AutoDock4.2.5_UserGuide20121105g Auto Dock4.2 User Guide
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 66

1
User Guide
AutoDock Version 4.2
Updated for version 4.2.5
Automated Docking of Flexible Ligands to Flexible Receptors
Garrett M. Morris, David S. Goodsell, Michael E. Pique, William “Lindy” Lindstrom, Ruth Huey, Stefano
Forli, William E. Hart, Scott Halliday, Rik Belew and Arthur J. Olson
Modification date: November 05, 2012 08:37 D11/P11
AutoDock, AutoGrid, AutoDockTools, Copyright © 1991-2009
2
Contents
Automated Docking
Introduction ...................................................................................................... 3
Getting Started with AutoDock .......................................................................... 3
Whatʼs New? ..................................................................................................... 5
Support.............................................................................................................. 7
Theory
Overview of the Free Energy Scoring Function ................................................ 8
Using AutoDock
STEP 1: Preparing Coordinates........................................................................ 12
Creating PDBQT files with AutoDockTools ................................................ 13
STEP 2: Running AutoGrid ............................................................................... 18
Creating grid parameter files with AutoDockTools ..................................... 19
STEP 3: Running AutoDock .............................................................................. 20
Choosing a protocol for your application .................................................... 21
Creating docking parameter files with AutoDockTools ............................... 23
STEP 4: Evaluating the Results of a Docking ................................................... 24
Information in the docking log file ............................................................... 24
Analyzing docking results with AutoDockTools .......................................... 24
Appendix I: AutoDock File Formats
PDBQT format for coordinate files .................................................................... 27
PDBQT format for flexible receptor sidechains ................................................. 29
AutoGrid Grid Parameter File: GPF .................................................................30
Atomic Parameter File....................................................................................... 33
Grid Map File .................................................................................................. 35
Grid Map Field File .......................................................................................... 36
AutoDock Docking Parameter File: DPF .......................................................... 37
Appendix II: Customizing the Docking Protocol
Introduction ....................................................................................................... 51
Docking parameter file examples...................................................................... 56
Appendix III: Docking Flexible Rings with AutoDock
Introduction ....................................................................................................... 61
Flexible RIngs ................................................................................................... 62
Reference.......................................................................................................... 65
Appendix IV: AutoDock References

3
Automated Docking
Introduction
AutoDock is an automated procedure for predicting the interaction of ligands with
biomacromolecular targets. The motivation for this work arises from problems in the design of
bioactive compounds, and in particular the field of computer-aided drug design. Progress in
biomolecular x-ray crystallography continues to provide important protein and nucleic acid
structures. These structures could be targets for bioactive agents in the control of animal and
plant diseases, or simply key to the understanding of fundamental aspects of biology. The precise
interaction of such agents or candidate molecules with their targets is important in the
development process. Our goal has been to provide a computational tool to assist researchers in
the determination of biomolecular complexes.
In any docking scheme, two conflicting requirements must be balanced: the desire for a robust
and accurate procedure, and the desire to keep the computational demands at a reasonable level.
The ideal procedure would find the global minimum in the interaction energy between the
substrate and the target protein, exploring all available degrees of freedom (DOF) for the system.
However, it must also run on a laboratory workstation within an amount of time comparable to
other computations that a structural researcher may undertake, such as a crystallographic
refinement. In order to meet these demands a number of docking techniques simplify the docking
procedure. AutoDock combines two methods to achieve these goals: rapid grid-based energy
evaluation and efficient search of torsional freedom.
The current version of AutoDock, using the Lamarckian Genetic Algorithm and empirical free
energy scoring function, typically will provide reproducible docking results for ligands with
approximately 10 flexible bonds. Our related software, AutoDock Vina (http://vina.scripps.edu),
uses a simpler scoring function that allows a faster search method, and provides reproducible
results for larger systems with upwards of 20 flexible bonds. A more complete discussion of
options is included below in the section "Choosing a Protocol for Your Application."
This guide includes information on the methods and files used by AutoDock and information on
use of AutoDockTools to generate these files and to analyze results.
Getting Started with AutoDock
AutoDock and AutoDockTools, the graphical user interface for AutoDock are available on the
WWW at:
http://autodock.scripps.edu/
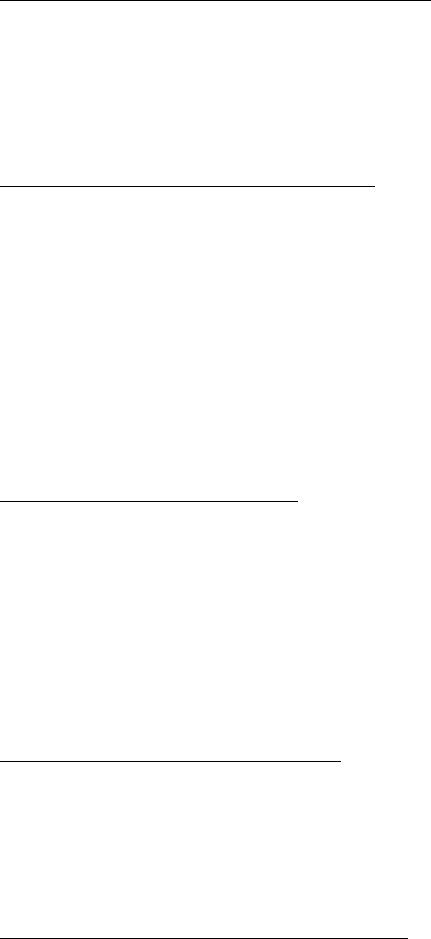
4
The WWW site also includes many resources for use of AutoDock, including detailed Tutorials
that guide users through basic AutoDock usage, docking with flexible rings, and virtual
screening with AutoDock. Tutorials may be found at:
http://autodock.scripps.edu/faqs-help/tutorial
AutoDock calculations are performed in several steps: 1) preparation of coordinate files using
AutoDockTools, 2) precalculation of atomic affinities using AutoGrid, 3) docking of ligands
using AutoDock, and 4) analysis of results using AutoDockTools.
Step 1—Coordinate File Preparation. AutoDock4.2 is parameterized to use a model of the
protein and ligand that includes polar hydrogen atoms, but not hydrogen atoms bonded to carbon
atoms. An extended PDB format, termed PDBQT, is used for coordinate files, which includes
atomic partial charges and atom types. The current AutoDock force field uses several atom types
for the most common atoms, including separate types for aliphatic and aromatic carbon atoms,
and separate types for polar atoms that form hydrogen bonds and those that do not. PDBQT files
also include information on the torsional degrees of freedom. In cases where specific sidechains
in the protein are treated as flexible, a separate PDBQT file is also created for the sidechain
coordinates. AutoDockTools, the Graphical User Interface for AutoDock, may be used for
creating PDBQT files from traditional PDB files.
Step2—AutoGrid Calculation. Rapid energy evaluation is achieved by precalculating atomic
affinity potentials for each atom type in the ligand molecule being docked. In the AutoGrid
procedure the protein is embedded in a three-dimensional grid and a probe atom is placed at each
grid point. The energy of interaction of this single atom with the protein is assigned to the grid
point. AutoGrid affinity grids are calculated for each type of atom in the ligand, typically carbon,
oxygen, nitrogen and hydrogen, as well as grids of electrostatic and desolvation potentials. Then,
during the AutoDock calculation, the energetics of a particular ligand configuration is evaluated
using the values from the grids.
Step 3—Docking using AutoDock. Docking is carried out using one of several search methods.
The most efficient method is a Lamarckian genetic algorithm (LGA), but traditional genetic
algorithms and simulated annealing are also available. For typical systems, AutoDock is run
several times to give several docked conformations, and analysis of the predicted energy and the
consistency of results is combined to identify the best solution.
Step 4—Analysis using AutoDockTools. AutoDockTools includes a number of methods for
analyzing the results of docking simulations, including tools for clustering results by
conformational similarity, visualizing conformations, visualizing interactions between ligands
and proteins, and visualizing the affinity potentials created by AutoGrid.

5
Whatʼs New?
AutoDock 4.2 includes several enhancements over the methods available in AutoDock 3.0.
Sidechain Flexibility. AutoDock 4.2 allows incorporation of limited sidechain flexibility into the
receptor. This is achieved by separating the receptor into two files, and treating the rigid portion
with the AutoGrid energy evaluation and treating the flexible portion with the same methods as
the flexible ligand.
Force Field. The AutoDock 4.2 force field is designed to estimate the free energy of binding of
ligands to receptors. It includes an updated charge-based desolvation term, improvements in the
directionality of hydrogen bonds, and several improved models of the unbound state.
Expanded Atom Types. Parameters have been generated for an expanded set of atom types
including halogens and common metal ions.
Desolvation Model. The desolvation model is now parameterized for all supported atom types
instead of just carbon. Because of this, the constant function in AutoGrid is no longer used,
since desolvation of polar atoms is treated explicitly. The new model requires calculation of a
new map in AutoGrid that holds the charge-based desolvation information.
Unbound State. Several models are available for estimating the energetics of the unbound state,
including an extended model and a model where the unbound state is assumed to be identical
with the protein-bound state.
For users of AutoDock 4.0, there are several changes in AutoDock 4.2:
Default Unbound State. The default model for the unbound state has been changed from
“extended” to “bound=unbound”. This is in response to persistent problems when docking
sterically-crowded ligands. The “extended” unbound state model is available in AutoDock 4.2
through use of the “unbound extended” keyword.
Backwards Compatibility. We have made every attempt to ensure that docking parameter files
generated for use in AutoDock 4.0 should be correctly run by AutoDock 4.2.
For users of AutoDock 4.2.3 or 4.2.4, there are several changes in AutoDock 4.2.5:
Finer Control of Output. In response to the widespread use of AutoDock in virtual screening, we
have modified the “outlev” command to allow more control over the level of output. The default
of “1” will now output primarily information on the docked conformations and analysis, and
higher levels will provide diagnostic information.
Program will Halt with Critical Errors. Several error conditions that previously gave warnings
will now cause AutoDock to halt. This is a response to use of AutoDock in virtual screening,

6
where the user may not examine each individual docking experiment, and critical errors may not
be noticed.
Ligand Internal Electrostatics are now ‘on’ by Default. The electrostatic interactions between
non-bonded atoms in flexible ligands are now considered ‘on’ by default. Previous releases of
AutoDock required the “intelec” command to turn this on. If you wish to ignore these
interactions and restore the previous default behavior, use “intelec off” in your DPF.
Force Field Consistency. Smoothing of potentials has been added to internal energy potentials, to
make them consistent with intermolecular energy potentials. This will have a small effect on
conformations when docking with rigid receptors, and possibly a significant effect on
conformations and predicted free energies when docking with flexible receptors. In addition, an
error in output of energies with flexible receptors has been corrected, and AutoDock’s intra-
ligand desolvation potential cutoff distance has been increased to match AutoGrid’s.

7
Support
AutoDock is distributed, with full source code, free of charge. There are some caveats, however.
Firstly, since we receive limited funding to support the academic community of users, we cannot
guarantee response to queries on installation and use. While there is documentation, it may
require at least some basic Unix abilities to install. If you still need help:
(1) Ask your local system administrator or programming guru for help about compiling, using
Unix/Linux, etc.
(2) Consult the AutoDock web site, where you will find a wealth of information and a FAQ
(Frequently Asked Questions) page with answers on AutoDock:
http://autodock.scripps.edu/faqs-help
(3) If you can’t find the answer to your problem, send your question to the AutoDock List
(ADL) or the AutoDock Forum. There are many seasoned users of computational chemistry
software and some AutoDock users who may already know the answer to your question. You can
find out more about the ADL on the WWW at:
http://mgldev.scripps.edu/mailman/listinfo/autodock
The Forum is available on the WWW at:
http://mgl.scripps.edu/forum
(4) If you have tried (1), (2) and (3), and you still cannot find an answer, send email to
goodsell@scripps.edu for questions about AutoGrid or AutoDock; or to rhuey@scripps.edu for
questions about AutoDockTools.
Thanks for your understanding!
E-mail addresses
Arthur J. Olson, Ph.D. olson@scripps.edu
David S. Goodsell, Ph.D. goodsell@scripps.edu
Ruth Huey, Ph.D. rhuey@scripps.edu
Fax: +1 (858) 784-2860
The Scripps Research Institute
Molecular Graphics Laboratory
Department of Molecular Biology, Mail Drop MB-5
10550 North Torrey Pines Road
La Jolla, CA 92037-1000, U.S.A.
8
Theory
Overview of the Free Energy Scoring Function
AutoDock 4.2 uses a semi-empirical free energy force field to evaluate conformations during
docking simulations. The force field was parameterized using a large number of protein-inhibitor
complexes for which both structure and inhibition constants, or Ki, are known.
The force field evaluates binding in two steps. The ligand and protein start in an unbound
conformation. In the first step, the intramolecular energetics are estimated for the transition from
these unbound states to the conformation of the ligand and protein in the bound state. The second
step then evaluates the intermolecular energetics of combining the ligand and protein in their
bound conformation.
The force field includes six pair-wise evaluations (V) and an estimate of the conformational
entropy lost upon binding (ΔSconf):
€
ΔG = (Vbound
L−L−Vunbound
L−L)+(Vbound
P−P−Vunbound
P−P)+(Vbound
P−L−Vunbound
P−L+ΔSconf )
where L refers to the “ligand” and P refers to the “protein” in a ligand-protein docking
calculation.
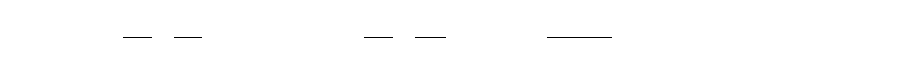
9
Each of the pair-wise energetic terms includes evaluations for dispersion/repulsion, hydrogen
bonding, electrostatics, and desolvation:
€
V = Wvdw
Aij
rij
12 −Bij
rij
6
i, j
∑+Whbond E(t) Cij
rij
12 −Dij
rij
10
i, j
∑+Welec
qiqj
e(r
ij )rij
i, j
∑+Wsol SiVj+SjVi
( )
i, j
∑e(−rij
2/2
σ
2)
The weighting constants W have been optimized to calibrate the empirical free energy based on a
set of experimentally determined binding constants. The first term is a typical 6/12 potential for
dispersion/repulsion interactions. The parameters are based on the Amber force field. The second
term is a directional H-bond term based on a 10/12 potential. The parameters C and D are
assigned to give a maximal well depth of 5 kcal/mol at 1.9Å for hydrogen bonds with oxygen
and nitrogen, and a well depth of 1 kcal/mol at 2.5Å for hydrogen bonds with sulfur. The
function E(t) provides directionality based on the angle t from ideal H-bonding geometry. The
third term is a screened Coulomb potential for electrostatics. The final term is a desolvation
potential based on the volume of atoms (V) that surround a given atom and shelter it from
solvent, weighted by a solvation parameter (S) and an exponential term with distance-weighting
factor σ=3.5Å. For a detailed presentation of these functions, please see our published reports,
included in Appendix IV.
By default, AutoGrid and AutoDock use a standard set of parameters and weights for the force
field. The parameter_file keyword may be used, however, to use custom parameter files.
The format of the parameter file is described in Appendix I.
Several methods for estimating the contribution of the unbound state are implemented in
AutoDock. In Autodock 3.0 and earlier versions, an assumption is made that the unbound form
of the ligand (VL-Lbound in the equation above) is the same as the final docked conformation of the
ligand (VL-Lunbound ), yielding a final contribution VL-Lbound -VL-Lunbound = 0. AutoDock 4.1
introduced a method of generating an extended form of the ligand to model the unbound state.
Reports from users, however, revealed that the method caused significant problems with
sterically-crowded molecules, and the default method was changed to the bound=unbound
assumption in AutoDock 4.2 and later. In addition, there is an option for a user-defined unbound
state.
10
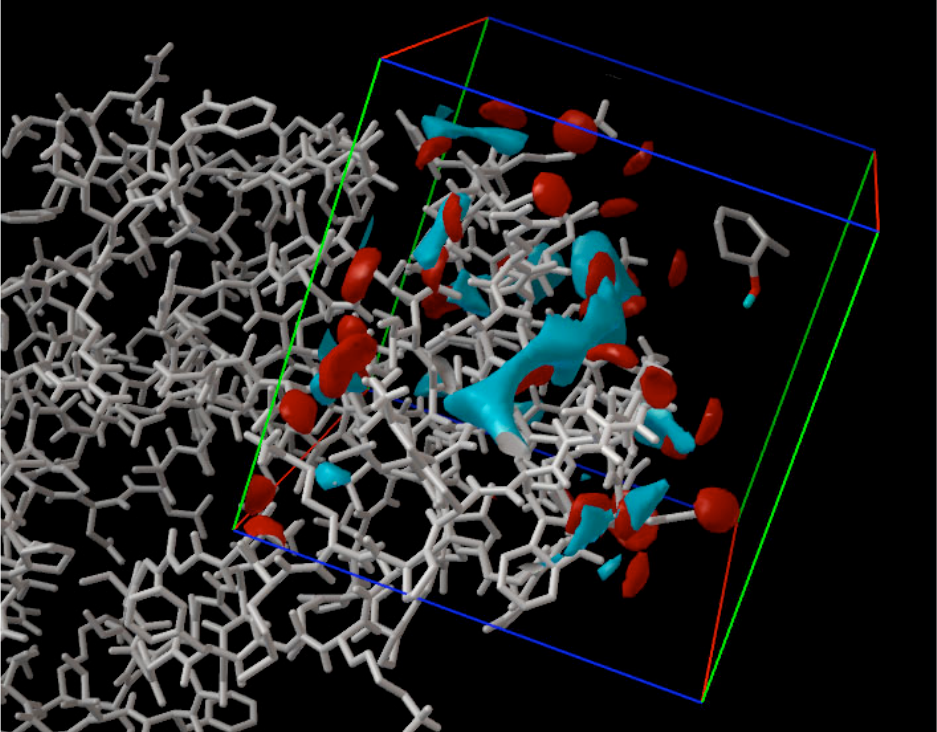
Viewing Grids in AutoDockTools. The protein is shown on the left in white bonds, and the grid
box is shown on the right side. The blue contours surround areas in the box that are most
favorable for binding of carbon atoms, and the red contours show areas that favor oxygen
atoms. A ligand is shown inside the box at upper right.
11
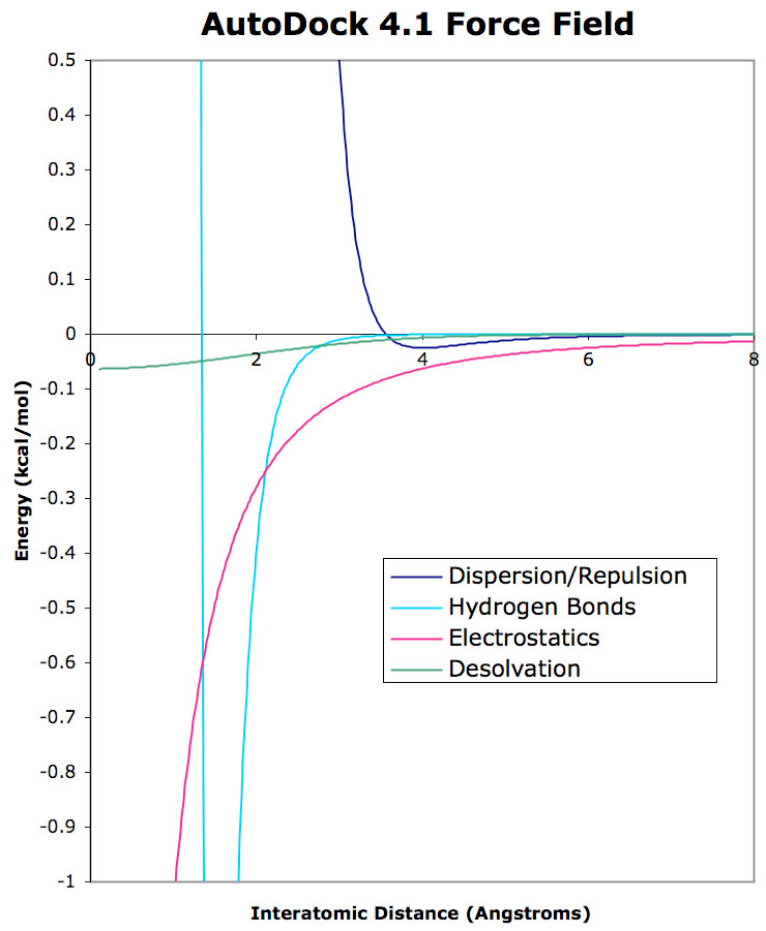
AutoDock Potentials. Examples of the four contributions to the AutoDock force field are shown
in this graph. The dispersion/repulsion potential is for interaction between two carbon atoms.
The hydrogen bond potential, which extends down to a minimum of about –2 kcal/mol, is shown
for an oxygen-hydrogen interaction. The electrostatic potential is shown for interaction of two
oppositely-charged atoms with a full atomic charge. The desolvation potential is shown for a
carbon atom, with approximately 10 atoms displacing water at each distance.
12
Using AutoDock
STEP 1: Preparing Coordinates
The first step is to prepare the ligand and receptor coordinate files to include the information
needed by AutoGrid and AutoDock. These coordinate files are created in an AutoDock-specific
coordinate file format, termed PDBQT, which includes:
1) Polar hydrogen atoms;
2) Partial charges;
3) Atom types;
4) Information on the articulation of flexible molecules.
For a typical docking calculation, you will create a file of coordinates for the receptor, and a
separate file of coordinates for the ligand. In dockings where selected amino acids in the receptor
are treated as flexible, you will create a third file that includes the coordinates of the atoms in the
flexible portions of the receptor.
In a typical study, the user prepares coordinate files in several steps using AutoDockTools. A
detailed tutorial is available on the AutoDock WWW site to guide you through this process. The
first two steps may be performed using the tools in the Edit menu of AutoDockTools, or with
other molecular modeling programs:
1) Add hydrogen atoms to the molecule.
2) Add partial charges.
Then, read the molecule into AutoDockTools using the Ligand (for the ligand) or Grid (for
the receptor) menus, and create the PDBQT file:
3) Delete non-polar hydrogens and merge their charges with the carbon atoms.
4) Assign atom types, defining hydrogen bond acceptors and donors and aromatic and aliphatic
carbon atoms.
5) Choose a root atom that will act as the root for the torsion tree description of flexibility.
6) Define rotatable bonds and build the torsion tree.
There are a few things to keep in mind during this process:
Be Critical. AutoDockTools and PMV currently use a modified version of Babel to add hydrogen
atoms and assign charges. Unfortunately the method has trouble with some molecules. In those
cases, hydrogen positions and charges may be assigned by the user’s preferred method, e.g. using
Reduce, InsightII, Quanta, Sybyl, AMBER or CHARMm.
Check Your Hydrogen Positions. In addition, most modeling systems add polar hydrogens in a
default orientation, typically assuming each new torsion angle is 0° or 180°. Without some form

13
of refinement, this can lead to spurious locations for hydrogen bonds. One option is to relax the
hydrogens and perform a molecular mechanics minimization on the structure. Another is to use a
program like “pol_h” which takes as input the default-added polar hydrogen structure, samples
favorable locations for each movable proton, and selects the best position for each. This
“intelligent” placement of movable polar hydrogens can be particularly important for tyrosines,
serines and threonines.
Watch for Disordered Residues. Care should be taken when the PDB file contains disordered
residues, where alternate location indicators (column 17) have been assigned. For each such
atom, the user must select only one of the possible alternate locations, making sure that a locally
consistent set is chosen.
Randomize Starting Coordinates When Redocking. In redocking experiments, where
coordinates from a known protein-ligand complex are separated and docked, it is important to
randomize the conformation of the ligand before it is docked. The techniques used to implement
rotation of the ligand may be biased to prefer values of zero rotation, so to obtain an unbiased
result, create a pdbqt file with the randomized ligand coordinates using the "Randomize"
command in the ADT Ligand menu, then use this randomized coordinate file for docking
experiments.
Please note: coordinate preparation is the most important step in the docking simulation. The
quality and accuracy of the docked results will only be as good as the quality of the starting
coordinates. Be critical and carefully examine hydrogen positions, atom type assignments, partial
charges, and articulation of the molecules to ensure that they make sense chemically. If you are
using the Babel method within AutoDockTools to add charges and hydrogens, carefully check
the results and make corrections if necessary—it often has trouble with molecules such as
nucleotides.
Creating PDBQT files in AutoDockTools
Overview of AutoDockTools
AutoDockTools is a set of commands implemented within the Python Molecular Viewer (PMV),
providing a Graphical User Interface for AutoGrid and AutoDock. It is available at:
http://autodock.scripps.edu/resources/adt.
14
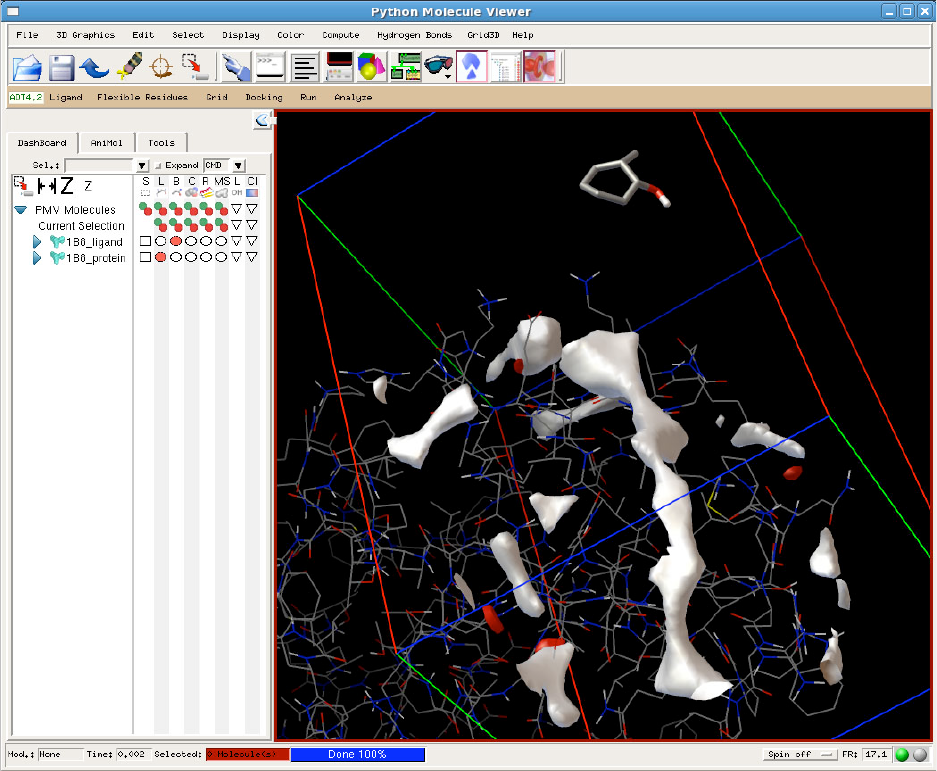
The AutoDockTools window has several parts:
1) at the top are menus that access the general methods available in PMV. These include tools for
reading and writing coordinates and images, for modifying coordinates, for selection, and for
visualization.
2) a row of buttons at the top allows quick access to the most popular tools of PMV
3) below the buttons, there are a series of menus that access the AutoDock-specific tools of
AutoDockTools.
4) the 3-D molecular viewer is at center right.
5) the Dashboard, located to the left of the viewer, allows quick selection, visualization, and
coloring of molecules currently displayed in the viewer.
15
Hydrogen Atoms and Charges
The tools available in PMV are used to read coordinates in PDB and other formats, to add
hydrogens, to select portions of the molecule, and to add partial charges. These functions are all
accessed through menus at the top of the PMV window. A few useful commands will be
described here—for more information on the many other functions of PMV, please see the PMV
documentation.
File>ReadMolecule: opens a browser that allows reading of PDB coordinate files.
Edit>Delete: several options for deleting entire molecules, selected sets of atoms, or
hydrogen atoms.
Edit>Hydrogens>Add: options for adding all hydrogens or polar hydrogens using Babel.
Edit>Charges: options for computing Gasteiger charges for arbitrary molecules using Babel.
Ligand PDBQT Files – the “Ligand” Menu
Once ligand coordinates are created with hydrogen atoms and charges, they can be processed in
the “Ligand” menu to create the ligand PDBQT file.
Ligand>Input>QuickSetup: uses defaults to create the PDBQT file. PDB files can be
read from the PMV viewer or from a file, and written directly to a new PDBQT file. Please note
that hydrogen atoms will not be added.
Ligand>Input>Open: reads coordinates from a file.
Ligand>Input>Choose: chooses a molecule already read into PMV.
Ligand>Input>OpenAsRigid: reads an existing PDBQT file and writes a new file with
NO active torsions.
Ligand>TorsionTree>ChooseRoot: manual selection of the root atom.
Ligand>TorsionTree>DetectRoot: automatic detection of the root that provides the
smallest largest subtree.
Ligand>TorsionTree>ShowRootExpansion: for molecules with several atoms in the
root, displays small spheres to show all atoms in the root, including atoms connected to each root
atom by rigid bonds.
16
Ligand>TorsionTree>ShowRootMarker: displays a sphere on the root atom.
Ligand>TorsionTree>ChooseTorsions: launches an interactive browser for choosing
rotatable bonds. Rotatable bonds are shown in green, and non-rotatable bonds are shown in red.
Bonds that are potentially rotatable but treated as rigid, such as amide bonds and bonds that are
made rigid by the user, are shown in magenta. Rotation of rotatable bonds may be switched on
and off by clicking on the bonds.
Ligand>TorsionTree>SetNumberOfTorsions: sets the number of rotatable bonds in
the ligand by leaving the specified number of bonds as rotatable. The two options will choose the
torsions that rotate either the fewest atoms in the ligand or the most atoms in the ligand.
Ligand>AromaticCarbon>SetNames: clicking on atom positions will switch carbon
atoms between aromatic and aliphatic. Aromatic carbons are shown in green. Click on the “Stop”
button when finished.
Ligand>AromaticCarbon>AromaticityCriterion: Sets the angular deviation from
planarity that AutoDockTools uses to identify aromatic rings.
Ligand>Output>RandomizethenSaveasPDBQT:randomizes the conformation of the
ligand and writes the formatted PDBQT file.
Ligand>Output:opens a browser to write the formatted PDBQT file.
Rigid Receptor PDBQT Files – the “Grid” Menu
For docking calculations using rigid receptor coordinates, add the hydrogen atoms and charges in
PMV, then read the coordinates into AutoDockTools using the “Grid” menu.
Grid>Macromolecule>Open: launches a browser to open an existing PDBQT file.
Grid>Macromolecule>Choose: chooses a molecule that has been previously read into
PMV. It will merge non-polar hydrogen atoms and charges, assign aromatic carbons, and prompt
the user to write a PDBQT file.
Flexible Receptor PDBQT Files – the “FlexibleResidues” Menu
For docking calculations with selected flexibility in the receptor, add the hydrogen atoms and
charges in PMV, then create two PDBQT files in AutoDockTools, one for the rigid portion of the
receptor and one for the flexible atoms.
FlexibleResidues>Input>OpenMacromolecule: launches a browser to open an
existing PDBQT file.
17
FlexibleResidues>Input>ChooseMacromolecule: chooses a molecule that has
been previously read into PMV. It will merge non-polar hydrogen atoms and charges, assign
aromatic carbons, and prompt the user to write a PDBQT file.
FlexibleResidues>ChooseTorsionsInCurrentlySelectedResidues:
flexible residues are chosen using the tools in the PMV “Select” menu, then this option is used to
assign these residues as flexible. As with the ligand, you can choose which bonds to keep
rotatable by clicking on the bonds.
FlexibleResidues>RedisplayMacromolecule: cleans up the display.
FlexibleResidues>Output>SaveRigidPDBQT:
FlexibleResidues>Output>SaveFlexiblePDBQT: these two commands launch a
browser to write PDBQT files for the rigid portion of the receptor and the flexible portion of the
receptor.
18
STEP 2: Running AutoGrid
AutoDock requires pre-calculated grid maps, one for each atom type present in the ligand being
docked. This helps to make the docking calculations fast. These maps are calculated by
AutoGrid. A grid map consists of a three-dimensional lattice of regularly spaced points, sur-
rounding (either entirely or partly) and centered on some region of interest of the macromolecule
under study. This could be a protein, enzyme, antibody, DNA, RNA or even a polymer or ionic
crystal. Typical grid point spacing varies from 0.2Å to 1.0Å, and the default is 0.375Å (roughly
a quarter of the length of a carbon-carbon single bond). Each point within the grid map stores the
potential energy of a ‘probe’ atom or functional group that is due to all the atoms in the
macromolecule.
AutoGrid requires a grid parameter file to specify the files and parameters used in the
calculation. The grid parameter file usually has the extension “.gpf”. As described below,
AutoDockTools may be used to create the grid parameter file. A full description of the grid
parameter file is included in Appendix I.
To run AutoGrid, the command is issued as follows:
% autogrid4 -p macro.gpf [-l macro.glg]
where ‘-p macro.gpf’ specifies the grid parameter file, and ‘-l macro.glg’ specifies the log file
written during the grid calculation. If no log file is specified, the output is written to the terminal.
AutoGrid writes out the grid maps in ASCII form, for readability and portability; AutoDock
expects ASCII format grid maps. For a description of the format of the grid map files, see
Appendix I. Check the minimum and maximum energies in each grid map: these are reported at
the end of the AutoGrid log file (here, it is “macro.glg”). Minimum van der Waals’ energies and
hydrogen bonding energies are typically -10 to -1 kcal/mol, while maximum van der Waals’
energies are clamped at +105 kcal/mol. Electrostatic potentials tend to range from around -103 to
+103 kcal/mol/e: if these are both 0, check to make sure that partial charges have been assigned
on the macromolecule.
As well as the grid maps, AutoGrid creates two files, with the extensions ‘.fld’, and ‘.xyz’. The
former is a field file summarizing the grid maps, and the latter describes the spatial extent of the
grids in Cartesian space.
19
Creating grid parameter files in AutoDockTools
The tools available in “grid” menu of AutoDockTools may be used to create grid parameter files.
Grid>OpenGPF: gets parameters from an existing grid parameter file.
Grid>Macromolecule: has options for opening an existing PDBQT file or choosing a
molecule that has been read using PMV.
Grid>SetMapTypes: tools to define the atom types for the grids that will be calculated.
Grids must be calculated for each type of atom in the ligand, and if flexible sidechains are used
in the receptor, their atom types must also be included. The option “Directly” allows the user to
input the list of atom types directly. Other options allow the user to define the atom types based
on a ligand or flexible residue that has been read by PMV, or to open ligand or flexible residue
PDBQT and use the atom types in these files.
Grid>SetMapTypes>SetUpCovalentMap: specifies parameters for creation of a
covalent map, which may be used in specialized applications to favor binding of a given ligand
atom in a single position. This is particularly useful for docking of covalent complexes between
ligands and proteins. This will calculate a separate grid with atom type “Z” with a favorable
Gaussian well at the coordinates given. The potential will have zero energy at the site, rising to
the energy barrier height in surrounding areas.
Grid>GridBox: launches interactive commands for setting the grid dimensions and center.
To enter numbers on the thumbwheel, place the cursor over the thumbwheel and type in the new
value. Right clicking on the thumbwheel gives more options. IMPORTANT: when finished, use
the “close saving current” option in the “File” menu on the Grid Options Panel. Options in the
“Center” menu on the browser provide different methods to choose the center of the grid box.
Grid>OtherOptions: allows specification and editing of an existing parameter file.
Grid>Output: writes a new grid parameter file.
Grid>EditGPF: interactive editor for grid parameter files, which allows viewing of the latest
grid parameter file written by AutoDockTools.
20
STEP 3: Docking with AutoDock
AutoDock uses one of several conformational search algorithms to explore the conformational
states of a flexible ligand, using the maps generated by AutoGrid to evaluate the ligand-protein
interaction at each point in the docking simulation. In a typical docking, the user will dock a
ligand several times, to obtain multiple docked conformations. The results may be clustered to
identify similar conformations—this is described in more detail in the section on Analysis (Step
4, below).
AutoDock requires: 1) grid maps for each atom type in the ligand, calculated by AutoGrid, 2) a
PDBQT file for the ligand, and 3) a docking parameter file that specifies the files and parameters
for the docking calculation. AutoDockTools may be used to generate the docking parameter file,
as described below, which typically has the extension “.dpf”. A full description of the docking
parameter file is included in Appendix I. AutoDock writes the final docked coordinates into the
docking log file. As described in Step 4 below, these docked conformations may be viewed using
AutoDockTools, they may be written as PDBQT files using AutoDockTools, or they may be
taken directly from the docking log file using a text editor.
An AutoDock calculation is started from the command line using the following command:
% autodock4 [-i][-u][-t] -p lig.dpf [-l lig.dlg]
Input parameters are specified by “-p lig.dpf”, and the log file containing the output and
results from the docking is defined by “-l lig.dlg”. This is the normal usage of AutoDock,
and performs a standard docking calculation.
-p dpf_filename
Specifies the docking parameter file.
-l dlg_filename
Specifies the docking log file. If this is omitted, output will be written to a file with the same root
name as the dpf.
-i
This is used to ignore any grid map header errors that may arise due to conflicting filenames.
This overrides the header checking that is normally performed to ensure compatible grid maps
are being used.
-u, -h
This returns a helpful message describing the command line usage of AutoDock.

21
-t
This instructs AutoDock to parse the PDBQT file to check the torsion definitions, and then stop.
-version
This returns a message describing the version of AutoDock being used, and then stops.
Choosing a protocol for your application
AutoDock provides a number of different methods for doing the docking simulation, and
different methods might be useful for different applications. This section includes some
guidelines for choosing the best approach.
1) Conformation Search. AutoDock provides several methods for doing the conformation
search. Currently, the Lamarckian Genetic Algorithm provides the most efficient search for
general applications, and in most cases will be the technique used. It is typically effective for
systems with about 10 rotatable bonds in the ligand. The Genetic Algorithm may also be run
without the local search, but this is typically less efficient than the Lamarckian GA-LS
combination. Simulated Annealing is also less efficient that the Lamarckian Genetic Algorithm,
but it can be useful in applications where search starting from a given point is desired. Local
Search may be used to optimize a molecule in its local environment.
2) Number of Evaluations. Each of the search methods includes parameters for determining the
amount of computational effort that will be used in the search. In the GA methods, this parameter
is ga_num_evals, and in simulated annealing, this is nacc and nrej. The defaults given for
these parameters are typically sufficient for docking systems with 10 or fewer rotatable bonds,
and shorter simulations may often be used for systems with very few rotatable bonds. For
complex systems with many more rotatable bonds that this, it is not generally effective simply to
increase the number of evaluations. Rather, it is best to look for simpler formulations of the
system, such as breaking a large ligand into two pieces and docking them separately, or freezing
some rotatable bonds in likely conformations.
3) Model for the Unbound Ligand. In order to estimate a free energy of binding, AutoDock
needs to estimate an energy for the unbound state of the ligand and protein. Several options are
available for this. By default, AutoDock4.2 uses the assumption that the conformation of the
unbound ligand and protein are the same as the conformation of the ligand and protein in the
complex. Because these two conformations are the same, the total contribution of the internal
energy (the interaction of atoms within the ligand or the interaction of atoms within the protein)
will be zero, and reported in line 4 of the energy breakdown in the docking log file.
22
AutoDock4.0 used a different model, where the ligand was assumed to be in an extended state in
solution, and an energy was calculated for this extended state before the docking simulation was
performed. This model may be used in AutoDock4.2 by using the key word
“unbound_model_extended.” This keyword will launch the calculation of the extended
model and then will report the difference between the internal energy of the unbound model and
the internal energy of the ligand when it is bound to the protein. In studies where many separate
dockings are performed with the same ligand, this energy for the extended ligand may be
precalculated and then used in the free energy calculation by using the key word
“unbound_model_extended_energy VALUE.”
The user may also use other methods to calculate the energy of the unbound ligand outside of
AutoDock. In this case, the keyword “unbound_energy VALUE” may be used to set the
internal energy of the unbound state to a desired value. This value will then be used in the
difference between the bound and unbound states to estimate the free energy.
4) Redocking Studies for Validation. Redocking studies may be used to validate the docking
methods being used for a particular system. A known system is chosen with a similar
conformational complexity--with a similar number of atoms and similar number of rotatable
bonds. The known complex is then separated and docked, to evaluate the success of the docking
method in reproducing the experimentally-know complex. These types of studies are also often
used when comparing different docking methods. It is essential to randomize the ligand
coordinates before the redocking simulation, to remove any bias that is caused by having the
desired docked complex exactly equivalent to the starting coordinates.
5) Special Cases. AutoDock4.2 includes a number of optional methods for use in specialized
applications. For instance, the keyword intnbp_r_eps may be used to override the standard
parameters for the internal energy calculation. This has been used to model flexible cyclic
molecules, but creating a special set of atom types to close rings during a docking simulation
(this method is described in more detail in a tutorial on the AutoDock WWW site). Other
optional features include methods for adding torsional constraints, and options for modifying the
force field and analysis.
23
Creating docking parameter files in AutoDockTools
The tools available in the “Docking” menu of AutoDockTools may be used to create docking
parameter files.
Docking>OpenDPF: gets parameters from an existing docking parameter file.
Docking>Macromolecule>SetRigidFilename:
Docking>Macromolecule>SetFlexibleResiduesFilename: these two commands
specify the PDBQT file name that will be used for the rigid receptor, and if flexible receptor
residues are used, specifies the PDBQT file name for the flexible portion of the receptor.
Docking>Ligand>Choose
Docking>Ligand>Open: These two commands allow the user to choose a ligand that is
already read into ADT, or open an existing ligand PDBQT file.
Docking>Ligand>Ligand_Parameters: opens a panel for setting various ligand
parameters, including the starting values for the translation, rotation, and torsion angles. For
details, see the full description of the docking parameter file in the Appendix I.
Docking>SearchParameters>GeneticAlgorithmParameters:
Docking>SearchParameters>SimulatedAnnealingParameters:
Docking>SearchParameters>LocalSearchParameters: these three commands
open a panel for setting the parameters used by each of the search algorithms, such as
temperature schedules in simulated annealing and mutation/crossover rates in genetic algorithms.
For details of each parameter, see the full description in the Appendix.
Docking>DockingParameters: opens a panel for setting the parameters used during the
docking calculation, including options for the random number generator, options for the force
field, step sizes taken when generating new conformations, and output options. For details of
each parameter, see the full description in the Appendix I.
Docking>OtherOptions: specifies the name of an external atomic parameter file, if used.
Docking>Output>LamarckianGA:
Docking>Output>GeneticAlgorithm:
Docking>Output>SimulatedAnnealing:
Docking>Output>LocalSearch: These four commands write the docking parameter file
using one of the four available search methods.
Docking>Edit: interactive editor for docking parameter files, which allows viewing of the
latest docking parameter file written by AutoDockTools.
24
STEP 4: Evaluating the Results of a Docking
At the end of a docking simulation, AutoDock writes the coordinates for each docked
conformation to the docking log file, along with information on clustering and interaction
energies. AutoDockTools provides options for analyzing the information stored in the docking
log file.
Information in the Docking Log
The analysis command in the docking parameter file causes AutoDock to perform a cluster
analysis of the different docked conformations – the minimum energy found in each run. The
results of this analysis are reported as a histogram, which may be found by searching for the
word “HISTOGRAM” (all in capital letters) in the docking log file. This is followed by a table of
RMSD values within each cluster.
AutoDock then writes coordinates for the conformation of best predicted energy in each cluster
(to write coordinates for the best conformation from each run, include the keyword write_all
in the docking parameter file). A header for each conformation includes information on the
predicted energy of binding, broken down into several components, along with information on
the state variables of the conformation. The coordinates are written in a modified PDB format,
with four real values appended after the x,y,z coordinates: the vdW+hbond+desolvation energy
of interaction of the atom, the electrostatic interaction of the atom, the partial charge, and the
RMSD from the reference conformation.
The amount of information included in the docking log may be controlled using the “outlev”
parameter, ranging from very minimal output for large virtual screens, to the default that is read
by ADT (with information on docked conformations and clustering), to more verbose files that
are useful for methods development.
Analyzing Docking Results with AutoDockTools
Options in the “Analyze” menu of AutoDockTools may be used to process and analyze the
results from a docking simulation.
Analyze>Dockings>Open: opens a docking log file.
Analyze>Dockings>OpenAutoDockVinaResult: opens a docking log file from
AutoDock Vina.
25
Analyze>Dockings>OpenAutoDockVirtualScreeningResult: opens an
expanded result file that includes information on the conformation, clustering, and interactions
with the receptor.
Analyze>Dockings>OpenAll: opens a set of docking log files in a directory.
Analyze>Dockings>Select: selects from a set of log files previously read into
AutoDockTools.
Analyze>Dockings>Clear: clears log files that have been read into AutoDockTools.
Analyze>Dockings>ShowAsSpheres: creates a sphere at the center of mass of each
docked conformation, which may be colored according to the predicted energy of interaction.
Analyze>Dockings>ShowInteractions: creates a specialized visualization to
highlight interactions between the docked conformation of the ligand and the receptor. By
default, the ligand is shown as ball-and-stick, surrounded by a molecular surface. The surface is
colored with atomic colors in regions that contact the receptor, and gray in regions that are not in
contact. Portions of the receptor that are in contact with the ligand are shown with ball-and-stick
and spacefilling spheres. Hydrogen bonds are shown as a string of small spheres. A dialogue box
is also launched that provides many other options for visualization.
Analyze>Dockings>WriteAutoDockVirtualScreeningResult: writes an
expanded result file that includes information on the conformation, clustering, and interactions
with the receptor.
Analyze>Macromolecule: options to open a macromolecule PDBQT file or choose a
macromolecule that is already read into PMV.
Analyze>Grids>Open
Analyze>Grids>OpenOther: Opens a grid map file and launches the AutoDockTools grid
visualizer. A dialogue box allows specification of the contour level and several rendering
options. The contour level slider and input box limits the range to favorable energies. The
“sampling” value is used to create coarse representations of complex maps—set to 1, it uses the
actual grid spacing, set to higher values, it decimates the map to coarser grid spacing. The
“Grid3D” tool is also available in the PMV menu for more advanced representation methods for
grid visualization. The “OpenOther” command allows opening of grid map files that are not
specified in the current docking log that is being displayed.
Analyze>Conformations>Play
Analyze>Conformations>PlayRankedByEnergy: Opens a window with controls for
stepping through conformations as a movie. “Play” will use the order of conformations as they
were found in the docking calculations, and “PlayRankedByEnergy” will order the
conformations from lowest energy to highest energy. The “&” button opens a window with
additional options:
26
ShowInfo opens a panel that displays information on the predicted energy of interaction,
RMSD, etc.
BuildHbonds and ColorbyATOM/vdW/elect/total allow visualization of hbonds
and interaction energies.
PlayMode and PlayParameters modify the parameters of the player.
BuildCurrent will build a new set of coordinates in the viewer for the conformation
currently specified in the player. This is useful for displaying multiple conformations in the
same view. BuildAll will build coordinates for all conformations in the player.
WriteCurrent will write a PDBQT file for the current conformation in the player.
WriteAll will write separate PDBQT files for all conformations in the player.
WriteComplex will write a PDBQT file for the current conformation of the ligand and the
receptor.
Analyze>Conformations>Load: Launches an interactive browser that allows selection of
clustered docked conformations. Information on the predicted interaction energy is shown at the
top, and individual conformations may be chosen in the bottom panel. The “rank” value gives the
cluster_rank—for instance, “1_3” is the third most favorable conformation in the best cluster.
Buttons at the bottom, which may be revealed by enlarging the window, will write the current
coordinates and dismiss the window.
Analyze>Conformations>ViewInitialPopulation: Tools to display all members
of the population in a genetic algorithm search. Requires a DLG from a run with a specialized
output level: “outlev runvvv”.
Analyze>Clusterings>Show: Tools to show an interactive histogram of clustered
conformations.
Analyze>Clusterings>Recluster: Reclusters docked conformations based on new
tolerances. Several values may be input in the dialogue window for use in reclustering. The
results may be analyzed using Clusterings>Show.
Analyze>Clusterings>ReclusterOnSubset: Reclusters docked conformations using
only a selected set of atoms. The selection is performed using the tools in the “select” menu of
PMV, and then using the “save current selection as a set” option.
27
Appendix I: AutoDock File Formats
PDBQT Format for Coordinate Files
Extension: .pdbqt
“ATOM%5d%-4s%1s%-3s%1s%4d%1s%8.3f%8.3f%8.3f%6.2f%6.2f%4s%6.3f%2s \n",
atom_serial_num, atom_name, alt_loc, res_name, chain_id, res_num, ins_code, x, y, z,
occupancy, temp_factor, footnote, partial_charge, atom_type
(The “” symbol is used to indicate one space.)
The PDBQT format adds four things to standard formatted PDB files:
1) partial charges are included in each ATOM or HETATM record, substituting for the fields in
columns 67-76.
2) AutoDock atom types (which may be one or two letters) are included in each ATOM or
HETATM record in columns 78-79.
3) To allow flexibility in the ligand, it is necessary to assign the rotatable bonds. AutoDock can
handle up to MAX_TORS rotatable bonds: this parameter is defined in “autodock.h”, and is
ordinarily set to 32. If this value is changed, AutoDock must be recompiled. Please note that
AutoDock4.2 is currently effective for systems with roughly 10 torsional degrees of freedom, and
systems with more torsional flexibility may not give consistent results. Torsions are defined in the
PDBQT file using the following keywords:
ROOT / ENDROOT
BRANCH / ENDBRANCH
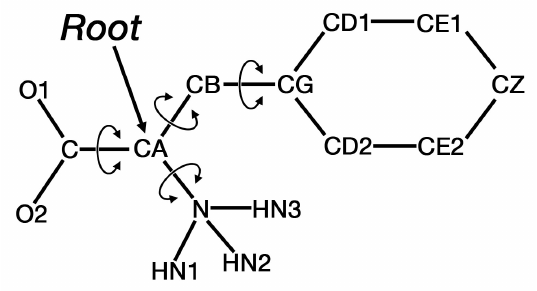
These keywords use the metaphor of a tree. See the diagram below for an example. The “root” is
defined as the central portion of the ligand, from which rotatable ‘branches’ sprout. Branches
within branches are possible. Nested rotatable bonds are rotated in order from the “leaves” to the
“root”. The PDBQT keywords must be carefully placed, and the order of the ATOM or
HETATM records often need to be changed in order to fit into the correct branches.
AutoDockTools is designed to assist the user in placing these keywords correctly, and in re-
ordering the ATOM or HETATM records in the ligand PDBQT file.
4) The number of torsional degrees of freedom, which will be used to evaluate the
conformational entropy, is specified using the TORSDOF keyword followed by the integer
number of rotatable bonds. In the current AutoDock 4.2 force field, this is the total number of
rotatable bonds in the ligand, including rotatable bonds in hydroxyls and other groups where
28
only hydrogen atoms are moved, but excluding bonds that are within cycles. The value in the
PDBQT file can be overridden in the AutoDock DPF using the “torsdof” statement.
Note: AutoDockTools, AutoGrid and AutoDock do not recognize or write out PDB “CONECT”
records.
Sample PDBQT file
REMARK 4 active torsions:
REMARK status: ('A' for Active; 'I' for Inactive)
REMARK 1 A between atoms: N_1 and CA_5
REMARK 2 A between atoms: CA_5 and CB_6
REMARK 3 A between atoms: CA_5 and C_13
REMARK 4 A between atoms: CB_6 and CG_7
ROOT
ATOM 1 CA PHE A 1 25.412 19.595 12.578 1.00 12.96 0.287 C
ENDROOT
BRANCH 1 2
ATOM 2 N PHE A 1 25.225 18.394 13.381 1.00 13.04 -0.065 N
ATOM 3 HN3 PHE A 1 25.856 17.643 13.100 1.00 0.00 0.275 HD
ATOM 4 HN2 PHE A 1 25.558 18.517 14.337 1.00 0.00 0.275 HD
ATOM 5 HN1 PHE A 1 24.247 18.105 13.350 1.00 0.00 0.275 HD
ENDBRANCH 1 2
BRANCH 1 6
ATOM 6 CB PHE A 1 26.873 20.027 12.625 1.00 12.45 0.082 C
BRANCH 6 7
ATOM 7 CG PHE A 1 27.286 20.629 13.923 1.00 12.96 -0.056 A
ATOM 8 CD2 PHE A 1 27.470 22.001 14.050 1.00 12.47 0.007 A
ATOM 9 CE2 PHE A 1 27.877 22.571 15.265 1.00 13.98 0.001 A
ATOM 10 CZ PHE A 1 28.108 21.754 16.360 1.00 13.84 0.000 A
ATOM 11 CE1 PHE A 1 27.919 20.380 16.242 1.00 13.77 0.001 A
ATOM 12 CD1 PHE A 1 27.525 19.821 15.027 1.00 11.32 0.007 A
ENDBRANCH 6 7
ENDBRANCH 1 6
BRANCH 1 13
ATOM 13 C PHE A 1 25.015 19.417 11.141 1.00 13.31 0.204 C
ATOM 14 O2 PHE A 1 24.659 20.534 10.507 1.00 12.12 -0.646 OA
ATOM 15 O1 PHE A 1 25.024 18.283 10.608 1.00 13.49 -0.646 OA
ENDBRANCH 1 13
TORSDOF 4
29
PDBQT Format for Flexible Receptor Sidechains
Flexible sidechains in the receptor are treated explicitly during AutoDock simulation. AutoDock
requires a separate PDBQT file with atomic coordinates of the sidechains that will be treated as
flexible. Atomic coordinates and branching keywords for each amino acid is placed between
BEGIN_RES and END_RES records. The atom linking the amino acid to the protein, which will
remain in fixed position during the simulation, is included as the root. The atoms included in the
flexible residue PDBQT must be omitted from the PDBQT for the rigid portions of the receptor.
For instance, in the example below, the CA atom of the PHE residue is used as the root of the
flexible residue. It is included in the flexible sidechain PDBQT file, and it will be omitted from
the rigid protein PDBQT file.
Sample flexible residue file, with two flexible amino acids
BEGIN_RES PHE A 53
REMARK 2 active torsions:
REMARK status: ('A' for Active; 'I' for Inactive)
REMARK 1 A between atoms: CA and CB
REMARK 2 A between atoms: CB and CG
ROOT
ATOM 1 CA PHE A 53 25.412 19.595 12.578 1.00 12.96 0.180 C
ENDROOT
BRANCH 1 2
ATOM 2 CB PHE A 53 26.873 20.027 12.625 1.00 12.45 0.073 C
BRANCH 2 3
ATOM 3 CG PHE A 53 27.286 20.629 13.923 1.00 12.96 -0.056 A
ATOM 4 CD1 PHE A 53 27.525 19.821 15.027 1.00 11.32 0.007 A
ATOM 5 CE1 PHE A 53 27.919 20.380 16.242 1.00 13.77 0.001 A
ATOM 6 CZ PHE A 53 28.108 21.754 16.360 1.00 13.84 0.000 A
ATOM 7 CE2 PHE A 53 27.877 22.571 15.265 1.00 13.98 0.001 A
ATOM 8 CD2 PHE A 53 27.470 22.001 14.050 1.00 12.47 0.007 A
ENDBRANCH 2 3
ENDBRANCH 1 2
END_RES PHE A 53
BEGIN_RES ILE A 54
REMARK 2 active torsions:
REMARK status: ('A' for Active; 'I' for Inactive)
REMARK 3 A between atoms: CA and CB
REMARK 4 A between atoms: CB and CG1
ROOT
ATOM 9 CA ILE A 54 24.457 20.591 9.052 1.00 12.30 0.180 C
ENDROOT
BRANCH 9 10
ATOM 10 CB ILE A 54 22.958 20.662 8.641 1.00 11.82 0.013 C
ATOM 11 CG2 ILE A 54 22.250 19.367 9.046 1.00 12.63 0.012 C
BRANCH 10 12
ATOM 12 CG1 ILE A 54 22.266 21.867 9.298 1.00 13.03 0.002 C
ATOM 13 CD1 ILE A 54 20.931 22.246 8.670 1.00 14.42 0.005 C
ENDBRANCH 10 12
ENDBRANCH 9 10
END_RES ILE A 54
30
AutoGrid Grid Parameter File: GPF
Extension: .gpf
The grid parameter file specifies an AutoGrid calculation, including the size and location of the
grid, the atom types that will be used, the coordinate file for the rigid receptor, and other
parameters for calculation of the grids. Unlike previous versions of AutoGrid, the pairwise
atomic parameters are now read from a separate file (described below) or taken from defaults in
AutoGrid.
All delimiters where needed are white spaces. Default values, where applicable, are given here in
square brackets [thus]. A comment must be prefixed by the “#” symbol, and can be placed after
a space at the end of a parameter line, or on a line of its own. Upper/lower case is ignored in
keywords but is significant in atom names and file names. File names cannot contain white space
or non-ASCII characters. Although ideally it should be possible to give AutoGrid keywords in
any order, not every possible combination has been tested, so it would be wise to stick to the
following order.
AutoGrid Keywords and Commands
parameter_file <existing atomic parameter file name>
(Optional) User-defined atomic parameter file (format described in the next section). By default,
AutoGrid uses internal parameters.
npts <integer> <integer> <integer>
[40, 40, 40]
Number of x-, y- and z-grid points. Each must be an even integer number. When added to the
central grid point, there will be an odd number of points in each dimension. The number of x-, y-
and z-grid points need not be equal.
gridfld <new grid file name>
The grid field filename, which will be written in a format readable by AutoDock. The filename
extension is ‘.fld’.
spacing <float>
[0.375 Å]
(Optional) The grid point spacing, in Å. Grid points are orthogonal and uniformly spaced in
AutoDock: this value is used in each dimension.
receptor_types <string>
[A C HD N OA SA]
31
Atom types present in the receptor, separated by spaces; e.g. for a typical protein, this will be, “A
C HD N OA SA”. Atom types are one or two letters, and several specialized types are used in the
AutoDock4.2 forcefield, including: C (aliphatic carbon), A (aromatic carbon), HD (hydrogen
that donates hydrogen bond), OA (oxygen that accepts hydrogen bond), N (nitrogen that doesn’t
accept hydrogen bonds), SA (sulfur that accepts hydrogen bonds).
ligand_types <string>
[A C HD N NA OA SA]
Atom types present in the ligand, separated by spaces, such as, “A C HD N NA OA SA”.
receptor <existing receptor file name>
Macromolecule filename, in PDBQT format.
gridcenter <float> <float> <float>
gridcenter auto
[auto]
The user can explicitly define the center of the grid maps, respectively the x, y and z coordinates
of the center of the grid maps (units: Å, Å, Å.) Or the keyword “auto” can be given, in which
case AutoGrid will center the grid maps on the center of the macromolecule.
smooth <float>
[0.5 Å]
(Optional) Smoothing parameter for the pairwise atomic affinity potentials (both van der Waals
and hydrogen bonds). For AutoDock4, the force field has been optimized for a value of 0.5 Å.
map <new map file name>
Filename of the grid map for each ligand atom type; the extension is usually “.X.map”, where
“X” is the atom type. One line must be included for each atom type in the ligand_types
command, in the order given in that command.
elecmap <new map file name>
Filename for the electrostatic potential energy grid map to be created; filename extension
‘.e.map’.
dsolvmap <new map file name>
Filename for the desolvation potential energy grid map to be created; filename extension
‘.d.map’.
dielectric <float>
[-0.1465]
(Optional) Dielectric function flag: if negative, AutoGrid will use distance-dependent dielectric
of Mehler and Solmajer; if the float is positive, AutoGrid will use this value as the dielectric
constant. AutoDock4 has been calibrated to use a value of –0.1465.
32
Sample Grid Parameter File (from tutorial)
npts 60 60 60 # num.grid points in xyz
gridfld 1hsg.maps.fld # grid_data_file
spacing 0.375 # spacing(A)
receptor_types A C HD N OA SA # receptor atom types
ligand_types A C NA OA N HD # ligand atom types
receptor 1hsg.pdbqt # macromolecule
gridcenter 2.5 6.5 -7.5 # xyz-coordinates or auto
smooth 0.5 # store minimum energy w/in rad(A)
map 1hsg.A.map # atom-specific affinity map
map 1hsg.C.map # atom-specific affinity map
map 1hsg.NA.map # atom-specific affinity map
map 1hsg.OA.map # atom-specific affinity map
map 1hsg.N.map # atom-specific affinity map
map 1hsg.HD.map # atom-specific affinity map
elecmap 1hsg.e.map # electrostatic potential map
dsolvmap 1hsg.d.map # desolvation potential map
dielectric -0.1465 # <0, AD4 distance-dep.diel;>0,
constant
33
Atomic Parameter File
Filename: AD4.1_bound.dat
Atomic parameters are assigned by default from values internal to AutoGrid and AutoDock, but
custom parameters may be read from a file. The file includes weighting parameters for each term
in the free energy function, and parameters for each atom type:
FE_coeff-vdW <float>
FE_coeff-hbond <float>
FE_coeff-estat <float>
FE_coeff-desolv <float>
FE_coeff-tors <float>
These are followed by pairwise atomic parameters for each type of atom:
atom_par <string> 6*<float> 4*<integer>
Each atom_par record includes:
1. Atom type.
2. Rii = sum of the vdW radii of two like atoms (Å).
3. epsii = vdW well depth (kcal/mol)
4. vol = atomic solvation volume (Å^3)
5. Rij_hb = H-bond distance between heteroatom and hydrogen (Å)
value is included in the heteroatom record and set to zero for hydrogens
6. epsij_hb = well depth for hydrogen bonds (kcal/mol)
7. hbond = integer indicating the type of hbond
0, no hbond
1, spherical H donor
2, directional H donor
3, spherical acceptor
4, directional N acceptor
5, directional O/S acceptor
8. rec_index = initialized to –1, used to hold number of atom types
9. map_index = initialized to –1, used to hold the index of the AutoGrid map
10. bond_index = used to detect bonds of different lengths, see “mdist.h” for information
34
Default Atomic Parameter File AD4.1_bound.dat
#
# Free Energy Coefficient
# ------
FE_coeff_vdW 0.1662
FE_coeff_hbond 0.1209
FE_coeff_estat 0.1406
FE_coeff_desolv 0.1322
FE_coeff_tors 0.2983
#
# Atom Rii Rij_hb rec_index
# Type epsii solpar epsij_hb map_index
# vol hbond bond_index
# -- ---- ----- ------- -------- --- --- - -- -- --
atom_par H 2.00 0.020 0.0000 0.00051 0.0 0.0 0 -1 -1 3 # Non H-bonding Hydrogen
atom_par HD 2.00 0.020 0.0000 0.00051 0.0 0.0 2 -1 -1 3 # Donor 1 H-bond Hydrogen
atom_par HS 2.00 0.020 0.0000 0.00051 0.0 0.0 1 -1 -1 3 # Donor S Spherical Hydrogen
atom_par C 4.00 0.150 33.5103 -0.00143 0.0 0.0 0 -1 -1 0 # Aliphatic Carbon
atom_par A 4.00 0.150 33.5103 -0.00052 0.0 0.0 0 -1 -1 0 # Aromatic Carbon
atom_par N 3.50 0.160 22.4493 -0.00162 0.0 0.0 0 -1 -1 1 # Non H-bonding Nitrogen
atom_par NA 3.50 0.160 22.4493 -0.00162 1.9 5.0 4 -1 -1 1 # Acceptor 1 H-bond Nitrogen
atom_par NS 3.50 0.160 22.4493 -0.00162 1.9 5.0 3 -1 -1 1 # Acceptor Spherical Nitrogen
atom_par OA 3.20 0.200 17.1573 -0.00251 1.9 5.0 5 -1 -1 2 # Acceptor 2 H-bonds Oxygen
atom_par OS 3.20 0.200 17.1573 -0.00251 1.9 5.0 3 -1 -1 2 # Acceptor Spherical Oxygen
atom_par F 3.09 0.080 15.4480 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Fluorine
atom_par Mg 1.30 0.875 1.5600 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Magnesium
atom_par MG 1.30 0.875 1.5600 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Magnesium
atom_par P 4.20 0.200 38.7924 -0.00110 0.0 0.0 0 -1 -1 5 # Non H-bonding Phosphorus
atom_par SA 4.00 0.200 33.5103 -0.00214 2.5 1.0 5 -1 -1 6 # Acceptor 2 H-bonds Sulphur
atom_par S 4.00 0.200 33.5103 -0.00214 0.0 0.0 0 -1 -1 6 # Non H-bonding Sulphur
atom_par Cl 4.09 0.276 35.8235 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Chlorine
atom_par CL 4.09 0.276 35.8235 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Chlorine
atom_par Ca 1.98 0.550 2.7700 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Calcium
atom_par CA 1.98 0.550 2.7700 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Calcium
atom_par Mn 1.30 0.875 2.1400 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Manganese
atom_par MN 1.30 0.875 2.1400 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Manganese
atom_par Fe 1.30 0.010 1.8400 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Iron
atom_par FE 1.30 0.010 1.8400 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Iron
atom_par Zn 1.48 0.550 1.7000 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Zinc
atom_par ZN 1.48 0.550 1.7000 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Zinc
atom_par Br 4.33 0.389 42.5661 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Bromine
atom_par BR 4.33 0.389 42.5661 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Bromine
atom_par I 4.72 0.550 55.0585 -0.00110 0.0 0.0 0 -1 -1 4 # Non H-bonding Iodine
atom_par Z 4.00 0.150 33.5103 -0.00143 0.0 0.0 0 -1 -1 0 # Non H-bonding covalent map
atom_par G 4.00 0.150 33.5103 -0.00143 0.0 0.0 0 -1 -1 0 # Ring closure Aliphatic C
atom_par GA 4.00 0.150 33.5103 -0.00052 0.0 0.0 0 -1 -1 0 # Ring closure Aromatic C
atom_par J 4.00 0.150 33.5103 -0.00143 0.0 0.0 0 -1 -1 0 # Ring closure Aliphatic C
atom_par Q 4.00 0.150 33.5103 -0.00143 0.0 0.0 0 -1 -1 0 # Ring closure Aliphatic C
35
Grid Map File
Extension: .map
The first six lines of each grid map describe the spatial features of the maps and the files used or
created. These headers are checked by AutoDock to ensure that they are appropriate for the
requested docking. The remainder of the file contains grid point energies, written as floating
point numbers, one per line. They are ordered according to the nested loops: z(y(x)), so x is
changing fastest. The coordinate system is right-handed, and the actual file contains one more
element than the NELEMENTS specified because the maps always have an odd number of
elements in each direction. The “CENTER” is the position of the middle coordinate point, in the
receptor (macromolecule) reference frame.
Sample Grid Map File
GRID_PARAMETER_FILE vac1.nbc.gpf
GRID_DATA_FILE 4phv.nbc_maps.fld
MACROMOLECULE 4phv.new.pdbq
SPACING 0.375
NELEMENTS 50 50 80
CENTER -0.026 4.353 -0.038
125.095596
123.634560
116.724602
108.233879
:
36
Grid Map Field File
Extension: .maps.fld
This is essentially two files in one. It is both an AVS field file, which may be read by a number
of scientific visualization programs, and an AutoDock input file with AutoDock-specific
information in the comments at the head of the file. AutoDock uses this file to check that all the
maps it reads in are compatible. For example, in this file, the grid spacing is 0.375 Angstroms,
there are 60 intervals in each dimension (and 61 actual grid points), the grid is centered near (16.,
39., 1.), it was calculated around the macromolecule ‘protein.pdbqt’, and the AutoGrid
parameter file used to create this (and the maps) was ‘protein.gpf’. This file also points to a
second file, ‘protein.maps.xyz’, which contains the minimum and maximum extents of the
grid box in each dimension, x, y, and z. Finally, it lists the grid map files that were calculated by
AutoGrid, here ‘protein.A.map’, ‘protein.C.map’, etc.
Sample Grid Map Field File
# AVS field file
#
# AutoDock Atomic Affinity and Electrostatic Grids
#
# Created by autogrid4.
#
#SPACING 0.375
#NELEMENTS 60 60 60
#CENTER 16.000 39.000 1.000
#MACROMOLECULE protein.pdbqt
#GRID_PARAMETER_FILE protein.gpf
#
ndim=3 # number of dimensions in the field
dim1=61 # number of x-elements
dim2=61 # number of y-elements
dim3=61 # number of z-elements
nspace=3 # number of physical coordinates per point
veclen=8 # number of affinity values at each point
data=float # data type (byte, integer, float, double)
field=uniform # field type (uniform, rectilinear, irregular)
coord 1 file=protein.maps.xyz filetype=ascii offset=0
coord 2 file=protein.maps.xyz filetype=ascii offset=2
coord 3 file=protein.maps.xyz filetype=ascii offset=4
label=A-affinity # component label for variable 1
label=C-affinity # component label for variable 2
label=HD-affinity # component label for variable 3
label=N-affinity # component label for variable 4
label=OA-affinity # component label for variable 5
label=SA-affinity # component label for variable 6
label=Electrostatics # component label for variable 7
label=Desolvation # component label for variable 8
#
# location of affinity grid files and how to read them
#
variable 1 file=protein.A.map filetype=ascii skip=6
variable 2 file=protein.C.map filetype=ascii skip=6
variable 3 file=protein.HD.map filetype=ascii skip=6
variable 4 file=protein.N.map filetype=ascii skip=6
variable 5 file=protein.OA.map filetype=ascii skip=6
variable 6 file=protein.SA.map filetype=ascii skip=6
variable 7 file=protein.e.map filetype=ascii skip=6
variable 8 file=protein.d.map filetype=ascii skip=6
37
AutoDock Docking Parameter File: DPF
Extension: .dpf
The docking parameter file specifies the files and parameters for an AutoDock calculation,
including the map files that will be used for the docking, the ligand coordinate files, and
parameters for the search. Unlike previous versions of AutoDock, the pairwise atomic
parameters used for the internal energy calculation may now read from a separate file (described
above), or taken from defaults in AutoDock.
All delimiters where needed are white spaces. Default values, where applicable, are given here in
square brackets [thus]. A comment must be prefixed by the “#” symbol, and can be placed after
a space at the end of a parameter line, or on a line of its own. Upper/lower case is ignored in
keywords but is significant in atom names and file names. File names cannot contain white space
or non-ASCII characters. Although ideally it should be possible to give AutoDock keywords in
any order, not every possible combination has been tested, so it would be wise to stick to the
following order.
Parameter to define the version
autodock_parameter_version <version>
[4.2]
Defines the version of the parameters that are going to follow in the rest of the DPF. By default,
this keyword is set to the value of '4.2' but it can be changed in the future to support several
compatibility modes among different versions of AutoDock.
Parameter to set the amount of output
outlev { <symbolic name> | <integer> }
[adt] or [1]
(Optional) Set the output level of data written into the docking log file. Either the symbolic
names or the numeric values can be used in the DPF. Values of "adt", "runv" and higher are
compatible with the result analysis tools in AutoDockTools. The highly verbose levels are
provided for development and debugging work, and the minimal levels are provided for data-
intensive applications such as virtual screening.
Symbolic
Numeric
Result
min
-2
minimal - energy breakdown and state of one result
minclust
-1
energy breakdown, states, coords of clustering results
38
basic
0
echoes input, parms used, final coords and state from each run
adt
1
ADT-compatible output during docking
default
1
default value is same as "adt"
runv
2
more verbose run - expanded output during docking
ligread
3
expanded output during ligand setup (e.g., the nonbond matrix)
recread
4
expanded output during receptor map setup
runvv
5
still more verbose run (e.g., min/max/median per "N" evals)
runvvv
6
most verbose supported run (e.g., state vars per "N" evals)
etables
7
energy table / scoring function atom-by-atom details
nbinte
8
report of nonbond internal energy table (see analysis.cc)
nbintev
9
detailed report of nonbond internal energy table (see analysis.cc)
Atomic parameters for pairwise energy evaluation
parameter_file <existing atomic parameter file name>
(Optional) Atomic parameter file used for pairwise energy evaluation in internal energies and
interactions between ligand and flexible sidechains. If this is not given, AutoDock uses default
parameters identical to values in the file AD4.1_bound.dat.
intelec [off]
(Optional) By default in AutoDock 4.2.5 and later, internal ligand electrostatic energies will be
calculated. To ignore those terms, include “intelec off” in the DPF. Note that this is only relevant
for flexible ligands.
intnbp_r_eps <float><float><integer><integer><string><string>
(Optional) This optional keyword allows the user to override manually the internal energy
potential for a given class of interactions. The parameters are: req, ε, n, m, and the two atom
types, where req is the equilibribium distance for the bottom of the energetic well, ε is the depth
of the well, n and m are the coefficients. For instance, the command “intnbp_r_eps 1.5 10. 12 6
OA FE” will set up a potential with well depth of 10 kcal/mol at a distance of 1.5 Å for
interaction between oxygen and iron atoms. The potential V(r) is calculated with the expression:
V(r)=Cn/rn – Cm/rm
Cn = m/(n-m) * ε *reqn
Cm = n/(n-m) * ε *reqm
39
A special atom type “G” has been created for using this feature for ring closure simulations.
Please see the tutorial for more information.
torsdof <integer>
(Optional) Overrides the number of torsional degrees of freedom appearing in the ligand PDBQT
file. This term affects the reported binding energy but not the conformational search because it is
constant for any particular run.
Command to set the seeds for the random number generator
seed { pid | time | <integer> } { pid | time | <integer> }
[ pid time ]
(Optional) AutoDock search operations use a pseudo-random number generator which, for each
docking job, must be ‘seeded’ with two integers, in any combination of explicit integers, the
keyword “time” or the keyword “pid”. The keyword “time” sets a seed based on the current
time, and “pid” sets a seed to the operating system’s process ID of the currently executing
AutoDock process. The default seed set (pid time) will yield different docking results for each
AutoDock run; to have instead completely repeatable results, use two explicit integers.
Parameters defining the grid maps to be used
ligand_types <string>
Atom names for all atom types present in ligand, using the same blank-separated, one or two
letter atom types used in AutoGrid.
fld <existing .fld file name>
Grid data field file created by AutoGrid (must have the extension “.fld”).
map <existing map file name>
Filename for the AutoGrid affinity grid maps. This keyword plus filename must be repeated for
all atom types in the order specified by the “ligand_types” command. In all map files a 6-line
header is required, and energies must be ordered according to the nested loops z( y( x ) ).
elecmap <existing map file name>
Filename for the electrostatics grid map. 6-line header required, and energies must be ordered
according to the nested loops z( y( x ) ).
desolvmap <existing map file name>
Filename for the desolvation grid map. 6-line header required, and energies must be ordered
according to the nested loops z( y( x ) ).
40
Parameters defining the state of the unbound ligand
(Optional) unbound_energy <float>
Sets the internal energy of the unbound state to the value.
(Optional) unbound_model_extended
Launches a calculation to find an extended conformation of the ligand, then uses this
conformation to calculate the internal energy of the unbound state. The AutoDock4.0 keyword
“compute_unbound_extended” will perform the same process.
(Optional) unbound_model_extended_energy <float>
Sets the internal energy of the unbound state to the value. This also sets the default atomic
parameters used for pairwise energy evaluate to be appropriate for the extended unbound model.
Parameters defining the ligand and its initial state
move <existing PDBQT file name>
Filename for the PDBQT coordinate file of the ligand to be docked.
about <float> <float> <float>
[automatically calculated]
(Optional) Use this keyword to specify the center of the ligand, about which rotations will be
made. (The coordinate frame of reference is that of the ligand PDBQT file.) The default rotation
center is the mean x,y,z-coordinates of the ligand root atoms. Units: Å, Å, Å.
tran0 <float> <float> <float>
tran0 random
[automatically calculated]
(Optional) Initial coordinates for the center of the ligand, in the same frame of reference as the
receptor grid maps. Every docking simulation specified in the docking parameter file starts the
ligand from this location.
Alternatively, the user can just give the keyword “random” and AutoDock will pick random
initial coordinates instead. Units: Å, Å, Å.
quaternion0 <float> <float> <float> <float>
axisangle0 <float> <float> <float> <float>
quat0 <float> <float> <float> <float> (synonym for “axisangle0”)
quaternion0 random
[random]
(Optional) Initial ligand rigid-body orientation
quaternion0: Qx, Qy, Qz, Qw. Specified as quaternion. (Units: none,none,none, none.) AutoDock
will automatically normalize. A ‘no rotation’ (identity) quaternion would be ‘0 0 0 1’.
41
axisangle0: Axisx, Axisy, Axisz, AngleΘ. Specified as axis-angle: Axisx, Axisy, Axisz define the unit
vector of rotation, and AngleΘ defines the angle of rotation about this vector. (Units:
none,none,none, degrees.) AutoDock will automatically normalize the vector. A ‘no rotation’
(identity) axis-angle would be ‘1 0 0 0’.
Alternatively, the user can just give the keyword “random” and AutoDock will pick a random 3-
D orientation. Each docking simulation specified in the docking parameter file will begin at this
same random rigid body rotation.
dihe0 <float> ...
dihe0 random
[random]
Initial relative dihedral angles; there must be a floating point number specified on this line for
each rotatable bond in the PDBQT file. Each value specified here will be added to the
corresponding torsion angle in the input PDBQT file, at the start of each run. Torsion angles are
specified by only two atoms, so the definition of rotations is relative to the input conformation of
the ligand, not an absolute conformation. Units: °.
Parameters defining ligand step sizes for simulated annealing [SA] calculations
tstep <float>
tstep <float> <float>
[2.0 Å]
(Optional, SA only) The first form, with one argument, defines the maximum translation jump
for the first cycle that the ligand may make in one simulated annealing step. When “trnrf” is less
than 1, the reduction factor is multiplied with the tstep at the end of each cycle, to give the new
value for the next cycle. The second form allows the user to specify the value for the first cycle
and the last cycle: AutoDock then calculates the reduction factor that satisfies these constraints.
Units: Å.
qstep <float>
[50.0°]
(Optional, SA only) Maximum angular step size for the orientational component. Units: °.
dstep <float>
[50.0°]
(Optional, SA only) Maximum dihedral (torsion) step size. Units: °.
Parameters defining optional ligand torsion constraints for simulated annealing
barrier <float>
[10000.0]
(Optional, SA only) This defines the energy-barrier height applied to constrained torsions. When
the torsion is at a preferred angle, there is no torsion penalty: this torsion’s energy is zero. If the
42
torsion angle falls within a disallowed zone, however, it can contribute up to the full barrier
energy. Since the torsion-energy profiles are stored internally as arrays of type ‘unsigned short’,
only positive integers between 0 and 65535 are allowed.
gausstorcon <integer> <float> <float>
(Optional, SA only) Adds a constraint to a torsion. The torsion number is identified by an integer.
This identifier comes from the list at the top of the AutoDockTools-generated input ligand
PDBQT file (on the REMARK lines). An energy profile will be calculated for this torsion. An
inverted Gaussian is added for each new constraint. To completely specify each Gaussian, two
floating point numbers are needed: the preferred angle and the half-width respectively (both in
degrees). Note that the preferred angle should be specified in the range -180° to +180°; numbers
outside this range will be wrapped back into this range. This angle, χ, is relative to the original
torsion angle in the input structure. The half-width is the difference between the two angles at
which the energy is half the barrier. The smaller the half-width, the tighter the constraint.
If you wish to constrain to absolute-valued torsion angles, it will be necessary to zero the initial
torsion angles in the ligand. The problem arises from the ambiguous 2-atom definition of the
rotatable bond B-C. To identify a torsion angle unambiguously, 4 atoms must be specified: A-B-
C-D. The sign convention for torsion angles is anti-clockwise (counter-clockwise) with positive
angles, clockwise negative.
There is no limit to the number of constraints that can be added to a given torsion. Each new tor-
sion-constraint energy profile is combined with the pre-existing one by selecting the minimum
energy of either the new or the existing profiles.
Please note that in our tests, torsion constrains are highly inefficient, and are only effective when
used in systems with few degrees of freedom in the ligand, and only a few torsion constraints.
showtorpen
(Optional, SA only) (Use only with “gausstorcon”) This switches on the storage and subsequent
output of torsion energies. During each energy evaluation, the penalty energy for each
constrained torsion, as specified by the “gausstorcon” command, will be stored in an array. At
the end of each run, the final docked conformation’s state variables are output, but with this
command, the penalty energy for each torsion will be printed alongside its torsion angle.
Parameters for cluster analysis of docked conformations
rmstol <float>
[2.0Å]
(Optional) When more than one run is carried out in a given job, cluster analysis or ‘structure
binning’ will be performed, based on all-atom root mean square deviation (RMSD), ranking the
resulting families of docked conformations in order of increasing energy. The lowest energy
representative from each cluster is written in PDBQT format to the log file. Normally, AutoDock
usues the ligand residue number from the input PDBQT file; to number the clustered
conformations incrementally from 1, use “output_resnum_as runnum”, see below. (Units: Å).
43
rmsref <existing PDB/PDBQT file name>
(Optional) If included, the RMSD of the docked conformations will be calculated with respect to
the coordinates in the PDB or PDBQT file specified here. This is useful when the experimentally
determined complex conformation of the ligand is known. The order of the atoms in this file
must match that in the input PDBQT file given by the move command. These values of RMSD
will be output in the last column of the final PDBQT records, after the clustering has been per-
formed. If this keyword is not included, the RMSD is calculated based on the starting position of
the ligand.
rmsnosym
(Optional) The default method for structure binning allows for atom similarity, as in a tertiary-
butyl which can be rotated by +/-120°, but in other cases it may be desirable to bypass this
similar atom type checking and calculate the RMSD on a one-for-one basis. The symmetry
checking algorithm scans all atoms in the reference structure, and selects the nearest atom of
identical atom type to be added to the sum of squares of distances. This works well when the two
conformations are very similar, but this assumption breaks down when the two conformations
are translated significantly. Symmetry checking can be turned off using the rmsnosym
command; omit this command if you still want symmetry checking.
rmsatoms all
(Optional) If this keyword is included, RMSD calculation will be performed using both ligand
and flexible receptor sidechain atoms. If an “rmsref” file is specified, it must include both ligand
and flexible receptor atom coordinates.
rmsmode { atype | unique_pair }
rmsmode heavy_atoms_only
(Optional; default “atype” with all atoms) If included, this keyword will modify the symmetry
checking used in the RMSD calculation. With “atype” (default), the algorithm will evaluate
RMSD based on the nearest atom between the two structures with the same atom type. With
“unique_pair”, each individual atom will be paired at most one time. If the “heavy_atoms_only”
keyword is included, in a separate DPF line, hydrogen atoms will be omitted from the
calculation, otherwise all atoms are included.
output_resnum_as { resnum | runnum }
(Optional; default “resnum”) Normally, AutoDock keeps the original residue number of the input
ligand PDBQT file. With “runnum”, AutoDock re-numbers the starting position to residue-
number 0, and any cluster-representatives are numbered incrementally from 1, according to their
rank (rank 1 is the lowest energy cluster). [This keyword replaces the obsolete “-k” flag.]
Parameter for AutoDock ʻCluster modeʼ run
cluster <existing PDBQT file name>
(Clustering multi-job output only.) AutoDock will go into ‘cluster mode’. Only use this
command to perform cluster analysis on the combined output, <PDBQTfilename>, of several
jobs. This command can be very useful when many jobs have been distributed to several
44
machines and run in ‘parallel’. The docking parameter file will need the following keywords:
rmstol and types; and optionally write_all_cluster_members and/or rmsnosym. You must first
extract the USER lines along with the ATOM records, since AutoDock parses these lines to
determine what the energy of that particular conformation was. For more information, see the
example DPF files given later.
Parameter for calculating energy of a ligand
epdb
This keyword will report the energy of the ligand included in the “move” command. This
command may be used to calculate the energy of a particular ligand conformation without (or
before) performing a docking. Note that all “about”, “tran0”, “quaternion0”, “dihe0” settings are
ignored in the epdb calculation.
Parameters for simulated annealing [SA] searches
e0max <float> <positive_integer>
[0., 10000]
(Optional, SA only) This keyword stipulates that the ligand’s initial state cannot have an energy
greater than the first value, nor can there be more than the second value’s number of retries.
Typical energy values range from 0 to 1000 kcal/mol. If the initial energy exceeds this value, a
new random state is generated and tested. This process is iterated until the condition is satisfied.
This can be particularly useful in preventing runs starting in exceptionally high energy regions.
In such cases, the ligand can get trapped because it is unable to take a long enough translational
jump. In those grids were the ligand is small enough to fit into the low energy regions with ease,
there will not be many iterations before a favorable location is found. But in highly constrained
grids, with large ligands, this initialization loop may run almost indefinitely.
rt0 <float>
[500. cal/mol].
(Optional, SA only) Initial “annealing temperature”; this is actually the absolute temperature
multiplied by the gas constant R. R = 8.314 J mol-1 K-1 = 1.987 cal mol-1 K-1. (Units: cal mol-1.)
linear_schedule ( synonyms: schedule_linear linsched schedlin )
geometric_schedule
[linear_schedule]
(Optional, SA only) The default “linear_schedule” uses a linear or arithmetic temperature
reduction schedule during Monte Carlo simulated annealing. The “geometric_schedule” keyword
uses instead a geometric reduction schedule, according to the rtrf parameter described next. If
the linear schedule is used, any rtrf parameters will be ignored. The first simulated annealing
cycle is carried out at the annealing temperature rt0. At the end of each cycle, the temperature
is reduced by (rt0/cycles). The advantage of the linear schedule is that the system samples
evenly across the temperature axis, which is vital in entropic calculations. Geometric temperature
reduction schedules, on the other hand, under-sample high temperatures and over-sample low
temperatures.
45
rtrf <float>
(Optional, SA geometric schedule only) Annealing temperature reduction factor, g [0.90 cycle-1].
At the end of each cycle, the annealing temperature is multiplied by this factor, to give that of the
next cycle. This must be positive but < 1 in order to cool the system. Gradual cooling is recom-
mended, so as to avoid “simulated quenching”, which tends to trap systems into local minima.
runs <integer>
[50]
Number of automated docking runs. Can instead be specified in the “simanneal” statement
following.
cycles <integer>
[50]
(Optional, SA only) Number of temperature reduction cycles.
accs <integer>
[30000]
(Optional, SA only) Maximum number of accepted steps per cycle.
rejs <integer>
[30000]
(Optional, SA only) Maximum number of rejected steps per cycle.
select <character>
[m]
(Optional, SA only) State selection flag. This character can be either m for the minimum state, or
l for the last state found during each cycle, to begin the following cycle.
trnrf <float>
[1.0]
(Optional, SA only) Per-cycle reduction factor for translation steps.
quarf <float>
[1.0]
(Optional, SA only) Per-cycle reduction factor for orientation steps.
dihrf <float>
(Optional, SA only) Per-cycle reduction factor for torsional dihedral steps [1.].
46
Parameters for genetic algorithm, Lamarckian GA and evolutionary programming
searches
ga_pop_size <positive_integer>
[150]
(Optional, GA/LGA/LS only) This is the number of individuals in the population. Each
individual is a coupling of a genotype and its associated phenotype. Typical values range from
50 to 200.
ga_num_evals <positive_integer>
[2500000]
(Optional, GA/LGA/LS only) Set the maximum number of energy evaluations performed during
each GA, LGA, or LS run.
ga_num_generations <positive_integer>
[27000]
(Optional, GA/LGA only) This is the maximum number of generations simulated during each
GA or LGA run.
ga_elitism <integer>
[1]
(Optional, GA/LGA only) This is used in the selection mechanism of the GA. This is the number
of top individuals that are guaranteed to survive into the next generation.
ga_mutation_rate <float>
[0.02]
(Optional, GA/LGA only) This is a floating point number from 0 to 1, representing the
probability that a particular gene is mutated. This parameter is typically small.
ga_crossover_rate <float>
[0.80]
(Optional, GA/LGA only) This is a floating point number from 0 to 1 denoting the crossover
rate. Crossover rate is the expected number of pairs in the population that will exchange genetic
material. Setting this value to 0 turns the GA into the evolutionary programming (EP) method,
but EP would probably require a concomitant increase in the ga_mutation_rate in order to be
effective.
ga_window_size <positive_integer>
[10]
(Optional, GA/LGA only) This is the number of preceding generations to take into consideration
when deciding the threshold for the worst individual in the current population.
47
Genetic algorithm parameters
set_ga
This command sets the global optimizer to be a genetic algorithm [GA]. This is required to per-
form a GA search. This passes any ’ga_’ parameters specified before this line to the global opti-
mizer object. If this command is omitted, or it is given before the ’ga_’ parameters, your choices
will not take effect, and the default values for the optimizer will be used.
To use the traditional (non-Lamarckian) genetic algorithm, do not specify the local search
parameters, and do not use the “set_sw1” or “set_psw1” commands.
To use the Lamarckian genetic algorithm, you must also specify the parameters for local
search, and then issue either the ’set_sw1’ or ’set_psw1’ command. The ’set_sw1’ command
uses the strict Solis and Wets local search algorithm, whereas ’set_psw1’uses the pseudo-Solis
and Wets algorithm (see below).
Parameters for local search
sw_max_its <positive_integer>
[300]
(Optional, LGA/LS only) This is the maximum number of iterations that the local search
procedure applies to the phenotype of any given individual, per generation.
sw_max_succ <positive_integer>
[4]
(Optional, LGA/LS only) This is the number of successes in a row before a change is made to
the “rho” parameter in Solis & Wets algorithms. This is an unsigned integer and is typically
around four.
sw_max_fail <positive_integer>
[4]
(Optional, LGA/LS only) This is the number of failures in a row before Solis & Wets algorithms
adjust “rho.” This is an unsigned integer and is usually four to eight.
sw_rho <float>
[1.0]
(Optional, LGA/LS only) This is a parameter of the Solis & Wets algorithms. It defines the
initial variance, and specifies the size of the local space to sample.
sw_lb_rho <float>
[0.01]
(Optional, LGA/LS only) This is the lower bound on rho, the variance for making changes to
genes (i.e. translations, orientation and torsions). rho can never be modified to a value smaller
than “sw_lb_rho”.
48
ls_search_freq <float>
[0.06 for LGA, 1.0 for LS]
(Optional, LGA only) This is the probability of any particular phenotype being subjected to local
search.
Commands to choose and set the local search method (LGA or LS)
Both of these commands, ’set_sw1’ and ’set_psw1’, pass any ’sw_’ parameters set before this
line to the local searcher. If you forget to use this command, or give it before the ’sw_’
keywords, your choices will not take effect, and the default values for the optimizer will be used.
Currently, the psw1 method has shown the best performance and is used as the default.
set_sw1
Instructs AutoDock to use the classical Solis and Wets local searcher, using the method of
uniform variances for changes in translations, orientations, and torsions.
set_psw1
Instructs AutoDock to use the pseudo-Solis and Wets local searcher. This method maintains the
relative proportions of variances for the translations in Å and the rotations in radians. These are
typically 0.2 Å and 0.087 radians to start with, so the variance for translations will always be
about 2.3 times larger than that for the rotations (i.e. orientation and torsions).
Commands to specify the search method
simanneal <integer>
[50]
This command instructs AutoDock to do the specified number of docking runs using the
simulated annealing (SA) search engine. All relevant parameters for the simulated annealing job
must be set first. These are indicated above by [SA] in each keyword description.
do_local_only <integer>
[50]
This keyword instructs AutoDock to carry out only the local search of a global-local search; the
genetic algorithm parameters are ignored, with the exception of the population size. This is an
ideal way of carrying out a minimization using the same force field as is used during a docking
calculation. The “ga_run” keyword should not be given. The number after the keyword
determines how many local search simulations will be performed.
do_global_only <integer>
[50]
This keyword instructs AutoDock to carry out dockings using only a global search, i.e. the tradi-
tional genetic algorithm. The local search parameters are ignored. The “ga_run” keyword should
not be given. The number after the keyword determines how many dockings will be performed.
49
ga_run <integer>
[50]
This command invokes the Lamarckian genetic algorithm search engine, and performs the
requested number of dockings. All appropriate parameters must be set first: these are listed
above by “ga_”.
Command to perform clustering of docked conformations
analysis
This performs a cluster analysis on results of a docking, and writes the results to the log file. The
docked conformations are sorted in order of increasing energy and then compared by root mean
square deviation. A histogram is printed showing the number in each cluster, and if more than
one member, the cluster’s mean energy. Furthermore, a table is printed to the docking log file of
cluster rmsd and reference rmsd values.
50
Sample Docking Parameter File (LGA method, from tutorial)
autodock_parameter_version 4.2 # used by autodock to validate parameter set
outlev ADT # diagnostic output level
seed pid time # seeds for random generator
unbound_model bound # state of unbound ligand
ligand_types A C NA OA N HD # atoms types in ligand
fld 1hsg.maps.fld # grid_data_file
map 1hsg.A.map # atom-specific affinity map
map 1hsg.C.map # atom-specific affinity map
map 1hsg.NA.map # atom-specific affinity map
map 1hsg.OA.map # atom-specific affinity map
map 1hsg.N.map # atom-specific affinity map
map 1hsg.HD.map # atom-specific affinity map
elecmap 1hsg.e.map # electrostatics map
desolvmap 1hsg.d.map # desolvation map
move ind.pdbqt # small molecule
about 0.3689 -0.2148 -4.9865 # small molecule root center
tran0 random # initial coordinates/A or random
quaternion0 random # initial orientation
dihe0 random # initial dihedrals (relative) or random
ga_pop_size 150 # number of individuals in population
ga_num_evals 2500000 # maximum number of energy evaluations
ga_num_generations 27000 # maximum number of generations
ga_elitism 1 # top individuals to survive to next generation
ga_mutation_rate 0.02 # rate of gene mutation
ga_crossover_rate 0.8 # rate of crossover
set_ga # set the above parameters for GA or LGA
sw_max_its 300 # iterations of Solis & Wets local search
sw_max_succ 4 # consecutive successes before changing rho
sw_max_fail 4 # consecutive failures before changing rho
sw_rho 1.0 # size of local search space to sample
sw_lb_rho 0.01 # lower bound on rho
ls_search_freq 0.06 # probability of performing local search
set_psw1 # set the above pseudo-Solis & Wets parameters
ga_run 10 # do this many hybrid GA-LS runs
rmstol 2.0 # cluster_tolerance/A
analysis # perform a ranked cluster analysis
Lines in bold are required for this job.
Lines in italics are optional and show default values.
51
Appendix II: Customizing the Docking Protocol
A variety of options are available within AutoDock for customizing the docking protocol
for a given application, by customizing the docking parameters in the DPF. A DPF can be
subdivided into sections, each specifying a different aspect of the calculation. Some of
these sections are common to every calculation and some are more specific. Usually, a
DPF contains a generic section, a ligand-specific section, a search-specific section, and
finally an analysis section. Hence a DPF can then be built in a modular fashion by
combining different sections.
For example, the typical simulated annealing DPF is composed of the following sections:
- generic section
- ligand-specific section
- simulated annealing section
- cluster analysis [optional]
whereas a Lamarckian Genetic Algorithm DPF will contain both genetic algorithm and
local search sections:
- generic section
- ligand-specific section
- genetic algoritm section
- local search section
- cluster analysis [optional]
The typical parameters used for the common AutoDock calculations will be described.
For each calculation type, refer to the DPF Examples section. For a detailed description
of each keyword, refer to the AutoDock Docking Parameter File section.
Generic section
In the first section are usually specified parameters controlling generic aspects of the
calculation that are independent from the type of calculation itself: All are optional.
autodock_parameter_version 4.2
outlev ADT
intelec
seed pid time
unbound_model bound
The first keyword autodock_parameter_version defines the version of the parameters that
are going to follow in the rest of the DPF. By default, this keyword is set to the value of
"4.2" but it can be changed to support several compatibility modes among different
versions of AutoDock.
52
If a customized forcefield (see Atomic Parameter File section) is going to be used, it can
be specified by adding a line similar to the following, after the AutoDock version
keyword :
parameter_file autodock_custom_forcefield.dat
Other options are also available. The outlev sets the amount of detail that is going to be
printed in the output DLG for every step of the calculation. intelec enable the inclusion of
the electrostatic term in the calculation of the internal energy; this is the default in
AutoDock4.2.5 and later. The search methods available in AutoDock are all stochastic
and are performed with a random initial state at every run. The random number generator
is by default seeded with the process identification number (PID) assigned by the
operating system to the AutoDock calculation (pid value) and the current time (time
value). The unbound_model keyword defines the energy model used to calculate the
energy of the unbound ligand before the docking. By default, this energy is set to be
equal to the bound state.
Ligand-specific section
This section contains the keywords that are specific to the ligand to be docked and its
properties:
receptor
fld receptor.maps.fld
map receptor.A.map
map receptor.C.map
map receptor.HD.map
map receptor.N.map
map receptor.OA.map
map receptor.S.map
elecmap receptor.e.map
desolvmap receptor.d.map
move ligand.pdbqt
Ligand atom types (ligand_types keyword) are declared first, then associated to the map
files (map keyword) for each type. The order of the map entries must match the one
specified in the ligand_types keyword. As explained in Appendix III, this section can be
used to set up special calculations. The atom types must include at least all those present
in the ligand to be docked (see below), but more can be specified allowing use of the
same DPF for different ligands. The limitations are the maximum number of atom types
of AutoDock and the amount of memory available on the computer where the calculation
is performed. Independently from the ligand atom types, an electrostatic interaction map
(elecmap keyword) and a desolvation map (desolvmap keyword) must be always specified.
Finally, the move keyword defines the ligand PDBQT filename to be read for the
calculation. If the calculation includes flexible receptor residues, the flexres keyword
must be used to specify the filename containing the flexible residues coordinates, adding
the following line after the ligand move keyword:
flexres flexres.pdbqt
53
Note that if flexible residues are going to be used, affinity maps must be calculated only
on the rigid part of the receptor; ADT has tools to help do this.
Search-specific section
In this section, the type of search method is going to be specified. Currently, AutoDock
supports the following search methods (see Choosing a protocol for your application
section): Simulated Annealing (SA), Local Search (LS), Genetic Algorithm (GA) and
Lamarckian Genetic Algorithm (LGA). Depending on the chosen search method,
different group of keywords will be used in the DPF.
Simulated annealing
(sample file: simulated_annealing/1dwd_simulated_annealing_long.dpf)
If the simulated annealing search method is going to be performed, keywords are used to
specify the initial states for the sampling (global search), and how the temperature
reduction and the ligand movement are simulated (local search). All are optional.
tstep 2.0
qstep 5.0
dstep 5.0
rt0 616.0
First, the size with which the search space is sampled is specified for ligand translation
(tstep keyword), orientation (qstep keyword) and torsions (dstep keyword). Optimal
values for these keywords are 0.2 Angstrom/step and 5 degrees/step. The initial annealing
temperature is also set (rt0 keyword) to 100 cal/mol.
linear_schedule
trnrf 1.0
quarf 1.0
dihrf 1.0
cycles 50
accs 25000
rejs 25000
select m
During the annealing, the step sizes are scaled The initial state is then modified during the
annealing phase. At each step, the simulated temperature is reduced linearly, while
translation (trnrf keyword), orientation (quarf keyword), and dihedral (dihrf keyword) steps
are not changed. The number of temperature reductions is set with the cycles keyword.
The number of accepted (accs keyword) and rejected (rejs keyword) steps at each cycle
are specified. The state selected (select keyword) to begin the following temperature
reduction cycle is the minimum result found so far ('m'). Finally, the simulated annealing
simulation may be started by the trigger keyword simanneal, with a total of 10 simulated
annealing runs to be performed:
Simanneal 10
Local search
(sample file: local_search_lig/1dwd_local_only_long.dpf)
With the local search, only the local space is sampled, performing a sort of
“minimization” of the ligand in the current position.
54
sw_max_its 300
sw_max_succ 4
sw_max_fail 4
sw_rho 1.0
sw_lb_rho 0.01
set_psw1
A local search consists of a number of iterations (sw_max_its keyword) during which
rotational, translational, and torsional degrees of freedom of the ligand are randomly
sampled by a given step. After a given number of successful (sw_max_succ keyword) or
unsuccessful (sw_max_fail keyword) attempts, the sampling step is enlarged or reduced,
respectively, within a rho factor range (sw_rho, sw_lb_rho keywords) and the local search
continued. The most efficient local search method in AutoDock, the pseudo-Solis-Wet
algorithm is selected with the set_psw1 keyword.
The local search method may be executed with 10 independent runs with the
do_local_only keyword:
do_local_only 10
Lamarckian Genetic Algorithm
(sample file: dock_flexlig/1dwd_1dwd.dpf)
With the genetic algorithm, a population of ligand poses is generated and optimized
iteratively, and the global search space is mainly sampled.
ga_pop_size 150
ga_num_evals 2500000
ga_num_generations 27000
ga_elitism 1
ga_mutation_rate 0.02
ga_crossover_rate 0.8
ga_window_size 10
ga_cauchy_alpha 0.0
ga_cauchy_beta 1.0
set_ga
The size of the initial population is defined by the ga_pop_size keyword. The GA search
is performed until either the maximum number of evaluations (ga_num_evals keyword) or
the maximum number of generations (ga_num_generations keyword) limits are reached.
At each generation step, one individual each generation will survive unmodified
(ga_elitism keyword), while chances of mutation (ga_mutation_rate keyword) and
crossover between individual chromosomes (ga_crossover_rate keyword) are allowed. The
last 10 generations are going to be considered when deciding how to rank the individuals
in the current population (ga_window_size keyword). The genetic algorithm search is
enabled (set_ga keyword) and 10 independent runs are performed with the ga_run
keyword:
ga_run 10
The LGA combines local search and genetic algorithm to provide both efficient global
space coverage and local search optimization. Consequently, there will be both GA and
LS keywords.
55
With the genetic algorithm, a population of ligand poses is generated and optimized
iteratively, and the global search space is mainly sampled.
Analysis section
In this section, all operations not necessarily related to docking can be specified (i.e.
results clustering, single-point energy estimation).
Energy estimation
(sample file: eval_lig/1dwd_epdb.dpf)
The energy estimation (epdb keyword) is used to calculate the energy of a ligand pose as
found in a complex, such as in an X-ray crystallographic structure, without performing
any searches. The coordinates of the ligand will not be changed, and grid maps will be
used only to estimate the different energy contributions. The epdb keyword is specified
without any options.
Clustering options
(sample file: recluster/recluster.dpf)
AutoDock provides tools for clustering the results either at the end of each docking or by
joining together multiple docking results and re-clustering them. First, the clustering
RMSD tolerance (rmstol keyword) is specified. By default, AutoDock takes into account
symmetry, which improves significantly the clustering results, but this can be turned off
(see rmsnosym keyword description). By default, only the single best score resulting from
each cluster is written in the output file, but it is possible to save all poses in all clusters,
the write_all keyword must be used. If the calculation will re-cluster poses previously
generated, the file containing them will be specified with the cluster keyword, followed
by the PDBQT filename containing all the poses (i.e. USER, ATOM and/or HETATM
entries extracted from the multiple DLG's). Finally, the actual clustering is performed by
specifying the analysis keyword.
56
Docking Parameter Files (DPF) Examples
Simulated Annealing Docking (sample file:
simulated_annealing/1dwd_simulated_annealing_long.dpf , default values in italics)
## GENERIC SECTION
autodock_parameter_version 4.2 # used by autodock to validate parameter set
outlev adt
intelec # calculate internal electrostatics
seed 28641 1106107140 # seeds for random generator
## LIGAND-SPECIFIC SECTION
ligand_types A C HD N OA S # atoms types in ligand
fld 1dwd_rec.maps.fld # grid_data_file
map 1dwd_rec.A.map # atom-specific affinity map
map 1dwd_rec.C.map # atom-specific affinity map
map 1dwd_rec.HD.map # atom-specific affinity map
map 1dwd_rec.N.map # atom-specific affinity map
map 1dwd_rec.OA.map # atom-specific affinity map
map 1dwd_rec.S.map # atom-specific affinity map
elecmap 1dwd_rec.e.map # electrostatics map
desolvmap 1dwd_rec.d.map # desolvation map
move 1dwd_lig.pdbqt # small molecule
## SIMULATED ANNEALING SEARCH PARAMETERS SECTION
tstep 0.2 # translation step/A
qstep 5.0 # quaternion step/deg
dstep 5.0 # torsion step/deg
rt0 100.0 # initial annealing temperature (times gas constant)
linear_schedule # use linear, arithmetic temperature reduction
cycles 50 # number of temperature reduction cycles
accs 30000 # maximum number of accepted steps per cycle
rejs 30000 # maximum number of rejected steps per cycle
select m # state selection flag: (m)inimum or (l)ast state
## PERFORM SEARCH SECTION
simanneal 10
## ANALYSIS SECTION
rmstol 0.5 # cluster_tolerance/A
analysis # perform a ranked cluster analysis
57
Local Search (sample file: local_search_lig/1dwd_local_only_long.dpf, default values in
italics)
## GENERIC SECTION
autodock_parameter_version 4.2 # used by autodock to validate parameter set
outlev basic # less verbose output than with "ADT"
intelec # calculate internal electrostatics
## LIGAND-SPECIFIC SECTION
ligand_types A C HD N OA S # atoms types in ligand
fld 1dwd_rec.maps.fld # grid_data_file
map 1dwd_rec.A.map # atom-specific affinity map
map 1dwd_rec.C.map # atom-specific affinity map
map 1dwd_rec.HD.map # atom-specific affinity map
map 1dwd_rec.N.map # atom-specific affinity map
map 1dwd_rec.OA.map # atom-specific affinity map
map 1dwd_rec.S.map # atom-specific affinity map
elecmap 1dwd_rec.e.map # electrostatics map
desolvmap 1dwd_rec.d.map # desolvation map
move 1dwd_lig.pdbqt # small molecule
about 32.34 14.05 24.92 # small molecule center
## FREE ENERGY ENTROPY ADJUSTMENT SECTION
torsdof 10 # torsional degrees of freedom
## LOCAL SEARCH PARAMETERS SECTION
sw_max_its 300 # iterations of Solis & Wets local search
sw_max_succ 4 # consecutive successes before changing rho
sw_max_fail 4 # consecutive failures before changing rho
sw_rho 1.0 # size of local search space to sample
sw_lb_rho 0.01 # lower bound on rho
ls_search_freq 1.00 # probability of performing local search on individual
set_psw1 # set the above psw Solis & Wets parameters
## PERFORM SEARCH SECTION
do_local_only 50 # do this many hybrid GA-LS runs
## ANALYSIS SECTION
rmstol 0.5 # cluster_tolerance/A
analysis # perform a ranked cluster analysis
58
Lamarckian Genetic Algorithm Docking (sample file: dock_flexlig/1dwd_1dwd.dpf,
default values in italics)
## GENERIC SECTION
autodock_parameter_version 4.2 # used by autodock to validate parameter set
outlev adt # diagnostic output level
seed pid time # seeds for random generator
## LIGAND-SPECIFIC SECTION
ligand_types A C HD N OA S # atoms types in ligand
fld 1dwd_rec.maps.fld # grid_data_file
map 1dwd_rec.A.map # atom-specific affinity map
map 1dwd_rec.C.map # atom-specific affinity map
map 1dwd_rec.HD.map # atom-specific affinity map
map 1dwd_rec.N.map # atom-specific affinity map
map 1dwd_rec.OA.map # atom-specific affinity map
map 1dwd_rec.S.map # atom-specific affinity map
elecmap 1dwd_rec.e.map # electrostatics map
desolvmap 1dwd_rec.d.map # desolvation map
move 1dwd_lig.pdbqt # small molecule
about 32.34 14.05 24.92 # small molecule center
## INITIAL SEARCH STATE SECTION
tran0 random # initial coordinates/A or random
quaternion random # initial quaternion
dihe0 random # initial dihedrals (relative) or random
## FREE ENERGY ENTROPY ADJUSTMENT SECTION
torsdof 10 # torsional degrees of freedom
## SEARCH-SPECIFIC SECTION
ga_pop_size 150 # number of individuals in population
ga_num_evals 2500000 # maximum number of energy evaluations
ga_num_generations 27000 # maximum number of generations
ga_elitism 1 # number of top individuals to survive to next
generation
ga_mutation_rate 0.02 # rate of gene mutation
ga_crossover_rate 0.8 # rate of crossover
set_ga # set the above parameters for GA or LGA
## LOCAL SEARCH PARAMETERS SECTION
sw_max_its 300 # iterations of Solis & Wets local search
sw_max_succ 4 # consecutive successes before changing rho
sw_max_fail 4 # consecutive failures before changing rho
sw_rho 1.0 # size of local search space to sample
sw_lb_rho 0.01 # lower bound on rho
ls_search_freq 0.06 # probability of performing local search on individual
set_psw1 # set the above pseudo Solis & Wets parameters
## PERFORM SEARCH SECTION
ga_run 10 # do this many hybrid GA-LS runs
## ANALYSIS SECTION
rmstol 0.5 # cluster_tolerance/A
analysis # perform a ranked cluster analysis
59
Single-point Energy Estimation (sample file: eval_lig/1dwd_epdb.dpf)
## GENERIC SECTION
autodock_parameter_version 4.2 # used by autodock to validate parameter
set
outlev ADT # diagnostic output level
## LIGAND-SPECIFIC SECTION
ligand_types A C HD N OA S # atoms types in ligand
fld 1dwd_rec.maps.fld # grid_data_file
map 1dwd_rec.A.map # atom-specific affinity map
map 1dwd_rec.C.map # atom-specific affinity map
map 1dwd_rec.HD.map # atom-specific affinity map
map 1dwd_rec.N.map # atom-specific affinity map
map 1dwd_rec.OA.map # atom-specific affinity map
map 1dwd_rec.S.map # atom-specific affinity map
elecmap 1dwd_rec.e.map # electrostatics map
desolvmap 1dwd_rec.d.map # desolvation map
move 1dwd_lig.pdbqt # small molecule
## EVALUATE LIGAND ENERGY in RECEPTOR SECTION
epdb # small molecule to be evaluated
60
Re-clustering of PDBQT Poses (sample file: recluster/recluster.dpf)
# GENERIC PARAMETERS
autodock_parameter_version 4.2 # used to validate parameter set
outlev ADT # diagnostic output level
ligand_types A C HD N OA S # atoms types in ligand
# OPTIONAL
rmsref 1dwd_lig_xray.pdbqt # reference for RMSD calculation
# RMS MODE
rmsmode unique_pair # set symmetry handling option
rmstol 2.0 # cluster rms tolerance
write_all # write all poses in each cluster
cluster poses.pdbqt # input PDBQT file containing all poses
# PERFORM ANALYSIS
analysis # perform a ranked cluster analysis
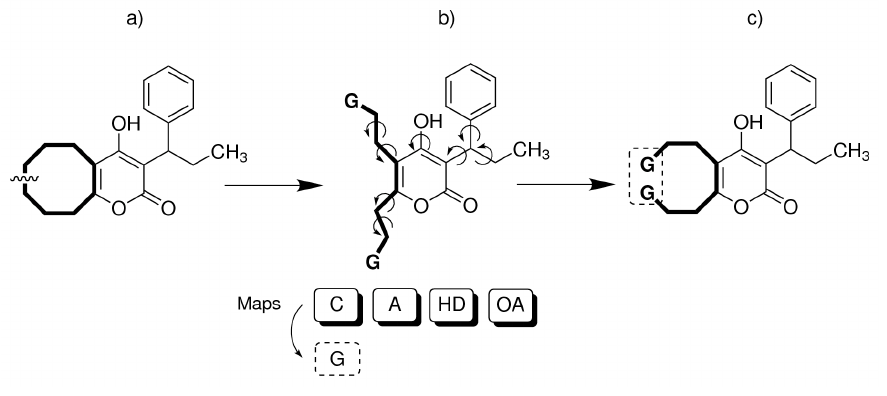
61
Appendix III: Docking Flexible Rings with AutoDock
1. Introduction
AutoDock is not able to manage directly the flexibility associated with bonds in cyclic
molecules, which leads to cyclic portions of the ligands to be considered as rigid. Different
approaches can be used to dock macrocyclic molecules, like identifying one or more low energy
conformations and docking them as different ligands, but generating them and docking them
separately can be a time-consuming task. As an alternative, an indirect method may be used to
manage the ring as a fully flexible entity and use the AutoDock conformation search to explore
its flexibility. The method was initially developed for version 3.05, and now is implemented in
version 4.2. The protocol converts the cyclic ligand into its corresponding acyclic form by
removing a bond, and then docks the fully flexible molecule in the open form. A special atom
type definition allows AutoDock to restore the original cycle structure during the calculation
while exploring the cycle conformations during the search. The protocol can be subdivided in
three main steps:
RING OPENING (a): by removing a bond, the ring is opened and the ligand is transformed to
an acyclic form.
LIGAND PRE-PROCESSING (b): the ligand is processed following the standard
AutoDockTools protocol, but the edge atoms are replaced with G atoms.
DOCKING AND RING CLOSURE (c): the ligand is docked applying a 12-2 pseudo-Lennard-
Jones potential to the G-atoms that restore the cyclic structure.
To restore the closed ring geometry a custom long range pseudo-Lennard-Jones 12-2 potential is
applied to these atoms during the docking calculation. This potential is effective at long range
distances and guarantees the ring closure even with large cycles.
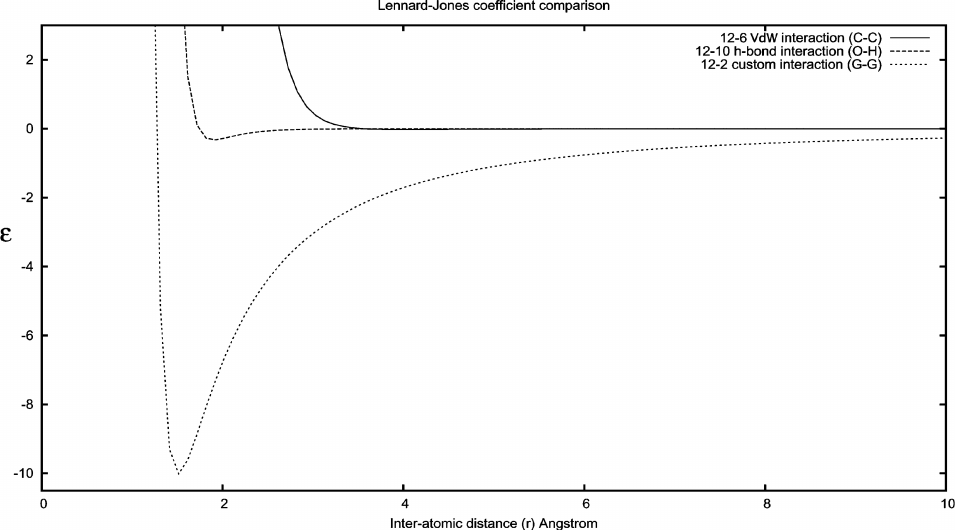
62
Ring closure parameters. Comparison between standard 12-6 van der Waals, 12-10 hydrogen
bond and 12-2 pseudo-Lennard-Jones potentials, before the AutoDock smoothing function is
applied.
No extra maps are calculated for the G atoms because, for sake of evaluation of ligand-protein
interaction, they are considered as normal carbon atoms. Therefore, C maps are used in their
place. During the docking process, the potential guides the edge atoms next to each other
resulting in an effective ring closure, while allowing the GA algorithm to explore the ring
conformations.
2. Flexible rings
Opening the ring
To convert the molecule into the acyclic form, the bond to be disrupted must be identified. The
way the acyclic form is obtained influences the subsequent the ring closure. The following
guidelines may help to choose which bond to remove while keeping the calculation simple and
improving the quality of the final results:
Keep number rotatable bonds low
The ring opening can dramatically increase the total number of rotatable bonds,
requiring longer calculation times. Therefore, when less flexible or partially rigid
regions are present they should not be broken. Bonds resulting in shorter chains
should be preferred.
Break carbon-carbon bonds
For sake of calculation consistency, the bond to be broken should be between two
identical atom types. AutoDock supports aliphatic and aromatic carbon atoms.
Atoms different than carbon can be used but they will require a special
parameterization (see 4.Extension and Limits).
63
Avoid chiral atoms (...whenever possible)
Due to the lack of directionality and the united atom description of hydrogens,
original chirality is not guaranteed if a bond between one or more chiral carbon
atoms is broken. When all ring carbons are chiral (e.g. natural compounds,
antibiotics) any bond can be suitable, while chirality in docking results should be
inspected and manually corrected if necessary.
Once the ring is disrupted, the previously connected atoms must be renamed as “G” in the atom
type column of the PDBQT file:
[...]
HETATM 21 CD4 UIN B 100 -2.919 22.061 19.604 1.00 19.90 0.005
G
[...]
HETATM 24 CD3 UIN B 100 -3.821 22.402 20.791 1.00 19.60 0.005
G
[...]
The 12-2 potential is defined in the DPF using the intnb_r_eps keyword to override the
AutoDock internal interaction parameter table, using the following syntax :
intnbp_r_eps 1.51 10.000000 12 2 G G
AutoDock, when a diagnostic outlev of "etables" is used, will acknowledge the new
parameterization in the DLG:
Ring closure distance potential found for atom type G :
Equilibrium distance = 1.51 Angstroms
Equilibrium potential = 10.000042 Kcal/mol
Pseudo-LJ coefficients = 12 - 2
Calculating internal non-bonded interaction energies for docking calculation;
Non-bonded parameters for G-G interactions, used in internal energy
calculations:
281.0 27.4
E = ----------- - -----------
G,G 12 2
r r
More than one ring
Multiple flexible rings can be docked by disrupting a bond for each ring and using a different
atom type for each edge atom pair. AutoDock includes four ring closure carbon atom types: G, J,
Q (aliphatic) and GA (aromatic), then up to four flexible rings can be docked simultaneously. For
example, if a second ring is opened then the next two edge atoms are renamed as J, and the DPF
64
will include an extra intnb_r_eps keyword and another C map reference. While there is no
actual limitation to the number of cycles that can be opened in the same molecule, there is the
implicit limit of the docking complexity, as well as the maximum number of rotatable bonds
allowed. If further atom types need to be defined (e.g., -S-S- disulfide bond), a customized
atomic parameter file must be generated and included in the DPF with the parameter_file
keyword.
Limitations
Using this approach for docking flexible rings can save a lot of time compared to rigid ring
docking of different conformations, but there are some limitations associated with the protocol
implementation:
Chirality. Hydrogen atoms bound to chiral edge atoms will be merged in the united-
atom model used in AutoDock, then chirality information is lost. In the docking
process G-atoms can eventually approach each other from directions different than
the original geometry, leading to potentially wrong chirality.
Bond distance. The pseudo-Lennard-Jones potential describes the ideal equilibrium
distance of the two G-atoms, corresponding to the equilibrium C-C bonding distance
(~1.5 Å). The final distance although can be slightly bigger, because of the van der
Waals repulsion between the two atoms preventing atomic volume overlaps.
Energy calculation. During the calculation the pseudo-Lennard-Jones potential
provides an extra energy contribution to the total energy sum to induce the ring
closure. This can result in an overall shift of the final energy to lower values. While
not being an actual limitation, it should be considered to avoid comparisons between
scores obtained with and without flexible rings.
For these reasons, the final docking result should be refined by inspecting the chirality and
performing a geometry refinement to correct bonding angles and distances.

65
This is the DPF corresponding to the example structure in the introduction:
autodock_parameter_version 4.1 # used by autodock to validate parameter set
outlev ADT # diagnostic output level
seed pid time # seeds for random generator
unbound_model bound # state of unbound ligand
ligand_types A C G HD OA # atoms types in ligand
fld protein.maps.fld # grid_data_file
map protein.A.map # atom-specific affinity map
map protein.C.map # atom-specific affinity map
map protein.C.map # C map for G atoms
map protein.HD.map # atom-specific affinity map
map protein.OA.map # atom-specific affinity map
elecmap protein.e.map # electrostatics map
desolvmap protein.d.map # desolvation map
intnbp_r_eps 1.51 10.000000 12 2 G G # pseudo-LJ potential
move ligandG.pdbqt # small molecule
about -0.8665 18.5882 20.1623 # small molecule center
tran0 random # initial coordinates/A or random
quat0 random # initial quaternion
dihe0 random # initial dihedrals (relative) or random
torsdof 8 # torsional degrees of freedom
ga_pop_size 350 # number of individuals in population
ga_num_evals 2500000 # maximum number of energy evaluations
ga_num_generations 27000 # maximum number of generations
ga_elitism 1 # number of top individuals to survive to next generation
ga_mutation_rate 0.02 # rate of gene mutation
ga_crossover_rate 0.8 # rate of crossover
set_ga # set the above parameters for GA or LGA
sw_max_its 300 # iterations of Solis & Wets local search
sw_max_succ 4 # consecutive successes before changing rho
sw_max_fail 4 # consecutive failures before changing rho
sw_rho 1.0 # size of local search space to sample
sw_lb_rho 0.01 # lower bound on rho
ls_search_freq 0.26 # probability of performing local search on individual
set_psw1 # set the above Solis & Wets parameters
ga_run 100 # do this many hybrid GA-LS runs
rmstol 2.0 # cluster_tolerance/A
analysis # perform a ranked cluster analysis
3. Reference
Forli, S., et al. J. Chem. Inf. Model., 2007, 47, 1481–1492
4. Tutorial
http://autodock.scripps.edu/faqs-help/tutorial/flexible-rings-docking
66
Appendix IV: AutoDock References
AutoDock 4.2
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S. and Olson,
A. J. (2009) J. Comput. Chem., 30: 2785-2791. “Autodock4 and AutoDockTools4: automated
docking with selective receptor flexiblity.”
Cosconati, S., Forli, S., Perryman, A. L., Harris, R., Goodsell, D. S. and Olson, A. J. (2010)
Expert Opin. Drug Discovery 5: 597-607. "Virtual screening with AutoDock: theory and
practice."
Forli, S. and Olson, A. J. (2012) J. Med. Chem. 55: 623-638. "A force field with discrete
displaceable waters and desolvations entropy for hydrated ligand docking."
AutoDock 4.0
Huey, R., Morris, G. M., Olson, A. J. and Goodsell, D. S. (2007) J. Comput. Chem. 28, 1145-
1152. “A semiempirical free energy force field with charge-based desolvation.”
Huey, R., Goodsell, D. S., Morris, G. M. and Olson, A. J. (2004) Letters in Drug Design and
Discovery 1, 178-183. “Grid-based hydrogen bond potentials with improved directionality”.
AutoDock 3.0
Morris, G. M., Goodsell, D. S., Halliday, R.S., Huey, R., Hart, W. E., Belew, R. K. and Olson,
A. J. (1998), J. Computational Chemistry, 19: 1639-1662. "Automated Docking Using a Lama-
rckian Genetic Algorithm and an Empirical Binding Free Energy Function".
AutoDock 2.4
Morris, G. M., Goodsell, D. S., Huey, R. and Olson, A. J. (1996), J. Computer-Aided Molecular
Design, 10: 293-304. "Distributed automated docking of flexible ligands to proteins: Parallel
applications of AutoDock 2.4".
Goodsell, D. S., Morris, G. M. and Olson, A. J. (1996), J. Mol. Recognition, 9: 1-5. "Docking of
Flexible Ligands: Applications of AutoDock".
AutoDock 1.0
Goodsell, D. S. and Olson, A. J. (1990), Proteins: Str. Func. and Genet., 8: 195-202. "Auto-
mated Docking of Substrates to Proteins by Simulated Annealing".
