1988:319.
d. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91-9.
e. Package inserts.
Copyright © 2005 by the AABB. All rights reserved.
prolonged PT or aPTT than on a mildly
prolonged PT or aPTT. For example, the
infusion of two units of FFP in a patient
with a PT of 14.5 seconds (normal: 11 to
13 seconds) is unlikely to provide any clin-
ical benefit and is also unlikely to correct
the PT to the normal range.
Guidelines typically cite a PT 1.5 times
the upper limit of normal3or the midpoint
of the normal range39 and an aPTT 1.5 times
the upper limit of normal3,39 as thresholds at
which therapeutic or prophylactic replace-
ment may be indicated in an appropriate
clinical setting. Of note, however, studies
have consistently shown that the PT and
aPTT, even when elevated to this degree,
have little predictive value for bleeding
complications of invasive procedures in-
cluding paracentesis or thoracentesis,25 liver
biopsy,26,27 angiography,23 or central venous
catheter placement.24
Components and Products Available for
Coagulation Factor Replacement
The plasma components that are avail-
able differ according to the timing of
freezing and/or thawing and variations in
preparation (see Chapter 8). FFP contains
all the clotting factors, including labile
Factors V and VIII. Other forms of plasma
have lower levels of labile factors but
could substitute for FFP for most of the
indications for which the latter is trans-
fused. Plasma Cryoprecipitate Reduced
has a decreased content of vWF and is
used specifically for treatment of TTP (see
Chapter 6). Pooled Plasma, Solvent/De-
tergent-Treated is no longer available in
the United States.
Cryoprecipitated AHF (CRYO) is a con-
centrate of high-molecular-weight plasma
proteins that precipitate in the cold, includ-
ing vWF, Factor VIII, fibrinogen, Factor XIII,
and fibronectin (see Chapter 8).
Plasma derivatives are concentrates of
specific plasma proteins from large pools of
plasma or cryoprecipitate. Cohn fraction-
ation, which relies on the precipitation of
various plasma proteins in cold ethanol-
water mixtures, was developed during
World War II and is still used with some
modifications.68 After fractionation, deriva-
tives undergo further processing to purify
and concentrate the proteins and inactivate
contaminating viruses. Virus-inactivation
procedures include heat treatment, micro-
filtration, the use of chemical solvents and
detergents, and affinity column purifica-
tion. Factors VIII, IX, VIIa, and anti-
thrombin are also produced by recombi-
nant DNA technology. These products
appear to be efficacious and are not known
to carry any infectious risk.
The specific activity (factor units/mg
protein) of presently available concentrates
has been dramatically increased in concen-
trates prepared with affinity columns or by
recombinant technology. Moreover, HIV,
HBV, and HCV transmission appear to be
absent in patients with hemophilia treated
exclusively with new preparations.69 Unfor-
tunately, their cost has also increased due
to the increased complexity of manufactur-
ing and the protein losses resulting from
extensive manipulation. Coagulation factor
concentrates are supplied in lyophilized
form and the factor activity is stated on the
label.69 Currently available products for re-
placement of Factors VIII and IX are listed
in Table 21-4.
Selection of ABO-Compatible Plasma
Because of its long shelf life, group-spe-
cificorcompatibleplasma(seeTable
21-5) is typically available. (Note that
platelet transfusions usually contain a
volume of plasma equivalent to one unit
of FFP, and limitations in availability may
496 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 21: Blood Transfusion Practice 497
Table 21-4. Available Coagulation Factor Concentrates
Product Type Product Approved Indications Comment
Factor VIII, fractionated Humate-P Factor VIII and vWF repl. Contains vWF
Koate-HP Factor VIII replacement Contains vWF
Koate-DVI Factor VIII replacement Contains vWF
Factor VIII, affinity puri-
fied
Alphanate Factor VIII replacement Contains vWF
Factor VIII, immunaffinity
purified
Hemofil-M Factor VIII replacement
Monarc M Factor VIII replacement
Monoclate-P Factor VIII replacement
Factor VIII, recombinant Kogenate FS
Helixate FS
Factor VIII replacement Contains trace amounts
of human albumin
Recombinate
(Bioclate)
Factor VIII replacement
ReFacto Factor VIII replacement Does not contain human
albumin
Factor VIII, porcine Hyate C Factor VIII inhibitor tx.
Factor IX, affinity
purified
AlphaNine-SD Factor IX replacement
Factor IX, immunaffinity
purified
Mononine Factor IX replacement
Factor IX, recombinant BeneFIX Factor IX replacement Does not contain human
albumin
Factor IX complex Bebulin VH Factor IX replacement Contains Factor II,
Factor X, trace
Factor VII
Konyne 80 Factor IX replacement
Factor VIII inhibitor tx.
Warfarin reversal
Contains Factor II,
Factor X, some
Factor VII
Profilnine SD Factor IX replacement Contains Factor II,
Factor X, some
Factor VII
Proplex T Factor IX and Factor VII
replacement
Factor VIII inhibitor tx.
Contains Factor II,
Factor VII, and
Factor X
Factor IX complex,
activated
Autoplex-T Factor VIII inhibitor tx. Contains Factor lIa,
Factor VIIa, and
Factor Xa
FEIBA VH Factor VIII inhibitor tx. Contains Factor IIa,
Factor VIIa, and
Factor Xa
Factor VIIa,
recombinant
NovoSeven Factor VIII inhibitor tx. Does not contain human
albumin
Copyright © 2005 by the AABB. All rights reserved.

require infusion of incompatible plasma
in this context.)
Indications for FFP
Guidelines exist for the appropriate use of
FFP.3,39,70 FFP is the only approved compo-
nent for clinically significant deficiency of
Factors II, V, X, and XI. Plasma is most of-
tenusedinpatientswithmultiplefactor
deficiencies, including those with liver
disease, dilutional and consumption
coagulopathy, or a need for rapid reversal
of warfarin treatment. It is of limited clini-
cal benefit in patients with inhibitors to
any coagulation factor. Plasma Cryopreci-
pitate Reduced may be more effective
than FFP for some patients receiving
plasma exchange treatment for TTP or
hemolytic-uremic syndrome71 (see Chap-
ter 6).
Vitamin K Deficiency and Warfarin Reversal
The most common cause of multiple co-
agulation factor abnormalities among
hospitalized patients is deficiency of the
vitamin-K-dependent factors due to treat-
ment with the vitamin K antagonist
warfarin (Coumadin) or nutritional defi-
ciency.67 Vitamin K is a fat-soluble vitamin
required for hepatocellular synthesis of
coagulation Factors II, VII, IX, and X, as
well as the anticoagulant proteins C and
S. Body stores of vitamin K last only 2
weeks, so deficiency may occur in hospi-
talized patients unable to tolerate normal
food intake. Absorption of vitamin K re-
quires precursor metabolism by bacteria
in the intestine and the action of bile salts;
therefore, deficiencies can occur with anti-
biotic use, obstructive jaundice, and fat
malabsorption syndromes.
VitaminKdepletionorwarfarinusually
cause a prolongation of the PT that is out of
proportion to the aPTT because Factor VII,
which has the shortest half-life of the vita-
min-K-dependent factors, has little effect
on the aPTT. Deficiency of vitamin K is best
managed by treatment of the underlying
condition and by administration of vitamin
K.67 If liver function is adequate, coagulation
factors will return to effective levels in
about 12 hours.
Although most patients with vitamin K
deficiency do not require plasma, plasma
transfusion is occasionally needed to treat
active bleeding or to prepare for emergency
invasive procedures.67 Transfusion of 10 to
15 mL of plasma per kg of body weight will
generally achieve hemostatic coagulation
factor levels in patients with warfarin-in-
duced coagulopathy. One form of Factor IX
complex concentrate is licensed for warfa-
rin reversal (see Table 21-4) but carries a sig-
nificant risk of thrombotic complications and
is rarely used for this indication. Concurrent
vitamin K supplementation should also be
given unless only transient correction is de-
498 AABB Technical Manual
Table 21-5. Suggested ABO Group Selection Order for Transfusion of Plasma
Recipient
ABO Group
Component ABO Group
1st Choice 2nd Choice 3rd Choice 4th Choice
AB AB (A) (B) (O)
AAAB(B)(O)
BBAB(A)(O)
OOABAB
(Blood groups in parentheses represent choices with incompatible plasma, listed in “least incompatible” order.)
Copyright © 2005 by the AABB. All rights reserved.
sired. Although the International Normalized
Ratio can provide useful information about
response to therapy, the need for additional
treatment should be guided by the clinical re-
sponse and not by the results of laboratory
tests. As discussed above, it is rarely neces-
sary to correct the PT or aPTT to normal to
achieve adequate hemostasis.
Liver Disease
Patients with liver disease have multiple
derangements that contribute to an in-
creased bleeding tendency. These abnor-
malities include portal hypertension and
engorgement of systemic collateral ve-
nous shunts; splenomegaly with second-
ary thrombocytopenia; decreased synthe-
sis of all coagulation factors except Factor
VIII; dysfibrinogenemia; decreased clear-
ance of fibrin, fibrinogen degradation
products, and fibrinolytic activators; and
decreased synthesis of inhibitors of the
fibrinolytic system. As in vitamin K defi-
ciency, the short half-life of Factor VII
causes the PT to be prolonged more than
the aPTT, and, for the same reason, FFP
infusion corrects the PT for only about 4
hours.72 Because the defect in hepato-
cellular disease is in primary protein syn-
thesis, supplemental vitamin K will not
usually correct the abnormality. However,
because it is one of the few treatable causes
of coagulopathy in liver disease, a trial of
replacement vitamin K may be indicated.
FFP corrects coagulation factor deficien-
cies found in severe liver disease, but is of-
ten used inappropriately. The most com-
mon error is to attribute all bleeding to
coagulopathy and to give systemic treat-
ment when the cause is localized bleeding.
For example, bleeding at the operative site
after cardiac surgery usually responds
better to local hemostatic measures than to
intravenous infusion of FFP. A second com-
mon error in treating liver-associated
coagulopathy is overdependence on the PT.
Again, a normal PT is rarely, if ever, re-
quired for the cessation of serious bleeding,
and the goal of FFP therapy in severe liver
disease should be to correct or prevent
bleeding complications. If FFP is to be used
prophylactically before an invasive proce-
dure, it should be given immediately before
the procedure.
Patients with liver disease may also have
abnormalities of platelet plug formation
and fibrinolysis. In addition, severe spleno-
megaly may impair the response to platelet
transfusions. Platelet function in some pa-
tients with liver disease can be enhanced by
administration of 1-deamino-8-D-arginine
vasopressin (desmopressin, DDAVP).73 Cryo-
precipitated AHF should be given if there is
severe hypofibrinogenemia or bleeding re-
lated to dysfibrinogenemia. The increase in
systemic fibrinolysis associated with severe
liver disease may not respond to FFP alone,
and antifibrinolytic agents in combination
with plasma therapy can be useful in these
patients (see below).
Dilutional Coagulopathy
Massive blood loss and replacement with
crystalloid and/or colloid solutions may
produce a dilutional coagulopathy,74 but
most patients can tolerate loss and re-
placement of at least one blood volume
without developing impaired hemo-
stasis.75 Shock accompanying traumatic
hemorrhage also contributes to the coa-
gulopathy (see Chapter 27). In the setting
of trauma, thrombocytopenia generally
develops before plasma clotting factors are
diluted to the point of causing impaired
hemostasis, and adequate platelet re-
placement generally has priority.75 However,
in elective surgical patients, coagulation
factor deficits may predominate.74 FFP may
be beneficial if the PT and/or aPTT are
greater than 1.5 times normal.3,39 If surgi-
Chapter 21: Blood Transfusion Practice 499
Copyright © 2005 by the AABB. All rights reserved.
cal hemostasis has not been achieved and
significant continued bleeding is expected,
FFP may be indicated.39
Patients undergoing plasmapheresis
without plasma replacement develop a va-
riety of coagulation factor deficits, particu-
larly hypofibrinogenemia, depending on
thevolumeandfrequencyoftheex
-
changes.76 Although these changes can be
striking, most authors have concluded that
routine supplements with FFP in patients
with normal liver function are unnecessary,
particularly for alternate-day regimens.
Other Conditions
Plasma exchange is lifesaving in TTP (see
Chapter 6). FFP may be an adjunct to treat-
ment of DIC. Hereditary angioneurotic
edema results from a congenital defi-
ciency of C1-esterase inhibitor, an inhibi-
tory protein that regulates complement
activation. Patients with this condition
develop localized edema and may experi-
ence lifethreatening obstruction of the
upper respiratory tract following comple-
ment activation. FFP or Liquid Plasma
contain normal levels of C1-esterase in-
hibitor and FFP transfusion appeared to
prevent attacks at the time of oral surgery
in one study.77 There are rare anecdotal re-
ports of exacerbation of angioneurotic
edema with FFP administration. The need
for treatment of isolated deficiency of
Factors II, V, VII, X, or XI is uncommon;
guidelines for initial therapy are given in
Table 21-3 (however, for a more complete
treatment, refer to one of the standard he-
matology or coagulation texts).
Plasma can also be used to replace pro-
teins C and S, and antithrombin; these are
discussed separately below.
Misuse of Fresh Frozen Plasma
Plasma should not be used as a volume
expander, as a nutritional source, or to
enhance wound healing.38,70,75 Transfusing
plasma for volume expansion carries a
risk of infectious disease transmission and
other transfusion reactions (eg, allergic)
that can be avoided by using crystalloid or
colloid solutions. Plasma is also not a
suitable source of immunoglobulins for
patients with severe hypogammaglobu-
linemia because more effective prepara-
tions exist (immunoglobulin for intrave-
nous or intramuscular use).
FFP is often given prophylactically to pa-
tients with mild to moderate prolongation
of the PT or aPTT before invasive proce-
dures, but there is little or no evidence that
this prevents bleeding complications. Be-
cause these tests do not accurately predict
the risk of bleeding when mildly prolonged,
there is little logic for a transfusion in-
tended to “improve” the results.
Cryoprecipitated AHF
Transfusion
CRYO is the only concentrated fibrinogen
product currently available for systemic
use, and intravenous supplementation of
fibrinogen is its primary clinical use, par-
ticularly in DIC. A second major use has
been in patients with severe von Wille-
brand disease, but there are Factor VIII
concentrates that contain vWF and are
more appropriate if available (see Table
21-4). CRYO can be used in isolated Fac-
tor XIII deficiency and to ameliorate the
platelet dysfunction associated with ure-
mia. It is also used topically as a fibrin
sealant, although a commercial prepara-
tion is available. CRYO is seldom used for
patients with hemophilia because Factor
VIII concentrates are available commer-
cially and have been processed to reduce
or eliminate the risk of blood-borne viral
infection; CRYO is used as a last resort for
this indication. Because CRYO contains
500 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
ABO antibodies, consideration should be
given to ABO compatibility when the in-
fused volume will be large relative to the
recipient’s red cell mass.
Calculating the CRYO Dose for Fibrinogen
Replacement
On average, one unit of CRYO contains
approximately 250 mg of fibrinogen; the
minimum required by AABB Standards is
150 mg.59(p30) The amount of transfused
CRYO required to correct the fibrinogen
level depends upon the nature of the
bleeding episode and the severity of the
initial deficiency and can be calculated as
follows:
1. Weight (kg) ×70 mL/kg = blood vol-
ume (mL).
2. Blood volume (mL) ×(1.0–hemato-
crit) = plasma volume (mL).
3. Mg of fibrinogen required = (Desired
fibrinogen level in mg/dL – initial
fibrinogen level in mg/dL) ×plasma
volume (mL) ÷ 100 mL/dL.
4. Bags of CRYO required = mg of fibrino-
gen required ÷ 250 mg fibrinogen/
bag of CRYO
This calculation assumes that 100% of
administered fibrinogen is recovered as
measurable intravascular fibrinogen, but,
because the content of CRYO is variable,
further refinements are unproductive.
von Willebrand Syndromes
von Willebrand syndromes are the most
common major inherited coagulation ab-
normalities.78 These conditions are usu-
ally autosomal dominant and represent a
collection of quantitative and qualitative
abnormalities of vWF, the most important
protein mediating platelet adhesion to
damaged endothelial surfaces. The pro-
tein also transports Factor VIII. As a re-
sult, patients with von Willebrand syn-
dromes have varying degrees of abnormal
platelet plug formation and partial defi-
ciency of Factor VIII. The former may
manifest as a prolonged bleeding time
and the latter as a prolonged aPTT, but
these abnormalities vary between syn-
dromes. vWF exists in the plasma as a
family of multimeric molecules with a
wide range of molecular weights. The
high-molecular-weight species of vWF are
the most hemostatically effective. Labora-
tory evaluation demonstrates a specific de-
ficiency in the level of vWF, often mea-
sured as ristocetin cofactor activity because
vWF is required for the platelet-agglutinat-
ing effect of ristocetin in vitro.
Mild cases of von Willebrand syndrome
can often be treated with DDAVP, which
causes a release of endogenous stores of
Factor VIII and vWF. Many Factor VIII con-
centrates do not contain therapeutic levels
of vWF, but several with satisfactory levels
are commercially available and one is li-
censed for this indication (see Table 21-4).
In the absence of a suitably therapeutic vi-
rus-inactivated concentrate, severe von
Willebrand syndrome can be treated with
CRYO. The quantity of CRYO required to
treat bleeding episodes or to prepare for
major surgery varies greatly among patients
with von Willebrand syndromes. In addi-
tion to the clinical response of the patient,
the template bleeding time, the level of Fac-
tor VIII, or the ristocetin cofactor activity
may help to guide therapy.
Fibrinogen Abnormalities
Hypofibrinogenemia may occur as a rare
isolated congenital deficiency, may be ac-
quired as part of the DIC syndrome, or may
be due to obstetric complications such as
abruptio placentae. Dysfibrinogenemias
may be congenital or acquired and repre-
sent conditions in which fibrinogen is
present as measured by immunoassays
but functionally defective as measured by
Chapter 21: Blood Transfusion Practice 501
Copyright © 2005 by the AABB. All rights reserved.
the thrombin time. Patients with severe
liver disease frequently exhibit a dys-
fibrinogenemia.
Disseminated Intravascular Coagulation
DIC occurs when circulating thrombin in-
duces widespread fibrin formation in the
microcirculation and consumption of pla-
telets and coagulation factors, particularly
fibrinogen,prothrombin,FactorV,and
Factor VIII. Fibrin strands in the micro-
circulation may cause mechanical damage
to red cells, a condition called microangio-
pathic hemolysis, manifest as schistocytes
(fragmented red cells) in the circulation.
Widespread microvascular thrombi pro-
mote tissue ischemia and release of tissue
factor, which further activates thrombin.
Lysis of microvascular fibrin causes in-
creased quantities of fibrin degradation
products to enter the bloodstream.
Several clinical conditions can initiate
DIC, including shock, tissue ischemia, sep-
sis, hemolytic transfusion reactions, dis-
seminated cancer (particularly adeno-
carcinoma), acute promyelocytic leukemia,
tumor lysis syndrome, and obstetric com-
plications such as amniotic fluid embolism.
The common precipitating event is a pro-
coagulant signal for thrombin production that
exceeds the normal physiologic defenses
against disseminated thrombin activity.
Treatment of DIC depends on correcting
the underlying problem and preventing
further hypotension and tissue ischemia.
Replacement therapy focuses on the build-
ing blocks of the thrombus (platelets and
fibrinogen), and secondarily on other coag-
ulation factors, including Factors VIII, XIII,
V, and II. Thus, in bleeding patients, platelet
transfusion is indicated when the platelet
count falls below 50,000/µL, and cryopre-
cipitate is supplemented if the fibrinogen
level is below 100 mg/dL. FFP is indicated
in the setting of hemorrhage that results
from DIC once the fibrinogen is above 100
mg/dL.
Topical Use
The fibrinogen in CRYO has been used
during surgery as a topical hemostatic
preparation, so-called fibrin sealant or fi-
brin glue, made from one or two units of
CRYO, which may be of autologous origin.
The fibrinogen is converted to fibrin by
the action of bovine thrombin at the site
that is bleeding or to be “glued.” Commer-
cial preparations of fibrin sealant are
available that have a higher fibrinogen
concentration than that of CRYO and in-
clude human thrombin and bovine apro-
tinin (see below) to decrease the lysis of
the resulting fibrin. These pooled, virus-
inactivated products have been licensed
for the reduction of bleeding in cardiovas-
cular surgery,79 for repairing splenic
trauma, and for colostomy closure. Fibrin
sealants have also been used for a variety
of indications in which it is desired to
bind two tissue surfaces together, includ-
ingrepairoftheduramateroreardrum.
Useofbovinethrombincanstimulate
the formation of antibodies against thrombin
and other contaminant proteins including
Factor V.80 These antibodies can cross-react
with human thrombin and Factor V, caus-
ing abnormal clotting times and, in some
cases, bleeding. For this reason, it has been
suggested that use of “homemade” fibrin
sealants be replaced by use of the commer-
cial product.80
Hemophilia A
Each unit of CRYO prepared from a single
blood donation should contain a minimum
of 80 international units (IU) of Factor
VIII.59(p30) Although no longer the compo-
nent of choice, CRYO can serve as replace-
502 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
ment therapy for patients with hemo-
philia A if virus-inactivated Factor VIII
concentrates are unavailable.39 If CRYO is
used, the amount required to provide a
therapeutic dose of Factor VIII is based on
calculations similar to those used for AHF.
Hemophilia A is a sex-linked recessive
trait (ie, affected males, carrier females)
causing deficiency of Factor VIII (anti-
hemophilic factor, AHF).81 The responsible
genes usually produce a protein with re-
duced activity, so immunologic measure-
ment of Factor VIII antigen gives normal re-
sults despite deficient Factor VIII coagulant
activity. In contrast, antigen is typically de-
pressed in von Willebrand disease. Charac-
teristic laboratory findings include a pro-
longed aPTT, normal PT and template
bleeding time, and decreased Factor VIII
activity.
The severity of hemophilia A depends on
the patient’s level of Factor VIII activity, and
this varies; patients with severe hemophilia
have Factor VIII levels below 1%, whereas
those with moderate disease typically have
1% to 5% activity, and severity is mild with
levels of 6% to 30%.81 One unit of Factor VIII
activity is defined as the Factor VIII content
of 1 mL of fresh, citrated, pooled normal
plasma. The measured level of Factor VIII
can be expressed as a concentration, as
percent activity, or as a decimal fraction.
For example, a patient with mild hemo-
philia with one-tenth the normal activity of
Factor VIII can be said to have a Factor VIII
level of 10 units/dL or 0.1 unit/mL or 10%
activity.
Patients with mild-to-moderate hemo-
philia can often be managed without re-
placement therapy.81 Careful attention to lo-
cal hemostasis and the use of topical
antifibrinolytics may prevent the need for
further replacement. Systemic levels of Fac-
torVIIIcanberaisedinmildhemophilia
with the use of DDAVP, which stimulates
the release of endogenous Factor VIII from
storage sites. However, DDAVP is ineffective
in patients with severe hemophilia A; in
such cases, Factor VIII replacement is re-
quired. The amount of Factor VIII infused
depends upon whether therapy is intended
to prevent bleeding or, if bleeding has oc-
curred, the nature of the bleeding episode
and the severity of the initial deficiency (see
Table 21-6). For example, treatment for
hemarthrosis ordinarily requires more Fac-
tor VIII than epistaxis.
Calculating the Dose of Factor VIII
When the desired result is determined
(see Table 21-6), the amount of Factor VIII
required for transfusion can be calculated
by one of the following formulas:
Factor VIII dose (IU/kg) =
Desired factor increase (%) ×0.5 (1
IU/kg typically raises the Factor VIII
level by 2%)
Total dose =
(Patient mass ×70mL/kg) ×(1 – Hct) ×
(Desired activity – current activity)
Example: A 70-kg patient with severe he-
mophilia has an initial Factor VIII level of
2% (0.02 unit/mL) and a hematocrit of 40%.
How many units of Factor VIII concentrate
should be given to raise his Factor VIII level
to 50% (0.5 unit/mL)?
(70 ×70) ×(1 – 0.4) ×(0.5 – 0.02) =
1411 units
The therapy of choice for severe hemo-
philia A is a Factor VIII concentrate (see Ta-
ble 21-4). CRYO could be used to supply
1411 units of Factor VIII, but, at 80 IU per
bag, this would require at least 18 bags (and
18 allogeneic donor exposures). The half-
life of Factor VIII is about 12 hours, so infu-
sions are repeated at 8- to 12-hour intervals
Chapter 21: Blood Transfusion Practice 503
Copyright © 2005 by the AABB. All rights reserved.

to maintain hemostatic levels. The duration
of treatment with Factor VIII infusions de-
pends upon the type and location of the
hemorrhage or the reason for prophylaxis,
and the clinical response of the patient (see
Table 21-6). After major surgery, the Factor
VIII level should be maintained above 40%
to 50% for at least 10 days. When elective
surgery is planned, Factor VIII assays
should be made available to serve as a
guide to therapy.
Treatment of Inhibitors of Factor VIII
About 10% to 35% of patients with hemo-
philia A, typically, those with severe dis-
ease or genetic defects involving large
portions of the molecule, develop a de-
tectable inhibitor to human Factor VIII.68,81
These antibodies are directed against the
active site of Factor VIII, rendering the pa-
tient relatively unresponsive to infusion.
Patients having an elective invasive pro-
cedure should be screened for such inhi-
bitors. Management is difficult; ap-
proaches have included attempts to
overwhelm the inhibitor with very large
doses of human Factor VIII; use of por-
cine Factor VIII, which has low cross-reac-
tivity with human Factor VIII antibody81;
use of Factor VIII bypassing agents in-
504 AABB Technical Manual
Table 21-6. General Factor Replacement Guidelines for the Treatment of Bleeding
in Hemophilia
Indication
Initial
Minimum
Desired
Factor Level
(%)
Factor VIII
Dose*
(IU/kg)
Factor IX
Dose*
(IU/kg)
Duration
(days)
Severe epistaxis, oral
mucosal bleeding†
20-30 10-15 20-30 1-2
Hemarthrosis, hematoma,
persistent hematuria,‡
gastrointestinal tract
bleeding, retroperitoneal
bleeding
30-50 15-25 30-50 1-3
Trauma without signs of
bleeding, tongue/
retropharyngeal
bleeding†
40-50 20-25 40-50 2-4
Trauma with bleeding,
surgery,§intracranial
bleeding§
100 50 100 10-14
Data from USP.69
*Dosing intervals are based on a half-life for Factor VIII of 8 to 12 hours (2 to 3 doses/day) and a half-life for Factor IX
of 18 to 24 hours (1 to 2 doses/day). Maintenance doses of one half the initial dose may be given at these intervals. The
frequency depends on the severity of bleeding, with more frequent dosing for serious bleeding.
†In addition to antifibrinolytics.
‡Painless spontaneous hematuria usually requires no treatment. Increased oral or intravenous fluids are necessary to
maintain renal output.
§Continuous factor infusion may be administered. Following the initial loading dose, a continuous infusion at a dose of 3
IU/kg per hour is given. Subsequent doses are adjusted according to the plasma factor levels.
Copyright © 2005 by the AABB. All rights reserved.
cluding Factor IX complex, activated Fac-
tor IX complex, and activated Factor VII
(see Table 21-4); and desensitization ther-
apy. The latter includes large daily doses
of AHF in conjunction with cortico-
steroids or Immunoglobulin Intravenous
(IGIV) and cyclophosphamide. Success rates
of 50% to 80% are reported. If hemorrhage
is life-threatening, intensive plasma-
pheresis to remove the inhibiting anti-
body, coupled with immunosuppression,
as well as infusions of Factor VIII and pos-
sibly antifibrinolytic therapy (see below),
can be employed.
Hemophilia B
Factor IX deficiency (hemophilia B, Christ-
mas disease) is clinically indistinguish-
able from Factor VIII deficiency in that
both are sex-linked disorders that cause a
prolonged aPTT in the presence of a nor-
mal PT and bleeding time.81 The disorder
is confirmed by specific measurement of
Factor IX activity. Factor IX complex con-
centrate has been used for treatment of
hemophilia B for the past 2 decades, but
the new, more pure forms of Factor IX
concentrate (see Table 21-4) are preferred
because they carry much less risk of in-
ducing thrombosis.82
The formula for calculating of Factor IX
dosage is similar to that for Factor VIII, but
the units to be given should be doubled be-
causeonlyhalfoftheinfusedFactorIX
dose is recovered in the vascular space. The
biologic half-life is 18 to 24 hours, so doses
are given 1 or 2 times/day. As with hemo-
philia A, recommended dose and treatment
schedules vary with the severity and type of
bleeding (see Table 21-6).
Immunoglobulin, Intravenous
IGIV is prepared from modified Cohn frac-
tion II and subjected to virus inactivation.
Preparations intended for intramuscular
administration contain aggregates that
may activate the complement and kinin
systems and produce hypotensive and/or
anaphylactoid reactions if administered
intravenously, but the intravenous prod-
uct contains almost exclusively mono-
meric IgG molecules.83
The indications for the use of IGIV are
evolving.83,84 Some conditions in which IGIV
is used are listed in Table 21-7. Infusion of
IGIV can induce such reactions as head-
ache, vomiting, volume overload, allergic
reactions, renal failure, and pulmonary re-
actions, but they can usually be prevented
by infusing slowly and pretreating with
diphenhydramine and/or hydrocortisone.
Passively transferred blood group allo- and
autoantibodies and/or therapy-induced
hypergammaglobulinemia may cause a
positive DAT result in recipients, but signif-
icant hemolysis is rarely noted.
Antiprotease Concentrates
Antithrombin, also known as heparin co-
factor, is a serine protease inhibitor syn-
thesized in the liver.85 It circulates in nor-
mal plasma at a concentration of 15 mg/
dL but is typically measured as % activity,
with a normal range of 84% to 116%. Anti-
thrombin inactivates serine proteases in-
cluding thrombin, and Factors IXa, Xa,
XIa, and XIIa by covalently bonding at the
serine site, followed by a conformation
change.86 This activity is greatly acceler-
ated by heparin, which induces a confor-
mation change in antithrombin and helps
approximate thrombin and antithrombin
as well.
Patients who are deficient in anti-
thrombin are prone to thromboembolic
complications.87 Such deficiency can be
congenital or acquired. Acquired deficiency
occurs in a wide variety of disease states,
including decreased synthesis due to liver
Chapter 21: Blood Transfusion Practice 505
Copyright © 2005 by the AABB. All rights reserved.

disease or malnutrition; losses due to
nephroticsyndromeandgastrointestinal
states; accelerated consumption as in DIC,
surgery or trauma, and preeclampsia; and
associated with pharmacotherapy includ-
ing heparin, L-asparaginase, and oral
contraceptives.
Antithrombin is stable in plasma so defi-
ciency can be treated with FFP or with Liq-
uid or Thawed Plasma. A heat-treated con-
centrate of antithrombin is also available;
recombinant and transgenic sources are
under investigation.
Clinical uses of antithrombin have re-
cently been reviewed.87 Antithrombin is ap-
proved for use in hereditary antithrombin
deficiency as part of the treatment for
thromboembolic episodes and for prophy-
lactic use in perioperative, postoperative,
and peripartum settings. Several off-label
uses of antithrombin exist, eg, patients with
low antithrombin levels and DIC, neonates
born to mothers with antithrombin defi-
ciency, and liver transplant recipients.
The half-life of purified antithrombin is
long,approximately60to90hours,
88 but is
abbreviated when replacement is for con-
sumptive states. The dose is calculated on
the basis of an expected increment of 1.4%
per U per kg, with an initial target of 120%.
Other available concentrates of antipro-
teases include alpha-1-proteinase inhibitor
(alpha-1-antitrypsin). C1-esterase inhibitor
is not available in the United States.
506 AABB Technical Manual
Table 21-7. Potential Indications and Clinical Uses for Intravenous Immunoglobulin
Preparations
Congenital immune deficiencies
Hypogammaglobulinemia and agammaglobulinemia
Selective antibody deficiency
IgG subclass deficiency and recurrent infection
Premature newborns
Acquired antibody deficiency
Malignancies with antibody deficiency and recurrent infection: multiple myeloma,
chronic lymphocytic leukemia
Protein-losing enteropathy
Drug- or radiation-induced humoral immunodeficiency
Prophylaxis or treatment of bacterial and viral diseases
Pediatric HIV infection for prevention of bacterial and secondary viral infections
Cytomegalovirus infection in transplant recipients
Neonatal sepsis
Other
HIV-related immune thrombocytopenic purpura
Immune cytopenias (ITP, NAIT, PTP, WAIHA)
Kawasaki syndrome
Guillain-Barré syndrome and chronic inflammatory demyelinating neuropathy
Acquired Factor VIII inhibitors
Myasthenia gravis
Multiple sclerosis
Copyright © 2005 by the AABB. All rights reserved.
Protein C and Protein S
Protein C and protein S are vitamin-K-de-
pendent proteins with anticoagulant ef-
fects.86 Protein S is a cofactor for activated
protein C, which, in turn, inactivates Fac-
tor Va and Factor VIIIa. Patients with defi-
ciencies of protein C or protein S have a
predisposition to thrombotic complications
and are often treated with anticoagulants.85
Warfarin treatment, however, can cause
these vitamin-K-dependent proteins to
decrease to dangerously low levels, lead-
ing to skin necrosis and exacerbating
thrombosis. Transfusion of FFP can serve
as an immediate source of supplemental
protein C or protein S for patients with se-
vere deficiencies, and a human protein C
concentrate is under development.
Patients with heterozygous protein C de-
ficiency have plasma levels 40% to 60% of
normal and characteristically have minimal
symptoms, rarely requiring treatment with
protein C supplementation.85 If treatment is
needed for a thrombotic episode, anticoag-
ulants suffice. Homozygous protein C defi-
ciency causes neonatal purpura fulminans,
which requires immediate administration of
protein C, along with complex regulation of
the rest of the coagulation cascade. The
half-life of infused protein C is 6 to 16 hours.
Colloid Solutions
Human albumin (5% and 25%) and plasma
protein fraction (PPF) provide volume ex-
pansion and colloid replacement without
risk of transfusion-transmitted viruses.89
PPF has a greater concentration of non-
albumin plasma proteins than 5% albumin.
Pharmacologic agents such as hydroxyethyl
starch or dextran are also commonly used
for volume expansion.
Physiology of Albumin
The total body albumin mass is about 300
g, of which 40% (120 g) is in the plasma.
Daily albumin synthesis in a normal adult
approximates 16 g. Albumin has complex
roles in normal physiology and disease in
addition to its obvious one of mainte-
nance of intravascular volume.90 Hypo-
albuminemia resulting from decreased
synthesis, increased catabolism or losses,
and shifts between different fluid com-
partmentsiscommoninacuteandchronic
illness.
Replacement
Albumin solutions are effective volume
expanders, with the promise of better
intravascular fluid retention than simpler
and less expensive crystalloids (eg, nor-
mal saline or lactated Ringer’s solution).
Because hypoalbuminemia is involved in
the pathogenesis of many disease states
and may correlate with their prognosis, it
has been tempting to try to alter their
course by exogenous supplementation,
particularly in view of the perceived low
risk status of albumin solutions. However,
this low-risk is in question.
Indications for albumin approved by a
consensus panel91 include:
1. Volume replacement in nonhemor-
rhagic shock unresponsive to crystal-
loid or in the presence of capillary
leak syndromes.
2. Volume replacement after the first
day in patients with extensive burns
(>50%) unresponsive to crystalloid.
3. Replacement after removal of large
volumes (>4 L) of ascitic fluid in pa-
tients unresponsive to crystalloid.
4. Replacement of ascitic fluid or post-
operative treatment of ascites and
peripheral edema in hypoalbumine-
mic liver transplant recipients.
5. Replacement during large-volume
plasma exchange.
6. Volume replacement in patients with
severe necrotizing pancreatitis.
Chapter 21: Blood Transfusion Practice 507
Copyright © 2005 by the AABB. All rights reserved.
7. Diarrhea (>2 L/day) in hypoalbum-
inemic (<2.0 g/dL) patients on enteral
feedings, unresponsive to short chain
peptide supplementation.
Nonalbumin colloid solutions were con-
sidered less expensive first alternatives in
several of these situations.
In spite of the conceptual attractiveness,
the long history of albumin replacement,
and the consensus on its use, a number of
prospective randomized trials have sug-
gested that albumin use was ineffective or
increased mortality. A meta-analysis of 30
randomized trials including 1419 patients
grouped according to indication (namely,
hypovolemia, burns, and hypoproteinemia)
demonstrated higher mortality with albu-
min treatment for each of the groups.92 In
response to this outcome, the authors of the
meta-analysis called for an immediate re-
view of albumin use in critically ill patients.
Special Transfusion
Situations
Thalassemia
Thalassemia and sickle cell disease are in-
herited syndromes characterized by defi-
cient or abnormal hemoglobin structures
and anemia. Thalassemia is caused by a
deficiency in alpha or beta chain produc-
tion that ranges from mild to severe. Total
absence of synthesis of one of the alpha
chains is lethal in utero; absence of beta
chain synthesis (thalassemia major) re-
sults in a progressive anemia in the new-
born period. In an attempt to compensate
for significant degrees of anemia, hemato-
poietic tissue expands, causing character-
istic bone abnormalities and enlargement
of the liver and spleen. Tissue iron accu-
mulates as a result of increased adsorp-
tion and transfusion (see Chapter 27). The
only current cure for thalassemia is hemato-
poietic transplantation, but because the
anemia can be controlled with red cell
transfusions and concurrent iron chela-
tion therapy, the use of this expensive and
potentially hazardous therapy is contro-
versial. Transfusion of thalassemia pa-
tients is discussed in Chapter 24.
Sickle Cell Disease
Sickle cell disease (SCD) results from a
single base substitution in the gene for
the beta chain of hemoglobin. The hemo-
globin of individuals homozygous for this
abnormality can irreversibly polymerize
and cause red cells to deform or “sickle.”
Such cells may initiate blockage of the
microvasculature directly or in associa-
tion with endothelial damage and throm-
bosis. They also have a decreased life
span, so SCD patients have a variably
compensated hemolytic anemia. Sickling,
which can be triggered by fever, infection,
or hypoxia, can lead to a variety of com-
plications or “crises,” including pain crisis
due to musculoskeletal or other tissue
ischemia, splenic or pulmonary seques-
tration crisis (chest syndrome), aplastic
crisis due to transient marrow suppression
by viruses (particularly parvoviruses), leg
ulcers, priapism, tissue infarction, and
stroke.
Most patients with sickle cell disease are
asymptomatic most of the time and do not
require routine transfusion. Although
sickling can be prevented or reversed by
maintaining the level of normal hemoglo-
bin above 50% to 70%, the risks from allo-
immunization, iron overload, and disease
transmission outweigh the benefits of pro-
phylactic transfusion in most patients.
Moreover, uncomplicated pain crises do not
respond well to simple transfusion. Simple
transfusion is indicated for symptomatic
anemia, aplastic crises, and blood loss.
Sometimes, patients with a history of stroke
508 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
or pulmonary or cardiac disease are some-
times treated with a hypertransfusion proto-
col or chronic red cell exchange program to
maintain their hematocrit at 25% to 30%
and the proportion of hemoglobin S below
about 30%. Care should be taken to avoid
raising the hematocrit above 35% because
of the risks of hyperviscosity. Red cell ex-
change is used to manage and/or prevent
life- or organ-threatening complications,
particularly stroke and pulmonary crisis.
Red cell exchange has also been used to
prepare patients for surgery, but a random-
ized controlled trial did not support this
measureoversimpletransfusiontoahe
-
moglobin of 10 g/dL.93
The clinical management of sickle cell
disease is complex and the reader is re-
ferred to recent reviews for more details.94,95
Patients with thalassemia and sickle cell
disease can receive standard red cell com-
ponents. However, leukocyte reduction is
generally offered to avoid febrile, non-
hemolytic transfusion reactions. Pheno-
typing the patient’s red cells and providing
antigen-matched units for transfusion
helps reduce alloimmunization to red cell
antigens and delayed hemolytic transfusion
reactions, although the cost and logistics of
such a program may be impractical for
many institutions. A frequent compromise
is to match for Rh system and K antigens.
Patients with SCD should be given hemo-
globin-S-negative RBC units. For more
details, see Chapter 24.
Transfusing Known Incompatible Blood
Clinicians must occasionally transfuse a
patient for whom no serologically com-
patible RBC units are available. This most
often occurs in patients with autoanti-
bodies, which typically react with all red
cells; however, once alloantibodies are
ruled out, the transfused cells are ex-
pected to survive as long as autologous
cells. Other situations in which all units
appear incompatible include the presence
of alloantibodies to high-incidence anti-
gens and multiple antibody specificities.
If serologic testing fails to resolve the
problem, or if the problem is identified but
time is not sufficient for acquisition of
compatible units, the physician must weigh
the risks and benefits of transfusion and
consider what alternative therapies are
suitable. If the need is sufficiently urgent,
incompatible red cells of the patient’s ABO
and Rh type may have to be given. Depend-
ing on the alloantibody, incompatible
transfusion does not always result in imme-
diatehemolysis,andtheincompatiblecells
mayremaininthecirculationlongenough
to provide therapeutic benefit.96
If time permits and if equipment is avail-
able, the survival of a radiolabeled aliquot
of the incompatible cells can be deter-
mined. Alternatively, an “in-vivo cross-
match” can be performed by cautiously
transfusing 25 to 50 mL of the incompatible
cells, watching the patient’s clinical re-
sponse, and checking a 30-minute post-
transfusion specimen for hemoglobin-
tinged serum. Such assessment does not
guarantee normal survival, but it can indi-
cate whether an acute reaction will occur. If
no adverse symptoms or hemolysis are ob-
served, the remainder of the unit can be
transfused slowly with careful clinical mon-
itoring. If the transfusion need is life-
threatening, RBC units may sometimes be
given without special testing, but clinical
staff should be prepared to treat any
reaction that may result.
Transfusing Patients with Autoimmune
Hemolytic Anemia
Because of the serologic difficulties that
accompany autoimmune hemolytic ane-
mia and the expected short red cell sur-
vival, a conservative approach to trans-
Chapter 21: Blood Transfusion Practice 509
Copyright © 2005 by the AABB. All rights reserved.
fusion is recommended. The presence of
underlying alloantibodies should be in-
vestigated before beginning transfusions,
time permitting. It is very helpful to es-
tablish the patient’s phenotype before
transfusion in order to simplify subse-
quent investigation for the presence of
possible alloantibodies. Chapter 20 con-
tains a more complete discussion.
Massive Transfusion
Massive transfusion is defined as replace-
ment approximating or exceeding the pa-
tient’s blood volume within a 24-hour in-
terval. The most important factor in
supporting tissue oxygenation is mainte-
nance of adequate blood flow and blood
pressure by infusing a sufficient volume
of crystalloid or blood components to
correct or prevent hypovolemic shock.
Emergency Issue
The transfusion service should establish a
standard operating procedure for emer-
gency provision of blood. Immunohema-
tologic testing is relatively time-consum-
ing, so it may have to be abbreviated in
trauma cases or other hemorrhagic emer-
gencies.BecauseABO-compatiblecom
-
ponents are entirely compatible in the
vast majority of cases, particularly if the
patient has never been transfused before,
group O RBCs are often used for emer-
gency transfusion before completion of
any compatibility tests. In this situation,
Rh-negativeRBCsshouldbeusedforfe
-
males of childbearing potential because
of the concern for immunizing such indi-
viduals and possibly causing hemolytic
disease of the newborn in the future. For
women beyond their childbearing years or
men, only the current presence of anti-D
is a concern. Because this antibody is no
more common than certain other blood
group alloantibodies, Rh-positive RBCs can
be used with similar safety.
The use of so-called “universal donor”
RBCs, as discussed above, has several draw-
backs. Group O RBCs are typically in short
supply because of demographic differences
between the donor and recipient popula-
tions in the United States, and such univer-
sal donor practices accentuate this prob-
lem. ABO typing can be performed very
quickly, and, in most emergencies, there is
time for ABO typing of the recipient and
provision of ABO-specific components.
Second, transfusion of large quantities of
group O RBCs before a recipient blood
sample is obtained may obscure subse-
quent immunohematologic testing. There-
fore, even if universal donor RBCs are to be
used, a blood sample should be obtained
before transfusion; this is possible in all but
the most dire cases of exsanguinating hem-
orrhage. Finally, unexpected blood group
antibodies can cause fatal transfusion reac-
tions, particularly in multitransfused pa-
tients such as those with SCD or liver dis-
ease who present to an emergency room
with severe anemia or bleeding. In such
situations, a call to another hospital trans-
fusion service at which the patient was pre-
viously transfused can be life-saving. Indi-
viduals caring for such patients must
understand the above principle concerning
the priority of volume deficits over anemia.
Emergency situations should not be
construed as justification for exceptions to
strict identification of recipient blood sam-
ples. On the contrary, increased attention to
patient identification is warranted, and one
indication for use of universal donor RBCs is
in situations when multiple trauma victims
arrive concurrently, when there is a high
risk of recipient misidentification.
AABB Standards requires physicians who
order transfusion before completion of
standard compatibility tests to document the
need by signing some type of “emergency
510 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
release form.”59(p46) This documentation should
be required only after the emergency is
over, typically within 24 hours, and signa-
tures on such forms should not be con-
strued as a release of the transfusion ser-
vice’s responsibility. The transfusion service
must insist on strict identification of sam-
ples, documentation of unit disposition,
and documentation of the emergency sta-
tus of the transfusion. However, it also has
the responsibility to perform testing on a
STAT basis, provide consultation, and avoid
unnecessarily restrictive practices.
Changing Blood Types
The transfusion service should establish
guidelines for switching blood types dur-
ing massive transfusion. An alternative to
ABO-identical RBCs is the use of ABO-
compatible units (see Table 21-1). The age
and sex of the patient are important con-
siderations. For example, when transfus-
ingayounggroupB,Rh-negativewoman,
it is preferable to switch to group O, Rh-
negative RBCs before switching to group
B, Rh-positive cells. The clinical situation
should be evaluated by the transfusion
service’s physician. If the continuing
transfusion requirement is expected to
exceed the available supply of Rh-nega-
tive blood, evaluation of the change to
Rh-positive blood should be made early,
to conserve blood for other recipients. Once
the patient receives one or more Rh-posi-
tive units, there may be little advantage in
returning to Rh-negative blood.
Coagulation Support During Massive
Transfusion
Massive transfusion is often associated with
coagulation abnormalities that may man-
ifest as microvascular bleeding (MVB) in
theformofoozingfrommultipleIVsites,
failure of blood shed into body cavities to
clot, and bleeding from tissue surfaces on
which hemostasis was previously obtained.
These situations have been attributed to
the dilution of platelets or coagulation
factors, but consumptive coagulopathy
also plays a role (see Chapter 27). Inade-
quate volume resuscitation and poor tis-
sue perfusion not only promote the re-
lease of tissue procoagulant material
leading to DIC but also result in lactic aci-
dosis, acidemia, and poor myocardial
performance. If MVB occurs, the results of
platelet counts, fibrinogen level, PT, and
aPTT ideally should guide the need for
hemostatic components. Empiric therapy
with platelets and/or plasma may be initi-
ated immediately after specimens are ob-
tained. Additional tests may be indicated
to evaluate the possibility of DIC. In this
situation, a platelet count less than
50,000/µL and a fibrinogen level less than
100 mg/dL are better predictors of hem-
orrhage than the PT and aPTT.97 PT results
below 1.5 times normal are usually associ-
ated with adequate hemostasis during
surgery.3In most adult patients, these lev-
els are encountered only after transfusion
of 15 to 20 RBC units (1.5 to 2 red cell vol-
umes). FFP or platelets should not be ad-
ministered in a fixed ratio to the number
of RBC units given.
Hypothermia, Tissue Oxygenation, and
2,3-DPG
Hypothermia as a complication of trans-
fusion is discussed in Chapter 27. In hypo-
volemic shock, the underlying patho-
physiologic defect is inadequate tissue
oxygenation. Oxygen supply to the tissues
is determined by many factors, the most
important of which are blood flow (perfu-
sion) and hemoglobin concentration. The
level of 2,3-DPG decreases in stored RBCs,
and this decrease has been suggested as a
potential cause of poor tissue oxygena-
tion after massive transfusion. Low 2,3-DPG
Chapter 21: Blood Transfusion Practice 511
Copyright © 2005 by the AABB. All rights reserved.
levels have not been shown to be detri-
mental to massively transfused patients,
although for infants undergoing exchange
transfusion, blood with near-normal
2,3-DPG levels is frequently requested.
Within 3 to 8 hours after transfusion, pre-
viously stored red cells regenerate 50% of
normal 2,3-DPG levels.98
Pharmacologic Alternatives
to Transfusion
Concern over the risks and limitations of
transfusion has led to examination of
pharmacologic alternatives. Such alterna-
tives might: 1) stimulate increased pro-
duction or release of blood elements that
otherwise would require replacement (eg,
erythropoietin); 2) substitute for a blood
component (eg, colloid solutions or oxy-
gen-carrying solutions including chemi-
cally modified hemoglobins); or 3) alter
physiologic mechanisms to reduce the
need for replacement (eg, fibrinolytic in-
hibitors).99
Recombinant Growth Factors
Growth factors are low-molecular-weight
protein hormones that regulate hemato-
poiesis by specific interaction with recep-
tors found on progenitor cells. The use of
growth factors to stimulate endogenous
blood cell production is an important al-
ternativetotheuseofblood.
100
Erythropoietin
Recombinant erythropoietin (EPO) is a
growth factor that stimulates RBC pro-
duction.100 It has been approved for pre-
surgical administration to increase preop-
erative hemoglobin and hematocrit levels;
typical dose regimens range from 300 to
600 U/kg by subcutaneous injection weekly.
The use of EPO has markedly reduced the
need for transfusion in patients with end-
stage renal disease and is indicated for
the treatment of anemia in patients in-
fected with human immunodeficiency vi-
rus. Intensive EPO treatment (40,000 units
weekly) reduced the transfusion require-
ment of ICU patients.101 It may also have a
role in treating anemia due to chronic dis-
ease or to receipt of other medications
that suppress the marrow.
Other Blood Cell Growth Factors
Granulocyte-macrophage colony-stimu-
lating factor (GM-CSF) and G-CSF stimu-
late marrow production of granulocytes.100
G-CSF is approved for the treatment of
chemotherapy-induced neutropenia, for
patients undergoing peripheral blood
progenitor cell collection and therapy,
and for patients with chronic neutropenia.
The use of these stimulants decreases the
duration of neutropenia, increases toler-
ance to cytotoxic drugs, and decreases the
need for granulocyte transfusions. GM-
CSF is approved for use in patients under-
going autologous marrow transplanta-
tion. Another potential use for GM-CSF
and G-CSF is support of patients under-
going allogeneic marrow transplantation
or patients receiving antiviral agents that
suppress the marrow. Recombinant inter-
leukin-11 is licensed for cancer patients
with thrombocytopenia, but other activa-
tors for thrombopoietin receptors have
been disappointing and none are avail-
able at this time.
Oxygen Therapeutics (Carriers)
Stroma-free hemoglobin solution, in which
free hemoglobin has been separated from
cell membranes, has several characteris-
tics that render it unsuitable as a blood
substitute, including a low p50, short cir-
culation time, high oncotic pressure, and
vasopressor/nephrotoxic properties.102,103
512 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
However, chemical modifications of he-
moglobin solutions may successfully
overcome these disadvantages and such
products are in Phase III clinical trials at
thetimeofthiswriting.Patientsinthese
trials have survived very severe anemia
(residual cellular hemoglobin as low as 1
g/dL) without significant toxicity when
supported by these agents.104 Bovine he-
moglobin is approved for veterinary use.
Hemoglobin produced by recombinant
DNA techniques has also been investi-
gated. Finally, fluorocarbon products that
bind oxygen have been extensively inves-
tigated.103 One of the latter, Fluosol, was
approved by the FDA for use during per-
cutaneous transluminal angioplasty, but
it is no longer available in the United
States.
DDAVP
DDAVP is a synthetic analogue of the hor-
mone vasopressin that lacks significant
pressor activity.99,105 First used in the treat-
ment of diabetes insipidus, DDAVP is also
useful in promoting hemostasis because
of its ability to cause the release of endog-
enous stores of high-molecular-weight
vWF from the vascular subendothelium
and the concomitant increase in Factor
VIII. Because of its effect on Factor VIII
and vWF, DDAVP is used as a hemostatic
agent in patients with mild-to-moderate
hemophilia A and in patients with some
von Willebrand syndromes. Because
platelet adhesion and the subsequent for-
mation of a platelet plug depend upon
vWF, DDAVP may also be beneficial in a
wide variety of platelet function disor-
ders, including uremia, cirrhosis, drug-in-
duced platelet dysfunction (including as-
pirin), primary platelet disorders, and
myelodysplastic syndromes.99,105,106
DDAVP can be administered intrave-
nously, subcutaneously, or intranasally. It is
usually given as a single injection (0.3 to 0.4
µg/kg) to treat bleeding or for prophylaxis
before a procedure. Doses are not usually
repeated within a 24- to 48-hour period be-
cause of tachyphylaxis (the loss of biologic
effect with repeated administration of an
agent) and the induction of water retention
and hyponatremia. Some patients experi-
ence facial flushing or mild hypotension,
but side effects are rare. Its effect on vWF
occurs within 30 minutes and lasts 4 to 6
hours.105
Fibrinolytic Inhibitors
Epsilon aminocaproic acid (EACA) and
tranexamic acid—synthetic analogues of
lysine—competitively inhibit fibrinolysis
by saturating the lysine binding sites at
which plasminogen and plasmin bind to
fibrinogen and fibrin. The drugs can be
used locally or systemically and can be
given orally. Aprotinin is a polypeptide
prepared from bovine lung that inhibits
proteinases including plasmin, kallikrein,
trypsin, and, to some extent, urokinase.
Thus, it has an antifibrinolytic action but
may also inhibit coagulation because
kallikrein activates Factor XII. Aprotinin is
used intravenously. Because it is a poly-
peptide, hypersensitivity reactions can
occur.99,107
Antifibrinolytic agents have been used
successfully in cardiac surgery, prostatec-
tomy, and liver transplantation. EACA and
tranexamic acid can also be used locally at
sites where fibrinolysis contributes to
bleeding, as from mucosal lesions of the
mouth and gastrointestinal tract, and are of
benefit in the control of hemorrhage fol-
lowing dental extractions in patients with
hemophilia and in the control of gastroin-
testinal bleeding. EACA and tranexamic
acid may be helpful in controlling bleeding
due to severe thrombocytopenia. Systemic
Chapter 21: Blood Transfusion Practice 513
Copyright © 2005 by the AABB. All rights reserved.
administration of fibrinolytic inhibitors has
been associated with serious thrombotic
complications, including ureteral obstruc-
tion due to clot formation and thrombosis
of large arteries and veins. When used in
excessive doses, fibrinolytic inhibitors can
prolongthebleedingtime.Thesedrugs
should be employed by physicians with
experience in their use.
All three of these antifibrinolytic agents,
as well as DDAVP, have been used in an at-
tempt to decrease blood use in cardiac sur-
gery, and meta-analyses have shown a
decrease in the proportion of patients re-
ceiving allogeneic RBCs, the number of units
transfused,108,109 estimated blood losses,108
and the number of patients requiring re-
operation for bleeding.108,109 Of the three
agents, a much larger data base exists for
aprotinin, which is the most frequently
used. DDAVP was not effective overall but
may be useful in patients taking aspirin.108
Of concern has been a trend toward in-
creased thrombotic complications (myo-
cardial infarction and graft thrombosis)
with aprotinin, but they do not appear to
be statistically significant.107,108 Unfortu-
nately, very large studies would be required
to demonstrate whether the risk profile of
these agents is superior to that of blood,
particularly in view of the decreasing risks
of transfusion.
Oversight of Transfusion
Practice
Of the various institutions that regulate or
accredit aspects of transfusion, the Joint
Commission for Accreditation of Health-
care Organizations (JCAHO) has histori-
cally emphasized oversight of transfusion
practice as a requirement for accredita-
tion. As part of its performance improve-
ment standards, the JCAHO requires col-
lection of data regarding the use of blood
and scores the institution on the appro-
priateness of the selected performance
measures and the size of the data sam-
ple.110 Moreover,theJCAHOstandardsre
-
quire that the medical staff take the lead-
ership role in measurement, assessment,
and improvement of clinical processes re-
lated to the use of blood and blood com-
ponents. This assessment process must
include peer review, and its findings must
be communicated to involved staff mem-
bers as well as being a part of the renewal
of their clinical privileges.
Typically, this function has been dele-
gated to a medical staff committee, often a
dedicated “Transfusion Committee.” The
medical director of the transfusion service
should be a member of the committee. This
committee should review blood bank activ-
ities and statistics, blood ordering, and
transfusion practices and should have a
process to review records of patients trans-
fused with blood or components. The com-
mittee should monitor significant develop-
ments in transfusion medicine that would
affect patients in the health-care institution
and take appropriate action regarding these
developments.
The College of American Pathologists
(CAP) laboratory accreditation program
also requires transfusion oversight. This is
mandated by its general standard on qual-
ity control and improvement, which states
that the blood bank director must evaluate
the appropriateness of any laboratory’s out-
put in a multidisciplinary fashion.111 More-
over, the CAP accreditation checklist for
blood banks seeks documentation that “. . .
the transfusion service medical director ac-
tively participates in establishing criteria
and in reviewing cases not meeting transfu-
sion audit criteria.”112
Finally, the AABB Standards requires that
there be a peer-review program that moni-
tors appropriateness of use of blood com-
ponents.59(p86)
514 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
References
1. WelchHG,MeehanKR,GoodnoughLT.Pru
-
dent strategies for elective red blood cell
transfusion. Ann Intern Med 1992;116:393-
402.
2. Wilkerson DK, Rosen AL, Gould SA, et al. Ox-
ygen extraction ratio: A valid indicator of
myocardial metabolism in anemia. J Surg
Res 1987; 42:629-34.
3. Practice guidelines for blood component
therapy: Report by the American Society of
Anesthesiologists Task Force on Blood Com-
ponent Therapy. Anesthesiology 1996;84:
732-47.
4. Laine E, Steadman R, Calhoun L, et al. Com-
parison of RBCs and FFP with whole blood
during liver transplant surgery. Transfusion
2003;43:322-7.
5. American Association of Blood Banks, Amer-
ica’s Blood Centers, American Red Cross. Cir-
cular of information for the use of human
blood and blood components. Bethesda,
MD: AABB, 2002.
6. Practice parameter for the use of red blood
cell transfusions: Developed by the Red
Blood Cell Administration Practice Guide-
lines Development Task Force of the College
of American Pathologists. Arch Pathol Lab
Med 1998; 122:130-8.
7. National Institutes of Health Consensus De-
velopment Conference. Perioperative red
blood cell transfusion. JAMA 1988;260:
2700-3.
8. HébertPC,SchweitzerI,CalderL.Reviewof
the clinical practice literature on allogeneic
red blood cell transfusion. Can Med Assoc J
1997;156(Suppl):S9-26.
9. Goodnough LT, Brecher ME, Kanter MH,
AuBuchon JP. Transfusion medicine: First of
two parts. N Engl J Med 1999;340:438-47.
10. WeiskopfRB,VieleMK,FeinerJ,etal.Hu
-
man cardiovascular and metabolic response
to acute, severe isovolemic anemia. JAMA
1998;279:217-21.
11. Carson JL, Noveck H, Berlin JA, Gould SA.
Mortality and morbidity in patients with
very low postoperative Hb levels who decline
blood transfusion. Transfusion 2002;42:812-
8.
12. Vamvakas EC. Epidemiology of red blood cell
transfusion. Transfus Med Rev 1996;10:44-61.
13. Bracey AW, Radovancevic R, Riggs SA, et al.
Lowering the hemoglobin threshold for
transfusion in coronary artery bypass proce-
dures: Effect on patient outcome. Transfu-
sion 1999; 39:1070-7.
14. Johnson RG, Thurer RL, Kruskall MS, et al.
Comparison of two transfusion strategies
after elective operations for myocardial
revascularization. J Thorac Cardiovasc Surg
1992;104:307-14.
15. Carson JL, Terrin ML, Barton FB, et al. A pilot
randomized trial comparing symptomatic
vs. hemoglobin-level-driven red blood cell
transfusions following hip fracture. Transfu-
sion 1998;38:522-9.
16. Blair SD, Janvrin SB, McMollum CN, et al. Ef-
fect of early blood transfusion on gastroin-
testinal hemorrhage. Br J Surg 1986;73:783-5.
17. Hébert PC, Wells G, Blajchman MA, et al. A
multicenter, randomized, controlled clinical
trial of transfusion requirements in critical
care. N Engl J Med 1999;340:409-17.
18. Hébert PC, Yetisir E, Martin C. Is a low trans-
fusion threshold safe in critically ill patients
with cardiovascular diseases? Critical Care
Med 2001;29:227-34.
19. Wu WC, Rathore SS, Wang Y, et al. Blood
transfusion in elderly patients with acute
myocardial infarction. N Engl J Med 2001;
345:1230-6.
20. Elwood PC, Waters WR, Green WJW, et al.
Symptoms and circulating hemoglobin level.
J Chronic Dis 1969;21:615-28.
21. Canadian Erythropoietin Study Group. Asso-
ciation between recombinant human eryth-
ropoietin and quality of life and exercise ca-
pacity of patients receiving hemodialysis. Br
Med J 1990;300:573-8.
22. Parise LV, Smyth SS, Collor BS. Platelet mor-
phology, biochemistry, and function. In:
Beutler E, Lichtman MA, Coller BS, et al. Wil-
liams’ hematology. 6th ed. New York:
McGraw-Hill, 2001:1357-408.
23. Darcy MD, Kanterman RY, Kleinhoffer MA, et
al. Evaluation of coagulation tests as predic-
tors of angiographic bleeding complications.
Radiology 1996;198:741-4.
24. Doerfler ME, Kaufman B, Goldenberg AS.
Central venous catheter placement in pa-
tients with disorders of hemostasis. Chest
1996;110:185-8.
25. McVay PA, Toy P. Lack of increased bleeding
after paracentesis and thoracentesis in pa-
tients with mild coagulopathy. Transfusion
1991;31:164-71.
26. Ewe K. Bleeding after liver biopsy does not
correlate with indices of peripheral coagula-
tion. Dig Dis Sci 1981;26:388-93.
27. McVay PA, Toy P. Lack of increased bleeding
after liver biopsy in patients with mild
hemostatic abnormalities. Am J Clin Pathol
1990;94: 747-53.
28. Lind SE. Review: The bleeding time does not
predict surgical bleeding. Blood 1991;77:
2547-52.
Chapter 21: Blood Transfusion Practice 515
Copyright © 2005 by the AABB. All rights reserved.
29. Rodgers RP, Levin J. A critical appraisal of the
bleeding time. Semin Thromb Hemost 1990;
16:1-20.
30. Despotis GJ, Goodnough LT. Management
approaches to platelet-related microvascular
bleeding in cardiothoracic surgery. Ann
Thorac Surg 2000;70:S20-32.
31. Hanson SR, Slichter SJ. Platelet kinetics in
patients with bone marrow hypoplasia: Evi-
dence for a fixed platelet requirement. Blood
1985;66:1105-9.
32. Aster RH. Effect of anticoagulant and ABO in-
compatibility on recovery of transfused hu-
man platelets. Blood 1965;26:732-43.
33. Heal JM, Masel D, Rowe JM, Blumberg N. Cir-
culating immune complexes involving the
ABO system after platelet transfusion. Br J
Haematol 1993;85:566-72.
34. Benjamin RB, Antin JH. ABO-incompatible
bone marrow transplantation: The transfu-
sion of incompatible plasma may exacerbate
regimen-related toxicity. Transfusion 1999;
39:1273-4.
35. Heal JM, Blumberg N. The second century of
ABO: And now for something completely dif-
ferent. Transfusion 1999;39:1155-9.
36. McManigal S, Sims KL. Intravascular hemoly-
sis secondary to ABO incompatible platelet
products; an underrecognized transfusion re-
action. Am J Clin Pathol 1999;111:202-6.
37. Norfolk DR, Ancliffe PJ, Contreras M, et al.
Synopsis of background papers (Consensus
Conference on Platelet Transfusion). Br J
Haematol 1998;101:609-14.
38. National Institutes of Health Consensus De-
velopment Conference. Platelet transfusion
therapy. Transfus Med Rev 1987;1:195-200.
39. Development Task Force of the College of
American Pathologists. Practice parameter
for the use of fresh frozen plasma, cryopreci-
pitate, and platelets. JAMA 1994;271:777-81.
40. Schiffer CA, Anderson KC, Bennett CL, et al.
Platelet transfusion for patients with cancer:
Clinical practice guidelines of the American
Society of Clinical Oncology. J Clin Oncol
2001;19:1519-38.
41. Gmur J, Burger J, Schanz U, et al. Safety of
stringent prophylactic platelet transfusion
policy for patients with acute leukemia. Lan-
cet 1991;338:1224-6.
42. Wandt H, Frank M, Ehninger G, et al. Safety
and cost effectiveness of a 10 ×109/L trigger
for prophylactic platelet transfusions com-
pared with the traditional 20 ×109/L trigger;
a prospective comparative trial in 105 pa-
tients with acute myeloid leukemia. Blood
1991;10:3601-6.
43. Rebulla P, Finazzi G, Morangoni F, et al. The
threshold of prophylactic platelet transfu-
sions in adults with acute myeloid leukemia.
N Engl J Med 1997;337:1870-5.
44. Murphy S, Litwin S, Herring LM, et al. Indica-
tions for platelet transfusion in children with
acute leukemia. Am J Hematol 1982;12:347-
56.
45. Reed RL, Ciavarella D, Heimbach DM, et al.
Prophylactic platelet administration during
massive transfusion; a prospective, random-
ized, double-blind clinical study. Ann Surg
1986;203:40-8.
46. Simon TL, Akl BF, Murphy W. Controlled trial
of routine administration of platelet concen-
trates in cardiopulmonary bypass surgery.
Ann Thorac Surg 1984;37:359-64.
47. Nguyen CM, Harrington RA. Glycoprotein
IIb/IIIa receptor antagonists; a comparative
review of their use in percutaneous coronary
intervention. Am J Cardiovasc Drugs 2003;3:
423-36.
48. McFarland JG. Matched apheresis platelets.
In: McLeod BC, Price TH, Weinstein R, eds.
Apheresis: Principles and practice. 2nd ed.
Bethesda, MD: AABB Press, 2003:199-220.
49. KicklerTS,HermanJH,eds.Currentissuesin
platelet transfusion therapy and platelet allo-
immunity. Bethesda, MD: AABB Press, 1999.
50. Warkentin TE. Management of immune
thrombocytopenia. In: Simon TL, Dzik WH,
Snyder EL, et al, eds. Rossi’s principles of
transfusion medicine. 3rd ed. Baltimore, MD:
Lippincott Williams and Wilkins, 2002:367-
95.
51. GordonLI,KwaanHC,RossiEC.Deleterious
effects of platelet transfusions and recovery
thrombocytosis in patients with thrombotic
microangiopathy. Semin Hematol 1987;24:
194-201.
52. Strauss RG. Granulocyte transfusion. In:
McLeodBC,PriceTH,WeinsteinR,eds.
Apheresis: Principles and practice. 2nd ed.
Bethesda, MD: AABB Press, 2003:237-52.
53. Price TH. The current prospects for neutro-
phil transfusions for the treatment of granulo-
cytopenic infected patients. Transfus Med
Rev 2000;14:2-11.
54. Vamvakas EC, Pineda A. Determinants of the
efficacy of prophylactic granulocyte transfu-
sions: A meta-analysis. J Clin Apheresis 1997;
12:74-81.
55. Gresens CJ, Paglieroni TG, Moss CB, et al.
WBC populations in thawed fresh frozen
plasma (abstract). Transfusion 1999;39(Suppl):
99S.
56. Stringham JC, Bull DA, Fuller TC, et al. Avoid-
ance of cellular blood product transfusions
in LVAD recipients does not prevent HLA
allosensitization. J Heart Lung Transplant
1999;18(2):160-5.
516 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
57. Food and Drug Administration. Memoran-
dum: Recommendations and license re-
quirements for leukocyte-reduced blood
products. (May 29, 1996) Rockville, MD:
CBER Office of Communication, Training,
and Manufacturers Assistance, 1996.
58. Food and Drug Administration. Draft guid-
ance for industry: Prestorage leukocyte re-
duction with whole blood and blood compo-
nents intended for transfusion. (January 23,
2001) Rockville, MD: CBER Office of Com-
munication, Training, and Manufacturers
Assistance, 2001.
59. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
60. Preiksaitis J. The cytomegalovirus-“safe”
blood product: Is leukoreduction equivalent
to antibody screening? Transfus Med Rev
2000;14:112-36.
61. The trial to reduce alloimmunization to pla-
telets study group. Leukocyte reduction and
ultraviolet B irradiation of platelets to pre-
vent alloimmunization and refractoriness to
platelet transfusions. N Engl J Med 1997;337:
1861-9.
62. Vamvakas EC, Dzik WH, Blajchman MA. Del-
eterious effects of transfusion associated
immunomodulation: Appraisal of the evi-
dence and recommendations for prevention.
In: Vamvakas EC, Blajchman MA, eds. Im-
munomodulatory effects of blood transfu-
sion. Bethesda, MD: AABB Press, 1999:
253-85.
63. KrishnanLA,BrecherME.Transfusiontrans
-
mitted bacterial infection. Hematol Oncol
Clin North Am 1995;9:167-85.
64. Dzik WH, Anderson JK, O’Neill EM, et al. A
prospective, randomized clinical trial of uni-
versal WBC reduction. Transfusion 2002;42:
1114-22.
65. Vamvakas EC, Blajchman MA. Universal
WBC reduction: The case for and against.
Transfusion 2001;41:691-712.
66. Akahoshi M, Takanashi M, Masuda H, et al.
Case reports: A case of transfusion-associ-
ated graft-versus-host disease not prevented
by white cell-reduction filters. Transfusion
1992; 32:169-72.
67. Roberts HR, Monroe DM III, Hoffman M.
Molecular biology and biochemistry of the
coagulation factors and pathways of hemos-
tasis. In: Beutler E, Lichtman MA, Coller BS,
et al. Williams’ hematology. 6th ed. New
York: McGraw-Hill, 2001:1409-34.
68. vanAkenWG.Preparationofplasmaderiva
-
tives.In:SimonTL,DzikWH,SnyderEL,et
al, eds. Rossi’s principles of transfusion med-
icine. 3rd ed. Baltimore, MD: Lippincott Wil-
liams and Wilkins, 2002:304-15.
69. The United States Pharmacopoeial Conven-
tion, Inc. Hemophilia management. Transfus
Med Rev 1998;12:128-40.
70. British Committee for Standards in Haema-
tology, Working Party of the Blood Transfu-
sion Task Force. Guidelines for the use of
fresh frozen plasma. Transfus Med 1992;2:
557-63.
71. Rock G, Shumak KH, Sutton DM, et al. Cryo-
supernatant as replacement fluid for plasma
exchange in thrombotic thrombocytopenic
purpura. Members of the Canadian Aphere-
sis Group. Br J Haematol 1996;94: 383-6.
72. SpectorI,CornM,TicktinHE.Effectof
plasma transfusions on the prothrombin
time and clotting factors in liver disease. N
Engl J Med 1966;275:1032-7.
73. Mannucci PM, Vicente V, Vianello L, et al.
Controlled trial of desmopressin in liver cir-
rhosis and other conditions associated with
a prolonged bleeding time. Blood 1986;67:
1148-53.
74. MurrayDJ,PennellBJ,WeinsteinSL,Olson
JD. Packed red cells in acute blood loss: Dilu-
tional coagulopathy as a cause of surgical
bleeding. Anesth Analg 1995;80:336-42.
75. National Institutes of Health Consensus De-
velopment Conference. Fresh frozen plasma;
indications and risks. JAMA 1985;253:551-3.
76. Weinstein R. Basic principles of therapeutic
blood exchange. In: McLeod BC, Price TH,
Weinstein R, eds. Apheresis: Principles and
practice.2nded.Bethesda,MD:AABBPress,
2003:295-320.
77. Jaffee CJ, Atkinson JP, Gelfand JA, Frank MM.
Hereditary angioedema: The use of fresh
frozen plasma for prophylaxis in patients
undergoing oral surgery. J Allergy Clin
Immunol 1975;545:386-93.
78. Ginsburg D. von Willebrand disease. In:
Beutler E, Lichtman MA, Coller BS, et al, eds.
Williams’ hematology. 6th ed. New York:
McGraw-Hill, 2001:1813-28.
79. Rousou J, Levitsky S, Gonzalez-Levin J, et al.
Randomized clinical trial of fibrin sealant in
patients undergoing resternotomy or re-
operation after cardiac operations. J Thorac
Cardiovasc Surg 1989;97:194-203.
80. Streiff MB, Ness PM. Acquired FV inhibitors:
A needless iatrogenic complication of bovine
thrombin exposure. Transfusion 2002;42:
18-26.
81. Roberts HR, Hoffman M. Hemophilia A and
hemophilia B. In: Beutler E, Lichtman MA,
Coller BS, et al, eds. Williams’ hematology.
6th ed. New York: McGraw-Hill, 2001:1639-
71.
Chapter 21: Blood Transfusion Practice 517
Copyright © 2005 by the AABB. All rights reserved.
82. Smith KJ. Factor IX concentrates: The new
products and their properties. Transfus Med
Rev 1992;6:124-36.
83. Nydegger UE, Mohacsi PJ. Immunoglobulins
in clinical medicine. In: Simon TL, Dzik WH,
Snyder EL, et al, eds. Rossi’s principles of
transfusion medicine. 3rd ed. Baltimore, MD:
Lippincott Williams and Wilkins, 2002:316-
32.
84. Kobayashi RH, Stiehm ER. Immunoglobulin
therapy. In: Petz LD, Swisher SN, Kleinman S,
etal,eds.Clinicalpracticeoftransfusion
medicine.3rded.NewYork:ChurchillLiving
-
stone, 1995:985-1010.
85. Goodnigh t SW, Gr iffin JH. He reditar y
thrombophilia. In: Beutler E, Lichtman MA,
Coller BS, et al, eds. Williams’ hematology.
6th ed. New York: McGraw-Hill, 2001:1697-
714.
86. Griffin JH. Control of coagulation reactions.
In: Beutler E, Lichtman MA, Coller BS, et al,
eds. Williams’ hematology. 6th ed. New York:
McGraw-Hill, 2001:1435-49.
87. Bucar SJ, Levy JH, Despotis GJ, et al. Uses of
antithrombin III concentrate in congenital
and acquired deficiency states. Transfusion
1998;38:481-98.
88. Friedman KE, Menitove JE. Preparation and
clinical use of plasma and plasma fractions.
In: Beutler E, Lichtman MA, Coller BS, et al,
eds. Williams’ hematology. 6th ed. New York:
McGraw-Hill, 2001:1917-34.
89. McClelland DBL. Safety of human albumin as
a constituent of biologic therapeutic prod-
ucts. Transfusion 1998;38:690-4.
90. Doweiko JP, Nompleggi DJ. The role of albu-
min in human physiology and pathophy-
siology. Part III: Albumin and disease states. J
Parenter Enteral Nutr 1991;15:476-83.
91. VermeulenLCJr.,RatkoTA,ErstadBL,etal.A
paradigm for consensus. The University Hos-
pital consortium guidelines for the use of al-
bumin, nonprotein colloid, and crystalloid
solutions. Arch Intern Med 1995;155:373-9.
92. Cochrane Injuries Group Albumin Reviewers.
Human albumin administration in critically
ill patients: Systemic review of randomized
controlled trials. Br Med J 1998;317:235-46.
93. Vichinsky EP, Haberkern CM, Newmayer L, et
al. A comparison of conservative and aggres-
sive transfusion regimens in the periopera-
tive management of sickle cell disease. N
Engl J Med 1995;333:206-13.
94. Rosse W, Telen M, Ware R. Transfusion sup-
port for patients with sickle cell disease.
Bethesda, MD: AABB Press, 1998.
95. The National Institutes of Health, National
Heart, Lung and Blood Institute, Division of
Blood Diseases and Resources. The manage-
ment of sickle cell disease. 4th ed. Bethesda,
MD: NHLBI, 2002. (Available at http://www.
nhlbi.nih.gov/health/prof/blood/sickle/
sc_mngt.pdf.)
96. Mollison PL, Engelfriet CP, Contreras M.
Blood transfusion in clinical medicine. 10th
ed. Oxford, England: Blackwell Scientific
Publications, 1998.
97. Ciavarella D, Reed RL, Counts RB, et al. Clot-
ting factors and the risk of diffuse microvas-
cular bleeding in the massively transfused
patient. Br J Haematol 1987;67:365-8.
98. Heaton A, Keegan T, Holme S, et al. In vivo re-
generation of red cell 2,3-diphosphoglycerate
following transfusion of DPG-depleted AS-1,
AS-3 and CPDA-1 red cells. Br J Haematol
1989;71:131-6.
99. Mannucci PM. Drug therapy: Hemostatic
drugs. N Engl J Med 1998;339:245-53.
100. Kruskall MS. Biologic response modifiers—
hematopoietic growth factors. In: Petz LD,
Swisher SN, Kleinman S, et al, eds. Clinical
practice of transfusion medicine. 3rd ed. New
York: Churchill Livingstone, 1996:1023-39.
101. Corwin HL, Gettinger A, Pearl RG, et al. Effi-
cacy of recombinant human erythropoietin
in critically ill patients: A randomized, con-
trolled trial. JAMA 2002;288:2827-35.
102. Spence RK. Blood substitutes. In: Petz LD,
Swisher SN, Kleinman S, et al, eds. Clinical
practice of transfusion medicine. 3rd ed. New
York: Churchill Livingstone, 1996:967-84.
103. Stowell CP, Levin J, Spiess BD, Winslow RM.
Progress in the development of RBC substi-
tutes. Transfusion 2001;41:287-99.
104. Gould S, Moore ED, Hoyt DB, et al. The life-
sustaining capacity of human polymerized
hemoglobin when red cells might be unavail-
able. J Am Cell Surg 2002;195:445-52.
105. Schulman S. DDAVP, the multipotent drug in
patients with coagulopathies. Transfus Med
Rev 1991;5:132-44.
106. Shattil SJ, Abrams CS, Bennett JS. Acquired
qualitative platelet disorders due to diseases,
drugs, and foods. In: Beutler E, Lichtman MA,
Coller BS, et al, eds. Williams’ hematology.
6th ed. New York: McGraw-Hill, 2001:1583-
602.
107. Bachman F. Disorders of fibrinolysis and use
of antifibrinolytic agents. In: Beutler E, Licht-
man MA, Coller BS, et al, eds. Williams’ he-
matology. 6th ed. New York: McGraw-Hill,
2001:1829-40.
108. Laupacis A, Fergusson D. Drugs to minimize
perioperative blood loss in cardiac surgery:
Meta-analysis using perioperative blood
transfusion as the outcome. Anesth Analg
1997;85:1258-67.
518 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
109. Munoz JJ, Birkmeyer NJ, Birkmeyer JD, et al.
Is epsilon-aminocaproic acid as effective as
aprotinin in reducing bleeding with cardiac
surgery? A meta-analysis. Circulation 1999;
99:81-9.
110. Joint Commission for Accreditation of
Healthcare Organizations. 2004 Comprehen-
sive accreditation manual for hospitals,
standard PI.3.1.1 and MS.8.1.3 and MS.8.3.
Oakbrook, IL: Joint Commission Resources,
2004.
111. Hamlin WB. Requirements for accreditation
by the College of American Pathologists Lab-
oratory Accreditation Program. Arch Pathol
Lab Med 1999;123:465-7.
112. CAP Transfusion Medicine Checklist. Revi-
sion date 12/31/2003. Northfield, IL: College
of American Pathologists, 2003.
Chapter 21: Blood Transfusion Practice 519
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 22: Administration of Blood and Components
Chapter 22
Administration of Blood and
Components
GOOD TRANSFUSION PRACTICE
requires that comprehensive poli-
cies and procedures for blood ad-
ministration be designed to prevent errors.
The development of these policies should
be a collaborative effort between the medi-
cal director of the transfusion service, the
directors of the clinical services, both nurs-
ing and medical, and all personnel involved
in blood administration. Policies and pro-
cedures must be accessible, periodically
reviewed for appropriateness, and moni-
tored for compliance. In addition to blood
administration policies and procedures,
this chapter discusses pretransfusion pre-
paration, issuing of blood components,
the equipment used in blood administra-
tion, and compatible intravenous solutions.
Pre-Issue Events
Patient Education and Consent
Patients who are aware of the steps in-
volved in a transfusion will experience less
anxiety. This is important not only for an
adult but also for any child who has the
ability to understand the process. In the
latter situation, it is appropriate to edu-
cate the parents, so that they are better
prepared to support their child through-
out the transfusion. The transfusionist
should explain how the transfusion will be
given, how long it will take, what the ex-
pected outcome is, what symptoms to re-
port, and that vital signs will be taken. The
physician has a responsibility to explain
the benefits and risks of transfusion ther-
apy as well as the alternatives in a fashion
that the patient can comprehend. Other
than in emergency situations, the patient
shouldbegivenanopportunitytoask
questions, and his or her informed choice
should be documented. State and local
laws govern the process of obtaining and
documenting the consent of the patient.
Some states have specific requirements
for blood transfusion consent. Institu-
tions should be careful to ensure that
their individual processes and procedures
comply with applicable laws.
521
22
Copyright © 2005 by the AABB. All rights reserved.
Individual institutions have different re-
quirements for obtaining and documenting
this interaction, as well as different policies
about how often it is necessary. Some facili-
tiesrequiretheuseofaformalconsent
form, which provides information in un-
derstandable language, signed by the pa-
tient. Others expect the physicians to make
a note in the medical record stating that the
risksof,andalternativesto,bloodtransfu
-
sions were explained and that the patient
consented. If a patient is unable to give
consent, a responsible family member
should be asked. If no family member is
available or if the emergency need for
transfusion leaves no time for consent, it is
prudent to note this in the medical record.1-3
Prescription and Special Instructions
There must be documentation of the or-
der by the physician for the blood compo-
nent(s).4Although a telephone order may
be acceptable during urgent situations, this
must be followed by a written request.
Special instructions should be indicated
regarding the transfusion relating to the:
■Component—eg, washed, irradiated,
leukocyte-reduced, cytomegalovirus
negative
■Patient—eg, premedication, timing
■Process—flowrates,rateofinfusion,
use of a blood warmer or electrome-
chanical pump
■Need for emergency release
Component Considerations
Some components require special prepa-
ration before release for transfusion. Be-
cause these steps are time-consuming and
may significantly shorten the shelf life of
the component, preparation should be
carefully coordinated with the anticipated
time of transfusion. The transfusion ser-
vice must strive to make every effort to
ensure that the component is ready when
needed, but not so early that it expires be-
fore administration.
Medical and nursing staffs need to be
aware of special requirements for prepara-
tion and to understand that these times
cannot be significantly shortened, even in
urgent situations. Close communication is
required. Some examples of pretransfusion
processing procedures are given in Table
22-1.
Patient Considerations
Patients with a history of recurrent aller-
gic transfusion reactions may benefit from
premedication with antihistamines or by
slowing the rate of transfusion. Routine
premedication with antipyretics should
be discouraged because delaying a rise in
temperature may mask one sign of a
hemolytic reaction and partly because
they may be ineffective.5-7
Antipyretics typically do not mask other
clinical features of hemolysis, such as
changes in blood pressure, pulse, or respi-
ration. Premedication orders should be
carefully timed with the anticipated admin-
istration of the unit. Medication ordered in-
travenously may be given immediately be-
fore the start of the transfusion, but orally
administered drugs need to be given 30 to
60 minutes before the start of the transfu-
sion.
Process Considerations
Blood warmers and electromechanical
pumps need to be available if required for
the transfusion.
Emergency Release
Blood may be released without complet-
ing pretransfusion testing if it is urgently
needed for a patient’s survival, provided
that: 1) the records properly document
522 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

the emergency request and 2) the issued
units are of an ABO group unlikely to
cause immediate harm to the recipient.
Venous Access
To avoid any delay in transfusion and po-
tential wastage of blood components, ve-
nous access should be established before
the component is issued. If a pre-existing
line is to be used, it should be checked for
patency; signs of infiltration, inflamma-
tion, or infection; and the compatibility of
any intravenous solutions (see below). Many
venous access devices can be used for
blood component transfusion. Selection
depends on the location, size, and integ-
rity of the patient’s veins; the type of med-
ication or solution to be infused; the type
of component to be transfused; the vol-
ume and timing of the administration; the
possibility of interactions among paren-
teral solutions; and expected duration of
intravenous therapy.
The lumen of needles or catheters used
for blood transfusion should be large
enough to allow appropriate flow rates
without damaging the vein. There are no
strict guidelines limiting the size of the
catheter or needles used for transfusion. An
18-gauge catheter provides good flow rates
for cellular components without excessive
discomfort to the patient, but patients with
small veins require much smaller catheters.
High-pressure flow through needles or
catheters with a small lumen may damage
red cells8-10 unless the transfusion compo-
nent is sufficiently diluted.10 Undiluted
preparations of red cells flow very slowly
through a 23-gauge needle, but dilution
with saline to increase the flow rate may
cause unwanted volume expansion. Even
in patients with cardiac disease or volume
expansion, transfusions should be able to
be given safely within 4 hours. For rare pa-
tients unable to tolerate a transfusion with-
in 4 hours, local policy should be developed
regarding whether to split units or to dis-
card the unused portion. Specific models of
infusion pumps have been approved for
use in blood transfusion. These pumps
maintain a constant delivery of blood, and
studies have indicated no significant evi-
dence of hemolysis as the needle size var-
ies.11
Chapter 22: Administration of Blood and Components 523
Table 22-1. Component Preparation Times
Component Minimum Time* Shelf Life
RBCs: saline-washed 45 minutes 24 hours
RBCs: thawed-deglycerolized 75 minutes 24 hours or 2 weeks†
Fresh Frozen Plasma: thawed 30 minutes 24 hours
Thawed Plasma — 5 days
Platelets: pooled 15 minutes 4 hours
CRYO: thawed (single unit) 15 minutes 6 hours
CRYO: pooled 15 minutes 4 hours
*Will vary with institutional procedures.
†Depends on the method used.
RBCs = Red Blood Cells; CRYO = Cryoprecipitated AHF.
Copyright © 2005 by the AABB. All rights reserved.
Central venous catheters are used for
medium- and long-term therapy or for the
administration of solutions potentially toxic
to a peripheral vein to allow the dilution
achieved with high-volume blood flow.
Some catheters are placed via special intro-
ducing needles and guide-wires; others are
surgically implanted. Catheters with a
multilumen design have separate infusion
ports for each lumen, permitting the simul-
taneous infusion of fluids without intermix-
ture in the infusion line, thereby avoiding
the potential for hemolysis from incompat-
ible fluids.
Need for Compatibility Testing
The transfusion service personnel deter-
mine what pretransfusion testing is re-
quired. Compatibility testing must be per-
formed for transfusion of Whole Blood,
components with a clinically significant
red cell content, and all red cell compo-
nents. For test and specimen require-
ments, refer to Chapter 18. Compatibility
testing other than ABO and Rh typing is
not required for platelet and plasma com-
ponents, but most facilities require that
the recipient’s ABO and D types be known
(onfile)beforesuchcomponentsarese
-
lected for issue. Whenever possible, plasma-
containing components should be com-
patible with the patient’s red cells (see
Chapter 21).
Blood Issue and
Transportation
Delivering Blood to the Patient Area
Institutions should define blood pick-up
and delivery policies and appropriate
training programs for the staff assigned to
these functions. Blood is not routinely
dispensed from the controlled environ-
ment of the blood bank until all testing is
completed, the patient is properly pre-
pared, and the transfusionist is ready to
begin the procedure. There must be a
mechanism to identify the intended re-
cipient and the requested component at
the time of issue. It is optimal to identify
the transporter.
Transfusion service personnel will re-
view identifying information, inspect the
appearance of the component before re-
lease, and ensure there is a system to main-
tain proper storage temperature during
transport.Thesafestpracticeistoissueone
unit to one patient at a time. For patients
who are rapidly bleeding or who have mul-
tiple venous access sites, multiple units
may be issued to a single patient. It is not
recommended that blood for two or more
patients be issued simultaneously to one
transporter because this could increase the
chance of transfusion error. However, logis-
tical considerations may make this imprac-
tical. The transporter should transport the
blood to the intended site of transfusion
without delay—preferably to the transfu-
sionist. It is preferable to place the unit of
blood in a protective container that would
contain any spillage in the event of inadver-
tent breakage during transport.
The responsibility for accurately identi-
fying a transfusion component rests with both
the transfusion service personnel who issue
the blood and the transfusionist who re-
ceives it. Before a unit of blood is issued,
transfusion service personnel complete the
following steps:
1. The records that identify the in-
tended recipient and the requested
component are reviewed.
2. The identifiers (Standards requires
at least two) of the intended recipi-
ent, the ABO and D type of the recip-
ient, the component unit number,
the ABO and D type of the donor
unit, and the interpretation of com-
patibility tests (if performed) are re-
524 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
corded on a transfusion form for
each unit. This form, or a copy, be-
comes a part of the patient’s perma-
nent medical record after comple-
tion of the transfusion. In some
institutions, the transfusion form is
attached to the unit and, therefore,
serves as the tie tag that is required
and described below. This form typi-
cally will have fields to identify the
transfusionist and co-identifier (if re-
quired) and other information, such
as pre- and posttransfusion vital signs,
amount of blood given, whether a re-
action occurred, etc.
3. A tie tag or label with the name and
identification number of the in-
tended recipient, the component unit
number, and the interpretation of
compatibility tests (if performed) must
be securely attached to the blood
container.
4. The appearance of the unit is checked
before issue and a record is made of
this inspection.
5. The expiration date (and time if ap-
plicable) is checked to ensure that the
unit is suitable for transfusion.
6. The name of the person issuing the
blood and the date and time of issue
are recorded. Recording the name of
the transporter to whom the blood is
issued is optimal.
Delay in Starting the Transfusion
Ideally, blood should be requested from
the blood bank only at the time when it is
intended to be administered. If the trans-
fusion cannot be initiated promptly, the
blood should be returned to the blood
bank for storage, unless the transfusion to
the originally intended recipient can be
completed within 4 hours. It should not
be left at room temperature or stored in
an unmonitored refrigerator. Units re-
turned to the blood bank after a period
outside monitored refrigeration will be
unsuitable for reissue if the sterility of the
container is compromised or if the tem-
perature has risen to 10 C or above, which
is generally considered to happen in less
than 30 minutes. If the units have been
kept in suitable conditions, such as iced
coolers that have been validated for sev-
eral hours of storage, longer periods are
acceptable. Requirements for blood col-
lected intraoperatively differ. See Table
5-8. Units that have been entered (punc-
tured) after release from the blood bank
cannot be accepted into general inven-
tory for later reissue.
Pre-Administration Events
Identifying the Recipient and Donor Unit
Accurate identification of the transfusion
component and the intended recipient
may be the single most important step
in ensuring transfusion safety.12-15 Most
hemolytic transfusion reactions and
deaths occur because of inadvertent ad-
ministration of ABO-incompatible red
cells.14-15 Plasma and platelets are also ca-
pable of causing serious transfusion reac-
tions.16 Identification and labeling of do-
nor blood are discussed in Chapter 7;
procedures to identify the patient’s speci-
men used for compatibility testing are
discussed in Chapter 18. The most impor-
tant steps in safe transfusion administra-
tion are clerical and occur when the
transfusion service issues blood for a spe-
cific patient and when the blood is ad-
ministered.
The transfusionist who administers the
blood represents the last point at which
identification errors can be detected before
transfusion of the component is initiated.
The transfusionist must check all identify-
ing information immediately before begin-
Chapter 22: Administration of Blood and Components 525
Copyright © 2005 by the AABB. All rights reserved.
ning the transfusion and record that this in-
formation has been checked and found to
be correct, typically, on the transfusion
form. Any discrepancy must be resolved
before the transfusion is started. It is com-
mon practice in many institutions that a
second person (co-identifier) confirms the
identity of the blood unit and of the patient.
Some institutions may require that the
transfusionist check for documentation of
patient consent before blood is given. It is
also common practice to check the ABO
and D type as written in the transfusion
form with a record of ABO and D type in
the patients’ chart. The following informa-
tion, however, must be reviewed and found
to be correct:
1. Physician’s order. The nature of the
blood or component should be
checked against the physician’s writ-
ten order to verify that the correct
component and dose (number of
units) are being given. All identifica-
tion attached to the container must
remain attached until the transfu-
sion has been completed.
2. Recipient identification. The patient’s
identifiers on the patient’s identificat-
ion band must be identical with the
identifiers attached to the unit. It is de-
sirable to ask the patient to state his or
her name, if capable of so doing, be-
cause the information on the identifi-
cation band may be in error.17
3. Unit identification. The unit identifi-
cation number on the blood con-
tainer, the transfusion form, and the
tie tag attached to the unit (if not the
same as the latter) must agree.
4. ABO and D. The ABO and D type on
the primary label of the donor unit
must agree with those recorded on
the transfusion form. The recipient’s
ABO and D type must be recorded
on the transfusion form. The pa-
tient’stypeandthetypeofthecom
-
ponent may not be identical (see
Chapter 21), but the information on
the transfusion form and that on the
container label must be the same.
5. Expiration. The expiration date and
time of the component should be
verified as acceptable.
6. Compatibility. The interpretation of
compatibility testing (if performed)
must be recorded on the transfusion
form and on the tag attached to the
unit (if not the same). If blood was
issued before compatibility tests were
completed, this must be conspicu-
ously indicated.
In certain clinical situations, such as the
operating room (OR), emergency room, or
in the outpatient setting, an identifying
band may not be attached to all patients at
thetimeoftransfusion.Wristbandsarefre-
quently removed in the OR for arterial line
insertions and identity confirmation may
not be possible in this manner. Further-
more, a co-identifier confirming identity,
although always desirable, may not func-
tion for all circumstances in this setting.
Institutions should address such clinical
situations, and site-specific transfusion
protocols should be developed.18
Starting the Transfusion
After checking all the identifying informa-
tion, the transfusionist (and co-identifier)
must indicate in the medical record that
the identification was correct (such as by
signing the transfusion form) to docu-
ment who started the transfusion and to
recordthedateandtime.Vitalsigns
should be taken and recorded, if not done
previously. A record of the date and time
of transfusion, the name and volume of
the component, and vital signs may be re-
quired on other parts of the medical re-
cord, such as intake/output records, anes-
526 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
thesia records, or intensive care flow
sheets, depending on the institution’s pol-
icy. This may suffice for documentation
purposes.
Several reports have documented the oc-
currence of errors at the point of blood ad-
ministration.19-22 In particular, Baele and
colleagues19 studied the charts and records
of 808 patients who received 3485 units of
blood over a period of 15 months, to deter-
mine if there had been errors in blood ad-
ministration. They detected 165 errors oc-
curring after blood units had left the blood
bank, 15 of which were considered to be
major. Seven of the major errors involved
patient misidentification that resulted in
blood being given to patients for whom it
was not intended, constituting 0.74% of pa-
tients and 0.2% of units. One error resulted
in an ABO-incompatible hemolytic reaction
that was not reported to the blood bank.
Eight other major errors occurred in four
patients (0.5%), including the administra-
tion of five allogeneic units to a patient for
whom autologous blood was available, and
the transfusion of one anemic patient
whose doctor had ordered only a cross-
match. The remaining 150 errors included
misrecording (n = 61), mislabeling (n = 6),
and failure to adequately document the
transfusion(n=83).
Both mechanical barrier systems23 and
electronic means of patient and product
identifications are marketed to supplement
(but not substitute for) the paper identifica-
tion process. In electronic systems, a bar
code or radio frequency identifiers are at-
tached to the blood component (and trans-
fusion form) that, when scanned/read, will
match the patient’s information on the
identification band.24 It is likely that these
electronic means will find more widespread
use in the future. Empiric experience with
medication administration, however, sug-
gests that unanticipated side effects of such
procedures may need consideration.25
Compliance with institutional blood ad-
ministration policies requires a Quality
Assurance/Continuous Improvement pro-
gram in which continued monitoring and
re-education of staff occur when variance
with procedures is observed. Such an ap-
proach has been initiated in some institu-
tions, resulting in improvement in transfu-
sion practice.26 Direct observation of
administration27 or educational videos28
may also be useful.
Administration
Infusion Sets
Any blood component must be adminis-
tered through a filter designed to retain
blood clots and particles potentially harm-
ful to the recipient.4,29(p48) All filters and in-
fusion devices must be used according to
the manufacturer’s directions.
Standard Sets
Standard blood infusion sets have inline
filters (pore size: 170 to 260 microns), drip
chambers, and tubing in a variety of con-
figurations. Sets should be primed ac-
cording to the manufacturer’s directions,
using either the component itself or a so-
lution compatible with blood (see the sec-
tion on Compatible IV Solutions). For op-
timal flow rates and performance, filters
should be fully wetted. Drip chambers
should be half-filled to allow observation
of blood flow.
Many institutions have a policy of chang-
ing sets after every transfusion or of limit-
ing their use to several units or several
hours in order to reduce the risks of bacte-
rial contamination. A reasonable time limit
is 4 hours; this is consistent with the 4-hour
outdate that the Food and Drug Adminis-
tration (FDA) places on blood in an open
system held at room temperature. Most
Chapter 22: Administration of Blood and Components 527
Copyright © 2005 by the AABB. All rights reserved.
standard filters are designed to filter 2 to 4
units of blood, but if the first unit required 4
hours for infusion, the filter should not be
reused. The filter traps cell aggregates, cel-
lular debris, and coagulated proteins, re-
sulting in a high protein concentration at
the filter surface. The high protein milieu
and room temperature conditions promote
growth of any bacteria that might be pres-
ent. Accumulated material also slows the
rate of flow.30
Special Sets
High flow sets for rapid transfusion have
large filter surface areas, large-bore tub-
ing, and may have an inline hand pump.
Sets designed for rapid infusion devices
also may have “prefilters” to retain parti-
cles over 300 microns in diameter and to
extend the life of standard blood filters
“downstream.” Gravity-drip sets for the
administration of platelets and cryopreci-
pitate have small drip chamber/filter ar-
eas, shorter tubing, and smaller priming
volumes. Syringe-push sets for compo-
nent administration have the smallest
priming volumes and an inline blood fil-
ter that may be inconspicuous.
Microaggregate Filters
Microaggregate filters are designed for the
transfusion of red cells. Microaggregates
(smaller than 170 microns in size) pass
through standard blood filters. Screen- or
depth-type filters have an effective pore
size of 20 to 40 microns and trap the
microaggregates composed of degenerat-
ing platelets, leukocytes, and fibrin strands
that form in blood after 5 or more days of
refrigerated storage.
Microaggregate filters may be used for
other components if this use is mentioned
in the manufacturer’s instructions; how-
ever, the large volume required for priming
causes a significant portion of these com-
ponents to be lost if the set is not flushed
with saline afterward. Depth-type micro-
aggregate filters, or any filters capable of re-
moving leukocytes, must not be used for
the transfusion of granulocyte concen-
trates.29(p48) Hemolysis of red cells has been
reported with microaggregate filters.30
Leukocyte Reduction Filters
Special “third-generation” blood filters
can reduce the number of leukocytes in
red cell or platelet components to less
than 5 × 106, a level that reduces the risk of
HLA alloimmunization and the transmis-
sionofcytomegalovirusaswellasthein-
cidence of febrile nonhemolytic transfu-
sion reactions31-35 (see Chapters 8, 21, and
27). These filters contain multiple layers
of synthetic nonwoven fibers that selec-
tively retain leukocytes but allow red cells
or platelets to pass, depending on the fil-
ter type. Selectivity is based on cell size,
surface tension characteristics, the differ-
ences in surface charge, density of the
blood cells, and, possibly, cell-to-cell in-
teractions and cell activation/adhesion
properties.36 Because filters for red cells
and filters for platelets do not use the
same technology for leukocyte removal
and may have strict priming and flow rate
requirements, they must be used only
with their intended component and only
according to the manufacturer’s direc-
tions.37
The use of these filters at the bedside is
more complex than the use of standard in-
fusion sets. The filters are expensive and
can be ineffective or may clog, if improp-
erly used.38-40 Those designed only for grav-
ity infusion should not be used with infu-
sion pumps or applied pressure. A quality
control program that measures the effec-
tiveness of leukocyte reduction is impor-
528 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
tant, but impractical, at the bedside; there-
fore, adherence to proper protocol is very
important.
Blood Warmers
It is desirable for the medical staff of the
transfusion service to participate in the
assessment and selection of transfusion
equipment and ensure that such items
are included in the facility’s quality assur-
ance program. The performance of de-
vices such as blood warmers or infusion
pumps must be validated before the
equipment is used and must be moni-
tored regularly throughout the facility to
identify malfunctions and ensure appro-
priate use. This characteristically requires
cooperation among personnel of several
hospital departments, including transfu-
sion medicine, nursing, anesthesiology,
quality assurance, and clinical engineer-
ing.
Patients who receive blood or plasma at
rates faster than 100 mL/minute for 30
minutes are at increased risk for cardiac ar-
rest unless the blood is warmed.41 Rapid in-
fusion of large volumes of cold blood can
lower the temperature of the sinoatrial
node to below 30 C, at which point an ar-
rhythmia can occur.
Transfusions at such rapid rates gener-
ally occur only in the OR or trauma settings.
There is no evidence that patients receiving
1 to 3 units of blood over several hours have
a comparable risk for arrhythmias; there-
fore, routine warming of blood is not rec-
ommended.42 Several types of blood
warmers are available: thermostatically
controlled waterbaths; dry heat devices
with electric warming plates; and high-vol-
ume countercurrent heat exchange with wa-
ter jackets.43 Blood warming devices must
not raise the temperature of blood to a level
that causes hemolysis.29(p6)
Devices should have a visible thermom-
eter and, ideally, an audible alarm that
sounds before the manufacturer’s desig-
nated temperature limit is exceeded. The
standard operating procedure for warming
blood should include guidelines on per-
forming temperature and alarm checks,
and instructions on what action to take
when warmers are out of range or the alarm
activates.44 Conventional microwave ovens
and microwave devices for thawing plasma
are not designed for warming other blood
components and can damage red cells.
Electromechanical Infusion Devices
Mechanical pumps that deliver infusions
at a controlled rate are useful, especially
for very slow rates of transfusion used for
pediatric, neonatal, and selected adult
patients. Some pumps use a mechanical
screw drive to advance the plunger of a
syringe filled with blood; others use roller
pumpsorotherformsofpressureapplied
to the infusion tubing. Although some can
be used with standard blood administra-
tion sets, many require special plastic
disposables or tubing supplied by the
manufacturer. Blood filters can be added
to the required setups upstream of the
pumps.
The manufacturer should be consulted
before blood is administered with an infu-
sion pump designed for crystalloid or
colloid solutions. Many induce hemolysis,
but of a magnitude that does not adversely
affect the patient.43 Red cells in components
with high hematocrit and high viscosity are
more likely to be hemolyzed when infused
under pressure than red cells in Whole
Blood or red cell components prepared in a
manner that reduces viscosity, such as ad-
ditive solutions.45 Platelets and granulocytes
appear to sustain no adverse effects when
infused with a pumping device.46,47 Proper
Chapter 22: Administration of Blood and Components 529
Copyright © 2005 by the AABB. All rights reserved.
training of personnel and appropriate poli-
cies for maintenance and quality control
should reduce the chances of damage to
transfused components.
Pressure Devices
Urgent transfusion situations may require
flow rates faster than gravity can provide.
The simplest method to speed infusion is
to use an administration set with an inline
pump that the transfusionist squeezes by
hand. Pressure bags specially designed as
compression devices are also available.
These devices operate much like blood
pressure cuffs except that they completely
encase the blood bag and apply pressure
more evenly to the bag’s surface. Such de-
vices should be carefully monitored dur-
ing use because pressures greater than
300 mm Hg may cause the seams of the
blood bag to rupture or leak and air em-
bolism is a concern. Large-bore needles
are traditionally recommended for venous
access when the use of external pressure
is anticipated, but recent data question
this practice.11 Manually forcing red cells
through a small-gauge line has been
shown to cause hemolysis.45
Devices for intraoperative and postoper-
ative blood collection are discussed in
Chapter 5.
Compatible IV Solutions
AABB Standards for Blood Banks and
Transfusion Services29(p48) and the Circular
of Information for the Use of Human
Blood and Blood Components48 are explicit
in stating that medications must not be
added to blood or components. If red cells
require dilution to reduce their viscosity
or if a component needs to be rinsed from
the blood bag or tubing, normal saline
(0.9% sodium chloride injection, USP)
can be used. Red cells prepared with an
additive solution (AS) ordinarily do not
require dilution. These red cell compo-
nents have a hematocrit of approximately
60%. Other solutions intended for intrave-
nous use may be added to blood or com-
ponents or may come into contact with
blood in an administration set only if they
have been approved for this use by the
FDA or if there is documentation to show
that their addition to blood is safe and ef-
ficacious.29(p6),48 Calcium-free, isotonic
electrolyte solutions that meet the above
requirements also may be used, but they
usually are more expensive than saline
and offer little benefit in routine transfu-
sion. Lactated Ringer’s solution, 5% dex-
trose, and hypotonic sodium chloride so-
lutions should not be added to blood.
Dextrose solution may cause red cells to
clump in the tubing and, more important,
to swell and hemolyze as dextrose and as-
sociated water diffuse from the medium
into the cells. Lactated Ringer’s solution
contains enough ionized calcium (3
mEq/L) to overcome the chelating agents
in anticoagulant-preservative or additive
solutions, which results in clot develop-
ment.49-51
Patient Care During Transfusion
The transfusionist should either remain
with, or be in a position to closely observe,
the patient for at least the first 15 minutes
of the infusion. The transfusion should be
started slowly at a rate of approximately 2
mL/minute except during urgent restora-
tion of blood volume. Catastrophic reac-
tions from acute hemolysis, anaphylaxis,
transfusion-related acute lung injury, or
bacterial contamination can become ap-
parent after a very small volume enters
the patient’s circulation (see Chapter 27).
After the first 15 minutes, some institu-
tions record vital signs, but this is unneces-
530 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
sary if the patient’s condition is satisfactory.
Therateofinfusioncanbeincreasedto
that specified in the order or to be consis-
tent with institutional practice (approxi-
mately 4 mL/minute). The desirable rate of
infusion depends upon the patient’s blood
volume, cardiac status, and hemodynamic
condition. No experimental or clinical data
exist to support a specific time restriction;
however, the Circular of Information48 gives
4 hours as the maximum duration for an
infusion. Maximum time should not be
confused with recommended time. Most
RBC units are transfused within 1 to 2
hours, whereas platelet or plasma transfu-
sions are commonly administered over a
shorter period (30 to 60 minutes). However,
there is no physiologic reason to administer
compatible red cells more slowly than
plasma or platelets and rapid infusion of
these products may increase the risk of ad-
verse events. If rapid transfusion is needed,
blood can be infused as rapidly as the pa-
tient’s circulatory system will tolerate and
the type of vascular access will allow.
If it is anticipated that an infusion time
of longer than 4 hours may be required, the
physician covering the transfusion service
should be notified to assess the specific
clinical situation. Administration rates are
calculated by counting the drops per min-
ute in the drip chamber and dividing this
number by the “drop/mL” rating of the in-
fusion system. Blood may flow more slowly
than desired as a result of obstruction of
the filter or when there is excessive viscos-
ity of the component. Steps to investigate
and correct the problem include the follow-
ing:
■Elevate the blood container to in-
crease hydrostatic pressure.
■Check the patency of the needle.
■Examine the filter of the administra-
tion set for excessive debris.
■Consider the addition of 50 to 100
mL of saline to a preparation of red
cells, if there is an order permitting
such addition.
Clinical personnel should continue to
observe the patient periodically throughout
the transfusion (eg, every 30 minutes) and
up to an hour after completion.
Action for Suspected Reactions
Most transfusions proceed without com-
plication, but when adverse reactions do
occur, medical and nursing staff must be
prepared to deal with them immediately.
Different types of reactions, their etiology,
symptoms, treatment, and prevention are
discussed in Chapter 27. Because severity
can vary significantly and symptoms are
not specific, all transfusions must be care-
fullymonitoredandstoppedassoonasa
reaction is suspected. It may be helpful to
summarize common symptoms and the
immediate steps to take on the transfu-
sion form that accompanies the unit. This
eliminates the need to search for instruc-
tions and helps standardize patient care
in an urgent situation.
Post-Administration Events
After each unit of blood has been infused,
personnel should measure vital signs; re-
cord the time, the volume, and the com-
ponent given; record the patient’s condi-
tion; and record the identity of the person
who stopped the transfusion (if not the
transfusionist), made the observations,
and measured and recorded vital signs.
Many transfusion services require that a
copy of the completed transfusion form
be returned to the laboratory to docu-
ment the unit’s disposition for laboratory
records. The empty blood bag need not be
returned after uncomplicated transfu-
sions, but bags, tubing, and attached so-
lutions should be returned to the transfu-
sion service if a complication occurs.
Chapter 22: Administration of Blood and Components 531
Copyright © 2005 by the AABB. All rights reserved.
Some transfusion services choose to have
all bags returned following all transfu-
sions in order to investigate reactions that
may not be evident at the time of transfu-
sion. Proper biohazard precautions
should be used in the handling of entered
containers and used administration sets.
The patient should remain under obser-
vation after the transfusion has been
completed, if this is practical. Because not
all severe reactions are immediately ap-
parent, all patients who receive transfu-
sionsinoutpatientorhomecaresettings,
or their caretakers, must be given clearly
written instructions outlining posttransfu-
sion care, a description of the signs and
symptoms of acute and delayed reactions,
and the appropriate action to take if such
are noted.
Quality Assurance
The process of blood administration should
begin and end with patient safety in
mind, starting with the generation of an
appropriate order; continuing through
collection of the patient’s pretransfusion
specimen, preparation and delivery of the
unit, identification of the unit with the re-
cipient, and selection and proper use of
equipment; and concluding with patient
care during the transfusion and mainte-
nance of appropriate records. Policies,
procedures, training, and assessment are
all critical to this process and must be
monitored as parts of blood usage review.
Periodic auditing of the blood adminis-
tration process and forms should be per-
formed in order to identify patterns of non-
conformance. Errors, regardless of clinical
outcome, should be subjected to root-cause
analysis because such errors are often in-
dicative of systematic problems.
References
1. Sazama K. Practical issues in informed con-
sent for transfusion. Am J Clin Pathol 1997;
107:572-4.
2. Holland PV. Consent for transfusion: Is it in-
formed? Transfus Med Rev 1997;11:274-85.
3. Williams FG. Consent for transfusion: A duty
of care. Br Med J 1997;315:380-1.
4. Code of Federal Regulations. 42 CFR
482.23(c)(2). Washington, DC: US Govern-
ment Printing Office, 2004 (revised annually).
5. Wang SE, Lara PN, Lee-Ow A, et al. Aceta-
minophen and diphenhydramine as pre-
medication for platelet transfusions: A pro-
spective randomized double-blind
placebo-controlled trial. Am J Hematol
2002;70: 191-4.
6. Agostini JV, Leo-Summers LS, Inouye SK.
Cognitive and other adverse effects of
diphenhydramine use in hospitalized older
patients. Arch Intern Med 2001;161:2091-7.
7. Patterson BJ, Freedman J, Blanchette V, et al.
Effect of pre-medication guidelines and
leukoreduction on the rate of febrile non-
hemolytic platelet transfusions. Transfus Med
2000;10:199-206.
8. WilcoxGJ,BarnesA,ModanlouH.Does
transfusion using a syringe infusion pump
and small gauge needle cause hemolysis?
Transfusion 1981;21:750-1.
9. Herrera AJ, Corless J. Blood transfusions: Ef-
fect of speed of infusion and of needle gauge
on hemolysis. J Pediatr 1981;99:757-8.
10. de la Roche MR, Gauthier L. Rapid transfu-
sion of packed red blood cells: Effects of dilu-
tion, pressure, and catheter size. Ann Emerg
Med 1993;22:1551-5.
11. Frelich R, Ellis MH. The effect of external
pressure, catheter gauge, and storage time on
hemolysis in RBC transfusion. Transfusion
2001;41:799-802.
12. AuBuchon JP, Kruskall MS. Transfusion
safety: Realizing efforts with risks. Transfu-
sion 1997;37:1211-15.
13. McClelland DBL, Phillips P. Errors in blood
transfusion in Britain: Survey of hospital
haematology departments. Br Med J 1994;
308:1205-6.
14. LindenJV,WagnerK,VoytovichAE,Sheehan
J.TransfusionerrorsinNewYorkState:An
analysis of 10 years’ experience. Transfusion
2000;40:1207-13.
15. Sazama K. Reports of 355 transfusion-associ-
ated deaths: 1976-1985. Transfusion 1990:30:
583-90.
16. McManigalS,SimmsKL.Intravascular
hemolysis secondary to ABO incompatible
532 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
platelet products. Am J Clin Pathol 1999;11:
202-6.
17. Renner SW, Howanitz PJ, Bachner P. Wrist
band identification error reporting in 712
hospitals. Arch Pathol Lab Med 1993;117:
573-7.
18. Vicki C, Bower J. Blood administration in
perioperative settings. AORN J 1997;66:133-
43.
19. Baele PL, De Bruyere M, Deneys V, et al. Bed-
side transfusion errors. Vox Sang 1994;66:117-
21.
20. Murphy MF, Atterbury CLJ, Chapman JF, et al.
The administration of blood and blood com-
ponents and the management of transfused
patients. Transfus Med 1999;9:227-38.
21. Ibojie J, Urbaniak SJ. Comparing near misses
with actual mistransfusion events: A more ac-
curate reflection of transfusion error. Br J
Haematol 2000;108:458-60.
22. GallowayM,WoodsR,WhiteheadS,etal.An
audit of error rates in a UK district hospital
transfusion laboratory. Transfus Med 1999;9:
199-203.
23. Wenz B, Burns ER. Improvement in transfu-
sion safety using a new blood unit and and
patient identification system as part of safe
transfusion practice. Transfusion 1991;31:
401-3.
24. Jensen NJ, Crosson JT. An automated system
for bedside verification of the match between
patient identification and blood unit identifi-
cation. Transfusion 1996;36:216-21.
25. Patterson ES, Cook RI, Render ML. Improving
patient safety by identifying side effects from
introducing bar coding in medication admin-
istration. J Am Med Inform Assoc 2002;9:
540-3.
26. Shulman IA, Lohr K, Derdiarian A, Picukaric
JM. Monitoring transfusionist practices: A
strategy for improving transfusion safety.
Transfusion 1994;34:11-15.
27. Whitsett CF, Robichaux MG. Assessment of
blood administration procedures: Problems
identified by direct observation and adminis-
trative incident reporting. Transfusion 2001;
41:581-6.
28. Broods JP, Combest TG. In-service training
with videotape is useful in teaching transfu-
sion medicine principles. Transfusion 1996;
36:739-42.
29. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
30. Schmidt WF, Kim HC, Tomassini N, Schwartz
E. Red blood cell destruction caused by a mi-
cro-pore blood filter. JAMA 1982;248:1629-32.
31. Stack G, Pomper GJ. Febrile allergic and non-
immune transfusion reactions. In: Simon TL,
DzikWH,StowellCP,etal,eds.Principlesof
transfusion medicine. 3rd ed. Baltimore, MD:
Williams and Wilkins, 2002:831-51.
32. Novotny VM, van Doorn R, Witvliet MD, et al.
Occurrence of allogeneic HLA and non-HLA
antibodies after transfusion of prestorage fil-
tered platelets and red blood cells: A prospec-
tive study. Blood 1995;85:1736-41.
33. Brand A, Claas FH, Voogt PJ, et al. Alloim-
munization after leukocyte-depleted multi-
ple random donor platelet transfusions. Vox
Sang 1998;54:160-6.
34. Bowden RA, Slichter SJ, Sayers M, et al. A
comparison of leukocyte-reduced and
cytomegalovirus (CMV) seronegative blood
products for the prevention of transfusion-
associated CMV infection after marrow trans-
plant. Blood 1995;86:3598-603.
35. Narvios AB, Przepiorka K, Tarrand J, et al.
Transfusion support using filtered un-
screened blood products for cytomegalovirus
negative allogeneic marrow transplant recipi-
ents. Bone Marrow Transplant 1998;22:575-7.
36. Buril A, Beugeling T, Feijen J, van Aken WG.
The mechanisms of leukocyte removal by fil-
tration. Transfus Med Rev 1995;9:145-66.
37. Dzik WH. Leukoreduced blood components:
Laboratory and clinical aspects. In: Simon
TL,DzikWH,StowellCP,etal,eds.Principles
of transfusion medicine. 3rd ed. Baltimore,
MD: Williams and Wilkins, 2002:270-87.
38. Sprogre-Jakobsen U, Saetre AM, Georgsen J.
Preparation of white cell-reduced red cells by
filtration: Comparison of a bedside filter and
two blood bank filter systems. Transfusion
1995;35:421-6.
39. Ledent E, Berlin G. Inadequate white cell re-
duction by bedside filtration of red cell con-
centrates. Transfusion 1994;34:765-8.
40. Kao KJ, Hudson S, Orsini LA, et al. Effect of in
vitro storage time of platelet concentrates on
clogging of white cell reduction filters. Trans-
fusion 1994;34:740-1.
41. Boyan CP, Howland WS. Cardiac arrest and
temperature of bank blood. JAMA 1963;183:
58-60.
42. Calhoun L. Blood product preparation and
administration. In: Petz LD, Swisher SN,
Kleinman S, eds. Clinical practice of transfu-
sionmedicine.3rded.NewYork:Churchill
Livingstone, 1996:305-33.
43. Iserson KV, Huestis DW. Blood warming: Cur-
rent applications and techniques. Transfu-
sion 1991;31:558-71.
44. UhlL,PaciniD,KruskallMS.Acomparative
study of blood warmer performance. Anes-
thesiology 1992;77:1022-8.
45. BurchKJ,PhelpsSJ,ConstanceTD.Effectof
an infusion device on the integrity of whole
Chapter 22: Administration of Blood and Components 533
Copyright © 2005 by the AABB. All rights reserved.
blood and packed red cells. Am J Hosp Pharm
1991;48:92-7.
46. Snyder EL, Ferri PM, Smith EO, Ezekowitz MD.
Use of electromechanical infusion pump for
transfusion of platelet concentrates. Transfu-
sion 1984;24:524-7.
47. Snyder EL, Malech HL, Ferri PM, et al. In vitro
function of granulocyte concentrates follow-
ing passage through an electromechanical in-
fusion pump. Transfusion 1986;26:141-4.
48. American Association of Blood Banks, Amer-
ica’s Blood Centers, American Red Cross. Cir-
cular of information for the use of human
blood and blood components. Bethesda, MD:
AABB, 2002.
49. Ryden SE, Oberman HA. Compatibility of
common intravenous solutions with CPD
blood. Transfusion 1975;15:250-5.
50. Dickson DN, Gregory MA. Compatibility of
blood with solutions containing calcium. S
Afr Med J 1980;57:785-7.
51. Strautz RL, Nelson JM, Meyer EA, Shulman
IA. Compatibility of ADSOL-stored red cells
with intravenous solutions. Am J Emerg Med
1989;7:162-4.
534 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 23: Perinatal Issues in Transfusion Practice
Chapter 23
Perinatal Issues in
Transfusion Practice
PREGNANCY PRESENTS SPECIAL
immunohematologic problems for
the transfusion service. The mother
may exhibit alloimmunization to antigens
on fetal cells, and the fetus may be affected
by maternal antibodies provoked by pre-
vious pregnancies, by previous or present
transfusions, or by the ongoing pregnancy.
This chapter discusses hemolytic disease
of the fetus and newborn (HDFN) and neona-
tal alloimmune thrombocytopenia (NAIT)—
the two primary immunohematologic
concerns during the perinatal period. Also
included is a brief discussion of neonatal
thrombocytopenia secondary to maternal
idiopathic thrombocytopenic purpura.
Hemolytic Disease of the
Fetus and Newborn
In HDFN, fetal red cells become coated
with IgG alloantibody of maternal origin,
directed against a paternally inherited an-
tigen present on the fetal cells that is
absent from maternal cells. The IgG-
coated cells may undergo accelerated de-
struction both before and after birth, but
the severity of the disease can vary from
serologic abnormalities detected in an
asymptomatic infant to intrauterine death.
Pathophysiology
Accelerated red cell destruction stimulates
increased production of red cells, many of
which enter the circulation prematurely as
nucleated cells, hence the term “erythro-
blastosis fetalis.” Severely affected fetuses
may develop generalized edema, called
“hydrops fetalis.” In HDFN resulting from
anti-D, erythropoiesis in the fetal liver
may be so extensive that portal circulation
is disrupted and albumin synthesis im-
paired, thereby reducing plasma colloid
osmotic pressure. The severe anemia may
cause cardiovascular failure, tissue hypo-
535
23
Copyright © 2005 by the AABB. All rights reserved.
xia, and death in utero. Intrauterine trans-
fusion may be lifesaving in these circum-
stances. If live-born, the severely affected
infant exhibits profound anemia and
heart failure.1Less severely affected in-
fants continue to experience accelerated
red cell destruction, which generates
large quantities of bilirubin. Unlike HDFN
due to anti-D, HDFN due to anti-K1 re-
sults from suppression of fetal erythropo-
iesis in addition to causing peripheral red
cell destruction.2
Before birth severs the communication
between maternal and fetal circulation, fe-
tal bilirubin is processed by the mother’s
liver. At birth, the infant’s immature liver is
incapable of conjugating the amount of bil-
irubin that results from destruction of anti-
body-coated red cells. Unconjugated biliru-
bin is toxic to the developing central
nervous system (CNS), causing brain dam-
age referred to as “kernicterus.” For the
live-born infant with HDFN and rising lev-
els of unconjugated bilirubin, kernicterus
may pose a greater clinical danger than the
consequences of anemia.3Prematurity, aci-
dosis, hypoxia, and hypoalbuminemia in-
crease the risk of CNS damage. Decisions
about undertaking exchange transfusion
are based primarily on the bilirubin level,
the rate of bilirubin accumulation, and, to a
lesser degree, on the severity of the anemia.
Recently, the American Academy of Pediat-
rics has published guidelines aimed toward
preventing and managing hyperbiliru-
binemia in infants ≥35 weeks of gestation.4
Mechanisms of Maternal Immunization
HDFN is often classified into three cate-
gories, on the basis of the specificity of the
causative IgG antibody. In descending or-
der of potential severity, they are:
1. D hemolytic disease caused by anti-D
alone or, less often, in combination
with anti-C or anti-E. (The Rh blood
group is discussed in greater detail
in Chapter 14.)
2. “Other” hemolytic disease caused by
antibodies against other antigens in
the Rh system or against antigens in
other systems; anti-c and anti-K1 are
most often implicated.5
3. ABOHDFNcausedbyanti-A,Bina
group O woman or by isolated anti-A
or anti-B.
In all but ABO HDFN, maternal antibod-
ies reflect alloimmunization by pregnancy
or transfusion. Rising titers of antibody can
be documented, at least in the first affected
pregnancy, and the infant may be symp-
tomatic at birth as a result of effects on the
fetus in utero. In contrast, ABO fetomater-
nal incompatibility cannot be diagnosed
during pregnancy and the infant is rarely
symptomatic at birth.
Pregnancy as the Immunizing Stimulus
Pregnancy causes immunization when fe-
tal red cells, possessing a paternal antigen
foreign to the mother, enter the maternal
circulation as a result of fetomaternal he-
morrhage (FMH). FMH occurs in the vast
majority of pregnancies, usually during the
third trimester and during delivery.6De-
livery is the most common immunizing
event, but fetal red cells can also enter the
mother’s circulation after amniocentesis,
spontaneous or induced abortion, chori-
onic villus sampling, cordocentesis, rup-
ture of an ectopic pregnancy, and blunt
trauma to the abdomen.
Immunogenic Specificities. The antigen
that most frequently induces immunization
is D, but, in theory, any red cell antigen
present on fetal cells and absent from the
mother can stimulate antibody production.
One retrospective study determined that
there was a 0.24% prevalence of production
of clinically significant antibodies other
than anti-D during pregnancy. Because
536 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
other red cell antigens are less immuno-
genic than D, sensitization is more likely to
result from exposure to a large volume of
red cells, such as during blood transfusion.
Immunization to D, on the other hand, can
occur with volumes of fetal blood less than
0.1 mL.7
Frequency of Immunization. The proba-
bility of immunization to D correlates with
thevolumeofD-positiveredcellsentering
the D-negative mother’s circulation.6The
overall incidence of D sensitization in un-
treated D-negative mothers of D-positive
infantsisabout16%;1.5%to2%become
sensitized at the time of their first delivery,
an additional 7% become sensitized within
6 months of the delivery, and the final 7%
become sensitized during the second af-
fected pregnancy.8The sensitization during
the second affected pregnancy probably re-
flects primary immunization during the
first D-positive pregnancy and delivery that
happened without production of detectable
levels of antibody. The small numbers of
D-positive fetal red cells entering the ma-
ternal circulation early during the second
affected pregnancy constitute a secondary
stimulus sufficient to elicit overt production
of IgG anti-D. In susceptible women not
immunized after two D-positive pregnan-
cies, later pregnancies may be affected but
with diminished frequency. The incidence
of the more common genotypes in D-posi-
tive individuals can be found in Table 14-4.
This can be used to get a general idea of the
likelihood of how often an infant with a
D-positive father and D-negative mother
will express the D antigen.
Once immunization has occurred, suc-
cessive D-positive pregnancies often mani-
fest HDFN of increasing severity, particu-
larly between the first and second affected
pregnancies. After the second affected
pregnancy, the history is predictive of out-
come, although, in rare instances, some
women have a stable or diminishing pat-
tern of clinical disease in subsequent preg-
nancies.
Effect of ABO Incompatibility. Rh im-
munization of untreated D-negative women
occurs less frequently after delivery of an
ABO-incompatible D-positive infant than
when the fetal cells are ABO-compatible
with the mother. ABO incompatibility be-
tween mother and fetus has a substantial
but not absolute protective effect against
maternal immunization by virtue of the in-
creased rate of red cell destruction by anti-A
or anti-B. The rate of immunization is de-
creased from 16% to between 1.5% and 2%.7
Transfusion as the Immunizing Stimulus
It is extremely important to avoid trans-
fusing D-positive whole blood or red cells
to D-negative females of childbearing
potential because anti-D stimulated by
transfusion characteristically causes severe
HDFN in subsequent pregnancies with a
D-positive fetus. Red cells present in pla-
telet or granulocyte concentrates can con-
stitute an immunizing stimulus; if com-
ponents from D-positive donors are
necessary for young D-negative female
recipients, Rh immunoprophylaxis should
be considered. This is discussed further in
Chapter 21.
The risk of immunization to a red cell
antigen other than D after an allogeneic red
cell transfusion has been estimated to be
1% to 2.5% in the general hospital popula-
tion.7This will endanger the fetus only if the
antibody is IgG and directed against an an-
tigen that is also present on the fetal red
cells. For a couple planning to have chil-
dren, the woman should not be transfused
with red cells from her sexual partner or his
blood relatives. This form of directed dona-
tion increases the risk that the mother will
be immunized to paternal red cell, leuko-
cyte, and/or platelet antigens, which could
cause alloimmune cytopenias in future
Chapter 23: Perinatal Issues in Transfusion Practice 537
Copyright © 2005 by the AABB. All rights reserved.
children who share the same paternal anti-
gens. Programs using parents as directed
donors for their sick newborns deserve spe-
cial consideration because of these unique
issues in the face of strong parental desires.9,10
ABO Antibodies
The IgG antibodies that cause ABO HDFN
nearly always occur in the mother’s circu-
lation without a history of prior exposure
to human red cells. ABO HDFN can occur
in any pregnancy, including the first. It is
restricted almost entirely to group A or B
infants born to group O mothers because
group O individuals make the IgG anti-
body, anti-A,B. Group A or B mothers with
an A- or B-incompatible fetus predomi-
nantly produce IgM antibody, with only
small amounts of IgG antibody capable of
crossing the placenta.
Prenatal Evaluation
Maternal History
Information about previous pregnancies or
blood transfusions is essential in evaluat-
ing fetal risk. Invasive tests, which carry
risk to the fetus, should be performed
only for pregnancies in which the fetus is
at risk for HDFN, by history and/or sero-
logic testing. For a woman with a history
of an infant with hydrops fetalis due to
anti-D, there is a 90% or more chance of a
subsequent fetus being similarly affected.5
In contrast, during the first sensitized
pregnancy, the risk of a hydropic fetus is
8% to 10%. Experience with other allo-
antibodies has not been as extensive as with
anti-D; in some series, anti-c and anti-K1
were by far the most common causes of
severe HDFN, other than anti-D.5,11
Serologic Studies
Alloantibodies capable of causing HDFN
can be detected during pregnancy. Initial
studies should be performed on all preg-
nant women as early in pregnancy as
possible; they should include tests for
ABO and D, and a screen for unexpected
red cell antibodies.12 If a woman’s red cells
are not directly agglutinated by anti-D,
testing for weak D is not required. When
testing for weak D is not performed,
women with weak D will be labeled as D
negative, although they are in fact D posi-
tive; the only potential negative outcome
is that these women will receive unneces-
sary Rh Immune Globulin (RhIG). Women
with some partial D phenotypes, such as
DVI, will also most likely type as D negative
in direct tests, and, in the absence of weak
D testing, these women are also candi-
dates for RhIG antenatal prophylaxis.
With the application of molecular tech-
niques, knowledge of the RHD gene is
evolving. Not all weak D red cells are the re-
sult of a decreased expression of the D anti-
gen, but, rather, some have altered RhD
proteins. Consequently, these patients are
at risk for immunization when exposed to
the D antigen, explaining formation of
anti-D in some patients classified as weak
D. As more is learned about the D antigen,
the distinction between partial D and weak
Disblurring.
13,14
Whether or not prophylaxis would be
successful in the setting of partial D remains
unknown.Theappropriatedoseisalsosub
-
ject to speculation. As a result, some practi-
tioners administer RhIG to these women,
whereas others consider it unnecessary.
Very rarely, a mother with partial D antigen
produces anti-D as a result of pregnancy.
If weak D testing is performed and the
test is clearly positive, the woman should
be regarded as D positive. If testing for
weak D is not performed, women whose
red cells do not react in direct tests with
anti-D can be considered candidates for
RhIG prophylaxis. Some laboratories con-
tinuetodoweakDtestingtoavoidconfu
-
538 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
sion in the interpretation of the FMH screen
during postpartum testing.12 Chapter 14
contains a more complete discussion of the
Rh blood group.
A woman should be classified as D posi-
tive if the test for either D or weak D is posi-
tive. If a D-negative woman has a negative
initial antibody screen, the test can be re-
peated at 28 weeks’ gestation before ad-
ministration of RhIG to detect immuniza-
tion that might have occurred before 28 weeks,
in accordance with AABB recommendations.
Because the incidence of immunization
during this period of pregnancy is extremely
low, the American College of Obstetricians
and Gynecologists (ACOG) points out that
no data exist that support the cost-effective-
ness of this practice.15 Repeat antibody
screening of D-positive women may be rec-
ommended if there is a history of clinically
significant red cell antibodies associated
with HDFN, previous blood transfusion, or
trauma to the abdomen.
Antibody Specificity. All positive screens
for red cell antibodies require identification
of the antibody.12 Themerepresenceofan
antibody, however, does not indicate that
HDFN will occur. Non-red-cell-stimulated
IgM antibodies, notably anti-Leaand anti-I,
are relatively common during pregnancy but
do not cross the placenta. In addition, the
fetal red cells may lack the antigen corre-
sponding to the mother’s antibody; the like-
lihood of fetal involvement can often be
predicted by typing the father’s red cell an-
tigens.16 The laboratory report on prenatal
antibody studies should include sufficient
information to aid the clinician in deter-
mining the clinical significance of the iden-
tified antibody.
Typing the Fetus. ThefetalDtypecanbe
established by using the polymerase chain
reaction (PCR) to amplify DNA obtained
from amniotic fluid, chorionic villus sam-
ples, or by serologic typing of fetal blood
obtained by cordocentesis.17 Chorionic villus
sampling is discouraged because it causes
FMH and has been associated with more
severe HDFN. Amniotic fluid samples are
recommended over cordocentesis because
cordocentesis has a fourfold or higher rate
of perinatal loss over amniocentesis.18 The
use of molecular techniques can help de-
tect variations in the RHD gene that might
go undetected using serology alone. It is
preferable that both paternal and maternal
blood samples accompany the fetal sam-
ples.19 Fetal DNA typing is also available for
Jka/Jkb,20 K1/K2,21 c,22 and E/e23 antigens. A
more recent development in fetal RhD typ-
ing involves the isolation of free fetal DNA
in the maternal serum. Although not rou-
tinely available in the United States at this
time, this will likely replace amniocentesis
for fetal genotyping in the near future.24
Maternal Antibody Titer
Antibody titrations can help in decisions
about the performance and the timing of
invasive procedures, especially if the anti-
body is anti-D. The antibody titer should
be established in the first trimester to
serve as a baseline, and the specimen
should be frozen for future comparisons
(see Method 5.3).7Because invasive tests
will not be undertaken before 16 to 18
weeks’ gestation, no further titration is in-
dicated until this time. The true signifi-
cance of an antibody titer in maternal se-
rum is controversial because some studies
have shown poor correlation between the
level of the titer and effects on the fetus.
For antibodies other than anti-D, critical
titers have not been identified, although a
critical titer similar to that used in cases
of anti-D alloimmunization is often uti-
lized.25 These techniques continue to be
performed because they represent a non-
invasive way to try to assess the presence
and severity of alloimmunization. When
performed, it is important that successive
Chapter 23: Perinatal Issues in Transfusion Practice 539
Copyright © 2005 by the AABB. All rights reserved.
titrations use the same methods and test
cells of the same red cell phenotype. Test-
ing previously frozen serum samples in
parallel with a current specimen mini-
mizes the possibility that changes in the
titer result from differences in technique.
The critical titer for anti-D (the level be-
low which HDFN and hydrops fetalis are
considered so unlikely that no further in-
vasive procedures will be undertaken)
should be selected at each facility and is
usually 16 or 32 in the antihuman globu-
lin phase.26,27 Follow-up testing is recom-
mended for any titer greater than 8.26 The
critical titer for anti-K1 may be lower than
anti-D, typically a value of 8.25,28 Currently,
it is not recommended that gel technol-
ogy be used for prenatal antibody titra-
tion because of the lack of data showing a
correlation between gel and tube aggluti-
nation titers.12
Other Measures of HDFN Severity
Numerous laboratory procedures have been
investigated to improve the accuracy of
predicting the severity of hemolysis.29 The
antibody titer discussed above is not al-
ways reliable, nor is the serial change in
titer. Functional assays, including measures
of adherence, phagocytosis, antibody-de-
pendent cytotoxicity, and chemilumines-
cence have been investigated, but their use
has been limited and remains controver-
sial. These procedures are usually per-
formed in referral centers and may be use-
ful when additional information is required
to manage difficult and complex cases.
Amniotic Fluid Analysis
A good index of intrauterine hemolysis
and fetal well-being is the level of biliru-
bin pigment found in amniotic fluid ob-
tained by amniocentesis. Amniocentesis
is usually performed in alloimmunized
women who have a history of previously
affected pregnancies or have an antibody
titer at or above the critical titer.28 Because
fetal anemia secondary to K1 alloimmuni-
zation is not always associated with ele-
vated levels of bilirubin in amniotic fluid,
it has been recommended that fetal blood
sampling be used instead of serial amnio-
centesis when anti-K1 is detected in a
pregnant woman.2
Amniotic fluid is obtained by inserting a
long needle through the mother’s abdomi-
nal wall and uterus into the uterine cavity
under continuous ultrasound guidance.
The aspirated fluid is scanned spectropho-
tometrically at wavelengths of 350 to 700
nm. Peak absorbance of bilirubin is at 450
nm. An increase in optical density from the
projected baseline at 450 nm (∆OD450)isa
measure of the concentration of bile pig-
ments.30,31 The ∆OD450 value is plotted on a
graph against the estimated length of gesta-
tion, because bile pigment concentration
has different clinical significance at differ-
ent gestational ages. Liley’s system31 (Fig
23-1) of predicting the severity of fetal dis-
ease based on the ∆OD450 has been used for
decades. It delineates three zones to esti-
mate severity of disease: a top zone (zone 3)
indicates severe disease, the bottom zone
(zone 1) indicates mild or no disease, and
mid-zone (zone 2) values require repeat de-
termination to establish a trend. This
method is applicable to pregnancies from
27 weeks through term. Queenan et al32
have proposed a system for managing
D-immunized pregnancies based on the
∆OD450 from as early as 14 weeks’ gestation.
They identified four zones (Fig 23-2), with
early invasive intervention recommended if
∆OD450 values fall in the highest zone. With
both systems, the severity of HDFN is more
accurately predicted with serial ∆OD450
measurements than with a single observa-
tion, to evaluate whether readings are fall-
ing, rising, or stable. In general, the higher
the pigment concentration, the more se-
540 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
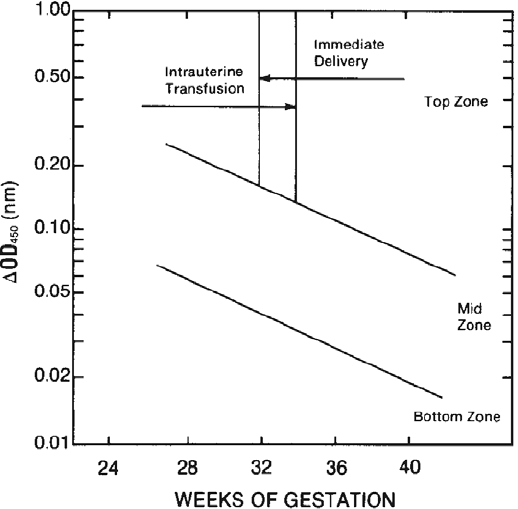
veretheintrauterinehemolysis.Itisimpor
-
tant to perform an ultrasound to establish
the correct gestational age so that the test
can be interpreted and the course of ther-
apy for a particular ∆OD450 will be appro-
priate.18 Alternatively, a ∆OD450 value in the
upper mid-zone of the Liley curve indicates
the need for fetal blood sampling.
Amniocentesis, particularly if the needle
goes through the placenta, or cordocentesis
may cause FMH, which can boost the titer
of existing red cell alloantibody, thereby in-
creasing the severity of HDFN, or inducing
immunization to additional antigens.18
Therefore, when amniocentesis or cordo-
centesis is performed for any reason on a
D-negative woman who does not have
anti-D, Rh immunoprophylaxis should be
given.
Percutaneous Umbilical Blood Sampling
In the early 1980s, the use of sophisticated
ultrasound equipment made it feasible to
direct a needle into an umbilical blood
vessel, preferably the vein at its insertion
into the placenta, and obtain a fetal blood
sample. Percutaneous umbilical blood
sampling (PUBS, or cordocentesis) allows
direct measurement of hematologic and
biochemical variables. Determination of the
fetal hematocrit provides an accurate as-
sessment of the severity of fetal hemolytic
disease.18 It is important to verify that the
sample has been obtained from the fetus.
Fetal and maternal red cells can be distin-
guished because of differences in red cell
size and red cell phenotyping, as well as
bythepresenceoffetalhemoglobin.
33
Chapter 23: Perinatal Issues in Transfusion Practice 541
Figure 23-1. Liley graph for collecting data from amniotic fluid studies. Intrauterine transfusion should
be done if the ∆OD450 value is in the top zone before 32 weeks’ gestation. After 34 weeks, top zone val-
ues indicate immediate delivery. Either intrauterine transfusion or immediate delivery may be indi-
cated for top zone ∆OD450 between 32 and 34 weeks, depending on studies of fetal maturity. Modified
from Liley.31
Copyright © 2005 by the AABB. All rights reserved.
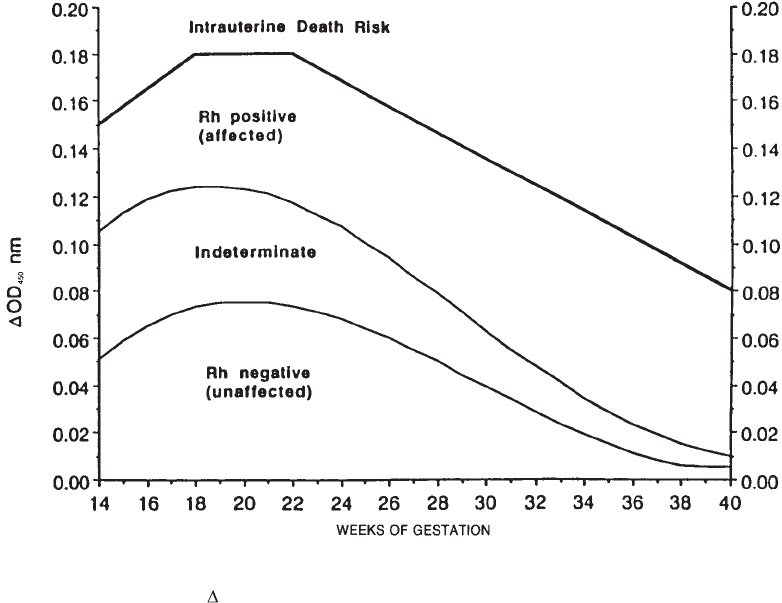
The fetal mortality of intrauterine fetal
blood sampling has been reported to be 1%
to 2%,34 and the procedure carries a high
risk of FMH. Its use is recommended only
for certain circumstances, such as when se-
rial amniotic fluid determinations indicate
severe HDFN, when hydrops is present,
when the D titer is high or rising, or when
HDFN occurred in previous pregnancies. In
addition to diagnosis, PUBS allows treat-
ment of the affected fetus.
Doppler Flow Studies
Because fetal anemia results in increased
cardiac output, several investigators have
measured various blood velocities in fetal
vessels using Doppler ultrasonography to
determine the clinical status of the fetus
in a noninvasive manner.18,35 Recent stud-
ies have found good correlation between
middle cerebral artery (MCA) peak veloc-
ity, fetal hemoglobin, and ∆OD450 read
-
ings.36 Many centers routinely use an MCA
peak systolic velocity value of greater than
1.5 multiples of the median to proceed
with cordocentesis to determine if the fe-
tus is anemic. In such centers, amniocen-
tesis is performed only after 35 weeks’
gestation when MCA Doppler is associ-
ated with a high false-positive rate for the
diagnosis of fetal anemia (Moise K, per-
sonal communication).
Suppression of Maternal
Alloimmunization
Several approaches to suppress maternal
alloimmunization have been attempted,
two of which have limited clinical benefit
542 AABB Technical Manual
Figure 23-2. Amniotic fluid OD450 management zones. (Reproduced with permission from Queenan et
al.32)
Copyright © 2005 by the AABB. All rights reserved.
in reducing maternal antibody levels: in-
tensive plasma exchange and the admin-
istration of immunoglobulin (intravenous)
(IGIV).5,18 Plasma exchange can reduce an-
tibody levels by as much as 75%. Unfortu-
nately, rebound usually follows because
the IgG antibody is mostly extravascular
and antigen exposure may be ongoing.
Plasma exchange has been proposed as a
way to delay the need for fetal interven-
tion, particularly when there is a previous
pregnancy complicated by early hydrops.37
In this setting, plasma exchange can delay
the need for more invasive procedures un-
til the second trimester. The AABB and
the American Society for Apheresis (ASFA)
categorize plasma exchange as treatment
Category III because its efficacy and safety
have not been proven for this indication
(see Chapter 6). With the increasing safety
of intrauterine transfusion through ultra-
sound guidance and the decreasing inci-
dence of HDFN due to anti-D, the use of
plasma exchange as a treatment modality
has declined.
IGIV infusion has also been shown to
stabilize anti-D titers, with best results ob-
tained when started before 28 weeks’ gesta-
tion and when the fetus is not hydropic.38 In
a small study assessing the efficacy of IGIV,
it was found to be well tolerated and there
was a decrease in hemolysis.39 The mecha-
nism of IGIV effect is not clear, although it
may work by saturating placental Fc recep-
tors and inhibiting the transplacental trans-
fer of maternal antibody or by suppressing
ingestion of IgG-coated red cells by the fetal
reticuloendothelial system. An alternative
explanation is that the introduction of
anti-idiotype antibodies modifies maternal
antibody production. IGIV has also been
used with plasma exchange to reduce the
antibody rebound that has been seen fol-
lowing plasma exchange.18 Until larger stud-
ies assessing safety and efficacy can be per-
formed, intrauterine transfusion remains
the mainstay of standard therapy.
Intrauterine Transfusion
Intrauterine transfusion can be performed
by the intraperitoneal route (IPT) or the
direct intravascular approach (IVT) by the
umbilical vein. In many instances, IVT is
the procedure of choice, but there may be
problems of access that make IPT prefera-
ble; a combination may also be used to
minimize peaks and troughs of fetal hema-
tocrit between procedures. Intrauterine
transfusion is seldom feasible before the
20th week of gestation; once initiated,
transfusions are usually administered pe-
riodically until delivery. It is important
that blood is available and ready at the
time of the first diagnostic, and subse-
quent, cordocenteses. If fetal anemia is
detected, an intrauterine transfusion can
be performed at once, minimizing fetal
risks. The interval between transfusions
depends on the presence or absence of
hydrops, the gestational age, and the
amount of blood infused. Good outcomes
areachievedover80%ofthetimeand
94% of nonhydropic fetuses survive.40 Be-
cause intrauterine transfusion carries a 1%
to 2% risk of perinatal loss, it should be
performed only after careful clinical eval-
uation.18,34,41,42 Other perinatal conditions
that have been treated with intrauterine
transfusion include parvovirus infection,
large FMH, and alpha thalassemia.43
Techniques
IPT is performed through a needle passed,
with ultrasonographic monitoring, through
the mother’s abdominal wall into the
abdominal cavity of the fetus. Transfused
red cells enter the fetal circulation through
lymphatic channels that drain the peri-
toneal cavity. In IVT, the umbilical vein is
penetrated under ultrasound guidance,
Chapter 23: Perinatal Issues in Transfusion Practice 543
Copyright © 2005 by the AABB. All rights reserved.

and a blood sample is taken to verify posi-
tioning in the fetal vasculature. Injection
of saline can also confirm correct place-
ment because it can be visualized by ul-
trasound. Blood is infused directly, as ei-
ther a simple transfusion or as a partial
exchange transfusion. IVT can be particu-
larly valuable for very severe cases of
HDFN associated with hydrops fetalis. In
hydropic infants, red cells administered
by IPT are not efficiently absorbed.
Selection of Red Cells
The red cells used should be group O,
D-negative, or negative for the antigen
corresponding to the mother’s antibody if
the specificity is not anti-D. Blood for
intrauterine transfusion should be irradi-
ated (see Chapter 27), and should be cyto-
megalovirus (CMV)-reduced-risk. It may
also be desirable to transfuse blood that is
knowntolackhemoglobinSinorderto
transfuse red cells with maximal oxygen-
transporting capacity, in the setting of low
oxygen tension. For optimal survival of the
transfused cells, blood used for intraute-
rine transfusion should be drawn as re-
cently as possible, generally less than 7
days old.
The hematocrit of the RBCs prepared for
exchange transfusion is usually high to
minimize the chance of volume overload in
the fetus. Washed, irradiated maternal
blood has also been used for intrauterine
transfusion.44 Toremovetheoffendinganti
-
body, the red cells are washed and resus-
pended in saline to a final hematocrit
between 75% and 85%. Washed or degly-
cerolized preparations have been used as a
means to remove plasma, anticoagulant/
preservative solutions, and excess electro-
lytes that might accumulate during pro-
longed storage. Blood for intrauterine
transfusions and all blood and cellular
components subsequently transfused in
the neonatal period should be irradiated to
prevent transfusion-associated graft-vs-host
diseasebecausethefetusisconsideredim
-
munologically naïve and tolerant.45,46
Volume Administered
Thevolumetransfusedvarieswiththe
technique used as well as the fetal size,
initial hematocrit, and gestational age.
For IPT, a volume calculated by the for-
mula V = (gestation in weeks – 20) ×10 mL
appears to be well tolerated by the fetus.
The volume of red cells transfused by IVT
can be calculated by the following for-
mula.47
Fetoplacental volume (mL) =
ultrasound estimated fetal weight
(g) ×0.14
Volume to transfuse (mL) =
Fetoplacental volume ×
(Hct after IVT – Hct before IVT)
Hct of donor cells
where Hct = hematocrit
Transfusion is repeated on the basis of
an estimated decline in fetal hematocrit of
approximately 1% per day in an effort to
maintain the fetal hematocrit in the range
of 27% to 30%.48
Postpartum Evaluation
It may be desirable to collect a sample of
cord blood, preferably by cannulation of
an umbilical vessel at delivery, from new-
bornswherethereisariskofHDFN(eg,
Rh-positive infants born to Rh-negative
mothers, type A and B infants born to
type O mothers). This sample should be
identified as cord blood and labeled in
the delivery suite with the mother’s name,
the date, and two unique forms of identi-
fication for the infant (eg, name and med-
ical record number).
544 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
In cases of suspected HDFN, samples of
both cord and maternal blood should be
tested. When the mother is known to have
antibodies capable of causing HDFN, the
hemoglobin or hematocrit and the biliru-
bin level of cord blood should also be de-
termined. If the mother is D negative and
the infant D positive, the mother’s blood
should be tested to determine the volume
of FMH, as discussed later. Testing cord
blood may present some special problems,
which are described below.
ABO Testing
ABO testing on newborns relies entirely on
red cell typing because ABO antibodies in
cord serum are nearly always of maternal
origin and are IgG. However, in the inves-
tigation of possible HDFN due to ABO in-
compatibility, cord serum should be tested
for antiglobulin-reactive ABO antibodies.
Also, if the infant will receive non-group-O
red cells, testing must be taken to the
antiglobulin phase.49(p42)
D Testing
Newborns who have had successful intra-
uterine transfusions often type at birth as
D negative or very weakly positive be-
cause over 90% of their circulating red
cells may be those of the donors. The ABO
and direct antiglobulin tests may also give
misleading results. If the infant’s red cells
are heavily coated with IgG antibodies, tests
with anti-D may give either false-positive
or false-negative results (see Chapter 14).
Antiglobulin Testing
The direct antiglobulin test (DAT) is usu-
ally strongly positive in HDFN resulting
from anti-D or antibodies to other blood
groups; reactions are much weaker or
even negative in HDFN resulting from
ABO antibodies. However, the strength of
the DAT does not correlate with the sever-
ity of hemolysis, especially in ABO HDFN.
Infants who have received an intrauterine
transfusion may have a weakly positive
DAT with a mixed-field pattern of aggluti-
nation. If the DAT on cord cells is positive,
the antibody can be eluted from the red
cells and tested for specificity. It is not
necessary to make and test an eluate if
the maternal serum has been shown to
contain a single red cell antibody. All clin-
ically significant red cell antibodies in the
maternal serum must be respected. (See
the section on Selection of Blood, later in
this chapter.) If the DAT is positive and
the maternal antibody screen is negative,
investigation should turn toward ABO an-
tibodies or HDFN caused by an antibody
directed against a low-incidence antigen
not present on reagent red cells.
Evaluation of ABO Antibodies. ABO
HDFN may be suspected on clinical
grounds even though the DAT is negative.
Testing the eluate from the cord cells
against A1and B red cells should establish
the diagnosis of ABO HDFN. Cord blood or
peripheral blood serum should be tested by
an indirect antiglobulin technique against
A1, B, and O red cells. The presence of anti-A,
anti-B, or anti-A,B confirms the potential
for ABO HDFN. It is often possible to elute
anti-A and/or anti-B from the infant’s red
cells despite a negative DAT, but this step is
not necessary for the presumptive diagno-
sis.IntherarecasesofABOHDFNthatre
-
quire transfusion, D-compatible group O
blood should be transfused, whether or not
the diagnosis has been serologically con-
firmed. Nonimmune causes of hyperbiliru-
binemia and hemolysis should still be con-
sideredinaninfantwithanegativeDAT
before concluding that it is due to ABO
HDFN because other hematologic disor-
ders might be present.50
Chapter 23: Perinatal Issues in Transfusion Practice 545
Copyright © 2005 by the AABB. All rights reserved.
Antibodies to Low-Incidence Antigens.
If ABO HDFN is ruled out, antibody against
a low-incidence red cell antigen should be
suspected. Testing an eluate or maternal
serum against the father’s red cells with an
antiglobulin technique may provide an an-
swer. Maternal serum must be ABO com-
patible, if it is used. If either or both of these
tests are positive, it indicates that the infant
has an antigen of paternal origin that the
mother lacks, causing her to make an IgG
antibody directed against this antigen. Un-
less the mother has been exposed to red cells
from the father or his blood relations, trans-
fusion would be an unlikely immunizing
event for a low-incidence antigen. Because
there should be no difficulty in obtaining
compatible blood, diagnostic studies can
be performed after initial clinical concerns
have been resolved. If the DAT is positive
and all attempts to characterize a coating
red cell antibody are consistently negative,
causes of a false-positive DAT should be
considered (see Chapter 20).
Exchange Transfusion
Exchange transfusion for HDFN achieves
several desired effects, including:
1. Removal of antibody-coated fetal red
cells.
2. Removalofmaternalantibody.
3. Removal of bilirubin.
4. Replacement of red cells, thereby
treating anemia.
The red cells used for replacement must
be compatible with the causative antibody.
FreshFrozenPlasmaisfrequentlyusedto
reconstitute whole blood because it pro-
vides coagulation factors. Platelet values
should also be monitored and transfusion
used as necessary. Plasma frozen within 24
hours and thawed plasma can be used too,
with the understanding that there might be
decreases in the activity of the labile
clotting Factors V and VIII.
Selection of Blood
In most cases, the mother’s serum is used
for crossmatching and the red cells se-
lected for transfusion are compatible with
her ABO antibodies as well as any addi-
tional antibody(ies) responsible for the
hemolytic process. Group O red cells re-
suspended in AB plasma are commonly
used. In ABO HDFN, the red cells used for
exchange must be group O. If the anti-
body is anti-D, the red cells must be D
negative, but not every exchange transfu-
sion requires group O RBCs. If mother
and infant are ABO-identical, group-spe-
cific red cells or whole blood can be used.
If the implicated antibody is not anti-D,
D-positive red cells may be given to a
D-positive infant.
Maternal serum or plasma is the speci-
men of choice for crossmatching in ex-
change transfusion; it is available in large
quantities, decreases the volume of blood
taken from the infant, has the red cell anti-
body present in high concentration, and
can be analyzed accurately and completely
before delivery. On the other hand, use of
maternal serum may be problematic if it
contains antibodies directed against anti-
gens not present on the infant’s red cells
because of other sources of sensitization, or
if it contains IgM antibodies that have not
crossed the placenta. These additional
antibodies could complicate the serologic
picture.
If maternal blood is not available or is
unsuitable for crossmatching, the infant’s
serum and/or, preferably, an eluate from
the infant’s red cells can be used for cross-
matching. The concentration of antibody in
the infant’s serum may be low, especially if
most of the molecules are bound to the red
cells. Use of the eluate or serum, or of both
together, may be indicated if attempts to
obtain a maternal specimen would delay
therapy. Blood used for exchange transfu-
546 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
sion should be irradiated. Typically, a vol-
umeoftwicetheinfant’sbloodvolumeis
used for exchange.51
Subsequent Transfusion
Bilirubin may reaccumulate rapidly after
a successful exchange transfusion despite
appropriate phototherapy. This occurs
because most bilirubin in extravascular
fluid will reequilibrate by entering the
intravascular space and also because re-
sidual antibody-coated cells continue to
hemolyze. If rising bilirubin levels make a
second or third exchange transfusion nec-
essary, the same considerations of red cell
selection and crossmatching apply.
Infants who have undergone intraute-
rine transfusion need to be followed closely
after birth because intrauterine transfusion
suppresses fetal erythropoiesis. Weekly
hematocrit and reticulocyte counts should
be performed on the neonate for a 1- to
3-month period.18 Many of these infants
will subsequently develop anemia and
need to be supported with red cell transfu-
sion until their own production begins, as
evidenced by reticulocytosis and age-ap-
propriate hemoglobin levels.18,52
Antibody Against a High-Incidence Antigen
Rarely, the mother’s antibody reacts with
a high-incidence antigen and no compati-
ble blood is available. If this problem is
recognized and identified before delivery,
the mother’s siblings can be evaluated for
compatibility and suitability, or compati-
ble donors can be sought through a rare
donor file. Maternal red cells can also be
collected and frozen. Any products collected
from blood relatives must be irradiated. If
this very rare event is not recognized until
after delivery, three choices are available:
1. Collect blood from the mother, if the
obstetrician agrees. Remove as much
plasma as possible, preferably by sa-
line washing, and resuspend the red
cells in compatible plasma to the de-
sired hematocrit.
2. If time permits, test the mother’s sib-
lings or other close relatives for
compatibility and eligibility.
3. Use incompatible donor blood for
theexchangetransfusioniftheclini
-
cal situation is sufficiently urgent.
The exchange will reduce the biliru-
bin load, the most heavily anti-
body-coated cells, and the number
of unbound antibody molecules.
However, residual antibody will at-
tach to the transfused cells, and one
or more additional exchanges will
probably be needed as bilirubin ac-
cumulates.
Rh Immune Globulin
RhIG is a concentrate of predominantly
IgG anti-D derived from pools of human
plasma. A full dose of anti-D (300 µg, 1500
IU, or the actual content of a “dose” as in-
dicated by the individual manufacturer53)
is sufficient to counteract the immunizing
effects of 15 mL of D-positive red cells;
this corresponds to approximately 30 mL
of fetal whole blood. RhIG is available in a
reduced dose, approximately 50 µg, which
is protective for up to 2.5 mL of D-positive
fetal red cells. This dose can be used for
first-trimester abortion or miscarriage, when
the total blood volume of the fetus is less
than 2.5 mL. However, because of fears of
miscalculating the length of pregnancy
and concerns of inadvertently mixing up
inventory resulting in undertreatment, a
full dose is usually administered and
these low doses are frequently not stocked.
The protective effect of RhIG on D-nega-
tive individuals exposed to D-positive
cells probably results from interference
with antigen recognition in the induction
phase of primary immunization.54
Chapter 23: Perinatal Issues in Transfusion Practice 547
Copyright © 2005 by the AABB. All rights reserved.
RhIG is available in two formulations: 1)
for intramuscular (IM) injection only and 2)
for either IM or intravenous (IV) adminis-
tration.Thedoseoftheintravenousprepa
-
ration is expressed in international units (5
IU is equivalent to 1 µg), with 1500 IU (300
µg) neutralizing 17 mL of D-positive red
cells, according to the package insert.
Antepartum Administration
Widespread postpartum use of Rh immuno-
prophylaxis has reduced pregnancy-asso-
ciated immunization to the D antigen to
1% to 2%.8This risk is further decreased to
0.1% if RhIG is also given antepartum at
28 weeks of gestation.7The ACOG recom-
mends antepartum RhIG prophylaxis at
28 weeks of gestation, based on the obser-
vation that, of women who develop anti-D
during pregnancy, 92% do so at or after 28
weeks.7,15
Blood obtained before injection of RhIG
should be tested for ABO and D. A D-nega-
tive woman who has antibodies other than
anti-D (eg, anti-G) is still a candidate for
anti-D immunoprophylaxis. When the
mother receives RhIG during pregnancy,
the infant may be born with a positive DAT,
but without hemolysis. The mother’s serum
will often exhibit anti-D reactivity. There
must be good communication between the
patient’s physician and the blood bank staff
at the institution where delivery takes
place, to ensure correct interpretation of
laboratory tests made at the time of deliv-
ery. The half-life of an injected dose of RhIG,
in the absence of significant FMH, is ap-
proximately 21 days. Therefore, of 300 µgof
anti-D given at 28 weeks, 20-30 µgcouldre
-
main at the time of delivery 12 weeks later.
Some practitioners will administer another
dose of RhIG if delivery is delayed beyond
40 weeks.18 Anti-D can be detected in the
maternal circulation for as long as 6 months.
Postpartum Administration
Cord blood from infants born to D-nega-
tive mothers should be tested for the D
antigen. A D-negative woman with a
D-positive infant should receive one full
dose of RhIG within 72 hours of delivery,
unless she is known to be alloimmunized
to D previously. The presence of residual
anti-D from antepartum RhIG does not
indicate ongoing protection.
Active vs Passive Antibody. In-vitro clues
can help distinguish passively administered
RhIG from the anti-D formed as a result of
active alloimmunization. Passively acquired
anti-D is entirely IgG; if a woman’s anti-D is
saline-reactive or can be completely or par-
tially inactivated by treating the serum with
2-mercaptoethanol or dithiothreitol, it has
an IgM component and probably repre-
sents active immunization. Passively ac-
quired anti-D rarely achieves an anti-
globulin titer above 4, so a high-titered
antibody or rising antibody titer is likely to
indicate active immunization. It is desirable
to obtain confirmation from the physician’s
records, but RhIG should always be given
when doubt cannot easily be resolved. It
should also be given if there is any problem
determining the Rh type.
Postpartum Evaluation. Asampleofthe
mother’s blood should be drawn, preferably
within 1 hour after delivery, and evaluated
for FMH of a quantity greater than that for
which 300 µg RhIG is immunosuppressive.
If the screening test demonstrates the pres-
ence of fetal cells, the extent of FMH must
be determined so that an appropriate dose
of RhIG can be administered.49(pp48,49),55
Postpartum RhIG should be given within
72hoursofdelivery.Ifprophylaxisisde
-
layed, the likelihood that alloimmunization
will be prevented decreases. Despite the de-
crease seen, the ACOG recommends that
treatment still be administered because
some studies have found partial protection
548 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
has occurred as late as 13 days after expo-
sure and, possibly, as late as 28 days.15
The following women are not candidates
for RhIG:
1. The D-negative woman whose infant
is D-negative.
2. Any D-positive woman. Very rare cases
of HDFN have been reported in in-
fants whose mothers had a weak/par-
tial D phenotype, but routine RhIG
prophylaxis is not routinely recom-
mended for women of the weak/
partial D phenotype.12
3. A D-negative woman known to be
immunized to D.
Other Indications for RhIG
RhIG should be given to a D-negative woman
after any obstetric event that might allow
fetal cells to enter the mother’s circula-
tion: spontaneous or therapeutic abor-
tion, ectopic pregnancy, amniocentesis,
chorionic villus sampling, molar preg-
nancy, cordocentesis, antepartum hemor-
rhage, blunt abdominal trauma, or fetal
death.55 As mentioned earlier, at 12 weeks
of gestation or earlier, a 50 µgdoseofRhIG
would be adequate to protect against the
small fetal blood volume during the first
trimester. From 13 weeks’ gestation until
term, a full dose of RhIG should be given.
At <20 weeks, the fetal blood volume is
rarely more than 30mL,56 small enough
that a single dose of 300 µgRhimmune
globulin will be sufficient for prophylaxis
for any FMH. Therefore, it is not neces-
sary to quantitate fetal red cells in the ma-
ternal circulation before 20 weeks of ges-
tation.57
Amniocentesis
Amniocentesis can cause FMH and con-
sequent Rh immunization. The D-negative
woman who has amniocentesis at 16 to 18
weeks’ gestation for genetic analysis should
receive a full dose of RhIG at that time, a
second full dose at 28 weeks of gestation,
and the usual postpartum dose if the in-
fant is D positive. If a nonimmunized
D-negative woman undergoes amniocen-
tesis for any reason in the second or third
trimester, a full dose of RhIG is indicated.
If the procedure is repeated more than 21
days later, an additional full dose should
be given.18 If amniocentesis is performed
to assess fetal maturity, and if delivery is
expected within 48 hours of the proce-
dure, RhIG can be withheld until the in-
fant is born and confirmed to be D posi-
tive. If more than 48 hours will elapse,
RhIG should be given following amnio-
centesis. If delivery occurs within 21 days
thereafter and there is no evidence of a
massive FMH,15 additional RhIG may not
be essential, but prudent management
suggests repeat RhIG administration at
delivery.
Screening for Large-Volume FMH
Postpartum administration of RhIG may
not prevent immunization if the quantity
of D-positive fetal red cells entering the
mother’s circulation exceeds the immuno-
suppressive capacity of RhIG. One 300-µg
dose protects against 15 mL of D-positive
red cells or 30 mL of fetal blood. Only 0.3%
of pregnancies are estimated to sustain
FMH greater than 30 mL, but large FMH
is an important and preventable cause of
failed immunoprophylaxis.7The ACOG
recommends postpartum testing for large
FMHonlyforhigh-riskpregnancies,
15 but
Ness and colleagues58 showed that testing
based only on the ACOG criteria would
miss 50% of mothers exposed to large-vol-
ume FMH. AABB Standards for Blood
Banks and Transfusion Services requires
examination of a postpartum specimen
from all D-negative women at risk of im-
Chapter 23: Perinatal Issues in Transfusion Practice 549
Copyright © 2005 by the AABB. All rights reserved.
munization, to detect the presence of FMH
that requires more than one dose of RhIG.49(p49)
In rare cases, a massive FMH can cause
fetal death and infuse enough Rh-positive
cells into the maternal circulation to simu-
late a weak D phenotype in an Rh-nega-
tive patient.15,59 Unless recognized and
treated with an adequate dose of RhIG, this
will likely lead to alloimmunization.
“Microscopic Weak D.” In the past, some
workers looked for D-positive red cells in
the mother’s D-negative blood by examin-
ing the antiglobulin phase of the test for D
microscopically (“microscopic weak D
test”); mixed-field reactivity indicated a
substantial admixture with D-positive cells.
This procedure should not be used to iden-
tify large FMH, however, because of its lack
of reliability.60
The Rosette Test. The rosette test dem-
onstrates small numbers of D-positive cells
in a D-negative suspension. The suspen-
sion is incubated with an anti-D reagent of
human origin, and antibody molecules at-
tach to sites on D-positive cells in the sus-
pension. Indicator D-positive cells are
added, which react with antibody mole-
cules bound to the surface of the al-
ready-present D-positive cells and form vis-
ible agglutinates (rosettes) around them
(see Method 5.1). This method will detect
FMHs of approximately 10 mL,60 asensitiv
-
ity that provides a desirable margin of
safety for a screening test. Weak D-positive
cells do not react as strongly in the rosette
procedure as normal D-positive cells. If the
newborn has a weak D phenotype, FMH
can be evaluated by the Kleihauer-Betke
acid-elution test (see below), which identi-
fies fetal hemoglobin, not a surface antigen.
In all cases, the rosette test gives only quali-
tative results and a positive result must be
followed by a quantitative test, such as an
acid-elution procedure. Other tests that can
be used to detect and/or quantify FMH are
flow cytometry, gel agglutination, and the
enzyme-linked antiglobulin test (ELAT,
which is in limited use). Each of these
methods has various advantages and disad-
vantages that must be evaluated by each in-
stitution. The use of nucleic acid amplifica-
tion techniques, designed to detect very
small amounts of fetal cells, remains a re-
search endeavor.61-63
Quantifying FMH
Historically, quantification of FMH has
been achieved by the Kleihauer-Betke
acid-elution test, which relies on the dif-
ferences between fetal and adult hemo-
globin in resistance to acid elution (see
Method 5.2). Results are reported as a per-
centage of fetal cells, but the precision
and accuracy of the procedure may be
poor. Because 300 µg of RhIG will protect
against FMH of 30 mL of D-positive fetal
blood, the number of doses of RhIG re-
quired is determined by dividing the esti-
mated volume of fetal blood present by 30.
For example:
1. Kleihauer-Betke test results reported
as 1.3%
2. (1.3/100) ×5000 mL* = 65 mL of fetal
blood
3. 65/(30 mL per dose) = 2.2 doses of
RhIG required
* = mother’s arbitrarily assigned blood volume
Because quantification by this procedure
is inherently inaccurate and because the
consequences of undertreatment can be
serious, it is desirable to provide a safety
margin in calculating RhIG dosage. One
approach is as follows:
1. When the number to the right of the
decimal point is less than 5, round
down and add one dose of RhIG (ex-
ample: If the calculation comes to
2.2 doses, give 3 doses).
550 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

2. When the number to the right of the
decimal point is 5 or greater, round
up to the next number and add one
dose of RhIG (example: If the calcu-
lation comes to 2.8 doses, give 4
doses). (See Table 23-1).
Not more than five doses of RhIG should
be injected intramuscularly at one time. For
larger quantities, injections can be spaced
over a 72-hour period for the patient’s com-
fort; an optimal time sequence has not
been established. The intravenous prepara-
tion of RhIG can be used when higher
doses are required. According to the pack-
age insert, a maximum dose of 300 IU
should be given at each injection, every 8
hours, until the total calculated dose has
been administered.
Neonatal Immune
Thrombocytopenia
Maternal IgG antibodies to platelets can
cross the placenta and cause severe ante-
natal and neonatal thrombocytopenia. Two
categories of immune thrombocytopenia
are recognized, and the distinction be-
tween them is therapeutically important.
Neonatal Alloimmune Thrombocytopenia
The mechanism of NAIT is similar to that
of HDFN. Fetal platelets, expressing a pa-
ternal antigen absent from the mother’s
cells, may enter the mother’s circulation
during gestation or delivery. If she becomes
immunized, the maternal IgG antibody
crosses the placenta and causes fetal and
neonatal thrombocytopenia. The maternal
platelet count remains normal. The inci-
dence of NAIT is approximately 1 in 1500
to 2000 live births.64,65 NAIT is the cause of
the majority of cases of intracranial hem-
orrhage due to thrombocytopenia, greater
than all other etiologies of thrombocyto-
penia combined.66
Unlike HDFN, NAIT often affects firstborn
children, with about 50% to 60% of cases
occurring in a woman’s first child. The
thrombocytopenia is self-limiting, normally
resolving in 2 to 3 weeks. NAIT varies in se-
verity from mild thrombocytopenia with no
clinical signs to overt clinical bleeding. The
incidence of intracranial hemorrhage has
been reported as 10% to 30%, with approxi-
mately half occurring in utero.65,67 Recur-
rence in subsequent pregnancies is fre-
quent, with equal or increasing severity, so
Chapter 23: Perinatal Issues in Transfusion Practice 551
Table 23-1. RhIG Dosage for Massive Fetomaternal Hemorrhage, Based on the Acid
Elution Test
% Fetal Cells Vials of RhIG to Inject
Dose
In gInIU
0.3 - 0.8 2 600 3000
0.9 - 1.4 3 900 4500
1.5 - 2.0 4 1200 6000
2.1 - 2.5 5 1500 7500
Notes:
1. Based on a maternal blood volume of 5000 mL.
2. 1 vial of 300 µg (1500 IU) is needed for each 15 mL fetal red cells or 30 mL fetal whole blood.
Copyright © 2005 by the AABB. All rights reserved.
a woman known to be alloimmunized must
receive skilled antenatal attention.
Serologic Testing
Serologic diagnosis should be sought in a
woman whose infant has had NAIT if fur-
ther pregnancies are planned. Several
platelet-specific antigen systems have
been associated with NAIT, with HPA-1a
antigen (PlA1) accounting for the vast ma-
jority of cases in Caucasians.64 Pregnancy,
rather than transfusion, is the usual im-
munizing event. Approximately 2% of the
population is HPA-1a negative; approxi-
mately 10% of HPA-1a-negative women
with HPA-1a-positive infants become im-
munized.68 Some studies have shown an
association between developing anti-HPA-
1a and possessing the HLA phenotype
DRw52a.66,68 Chapter 16 contains more in-
formation about platelet antigens. Al-
though antibodies to HLA Class I antigens
are frequently encountered in pregnancy,
and platelets express Class I antigens, this
isararecauseofNAIT,andthetrueroleof
HLA antibodies in this setting remains
controversial.69,70
Anyfamilywithahistoryofaninfant
born with a platelet count of <50,000/µL
should be evaluated. Ideally, serologic test-
ing uses maternal serum and maternal and
paternal whole blood from which platelets
are isolated. Maternal serum is screened for
both platelet-nonspecific and platelet-spe-
cific antibodies against paternal cells, as
well as panels of phenotyped platelets. Ma-
ternal and paternal platelet typing can be
performed using serologic and/or molecu-
lar typing methods. Some clinicians have
proposed screening pregnant women for
HPA-1a antigen because it is the most com-
monly implicated antigen causing incom-
patibility in Caucasians. Because only 10%
of HPA-1a-negative women are truly at risk
for forming antibody, and, of those, only
about 33% will have neonates with clini-
cally important thrombocytopenia, this has
not been widely adopted. In addition, the
cost and logistics of performing platelet an-
tigen typing are impediments to broad im-
plementation.65,71
Prenatal Considerations
With knowledge of antibody specificity
and gene frequencies, the likelihood of
subsequent offspring being affected can
be predicted (see Table 16-1). The recog-
nized platelet-specific antigens occur in
diallelic systems, so typing the father’s
platelets indicates zygosity. If the father is
homozygous for the expression of the an-
tigen, there is no need to determine the
fetal antigen status because all offspring
will be affected. If the father is heterozy-
gous for the expression of the antigen,
then there is a 50% chance that subse-
quent offspring will have the offending
antigen. In an at-risk pregnancy, the ge-
notype of the fetus (and by inference the
platelet phenotype) can be determined by
DNA typing on fetal cells obtained by am-
niocentesis.
When the risk of NAIT is high, a fetal
blood sample for platelet count determina-
tion can be obtained by cordocentesis as
early as 20 weeks’ gestation. Because
cordocentesis carries a risk of serious
bleeding in a thrombocytopenic fetus,
compatible platelets must be available at
thetimeoftheprocedureandareoftenin
-
fused if the platelet count is low. Some in-
stitutions will infuse platelets during the
procedure, before knowing the platelet
count, because of the risk of bleeding dur-
ing the cannulation itself. When the fetus
is found to be thrombocytopenic, the
mother is often given infusions of IGIV in
weekly doses of 1 g/kg, with or without ste-
roids, until delivery.64-67,72 Alternatively, some
would recommend empiric treatment with
552 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
IGIV in cases of a homozygous paternal ge-
notype for the specific platelet antigen or
in situations when PCR performed on
amniotic fluid reveals that the fetus carries
that platelet antigen. Many centers will pro-
ceed with elective cesarean section instead
of cordocentesis near term to determine
the fetal platelet count.
Sources of Platelets. Maternal platelets
are often prepared for use at cordocentesis
or delivery. The mother will undergo re-
quired testing for infectious disease mark-
ers. Prior administration of high-dose IGIV
may cause false-positive immunoassays;
therefore, it is desirable to test the mother
before initiating IGIV therapy. Those with
confirmed positive results (eg, hepatitis C
virus, which is transmitted more efficiently
by transfusion than perinatally) should not
be used as a source of platelets because
these results are more likely to represent
maternal infection. Of note, pregnancy it-
self can also cause false-positive results on
serologic infectious disease tests.
Platelets can be collected either from the
mother or from another donor whose
platelets lack the corresponding antigen
and whose plasma is compatible with the
fetal red cells. If maternal platelets are used,
the antibody-containing plasma should be
removed or reduced and the platelets re-
suspended in compatible plasma or saline
with reduced volume (see Method 6.15). All
components for intrauterine transfusion must
be irradiated (see Chapter 27) and should
be CMV-reduced risk.73
Scheduling Therapy. Various strategies
have been used in the management of fetal
thrombocytopenia. Although weekly plate-
let transfusions have been used in the past,
the inherent risk of repeated cordocentesis
makes the administration of IGIV to the
mother the preferred treatment in the
United States. Practice is different in Eu-
rope, where weekly platelet transfusions are
still performed. Platelet transfusion and re-
peated cordocentesis are reserved for pa-
tients when noninvasive forms of therapy
are not effective. Another approach is ad-
ministration of a single platelet transfusion
just before delivery if cordocentesis reveals
severe thrombocytopenia. This approach is
usually reserved for pregnancies at extreme
risk for intracranial hemorrhage.
Management After Delivery
Platelet counts can continue to decrease
after birth and should be monitored. For
patients at increased risk of bleeding due
to severe thrombocytopenia, compatible
platelets should be given prophylactically.
If compatible platelets are not available,
the use of high-dose IGIV should be con-
sidered, but response to this treatment is
variable. In patients who do respond, pla-
telet counts usually start increasing with-
in 24 to 48 hours, although it may take
longer in some patients.64 Because re-
sponse is slow, the neonate with an urgent
need for transfusion and no available
compatible platelets can receive platelets
from random donors, frequently resulting
in an adequate response. For patients re-
quiring platelet transfusion, giving con-
current IGIV can accelerate the recovery
of the patient’s own platelets and shorten
the period of transfusion dependency. If
the patient has mild thrombocytopenia
without bleeding, it can be managed
without specific therapeutic intervention.
Thrombocytopenia Secondary to
Maternal ITP
Infants born to mothers with active idio-
pathic (immune) thrombocytopenic purpura
(ITP) are often not profoundly thrombo-
cytopenic and have a smaller risk of hem-
orrhage than infants with NAIT.69,74 The
antibody in ITP is usually IgG, which
readily crosses the placenta. Occasionally,
delivery of a severely thrombocytopenic
Chapter 23: Perinatal Issues in Transfusion Practice 553
Copyright © 2005 by the AABB. All rights reserved.
infant has led to the diagnosis of previ-
ously unsuspected ITP in a moderately af-
fected mother (postpartum platelet count
75,000-100,000/µL). Such cases of mild ITP
should be distinguished from gestational
thrombocytopenia, in which a mother
with no history of autoimmune thrombo-
cytopenia has a platelet count less than
150,000/µL. In cases of maternal ITP, the
risk of severe fetal thrombocytopenia
(usually defined as a platelet count less
than 50,000/µL) is 7% to 10%.74,75 The risk
of intracranial hemorrhage in infants
born to mothers with ITP is low (≤1%),
with only a few cases reported in the liter-
ature. This is lower than the rate in NAIT
because, infants born to mothers with ITP
are generally born with higher platelet
counts and their platelet function is not
impaired, as it seems to be in NAIT. Rou-
tine fetal platelet assessment is not rec-
ommended and cesarean section is re-
served for obstetric indications only.74,76
The antibody in ITP has broad reactivity
against platelets. If the infant has a high
concentration of antibody, there will be
uniformly short survival of platelets from
random donors, from the mother, or from
other family members. Responses do some-
times occur, and, in the presence of hemor-
rhage, platelet transfusions will be used as
emergency therapy.69 IGIV therapy may also
be effective for severe autoimmune throm-
bocytopenia.66,72,74,76
References
1. Goodstein M. Neonatal red cell transfusion.
In:HermanJH,MannoCS,eds.Pediatric
transfusion therapy. Bethesda, MD: AABB
Press, 2002:39-91.
2. Vaughn JI, Manning M, Warwick RM, et al. In-
hibition of erythroid progenitor cells by anti-
Kell antibodies in fetal alloimmune anemia.
N Engl J Med 1998;338:798-803.
3. Dennery PA, Seidman DS, Stevenson DK.
Neonatal hyperbilirubinemia. N Engl J Med
2001;344:581-90.
4. American Academy of Pediatrics Subcommit-
tee on Hyperbilirubinemia. Management of
hyperbilirubinemia in the newborn infant 35
or more weeks of gestation. Clinical practice
guideline. Pediatrics 2004;114:297-316.
5. Bowman JM. Intrauterine and neonatal
transfusion. In: Anderson KC, Ness PM, eds.
Scientific basis of transfusion medicine: Im-
plications for clinical practice. Philadelphia:
WB Saunders, 2000:307-20.
6. Bowman JM. Treatment options for the fetus
with alloimmune hemolytic disease. Transfus
Med Rev 1990;4:191-207.
7. Bowman JM. The prevention of Rh immuni-
zation. Transfus Med Rev 1988;2:129-50.
8. Bowman JM. Controversies in Rh prophy-
laxis. Who needs Rh immune globulin and
when should it be given? Am J Obstet Gynecol
1985;151:289-94.
9. Strauss RG, Burmeister LE, Johnson K, et al.
Randomized trial assessing the feasibility and
safety of biologic parents as RBC donors for
their preterm infants. Transfusion 2000;40:450-6.
10. ElbertC,StraussRG,BarrettF,etal.Biological
mothers may be dangerous blood donors for
their neonates. Acta Hematol 1991;85:189-91.
11. Geifman-Holtzman O, Wojtowycz M, Kosmas
K, Artal R. Female alloimmunization with an-
tibodies known to cause hemolytic disease.
Obstet Gynecol 1997;89:272-5.
12. Judd WJ. Practice guidelines for prenatal and
perinatal immunohematology, revisited.
Transfusion 2001;41:1445-52.
13. LeglerTJ,MaasJH,KöhlerM,etal.RHD se-
quencing: A new tool for decision making on
transfusion therapy and provision of Rh pro-
phylaxis. Transfus Med 2001;11:383-8.
14. Wagner FF, Frohmajer A, Ladewig B, et al. Weak
D alleles express distinct phenotypes. Blood
2000;95:2699-708.
15. Prevention of Rh D alloimmunization. ACOG
Practice Bulletin Number 4. Washington, DC:
American College of Obstetricians and Gyne-
cologists, May 1999.
16. Kanter MH. Derivation of new mathematic
formulas for determining whether a D-posi-
tive father is heterozygous or homozygous for
the D antigen. Am J Obstet Gynecol 1992;166:
61-3.
17. Bennett PR, Le Van Kim C, Colon Y, et al. Pre-
natal determination of fetal RhD type by DNA
amplification. N Engl J Med 1993;329:607-10.
18. Moise KJ. Management of Rhesus alloim-
munization in pregnancy. Obstet Gynecol
2002;100:600-11.
554 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
19. SingletonBK,GreenCA,AventND,etal.The
presence of an RHD pseudogene containing a
37 base pair duplication and a nonsense mu-
tation in Africans with the Rh D-negative
blood group phenotype. Blood 2000;95:12-8.
20. Hessner MJ, Pircon RA, Luhm RA. Develop-
ment of an allele specific polymerase chain
reaction assay of prenatal genotyping of Jka
and Jkbof the Kidd blood group system (ab-
stract). Am J Obstet Gynecol 1998;178(Suppl):
52S.
21. Lee S, Bennett PR, Overton T, et al. Prenatal
diagnosis of Kell blood group genotypes:
KEL1 and KEL2. Am J Obstet Gynecol 1996;
175:445-9.
22. LeVanKimC,MouroI,BrossardY,etal.PCR-
based determination of the Rhc and RhE sta-
tus of the fetus at risk for Rhc and RhE
hemolytic disease. Br J Haematol 1994;88:
193-5.
23. Spence WC, Potter P, Maddalena A, et al. DNA-
based prenatal determination of the RhEe ge-
notype. Obstet Gynecol 1995;86:670-2.
24. Randen I, Hauge R, Kjeldsen-Kragh J,
Fagerhol MK. Prenatal genotyping of RHD
and SRY using maternal blood. Vox Sang
2003;85:300-6.
25. Issitt PD, Anstee DJ. Applied blood group se-
rology. 4th ed. Durham, NC: Montgomery
Scientific Publications, 1998:1067-9.
26. Management of isoimmunization in preg-
nancy. ACOG Educational Bulletin Number
227. Washington, DC: American College of
Obstetricians and Gynecologists, 1996.
27. Gottvall T, Hilden JO. Concentration of anti-D
antibodies in Rh(D) alloimmunized pregnant
women as a predictor of anemia and/or
hyperbilirubinemia in their newborn infants.
Acta Obstet Gynecol Scand 1997;76:733-8.
28. Bowman JM, Pollock JM, Manning FA, et al.
Maternal Kell blood group alloimmunization.
Obstet Gynecol 1992;79:239-44.
29. Hadley AG. Laboratory assays for predicting
the severity of haemolytic disease of the fetus
and newborn. Transpl Immunol 2002;10:191-
8.
30. Hume HA. Fetal and neonatal transfusion
therapy. In: Pomphilon DH, ed. Modern
transfusion medicine. Boca Raton, FL: CRC
Press, 1995:193-215.
31. Liley AW. Liquor amnii analysis in the man-
agement of the pregnancy complicated by
rhesus sensitization. Am J Obstet Gynecol
1961;82:1359-70.
32. QueenanJT,TomaiTP,UralSH,KingJC.Devi
-
ation in amniotic fluid optical density at a
wavelength of 450 nm in Rh-immunized
pregnancies from 14 to 40 weeks’ gestation: A
proposal for clinical management. Am J
Obstet Gynecol 1993;168:1370-6.
33. Steiner EA, Judd WJ, Oberman HA, et al. Per-
cutaneous umbilical blood sampling and
umbilical vein transfusions: Rapid serologic
differentiation of fetal blood from maternal
blood. Transfusion 1990;30:104-8.
34. Ludomirsky A. Intrauterine fetal blood sam-
pling—a multicenter registry evaluation of
7462 procedures between 1987-1991 (ab-
stract). Am J Obstet Gynecol 1993;168:318.
35. Mari G, Deter RL, Carpenter FL, et al. Non-
invasive diagnosis by Doppler ultrasono-
graphy of fetal anemia due to maternal
red-cell alloimmunization. N Engl J Med
2000;342:9-14.
36. Nishie EN, Brizot ML, Liao AW, et al. A com-
parison between middle cerebral artery peak
systolic velocity and amniotic fluid optical
density at 450 nm in the prediction of fetal
anemia. Am J Obstet Gynecol 2003;188:214-9.
37. QuillenK,BerkmanEM.Introductiontother
-
apeutic apheresis. In: McLeod BC, Price TH,
Weinstein R, eds. Apheresis: Principles and
practice.2nded.Bethesda,MD:AABBPress,
2003:49-69.
38. MarguliesM,VotoLS,MathetE,MarguliesM.
High dose intravenous IgG for the treatment
of severe Rhesus alloimmunization. Vox Sang
1991;61:181-9.
39. UlmB,KirchnerG,SvolbaG,etal.Immuno-
globulin administration to fetuses with ane-
mia due to alloimmunization to D. Transfu-
sion 1999;39:1235-8.
40. Ghidini A, Sepulveda W, Lockwood CJ,
Romero R. Complications of fetal blood sam-
pling. Am J Obstet Gynecol 1993;168:1339-44.
41. Weiner CP, Okamura K. Diagnostic fetal blood
sampling-technique related losses. Fetal
Diagn Ther 1996;11:169-75.
42. Schumacher B, Moise KJ Jr. Fetal transfusion
for red blood alloimunization in pregnancy
(review). Obstet Gynecol 1996;88:137-50.
43. Skupski DW, Wolf CF, Bussel JB. Fetal transfu-
sion therapy. Obstet Gynecol Surg 1996;51:181-
92.
44. Gonsoulin WJ, Moise KJ, Milam JD, et al. Se-
rial maternal blood donations for intra-
uterine transfusion. Obstet Gynecol 1990;75:
158-62.
45. Sanders MR, Graeber JE. Posttransfusion
graft-versus-host disease in infancy. J Pediatr
1990;117:159-63.
46. Linden JV, Pisciotto PT. Transfusion-associ-
ated graft-versus-host disease and blood irra-
diation. Transfus Med Rev 1992;6:116-23.
47. Mandelbrot L, Daffos F, Forestier F, et al. As-
sessment of fetal blood volume for computer-
Chapter 23: Perinatal Issues in Transfusion Practice 555
Copyright © 2005 by the AABB. All rights reserved.
assisted management of in utero transfusion.
Fetal Ther 1988;3:60-6.
48. Moise KJ, Carpenter RJ, Kirshon B, et al. Com-
parison of four types of intrauterine transfu-
sion: Effect on fetal hematocrit. Fetal Ther
1989;4:126-37.
49. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
50. Herschel M, Karrison T, Wen M, et al. Isoim-
munization is unlikely to be the cause of
hemolysis in ABO-incompatible but direct
antiglobulin test-negative neonates. Pediat-
rics 2002;110:127-30.
51. Koenig JM. Evaluation and treatment of
erythroblastosis fetalis in the neonate. In:
Christensen RD, ed. Hematologic problems
of the neonate. Philadelphia: WB Saunders
Company, 2000:185-207.
52. Saade GR, Moise KJ, Belfort MA, et al. Fetal
and neonatal hematologic parameters in red
cell alloimmunization: Predicting the need
for late neonatal transfusions. Fetal Diagn
Ther 1993;8:161-4.
53. MoiseKJ,BrecherME.Packageinsertfor
Rhesus immune globulin (letter). Obstet
Gynecol 2004;103:998-9.
54. Mollison PL, Engelfriet CP, Contreras M.
Blood transfusion in clinical medicine. 10th
ed. Oxford, England: Blackwell Scientific Pub-
lications, 1997.
55. Hartwell EA. Use of Rh immune globulin.
ASCP Practice Parameter. Am J Clin Pathol
1998;110:281-92.
56. Nicolaides KH, Clewell WH, Rodeck CH. Mea-
surement of human fetoplacental blood vol-
ume in erythroblastosis fetalis. Am J Obstet
Gynecol 1987;157:50-3.
57. Vengelen-Tyler V, Telen MJ. Collected ques-
tions and answers. 5th ed. Bethesda, MD:
AABB, 1997:52-3.
58. Ness PM, Baldwin ML, Niebyl JR. Clinical
high risk designation does not predict excess
fetal maternal hemorrhage. Am J Obstet
Gynecol 1987;156:154-8.
59. Owen J, Stedman CH, Tucker TL. Comparison
of predelivery versus postdelivery Kleihauer-
Betke stains in cases of fetal death. Am J
Obstet Gynecol 1989;161:663-6.
60. Sebring ES. Fetomaternal hemorrhage—inci-
dence and methods of detection and quan-
titation. In: Garratty G, ed. Hemolytic disease
of the newborn. Arlington, VA: AABB, 1984:87-117.
61. Duguid JKM, Bromilow IM. Laboratory mea-
surement of fetomaternal hemorrhage and
its clinical relevance. Transfus Med Rev 1999;
13:43-8.
62. Riley JZ, Ness PM, Taddie SJ, et al. The detec-
tion and quantitation of fetal-maternal hem-
orrhage using an enzyme-linked antiglobulin
test (ELAT). Transfusion 1982;22:472-4.
63. Bayliss KM, Kueck BD, Johnson ST, et al. De-
tecting fetomaternal hemorrhage: A compari-
son of five methods. Transfusion 1991;31:303-7.
64. Uhrynowska M, Maslanka K, Zupanska B.
Neonatal thrombocytopenia: Incidence,
serological and clinical observations. Am J
Perinatol 1997;14:415-18.
65. Williamson LM, Hackett G, Rennie J, et al.
The natural history of fetomaternal alloim-
munization to the platelet-specific antigen
HPA-1a (PlA1,Zw
a) as determined by antena-
tal screening. Blood 1998;92:2280-7.
66. Bussel JB. Alloimmune thrombocytopenia in
the fetus and newborn. Semin Thromb
Hemost 2001;27:245-52.
67. Johnson JA, Ryan G, al-Musa A, et al. Prenatal
diagnosis and management of neonatal allo-
immune thrombocytopenia. Semin Perinatol
1997;21:45-52.
68. WatersAW,MurphyM,HambleyH,Nicolai
-
des K. Management of alloimmune thrombo-
cytopenia in the fetus and neonate. In: Nance
SJ, ed. Clinical and basic science aspects of
immunohematology. Arlington, VA: AABB,
1991:155-77.
69. Blanchette VS, Kuhne T, Hume H, Hellman J.
Platelet transfusion therapy in newborn in-
fants. Transfus Med Rev 1995;9:215-30.
70. Maes LY, Gautreaux M, Southgate WM, Lazar-
chick J. Evidence of neonatal alloimmune
thrombocytopenia mediated by anti-HLA an-
tibodies (abstract). Transfusion 2001;41
(Suppl):20S.
71. Murphy MF, Williamson LM, Urbaniak SJ. An-
tenatal screening for fetomaternal allo-
immune thrombocytopenia; should we be
doing it? Vox Sang 2002;83(Suppl 1):409-16.
72. Gaddipati S, Berkowitz RL, Lembet AA, et al.
Initial fetal platelet counts predict the re-
sponse to intravenous gammaglobulin ther-
apy in fetuses that are affected by PLA1 in-
compatibility. Obstet Gynecol 2001;185:976-80.
73. Leukocyte reduction. Association Bulletin
99-7. Bethesda, MD: AABB, 1999.
74. Bussel JB. Immune thrombocytopenia in
pregnancy: Autoimmune and alloimmune. J
Reprod Immunol 1997;37:35-61.
75. PayneSD,ResnikR,MooreTR,etal.Maternal
characteristics and risk of severe neonatal
thrombocytopenia and intracranial hemorrhage
in pregnancies complicated by autoimmune
thrombocytopenia. Am J Obstet Gynecol
1997;177:149-55.
76. BlanchetteVS,KirbyMA,TurnerC.Roleofin
-
travenous immunoglobulin G in autoim-
mune hematologic disorders. Semin Hematol
1992;29:72-82.
556 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 24: Neonatal and Pediatric Transfusion Practice
Chapter 24
Neonatal and Pediatric
Transfusion Practice
MANY PHYSIOLOGIC CHANGES
accompany the transitions from
fetus to neonate, neonate to
infant, and throughout childhood. Hema-
tologic values, blood volume, and physio-
logic responses to stresses such as hypo-
volemia and hypoxia vary widely. The
most rapid changes occur during early in-
fancy. Consequently, discussions of pedi-
atric transfusion are usually divided into
two periods: neonates from birth through
4 months, and older infants (>4 months)
and children. Some concerns in neonatal
transfusion practice overlap with those of
the perinatal period and are discussed in
Chapter 23.
Advances in medical care now permit
the survival of extremely premature neo-
nates. Blood providers must be capable of
furnishing blood components that are tai-
lored to satisfy the specific needs of very
low birthweight (VLBW <1500 g) and ex-
tremely low birthweight (ELBW <1000 g)
patients, whose small blood volumes and
impaired or immature organ functions pro-
vide little margin for safety. Ill neonates are
more likely than hospitalized patients of
any other age group to receive red cell
transfusions.1Advances in critical-care neo-
natology, such as surfactant therapy, nitric
oxide therapy, use of high-frequency venti-
lators, and adherence to transfusion prac-
tice guidelines, have diminished the num-
ber of blood transfusions given; most are
now given to infants with birthweights less
than 1000 g.1The doses of various compo-
nents used for simple, small-volume trans-
fusionsaregiveninTable24-1.
2
Fetal and Neonatal
Erythropoiesis
The predominant sites of hematopoiesis
in the developing embryo shift from the
wall of the yolk sac to the liver to the mar-
557
24
Copyright © 2005 by the AABB. All rights reserved.

row in the first 24 weeks.3Hematopoiesis
is regulated by gradually increasing eryth-
ropoietin (EPO) levels stimulated by low
oxygen tensions during intrauterine life.
Fetal red cells, rich in hemoglobin F, are
well adapted to low intrauterine oxygen
tensions. The high oxygen affinity of fetal
hemoglobin enhances transfer of oxygen
from maternal erythrocytes to fetal eryth-
rocytes throughout pregnancy.
The “switch” from fetal to adult hemo-
globin begins at about 32 weeks’ gestation;
at birth, 60% to 80% of the total hemoglo-
bin is hemoglobin F. Preterm neonates,
therefore, are born with higher levels of fe-
talhemoglobinthanthosebornatterm.
The mean cord hemoglobin of healthy term
neonates is 16.9 ± 1.6 g/dL, and that of
preterm neonates is 15.9 ± 2.4 g/dL.4He-
moglobin concentration gradually falls in
the first few weeks of life. This has been
called “physiologic anemia of infancy” in
term newborns and “physiologic anemia of
prematurity” in preterm newborns. The
anemia is considered physiologic because
it is self-limited, is usually well tolerated,
and is not associated with any deleterious
effects to the infant. Erythropoietic activity
diminishes secondary to an increase in pul-
monary blood flow and a rise in arterial
pO2, as well as the increase in red cell con-
tent of 2,3-diphosphoglycerate (2,3-DPG)
and hemoglobin A, which enhance the re-
lease of oxygen to the tissues. As tissue oxy-
genation improves, levels of EPO decline
and erythropoiesis diminishes. This, along
with decreased survival of fetal red cells
and expansion of the blood volume due to
rapid growth, causes the hemoglobin con-
centration to decline. The rate of decline is
dependent on gestational age at birth; he-
moglobin may drop to as low as 8.0 g/dL at
4 to 8 weeks of age in preterm infants with
birthweights of 1000 to 1500 g, and 7.0 g/dL
in neonates with birthweights less than
1000 g.3
Despite hemoglobin levels that would
indicate anemia in older children and
adults, the normally developing infant usu-
ally maintains adequate tissue oxygenation.
Physiologic anemia requires treatment only
if the degree or timing of the anemia causes
symptoms in the patient.
Unique Aspects of Neonatal
Physiology
Infant Size and Blood Volume
Full-term newborns have a blood volume
ofapproximately85mL/kg;pretermlow
558 AABB Technical Manual
Table 24-1. Volumes for Simple, Small-Volume Transfusions of Neonates
Component Volume Estimated Change
RBC 10-15 mL/kg Hemoglobin ↑2-3 g/dL
Platelet 5-10 mL/kg Platelet ↑50,000-100,000/µL
Granulocyte ≥1×109neutrophils/kg in
volume of 10-15 mL/kg
Repeat until clinical response
FFP 10-15 mL/kg Factor activity ↑15-20%
Cryoprecipitate 1-2 units/10 kg ↑60-100 mg/dL fibrinogen (infant)
↑5-10 mg/dL fibrinogen (larger child)
Adapted with permission from Roseff.2
Copyright © 2005 by the AABB. All rights reserved.
birthweight newborns have an average
blood volume of 100 mL/kg. As survival
rates continue to improve for infants
weighing 1000 g or less at birth, blood
banks are being asked to provide blood
components for patients whose total
blood volume is less than 100 mL on a
more frequent basis. The need for fre-
quent laboratory tests has made replace-
ment of iatrogenic blood loss the most
common indication for transfusion of low
birthweight preterm neonates. However,
the previous practice of replacing blood
mL for mL is giving way to replacement as
needed in order to maintain a target hema-
tocrit in certain clinical situations.1
Newborns do not compensate for hypo-
volemia as well as adults. After 10% volume
depletion in a newborn, left ventricular
stroke volume is diminished without in-
creasing heart rate. To maintain systemic
blood pressure, peripheral vascular resis-
tance increases, and this, combined with a
decreased cardiac output, results in poor
tissue perfusion, low tissue oxygenation,
and metabolic acidosis.5
Erythropoietin Response
Erythropoietin response in newborns dif-
fers from that in adults and older chil-
dren. In older children and adults, oxygen
sensors in the kidney recognize dimin-
ished oxygen delivery and release EPO
into the circulation. In the fetus, the oxy-
gen sensor that stimulates EPO produc-
tion is believed to be the liver, which ap-
pears to be programmed for the hypoxic
intrauterine environment.
This hyporesponsiveness to hypoxia pro-
tects the fetus from becoming poly-
cythemic in utero. Eventually, EPO produc-
tion shifts from the liver to the kidneys, a
developmental change thought to be regu-
lated based on the time of conception, not
birth, and possibly not beginning until
term. The most premature infants produce
the least amount of EPO for any degree of
anemia; this may reflect the absence of the
developmental shift of erythropoietin pro-
duction from the liver to the kidneys.6Sick
preterm neonates who receive many trans-
fusions shortly after birth have reduced cir-
culating levels of fetal hemoglobin. Circu-
lating EPO levels are lower, for a given
hematocrit, in preterm neonates with
higher proportions of hemoglobin A rela-
tive to hemoglobin F, which favors release
of oxygen to the tissues.6Erythroid progeni-
tor cells in the hypoproliferative marrow of
these preterm infants show normal intrin-
sic sensitivity to EPO. Clinical trials of re-
combinant human erythropoietin (rHuEPO)
in premature neonates show that the num-
ber of transfusions and severity of anemia
can be lessened.7Adverse effects of rHuEPO
in this age group are also different from
those seen in older children and adults and
include transient, reversible neutropenia.
Since the adherence to strict transfusion
guidelines and the decrease in phlebotomy
in VLBW infants, the ultimate role of
rHuEPO in the management of “anemia of
prematurity” has remained unclear. A re-
cent multicenter study in Europe showed
decreased need for transfusion when ad-
ministering EPO to ELBW infants within 3
to 5 days of life (early EPO administration)
and continuing for 9 weeks.8The mean
number of transfusions dropped from 2.66
to 1.86, with a reduction in donor expo-
sures from 2 to 1. Questions regarding opti-
mal dosing, route of administration, and
use of supplemental iron remain to be an-
swered.1,7-10 In any event, with the tremen-
dous strides made in decreasing donor ex-
posure in transfused newborns by using
restrictive transfusion practices alone, the
role of EPO for this purpose may no longer
be relevant.
Chapter 24: Neonatal and Pediatric Transfusion Practice 559
Copyright © 2005 by the AABB. All rights reserved.
Cold Stress
Hypothermiainthenewborncausesex
-
aggerated effects, including increased
metabolic rate, hypoglycemia, metabolic
acidosis, and a tendency toward apneic
episodes that may lead to hypoxia, hypo-
tension, and cardiac arrest. Blood used for
exchange transfusion should be warmed
because blood at room temperature may
decrease a newborn’s core temperature by
0.7 to 2.5 C. The usual method is to use an
inline warmer. Blood, either large or small
volumes, should not be warmed under
a radiant heater because of the risk of
hemolysis in an unmonitored apparatus.
When transfusions are given to infants
undergoing phototherapy, the tubing
should be positioned to minimize expo-
sure to the phototherapy light in order to
prevent hemolysis.11
Immunologic Status
Infants have an immature and inexperi-
enced cellular and humoral immune system.
Antibodies present derive almost entirely
from the maternal circulation. Trans-
placental transfer of immunoglobulin and
other proteins is independent of molecu-
lar size; IgG (150 kD) is transferred much
more readily than albumin (64 kD). In hu-
mans, maternal IgM does not reach the
fetus and IgA is not readily transferred, al-
though low levels have been found in the
newborn.
All four subclasses of IgG are transported
acrosstheplacenta,buttheratevariesbe
-
tween individual mother-fetus pairs. Early
in pregnancy (approximately 12 weeks), IgG
probably passes from mother to fetus by
diffusion, and concentration in fetal serum
is low for all subgroups.12 Between 20 and
33 weeks of gestation, fetal IgG levels rise
markedly, apparently because of matura-
tion of a selective transport system that in-
volves, in part, specific protein receptors on
the membrane of placental cells. IgG1, the
predominant subclass in maternal blood,
crosses the placenta first and is transported
in greatest quantity. Cord blood has higher
antibody concentrations than maternal
blood. Catabolism of IgG occurs more
slowly in the fetus than in the mother, so
that transplacental maternal antibody is
conserved during the neonatal period.
A fetus exposed to an infectious process
in utero or an infant exposed shortly after
birth may produce small amounts of IgM
detectable by sensitive techniques, but un-
expected red cell alloantibodies of either
IgG or IgM class are rarely formed during
the neonatal period. The mechanisms
responsible for the lack of alloantibody pro-
duction in the neonate are not clearly un-
derstood and are most likely multifactorial,
including deficient T helper function, en-
hanced T suppressor activity, and poor an-
tigen-presenting cell function.13
The cellular immune response is critical
to the occurrence of transfusion-associated
graft-vs-host disease (TA-GVHD). In the
newborn, TA-GVHD has been reported
most often in the clinical setting of con-
firmed or suspected congenital immunode-
ficiency. It is recommended that infants
with suspected and/or documented T-cell
immunodeficiency receive irradiated blood
components. The majority of TA-GVHD cases
reported in nonimmunocompromised hosts
have occurred in infants who received intra-
uterine transfusion followed by postnatal
exchange transfusion.14 Aproposedexpla
-
nation is that lymphocytes given during
intrauterine transfusion could induce host
tolerance, impairing rejection of lymphocytes
given in the subsequent exchange transfu-
sions. There have also been rare cases of
TA-GVHDreportedinassociationwithex
-
treme prematurity, neonatal alloimmune
thrombocytopenia, and the use of extracor-
poreal membrane oxygenation (ECMO).14,15
Neonates present with TA-GVHD after a
560 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
longer latent period than adults, with fever
occurring at an average of 28 days after
exposure, rather than 10 days for immuno-
competent adults. There may be several
risk factors other than the immune status of
the recipient that predispose to TA-GVHD,
such as the number and viability of lym-
phocytes in the transfused component and
donor-recipient HLA compatibility. The
true incidence of TA-GVHD in the neonatal
setting is not known. However, on the basis
of data from Japan, it appears that the inci-
dence of reported TA-GVHD is far lower
than would be expected.15 The postulated
mechanism for this apparent decreased
susceptibility of newborns to TA-GVHD is
thought to be extrathymic and/or thymic
semitolerance of allogeneic donor T lym-
phocytes. As for all patients, directed-donor
units from biologic relatives must be irradi-
ated.15,16 There are no data to support the
practice of universal irradiation of blood
transfused to all infants or children.
Metabolic Problems
Acidosis or hypocalcemia may occur after
large-volume whole blood or plasma
transfusion because the immature liver of
the newborn metabolizes citrate ineffi-
ciently. Immature kidneys have reduced
glomerular filtration rate and concentrat-
ing ability, and newborns may have diffi-
culty excreting excess potassium, acid,
and/or calcium.
Potassium
Although potassium levels increase rapidly
in the plasma of stored red cells, small-
volume, simple transfusions administered
slowly have little effect on serum potas-
sium concentration in newborns. It has
been calculated that transfusion of 10
mL/kg of red cells (hematocrit 80%) ob-
tained from a unit of blood stored for 42
days in extended storage medium would
deliver 2 mL of plasma containing only
0.1mmol/Lofpotassium.Thisismuch
less than the daily potassium requirement
of 2 to 3 mmol/L for a 1-kg patient.17 How-
ever, serum potassium may, rise rapidly
after infusion of large volumes of red cells
in such circumstances as surgery, ex-
change transfusion, or extracorporeal cir-
culation, depending upon the plasma
potassium levels in the blood and manip-
ulation of the blood component.18,19 Of
interest, a unit of Red Blood Cells (RBCs)
preserved in AS-1 will deliver less extra-
cellular potassium compared to the
amount in RBCs stored in CPDA-1.16,20 In
stored irradiated blood, the problem of
potassium leak is potentiated; for selected
patients, it may be desirable to wash irra-
diated cells, if they have subsequently
been stored >24 hours.19 There are in-
creasing anecdotal reports of infants who
receive either older RBC units or units ir-
radiated more than 1 day before transfu-
sion having severe adverse effects (eg,
cardiac arrest, death) after transfusion of
these products into central lines or intra-
cardiac lines.21,22
The untoward consequences of washing
blood, such as reducing its shelf life and the
possible introduction of bacteria, must be
considered. It is preferable to perform irra-
diation as close to the time of administra-
tion as possible, obviating the concern for
high levels of potassium in the transfused
product.
2,3-Diphosphoglycerate
Neonates with respiratory distress syn-
drome or septic shock have decreased
levels of intracellular red cell 2,3-DPG.
Alkalosis and hypothermia may further
increase the oxygen affinity of hemoglo-
bin, shifting the dissociation curve to the
left and making oxygen even less available
Chapter 24: Neonatal and Pediatric Transfusion Practice 561
Copyright © 2005 by the AABB. All rights reserved.
to the tissues. Arterial oxygenation may
be further compromised by respiratory
distress syndrome or other pulmonary
disease. Mechanisms that compensate for
hypoxia in adults, such as increased heart
rate, are limited in newborns. If a large
proportion of an infant’s blood volume has
come from transfusion of 2,3-DPG-de-
pleted blood, this may cause problems
that would not affect older children or
adults. Because 2,3-DPG levels decline
rapidly after the first week of storage, the
freshest blood conveniently available (up
to14days)shouldbeusedforexchange
transfusion in newborns. For small-volume
transfusions, the medical necessity for
fresh blood has never been demonstrated
and arguments have been raised to sug-
gest it is unnecessary.1,16,19
Cytomegalovirus Infection
Perinatal infection with cytomegalovirus
(CMV) may occur, acquired either in utero
or during the birth process. Neonates can
be infected during breast-feeding or by
close contact with mothers or nursery
personnel. CMV can also be transmitted
by transfusion, although the risk from the
current blood supply is small.23,24
CMV infection in newborns has ex-
tremely variable manifestations, ranging
from asymptomatic seroconversion to death.
Studies of CMV in neonatal transfusion re-
cipients reveal the following observations:
1. The overall risk of symptomatic post-
transfusion CMV infection seems to
be inversely related to the seropo-
sitivity rate in the community. Al-
though many adults are positive for
CMV antibodies, the rate of symp-
tomatic CMV infection in newborns
is low.
2. Symptomatic CMV infection during
the neonatal period is uncommon in
children born to seropositive moth-
ers.25
3. The risk of symptomatic posttrans-
fusion infection is high in multi-
transfused preterm infants weighing
less than 1200 g who are born to
seronegative mothers.17,26
4. The risk of acquiring CMV infection
is directly proportional to the cumu-
lative number of donor exposures
incurred via transfusion.
5. Cytomegalovirus in blood is associ-
ated with leukocytes. The risk of vi-
rus transmission can be reduced by
transfusing CMV-reduced-risk blood
from seronegative donors or by us-
ing leukocyte-reduced components.
Although deglycerolized and washed
red cells also have a reduced risk of
CMV infection, leukocyte reduction
by filtration is the technique of
choice.16,27-30
Red Cell Transfusions in
Infants Less than 4 Months
of Age
RBCs are the component most often
transfused during the neonatal period.
Many of the physiologic considerations
mentioned above directly affect decisions
regarding indications for transfusion, se-
lection, and administration of red cell
components, as well as the requirements
for compatibility testing.
Compatibility Testing
Because the neonate and young infant are
immunologically immature, alloimmuni-
zation to red cell antigens is rare during
the neonatal period. A study of 90 neo-
nates who received 1269 transfusions from
different donors found no instances of
antibody production even with use of
562 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
very sensitive detection techniques.31 Other
investigators confirm the relative infre-
quency of alloantibodies directed against
red cell, as well as HLA, antigens.13,28
Because alloimmunization is extremely
rare and repeated testing increases iatro-
genic blood loss, AABB Standards for Blood
Banks and Transfusion Services32(p42) requires
only limited pretransfusion serologic test-
ing for infants under 4 months old. Initial
testing must include ABO and D typing of
red cells and a screen for red cell antibod-
ies, using either serum or plasma, from the
mother or the infant.
During any one hospitalization, compat-
ibility testing and repeat ABO and D typing
may be omitted, provided that the screen
for red cell antibodies is negative; that all
red cells transfused are group O or ABO-
identical or ABO-compatible; and that red
cellsareeitherDnegativeorthesameD
type as the patient. It is unnecessary to test
the infant’s serum for anti-A and/or anti-B
as a component of blood typing. Before giv-
ing non-group-O red cells, the neonate’s se-
rum must be checked for passively ac-
quired maternal anti-A or anti-B and must
include the antiglobulin phase. If the anti-
body is present, ABO-compatible red cells
lacking the corresponding A or B antigen
must be used until the antibody is no lon-
ger detected. In this setting, it is not neces-
sary to perform crossmatches. If an unex-
pected non-ABO red cell antibody is
detected in the infant’s specimen or the
mother’s serum contains a clinically signifi-
cant red cell antibody, the infant should be
given either RBC units tested and found to
lack the corresponding antigen(s) or units
compatible by antiglobulin crossmatch.
This practice should continue for as long as
maternal antibody persists in the infant’s
blood. The institution’s policy will deter-
mine how frequently to recheck the screen
for red cell antibodies; once a negative re-
sult is obtained, subsequent crossmatches
and/or provision of blood lacking the target
antigen are unnecessary. It is important to
avoid transfusion of any component that
may transfer unexpected antibody or ABO-
incompatible antibodies to the infant.
Indications for Red Cell Transfusion
Certain events in the perinatal period cause
anemia, for which the benefits of red cell
transfusion are unquestioned. These in-
clude spontaneous fetomaternal or feto-
placental hemorrhage, twin-twin transfu-
sion, obstetric accidents, and internal
hemorrhage. A venous hemoglobin of less
than 13 g/dL in the first 24 hours of life in-
dicates significant anemia.33 For severely
anemic neonates with congestive heart
failure, it may be necessary to remove ali-
quots of their dilute blood and transfuse
concentrated red cells. This “partial ex-
change” transfusion will prevent intravas-
cular volume overload. Most red cell
transfusions in the neonatal period, how-
ever, are given either to replace iatrogenic
blood loss or to treat the physiologic de-
cline in hemoglobin (anemia of pre-
maturity) when it complicates clinical
problems.
Because tissue demand for oxygen can-
not be measured directly and because so
many variables determine oxygen availabil-
ity, no universally accepted criteria exist for
transfusion of preterm or term neonates.
Despite the widespread use of micro-
methods for laboratory tests and growing
use of bedside or noninvasive monitoring
devices, infants still sustain significant cu-
mulative blood loss from laboratory sam-
pling. In a sick neonate, red cell replacement
is usually considered when approximately
10% of the blood volume has been re-
moved. The decision to transfuse a new-
born for anemia should include evaluation
of the hemoglobin levels expected for the
patient’s age and clinical status, as well as
Chapter 24: Neonatal and Pediatric Transfusion Practice 563
Copyright © 2005 by the AABB. All rights reserved.
the amount of blood loss over time. Trans-
fusionmaybemoreaggressiveintheinfant
in respiratory distress who is hypoxic and
more vulnerable to cerebral hemorrhage.
Considerable controversy surrounds the
correlation of the “signs of anemia” in the
preterm infant (tachycardia, tachypnea,
bradycardia, recurrent apnea, and poor
weight gain) with response to red cell trans-
fusions.1When red cells are transfused, they
are usually given in small volumes of 10 to
15 mL/kg (or less if the infant cannot toler-
ate this volume). The hematocrit of the red
cell component transfused will depend on
the anticoagulant/preservative used and
how the original unit is processed to pro-
vide small component transfusions for neo-
nates. A transfusion of 10 mL/kg of red cells
adjusted to a hematocrit greater than 80%
just before release for transfusion should
raise the hemoglobin concentration by ap-
proximately 3 g/dL. A transfusion of 10
mL/kg of red cells in additive solution,
which have a hematocrit of approximately
65%, will result in a posttransfusion hemo-
globin increment less than 3 g/dL.
Red Cell Components Used for Neonatal
Transfusion
The small-volume requirements of trans-
fusion to neonatal recipients make it pos-
sible to prepare several aliquots from a
single donor unit, thus limiting donor ex-
posure and decreasing donor-related risks.
Several technical approaches are avail-
able to realize this advantage and to mini-
mize wastage.34
Aliquoting for Small-Volume Transfusion
A multiple-pack system is a common tech-
nique for providing small-volume red cell
transfusions.34,35 Quad packs, where a sin-
gle unit of Whole Blood is collected into a
bag with four integrally attached contain-
ers, can be used to increase the number of
transfusions an infant can receive from
one donor. Because the original seal re-
mains intact, each container has the expi-
ration date of the original unit. With use
of a sterile connecting device, multiple
bags called pedi-packs or specially de-
signed syringe systems can be integrally
attached to a unit of RBCs after compo-
nent preparation. This maintains a closed
system and further increases the number
of small-volume transfusions obtained
from a single donor. Sterile connecting
devices can be used to prepare small
aliquots for transfusion in the blood bank.
If aliquots are prepared by entering the
bag through a port, the unit and the
aliquot are assigned a 24-hour shelf life, if
refrigerated.
Each aliquot must be fully labeled as it is
prepared, including the time it outdates.
The origin and disposition of each aliquot
must be recorded. Using these techniques,
a recipient can receive multiple small-vol-
ume transfusions from a single donation
until the expiration of the original unit,
thereby reducing donor exposure.34,36 The
disadvantage of any method that creates
aliquots from a single donation is that any
undetected, transmissible pathogen that
might be present in the primary unit can be
disseminated to multiple recipients.
In order to prevent waste, many transfu-
sion services assign a unit of RBCs to one or
more infants based on their weight. As an
example, 1 or 2 lower birthweight infants
may be assigned to one unit because they
will most likely require the greatest number
of transfusions. On the other hand, four
larger infants may be assigned to one unit
because their transfusion needs will not be
as great.37,38
Red Cells with Additive Solution
RBCs used for pediatric transfusions were
traditionally stored in CPDA-1.35 Additive
564 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
solutions (AS) used as anticoagulants/
preservatives contain additional adenine
and dextrose and some contain mannitol.
There has been concern about the poten-
tial side effects of these additives because
large amounts of adenine and mannitol
have been associated with renal toxicity.
Mannitol is also a potent diuretic and, be-
cause of its effect on the fluid dynamics in
preterm infants, may cause unacceptable
fluctuations in cerebral blood flow. The
different constituents of these solutions
are found in Chapter 8. However, when
the dose of transfused red cells is small (5
to 15 mL/kg), the recipient is exposed to
relatively small amounts of the preserva-
tive solutions. Clinical studies comparing
red cells stored in AS-1 and AS-3 solutions
have shown no apparent detrimental ef-
fects in neonates receiving simple trans-
fusions, and, after adjustment for the
lower hematocrit of the component, they
are as effective as CPDA-1 cells in increas-
ing hemoglobin. Furthermore, the addi-
tional sugars present have been shown to
benefit glucose homeostasis in compari-
son with CPDA-1.39 Studies on the safety
of AS-3-preserved red cells in neonates have
been published.38,40 Because the compo-
nents of AS-5 are the same as those found
in the other additive solutions, it is con-
sidered acceptable for use in neonates.
Using theoretical calculations in a vari-
ety of clinical situations, Luban et al41 dem-
onstrated that red cells preserved in
extended-storage media present no sub-
stantive risks when used for small-volume
transfusions. For preterm infants with se-
vere hepatic or renal insufficiency, however,
removing the additive-containing plasma
may be beneficial. This is particularly im-
portant if there will be multiple transfu-
sions that could have a cumulative effect.
The safety of red cells stored in additive so-
lutions and used for massive transfusions,
such as cardiac surgery or exchange trans-
fusion, has not been studied. Concern still
remains regarding the use of blood stored
in additive solutions being used outside the
setting of simple, small-volume transfu-
sion.40,41 However, with extensive anecdotal
use in large transfusion services, there have
not been reports of deleterious effects from
the infusion of additive solutions.42,43
Whether or not placental/umbilical cord
blood will become an acceptable form of
neonatal transfusion remains to be seen.
Althoughuseofthistypeof“autologous”
blood would eliminate some infectious dis-
ease risks, questions regarding quantity,
quality, and sterility raise concerns about
its safety and efficacy.18
Transfusion Administration
Vascular access is often difficult in the tiny
newborn and in any infant requiring
long-term or repeated intravenous infu-
sions. Within a short time after birth, the
umbilical artery may be cannulated. Trans-
fusion through a needle as small as 25-
gauge or a vascular catheter as small as
24-gauge has been shown to cause little
hemolysis and to be safe when constant
flow rates are used. Transfusion through
smaller gauge catheters has not been
thoroughly evaluated.
It is not usually necessary to warm small-
volume transfusions that are given slowly,
but it is important to be able to control the
volume and rate of infusion. Constant-rate
electromechanical syringe delivery pumps
provide this control and cause minimal
hemolysis, even when used with inline leu-
kocyte reduction filters.44,45
The length of the plastic tubing used can
add significantly to the volume required for
transfusion. Infusion sets identified as suit-
able for platelets or components have less
dead space than standard sets because they
have short tubing and a small 170-micron
filter. Pediatric microaggregate filters (20-
Chapter 24: Neonatal and Pediatric Transfusion Practice 565
Copyright © 2005 by the AABB. All rights reserved.
or 40-micron) are often used for their small
priming volume, not for the removal of
microaggregates. Hemolysis may occur
when stored blood is given by negative
pressure filtration through these filters.46
Administration rates for RBCs have not
been extensively studied, nor are there
standard practices; rates of transfusion as
well as devices used vary with institutions.
The rate of administration of blood and
blood components in neonates and infants
should be individualized on the basis of the
patient’s clinical needs. Theoretical con-
cerns regarding rapid changes in intra-
vascular volume and electrolyte changes in
these small, labile patients have focused on
an increased risk for intracranial hemor-
rhage. This has not been clearly demon-
strated. For simple RBC transfusions, trans-
fusing products over 2 to 4 hours is usually
adequate. When there is an urgent need be-
cause of shock or severe bleeding, infusion
should be as rapid as possible. Products
can be transfused safely using a variety of
devices. It is important that mechanical
systems be tested and validated for use
with blood and blood components.
Exchange Transfusion for
Hyperbilirubinemia
The fetal liver has limited capacity to con-
jugate bilirubin. In utero, unconjugated
bilirubin crosses the placenta for excre-
tion through the mother’s hepatobiliary
system. After birth, transient mild hyper-
bilirubinemia normally occurs during the
first week of life and is referred to as “phy-
siologic jaundice.” Liver function is less ma-
ture, and jaundice worsens in premature
neonates. When the level of unconjugated
bilirubin is excessive, bilirubin may cross
the blood-brain barrier and concentrate
in the basal ganglia and cerebellum; the
resulting damage to the central nervous
system (CNS) is called kernicterus. Photo-
therapy with fluorescent blue lights is the
most common treatment for hyperbiliru-
binemia; exchange transfusion is reserved
for phototherapy failures. The most com-
mon reason, however, for an exchange to
be performed in a neonate is to correct
hyperbilirubinemia.
Pathologic processes that may result in
excessively high unconjugated bilirubin
levels in neonates include immune-medi-
ated hemolysis, nonimmune hemolysis,
bile excretion defects and impaired albu-
min binding. Exchange transfusion re-
moves unconjugated bilirubin and provides
additional albumin to bind residual biliru-
bin. If hyperbilirubinemia is due to anti-
body-mediated hemolysis, exchange trans-
fusion is of additional benefit by removing
free antibody and antibody-coated red cells
while providing antigen-negative red cells
that will survive normally.
Exchange transfusion should be per-
formed before bilirubin rises to levels at
which CNS damage occurs. Several factors
affect the threshold for toxicity. CNS dam-
age occurs at lower levels if there is pre-
maturity, decreased albumin binding capac-
ity,orthepresenceofsuchcomplicating
conditions as sepsis, hypoxia, acidosis, hy-
pothermia, or hypoglycemia. In full-term
infants, kernicterus rarely develops at indi-
rect bilirubin levels less than 25 mg/dL, but,
in sick VLBW infants, kernicterus has oc-
curred at levels as low as 8 to 12 mg/dL.47
Therateatwhichbilirubinrisesismore
predictive of imminent need for exchange
transfusion than the absolute level attained.
Neonates with severe anemia and a rapid
rise in bilibrubin, despite phototherapy, re-
quire exchange transfusion. A two-volume
exchange transfusion removes approxi-
mately 70% to 90% of circulating erythro-
cytes and about 25% of the total bilirubin.
Because of reequilibration between the
extravascular tissue and plasma bilirubin,
levels may again rise, resulting in the need
566 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
for a second exchange transfusion.48 Indica-
tions for repeat exchange are similar to
those for the initial exchange.
The American Academy of Pediatrics re-
cently released new guidelines for the man-
agement of newborn infants born ≥35
weeks of gestation with hyperbilirubine-
mia. It is hoped that raising awareness
about the potential for hyperbilirubinemia
in this patient group will reduce its fre-
quency and provide a framework for opti-
mal treatment, to include the use of photo-
therapy, exchange transfusion, and Immune
Globulin, Intravenous (IGIV).49
Exchange Transfusion for Other Causes
The safety and efficacy of exchange trans-
fusion in the neonatal period for other in-
dications should be evaluated on a case-
by-case basis, using guidelines in pub-
lished literature. Treatment categories for
disorders based on proven vs theoretical
benefits have been recommended (see
Table6-1).Thetreatmentofdisseminated
intravascular coagulation (DIC) using ex-
change transfusion has yielded variable
results, perhaps because only the sickest
infants have been selected to receive this
therapy. The most important aspect of
therapy for neonatal DIC is to treat the
underlying disease.
Exchange transfusion is occasionally
used to remove other toxins, such as drugs
or chemicals given to the mother near the
time of delivery, drugs given in toxic doses
to the neonate/infant, or substances such
as ammonia that accumulate in the new-
born because of prematurity or inherited
metabolic diseases.50,51
Technique of Exchange Transfusion
Choice of Components
Red cells are resuspended in compatible
thawed Fresh Frozen Plasma (FFP) for ex-
change transfusion. If AS-RBC units are
used, depending on the clinical situation,
some institutions would choose to remove
the additive-containing plasma, to reduce
the volume transfused. As discussed ear-
lier, washing components may not be
necessary or desirable. Many transfusion
services use red cells that have been
screened and found to lack hemoglobin S
for exchange transfusion, to avoid the
possibility of intravascular sickling.
The glucose load administered during
exchange transfusion can be extremely
high. This stimulates the infant to secrete
insulin, which may lead to rebound hypo-
glycemia. It is important to monitor blood
glucose levels for the first few hours after
the procedure.
Because unconjugated bilirubin binds to
albumin, albumin is frequently used to in-
crease intravascular binding. With addi-
tional albumin in the circulation, bilirubin
from the extravascular space diffuses out to
the intravascular space. This, in turn, in-
creases the total quantity of bilirubin re-
moved during the exchange. There have
been conflicting results, however, about the
efficacy of administering albumin either
before or during exchange to enhance bili-
rubin removal. A study that compared 15
hyperbilirubinemic neonates given albu-
min with 27 who received none found simi-
lar efficiency of bilirubin removal in both
groups.52 Infusing albumin raises the colloid
osmotic pressure and increases intravas-
cular volume. Therefore, it should be given
cautiously, if at all, to neonates or infants
who are severely anemic, have increased
central venous pressure, or are in renal or
congestive heart failure.
Exchange transfusion may cause dilu-
tional thrombocytopenia and/or coagulo-
pathy that require transfusion of platelets
and/or other components containing coag-
ulation factors. Platelet counts and coagu-
lation parameters should be monitored af-
ter exchange transfusion.
Chapter 24: Neonatal and Pediatric Transfusion Practice 567
Copyright © 2005 by the AABB. All rights reserved.
Volume and Hematocrit
An exchange transfusion equal to twice the
patient’s blood volume is typically recom-
mended for newborns; rarely is more than
one full unit of donor blood required. In
practice, the volume calculated for ex-
change is an estimate. The final hemato-
crit of transfused blood should be approx-
imately 40% to 50%, with sufficient plasma
to provide clotting factors, if needed. In
the unusual event that the infant’s condi-
tion demands a high postexchange hemato-
crit, a small-volume transfusion of red cells
can be given after the exchange, or units
with a higher hematocrit used for ex-
change. It is important to keep the blood
mixed during the exchange; if it settles in
the container, the final aliquots will not
have the intended hematocrit. The in-
fant’s hematocrit and bilirubin level
should be measured using the last aliquot
removed in the exchange.
Vascular Access
Exchange transfusions in the newborn
period are usually accomplished via cath-
eters in the umbilical vessels. Catheteriza-
tion is easiest within hours of birth, but it
may be possible to achieve vascular ac-
cess at this site for several days. The cath-
eters should be radio-opaque to facilitate
radiographic monitoring during and after
placement. If umbilical catheters are not
available for exchange transfusion, small
central venous or saphenous catheters
may be used.
Methods Used
Two methods of exchange transfusion are
incommonuse.Intheisovolumetric
method, there is vascular access through
two catheters of identical size. Withdrawal
and infusion occur simultaneously, regu-
lated by a single peristaltic pump. The
umbilical artery is usually used for with-
drawal and the umbilical vein for infu-
sion.
The manual push-pull technique can be
accomplished through a single vascular ac-
cess. A three-way stopcock joins the unit of
blood, the patient, and an extension tube
that leads to the graduated discard con-
tainer. An inline blood warmer and a stan-
dard blood filter should be incorporated in
the administration set. The maximum vol-
ume of each withdrawal and infusion will
depend on the infant’s size and hemo-
dynamic status. The rate at which exchange
transfusion occurs may alter the infant’s
hemodynamic status. It is important to
maintain careful records during an ex-
change transfusion. The procedure should
take place over 1 to 1.5 hours.
Transfusion of Other
Components
Although the percentage of VLBW and
ELBW infants being transfused has de-
creased significantly since the 1980s, be-
tween 61% and 94% of these neonatal pa-
tients can be expected to receive multiple
red cell transfusions. The smallest pa-
tients will receive the greatest number of
transfusions. It is estimated that a much
lower percentage of infants receive other
components.1,9,53,54
Platelet Transfusion
The normal platelet count of newborns is
similar to that of adults. A platelet count
less than 150,000/µL in a full-term or pre-
mature infant is abnormal. Approximately
20% of infants in neonatal intensive care
units have mild-to-moderate thrombo-
cytopenia, which is the most common
hemostatic abnormality in the sick in
-
fant.55 Neonatal thrombocytopenia may
result from impaired production or in-
568 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
creased destruction of platelets, abnormal
distribution, or a dilutional effect second-
ary to massive transfusion such as ex-
change transfusion. Increased destruction
is the most common cause; it may be as-
sociated with a multitude of conditions and
is usually transient. Neonatal alloimmune
thrombocytopenia is discussed in Chap-
ter 23.
Indications
Platelet transfusion is indicated in neo-
nates and young infants with platelet
counts below 50,000/µLwhoareexperi
-
encing bleeding.55 The use of prophylactic
platelet transfusions in the newborn re-
mains controversial. In thrombocyto-
penic adults, the risk of severe bleeding is
rare unless the platelet count is less than
10,000/µL.56 Conversely, preterm neonates
and infants with other complicating ill-
nesses may bleed at higher platelet counts.
Factors contributing to this increased risk
of bleeding include a quantitatively lower
concentration of plasma coagulation fac-
tors; circulation of an anticoagulant that
enhances inhibition of thrombin; intrin-
sic or extrinsic platelet dysfunction; and
increased vascular fragility.57 Of major
concern is intraventricular hemorrhage,
which occurs in up to 40% of preterm ne-
onates in the first 72 hours. Although pro-
phylactic platelet transfusions increase
platelet counts and shorten the bleeding
time in these infants, the incidence or ex-
tent of intraventricular hemorrhage is not
reduced.57 Because of the apparent lack of
clinical benefit, controversy exists about
the use of platelet transfusion in this set-
ting, as well as the selection of an optimal
dose. After a platelet transfusion, a post-
transfusion platelet count soon after
transfusion can be used to evaluate sur-
vivalinthecirculationbutmaynotpre
-
dict hemostatic efficacy. Repeated platelet
transfusions, without an appropriate rise,
may not be beneficial.
Platelet Components
A platelet dose of 5 to 10 mL/kg body
weight should raise the platelet count of
an average full-term newborn by 50,000
to 100,000/µL, depending on the platelet
concentration in the component used.17,57
The platelet component should be group
specific, if possible, and should not con-
tain clinically significant unexpected red
cell antibodies. Transfusion of ABO-in-
compatible plasma is more dangerous in
infants than in adults because of their
very small blood volumes. If it is neces-
sarytogiveaplateletunitthatcontains
incompatible plasma (due to antibodies
in the ABO or other blood groups), plasma
can be removed (see Method 6.15) and
the platelets resuspended in saline. FFP
can be substituted as the resuspending
medium if the patient also requires clot-
ting factors, but it carries the risk of infec-
tious disease transmission. Routine cen-
trifugation of platelets to reduce the
volume of transfusion is not necessary.53,55
If platelets have been volume reduced
and placed in a syringe, the pH declines
rapidly, a potential problem for an already
ill, acidotic patient.58 Therefore, if there is
a need to reduce the volume of platelets,
it should be done just before transfusion
and the component must be infused
within4hours,ifdoneinanopensystem.
Granulocyte Transfusion
Neonates are more susceptible to severe
bacterial infection than older children be-
cause of both the quantitative and quali-
tative defects of neutrophil (polymorpho-
nuclear cell or PMN) function and, in the
Chapter 24: Neonatal and Pediatric Transfusion Practice 569
Copyright © 2005 by the AABB. All rights reserved.
absence of pathogen-specific maternal
antibody, to deficiency of humoral immu-
nity. Group B streptococcus is the most
frequent cause of early-onset neonatal
sepsis, and, despite improvement in
antimicrobial therapy and intensive care,
it is still associated with a high mortality
rate. Controversy surrounds several issues
in granulocyte transfusions for neonates,
including dose, neutrophil level at which
to transfuse, and efficacy as compared to
other forms of therapy.59 Because it ap-
pears that efficacy is related to dose, the
smaller blood volume of infants and
young children may result in these pa-
tients having a better response to this
form of therapy. A meta-analysis pub-
lished in 1996 concluded that granulocyte
doses greater than 1 ×109PMN/kg re-
sulted in a better clinical response.60
Granulocyte concentrates prepared by
apheresis are more desirable than those
prepared from buffy coats because they
produce a higher yield.17,60 Products col-
lected from donors on a regimen of
granulocyte colony-stimulating factor
(G-CSF) and steroid mobilization can
yield higher numbers of granulocytes
than those collected from an unstimulat-
ed donor. There have been some encour-
aging observations on the use of IGIV in
the treatment of early neonatal sepsis, al-
though conflicting data result in a lack of
consensus on its use.59,61-64 Preliminary stud-
ies of hematopoietic growth factors (eg,
G-CSF) show promise in the treatment of
overwhelming bacterial infection in the
newborn.59,61,65,66 For adults and larger chil-
dren, a minimum dose of 1 ×1010 PMN/kg
is recommended.60,67,68
Indications
Although the precise role of granulocyte
transfusion for neonatal sepsis is unclear,
certain clinical situations exist in which
granulocyte transfusion may be consid-
ered as an adjunct to antibiotic therapy.
Candidates for possible granulocyte
transfusion are infants with strong evi-
dence of bacterial septicemia, an absolute
neutrophil count below 3000/µL, and a
diminished marrow storage pool, such
that less than 7% of their nucleated cells
in the marrow are granulocytes at the
stage of metamyelocytes or more mature
forms.18,69
Granulocyte Components
Granulocytes are harvested by standard
apheresis techniques. For infants, a dose
of 10 to 15 mL/kg is recommended, which
is about 1 ×109to 2 ×109PMN/kg.17,60 Be-
cause neonatal neutrophil function is of-
ten abnormal, the use of granulocyte
transfusions in this age group may be
beneficial.17,60,67,70-72 Administration should
be continued daily until an adequate
white cell mass is achieved or the patient
has clinically improved. Based on the fact
that granulocyte concentrates contain
large numbers of lymphocytes, there is
general consensus that all granulocyte
transfusions should be irradiated to pre-
vent graft-vs-host disease. For granulo-
cyte transfusions to neonates, donors are
usually selected to be CMV seronegative
and must be ABO compatible with the in-
fant, in accordance with Standards,be
-
cause there is a significant volume of red
cells in these products.32(p40) Many institu-
tions also provide products that are
D-compatible (see Chapter 21).
Transfusion to Enhance Hemostasis
The elements of the hemostatic system of
the newborn are similar to those of older
children and adults, but the concentration
of many plasma proteins is decreased.
Coagulation factors do not cross the pla-
centa, but are independently synthesized
570 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
by the fetus. Plasma levels of coagulation
proteins increase progressively with ges-
tational age. At birth, the infant’s pro-
thrombin time and partial thrombo-
plastin time are prolonged, compared to
older children and adults, primarily the
result of physiologically low levels of the
vitamin-K-dependent factors (II, VII, IX,
and X) and contact factors (XI, XII, pre-
kallikrein, and high-molecular-weight
kininogen).73 Proteins C and S and anti-
thrombin inhibitors of coagulation are
also at low levels. These two systems usu-
ally balance each other, so that spontane-
ous bleeding and thrombosis in the healthy
newborn are rare, but very little reserve
capacity exists for response to pathologic
insults. Therefore, serious bleeding may
occur in the first week of life in the sick
premature infant as a result of hemostatic
immaturity coupled with an acquired dis-
order of hemostasis.
In addition to having physiologically low
levels of the vitamin-K-dependent factors,
neonates may also become vitamin-K-defi-
cient during the first 2 to 5 days of life, plac-
ing them at risk of bleeding. This “hemor-
rhagic disease of the newborn” is rare in
developed countries because intramuscular
vitamin K is routinely given at birth. If vita-
min K therapy is omitted, especially if the
neonate is breast fed, life-threatening hem-
orrhage may occur. This should be treated
with FFP.2,74
Although hereditary deficiencies of coag-
ulation factors may be apparent in the
newborn, significant bleeding is rare.
Coagulopathy more often results from an
acquired defect such as liver disease or
DIC.74 Although component therapy re-
placement may temporarily correct the
hemostatic problem, treatment of the un-
derlying disease will ultimately reduce the
need to treat the acquired defect.
Newborns who are heterozygous for de-
ficiencies of inhibitory proteins rarely expe-
rience complications in the absence of an-
other pathologic insult. However, the ho-
mozygous form of protein C deficiency has
caused life-threatening thrombotic compli-
cations in the newborn period. In countries
where it is available, protein C concentrates
prepared from human plasma should be
used as the initial treatment for neonates
with homozygous protein C deficiency pre-
senting with purpura fulminans. Protein C
concentrates may be available on a com-
passionate use basis in the United States.
Otherwise, plasma infusion is used as the
initial treatment during the acute event,
with subsequent anticoagulant therapy for
long-term management.74,75
Fresh Frozen Plasma
Freshfrozenplasmamaybeusedtore-
place coagulation factors in newborns,
particularly if multiple factors are in-
volved, such as in vitamin K deficiency.
The usual dose is 10 to 15 mL/kg, which
should increase factor activity by 15% to
20% unless there is marked consumptive
coagulopathy.74 As with red cell transfu-
sions, there are several methods to pro-
vide small-volume FFP infusions while
limiting donor exposure and wastage of
components. Blood can be collected into
a system with multiple integrally attached
bags, creating aliquots that can be pre-
pared for freezing.34 Once thawed, these
aliquots can be further divided and used
for several patients within a 24-hour pe-
riod. If not used within 24 hours as aliquots,
the thawed plasma (stored at 1 to 6 C) can
still be used as a means to decrease donor
exposure. As with all patients, newborns
must receive FFP that is ABO compatible
and free of clinically significant unex-
pected antibodies. Group AB FFP is often
used because a single unit provides com-
patible small aliquots for several neonates
requiring FFP simultaneously.
Chapter 24: Neonatal and Pediatric Transfusion Practice 571
Copyright © 2005 by the AABB. All rights reserved.

Cryoprecipitate
Cryoprecipitate is rich in fibrinogen, co-
agulation Factors VIII and XIII, and von
Willebrand factor. This component is of-
ten used in conjunction with platelet
transfusions to treat DIC in the newborn.
In DIC, fibrinogen and platelets are the el-
ements most often severely depleted. The
plasma in which the platelets are sus-
pended is a source of stable coagulation
factors. Cryoprecipitate provides concen-
trated levels of additional fibrinogen and
storage-labile Factor VIII. For an infant, one
bag is sufficient to achieve hemostatic
levels. As with FFP and platelets, the
cryoprecipitate should be ABO compati-
ble with the neonatal recipient. Directed
donor cryoprecipitate is not recommended
as first-line therapy in the newborn with
hemophilia A because safer alternatives are
available. Recombinant Factor VIII prod-
ucts or virus-inactivated, monoclonal-an-
tibody-purified, plasma-derived products
are the standard treatment.76 Cryoprecipi-
tate should be used to treat von Wille-
brand disease only as a last resort, when
safer products are not readily available.
More information on the use of cryopreci-
pitate can be found in Chapter 21.
Neonatal Polycythemia
A venous hematocrit greater than 65% or
hemoglobin in excess of 22 g/dL any time
in the first week of life defines polycythemia,
a condition that occurs in approximately
5% of all newborns. Small-for-gestational-
age infants and infants of diabetic moth-
ers are at increased risk for developing
polycythemia. As the hematocrit rises
above 65%, the viscosity of blood in-
creases exponentially and oxygen trans-
port decreases. For neonates, the expo-
nential rise may occur at a hematocrit
closer to 40%.77 Infants have a limited
ability to increase cardiac output to com-
pensate for hyperviscosity and may
develop congestive heart failure. Impair-
ment of blood flow can cause CNS abnor-
malities, pulmonary and renal failure, and
necrotizing enterocolitis. Phlebotomy can
be used to normalize the hematocrit to
55% to 60% and improve tissue perfusion,
while maintaining the blood volume.
To treat polycythemia, whole blood is re-
moved and the volume replaced with
crystalloid (such as normal saline), the
choice being based on the quantity needed
and on the infant’s clinical condition.
Plasma is not recommended because the
volume administered will be insufficient to
correct any coagulopathy and because
necrotizing enterocolitis has been reported
when it is used in this procedure.78 Afor-
mula to approximate the volume of colloid
replacement required (and the volume of
blood to be drawn) for the exchange is47:
Volume of replacement fluid =
blood volume ×
(observed hematocrit – desired hematocrit)
observed hematocrit
Extracorporeal Membrane
Oxygenation
ECMO is a modified cardiopulmonary by-
pass technique that has been used for
short-term support for cardiac or respira-
tory failure. It is performed in specialized
centers and only for patients in whom
conventional medical therapy has failed
and anticipated survival with such ther-
apy is limited (see Table 24-2). The use of
ECMO in patients other than neonates is
not widespread. The therapy is more suc-
cessful in infants, whose small blood vol-
ume allows for total cardiorespiratory
support and whose primary respiratory
problem often resolves after 1 to 2 weeks
572 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

of support. ECMO provides gas exchange
independent of the patient’s lungs, allow-
ing them time to improve or heal without
exposure to aggressive ventilator support
and the secondary lung damage this may
cause.79
Individual ECMO centers establish their
own specific criteria for transfusion and
blood component selection, and standard
transfusion practices are lacking. Because
of the combination of factors present (in-
cluding systemic heparinization, platelet
dysfunction, thrombocytopenia, and other
coagulation defects) as well as the ECMO
circuitry itself, bleeding complications are
frequent. The ECMO team should be in
close communication with the blood bank
or transfusion service staff, and there
should be mutual agreement on protocols
to ensure consistency of care. Many infants
requiring ECMO have been transferred
from other hospitals, where they may al-
ready have received numerous transfu-
sions. The amount of red cell, platelet, and
FFP support required to maintain hemato-
logic and hemodynamic equilibrium will
vary depending on the clinical situation
and in accordance with the institutions’
practices.68 When platelet transfusion is re-
quired, some practitioners think it is im-
portant to transfuse through peripheral ac-
cess to avoid platelet damage, whereas
others will transfuse directly into the ECMO
circuitry because all blood will eventually
flow through the equipment. It is also im-
portant to monitor ionized calcium levels
and supplement them as needed.47,79 (See
Tables 24-3 and 24-4.)
Leukocyte Reduction
The benefit of leukocyte reduction of
components transfused to infants re-
mains controversial. Difficulty in identify-
ing transfusion reactions in this patient
population makes this question hard to
study. In addition, infants are rarely alloi-
mmunized because of the immaturity of
their immune system during this period
of development. The reduction of risk of
CMVtransmissiontoinfantsistheonly
benefit of leukocyte reduction that has
been well documented, as discussed ear-
lier.16,30,80
Of interest, a recent study in Canada
compared the clinical outcomes of prema-
ture infants weighing <1250 g before and
after implementation of universal leukocyte
reduction. Although neither mortality nor
bacteremia were reduced in the setting of
universal leukocyte reduction, other sec-
ondary clinical outcomes, such as retino-
pathy of prematurity and bronchopul-
monary dysplasia, were improved. Length
Chapter 24: Neonatal and Pediatric Transfusion Practice 573
Table 24-2. Disorders Treated by ECMO
■Meconium aspiration
■Diaphragmatic hernia
■Persistent pulmonary hypertension of the
newborn
■Severe group B streptococcal sepsis
Table 24-3. Risks of ECMO
■Bleeding
■Thrombosis
■Thrombocytopenia
■Neutropenia
■Platelet dysfunction
■Stroke
■Seizure
■Air embolism
■Hemolysis
■Systemic hypertension
■Cannulization of carotid artery
■Infectious complications of blood
transfusion
Copyright © 2005 by the AABB. All rights reserved.
of stay was decreased with universal leuko-
cyte reduction.81
Transfusion Practices in
Older Infants and Children
The indications for transfusion of red cells
and other components in older infants
(>4 months) and children are similar to
those for adults but must take into ac-
count differences in blood volume, ability
to tolerate blood loss, and age-appropri-
ate hemoglobin and hematocrit levels.
The most common indication for red cell
transfusioninchildrenistoreverseor
prevent tissue hypoxia resulting from de-
creased red cell mass associated with sur-
gical procedures or in response to anemia
of chronic diseases or hematologic malig-
nancies. It is important to remember that
normal hemoglobin and hematocrit levels
are lower in children than adults. Pediat-
ric patients may remain asymptomatic
despite extremely low levels of hemoglo-
bin, particularly if the anemia has devel-
oped slowly.
The decision to transfuse should be
based not only on the hemoglobin level but
also on the presence or absence of symp-
toms, the functional capacity of the child,
the etiology of the anemia, the possibility of
using alternative therapies, and the pres-
ence or absence of additional clinical con-
ditions that increase the risk for developing
hypoxia. If small-volume transfusions are
required, many of the methods described
to provide small-volume transfusions to ne-
onates could be applied. All pediatric pa-
tients over 4 months of age, however, must
be tested for ABO and D type as well as for
the presence of clinically significant antibod-
ies before red cell transfusions. Compatibil-
ity testing must be done in accordance with
Standards.32(pp37-41) Of note, a published ab-
stract reported that leukocyte reduction
does decrease the occurrence of febrile non-
hemolytic transfusion reactions in pediatric
hematology/oncology patients.82
Red Cell Support for Children with
Hemoglobinopathies
In certain childhood conditions, chronic
red cell transfusions are given not only to
treat tissue hypoxia but also to suppress
endogenous hemoglobin production. Ap-
proximately 6% to 10% of children with
sickle cell disease suffer a stroke, with
two-thirds experiencing a recurrence.83
The goal of transfusion for these patients
is to reduce the risk of stroke by decreas-
ing the percentage of circulating red cells
capable of sickling, while simultaneously
avoiding an increase in blood viscosity. It
is important to remember that raising the
hematocrit, without significantly reduc-
ing the percent of sickle cells, could in-
crease viscosity and negate any beneficial
effects of the transfusion.83,84 Therateof
recurrent stroke can be reduced to less
than 10% by maintaining a hemoglobin
level of 8 to 9 g/dL, with a hemoglobin S
level less than 30%, in children who have
had a cerebrovascular accident. This can
usually be achieved with a simple or par-
tial exchange transfusion every 3 to 4 weeks.
Therapy is continued indefinitely, as ces-
sation can lead to subsequent stroke.83,84
Because of concern about iron overload,
some workers follow several uneventful
years of transfusions to keep hemoglobin
574 AABB Technical Manual
Table 24-4. Contraindications for ECMO
■High risk for intraventricular hemorrhage
■Irreversible lung disease
■Systemic bleeding
■Severe asphyxia
■Presence of lethal malformations
Copyright © 2005 by the AABB. All rights reserved.
S below 30% with a less aggressive proto-
col that maintains hemoglobin S between
40% and 50%.84 Erythrocytapheresis has
been shown to improve iron balance in a
small cohort of patients.85 Adams et al
showed that transfusion to maintain a he-
moglobin S level less than 30% in children
who have abnormal results on trans-
cranial Doppler ultrasound reduced the
risk of a first stroke.86 The benefits of this
transfusion therapy, however, must be
weighed against the complications of
transfusion, such as iron overload and
alloimmunization, as well as the risks of
increased donor exposure during erythro-
cytapheresis. Although simple transfusion
increases blood viscosity, exchange trans-
fusion does not. Blood for transfusion to a
patient with sickle cell disease should ide-
ally be screened for hemoglobin S. In ad-
dition, most centers now provide leuko-
cyte-reduced blood for patients with sickle
cell disease to prevent alloimmunization
to platelets, which could complicate trans-
plantation.87
Red cell transfusions are also used to
treat acute complications associated with
sickle cell disease, such as splenic seques-
tration and aplastic crisis.88 Acute chest syn-
drome, a new pulmonary infiltrate in a pa-
tient with sickle cell disease, other than
atelectasis, with additional respiratory
symptoms and fever, carries a poor progno-
sis if untreated. Simple transfusion can be
used as a first-line therapy to improve oxy-
genation. For those patients who continue
to deteriorate or who do not improve, red
cell exchange transfusion should be per-
formed. There are no randomized con-
trolled trials comparing exchange and sim-
ple transfusion in this setting.89 Astudy
evaluating preoperative transfusion proto-
cols found that a conservative protocol, in
which the hemoglobin was raised to 10 g/
dL, was as effective in preventing perio-
perative complications as an aggressive ap-
proach to decrease hemoglobin S levels to
below 30%.90
Patients with sickle cell disease might
also be at risk for severe delayed hemolytic
transfusion reactions that could be life-
threatening because of the coincident sup-
pression of erythropoiesis. When a patient’s
hemoglobin level decreases after transfu-
sion, this “hyperhemolytic” syndrome—
wherein it appears that autologous red cells
are destroyed through an innocent by-
stander mechanism—should be suspected.
In these circumstances, transfusion should
be stopped and corticosteroid therapy, or a
combination of corticosteroid therapy
and IGIV, considered, based on reports of
efficacy in case studies.91,92 Autoantibody
formation also occurs in these patients af-
ter transfusion.93 In the hope of decreasing
the need for transfusion, medical interven-
tions are being explored. One therapy uses
hydroxyurea to increase the percentage of
hemoglobin F. By having a higher percent-
age of cells that are hemoglobin F, the for-
mation of hemoglobin S polymers is re-
duced. In addition, the concomitant decrease
in neutrophil counts that accompanies
hydroxyurea administration was found to
be independently associated with a reduc-
tion in the rate of crisis.94 Marrow trans-
plantation has also been used in some pa-
tients and may have a role in the future
treatment of patients with sickle cell dis-
ease.95,96
For children with thalassemia and severe
anemia, transfusion not only improves tis-
sue oxygenation but also suppresses
erythropoiesis. By suppressing ineffective
erythropoiesis, many of the complications
associated with the disease are amelio-
rated. So-called hypertransfusion, in which
the pretransfusion hemoglobin is kept be-
tween 8 and 9 g/dL, allows normal growth
and development, as well as normal levels
of activity for the child’s age. Supertransfu-
sion programs aim to maintain a pre-
Chapter 24: Neonatal and Pediatric Transfusion Practice 575
Copyright © 2005 by the AABB. All rights reserved.
transfusionhemoglobinconcentrationbe
-
tween 11 and 12 g/dL, in order to decrease
iron absorption from the gastrointestinal
tract. The results of maintaining near-nor-
mal hemoglobin levels are still controver-
sial. Iron overload is a complication of
treatment, requiring chelation therapy be-
ginning early in childhood.97
Antibody Production in Sickle Cell and
Thalassemia Patients
The frequency of red cell alloimmuni-
zation in chronically transfused children
varies with the disease, the age of first
transfusion, the number of transfusions
given, and the ethnic background of do-
nors and recipients, although patients
with sickle cell disease have the highest
rates of alloimmunization of any patient
group.98-100 Antibodies to the common an-
tigens of the Rh, Kell, Duffy, and Kidd
systems are often identified. It may be
prudent, therefore, to phenotype the pa-
tient’s red cell antigens as completely as
possible before beginning transfusion
therapy and to maintain a permanent re-
cord of the results. This can be helpful in
selecting compatible blood if alloim-
munization occurs. There is evidence that
transfusing K, C, and E antigen-negative
red cells can significantly reduce the rate
of alloimmunization.101 However, the
practice of transfusing only phenotypi-
cally matched units is controversial, espe-
cially in patients who have not yet devel-
oped the corresponding antibody, because
many of these units are difficult to ob
-
tain.83,102 A recent survey of 50 academic
medical centers in the United States and
Canada found that the most common
practice is to perform pretransfusion
phenotypicmatchingforC,E,andK.
103 In
patients who have already become immu-
nized and who are at high risk of develop-
ing additional antibodies, use of pheno-
typically matched units may be a reason-
able approach to prevent further allo-
immunization.102 Method 2.16 can be used
to perform red cell phenotyping on auto-
logous red cells of recently transfused pa-
tients.
African Americans with sickle cell dis-
ease frequently become immunized be-
cause the majority of red cell transfusions
are obtained from Caucasian donors, with
major differences in antigen exposure. In
some areas, blood collectors have devel-
oped programs to specifically recruit Afri-
can American donors to supply blood for
patients with sickle cell disease, in order to
reduce rates of alloimmunization.101 Leuko-
cyte-reduced blood components may be of
particular value for these chronically trans-
fused patients. One benefit would be to di-
minish the development of alloimmuniza-
tion to HLA antigens, in light of the prospect
of future marrow transplantation.87 There
are conflicting data whether leukocyte re-
duction can prevent alloimmunization to
red cell transfusion.104,105 Preventing febrile
transfusion reactions is also important for
patients who may be receiving a unit of
phenotypically matched blood. Discarding
such a unit would be both wasteful and
may have an impact on the ability to ade-
quately transfuse the patient.106
Platelets and Plasma
The indications for FFP and platelet
transfusions in older infants and children
parallel those for adults. Platelet transfu-
sions are most often given as prophylaxis
to children receiving chemotherapy. Pro-
phylactic platelet transfusions are seldom
givenwhenplateletcountsareabove
10,000 to 20,000/µL, but, as with red cell
transfusion and hemoglobin, the indica-
tion for platelet transfusion should not be
based solely on the platelet count. When
additive risk factors such as fever, sepsis,
576 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
DIC, or clotting abnormalities are present,
the platelet count may need to be higher
to prevent spontaneous hemorrhage. In the
absence of such factors, a much lower level
may be safe.56 In recent studies, the use of
ABO-compatible platelets has been asso-
ciated with better clinical outcomes.107-109
References
1. Hume H, Bard H. Small volume red blood cell
transfusions for neonatal patients. Transfus
Med Rev 1995;9:187-99.
2. Roseff SD, ed. Pediatric transfusion: A physi-
cian’shandbook.1sted.Bethesda,MD:
AABB, 2003.
3. Brugnara C, Platt OS. The neonatal erythro-
cyte and its disorders. In: Nathan DG, Orkin
SH, eds. Nathan and Oski’s hematology of in-
fancy and childhood. 5th ed. Philadelphia:
WB Saunders, 1998:19-52.
4. Blanchette V, Doyle J, Schmidt B, Zipursky A.
Hematology. In: Avery GB, Fletcher MA, Mac-
DonaldMG,eds.Neonatology:Pathophy-
siology and management of the newborn.
4th ed. Philadelphia: JB Lippincott, 1994:
952-99.
5. Wallgren G, Hanson JS, Lind J. Quantitative
studies of the human neonatal circulation.
Acta Paediatr Scand 1967;179(Suppl):43-54.
6. Ohls RK. Evaluation and treatment of ane-
mia in the neonate. In: Christensen RD, ed.
Hematologic problems of the neonate. Phila-
delphia: WB Saunders, 2000:137-69.
7. Ohls RK. Erythropoietin to prevent and treat
the anemia of prematurity. Curr Opin Pediatr
1999;11:108-14.
8. Maier RE, Obladen M, Müller-Hansen I, et al.
Early treatment with erythropoietin βame-
liorates anemia and reduces transfusion re-
quirements in infants with birth weights be-
low 1000 g. J Pediatr 2002;141:8-15.
9. Widness JA, Seward VJ, Kromer IJ, et al.
Changing patterns of red cell transfusion in
very low birth weight infants. J Pediatr 1996;
129:680-7.
10. Vamvakas EC, Strauss RG. Meta-analysis of
controlled clinical trials studying the effi-
cacy of rHuEPO in reducing blood transfu-
sions in the anemia of prematurity. Transfu-
sion 2001;41:406-15.
11. Luban NLC, Mikesell G, Sacher RA. Techni-
ques for warming red blood cells packaged
in different containers for neonatal use. Clin
Pediatr 1985;24:642-5.
12. Simister NE. Placental transport of immuno-
globulin G. Vaccine 2003;21:3365-9.
13. DePalma L. Red cell alloantibody formation
in the neonate and infant: Considerations
for current immunohematologic practice.
Immunohematology 1992;8:33-7.
14. Sanders MR, Graeber JE. Posttransfusion graft-
versus-host disease in infancy. J Pediatr 1990;
117:159-63.
15. Ohto H, Anderson KC. Posttransfusion graft-
versus-host disease in Japanese newborns.
Transfusion 1996;36:117-23.
16. Stauss RG. Data-driven blood banking prac-
tices for neonatal RBC transfusions. Transfu-
sion 2000;40:1528-40.
17. Strauss RG. Transfusion therapy in neonates.
Am J Dis Child 1991;145:904-11.
18. Strauss RG. Neonatal transfusion. In: Ander-
son KC, Ness PM, eds. Scientific basis of
transfusion medicine. Implications for clini-
cal practice. 2nd ed. Philadelphia: WB
Saunders, 2000:321-6.
19. Strauss RG. Routinely washing irradiated red
cells before transfusion seems unwarranted.
Transfusion 1990;30:675-7.
20. McDonaldTB,BerkowitzRA.Massivetrans-
fusion in children. In: Jeffries LC, Brecher
ME,eds.Massivetransfusion.Bethesda,MD:
AABB, 1994:97-119.
21. Harris B, Lumadue J, Luban NLC, Pollack M.
Transfusion-related hyperkalemic arrest
from irradiated packed red blood cells (ab-
stract). Transfusion 1998;38(Suppl):69S.
22. HallTL,BarnesA,MillerJR,etal.Neonatal
mortality following transfusion of red cells
with high plasma potassium levels. Transfu-
sion 1993;33:606-9.
23. Pisciotto PT. Cytomegalovirus safety issues
in neonatal transfusion. In Hermann JH,
Manno CS, eds. Pediatric transfusion ther-
apy. Bethesda, MD: AABB Press, 2002:171-92.
24. Delage G. Transfusion-transmitted infec-
tions in the newborn. Transfus Med Rev
1995;9:271-6.
25. Hamprecht K, Maschmann J, Vochem M, et
al. Epidemiology of transmission of cytome-
galovirus from mother to preterm infant by
breastfeeding. Lancet 2001;357:513-8.
26. Adler SP, Chandrika T, Lawrence L, Baggett J.
Cytomegalovirus infections in neonates ac-
quired by blood transfusions. Pediatr Inf Dis
1983;2:114-8.
27. Brady MT, Milam JD, Anderson DC. Use of
deglycerolized red blood cells to prevent post-
transfusion infection with cytomegalovirus
in neonates. J Infect Dis 1984;150:334-9.
28. Strauss RG. Selection of white cell-reduced
blood components for transfusions during
infancy. Transfusion 1993;33:352-7.
Chapter 24: Neonatal and Pediatric Transfusion Practice 577
Copyright © 2005 by the AABB. All rights reserved.
29. Bowden RA, Slichter SJ, Sayers M, et al. A
comparison of filtered leukocyte-reduced
and cytomegalovirus (CMV) seronegative
blood products for the prevention of transfu-
sion-associated CMV infection after marrow
transplant. Blood 1995;86:3598-603.
30. Gilbert GL, Hayes K, Hudson H, et al. Preven-
tion of transfusion-associated cytomegalo-
virus infection in infants by blood filtration
to remove leukocytes. Lancet 1989;i:1228-31.
31. LudvigsenC,SwansonJL,ThompsonTR,
McCullough J. The failure of neonates to
form red cell alloantibodies in response to
multiple transfusions. Am J Clin Pathol 1987;
87:250-1.
32. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
33. Pridjian G. Fetomaternal interactions: Pla-
cental physiology and its role as a go-be-
tween. In: Avery GB, Fletcher MA, MacDonald
MG, eds. Neonatology: Pathophysiology and
management of the newborn. 5th ed. Phila-
delphia: JB Lippincott, 1999:125-41.
34. Roseff SD. Pediatric blood collection and
transfusion technology. In: Herman JK,
Manno CS, eds. Pediatric transfusion therapy.
Bethesda, MD: AABB Press, 2002:217-47.
35. LevyGJ,StraussRG,HumeH,etal.National
survey of neonatal transfusion practices, I:
Red blood cell therapy. Pediatrics 1993;91:
523-9.
36. Cook S, Gunter J, Wissel M. Effective use of a
strategy using assigned red cell units to limit
donor exposure for neonatal patients. Trans-
fusion 1993;33:379-83.
37. Wang-Rodriguez J, Mannino FL, Liu D, Lane
TA. A novel strategy to limit blood donor ex-
posure and blood waste in multiply transfused
premature infants. Transfusion 1996;36:64-
70.
38. Strauss RG, Burmeister LF, Johnson K, et al.
Feasibility and safety of AS-3 red blood cells
for neonatal transfusions. J Pediatr 2000;136:
215-9.
39. Goodstein MH, Herman JH, Smith JF, et al.
Metabolic consequences in very low birth
weight infants transfused with older AS-1
preserved erythrocytes. Pediatr Pathol Lab
Med 1999;18:173-85.
40. Rock G, Poon A, Haddad R, et al. Nutricel as
an additive solution for neonatal transfusion.
Transfus Sci 1999;20:29-36.
41. Luban NLC, Strauss RG, Hume HA. Commen-
tary on the safety of red cells preserved in ex-
tended-storage media for neonatal transfu-
sions. Transfusion 1991;31:229-35.
42. TuchschmidP,MiethD,BurgerR,etal.Po
-
tential hazard of hypoalbuminemia in new-
born babies after exchange transfusions with
Adsol red blood cell concentrates (letter). Pe-
diatrics 1990;85:234-5.
43. Brecher ME, ed. Collected questions and an-
swers. 6th ed. Bethesda, MD: AABB, 2000:
73-5.
44. BurchKJ,PhelpsSJ,ConstanceTD.Effectof
an infusion device on the integrity of whole
blood and packed red blood cells. Am J Hosp
Pharm 1991;48:92-7.
45. Criss VR, DePalma L, Luban NLC. Analysis of
a linear peristaltic infusion device for the
transfusion of red cells to pediatric patients.
Transfusion 1993;33:842-4.
46. Longhurst DM, Gooch W, Castillo RA. In vitro
evaluation of a pediatric microaggregate
blood filter. Transfusion 1983;23:170-2.
47. Behrman RE, Kleigman RM, Jenson HB, eds.
Nelson’s textbook of pediatrics. 16th ed. Phil-
adelphia: WB Saunders, 2000.
48. Koenig JM. Evaluation and treatment of
erythroblastosis fetalis in the neonate. In:
Christensen RD, ed. Hematologic problems
of the neonate. Philadelphia: WB Saunders,
2000:185-207.
49. American Academy of Pediatrics Subcommit-
tee on Hyperbilirubinemia. Management of
hyperbilirubinemia in the newborn infant 35
or more weeks of gestation. Clinical Practice
Guideline. Pediatrics 2004;114:297-316.
50. Ballard RA, Vincour B, Reynolds JW, et al.
Transient hyperammonemia of the preterm
infant. N Engl J Med 1978;299:920-5.
51. Leonard JV. The early detection and manage-
ment of inborn errors presenting acutely in
the neonatal period. Eur J Pediatr 1985;143:
253-7.
52. Chan G, Schoff D. Variance in albumin load-
ing in exchange transfusions. J Pediatr 1976;
88:609-13.
53. Strauss RG, Levy GJ, Sotelo-Avila C, et al. Na-
tional survey of neonatal transfusion prac-
tices: II. Blood component therapy. Pediat-
rics 1993;91:530-6.
54. Maier RF, Sonntag J, Walka MW, et al. Chang-
ing practices of red blood cell transfusions in
infants with birth weights less than 1000 g. J
Pediatr 2000;136:220-4.
55. Blanchette VS, Kuhne T, Hume H, Hellman J.
Platelet transfusion therapy in newborn in-
fants. Transfus Med Rev 1995;9:215-30.
56. Beutler E. Platelet transfusions: The 20,000/
µL trigger. Blood 1993;81:1411-13.
57. Andrew M, Vegh P, Caco C, et al. A random-
ized, controlled trial of platelet transfusions
in thrombocytopenic premature infants. J
Pediatr 1993;123:285-91.
58. Pisciotto P, Snyder EL, Snyder JA, et al. In vi-
tro characteristics of leukocyte-reduced sin-
578 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
gle unit platelet concentrates stored in sy-
ringes. Transfusion 1994;34:407-11.
59. Sweetman RW, Cairo MS. Blood component
and immunotherapy in neonatal sepsis.
Transfus Med Rev 1995;9:251-8.
60. Vamvakas EC, Pineda AA. Meta-analysis of
clinical studies of the efficacy of granulocyte
transfusions in the treatment of bacterial
sepsis. J Clin Apheresis 1996;11:1-9.
61. RosenthalJ,CairoMS.Neonatalmyelo
-
poiesis and immunomodulation of host de-
fenses.In:PetzLD,SwisherSN,KleinmanS,
etal,eds.Clinicalpracticeoftransfusion
medicine.3rded.NewYork:Churchill
Livingstone, 1996:685-703.
62. Jenson HB, Pollock BH. The role of intrave-
nous immunoglobulins for the prevention
and treatment of neonatal sepsis. Semin
Perinatol 1998;22:50-63.
63. Sandberg K, Fasth A, Berger A, et al. Preterm
infants with low immunoglobulin G levels
have increased risk of neonatal sepsis but do
not benefit from prophylactic immunoglob-
ulin G. J Pediatr 2000;137:623-8.
64. Hill HR. Additional confirmation of the lack
of effect of intravenous immunoglobulin in
the prevention of neonatal infection (edito-
rial). J Pediatr 2000;137:595-7.
65. SchiblerKR,OsborneKA,LeungLY,etal.A
randomized, placebo-controlled trial of
granulocyte colony-stimulating factor ad-
ministration to newborn infants with
neutropenia and clinical signs of early-onset
sepsis. Pediatrics 1998;102:6-13.
66. CalhounDA,LunoeM,DuY,etal.Granulo
-
cyte colony-stimulating factor serum and
urine concentrations in neutropenic neo-
nates before and after intravenous adminis-
tration of recombinant granulocyte colony-
stimulating factor. Pediatrics 2000;105:392-7.
67. Price TH. The current prospects for neutro-
phil transfusion for the treatment of granu-
locytopenic infected patients. Transfus Med
Rev 2000;14:2-11.
68. Roseff SD, Luban NLC, Manno CS. Guide-
lines for assessing appropriateness of pedi-
atric transfusion. Transfusion 2002;42:1398-
413.
69. Christensen RD, Bradley PP, Rothstein G. The
leukocyte left shift in clinical and experi-
mental neonatal sepsis. J Pediatr 1981;98:
101-5.
70. Cairo MS, Rucker R, Bennetts GA, et al. Im-
proved survival of newborns receiving leuko-
cyte transfusions for sepsis. Pediatrics 1984;
74:887-92.
71. Cairo MS, Worcester CC, Rucker RW, et al.
Randomized trial of granulocyte transfu-
sions versus intravenous immune globulin
therapy for neutropenia and sepsis. J Pediatr
1992;120:281-5.
72. Hübel K, Dale DC, Liles WC. Granulocyte
transfusion therapy: Update on potential
clinical applications. Curr Opin Hematol
2001;8:161-4.
73. Andrew M, Paes B, Johnston M. Develop-
ment of the hemostatic system in the neo-
nate and young infant. Am J Pediatr Hematol
Oncol 1990;12:95-104.
74. Andrew M. Transfusion in the newborn:
Plasma products. In: Kennedy M, Wilson S,
Kelton J, eds. Perinatal transfusion medicine.
Arlington, VA: AABB, 1990:145-77.
75. MonagleP,MichelsonAD,BovillE,Andrew
M. Antithrombotic therapy in children.
Chest 2001;119(Suppl):344-70S.
76. Pressey JG, Manno CS. Therapy for hemo-
philia and von Willebrand disease. In:
HermanJK,MannoCS,eds.Pediatrictrans
-
fusion therapy. Bethesda, MD: AABB Press,
2002:355-82.
77. Lindemann R, Haga P. Evaluation and treat-
ment of polycythemia in the neonate. In:
Christensen RD, ed. Hematologic problems
of the neonate. Philadephia: WB Saunders,
2000:171-83.
78. Black VD, Rumack CM, Lubchenco LD,
Koops BL. Gastrointestinal injury in poly-
cythemic term infants. Pediatrics 1985;76:
225-31.
79. Kevy SV. Extracorporeal therapy for infants
and children. In: Petz LD, Swisher SN,
Kleinman S, et al, eds. Clinical practice of
transfusion medicine. 3rd ed. New York:
Churchill Livingstone, 1996:733-55.
80. Strauss RG. Selection of white cell-reduced
blood components for transfusions during
early infancy. Transfusion 1993;33:352-7.
81. Fergusson D, Hébert PC, Lee SK, et al. Clini-
cal outcomes following institution of univer-
sal leukoreduction of blood transfusions in
premature infants. JAMA 2003;289:1950-6.
82. Young G, Jubran RF, Luban NLC. Febrile
transfusion reactions in pediatric hematol-
ogy/oncology patients: The effect of leuko-
depletion (abstract). Blood 1999;94(Suppl 1):
3371a.
83. Sharon BI, Honig GR. Management of con-
genital hemolytic anemias. In: Simon TL,
DzikWH,SnyderES,etal,eds.Rossi’sprinci
-
ples of transfusion medicine. 3rd ed. Phila-
delphia: Lippincott Williams and Wilkins,
2002:463-82.
84. Cohen AR, Norris CF, Smith-Whitley K.
Transfusion therapy for sickle cell disease.
In: Capon SM, Chambers LA, eds. New direc-
tions in pediatric hematology. Bethesda,
MD: AABB, 1996:39-88.
Chapter 24: Neonatal and Pediatric Transfusion Practice 579
Copyright © 2005 by the AABB. All rights reserved.
85. Adams DM, Schultz WH, Ware RF, Kinney TR.
Erythrocytapheresis can reduce iron over-
load and prevent the need for chelation ther-
apy in chronically transfused pediatric pa-
tients. J Pediatr Hematol Oncol 1996;18:3-7.
86. Adams RJ, McKie VC, Hsu L, et al. Prevention
of a first stroke by transfusions in children
with sickle cell anemia and abnormal results
on transcranial Doppler ultrasonography. N
Engl J Med 1998;339:5-11.
87. Friedman DF, Lukas MB, Jawad A, et al. Allo-
immunization to platelets in heavily trans-
fused patients with sickle cell disease. Blood
1996;88:3216-22.
88. National Heart Lung and Blood Institute. The
management of sickle cell disease. 4th ed.
No. 02-2117. Bethesda, MD: National Insti-
tutes of Health, 2002.
89. Platt OS. The acute chest syndrome of sickle
cell disease. N Engl J Med 2000;342:1904-7.
90. Vichinsky EP, Heberkern CM, Neumayr L, et
al. A comparison of conservative and aggres-
sive transfusion regimens in the periopera-
tive management of sickle cell disease. N
Engl J Med 1995;333:206-13.
91. Petz LD, Calhoun L, Shulman IA, et al. The
sickle cell hemolytic transfusion reaction
syndrome. Transfusion 1997;37:382-92.
92. Win N, Doughty H, Telfer P, et al. Hyper-
hemolytic transfusion reaction in sickle cell
disease. Transfusion 2001;41:323-8.
93. Garratty G. Autoantibodies induced by blood
transfusion (editorial). Transfusion 2004;44:
5-9.
94. Charash S, Barton FB, Moore RD, et al. Hydroxy-
urea and sickle cell anemia. Clinical utility of
a myelosuppressive “switching” agent. The
Multicenter Study of Hydroxyurea in Sickle
Cell Anemia. Medicine (Baltimore) 1996;75:
300-26.
95. Steinberg MH. Management of sickle cell dis-
ease. N Engl J Med 1999;340:1021-30.
96. Woodard P, Jeng M, Handgretinger R, et al.
Summary of symposium: The future of stem
cell transplantation for sickle cell disease. J
Pediatr Hematol Oncol 2002;24:512-4.
97. Hoffbrand AV, Al-Refaie F, Davis B, et al.
Long-term trial of deferiprone in 51 transfu-
sion-dependent iron overload patients. Blood
1998;91:295-300.
98. Rosse WF, Gallagher D, Kinney TR, et al.
Transfusion and alloimmunization in sickle
cell disease. Blood 1990;76:1431-7.
99. Spanos T, Karageorge M, Ladis V, et al. Red
cell alloantibodies in patients with thalas-
semia. Vox Sang 1990;58:50-5.
100. Rosse WF, Telen M, Ware RE. Transfusion
support for patients with sickle cell disease.
Bethesda, MD: AABB Press, 1998.
101. Smith-Whitley K. Alloimmunization in pa-
tients with sickle cell disease. In: Herman JK,
Manno CS, eds. Pediatric transfusion therapy.
Bethesda, MD: AABB Press, 2002:249-82.
102. Tahhan HR, Holbrook CT, Braddy LR, et al.
Antigen-matched donor blood in the transfu-
sion management of patients with sickle cell
disease. Transfusion 1994;34:562-9.
103. Afenyi-Annan A, Brecher ME. Pre-transfusion
phenotype matching for sickle cell disease
patients (letter). Transfusion 2004;44:619-20.
104. Blumberg N, Heal JM, Gettings KF. Leuko-
reduction of red cell transfusions is associ-
ated with a decreased incidence of red cell
alloimmunization. Transfusion 2003;43:945-
52.
105. Van de Watering L, Jermans J, Witvliet M, et
al. HLA and RBC immunization after filtered
and buffy coat-depleted blood transfusion in
cardiac surgery: A randomized controlled
trial. Transfusion 2003;43:765-71.
106. Lane TA, Anderson KC, Goodnough LT, et al.
Leukocyte reduction in blood component
therapy. Ann Intern Med 1992;117:151-62.
107. Larsson LG, Welsh VJ, Ladd DJ. Acute intra-
vascular hemolysis to out-of-group platelet
transfusion. Transfusion 2000;40:902-6.
108. Heal JM, Blumberg N. The second century of
ABO: And now for something completely dif-
ferent. Transfusion 1999;39:1155-9.
109. Blumberg N, Heal JM, Hicks GL, Risher WH.
Association of ABO-mismatched platelet
transfusions with morbidity and mortality in
cardiac surgery. Transfusion 2001;41:790-3.
580 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 25: Cell Therapy and Cellular Product Transplantation
Chapter 25
Cell Therapy and Cellular
Product Transplantation
THERAPEUTIC CELLS INCLUDE cell
populations collected and proces-
sed to provide a special therapeu-
tic effect. The classic example of cell
therapy is the use of pluripotent stem
cells, which are capable of self-renewal
and differentiation into all blood-cell lin-
eages. These stem cells—transplanted in
preparations of marrow, stimulated pe-
ripheral blood mononuclear preparations,
or cord blood—also give rise to other re-
generative tissues such as hepatocytes,
endothelial cells, and other tissue under
the proper microenvironmental circum-
stances.1Other cell populations may serve
therapeutic purposes such as immune
modulation in posttransplant donor lym-
phocyte infusion (DLI). The hemato-
poietic progenitor cells (HPCs), which are
committed to a blood-cell lineage, were
first studied in the laboratory for their
power to give rise to complete, sustained
hematopoietic engraftment when given in
sufficient numbers. This chapter primarily
discusses the hematopoietic repopulation
cells. Cell preparations for HPC transplan-
tation are thought to contain both hemato-
poietic stem cells (HSCs) capable of
self-renewal and HPCs committed to a
blood-cell lineage. However, committed
progenitor cells lack capacity for sus-
tained self-renewal or the ability to differ-
entiate into other blood-cell lineages. The
committed cells are important for the
speed of engraftment.2Current measure-
ment methods cannot easily separate the
earlier and more committed cell popula-
tions. Both cell populations are referred to
as HPCs in this chapter. HPCs collected
from peripheral blood via apheresis are
referred to as HPC-A. Those collected by
harvesting marrow are termed HPC-M and
those from cord blood are called HPC-C.
HPC transplantation has advanced from
a research procedure performed in a few
centers to a common medical procedure
581
25
Copyright © 2005 by the AABB. All rights reserved.
performed in many tertiary care centers.
HPC transplantation can be classified ac-
cording to the source of the HPCs used for
engraftment as autologous, allogeneic,
syngeneic, and xenogeneic.
Autologous HPC transplantation is tech-
nically not a transplant but, rather, the “res-
cue” of a patient with the patient’s own
HPCs, which are removed and stored frozen
or in nonfrozen conditions for later infusion
as protection from the lethal effects of ther-
apeutic or ablative irradiation or chemo-
therapy. The autologous graft may be ma-
nipulated to retain HPCs and leave behind
or exclude tumor cells or damaging im-
mune cells. Autologous HPCs are reinfused
to repopulate the patient’s marrow after an
otherwise lethal or near-lethal dose of radi-
ation or chemotherapy given to treat malig-
nancies of the marrow and metastatic or re-
current solid tumors. In the special case of
an identical twin donor and recipient, such
transplants are referred to as syngeneic.
Allogeneic HPC transplantation involves
the infusion of HPCs from another human
(an HLA-matched related or unrelated
donor) in order to establish donor cell
chimerism to rescue the patient from dose-
intense therapy and/or as an active immuno-
therapy against a disease after a potentially
lethal dose of radiation or chemotherapy.
Such transplants are preferred in patients
who have acute myelogenous leukemia
(AML), acute lymphocytic leukemia (ALL),
severe immunodeficiency, aplastic anemia,
marrow involvement with their malignancy,
or those who are incapable of supplying
their own autologous “normal” HPCs, as
with hemoglobinopathies (thalassemias or
sickle cell disease). As new indications for
transplantation are developed, some others
are drastically changed by new discoveries.
For instance, patients with chronic myelo-
genous leukemia (CML) used to receive the
largest number of adult hematopoietic
transplants; however, imatinib mesylate
has been such a successful treatment for
this disease that the number of CML trans-
plants has dropped sharply since licensure
of this drug.3
Another advance in marrow transplanta-
tion has been the recognition and exploita-
tion of the effects of transplanted allogeneic
immune cells on the malignancy of the pa-
tient. Because the transplanted cells are the
treatment modality for the patient, the
preparation for transplantation needs only
to modify the patient’s immune system to
allow the new cells to engraft. This non-
myeloablative transplantation expands the
number of patients eligible for transplant
by accepting patients of more advanced age
and with more health problems. These pa-
tients would not be candidates to enter
more toxic myeloablative regimens because
the risk of the treatment would be so great.
In this setting, chemotherapy and/or low-
dose total body irradiation targets the re-
cipient’s T lymphocytes to allow tolerance
for the healthy, allogeneic graft.
In successful marrow transplants, the
cells gradually produce full chimerism in
the patient’s marrow and attack and elimi-
nate the tumor cells immunologically.4-6 In
patients where healthy cell replacement is
the intended result of treatment, such as in
children with immune deficiencies, this
method allows replacement cells to engraft
without putting the child through the dan-
ger of full myeloablation. The patient does
not have to undergo extensive periods of
cytopenia with exposure to infection and
bleeding risk. The engrafted cells become
the treatment agent, allowing the chemo-
therapy and/or radiation to be low-dose
and relatively low toxicity. This approach
uses the immune reconstitution as the tool
to control disease, but it is not without risk
because the immune cells can also attack
the healthy tissues of the patient, causing
graft-vs-host disease (GVHD).7The number
of nonmyeloablative transplants has in-
582 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
creased from 20 in 1997 to 870 in 2001, ac-
cording to the Center for International
Blood and Marrow Transplant Research
(CIBMTR) reporting system.3
Xenogeneic HPC transplantation would
involve HPC transplants derived from a
nonhuman species. However, because of
currently insurmountable immunologic
barriers and disease concerns, these trans-
plants are not now clinically feasible.
Sources of progenitor cells include mar-
row, peripheral blood, umbilical cord
blood, and fetal liver (although this source
is experimental and not in routine clinical
use).1Once collected, the HPCs may be
subjected to ex-vivo processing, eg, re-
moval of incompatible red cells or plasma
and/or cell selection (purging) before the
transplantation procedure. In some cases,
certain cell populations are “positively” se-
lected (selectively isolated) for their special
therapeutic effects. T lymphocytes may be
isolated and later infused in measured
doses for antitumor effects, thus minimiz-
ing GVHD. Separation of cell populations
can be accomplished using a cell separa-
tion device approved by the Food and Drug
Administration (FDA), such as the Isolex
system (Baxter Healthcare, Deerfield, IL).
Alternatively, some cell populations may be
“negatively selected” (culled out or de-
stroyed), as by antibody-mediated lysis of
malignant cells or by flow cytometry sepa-
ration of cell populations in clinical trial sit-
uations.
Diseases Treated with
Hematopoietic Cell
Transplantation
Many diseases have been treated with
HPC transplantation.8Malignant diseases
are the most frequent indications for HPC
transplantation. Although many nonma-
lignant diseases have also been treated with
life-saving transplantation, as a group,
they represent less than 15% of all trans-
plants.3,9 The success rate of HPC transplan-
tation depends on the condition and age
of the patient, type and stage of the dis-
ease being treated, the cell dose, and de-
gree of HLA matching between the donor
and the patient. Overall, long-term sur-
vival rates are generally 30% to 60% for
otherwise fatal diseases. Table 25-1 de-
scribes general outcomes.10
Sources of Hematopoietic
Progenitor Cells
Historically, marrow was the primary source
of hematopoietic cells for transplantation.
However, HPC-A transplants constituted
approximately 90% of adult autologous
and about half of allogeneic transplanta-
tion procedures in the 1998-2000 Interna-
tional Bone Marrow Transplant Registry
report.3Data on umbilical cord blood
transplants have shown promising results
in pediatric patients for whom a matched
unrelated allogeneic HPC-M or HPC-A do-
nor is unavailable. Clinical studies are in
progress to determine 1) the safety and ef-
ficacy of cord blood transplantation, 2)
whether adults can be successfully trans-
planted, and 3) the extent of HLA mis-
match that can safely and effectively pro-
vide durable engraftment.
HPCs from Marrow (Autologous)
Currently, only about 5% of autologous
transplants are performed with HPC-M.
This source has been largely replaced by
mobilized autologous HPC-A.
HPCs from Marrow (Allogeneic)
Allogeneic transplants address the prob-
lem of the inability to obtain a tumor-free
Chapter 25: Cell Therapy and Cellular Product Transplantation 583
Copyright © 2005 by the AABB. All rights reserved.

584 AABB Technical Manual
Table 25-1. Examples of Diseases Responsive to Hematopoietic Stem Cell
Transplantation10
Disease Type of Transplant Timing Clinical Results
AML Allogeneic First CR OS 40%-50%
ALL (children) Allogeneic Second CR OS 40%-65%
ALL (high risk) Allogeneic First CR OS 50%
Chronic phase CML Allogeneic Chronic phase (CP) OS 50%-80%
Accelerated phase CML Allogeneic Individualized OS 30%-40%
Blast phase CML Allogeneic Second CP OS 15%-25%
Myelodysplastic
syndrome
Allogeneic Age <60 OS 40%
Aplastic anemia Allogeneic Individualized OS 70%-90%
CLL Allogeneic or
autologous
Participation in
clinical trial
Small series of patients
with durable CR;
nonablative transplants
under investigation
Intermediate-grade NHL Autologous Chemosensitive
relapse
OS 40%-50%
High-risk NHL Autologous First CR OS 50%-60%
Low-grade NHL Allogeneic or autologous Chemosensitive
relapse
DFS 25%-50% at 5 years
Mantle cell lymphoma Allogeneic or autologous
clinical trial
First CR Small series with durable
CR rates of 25%-50%
Lymphoblastic lymphoma Allogeneic or autologous
clinical trial
Chemosensitive
relapse or first CR
Small series with durable
CR
NHL or Hodgkin's disease Allogeneic clinical trial Advanced refractory
disease
DFS 15%-25%
Multiple myeloma Autologous Chemosensitive re-
lapse or first CR
OS 50% at 5 years; DFS
20%
High-risk breast,
testicular, or ovarian
cancer
Autologous clinical trial Chemosensitive
disease
Improved survival over
historical controls not
confirmed in
randomized trials
Renal cell carcinoma Nonablative allogeneic Clinical trial Small series with durable
CR
Thalassemia Allogeneic Clinical trial OS 75% for patients
without cirrhosis
Sickle cell anemia Allogeneic Clinical trial OS 75%
Autoimmune disorders Allogeneic Clinical trial Small series of remissions
CR = complete response; DFS = disease-free survival; OS = overall survival; AML = acute myelogenous leukemia; CML =
chronic myelogenous leukemia; ALL = acute lymphocytic leukemia; CP = chronic phase; NHL = non-Hodgkin's lymphoma.
Netter examples used with permission from Icon Learning Systems, a division of MediMedia USA, Inc. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.
graft with an autologous transplant for a
patient with malignant disease and pro-
vide possible immune help after trans-
plantation from graft-vs-tumor response.
For other patients, such as those with
marrow failure, immunodeficiency, in-
born errors of metabolism, or hemoglob-
inopathy, an allogeneic transplant is the
only appropriate type of graft. Sources of
allogeneic HPC-M may be from matched
or partially matched related or unrelated
donors.
Matched, Unrelated Donor Transplantation
Matched, unrelated donor searches can
be initiated for the 60% to 70% of candi-
dates without an HLA-identical related
(usually sibling) donor. Several marrow
donor databases are available worldwide.
The largest is the National Marrow Donor
Program (NMDP) database. Since its
founding in 1986, the NMDP has facili-
tated approximately 12,000 transplant
procedures with a total of over 2000 trans-
plants per year. A directory of transplant
centers, outcome results, and charges is
available from the NMDP. (See Appendix
11 at the end of the book).
Upon initial search, 80% of transplanta-
tion candidates usually find an HLA pheno-
typic match. However, patients from racial
or ethnic minorities have a lower chance of
success in such a donor search (Caucasian,
81%; Hispanic, 64%; Asian/Pacific Islander,
55%; African American, 47%; and American
Indian/Alaska Native, 50%; according to the
NMDP). The median time from initiating a
search to receiving a transplant has been
120 days, but a new expedited search and
donor preparation program is being imple-
mented to shorten this time.
The NMDP is conducting a clinical trial
of HPC-A collections from unrelated, HLA-
matched volunteers stimulated with granu-
locyte colony-stimulating factor (G-CSF or
filgrastim).11 The donors’ physical symp-
toms as well as their attitudes and feelings
about the process are being monitored for
thetrial.TheuseofHPC-Aoffersthepoten
-
tial advantage of improved engraftment ki-
netics and enhanced graft-vs-leukemia
(GVL) effect. In allogeneic, related trans-
plant settings clinical trials comparing
HPC-M to HPC-A, the latter showed im-
proved survival. Although chronic GVHD
was a problem in the HPC-A study arm, it
was manageable in a sufficient number of
cases to give those patients a survival ad-
vantage.12
Initially, the most common diagnosis in
patients undergoing matched, unrelated
donor transplantation was CML. The dis-
covery of imatinib mesylate has revolution-
ized the treatment of CML such that the
most common indication for allogeneic
transplantation is acute leukemia followed
by non-Hodgkin’s lymphomas, multiple
myeloma, and other diseases of marrow
failure such as aplastic anemia.13
Traditionally, HLA typing for Class I
(HLA-A and HLA-B) has been dependent
on serologic techniques. However, it is
likely that the posttransplant complications
of GVHD and failure to engraft result from
use of phenotypically matched, unrelated
donors with significant disparities in allo-
antigens that were not identified through
serologic matching techniques.14 HLA-A
and HLA-B molecular Class I typing, inter-
mediate resolution, and high-resolution
typing of Class II alleles are the current
standard of care, particularly in unrelated
transplants.
The risk of GVHD is greater with HLA
Class II disparity than with Class I dispar-
ity.15 HLA typing for HLA-DR and HLA-DQ
is routinely performed by DNA-based tech-
niques. Molecular technology provides
greater resolution, including subtypes of al-
leles identified as cross-reactive groups us-
ing conventional serologic techniques. Mis-
Chapter 25: Cell Therapy and Cellular Product Transplantation 585
Copyright © 2005 by the AABB. All rights reserved.
matching for a single Class I or Class II
antigen has no effect on survival, but mor-
tality increases with more than one Class I
mismatch or simultaneous mismatches in
Class I and Class II antigens.15 Recent stud-
ies demonstrated the importance of recipi-
ent HLA-DRB1 and HLA-DQB1 allele dis-
parity in the development of GVHD.16,17
With further experience in molecular typ-
ing and transplant outcomes, the extent to
which successfully transplanted cells can
tolerate disparities in specific alleles will be
elucidated. Although the importance of
HLA-A, -B, and -DR disparities is well
known, the significance of HLA-C disparity
is being investigated. HLA-C typing has been
hampered by poor serologic identification,
and its significance has been thought, until
recently, to play a minor role in the T-cell
immune response because of its reduced
polymorphism and low level of cell surface
expression. Early studies showed that
HLA-C antigens can be recognized by
alloreactive cytotoxic T lymphocytes and
natural killer cells, which may be associated
with an increased risk of graft failure.18
Graft-vs-Host Disease
The negative outcomes of HLA mismatch-
ing are graft rejection, host-vs-graft reac-
tions, and graft-vs-host reactions. In acute
GVHD, the transplanted cells may attack
the tissues of the recipient early in the
engraftment—within 100 days after the
initial engraftment-associated events. The
skin, the gastrointestinal tract, and the
liver are most commonly involved, al-
though usually not concurrently. The site
and severity determine the clinical grade
ofacuteGVHD.TheriskofGVHDis
greater with HLA-mismatched, unrelated
and related transplants than with HLA-
identical transplants.19
Chronic GVHD characteristically occurs
spontaneously months after transplanta-
tion or after acute GVHD (generally after
posttransplant day 50) and may severely af-
fect the patient’s quality of life. In addition
to the symptoms found in the acute form,
chronic autoimmune-type disorders such
as biliary cirrhosis, Sjogren’s syndrome, and
systemic sclerosis may develop as the
transplanted immune cells attack the secre-
tory epithelial cells in the saliva glands, the
biliary tree, or the patient’s connective tis-
sue. Chronic GVHD was reported in 55% to
65% of allogeneic transplant patients who
survived beyond day 100 in a large study.2
Both forms of GVHD impair the patient’s
immune response and predispose the pa-
tient to infections. To decrease or eliminate
GVHD in these transplants, HPCs can
undergo procedures for T-cell reduction
(depletion) and the patient can be treated
prophylactically with a variety of immuno-
suppressive drug therapies.
GVHD has been associated with both a
decreased disease relapse and an improved
overallsurvivalinleukemiapatientsifthe
GVHD is relatively mild. Such a GVL effect
is believed to be secondary to the graft at-
tacking residual malignant cells. A major
clinical challenge is to maximize the GVL
effect while minimizing the adverse se-
quelae of GVHD. The mechanism of the
GVL effect is incompletely understood, but
donor-derived cytotoxic T lymphocytes
specific for the patient’s minor histocom-
patibility antigens may contribute to the ef-
fect. Donor lymphocyte infusion of thera-
peutic T cells in patients with leukemic
relapse after allogeneic transplantation has
been attempted to induce a GVL effect.20
Conflicting data exist about whether a GVL
effect can occur independently of GVHD. A
strategy to maximize the GVL effect and
minimize GVHD has been to titrate the
number of donor lymphocytes until GVHD
Grade II-III occurs in order to create a GVL
effect without severe GVHD. However, the
optimal number of donor lymphocytes ca-
586 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
pableofinducingaGVLeffectwithoutsig
-
nificant GVHD is not known. Immunosup-
pression, number of T cells transfused,
T-cell phenotype, myelosuppression, and
thetimingoftheDLIoftherapeuticTcells
all play a role in the balance between the
GVL effect and GVHD. The GVL effect is best
documented in CML and less well shown in
AML, ALL, and other lymphoid malignancies.
However, because it is difficult to identify
a good HLA match, allogeneic transplanta-
tion is associated with a major risk that
immunocompetent donor T cells reacting
against recipient tissues will cause GVHD.
Even in HLA-identical (six-antigen match)
donor/recipient pairs, up to 6% of the grafts
will fail and GVHD will occur in 20% to 60%
of cases as a result of nucleated cells exhib-
iting minor histocompatibility antigens that
are not linked to the major histocompati-
bility complex antigens.21 Improved HLA
typing techniques now employ molecular
techniques that give a fuller picture of the
antigen mapping and match/mismatch
picture, but serologic terms of estimation of
number of matches are still frequently used
in clinical descriptions for reference. This
occurs despite immunosuppressive therapy
administered for several months after the
procedure.22 New and emerging immune
cell manipulations are being explored to
exploit the cancer-controlling effects of
these cells while seeking to avoid the un-
wanted consequences of GVHD. Extracor-
poreal photopheresis is being used to treat
patients with GVHD and to reduce the dose
of immunosuppressive medications re-
quired.23
HPCs by Apheresis (Autologous)
Autologous HPC-A collection involves
mobilizing the hematopoietic cells from
the patient’s marrow compartment into
the peripheral blood with hematopoietic
growth factors, most commonly filgrastim,
with or without treatment with chemo-
therapy before collection. Once in the
circulation, the HPCs are collected by
leukapheresis. HPC-A collection carries
no anesthesia risk, is less invasive, and
contains fewer tumor cells than marrow
harvests.
HPCs by Apheresis (Allogeneic)
Related Transplantation
For adult allogeneic transplantation, the
best clinical results are obtained with a
completely HLA-matched, related donor.
The best chance of finding a six-antigen
HLA match is among the patient’s genetic
sisters and brothers. Parents and children
will be at least a haplotype match.
Genetically, there is a 25% chance of a
sibling being a complete match, a 50%
chance of a haplotype match, and a 25%
chance of a complete mismatch. Pediatric
patients are more tolerant of partially mis-
matched grafts and, therefore, have a larger
available donor pool.24 Intherareinstancea
recipient has an identical twin, a syngeneic
transplant may be optimal because the do-
nor and recipient cells are genotypically
identical and the risk of GVHD is reduced.
However, syngeneic grafts do not provide
the graft-vs-tumor effect found in allo-
geneic transplants.
HPC-A has largely replaced HPC-M for
related transplantation.25 The use of G-CSF
in healthy donors has been shown to mobi-
lize sufficient HSCs for allogeneic trans-
plantation,26 thus avoiding the need for an-
esthesia and a marrow harvest. Clinical
data comparing allogeneic HPC-A vs allo-
geneic HPC-M from HLA-identical siblings
have shown that HPC-A recipients have
faster engraftment, improved immune re-
constitution, lower transplant-related mor-
bidity, and a similar incidence of acute
GVHD.2,27 Retrospective analyses reported a
higher incidence of chronic GVHD in re-
Chapter 25: Cell Therapy and Cellular Product Transplantation 587
Copyright © 2005 by the AABB. All rights reserved.
lated allogeneic HPC-A recipients.28 How-
ever, a recent prospective randomized
study found no difference in the risk of
chronic GVHD.12
HPCs from Cord Blood
Despite the fact that over 3 million indi-
viduals are registered with the NMDP, pa-
tients in need of an allogeneic HPC trans-
plant have ≤85% chance of finding a
matched donor. Finding and qualifying a
willing donor for HPC-A or HPC-M collec-
tion typically takes weeks. Because of the
time and availability constraints, atten-
tion has turned to the third available
source—HPC-C. Umbilical cord blood,
which in clinical practice is routinely dis-
carded, is being banked as an alternative
source of HPCs, especially for children,
for whom smaller collection numbers of
HPCs are adequate for successful engraft-
ment. The advantages of cord blood as a
source of HPCs for transplantation in-
clude: no risk to the donor, potential
availability of cord blood from donor pop-
ulations underrepresented in the NMDP,
more rapid availability, and possibly
lower risk of viral infection and GVHD. Ar-
eas of concern regarding HPC-C include
ethical and informed consent issues, speed
of engraftment, higher mortality in the
posttransplant period, and ability to
achieve engraftment in adults as a result
of the limited number of nucleated cells
in cord blood.
Cord blood obtained from a delivered
placenta is known to be rich in early and
committed progenitor cells.29 Since the first
cord blood transplant was reported in 1989
for Fanconi anemia,30 more than 2500 pa-
tients have received cord blood for a variety
of hematologic malignant and nonmalig-
nant conditions.31
Cord blood is collected from the placenta
at the time of delivery using a variety of
techniques, but the preferred system uses a
small collection bag (150 to 250 mL) with
appropriate anticoagulant. In-vitro data
have suggested that placental blood has an
increased capacity for proliferation.32 The
volume of units retained for processing is
typically 80 to 100 mL (range, 40-240 mL),
with a median nucleated cell count of 1.2 ×
109in the units chosen for transplantation
and a median CD34+ cell count of 2.7 ×
105/kg.33 Clinical studies have reported suc-
cessful engraftment in children.34,35 The me-
dian time to neutrophil engraftment (500/
µL) is 30 days; median time to platelet
engraftment (50,000/µL) is 56 days.36 Com-
pared with engraftment observed after allo-
geneic marrow transplantation, neutrophil
and platelet engraftment appear to be de-
layed. Clinical studies have also suggested
that unrelated HPC-C transplants are asso-
ciated with a lower risk of GVHD compared
with unrelated HPC-M transplants in chil-
dren, even considering the lower risk sus-
ceptibility of pediatric patients to GVHD.31,34,36,37
Two studies have compared engraftment
and outcomes of transplantation of adults
with one- or two-antigen mismatched
HPC-C, matched HPC-M, and one-antigen
mismatched HPC-M. The findings in the
two studies were very similar: HPC-M
transplant recipients recovered neutrophils
on day 18 to 19, and HPC-C recipients re-
covered neutrophils on day 26 to 27.38,39
Platelet engraftment in these studies re-
peated the finding that HPC-M transplan-
tation provided more rapid recovery at 29
days compared with 60 days for HPC-C.
Both of these studies found that the HPC-C
transplantation compares favorably with
one-antigen mismatched HPC-M trans-
plantation, with equal advantage for sur-
vival for each group. A matched HPC-M
transplant was superior to HPC-C or mis-
matched HPC-M, but the advantage was
small. This is encouraging news for adults
who need an HPC-M transplant but have
588 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
difficulty finding a suitably matched donor.
Current clinical trials are testing whether
multiple HPC-C units received by adults
will facilitate faster engraftment. A study of
23 such recipients with advanced-stage dis-
ease showed a mean neutrophil engraft-
ment of 23 days and a 57% chance of dis-
ease-free survival after 1 year. In these
two-unit transplants, the cells of one of the
units prevailed and provided the lasting
engraftment population.40
Related Cord Blood Donors
Sibling-derived cord blood has been used
as a source of hematopoietic engraftment
in more than 250 allogeneic transplants in
Europe and North America, representing
15% of the allogeneic cord blood trans-
plants.31,35 The kinetics of hematopoietic
recovery are similar to those observed with
unrelated cord blood recipients.31 Times to
both platelet and neutrophil engraftment
are slower than those observed in HLA-
matched sibling marrow transplants. How-
ever, recipients of cord blood from HLA-
identical siblings have a lower incidence
of acute and chronic GVHD compared with
marrow from HLA-identical siblings.31
Autologous Cord Blood
Companies are marketing the freezing and
long-term storage of an infant’s cord blood
cells to parents, in case the child may need
them.41,42 The chance of an individual
needing a cord blood transplant by age 18
is estimated to be 1 in 200,000.42 The first
successful autologous cord blood transplan-
tation procedure was reported in a 14-
month-old patient with neuroblastoma.43
Cord Blood Banks
The first large-scale community cord
blood program was the Placental Blood
Program at the New York Blood Center,
which began collecting placental prod-
ucts in 1992. The program has stored in
excess of 14,000 cord blood units and has
provided cells for more than 1500 trans-
plant procedures. The outcomes of the
first 562 procedures have been reported
in detail.34 The NMDP has developed a
registry of participating cord blood banks.
The National Cord Blood Bank of the New
York Blood Center, NetCord, Bone Marrow
Donors Worldwide, and the Caitlin Ray-
mond Registry are additional search sites
available today.
Donor Eligibility
Donor Evaluation: Autologous Setting
In the autologous setting, the major con-
cern regarding eligibility for transplanta-
tion arises from the condition of the pa-
tient. For HPC-M, the patient’s marrow
first should be assessed for residual ma-
lignancy and marrow cellularity. Patients
scheduled for an autologous transplant
should undergo an extensive history and
physical examination to identify any risks
from the marrow harvest and/or aphere-
sis procedures. For HPC-A, the patient/
donor is evaluated for the likelihood that
he or she can undergo successful mobili-
zation and collection.
Donor Evaluation: Allogeneic Setting
Allogeneic donors must be selected ac-
cording to their degree of HLA match;
qualifications according to the FDA regu-
lations; standards of the AABB, NMDP,
and Foundation for the Accreditation of
Cellular Therapy (FACT); and physical
ability and willingness of the donor to un-
dergo the collection procedure. Ideally, a
full six-antigen match should be found;
however, transplant procedures have been
performed successfully using one-antigen
mismatched and haploidentical donors.
Chapter 25: Cell Therapy and Cellular Product Transplantation 589
Copyright © 2005 by the AABB. All rights reserved.
Cytomegalovirus (CMV) status of the do-
nor is also a deciding factor in the selec-
tion process in the case of CMV-negative
recipients. The use of parous females or
gender-mismatched individuals as donors
is associated with an increased risk of
GVHD,asisahistoryoftransfusioninthe
donor.44-46 Therefore, the preferred HLA-
identical donor would be CMV negative
(in the case of CMV-negative recipients);
of the same gender as the recipient; if fe-
male, nonparous; and untransfused.
Infectious Disease Testing
Autologous and tested allogeneic donors
must be screened and tested for certain
infectious diseases.47 In the Code of Fed-
eral Regulations (CFR) Title 21, sections
of the regulations relevant to cell therapy
donors are Donor Screening (1271.75),
Donor Testing: General (1271.80), and Do-
nor Testing: Specific Requirements
(1271.85). The rules are described by the
FDA as “comprehensive, but flexible.” The
donor screening rules are consistent with
the 1994 Centers for Disease Control and
Prevention (CDC) guidelines to prevent
transmission of human immunodeficiency
virus (HIV), and 1993 FDA guidelines for
prevention of transmission of hepatitis B
virus (HBV) and hepatitis C virus (HCV).
Testing for syphilis is required, and tissue
rich in leukocytes must be tested for hu-
man T-cell lymphotropic virus (HTLV)
and CMV. They also include requirements
for questioning donors about risk factors
for variant Creutzfeldt-Jakob disease and
new or emerging pathogens such as West
Nile virus and severe acute respiratory
syndrome, and requirements for tests for
carriers of these illnesses as soon as they
are practically available. The regulations
and guidance are extensive and fit the
FDA’s stated “layered approach” that in-
tensifies the amount of screening and test-
ing of the donor to the level of risk to
which the recipient may be subjected.
Thus, unrelated allogeneic donors repre-
sent the highest level of risk, whereas
family members or regular sexual partners
are considered a lower risk source of new
exposure to patients.
The donor must be screened in order to
minimize the risk of disease transmission
to an already immunocompromised recipi-
ent. FDA regulations specify that the HPC
donor sample should normally be tested up
to7daysbeforecollection,oratthetimeof
collection but before release of the product.
HPC-A donor samples may be obtained up
to 30 days before collection in order to have
infectious disease testing completed before
the patient begins myeloablative chemo-
therapy [21 CFR 1271.80(b)]. For purposes
of optimal donor selection, it may be advis-
able to test the donor earlier in the trans-
plantation workup period as well.48,49
The use of approved nucleic acid tests
for HIV and HCV has considerably short-
ened the possible “window period” in do-
nors who may have contracted either of
these infections but who do not yet test
positive on antibody tests. Donors being
stimulated with filgrastim develop higher
sample-to-cutoff ratios. This makes false-
positive reactions more likely in the
immunoassays currently used for infectious
disease. False-positive test results for hepa-
titis B surface antigen and HCV antibody
have been associated with G-CSF adminis-
tration in normal donors.50,51
Donors who are confirmed positive for
HIV should not be used as a source for the
transplant. Other positive disease markers
do not necessarily prohibit use of collec-
tions from a particular donor (eg, anti-HBc
positive), and special evaluation of donors
with this marker is under way by the NMDP
at this time. The allograft material from
such donors may be used when the pa-
tient’s transplantation physician informs
590 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
him or her of the status of the donor and
documents consent of the patient. Segre-
gated methods of storage must be used for
the biohazardous material collected until it
is infused so that cross-contamination of
otherstoredproductsisprevented.
48(p78),49,52
In cases of allogeneic HPC-C transplants,
a sample for infectious disease studies
should be obtained from the donor’s
mother within 48 hours of collection of the
cord blood and tested. Any positive results
should be reported to the mother and the
mother’s physician. The HPC-C donation
involving a mother or a child who has a
positive infectious disease history or results
should either be discarded or the patient
must go through a special notification and
consent process.52 In cases of allogeneic
transplantation, CMV status of the donor
and the recipient should be carefully con-
sidered.
A recipient may develop primary CMV
infection if he or she is CMV negative and
receives a CMV-positive graft but may have
some protection from the immunity in the
graft. CMV-positive individuals receiving a
CMV-negative graft may have a severe pri-
mary infection of the graft from virus resi-
dent in the recipient’s tissue.46
Collection of Products
HPC-M Collection
A marrow harvest is the same for an allo-
geneic donor as for an autologous patient.
The procedure is performed under sterile
conditions in the operating room. The
posterior iliac crest provides the richest
site of marrow. In the autologous patient,
prior radiation therapy to an aspiration
site may result in hypocellular yields. In
general,thesesitesareunsuitableforhar
-
vestandshouldbeavoided.Onceaspi
-
rated, the marrow should be mixed and
diluted with an anticoagulant (usually
preservative-free heparin and/or ACD).
The marrow aliquots are pooled into a
sterile vessel or a harvest collection bag
equipped with filters of graduated pore
size. The marrow is then transferred to a
sterile blood bag and transported to the
processing laboratory, where samples are
removed for graft evaluation, quality as-
surance testing, possible manipulation of
the product, final labeling, and/or cryo-
preservation.
Collection Targets
The recipient’s body weight and type of
manipulation of the collection, if any, will
determine the volume of marrow to be
collected. A frequently used minimum
target (after processing), for both autologous
and allogeneic transplants, is 2.0 ×108nu-
cleated cells per kilogram of recipient
body weight. Marrow harvests in autolo-
gous patients who have received alkylat-
ing agents as therapy may yield fewer pro-
genitor cells relative to the total nucleated
cells collected. In these cases, extra mar-
row should be obtained if possible.53 If the
harvested product is to be processed (ie,
T-cell depletion, ex-vivo tumor purging),
additional marrow (up to double the re-
infusion target) should be collected. The
NMDP limits the volume harvested from
its donors to a maximum of 1500 mL. Many
centers use the cell number requested for
transplant and the cell count on the prod-
uct during collection to determine the fi-
nal collection volume.
Clinical Considerations
Theageofthedonorand,intheautolo
-
gous setting, the underlying disease and
previous treatment regimen influence the
HPC yield (Table 25-2).54 Autologous or allo-
geneic donors may require RBC transfu-
sions to replace blood taken with marrow
Chapter 25: Cell Therapy and Cellular Product Transplantation 591
Copyright © 2005 by the AABB. All rights reserved.
collection. Frequently, allogeneic marrow
donors will have an autologous RBC unit
collected 2 to 3 weeks before the harvest.
Most RBC transfusions occur after the har-
vest to avoid marrow dilution. Allogeneic
blood components must be irradiated if
given before or during the procedure, to
incapacitate donor lymphocytes from re-
plication and attack of the transplant re-
cipient, possibly causing GVHD. In trans-
plants that require marrow manipulation,
such as red cell depletion, the recovered
red cells may be returned to the donor.
HPC-A Collection
HPCs can be mobilized into the periph-
eral blood with the use of recombinant
colony-stimulating factors and collected
in sufficient numbers to achieve long-
term hematopoietic engraftment in a trans-
plant recipient.
CD34
CD34 is the cluster designation given to a
transmembrane glycoprotein present on
immature hematopoietic cells, some ma-
ture endothelial cells, and stromal cells.55
The antigen has an approximate molecu-
lar weight of 110 kD and carries a negative
charge. Cells expressing the CD34 antigen
encompass the lineage-committed cells as
well as the pluripotent stem cells. CD34+
cells purified from marrow are capable of
trilineage reconstitution in humans.55 One
to three percent of normal marrow cells
express this antigen. In normal peripheral
blood, CD34+ cells circulate in low num-
bers (0.01-0.1%).56 Monoclonal antibodies
(MoAbs) to CD34 have been developed
and are used in flow cytometric assays to
measure the stem/progenitor content of
HPC collections and CD34+ cell concen-
tration in donor peripheral blood.57
HPC-A Collection (Autologous)
In the autologous setting, HPC-A donors
are routinely mobilized with hematopo-
ietic growth factors (with or without che-
motherapy), which include G-CSF and
granulocyte-macrophage colony-stimu-
lating factor (GM-CSF) or a combination
of the two. Mobilization with chemother-
apy or growth factors alone results in a
10- to 30-fold increase in the concentra-
tion of HPCs over the steady state.8Com-
bined mobilization with growth factors
and chemotherapy enriches HPC concen-
trations 50- to 200-fold.58 Transplantation
of autologous HPC-A results in a reduced
time to hematopoietic recovery compared
with autologous HPC-M.59 Platelet recon-
stitution is most striking and patient hos-
592 AABB Technical Manual
Table 25-2. Factors Reported to Affect the Mobilization of Hematopoietic Cells
1. Mobilization technique
a. Chemotherapy—degree of transient myelosuppression
b. Growth factors—type, schedule, dose
c. Use of combined chemotherapy and growth factors
2. Extent of prior chemotherapy/radiation
3. Type of underlying disease
4. Age of patient
5. Presence of marrow metastases
Adapted with permission from Lane.54
Copyright © 2005 by the AABB. All rights reserved.
pital stays are reduced by 7 to 10 days.
Schwella and colleagues60 demonstrated
that the median time to an unsupported
platelet count of ≥20,000/µLwas10days
in the group receiving chemotherapy plus
G-CSF-mobilized HPCs vs 17 days in the
group receiving autologous HPC-M. Ef-
fective mobilization in the autologous pa-
tient is influenced by previous chemo-
therapy or radiation.
There is a linear relation between the
precollection peripheral blood CD34 count
and the volume of blood that needs to be
processed to achieve a target dose. Graphs
and predictive models would be expected
to vary based on the efficiency of CD34+
collection related to the specific equipment
and software employed. It would also be
expected to be least predictive at low pe-
ripheral blood CD34+ concentrations, where
the enumeration is least accurate. White
cell counts, the number of mononuclear
cells, and the number of circulating CD34+
cells have all been used as surrogate mark-
ers for the timing of the HPC-A collec-
tion.59,61-63 It has been suggested that the op-
timal time to begin HPC collection in pa-
tients who have been primed with chemo-
therapy is when the white cell count first
exceeds 5 ×109/L.59 However, the white cell
concentration does not correlate with the
number of HPCs in the peripheral blood.
Phenotypic analysis of CD34+ cells by flow
cytometry provides a more real-time mea-
surement of CD34+ cell content in an HPC
collection or hematopoietic graft. The tech-
nique can be used to indicate the timing of
the first collection and the total number of
CD34+ cells/kg finally collected (as a func-
tion of the volume of blood processed).56,57,64-66
The length of time for engraftment corre-
lates with the number of CD34+ cells in the
collection.65,67-69 In general, a peripheral blood
CD34+ cell concentration of 10/µLcanbe
expected to result in a yield of at least
1.0 ×106CD34+ cells/kg.60,61,64
In most autologous patients, venous ac-
cess is obtained through a dual- or triple-
lumen central venous apheresis catheter.
Operators of blood cell separators generally
process two to three blood volumes per
procedure. In the pediatric setting, it may
be necessary to prime the cell separator
with compatible, irradiated red cells. The
donor will undergo daily procedures of ap-
proximately 2 to 5 hours each. Large-vol-
ume leukapheresis procedures (processing
at least three blood volumes or 15-20 liters)
are performed at many centers to reduce
the overall number of collections. Investiga-
tional studies have suggested that there is
equilibration of noncirculating HPCs into the
peripheral circulation with large-volume
collections.70-72 Hillyer73 reported a 2.5-fold
increase in colony-forming units–granulo-
cyte-macrophage (CFU-GM) per mL pro-
cessed in collection when the collection
volumewas15literscomparedtotheyield
in the first blood volume of the collection.
Collection Targets. The adequacy of
autologous HPC collection is gauged by the
CD34+ dose (the number of CD34+ cells per
kilogram of recipient body weight). The re-
ported minimum threshold of CD34+ cells
necessary for neutrophil and platelet engraft-
ment in the autologous patient has ranged
from 0.75 to 1.0 ×106/kg.8,64 This minimum
target is a broad guideline and higher doses
have been associated with accelerated
platelet engraftment,74 reduced febrile com-
plications, and use of antibiotic therapy af-
ter transplantation.75 Recent data support
an economic benefit associated with greater
CD34+ cell collections (greater or equal to
5.0 ×106/kg) compared to the minimum ac-
ceptable collections required for engraft-
ment (1.0 ×106/kg).76 Thetypeofprocessing
will also influence the volume necessary for
collection.
Poor Autologous HPC Mobilization. Pa-
tients who have been heavily pretreated
with chemotherapy and/or radiation ther-
Chapter 25: Cell Therapy and Cellular Product Transplantation 593
Copyright © 2005 by the AABB. All rights reserved.
apy may fail to mobilize enough HPCs after
stimulation with growth factors/chemo-
therapy. The best way to obtain an ade-
quate collection from poorly mobilized pa-
tients is unknown. Collection of HPC-M in
combination with, or in place of, HPC-A is
frequently ineffective in improving engraft-
ment.77 Second attempts at mobilization
with G-CSF alone have been successful in
obtaining adequate CD34+ cell counts and
can be as effective as G-CSF and chemo-
therapy.78 Increasing the dose of G-CSF or
combining with GM-CSF has also success-
fully mobilized some autologous donors af-
ter prior failed attempts.79
Mobilization can be very difficult in some
patients, particularly if they have been
heavily pretreated or are older. Another ap-
proach to increase production of stem/pro-
genitor cells by growth factors is manipula-
tion of the chemokines that attach the cells
to their microenvironment. The interaction
of stromal-derived growth factor-1 (SDF-1)
and CXCR4, its ligand, controls mobiliza-
tion. The molecule AMD 3100 was devel-
oped originally as a potent and selective
inhibitor of CXCR4 in order to control repli-
cation of HIV-containing cells. An observed
side effect of the drug was a rapid increase
in white cells. Broxmeyer and colleagues
measured a 40-fold increase in HPCs after
injection of AMD 3100 into human volun-
teers.80 Phase I and II clinical trials are un-
der way in conjunction with G-CSF to en-
hance mobilization and HPC-A yields in
difficult-to-mobilize patients.81
Clinical Considerations for Autologous
HPC-A Collection. The frequency and length
of HPC-A collections may result in donor
discomfort and side effects. Complete blood
counts are performed before and after each
apheresis procedure to monitor the hemato-
crit and platelet values. Red cell and/or
platelet transfusions may be required. Red
cell loss should be minimal in HPC-A col-
lection, but platelet counts typically de-
crease from 25% to 40% because the HPCs
lie close to the platelet layer in the buffy
coat. Collection can be performed via pe-
ripheral or central venous access. Patients
who have received prior chemotherapy are
more likely to need a central venous cathe-
ter because of poor peripheral access. Cath-
eter-associated thrombosis (either in the
catheter or in the vein surrounding it) is the
most common complication associated
with HPC-A collection.82
HPC-A Collection (Allogeneic)
Collection of allogeneic HPCs from HLA-
matched relatives is primarily performed
using G-CSF mobilization. Typically, do-
nors are given 10 to 16 µg/kg as a subcu-
taneous injection once or twice daily. The
concentration of CD34+ cells in the pe-
ripheral blood begins to rise after 3 days
of G-CSF administration and peaks after 5
to 6 days. Standard volume leukapheresis
(processing 8-9 L) results in a component
with the following median values: white
cells, 32.4 ×109; mononuclear cell count,
31.4 ×109; CD34+ cells, 330 ×106;plate-
lets, 470 ×109;andRBCs,7.6mL.
83 Clinical
trials have suggested a minimum dose of
2.0 ×106/kg of CD34+ cells to be adequate
for allogeneic HPC-A transplants.84
Allogeneic donors with poor venous ac-
cess may require central venous catheters.
In the NMDP report on normal donor col-
lections of HPCs, 5% of men and 20% of
women require central venous access for
collection.85 There is a 1% risk of complica-
tions associated with these catheters includ-
ing infection, hemorrhage, and pneumo-
thorax. Thrombocytopenia has been reported
as a complication of G-CSF administration
and HPC-A collection in healthy donors.86,87
There is also a risk in exposing normal
donors to G-CSF. Ninety percent of donors
experience side effects.88 The most com-
mon complaint is bone pain followed by
594 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
headaches, body aches, fatigue, nausea,
and/or vomiting. Bone pain, headaches,
and body aches can be successfully man-
aged with nonsteroidal analgesics such as
acetaminophen. As mentioned earlier,
false-positive infectious disease serologies
have been reported.51 Serious adverse ef-
fects are rare but include reports of splenic
rupture,89,90 neutrophilic infiltration of the
skin (Sweet’s syndrome),91 arterial thrombo-
sis,92 and an anaphylactoid reaction.93 Do-
nors with complex hemoglobinopathies
(eg, sickle trait and β+ thalassemia) have
been observed to have complications with
G-CSF stimulation for the purpose of be-
coming an HPC donor.94,95
HPC-C Collection
Umbilical cord blood can be collected from
either a delivered or an undelivered pla-
centa.96 Cord blood collection should not
interfere with obstetric care of the mother
or the infant. If the birth provider intends
to collect the cord blood, plans should be
made in advance to abandon the collec-
tion if care of the mother or the infant re-
quires the care provider’s attention in-
stead. Cord blood collected after delivery
of the placenta is preferably initiated
within 15 minutes of parturition, if ade-
quatevolumesaretobeobtained.
97 To
minimize the risk of bacterial contamina-
tion, the surface of the cord should be
cleaned with alcohol, then disinfected, in
a similar manner to the preparation of skin
for a blood donation. A large-bore needle
connected to a blood collection bag con-
taining CPDA, CPD, or ACD anticoagulant
is inserted into the umbilical vein so that
the placental blood drains by gravity into
the bag. Alternatively, a delivered pla-
centa can be suspended above the collec-
tion bag. CPDA or CPD are the preferred
anticoagulants because they are isotonic
and have a neutral pH. However, other
closed system arrangements using ACD or
heparinareinuseinsomecenters.In
-
formed consent from the biologic mother
or legal representative of the child must
be obtained, preferably before delivery.
A personal and family medical history of
the biologic mother (and, if available, of the
infant donor) must be obtained and docu-
mented before, or within 48 hours of, the
collection. If available, his medical history
should be obtained from the biologic fa-
ther. HPC-C collections are not acceptable
for allogeneic use if there is a family history
(biologic mother, father, or sibling) of ge-
netic disorders that may affect the graft’s ef-
fectiveness in the recipient or otherwise ex-
pose the recipient to a genetic disorder
through the transplant.
Red cells from the HPC-C collection or
from the infant donor must be typed for
ABO/Rh and a screen for unexpected red
cell antibodies must be performed using ei-
ther the mother’s serum or plasma or a
sample from either the infant donor or the
cord blood collection.52,56 White cells from the
cord blood must be typed for HLA (HLA-A
and -B antigen testing, and DNA-based
Class II typing) by a laboratory accredited
by the American Society of Histocompati-
bility and Immunogenetics (ASHI) or an
equivalent accrediting organization.49,52 In
order for HPC-C to be a feasible alternative
to allogeneic HPC-M or HPC-A transplants,
it is essential to have a frozen inventory of
ready-to-use HLA-typed products.
Samples, preferably from an attached
segment, should be frozen for pretrans-
plant confirmation of HLA type by the
transplant facility. Many centers freeze a
sample of the mother’s serum to allow new
infectious disease tests to be done if they
become available after the HPC-C has been
collected and are thought to be important
at the time the transplant is planned. The
emergence of West Nile virus as a threat to
Chapter 25: Cell Therapy and Cellular Product Transplantation 595
Copyright © 2005 by the AABB. All rights reserved.
mothers, infants, and transplant recipients
is an example. A sample of the mother’s
blood containing cells or DNA (a dried filter
paper sample is adequate) may be useful if
questions concerning the child’s HLA type
arise. These samples from the mother were
more useful when HLA typing was done by
serologic methods to investigate ambigu-
ous types in the infant. CD34+ cell enumer-
ation at the time of thawing may also be
performed to estimate the stem cell con-
tent of the HPC-C graft but is useful only if
a viability marker is employed so that only
viable cells are counted.
Thedesiretoincreasethedoseofstem
cells in HPC-C has fueled interest in ex-vivo
expansion of the cells in a laboratory set-
ting.98 Early clinical trials with expanded
cord blood progenitors are promising, hav-
ing shown successful engraftment; how-
ever, no improvement in patient survival
has been reported.99,100 Ex-vivo expansion of
cord blood is an area of active experimental
investigation.
Processing of Hematopoietic
Progenitor Cells
Techniques for Cell Selection and/or
Purging of Hematopoietic Progenitor Cells
Selection of the CD34+ Cells
As an HPC undergoes differentiation and
maturation, CD34 antigen levels decrease.
This property, coupled with the use of MoAbs
specific for the different epitopes of the
CD34 molecule, permits physical separa-
tion procedures. There are several meth-
ods of immunoselection available, such as
fluorescence-activated cell sorting (FACS)
and immunomagnetic beads.101 Selection
of CD34+ HPCs may be associated with a
reduction of tumor cells (autologous grafts)
or T cells (allogeneic grafts). The clinical
utility of CD34 and AC133 selection for tu-
mor or T-cell reduction is under investiga-
tion.102
Immunomagnetic Separation. Various
immunomagnetic separation techniques,
direct or indirect, are available. Typically, a
CD34 antibody is coupled to a magnetic
bead. This complex is incubated with mono-
nuclear cells, and the cells expressing the
CD34 antigen bind to the antibody-coated
beads, forming rosettes. A magnet is ap-
plied to separate the rosetting CD34+ cells
from the nonrosetting cells. Bead detach-
ment, which varies among methods, may
be accomplished through anti-Fab frag-
ments or enzymatic treatment (eg, chymo-
papain).103 The Isolex 300 system (Baxter
Healthcare Immunotherapy Division, Irvine,
CA), a magnetic cell separator, is a semi-
automated instrument for clinical-scale
CD34+ selection applications. This method
uses antibody-coated Dynal paramagnetic
beads to rosette the CD34+ cells. A fully au-
tomated device (Isolex 300i, Baxter) and a
peptide release agent are available for clini-
cal use.104 Other techniques of magnetic cell
separation employ superparamagnetic micro-
beads that remain attached to the HPC sur-
face.105 One such method, magnetic cell
sorting (MACS, Miltenyi Biotec, Bergisch
Gladbach, Germany), was first introduced
on a small scale by Miltenyi.106 The
CliniMACS (Miltenyi Biotec) is a clinical-
scale version of the MACS system and is
available in the United States for investiga-
tional use only. This system employs anti-
body-conjugated iron-dextran microbeads.
The magnetically stained cells are sepa-
rated over a high gradient magnetic column
and a microprocessor controls the elution
of the CD34+ cells.
Counterflow Centrifugal Elutriation.
Counterflow centrifugal elutriation (CCE) is
a method of separating cells based on their
size and density. A continuous-flow centri-
fuge (Beckman Instruments, Palo Alto, CA,
and Gambro, Golden, CO) and unique
596 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
chamber design allow for the basic separa-
tion principle: two opposing forces (centrif-
ugal force and counter media flow) acting
upon cells at the same time. As cells are
pumped into the chamber (centripetal di-
rection), they align according to their sedi-
mentation properties. With adjustment of
the counterflow rate, the centrifugal and
counterflow forces are balanced, and a gra-
dient of flow rates exists across the cham-
ber. By gradually increasing the flow rate of
the medium or decreasing the speed of the
rotor, cells can be eluted out of the cham-
ber and collected. Smaller, slower sedi-
menting cells elute first. Although this tech-
nique is used primarily for T-cell depletion,
CCE has been applied to CD34+ cell selec-
tion. CD34+ cells are heterogeneous and
will elute in subset fractions that are useful
for repopulation experiments and ex-vivo
expansion trials.107
Autologous Tumor Purging
Purging or negative selection refers to the
removal of tumor cells that may contami-
nate the autologous graft. In patients with
hematopoietic disease or malignancies
that frequently involve the marrow (eg,
lymphomas), minimal residual disease
contributes to relapse.108 Although autolo-
gous HPC-A collections have a lower prob-
ability than HPC-M of tumor contamina-
tion and fewer tumor cells/mL, they still
may contain large numbers of viable tu-
mor cells. Studies have demonstrated that
tumor cells may be mobilized from the
marrow into the peripheral circula
-
tion.109,110 Whether tumor purging de-
creases the likelihood of relapse is contro-
versial. Because some purging methods
may damage HPCs, the probability of re-
sidual disease or relapse should be care-
fully balanced against the higher graft
failure rate or increased mortality from
prolonged aplasia that may be associated
with tumor purging.111 Numerous tech-
niques for autologous purging are
available. The goal of all purging meth-
ods— whether physical, immunologic, or
pharmacologic—is the destruction or re-
moval of the malignant clone while main-
taining the efficacy of the HPCs necessary
for engraftment.112
Pharmacologic Techniques. In-vivo
antineoplastic therapy produces greater tu-
mor cell kill because of the differential sen-
sitivity of malignant cells over normal cells.
In-vitro pharmacologic purging is an effort
to expand this therapeutic ratio. In-vitro
purgingallowsfordoseandexposurein
-
tensification without concern for organ tox-
icity because higher drug concentrations
can be used on isolated hematopoietic
grafts ex vivo, which then can be adminis-
tered in vivo. However, the drug concentra-
tion must be at nontoxic levels before re-
infusion. Activated oxazaphosphorines
(4-hydroperoxycyclophosphamide, mafos-
famide) were the most frequently used
compounds but are generally no longer in
use in the United States. Other chemo-
therapeutic agents have been investigated
in preclinical models but are not in clinical
use. Results from purged autologous trans-
plants for AML with either of these drugs
showed prolonged aplasia with less than
1% CFU-GM survival.112
Physical Techniques. Separation meth-
ods based on cell size and density through
gradient-generating reagents or CCE do not
achieve adequate tumor cell depletion.111
However, the combined use of physical and
immunologic techniques requires further
study.
Immunologic Techniques. The develop-
ment of MoAbs coupled with the discovery
of tumor-associated antigens opened the
field for immunologic purging.113 MoAbs
may be directed at tumor-specific antigens
or cell-differentiation antigens.111 The im-
munologic techniques differ primarily by
Chapter 25: Cell Therapy and Cellular Product Transplantation 597
Copyright © 2005 by the AABB. All rights reserved.
the method of target cell removal. MoAbs
are used in conjunction with complement,
bound to toxins, or coupled to magnetic
beads.ThechoiceofMoAbandthehetero
-
geneity of antigen expression on the target
cell affect the success of the purge or the
level of depletion. Many investigators em-
ploy a cocktail of MoAbs in an effort to en-
hance the purging efficiency.114,115 Comple-
ment-mediated cytotoxicity is frequently used
if the antibody is of IgM isotype or if the ex-
pected level of antigen density is high.111
Immunomagnetic cell separation, either
by direct or indirect method, employs an
antibody-coated magnetic bead to target
the antigen or antigens of interest. A recent
study described a 5-log tumor cell deple-
tion with two cycles using the indirect
method for B-cell lymphoma. Colony assays
showed only a 20% reduction in CFU-GM
and multipotential CFU (CFU-GEMM).114
T-Cell Depletion
GVHD is a significant complication in
allogeneic HPC transplantation. Because
the disease process is mediated by donor
T cells that are reactive against the host,
depletion strategies are used to target and
remove these cells in an effort to decrease
the incidence or at least lessen the sever-
ity of GVHD. Many of the techniques out-
lined for positive selection and tumor purg-
ing are applicable to T-cell depletion (Table
25-3).17
In T-cell-depleted grafts, two major areas
of concern are graft failure and recurrent
leukemia. Most instances of graft failure
(initial or late) are caused by immunologic
rejection. Early transplants involving de-
pleted grafts in patients with CML showed
a 50% or greater relapse rate. In contrast,
patients who developed GVHD had a lower
relapse rate. Subsequent clinical and inves-
tigational data provided evidence of an im-
mune-mediated GVL effect.116
Research and developmental efforts have
focused on determining the optimal level of
T-cell depletion. Dreger et al115 reported a
comparison of T-cell depletion methods
(CAMPATH-1 plus complement, immuno-
magnetic CD34+ selection, and biotin-
avidin-mediated CD34+ selection). The
immunomagnetic method provided a 4-log
reduction in T cells vs a 3.1-log reduction
with the biotin-avidin method. MoAb treat-
ment with autologous complement yielded
a 2.1-log reduction. The challenge of mini-
mizing the severity of GVHD and maintain-
ing the GVL effect is ongoing. Additional
studies are examining the role of sub-
populations of T cells and/or cytokines in
potentiating the GVL effect.117
ABO Incompatibility
Although HLA compatibility is crucial in
the successful engraftment of myelosup-
pressed or ablated patients, ABO compat-
ibility is not. Pluripotent and very early
committed HPCs do not possess ABH anti-
gens, allowing engraftment to occur suc-
cessfully regardless of the ABO compati-
bility between the recipient and donor.
ABO incompatibility does not affect neutro-
phil or platelet engraftment, graft failure,
or rejection. However, delayed red cell
engraftment may occur after a major
ABO-incompatible transplant and delayed
hemolysis may occur after a minor ABO-
incompatible transplant where the donor
598 AABB Technical Manual
Table 25-3. Methods of T-Cell Depletion
Nonimmunologic
Counterflow centrifugal elutriation
Pharmacologic/cytotoxic drugs
Immunologic
Monoclonal antibodies with or without
complement
Immunotoxins
Immunomagnetic beads
Copyright © 2005 by the AABB. All rights reserved.

has antibodies to the recipient’s blood
cells (Table 25-4).118
Major ABO Incompatibility
Major incompatibility occurs when the
recipient has ABO antibodies or other red
cell antibodies, such as anti-D or anti-
Kell, against the donor red cell antigens.
The HPC preparation can be processed to
remove mature red cells. The group O re-
cipient who receives a group A graft may
continue to produce anti-A and anti-B for
3 to 4 months or longer in rare instances,
and the presence of anti-A will delay
erythropoiesis by the group A graft; group
A red cells appear in the circulation when
the recipient’s anti-A disappears. Granulo-
cyte and platelet production are not
affected.119 Red cell engraftment may be
delayed to ≥40 days after the transplanta-
tion.120 Red cells used for transfusion of the
recipient must be compatible with both the
donor and the recipient. In some centers,
group O red cells are given to all major
ABO-incompatible transplant recipients
in order to avoid confusion. Recommen-
dations for optimal blood component se-
lectionareshowninTable25-5.
Minor ABO Incompatibility
Minor ABO incompatibility occurs when
the donor has antibodies against the re-
cipient red cell antigens (such as a group
O donor to a non-group O recipient). Be-
Chapter 25: Cell Therapy and Cellular Product Transplantation 599
Table 25-4. Potential Problems with ABO- and Rh-Incompatible HPC
Transplantation
Example
Incompatibility Donor Patient Potential Problems
ABO (major) Group A Group O Hemolysis of infused donor RBCs, delay
of RBC engraftment, hemolysis at the
time of donor RBC engraftment
ABO (minor) Group O Group A Hemolysis of patient RBCs from infused
donor plasma; in pediatric cases,
hemolysis of patient RBCs 7-10 days
after transplant caused by passenger
lymphocyte-derived
isohemagglutinins
Rh Negative Positive Hemolysis of patient RBCs by donor
anti-D produced after engraftment
Positive Negative
(with
anti-D)
Hemolysis of donor RBCs from newly
engrafted HPCs (rare), or delayed
RBC recovery
Other RBC
antigens
Negative Positive Hemolysis of patient RBCs by donor an-
tibodies in plasma produced after
engraftment
Positive Negative
(with
antibody)
Hemolysis of donor RBCs from newly
engrafted HPCs (rare), or delayed
RBC recovery
Copyright © 2005 by the AABB. All rights reserved.

600 AABB Technical Manual
Table 25-5. Transfusion Support for Patients Undergoing ABO-Mismatched Allogeneic HPC Transplantation
Phase I Phase II Phase III
Recipient Donor
Mismatch
Type
All
Components RBCs
First
Choice
Platelets
Next Choice
Platelets* FFP
All
Components
A O Minor Recipient O A, AB AB; B; O A, AB Donor
B O Minor Recipient O B, AB AB; A; O B, AB Donor
AB O Minor Recipient O AB A; B; O AB Donor
AB A Minor Recipient A, O AB A; B; O AB Donor
AB B Minor Recipient B, O AB B; A; O AB Donor
O A Major Recipient O A AB; B; O A, AB Donor
O B Major Recipient O B AB; A; O B, AB Donor
O AB Major Recipient O AB A; B; O AB Donor
A AB Major Recipient A, O AB A; B; O AB Donor
B AB Major Recipient B, O AB B; A; O AB Donor
A B Minor & major Recipient O AB A; B; O AB Donor
B A Minor & major Recipient O AB B; A; O AB Donor
*Platelet concentrates should be selected in the order presented. Modified from Friedberg et al.121
Phase I = the time when the patient/recipient is prepared for HPC transplantation.
Phase II = from the initiation of myeloablative therapy until:
For RBC—DAT is negative and antidonor isohemagglutinins are no longer detectable (ie, the reverse typing is donor type).
For FFPs—recipient’s erythrocytes are no longer detectable (ie, the forward typing is consistent with donor’s ABO group).
Phase III = after the forward and reverse type of the patient are consistent with donor’s ABO group.
Beginning from Phase I, all cellular components should be irradiated and leukocyte reduced.
Copyright © 2005 by the AABB. All rights reserved.
fore infusion of a graft from a plasma-in-
compatible donor, the plasma may be re-
moved to avoid infusion of preformed
anti-A and/or anti-B. Minor ABO-incom-
patible transplants can be characterized by
rather abrupt onset of immune hemolysis,
which begins about 7 to 10 days after trans-
plantation and may last for 2 weeks. The
direct antiglobulin test (DAT) is positive;
anti-A and/or anti-B can be recovered in
theeluateandhemoglobinemiaand/or
hemoglobinuria may occur. An additional
30% of such transplant recipients develop
a positive DAT without experiencing gross
hemolysis. This phenomenon is the result
of red cell antibodies produced by pas-
senger B lymphocytes in the stem cell
graft.121 Non-ABO antibodies from passen-
ger lymphocytes in HPC transplants have
also been reported.122 Although transient,
the hemolysis may persist for up to 2 weeks
and may require transfusion with group O
red cells. Minor ABO-mismatched HPC
transplants may be associated with a
higher risk of hemolysis, possibly caused
by greater B-cell content.123 In all cases,
plasma used for transfusion should be
compatible with both the donor and the
recipient. In some centers, group AB plasma
products are given to all minor ABO-in-
compatible transplant recipients in order
to avoid confusion. RBC transfusions, be-
ginning with the conditioning regimen,
should be of donor type. Recommenda-
tions for optimal blood component selec-
tion are shown in Table 25-5.
Chimerism
In spite of intensive pretransplant chemo-
therapy and irradiation, some of the host’s
hematopoietic cells may survive and sub-
sequently coexist with cells produced by
the transplanted HPCs. This dual cell pop-
ulation, called partial hematopoietic
chimerism, may have an effect on immu-
nologic tolerance.124 This tolerance devel-
ops in successful transplants and allows
normal immune reconstitution. Full chim-
erism (replacement of host marrow cells
entirely by donor cells) usually occurs in
successful HPC-M transplantation.
Processing in the Presence of ABO
Incompatibilities
Major ABO Incompatibility. Two ap-
proaches have been employed with major
ABO incompatibilities: 1) removal of or
decrease in the isoagglutinin level in the
recipient or 2) removal of the red cells in
the HPC-M collection. Processing is not
generally required with HPC-A because
such a small volume of red cells is usually
included.
Attempts to remove or decrease the iso-
agglutinin titer in recipients involve the use
of one or more large-volume plasma ex-
changes with or without the subsequent in-
fusion of donor-type red cells as a secondary
effort to absorb any additional isoaggluti-
nins.125,126
Other approaches to ABO-incompatible
products include red cell depletion. The
most prevalent method in use is red cell
sedimentation.
Some institutions accomplish red cell
depletion using density gradient separation
and cell washers (Gambro BCT, Lakewood,
CO); others use continuous-flow apheresis
machines such as the Spectra (Gambro) and
the Fenwal CS-3000 (Baxter Healthcare),
with mononuclear cell recoveries of 94%
and 87% and red cell depletion of 99% and
98%, respectively.
Minor ABO Incompatibility. Theoreti-
cally, the level of hemolysis is dependent on
thevolumeofHPC-Minfusedrelativeto
the recipient’s plasma volume and the IgM
titer of the donor. Incompatible plasma can
be easily removed by centrifugation.
Plasma removal using this method removes
Chapter 25: Cell Therapy and Cellular Product Transplantation 601
Copyright © 2005 by the AABB. All rights reserved.

approximately 75% of the plasma volume
while recovering >70% of the initial nucle-
ated cell count. Plasma removal is not gen-
erally required in HPC-A because the vol-
ume of incompatible plasma is usually small.
Umbilical Cord Blood Processing
Advances in the processing of umbilical
cord blood cells have solved the early
problems of unacceptably high progeni-
tor cell losses with manipulation.127 Unlike
cryopreservation laboratories that store
relatively few autologous or allogeneic
HPC-M or HPC-A products (typically, less
than a few hundred products at any one
time), a successful cord bank would be
expected to store thousands of products
for prolonged periods. Therefore, attempts
to reduce umbilical cord blood bulk (and,
therefore, storage space, liquid nitrogen
requirements, and cost) have been ac-
tively pursued. Currently, many centers
arefollowingtheapproachusedbythe
New York Blood Center, which involves
sedimentation and volume reduction be-
fore cryopreservation.128
Cultures for Microbial Contamination
Sterility is essential in blood products for
infusion to immunosuppressed patients.
This is particularly important with HPC-M,
which is frequently collected in an open
system, and with products that require
multiple manipulations (Table 25-6).129-134
Culture growth can result from contami-
nation during collection or product pro-
cessing, or as a result of an infected cathe-
ter or the patient’s sepsis. Skin commensals
are the predominant isolates from these
cultures. In all cases it is important to
identify the source, the degree of contam-
ination, and the causative organism,
given the fact that this product is in-
tended for transplantation in an immuno-
compromised recipient. Each product
must be tested for microbial contamina-
tion at least once during the course of
processing, usually just before freezing or
infusion with allogeneic transplants.48 A
positive culture does not necessitate im-
mediate discard of the product because
these are frequently irreplaceable cells.135
Thus, positive results need to be reviewed
by the appropriate physician on a case-
by-case basis and sensitivity tests ordered
to better define the organism and to allow
for appropriate clinical management at
the time of transplant. This should also
serve as the impetus to review all proce-
dures and techniques to ensure that they
602 AABB Technical Manual
Table 25-6. Percent Microbial Contamination of Cell Therapy Products
Source of HPCs No. of Products Tested % Contaminated Reference
Marrow 291 0 129
227 1.3 130
317 6.0 131
194 <0.1 132
Peripheral blood 1380 0.65 129
560 0.7 130
576 0.5 131
1040 0.2 133
Cord blood 1000 <1.0* 134
*Initial rates were as high as 28%; however, with improved donor selection and technique, the rate fell to <1%.
Copyright © 2005 by the AABB. All rights reserved.
provide safeguards to prevent contamina-
tion.
Stem Cell Enumeration: CD34 Analysis,
Aldehyde Dehydrogenase Activity, and Others
As stem cells differentiate into hemato-
poietic cells, CD34 surface markers ap-
pear. As the cells continue to differentiate
and acquire other surface markers, CD34
expression diminishes. Stages of differen-
tiation may be studied by the expression
or co-expression of other antigens or
properties. There are currently two assays
for CD34 that are FDA-cleared for in-vitro
diagnostic use. These CD34+ measure-
ment methods are the most widely used
for the evaluation of grafts, have the lon-
gest history of use, and show the most
clinical correlations in the marrow trans-
plantation literature.
The principle of gain and later loss of
CD34 expression has been applied to evalu-
ate the quality of HPC grafts. The current
approach is to use a method based on
multiparameter flow cytometry to deter-
mine the number of CD34+ cells and to cal-
culate a subsequent CD34+ dose based on
the recipient’s weight. Numerous flow
cytometric analysis methods for CD34 enu-
meration exist.56,136,137 Because these meth-
ods differ, correlation of CD34 dose values
from site to site is often unreliable. Multiple
studies have documented the variability of
CD34 analysis methods and results and
have examined possible causes of the vari-
ability.56,57,138,139 Becauseofthe“rareevent”
nature of CD34 enumeration, several pro-
cedural components play a critical role in
the assay. Selection of the CD34 antibody
clone, fluorescent conjugate, lysing solu-
tion, lyse-wash format, gating strategy, and
the number of events analyzed are some of
the factors that influence the end result.140
Growing awareness of, and concern
about, the need for a standardized ap-
proach to CD34 analysis have prompted
several collaborative groups such as the In-
ternational Society for Cellular Therapy
(ISCT) to propose guidelines for CD34+ cell
determination by flow cytometry.56,141 Alter-
nate approaches are available and will con-
tinue to be developed and investigated as
more is discovered about the CD34 antigen
and other markers for the pluripotent stem
cell.142
A second method for enumeration of
early cells has gained FDA clearance. The
cytosolic expression of aldehyde dehydro-
genase (ALDH) in stem cells was found to
betheprotectivepropertyofHSCsincyclo
-
phosphamide treatment of patients and in
preservation of stem cells in graft purging
with 4HC. Flow cytometric analysis of
ALDH-positive cells with the enzymatic ex-
citation and trapping inside the stem cells
of a fluorescent substrate (because the
fluorophore becomes charged and does not
diffuse freely) allows measurement of a via-
ble population enriched in colony-forming
progenitor cells. Both long-term repopulat-
ing and committed progenitor cells are
enriched and the ALDHbr contains the pop-
ulation responsible for hematopoietic engraft-
ment of NOD-SCID mice.143,144 This method
to enumerate ALDHbr SSclo cells identifies
only cells with intact membranes and is
well correlated with engraftment in post-
thaw HPC-A grafts.145 Another measure-
ment technique involving the use of Flk-2
and Thy 1.1lo showed that Flk-2–, Thy 1.1lo
cells represented long-term repopulating
cells, whereas Flk-2+, Thy 1.1lo cells were
short-term engraftment-enhancing cells.
These tools may be developed for use in fu-
ture graft engineering approaches to finely
separate early cell populations.146
Colony-Forming Cell Assays
Culture systems are available that can
demonstrate in-vitro proliferative capacity
Chapter 25: Cell Therapy and Cellular Product Transplantation 603
Copyright © 2005 by the AABB. All rights reserved.
of a hematopoietic sample. It is thought
that short-term (generally within 2 weeks)
repopulating potential is produced from
the committed HPCs. Long-term repopu-
lating ability is thought to be a result of
the pluripotential HPCs that are princi-
pally necessary for a complete and sus-
tained engraftment. Cultures may be use-
ful to assess engraftment potential; however,
because media, culture techniques, and
colony identification are quite variable,
interinstitutional comparisons are diffi-
cult. Reported CFU-GM doses below
which engraftment may be delayed range
from 1.5 ×105to 5.0 ×105/kg.147
Long-term cultures are not routinely used
clinically because of the 5- to 8-week incu-
bation requirements. However, they may be
useful in evaluating developmental proce-
dures involving product manipulation.
Freezing and Storage
Although easily performed by many trans-
plant centers, HPC cryopreservation and
reinfusion are not without risk of loss of
HPCs. The infusion of cryopreserved cells
also results in risks to the recipient, many
of which are life-threatening, and some of
which are not well understood. The loss of
HPCs from cryopreservation and the ef-
fect of this loss on engraftment speed have
not been quantified. There are multiple
aspects to successful cryopreservation of
HPCs. Each of these variables affects the
recovery of HPCs during the cryopreser-
vationstepsand,aswithanymanufacturing
process, must be rigidly controlled for re-
producible results. The question that still
faces cryopreservation facilities is whether
other solutions or processing techniques
will provide better cryosurvival of cells, at
less cost, with greater simplicity, or with
less toxicity to either the recipient or to
the cell type being frozen.
HPC-A, HPC-M, and HPC-C are frozen
and stored using the same techniques. The
general parameters include cryopreservation
in dimethylsulfoxide (DMSO) and a source
of plasma protein with or without hydrox-
yethyl starch (HES), cooling at 1 to 3 C/
minute, and storage at –80 C or colder. Vari-
ations on this technique include the con-
centration at which the cells are frozen, the
amount and source of the plasma protein,
and the cooling techniques used.148 Most of
these variations probably have little effect
on the survival of the HPCs as shown by the
consistent engraftment of cryopreserved
components. However, cryopreservation
results in the loss of an undefined but po-
tentially substantial proportion of HPCs, and
delay in engraftment can occur if the com-
ponent being frozen has borderline quanti-
ties of HPCs. There is also considerable but
generally minor toxicity associated with the
infusion of cryopreserved cells that must be
considered when developing cryopreserva-
tion techniques.
Allogeneic Products
The collection of allogeneic marrow is
generally timed to coincide with the com-
pletion of the recipient’s preparative regi-
men. Because of the brief storage period
required, the product is maintained in the
liquid state. Unseparated HPC-M can be
stored in the liquid state for up to 3 days
at either 4 C or 22 C without any signifi-
cant loss in viability of either uncommit-
ted or committed progenitors. The ability
to store unmanipulated marrow under
these conditions is vital in the context of
the NMDP or other transplant registries
where unrelated HPC-M units are col-
lected and then transported great dis-
tances for transplantation. Similarly,
HPC-A units may be stored in the liquid
stateunderthesameconditionsasHPC-M
for up to 3 days at 4 C.149,150 The cell con-
604 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
centration is important in determining
the storage time and temperature that the
cells will tolerate without damage. High
concentrations of white cells in liquid
storage at room temperature quickly ex-
pend the supplies of oxygen and glucose
and cause a decrease in pH in the storage
medium that is toxic to the cells. A lower
concentration of cells (higher volume of
suspending medium, often autologous
plasma) and refrigerated storage are pro-
tective.
Autologous Products
Because of the length of time required for
treatment and/or HPC product collection,
autologous products are usually cryopre-
servedandstoreduntilthetimeofinfu-
sion. Cryopreservation also allows HPCs to
be collected while the patient is in remis-
sion and stored for use in case a relapse
occurs (prophylactic storage). At present,
no expiration date has been defined for
these products; however, HPC-M stored
for 11 years has been used for transplanta-
tion, with sustained engraftment.151
Computer-Controlled Cryopreservation
The purpose of cryopreservation is to freeze
cells in such a way as to allow for their
long-term storage with a minimal loss of
cell viability or reconstitutive ability upon
thaw. The main obstacle to maintaining
viability during cryopreservation and
storage is the formation of intracellular
ice crystals along with an increase in ex-
ternal osmolarity, causing exit of water from
the cell, both resulting in cell lysis. Ther-
mal shock, the time required for phase
change (liquid state to solid state), and
the posttransition freezing rate also pres-
ent specific conditions to be managed for
cryopreservation.152
The adverse effects caused by the forma-
tion of intracellular ice crystals or by cell
dehydration can be minimized by a slow
cooling rate and the addition of a cryo-
protective agent such as DMSO. Cryo-
protectants such as DMSO prevent the for-
mation of large ice crystals within the cells
by penetrating the cell and providing some
balance of the external hyperosmolar sol-
ute conditions in the freezing medium sur-
rounding the cell.153 A commonly used
cryoprotectant consists of 20% DMSO and
20% plasma or albumin prepared in a buf-
fered electrolyte solution or tissue culture
media. The plasma or albumin provides a
proteinsource,whichaidsinpreventingcell
damage during freezing and thawing. The
cryoprotectant is combined with an equal
volume of product immediately before
freezing, resulting in a final cell suspension
containing 10% DMSO and 10% plasma.
Phase transition time is a physical phe-
nomenon of freezing water where forming
ice crystals release heat energy into the cell
solution that needs to be offset by contin-
ued cooling to prevent crystallization of ice,
thawing, and recrystallization during this
energy exchange, with damage to the cell
membrane.152 This method of freezing re-
quires physical conditions that support this
process. This may be accomplished by a
computerized programmable freezing
chamber that adds liquid nitrogen in suffi-
cient amounts to push the freezing process
through the phase change smoothly. The
freezer is programmed to cool cells at an
optimal rate of 1 to 3 C/minute until a tem-
perature of –90 to –100 C is reached.154-156 With
the combination of a cryoprotectant and a
programmed rate of freezing, cryopreserva-
tion and long-term storage of HPCs are
possible, with minimal damage to the cells.
Passive Controlled-Rate Freezing
Mechanical, methanol bath immersion,
or “passive controlled-rate” freezing is ad-
vocated by some investigators as a simple,
Chapter 25: Cell Therapy and Cellular Product Transplantation 605
Copyright © 2005 by the AABB. All rights reserved.
reliable, cost-effective, and validated
method of managing the physical condi-
tions of cooling as an alternative to a con-
trolled-rate device.157-159 The principle is
that products can be cooled without the
aid of a programmable freezer or liquid
nitrogen if the conditions of freezing are
predetermined, measured, and performed
according to standard operating proce-
dures. The products are stored in a –80 C
mechanical freezer or colder after the ini-
tial freeze. HPCs stored in this fashion
have successfully engrafted after as long
as 2 years of storage.159 In some cases, the
combination of the metal canisters the
bags are placed in, the bag itself, and the
volume of product frozen produces a
freezing rate of approximately 3 C/min-
ute, which falls within the optimal range
previously discussed.160 The major deter-
rent to use of mechanical freezers is fear
of mechanical failure and the lack of data
on long-term storage and engraftment.
Frozen Storage
Although products frozen in a mechanical
freezer are stored at –80 to –150 C, prod-
ucts cryopreserved using a programmable
freezer generally are stored in a liquid ni-
trogen freezer. The storage temperature
achieved with vapor phase, although not
as cold as liquid phase, averages –140 C, a
temperature that has been shown to allow
for viable long-term marrow storage.151
The major drawbacks to vapor phase stor-
age include the potential for large fluctua-
tion in temperature when the freezer is
entered as well as the variation in storage
temperature throughout the freezer itself,
dependent on the design of the storage
chamber and the procedure for entering
and retrieving units. The time to rising
temperatures of the stored products in
cases of electrical or liquid nitrogen sup-
ply emergencies is significantly shorter
than with liquid phase. The advantage of
vapor phase storage is the possibly de-
creased risk of cross-contamination that
has become an issue with liquid phase
storage.161
Storage of Untested or Infectious Products
Regulations [21 CFR 1271.60(a) and
1271.65(a)] and standards48(p35),49 require al-
ternative storage for products that are un-
tested or have a positive disease marker
test result. Some institutions comply by
storing all products in vapor phase and
physical separation by category of prod-
uct. Other institutions use an overwrap,
placing the component in an outer plastic
bag that is sealed before storage. A third
alternative is a separate storage compart-
ment for units that are untested or posi-
tive for infectious disease markers.
Final Suitability Criteria
Before each autologous or allogeneic unit
is released, it must undergo review and
meet the predetermined release criteria
described in the facility’s quality plan.
The pertinent facts describing donor se-
lection, product collection, processing,
and storage must be reviewed. The con-
tainer label and associated information
must accurately reflect the product classi-
fication, storage/preservative medium in
the final container, the product content
(usually cell content), and results of re-
lease testing including infectious disease
tests. If exceptions to standard practice
were made, they must be explained either
on the label or in the accompanying re-
lease material.
Transportation and Shipping
In some cases, a hematopoietic component
must be transported from one center to
another. The product must be positively
606 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
identified upon its removal from inven-
tory in preparation for shipping. In all
cases, precautions should be taken to pro-
tect the component from rough handling,
out-of-range temperatures, X-ray exami-
nation, breakage, and spillage. The ship-
ping container must undergo quality con-
trol to ensure that it is capable of holding
the expected temperature during ship-
ping. In the case of cryopreserved compo-
nents, the use of a liquid nitrogen “dry
shipper” is desirable. Such “dry shippers”
have liquid nitrogen absorbent material
between the walls of the container that al-
lows the inside of the container to main-
tain temperatures in the range of –180 C
forupto10to14daysiftheyareproperly
filled with liquid nitrogen and shipped in
the upright position. Tipping or inversion
of the container during shipping permits
the liquid nitrogen to drain out, allowing
the container to warm toward ambient
temperature.
Thawing and Infusion
For all components, final identification is
done by the nurse or physician perform-
ing the infusion. Flow through the central
venous catheter is confirmed and the cells
are infused by gravity drip, calibrated
pump, or manual push with or without an
in-line filter (a standard 170-micron red
cell infusion filter is acceptable). Although
DMSO was thought to be toxic to HPCs, it
is now known to be nontoxic after short-
term exposure (up to 1 hour) at the con-
centration used for cryopreservation of
HPCs.162 However, prolonged exposure to
DMSO ex vivo at 22 to 37 C may be harm-
ful to HPCs. To minimize the exposure of
thawed cells to DMSO, many centers rap-
idly thaw one bag at a time near the bed-
side. Some centers place product bags in
secondary containment bags before thaw-
ing; others immerse the bag (all but the
access ports) directly into sterile water or
salineat37to40C.
152 The bag is kneaded
gently until all solid clumps have thawed.
The cells are then infused (usually 10-15
mL per minute). Products may also be
washed and resuspended in the laboratory
before infusion to prevent cell aggregation.152
Side effects associated with infusions in-
clude nausea, diarrhea, flushing, brady-
cardia, hypertension, and abdominal pain.
In general, such side effects may justify
slowing, but not halting, the infusion until
the symptoms pass. A limit of 1 gram of
DMSO per kilogram of body weight of the
patient at one infusion is recommended to
allow the patient to tolerate both the DMSO
and the volume infusion effects of the ad-
ministration. Sudden and severe hypoten-
sion can occur in the absence of adequate
antihistamine premedication. The patient
should receive fluids and treatment to en-
sure that the urine is “alkalinized.” This fa-
cilitates the clearance of hemoglobin caused
by red cell lysis, which occurs during freez-
ing and reduces the risk of renal complica-
tions. If the total infusion volume exceeds
10 mL/kg of recipient body weight, many
centers divide the volume over a morning
and an afternoon infusion or over 2 consec-
utive days.
It is best to collect postthaw laboratory
samples directly from the patient’s infusion
bags instead of freezing separate individual
specimens because these samples are iden-
tical to the infused product.163
Evaluation and Quality Control
of Hematopoietic Products
Cell Counts
Each hematopoietic product is analyzed
to determine the total cell concentration
and the mononuclear cell concentration,
Chapter 25: Cell Therapy and Cellular Product Transplantation 607
Copyright © 2005 by the AABB. All rights reserved.
which are used to calculate the number of
cells per kilogram (of the recipient) or cell
dose for each product.48(p55),49 These doses,
in combination with other assays, deter-
mine the number of collections necessary
to achieve engraftment.135 In addition,
they are used to calculate the percent re-
covery, providing a quality control mea-
sure for processing procedures and
equipment.164
In general, automated cell counts provide
the most rapid and accurate value. How-
ever, platelet/cellular aggregates in HPC-A
or fat globules in HPC-M specimens can
falsely decrease or increase cell counts.135,165
In such cases, manual cell counts may be
preferable. It is important that cell counts
are not overestimated because this may re-
sult in inaccurate estimation of time to
engraftment or graft failure.
Engraftment Data
Ultimately, engraftment of neutrophils,
platelets, and red cells is the primary de-
terminant of graft quality. Monitoring and
documenting days to engraftment for
neutrophil and platelet lineage are re-
quired by FACT49 and AABB48(p41) for ac-
creditation.
Tumor Cell Detection
Tumor cell detection techniques have been
developed to screen products suspected
of tumor cell contamination and to evalu-
ate purged products. The majority of these
assays use MoAbs that specifically bind
tumor antigens. Detection and quantita-
tioncanbedonebyflowcytometry,immuno
-
fluorescence, or immunohistochemical
staining. Sensitivity varies with technique
from 0.1% to 0.0004% of cells examined.166
Preliminary studies indicate that the pres-
ence of tumor cells may be associated
with a reduced disease-free survival.167
Regulations
In 1997, the FDA announced a new com-
prehensive approach to the regulation of
cellular and tissue-based products.168
HPCs from placental/umbilical cord
blood and peripheral blood were covered
by this proposal. Many of the policies,
proposed regulations, and guidance doc-
uments needed to implement this ap-
proach have been published.47,169-173
These regulations require establishment
registration and listing of facilities collect-
ing, processing, or distributing tissue or cell
therapy products. Although the regulations
are similar to those for blood donor qualifi-
cation, there are differences appropriate to
the specific tissue source under consider-
ation. The cGTP regulations (found in Title
21 CFR 1271.145 to 320) are specific in-
structions intended to ensure that facilities
establish and maintain a quality program
that documents that personnel, proce-
dures, facilities, equipment, supplies, and
reagents are set up and maintained in an
acceptable and a standard manner. Process
control is required: there must be written
procedures and documented validation.
Products are required to be collected,
tested, labeled, and stored in ways that pre-
serve their identity and prevent contamina-
tion and cross-contamination. It may be
necessary to demonstrate regulatory com-
pliance during FDA inspection visits. Re-
porting of adverse events (when applicable)
is also required within 15 days of receipt of
the information if the event is serious as de-
fined in 21 CFR 1271.350. This body of reg-
ulations represents a major effort on the part
of the FDA to ensure that tissue and cell
therapy products are safe for the recipient.
608 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Standards
The AABB and FACT have published sepa-
rate, but substantially similar, standards
for HPCs.48,49 The AABB Standards for Cel-
lular Therapy Product Services addresses
the collection, processing, storage, and
distribution of HPCs. Other organizations
such as the NMDP also publish voluntary
standards. FACT standards address clini-
cal issues provided by HPC clinical trans-
plant programs as well as laboratory ser-
vices performing the collection, processing,
storage, and distribution of HPCs. Both
AABB and FACT provide an inspection
and certification program for requesting
members. These program reviews are an
important, external, and impartial look at
program organization and how the pro-
gram’s organization affects patient out-
come. As an outgrowth of the standards
writing process, representatives from the
constituent organizations of AABB and
FACT cooperated to produce a uniformly
endorsed Circular of Information for the
Use of Cellular Therapy Products.174 In this
circular, the organizations agreed upon a
number of principles, including a com-
mon set of names for cell therapy prod-
ucts. This agreement on product names
allowed the International Committee on
Commonality of Blood Bank Automation’s
(ICCBBA) North American Task Force to
approve progenitor cell label designs, in-
cluding ICCBBA’s quality design require-
ments.
References
1. Korbling M, Estrov Z. Adult stem cells for tis-
sue repair—a new therapeutic concept? N
Engl J Med 2003;349:570-82.
2. Champlin RE, Schmitz N, Horowitz MM, et
al. Blood stem cells compared with bone
marrow as a source of hematopoietic cells
for allogeneic transplantation. IBMTR Histo-
compatibility and Stem Cell Sources Work-
ing Committee and the European Group for
Blood and Marrow Transplantation (EBMT).
Blood 2000;95:3702-9.
3. Report on state of the art in blood and mar-
row transplantation—the IBMTR/ABMTR
summary slides with guide (2002). IBMTR/
ABMTR Newsletter 2002;9(1):4-11. [Available
at http://www.ibmtr.org/newsletter/pdf/
2002Feb.pdf.]
4. Champlin R, Khouri I, Anderlini P, et al. Non-
myeloablative preparative regimens for allo-
geneic hematopoietic transplantation. Biology
and current indications. Oncology (Hunt-
ing), 2003;17:94-100.
5. Sandmaier BM, McSweeney P, Yu C, Storb R.
Nonmyeloablative transplants: Preclinical
and clinical results. Semin Oncol 2000;27:
78-81.
6. Storb RF, Champlin R, Riddell SR, et al. Non-
myeloablative transplants for malignant dis-
ease. Hematology (Am Soc Hematol Educ
Program) 2001;375-91.
7. Giralt S, Anagnostopoulos A, Shahjahan M,
et al. Nonablative stem cell transplantation
for older patients with acute leukemias and
myelodysplastic syndromes. Semin Hematol
2002;39:57-62.
8. ToLB,HaylockDN,SimmonsPJ,JuttnerCA.
The biology and clinical uses of blood stem
cells. Blood 1997;89:2233-58.
9. National Marrow Donor Program Overview
(2001 slide presentation). [Available at http://
www.nmdp.org/nmdp/slide_presentation.
html.]
10. Powderly J, Shea T. Hematopoietic cell trans-
plantation. In: Runge MS, Greganti MA, eds.
Netter’s internal medicine. Teterboro, NJ:
Icon Learning Systems, 2003:401-05.
11. Stroncek DF, Confer DL, Leitman SF. Periph-
eral blood progenitor cells for HPC trans-
plants involving unrelated donors. Transfu-
sion 2000;40:731-41.
12. Bensinger WI, Martin PJ, Storer B, et al.
Transplantation of bone marrow as com-
pared with peripheral-blood stem cells from
HLA-identical relatives in patients with
hematologic malignancy. N Engl J Med 2001;
344:175-81.
13. Beatty PG, Kollman C, Howe CW. Unrelated-
donor marrow transplants: The experience
of the National Marrow Donor Program. Clin
Transpl 1995:271-7.
14. Scott I, O’Shea J, Bruce M, et al. Molecular
typingshowsahighlevelofHLAclassIin
-
compatibility in serologically well matched
donor/patient pairs: Implications for unre-
lated bone marrow donor selection. Blood
1998;92:4864-71.
Chapter 25: Cell Therapy and Cellular Product Transplantation 609
Copyright © 2005 by the AABB. All rights reserved.
15. Petersdorf EW, Gooley TA, Anasetti C, et al.
Optimizing outcome after unrelated marrow
transplantation by comprehensive matching
of HLA class I and II alleles in the donor and
recipient. Blood 1998;92:3515-20.
16. Petersdorf EW, Longton GM, Anasetti C, et al.
Definition of HLA-DQ as a transplantation
antigen. Proc Natl Acad Sci U S A 1996;93:
15358-63.
17. Gajewski J, Gjertson D, Cecka M, et al. The
impact of T-cell depletion on the effects of
HLA DR beta 1 and DQ beta allele matching
in HLA serologically identical unrelated bone
marrow transplantation. Biol Blood Marrow
Transplant 1997;3:76-82.
18. Petersdorf EW, Longton GM, Anasetti C, et al.
Association of HLA-C disparity with graft fail-
ure after marrow transplantation from unre-
lated donors. Blood 1997;89:1818-23.
19. Kanfer E. Graft-versus-host disease. In:
Treleaven J, Wiernik P, eds. Color atlas and
text of bone marrow transplantation. Lon-
don: Mosby-Wolfe, 1995:185-8.
20. Kolb HJ, Schattenberg A, Goldman JM, et al.
Graft-versus-leukemia effect of donor lym-
phocyte transfusion in marrow grafted pa-
tients. Blood 1995;86:2041-50.
21. Champlin R. Bone marrow transplantation
from HLA-matched unrelated donors as treat-
ment for leukemia. J Hematother 1993;2:
323-7.
22. Champlin R, Kyoung L. T-cell depletion to
prevent graft-versus-host disease following
allogeneic bone marrow transplantation. In:
Areman EM, Deeg HJ, Sacher RA, eds. Bone
marrow and stem cell processing: A manual
of current techniques. Philadelphia: FA Da-
vis, 1992:163-217.
23. ApisarnthanaraxN,DonatoM,KorblingM,et
al. Extracorporeal photopheresis therapy in
the management of steroid-refractory or ste-
roid-dependent cutaneous chronic graft-ver-
sus-host disease after allogeneic stem cell
transplantation: Feasibility and results. Bone
Marrow Transplant 2003;31:459-65.
24. Chan KW, Wadsworth LD. Pediatric bone
marrow transplantation and processing. In:
Areman EM, Deeg HJ, Sacher RA, eds. Bone
marrow and stem cell processing: A manual
of current techniques. Philadelphia: FA Da-
vis, 1992:363-85.
25. Stroncek D, Anderlini P. Mobilized PBPC con-
centrates: A maturing blood component (edi-
torial). Transfusion 2001;41:168-71.
26. Bensinger WI, Clift R, Martin P, et al. Allo-
geneic peripheral blood stem cell transplan-
tation in patients with advanced hematologic
malignancies: A retrospective comparison
with marrow transplantation. Blood 1996;88:
2794-800.
27. Ottinger HD, Beelen DW, Scheulen B, et al.
Improved immune reconstitution after allo-
transplantation of peripheral blood stem
cells instead of bone marrow. Blood 1996;88:
2775-9.
28. Storek J, Gooley T, Siadak M, et al. Allogeneic
peripheral blood stem cell transplantation
may be associated with a high risk of chronic
graft-versus-host disease. Blood 1997;90:
4705-9.
29. Broxmeyer HE, Gluckman E, Auerbach AD, et
al. Human umbilical cord blood: A clinically
useful source of transplantable hematopo-
ietic stem/progenitor cells. Int J Cell Cloning
1990;8:76.
30. Gluckman E, Broxmeyer HE, Auerbach AD, et
al. Hematopoietic reconstitution in a patient
with Fanconi anemia by means of umbili-
cal-cord blood from an HLA-identical sib-
ling. N Engl J Med 1989;321:1174-8.
31. Rocha V, Wagner JE, Sobocinski KA, et al.
Graft-versus-host disease in children who
have received a cord-blood or bone marrow
transplant from an HLA-identical sibling. N
Engl J Med 2000;342:1846-54.
32. Laughlin MJ, Barker J, Bambach B, et al.
Hematopoietic engraftment and survival in
adult recipients of umbilical-cord blood from
unrelated donors. N Engl J Med 2001;
344:1815-22.
33. Wagner J, Barker J, DeFor T, et al. Transplan-
tation of unrelated donor umbilical cord
blood in 102 patients with malignant and
non-malignant diseases: Influence of CD34
cell dose and HLA disparity on treatment-re-
lated mortality and survival. Blood 2002;
100:1611-18.
34. Rubinstein R, Carrier C, Scaradavou A, et al.
Outcomes among 562 recipients of placen-
tal-blood transplants from unrelated donors.
N Engl J Med 1998;339:1565-77.
35. Wagner JE, Kernan NA, Steinbuch M, et al.
Allogeneic sibling umbilical-cord-blood
transplantationinchildrenwithmalignant
and non-malignant disease. Lancet 1995;
346:214-9.
36. Locatelli F, Rocha V, Chastang C, et al. Factors
associated with outcome after cord blood
transplantationinchildrenwithacuteleuke
-
mia. Eurocord-Cord Blood Transplant Group.
Blood 1999;93:3662-71.
37. Rocha V, Cornish J, Sievers EL, et al. Compari-
sonofoutcomesofunrelatedbonemarrow
and umbilical cord blood transplants in chil-
dren with acute leukemia. Blood 2001;97:
2962-71.
610 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
38. Laughlin MJ, Eapen M, Rubinstein P, et al.
Outcomes after transplantation of cord blood
or bone marrow from unrelated donors in
adults with leukemia. N Engl J Med 2004;351:
2265-75.
39. Rocha V, Labopin M, Sanz G, et al. Transplants
of umbilical-cord blood or bone marrow
from unrelated donors in adults with acute
leukemia. N Engl J Med 2004;351:2276-85.
40. Barker JN, Weisdorf DJ, DeFor TE, et al. Trans-
plantation of two partially HLA-matched
umbilical cord blood units to enhance engraft-
ment in adults with hematologic malignancy.
Blood 2005;105:1343-7.
41. Mehta SN. Umbilical-blood storage: Uncer-
tain benefits, high price. The Wall Street
Journal 1995 Nov 14;Sect B:1-2.
42. Rubin R. A hard sell to bank your baby’s
blood. US News & World Report 1996 July
29:60-1.
43. Ferreira E, Pasternak J, Bacal N, et al. Autolo-
gous cord blood transplantation (letter). Bone
Marrow Transplant 1999;24:1041.
44. Flowers MED, Pepe MS, Longton G, et al.
Previous donor pregnancy as a risk factor for
acute graft-versus-host disease in patients
with aplastic anemia treated by allogeneic
marrow transplantation. Br J Haematol 1990;
74:492-6.
45. Gale RP, Bortin MM, Van Bekkum DW, et al.
Risk factors for acute graft-versus-host dis-
ease. Br J Haematol 1987;67:397-406.
46. Bensinger WI, Deeg JH. Transfusion support
and donor considerations in marrow trans-
plantation. In: Sacher RA, AuBuchon JP, eds.
Marrow transplantation: Practical and tech-
nical aspects of stem cell reconstitution.
Bethesda, MD: AABB, 1992:157-84.
47. Food and Drug Administration. Eligibility
determination for donors of human cells, tis-
sues, and cellular and tissue-based products;
final rule (May 25, 2004). Fed Regist 2004;69:
29786-834.
48. Szczepiorkowski ZM, ed. Standards for cellu-
lar therapy product services. Bethesda, MD:
AABB, 2004.
49. Warkentin PI, ed. Standards for hematopoi-
etic progenitor cell collection, processing
and transplantation. 2nd ed. Omaha, NE:
Foundation for the Accreditation of Cellular
Therapy, 2002.
50. MairDC,BrecherME,HomE,etal.False-
positive hepatitis B surface antigen screen-
ing test results in patients receiving granulo-
cyte-colony-stimulating factor. Transfusion
1996;36:948-51.
51. Kaufman RM, Goodnough LT, Lublin DM, et
al. False-positive hepatitis C screening tests
results in healthy allogeneic peripheral blood
stem cell donors (abstract). Transfusion 2000;
40(Suppl):S34.
52. International standards for cord blood col-
lection, processing, testing, banking, selec-
tion and release. 2nd ed. Omaha, NE: Foun-
dation for the Accreditation of Cellular
Therapy, 2001.
53. Treleaven JG. Bone marrow harvesting and
reinfusion.In:GeeAP,ed.Bonemarrowpro
-
cessing and purging. Boca Raton, FL: CRC
Press, 1991:32-4.
54. Lane TA. Mobilization of hematopoietic pro-
genitor cells. In: Brecher ME, Lasky LC,
Sacher RA, Issitt LA, eds. Hematopoietic pro-
genitor cells: Processing, standards and
practice. Bethesda, MD: AABB, 1995:59-108.
55. Krause DS, Fackler MJ, Civin CI, et al. CD34:
Structure, biology, and clinical utility. Blood
1996;87:1-13.
56. Sutherland DR, Anderson L, Keeney M, et al.
The ISHAGE guidelines for CD34+ cell deter-
mination by flow cytometry. J Hematother
1996;5:213-26.
57. Brecher ME, Sims L, Schmitz J, et al. North
American multicenter study on flow cyto-
metric enumeration of CD34+ hemato-
poietic stem cells. J Hematother 1996;5:227-
36.
58. Lie AK, Rawlings TP, Bayly JL, To LB. Progeni-
tor cell yield in sequential blood stem cell
mobilization in the same patients: Insights
into chemotherapy dose escalation and
combination of haemopoietic growth factor
and chemotherapy. Br J Haematol 1996;95:
39-44.
59. Leitman SF, Read SJ. Hematopoietic progeni-
tor cells. Semin Hematol 1996;33:341-58.
60. Schwella N, Beyer J, Schwaner I, et al. Impact
of preleukapheresis cell counts on collection
results and correlation of progenitor-cell
dose with engraftment after high-dose che-
motherapy in patients with germ cell cancer.
Blood 1994;83:3787-94.
61. Elliot C, Samson DM, Armitage S, et al. When
to harvest peripheral blood stem cells after
mobilization therapy: Prediction of CD34
positive cell yield by preceding day CD34
positive concentration cell in peripheral
blood. J Clin Oncol 1996;14:970-3.
62. Owen HG, Brecher ME. Paired randomized
comparative study of PBSC collection (ab-
stract). J Clin Apheresis 1997;12:29.
63. Bandarenko N, Sims LC, Brecher ME. Circu-
lating CD34+ counts are predictive of PBSC
CD34+ yields (letter). Transfusion 1997;37:
1218.
64. Perez-Simon JA, Caballero MD, Corral M, et
al. Minimum number of circulating CD34+
cells to ensure successful leukapheresis and
Chapter 25: Cell Therapy and Cellular Product Transplantation 611
Copyright © 2005 by the AABB. All rights reserved.
engraftment in autologous peripheral blood
progenitor cell transplantation. Transfusion
1998;38:385-91.
65. Green RHA, Morrison AE, Watson D, et al.
The use of peripheral blood CD34 measure-
ments in determining the optimal timing of
peripheral blood progenitor cell (PBPC) col-
lections (abstract). J Clin Apheresis 1997;12:
47.
66. Yu J, Leisenring W, Bensinger WI, et al. The
predictive value of white cell or CD34+ cell
count in the peripheral blood for timing
apheresis and maximizing yield. Transfusion
1999;39:442-50.
67. Shpall EJ, Champlin R, Glaspy JA. Effect of
CD34+ peripheral blood progenitor cell dose
on hematopoietic recovery. Biol Blood Mar-
row Transplant 1998;4:84-92.
68. Schulman KA, Birch R, Zhen B, et al. Effect of
CD34+ cell dose on resource utilization in pa-
tients after high-dose chemotherapy with pe-
ripheral-blood stem-cell support. J Clin
Oncol 1999;17:1227.
69.BensingerW,AppelbaumF,RowleyS,etal.
Factors that influence collection and engraft-
ment of autologous peripheral-blood stem
cells. J Clin Oncol 1995;13:2547-55.
70. Malachawski ME, Comenzo RL, Hillyer CD, et
al. Large-volume leukapheresis for periph-
eral blood stem cell collection in patients
with hematologic malignancies. Transfusion
1992;32:732-5.
71. Passos-Coelho JL, Braine HG, Davis JM, et al.
Predictive factors for peripheral-blood pro-
genitor-cell collections using a single large-
volume leukapheresis after cyclophospha-
mide and granulocyte-macrophage col-
ony-stimulating factor mobilization. J Clin
Oncol 1995;13:705-14.
72. Hillyer CD, Lackey DA, Hart KK, et al. CD34+
progenitors and colony-forming units-granu-
locyte macrophage are recruited during
large-volume leukapheresis and concen-
trated by counterflow centrifugal elutriation.
Transfusion 1993;33:316-21.
73. Hillyer CD. Large volume leukapheresis to
maximize peripheral blood stem cell collec-
tion. J Hematother 1993;2:529-32.
74. Glaspy JA, Shpall EJ, LeMaistre CF, et al. Pe-
ripheral blood progenitor cell mobilization
using stem cell factor in combination with
filgrastim in breast cancer patients. Blood
1997;90:2939-51.
75. Scheid C, Draube A, Reiser M, et al. Using at
least 5 ×106/kg CD34+ cells for autologous
stem cell transplantation significantly re-
duces febrile complications and use of anti-
biotics after transplantation. Bone Marrow
Transplant 1999;23:1177-81.
76. Glaspy JA. Economic considerations in the
use of peripheral blood progenitor cells to
support high-dose chemotherapy. Bone Mar-
row Transplant 1999;23(Suppl 2):S21-7.
77. Watts MJ, Sullivan AM, Leverett D, et al.
Back-up bone marrow is frequently ineffec-
tive in patients with poor peripheral stem cell
mobilization. J Clin Oncol 1998;16:1554-60.
78. Gazitt Y, Shaughnessy P, Liu Q. Differential
mobilization of CD34+cells and lymphoma
cells in non-Hodgkin’s lymphoma patients
mobilized with different growth factors. J
Hematother Stem Cell Res 2001;10:167-76.
79.JennisA,PecoraA,PretiR,etal.Highdose
G-CSF stem cell mobilization after initial
stem cell mobilization failure (abstract).
Blood 1998;92(Suppl 1):271a.
80. Broxmeyer HE, Hangoc G, Cooper S, Bridger
G. Interference of the SDF-1/CXCR4 axis in
mice with AMD 3100 induces rapid high level
mobilization of hematopoietic progenitor
cells and AMD 3100 acts synergistically with
G-CSF and MIP-1 alpha to mobilize progeni-
tors (abstract). Blood 2001;96(Suppl 1):3371a.
81. Devine S, Flomenberg N, Vesole D, et al.
Safety and effect on WBC count and CD34
cell mobilization following administration of
AMD 3100 to patients with multiple myeloma
or non-Hodgkin’s lymphoma. Presented at
the Eighth Congress of the European Hema-
tology Association, Rotterdam, The Nether-
lands, July 5-9, 2003.
82. Goldberg SL, Mangan KF, Klumpp TR, et al.
Complications of peripheral blood stem cell
harvesting: Review of 554 PBSC leukaphere-
ses. J Hematother 1995;4:85-90.
83. Stroncek DF, Clay ME, Smith J, et al. Compo-
sition of peripheral blood progenitor cell
components collected from healthy donors.
Transfusion 1997;37:411-7.
84.IlhanO,ArslanO,AratM,etal.Theimpactof
the CD34+ cell dose on engraftment in allo-
geneic peripheral stem cell transplantation.
Transfus Sci 1999;20:69-71.
85.AnderliniP,RizzoJD,NugentML,etal.Pe
-
ripheral blood stem cell donation: An analy-
sis from the International Bone Marrow
Transplant Registry (IBMTR) and European
Group for Blood and Marrow Transplant
(EBMT) databases. Bone Marrow Transplant
2001;27:689-92.
86. Stroncek DF, Clay ME, Smith J, et al. Changes
in blood counts after the administration of
granulocyte-colony-stimulating factor and
the collection of peripheral blood stem cells
from healthy donors. Transfusion 1996;36:
596-600.
87. Bandarenko N, Brecher ME, Owen H, et al.
Thrombocytopenia in allogeneic peripheral
612 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
blood stem cell collections (letter). Transfu-
sion 1996;36:668.
88. StroncekDF,ClayME,PetzoldtML,etal.
Treatment of normal individuals with granulo-
cyte colony-stimulating factor: Donor expe-
riences and the effects on peripheral blood
CD34+ cell counts and on the collection of
peripheral blood stem cells. Transfusion
1996;36:601-10.
89. Becker PS, Wagle M, Matous S, et al. Sponta-
neous splenic rupture following administra-
tion of granulocyte colony-stimulating fac-
tor (G-CSF): Occurrence in an allogeneic
donor of peripheral blood stem cells. Biol
Blood Marrow Transplant 1997;3:45-9.
90. Falzetti F, Aversa F, Minelli O, Tabilil A. Spon-
taneous rupture of spleen during peripheral
blood stem cell mobilization in a healthy do-
nor (letter). Lancet 1999;353:555.
91. Arbetter KR, Hubbard KW, Markovic SN, et
al. Case of granulocyte colony-stimulating
factor-induced Sweet’s syndrome. Am J
Hematol 1999;61:126-9.
92. Kawachi Y, Watanabe A, Uchida T, et al.
Acute arterial thrombosis due to platelet ag-
gregation in a patient receiving granulocyte
colony-stimulating factor. Br J Haematol
1996;94:413-6.
93. Adkins DR. Anaphylactoid reaction in a nor-
mal donor given granulocyte colony-stimu-
lating factor (letter). J Clin Oncol 1998;16:
812-3.
94. Blau CA. Adverse effects of G-CSF in sickle
cell syndromes (letter). Blood 2001;97:3682.
95. Wei A, Grigg A. Granulocyte colony-stimulat-
ing factor-induced sickle cell crisis and
multiorgan dysfunction in a patient with
compound heterozygous sickle cell/β+tha-
lassemia. Blood 2001;97:3998-9.
96. Rubinstein P, Carrier C, Taylor P, Stevens CE.
Placental and umbilical cord blood banking
for unrelated marrow reconstitution. In:
Brecher ME, Lasky LC, Sacher RA, Issitt LA,
eds. Hematopoietic progenitor cells: Pro-
cessing, standards and practice. Bethesda,
MD: AABB, 1995:1-17.
97. Yao AC, Minian M, Lind J. Distribution of
blood between infant and placenta after
birth. Lancet 1969;ii:871-3.
98. Di Gusto DL, Lee R, Moon J, et al. Hemato-
poietic potential of cryopreserved and ex
vivo manipulated umbilical cord blood pro-
genitor cells evaluated in vitro and in vivo.
Blood 1996;87:1261-71.
99. PecoraAL,StiffP,JennisA,etal.Promptand
durable engraftment in two older adult pa-
tients with high risk chronic myelogenous
leukemia (CML) using ex vivo expanded and
unmanipulated unrelated umbilical cord
blood. Bone Marrow Transplant 2000;25:
797-9.
100. Kogler G, Nurnberger W, Fuscher J, et al. Si-
multaneous cord blood transplantation of ex
vivo expanded together with non-expanded
cellsforhighriskleukemia.BoneMarrow
Transplant 1999;24:397-403.
101. Silvestri F. CD34+ cell purification by im-
munomagnetic beads/chymopapain system
from normal and myeloid leukemias bone
marrow and peripheral blood for research
use.In:WunderE,SovalatH,HenonP,Serke
S, eds. Hematopoietic stem cells: The Mul-
house manual. Dayton, OH: AlphaMed Press,
1995:161-9.
102. Gallacher L, Murdoch B, Wu DM, et al. Isola-
tion and characterization of human
CD34–Lin–and CD34+Lin–hematopoietic
stem cells using cell surface markers AC133
and CD7. Blood 2000;95:2813-20.
103. Wunder E, deWynter E. Purification of
CD34+ cells. In: Wunder E, Sovalat H, Henon
P,SerkeS,eds.Hematopoieticstemcells:
The Mulhouse manual. Dayton, OH:
AlphaMed Press, 1995:125-82.
104. Marolleau JP, Brice P, Cortivo LD, et al.
CD34+ selection by immunomagnetic selec-
tion (Isolex 300) for patients with malignan-
cies (abstract). Blood 1996;88(Suppl 1):110a.
105. Devernardi N, Camilla C, Traore Y, et al. Anti-
body-coated magnetic particles for CD34+
cell separation. In: Wunder E, Sovalat H,
Henon P, Serke S, eds. Hematopoietic stem
cells:TheMulhousemanual.Dayton,OH:
AlphaMed Press, 1995:183-200.
106. Miltenyi S, Guth S, Radbruch A, et al. Isola-
tion of CD34+ hematopoietic progenitor
cells by high-gradient magnetic cell sorting
(MACS). In: Wunder E, Sovalat H, Henon P,
Serke S, eds. Hematopoietic stem cells: The
Mulhouse manual. Dayton, OH: AlphaMed
Press, 1995:201-13.
107. Wunder E, Herbein G, Sovalat H, et al. Sepa-
ration of hematopoietic cells of different ma-
turity in mononuclear cell subsets generated
by elutriation. In: Wunder E, Sovalat H,
Henon P, Serke S, eds. Hematopoietic stem
cells:TheMulhousemanual.Dayton,OH:
AlphaMed Press, 1995:271-9.
108. Brenner MK, Rill DR, Moen RC, et al. Gene-
marking to trace origin of relapse after auto-
logous bone-marrow transplantation. Lan-
cet 1993;341:85-6.
109. Ross AA, Cooper BW, Lazarus HM, et al. De-
tection and viability of tumor cells in periph-
eral blood stem cell collections from breast
cancer patients using immunocytochemical
and clonogenic assay techniques. Blood
1993;82:2605-10.
Chapter 25: Cell Therapy and Cellular Product Transplantation 613
Copyright © 2005 by the AABB. All rights reserved.
110. Brugger W, Bross KJ, Glatt M, et al. Mobiliza-
tion of tumor cells and hematopoietic pro-
genitor cells into peripheral blood of patients
with solid tumors. Blood 1994;83:636-40.
111. Rowley SD, Davis JM. Purging techniques in
autologous transplantation. In: Areman EM,
Deeg HJ, Sacher RA, eds. Bone marrow and
stem cell processing: A manual of current
techniques. Philadelphia: FA Davis, 1992:218-
35.
112. Rowley SD, Davis JM. The use of 4-HC in
autologous purging. In: Gee AP, ed. Bone
marrow processing and purging. Boca Raton,
FL: CRC Press, 1991:248-62.
113. Ball ED. Monoclonal antibodies and comple-
ment for autologous marrow purging. In: Gee
AP, ed. Bone marrow processing and purging.
Boca Raton, FL: CRC Press, 1991:281-8.
114. Kvalheim G, Wang MY, Pharo A, et al. Purging
of tumor cells from leukapheresis products:
Experimental and clinical aspects. J Hemato-
ther 1996;5:427-36.
115. Dreger P, Viehmann K, Steinmann J, et al.
G-CSF mobilized peripheral blood progeni-
tor cells for allogeneic transplantation: Com-
parison of T cell depletion strategies using
different CD34+ selection systems or
CAMPATH-1. Exp Hematol 1995;23:147-54.
116. van der Straaten HM, Fijnheer R, Dekker AW,
et al. Relationship between graft-versus-host
disease and graft-versus-leukaemia in partial
T cell-depleted bone marrow transplanta-
tion. Br J Haematol 2001;114:31-5.
117. Krenger W, Ferrara J. Dysregulation of cyto-
kines during graft-versus-host disease. J
Hematother 1996;5:3-14.
118. Lopez-Plaza I, Triulzi DJ. Transfusion sup-
portinHSCT.In:BallED,ListerJ,LawP,eds.
Hematopoietic stem cell therapy. Philadel-
phia: Churchill Livingstone, 2000:589-97.
119. Long GD, Blume KG. Allogeneic and autolo-
gous bone marrow transplantation. In: Beutler
E, Lichtman MA, Coller BS, Kipps TL, eds.
Williams’ hematology. 5th ed. New York:
McGraw-Hill, 1995:172-94.
120. Worel N, Greinix HT, Schneider B, et al. Re-
generation of erythropoiesis after related and
unrelated donor BMT or peripheral blood
HPC transplantation: A major ABO mismatch
means problems. Transfusion 2000;40:543-
60.
121. Friedberg RC, Andrzejewski C. Transfusion
therapy in hematopoietic stem cell trans-
plantation. In: Mintz PD, ed. Transfusion
therapy: Clinical principles and practice. 2nd
ed. Bethesda, MD: AABB Press, 2005:279-96.
122. Leo A, Mytilineos J, Voso MT, et al. Passenger
lymphocyte syndrome with severe hemolytic
anemia due to an anti-Jkaafter allogeneic
PBPC transplantation. Transfusion 2000;40:
632-6.
123. Salmon JP, Michaux S, Hermanne JP, et al. De-
layed massive immune hemolysis mediated
by minor ABO incompatibility after allo-
geneic peripheral blood progenitor cell trans-
plantation. Transfusion 1999;39:824-7.
124. Good R. Mixed chimerism and immunologi-
cal tolerance. N Engl J Med 1993;328:801-2.
125. Buckner CD, Clift RA, Sanders JE, et al. ABO
incompatible marrow transplants. Trans-
plantation 1978;26:233-8.
126. Hershko C, Gale RP, Ho W, Fitchen J. ABH an-
tigens and bone marrow transplantation. Br J
Haematol 1980;44:65-73.
127. Broxmeyer HE, Douglas GW, Hangoc G, et al.
Human umbilical cord blood as a potential
source of transplantable hematopoietic stem/
progenitor cells. Proc Natl Acad Sci U S A
1989;86:3828-32.
128. Rubinstein P, Dobrila L, Rosenfield RE, et al.
Processing and cryopreservation of placen-
tal/umbilical cord blood for unrelated bone
marrow reconstitution. Proc Natl Acad Sci
U S A 1995;92:10119-22.
129. Webb IJ, Coral FS, Andersen JW, et al. Sources
and sequelae of bacterial contamination of
hematopoietic stem cell components: Implica-
tions for the safety of hematotherapy and
graft engineering. Transfusion 1996;36:
782-8.
130. Cohen A, Tepperberg M, Waters-Pick B, et al.
The significance of microbial cultures of the
hematopoietic support for patients receiving
high-dose chemotherapy. J Hematother
1996;5:289-94.
131. Padley D, Koontz F, Trigg ME, et al. Bacterial
contamination rates following processing of
bone marrow and peripheral blood progeni-
tor cell preparations. Transfusion 1996;36:
53-6.
132. Lazarus HM, Mogalhaes-Silverman M, Fox
RM, et al. Contamination during in vitro pro-
cessing of bone marrow for transplantation:
Clinical significance. Bone Marrow Trans-
plant 1991;7:241-6.
133. Espinosa MT, Fox R, Creger RJ, Lazarus HM.
Microbiologic contamination of peripheral
blood progenitor cells collected for hemato-
poietic cell transplantation. Transfusion
1996;36:789-93.
134. Armitage D, Warwick R, Fehily D, et al. Cord
blood banking in London: The first 1000 col-
lections. Bone Marrow Transplant 1999;24:
130-45.
135. Davis JM, Schepers KG. Quality control of
hematopoietic progenitor cell products. In:
Brecher ME, Lasky LC, Sacher RA, Issitt LA,
eds. Hematopoietic progenitor cells: Process-
614 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
ing, standards and practice. Bethesda, MD:
AABB, 1995:159-81.
136. Siena S, Bregni M, Brando B, et al. Flow
cytometry for clinical estimation of circulat-
ing hematopoietic progenitors for autolo-
gous transplantation in cancer patients.
Blood 1991;77:400-9.
137. Roscoe RA, Rybka WB, Winkelstein A, et al.
Enumeration of CD34+ hematopoietic stem
cells for reconstitution following myelo-
ablative therapy. Cytometry 1994;16:74-9.
138. Johnsen HE. Report from a Nordic Workshop
on CD34+ cell analysis: Technical recom-
mendations for progenitor cell enumeration
in leukapheresis from multiple myeloma pa-
tients. J Hematother 1995;4:21-8.
139. Chang A, Ma DDF. The influence of flow
cytometric gating strategy on the standard-
ization of CD34+ cell quantitation: An Aus-
tralian multicenter study. J Hematother 1996;
5:605-16.
140. Säberlich S, Kirsch A, Serke S. Determination
of CD34+ hematopoietic cells by multi-
parameter flow cytometry: Technical re-
marks.In:WunderE,SovalatH,HénonP,
Serke S, eds. Hematopoietic stem cells: The
Mulhouse manual. Dayton, OH: AlphaMed
Press, 1994:45-60.
141. Johnsen HE. Toward a worldwide standard for
CD34+ enumeration? (letter) J Hematother
1997;6:83-4.
142. Sims LC, Brecher ME, Gertis K, et al. Enu-
meration of CD34 positive stem cells: Evalu-
ation and comparison of three methods. J
Hematother 1997;6:213-26.
143. Storms RW, Trujillo AP, Springer JB, et al. Iso-
lation of primitive human hematopoietic
progenitors on the basis of aldehyde de-
hydrogenase activity. Proc Natl Acad Sci
U S A 1999;96:9118-23.
144. Hess DA, Meyerrose TE, Wirthlin L, et al.
Functional characterization of highly purified
human hematopoietic repopulating cells
isolated according to aldehyde dehydro-
genase activity. Blood 2004;104:1648-55.
145. Fallon P, Gentry T, Balber AE, et al. Mobilized
peripheral blood SSClo ALDHbr cells have the
phenotypic and functional properties of
primitive hematopoietic progenitor cells and
their number correlates with engraftment
following autologous transplantation. Br J
Haematol 2003;122:99-108.
146. Christensen JL, Weissman IL. Flk-2 is a
marker in hematopoietic stem cell differenti-
ation: A simple method to isolate long-term
stem cells. Proc Natl Acad Sci U S A 2001;98:
14541-6.
147. Wunder E, Sovalat H, Henon P, Serke S, eds.
Hematopoietic stem cells: The Mulhouse
Manual; Dayton, OH: AlphaMed Press, 1995:
126-82.
148. Moroff G, Seetharman S, Kurtz JW, et al. Re-
tention of cellular properties of PBPCs fol-
lowing liquid storage and cryopreservation.
Transfusion 2004;44:245-52.
149. Hechler G, Weide R, Heymanns J, et al. Stor-
age of noncryopreserved peripheral blood
stem cells for transplantation. Ann Hematol
1996;72:303-6.
150. Sugrue SR, Hubel AH, McCullough J, et al.
The effect of overnight storage of leuka-
pheresis stem cell products on cell viability,
recovery and cost. J Hematother 1998;7:431-6.
151. Aird W, Laborpin M, Gorin NC, Anten JH.
Long term cryopreservation of human stem
cells. Bone Marrow Transplant 1992;9:487-
90.
152. Gorin NC. Cryopreservation and storage of
stem cells. In: Areman EM, Deeg JH, Sacher
RA,eds.Bonemarrowandstemcellprocess
-
ing: A manual of current techniques. Phila-
delphia: FA Davis, 1992:138-41.
153. Meryman HT. Cryoprotective agents. Cryo-
biology 1971;8:173-83.
154. Leibo SP, Farrant J, Mazur P, et al. Effects of
freezing on marrow stem cell suspensions:
Interactions of cooling and warming rates in
thepresenceofPVP,sucroseorglycerol.
Cryobiology 1970;6:15-32.
155. Mazur P. Theoretical and experimental ef-
fects of cooling and warming velocity in the
survival of frozen and thawed cells. Cryobio-
logy 1966;2:181-92.
156. Lewis JP, Passovoy M, Trobaugh FE. The ef-
fect of cooling regimens on the transplanta-
tion potential of marrow. Transfusion
1967;7:17-32.
157. Hernandez-Navarro F, Ojeda E, Arrieta R, et
al. Hematopoietic cell transplantation using
plasma and DMSO without HES, with non-
programmed freezing by immersion in a
methanol bath: Results in 213 cases. Bone
Marrow Transplant 1998;21:511-7.
158. Galmes A, Besalduch J, Bargay J, et al. Cryo-
preservation of hematopoietic progenitor
cells with 5-percent dimethyl sulfoxide at
–80 degrees C without rate-controlled freez-
ing. Transfusion 1996;36:794-7.
159. Stiff PJ, Murgo AJ, Zaroules CG, et al. A sim-
plified bone marrow cryopreservation method.
Blood 1988;71:1102-3.
160. Stiff PJ, Murgo AJ, Zaroules CG, et al. Un-
fractionated marrow cell cryopreservation
using dimethylsulfoxide and hydroxyethyl
starch. Cryobiology 1983;21:17-21.
161. Tedder RS, Zuckerman MA, Goldstone AH, et
al. Hepatitis B transmission from contami-
Chapter 25: Cell Therapy and Cellular Product Transplantation 615
Copyright © 2005 by the AABB. All rights reserved.
nated cryopreservation tank. Lancet 1995;
346:137-40.
162. Rowley SD, Anderson GL. Effect of DMSO ex-
posure without cryopreservation on hemato-
poietic progenitor cells. Bone Marrow Trans-
plant 1993;11:389-93.
163. Gee AP. Quality control in bone marrow pro-
cessing. In: Gee AP, ed. Bone marrow process-
ing and purging: A practical guide. Boca
Raton, FL: CRC Press, 1991:19-27.
164. Lasky LC, Johnson NL. Quality assurance in
marrow processing. In: Areman EM, Deeg HJ,
SacherRA,eds.Bonemarrowandstemcell
processing: A manual of current techniques.
Philadelphia: FA Davis, 1992:386-443.
165. Bentley SA, Taylor MA, Killian DE, et al. Cor-
rection of bone marrow nucleated cell counts
for the presence of fat particles. Am J Clin
Pathol 1995;140:60-4.
166. Moss TJ. Detection of metastatic tumor cells
in bone marrow. In: Gee AP, ed. Bone marrow
processing and purging: A practical guide.
Boca Raton, FL: CRC Press, 1991:121-35.
167. Pecora AL, Lazarus EM, Cooper B, et al.
Breast cancer contamination in peripheral
blood cell (PBPC) collections association
with bone marrow disease and type of mobi-
lization (abstract). Blood 1997;89(Suppl):99a.
168. Food and Drug Administration. A proposed
approach to the regulation of cellular and tis-
sue-based products (March 4, 1997). Fed
Regist 1997;62:9721-2.
169. Food and Drug Administration. Human tissue
intended for transplantation; final rule, 21
CFR 16 and 1270 (July 29, 1997). Fed Regist
1997;62:40429-47.
170. Food and Drug Administration. Guidance for
industry: Screening and testing of donors of
human tissue intended for transplantation.
(July 29, 1997) Rockville, MD: CBER Office of
Communication, Training, and Manufactur-
ers Assistance, 1997.
171. Food and Drug Administration. Draft guid-
ance for industry: Preventive measures to re-
duce possible risk of transmission of Creutz-
feldt-Jakob disease (CJD) and variant
Creutzfeldt-Jakob disease (vCJD) by human
cells, tissues, and cellular and tissue-based
products (HCT/Ps) (June 25, 2002). Fed
Regist 2002;67:42789-90.
172. Food and Drug Administration. Request for
proposed standards for unrelated allogeneic
peripheral and placental/umbilical cord
blood hematopoietic stem/progenitor cell
products; request for comments (January 20,
1998). Fed Regist 1998;63:2985-8.
173. Food and Drug Administration. Current good
tissue practice for human cell, tissue, and
cellular and tissue-based product establish-
ments; inspection and enforcement (Novem-
ber 24, 2004). Fed Regist 2004;69:68612-88.
174. AABB, American Red Cross, America’s Blood
Centers, American Society for Blood and
Marrow Transplantation, Foundation for the
Accreditation of Cellular Therapy, Interna-
tional Society for Cellular Therapy, National
MarrowDonorProgram.Circularofinforma-
tion for the use of cellular therapy products.
Bethesda, MD: AABB, 2003.
616 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 26: Tissue and Organ Transplantation
Chapter 26
Tissue and Organ
Transplantation
IN RECENT YEARS, the numbers of
cornea, bone, skin, heart valve, and other
tissue donations and transplants1-3 have
exceeded those of solid organs,4such as
kidneys, livers, hearts, and lungs. The
hospital blood bank, transfusion service,
community blood center, and regional
blood center are uniquely qualified to
provide essential support for organ and
tissue transplantation and to serve as tis-
sue banks (Table 26-1).5It is common for
hospital blood banks to provide transfu-
sion support for organ and tissue recipi-
ents and, in some cases, to store, keep records
of, and dispense tissue for allografts. AABB
Standards for Blood Banks and Transfu-
sion Services6(pp8,14,15,60,79) addresses the re-
ceipt, storage, transportation, and records
of tissue allografts. Additional guidance for
the collection and preparation of tissue
and organ allografts is available from fed-
eral and state regulations, Public Health
Service Guidelines, and the standards,
guidelines, and technical manuals of other
national or local organizations.7-14
Transplant-Transmitted
Diseases and Preventive
Measures
The widened availability of tissue and or-
gan grafts has encouraged new clinical
uses and highlighted not only their effec-
tiveness and advantages but also their
drawbacks, side effects, and complications.
Organs and tissues can transmit bacterial,
fungal, viral, and prion diseases from the
donor to the recipient3,15-26 (Table 26-2),
but careful donor screening and testing,
along with disinfection and sterilization
steps for specific tissues, can markedly re-
duce the risk.
617
26
Copyright © 2005 by the AABB. All rights reserved.

Risk Reduction for Tissues
Crucial to the safety of transplanted tissue
is an evaluation of the potential donor’s
eligibility. Listed below are the questions,
examinations, and tests undertaken to en-
sure that material from the potential donor
poses a low threat of disease transmission.
Additional measures apply to reproductive
tissue donors.10,11
1. Review of health history, through in-
terviews with next of kin and/or sig-
nificant other, and possibly health-
care provider, and review of medical
records. Review includes an evalua-
tion (although not necessarily rejec-
tion for asterisked items*) of the po-
tential donor for:
a. History of infection,* malignant
disease,* or neurodegenerative
disease
b. History of autoimmune processes*
c. History of exposure to hormone
derived from human pituitary
gland or dura mater transplant
618 AABB Technical Manual
Table 26-1. Skills and Experience Appropriate for Institutions Undertaking
Tissue Banking
■Community support
■Public accountability
■Public education with a broad-based public information system
■Donor recruitment
■Counseling
■Medical overview
■Donor selection
■Donor testing including automated virology testing to avoid transcription errors
■Cellular cryopreservation
■Temperature-controlled and monitored storage
■Regulatory compliance
■Transportation infrastructure
■Financial relations with hospitals
■Computerized inventory control
■Record-keeping
■Logistics management
■Investigation of adverse reactions
■Peer review of medical, scientific, and operational practice
■Recipient matching
■Concern over the balance of the adequacy and safety of supply
■Reputation for dependable service
■Commitment to research and development
■24-hours-per-day, 7-days-per-week operation
Reproduced with permission from Warwick et al.5
Copyright © 2005 by the AABB. All rights reserved.

Chapter 26: Tissue and Organ Transplantation 619
Table 26-2. Infectious Diseases Reported to Have Been Transmitted by Organ and
Tissue Allografts15-26
Allograft Infectious Disease/Disease Agent
Bone HIV-1
Hepatitis C
Hepatitis, unspecified type
Bacteria
Tuberculosis
Cornea Hepatitis B
Creutzfeldt-Jakob disease
Rabies
Cytomegalovirus (?)
Bacteria
Fungus
Dura mater Creutzfeldt-Jakob disease
Heart valve Hepatitis B
Tuberculosis
Skin Bacteria
Cytomegalovirus (?)
HIV-1 (?)
Pericardium Creutzfeldt-Jakob disease
Bacteria
Solid organ (eg, kidney, liver,
heart)
HIV-1
Hepatitis B
Hepatitis C
Cytomegalovirus
Epstein-Barr virus
Parvovirus
Toxoplasmosis
Chagas’ disease
Malaria
Bacteria
Tuberculosis
HHV-8
Strongyloidiasis
Sarcoidosis
West Nile virus
Rabies
Lymphocytic choriomeningitis
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

2. Review for evidence of high-risk be-
havior (exclusion for any of the do-
nor-deferral criteria included in items
9c, 9e, and 10 of Standard 5.4.1A of
Standards for Blood Banks and Trans-
fusion Services.6(pp62-65)
3. Serologic testing on suitable blood
specimens10 (see below for more de-
tail).
a. Hepatitis B surface antigen
(HBsAg)
b. Antibodies to human immuno-
deficiency viruses 1 and 2 (anti-
HIV-1 and -2)
c. Antibody to hepatitis C virus
(anti-HCV)
d. Antibody to human T-cell lym-
photropic virus types I and II
(anti-HTLV-I and -II)
e. Serologic test for syphilis
4. Physical examination to detect:
a. Evidence of intravenous drug use
b. Jaundice
c. External signs of infection
d. Signs of AIDS
5. Review of results of autopsy exami-
nation, if performed.
Consent and Donor Eligibility
Written consent for clinical use of any tis-
sue or organ must be obtained from a liv-
ing donor or from the next of kin of a de-
ceased donor (formerly referred to as a
cadaveric donor), except when corneas are
procured under statutory consent. State
and federal referral statutes and regula-
tions exist, mandating that hospitals 1)
maintain policies for notifying organ pro-
curement organizations and appropriate
620 AABB Technical Manual
Table 26-2. Infectious Diseases Reported to Have Been Transmitted by Organ and
Tissue Allografts15-26 (cont'd)
Allograft Infectious Disease/Disease Agent
Pancreatic islet Bacteria
Semen Hepatitis B
Hepatitis C (?)
Gonorrhea
Syphilis (?)
HIV-1
HTLV-I (?)
Human papilloma virus (?)
Trichomonas vaginalis
Chlamydia trachomatis
Cytomegalovirus (?)
Ureaplasma urealyticum
HSV-2
Mycoplasma hominis
Group B streptococcus
Copyright © 2005 by the AABB. All rights reserved.
tissue banks, including eye banks, when
death of a patient has occurred or is im-
minent and 2) designate requestors to ap-
proach families about organ and tissue
donation. All applicable federal, state, and
local laws concerning the consent of next
of kin must be obeyed. When the next of
kin signs consent for tissue donation, he
or she should specify which tissues may
be donated and whether the permission
includes tissue to be used for research or
other specific uses.
Living donors and the families of de-
ceased donors are not responsible for ex-
penses involved in recovering and process-
ing donated tissues and organs. Donors do
not receive compensation for the donation,
although donors of reproductive tissue are
often compensated for their time, risk, and
inconvenience, and donor families may re-
ceive incentives, such as a contribution to-
ward funeral expenses, for donation from
deceased donors. Tissues can be recovered
up to 24 hours after death if the body is re-
frigerated within 12 hours of death. Tissues
can be recovered up to 15 hours after death
if not refrigerated. Organs must be recov-
ered from a donor whose neurologic death
has been declared and whose circulation
has been maintained (see Table 26-3). Eyes
as a source of corneal allografts should be
removed as soon as possible after death to
ensure viability of endothelial cells.
Each prospective deceased donor must
be evaluated against eligibility criteria for
the specific tissue(s) and organ(s) to be col-
lected; eg, deceased newborns are not suit-
able for bone donation because of their
cartilaginous skeletal structure but may be
candidates for heart valve donation. Al-
though exceptions may be made in specific
cases, the medical eligibility of a donor is
determined on the basis of absence of in-
fection and malignancy as revealed by the
medical history, physical examination, lab-
oratory tests, and autopsy, if performed.
Donation of organs and tissues does not or-
dinarily cause delays in funerals or prevent
family viewing of the body.
Serologic Testing
Federal regulations require that donors of
tissues be tested for HBsAg, anti-HIV-1/2,
and anti-HCV, and given a syphilis screen-
ing test, with tests licensed by the Food
and Drug Administration (FDA). Testing
must be performed by a laboratory that is
certified under the provisions of the Clini-
cal Laboratory Improvement Amendments
of 19887,8 or that has met equivalent re-
quirements as determined by the Centers
for Medicare and Medicaid Services. A
screening test approved for cadaveric
specimens must be used when available.27
National standard-setting organizations,
such as the American Association of Tis-
sue Banks (AATB),10 Eye Bank Association
of America (EBAA),12 and the United Net-
work for Organ Sharing (UNOS),13 may re-
quire additional tests for infectious dis-
ease markers. The American Society for
Reproductive Medicine also has issued
recommendations for gamete and embryo
donation.11 For anonymous semen do-
nors, tests for anti-HIV-1/2 and anti-HCV
must be repeated on a sample obtained 6
months after donation and the results
found negative before semen can be re-
leased for use.9,10 Testing for anti-HBc
must also be performed on this sample to
rule out hepatitis B infection at the time
of collection. ABO typing is required for
organ grafts. HLA typing is essential for
organ grafts and hematopoietic progeni-
tor cells13 but not for tissues.28 Other tests,
such as antibody to cytomegalovirus (anti-
CMV), are also generally performed on or-
gan donors. For deceased donors, tests are
optimally performed on a blood sample
obtained before administration of transfu-
sions or fluids; these patients may have re-
Chapter 26: Tissue and Organ Transplantation 621
Copyright © 2005 by the AABB. All rights reserved.

ceived large volumes of replacement
fluids shortly before death, and the conse-
quent plasma dilution may cause false-
negative results.19,20
If donor serum collected before blood
transfusion or intravenous fluid adminis-
tration is not available from other sources, a
pretransfusion sample is often available
from the blood bank because blood banks
hold specimens collected for compatibility
for at least 7 days. For samples obtained af-
ter transfusion or infusion of intravenous
fluids, algorithms for determining the suit-
ability of a donor sample are available.7,8 For
example, the sample is not suitable for in-
fectious disease marker testing if either one
of the following situations exists:
1. The total volume of colloid (plasma,
dextran, platelets, or hetastarch) trans-
fused in 48 hours plus the total vol-
ume of crystalloid infused in the
hour before the sample is obtained
exceed the patient’s plasma volume.
2. The sum of the volume of blood
transfused (RBCs, whole blood) and
colloid transfused in 48 hours plus
the total volume of crystalloid infused
in the hour before the sample is ob-
tained exceed the patient’s blood
volume.
Testing of cadaveric blood specimens can
be complicated by postmortem hemolysis,
which can cause misleading test results (eg,
false-positive HBsAg).15,29 Many tests li-
622 AABB Technical Manual
Table 26-3. Kinds of Donors Providing Organs and Tissue for Transplantation
Living Donor
Deceased Donor
(Neurologic Death)
Deceased Donor
(Cardiorespiratory and
Neurologic Death)
Amniotic membrane
Bone
Fetal tissues (mother is the
donor)
Foreskin
Kidney
Liver
Lung
Marrow
Milk
Pancreas
Parathyroid (frequently
autologous)
Peripheral blood progenitor
cells
Reproductive tissue
Umbilical cord blood
Umbilical vein
Heart
Kidney
Liver
Lung
Pancreas (with or without
small intestine)
Bone
Cartilage
Cornea
Dura mater
Fascia lata
Heart valve
Marrow
Pericardium
Skin
Tendon
Vascular tissue
Copyright © 2005 by the AABB. All rights reserved.
censed for use on blood donor specimens
are not licensed for use with postmortem
specimens, but licensed assays for post-
mortem specimens are available for anti-
HIV-1/2, HBsAg, and HIV-1/HCV nucleic
acid.27
Bone Banking
Except for blood cells, bone is the most
commonly transplanted tissue1or organ.
When bone grafting is needed, fresh auto-
logous bone, usually removed from the
iliac crest during surgery, is generally con-
sidered the most effective graft material.
As with blood, the use of autologous bone
for graft material is not risk free and there
may be morbidity and infectious compli-
cations. The quantity of bone graft needed
for some surgical procedures may make
the use of autologous bone impractical.
Allografts are used for these patients and
for patients in whom the prolongation of
surgery, extra bleeding, and potential
complications of autograft collection are
considered undesirable. Bone allografts
have achieved widespread clinical appli-
cation for acetabular and proximal femur
support in revisions of failed hip prosthe-
ses; packing of benign bone cysts; spinal
fusion to treat disc disease or scoliosis; re-
construction of maxillo-facial defects; and
correction of healed fractures. Demineral-
ized bone powder is commonly used by
periodontal surgeons to restore alveolar
bone in periodontal pockets.
The surgeon today has access to a wide
choice of processed bone allografts: freeze-
dried or frozen, cancellous or cortical, with
or without treatment with sterilants. Com-
mon preparations include frozen or freeze-
dried cancellous cubes or chips, cortical
struts, and cortical-cancellous blocks and
dowels. Bone can be stored frozen or, if
freeze-dried to a low residual moisture con-
tent (6% or less by gravimetric analysis or
8% by nuclear magnetic resonance spec-
trometry), at room temperature for 5 years
(Table 26-4). Frozen bone is processed
aseptically, some without a microbial inac-
tivation step. Such tissue carries a greater
risk of bacterial contamination. A series of
infection cases, including one death, linked
to aseptically processed musculoskeletal allo-
grafts underscores the importance of recov-
ery cultures of donated tissue.24 Freeze-
dried tissue has undergone extensive pro-
cessing to remove blood and marrow and
may have been exposed to alcohol. Thus,
the risk of disease transmission is reduced.
Most bone allografts used in the United
States are freeze-dried to simplify storage.
Some grafts, whether frozen or freeze-dried,
may be treated with gamma irradiation or
ethylene oxide to reduce the risk of infec-
tious disease transmission. Demineraliza-
tion of bone is believed to make its proteins
and growth factors more readily available,
thereby enhancing its capacity to promote
healing and bone formation.
The implantation of frozen bone allo-
grafts has stimulated blood group antibod-
ies that have been implicated in hemolytic
disease of the newborn. Frozen, unpro-
cessed bone allografts contain sufficient red
cells to stimulate production of Rh and
other red cell antibodies. When indicated,
the risk of blood group sensitization can be
avoided by using processed frozen or freeze-
dried bone allograft, both of which are de-
void of blood cells and marrow. With these
grafts, matching blood groups of donors
and recipients is not necessary. When using
unprocessedfrozenboneinRh-negative
females with childbearing potential, it is
advisable to use bone from Rh-negative do-
nors to prevent alloimmunization or ad-
minister Rh immune globulin prophylacti-
cally. There is no evidence to date that ABO
or Rh incompatibility between the bone
Chapter 26: Tissue and Organ Transplantation 623
Copyright © 2005 by the AABB. All rights reserved.

624 AABB Technical Manual
Table 26-4. Recommended Preservation Conditions and Dating Periods for
Human Tissue and Organs
Storage Condition Dating Period
Tissue
Bone –40 C 5 years*
–20 C 6 months
1-10 C 5 days
Liquid nitrogen Not defined
Lyophilized, room tempera-
ture
5 years*
Tendon –40 C 5 years*
Fascia lata Lyophilized, room tempera-
ture
5 years*
–40 C 5 years*
Articular cartilage –40 C 5 years*
1-10 C 5 days
Liquid nitrogen, immersed Not defined
Skin 1-10 C 14 days
–40 C Not defined
Lyophilized, room tempera-
ture
Not defined
Cornea 2-6 C 14 days
Hematopoietic progenitor
cells
Liquid nitrogen, immersed Not defined
Liquid nitrogen, vapor phase Not defined
Semen Liquid nitrogen, immersed Not defined
Liquid nitrogen, vapor phase Not defined
Heart valve, vein, artery –100 C Not defined
Dura mater Lyophilized, room tempera-
ture
Not defined
Organ
Kidney Refrigerated 48-72 hours
Liver Refrigerated 8-24 hours
Heart Refrigerated 3-5 hours
Heart-lung Refrigerated 3-5 hours
Pancreas Refrigerated 12-24 hours
*Unless a longer dating period has been validated by the processor
Copyright © 2005 by the AABB. All rights reserved.
donor and recipient has an adverse effect
on the success of the bone graft.30
Skin Banking
A human skin allograft is the dressing of
choice for deep burn wounds if sufficient
amounts of skin for autografting are un-
available. A skin allograft provides tempo-
rary coverage; speeds reepithelialization;
acts as a metabolic barrier against loss of
water, electrolytes, protein, and heat; and
provides a physical barrier to bacterial in-
fection. Skin allografts are replaced peri-
odically until sufficient autograft skin can
be obtained. A skin allograft may also be
used for donor sites for pedicle flaps and
skin autografts, and for traumatically de-
nuded areas or unhealed areas of chronic
injury, such as decubitus ulcers.
Following preparation, skin donation in-
volves removing a layer of skin approxi-
mately 0.015 inch thick. After collection, re-
frigerated skin can be stored at 1 to 10 C for
up to 14 days. For refrigerated storage, stan-
dard tissue-culture nutrient media are
used, with added antibiotics. Skin can be
frozen soon after collection, usually with
10% to 15% glycerol, although dimethyl
sulfoxide (DMSO) is an acceptable alterna-
tive.31 Skin is often cryopreserved on fine-
mesh gauze in flat cryopreservation bags.
Cryogenic damage is minimized by con-
trolled-rate freezing at about 1 C per min-
ute, or by freezing, using a validated heat
sinking method, followed by storage in liq-
uid nitrogen or in a mechanical freezer at a
temperature colder than –40 C. Alternatively,
skin placed in aluminum plates inside in-
sulated boxes can be placed directly into a
–40 C mechanical freezer. This simple pro-
cess also provides a slow, predictable freez-
ing rate and maintains cellular viability. The
optimal freezing procedure and the maxi-
mal storage period that maintain viability
and structural integrity in the frozen state
have not been determined. Skin should be
transported to the operating room on wet
ice if stored at 4 C, or on dry ice if cryo-
preserved. A variety of bioengineered skin
substitutes, such as cultured keratinocyte
allografts and autografts, are available for
treatment of acute and chronic wounds.
Keratinocytes extracted from foreskin can
be seeded onto biodegradable platforms to
create bioengineered tissue products used
in wound healing or as cadaveric skin sub-
stitutes for burns.32
Heart Valves
Human heart valve allografts provide long-
term function for valve replacement—su-
perior to that of mechanical or porcine
valves. Recipients of human heart valve
allografts do not require anticoagulation
and the incidence of thromboembolism is
low. These allografts may rarely transmit
bacterial or fungal infection. They are the
graft of choice for children, to avoid long-
term anticoagulation; for pregnant women,
to avoid teratogenic risks of anticoagulants;
and for patients with infection at the aor-
tic root. Despite their advantages, wide-
spread use of human valve allografts has
been slow because implantation is techni-
cally difficult and appropriate size valve
allografts are not always readily available.
To obtain allograft valves, hearts are
aseptically collected in an operating room,
autopsy room, or morgue. Subsequently, in
thetissuebank,thepulmonicandaortic
valves are dissected out, cryopreserved with
DMSO, and stored in liquid nitrogen. Com-
pared with valves stored at 5 C in antibiot-
ics and culture medium, cryopreserved heart
valves are associated with increased cell vi-
ability; reduced incidence of valve degener-
ation, rupture, and leaflet perforation; and
reduced occurrence of valve-related death.33
Chapter 26: Tissue and Organ Transplantation 625
Copyright © 2005 by the AABB. All rights reserved.
Records of Stored Tissue
Allografts
In two cases in which HIV and HCV were
transmitted (through unprocessed frozen
bone or organs and tendon allografts, re-
spectively) from two deceased donors to
multiple recipients,22,23 investigations re-
vealed that several hospitals had insuffi-
cient records to identify recipients of other
tissue from the same infected donors. Vol-
untary standards of national professional
associations and government regulations
requiretissuebankstohavearecord-
keeping system that identifies the donor
and allows tracking of any tissue from the
donor (or supplier source) to the con-
signee.6(pp12,47,79),10,14,34 Records must show the
source facility, the identification number
of the donor or lot, storage temperatures,
and final disposition of each tissue. These
records must be retained at least 10 years
beyond the distribution date, transplanta-
tion date, disposition date, or expiration
date, whichever is latest. Donor eligibility
records for dura mater must be retained
indefinitely. Hospitals should also have
records that identify recipients who re-
ceived tissue from a specific donor or tis-
sue lot. Hospitals should have procedures
in place to recognize adverse outcomes of
tissue use and to report them to the tissue
bank supplying the tissue.
FDA Regulation of Tissue
The FDA regulates human tissue collected
for transplantation.7,8 There are require-
ments for infectious disease testing, do-
nor screening, and record-keeping. A tis-
sue establishment must have and follow
written, validated procedures to prevent
contamination and cross-contamination
during processing.7,35 The FDA has pub-
lished rules for determining eligibility for
donation of human cells, tissues, and cel-
lular and tissue-based products (HCT/
Ps)36 that became effective May 25, 2005.
Federal rules also require all facilities that
recover, process, store, or distribute HCT/Ps
or screen or test the donor to register with
the agency and list their products by mail-
ing or faxing Form FDA 3356.37 Informa-
tion may be submitted electronically at
the FDA website www.fda.gov/cber/tis-
sue/tisreg.htm. Products not covered by
these regulations include xenogeneic tis-
sue, vascularized organs, transfusable
blood products, products used in the
propagation of cells or tissues, and prod-
ucts that are secreted or extracted from
cells or tissues. Minimally manipulated
marrow, such as marrow that undergoes
cell separation, the relevant characteris-
tics of which are not altered by the pro-
cessing, is not covered.
Under the federal regulations, infectious
disease testing is required for allogeneic tis-
sues, except reproductive tissue from sexu-
ally intimate partners. Current good tissue
practice rules to address concerns about
proper handling, storage and processing of
tissuehavebeenfinalized.
34 Until such time
that the comprehensive regulatory frame-
work for human cells, tissues, and cellular
and tissue-based products is effective, in-
cluding donor eligibility requirements,
good tissue practice regulations, and ap-
propriate enforcement provisions, human
dura mater and human heart valves will re-
main subject to the medical devices re-
quirements under the Federal Food, Drug,
and Cosmetic Act.38 Federal regulation of
tissue banks, formerly under the purview of
the FDA’s Center for Biologics Research and
Review, Office of Blood Research and Re-
view, is now the responsibility of the Center
for Biologics Evaluation and Research, Of-
fice of Cellular, Tissue, and Gene Therapies’
Division of Human Tissues.
626 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
The Importance of ABO
Compatibility
The ABO antigens are important in trans-
plantation practice because they constitute
very strong histocompatibility antigens that
are expressed on vascular endothelium.
Major ABO mismatching can cause rapid
graft rejection due to endothelial damage
by ABO antibodies and subsequent wide-
spread thrombosis within the graft. ABO
matching is important to the success of
vascularized grafts (ie, kidney,39 heart,
liver, and pancreas), but ABO matching is
not important in tissue grafts (ie, fascia,
bone, heart valves, skin, and cornea).40
The definition of an ABO-compatible
graft is the same as that for a red cell trans-
fusion. A group O donor of tissue or organ
is a universal donor whose graft can be
transplanted into recipients of all blood
groups. Case reports document rare suc-
cessful organ transplants with major ABO
incompatibility, but these are few in num-
ber.41 In some cases, A2donor kidneys can
be successfully transplanted into group O
recipients with survival comparable to that
of group O donor kidneys.42 ABO-incompat-
ible transplants have occurred, often with
fatal results, due to errors of record-keeping
or labeling. It has been estimated that inad-
vertent ABO-incompatible heart or kidney
transplants occur with a frequency of 1 per
1000.43 This underscores the importance of
a final ABO check of donor and recipient
blood at the transplant facility to reduce
this risk.
The Role of Transfusion in
Kidney Transplants
In 1973, Opelz et al44 noted decreasing kid-
ney allograft survival when hemodialysis
staff attempted to avoid priming the dial-
ysis equipment with blood and to limit
pretransplant blood transfusions. The as-
sociation between fewer transfusions and
declining renal allograft survival led
transplant centers to initiate deliberate
pretransplant transfusion protocols in the
mid-1970s. Subsequent studies have sup-
ported the theory that pretransplant blood
transfusions enhance renal allograft sur-
vival through mechanisms of inducing
tolerance that remain imperfectly under-
stood.45,46 However, with the availability of
cyclosporine and other immunosuppres-
sive agents, interest in this approach has
waned.47 If the controversial practice of
transfusion to induce tolerance in patients
before transplantation is begun, nonleuko-
cyte-reduced Red Blood Cells (RBCs) should
be used.48,49 The introduction of erythro-
poietin has reduced the need for red cell
transfusions in patients awaiting a renal
transplant.
Liver Transplants
A liver transplant program presents one of
the greatest challenges to the donor cen-
ter and hospital transfusion service, de-
manding maximal support in terms of
preparedness, supply, and responsive-
ness. Massive blood loss and hypocoagu-
lability due to preexisting liver disease
and/or the anhepatic interval during the
procedure create complex problems for the
transfusion service. The liver is the major
site for synthesis of clotting factors and
other essential proteins and is a prime reg-
ulator of acid-base, electrolyte, and glucose
homeostasis. The three surgical phases of
the procedure—recipient hepatectomy,
anhepatic interval, and biliary reconstruc-
tion—seriously derange these functions.
Support Required
To achieve a successful liver transplant
program, a major commitment to this sup-
Chapter 26: Tissue and Organ Transplantation 627
Copyright © 2005 by the AABB. All rights reserved.
port is required. A successful program re-
quires cooperation and communication
among the hospital administration; oper-
ating room and intensive care unit; respi-
ratory therapy, radiology, gastroentero-
logy, and anesthesiology services; the
coagulation and transfusion laboratories;
and the regional donor center. The insti-
tutional commitment must extend 24
hours a day, 365 days a year, because there
may be no more than a few hours’ ad-
vance notice of a liver transplant. Consis-
tent availability of blood bank staff on
short notice is essential to meet the trans-
fusion requirements. The surgical proce-
dure frequently takes place at night or on
weekends because of the availability of the
donor organ and in order to avoid dis-
rupting the operating room schedule. The
surgical procedure takes an average of 6
to 8 hours but may take up to 24 hours
and involve massive blood use and sev-
eral surgical teams.
The blood bank should be notified as
soon as the donor organ becomes available
and the decision for transplantation is
made. The blood bank obtains a generous
blood sample from the recipient for cross-
matching, but there may be more than one
patient waiting for a liver and the surgeons
may be undecided about the specific recip-
ient. Therefore, the blood bank may have to
perform numerous crossmatches for pa-
tients who may have different ABO groups
and/or Rh types. Liver transplant programs
initially used hundreds of units of blood
and components per patient. Although
blood use has steadily declined over the
years, liver transplant procedures fre-
quently use a volume of blood components
equal to one whole-body blood volume and
sometimes several blood volumes. Intra-
operative blood recovery frequently plays a
major role in the conservation of red cells
in such cases.
Considerations of ABO and Rh
Except in emergencies, donor livers
should be ABO-compatible with the re-
cipient. ABO-identical RBCs and Fresh
Frozen Plasma (FFP) are generally used
for transfusion support of group O and
group A recipients. Group B recipients
who need large quantities of red cells can
be switched to group O RBCs. Group AB
recipients needing massive transfusion are
often switched to group A RBCs to con-
serve group O RBCs for other patients. If
the supply of AB FFP is insufficient, early
use of group A RBCs followed by a switch
to group A FFP is appropriate. A general
rule for massive transfusions is to switch
red cells first, then switch plasma, and re-
verse the order when returning to the pa-
tient’s original blood group.46
Special considerations apply to the re-
cipient of an out-of-group but ABO-com-
patible liver transplant. In a group A patient
receiving a group O liver, lymphocytes of
donor origin may produce ABO antibodies
that cause hemolysis that begins several
days after the procedure and may continue
for 2 weeks or longer.50 Although passenger
lymphocytes may produce antibodies in
any out-of-group but compatible combina-
tion, significant hemolysis is seen most
often in the recipient of a group O liver.
Transfusion support of Rh-negative pa-
tients not immunized to the D antigen is
not standardized.46 Because successful
pregnancy has occurred after liver trans-
plantation, most programs consider it pref-
erable to provide D-negative units to
D-negative females with childbearing po-
tential, if needs are expected to be moder-
ate. Should massive blood loss occur, the
patient could be switched intraoperatively
to D-positive blood, if necessary. For pre-
menopausal females without anti-D, some
programs reserve 10 units of D-negative
RBCs; if more than 10 units are required,
628 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
they switch to D-positive blood.46 Produc-
tion of anti-D occurs less frequently in
D-negative liver transplant patients ex-
posed to the D antigen than in other
D-negative patients.51 In some programs,
patients without anti-D who are D-negative
postmenopausal females or D-negative
males are transfused exclusively with
D-positive blood.
Red Cell Alloantibodies
Liver transplant patients with clinically
significant red cell alloantibodies repre-
sent a special challenge to blood banks.
Sometimes, a sufficient quantity of anti-
gen-negative blood can be secured before
surgery. Some programs reserve a limited
number of antigen-negative units for use
at the beginning of surgery, when allo-
antibody is present, and at the end of
massive blood loss, when transfused cells
are expected to remain in circulation. An-
tibody screening during the interval of
massive blood loss can help guide use of
antigen-positive units during surgery.
Coagulation Considerations
During surgery, hemodilution, platelet
consumption, disordered thrombin regu-
lation, and fibrinolysis derange the hemo-
static process. The coagulopathy is espe-
cially severe during the anhepatic and
early reperfusion stage. The following tests
are useful: the hematocrit guides the use
of red cells, colloids, and crystalloids; the
platelet count guides transfusion of plate-
lets; the prothrombin time and activated
partial thromboplastin time guide FFP
use; and fibrinogen determinations guide
use of Cryoprecipitated AHF and anti-
fibrinolytic agents.46,52,53
Other Organ Transplants
Blood bank support for cardiac transplan-
tation is very similar to that routinely
used for other surgical procedures in
which cardiopulmonary bypass is em-
ployed. The blood bank may also provide
ABO testing and assist in the release
of ABO-compatible organs to prevent
ABO-mismatched organ transplantation.
Pancreatic transplants have compara-
tively low transfusion requirements, but a
specimen from the recipient should rou-
tinely be examined for clinically signifi-
cant unexpected red cell antibodies; in
some institutions, the protocol calls for
crossmatching several units.
Transfusion Service Support
for Organ Transplantation
The blood bank provides vital support for
a clinical transplantation program. Close
communication with the surgeons and
other professionals involved in the pro-
gram is essential. Transfusion practices in
the peritransplant period have a major ef-
fect on morbidity, mortality, and graft sur-
vival rates.
Potential recipients of solid organ trans-
plants are generally available well before
the procedure, so there is ample time to ob-
tain a history and perform laboratory tests.
It is important for the transfusion service to
know if there have been previous pregnan-
cies, transplants, or transfusions.
Laboratory tests routinely performed in-
clude: ABO group and Rh type, the direct
antiglobulin test (DAT), a screen for unex-
pected red cell antibodies, and determina-
tion of CMV serostatus. HLA typing and
HLA antibody studies are routine for organ
recipients.
Passenger lymphocyte hemolysis (typi-
cally “ABO”-incompatible hemolysis), as
Chapter 26: Tissue and Organ Transplantation 629
Copyright © 2005 by the AABB. All rights reserved.
discussed previously in regard to liver
transplantation, can occur with other solid
organ transplants, such as lung, heart, and
kidney. In the case of a recipient receiving
an ABO-compatible but non-group-identi-
cal organ, prophylactic use of mutually
ABO-compatible erythrocytes (compatible
for the donor and the recipient) has been
suggested for intraoperative and postopera-
tive infusions, during the first postoperative
month, or at the appearance of an anti-
body. At present, there is no consensus on
this issue. It is important to remember that
if immediate-spin or computer cross-
matching is routinely performed after an
ABO-unmatched transplant, ABO incom-
patibility due to these IgG antibodies may
be missed. In such cases, the routine use of
a crossmatch with an antihuman globulin
phaseortheuseofaDAT(whichmayde-
tect such cases earlier than a crossmatch) is
recommended. If ABO hemolysis is present,
the patient should be transfused with group
ORBCs.
CMV infection, a serious and often fatal
complication in transplant recipients, is re-
lated to the presence of CMV in the donor
and recipient and the degree to which the
recipient is immunosuppressed. The pri-
mary test used to determine CMV status is
the demonstration of circulating antibody.
CMV-seronegative recipients of CMV-sero-
negative transplants characteristically re-
ceive transfusion components processed to
reduce risk of CMV transmission, either by
preparation from seronegative donors or by
leukocyte reduction to 5 × 106cells/compo-
nent or below.
References
1. Strong M, Eastlund T, Mowe J. Tissue bank
activity in the United States: 1992 survey of
AATB-inspected tissue banks. Tissue Cell Rep
1996;3:15-18.
2. 2002 EBAA statistical report. Washington, DC:
Eye Bank Association of America, 2002.
3. Yap PL. Viral transmission by blood, organs
and tissues. J Hosp Infect 1999;43(Suppl):S137-
44.
4. The Organ Procurement and Transplantation
Network data reports, 2003. Richmond, VA:
United Network for Organ Sharing, 2003.
5. Warwick RM, Eastlund T, Fehily D. Role of the
blood transfusion service in tissue banking.
Vox Sang 1996;71:71-7.
6. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
7. Food and Drug Administration. Human tissue
intended for transplantation. Fed Regist 1997;
62:40429-47.
8. Food and Drug Administration. Guidance for
industry: Screening and testing of donors of
human tissue intended for transplantation.
(July 1997) Rockville, MD: CBER Office of
Communication, Training, and Manufactur-
ers Assistance, 1997.
9. Centers for Disease Control and Prevention.
Guidelines for preventing HIV transmission
through organ and tissue transplantation.
MMWR 1994;43(RR-8):1-17.
10. Woll J, Kasprisin D, eds. Standards for tissue
banking. McLean, VA: American Association
of Tissue Banks, 2002.
11. American Society for Reproductive Medicine.
Guidelines for gamete and embryo donation.
Fertil Steril 2002;77:Suppl 5.
12. Medical standards of EBAA. Washington, DC:
Eye Bank Association of America, 2002.
13. Articles of incorporation, by-laws, and poli-
cies of UNOS. Richmond, VA: United Network
of Organ Sharing, 2004 (revised at least annu-
ally).
14. New York State Department of Health. Tissue
banks and nontransplant anatomic banks,
Part 52. New York State Codes, Rules, and Reg-
ulations, Title 10. Albany, NY: New York State
Department of Health, 2004. (Available at
http://www.wadsworth.org/labert/blood_tissue.)
15. Eastlund T. Infectious disease transmission
through cell, tissue and organ transplanta-
tion. Reducing the risk through donor selec-
tion. Cell Transplant 1995;4:455-77.
16. Eastlund T, Strong DM. Infectious disease trans-
mission through tissue transplantation. In:
Phillips GO, ed. Advances in tissue banking.
Vol. 7. Singapore: World Scientific, 2004:51-132.
17. CJD and the eye. Monograph 7. London:
Royal College of Ophthalmologists, 1998.
18. Hogan RN, Brown P, Heck E, Cavanagh HD.
Risk of prion disease transmission from ocu-
lar tissue transplantation. Cornea 1999;18:2-11.
630 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
19. Human immunodeficiency virus transmitted
from an organ donor screened for HIV anti-
body—North Carolina. MMWR Morb Mortal
Wkly Rep 1987;36:306-8.
20. Eastlund T. Hemodilution due to blood loss
and transfusion and reliability of cadaver tis-
sue donor infectious disease testing. Cell and
Tissue Banking 2000;1:121-7.
21. Linden JV, Critser JK. Therapeutic insemina-
tion by donor II: A review of the known risks.
Reprod Med Rev 1995;4:19-29.
22. SimondsRJ,HolmbergSD,HurwitzRL,etal.
Transmission of human immunodeficiency
virus type 1 from a seronegative organ and
tissue donor. N Engl J Med 1992;326:726-32.
23. Conrad EU, Gretch D, Obermeyer K, et al. The
transmission of hepatitis C virus by tissue
transplantation. J Bone Joint Surg 1995;77A:
214-23.
24. Kainer MA, Linden JV, Whaley DN, et al.
Clostridium infections associated with
musculoskeletal-tissue allografts. N Engl J
Med 2004;350:2564-71.
25. Centers for Disease Control and Prevention.
Investigation of rabies infections in organ do-
nor and transplant recipients—Alabama, Ar-
kansas, Oklahoma, and Texas, 2004. MMWR
Morb Mortal Wkly Rep 2004;53:586-9.
26. Centers for Disease Control and Prevention.
Lymphocytic choriomeningitis virus infec-
tion in organ transplant recipients—Massa-
chusetts, Rhode Island, 2005. MMWR Morb
Mortal Wkly Rep 2005;54:537-9.
27. Food and Drug Administration. Guidance for
industry: Availability of licensed donor
screening tests for use with cadaveric blood
specimens. (June 23, 2000) Rockville, MD:
CBER Office of Communication, Training, and
Manufacturers Assistance, 2000.
28. Choo SY, Eastlund T. Tissue transplantation
and HLA typing. Tissue Cell Rep 1995;2:3-4.
29. LeFor WM, McGonigle AF, Wright CE, Shires
DL. The frequency of false positive HBsAg
screening test results with cadaver tissue do-
nors is dependent upon the assay procedure
used. Tissue Cell Rep 1996;3:6-170.
30. Eastlund T. Bone transplantation and bone
banking.In:LonsteinJE,BradfordDS,Winter
RB, Ogilvie JW, eds. Moe’s textbook of scoliosis
and other spinal deformities. 3rd ed. Phila-
delphia: WB Saunders, 1995:581-95.
31. Bravo D, Rigley TH, Gibran N, et al. Effect of
storage and preservation methods on viabil-
ity of transplantable human skin allografts.
Burns 2000;26:367-78.
32. Phillips TJ. New skin for old: Developments
in biological skin substitutes. Arch Dermatol
1998;134:344-9.
33. O’Brien MF, Stafford EG, Gardner MAH, et al.
Cryopreserved viable allograft aortic valves.
In:YankohAC,HetzerR,MillerDC,etal,eds.
Cardiac valve allografts 1972-1987. New York:
Springer-Verlag, 1988:311-21.
34. Food and Drug Administration. Current good
tissue practice for human cell, tissue, and cel-
lular and tissue-based product establishments;
inspection and enforcement; final rule. (No-
vember 4, 2004) Fed Regist 2004;69:68611-88.
35. Food and Drug Administration. Guidance for
Industry. Validation of procedures for pro-
cessing human tissues intended for trans-
plantation. (March 2002) Rockville, MD:
CBER Office of Communications, Training,
and Manufacturers Assistance, 2002.
36. Food and Drug Administration. Eligibility de-
termination for donors of human cells, tissues,
and cellular and tissue-based products; final
rule. Fed Regist 2004;69:29786-834.
37. Food and Drug Administration. Human cells,
tissues, and cellular and tissue-based prod-
ucts; establishment registration and listing;
final rule. Fed Regist 2001;66:5447-69.
38. Food and Drug Administration. Human cells,
tissues, and cellular and tissue-based prod-
ucts; establishment registration and listing;
interim final rule; opportunity for public
comment. Fed Regist 2004;69:3823-6.
39. Alkhunaizi AM, de Mattos AM, Barry JM, et al.
Renal transplantation across the ABO barrier
using A2kidneys. Transplantation 1999;67:
1319-24.
40. Eastlund T. The histo-blood group ABO sys-
tem and tissue transplantation. Transfusion
1998;38:975-88.
41. Alexandre GPJ, Squifflet JP, DeBruyere M, et
al. ABO-incompatible related and unrelated
living donor renal allografts. Transplant Proc
1986;18:1090-2.
42. Breimer ME, Brynger H, Rydberg L, et al.
Transplantation of blood group A2kidneys to
O recipients. Biochemical and immunologi-
cal studies of group A antigens in human kid-
neys. Transplant Proc 1985;17:2640-3.
43. Terasaki PI. Red-cell crossmatching for heart
transplants (letter). N Engl J Med 1991;325:
1748-9.
44. Opelz G, Sengar DPS, Mickey MR, Terasaki P.
Effect of blood transfusion on subsequent
kidney transplants. Transplant Proc 1973;5:
253-9.
45. Blumberg N, Heal JM. Transfusion immuno-
modulation. In: Anderson KC, Ness PM, eds.
Scientific basis of transfusion medicine. 2nd
ed. Philadelphia:WB Saunders, 2000:427-43.
46. Dzik WH. Solid organ transplantation. In:
PetzLD,SwisherSN,KleinmanS,etal,eds.
Clinical practice of transfusion medicine. 3rd
Chapter 26: Tissue and Organ Transplantation 631
Copyright © 2005 by the AABB. All rights reserved.
ed. New York: Churchill Livingstone, 1996:
783-806.
47. Lundgren G, Groth CG, Albrechtsen D, et al.
HLA matching and pretransplant blood trans-
fusions in cadaveric renal transplantation—a
changing picture with cyclosporin. Lancet
1986;ii:66-9.
48. Iwaki Y, Cecka JM, Terasaki PI. The transfu-
sion effect in cadaver kidney transplants, yes
or no. Transplantation 1990;49:56-9.
49. Opelz G, Vanrenterghem Y, Kirste G, et al.
Prospective evaluation of pretransplant
blood transfusions in cadaver kidney recipi-
ents. Transplantation 1997;63:964-7.
50. Triulzi DJ, Shirey RS, Ness PM, Klein AS. Im-
munohematologic complications of ABO-
unmatched liver transplants. Transfusion
1992;32:829-33.
51. Casanueva M, Valdes MD, Ribera MC. Lack of
alloimmunization to D antigen in D-negative
immunosuppressed liver transplant recipi-
ents. Transfusion 1994;34:570-2.
52. Triulzi DJ, Bontempo FA, Kiss JE, Winkelstein
A. Transfusion support in liver transplanta-
tion. Transfus Sci 1993;14:345-52.
53. Motschman TL, Taswell HF, Brecher ME, et al.
Blood bank support of a liver transplantation
program. Mayo Clin Proc 1989;64:103-11.
632 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Chapter 27: Noninfectious Complications of Blood Transfusion
Chapter 27
Noninfectious
Complications of Blood
Transfusion
THIS CHAPTER ADDRESSES four
broad categories of transfusion re-
actions: 1) acute immunologic, 2)
acute nonimmunologic, 3) delayed immu-
nologic, and 4) delayed nonimmunologic
complications, as shown in Table 27-1.1,2
For each individual type of reaction, the
pathophysiology, treatment, and preven-
tion are discussed. More detailed cover-
age is available elsewhere.1Infectious
risks of transfusion are discussed in
Chapter 28.
Manifestations
All personnel involved in ordering and
administering transfusions must be able
to recognize a transfusion reaction so that
appropriate actions can be taken promptly.
Listed below are signs and symptoms that
are typically associated with acute trans-
fusion reactions and can aid in their rec-
ognition. In general, one should consider
any adverse manifestation occurring at
thetimeofthetransfusiontobeatransfu
-
sion reaction until proven otherwise.
■Fever with or without chills [gener-
ally defined for surveillance pur-
poses as a 1 C (2 F) increase in body
temperature] associated with trans-
fusion. Fever is the most common
symptom of a hemolytic transfusion
reaction (HTR),3but more frequently
it has other causes.
■Shaking chills (rigors) with or with-
out fever.
■Pain at the infusion site or in the
chest, abdomen, or flanks.
■Blood pressure changes, usually acute,
either hypertension or hypotension.
Circulatory shock in combination
with fever, severe chills, and high-
output cardiac failure suggests acute
633
27
Copyright © 2005 by the AABB. All rights reserved.

634 AABB Technical Manual
Table 27-1. Categories and Management of Adverse Transfusion Reactions*
Type Incidence Etiology Presentation Diagnostic Testing
Therapeutic/Prophylactic
Approach
Acute (<24 hours) Transfusion Reactions–Immunologic
Hemolytic 1:38,000-
1:70,000
Red cell incompatibility Chills, fever,
hemoglobinuria,
hypotension, renal failure
with oliguria, DIC (oozing
from IV sites), back pain,
pain along infusion vein,
anxiety
■Clerical check
■DAT
■Visual inspection (free
Hb)
■Repeat patient ABO, pre-
and posttransfusion
sample
■Further tests as
indicated to define
possible incompatibility
■Further tests as
indicated to detect
hemolysis (LDH,
bilirubin, etc)
■Keep urine output >100
mL/hr with fluids and IV
diuretic (furosemide)
■Analgesics (may need
morphine)
■Pressors for
hypotension (low-dose
dopamine)
■Hemostatic components
(platelets, cryo, FFP) for
bleeding
Fever/chill,
nonhemoly-
tic
RBCs:
1:200-1:17
(0.5-6%)
Plts:
1:100-1:3
(1-38%)
■Antibody to donor WBCs
■Accumulated cytokines
in platelet unit
Fever, chills/rigors, head-
ache, vomiting
■Rule out hemolysis
(DAT, inspect for
hemoglobinemia, repeat
patient ABO)
■Rule out bacterial
contamination
■WBC antibody screen
■Antipyretic
premedication
(acetaminophen, no
aspirin)
■Leukocyte-reduced
blood
Urticarial 1:100-1:33
(1-3%)
Antibody to donor plasma
proteins
Urticaria, pruritis, flushing ■Rule out hemolysis
(DAT, inspect for
hemoglobinemia, repeat
patient ABO)
■Antihistamine, treatment
or premedication (PO or
IV)
■May restart unit slowly
after antihistamine if
symptoms resolve
Copyright © 2005 by the AABB. All rights reserved.

Chapter 27: Noninfectious Complications of Blood Transfusion 635
Anaphylactic 1:20,000-
1:50,000
Antibody to donor plasma
proteins (includes IgA,
haptoglobin, C4)
Hypotension, urticaria,
bronchospasm (respira-
tory distress, wheezing),
local edema, anxiety
■Rule out hemolysis
(DAT, inspect for
hemoglobinemia, repeat
patient ABO)
■Anti-IgA
■IgA, quantitative
■Trendelenberg (feet up)
position
■Fluids
■Epinephrine (adult dose:
0.3-0.5 mL of 1:1000
solution SC or IM; in
severe cases, 1:10,000
IV)
■Antihistamines,
corticosteroids, beta-2
agonists
■IgA-deficient blood
components
Transfusion-
related acute
lung injury
1:5,000-
1:190,000
WBC antibodies in donor
(occasionally in recipient),
other WBC-activating
agents in components
Hypoxemia, respiratory
failure, hypotension, fever,
bilateral pulmonary edema
■Rule out hemolysis
(DAT, inspect for
hemoglobinemia, repeat
patient ABO)
■WBC antibody screen in
donor and recipient. If
positive, antigen typing
may be indicated
■WBC crossmatch
■Chest X-ray
■Supportive care until
recovery
■Defer implicated donors
Acute (<24 hours) Transfusion Reactions—Nonimmunologic
Transfusion-
associated
sepsis
Varies by
component
(see Chapter
28)
Bacterial contamination Fever, chills, hypotension ■Gram’s stain
■Culture of component
■Patient culture
■Rule out hemolysis
(DAT, inspect for
hemoglobinemia, repeat
patient ABO)
■Broad spectrum
antibiotics (until
sensitivities completed)
■Treat complications (eg,
shock)
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

636 AABB Technical Manual
Table 27-1. Categories and Management of Adverse Transfusion Reactions* (cont’d)
Type Incidence Etiology Presentation Diagnostic Testing
Therapeutic/Prophylactic
Approach
Hypotension
associated with
ACE inhibition
Dependent
on clinical
setting
Inhibited metabolism of
bradykinin with infusion of
bradykinin (negatively
charged filters) or activa-
tors of prekallikrein
Flushing, hypotension ■Rule out hemolysis
(DAT, inspect for
hemoglobinemia, repeat
patient ABO)
■Withdraw ACE inhibition
■Avoid albumin volume
replacement for
plasmapheresis
■Avoid bedside leukocyte
filtration
Circulatory
overload
<1% Volume overload Dyspnea, orthopnea,
cough, tachycardia, hyper-
tension, headache
■Chest X-ray ■Upright posture
■Oxygen
■IV diuretic (furosemide)
■Phlebotomy (250-mL
increments)
Nonimmune
hemolysis
Rare Physical or chemical de-
struction of blood (heat-
ing, freezing, hemolytic
drug or solution added to
blood)
Hemoglobinuria,
hemoglobinemia
■Rule out patient
hemolysis (DAT, inspect
for hemoglobinemia,
repeat patient ABO)
■Test unit for hemolysis
■Identify and eliminate
cause
Air embolus Rare Air infusion via line Sudden shortness of
breath, acute cyanosis,
pain, cough, hypotension,
cardiac arrythmia
■X-ray for intravascular
air
■Place patient on left side
with legs elevated above
chest and head
Hypocalcemia
(ionized cal-
cium)
Dependent
on clinical
setting
Rapid citrate infusion
(massive transfusion of
citrated blood, delayed
metabolism of citrate,
apheresis procedures)
Paresthesia, tetany,
arrhythmia
■Ionized calcium
■Prolonged Q-T interval
on electrocardiogram
■Slow calcium infusion
while monitoring ionized
calcium levels in severe
cases
■PO calcium supplement
for mild symptoms
during apheresis
procedures
Copyright © 2005 by the AABB. All rights reserved.

Chapter 27: Noninfectious Complications of Blood Transfusion 637
Hypothermia Dependent
on clinical
setting
Rapid infusion of cold
blood
Cardiac arrhythmia Central body temperature ■Employ blood warmer
Delayed (>24 hours) Transfusion Reactions–Immunologic
Alloimmuni-
zation, RBC
antigens
Alloimmuni-
zation, HLA
antigens
1:100 (1%)
1:10 (10%)
Immune response to
foreign antigens on
RBCs, or WBCs and
platelets (HLA)
Positive blood group
antibody screening test
Platelet refractoriness,
delayed hemolytic
reaction, hemolytic dis-
ease of the newborn
■Antibody screen
■DAT
■Platelet antibody screen
■Lymphocytotoxicity test
■Avoid unnecessary
transfusions
■Leukocyte-reduced
blood
■Avoid unnecessary
transfusions
■Leukocyte-reduced
blood
Hemolytic 1:11,000-
1:5000
Anamnestic immune
response to red cell
antigens
Fever, decreasing
hemoglobin, new
positive antibody screen-
ing test, mild jaundice
■Antibody screen
■DAT
■Tests for hemolysis
(visual inspection for
hemoglobinemia, LDH,
bilirubin, urinary
hemosiderin as clinically
indicated)
■Identify antibody
■Transfuse compatible
red cells as needed
Graft-vs-host
disease
Rare Donor lymphocytes en-
graft in recipient and
mount attack on host tis-
sues
Erythroderma,
maculopapular rash, an-
orexia, nausea, vomiting,
diarrhea, hepatitis,
pancytopenia, fever
■Skin biopsy
■HLA typing
■Corticosteroids,
cytotoxic agents
■Irradiation of blood
components for patients
at risk (including related
donors and HLA-
selected components)
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.

638 AABB Technical Manual
Table 27-1. Categories and Management of Adverse Transfusion Reactions* (cont’d)
Type Incidence Etiology Presentation Diagnostic Testing
Therapeutic/Prophylactic
Approach
Posttransfusion
purpura
Rare ■Recipient platelet
antibodies (apparent
alloantibody, usually
anti-HPA-1) destroy
autologous platelets
Thrombocytopenic
purpura, bleeding, 8-10
days after transfusion
■Platelet antibody screen
and identification
■IGIV
■HPA-1-negative platelets
■Plasmapheresis
Immunomo-
dulation
Unknown Incompletely understood
interaction of donor WBC
or plasma factors with re-
cipient immune system
Increased renal graft sur-
vival, infection rate,
postresection tumor recur-
rence rate (controversial)
■None specific ■Avoid unnecessary
transfusions
■Autologous transfusion
■Leukocyte-reduced red
cells and platelets
Delayed (>24 hours) Transfusion Reactions–Nonimmunologic
Iron overload Typically af-
ter >100 RBC
units
Multiple transfusions with
obligateironloadintrans-
fusion-dependent patient
Diabetes, cirrhosis,
cardiomyopathy
■Serum ferritin
■Liver enzymes
■Endocrine function tests
■Desferioxamine (iron
chelator)
*For platelet refractoriness, see Chapter 16; for septic transfusion reactions, see Table 28-1; for a recent summary of transfusion reactions, see Popovsky.1
ACE = angiotensin-converting enzyme; antibody screen = blood group antibody screening test; DAT = direct antiglobulin test; DIC = disseminated intravascular coagulation; FFP
= Fresh Frozen Plasma; Hb = hemoglobin; IV = intravenous; IGIV = intravenous immunoglobulin; IM = intramuscular; LDH = lactate dehydrogenase; PO = by mouth; RBC = Red
Blood Cell; SC = subcutaneous; WBC = White Blood Cell.
Copyright © 2005 by the AABB. All rights reserved.
sepsis but may also accompany an
acute HTR. Circulatory collapse with-
out fever and chills may be the most
prominent finding in anaphylaxis.
■Respiratory distress, including dyspnea,
tachypnea, wheezing, or hypoxemia.
■Skin changes, including urticaria,
pruritis (itching), flushing, or local-
ized edema (angioedema).
■Nausea with or without vomiting.
■Darkened urine or jaundice. Dark urine
may be the earliest indication of an
acute hemolytic reaction in anesthe-
tized patients.
■Bleeding or other manifestations of
a consumptive coagulopathy.
Acute Transfusion Reactions
Immune-Mediated Hemolysis
Pathophysiology and Manifestations
The most severe hemolytic reactions oc-
cur when transfused red cells interact
with preformed antibodies in the recipi-
ent. In contrast, the interaction of trans-
fused antibodies with the recipient’s red
cells rarely causes symptoms. However,
there may be accelerated red cell destruc-
tion, and plasma-containing products with
high-titer ABO antibodies can cause acute
hemolysis. The interaction of antibody
with antigen on the red cell membrane
can initiate a sequence of complement
activation (see Chapter 11), cytokine and
coagulation effects, and other elements of
a systemic inflammatory response4that
result in the clinical manifestations of a
severe acute HTR. Severe symptoms can
occur after the infusion of as little as 10 to
15 mL of ABO-incompatible red cells. In
anesthetized patients who cannot report
symptoms, the initial manifestations of
an acute HTR may be hemoglobinuria,
hypotension, or diffuse bleeding at the
surgical site.
Such severe acute HTRs today are usu-
ally caused by ABO incompatibility5but oc-
casionally may be caused by antibodies
with other specificities.6In contrast,
hemolysis of an entire unit of blood can oc-
cur in the virtual absence of symptoms7
and may be a relatively slow process. In
such cases, hemolysis is typically extra-
vascular, without generation of significant
systemic levels of inflammatory mediators.
Complement Activation. The binding of
antibody to blood group antigens may acti-
vate complement, depending on the char-
acteristics of both the antibody and the an-
tigen, including antibody specificity, class,
subclass, titer, and antigen density (see
Chapter 11). C3 activation releases the ana-
phylatoxin C3a (see Chapter 11), and red
cells coated with C3b are removed by
phagocytes with complement receptors,
more rapidly than if antibody is present
alone. If the enzymatic cascade proceeds to
completion and a membrane attack com-
plex is assembled, intravascular hemolysis
results, with the production of C5a, which
is100timesaspotentananaphylatoxinas
C3a.3This sequence is characteristic of ABO
incompatibility and causes the cardinal
manifestations of hemoglobinemia and, if
the renal threshold for hemoglobin is ex-
ceeded, hemoglobinuria.8(p182) Anaphylatoxins
interact with a wide variety of cells, includ-
ing monocytes/macrophages, granulocytes,
platelets, vascular endothelial cells, and
smooth muscle cells, the latter leading to
hypotension and bronchospasm. Anaphy-
latoxins also cause the release or produc-
tion of multiple local and systemic media-
tors, including granule enzymes, histamine
and other vasoactive amines, kinins, oxy-
gen radicals, leukotrienes, nitric oxide, and
cytokines.3These mechanisms may cause
manifestations that mimic allergy, such as
flushing and rarely urticaria, wheezing and
Chapter 27: Noninfectious Complications of Blood Transfusion 639
Copyright © 2005 by the AABB. All rights reserved.
chest pain or tightness, and abdominal
pain, nausea, and vomiting.
With most non-ABO blood group anti-
bodies, complement activation is usually
incomplete; hemoglobinemia is absent or
mild, but the consequences of complement
activation, most notably the release of ana-
phylatoxins and opsonization of red cells,
may still have adverse effects.
Cytokines. The role of cytokines in in-
flammatory responses (see Chapter 11), in-
cluding acute HTRs, is increasingly recog-
nized.9,10 The known activities of inflammatory
cytokines, such as tumor necrosis factor α
(TNFα), interleukin-1βand -6 (IL-1β,IL-6),
and chemokines such as IL-8 and mono-
cyte chemoattractant protein (MCP), sug-
gest that they mediate some of the effects of
alloimmune hemolysis. IL-1 and TNF cause
fever and hypotension (particularly in syn-
ergy), stimulation of endothelial cells to in-
crease expression of adhesion molecules
and procoagulant activity, and recruitment
and activation of neutrophils and platelets,
perhaps through the induction of IL-8 and
MCP. Incubation of whole blood with
washed, ABO-incompatible red cells in vi-
tro has been shown to cause dramatic in-
creases in TNF, IL-8, and MCP. This cyto-
kine response is complement dependent. A
similar model of hemolysis resulting from
anti-D (IgG) showed a different pattern of
cytokine production with “high-level” re-
sponses of IL-8 and MCP and “low-level”
responses of IL-1β,IL-6,andTNFα.9,10
The relevance of these in-vitro models to
HTRs in vivo is suggested by a case in
which TNFαand neutrophil elastase levels
werefoundtobeelevatedwhenagroupO
patient received 100 mL of group A red
cells; elevation of neutrophil elastase is
consistent with IL-8 activity.11 These find-
ings may lead to new therapeutic options
forpatients.However,thecompleteroleof
cytokines in the consequences of immune
hemolysis remains to be defined.
Coagulation Activation. Several mecha-
nisms, including those listed above, may be
responsible for abnormalities of coagula-
tion in HTRs.4The antigen-antibody inter-
action may activate the “intrinsic” clotting
cascade through Hageman factor. In addi-
tion, activated Hageman factor (Factor XIIa)
acts on the kinin system to generate brady-
kinin; bradykinin increases capillary per-
meability and dilates arterioles, causing a
decrease in systemic arterial pressure. Sev-
eral factors cited above may increase the
expression of tissue factor by leukocytes
and endothelial cells, including activated
complement components, TNFα, and
IL-1β. Tissue factor activates the “extrinsic”
coagulation pathway, and its release is as-
sociated with disseminated intravascular
coagulation (DIC), which may, in turn, cause:
1) formation of thrombi within the micro-
vasculature and ischemic damage to tissues
and organs; 2) consumption of fibrinogen,
platelets, and other coagulation factors; 3)
activation of the fibrinolytic system and
generation of fibrin degradation products.
The outcome can be a hemorrhagic diathesis
characterized by generalized oozing or un-
controlled bleeding.
Shock and Renal Failure. Considering
the absolute mass of antigen and anti-
body—and the list of mediators that may
be involved in HTRs, including anaphyla-
toxins, vasoactive amines, kinins, and cyto-
kines—it may not be surprising that shock
can occur. Hypotension provokes a com-
pensatory sympathetic nervous system re-
sponse that produces vasoconstriction in
organs and tissues with a vascular bed rich
in alpha-adrenergic receptors, notably, the
renal, splanchnic, pulmonary, and cutane-
ous capillaries, aggravating ischemia in
these sites.
Renal failure is another sequel of an
acute HTR. Although free hemoglobin, his-
torically considered the cause of renal fail-
ure, does impair renal function,12 current
640 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
thought attributes renal failure largely to
hypotension, renal vasoconstriction, anti-
gen-antibody complex deposition, and for-
mation of thrombi in the renal vasculature,
all of which compromise renal cortical
blood supply.
Frequency
Clerical and other human errors leading
to mistaken identity are the most common
causes of ABO-incompatible transfusion,
occurring either at pretransfusion sample
collection, within the transfusion service,
or at the time of blood administration. A
study of reported transfusion errors in
New York State over a 10-year period (the
1990s) estimated the incidence of ABO-
incompatible red cell transfusions at
1:38,000. Correction for the expected rate
of fortuitously compatible transfusions
led to an estimate of the rate of mis-
transfusion of 1:14,000.7A survey of 3601
institutions by the College of American
Pathologists found 843 acute HTRs re-
ported over a 5-year period, of which 50
(6%) were fatal.13 The Serious Hazards of
Transfusion (SHOT) initiative in the United
Kingdom and Republic of Ireland re-
ported 161 cases of ABO-incompatible
transfusion, with nine fatal cases (five def-
initely related deaths, one probably re-
lated death, and three possibly related
deaths) in 5 years.14 Although no precise
denominator is available for these confi-
dential reports, it is believed that over
90% of the total transfusions were re-
viewed and approximately 2.5 million
RBC units were issued each year, for a rate
of no less than 1 in 78,000. These values
probably underestimate the true fre-
quency, because even acute HTRs go un-
recognized or unreported. Estimates of
mortality rates from acute HTRs are gener-
ally in the range of 1 in 1,000,000 transfu-
sions.5,7,14
Treatment
The treatment of an acute HTR depends
on its severity.4Vigorous treatment of hypo-
tension and promotion of adequate renal
blood flow are the primary concerns. If
shock can be prevented or adequately
treated, progression to renal failure may
be avoided. Adequacy of renal perfusion
can be monitored by measurement of
urine output, with a goal of maintaining
urine flow rates above 100 mL/hour in
adults for at least 18 to 24 hours. The
usual first support is intravenous normal
saline, but underlying cardiac and/or re-
nal disease may complicate therapy, and
it is important to avoid overhydration. In-
vasive monitoring of pulmonary capillary
wedge pressure is recommended in guid-
ing fluid therapy in the face of hemo-
dynamic instability. Diuretics help to im-
prove blood flow to the kidneys and
increase urine output. Intravenous furo-
semide at a dose of 40 to 80 mg for an
adult or 1 to 2 mg/kg for a child not only
has a diuretic effect but also improves
blood flow to the renal cortex. This dose
may be repeated once, and the patient
should be adequately hydrated. Mannitol,
an osmotic diuretic, has been used in the
past, but furosemide is better for main-
taining renal cortical blood flow. If no di-
uretic response occurs within a few hours
of instituting fluid and diuretic therapy,
there is a strong likelihood that acute tu-
bular necrosis has occurred, and further
fluid administration may be harmful.
Treatment of hypotension with pressor
agents that decrease renal blood flow, such
as dopamine in higher doses, should be
avoided if possible. The use of low-dose do-
pamine (2-5 µg/kg/minute), as an agent to
protect renal function, has been recom-
mended in the management of acute HTRs.4
However, evidence suggests that it is not ef-
Chapter 27: Noninfectious Complications of Blood Transfusion 641
Copyright © 2005 by the AABB. All rights reserved.
fective in this role, and it has many toxici-
ties.15
Consumptive coagulopathy, with resul-
tant bleeding or generalized oozing, may be
a prominent clinical finding in some HTRs
and may be the initial presentation in an
anesthetized patient. Heparin has been rec-
ommended by some, both to forestall DIC
when an ABO incompatibility is first dis-
covered and to treat the established coa-
gulopathy. Others believe the dangers of
heparin outweigh its potential benefits, es-
pecially because the immune event that
provoked the DIC is self-limited. Adminis-
tration of Platelets, Fresh Frozen Plasma
(FFP), and Cryoprecipitated AHF, a source
of fibrinogen and Factor VIII, may be nec-
essary. Red cell exchange may be consid-
ered in patients with a significant load of
circulating incompatible red cells.
Acute hemolytic reactions are rare and
few clinicians have first-hand experience
with their treatment. Because medical
management of an acute HTR is often com-
plicated and may require aggressive inter-
ventions such as hemodialysis, consultation
with physicians experienced in the organ
systems most damaged or specialists in
critical care medicine may be prudent when
treating a patient with a severe acute HTR.
Prevention
Because clerical errors cause the majority
of acute, immune-mediated HTRs, the
best hope for prevention lies in prevent-
ingordetectingerrorsineveryphaseof
the transfusion process. In each institu-
tion, there should be systems designed to
prevent and detect errors in patient and
unit identification at the time of phlebot-
omy (sample acquisition), at all steps in
laboratory testing, at the time of issue,
and when the transfusions are given. The
SHOT reports document multiple errors
in a majority of mistransfusion incidents
and particularly emphasize the impor-
tanceofthebedsidecheckatthetimeof
transfusion.14 Ensuring that all clinical staff
recognize the signs of acute reactions and
stop the transfusion before a critical vol-
ume of blood has been administered is es-
sential to preventing harm to the patient.
Crucial in the prevention of transfusion
mishaps are training and assessment of
personnel performing transfusions. Active
participation by physicians and manage-
ment, as well as by nursing, technical, and
clinical personnel, is essential.
Nonimmune-Mediated Hemolysis
Causes
Red cells may undergo in-vitro hemolysis
if the unit is exposed to improper temper-
atures during shipping or storage or is
mishandled at the time of administration.
Malfunctioning blood warmers, use of
microwave ovens or hot waterbaths, or in-
advertentfreezingmaycausetempera-
ture-related damage. Mechanical hemolysis
maybecausedbytheuseofrollerpumps
(such as those used in cardiac bypass sur-
gery), pressure infusion pumps, pressure
cuffs, or small-bore needles.16 Osmotic
hemolysis in the blood bag or infusion set
may result from the addition of drugs or
hypotonic solutions. Inadequate degly-
cerolization of frozen red cells may cause
the cells to hemolyze after infusion. Finally,
hemolysis may be a sign of bacterial
growth in blood units. In a patient with
transfusion-associated hemolysis for which
both immune and nonimmune causes
have been eliminated, the possibility might
be considered that the patient or donor has
an intrinsic red cell defect, such as glu-
cose-6-phosphatase dehydrogenase defi-
ciency, causing coincidental hemolysis.
642 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Treatment
Treatment depends on the severity of the
reaction. If the patient develops a severe
reaction with hypotension, shock, and re-
nal dysfunction, intensive clinical man-
agement is required even before the cause
of the mishap is investigated. If the pa-
tient exhibits only hemoglobinemia and
hemoglobinuria, supportive therapy may
be sufficient.
Prevention
There should be written procedures for all
aspects of procuring, processing, and is-
suing blood, and administering transfusions.
All staff should be trained in the proper
use of equipment, intravenous solutions,
and drugs used during the administration
of blood and blood components. Equip-
ment must be properly maintained and
records kept of how and when items are
used. Intravenous medications shall not
be injected into blood bags, unless ap-
proved by the Food and Drug Administra-
tion (FDA) or documented to be safe for
that purpose,17(p48) and care must be exer-
cised in the selection and use of intrave-
nous access devices. Chapter 22 discusses
the details of administering transfusions.
Transfusion-Associated Sepsis
Bacterial contamination of transfused blood
should be considered if the patient expe-
riences severe rigors, especially if they are
accompanied by cardiovascular collapse
and/or fever over 40 C.18 For a more de-
tailed discussion of this potentially life-
threatening transfusion complication, see
Chapter 28.
Febrile Nonhemolytic Reactions
Pathophysiology and Manifestations
A febrile nonhemolytic transfusion reac-
tion (FNHTR) is often defined as a tem-
perature increase of >1 C associated with
transfusion and without any other expla-
nation. Such reactions are often associated
with chills or rigors. The 1 C definition is
arbitrary; the same events might cause
smaller temperature increments. Indeed,
some authors discuss reactions character-
ized by rigors or other symptoms, in the
absence of fever, as FNHTRs because of a
presumed common mechanism.2In one
study of 108 reactions characterized by
chills, cold, or rigors, only 18 involved a
rise in temperature.19 Febrile reactions
complicate 0.5% to 6% of nonleukocyte-
reduced red cell transfusions. Previous
opportunities for alloimmunization, es-
pecially pregnancies and multiple trans-
fusions, increase the frequency of FNHTRs
to red cells. The rate of such reactions is
much higher after platelet transfusion
(1-38%). Most FNHTRs are benign, al-
though some may cause significant dis-
comfort and hemodynamic or respiratory
changes. The temperature increase may
begin early in the transfusion or be de-
layed in onset for hours after completion
of the transfusion.
Many febrile reactions are thought to re-
sult from an interaction between antibod-
ies in the recipient’s plasma and antigens
present on transfused lymphocytes, granu-
locytes, or platelets, most frequently HLA
antigens. There is also evidence that febrile
reactions, particularly those due to plate-
lets, may be caused by the infusion of bio-
logic response modifiers, including cyto-
kines, that accumulate in the blood bag
during storage. Cytokine release in the re-
cipient undoubtedly contributes to those
reactions that begin with recipient antibody
against donor leukocytes.2,20-22 Because fever
maybeaninitialmanifestationofanacute
HTR or a reaction to transfusion of blood
contaminated with bacteria, any observa-
tion of an increase in temperature associ-
ated with transfusion warrants prompt at-
Chapter 27: Noninfectious Complications of Blood Transfusion 643
Copyright © 2005 by the AABB. All rights reserved.
tention. The diagnosis of an FNHTR is
made after excluding other possible expla-
nations for the fever, particularly a
hemolytic or septic reaction. Guidelines for
evaluating a suspected acute transfusion
reaction are presented later in this chapter.
Treatment
Traditionally, transfusion was discontin-
ued when an FNHTR occurred.23 However,
some clinicians believe that fever should
not routinely cause discontinuation of a
transfusion,2,24 depending on whether the
patient has symptoms, signs, or labora-
tory data that suggest hemolysis, transfu-
sion-related acute lung injury (TRALI), or
bacterial contamination. The fever of an
FNHTR usually responds to antipyretics.
Acetaminophen is preferred to the use of
salicylates because the former drug does
not affect platelet function. Meperidine
injection may be useful in patients with
severe shaking chills. Antihistamines are
not indicated because most FNHTRs do
not involve histamine release.
Prevention
Febrile reactions in an alloimmunized
individual can often be prevented by
transfusion of leukocyte-reduced blood
components. Prevention of reactions
caused by cytokine accumulation during
storage requires that the leukocyte reduc-
tion be performed before storage,2,19 but
some patients will still react. With non-
leukocyte-reduced platelets, cytokine-
mediated reactions may be less frequent
when the component(s) are less than or
equal to 3 days old. Plasma removal may
also decrease febrile reactions. Aceta-
minophen is commonly given before
transfusion, but there is no evidence that
the premedication lessens the incidence
of FNHTR symptoms due to prestorage
leukocyte-reduced platelets.25
Allergy; Urticaria (Hives) to Anaphylaxis
Pathophysiology and Manifestations
Allergic reactions to transfusion form a
continuum, with the vast majority clus-
tered at the mild end, in the form of urti-
caria or “hives”—erythematous, sharply
circumscribed raised lesions, most often
present over the upper trunk and neck,
which may itch and which are not usually
accompanied by fever or other adverse
findings. At the other end of the spectrum
are anaphylactic reactions, in which there
are systemic symptoms including hypo-
tension, loss of consciousness, shock,
and, in rare cases, death. The latter may
begin after infusion of only a few millili-
ters, but less severe reactions tend to take
longer to develop. The term “anaphylac-
toid” is used in transfusion medicine to
denote reactions in between these ex-
tremes, but it is also used to denote reac-
tions that have clinical similarities to
anaphylaxis but different mechanisms.
Manifestations of these reactions may in-
volve one or several systems, notably, the
skin (urticaria, generalized flushing or
rash, localized swelling or “angioedema”),
respiratorytract(upperorlowerrespira
-
tory tract obstruction with cough, hoarse-
ness, stridor, wheezing, chest tightness or
pain, dyspnea), the gastrointestinal tract
(cramps, nausea, vomiting, diarrhea), or
the circulatory system (tachycardia and
other arrhythmias including cardiac ar-
rest).26 Fever is characteristically absent, a
feature that aids in differentiating these
reactions from hypotension due to a hemoly-
tic reaction or bacterial contamination,
and from respiratory compromise caused
by TRALI (see below). The severity of al-
lergic transfusion reactions may increase
with successive transfusions.
Allergic reactions are attributed to expo-
sure to a soluble substance in donor plasma
that binds to preformed IgE antibodies on
644 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
mast cells, resulting in the activation and
release of histamine. This presumption is
based on the facts that reactions tend to re-
cur in an affected recipient and that they
can be prevented by removal of the plasma
from cellular components or, in the case of
urticaria, by antihistamines. Anaphylactic
and anaphylactoid reactions are sometimes
associated with class, subclass, and allo-
type-specific antibodies against IgA, partic-
ularly in IgA-deficient patients.27 IgE anti-
IgA has been demonstrated in two patients
with common variable immunodeficiency
having reactions to immunoglobulin prep-
arations.28 However, most of the IgA anti-
bodies to which anaphylactic reactions are
attributed are of the IgG or IgM class,27 and
these antibodies are demonstrable in only a
minority of the anaphylaxis cases referred
for study (17.5% in the series of Sandler et
al27). Moreover, IgA antibodies are common
but anaphylactic reactions are not. There-
fore, demonstration of anti-IgA in an indi-
vidual who has not been transfused does
not predict anaphylaxis. Other allergens or
other mechanisms are likely.
Severe allergic reactions have been re-
ported in patients with antibodies directed
against C4 determinants,29,30 haptoglobin,31
and elements of nonbiologic origin such as
ethylene oxide used for sterilizing tubing
sets.32 However, the causative antigens have
not been identified in the vast majority of
cases. Reactions caused by passively trans-
ferred donor antibody have rarely been
documented.33,34
Hypotensive reactions mimicking ana-
phylaxis have been observed in patients
taking angiotensin-converting enzyme
(ACE) inhibitors who receive albumin dur-
ing plasma exchange.35 They were thought
to be due to inhibition of bradykinin catab-
olism by the ACE inhibitors combined with
bradykinin activation by low levels of pre-
kallikrein activator (a Hageman factor frag-
ment) in the albumin used for replacement.
Similarly, bradykinin activation by pre-
kallikrein activity in plasma protein fraction
has also been implicated in hypotensive re-
actions,36 and a similar mechanism is prob-
ably responsible for the many patients
taking ACE inhibitors reported to have
hypotensive reactions when receiving
blood components via bedside leukocyte
reduction filters.37-39 Similar reactions have
been observed in association with the con-
tact of plasma with charged dialysis mem-
branes, low-density lipoprotein adsorption
columns, and staphylococcal protein A
immunoadsorption columns. Other mech-
anisms that have been proposed include the
infusion of complement-derived anaphyla-
toxins and histamine.26 The differentiation
and appropriate classification of these dif-
ferent reactions will require additional re-
search and refined diagnostic tools.
Frequency
Urticaria may complicate as many as 1%
to 3% of transfusions, the observed fre-
quency depending on how vigorously it is
sought. The incidence of anaphylactic re-
actions fortunately is low, estimated to be
1 in 20,000 to 50,000 units. The SHOT data
suggest that anaphylaxis is much more
common as a complication of plasma and
platelet transfusions, than of red cells14;
although these reactions may have con-
tributed to the death of a few severely ill
patients, they were not a primary cause of
death. The mortality rate reported to the
FDA is about 1 per year.26
Treatment
If urticaria is the only adverse event noted,
the transfusion may be temporarily inter-
rupted while an antihistamine (eg, di-
phenhydramine, 25-50 mg) is adminis-
tered orally or parenterally. If symptoms
Chapter 27: Noninfectious Complications of Blood Transfusion 645
Copyright © 2005 by the AABB. All rights reserved.
are mild and promptly relieved, the trans-
fusion may be resumed, provided the
interrupted infusion can be completed
within the acceptable time (see Chapter
22). If the patient develops severe urticaria,
a significant local swelling, respiratory or
gastrointestinal symptoms, or hypotension,
the transfusion should be discontinued.26
The immediate treatment of an anaphy-
lactic transfusion reaction should be to stop
the transfusion and treat hypotension by
placing the patient in the Trendelenberg
(feet up) position and administering a fluid
challenge. If the blood pressure does not
improve immediately, epinephrine should
be given. In mild to moderate cases, epi-
nephrine (1:1000) should be delivered sub-
cutaneously or intramuscularly in a starting
dose of 0.3 to 0.5 mL in adults, or 0.01
mL/kg in children. This dose may be re-
peated a second and third time at 5- to
15-minute intervals. In severe reactions (eg,
systolic blood pressure below 80 mm Hg,
laryngeal edema with upper airway com-
promise, or respiratory failure), the drug
should be given intravenously (1:10,000) for
the most rapid effect because drug absorp-
tion is unreliable in hypotensive patients.
Aerosolized or intravenous beta-2 agonists
and theophylline may be required in se-
lected patients in whom bronchospasm is
unresponsive to epinephrine treatment, or
in whom epinephrine is ineffective because
of pre-existing beta-blocker therapy. Oxy-
gen therapy should be administered as re-
quired, with endotracheal intubation if
there is significant upper airway obstruc-
tion. Continued hemodynamic instability
may require invasive hemodynamic moni-
toring. Under no circumstances should the
transfusion be restarted. Coincidental oc-
currence of myocardial infarction, pulmo-
nary embolism, or other medical catastro-
phes could present with hypotension and
respiratory compromise and should be
considered.26
Prevention
Recipients who have frequent transfusion-
associated urticarial reactions may re-
spond well to administration of antihista-
mine (eg, 25-50 mg of diphenhydramine)
one-half hour before transfusion. However,
diphenhydramine should not be given
routinely without a history of previous al-
lergic reactions, particularly to elderly pa-
tients.40 If antihistamine administration is
insufficient, 100 mg of hydrocortisone given
1 hour before transfusion may be useful.
If reactions are recurrent and severe or as-
sociated with other allergic manifestations
in spite of adequate premedication, trans-
fusion of washed red cell or platelet com-
ponents, or red cells that have been frozen,
thawed, and deglycerolized will usually be
tolerated.
Patients who have had a prior life-threat-
eninganaphylacticreactionandwhoare
IgA-deficient or have a demonstrated IgA
antibody should receive blood components
that lack IgA, either by washing or prepara-
tion of components from IgA-deficient
blood donors. Severe reactions that are not
caused by anti-IgA can be prevented only
by maximal antiallergy immunosuppres-
sion or washing. A need for red cells may be
met by the use of washed or frozen, thawed,
and deglycerolized units.26 Washed platelets
are generally not readily available and may
result in decreased platelet recovery, func-
tion, and survival41; therefore, after the first
reaction, if there is no evidence that the re-
action is mediated by IgA, some centers
elect to rechallenge the patient under
closely controlled circumstances. Preven-
tion of anaphylactoid reactions in patients
such as those with thrombotic thrombocy-
topenic purpura who absolutely require
plasma components may be a tremendous
challenge if IgA-deficient donors will not
suffice. Pretreatment with antihistamines,
corticosteroids (starting with 100 mg of hy-
646 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
drocortisone), and ephedrine may help. Fi-
nally, it may be possible to collect and store
autologous blood components from patients
known to have experienced anaphylactic
reactions.
Transfusion-Related Acute Lung Injury
Pathophysiology and Manifestations
TRALI should be considered whenever a
transfusion recipient experiences acute
respiratory insufficiency and/or X-ray
findings are consistent with bilateral pul-
monary edema but has no other evidence
of cardiac failure or a cause for respiratory
failure. The severity of the respiratory dis-
tress is usually disproportionate to the
volume of blood infused. The reaction
typically includes fever, chills, and hypo-
tension, usually occurring during or with-
in 1 to 2 after transfusion, often with an
immediate and dramatic onset. Implicated
components always have contained plasma,
but the volume may be as small as that of
a unit of cryoprecipitate or RBCs in an ad-
ditive solution.42,43
Because the manifestations of TRALI are
variable and may overlap with those of the
patient’s underlying medical problems, it is
useful to define the syndrome, particularly
for the purpose of conducting studies of its
epidemiology and pathogenesis. A consen-
sus conference of the blood services in
Canada developed such a definition.44 The
panel defined acute lung injury (ALI) as a
syndrome of: 1) acute onset; 2) hypoxemia
(PaO2/FIO2<300 mm Hg, or O2saturation
<90% on room air, or other clinical evi-
dence); 3) bilateral lung infiltrates on a
chest x-ray; and 4) no evidence of circula-
tory overload. TRALI is then defined as: 1)
new ALI occurring during transfusion or
within 6 hours of completion; and 2) no
other temporally associated ALI risk factors.
If the latter are present, the case is consid-
ered “Possible TRALI.” Risk factors for ALI
include aspiration, pneumonia, toxic inha-
lation, lung contusion, near drowning,
severe sepsis, shock, multiple trauma, burn
injury, acute pancreatitis, cardiopulmonary
bypass, and drug overdose. It was noted
that such a definition will not include cases
of mild respiratory embarrassment having
a similar pathogenesis, cases of ALI in pa-
tients with circulatory overload, and cases
in which a transfusion-related process causes
worsening of pre-existing ALI.44
TRALI may result from multiple mecha-
nisms. Donor antibodies directed against
recipient HLA class I or II antigens, or
neutrophil antigens of the recipient, have
been demonstrated45-47 and are thought to
cause a sequence of events that increase the
permeability of the pulmonary microcir-
culation so that high-protein fluid enters
the interstitium and alveolar air spaces. In-
frequently, antibodies in the recipient’s cir-
culation against HLA or granulocyte anti-
gens initiate the same events.45,46,48 Although
one would expect causative antibodies to
be far more common in recipients than do-
nors, the rarity of TRALI due to recipient
antibody might be due to the fact that the
pool of target leukocytes is much smaller in
a cellular blood component than in a recip-
ient’s circulation. Monocyte activation, with
expression of cytokines including IL-1β,
TNFα, and tissue factor, has been demon-
strated, and these reactions were highly
specific for cells bearing the target antigens.48
Perfusion of neutrophils, complement, and
neutrophil-specific antibody into an ex-vivo
rabbit lung preparation causes severe edema,49
and autopsy studies demonstrate neutro-
phil aggregation in the lungs of patients
who have died of TRALI.50 These and other
observations suggest that pulmonary edema
in TRALI is caused by neutrophil-mediated
endothelial damage, initiated by antibodies
activating neutrophils directly or via activa-
tion of monocytes, pulmonary macrophages,
and/or endothelial cells.
Chapter 27: Noninfectious Complications of Blood Transfusion 647
Copyright © 2005 by the AABB. All rights reserved.
As the spectrum of antibodies implicated
in cases of TRALI broadens, more cases will
appear to be antibody-mediated. However,
other mechanisms have been proposed as
causes for transfusion-related respiratory
failure. Severe pulmonary reactions are re-
ported after granulocyte transfusions, par-
ticularly in patients with known or unap-
parent lung infections or with conditions
likely to promote prompt complement acti-
vation.51 Other factors may include anaphy-
latoxins C3a and C5a, aggregation of granu-
locytes into leukoemboli that lodge in the
pulmonary microvasculature, or transfusion
of cytokines that have accumulated in
stored blood components. Recently, reac-
tive lipid products from donor blood cell
membranes have been implicated as po-
tential granulocyte activators in the patho-
genesis of TRALI.52 These substances accu-
mulate during blood bank storage and
prime neutrophils to produce vasoactive
mediators in response to a second stimulus
such as infection. One nested case-control
study found that component age and levels
of bioactive lipids, but not leukocyte anti-
bodies, were associated with TRALI.53
The incidence rate of TRALI is not known,
but data from one institution in the 1980s
suggest that this complication may occur as
frequently as 1 in 5000 transfusions.45 The
passive surveillance data of 5 years of SHOT
reports14 include 70 TRALI cases (approxi-
mately 1 per 250,000 total components), of
which 18 cases were fatal (includes six defi-
nite,twoprobable,and10possibleTRALI-
related fatalities). In this series, TRALI was
the most common cause of morbidity and
mortality, ahead of transfusion-associated
graft-vs-host disease (TA-GVHD), ABO in-
compatibility, and bacterial contamination.
The SHOT data suggest that the rate of
TRALI is higher after plasma and platelet
transfusion. Conclusions regarding incidence
and fatality rates will, of course, depend on
the definition of TRALI used.
Treatment
If any kind of acute pulmonary reaction is
suspected, the transfusion should be
stopped immediately and the same unit
should not be restarted even if symptoms
abate. Clinical management focuses on
reversing progressive hypoxemia with ox-
ygen therapy and ventilatory assistance, if
necessary. The role of intravenous ste-
roids is unproved. Unlike other forms of
acute respiratory distress syndrome, most
patients recover adequate pulmonary
function within 2 to 4 days,42,45 and the ob-
served mortality is less than 6% to 23%
(Holness L, personal communication).
Prevention
If antibody in donor plasma can be shown
to have caused an acute pulmonary reac-
tion, blood from that donor should not be
used for plasma-containing components.
No special precautions are needed for the
patient if the problem was donor-specific
and components from other donors are
available. Current policy in the United
Kingdom is not to prepare plasma from
female donors.
Circulatory Overload
Pathophysiology and Manifestations
Transfusion therapy may cause acute pul-
monary edema due to volume overload,
and this can have severe consequences,
including death. Few data are available on
the incidence rate of transfusion-induced
circulatory overload in the general popu-
lation, but young children and the elderly
are considered most at risk, and incidence
rates of up to 1% have been observed in a
study of elderly orthopedic patients.54
Rapidincreasesinbloodvolumearees
-
pecially poorly tolerated by patients with
compromised cardiac or pulmonary sta-
tus and/or chronic anemia with expanded
648 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
plasma volume. The infusion of 25% albu-
min, which shifts large volumes of extra-
vascular fluid into the vascular space, may
also cause circulatory overload. Hyper-
volemia must be considered if dyspnea,
cyanosis, orthopnea, severe headache, hy-
pertension, or congestive heart failure oc-
curduringorsoonaftertransfusion.Ele
-
vated levels of brain natriuretic peptide
may be seen in cases of circulatory over-
load,55 and this test may be useful in sepa-
rating such cases from cases of TRALI.
Treatment
Symptoms usually improve when the in-
fusion is stopped, and it should not be re-
started until volume overload has been
addressed. Placing the patient in a sitting
position may help. Diuretics and oxygen
are often indicated and, if symptoms are
not relieved, multiple medical interven-
tions may be required, including phlebot-
omy.
Prevention
Except in conditions of ongoing, rapid
blood loss, anemic patients should re-
ceive blood transfusions slowly, with at-
tention to total fluid input and output.
The administration of diuretics before
and during the transfusion may be help-
ful.
Complications of Massive Transfusion
Among the numerous complications that
may accompany massive transfusion,
metabolic and hemostatic abnormalities
are matters of particular concern. Some
or all of the following metabolic derange-
ments can depress left ventricular func-
tion: hypothermia from refrigerated blood,
citrate toxicity, and lactic acidosis from
systemic underperfusion and tissue
ischemia, often complicated by hyper-
kalemia. Metabolic alkylosis due to me-
tabolism of citrate can occur after mas-
sive transfusion but is probably not clini-
cally significant. Patients who are losing
blood rapidly may have pre-existing or
coexisting hemostatic abnormalities or
develop them during resuscitation.
Hemostatic abnormalities may include
dilutional coagulopathy, DIC, and liver
and platelet dysfunction.
Citrate Toxicity
Pathophysiology and Manifestations.
When large volumes of FFP, Whole Blood,
or Platelets are transfused rapidly, partic-
ularlyinthepresenceofliverdisease,
plasma citrate levels may rise, binding
ionized calcium and causing symptoms.
Citrate is rapidly metabolized, however,
so these manifestations are transient.56
Hypocalcemia is more likely to cause clin-
ical manifestations in patients who are in
shock or are hypothermic. Prolonged
apheresis procedures put patients, and
occasionally blood donors, at some risk.
Exchange transfusion, especially in tiny
infants who are already ill, requires care-
ful attention to all electrolytes.
A decrease in ionized calcium increases
neuronal excitability, leading, in the awake
patient or apheresis donor, to symptoms of
perioral and peripheral tingling, shivering,
and lightheadedness, followed by a diffuse
sense of vibration, tetanic symptoms such
as muscle cramps, fasciculations and
spasm, and nausea. In the central nervous
system, hypocalcemia is thought to in-
crease the respiratory center’s sensitivity to
CO2, causing hyperventilation. Because
myocardial contraction is dependent on
the intracellular movement of ionized cal-
cium, hypocalcemia also depresses cardiac
function.57
Treatment and Prevention. Massively
transfused patients, particularly those with
severe liver disease or those undergoing
Chapter 27: Noninfectious Complications of Blood Transfusion 649
Copyright © 2005 by the AABB. All rights reserved.
rapid apheresis procedures such as periph-
eral blood progenitor cell collections, may
benefit from calcium replacement. It should
be noted, however, that empiric replace-
ment therapy in the era before accurate
monitoring of ionized calcium was avail-
able was associated with iatrogenic mortal-
ity.58 Usually, however, unless a patient or
donor has a predisposing condition that
hinders citrate metabolism, hypocalcemia
due to citrate overload requires no treat-
ment other than slowing the infusion. Cal-
cium must never be added directly to the
blood container because the blood will clot.
Hypothermia
Pathophysiology and Manifestations.
Ventricular arrhythmias may occur in pa-
tients who receive rapid infusions of large
volumes of cold blood, and they can be
prevented by blood warming.59 The effect
of cold blood is presumed to be more
likely if the blood is administered via cen-
tral catheters positioned close to the car-
diac conduction system.60 Hypothermia
increases the cardiac toxicity of hypo-
calcemia or hyperkalemia and can result
in poor left ventricular performance.
Other complications of hypothermia in-
clude impaired hemostasis61 and in-
creased susceptibility to wound infec-
tions.62 Blood warming is a must during
massive transfusion of cold blood.
Treatment and Prevention. Hypother-
mia-induced arrhythmias are reduced by
avoiding rapid infusion of cold blood into
the cardiac atrium. Generalized effects of
hypothermia can be prevented by using
blood warmers. AABB Standards for Blood
Banks and Transfusion Services mandates
that warmers have a temperature monitor
and a warning system to detect malfunc-
tion and prevent hemolysis.17(p6) Attention to
proper protocol is critical during the use of
blood warming devices because overheat-
ing destroys red cells and has caused fatali-
ties.5
Hyperkalemia and Hypokalemia
Pathophysiology. When red cells are stored
at 1 to 6 C, the potassium level in the super-
natant plasma or additive solution in-
creases. Although the concentration in the
plasma/anticoagulant portion of an RBC
unit may be high (see Chapter 8), because
of the small volume, the total extracellular
potassium load is less than 0.5 mEq for
fresh units and only 5 to 7 mEq for units at
their outdate. This rarely causes hyperkale-
mic problems in the recipient because
rapid dilution, redistribution into cells, and
excretion blunt the effect. Hypokalemia is
probably more often observed63 because
potassium-depleted red cells reaccumulate
this intracellular ion, and citrate metabo-
lism causes movement of potassium into
the cells in response to the consumption
of protons. Hyperkalemia may be a prob-
lem in patients with renal failure and in
premature infants and newborns receiv-
ing relatively large transfusions, such as in
cardiac surgery or exchange transfusion;
otherwise, it can be demonstrated only as
a transient effect in very rapid transfusion.
Treatment and Prevention. No treat-
ment or preventive strategy is usually nec-
essary, provided the patient is adequately
resuscitated from whatever condition re-
quired the massive transfusion.63 For large-
volume transfusion to sick infants or adults
at risk, many professionals prefer red cells
that are no more than 5 to 14 days old or
washed units. However, for infants receiv-
ing small-volume transfusions infused
slowly, units may be used safely until their
expiration date.64 Thereisnoevidencethat
routine red cell transfusions require manip-
ulation to lower potassium levels, even in
patients with no renal function.
650 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Coagulopathy in Massive Transfusion
Pathophysiology. Of greater concern is the
occurrence of coagulopathy during mas-
sive transfusion. Classically, this coagulo-
pathy is ascribed to dilution of platelets
and clotting factors, which occurs as pa-
tients lose hemostatically active blood. The
lost blood is initially replaced with red
cells and asanguinous fluids. Classic stud-
ies of military65 and civilian66 trauma
patients receiving stored Whole Blood
demonstrated a progressive increase in the
incidence of “microvascular bleeding” (MVB)
characteristic of a coagulopathy with in-
creasing transfusion, typically occurring af-
ter replacement of two to three blood vol-
umes (20 to 30 Whole Blood units). Although
platelet counts, coagulation times, and lev-
els of selected clotting factors all correlated
with volume transfused, contrary to ex-
pectations from a simple dilutional model,
the relationship was marked by tremen-
dous variability. Inspection of laboratory
parameters in the patients developing a
bleeding diathesis, as well as the response
to various hemostatic components, sug-
gested that platelet deficits were more im-
portant in causing the bleeding than were
coagulation factor deficiencies. MVB typi-
cally occurred when the platelet count fell
below 50,000 to 60,000/µL. On the other
hand, no simple relationship could be de-
termined between a patient’s coagulation
tests and the onset of bleeding.
Subsequent studies have refined these
observations. Significant platelet dysfunc-
tion has been demonstrated in massively
transfused trauma patients.67,68 In the stud-
iesofCountsandcoworkers,
66,69 low fibrino-
gen and platelet levels were better pre-
dictors of hemostatic failure than elevations
of prothrombin time (PT) and partial
thromboplastin time (PTT), suggesting that
consumption coagulopathy was an impor-
tant factor in addition to dilution. A similar
conclusion was reached by Harke and
Rahman,70 who showed that the degree of
platelet and clotting abnormalities corre-
lated with the length of time the patient
washypotensive,ingroupsofpatientsre
-
ceiving similar transfusion volumes, also
suggesting that the most important cause
wasDICduetoshock.Takingthesedatato
-
gether, Collins71 concluded that “...coagulo-
pathy in heavily transfused patients was due
to hypoperfusion, not transfusion.”
These data may not be generalizable to
patients undergoing massive transfusion in
the “clean” setting of the operating room,
where hypotension due to volume loss is
prevented. In this setting, coagulation fac-
tor levels may indeed have priority over
platelet problems.72
Treatment and Prevention. The dilu-
tional model of coagulopathy in massive
transfusion would suggest that prophylactic
replacement of hemostatic components
based on the volume of red cells or whole
blood transfused would prevent develop-
ment of a bleeding diathesis. However, pro-
spective studies have consistently shown
that such regimens do not work,73 perhaps
due to patient variability. Instead, replace-
ment of platelets and coagulation factors in
the massively transfused trauma or surgical
patient should be based on characteriza-
tion of the specific abnormality by use of
platelet counts, the PT (international nor-
malized rate), aPTT, and fibrinogen levels.
Thromboelastography may also be useful.
It is imperative that the laboratory rapidly
complete testing. Empiric therapy with
platelets and/or plasma may be initiated
immediately after specimens are obtained.
Air Embolism
Air embolism can occur if blood in an open
system is infused under pressure or if air
enters a central catheter while containers
or blood administration sets are being
Chapter 27: Noninfectious Complications of Blood Transfusion 651
Copyright © 2005 by the AABB. All rights reserved.
changed. It has been reported in associa-
tion with intraoperative and perioperative
blood recovery systems that allow air into
the infusion bag.7The minimum volume
of air embolism that is potentially fatal for
an adult is approximately 100 mL.74 Symp-
toms include cough, dyspnea, chest pain,
and shock.
If air embolism is suspected, the patient
should be placed on the left side with the
head down, to displace the air bubble from
the pulmonic valve. Aspiration of the air is
sometimes attempted. However, proper use
of infusion pumps, equipment for blood re-
covery or apheresis, and tubing couplers is
still essential to prevent this complication.
Evaluation of a Suspected
Acute Transfusion Reaction
The Role of Clinical Personnel Attending
the Patient
Medical personnel attending the patient
are generally the first to suspect that a
transfusion reaction has occurred and the
firsttotakeaction.Theappropriateactions
should be specified in the institution’s pa-
tient care procedures manual, and trans-
fusion service personnel should be pre-
pared to act as consultants.
1. If a transfusion reaction is suspected,
the transfusion should be stopped to
limit the volume of blood infused.
2. All labels, forms, and patient identi-
fication should be checked to deter-
mine whether the transfused compo-
nent was intended for the recipient.
3. An intravenous line should be main-
tained with normal saline (0.9% so-
dium chloride), at least until a medi-
cal evaluation of the patient has been
completed.
4. Thetransfusionserviceandthepa
-
tient’s physician should be notified
immediately. A responsible physician
should evaluate the patient to deter-
mine whether a transfusion reaction
is a possibility, what kind it might be,
and what immediate actions should
be undertaken. The possibilities of acute
hemolytic reaction, anaphylaxis, trans-
fusion-induced sepsis, and TRALI
should be kept in mind because these
conditions require aggressive medical
management and must be reported
promptly to the laboratory.
5. If the observed events are limited to
urticaria or circulatory overload, the
transfusion service need not evalu-
ate postreaction blood samples. If there
are signs and symptoms other than
urticaria or circulatory overload,
particularly if there is any possibility
of acute HTR, anaphylaxis, TRALI,
transfusion-induced sepsis, or other
serious problem, a postreaction blood
sample(s) should be sent to the labo-
ratory for evaluation. The speci-
men(s) must be carefully drawn to
avoid mechanical hemolysis and
must be properly labeled. In addi-
tion, the transfusion container with
whatever contents remain, the ad-
ministration set (without the nee-
dle), and the attached intravenous
solutions should be sent to the labo-
ratory, following standard precau-
tions. In some cases, a postreaction
urine sample will be useful.
The Role of the Laboratory
Whenever hemolysis is a possibility, the
laboratory should perform three steps as
soon as possible after receiving notifica-
tion and the clinical material: check for
clerical errors; perform a visual check for
hemolysis; and check for evidence of blood
group incompatibility by performing a di-
rect antiglobulin test (DAT) and a recon-
652 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
firmation of the recipient’s ABO type. Some
laboratories do not follow this sequence
when the only manifestations are urtica-
rial or febrile reactions to ABO-compati-
ble platelets.
Check for Identification Errors
The identification of each patient’s sam-
ple and the blood component(s) must be
checked for errors. If an error is discov-
ered, the patient’s physician or other re-
sponsible health-care professional must
be notified immediately, and a search of
appropriate records should be initiated to
determine whether misidentification or
incorrect issue of other specimens or
components has put other patients at risk.
Once the acute crisis has passed, each step
of the transfusion process should be re-
viewed to find the source of error.
Visual Check for Hemolysis
The serum or plasma in a postreaction
blood specimen must be inspected for ev-
idence of hemolysis and compared with a
prereaction sample, if available. Pink or
red discoloration after, but not before, the
reaction suggests destruction of red cells
and release of free hemoglobin. Intravas-
cular hemolysis of as little as 2.5 mL of red
cells may produce visible hemoglobinemia.75
Hemolysis resulting from poor collection
technique or other medical interventions
can cause hemoglobinemia; if faulty sam-
pling is suspected, examination of a sec-
ond specimen should resolve the question.
Myoglobin, released from injured muscle,
may also cause pink or red plasma and
might be suspected if a patient has suf-
fered severe trauma or muscle injury.76(p369)
Ifthesampleisnotdrawnuntil5to7
hours after an episode of acute hemolysis,
hemoglobin degradation products, espe-
cially bilirubin, may be in the blood-
stream and cause yellow or brown discol-
oration. An increase in bilirubin may begin
as early as 1 hour after the reaction, peak
at 5 to 7 hours, and disappear within 24
hours if liver function is normal.
In examining a postreaction urine speci-
men, it is important to differentiate among
hematuria (intact red cells in the urine),
hemoglobinuria (free hemoglobin in the
urine), and myoglobinuria (free myoglobin
in the urine). In acute HTRs, free hemoglo-
bin released from damaged cells can cross
the renal glomeruli and enter the urine, but
hematuria and myoglobinuria would not
be expected. Urine examination should be
done on the supernatant fluid after centri-
fugation of a freshly collected specimen;
misinterpretation can occur if free hemo-
globin is released when previously intact
red cells in a specimen undergo in-vitro
hemolysis during transportation or storage.
Serologic Check for Incompatibility
A DAT must be performed on a postreac-
tion specimen, preferably one anticoagu-
lated with a chelating agent (such as EDTA)
to avoid in-vitro coating of red cells by
complement proteins. If the postreaction
DAT is positive, a DAT should be performed
on red cells from the pretransfusion spec-
imen (unless this was already done as part
of pretransfusion testing) and compared.
If transfused incompatible cells have been
coated with antibody but not immediately
destroyed, the postreaction specimen DAT
is likely to be positive, often with a mixed-
field agglutination pattern. If the trans-
fused cells have been rapidly destroyed,
the postreaction DAT may be negative,
particularly if the specimen is drawn sev-
eral hours later. If both the pre- and post-
reaction DATs are positive, further work-
up is required to rule out incompatibility.
Comparison of the graded strength of these
two tests is not a reliable method to rule
this out. Nonimmune hemolysis (eg, from
Chapter 27: Noninfectious Complications of Blood Transfusion 653
Copyright © 2005 by the AABB. All rights reserved.
thermal damage or mechanical trauma)
causes hemoglobinemia but not a posi-
tive DAT. The recipient’s ABO type must
also be confirmed on the postreaction
specimen.
Additional Laboratory Evaluation
If any of the three initial checks and tests
(error check, visual inspection for hemo-
globinemia, DAT and ABO confirmation)
gives positive or suspicious results, the di-
agnosis of an acute HTR should be vigor-
ously pursued. Even if no error or apparent
incompatibility is found, the possibility of
an acute HTR should still be considered if
the patient’s clinical presentation is con-
sistent with such a reaction. The tests
listed below help characterize the cause
of the HTR, if one has occurred, or help
clarify the immunologic and serologic sta-
tusofpatientsinwhomthediagnosisis
unclear. Some or all may be performed
following a written institutional protocol
or at the discretion of the physician in
charge of the transfusion service.
1. If ABO and Rh typing on the prereac-
tion and postreaction samples do not
agree, there has been an error in pa-
tient or sample identification, or in
testing. If sample mix-up or misla-
beling has occurred, another pa-
tient’s specimen may also have been
incorrectly labeled; it is important to
check the records of all specimens
received at approximately the same
time.
2. Perform ABO and Rh testing on blood
from the unit or an attached seg-
ment. If blood in the bag is not of the
ABO type noted on the bag label,
there has been an error in unit label-
ing.
3. Perform antibody detection tests on
the prereaction and postreaction
samples and on the donor blood. If a
previously undetected antibody is
discovered, it should be identified.
Once the antibody has been identi-
fied, retained samples from transfused
donor units should be tested for the
corresponding antigen. If a previ-
ously undiscovered antibody is pres-
ent in a postreaction specimen but
not in a prereaction sample, the rea-
son may be 1) a sample identifica-
tion error, 2) anamnestic antibody
production after a recent transfusion,
or, less likely, 3) passive transfer of
antibody from a recently transfused
component. It may be desirable to use
enhancement techniques, such as an
increased serum-to-cell ratio, low-
ionic-strength saline, Polybrene, poly-
ethylene glycol, or enzyme techni-
ques, when retesting the prereaction
specimen.
4. Repeat crossmatch tests, with pre-
reaction and postreaction samples in
parallel using the antiglobulin tech-
nique. A positive crossmatch in the
face of a negative antibody screen-
ingtestmayindicatethepresenceof
an antibody directed against a low-
incidence blood group antigen.
5. Perform DAT and antibody detection
tests on additional specimens ob-
tained at intervals after the transfu-
sion reaction. A first postreaction
sample may have serologically un-
detectable levels of a significant allo-
antibody, especially if all the anti-
body molecules have attached to the
incompatible transfused cells. In this
event, antibody levels would rise
rapidly, and antibody detection and
identification would become possi-
ble within a few days.
6. Perform frequent checks of the pa-
tient’s hemoglobin values, to see
whether the transfused cells produce
the expected therapeutic rise, or
654 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
whether a decline occurs after an
initial increase. In patients with sickle
cellanemia,thesurvivaloftrans
-
fused red cells can be followed by
evaluation of the levels of hemoglo-
bin A. In complex cases, phenotypic
differences between autologous and
transfused cells quantitated by flow
cytometry have been used to follow
survival.77
7. In-vivo red cell survival studies have
been used to demonstrate the rare
occurrence of an acute HTR in the
absence of detectable alloantibody.78
When the patient is phenotyped in
preparation for such studies, it is im-
portant that the sample be one that
contains only the patient’s red cells.
This may be difficult if the patient
has received transfusions within the
previous several weeks. Method 2.15
gives a technique for obtaining auto-
logous red cells from a patient who
has been transfused. If an antigen is
present on the donor’s red cells and
absent from those of the patient, its
presence or absence in postreaction
samples indicates whether the trans-
fused cells have survived and re-
mained in the circulation.
8. Markers of hemolysis, including lac-
tate dehydrogenase, unconjugated
bilirubin, and haptoglobin levels,
may be useful, particularly if pre-
and multiple postreaction measure-
ments are available.
9. Examine the blood remaining in the
unit and the administration tubing
for evidence of hemolysis, especially
if no immune explanation for hemo-
lysis can be demonstrated. Depending
on how the blood was handled and
administered, hemolysis may be
present in the container and the ad-
ministration tubing, or only in the
administration tubing. For example,
if the unit had been inappropriately
heated in the container, both the
blood in the container and in the
administration tubing would be
hemolyzed. If a faulty infusion de-
vice had been used during blood
administration, hemolysis might be
present in the administration tubing,
but not in the container.
Additional testing may also be useful for
significant nonhemolytic reactions.
1. If the presentation suggests an ana-
phylactic reaction, test the patient’s
serum for the presence of anti-IgA.
Preliminary information can be ob-
tained by quantitation of IgA because
most patients with IgA-related ana-
phylaxis have been IgA deficient.27
Note, however, that subclass- or allo-
type-specific antibodies may de-
velop in patients with normal IgA
levels.26 If additional transfusions are
required, cellular components can
be washed; if plasma (or platelets;
see above) is required, IgA-deficient
plasma can be used.
2. Examine the returned unit for any
abnormal appearance, including clots
or any brownish, opaque, muddy, or
purple discoloration. If the clinical
presentation suggests bacterial sep-
sis, a Gram stain and bacterial cul-
tures of the contents should be per-
formed, even if the unit looks normal.
Blood cultures should also be per-
formed on the patient’s blood.79
Treatment for suspected bacterial
contamination should be based on
clinical considerations because a de-
lay in therapy may result in severe
morbidity or death. Treatment in-
cludes prompt intravenous adminis-
tration of broad-spectrum antibiot-
ics after blood and other appropriate
cultures have been obtained, combined
with therapy for shock, if present.
Chapter 27: Noninfectious Complications of Blood Transfusion 655
Copyright © 2005 by the AABB. All rights reserved.
3. If the clinical presentation suggests
TRALI, test the patient’s pretransfu-
sion sample and a sample of the do-
nor’s plasma for antibodies to HLA and
neutrophil antigens. Crossmatching
recipient lymphocytes or granulocytes
with implicated donor sera can pro-
vide supportive evidence for TRALI.
Delayed Consequences of
Transfusion
Alloimmunization to Red Cell Antigens
Pathophysiology
Primary alloimmunization, evidenced by
the appearance of newly formed antibod-
ies to red cell antigens, becomes apparent
weeks or months after transfusion. It has
been estimated that alloimmunization
occurs in unselected immunocompetent
recipients with a risk of 1% to 1.6% per
RBC unit, provided that D-negative recip-
ients receive D-negative cellular compo-
nents.80 Hemolysis has been reported in
cases of primary immunization, but these
reports are controversial,81 and, even if it
occurs, the phenomenon must be very
rare and usually subclinical.
Serologic Observations. Once allo-
immunization has occurred, blood group
antibodies can become undetectable, espe-
cially those of the Kidd system. One investi-
gator reported that this occurred to 29% of
antibodies after a median of 10 months82
and to 41% of antibodies after 5 or more
years.83 If red cells that express the antigen
are subsequently transfused, however, an
anamnestic response may cause the ap-
pearance, within hours or days, of IgG anti-
bodies that react with the transfused red
cells. In a prospective study, previously un-
detected alloantibodies were found in 58 of
2082 (2.8%) recipients (37% known previ-
ously transfused, 36% previously pregnant)
within 7 days of transfusion.84 In two of the
58, only the DAT was positive, but, in all
others, repeat antibody screening would
have detected the new antibody. Regard-
less, characterization of an eluate is neces-
sary because alloantibodies may be present
on the red cells that are not in the serum. If
the clinical laboratory discovers an anam-
nestic response, both the transfusion ser-
vice director and the patient’s clinician
should be notified and the possibility of a
delayed HTR (DHTR) should be investigated.
Delayed Reactions. In most cases, ana-
mnestic antibody production does not cause
detectable hemolysis, leading to the desig-
nation “delayed serologic transfusion reac-
tion” (DSTR).85 However, in some patients,
clinically apparent hemolysis will result
from the combination of significant levels
of antibody with hemolytic potential and
large numbers of transfused red cells in the
circulation; in the study cited above, only
one of the 58 recipients with a new anti-
body within 7 days of transfusion had clini-
cally evident hemolysis.84 This translated
into a DHTR rate of one per 2082 recipients,
or one for every 11,328 units transfused. As
would be expected, retrospective studies,
which would be similar to the routine expe-
rience of a transfusion service, yield a lower
rate of DSTRs, but the rate of clinically de-
tectable hemolysis may be roughly equiva-
lent.85-87 Themostcommonpresentationof
a DHTR is a declining hemoglobin and a
newly positive antibody screen, but fever,
leukocytosis, and mild jaundice may be
present. Some DHTRs present as the ab-
sence of the anticipated increase in hemo-
globin after transfusion. Other clinical
problems are infrequent; hemoglobinuria is
occasionally noted, but acute renal failure
is uncommon. However, DHTRs may be
particularly problematic in patients with
sickle cell disease. In these patients,
hemolysis may include autologous red
cells, a phenomenon termed sickle cell
656 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
hemolytic transfusion reaction syndrome
(see Chapter 24).88
If a DHTR is suspected, a freshly ob-
tained blood sample may be tested for un-
expected alloantibodies, both in the serum
and, by DAT, on the red cells. Discovery of a
new red cell alloantibody in a recently
transfused patient with hemolysis strongly
suggests a DHTR, and the diagnosis is sup-
ported by demonstration of the corre-
sponding antigen on the red cells from a
retained segment from one or more trans-
fused units. Antigen typing of the red cells
circulating in the patient may also suggest
whether the newly incompatible cells have
been eliminated, or whether some are still
circulating. Repeat antibody screening on
the patient’s previous specimen will rule
out technical errors.
Treatment
Specific treatment is rarely necessary, al-
though it may be prudent to monitor the
patient’s urine output and renal function
and observe for changes in coagulation
function. If transfusion is still necessary,
donor red cells should lack the antigen
corresponding to the newly discovered
antibody. Passenger lymphocyte hemolysis,
seen after solid organ transplantation, is a
variant of DHTR and is covered in Chap-
ter 26.
Prevention
Future transfusions for the patient should
lack the antigen(s) responsible for the
anamnestic response, even if the antibody
again becomes undetectable. Some facili-
ties issue a medical alert card with this in-
formation for the patient to carry and
present at the time of hospitalization or
transfusion in a different facility. It is to
prevent these problems that Standards for
Blood Banks and Transfusion Services
mandates permanent preservation of re-
cords of clinically significant antibodies,
and review of previous records before red
cells are issued for transfusion.17(pp39,72) Pro-
spective antigen matching may prevent
DHTRs in selected patients, particularly
those with sickle cell disease (see Chap-
ters 21 and 24).89
Posttransfusion Autoantibody
Occasionally, transfusion of allogeneic red
cells and platelets stimulates production
of autoantibodies; in some of these pa-
tients, hemolytic anemia or thrombocyto-
penia may occur.90 See Chapter 20 for more
details.
Alloimmunization to Leukocyte Antigens
and Refractoriness to Platelet
Transfusions
See Chapter 16 and Chapter 17.
Transfusion-Associated Graft-vs-Host
Disease
Transfusion-associated graft-vs-host dis-
ease is a usually fatal immunologic trans-
fusion complication caused by engraft-
ment and proliferation of donor lympho-
cytes in a susceptible host.91 The engrafted
lymphocytes mount an immunologic at-
tack against the recipient tissues, includ-
ing hematopoietic cells, leading to refrac-
tory pancytopenia with bleeding and
infectious complications, which are pri-
marily responsible for the 90% to 100%
mortality rate in afflicted patients. TA-
GVHD is rare in US transfusion recipients
and has been observed almost exclusively
in immunocompromised patients. In con-
trast, over 200 cases of TA-GVHD have
been described in Japan,92 with incidence
rates reaching 1:660 in patients undergo-
ing cardiovascular surgery.93 Greater ge-
netic homogeneity of the Japanese popu-
lation and frequent use of fresh Whole
Chapter 27: Noninfectious Complications of Blood Transfusion 657
Copyright © 2005 by the AABB. All rights reserved.
Blood from related donors are thought to
be the primary reasons for the surpris-
ingly frequent occurrence of TA-GVHD in
that country. The SHOT data14 demon-
strate a significant decline in the inci-
dence of TA-GVHD since the introduction
of universal leukocyte reduction, but two
cases occurred despite leukocyte reduction.
In the first 3 years of this study, the rate of
TA-GVHD was approximately 1 per 600,000
cellular components transfused and, in
the first 5 years, accounted for a greater
number of deaths than acute HTRs and
only slightly fewer than did TRALI.
Pathophysiology and Manifestations
The pathophysiology of TA-GVHD is com-
plex and incompletely understood. The
overall mechanism includes the escape of
donor T lymphocytes present in cellular
blood components from immune clearance
in the recipient and subsequent prolifera-
tion of these cells, which then mount an
immune attack on host tissues. Manifes-
tations include fever, enterocolitis, rash,
hepatitis, and pancytopenia. The rash
typically begins as a blanching, macu-
lopapular erythema of the upper trunk,
neck,palms,soles,andearlobes,which
becomes confluent with additional find-
ings ranging from edema to widespread
blistering. Skin biopsy reveals infiltration
of the upper dermis by mononuclear cells
and damage to the basal layer of epithe-
lial cells. Hepatitis manifests as elevations
in alanine and aspartate aminotrans-
ferases, alkaline phosphatase, and biliru-
bin. Enterocolitis causes anorexia, nausea,
and up to 3 to 4 liters per day of secretory
diarrhea. Pancytopenia is associated with
a hypocellular marrow. Symptoms typi-
cally appear within 8 to 10 days of the
transfusion but may occur as early as 3
days and as late as 30 days. The diagnosis
is proven by demonstration of donor-de-
rived lymphocytes in the recipient’s pe-
ripheral blood or tissues by HLA typing.91
Factors that determine an individual pa-
tient’s risk for TA-GVHD include whether and
to what degree the recipient is immunode-
ficient, the degree of HLA similarity be-
tween donor and recipient, and the num-
ber and type of T lymphocytes transfused
that are capable of multiplication.91 TA-GVHD
may occur in an immunologically normal
recipient if the donor is homozygous for an
HLA haplotype for which the recipient is
heterozygous, a so-called “one-way” HLA
match, and if the component contains via-
ble T cells (the fresher the unit, the higher
the risk). Cytokine dysfunction, recruitment
of host cells into the immune reaction, and
release of biologic mediators, in particular
nitric oxide, all play a role in the pathogene-
sis.94 Of interest is the fact that TA-GVHD
has not been reported in an AIDS patient.
Treatment and Prevention
Treatment of TA-GVHD with immunosup-
pressive agents has been attempted but
rarely succeeds, so prevention is neces-
sary. Irradiation of cellular blood compo-
nents is the accepted standard method to
prevent TA-GVHD. The dose mandated by
the FDA is a minimum of 25 Gy targeted
to the midline of the container and a min-
imum dose of 15 Gy delivered to all other
parts of the component.95 This renders T
lymphocytes incapable of replication
without substantially affecting the function
of red cells, platelets, and granulocytes.
AABB Standards for Blood Banks and
Transfusion Services requires routine irradi-
ation of cellular components from units
collected from the recipient’s blood rela-
tives, and donors selected for HLA compat-
ibility by typing or crossmatching.17(p43) Poli-
cies should be in place to define the other
groups of patients who should receive irra-
diated cellular components, and there must
658 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
be a process for ensuring that once a pa-
tient has been determined to be at risk for
TA-GVHD, all cellular components will be irra-
diated as long as clinically indicated.
Published guidelines96 additionally rec-
ommend component irradiation for: 1)
hematopoietic progenitor cell (HPC) trans-
plant recipients (this includes allogeneic
and autologous HPC transplants), 2) pa-
tients with hematologic disorders who will
be undergoing allogeneic HPC transplanta-
tion imminently, 3) intrauterine transfu-
sions, 4) neonates undergoing exchange
transfusion or use of extracorporeal mem-
brane oxygenation, 5) patients with Hodg-
kin’s disease, and 6) patients with congeni-
tal cellular immunodeficiencies. TA-GVHD
has also been reported in patients with
acute lymphoid and myeloid leukemias,
chronic lymphocytic leukemia particularly
in patients receiving fludarabine phos-
phate,91 patients with B-cell malignancies
including non-Hodgkin’s lymphoma,
myeloma, and Waldenstrom’s macroglobu-
linemia,14 premature or low-birthweight in-
fants without specific immunodeficiency
disorders, and children being treated for
neuroblastoma and rhabdomyosarcomas.91
Posttransfusion Purpura
Pathophysiology and Manifestations
Posttransfusion purpura (PTP) is an un-
common event, although over 200 cases
have been published. It is characterized by
the abrupt onset of severe thrombocyto-
penia (platelet count usually <10,000/µL)
an average of 9 days after transfusion
(range, 1-24 days).97 Components provok-
ing the reaction have usually been RBCs
or Whole Blood, but PTP has also been
reported after platelet and plasma trans-
fusion, and after transfusion of frozen
deglycerolized RBCs. Most patients have
previously been pregnant or transfused.
“Wet purpura” is common, and fatal intra-
cranial hemorrhage can occur. The ratio
of affected patients is five women to one
man, and the median age is 51 years
(range, 16-83). Most cases (68%) involve
patients whose platelets lack the HPA-1a
(PlA1) antigen (<2% of the population) and
who form the corresponding antibody.
However, immunization to HPA-1b is re-
ported in 10%, and other platelet antibod-
ies, including HLA antibodies, have been
associated with the syndrome as well. PTP
is usually self-limited, with full recovery
within 21 days. Historically, 10% to 15% of
patients have been reported to die from
PTP, typically from intracranial bleeding,
so treatment is desirable.
The reason for destruction of the pa-
tient’s own platelets by what appears to be
a platelet alloantibody is controversial.
Three mechanisms have been proposed,
including: 1) formation of immune com-
plexes of patient antibody and soluble do-
nor antigen that bind to Fc receptors on the
patient’s platelets and mediate their de-
struction, 2) conversion of antigen-negative
autologous platelets to antibody targets by
soluble antigen in the transfused compo-
nent, and 3) cross-reactivity of the patient’s
antibodies with autologous platelets (ie, the
presence of an autoantibody component).
The last of these theories has received the
most support.
Treatment
Because PTP remits spontaneously, treat-
ment may appear falsely efficacious. Ste-
roids are frequently given but their role is
controversial. Plasma exchange can achieve
platelet counts of 20,000/µLin1to2days,
98
but the use of high-dose Immune Globu-
lin Intravenous (IGIV) is now supplanting
this therapy.98,99 With the use of IGIV, re-
covery to platelet counts of 100,000/µLis
typically achieved within 3 to 5 days. As it
does in other disorders such as immune
Chapter 27: Noninfectious Complications of Blood Transfusion 659
Copyright © 2005 by the AABB. All rights reserved.
thrombocytopenic purpura, IGIV appears
to block antibody-mediated clearance of
the target cells, although the mechanism
of action has not been established. If ran-
domly selected platelets are transfused,
patients may experience a febrile transfu-
sion reaction, and, in the vast majority of
cases, such transfusions have not been ef-
ficacious. Antigen-negative platelets can
be of benefit in PTP, and, in conjunction
with IGIV, reversal of this disorder in 1 day
is now possible.100,101 Unfortunately, the
time necessary to procure such platelets
often limits their usefulness. After recov-
ery, some authors have suggested that fu-
ture transfusions should be from donors
lacking the offending antigen, or, if they
are not available, washed platelet units.97
Immunomodulatory Effects of Transfusion
Transfusion has been known to modulate
immune responses since the 1973 obser-
vation by Opelz and coworkers102 of im-
provedrenalallograftsurvivalintrans-
fused patients. This beneficial tolerance-
inducing effect of transfusion raised con-
cerns that transfusion may have other ad-
verse effects in different clinical settings,
including increased rates of postoperative
solid tumor recurrence and bacterial in-
fection.103 Despite numerous retrospective
and several large prospective studies, the
clinical significance of transfusion-associ-
ated immunomodulation and the useful-
ness of preventive strategies, such as leu-
kocyte reduction of transfused components,
remain controversial.104,105
Iron Overload
Every RBC unit contains approximately
200 mg of iron. Chronically transfused
patients, especially those with hemoglo-
binopathies, have progressive and contin-
uous accumulation of iron and no phy-
siologic means of excreting it. Storage oc-
curs initially in reticuloendothelial sites,
but when they are saturated, there is de-
position in parenchymal cells. The
threshold for clinical damage is lifetime
exposure to greater than 50 to 100 RBC
units in a nonbleeding person.106 Iron de-
position interferes with function of the
heart, liver, and endocrine glands (eg,
pancreatic islets, pituitary); hepatic fail-
ure and cancer, diabetes mellitus, and car-
diac toxicity cause most of the morbidity
and mortality. Elevated ferritin levels
demonstrate increased iron stores, and tis-
sue damage can be shown with organ-spe-
cific assays such as liver enzyme levels or
endocrine function tests (eg, glucose, thy-
roid-stimulating hormone).
Treatment is directed at removing iron
without reducing the patient’s circulating
hemoglobin. Metered subcutaneous infu-
sion of desferoxamine, an iron-chelating
agent, can reduce body iron stores in such
patients, but the regimen of nightly subcu-
taneous infusion by pump is arduous and
expensive, and compliance is often poor. In
transfusion-dependent patients with hemo-
globinopathies, red cell exchange can mini-
mize additional iron loads and can reduce
the total iron burden.107 An oral iron che-
lator has been studied but is not yet avail-
able in the United States.
Records of Transfusion
Complications
Each transfusion service must maintain
indefinitely the records of patients who
have had transfusion complications or ev-
idence of alloimmunization. Possible cases
of contaminated blood must be reported
to the institution where the blood was drawn.
Records must be kept, and consulted, to
prevent patients who have had a transfu-
sion reaction from having a recurrence with
660 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
subsequent transfusions. For example, pa-
tients with a history of IgA-related anaphy-
lactic reactions should be transfused with
plasma products that lack IgA. A history of
repeated or severe FNHTRs might prompt
the use of leukocyte-reduced cellular blood
components. Red cell alloantibodies may
become undetectable over time as dis-
cussed above,82,83 so records should be
checked and compatible blood issued in
order to prevent a DHTR. Routine checking
of previous results of ABO and Rh testing
may disclose an error in testing or in the
identification of a current sample.
Records of Patients with Special Needs
In addition to records of transfusion reac-
tions, transfusion services should maintain
records of patients who need specially
prepared or manipulated components.
This is especially important in institu-
tions where physicians rotate frequently,
and the need for irradiated, leukocyte-re-
duced, or IgA-deficient components may
not be known to a particular physician
writing an individual order.
Reporting Transfusion Fatalities
When a complication of blood transfusion
has been confirmed to be fatal, it must be
reported to the Director, Office of Compli-
ance, Center for Biologics Evaluation and
Research, FDA, as soon as possible, with a
written report within 7 days (see Chapter
28 for reporting information). Patients
who are critically ill and near death often
receive transfusions in close temporal
proximity to death, and clinical suspicion
of cause and effect may occasionally be
raised. The overwhelming majority of
such deaths are unrelated to transfusion,
but if there is a suggestion that a transfu-
sion might have contributed to death, it
may be prudent to pursue an investiga-
tion.
In the absence of such errors as adminis-
tration of ABO-incompatible blood or of
physiologic events clearly attributable to
acutehemolysis,anaphylaxis,TRALI,or
sepsis, transfusion is highly unlikely to be
acutely responsible for death. The review
should include all available medical and
laboratory records and the results of an au-
topsy, if performed. On the other hand, if
an investigation does reveal evidence or the
possibility of hemolysis, anaphylactic or
pulmonary events, unexplained sepsis, or
ambiguous identification records, the case
may warrant more extensive inquiry.
References
1. Popovsky MA, ed. Transfusion reactions. 2nd
ed. Bethesda, MD: AABB Press, 2001.
2. Heddle NM, Kelton JG. Febrile nonhemolytic
transfusion reactions. In: Popovsky MA, ed.
Transfusion reactions. 2nd ed. Bethesda, MD:
AABB Press, 2001:45-82.
3. Davenport RD. Hemolytic transfusion reac-
tions. In: Popovsky MA, ed. Transfusion reac-
tions. 2nd ed. Bethesda, MD: AABB Press, 2001:
1-44.
4. Capon SM, Goldfinger D. Acute hemolytic
transfusion reaction, a paradigm of the sys-
temic inflammatory response: New insights
into pathophysiology and treatment. Trans-
fusion 1995;35:513-20 [Erratum in Transfusion
1995;35:794].
5. Sazama K. Report of 355 transfusion-associ-
ated deaths: 1976-1985. Transfusion 1990;30:
583-90.
6. Del Greco F, Kurtides ES. Kell incompatibility
with acute renal failure. Arch Intern Med 1963;
112:727-30.
7. LindenJV,WagnerK,VoytovichAE,Sheehan
J.TransfusionerrorsinNewYorkState:An
analysis of 10 years’ experience. Transfusion
2000;40:1207-13.
8. Jandl JH. Blood: Textbook of hematology.
2nd ed. Boston: Little, Brown & Co, 1996.
9. Davenport RD, Kunkel SL. Cytokine roles in
hemolytic and non-hemolytic transfusion
reactions. Transfus Med Rev 1994;8:157-68.
10. Davenport RD. Inflammatory cytokines in
hemolytic transfusion reactions. In: Daven-
port RD, Snyder EL, eds. Cytokines in trans-
fusionmedicine:Aprimer.Bethesda,MD:
AABB Press, 1996:85-97.
Chapter 27: Noninfectious Complications of Blood Transfusion 661
Copyright © 2005 by the AABB. All rights reserved.
11. Butler J, Parker D, Pillai R, et al. Systemic re-
lease of neutrophil elastase and tumour ne-
crosis factor alpha following ABO incompati-
ble blood transfusion. Br J Haematol 1991;79:
525-6.
12. Savitsky JP, Doczi J, Black J, Arnold JD. A clini-
cal safety trial of stroma-free hemoglobin.
Clin Pharmacol Ther 1978;23:73-80.
13. Simon T. Proficiency testing program. CAP
Survey 1991 J-C. Northfield, IL: College of
American Pathologists, 1991.
14. Asher D, Atterbury CLJ, Chapman C, et al. Se-
rious Hazards of Transfusion (SHOT). Annual
Report, 2000-2001. Manchester, UK: Serious
Hazards of Transfusion Steering Group, 2002.
15. Marik PE. Low-dose dopamine: A systematic
review. Intensive Care Med 2002;28:877-83.
16. Beauregard P, Blajchman MA. Hemolytic and
pseudo-hemolytic transfusion reactions: An
overview of the hemolytic transfusion reac-
tions and the clinical conditions that mimic
them. Transfus Med Rev 1994;8:184-99.
17. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
18. Blajchman MA. Transfusion-associated bac-
terial sepsis: The phoenix rises yet again.
Transfusion 1994;34:940-2.
19. Heddle NM, Blajchman MA, Meyer RM, et al.
A randomized controlled trial comparing the
frequency of acute reactions to plasma-reduced
platelets and prestorage WBC-reduced plate-
lets. Transfusion 2002;42:556-66.
20. Ferrara JLM. The febrile platelet transfusion
reaction: A cytokine shower. Transfusion 1995;
35:89-90.
21. Davenport RD, Burdick M, Moore SA, Kunkel
SL. Cytokine production in IgG-mediated red
cell incompatibility. Transfusion 1993;33:19-
24.
22. Brand A. Passenger leukocytes, cytokines, and
transfusion reactions. N Engl J Med 1994;331:
670-1.
23. WidmannFK.Controversiesintransfusion
medicine: Should a febrile transfusion re-
sponse occasion the return of the blood com-
ponent to the blood bank? Pro. Transfusion
1994;34:356-8.
24. ObermanHA.Controversiesintransfusion
medicine: Should a febrile transfusion re-
sponse occasion the return of the blood com-
ponent to the blood bank? Con. Transfusion
1994;34:353-5.
25. Wang SE, Lara PN, Lee-Ow A, et al. Acetamino-
phen and diphenhydramine as premedi-
cation for platelet transfusions: A prospective
randomized double-blind placebo-con-
trolled trial. Am J Hematol 2001;70:191-4.
26. Vamvakas EC, Pineda AA. Allergic and ana-
phylactic reactions. In: Popovsky MA, ed.
Transfusion reactions. 2nd ed. Bethesda, MD:
AABB Press, 2001:83-128.
27. Sandler SG, Mallory D, Malamut D, Eckrich R.
IgA anaphylactic transfusion reactions. Trans-
fus Med Rev 1995;9:1-8.
28. Burks AW, Sampson HA, Buckley RH. Ana-
phylactic reactions after gamma globulin ad-
ministration in patients with hypogamma-
globulinemia. N Engl J Med 1986;314:560-4.
29. Lambin P, LePennec PY, Hauptmann G, et al.
Adverse transfusion reactions associated with
a precipitating anti-C4 antibody of anti-
Rodgers specificity. Vox Sang 1984;47:242-9.
30. WesthoffCM,SipherdBD,WylieDE,Toalson
LD. Severe anaphylactic reactions following
transfusions of platelets to a patient with
anti-Ch. Transfusion 1992;32:576-9.
31. Shimada E, Tadokoro K, Watamabe Y, et al.
Anaphylactic transfusion reactions in hapto-
globin-deficient patients with IgE and IgG
haptoglobin antibodies. Transfusion 2002;42:
766-73.
32. Leitman SF, Boltansky H, Alter HJ, et al. Aller-
gic reactions in healthy plateletpheresis do-
nors caused by sensitization to ethylene ox-
ide gas. N Engl J Med 1986;315:1192-6.
33. Ramirez MA. Horse asthma following blood
transfusion; report of case. JAMA 1919;73:985.
34. Routledge RC, De Kretser DMH, Wadsworth
LD. Severe anaphylaxis due to passive sensiti-
zation to donor’s blood. Br Med J 1976;1:434.
35. Owen HG, Brecher ME. Atypical reactions as-
sociated with use of angiotensin-converting
enzyme inhibitors and apheresis. Transfusion
1994;34:891-4.
36. AlvingBM,HojimaY,PisanoJJ,etal.Hypo
-
tension associated with pre-kallikrein activa-
tor (Hageman factor fragments) in plasma
protein fraction. N Engl J Med 1978;299:66-70.
37. Shiba M, Tadokoro K, Sawanobori M, et al.
Activation of the contact system by filtration
of platelet concentrates with a negatively
charged white cell-removal filter and mea-
surement of venous blood bradykinin level in
patients who received filtered platelets.
Transfusion 1997;37:457-62.
38. Hume HA, Popovsky MA, Benson K, et al. Hypo-
tensive reactions: A previously uncharac-
terized complication of platelet transfusion?
Transfusion 1996;36:904-9.
39. Mair B, Leparc GF. Hypotensive reactions as-
sociated with platelet transfusions and an-
giotensin-converting enzyme inhibitors. Vox
Sang 1998;74:21-30.
40. Agostini JV, Leo-Summers LS, Inonye SK.
Cognitive and other adverse effects of
662 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
diphenhydramine use in hospitalized older
patients. Arch Intern Med 2001;161:2091-7.
41. Pineda AA, Zylstra VW, Clare DE, et al. Viabil-
ity and functional integrity of washed plate-
lets. Transfusion 1989;29:524-7.
42. Popovsky MA. Transfusion-related acute lung
injury (TRALI). In: Popovsky MA, ed. Trans-
fusion reactions. 2nd ed. Bethesda, MD: AABB
Press, 2001:155-70.
43. Zoon KC. Transfusion related acute lung in-
jury (“Dear Colleague” letter). (October 19,
2001) Rockville, MD: CBER Office of Com-
munication, Training, and Manufacturers
Assistance, 2001.
44. Kleinman S, Caulfield T, Chan P, et al. Toward
an understanding of transfusion-related acute
lung injury: Statement of a consensus panel.
Transfusion 2004;44:1774-89.
45. Popovsky MA, Moore SB. Diagnostic and patho-
genetic considerations in transfusion-re-
lated acute lung injury. Transfusion 1985;25:
573-7.
46. Popovsky MA, Haley NR. Further character-
ization of transfusion-related acute lung in-
jury: Demographics, clinical and laboratory
features, and morbidity. Immunohematology
2000;16:157-9.
47. Kao GS, Wood IG, Dorfman DM, et al. Inves-
tigations into the role of anti-HLA class II
antibodies in TRALI. Transfusion 2003;43:
185-91.
48. Kopko PM, Paglieroni TG, Popovsky MA, et
al. TRALI: Correlation of antigen-antibody and
monocytes activation in donor-recipient pairs.
Transfusion 2003;43:177-84.
49. Seeger W, Schneider U, Kreusler B, et al. Re-
production of transfusion-related acute lung
injury in an ex vivo lung model. Blood 1990;
76:1438-44.
50. Dry SM, Bechard KM, Milford EL, et al. The
pathology of transfusion-related acute lung
injury. Am J Clin Pathol 1999;112:216-21.
51. McCullough J. Granulocyte transfusion. In:
PetzLD,SwisherSN,KleinmanS,etal,eds.
Clinical practice of transfusion medicine. 3rd
ed. New York: Churchill Livingstone, 1996:413-
32.
52. Silliman CC, Paterson AJ, Dickey WO, et al.
The association of biologically active lipids
with the development of transfusion-related
acute lung injury: A retrospective study.
Transfusion 1997;37:719-26.
53. Silliman CC, Boshkov LK, Mehdizadehkashi
Z, et al. Transfusion-related acute lung injury:
Epidemiology and a prospective analysis of
etiologic factors. Blood 2003;101:454-62.
54. Audet AM, Popovsky MA, Andrzejewski C.
Transfusion-associated circulatory overload
in orthopedic surgery patients: A multi-insti-
tutional study. Immunohematology 1996;12:
87-9.
55. Bowman RJ, Laudi N, Aslan D, Burgher A. Use
of BNP to evaluate TRALI (abstract). Transfu-
sion 2004;44(Suppl):24A.
56. Dzik WH, Kirkley SA. Citrate toxicity during
massive blood transfusion. Transfus Med Rev
1988;2:76-94.
57. Olinger GN, Hottenrott C, Mulder DG, et al.
Acute clinical hypocalcemic myocardial de-
pression during rapid blood transfusion and
postoperative hemodialysis: A preventable
complication. J Thorac Cardiovasc Surg 1976;
72:503-11.
58. Howland WS, Jacobs RG, Goulet AH. An eval-
uation of calcium administration during rapid
blood replacement. Anesth Analg 1960;39:
557-63.
59. Boyan CP, Howland WS. Cardiac arrest and
temperature of bank blood. JAMA 1963;183:
58-60.
60. Iserson KV, Huestis DW. Blood warming: Cur-
rent applications and techniques. Transfusion
1991;31:558-71.
61. ValeriCR,FeingoldH,CassidyG,etal.Hypo-
thermia-induced reversible platelet dysfunc-
tion. Ann Surg 1987;205:175-81.
62. Sessler DI. Current concepts: Mild periopera-
tive hypothermia. N Engl J Med 1997;336:
1730-7.
63. Collins JA. Problems associated with the
massive transfusion of stored blood. Surgery
1974;174:274-95.
64. Liu EA, Manino FL, Lane TA. Prospective,
randomized trial of the safety and efficacy of
a limited donor exposure transfusion pro-
gram for premature neonates. J Pediatr 1994;
125:92-6.
65. Miller RD, Robbins TO, Tong MJ, Barton SL.
Coagulation defects associated with massive
blood transfusion. Ann Surg 1971;174:794-801.
66. CountsRB,HaischC,SimonTL,etal.Hemo
-
stasis in massively transfused trauma patients.
Ann Surg 1979;190:91-9.
67. Lim RC, Olcott C, Robinson AJ, Blaisdell FW.
Platelet response and coagulation changes
following massive blood replacement. J
Trauma 1973;13:577-82.
68. Harrigan C, Lucas CE, Ledgerwood KAM, et
al. Serial changes in primary hemostasis af-
ter massive transfusion. Surgery 1985;98:
836-43.
69. Ciavarella D, Reed RL, Counts RB, et al. Clot-
ting factor levels and the risk of microvascu-
lar bleeding in the massively transfused pa-
tient. Br J Haematol 1987;67:365-8.
70. Harke H, Rahman S. Haemostatic disorders
in massive transfusion. Bibl Haematol 1980;
46:179-88.
Chapter 27: Noninfectious Complications of Blood Transfusion 663
Copyright © 2005 by the AABB. All rights reserved.
71. Collins JA. Recent developments in the area
of massive transfusion. World J Surg 1987;11:
75-81.
72. MurrayDJ,PennellBJ,WeinsteinSL,Olson
JD. Packed red cells in acute blood loss: Dilu-
tional coagulopathy as a cause of surgical
bleeding. Anesth Analg 1995;80:336-42.
73. Reed RL, Ciavarella D, Heimbach DM, et al.
Prophylactic platelet administration during
massive transfusion: A prospective, random-
ized, double-blind clinical study. Ann Surg 1986;
203:40-8.
74. O’QuinRJ,LakshminarayanS.Venousair
embolism. Arch Intern Med 1982;142:2173-6.
75. Elliott K, Sanders J, Brecher, ME. Transfusion
medicine illustrated. Visualizing the hemoly-
tic transfusion reaction. Transfusion 2003;43:
297.
76. Henry JB. Clinical diagnosis and management
by laboratory methods. 20th ed. Philadel-
phia: WB Saunders, 2001.
77. Nance ST. Flow cytometry in transfusion
medicine. In: Anderson KC, Ness PM, eds.
Scientific basis of transfusion medicine. Phil-
adelphia: WB Saunders, 1994:707-25.
78. Baldwin ML, Barrasso C, Ness PM, Garratty
G. A clinically significant erythrocyte antibody
detectable only by 51Cr survival studies.
Transfusion 1983;23:40-4.
79. Sazama K. Bacteria in blood for transfusion:
A review. Arch Pathol Lab Med 1994;118:350-
65.
80. Lostumbo MM, Holland PV, Schmidt PJ. Iso-
immunization after multiple transfusions. N
Engl J Med 1966;275:141-4.
81. Issitt PD, Anstee DJ. Applied blood group se-
rology. 4th ed. Durham, NC: Montgomery
Scientific Publications, 1998.
82. Ramsey G, Larson P. Loss of red cell antibod-
ies over time. Transfusion 1988;28:162-5.
83. Ramsey G, Smietana SJ. Long-term follow-up
testing of red cell alloantibodies. Transfusion
1994;34:122-4.
84. Heddle NM, Soutar RL, O’Hoski PL, et al. A
prospective study to determine the frequency
and clinical significance of alloimmunization
post-transfusion. Br J Haematol 1995;91:
1000-5.
85. Ness PM, Shirey RS, Thoman SK, Buck SA.
The differentiation of delayed serologic and
delayed hemolytic transfusion reactions: In-
cidence, long-term serologic findings, and
clinical significance. Transfusion 1990;30:
688-93.
86. Pinkerton PH, Coovadia AS, Goldstein J. Fre-
quency of delayed haemolytic transfusion re-
actions following antibody screening and im-
mediate-spin crossmatching. Transfusion 1992;
32:814-7.
87. Vamvakas EC, Pineda AA, Reisner R, et al. The
differentiation of delayed hemolytic and se-
rologic transfusion reactions: Incidence and
predictors of hemolysis. Transfusion 1995;35:
26-32.
88. Garratty G. Severe reactions associated with
transfusions of patients with sickle cell dis-
ease. Transfusion 1997;37:357-61.
89. Vichinsky EP, Luban NL, Wright E, et al. Pro-
spective RBC phenotype matching in a
stroke-prevention trial in sickle cell anemia:
A multicenter transfusion trial. Transfusion 2001;
41:1086-92.
90. Petz LD, Garratty G. Immune hemolytic
anemias. 2nd ed. Philadelphia: Churchill
Livingstone, 2004:335-40.
91. Webb IJ, Anderson KC. Transfusion-associated
graft-vs-host disease. In: Popovsky MA, ed.
Transfusion reactions. 2nd ed. Bethesda, MD:
AABB Press, 2001:171-86.
92. Ohto H, Anderson KC. Survey of transfusion-
associated graft-versus-host disease in im-
munocompetent recipients. Transfus Med
Rev 1996;10:31-43.
93. Juji T, Takahashi K, Shibata Y. Post-transfu-
sion graft versus host disease as a result of di-
rected donations from relatives (letter). N
Engl J Med 1989;321:56.
94. Ferrara JL, Krenger W. Graft-vs-host disease:
The influence of type 1 and type 2 cell cyto-
kines. Transfus Med Rev 1998;12:1-17.
95. Food and Drug Administration. Memorandum.
Recommendations regarding license amend-
ments and procedures for gamma irradiation
of blood products. (July 22, 1993) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 1993.
96. Przepiorka D, LeParc GF, Stovall MA, et al. Use
of irradiated blood components. Practice pa-
rameter. Am J Clin Pathol 1996;106:6-11.
97. McFarland JG. Postransfusion purpura. In:
Popovsky MA, ed. Transfusion reactions. 2nd
ed. Bethesda, MD: AABB Press, 2001:187-212.
98. McLeod BC, Strauss RG, Ciavarella D, et al.
Management of hematological disorders and
cancers. J Clin Apheresis 1996;11:211-30.
99. Mueller-Eckhardt C, Kiefel V. High-dose IgG
for post-transfusion purpura revisited. Blut 1988;
57:163-7.
100. Brecher ME, Moore SB, Letendre L. Post-
transfusion purpura: The therapeutic value of
PlA1-negative platelets. Transfusion 1990;30:
433-5.
101. Win N, Matthey F, Slater NGP. Blood compo-
nents—Transfusion support in post-transfu-
sion purpura due to HPA-1a immunization.
Vox Sang 1996;71:191-3.
102. Opelz G, Senger DP, Mickey MR, Terasaki PI.
Effect of blood transfusions on subsequent
664 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
kidney transplants. Transplant Proc 1973;5:
253-9.
103. Blumberg N, Heal JM. Effects of transfusion
on immune function: Cancer recurrence and
infection. Arch Pathol Lab Med 1994;118:
371-9.
104. Blajchman MA. Allogeneic blood transfusions,
immunomodulation, and postoperative bac-
terial infection: Do we have the answers yet?
Transfusion 1997;37:121-5.
105. Vamvakas EC, Blajchman MA, eds. Immuno-
modulatory effects of blood transfusion.
Bethesda, MD: AABB Press, 1999.
106. Sharon BI, Honig GR. Management of con-
genital hemolytic anemias. In: Simon TL,
DzikWH,SnyderEL,etal,eds.Rossi’sprinci
-
ples of transfusion medicine. 3rd ed. Balti-
more, MD: Lipincott Williams and Wilkins,
2002:463-82.
107. Adams DM, Schultz WH, Ware RE, Kinney
TR. Erythrocytapheresis can reduce iron
overload and prevent the need for chelation
therapy in chronically transfused pediatric
patients. J Pediatr Hematol Oncol 1996;18:
46-50.
Chapter 27: Noninfectious Complications of Blood Transfusion 665
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Chapter 28: Transfusion-Transmitted Diseases
Chapter 28
Transfusion-Transmitted
Diseases
MANY ADVANCES HAVE been
made in the testing of blood do-
nations for infectious diseases.
However, the risk of transmitting viral,
bacterial, and parasitic diseases via trans-
fusion still exists, and new agents may
appear at any time. Thus, infectious com-
plications of transfusion remain an im-
portant area of concern in transfusion
medicine.
Hepatitis
Hepatitis is inflammation of the liver that
can be caused by many different toxins,
immunologic processes, or infectious agents.
Hepatitis linked to transfusion is almost
exclusively caused by viruses. These viruses
include hepatitis viruses A-E (HAV, HBV,
HCV, HDV, HEV), cytomegalovirus (CMV),
Epstein-Barr virus (EBV), and possibly
newly described putative hepatitis viruses
(such as TTV and SEN-V). Infectious agents
pose a serious threat to transfusion recipi-
ents if they persist in the circulation of
asymptomatic blood donors and can
cause clinically significant acute or chronic
disease manifestations in recipients.
The vast majority of posttransfusion
hepatitis in the past was attributable to
HBV and HCV, both of which can establish
prolonged carrier states in donors charac-
terized by high-titer viremia in the absence
of symptoms. HBV and HCV also cause sig-
nificant long-term liver-related morbidity
and mortality.1,2 These viruses are consid-
ered in detail below.
HAV and HEV, which are enterically trans-
mitted viruses, circulate only transiently
during the acute phase of infection. Be-
cause the viremic individual is usually clini-
cally ill and not a candidate for donation,
HAV and HEV are not serious threats to trans-
fusion recipients. However, HAV viremia
667
28
Copyright © 2005 by the AABB. All rights reserved.
maybepresentforupto28daysbefore
symptoms develop, and isolated cases have
been reported associated with transfusion
of cellular components3and outbreaks with
Factor VIII concentrate.4Because HAV lacks
a lipid envelope, it is not inactivated by sol-
vent/detergent treatment; additional inac-
tivation methods are under development to
prevent recurrence of such outbreaks. HEV
is rare in the United States, and there have
been no documented cases of transfusion
transmission in this country.
HDV, formerly called the delta agent, can
cause infection and serious hepatitis after
transfusion or other parenteral exposure.
However, because HDV is a defective virus
found only in HBV carriers, screening do-
nors for HBV infection simultaneously
eliminates the risk of HDV.5HGV, also called
GBV-C, is distantly related to HCV and has
ahighprevalencerate(>1%)amongasymp-
tomatic donors. Although HGV is unequiv-
ocally transfusion-transmissible,6acausal
relationship has not been established be-
tween HGV infection and hepatitis or any
other disease manifestation, despite inten-
sive study.
TTV appears similar to HGV with respect
to prevalence, transmissibility, and the lack
of clinical disease significance. Thus,
screening blood donors for HGV or TTV is
not currently recommended. Hepatitis as-
sociated with CMV or EBV is generally mild
in the absence of severe immunosuppres-
sion. The frequency and severity of such
hepatitis cases do not justify routine
screening measures.7SEN-V has been asso-
ciated with transfusion-associated non-A
through non-E hepatitis in one study,8but a
causal association has not been estab-
lished, nor has SEN-V been significantly as-
sociated with chronic non-A through non-E
hepatitis. SEN-V appears to be distantly re-
latedtoTTVandtobeamemberofafam
-
ily of small, circular DNA viruses called
Circoviridae. Screening for these agents is
not currently recommended because disease
associations have not been established.
Clinical Manifestations of Hepatitis
Most individuals who acquire HBV or
HCV infection have a subclinical primary
infection without obvious symptoms or
physical evidence of disease. Some de-
velop overt hepatitis with jaundice, nau-
sea, vomiting, abdominal discomfort, fa-
tigue, dark urine, and elevation of liver
enzymes. Signs and symptoms usually re-
solve spontaneously. Acute hepatitis C
tends to be milder than hepatitis B. Un-
commonly, the clinical course of HBV and,
rarely, HCV infections may be compli-
cated by fulminant hepatitis. Of greater
concern is a propensity of hepatitis C to
evolve to chronic hepatitis (75% to 85% of
affected individuals), with a significant
number demonstrating long-term pro-
gression to cirrhosis, liver failure, or hepa-
tocellular carcinoma. Hepatitis A tends to
be clinically mild in otherwise healthy
hosts and is not known to progress to
chronic hepatitis or a chronic carrier
state.9HEV infection may lead to severe
disease in pregnant women.10 Vaccination
is available for hepatitis A and for hepati-
tis B, and hepatitis B immune globulin
(HBIG) has proven useful for post-expo-
sure prophylaxis for hepatitis B and im-
mune serum globulin (ISG) for hepatitis
A.
Chronic Carriers of HBV
After initial HBV infection, a proportion of
patients fail to clear infectious virus from
the bloodstream and become chronic car-
riers for years or life. HBV carriers produce,
in addition to the infectious viral particle,
large amounts of noninfectious envelope
protein detected by the assay for hepatitis
B surface antigen (HBsAg). The risk of be-
coming an HBsAg carrier is strongly age-
668 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
dependent; ≤5% of those infected with
HBV as adults become chronic HBsAg
carriers, whereas ≥95% recover completely
and develop protective antibody against
HBsAg (anti-HBs). In contrast, before rou-
tine prophylactic immune globulin infu-
sion and immunization, 90% or more of
infants infected perinatally became carri-
ers and became at risk for progressing to
cirrhosis and hepatocellular carcinoma.
According to World Health Organization
estimates, the number of HBsAg carriers is
approximately 400 million worldwide,11
withaprevalenceofupto10%insome
Asian countries, 0.1% to 0.5% in the gen-
eral US population, and 0.02% to 0.04% in
US blood donors. Small proportions (<10%)
of HBsAg carriers develop clinical mani-
festations, such as hepatic insufficiency,
cirrhosis, or hepatocellular carcinoma.
Chronic Carriers of HCV
Most people who are initially infected with
HCV become chronic HCV carriers, with
75% to 85% having persistent HCV RNA in
the serum and liver for years to decades.
At least 50% of such HCV carriers have
biochemical and histologic evidence of
chronic liver inflammation.12 Despite this
chronic inflammatory process, most HCV-
infected individuals remain asymptom-
atic. During the first 20 years after infec-
tion, HCV is usually indolent and associ-
ated with low mortality and morbidity.
Those with clinical liver disease repre-
sented about 10% of the entire infected
cohort in one study.13 The risk that and the
rate at which chronic hepatitis by itself
will progress to cirrhosis is unknown. It is
thought that alcohol may play a synergis-
tic role in exacerbating chronic hepatitis
C. Posttransfusion HCV in children in-
fected after cardiac surgery appears to re-
solvemorefrequentlythanthatinadults
and to be a mild infection at almost 20
years of follow-up.14
Recommendations for clinical manage-
ment of persons with chronic HCV infec-
tion were developed by a National Insti-
tutes of Health consensus development
conference15 and have been updated and
expanded by others.16
Markers of Viral Infection
Laboratory tests can identify markers of
previous exposure and probable current
infectivity for HBV and HCV, which are
useful for screening and diagnostic appli-
cations. Table 28-1 lists the molecular and
serologic markers commonly used in the
diagnosis of hepatitis. Figure 28-1 illus-
trates the sequence of test results typical
of individuals with acute HBV infection
that completely resolves.
The period between exposure to HBV
and emergence of circulating markers of in-
fection (HBV DNA or HBsAg) is usually
about 6 weeks.3,9 HBV DNA, detectable by
pooled nucleic acid amplification testing
(NAT) techniques, is the first marker to ap-
pear, followed by detectable HBsAg. Indi-
vidual donor NAT (ID-NAT) is able to detect
HBV DNA approximately 19 days before
minipooled NAT (MP-NAT).17 Antibody to
the HBV core protein (anti-HBc) usually ap-
pears several weeks later, first as IgM and
then as IgG. The clearance of HBsAg and
appearance of anti-HBs signal resolution of
infection. However, there has been a report
of a fatality, after reactivation of HBV in a
patient treated with rituximab, who was
previously anti-HBs-reactive.18 The specific
virus showed multiple mutations in major
antigenic sites and was thought to escape
the patient’s endogenous immunity.
Two additional HBV markers, HBeAg or
its antibody (anti-HBe), are useful diagnos-
tic and prognostic markers but are not em-
ployed in donor screening. An asymptom-
Chapter 28: Transfusion-Transmitted Diseases 669
Copyright © 2005 by the AABB. All rights reserved.

670 AABB Technical Manual
Table 28-1. Molecular and Serologic Tests in the Diagnosis of Viral Hepatitis
Virus Test Reactivity Interpretation
HBV DNA HBsAg Anti-HBc
Total
IgM Anti-HBs HBeAg Anti-HBe
+ – – – – – – Window period
+ + +/– +/– – +/– – Early acute HBV infection/chronic carrier
+ + + + – + – Acute infection
+/– – + + – +/– +/– Early convalescent infection/possible early
chronic carrier
+/– + + – – +/– +/– Chronic carrier*
– – + – + – +/– Recovered infection
– – – – + – – Vaccinated or recovered infection
– – + – – – – Recovered infection? False positive?
HDV RNA HBsAg Anti-HBc Anti-HBs Anti-Delta
+ + + – + Acute or chronic HDV infection
– – + + + Recovered infection
HCV RNA Anti-HCV
(Screening EIA)
Recombinant Antigens (RIBA)
5-1-1 c100-3 c33c c22-3
+/– + Not available Probable acute or chronic HCV infection (if RNA
is positive)
– + – – – – False positive
Copyright © 2005 by the AABB. All rights reserved.

Chapter 28: Transfusion-Transmitted Diseases 671
+/– + + + – – Probable false positive (if RNA is negative); pos-
sible acute infection (if RNA is positive)†
+/– + – – + + Early acute or chronic infection (if RNA is posi-
tive); false positive or late recovery (if RNA is
negative)†
+ + + + + + Acute or chronic infection
– + +/– +/– + + Recovered HCV†
HAV RNA Anti-HAV
Total IgM
++ + AcuteHAV
– + – Recovered HAV/vaccinated
HEV RNA Anti-HEV
Total IgM
++ + AcuteHEV
– + – Recovered HEV
*Those with HBeAg are more infectious and likely to transmit vertically.
†Anti-5-1-1 and anti-c100-3 generally appear later than anti-c22-3 and anti-c33c during seroconversion and may disappear spontaneously, during immunosuppression or after
successful antiviral therapy.
HBsAg = hepatitis B surface antigen; anti-HBc = antibody to hepatitis B core antigen; anti-HBs = antibody to HBsAg; HBeAg = hepatitis B e antigen; anti-delta = antibody to delta
antigen; anti-HAV = antibody to hepatitis A virus; anti-HCV = antibody to hepatitis C virus; anti-HEV = antibody to hepatitis E virus.
Copyright © 2005 by the AABB. All rights reserved.
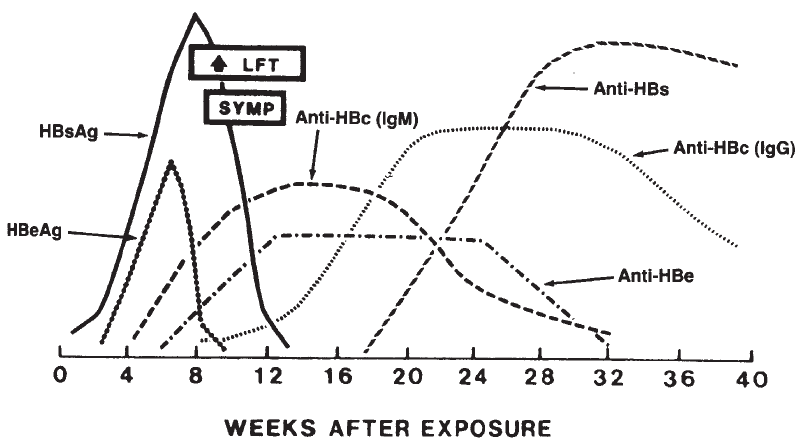
atic HBsAg-positive individual may either
be in the early phase of acute HBV infection
(without anti-HBc or with IgM anti-HBc) or
a chronic HBV carrier (with IgG anti-HBc).
HBsAg particles are produced in excess
during acute and chronic infection; blood
from individuals with circulating HBsAg
can infect others. Current screening im-
munoassays detect approximately 0.2 to 0.7
ng/mL HBsAg or ≥3×107particles.3The
number of HBsAg particles in most acute
and chronic infections exceeds this level,
but transmission of HBV from HBsAg
seronegative donors has been described.
NAT testing allows the detection of as few
as 10 genomic copies of HBV DNA.3The
value of NAT for the detection of sero-
negative donors infected with HBV is under
study but may be reduced by high sensitiv-
ity of current assays for HBsAg and the rela-
tively slow rise in HBV DNA levels con-
trasted with HIV and HCV. It is possible that
effective detection of seronegative HBV in-
fected donors by NAT will require testing at
the single donation level.19 HBV vaccines
contain noninfectious HBsAg protein, which
may result in false-positive HBsAg screen-
ing test results for a few days after the inoc-
ulation. Resulting protective antibodies are
directed against HBsAg; vaccination does
not produce anti-HBc.
Tests for antibodies to HCV are enzyme
immunoassays (EIAs) using recombinant
antigens of HCV coated on a solid phase as
the capture reagent. Current assays detect
antibodies to c200 (including c33c and
c100-3), c22-3, and NS-5. Anti-HCV is de-
tectable by third-generation EIAs approxi-
mately 10 weeks after infection. HCV RNA
is present at high concentrations in plasma
during most of the period from exposure to
antibody seroconversion. Anti-HCV is de-
tected in 40% to 50% of samples from pa-
tients at initial diagnosis of acute hepatitis,
672 AABB Technical Manual
Figure 28-1. Serologic markers in hepatitis B virus infection that resolved without complications. In the
acute phase, markers often appear before onset of liver function test (LFT) abnormalities and symptoms
(SYMP). Anti-HBs and anti-HBc persist after recovery and indicate immunity. In chronic carriers (not
shown), HBsAg persists and anti-HBc is usually present, but anti-HBs is absent (HBeAg and anti-HBe
may be present, see Table 28-1). HBV DNA (not shown) may be detected approximately 1 to 2 weeks be-
fore HBsAg.
Copyright © 2005 by the AABB. All rights reserved.
either transfusion-transmitted or community-
acquired.20
The clinical significance of a positive
screening test for anti-HCV in healthy blood
donors is unclear without supplemental
testing. Approximately 0.21% of US blood
donors have repeatedly reactive EIA re-
sults.21 Several generations of recombinant
immunoblot assays (RIBA) have been li-
censedbytheFoodandDrugAdministra
-
tion (FDA) for further elucidation of repeat-
edly reactive EIA results. An individual who
is positive by RIBA is considered to have true
HCV antibody; in 70% to 90% of these
cases, HCV nucleic acid is detectable by
NAT methods. The infectivity of units that
are positive for HCV RNA approaches
100%.22 In contrast, EIA repeatedly reactive
donors with negative or indeterminate
RIBA 3.0 results, representing 37% of EIA
repeatedly reactive donors, are rarely in-
fected or infectious. Regardless of RIBA re-
sults, a donation with a repeatedly reactive
EIA result cannot be used for transfusion.
Donors with negative RIBA results may be
considered for reentry (Table 28-2).
In 1999, NAT for HCV RNA was imple-
mented as a donor screening assay under
FDA-sanctioned investigational new drug
(IND) protocols. The testing was performed
in minipools of samples from 16 to 24 whole
blood donations. The rapid increase in
viremia and high viral load of seronegative,
acutely infected donors allows sensitive de-
tectionofHCVRNAeveninthesediluted
pools. The window period for HCV detec-
tion with pooled NAT is reduced to 10 to 30
days.17 After testing over 39 million donors,
approximately 1:270,000 donors had been
identified as being in the seronegative win-
dow with a positive NAT result.23
NAT results can be used in lieu of sup-
plemental testing in specific circumstances.
An FDA variance is required.24 It is antici-
patedthatNATHCVtestingwillbeusedin
the future for reentry.
Surrogate Markers
Before HCV was identified and anti-HCV
testing became feasible, two nonspecific
or “surrogate” tests on donor blood were
introduced to reduce the risk of non-A, non-B
(NANB) hepatitis after transfusion. In 1986
and 1987, the AABB called for testing of
whole blood donations for alanine amino-
transferase (ALT) and anti-HBc as surro-
gates for the direct detection of the NANB
agent. Current very sensitive tests for anti-
HCV have essentially eliminated the value
of surrogate tests in preventing hepatitis,25
but anti-HBc testing continues as recom-
mended by FDA to prevent HBV transmis-
sion. HBV from liver transplant donors with
reactive anti-HBc and negative HBsAg test
results has been transmitted to their re-
cipients, and reports in the literature
show that transfusions of blood reactive
for anti-HBc and negative for HBsAg have
been associated with development of
hepatitis B in some recipients. There may
also be a small number of potential do-
nors infected with HBsAg mutants of HBV
that may not be optimally detected by
currently licensed HBsAg tests.26 Donors
who test repeatedly reactive for anti-HBc
on two occasions or who test repeatedly
reactive on tests from two different manu-
facturers should be deferred.
Current Risk of Posttransfusion Hepatitis
The risk of posttransfusion HBV or HCV
infection decreased dramatically, to an es-
timated 1 in 60,000 to 1 in 100,000 risk be-
fore implementation of HCV NAT.27 Not a
single new case of transfusion-associated
HCV has been detected by the CDC Senti-
nel Counties Viral Hepatitis Surveillance
System since 1994 (M. Alter, personal com-
munication, 3/04).1The development of
progressively improved HCV antibody
tests and stringent selection measures for
donors have contributed to this remark-
Chapter 28: Transfusion-Transmitted Diseases 673
Copyright © 2005 by the AABB. All rights reserved.

674 AABB Technical Manual
Table 28-2. Reentry of Donors with Repeatedly Reactive Screening Tests
Repeatedly Reactive for
Reentry
Status
Anti-HIV-1
or -1/2 Anti-HIV-2 HIV-1-Ag HBsAg Anti-HCV
Initial sample
Licensed West-
ern blot posi-
tive or inde-
terminate or
IFA reactive
Different HIV-2
EIA RR
Confirmed by
neutraliza-
tion
Confirmed by
neutraliza-
tion or
anti-HBc
RR
RIBA indeter-
minate or
positive
Not eligible
for reentry
Licensed West-
ern blot or IFA
NR
Different HIV-2
EIA NR and li-
censed West-
ern blot or IFA
NR
Not confirmed
by neutral-
ization
HBsAg speci-
ficity not
confirmed
by neutral-
ization and
anti-HBc
NR
RIBA negative Evaluate for
reentry
Follow-up sample
(Drawn 6
months
later)
(Drawn 6
months
later)
(Drawn 8
weeks
later)
(Drawn 8
weeks
later)
(Drawn 6
months
later)
EIA RR or West-
ern blot posi-
tive or inde-
terminate or
IFA reactive
RR HIV-1 or dif-
ferent HIV-2
EIA RR or a li-
censed West-
ern blot or IFA
reactive or in-
determinate
HIV-1-Ag RR,
neutraliza-
tion con-
firmed or
not con-
firmed
HBsAg RR or
anti-HBc
RR
EIA RR or
RIBA inde-
terminate
or positive
Not eligible
for reentry
Original EIA
method NR
and whole vi-
rus lysate
anti-HIV-1 EIA
NR and li-
censed West-
ern blot or IFA
NR
Screening test
and a differ-
ent HIV-2 EIA
NR and li-
censed West-
ern blot or
IFA NR
HIV-1-Ag and
anti-HIV-1
EIA NR or
HIV-1-Ag
RR, not
confirmed
(temporary
deferral for
additional
8 weeks)
HBsAg NR
and
anti-HBc
NR
Licensed
multiantigen
EIA
method NR
and RIBA
negative
Eligible for
reentry
NR = nonreactive; RR = repeatedly reactive; RIBA = recombinant immunoblot assay; IFA = immunofluorescence assay;
EIA = enzyme immunoassay; anti-HIV-1 = antibody to human immunodeficiency virus, type 1; anti-HIV-2 = antibody to
human immunodeficiency virus, type 2; HIV-1-Ag = HIV-1 antigen; HBsAg = hepatitis B surface antigen; anti-HCV = anti-
body to hepatitis C virus; anti-HBc = antibody to hepatitis B core antigen.
Copyright © 2005 by the AABB. All rights reserved.
able decline, and transfusion is no longer
considered a major risk factor for HCV
transmission.28 Nucleic acid screening as-
says for HCV were implemented by blood
centers in 1999. In 3 years of NAT testing,
170 HCV NAT-positive, seronegative do-
nations were identified in the United
States among 39.7 million screened dona-
tions.23 NAT has probably reduced the re-
sidual risk for HCV transmission to ≤1in
2,000,000 components transfused.29,30
Quarantine and Recipient Tracing
Donations with repeatedly reactive screen-
ing test results (HBsAg, anti-HBc, and/or
anti-HCV) cannot be used for transfusion.
In addition, in-date components from
collections preceding the current unsuit-
able donation may need to be quaran-
tined as follows, and consignee notifica-
tion for the purpose of recipient tracing
(ie, look-back) may be required31,32:
For HCV:
■Extending back indefinitely, to the
extent that computerized electronic
records exist for anti-HCV repeat-
edly reactive and confirmed dona-
tions or repeatedly reactive donations
for which supplemental tests were
not performed.
■Extending back to January 1, 1988 if
computerized electronic records are
not available.
For HBsAg and anti-HBc:
■In-date components, extending back
5years,or12monthsfromthemost
recent negative test result for units
that were repeatedly reactive or con-
firmed, or for which confirmatory
testing was not performed.
Depending on the results of licensed
supplemental tests and prior screening
tests, the quarantined units may be re-
leased for transfusion or further manufac-
ture, or may have to be destroyed. Recipi-
ents notified as a result of the HCV look-back
should be counseled regarding the nature
of the subsequent donor test results and of-
fered appropriate testing. If they test posi-
tive, life style changes (eg, abstinence from
alcohol consumption) and evaluation for
chronic liver disease justifying antiviral
therapy to reduce the likelihood of disease
progression may be warranted. Earlier ex-
periences from Canada and several Euro-
pean countries indicate that the number of
transfusion recipients who ultimately bene-
fit from look-back efforts is small.33 Asurvey
of US blood collection facilities and hospi-
tal transfusion services after implementa-
tion of targeted HCV look-back resulted in
an estimate that notification of the recipi-
ents of 98,484 components would result in
the identification of 1520 infected persons
who were previously unaware of their in-
fection.34 This would represent less than 1%
of the 300,000 still-living recipients who
may have acquired infection by blood
transfusion. More recently, 0.9% to 5.0% of
patients tested for hepatitis C were found to
be positive in a review of look-back studies
in Canada that notified recipients of any
previous transfusions of the risk of HCV
and then provided testing; 42% to 58% of
the cases were newly identified.35
Human Immunodeficiency
Viruses
The human immunodeficiency viruses type
1 (HIV-1) and type 2 (HIV-2) are the
etiologic agents of AIDS. The AIDS syn-
drome was recognized in 1981, well be-
fore the discovery of the causative virus in
1984. Wider implications of the immune
disorder were noted when, in 1982, AIDS
was reported in three patients with hemo-
philia,36 and in a 17-month-old infant
whosemultipletransfusionsatbirthin
-
cluded a unit of platelets from a donor
Chapter 28: Transfusion-Transmitted Diseases 675
Copyright © 2005 by the AABB. All rights reserved.
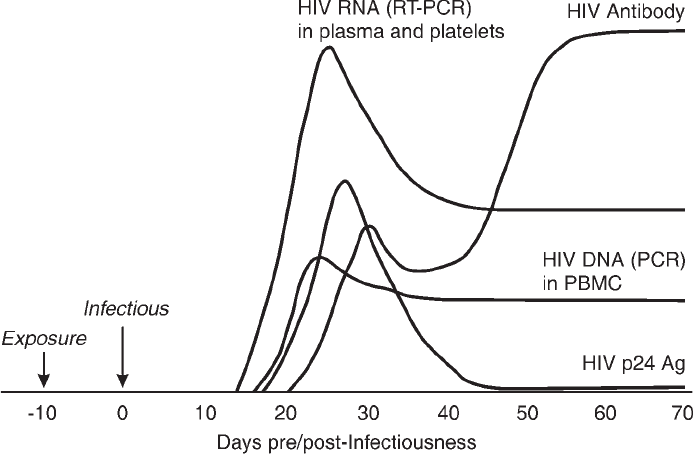
who subsequently developed AIDS.37 With-
in a few years, studies established that well
over 50% of patients with hemophilia who
received clotting factor concentrates in
the early 1980s developed HIV-1 infec-
tion.38 In some regions of the United States,
up to 1% of single-donor unit transfusions
were infected with HIV in the early 1980s.39
Clinical Manifestations of HIV Infection
HIV is a cytopathic retrovirus that prefer-
entially infects CD4-positive T lympho-
cytes (helper T cells) in lymph nodes and
other lymphoid tissue.40 After primary in-
fection, HIV replicates and disseminates
initially as cell-free virions, and 10 days to
3 weeks after infection, viremia is first de-
tectable in the plasma. During this time,
about 60% of acutely infected persons de-
velop an acute retroviral syndrome, char-
acterized by a flu-like illness with fever,
enlarged lymph nodes, sore throat, rash,
joint and muscle pain—with or without
headache, diarrhea, and vomiting. As HIV-1
antibodies appear, the disease enters a
clinically latent stage; however, viral repli-
cation and dissemination continue. Dur-
ing this phase, the virus can be transmit-
ted by blood or genital secretions (Fig
28-2).
Persistent infection with an asymptom-
atic clinical status has been estimated to
lastamedianof10to12yearsintheab
-
sence of treatment.41 After years of asymp-
tomatic infection, both plasma viremia and
the percentage of infected T lymphocytes
676 AABB Technical Manual
Figure 28-2. Virologic events during primary HIV infection. After initial infection and propagation of
HIV in lymph nodes, a blood donor becomes infectious (defined as day 0), with HIV RNA being detect-
able in plasma on days 14-15, HIV DNA detectable in leukocytes at days 17-20, and HIV antibodies de-
tectable between days 20 and 25. Anti-HIV persists indefinitely but may be lost in the preterminal
stage of the disease, in parallel with a surge in viral burden, indicating collapse of the immune sys-
tem. HIV = human immunodeficiency virus; RT-PCR = reverse transcriptase polymerase chain reaction;
PBMC = peripheral blood mononuclear cells.
Copyright © 2005 by the AABB. All rights reserved.
increase. Loss of the immune functions
served by helper T cells impairs immune
reactivity, and there may be inappropriate
immune activation and cytokine secretion.
Eventually, there is a sharp decline in the
number of CD4+ T lymphocytes, and the
vast majority of infected individuals suc-
cumb to opportunistic illnesses fostered by
profound immunosuppression.
Enumeration of viral load and CD4+ cells
is used to guide clinical and therapeutic
management of HIV-infected persons. The
AIDS classification system devised by the
Centers for Disease Control and Prevention
(CDC) is based on the number of CD4+ T
cells (<200/µL defines AIDS), the presence
orabsenceofsystemicsymptoms,andex
-
istence of any of the 26 clinical conditions
considered to be AIDS-defining illnesses.42
Among these conditions are otherwise un-
usual malignancies, such as Kaposi’s sar-
coma, central nervous system lymphoma,
and a wide array of devastating, potentially
lethal opportunistic infections with fungi
and parasites, the most common being
Pneumocystis carinii pneumonia.
Advances in treatment of HIV and op-
portunistic infections have dramatically en-
hanced the survival of infected persons.
Unfortunately, worldwide, the disease is
still spreading rapidly and, for the majority
of HIV-infected individuals in developing
countries, effective therapy is either not
available or not affordable.
Risk Factors for HIV Infection
HIV can be transmitted through sexual
contact, childbirth, breast-feeding, and
parenteral exposure to blood. Those iden-
tified early as being at highest risk were
men who had sex with other men; com-
mercial sex workers and their contacts;
needle-sharing drug users; patients with
hemophilia who received human-derived
clotting factor concentrates; and, to a les-
ser extent, recipients of blood transfusions.
By 1989, the rate of infection in each
group was no longer increasing exponen-
tially and appeared to have reached a pla-
teau in the populations most at risk.43 HIV
seroprevalence had stabilized in most US
cities. Heterosexual transmission of HIV
represented a progressively larger propor-
tion of US HIV infections and AIDS cases
reported in the 1990s.44 This is of impor-
tance in transfusion medicine because
screening for heterosexual high-risk be-
havior is more problematic than screen-
ing for male-to-male sex and parenteral
drug use.45
HIV-2 and HIV-1, Group O
First discovered in 1985, HIV-2 causes en-
demic infection in many countries in
West Africa. Although HIV-2 was initially
restricted to West Africa, recent studies in
European countries such as Great Britain
and France (which have significant immi-
gration from West Africa) have observed
increasing rates of HIV-2 and other HIV
subtypes.46,47
The first case of HIV-2 infection in the
United States was reported in March 1988
in a young West African who had recently
immigrated to the United States.48 The
spectrum of disease attributable to HIV-2 is
similar to that caused by HIV-1; however,
there appears to be a longer incubation pe-
riod and lower incidence of progression to
AIDS. HIV-2 is spread both sexually and
from mother to child, but transmission is
less efficient than for HIV-1.
Tests in the United States on parenteral
drug users, persons with sexually transmit-
ted diseases, newborn infants, and homo-
sexual men confirm the very limited preva-
lence and transmission of the agent. HIV-1/
HIV-2 combination tests were implemented
in the United States in 1992. Since then,
three HIV-2-infected donors have been
Chapter 28: Transfusion-Transmitted Diseases 677
Copyright © 2005 by the AABB. All rights reserved.
identified; none appeared to have been in-
fected in the United States.49
To date, three groups of HIV-1 viruses
have been identified: group M (major
group); group O (outlier group); and, most
recently, group N. Further, there are 10 sub-
types (A-J) of group M. None of 97 donors
retrospectively identified as being HIV-1 in-
fected in 1985 and three (1%) of 383 donors
prospectively identified between 1993 and
1996 were found to have non-B subtypes
(two subtype As and one subtype C).50 Of
note, this study did find an increase in env
gene diversity among HIV-1 group B strains
overtimeandcalledforcontinuedsurveil
-
lance for emergence of non-B subtypes and
development of test systems for their de-
tection. A follow-up study from these same
investigators documented characterized
HIV subtypes in 291 infected US donors
identified from 1997 through 2000 and
identified that six (2%) were non-B sub-
types of HIV-1 and one was HIV-2.51 In
Cameroon and surrounding West African
countries, an estimated 1% to 2% of HIV in-
fections are caused by group O viral strains.52
As with HIV-2, group O isolates have rarely
been seen outside this geographic area.
Concern arose when studies demonstrated
that some group O viral isolates were not
reliably detected by several EIA tests used
for blood donor screening. Of the two
FDA-licensed tests for NAT, both were eval-
uated by the manufacturer for the detection
of non-B subtypes including group O and
N, using a limited number of specimens.
(although neither test detects HIV-2). How-
ever, until reliable detection of group O in-
fections is established, the FDA recom-
mends indefinite deferral of blood and
plasma donors who were born, resided, or
traveled in West Africa since 1977, or had
sexual contact with someone identified by
these criteria.53 The risk of group O infec-
tion in the United States is very low. In a
survey of HIV subtypes in US blood donors
over a period spanning two decades, only
three non-B subtypes were identified. Two
of these three donors were born in Africa.50
Transfusion Considerations
Transfusion-Transmitted HIV-1
All blood components can transmit HIV-1.
Although approximately 1% of all AIDS
exposures have resulted from transfusion
or organ or tissue transplantation, the in-
troduction of MP-NAT in 1999 virtually
eliminated the risk of transfusion-trans-
mitted HIV.54 The few cases of HIV infec-
tion that have been documented since
1999 were attributed to low-level viremic
units that likely would have been detected
by ID-NAT.55-57
Most but not all recipients of HIV-in-
fected blood transfusions become infected.
In one large study, HIV infection developed
in 89.5% of recipients who received blood
from anti-HIV-positive donors.58 Transmis-
sion rates correlated with component type
and viral load in the donation. With the ex-
ception of coagulation factor concentrates,
plasma derivatives (such as albumin and
immune globulins) have not been reported
to transmit HIV infection. No transmission
of HIV attributable to coagulation factors has
been documented in the United States
since implementation of full donor screening
and virus inactivation techniques in 1987.59
Transfusion-Transmitted HIV-2 and HIV-1,
Group O
There have been two reports of possible
HIV-2 transmission through blood com-
ponent use, both in Europe. Two women
were infected by Whole Blood obtained from
a donor who developed AIDS at least 16
years after becoming infected with HIV-2;
both women were asymptomatic 14 years
after transfusion.60 Two hemop hili a p a-
tients who received clotting factors were
also infected. Because of their extremely
678 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
low prevalence, no HIV-2 or HIV-1 group
O transmissions have been reported in
the United States by blood transfusion or
any other transmission route.
Current Risk of Posttransfusion HIV
With screening tests available before 1992,
the seronegative interval (“window pe-
riod”) averaged 45 days. More sensitive
screening tests for HIV antibody closed
the antibody-negative window to approxi-
mately 22 days.17 Introduced in 1996, p24
antigen screening further reduced the po-
tentially infectious window by an esti-
mated 6 days,61 although it appears that
fewer HIV-infected units were intercepted
by the introduction of this test than had
been expected based on the calculated re-
duction of the infectious window period.
Risk from seronegative donations will
vary in proportion to the incidence of HIV
infection in the donor community. Overall
estimates of posttransfusion HIV risk in the
United States since the implementation of
HIV NAT are approximately 1 in 2 million
screened donations.23,30
HIV Testing of Blood Donors
AABB Standards for Blood Banks and Trans-
fusion Services62(pp33,34) and FDA regulations63
require that all units of blood and compo-
nents be nonreactive for anti-HIV-1 and
anti-HIV-2 before they are issued for
transfusion. HIV-1-antigen (HIV-1-Ag)
testingisnomorerequiredbytheFDAor
AABBaslongaslicensedHIV-1NATisin
place.Figure28-3showsthesequenceof
screening and confirmatory testing for anti-
HIV-1/2.
Because the consequence of missing even
onetruepositiveisgreat,screeningtests
are designed to have high sensitivity both
to immunovariant viruses and to low-titer
antibody during seroconversion. EIA-de-
tectable antibody develops 2 to 4 weeks
after exposure,58 days to a week after the on-
set of symptoms in those who have any rec-
ognized acute illness,64 and about 12 days
after detectable viremia by ID-NAT or 9 to
10 days by MP-NAT.17,65
A few days later, HIV-1 antibodies become
detectable by the HIV-1 Western immuno-
blot. With very rare exceptions, all persons
infected with HIV develop anti-HIV reactiv-
ity detectable by EIA and Western blot that
persists for life.
More sensitive tests using NAT technol-
ogies have been shown to detect additional
potentially infectious donors. In February
2002, the FDA approved the first NAT sys-
tem for screening whole blood donors and
issued draft guidance for blood establish-
ments; the final guidance was issued in
October 2004.66,67 It has been estimated that
HIV NAT has reduced the window period
for HIV from 16 days to 10 days.17 The use
of NAT for donor testing has not only in-
creased sensitivity, but has also decreased
the number of false-positive tests, increas-
ing specificity. During the 3 years of inves-
tigational HIV NAT, 12 confirmed HIV-1
RNA-positive antibody-negative donors
were detected in 37 million donations
screened, or 1 in 3.1 million, of which only
two were detected by HIV-1 p24 antigen.23
Confirmatory Testing for Antibodies to
HIV-1/2
If a disease has low prevalence in the
tested population, the likelihood is high
that most positive screening test results will
be false positive. More specific supplemen-
tal tests are then required to confirm the
screening test results. The most commonly
used of these tests for antibodies to HIV-1/2
is the Western blot (see Chapter 7).
According to current FDA and CDC cri-
teria, a sample is defined as anti-HIV-posi-
tive if at least two of the following bands are
Chapter 28: Transfusion-Transmitted Diseases 679
Copyright © 2005 by the AABB. All rights reserved.
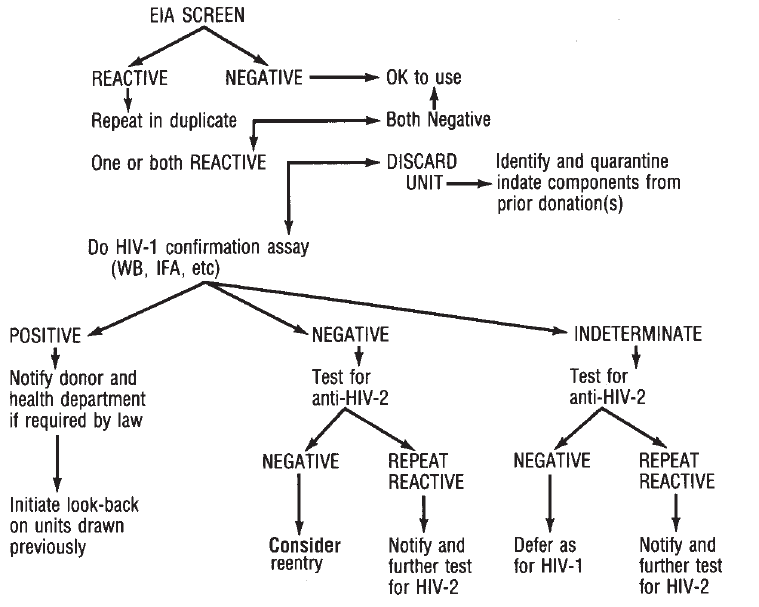
present: p24, gp41, and/or gp120/160.68
Negative Western blot results have no
bands present. Western blot results classi-
fied as indeterminate have some bands
present but do not have the pattern defin-
ing HIV positivity. Individuals infected with
HIV may have indeterminate patterns when
initially tested but develop additional bands
within 6 weeks. Healthy individuals with
initial indeterminate patterns continue to
have negative or indeterminate results on
repeat samples and are negative on clinical
examination and additional tests, including
viral cultures and NAT. Healthy donors who
continue to show the same indeterminate
pattern for more than 3 months can be reas-
sured that they are unlikely to have HIV in-
fection, but they are not currently eligible
to donate blood. Several groups have iden-
tified Western blot patterns in blood donors
that were identified as false-positive results;
for these, testing (using NAT) is recom-
mended to resolve the infectious status of
the donor.69
Approximately 50% of all HIV screening
EIA repeatedly reactive donors test indeter-
minate by licensed HIV-1 Western blot as-
says. However, when these donations were
tested by ID-NAT, less than 0.1% were
shown to contain HIV-1 RNA (1:1450 RNA-
positive, indeterminate donors). When com-
bined with those donations that tested
Western blot negative, the frequency of a
donor testing HIV repeatedly reactive and
680 AABB Technical Manual
Figure 28-3. Decision tree for anti-HIV-1/HIV-2 testing of blood donors. IFA = immunofluorescence as-
say; WB = Western blot. If EIA testing is nonreactive, NAT testing must also be nonreactive before re-
lease of a donation.
Copyright © 2005 by the AABB. All rights reserved.
then Western blot indeterminate or nega-
tive and demonstrating RNA was 1:4.27
million (S. Stramer, personal communica-
tion, 3/17/04).
The FDA has approved reentry protocols
to qualify donors with negative confirma-
tory test results as eligible for subsequent
donations (see Table 28-2).70 Reentry currently
requires retesting at least 6 months later, to
detect delayed seroconversion; the use of
EIA tests based on whole-virus lysate; and
use of either a licensed Western blot to en-
sure appropriate sensitivity of the methods
or an FDA-licensed immunofluorescence
assay.71 The later sample must also be
nonreactive in an EIA test for anti-HIV-2, if
standard testing does not include HIV-2.
The FDA recently published draft reentry
guidelines that will allow reinstatement of
donors with indeterminate Western blot re-
sults, eliminate the need for viral lysate EIA
testing, and include testing by HIV-1 NAT.
NAT results can be used in lieu of sup-
plemental testing in specific circumstances.
An FDA variance is required.24 It is antici-
pated that NAT HIV-1 testing will be used in
the future for reentry.
Positive Tests in Autologous Donors
Whether HIV EIA repeatedly reactive or
NAT-positive autologous donations should
be withheld from transfusion is contro-
versial.72 Theseunitsmaybesuppliedfor
autologous use if the following conditions
are met: 1) there is a written, signed, and
dated request from the patient’s physician
authorizing this shipment, 2) there is a
written statement from the transfusion
service indicating willingness to receive this
product, and 3) the transfusion service
takes responsibility for ensuring that there
is documented verification of the accurate
identity of the transfusion recipient. These
units must be labeled “BIOHAZARD” and
“FOR AUTOLOGOUS USE ONLY.”
Whether a facility elects to offer autolo-
gous services is an internal decision. Insti-
tutions should consider, however, that
where feasible for a patient, it is generally
accepted that the patient should have the
option to use his or her blood. In addition,
aUSSupremeCourtdecisionintheBrag-
don v Abbott case ruled that HIV-positive
individuals are protected under the Ameri-
cans with Disabilities Act.73 Whether this
would apply to HIV-positive donations that
could represent a risk to hospitalized trans-
fused patients remains controversial.
Recipient Tracing (Look-Back)
Identification of persons who have received
seronegative or untested blood from a do-
nor later found to be infected by HIV is
referred to as “look-back.” Because the in-
terval between receipt of an infected
transfusion and onset of AIDS can be very
long, recipients are usually unaware of their
infection and may be infectious to others.
To identify these individuals, blood cen-
ters must have procedures to notify recip-
ients of previous donations from any do-
nor later found to have a confirmed
positive test for anti-HIV or a confirmed
positive test for HIV using licensed NAT. If
a patient with AIDS is known to have do-
nated previously, recipients of blood or
blood components from these donations
should be traced and notified. Recipient
tracing and testing are usually accom-
plished through the patient’s physician, not
through direct contact with the patient. In
companion rules, the FDA and the Cen-
ters for Medicare and Medicaid Services
established timelines and standards de-
fining look-back.74-77 If recipients of units
that were donated at least 12 months be-
fore the last known negative test are tested
and found negative, earlier recipients are
probably not at risk because infectivity
Chapter 28: Transfusion-Transmitted Diseases 681
Copyright © 2005 by the AABB. All rights reserved.
earlier than 12 months before a negative
screening test is extremely unlikely.
Human T-Cell Lymphotropic
Viruses
HTLV, Type I
Human T-cell lymphotropic virus, type I
(HTLV-I) was the first human retrovirus
isolated and the first to be causally associ-
ated with a malignant disease of humans,
adult T-cell lymphoma-leukemia (ATL).58
HTLV-I is also associated with the neuro-
logic condition HTLV-associated myelo-
pathy (HAM), often called tropical spastic
paraparesis (TSP). ATL was described be-
fore HAM. Both these conditions occur in
a small minority (no more than 2-4%) of
persons harboring the virus. Infection
during childhood is an important aspect
of, and possibly a requirement for, devel-
oping ATL many years later, whereas
childhood or adult infection can cause
HAM, with a variable latent period.
Prevalence of HTLV-I infection shows
striking geographic clustering, with pockets
of high endemicity in parts of southern
Japan and certain Pacific Islands; sub-Saha-
ran Africa; and the Caribbean basin, Cen-
tral America, and South America. Transmis-
sion is by mother to child through breast
milk, by sexual contact (predominantly
male-to-female), and by exposure to blood.
HTLV, Type II
Human T-cell lymphotropic virus, type II
(HTLV-II) was described several years af-
ter HTLV-I. There is at least 60% similarity
of genetic sequences to those of HTLV-I;
antibodies to either show strong cross-re-
activity in tests with viral lysates. HTLV-II
also shows clustering, but in different
populations. High prevalence has been
noted among some Native American pop-
ulations and in intravenous drug users in
the United States, in whom seropreva-
lence is 1% to 20%. Rare disease associa-
tions with HTLV-II include HAM; its
occurrence seems to be somewhat less
frequent than with HTLV-I.58
Epidemiologic data suggest that there is
an excess of infectious syndromes (eg,
bronchitis, urinary infections, and pneu-
monia) among blood donors infected with
HTLV-I or -II.78-81
Clinical Observations
For both HTLV-I and -II, infection persists
lifelong, as does the presence of antibody.
Studies of prevalence and transmission
use seroconversion as the endpoint for di-
agnosis. Infection does not cause any rec-
ognizable acute events, and with the ex-
ception of those developing ATL or HAM,
infected individuals experience few, if
any, health consequences. Most carriers
are asymptomatic and completely un-
aware of the infection.
Transmission
Both viruses are very strongly cell-associ-
ated. Contact with infected viable lym-
phocytes can cause infection, but plasma
appears to be not, or much less, infective.
Cellular components from infected donors
cause seroconversion in at least 50% of
recipients in Japan, but apparently in a
much smaller proportion of US recipi-
ents.58 After refrigerated storage for 10
days or more, red cells transfused from an
infected donor are far less likely to result
in seroconversion, presumably due to de-
gradation of lymphocytes that transmit
the virus.82,83 Transfusion-transmitted
HTLV-I infection has been associated with
HAM of rather rapid onset and at least
onecaseofATL.
682 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Donor Tests
Donor screening for anti-HTLV-I began in
the United States in late 1988; at that time,
the rate of confirmed positive tests was
approximately 0.02%, or 1 in 5000 units
collected, a figure that has since declined
at least 10-fold as seropositive persons
have been removed from the donor pool.84
The combined risk of transfusion-trans-
mitted HTLV-I/II infection has been esti-
mated as about 1 in 641,000 units.84 Fur-
ther risk reduction can be expected from
the implementation of combination
HTLV-I/II EIA tests in blood donor screen-
ing. The first such test was licensed in
1998 in the United States. By using viral
lysatesfrombothHTLV-IandHTLV-IIvi-
ruses, the test offers sensitive detection of
both anti-HTLV-I and anti-HTLV-II. The
originally licensed anti-HTLV-I EIA
screening tests might have missed up to
50% of HTLV-II infections.85
A donation that is repeatedly reactive on
EIA may not be used for transfusion. If a
donor tests repeatedly reactive by the EIA
screening assay on two or more occasions,
he or she should be notified and indefi-
nitely deferred.85 Another recommended
approach is to test donor serum with a sec-
ond manufacturer’s EIA test kit.86 If that test
is also repeatedly reactive, then the donor is
indefinitely deferred on the basis of that
single donation. Further testing of serum
that is repeatedly reactive for anti-HTLV-I/II
against antigen preparations specific for
the two agents (HTLV-I or HTLV-II), or by
NAT on material from peripheral blood
mononuclear cells, can characterize the in-
fecting agent. Half or more of US blood do-
nors confirmed infected after EIA screening
provetohaveHTLV-IIinfections.Although
there is no FDA requirement to perform ad-
ditional testing (no confirmatory test for
HTLV is licensed), most centers do so using
an investigational supplemental test or se-
ries of tests in an algorithm if the donation
tests repeatedly reactive by both the test-
of-record and second HTLV-I/II EIAs; if
supplemental tests are positive, the donor
is indefinitely deferred (see Table 28-3). Re-
gardless of the results of these investiga-
tional supplemental tests, a donor meeting
the EIA criteria for indefinite deferral de-
scribed above must still be indefinitely de-
ferred.
Quarantine and Look-Back
In-date prior collections of blood or com-
ponents from donors who subsequently
are found repeatedly reactive for anti-
HTLV-I/II need to be quarantined. Be-
cause recipients of units from seroposi-
tive donors do not consistently sero-
convert and because many seropositive
donors have lifelong infection, the time
frame for look-back is not self-evident. No
requirement for recipient tracing and no-
tification has been established.85 Screen-
ing, in place since 1988, has probably re-
moved from the active donor pool most
donors with lifelong infection.
West Nile Virus
West Nile virus (WNV) is a flavivirus pri-
marily transmitted in birds through mos-
quito bites; humans are incidental hosts.
In humans, symptoms range from a mild
febrile illness to encephalitis, coma, and
death, although about 80% of infected in-
dividuals remain asymptomatic. WNV
outbreaks have been reported in Europe,
the Middle East, and Russia over the past
decade and have been associated with
human encephalitis and meningitis (in
<1%), although no transfusion-associated
cases were reported. WNV was observed
in the United States, in the metropolitan
Chapter 28: Transfusion-Transmitted Diseases 683
Copyright © 2005 by the AABB. All rights reserved.

684 AABB Technical Manual
Table 28-3. Recommended Actions for HTLV-I/II Testing
First Donation to Be Tested for HTLV-I/II Antibodies Subsequent Donation(s)
EIA WB/RIPA Donation Donor EIA
Second
Manufacturer WB/RIPA Donation Donor
Repeatedly
reactive
Positive Destroy all
compo-
nents*
Defer and
counsel
Not applicable/Donor deferred
Repeatedly
reactive
Negative or
indeterm.
Destroy all
compo-
nents*
No action Repeatedly
reactive
Pos Any result Destroy all
compo-
nents*
Defer and
counsel
Nonreactive†Neg Not done All compo-
nents ac-
ceptable
No action
*Destroyed unless appropriately labeled as positive for HTLV-I/II antibodies, and labeled for laboratory research use or further manufacture into in-vitro diagnostic reagents.
†Assuming that separate prior donations have been repeatedly reactive for HTLV-I/II antibody no more than once. If separate prior donations had been repeatedly reactive for
HTLV-I/II antibodies on
two or more
occasions, the donor should have been indefinitely deferred.
HTLV-I = human T-cell lymphotropic virus, type I; HTLV-II = human T-cell lymphotropic virus, type II; EIA = enzyme immunoassay; WB = Western blot; RIPA = recombinant
immunoprecipitation assay.
Copyright © 2005 by the AABB. All rights reserved.
New York City area, in 1999. Subsequently,
it dispersed rapidly westward throughout
the country, spread by infected birds. In
2001, 66 human cases occurred in 10
states. In 2002, 4161 cases of WNV illness
were reported in 39 states, including 284
deaths. In 2002, 23 cases were associated
with transfusion (with an infected donor
identified); Red Blood Cells (RBCs), Fresh
Frozen Plasma, and Platelets were impli-
cated.87 The implicated donations oc-
curred between July 22 and October 6,
2002. Statistical resampling of data avail-
able regarding case onset dates during the
2002 epidemic was used to generate esti-
mates of the mean risk of transfusion-as-
sociated WNV transmission (per 10,000
donations) for six states and six selected
metropolitan areas, with results ranging
from a mean of 2.12 to 4.76 and 1.46 to
12.33, respectively.88
An FDA guidance document in May
2003 recommended deferral of donors with
a diagnosis of WNV infection for 28 days af-
ter onset of symptoms or 14 days after reso-
lution, whichever is later, and recom-
mended inquiring whether donors had
experienced a fever with headache in the
week before donation.89 However, because
most infected persons are asymptomatic,
the yield of such measures would be ex-
pected to be modest, and testing was
sought as the best method to identify in-
fected donors. During the summer and fall
of 2003, almost 5 million donations consti-
tuting over 95% of collections in the United
States were tested for WNV by NAT in
minipools of six or 16 samples under one of
two IND protocols—one using a polymer-
ase chain reaction method and one using a
transcription-mediated amplification
method. During 2003, approximately 1000
donors were confirmed as viremic for WNV
and approximately 1500 likely infected
components were interdicted.90 However,
six probable or confirmed transfusion-as-
sociated WNV cases were reported in 2003;
four recipients had WNV encephalitis, one
had West Nile fever, and one critically ill
patient did not have discernible WNV-com-
patible illness despite confirmed WNV in-
fection.91 Some of the donors were identi-
fied through retrospective testing of
individual samples, and it appears that all
were related to specimens with very low vi-
ral titers. The number of transfusion-asso-
ciated cases undoubtedly would have been
much higher had widespread testing under
IND protocols not been initiated. There
were 9862 WNV cases and 264 deaths in the
United States overall during this time.
The persistent low-level transmission of
WNV by transfusion in 2003 led to the im-
plementation of targeted ID-NAT in high
epidemic regions in 2004. This effort suc-
cessfully interdicted low-level viremic units
that would have been missed by MP-
NAT.92,93
In 2004, the virus appeared predomi-
nantly in western states, with California ac-
counting for 31% of cases.94 Only three
states (Hawaii, Alaska, and Washington) re-
mained free of reports of infection in either
humans or animals (mammalian and
avian). A total of 2470 cases of human WNV
infection were reported in 40 states plus the
District of Columbia, including 88 fatal
cases—far fewer than the previous year. A
total of 192 WNV-positive donors were
identified, almost all from states west of the
Mississippi River.94 Of these donors, three
subsequently reported neuroinvasive WNV
illness and 55 subsequently developed
WNV fever.94 One probable transfusion-as-
sociated case was reported.95
Theexperiencetodateindicatesthat
blood screening for WNV has improved
blood safety. However, a small risk of WNV
transfusion-associated transmission re-
mains. If the number of WNV cases contin-
ues to dramatically decline, the need for
WNV testing will be reassessed. The FDA
Chapter 28: Transfusion-Transmitted Diseases 685
Copyright © 2005 by the AABB. All rights reserved.
has agreed that asking the question con-
cerning fever with headache in the week
before donation may be discontinued (K
Gregory, personal communication,
3/30/05).
Herpesviruses and Parvovirus
Cytomegalovirus
CMV, a member of the human herpes-
virus family, is a ubiquitous DNA virus
that causes widespread infection; trans-
mission can occur through infectious
body secretions, including urine, oro-
pharyngeal secretions, breast milk, blood,
semen, and cervical secretions. About 1%
of newborns are infected, transplacentally
or through exposure to infected cervical
secretions at delivery or by breast milk. In
early childhood, CMV is often acquired
through close contact, especially in day-
care settings; in adulthood, through sex-
ual intercourse. The prevalence of CMV
antibodies ranges from 50% to 80% in the
general population.96 Therateincreases
with age and is generally higher in lower
socioeconomic groups, in urban areas,
and in developing countries.
Clinical Observations
Inpersonswithanintactimmunesystem,
CMV infection may be asymptomatic and
remain latent in tissues and leukocytes
for many years. Infection, either primary
or reactivation of latent infection, can be
associated with a mononucleosis-like
syndrome of sore throat, enlarged lymph
nodes, lymphocytosis, fever, viremia,
viruria, and hepatitis. Intrauterine infec-
tion may cause jaundice, thrombocyto-
penia, cerebral calcifications, and motor
disabilities; the syndrome of congenital
infection causes mental retardation and
deafness and may be fatal.
CMV infection can progress to CMV dis-
ease and cause serious morbidity and mor-
tality in premature infants, recipients of
organ, marrow, or peripheral blood progen-
itor cell transplants, and in AIDS patients.96
Pneumonitis, hepatitis, retinitis, and multi-
system organ failure are manifestations of
CMV disease. CMV infection can result from
blood transfusions. Other sources of infec-
tion, however, such as organ transplants
from CMV-positive donors or reactivation
of latent virus, may be as much or more of a
risk than transfusion.
Transfusion-Transmitted CMV
Infection with CMV varies greatly accord-
ing to socioeconomic status and geo-
graphic region. Although approximately
50% of blood donors can be expected to
be CMV seropositive, it has been esti-
mated that, currently, less than 1% of se-
ropositive cellular blood components are
able to transmit the virus.96,97 Rarely, post-
transfusion hepatitis may be due to CMV.
The postperfusion mononucleosis syn-
drome that first focused attention on
CMV in transfused components in the
early 1960s is now rarely seen. Posttrans-
fusion CMV infection is generally of no
clinical consequence in immunocompe-
tent recipients, and intentional selection
of CMV-reduced-risk blood (see below) is
not warranted.
In light of the potential for severe CMV
disease in immunocompromised patients,
several categories of recipients have been
identified who should be protected from
transfusion-transmitted CMV.7These in-
clude low-birthweight premature infants
born to seronegative mothers; seronegative
recipients of hematopoietic progenitor cells
from CMV-negative donors; seronegative
pregnant women, because the fetus is at
risk of transplacental infection; and recipi-
686 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
ents of intrauterine transfusions. In some
cases, seronegative recipients of organ
transplants from a seronegative donor;
seronegative individuals who are candi-
dates for autologous or allogeneic hemato-
poietic progenitor cell transplants; and
those few patients with AIDS who are free
of CMV infection are also included.
Preventive Measures
Blood from donors who test negative for
CMV antibody has very little risk of trans-
mitting CMV, but the supply of seronega-
tive blood is limited.96,97 Another approach
to reduce risk is to remove leukocytes
from donated blood (because leukocytes
are the principal reservoir for CMV).98-100
Although the precise leukocyte population
that harbors the virus has not been de-
fined, leukocyte removal with high-effi-
ciency filters, to 5 ×106leukocytes per
component or fewer, can significantly re-
duce, if not prevent, posttransfusion CMV
in high-risk neonates and transplant re-
cipients. Effectively leukocyte-reduced
cellular components are considered equiv-
alent to serologically screened compo-
nents by many experts, although this is
controversial.98-100 The incremental benefit
of serologic testing when added to leuko-
cyte reduction has not been established.
Prophylactic therapy with CMV immune
globulin and prophylactic use of antiviral
agents are being investigated as options
for high-risk immunosuppressed organ
transplant recipients.96,97
Epstein-Barr Virus
EBV causes most cases of infectious mono-
nucleosis and is closely associated with
the endemic form of Burkitt’s lymphoma
in Africa and with nasopharyngeal carci-
noma. Most persons have been infected
by the time they reach adulthood; al-
though usually asymptomatic, infection
persists. Infection is spread by contact
with infected saliva. Primary infection in
children is either asymptomatic or is
characterized by a sore throat and en-
larged lymph nodes. Primary infection in
older, immunologically mature persons
usually causes a systemic syndrome, infec-
tious mononucleosis, with fever; tonsillar
infection, sometimes with necrotic ulcers;
enlarged lymph nodes; hematologic and
immunologic abnormalities; and some-
times hepatitis or other organ involvement.
EBV infection targets B lymphocytes,
which undergo polyclonal proliferation and
then induce a T-lymphocyte response, ob-
served as “atypical lymphocytes.”
Transfusion-transmitted EBV infection is
usually asymptomatic, but it has been a
rare cause of the postperfusion syndrome
that follows massive transfusion of freshly
drawn blood during cardiac surgery and is
a rare cause of posttransfusion hepatitis.101
EBV plays a role in the development of na-
sopharyngeal carcinoma and at least one
form of Burkitt’s lymphoma and has the
in-vitro capacity to immortalize B lympho-
cytes. EBV contributes to the development
of lymphoproliferative disorders in im-
munosuppressed recipients of hemato-
poietic and organ transplants. Given a 90%
seropositivity rate for EBV among blood do-
nors and essentially no risk for clinical dis-
ease from transfusion-transmitted EBV in
immunocompetent recipients, serologic
screening for this virus has not been con-
sidered helpful. As is the case for CMV, leu-
kocyte reduction of cellular blood compo-
nents would be expected to reduce the risk
of EBV infection in severely immunosup-
pressed seronegative patients who may be
at risk for clinical disease. However, there
have been no studies to verify such a
reduced risk.
Chapter 28: Transfusion-Transmitted Diseases 687
Copyright © 2005 by the AABB. All rights reserved.
Human Herpesviruses 6 and 8
As with CMV and EBV, human herpesvirus
6 (HHV-6) is a cell-associated virus that
integrates with the genome of lympho-
cytes.Theseroprevalenceinsomeadult
populations approaches 100%. Primary
infection in immunocompetent children
is recognized as exanthem subitum, a fe-
brile illness characterized by a rash; it is
rarely complicated by involvement of other
organ systems. Immunocompromised pa-
tients (eg, those with transplants or AIDS)
may experience manifestations of reacti-
vation of HHV-6 infection in multiple or-
gan systems. Studies have sought an asso-
ciation between multiple sclerosis and
HHV-6 infection, but this remains contro-
versial. With the ubiquity of antibodies to
HHV-6 and absence of disease associations
after transfusion transmission, no recom-
mendations have been made for protec-
tion of seronegative blood recipients from
transmission by blood components.102
HHV-8 (also known as Kaposi’s sarcoma-
associated herpesvirus or KSHV) is causally
associated with both Kaposi’s sarcoma and
body cavity-based lymphomas. It has been
found in apparently healthy blood donors,
but spread appears to be primarily by the
venereal route. Low titers of HHV-8 antibodies
were found in 11% of 91 healthy US blood
donors.103 Among HHV-8-seropositive women,
injection drug use and indices of sexual ac-
tivity were independent risk factors for
HHV-8 infection.104 In this study, the associ-
ation with injection drug use suggests that
transmission by infected blood is possible.
Transmission by organ donation has been
documented.105-107 Epidemiologic studies
suggest that blood transfusion is associated
with a small risk of HHV-8 transmission.108
Parvovirus
Parvovirus B19 is the cause of erythema
infectiosum or “fifth disease,” a contagious
febrile illness of early childhood. Infections
in adults may be associated with arthritis
but are generally benign. More ominou-
sly, parvovirus B19 can infect and lyse red
cell precursors in the marrow.109 This may
result in sudden and severe anemia in pa-
tients with underlying chronic hemolytic
disorders who depend on active erythro-
poiesis to compensate for shortened red
cell survival. Patients with cellular immu-
nodeficiency, including those infected with
HIV, are at risk for chronic viremia and as-
sociated hypoplastic anemia. Infection dur-
ing pregnancy predisposes to spontaneous
abortion, fetal malformation, and hydrops
from severe anemia and circulatory failure.110
The red cell P antigen is the cellular re-
ceptor for parvovirus B19, and people who
do not have the P antigen are naturally re-
sistant to infection.111 About 30% to 60% of
normal blood donors have antibodies to par-
vovirus B19, which indicates immunity
rather than chronic persistent infection.109
Viremia occurs only in the early phases of
infection and there is no evidence for a car-
rier state; the incidence of viremia in blood
donors has been estimated to range from 1
in 3300 to 1 in 40,000.110 Parvovirus B19 lacks
a lipid envelope and is therefore not inacti-
vated by solvent/detergent treatment or heat
inactivation using temperatures below 100
C.109 The virus has been found regularly in
clotting factor concentrates and has been
transmitted to persons with hemophilia. Rare
transmission through cellular blood compo-
nents and plasma, but not intravenous im-
munoglobulin and albumin, has been re-
ported.110
After the description of parvovirus B19
seroconversion (without clinical illness) in
volunteers during Phase IV clinical evalua-
tion of solvent/detergent-treated plasma,
derivative manufacturers (with the concur-
rence of FDA) began development and im-
plementation of NAT screening for high-ti-
ter parvovirus B19 viremic donations in
688 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
minipools. The rationale, supported by ob-
servations from the Phase IV evaluation, is
that high-titer donations overwhelm neu-
tralizing antibody in plasma pools, allowing
transmission of this highly resistant virus
by some derivatives. The FDA has classified
this MP-NAT as an in-process manufactur-
ing control rather than a donor screening
test. Screening of whole blood donations has
not been a high priority because of the be-
nign and/or transient nature of most par-
vovirus disease, the availability of effective
treatment (intravenous immunoglobulin)
for chronic hematologic sequelae, and the
extreme rarity of reports of parvovirus B19
transmission by individual components.112
Transmissible Spongiform
Encephalopathies
The transmissible spongiform encephalo-
pathies (TSEs) are degenerative brain dis-
orders caused by agents often called prions,
postulated to be infectious proteins. They
are characterized by long incubation peri-
ods, measured in years to decades, and by
the extreme resistance of the pathogens
to inactivation by physical and chemical
methods sufficient for classic pathogens.
Two TSEs, Creutzfeldt-Jakob disease (CJD)
and variant Creutzfeldt-Jakob disease
(vCJD), are of particular interest in trans-
fusion medicine.
Classic CJD
CJD is a degenerative brain disorder that
is rapidly fatal once symptoms of progres-
sive dementia and motor disturbances
develop. Approximately 85% of cases are
sporadic. Symptoms do not develop until
many years to several decades after the
initial infection. Ten to fifteen percent of
cases are familial, associated with inheri-
tance of one of at least 20 described mu-
tations in the prion gene that, in its non-
mutated form, encodes for a normal cel-
lular protein. Worldwide, there is about
one case of CJD per million people per
year, nearly all in older individuals. In
sporadic CJD, the vast majority of cases,
themodeofacquisitionisunknown.The
agent causing CJD is resistant to commonly
used disinfectants and sterilants. Iatro-
genic CJD has been transmitted by ad-
ministration of growth hormone and go-
nadotropic hormone derived from pooled
human pituitary tissue, through allografts
of dura mater, and through reuse of intra-
cerebral electroencephalographic elec-
trodes from infected patients.113
Early experimental studies in animals
raised the possibility that CJD could be
transmitted by blood transfusion. Addition-
ally, iatrogenic transmission from periph-
eral injection of human pituitary-derived
hormones has been observed. Neverthe-
less, several population-based, case-con-
trolled studies have shown no evidence that
blood transfusion is a risk factor for the de-
velopment of CJD.114-116
Individuals at increased risk for CJD are
excluded from donating blood; this group
includes persons who have received tissue
or tissue derivatives known to be a source
of the CJD agent (eg, dura mater allografts,
pituitary growth hormone of human origin)
and persons with a family history of
CJD.117-119 For the purposes of donor exclu-
sion, unit quarantine, and unit destruction,
family history has been defined as having
one blood relative who has had this diagno-
sis. However, a donor with CJD in a family
member may be accepted if gene se-
quences have been tested and found to be
normal.
Variant CJD
In 1996, the first cases of an unusual CJD
outbreak or cluster were described in the
Chapter 28: Transfusion-Transmitted Diseases 689
Copyright © 2005 by the AABB. All rights reserved.
United Kingdom (UK). These cases were
later termed variant CJD (vCJD) and ap-
peared to be caused by the same prion
responsible for bovine spongiform en-
cephalopathy (BSE). This prion is distinct
from the prion found in classical CJD. A
donor infected by dietary exposure to BSE
during the incubation period of vCJD might
theoretically infect a transfusion recipi-
ent. Consequently, UK health authorities
prohibited the use of UK plasma for fur-
ther manufacture, restricted the use of UK
plasma for children (born on or after 1
January 1996), and implemented univer-
sal leukocyte reduction of cellular compo-
nents (to reduce the prions known to be
present in white cells).
Experimental transfusion transmission
of BSE to two sheep (and four cases of
transmission of natural scrapie—a sheep
prion illness) have been reported.120 Positive
transmissions occurred with blood taken at
preclinical and clinical stages of infection.
Two cases of transfusion-transmitted BSE
in humans have been observed121,122 as a re-
sult of surveillance in the UK of 48 individ-
uals identified as having received a labile
blood component from a total of 15 donors
who later developed vCJD. In the first possi-
ble case, the recipient developed symptoms
of vCJD 6.5 years after receiving a transfu-
sion of red cells donated by an individual
3.5 years before the donor developed
symptoms of vCJD. Although the source of
the infection could have been caused by
past dietary exposure to the BSE agent, the
age of the patient was well beyond that of
most vCJD cases, and the chance of observ-
ing a case of vCJD in a recipient in the ab-
sence of transfusion-transmitted infection
was estimated to be about 1 in 15,000 to 1
in 30,000, making dietary transmission un-
likely in this case.121 In the second possible
case, the person received a blood transfu-
sion in 1999 from a donor who later devel-
oped vCJD. This patient died of causes
unrelated to vCJD, but a postmortem exam-
ination revealed the presence of the
abnormal prion protein in the patient’s
spleenandinalymphnode.
122 Notably, un-
like previous cases of vCJD (by any method
of transmission), in which all involved peo-
ple were homozygous for methionine (MM)
at codon 129 of the prion protein gene
(PRNP), this individual was heterozygous
(methionine valine— MV). In the UK, the
population distribution of this gene is MM,
MV, or VV in 42%, 47%, and 11%, respec-
tively.123
In the United States, blood donors who
wereintheUKorEuropeduringtheyears
of potential exposure to the BSE agent are
deferred based on the duration of residence
there. Balancing the theoretical risk against
considerations of the adequacy of the blood
supply, the recommended deferral is for 3
months of cumulative residence in the UK
between 1980 and 1996 (and current and
former US military personnel, civilian mili-
tary employees, and their dependents who
were stationed at European bases for 6
months or more during this period) or 5
years of cumulative residence in Europe.
Potential donors who may have injected
bovine insulin from the UK or received
transfusions in the UK during the BSE epi-
demic are also excluded.119
Bacterial Contamination
Bacterial contamination remains an im-
portant cause of transfusion morbidity
and mortality. Bacterial contamination of
blood components accounted for 29 (16%)
of the transfusion fatalities reported to
the FDA between 1986 and 1991. How-
ever, in 2002 alone, there were 17 deaths
reported to the FDA from bacterial con-
tamination of blood components, most
commonly caused by contaminated
apheresis platelets and whole-blood-de-
690 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
rived platelets.124,125 Although the hepatitis
viruses, HIV, and WNV have been more
prominently featured in the media and
remain a primary concern of the public,
bacterial contamination is believed to be
the most common infectious source of
morbidity and mortality related to trans-
fusion. To place the risk of bacterial con-
tamination into some perspective, in 2002,
there were 23 transfusion-transmitted cases
of WNV identified in the United States.87
Of these 23 recipients, seven died, but
only five of these deaths were associated
with WNV meningoencephalitis. Thus, the
deaths from bacterial contamination were
more than three times more common than
those from WNV.
No matter how carefully blood is drawn,
processed, and stored, complete elimina-
tion of microbial agents is impossible. Bac-
teria are most often believed to originate
with the donor, either from the venipunc-
ture site or from unsuspected bacteremia.126
Bacterial multiplication is more likely in
blood components stored at room temper-
ature than in refrigerated components.126
Organisms that multiply in refrigerated
blood components are often psychrophilic
gram-negative organisms (such as Yersinia
enterocolitica, Serratia liquifaciens, and
Pseudomonas fluorescens). Gram-positive
organisms are more often seen in platelets
stored at 20 to 24 C.
For RBCs, the CDC estimates a sympto-
matic contamination rate of approximately
1 case per million units, primarily with Y.
enterocolitica, followed by S. liquifaciens.127
InNewZealand,theincidenceofsymp
-
tomatic Yersinia contamination of RBC
units has been reported to be as high as
one in 65,000 units, with a fatality rate of
one in 104,000.128 TransfusionofanRBC
unit heavily contaminated with a gram-
negative organism is often a rapid and cata-
strophic event, with a quick onset of sepsis
and a greater than 60% mortality rate.129,130
Because platelets are stored at 20 to 24 C
to retain their viability and function, they
serve as an excellent growth medium for
bacteria. Sepsis resulting from transfusion
of contaminated platelets is believed to be
both underrecognized and underreported.
Sepsis occurring after transfusion of con-
taminated platelets is usually not a cata-
strophic event, but it can occur several
hoursorlongeraftertransfusion,makingit
more difficult to connect the transfusion to
the sepsis. Because many of the patients in-
fected by bacteria from a platelet transfu-
sion are immunocompromised by their
underlying condition and treatment (eg,
chemotherapy), the event is frequently at-
tributed to other causes, such as an in-
fected catheter, which often involves the
same organisms.
In the United States, 4 million platelet
units are transfused annually (1 million
apheresis platelets and 3 million whole-
blood-derived platelet concentrates).130
Given that approximately 1:1000 to 1:2000
platelet units are contaminated with bacte-
ria (as measured by aerobic cultures done
in multiple studies before 2002), it would
be expected that 2000 to 4000 bacterially
contaminated units would be transfused.131
Estimates of the fraction of such units that
would result in signs or symptoms have
been as low as 1 in 10 cases. However, in
the only study that has prospectively cul-
tured platelets that were transfused, symp-
toms occurred in 3 of 8 (35.8%) patients
who received culture-positive but Gram’s-
stain-negative platelet pools.132 Notably, six
Gram’s-stain-positive pools were inter-
dicted and never transfused. Thus, of con-
taminated products, perhaps 1/10 to 2/5
would be expected to result in clinical sep-
sis (200 to 1600 cases) if transfused. Data
from national passive reporting studies in
the United States, Great Britain, and France
(Table 28-4) suggest that perhaps 1/5 to 1/3
would result in death (40 to 533 deaths per
Chapter 28: Transfusion-Transmitted Diseases 691
Copyright © 2005 by the AABB. All rights reserved.

year).127,133,134 This translates to a risk of death
from a transfusion of a platelet unit con-
taminated with bacteria of between 1:7500
to 1:100,000. Clinical observations from
university hospitals with heightened aware-
ness of platelet-related sepsis confirm such
estimates. A fatality rate of 1:17,000 has
been reported by Ness et al, from Johns
Hopkins, with pooled whole-blood-derived
platelets and 1:61,000 with apheresis plate-
lets.135 University Hospitals of Cleveland
similarly observed a fatality rate of approxi-
mately 1:48,000 per whole-blood-derived
platelet concentrate.136 With the implemen-
tation of bacteria detection of platelets (see
below), it is anticipated that this rate will be
greatly reduced.
Clinical Considerations
Severe reactions are characterized by fe-
ver, shock, and disseminated intravascular
692 AABB Technical Manual
Table 28-4. Summary of Organisms Identified in the BaCon, SHOT, and BACTHEM
Studies*
Organism
United
States127
United
Kingdom133 France134 Total
Gram positive
Bacillus cereus
14(1)27(1)
Coagulase-negative
Staphylococci
9 6 (1) 5 20 (1)
Streptococcus
sp
.
3(1) 2 5(1)
Staphylococcus aureus
4 2 (1) 6 (1)
Propionibacterium
acnes
33
Subtotal 17 (1 = 6%)†14 (3 = 21%) 10 (0 = 0%) 41 (4 = 10%)
Gram negative
Klebsiella
sp
.
2(1) 2(1)
Serratia
sp
.
2(2) 1(1) 3(3)
Escherichia coli
5(1) 2(1) 1 8(2)
Acinetobacter
11
Enterobacter
sp
.
2(1) 1(1) 1 4(2)
Providencia rettgeri
1(1) 1(1)
Yersinia enterocolitica
11
Subtotal 11 (5 = 45%) 3 (2 = 67%) 6 (2 = 33%) 20 (9 = 45%)
Total 28 (6 = 21%) 17 (5 = 29%) 16 (2 = 13%) 58 (13 = 14%)
*Number of cases (fatalities) and the percent of the subtotal and total cases are listed. This table illustrates that although
gram-positive organisms are associated with the majority of reported cases (41/58 = 71/%), gram-negative organisms
account for the majority of deaths (9/11 = 82%). Modified with permission from Brecher and Hay.131
†There were 17 cases of gram-positive organisms identified in the US study; however, only one case (1/17 = 6%) re-
sulted in a fatality.
Copyright © 2005 by the AABB. All rights reserved.
coagulation (DIC). If bacterial contamina-
tion is suspected, the transfusion should
be stopped immediately and a Gram’s stain
and blood culture should be obtained
from the unit (not an attached segment of
tubing because the bag may be contami-
nated but an isolated segment of tubing
may be sterile) and recipient as promptly
as possible after the reaction is observed.
Bacterial multiplication may cause the
oxygen in an RBC unit to be consumed,
resulting in hemoglobin desaturation and
erythrocyte lysis, both of which contrib-
ute to a darkening of the unit compared
to the color of the blood in the attached
sealed segments. Color change (to dark pur-
ple or black), clots in the bag, or hemoly-
sis suggest contamination, but the ap-
pearance of the blood in the bag is often
unremarkable. The presence of bacteria
on a Gram’s stain of the component is
confirmatory, but absence of visible or-
ganisms does not exclude the possibility.
Gram’s stain has a sensitivity of only 106to
107CFU/mL.Thepatient’sblood,thesus-
pect component, and intravenous solu-
tions in all the administration tubing used
should be cultured.
Treatment should not await the results of
these investigations and should include im-
mediate intravenous administration of an-
tibiotics combined with therapy for shock,
renal failure, and DIC, if present.
Preventive Measures
Prevention of septic reactions depends upon
reducing or preventing contamination of
components with bacteria. Careful selec-
tion of healthy blood donors is the first
and most important step.
The donor’s present appearance and re-
cent medical history should indicate good
health; additional questioning may be
needed if there is a present or recent his-
tory of antibiotic use, of medical or surgical
interventions, or of any constitutional
symptoms. Questions to elicit the possibil-
ity of bacteremia are especially important
for autologous donors, who may have un-
dergone recent hospitalization, antibiotic
therapy, or invasive diagnostic or therapeu-
tic procedures; there have been several re-
ports of Yersinia sepsis complications after
the infusion of stored autologous blood.
Scrupulous attention must be paid to se-
lecting and cleansing the donor’s phlebot-
omy site. Skin preparation reduces but does
not completely abrogate the contamination
of components by bacteria. Scarred or dim-
pled areas associated with previous derma-
titis or repeated phlebotomy can harbor
bacteria and should be avoided. Green
soap must not be used to prepare the
phlebotomy site.
Discarding the first aliquot of donor blood
removed (“diversion”) has been proposed
as a measure to reduce bacterial contami-
nation of blood components. This measure
would remove the skin core that may enter
the collection from the hollow bore needle
used in the phlebotomy. Systems have been
developed to facilitate the application of
this approach and would be expected to re-
duce skin contaminants (mostly gram-pos-
itive organisms).
Phagocytosis of contaminating bacteria
by donor white cells in blood components
may be important for the minimization of
clinical bacterial contamination. Leukocyte
removal, with coincident removal of adher-
ent or engulfed bacteria, has been advo-
cated as an approach to reducing Yersinia
contamination of RBCs.137,138
Care in the preparation of components
and handling of materials used in blood ad-
ministration is essential. If a waterbath is
used, components should be protected by
overwrapping, outlet ports should be in-
spected for absence of trapped fluid, and
the waterbath should be frequently emp-
tied and disinfected.
Chapter 28: Transfusion-Transmitted Diseases 693
Copyright © 2005 by the AABB. All rights reserved.
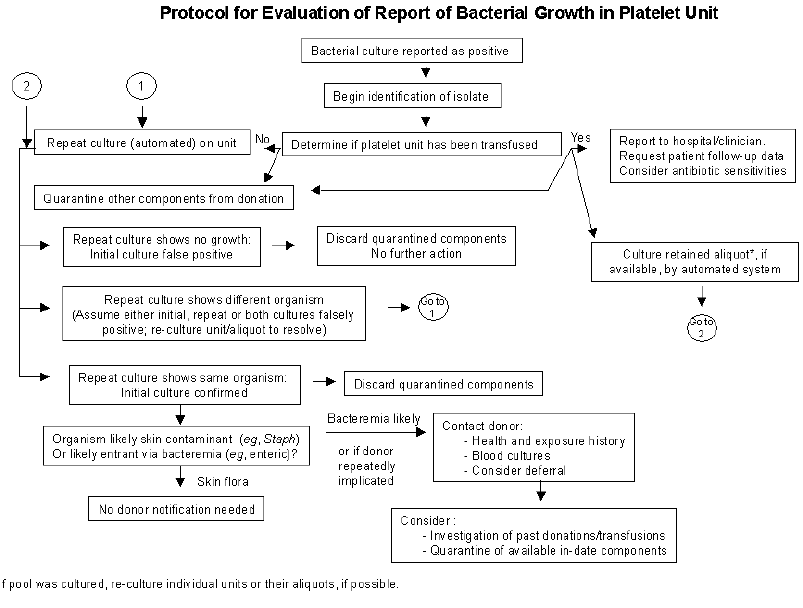
Worldwide, screening of platelets for
bacteria is being implemented. Screening is
mandatory in several countries [eg, Bel-
gium (Flemish Red Cross), the Netherlands,
Hong Kong (Red Cross), and Wales].139
In the United States, the College of
American Pathologists (CAP) Commission
on Laboratory Accreditation has added a
question to the Transfusion Medicine
Checklist to assess the presence of a labora-
tory system to detect bacteria in platelet
components (TRM.44955).140 Similarly, the
AABB requires bacteria detection.62(p11) Cur-
rently, two culture techniques are approved
by the FDA for quality control of leuko-
cyte-reduced platelets and are available in
the United States. Because of expense and
logistics, whole-blood-derived platelets are
often tested in the United States with less
sensitive but more rapid detection strate-
gies, such as staining or the use of surrogate
markers of bacterial metabolism (eg, pH
and glucose). Several other more rapid and
sensitive detection strategies are under de-
velopment or are not readily available. One
possible investigative strategy after detect-
ing a confirmed culture-positive platelet
unit is outlined in Fig 28-4.
Prospect for Extended Storage
The extent of bacterial growth in platelet
components correlates with the duration
of storage. In 1983, in recognition of tech-
694 AABB Technical Manual
Figure 28-4. Possible investigative strategy for a positive culture in a platelet unit. Modified with per-
mission.141
Copyright © 2005 by the AABB. All rights reserved.
nically improved storage conditions, the
FDA increased storage limits of platelets
at room temperature from 3 to 7 days.
However, it reduced the limits to a maxi-
mum of 5 days in 1986, responding to re-
ports of bacterial contamination after
more than 5 days of storage.142 The use of
bacteria detection systems has been given
as the rationale for an extension of plate-
let storage to 7 days in several European
countries and is being implemented in the
United States.139
Syphilis
Syphilis is caused by the spirochete Tre-
ponema pallidum and is characteristically
spread by sexual contact. The phase of
spirochetemia is brief and the organisms
survive only a few days at 4 C. Although
transmission by transfusion is possible,
its occurrence is exceedingly rare (the last
case reported in the United States oc-
curred in 1965). Syphilis transmission by
transfusion may not be effectively pre-
vented by subjecting the donor blood to
standard serologic tests for syphilis (STS)
because seroconversion often occurs after
the phase of spirochetemia. Most positive
STS results reflect immunologic abnor-
malities unrelated to syphilis (biologic
false-positives), inadequately treated non-
infectious syphilis that is more of a threat
to the individual being tested than to a
transfusion recipient, or the serologic re-
sidual of an effectively treated infection.
A recent series described T. pallidum
DNAandRNAtestingof169aliquotsfrom
platelet concentrates that were reactive by
STS and confirmed positive by fluorescent
treponemal antibody absorption.143 This se-
ries included 48 donors who were positive
by rapid plasma reagin tests (compatible
with recent or active disease). No sample
contained T. pallidum DNA or RNA, sug-
gesting a low probability that the blood of
donors who have a confirmed positive
syphilis test result is infectious for syphilis.
Nevertheless, a study from the CDC showed
that from 1995-2000, 22 primary, 81 sec-
ondary, and 413 early latent syphilis cases
were identified through blood or plasma
donor screening in the United States.144
Thus, screening of blood donors for syphilis
may have broader public health implica-
tions. Currently, performance of a STS is
still required.62(pp33,34)
Tick-Borne Infections
Because many tick-borne infectious agents
circulate in the blood, it is theoretically
possible that they will be transmitted by
transfusion of blood components.
Babesia
Clinical Events
In the United States, the most frequently
recognized transfusion-associated tick-borne
infection is babesiosis.145 Babesiosis is
usually transmitted by the bite of an in-
fected deer (black-legged) tick and is re-
ported most frequently in the coastal lands
and islands of northeastern United States,
including Martha’s Vineyard, Cape Cod, and
Long Island. Geographic areas of the hosts
and the vectors appear to be expanding.146
Transfusion-associated babesiosis has been
documented in more than 50 cases, caused
mostly by Babesia microti from the North-
east, but also by the recently recognized
WA1-type Babesia parasite, from asymp-
tomatic infected blood donors.145-147 As hu-
mans continue to encroach on the habitat
of vectors and natural reservoirs of infec-
tion [eg, deer (and other Cervidae)and
mice populations in the northeastern
United States], the incidence of transfu-
sion-transmitted babesiosis may increase.
Chapter 28: Transfusion-Transmitted Diseases 695
Copyright © 2005 by the AABB. All rights reserved.
The vector and reservoir of the Babesia
more recently found in the northwestern
and western United States remain to be
defined. Babesia species survive blood
bank storage for up to at least 35 days and
canbetransmittedbybothRBCsand
platelet concentrates. Babesiosis classi-
cally causes a febrile illness with hemoly-
tic anemia, but infection can also cause
chronic asymptomatic or mildly symp-
tomatic parasitemia. Studies suggest that
untreated persons can harbor B. microti
DNA for long periods, despite mild or ab-
sent symptoms, and may transmit infec-
tion for months or possibly longer.148 Sym-
ptoms are often so mild that the infection
is not recognized, which likely explains
the low rate of reported transfusion-trans-
mitted babesiosis. Symptomatic patients
develop fever 2 to 8 weeks after transfu-
sion, sometimes associated with chills,
headache, hemolysis, or hemoglobinuria.
Rarely, life-threatening hemolytic anemia,
renal failure, and coagulopathy develop,
particularly in asplenic or severely im-
munocompromised patients.149
Preventive Measures
The Babesia carrier state may be asymp-
tomatic and may exceed a year in dura-
tion. Persons with a history of babesiosis
are indefinitely deferred, because lifelong
parasitemia can follow recovery from
symptomatic illness. Restrictive policies,
such as not collecting blood in areas
where the disease vectors are endemic
during spring and summer months when
tick bites are more common, are in prac-
tice in some locations but probably are of
limited value. No test is available for mass
screening to detect asymptomatic carriers
of Babesia species.
Other Agents
One case of transfusion transmission of
Rocky Mountain spotted fever (Rickettsia
rickettsii) and no cases of human mono-
cytic ehrlichiosis (caused by Erlichia chaf-
feensis) have been documented.150 Asingle
possible transfusion transmission of the
unnamed agent of human granulocytic
erlichiosis has been reported.151 In 1997,
Rocky Mountain spotted fever and/ or hu-
man monocytic erlichiosis developed in
National Guard trainees at Fort Chaffee,
AR. Ten components donated by infected
trainees had been transfused before a re-
call; however, none of the persons who re-
ceived blood from infected donors became
clinically ill.152
Lyme disease is the most common tick-
borne infection in the United States.
Borrelia burgdorferi, the causative spiro-
chete, is transmitted through bites of the
deer (black-legged) tick. No transfusion-
related cases have been reported, but
chronic subclinical infections do occur
and experimentally inoculated organisms
can survive conditions of frozen, refriger-
ated, and room temperature storage.153 On
the other hand, the phase of spirochete-
mia seems to be associated with symp-
toms that would render a potential donor
ineligible, and in two reported cases where
the donor became ill shortly after dona-
tion, the recipient did not develop infec-
tion.149 Potential donors who give a history
ofLymediseaseshouldbecompletely
asymptomatic and should have com-
pleted a full course of antibiotic therapy
before they are permitted to donate. Trans-
fusion transmission of tick-borne agents is
biologically plausible and, for some agents,
has been demonstrated. Nevertheless,
modifications to current donor screening
are not likely to be useful because of their
low predictive value and the potential
for nonspecific questions to defer large
696 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
numbers of donors for a small increment in
transfusion safety.153
Other Nonviral Infectious
Complications of Blood
Transfusion
Malaria
Malaria is caused by several species of
the intraerythrocytic protozoan genus
Plasmodium. Transmission usually results
fromthebiteofananophelesmosquito,but
infection can follow transfusion of para-
sitemicblood.Althoughveryrareinthe
United States, malaria is probably the most
commonly recognized parasitic complica-
tion of transfusion; the risk in the United
States is estimated to be <0.3 case per mil-
lion transfusions.154,155 From 1963 to 1999, 93
cases of transfusion-transmitted malaria (10
fatal) in the United States were reported to
CDC.155
The species involved in transfusion-
transmitted malaria in the United States are
P. f a l c i p a r u m (35%), P. m a l a r i a e (27%), P.
vivax (27%), and P. o v a l e (5%).155 Three per-
cent were mixed infections, and 2% were
caused by unidentified species. Fever,
chills, headache, and hemolysis occur a
week to several months after the infected
transfusion; morbidity varies but can be se-
vere, and deaths have occurred, especially
from P. f a l c i p a r u m . Adding to the risk of a
fatal outcome may be a lack of immunity in
the recipient, the patient’s underlying con-
dition(s), and delay in the diagnosis be-
cause of lack of suspicion and unfamiliarity
with the disease in areas where the parasite
is not endemic.
Malaria parasites survive for at least a
week in components stored at room tem-
perature or at 4 C. The parasites can also
survive cryopreservation with glycerol and
subsequent thawing. Any component that
contains red cells can transmit infection,
via the asexual form of the intraerythrocytic
parasite.
Asymptomatic carriers are generally the
source of transfusion-transmitted malaria,
although their parasite density is very low.
Asymptomatic infections rarely persist
more than 3 years, but asymptomatic P.
falciparum and P. v i v a x infections may per-
sist for 5 years, P. o v a l e for 7 years, and P.
malariae can remain transmissible for the
lifetime of the asymptomatic individual. In
extreme cases, transmission of P. v i v a x ,P.
ovale,P. f a l c i p a r u m , and P. m a l a r i a e have
been reported at 27, 7, 13, and 53 years, re-
spectively.156 There are no practical sero-
logic tests to detect transmissible malaria in
asymptomatic donors. Malaria transmis-
sion is prevented by deferral of prospective
donors with increased risk of infectivity,
based on their medical and travel history.
The AABB requires that prospective donors
who have had a diagnosis of malaria, or
who have traveled or lived in a malaria-en-
demic area and have had unexplained
symptoms suggestive of malaria, be de-
ferred for 3 years after becoming asymp-
tomatic.62(p65) Individuals who have lived for
at least 5 consecutive years in areas in
which malaria is considered endemic by
the CDC Malarial Branch shall be deferred
for 3 years after departure from that area.
Individuals who have traveled to an area
where malaria is endemic shall be deferred
for 12 months after departing that area.
These deferral periods apply irrespective of
the receipt of antimalarial prophylaxis. Up-
dated information on malaria risks world-
wide is available from the CDC, including
an on-line resource (http://www.cdc.gov/
travel/yb/outline.htm#2).
Chagas’ Disease
American trypanosomiasis, or Chagas’
disease, is endemic in South and Central
Chapter 28: Transfusion-Transmitted Diseases 697
Copyright © 2005 by the AABB. All rights reserved.
America and is caused by the protozoan
parasite Trypanosoma cruzi. The human
host sustains infection after the bite of
reduviid bugs (called cone-nosed or “kiss-
ing” bugs), which usually exist in hollow
trees, palm trees, and in thatched-roofed
mud or wooden dwellings. Naturally ac-
quired Chagas’ disease in the United States
is exceedingly rare. Five such cases have
been recognized in the United States since
1955, the most recent in 1998 in Tennes-
see.157
Clinical Events
T. cruzi infectshumanswhoseskinormu
-
cosa comes in contact with feces of in-
fected reduviid bugs, usually as the result
of a bite. Recent infections are usually ei-
ther asymptomatic or the very mild signs,
and symptoms go undetected. Rarely, the
site of entry evolves into an erythematous
nodule called a chagoma, which may be
accompanied by lymphadenopathy. Fever
and enlargement of the spleen and liver
may follow. Recently infected young chil-
dren may experience acute myocarditis or
meningoencephalitis. Acute infection
usually resolves without treatment, but
persisting low-level parasitemia is usual
and up to one-third of infected individu-
als develop a chronic form associated with
cardiac or gastrointestinal symptoms
years or decades later.158
Transfusion Considerations
Blood transfusion has been a major source
of infection with T. cruzi in South Ameri-
can urban centers that receive large num-
bers of immigrants from rural areas where
the parasite is endemic. However, in many
countries, serologic screening has been
effective in reducing the risk of transfu-
sion-transmitted Chagas’ disease. Four
cases of transfusion-transmitted Chagas’
diseasehavebeenreportedintheUnited
States158:inNewYork,LosAngeles,Texas,
and Florida. All occurred in immuno-
compromised patients. Additionally, two
cases were reported in Manitoba, Canada.
In one interesting study, postoperative
blood specimens from 11,430 cardiac sur-
gery patients were tested by EIA and, if
repeatedly reactive, were confirmed by
radioimmunoprecipitation. Six postoper-
ative specimens (0.05%) were confirmed
positive. All six seropositive patients ap-
parently were infected with T. cruzi before
surgery; however, a diagnosis of Chagas’
disease was not known or even consid-
ered in any of these patients. No evidence
for transfusion-transmitted T. cruzi was
found.159
Reasonably sensitive and specific EIA
screening tests for antibodies to T. cruzi,as
well as confirmatory Western blot and
radioimmunoprecipitation assays, have
been developed.158 Testing in several US
blood centers located in geographic areas
with a large immigrant population from
Central or South America found a sero-
prevalence of 0.1% to 0.2% among at-risk
donors, who were identified by question-
naire.160,161 However, look-back studies iden-
tified no infected recipients; it is also likely
that not all at-risk donors can be identified
by questionnaire.160 As a consequence, if blood
donor screening were to be implemented,
testing of all donors might be necessary.
Other Parasites
Toxoplasmosis is caused by the ubiquitous
parasite Toxoplasma gondii, and infection
has been reported as an unusual transfu-
sion complication in immunocompromi-
sed patients.162 The disease has not been
considered a problem in routine transfu-
sion practice.
There have been occasional reports of
parasitic worm infections transmitted by
transfusion in countries other than the
698 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
United States.162 Microfilariasis is a potential
transfusion risk in tropical zones, acquired
by donors through bites by insects carrying
Wuchereria bancrofti.Transfusiontrans
-
mission of Leishmania species is a rare risk
in countries where such organisms are en-
demic. Currently, the AABB defers potential
donors who have been to Iraq in the previ-
ous12monthsasaresultofpossibleLeish-
mania exposure.163
Reducing the Risk of
Infectious Disease
Transmission
Overall, the risk per unit of transfusion-
transmitted disease is remarkably low
(Table 28-5). This low incidence is due to
both donor screening and specific disease
testing. Nevertheless, in pooled compo-
nents, which may contain elements from
thousands of donors, the risk of disease
transmission is increased. Therefore, sev-
eral strategies have been developed and
implemented to further reduce the risk of
disease transmission in pooled acellular
components and, in some cases, cellular
components.
Inactivation/Destruction of Agents in
Derivatives or Plasma Products
The first intervention specifically added
to reduce the risk of hepatitis transmis-
sion was heating (to 60 C for 10 hours),
which has been used for albumin prod-
ucts since at least 1948.167 In those rare in-
stances when infections occurred with
plasma protein fractions prepared with
this step, the processing had been com-
promised.
Immunoglobulins
The plasma fractionation process used for
most immunoglobulin products employs
cold ethanol precipitation after removal
of cryoprecipitate. Historically, when anti-
bodies to HCV were present in the plasma,
this process concentrated HCV in the Fac-
tor VIII-rich cryoprecipitate and other
fractions and left little in the immuno-
globulin fraction. The immunoglobulin
fraction also has a high concentration of
virus-neutralizing antibodies and the re-
sulting product for intramuscular appli-
cation has a remarkably low risk of virus
transmission.168
Preparations of immunoglobulin intended
for intravenous administration (IGIV) were
expected to be similarly free of disease
transmission. However, NANB hepatitis
transmission did occur in the 1980s during
early clinical trials of IGIV products in the
United States and with routinely manufac-
tured IGIV products in Europe.169 In late
1993 and early 1994, a worldwide outbreak
with more than 200 reported HCV infec-
tions was traced to a single IGIV prepara-
tion licensed in the United States.170,171 In
this case, transmission apparently occurred
because of lack of virus inactivation steps in
the specific manufacturing process for this
product172 and absence of complexing and
neutralizing anti-HCV subsequent to anti-
HCV screening of plasma donors, with re-
sultant accumulation of virus particles in
the immunoglobulin fraction.170 Anti-HCV-
positive source plasma has been excluded
from the manufacture of IGIV since 1992.
Theimportanceofthemanufacturing
method is underscored by outbreaks of HCV
infection from intravenous anti-D immu-
noglobulin in Germany in the late 1970s
and in Ireland from the late 1970s to the
early 1990s.173,174 Both products were pre-
pared by anion exchange chromatography
rather than cold-ethanol (Cohn) fraction-
ation.175 To prevent further HCV outbreaks,
the FDA has required, since 1994, virus
clearance steps in the manufacturing pro-
cess of immunoglobulin or proof of ab-
Chapter 28: Transfusion-Transmitted Diseases 699
Copyright © 2005 by the AABB. All rights reserved.

700 AABB Technical Manual
Table 28-5. Infectious Risks of Blood Transfusion in the United States
Infectious Agent
or Outcome
Estimated Risk per
Unit Transfused
Estimated % of
Infected Units that
Transmit or Cause
Clinical Sequelae* Reference
Viruses
HIV-1 and -2 1:1,400,000-
1:2,400,000
90 23, 29, 30
HTLV-I and -II 1:256,000-
1:2,000,000
30 84
HAV 1:1,000,000 90 164
HBV 1:58,000-1:147,000 70 29
HCV 1:872,000-1:
1,700,000
90 23, 29, 30
B19 parvovirus 1:3,300-1:40,000 Low 110
Bacteria
RBCs 1:1000 1:10,000,000 fatal 165
Platelets
(screened with
Gram’s stain, pH,
or glucose con-
centration)
1:2000-1:4000 >40% result in clini-
cal sequelae
30, 132, 166
(screened with
early aerobic cul-
ture)
<1:10,000 Unknown
Parasites
Babesia and malaria <1:1,000,000†Unknown 145, 156, 164
Trypanosoma cruzi
Unknown <20 158
*Units that were confirmed test positive for the infectious agent.
Note: West Nile virus is not included in this table because of regional, temporal, and testing (eg, minipool vs individual
donation testing) variation; decreasing rates of infection; and the fact that all testing in the United States is being con-
ducted under an investigational new drug protocol.
†Risk is higher in areas where
Babesia
is endemic.
Copyright © 2005 by the AABB. All rights reserved.
senceofHCVfromthefinalproductby
NAT. In addition, NAT technology is now
applied to screening of source plasma as an
additional layer of safety.
Coagulation Factors
Until the early 1980s, clotting factor con-
centrates frequently transmitted viral in-
fections. As the significance of HIV trans-
mission was recognized, virus inactivation
steps were applied more rigorously to
Factor VIII and other clotting factor con-
centrates, even though these steps were
initially introduced in the hope of reduc-
ing hepatitis transmission. Unfortunately,
a large proportion (over 50%) of the hemo-
philic population receiving concentrates
before processing was improved became
infected with HIV. Chronic hepatitis was
an additional complication in almost all
patients with hemophilia receiving older
clotting factor products.176
The thermal instability of Factor VIII
made it difficult to develop an effective heat
treatment, until a practical approach was
widely adopted in 1985. Since then, many
virus inactivation steps have been introduced,
and factor concentrates are now, in general,
very safe products. Each process has its
own set of advantages and disadvantages.
Application of organic solvents and deter-
gents inactivates viruses with a lipid-con-
taining envelope (eg, HIV, HBV, HCV, HTLV,
EBV, CMV, HHV-6, HHV-8) but is ineffective
against nonenveloped agents such as HAV
and parvovirus B19. Virus inactivation steps
have the potential drawback of reducing the
potency and biologic effectiveness of the
product. Another concern is whether virus
inactivation steps affect immunogenicity,
especially the induction of Factor VIII in-
hibitors in patients with hemophilia.
Current Risks of Human Plasma Deriva-
tives. Many methods are highly effective
against enveloped viruses, but sporadic re-
ports of viral transmission continue to
occur, possibly resulting from accident or
error during the manufacturing process.
The combination of heat treatment, sol-
vent/detergent treatment, and purification
steps with monoclonal antibodies provides
clotting factor concentrates with a risk of
transmitting hepatitis and HIV that is lower
than the risk associated with use of Cryo-
precipitated Antihemophilic Factor derived
from individual voluntary whole blood do-
nations. Documented transmission of HBV,
HCV, or HIV by US-licensed plasma deriva-
tivesisraresincetheintroductionofeffec
-
tive virus inactivation procedures and im-
proved viral screening. Although absolute
safety of products derived from human
plasma cannot be guaranteed, starting with
the safest possible donated plasma reduces
the viral load and has contributed to the ex-
cellent safety record of the products sub-
jected to virus inactivation/removal.177
Avoiding Human Plasma. Factor VIII
concentrates produced by recombinant
DNA technology are licensed for use and
have become the preparation of choice for
previously untreated patients with hemo-
philia.178 Batches are produced by culture of
mammalian cells engineered to secrete
Factor VIII into the supernatant medium,
which is purified by ion-exchange chroma-
tography and immunoaffinity chromatog-
raphy using a mouse monoclonal antibody.
Exceptforthefactthatexcipienthumanal
-
bumin is sometimes added to stabilize Fac-
tor VIII, the product is free of human pro-
teins, HIV, hepatitis viruses, and other
unwanted agents, thus avoiding many of the
risks associated with using human plasma.
On the other hand, recombinant products
have a relatively short history of use and
there is no guarantee that they are risk-free.
Plasma
Virus reduction steps, originally developed
for purified plasma protein fractions, have
Chapter 28: Transfusion-Transmitted Diseases 701
Copyright © 2005 by the AABB. All rights reserved.
also been applied to plasma intended for
transfusion. Alternative approaches being
studied include organic solvents and de-
tergents and use of photochemicals. Sol-
vent/detergent treatment, which is effec-
tive against lipid-enveloped viruses, involves
addition of 1% Triton X-100 and 1% tri-n-
butyl phosphate (TNBP) to pooled plasma,
followed by oil extraction of the TNBP and
chromatographic adsorption of the Triton
X-100. After several years of experience
with this method in Europe, solvent/de-
tergent-treated plasma was transiently
available in the United States. However,
concern with the use of a pooled product,
expense, poor market penetration, and
possible thrombotic (or excessive bleed-
ing) events led to the discontinuation of
this product. A psoralen (S59) activated by
ultraviolet A light is undergoing US clini-
cal trials for pathogen inactivation in
platelets and plasma and is available in
some countries in Europe.
Processing Cellular Components
Photochemical and chemical pathogen
inactivation methods are theoretically ap-
plicable to cellular blood components;
another organic chemical (S-303) and a
nucleic acid targeting compound (PEN
110) have undergone initial clinical evalu-
ation for pathogen inactivation in red cells.
These methods have the potential for reli-
able inactivation of bacteria, viruses, and
parasites, including intracellular forms.179,180
However, both products have been shown
to result in the formation of antibodies in
recipients. Pathogen reduction trials in
platelets (eg, with S-59) have been associ-
ated with decreased cell recovery and sur-
vival and the need for increased platelet
transfusions. Such unintended consequences
of pathogen reduction have resulted in
some trials being halted and may ulti-
mately eliminate such molecules from
widespread clinical application.
Reporting Transfusion-Associated
Infections
Unexplained infectious disease reported
in a transfusion recipient must be investi-
gated for the possibility of transfusion-
transmitted illness.62(pp83,85) Hepatitis is ex-
pected to become apparent within 2 weeks
to 6 months if it resulted from transfu-
sion, but, even within this interval, the
cause need not necessarily have been
blood-borne infection. Blood centers and
transfusion services must have a mecha-
nism to encourage recognition and re-
porting of possible transfusion-associated
infections. HIV infection thought to be a
result of transfusion should also be re-
ported to the blood supplier, although the
interval between transfusion and the rec-
ognition of infection or symptoms may be
years.
Infectioninarecipientshouldbere-
ported to the collecting agency so that do-
nors shown or suspected of being infec-
tious can be evaluated and recipients of
other components from the implicated or
other donations can be contacted and, if
necessary, tested. A donor who proves to
have positive results on tests during the in-
vestigation must be placed on an appropri-
ate deferral list.
Reporting Fatalities
The Code of Federal Regulations [21 CFR
606.170(b)] requires that fatalities attrib-
uted to transfusion complications (eg,
hepatitis, AIDS, and hemolytic reactions)
be reported to the Director, Center for
Biologics Evaluation and Research
(CBER), Office of Compliance and
Biologics Quality, Attn: Fatality Program
Manager (HFM-650), 1401 Rockville Pike,
Suite 200N, HFM-650, Rockville, MD
702 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
20852-1448. A report should be made as
soon as possible by telephone
(301-827-6220), fax (301-827-6748), or
email to fatalities2@cber.fda.gov and a
written report should be submitted within
7 days. Current information can be found
on the Internet at http://www.fda.gov/
cber/gdlns/bldfatal.pdf.
Management of Posttransfusion Infections
Implicated Donors
If documented transfusion-associated
hepatitis, HIV, or HTLV-I/II occurs in a pa-
tient who received only a single unit, that
donor must be permanently excluded
from future donations, and the name
placed in a file of permanently deferred
individuals. If posttransfusion viral infec-
tion occurs after exposure to blood from
several donors, it is not necessary to ex-
clude all of the potentially implicated do-
nors.Ifonlyafewdonorsareinvolved,it
may be desirable to recall them to obtain
an interim medical history and to per-
form additional tests. Donors found to
have been implicated in more than one
case of transfusion-associated viral infec-
tion should be appropriately investigated
and possibly deferred permanently ac-
cording to procedures established by the
collecting agency.
Notification
A donor who will be permanently exclu-
ded as a future blood donor because of a
positive test implication in posttransfu-
sion viral infection must be notified of
this fact. Follow-up testing should, ideally,
be done by the donor’s own physician,
and the collecting agency should obtain
the donor’s consent to release available
information to a designated health-care
provider. If the donor does not have a
physician, a blood bank physician or
other trained staff member should pro-
vide initial counseling and appropriate
medical referral. The notification process
andcounselingmustbedonewithtact
and understanding, and the concerns of
the donor should be addressed. The do-
nor should be told clearly why he or she is
deferred and, when appropriate, about
the possibility of being infectious to oth-
ers. Notification should occur promptly
because a delay in notification can delay
initiation of treatment or institution of
measures to prevent the spread of infec-
tion to others.
Use of Immunoglobulins
It is not recommended practice to give in-
tramuscular or intravenous immune serum
globulin or HBIG prophylactically to pre-
vent posttransfusion hepatitis181;these
agents have not been shown to prevent
posttransfusion hepatitis B, and the avail-
able evidence is conflicting about their ef-
fect on posttransfusion hepatitis C.182,183 If
there has been inadvertent transfusion of
known marker-positive blood or needle-
stick exposure to infectious material, HBIG
may prevent or attenuate HBV infection.184
Prophylaxis with immunoglobulin is inef-
fective in preventing HCV transmission
following occupational exposures and is
not recommended for this indication.185
References
1. Alter MJ. Epidemiology and prevention of
hepatitis B. Semin Liver Dis 2003;23:39-46.
2. Centers for Disease Control and Prevention.
Recommendations for prevention and con-
trol of hepatitis C virus (HCV) infection and
HCV-related chronic disease. MMWR Morb
Mortal Wkly Rep 1998;47(No. RR019):1-39.
3. Conry-Cantilena C, Menitove J. Hepatitis. In:
Anderson K, Ness P, eds. Scientific basis of
transfusion medicine. Implications for clini-
Chapter 28: Transfusion-Transmitted Diseases 703
Copyright © 2005 by the AABB. All rights reserved.
cal practice. Philadelphia: WB Saunders,
2000:472-87.
4. Dodd R. Hepatitis. In: Petz LD, Swisher SN,
Kleinman S, et al, eds. Clinical practice of
transfusion medicine. 3rd ed. New York:
Churchill Livingstone, 1996:847-73.
5. Dienstag JL. Transfusion-transmitted hepati-
tisB,A,andD.In:SimonTL,DzikWH,
Snyder EL, et al, eds. Rossi’s principles of
transfusion medicine. 3rd ed. Philadelphia:
Lippincott Williams and Wilkins, 2002:733-
41.
6. Alter HJ, Nakatsuji Y, Melpolder J, et al. The
incidence of transfusion-associated hepatitis
G virus infection and its relation to liver dis-
ease. N Engl J Med 1997;336:747-54.
7. Sayers M. Cytomegalovirus and other herpes-
viruses. In: Petz LD, Swisher SN, Kleinman S,
etal,eds.Clinicalpracticeoftransfusion
medicine.3rded.NewYork:ChurchillLiving
-
stone, 1996:875-89.
8. Unemura T, Yeo AET, Sottini A, et al. SEN vi-
rus infection and its relationship to transfu-
sion-associated hepatitis. Hepatology 2001;
33:1303-11.
9. Alter HJ. Transfusion-transmitted hepatitis C
and non-A, non-B, non-C virus infections. In:
Simon TL, Dzik WH, Snyder EL, et al, eds.
Rossi’s principles of transfusion medicine.
3rd ed. Philadelphia: Lippincott Williams and
Wilkins, 2002:718-32.
10. KumarA,BeniwalB,KarP,etal.HepatitisEin
pregnancy. Obstet Gynecol Surv 2005;60: 7-8.
11. Lee WM. Hepatitis B virus infection. N Engl J
Med 1997;337:1733-45.
12. Alter HJ. To C or not to C: These are the ques-
tions. Blood 1995;85:1681-95.
13. Seeff LB, Buskell-Bales Z, Wright EC, et al.
Long-term mortality after transfusion-asso-
ciated non-A, non-B hepatitis. The National
Heart, Lung, and Blood Institute Study
Group. N Engl J Med 1992;327:1906-11.
14. Vogt M, Lang T, Frosner G, et al. Prevalence
and clinical outcome of hepatitis C infection
in children who underwent cardiac surgery
before the implementation of blood-donor
screening. N Engl J Med 1999;341:866-70.
15. National Institutes of Health. National Insti-
tutes of Health consensus development con-
ference panel statement: Management of
hepatitis C. Hepatol 1997;26:2S-10S.
16. ShermanM,BainV,VilleneuveJP,etal.The
management of chronic viral hepatitis: A Ca-
nadian consensus conference 2004. Can J
Gastroenterol 2004;18:715-28.
17. Busch MP. Closing the windows on viral trans-
mission by blood transfusion. In: Stramer S,
ed. Blood safety in the new millennium.
Bethesda MD: AABB, 2000:33-54.
18. Westhoff TH, Jochimsen F, Schmittel A, et al.
Fatal hepatitis B virus reactivation by an es-
cape mutant following rituximab therapy
(letter). Blood 2003;102:1930.
19. Busch MP, Kleinman SH, Jackson B, et al. Nu-
cleic acid amplification testing of blood do-
nors for transfusion-transmitted diseases:
Report of the Interorganizational Task Force
on Nucleic Acid Amplification Testing of
Blood Donors. Transfusion 2000;40:143-59.
20. Alter HJ, Purcell RH, Shih JW, et al. Detection
of antibody to hepatitis C virus in prospec-
tively followed transfusion recipients with
acute and chronic non-A, non-B hepatitis. N
Engl J Med 1989;321:1494-500.
21. Brecher ME, ed. Collected questions and an-
swers. 6th ed. Bethesda, MD: AABB, 2000:21.
22. Van der Poel CL, Cuypers HT, Reesink HW, et
al. Confirmation of hepatitis C virus infection
by new four-antigen recombinant immuno-
blot assay. Lancet 1991;337:317-9.
23. Stramer SL, Glynn SA, Kleinman SH, et al for
the National Heart, Lung, and Blood Institute
Nucleic Acid Test Study Group. Detection of
HIV-1 and HCV infections among antibody-
negative blood donors by nucleic acid-ampli-
fication testing. N Engl J Med 2004;351:760-8.
24. Variance request to use NAT results and be
exempted from HIV and HCV supplemental
testing in specific circumstances (Associa-
tion Bulletin #05-03). Bethesda, MD: AABB,
2005.
25. Busch MP, Korelitz JJ, Kleinman SH, et al. De-
clining value of alanine aminotransferase in
screening of blood donors to prevent post-
transfusion hepatitis B and C virus infection.
The Retrovirus Epidemiology Donor Study.
Transfusion 1995;35:903-10.
26.JongeriusJM,WebsterM,CuypersHT,etal.
New hepatitis B virus mutant form in a blood
donor that is undetectable in several hepati-
tis B surface antigen screening assays. Trans-
fusion 1998;38:56-9.
27. Alter H. Transfusion-transmitted non-A,
non-B and hepatitis C infections. In: Rossi
EC, Simon TL, Moss GL, Gould S, eds. Princi-
ples of transfusion medicine. 2nd ed. Balti-
more, MD: Williams and Wilkins, 1996:687-98.
28. Armstrong GL, Alter MJ, McQuillan GM,
Margolis HS. The past incidence of hepatitis
C virus infection: Implications for the future
burden of chronic liver disease in the United
States. Hepatology 2000;31:777-82.
29.GoodnoughLT,ShanderA,BrecherME.
Transfusion medicine: Looking to the future.
Lancet 2003;361:161-9.
30. Busch MP, Glynn SA, Stramer S, et al. A new
strategy for estimating risks of transfusion-
transmitted viral infections based on rates of
704 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
detection of recently infected donors. Trans-
fusion 2005;45:254-64.
31. 65th Blood Product Advisory Committee
Meeting (transcripts). March 16, 2000. [Avail-
able at http://www.fda.gov/ohrms/dockets/
ac/00/transcripts/3603t1.rtp.]
32. Food and Drug Administration. Draft guid-
ance for industry: Current good manufactur-
ing practice for blood and blood compo-
nents: (1) quarantine and disposition of
units from prior collections from donors
with repeatedly reactive screening tests for
antibody to hepatitis C virus (anti-HCV); (2)
supplemental testing and the notification of
consignees and blood recipients of donor
test results for anti-HCV. (June 17, 1999)
Rockville, MD: CBER Office of Communica-
tion, Training, and Manufacturers Assis-
tance, 1999.
33. GoldmanM,JuodvalkisS,GillP,SpurliG.
Hepatitis C lookback. Transfus Med Rev
1998; 12:84-93.
34. Culver DH, Alter MJ, Mullan RJ, Margolis HS.
Evaluation of the effectiveness of targeted
lookback for HCV infection in the United
States—interim results. Transfusion 2000;40:
1176-81.
35. Bowker SL, Smith LJ, Rosychuk RJ, Preiksaitis
JK. A review of general hepatitis C virus look-
backs in Canada. Vox Sang 2004;86:21-7.
36. Centers for Disease Control. Pneumocystis
carinii pneumonia among persons with he-
mophilia A. MMWR Morb Mortal Wkly Rep
1982;31:365-7.
37. Centers for Disease Control. Possible trans-
fusion-associated acquired immune defi-
ciency syndrome (AIDS)—California. MMWR
Morb Mortal Wkly Rep1982;31:652-4.
38. Ragni MV, Winkelstein A, Kingsley L, et al.
1986 update of HIV seroprevalence, sero-
conversion, AIDS incidence, and immuno-
logic correlates of HIV infection in patients
with hemophilia A and B. Blood 1987;70:
786-90.
39. Busch MP, Young MJ, Samson SM, et al. Risk
of human immunodeficiency virus (HIV)
transmission by blood transfusions before
the implementation of HIV-1 antibody
screening. The Transfusion Safety Study
Group. Transfusion 1991;31:4-11.
40. Levine A, Liebman H. The acquired immu-
nodeficiency syndrome (AIDS). In: Beutler E,
Lichtman M, Coller B, Kipps T, eds. Williams’
hematology. New York: McGraw-Hill, 1995:
975-97.
41. Mayer A, Busch M. Transfusion-transmitted
HIV infection. In: Anderson K, Ness P, eds.
Scientific basis of transfusion medicine. Im-
plications for clinical practice. Philadelphia:
WB Saunders, 1994:659-68.
42. Centers for Disease Control. 1993 revised
classification system for HIV infection and
expanded surveillance case definition for
AIDS among adolescents and adults. MMWR
Morb Mortal Wkly Rep 1992;41:1-19.
43. Brookmeyer R. Reconstruction and future
trends of the AIDS epidemic in the United
States. Science 1991;253:37-42.
44. Centers for Disease Control and Prevention.
US HIV and AIDS cases reported through De-
cember 2000. HIV/AIDS Surveillance Report
2000;12(2):1-44. [Available at http://www.
cdc.gov/hiv/stats/hasr1202.pdf.]
45. Petersen LR, Doll LS, White CR, et al. Hetero-
sexually acquired HIV infection and the US
blood supply: Consideration for screening of
potential donors. Transfusion 1993;33:552-7.
46. Simon F, Loussert-Ajaka I, Damond F, et al.
HIVtype1diversityinnorthernParis,France.
AIDS Res Hum Retroviruses 1996;12:1427-33.
47. Parry JV, Murphy G, Barlow KL, et al. Na-
tional surveillance of HIV-1 subtypes for
England and Wales: Design, methods, and
initial findings. J Acquir Immune Defic Syndr
2001;26:381-8.
48. Leads from the MMWR. AIDS due to HIV-2
infection—New Jersey. JAMA 1988;259:969,
972.
49. Sullivan MT, Guido EA, Metler RP, et al. Iden-
tification and characterization of an HIV-2
antibody-positive blood donor in the United
States. Transfusion 1998;38:189-93.
50. de Oliveira CF, Diaz RS, Machado DM, et al.
Surveillance of HIV-1 genetic subtypes and
diversity in the US blood supply. Transfusion
2000;40:1399-406.
51. Delwart EL, Orton S, Parekh B, et al. Two per-
cent of HIV-positive US blood donors are in-
fected with non-subtype-B strains. AIDS Res
Hum Retroviruses 2003;19:1065-70.
52. Peeters M, Gueye A, Mboup S, et al. Geo-
graphical distribution of HIV-1 group O vi-
ruses in Africa. AIDS 1997;11:493-8.
53. Food and Drug Administration. Memoran-
dum: Interim recommendations for deferral
of donors at increased risk for HIV-1 group O
infection. (December 11, 1996) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 1996.
54. Kaiser Family Foundation. Cumulative adult/
adolescent HIV infection cases reported
through 2003 among states with confidential
name-based reporting. [Available at http://
www.statehealthfacts.kff.org.]
55. Delwart EL, Kalmin ND, Jones TS, et al. First
case of HIV transmission by an RNA-
Chapter 28: Transfusion-Transmitted Diseases 705
Copyright © 2005 by the AABB. All rights reserved.
screened blood donation. Vox Sang 2004;86:
771-7.
56. Phelps R, Robbins K, Liberti T, et al. Window-
period human immunodeficiency virus
transmission to two recipients by an adoles-
cent blood donor. Transfusion 2004;44:929-
33.
57. LingAE,RobbinsKE,BrownTM,etal.Failure
of routine HIV-1 tests in a case involving
transmission with preseroconversion blood
components during the infectious window
period. JAMA 2000;284:210-4 [comment in
JAMA 2000;284:238-40].
58. Fiebig E, Busch MP. Retroviral infections. In:
Simon TL, Dzik WH, Snyder EL, et al, eds.
Rossi’s principles of transfusion medicine.
3rd ed. Philadelphia: Lippincott Williams and
Wilkins, 2002:742-56.
59. Tabor E. The epidemiology of virus transmis-
sion by plasma derivatives: Clinical studies
verifying the lack of transmission of hepatitis
B and C viruses and HIV type 1. Transfusion
1999;39:1160-8.
60. Dufoort G, Courouce AM, Ancelle-Park R,
Bletry O. No clinical signs 14 years after HIV-2
transmission via blood transfusion (letter).
Lancet 1988;ii:510.
61. Kleinman S, Busch MP, Korelitz JJ, Schreiber
GB. The incidence/window period model and
its use to assess the risk of transfusion-trans-
mitted human immunodeficiency virus and
hepatitis C virus infection. Transfus Med Rev
1997;11:155-72.
62. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
63. Code of federal regulations. Title 21 CFR
610.40. Washington, DC: US Government
Printing Office, 2004 (revised annually).
64. Busch MP. HIV and blood transfusions: Focus
on seroconversion. Vox Sang 1994;67 (Suppl
3):13-8.
65. Fiebig EW, Wright DJ, Rawal BD, et al. Dyna-
mics of HIV viremia and antibody serocon-
version in plasma donors: Implications for
diagnosis and staging of primary HIV infec-
tion. AIDS 2003;17:1871-9.
66. Food and Drug Administration. Talk paper:
FDA approves first nucleic acid test (NAT)
system to screen whole blood donors for in-
fections with human immunodeficiency vi-
rus (HIV) and hepatitis C virus (HCV). (Feb-
ruary 28, 2002) [Available at http://fda.
gov/cber/products/hivhcvgen022702.htm.]
67. Food and Drug Administration. Guidance for
industry: Use of nucleic acid tests on pooled
and individual samples from donations of
whole blood and blood components (includ-
ing Source Plasma and Source Leukocytes) to
adequately and appropriately reduce the risk
of transmission of HIV-1 and HCV. (October
21, 2004) Rockville, MD: CBER Office of Com-
munication, Training, and Manufacturers As-
sistance, 2004. [Available at http://www.fda.
gov/cber/gdlns/hivhcvnatbld.htm.]
68. Centers for Disease Control. Interpretive cri-
teria used to report Western blot results for
HIV-1-antibody testing: United States. MMWR
Morb Mortal Wkly Rep 1991;40:692-5.
69. Kleinman S, Busch MP, Hall L, et al. False-
positive HIV-1 test results in a low-risk
screening setting of voluntary blood dona-
tion. Retrovirus Epidemiology Donor Study.
JAMA 1998;280:1080-5.
70. Food and Drug Administration. Memoran-
dum: Revised recommendations for the pre-
vention of human immunodeficiency (HIV)
transmission by blood and blood products.
(April 23, 1992) Rockville, MD: CBER Office of
Communication, Training, and Manufactur-
ers Assistance, 1992.
71. Food and Drug Administration. Memorandum:
Use of fluorognost HIV-1 immunofluorescent
assay (IFA). (April 23, 1992) Rockville, MD:
CBER Office of Communication, Training,
and Manufacturers Assistance, 1992.
72. Mintz P. Participation of HIV-infected pa-
tients in autologous blood programs. JAMA
1993;269:2892-4.
73. Pub. Law. 101-336, 104 Stat. 327 (1990). Codi-
fied at 42 U.S.C. §12101-12213.
74. Food and Drug Administration. Current good
manufacturing practice for blood and blood
components: Notification of consignees re-
ceiving blood and blood components at in-
creased risk for transmitting HIV infection.
(September 9, 1996) Fed Regist 1996;61:
47413-23.
75. Health Care Financing Administration. Medi-
care and Medicaid programs; hospital stan-
dard for potentially HIV infectious blood and
blood products. (September 9, 1996) Fed Regist
1996;61:47423-34.
76. Code of federal regulations. 42 CFR 610.46-47.
Washington, DC: US Government Printing
Office, 2004 (revised annually).
77. Code of federal regulations. 21 CFR 482.27(c).
Washington, DC: US Government Printing
Office, 2004 (revised annually).
78. MurphyEL,GlynnSA,FrideyJ,etal.In
-
creased incidence of infectious diseases dur-
ing prospective follow-up of human T-lym-
photropic virus type II- and I-infected blood
donors. Retrovirus Epidemiology Donor
Study. Arch Intern Med 1999;159:1485-91.
79. OrlandJR,EngstromJ,FrideyJ,etal.Preva
-
lence and clinical features of HTLV neuro-
706 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
logic disease in the HTLV Outcomes Study.
Neurology 2003;61:1588-94.
80. Murphy EL, Wang B, Sacher RA, et al. Respi-
ratory and urinary tract infections, arthritis,
and asthma associated with HTLV-II and
HTLV-I infection. Emerg Infect Dis 2004;10:
109-16.
81. OrlandJR,WangB,WrightDJ,etal.In
-
creased mortality associated with HTLV-II
infection in blood donors: A prospective co-
hort study. Retrovirology 2004;1:4.
82. Levine PH, Manns A. Transfusion transmis-
sion of human T-lymphotropic virus types I
and II: Lessons to be learned from look-back
investigations and implications for patient
counseling (editorial). Transfusion 1993;33:
4-6.
83. Kleinman S, Swanson P, Allain JP, Lee H.
Transfusion transmission of human T-lym-
photropic virus types I and II: Serologic and
polymerase chain reaction results in recipi-
ents identified through look-back investiga-
tions. Transfusion 1993;33:14-8.
84.SchreiberGB,BuschMP,KleinmanSH,
Korelitz JJ. The risk of transfusion-transmit-
ted viral infections. The Retrovirus Epidemi-
ology Donor Study. N Engl J Med 1996;334:
1685-90.
85. Food and Drug Administration. Guidance for
industry: Donor screening for antibodies to
HTLV-II. (August 15, 1997) Rockville, MD:
CBER Office of Communication, Training,
and Manufacturers Assistance, 1997.
86. Dual enzyme immunoassay (EIA) approach
for deferral and notification of anti-HTLV-
I/II EIA reactive donors. Association Bulletin
99-9. Bethesda, MD: AABB, 1999.
87. Pealer LN, Marfin AA, Petersen LR, et al for
the West Nile Virus Transmission Investiga-
tion Team. Transmission of West Nile virus
through blood transfusion in the United
States in 2002. N Engl J Med 2003;349:1236-
45.
88. Biggerstaff BJ, Petersen LR. Estimated risk of
transmission of the West Nile virus through
blood transfusion in the US, 2002. Transfu-
sion 2003;43:1007-17.
89. Food and Drug Administration. Guidance for
industry: Revised recommendations for the
assessment of donor suitability and blood
and blood product safety in cases of known
or suspected West Nile virus infection. (May
1, 2003) Rockville, MD: CBER Office of Com-
munication, Training, and Manufacturers
Assistance, 2003.
90. Kleinman S, Glynn SA, Busch M, et al for the
NHLBI Retrovirus Epidemiology Study
(REDS). The 2003 West Nile virus United
States epidemic: The America's Blood Cen-
ters experience. Transfusion 2005;45:469-79.
91. Centers for Disease Control and Prevention.
Update: West Nile virus screening of blood
donations and transfusion-associated trans-
mission—United States, 2003. MMWR Morb
Mortal Wkly Rep 2004;53:281-4.
92. Busch MP, Caglioti S, Robertson GF, et al.
Screening the blood supply for West Nile vi-
rus RNA by nucleic acid amplification test-
ing. N Engl J Med 2005 (in press).
93. Update on WNV-related activities and con-
siderations, 2004; a summary of the WNV
Task Force meeting. Association Bulletin
#04-03. Bethesda, MD: AABB, 2004.
94. Centers for Disease Control and Prevention.
2004 West Nile virus activity in the United
States (reported as of January 11, 2005).
[Available at http://www.cdc.gov/ncidod/
dvbid/westnile/surv&controlCaseCount04_
detailed.htm.]
95. Centers for Disease Control and Prevention.
Transfusion-associated transmission of West
Nile virus—Arizona, 2004. MMWR Morb
Mortal Wkly Rep 2004;53:842-4.
96. Roback JD. Human herpes infections. In:
Hillyer CD, Silberstein LE, Ness PM, Ander-
son KC, eds. Blood banking and transfusion
medicine.NewYork:ChurchillLivingstone,
2003:465-85.
97. Roback JD. CMV and blood transfusions. Rev
Med Virol 2002;12:211-19.
98. Leukocyte reduction for the prevention of
transfusion-transmitted cytomegalovirus.
Association Bulletin #97-2. Bethesda, MD:
AABB, 1997.
99. Bowden RA, Slichter SJ, Sayers M, et al. A
comparison of filtered leukocyte-reduced
and cytomegalovirus (CMV) seronegative
blood products for the prevention of transfu-
sion-associated CMV infection after marrow
transplant. Blood 1995;86:3598-603.
100. Preiksaitis JK. The cytomegalovirus-“safe”
blood product: Is leukoreduction equivalent
to antibody screening? Transfus Med Rev
2000;14:112-36.
101. McMonigal K, Horwitz CA, Henle W. Post-
perfusion syndrome due to Epstein-Barr vi-
rus. Report of two cases and review of the lit-
erature. Transfusion 1983;23:331-5.
102. Campadelli-Fiume G, Mirandola P, Menotti
L. Human herpesvirus 6: An emerging patho-
gen. Emerg Infect Dis 1999;5:353-66.
103. Chatlynne LG, Lapps W, Handy M, et al. De-
tection and titration of human herpesvirus-
8-specific antibodies in sera from blood do-
nors, acquired immunodeficiency syndrome
patients, and Kaposi’s sarcoma patients us-
Chapter 28: Transfusion-Transmitted Diseases 707
Copyright © 2005 by the AABB. All rights reserved.
ing a whole virus enzyme-linked immunosor-
bent assay. Blood 1998;92:53-8.
104. Cannon MJ, Dollard SC, Smith DK, et al.
Blood-borne and sexual transmission of hu-
man herpesvirus 8 in women with or at risk
for human immunodeficiency virus infec-
tion. N Engl J Med 2001;344:637-43.
105. Luppi M, Barozzi P, Santagostino G, et al. Mo-
lecular evidence of organ-related transmission
of Kaposi sarcoma-associated herpesvirus or
human herpesvirus-8 in transplant patients.
Blood 2000;96:3279-81.
106. Regamey N, Tamm M, Wernli M, et al. Trans-
mission of human herpesvirus 8 infection
from renal-transplant donors to recipients. N
Engl J Med 1998;339:1358-63.
107. Parravicini C, Olsen SJ, Capra M, et al. Risk of
Kaposi’s sarcoma-associated herpes virus
transmission from donor allografts among
Italian posttransplant Kaposi’s sarcoma pa-
tients. Blood 1997;90:2826-9.
108. Mbulaiteye SM, Biggar RJ, Bakaki PM, et al.
Human herpesvirus 8 infection and transfu-
sion history in children with sickle-cell dis-
ease in Uganda. J Natl Cancer Inst 2003;95:
1330-5.
109. Luban NL. Human parvoviruses: Implica-
tions for transfusion medicine. Transfusion
1994;34:821-7.
110. Prowse C, Ludlam CA, Yap PL. Human par-
vovirus B19 and blood products. Vox Sang
1997;72:1-10.
111. Brown KE, Hibbs JR, Gallinella G, et al. Resis-
tance to parvovirus B19 infection due to lack
of virus receptor (erythrocyte P antigen). N
Engl J Med 1994;330:1192-6.
112. Nucleic acid testing of blood donors for hu-
man parvovirus B19 (presentations, public
hearing, discussion, and recommendations).
64th meeting of the Blood Products Advisory
Committee, Bethesda, MD, September 16,
1999. [Available at http://www.fda.gov/cber/
advisory/bp/bpmain.htm.]
113. Manuelidis L. The dimensions of Creutzfeldt-
Jakob disease. Transfusion 1994;34:915-28.
114. Ricketts MN, Cashman NR, Stratton EE, El
Saadany S. Is Creutzfeldt-Jakob disease
transmitted in blood? Emerg Infect Dis 1997;
3:155-63.
115. Evatt B, Austin H, Barnhart E, et al. Surveil-
lance for Creutzfeldt-Jakob disease among
persons with hemophilia. Transfusion 1998;
38:817-20.
116. van Duijn CM, Delasnerie-Laupretre N,
Masullo C, et al. Case-control study of risk
factors of Creutzfeldt-Jakob disease in Eu-
rope during 1993-95. European Union (EU)
Collaborative Study Group of Creutzfeldt-
Jakob Disease (CJD). Lancet 1998;351:1081-5.
117. Food and Drug Administration. Memoran-
dum: Revised precautionary measures to re-
duce the possible risk of transmission of
Creutzfeldt-Jakob disease (CJD) by blood and
blood products. (August 5, 1995) Rockville,
MD: CBER Office of Communication, Train-
ing, and Manufacturers Assistance, 1995.
118. Food and Drug Administration. Memoran-
dum: Revised precautionary measures to re-
duce the possible risk of transmission of
Creutzfeldt-Jakob disease (CJD) by blood and
blood products. (December 11, 1996) Rock-
ville, MD: CBER Office of Communication,
Training, and Manufacturers Assistance, 1996.
119. Food and Drug Administration. Draft guid-
ance for industry: Preventive measures to re-
duce the possible risk of transmission of
Creutzfeldt-Jakob disease (CJD) and variant
Creutzfeldt-Jakob disease (vCJD) by human
cells, tissues, and cellular and tissue-based
products (HCT/Ps). (June 25, 2002) Fed
Regist 2002;67:42789-90.
120. Hunter N, Foster J, Chong A, et al. Transmis-
sion of prion diseases by blood transfusion. J
Gen Virol 2002;83:2897-905.
121. Llewelyn CA, Hewitt PE, Knight RS, et al. Pos-
sible transmission of variant Creutzfeldt-
Jakob disease by blood transfusion. Lancet
2004;363:417-21.
122. Peden AH, Head MW, Ritchie DL, et al. Pre-
clinical vCJD after blood transfusion in a
PRNP codon 129 heterozygous patient. Lan-
cet 2004;364:521-9.
123. Nurmi MH, Bishop M, Strain L, et al. The nor-
mal population distribution of PRNP codon
129 polymorphism. Acta Neurol Scand 2003;
108:374-8.
124. Sazama K. Bacteria in blood for transfusion.
A review. Arch Pathol Lab Med 1994;118:350-65.
125. Food and Drug Administration. CBER annual
report FY 2002 (October 1, 2001 through Sep-
tember 30, 2002). Rockville, MD: CBER Office
of Communication, Training, and Manufac-
turers Assistance, 2002. [Available at http://
www. fda.gov/cber/inside/annrptpart3.htm.]
126. Morrow JF, Braine HG, Kickler TS, et al. Septic
reactions to platelet transfusions. A persistent
problem. JAMA 1991;266:555-8.
127. Kuehnert MJ, Roth VR, Haley NR, et al. Trans-
fusion-transmitted bacterial infection in the
United States, 1998 through 2000. Transfu-
sion 2001;41:1493-9.
128. Theakston EP, Morris AJ, Streat SJ, et al.
Transfusion transmitted Yersinia entero-
colitica infection in New Zealand. Aust N Z J
Med 1997;27:62-7.
129. Cookson ST, Arduino MJ, Aguero SM, Jarvis
WR, and the Yersinia Study Group. Yersinia
enterocolitica-contaminated Red Blood Cells
708 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
(RBCs)—an emerging threat to blood safety
(abstract). In: Program and abstracts of the
36th Interscience Conference on Antimicro-
bial Agents and Chemotherapy, New Or-
leans, September 15-18, 1996. Washington,
DC: American Society for Microbiology,
1996:237.
130. National Blood Data Resource Center. Com-
prehensive report on blood collection and
transfusion in the United States in 2001.
Bethesda, MD: AABB, 2003.
131. Brecher ME, Hay SN. Improving platelet
safety: Bacterial contamination of platelets.
Curr Hematol Rep 2004;3:121-7.
132. Dykstra A, Hoeltge G, Jacobs M, et al. Platelet
bacterial contamination (PBC) rate is sur-
veillance method (SM) dependent (abstract).
Transfusion 1998;38(Suppl):104S.
133. Serious Hazards of Transfusion (SHOT). An-
nual report 2000-2001. Manchester, UK:
SHOT Office, 2002. [Available at http://www.
shot.demon.co.uk/toc.htm.]
134. Perez P, Salmi LR, Follea G, et al. Determi-
nants of transfusion-associated bacterial
contamination: Results of the French
BACTHEM case-control study. Transfusion
2001;41:862-71.
135. Ness PM, Braine HG, King K, et al. Single do-
nor platelets reduce the risk of septic trans-
fusion reactions. Transfusion 2001;41:857-61.
136. Engelfriet CP, Reesink HW, Blajchman MA, et
al. Bacterial contamination of blood compo-
nents. Vox Sang 2000;78:59-67.
137. Kim DM, Brecher ME, Bland LA, et al. Pre-
storage removal of Yersinia enterocolitica
from red cells with white cell-reduction fil-
ters. Transfusion 1992;32:658-62.
138. Buchholz DH, AuBuchon JP, Snyder EL, et al.
Removal of Yersinia enterocolitica from AS-1
red cells. Transfusion 1992;32:667-72.
139. Pietersz RNI, Engelfriet CP, Reesink HW, et
al. Detection of bacterial contamination of
platelet concentrates. International Forum.
Vox Sang 2003;85:224-39.
140. Commission on Laboratory Accreditation.
Transfusion medicine checklist. TRM.44955
Phase I. Northfield, IL: College of American
Pathologists, January 2003. [Available at
http://www.cap.org/html/checklist_html/
transfusionmedicine_1202.html.]
141. Brumit MC, Hay SN, Brecher ME. Bacteria
detection. In: Brecher ME, ed. Bacterial and
parasitic contamination of blood compo-
nents. Bethesda, MD: AABB Press, 2003:57-
82.
142. Anderson KC, Lew MA, Gorgone BC, et al.
Transfusion-related sepsis after prolonged
platelet storage. Am J Med 1986;81:405-11.
143. Orton SL, Liu H, Dodd RY, Williams AE. Prev-
alence of circulating Treponema pallidum
DNA and RNA in blood donors with con-
firmed positive syphilis tests. Transfusion
2002;42:94-9.
144. Gardella C, Marfin AA, Kahn RH, et al. Per-
sons with early syphilis identified through
blood or plasma donation screening in the
United States. J Infect Dis 2000;185:545-9.
145. Leiby DA. Babesia and other parasites. In:
Brecher ME, ed. Bacterial and parasitic con-
tamination of blood components. Bethesda,
MD: AABB Press, 2003:179-200.
146. Herwaldt BL, Springs FE, Roberts PP, et al.
Babesiosis in Wisconsin: A potentially fatal
disease. Am J Trop Med Hyg 1995;53:146-51.
147. Herwaldt BL, Kjemtrup AM, Conrad PA, et al.
Transfusion-transmitted babesiosis in Wash-
ington State: First reported case caused by a
WA1-type parasite. J Infect Dis 1997;175:
1259-62.
148. Krause PJ, Spielman A, Telford SR 3rd, et al.
Persistent parasitemia after acute babesiosis.
N Engl J Med 1998;339:160-5.
149. Cable R, Trouern-Trend J. Tick-borne infec-
tions. In: Linden JV, Bianco C, eds. Blood
safety and surveillance. New York: Marcel
Dekker, 2001:399-422.
150. Wells GM, Woodward TE, Fiset P, Hornick RB.
Rocky Mountain spotted fever caused by
blood transfusion. JAMA 1978;239:2763-5.
151. Eastlund T, Persing D, Mathiesen D, et al.
Human granulocytic ehrlichiosis after red
cell transfusion (abstract). Transfusion 1999;
39(Suppl):117S.
152. Arguin PM, Singleton J, Rotz LD, et al. An in-
vestigation into the possibility of transmis-
sion of tick-borne pathogens via blood
transfusion. Transfusion-Associated Tick-
Borne Illness Task Force. Transfusion 1999;
39:828-33.
153. McQuiston JH, Childs JE, Chamberland ME,
TaborEfortheWorkingGrouponTransfu
-
sion Transmission of Tick-Borne Diseases.
Transmission of tick-borne agents of disease
by blood transfusion: A review of known and
potential risks in the United States. Transfu-
sion 2000;40:274-84.
154. Mungai M, Tegtmeier G, Chamberland M,
Parise M. Transfusion-transmitted malaria
in the United States from 1963 through 1999.
N Engl J Med 2001;344:1973-8.
155. Centers for Disease Control and Prevention.
Probable transfusion-transmitted malaria—
Houston, Texas, 2003. MMWR Morb Mortal
Wkly Rep 2003;52:1075-6.
156. Katz LM. Transfusion-induced malaria. In:
Brecher ME, ed. Bacterial and parasitic con-
Chapter 28: Transfusion-Transmitted Diseases 709
Copyright © 2005 by the AABB. All rights reserved.
tamination of blood components. Bethesda,
MD: AABB Press, 2003:127-55.
157. Herwaldt BL, Grijalva MJ, Newsome AL, et al.
Use of polymerase chain reaction to diagnose
the fifth reported US case of autochthonous
transmission of Trypanosoma cruzi,inTen
-
nessee, 1998. J Infect Dis 2000; 181:395-9.
158. Shulman IA. Transfusion of T. cruzi infection
bybloodtransfusion.In:BrecherME,ed.
Bacterial and parasitic contamination of
blood components. Bethesda, MD: AABB
Press, 2003:157-78.
159. Leiby DA, Rentas FJ, Nelson KE, et al. Evi-
dence of Trypanosoma cruzi infection (Cha-
gas’ disease) among patients undergoing car-
diac surgery. Circulation 2000;102:2978-82.
160. Leiby DA, Read EJ, Lenes BA, et al. Seroepi-
demiology of Trypanosoma cruzi, etiologic
agent of Chagas’ disease, in US blood donors.
J Infect Dis 1997;176:1047-52.
161. Shulman IA, Appleman MD, Saxena S, et al.
Specific antibodies to Trypanosoma cruzi
among blood donors in Los Angeles, Califor-
nia. Transfusion 1997;37:727-31.
162. Shulman I, Haimowitz MD. Transmission of
parasitic infections by blood transfusion. In:
Simon TL, Dzik WH, Snyder EL, et al, eds.
Rossi’s principles of transfusion medicine.
3rd ed. Philadelphia: Lippincott Williams and
Wilkins, 2002:774-83.
163. Deferral for risk of Leishmaniasis exposure.
Association Bulletin #03-14. Bethesda, MD:
AABB, 2003.
164. US General Accounting Office. Blood supply:
Transfusion-associated risks. GAO/PEMD-97-1.
Washington, DC: US Government Printing
Office, 1997.
165. Brecher ME. Bacterial contamination of
blood products. In: Simon TL, Dzik WH,
Snyder EL, et al, eds. Rossi’s principles of
transfusion medicine. 3rd ed. Philadelphia:
Lippincott Williams and Wilkins, 2002:
789-801.
166. Rock G, Neurath D, Toye B, et al. The use of a
bacteria detection system to evaluate bacte-
rial contamination in PLT concentrates.
Transfusion 2004;44:337-42.
167. Suomela H. Inactivation of viruses in blood
and plasma products. Transfus Med Rev 1993;
7:42-57.
168. Centers for Disease Control. Safety of thera-
peutic immune globulin preparations with
respect to transmission of human T-lympho-
tropic virus type III/lymphadenopathy-asso-
ciated virus infection. MMWR Morb Mortal
Wkly Rep 1986;35:231-3. [Erratum in MMWR
Morb Mortal Wkly Rep 1986;35:607.]
169. Williams PE, Yap PL, Gillon J, et al. Non-A,
non-B hepatitis transmission by intravenous
immunoglobulin (letter). Lancet 1988;ii:501.
[Erratum in Lancet 1988;ii:584.]
170. Yu MW, Mason BL, Guo ZP, et al. Hepatitis C
transmission associated with intravenous
immunoglobulins (letter). Lancet 1995;345:
1173-4.
171. Centers for Disease Control. Outbreak of
hepatitis C associated with intravenous im-
munoglobulin administration: United States,
October 1993-June 1994. MMWR Morb Mor-
tal Wkly Rep 1994;43:505-9.
172. Farrugia A, Walker E. Hepatitis C virus trans-
mission by intravenous immunoglobulin
(letter). Lancet 1995;346:373-5.
173. Meisel H, Reip A, Faltus B, et al. Transmission
of hepatitis C virus to children and husbands
by women infected with contaminated anti-D
immunoglobulin. Lancet 1995;345:1209-11.
174. Power JP, Lawlor E, Davidson F, et al. Hepati-
tis C viraemia in recipients of Irish intrave-
nous anti-D immunoglobulin (letter). Lancet
1994;344:1166-7.
175. Foster PR, McIntosh RV, Welch AG. Hepatitis
C infection from anti-D immunoglobulin
(letter). Lancet 1995;346:372.
176. Makris M, Preston FE. Chronic hepatitis in
haemophilia. Blood Rev 1993;7:243-50.
177. Prowse C. Kill and cure. The hope and reality
of virus inactivation. Vox Sang 1994;67(Suppl
3):191-6.
178. Lusher JM, Arkin S, Abildgaard CF, Schwartz
RS. Recombinant factor VIII for the treatment
of previously untreated patients with hemo-
philia A. Safety, efficacy, and development of
inhibitors. Kogenate Previously Untreated
Patient Study Group. N Engl J Med 1993;328:
453-9.
179. Ben-Hur E, Moor AC, Margolis-Nunno H, et
al. The photodecontamination of cellular
blood components: Mechanisms and use of
photosensitization in transfusion medicine.
Transfus Med Rev 1996;10:15-22.
180. Zhang Q-X, Edson C, Budowsky E, Purmal A.
InactineTM—a method for viral inactivation
in red blood cell concentrates (abstract).
Transfusion 1998;38(Suppl):75S.
181. Seeff L. The efficacy of and place for HBIG in
the prevention of type B hepatitis. In:
Szmuness W, Alter H, Maynard J, eds. Viral
hepatitis: 1981 International Symposium.
Philadelphia: The Franklin Institute Press,
1982:585-95.
182. Sanchez-Quijano A, Pineda JA, Lissen E, et al.
Prevention of post-transfusion non-A, non-B
hepatitis by non-specific immunoglobulin in
heart surgery patients. Lancet 1988;i:1245-9.
183. Conrad ME. Prevention of post-transfusion
hepatitis (letter). Lancet 1988;ii:217.
710 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
184. Kobayashi R, Stiehm E. Immunoglobulin
therapy. In: Petz LD, Swisher SN, Kleinman S,
etal,eds.Clinicalpracticeoftransfusionmed
-
icine. 3rd ed. New York: Churchill Living-
stone, 1996:985-1010.
185. Centers for Disease Control. Recommenda-
tions for follow-up of health-care workers af-
ter occupational exposure to hepatitis C vi-
rus. MMWR Morb Mortal Wkly Rep 1997;46:
603-6.
Suggested Reading
Actions following an initial positive test for possi-
ble bacterial contamination of a platelet unit (As-
sociation Bulletin #04-07). Bethesda, MD: AABB,
2004.
Criteria for donor deferral in known or suspected
common source outbreaks of hepatitis A virus in-
fection (Association Bulletin #04-08). Bethesda,
MD: AABB, 2004.
Guidance on management of blood and platelet
donors with positive or abnormal results on bac-
terial contamination tests (Association Bulletin
#05-02). Bethesda, MD: AABB, 2005.
Chapter 28: Transfusion-Transmitted Diseases 711
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Methods
The inclusion of methods in this edition
of the Technical Manual is a subjective
decision of the Technical Manual Pro-
gram Unit. Readers are encouraged to re-
fer to previous editions of the manual for
methods not appearing in this edition be-
cause exclusion from the current edition
does not necessarily indicate that their
use is prohibited. However, some proce-
dures, such as xylene and chloroform elu-
tion techniques, were removed because
the chemicals used in the procedures
could present a safety risk. Thus, readers
are cautioned when referring to proce-
duresinpreviouseditionsbecausethey
have not been reviewed for content and
safety.
There are often many different ways to
perform the same test procedure. Although
some workers may prefer other methods,
those given here are reliable, straightfor-
ward, and of proven value. Although the in-
vestigation of unusual serologic problems
often requires flexibility in thought and
methodology, the adoption of uniform
methods for routine procedures in the lab-
oratory is imperative. In order for labora-
tory personnel to have reproducible and
comparable results in a test procedure, it is
essential that everyone in the laboratory
perform the same procedure in the same
manner.
713
Methods
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.
Methods Section 1: General Laboratory Methods
Methods Section 1
General Laboratory Methods
Introduction
The methods outlined in the following
sections are examples of acceptable pro-
cedures. Other acceptable procedures
may be used by facilities if desired. To the
greatest extent possible, the written pro-
cedures conform to the Guidelines for
Clinical Laboratory Technical Procedure
Manuals developed by the National Com-
mittee for Clinical Laboratory Standards.
As indicated in Title 21 of the Code of
Federal Regulations (CFR) Part 606.65(e),
the manufacturer’s instructions (eg, prod-
uct insert) for reagents and supplies li-
censed by the Food and Drug Administra-
tion (FDA) should be followed. Any deviation
should be validated using appropriate
controls and incorporated into a standard
operating procedure before approval by
the medical director. (Note: Deviations
may also require concurrence from the
FDA.) It is important to use Standard Pre-
cautions when appropriate (see Chapter
2).
Reagent Preparation
Many procedures include formulas for re-
agent preparation. Labels for reagents pre-
pared in-house must contain the follow-
ing:
■Name of solution.
■Date of preparation.
■Expiration date (if known).
■Storage temperature and/or conditions.
■Mechanism to identify the person pre-
paring the solution.
■Universal hazardous substance label.
Temperatures
Whenever specific incubation or storage
temperatures are given, the following ranges
are considered satisfactory:
Stated Temperature Acceptable Range
4C 2-8C
Room temperature 20-24 C
37 C 36-38 C
56 C 54-58 C
715
Section 1
Copyright © 2005 by the AABB. All rights reserved.
Centrifugation Variables
Centrifugation speeds (relative centrifugal
force) and times should be standardized
for each piece of equipment. (See Meth-
ods Section 8.)
Reference
Guidelines for clinical laboratory technical proce-
dure manuals. 3rd ed. (NCCLS Document GP2-A3,
Vol. 12, No. 10.) Wayne, PA: National Committee
for Clinical Laboratory Standards, 1996.
Method 1.1. Transportation
and Shipment of Dangerous
Goods
Several agencies specify packaging and
shipping requirements for dangerous or
hazardous materials, depending on how
the material is shipped (mail, ground, or
air). For transport by mail of infectious
materials, clinical specimens, or biologic
products, the United States Postal Service
(USPS) Dangerous Goods Regulations
must be followed.1,2 For interstate trans-
port of infectious materials by ground or
air, the United States Department of
Transportation (DOT) regulations apply.3
Most air carriers apply the International
Air Transport Association (IATA)4regula-
tions and the technical instructions of the
International Civil Aviation Organization
(ICAO).5These agencies adopt the recom-
mendations of the United Nations (UN)
Committee of Experts on the Transport of
Dangerous Goods for the international
transport of infectious substances and
clinical specimens.
The Centers for Disease Control and Pre-
vention (CDC)6and the IATA7provide pack-
ing and labeling requirements for ship-
ments of infectious materials in order to
protect the public health by minimizing the
potential for direct contact with such mate-
rials, contamination of the environment,
and the spread of disease. The CDC also
serves as a Center for Applied Biosafety and
Training for the World Health Organization
(WHO)8andfortheUN.TheOccupational
Safety and Health Administration (OSHA)9
regulates worker safety related to handling,
packing, and transport. Both the DOT and
the IATA require active training for anyone
who packages infectious or toxic materials
for shipment.3,4 DOT and IATA documents
offer general shipping advice for all hazard-
ous materials because the regulations are
similar and most carriers follow the ship-
ping guidelines set forth in the IATA Dan-
gerous Goods Regulations4and the Infec-
tious Substances Shipping Guidelines.7
Facilities should also consult their local car-
riers for additional requirements. In gen-
eral, all dangerous goods regulations are
based on public risk from the materials and
therefore have specific packaging, labeling,
and documentation requirements. It is the
responsibility of the shipper of biologic or
infectious material to properly classify,
package, label, and document the sub-
stance being shipped.
Dangerous Goods Classifications
IATA classifies hazards into nine catego-
ries:
1. Explosives.
2. Compressed gases.
3. Flammable liquids.
4. Other flammable hazards.
5. Oxygen-rich material, oxidizers, and
organic peroxides.
6. Material affecting health, poisons,
and infectious substances.
7. Radioactive materials.
8. Corrosive material.
9. Miscellaneous hazards.4
The infectious category includes:
■Infectious substances: microbiologic
agents or their toxins that cause, or
716 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
may cause, disease; also called etio-
logic agents.
■Biologic products: products that are
prepared and shipped in compli-
ance with the provisions of 9 CFR
part 102 (Licensed Veterinary Bio-
logical Products), 9 CFR part 103 (Bi-
ological Products for Experimental
Treatment of Animals), 9 CFR part
104 (Imported Biological Products),
21 CFR part 312 (Investigational
New Drug Application), 21 CFR parts
600-680 (Biologics), 29 CFR Part
1910.1030 (Occupational Exposure
to Bloodborne Pathogens), 42 CFR
Part 72 (Interstate Shipment of Etio-
logical Agents), or 49 CFR Parts 171-
178 (Hazardous Materials Regula-
tions).
■Clinical or diagnostic specimens: hu-
man or animal material (including
excreta, secreta, blood and its com-
ponents, body fluids, tissue) being
shipped for the purpose of diagno-
sis.
■Genetically modified organisms.
■Clinical and medical waste.
Blood Bank Applications
Although the USPS regulates some bio-
logic products, such as live poliovirus vac-
cine, the DOT and IATA take the position
that biologic products and diagnostic
specimens may be shipped with less
stringent packaging and labeling require-
ments, unless they contain, or are reason-
ably believed to contain, an infectious
substance. The CDC has proposed regula-
tions to harmonize these requirements
with other federal and international re-
quirements.10 Units of blood that meet
disease testing requirements and un-
screened blood from healthy donors
meeting all FDA donor eligibility criteria
including specimens for screening may
be shipped without hazard restrictions.
Specimens being shipped for initial diag-
nosis purposes and with a low probability
that infectious agents are present are
packaged as diagnostic samples (IATA
packing instruction 650); specimens from
individuals known, or thought likely, to
have an infectious disease are shipped as
infectious substances (IATA packing in-
struction 602). The IATA Dangerous
Goods Regulations should be consulted
for the most recent amendments to ship
biologic specimens.
Method 1.1.1. Shipping Diagnostic
Specimens and Infectious Substances
Principle
The safe transport of hazardous materials
requires that they be packaged in a way to
protect the materials and those who han-
dle them. Prevention from leakage and
from being crushed in unexpected acci-
dents are required. Primary containers
should be leakproof and securely closed.
The primary container should be placed
in a watertight secondary container. Ab-
sorbent material capable of absorbing the
entire liquid contents in the package is
placed between the primary and second-
ary containers. The outer packaging should
have adequate strength for its intended
use and capacity and be labeled in accor-
dance with applicable regulations. Pack-
aging requirements for clinical specimens
are similar to those for infectious sub-
stances except that performance stan-
dards of the packaging materials are less
rigorous.
Materials
1. Leakproof, watertight primary con-
tainer (eg, test tube), heat sealed,
crimped, or otherwise reinforced to
prevent cap slippage.
Methods Section 1: General Laboratory Methods 717
Copyright © 2005 by the AABB. All rights reserved.
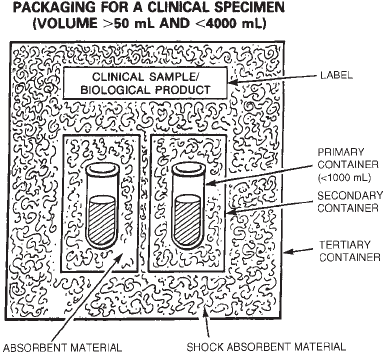
2. Leakproof, watertight secondary pack-
age (eg, sealable plastic bag or con-
tainer with screw cap).
3. Nonparticulate absorbent material (eg,
paper towels, gauze, disposable dia-
per).
4. For infectious substances and for to-
tal shipment volumes greater than
50 mL (but less than 4000 mL or 4
kg), shock-absorbent material equal
in volume to the nonparticulate-ab-
sorbent material.
5. Outer packaging.
a. For infectious substances with a
shipping volume greater than 50
mL (but less than 4000 mL or 4
kg), an outer package of corru-
gated cardboard, fiberboard,
wood, metal, or rigid plastic meet-
ing UN strength requirements.
b. For biologic and diagnostic
specimens with a shipping vol-
ume less than 50 mL, a non-
certified outer container.
6. Coolant material (if necessary).
a. Wet ice, enclosed in sealed bags
to prevent leakage.
b. Dry ice, not to exceed 5 lb (for
air shipments).
7. Itemized listing of package contents.
8. Address label that includes the names,
addresses, and contact names and
telephone numbers of both the sender
and intended recipient.
9. Special labels, as applicable to cir-
cumstances (see Table 1.1.1-1).
a. Diagnostic specimens.
b. Infectious substances.
c. Dry ice.
d. Liquid nitrogen.
e. Cargo aircraft only.
10. Carrier-specific documents, such as
airbills or dangerous goods declara-
tions.
Procedure
1. Place the sealed primary container
into the secondary container.
2. Add sufficient absorbent material
around the primary container (be-
tween the primary and secondary
containers) to cover all sides of the
primary container and to absorb the
entire contents of the primary con-
tainer should breakage occur.
3. Seal the secondary container securely.
4. Place the secondary container into
the applicable outer package.
5. Forinfectioussubstanceswitha
shipping volume greater than 50 mL,
place shock-absorbent materials at
the top, bottom, and sides between
the secondary container and the
outer packaging. See Fig 1.1.1-1.
6. Place any necessary coolant material
(eg, wet ice or dry ice) between the
secondary container and the appli-
cable outer packaging. Provide inte-
rior support to secure the secondary
packaging in the original position
becausetheiceordryicemeltsor
dissipates.
718 AABB Technical Manual
Figure 1.1.1-1. Appropriate packaging of clinical
specimen material.
Copyright © 2005 by the AABB. All rights reserved.

Methods Section 1: General Laboratory Methods 719
Table 1.1.1-1. Special Labels for Shipping
Clinical (Diagnostic) Specimens
Specimens collected for diagnosis, research, or other purposes must be labeled as biohazardous. Both the
primary container and the outer packaging must contain a biohazard label. The outer packaging label must
appear as follows:
1. The color of the material on which the label is printed must be
bright orange
, with the biohazard symbol
and printing in black.
2. The label must be a
rectangle
measuring 2 inches high by 4 inches long.
3. The biohazard symbol, measuring 1.56 inches in diameter, must be centered on a square measuring 2
inches on each side.
4. The size of the letters, printed in Helvetica, must be:
Biohazard 16 pt.
Clinical specimens 14 pt.
Packaged in compliance with 42 CFR part 72 6 pt.
In case of damage or leakage, notify 10 pt.
Shipper and receiver 10 pt.
Infectious Substances (Etiologic Agents)
For substances known to contain infectious agents or reasonably anticipated to contain such agents,
biohazardous labels must be placed on the primary container and outer packaging. The outer packaging
label must appear as follows:
1. The color of the material on which the label is printed must be
white
, with the biohazard symbol and
printing in black.
2. The label must be a
diamond-on-point
measuring, at a minimum, 4 inches on each side.
3. The biohazard symbol, measuring 0.81 inches in diameter, must be centered on a square measuring 2
inches on each side.
4. The Class 6 symbol for infectious substances must be centered at the bottom of the label.
5. The size of the letters, printed in Helvetica, must be:
Infectious substance 16 pt.
Packaged in compliance with 42 CFR part 72 5 pt.
In case of damage or leakage, notify 7 pt.
Immediately notify 7 pt.
Public Health Authority 7 pt.
In the United States 5 pt.
Centers for Disease Control and Prevention 5 pt.
Atlanta, GA 5 pt.
1-800-232-0124 5 pt.
624pt.
Dry Ice
Packages with dry ice must be labeled with the following information:
1. The diamond-shaped Class 9 symbol for miscellaneous hazardous materials.
2. The words “Dry Ice” or “Carbon Dioxide, Solid.”
3. “UN 1845,” the United Nations hazardous material category for dry ice (if shipped out of the country).
4. The name of the contents being cooled.
5. The weight of the dry ice in kg (not to exceed 5 lb if transported by air).
Liquid Nitrogen
Packages with liquid nitrogen must be labeled with a green IATA label stating “Contains Cryogenic Liquid.”
Cargo Aircraft Only
If the mode of transport is air and more than 50 mL of infectious substance is enclosed, the package must
have a warning label “Cargo Aircraft Only” to preclude the carrier from transporting the package on a
passenger plane.
Copyright © 2005 by the AABB. All rights reserved.
7. Enclose the itemized listing of con-
tainer contents between the second-
ary and outer packaging.
8. Seal the outer packaging and label
with address label and applicable
special labels.
9. Complete applicable shipping forms
andsendthemwiththepackage.
Notes
1. Package specimens within a con-
tainer such that they will remain in
an upright position to help prevent
leakage.
2. Closures on primary containers can
be reinforced with adhesive tape or
paraffin. It is not necessary to rein-
force unopened evacuated specimen
collecting tubes.
3. For infectious substances, the name,
address, and telephone number of
the shipper must appear on both the
secondary and outer shipping con-
tainers.
4. Shipments of infectious substances
may contain multiple secondary con-
tainers, but the total volume of the
shipment may not exceed 4 L or 4
kg, excluding the packaging and
coolant weights.
5. Primary containers or secondary
packaging must be capable of with-
standing an internal pressure differ-
ential of 95 kPA (0.95 bar, 13.8 lb/in2)
between the temperatures of –40 C
and 55 C.
6. Styrofoam, plastic bags, and paper
envelopes are unacceptable for outer
packaging.
7. UN-certified containers must be able
to withstand a 30-foot “drop test,” as
specified in 49 CFR 178.609, without
breaking enclosed tubes. The UN
certification number must appear
on the container. Noncertified con-
tainersmustbeabletowithstandat
least a 1.2-meter drop on a hard un-
yielding surface without release of
the container’s contents.
8. Ensure that all state and local regu-
lations are followed.
9. Some exceptions may apply for
ground transportation.
10. If unsure about how to package and
ship materials, contact the CDC.
11. If breakage occurs during shipment,
the package should be handled with
extreme caution (wear personal pro-
tective equipment) and the entire
package (including containers, con-
tents, and packaging materials) should
be autoclaved before discard. Super-
visory personnel should be notified
if the contents are lost in transit or if
the damage appears related to inade-
quate packaging by the sender.
12. The carrier, the receiver, or anyone
handling damaged or leaking pack-
ages must isolate the package and
notify the shipper and intended re-
cipient immediately. In addition, for
infectious substances, notify the CDC
as soon as possible (1-800-232-0124).
When notifying the CDC, the caller
should provide a description of the
condition of the package, the name,
address, and telephone number of
the shipper, and any other pertinent
information, so that decontamina-
tion and disposal procedures can be
provided.
Additional Considerations with the Use of
Dry Ice
1. Solid carbon dioxide or “dry ice” is
classified as a hazardous material
because it can cause burns on con-
tact and gives off carbon dioxide gas
as it volatilizes.
720 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
2. Insulated gloves must be worn when
handling dry ice; eye protection must
be worn when breaking up chunks
of solid ice or when breaking apart
ice pellets.
3. Dry ice should be handled in a well-
ventilated area; its gas can cause light-
headedness and, in extreme cases, as-
phyxiation.
4. When kept in a tightly sealed ship-
ping container, dry ice could rupture
thepackaging.Ifdryiceisused,it
must be placed outside the second-
ary packaging or in an overpack with
one or more complete packages. The
outer packaging must be leakproof.
Procedures for packing with dry ice
must include instructions for sealing
the outer container in a manner that
allows the gas to escape and pre-
vents loosening of secondary con-
tainers as the dry ice dissipates.
Consult the Domestic Mail Manual
(section C023),249 CFR 173.217 and
175.10 (a)(13),3IATA Dangerous Goods
Regulations,4and IATA packing in-
struction 904.
5. HAZMAT training requirements for
dry ice are found in 49 CFR 172.704.3
Training for staff who come in con-
tact with dry ice should include in-
formation on:
■Potential hazards.
■Personal protective equipment
tousewhenhandlingorchip
-
ping.
■Procedures for packing, seal-
ing, labeling, and handling
boxes containing dry ice.
■Special considerations in oper-
atingavehicleusedtotrans
-
port boxes containing dry ice.
■Procedures for storing or dis-
posing of dry ice (eg, allowing
the gas to dissipate by natural
means in a well-ventilated
area) after a shipment is re-
ceived.
Additional Considerations with the Use of
Liquid Nitrogen
1. Liquid nitrogen is classified as a cryo-
genic liquid.
2. Insulated gloves, eye protection, and
a protective laboratory coat must be
worn when working with liquid ni-
trogen.
3. A watertight material capable of with-
standing cryogenic temperatures must
be used for the primary container.
This container must maintain its con-
tainment integrity at the tempera-
ture of the refrigerant as well as at
the temperature and pressure of air
transport if refrigeration is lost.
4. The secondary packaging must be
designed to withstand cryogenic tem-
peratures.
5. The design of the outer packaging
must allow for relief of pressure when
the liquid nitrogen evaporates. A
loose-fitting, but secured, container
cover or pressure relief valve may be
used.
References
1. Code of Federal Regulations. Title 39 CFR.
Washington, DC: US Government Printing
Office, 2004 (revised annually).
2. Etiologic agent preparations, clinical speci-
mens, and biological products. Domestic
Mail Manual (section C023), issue 57, July 10,
2003.
3. Code of Federal Regulations. Title 49 CFR Part
171-180. Washington, DC: US Government
Printing Office, 2004 (revised annually).
4. Dangerous goods regulations. 45th ed. Mon-
treal, Canada: International Air Transport As-
sociation, 2004 (revised annually).
5. Technical instructions for the safe transport
of dangerous goods by air. 2002-2004 ed.
Montreal, Canada: International Civil Avia-
tion Organization, Documents 9284 and
9284SU, 2002.
Methods Section 1: General Laboratory Methods 721
Copyright © 2005 by the AABB. All rights reserved.
6. Code of Federal Regulations. Title 42 CFR Part
72. Washington, DC: US Government Printing
Office, 2004 (revised annually).
7. Infectious substances and diagnostic speci-
mens shipping guidelines. 4th ed. Montreal,
Canada: International Air Transport Associa-
tion, 2003.
8. World Health Organization. Guidelines for
thesafetransportofinfectioussubstances
and diagnostic specimens. Geneva, Switzer-
land: World Health Organization, 1997.
9. Code of Federal Regulations. Title 29 CFR
1910.1030. Washington, DC: US Government
Printing Office, 2004 (revised annually).
10. Centers for Disease Control and Prevention.
Packaging and handling of infectious sub-
stances and select agents, notice of proposed
rulemaking. Fed Regist 1999;64(208):58022-
31.
Method 1.1.2. Monitoring Temperature
During Shipment of Blood
Principle
Some form of temperature indication or
monitoring is desirable when shipping
blood. The temperature of the contents of
a shipping container used for whole blood
or liquid-stored red cell components can
be ascertained when the shipment is re-
ceived, as follows:
Procedure
1. Open the shipping container and
promptly place the sensing end of a
calibrated liquid-in-glass or elec-
tronic thermometer between two
bags of blood or components (labels
facing out) and secure the “sand-
wich” with two rubber bands.
2. Close the shipping container.
3. After approximately 3 to 5 minutes,
read the temperature.
4. If the temperature of red-cell-con-
taining components exceeds 10 C,
quarantine the units until their ap-
propriate disposition can be deter-
mined.
Notes
Other suitable methods for monitoring
shipments are:
1. Use time/temperature indicators,
one such indicator per shipping car-
ton. These indicators will change
colororshowanothervisibleindica
-
tion if the temperature has exceeded
10 C.
2. Place a “high-low” thermometer in
the shipping container. This simple,
reusable thermometer measures and
records the highest and lowest tem-
peratures during any period.
Method 1.2. Treatment of
Incompletely Clotted Specimens
Principle
Fibrin generation may continue in serum
separated from incompletely clotted blood,
especially during incubation at 37 C. This
produces strands of protein that entrap
red cells and make it difficult to evaluate
agglutination. Blood from patients who
have recently received heparin may not
clot at all, and blood from patients with
excessive fibrinolytic activity may relique-
fy or may contain protein fragments that
interfere with examination for agglutina-
tion.
Materials
1. Thrombin: dry human/bovine thrombin
or thrombin solution (50 units/mL
in saline).
2. Glass beads.
3. Protamine sulfate: 10 mg/mL in sa-
line.
4. Epsilon aminocaproic acid (EACA):
0.25 g/mL in saline.
722 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Procedure
1. To accelerate clotting: Either of the
following techniques may be used:
a. Add to the specimen the amount
of dry thrombin that adheres to
the tip of an applicator stick or
1 drop of thrombin solution
permLofwholebloodorse
-
rum. Allow 10 to 15 minutes for
the clot to form. Use standard
centrifugation to separate the
clot and serum.
b. Gently agitate the separated se-
rum with small glass beads, at
37 C, for several minutes. Then,
use low speed centrifugation to
pellet the glass beads. Transfer
the serum to another tube.
2. To neutralize heparin: Protamine
sulfate can be added to the speci-
men to neutralize heparin; however,
excess protamine promotes rouleaux
formation and, in great excess, will
inhibit clotting. Add 1 drop of prota-
mine sulfate solution to 4 mL of
whole blood and wait 30 minutes to
evaluate the effect on clotting. If
clotting does not occur, add addi-
tional protamine sparingly. Note:
protamine sulfate may work more
rapidly when briefly incubated (5-10
minutes) at 37 C.
3. To inhibit fibrinolytic activity: Add 0.1
mL of EACA to 4 mL of whole blood.
Notes
1. The use of anticoagulated (eg, ACD
or EDTA) collection tubes may help
to avoid the problem of incom-
pletely clotted specimens. The use of
anticoagulated specimens must be
validated in accordance with each
standard operating procedure.
2. Because preparations of human
thrombin may contain red cell anti-
bodies, test results must be carefully
observed for false-positive reactions.
Quality control should be performed
on thrombin reagents before or con-
current with their use to identify
those with contaminating antibod-
ies.
Method 1.3. Solution
Preparation—Instructions
Principle
The basic definitions, calculations, and
instructions given below serve as a review
of simple principles necessary for solu-
tion preparation.
1. Mole, gram-molecular weight: Weight,
expressed in grams equal to the
atomic or molecular weight of the
substance.
2. Molar solution: A one molar (1 M)
solution contains one mole of solute
in a liter of solvent. The solvent is as-
sumedtobedistilledordeionized
water unless otherwise indicated.
3. Gram-equivalent weight: Weight, in
grams, of a substance that will pro-
duce or react with 1 mole of hydro-
gen ion.
4. Normal solution: A one normal (1 N)
solution contains one gram-equiva-
lent weight of solute in a liter of
solution.
5. Percentage solutions: The percent
designation of a solution gives the
weight or volume of solute present
in 100 units of total solution. Percent
can be expressed as:
a. Weight/weight (w/w), indicating
gramsofsolutein100gofsolu
-
tion.
b. Volume/volume (v/v), indicating
milliliters of solute present in
100 mL of solution.
Methods Section 1: General Laboratory Methods 723
Copyright © 2005 by the AABB. All rights reserved.
c. Weight/volume (w/v), indicating
grams of solute in 100 mL of so-
lution. Unless otherwise speci-
fied, a solution expressed in
percentage can be assumed to
be w/v.
6. Water of crystallization, water of hy-
dration: Molecules of water that form
an integral part of the crystalline
structure of a substance. A given
substance may have several crystal-
line forms, with different numbers of
water molecules intrinsic to the en-
tire molecule. The weight of this wa-
ter must be included in calculating
molecular weight of the hydrated
substance.
7. Anhydrous: The salt form of a sub-
stance with no water of crystallization.
8. Atomic weights (rounded to whole
numbers): H, 1; O, 16; Na, 23; P, 31; S,
32; Cl, 35; K, 39.
9. Molecular weights:
HCl: 1 + 35 = 36; NaCl: 23 + 35 = 58
KCl: 39 + 35 = 74
H2O: (2 ×1) + 16 = 18
NaH2PO4:23+(2×1) + 31 + (4 ×16) =
120
NaH2PO4•H2O: 23 + (2 ×1) + 31 +
(4 ×16) + (2 ×1) + 16 = 138
KH2PO4:39+(2×1) + 31 + (4 ×16) =
136
H2SO4:(2×1) + 32 + (4 ×16) = 98
Examples
1. Molar solutions:
1MKH
2PO4=136gofsolutemadeup
to 1 L.
0.15 M KH2PO4= (136 ×0.15) = 20.4 g
of solute made up to 1 L.
0.5 M NaH2PO4= (120 ×0.5) = 60 g of
solute made up to 1 L.
2. Molar solution with hydrated salt:
0.5 M NaH2PO4•H2O = (138 ×0.5) =
69 g of the monohydrate crystals
made up to 1 L.
3. Normal solutions:
1NHCl=36gofsolutemadeupto1
L. One mole HCl dissociates into one
mole H+, so gram-equivalent weight
and gram-molecular weight are the
same.
12 N HCl = (36 ×12) = 432 g of solute
made up to 1 L.
1NH
2SO4=(98÷2)=49gofsolute
made up to 1 L. One mole H2SO4dis-
sociates to give two moles of H+,so
the gram-equivalent weight is half
the gram-molecular weight.
4. Percent solution:
0.9% NaCl (w/v) = 0.9 g of solute
made up to 100 mL of solution.
Notes
Accurate results require accurate prepara-
tion of reagents. It is important to care-
fully read and follow all instructions and
labels.
1. Weigh only quantities appropriate
for the accuracy of the equipment.
The operator’s manual should give
these specifications.
2. Prepare the largest volume that is
practical. There is greater accuracy
in measuring larger volumes than
smaller volumes. If a reagent bal-
ance is accurate to ±0.01 g, the po-
tential error in weighing 0.05 g (50
mg) will be 20%, whereas the poten-
tial error in weighing 0.25 g (250 mg)
will be only 4%. If the solution re-
tains its activity when stored appro-
priately, it is usually preferable to
preparealargevolume.Ifthesolu
-
tion deteriorates rapidly, smaller vol-
umes may be preferred to reduce
waste.
3. Note whether a substance is in the
hydrated or anhydrous form. If the
724 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

instructions give solute weight for
one form, and the available reagent
is in another form, be sure to adjust
the measurements appropriately.
For example, if instructions for 0.5 M
NaH2PO4call for 60 g, and the re-
agent is NaH2PO4H2O, find the ra-
tio between the weights of the
two forms. The molecular weight of
NaH2PO4H2O is 138 and the mo-
lecular weight of NaH2PO4is 120.
Therefore, the ratio is 138 ÷ 120 =
1.15. Multiply the designated weight
by the ratio (60 g ×1.15 = 69 g) to ob-
tain the final weight needed.
4. Dissolve the solute completely be-
fore making the solution to the final
volume. This is especially important
for substances, such as phosphates,
that dissolve slowly. For example, to
make 500 mL of 0.15 M KH2PO4:
a. Weigh 10.2 g of solute in a weigh-
ing boat or glass [(0.15 ×136) ÷
2] because only 500 mL will be
made.
b. Place 350 mL of water in a
500-mL volumetric flask on a
magnetic stirrer. Add the stir-
ring bar and adjust it to a slow,
steady stirring speed.
c. Add 10.2 g of salt, then rinse the
boat with several aliquots of
water until no salt remains. Nu-
merous small-volume rinses
remove adherent material more
effectively than a few larger
volumes. Add the rinse water to
the material in the flask and stir
until the salt has completely
dissolved.
d. If pH measurement is unneces-
sary, add water to the 500-mL
mark, adjusting the volume for
the stirring bar, and mix thor-
oughly. For solutions needing pH
adjustment, see the next step.
5. AdjustthepHofthesolutionbefore
bringing it to its final volume so that
the addition of water (or other sol-
vent) does not markedly change the
molarity. For example, to bring 500
mL of 0.1 M glycine to a pH of 3:
a. Add 3.75 g of glycine
(H2NCH2COOH: molecular
weight, 75) to 400-475 mL of
water in a beaker. Dissolve
completely, using a magnetic
stirrer.
b. Add a few drops of concentrated
(12 N) HCl and measure pH af-
ter acid is thoroughly mixed.
Continue adding HCl until pH
is 3.0.
c. Transfer the solution to a 500-mL
volumetric flask. Rinse beaker
and stirring bar with aliquots of
water, adding the rinse water to
the flask. Use the rinses to con-
tribute to the total 500-mL vol-
ume.
d. Measure the pH of the solution
at final volume.
References
1. Remson ST, Ackerman PG. Calculations for
the medical laboratory. Boston, MA: Little,
Brown & Co., 1977.
2. Henry JB, ed. Clinical diagnosis and manage-
ment by laboratory methods. 18th ed. Phila-
delphia: WB Saunders, 1991.
Method 1.4. Serum Dilution
Principle
Serum is sometimes diluted in saline or
other diluents to determine its relative
antibody concentration. It is customary to
express the dilution as 1 part of serum
contained in the total number of parts of
the dilution. For example, to test the se-
rum at one-tenth its original concentra-
tion, a dilution of 1 part in 10 may be
Methods Section 1: General Laboratory Methods 725
Copyright © 2005 by the AABB. All rights reserved.
made by mixing 1 mL of serum with 9 mL
of saline. The final volume is 10,andthe
dilution is expressed as a 1 in 10 dilution.
The diluted material contains one-tenth
(1/10 or 0.1) of the unmodified serum. It
is often customary to report the titer of an
antibody as the reciprocal of the highest
dilution that retains a 1+ agglutination.
Therefore, serum that reacts at a dilution
of 1/32 is considered to have a titer of 32.
Note:A1in10dilutionis1partin9parts,
whereasa1to10or1:10is1partin10parts.
Procedure
1. Diluting an existing dilution:
a. A new higher dilution can be
prepared from diluted material
by adding more diluent. The
formula for calculating either
the new higher final dilution or
theamountofdiluenttoaddto
obtain a higher final dilution is:
reciprocal of present serum dilution
volume of serum dilution used
= reciprocal of new final dilution
total final volume
b. Example: Serum dilution is one
in two and volume of serum di-
lution is 1.0 mL. If 4.0 mL of sa-
line is added, what will be the
new final dilution?
2=X
15
X = 10 or 1 in 10 dilution
2. Diluting a dilution to a specified volume:
a. The formula for calculating the
volume of diluent to add to a
dilution to achieve a certain
quantity of a new higher final
dilution is:
reciprocal of present dilution
volume of present dilution needed
= reciprocal of final dilution
total final volume required
b. Example: Present serum dilu-
tion is one in two, total final
volume is 100 mL, and new fi-
nal serum dilution is 1 in 10.
How much serum (diluted one
in two) will have to be added to
make up a final volume of 100
mL of a 1 in 10 dilution?
2=10
X100
X = 20 or 20 mL of serum (dilu-
tion of one in two) must be added
to 80 mL of diluent to obtain
100 mL of a 1 in 10 dilution.
Method 1.5. Dilution of %
Solutions
Procedure
1. Dilutions can be prepared from more
concentrated solutions by use of the
following formula:
(Volume1×Concentration1)=
(Volume2×Concentration2)
V1×C1=V
2×C2
where V1and C1represent original
volume and concentration, and V2
and C2represent final desired vol-
ume and concentration.
726 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
2. Example: 30% albumin is available,
but 2 mL of 6% albumin is needed.
How should the albumin be diluted?
V1×30 = 2 ×6
30V1=12
V1=12÷30 = 0.4
Therefore, mix 0.4 mL of 30% albu-
minwith1.6mLsalinetoobtain2.0
mL of 6% albumin, or for small-vol-
ume use, mix 4 drops 30% albumin
with 16 drops saline to obtain 20
drops of 6% albumin.
Method 1.6. Preparation of a
3% Red Cell Suspension
Principle
A 3% red cell suspension is a common re-
agent in many serologic procedures. The
suspension need not be exactly 3%; an
approximation achieves the appropriate
serum-to-cell ratio for most test proce-
dures and an adequate number of red
cells to read and grade the reactions. The
following steps are intended to help an in-
dividual gain confidence in approximat-
ing a 3% red cell suspension visually, both
as a suspension of cells and in the appro-
priate size of the cell pellet achieved after
centrifugation.
Materials
1. Whole blood sample.
2. Test tubes.
3. Disposable pipettes (1 mL and 10 mL
serologic).
4. Saline.
5. Centrifuge (3000 rpm or equivalent).
6. Commercially prepared 3% reagent
red cell suspension.
Procedure
To prepare 10 mL of a 3% red cell suspen-
sion:
1. Transfer at least 1 mL of whole blood
to a 10-mL tube.
2. Wash the red cells in saline or phos-
phate-buffered saline (PBS), centri-
fuging for 5 minutes to pellet the
cells. Repeat two or three times. The
final supernate should be clear and
should be completely removed by
aspiration.
3. Transfer 0.3 mL of the washed red
cells to a tube with 9.7 mL of saline,
PBS, or Alsever’s solution.
4. Cap or cover the tube with parafilm.
Thoroughly mix the red cells and sa-
line by gently inverting the tube sev-
eral times.
5. To compare the color and density of
the suspension by eye, transfer a vol-
ume of the prepared suspension to a
10 ×75 mm tube. Also transfer a sim-
ilar volume of a known 3% red cell
suspension (eg, commercial reagent
red cell suspension) to another 10 ×
75 mm tube. Hold the two tubes in
front of a light source to compare them.
6. To compare the size of the cell pellet
expected from a 3% red cell suspen-
sion, transfer one drop of the pre-
pared suspension to a 10 ×75 mm
tube. Similarly, transfer one drop of
a known 3% commercial reagent red
cell suspension to another 10 ×75 mm
tube. Centrifuge the tubes in a sero-
logic centrifuge, using the spin time
designated for “saline.” The size of
the two cell pellets should be similar.
Note
For best results use red cell suspensions
on the day of preparation only, unless sta-
bility for a longer time has been validated.
Methods Section 1: General Laboratory Methods 727
Copyright © 2005 by the AABB. All rights reserved.

Method 1.7. Preparation and
Use of Phosphate Buffer
Principle
Mixtures of acids and bases can be pre-
pared at specific pH values and used to
buffer (render) other solutions to that pH.
Thefollowingprocedureincludesamethod
for preparing phosphate-buffered saline
(PBS), which can be used as a diluent in
serologic tests.
Reagents
1. Prepare acidic stock solution (solu-
tion A) by dissolving 22.16 g of
NaH2PO4•H2O in 1 L of distilled wa-
ter. This 0.16 M solution of the mono-
basicphosphatesalt(monohydrate)
has a pH of 5.0.
2. Prepare alkaline stock solution (so-
lution B) by dissolving 22.7 g of
Na2HPO4in 1 L of distilled water.
This 0.16 M solution of the dibasic
phosphate salt (anhydrous) has a pH
of 9.0.
Procedure
1. Prepare working buffer solutions of
the desired pH by mixing appropri-
ate volumes of the two solutions. A
few examples are:
pH Solution A Solution B
5.5 94 mL 6 mL
7.3 16mL 84mL
7.7 7 mL 93 mL
2. CheckthepHoftheworkingsolu
-
tion before using it. If necessary, add
small volumes of acid solution A or
alkaline solution B to achieve the de-
sired pH.
3. To prepare PBS of a desired pH, add
one volume of phosphate buffer at
that pH to nine volumes of normal
saline.
References
1. Hendry EB. Osmolarity of human serum and
of chemical solutions of biologic importance.
Clin Chem 1961;7:156-64.
2. Dacie JV, Lewis SM. Practical haematology. 4th
ed. London, England: J and A Churchill, 1968:
540-1.
Method 1.8. Reading and
Grading Tube Agglutination
Principle
The purpose of grading reactions is to al-
low comparison of reaction strengths.
This is beneficial in detecting multiple
antibody specificities or antibodies exhib-
iting dosage. The grading of agglutination
reactions should be standardized among
all members of the laboratory staff, in the
interest of uniformity and reproducibility
of test results. Most laboratories define
their own version of a grading system,
which is described in a written procedure
available to all staff. Some systems use as-
signed numeric values (scores) for the
observed reactions.
Materials
1. Centrifuged serologic tests for agglu-
tination.
2. Agglutination viewer.
Procedure
1. Gentlyshakeortiltthetubeandre
-
suspend the red cell button in the
tube. The tilt technique uses the me-
niscus to gently dislodge the red cell
button from the wall of the tube.
2. Observe the way that cells are dis-
persed from the red cell button.
3. Record reactivity by comparing the
agglutinates to the descriptions in
Table 1.8-1. The reactivity should be
assessed when the red cells have been
728 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

completely resuspended from the
button.
Interpretation
Refer to Table 1.8-1.
Notes
1. An agglutination viewer may facilitate
the reading of tube tests. However,
the manufacturer’s test recommen-
dation must be followed for inter-
preting the test results.
2. Serum overlying the centrifuged cell
button must be inspected for
hemolysis, which is a positive sign of
an antigen-antibody reaction, pro-
vided the pretest serum was not
hemolyzed and no hemolytic agent
was added to the test.
3. The character of the agglutination
should be noted and recorded. This
information provides valuable clues
in the investigation such as the char-
acteristic refractile agglutination of
anti-Sda.
4. Mixed-field agglutination is ex-
pected when using pooled cells for
donor antibody detection and add-
ing check cells to negative antiglo-
bulin tests.
Reference
Race RR, Sanger R. Blood groups in man. 6th ed.
Oxford: Blackwell Scientific Publications, 1975.
Methods Section 1: General Laboratory Methods 729
Table 1.8-1. Interpretation of Agglutination Reactions
Macroscopically Observed Findings Designation Score
One solid agglutinate
Several large agglutinates
Medium-size agglutinates, clear background
Small agglutinates, turbid background
Very small agglutinates, turbid background
Barely visible agglutination, turbid background
No agglutination
Mixtures of agglutinated and unagglutinated red cells
(mixed field)
Complete hemolysis
Partial hemolysis, some red cells remain
4+
3+
2+
1+
1w
w+ or +/–
0
mf
H
PH
12
10
8
5
4
2
0
Copyright © 2005 by the AABB. All rights reserved.

Methods Section 2: Red Cell Typing
Methods Section 2
Red Cell Typing
Method 2.1. Slide Test for
Determination of ABO Type
of Red Cells
Principle
See Chapter 13 for a discussion of the
principles of testing for ABO groups.
Specimen
The reagent manufacturer’s instructions
must be consulted before slide tests are
performed; some manufacturers recom-
mend performing slide tests with whole
blood, whereas others specify the use of
red cell suspensions of lighter concentra-
tions prepared in saline, serum, or plasma.
Reagents
1. Anti-A.
2. Anti-B.
3. Anti-A,B (optional).
All reagents must be used in accordance
with the manufacturer’s instructions.
Procedure
1. Place 1 drop of anti-A on a clean, la-
beled glass slide.
2. Place 1 drop of anti-B on a separate
clean, labeled glass slide.
3. Place 1 drop of anti-A,B on a third
slide, if parallel tests are to be per-
formed with this reagent, or on a
single clean, labeled slide if this is
the only test performed.
4. Add to each drop of reagent on the
slides 1 drop of well-mixed suspen-
sion (in saline, serum, or plasma) of
the red cells to be tested. (Consult
the reagent manufacturer’s instruc-
tions to determine the correct cell
concentration to be used.)
5. Mix the reagents and red cells thor-
oughly, using a clean applicator stick
for each reagent. Spread the mixture
731
Section 2
Copyright © 2005 by the AABB. All rights reserved.
over an area approximately 20 mm ×
40 mm.
6. Gently tilt the slide continuously for
up to 2 minutes. Do not place the slide
over a heated surface, such as an Rh
viewbox, during this period.
7. Read, interpret, and record the re-
sults of the reactions on all slides.
Interpretation
1. Strong agglutination of red cells in
thepresenceofanyABOtypingre
-
agent constitutes a positive result.
2. A smooth suspension of red cells at
the end of 2 minutes is a negative re-
sult.
3. Samples that give weak or doubtful
reactions should be retested using
Method 2.2.
Notes
1. Slide testing imposes a greater risk
of exposure to infectious samples.
Personnel should follow safety mea-
sures detailed in the facility’s proce-
dures manual.
2. Slide testing is not suitable for detec-
tion of ABO antibodies in serum/
plasma.
Method 2.2. Tube Tests for
Determination of ABO Group
of Red Cells and Serum
Principle
See Chapter 13 for a discussion of the
principles of testing for ABO groups. The
following procedure is an acceptable rep-
resentative method, but the manufacturer’s
instructions for the specific reagents must
be consulted.
Specimen
The reagent manufacturer’s package in-
sert must be consulted to determine spe-
cific specimen requirements. Generally,
clotted or anticoagulated blood samples
maybeusedforABOtesting.Theredcells
may be suspended in autologous serum,
plasma, or saline, or may be washed and
resuspended in saline.
Reagents
1. Anti-A.
2. Anti-B.
3. Anti-A,B. Note: Use of this reagent is
optional.
4. A1,A
2, and B red cells. They can be
obtained commercially or the testing
laboratory can prepare a 2% to 5%
suspension on each day of use. (Note:
The use of A2cells is optional.)
All reagents must be used in accordance
with the manufacturer’s instructions.
Procedures
Testing Red Cells
1. Place 1 drop of anti-A in a clean, la-
beled test tube.
2. Place 1 drop of anti-B in a clean, la-
beled tube.
3. Place 1 drop of anti-A,B in a clean,
labeled tube, if tests are to be per-
formed with this reagent.
4. Add to each tube 1 drop of a 2% to
5% suspension (in saline, serum, or
plasma) of the red cells to be tested.
Alternatively, the equivalent amount
of red cells can be transferred to each
tube with clean applicator sticks.
5. Mix the contents of the tubes gently
and centrifuge them for the cali-
brated spin time.
6. Gently resuspend the cell buttons
and examine them for agglutination.
732 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
7. Read, interpret, and record the test re-
sults. Compare the red cell test re-
sults with those obtained in the se-
rum/plasma tests (see below).
Testing Serum/Plasma
1. Label two clean test tubes as A1and
B. (Note: Label an additional tube if
an optional test with A2red cells is to
be performed.)
2. Add 2 or 3 drops of serum/plasma to
each tube.
3. Add 1 drop of A1reagent cells to the
tube labeled A1.
4. Add 1 drop of B reagent cells to the
tube labeled B.
5. Add A2cells to the appropriate tube,
if this optional test is being per-
formed.
6. Mix the contents of the tubes gently
and centrifuge them for the cali-
brated spin time.
7. Examinetheserumoverlyingthecell
buttons for evidence of hemolysis.
Gently resuspend the cell buttons
and examine them for agglutination.
8. Read, interpret, and record test results.
Compare serum test results with
those obtained in testing red cells
(see above).
Interpretation
1. Agglutination of tested red cells and
either hemolysis or agglutination in
tests with serum constitute positive
test results.
2. A smooth cell suspension after re-
suspension of the cell button is a
negative test result.
3. Interpretation of serum/plasma and
cell tests for ABO is given in Table
13-1.
4. Any discrepancy between the results
of the tests with serum/plasma and
cells should be resolved before an
interpretation is recorded for the
patient’s or donor’s ABO group (see
Chapter 13).
Note
Positive reactions characteristically show
3+ to 4+ agglutination by reagent ABO an-
tibodies; reactions between test serum
and reagent red cells are often weaker.
The serum tests may be incubated at
room temperature for 5 to 15 minutes to
enhance weak reactions. See Chapter 13
for a discussion of weakly reactive sam-
ples.
Method 2.3. Microplate Test
for Determination of ABO
Group of Red Cells and
Serum
Principle
See Chapter 13 for a discussion of the
principles of testing for ABO blood group.
Microplate techniques can be used to test
for antigens on red cells and for antibod-
ies in serum.
A microplate can be considered as a ma-
trix of 96 “short” test tubes; the principles
that apply to hemagglutination in tube tests
also apply to tests in microplates.
Microplates may be rigid or flexible, with
either U-shaped or V-shaped bottoms.
U-shaped bottom plates are more widely
used because results can be read either af-
ter centrifuging the plate and observing the
characteristics of resuspended red cells or
by observing the streaming pattern of the
cells when the plate is placed at an angle.
Either reading technique permits estima-
tion of the strength of agglutination.
Specimen
Refer to Method 2.2.
Methods Section 2: Red Cell Typing 733
Copyright © 2005 by the AABB. All rights reserved.
Equipment
1. Dispensers (optional): Semiautoma-
ted devices are available for dispens-
ing equal volumes to a row of wells.
Special plate carriers can be pur-
chased to fit common table-top cen-
trifuges.
2. Microplate readers (optional): Auto-
mated photometric devices are avail-
able that read microplate results by
the light absorbance in U-shaped
bottom wells to differentiate be-
tween positive and negative tests.
The microprocessor component of
the reader interprets the reactions
and prints the blood testing results.
The manufacturer’s instructions for
the collection and preparation of se-
rum/plasma and cell specimens
must be followed.
3. Centrifuges: Appropriate conditions
must be established for each centri-
fuge. The following times and rela-
tive centrifugal forces, expressed as
g, are suggested. Consult the manu-
facturer’s directions for specific in-
formation.
For a flexible U-shaped bottom
microplate: 700 ×gfor 5 seconds for
red cell testing and serum/plasma
testing.
ForarigidU-shapedbottommicro
-
plate: 400 ×gfor 30 seconds for red
cell testing and serum/plasma testing.
Reagents
Many manufacturers supply ABO or Rh
typing reagents that are licensed by the
Food and Drug Administration (FDA) for
use as undiluted reagents in microplate
tests.
1. Anti-A.
2. Anti-B.
3. Anti-A,B. Note: Use of this reagent is
optional.
4. Group A1,A
2, and B red cells. They
can be obtained commercially or the
testing laboratory can prepare a 2%
to 5% suspension on each day of use.
(Note: The use of A2cells is optional.)
Procedure
Testing Red Cells
1. Place 1 drop of anti-A and anti-B in
separate clean wells of a U-bottom
microplate. If tests with anti-A,B are
to be performed, add this reagent to
a third well.
2. Add1dropofa2%to5%salinesus
-
pension of red cells to each well con-
taining blood typing reagent.
3. Mix the contents of the wells by
gently tapping the sides of the plate.
4. Centrifuge the plate at the appropri-
ate conditions established for the
centrifuge.
5. Resuspend the cell buttons by man-
ually tapping the plate or with the
aid of a mechanical shaker, or place
the plate at an angle for the tilt and
stream method.
6. Read, interpret, and record results.
Compare red cell test results with those
obtained in testing serum/plasma.
Testing Serum/Plasma
1. Add 1 drop of a 2% to 5% suspension
of reagent A1andBredcellstosepa
-
rate clean wells of a U-bottom micro-
plate. (Note: If an optional test on A2
cells will be performed, add A2cells
to a third well.)
2. Add 1 drop of serum or plasma un-
der test to each well.
3. Mix the contents of the wells by gently
tapping the sides of the plate.
4. Centrifuge the plate at the appropri-
ate conditions established for the
centrifuge.
734 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
5. Resuspend the cell buttons by man-
ually tapping the plate or with aid of
a mechanical shaker, or place the plate
at an angle for the tilt and stream
method.
6. Read, interpret, and record results.
Compare test results on serum/plasma
with those obtained in testing red
cells.
Note
To enhance weak serum/plasma reac-
tions, the plates may be incubated at
room temperature for 5 to 10 minutes,
then the centrifugation, reading, and re-
cording steps may be repeated.
Interpretation
1. Agglutination in any well of red cell
tests and hemolysis or agglutination
in any well of a serum test constitute
positive results.
2. A smooth suspension of red cells af-
ter resuspension of the cell button is
a negative test.
3. The interpretation of ABO tests is
given in Table 13-1.
4. Any discrepancy between results on
cell and serum/plasma tests should
be resolved before an interpretation
is recorded for the patient’s or donor’s
ABO group (see Chapter 13).
Method 2.4. Confirmation of
Weak A or B Subgroup by
Adsorption and Elution
Principle
See Chapter 13 for a discussion of the
principles of testing for ABO groups.
Specimen
Refer to Method 2.2.
Reagents
1. Human anti-A and/or anti-B. Be-
causesomemonoclonalABOtyping
reagents are sensitive to changes in
pH and osmolarity, they may not be
suitable for use in adsorption/elution
tests.
2. Eluting agent: See Methods Section
4.
Procedure
1. Wash1mLoftheredcellstobe
tested at least three times with saline.
Remove and discard the supernatant
salineafterthelastwash.
2. Add 1 mL of reagent anti-A (if a weak
variant of A is suspected) or 1 mL of
anti-B (if a weak variant of B is sus-
pected) to the washed cells.
3. Mix the red cells with the reagent
antibody and incubate them at 4 C
for 1 hour, mixing occasionally.
4. Centrifuge the mixture to pack the
red cells. Remove all supernatant re-
agent.
5. Transfer the red cells to a clean test
tube.
6. Wash the cells at least eight times
with large volumes (10 mL or more)
of cold (4 C) saline. Save an aliquot
of the final wash supernatant fluid
and test it in parallel with the eluate.
7. Use an elution method suitable for
recovery of ABO antibodies, eg, heat
or Lui freeze-thaw elution techni-
ques can be used to remove anti-
body from the cells (see Methods
Section 4).
8. Centrifuge to pack the cells and trans-
fer the supernatant eluate to a clean
test tube.
Methods Section 2: Red Cell Typing 735
Copyright © 2005 by the AABB. All rights reserved.
9. Testtheeluateandthefinalwash
solution (from step 6), in parallel,
against two examples of group O
cells and two examples of cells ex-
pressing the relevant antigen (A1
cells for suspected anti-A, B cells for
anti-B). Add 2 drops of eluate or
wash to 1 drop of cells and examine
them for agglutination after immedi-
ate centrifugation; if negative, incu-
bate 15 to 30 minutes at room tem-
perature. If these phases are both
negative, a 15-minute incubation at
37 C and the indirect antiglobulin
test may also be performed.
Interpretation
1. The presence of anti-A or anti-B in
the eluate, hence the presence of A
or B antigen on the test cells, is con-
firmed if: a) the eluate reacts with
both antigen-positive cells, at any
phase; b) the eluate is nonreactive at
all phases with all group O cells; and
c) the final wash solution is non-
reactive with all four cells.
Iftheeluatedoesnotreactwith
the A or B cells, it may indicate that
the test cells do not express the anti-
gen and cannot adsorb the relevant
antibody; alternatively, it could re-
flect failure to prepare the eluate
correctly.
Iftheeluatereactswithoneor
both of the A or B cells and also with
one or both or all of the O cells, it in-
dicates recovery of some other or
additional antibody in the adsorp-
tion/elution process.
2. If the final wash solution reacts with
the A or B cells, tests on the eluate
cannot be considered valid. This can
occur if unbound reagent antibody
was not adequately removed before
beginning the elution, if the cells
were not adequately washed, or if
there was dissociation of bound an-
tibody during the wash process.
3. A and B cells can be used in the ad-
sorption/elution procedure as posi-
tive/negative controls and tested in
parallel. Group O cells can also be
used as a negative control.
Note
The eluate may be stained by hemoglobin
and be difficult to read except at the indi-
rect antiglobulin phase.
Reference
Beattie KM. Identifying the causes of weak or
“missing” antigens in ABO grouping tests. In: The
investigation of typing and compatibility prob-
lems caused by red blood cells. Washington, DC:
AABB, 1975:15-37.
Method 2.5. Saliva Testing
for A, B, H, Lea, and Leb
Principle
Approximately 78% of all individuals pos-
sess the Se gene that governs the secretion
of water-soluble ABH antigens into all
body fluids with the exception of cere-
brospinal fluid. These secreted antigens
can be demonstrated in saliva by inhibi-
tion tests with ABH and Lewis antisera
(see Chapter 13).
Specimen
1. Collect 5 to 10 mL of saliva in a small
beaker or wide-mouthed test tube.
Most people can accumulate this
amount in several minutes. To en-
courage salivation, the subject may
be asked to chew wax, paraffin, or a
clean rubber band, but not gum or
736 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
anything else that contains sugar or
protein.
2. Centrifuge saliva at 900 to 1000 ×g
for 8 to 10 minutes.
3. Transfer supernatant to a clean test
tube and place it in a boiling water-
bath for 8 to 10 minutes to inactivate
salivary enzymes.
4. Recentrifuge at 900 to 1000 ×gfor 8
to 10 minutes, remove clear or slightly
opalescent supernatant fluid, and
discard the opaque or semisolid ma-
terial. Dilute the supernatant fluid
with an equal volume of saline.
5. Refrigerate, if testing is to be done
within several hours. If testing will
notbedoneonthedayofcollection,
freezethesampleandstoreitat–20
C. Frozen samples retain activity for
several years.
Reagents
1. Human (polyclonal) anti-A and anti-B.
Note: Some monoclonal reagents
may not be appropriate for use;
therefore, appropriate controls are
essential.
2. Anti-H lectin from Ulex europaeus
obtained commercially or prepared
by saline extraction of Ulex euro-
paeus seeds.
3. Polyclonal (rabbit/goat/human)
anti-Lea.Therearenopublisheddata
on the suitability of monoclonal
Lewis antibodies.
4. A1andBredcells,asusedinMethod
2.2.
5. Group O, Le(a+b–) red cells, as used
for antibody detection or identifica-
tion (see Chapter 19).
6. Specimens, frozen or fresh saliva,
from persons known to be secretors
or nonsecretors, to use as positive
and negative controls.
Procedures
Selection of Blood Grouping Reagent
Dilution
1. Prepare doubling dilutions of the ap-
propriate blood typing reagent.
2. To 1 drop of each reagent dilution,
add 1 drop of 2% to 5% saline sus-
pension of red cells. Use A, B, or O
cells to determine, respectively, A, B,
or H secretor status. Use Le(a+b–)
red cells to determine Lewis secretor
status.
3. Centrifuge each tube and examine
macroscopically for agglutination.
4. Select the highest reagent dilution
that gives 2+ agglutination.
Inhibition Test for Secretor Status
1. Add 1 drop of appropriately diluted
blood grouping reagent to each of
four tubes. For ABH studies, the
tubes should be labeled “Secretor,”
“Nonsecretor,” “Saline,” and “Un-
known.” For Lewis studies, they will
be “Lewis-positive,” “Lewis-nega-
tive,” “Saline,” and “Unknown.”
2. Add 1 drop of the appropriate saliva
to the “Secretor,” “Nonsecretor,” and
“Unknown” tubes, and 1 drop of sa-
line to the tube marked “Saline.”
3. Mix the contents of the tubes. Incu-
bate the tubes for 8 to 10 minutes at
room temperature.
4. Add 1 drop of 2% to 5% saline sus-
pension of washed indicator cells to
each tube, group A, B, or O for ABH
secretor status, as appropriate, or
Le(a+) for Lewis testing.
5. Mix the contents of the tubes. Incu-
bate the tubes for 30 to 60 minutes
at room temperature.
Methods Section 2: Red Cell Typing 737
Copyright © 2005 by the AABB. All rights reserved.

6. Centrifuge each tube and inspect
each cell button macroscopically for
agglutination.
Interpretation
1. Agglutination of indicator cells by
antibody in tubes containing saliva
indicates that the saliva does not
contain the corresponding antigen.
2. The failure of known antibody to ag-
glutinate indicator cells after incu-
bation with saliva indicates that the
saliva contains the corresponding
antigen.
3. The failure of antibody in the saline
control tube to agglutinate indicator
cells invalidates the results of saliva
tests; this usually reflects use of re-
agents that are too dilute. Redeter-
mine the appropriate reagent dilu-
tion, as described above, and repeat
the testing.
4. For further interpretation, see Table
2.5-1.
Notes
1. Include, as controls, saliva from a
known secretor and nonsecretor. For
ABH status, use saliva from previ-
ously tested Se and sese persons. For
Lewis testing, use saliva from a per-
son whose red cells are Le(a+b–) or
Le(a–b+) as the positive control; use
saliva from a Le(a–b–) person as the
negative control. Aliquots of saliva
from persons of known secretor sta-
tus may be frozen for later use.
2. This screening procedure can be
adapted for the semiquantitation of
blood group activity by testing serial
saline dilutions of saliva. The higher
the dilution needed to remove inhib-
itory activity, the more blood group
substance is present in the saliva.
738 AABB Technical Manual
Table 2.5-1. Interpretation of Saliva Testing
Testing with Anti-H
Unknown
Saliva
Se
Saliva
(H Substance
Present)
Non-
Se
Saliva
(H Substance Not
Present)
Saline
(Dilution Control) Interpretation
2+ 0 2+ 2+ Nonsecretor of H
0 0 2+ 2+ Secretor of H
Testing With Anti-Lea
Unknown
Saliva Le-positive Saliva
Le-negative
Saliva
Saline
(Dilution Control) Interpretation
2+ 0 2+ 2+ Lewis-negative
0 0 2+ 2+ Lewis-positive*
*A Lewis-positive person shown to be a secretor of ABH can be assumed to have Lebas well as Leain saliva. A Le(a+)
person who is
sese
and does not secrete ABH substance will have only Leain saliva.
Copyright © 2005 by the AABB. All rights reserved.
Saliva should be diluted before it is
incubated with antibody. To detect
or to measure salivary A or B sub-
stance in addition to H substance,
thesameprocedurecanbeused
with diluted anti-A and anti-B re-
agents. The appropriate dilution of
anti-A or anti-B is obtained by titrat-
ing the reagent against A1or B red
cells, respectively.
3. A Lewis-positive person shown to be
a secretor of A, B, and H can be as-
sumed to have Lebas well as Leain
the saliva. A Le(a+) person who does
not secrete A, B, or H substances
lacks the Se gene and will have only
Leain the saliva.
4. Specimens with a high concentra-
tion of soluble antigen may give a
false-negative result and require di-
lution before testing.
Method 2.6. Slide Test for
Determination of Rh Type
Principle
See Chapter 14 for a discussion of the
principles of Rh typing.
Specimen
Refer to Method 2.2.
Equipment
View box.
Reagents
1. Reagent anti-D: Suitable reagents in-
clude polyclonal high-protein or low-
protein (eg, monoclonal) reagents.
Follow the instructions from the
manufacturer of the anti-D in use
before performing slide tests; the
method presented here is a repre-
sentative procedure.
2. Rh control reagent: The manufac-
turer’s instructions will indicate the
type of reagent to use, if needed.
Procedure
1. Place 1 drop of anti-D onto a clean,
labeled slide.
2. Place 1 drop of the appropriate con-
trol reagent, if needed, onto a sec-
ond labeled slide.
3. To each slide, add 2 drops of a well-
mixed 40% to 50% suspension (in auto-
logous or group-compatible serum
or plasma) of the red cells to be tested.
4. Thoroughly mix the cell suspension
and reagent. Using a clean applica-
tor stick for each test, spread (mix)
the reaction mixture over an area ap-
proximately20mm×40 mm.
5. Place both slides on the viewbox and
tilt the slides gently and continu-
ously to observe them for agglutina-
tion (see note 1). Most manufactur-
ers stipulate that the test must be
read within 2 minutes because dry-
ing of the reaction mixture may
cause the formation of rouleaux,
which may be mistaken for aggluti-
nation.
6. Interpret and record the results of
the reactions on both slides.
Interpretation
1. Agglutination with anti-D and a
smooth suspension on the control
slide constitute a positive test result
and indicate that the cells being
tested are D+.
2. No agglutination with either anti-D
or the Rh control suggests that the
cells are D–. Testing by the anti-
globulin procedure (see Method 2.9)
will show weak expression of D on
cells that are not agglutinated on
slide testing.
Methods Section 2: Red Cell Typing 739
Copyright © 2005 by the AABB. All rights reserved.
3. If there is agglutination on the con-
trol slide, results of the anti-D test
must not be interpreted as positive
without further testing.
4. Drying around the edges of the reac-
tion mixture must not be confused
with agglutination.
Notes
1. Slide testing imposes a much greater
risk of biohazardous exposure. Per-
sonnel should follow safety mea-
sures detailed in the facility’s proce-
dures manual.
2. For slide tests using low-protein
anti-D, a negative result on slide
testing with either anti-A or anti-B
serves as the control reaction.
Method 2.7. Tube Test for
Determination of Rh Type
Principle
See Chapter 14 for a discussion of the
principles of Rh typing.
Specimen
Refer to Method 2.2.
Reagents
1. Reagent anti-D: Suitable reagents in-
clude polyclonal high-protein or low-
protein (eg, monoclonal) reagents.
Follow the instructions from the
manufacturer of the anti-D in use
before performing tube tests. The
method presented here is a repre-
sentative procedure.
2. Rh control reagent: The manufac-
turer’s instructions will indicate the
type of control to use, if needed.
Procedure
1. Place 1 drop of anti-D in a clean, la-
beled test tube.
2. Place 1 drop of the appropriate con-
trol reagent, if needed, in a second
labeled tube.
3. Add to each tube 1 drop of a 2% to
5% suspension (in saline, serum or
plasma) of the red cells to be tested;
alternatively, the equivalent amount
of red cells can be transferred to each
tube with clean applicator sticks.
4. Mix gently and centrifuge for the
time and at the speed specified by
the manufacturer.
5. Gently resuspend the cell button and
examine it for agglutination. If a
stick was used to transfer the red
cells, adding 1 drop of saline to each
tube will make it easier to resuspend
the cell button.
6. Grade reactions and record test and
control results.
Interpretation
1. Agglutination in the anti-D tube,
combined with a smooth suspension
in the control tube, indicates that
the red cells under investigation are
D+.
2. A smooth suspension of red cells in
both the anti-D and the control
tubes is a negative test result. Speci-
mens from patients may be desig-
nated as D– at this point. Donor
blood must be further tested for the
presence of weak D antigen. The se-
rum-and-cell mixture used in steps 1
through 5, above, may be used to test
for weak D, providing the manufac-
turer’s directions state that the re-
agent is suitable for the test for weak
D.
740 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Notes
1. Most commercially prepared antisera
provide a 2+ or greater agglutination
with D+ cells. A facility may choose
to do additional testing on results
with an agglutination of less than 2+.
Required testing must be defined in
the facility’s procedures manual.
2. A negative tube test with anti-A and/
or anti-B serves as a valid control
when a low-protein anti-D reagent
has been used.
Method 2.8. Microplate Test
for Determination of Rh Type
Principle
See Chapter 14 for a discussion of the
principles of Rh typing
Specimen
RefertoMethod2.2.Clottedoranticoagu-
latedsamplesmaybeusedforRhtesting.
Follow the manufacturer’s instructions for
specimen preparation when using semi-
automated microplate readers.
Reagents
Use only anti-D approved for use in micro-
plate tests (see the discussion in Method
2.3).
Procedure
The following is a representative method;
the manufacturer’s instructions should be
followed for specific reagents and equip-
ment.
1. Place 1 drop of the Rh reagent in a
clean well of the microplate. If the
reagent requires the use of an Rh
control, add 1 drop of the control to
a second well.
2. Add 1 drop of a 2% to 5% saline sus-
pension of red cells to each well.
3. Mix the contents of the wells by
gently tapping the sides of the plate.
4. Centrifuge the plate at the appropri-
ate conditions established for the
centrifuge.
5. Resuspend the cell buttons by man-
ually tapping the plate or with the
aid of a mechanical shaker, or place
the plate at an angle for the tilt and
stream method.
6. Read, interpret, and record the results.
7. Incubate negative tests at 37 C for 15
minutes.
8. Centrifuge the plate at the appropri-
ate conditions established for the
centrifuge.
9. Resuspend the cell buttons by man-
ually tapping the plate or with the
aid of a mechanical shaker, or place
the plate at an angle for the tilt and
stream method.
10. Read, interpret, and record the re-
sults.
Interpretation
Agglutination with anti-D reagent after
the immediate-spin or 37 C incubation
phase indicates a positive test provided
there is no agglutination with the control
reagent. See Table 14-3 for determining
Rh phenotypes from reactions obtained
with Rh blood typing reagents.
Note
Refer to the manufacturer’s instructions
for the necessity for weak D testing.
Method 2.9. Test for Weak D
Principle
Some red cells express the D antigen so
weakly that most anti-D reagents do not
Methods Section 2: Red Cell Typing 741
Copyright © 2005 by the AABB. All rights reserved.
directly agglutinate the cells. Weak D ex-
pression can be recognized most reliably
by an indirect antiglobulin procedure af-
ter incubation of the test red cells with
anti-D.
Specimen
Refer to Method 2.2.
Reagents
1. Reagent anti-D: Suitable reagents in-
clude polyclonal high-protein or low-
protein (eg, monoclonal blend) re-
agents, but the manufacturer’s pack-
age insert should be consulted be-
fore any anti-D reagent is used for
this purpose.
2. Antihuman globulin reagent, either
anti-IgG or polyspecific.
3. IgG-coated red cells.
Procedure
If the original, direct test with anti-D was
performed by tube testing, the same tube
may be used for the weak D test, provid-
ing the manufacturer’s directions so state.
In this case, proceed directly to step 4, af-
ter recording the original anti-D tube test
as negative.
1. Place 1 drop of anti-D in a clean, la-
beled test tube.
2. Place 1 drop of the appropriate con-
trol reagent in a second labeled test
tube.
3. To each tube, add 1 drop of a 2% to
5% suspension in saline of the red
cells to be tested. It is permissible to
use a direct antiglobulin test (DAT)
on the test cells as a control, but an
indirect antiglobulin procedure with
an Rh control reagent is preferable
because this ensures that all reagent
components that might cause a
false-positive result are represented.
4. Mix and incubate both tubes accord-
ing to the reagent manufacturer’s di-
rections. Typically, this is 15 to 30
minutes at 37 C.
5. If a reading is desired after the 37 C
incubation phase, centrifuge the tubes
according to the reagent manufac-
turer’s directions.
6. Gently resuspend the cell buttons
and examine the tubes for agglutina-
tion. If the test red cells are strongly
agglutinated in the anti-D tube but
not in the control tube, record the test
sample as D+ and do not proceed with
the antiglobulin phase of the test.
7. If the test cells are not agglutinated
or the results are doubtful, wash the
cells three or four times with large
volumes of saline.
8. Add antiglobulin reagent, according
to the manufacturer’s directions.
9. Mix gently and centrifuge according
to the calibrated spin times.
10. Gently resuspend each cell button,
examine the tubes for agglutination,
and grade and record the test result.
11. If the test result is negative, add IgG-
coated red cells.
Interpretation
1. Either a diluent control or a direct
antiglobulin test (DAT) must accom-
pany the test for weak D. Agglutina-
tionintheanti-Dtubeandnonein
the control tube constitutes a posi-
tive test result. If the facility chooses
to perform the test for weak D, and
the result is clearly positive, the
blood should be classified as D+. It is
incorrect to report such red cells as
being “D–, weak D” or “D–, Du.”
2. Absence of agglutination in the tube
with anti-D is a negative result, indi-
cating that the cells do not express D
andshouldbeclassifiedasD–.
742 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

3. If there is agglutination at any phase
in the control tube, no valid inter-
pretation of the weak D test can be
made. If the specimen is from a po-
tential transfusion recipient, Rh-
negative blood should be given until
the D type can be resolved. If the
specimen is from a donor, the unit
should not be used for transfusion.
Note
Some facilities may elect to do an addi-
tional reading after the 37 C incubation
and before completing the antiglobulin
phase of testing. Refer to the manufac-
turer’s instructions. If this optional read-
ing is performed, the facility’s procedures
manual should indicate its policy on the
interpretation of this result and on the ad-
ditional testing requirements.
Method 2.10. Preparation
and Use of Lectins
Principle
Saline extracts of seeds react with specific
carbohydrates on red cell membranes and
make useful typing reagents that are
highly specific at appropriate dilutions.
Diluted extract of Dolichos biflorus agglu-
tinates A1red cells but not A2.Ulex
europaeus extract reacts with the H deter-
minant; it agglutinates in a manner pro-
portional to the amount of H present
(O>A2>B>A1>A1B red cells). Other lectins
useful for special purposes include
Arachis hypogaea (anti-T), Glycine max
(anti-T, -Tn), Vicia graminea (anti-N), and
the Salvia lectins (S. horminum,anti-
Tn/Cad; S. sclarea, anti-Tn). To investi-
gate red cell polyagglutination, prepare
and test the cells with Arachis,Glycine,
Salvia,andDolichos lectins. The antici-
pated reactions with various types of
polyagglutinable red cells are shown in
Table 2.10-1. If commercially made lectins
are used, follow the manufacturer’s in-
structions.
Reagents
Seeds may be obtained from health-food
stores, pharmacies, or commercial seed
companies. The seeds should be raw.
Procedure
1. Grind the seeds in a food processor
or blender until the particles look
like coarse sand. A mortar and pestle
may be used, or seeds can be used
whole.
2. In a large test tube or small beaker,
placegroundseedsandthreetofour
times their volume of saline. (Seeds
vary in the quantity of saline they
absorb.)
3. Incubate at room temperature for 4
to 12 hours, stirring or inverting oc-
casionally.
4. Transfer supernatant fluid to a cen-
trifuge tube and centrifuge it for 5
minutes, to obtain clear superna-
Methods Section 2: Red Cell Typing 743
Table 2.10-1. Reactions Between Lectins
and Polyagglutinable Red Cells
TThTkTnCad
Arachis hypogaea*
+++00
Dolichos biflorus
†000++
Glycine max (soja)
+00++
Salvia sclarea
000+0
Salvia horminum
000++
*T and Th cells give weaker reactions with
Arachis
after
protease treatment; Tk reactivity is enhanced after prote-
ase treatment.
†A and AB cells may react due to anti-A reactivity of
Doli-
chos
lectin.
Copyright © 2005 by the AABB. All rights reserved.
tant. Collect and filter the super-
natant fluid and discard seed residue.
5. Test dilutions of the extract to find
dilution for the desired activity. De-
termine the activity of the extract
with the appropriate red cells, as be-
low.
For Dolichos biflorus:
a. Add 1 drop of 2% to 5% saline
suspension of known A1,A
2,
A1B, A2B, B, and O red cells to
appropriately labeled tubes.
b. Add 1 drop of the extract to each
tube.
c. Centrifuge for calibrated time.
d. Inspect for agglutination and
record results.
e. The lectin should agglutinate
A1and A1B cells but not A2,A
2B,
B, or O cells. The native extract
often agglutinates all the cells
tested. To make the product
useful for reagent purposes, add
enough saline to the extract so
that there is 3+ or 4+ agglutina-
tion of A1and A1B cells, but not
of A2,A
2B, B, or O cells.
For Ulex europaeus:
a. Add 1 drop of 2% to 5% saline
suspension of known A1,A
2,
A1B, B, and O cells to appropri-
ately labeled tubes.
b. Add 1 drop of extract to each tube.
c. Centrifuge for the calibrated
time.
d. Inspect for agglutination and
record results.
e. The strength of the agglutina-
tion should be in the order of
O>A2>B>A1>A1B.
f. Dilute extract with saline, if
necessary,toapointthatO
cells show 3+ or 4+ agglutina-
tion, A2and B cells show 1+ to
2+ agglutination, and A1or A1B
cells are not agglutinated.
Notes
1. To facilitate grinding hard seeds, the
seeds can be covered with saline and
soaked for several hours before
grinding. The container used for
soaking should not be tightly closed
becausesomebeansreleasegasdur
-
ing the soaking process, which could
cause the container to explode.
2. The saline extracts may be stored in
the refrigerator for several days; they
may be stored indefinitely if frozen.
3. Tests should include a positive and
negative control.
Method 2.11. Use of
Sulfhydryl Reagents to
Disperse Autoagglutination
Principle
See Chapter 20 for a discussion of auto-
agglutination dispersion.
Specimen
Immunoglobulin-coated red cells to be
evaluated.
Reagents
1. 0.01 M dithiothreitol (DTT): 0.154 g
of DTT dissolved in 100 mL of phos-
phate-buffered saline (PBS) at pH
7.3, or 0.1 M 2-mercaptoethanol
(2-ME), 0.7 mL of stock solution of
14 M 2-ME diluted in 100 mL of PBS
at pH 7.3.
2. PBS at pH 7.3.
Procedure
1. Dilute red cells to a 50% concentra-
tion in PBS.
2. Add an equal quantity of 0.01 M DTT
in PBS, or 0.1 M 2-ME in PBS, to the
cells.
744 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
3. Incubate at 37 C for 10 minutes (2-ME)
or 15 minutes (DTT).
4. Wash cells three times in saline and
resuspend them.
5. Dilute the treated red cells to a 2% to
5% concentration in saline for use in
blood grouping tests. Verify that red
cells do not spontaneously aggluti-
nate before typing or use.
Note
This procedure is normally used only for
ABO forward cell typing, Rh testing, and
the direct antiglobulin test. At this con-
centration of DTT, some antigens, in par-
ticular Jsaand Jsb, may be weakened or de-
stroyed by 0.01M DTT.
Reference
Reid ME. Autoagglutination dispersal utilizing
sulfhydryl compounds. Transfusion 1978;18:353-5.
Method 2.12. Gentle Heat
Elution for Testing Red Cells
with a Positive DAT
Principle
Red cells heavily coated with IgG may
spontaneously agglutinate in high-pro-
tein reagents and will cause false-positive
AHG test results. To perform red cell anti-
gen typing, it may be necessary to dissoci-
ate antibody from the cells by elution
without damaging membrane integrity or
altering antigen expression. The gentle
heat elution procedure employed to pre-
pare immunoglobulin-free red cells dif-
fers from procedures intended to recover
active antibody.
Reagent
Antihuman globulin.
Specimen
Test cells with a positive direct antiglo-
bulin test (DAT) result.
Procedure
1. Place one volume of washed anti-
body-coated red cells and three vol-
umes of normal saline in a test tube
of appropriate size. In another tube,
place the same volumes of saline
and washed red cells positive for the
antigen under test. This will provide
a check that the elution technique
does not destroy the antigen reactiv-
ity.
2. Incubate the contents of both tubes
atapproximately45Cfor10to15
minutes. The tubes should be agi-
tated frequently. The time of incuba-
tion should be roughly proportional
to the degree of antibody coating, as
indicated by strength of antiglobulin
reactivity.
3. Centrifuge the tubes and discard the
supernatant saline.
4. Test the person’s cells for degree of
antibody removal by comparing a
DAT on the treated cells with the
antiglobulin results on untreated red
cells. If the antibody coating is re-
duced but still present, steps 1
through 3 can be repeated; the con-
trol cells should be subjected to a
similar second treatment.
5. Test the treated cells for the desired
antigen.
Notes
1. This procedure may be unnecessary
if IgM monoclonal reagents are
available; these reagents cause di-
rect agglutination and are not usu-
ally affected by bound immunoglob-
ulin.
Methods Section 2: Red Cell Typing 745
Copyright © 2005 by the AABB. All rights reserved.
2. As with untreated patient cells, re-
sults of antigen testing in recently
transfused patients should be inter-
preted with caution because of the
potential presence of donor cells.
Method 2.13. Dissociation of
IgG by Chloroquine for Red
Cell Antigen Testing of Red
Cells with a Positive DAT
Principle
Red cells with a positive direct antiglobulin
test (DAT) cannot be tested accurately
with blood typing reagents that require an
indirect antiglobulin technique. Under
controlled conditions, chloroquine dip-
hosphate dissociates IgG from the red cell
membrane with little or no damage to its
integrity. Use of this procedure permits
complete phenotyping of red cells coated
with warm-reactive autoantibody, includ-
ing tests with reagents solely reactive by
indirect antiglobulin techniques.
Specimen
Red cells with a positive DAT due to IgG
coating.
Reagents
1. Chloroquine diphosphate solution
prepared by dissolving 20 g of chloro-
quine diphosphate in 100 mL of sa-
line. Adjust to pH 5.1 with 1 N NaOH,
and store at 2 to 6 C.
2. Control red cells carrying a single-dose
expression of antigens for which the
test samples are to be phenotyped.
3. Anti-IgG antiglobulin reagent.
Procedure
1. To 0.2 mL of washed IgG-coated cells,
add 0.8 mL of chloroquine diphos-
phate solution. Similarly treat the
control sample.
2. Mix and incubate at room tempera-
ture for 30 minutes.
3. Remove a small aliquot (eg, 1 drop)
of the treated test cells and wash
them four times with saline.
4. Test the washed cells with anti-IgG.
5. If this treatment has rendered the
cells nonreactive with anti-IgG, wash
the total volumes of treated test cells
and control cells three times in sa-
lineandmakea2%to5%suspen
-
sion in saline to use in subsequent
blood typing tests.
6. If the treated red cells react with
anti-IgG after 30 minutes of incuba-
tion with chloroquine diphosphate,
steps 3 and 4 should be repeated at
30-minute intervals (for a maximum
incubation period of 2 hours), until
the sample tested is nonreactive
with anti-IgG. Then proceed as de-
scribed in step 5.
Notes
1. Chloroquine diphosphate does not
dissociate complement proteins
from the cell membrane. If red cells
are coated with both IgG and C3,
only anti-IgG should be used in tests
performed after chloroquine treat-
ment.
2. Incubation with chloroquine diphos-
phate should not be extended be-
yond 2 hours. Prolonged incubation
at room temperature or incubation
at 37 C may cause hemolysis and
loss of red cell antigens.
3. Some denaturation of Rh antigens
may occur.
4. Many serologists run chloroquine-
treated control cells for each antigen
tested. Select control cells that are
positive for the antigen correspond-
746 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
ing to the antisera that will be used
to type the patient’s cells.
5. Chloroquine diphosphate may not
completely remove antibody from
sensitized red cells. DAT results on
red cells from some persons, partic-
ularly those with a strongly positive
initial test, may only be diminished
in strength.
6. In addition to its use for removal of
autoantibodies, this method can be
used for removal of Bg (HLA)-related
antigens from red cells. Appropriate
Bg controls should be used.
7. If a commercial kit is used, manufac-
turer’s instructions should be followed
for testing and controls.
References
1. Edwards JM, Moulds JJ, Judd WJ. Chloro-
quine diphosphate dissociation of antigen-
antibody complexes: A new technique for
phenotyping rbcs with a positive direct
antiglobulin test. Transfusion 1982;22:59-61.
2. Swanson JL, Sastamoinen R. Chloroquine
stripping of the HLA-A,B antigens from red
cells (letter). Transfusion 1985;25:439-40.
Method 2.14. Acid Glycine/
EDTA Method to Remove
Antibodies from Red Cells
Principle
Acid glycine/EDTA can be used to dissoci-
ate antibody molecules from red cell
membranes. This procedure is routinely
used for blood typing tests or adsorption
procedures. All common red cell antigens
can be detected after treatment with acid
glycine/EDTA except antigens of the Kell
system, Bg antigens, and Er antigens.
Thus, cells treated in this manner cannot
be used to determine these phenotypes.
Specimen
Red cells with a positive direct antiglobulin
test (DAT).
Reagents
1. 10% EDTA prepared by dissolving 2 g
of disodium ethylenediamine tetra-
acetic acid (Na2EDTA) in 20 mL of
distilled or deionized water.
2. 0.1 M glycine-HCl buffer (pH 1.5)
prepared by diluting 0.75 g of glycine
to 100 mL with isotonic (unbuffered)
saline. Adjust the pH to 1.5 using
concentrated HCl.
3. 1.0 M TRIS-NaCl prepared by dis-
solving 12.1 g of tris (hydroxymethyl)
aminomethane (TRIS) and 5.25 g of
sodium chloride (NaCl) to 100 mL
with distilled or deionized water.
Procedure
1. Wash the red cells to be treated six
times with isotonic saline.
2. In a test tube, mix together 20 vol-
umes of 0.1 M acid glycine-HCl (pH
1.5) with five volumes of 10% EDTA.
This is the acid glycine/EDTA reagent.
3. Place 10 volumes of washed red cells
in a clean tube.
4. Add 20 volumes of acid glycine/EDTA.
5. Mix the contents of the tube thor-
oughly.
6. Incubate the mixture at room tem-
perature for no more than 2 to 3
minutes.
7. Add one volume of 1.0 M TRIS-NaCl
and mix the contents of the tube.
8. Centrifuge at 900 to 1000 ×gfor 1 to
2 minutes, then aspirate, and dis-
card the supernatant fluid.
9. Wash the red cells four times with
saline.
10. Test the washed cells with anti-IgG;
if nonreactive with anti-IgG, the cells
Methods Section 2: Red Cell Typing 747
Copyright © 2005 by the AABB. All rights reserved.
are ready for use in blood typing or
adsorption procedures. If the DAT is
still positive, one additional treatment
can be performed.
Notes
1. Overincubation of red cells with acid
glycine/EDTA causes irreversible
damage to cell membranes.
2. Include a parallel control reagent,
such as 6% bovine albumin or inert
plasma, when typing treated red cells.
3. Use anti-IgG, not a polyspecific anti-
globulin reagent, in step 10.
4. Many serologists run acid glycine/
EDTA treated control cells for each
antigen tested. Select control cells that
arepositivefortheantigencorre-
sponding to the antisera that will be
used to type the patient’s cells.
5. If a commercial kit is used, manufac-
turer’s instructions should be fol-
lowed for testing and controls.
References
1. Louie JE, Jiang AF, Zaroulis CG. Preparation of
intact antibody-free red cells in autoimmune
hemolytic anemia (abstract). Transfusion
1986;26:550.
2. Champagne K, Spruell P, Chen J, et al. EDTA/
glycine-acid vs. chloroquine diphosphate
treatment for stripping Bg antigens from red
blood cells (abstract). Transfusion 1996;36
(Suppl):21S.
3. Reid ME, Lomas-Francis C. The blood group
antigen factsbook. New York: Academic Press,
2004.
Method 2.15. Separation of
Transfused from Autologous
Red Cells by Simple
Centrifugation
Principle
Newly formed autologous red cells gener-
ally have a lower specific gravity than
transfused red cells and may be separated
from the transfused population by simple
centrifugation. Newly formed autologous
cells concentrate at the top of the column
of red cells when blood is centrifuged in a
microhematocrit tube, providing a simple
method for recovering autologous cells in
a blood sample from recently transfused
patients. Note: Red cells from patients
with hemoglobin S or spherocytic disor-
ders are not effectively separated by this
method (see Method 2.16 for an alterna-
tive procedure).
Specimen
Red cells from whole blood collected into
EDTA.
Materials
1. Microhematocrit centrifuge.
2. Plain (not heparinized) glass or plas-
tic hematocrit tubes.
3. Sealant.
Procedure
1. Wash the red cells three times in sa-
line. For the last wash, centrifuge
them at 900 to 1000 gfor 5 to 15
minutes. Remove as much of the
supernatant fluid as possible with-
out disturbing the buffy coat. Mix
thoroughly.
2. Fill 10 microhematocrit tubes to the
60-mm mark with well-mixed washed
red cells.
3. Seal the ends of the tubes by heat or
with sealant.
4. Centrifuge all tubes in a microhema-
tocrit centrifuge for 15 minutes.
5. Cut the microhematocrit tubes 5
mm below the top of the column of
red cells. This 5-mm segment con-
748 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
tains the least dense, hence youn-
gest, circulating red cells.
6. Place the cut microhematocrit tubes
into larger test tubes (10 or 12 ×75
mm), add saline, and mix well to
flush the red cells from the micro-
hematocrit tubes. Then, either a)
centrifuge them at 1000 ×gfor 1
minute and remove the empty hema-
tocrit tubes or b) transfer the saline-
suspended red cells to a clean test
tube.
7. Wash the separated red cells three
times in saline before resuspending
them to 2% to 5% in saline for test-
ing.
Notes
1. Separation is better if 3 or more days
have elapsed since transfusion than
if the sample has been obtained
shortly after transfusion.
2. The red cells should be mixed con-
tinuously while the microhematocrit
tubes are being filled.
3. Separation techniques are only ef-
fective if the patient is producing
normal or above-normal numbers of
reticulocytes. This method will be
ineffective in patients with inade-
quate reticulocyte production.
4. Some red cell antigens may not be as
strongly expressed on reticulocytes
as on older cells. Particular attention
should be given to determinations of
theE,e,c,Fy
a,Jk
a, and Ge antigens.
References
1. Reid ME, Toy P. Simplified method for recov-
ery of autologous red blood cells from trans-
fused patients. Am J Clin Pathol 1983;79:364-6.
2. Vengelen-Tyler V, Gonzales B. Reticulocyte
rich RBCs will give weak reactions with many
blood typing antisera (abstract). Transfusion
1985;25:476.
Method 2.16. Separation of
Transfused Red Cells from
Autologous Red Cells in
Patients with Hemoglobin S
Disease
Principle
Red cells from patients with sickle cell
disease, either hemoglobin SS or SC, are
resistant to lysis by hypotonic saline, in
contrast to red cells from normal persons
and those with hemoglobin S trait. This
procedure permits isolation of autologous
red cells from patients with hemoglobin
SS or SC disease who have recently been
transfused.
Specimen
Red cells to be evaluated.
Reagents
1. Hypotonicsaline(0.3%w/vNaCl):
NaCl, 3 g; distilled water to 1 L.
2. Normal saline (0.9% w/v NaCl):
NaCl, 9 g; distilled water to 1 L.
Procedure
1. Place 4 or 5 drops of red cells into a
10 or 12 ×75-mm test tube.
2. Wash the cells six times with 0.3%
NaCl, or until the supernatant fluid
no longer contains grossly visible
hemoglobin. For each wash, centri-
fuge at 1000 ×gfor 1 minute.
3. Wash the cells twice with 0.9% NaCl
to restore tonicity. For each wash,
centrifuge at 200 ×gfor 2 minutes to
facilitate removal of residual stroma.
4. Resuspend the remaining intact red
cells to a 2% to 5% concentration for
phenotyping.
Methods Section 2: Red Cell Typing 749
Copyright © 2005 by the AABB. All rights reserved.
Note
Larger volumes, for use in adsorption stud-
ies, can be processed in a 16 ×100-mm
test tube.
Reference
Brown D. A rapid method for harvesting autolo-
gous red cells from patients with hemoglobin S
disease. Transfusion 1988;28:21-3.
750 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Methods Section 3: Antibody Detection/Identification and Compatibility Testing
Methods Section 3
Antibody Detection,
Antibody Identification, and
Serologic Compatibility
Testing
Method 3.1. Immediate-Spin
Compatibility Testing to
Demonstrate ABO
Incompatibility
Principle
See Chapter 18 for a discussion of the prin-
ciples of compatibility testing.
Specimen
Patient’s serum or plasma may be used.
The age of the specimen must comply with
the pretransfusion specimen requirements
in AABB Standards for Blood Banks and
Transfusion Services.1(p38)
Reagents
1. Normal saline.
2. Donor red cells, 2% to 5% suspen-
sion in normal saline or EDTA saline.
Some serologists prefer to suspend
the donor red cells in EDTA saline be-
cause high-titered anti-A or -B can
initiate complement coating, which can
cause steric hindrance of agglutina-
tion.2The use of a patient’s sample
collected in EDTA is an alternative
approach to prevent this phenome-
non.
Procedure
1. Label a tube for each donor red cell
suspension being tested with the pa-
tient’s serum.
2. Add2dropsofthepatient’sserumor
plasma to each tube.
3. Add 1 drop of the suspension of do-
nor red cells to the appropriate test
tube.
4. Mix the contents of the tube(s) and
centrifuge according to the calibra-
tion of the centrifuge.
751
Section 3
Copyright © 2005 by the AABB. All rights reserved.
5. Examine the tube(s) for hemolysis,
gently resuspend the red cell button(s),
and examine for agglutination.
6. Read, interpret, and record test results.
Interpretation
1. Agglutination or hemolysis constitutes
a positive (incompatible) test result.
2. A smooth suspension of red cells af-
ter resuspension of the red cell but-
ton constitutes a negative result and
indicates a compatible immediate-
spin crossmatch.
References
1. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
2. JuddWJ,SteinerEA,O’DonnellDB,Oberman
HA. Discrepancies in ABO typing due to
prozone; how safe is the immediate-spin
crossmatch? Transfusion 1988;28:334-8.
Method 3.2. Indirect
Antiglobulin Test (IAT) for
the Detection of Antibodies
to Red Cell Antigens
Principle
For a discussion of the principles of sa-
line, albumin, low-ionic-strength saline
(LISS), and polyethylene glycol (PEG) in-
direct antiglobulin testing, see Chapters 12,
18, and 19.
Specimen
Serum or plasma may be used. The age of
thespecimenmustcomplywithpretrans
-
fusion specimen requirements in AABB
Standards for Blood Banks and Transfusion
Services.
Reagents
1. Normal saline.
2. Bovine albumin (22% or 30%).
3. LISS made as follows:
a. Add 1.75 g of NaCl and 18 g of
glycine to a 1-liter volumetric flask.
b. Add 20 mL of phosphate buffer
prepared by combining 11.3 mL
of 0.15 M KH2PO4and 8.7 mL of
0.15 M Na2HPO4.
c. Add distilled water to the 1-liter
mark.
d. Adjust the pH to 6.7 ± 0.1 with
NaOH.
e. Add 0.5 g of sodium azide as a
preservative.
Note: LISS may be used as an addi-
tive (Method 3.2.2) or for the suspen-
sion of test red cells (Method 3.2.3).
LISS preparations are also available
commercially.
4. PEG, 20% w/v: To 20 g of 3350 MW
PEG, add phosphate-buffered saline
(PBS) pH 7.3 (see Method 1.7) to 100 mL.
PEG is also available commercially.
5. Antihuman globulin (AHG) reagent.
Polyspecific or anti-IgG may be used
unless otherwise indicated.
6. Commercially available group O an-
tibody detection cells. Pooled group
O antibody detection cells may be
used only for donor testing. Testing
of patients’ samples must be per-
formed with unpooled cells.
7. IgG-coated red cells.
Method 3.2.1. Saline Indirect Antiglobulin
Test
Procedure
1. Add 2 drops of serum or plasma to
properly labeled tubes.
2. Add 1 drop of 2% to 5% saline-sus-
pended reagent group O cells or do-
nor red cells to each tube and mix.
3. Centrifuge and observe for hemoly-
sis and agglutination. Grade and re-
cord the results.
752 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
4. Incubate at 37 C for 30 to 60 minutes.
5. Centrifuge and observe for hemoly-
sis and agglutination. Grade and re-
cord the results.
6. Wash the cells three or four times with
saline and completely decant the fi-
nal wash.
7. Add AHG to the dry cell button ac-
cording to the manufacturer’s direc-
tions. Mix well.
8. Centrifuge and observe for aggluti-
nation. Grade and record the results.
9. Confirm the validity of negative tests
by adding IgG-coated red cells.
Method 3.2.2. Albumin or LISS-Additive
Indirect Antiglobulin Test
Procedure
1. Add 2 drops of serum or plasma to
properly labeled tubes.
2. Add an equivalent volume of 22% or
30% bovine albumin or LISS additive
(unless the manufacturer’s directions
state otherwise.)
3. Add 1 drop of a 2% to 5% saline-sus-
pended reagent or donor red cells to
each tube and mix.
4. For albumin, incubate at 37 C for 15
to 30 minutes. For LISS, incubate for
10 to 15 minutes or follow the manu-
facturer’s directions.
5. Centrifuge and observe for hemoly-
sis and agglutination. Grade and re-
cord the results.
6. Perform the test described in Method
3.2.1, steps 6 through 9.
Method 3.2.3. LISS Indirect Antiglobulin Test
Procedure
1. Wash reagent or donor red cells three
times in normal saline and comple-
tely decant saline.
2. Resuspend the cells to a 2% to 3%
suspension in LISS.
3. Add 2 drops of serum to a properly
labeled tube.
4. Add 2 drops of LISS-suspended red
cells, mix, and incubate at 37 C 10 to
15 minutes or follow the manufac-
turer’s directions.
5. Centrifuge and observe for hemoly-
sis and agglutination by gently re-
suspending the cell button. Grade and
record results.
6. Perform the test described in Method
3.2.1, steps 6 through 9.
Method 3.2.4. PEG Indirect Antiglobulin
Test
Procedure
1. For each cell sample to be tested, mix
2 drops of test serum, 4 drops of 20%
PEG in PBS, and 1 drop of a 2% to
5% suspension of red cells.
2. Incubate at 37 C for 15 minutes.
3. DO NOT CENTRIFUGE.
4. Wash the cells four times with saline
and completely decant the final wash.
5. Perform the AHG test, using anti-IgG,
described in Method 3.2.1, steps 7
through 9.
Note: The manufacturer’s instructions
should be followed for the proper use
of commercial PEG solutions.
Interpretation (for Antiglobulin Tests,
Methods 3.2.1 through 3.2.4)
1. Thepresenceofagglutination/hem
-
olysis after incubation at 37 C con-
stitutes a positive test.
2. Thepresenceofagglutinationafter
addition of AHG constitutes a posi-
tive test.
3. Antiglobulin tests are negative when
no agglutination is observed after
initial centrifugation and the IgG-
coated red cells added afterward are
agglutinated. If the IgG-coated red
cells are not agglutinated, the nega-
Methods Section 3: Antibody Detection/Identification and Compatibility Testing 753
Copyright © 2005 by the AABB. All rights reserved.
tive result is invalid and the test must
be repeated.
Controls
The procedure used for the detection of
unexpected antibodies in pretransfusion
testing should be checked daily with weak
examples of antibody. Control sera can be
prepared from reagent grade typing sera
diluted with 6% bovine albumin to give 2+
reactions by an IAT. Human sources of IgG
antibodies are also acceptable.
Notes
1. The incubation times and the volume
and concentration of red cells indi-
cated are those given in the literature.
Individual laboratories may choose to
standardize techniques with somewhat
different values. See Chapter 12 for
other limitations when modifying
procedures. In all cases, the manu-
facturer’s package insert should be
consulted before modifying a proce-
dure.
2. For the saline procedure, step 3 may
be omitted to avoid the detection of
antibodies reactive at room temper-
ature.
3. For the PEG procedure:
a. Omit centrifugation after 37 C
incubation because red cells will
not resuspend readily.
b. Use anti-IgG rather than poly-
specific AHG to avoid unwanted
positive reactions due to C3-
binding autoantibodies.
4. Steps 6 through 9 of the IAT (Method
3.2.1) must be performed without in-
terruption.
Reference
Silva MA, ed. Standards for blood banks and trans-
fusion services. 23rd ed. Bethesda, MD: AABB, 2005:
38.
Method 3.3. Prewarming
Technique
Principle
Prewarming may be useful in the detec-
tion and identification of red cell antibod-
ies that bind to antigen only at 37 C. This
test is particularly useful for testing sera
of patients with cold-reactive autoantibody
activity that may mask the presence of
clinically significant antibodies. However,
use of the prewarming technique for this
application has become controversial.1-2 It
has been shown to result in decreased
reactivity of some potentially significant
antibodies and weak antibodies can be
missed.3The technique should be used
with caution and not used to eliminate
unidentified reactivity.
Strong cold-reactive autoantibodies may
react in prewarmed tests; other techniques
such as cold allo- or autoadsorption or
dithiothreitol treatment of plasma may be
required to detect underlying clinically sig-
nificant antibodies.
Specimen
Serum or plasma may be used. The age of
thespecimenmustcomplywithpretrans
-
fusion specimen requirements in AABB
Standards for Blood Banks and Transfusion
Services.4(p38)
Reagents
1. Normal saline.
2. Anti-IgG.
3. Commercially available group O an-
tibody detection cells. Pooled group
O antibody detection cells may be used
only for donor testing. Testing of pa-
tients’ samples must be done with
unpooled cells.
4. IgG-coated red cells.
754 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Procedure
1. Prewarmabottleofsalineto37C.
2. Label one tube for each reagent or
donorsampletobetested.
3. Add 1 drop of 2% to 5% saline-sus-
pended red cells to each tube.
4. Place the tubes containing red cells
and a tube containing a small volume
of the patient’s serum and a pipette
at 37 C; incubate for 5 to 10 minutes.
5. Using the prewarmed pipette, trans-
fer 2 drops of prewarmed serum to
each tube containing prewarmed red
cells. Mix without removing tubes
from the incubator.
6. Incubate at 37 C for 30 to 60 min-
utes.
7. Without removing the tubes from the
incubator, fill each tube with pre-
warmed (37 C) saline. Centrifuge
and wash three or four times with 37
C saline.
8. Add anti-IgG, according to the man-
ufacturer’s directions.
9. Centrifuge and observe for reaction.
Grade and record the results.
10. Confirm the validity of negative tests
by adding IgG-coated red cells.
Notes
1. The prewarming procedure described
above will not detect alloantibodies
that agglutinate at 37 C or lower and
are not reactive in the antiglobulin
phase. If detection of these antibod-
ies is desired, testing and centrifuga-
tion at 37 C are required. If time per-
mits, a tube containing a prewarmed
mixture of serum and cells can be
incubatedat37Cfor60to120min
-
utes, and the settled red cells exam-
ined for agglutination by resuspend-
ing the button without centrifugation.
2. Cold-reactive antibodies may not be
detectable when room-temperature
saline instead of 37 C saline is used
in the wash step.2The use of room-
temperature saline may avoid the
elution of clinically significant anti-
body(ies) from reagent red cells that
can occur with the use of 37 C saline.
Some strong cold-reactive autoanti-
bodies, however, may still react and
therefore require the use of 37 C sa-
line to avoid their detection.
References
1. Judd WJ. Controversies in transfusion medi-
cine. Prewarmed tests: Con. Transfusion 1995;
35:271-7.
2. Mallory D. Controversies in transfusion med-
icine. Prewarmed tests: Pro—why, when, and
how—not if. Transfusion 1995;35:268-70.
3. Leger RM, Garratty G. Weakening or loss of
antibody reactivity after prewarm technique.
Transfusion 2003;43:1611-14.
4. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
Method 3.4. Saline
Replacement to Demonstrate
Alloantibody in the Presence
of Rouleaux
Principle
Rouleaux are aggregates of red cells that,
characteristically, adhere to one another
on their flat surface, giving a “stack of coins”
appearance when viewed microscopically.
Rouleaux formation is an in-vitro phe-
nomenon resulting from abnormalities of
serum protein concentrations. The pa-
tient is often found to have liver disease,
multiple myeloma, or another condition
associated with abnormal globulin levels.
It may be difficult to detect antibody-as-
sociated agglutination in a test system
containing rouleaux-promoting serum. In
the saline replacement technique, serum
and cells are incubated to allow antibody
Methods Section 3: Antibody Detection/Identification and Compatibility Testing 755
Copyright © 2005 by the AABB. All rights reserved.
attachment, but the serum is removed
and saline is added as the resuspending
medium.
Reagents
Saline.
Procedure
After routine incubation and resuspension,
proceed with the following steps if the ap-
pearance of the resuspended cells suggests
rouleaux formation:
1. Recentrifuge the serum/cell mixture.
2. Remove the serum, leaving the cell
button undisturbed.
3. Replacetheserumwithanequal
volume of saline (2 drops).
4. Resuspend the cell button gently and
observe for agglutination. Rouleaux
will disperse when suspended in saline,
whereas true agglutination will remain.
Reference
Issitt PD, Anstee DJ. Applied blood group serology.
4th ed. Durham, NC: Montgomery Scientific Pub-
lications, 1998:1135.
Method 3.5. Enzyme
Techniques
For a discussion of the principles of en-
zyme testing, see Chapter 19.
Method 3.5.1. Preparation of Ficin
Enzyme Stock, 1% w/v
Principle
The enzyme preparations used in blood
banking differ from lot to lot; each time a
stock enzyme solution is prepared, its re-
activity should be tested and incubation
periods standardized for optimal effective-
ness. See Method 3.5.3.
Reagents
1. Dryenzymepowder,1g.
2. Phosphate-buffered saline (PBS), pH
7.3: see Method 1.7.
3. Phosphate buffer, pH 5.4.
Procedure
1. Place 1 g of powdered ficin in a 100-
mL volumetric flask. Handle the
ficin carefully; it is harmful if it gets
intheeyesorisinhaled.Itisdesir
-
able to wear gloves, mask, and apron,
or to work under a hood.
2. AddPBS,pH7.3to100mL,todis
-
solve the ficin. Agitate vigorously by
inversion, rotate for 15 minutes, or
mix with a magnetic stirrer until
mostly dissolved. The powder will not
dissolve completely.
3. Collect clear fluid, either by filtration
or centrifugation, and prepare small
aliquots. Store the aliquots at –20 C
or colder. Do not refreeze a thawed
solution.
Method 3.5.2. Preparation of Papain
Enzyme Stock, 1% w/v
Principle
The enzyme preparations used in blood
banking differ from lot to lot; each time a
stock enzyme solution is prepared, its re-
activity should be tested and incubation
periods standardized for optimal effective-
ness. See Method 3.5.3.
Reagents
1. L-cysteine hydrochloride 0.5 M, 0.88
g in 10 mL distilled water.
2. Dryenzymepowder,2g.
3. Phosphate buffer 0.067 M at pH 5.4,
prepared by combining 3.5 mL of
Na2HPO4and 96.5 mL of KH2PO4.
756 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Procedure
1. Add 2 g of powdered papain to 100
mL of phosphate buffer (pH 5.4).
Handle papain carefully; it is harm-
ful to mucous membranes. Use ap-
propriate protective equipment.
2. Agitate enzyme solution for 15 min-
utes at room temperature.
3. Collect clear fluid by filtration or
centrifugation.
4. Add L-cysteine hydrochloride and
incubate solution at 37 C for 1 hour.
5. Add phosphate buffer (pH 5.4) to fi-
nal volume of 200 mL. Store aliquots
at –20 C or colder. Do not refreeze
aliquots.
Method 3.5.3. Standardization of Enzyme
Procedures
Principle
Foratwo-stageenzymeprocedure,the
optimal treatment time must be deter-
mined for each new lot of stock solution.
The technique given below for ficin can
be modified for use with other enzymes.
Reagents
1. 1% stock solution of ficin in PBS, pH
7.3.
2. Several sera known to lack unex-
pected antibodies.
3. Anti-D that agglutinates only en-
zyme-treated D+ red cells and does
not agglutinate untreated D+ cells.
4. Anti-Fyaof moderate or strong reac-
tivity.
5. D+ and Fy(a+b–) red cell samples.
6. Antihuman globulin (AHG) reagent.
Polyspecific or anti-IgG may be used
unless otherwise indicated.
7. IgG-coated red cells.
Procedure
1. Prepare 0.1% ficin by diluting one
volume of stock ficin solution with
nine volumes of PBS, pH 7.3.
2. Label three tubes: 5 minutes, 10
minutes, and 15 minutes.
3. Add equal volumes of washed red cells
and 0.1% ficin to each tube.
4. Mix and incubate at 37 C for the time
designated. Incubation times are
easily controlled if the 15-minute
tube is prepared first, followed by
the 10- and 5-minute tubes at
5-minute intervals. Incubation will
be complete for all three tubes at the
same time.
5. Immediately wash the red cells three
times with large volumes of saline.
6. Resuspend treated cells to 2% to 5%
in saline.
7. Label four tubes for each serum to
be tested: untreated, 5 minutes, 10
minutes, 15 minutes.
8. Add 2 drops of the appropriate se-
rum to each of the four tubes.
9. Add 1 drop of the appropriate red cell
suspension to each of the labeled
tubes.
10. Mix and incubate at 37 C for 15 min-
utes.
11. Centrifuge and examine for aggluti-
nation by gently resuspending the red
cell button.
12. Proceed with the AHG test described
in Method 3.2.1, steps 6 through 9.
Interpretation
Table 3.5.3-1 shows possible results with
D+, Fy(a+b–) cells and the sera indicated.
In this case, the optimal incubation time
would be 10 minutes. Incubation for only
5 minutes does not completely abolish Fya
activity or maximally enhance anti-D re-
activity. Incubation for 15 minutes causes
Methods Section 3: Antibody Detection/Identification and Compatibility Testing 757
Copyright © 2005 by the AABB. All rights reserved.

false-positive antiglobulin reactivity with
inert serum.
If incubation for 5 minutes overtreats the
cells,itispreferabletouseamoredilute
working solution of enzyme than to reduce
incubation time because it is difficult to ac-
curately monitor very short incubation
times. Additional tests can evaluate a single
dilution at different incubation times, or a
single incubation time can be used for
different enzyme dilutions.
Method 3.5.4. Evaluating Enzyme-Treated
Red Cells
Principle
After optimal incubation conditions have
been determined for a lot of enzyme solu-
tion, treated red cells should be evaluated
before use to demonstrate that they are
adequately, but not excessively, modified.
Satisfactory treatment produces cells that
are agglutinated by an antibody that causes
only indirect antiglobulin test reactivity of
unmodified cells but are not agglutinated
or aggregated by inert serum.
Specimen
Enzyme-treated red cells.
Reagents
1. Sera known to contain antibody that
will agglutinate enzyme-treated cells.
2. Sera free of any unexpected antibod-
ies.
3. Antihuman globulin (AHG) reagent.
Polyspecific or anti-IgG may be used
unless otherwise indicated.
4. IgG-coated red cells.
Procedure
1. Select an antibody that agglutinates
enzyme-treated red cells positive for
the antigen but gives only AHG reac-
tions with unmodified cells. Many
examples of human source anti-D
behave in this way.
2. Add 2 drops of the selected antibody-
containing serum to a tube labeled
“positive.”
3. Add2dropsofaserumfreeofunex
-
pected antibodies to a tube labeled
“negative.”
4. Add 1 drop of 2% to 5% suspension
of enzyme-treated red cells to each
tube.
5. Mix and incubate 15 minutes at 37 C.
6. Centrifuge and resuspend the cells by
gentle shaking.
758 AABB Technical Manual
Table 3.5.3-1. Hypothetical Results with D+, Fy(a+b–) Red Cells
Cells and Enzyme Inert Serum Anti-D Anti-Fya
Untreated 37 C incubation 0 0 0
antihuman globulin test 0 1+ 3+
5 minutes 37 C incubation 0 1+ 0
antihuman globulin test 0 2+ 1+
10 minutes 37 C incubation 0 2+ 0
antihuman globulin test 0 2+ 0
15 minutes 37 C incubation 0 2+ 0
antihuman globulin test w+ 2+ w+
Copyright © 2005 by the AABB. All rights reserved.
7. Examine macroscopically for the pre-
sence of agglutination.
8. Perform the AHG test described in
Method 3.2.1, steps 6 through 9, on
the tube labeled “negative.”
Interpretation
Thereshouldbeagglutinationinthe“pos
-
itive” tube and no agglutination in the
“negative” tube. If agglutination occurs in
the “negative” tube, the cells have been
overtreated; if agglutination does not oc-
cur in the “positive” tube, treatment has
been inadequate.
Method 3.5.5. One-Stage Enzyme
Technique
Specimen
Serum or plasma to be tested.
Reagent
1. Reagent red cells.
2. Antihuman globulin (AHG) reagent.
Polyspecific or anti-IgG may be used
unless otherwise indicated.
3. IgG-coated red cells.
Procedure
1. Add 2 drops of serum to an appro-
priately labeled tube.
2. Add 2 drops of a 2% to 5% saline sus-
pension of reagent red cells.
3. Add 2 drops of 0.1% papain solution
and mix well.
4. Incubate at 37 C for 15 minutes.
5. Centrifuge; gently resuspend the cells
and observe for agglutination. Grade
and record the results.
6. Proceed with the AHG test described
in Method 3.2.1, steps 6 through 9.
Method 3.5.6. Two-Stage Enzyme
Technique
Specimen
Serum or plasma to be tested.
Reagent
1. Reagent red cells.
2. Antihuman globulin (AHG) reagent.
Polyspecific or anti-IgG may be used
unless otherwise indicated.
3. IgG-coated red cells.
Procedure
1. Prepare a diluted enzyme solution
(papain or ficin) by adding 9 mL of
PBS, pH 7.3, to 1 mL of stock enzyme.
2. Add one volume of diluted enzyme
to one volume of packed, washed re-
agent red cells.
3. Incubate at 37 C for the time deter-
mined to be optimal for that enzyme
solution.
4. Wash treated cells at least three times
with large volumes of saline and re-
suspend the cells to a 2% to 5% con-
centration in saline.
5. Add 2 drops of serum or plasma to
be tested to an appropriately labeled
tube.
6. Add 1 drop of 2% to 5% suspension
of enzyme-treated cells.
7. Mix and incubate for 15 minutes at
37 C.
8. Centrifuge; gently resuspend the cells
and observe for agglutination. Grade
and record the results.
9. Proceed with the AHG test described
in Method 3.2.1, steps 6 through 9.
Notes
1. An alternative method for steps 4 and
5 (Method 3.5.5) or steps 7 and 8
(Method 3.5.6) is to incubate the se-
rum and enzyme-treated cells at 37
Methods Section 3: Antibody Detection/Identification and Compatibility Testing 759
Copyright © 2005 by the AABB. All rights reserved.
C for 60 minutes and examine the
settled cells for agglutination with-
out centrifugation. This can be use-
ful for serum with strong cold-reac-
tive agglutinins and can sometimes
prevent the occurrence of false-posi-
tive results.
2. Microscopic examination is not rec-
ommended for routine use and is
particularly inappropriate with en-
zyme enhanced tests; false-positive
reactions will often be detected.
3. Either papain or ficin may be used in
a two-stage procedure.
4. Enzyme preparations are available
commercially. The manufacturer’s
directions should be followed for ap-
propriate use and quality control.
References
1. Issitt PD, Anstee DJ. Applied blood group se-
rology,4thed.Durham,NC:Montgomery
Scientific, 1998.
2. Judd WJ. Methods in immunohematology. 2nd
ed. Durham, NC: Montgomery Scientific, 1994.
Method 3.6. Direct
Antiglobulin Test (DAT)
Principle
See Chapter 20 for a discussion of the
principles of direct antiglobulin testing.
Specimen
Red cells from an anticoagulated blood
sample.
Reagents
1. Antihuman globulin (AHG) reagent:
polyspecific antiglobulin reagent,
anti-IgG, anti-complement antisera.
2. A control reagent (eg, PBS) is re-
quired when all antisera tested give
a positive result.
3. IgG-coated red cells.
Procedure
1. Dispense 1 drop of a 2% to 5% sus-
pension of red cells into each tube.
2. Wash each tube three or four times
with saline. Completely decant the fi-
nal wash.
3. Immediately add antisera and mix. For
the amount of antisera required, re-
fer to the manufacturer’s directions.
4. Centrifuge according to the manu-
facturer’s directions.
5. Examine the cells for agglutination.
Grade and record the reaction.
6. IfusingpolyspecificAHGoranti-
C3d, incubate nonreactive tests at
room temperature for 5 minutes, then
centrifuge, and read again.
7. Confirm the validity of negative tests
by adding IgG-coated red cells to tests
containing anti-IgG.
8. Centrifuge according to the manu-
facturer’s directions.
9. Examine the cells for agglutination and
record the reaction.
Interpretation
1. The DAT is positive when agglutina-
tion is observed either after im-
mediate centrifugation or after the
centrifugation that followed room-
temperature incubation. IgG-coated
red cells usually give immediate re-
actions, whereas complement coat-
ing may be more easily demonstra-
ble after incubation.1,2 Monospecific
AHG reagents are needed to confirm
which globulins are present.
2. The DAT is negative when no agglu-
tination is observed at either test phase
and the IgG-coated cells added in step
7 are agglutinated. If the IgG-coated
cells are not agglutinated, the nega-
tive DAT result is considered invalid
and the test must be repeated. A
negative DAT does not necessarily
760 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
mean that the red cells have no at-
tached globulin molecules. Poly-
specific and anti-IgG reagents detect
as few as 200 to 500 molecules of IgG
per cell,1but patients may experi-
ence autoimmune hemolytic ane-
mia when IgG coating is below this
level.2
3. No interpretation can be made if the
results with all antisera used to per-
form a DAT and the control are reac-
tive. This indicates spontaneous ag-
glutination, which must be resolved
before further testing is performed.
Notes
1. Steps 2 through 7 must be performed
without interruption.
2. Initial testing may be performed with
polyspecific reagent only. If the DAT
is negative with polyspecific reagent,
no further testing is necessary. If the
DAT is positive with polyspecific re-
agent, perform the DAT test with
monospecific reagents, anti-IgG, and
anticomplement, to determine which
globulins are present.
3. Verification of negative results with
anti-C3d is recommended. Refer to
the manufacturer’s instructions to
determine appropriate controls.
References
1. Mollison PL, Engelfriet CP, Contreras M, eds.
Blood transfusion in clinical medicine. 10th
ed. Oxford, England: Blackwell Scientific Pub-
lications, 1997.
2. Petz LD, Garratty G. Immune hemolytic ane-
mia. Philadelphia: Churchill-Livingstone, 2004.
Method 3.7. Antibody Titration
Principle
Titration is a semiquantitative method used
to determine the concentration of antibody
in a serum sample or to compare the
strength of antigen expression on differ-
ent red cell samples. The usual applica-
tions of titration studies are: 1) estimating
antibody activity in alloimmunized preg-
nant women to determine whether and
when to perform more complex invasive
investigation of the fetal condition (see
Chapter 23); 2) elucidating autoantibody
specificity (see Chapter 20); 3) character-
izing antibodies as high-titer, low-avidity,
traits common in antibodies to antigens
of the Knops and Chido/ Rodgers sys-
tems, Csa,andJMH(seeChapter15);and
4) observing the effect of sulfhydryl re-
agents on antibody behavior, to deter-
mine immunoglobulin class (IgG or IgM).
See Method 5.3 for titration studies spe-
cifically to assist in monitoring clinically
significant antibodies in the pregnant
woman.
Specimen
Serum or plasma antibody to be titrated.
Reagents
1. Red cells that express the antigen(s)
corresponding to the antibody spec-
ificity (ies), in a 2% to 5% saline sus-
pension. Uniformity of cell suspen-
sionsisveryimportanttoensure
comparability of results.
2. Saline.(Note:Dilutionsmaybemade
with albumin if desired.)
Procedure
The master dilution technique for titration
studiesisasfollows:
1. Label 10 test tubes according to the
serum dilution (eg, 1 in 1, 1 in 2, etc).
A 1 in 1 dilution means one volume
of serum undiluted; a 1 in 2 dilution
means one volume of serum in a fi-
Methods Section 3: Antibody Detection/Identification and Compatibility Testing 761
Copyright © 2005 by the AABB. All rights reserved.
nal volume of two, or a 50% solution
of serum in the diluent. See Methods
1.4 and 1.5.
2. Deliver one volume of saline to all test
tubes except the first (undiluted 1 in
1) tube.
3. Add an equal volume of serum to
each of the first two tubes (undiluted
and 1 in 2).
4. Using a clean pipette, mix the con-
tents of the 1 in 2 dilution several
times and transfer one volume into
the next tube (the 1 in 4 dilution).
5. Continue the same process for all di-
lutions, using a clean pipette to mix
and transfer each dilution. Remove
one volume of diluted serum from
the final tube and save it for use if
further dilutions are required.
6. Label 10 tubes for the appropriate
dilutions.
7. Using separate pipettes for each di-
lution, transfer 2 drops of each di-
luted serum into the appropriately
labeled tubes and add 2 drops of a
2% red cell suspension. Alterna-
tively, for convenience, add 1 drop of
a 3%-4% suspension of red cells as
supplied by the reagent manufac-
turer, although this method is less
precise.
8. Mix well and test by a serologic tech-
nique appropriate to the antibody
(see Chapter 19).
9. Examine test results macroscopically;
grade and record the reactions. The
prozone phenomenon (see Chapter
12) may cause reactions to be weaker
in the more concentrated serum pre-
parations than in higher dilutions.
To avoid misinterpretation of results,
itmaybepreferabletoexaminefirst
the tube containing the most dilute
serum and proceed through the more
concentrated samples to the undiluted
specimen.
Interpretation
1. Observe the highest dilution that
produces 1+ macroscopic agglutina-
tion. The titer is reported as the re-
ciprocal of the dilution level, eg,
32—not 1in32or1:32(seeTable
3.7-1). If there is agglutination in the
tube containing the most dilute se-
rum, the endpoint has not been
reached, and additional dilutions
should be prepared and tested.
2. In comparative studies, a significant
difference in titer is three or more di-
lutions. Variations in technique and
inherent biologic variability can cause
duplicate tests to give results that
differ by one dilution in either direc-
tion. Serum containing antibody at a
true titer of 32 may show, on repli-
cate tests, the endpoint in the 1:32
tube, the 1:64 tube, or the 1:16 tube.
3. Titer values alone can be misleading
without also evaluating the strength
of agglutination. The observed strength
of agglutination can be assigned a
number and the sum of these num-
bers for all tubes in a titration study
represents the score, another semi-
quantitative measurement of antibody
reactivity. The arbitrarily assigned
threshold for significance in com-
paring scores is a difference of 10 or
more between different test samples.
See Table 3.7-1.
4. Antibodies with high-titer, low-avid-
ity characteristics generally have a ti-
ter greater than 64, with most tubes
showing consistently weak reactivity.
Table 3.7-1 shows the results obtained
with three sera, each of which shows no
more agglutination after 1:256. The differ-
ences in score, however, indicate consider-
able variation in strength of reactivity.
762 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Notes
Titration is a semiquantitative technique.
Technical variables greatly affect the re-
sults and care should be taken to achieve
the most uniform possible practices.
1. Careful pipetting is essential. Pi-
pettes with disposable tips that can
be changed after each dilution are
recommended.
2. Optimal time and temperature of in-
cubation and time and force of centri-
fugation must be used consistently.
3. The age, phenotype, and concentra-
tion of the test red cells will influence
the results. When the titers of several
antibody-containing sera are to be
compared, all of them should be
tested against red cells (preferably
freshly collected) from the same do-
nor. If this is not possible, the tests
should use a pool of reagent red cells
from donors of the same phenotype.
When a single serum is to be tested
against different red cell samples, all
samples should be collected and pre-
servedinthesamemanneranddi
-
luted to the same concentration be-
fore use.
4. Completely reproducible results are
virtually impossible to achieve. Com-
parisons are valid only when speci-
mens are tested concurrently. In tests
with a single serum against different
red cell samples, material from the
master dilution must be used for all
the tests.
5. Measurements are more accurate with
large volumes than with small vol-
umes; a master dilution technique
(see above) gives more reliable re-
sults than individual dilutions for a
single set of tests. The volume needed
for all planned tests should be calcu-
Methods Section 3: Antibody Detection/Identification and Compatibility Testing 763
Table 3.7-1. Examples of Antibody Titers, Endpoints, and Scores
Reciprocal of Serum Dilution
1248163264128256512 Titer* Score
Strength: 3+ 3+ 3+ 2+ 2+ 2+ 1+ ± ± 0 64(256)
Sample 1
Score: 10101088853 20 64
Strength: 4+ 4+ 4+ 3+ 3+ 2+ 2+ 1+ ± 0 128(256)
Sample 2
Score: 12 12 12 10 10 8 8 5 3 0 80
Strength: 1+1+1+1+±±±±±0 8(256)
Sample 3
Score: 55553332 20 33
*The titer is often determined from the highest dilution of serum that gives a reaction ≥1+ (score 5). This may differ sig-
nificantly from the titration endpoint (shown in parentheses), as with the reactions of an antibody with high-titer,
low-avidity characteristics, manifested by Sample 3.
Copyright © 2005 by the AABB. All rights reserved.

lated and an adequate quantity of
each dilution prepared.
6. When performing a titration for
anti-D for HDFN, see Method 5.3.
Method 3.8. Use of Sulfhydryl
Reagents to Distinguish IgM
from IgG Antibodies
Principle
Treating IgM antibodies with sulfhydryl
reagents abolishes both agglutinating and
complement-binding activities. Observa-
tions of antibody activity before and after
sulfhydryl treatment are useful in deter-
mining immunoglobulin class. Sulfhydryl
treatment can also be used to abolish IgM
antibody activity to permit detection of co-
existing IgG antibodies. For a discussion of
IgM and IgG structures, see Chapter 11.
Specimen
2 mL of serum or plasma to be treated.
Reagents
1. Phosphate-buffered saline (PBS) at
pH 7.3.
2. 0.01 M dithiothreitol (DTT), prepared
by dissolving 0.154 g of DTT in 100
mL of pH 7.3 PBS. Store at –18 C or
lower.
Procedure
1. Dispense 1 mL of serum or plasma
into each of two test tubes.
2. To one tube, labeled dilution control,
add 1 mL of pH 7.3 PBS.
3. To the other tube, labeled test, add 1
mL of 0.01 M DTT.
4. Mix and incubate at 37 C for 30 to 60
minutes.
5. Use the DTT-treated and dilution con-
trol samples in standard procedures.
Interpretation
1. Reactivity in the dilution control
serum and no reactivity in the DTT-
treated serum indicates an IgM anti-
body.
2. Reactivity in the dilution control se-
rum and the DTT-treated serum in-
dicates an IgG antibody or an IgG
and IgM mixture. Titration studies
may be necessary to distinguish be-
tween them. See Table 3.8-1.
764 AABB Technical Manual
Table 3.8-1. Effect of Dithiothreitol on Blood Group Antibodies
Dilution
Test Sample 1/2 1/4 1/8 1/16 1/32 Interpretation
Serum+DTT 3+2+2+1+ 0 IgG
Serum + PBS 3+ 2+ 2+ 1+ 0
Serum+DTT 0000 0 IgM
Serum + PBS 3+ 2+ 2+ 1+ 0
Serum+DTT 2+1+0 0 0 IgG + IgM*
Serum + PBS 3+ 2+ 2+ 1+ 0
*May also indicate only partial inactivation of IgM.
Copyright © 2005 by the AABB. All rights reserved.
3. No reactivity in the dilution control
serum indicates dilution of weak an-
tibody reactivity and an invalid test.
Control
A serum or plasma sample known to con-
tain an IgM antibody should be treated and
tested in parallel.
Notes
1. 2-mercaptoethanol can also be used
for this purpose. See Method 2.11 for
preparation.
2. Sulfhydryl reagents used at low con-
centration may weaken antigens of
the Kell system. For investigation of
antibodies in the Kell system, it may
be necessary to use other methods.
3. Gelling of a serum or plasma sample
may be observed during treatment with
DTT. This can occur if the DTT has
been prepared incorrectly and has a
concentration above 0.01 M. Gelling
may also occur if serum and DTT are
incubated too long. An aliquot of the
sample undergoing treatment can be
tested after 30 minutes of incubation;
if the activity thought to be due to IgM
has disappeared, there is no need to
incubate further. Gelled samples can-
not be tested for antibody activity be-
cause overtreatment with DTT causes
the denaturation of all serum proteins.
Reference
Mollison PL, Engelfriet CP, Contreras M, eds. Blood
transfusion in clinical medicine. 10th ed. Oxford,
England: Blackwell Scientific Publications, 1997.
Method 3.9. Plasma Inhibition
to Distinguish Anti-Ch and
-Rg from Other Antibodies
with HTLA Characteristics
Principle
For a discussion of the principles of plasma
inhibition of anti-Ch and -Rg, see Chapter 19.
Specimen
Serum or plasma to be tested.
Reagents
1. Reactive red cell samples.
2. A pool of six or more normal plasma
samples.
3. 6% bovine albumin, see Method 1.5.
4. Anti-IgG.
5. IgG-coated red cells.
Procedure
1. Prepare serial twofold dilutions of test
serum in saline. The dilution range
should be from 1 in 2 to 1 in 512, or
toonetubebeyondtheknowntiter
as determined above (Method 3.7). The
volume prepared should be not less
than 0.3 mL for each red cell sample
to be tested.
2. For each red cell sample to be tested,
place 2 drops of each serum dilution
into each of two sets of appropriately
labeled 10 or 12 ×75-mm test tubes.
3. To one set, add 2 drops of pooled
plasma to each tube.
4. To the other set, add 2 drops of 6%
albumin to each tube.
5. Gently agitate the contents of each
tube and incubate the tubes at room
temperature for at least 30 minutes.
6. Add1dropofa2%to5%suspension
of red cells to each tube.
Methods Section 3: Antibody Detection/Identification and Compatibility Testing 765
Copyright © 2005 by the AABB. All rights reserved.
7. Gently agitate the contents of each
tube and incubate the tubes at 37 C
for 1 hour.
8. Wash the cells four times in saline,
add anti-IgG, and centrifuge accord-
ing to the manufacturer’s directions.
9. Resuspend the cell buttons and ex-
amine for agglutination; confirm all
nonreactive tests microscopically.
Grade and record the results.
10. Confirm the validity of negative tests
by adding IgG-coated red cells.
Interpretation
1. Inhibition of antibody activity in the
tubes to which plasma has been added
suggests anti-Ch or anti-Rg specific-
ity; this inhibition is often complete.
2. Thepresenceofpartialinhibition
suggests the possibility of additional
alloantibodies. This can be tested by
preparingalargevolumeofinhibited
serum and testing it against a re-
agent red cell panel to see if the non-
neutralizable activity displays anti-
genic specificity.
3. Lack of reactivity in the control (6%
albumin) indicates dilution of weakly
reactive antibody and an invalid test.
Notes
1. Antibodies to other plasma antigens
may also be partially inhibited by
plasma.1
2. Adsorption with C4-coated red cells
is an alternative procedure that may
be used for identifying anti-Ch or
anti-Rg and for detecting underlying
alloantibodies.2
References
1. Reid ME, Lomas-Francis C. The blood group
antigenfactsbook.2nded.NewYork:Aca
-
demic Press, 2004.
2. Ellisor SS, Shoemaker MM, Reid ME. Adsorp-
tion of anti-Chido from serum using autolo-
gous red blood cells coated with homologous
C4. Transfusion 1982;22:243-5.
Method 3.10. Dithiothreitol
(DTT) Treatment of Red Cells
Principle
DTT is an efficient reducing agent that
can disrupt the tertiary structure of pro-
teins by irreversibly reducing disulfide
bonds to free sulfhydryl groups. Without
tertiary structure, protein-containing an-
tigens can no longer bind antibodies that
are specific for them. Red cells treated
with DTT will not react with antibodies in
the Kell blood group system, most anti-
bodies in the Knops system, or most ex-
amples of anti-LWa,-Yt
a,-Yt
b,-Do
a,-Do
b,
-Gya,-Hy,and-Jo
a. This inhibition tech-
nique may be helpful in identifying some
of these antibodies or in determining if a
serum contains additional underlying allo-
antibodies.
Specimen
Red cells to be tested.
Reagents
1. Prepare 0.2 M DTT by dissolving 1 g
of DTT powder in 32 mL of phos-
phate-buffered saline (PBS), pH 8.0.
Divide it into 1-mL volumes and
freeze aliquots at –18 C or colder.
2. PBS at pH 7.3, see Method 1.7.
3. Red cells known to be positive for the
antigen in question and, as a control,
red cells known to be positive for K,
which is consistently disrupted by
DTT.
4. Anti-K, either in reagent form or
strongly reactive in a serum specimen.
766 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Procedure
1. Wash one volume of the test cells and
the control cells with PBS. After de-
canting, add four volumes of 0.2 M
DTT, pH 8.0.
2. Incubate at 37 C for 30 to 45 min-
utes.
3. Wash four times with PBS. Slight
hemolysis may occur; if hemolysis is
excessive, repeat the procedure us-
ing fresh red cells and a smaller vol-
ume of DTT, eg, two or three volumes.
4. Resuspend the cells to a 2% to 5%
suspension in PBS.
5. Test DTT-treated cells with serum
containing the antibody in question.
Test K+ red cells with anti-K.
Interpretation
1. The control K+ red cells should give
negative reactions when tested with
anti-K; if not, the DTT treatment has
been inadequate. Other antigens in
the Kell system can also serve as the
control.
2. If reactivity of the test serum is elim-
inated, the suspected antibody spec-
ificity may be confirmed. Enough red
cell samples should be tested to ex-
clude most other clinically signifi-
cant alloantibodies.
Note
Treatment of red cells with 0.2 M DTT, pH
8.0, is optimal for denaturation of all anti-
gens of the Kell, Cartwright, LW, and
Dombrock systems, and most antigens of
the Knops system. Lower concentrations
of DTT may selectively denature particu-
lar blood group antigens (ie, 0.002 M DTT
will denature only Jsaand Jsbantigens). This
property may aid in certain antibody in-
vestigations.
Reference
BranchDR,MuenschHA,SySiokHianS,PetzLD.
DisulfidebondsarearequirementforKelland
Cartwright (Yta) blood group antigen integrity. Br J
Haematol 1983;54:573-8.
Method 3.11. Urine
Neutralization of Anti-Sda
Principle
For a discussion of anti-Sdaneutralization
by urine, see Chapter 15.
Specimen
Serum or plasma suspected of containing
anti-Sda.
Reagents
1. Urine from a known Sd(a+) individ-
ual, or from a pool of at least six in-
dividuals of unknown Sdatype, pre-
pared as follows: Collect urine and
immediately boil it for 10 minutes.
Dialyze it against phosphate-buf-
fered saline (PBS), pH 7.3, at 4 C for
48 hours. Change PBS several times.
Centrifuge. Dispense supernatant into
aliquots, which can be stored at –20
Cuntilthawedforuse.
2. PBS, pH 7.3. See Method 1.7.
Procedure
1. Mix equal volumes of thawed urine
and test serum.
2. Prepare a dilution control tube con-
taining equal volumes of serum and
PBS.
3. Prepare a urine control tube by mix-
ing equal volumes of thawed urine
and PBS.
4. Incubate all tubes at room tempera-
ture for 30 minutes.
5. Mix 1 drop of each test red cell sam-
plewith4dropsfromeachofthe
Methods Section 3: Antibody Detection/Identification and Compatibility Testing 767
Copyright © 2005 by the AABB. All rights reserved.
tubes: neutralized serum, serum with
PBS, and urine with PBS. Test each
one using standard procedures.
Interpretation
1. Persistent agglutination in the serum
sample incubated with urine means
either that partial or no neutraliza-
tion was achieved or that underlying
antibodies are present. Microscopic
examination may be helpful; aggluti-
nation due to anti-Sdahas a refractile,
mixed-field appearance on micro-
scopic examination.
2. No agglutination in the neutralized
tube with persistent agglutination in
the dilution control tube and absence
of hemolysis and agglutination in
the urine control tube indicate that
the antibody has been neutralized
and is quite probably anti-Sda.
3. The absence of agglutination in the
dilution control tube means that the
dilution in the neutralization step
was too great for the antibody pres-
ent and the results of the test are in-
valid. The urine control tube provides
assurance that no substances in the
urine are agglutinating or damaging
the red cells.
Note
Urine may also contain ABO and Lewis
blood group substances, depending upon
the ABO, Lewis, and secretor status of the
donor.
Reference
Judd WJ. Methods in immunohematology. 2nd ed.
Durham, NC: Montgomery Scientific Publications,
1994.
Method 3.12. Adsorption
Procedure
Principle
See Chapter 19.
Specimen
Serum or plasma containing antibody to
be adsorbed.
Reagents
Red cells (eg, autologous or allogeneic) that
carry the antigen corresponding to the an-
tibody specificity to be adsorbed.
Procedure
1. Wash the selected red cells at least
three times with saline.
2. After the last wash, centrifuge the red
cells at 800 to 1000 ×gfor at least 5
minutes and remove as much of the
supernatant saline as possible. Addi-
tional saline may be removed by
touching the red cell mass with a
narrow piece of filter paper.
3. Mix appropriate volumes of the pack-
ed red cells and serum and incubate
at the desired temperature for 30 to
60 minutes.
4. Mix the serum/cell mixture periodi-
cally throughout the incubation
phase.
5. Centrifuge the red cells at 800 to
1000 ×gfor 5 minutes to pack cells
tightly. Centrifuge at the incubation
temperature, if possible, to avoid
dissociation of antibody from the red
cell membranes.
6. Transfer the supernatant fluid, which
is the adsorbed serum, to a clean test
tube.Ifaneluateistobeprepared,
save the red cells.
7. Test an aliquot of the adsorbed se-
rum, preferably against an addi-
768 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
tional aliquot of the cells used for
adsorption, to see if all antibody has
been removed.
Interpretation
If reactivity remains, the antibody has not
been completely removed. No reactivity
signifies that antibody has been com-
pletely adsorbed.
Notes
1. Adsorption is more effective if the
area of contact between the red cells
and serum is large; use of a large-
bore test tube (13 mm or larger) is
recommended.
2. Multiple adsorptions may be neces-
sary to completely remove an anti-
body, but each successive adsorp-
tion increases the likelihood that the
serum will be diluted and un-
adsorbed antibodies weakened.
3. Repeat adsorptions should use a
fresh aliquot of cells and not the
cells from the prior adsorption.
4. Enzyme pretreatment of adsorbing
cells can be performed to increase an-
tibody uptake for enzyme-resistant
antigens.
Reference
Judd WJ. Methods in immunohematology. 2nd ed.
Durham, NC: Montgomery Scientific Publications,
1994.
Method 3.13. Using the
American Rare Donor
Program
Principle
The American Rare Donor Program (ARDP)
helps to locate blood products for pa-
tients requiring rare or unusual blood.
The ARDP maintains a database of rare
donors submitted by immunohematology
reference laboratories that are accredited
by the AABB or the American Red Cross
(ARC). Donors are considered rare due to
the absence of a high-incidence antigen,
absence of multiple common antigens, or
IgA deficiency.
All requests to the ARDP must originate
from an AABB- or ARC-accredited im-
munohematology reference laboratory to
ensure that the patient in question has
been accurately evaluated and reported. All
shipping and rare unit fees are established
by the shipping institution.
Procedure
1. A hospital blood bank, transfusion
service, or blood center identifies a
patient who needs rare blood.
2. The institution contacts the nearest
AABB- or ARC-accredited immuno-
hematology reference laboratory to
supply the needed blood.
3. If the laboratory cannot supply the
blood, it contacts the ARDP. All re-
quests to the ARDP must come from
an AABB- or ARC-accredited labora-
tory (or another rare donor program).
Requests received directly from a non-
accredited facility will be referred to
the nearest accredited institution.
4. The institution contacting the ARDP
(requesting institution) must confirm
the identity of the antibody(ies) by
serologic investigation or by exam-
ining the serologic work performed
by another institution.
5. ARDP staff search their database for
centers that have identified donors
with the needed phenotype and con-
tact the centers for availability of
units. ARDP staff give the name(s) of
the shipping center(s) to the request-
ing institution.
Methods Section 3: Antibody Detection/Identification and Compatibility Testing 769
Copyright © 2005 by the AABB. All rights reserved.
6. The requesting and shipping institu-
tions should discuss and agree on
charges and testing requirements
before units are shipped.
7. If an initial search does not result in
a sufficient number of units, the fol-
lowing mechanisms can be used by
ARDP staff to obtain needed units: 1)
communication to all ARDP partici-
pating centers alerting them to search
their inventories and/or recruit do-
nors matching the needed pheno-
type, 2) contacting other rare donor
filessuchasthoseadministeredby
the World Health Organization, Jap-
anese Red Cross, etc.
770 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Methods Section 4: Investigation of a Positive DAT
Methods Section 4
Investigation of a Positive
Direct Antiglobulin Test
Elution Techniques
The objective of all elution techniques is
to interfere with the noncovalent binding
forces that hold antibody-antigen complexes
together on the red cell surface. The cell
membrane can be physically disrupted by
heat, ultrasound, freezing and thawing,
detergents, or organic solvents. The bind-
ing forces of antigen-antibody complexes
can be interrupted by alterations in pH or
salt concentration. For a comparison of the
advantages and disadvantages of various
elution methods, see Chapter 20. Selected
elution methods follow, including one ex-
ample of an organic solvent method. The
cold-acid elution method (Method 4.1) is
the basis of the commercially available acid
elution kits commonly used in the United
States. Because no single elution method
will result in the identification of all
antibodies, use of an alternative elution
method (eg, organic solvent) may be indi-
cated when a nonreactive eluate is not in
agreement with clinical data. The reader
should refer to Chapter 2 for the proper
handling of hazardous chemicals that are
sometimes used in these techniques. Ac-
cess to a chemical fume hood is desirable
when organic solvents are in use.
Very thorough washing of the red cells
before elution is essential to ensure that an-
tibody in the eluate is only red cell-bound
and does not represent free antibody, eg,
from plasma. A control to show that all free
antibody has been removed by washing can
be obtained by saving the saline from the
last wash and testing it in parallel with the
eluate. Additionally, transferring the red
cells into a clean test tube just before the
elution step eliminates the possibility of
dissociating antibody that may have non-
specifically bound to the glass test tube
during an adsorption or the initial eluate
preparation steps.
771
Section 4
Copyright © 2005 by the AABB. All rights reserved.
Method 4.1. Cold-Acid Elution
Principle
Dissociation of antibodies from red cells
enables the identification of auto- or allo-
antibodies. Elution methods used in con-
junction with adsorption techniques are
also useful in detecting weak antigen ex-
pression on the adsorbing cells and in
separating mixtures of antibodies against
red cell antigens.
Specimen
Red cells positive by the direct antiglobulin
test (DAT) washed six times with large
volumes of saline (save the last wash).
Reagents
1. Glycine-HCl (0.1 M, pH 3.0), prepared
by dissolving 3.75 g of glycine and
2.922 g of sodium chloride in 500 mL
of deionized or distilled water. Adjust
the pH to 3.0 with 12 N HCl. Store at
4C.
2. Phosphate buffer (0.8 M, pH 8.2),
prepared by dissolving 109.6 g of
Na2HPO4and 3.8 g of KH2PO4in ap-
proximately 600 mL of deionized or
distilled water and adjusting the fi-
nalvolumeto1L.AdjustthepH,if
necessary,witheither1NNaOHor1
N HCl. Store at 4 C (see note 2).
3. NaCl, 0.9%, at 4 C.
4. Supernatant saline from the final
wash of red cells to be tested.
Procedure
1. Place 1 mL of red cells in a 13 ×
100-mm test tube and chill in an ice
water bath for 5 minutes before add-
ing the glycine-HCl.
2. Add 1 mL of chilled saline and 2 mL
of chilled glycine-HCl to the red cells.
3. Mix and incubate the tube in an ice
water bath (0 C) for 1 minute.
4. Quickly centrifuge the tube at 900 to
1000 ×gfor 2 to 3 minutes.
5. Transfer the supernatant eluate into
a clean test tube and add 0.1 mL of
pH 8.2 phosphate buffer for each 1
mL of eluate (see note 3).
6. Mix and centrifuge at 900 to 1000 ×g
for 2 to 3 minutes.
7. Transfer the supernatant eluate into
a clean test tube and test it in paral-
lel with the supernatant saline from
the final wash.
Notes
1. Keep glycine-HCl in an ice bath dur-
ing use, to maintain the correct pH.
2. Phosphate buffer will crystallize dur-
ing storage at 4 C. Redissolve it at 37
Cbeforeuse.
3. The addition of phosphate buffer re-
stores neutrality to the acidic eluate.
Acidity may cause hemolysis of the
reagent red cells used in testing the
eluate. The addition of 22% bovine
albumin (one part to four parts of
eluate) may reduce such hemolysis.
References
1. Judd WJ. Methods in immunohematology. 2nd
ed. Durham, NC: Montgomery Scientific Pub-
lications, 1994.
2. Rekvig OP, Hannestad K. Acid elution of blood
group antibodies from intact erythrocytes.
Vox Sang 1977;33:280-5.
Method 4.2.
Glycine-HCl/EDTA Elution
Principle
See Method 4.1.
Specimen
Red cells positive by the direct antiglobulin
test (DAT) washed six times with large
volumes of saline (save the last wash).
772 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Reagents
1. Disodium EDTA (10% w/v): Na2EDTA,
10 g; distilled water to 100 mL.
2. Glycine-HCl (0.1 M at pH 1.5): 0.75 g
glycine diluted to 100 mL with 0.9%
NaCl;adjusttopH1.5with12NHCl.
3. TRIS-NaCl (1 M): Tris(hydroxymethyl)
aminomethane [TRIS] or TRIZMA
BASE, 12.1 g; 5.25 g NaCl; distilled
water to 100 mL.
4. Supernatant saline from the final wash
of the red cells to be tested.
Procedure
1. In a test tube, mix together 20 vol-
umes (eg, drops) of 0.1 M glycine-
HCl buffer and 5 volumes of 10%
EDTA. This is the eluting solution.
2. In a 12 ×75-mm tube, place 10 vol-
umes of packed red cells.
3. Add 20 volumes of the eluting solu-
tion to the red cells, mix well, and in-
cubate at room temperature for 2
minutes. Do not overincubate.
4. Add 1 volume of TRIS-NaCl, mix,
and immediately centrifuge the tube
at 900 to 1000 ×gfor 60 seconds.
5. Transfer the supernatant eluate into
a clean test tube and adjust it care-
fully dropwise to pH 7.0 to 7.4 with
1 M TRIS-NaCl. The pH can be
checked with pH paper.
6. Centrifuge at 900 to 1000 ×gfor 2 to
3 minutes to remove the precipitate.
7. Transfer the supernatant eluate into
a clean test tube and test it in paral-
lel with the supernatant saline from
the final wash.
Notes
1. Once the red cells have been rendered
DAT negative, they may be tested for
the presence of blood group anti-
gens, except those in the Kell system.
Treatment with glycine-HCl/EDTA
denatures Kell system antigens and
Era. Wash the red cells at least three
timesinsalinebeforeuse.
2. Red cells modified with glycine-
HCl/EDTA may be treated with a
protease and used in autologous ad-
sorption studies.
3. Overincubation with the eluting so-
lution (step 3) will irreversibly dam-
age the red cells.
4. TRIS-NaCl is very alkaline and only a
few drops should be required to at-
tain the desired pH (step 5).
5. Aliquots of the reagents can be stored
frozen and one tube of each can be
thawed just before use. The 10% EDTA
may precipitate when stored at 2 to 8 C.
6. Stored eluate (4 C or frozen) may be
more stable if albumin is added (1
volume of 30% bovine albumin for
every 10 volumes of eluate). If albu-
min is added to the eluate, it should
be added to the last wash.
Reference
ByrnePC.Useofamodifiedacid/EDTAelution
technique. Immunohematology 1991;7:46-7. [Cor-
rection note: Immunohematology 1991;7:106.]
Method 4.3. Heat Elution
Principle
Heatelutionusesanincreaseintempera
-
ture to dissociate antibodies from red cells.
This method is best suited for the investi-
gation of ABO hemolytic disease of the fe-
tus and newborn and for the elution of IgM
antibodies from red cells. It should not
routinely be used for the investigation of
abnormalities caused by IgG auto- or allo-
antibodies.
Specimen
Red cells positive by the direct antiglobulin
test (DAT) washed six times with large
Methods Section 4: Investigation of a Positive DAT 773
Copyright © 2005 by the AABB. All rights reserved.
volumes of saline; save the last wash (see
note).
Reagents
1. 6% bovine albumin (see Method 1.5).
2. Supernatant saline from the final
wash of the red cells to be tested.
Procedure
1. Mix equal volumes of washed packed
cells and 6% bovine albumin in a
13 ×100-mm test tube.
2. Place the tube at 56 C for 10 min-
utes. Agitate the tube periodically
during this time.
3. Centrifuge the tube at 900 to 1000 ×
gfor 2 to 3 minutes, preferably in a
heated centrifuge.
4. Immediately transfer the supernatant
eluate into a clean test tube and test
in parallel with the supernatant sa-
line from the final wash.
Note
For optimal recovery of cold-reactive anti-
bodies, the red cells should be washed in
ice-cold saline to prevent dissociation of
bound antibody before elution.
References
1. Judd WJ. Methods in immunohematology. 2nd
ed. Durham, NC: Montgomery Scientific Pub-
lications, 1994.
2. Landsteiner K, Miller CPJr.Serologicalstud
-
ies on the blood of primates. II. The blood
groups in anthropoid apes. J Exp Med 1925;
42:853-62.
Method 4.4. Lui
Freeze-Thaw Elution
Principle
As red cells freeze, extracellular ice crys-
tals form that attract water from their sur-
roundings. This increases the osmolarity
of the remaining extracellular fluid, which
then extracts water from the red cells. The
red cells shrink, resulting in lysis. As the
membranes are disrupted, antibody is
dissociated. This method is used primar-
ily for the investigation of ABO hemolytic
disease of the fetus and newborn.
Specimen
1. Red cells washed six times with large
volumes of saline.
2. Supernatant saline from the final
wash of the red cells to be tested.
Procedure
1. Mix 0.5 mL of the red cells to be tested
with 3 drops of saline in a test tube.
2. Cap the tube, then rotate the tube to
coat the tube wall with cells.
3. Place the tube in a horizontal posi-
tioninafreezerat–6Cto–70Cfor
10 minutes.
4. Removethetubefromthefreezerand
thaw it quickly with warm, running
tap water.
5. Centrifuge for 2 minutes at 900 to
1000 ×g.
6. Transfer the supernatant to a clean
test tube and test it in parallel with
the supernatant saline from the final
wash.
References
1. Judd WJ. Methods in immunohematology. 2nd
ed. Durham, NC: Montgomery Scientific Pub-
lications, 1994.
2. FengCS,KirkleyKC,EicherCA,etal.TheLui
elution technique: A simple and efficient
method for eluting ABO antibodies. Transfu-
sion 1985;25:433-4.
774 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Method 4.5. Methylene
Chloride Elution
Principle
Organic solvents can influence antigen-
antibody dissociation by several mecha-
nisms, including alteration of the tertiary
structure of antibody molecules and dis-
ruption of the red cell membrane. This
method is suitable for elution of IgG auto-
and alloantibodies.
Specimen
DAT-positive red cells washed six times with
large volumes of saline (save last wash).
Reagents
1. Methylene chloride (dichloromethane).
2. Supernatant saline from final wash
of the red cells to be tested.
Procedure
1. Mix 1 mL of red cells, 1 mL of saline,
and 2 mL of methylene chloride in a
test tube, eg, 13 ×100 mm.
2. Stopper the tube and mix by gentle
agitation for 1 minute.
3. Remove the stopper and centrifuge
the tube at 1000 ×gfor 10 minutes.
4. Remove the lower layer of methylene
chloride with a transfer pipette and
discard it.
5. Place the tube at 56 C for 10 min-
utes. Stir the eluate constantly with
wooden applicator sticks in the first
several minutes to avoid it boiling
over; thereafter, stir it periodically.
6. Centrifuge at 1000 ×gfor 10 min-
utes.
7. Transfer the supernatant eluate into
a clean test tube and test it in paral-
lel with the supernatant saline from
the final wash.
Reference
Judd WJ. Methods in immunohematology. 2nd ed.
Durham, NC: Montgomery Scientific Publications,
1994.
Immune Hemolytic Anemia
Serum/Plasma Methods
Included in this section are methods used
to remove warm or cold autoantibody re-
activity (eg, adsorptions) so that alloanti-
body detection tests and diagnostic tests
for differentiating the immune hemolytic
anemias can be performed. See Chapter
20 for a discussion of the immune hemoly-
tic anemias.
Method 4.6. Cold
Autoadsorption
Principle
Although most cold autoantibodies do not
cause a problem in serologic tests, some
potent cold-reactive autoantibodies may
mask the concomitant presence of clini-
cally significant alloantibodies. In these
cases, adsorbing the serum in the cold
with autologous red cells can remove the
autoantibody, permitting detection of un-
derlying alloantibodies. In the case of most
nonpathologic cold autoantibodies, a
simple quick adsorption of the patient’s
serum with enzyme-treated autologous
red cells will remove most cold antibody.
See Method 3.5.6. A more efficient method
of removing immunoglobulins is the use
of ZZAP reagent, a combination of pro-
teolytic enzyme and a powerful reducing
agent. ZZAP treatment removes IgM and
complement from autologous red cells
and uncovers antigen sites that can be
used to bind free autoantibody in the se-
rum.
Methods Section 4: Investigation of a Positive DAT 775
Copyright © 2005 by the AABB. All rights reserved.
Specimen
1. 1 mL of serum or plasma to be ad-
sorbed.
2. Two 1-mL aliquots of autologous red
cells.
Reagents
1. 1% cysteine-activated papain or 1%
ficin (see Methods 3.5.1 and 3.5.2).
2. Phosphate-buffered saline (PBS), pH
7.3 (see Method 1.7).
3. 0.2 M dithiothreitol (DTT) prepared
by dissolving 1 g of DTT in 32.4 mL
of pH 7.3 PBS. Dispense into 3-mL
aliquots and store at –18 C or colder.
Procedure
1. Prepare ZZAP reagent by mixing 0.5
mL of 1% cysteine-activated papain
with2.5mLof0.2MDTTand2mL
of pH 7.3 PBS. Alternatively, use 1
mL of 1% ficin, 2.5 mL of 0.2 M DTT,
and 1.5 mL of pH 7.3 PBS. The pH
should be between 6.0 and 6.5.
2. Add 2 mL of ZZAP reagent to 1 mL of
autologous red cells. Mix and incu-
bate at 37 C for 30 minutes.
3. Wash the cells three times in saline.
Centrifuge the last wash for at least 5
minutes at 900 to 1000 ×gand re-
move as much of the supernatant sa-
lineaspossible(seenote1).
4. To the tube of ZZAP-treated red cells,
add 1 mL of the autologous serum.
Mixandincubateat4Cfor30min
-
utes.
5. Centrifuge at 900 to 1000 ×gfor 4 to
5 minutes and transfer the serum into
acleantube.
6. Steps 2 through 5 may be repeated if
the first autoadsorption does not sat-
isfactorily remove the autoantibody
activity.
7. After the final adsorption, test the
serum with reagent red cells for allo-
antibody activity.
Notes
1. To avoid dilution of the serum and
possible loss of weak alloantibody
activity, it is important in step 3 to
remove as much of the residual sa-
line as possible.
2. If the reactivity of the autoantibody
is not diminished, the target auto-
antigen may have been destroyed by
either the enzyme or the DTT. The
adsorption should be repeated against
untreated autologous red cells washed
several times in warm saline.
References
1. Branch DR. Blood transfusion in autoimmune
hemolytic anemias. Lab Med 1984;15:402-8.
2. Branch DR, Petz LD. A new reagent (ZZAP)
having multiple applications in immuno-
hematology. Am J Clin Pathol 1982;78:161-7.
Method 4.7. Determining the
Specificity of Cold-Reactive
Autoagglutinins
Principle
For a discussion of specificity of cold-re-
acting autoantibodies, see Chapter 20.
Specimen
1. Serum, separated at 37 C from a blood
sample allowed to clot at 37 C, or
plasma, separated from an anti-
coagulated sample after periodic in-
version at 37 C for approximately 15
minutes.
2. Autologous red cells.
Reagents
Test red cells of the following phenotypes:
776 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

1. A pool of two examples of adult
group O I adult red cells; they can be
the reagent cells routinely used for
alloantibody detection.
2. Group O i cord red cells.
3. The patient’s own (autologous) red
cells, washed at least three times
with 37 C saline.
4. Red cells of the same ABO group as
the patient, if the patient is not
group O. If the patient is group A or
AB, use both A1and A2cells.
5. Saline or phosphate-buffered saline
(PBS), pH 7.3 (see Method 1.7).
Procedure
1. Prepare serial twofold dilutions of
the serum or plasma in saline or
PBS. The dilution range should be
from 1 in 2 to 1 in 4096 (12 tubes),
andthevolumespreparedshouldbe
more than the total volume needed
to test all of the desired red cells. See
Method 3.7.
2. Label a set of 12 tubes with the dilu-
tion (eg, 2, 4, 8, etc) for each of the
red cells to be tested (eg, adult, cord,
autologous).
3. Dispense 2 drops of each dilution
into the appropriate tubes.
4. Add 1 drop of a 3% to 5% saline sus-
pension of each red cell sample to
the appropriate set of tubes.
5. Mix and incubate at room tempera-
turefor30to60minutes.
6. Centrifuge for 15 to 20 seconds at
900 to 1000 ×g. Examine the tubes
one by one macroscopically for agglu-
tination, starting with the set of tubes
at the highest dilution for each cell
tested (ie, read all the tubes for each
dilution as a set). Grade and record
the results.
7. Transfer the tubes to 4 C and incu-
bate them at this temperature for 1
to 2 hours.
8. Centrifuge for 15 to 20 seconds at
900 to 1000 ×g. Immediately place
thetubesinarackinanicewater
bath. Examine the tubes as in step 6.
Grade and record the results.
Interpretation
Table4.7-1summarizesthereactionsof
the commonly encountered cold-reactive
autoantibodies. In cold agglutinin syn-
Methods Section 4: Investigation of a Positive DAT 777
Table 4.7-1. Typical Relative Reactivity Patterns of Cold Autoantibodies
Red Cells
Antibody Specificity
Anti-I Anti-i Anti-ITAnti-IH Anti-Pr
O I adult + 0/↓0/↓++
Oicord 0/↓++↓+
O i adult 0/↓+0/↓↓ +
A1I adult + 0/↓0/↓↓ +
Autologous + 0/↓0/↓↓ +
O I enzyme-treated ↑↑↑↑0
+ = reactive; 0 = nonreactive; ↓= weaker reaction; ↑= stronger reaction.
Copyright © 2005 by the AABB. All rights reserved.
drome, anti-I is seen most frequently, but
anti-i may also be encountered. When
cord cells react stronger than adult cells,
the specificity may be anti-i, but adult i
red cells need to be tested to confirm that
these reactions are due to anti-i and not
anti-IT. Some examples of anti-I react
more strongly with red cells that have a
strong expression of H antigen (eg, O and
A2cells); such antibodies are called anti-
IH. Rarely, the specificity may be anti-Pr,
which should be suspected if all the cells
tested react equally. Anti-Pr can be con-
firmed by testing enzyme-treated cells;
anti-Pr does not react with enzyme-
treated cells, whereas anti-I and anti-i
react better with enzyme-treated cells.
Anti-Pr reacts equally with untreated red
cells of I or i phenotypes.
Notes
1. It is important to use separate pi-
pettes or pipette tips for each tube
when preparing serum dilutions be-
cause the serum carried from one
tube to the next when a single pi-
pette is used throughout may cause
falsely high titration endpoints. The
difference can convert a true titer of
4000 to an apparent titer of 100,000,
when the use of separate pipettes is
compared with the use of a single pi-
pette.
2. Serum dilutions can be prepared
more accurately with large volumes
(eg, 0.5 mL) than with small vol-
umes.
3. Potent examples of cold-reactive
autoantibodies generally do not
show apparent specificity until titra-
tion studies are performed; this
specificity may not even be apparent
with dilutions at room temperature
or 4 C. In such circumstances, tests
can be incubated at 30 to 37 C. Dif-
ferential reactivity may be more
apparent if incubation times are
prolonged and agglutination is eval-
uated after settling, without centrifu-
gation. Settled readings are more ac-
curate after a 2-hour incubation.
4. This procedure can be used to deter-
mine both the titer and the specific-
ity. If incubations are started at 37 C
(set up prewarmed, and readings are
taken sequentially after incubation
at each temperature—eg, 37 C, 30 C,
room temperature, 4 C), the specific-
ity, titer, and thermal amplitude of
the autoantibody can be determined
with a single set of serum dilutions.
5. If testing will also be performed at
30 C and 37 C, include a test of the
neat (undiluted) serum.
Reference
Petz LD, Garratty G. Immune hemolytic anemias.
2nd ed. Philadelphia: Churchill Livingstone, 2004.
Method 4.8. Cold Agglutinin
Titer
Principle
Cold-reactive autoantibodies, if present at
very high titers, may suggest a pathologic
cold agglutinin disease. This may result in
overt hemolysis and systemic symptoms
and may indicate underlying B-cell hema-
tologic neoplasia.
Specimen
Serum, separated at 37 C from a sample
allowed to clot at 37 C, or plasma, sepa-
rated from an anticoagulated sample after
periodic inversion at 37 C for approxima-
tely 15 minutes.
778 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Reagents
1. A pool of 2 examples of washed group
O I adult red cells, eg, antibody de-
tection cells.
2. Phosphate-buffered saline (PBS), pH
7.3 (see Method 1.7).
Procedure
1. Prepare serial twofold dilutions of the
patient’s serum or plasma in PBS.
Thedilutionrangeshouldbefrom1
in 2 to 1 in 4096 (12 tubes). See
Method 3.7.
2. Mix 2 drops of each dilution with 1
dropofa3%to5%cellsuspensionof
red cells.
3. Mix and incubate at 4 C for 1 to 2
hours.
4. Centrifuge the tubes for 15 to 20 sec-
onds at 900 to 1000 ×g,thenplace
thetubesinarackinanicewater
bath. Examine the tubes one by one
macroscopically for agglutination,
starting with the tube at the highest
dilution. Grade and record the re-
sults.
Interpretation
Thetiteristhereciprocalofthehighest
serum dilution at which macroscopic ag-
glutination is observed. Titers above 64
are considered elevated, but hemolytic
anemia resulting from cold-reactive auto-
agglutinins rarely occurs unless the titer is
>1000. Titers below 1000 may be obtained
when the autoantibody has a different
specificity (eg, anti-i), or if the cold agglu-
tinin is of the less common low-titer,
high-thermal-amplitude type. If the pa-
tient has a positive direct antiglobulin test
(DAT) because of complement only and
has clinical signs of hemolytic anemia,
specificity and thermal amplitude studies
should be performed (see Method 4.7).
Notes
1. It is important to use separate pi-
pettes for each tube when preparing
serum dilutions because the serum
carried from one tube to the next when
a single pipette is used throughout
may cause falsely high titration end-
points.
2. Serum dilutions can be prepared
more accurately with large volumes
(eg, 0.5 mL) than with small vol-
umes.
Reference
Petz LD, Garratty G. Immune hemolytic anemias.
2nd ed. Philadelphia: Churchill Livingstone, 2004.
Method 4.9. Autologous
Adsorption of
Warm-Reactive
Autoantibodies
Principle
Warm-reactive autoantibodies in serum
maymasktheconcomitantpresenceof
clinically significant alloantibodies. Ad-
sorption of the serum with autologous red
cells can remove autoantibody from the
serum, permitting detection of underly-
ing alloantibodies. However, autologous
red cells in the circulation are coated with
autoantibody. Autologous adsorption of
warm-reactive autoantibodies can be fa-
cilitated by dissociating autoantibody from
the red cell membrane, thereby uncover-
ing antigen sites that can bind free auto-
antibody to remove it from the serum.
Some autoantibody can be dissociated by
a gentle heat elution for 3 to 5 minutes at
56 C. Subsequent treatment of the cells
withenzymesenhancestheadsorption
process by removing membrane struc-
tures that otherwise hinder the associa-
Methods Section 4: Investigation of a Positive DAT 779
Copyright © 2005 by the AABB. All rights reserved.
tion between antigen and antibody. The
most effective procedure involves the use
of ZZAP reagent, a mixture of a proteolytic
enzyme and a sulfhydryl reagent. ZZAP
removes immunoglobulins and comple-
ment from the red cells and enhances the
adsorption process. Red cells from pa-
tients transfused within the last 3 months
should not be used for autoadsorption
because transfused red cells present in
the circulation are likely to adsorb the
alloantibodies that are being sought (see
Chapter 20).
Specimen
1. 1 mL of serum or plasma (or eluate)
to be adsorbed.
2. 2 mL of autologous red cells.
Reagents
1. 1% cysteine-activated papain or 1%
ficin (see Methods 3.5.1 and 3.5.2).
2. Phosphate-buffered saline (PBS), pH
7.3 (see Method 1.7).
3. 0.2 M DTT prepared by dissolving 1
gofDTTin32.4mLofpH7.3PBS.
Dispense into 3-mL aliquots and
store at –18 C or colder.
Procedure
1. Prepare ZZAP reagent by mixing 0.5
mL of 1% cysteine-activated papain
with2.5mLof0.2MDTTand2mL
of pH 7.3 PBS. Alternatively, use 1
mL of 1% ficin, 2.5 mL of 0.2 M DTT,
and 1.5 mL of pH 7.3 PBS. The pH
should be between 6.0 and 6.5.
2. To each of two tubes containing 1
mL of red cells, add 2 mL of ZZAP re-
agent. Mix and incubate at 37 C for
30 minutes with periodic mixing.
3. Wash the red cells three times in sa-
line. Centrifuge the last wash for at
least 5 minutes at 900 to 1000 ×g
and remove as much supernatant
saline as possible.
4. Add serum to an equal volume of
ZZAP-treated red cells, mix, and in-
cubate at 37 C for approximately 30
to 45 minutes.
5. Centrifuge and carefully remove serum.
6. If the original serum reactivity was
only 1+, proceed to step 7; otherwise,
repeat steps 4 and 5 once more us-
ing the once-adsorbed patient’s se-
rum and the second aliquot of ZZAP-
treated cells.
7. Test the serum against a specimen of
group O reagent cells. If reactivity
persists, repeat steps 4 and 5.
Interpretation
One or two adsorptions ordinarily remove
sufficient autoantibody so that alloanti-
body reactivity, if present, is readily ap-
parent. If the twice-autoadsorbed serum
reacts with defined specificity, as shown
by testing against a small antibody identi-
fication panel, then the defined specificity
of the antibody is probably an alloantibody.
If the serum reacts with all cells on the
panel, either additional autoadsorptions
are necessary, the serum contains anti-
body to a high-incidence antigen, or the
serum contains an autoantibody (eg,
anti-Kpb) that does not react with ZZAP-
treated cells and thus will not be adsorbed
by this procedure. To check this latter
possibility, test the reactive autoadsorbed
serum against reagent cells that have
been pretreated with the ZZAP reagent.
Notes
1. ZZAP treatment destroys all Kell sys-
tem antigens and all other antigens
that are destroyed by proteases, eg,
M, N, Fya,andFy
b.ZZAPreagentalso
denatures the antigens of the LW,
Cartwright, Dombrock, and Knops
780 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
systems. If the autoantibody is sus-
pected to have specificity in any of
these latter blood groups, an alter-
native procedure is to perform auto-
adsorption with untreated autologous
cells or autologous cells treated only
with 1% ficin or 1% cysteine-activated
papain.
2. There is no need to wash packed red
cells before treatment with ZZAP.
3. Cold autoantibodies reactive at room
temperature can also be present in
the serum of about 30% of patients
with warm-reactive autoantibodies.
Removal of these cold antibodies can
be facilitated by placing the serum
and cell mixture at 4 C for about 15
minutes after incubation at 37 C.
4. As a guide, when the original serum
reactivity is 1+ in the low-ionic-
strength saline indirect antiglobulin
test (LISS-IAT), usually only one ad-
sorption would be required. Antibod-
ies with 2+ to 3+ reactivity will gen-
erally be removed in two to three
adsorptions. Performing greater
than four adsorptions increases the
risk of diluting alloantibody reactiv-
ity.
Reference
Branch DR, Petz LD. A new reagent (ZZAP) having
multiple applications in immunohematology. Am
J Clin Pathol 1982;78:161-7.
Method 4.10. Differential
Warm Adsorption Using
Enzyme- or ZZAP-Treated
Allogeneic Red Cells
Principle
Adsorption of serum with selected red cells
of known phenotypes will remove auto-
antibody and leave antibodies to most
blood group systems. The specificity of the
antibodies that remain after adsorption
can be confirmed by testing against a
panel of reagent red cells. This procedure
can be used to detect underlying alloanti-
bodies if the patient has been recently
transfused, or if insufficient autologous
red cells are available and the patient’s
phenotype is unknown.
Treating the adsorbing cells with enzyme
or ZZAP typically enhances the adsorption
process. In addition, the treated red cells will
lack the antigens destroyed by dithiothrei-
tol (DTT) and/or enzymes (see Chapter 19).
Specimen
Serum/plasma containing warm-reactive
autoantibodies or eluate from direct anti-
globulin test (DAT)-positive cells.
Reagents
1. 1% cysteine-activated papain or 1%
ficin (see Methods 3.5.1 and 3.5.2).
2. ZZAP reagent (papain or ficin plus
0.2 M DTT). See Method 4.9.
3. Phosphate-buffered saline (PBS), pH
7.3 (see Method 1.7).
4. Group O red cells of the phenotypes
R1R1,R
2R2, and rr; one of these cells
should be Jk(a–b+) and one should
be Jk(a+b–). Additionally, if the red
cells are to be only enzyme-treated,
at least one of the cells should also
be K–. They can be reagent cells or
from any blood specimen that will
yield a sufficient volume of red cells.
Procedure
1. Wash 1 mL of each red cell specimen
once in a large volume of saline,
centrifuge to pack the cells, and re-
move the supernatant saline.
2. To each volume of washed packed
cells,addonevolumeof1%enzyme
solution or two volumes of working
Methods Section 4: Investigation of a Positive DAT 781
Copyright © 2005 by the AABB. All rights reserved.
ZZAP reagent. Invert several times to
mix them.
3. Incubate at 37 C: 15 minutes for en-
zyme or 30 minutes for ZZAP. Mix
periodically throughout incubation.
4. Wash the red cells three times with
large volumes of saline. Centrifuge
at 900 to 1000 ×gfor at least 5 min-
utes and remove the last wash as
completely as possible to prevent di-
lution of the serum.
5. For each of the three red cell speci-
mens, mix one volume of treated
cells with an equal volume of the pa-
tient’s serum and incubate at 37 C
for 30 minutes, mixing occasionally.
6. Centrifuge at 900 to 1000 ×gfor ap-
proximately 5 minutes and harvest
the supernatant serum.
7. Test the three samples of adsorbed
serum against the cells (untreated)
used for adsorption, respectively. If
reactivity is present, repeat steps 5
through 7 with a fresh aliquot of treat-
ed red cells until no reactivity re-
mains. The three samples of adsorbed
serum can then be tested against an-
tibody detection/panel cells and the
results compared for demonstration
of persisting and removed alloanti-
body activity. See section on allo-
geneic adsorption in Chapter 20.
Notes
1. If the autoantibody is very strong,
three or more aliquots of adsorbing
cells should be prepared. If the first
adsorption is unsuccessful, the use
of a higher proportion of cells to se-
rum/eluate may enhance effective-
ness.
2. The adsorbing red cells should be
tightly packed to remove residual sa-
line that might dilute the antibodies
remaining in the serum/eluate.
3. Agitate the serum/cell mixture dur-
ing incubation to provide maximum
surface contact.
4. A visible clue to the effectiveness of
adsorption is clumping of the en-
zyme- or ZZAP-treated cells when they
are mixed with the serum, especially
when strong antibodies are present.
5. As a guide, when the original serum
reactivity is 1+ in the low-ionic-
strength saline indirect antiglobulin
test (LISS-IAT), usually only one ad-
sorption would be required. Anti-
bodies with 2+ to 3+ reactivity will
generally be removed in two to three
adsorptions. Performing greater than
four adsorptions increases the risk of
diluting alloantibody reactivity.
6. If adsorption with enzyme- or ZZAP-
treated cells has no effect on the
autoantibody, adsorption with un-
treated red cells may be tried.
References
1. Branch DR, Petz LD. A new reagent (ZZAP)
having multiple applications in immuno-
hematology. Am J Clin Pathol 1982;78:161-7.
2. Judd WJ. Methods in immunohematology.
2nd ed. Durham, NC: Montgomery Scientific
Publications, 1994.
Method 4.11. One-Cell
Sample Enzyme or ZZAP
Allogeneic Adsorption
Principle
If the Rh and Kidd phenotypes of a re-
cently transfused patient are known or can
be determined, autoantibody activity can
be adsorbed from the serum onto a single
allogeneic red cell sample, leaving serum
that can be evaluated for the presence of
alloantibodies. The red cells used should
have the same Rh and Kidd phenotypes as
the patient; they can be treated with en-
782 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
zyme or ZZAP to denature antigens (see
Chapter 19). This method is a simplified
version of the previous adsorption proce-
dure, but it should be used only if the pa-
tient’s Rh and Kidd phenotypes are known
(see the note).
Specimen
Serum, plasma, or eluate to be tested.
Reagents
1. 1% cysteine-activated papain or 1%
ficin (see Methods 3.5.1 and 3.5.2).
2. ZZAP reagent (papain or ficin plus
0.2 M DTT). See Method 4.9.
3. ABO-compatible red cells of the pa-
tient’s Rh and Kidd phenotypes; they
can be reagent cells or cells from any
blood specimen that will yield suffi-
cient cells.
Procedure
1. Wash the selected allogeneic red cells
once in a large volume of saline and
centrifuge to pack them.
2. Add one volume of 1% enzyme solu-
tion or two volumes of ZZAP reagent
to one volume of these packed cells.
Invert several times to mix them.
3. Incubate at 37 C: 15 minutes for en-
zyme or 30 minutes for ZZAP. Mix
periodically throughout incubation.
4. Wash the cells three times with sa-
line. Centrifuge at 900 to 1000 ×gfor
at least 5 minutes and remove the
last wash as completely as possible
to prevent dilution of the serum.
5. To one volume of treated cells, add
an equal volume of the patient’s se-
rum, mix, and incubate at 37 C for 30
minutes, mixing occasionally.
6. Centrifuge at 900 to 1000 ×gfor ap-
proximately 5 minutes and harvest
the supernatant serum.
7. Test the adsorbed serum against the
cells (untreated) used for adsorption.
If reactivity persists, repeat steps 5
through 7 with a fresh aliquot of
treated cells until the serum is no
longer reactive.
Note
The s antigen may not be denatured by a
particular enzyme or ZZAP solution. The s
antigen status of the adsorbing red cells
may need to be considered.
Method 4.12. Polyethylene
Glycol Adsorption
Principle
Polyethylene glycol (PEG) enhances the
adsorption of antibody by untreated red
cells. Testing the adsorbed aliquot against
a panel of red cells can identify the speci-
ficity of antibodies that remain after ad-
sorption. This method can be used for both
autologous and allogeneic adsorption.
Specimen
Serum or plasma to be tested.
Reagents
1. PEG, 20% (20 g PEG, 3350 MW, in
100 mL of PBS, pH 7.3) or commer-
cial PEG enhancement reagent.
2. Autologous red cells or ABO-com-
patible allogeneic red cells of known
phenotype.
Procedure
1. Wash aliquots of red cells in large
volumes of saline three times and
centrifuge for 5 to 10 minutes at
1000 ×g. Remove all residual saline.
2. To 1 volume (eg, 1 mL) of red cells,
add 1 volume of serum and 1 vol-
Methods Section 4: Investigation of a Positive DAT 783
Copyright © 2005 by the AABB. All rights reserved.
ume of PEG. Mix well and incubate
at 37 C for 15 minutes.
3. Centrifuge the serum/PEG/cell mix-
ture for 5 minutes and harvest the
adsorbed serum/PEG mixture.
4. To test the adsorbed serum, add 4
drops of serum to 1 drop of test red
cells, incubate for 15 minutes at 37
C, and proceed to the antiglobulin
test with anti-IgG. The larger volume
of serum tested (4 drops) is required
to account for the dilution of the se-
rum by the PEG. See the notes.
5. To check for completeness of ad-
sorption, test the adsorbed serum
against the red cells used for the ad-
sorption. If positive, repeat the ad-
sorption by adding the adsorbed se-
rum to a fresh aliquot of red cells but
do not add additional PEG. If the test
was negative, test the adsorbed se-
rum with a panel of cells.
Notes
1. Red cells for adsorption may be chem-
ically modified (eg, with enzymes or
ZZAP) before adsorption if denatur-
ation of antigens is desired.
2. The adsorbing cells should be thor-
oughly packed to remove any resid-
ual saline that could result in dilu-
tion of the antibodies remaining in
the serum.
3. Test the adsorbed serum on the day
it was adsorbed. Weak antibody re-
activity may be lost upon storage of
PEG-adsorbed sera, possibly due to
precipitation of the protein noticeable
after 4 C storage.
4. Although many laboratories suc-
cessfully use the PEG adsorption
method, some serologists have re-
ported a weakening or loss of anti-
body reactivity in some samples
when compared with results ob-
tained using a different technique.
To accommodate this potential
weakening of antibody reactivity,
some serologists test 6 drops of the
PEG-adsorbed serum.
5. Agglutination of the adsorbing red
cells does not occur when PEG is
used; therefore, there is no visible
clue to the efficiency of the adsorp-
tion process. As a guide, when the
original serum reactivity is 1+ in
low-ionic-strength saline indirect
antiglobulin test (LISS-IAT), usually
only one adsorption would be re-
quired. Antibodies with 2 to 3+ reac-
tivity will generally be removed in
two adsorptions.
References
1. Leger RM, Garratty G. Evaluation of methods
for detecting alloantibodies underlying warm
autoantibodies. Transfusion 1999;39:11-6.
2. Leger RM, Ciesielski D, Garratty G. Effect of
storage on antibody reactivity after adsorp-
tion in the presence of polyethylene glycol.
Transfusion 1999;39:1272-3.
Method 4.13. The
Donath-Landsteiner Test
Principle
IgG autoantibodies that cause paroxysmal
cold hemoglobinuria (PCH) act as bi-
phasic hemolysins in vitro. The IgG auto-
antibodies bind to the red cells at cold
temperatures, and, as the test is warmed
to 37 C, complement is activated and lysis
of the red cells occurs. The patient for whom
this procedure should be considered is
one with a positive direct antiglobulin test
(DAT) resulting from C3; demonstrable
hemoglobinemia, hemoglobinuria, or
both; and no evidence of autoantibody
activity in the serum or the eluate made
from the DAT-positive cells. For a discus-
sion of PCH, see Chapter 20.
784 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Specimen
Serum separated from a freshly collected
blood sample maintained at 37 C.
Reagents
1. Freshly collected pooled normal sera
known to lack unexpected antibod-
ies, to use as a source of complement.
2. 50% suspension of washed group O
red cells that express the P antigen,
eg, antibody detection cells.
Procedure
1. Label three sets of three 10 ×75-mm
test tubes as follows: A1-A2-A3;
B1-B2-B3; C1-C2-C3.
2. To tubes 1 and 2 of each set, add 10
volumes (eg, drops) of the patient’s
serum.
3. To tubes 2 and 3 of each set, add 10
volumes of fresh normal serum.
4. To all tubes, add one volume of the
50% suspension of washed P-posi-
tive red cells and mix well.
5. Place the three “A” tubes in a bath of
melting ice for 30 minutes and then
at 37 C for 1 hour.
6. Place the three “B” tubes in a bath of
melting ice and keep them in melt-
ing ice for 90 minutes.
7. Placethethree“C”tubesat37Cand
keep them at 37 C for 90 minutes.
8. Centrifuge all tubes and examine the
supernatant fluid for hemolysis.
Interpretation
The Donath-Landsteiner test is considered
positive when the patient’s serum, with or
without added complement, causes
hemolysis in the tubes that were incu-
bated first in melting ice and then at 37 C
(ie, tubes A1 and A2), and there is no
hemolysis in any of the tubes maintained
throughout at 37 C (ie, tubes C1, C2) or in
melting ice (ie, tubes B1, B2). The A3, B3,
and C3 tubes serve as a control of the nor-
mal sera complement source and should
notmanifesthemolysis.
Notes
1. Thebiphasicnatureofthehemoly
-
sin associated with PCH requires
that serum be incubated with cells at
a cold temperature first (eg, melting
ice bath) and then at 37 C.
2. Active complement is essential for
demonstration of the antibody. Be-
cause patients with PCH may have
low levels of serum complement,
fresh normal serum should be in-
cluded in the reaction medium as a
source of complement.
3. To avoid loss of antibody by cold
autoadsorption before testing, the
patient’s blood should be allowed to
clot at 37 C, and the serum sepa-
rated from the clot at this tempera-
ture.
4. If a limited amount of blood is avail-
able (eg, from young children), set
up tubes A-1, A-2, A-3 and C-1, C-2;
if there is only enough serum for two
tests (ie, 20 drops), set up tubes A-2,
A-3, and C-2.
5. To demonstrate the P specificity of
the Donath-Landsteiner antibody,
ABO-compatible p red cells should
be tested in a second set of tubes
A-1, A-2, and A-3. No lysis should
develop in these tubes, confirming
the P specificity of the antibody.
References
1. Judd WJ. Methods in immunohematology.
2nd ed. Durham, NC: Montgomery Scientific
Publications, 1994.
2. Dacie JV, Lewis SM. Practical hematology. 7th
ed. New York: Churchill Livingstone, 1991:
500-1.
Methods Section 4: Investigation of a Positive DAT 785
Copyright © 2005 by the AABB. All rights reserved.
Method 4.14. Detection of
Antibodies to Penicillin or
Cephalosporins by Testing
Drug-Treated Red Cells
Principle
See Chapter 20 for a discussion of the
mechanisms by which drugs cause a posi-
tive direct antiglobulin test (DAT). The
preparations of drugs used should, to the
extent possible, be the same as those
given to the patient. For drugs other than
penicillin and the cephalosporins, refer to
published reports for the method used to
treat the red cells.
Specimen
Serum or plasma and eluate (and last
wash) to be studied.
Reagents
1. 0.1 M sodium barbital-buffer (BB) at
pH 9.6 to 9.8, prepared by dissolving
2.06 g of sodium barbital in 80 mL of
distilled or deionized H20. Adjust the
pH to between 9.6 and 9.8 with 0.1 N
HCl. Bring total volume to 100 mL.
Store at 2 to 8 C.
2. Phosphate-buffered saline (PBS), pH
7.3 (see Method 1.7).
3. Drug, eg, penicillin, cephalosporin.
4. Washed, packed, group O red cells.
5. Normal sera/plasma.
6. IgG-coated red cells.
Procedure
1. For penicillin-treated cells, dissolve
600 mg of penicillin in 15 mL of BB.
This high pH is optimal, but if the
buffer is unavailable, PBS, pH 7.3,
can be used. Add 1 mL of red cells. In
a separate tube, prepare control cells
by adding 1 mL of untreated red
cells (without the drug) to 15 mL of
the same buffer. Incubate both tubes
for 1 hour at room temperature with
occasional mixing. Wash three times
in saline and store in PBS at 2 to 8 C
for up to 1 week. See note 1.
2. For cephalosporin-treated cells, dis-
solve400mgofthedrugin10mLof
PBS, pH 7.3. Add 1 mL of red cells. In
a separate tube, prepare control cells
by adding 1 mL of untreated red cells
(without the drug) to 10 mL of PBS.
Incubate both tubes for 1 hour at 37
C with occasional mixing. Wash three
timesinsalineandstoreinPBSfor
up to 1 week at 2 to 8 C. See notes 1
and 2.
3. Mix 2 or 3 drops of each specimen
(serum, eluate, and last wash) and
controls with 1 drop of 5% saline
suspension of drug-treated red cells.
4. In parallel, test each specimen and
control with the untreated red cells.
See notes 3 and 4.
5. Incubate the tests at 37 C for 60 min-
utes. Centrifuge and examine for
hemolysis and agglutination. Record
the results.
6. Wash the cells four times in saline
and test by an indirect antiglobulin
technique using polyspecific anti-
human globulin or anti-IgG reagent.
Centrifuge and examine for aggluti-
nation. Record the results.
7. Confirm the validity of negative tests
by adding IgG-coated red cells.
Interpretation
Reactivity (hemolysis, agglutination, and/
or positive indirect antiglobulin test) with
drug-treated cells, but not with untreated
cells, indicates that drug antibodies are
present (see notes 3 and 4). No hemolysis
786 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
will be seen in tests with plasma or the
eluate. Antibodies to either penicillin or
cephalothin may cross-react with cells
treated with the other drug (ie, penicillin
antibodies may attach to cephalothin-
treated cells and vice versa). Antibodies to
other cephalosporins may react with
cephalothin-treated cells. It is best to treat
cells with the drug that is suspect.
Negative results without a positive con-
trol can only be interpreted to mean that
drug antibodies were not detected. The
drug may or may not be bound to the test
red cells.
Notes
1. The volume of drug-treated red cells
can be scaled down as long as the ra-
tio of the 40 mg/mL drug solution to
red cells is constant; eg, 120 mg pen-
icillin in 3 mL BB plus 0.2 mL red
cells or 100 mg cephalosporin in 2.5
mL PBS plus 0.25 mL red cells.1
2. Cephalosporins do not require a
high pH for optimal coating of red
cells. In fact, a lower pH, ie, pH 6 to
7, decreases nonspecific protein ad-
sorption seen when a high pH buffer
is used. The least amount of nonspe-
cific protein adsorption by drug-
treated red cells will occur if a pH 6.0
buffer is used, but this leads to a
slight decrease in coating by the
drug.
3. Test normal pooled serum and PBS
as negative controls and, when avail-
able, a specimen known to contain
an antibody to the drug being inves-
tigated as a positive control.
4. To control for nonspecific protein
adsorption and nonspecific aggluti-
nation of normal sera observed with
some cephalosporins (eg, cephalo-
thin), test the normal serum and the
test serum at a 1 in 20 dilution in PBS.
Normal sera diluted 1 in 20 generally
do not react nonspecifically. Thus,
reactivity of the diluted serum with
the drug-treated cells but not with
the untreated cells indicates that
drug antibody is present.
5. When testing cefotetan-treated red
cells, test the serum at a 1 in 100 di-
lutioninPBStopreventafalse-posi
-
tive test result. In addition to the
nonspecific uptake of protein onto
cefotetan-treated red cells, some
normal sera appear to have a “natu-
rally occurring” anticefotetan,2afew
of which react weakly at a 1 in 20 di-
lution. Cases of cefotetan-induced
immune hemolytic anemia are asso-
ciated with very high antibody titers
(eg, mean antiglobulin test titer =
16,000).3
6. About 30% of patients with antice-
fotetan also have a drug-independ-
ent antibody3; in these cases, the
serum and/or eluate, when tested
undilute, may react with both the
cefotetan-treated and untreated red
cells.
7. The last wash control can some-
times react with cefotetan-treated
red cells when antibodies to cefo-
tetan are present, regardless of the
wash solution used (commercial
acid eluate kit wash solution, 4 C
LISS, or 4 C PBS) or the number of
washes performed.3
8. Prepare drug solutions just before
use.
9. Drug-treated red cells may be kept
in PBS at 4 C for up to 1 week; how-
ever, there may be some weakening
of drug coating upon storage. Drug-
treated and untreated red cells may
also be stored frozen.
10. When antibodies are not detected
with drug-treated red cells, test by
Methods Section 4: Investigation of a Positive DAT 787
Copyright © 2005 by the AABB. All rights reserved.
the immune complex method (Method
4.15). Antibodies to some third-gen-
eration cephalosporins (eg, ceftriaxone)
do not react with drug-treated red cells.
References
1. Petz LD, Garratty G. Immune hemolytic
anemias. 2nd ed. Philadelphia: Churchill
Livingstone, 2004.
2. Arndt P, Garratty G. Is severe immune
hemolytic anemia, following a single dose of
cefotetan, associated with the presence of
“naturally occurring” anti-cefotetan? (ab-
stract) Transfusion 2001;41(Suppl):24S.
3. Arndt PA, Leger RM, Garratty G. Serology of
antibodies to second- and third-generation
cephalosporins associated with immune
hemolytic anemia and/or positive direct
antiglobulin tests. Transfusion 1999;39:1239-
46.
Method 4.15. Demonstration
of Immune-Complex
Formation Involving Drugs
Principle
For a discussion of the mechanism of
drug-induced immune-complex forma-
tion, see Chapter 20.
Specimen
The patient’s serum.
Reagents
1. The drug under investigation, in the
same form (powder, tablet, capsules)
that the patient is receiving.
2. Phosphate-buffered saline (PBS) at
pH 7.3 (see Method 1.7).
3. Fresh, normal serum known to lack
unexpected antibodies, as a source
of complement.
4. Pooled group O reagent red cells, 5%
suspension, one aliquot treated with
a proteolytic enzyme (see Method
3.5.6) and one untreated.
5. Polyspecific antihuman globulin re-
agent.
6. IgG-coated red cells.
Procedure
1. Prepare a 1 mg/mL solution of the
drug in PBS. Centrifuge to remove
any particulate matter and adjust
the pH of the supernatant fluid to
approximately 7 with either 1 N
NaOH or 1 N HCl, as required, if the
pH is below 5 or above 8.
2. Label two sets of tubes for the fol-
lowing test mixtures:
a. Patient’s serum + drug.
b. Patient’s serum + PBS.
c. Patient’s serum + complement
(normal serum) + drug.
d. Patient’s serum + complement
(normal serum) + PBS.
e. Normal serum + drug.
f. Normal serum + PBS.
3. Add 2 volumes (eg, 2 drops) of each
component in the appropriate tubes
(eg,2dropsofserum+2dropsof
drug).
4. Add 1 drop of a 5% saline suspen-
sion of untreated group O reagent
red cells to one set of tubes. Add 1
drop of a 5% saline suspension of
enzyme-treated group O reagent red
cells to the second set of tubes.
5. Mix and incubate at 37 C for 1 to 2
hours, with periodic gentle mixing.
6. Centrifuge, examine for hemolysis
and agglutination, and record the re-
sults.
7. Wash the cells four times in saline
and test with a polyspecific antiglo-
bulin reagent.
8. Centrifuge, examine for agglutina-
tion, and record the results.
9. Confirm the validity of negative tests
by adding IgG-coated red cells.
788 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Interpretation
Hemolysis, direct agglutination, or posi-
tive indirect antiglobulin tests can occur
together or separately. Reactivity in any of
the tests containing the patient’s serum to
which the drug was added, and absence
of reactivity in the corresponding control
tests containing PBS instead of the drug,
indicate that antibody to the drug is pres-
ent.Seenote4.
Notes
1. Thedrugmaybemoreeasilydis
-
solved by incubation at 37 C and vig-
orous shaking of the solution. If the
drug is in tablet form, crush it with a
mortar and pestle and remove any
visible outer tablet coating material
before adding PBS.
2. Not all drugs will dissolve com-
pletely in PBS. Consult the manufac-
turer or a reference such as the Merck
Index for the solubility of the drug in
question. A previous report of drug-
induced immune hemolytic anemia
resulting from the drug in question
mayprovideinformationonthe
drug solution preparation.
3. When available, a serum/plasma
known to contain antibody with the
drug specificity being evaluated
should be included as a positive
control.
4. Tests without the drug added may be
positive if autoantibodies or circu-
lating drug-antibody immune com-
plexes are present in the patient’s
sample. Autoantibody reactivity would
be persistent over time, whereas cir-
culating immune complexes are
transient.
5. Testing with enzyme-treated red
cells and the addition of fresh nor-
mal serum as a source of comple-
ment may increase the sensitivity of
the test.
6. If tests for drug antibodies by the
immune-complex method and drug
adsorption method are noninforma-
tive, consider testing with ex-vivo
antigen (see Method 4.16).
Reference
Petz LD, Garratty G. Immune hemolytic anemias.
2nd ed. Philadelphia: Churchill Livingstone, 2004.
Method 4.16. Ex-Vivo
Demonstration of
Drug/Anti-Drug Complexes
Principle
Immune drug/anti-drug complexes can
activate complement and cause hemolysis
in vivo. These immune complexes may be
demonstrable by serologic testing in the
presence of the drug, but with some drugs
(notably nomifensine), antibodies are di-
rected against metabolites of the drug,
rather than the native drug. Serum and/or
urine from volunteers who have ingested
therapeutic levels of the drug can be used
as a source of these metabolites. See note
1.
This procedure is used to investigate
drug-associated immune hemolysis, partic-
ularly when use of the preceding methods
has been noninformative.
Specimen
Patient’s serum.
Reagents
1. Polyspecific antihuman globulin
(AHG) reagent.
2. Drug metabolites from volunteer
drug recipients. See note 2.
Methods Section 4: Investigation of a Positive DAT 789
Copyright © 2005 by the AABB. All rights reserved.
a. Volunteer serum (VS) obtained
immediately before (VS0), at 1
hour (VS1), and 6 hours (VS6)
after drug administration. Di-
vide serum into 1-mL aliquots
and store them at 2 to 8 C for a
few hours or at –20 C or colder
until use.
b. Volunteer urine (VU) obtained
immediately before (VU0), at 1
hour (VU1), 3.5 hours (VU3.5), 7
hours (VU7), and 16 hours
(VU16)afterdrugadministra
-
tion. Divide into 1-mL aliquots
and store them at 2 to 8 C for a
few hours or at –20 C or colder
until use.
3. Fresh normal serum, known to lack
unexpected antibodies, as a source of
complement.
4. Phosphate-buffered saline (PBS), pH
7.3 (see Method 1.7).
5. Pooled group O reagent red cells
washed three times with saline and
resuspended to a 5% concentration
with PBS.
6. Pooled, enzyme-treated, group O red
cells, 5% suspension in PBS.
7. IgG-coated red cells.
Procedure
1. For each volunteer serum and/or
volunteer urine sample collected, la-
bel two sets of the following test
mixtures:
a. Patient’s serum + VS (or VU).
b. Patient’s serum + PBS.
c. Patient’s serum + complement
+VS(orVU).
d. Patient’s serum + complement
+PBS.
e. Complement + VS (or VU).
f. Complement + PBS.
2. Add 0.1 mL of each component to the
appropriate tubes.
3. Add 1 drop of a 5% saline suspen-
sion of the untreated group O re-
agent red cells to one set of tubes.
Add 1 drop of a 5% saline suspen-
sion of enzyme-treated reagent red
cells to the second set of tubes.
4. Mix the contents of each tube and
incubate at 37 C for 1 to 2 hours, with
periodic mixing.
5. Centrifuge, examine for agglutina-
tion and/or hemolysis, and record
the results.
6. Wash the cells four times with saline
and test with a polyspecific antiglo-
bulin reagent.
7. Centrifuge, examine for agglutina-
tion, and record the results.
8. Confirm the validity of negative tests
by adding IgG-coated red cells.
Interpretation
Hemolysis, direct agglutination, or reac-
tivity with AHG in any of the tubes con-
taining test serum and VS or VU, and ab-
sence of reactivity in all the control tubes,
indicate antibody against a metabolite of
thedruginquestion.
Notes
1. Approval of the institutional ethics
committeeshouldbeobtainedfor
the use of volunteers for obtaining
drug metabolites.
2. The urine sample collection times
given are those optimal for antibod-
ies to nomifensine metabolites; dif-
ferent collection times may be re-
quired for other drugs.
3. Complement may be omitted from
step 1 if the VS samples have been
keptoniceandareusedfortesting
within 8 hours of collection.
790 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
4. Testing with enzyme-treated red
cells and the addition of fresh nor-
mal serum as a source of complement
may increase the sensitivity of the
test.
References
1. Judd WJ. Methods in immunohematology.
2nd ed. Durham, NC: Montgomery Scientific
Publications, 1994.
2. Salama A, Mueller-Eckhardt C. The role of me-
tabolite-specific antibodies in nomifensine-
dependent immune hemolytic anemia. N
Engl J Med 1985;313:469-74.
Methods Section 4: Investigation of a Positive DAT 791
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Methods Section 5: Hemolytic Disease of the Fetus and Newborn
Methods Section 5
Hemolytic Disease of the
Fetus and Newborn
Method 5.1. Indicator Cell
Rosette Test for
Fetomaternal Hemorrhage
Principle
This test detects D+ red cells in the blood
of a D– woman whose fetus or recently
delivered infant is D+. When reagent
anti-D is added to maternal blood con-
taining D+ fetal cells, fetal red cells be-
come coated with anti-D. When D+ re-
agent cells are subsequently added, easily
visible rosettes are formed, with several
redcellsclusteredaroundeachanti
-
body-coated D+ red cell.
Although the number of rosettes is
roughly proportional to the number of D+
red cells present in the original mixture,
this test provides only qualitative informa-
tion about fetal-maternal admixture. Speci-
mens giving a positive result should be sub-
jected to further testing to quantify the
number of fetal cells. The acid-elution pro-
cedure given below and flow cytometry are
acceptable choices. If a commercial test is
available, the directions in the package in-
sert should be followed.
Specimen
A 2% to 5% saline suspension of washed
red cells from a maternal blood sample.
Reagents
Prepared reagents are commercially avail-
able. The steps below can be used for in-
house preparation.
1. Negative control: a 2% to 5% saline
suspension of washed red cells known
to be D–.
2. Positivecontrol:a2%to5%saline
suspension of a mixture containing
approximately 0.6% D+ red cells and
99.4% D– red cells. The positive con-
trol can be prepared by adding 1 drop
793
Section 5
Copyright © 2005 by the AABB. All rights reserved.
ofa2%to5%suspensionofD+con
-
trol cells to 15 drops of a 2% to 5%
suspension of washed D– control cells.
Mix well, then add 1 drop of this cell
suspension to 9 drops of the 2% to
5% suspension of D– red cells. Mix well.
3. Indicator red cells: a 2% to 5% saline
suspension of group O, R2R2red cells.
Either enzyme-treated or untreated
cells in an enhancing medium can be
used.
4. Chemically modified or high-protein
reagent anti-D serum. Some mono-
clonal/polyclonal blended reagents
areunsuitableforuseinthismethod.
The antisera selected for use should
be evaluated for suitability before in-
corporation into the test procedure.
Procedure
1. To each of three test tubes, add 1 drop
(or volume specified in the manufac-
turer’s instructions) of reagent anti-D.
2. Add 1 drop of maternal cells, nega-
tive control cells, and positive control
cells to the appropriately labeled tubes.
3. Incubate at 37 C for 15 to 30 min-
utes, or as specified by the manufac-
turer’s instructions.
4. Wash cell suspensions at least four
times with large volumes of saline, to
remove all unbound reagent anti-D.
Decant saline completely after last
wash.
5. To the dry cell button, add 1 drop of
indicator cells and mix thoroughly to
resuspend them.
6. Centrifuge the tubes for 15 seconds
at 900 to 1000 ×g.
7. Resuspend cell button and examine
the red cell suspension microscopi-
cally at 100 to 150 ×magnification.
8. Examine at least 10 fields and count
the number of red cell rosettes in each
field.
Interpretation
The absence of rosettes is a negative re-
sult. With enzyme-treated indicator cells,
up to one rosette per three fields may oc-
cur in a negative specimen. With un-
treated indicator cells and an enhancing
medium, there may be up to six rosettes
per five fields in a negative test. The pres-
ence of more rosettes than these allow-
able maxima constitutes a positive result,
and the specimen should be examined
using a test that quantifies the amount of
fetal blood present.
Thepresenceofrosettesoragglutination
in the negative control tube indicates inad-
equate washing after incubation, allowing
residual anti-D to agglutinate the D+ indi-
cator cells. A strongly positive result is seen
with red cells from a woman whose Rh
phenotype is weak D rather than D–; mas-
sive fetomaternal hemorrhage may pro-
duce an appearance difficult to distinguish
from that caused by a weak D phenotype,
and a quantitative test for fetal cells should
be performed. If the infant’s cells are shown
to be weak D, a negative result on the
mother’s specimen should be interpreted
with caution. In this situation, a quantita-
tive test that does not rely on D antigen
expression should be performed.
Reference
Sebring ES, Polesky HF. Detection of fetal mater-
nal hemorrhage in Rh immune globulin candi-
dates. Transfusion 1982;22:468-71.
Method 5.2. Acid-Elution Stain
(Modified Kleihauer-Betke)
Principle
Fetal hemoglobin resists elution from red
cells under acid conditions, whereas adult
hemoglobin is eluted. When a thin blood
smear is exposed to an acid buffer, hemo-
794 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
globin from adult red cells is leached into
the buffer so that only the stroma remains;
fetal cells retain their hemoglobin and
can be identified by a positive staining
pattern. The approximate volume of feto-
maternal hemorrhage can be calculated
from the percentage of fetal red cells in
the maternal blood film.
Specimen
Maternal anticoagulated whole blood
sample.
Reagents
Prepared reagents are commercially avail-
able in kits. The steps below can be used
for in-house preparations.
1. Stock solution A (0.1 M of citric acid).
C6H8O7•H2O, 21.0 g, diluted to 1 liter
with distilled water. Keep it in the re-
frigerator.
2. Stock solution B (0.2 M of sodium
phosphate). Na2HPO4•7H2O, 53.6 g,
diluted to 1 liter with distilled water.
Keep it in the refrigerator.
3. McIlvaine’s buffer, pH 3.2. Add 75
mL of stock solution A to 21 mL of
stock solution B. Prepare fresh mix-
ture for each test. This buffer mix-
ture should be brought to room tem-
perature or used at 37 C.
4. Erythrosin B, 0.5% in water.
5. Harris hematoxylin (filtered).
6. 80% ethyl alcohol.
7. Positive control specimen. Ten parts
of anticoagulated adult blood, mixed
with one part of anticoagulated ABO-
compatible cord blood.
8. Negative control specimen. Anticoa-
gulated adult blood.
Procedure
1. Prepare very thin blood smears, di-
luting blood with an equal volume of
saline. Air dry.
2. Fix the smears in 80% ethyl alcohol
for 5 minutes.
3. Wash the smears with distilled wa-
ter.
4. Immerse the smears in McIlvaine’s
buffer, pH 3.2, for 11 minutes at room
temperature or 5 minutes at 37 C. This
reaction is temperature-sensitive.
5. Wash the smears in distilled water.
6. Immerse the smears in erythrosin B
for 5 minutes.
7. Wash the smears completely in dis-
tilled water.
8. Immerse the smears in Harris hema-
toxylin for 5 minutes.
9. Wash the smears in running tap wa-
ter for 1 minute.
10. Examine dry using 40×magnifica-
tion, count a total of 2000 red cells,
and record the number of fetal cells
observed.
11. Calculate the percent of fetal red cells
in the total counted.
Interpretation
1. Fetal cells are bright pink and re-
fractile; normal adult red cells ap-
pear as very pale ghosts.
2. The conversion factor used to indi-
cate the volume (as mL of whole
blood) of fetomaternal hemorrhage
is the percent of fetal red cells ob-
served times 50.
Note
The accuracy and precision of this proce-
dure are poor, and decisions regarding Rh
Immune Globulin (RhIG) dosage in mas-
sive fetomaternal hemorrhage should be
made accordingly. If there is a question
regarding the need for additional RhIG, it
is preferable to administer another dose
to prevent the risks of undertreatment. (See
Table 23-1 for dosage.)
Methods Section 5: Hemolytic Disease of the Fetus and Newborn 795
Copyright © 2005 by the AABB. All rights reserved.
Reference
Sebring ES. Fetomaternal hemorrhage—incidence
and methods of detection and quantitation. In:
Garratty G, ed. Hemolytic disease of the newborn.
Arlington, VA: AABB, 1984:87-118.
Method 5.3. Antibody
Titration Studies to Assist in
Early Detection of Hemolytic
Disease of the Fetus and
Newborn
Principle
Antibody titration is a semiquantitative
method of determining antibody concen-
tration. Serial twofold dilutions of serum
are prepared and tested for antibody ac-
tivity. The reciprocal of the highest dilu-
tion of plasma or serum that gives a 1+ re-
action is referred to as the titer (ie, 1 in
128 dilution; titer = 128).
In pregnancy, antibody titration is per-
formed to identify women with significant
levels of antibodies that may lead to
hemolytic disease of the fetus and newborn
(HDFN) and, for low-titer antibodies, to es-
tablish a baseline for comparison with
titers found later in pregnancy. Titration of
non-Rh antibodies should be undertaken
only after discussion with the obstetrician
about how the data will be used in the clini-
cal management of the pregnancy. The sig-
nificance of titers has been sufficiently es-
tablished only for anti-D (using a saline
technique).
Specimen
Serum for titration (containing potentially
significant unexpected antibodies to red
cell antigens, 1 mL). If possible, test the
current sample in parallel with the most
recent previously submitted (preceding)
sample from the current pregnancy.
Materials
1. Antihuman IgG: need not be heavy-
chain-specific.
2. Isotonic saline.
3. Volumetric pipettes, or equivalent:
0.1- to 0.5-mL delivery, with dispos-
able tips.
4. Red cells: group O reagent red cells,
2% suspension. (See note 1 regard-
ing the selection of red cells for test-
ing.) Avoid using Bg+ red cells be-
cause they may result in falsely high
values, especially with sera from
multiparous women.
5. IgG-coated red cells.
Quality Control
1. Test the preceding sample in parallel
with the most recent sample.
2. Prepare the dilutions using a sepa-
rate pipette for each tube. Failure to
do so will result in falsely high titers
because of carryover.
3. Confirm all negative reactions with
IgG-coated red cells (see step 9 be-
low).
Procedure
1. Using 0.5-mL volumes, prepare serial
twofold dilutions of serum in saline.
The initial tube should contain un-
diluted serum and the doubling di-
lutionrangeshouldbefrom1in2to
1 in 2048 (total of 12 tubes). (See
Method 3.7.)
2. Place 0.1 mL of each dilution into
appropriately labeled test tubes.
3. Add 0.1 mL of the 2% suspension of
red cells to each dilution. Alterna-
tively, for convenience, add 1 drop of
a solution of a 3% to 4% suspension
of red cells as supplied by the re-
agent manufacturer, although this
method is less precise.
796 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
4. Gently agitate the contents of each
tube; incubate at 37 C for 1 hour.
5. Wash the red cells four times with
saline; completely decant the final
wash supernatant.
6. To the dry red cell buttons thus ob-
tained, add anti-IgG according to the
manufacturer’s directions.
7. Centrifuge as for hemagglutination
tests.
8. Examine the red cells macroscopically;
grade and record the reactions.
9. Add IgG-coated red cells to all nega-
tive tests; recentrifuge and examine
the tests for macroscopic agglutina-
tion; repeat the testing if the tests with
IgG-coated red cells are nonreactive.
Interpretation
The titer is reported as the reciprocal of
the highest dilution of serum at which 1+
agglutination is observed. A titer ≥16 (this
value may vary according to the labora-
tory) is considered significant and may
warrant further monitoring for HDFN.
Notes
1. The selection of the most suitable
phenotype of red cells to use when
performing titration studies for HDFN
is controversial. Some workers select
red cells that have the strongest ex-
pression of antigen, such as R2R2for
anti-D. Others select red cells with the
phenotype that would be expected
in fetal circulation—ie, red cells that
express a single dose of the antigen,
such as R1r for testing for anti-D.
Whichever viewpoint is followed, it
is important that the laboratory be
consistent and use red cells of the
same phenotype for future titrations
to test the same patient’s serum.
2. Titration studies should be performed
upon initial detection of the anti-
body; save an appropriately labeled
aliquot of the serum (frozen at –20 C
or colder) for comparative studies
with the next submitted sample.
3. When the titer (eg, >16) and the anti-
body specificity have been associ-
ated with HDFN, it is recommended
that repeat titration studies be per-
formed every 2 to 4 weeks, beginning
at 18 weeks’ gestation; save an ali-
quot of the serum (frozen at –20 C or
colder) for comparative studies with
the next submitted sample.
4. When invasive procedures (eg, am-
niocentesis) have demonstrated fetal
compromise and are being used to
monitor the pregnancy, use the opti-
mal method for follow-up of fetal
well-being. However, if initial studies
do not show fetal compromise or the
Liley curve result is borderline, addi-
tional titrations may be helpful as a
means of following the pregnancy in
a less invasive manner.
5. Each institution should develop a
policy to ensure a degree of unifor-
mity in reporting and interpreting
antibody titers.
6. For antibodies to low-incidence an-
tigens, consider using putative pa-
ternal red cells, having established
that they express the antigen in ques-
tion.
7. Do not use enhancement techniques
[albumin, polyethylene glycol, low-
ionic-strength saline (LISS)] or en-
zyme-treated red cells because falsely
elevated titers may be obtained. Gel
testing is not recommended.
8. LISS should not be used as a diluent
in titration studies; nonspecific up-
take of globulins may occur in serum-
LISS dilutions.
9. Failure to obtain the correct results
may be caused by 1) incorrect tech-
nique, notably, failure to use sepa-
Methods Section 5: Hemolytic Disease of the Fetus and Newborn 797
Copyright © 2005 by the AABB. All rights reserved.
rate pipette tips for each dilution or
2) failure to adequately mix thawed
frozen serum.
References
1. Issitt PD, Anstee DJ. Applied blood group se-
rology. 4th ed. Durham, NC: Montgomery
Scientific Publications, 1998:1067-9.
2. Judd WJ, Luban NLC, Ness PM, et al. Prenatal
and perinatal immunohematology: Recom-
mendations for serologic management of the
fetus, newborn infant, and obstetric patient.
Transfusion 1990;30:175-83.
3. Judd WJ. Methods in immunohematology.
2nd ed. Durham, NC: Montgomery Scientific
Publications, 1994.
4. Judd WJ. Practice guidelines for prenatal and
perinatal immunhematology, revisited. Trans-
fusion 2001;41:1445-52.
798 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Methods Section 6: Blood Collection, Storage, and Component Preparation
Methods Section 6
Blood Collection, Storage,
and Component Preparation
Method 6.1. Copper Sulfate
Method for Screening Donors
for Anemia
Principle
This method estimates the hemoglobin
content of blood from its specific gravity.
A drop of blood in contact with copper
sulfate solution of specific gravity 1.053
becomes encased in a sac of copper pro-
teinate, which prevents dispersion of the
fluid or any change in specific gravity for
about 15 seconds. If the specific gravity of
the blood is higher than that of the solu-
tion, the drop will sink within 15 seconds;
if not, the drop will hesitate and remain
suspended or rise to the top of the solu-
tion. A specific gravity of 1.053 corre-
sponds to a hemoglobin concentration of
12.5 g/dL.
This is not a quantitative test; it shows
only whether the prospective donor’s he-
moglobin is below or above the acceptable
level of 12.5 g/dL. False-positive reactions
are rare; donors whose drop of blood sinks
nearly always have an acceptable hemoglo-
bin level. False-negative reactions occur
fairly commonly and can cause inappropri-
ate deferral.1,2 Measuring hemoglobin by
another method or determining hematocrit
sometimes reveals that the prospective do-
nor is acceptable.
Reagents and Materials
1. Copper sulfate solution at specific
gravity 1.053, available commer-
cially. Store it in tightly capped con-
tainers to prevent evaporation. The
solution should be kept at room tem-
perature or brought to room temper-
ature before it is used.
2. Sterile gauze, antiseptic wipes, and
sterile lancets.
3. Containers for the disposal of sharps
and other biohazardous materials.
4. Capillary tubes and dropper bulbs or
a device to collect capillary blood
without contact.
799
Section 6
Copyright © 2005 by the AABB. All rights reserved.
Procedure
1. Into a labeled, clean, dry tube or
bottle, dispense a sufficient amount
(atleast30mL)ofcoppersulfateso
-
lution to allow the drop to fall ap-
proximately 3 inches. Change the so-
lution daily or after 25 tests. Be sure
that the solution is adequately mixed
before beginning each day’s deter-
minations.
2. Clean the site of the skin puncture
thoroughly with antiseptic solution
and wipe it dry with sterile gauze.
3. Puncture the finger firmly, near the
end but slightly to the side, with a
sterile, disposable lancet or spring-
loaded, disposable needle system. A
good free flow of blood is important.
Donotsqueezethepuncturesitere-
peatedly because this may dilute the
drop of blood with tissue fluid and
lower the specific gravity.
4. Collect the blood in a capillary tube
without allowing air to enter the
tube.
5. Let one drop of blood fall gently
from the tube at a height about 1 cm
above the surface of the copper sul-
fate solution.
6. Observe for 15 seconds.
7. Dispose of lancets and capillary tubes
in appropriate biohazard containers.
Dispose of gauze appropriately;
gauze contaminated with droplets of
blood that subsequently dry such
that the item is stained but not
soaked or caked may be considered
nonhazardous.
Interpretation
1. If the drop of blood sinks, the do-
nor’s hemoglobin is at an acceptable
level for blood donation.
2. If the drop of blood does not sink,
the donor’s hemoglobin may not be
at an acceptable level for blood do-
nation. If time and equipment permit,
it is desirable to perform a quantita-
tive measurement of hemoglobin or
hematocrit.
Notes
1. A certificate of analysis from the man-
ufacturer should be obtained with
each new lot of copper sulfate solu-
tion.
2. Used solution should be disposed of
as biohazardous or chemical mate-
rial because of the blood in the con-
tainer. Refer to local and state laws
regarding disposal.
3. Usecaretopreventbloodfromcon-
taminating work surfaces, the donor’s
clothing, or other persons or equip-
ment.
4. Cover the container between uses to
prevent evaporation.
References
1. Lloyd H, Collins A, Walker W, et al. Volunteer
blood donors who fail the copper sulfate
screening test: What does failure mean, and
what should be done? Transfusion 1988;28:
467-9.
2. Morris MW, Davey FR. Basic examination of
blood. In: Henry JB, ed. Clinical diagnosis
and management by laboratory methods.
20th ed. Philadelphia: WB Saunders, 2001:
479-519.
Method 6.2. Arm Preparation
for Blood Collection
Detailed instructions are specific to each
manufacturerandshouldbefollowedas
indicated. The following procedure is
writteningeneraltermsasanexample.
800 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Principle
Iodophor compounds, or other sterilizing
compounds, are used to sterilize the veni-
puncture site before blood collection.
Materials
1. Scrub solution: Disposable povidone-
iodine scrub 0.75% or disposable
povidone-iodine swabstick 10%; avail-
able in prepackaged single-use form.
2. Preparation solution: 10% povidone-
iodine; available in prepackaged sin-
gle-use form.
3. Sterile gauze.
Procedure
1. Apply tourniquet or blood pressure
cuff; identify venipuncture site, then
release tourniquet or cuff.
2. Scrub area at least 4 cm (1.5 inches)
in all directions from the intended
site of venipuncture (ie, 8 cm or 3
inches in diameter) for a minimum
of 30 seconds with 0.7% aqueous so-
lution of iodophor compound. Excess
foam may be removed, but the arm
need not be dry before the next step.
3. Starting at the intended site of veni-
puncture and moving outward in a
concentric spiral, apply “prep” solu-
tion; let stand for 30 seconds or as
indicated by manufacturer.
4. Cover the area with dry, sterile gauze
until the time of venipuncture. After
the skin has been prepared, it must
not be touched again. Do not repal-
pate the vein at the intended veni-
puncture site.
Notes
1. For donors sensitive to iodine (tinc-
ture or povidone preparations), an-
other method (eg, ChloraPrep 2%
chlorhexidine and 70% isopropyl al-
cohol) should be designated by the
blood bank physician. Green soap
should not be used.
2. For donors sensitive to both iodine
and chlorhexidine, a method using
only isopropyl alcohol could be con-
sidered. The preferred procedure is
the use of a 30-second up-and-down
scrub, followed by enough time for
the skin to dry. A second scrub is
then applied. This method may re-
quire a variance from the Food and
Drug Administration.
3. Arm preparation methods approved
bytheFoodandDrugAdministra
-
tion are available at http://www.fda.
gov/cber/infosheets/armprep.htm.
Reference
Goldman M, Roy G, Frechette N, et al. Evaluation
of donor skin disinfection methods. Transfusion
1997;37:309-12.
Method 6.3. Phlebotomy and
Collection of Samples for
Processing and Compatibility
Tests
Principle
Blood for transfusion and accompanying
samples is obtained from prominent veins
on the donor’s arm, usually in the area of
the antecubital fossa.
Materials
1. Sterile collection bag containing an-
ticoagulant, with integrally attached
tubing and needle.
2. Metal clips and hand sealers.
3. Balance system to monitor volume
of blood drawn.
4. Sterile gauze and clean instruments
(scissors, hemostats, forceps).
Methods Section 6: Blood Collection, Storage, and Component Preparation 801
Copyright © 2005 by the AABB. All rights reserved.
5. Test tubes for sample collection.
6. Device for stripping blood in the
tubing.
7. Dielectric sealer (optional).
Procedure
1. Ask donor to confirm his or her iden-
tification.
2. Ensure that all labeling on blood con-
tainer, processing tubes, retention
segment, and donor records is cor-
rect.
3. Prepare donor’s arm as described in
Method 6.2.
4. Inspect bag for any defects and dis-
coloration. The anticoagulant and
additive solutions should be in-
spected for particulate contaminants.
5. Position bag below the level of the
donor’s arm.
a. If a balance system is used, be
sure the counterbalance is level
andadjustedfortheamountof
blood to be drawn. Unless metal
clips and a hand sealer are used,
make a very loose overhand knot
in the tubing. Hang the bag and
route the tubing through the
pinch clamp. A hemostat
should be applied to the tubing
before the needle is uncapped
to prevent air from entering the
line.
b. If a balance system is not used,
be sure to monitor the volume
of blood drawn.
6. Reapply tourniquet or inflate blood
pressure cuff. Ask the donor to open
and close hand until previously se-
lected vein is again prominent.
7. Uncover sterile needle and perform
the venipuncture immediately. A
clean, skillful venipuncture is essen-
tial for collection of a full, clot-free
unit. Once the bevel has penetrated
the skin, palpation of the skin above
the needle stem may be performed
with a gloved finger, provided the
needle is not touched. When the
needle position is acceptable, tape
the tubing to the donor’s arm to hold
the needle in place and cover the
site with sterile gauze.
8. Release the hemostat. Open the tem-
porary closure between the interior
of the bag and the tubing.
9. Ask the donor to open and close hand
slowly every 10 to 12 seconds during
collection.
10. Keep the donor under observation
throughout the donation process.
The donor should never be left unat-
tended during or immediately after
donation.
11. Mix blood and anticoagulant gently
and periodically (approximately ev-
ery 45 seconds) during collection.
Mixingmaybedonebyhandorby
continuous mechanical mixing.
12. Be sure blood flow remains fairly
brisk, so that coagulation activity is
not triggered. If there is continuous,
adequate blood flow and constant
agitation, rigid time limits are not
necessary. However, units requiring
more than 15 minutes to draw may
notbesuitableforpreparationof
Platelets, Fresh Frozen Plasma, or
Cryoprecipitated AHF. The time re-
quired for collection can be moni-
tored by indicating the time of phle-
botomy or the maximal allowable
time (start time plus 15 minutes) on
the donor record.
13. Monitor volume of blood being drawn.
If a balance is used, the device will
interrupt blood flow after the proper
amount has been collected. One mL
of blood weighs at least 1.053 g, indi-
cated by the minimum allowable
specific gravity for donors. A conve-
802 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
nient figure to use is 1.06 g/mL; a unit
containing 405 to 550 mL should
weigh 429 to 583 g plus the weight of
the container and anticoagulant. For
a 500-mL bag, this is 565 to 671 g.
14. Clamp the tubing near the veni-
puncture using a hemostat, metal
clip, or other temporary clamp. Re-
lease the blood pressure cuff/tourni-
quet to 20 mm Hg or less and fill the
tube(s) for blood processing sam-
ple(s) by a method that prevents
contamination of the contents of the
bag. This can be done in several ways.
a. If the blood collection bag con-
tains an inline needle, make an
additional seal with a hemo-
stat, metal clip, hand sealer, or
a tight knot made from previ-
ously prepared loose knot just
distal to the inline needle. Open
the connector by separating
the needles. Insert the proximal
needle into a processing test
tube, remove the hemostat, al-
low the tube to fill, and re-
clamp the tubing. The donor
needle is now ready for removal.
b. If the blood collection bag con-
tains an inline processing tube,
be certain that the processing
tube,orpouch,isfullwhenthe
collection is complete and the
original clamp is placed near
the donor needle. The entire
assembly may now be removed
from the donor.
c. If a straight-tubing assembly
set is used, the following proce-
dure should be followed. Place
a hemostat on the tubing, al-
lowing about four segments be-
tween the hemostat and the
needle. Pull tight the loose
overhand knot made in step 3.
Release the hemostat and strip
asegmentofthetubingfreeof
blood between the knot and
the needle (about 1 inch in
length). Reapply the hemostat
and cut the tubing in the strip-
ped area between the knot and
the hemostat. Fill the required
tube(s) by releasing the hemo-
stat and then reclamp the tub-
ing with the hemostat. Because
this system is open, Biosafety
Level 2 precautions should be
followed.
15. Deflate the cuff and remove the
tourniquet. Remove the needle from
the donor’s arm, if not already re-
moved. Apply pressure over the gauze
and ask the donor to raise his or her
arm (elbow straight) and hold the
gauze firmly over the phlebotomy
site with the other hand.
16. Discard the needle assembly into a
biohazard container designed to pre-
vent accidental injury to, and con-
tamination of, personnel.
17. Strip donor tubing as completely as
possible into the bag, starting at the
seal.Workquickly,topreventthe
blood from clotting in the tubing. In-
vert the bag several times to mix the
contents thoroughly; then allow the
tubing to refill with anticoagulated
blood from the bag. Repeat this pro-
cedure a second time.
18. Seal the tubing attached to the col-
lection bag into segments, leaving a
segment number clearly and com-
pletely readable. Attach a unit iden-
tification number to one segment to
be stored as a retention segment.
Knots, metal clips, or a dielectric
sealer may be used to make seg-
ments suitable for compatibility
testing. It must be possible to sepa-
rate segments from the unit without
breaking sterility of the bag. If a di-
Methods Section 6: Blood Collection, Storage, and Component Preparation 803
Copyright © 2005 by the AABB. All rights reserved.
electric sealer is used, the knot or
clip should be removed from the dis-
tal end of the tubing after creating a
hermetic seal.
19. Reinspect the container for defects.
20. Recheck numbers on the container,
processing tubes, donation record,
and retention segment.
21. Place blood at appropriate tempera-
ture. Unless platelets are to be re-
moved, whole blood should be placed
at 1 to 6 C immediately after collec-
tion. If platelets are to be prepared,
blood should not be chilled but
should be stored in a manner in-
tended to reach a temperature of 20
to 24 C until platelets are separated.
Platelets must be separated within 8
hours after collection of the unit of
Whole Blood.
Notes
1. If the needle is withdrawn and veni-
puncture is attempted again, prepa-
rationofthesitemustberepeatedas
in Method 6.2.
2. In addition to routine blood donor
phlebotomy, this procedure may be
adapted for use in therapeutic phle-
botomy.
References
1. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
2. Smith LG. Blood collection. In: Green TS,
Steckler D, eds. Donor room policies and pro-
cedures. Arlington, VA: AABB, 1985:25-45.
3. Huh YO, Lightiger B, Giacco GG, et al. Effect
of donation time on platelet concentrates
and fresh frozen plasma. Vox Sang 1989;56:
21-4.
4. Sataro P. Blood collection. In: Kasprisin CA,
Laird-Fryer B, eds. Blood donor collection
practices. Bethesda, MD: AABB, 1993:89-103.
Method 6.4. Preparation of
Red Blood Cells
Principle
Red Blood Cells (RBCs) are obtained by
removal of supernatant plasma from cen-
trifuged Whole Blood. The volume of
plasma removed determines the hema-
tocrit of the component. When RBCs are
preserved in CPDA-1, maximal viability
during storage requires an appropriate ra-
tio of cells to preservative. A hematocrit of
80% or lower ensures the presence of ade-
quate glucose for red cell metabolism for
up to 35 days of storage.
Materials
1. Freshly collected Whole Blood, ob-
tained by phlebotomy as described
in Method 6.3. Collect blood in a col-
lection unit with integrally attached
transfer container(s).
2. Plasma extractor.
3. Metal clips and hand sealer.
4. Clean instruments (scissors, hemo-
stats).
5. Dielectric sealer (optional).
6. Refrigerated centrifuge.
7. Scale.
Procedure
1. Centrifuge whole blood using a “heavy”
spin (see Method 7.4), with a tem-
perature setting of 4 C.
2. Place the primary bag containing
centrifuged blood on a plasma ex-
pressor, and release the spring, al-
lowing the plate of the expressor to
contact the bag.
3. Temporarily clamp the tubing be-
tween the primary and satellite bags
with a hemostat or, if a mechanical
804 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

sealer will not be used, make a loose
overhand knot in the tubing.
4. If two or more satellite bags are at-
tached, apply the hemostat to allow
plasma to flow into only one of the
satellite bags. Penetrate the closure
of the primary bag. A scale, such as a
dietary scale, may be used to mea-
sure the expressed plasma. Remove
the appropriate amount of plasma
to obtain the desired hematocrit.
5. Reapply the hemostat when the de-
sired amount of supernatant plasma
has entered the satellite bag. Seal the
tubing between the primary bag and
the satellite bag in two places.
6. Check that the satellite bag has the
same donor number as that on the
primary bag and cut the tubing be-
tween the two seals.
Notes
1. If blood was collected in a single bag,
modify the above directions as fol-
lows: after placing the bag on the
expressor, apply a hemostat to the
tubing of a sterile transfer bag, asep-
tically insert the cannula of the trans-
fer bag into the outlet port of the bag
of blood, release the hemostat, and
continue as outlined above. The ex-
piration date will change, however.
2. Collection of blood in an additive so-
lution allows removal of a greater
volume of plasma in step 4. After the
plasma has been removed, the addi-
tive solution is allowed to flow from
the attached satellite bag into the red
cells. This will result in a hematocrit
of 55% to 65%. Be sure that an ap-
propriate label and dating period are
used.
3. The removal of 230 to 256 g (225 to
250 mL) of plasma and preservation
of the red cells in the anticoagulant-
preservative solution will generally
result in a red cell component with a
hematocrit between 70% and 80%.
4. See Table 6.4-1 to prepare Red Blood
Cells with a specific (desired) hema-
tocrit.
Method 6.5. Preparation of
Prestorage Red Blood Cells
Leukocytes Reduced
Principle
The general principle and materials of
Method 6.4 apply, except that the red cells
are filtered using a special leukocyte re-
duction filter. All red cell leukocyte reduc-
tion filters licensed in the United States
remove platelets to some degree. Anti-
coagulated whole blood may be filtered,
from which only platelet-poor plasma
Methods Section 6: Blood Collection, Storage, and Component Preparation 805
Table 6.4-1. Removing Plasma from
Units of Whole Blood (To Prepare RBCs
in Anticoagulant-Preservative with a
Known Hematocrit)
Hematocrit of
Segment from
Whole Blood
Unit
Volume of
Plasma
to Be
Removed
Final
Hematocrit of
Red Blood Cell
Unit
40% 150 mL 56%
39% 150 mL 55%
38% 160 mL 55%
37% 165 mL 54%
36% 170 mL 54%
35% 180 mL 54%
34% 195 mL 55%
33% 200 mL 55%
Copyright © 2005 by the AABB. All rights reserved.
(leukocyte reduced) and red cells may be
made. Alternatively, the red cells may be
filtered in additive solution, potentially
allowing the preparation of platelets,
plasma, and red cells. Nonleukocyte-re-
duced red cells may also undergo leuko-
cyte reduction after preparation by at-
taching a leukocyte reduction filter con-
nected to a storage container using a ster-
ile connection device.
Procedure
1. Before centrifugation, the antico-
agulated Whole Blood may be fil-
tered by hanging the container up-
side down and allowing the blood to
flow through an in-line filter by grav-
ity into a secondary container. The
steps in Method 6.4 are then fol-
lowed (see note 2, for addition of ad-
ditive solution).
2. The anticoagulated Whole Blood
may be centrifuged with the in-line
filter attached. After centrifugation,
the plasma is expressed. The addi-
tive solution is added, and the red
cells in the additive solution are fil-
tered by gravity, as in step 1 above.
3. A red cell component prepared us-
ing Method 6.4 either in residual
anticoagulated plasma or in additive
solution (AS-1, AS-3, AS-5) may have
a secondary container with an in-
line filter attached using a sterile
connection device. Filtration can
proceed according to the manufac-
turer’s directions using gravity, as in
step 1. The timing of this filtration is
often within 24 hours of collection
but can be up to 5 days.
4. Red cells that are leukocyte reduced
are labeled “Red Blood Cells Leuko-
cytes Reduced.” There is no specific
label for prestorage leukocyte reduc-
tion.
Notes
1. If the collection system does not in-
clude an in-line filter, a sterile con-
nection device can be used to attach
a leukocyte reduction filter to the
collection system. The filter should
be used according to the manufac-
turer’s directions.
2. Whole-blood-derived platelets can
be manufactured only before leuko-
cyte reduction (see Method 6.14).
Method 6.6. Rejuvenation of
Red Blood Cells
Principle
Rejuvenation is a process to restore de-
pleted metabolites and improve the func-
tion and posttransfusion survival of stored
red cells. The rejuvenating solution is not
intended for intravenous administration;
after warm incubation with the solution,
the red cells are washed and either gly-
cerolized for frozen storage or kept at 1 to
6 C for transfusion within 24 hours.
The rejuvenating solution approved by
the Food and Drug Administration contains
pyruvate, inosine, phosphate, and adenine.
Its use is permitted only with RBCs pre-
pared from Whole Blood collected into
CPD, CP2D, or CPDA-1, and it may be
added at any time between 3 days after col-
lection of the blood and 3 days after the ex-
piration of the unit. However, the use of the
rejuvenation solution with RBC units be-
fore 14 days of storage is not routinely ac-
cepted because the treated cells may develop
supranormal levels of 2,3-diphospho-
glycerate, which impairs oxygen uptake.
Reagents and Materials
1. RBCs stored at 1 to 6 C and prepared
from Whole Blood collected in CPD
or CPDA-1. After collection, RBCs
806 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
suspended in CPD from day 3 to day
24(orinCPDA-1fromday3today
38)maybeused.Thesolutionisnot
approved for use with cells stored in
additive solutions.
2. Red Blood Cell rejuvenation solu-
tion, in 50-mL sterile vial (Rejuvesol,
Cytosol Laboratories, Braintree, MA);
also called rejuvenating solution.
3. Waterproof plastic bag.
4. Metal clips and hand sealer.
5. Sterile airway.
Procedure
1. Connect the container of rejuvenat-
ing solution to the RBCs, using a
transfer set and aseptic technique.
2. Allow 50 mL of rejuvenating solution
to flow by gravity into the container
of red cells. Gently agitate the cell/
solution mixture during this addition.
Note: A sterile airway is required if
the solution is in a bottle.
3. Sealthetubingnearthebloodbag
and incubate the mixture for 1 hour
at 37 C. Either a dry incubator or cir-
culating waterbath can be used. If
placed in a waterbath, the container
should be completely immersed; use
of a waterproof overwrap is essential
to prevent contamination.
4. For use within 24 hours, wash the re-
juvenated cells with saline (2 L un-
buffered 0.9% NaCl) by the use of an
approved protocol. Storage of the
washed cells from the start of the
wash procedure should be at 1 to 6 C
fornolongerthan24hours.
5. If the rejuvenated cells are to be cryo-
preserved, the standard glyceroli-
zation protocol adequately removes
the rejuvenation solution from the
processed cells. Expiration date re-
mains 10 years from the date of col-
lection.
6. Be sure that units are appropriately
labeled and that all applicable re-
cords are complete.
Reference
Valeri CR, Zaroules CG. Rejuvenation and freezing
of outdated stored human red cells. N Engl J Med
1972;287:1307-13.
Method 6.7. Red Cell
Cryopreservation Using
High-Concentration
Glycerol—Meryman Method
Principle
Cryoprotectiveagentsmakepossiblethe
long-term (10 or more years) preservation
of red cells in the frozen state. High-con-
centration glycerol is particularly suitable
for this purpose. A practical method for
RBCs collected in a 450-mL bag is de-
scribed below.
Materials
(See Chapter 8 for additional information
on frozen cellular components.)
1. Donor blood, collected into CPD,
CD2D, CPDA-1, or AS.
a. Complete all blood processing
on units intended for freezing.
b. RBCs preserved in CPD or CPDA-1
may be stored at 1 to 6 C for up
to 6 days before freezing.
c. RBCs preserved in AS-1 and AS-3
may be stored at 1 to 6 C for up
to 42 days before freezing.
d. RBCs that have undergone re-
juvenation (see Method 6.6) may
be processed for freezing up to
3 days after their original expi-
ration.
e. RBCs in any preservative solu-
tion that have been entered for
Methods Section 6: Blood Collection, Storage, and Component Preparation 807
Copyright © 2005 by the AABB. All rights reserved.
processing must be frozen with-
in 24 hours of puncturing the
seal.
2. Storage containers, either polyvinyl
chloride or polyolefin bags.
3. 6.2 M of glycerol lactate solution
(400 mL).
4. Cardboard or metal canisters for
freezing.
5. Hypertonic (12%) sodium chloride
solution.
6. 1.6% NaCl, 1 liter for batch wash.
7. Isotonic (0.9%) NaCl with 0.2% dex-
trose solution.
8. 37 C waterbath or 37 C dry warmer.
9. Equipment for batch or continuous-
flow washing, to deglycerolize cells
frozen in high-concentration glyc-
erol.
10. Freezer tape.
11. Freezer (–65 C or colder).
Procedure
Preparing RBCs for Glycerolization
1. Prepare RBCs from Whole Blood
units by removal of supernatant an-
ticoagulant-preservative or additive
solution. Weigh the RBC unit to be
frozen and obtain the net weight of
the RBCs. The combined weight of
the cells and the collection bag
should be between 260 g and 400 g.
2. Underweight units can be adjusted
to approximately 300 g either by the
addition of 0.9% NaCl or by the re-
moval of less plasma than usual. Re-
cord the weight and, if applicable,
document the amount of NaCl added.
3. Record the Whole Blood number,
ABO group and Rh type, anticoagu-
lant, date of collection, date frozen,
expiration time, and the identifica-
tion of the person performing the
procedure. If applicable, document
the lot number of the transfer bag.
4. Warm the red cells and the glycerol
to at least 25 C by placing them in a
dry warming chamber for 10 to 15
minutes or by allowing them to re-
main at room temperature for 1 to 2
hours. The temperature must not
exceed 42 C.
5. Apply a “Red Blood Cells Frozen” la-
bel to the freezing bag in which the
unit will be frozen. The label must
also include: name of the facility
freezing the unit; Whole Blood num-
ber; ABO group and Rh type; date
collected; date frozen; the cryopro-
tective agent used; and the expira-
tion date.
Glycerolization
1. Document the lot numbers of the
glycerol, the freezing bags, and, if
used, the 0.9% NaCl.
2. Place the container of red cells on a
shaker and add approximately 100
mL of glycerol as the red cells are
gently agitated.
3. Turn off the shaker and allow the
cells to equilibrate, without agita-
tion, for 5 to 30 minutes.
4. Allow the partially glycerolized cells
to flow by gravity into the freezing bag.
5. Add the remaining 300 mL of glyc-
erol slowly in a stepwise fashion,
with gentle mixing. Add smaller vol-
umes of glycerol for smaller volumes
of red cells. The final glycerol con-
centration is 40% w/v. Remove any
air from the bag.
6. Allow some glycerolized cells to flow
back into the tubing so that seg-
ments can be prepared.
7. Maintain the glycerolized cells at
temperatures between 25 and 32 C
until freezing. The recommended in-
808 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
terval between removing the RBC
unit from refrigeration and placing
the glycerolized cells in the freezer
should not exceed 4hours.
Freezing and Storage
1. Place the glycerolized unit in a card-
board or metal canister and place it
in a freezer at –65 C or colder.
2. Label the top edge of the canister
with freezer tape marked with the
Whole Blood number, ABO group
and Rh type, the date frozen, and the
expiration date.
3. Do not bump or handle the frozen
cells roughly.
4. Thefreezingrateshouldbelessthan
10 mL/minute.
5. Store the frozen RBCs at –65 C or
colder for up to 10 years. For blood
of rare phenotypes, a facility’s medi-
cal director may wish to extend the
storage period. The unusual nature
of such units and the reason for re-
taining them past the routine 10-
year storage period must be docu-
mented.
Thawing and Deglycerolizing
1. Putanoverwrapontheprotective
canister containing the frozen cells
and place it in either a 37 C water-
bath or 37 C dry warmer.
2. Agitate it gently to speed thawing.
The thawing process takes at least 10
minutes. Thawed cells should be at
37 C.
3. After the cells have thawed, use a
commercial instrument for batch or
continuous-flow washing to degly-
cerolize cells. Follow the manufac-
turer’s instructions.
4. Record the lot numbers and manu-
facturer of all the solutions and soft-
ware used. Apply a “Red Blood Cells
Deglycerolized” label to the transfer
packandbesurethatthelabelin
-
cludes identification of the collect-
ing facility, the facility preparing the
deglycerolized cells, the ABO group
and Rh type of the cells, the Whole
Blood number, and the expiration
date and time.
5. Dilute the unit with a quantity of
hypertonic (12%) NaCl solution ap-
propriate for the size of the unit. Al-
low it to equilibrate for approxi-
mately5minutes.
6. Wash the cells with 1.6% NaCl until
deglycerolization is complete. Ap-
proximately 2 liters of wash solution
are required. To check for residual
glycerol, see Method 6.8.
7. Suspend the deglycerolized cells in
isotonic (0.9%) saline with 0.2% dex-
trose.
8. Fill the integrally attached tubing
with an aliquot of cells sealed in
such a manner that it will be avail-
able for subsequent compatibility
testing.
9. Deglycerolized RBCs must be stored
at 1 to 6 C for no longer than 24 hours.
(A closed system has been licensed
that allows storage of deglycerolized
RBCsat1to6Cfor2weeks.)
Notes
1. An aliquot of the donor’s serum or
plasma should be frozen and stored
at –65 C or colder for possible future
use if new diagnostic tests are imple-
mented.
2. When new diagnostic tests have
been implemented and stored units
do not have aliquots available for
testing, the units may have to be is-
sued with a label stating that the test
has not been performed. The reason
for distributing an untested compo-
Methods Section 6: Blood Collection, Storage, and Component Preparation 809
Copyright © 2005 by the AABB. All rights reserved.
nent should be documented. If a
specimen from the donor is obtained
and tested after the unit was stored,
the date of testing should be noted
ontheunitwhenitisissued.
Reference
Meryman HT, Hornblower M. A method for freez-
ing and washing RBCs using a high glycerol con-
centration. Transfusion 1972;12:145-56.
Method 6.8. Red Cell
Cryopreservation Using
High-Concentration
Glycerol—Valeri Method
Principle
RBCs collected in an 800-mL primary col-
lection bag in CPDA-1 and stored at 1 to 6
C for 3 to 38 days can be biochemically re-
juvenated and frozen with 40% w/v glyc-
erol in the 800-mL primary container. See
Method 6.6 for additional information.
Materials
1. Quadruple plastic bag collection
system with 800-mL primary bag.
2. Hand sealer clips.
3. Empty 600-mL polyethylene cryo-
genic vials (eg, Corning 25702 or
Fisher 033746).
4. Sterile connection device with wa-
fers.
5. Freezer tape.
6. 600-mL transfer bag.
7. 50 mL of Red Blood Cell Processing
Solution (Rejuvesol, Cytosol Labora-
tories, Braintree, MA).
8. Heat-sealable 8" × 12" plastic bags.
9. Rejuvenation harness (Fenwal 4C1921
or Cutter 98052).
10. Sterile filtered airway needle (BD
5200), for Fenwal rejuvenation har-
ness only.
11. 500 mL of glycerolyte 57 solution
(Fenwal 4A7833) or 500 mL of 6.2 M
glycerolization solution (Cytosol
PN5500).
12. Labels—Red Blood Cells Frozen Re-
juvenated.
13. Corrugated cardboard storage box
(7" × 5.5" × 2" outside dimensions).
14. Heat sealing device.
15. Plastic bag for overwrapping.
Procedure
Preparing RBCs for Glycerolization
1. Collect 450 mL of Whole Blood in
the primary bag. Invert the bag, fold
it about 2 inches from the base, se-
cure the fold with tape, and place
the bag upright in a centrifuge. Cen-
trifuge and remove all visible super-
natant plasma. The hematocrit of
the RBC unit must be 75% ± 5%.
2. Store RBCs at 1 to 6 C in the 800-mL
primary bag, along with the adapter
port on the tubing that connects the
primary bag and transfer pack.
3. Centrifuge the stored cells to remove
all visible plasma before undertaking
rejuvenation. The gross and net
weights of the RBCs should not ex-
ceed 352 g and 280 g, respectively.
4. Transfer the plasma to the integrally
connected transfer pack, fold the in-
tegral tubing, and replace the hand
sealer clip (not crimped).
5. Attach an empty 600-mL transfer
pack to the integral tubing of the pri-
mary collection bag, using a sterile
connection device.
6. Transfer 1 mL of plasma to each of
threecryogenicvialstobeusedfor
future testing.
810 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Biochemical Modification of the Cells
1. Using the Fenwal Rejuvenation Har-
ness: Aseptically insert the needle of
the Y-type Fenwal Harness into the
rubberstopperofa50-mLRed
Blood Cell Processing Solution bot-
tle and the coupler of the set into the
adapter port of the primary collec-
tion bag. Insert the filtered airway
needle into the rubber stopper of the
Red Blood Cell Processing Solution
bottle.
2. Using the Cutter Rejuvenation Har-
ness: Aseptically insert the vented
white spike with the drip chamber
into the rubber stopper of the Red
Blood Cell Processing Solution bot-
tle and the nonvented spike into the
special adapter port on the primary
collection bag.
3. With gentle manual agitation, allow
50 mL of Red Blood Cell Processing
Solution to flow directly into the red
cells.
4. Heat-seal the tubing of the harness
setthatconnectstheRedBloodCell
Processing Solution to the adapter
port. The second tubing of the har-
ness Y-set is used to add glycerol
(see below).
5. Completely overwrap the 800-mL
primary bag, the integrally con-
nected empty transfer pack, and the
coupler of the Y-type harness and
incubate them in a 37 C waterbath
for 1 hour.
Glycerolization
1. Remove the numbered crossmatch
segments, leaving the initial seg-
ment and number attached to the
collection bag. Weigh the unit.
2. Determine the amount of glycerol to
be added, based on the gross or net
weight of the unit, from the values
showninTable6.8-1.
3. Aseptically insert the coupler of the
rejuvenation harness into the outlet
port of the rubber stopper on the
glycerol solution bottle. For the
Fenwal harness only, insert a filtered
airway needle into the vent portion
of the glycerol bottle stopper.
4. Placethebagonashaker.Addthe
amount of glycerol shown in Table
6.8-1 for the first volume while the
bag is shaking at low speed (180 os-
cillations/minute).
5. Equilibrate the mixture for 5 min-
utes without shaking and add the
second volume. Equilibrate it for 2
minutes. Add the third volume of
glycerol, using vigorous manual
shaking.
Methods Section 6: Blood Collection, Storage, and Component Preparation 811
Table 6.8-1. Amount of Glycerol Needed for Different Weights of Red Cell Units
Gross Weight
of Unit
(grams)*
Net Weight
of Unit
(grams)
Initial
Addition of
Glycerol (mL)
Second
Addition of
Glycerol (mL)
Third
Addition of
Glycerol (mL)
Total
Glycerol
Added (mL)
222-272 150-200 50 50 250 350
273-312 201-240 50 50 350 450
313-402 241-330 50 50 400 500
*Weight of the empty 800-mL primary bag with the integrally attached transfer pack and the adapter port is 72 grams
(average).
Copyright © 2005 by the AABB. All rights reserved.
6. Heat-seal the tubing between the
empty bottle of glycerol and the tub-
ing proximal to the adapter port. En-
sure that the transfer pack remains
integrally attached to the primary
collection bag.
7. Centrifuge the mixture of red cells
and glycerol and transfer all visible
supernatant glycerol to the transfer
pack, resuspend, and mix. Note: This
step differs from Method 6.7.
8. Sealthetubing4"fromtheprimary
collection bag, detach the transfer
pack containing the supernatant fluid,
and discard it.
9. Affix an overlay blood component
label, the facility label, and an ABO/
Rh label. Record the expiration date
on the label.
10. Weigh the unit just before freezing
and record the weight.
11. Fold over the top portion of the pri-
mary bag (approximately 2"). Place
theprimarybagintoaplasticbag
overwrap and heat-seal the outer
bag across the top so that there is as
littleairaspossiblebetweenthebags.
12. Place one vial of plasma and the
plastic bag containing the glyceroli-
zed red cells in the cardboard box.
Store the other two vials, suitably
identified, at –65 C or colder for fu-
ture testing, if needed.
13. Affix a “Red Blood Cells Frozen Reju-
venated” label, an ABO/Rh label, a
facility label, and the original unit
number on the outside of the box.
Record separately or affix on the
cardboard box the collection, freez-
ing, and expiration dates.
14. Freeze the unit in a –80 C freezer. No
more than 4 hours should be al-
lowed to elapse between the time
the unit was removed from the 4 C
refrigerator and the time the cells
are placed in the –80 C freezer.
Thawing and Deglycerolization
See Method 6.7. Note, however, that the
supernatant glycerol is removed before
freezing. Therefore, only two salt solu-
tions (the hypertonic 12% saline and the
0.9% saline-0.2% dextrose solution) are
used in the deglycerolization process.
References
1. Rejuvesol Package insert. Braintree, MA:
Cytosol Laboratories, 2002.
2. Valeri CR, Ragno G, Pivacek LE, et al. A
multicenter study of in vitro and in vivo val-
ues in human RBCs frozen with 40% (wt/vol)
glycerol and stored after deglycerolization for
15 days at 4 C in AS-3: Assessment of RBC
processing in the ACP 215. Transfusion 2001;
41:933-9.
Method 6.9. Checking the
Adequacy of
Deglycerolization of Red
Blood Cells
Principle
Glycerolization of red cells for frozen stor-
age creates a hyperosmolar intracellular
fluid, which must be restored to physio-
logically compatible levels before the cells
are transfused. Inadequately deglycerolized
red cells will be hemolyzed by contact
with normal saline, or with serum or
plasma if subjected to crossmatching.
During deglycerolization, the last solution
in contact with the cells is normal saline.
The easiest way to determine adequacy of
glycerol removal is to determine the level
of free hemoglobin (mg/dL) in the final
wash. An adequate estimate of hemolysis
can be achieved by comparing the color
of the final wash fluid with the blocks in a
commercially available color comparator.
Alternatively, normal saline can be added
to an aliquot of deglycerolized cells and
812 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
the color of the supernatant fluid evalua-
ted against the color comparator.
Materials and Equipment
1. Semiautomated instrument for de-
glycerolizing cryopreserved RBCs.
2. Transparent tubing, as part of dis-
posable material used to deglycerol-
ize individual unit.
3. Color comparator, available com-
mercially.
Procedure
1. Interrupt the last wash cycle at a
point when wash fluid is visible in
the tubing leading to the disposal bag.
2. Hold the comparator block next to
an accessible segment of tubing,
against a well-lighted white back-
ground.
3. Note coloration of the wash fluid,
whichshouldbenostrongerthan
the block, indicating 3% hemolysis
(3% of the red cells are hemolyzed).
4. If the level of hemolysis is excessive,
continue the wash process until the
color is within acceptable limits.
5. Record observation for the individ-
ual unit and for the quality assurance
program.
6. If unacceptable hemolysis occurs re-
peatedly, document corrective action.
Method 6.10. Preparation of
Fresh Frozen Plasma from
Whole Blood
Principle
Plasma is separated from cellular blood
elements and frozen to preserve the activ-
ity of labile coagulation factors. Plasma
must be placed in the freezer within the
time frame required for the anticoagulant
or collection process.
Materials
1. Freshly collected Whole Blood, ob-
tained by phlebotomy as described
in Method 6.3, in a collection unit
with integrally attached transfer con-
tainer(s).
2. Metal clips and hand sealer.
3. Clean instruments (scissors, hemo-
stats).
4. Dielectric sealer (optional).
5. Plasma extractor.
6. Freezing apparatus.
7. Refrigerated centrifuge.
8. Scale.
Procedure
1. Centrifuge blood soon after collection,
usinga“heavy”spin(seeMethod7.4).
Use a refrigerated centrifuge at 1 to 6
C unless also preparing platelets (see
Method 6.13).
2. Place the primary bag containing
centrifuged blood on a plasma ex-
tractor and place the attached satel-
lite bag on a scale adjusted to zero.
Express the plasma into the satellite
bag and weigh the plasma.
3. Sealthetransfertubingwithadi
-
electric sealer or metal clips but do
not obliterate the segment numbers
ofthetubing.Placeanotherseal
nearer the transfer bag.
4. Label the transfer bag with the unit
number before it is separated from
the original container. Record the
volume of plasma on the label.
5. Cut the tubing between the two seals.
The tubing may be coiled and taped
against the plasma container, leaving
thesegmentsavailableforanytest
-
ing desired.
Methods Section 6: Blood Collection, Storage, and Component Preparation 813
Copyright © 2005 by the AABB. All rights reserved.
6. Place the plasma at –18 C or colder
within the time frame required for the
anticoagulant or collection process.
Method 6.11. Preparation of
Cryoprecipitated AHF from
Whole Blood
Principle
Coagulation Factor VIII (antihemophilic
factor, AHF) can be concentrated from
freshly collected plasma by cryoprecipita-
tion. Cryoprecipitation is accomplished
by slow thawing, at 1 to 6 C, plasma that
has been prepared for freezing within the
time frame required for the anticoagulant
or collection process.
Materials
1. Freshly collected Whole Blood, ob-
tained by phlebotomy as described
in Method 6.3, in a collection unit
with at least two integrally attached
transfer containers.
2. Metal clips and hand sealer.
3. Clean instruments (scissors, hemo-
stats).
4. Dielectric sealer (optional).
5. Plasma extractor.
6. Refrigerated centrifuge.
7. Freezing apparatus: suitable freezing
devices include blast freezers or me-
chanical freezers capable of main-
taining temperatures of –18 C or
colder; dry ice; or an ethanol dry ice
bath. In a bath of 95% ethanol and
chipped dry ice, freezing will be
complete in about 15 minutes.
8. 1 to 6 C circulating waterbath or re-
frigerator.
9. Scale.
Procedure
1. Collect blood in a collection unit
with two integrally attached transfer
containers.
2. Centrifuge blood shortly after collec-
tion at 1 to 6 C, using a “heavy” spin
(see Method 7.4). Collect at least 200
mL (205 g) of cell-free plasma for
processing into cryoprecipitate.
3. Promptly place plasma in a freezing
device so that freezing is started
within the time frame required for
the anticoagulant or collection pro-
cess. Plasma containers immersed in
liquid must be protected with a plas-
tic overwrap.
4. Allow the frozen plasma to thaw at 1
to6Cbyplacingthebagina1to6C
circulating waterbath or in a refrig-
erator. If thawed in a waterbath, use
a plastic overwrap (or other means)
to keep container ports dry.
5. Whentheplasmahasaslushycon-
sistency, separate liquid plasma from
the cryoprecipitate by one of the
procedures below:
a. Centrifuge the plasma at 1 to 6
C using a “heavy” spin. Hang the
baginaninvertedpositionand
allow the separated plasma to
flow rapidly into the transfer
bag, leaving the cryoprecipitate
adhering to the sides of the pri-
mary bag. Separate the cryo-
precipitate from the plasma
promptly, to prevent the cryo-
precipitate from dissolving and
flowingoutofthebag.Tento
15 mL of supernatant plasma
maybeleftinthebagforresus
-
pension of the cryoprecipitate
after thawing. Refreeze the cryo-
precipitate immediately.
b. Place the thawing plasma in a
plasma expressor when appro-
814 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
ximately one-tenth of the con-
tents is still frozen. With the
baginanuprightposition,al
-
low the supernatant plasma to
flow slowly into the transfer
bag, using the ice crystals at
the top as a filter. The cryo-
precipitate paste will adhere to
the sides of the bag or to the ice.
Seal the bag when about 90% of
the cryoprecipitate-reduced
plasma has been removed and
refreeze the cryoprecipitate im-
mediately.
6. The cryoprecipitate should be re-
frozen within 1 hour of thawing.
Store at –18 C or colder, preferably
–30 C or colder, for up to 12 months
from the date of blood collection.
Note
Cryoprecipitated AHF may be prepared
fromFreshFrozenPlasmaatanytime
within 12 months of collection. The expi-
ration date of Cryoprecipitated AHF is 12
months from the date of phlebotomy, not
from the date it was prepared.
Method 6.12. Thawing and
Pooling Cryoprecipitated
AHF
Principle
Cryoprecipitated AHF should be rapidly
thawed at 30 to 37 C but should not re-
main at this temperature once thawing is
complete. The following method permits
rapid thawing and pooling of this product.
Materials
1. Circulating waterbath at 37 C (water-
baths designed for thawing plasma
are available commercially, as are
specially designed dry heat devices).
2. Medication injection ports.
3. Sterile 0.9% sodium chloride for in-
jection.
4. Syringes and needles.
Procedure
1. Cover the container with a plastic
overwrap to prevent contamination
of the ports with unsterile water, or
use a device to keep the containers
upright with the ports above water.
2. Resuspend the thawed precipitate
carefully and completely, either by
kneading it into the residual 10 to 15
mL of plasma or by adding approxi-
mately 10 mL of 0.9% sodium chlo-
ride and gently resuspending.
3. Pool by inserting a medication injec-
tion site into a port of each bag. As-
pirate contents of one bag into a sy-
ringe and inject into the next bag. Use
the ever-increasing volume to flush
each subsequent bag of as much dis-
solved cryoprecipitate as possible,
until all contents are in the final bag.
4. Thawed Cryoprecipitated AHF must
be stored at room temperature. If
pooled,itmustbeadministered
within 4 hours. Thawed single units,
if not entered, must be administered
within 6 hours of thawing if intend-
ed for replacement of Factor VIII.
Pools of thawed individual units may
not be refrozen.
Method 6.13. Preparation of
Platelets from Whole Blood
Principle
Platelet-rich plasma is separated from
Whole Blood by “light-spin” centrifugation
and the platelets are concentrated by
Methods Section 6: Blood Collection, Storage, and Component Preparation 815
Copyright © 2005 by the AABB. All rights reserved.
“heavy-spin” centrifugation, with subse-
quent removal of supernatant plasma (see
Method 7.4).
Materials
1. Freshly collected Whole Blood, ob-
tained by phlebotomy as described
in Method 6.3, in a collection unit
with two integrally attached transfer
containers. The final container must
be a plastic approved for platelet
storage. Keep blood at room temper-
ature (20 to 24 C) before separating
platelet-rich plasma from the red cells.
This separation must take place
within 8 hours of phlebotomy.
2. Metal clips and hand sealer.
3. Scissors, hemostats.
4. Plasma extractor.
5. Dielectric sealer (optional).
6. Centrifuge, calibrated as in Method
7.4.
7. Scale.
8. Rotator.
Procedure
1. Do not chill the blood at any time
before or during platelet separation.
If the temperature of the centrifuge
is 1 to 6 C, set the temperature con-
trol of the refrigerated centrifuge at
20 C and allow the temperature to
risetoapproximately20C.Centri
-
fuge the blood using a “light” spin
(see Method 7.4).
2. Express the platelet-rich plasma into
the transfer bag intended for platelet
storage. Seal the tubing twice be-
tween the primary bag and Y con-
nector of the two satellite bags and
cut between the two seals. Place the
red cells at 1 to 6 C.
3. Centrifuge the platelet-rich plasma
at 20 C using a “heavy” spin (see
Method 7.4).
4. Express the platelet-poor plasma
into the second transfer bag and seal
the tubing. Some plasma should re-
main on the platelet button for stor-
age, but no exact volume can be des-
ignated. AABB Standards for Blood
Banks and Transfusion Services re-
quires that sufficient plasma remain
with the platelet concentrate to main-
tain the pH at 6.2 or higher for the
entire storage period. This usually
requires a minimum of 35 mL of
plasma when storage is at 20 to 24 C,
but50to70mLispreferable.
5. The platelet concentrate container
should be left stationary, with the la-
bel side down, at room temperature
for approximately 1 hour.
6. Resuspend the platelets in either of
the following ways:
a. Manipulate the platelet con-
tainer gently by hand to achieve
uniform resuspension.
b. Place the container on a rotator
at room temperature. The slow,
gentle agitation should achieve
uniform resuspension within 2
hours.
7. Maintain the platelet suspensions at
20 to 24 C with continuous gentle
agitation.
8. Platelets should be inspected before
issue to ensure that no platelet ag-
gregates are visible.
Notes
The platelet-reduced plasma may be frozen
promptlyandstoredasFreshFrozen
Plasma (FFP), if the separation and freez-
ing are completed within the time frame
required for the anticoagulant or collec-
tion process. The volume of FFP prepared
after platelet preparation will be substan-
tially less than that prepared directly from
Whole Blood.
816 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Reference
Silva MA, ed. Standards for blood banks and trans-
fusion services. 23rd ed. Bethesda, MD: AABB,
2005:30.
Method 6.14. Preparation of
Prestorage Platelets
Leukocytes Reduced from
Whole Blood
Principle
Prestorage leukocyte-reduced platelets
may be prepared from whole blood using
in-line filtration of the platelet-rich plasma
(PRP). The resulting intermediate product
is a filtered PRP, from which a leukocyte-
reduced platelet concentrate and leuko-
cyte-reduced plasma may be manufac-
tured. Materials and procedures are the
same as for Method 6.13, except that the
PRP is expressed through an in-line filter.
References
1. Sweeney JD, Holme S, Heaton WAL, Nelson E.
Leukodepleted platelet concentrates pre-
pared by in-line filtration of platelet rich
plasma. Transfusion 1995;35:131-6.
2. Sweeney JD, Kouttab N, Penn LC, et al. A
comparison of prestorage leukoreduced
whole blood derived platelets with bedside
filtered whole blood derived platelets in
autologous stem cell transplant. Transfusion
2000;40:794-800.
Method 6.15. Removing
Plasma from Platelet
Concentrates (Volume
Reduction)
Principle
Although optimal storage of platelets re-
quires an adequate volume of plasma, a
few patients may not tolerate large-vol-
ume infusion. Stored platelets may be
centrifuged and much of the plasma re-
moved shortly before transfusion, but
appropriate resuspension is essential.
The platelets must remain at room tem-
perature, without agitation, for 20 to 60
minutes before resuspension into the re-
maining plasma. Transfusion must take
place within 4 hours of the time the
platelet bag was entered. Volume reduc-
tion can be performed on individual or
pooled units.
No consensus exists regarding the opti-
mal centrifugation rate. One study1found
35% to 55% platelet loss in several units
centrifuged at 500 × gfor 6 minutes, com-
pared with 5% to 20% loss in units centri-
fuged at 5000 × gfor 6 minutes or 2000 × g
for 10 minutes. The authors recommend
2000 × gfor 10 minutes, to avoid any risk
that a higher centrifugal force might inflict
on the plastic container. A study by Moroff
et al2found mean platelet loss to be less
than 15% in 42 units centrifuged at 580 × g
for 20 minutes. High gforces are of theoret-
ical concern because they may damage the
platelets when they are forced against the
wall of the container and also increase the
possibility of container breakage.
Materials
1. Platelet concentrate(s), prepared as
described in Method 6.13.
2. Metal clips and hand sealer.
3. Scissors, hemostats.
4. Dielectric sealer (optional).
5. Centrifuge, calibrated as in Method
7.4.
6. Plasma extractor.
Procedure
1. Pool platelets, if desired, into a trans-
fer pack, using standard technique.
Single platelet concentrates may
need volume reduction for pediatric
Methods Section 6: Blood Collection, Storage, and Component Preparation 817
Copyright © 2005 by the AABB. All rights reserved.
recipients. Apheresis components may
be processed directly.
2. Centrifuge at 20 to 24 C, using one of
the following protocols:
a. 580 ×gfor 20 minutes.
b. 2000 ×gfor 10 minutes.
c. 5000 ×gfor 6 minutes.
3. Without disturbing the contents,
transfer the bag to a plasma extrac-
tor. Remove all but 10 to 15 mL plasma
from single units, or somewhat more
volume, proportionately, from a pool
or from a component prepared by
apheresis.
4. Mark expiration time on bag as 4
hoursafterthetimetheunitwasen
-
tered.
5. Leave bag at 20 to 24 C without agi-
tation for 20 minutes if centrifuged
at 580 ×g, or for 1 hour if centrifuged
at 2000 or 5000 ×g.
6. Resuspend platelets as described in
Method 6.13.
Notes
1. If a sterile connection device is used
for removing plasma from a hema-
pheresis component or individual
platelet concentrate, the unit can be
considered sterile and it is not nec-
essary to impose the 4-hour expira-
tion interval required for entered
Platelets. However, no data exist to
support storage of reduced volume
platelet concentrates; therefore, it is
preferable to transfuse them as soon
as possible.
2. Reduced-volume platelet concentrates
may not be distributed as a licensed
product.
3. Platelets that have been pooled must
be used within 4 hours of entering
the units, whether or not they have
been volume-reduced. Pooled plate-
lets may not be distributed as a li-
censed product.
References
1. Simon TL, Sierra ER. Concentration of plate-
let units into small volumes. Transfusion
1984;24:173-5.
2. Moroff G, Friedman A, Robkin-Kline L, et al.
Reduction of the volume of stored platelet
concentrates for use in neonatal patients.
Transfusion 1984;24:144-6.
818 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Methods Section 7: Quality Control
Methods Section 7
Quality Control
Method 7.1. Quality Control
for Copper Sulfate Solution
Either of the two methods presented be-
low is acceptable for quality control of cop-
per sulfate solution.
Method 7.1.1. Functional Validation of
Copper Sulfate Solution
Principle
Copper sulfate solution can be checked for
suitability in donor screening by observing
the behavior (sinking or floating) of drops
of blood of known hemoglobin concentra-
tion.
Materials
1. Copper sulfate—specific gravity 1.053.
2. Capillary tubes.
3. Worksheet for recording results.
Procedure
1. Obtain several (three to six, if possi-
ble) blood samples with known he-
moglobin levels. Samples should in-
clude hemoglobin levels slightly
above and below 12.5 g/dL.
2. Gently place a drop of each blood
sample into a vial of copper sulfate
solution of stated specific gravity of
1.053.
3. Drops of all blood samples with he-
moglobin at or above 12.5 g/dL must
sink and those with hemoglobin lev-
els below 12.5 g/dL must float.
4. Record the date of testing; the man-
ufacturer, lot number, and expiration
date of the copper sulfate; sample
identity; the results; and the identity
of the person performing the test.
5. Document the corrective action taken
if the results are outside acceptable
limits.
819
Section 7
Copyright © 2005 by the AABB. All rights reserved.
Method 7.1.2. Use of Measurement
Instruments for Specific Gravity, Density,
or Refractive Index of Copper Sulfate
Solution
Principle
The specific gravity, density, or refractive
index of the copper sulfate solution can
be measured directly and the result com-
pared with the value stated by the manu-
facturer. An error in the specific gravity
reading of ±0.0001 corresponds to ±0.06
g/dL hemoglobin in whole blood.1There-
fore, a copper sulfate solution with a spe-
cific gravity of 1.053 ± 0.0003 would result
in a corresponding hemoglobin range of
12.5 ± 0.18 g/dL.
Materials
1. Copper sulfate—specific gravity 1.053.
2. Measurement instruments
•For specific gravity—high-preci-
sion hydrometer, with gradations
of 0.0005 or smaller.
•For refractive index—refracto-
meter.
3. Pipette.
4. For specific gravity method—gradu-
ated cylinder or other container suit-
able for use with a hydrometer.
5. Alcohol for cleaning hydrometer.
6. Worksheet for recording results.
Procedure
Hydrometer method:
1. Wipe the hydrometer clean with al-
cohol and dry it.
2. Gently lower the hydrometer into the
solution until it floats on its own.
Drops of solution on the stem will
cause inaccurate readings.
3. To read, observe a point just below
the plane of the liquid surface and
then raise the line of vision until the
surface is seen as a straight line in-
stead of an ellipse. Read at the point
where the line intersects the scale on
the instrument.
Refractometer method:
1. Follow manufacturer’s instructions for
operation and maintenance of the in-
strument.
2. Add a drop of copper sulfate to the
refractometer.
3. Observe refractive index.
Note: Some refractometers designed
for urine analysis provide both a re-
fractive index scale and a specific
gravity scale. Do not read specific
gravity directly from this type of
refractometer.
4. A copper sulfate solution with a spe-
cific gravity of 1.053 will have a re-
fractive index of about 1.3425 at room
temperature.2
Both methods:
1. Record the results; the date of test-
ing; the manufacturer, lot number,
and expiration date of the copper
sulfate solution; the identity of the
person performing the test; and the
identification of the instrument used
for measurement.
2. Document the corrective action taken
if the results are outside the accept-
able limits.
Note
A liquid densitometer may be used to
measure density directly. At 4 C, the den-
sity of the solution will be exactly the
same as the specific gravity (specific
gravity = density of solution/density of
water; density of water at 4 C = 1 g/mL).
When density is measured at room tem-
perature, a conversion factor of 0.9970
(the density of water at 25 C) is used to
calculate specific gravity.2Specific gravity
of copper sulfate solution at 25 C = the
820 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
observed density in g/mL divided by
0.9970 g/mL.
References
1. Phillips RA, Van Slyke DD, Hamilton PB, et al.
Measurement of specific gravities of whole
blood and plasma by standard copper sulfate
solutions. J Biol Chem 1950;183:305-30.
2. Blood hemoglobin screening (specific gravity
method). Technical Reference Document 12.
Arlington, TX: Ricca Chemical Company,
2002.
Method 7.2. Standardization
and Calibration of
Thermometers
Principle
Thermometers used during laboratory
testing and in the collection (donor suit-
ability), processing, and storage of blood
components and reagents should be cali-
brated and standardized to ensure accu-
rate indication of temperatures. Calibra-
tion should be performed at temperatures
close to the temperature at which the
thermometers will be used. Over time,
liquid-in-glass thermometers may give a
different reading at a given temperature
because of permanent changes in the vol-
ume of the bulb related to relaxation of
the glass.1Each thermometer should be
calibrated before initial use and periodi-
cally thereafter, as well as any time there
is reason to suspect change or damage.
Calibration must be verified for all elec-
tronic thermometers, even those describ-
ed as “self-calibrating.”
Materials
1. National Institute of Standards and
Technology (NIST)-certified thermo-
meterorthermometerwithNIST-
traceable calibration certificate.
2. Thermometer to be calibrated.
3. Suitable container, eg, 250-500 mL
beaker.
4. Water.
5. Crushed ice.
6. 37 C waterbath.
7. Worksheet for recording results.
Method 7.2.1. Liquid-in-Glass Laboratory
Thermometers
Procedure
1. Before choosing a thermometer for a
particular application, consider all
the governing factors; be sure that
the thermometer will be used at its
proper immersion; and follow the
manufacturer’s instructions for its
proper use. When using a certified
thermometer, read and follow the
applicable notes. Be sure to include
any correction factors noted on the
certificate for the NIST-traceable
thermometer and apply them in
calculations.
2. Categorize the thermometers by key
factors, such as immersion, incre-
ments, and temperature of intended
use. Test them in groups, comparing
similar thermometers. Do not attempt
to compare dissimilar thermometers
in a single procedure.
3. Number each thermometer being
tested (eg, place a numbered piece
of tape around the top of each ther-
mometer or use the manufacturer’s
serial number).
4. Perform calibration with water at a
temperature close to that which the
thermometer will monitor.
a. To calibrate at 37 C, place the
thermometers to be tested and
the NIST thermometer at a uni-
form depth in a standard 37 C
waterbath, making sure that the
tips of all devices are at the same
level in the liquid.
Methods Section 7: Quality Control 821
Copyright © 2005 by the AABB. All rights reserved.
b. To calibrate at 1 to 6 C, fill a
suitable container with water.
Add crushed ice until the ap-
proximate desired temperature
is reached. Place the thermom-
eters to be tested and the NIST
thermometer at a uniform depth
in the water/ice mixture, mak-
ingsurethatthetipsofallde
-
vices are at the same level and
are in the liquid, not the upper
ice.
5. Stir constantly in a random motion
until the temperature equilibrates,
approximately 3 to 5 minutes.
6. Observe temperatures. Record each
thermometer’s identification and re-
sults. Acceptance criteria depend on
the level of precision required, but,
for most blood banking applications,
agreement within 1 C between the two
thermometers may be considered
acceptable. If the reading varies by
more than one degree from the stan-
dard, the thermometer may be re-
turned to the distributor (if newly
purchased), labeled with the correc-
tion factor (degrees different from
theNISTthermometer)thatmustbe
applied to each reading, or dis-
carded.
7. Complete the calibration record
with the date of testing and identity
of the person who performed the
test.
Notes
1. If a thermometer is to be used for
temperatures over a range greater
than a few degrees (eg, 10 degrees), a
three-point calibration should be
performed. Use water of appropriate
temperature. Test at temperatures
just below, just above, and at the
midway point of intended use. All
results should be within 1 C of the
NIST thermometer to be considered
acceptable.
2. Thermometers should be observed
routinely for any split in the column
because this will cause inaccurate
readings. The methods for reuniting
the separation can be found in NCCLS
Standard I2-A2.2When this occurs,
document corrective action and
recalibrate the thermometer.
References
1. WiseJA.Aprocedurefortheeffectiverecali
-
bration of liquid-in-glass thermometers.
NIST Special Publication 819. Gaithersburg,
MD: National Institute of Standards and
Technology, 1991.
2. Temperature calibration of water baths, in-
struments, and temperature sensors. 2nd ed;
approved standard I2-A2 Vol. 10 No. 3. Wayne,
PA: National Committee for Clinical Labora-
tory Standards, 1990.
Method 7.2.2. Electronic Oral
Thermometers
Procedure
1. Useanyofthefollowingmethodsto
verify calibration:
a. Follow manufacturer’s instruc-
tions for verifying calibration.
b. Use a commercially available
calibration device by following
the instructions provided by
the device’s manufacturer.
c. Calibrate the thermometer by
inserting the probe in a 37 C
waterbath with a NIST-certified
thermometer.
2. A result is acceptable if the reading
on the thermometer agrees with the
NIST thermometer within 0.1 C. If
expected results are not achieved,
unsatisfactory thermometers should
be returned to the distributor. Docu-
ment corrective actions.
822 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
3. Record the date of testing, thermom-
eter identification numbers, temper-
ature readings, and the identity of the
person performing the test.
Method 7.3. Testing Blood
Storage Equipment Alarms
Blood storage refrigerators and freezers
must be equipped with a system for con-
tinuous temperature monitoring and an
audible alarm. If a storage unit goes into
alarm, it is essential that personnel know
the appropriate actions to take. Direc-
tions for such events should be available
in a conspicuous location, and personnel
should be trained to initiate these actions
if the temperature cannot be corrected
rapidly. The alarm on each storage unit
should be checked periodically for proper
functioning. Monthly checks are appro-
priate until consistent behavior of a par-
ticular storage unit has been demon-
strated. Thereafter, alarms should be
tested regularly and frequently enough to
achieve and maintain personnel compe-
tency as well as to detect malfunctions.
For equipment in good condition, quar-
terly checks are usually sufficient. Be-
cause alarms may be disconnected or si-
lenced during repairs, it is also prudent to
verify alarm functioning after repairs.
The high and low temperatures of activa-
tion must be checked and the results re-
corded. AABB Standards for Blood Banks
and Transfusion Services1(p5) requires that
thealarmbesettoactivateatatempera
-
ture that will allow appropriate interven-
tion before blood or components reach an
undesirable temperature. Because of the di-
versity of equipment available, it is not pos-
sibletogivespecificinstructions for all ap-
plicable alarm systems. If the equipment
user’s manual does not provide suitable di-
rections for testing the alarm, consult the
manufacturer or other equipment storage
expert. The facility procedures manual
must include a detailed description of the
method(s) in local use. (See Appendix 10 for
quality control testing intervals.)
Method 7.3.1. Testing Refrigerator Alarms
Principle
Refrigerator temperatures may increase
beyond acceptable limits for several rea-
sons, including:
1. Improperly closed door.
2. Insufficient refrigerant.
3. Compressor failure.
4. Dirty or blocked heat exchanger.
5. Loss of electrical power.
Materials
1. Calibrated thermometer.
2. Pan large enough to hold the ther-
mocouple container.
3. Water.
4. Crushed ice.
5. Table salt.
6. Worksheet for recording results.
Procedure
1. Verify that the alarm circuits are op-
erating, the alarm is switched on, and
the starting temperature is 1 to 6 C.
Immerse an easy-to-read calibrated
thermometer in the container with
the alarm thermocouple.
For low activation:
2. Place the container with the thermo-
couple and thermometer in a pan
containing an ice and water slush at
a temperature of –4 C or colder. To
achieve this temperature, add several
spoonfuls of table salt to the slush.
3. Close the refrigerator door to avoid
changing the temperature of the
storage compartment. Keep the con-
tainer in the pan of cold slush, and
Methods Section 7: Quality Control 823
Copyright © 2005 by the AABB. All rights reserved.
gently agitate it periodically until the
alarm sounds.
4. Record this temperature as the low-
activation temperature.
For high activation:
5. Place the container with thermocou-
ple and thermometer in a pan con-
taining cool water (eg, 12 to 15 C).
6. Close the refrigerator door. Allow the
fluid in the container to warm slowly,
with occasional agitation.
7. Record the temperature at which the
alarm sounds as the high-activation
temperature.
8. Record the date of testing, the refrig-
erator identification, the thermome-
ter identification, and the identity of
the person performing the test.
9. If temperatures of activation are too
low or too high, take appropriate
corrective actions such as those sug-
gested by the manufacturer, record
the nature of the corrections, and re-
peat the alarm check to document
that the corrections were effective.
Notes
1. The thermocouple for the alarm should
be easily accessible and equipped
with a cord long enough so that it
can be manipulated easily.
2. Thethermocoupleforthecontinu
-
ous temperature monitor need not
be in the same container as that of
the alarm. If it is in the same con-
tainer, a notation should be made in
the records that explains any out-
of-range temperature registered as a
result of the alarm check.
3. When the temperatures of alarm ac-
tivation are checked, the temperature
change should occur slowly enough
so that the measurements and re-
cording are accurate. Too rapid a
change in temperature may give the
false impression that the alarm does
not sound until an inappropriate
temperature is registered.
4. Thelowtemperatureofactivation
should be greater than 1 C (eg, 1.5 C);
the high temperature of activation
should be less than 6 C (eg, 5.5 C).
5. Alarms should sound simultaneou-
sly at the site of the refrigerator and
at the location of remote alarms,
when employed. If remote alarms
are used, the alarm check should in-
clude a verification that the alarm
sounded at the remote location.
6. The amount of fluid in which the
thermocouple is immersed must be
no larger than the volume of the
smallest component stored in that
refrigerator. The thermocouple may
be immersed in a smaller volume,
but this means that the alarm will
go off with smaller temperature
changes than those registered in a
larger volume of fluid. Excessive sen-
sitivity may create a nuisance.
7. With the one-time assistance of a
qualified electrician, the required re-
frigerator alarm checks of units with
virtually inaccessible temperature
probes can be performed with an
electrical modification cited by Wenz
and Owens.2
References
1. Silva MA, ed. Standards for blood banks and
transfusion services. 23rd ed. Bethesda, MD:
AABB, 2005.
2. Wenz B, Owens RT. A simplified method for
monitoring and calibrating refrigerator alarm
systems. Transfusion 1980;20:75-8.
Method 7.3.2. Testing Freezer Alarms
Principle
Freezer temperatures may rise to unac-
ceptable levels for a variety of reasons.
824 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Common causes of rising temperatures
include:
1. Improperly closed freezer door or lid.
2. Low level of refrigerant.
3. Compressor failure.
4. Dirty or blocked heat exchanger.
5. Loss of electrical power.
Materials
1. Protection for the freezer contents,
eg, a blanket.
2. Calibrated thermometer or thermo-
couple independent from that built
into the system.
3. Warm water or an oven mitt.
4. Worksheet for recording results.
Procedure
1. Protect frozen components from ex-
posure to elevated temperatures dur-
ing the test.
2. Use a thermometer or thermocou-
ple, independent from that built into
the system, that will accurately indi-
cate the temperature of alarm acti-
vation. Compare these readings with
the temperatures registered on the
recorder.
3. Warm the alarm probe and thermo-
meter slowly (eg, in warm water, by
an oven-mitt-covered hand, expo-
sure to air). The specific temperature
of activation cannot be determined
accurately during rapid warming,
and the apparent temperature of ac-
tivation will be too high.
4. Record the temperature at which the
alarm sounds, the date of testing,
the identity of the person perform-
ing the test, the identity of the
freezer and calibrating instrument,
and any observations that might
suggest impaired activity.
5. Return the freezer and the alarm sys-
tem to their normal conditions.
6. If the alarm sounds at too high a
temperature, take appropriate cor-
rective actions such as those sug-
gested by the manufacturer, record
the nature of the correction, and re-
peat the alarm check to document
that the corrections were effective.
Notes
1. Alarms should sound simultaneou-
sly at the site of the freezer and at
the location of the remote alarms,
when employed. If remote alarms
are used, the alarm check should in-
clude a verification that the alarm
sounded at the remote location.
2. Test battery function, electrical cir-
cuits, and power-off alarms more
frequently than the activation tem-
perature. Record function, freezer
identification, date, and identity of
person performing the testing.
3. For units with the sensor installed in
the wall or in air, apply local warmth
to the site or allow the temperature
of the entire compartment to rise to
the point at which the alarm sounds.
Remove the frozen contents or pro-
tect them with insulation while the
temperature rises.
4. For units with the thermocouple lo-
cated in antifreeze solution, pull the
container and the cables outside the
freezer chest for testing, leaving the
door shut and the contents protected.
5. For units with a tracking alarm that
sounds whenever the temperature
reaches a constant interval above
the setting on the temperature con-
troller, set the controller to a warmer
setting and note the temperature in-
terval at which the alarm sounds.
Methods Section 7: Quality Control 825
Copyright © 2005 by the AABB. All rights reserved.
6. Liquid nitrogen freezers must have
alarm systems that activate at an un-
safe level of contained liquid nitro-
gen.
Method 7.4. Functional
Calibration of Centrifuges for
Platelet Separation
Principle
Successful preparation of platelet concen-
trates requires adequate but not excessive
centrifugation; the equipment used must
perform in a consistent and dependable
manner. Each centrifuge used to prepare
platelets should be calibrated upon re-
ceipt and after adjustment or repair.
Functional calibration of the centrifuge
for both the preparation of platelet-rich
plasma (PRP) from whole blood and sub-
sequent preparation of platelet concen-
trate from PRP can be performed during
thesameprocedure.
Materials
1. Freshly collected whole blood, ob-
tained by phlebotomy into a bag with
two integrally attached transfer con-
tainers.
2. A specimen of blood from the donor,
anticoagulated with EDTA and col-
lected in addition to the specimens
drawn for routine processing.
3. Metal clips and hand sealer or di-
electric sealer.
4. Clean instruments (scissors, hemo-
stats, tubing stripper).
5. Plasma extractor.
6. Centrifuge suitable for preparation
of platelet concentrates.
7. Worksheet for recording results.
Procedure
Preparation of Platelet-Rich Plasma
1. Perform a platelet count on the anti-
coagulated specimen. If the platelet
count is below 133,000/µL, do not
use this donor’s blood for calibration.
2. Calculateandrecordthenumberof
platelets in the Whole Blood unit: pla-
telets/µL×1000 ×mL of whole blood
= number of platelets in whole blood.
3. Prepare PRP at a selected speed and
time. (See “light spin” in Table 7.4-1
or guidance provided by the centri-
fuge manufacturer.)
4. Place a temporary clamp on the tub-
ing so that one satellite bag is closed
off. Express the PRP into the other
satellite bag. Seal the tubing close to
the primary bag, leaving a long sec-
tion of tubing, the “tail.” Disconnect
the two satellite bags from the pri-
mary bag. Do not remove the tem-
porary clamp between the satellite
bags until the platelets are prepared
(see next section).
5. Strip the tubing and “tail” several
times so that they contain a repre-
sentative sample of PRP.
6. Seal off a segment of the “tail” and
disconnect it, so that the bag of PRP
remains sterile.
7. Perform a platelet count on the sam-
ple of PRP in the sealed segment.
Calculateandrecordthenumberof
platelets in the bag of PRP: platelets/
µL×1000 ×mL of PRP = number of
platelets in PRP.
8. Calculate and record percent yield:
(number of platelets in PRP ×100)
divided by (number of platelets in
whole blood) = % yield.
9. Repeat the above process three or four
times with different donors, using
different speeds and times of centri-
fugation, and compare the yields
826 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
achieved under each set of test con-
ditions.
10. Select the shortest time/lowest speed
combination that results in the high-
est percent of platelet yield without
unacceptable levels of red cell con-
tent in the PRP.
11. Record the centrifuge identification,
the calibration settings selected, the
date, and the identity of the person
performing the calibration.
Preparation of Platelets
1. Centrifuge the PRP (as prepared
above) at a selected time and speed
to prepare platelets. (See “heavy spin”
in Table 7.4-1 or guidance provided
by the centrifuge manufacturer.)
2. Remove the temporary clamp be-
tween the two satellite bags and ex-
press the platelet-reduced plasma
into the second attached satellite
bag, leaving approximately 55 to 60
mL volume in the platelet bag. Seal
the tubing, leaving a long section of
tubing attached to the platelet bag.
3. Allow the platelets to rest for approx-
imately 1 hour.
4. Place the platelets on an agitator for
at least 1 hour to ensure that they are
evenly resuspended. Platelet counts
performed immediately after centri-
fugation will not be accurate.
5. Strip the tubing several times, mix-
ing tubing contents well with the
contents of the platelet bag. Seal off
asegmentofthetubinganddiscon
-
nect it, so that the platelet bag re-
mains sterile.
6. Perform a platelet count on the con-
tents of the segment.
7. Calculateandrecordthenumberof
platelets in the concentrate: platelets/
µL×1000 ×mL of platelets = number
of platelets in platelet concentrate.
8. Calculate and record percent yield.
9. Repeat the above process with PRP
from different donors, using differ-
ent speeds and times of centrifuga-
tion, and compare the yields achieved
under each set of test conditions.
Methods Section 7: Quality Control 827
Table 7.4-1. Centrifugation for Component Preparation
Heavy Spin
}
Red cells
Platelets from whole blood 55000 ×
g
, 5 minutes*
}
Cell- free plasma
Cryoprecipitate 55000 ×
g
, 7 minutes*
Light Spin
Platelet-rich plasma 2000 ×
g
, 3 minutes*
To calculate relative centrifugal force in
g
:
rcf (in
g
) = 28.38 ×R†×(rpm/1000)2
*Times include acceleration but not deceleration times. Times given are approximations only. Each individual centrifuge
must be evaluated for the preparation of the various components.
†R = radius of centrifuge rotor in inches.
Copyright © 2005 by the AABB. All rights reserved.
10. Select the shortest time/lowest speed
combination that results in the high-
est percent of platelet yield in the
platelet concentrate.
11. Record the centrifuge identification,
the calibration settings selected, the
date, and the identity of the person
performing the calibration.
Notes
1. It is not necessary to perform func-
tional recalibration of a centrifuge
unless the instrument has under-
gone adjustments or repairs, or
component quality control indicates
that platelet counts have fallen be-
low acceptable levels. However,
timer, speed, and temperature cali-
brations of the centrifuge should oc-
cur on a regularly scheduled basis
(see Appendix 10).
2. Each centrifuge used for preparing
platelets must be calibrated individ-
ually. Use the conditions determined
to be optimal for each instrument.
3. When counting platelet samples on
an instrument intended for whole
blood, it may be necessary to use a
correction factor to obtain accurate
results.
4. When determining the appropriate
time and speed of centrifugation,
consideration should also be given to
other products that will be prepared
from the whole blood. Final size and
hematocrit of red cell and plasma
volume made available for further
processing are important factors to
consider.
References
1. Kahn R, Cossette I, Friedman L. Optimum
centrifugation conditions for the preparation
of platelet and plasma products. Transfusion
1976;16:162-5.
2. McShine R, Das P, Smit Sibinga C, Brozovic B.
Effect of EDTA on platelet parameters in
blood and blood components collected with
CPDA-1. Vox Sang 1991;61:84-9.
Method 7.5. Functional
Calibration of a Serologic
Centrifuge
Principle
Each centrifuge should be calibrated
upon receipt, after adjustments or repairs,
and periodically. Calibration evaluates the
behavior of red cells in solutions of differ-
ent viscosities, not the reactivity of differ-
ent antibodies.
For Immediate Agglutination
Materials
1. Test tubes, 10 ×75 mm or 12 ×75
mm (whichever size is routinely
used in the laboratory)
2. Worksheet for recording results.
3. For saline-active antibodies:
■SerumfromagroupAperson
(anti-B) diluted with 6% albu-
min to give 1+ macroscopic ag-
glutination (3 mL of 22% bovine
albumin + 8 mL of normal sa-
line = 6% bovine albumin). See
Method 1.5.
■Positive control: Group B red cells
in a 2% to 5% saline suspension.
■Negative control: Group A red cells
in a 2% to 5% saline suspension.
4. For high-protein antibodies:
■Anti-D diluted with 22% or 30%
albumin to give 1+ macro-
scopic agglutination.
■Positive control: D+ red cells in
a2%to5%salinesuspension.
■Negative control: D– red cells in
a2%to5%salinesuspension.
828 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Procedure
1. For each set of tests (saline and high-
protein antibodies), label five test
tubes for positive reactions and five
for negative reactions.
2. In quantities that correspond to rou-
tine use, add diluted anti-B to each
of 10 tubes for the saline test and
add diluted anti-D to each of 10 tubes
for the high-protein test. Add serum
and reagents in quantities that cor-
respond to routine use.
3. Add the appropriate control cell sus-
pension to one set of tubes (one pos-
itiveandonenegativetubeforthe
saline test, and one positive and one
negative tube for the high-protein
antibody test). Centrifuge immedi-
ately for the desired time interval
(eg, 10 seconds).
4. Observe each tube for agglutination
and record observations. (See exam-
ple in Table 7.5-1.)
5. Repeat steps 2 and 3 for each time
interval (eg, 15, 20, 30, and 45 sec-
onds). Do not allow cells and sera to
incubate before centrifugation.
6. Select the optimal time of centrifu-
gation, which is the shortest time re-
quired to fulfill the following criteria:
a. Agglutination in the positive
tubesisasstrongasdetermined
in preparing reagents.
b. There is no agglutination or
ambiguity in the negative tubes.
c. The cell button is clearly delin-
eated and the periphery is
sharply defined, not fuzzy.
d. The supernatant fluid is clear.
e. The cell button is easily resus-
pended.
IntheexampleshowninTa
-
ble 7.5-1, these criteria are met
by the 30-second and the 45-
second spins; the optimal time
for these tests in this centrifuge
is 30 seconds.
7. Record centrifuge identification, the
times selected, the date, and the
identity of the person performing
the calibration.
For Washing and Antiglobulin Testing
Tests in which antihuman globulin (AHG)
serum is added to red cells may require
Methods Section 7: Quality Control 829
Table 7.5-1. Example of Serologic Centrifuge Test Results*
Time in Seconds
Criteria 10152030 45
Supernatant fluid is clear No No Yes Yes Yes
Cell button is clearly delineated No No No Yes Yes
Cells are easily resuspended Yes Yes Yes Yes Yes
Agglutination is observed ± ± 1+ 1+ 1+
Negative tube is negative Yes Yes Yes Yes Resuspends
roughly
*The optimal time for centrifugation in this example is 30 seconds.
Copyright © 2005 by the AABB. All rights reserved.
centrifugation conditions different from
those for immediate agglutination. Cen-
trifugation conditions appropriate for
both washing and AHG reactions can be
determined in one procedure. Note that
this procedure does not monitor the com-
pleteness of washing; use of IgG-coated cells
to control negative AHG reactions provides
this check. The following procedure ad-
dresses only the mechanics of centrifu-
gation.
Materials
1. AHG reagent, unmodified.
2. Saline, large volumes.
3. Test tubes, 10 ×75 mm or 12 ×75
mm (whichever size is routinely used
in the laboratory).
4. Worksheet for recording results.
5. Positive control: a2%to5%saline
suspension of D+ red cells incubated
for 15 minutes at 37 C with anti-D
diluted to give 1+ macroscopic ag-
glutination after addition of AHG.
6. Negative control: a2%to5%suspen-
sion of D+ red cells incubated for 15
minutes at 37 C with 6% albumin.
[Note: D– red cells incubated with
diluted anti-D may also be used as a
negative control.]
Procedure
1. Prepare five test tubes containing 1
drop of positive cells and five tubes
containing 1 drop of negative con-
trol cells.
2. Fill tubes with saline and centrifuge
them in pairs, one positive and one
negative, for different times (eg, 30,
45, 60, 90, and 120 seconds). The red
cells should form a clearly delin-
eated button, with minimal cells
trailing up the side of the tube. After
the saline has been decanted, the
cell button should be easily resus-
pended in the residual fluid. The
optimal time for washing is the
shortest time that accomplishes
these goals.
3. Repeat washing process on all pairs
three more times, using time deter-
mined to be optimal.
4. Decant supernatant saline thoroughly.
5. Add AHG to one positive control test
tube and one negative control test tube.
Centrifuge immediately for the de-
sired interval (eg, 10 seconds).
6. Observe each tube for agglutination
and record observations.
7. Repeat steps 5 and 6 for each inter-
val (eg, 15, 20, 30, and 45 seconds).
Do not allow cells and AHG to incu-
bate before centrifugation.
8. Select optimal time as in immediate
agglutination procedure.
9. Record centrifuge identification, the
times selected, the date, and the
identity of the person performing
the calibration.
Notes
Periodic recalibration is performed to ver-
ify that the timing in use continues to be
the optimal timing. This may be accom-
plished by using a shortened version of
the procedures outlined above. For exam-
ple, use the current timing for a particular
centrifuge and each medium and those
times just above and just below the cur-
rent timing.
Method 7.6. Performance
Testing of Automatic Cell
Washers
Principle
Antihuman globulin (AHG) is inactivated
readily by unbound immunoglobulin. The
red cells to which AHG will be added
830 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
must be washed free of all proteins and
suspended in a protein-free medium. A
properly functioning cell washer must add
large volumes of saline to each tube, re-
suspend the cells, centrifuge them ade-
quately to avoid excessive red cell loss, and
decant the saline to leave a dry cell button.
Materials
1. Test tubes routinely used in the lab-
oratory, 10 ×75 mm or 12 ×75 mm.
2. Additive routinely used to potentiate
antigen-antibody reactions.
3. Human serum, from patient or do-
nor.
4. IgG-coated red cells, known to give a
1 to 2+ reaction in antiglobulin test-
ing.
5. Normal saline.
6. AHG reagent, anti-IgG or polyspeci-
fic.
7. Worksheet for recording results.
Procedure
1. To each of 12 tubes, add potentiator
and human serum in quantities that
correspond to routine use and 1 drop
of IgG-coated red cells.
2. Place the tubes in a centrifuge car-
rier, seat the carrier in the cell washer,
and start the wash cycle.
3. After addition of saline in the second
cycle, stop the cell washer. Inspect
the contents of all tubes. There
should be an approximately equal
volume of saline in all tubes; some
variation is acceptable. Tubes should
be approximately 80% full, to avoid
splashing and cross-contamination.
(Refer to manufacturer’s instructions
for specific requirements.) Record
observations.
4. Observe all tubes to see that the red
cells have been completely resus-
pended. Record observations.
5. Continue the washing cycle.
6. After addition of saline in the third
cycle, stop the cell washer and in-
spect tubes as above. Record obser-
vations.
7. Complete the wash cycle.
8. At the end of the wash cycle, inspect
all tubes to see that saline has been
completely decanted and that each
tube contains a dry cell button. Re-
cord observations.
9. Add AHG according to the manufac-
turer’s directions, centrifuge, and ex-
amine all tubes for agglutination. If
the cell washer is functioning prop-
erly, the size of the cell button
shouldbethesameinalltubes.All
tubes should show the same degree
of agglutination. Record observa-
tions.
10. Record identity of centrifuge, the date
of testing, and the identity of the
person performing the testing.
Notes
1. Further investigation is needed if:
a. The amount of saline varies
significantly from tube to tube
or cycle to cycle.
b. The cell button is not resus-
pended completely after being
filled with saline.
c. Any tube has weak or absent
agglutination in the antiglobu-
lin phase.
d. Any tube has a significant de-
crease in the size of the cell
button.
2. Cell washers that automatically add
AHG should also be checked for uni-
form addition of AHG. In step 9
above, AHG would be added auto-
matically, and failure of addition
would be apparent by absence of ag-
glutination. The volume of AHG
Methods Section 7: Quality Control 831
Copyright © 2005 by the AABB. All rights reserved.
should be inspected and found to be
equal in all tubes. The volume of
AHG delivered automatically by cell
washers should be checked monthly
to ensure that it is as specified in the
manufacturer’s directions and that
delivery is uniform in all tubes.
3. Some manufacturers market AHG
colored with green dye for use in au-
tomated cell washers so that it will
be immediately obvious if no re-
agent has been added.
Method 7.7. Monitoring Cell
Counts of Apheresis
Components
Principle
When cellular components are prepared
by apheresis, it is essential to determine
cell yields without compromising the ste-
rility of the component.
Materials
1. Component collected by apheresis.
2. Metal clips and hand sealer or di-
electric sealer.
3. Tubing stripper.
4. Clean instruments (scissors, hemo-
stats).
5. Test tubes.
6. Cell-counting equipment.
7. Worksheet for recording results.
Procedure
1. Ensure that the contents of the aphere-
sis component bag are well mixed.
2. Strip the attached tubing at least four
times, mixing the contents of the
tubing with the contents of the bag,
to ensure that the contents of the
tubing accurately represent the en-
tire contents of the bag.
3. Seal a 5- to 8-cm (2- to 3-inch) seg-
ment distal to the collection bag.
There should be approximately 2 mL
of fluid in the segment. Double-seal
theendofthetubingnexttothe
component bag and detach the seg-
ment.
4. Empty the contents of the segment
into a suitably labeled tube.
5. Determine and record cell counts in
cells/mL.
a. For results reported as cells/µL,
change values to cells/mL by
multiplying by 1000 (or 103).
b. For results reported as cells/L,
change values to cells/mL by
dividing by 1000 (or 103).
6. Multiply cells/mL by the volume of
the component, in mL, to obtain to-
tal cell count in the component.
7. Record component’s identity, the date,
and the identity of the person per-
forming the testing.
Note
Refer to manufacturer’s directions for any
additional requirements.
Method 7.8. Manual Method
for Counting Residual White
Cells in Leukocyte-Reduced
Blood and Components
Principle
The residual white cell content of leuko-
cyte-reduced whole blood and components
can be determined using a large-volume
hemocytometer. For red-cell-containing
components, the red cells in the aliquot to
be counted are first lysed. Crystal violet is
used to stain the leukocyte nuclei. The
Nageotte counting chamber has a volume
56 times that of the standard hemocyto-
832 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
meter. Accuracy of counting is improved
by examining a larger volume of mini-
mally diluted specimen, compared with
standard counting techniques.
Materials
1. Hemocytometer chamber with 50 µL
counting volume (eg, Nageotte Brite
Line Chamber).
2. Crystal violet stain: 0.01% w/v crys-
tal violet in 1% v/v acetic acid (eg,
Turks solution).
3. Red cell lysing agent (eg, Zapoglobin,
Coulter Electronics, Hialeah, FL), for
red-cell-containing components
only.
4. Pipettor (40 µL and 100 µL) with dis-
posable tips.
5. Talc-free gloves, clean plastic test
tubes, plastic petri dish, filter paper.
6. Light microscope with 10×ocular
lens and 20×objective.
7. Worksheet for recording results.
Procedure
1. Dilute and stain leukocyte-reduced
blood and component samples as
follows:
a. For Red-Cell-Containing Com-
ponents
1) Pipette 40 µLoflysing
agent into a clean test tube.
2) Place a representative
sample of the component
to be tested in a clean test
tube. The hematocrit of the
sample to be tested should
not exceed 60%.
3) Pipette 100 µLofthesam
-
ple into the tube contain-
ing 40 µLoflysingagent.
Rinse the pipette several
times to mix the two flu-
ids, until the pipette tip is
no longer coated with in-
tact red cells.
4) Pipette 360 µLofcrystal
violet stain into the mix-
ture and mix fluids by
pipetting up and down
several times. The final
volume is now 500 µL.
b. For Platelets
1) Place a representative sam-
ple of the platelet in a clean
test tube.
2) Pipette 100 µLofthepla
-
telet sample into a clean
test tube.
3) Pipette 400 µLofcrystal
violet stain into the 100 µL
of platelets and mix fluids
by pipetting up and down
several times. The final
volume is now 500 µL.
2. Fit the hemocytometer with a cover-
slip and, using a pipette, load the
mixtureuntilthecountingareais
completely covered but not over-
flowing.
3. Cover the hemocytometer with a
moist lid to prevent evaporation (a
plastic petri dish into which a piece
of damp filter paper has been placed
works well) and let it rest undisturbed
for10to15minutes,toallowthe
white cells to settle in the counting
area of the chamber.
4. Removethemoistlid,placethe
hemocytometer on the microscope
and, using a 20×objective, count the
white cells present in the entire
50-µL volume of the counting cham-
ber.
5. Calculate and record results.
a. White cell concentration:
leukocytes/µL = (cells counted/50 µL) ×5
= cells counted/10
Methods Section 7: Quality Control 833
Copyright © 2005 by the AABB. All rights reserved.
where 50 µListhevolume
counted and 5 is the dilution
factor resulting from the addi-
tion of lysing agent and stain.
b. Total white cell content of the
leukocyte-reduced component:
leukocytes/
component
leukocytes/ L
1000 L/mL
vo
=
×
×
µ
µ
lume in mL
of the component
6. Record the component’s identity, the
date, and the identity of the person
performing the testing.
Notes
1. White cells deteriorate during refrig-
erated storage; counts on stored blood
or red cell components may give in-
accurate results.
2. Use of talc-free gloves is recom-
mended because talc particles that
contaminate the counting chamber
can be misread as white cells.
3. Experience identifying crystal-violet-
stained white cells can be obtained
by examining samples from compo-
nents that have not been leukocyte
reduced.
4. The accuracy of the counting method
can be validated from a reference
sample with a high white cell con-
tent that has been quantified by an-
other means. This reference sample
can be used for serial dilutions in
blood or a component that has been
rendered extremely leukocyte re-
duced by two passages through a
leukocyte reduction filter. Counts
obtained on the serially diluted sam-
ples can be compared with the ex-
pected concentration derived by cal-
culation.
5. This counting technique is not known
to be accurate at concentrations lower
than 1 white cell/µL.
References
1. Lutz P, Dzik WH. Large-volume hemocyto-
meter chamber for accurate counting of
white cells (WBCs) in WBC-reduced platelets;
validation and application for quality control
of WBC-reduced platelets prepared by
apheresis and filtration. Transfusion 1993;33:
409-12.
2. Dzik WH, Szuflad P. Method for counting
white cells in white cell-reduced red cell con-
centrates (letter). Transfusion 1993;33:272.
834 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.

Appendices
Appendices
835
Appendices
Appendix 1. Normal Values in Adults
Determination SI Units Conventional Units
Alanine aminotransferase 4-36 U/L at 37 C 4-36 U/L at 37 C
Bilirubin, total 2-21 µmol/L 0.1-1.2 mg/dL
Haptoglobin 0.6-2.7 g/L 60-270 mg/dL
Hematocrit
Males 0.40-0.54 40-54%
Females 0.38-0.47 38-47%
Hemoglobin
Males 135-180 g/L 13.5-18.0 g/dL
Females 120-160 g/L 12.0-16.0 g/dL
Hemoglobin A20.015-0.035 total Hb 1.5-3.5% total Hb
Hemoglobin F 0-0.01 total Hb <1% total Hb
Hemoglobin (plasma) 5-50 mg/L 0.5-5.0 mg/dL
Immunoglobulins
IgG 8.0-18.0 g/L 800-1801 mg/dL
IgA 1.1-5.6 g/L 113-563 mg/dL
IgM 0.5-2.2 g/L 54-222 mg/dL
IgD 5.0-30 mg/L 0.5-3.0 mg/dL
IgE 0.1-0.4 mg/L 0.01-0.04 mg/dL
Methemoglobin <0.01 total Hb <1% total Hb
Platelet count 150-450 ×109/L 150-450 ×103/µL
Red cells
Males 4.6-6.2 ×1012/L 4.6-6.2 ×106/µL
Females 4.2-5.4 ×1012/L 4.2-5.4 ×106/µL
Reticulocyte count 25-75 ×109/L 25-75 ×103/µL
Viscosity, relative 1.4-1.8 ×water 1.4-1.8 ×water
White cells 4.5-11.0 ×109/L 4.5-11.0 ×103/µL
Copyright © 2005 by the AABB. All rights reserved.

836 AABB Technical Manual
Appendix 2. Selected Normal Values in Children
SI Units Conventional Units
Bilirubin (total)
Cord Preterm <30 mmol/L <1.8 mg/dL
Term <30 mmol/L <1.8 mg/dL
0-1 day Preterm <137 mmol/L <8 mg/dL
Term <103 mmol/L <6 mg/dL
1-2 days Preterm <205 mmol/L <12 mg/dL
Term <137 mmol/L <8 mg/dL
3-7 days Preterm <274 mmol/L <16 mg/dL
Term <205 mmol/L <12 mg/dL
7-30 days Preterm <205 mmol/L <12 mg/dL
Term <120 mmol/L <7 mg/dL
Thereafter Preterm <34 mmol/L <2 mg/dL
Term <17 mmol/L <1 mg/dL
Hemoglobin WBC Platelets
26-30 weeks’ gestation 11.0-15.8 g/dL 1.7-7.1 ×10 /L 180,000-327,000/µL
Term 13.5-19.5 g/dL 9-30 ×10 /L 192,000/µL (mean)
1-3 days 14.5-22.5 g/dL 9.4-34 ×10 /L 252,000/µL (mean)
2 weeks 13.4-19.8 g/dL 5-20 ×10 /L
1 month 10.7-17.1 g/dL 4-19.5 ×10 /L
2 months 9.4-13.0 g/dL
6 months 11.1-14.1 g/dL 6-17.5 ×10 /L
6 months-2 years 10.5-13.5 g/dL 6-17 ×10 /L 150,000-350,000/µL
2-6 years 11.5-13.5 g/dL 5-15.5 ×10 /L 150,000-350,000/µL
6-12 years 11.5-15.5 g/dL 4.5-13.5 ×10 /L 150,000-350,000/µL
12-18 years
Male 13.0-16.9 g/dL 4.5-13.5 ×10 /L 150,000-350,000/µL
Female 12.0-16.0 g/dL 4.5-13.5 ×10 /L 150,000-350,000/µL
Copyright © 2005 by the AABB. All rights reserved.

Appendices 837
Appendix 2. Selected Normal Values in Children (cont’d)
IgG IgM IgA
Newborn 831-1231 mg/dL 6-16 mg/dL <3 mg/dL
1-3 months 312-549 mg/dL 19-41 mg/dL 8-34 mg/dL
4-6 months 241-613 mg/dL 26-60 mg/dL 10-46 mg/dL
7-12 months 442-880 mg/dL 31-77 mg/dL 19-55 mg/dL
13-24 months 553-971 mg/dL 35-81 mg/dL 26-74 mg/dL
25-36 months 709-1075 mg/dL 42-80 mg/dL 34-108 mg/dL
3-5 years 701-1157 mg/dL 38-74 mg/dL 66-120 mg/dL
6-8 years 667-1179 mg/dL 40-80 mg/dL 79-169 mg/dL
9-11 years 889-1359 mg/dL 46-112 mg/dL 71-191 mg/dL
12-16 years 822-1070 mg/dL 39-79 mg/dL 85-211 mg/dL
Activated Partial Thromboplastin Time
Preterm 70 seconds
Full-term 45-65 seconds
Prothrombin Time
Preterm 12-21 seconds
Full-term 13-20 seconds
Reprinted with permission from The Harriet Lane Handbook. 15th ed. St. Louis, MO: Mosby, 2000.
Appendix 3. Typical Normal Values in Tests of Hemostasis and Coagulation
(Adults)
Test Normal Value
Activated partial thromboplastin time 25-35 seconds
Bleeding time 2-8 minutes
Coagulation factors 500-1500 U/L
Fibrin degradation products <10 mg/L
Fibrinogen 2.0-4.0 g/L
Plasma D-dimers <200 mg/L
Protein C 70-1400 U/L
Protein S (total) 70-1400 U/L
Prothrombin time 10-13 seconds
Thrombin time 17-25 seconds
Reprinted with permission from Henry JB. Clinical diagnosis and management by laboratory methods. 20th ed. Phila-
delphia: WB Saunders, 2001.
Copyright © 2005 by the AABB. All rights reserved.

838 AABB Technical Manual
Appendix 4. Coagulation Factor Values in Platelet Concentrates
Factor/
Protein
Normal
Range Day 0 Day 1 Day 2 Day 3 Day 4 Day 5
II % 78-122 104 91-96 96 85-94 90 90
V % 47-153 78-98 69-78 50 36-47 28 24-35
VII % 51-168 108 93-117 88 80-103 75 72
VIII % 48-152 68-126 85-99 76 68-76 75 39-70
IX % 62-138 72-105 100-106 95 91-98 93 63-97
X % 58-142 66-101 93-94 92 85-88 84 60-83
XI % 52-148 91-111 106-108 103 96-98 101 86-110
XII % 46-126 117 107-112 116 106-123 123 131
C % 57-128 106 102 101 98 99 100
S % 83-167 95 75 61 40 32 31
Antithrombin % 88-126 103 99 101 102 103 97
Plasminogen % 60-140 140 133 126 122 124 117
Fibrinogen
mg/dL
198-434 217-308 278-313 310 265-323 302 221-299
Ristocetin
cofactor %
50-150 106 124 125 133 116 127
Note: Coagulation factor % = 100 x coagulation factor units/mL.
Reproduced with permission from Brecher ME, ed. Collected questions and answers. 6th ed. Bethesda, MD:
AABB, 2000.
Copyright © 2005 by the AABB. All rights reserved.
Appendices 839
Appendix 5. Approximate Normal Values for Red Cell, Plasma, and Blood
Volumes
Infant1Adult2
Premature
Term Birth at
72 hours Male Female
Red Cell Volume mL/kg 50 40 26 24
Plasma Volume mL/kg 58 47 40 36
Blood Volume mL/kg 108 87 66 60
The adult values should be modified to correct
for:
1. Below age 18: increase values by 10%.
2. Weight loss:
a. Marked loss within 6 months—calcula-
tions made at original weight.
b. Gradual loss over a longer time—calcu-
lations made at present weight and
raised 10% to 15%.
3. Obese and short: values are reduced by
10%.
4. Elderly: values are reduced by 10%.
5. Pregnancy3:
Estimation of Body Surface Area4:
BSA(m ) Ht(cm) Wt(kg)
3600 or Ht(in) Wt(lb)
3131
2=××
Blood Volume (BV)5:
BV = 2740 mL/m2—males
BV = 2370 mL/m2—females
Hematocrit6:
Venous hematocrit = Hv(blood obtained by
vein or finger puncture)
Whole-body hematocrit = HB
HB=(Hv)×(0.91)
References
1. Miller D. Normal values and examination of the
blood: Perinatal period, infancy, childhood and ado-
lescence. In: Miller DR, Baehner RL, McMillan CW,
Miller LP, eds. Blood diseases of infancy and child-
hood. St. Louis: C.V. Mosby, 1984:21,22.
2. Albert SN. Blood volume. Springfield, IL: Charles C.
Thomas, 1963:26.
3. Peck TM, Arias F. Hematologic changes associated
with pregnancy. Clin Obstet Gynecol 1979;22:788.
4. Mosteller RD. Simplfied calculation of body-surface
area. N Engl J Med 1987;317:1098.
5. Shoemaker WC. Fluids and electrolytes in the
acutely ill adult. In: Shoemaker WC, Ayres S,
Grenvik A, et al, eds. Textbook of critical care. 2nd
ed. Philadelphia: WB Saunders Co., 1989:1130.
6. Mollison PL, Englefriet CP, Contreras M. Blood
transfusion in clinical medicine. 9th ed. Oxford:
Blackwell Scientific Publications, 1993.
Copyright © 2005 by the AABB. All rights reserved.

840 AABB Technical Manual
Appendix 6. Blood Group Antigens Assigned to Systems
In 1980, the International Society of Blood Transfusion (ISBT) formed a Working Party on Terminology
for Red Cell Surface Antigens. The task of this group was to devise a uniform nomenclature that would
be both eye- and machine-readable. The numeric system proposed by this group was not intended to re-
place traditional terminology but, instead, to enable communication using computer systems where
numbers are necessary. ISBT terminology uses uppercase letters and Arabic numbers for system and
antigen codes. Each system, collection, or series of antigens is given a number (eg, ABO system = 001),
and each antigen within the system is given a number (eg, A = 001, B = 002). Sinistral zeros may be
omitted. Thus, in ISBT terminology, the A antigen would be written using computer code as 001001 or
1.1, or using the system symbol, as ABO1.
Periodically, the Working Party meets to update assignment of antigens to systems, collections, and se-
ries. The table below lists the blood group systems and the antigens assigned to those systems. Other
red cell antigens are assigned to collections and to series of high- and low-incidence antigens. Although
all terms in the table are acceptable, the
Technical Manual
and
TRANSFUSION
choose to use traditional
terminology in most cases. Further information on blood group terminology, which antigens are assigned
to the collections, and the series of high- and low-incidence antigens can be found in the references.
System (ISBT
Symbol/Number) Antigen (ISBT Number)
ABO (ABO/001) A(ABO1)
B (ABO2)
A,B (ABO3)
A1(ABO4)
MNSs or MNS or M(MNS1) M
e(MNS13) Dantu (MNS25) ERIK (MNS37)
MN (MNS/002) N (MNS2) Mta(MNS14) Hop (MNS26) Osa(MNS38)
S(MNS3) St
a(MNS15) Nob (MNS27) ENEP (MNS39)
s(MNS4) Ri
a(MNS16) Ena(MNS28) ENEH (MNS40)
U(MNS5) Cl
a(MNS17) ENKT (MNS29) HAG (MNS41)
He (MNS6) Nya(MNS18) ‘N’ (MNS30) ENAV (MNS42)
Mia(MNS7) Hut (MNS19) Or (MNS31) MARS (MNS43)
Mc(MNS8) Hil (MNS20) Dane (MNS32)
Vw (MNS9) Mv(MNS21) TSEN (MNS33)
Mur (MNS10) Far (MNS22) MINY (MNS34)
Mg(MNS11) sD(MNS23) MUT (MNS35)
Vr (MNS12) Mit (MNS24) SAT (MNS36)
P (P/003) P1(P1)
Copyright © 2005 by the AABB. All rights reserved.

Appendices 841
(cont’d)
Rh (RH/004) D (RH1) Hro(RH17) hrB(RH31) Nou (RH44)
C (RH2) Hr (RH18) Rh32 (RH32) Riv (RH45)
E (RH3) hrS(RH19) Rh33 (RH33) Sec (RH46)
C (RH4) VS (RH20) HrB(RH34) Dav (RH47)
e (RH5) CG(RH21) Rh35 (RH35) JAL (RH48)
f (RH6) CE (RH22) Bea(RH36) STEM (RH49)
Ce (RH7) DW(RH23) Evans (RH37) FPTT (RH50)
CW(RH8) c-like (RH26) Rh39 (RH39) MAR (RH51)
CX(RH9) cE (RH27) Tar (RH40) BARC (RH52)
V (RH10) hrH(RH28) Rh41 (RH41) JAHK (RH53)
EW(RH11) Rh29 (RH29) Rh42 (RH42) DAK (RH54)
G (RH12) Goa(RH30) Crawford (RH43) LOCR (RH55)
CENR (RH56)
Lutheran (LU/005) Lua(LU1) Lu6 (LU6) Lu12 (LU12) Aua(LU18)
Lub(LU2) Lu7 (LU7) Lu13 (LU13) Aub(LU19)
Lu3 (LU3) Lu8 (LU8) Lu14 (LU14) Lu20 (LU20)
Lu4 (LU4) Lu9 (LU9) Lu16 (LU16) Lu21 (LU21)
Lu5 (LU5) Lu11 (LU11) Lu17 (LU17)
Kell (KEL/006) K (KEL1) Ula(KEL10) K18 (KEL18) VLAN (KEL25)
k (KEL2) K11 (KEL11) K19 (KEL19) TOU (KEL26)
Kpa(KEL3) K12 (KEL12) Km (KEL20) RAZ (KEL27)
Kpb(KEL4) K13 (KEL13) Kpc(KEL21) VONG (KEL28)
Ku (KEL5) K14 (KEL14) K22 (KEL22)
Jsa(KEL6) K16 (KEL16) K23 (KEL23)
Jsb(KEL7) Wka(KEL17) K24 (KEL24)
Lewis (LE/007) Lea(LE1) LebH (LE4)
Leb(LE2) ALeb(LE5)
Leab (LE3) BLeb(LE6)
Duffy (FY/008) Fya(FY1) Fy4 (FY4)
Fyb(FY2) Fy5 (FY5)
Fy3 (FY3) Fy6 (FY6)
Kidd (JK/009) Jka(JK1)
Jkb(JK2)
Jk3 (JK3)
Appendix 6. Blood Group Antigens Assigned to Systems (cont’d)
System (ISBT
Symbol/Number) Antigen (ISBT Number)
Copyright © 2005 by the AABB. All rights reserved.

842 AABB Technical Manual
Diego (DI/010) Dia(DI1) WARR (DI7) Vga(DI13) Fra(DI20)
Dib(DI2) ELO (DI8) Swa(DI14) SW1 (DI21)
Wra(DI3) Wu (DI9) BOW (DI15)
Wrb(DI4) Bpa(DI10) NFLD (DI16)
Wda(DI5) Moa(DI11) Jna(DI17)
Rba(DI6) Hga(DI12) KREP (DI18)
Tra(DI19)*
Yt or Cartwright Yta(YT1)
(YT/011) Ytb(YT2)
Xg (XG/012) Xga(XG1) CD99 (XG2)
Scianna (SC/013) Sc1 (SC1) Rd (SC4)
Sc2 (SC2) STAR (SC5)
Sc3 (SC3)
Dombrock (DO/014) Doa(DO1)
Dob(DO2)
Gya(DO3)
Hy (DO4)
Joa(DO5)
Colton (CO/015) Coa(CO1)
Cob(CO2)
Co3 (CO3)
LW or Landsteiner- LWa(LW5)
Wiener (LW/016) LWab (LW6)
LWb(LW7)
Chido/Rodgers Ch1 (CH/RG1) Rg1 (CH/RG11)
(CH/RG /017) Ch2 (CH/RG2) Rg2 (CH/RG12)
Ch3 (CH/RG3)
Ch4 (CH/RG4)
Ch5 (CH/RG5)
Ch6 (CH/RG6)
WH (CH/RG7)
H (H/018) H (H1)
Kx (XK/019) Kx (XK1)
Appendix 6. Blood Group Antigens Assigned to Systems (cont’d)
System (ISBT
Symbol/Number) Antigen (ISBT Number)
Copyright © 2005 by the AABB. All rights reserved.

Appendices 843
Gerbich (GE/020) Ge2 (GE2) Wb (GE5) Dha(GE8)
Ge3 (GE3) Lsa(GE6) GEIS (GE9)
Ge4 (GE4) Ana(GE7)
Cromer (CR/021) Cra(CROM1) Dra(CROM5) WESb(CROM9) ZENA (CROM13)
Tca(CROM2) Esa(CROM6) UMC (CROM10)
Tcb(CROM3) IFC (CROM7) GUTI (CROM11)
Tcc(CROM4) WESa(CROM8) SERF (CROM12)
Knops (KN/022) Kna(KN1) McCa(KN3) Yka(KN5) Sl2 (KN7)
Knb(KN2) Sla(KN4) McCb(KN6) Sl3 (KN8)*
Indian (IN/023) Ina(IN1)
Inb(IN2)
Ok (OK/024) Oka(OK1)
Raph (RAPH/025) MER2 (RAPH1)
JMH or John Milton
Hagen
(JMH/026)
JMH (JMH1)
I (I/027) I(I1)
Globoside
(GLOB/028)
P (GLOB1)
GIL (GIL/029) GIL (GIL1)
*Provisional.
Daniels GL, Anstee DJ, Cartron JP, et al. International Society of Blood Transfusion working party on terminol-
ogy for red cell surface antigens. Vox Sang 2001;80:193-6.
Daniels GL, Fletcher A, Garratty G, et al. Blood group terminology 2004. From the ISBT committee on terminol-
ogy for red cell surface antigens. Vox Sang 2004;87:304-16.
Garratty G, Dzik W, Issitt PD, et al. Terminology for blood group antigens and genes—historical origins and
guidelines in the new millennium. Transfusion 2000;40:477-89.
Issitt PD, Anstee DJ. Applied blood group serology. 4th ed. Durham, NC: Montgomery Scientific, 1998.
Appendix 6. Blood Group Antigens Assigned to Systems (cont’d)
System (ISBT
Symbol/Number) Antigen (ISBT Number)
Copyright © 2005 by the AABB. All rights reserved.

844 AABB Technical Manual
Appendix 7. Examples of Gene, Antigen, and Phenotype Terms
System Genes Antigens Phenotypes
ABO
AA
1
A
2
B
AA
1A2BAA
1A2B
Rh
DCEce
D C E c e D+C+E–c+e+
MN
MNSs
MNSs M+N+S–s+
P
P1
P1P1+ P1–
Lewis
Le le
LeaLebLe(a+)Le(a–b+)
Kell
KkKp
a
Js
a
KkKp
aJsaK–k+Kp(a+)Js(a–)
Kell
K
1
K
2
K
3
K1 K2 K3 K:–1,2,–3
Scianna
Sc
1
Sc
2
Sc
Sc1 Sc2 Sc:–1,–2,–3
Kidd
Jk
a
Jk
b
Jk
3
JkaJkbJk3 Jk(a+)Jk(a+b+)Jk:3
Modified from:
Denomme G, Lomas-Francis C, Storry J, Reid ME. Approaches to blood group molecular genotyping and its ap-
plications. In: Stowell C, Dzik W, eds. Emerging diagnostic and therapeutic technologies in transfusion medi-
cine. Bethesda, MD: AABB Press, 2003:95-129.
Garratty G, Dzik W, Issitt PD, et al. Terminology for blood group antigens and genes—historical origins and
guidelines in the new millennium. Transfusion 2000;40:477-89.
Zelinski T. Chromosomal localization of human blood group genes. In: Silberstein LE, ed. Molecular and func-
tional aspects of blood group antigens. Bethesda, MD: AABB, 1995:41-73.
Appendix 8. Examples of Correct and Incorrect Terminology*
Term Description Correct Terminology Incorrect Terminology
Phenotype Fy(a+) Fya+,Fy
(a+), Fya(+),Fy
a+, Fya(+), Duffya+,
Duffya-positive
Phenotype Fy(a+b–) Fya+b–,Fy
(a+b–),Fy
a(+)b(–), Fya(+)b(–)
Antibody Anti-FyaAnti Fya, Anti-Duffy
Antigen K Kell (name of system)
Antibody Anti-k Anti-Cellano
Phenotype K:1, K:–1 K1+, K:1+, K(1), K:(1), K1–, K:1–, K1-negative
Phenotypes A Rh+, B Rh– A+ (means positive for A antigen)
B– (means negative for B antigen)
Phenotype M+N– M(+), MM (implies unproved genotype)
Phenotype Rh:–1,–2,–3,4,5 Rh:–1,–2,–3,+4,+5
Rh:1–,2–,3–,4+,5+
Note: The examples shown may not represent the only correct terminologies. In the Rh system, for example,
use of CDE terminology is also acceptable and is more commonly used. The example demonstrates the correct
usage if numeric terminology is used.
*Issitt L. Blood group nomenclature. In: Blood groups: Refresher and updates. Bethesda, MD: AABB, 1995.
Copyright © 2005 by the AABB. All rights reserved.

Appendices 845
Appendix 9. Distribution of ABO/Rh Phenotypes by Race or Ethnicity*
Phenotype Distribution (%)†
Race or
Ethnicity Number O Rh+ O Rh– A Rh+ A Rh– B Rh+ B Rh– AB Rh+ AB Rh–
White non-Hispanic 2,215,623 37.2 8.0 33.0 6.8 9.1 1.8 3.4 0.7
Hispanic‡259,233 52.6 3.9 28.7 2.4 9.2 0.7 2.3 0.2
Black non-Hispanic 236,050 46.6 3.6 24.0 1.9 18.4 1.3 4.0 0.3
Asian§126,780 39.0 0.7 27.3 0.5 25.0 0.4 7.0 0.1
North American Indian 19,664 50.0 4.7 31.3 3.8 7.0 0.9 2.2 0.3
All donors 3,086,215 39.8 6.9 31.5 5.6 10.6 1.6 3.5 0.6
*Used with permission from Garratty G, Glynn SA, McEntire R, et al for the Retrovirus Epidemiology Donor Study. ABO and Rh(D) phenotype frequencies of different racial/ethnic
groups in the United States. Transfusion 2004;44:703-6.
†Percentages may not add up to 100.0% because of rounding.
‡Hispanic includes Mexican (68.8%), Puerto Rican (5.0%), Cuban (1.6%), and other Hispanic donors (24.6%).
§Asian includes Chinese (29.8%), Filipino (24.1%), Indian (13.8%), Japanese (12.7%), Korean (12.5%), and Vietnamese (7.1%) donors.
Copyright © 2005 by the AABB. All rights reserved.

846 AABB Technical Manual
Appendix 10. Suggested Quality Control Performance Intervals
Equipment and Reagents Frequency
I. Refrigerators/Freezers/Platelet Incubators
A. Refrigerators
1. Recorder Daily
2. Manual temperature Daily
3. Alarm system board (if applicable) Daily
4. Temperature charts (review daily) Weekly
5. Alarm activation Quarterly
B. Freezers
1. Recorder Daily
2. Manual temperature Daily
3. Alarm system board (if applicable) Daily
4. Temperature charts (review daily) Weekly
5. Alarm activation Quarterly
C. Platelet incubators
1. Recorder Daily
2. Manual temperature Daily
3. Temperature charts (review daily) Weekly
4. Alarm activation Quarterly
D. Ambient platelet storage area Every 4 hours
II. Laboratory Equipment
A. Centrifuges/cell washers
1. Speed Quarterly
2. Timer Quarterly
3. Function Yearly
4. Tube fill level (serologic) Day of use
5. Saline fill volume (serologic) Weekly
6. Volume of antihuman globulin dispensed
(if applicable)
Monthly
7. Temperature check (refrigerated centrifuge) Day of use
8. Temperature verification (refrigerated centrifuge) Monthly
B. Heating blocks/Waterbaths/View boxes
1. Temperature Day of use
2. Quadrant/area checks Periodically
C. Component thawing devices Day of use
D. pH meters Day of use
E. Blood irradiators
1. Calibration Yearly
2. Turntable (visual each time of use) Yearly
3. Timer Monthly/quarterly
4. Source decay Dependent on source type
5. Leak test Twice yearly
6. Dose delivery check (with indicator) Each irradiator use
7. Dose delivery verification
a. Cesium-137 Yearly
b. Cobalt-60 Twice yearly
c. Other source As specified by manufacturer
Copyright © 2005 by the AABB. All rights reserved.

Appendices 847
Appendix 10. Suggested Quality Control Performance Intervals (cont’d)
Equipment and Reagents Frequency
F. Thermometers (vs NIST-certified or traceable)
1. Liquid-in-glass Yearly
2. Electronic Monthly
G. Timers/clocks Yearly
H. Pipette recalibration Yearly
I. Sterile connecting device
1. Weld check Each use
2. Function Yearly
J. Blood warmers
1. Effluent temperature Quarterly
2. Heater temperature Quarterly
3. Alarm activation Quarterly
III. Blood Collection Equipment
A. Whole blood equipment
1. Agitators Day of use
2. Balances/scales Day of use
3. Gram weight (vs NIST-certified) Yearly
B. Microhematocrit centrifuge
1. Centrifuge timer check Quarterly
2. Calibration Yearly
C. Cell counters/hemoglobinometers Day of use
D. Blood pressure cuffs Periodically
E. Apheresis equipment
Checklist requirements As specified by the
manufacturer
IV. Reagents
A. Red cells Day of use
B. Antisera Day of use
C. Antiglobulin serum Day of use
D. Transfusion-transmissible disease marker testing Each test run
V. Miscellaneous
A. Copper sulfate specific gravity Day of use
B. Shipping containers for blood transport
(usually at temperature extremes)
Twice yearly
Note: The frequencies listed above are suggested intervals, not requirements. For any new piece of equipment,
installation, operational, and process qualification must be performed. After the equipment has been suitably
qualified for use, ongoing quality control (QC) testing should be performed. Depending upon the operational
and process qualification methodology, the ongoing QC may initially be performed at a greater frequency than
one ultimately wishes to use. Once a track record of appropriate in-range QC results has been established (ei-
ther during equipment qualification or the ongoing QC), the frequency of testing can be reduced, but, at a mini-
mum, the frequency must comply with the manufacturer’s suggested intervals. If no such guidance is provided
by the manufacturer, the intervals given in this table would be appropriate to use.
Copyright © 2005 by the AABB. All rights reserved.

848 AABB Technical Manual
Appendix 11. Directory of Organizations
AABB
8101 Glenbrook Road
Bethesda, MD 20814-2749
(301) 907-6977
FAX: (301) 907-6895
www.aabb.org
American Association of Tissue Banks (AATB)
1320 Old Chain Bridge Road, Suite 450
McLean, VA 22101
(703) 827-9582
FAX: (703) 356-2198
www.aatb.org
American Medical Association (AMA)
515 N. State Street
Chicago, IL 60610
(800) 621-8335
www.ama-assn.org
American Red Cross National Headquarters (ARC)
2025 E Street, NW
Washington, DC 20006
(202) 303-4498
Disaster Assistance: (866) 438-4636
www.redcross.org
American Society for Apheresis (ASFA)
570 West 7th Avenue, Suite 402
Vancouver, BC, Canada V5Z 1B3
(604) 484-2851
FAX: (604) 874-4378
www.apheresis.org
American Society for Clinical Pathology (ASCP)
2100 West Harrison Street
Chicago, IL 60612-3798
(312) 738-1336
Outside Illinois: (800) 621-4142
FAX: (312) 738-1619
www.ascp.org
American Society for Histocompatibility
and Immunogenetics (ASHI)
15000 Commerce Parkway, Suite C
Mount Laurel, NJ 08054
(856) 638-0428
FAX: (856) 439-0525
www.ashi-hla.org
American Society of Anesthesiologists (ASA)
520 N. Northwest Highway
Park Ridge, IL 60068-2573
(847) 825-5586
FAX: (847) 825-1692
www.asahq.org
American Society of Hematology (ASH)
1900 M Street, NW, Suite 200
Washington, DC 20036
(202) 776-0544
FAX: (202) 776-0545
www.hematology.org
America’s Blood Centers (ABC)
725 15th Street, NW
Suite 700, The Folger Building
Washington, DC 20005
(202) 393-5725
FAX: (202) 393-1282
www.americasblood.org
Armed Services Blood Program Office (ASBPO)
5109 Leesburg Pike, Suite 698
Falls Church, VA 22041-3258
(703) 681-8024
FAX: (703) 681-7541
www.tricare.osd.mil/asbpo
Association of Donor Recruitment Professionals
(ADRP)
P.O. Box 540524
Grand Prairie, TX 75054-0524
www.adrp.org
College of American Pathologists (CAP)
325 Waukegan Road
Northfield, IL 60093-2750
(800) 323-4040
FAX: (847) 832-8000
www.cap.org
Foundation for the Accreditation of Cellular Therapy
(FACT)
University of Nebraska Medical Center
986065 Nebraska Medical Center
Omaha, NE 68198-6065
(402) 559-1950
FAX: (402) 559-1951
www.factwebsite.org
ICCBBA, Inc.
204 St. Charles Way, Unit 179E
York, PA 17402
(717) 845-4790
FAX: (717) 845-9727
www.iccbba.com
Copyright © 2005 by the AABB. All rights reserved.

850 AABB Technical Manual
Appendix 12. Resources for Safety Information
Centers for Disease Control and Prevention (CDC)
Office of Health and Safety, Biosafety Branch
Mail Stop F-05
1600 Clifton Road
Atlanta, GA 30333
(404) 639-2453
FAX: (404) 639-2294
www.cdc.gov
Clinical and Laboratory Standards Institute (CLSI)
940 West Valley Road, Suite 1400
Wayne, PA 19087-1898
(610) 688-0100
FAX: (610) 688-0700
www.clsi.org
Department of Transportation (DOT)
Office of Hazardous Materials Standards
Research and Special Programs Administration
DHM-10
400 7th Street, SW
Washington, DC 20590-0001
(202) 366-8553
FAX: (202) 366-3012
www.dot.gov
Environmental Protection Agency (EPA)
Chemical Emergency Preparedness and Prevention
Office (5104A)
1200 Pennsylvania Avenue, NW
Washington, DC 20460
(800) 424-9346
In Washington, DC metropolitan area: (703) 412-9810
www.epa.gov
Environmental Protection Agency (EPA)
Office of Solid Waste (5305W)
1200 Pennsylvania Avenue, NW
Washington, DC 20460
(800) 424-9346
www.epa.gov/osw
Food and Drug Administration (FDA)
Center for Biologics Evaluation and Research
Division of Blood Applications, HFM-370
1401 Rockville Pike
Rockville, MD 20852-1448
(301) 827-3524
FAX: (301) 827-3535
www.fda.gov/cber
International Air Transport Association (IATA)
1776 K Street, NW, Suite 400
Washington, DC 20006
(202) 293-9292
FAX: (202) 293-8448
www.iata.org
International Civil Aviation Organization (ICAO)
999 University Street
Montreal, Quebec
Canada H3C 5H7
(514) 954-8220
FAX: (514) 954-6376
www.icao.int
National Fire Protection Association (NFPA)
1 Batterymarch Park
Quincy, MA 02169-7471
(617) 770-3000
FAX: (617) 770-0700
www.nfpa.org
National Institute for Occupational Safety and
Health (NIOSH)
Education and Information Division
4676 Columbia Parkway
Cincinnati, OH 45226-1998
(800) 356-4674
Outside the US: (513) 533-8328
Clinicians’ Post-Exposure Prophylaxis Hotline:
(888) 448-4911
FAX: (513) 533-8588
www.cdc.gov/niosh
National Institutes of Health (NIH)
Division of Safety
Building 13, Room 3K04
Bethesda, MD 20892
(301) 496-2346
FAX: (301) 402-0313
www.nih.gov
Nuclear Regulatory Commission (NRC)
Office of Public Affairs
Washington, DC 20555
(800) 368-5642
In Washington, DC metropolitan area: (301) 415-8200
FAX: (301) 415-2234
www.nrc.gov
Copyright © 2005 by the AABB. All rights reserved.

Appendices 849
Appendix 11. Directory of Organizations (cont'd)
International Society for Cellular Therapy (ISCT)
570 West 7th Avenue, Suite 402
Vancouver, BC, Canada V5Z 1B3
(604) 874-4366
FAX: (604) 874-4378
www.celltherapy.org
International Society of Blood Transfusion (ISBT)
Central Office
C/O Jan van Goyenkade 11
1075 HP Amsterdam
The Netherlands
+ 31 (0) 20 679 3411
FAX: + 31 (0) 20 673 7306
www.isbt-web.org
Joint Commission on Accreditation of Healthcare
Organizations (JCAHO)
One Renaissance Boulevard
Oakbrook Terrace, IL 60181
(630) 792-5000
FAX: (630) 792-5005
www.jcaho.org
National Hemophilia Foundation (NHF)
116 West 32nd Street, 11th Floor
New York, NY 10001
(212) 328-3700
FAX: (212) 328-3777
www.hemophilia.org
National Marrow Donor Program (NMDP)
3001 Broadway Street NE, Suite 500
Minneapolis, MN 55413-1753
(800) 627-7692
Outside the US: (612) 627-5800
www.marrow.org
Plasma Protein Therapeutics Association (PPTA)
147 Old Solomons Island Road, Suite 100
Annapolis, MD 21401
(410) 263-8296
FAX: (410) 263-2298
www.plasmainfo.org
United Network for Organ Sharing (UNOS)
700 North 4th Street
P.O. Box 2484
Richmond, VA 23218
(804) 782-4800
FAX: (804) 782-4817
www.unos.org
Note: Contact information changes rapidly. Therefore, the data listed above may not be current for the entire life of this publi-
cation.
Copyright © 2005 by the AABB. All rights reserved.
Appendices 851
Note: Contact information changes rapidly. Therefore, the data listed above may not be current for the entire life of this publi-
cation.
Occupational Safety and Health Administration (OSHA)
Office of Communications
Room N-3647
200 Constitution Avenue, NW
Washington, DC 20210
(202) 693-1999
Workplace safety and health-related questions:
(800) 321-6742
www.osha.gov
US Postal Service (USPS)
Headquarters, Room 9301
475 L’Enfant Plaza, SW
Washington, DC 20260
(800) 275-8777
www.usps.com
Appendix 12. Resources for Safety Information (cont'd)
Copyright © 2005 by the AABB. All rights reserved.
Copyright © 2005 by the AABB. All rights reserved.

Index
Index
Page numbers in italics refer to tabular or illustrative material.
A
A, B, H substances, 290-291, 298, 301, 445,
736-739
ABO compatibility
of bone grafts, 623-624
of Cryoprecipitated AHF, 411, 500-501
of Granulocytes Pheresis, 411
of HPC transplants, 598-599, 600, 601-602
of kidney transplants, 401
of liver transplants, 402, 628
of plasma products, 411, 496, 498
of platelet components, 361-362, 411,
489-490, 569
of Red Blood Cells, 411, 418, 486
of solid organ transplants, 402, 627, 629-630
of Whole Blood, 411, 486
ABO hemolytic disease of the fetus and new-
born, 536, 537, 538, 545
ABO system (ISBT 001), 289-303
acquired B phenotype, 298, 301-302
antibodies, 294-296
age variations, 295, 298, 299
anti-A and anti-B, 294-296
anti-A1, 295-296, 302
anti-A,B, 295, 296
in HDFN, 536, 538, 545
passively acquired, 563
reactivity, 295
antigens, 293
in newborns, 293
nomenclature, 226, 840, 844
on platelets, 361-362, 397
weakly expressed, 297-298, 300-301
B(A) phenotype, 298, 301
discovery of, 289
genetics and biochemistry, 229-230, 235,
290-293, 844
phenotypes, 844, 845
soluble substances, 290-291, 298, 301, 445,
736-739
subgroups, 293-294
in ABO discrepancies, 302
confirmation of by adsorption/elution,
735-736
testing for, 296
ABO testing
of autologous blood, 122
of blood components, 164, 165
in children, 296, 415, 545, 563, 574
with cold autoagglutinins, 299, 302-303, 469
comparison with previous records, 413, 526
confirmationofweakAorBsubgroup,
735-736
853
Index
Copyright © 2005 by the AABB. All rights reserved.
of cord blood, 545, 595
discrepancies, 296-303, 469
absence of expected antibodies, 301
absence of expected antigens, 300-301
causes, 297
mixed-field agglutination, 298, 299
resolving, 300-303
specimen-related problems, 297-299
technical errors, 299-300
unexpected red cell reactions, 301-302
unexpected serum reactions, 302-303
hemolysis in, 295
microplate tests, 733-735
for organ transplantation, 621, 629
of Platelets Pheresis, 142
in prenatal studies, 538
reagents, 296
of recipients, 122, 410, 411
routine, 290, 296
slide tests, 731-732
in transfusion reaction evaluation, 654
tube tests, 732-733
ABTI antigen, 355
Accidents, 45-46
ACE (angiotensin-converting enzyme) inhibi-
tion, 636, 645
Acid-elution stain (Kleihauer-Betke), 550-551,
794-796
Acid glycine/EDTA, 446, 469, 747-748
Acidosis, in neonates, 561
Acquired antigens, 298, 301-302
Activated partial thromboplastin time (aPTT)
in massive transfusion, 511
monitoring hemostasis with, 494, 496
normal values, 837
in vitamin K deficiency, 498
Active failures, 26
Acute lung injury (ALI), 647
Acute normovolemic hemodilution (ANH), 117,
126-130
clinical studies, 128, 129, 130
physiologic considerations, 127-128
practical considerations, 128, 130
selection of patients, 131
Acute transfusion reactions. See Transfusion re-
actions
Adaptive (acquired) immunity, 243-244
ADCC (antibody-dependent cellular
cytotoxicity), 255, 441
Additives
anticoagulants-preservatives for red cells,
176, 186, 564-565
in serologic testing, 276-277, 437-438, 443,
753-754
Adhesion molecules, 246, 269
Adsol (AS-1), 176, 186, 565
Adsorption
allogeneic, 471-472, 781-782
autologous cold, 438, 466, 472, 775-776
autologous warm, 470-471, 779-781
differential, 781-782
and elution, 300-301, 448, 735-736
methods, 768-769, 775-776, 779-784
polyethylene glycol, 783-784
purposes, 446-447
with rabbit red cells, 439, 466
Adsorption separation, 140
Adult T-cell lymphoma/leukemia (ATL), 682
Adverse reactions
to apheresis, 141, 150-153
to DMSO, 607
in donors, 19, 107-109
to G-CSF, 594-595
management, 19, 20, 21
to transfusions
action for, 531
autologous, 123
infectious, 667-703
noninfectious, 633-661
records, 660-661
reporting, 19, 20, 21 , 661, 702-703
AET (2-aminoethylisothiouronium bromide),
342, 446
Affinity constant (Ko), 273
Age
of blood samples, 410, 431, 433
of donor, 98
effect on ABO antigens/antibodies, 293, 295,
298, 299
Agglutination
defined, 271
factors affecting, 272-275
antigen-antibody proportions, 274-
275
chemical bonding, 273
equilibrium (affinity) constant, 273
incubation time, 274, 444
ionic strength, 274
854 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
pH, 274, 444
temperature, 273-274, 336, 443
grading reactions, 412, 728-729
inhibition, 275-276, 444-445
mixed-field, 298, 299, 348, 357
stages, 272-275
tests for granulocyte antibodies, 380
Agreements, 10, 177
AHF. See Cryoprecipitated Antihemophilic Fac-
tor
AHG test. See Antiglobulin test
AIDS (acquired immunodeficiency syndrome),
675-676, 677. See also HIV
AIHA. See Autoimmune hemolytic anemia
Air embolism, 636, 651-652
AITP (autoimmune thrombocytopenic
purpura), 158, 373-374, 492, 553-554
Alanine aminotransferase (ALT), 164, 673, 835
Alarm systems, 185, 198-199, 823-826, 846
Albumin
antibodies to ingredients in, 437-438
as colloid replacement, 507-508
physiology, 507
recombinant, 219
as replacement fluid in apheresis, 150, 151
in serologic testing, 276, 427, 753
use in exchange transfusion, 567
Aldehyde dehydrogenase (ALDH), 603
ALI (acute lung injury), 647
Aliquoting components, 193-194, 564, 571
Allele-specific oligonucleotide (ASO) hybridiza-
tion. See Sequence-specific oligonucleotide
probes
Alleles, 225, 227. See also specific blood groups
blank, 389
dosage effect, 225, 227
frequencies, 227-229
rare, 233, 234
Allelic, defined, 241
Allergic reactions
in apheresis, 152
to latex, 46-47
to transfusions, 522, 634, 644-647
Alloantibodies, red cell. See also Antibody detec-
tion; Antibody identification; specific blood
groups
in ABO discrepancies, 299, 303
with autoantibodies, 438-439, 470-472
in bone graft patients, 623
clinical significance, 411, 418, 423-424, 439, 441
dosage effect, 225, 227, 327, 345, 425, 431
to high-incidence antigens, 356-357,
435-436, 441-442, 547
in liver transplant patients, 629
to low-incidence antigens, 340, 358, 436-437,
546
multiple, 236, 434-435, 441-442, 449
in selection of units, 418, 439-443
serologic behavior, 336
in sickle cell disease, 576
temperature of reaction, 273-274, 336, 443
in thalassemia patients, 576
titration, 449, 539-540, 761-764, 796-798
Allogeneic adsorption, 471-472, 781-782
Allogeneic HPC transplantation
ABO incompatibilities, 598-599, 600, 601-602
cell processing, 596-597, 598, 601-604
collection of products, 591-596
defined, 582
donor evaluation, 589-591
evaluation and QC of products, 607-608
freezing and storage of products, 604-606
graft-vs-host disease in, 586-587
of HPC-A (apheresis), 587-588, 592, 594-595
of HPC-C (cord blood), 588-589, 595-596
of HPC-M (marrow), 583, 585-587, 591-592
indications for, 582
infectious disease testing, 590-591
matched, unrelated transplantation,
585-586, 589
mobilization of HPCs, 587, 594-595
nonmyeloablative, 582-583
positive DAT after, 455
regulations, 608
related transplantation, 587-588, 589
standards, 609
thawing and infusion of products, 607
transfusion support in, 591-592, 599, 600,
601
transportation and shipping of products,
606-607
Allografts. See Organ donation and transplanta-
tion; Tissue transplantation
Alloimmune hemolytic anemia. See Hemolytic
disease of the fetus and newborn; Hemolytic
transfusion reactions
Alloimmunization. See also Alloantibodies, red cell
direct/indirect allorecognition, 265
Index 855
Copyright © 2005 by the AABB. All rights reserved.
HLA, 265, 266, 362-366, 397-398, 637
in sickle cell disease, 575, 576
as transfusion complication, 265, 637,
656-657
Allotypic, defined, 269
α-methyldopa, 455, 475-476
ALT (alanine aminotransferase), 164, 673, 835
AMD 3100, 594
American Rare Donor Program (ARDP), 441-442,
769-770
American trypanosomiasis (Chagas’ disease),
697-698, 700
2-aminoethylisothiouronium bromide (AET),
342, 446
Amniocentesis, 539, 540, 541, 549
Amniotic fluid analysis, 540-541, 542
Ana(Gerbich) antigen, 352, 843
Anaphylactic transfusion reactions, 644-647
evaluation, 655
frequency, 645
management, 635
pathophysiology, 635, 644-645
prevention, 646-647
symptoms, 639, 644-645
treatment, 645-646
Anaphylactoid transfusion reactions, 644, 645
Anaphylatoxins, 262, 639
Anemia. See also Hemolytic disease of the fetus
and newborn
alloimmune hemolytic, 459
autoimmune hemolytic, 458-477, 509-510
cold agglutinin syndrome, 459, 460, 464-
466
DAT-negative AIHA, 459, 468
drug-induced hemolytic, 459, 460, 472-477,
481-482
IgM warm AIHA, 464
mixed-type AIHA, 459, 460, 466-467
in neonates, 558, 559, 563-564
oxygen compensation in, 484
paroxysmal cold hemoglobinuria, 459, 460,
467-468
screening donors for, 102, 799
sickle-cell disease, 119, 155-156, 508-509,
574-576
in thalassemia, 508
treatment, 487-488
warm autoimmune hemolytic anemia, 459,
460, 461-464
Angiotensin-converting enzyme (ACE) inhibi-
tion, 636, 645
ANH. See Acute normovolemic hemodilution
Anhydrous, defined, 724
Ankylosing spondylitis, 403
Anti-A and anti-B, 294-296
Anti-A1, 293, 295-296, 302
Anti-A,B, 295, 296
Anti-C3b, -C3d, 279, 280, 456
Anti-CMV, 170
Anti-D
active vs passive antibody, 548
antibody titrations, 539-540
in D+ individuals, 327
discovery of, 315-316
dosage effect, 327
in HDFN, 330, 535, 536-537, 539-540
in liver transplantation, 628-629
partial D, 322-323
reagents, 328-330
and weak D, 323
Anti-Delta, 670
Anti-HAV, 671
Anti-HBc (antibody to hepatitis B core antigen)
blood component testing, 164, 166, 675
look-back for, 675
marker of infection, 669, 670
reentry of donors with, 675
as surrogate marker, 672
testing transplant donors for, 621
Anti-HBe (antibody to hepatitis B e antigen),
669, 670
Anti-HBs (antibody to hepatitis B surface anti-
gen), 669, 670
Anti-HCV (antibody to hepatitis C virus)
blood component testing, 164, 166, 170
marker of infection, 670-671, 672-673
reentry of donors with, 673, 674, 675
supplemental testing, 170
testing organ donors for, 620, 621, 623
Anti-HEV, 671
Anti-HIV-1, 2
reentry of donors with positive tests, 674,
681
supplemental testing, 169-170
testing blood donors for, 164, 166, 169-170,
676, 679-681
testing organ donors for, 620, 621,
623
856 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Anti-HTLV-I, II
blood component testing, 120, 164, 166, 170,
683
donors for organ transplants, 620
supplemental testing, 170
Anti-IgA, 645, 646, 655
Anti-IgG, 279, 280, 427, 438, 456
Anti-K1, 536, 538, 540
Anti-Ku, 343
Anti-Pr, 777, 778
Antibodies. See also Alloantibodies; Antigen-
antibody reactions; Autoantibodies; other
Antibody entries; specific blood groups
complement-binding, 280
concomitant, 327-328, 441
defined, 269
distinguishing IgG and IgM, 764-765
drug-induced, 374-377, 472-477, 481-482,
786-791
equilibrium constant (Ko), 273
high-titer, low avidity, 449, 762, 765-
766
HLA
in antibody identifications, 433
and Bg antigens, 358
detection, 365-366, 397
in platelet refractoriness, 362-365,
397-398, 491
in transfusion reactions, 398-400, 637,
647, 656
monoclonal
in ABO testing, 296
anti-D, 328, 329-330
assays utilizing, 284, 373, 380
in autologous tumor purging, 597-598
for reagent use, 266-267
in neonates, 560
platelet
autoantibodies, 373-374
clinical importance, 370-373
detecting, 370-373, 374, 375, 377, 552
drug-induced, 374-377
HLA, 266, 362-366, 397-398
platelet-specific, 366, 367-368, 370-373,
552
production, 249
reagent, 266-267
to reagent components, 298, 437-438
temperature of reaction, 273-274, 443
Antibody-dependent cellular cytotoxicity
(ADCC), 255, 441
Antibody detection
antiglobulin test, 277-281, 282, 283
with autoantibodies, 438-439, 463-464,
470-472
in autologous blood, 122
autologous control, 412, 427, 438-439
automated testing, 285
in blood components, 165
in children, 415, 563, 574
column agglutination for, 278, 284-285
in cord blood, 595
ELISA for, 286
enhancement, 276-277, 427, 443-444,
753-754
enzyme-linked immunosorbent assay, 286
frequency of testing, 442-443, 463-464
of granulocyte autoantibodies, 380
of HLA antibodies, 365-366, 397
immunofluorescence, 285-286
indirect antiglobulin test, 277, 278, 752-754
interpreting reactions, 415-416, 417
MAIEA assay, 284, 286
in organ transplantation, 629
of platelet antibodies, 370-373, 374, 375, 377
in Platelets Pheresis, 142
in positive DAT evaluation, 456
practical considerations, 412
in prenatal evaluations, 539
in pretransfusion testing, 411-413, 417
prewarming technique, 308, 438, 754-755
reagents, 278-280, 425, 427, 468
solid-phase red cell adherence tests, 283-284
specimen requirements, 424
in transfusion reaction evaluation, 654
Antibody identification, 427-450
ABO type of red cells tested, 434
adsorption, 446-447, 448, 470-472
anomalous reactions, 437-438
antibodies to high-incidence antigens,
356-357, 435-436, 441-442, 547
antibodies to low-incidence antigens, 340,
358, 436-437, 546
with antibodies to reagent components,
437-438
with autoantibodies, 438-439, 463-464,
470-472
autologous control, 427, 429-430, 438-439
Index 857
Copyright © 2005 by the AABB. All rights reserved.
elution, 447-448, 456-458
enhancement techniques, 276-277, 427,
443-444, 753-754
flowchart for, 432
frequency of testing, 442-443, 463-464
inactivation of antigens, 445-446, 766-767
inhibition tests, 444-445
interpreting results, 428-429
medical history in, 424
multiple antibodies, 236, 434-435, 441-442,
449
no discernible specificity, 433-434
phenotyping autologous red cells, 429-430,
439
with positive DAT, 427, 436, 438-439, 456
in prenatal evaluations, 539
prewarming technique, 308, 438, 754-755
probability values, 429, 430
reagents, 278-280, 425, 426, 427, 468
and selection of blood, 418, 439-443, 463
specimen requirements, 424
sulfhydryl reagents in, 446, 448-449, 744-745,
766-767
titration studies, 449, 539-540, 761-764,
796-798
in transfusion reaction evaluation, 654
using immunohematology reference labs,
439
variations in antigen expression, 431, 433,
468
Antibody screens. See Antibody detection
Antibody specificity prediction (ASP) method,
365
Anticoagulant-preservative solutions, 178-179
CPD, CP2D, CPDA-1, 176, 178, 186, 564,
565
red cell changes during storage, 185, 186,
187, 431, 433
shelflifeofcomponents,178, 188, 189-190
Antifibrinolytic agents, 513-514
Antigen-antibody reactions. See also Antibody
detection; Antibody identification
agglutination, 271, 272-276
hemolysis, 271-272
precipitation, 272
prozone, 272, 275
Antigen-presenting cells (APCs), 269
Antigens. See also specific blood groups
acquired, 298, 301-302
blood group nomenclature, 238-239,
318-319, 840-844
CD, 246, 247-248
collections, 335, 355-356
defined, 269
depressed, 341-342, 468
distribution in population, 335, 337
granulocyte/neutrophil, 377-380
high-incidence
antibodies to, 356-357, 435-436, 441-442,
547
of Cromer system, 353, 354
defined, 335
of Gerbich system, 352
GIL, 355
of Knops system, 354
of Lutheran system, 347
not assigned to a system or collection,
356-357
with positive DAT, 436
selecting blood negative for, 441-442
of Vel collection, 355-356
HLA, 362, 385, 388-389, 390-391
inactivation, 342, 445-446, 766-767
low-incidence, 357-358, 436-437, 546
antibodies to, 340, 358, 436-437, 546
of Cromer system, 353, 354
defined, 335
of Gerbich system, 352
of Lutheran system, 347
of MNS system, 227, 228, 337-338, 340
not assigned to a system or collection,
357
of Scianna system, 350
platelet, 361-363, 366, 367-368, 397, 552
public, 392
typing donor units for, 418, 440-441, 657,
745-748
variations in expression of, 431, 433, 468
Antiglobulin (AHG) test. See also Direct
antiglobulin test
in antibody detection/identification,
277-281, 282, 283
complement in, 280-281, 453
in crossmatching, 413, 414
direct, 278, 454-458, 760-761
indirect, 278, 752-754
principles, 277-278
reagents, 278-280, 427, 454, 468
858 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
sources of error, 282, 283
use of IgG-coated cells, 281, 412
Antiprotease concentrates, 505-506
Antipyretics, 522
Antithrombin, 500, 505-506
AnWj antigen, 356-357
APCs (antigen-presenting cells), 269
Apheresis
complications, 141, 150-153
for component collection, 140-144
donation intervals, 141, 143
equipment, 139-140, 152, 847
HPCs collected by, 581, 587-588, 592-595
monitoring cell counts, 832
records, 142
separation techniques, 139-140
therapeutic, 144-158
indications for, 146-148, 153-158
photopheresis, 158
plasma volumes exchanged, 145, 149
removal of normal plasma constituents,
149-150
removal of pathologic substances, 145,
148-149
replacement fluids, 150, 151
SPA immunoadsorption, 158
vascular access, 145, 150
Aprotinin, 502, 513-514
aPTT. See Activated partial thromboplastin time
Arachis hypogaea (anti-T), 743
ARDP (American Rare Donor Program), 441-442,
769-770
Arrhythmias, 650
Arterial transplants, 624
Articular cartridge transplants, 624
AS (additive solutions), 176, 186, 564-565
ASP (antibody specificity prediction) method,
365
Assessments, 22-24
blood utilization, 23, 36-38, 514
competency, 8
external, 23-24
internal, 22
proficiency testing, 24
quality indicators, 22-23, 31
transfusion oversight, 514
Ataantigen, 356
ATL (adult T-cell lymphoma/leukemia), 682
Audits, transfusion, 23
Auto controls. See Autologous controls
Autoadsorption. See Adsorption
Autoantibodies
cold
ABO discrepancies with, 299, 302-303,
469
adsorption, 438, 466, 472, 775-776
with alloantibodies, 438-439, 472
anti-I/i, 306, 307-308, 438, 465-466
anti-IH, 308, 434, 777, 778
in cold agglutinin syndrome, 281, 307,
465-466
and complement, 281
determining specificity, 776-778
in mixed AIHA, 466, 467
in paroxysmal cold hemoglobinuria,
467
in Rh testing, 329, 469-470
titration, 458, 465, 778-779
use of sulfhydryl reagents with, 469-470,
744-745
in warm autoimmune hemolytic anemia,
461
in DAT-negative AIHA, 468
granulocyte, 380
platelet-specific, 373-374
posttransfusion, 657
warm
adsorption, 470-472, 779-781
with alloantibodies, 439, 470-472
drug-induced, 475-476
frequency of testing, 463-464
mimicking alloantibodies, 472
transfusion-stimulated, 462
transfusion with, 462-464
in warm autoimmune hemolytic anemia,
461-462
Autoclaving biohazardous waste, 57
Autoimmune hemolytic anemia (AIHA)
classification, 458, 459
cold agglutinin syndrome, 459, 460, 464-466
complement coating cells, 281, 460, 465
DAT-negative AIHA, 459, 468
IgM warm AIHA, 464
mixed-type AIHA, 459, 460, 466-467
paroxysmal cold hemoglobinuria, 459, 460,
467-468
serologic findings in, 460
transfusion in, 462-464, 467, 468, 509-510
Index 859
Copyright © 2005 by the AABB. All rights reserved.
warm autoimmune hemolytic anemia, 459,
460, 461-464
Autoimmune neutropenia, 379-380
Autoimmune platelet disorders, 373-374
Autoimmune thrombocytopenic purpura
(AITP), 158, 373-374, 492, 553-554
Autologous adsorption
cold, 438, 466, 472, 775-776
warm, 470-471, 779-781
Autologous blood
acute normovolemic hemodilution, 117,
126-130
clinical studies, 128, 129, 130
physiologic considerations, 127-128
practical considerations, 128, 130
selection of patients, 131
bacterial contamination of, 693
categories, 117
for HIV-positive donors, 117, 681
intraoperative blood collection, 117, 130-133
clinical studies, 131-132
controversies in, 132-133
direct reinfusion, 133
equipment for, 132-133
practical considerations, 132
processing, 132-133
requirements and recommendations, 133
inventory management, 94-95
for patients needing rare blood types, 442
postoperative blood collection, 117, 133-135
preoperative blood donation, 117, 118-126
advantages/disadvantages, 118
adverse reactions, 123
aggressive phlebotomy, 125
collection, 121, 126
compliance requirements, 119-120
continuous quality improvement,
123-124
contraindications, 119
cost-effectiveness, 125, 127
donor deferrals, 120
donor screening, 121-122, 124-125
erythropoietin use, 125
establishing program, 120-124
labeling, 120, 122-123
medical interview, 122
in pediatric patients, 118-119
physician responsibility, 120-121
records, 111, 123
shipping, 120
storage, 123
supplemental iron use, 121
testing, 119-120, 122
timing and red cell regeneration during,
122
transfusionofunits,123
transfusion trigger, 125
volume collected, 122
voluntary standards, 119
weakDindonor,324
separation from transfused cells, 748-750
Autologous controls
in antibody identification, 427, 429-430,
438-439
positive, 417, 438-439
in pretransfusion testing, 412, 417, 454
Autologous HPC transplantation
autologous tumor purging, 597-598
cell processing, 596-604
collection of products, 591-596
defined, 582
donor evaluation, 589, 590-591
evaluation and QC of products, 607-608
freezing and storage of products, 604,
605-606
of HPC-A (apheresis), 587, 592-595
of HPC-C (cord blood), 589, 595-596
of HPC-M (marrow), 583, 591-592
infectious disease testing, 590-591
mobilization of HPCs, 592-594
regulations, 608
standards, 609
thawing and infusion of products, 607
transportation and shipping, 606-607
Autologous tumor purging, 597-598
Automated testing platforms, 285
Autosomal dominant and recessive traits, 233,
234
B
5b antigen, 378
B-cell receptor (BCR), 249
B lymphocytes, 249-250, 252-253
receptors/markers on, 249-253
stimulation of, 254-255
β2-microglobulin, 245
B(A) phenotype, 298, 301
Babesiosis, 695-696, 700
860 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Bacterial contamination, 690-695
clinical considerations, 692-693
fatalities associated with, 690-692
of HPCs, 602-603
organisms involved, 691, 692
platelet-associated, 690-692, 694-695
preventive measures for, 693-694
of reagents, 331, 332
of Red Blood Cells, 691
testing components for, 195, 655, 693, 694
in transfusion-associated sepsis, 635, 643,
655, 691-692
transfusion risk of, 700
Bacteriologic studies
of components, 195, 655, 693, 694
of hematopoietic products, 602-603
in transfusion reaction evaluation, 655
Bags, blood, 104-105
centrifugation of, 179-180
for frozen storage, 182
returning to lab, 531-532
Barcode labels, 171
Basophils, 255
BCR (B-cell receptor), 249
Bennett-Goodspeed (Bg) antigens, 358, 391
Bilirubin
in HDFN, 536, 540-541, 542, 547
hyperbilirubinemia, exchange transfusion
for, 566-567
total, normal values, 835, 836
Bioassays, 65
Biohazardous waste
defined, 55-56
disposal, 56-57
treating, 57
Biologic product deviations, 19, 21
Biological products, 717
Biological safety cabinets (BSCs), 50, 51, 52-53
Biosafety, 49-57
biosafety levels and precautions, 49-50,
54-55, 77
Bloodborne Pathogen Standard, 49
decontamination, 51, 54
emergency response plan, 55-57
engineering controls, 50-51
hazard identification and communication,
50
labeling, 719
personal protective equipment, 54
safe work practices, 54-55
shipping dangerous goods, 716-722
Standard Precautions, 49
storage, 54
training, 50
waste management, 55-57
Biphasic hemolysin of paroxysmal cold
hemoglobinuria, 467
Blank alleles, 364
Bleeding
indication for transfusion, 486
microvascular, 511, 651
with platelet defects, 490
transfusion-related, 639
Bleeding time, 488, 489, 837
Blocking phenomenon, 330
Blood administration
assessment, 37-38
blood warmers for, 522, 529, 560, 650, 847
compatibility testing, 524
component preparation in, 522, 523
delays in starting, 525
delivering blood to transfusion area, 524-
525
electromechanical pumps for, 522, 523,
529-530, 565
emergency release, 522-523
errors, 525-526, 527, 641, 642, 653, 654
filters for, 528-529, 565-566
identification procedures, 525-526
infusion sets for, 527-528, 565-566
IV solutions, 530
in neonates, 565-566
patient care during, 530-531
patient consent, 521-522
patient considerations in, 522
patient education, 521-522
post-administration events, 531-532
prescription for, 522-523
pressure devices for, 530
quality assurance, 532
reaction evaluation, 531, 652-656
special instructions for, 522-523
starting the transfusion, 526-527
venous access, 523-524, 565-566
Blood bags, 104-105
centrifugation of, 179-180
for frozen storage, 182
returning to lab, 531-532
Index 861
Copyright © 2005 by the AABB. All rights reserved.
Blood collection
adverse donor reactions, 19, 107-109
anticoagulants and preservatives, 178-179,
186
blood containers, 104-105
of blood samples, 105-106, 408-409, 801-804
care of donor, 106
of components by apheresis, 140-144
equipment quality control, 847
fatalities related to, 19
identification in, 98, 105
intraoperative, 117, 130-133
phlebotomy, 105-106, 178, 693, 801-804
postoperative, 117, 133-135
preoperative, 126
preoperative autologous, 118-126
preparing venipuncture site, 105, 800-801
records, 97-98, 165
safety, 41, 51
volume collected, 101, 122, 178-179
of whole blood, 178-179
Blood component selection
ABO/Rh compatibility in, 411, 418, 486,
489-490, 496, 498
after non-group-specific transfusions,
419-420
with alloantibodies, 418, 439-443
for exchange transfusions, 546-547, 567
for intrauterine transfusions, 544
for massive transfusions, 419, 510-511
platelets, 363-365, 489-490, 569
rare types, 441-442
red cell components, 411, 486
in urgent situations, 419, 510-511
in warm autoimmune hemolytic anemia,
463
Blood components. See also Blood collection;
Blood component selection; specific compo-
nents
aliquoting, 193-194, 564, 571
appearance, 194, 525, 655
bacterial contamination, 195, 643, 655,
690-695
for children, 564-572, 574-577
CMV-negative, 95, 172
disposition, 196
distribution, handling and dispensing, 36-37
for exchange transfusion, 567
expiration and shelf life, 178, 188, 189-190
freezing, 180-183, 807-812
for hemostasis, 496, 497, 498-507, 570-572
identification, 105, 416, 524-526, 527
inspection, 93-94, 194-195, 416, 418, 525
irradiated, 183, 192-193, 493
issuing, 196-197, 416, 418, 524-525
labeling, 15, 170-172, 416, 440
leukocyte reduction, 180, 190, 492-493,
573-574
ordering, 36, 91-94, 522-523, 526
outdating, factors affecting, 90-91
pathogen inactivation, 702
phenotyping, 440-441
pooling, 183-184, 193, 815
preparation, 173, 179-184, 522, 523, 804-818
quality control, 197-199, 202
quarantine, 173-174, 194
records, 165, 172-173, 416, 418
reissuing, 197, 525
rejuvenation, 194, 806-807
returning, 525
storage, 93-94, 184-185, 186, 187
testing
ABO and D, 164, 165
alanine aminotransferase, 164
antibody screening, 165
cytomegalovirus, 170
equipment requirements, 164-165
general requirements, 163-164
hepatitis, 669-673
HIV, 164, 166, 169-170, 679-681
HTLV, 120, 164, 166, 170, 683
records, 165
syphilis, 165-166, 695
viral markers, 166-170
thawing, 191-192, 809, 815
transportation and shipping, 66, 179,
195-196, 717, 722
volume reducing, 193, 490, 569, 817-818
washed, 193
Blood containers, 104-105, 179-180, 182, 564
Blood donation. See Blood collection
Blood exposure, 45, 54-55
Blood Group Antigen Mutation Database, 236
Blood group sugars, 445
Blood group systems. See also specific blood
groups
chromosomal assignments, 225, 226,
235-236
862 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
co-dominant traits in, 234-235
defined, 335
distribution of antigens, 335, 337
genetics
basic principles, 223-225
and heredity, 225-232
patterns of inheritance, 232-236
population genetics, 236-237
nomenclature, 238-239, 318-319, 840-
844
serologic behavior of antibodies, 336
Blood inspections
before release, 194-195, 416, 418, 525
of inventory, 93-94
Blood loss
dilutional coagulopathy in, 499-500, 651
due to platelet dysfunction, 490
in preterm neonates, 559
transfusion in, 486-487
Blood ordering practices
assessment, 36
improving, 91-94
physicians’ orders, 522-523, 526
for routine vs emergency orders, 93, 414
Blood pressure, 633
of donors, 102
hypertension, 633
hypotension
in donors, 107
in recipients, 585, 586, 591, 595
in therapeutic apheresis, 152
treatment, 592
Blood pressure cuffs, 847
Blood samples, 409-410
age of, 410, 431, 433
appearance, 410
frozen, 184-185
hemolyzed, 410
identification and collection, 105-106,
408-409, 801-804
incompletely clotted, 409, 722-723
labeling, 409, 719
lipemic, 410
packaging, 717-718, 720-721
requirements for testing, 424, 454
retention and storage, 410
transportation and shipment, 66, 717-721
Blood spills, 55, 56
Blood transfusions. See Transfusions
Blood utilization
assessments, 23, 36-38
blood ordering practices, 36, 91-94, 414
inventory levels, 89-90, 93
maximum surgical blood order schedules,
92-93, 414
outdating factors, 90-91
routine vs emergency orders, 93
of special components, 94-95
units processed, transfused and outdated in
2001, 91
Blood volume, 558-559, 839
Blood warmers, 522, 529, 560, 650, 847
Bloodborne Pathogen Standard, 49
Bombay (Oh) phenotype, 304
Bonds, chemical, 273
Bone banking, 623, 625
infectious disease transmission in, 619
preservation and dating periods for, 624
Bone marrow. See Marrow transplantation
Bovine aprotinin, 502, 513-514
Bovine spongiform encephalopathy (BSE), 690
Burkitt’s lymphoma, 687
C
C/c antigens and antibodies (Rh system), 316,
841
cis product antigens, 324
concomitant antibodies, 327-328, 441
dosage in, 317
expression of, 320
gene complexes, 319
in HDFN, 536
incidence of, 317, 321
phenotypes, 320
testing for, 330-331, 441
C1-esterase inhibitor, 500
Calcium, 151-152, 561, 636, 649-650
Calculations
for CRYO dose, 501
for Factor VIII dose, 503-504
for multiple alloantibodies, 236, 441
Calibration
of cell washers, 830-832
defined, 30
of platelet separation centrifuges, 179,
826-828
of serologic centrifuges, 828-830
of thermometers, 198, 821-823
Index 863
Copyright © 2005 by the AABB. All rights reserved.
CAP (College of American Pathologists), 514
Capture-P, 372
Cardiovascular system. See Heart and heart valves
Carriers, defined, 233
Cartwright (Yt) system (ISBT 011), 226, 336, 348,
349, 842
CAS. See Cold agglutinin syndrome
Catheters
for apheresis, 145
for HPC collection, 593, 594
for neonates, 565, 568
for transfusions, 523-524
Cause-and-effect diagrams, 26
CBER (FDA Center for Biologics Evaluation and
Research), 19, 702-703
CCE (counterflow centrifugal elutriation),
596-597
CCI (corrected platelet count increment), 362,
363, 489
CD (cluster of differentiation) markers, 246,
247-248, 250
CD34
analysis/enumeration, 603
collection targets, 593-594
defined, 592
selection of, 596-597
Cefotetan antibodies, 477
Cell adhesion molecules, 246, 269
Cell counters, 847
Cell counts
in apheresis components, 832
in HPC products, 607-608
Cell selection techniques, 596-598
Cell therapy. See Hematopoietic progenitor cell
transplantation
Cell washers, 830-832, 846
Center for Biologics Evaluation and Research
(CBER), 19, 702-703
Central venous catheters, 524, 593
Centrifugation
apheresis separation by, 139-140
in component preparation, 179-180
counterflow elutriation, 596-597
in separating autologous cells, 748-749
standardization of variables, 716
Centrifuges
calibrating, 179, 826-830
continuous-flow, 596-597
QC performance intervals, 846, 847
Centromere, 224, 241
Cephalosporins, 476
detection of antibodies to, 786-788
in positive DAT, 455, 474, 476, 477
Cephalothin (Keflin), 476
CFR (Code of Federal Regulations), 1, 32
CGD (chronic granulomatous disease), 343
Ch (Chido/Rodgers) antigens, 351-352, 842
Chagas’ disease, 697-698, 700
Change control, defined, 30
Charts, run and control, 23
Check cells, 281, 412
Chemical bonds, 273
Chemical Hygiene Plan (CHP), 57-58
Chemical safety, 57-63
chemical categories, 82-83
chemical data sheet, 78-79
chemical hygiene officer, 58
chemical spills, 61-62, 84-88
chemicals found in blood banks, 80-81
engineering controls, 61
general principles, 57-58
hazard communication, 59
hazardous vapors, 62, 63
health hazards categories, 58, 59
labeling and signs, 59-60
material data safety sheets, 59, 60-61, 78-79
personal protective equipment, 61
safe work practices, 61, 82-83
spills, 61-62, 84-88
training, 58-59
waste disposal, 63
Chemiluminescence assay, 380, 441
Chemokines, 594
Chido/Rodgers system (ISBT 017), 351-352
antibodies, 352
antigens, 351-352, 842
genes, 226
plasma inhibition studies, 445, 765-766
Children. See also Hemolytic disease of the fetus
and newborn
ABO antigens/antibodies in, 293, 295, 298,
420
ABO discrepancies in, 298
anemia in, 558, 559, 563-564
autologous collection from, 118-119
blood volume of, 558-559, 839
compatibility testing in, 415, 562-563, 574
cytomegalovirus in, 562
864 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
DIC in, 567, 572
erythropoiesis in, 557-558, 559
extracorporeal membrane oxygenation in,
572-573, 574
graft-vs-host disease in, 560-561
hematopoietic transplantation in, 587, 593
hypothermia in, 560
immunologic status, 560-561
leukocyte reduction for, 573-574, 576
Lewis antigens in, 306
metabolic problems in, 561-562
normal laboratory values in, 836-837
paroxysmal cold hemoglobinuria in, 468
plasma volume of, 839
polycythemia in, 572
red cell volume of, 839
size of, 558-559
transfusions in
administration, 565-566
aliquoting for small volumes, 193-194,
564, 571
Cryoprecipitated AHF, 572
education and consent, 521
to enhance hemostasis, 570-572
exchange, 546-547, 560, 566-568
FFP, 571, 576-577
Granulocytes, 569-570
with hemoglobinopathies, 574-576
indications for, 563-564
older infants and children, 574-577
Platelets, 490, 552-553, 568-569, 576-577
Red Blood Cells, 564-568
with sickle cell disease, 574-576
with thalassemia, 575-576
volumes for, 558, 564, 568
variations in antigen expression, 431
Chills with transfusions, 633, 634, 643
Chimerism
blood group, 237, 299
hematopoietic, 298, 601
and posttransfusion GVHD, 399
transfusion, 298, 399
Chloroquine diphosphate, 446, 469, 746-747
Chorionic villus sampling, 539
CHP (chemical hygiene plan), 57-58
Christmas disease, 505
Chromatid, 241
Chromatin, 223, 241
Chromosomes. See also Genes
defined, 223-224, 241
DNA structure in, 203-204
locations of blood group genes, 225, 226,
235-236
Chronic granulomatous disease (CGD), 343
Chronic inflammatory demyelinating
polyneuropathy (CIDP), 156
Circulatory overload, 636, 648-649
Circulatory shock, 633
Cis product antigens, 319-320, 324, 342
Citrate toxicity, 151, 636, 649-650
CJD (Creutzfeldt-Jakob disease), 689-690
Class I, II, III antigens (HLA), 244-246, 386-394
biochemistry and structure, 390-391
biologic function, 393-394
defined, 269, 385
genetics, 386-387
nomenclature, 392-393
Clinical specimens
defined, 717
labeling, 718, 719
shipping, 717-718, 720-722
Clocks, 847
Clone, defined, 269
Cluster of differentiation (CD) markers, 246,
247-248
CMV. See Cytomegalovirus
Co (Colton) antigens and antibodies, 336, 349,
351
Co-dominant traits, 233, 234-235, 241
Coagulation
in hemolytic transfusion reactions, 640, 642
in liver transplants, 629
in massive transfusion, 511, 651
monitoring hemostasis, 494, 496
normal test values, 837, 838
physiologic principles, 493-494
role of platelets, 488-489
Coagulation factors
changes in red cell storage, 187-188
concentrates, 496, 497
in Cryoprecipitated AHF, 500-502
Factor VIIa concentrates, 497
Factor VIII concentrates, 497, 503-504
Factor IX concentrates, 497, 505
minimum levels needed for hemostasis, 495
in neonates, 571
normal values, 837
in plasma, 496, 498-500
Index 865
Copyright © 2005 by the AABB. All rights reserved.
in platelet concentrates, 838
replacement, 496, 497, 498-508
support during massive transfusion, 511
virus inactivation in concentrates, 701
for vitamin K deficiency, 497, 498-499
for warfarin reversal, 497, 498-499
Coagulopathy
dilutional, 499-500, 567, 651
in hemolytic transfusion reactions, 640,
642
in liver disease, 499
in liver transplantation, 629
in massive transfusions, 511, 651
in neonates, 571
treatment with heparin, 642
in vitamin K deficiency, 498-499
in warfarin reversal, 498-499
Code of Federal Regulations (CFR), 1, 32
Codons, 206-207
Cold-acid elutions, 457, 772
Cold agglutinin syndrome (CAS), 464-466
adsorption procedures, 466
classification, 459
complement in, 281
pretransfusion testing in, 466
serologic findings in, 460, 465
specificity of autoantibodies, 307, 465-466
Cold agglutinin titer, 458, 465, 778-779
Cold autoadsorption
in cold agglutinin syndrome, 308, 466
for detection of alloantibodies, 438, 472
method, 775-776
in resolving ABO discrepancies, 303
Cold autoantibodies
ABO discrepancies with, 299, 302-303, 469
adsorption, 438, 466, 472, 775-776
with alloantibodies, 438-439, 472
anti-I/i, 306, 307-308, 438, 465-466
anti-IH, 308, 434, 777, 778
in cold agglutinin syndrome, 281, 307,
465-466
and complement, 281
determining specificity, 776-778
in mixed AIHA, 466, 467
in paroxysmal cold hemoglobinuria, 467
in Rh testing, 329, 469-470
titration, 458, 465, 778-779
use of sulfhydryl reagents with, 469-470,
744-745
in warm autoimmune hemolytic anemia,
461
Cold hemagglutinin disease (CHD). See Cold ag-
glutinin syndrome
Cold reactive alloantibodies, 273-274, 299, 303
Cold stress. See Hypothermia
Collectionofblood.See Blood collection
Collections, antigen, 335, 355-356
College of American Pathologists (CAP), 514
Colloid solutions, 486, 507-508, 572
Colony-forming cell assays, 603-604
Colony-stimulating factors, 264
Colton system (ISBT 015), 226, 336, 349, 351, 842
Column agglutination technology, 278, 284-285
Compatibility testing. See Pretransfusion testing
Competency assessments, 8
Complement, 259-262
in acute transfusion reactions, 639-640
alternative pathway, 260, 261
in antiglobulin testing, 280-281, 453
in autoimmune hemolytic anemia, 281, 460,
465
C1-esterase inhibitor, 500
classical pathway, 259-261
complement receptors, 262, 354
membrane attack complex, 261-262, 263
physiologic effects of activation, 262
and positive DAT, 453
regulation of activation, 262, 263
Complications. See Adverse reactions
Computer crossmatch, 413, 414-415
Computer systems
alternative systems, 18-19
backups, 19
for record storage, 17-19
security, 18
validation, 13-14, 415
Confidential unit exclusion, 101
Confidentiality of records, 18
Consent
for apheresis, 140, 142
for blood donation, 103
for transfusion, 521-522
for transplantation, 620-621
Containers
for biological specimens, 717
for blood collection, 104-105, 179-180
damaged or leaking, 720
freezer storage bags, 182
866 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
quality control, 847
for shipments, 195, 607, 717-718
Contamination. See Bacterial contamination
Continuous-flow centrifuges, 596-597
Continuous Quality Improvement, 123-124
Contracts, 10
Control charts, 23, 30
Controls
autologous, 412, 427, 438-439, 454
for high-protein reagents, 328-329
IgG-coated cells, 281, 412
for indirect antiglobulin tests, 754
for low-protein reagents, 330
in viral marker testing, 167-169
Convulsions in donors, 108
Copper sulfate
quality control, 819-821, 847
in screening donors, 102, 799-800
Cord blood
antigen expression on, 431, 433
HPC-C transplantation, 588-589
autologous, 589
collection, 588, 595-596
colony-forming cell assays, 603-604
cord blood banks, 589
diseases treated with, 583, 584
engraftment in, 588-589
evaluation and quality control, 607-
608
freezing and storage, 604, 605-606
infectious disease testing, 591
microbial cultures, 602-603
processing, 602
regulations, 608
related donors, 589
standards, 609
stem cell enumeration, 603
thawing and infusion, 607
transportation and shipping, 606-607
serologic testing, 544-546, 595
Cordocentesis, 539, 541-542, 552
Corneal transplants, 619, 621, 624
Corrected platelet count increment (CCI), 362,
363, 489
Corrective action, 24-25
Corticosteroids, 144
Cost blood group collection, 352, 355
Counterflow centrifugal elutriation (CCE),
596-597
CP2D (citrate-phosphate-dextrose-dextrose),
176, 178
CPD (citrate-phosphate-dextrose), 176, 178, 186
CPDA-1 (citrate-phosphate-dextrose-adenine),
176, 178, 564-565
CR (complement receptors), 262, 354
Cra(Cromer) antigen/antibody, 353, 354, 843
CREG (cross-reactive groups), 392
Creutzfeldt-Jakob disease (CJD), 689-690
Cromer system (ISBT 021), 226, 353, 354, 843
Cross-reactive groups (CREGs), 392
Crossmatch-to-transfusion (C:T) ratios, 92, 93
Crossmatching
antiglobulin test in, 413, 414
for children, 415, 546, 563
computer, 413, 414-415
HLA, 397, 398, 401, 402, 492
immediate-spin, 413, 414, 751-752
“in-vivo,” 509
interpretation of results, 415-416, 417
platelets, 364, 365, 398
in pretransfusion testing, 412, 413-415, 417
in transfusion reaction evaluation, 654, 655
Crossover, gene, 208, 209, 230-231, 232, 241, 389
CRYO. See Cryoprecipitated Antihemophilic Fac-
tor
Cryoglobulinemia, 156
Cryoprecipitate Reduced Plasma, 177, 190, 496
Cryoprecipitated Antihemophilic Factor (CRYO)
ABO compatibility, 411, 500-501
calculating dose, 501
for coagulation factor replacement, 496
description, 177
for disseminated intravascular coagulation, 502
expiration dates, 189
for fibrinogen abnormalities, 501-502
for hemophilia A, 502-503
indications for use, 499, 500-503
inspection, 194
inventory management, 94
for neonates, 558, 572
pooled, 183-184, 191, 193, 815
preparation, 814-815
quality control, 199, 202
room temperature storage, 185
thawing, 191, 815
topical use, 502
transportation and shipping, 196
for von Willebrand syndromes, 501
Index 867
Copyright © 2005 by the AABB. All rights reserved.
Cryopreservation
agents, 181, 604
computer-controlled, 605
effects of, 181
of hematopoietic progenitor cells, 604-606
passive controlled-rate, 605-606
of platelets, 183
of red cells, 181-183, 807-812
storage bags, 182
Crystalloids, 150, 151, 486, 572
Cs (Cost) antigens and antibodies, 352, 355
C:T (crossmatch-to-transfusion) ratios, 92, 93
Cultures, bacterial, 195, 602-603, 655, 693, 694
Customer relations, 9-10
CXCR4, 594
Cytokines
defined, 269
in immune response, 251, 254, 262-263, 264
in transfusion reactions, 640, 643
Cytomegalovirus (CMV), 686-687
associated with hepatitis, 667, 668
blood component testing for, 170
clinical manifestations, 686
and leukocyte reduction, 687
in neonates, 562
prevention, 687
transfusion-transmitted, 686-687
in transplantation, 590, 591, 621, 629, 630
Cytomegalovirus (CMV)-negative products, 95
granulocytes, 492, 570
for intrauterine transfusion, 544, 553
labeling, 172
D
Dantigen.See also Rh system; Rh testing
clinical significance, 316
discovery of, 315-316
expression of, 319-320
in HDFN, 330, 536-537
ISBT classification, 841
partial D, 322-323, 538
testing, 165, 328-330, 414
weak D, 322-324
in autologous donations, 324
in donors, 165, 323
“microscopic,” 550
partial D, 322-323
as position effect, 302
in pregnancy, 538-539
quantitative, 322
in recipients, 323-324, 411
Dangerous goods, 716-722
DAT. See Direct antiglobulin test
DAT-negative autoimmune hemolytic anemia,
459, 468
DDAVP (desmopressin), 513
as fibrinolytic inhibitor, 514
in hemophilia A, 503, 513
indications for, 513
in liver disease, 499
in von Willebrand syndrome, 501, 513
Decay-accelerating factor (DAF), 353
Decontamination of biohazardous waste, 51, 54,
57
Deferrals, donor
of autologous donors, 120
for babesiosis, 696
for Creutzfeldt-Jakob disease, 689, 690
for drugs taken by donor, 100-101, 113
for hepatitis, 674, 703
for HIV, 674, 680, 681, 703
for HTLV, 683, 684, 685, 703
implicated in posttransfusion infections, 703
for Lyme disease, 696-697
for malaria, 697
records of, 98, 173
Deglycerolized Red Blood Cells
checking adequacy of deglycerolization,
812-813
expiration date, 189
preparation, 191-192, 809
refreezing, 183
of sickle cell trait, 192
storage, 192
Delayed hemolytic transfusion reactions
(DHTR), 346, 575, 637, 656-657
Delayed serologic transfusion reaction (DSTR),
656
Delayed transfusion reactions. See also Transfu-
sion reactions
alloimmunization, 637, 656-657
graft-vs-host disease, 399, 560, 637, 657-659
hemolytic, 346, 575, 637, 656-657
immunomodulation, 638, 660
iron overload, 638, 660
Kidd antibodies in, 346, 656
posttransfusion purpura, 366, 370, 638, 659-660
Density gradient separation, 601
868 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Derivatives. See also specific derivatives
for coagulation factor replacement, 496
virus inactivation, 496, 699, 701
Desensitization therapy, 505
Designoutput,defined,4,30
Designated donations, 98, 103-104
Desmopressin. See DDAVP
Deviations, 19, 20, 21
Dextrose solution, 530
Dha(Gerbich) antigen, 352, 843
DHTR (delayed hemolytic transfusion reac-
tions), 346, 575, 637, 656-657
Di (Diego) antigens and antibodies, 336, 348, 349
Diagnostic specimens
defined, 717
labeling, 718, 719
shipping, 717-718, 719, 720-722
DIC. See Disseminated intravascular coagula-
tion
Dichloromethane/methylene chloride elutions,
457, 775
Diego system (ISBT 010), 348
antigens/antibodies, 336, 348, 842
chromosomal location of genes, 226
and En(a–), 338
in HDFN, 348
phenotype frequencies, 349
in transfusion reactions, 348
Differential centrifugation, 179-180
Differential warm adsorption, 781-782
Digitonin acid elutions, 457
Dilution
of % solutions, 726-727
of serum, 725-726
Dilutional coagulopathy, 499-500, 567, 651
Dimethylsulfoxide (DMSO), 604, 607
2,3-Diphosphoglycerate. See 2,3-DPG
Direct antiglobulin test (DAT)
in classifying AIHA, 458
complement in, 280-281
method, 760-761
positive test
in antibody identification, 427, 436,
438-439
causes, 453-454
in cold agglutinin syndrome, 460, 465
drug-induced, 455, 472-477, 481-482
elution with, 456-458, 745-746
evaluation, 454-458, 480
in HDFN, 545-546
in IgM warm AIHA, 464
medical history in, 454-456
in mixed-type AIHA, 460, 466
in paroxysmal cold hemoglobinuria, 460,
467
red cell testing with, 745-747
serologic studies, 456
in warm autoimmune hemolytic anemia,
459, 460, 461
in pretransfusion testing, 412, 454
principles, 278
reagents, 278-280 , 427, 454, 468
specimens, 454
in transfusion reaction evaluation, 653-654
in transplantation, 455, 629
vs autologous control, 427
Directed donations, 94-95, 537-538
Disaster planning, 67-68
Disinfectants, 51, 54
Disposal
of biohazardous waste, 56-57
of blood components, 196
of chemical waste, 63
of radioactive waste, 66
Disposition of blood components, 196
Disseminated intravascular coagulation (DIC)
in neonates, 567, 572
in transfusion reactions, 640, 642
treatment for, 502, 567
Dithiothreitol. See DTT
Diversion, 693
DMSO (dimethylsulfoxide), 604, 607
DNA (deoxyribonucleic acid)
libraries, 217
molecular techniques
cloning, 217, 222
isolation of nucleic acids, 209-211
microarrays, 218
polymerase chain reaction technique,
211, 212, 213-214, 222, 394-396
profiling (typing or fingerprinting), 216, 222
recombinant proteins, 218-219, 222
restriction endonucleases, 214, 215
restriction fragment length polymor-
phism analysis, 214, 215, 222
sequencing, 217-218, 222, 396
structure, 203-204
transcription, 204, 205
Index 869
Copyright © 2005 by the AABB. All rights reserved.
Do (Dombrock) antigens and antibodies, 336,
349, 350-351, 842
Documents, 14-17. See also Records
management, 14-15, 17
quality system, 14, 16
types, 15
Dolichos biflorus lectin, 293, 296, 302, 743-744
Dombrock system (ISBT 014)
antibodies, 336, 350-351
antigens, 350, 842
genes, 226
phenotype frequencies, 349
Dominant traits, 232-233, 234, 241
Donath-Landsteiner test, 458, 467, 784-785
Donation intervals for apheresis, 141, 143
Donation process. See Blood collection
Donor area, 55, 97
Donor deferrals
of autologous donors, 120
for babesiosis, 696
for Creutzfeldt-Jakob disease, 689, 690
for drugs taken by donor, 100-101, 113
for hepatitis, 674, 703
for HIV, 674, 680, 681, 703
for HTLV, 683, 684, 685, 703
implicated in posttransfusion infections, 703
for Lyme disease, 696-697
for malaria, 697
for positive infectious disease tests, 680, 683,
684, 685
records, 98, 173
Donor history questionnaire, 110-112, 114-118
Donors
adverse reactions in, 19, 107-109
autologous, 103, 121-122, 124-125, 131, 135,
693
care of, after phlebotomy, 106
confidential unit exclusion, 101
consent, 103, 620-621
designated donations, 98, 103-104
directed donations, 104, 537-538
drugs taken by, 100-101, 113, 115
eligibility, 99-100
family members as, 442
hematocrit, 102
for hematopoietic transplantation, 589-591
hemoglobin levels, 102, 799-800
for HLA-matched platelets, 398
hypotension in, 107
implicated in posttransfusion infection, 703
information provided to, 98-99, 114
medical history, 100-101, 110-112, 693
notification of abnormal tests, 99, 103, 703
for organ and tissue transplantation, 618,
620-623
physical examination of, 100, 101-103, 620
for plateletpheresis, 141
records, 165, 173
red cell density, 102
reentry with positive screening tests, 673,
674, 675, 681-682
registration, 97-98
report of illness after donation, 103
testing
ABO, 165, 290, 296, 413
alanine aminotransferase, 164
antibody detection, 165
for blood group antigens, 418, 440-441,
745-748
cytomegalovirus, 170
equipment requirements, 164-165
general requirements, 163-164
hepatitis, 120, 166-170, 669-673
HIV, 120, 164, 166-170, 679-681
HTLV, 120, 683
repeat, 413
Rh, 165, 328-332, 413
syphilis, 120, 165-166, 695
viral markers, 166-170
weak D, 165, 323, 328
weight, 101, 143
Doppler flow studies, 542
Dosage effect
in antibody identification, 425, 431
in Duffy system, 345
genetics, 225, 227
in Rh system, 327
Dosimeters, 64-65
Dot blot, 213, 395
2,3-DPG
in neonatal transfusions, 561-562
in red cell storage lesion, 185, 187
in tissue oxygenation, 484, 511-512
Dra(Cromer) antigen, 353, 354, 843
Drug-induced immune hemolytic anemia,
472-477
autoantibodies, 474, 475-476
classifications, 473-476
870 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
drug adsorption mechanism, 473, 474, 475
drug-dependent antibodies, 473, 474-475
drugs associated with, 476, 481-482
laboratory investigation, 460, 476-477,
786-791
metabolites, 477
nonimmunologic protein adsorption, 476
theories of, 472-473
Drugs
administered for leukapheresis, 143-144
alteration of pharmacodynamics in
apheresis, 150-151
for autologous tumor purging, 597
as cause of thrombocytopenia, 374-377
for febrile transfusion reactions, 644
in positive DAT, 455, 472-477, 481-482
to prevent allergic transfusion reactions, 646
taken by donors, 100-101, 113, 115
Dry ice, 196, 719, 720-721
Dry shippers, 607
DSTR (delayed serologic transfusion reaction),
656
DTT (dithiothreitol)
applications for, 448-449
differentiating IgG from IgM, 764-765
to disperse autoagglutination, 302, 469, 470,
744-745
inactivating blood group antigens, 342, 445,
446, 766-767
Duffy system (ISBT 008), 343-345
antibodies, 336, 345, 844
antigens, 343-344, 841
biochemistry, 344-345
chromosomal locations of genes, 226
effect of enzymes on, 276, 336, 345
in HDFN, 345
and malaria, 337, 344
phenotypes and frequencies, 344, 844
in transfusion reactions, 345
Dura mater transplants, 619, 624
Dysfibrinogenemias, 501-502
E
E/e antigens and antibodies
cis product antigens, 324
concomitant antibodies, 327-328, 441
dosage in, 317
expression of, 320
gene complexes, 319
in HDFN, 536
incidence of, 317, 321
phenotypes, 320
testing for, 330-331, 441
E/e antigens and antibodies (Rh system), 316,
841
EACA (epsilon aminocaproic acid), 513-514
EBV (Epstein-Barr virus), 667, 668, 687-689
ECMO (extracorporeal membrane oxygenation),
572-573, 574
Education
donor, 98-99, 114, 117-118
patient, 521-522
EIA. See Enzyme-linked immunosorbent assay
ELBW (extremely low birthweight) neonates,
557, 568. See also Neonates
Elderly people, 295, 299
Electrical safety, 48-49
Electromechanical infusion devices, 522,
529-530, 565
ELISA. See Enzyme-linked immunosorbent as-
say
Elutions
with adsorption, 300-301, 448, 735-736
in antibody identification, 447-448
in cold agglutinin syndrome, 460, 465
in IgM warm AIHA, 464
methods, 457, 771-775
cold-acid, 457, 772
dichloromethane/methylene chloride,
457, 775
digitonin-acid, 457
gentle-heat, 745-746
glycine-HCl/EDTA, 772-773
heat, 457, 773-774
Lui freeze thaw, 457, 774
in mixed-type AIHA, 460, 467
in paroxysmal cold hemoglobinuria, 460, 467
in positive DAT evaluation, 456-458
technical factors in, 447-448
uses, 448
in warm antibody AIHA, 460, 461
Embolism, air, 636, 651-652
Emergency procedures
blood orders, 93
identification of patients, 408
issue of blood, 419, 510-511, 522-523
in pretransfusion testing, 419
Emergency release, 510-511, 522-523
Index 871
Copyright © 2005 by the AABB. All rights reserved.
Emergency response plans, 44
for biohazardous waste, 55-57
for chemicals, 61-62, 63
for electrical emergency, 49
for fires, 48
for radiation safety, 66
Emergency showers, 61
Emergency supplies for donor area, 108
Employees. See Personnel
En(a–) red cells, 338
Engineering controls
for biosafety, 50-51, 52-53, 54
for chemical safety, 61
for electrical safety, 48
for fire prevention, 47
general guidelines, 43-44, 73-76
for radiation safety, 65-66
Engraftment data, 608
Enhancement of antibodies
albumin additives, 276, 427, 753
alteration of pH, 274, 444
in antibody detection/identification,
276-277, 427, 443-444, 753-754
combined adsorption/elution, 300-301, 448,
735-736
effect of incubation times, 274, 444
enzymes, 276, 443, 445 , 446, 756-760
increased serum-to-cell ratio, 444
LISS and LISS additives, 274, 275, 277, 427,
443, 753, 754
polyethylene glycol, 276-277, 427, 443, 753
temperature reduction, 443
Environmental chambers, 185, 197-198, 846
Enzyme-linked immunosorbent assay
(EIA/ELISA)
in anti-HTLV testing, 683
in antibody detection, 286
in antigen-antibody detection, 286
for Chagas’ disease testing, 698
for heparin-dependent antibodies, 377
in hepatitis testing, 672-673
in HIV testing, 679
in viral marker testing, 166
Enzymes, proteolytic
in adsorption, 470, 471, 781-782
in antibody detection/identification, 276,
443, 445, 446
effect on blood group antigens, 276, 336,
339-340, 345
evaluating treated red cells, 758-759
ficin preparation, 756
one-stage technique, 759
papain preparation, 756-757
in resolving ABO discrepancies, 300, 301
standardization of procedures, 757-758
two-stage technique, 759-760
Eosinophils, 255
Epistasis, 236
Epitope, defined, 269
EPO. See Erythropoietin
Epsilon aminocaproic acid (EACA), 513-514
Epstein-Barr virus (EBV), 667, 668, 687-688
Equilibrium constant (Ko), 273
Equipment. See also specific equipment
for apheresis, 139-140, 152
calibration, 821-823, 826-830
for component testing, 164-165
critical, 10
management, 10-11
personal protective, 43-44, 73-75
quality control
automatic cell washers, 830-832
centrifuge calibration, 826-830
continuous temperature monitoring sys-
tems, 197-198
freezer alarms, 198-199, 824-826, 846
performance intervals, 846-847
refrigerator alarms, 198-199, 823-824, 846
thermometers, 198, 821-823, 847
for transfusions, 523-524, 527-530
validation, 11, 13
Er blood group collection, 355
Ergonomics, 41-42
Errors
in ABO typing, 299-300
in antiglobulin testing, 282, 283
identification, 525-526, 527, 641, 642, 653,
654
Erythroblastosis fetalis. See Hemolytic disease of
the fetus and newborn
Erythropoiesis, 557-558, 559
Erythropoietin (EPO)
in anemia treatment, 219, 488
in autologous donations, 125, 219
in newborns and infants, 558, 559
recombinant, 125, 219, 488, 512, 559
as transfusion alternative, 512
Esa(Cromer) antigen, 353, 354, 843
872 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Ethnic groups, differences in
in ABO system, 845
in Cromer system, 353
in distribution of antigens, 335, 337
in Duffy system, 337, 344, 345
in HLA system, 585
in Kell system, 341, 343
in Kidd system, 337
in Lutheran system, 347
in Rh system, 316, 317, 320, 321, 325
Etiologic agents, 719
Exchange transfusions
with antibody against high-incidence anti-
gen, 547
blood selection for, 546-547, 567
blood warming in, 560
in DIC, 567
in HDFN, 546-547
for hyperbilirubinemia, 566-567
methods used, 568
“partial,” 563
for removal of toxins, 567
in sickle cell disease, 574
vascular access, 568
volume and hematocrit, 568
Executive management, 6
Exons, 206
Expiration dates
checking before issue, 525, 526
of components, 188, 189-190
of tissues and organs, 624
Exposure control plan, 49, 50
External assessments, 23-24
External controls in viral marker testing, 167-169
Extracorporeal membrane oxygenation (ECMO),
572-573, 574
Extracorporeal photochemotherapy, 158
Eye washes, 50, 75
F
Face shields, 74
Facilities
design and workflow, 40
ergonomics, 41-42
housekeeping, 40-41
mobile sites, 41
organizational management, 6-7
restricted areas, 41
safety, 27, 39-42
FACT standards, 609
Factor V, 188
Factor VIIa, 497, 505
Factor VIII
calculating dose, 503-504
concentrates, 497, 503-504
deficiency, 503
for hemophilia A, 502-504
inhibitors to, 504-505
in stored blood, 188
virus inactivation, 710
for von Willebrand syndromes, 501
Factor IX, 497, 498, 504, 505
Failures, active and latent, 26-27
Fainting in donors, 107
False-positive/false-negative results
in antiglobulin testing, 282, 283
in Rh testing, 329, 330, 331-332
Fascialatatransplants,624
Fatalities
during apheresis, 153
due to bacterial contamination, 690-692
due to transfusions, 641, 645, 661, 691,
702-703
employees, 46
reporting, 19, 46, 661, 702-703
FDA. See Food and Drug Administration
Febrile nonhemolytic transfusion reactions
(FNHTR), 643-644
granulocyte antibodies in, 379
HLA antibodies in, 398-399
management, 634
manifestations, 643-644
pathophysiology, 634, 643-644
preventing, 576, 644
treatment, 644
Fetal hemoglobin (hemoglobin F), 550, 558, 835
Fetomaternal hemorrhage (FMH)
as immunizing event, 536
Kleihauer-Betke acid-elution test, 550-551,
794-796
microscopic weak D test, 550
postpartum evaluation, 548, 549-551
rosette test, 550, 793-794
Fetus. See also Hemolytic disease of the fetus
and newborn
erythropoiesis in, 557-558, 559
hematocrit, 541
Rh typing, 539
Index 873
Copyright © 2005 by the AABB. All rights reserved.
Fever, transfusion-associated, 398-399, 633, 634,
643
FFP. See Fresh Frozen Plasma
Fibrin degradation products, 837
Fibrin sealant, 502
Fibrinogen
abnormalities, 501-502
CRYO for replacement of, 500, 501
in monitoring coagulation, 494, 511
normal values, 837
Fibrinolytic inhibitors, 513-514
Ficin, preparation of, 756. See also Enzymes,
proteolytic
Filgrastim. See Granulocyte colony-stimulating
factor
Filters
leukocyte-reduction, 180, 190, 528-529
microaggregate, 528, 565-566
standard in-line, 527-528
Fire safety, 47-48
Fish-bone diagrams, 26
Fisher/Race Rh terminology, 319
Flow cytometry
in CD34 analysis/enumeration, 603
in granuloctye autoantibody testing, 380
in HLA crossmatch, 397, 401
in platelet antibody detection, 372, 375, 376
to predict clinical significance of antibodies,
441
Flow PRA, 366
Fluids, replacement, 150, 151
Fluosol, 513
FMH. See Fetomaternal hemorrhage
FNHTR. See Febrile nonhemolytic transfusion
reactions
Food and Drug Administration (FDA)
cGMP training, 8
quality assurance regulations, 1, 2
regulations for autologous blood, 119-120
regulations for tissue transplantation, 626
reporting adverse events to, 19, 20, 21, 661,
702-703
Forensic testing, 402
Forms, 15, 17. See also Documents; Records
FoundationfortheAccreditationofCellTherapy
(FACT), 609
Freeze-thaw elutions, 457, 774
Freezers, 185
alarm systems for, 185, 198-199, 824-826
quality control, 197-199, 846
temperature monitoring systems for,
197-198
thermometers for, 198
Freezing
blood samples, 184-185
cryoprotective agents, 181, 604
freeze-thaw damage, 181
Fresh Frozen Plasma, 180-181, 814
hematopoietic progenitor cells, 604-606
Platelets, 183
Red Blood Cells, 181-183, 807-812
refreezing deglycerolized cells, 183
skin grafts, 624, 625
storage bags for, 182
Fresh Frozen Plasma (FFP)
ABO compatibility, 411, 496, 498
coagulation factors in, 496
collection by apheresis, 141, 142
description, 177
expiration dates, 189-190
indications for, 496, 498-500
inspection, 194
inventory management, 94
misuse, 500
for neonates, 558, 571
in older infants and children, 576
preparation, 180-181, 813-814
small-volume, 571
thawing, 191
transfusion thresholds, 494, 496
transportation and shipping, 196
virus inactivation, 701-702
Frozen components. See specific components
Fume hoods, 61
Fy (Duffy) antigens and antibodies, 336,
343-344, 345, 841
G
G antigen (Rh system), 324-325, 841
G-CSF. See Granulocyte colony-stimulating fac-
tor
GBV-C virus, 668
Ge (Gerbich) antigens and antibodies, 352-353,
843
GEIS antigen, 352, 843
Gel test, 285
Gene chips, 218
Gene therapy, 217, 219-220
874 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Genes
blood group terminology, 844
chromosomal assignment, 225, 226, 235-236
defined, 241
DNA structure, 203-204
frequencies, 227-229, 320-321
gene conversion, 209, 210
major histocompatibility complex, 244-246,
386-389
modifier, 235-236
nucleotide insertions and deletions, 208
nucleotide substitutions in, 207-208, 228
obligatory, 237
single crossover, 208, 209, 230-231, 232, 241, 389
suppressor, 235-236
variability, 208-209, 210
Genetics of blood groups
of ABO system, 229-230, 235, 290-293
alleles, 225, 227-229
basic principles, 223-225
definitions of terms, 241
of H system, 290-293
and heredity, 225-232
of HLA system, 386-389
Lewis system, 305
of MNS system, 338
nomenclature, 238-239, 318-319, 840-844
patterns of inheritance, 232-236
polymorphism in, 207-208
population genetics, 236-237
of Rh system, 316-318
Genotypes. See also specific blood groups
defined, 233
frequencies, 227-228
nomenclature for, 238-239
Gentle heat elutions, 745-746
Gerbich system (ISBT 020), 226, 352-353, 843
GIL system (ISBT 029), 226, 355, 843
Glanzmann’s thrombasthenia Type I, 366, 369
Gleevec (imatinib mesylate), 220
Globoside (GLOB) blood group, 226, 308, 843
Gloves, 46, 55, 73-74, 106
Glycerolization of red cells, 181-182, 808-809,
810-812
Glycine-HCl/EDTA
to disperse autoagglutination, 469
to dissociate IgG from red cells, 469, 747-748
elutions, 772-773
to inactivate red cell antigens, 446
Glycine max (anti-T, -Tn), 743
Glycophorins, 338, 339
GM-CSF (granulocyte-macrophage col-
ony-stimulating factor), 264, 512, 592, 594
Goggles, safety, 74
Gov antigens, 370
Grading test results, 412, 728-729
Graft failure, 598
Graft-vs-host disease (GVHD)
acute/chronic, 586
and graft-vs-leukemia effect, 586-587
in hematopoietic transplantation, 585-588
HLA system in, 399, 400
in neonates, 560-561
transfusion-associated, 399, 400, 560-561,
637, 657-659
Graft-vs-leukemia (GVL) effect, 586-587
Gram-equivalent weight, 723
Gram-molecular weight, 723
Gram’s stain, 693
Granulocyte colony-stimulating factor (G-CSF)
and false-positive test results, 590
for HPC mobilization, 587, 592-595
in leukapheresis, 144
in neonatal sepsis, 570
production and function, 264
side effects of, 594-595
uses for, 218, 222, 512
Granulocyte-macrophage colony-stimulating
factor (GM-CSF), 264, 512, 592, 594
Granulocytes
alloantigens, 377-380, 647
antibodies to
in neonatal alloimmune neutropenia, 379
testing for, 380
in TRALI, 379-380, 647, 656
in immune system, 255-256
Granulocytes Pheresis
ABO compatibility, 411
collection, 143-144
description, 177-178
expiration dates, 189
indications/contraindications for use, 492,
570
irradiation, 144, 492
laboratory testing, 144
for neonates, 558, 569-570
quality control, 202
storage, 144
Index 875
Copyright © 2005 by the AABB. All rights reserved.
transfusion, 143, 492, 569-570
transportation and shipping, 196
validation, 35
Group O serum (anti-A,B), 295
Growth factors, recombinant
for HPC mobilization, 587, 592-595
in leukapheresis, 144
as transfusion alternative, 512, 559
uses for, 219, 222
GTI PAKAUTO, 374
Guillain-Barré syndrome, 156
GUTI (Cromer) antigen/antibody, 353, 354, 843
GVHD. See Graft-vs-host disease
GVL (graft-vs-leukemia) effect, 586-587
Gya(Dombrock) antigen, 350, 842
H
H system (ISBT 018), 303-304
anti-H, 304, 434
anti-IH, 308, 434, 777, 778
biochemistry and genetics, 290-293
chromosomal locations of genes, 226
H antigen, 290, 303-304, 361, 842
Ohphenotype (Bombay), 304
para-Bombay phenotypes, 304
saliva testing for substances, 301, 736-739
HAM (HTLV-associated myelopathy), 682
Hand-washing, 50, 75
Haplotypes
ancestral, 389
defined, 231
of HLA system, 387-388, 389, 400, 402
of Rh system, 318-319
Haptoglobin, 835
Hardy-Weinberg law, 227-229
HAV (hepatitis A virus), 667-668, 671, 700
Hazard Communication Standard, 59-60
Hazard identification and communication, 43
for biosafety, 50
for chemical safety, 59-61
for electrical safety, 48
for fire prevention, 47
Hazardous areas of facilities, 41
Hazardous materials
biohazards, 49-57
chemicals, 57-63
radioactive, 63-66
safety plan for, 42-47
transportation and shipping, 66, 716-722
waste management, 55-57, 67
HBeAg (hepatitis B e antigen), 669, 670
HBsAg. See Hepatitis B surface antigen
HBV. See Hepatitis B virus
HCV. See Hepatitis C virus
HDFN. See Hemolytic disease of the fetus and
newborn
HDV (hepatitis D virus), 668, 670
Heart and heart valves
arrhythmias in recipients, 650
cardiac arrest in donors, 108
cardiac output, 485-486
disease of, 119, 487-488
infectious disease transmission, 619
preservation, 624
transplantation, 402, 625, 629
Heat elutions, 457, 745-746, 773-774
Heating blocks, 846
Hemapheresis practitioner (HP), 139
Hematocrit
in autologous blood donors, 125, 126
in blood donors, 102
of blood in exchange transfusions, 568
fetal, 541
in neonates, 572
normal values, 835
of red cell components, 805
transfusion trigger, 487
Hematoma, in donors, 108
Hematopoietic growth factors
for HPC mobilization, 587, 592-595
for leukapheresis, 144
as transfusion alternative, 512, 559
uses for, 218-219, 222
Hematopoietic progenitor cell (HPC) transplan-
tation, 581-609
ABO incompatibility, 598-599, 600, 601-602
ABO typing discrepancies in, 299
allogeneic, 582, 583, 585-588, 589-590,
594-595
autologous, 582, 583, 587, 589, 592-594, 597
autologous tumor purging, 597-598
cell processing, 596-604
chimerism in, 298, 601
collection of products, 144, 591-596
colony-forming cell assays, 603-604
diseases treated with, 583, 584
donor eligibility, 589-591
876 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
evaluation and QC of products, 607-608
freezing and storage of cells, 604-606, 624
and graft-vs-host disease, 585, 586-587
HLA testing, 400-401, 585-587
of HPC-A (apheresis), 581, 587-588, 592-595
of HPC-C (cord blood), 588-589, 595-596,
602
of HPC-M (marrow), 583, 585-587, 591-592
indications for, 582
infectious disease testing, 590-591
matched, unrelated donors, 585-586, 589
microbial cultures, 602-603
mobilization of HPCs, 587, 592-595
nonmyeloablative, 582-583
positive DAT after, 455
processing, 596-598, 601-604
red cell depletion, 601
regulations, 608
related donors, 587-588, 589
selection of CD34+ cells, 596-597
sources of cells for, 583, 584-589
standards, 609
stem cell enumeration, 603
suitability criteria, 606
syngeneic, 582, 587
T-cell depletion, 586, 598
thawing and infusion of products, 607
transfusion support, 591-592, 599, 600, 601
transportation and shipping, 606-607
types, 581-582
xenogeneic, 582, 583
Hemoglobin
in autologous donors, 125
in blood donors, 102
bovine, 513
fetal (F), 550, 558, 835
in neonates, 558, 563, 572, 836
normal values, 835, 836
and oxygen, 185, 187, 483-485
recombinant, 219, 513
S, 544, 567, 574, 575
in transfusion reaction evaluation, 654-655
as transfusion trigger, 483, 486-488
Hemoglobin solutions, 512-513
Hemoglobinometers, 847
Hemoglobinopathies, 508-509, 574-576. See also
specific hemoglobinopathies
Hemolysis
in ABO testing, 295
in antibody detection, 412
defined, 271-272
immune-mediated
ABO/Rh typing problems with, 469-470
alloimmune hemolytic anemia, 459
antibodies associated with, 336
antibody detection with, 470-472
classification of anemias, 458, 459
cold agglutinin syndrome, 459, 460,
464-466
DAT-negative AIHA, 459, 468
defined, 458-459
drug-induced, 459, 460, 472-477, 481-482
in hemolytic transfusion reactions,
639-642
IgM warm AIHA, 464
intravascular/extravascular, 265-266, 267,
458
mixed-type AIHA, 459, 460, 466-467
paroxysmal cold hemoglobinuria, 459,
460, 467-468
warm antibody autoimmune hemolytic
anemia, 459, 460, 461-464
non-immune mediated, 152, 636, 642-643
passenger lymphocyte, 628, 629-630, 657
in patient samples, 410
and positive DAT evaluation, 455
in transfusion reaction evaluation, 653, 655
Hemolytic anemias. See also Hemolytic disease
of the fetus and newborn
alloimmune, 459
classification of, 458, 459
cold agglutinin syndrome, 459, 460, 464-466
DAT-negative AIHA, 459, 468
drug-induced, 459, 460, 472-477, 481-482
IgM warm AIHA, 426, 427, 428, 429-431
mixed-type AIHA, 459, 460, 466-467
paroxysmal cold hemoglobinuria, 459, 460,
467-468
serologic findings in, 460
warm autoimmune hemolytic anemia, 459,
460, 461-464
Hemolytic disease of the fetus and newborn
(HDFN), 535-551
ABO, 536, 537, 538, 545
ABO discrepancies in, 302
antibodies associated with, 336, 536-537,
545-546
elutions in, 456
Index 877
Copyright © 2005 by the AABB. All rights reserved.
exchange transfusion in, 546-547
intrauterine transfusion in, 541, 543-544
maternal immunization in, 536-538, 542-543
measuring severity of
amniotic fluid analysis, 540-541, 542
Doppler flow studies, 542
maternal antibody titer, 449, 539-540,
761-764, 796-798
percutaneous umbilical blood sampling,
541-542
pathophysiology, 535-536
postpartum evaluation, 544-546
prenatal evaluation, 538-540
Rh testing in, 330, 538-539, 545
use of Rh Immune Globulin, 547-551
Hemolytic transfusion reactions (HTR)
antibodies associated with, 336
coagulation activation in, 640, 642
complement activation in, 639-640
cytokines in, 640
delayed, 346, 575, 637, 656-657
due to ABO incompatibility, 639, 640, 641
elutions in, 456
frequency, 641
HLA antibodies in, 399-400
management, 634
non-immune, 642-643
pathophysiology, 639-641
prevention, 642
renal failure in, 640-641
shock in, 640
signs and symptoms of, 633, 639-641
treatment, 641-642
Hemolytic-uremic syndrome (HUS), 154-155
Hemophilia A, 502-505
calculating Factor VIII dose, 503-504
factor replacement for, 504
inheritance of, 233-234
inhibitors of Factor VIII, 504-505
Hemophilia B, 505
Hemorrhage, intraventricular, 569
Hemorrhagic disease of the newborn, 571
Hemostasis
in acute normovolemic hemodilution, 128
coagulation factors needed for, 495
components for, 496, 497, 498-508, 570-572
during massive transfusion, 511, 651
monitoring, 494, 496
in neonates, 570-571
normal test values, 837
physiologic principles, 488-489 , 493-494
Heparin, 375-377, 505, 642, 723
Heparin co-factor, 505-506
Heparin-induced thrombocytopenia (HIT),
375-377, 492
Hepatitis, 667-675
associated with CMV or EBV, 668
chronic carriers, 668-669
clinical manifestations, 668-669
donors implicated in, 703
markers, 669, 670-671, 672-673, 674
non A, non B (NANB), 673, 699
prophylaxis, 45, 668, 672, 703
quarantine and recipient tracing, 675
reentry of donors with, 673, 674, 675
reporting cases, 702
risk of, 673, 675, 700
surrogate markers, 673
vaccination for, 668
viruses, 667-668
Hepatitis A virus (HAV), 667-668, 671, 700
Hepatitis B core antigen, antibody to (anti-HBc)
blood component testing, 164, 166, 675
look-back for, 675
marker of infection, 669, 670
reentry of donors with, 675
as surrogate marker, 672
testing transplant donors for, 621
Hepatitis B e antigen (HBeAg), 669, 670
Hepatitis B immune globulin (HBIG), 45, 668,
703
Hepatitis B surface antigen (HBsAg)
antibody to (anti-HBs), 669, 670
blood component testing, 164, 166, 169
chronic carriers, 668-669
look-back for, 675
marker of infection, 669, 670, 672
reentry of donors with positive tests, 674, 675
testing organ and tissue donors for, 620, 621,
623
Hepatitis B virus (HBV)
chronic carriers, 668-669
clinical manifestations, 667, 668-669
employee exposure to, 45
HBV DNA, 669, 672
look-back for, 675
markers of infection, 120, 669, 670, 672, 674
NAT testing, 669, 672
878 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
prophylaxis for, 45, 668, 672, 703
reentry of donors with, 674, 675
testing transplantation donors for, 590
transfusion risk of, 672, 679, 700
Hepatitis C virus, antibody to (anti-HCV)
blood component testing, 164, 166, 170
marker of infection, 670-671, 672-673
reentry of donors with, 673, 674, 675
supplemental tests, 170
testing organ donors for, 620, 621, 623
Hepatitis C virus (HCV)
autologous blood testing, 120
blood component testing, 164, 166-167, 170
chronic carriers, 669
clinical manifestations, 667, 668, 669
employee exposure to, 45
false-positive test results, 590
infections due to IGIV, 699
look-back for, 675
markers of infection, 669, 670-671, 672-673,
674
NAT testing, 164, 166, 170, 213, 590, 673, 675
reentry of donors with, 673, 674, 675
testing transplantation donors for, 590, 620,
621, 623
transfusion risk of, 673, 675, 699, 700, 701
Hepatitis D virus (HDV), 668, 670
Hepatitis E virus (HEV), 667-668, 671
Hepatitis G virus (HGV), 668
Hereditary angioneurotic edema, 500
Hereditary hemochromatosis (HH), 387
Heredity, genetics of
alleles, 225, 227-229
Hardy-Weinberg Law, 227-229
independent assortment, 229-230, 231
linkage, 230-231, 232
linkage disequilibrium, 231-232, 337, 389
segregation, 229, 230
Herpesviruses, 686-688. See also
Cytomegalovirus
HES (hydroxyethyl starch), 143-144
Heterozygous, defined, 225
HEV (hepatitis E virus), 667-668, 671
HGV (hepatitis G virus), 668
HH (hereditary hemochromatosis), 387
HHV-6/HHV-8 (human herpesviruses), 688
High-incidence antigens
antibodies to, 356-357, 435-436, 441-442,
547
of Cromer system, 353, 354
defined, 335
of Gerbich system, 352
GIL, 355
of Knops system, 354
of Lutheran system, 347
not assigned to a system or collection,
356-357
selecting blood negative for, 441-442
of Vel collection, 355-356
High-titer, low-avidity antibodies, 449, 762,
765-766
HIT (heparin-induced thrombocytopenia),
375-377, 492
HIV (human immunodeficiency virus), 675-682
in AIDS, 675-676
in autologous donors, 117, 681
clinical manifestations, 99, 676-677
confirmatory tests for, 169-170, 679-681
donors implicated in, 703
employee exposure to, 45
false-positive test results, 590
HIV-1, 675, 676, 678
HIV-2, and HIV-1, group O, 675, 677-679
information provided to donors about, 99
nucleic acid testing, 164, 166-167, 213, 590,
678-681
recipient tracing (look-back), 681-682
reentry of donors with positive screens, 674,
681
reporting cases, 702
risk factors for, 677
testing components for, 164, 166-167,
169-170, 213, 679-681
testing transplantation donors for, 590, 620,
621, 623
transfusion considerations, 678-679
transfusion risks of, 679, 700, 701
Hives, 634, 644-647
HLA-B27, 403
HLA Matchmaker, 365
HLA system, 385-404
alloimmunization to platelets, 265, 266,
362-366, 397-398, 637
antibody detection, 365-366, 397
antigens, 362, 385, 388-389, 390-391
and Bg antigens, 358
biochemistry, 390-391
biologic function, 393-394
Index 879
Copyright © 2005 by the AABB. All rights reserved.
and chimerism, 399
crossmatching, 397, 398, 401, 402, 492
disease associations, 402-404
in forensic testing, 402
genetics, 386-389
in graft-vs-host disease, 399, 400
major histocompatibility complex, 244-246,
386-389
matching
granulocytes, 492
in HPC transplantation, 388, 400-401,
585-586
in organ transplantation, 387-388,
401-402
of platelets, 363-365, 398
nomenclature, 391-393
in parentage testing, 402
in platelet refractoriness, 362-365, 397-398,
491
structure of molecules, 244-246, 390-391
tissue distribution, 390-391
and transfusion, 397-400
in transfusion reactions, 398-400, 637, 643,
656
in transfusion-related acute lung injury, 399,
647
and transplantation, 388, 400-402
typing, 394-397, 595, 621
HMIWI (hospital/medical/infectious waste in-
cinerators), 57
HNA antigens, 378
Homozygous, defined, 225
Homozygous type II familial hypercholesterol-
emia, 140, 157
Housekeeping, 40-41
HPA antigens, 366, 367-368, 369-370, 552
HPCs. See Hematopoietic progenitor cell trans-
plantation
HTLA (high-titer, low-avidity antibodies), 449,
762, 765-766
HTLV-associated myelopathy (HAM), 682
HTLV (human T-cell lymphotropic virus),
682-683
autologous blood testing, 120
blood component testing, 164, 166, 170, 674,
683
clinical manifestations, 682
donors implicated in, 703
quarantine and look-back, 683
testing transplantation donors for, 590, 620
transfusion risk, 700
transmission, 682
Type I, II, 682
HTR. See Hemolytic transfusion reactions
Human albumin (5% and 25%), 507
Human Genome Project, 217
Human granulocytic erlichiosis, 696
Human herpesviruses, 688
Human immunodeficiency virus. See HIV
Human monocytic erlichiosis, 696
Human resources, 7-9
Human T-cell lymphotropic virus. See HTLV
Human thrombin, 502
Hy (Dombrock) antigen, 350, 842
Hydatid cyst fluid, 310, 445
Hydroxyethyl starch (HES), 143-144
Hydroxyurea, 575
Hyper-IgM immune deficiency, 255
Hyperbilirubinemia, 536, 566-567
Hypercholesterolemia, familial, 140, 157
Hyperhemolytic syndrome, 575
Hyperkalemia, 650
Hyperleukocytosis, 154
Hypertension, 633
Hypertransfusion programs, 575-576
Hyperventilation, in donors, 107
Hyperviscosity, serum, 153-154
Hypoalbuminemia, 507
Hypocalcemia, 561, 636, 649-650
Hypofibrinogenemia, 500, 501
Hypokalemia, 650
Hypotension
associated with ACE inhibition, 636, 645
in donors, 107
in recipients, 633, 640, 641, 645
in therapeutic apheresis, 152
treatment, 646
Hypothermia, 511, 560, 637, 650
Hypovolemia, 141, 152
I
I/i antigens and antibodies, 306-308
in ABO discrepancies, 302
chromosomal locations of genes, 226
in cold agglutinin syndrome, 307, 465-466
complex reactivity, 308
ISBT nomenclature, 843
880 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
in pregnancy, 539
serologic behavior, 307-308, 777, 778
IAT. See Indirect antiglobulin test
ID-NAT (individual donor NAT), 669, 678, 680, 685
Identification
of blood samples, 409
of components, 105, 416, 524-526, 527
of donors, 98, 105
of equipment, 11
errors in, 525-526, 527, 641, 642, 653, 654
of persons issuing blood, 416, 525
of phlebotomists, 409
of recipients, 407, 408-409, 418, 524-526, 527
traceability of components, 173
Idiopathic thrombocytopenic purpura (ITP),
158, 373-374, 492, 553-554
Idiotypes, 256, 269
IFC (Cromer) antigen, 353, 354, 843
IgA, 259
antibodies to, 645, 646, 655
normal values, 835, 837
polymers, 258, 259
properties, 253
secretory, 258, 259
IgD, 253, 259, 835
IgE, 259
in allergic transfusion reactions, 644-645
normal values, 835
properties, 253
IgG, 258
anti-A and -B, 295
in autoimmune hemolytic anemias,
458-459, 460
complement activation by, 260
dissociation of, by chloroquine, 469, 746-747
distinguishing from IgM, 764-765
in neonates, 560
normal values, 835, 837
in positive DAT, 280, 453, 461
properties, 253
subclasses, 258, 458-459
temperature of reaction, 274
IgG-coated cells (check cells), 281, 412
IGIV. See Immunoglobulin, Intravenous
IgM, 258
anti-A and -B, 295
in antiglobulin testing, 281
cold-reactive autoagglutinins, 302-303, 465,
466, 467, 469, 776-778
complement activation by, 260
dispersing autoagglutination, 302, 469-470,
744-745
distinguishing from IgG, 764-765
normal values, 835, 837
polymers, 257, 258
properties, 253
temperature of reactions, 274
IgM warm AIHA, 464
IgSF (immunoglobulin superfamily), 244, 245,
246
IL (interleukins), 264, 512, 640
Imatinib mesylate (Gleevec), 220
Immediate-spin crossmatch, 413, 414, 751-752
Immune complexes
in antigen-antibody reactions, 280, 281
demonstration of, 788-789
mechanism of drug-induced antibodies,
473, 474-475
Immune-mediated hemolysis. See also
Hemolytic disease of the fetus and newborn
ABO/Rh typing problems with, 469-470
alloimmune hemolytic anemia, 459
classification of anemias, 458, 459
cold agglutinin syndrome, 459, 460, 464-466
DAT-negative AIHA, 459, 468
defined, 458-459
drug-induced, 459, 460, 472-477, 481-482
in hemolytic transfusion reactions, 639-642
intravascular/extravascular, 265-266, 267,
458
mixed-type AIHA, 459, 460, 466-467
paroxysmal cold hemoglobinuria, 459, 460,
467-468
warm autoimmune hemolytic anemia, 459,
460, 461-464
Immune serum globulin (ISG), 668
Immune system
cell adhesion molecules in, 246
cells of, 249-250, 251, 252-256
cluster of differentiation molecules in, 246,
247-248
complement in, 259-262, 263
cytokines in, 262-263, 264
defined, 269
and drug-dependent antibodies, 472-473
in HLA alloimmunization to platelets, 265,
266
immune response, 243-246, 247-248
Index 881
Copyright © 2005 by the AABB. All rights reserved.
immunoglobulin superfamily in, 244, 245
immunoglobulins in, 253, 256-259
innate and adaptive immunity, 243-244
lymphocytes in, 249-250, 251, 252-255
major histocompatibility complex in,
244-246
of neonates, 560-561
organs of, 249
phagocytic cells in, 255-256
in production of reagent antibodies, 266-267
receptors/markers, 250
in red cell alloimmunization, 265
in red cell destruction, 265-266, 267
signal transduction in, 246
soluble components, 256-263
Immune thrombocytopenic purpura (ITP), 158,
373-374, 492, 553-554
Immune tolerance, 237
Immunity. See Immune system
Immunization, maternal
antibody titers, 449, 539-540, 761-764,
796-798
mechanisms, 536-538
prenatal evaluation, 538-542
suppression, 542-543
Immunocompromised patients, 298-299
Immunofluorescence tests
in antigen-antibody detection, 285-286
in confirming HIV tests, 169
for granulocyte antibodies, 380
for platelet antibody detection, 372
Immunogen, defined, 269
Immunoglobulin, Intravenous (IGIV), 505
ABO discrepancies with, 299
for idiopathic thrombocytopenic purpura, 554
indications for, 505, 506
for neonatal immune thrombocytopenia,
552-553
positive DAT with, 456
for posttransfusion purpura, 659-660
to suppress maternal alloimmunization, 543
virus inactivation in, 699
Immunoglobulin products
hepatitis B immune globulin (HBIG), 45,
668, 703
immune serum globulin (ISG), 668
immunoglobulin, intravenous (IGIV), 505,
506, 543
virus inactivation in, 699, 701
Immunoglobulin superfamily (IgSF), 244, 245,
246
Immunoglobulins, 256-259. See also specific
immunoglobulins
classes, 258-259
Fab and Fc fragments, 256-257
functions, 256
interchain bonds, 256
J chains, 258
normal values, 835, 837
polymers, 257-258
properties, 253
secretory component, 258
structure, 256, 257
Immunohematology reference laboratories, 439
Immunomagnetic cell separation, 596, 598
Immunomodulation, 638, 660
In (Indian) antigens and antibodies, 349, 354,
843
“In-vivo” crossmatch, 441, 509
Incinerators, hospital/medical/infectious waste
(HMIWI), 57
Incompletely clotted specimens, 409, 722-723
Incubation
temperatures, 443, 715
times, 274, 444
Independent assortment, 229-230, 231
Indian system (ISBT 023), 226, 349, 354, 843
Indirect antiglobulin test (IAT)
false-positive/false-negative results, 282, 283
methods, 752-754
principles, 278
reagents, 278-280, 425, 427, 468
role of complement, 280-281
use of additives, 276-277, 443, 753-754
use of IgG-coated cells, 281, 412
Infants. See Children; Neonates
Infectious disease testing
of autologous blood, 119-120
of blood components, 166-170
external controls in, 167-169
of granulocytes, 144
for hepatitis, 669, 670-671, 672-673, 674
for HIV, 164, 166-167, 169-170, 213, 679-681
of HPC donors, 590-591
for HTLV, 120, 164, 166, 170, 683
invalidation of results, 167-169
neutralization, 169
notification of abnormal tests, 703
882 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
in organ and tissue transplantation, 621-623
of Platelets Pheresis, 142
records of, 173
supplemental tests, 169-170
surrogate markers, 673
for syphilis, 120, 165-166, 695
Infectious diseases. See also Transfusion-trans-
mitted diseases
as apheresis complication, 152
in blood donors, 99
safety precautions for, 49-57
transfusion-transmitted, 667-699, 700,
701-703
transmitted by allografts, 617, 619-620
Infectious mononucleosis, 687
Infectious substances
defined, 716-717
labeling, 718, 719
shipping, 717-718, 720-722
waste, 55-57
Inflammation, 262
Informed consent
for apheresis, 140, 142
of blood donors, 103
for transfusion, 521-522
for transplantation, 620-621
Infusions. See also Transfusions
of hematopoietic components, 607
infusion pumps, 522, 523, 529-530, 565
infusion sets, 527-529, 565-566
rates for, 531, 566
Inheritance patterns
autosomal dominant, 233, 234
autosomal recessive, 233, 234
blood group co-dominant, 234-235
chromosomal assignment, 226, 235-236
dominant and recessive, 232-233
of major histocompatibility complex,
387-389
sex-linked dominant or co-dominant, 233,
234
sex-linked recessive, 233-234
Inhibition tests
agglutination, 275-276
in antibody identification, 444-445
for Chido/Rodgers, 445, 765-766
for secretor status, 737-739
Inhibitors
of complement activation, 262, 263
to Factor VIII, 504-505
fibrinolytic, 513-514
Injuries, 45-46, 51
Innate immunity, 243-244
Inspections
of components before release, 194-195, 416,
418, 525
external assessments, 23-24
of incoming supplies, 10
of inventory, 93-94
Integrins, 246, 369
Interferon γ(IFNγ), 264
Interleukins, 264, 512, 640
Internal assessments, 22
Internal controls, 167
Internal event report, 19, 20
International Organization for Standardization
(ISO), 1-2, 3
Intervening sequences, 206
Intraoperative blood collection, 117, 130-133
clinical studies, 131-132
controversies in, 132-133
direct reinfusion, 133
equipment for, 132-133
practical considerations, 132
processing, 132-133
requirements and recommendations, 133
Intrauterine transfusions, 541, 543-544
Intraventricular hemorrhage, 569
Introns, 206
Inventory
counts and inspection, 93-94
determining levels, 89-90
minimum and ideal levels, 89
and outdating, 90-91
routine vs emergency orders, 93
of special products, 94-95
Ionic strength, 274
Iron, supplemental, 121
Iron overload, 638, 660
Irradiated blood components, 192-193
expiration dates, 183, 189, 192
Granulocytes, 144, 492
HPC products, 592
indications for, 183, 193, 493, 658-659
for intrauterine transfusion, 544
inventory management, 95
labeling, 172
platelet products, 144, 189, 192-193, 492, 553
Index 883
Copyright © 2005 by the AABB. All rights reserved.
potassium leak in, 561
to prevent TA-GVHD, 183, 192, 493, 658-659
quality control, 202
Red Blood Cells, 189, 192, 193, 493
to reduce platelet alloimmunization, 365, 398
ultraviolet B, 365, 398
washing, 561
Irradiators, blood, 65-66, 192, 846
ISBT (International Society of Blood Transfu-
sion)
128 barcode labeling, 171
nomenclature
for platelet antigens, 366, 367-368
for red cell antigens, 226, 238-239,
840-843
ISG (immune serum globulin), 668
Ishikawa diagrams, 26
ISO 9001 standards, 1-2, 3
Isotype switching, 252
Issuing blood
delivering blood to patient area, 524-525
identification of recipient and component,
418, 524-526
policies and procedures of, 416, 418, 524-525
reissue, 197, 525
from transfusion service, 196-197
in urgent situations, 419, 510-511, 522-523
ITP (idiopathic thrombocytopenic purpura),
158, 373-374, 492, 553-554
IV solutions, 530
J
J chains, 258
Jaundice, 566, 639
JCAHO (Joint Commission on Accreditation of
Healthcare Organizations), 25, 514
Jk (Kidd) antigens and antibodies, 336, 345-346,
841
JMH antigen/antibodies, 355, 843
Joa(Dombrock) antigen, 350, 842
Job descriptions, 7
John Milton Hagen (JMH) system (ISBT 026),
226, 355, 843
Joint Commission on Accreditation of
Healthcare Organizations (JCAHO), 25, 514
Jraantigen, 356
Js (Kell) antigens and antibodies, 336, 341, 343,
841
Juran’s Quality Trilogy, 2, 4
K
K/k (Kell) antigens and antibodies, 336, 340-342,
343, 841
Kaposi’s sarcoma-associated herpesvirus, 688
Kell system (ISBT 006), 340-343
anti-Ku, 343
antibodies, 336, 343, 844
antigens, 340-342, 468, 841, 844
biochemistry, 342
genes, 226, 844
in HDFN, 343, 536, 540
inheritance, 229-230
Kx antigen, 226, 342, 842
McLeod phenotype, 342-343
phenotypes and frequencies, 341, 844
in transfusion reactions, 343
Kernicterus, 536, 566
Kidd system (ISBT 009), 345-346
allele frequencies in, 227-229
antibodies, 336, 346
antigens, 345-346, 841, 844
chromosomal locations of genes, 226
genes, 844
in HDFN, 346
inheritance patterns of, 235
phenotypes and frequencies, 345, 346, 844
in transfusion reactions, 346, 656
Kidney
diseases treated with apheresis, 156-157
failure, 640-641
transplantation, 401-402, 619, 624, 627
Kinetic PCR, 213
Kleihauer-Betke acid-elution test, 550-551,
794-796
Kn (Knops) antigens, 354, 843
Knops system (ISBT 022), 226, 352, 354, 843
Kp (Kell) antigens and antibodies, 336, 341, 343,
841
Kx antigen (ISBT 019), 226, 342, 842
L
Labeling
aliquots, 564
for antigen typing, 440
autologous blood, 120, 122-123
biohazardous materials, 50, 719
blood components, 15, 170-172, 416, 440
blood samples, 409
884 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
dry ice, 719
hazardous chemicals, 59-60
ISBT 128 system, 171
reagents, 715
shipments, 718, 719
Laboratory coats, 73
Lactated Ringer’s solution, 530
Lan antigen, 356
Landsteiner-Wiener blood group (ISBT 016),
226, 327, 351, 842
Latent failures, 26-27
Latex allergies, 46-47
Law of mass action, 273
LCR (ligase chain reaction), 213, 214
LDL apheresis, 140, 157
Le (Lewis) antigens/antibodies, 304-306, 336,
844
Leach phenotype, 352
Lectins
Dolichos biflorus (anti-A1), 293, 296, 302,
743-744
preparation and use, 743-744
Ulex europaeus, 304, 743-744
Leishmania sp., 699
Leukapheresis, 143-144, 154, 587, 593
Leukocyte-reduction filters, 180, 190, 528-529
Leukocytes-reduced components
for children, 573-574, 576
expiration dates, 189
indications for, 493
labeling, 172
leukocyte content in, 492-493
Platelets, 180, 184, 189, 199, 202, 365, 493,
817
Platelets Pheresis, 35, 202, 493
poststorage, 190
prestorage, 180, 184, 199, 805-806, 817
to prevent bacterial contamination, 693
to prevent CMV infection, 687
quality control, 202, 832-834
Red Blood Cells, 180, 189, 199, 202, 493,
805-806
to reduce platelet alloimmunization, 265,
365
Whole Blood, 180
Lewis blood group (ISBT 007), 304-306
antibodies, 305-306, 336, 434, 539
antigens, 304-305, 841
in children, 306
genes, 226, 305
Lewis substance, 445
phenotypes, 305
saliva testing for, 445, 736-739
transfusion practices, 306
Ligands, 269
Ligase chain reaction (LCR), 213, 214
Liley graphs, 540, 541
Linkage, 230-231, 232
Linkage disequilibrium, 231-232, 337, 389
Lipemic samples, 410
Liquid-in-glass thermometers, 821-822
Liquid nitrogen
shipping, 718, 719
storage, 185, 189, 606
Liquid Plasma, 177, 189
LISS (low-ionic-strength saline)
antibodies to ingredients in, 437-438
and antigen-antibody proportions, 275, 277
to enhance antigen-antibody reactions, 277,
427, 443, 753, 754
and incubation time, 274
Liver
disease, 499
transplantation, 402, 619, 624, 627-629
LKE (Luke) antigen, 308, 310, 311
Locus, defined, 241
Long distance PCR, 213, 214
Look-back
for hepatitis, 675
for HIV, 681-682
for HTLV, 683
Low-incidence antigens
antibodies to, 340, 358, 436-437, 546
of Cromer system, 353, 354
defined, 335
of Diego system, 348
of Gerbich system, 352
of Lutheran system, 347
of MNS system, 227, 228, 337-338, 340
not assigned to a system or collection, 357
of Scianna system, 350
Low-ionic-strength saline. See LISS
Low-volume units, 101, 122, 179
Lsa(Gerbich) antigen, 352, 843
Lu (Lutheran) antigens and antibodies, 336,
347-348, 841
Lui freeze-thaw elution, 457, 774
Luke (LKE) antigen, 308, 310, 311
Index 885
Copyright © 2005 by the AABB. All rights reserved.
Lung transplants, 402
Lutheran system (ISBT 005), 347-348
antibodies, 336, 347-348
antigens, 347, 841
biochemistry, 347
chromosomal location of genes, 226
in HDFN, 348
phenotypes and frequencies, 347
LW system (ISBT 016), 226, 327, 351, 842
Lyme disease, 696-697
Lymphocytes
B cells, 249-250, 251, 252-253
NK (natural killer) cells, 255
passenger, 628, 629-630, 657
T cells, 253-254
Lymphocytotoxicity assays, 365-366, 395, 396-397
Lyonization, 224, 241
M
Macrophage chemoattractant protein (MCP),
640
Macrophage colony-stimulating factor (M-CSF),
264
Macrophages, 250, 255, 256
Magnetic cell separation, 596
MAIEA (monoclonal antibody-specific immobi-
lization of erythrocyte antigens) assay, 284,
286
MAIGA (monoclonal antibody-specific immobi-
lization of granulocyte antigens), 380
MAIPA (monoclonal antibody-specific immobi-
lization of platelet antigen), 373
Major histocompatibility complex (MHC). See
also HLA system
Class I, II, III molecules, 244-246, 390-391
defined, 269
organization of regions, 386-387
patterns of inheritance, 387-389
restriction, 393
Malaria, 697, 700
Management
assessment, 38
of critical supplies and services, 9-10
of documents and records, 14-19
of equipment, 10-11
of facilities, 6-7
of organization, 6-7
of personnel, 7-9
of safety program, 45
Manual, safety, 45
Marrow transplantation
ABO incompatibilities, 598-599, 600, 601-
602
ABO typing discrepancies, 299
allogeneic, 582, 583, 585-587
autologous, 583
bacterial contamination, 602-603
collection of marrow, 591-592
diseases treated with, 583, 584
donor databases, 585
donor evaluation, 589-591
engraftment in, 588-589
evaluation and quality control, 607-608
freezing and storage of product, 604-606
and graft-vs-host disease, 585-587
HLA testing, 400-401, 585-586
infectious disease testing, 590-591
matched, unrelated donor, 585-586
nonmyeloablative, 582-583
positive DAT after, 455
processing, 596-598, 601-604
red cell depletion, 601
regulations, 608
standards, 609
thawing and infusion, 607
transportation and shipping product,
606-607
MART antigen, 378, 379
Masks, 75
Massive transfusions
2,3-DPG levels in, 511-512
changing blood types, 511, 628-629
citrate toxicity in, 649-650
coagulopathy in, 511, 651
emergency issue, 510-511
hyper- and hypokalemia in, 650
hypothermia in, 511, 650
selection of blood, 419, 510-511
tissue oxygenation in, 511
Material data safety sheets (MSDS), 59, 60-61,
78-79
Materials, critical, 9
Maternal immunization
antibody titers, 449, 539-540, 761-764,
796-798
mechanisms, 536-538
prenatal evaluation, 538-542
suppression, 542-543
886 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Maximum surgical blood order schedules
(MSBOS), 92-93, 414
McCoy (McC) antigens, 352, 354
McLeod phenotype, 342-343
MCP (macrophage chemoattractant protein),
640
2-ME (2-mercaptoethanol)
applications for, 448-449
to disperse autoagglutination, 469, 470,
744-745
inactivation of Kell antigens, 342
Mechanical barrier systems, 527
Mechanical hemolysis, 152, 642
Medical history
in allogeneic donor selection, 100-101,
110-112
in antibody identification, 424
in autologous donations, 122
in cord blood collection, 595
in evaluation of positive DAT, 454-456
in prenatal evaluations, 538
in tissue and organ transplantation, 618,
620
Medical waste, 55, 56, 57
Medications. See Drugs
Meiosis, 224-225, 241
Membrane attack complex (MAC), 261-262, 263
MER2 (Raph) antigen, 354-355, 843
2-mercaptoethanol. See 2-ME
Meryman method of red cell cryopreservation,
807-810
Messenger RNA (mRNA)
isolation of, 210-211
processing, 204-206
translation, 206-207
Metabolic abnormalities, in neonates, 561-562
Methemoglobin, 835
Methylene chloride (dichloromethane) elutions,
457, 775
MHC. See Major histocompatibility complex
MHO4 clone, 298, 301
Microaggregate filters, 528, 565-566
Microangiopathic hemolysis, 502
Microarrays, 218
Microbead array assay, 395-396
Microfilariasis, 699
Microhematocrit centrifuges, 847
Microlymphocytotoxicity tests, 365-366, 395,
396-397
Microplate tests
for ABO group, 733-735
in antigen-antibody detection, 283-284
for Rh typing, 328, 741
Microscopic weak D test, 550
Microvascular bleeding (MVB), 511, 651
Miltenberger system, 338
Missense responses (mutations), 208, 227
Mitosis, 224, 225, 241
Mixed-field agglutination, 298, 299, 348, 357
Mixed lymphocyte culture (reaction)
(MLC/MLR), 397
Mixed type AIHA, 466-467
classification, 459
serologic findings in, 460, 466-467
specificity of autoantibodies, 467
transfusion in, 467
MLC (mixed lymphocyte culture), 397
MLR (mixed lymphocyte reaction), 397
MNS system (ISBT 002), 337-340
antibodies, 303, 336, 340, 444
antigens
linkage disequilibrium in, 231, 232, 337
linkage in, 232
low-incidence, 227, 228, 337-338, 340
M, N, S, s, U, 337
nomenclature, 840, 844
phenotypes and frequencies, 337, 844
biochemistry, 338-339
effect of enzymes on, 276, 339-340
genes, 226, 227, 228, 338, 844
in HDFN, 340
hybrid molecules, 338
in transfusion reactions, 340
MoAb. See Monoclonal antibodies
Mobile blood collection, 41, 179
Mobilization of hematopoietic progenitor cells,
587, 592-595
Modifier genes, 235-236
Molar solutions, 723
Mole, defined, 723
Molecular biology, 203-220
DNA and mRNA, 203-207
genetic variability, 208-209, 210
polymorphism, genetic mechanisms in,
207-208
techniques
DNA cloning, 217, 222
DNA microarrays, 218
Index 887
Copyright © 2005 by the AABB. All rights reserved.
DNA profiling, 216-217, 222
DNA sequencing, 217-218, 222, 396
gene therapy, 219-220
isolation of nucleic acids, 209-211
phage display/repertoire cloning, 222
polymerase chain reaction, 211, 212,
213-214, 215, 222, 394-396
protein and RNA targeted inactivation, 220
recombinant proteins, 218-219, 222
restriction endonucleases, 214, 216
restriction fragment length polymor-
phism analysis, 214, 216, 222
Monoclonal antibodies
in ABO testing, 296
anti-D, 328, 329-330
assays utilizing, 284, 373, 380
in autologous tumor purging, 597-598
for reagent use, 266-267
Monoclonal antibody-specific immobilization
of granulocyte antigens (MAIGA), 380
Monoclonal antibody-specific immobilization
of platelet antigens assay (MAIPA), 373
Monoclonal antibody-specific immobilization
of erythrocyte antigens (MAIEA) assay, 284,
286
Monocyte monolayer assay, 441
Monocytes, 250, 255, 256
Mononuclear phagocytic system, 255
Monospecific AHG reagents, 280
MP-NAT (minipooled NAT)
for HCV, 669
for HIV, 678, 679
for parvovirus, 689
for West Nile virus, 685
MPHA (mixed passive hemagglutination assay),
371-372
mRNA. See Messenger RNA
MSBOS (maximum surgical blood order sched-
ules), 92-93, 414
MSDS (material data safety sheets), 59, 60-61,
78-79
Multiplex PCR, 213
Muscular spasms, in donors, 108
Mutations, genetic, 208, 227, 228
Myasthenia gravis, 156
N
N antigen/antibody, 336, 337, 340, 840
NA/NB neutrophil antigens, 378
NAIT. See Neonatal alloimmune
thrombocytopenia
NAN (neonatal alloimmune neutropenia), 379
Nasopharyngeal carcinoma, 687
NAT. See Nucleic acid amplification test
National Marrow Donor Program (NMDP), 585,
589
Natural killer (NK) cells, 255
Nausea, 107-108, 639
Needles, 51, 523, 565
Negative selection, 597-598
Neonatal alloimmune neutropenia (NAN), 379
Neonatal alloimmune thrombocytopenia
(NAIT), 551-553
management after delivery, 553
platelet-specific antigens in, 366, 552
prenatal considerations for, 552-553
scheduling therapy, 553
serologic testing in, 552
sources of platelets for, 553
Neonatal immune thrombocytopenia (NIT)
alloimmune (NAIT), 366, 551-553
secondary to maternal ITP, 553-554
Neonatal polycythemia, 572
Neonates. See also Hemolytic disease of the fetus
and newborn
ABO antigens/antibodies in, 293, 295, 298
ABO discrepancies in, 299
ABO/Rh typing in, 415, 545
anemia in, 558, 559, 563-564
antigen variations in, 431
blood volume, 558-559, 839
compatibility testing in, 415, 562-563
cytomegalovirus infection in, 562
DIC in, 567, 572
direct antiglobulin testing in, 545-546
erythropoiesis in, 557-558, 559
extracorporeal membrane oxygenation in,
572-573, 574
graft-vs-host disease in, 560-561
hypothermia in, 560
immune thrombocytopenia in, 551-554
immunologic status, 560-561
leukocyte reduction for, 573-574
Lewis antigens in, 306
low birthweight, 557
metabolic problems in, 561-562
neonatal alloimmune neutropenia, 379
normal laboratory values in, 836-837
888 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
plasma volume in, 839
polycythemia in, 572
red cell volume in, 839
size of, 558-559
transfusions in
administration, 565-566
Cryoprecipitated AHF, 558, 572
effect of additive/preservative solutions,
564-565
to enhance hemostasis, 570-572
exchange, 546-547, 560, 566-568
of Fresh Frozen Plasma, 558, 571
Granulocytes, 558, 569-570
indications for, 563-564
Platelets, 490, 552-553, 558, 568-569
Red Blood Cells, 558, 564-568
volumes for, 558, 564, 568, 571
Neutralization
in antibody identification, 445
of Sdaantigen, 357, 445, 767-768
in viral marker testing, 169
Neutropenia, 379-380
Neutrophils
alloantigens, 377-380, 647
antibodies to
in neonatal alloimmune neutropenia, 379
testing for, 380
in TRALI, 379-380, 647, 656
in immune system, 255
NIT. See Neonatal immune thrombocytopenia
NK (natural killer) cells, 255
Nomenclature
of blood group systems, 238-239, 318-319,
840-844
CD (clusters of differentiation), 246, 247-248
of HLA system, 391-393
ISBT, 226, 238-239, 840-843
of platelet antigens, 366, 367-368
of Rh system, 315, 317, 318-319
Non-A, non-B hepatitis (NANB), 673, 699
Non-immune hemolysis, 636, 642-643
Nonconforming products or services
management, 19, 20, 21
quarantine, 173-174, 194
Nonimmunologic protein adsorption, 476
Nonsense response (mutations), 208, 227
Normal solutions, 723
Nuclear Regulatory Commission (NRC), 41, 64,
65, 66
Nucleic acid amplification test (NAT)
in HBV testing, 669, 672
in HCV testing, 164, 166, 213, 590, 672, 673,
675, 701
in HIV testing, 164, 166-167, 213, 590,
678-681
in HTLV testing, 683
in parvovirus testing, 688-689
in viral maker testing, 166-167
in West Nile virus testing, 213
Nucleic acids
isolation of, 209-211
sequencing, 217-218, 222
Nucleotides
defined, 203
insertion and deletion, 208
sequences, 204, 205
substitution, 207-208, 228
Nutricel (AS-3), 176, 186, 565
O
Obligatory gene, 237
Occupational Safety and Health Administration
(OSHA), 41
Officers
chemical hygiene, 58
safety, 42-43
Ohphenotype (Bombay), 304
Ok system (ISBT 024), 226, 354, 843
Oligonucleotide probes, sequence-specific
(SSOP), 214, 373, 395-396
OND antigen, 378, 379
“Open” system, 189, 192
Opsonization, 262
Optisol (AS-5), 176, 186, 565
Oral thermometers, electronic, 822-823
Ordering policies, 36, 91-94, 414
Organ donation and transplantation
ABO compatibility in, 401, 402, 627, 629-630
consent for, 620-621
cytomegalovirus in, 629, 630
disease transmission in, 617, 619-620
donor eligibility, 620-621
of heart, 629
HLA testing, 401-402
of kidney, 401-402, 619, 624, 627
of liver, 402, 627-629
of pancreas, 401-402, 629
positive DAT after, 455
Index 889
Copyright © 2005 by the AABB. All rights reserved.
preservation conditions and dating periods,
624
records, 626
recovery of tissue, 621
regulations, 626
risk reduction in, 618, 620
serologic testing for, 621-623, 629
skills and experience appropriate for, 618
transfusion support, 627, 629-630
types of donors for, 622
Organisms
in bacterial contamination, 691, 692
genetically modified, 717
Organizational management, 6-7
Organizations, 848-850. See also specific organi-
zations
regulating quality systems, 1-2
regulating safety, 39-40, 71-72
Organs of immune system, 249
Orientation program, 8
OSHA (Occupational Safety and Health Admin-
istration), 41
Outdating, 90-91
Outpatients, 408-409
Oxygen
compensation for anemia, 484
delivery of, in ANH, 127-128
dissociation in red cell storage, 185, 187
in massive transfusion, 511
measuring adequacy of supply, 484-485
supply and demand, 483-484
treating inadequate supply of, 485
Oxygen therapeutics, 512-513
P
P blood group (ISBT 003), 308-311
anti-P1, 303, 310-311
antibodies, 311, 468
antigens, 308-309, 310, 311, 840, 844
association with PCH, 468
biochemistry and genetics, 309-310, 844
chromosomal locations of genes, 226
hydatid cyst fluid/P1 substance, 310, 445
phenotypes, 309, 844
P1A1 and P1A2 (HPA-1) antigens, 366, 367, 369,
552
p24 antigen, 679
Packaging biological materials, 717-718, 720-721
PAD. See Preoperative blood donation
Pain at infusion site, 633
Pancreatic transplantation, 401-402, 620, 624,
629
Panel-reactive antibody (PRA), 362, 397, 401
Panels, red cell, 425, 426
Papain, preparation of, 756-757. See also En-
zymes, proteolytic
Para-Bombay phenotype, 304
Parasitic worms, 698-699
Parentage testing
genetics, 236-237
HLA testing in, 402
linkage disequilibrium in, 389
Paresthesias, 141, 151
Pareto analysis, 27
Paroxysmal cold hemoglobinuria (PCH),
467-468
classification, 459
serologic findings in, 460, 467
specificity of autoantibodies, 469
transfusion in, 467
Paroxysmal nocturnal hemoglobinuria, 378
Partial D, 322-323
Parvovirus B19, 688-689, 700
Passenger lymphocyte hemolysis, 628, 629-630,
657
Paternity testing. See Parentage testing
Pathogen inactivation, 702
Patients. See Recipients
PCH. See Paroxysmal cold hemoglobinuria
PCR. See Polymerase chain reaction
PCR-SSO/PCR-SSOP. See Sequence-specific
oligonucleotide probes
Pedi packs, 564
Pediatric patients. See Children; Neonates
Peer review, 23, 514
PEG. See Polyethylene glycol
Penicillin, 455, 474, 477, 786-788
Percentage solutions, 723, 726-727
Percutaneous umbilical blood sampling (PUBS),
541-542
Performance improvement standards, 25
Pericardium transplants, 619
Personal protective equipment (PPE), 43-44
for biosafety, 54
for chemical safety, 61
face shields, 74
gloves, 46, 55, 73-74, 106
laboratory coats, 73
890 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
masks, 75
safety goggles, 74
uniforms, 73
Personnel
accidents and injuries in, 45-46
competency assessment, 8
hepatitis prophylaxis for, 45, 703
job descriptions for, 7
latex allergies in, 46-47
medical first aid and follow-up, 45
orientation program, 8
protective equipment for, 43-45, 73-75
records, 19
safety monitoring programs, 45
selection of, 7
staffing levels, 8-9
training
biosafety, 50
chemical safety, 58-59
computer systems, 32
electrical safety, 48
FDA cGMP, 8
fire safety, 47
general safety, 43, 44
new employee, 8
radiation safety, 65
PF4 ELISA, 377
pH, 274, 431, 444
pH meters, 846
Phage display/repertoire cloning, 222, 267
Phagocytic cells, 255-256, 262
Phagocytosis, 262, 269
Pharmacologic alternatives to transfusion,
512-514
Pharmacologic purging, 597
Phenotypes.Seealsospecific blood groups
of autologous red cells, 429-430, 439
calculations for combined phenotypes, 236,
441
defined, 233
of donor units, 440-441
frequencies, 236
nomenclature for, 238-239, 844
Phlebotomy
adverse reactions to, 19, 107-109
aggressive, in autologous donations, 125
care of donors after, 106
collection of blood samples, 105-106,
408-409, 801-804
of donors, 105-106, 800-804
prevention of contamination in, 693
Phosphate buffer, 728
Photochemical inactivation, 702
Photopheresis, 158
Physical examination of donors, 100, 101-103, 620
Physicians’ orders, 522-523, 526
Physician’s responsibilities, in autologous pro-
gram, 120-121
Physiologic anemia of infancy, 558, 559, 563-564
Physiologic jaundice, 566
Phytanic acid disease, 157-158
Pipettes, recalibration, 847
PlAantigens, 366, 552
Plasma
ABO compatibility, 411, 496, 498
avoiding use, 701
coagulation factor replacement with, 496,
498-500
collection by apheresis, 141, 142-143
components
derivatives/substitutes, 496, 503-508, 699,
701
Fresh Frozen Plasma, 142, 177, 496,
498-500
inventory management, 94
Liquid Plasma, 177, 189
Plasma, 177
Plasma, Cryoprecipitate Reduced, 177,
190, 496, 498
Plasma Frozen within 24 Hours after
Phlebotomy, 181, 189
Pooled Plasma, Solvent/deter-
gent-treated, 496, 702
Recovered Plasma (for manufacture), 177
Source Plasma, 142
Thawed Plasma, 189, 191
constituents removed in apheresis, 149-150
description, 177
expiration dates, 189-190
for pretransfusion testing, 409, 424
as replacement fluid in apheresis, 150, 151
use in children, 571
virus inactivation, 701-702
volume, normal value, 839
Plasma D-dimers, 837
Plasma exchange. See Therapeutic apheresis
Plasma inhibition of anti-Ch and-Rg, 445,
765-766
Index 891
Copyright © 2005 by the AABB. All rights reserved.
Plasma protein fraction (PPF), 507
Plasmapheresis, 142-143, 500
Plasmids, 217
Platelet chambers, 185, 197-198, 846
Platelet concentrates (Platelets)
aliquoting, 193-194
bacterial growth in, 690-692, 694-695
biochemical changes during storage, 188
coagulation factors in, 838
description, 176-177, 489
expiration dates, 188, 189
freezing, 183
infectious risks of, 700
inspection, 194-195
inventory management, 94, 95
irradiated, 189, 192-193, 553
leukocyte-reduced, 184, 189, 190, 199, 202,
365, 493, 817
pooled, 184, 189, 193
preparation, 176-177, 815-817, 827-828
quality control, 199, 202
storage, 185, 188, 694-695
transfusions, 488-492
ABO/Rh matching, 361-362, 489-490, 569
in children, 490, 552-553, 558, 568-569,
576-577
contraindications to, 491-492
HLA alloimmunization to, 265, 266,
362-366, 397-398, 637
indications for, 490-491, 569
physiologic principles, 488-489
prophylactic, 490-491
refractoriness to, 362-365, 397-398, 491
therapeutic, 490
transportation and shipping, 196
volume reduction, 193, 490, 569, 817-818
washed, 193, 646
Platelet counts
in assessing hemostasis, 488
corrected platelet count increment, 362, 363,
489
in idiopathic thrombocytopenic purpura, 554
in massive transfusion, 511, 651
in NAIT, 552, 553
in neonates, 568-569
normal values, 835, 836
predicted platelet count increment, 362, 363,
489
transfusion triggers, 490-491, 569, 576
Platelet disorders
DDAVP in, 513
in liver disease, 499
thrombocytopenia
in blood loss, 499
drug-induced, 374-377
heparin-induced, 375-377, 492
idiopathic thrombocytopenic purpura,
158, 373-374, 492, 553-554
neonatal alloimmune thrombocytopenia,
511-513, 551-553
posttransfusion purpura, 366, 370, 638,
659-660
secondary to maternal ITP, 553-554
thrombotic thrombocytopenic purpura,
154-155, 158, 492, 500
treatment, 158, 490-491
Platelet incubators, 185, 197-198, 846
Platelet-rich plasma (PRP), 176, 177
Plateletpheresis, 140-142, 154
Platelets. See also Platelet concentrates; Platelet
disorders; Platelets Pheresis
antigens/antibodies
ABH, 361-362, 397
autoantibodies, 373-374
clinical importance, 370-373
detecting, 370-373, 374, 375, 377, 552
drug-induced, 374-377
HLA, 265, 362-366, 397-398
platelet-specific, 366, 367-368, 369-373, 552
assessing function, 488-489
corrected count increment, 362, 363, 489
life span and kinetics, 489
membrane glycoproteins, 369-370
normal values, 835, 836
predicted platelet count increment, 362, 363,
489
refractoriness, 362-365
antibody specificity prediction method
for, 365
causes, 362-363, 364, 397-398, 491
defined, 362
finding compatible donors, 398
HLA-matched platelets for, 363-365, 398
preventing alloimmunization, 365-366
selecting platelets with, 363-365
Platelets Pheresis
ABO/Rh matching, 361-362, 411, 489-490, 590
bacterial growth in, 690-692, 694-695
892 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
collection, 140-141
crossmatching, 364, 365, 398
described, 489
donor selection and monitoring, 141
HLA matched, 95, 363-365, 398
infectious risks of, 700
inspection, 194-195
inventory management, 94, 95
laboratory testing, 142
leukocytes reduced, 142, 202, 493
product validation, 35
quality control, 202
records, 141
storage, 185, 694-695
transfusions, 488-492
ABO/Rh matching, 361-362, 411, 489-490
contraindications to, 491-492
HLA alloimmunization to, 265, 266,
362-366, 397-398, 637
physiologic principles, 488-489
prophylactic, 490-491
refractoriness to, 362-365, 397-398, 491
therapeutic, 490
transportation and shipping, 196
Pluripotent hematopoietic stem cell, 249, 251
Policies and procedures, 7, 11, 15
Polyagglutination, 298, 331, 743
Polycythemia, 572
Polyethylene glycol (PEG)
in antibody detection/identification,
276-277, 427, 443, 753-754
effect on incubation times, 274
use in adsorption, 783-784
Polymerase chain reaction (PCR)
applications, 213-214, 215, 222
in HLA typing, 394-396
kinetic, 213
ligase chain reaction, 213, 214
long distance, 213, 214
multiplex, 213
oligonucleotide probes, 214, 373, 395-396
in platelet typing, 373
reaction procedure, 211, 212, 213
reverse transcriptase, 214, 217
sequence-specific primers, 214, 373, 395, 396
in testing fetus for D antigen, 538
variations, 213
Polymorphisms, 207-209
Polymorphonuclear granulocytes, 255
Polyspecific AHG, 279-280, 427
Pooled components
Cryoprecipitated AHF, 183-184, 191, 193, 815
expiration, 183, 189
labeling, 172, 183
Platelets, 184, 189, 193
preparation, 183
Pooled plasma, solvent-detergent-treated, 496, 702
Population genetics, 236-237
Position effect, 319-320, 324
Postoperative blood collection, 117, 133-135
Posttransfusion platelet recovery (PPR), 362,
363, 489
Posttransfusion purpura (PTP), 366, 370, 638,
659-660
Potassium, 187, 561, 650
PPE. See Personal protective equipment
PPF (plasma protein fraction), 507
PPR (posttransfusion platelet recovery), 362,
363, 489
PRA (panel-reactive antibody), 362, 397, 401
Preadmission testing, 409
Precipitation, 272
Pregnancy. See also Hemolytic disease of the fe-
tus and newborn; Prenatal studies
autologous collection in, 119
as immunizing stimulus, 536-537, 552
Lewis antibodies in, 305
Premedication in transfusions, 522, 646-647
Prenatal studies
amniotic fluid analysis, 540-541, 542
antibody titration, 449, 539-540, 761-764,
796-798
Doppler flow studies, 542
maternal history, 538
in neonatal immune thrombocytopenia, 552
percutaneous umbilical blood sampling,
541-542
serologic studies, 538-539
typing the fetus, 539
Preoperative blood donation (PAD), 117,
118-126
advantages/disadvantages, 118
adverse reactions, 123
aggressive phlebotomy, 125
collection, 121
compliance requirements, 119-120
component collection, 126
continuous quality improvement, 123-124
Index 893
Copyright © 2005 by the AABB. All rights reserved.
contraindications, 119
cost-effectiveness, 125, 127
donor deferrals, 120
donor screening, 121-122, 124-125
erythropoietin use, 125
establishing program, 120-124
labeling, 120, 122-123
medical interview, 122
in pediatric patients, 118-119
physician responsibility, 120-121
records, 123
shipping, 120
storage, 123
supplemental iron in, 121
testing, 119-120, 122
timing and red cell regeneration during, 122
transfusion, 123
transfusion trigger, 125
volume collected, 122
voluntary standards, 119
weakDindonor,324
Prescriptions for blood orders, 522-523, 526
Preservatives
anticoagulant-preservative solutions,
178-179
CPD, CP2D, CPDA-1, 178-179, 186, 564, 565
red cell changes during storage, 185, 186,
187, 431, 432
shelflifeofcomponents,178, 188,
189-190
reagent, antibodies to, 437
Pressure devices, 530
Pretransfusion testing, 407-420
after non-group-specific transfusions, 419-420
with autoantibodies, 469-472
of autologous blood, 122
blood labeling and release, 416, 418
blood samples, 409-410, 424
in children, 415, 562-563, 574
in cold agglutinin syndrome, 466
comparison with previous records, 413, 526
crossmatching, 413-415, 417
interpretation of results, 415-416, 417
in massive transfusions, 419, 510-511
patient identification, 407, 408-409
procedures included, 524
selection of units, 418-420
serologic testing, 410-413, 417
surgical blood orders, 91-93, 414
transfusion requests, 407-408, 522-523
type and screen, 91-93, 414
in urgent situations, 419, 510-511
Preventive action, 24-25
Prewarming technique, 308, 438, 754-755
Probability values in antibody identification,
429, 430
Problem identification and resolution, 25-27
Procainamide, 455
Procedures and policies, 7, 11, 15
Process capability, defined, 30
Process control, 30
Process flowcharting, 26, 27
Process improvement, 24-27
Process management, 11-14
computer system validation, 13-14
concepts, 4-5, 12
equipment validation, 13
process validation, 11-12
quality control, 14
validation plan, 12-13
Processes, 7, 11-12, 15
Production, principles of, 5
Products, nonconforming, 19, 20, 21
Proficiency testing (PT), 24
Promoter sequence, 204, 205
Protein C, 500, 507, 571, 837
Protein inactivation, 220
Protein S, 500, 507, 837
Protein synthesis, 204-207
Proteolytic enzymes. See Enzymes, proteolytic
Prothrombin time (PT)
in liver disease, 499
in massive transfusion, 511
in monitoring hemostasis, 494, 496
normal value of, 837
in vitamin K deficiency, 498
Prozone, 272, 275
PRP (platelet-rich plasma), 176, 177
Psoralen (S59), 702
PT. See Prothrombin time
PTP (posttransfusion purpura), 366, 370, 638,
659-660
Public antigens, 392
PUBS (percutaneous umbilical blood sampling),
541-542
Pulmonary edema, 648-649
Pulse, of donor, 102
Pumps, infusion, 522, 523, 529-530, 565
894 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Q
QSE(QualitySystemEssentials),3
Quad packs, 564
Qualification, defined, 30
Quality assurance (QA)
of blood administration, 527, 532
defined, 2, 30
Quality control (QC)
of blood components, 197-199, 202
apheresis, 832
Cryoprecipitated AHF, 199
leukocyte-reduced components, 199,
832-834
Platelets, 199, 694
Red Blood Cells, 199
Red Blood Cells, Deglycerolized, 812-813
of copper sulfate solution, 819-821, 847
defined, 2, 31
of equipment
automatic cell washers, 830-832
centrifuge calibration, 826-830
continuous temperature monitoring sys-
tems, 197-198
freezer alarms, 198-199, 824-826, 846
frequency of, 14, 846-847
refrigerator alarms, 198-199, 823-824, 846
thermometers, 198, 821-823, 847
of hematopoietic products, 607-608
in process control, 14
records, 14
unacceptable results for, 14
Quality improvement, 4
Quality indicators, 22-23, 31
Quality management, 2, 4-6, 31
Quality oversight, 6-7, 8
Quality System Essentials (QSEs), 3
Quality systems, 1-38
application of principles, 6-28
Code of Federal Regulations references, 32
common terms, 30-31
customer and supplier relations, 9-10
defined, 1-2
deviations and nonconforming products or
services, 19, 20, 21
documents and records, 14-15, 16, 17-19
equipment management, 10-11
human resources, 7-9
monitoring and assessment, 22-24, 36-38
organizational management, 6-7
process improvement, 24-27
process management, 11-14
quality assurance, 2
quality concepts, 2, 4-6
quality control, 2
quality management, 2
work environment, 27-28
Quarantine
of nonconforming components, 173-174
of repeatedly reactive units, 673, 674, 675,
683
of unusable products, 93-94
R
Race of donor, 98
Radiation safety, 63-66
blood irradiators, 65-66
effects of radiation, 63-64
emergency response plan, 66
exposure limits, 64
monitoring radiation, 64-65
radiation measurement units, 63
regulations, 64
safe work practices, 66
training, 65
waste management, 66
Raph system (ISBT 025), 226, 354, 843
Rapidly progressive glomerulonephritis (RPGN),
156-157
Rare Donor Program, 441-442, 769-770
Rd (Scianna) antigen, 349, 350, 842
Reagents
in ABO testing, 296
albumin additives, 276, 427, 753
antibodies to components of, 298, 437-438
antiglobulin, 278-280 , 427, 454, 468
chloroquine diphosphate, 446, 469, 746-
747
contamination of, 331, 332
for elutions, 457, 772-773, 775
enzymes, 276, 443, 445, 446, 756-760
glycine-HCl/EDTA, 446, 469, 747-748
labeling, 715
LISS and LISS additives, 274, 275, 277, 427,
443, 753, 754
monoclonal, 266-267, 296, 328, 329-330
for phenotyping, 440-441
polyethylene glycol (PEG), 276-277, 427, 443,
753-754
Index 895
Copyright © 2005 by the AABB. All rights reserved.
preparation of, 715, 743-744, 756-757
quality control intervals, 847
red cells, 425, 426, 438
in Rh testing, 328-331
sulfhydryl, 448-449, 744-745, 764-765,
766-767
use of manufacturer’s directions, 11
ZZAP, 445, 446, 781-783
Receptors
on B cells, 249-253
complement, 262, 354
defined, 269
immunoglobulin superfamily, 244, 245
on macrophages and monocytes, 250,
255-256
for pathogens, P antigens as, 311
on phagocytic cells, 255-256
on T cells, 245, 254, 393, 394
Recessive traits, 233-234, 241
Recipients
ABO and Rh testing in, 122, 410, 411
care during transfusions, 530-531
education and consent, 521-522
identification, 407, 408-409, 416, 524-526,
527
phenotyping, 429-430, 439
records, 413, 416, 660-661
tracing (look-back), 675, 681-682, 683
weak D in, 323-324, 411
Recombinant human erythropoietin (rHuEPO),
219
in anemia treatment, 488
in autologous donations, 125
in neonates, 559
Recombinant immunoblot assay (RIBA), 170,
670-671, 672
Recombinant interleukin-11, 512
Recombinant proteins
for HPC mobilization, 587, 592-595
in leukapheresis, 144
as transfusion alternative, 512, 559
uses for, 218-219, 222
Records
archiving, 17-18
autologous donation/transfusion, 123
blood component, 165, 172-173, 416, 418
changing, 18
checking before blood release, 416, 526
confidentiality, 18
donor, 97-98, 103, 165, 173
electronic, 17-18
infectious disease testing, 173
linking personnel to, 19
management, 17-19
of patients with special needs, 661
plateletpheresis, 142
pretransfusion testing, 413
storage, 18
stored tissue allograft, 626
transfer, 173
transfusion, 416
transfusion complication, 660-661
Recovered Plasma, 177
Red blood cells
abnormalities in Rhnull, 326
alloimmunization to, 265, 637, 656-657
antigen nomenclature, 238-239, 318-319,
840-844
changes in storage, 185, 186, 187-188, 431,
433
immune-mediated destruction, 265-266
membrane components of, 290-291
normal values, 835
preparation of 3% suspension, 727
reagents, 425, 426, 438
removal from marrow, 601
separating autologous and transfused,
748-750
survival studies, 441, 509, 655
volume, normal value, 839
Red Blood Cells, Deglycerolized
adequacy of deglycerolization, 812-813
expiration date, 189
preparation, 191-192, 809
refreezing, 183
storage, 192
Red Blood Cells, Frozen
cryoprotective agents for, 181
expiration dates, 184, 189
freeze-thaw damage in, 181
preparation, 181-183, 807-812
storage, 184-185, 809
thawing and deglycerolizing, 191-192, 809
transportation and shipping, 196
Red Blood Cells, Pheresis, 35, 144
Red Blood Cells (RBCs)
ABO/Rh compatibility, 411, 418, 486
antigen-matched, 95
896 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
bacterial contamination, 691
collection by apheresis, 35, 143
description, 176
expiration dates, 189
freezing, 181-183, 807-812
infectious risks of, 700
irradiated, 189, 192, 193, 493
leukocytes-reduced, 189, 190, 199, 202, 493,
805
low volume, 101, 122, 179
open system, 189
preparation, 176, 804-807
quality control, 199, 202
rejuvenated, 189, 194, 806-807
storage, 185, 186, 187-188
substitutes, 512-513
transfusion, 483-488
in HPC transplantation, 591-592, 599,
600, 601
indications for, 485-488, 563-564
in neonates, 562-568, 588
physiologic principles, 483-485
selection of components, 411, 486
transportation and shipping, 195-196
washed, 189, 193
Red cell depletion, 601
Reentry protocols, 673, 674, 675, 681-682
Reference laboratories, 439
Refractoriness to platelets, 362-365
antibody specificity prediction method for,
365
causes, 362-363, 364, 397-398, 491
defined, 362
finding compatible donors, 398
HLA-matched platelets for, 363-365, 398
platelet crossmatching for, 365, 398
preventing alloimmunization, 365-366
selecting platelets with, 363-365
Refrigerators, 184
alarm systems for, 198-199, 823-824, 846
quality control, 197-199, 846
temperature monitoring systems for,
197-198
thermometers for, 198
Refsum’s disease, 157-158
Registration of donors, 97-98
Regulations
for autologous donations, 119-120
for hematopoietic transplantation, 608
quality-related CFR regulations, 32
for radiation safety, 64
for safety, 28, 39-40, 64, 71-72
for tissue transplantation, 626
for transport and shipping dangerous goods,
716-721
Reissuing blood products, 197, 525
Rejuvenated RBCs, 189, 194, 806-807
Relative risk (RR), 403-404
Release of blood
identification of recipient and component,
524-526
policies of, 416, 418
reissue, 197, 525
in urgent situations, 419, 510-511, 522-523
Remedial action, 25
Renal system
diseases treated with apheresis, 156-157
failure in transfusion reactions, 640-641
kidney transplantation, 401-402, 619, 624,
627
Reports
of deviations, nonconformances and com-
plications, 19, 20, 21
of fatalities, 19, 46, 661, 702-703
of injuries, 45-46
internal event, 19, 20
Requests
for autologous blood collection, 121
for transfusion, 407-408, 522-523
Requirement, defined, 31
Respiratory distress, 152, 639, 647-648
Restricted work areas, 41
Restriction endonucleases, 214, 215
Restriction fragment length polymorphism
analysis (RFLP), 214, 215, 222, 373
Reticulocyte counts, normal value, 835
Reverse line technique, 395
Reverse transcriptase PCR (RT-PCR), 214, 217
RFLP (restriction fragment length polymor-
phism analysis), 214, 215, 222, 373
Rg (Chido/Rodgers) antigens/antibodies,
351-352, 842
Rh Immune Globulin (RhIG), 547-551
in amniocentesis, 541, 549
antepartum administration of, 538, 541,
548
contraindications for, 549
dosage for, 547, 549, 550-551
Index 897
Copyright © 2005 by the AABB. All rights reserved.
for fetomaternal hemorrhage, 549-551
in platelet transfusions, 490
in positive DAT, 456
postpartum administration of, 548-549
Rh system (ISBT 004), 315-331
antibodies, 327-329
anti-D in D+ individuals, 327
concomitant, 327-328, 441
directed at cis products, 324
dosage effect, 327
antigens
C, c, E, e, 316, 319, 320, 330-331
cis product, 324
D antigen, 315-316, 319-320
deleted phenotypes, 326
G antigen, 324-325
incidence, 317
nomenclature, 841, 844
partial D, 322-323
Rhmod, 326
Rhnull, 325-326
variants, 325
weak D, 322-324
association with LW, 351
in component selection, 418, 441, 486, 490,
511
ethnic differences in, 316, 317, 320, 321,
325
genes, 226, 320-321
genetics and biochemistry, 316-318
genotypes, 321-322, 844
in HDFN, 536
phenotypes and haplotypes, 319, 320, 844
position effect in, 319-320, 324
red cell abnormalities in, 326
RhAG (Rh-associated glycoprotein), 318
terminology, 315, 317, 318-319
in transplantation, 599, 628-629
Rh testing, 328-332
with autoagglutinins, 329, 331, 469-470
of autologous blood, 122
of blood components, 164, 165
of C, c, E, e, 320, 330-331
in children, 330, 415, 545, 563, 574
comparison with previous records, 413,
526
controls, 328-329, 330
of cord blood, 545
for D, 319-320, 328-330
of donors and recipients, 328-332, 410, 411,
413, 441
false-positive/false-negative results, 329,
330, 331-332
of Granulocytes, 144
in HDFN, 330, 539, 545
high-protein reagents in, 328-329, 330
low-protein reagents in, 329-330
microplate test, 328, 741
of Platelets Pheresis, 142
in prenatal evaluation, 538-539
slide test, 328, 739-740
in transfusion reaction evaluation, 654
for transplantation, 629
tube test, 740-741
of umbilical cord blood, 595
for weak D, 165, 323, 328, 411, 538-539,
741-744
RIBA (recombinant immunoblot assay), 170,
670-671, 672
Rigors, 633, 643
Risks of disease transmission
HIV infection, 677
with plasma derivatives, 699, 701
posttransfusion hepatitis, 673, 675
reducing, 618, 620, 699, 701-703
with tissue transplantation, 617-618,
619-620, 620
with transfusions, 700
RNA interference, 220
RNA (ribonucleic acid)
isolation of, 209-211
mRNA processing, 204-206
mRNA translation, 206-207
splicing, 206
transfer, 207
Rocky Mountain spotted fever, 696
Rodgers blood group. See Chido/Rodgers
Room temperature antibodies, 302-303
Room temperature storage, 185
Rosette test, 550, 793-794
Rouleaux
in ABO discrepancies, 298, 299, 303
in antibody detection, 412
in Rh typing, 329, 330
saline replacement technique, 303, 755-
756
RT-PCR. See Reverse transcriptase PCR
Run charts, 23
898 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
S
S/santigensandantibodies,336, 337, 340, 840
Safe work practices
for biosafety, 54-55
for electrical safety, 48-49
for fire prevention, 47-48
general guidelines, 44, 73-76
for radiation safety, 66
Safety goggles, 74
Safety program
accidents and injuries, 45-46
biosafety, 49-57, 77
chemical safety, 57-63, 78-88
disaster planning, 67-68
electrical safety, 48-49
emergency response plans, 44
employee monitoring programs, 45
engineering controls, 43
fire prevention, 47-48
first aid and follow-up, 45
general elements, 42-47
hazard identification and communication,
43
hepatitis prophylaxis, 45, 668, 672, 703
latex allergies, 46-47
management controls, 45
personal protective equipment, 43-44
policy manual, 45
radiation safety, 63-66
regulations and recommendations, 71-72
resources for information, 850
safe work practices, 44
safety officer, 42-43
shipping hazardous materials, 66
training, 43, 44
waste management, 67
in work environment, 27-28
Saline replacement technique, 303, 755-756
Saliva
ABH substances in, 290-291, 301, 736-739
Lewis substance in, 445, 736-739
Salvia lectins, 743
Samples. See Blood samples
SBO (standard blood orders), 92-93
Scianna system (ISBT 013), 226, 336, 349-350,
842
Sdaantigen, 357
agglutination pattern of antibody, 357, 412
neutralization of, 357, 445, 767-768
SDF-1 (stromal-derived growth factor-1), 594
Secretor gene (Se), 290, 291, 304-305, 737-739
Secretory component, 258
Segregation, 229, 230
Selectins, 246
Semen, 620, 621, 624
SEN-V virus, 668
Sensitization, 272-275
Separation techniques
in apheresis, 139-140
immunomagnetic, 596, 598
of multiple antibodies, 449
for transfused and autologous cells, 748-750
Sepsis
neonatal, 569-570
and positive DAT, 456
prevention, 693-694
transfusion-associated, 635, 643, 655,
691-692
Sequence-specific oligonucleotide probes
(SSOP), 214, 373, 395-396
Sequence-specific primers (SSPs), 214, 373, 395,
396
SERF (Cromer) antigen, 353, 354, 843
Serious Hazards of Transfusion (SHOT) initia-
tive, 641
Serologic centrifuges, calibration, 828-830
Serologic testing
additives in, 276-277, 443, 753-754
of autologous blood, 120, 122
of blood components, 165-166
in cold agglutinin syndrome, 460, 465
comparison with previous results, 413
of cord blood, 595
of Granulocytes, 144
in HDFN, 538-539, 544-546
for HLA antigens, 396-397
in IgM warm AIHA, 464
incubation temperatures, 443, 715
in mixed-type AIHA, 460, 466-467
in neonatal immune thrombocytopenia, 552
for organ and tissue transplantation, 620,
621-623, 629
in paroxysmal cold hemoglobinuria, 460,
467
for platelet antibodies, 375
of Platelets Pheresis, 142
with positive DAT, 456
in pretransfusion testing, 410-413, 562-563
Index 899
Copyright © 2005 by the AABB. All rights reserved.
records of, 165
for syphilis, 165-166, 695
in transfusion reaction evaluations, 653-654
in warm autoimmune hemolytic anemia,
460, 461
in warm IgM AIHA, 464
Serotonin release assay, 377
Serum
dilution, 725-726
for pretransfusion testing, 409, 424
Serum hyperviscosity syndrome, 153-154
Serum proteins in typing discrepancies, 298,
299, 303, 331
Serum-to-cell ratio, 444
Services
critical, 6-7
nonconforming, 19, 20, 21
quality principles, 5
Severe acute respiratory syndrome (SARS), 590
Sex-linked traits, 233-234, 241
SH antigen, 378
Shelflifeofcomponents,178, 188, 189-190
Shipments
of autologous units, 120
of blood components, 66, 179, 195-196
cargo aircraft only, 719
of clinical specimens, 66, 717-718, 720-722
containers for, 195, 847
of dangerous goods, 66, 716-722
of hematopoietic components, 606-607
labeling, 718, 719
monitoring temperature during, 722
packaging with dry ice, 196, 719, 720-721
packaging with liquid nitrogen, 718, 719
Shock, 633, 640
Short supply agreement, 177
SHOT (Serious Hazards of Transfusion) initia-
tive, 641
Showers, emergency, 61
Sickle cell disease (SCD)
alloantibody production in, 576
autologous donation with, 119
in children, 574-576
delayed transfusion reactions in, 656-657
separation of transfused from autologous
cells, 749-750
transfusion in, 508-509, 574-576
treatment with apheresis, 155-156
Signal transduction, 246
Signs, safety, 47, 51, 60
Silent mutations, 207
Single-donor platelets. See Platelets Pheresis
Skin appearance
in donors, 102-103
in transfusion reactions, 639, 644-646
Skin banking, 619, 624, 625
Sl (Knops) antigens, 354, 843
Slide tests
for ABO group, 731-732
for Rh typing, 328, 739-740
Sodium, 187
Software, computer, 17
Solid organ transplantation. See Organ donation
and transplantation
Solid-phase red cell adherence assay (SPRCA)
in detecting antigen-antibody reactions,
283-284
in platelet antibody detection, 371, 372, 375
in platelet crossmatching, 364
Soluble antigens
ABH, 290-291, 298, 301, 445, 736-739
Chido/Rodgers, 445, 765-766
HLA, 391
Lewis, 445, 736-739
P1, 310, 445
Sda, 357, 445, 767-768
testing, 301, 445, 736-739
Solutions
additive, 176, 186, 564-565
colloid, 486, 507-508, 572
dilution, 726-727
hemoglobin, 512-513
IV, 530
phosphate buffer, 728
preparation, 723-725
Solvent/detergent-treated pooled plasma, 496,
702
Somatic cells, 223, 241
Source Plasma, 142
Southern blotting, 214, 216
SPA immunoadsorption, 158
Specification, defined, 31
Spills
blood, 55, 56
chemical, 61-62, 84-88
radioactive, 66
SPRCA. See Solid-phase red cell adherence assay
SRA (C-serotonin release assay), 377
900 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
SSOP (sequence-specific oligonucleotide
probes), 214, 373, 395-396
SSP (sequence-specific primers), 214, 373, 395,
396
Staffing, 8-9
Standard blood orders (SBO), 92-93
Standard Precautions, 49, 54
Standards
for autologous donations, 119
for hematopoietic transplantation, 609
ISO 9001, 1- 2, 3
performance improvement, 25
STAR (Scianna) antigen, 349, 350, 842
Statistical tables for binomial distribution,
33-35
Stem cells, pluripotent, 249, 251, 581
Sterile connection devices, 847
Sterility testing, 195
Storage
acceptable temperatures for, 184, 185, 715
antigen deterioration with, 431, 433
of autologous blood, 94-95, 123
biochemical changes with, 185, 186, 187
of biohazardous material, 57
of blood components, 93-94, 184-185, 186,
187-188
of blood samples, 184-185, 410
of directed donor units, 94-95
frozen, 184-185, 606
of Granulocytes, 144
of hazardous chemicals, 61
of hematopoietic progenitor cells, 604-606
liquid, 185, 186, 187-188
liquid nitrogen, 185, 606
of organs and tissue, 624
of Platelets, 185, 188, 694-695
of records, 18-19
of Red Blood Cells, 185, 186, 187-188
of Red Blood Cells, Deglycerolized, 192
of Red Blood Cells, Frozen, 184-185, 809
refrigerated, 184
room temperature, 185
of untested or infectious products, 606
vapor-phase, 606
Storage lesion
of platelets, 188
of red cells, 185, 186, 187-188
Stroma-free hemoglobin solution, 512-513
Stromal-derived growth factor-1 (SDF-1), 594
STS (serologic testing for syphilis), 165-166,
695
Subgroups of ABO system, 293-294
in ABO discrepancies, 302
confirmation of by adsorption/elution,
735-736
testing for, 296
Sulfhydryl reagents, 448-449
to alter blood group antigens, 342, 445, 446,
766-767
applications for, 448-449
in dispersing autoagglutination, 469-470,
744-745
to distinguish IgM from IgG, 764-765
effect on Kell antigens, 342
Supertransfusion programs, 575-576
Supplier relations, 9-10
Supplies, critical, 9, 10, 11
Suppressor genes, 235-236
Surgical blood order systems, 91-93, 414
Survey meters, 65
Survival studies of red cells, 441, 509, 655
Syncope, in donors, 107
Syngeneic HPC transplantation, 582, 587
Syphilis, 695
testing autologous blood, 120
testing blood components, 164, 165-166, 695
testing transplantation donors for, 590, 620,
621
T
T-cell receptor (TCR), 245, 254, 393, 394
T-cell reduction (depletion), 586, 598
T lymphocytes, 249, 253-255
cytotoxic T cells, 249, 253-254
depletion methods, 598
helper T cells, 249, 253-254
receptors (TCR), 245, 254, 393, 394
recognition by T cytotoxic cells, 254
stimulation of B cells, 254-255
subpopulations of, 249, 253
TA-GVHD. See Transfusion-associated
graft-vs-host disease
Tc (Cromer) antigens/antibodies, 353, 354, 843
TCR (T-cell receptor), 245, 254, 393, 394
Temperature
of donor, 101-102
effect on agglutination, 273-274, 336, 443
incubation, 443, 715
Index 901
Copyright © 2005 by the AABB. All rights reserved.
monitoring systems, 197-198, 823-826
during shipments, 195-196, 722
storage, 184, 185, 715
Tendon transplants, 624
Test results, grading, 412, 728-729
Thalassemia, 508, 575-576
Thawed Plasma, 189, 191
Thawing
Cryoprecipitated AHF, 191, 815
devices for, 846
effects of, 181
Fresh Frozen Plasma, 191
hematopoietic components, 607
Red Blood Cells, Frozen, 191-192, 809
Therapeutic apheresis, 144-158
complications, 150-153
in HDFN, 145, 149
indications for, 146-148, 153-158, 500
photopheresis, 158
plasma volumes exchanged, 145, 149
removal of normal plasma constituents,
149-150
removal of pathologic substances, 145,
148-149
replacement fluids in, 150, 151
SPA immunoadsorption, 158
vascular access in, 145, 150
Therapeutic cells, 581. See also Hematopoietic
progenitor cell transplantation
Therapeutic plasma exchange (TPE), 145. See
also Therapeutic apheresis
Thermal amplitude studies, 441, 458, 465
Thermometers, 198
electronic oral, 822-823
liquid-in-glass, 821-822
standardization and calibration, 198,
821-823, 847
Thrombin, 502
Thrombin time, normal value, 837
Thrombocythemia, 154
Thrombocytopenia
in blood loss, 499
drug-induced, 374-377
and exchange transfusion, 567
heparin-induced, 375-377, 492
idiopathic thrombocytopenic purpura, 158,
373-374, 492, 553-554
neonatal alloimmune thrombocytopenia,
551-553, 568-569
posttransfusion purpura, 366, 370, 638,
659-660
secondary to maternal ITP, 553-554
thrombotic thrombocytopenic purpura,
154-155, 158, 492, 500
treatment, 158, 490-491
Thrombopoietin, recombinant human, 219
Thrombotic thrombocytopenic
purpura/hemolytic-uremic syndrome
(TTP/HUS), 154-155, 158, 492, 500
Tick-borne infections, 695-697
Timers/clocks, 847
Tissue antigens. See HLA system
Tissue oxygenation, 484-485, 511-512
Tissue transplantation
ABO compatibility in, 627
bone banking, 623, 625
consent for, 620-621
disease transmission in, 617, 619-620
donor eligibility, 620-621
heart valves, 625
preservation conditions and dating periods,
624
records, 626
recovery of tissue, 621
regulation, 626
risk reduction in, 618, 620
serologic testing for, 621-623
skills and experience appropriate for, 618
skin banking, 625
types of donors for, 622
Titration of antibodies
applications, 449
cold agglutinins, 458, 465, 778-779
maternal antibodies, 449, 539-540, 730-731,
796-798
methods for, 761-764, 796-798
TNF (tumor necrosis factor), 264, 640
Toxoplasmosis, 698
TPE (therapeutic plasma exchange), 145. See
also Therapeutic apheresis
Training
biosafety, 50
chemical safety, 58-59
for dry ice shipments, 721
electrical safety, 48
FDA cGMP, 8
fire safety, 47
general safety, 43, 44
902 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
new employee, 8
radiation safety, 65
Traits, 232-236
TRALI. See Transfusion-related acute lung injury
Tranexamic acid, 513-514
Trans effect, 319, 320
Transforming growth factor-β(TNF-β), 264
Transfusion-associated graft-vs-host disease
(TA-GVHD), 657-659
HLA system in, 399, 400
management, 637
manifestations, 658
in neonates, 560-561
pathophysiology, 637, 658
treatment and prevention, 658-659
Transfusion-associated sepsis, 635, 643, 655,
691-692
Transfusion Committee, 514
Transfusion reactions
acute
air embolism, 636, 651-652
allergic (urticarial), 522, 634, 644-647
anaphylactic, 635, 644-647
circulatory overload, 636, 648-649
citrate toxicity, 649-650
coagulopathy, 651
evaluation, 531, 652-656
febrile nonhemolytic, 379, 398-399, 576,
634, 643-644
hemolytic, 336, 399-400, 634, 639-642
hyper- and hypokalemia, 650
hypocalcemia, 561, 636, 649-650
hypotension associated with ACE inhibi-
tion, 636, 645
hypothermia, 511, 637, 650
nonimmune hemolytic, 636, 642-643
sepsis, 635, 643, 655, 691-692
signs and symptoms, 633, 639
transfusion-related acute lung injury,
379-380, 399, 635, 647-648
classification, 633, 634-638
delayed
alloimmunization, 637, 656-657
graft-vs-host disease, 399, 400, 560-561,
637, 657-659
hemolytic, 346, 575, 637, 656-657
immunomodulation, 638, 660
iron overload, 638, 660
Kidd antibodies in, 346, 656
posttransfusion purpura, 366, 370, 638,
659-660
management, 634-639
records, 660-661
reporting, 19, 661
Transfusion-related acute lung injury (TRALI),
647-648
evaluation, 656
and granulocyte transfusions, 648
HLA antibodies in, 399, 647
management, 635
neutrophil antibodies in, 379-380, 647
pathophysiology, 635, 647
prevention, 648
symptoms, 634, 647-648
treatment, 648
Transfusion-transmitted diseases
babesiosis, 695-696
bacterial sepsis, 635, 643, 655, 691-692
Chagas’ disease, 697-698
cytomegalovirus, 686-687
Epstein-Barr virus, 687
erlichiosis, 696
fatalities from, 702-703
hepatitis, 667-675
herpesviruses, 686-689
human immunodeficiency viruses, 675-682
human T-cell lymphotropic viruses, 682-683,
684
Lyme disease, 696-697
malaria, 697
management, 703
parasitic worms, 698-699
parvovirus, 688-689
reducing risks of, 699, 700, 701-702
reporting, 702
Rocky Mountain spotted fever, 696
syphilis, 695
tick-borne infections, 695-697
toxoplasmosis, 698
transmissible spongiform encephalopathies,
689-690
West Nile virus, 683, 685-686
Transfusion triggers
for autologous transfusions, 125
for platelet transfusions, 490-491, 569, 576
for red cell transfusions, 483, 486-488
Transfusions
administration procedures, 521-532, 565-566
Index 903
Copyright © 2005 by the AABB. All rights reserved.
alternatives to, 512-514
of antiprotease concentrates, 505-506
assessments of, 23, 38, 514
in autoimmune hemolytic anemias,
462-464, 467, 468, 509-510
of autologous blood, 123
cause of ABO discrepancies, 298
cause of positive DAT, 455, 462
in children, 562-572, 574-577
of coagulation factors, 493-494, 495, 496,
497, 498-505
of colloid solutions, 486, 507-508, 572
consent for, 521-522
of Cryoprecipitated AHF, 500-502, 572
delays in starting, 525
equipment for, 523-524, 527-530
events following, 531-532
exchange, 546-547, 560, 566-568
fatalities from, 19, 641, 645, 661, 690-691,
702-703
of Fresh Frozen Plasma, 496, 498-500, 571
of Granulocytes, 144, 492, 569-570
and HLA system, 397-400
in HPC transplantation, 591-592, 599, 600,
601
identification of recipient and components,
524-526, 527
of Immune Globulin, Intravenous,
505, 506
as immunizing stimulus in HDFN,
537-538
of incompatible blood, 509, 641
indications for, 486-488, 563-564
infusion rates, 531, 566
infusion sets for, 527-529, 565-566
intrauterine, 541, 543-544
of irradiated components, 493
IV solutions for, 530
of leukocyte-reduced components, 492-493
massive, 419, 510-512, 628-629, 649-651
in neonates, 562-572
non-group-specific, 419-420, 510-511
in older infants and children, 574-577
patient care during, 530
perioperative, 486-487
of plasma derivatives and substitutes, 496,
503-508
of Platelets, 488-492, 553, 568-569
premedication in, 522
of protein C and protein S, 507
quality assurance, 514, 527, 532
of RBCs, 483-488, 558, 562-568
of recombinant proteins, 512
requests for, 407-408, 522-523
selection of components, 411, 486
in sickle cell disease, 508-509, 574-576
starting, 526-527
in thalassemia, 508, 575-576
in transplantation, 627, 629-630
in urgent situations, 419, 510-511,
522-523
of Whole Blood, 485, 486
Transmissible spongiform encephalopathies
(TSEs), 689-690
Transplantation. See Hematopoietic progenitor
cell transplantation; Organ donation and
transplantation; Tissue transplantation
Transplantation antigens. See HLA system
Transportation
of autologous units, 120
of blood components, 66, 179, 195-196
of blood to patient area, 524-525
cargo aircraft only, 719
of clinical specimens, 66, 717-718, 720-722
containers for, 195, 847
of dangerous goods, 66, 716-722
of hematopoietic components, 606-607
labeling, 718, 719
monitoring temperatures during, 722
packaging with dry ice, 196, 719, 720-721
packaging with liquid nitrogen, 718, 719
Trial to Reduce Alloimmunization to Platelets
(TRAP) Study Group, 365
Tropical spastic paraparesis (TSP), 682-683
Trypanosoma cruzi, 698, 700
TSEs (transmissible spongiform
encephalopathies), 689-690
TTP/HUS. See Thrombotic thrombocytopenic
purpura/hemolytic-uremic syndrome
TTV virus, 668
Tube tests
for ABO group, 732-733
for Rh type, 740-741
Tumor cell detection, 608
Tumor necrosis factor (TNF), 264, 640
Tumor purging, 597-598
Twitching, in donors, 108
Type and screen (T/S), 91-93, 414
904 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
U
U antigen/antibody (MNS system), 336, 337,
340, 840
Ulex europaeus (anti-H), 304, 743-744
Ultraviolet B (UVB) irradiation, 365
Umbilical cord blood. See Cord blood
UMC (Cromer) antigen, 353, 354, 843
Unasyn, 455
Uniforms, 73
Urgent release of blood, 419, 510-511, 522-523
Urine
examinations in transfusion reactions, 639,
653
neutralization of Sda, 357, 445, 767-768
Urticaria (hives), 634, 644-647
Utilization of blood. See Blood utilization
UVB (Ultraviolet B) irradiation, 365, 398
V
Vaccines, hepatitis, 45, 668, 672, 703
Valeri method for red cell cryopreservation,
810-812
Validation
of computer systems, 13-14, 415
defined, 31
of equipment, 13
of processes, 11-12
statistical tables for, 33-35
validation plans, 12-13
variances to, 11
Vapors, hazardous, 62, 63
Variant Creutzfeldt-Jakob disease (vCJD), 590,
689-690
Vascular access
in apheresis, 145, 150
in HPC collection, 593, 594
in neonates, 565-566, 568
for transfusions, 523-524
Vasovagal syndrome, 107, 141
vCJD (variant Creutzfeldt-Jakob disease), 590,
689-690
Vel collection, 355-356
Venipuncture, 105-106, 800-804
Venous access. See Vascular access
Venous transplants, 624
Verification, defined, 31
Vicia graminea (anti-N), 743
View boxes, 846
Vil antigen, 354
Viral marker testing. See Infectious disease test-
ing
Viruses
inactivation of, 699, 701-702
transmitted by transfusion
cytomegalovirus, 686-687
Epstein-Barr, 687
hepatitis, 667-675
HIV, 675-682
HTLV, 682-683, 684
human herpesviruses, 688
parvovirus, 688-689
transmissible spongiform
encephalopathies, 689-690
West Nile, 683, 685-686
Viscosity, relative, 835
Vital signs, 526-527, 531
Vitamin K, 498-499, 571
VLBW (very low birthweight) neonates, 557, 568.
See also Neonates
Volume of blood
administered in intrauterine transfusions,
544
administered in neonatal transfusions, 558,
564, 568, 571
collections, 101, 122, 178-179
normal values, 558-559, 839
Volume of marrow collections, 591
Volume of plasma
exchanged in TPE, 145, 149
normal values, 839
Volume of red cells, 839
Volume reduction of platelets, 193, 490, 569,
817-818
Vomiting, 107-108, 639
von Willebrand syndromes, 501
W
WAIHA. See Warm autoimmune hemolytic ane-
mia
Warfarin, 376, 498-499
Warm autoantibodies
adsorption, 470-472, 779-781
alloantibodies with, 439, 470-472
drug-induced, 475-476
frequency of testing, 463-464
mimicking alloantibodies, 472
transfusion-stimulated, 462
Index 905
Copyright © 2005 by the AABB. All rights reserved.
transfusion with, 462-464
in warm autoimmune hemolytic anemia,
461-462
Warm autoimmune hemolytic anemia (WAIHA),
459, 461-464
antiglobulin testing in, 281
chronic, 462-463
classification, 459
serologic findings in, 459, 460, 461
specificity of autoantibody, 461-462
transfusion in, 462-464
Warm-reactive alloantibodies, 273-274
Washed blood components, 189, 193, 561, 646
Wastage of autologous blood, 123-124
Waste
biohazardous, 55-57
chemical, 63
defined, 717
radioactive, 63
reduction program, 67
treating, 57
Water of crystallization, 724
Water of hydration, 724
Waterbaths, 693, 846
Wb (Gerbich) antigen, 352, 843
Weak D, 322-324
in autologous donations, 324
in donors, 165, 323
“microscopic,” 550
partial D, 322-323
in pregnancy, 538-539
quantitative, 322
in recipients, 323-324, 411
testing for, 165, 323, 328, 741-743
Weight, donor, 101, 143
WES (Cromer) antigens/antibodies, 353, 354,
843
West Nile virus (WNV), 683, 685-686
fatalities due to, 691
preventive measures, 590, 685
testing for, 166, 213, 595-596, 685
Western immunoblot, 169-170, 679-681, 698
WH (Chido/Rodgers) antigen, 352, 842
White cells
normal values, 835, 836
residual, in leukocyte-reduced components,
493, 832-834
Whole blood
ABO/Rh compatibility, 411, 486
biochemical changes during storage, 185,
186, 187-188
collection, 178-179
description, 175
equipment quality control, 847
expiration date, 189
leukocyte reduction, 180
processing, 179-180
transfusion, 485, 486
transportation and shipping, 179,
195-196
Wipe tests, 65
Work environment
design and workflow, 40
housekeeping, 40-41
restricted areas, 41
safety, 27-28, 54-55
Wr (Diego) antigens/antigens, 348, 842
Wristbands, patient, 408, 409, 526
X
X-linked traits, 233-234, 241
Xenogeneic HPC transplantation, 582
Xg system (ISBT 012), 226, 336, 348-349, 350,
842
Y
York (Yk) antigens, 352, 354, 843
Yt (Cartwright) system (ISBT 011), 226, 336, 348,
349, 842
Z
ZENA (Cromer) antigen, 353, 354, 843
Zygosity, 431. See also Dosage effect
ZZAP
in allogeneic adsorption, 471-472, 781-783
to alter antigens, 342, 445, 446
in autologous adsorption, 469, 470, 775-776,
779-781
in dispersing autoagglutination, 469
906 AABB Technical Manual
Copyright © 2005 by the AABB. All rights reserved.
Glossary of Abbreviations
AATB American Association of Tissue
Banks
ACD acid-citrate-dextrose
ACE angiotensin-converting enzyme
AChE acetylcholinesterase
ACOG American College of Obstetricians
and Gynecologists
ADCC antibody-dependent cellular
cytotoxicity
AET 2-aminoethylisothiouronium
AHF antihemophilic factor
AHG antihuman globulin
AHTR acute hemolytic transfusion reaction
AICC anti-inhibitor coagulation complex
AIDS acquired immune deficiency
syndrome
AIHA autoimmune hemolytic anemia
ALG antilymphocyte globulin
ALT alanine aminotransferase
ANCA antineutrophil cytoplasmic
antibodies
ANH acute normovolemic hemodilution
APC antigen-presenting cell
aPTT activated partial thromboplastin
time
AS additive solution
ASO allele-specific oligonucleotide
ATG antithymocyte globulin
ATL adult T-cell lymphoma/leukemia
ATP adenosine triphosphate
BFU burst-forming unit
BPO benzyl-penicilloyl
BSA bovine serum albumin or body
surface area
BSC biological safety cabinet
BUN blood urea nitrogen
C:T crossmatch-to-transfusion
CAD cold antibody (agglutinin) disease
CAP College of American Pathologists
CAS cold antibody (agglutinin) syndrome
CBER Center for Biologics Evaluation and
Research
CCE counterflow centrifugal elutriation
CCI corrected count increment
CD clusters of differentiation
CDC Centers for Disease Control and
Prevention
CFR Code of Federal Regulations
CFU colony-forming unit
CGD chronic granulomatous disease
cGMP current good manufacturing
practice
cGy centiGray
CHD cold hemagglutinin disease
CI continuous improvement or
confidence interval
CIDP chronic inflammatory demyelinating
polyneuropathy
CJD Creutzfeldt-Jakob disease
CLIA Clinical Laboratory Improvement
Amendments
CLSI Clinical and Laboratory Standards
Institute
CML cell-mediated lympholysis
CMS Centers for Medicare and Medicaid
Services
CMV cytomegalovirus
CNS central nervous system
CPD citrate-phosphate-dextrose
CPDA-1 citrate-phosphate-dextrose-adenine-1
CR complement receptor
CREG cross-reactive group
CRYO cryoprecipitated AHF
CUE confidential unit exclusion
DAF decay-accelerating factor
DAT direct antiglobulin test
DDAVP 1-deamino-8-d-arginine
vasopressin
DDR donor deferral registry
DHTR delayed hemolytic transfusion
reaction
DIC disseminated intravascular
coagulation
DMSO dimethylsulfoxide
DNA deoxyribonucleic acid
2,3-DPG 2,3-diphosphoglycerate
DRG diagnosis-related group
Copyright © 2005 by the AABB. All rights reserved.
DTT dithiothreitol
EACA epsilon aminocaproic acid
EBAA Eye Bank Association of America
EBV Epstein-Barr virus
ECMO extracorporeal membrane
oxygenation
EDTA ethylenediaminetetraacetic acid
EIA enzyme immunoassay
ELAT enzyme-linked antiglobulin test
ELBW extremely low birthweight
ELISA enzyme-linked immunosorbent
assay
EPO erythropoietin
ESR erythrocyte sedimentation rate
FACT Foundation for the Accreditation of
Cellular Therapy
FDA Food and Drug Administration
FFP Fresh Frozen Plasma
FMH fetomaternal hemorrhage
FNHTR febrile nonhemolytic transfusion
reaction
FTA-ABS fluorescent treponemal antibody
absorption test
5-FU 5-fluorouracil
G-CSF granulocyte colony-stimulating
factors
GalNAc N-acetylgalactosamine
GM-CSF granulocyte macrophage
colony-stimulating factors
GMP good manufacturing practice
Gp glycoprotein
GPA glycophorin A
GPB glycophorin B
GPC glycophorin C
GPD glycophorin D
GVHD graft-vs-host disease
Gy Gray
HAM HTLV-associated myelopathy
HAV hepatitis A virus
HAZMAT hazardous material
Hb hemoglobin
HBc hepatitis B core antigen
HBIG hepatitis B immunoglobulin
HBsAg hepatitis B surface antigen
HBV hepatitis B virus
Hct hematocrit
HCV hepatitis C virus
HDFN hemolytic disease of the fetus and
newborn
HDV hepatitis D virus
HES hydroxyethyl starch
HEV hepatitis E virus
HIV human immunodeficiency virus
HPC hematopoietic progenitor cell
HTLV-I human T-cell lymphotropic virus
type I
HTR hemolytic transfusion reaction
HUS hemolytic uremic syndrome
IAT indirect antiglobulin test
Ig immunoglobulin
IGIV Immunoglobulin Intravenous
IHA immune hemolytic anemia
IL-1αinterleukin 1 alpha
IL-1ß interleukin 1 beta
IL-2 interleukin 2
IPT intraperitoneal transfusion
IS immediate spin
ISBT International Society of Blood
Transfusion
ISCT International Society for Cellular
Therapy
ITP idiopathic thrombocytopenic
purpura
IUT intrauterine transfusion
IVT intravascular transfusion
JCAHO Joint Commission on Accreditation
of Healthcare Organizations
L/S lecithin to sphingomyelin
LDH lactate dehydrogenase
LDL low-density lipoproteins
LISS low ionic strength saline
LT-CIC long-term culture-initiating cells
MAC membrane attack complex
2-ME 2-mercaptoethanol
MF mixed field
MHC major histocompatibility complex
MLC mixed lymphocyte (leukocyte)
culture
(cont’d)
Copyright © 2005 by the AABB. All rights reserved.
MLR mixed lymphocyte (leukocyte)
reaction
MoAb monoclonal antibody
mRNA messenger ribonucleic acid
MSBOS maximum surgical blood order
schedule
MSDS material safety data sheets
NAIT neonatal alloimmune
thrombocytopenia
NAT nucleic acid testing
NIH National Institutes of Health
NK natural killer
NMDP National Marrow Donor Program
NRC Nuclear Regulatory Commission
NT not tested
OSHA Occupational Safety and Health
Administration
p probability
PAD preoperative autologous (blood)
donation
PBPC peripheral blood progenitor cell
PBS phosphate-buffered saline
PCH paroxysmal cold hemoglobinuria
PCR polymerase chain reaction
PEG polyethylene glycol
PHA phytohemagglutinin
PI paternity index
PPE personal protective equipment
PPF plasma protein fraction
PPTA Plasma Protein Therapeutics
Association
PRA panel reactive antibody
PT prothrombin time or proficiency test
PTP posttransfusion purpura
PUBS percutaneous umbilical blood
sampling
PVC polyvinyl chloride
QA quality assessment or quality
assurance
QC quality control
QSE quality system essential
RBCs Red Blood Cells (blood donor unit)
RCA regulators of complement activation
RES reticuloendothelial system
RFLP restriction fragment length
polymorphism
Rh Rhesus factor
RhIG Rh Immune Globulin
RIBA recombinant immunoblot assay
RNA ribonucleic acid
RPGN rapidly progressive
glomerulonephritis
RPR rapid plasma reagin (serologic test
for syphilis)
RR repeatedly reactive or relative risk
RT room temperature or reverse
transcriptase
SBO standard blood order
SCF stem cell factor
SGP sialoglycoprotein
SOP standard operating procedure
SPA staphylococcal protein A
SSO sequence-specific oligonucleotide
STS serologic test for syphilis
TA transfusion-associated
TCR T-cell receptor
TNF-αtumor necrosis factor alpha
TPE therapeutic plasma exchange
TRALI transfusion-related acute lung
injury
tRNA transfer ribonucleic acid
TTP thrombotic thrombocytopenic
purpura
UNOS United Network for Organ Sharing
VLBW very low birthweight
vWD von Willebrand disease
vWF von Willebrand factor
WAIHA warm autoimmune hemolytic
anemia
WB Whole Blood or Western blot
XM crossmatch
Copyright © 2005 by the AABB. All rights reserved.
