CLASS MANUAL
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 257 [warning: Documents this large are best viewed by clicking the View PDF Link!]
- 1 CLASS: Cosmic Linear Anisotropy Solving System
- 2 Where to find information and documentation on CLASS?
- 3 CLASS overview (architecture, input/output, general principles)
- 4 Data Structure Documentation
- 4.1 nonlinear Struct Reference
- 4.1.1 Detailed Description
- 4.1.2 Field Documentation
- 4.1.2.1 method
- 4.1.2.2 pk_size
- 4.1.2.3 k_size
- 4.1.2.4 k
- 4.1.2.5 tau_size
- 4.1.2.6 tau
- 4.1.2.7 nl_corr_density [1/2]
- 4.1.2.8 k_nl [1/2]
- 4.1.2.9 index_tau_min_nl
- 4.1.2.10 has_pk_eq
- 4.1.2.11 index_pk_eq_w
- 4.1.2.12 index_pk_eq_Omega_m
- 4.1.2.13 pk_eq_size
- 4.1.2.14 pk_eq_tau_size
- 4.1.2.15 pk_eq_tau
- 4.1.2.16 pk_eq_w_and_Omega
- 4.1.2.17 pk_eq_ddw_and_ddOmega
- 4.1.2.18 nonlinear_verbose
- 4.1.2.19 error_message
- 4.1.2.20 nl_corr_density [2/2]
- 4.1.2.21 k_nl [2/2]
- 4.1 nonlinear Struct Reference
- 5 File Documentation
- 5.1 background.c File Reference
- 5.1.1 Detailed Description
- 5.1.2 Function Documentation
- 5.1.2.1 background_at_tau()
- 5.1.2.2 background_tau_of_z()
- 5.1.2.3 background_functions()
- 5.1.2.4 background_w_fld()
- 5.1.2.5 background_init()
- 5.1.2.6 background_free()
- 5.1.2.7 background_free_noinput()
- 5.1.2.8 background_free_input()
- 5.1.2.9 background_indices()
- 5.1.2.10 background_ncdm_distribution()
- 5.1.2.11 background_ncdm_test_function()
- 5.1.2.12 background_ncdm_init()
- 5.1.2.13 background_ncdm_momenta()
- 5.1.2.14 background_ncdm_M_from_Omega()
- 5.1.2.15 background_solve()
- 5.1.2.16 background_initial_conditions()
- 5.1.2.17 background_find_equality()
- 5.1.2.18 background_output_titles()
- 5.1.2.19 background_output_data()
- 5.1.2.20 background_derivs()
- 5.1.2.21 V_e_scf()
- 5.1.2.22 V_p_scf()
- 5.1.2.23 V_scf()
- 5.2 background.h File Reference
- 5.3 class.c File Reference
- 5.4 common.h File Reference
- 5.5 input.c File Reference
- 5.5.1 Detailed Description
- 5.5.2 Function Documentation
- 5.5.2.1 input_init_from_arguments()
- 5.5.2.2 input_init()
- 5.5.2.3 input_read_parameters()
- 5.5.2.4 input_default_params()
- 5.5.2.5 input_default_precision()
- 5.5.2.6 get_machine_precision()
- 5.5.2.7 class_fzero_ridder()
- 5.5.2.8 input_try_unknown_parameters()
- 5.5.2.9 input_get_guess()
- 5.5.2.10 input_find_root()
- 5.5.2.11 input_prepare_pk_eq()
- 5.6 input.h File Reference
- 5.7 lensing.c File Reference
- 5.7.1 Detailed Description
- 5.7.2 Function Documentation
- 5.7.2.1 lensing_cl_at_l()
- 5.7.2.2 lensing_init()
- 5.7.2.3 lensing_free()
- 5.7.2.4 lensing_indices()
- 5.7.2.5 lensing_lensed_cl_tt()
- 5.7.2.6 lensing_addback_cl_tt()
- 5.7.2.7 lensing_lensed_cl_te()
- 5.7.2.8 lensing_addback_cl_te()
- 5.7.2.9 lensing_lensed_cl_ee_bb()
- 5.7.2.10 lensing_addback_cl_ee_bb()
- 5.7.2.11 lensing_d00()
- 5.7.2.12 lensing_d11()
- 5.7.2.13 lensing_d1m1()
- 5.7.2.14 lensing_d2m2()
- 5.7.2.15 lensing_d22()
- 5.7.2.16 lensing_d20()
- 5.7.2.17 lensing_d31()
- 5.7.2.18 lensing_d3m1()
- 5.7.2.19 lensing_d3m3()
- 5.7.2.20 lensing_d40()
- 5.7.2.21 lensing_d4m2()
- 5.7.2.22 lensing_d4m4()
- 5.8 lensing.h File Reference
- 5.9 nonlinear.c File Reference
- 5.10 nonlinear.h File Reference
- 5.11 output.c File Reference
- 5.12 output.h File Reference
- 5.13 perturbations.c File Reference
- 5.13.1 Detailed Description
- 5.13.2 Function Documentation
- 5.13.2.1 perturb_sources_at_tau()
- 5.13.2.2 perturb_init()
- 5.13.2.3 perturb_free()
- 5.13.2.4 perturb_indices_of_perturbs()
- 5.13.2.5 perturb_timesampling_for_sources()
- 5.13.2.6 perturb_get_k_list()
- 5.13.2.7 perturb_workspace_init()
- 5.13.2.8 perturb_workspace_free()
- 5.13.2.9 perturb_solve()
- 5.13.2.10 perturb_prepare_output()
- 5.13.2.11 perturb_find_approximation_number()
- 5.13.2.12 perturb_find_approximation_switches()
- 5.13.2.13 perturb_vector_init()
- 5.13.2.14 perturb_vector_free()
- 5.13.2.15 perturb_initial_conditions()
- 5.13.2.16 perturb_approximations()
- 5.13.2.17 perturb_timescale()
- 5.13.2.18 perturb_einstein()
- 5.13.2.19 perturb_total_stress_energy()
- 5.13.2.20 perturb_sources()
- 5.13.2.21 perturb_print_variables()
- 5.13.2.22 perturb_derivs()
- 5.13.2.23 perturb_tca_slip_and_shear()
- 5.14 perturbations.h File Reference
- 5.15 primordial.c File Reference
- 5.15.1 Detailed Description
- 5.15.2 Function Documentation
- 5.15.2.1 primordial_spectrum_at_k()
- 5.15.2.2 primordial_init()
- 5.15.2.3 primordial_free()
- 5.15.2.4 primordial_indices()
- 5.15.2.5 primordial_get_lnk_list()
- 5.15.2.6 primordial_analytic_spectrum_init()
- 5.15.2.7 primordial_analytic_spectrum()
- 5.15.2.8 primordial_inflation_potential()
- 5.15.2.9 primordial_inflation_hubble()
- 5.15.2.10 primordial_inflation_indices()
- 5.15.2.11 primordial_inflation_solve_inflation()
- 5.15.2.12 primordial_inflation_analytic_spectra()
- 5.15.2.13 primordial_inflation_spectra()
- 5.15.2.14 primordial_inflation_one_wavenumber()
- 5.15.2.15 primordial_inflation_one_k()
- 5.15.2.16 primordial_inflation_find_attractor()
- 5.15.2.17 primordial_inflation_evolve_background()
- 5.15.2.18 primordial_inflation_check_potential()
- 5.15.2.19 primordial_inflation_check_hubble()
- 5.15.2.20 primordial_inflation_get_epsilon()
- 5.15.2.21 primordial_inflation_find_phi_pivot()
- 5.15.2.22 primordial_inflation_derivs()
- 5.15.2.23 primordial_external_spectrum_init()
- 5.16 primordial.h File Reference
- 5.17 spectra.c File Reference
- 5.17.1 Detailed Description
- 5.17.2 Function Documentation
- 5.17.2.1 spectra_cl_at_l()
- 5.17.2.2 spectra_pk_at_z()
- 5.17.2.3 spectra_pk_at_k_and_z()
- 5.17.2.4 spectra_pk_nl_at_z()
- 5.17.2.5 spectra_pk_nl_at_k_and_z()
- 5.17.2.6 spectra_tk_at_z()
- 5.17.2.7 spectra_tk_at_k_and_z()
- 5.17.2.8 spectra_init()
- 5.17.2.9 spectra_free()
- 5.17.2.10 spectra_indices()
- 5.17.2.11 spectra_cls()
- 5.17.2.12 spectra_compute_cl()
- 5.17.2.13 spectra_k_and_tau()
- 5.17.2.14 spectra_pk()
- 5.17.2.15 spectra_sigma()
- 5.17.2.16 spectra_matter_transfers()
- 5.17.2.17 spectra_output_tk_data()
- 5.17.2.18 spectra_fast_pk_at_kvec_and_zvec()
- 5.18 spectra.h File Reference
- 5.19 thermodynamics.c File Reference
- 5.19.1 Detailed Description
- 5.19.2 Function Documentation
- 5.19.2.1 thermodynamics_at_z()
- 5.19.2.2 thermodynamics_init()
- 5.19.2.3 thermodynamics_free()
- 5.19.2.4 thermodynamics_indices()
- 5.19.2.5 thermodynamics_helium_from_bbn()
- 5.19.2.6 thermodynamics_onthespot_energy_injection()
- 5.19.2.7 thermodynamics_energy_injection()
- 5.19.2.8 thermodynamics_reionization_function()
- 5.19.2.9 thermodynamics_get_xe_before_reionization()
- 5.19.2.10 thermodynamics_reionization()
- 5.19.2.11 thermodynamics_reionization_sample()
- 5.19.2.12 thermodynamics_recombination()
- 5.19.2.13 thermodynamics_recombination_with_hyrec()
- 5.19.2.14 thermodynamics_recombination_with_recfast()
- 5.19.2.15 thermodynamics_derivs_with_recfast()
- 5.19.2.16 thermodynamics_merge_reco_and_reio()
- 5.19.2.17 thermodynamics_output_titles()
- 5.20 thermodynamics.h File Reference
- 5.21 transfer.c File Reference
- 5.21.1 Detailed Description
- 5.21.2 Function Documentation
- 5.21.2.1 transfer_functions_at_q()
- 5.21.2.2 transfer_init()
- 5.21.2.3 transfer_free()
- 5.21.2.4 transfer_indices_of_transfers()
- 5.21.2.5 transfer_get_l_list()
- 5.21.2.6 transfer_get_q_list()
- 5.21.2.7 transfer_get_k_list()
- 5.21.2.8 transfer_get_source_correspondence()
- 5.21.2.9 transfer_source_tau_size()
- 5.21.2.10 transfer_compute_for_each_q()
- 5.21.2.11 transfer_interpolate_sources()
- 5.21.2.12 transfer_sources()
- 5.21.2.13 transfer_selection_function()
- 5.21.2.14 transfer_dNdz_analytic()
- 5.21.2.15 transfer_selection_sampling()
- 5.21.2.16 transfer_lensing_sampling()
- 5.21.2.17 transfer_source_resample()
- 5.21.2.18 transfer_selection_times()
- 5.21.2.19 transfer_selection_compute()
- 5.21.2.20 transfer_compute_for_each_l()
- 5.21.2.21 transfer_integrate()
- 5.21.2.22 transfer_limber()
- 5.21.2.23 transfer_limber_interpolate()
- 5.21.2.24 transfer_limber2()
- 5.22 transfer.h File Reference
- 5.1 background.c File Reference
- 6 The `external_Pk` mode
- 7 Updating the manual
- Index
CLASS MANUAL
Last updated September 10, 2018
Generated by Doxygen 1.8.13
Contents
1 CLASS: Cosmic Linear Anisotropy Solving System 1
2 Where to find information and documentation on CLASS? 3
3 CLASS overview (architecture, input/output, general principles) 5
4 Data Structure Documentation 15
4.1 nonlinear Struct Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
4.1.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
4.1.2 Field Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
4.1.2.1 method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
4.1.2.2 pk_size . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
4.1.2.3 k_size . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
4.1.2.4 k . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
4.1.2.5 tau_size . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
4.1.2.6 tau . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.1.2.7 nl_corr_density [1/2] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.1.2.8 k_nl [1/2] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.1.2.9 index_tau_min_nl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.1.2.10 has_pk_eq . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.1.2.11 index_pk_eq_w . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.1.2.12 index_pk_eq_Omega_m . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.1.2.13 pk_eq_size . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
4.1.2.14 pk_eq_tau_size . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
4.1.2.15 pk_eq_tau . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
4.1.2.16 pk_eq_w_and_Omega . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
4.1.2.17 pk_eq_ddw_and_ddOmega . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
4.1.2.18 nonlinear_verbose . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
4.1.2.19 error_message . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
4.1.2.20 nl_corr_density [2/2] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
4.1.2.21 k_nl [2/2] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18

ii CONTENTS
5 File Documentation 19
5.1 background.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
5.1.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
5.1.2 Function Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
5.1.2.1 background_at_tau() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
5.1.2.2 background_tau_of_z() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
5.1.2.3 background_functions() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
5.1.2.4 background_w_fld() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
5.1.2.5 background_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
5.1.2.6 background_free() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
5.1.2.7 background_free_noinput() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
5.1.2.8 background_free_input() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
5.1.2.9 background_indices() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
5.1.2.10 background_ncdm_distribution() . . . . . . . . . . . . . . . . . . . . . . . . . . 28
5.1.2.11 background_ncdm_test_function() . . . . . . . . . . . . . . . . . . . . . . . . . 28
5.1.2.12 background_ncdm_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
5.1.2.13 background_ncdm_momenta() . . . . . . . . . . . . . . . . . . . . . . . . . . 29
5.1.2.14 background_ncdm_M_from_Omega() . . . . . . . . . . . . . . . . . . . . . . . 30
5.1.2.15 background_solve() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
5.1.2.16 background_initial_conditions() . . . . . . . . . . . . . . . . . . . . . . . . . . 32
5.1.2.17 background_find_equality() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
5.1.2.18 background_output_titles() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
5.1.2.19 background_output_data() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
5.1.2.20 background_derivs() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
5.1.2.21 V_e_scf() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
5.1.2.22 V_p_scf() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
5.1.2.23 V_scf() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
5.2 background.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
5.2.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
5.2.2 Data Structure Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
Generated by Doxygen

CONTENTS iii
5.2.2.1 struct background . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
5.2.2.2 struct background_parameters_and_workspace . . . . . . . . . . . . . . . . . 42
5.2.2.3 struct background_parameters_for_distributions . . . . . . . . . . . . . . . . . 42
5.2.3 Enumeration Type Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
5.2.3.1 spatial_curvature . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
5.2.3.2 equation_of_state . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
5.3 class.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
5.3.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
5.4 common.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
5.4.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
5.4.2 Data Structure Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
5.4.2.1 struct precision . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
5.4.3 Enumeration Type Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
5.4.3.1 evolver_type . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
5.4.3.2 pk_def . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
5.4.3.3 file_format . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
5.5 input.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
5.5.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
5.5.2 Function Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
5.5.2.1 input_init_from_arguments() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
5.5.2.2 input_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
5.5.2.3 input_read_parameters() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
5.5.2.4 input_default_params() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
5.5.2.5 input_default_precision() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
5.5.2.6 get_machine_precision() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
5.5.2.7 class_fzero_ridder() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
5.5.2.8 input_try_unknown_parameters() . . . . . . . . . . . . . . . . . . . . . . . . . 61
5.5.2.9 input_get_guess() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
5.5.2.10 input_find_root() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
5.5.2.11 input_prepare_pk_eq() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
Generated by Doxygen

iv CONTENTS
5.6 input.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
5.6.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
5.6.2 Enumeration Type Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
5.6.2.1 target_names . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
5.7 lensing.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
5.7.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
5.7.2 Function Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
5.7.2.1 lensing_cl_at_l() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
5.7.2.2 lensing_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
5.7.2.3 lensing_free() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
5.7.2.4 lensing_indices() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
5.7.2.5 lensing_lensed_cl_tt() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
5.7.2.6 lensing_addback_cl_tt() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
5.7.2.7 lensing_lensed_cl_te() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
5.7.2.8 lensing_addback_cl_te() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
5.7.2.9 lensing_lensed_cl_ee_bb() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
5.7.2.10 lensing_addback_cl_ee_bb() . . . . . . . . . . . . . . . . . . . . . . . . . . . 72
5.7.2.11 lensing_d00() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72
5.7.2.12 lensing_d11() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73
5.7.2.13 lensing_d1m1() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73
5.7.2.14 lensing_d2m2() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
5.7.2.15 lensing_d22() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
5.7.2.16 lensing_d20() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
5.7.2.17 lensing_d31() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
5.7.2.18 lensing_d3m1() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
5.7.2.19 lensing_d3m3() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
5.7.2.20 lensing_d40() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
5.7.2.21 lensing_d4m2() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
5.7.2.22 lensing_d4m4() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
5.8 lensing.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
Generated by Doxygen

CONTENTS v
5.8.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
5.8.2 Data Structure Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
5.8.2.1 struct lensing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
5.9 nonlinear.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
5.9.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
5.9.2 Function Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
5.9.2.1 nonlinear_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
5.9.2.2 nonlinear_halofit() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
5.10 nonlinear.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
5.10.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
5.10.2 Macro Definition Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
5.10.2.1 _M_EV_TOO_BIG_FOR_HALOFIT_ . . . . . . . . . . . . . . . . . . . . . . . 83
5.11 output.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
5.11.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
5.11.2 Function Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
5.11.2.1 output_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
5.11.2.2 output_cl() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
5.11.2.3 output_pk() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
5.11.2.4 output_pk_nl() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
5.11.2.5 output_tk() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
5.11.2.6 output_print_data() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88
5.11.2.7 output_open_cl_file() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88
5.11.2.8 output_one_line_of_cl() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
5.11.2.9 output_open_pk_file() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
5.11.2.10 output_one_line_of_pk() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
5.12 output.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
5.12.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
5.12.2 Data Structure Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
5.12.2.1 struct output . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
5.12.3 Macro Definition Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
Generated by Doxygen

vi CONTENTS
5.12.3.1 _Z_PK_NUM_MAX_ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
5.13 perturbations.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
5.13.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
5.13.2 Function Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
5.13.2.1 perturb_sources_at_tau() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
5.13.2.2 perturb_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
5.13.2.3 perturb_free() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 96
5.13.2.4 perturb_indices_of_perturbs() . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
5.13.2.5 perturb_timesampling_for_sources() . . . . . . . . . . . . . . . . . . . . . . . 98
5.13.2.6 perturb_get_k_list() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99
5.13.2.7 perturb_workspace_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100
5.13.2.8 perturb_workspace_free() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
5.13.2.9 perturb_solve() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
5.13.2.10 perturb_prepare_output() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102
5.13.2.11 perturb_find_approximation_number() . . . . . . . . . . . . . . . . . . . . . . . 103
5.13.2.12 perturb_find_approximation_switches() . . . . . . . . . . . . . . . . . . . . . . 103
5.13.2.13 perturb_vector_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
5.13.2.14 perturb_vector_free() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 106
5.13.2.15 perturb_initial_conditions() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
5.13.2.16 perturb_approximations() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111
5.13.2.17 perturb_timescale() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
5.13.2.18 perturb_einstein() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113
5.13.2.19 perturb_total_stress_energy() . . . . . . . . . . . . . . . . . . . . . . . . . . . 114
5.13.2.20 perturb_sources() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
5.13.2.21 perturb_print_variables() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 116
5.13.2.22 perturb_derivs() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
5.13.2.23 perturb_tca_slip_and_shear() . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
5.14 perturbations.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
5.14.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
5.14.2 Data Structure Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
Generated by Doxygen

CONTENTS vii
5.14.2.1 struct perturbs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
5.14.2.2 struct perturb_vector . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 129
5.14.2.3 struct perturb_workspace . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 130
5.14.2.4 struct perturb_parameters_and_workspace . . . . . . . . . . . . . . . . . . . . 132
5.14.3 Macro Definition Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132
5.14.3.1 _SELECTION_NUM_MAX_ . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132
5.14.3.2 _MAX_NUMBER_OF_K_FILES_ . . . . . . . . . . . . . . . . . . . . . . . . . 132
5.14.4 Enumeration Type Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132
5.14.4.1 tca_flags . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132
5.14.4.2 tca_method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 133
5.14.4.3 possible_gauges . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 133
5.15 primordial.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 133
5.15.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 134
5.15.2 Function Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 135
5.15.2.1 primordial_spectrum_at_k() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 135
5.15.2.2 primordial_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 136
5.15.2.3 primordial_free() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 137
5.15.2.4 primordial_indices() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 137
5.15.2.5 primordial_get_lnk_list() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 137
5.15.2.6 primordial_analytic_spectrum_init() . . . . . . . . . . . . . . . . . . . . . . . . 138
5.15.2.7 primordial_analytic_spectrum() . . . . . . . . . . . . . . . . . . . . . . . . . . 138
5.15.2.8 primordial_inflation_potential() . . . . . . . . . . . . . . . . . . . . . . . . . . . 139
5.15.2.9 primordial_inflation_hubble() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 139
5.15.2.10 primordial_inflation_indices() . . . . . . . . . . . . . . . . . . . . . . . . . . . 140
5.15.2.11 primordial_inflation_solve_inflation() . . . . . . . . . . . . . . . . . . . . . . . . 140
5.15.2.12 primordial_inflation_analytic_spectra() . . . . . . . . . . . . . . . . . . . . . . . 141
5.15.2.13 primordial_inflation_spectra() . . . . . . . . . . . . . . . . . . . . . . . . . . . 142
5.15.2.14 primordial_inflation_one_wavenumber() . . . . . . . . . . . . . . . . . . . . . . 142
5.15.2.15 primordial_inflation_one_k() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 143
5.15.2.16 primordial_inflation_find_attractor() . . . . . . . . . . . . . . . . . . . . . . . . 144
Generated by Doxygen

viii CONTENTS
5.15.2.17 primordial_inflation_evolve_background() . . . . . . . . . . . . . . . . . . . . . 144
5.15.2.18 primordial_inflation_check_potential() . . . . . . . . . . . . . . . . . . . . . . . 145
5.15.2.19 primordial_inflation_check_hubble() . . . . . . . . . . . . . . . . . . . . . . . . 146
5.15.2.20 primordial_inflation_get_epsilon() . . . . . . . . . . . . . . . . . . . . . . . . . 146
5.15.2.21 primordial_inflation_find_phi_pivot() . . . . . . . . . . . . . . . . . . . . . . . . 148
5.15.2.22 primordial_inflation_derivs() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 149
5.15.2.23 primordial_external_spectrum_init() . . . . . . . . . . . . . . . . . . . . . . . . 150
5.16 primordial.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 150
5.16.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
5.16.2 Data Structure Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
5.16.2.1 struct primordial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
5.16.3 Enumeration Type Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 156
5.16.3.1 primordial_spectrum_type . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 156
5.16.3.2 linear_or_logarithmic . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 156
5.16.3.3 potential_shape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 156
5.16.3.4 target_quantity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
5.16.3.5 integration_direction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
5.16.3.6 time_definition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
5.16.3.7 phi_pivot_methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
5.16.3.8 inflation_module_behavior . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
5.17 spectra.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
5.17.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 159
5.17.2 Function Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 159
5.17.2.1 spectra_cl_at_l() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 159
5.17.2.2 spectra_pk_at_z() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
5.17.2.3 spectra_pk_at_k_and_z() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
5.17.2.4 spectra_pk_nl_at_z() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
5.17.2.5 spectra_pk_nl_at_k_and_z() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
5.17.2.6 spectra_tk_at_z() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
5.17.2.7 spectra_tk_at_k_and_z() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
Generated by Doxygen

CONTENTS ix
5.17.2.8 spectra_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
5.17.2.9 spectra_free() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
5.17.2.10 spectra_indices() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
5.17.2.11 spectra_cls() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 169
5.17.2.12 spectra_compute_cl() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
5.17.2.13 spectra_k_and_tau() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
5.17.2.14 spectra_pk() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
5.17.2.15 spectra_sigma() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
5.17.2.16 spectra_matter_transfers() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
5.17.2.17 spectra_output_tk_data() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
5.17.2.18 spectra_fast_pk_at_kvec_and_zvec() . . . . . . . . . . . . . . . . . . . . . . . 174
5.18 spectra.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 175
5.18.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 176
5.18.2 Data Structure Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 176
5.18.2.1 struct spectra . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 176
5.19 thermodynamics.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 181
5.19.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182
5.19.2 Function Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 183
5.19.2.1 thermodynamics_at_z() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 183
5.19.2.2 thermodynamics_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184
5.19.2.3 thermodynamics_free() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 185
5.19.2.4 thermodynamics_indices() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 186
5.19.2.5 thermodynamics_helium_from_bbn() . . . . . . . . . . . . . . . . . . . . . . . 186
5.19.2.6 thermodynamics_onthespot_energy_injection() . . . . . . . . . . . . . . . . . . 187
5.19.2.7 thermodynamics_energy_injection() . . . . . . . . . . . . . . . . . . . . . . . . 188
5.19.2.8 thermodynamics_reionization_function() . . . . . . . . . . . . . . . . . . . . . 188
5.19.2.9 thermodynamics_get_xe_before_reionization() . . . . . . . . . . . . . . . . . . 189
5.19.2.10 thermodynamics_reionization() . . . . . . . . . . . . . . . . . . . . . . . . . . 189
5.19.2.11 thermodynamics_reionization_sample() . . . . . . . . . . . . . . . . . . . . . . 190
5.19.2.12 thermodynamics_recombination() . . . . . . . . . . . . . . . . . . . . . . . . . 192
Generated by Doxygen

x CONTENTS
5.19.2.13 thermodynamics_recombination_with_hyrec() . . . . . . . . . . . . . . . . . . . 192
5.19.2.14 thermodynamics_recombination_with_recfast() . . . . . . . . . . . . . . . . . . 193
5.19.2.15 thermodynamics_derivs_with_recfast() . . . . . . . . . . . . . . . . . . . . . . 194
5.19.2.16 thermodynamics_merge_reco_and_reio() . . . . . . . . . . . . . . . . . . . . . 195
5.19.2.17 thermodynamics_output_titles() . . . . . . . . . . . . . . . . . . . . . . . . . . 195
5.20 thermodynamics.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 196
5.20.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 197
5.20.2 Data Structure Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 197
5.20.2.1 struct thermo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 197
5.20.2.2 struct recombination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 200
5.20.2.3 struct reionization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 201
5.20.2.4 struct thermodynamics_parameters_and_workspace . . . . . . . . . . . . . . . 202
5.20.3 Macro Definition Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 202
5.20.3.1 f1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
5.20.3.2 f2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
5.20.3.3 _YHE_BIG_ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
5.20.3.4 _YHE_SMALL_ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
5.20.4 Enumeration Type Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
5.20.4.1 recombination_algorithm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
5.20.4.2 reionization_parametrization . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
5.20.4.3 reionization_z_or_tau . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 204
5.21 transfer.c File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 204
5.21.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 205
5.21.2 Function Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 206
5.21.2.1 transfer_functions_at_q() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 206
5.21.2.2 transfer_init() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 207
5.21.2.3 transfer_free() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 208
5.21.2.4 transfer_indices_of_transfers() . . . . . . . . . . . . . . . . . . . . . . . . . . . 209
5.21.2.5 transfer_get_l_list() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
5.21.2.6 transfer_get_q_list() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
Generated by Doxygen

CONTENTS xi
5.21.2.7 transfer_get_k_list() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 211
5.21.2.8 transfer_get_source_correspondence() . . . . . . . . . . . . . . . . . . . . . . 211
5.21.2.9 transfer_source_tau_size() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 212
5.21.2.10 transfer_compute_for_each_q() . . . . . . . . . . . . . . . . . . . . . . . . . . 212
5.21.2.11 transfer_interpolate_sources() . . . . . . . . . . . . . . . . . . . . . . . . . . . 213
5.21.2.12 transfer_sources() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 214
5.21.2.13 transfer_selection_function() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 215
5.21.2.14 transfer_dNdz_analytic() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 215
5.21.2.15 transfer_selection_sampling() . . . . . . . . . . . . . . . . . . . . . . . . . . . 216
5.21.2.16 transfer_lensing_sampling() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 217
5.21.2.17 transfer_source_resample() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 217
5.21.2.18 transfer_selection_times() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 218
5.21.2.19 transfer_selection_compute() . . . . . . . . . . . . . . . . . . . . . . . . . . . 219
5.21.2.20 transfer_compute_for_each_l() . . . . . . . . . . . . . . . . . . . . . . . . . . 219
5.21.2.21 transfer_integrate() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 220
5.21.2.22 transfer_limber() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223
5.21.2.23 transfer_limber_interpolate() . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223
5.21.2.24 transfer_limber2() . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 224
5.22 transfer.h File Reference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 225
5.22.1 Detailed Description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 226
5.22.2 Data Structure Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 226
5.22.2.1 struct transfers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 226
5.22.2.2 struct transfer_workspace . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 230
5.22.3 Enumeration Type Documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 230
5.22.3.1 radial_function_type . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 230
6 The ‘external_Pk‘ mode 231
7 Updating the manual 235
Index 237
Generated by Doxygen
Chapter 1
CLASS: Cosmic Linear Anisotropy Solving System
Authors: Julien Lesgourgues and Thomas Tram
with several major inputs from other people, especially Benjamin Audren, Simon Prunet, Jesus Torrado, Miguel
Zumalacarregui, Francesco Montanari, etc.
For download and information, see http://class-code.net
Compiling CLASS and getting started
(the information below can also be found on the webpage, just below the download button)
Download the code from the webpage and unpack the archive (tar -zxvf class_vx.y.z.tar.gz), or clone it from
https://github.com/lesgourg/class_public. Go to the class directory (cd class/ or class_public/ or
class_vx.y.z/) and compile (make clean; make class). You can usually speed up compilation with the option -j: make
-j class. If the first compilation attempt fails, you may need to open the Makefile and adapt the name of the compiler
(default: gcc), of the optimization flag (default: -O4 -ffast-math) and of the OpenMP flag (default: -fopenmp; this flag
is facultative, you are free to compile without OpenMP if you don't want parallel execution; note that you need the
version 4.2 or higher of gcc to be able to compile with -fopenmp). Many more details on the CLASS compilation are
given on the wiki page
https://github.com/lesgourg/class_public/wiki/Installation
(in particular, for compiling on Mac >= 10.9 despite of the clang incompatibility with OpenMP).
To check that the code runs, type:
./class explanatory.ini
The explanatory.ini file is THE reference input file, containing and explaining the use of all possible input parameters.
We recommend to read it, to keep it unchanged (for future reference), and to create for your own purposes some
shorter input files, containing only the input lines which are useful for you. Input files must have a ∗.ini extension.
If you want to play with the precision/speed of the code, you can use one of the provided precision files (e.g.
cl_permille.pre) or modify one of them, and run with two input files, for instance:
./class test.ini cl_permille.pre
The files ∗.pre are suppposed to specify the precision parameters for which you don't want to keep default values. If
you find it more convenient, you can pass these precision parameter values in your ∗.ini file instead of an additional
∗.pre file.
The automatically-generated documentation is located in
doc/manual/html/index.html
doc/manual/CLASS_manual.pdf
On top of that, if you wish to modify the code, you will find lots of comments directly in the files.

2 CLASS: Cosmic Linear Anisotropy Solving System
Python
To use CLASS from python, or ipython notebooks, or from the Monte Python parameter extraction code, you need
to compile not only the code, but also its python wrapper. This can be done by typing just 'make' instead of 'make
class' (or for speeding up: 'make -j'). More details on the wrapper and its compilation are found on the wiki page
https://github.com/lesgourg/class_public/wiki
Plotting utility
Since version 2.3, the package includes an improved plotting script called CPU.py (Class Plotting Utility), written by
Benjamin Audren and Jesus Torrado. It can plot the Cl's, the P(k) or any other CLASS output, for one or several
models, as well as their ratio or percentage difference. The syntax and list of available options is obtained by typing
'pyhton CPU.py -h'. There is a similar script for MATLAB, written by Thomas Tram. To use it, once in MATLAB, type
'help plot_CLASS_output.m'
Developing the code
If you want to develop the code, we suggest that you download it from the github webpage
https://github.com/lesgourg/class_public
rather than from class-code.net. Then you will enjoy all the feature of git repositories. You can even develop your
own branch and get it merged to the public distribution. For related instructions, check
https://github.com/lesgourg/class_public/wiki/Public-Contributing
Using the code
You can use CLASS freely, provided that in your publications, you cite at least the paper CLASS II←-
: Approximation schemes <http://arxiv.org/abs/1104.2933>. Feel free to cite more C←-
LASS papers!
Support
To get support, please open a new issue on the
https://github.com/lesgourg/class_public
webpage!
Generated by Doxygen
Chapter 2
Where to find information and documentation on
CLASS?
Author: Julien Lesgourgues
•For what the code can actually compute: all possible input parameters, all coded cosmological models,
all functionalities, all observables, etc.: read the file explanatory.ini in the main CLASS directory: it
is THE reference file where we keep track of all possible input and the definition of all input parameters. For
that reason we recommend to leave it always unchanged and to work with copies of it, or with short input files
written from scratch.
•For the structure, style, and concrete aspects of the code: this documentation, especially the CLASS
overview chapter (the extensive automatically-generated part of this documentation is more for advanced
users); plus the slides of our CLASS lectures, for instance those from Tokyo 2014 available at
http://lesgourg.github.io/class-tour-Tokyo.html
or the more recent and concise summary from the Narbonne 2016 lecture available at
http://lesgourg.github.io/class-tour/Narbonne.pdf
An updated overview of available CLASS lecture slides is always available at
http://lesgourg.github.io/courses.html
in the section Courses on numerical tools.
•For the python wrapper of CLASS: at the moment, the best are the last slides (pages 75-96) of the Nar-
bonne 2016 lectures
http://lesgourg.github.io/class-tour/Narbonne.pdf
Later we will expand the wrapper documentation with a dedicated chapter here.
•For the physics and equations used in the code: mainly, the following papers:
–Cosmological perturbation theory in the synchronous and conformal Newtonian gauges
C. P. Ma and E. Bertschinger.
http://arxiv.org/abs/astro-ph/9506072
10.1086/176550
Astrophys. J. 455, 7 (1995)
–The Cosmic Linear Anisotropy Solving System (CLASS) II: Approximation schemes
D. Blas, J. Lesgourgues and T. Tram.
http://arxiv.org/abs/1104.2933 [astro-ph.CO]
10.1088/1475-7516/2011/07/034
JCAP 1107, 034 (2011)

4 Where to find information and documentation on CLASS?
–The Cosmic Linear Anisotropy Solving System (CLASS) IV: efficient implementation of non-cold relics
J. Lesgourgues and T. Tram.
http://arxiv.org/abs/1104.2935 [astro-ph.CO]
10.1088/1475-7516/2011/09/032
JCAP 1109, 032 (2011)
–Optimal polarisation equations in FLRW universes
T. Tram and J. Lesgourgues.
http://arxiv.org/abs/1305.3261 [astro-ph.CO]
10.1088/1475-7516/2013/10/002
JCAP 1310, 002 (2013)
–Fast and accurate CMB computations in non-flat FLRW universes
J. Lesgourgues and T. Tram.
http://arxiv.org/abs/1312.2697 [astro-ph.CO]
10.1088/1475-7516/2014/09/032
JCAP 1409, no. 09, 032 (2014)
–The CLASSgal code for Relativistic Cosmological Large Scale Structure
E. Di Dio, F. Montanari, J. Lesgourgues and R. Durrer.
http://arxiv.org/abs/1307.1459 [astro-ph.CO]
10.1088/1475-7516/2013/11/044
JCAP 1311, 044 (2013)
plus also some latex notes on specific sectors:
–Equations for perturbed recombination
(can be turned on optionally by the user since v2.1.0)
L. Voruz.
http://lesgourg.github.io/class_public/perturbed_recombination.pdf
–PPF formalism in Newtonian and synchronous gauge
(used by default for the fluid perturbations since v2.6.0)
T. Tram.
http://lesgourg.github.io/class_public/PPF_formalism.pdf
Generated by Doxygen
Chapter 3
CLASS overview (architecture, input/output, general
principles)
Author: Julien Lesgourgues
Overall architecture of class
Files and directories
After downloading CLASS, one can see the following files in the root directory contains:
• some example of input files, the most important being explanatory.ini. a reference input file containing
all possible flags, options and physical input parameters. While this documentation explains the structure and
use of the code, explanatory.ini can be seen as the physical documentation of CLASS. The other
input file are alternative parameter input files (ending with .ini) and precision input files (ending with .pre)
• the Makefile, with which you can compile the code by typing make clean; make; this will create
the executable class and some binary files in the directory build/. The Makefile contains other
compilation options that you can view inside the file.
•CPU.py is a python script designed for plotting the CLASS output; for documentation type python CP←-
U.py --help
•plot_CLASS_output.m is the counterpart of CPU.py for MatLab
• there are other input files for various applications: an example of a non-cold dark matter distribution func-
tions (psd_FD_single.dat), and examples of evolution and selection functions for galaxy number count
observables (myevolution.dat, myselection.dat).
Other files are split between the following directories:
•source/ contains the C files for each CLASS module, i.e. each block containing some part of the physical
equations and logic of the Boltzmann code.
•tools/ contains purely numerical algorithms, applicable in any context: integrators, simple manipulation of
arrays (derivation, integration, interpolation), Bessel function calculation, quadrature algorithms, parser, etc.

6 CLASS overview (architecture, input/output, general principles)
•main/ contains the main module class.c with the main routine class(...), to be used in interactive
runs (but not necessarily when the code is interfaced with other ones).
•test/ contains alternative main routines which can be used to run only some part of the code, to test its
accuracy, to illustrate how it can be interfaced with other codes, etc.
•include/ contains all the include files with a .h suffix.
•output/ is where the output files will be written by default (this can be changed to another directory by
adjusting the input parameter root = <...>)
•python/ contains the python wrapper of CLASS, called classy (see python/README)
•cpp/ contains the C++ wrapper of CLASS, called ClassEngine (see cpp/README)
•doc/ contains the automatic documentation (manual and input files required to build it)
•external_Pk/ contains examples of external codes that can be used to generate the primordial spectrum
and be interfaced with CLASS, when one of the many options already built inside the code are not sufficient.
•bbn/ contains interpolation tables produced by BBN codes, in order to predict e.g. YHe(ωb,∆Neff ).
•hyrec/ contains the recombination code HyRec of Yacine Ali-Haimoud and Chris Hirata, that can be used
as an alternative to the built-in Recfast (using the input parameter recombination = <...>).
The ten-module backbone
Ten tasks
The purpose of class consists in computing some background quantities, thermodynamical quantities, perturba-
tion transfer functions, and finally 2-point statistics (power spectra) for a given set of cosmological parameters. This
task can be decomposed in few steps or modules:
1. set input parameter values.
2. compute the evolution of cosmological background quantities.
3. compute the evolution of thermodynamical quantities (ionization fractions, etc.)
4. compute the evolution of source functions S(k, τ )(by integrating over all perturbations).
5. compute the primordial spectra.
6. eventually, compute non-linear corrections at small redshift/large wavenumber.
7. compute transfer functions in harmonic space ∆l(k)(unless one needs only Fourier spectra P(k)'s and no
harmonic spectra Cl's).
8. compute the observable power spectra Cl's (by convolving the primordial spectra and the harmonic transfer
functions) and/or P(k)'s (by multiplying the primordial spectra and the appropriate source functions S(k, τ)).
9. eventually, compute the lensed CMB spectra (using second-order perturbation theory)
10. write results in files (when CLASS is used interactively. The python wrapper does not go through this step,
after 1.-9. it just keeps the output stored internally).
Generated by Doxygen

7
Ten structures
In class, each of these steps is associated with a structure:
1. struct precision for input precision parameters (input physical parameters are dispatched among the
other structures listed below)
2. struct background for cosmological background,
3. struct thermo for thermodynamics,
4. struct perturbs for source functions,
5. struct primordial for primordial spectra,
6. struct nonlinear for nonlinear corrections,
7. struct transfers for transfer functions,
8. struct spectra for observable spectra,
9. struct lensing for lensed CMB spectra,
10. struct output for auxiliary variable describing the output format.
A given structure contains "everything concerning one step that the subsequent steps need to know" (for instance,
struct perturbs contains everything about source functions that the transfer module needs to know). In par-
ticular, each structure contains one array of tabulated values (for struct background, background quantities
as a function of time, for struct thermo, thermodynamical quantities as a function of redshift, for struct
perturbs, sources S(k, τ ), etc.). It also contains information about the size of this array and the value of the
index of each physical quantity, so that the table can be easily read and interpolated. Finally, it contains any derived
quantity that other modules might need to know. Hence, the communication from one module A to another module
B consists in passing a pointer to the structure filled by A, and nothing else.
All "precision parameters" are grouped in the single structure struct precision. The code contains no other
arbitrary numerical coefficient.
Ten modules
Each structure is defined and filled in one of the following modules (and precisely in the order below):
1. input.c
2. background.c
3. thermodynamics.c
4. perturbations.c
5. primordial.c
6. nonlinear.c
7. transfer.c
8. spectra.c
9. lensing.c
10. output.c
Generated by Doxygen

8 CLASS overview (architecture, input/output, general principles)
Each of these modules contains at least three functions:
•module_init(...)
•module_free(...)
•module_something_at_somevalue
where module is one of input, background, thermodynamics, perturb, primordial,
nonlinear, transfer, spectra, lensing, output.
The first function allocates and fills each structure. This can be done provided that the previous structures in the
hierarchy have been already allocated and filled. In summary, calling one of module_init(...) amounts in
solving entirely one of the steps 1 to 10.
The second function deallocates the fields of each structure. This can be done optionally at the end of the code (or,
when the code is embedded in a sampler, this must be done between each execution of class, and especially
before calling module_init(...) again with different input parameters).
The third function is able to interpolate the pre-computed tables. For instance, background_init() fills a
table of background quantities for discrete values of conformal time τ, but background_at_tau(tau, ∗
values) will return these values for any arbitrary τ.
Note that functions of the type module_something_at_somevalue are the only ones which are called from
another module, while functions of the type module_init(...) and module_free(...) are the only one
called by the main executable. All other functions are for internal use in each module.
When writing a C code, the ordering of the functions in the ∗.c file is in principle arbitrary. However, for the sake of
clarity, we always respected the following order in each CLASS module:
1. all functions that may be called by other modules, i.e. "external functions", usually named like module_←-
something_at_somevalue(...)
2. then, module_init(...)
3. then, module_free()
4. then, all functions used only internally by the module
The main() function(s)
The main.c file
The main executable of class is the function main() located in the file main/main.c. This function consist
only in the following lines (not including comments and error-management lines explained later):
main() {
struct precision pr;
struct background ba;
struct thermo th;
struct perturbs pt;
struct primordial pm;
struct nonlinear nl;
Generated by Doxygen

9
struct transfers tr;
struct spectra sp;
struct lensing le;
struct output op;
input_init_from_arguments(argc, argv,&pr,&ba,&th,&pt,&tr,&pm,&sp,&nl,&le,&op,errmsg);
background_init(&pr,&ba);
thermodynamics_init(&pr,&ba,&th);
perturb_init(&pr,&ba,&th,&pt);
primordial_init(&pr,&pt,&pm);
nonlinear_init(&pr,&ba,&th,&pt,&pm,&nl);
transfer_init(&pr,&ba,&th,&pt,&nl,&tr);
spectra_init(&pr,&ba,&pt,&pm,&nl,&tr,&sp);
lensing_init(&pr,&pt,&sp,&nl,&le);
output_init(&ba,&th,&pt,&pm,&tr,&sp,&nl,&le,&op)
/****** done ******/
lensing_free(&le);
spectra_free(&sp);
transfer_free(&tr);
nonlinear_free(&nl);
primordial_free(&pm);
perturb_free(&pt);
thermodynamics_free(&th);
background_free(&ba);
We can come back on the role of each argument. The arguments above are all pointers to the 10 structures of the
code, excepted argc, argv which contains the input files passed by the user, and errmsg which contains the
output error message of the input module (error management will be described below).
input_init_from_arguments needs all structures, because it will set the precision parameters inside the
precision structure, and the physical parameters in some fields of the respective other structures. For in-
stance, an input parameter relevant for the primordial spectrum calculation (like the tilt ns) will be stored in the
primordial structure. Hence, in input_init_from_arguments, all structures can be seen as output
arguments.
Other module_init() functions typically need all previous structures, which contain the result of the previous
modules, plus its own structures, which contain some relevant input parameters before the function is called, as
well as all the result form the module when the function has been executed. Hence all passed structures can
be seen as input argument, excepted the last one which is both input and output. An example is perturb_←-
init(&pr,&ba,&th,&pt).
Each function module_init() does not need all previous structures, it happens that a module does not depend
on a all previous one. For instance, the primordial module does not need information on the background and ther-
modynamics evolution in order to compute the primordial spectra, so the dependency is reduced: primordial←-
_init(&pr,&pt,&pm).
Generated by Doxygen

10 CLASS overview (architecture, input/output, general principles)
Each function module_init() only deallocates arrays defined in the structure of their own module, so they
need only their own structure as argument. (This is possible because all structures are self-contained, in the
sense that when the structure contains an allocated array, it also contains the size of this array). The first and last
module, input and output, have no input_free() or output_free() functions, because the structures
precision and output do not contain arrays that would need to be de-allocated after the execution of the
module.
The test_<...>.c files
For a given purpose, somebody could only be interested in the intermediate steps (only background quantities, only
the thermodynamics, only the perturbations and sources, etc.) It is then straightforward to truncate the full hierarchy
of modules 1, ... 10 at some arbitrary order. We provide several "reduced executables" achieving precisely this.
They are located in test/test_module_.c (like, for instance, test/test_perturbations.c) and they
can be complied using the Makefile, which contains the appropriate commands and definitions (for instance, you
can type make test_perturbations).
The test/ directory contains other useful example of alternative main functions, like for instance test_←-
loops.c which shows how to call CLASS within a loop over different parameter values. There is also a version
test/test_loops_omp.c using a double level of openMP parallelisation: one for running several CLASS
instances in parallel, one for running each CLASS instance on several cores. The comments in these files are
self-explanatory.
Input/output
Input
There are two types of input:
• "precision parameters" (controlling the precision of the output and the execution time),
• "input parameters" (cosmological parameters, flags telling to the code what it should compute, ...)
The code can be executed with a maximum of two input files, e.g.
./class explanatory.ini cl_permille.pre
The file with a .ini extension is the cosmological parameter input file, and the one with a .pre extension is the
precision file. Both files are optional: all parameters are set to default values corresponding to the "most usual
choices", and are eventually replaced by the parameters passed in the two input files. For instance, if one is happy
with default accuracy settings, it is enough to run with
./class explanatory.ini
Input files do not necessarily contain a line for each parameter, since many of them can be left to default value. The
example file explanatory.ini is very long and somewhat indigestible, since it contains all possible parameters,
together with lengthy explanations. We recommend to keep this file unchanged for reference, and to copy it in e.g.
test.ini. In the latter file, the user can erase all sections in which he/she is absolutely not interested (e.g., all
the part on isocurvature modes, or on tensors, or on non-cold species, etc.). Another option is to create an input file
from scratch, copying just the relevant lines from explanatory.ini. For the simplest applications, the user will
just need a few lines for basic cosmological parameters, one line for the output entry (where one can specifying
which power spectra must be computed), and one line for the root entry (specifying the prefix of all output files).
The syntax of the input files is explained at the beginning of explanatory.ini. Typically, lines in those files
look like:
Generated by Doxygen

11
parameter1 = value1
free comments
parameter2 = value2 # further comments
# commented_parameter = commented_value
and parameters can be entered in arbitrary order. This is rather intuitive. The user should just be careful not to put
an "=" sign not preceded by a "#" sign inside a comment: the code would then think that one is trying to pass some
unidentified input parameter.
The syntax for the cosmological and precision parameters is the same. It is clearer to split these parameters in
the two files .ini and .pre, but there is no strict rule about which parameter goes into which file: in principle,
precision parameters could be passed in the .ini, and vice-versa. The only important thing is not to pass the
same parameter twice: the code would then complain and not run.
The CLASS input files are also user-friendly in the sense that many different cosmological parameter bases can
be used. This is made possible by the fact that the code does not only read parameters, it "interprets them" with
the level of logic which has been coded in the input.c module. For instance, the Hubble parameter, the photon
density, the baryon density and the ultra-relativistic neutrino density can be entered as:
h = 0.7
T_cmb = 2.726 # Kelvin units
omega_b = 0.02
N_eff = 3.04
(in arbitrary order), or as
H0 = 70
omega_g = 2.5e-5 # g is the label for photons
Omega_b = 0.04
omega_ur = 1.7e-5 # ur is the label for ultra-relativistic species
or any combination of the two. The code knows that for the photon density, one should pass one (but not more
than one) parameter out of T_cmb,omega_g,Omega_g (where small omega's refer to ωi≡Ωih2). It searches
for one of these values, and if needed, it converts it into one of the other two parameters, using also other input
parameters. For instance, omega_g will be converted into Omega_g even if his written later in the file than
omega_g: the order makes no difference. Lots of alternatives have been defined. If the code finds that not enough
parameters have been passed for making consistent deductions, it will complete the missing information with in-built
default values. On the contrary, if it finds that there is too much information and no unique solution, it will complain
and return an error.
In summary, the input syntax has been defined in such way that the user does not need to think too much, and can
pass his preferred set of parameters in a nearly informal way.
Let us mention a two useful parameters defined at the end of explanatory.ini, that we recommend setting to
yes in order to run the code in a safe way:
write parameters = [yes or no] (default:no)
When set to yes, all input/precision parameters which have been read are written in a file <root>parameters.←-
ini, to keep track all the details of this execution; this file can also be re-used as a new input file. Also, with this
option, all parameters that have been passed and that the code did not read (because the syntax was wrong,
Generated by Doxygen

12 CLASS overview (architecture, input/output, general principles)
or because the parameter was not relevant in the context of the run) are written in a file <root>unused_←-
parameters. When you have doubts about your input or your results, you can check what is in there.
write warnings = [yes or no] (default:no)
When set to yes, the parameters that have been passed and that the code did not read (because the syntax was
wrong, or because the parameter was not relevant in the context of the run) are written in the standard output as
[Warning:]....
There is also a list of "verbose" parameters at the end of explanatory.ini. They can be used to control the
level of information passed to the standard output (0 means silent; 1 means normal, e.g. information on age of the
universe, etc.; 2 is useful for instance when you want to check on how many cores the run is parallelised; 3 and
more are intended for debugging).
CLASS comes with a list of precision parameter files ending by .pre. Honestly we have not been updating all these
files recently, and we need to do a bit of cleaning there. However you can trust cl_ref.pre. We have derived
this file by studying both the convergence of the CMB output with respect to all CLASS precision parameters, and
the agreement with CAMB. We consider that this file generates good reference CMB spectra, accurate up to the
hundredth of per cent level, as explained in the CLASS IV paper and re-checked since then. You can try it with e.g.
./class explanatory.ini cl_ref.pre
but the run will be extremely long. This is an occasion to run a many-core machine with a lot of RAM. It may work
also on your laptop, but in half an hour or so.
If you want a reference matter power spectrum P(k), also accurate up to the hundredth of percent level, we recom-
mend using the file pk_ref.pre, identical to cl_ref.pre excepted that the truncation of the neutrino hierarchy
has been pushed to l_max_ur=150.
In order to increase moderately the precision to a tenth of percent, without prohibitive computing time, we recom-
mend using cl_permille.pre.
Output
The input file may contain a line
root = <root>
where <root>is a path of your choice, e.g. output/test_. Then all output files will start like this, e.←-
g. output/test_cl.dat,output/test_cl_lensed.dat, etc. Of course the number of output files
depends on your settings in the input file. There can be input files for CMB, LSS, background, thermodynamics,
transfer functions, primordial spectra, etc. All this is documented in explanatory.ini.
If you do not pass explicitly a root = <root>, the code will name the output in its own way, by concatenating
output/, the name of the input parameter file, and the first available integer number, e.g.
output/explanatory03_cl.dat, etc.
Generated by Doxygen

13
General principles
Error management
Error management is based on the fact that all functions are defined as integers returning either _SUCCESS_ or
_FAILURE_. Before returning _FAILURE_, they write an error message in the structure of the module to which
they belong. The calling function will read this message, append it to its own error message, and return a _FAIL←-
URE_; and so on and so forth, until the main routine is reached. This error management allows the user to see the
whole nested structure of error messages when an error has been met. The structure associated to each module
contains a field for writing error messages, called structure_i.error_message, where structure_i
could be one of background,thermo,perturbs, etc. So, when a function from a module iis called within
module jand returns an error, the goal is to write in structure_j.error_message a local error message,
and to append to it the error message in structure_i.error_message. These steps are implemented in a
macro class_call(), used for calling whatever function:
class_call(module_i_function(...,structure_i),
structure_i.error_message,
structure_j.error_message);
So, the first argument of call_call() is the function we want to call; the second argument is the location of
the error message returned by this function; and the third one is the location of the error message which should
be returned to the higher level. Usually, in the bulk of the code, we use pointer to structures rather than structure
themselves; then the syntax is
class_call(module_i_function(...,pi),
pi->error_message,
pj->error_message);‘
where in this generic example, pi and pj are assumed to be pointers towards the structures structure_i and
structure_j.
The user will find in include/common.h a list of additional macros, all starting by class_...(), which are all
based on this logic. For instance, the macro class_test() offers a generic way to return an error in a standard
format if a condition is not fulfilled. A typical error message from CLASS looks like:
Error in module_j_function1
module_j_function1 (L:340) : error in module_i_function2(...)
module_i_function2 (L:275) : error in module_k_function3(...)
...
=>module_x_functionN (L:735) : your choice of input parameter blabla=30
is not consistent with the constraint blabla<1
where the L's refer to line numbers in each file. These error messages are very informative, and are built almost
entirely automatically by the macros. For instance, in the above example, it was only necessary to write inside the
function module_x_functionN() a test like:
class_test(blabla >= 1,
px->error_message,
"your choice of input parameter blabla=%e
is not consistent with the constraint blabla<%e",
blabla,blablamax);
All the rest was added step by step by the various class_call() macros.
Generated by Doxygen

14 CLASS overview (architecture, input/output, general principles)
Dynamical allocation of indices
On might be tempted to decide that in a given array, matrix or vector, a given quantity is associated with an explicit
index value. However, when modifying the code, extra entries will be needed and will mess up the initial scheme;
the user will need to study which index is associated to which quantity, and possibly make an error. All this can be
avoided by using systematically a dynamical index allocation. This means that all indices remain under a symbolic
form, and in each, run the code attributes automatically a value to each index. The user never needs to know this
value.
Dynamical indexing is implemented in a very generic way in CLASS, the same rules apply everywhere. They are
explained in these lecture slides:
https://www.dropbox.com/sh/ma5muh76sggwk8k/AABl_DDUBEzAjjdywMjeTya2a?dl=0
in the folder CLASS_Lecture_slides/lecture5_index_and_error.pdf.
No hard coding
Any feature or equation which could be true in one cosmology and not in another one should not be written explicitly
in the code, and should not be taken as granted in several other places. Discretization and integration steps are
usually defined automatically by the code for each cosmology, instead of being set to something which might be
optimal for minimal models, and not sufficient for other ones. You will find many example of this in the code.
As a consequence, in the list of precision parameter, you rarely find actual stepsize. You find rather parameters
representing the ratio between a stepsize and a physical quantity computed for each cosmology.
Modifying the code
Implementing a new idea completly from scratch would be rather intimidating, even for the main developpers of C←-
LASS. Fortunately, we never have to work from scratch. Usually we want to code a new species, a new observable,
a new approximation scheme, etc. The trick is to think of another species, observable, approximation scheme, etc.,
looking as close as possible to the new one.
Then, playing with the grep command and the search command of your editor, search for all occurences of this
nearest-as-possible other feature. This is usually easy thanks to our naming scheme. For each species, observable,
approximation scheme, etc., we usually use the same sequence of few letters everywhere (fo instance, fld for the
fluid usually representing Dark Energy). Grep for fld and you'll get all the lines related to the fluid. There is another
way: we use everywhere some conditional jumps related to a given feature. For instance, the lines related to the fluid
are always in between if (pba->has_fld == _TRUE_) { ... } and the lines related to the cosmic
shear observables are always in between if (ppt->has_lensing_potential == _TRUE_) { ...
}. Locating these flags and conditional jumps shows you all the parts related to a given feature/ingredient.
Once you have localised your nearest-as-possible other feature, you can copy/paste these lines and adapt them to
the case of your new feature! You are then sure that you didn't miss any step, even the smallest technical steps
(definition of indices, etc.)
Units
Internally, the code uses almost everywhere units of Mpc to some power, excepted in the inflation module, where
many quantities are in natural units (wrt the true Planck mass).
Generated by Doxygen
Chapter 4
Data Structure Documentation
4.1 nonlinear Struct Reference
#include <nonlinear.h>
Data Fields
- input parameters initialized by user in input module
(all other quantitites are computed in this module, given these parameters and the content of the 'precision',
'background', 'thermo', 'primordial' and 'spectra' structures)
• enum non_linear_method method
- table non-linear corrections for matter density, sqrt(P_NL(k,z)/P_NL(k,z))
• int pk_size
• int index_pk_m
• int index_pk_cb
• short has_pk_cb
• int k_size
• double ∗k
• int tau_size
• double ∗tau
• double ∗∗ nl_corr_density
• double ∗∗ k_nl
• int index_tau_min_nl
• double ∗nl_corr_density
• double ∗k_nl
- parameters for the pk_eq method
• short has_pk_eq
• int index_pk_eq_w
• int index_pk_eq_Omega_m
• int pk_eq_size
• int pk_eq_tau_size
• double ∗pk_eq_tau
• double ∗pk_eq_w_and_Omega
• double ∗pk_eq_ddw_and_ddOmega
- technical parameters
• short nonlinear_verbose
• ErrorMsg error_message

16 Data Structure Documentation
4.1.1 Detailed Description
Structure containing all information on non-linear spectra.
Once initialized by nonlinear_init(), contains a table for all two points correlation functions and for all the ai,bj func-
tions (containing the three points correlation functions), for each time and wave-number.
Structure containing all information on non-linear spectra.
Once initialised by nonlinear_init(), contains a table for all two points correlation functions and for all the ai,bj func-
tions (containing the three points correlation functions), for each time and wave-number.
4.1.2 Field Documentation
4.1.2.1 method
enum non_linear_method nonlinear::method
method for computing non-linear corrections (none, Halogit, etc.)
4.1.2.2 pk_size
int nonlinear::pk_size
k_size = total number of pk: 1 (P_m) if no massive neutrinos, 2 (P_m and P_cb) if massive neutrinos are present
4.1.2.3 k_size
int nonlinear::k_size
calculate P(k) with only cold dark matter and baryons k_size = total number of k values
k_size = total number of k values
4.1.2.4 k
double ∗nonlinear::k
k[index_k] = list of k values
4.1.2.5 tau_size
int nonlinear::tau_size
tau_size = number of values
Generated by Doxygen

4.1 nonlinear Struct Reference 17
4.1.2.6 tau
double ∗nonlinear::tau
tau[index_tau] = list of time values
4.1.2.7 nl_corr_density [1/2]
double ∗nonlinear::nl_corr_density
nl_corr_density[index_pk][index_tau ∗ppt->k_size + index_k]
nl_corr_density[index_tau ∗ppt->k_size + index_k]
4.1.2.8 k_nl [1/2]
double ∗nonlinear::k_nl
wavenumber at which non-linear corrections become important, defined differently by different non_linear_method's
4.1.2.9 index_tau_min_nl
int nonlinear::index_tau_min_nl
index of smallest value of tau at which nonlinear corrections have been computed (so, for tau<tau_min_nl, the array
nl_corr_density only contains some factors 1
4.1.2.10 has_pk_eq
short nonlinear::has_pk_eq
flag: will we use the pk_eq method?
4.1.2.11 index_pk_eq_w
int nonlinear::index_pk_eq_w
index of w in table pk_eq_w_and_Omega
4.1.2.12 index_pk_eq_Omega_m
int nonlinear::index_pk_eq_Omega_m
index of Omega_m in table pk_eq_w_and_Omega
Generated by Doxygen

18 Data Structure Documentation
4.1.2.13 pk_eq_size
int nonlinear::pk_eq_size
number of indices in table pk_eq_w_and_Omega
4.1.2.14 pk_eq_tau_size
int nonlinear::pk_eq_tau_size
number of times (and raws in table pk_eq_w_and_Omega)
4.1.2.15 pk_eq_tau
double∗nonlinear::pk_eq_tau
table of time values
4.1.2.16 pk_eq_w_and_Omega
double∗nonlinear::pk_eq_w_and_Omega
table of background quantites
4.1.2.17 pk_eq_ddw_and_ddOmega
double∗nonlinear::pk_eq_ddw_and_ddOmega
table of second derivatives
4.1.2.18 nonlinear_verbose
short nonlinear::nonlinear_verbose
amount of information written in standard output
4.1.2.19 error_message
ErrorMsg nonlinear::error_message
zone for writing error messages
4.1.2.20 nl_corr_density [2/2]
double∗nonlinear::nl_corr_density
nl_corr_density[index_tau ∗ppt->k_size + index_k]
4.1.2.21 k_nl [2/2]
double∗nonlinear::k_nl
wavenumber at which non-linear corrections become important, defined differently by different non_linear_method's
The documentation for this struct was generated from the following files:
•nonlinear.h
• nonlinear_conflict-20170920-150212.h
• nonlinear_exp.h
• nonlinear_test.h
Generated by Doxygen
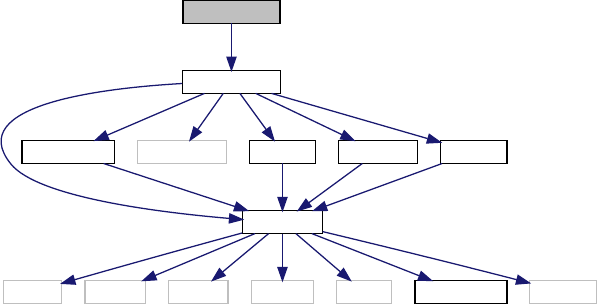
Chapter 5
File Documentation
5.1 background.c File Reference
#include "background.h"
Include dependency graph for background.c:
background.c
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.h parser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
Functions
• int background_at_tau (struct background ∗pba, double tau, short return_format, short intermode, int ∗last←-
_index, double ∗pvecback)
• int background_tau_of_z (struct background ∗pba, double z, double ∗tau)
• int background_functions (struct background ∗pba, double ∗pvecback_B, short return_format, double
∗pvecback)
• int background_w_fld (struct background ∗pba, double a, double ∗w_fld, double ∗dw_over_da_fld, double
∗integral_fld)
• int background_init (struct precision ∗ppr, struct background ∗pba)
• int background_free (struct background ∗pba)
• int background_free_noinput (struct background ∗pba)
• int background_free_input (struct background ∗pba)

20 File Documentation
• int background_indices (struct background ∗pba)
• int background_ncdm_distribution (void ∗pbadist, double q, double ∗f0)
• int background_ncdm_test_function (void ∗pbadist, double q, double ∗test)
• int background_ncdm_init (struct precision ∗ppr, struct background ∗pba)
• int background_ncdm_momenta (double ∗qvec, double ∗wvec, int qsize, double M, double factor, double z,
double ∗n, double ∗rho, double ∗p, double ∗drho_dM, double ∗pseudo_p)
• int background_ncdm_M_from_Omega (struct precision ∗ppr, struct background ∗pba, int n_ncdm)
• int background_solve (struct precision ∗ppr, struct background ∗pba)
• int background_initial_conditions (struct precision ∗ppr, struct background ∗pba, double ∗pvecback, double
∗pvecback_integration)
• int background_find_equality (struct precision ∗ppr, struct background ∗pba)
• int background_output_titles (struct background ∗pba, char titles[_MAXTITLESTRINGLENGTH_])
• int background_output_data (struct background ∗pba, int number_of_titles, double ∗data)
• int background_derivs (double tau, double ∗y, double ∗dy, void ∗parameters_and_workspace, ErrorMsg
error_message)
• double V_e_scf (struct background ∗pba, double phi)
• double V_p_scf (struct background ∗pba, double phi)
• double V_scf (struct background ∗pba, double phi)
5.1.1 Detailed Description
Documented background module
• Julien Lesgourgues, 17.04.2011
• routines related to ncdm written by T. Tram in 2011
Deals with the cosmological background evolution. This module has two purposes:
• at the beginning, to initialize the background, i.e. to integrate the background equations, and store all back-
ground quantities as a function of conformal time inside an interpolation table.
• to provide routines which allow other modules to evaluate any background quantity for a given value of the
conformal time (by interpolating within the interpolation table), or to find the correspondence between redshift
and conformal time.
The overall logic in this module is the following:
1. most background parameters that we will call {A} (e.g. rho_gamma, ..) can be expressed as simple analytical
functions of a few variables that we will call {B} (in simplest models, of the scale factor 'a'; in extended
cosmologies, of 'a' plus e.g. (phi, phidot) for quintessence, or some temperature for exotic particles, etc...).
2. in turn, quantities {B} can be found as a function of conformal time by integrating the background equations.
3. some other quantities that we will call {C} (like e.g. the sound horizon or proper time) also require an integra-
tion with respect to time, that cannot be inferred analytically from parameters {B}.
So, we define the following routines:
•background_functions() returns all background quantities {A} as a function of quantities {B}.
Generated by Doxygen

5.1 background.c File Reference 21
•background_solve() integrates the quantities {B} and {C} with respect to conformal time; this integration re-
quires many calls to background_functions().
• the result is stored in the form of a big table in the background structure. There is one column for conformal
time 'tau'; one or more for quantities {B}; then several columns for quantities {A} and {C}.
Later in the code, if we know the variables {B} and need some quantity {A}, the quickest and most precise way is to
call directly background_functions() (for instance, in simple models, if we want H at a given value of the scale factor).
If we know 'tau' and want any other quantity, we can call background_at_tau(), which interpolates in the table and
returns all values. Finally it can be useful to get 'tau' for a given redshift 'z': this can be done with background_←-
tau_of_z(). So if we are somewhere in the code, knowing z and willing to get background quantities, we should call
first background_tau_of_z() and then background_at_tau().
In order to save time, background_at_tau() can be called in three modes: short_info, normal_info, long_info (return-
ing only essential quantities, or useful quantities, or rarely useful quantities). Each line in the interpolation table is a
vector whose first few elements correspond to the short_info format; a larger fraction contribute to the normal format;
and the full vector corresponds to the long format. The guideline is that short_info returns only geometric quanti-
ties like a, H, H'; normal format returns quantities strictly needed at each step in the integration of perturbations;
long_info returns quantities needed only occasionally.
In summary, the following functions can be called from other modules:
1. background_init() at the beginning
2. background_at_tau(),background_tau_of_z() at any later time
3. background_free() at the end, when no more calls to the previous functions are needed
5.1.2 Function Documentation
5.1.2.1 background_at_tau()
int background_at_tau (
struct background ∗pba,
double tau,
short return_format,
short intermode,
int ∗last_index,
double ∗pvecback )
Background quantities at given conformal time tau.
Evaluates all background quantities at a given value of conformal time by reading the pre-computed table and
interpolating.
Parameters
pba Input: pointer to background structure (containing pre-computed table)
tau Input: value of conformal time
return_format Input: format of output vector (short, normal, long)
intermode Input: interpolation mode (normal or closeby)
last_index Input/Output: index of the previous/current point in the interpolation array (input only for
closeby mode, output for both)
pvecback Output: vector (assumed to be already allocated)
Generated by Doxygen

22 File Documentation
Returns
the error status
Summary:
• define local variables
• check that tau is in the pre-computed range
• deduce length of returned vector from format mode
• interpolate from pre-computed table with array_interpolate() or array_interpolate_growing_closeby() (depend-
ing on interpolation mode)
5.1.2.2 background_tau_of_z()
int background_tau_of_z (
struct background ∗pba,
double z,
double ∗tau )
Conformal time at given redshift.
Returns tau(z) by interpolation from pre-computed table.
Parameters
pba Input: pointer to background structure
zInput: redshift
tau Output: conformal time
Returns
the error status
Summary:
• define local variables
• check that zis in the pre-computed range
• interpolate from pre-computed table with array_interpolate()
Generated by Doxygen

5.1 background.c File Reference 23
5.1.2.3 background_functions()
int background_functions (
struct background ∗pba,
double ∗pvecback_B,
short return_format,
double ∗pvecback )
Background quantities at given a.
Function evaluating all background quantities which can be computed analytically as a function of {B} parameters
such as the scale factor 'a' (see discussion at the beginning of this file). In extended cosmological models, the
pvecback_B vector contains other input parameters than just 'a', e.g. (phi, phidot) for quintessence, some tempera-
ture of exotic relics, etc...
Parameters
pba Input: pointer to background structure
pvecback_B Input: vector containing all {B} type quantities (scale factor, ...)
return_format Input: format of output vector
pvecback Output: vector of background quantities (assumed to be already allocated)
Returns
the error status
Summary:
• define local variables
• initialize local variables
• pass value of ato output
• compute each component's density and pressure
• compute expansion rate H from Friedmann equation: this is the only place where the Friedmann equation is
assumed. Remember that densities are all expressed in units of [3c2/8πG], ie ρclass = [8πGρphysical/3c2]
• compute derivative of H with respect to conformal time
• compute relativistic density to total density ratio
• compute other quantities in the exhaustive, redundant format
• compute critical density
• compute Omega_m
Generated by Doxygen

24 File Documentation
5.1.2.4 background_w_fld()
int background_w_fld (
struct background ∗pba,
double a,
double ∗w_fld,
double ∗dw_over_da_fld,
double ∗integral_fld )
Single place where the fluid equation of state is defined. Parameters of the function are passed through the back-
ground structure. Generalisation to arbitrary functions should be simple.
Generated by Doxygen

5.1 background.c File Reference 25
Parameters
pba Input: pointer to background structure
aInput: current value of scale factor
w_fld Output: equation of state parameter w_fld(a)
dw_over_da_fld Output: function dw_fld/da
integral_fld Output: function Ra0
ada3(1 + wfld)/a
Returns
the error status
• first, define the function w(a)
• then, give the corresponding analytic derivative dw/da (used by perturbation equations; we could compute it
numerically, but with a loss of precision; as long as there is a simple analytic expression of the derivative of
the previous function, let's use it!
• finally, give the analytic solution of the following integral: Ra0
ada3(1 + wfld)/a. This is used in only one
place, in the initial conditions for the background, and with a=a_ini. If your w(a) does not lead to a simple
analytic solution of this integral, no worry: instead of writing something here, the best would then be to leave
it equal to zero, and then in background_initial_conditions() you should implement a numerical calculation of
this integral only for a=a_ini, using for instance Romberg integration. It should be fast, simple, and accurate
enough.
note: of course you can generalise these formulas to anything, defining new parameters pba->w..._fld. Just re-
member that so far, HyRec explicitely assumes that w(a)= w0 + wa (1-a/a0); but Recfast does not assume anything
5.1.2.5 background_init()
int background_init (
struct precision ∗ppr,
struct background ∗pba )
Initialize the background structure, and in particular the background interpolation table.
Parameters
ppr Input: pointer to precision structure
pba Input/Output: pointer to initialized background structure
Returns
the error status
Summary:
• define local variables
Generated by Doxygen
26 File Documentation
• in verbose mode, provide some information
• if shooting failed during input, catch the error here
• assign values to all indices in vectors of background quantities with background_indices()
• control that cosmological parameter values make sense
• this function integrates the background over time, allocates and fills the background table
• this function finds and stores a few derived parameters at radiation-matter equality
5.1.2.6 background_free()
int background_free (
struct background ∗pba )
Free all memory space allocated by background_init().
Parameters
pba Input: pointer to background structure (to be freed)
Returns
the error status
Here is the call graph for this function:
background_free background_free_input
5.1.2.7 background_free_noinput()
int background_free_noinput (
struct background ∗pba )
Free only the memory space NOT allocated through input_read_parameters()
Generated by Doxygen
5.1 background.c File Reference 27
Parameters
pba Input: pointer to background structure (to be freed)
Returns
the error status
5.1.2.8 background_free_input()
int background_free_input (
struct background ∗pba )
Free pointers inside background structure which were allocated in input_read_parameters()
Parameters
pba Input: pointer to background structure
Returns
the error status
Here is the caller graph for this function:
background_free_input background_free
5.1.2.9 background_indices()
int background_indices (
struct background ∗pba )
Assign value to each relevant index in vectors of background quantities.
Parameters
pba Input: pointer to background structure
Generated by Doxygen
28 File Documentation
Returns
the error status
Summary:
• define local variables
• initialize all flags: which species are present?
• initialize all indices
5.1.2.10 background_ncdm_distribution()
int background_ncdm_distribution (
void ∗pbadist,
double q,
double ∗f0 )
This is the routine where the distribution function f0(q) of each ncdm species is specified (it is the only place to
modify if you need a partlar f0(q))
Parameters
pbadist Input: structure containing all parameters defining f0(q)
qInput: momentum
f0 Output: phase-space distribution
• extract from the input structure pbadist all the relevant information
• shall we interpolate in file, or shall we use analytical formula below?
• a) deal first with the case of interpolating in files
• b) deal now with case of reading analytical function
Next enter your analytic expression(s) for the p.s.d.'s. If you need different p.s.d.'s for different species, put each
p.s.d inside a condition, like for instance: if (n_ncdm==2) {∗f0=...}. Remember that n_ncdm = 0 refers to the first
species.
This form is only appropriate for approximate studies, since in reality the chemical potentials are associated with
flavor eigenstates, not mass eigenstates. It is easy to take this into account by introducing the mixing angles. In the
later part (not read by the code) we illustrate how to do this.
5.1.2.11 background_ncdm_test_function()
int background_ncdm_test_function (
void ∗pbadist,
double q,
double ∗test )
This function is only used for the purpose of finding optimal quadrature weights. The logic is: if we can accurately
convolve f0(q) with this function, then we can convolve it accurately with any other relevant function.
Generated by Doxygen

5.1 background.c File Reference 29
Parameters
pbadist Input: structure containing all background parameters
qInput: momentum
test Output: value of the test function test(q)
Using a + bq creates problems for otherwise acceptable distributions which diverges as 1/r or 1/r2for r→0
5.1.2.12 background_ncdm_init()
int background_ncdm_init (
struct precision ∗ppr,
struct background ∗pba )
This function finds optimal quadrature weights for each ncdm species
Parameters
ppr Input: precision structure
pba Input/Output: background structure
Automatic q-sampling for this species
- in verbose mode, inform user of number of sampled momenta
for background quantities
Manual q-sampling for this species. Same sampling used for both perturbation and background sampling, since this
will usually be a high precision setting anyway
• in verbose mode, inform user of number of sampled momenta for background quantities
5.1.2.13 background_ncdm_momenta()
int background_ncdm_momenta (
double ∗qvec,
double ∗wvec,
int qsize,
double M,
double factor,
double z,
double ∗n,
double ∗rho,
double ∗p,
double ∗drho_dM,
double ∗pseudo_p )
For a given ncdm species: given the quadrature weights, the mass and the redshift, find background quantities by
a quick weighted sum over. Input parameters passed as NULL pointers are not evaluated for speed-up
Generated by Doxygen

30 File Documentation
Parameters
qvec Input: sampled momenta
wvec Input: quadrature weights
qsize Input: number of momenta/weights
MInput: mass
factor Input: normalization factor for the p.s.d.
zInput: redshift
nOutput: number density
rho Output: energy density
pOutput: pressure
drho_dM Output: derivative used in next function
pseudo←-
_p
Output: pseudo-pressure used in perturbation module for fluid approx
Summary:
• rescale normalization at given redshift
• initialize quantities
• loop over momenta
• adjust normalization
Here is the caller graph for this function:
background_ncdm_momenta background_ncdm_M_from
_Omega
5.1.2.14 background_ncdm_M_from_Omega()
int background_ncdm_M_from_Omega (
struct precision ∗ppr,
struct background ∗pba,
int n_ncdm )
When the user passed the density fraction Omega_ncdm or omega_ncdm in input but not the mass, infer the mass
with Newton iteration method.
Parameters
ppr Input: precision structure
pba Input/Output: background structure
n_ncdm Input: index of ncdm species Generated by Doxygen

5.1 background.c File Reference 31
Here is the call graph for this function:
background_ncdm_M_from
_Omega background_ncdm_momenta
5.1.2.15 background_solve()
int background_solve (
struct precision ∗ppr,
struct background ∗pba )
This function integrates the background over time, allocates and fills the background table
Parameters
ppr Input: precision structure
pba Input/Output: background structure
Summary:
• define local variables
• allocate vector of quantities to be integrated
• initialize generic integrator with initialize_generic_integrator()
• impose initial conditions with background_initial_conditions()
• create a growTable with gt_init()
• loop over integration steps: call background_functions(), find step size, save data in growTable with gt_add(),
perform one step with generic_integrator(), store new value of tau
• save last data in growTable with gt_add()
• clean up generic integrator with cleanup_generic_integrator()
• retrieve data stored in the growTable with gt_getPtr()
• interpolate to get quantities precisely today with array_interpolate()
• deduce age of the Universe
• allocate background tables
• In a loop over lines, fill background table using the result of the integration plus background_functions()
• free the growTable with gt_free()
Generated by Doxygen
32 File Documentation
• fill tables of second derivatives (in view of spline interpolation)
• compute remaining "related parameters"
–so-called "effective neutrino number", computed at earliest time in interpolation table. This should be
seen as a definition: Neff is the equivalent number of instantaneously-decoupled neutrinos accounting
for the radiation density, beyond photons
• done
5.1.2.16 background_initial_conditions()
int background_initial_conditions (
struct precision ∗ppr,
struct background ∗pba,
double ∗pvecback,
double ∗pvecback_integration )
Assign initial values to background integrated variables.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pvecback Input: vector of background quantities used as workspace
pvecback_integration Output: vector of background quantities to be integrated, returned with proper initial values
Returns
the error status
Summary:
• define local variables
• fix initial value of a
If we have ncdm species, perhaps we need to start earlier than the standard value for the species to be
relativistic. This could happen for some WDM models.
• We must add the relativistic contribution from NCDM species
–f is the critical density fraction of DR. The exact solution is:
f = -Omega_rad+pow(pow(Omega_rad,3./2.)+0.5∗pow(a/pba->a_today,6)∗pvecback←-
_integration[pba->index_bi_rho_dcdm]∗pba->Gamma_dcdm/pow(pba->H0,3),2./3.);
but it is not numerically stable for very small f which is always the case. Instead we use the Taylor expansion of this
equation, which is equivalent to ignoring f(a) in the Hubble rate.
There is also a space reserved for a future case where dr is not sourced by dcdm
Generated by Doxygen
5.1 background.c File Reference 33
• Fix initial value of φ, φ0set directly in the radiation attractor =>fixes the units in terms of rho_ur
TODO:
• There seems to be some small oscillation when it starts.
• Check equations and signs. Sign of phi_prime?
• is rho_ur all there is early on?
• –>If there is no attractor solution for scf_lambda, assign some value. Otherwise would give a nan.
• –>If no attractor initial conditions are assigned, gets the provided ones.
• compute initial proper time, assuming radiation-dominated universe since Big Bang and therefore t= 1/(2H)
(good approximation for most purposes)
• compute initial conformal time, assuming radiation-dominated universe since Big Bang and therefore τ=
1/(aH)(good approximation for most purposes)
• compute initial sound horizon, assuming cs= 1/√3initially
• set initial value of D and D' in RD. D will be renormalised later, but D' must be correct.
5.1.2.17 background_find_equality()
int background_find_equality (
struct precision ∗ppr,
struct background ∗pba )
Find the time of radiation/matter equality and store characteristic quantitites at that time in the background structure..
Parameters
ppr Input: pointer to precision structure
pba Input/Output: pointer to background structure
Returns
the error status
5.1.2.18 background_output_titles()
int background_output_titles (
struct background ∗pba,
char titles[_MAXTITLESTRINGLENGTH_] )
Subroutine for formatting background output
• Length of the column title should be less than OUTPUTPRECISION+6 to be indented correctly, but it can be
as long as .
Generated by Doxygen

34 File Documentation
5.1.2.19 background_output_data()
int background_output_data (
struct background ∗pba,
int number_of_titles,
double ∗data )
Stores quantities
5.1.2.20 background_derivs()
int background_derivs (
double tau,
double ∗y,
double ∗dy,
void ∗parameters_and_workspace,
ErrorMsg error_message )
Subroutine evaluating the derivative with respect to conformal time of quantities which are integrated (a, t, etc).
This is one of the few functions in the code which is passed to the generic_integrator() routine. Since generic_←-
integrator() should work with functions passed from various modules, the format of the arguments is a bit special:
• fixed input parameters and workspaces are passed through a generic pointer. Here, this is just a pointer to
the background structure and to a background vector, but generic_integrator() doesn't know its fine structure.
• the error management is a bit special: errors are not written as usual to pba->error_message, but to a generic
error_message passed in the list of arguments.
Parameters
tau Input: conformal time
yInput: vector of variable
dy Output: its derivative (already allocated)
parameters_and_workspace Input: pointer to fixed parameters (e.g. indices)
error_message Output: error message
Summary:
• define local variables
• calculate functions of awith background_functions()
• Short hand notation
• calculate a0=a2H
• calculate t0=a
• calculate rs0=cs
• solve second order growth equation [D00(τ) = −aHD0(τ)+3/2a2ρMD(τ)
• compute dcdm density ρ0=−3aHρ −aΓρ
Generated by Doxygen

5.1 background.c File Reference 35
• Compute dr density ρ0=−4aHρ −aΓρ
• Compute fld density ρ0=−3aH(1 + wfld(a))ρ
• Scalar field equation: φ00 + 2aHφ0+a2dV = 0 (note H is wrt cosmic time)
5.1.2.21 V_e_scf()
double V_e_scf (
struct background ∗pba,
double phi )
Scalar field potential and its derivatives with respect to the field _scf For Albrecht & Skordis model: 9908085
•V=Vpscf ∗Vescf
•Ve= exp(−λφ)(exponential)
•Vp= (φ−B)α+A(polynomial bump)
TODO:
• Add some functionality to include different models/potentials (tuning would be difficult, though)
• Generalize to Kessence/Horndeski/PPF and/or couplings
• A default module to numerically compute the derivatives when no analytic functions are given should be
added.
• Numerical derivatives may further serve as a consistency check.
The units of phi, tau in the derivatives and the potential V are the following:
• phi is given in units of the reduced Planck mass mpl = (8πG)(−1/2)
• tau in the derivative is given in units of Mpc.
• the potential V(φ)is given in units of m2
pl/Mpc2. With this convention, we have ρclass =
(8πG)/3ρphysical = 1/(3m2
pl)ρphysical = 1/3∗[1/(2a2)(φ0)2+V(φ)] and ρclass has the proper
dimension Mpc−2.
Here is the caller graph for this function:
V_e_scf V_scf
Generated by Doxygen

36 File Documentation
5.1.2.22 V_p_scf()
double V_p_scf (
struct background ∗pba,
double phi )
parameters and functions for the polynomial coefficient Vp= (φ−B)α+A(polynomial bump)
double scf_alpha = 2;
double scf_B = 34.8;
double scf_A = 0.01; (values for their Figure 2) Here is the caller graph for this function:
V_p_scf V_scf
5.1.2.23 V_scf()
double V_scf (
struct background ∗pba,
double phi )
Fianlly we can obtain the overall potential V=Vp∗VeHere is the call graph for this function:
V_scf
V_e_scf
V_p_scf
Generated by Doxygen
5.2 background.h File Reference 37
5.2 background.h File Reference
#include "common.h"
#include "quadrature.h"
#include "growTable.h"
#include "arrays.h"
#include "dei_rkck.h"
#include "parser.h"
Include dependency graph for background.h:
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.h parser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
This graph shows which files directly or indirectly include this file:
background.h
input.h
class.h
thermodynamics.h background.c
input.c
class.c
perturbations.hthermodynamics.c
primordial.h perturbations.c
nonlinear.h
nonlinear_conflict
-20170920-132422.h
nonlinear_conflict
-20170920-150212.h nonlinear_exp.h nonlinear_test.h primordial.c
transfer.h nonlinear.c
spectra.h transfer.c
lensing.h spectra.c
output.hlensing.c
output.c
Data Structures
• struct background
• struct background_parameters_and_workspace
• struct background_parameters_for_distributions
Generated by Doxygen
38 File Documentation
Enumerations
• enum spatial_curvature
• enum equation_of_state
5.2.1 Detailed Description
Documented includes for background module
5.2.2 Data Structure Documentation
5.2.2.1 struct background
All background parameters and evolution that other modules need to know.
Once initialized by the backgound_init(), contains all necessary information on the background evolution (except
thermodynamics), and in particular, a table of all background quantities as a function of time and scale factor, used
for interpolation in other modules.
Data Fields
double H0 H0: Hubble parameter (in fact, [ H0/c]) in Mpc−1
double Omega0_g Ω0γ: photons
double T_cmb Tcmb: current CMB temperature in Kelvins
double Omega0_b Ω0b: baryons
double Omega0_cdm Ω0cdm: cold dark matter
double Omega0_lambda Ω0Λ: cosmological constant
double Omega0_fld Ω0de: fluid
enum equation_of_state fluid_equation_of_state parametrisation scheme for fluid equation of state
double w0_fld w0DE : current fluid equation of state parameter
double wa_fld waDE : fluid equation of state parameter derivative
double Omega_EDE waDE : Early Dark Energy density parameter
double cs2_fld c2
s DE : sound speed of the fluid in the frame
comoving with the fluid (so, this is not [delta p/delta
rho] in the synchronous or newtonian gauge!!!)
short use_ppf flag switching on PPF perturbation equations
instead of true fluid equations for perturbations. It
could have been defined inside perturbation
structure, but we leave it here in such way to have
all fld parameters grouped.
double c_gamma_over_c_fld ppf parameter defined in eq. (16) of 0808.3125
[astro-ph]
double Omega0_ur Ω0νr : ultra-relativistic neutrinos
double Omega0_dcdmdr Ω0dcdm + Ω0dr: decaying cold dark matter (dcdm)
decaying to dark radiation (dr)
double Gamma_dcdm Γdcdm: decay constant for decaying cold dark
matter
double Omega_ini_dcdm Ωini,dcdm: rescaled initial value for dcdm density
(see 1407.2418 for definitions)
double Omega0_scf Ω0scf : scalar field
Generated by Doxygen
5.2 background.h File Reference 39
Data Fields
short attractor_ic_scf whether the scalar field has attractor initial
conditions
double phi_ini_scf φ(t0): scalar field initial value
double phi_prime_ini_scf dφ(t0)/dτ: scalar field initial derivative wrt
conformal time
double ∗scf_parameters list of parameters describing the scalar field
potential
int scf_parameters_size size of scf_parameters
int scf_tuning_index index in scf_parameters used for tuning
double Omega0_k Ω0k: curvature contribution
int N_ncdm Number of distinguishable ncdm species
double ∗M_ncdm vector of masses of non-cold relic: dimensionless
ratios m_ncdm/T_ncdm
double ∗Omega0_ncdm
double Omega0_ncdm_tot Omega0_ncdm for each species and for the total
Omega0_ncdm
double ∗deg_ncdm
double deg_ncdm_default vector of degeneracy parameters in factor of p-s-d:
1 for one family of neutrinos (= one neutrino plus its
anti-neutrino, total g∗=1+1=2, so deg = 0.5 g∗); and
its default value
double ∗T_ncdm
double T_ncdm_default list of 1st parameters in p-s-d of non-cold relics:
relative temperature T_ncdm1/T_gamma; and its
default value
double ∗ksi_ncdm
double ksi_ncdm_default list of 2nd parameters in p-s-d of non-cold relics:
relative chemical potential ksi_ncdm1/T_ncdm1;
and its default value
double ∗ncdm_psd_parameters list of parameters for specifying/modifying ncdm
p.s.d.'s, to be customized for given model (could be
e.g. mixing angles)
int ∗got_files list of flags for each species, set to true if p-s-d is
passed through file
char ∗ncdm_psd_files list of filenames for tabulated p-s-d
double h reduced Hubble parameter
double age age in Gyears
double conformal_age conformal age in Mpc
double K K: Curvature parameter K=−Ω0k∗a2
today ∗H2
0;
int sgnK K/|K|: -1, 0 or 1
double ∗m_ncdm_in_eV list of ncdm masses in eV (inferred from M_ncdm
and other parameters above)
double Neff so-called "effective neutrino number", computed at
earliest time in interpolation table
double Omega0_dcdm Ω0dcdm: decaying cold dark matter
double Omega0_dr Ω0dr: decay radiation
double a_eq scale factor at radiation/matter equality
double H_eq Hubble rate at radiation/matter equality [Mpc∧-1]
double z_eq redshift at radiation/matter equality
Generated by Doxygen

40 File Documentation
Data Fields
double tau_eq conformal time at radiation/matter equality [Mpc]
double a_today scale factor today (arbitrary and irrelevant for most
purposes)
int index_bg_a scale factor
int index_bg_H Hubble parameter in Mpc−1
int index_bg_H_prime its derivative w.r.t. conformal time
int index_bg_rho_g photon density
int index_bg_rho_b baryon density
int index_bg_rho_cdm cdm density
int index_bg_rho_lambda cosmological constant density
int index_bg_rho_fld fluid density
int index_bg_w_fld fluid equation of state
int index_bg_rho_ur relativistic neutrinos/relics density
int index_bg_rho_dcdm dcdm density
int index_bg_rho_dr dr density
int index_bg_phi_scf scalar field value
int index_bg_phi_prime_scf scalar field derivative wrt conformal time
int index_bg_V_scf scalar field potential V
int index_bg_dV_scf scalar field potential derivative V'
int index_bg_ddV_scf scalar field potential second derivative V''
int index_bg_rho_scf scalar field energy density
int index_bg_p_scf scalar field pressure
int index_bg_rho_ncdm1 density of first ncdm species (others contiguous)
int index_bg_p_ncdm1 pressure of first ncdm species (others contiguous)
int index_bg_pseudo_p_ncdm1 another statistical momentum useful in ncdma
approximation
int index_bg_Omega_r relativistic density fraction ( Ωγ+ Ωνr)
int index_bg_rho_crit critical density
int index_bg_Omega_m non-relativistic density fraction (
Ωb+ Ωcdm + Ωνnr )
int index_bg_conf_distance conformal distance (from us) in Mpc
int index_bg_ang_distance angular diameter distance in Mpc
int index_bg_lum_distance luminosity distance in Mpc
int index_bg_time proper (cosmological) time in Mpc
int index_bg_rs comoving sound horizon in Mpc
int index_bg_D scale independent growth factor D(a) for CDM
perturbations
int index_bg_f corresponding velocity growth factor [dlnD]/[dln a]
int bg_size_short size of background vector in the "short format"
int bg_size_normal size of background vector in the "normal format"
int bg_size size of background vector in the "long format"
int bt_size number of lines (i.e. time-steps) in the array
double ∗tau_table vector tau_table[index_tau] with values of τ
(conformal time)
double ∗z_table vector z_table[index_tau] with values of z(redshift)
Generated by Doxygen

5.2 background.h File Reference 41
Data Fields
double ∗background_table table background_table[index_tau∗pba->bg_←-
size+pba->index_bg] with all other quantities (array
of size bg_size∗bt_size)
double ∗d2tau_dz2_table vector d2tau_dz2_table[index_tau] with values of
d2τ/dz2(conformal time)
double ∗d2background_dtau2_table table d2background_dtau2_table[index_tau∗pba-
>bg_size+pba->index_bg] with values of d2bi/dτ2
(conformal time)
int index_bi_a {B} scale factor
int index_bi_rho_dcdm {B} dcdm density
int index_bi_rho_dr {B} dr density
int index_bi_rho_fld {B} fluid density
int index_bi_phi_scf {B} scalar field value
int index_bi_phi_prime_scf {B} scalar field derivative wrt conformal time
int index_bi_time {C} proper (cosmological) time in Mpc
int index_bi_rs {C} sound horizon
int index_bi_tau {C} conformal time in Mpc
int index_bi_D {C} scale independent growth factor D(a) for CDM
perturbations.
int index_bi_D_prime {C} D satisfies
[D00(τ) = −aHD0(τ)+3/2a2ρMD(τ)
int bi_B_size Number of {B} parameters
int bi_size Number of {B}+{C} parameters
short has_cdm presence of cold dark matter?
short has_dcdm presence of decaying cold dark matter?
short has_dr presence of relativistic decay radiation?
short has_scf presence of a scalar field?
short has_ncdm presence of non-cold dark matter?
short has_lambda presence of cosmological constant?
short has_fld presence of fluid with constant w and cs2?
short has_ur presence of ultra-relativistic neutrinos/relics?
short has_curvature presence of global spatial curvature?
int ∗ncdm_quadrature_strategy Vector of integers according to quadrature strategy.
int ∗ncdm_input_q_size Vector of numbers of q bins
double ∗ncdm_qmax Vector of maximum value of q
double ∗∗ q_ncdm_bg Pointers to vectors of background sampling in q
double ∗∗ w_ncdm_bg Pointers to vectors of corresponding quadrature
weights w
double ∗∗ q_ncdm Pointers to vectors of perturbation sampling in q
double ∗∗ w_ncdm Pointers to vectors of corresponding quadrature
weights w
double ∗∗ dlnf0_dlnq_ncdm Pointers to vectors of logarithmic derivatives of
p-s-d
int ∗q_size_ncdm_bg Size of the q_ncdm_bg arrays
int ∗q_size_ncdm Size of the q_ncdm arrays
double ∗factor_ncdm List of normalization factors for calculating energy
density etc.
short short_info flag for calling background_at_eta and return little
information
Generated by Doxygen
42 File Documentation
Data Fields
short normal_info flag for calling background_at_eta and return
medium information
short long_info flag for calling background_at_eta and return all
information
short inter_normal flag for calling background_at_eta and find position
in interpolation table normally
short inter_closeby flag for calling background_at_eta and find position
in interpolation table starting from previous position
in previous call
short shooting_failed flag is set to true if shooting failed.
ErrorMsg shooting_error Error message from shooting failed.
short background_verbose flag regulating the amount of information sent to
standard output (none if set to zero)
ErrorMsg error_message zone for writing error messages
5.2.2.2 struct background_parameters_and_workspace
temporary parameters and workspace passed to the background_derivs function
5.2.2.3 struct background_parameters_for_distributions
temporary parameters and workspace passed to phase space distribution function
5.2.3 Enumeration Type Documentation
5.2.3.1 spatial_curvature
enum spatial_curvature
list of possible types of spatial curvature
5.2.3.2 equation_of_state
enum equation_of_state
list of possible parametrisations of the DE equation of state
Generated by Doxygen
5.3 class.c File Reference 43
5.3 class.c File Reference
#include "class.h"
Include dependency graph for class.c:
class.c
class.h
stdio.h stdlib.h math.h string.h float.h
quadrature.h
common.h
growTable.h arrays.h dei_rkck.h parser.h
input.h
background.h
thermodynamics.h
perturbations.h
transfer.h
nonlinear.h
primordial.h
spectra.h
lensing.h
output.h
svnversion.hstdarg.h
evolver_ndf15.h
evolver_rkck.h
sparse.h
hyperspherical.h
sys/shm.hsys/stat.h errno.h
5.3.1 Detailed Description
Julien Lesgourgues, 17.04.2011
5.4 common.h File Reference
#include "stdio.h"
#include "stdlib.h"
#include "math.h"
#include "string.h"
#include "float.h"
#include "svnversion.h"
#include <stdarg.h>
Include dependency graph for common.h:
common.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
Generated by Doxygen
44 File Documentation
This graph shows which files directly or indirectly include this file:
common.h
arrays.h
background.h
input.h
class.h
output.h
quadrature.h dei_rkck.hparser.h
evolver_ndf15.h
sparse.h
hyperspherical.hthermodynamics.h background.c
input.c
class.c
perturbations.hthermodynamics.c
primordial.h perturbations.c
nonlinear.h
nonlinear_conflict
-20170920-132422.h
nonlinear_conflict
-20170920-150212.h nonlinear_exp.h nonlinear_test.h primordial.c
transfer.hnonlinear.c
spectra.htransfer.c
lensing.hspectra.c
lensing.c
output.c
evolver_rkck.h
Data Structures
• struct precision
Enumerations
• enum evolver_type
• enum pk_def {delta_m_squared,delta_tot_squared,delta_bc_squared,delta_tot_from_poisson_squared }
• enum file_format
5.4.1 Detailed Description
Generic libraries, parameters and functions used in the whole code.
5.4.2 Data Structure Documentation
5.4.2.1 struct precision
All precision parameters.
Includes integrations steps, flags telling how the computation is to be performed, etc.
Data Fields
double a_ini_over_a_today_default default initial value of scale factor in
background integration, in units of scale
factor today
Generated by Doxygen

5.4 common.h File Reference 45
Data Fields
double back_integration_stepsize default step d tau in background
integration, in units of conformal Hubble
time ( dτ = back_integration_stepsize / aH
)
double tol_background_integration parameter controlling precision of
background integration
double tol_initial_Omega_r parameter controlling how deep inside
radiation domination must the initial time
be chosen
double tol_M_ncdm parameter controlling relative precision of
ncdm mass for given ncdm current density
double tol_ncdm_newtonian parameter controlling relative precision of
integrals over ncdm phase-space
distribution during perturbation
calculation: value to be applied in
Newtonian gauge
double tol_ncdm_synchronous parameter controlling relative precision of
integrals over ncdm phase-space
distribution during perturbation
calculation: value to be applied in
synchronous gauge
double tol_ncdm parameter controlling relative precision of
integrals over ncdm phase-space
distribution during perturbation
calculation: value actually applied in
chosen gauge
double tol_ncdm_bg parameter controlling relative precision of
integrals over ncdm phase-space
distribution during background evolution
double tol_ncdm_initial_w parameter controlling how relativistic must
non-cold relics be at initial time
double safe_phi_scf parameter controlling the initial scalar field
in background functions
double tol_tau_eq parameter controlling precision with which
tau_eq (conformal time at radiation/matter
equality) is found (units: Mpc)
double recfast_z_initial initial redshift in recfast
int recfast_Nz0 number of integration steps
double tol_thermo_integration precision of each integration step
int recfast_Heswitch recfast 1.4 parameter
double recfast_fudge_He recfast 1.4 parameter
int recfast_Hswitch recfast 1.5 switching parameter
double recfast_fudge_H H fudge factor when recfast_Hswitch set to
false (v1.4 fudging)
double recfast_delta_fudge_H correction to H fudge factor in v1.5
double recfast_AGauss1 Amplitude of 1st Gaussian
double recfast_AGauss2 Amplitude of 2nd Gaussian
double recfast_zGauss1 ln(1+z) of 1st Gaussian
double recfast_zGauss2 ln(1+z) of 2nd Gaussian
double recfast_wGauss1 Width of 1st Gaussian
double recfast_wGauss2 Width of 2nd Gaussian
Generated by Doxygen
46 File Documentation
Data Fields
double recfast_z_He_1 down to which redshift Helium fully ionized
double recfast_delta_z_He_1 z range over which transition is smoothed
double recfast_z_He_2 down to which redshift first Helium
recombination not complete
double recfast_delta_z_He_2 z range over which transition is smoothed
double recfast_z_He_3 down to which redshift Helium singly
ionized
double recfast_delta_z_He_3 z range over which transition is smoothed
double recfast_x_He0_trigger value below which recfast uses the full
equation for Helium
double recfast_x_He0_trigger2 a second threshold used in derivative
routine
double recfast_x_He0_trigger_delta x_He range over which transition is
smoothed
double recfast_x_H0_trigger value below which recfast uses the full
equation for Hydrogen
double recfast_x_H0_trigger2 a second threshold used in derivative
routine
double recfast_x_H0_trigger_delta x_H range over which transition is
smoothed
double recfast_H_frac governs time at which full equation of
evolution for Tmat is used
double reionization_z_start_max maximum redshift at which reionization
should start. If not, return an error.
double reionization_sampling control stepsize in z during reionization
double reionization_optical_depth_tol fractional error on optical_depth
double reionization_start_factor parameter for CAMB-like parametrization
int thermo_rate_smoothing_radius plays a minor (almost aesthetic) role in the
definition of the variation rate of
thermodynamical quantities
enum evolver_type evolver which type of evolver for integrating
perturbations (Runge-Kutta? Stiff?...)
double k_min_tau0 number defining k_min for the computation
of Cl's and P(k)'s (dimensionless): (k_min
tau_0), usually chosen much smaller than
one
double k_max_tau0_over_l_max number defining k_max for the
computation of Cl's (dimensionless):
(k_max tau_0)/l_max, usually chosen
around two
double k_step_sub step in k space, in units of one period of
acoustic oscillation at decoupling, for
scales inside sound horizon at decoupling
double k_step_super step in k space, in units of one period of
acoustic oscillation at decoupling, for
scales above sound horizon at decoupling
double k_step_transition dimensionless number regulating the
transition from 'sub' steps to 'super' steps.
Decrease for more precision.
double k_step_super_reduction the step k_step_super is reduced by this
amount in the k–>0 limit (below scale of
Hubble and/or curvature radius)
Generated by Doxygen
5.4 common.h File Reference 47
Data Fields
double k_per_decade_for_pk if values needed between kmax inferred
from k_oscillations and k_kmax_for_pk,
this gives the number of k per decade
outside the BAO region
double k_per_decade_for_bao if values needed between kmax inferred
from k_oscillations and k_kmax_for_pk,
this gives the number of k per decade
inside the BAO region (for finer sampling)
double k_bao_center in ln(k) space, the central value of the BAO
region where sampling is finer is defined
as k_rec times this number
(recommended: 3, i.e. finest sampling
near 3rd BAO peak)
double k_bao_width in ln(k) space, width of the BAO region
where sampling is finer: this number gives
roughly the number of BAO oscillations
well resolved on both sides of the central
value (recommended: 4, i.e. finest
sampling from before first up to 3+4=7th
peak)
double start_small_k_at_tau_c_over_tau_h largest wavelengths start being sampled
when universe is sufficiently opaque. This
is quantified in terms of the ratio of thermo
to hubble time scales, τc/τH. Start when
start_largek_at_tau_c_over_tau_h equals
this ratio. Decrease this value to start
integrating the wavenumbers earlier in
time.
double start_large_k_at_tau_h_over_tau_k largest wavelengths start being sampled
when mode is sufficiently outside Hubble
scale. This is quantified in terms of the
ratio of hubble time scale to wavenumber
time scale, τh/τkwhich is roughly equal to
(k∗tau). Start when this ratio equals
start_large_k_at_tau_k_over_tau_h.
Decrease this value to start integrating the
wavenumbers earlier in time.
double tight_coupling_trigger_tau_c_over_tau_h when to switch off tight-coupling
approximation: first condition: τc/τH>
tight_coupling_trigger_tau_c_over_tau_h.
Decrease this value to switch off earlier in
time. If this number is larger than
start_sources_at_tau_c_over_tau_h, the
code returns an error, because the source
computation requires tight-coupling to be
switched off.
double tight_coupling_trigger_tau_c_over_tau_k when to switch off tight-coupling
approximation: second condition:
τc/τk≡kτc<
tight_coupling_trigger_tau_c_over_tau_k.
Decrease this value to switch off earlier in
time.
Generated by Doxygen
48 File Documentation
Data Fields
double start_sources_at_tau_c_over_tau_h sources start being sampled when
universe is sufficiently opaque. This is
quantified in terms of the ratio of thermo to
hubble time scales, τc/τH. Start when
start_sources_at_tau_c_over_tau_h
equals this ratio. Decrease this value to
start sampling the sources earlier in time.
int tight_coupling_approximation method for tight coupling approximation
int l_max_g number of momenta in Boltzmann
hierarchy for photon temperature (scalar),
at least 4
int l_max_pol_g number of momenta in Boltzmann
hierarchy for photon polarization (scalar),
at least 4
int l_max_dr number of momenta in Boltzmann
hierarchy for decay radiation, at least 4
int l_max_ur number of momenta in Boltzmann
hierarchy for relativistic neutrino/relics
(scalar), at least 4
int l_max_ncdm number of momenta in Boltzmann
hierarchy for relativistic neutrino/relics
(scalar), at least 4
int l_max_g_ten number of momenta in Boltzmann
hierarchy for photon temperature (tensor),
at least 4
int l_max_pol_g_ten number of momenta in Boltzmann
hierarchy for photon polarization (tensor),
at least 4
double curvature_ini initial condition for curvature for adiabatic
double entropy_ini initial condition for entropy perturbation for
isocurvature
double gw_ini initial condition for tensor metric
perturbation h
double perturb_integration_stepsize default step dτ in perturbation integration,
in units of the timescale involved in the
equations (usually, the min of 1/k,1/aH,
1/˙κ)
double perturb_sampling_stepsize default step dτ for sampling the source
function, in units of the timescale involved
in the sources: ( ˙κ−¨κ/ ˙κ)−1
double tol_perturb_integration control parameter for the precision of the
perturbation integration
double tol_tau_approx precision with which the code should
determine (by bisection) the times at
which sources start being sampled, and at
which approximations must be switched
on/off (units of Mpc)
int radiation_streaming_approximation method for switching off photon
perturbations
Generated by Doxygen
5.4 common.h File Reference 49
Data Fields
double radiation_streaming_trigger_tau_over_tau_k when to switch off photon perturbations, ie
when to switch on photon free-streaming
approximation (keep density and thtau, set
shear and higher momenta to zero): first
condition: kτ > radiation_streaming_←-
trigger_tau_h_over_tau_k
double radiation_streaming_trigger_tau_c_over_tau when to switch off photon perturbations, ie
when to switch on photon free-streaming
approximation (keep density and theta, set
shear and higher momenta to zero):
second condition:
int ur_fluid_approximation method for ultra relativistic fluid
approximation
double ur_fluid_trigger_tau_over_tau_k when to switch off ur (massless neutrinos /
ultra-relativistic relics) fluid approximation
int ncdm_fluid_approximation method for non-cold dark matter fluid
approximation
double ncdm_fluid_trigger_tau_over_tau_k when to switch off ncdm (massive
neutrinos / non-cold relics) fluid
approximation
double neglect_CMB_sources_below_visibility whether CMB source functions can be
approximated as zero when visibility
function g(tau) is tiny
double k_per_decade_primordial logarithmic sampling for primordial spectra
(number of points per decade in k space)
double primordial_inflation_ratio_min for each k, start following wavenumber
when aH = k/primordial_inflation_ratio_min
double primordial_inflation_ratio_max for each k, stop following wavenumber, at
the latest, when aH =
k/primordial_inflation_ratio_max
int primordial_inflation_phi_ini_maxit maximum number of iteration when
searching a suitable initial field value
phi_ini (value reached when no
long-enough slow-roll period before the
pivot scale)
double primordial_inflation_pt_stepsize controls the integration timestep for
inflaton perturbations
double primordial_inflation_bg_stepsize controls the integration timestep for
inflaton background
double primordial_inflation_tol_integration controls the precision of the ODE
integration during inflation
double primordial_inflation_attractor_precision_pivot targeted precision when searching
attractor solution near phi_pivot
double primordial_inflation_attractor_precision_initial targeted precision when searching
attractor solution near phi_ini
int primordial_inflation_attractor_maxit maximum number of iteration when
searching attractor solution
double primordial_inflation_tol_curvature for each k, stop following wavenumber, at
the latest, when curvature perturbation R
is stable up to to this tolerance
double primordial_inflation_aH_ini_target control the step size in the search for a
suitable initial field value
Generated by Doxygen
50 File Documentation
Data Fields
double primordial_inflation_end_dphi first bracketing width, when trying to
bracket the value phi_end at which
inflation ends naturally
double primordial_inflation_end_logstep logarithmic step for updating the
bracketing width, when trying to bracket
the value phi_end at which inflation ends
naturally
double primordial_inflation_small_epsilon value of slow-roll parameter epsilon used
to define a field value phi_end close to the
end of inflation (doesn't need to be exactly
at the end):
epsilon(phi_end)=small_epsilon (should
be smaller than one)
double primordial_inflation_small_epsilon_tol tolerance in the search for phi_end
double primordial_inflation_extra_efolds a small number of efolds, irrelevant at the
end, used in the search for the pivot scale
(backward from the end of inflation)
int l_linstep factor for logarithmic spacing of values of l
over which bessel and transfer functions
are sampled
double l_logstep maximum spacing of values of l over which
Bessel and transfer functions are sampled
(so, spacing becomes linear instead of
logarithmic at some point)
double hyper_x_min flat case: lower bound on the smallest
value of x at which we sample Φν
l(x)or
jl(x)
double hyper_sampling_flat flat case: number of sampled points x per
approximate wavelength 2π
double hyper_sampling_curved_low_nu open/closed cases: number of sampled
points x per approximate wavelength
2π/ν, when νsmaller than
hyper_nu_sampling_step
double hyper_sampling_curved_high_nu open/closed cases: number of sampled
points x per approximate wavelength
2π/ν, when νgreater than
hyper_nu_sampling_step
double hyper_nu_sampling_step open/closed cases: value of nu at which
sampling changes
double hyper_phi_min_abs small value of Bessel function used in
calculation of first point x ( Φν
l(x)equals
hyper_phi_min_abs)
double hyper_x_tol tolerance parameter used to determine
first value of x
double hyper_flat_approximation_nu value of nu below which the flat
approximation is used to compute Bessel
function
double q_linstep asymptotic linear sampling step in q
space, in units of 2π/ra(τrec)(comoving
angular diameter distance to
recombination)
Generated by Doxygen

5.4 common.h File Reference 51
Data Fields
double q_logstep_spline initial logarithmic sampling step in q space,
in units of 2π/ra(τrec)(comoving angular
diameter distance to recombination)
double q_logstep_open in open models, the value of
q_logstep_spline must be decreased
according to curvature. Increasing this
number will make the calculation more
accurate for large positive Omega_k
double q_logstep_trapzd initial logarithmic sampling step in q space,
in units of 2π/ra(τrec)(comoving angular
diameter distance to recombination), in the
case of small q's in the closed case, for
which one must used trapezoidal
integration instead of spline (the number of
q's for which this is the case decreases
with curvature and vanishes in the flat
limit)
double q_numstep_transition number of steps for the transition from
q_logstep_trapzd steps to
q_logstep_spline steps (transition must be
smooth for spline)
double transfer_neglect_delta_k_S_t0 for temperature source function T0 of
scalar mode, range of k values (in 1/Mpc)
taken into account in transfer function: for l
<(k-delta_k)∗tau0, ie for k >(l/tau0 +
delta_k), the transfer function is set to zero
double transfer_neglect_delta_k_S_t1 same for temperature source function T1
of scalar mode
double transfer_neglect_delta_k_S_t2 same for temperature source function T2
of scalar mode
double transfer_neglect_delta_k_S_e same for polarization source function E of
scalar mode
double transfer_neglect_delta_k_V_t1 same for temperature source function T1
of vector mode
double transfer_neglect_delta_k_V_t2 same for temperature source function T2
of vector mode
double transfer_neglect_delta_k_V_e same for polarization source function E of
vector mode
double transfer_neglect_delta_k_V_b same for polarization source function B of
vector mode
double transfer_neglect_delta_k_T_t2 same for temperature source function T2
of tensor mode
double transfer_neglect_delta_k_T_e same for polarization source function E of
tensor mode
double transfer_neglect_delta_k_T_b same for polarization source function B of
tensor mode
double transfer_neglect_late_source value of l below which the CMB source
functions can be neglected at late time,
excepted when there is a Late ISW
contribution
double l_switch_limber when to use the Limber approximation for
project gravitational potential cl's
Generated by Doxygen

52 File Documentation
Data Fields
double l_switch_limber_for_nc_local_over_z when to use the Limber approximation for
local number count contributions to cl's
(relative to central redshift of each bin)
double l_switch_limber_for_nc_los_over_z when to use the Limber approximation for
number count contributions to cl's
integrated along the line-of-sight (relative
to central redshift of each bin)
double selection_cut_at_sigma in sigma units, where to cut gaussian
selection functions
double selection_sampling controls sampling of integral over time
when selection functions vary quicker than
Bessel functions. Increase for better
sampling.
double selection_sampling_bessel controls sampling of integral over time
when selection functions vary slower than
Bessel functions. Increase for better
sampling
double selection_sampling_bessel_los controls sampling of integral over time
when selection functions vary slower than
Bessel functions. This parameter is
specific to number counts contributions to
Cl integrated along the line of sight.
Increase for better sampling
double selection_tophat_edge controls how smooth are the edge of
top-hat window function (<<1 for very
sharp, 0.1 for sharp)
double halofit_min_k_nonlinear parameters relevant for HALOFIT
computation value of k in 1/Mpc below
which non-linear corrections will be
neglected
double halofit_min_k_max when halofit is used, k_max must be at
least equal to this value (otherwise halofit
could not find the scale of non-linearity).
Calculations are done internally until this
k_max, but the output is still controlled by
P_k_max_1/Mpc or P_k_max_h/Mpc even
if they are smaller
double halofit_k_per_decade halofit needs to evalute integrals (linear
power spectrum times some kernels).
They are sampled using this logarithmic
step size.
double halofit_sigma_precision a smaller value will lead to a more precise
halofit result at the highest redshift at
which halofit can make computations, at
the expense of requiring a larger k_max;
but this parameter is not relevant for the
precision on P_nl(k,z) at other redshifts, so
there is normally no need to change it
double halofit_tol_sigma tolerance required on sigma(R) when
matching the condition sigma(R_nl)=1,
whcih defines the wavenumber of
non-linearity, k_nl=1./R_nl
Generated by Doxygen

5.4 common.h File Reference 53
Data Fields
double pk_eq_z_max Maximum z until which the Pk_equal
method of 0810.0190 and 1601.07230 is
used
double pk_eq_tol tolerance for finding the equivalent models
of the pk_equal method
int accurate_lensing switch between Gauss-Legendre
quadrature integration and simple
quadrature on a subdomain of angles
int num_mu_minus_lmax difference between num_mu and l_max,
increase for more precision
int delta_l_max difference between l_max in unlensed and
lensed spectra
double tol_gauss_legendre tolerance with which quadrature points are
found: must be very small for an accurate
integration (if not entered manually, set
automatically to match machine precision)
double smallest_allowed_variation machine-dependent, assigned
automatically by the code
ErrorMsg error_message zone for writing error messages
5.4.3 Enumeration Type Documentation
5.4.3.1 evolver_type
enum evolver_type
parameters related to the precision of the code and to the method of calculation list of evolver types for integrating
perturbations over time
5.4.3.2 pk_def
enum pk_def
List of ways in which matter power spectrum P(k) can be defined. The standard definition is the first one (delta_←-
m_squared) but alternative definitions can be useful in some projects.
Enumerator
delta_m_squared normal definition (delta_m includes all non-relativistic species at late times)
delta_tot_squared delta_tot includes all species contributions to (delta rho), and only
non-relativistic contributions to rho
delta_bc_squared delta_bc includes contribution of baryons and cdm only to (delta rho) and
to rho
delta_tot_from_poisson_squared use delta_tot inferred from gravitational potential through Poisson equation
Generated by Doxygen
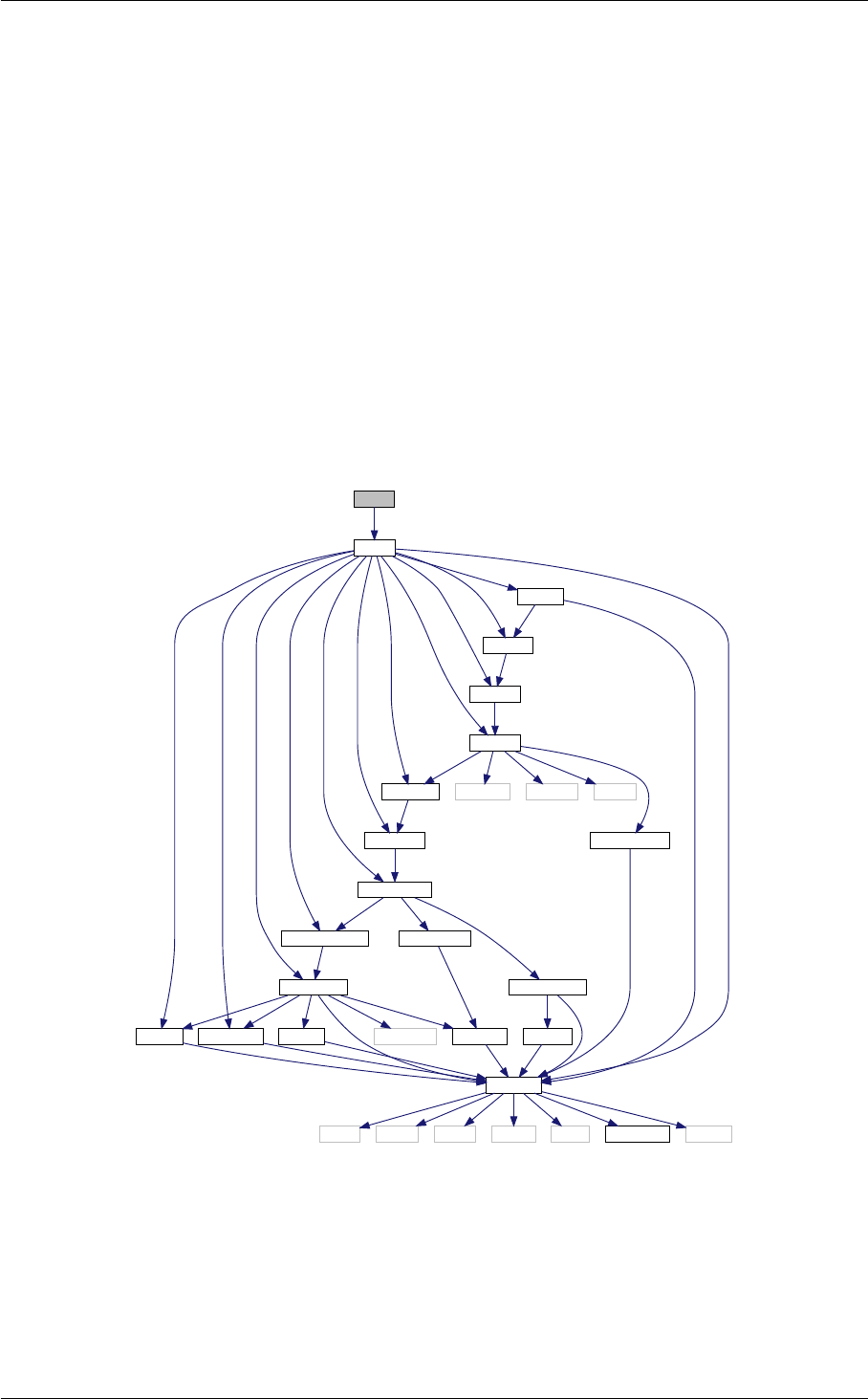
54 File Documentation
5.4.3.3 file_format
enum file_format
Different ways to present output files
5.5 input.c File Reference
#include "input.h"
Include dependency graph for input.c:
input.c
input.h
common.h
parser.h quadrature.h
background.h
thermodynamics.h
perturbations.h
transfer.h
nonlinear.h
primordial.h
spectra.h
lensing.h
output.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
growTable.harrays.h dei_rkck.h
evolver_ndf15.h
evolver_rkck.h
sparse.h
hyperspherical.h
sys/shm.h sys/stat.h errno.h
Functions
• int input_init_from_arguments (int argc, char ∗∗argv, struct precision ∗ppr, struct background ∗pba, struct
thermo ∗pth, struct perturbs ∗ppt, struct transfers ∗ptr, struct primordial ∗ppm, struct spectra ∗psp, struct
nonlinear ∗pnl, struct lensing ∗ple, struct output ∗pop, ErrorMsg errmsg)
Generated by Doxygen

5.5 input.c File Reference 55
• int input_init (struct file_content ∗pfc, struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct
perturbs ∗ppt, struct transfers ∗ptr, struct primordial ∗ppm, struct spectra ∗psp, struct nonlinear ∗pnl, struct
lensing ∗ple, struct output ∗pop, ErrorMsg errmsg)
• int input_read_parameters (struct file_content ∗pfc, struct precision ∗ppr, struct background ∗pba, struct
thermo ∗pth, struct perturbs ∗ppt, struct transfers ∗ptr, struct primordial ∗ppm, struct spectra ∗psp, struct
nonlinear ∗pnl, struct lensing ∗ple, struct output ∗pop, ErrorMsg errmsg)
• int input_default_params (struct background ∗pba, struct thermo ∗pth, struct perturbs ∗ppt, struct transfers
∗ptr, struct primordial ∗ppm, struct spectra ∗psp, struct nonlinear ∗pnl, struct lensing ∗ple, struct output ∗pop)
• int input_default_precision (struct precision ∗ppr)
• int get_machine_precision (double ∗smallest_allowed_variation)
• int class_fzero_ridder (int(∗func)(double x, void ∗param, double ∗y, ErrorMsg error_message), double x1,
double x2, double xtol, void ∗param, double ∗Fx1, double ∗Fx2, double ∗xzero, int ∗fevals, ErrorMsg error←-
_message)
• int input_try_unknown_parameters (double ∗unknown_parameter, int unknown_parameters_size, void
∗voidpfzw, double ∗output, ErrorMsg errmsg)
• int input_get_guess (double ∗xguess, double ∗dxdy, struct fzerofun_workspace ∗pfzw, ErrorMsg errmsg)
• int input_find_root (double ∗xzero, int ∗fevals, struct fzerofun_workspace ∗pfzw, ErrorMsg errmsg)
• int input_prepare_pk_eq (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct nonlinear
∗pnl, int input_verbose, ErrorMsg errmsg)
5.5.1 Detailed Description
Documented input module.
Julien Lesgourgues, 27.08.2010
5.5.2 Function Documentation
5.5.2.1 input_init_from_arguments()
int input_init_from_arguments (
int argc,
char ∗∗ argv,
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
struct transfers ∗ptr,
struct primordial ∗ppm,
struct spectra ∗psp,
struct nonlinear ∗pnl,
struct lensing ∗ple,
struct output ∗pop,
ErrorMsg errmsg )
Use this routine to extract initial parameters from files 'xxx.ini' and/or 'xxx.pre'. They can be the arguments of the
main() routine.
If class is embedded into another code, you will probably prefer to call directly input_init() in order to pass input
parameters through a 'file_content' structure. Summary:
Generated by Doxygen

56 File Documentation
• define local variables
• –>the final structure with all parameters
• –>a temporary structure with all input parameters
• –>a temporary structure with all precision parameters
• –>a temporary structure with only the root name
• –>sum of fc_inoput and fc_root
• –>a pointer to either fc_root or fc_inputroot
• Initialize the two file_content structures (for input parameters and precision parameters) to some null content.
If no arguments are passed, they will remain null and inform init_params() that all parameters take default
values.
• If some arguments are passed, identify eventually some 'xxx.ini' and 'xxx.pre' files, and store their name.
• if there is an 'xxx.ini' file, read it and store its content.
• check whether a root name has been set
• if root has not been set, use root=output/inputfilennameN_
• if there is an 'xxx.pre' file, read it and store its content.
• if one or two files were read, merge their contents in a single 'file_content' structure.
• Finally, initialize all parameters given the input 'file_content' structure. If its size is null, all parameters take
their default values.
5.5.2.2 input_init()
int input_init (
struct file_content ∗pfc,
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
struct transfers ∗ptr,
struct primordial ∗ppm,
struct spectra ∗psp,
struct nonlinear ∗pnl,
struct lensing ∗ple,
struct output ∗pop,
ErrorMsg errmsg )
Initialize each parameter, first to its default values, and then from what can be interpreted from the values passed
in the input 'file_content' structure. If its size is null, all parameters keep their default values. These two arrays must
contain the strings of names to be searched for and the corresponding new parameter
• Do we need to fix unknown parameters?
• –>input_auxillary_target_conditions() takes care of the case where for instance Omega_dcdmdr is set to
0.0.
• case with unknown parameters
Generated by Doxygen

5.5 input.c File Reference 57
• –>go through all cases with unknown parameters:
• –>Read all parameters from tuned pfc
• –>Set status of shooting
• –>Free arrays allocated
• case with no unknown parameters
• –>just read all parameters from input pfc:
• eventually write all the read parameters in a file, unread parameters in another file, and warnings about unread
parameters
5.5.2.3 input_read_parameters()
int input_read_parameters (
struct file_content ∗pfc,
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
struct transfers ∗ptr,
struct primordial ∗ppm,
struct spectra ∗psp,
struct nonlinear ∗pnl,
struct lensing ∗ple,
struct output ∗pop,
ErrorMsg errmsg )
Summary:
• define local variables
• set all parameters (input and precision) to default values
• if entries passed in file_content structure, carefully read and interpret each of them, and tune the relevant
input parameters accordingly
Knowing the gauge from the very beginning is useful (even if this could be a run not requiring perturbations at all:
even in that case, knowing the gauge is important e.g. for fixing the sampling in momentum space for non-cold dark
matter)
(a) background parameters
• scale factor today (arbitrary)
• h (dimensionless) and [ H0/c] in Mpc−1=h/2997.9... =h∗105/c
• Omega_0_g (photons) and T_cmb
• Omega0_g = rho_g / rho_c0, each of them expressed in Kg/m/s2
• rho_g = (4 sigma_B / c) T4
Generated by Doxygen

58 File Documentation
• rho_c0 = 3c2H2
0/(8πG)
• Omega_0_b (baryons)
• Omega_0_ur (ultra-relativistic species / massless neutrino)
• Omega_0_cdm (CDM)
• Omega_0_dcdmdr (DCDM)
• Read Omega_ini_dcdm or omega_ini_dcdm
• Read Gamma in same units as H0, i.e. km/(s Mpc)
• non-cold relics (ncdm)
• Omega_0_k (effective fractional density of curvature)
• Set curvature parameter K
• Set curvature sign
• Omega_0_lambda (cosmological constant), Omega0_fld (dark energy fluid), Omega0_scf (scalar field)
• –>(flag3 == FALSE)|| (param3 >= 0.) explained: it means that either we have not read Omega_scf
so we are ignoring it (unlike lambda and fld!) OR we have read it, but it had a positive value and should
not be used for filling. We now proceed in two steps: 1) set each Omega0 and add to the total for each
specified component. 2) go through the components in order {lambda, fld, scf} and fill using first unspecified
component.
• Test that the user have not specified Omega_scf = -1 but left either Omega_lambda or Omega_fld
unspecified:
• Read parameters describing scalar field potential
• Assign shooting parameter
(b) assign values to thermodynamics cosmological parameters
• primordial helium fraction
• recombination parameters
• reionization parametrization
• reionization parameters if reio_parametrization=reio_camb
• reionization parameters if reio_parametrization=reio_bins_tanh
• reionization parameters if reio_parametrization=reio_many_tanh
• reionization parameters if reio_parametrization=reio_many_tanh
• energy injection parameters from CDM annihilation/decay
(c) define which perturbations and sources should be computed, and down to which scale
(d) define the primordial spectrum
(e) parameters for final spectra
(f) parameter related to the non-linear spectra computation
(g) amount of information sent to standard output (none if all set to zero)
(h) all precision parameters
Generated by Doxygen

5.5 input.c File Reference 59
• (h.1.) parameters related to the background
• (h.2.) parameters related to the thermodynamics
• (h.3.) parameters related to the perturbations
• —>Include ur and ncdm shear in tensor computation?
• —>derivatives of baryon sound speed only computed if some non-minimal tight-coupling schemes is re-
quested
• (h.4.) parameter related to the primordial spectra
• (h.5.) parameter related to the transfer functions
• (h.6.) parameters related to nonlinear calculations
• (h.7.) parameter related to lensing
(i) Write values in file
• (i.1.) shall we write background quantities in a file?
• (i.2.) shall we write thermodynamics quantities in a file?
• (i.3.) shall we write perturbation quantities in files?
• (i.4.) shall we write primordial spectra in a file?
• (i.5) special steps if we want Halofit with wa_fld non-zero: so-called "Pk_equal method" of 0810.0190 and
1601.07230
5.5.2.4 input_default_params()
int input_default_params (
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
struct transfers ∗ptr,
struct primordial ∗ppm,
struct spectra ∗psp,
struct nonlinear ∗pnl,
struct lensing ∗ple,
struct output ∗pop )
All default parameter values (for input parameters)
Parameters
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfer structure
ppm Input: pointer to primordial structure
psp Input: pointer to spectra structure
pnl Input: pointer to nonlinear structure
ple Input: pointer to lensing structure
pop Input: pointer to output structure
Generated by Doxygen
60 File Documentation
Returns
the error status
Define all default parameter values (for input parameters) for each structure:
• background structure
• thermodynamics structure
• perturbation structure
• primordial structure
• transfer structure
• output structure
• spectra structure
• nonlinear structure
• lensing structure
• nonlinear structure
• all verbose parameters
5.5.2.5 input_default_precision()
int input_default_precision (
struct precision ∗ppr )
Initialize the precision parameter structure.
All precision parameters used in the other modules are listed here and assigned here a default value.
Parameters
ppr Input/Output: a precision_params structure pointer
Returns
the error status
Initialize presicion parameters for different structures:
• parameters related to the background
• parameters related to the thermodynamics
• parameters related to the perturbations
• parameter related to the primordial spectra
Generated by Doxygen

5.5 input.c File Reference 61
• parameter related to the transfer functions
• parameters related to spectra module
• parameters related to nonlinear module
• parameter related to lensing
• automatic estimate of machine precision
5.5.2.6 get_machine_precision()
int get_machine_precision (
double ∗smallest_allowed_variation )
Automatically computes the machine precision.
Parameters
smallest_allowed_variation a pointer to the smallest allowed variation
Returns the smallest allowed variation (minimum epsilon ∗TOLVAR)
5.5.2.7 class_fzero_ridder()
int class_fzero_ridder (
int(∗)(double x, void ∗param, double ∗y, ErrorMsg error_message) func,
double x1,
double x2,
double xtol,
void ∗param,
double ∗Fx1,
double ∗Fx2,
double ∗xzero,
int ∗fevals,
ErrorMsg error_message )
Using Ridders' method, return the root of a function func known to lie between x1 and x2. The root, returned as
zriddr, will be found to an approximate accuracy xtol.
5.5.2.8 input_try_unknown_parameters()
int input_try_unknown_parameters (
double ∗unknown_parameter,
int unknown_parameters_size,
void ∗voidpfzw,
double ∗output,
ErrorMsg errmsg )
Summary:
Generated by Doxygen

62 File Documentation
• Call the structures
• Optimise flags for sigma8 calculation.
• Do computations
• In case scalar field is used to fill, pba->Omega0_scf is not equal to pfzw->target_value[i].
• Free structures
• Set filecontent to unread
5.5.2.9 input_get_guess()
int input_get_guess (
double ∗xguess,
double ∗dxdy,
struct fzerofun_workspace ∗pfzw,
ErrorMsg errmsg )
Summary:
• Here we should write reasonable guesses for the unknown parameters. Also estimate dxdy, i.e. how the
unknown parameter responds to the known. This can simply be estimated as the derivative of the guess
formula.
• Update pb to reflect guess
–This guess is arbitrary, something nice using WKB should be implemented.
• Version 2: use a fit: xguess[index_guess] = 1.77835∗pow(ba.Omega0_scf,-2./7.);
dxdy[index_guess] = -0.5081∗pow(ba.Omega0_scf,-9./7.);
• Version 3: use attractor solution
• This works since correspondence is Omega_ini_dcdm ->Omega_dcdmdr and omega_ini_dcdm ->omega←-
_dcdmdr
• Deallocate everything allocated by input_read_parameters
5.5.2.10 input_find_root()
int input_find_root (
double ∗xzero,
int ∗fevals,
struct fzerofun_workspace ∗pfzw,
ErrorMsg errmsg )
Summary:
• Fisrt we do our guess
• Do linear hunt for boundaries
• root has been bracketed
• Find root using Ridders method. (Exchange for bisection if you are old-school.)
Generated by Doxygen
5.6 input.h File Reference 63
5.5.2.11 input_prepare_pk_eq()
int input_prepare_pk_eq (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct nonlinear ∗pnl,
int input_verbose,
ErrorMsg errmsg )
Perform preliminary steps fur using the method called Pk_equal, described in 0810.0190 and 1601.07230, extending
the range of validity of HALOFIT from constant w to (w0,wa) models. In that case, one must compute here some
effective values of w0_eff(z_i) and Omega_m_eff(z_i), that will be interpolated later at arbitrary redshift in the non-
linear module.
Returns table of values [z_i, tau_i, w0_eff_i, Omega_m_eff_i] stored in nonlinear structure.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
pnl Input/Output: pointer to nonlinear structure
input_verbose Input: verbosity of this input module
errmsg Input/Ouput: error message
Summary:
• define local variables
• store the true cosmological parameters (w0, wa) somwhere before using temporarily some fake ones in this
function
• the fake calls of the background and thermodynamics module will be done in non-verbose mode
• allocate indices and arrays for storing the results
• call the background module in order to fill a table of tau_i[z_i]
–loop over z_i values. For each of them, we will call the background and thermodynamics module for fake
models. The goal is to find, for each z_i, and effective w0_eff[z_i] and Omega_m_eff{z_i], such that:
the true model with (w0,wa) and the equivalent model with (w0_eff[z_i],0) have the same conformal
distance between z_i and z_recombination, namely chi = tau[z_i] - tau_rec. It is thus necessary to call
both the background and thermodynamics module for each fake model and to re-compute tau_rec for
each of them. Once the eqauivalent model is found we compute and store Omega_m_effa(z_i) of the
equivalent model
• restore cosmological parameters (w0, wa) to their true values before main call to CLASS modules
• spline the table for later interpolation
5.6 input.h File Reference
#include "common.h"
#include "parser.h"
Generated by Doxygen
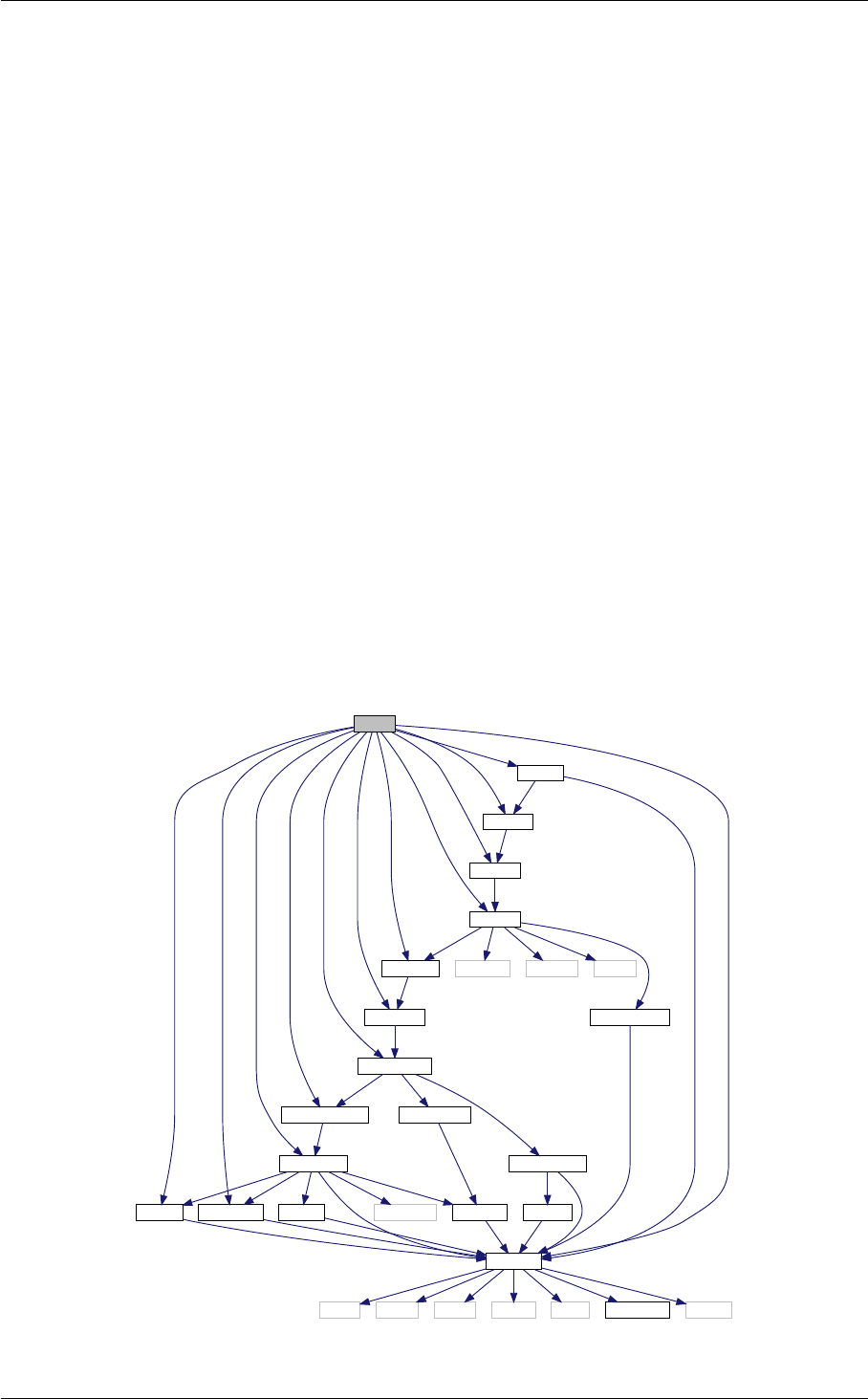
64 File Documentation
#include "quadrature.h"
#include "background.h"
#include "thermodynamics.h"
#include "perturbations.h"
#include "transfer.h"
#include "primordial.h"
#include "spectra.h"
#include "nonlinear.h"
#include "lensing.h"
#include "output.h"
Include dependency graph for input.h:
input.h
common.h
parser.h quadrature.h
background.h
thermodynamics.h
perturbations.h
transfer.h
nonlinear.h
primordial.h
spectra.h
lensing.h
output.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
growTable.harrays.h dei_rkck.h
evolver_ndf15.h
evolver_rkck.h
sparse.h
hyperspherical.h
sys/shm.h sys/stat.h errno.h
Generated by Doxygen
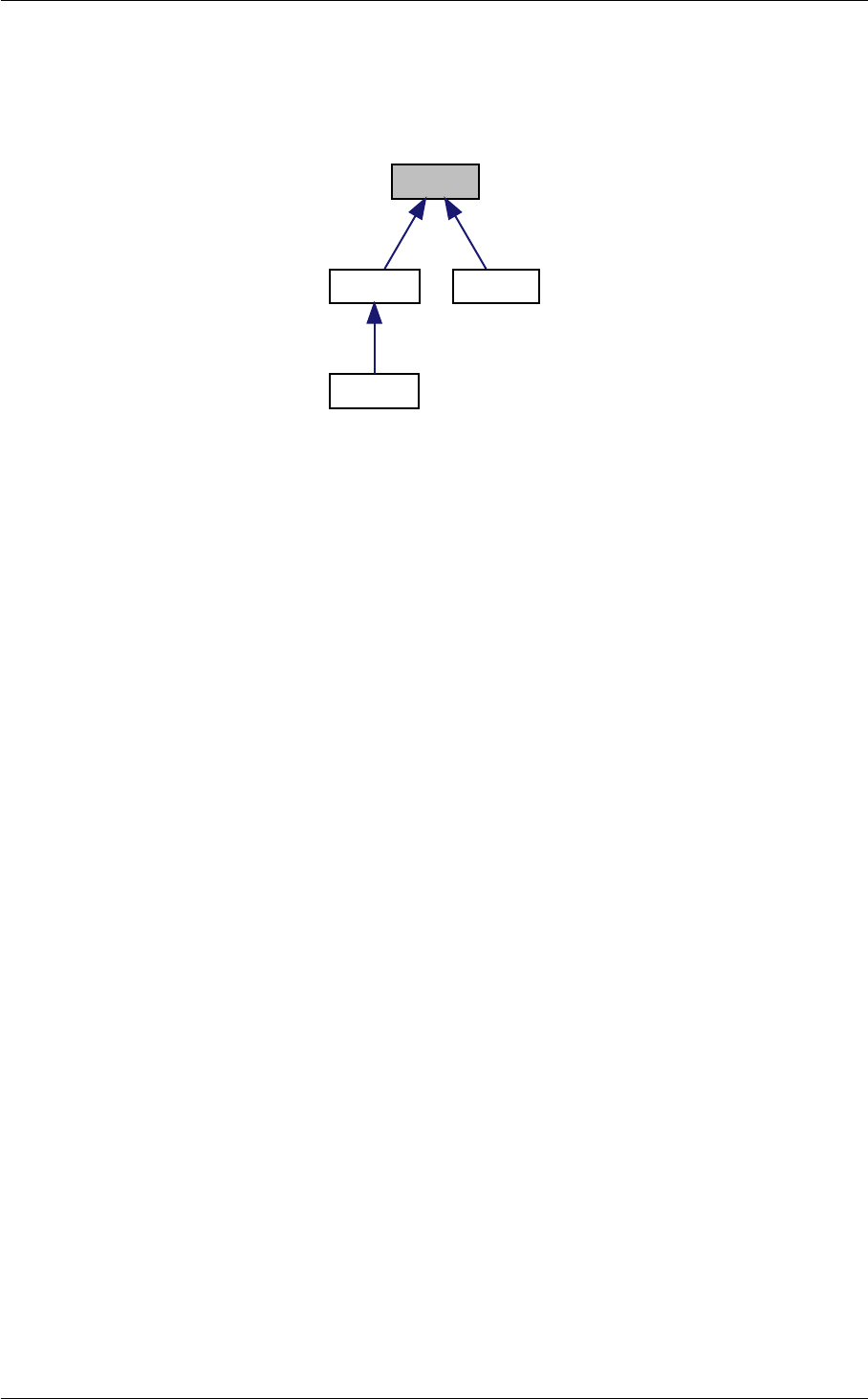
5.6 input.h File Reference 65
This graph shows which files directly or indirectly include this file:
input.h
class.h input.c
class.c
Enumerations
• enum target_names
5.6.1 Detailed Description
Documented includes for input module
5.6.2 Enumeration Type Documentation
5.6.2.1 target_names
enum target_names
temporary parameters for background fzero function
Generated by Doxygen
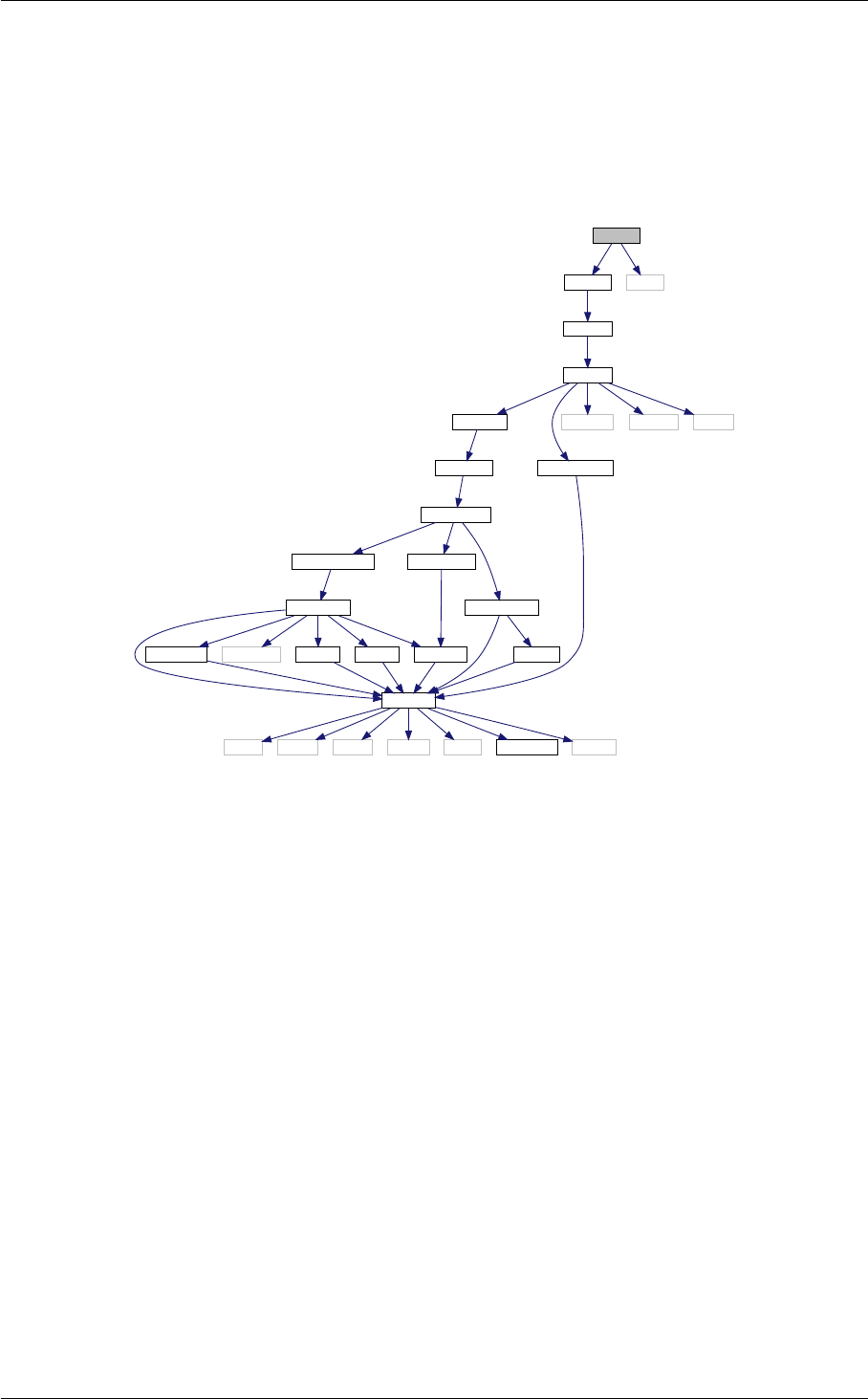
66 File Documentation
5.7 lensing.c File Reference
#include "lensing.h"
#include <time.h>
Include dependency graph for lensing.c:
lensing.c
lensing.h time.h
spectra.h
transfer.h
nonlinear.h
hyperspherical.h
sys/shm.h sys/stat.h errno.h
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
Functions
• int lensing_cl_at_l (struct lensing ∗ple, int l, double ∗cl_lensed)
• int lensing_init (struct precision ∗ppr, struct perturbs ∗ppt, struct spectra ∗psp, struct nonlinear ∗pnl, struct
lensing ∗ple)
• int lensing_free (struct lensing ∗ple)
• int lensing_indices (struct precision ∗ppr, struct spectra ∗psp, struct lensing ∗ple)
• int lensing_lensed_cl_tt (double ∗ksi, double ∗∗d00, double ∗w8, int nmu, struct lensing ∗ple)
• int lensing_addback_cl_tt (struct lensing ∗ple, double ∗cl_tt)
• int lensing_lensed_cl_te (double ∗ksiX, double ∗∗d20, double ∗w8, int nmu, struct lensing ∗ple)
• int lensing_addback_cl_te (struct lensing ∗ple, double ∗cl_te)
• int lensing_lensed_cl_ee_bb (double ∗ksip, double ∗ksim, double ∗∗d22, double ∗∗d2m2, double ∗w8, int
nmu, struct lensing ∗ple)
• int lensing_addback_cl_ee_bb (struct lensing ∗ple, double ∗cl_ee, double ∗cl_bb)
• int lensing_d00 (double ∗mu, int num_mu, int lmax, double ∗∗d00)
• int lensing_d11 (double ∗mu, int num_mu, int lmax, double ∗∗d11)
• int lensing_d1m1 (double ∗mu, int num_mu, int lmax, double ∗∗d1m1)
• int lensing_d2m2 (double ∗mu, int num_mu, int lmax, double ∗∗d2m2)
• int lensing_d22 (double ∗mu, int num_mu, int lmax, double ∗∗d22)
• int lensing_d20 (double ∗mu, int num_mu, int lmax, double ∗∗d20)
• int lensing_d31 (double ∗mu, int num_mu, int lmax, double ∗∗d31)
Generated by Doxygen

5.7 lensing.c File Reference 67
• int lensing_d3m1 (double ∗mu, int num_mu, int lmax, double ∗∗d3m1)
• int lensing_d3m3 (double ∗mu, int num_mu, int lmax, double ∗∗d3m3)
• int lensing_d40 (double ∗mu, int num_mu, int lmax, double ∗∗d40)
• int lensing_d4m2 (double ∗mu, int num_mu, int lmax, double ∗∗d4m2)
• int lensing_d4m4 (double ∗mu, int num_mu, int lmax, double ∗∗d4m4)
5.7.1 Detailed Description
Documented lensing module
Simon Prunet and Julien Lesgourgues, 6.12.2010
This module computes the lensed temperature and polarization anisotropy power spectra CX
l, P (k), ...'s given the
unlensed temperature, polarization and lensing potential spectra.
Follows Challinor and Lewis full-sky method, astro-ph/0502425
The following functions can be called from other modules:
1. lensing_init() at the beginning (but after spectra_init())
2. lensing_cl_at_l() at any time for computing Cl_lensed at any l
3. lensing_free() at the end
5.7.2 Function Documentation
5.7.2.1 lensing_cl_at_l()
int lensing_cl_at_l (
struct lensing ∗ple,
int l,
double ∗cl_lensed )
Anisotropy power spectra Cl's for all types, modes and initial conditions. SO FAR: ONLY SCALAR
This routine evaluates all the lensed Cl's at a given value of l by picking it in the pre-computed table. When relevant,
it also sums over all initial conditions for each mode, and over all modes.
This function can be called from whatever module at whatever time, provided that lensing_init() has been called
before, and lensing_free() has not been called yet.
Parameters
ple Input: pointer to lensing structure
lInput: multipole number
cl_lensed Output: lensed Cl's for all types (TT, TE, EE, etc..)
Generated by Doxygen
68 File Documentation
Returns
the error status
5.7.2.2 lensing_init()
int lensing_init (
struct precision ∗ppr,
struct perturbs ∗ppt,
struct spectra ∗psp,
struct nonlinear ∗pnl,
struct lensing ∗ple )
This routine initializes the lensing structure (in particular, computes table of lensed anisotropy spectra CX
l)
Parameters
ppr Input: pointer to precision structure
ppt Input: pointer to perturbation structure (just in case, not used in current version...)
psp Input: pointer to spectra structure
pnl Input: pointer to nonlinear structure
ple Output: pointer to initialized lensing structure
Returns
the error status
Summary:
• Define local variables
• check that we really want to compute at least one spectrum
• initialize indices and allocate some of the arrays in the lensing structure
• put all precision variables hare; will be stored later in precision structure
• Last element in µwill be for µ= 1, needed for sigma2. The rest will be chosen as roots of a Gauss-Legendre
quadrature
• allocate array of µvalues, as well as quadrature weights
• Compute dl
mm0(µ)
• Allocate main contiguous buffer
• compute Cgl(µ),Cgl2(µ)and sigma2( µ)
• Locally store unlensed temperature cltt and potential clpp spectra
• Compute sigma2 (µ)and Cgl2( µ)
• compute ksi, ksi+, ksi-, ksiX
• –>ksi is for TT
Generated by Doxygen
5.7 lensing.c File Reference 69
• –>ksiX is for TE
• –>ksip, ksim for EE, BB
• compute lensed Cl's by integration
• spline computed Cl's in view of interpolation
• Free lots of stuff
• Exit
5.7.2.3 lensing_free()
int lensing_free (
struct lensing ∗ple )
This routine frees all the memory space allocated by lensing_init().
To be called at the end of each run, only when no further calls to lensing_cl_at_l() are needed.
Parameters
ple Input: pointer to lensing structure (which fields must be freed)
Returns
the error status
5.7.2.4 lensing_indices()
int lensing_indices (
struct precision ∗ppr,
struct spectra ∗psp,
struct lensing ∗ple )
This routine defines indices and allocates tables in the lensing structure
Parameters
ppr Input: pointer to precision structure
psp Input: pointer to spectra structure
ple Input/output: pointer to lensing structure
Returns
the error status
Generated by Doxygen
70 File Documentation
5.7.2.5 lensing_lensed_cl_tt()
int lensing_lensed_cl_tt (
double ∗ksi,
double ∗∗ d00,
double ∗w8,
int nmu,
struct lensing ∗ple )
This routine computes the lensed power spectra by Gaussian quadrature
Parameters
ksi Input: Lensed correlation function (ksi[index_mu])
d00 Input: Legendre polynomials ( dl
00[l][index_mu])
w8 Input: Legendre quadrature weights (w8[index_mu])
nmu Input: Number of quadrature points (0<=index_mu<=nmu)
ple Input/output: Pointer to the lensing structure
Returns
the error status
Integration by Gauss-Legendre quadrature.
5.7.2.6 lensing_addback_cl_tt()
int lensing_addback_cl_tt (
struct lensing ∗ple,
double ∗cl_tt )
This routine adds back the unlensed cltt power spectrum Used in case of fast (and BB inaccurate) integration of
correlation functions.
Parameters
ple Input/output: Pointer to the lensing structure
cl←-
_tt
Input: Array of unlensed power spectrum
Returns
the error status
Generated by Doxygen

5.7 lensing.c File Reference 71
5.7.2.7 lensing_lensed_cl_te()
int lensing_lensed_cl_te (
double ∗ksiX,
double ∗∗ d20,
double ∗w8,
int nmu,
struct lensing ∗ple )
This routine computes the lensed power spectra by Gaussian quadrature
Parameters
ksiX Input: Lensed correlation function (ksiX[index_mu])
d20 Input: Wigner d-function ( dl
20[l][index_mu])
w8 Input: Legendre quadrature weights (w8[index_mu])
nmu Input: Number of quadrature points (0<=index_mu<=nmu)
ple Input/output: Pointer to the lensing structure
Returns
the error status
Integration by Gauss-Legendre quadrature.
5.7.2.8 lensing_addback_cl_te()
int lensing_addback_cl_te (
struct lensing ∗ple,
double ∗cl_te )
This routine adds back the unlensed clte power spectrum Used in case of fast (and BB inaccurate) integration of
correlation functions.
Parameters
ple Input/output: Pointer to the lensing structure
cl←-
_te
Input: Array of unlensed power spectrum
Returns
the error status
5.7.2.9 lensing_lensed_cl_ee_bb()
int lensing_lensed_cl_ee_bb (
double ∗ksip,
Generated by Doxygen

72 File Documentation
double ∗ksim,
double ∗∗ d22,
double ∗∗ d2m2,
double ∗w8,
int nmu,
struct lensing ∗ple )
This routine computes the lensed power spectra by Gaussian quadrature
Parameters
ksip Input: Lensed correlation function (ksi+[index_mu])
ksim Input: Lensed correlation function (ksi-[index_mu])
d22 Input: Wigner d-function ( dl
22[l][index_mu])
d2m2 Input: Wigner d-function ( dl
2−2[l][index_mu])
w8 Input: Legendre quadrature weights (w8[index_mu])
nmu Input: Number of quadrature points (0<=index_mu<=nmu)
ple Input/output: Pointer to the lensing structure
Returns
the error status
Integration by Gauss-Legendre quadrature.
5.7.2.10 lensing_addback_cl_ee_bb()
int lensing_addback_cl_ee_bb (
struct lensing ∗ple,
double ∗cl_ee,
double ∗cl_bb )
This routine adds back the unlensed clee,clbb power spectra Used in case of fast (and BB inaccurate) integration of
correlation functions.
Parameters
ple Input/output: Pointer to the lensing structure
cl_ee Input: Array of unlensed power spectrum
cl_bb Input: Array of unlensed power spectrum
Returns
the error status
5.7.2.11 lensing_d00()
int lensing_d00 (
double ∗mu,
Generated by Doxygen

5.7 lensing.c File Reference 73
int num_mu,
int lmax,
double ∗∗ d00 )
This routine computes the d00 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d00 Input/output: Result is stored here
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.12 lensing_d11()
int lensing_d11 (
double ∗mu,
int num_mu,
int lmax,
double ∗∗ d11 )
This routine computes the d11 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d11 Input/output: Result is stored here
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.13 lensing_d1m1()
int lensing_d1m1 (
double ∗mu,
int num_mu,
int lmax,
double ∗∗ d1m1 )
This routine computes the d1m1 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d1m1 Input/output: Result is stored here
Generated by Doxygen
74 File Documentation
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.14 lensing_d2m2()
int lensing_d2m2 (
double ∗mu,
int num_mu,
int lmax,
double ∗∗ d2m2 )
This routine computes the d2m2 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d2m2 Input/output: Result is stored here
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.15 lensing_d22()
int lensing_d22 (
double ∗mu,
int num_mu,
int lmax,
double ∗∗ d22 )
This routine computes the d22 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d22 Input/output: Result is stored here
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.16 lensing_d20()
int lensing_d20 (
double ∗mu,
int num_mu,
Generated by Doxygen

5.7 lensing.c File Reference 75
int lmax,
double ∗∗ d20 )
This routine computes the d20 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d20 Input/output: Result is stored here
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.17 lensing_d31()
int lensing_d31 (
double ∗mu,
int num_mu,
int lmax,
double ∗∗ d31 )
This routine computes the d31 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d31 Input/output: Result is stored here
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.18 lensing_d3m1()
int lensing_d3m1 (
double ∗mu,
int num_mu,
int lmax,
double ∗∗ d3m1 )
This routine computes the d3m1 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d3m1 Input/output: Result is stored here
Generated by Doxygen
76 File Documentation
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.19 lensing_d3m3()
int lensing_d3m3 (
double ∗mu,
int num_mu,
int lmax,
double ∗∗ d3m3 )
This routine computes the d3m3 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d3m3 Input/output: Result is stored here
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.20 lensing_d40()
int lensing_d40 (
double ∗mu,
int num_mu,
int lmax,
double ∗∗ d40 )
This routine computes the d40 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d40 Input/output: Result is stored here
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.21 lensing_d4m2()
int lensing_d4m2 (
double ∗mu,
int num_mu,
Generated by Doxygen
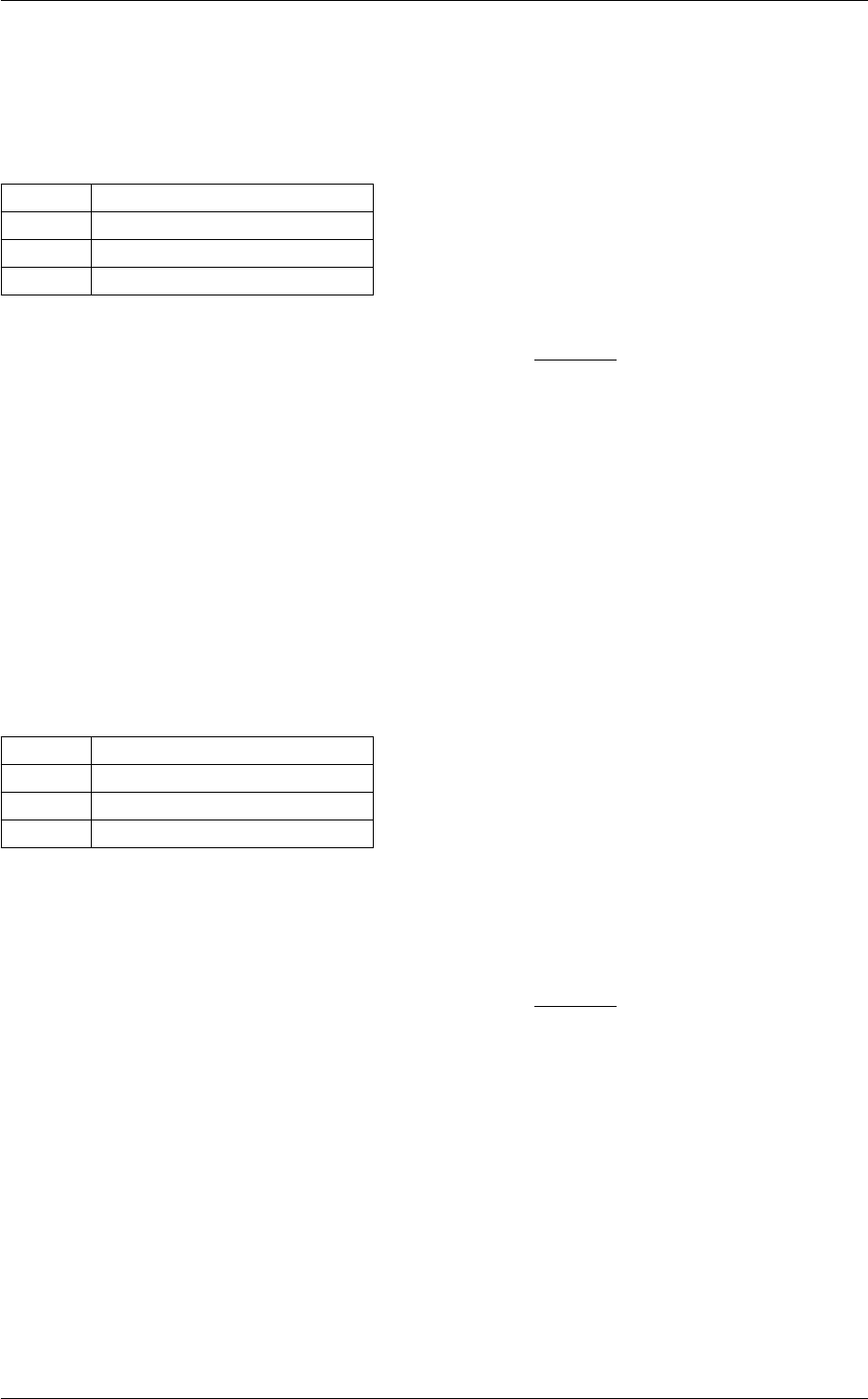
5.8 lensing.h File Reference 77
int lmax,
double ∗∗ d4m2 )
This routine computes the d4m2 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d4m2 Input/output: Result is stored here
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.7.2.22 lensing_d4m4()
int lensing_d4m4 (
double ∗mu,
int num_mu,
int lmax,
double ∗∗ d4m4 )
This routine computes the d4m4 term
Parameters
mu Input: Vector of cos(beta) values
num_mu Input: Number of cos(beta) values
lmax Input: maximum multipole
d4m4 Input/output: Result is stored here
Wigner d-functions, computed by recurrence actual recurrence on p(2l+ 1)/2dl
mm0for stability Formulae from
Kostelec & Rockmore 2003
5.8 lensing.h File Reference
#include "spectra.h"
Generated by Doxygen
78 File Documentation
Include dependency graph for lensing.h:
lensing.h
spectra.h
transfer.h
nonlinear.h
hyperspherical.h
sys/shm.h sys/stat.h errno.h
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
This graph shows which files directly or indirectly include this file:
lensing.h
input.h
class.h
output.h lensing.c
input.c
class.c
output.c
Generated by Doxygen

5.8 lensing.h File Reference 79
Data Structures
• struct lensing
5.8.1 Detailed Description
Documented includes for spectra module
5.8.2 Data Structure Documentation
5.8.2.1 struct lensing
Structure containing everything about lensed spectra that other modules need to know.
Once initialized by lensing_init(), contains a table of all lensed Cl's for the all modes (scalar/tensor), all types (TT,
TE...), and all pairs of initial conditions (adiabatic, isocurvatures...). FOR THE MOMENT, ASSUME ONLY SCALAR
& ADIABATIC
Data Fields
short has_lensed_cls do we need to compute lensed Cl's at all ?
int has_tt do we want lensed CT T
l? (T = temperature)
int has_ee do we want lensed CEE
l? (E = E-polarization)
int has_te do we want lensed CT E
l?
int has_bb do we want CBB
l? (B = B-polarization)
int has_pp do we want Cφφ
l? ( φ= CMB lensing potential)
int has_tp do we want CT φ
l?
int has_dd do we want Cdd
l? (d = matter density)
int has_td do we want CT d
l?
int has_ll do we want Cll
l? (l = lensing potential)
int has_tl do we want CT l
l?
int index_lt_tt index for type CT T
l
int index_lt_ee index for type CEE
l
int index_lt_te index for type CT E
l
int index_lt_bb index for type CBB
l
int index_lt_pp index for type Cφφ
l
int index_lt_tp index for type CT φ
l
int index_lt_dd index for type Cdd
l
int index_lt_td index for type CT d
l
int index_lt_ll index for type Cdd
l
int index_lt_tl index for type CT d
l
int lt_size number of Cltypes requested
int l_unlensed_max last multipole in all calculations (same as in spectra module)
int l_lensed_max last multipole at which lensed spectra are computed
Generated by Doxygen
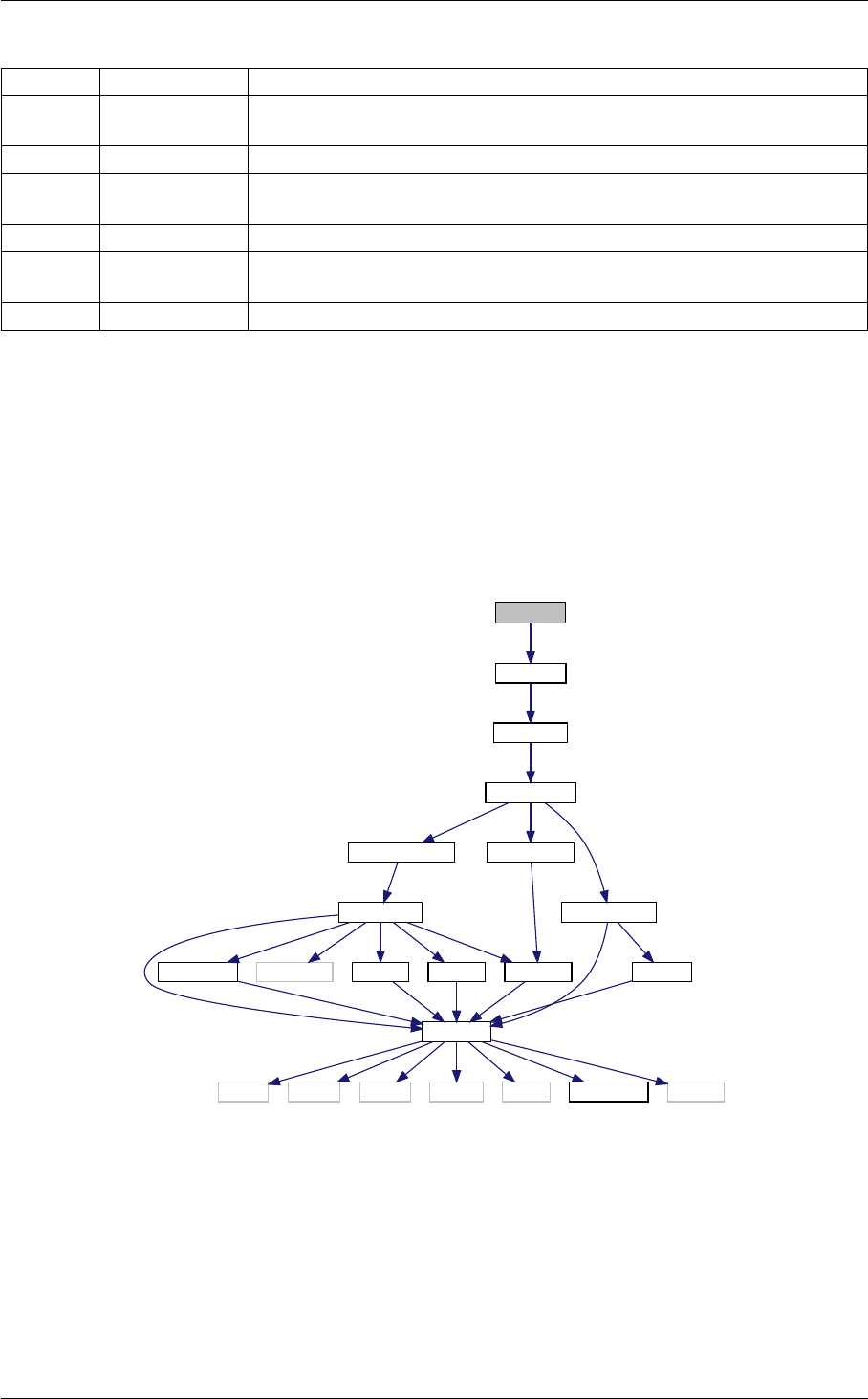
80 File Documentation
Data Fields
int l_size number of l values
int ∗l_max_lt last multipole (given as an input) at which we want to output Cl's for a given
mode and type
double ∗l table of multipole values l[index_l]
double ∗cl_lens table of anisotropy spectra for each multipole and types, cl[index_l ∗
ple->lt_size + index_lt]
double ∗ddcl_lens second derivatives for interpolation
short lensing_verbose flag regulating the amount of information sent to standard output (none if set to
zero)
ErrorMsg error_message zone for writing error messages
5.9 nonlinear.c File Reference
#include "nonlinear.h"
Include dependency graph for nonlinear.c:
nonlinear.c
nonlinear.h
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
Functions
• int nonlinear_init (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct perturbs ∗ppt,
struct primordial ∗ppm, struct nonlinear ∗pnl)
• int nonlinear_halofit (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, struct primordial
∗ppm, struct nonlinear ∗pnl, int index_pk, double tau, double ∗pk_l, double ∗pk_nl, double ∗lnk_l, double
∗lnpk_l, double ∗ddlnpk_l, double ∗k_nl, short ∗halofit_found_k_max)
Generated by Doxygen

5.9 nonlinear.c File Reference 81
5.9.1 Detailed Description
Documented nonlinear module
Julien Lesgourgues, 6.03.2014
New module replacing an older one present up to version 2.0 The new module is located in a better place in the
main, allowing it to compute non-linear correction to Cl's and not just P(k). It will also be easier to generalize to
new methods. The old implementation of one-loop calculations and TRG calculations has been dropped from this
version, they can still be found in older versions.
5.9.2 Function Documentation
5.9.2.1 nonlinear_init()
int nonlinear_init (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
struct primordial ∗ppm,
struct nonlinear ∗pnl )
Summary
(a) First deal with the case where non non-linear corrections requested
(b) Compute for HALOFIT non-linear spectrum
• copy list of (k,tau) from perturbation module
• loop over time
5.9.2.2 nonlinear_halofit()
int nonlinear_halofit (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
struct primordial ∗ppm,
struct nonlinear ∗pnl,
int index_pk,
double tau,
double ∗pk_l,
double ∗pk_nl,
double ∗lnk_l,
double ∗lnpk_l,
double ∗ddlnpk_l,
double ∗k_nl,
short ∗halofit_found_k_max )
Determine non linear ratios (from pk)
Generated by Doxygen
82 File Documentation
5.10 nonlinear.h File Reference
#include "primordial.h"
Include dependency graph for nonlinear.h:
nonlinear.h
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
This graph shows which files directly or indirectly include this file:
nonlinear.h
transfer.h
input.h
class.h
nonlinear.c
spectra.h transfer.c
input.c
class.c
lensing.hspectra.c
output.h lensing.c
output.c
Generated by Doxygen
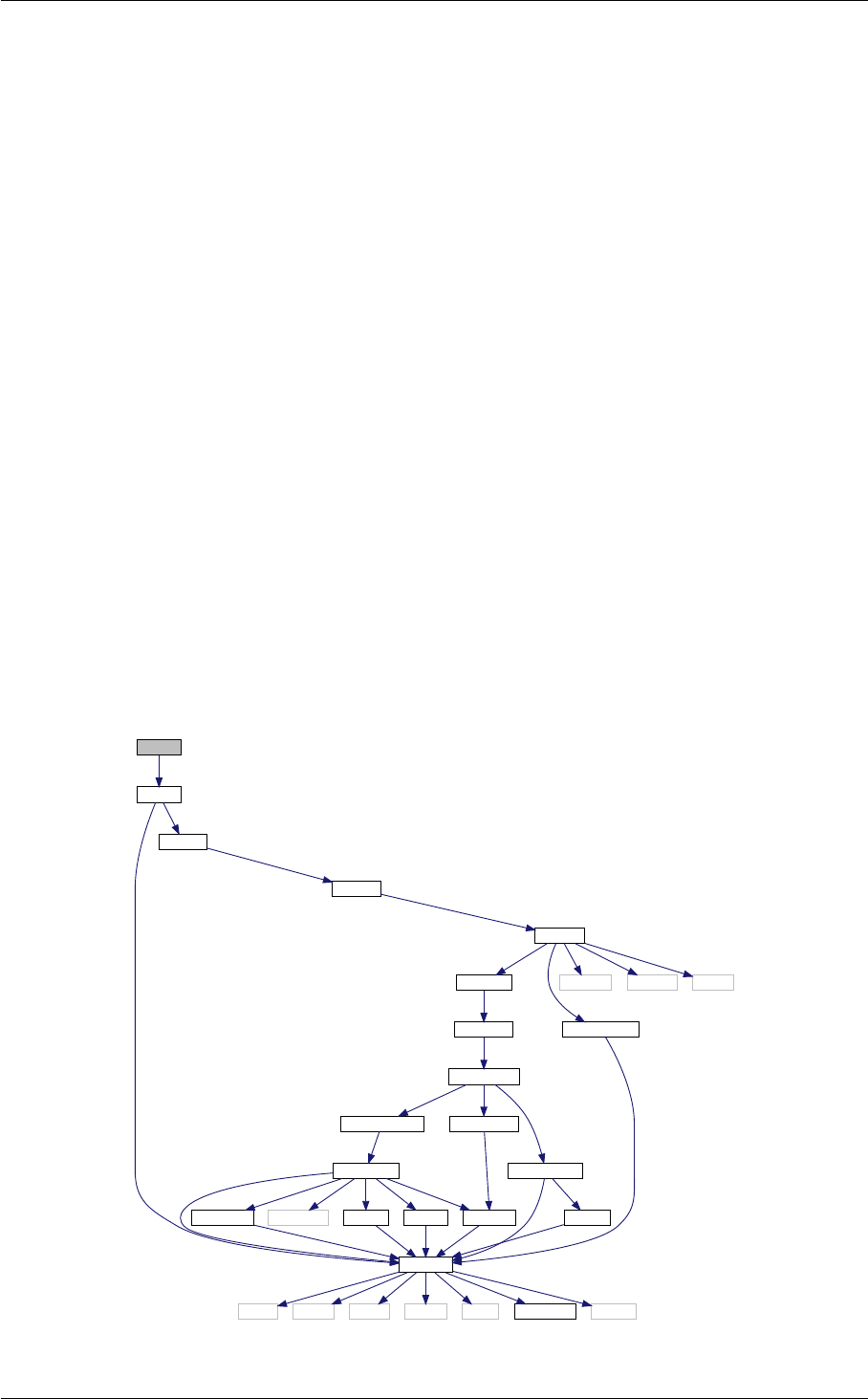
5.11 output.c File Reference 83
Data Structures
• struct nonlinear
Macros
• #define _M_EV_TOO_BIG_FOR_HALOFIT_ 10.
5.10.1 Detailed Description
Documented includes for trg module
5.10.2 Macro Definition Documentation
5.10.2.1 _M_EV_TOO_BIG_FOR_HALOFIT_
#define _M_EV_TOO_BIG_FOR_HALOFIT_ 10.
above which value of non-CDM mass (in eV) do we stop trusting halofit?
5.11 output.c File Reference
#include "output.h"
Include dependency graph for output.c:
output.c
output.h
common.h
lensing.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
spectra.h
transfer.h
nonlinear.h
hyperspherical.h
sys/shm.h sys/stat.h errno.h
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h sparse.h
Generated by Doxygen

84 File Documentation
Functions
• int output_init (struct background ∗pba, struct thermo ∗pth, struct perturbs ∗ppt, struct primordial ∗ppm, struct
transfers ∗ptr, struct spectra ∗psp, struct nonlinear ∗pnl, struct lensing ∗ple, struct output ∗pop)
• int output_cl (struct background ∗pba, struct perturbs ∗ppt, struct spectra ∗psp, struct lensing ∗ple, struct
output ∗pop)
• int output_pk (struct background ∗pba, struct perturbs ∗ppt, struct spectra ∗psp, struct output ∗pop)
• int output_pk_nl (struct background ∗pba, struct perturbs ∗ppt, struct spectra ∗psp, struct output ∗pop)
• int output_tk (struct background ∗pba, struct perturbs ∗ppt, struct spectra ∗psp, struct output ∗pop)
• int output_print_data (FILE ∗out, char titles[_MAXTITLESTRINGLENGTH_], double ∗dataptr, int size_←-
dataptr)
• int output_open_cl_file (struct spectra ∗psp, struct output ∗pop, FILE ∗∗clfile, FileName filename, char ∗first←-
_line, int lmax)
• int output_one_line_of_cl (struct background ∗pba, struct spectra ∗psp, struct output ∗pop, FILE ∗clfile, dou-
ble l, double ∗cl, int ct_size)
• int output_open_pk_file (struct background ∗pba, struct spectra ∗psp, struct output ∗pop, FILE ∗∗pkfile, File←-
Name filename, char ∗first_line, double z)
• int output_one_line_of_pk (FILE ∗pkfile, double one_k, double one_pk)
5.11.1 Detailed Description
Documented output module
Julien Lesgourgues, 26.08.2010
This module writes the output in files.
The following functions can be called from other modules or from the main:
1. output_init() (must be called after spectra_init())
2. output_total_cl_at_l() (can be called even before output_init())
No memory needs to be deallocated after that, hence there is no output_free() routine like in other modules.
5.11.2 Function Documentation
5.11.2.1 output_init()
int output_init (
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
struct primordial ∗ppm,
struct transfers ∗ptr,
struct spectra ∗psp,
struct nonlinear ∗pnl,
struct lensing ∗ple,
struct output ∗pop )
This routine writes the output in files.
Generated by Doxygen

5.11 output.c File Reference 85
Parameters
pba Input: pointer to background structure (needed for calling spectra_pk_at_z())
pth Input: pointer to thermodynamics structure
ppt Input: pointer perturbation structure
ppm Input: pointer to primordial structure
ptr Input: pointer to transfer structure
psp Input: pointer to spectra structure
pnl Input: pointer to nonlinear structure
ple Input: pointer to lensing structure
pop Input: pointer to output structure
Summary:
• check that we really want to output at least one file
• deal with all anisotropy power spectra Cl's
• deal with all Fourier matter power spectra P(k)'s
• deal with density and matter power spectra
• deal with background quantities
• deal with thermodynamics quantities
• deal with perturbation quantities
• deal with primordial spectra
5.11.2.2 output_cl()
int output_cl (
struct background ∗pba,
struct perturbs ∗ppt,
struct spectra ∗psp,
struct lensing ∗ple,
struct output ∗pop )
This routines writes the output in files for anisotropy power spectra Cl's.
Parameters
pba Input: pointer to background structure (needed for Tcmb)
ppt Input: pointer perturbation structure
psp Input: pointer to spectra structure
ple Input: pointer to lensing structure
pop Input: pointer to output structure
Summary:
Generated by Doxygen

86 File Documentation
• define local variables
• first, allocate all arrays of files and Cl's
• second, open only the relevant files, and write a heading in each of them
• third, perform loop over l. For each multipole, get all Cl's by calling spectra_cl_at_l() and distribute the results
to relevant files
• finally, close files and free arrays of files and Cl's
5.11.2.3 output_pk()
int output_pk (
struct background ∗pba,
struct perturbs ∗ppt,
struct spectra ∗psp,
struct output ∗pop )
This routines writes the output in files for Fourier matter power spectra P(k)'s.
Parameters
pba Input: pointer to background structure (needed for calling spectra_pk_at_z())
ppt Input: pointer perturbation structure
psp Input: pointer to spectra structure
pop Input: pointer to output structure
Summary:
• define local variables
• first, check that requested redshift z_pk is consistent
• second, open only the relevant files and write a heading in each of them
• third, compute P(k) for each k (if several ic's, compute it for each ic and compute also the total); if z_pk = 0,
this is done by directly reading inside the pre-computed table; if not, this is done by interpolating the table at
the correct value of tau.
• fourth, write in files
• fifth, free memory and close files
5.11.2.4 output_pk_nl()
int output_pk_nl (
struct background ∗pba,
struct perturbs ∗ppt,
struct spectra ∗psp,
struct output ∗pop )
This routines writes the output in files for Fourier non-linear matter power spectra P(k)'s.
Generated by Doxygen

5.11 output.c File Reference 87
Parameters
pba Input: pointer to background structure (needed for calling spectra_pk_at_z())
ppt Input: pointer perturbation structure
psp Input: pointer to spectra structure
pop Input: pointer to output structure
Summary:
• define local variables
• first, check that requested redshift z_pk is consistent
• second, open only the relevant files, and write a heading in each of them
• third, compute P(k) for each k (if several ic's, compute it for each ic and compute also the total); if z_pk = 0,
this is done by directly reading inside the pre-computed table; if not, this is done by interpolating the table at
the correct value of tau.
• fourth, write in files
• fifth, free memory and close files
5.11.2.5 output_tk()
int output_tk (
struct background ∗pba,
struct perturbs ∗ppt,
struct spectra ∗psp,
struct output ∗pop )
This routines writes the output in files for matter transfer functions Ti(k)'s.
Parameters
pba Input: pointer to background structure (needed for calling spectra_pk_at_z())
ppt Input: pointer perturbation structure
psp Input: pointer to spectra structure
pop Input: pointer to output structure
Summary:
• define local variables
• first, check that requested redshift z_pk is consistent
• second, open only the relevant files, and write a heading in each of them
• free memory and close files
Generated by Doxygen
88 File Documentation
5.11.2.6 output_print_data()
int output_print_data (
FILE ∗out,
char titles[_MAXTITLESTRINGLENGTH_],
double ∗dataptr,
int size_dataptr )
Summary
• First we print the titles
• Then we print the data
5.11.2.7 output_open_cl_file()
int output_open_cl_file (
struct spectra ∗psp,
struct output ∗pop,
FILE ∗∗ clfile,
FileName filename,
char ∗first_line,
int lmax )
This routine opens one file where some Cl's will be written, and writes a heading with some general information
concerning its content.
Parameters
psp Input: pointer to spectra structure
pop Input: pointer to output structure
clfile Output: returned pointer to file pointer
filename Input: name of the file
first_line Input: text describing the content (mode, initial condition..)
lmax Input: last multipole in the file (the first one is assumed to be 2)
Returns
the error status
Summary
• First we deal with the entries that are dependent of format type
• Next deal with entries that are independent of format type
Generated by Doxygen
5.11 output.c File Reference 89
5.11.2.8 output_one_line_of_cl()
int output_one_line_of_cl (
struct background ∗pba,
struct spectra ∗psp,
struct output ∗pop,
FILE ∗clfile,
double l,
double ∗cl,
int ct_size )
This routine write one line with l and all Cl's for all types (TT, TE...)
Parameters
pba Input: pointer to background structure (needed for Tcmb)
psp Input: pointer to spectra structure
pop Input: pointer to output structure
clfile Input: file pointer
lInput: multipole
cl Input: Cl's for all types
ct_size Input: number of types
Returns
the error status
5.11.2.9 output_open_pk_file()
int output_open_pk_file (
struct background ∗pba,
struct spectra ∗psp,
struct output ∗pop,
FILE ∗∗ pkfile,
FileName filename,
char ∗first_line,
double z)
This routine opens one file where some P(k)'s will be written, and writes a heading with some general information
concerning its content.
Parameters
pba Input: pointer to background structure (needed for h)
psp Input: pointer to spectra structure
pop Input: pointer to output structure
pkfile Output: returned pointer to file pointer
filename Input: name of the file
first_line Input: text describing the content (initial conditions, ...)
zInput: redshift of the output
Generated by Doxygen

90 File Documentation
Returns
the error status
5.11.2.10 output_one_line_of_pk()
int output_one_line_of_pk (
FILE ∗pkfile,
double one_k,
double one_pk )
This routine writes one line with k and P(k)
Parameters
pkfile Input: file pointer
one_k Input: wavenumber
one_pk Input: matter power spectrum
Returns
the error status
5.12 output.h File Reference
#include "common.h"
#include "lensing.h"
Generated by Doxygen
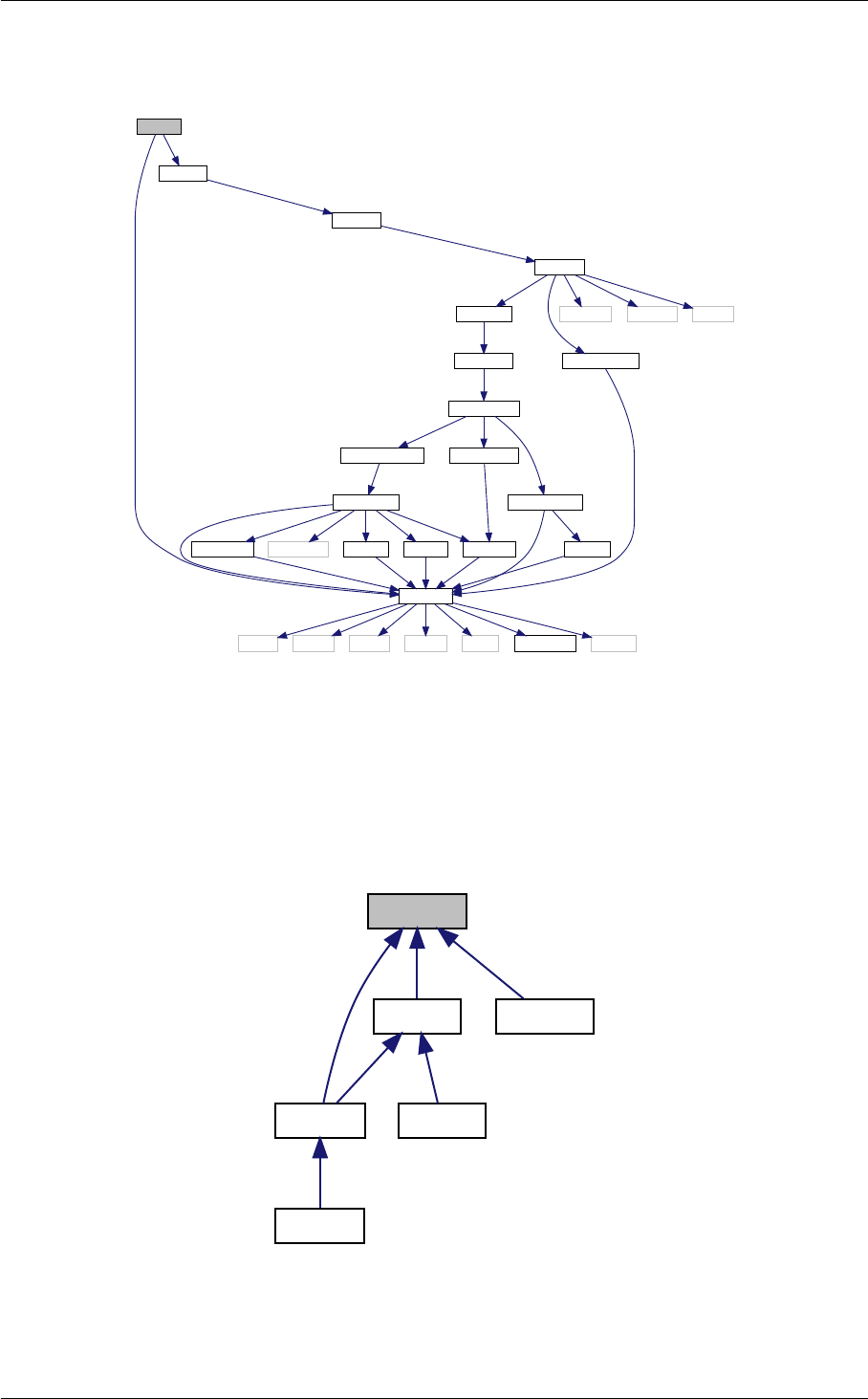
5.12 output.h File Reference 91
Include dependency graph for output.h:
output.h
common.h
lensing.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
spectra.h
transfer.h
nonlinear.h
hyperspherical.h
sys/shm.h sys/stat.h errno.h
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h sparse.h
This graph shows which files directly or indirectly include this file:
output.h
input.h
class.h
output.c
input.c
class.c
Generated by Doxygen
92 File Documentation
Data Structures
• struct output
Macros
• #define _Z_PK_NUM_MAX_ 100
5.12.1 Detailed Description
Documented includes for output module
5.12.2 Data Structure Documentation
5.12.2.1 struct output
Structure containing various informations on the output format, all of them initialized by user in input module.
Data Fields
FileName root root for all file names
int z_pk_num number of redshift at which P(k,z) and T_i(k,z) should be
written
double z_pk[_Z_PK_NUM_MAX_] value(s) of redshift at which P(k,z) and T_i(k,z) should be
written
short write_header flag stating whether we should write a header in output files
enum file_format output_format which format for output files (definitions, order of columns,
etc.)
short write_background flag for outputing background evolution in file
short write_thermodynamics flag for outputing thermodynamical evolution in file
short write_perturbations flag for outputing perturbations of selected wavenumber(s) in
file(s)
short write_primordial flag for outputing scalar/tensor primordial spectra in files
short output_verbose flag regulating the amount of information sent to standard
output (none if set to zero)
ErrorMsg error_message zone for writing error messages
5.12.3 Macro Definition Documentation
5.12.3.1 _Z_PK_NUM_MAX_
#define _Z_PK_NUM_MAX_ 100
Maximum number of values of redshift at which the spectra will be written in output files
Generated by Doxygen
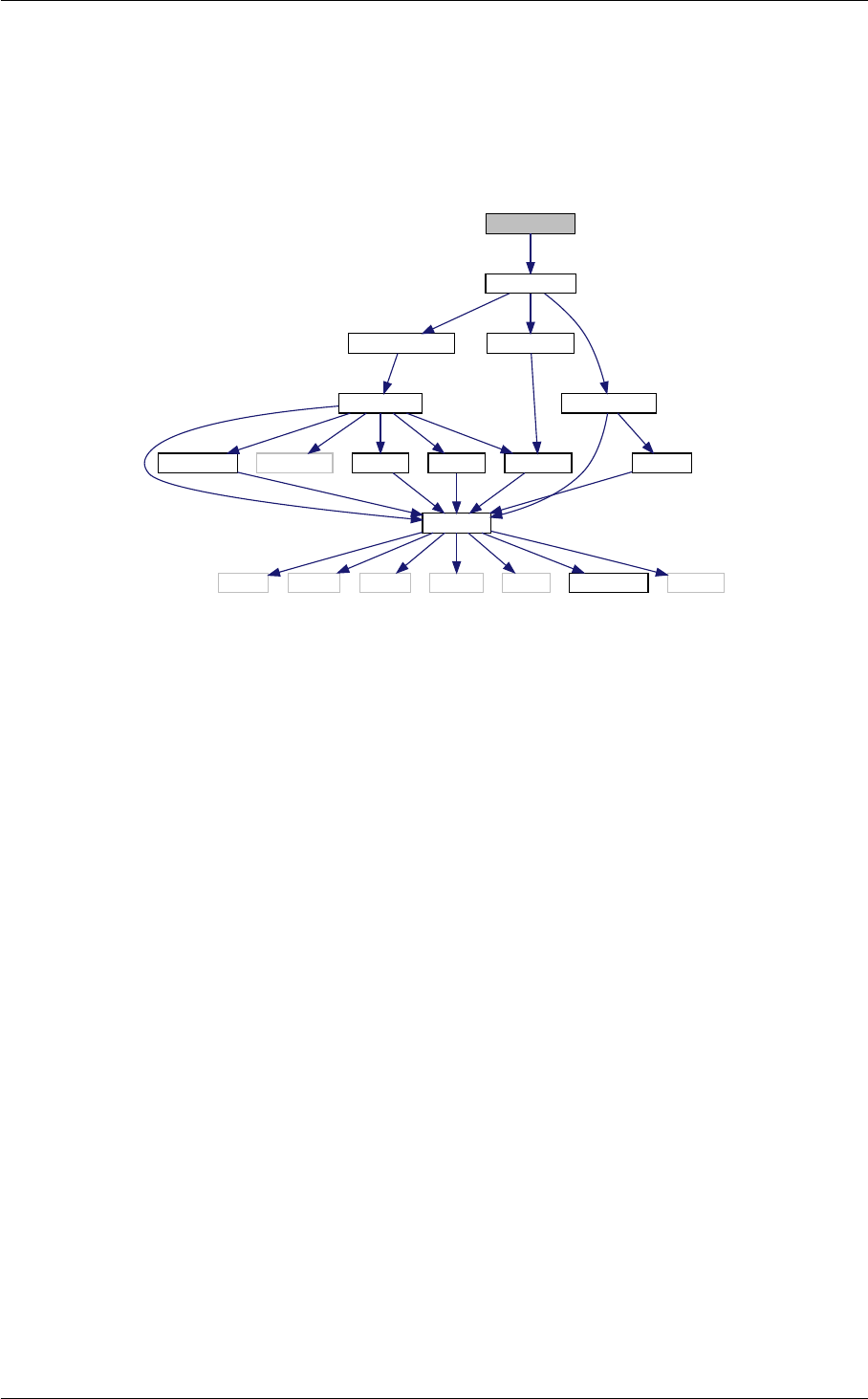
5.13 perturbations.c File Reference 93
5.13 perturbations.c File Reference
#include "perturbations.h"
Include dependency graph for perturbations.c:
perturbations.c
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
Functions
• int perturb_sources_at_tau (struct perturbs ∗ppt, int index_md, int index_ic, int index_type, double tau, double
∗psource)
• int perturb_init (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct perturbs ∗ppt)
• int perturb_free (struct perturbs ∗ppt)
• int perturb_indices_of_perturbs (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct
perturbs ∗ppt)
• int perturb_timesampling_for_sources (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth,
struct perturbs ∗ppt)
• int perturb_get_k_list (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct perturbs ∗ppt)
• int perturb_workspace_init (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct perturbs
∗ppt, int index_md, struct perturb_workspace ∗ppw)
• int perturb_workspace_free (struct perturbs ∗ppt, int index_md, struct perturb_workspace ∗ppw)
• int perturb_solve (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct perturbs ∗ppt, int
index_md, int index_ic, int index_k, struct perturb_workspace ∗ppw)
• int perturb_prepare_output (struct background ∗pba, struct perturbs ∗ppt)
• int perturb_find_approximation_number (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth,
struct perturbs ∗ppt, int index_md, double k, struct perturb_workspace ∗ppw, double tau_ini, double tau_end,
int ∗interval_number, int ∗interval_number_of)
• int perturb_find_approximation_switches (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth,
struct perturbs ∗ppt, int index_md, double k, struct perturb_workspace ∗ppw, double tau_ini, double tau_end,
double precision, int interval_number, int ∗interval_number_of, double ∗interval_limit, int ∗∗interval_approx)
• int perturb_vector_init (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct perturbs ∗ppt,
int index_md, int index_ic, double k, double tau, struct perturb_workspace ∗ppw, int ∗pa_old)
• int perturb_vector_free (struct perturb_vector ∗pv)
• int perturb_initial_conditions (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, int index←-
_md, int index_ic, double k, double tau, struct perturb_workspace ∗ppw)
Generated by Doxygen

94 File Documentation
• int perturb_approximations (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct perturbs
∗ppt, int index_md, double k, double tau, struct perturb_workspace ∗ppw)
• int perturb_timescale (double tau, void ∗parameters_and_workspace, double ∗timescale, ErrorMsg error_←-
message)
• int perturb_einstein (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct perturbs ∗ppt,
int index_md, double k, double tau, double ∗y, struct perturb_workspace ∗ppw)
• int perturb_total_stress_energy (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct
perturbs ∗ppt, int index_md, double k, double ∗y, struct perturb_workspace ∗ppw)
• int perturb_sources (double tau, double ∗y, double ∗dy, int index_tau, void ∗parameters_and_workspace,
ErrorMsg error_message)
• int perturb_print_variables (double tau, double ∗y, double ∗dy, void ∗parameters_and_workspace, ErrorMsg
error_message)
• int perturb_derivs (double tau, double ∗y, double ∗dy, void ∗parameters_and_workspace, ErrorMsg error_←-
message)
• int perturb_tca_slip_and_shear (double ∗y, void ∗parameters_and_workspace, ErrorMsg error_message)
5.13.1 Detailed Description
Documented perturbation module
Julien Lesgourgues, 23.09.2010
Deals with the perturbation evolution. This module has two purposes:
• at the beginning; to initialize the perturbations, i.e. to integrate the perturbation equations, and store tem-
porarily the terms contributing to the source functions as a function of conformal time. Then, to perform a few
manipulations of these terms in order to infer the actual source functions SX(k, τ), and to store them as a
function of conformal time inside an interpolation table.
• at any time in the code; to evaluate the source functions at a given conformal time (by interpolating within the
interpolation table).
Hence the following functions can be called from other modules:
1. perturb_init() at the beginning (but after background_init() and thermodynamics_init())
2. perturb_sources_at_tau() at any later time
3. perturb_free() at the end, when no more calls to perturb_sources_at_tau() are needed
5.13.2 Function Documentation
5.13.2.1 perturb_sources_at_tau()
int perturb_sources_at_tau (
struct perturbs ∗ppt,
int index_md,
int index_ic,
int index_type,
double tau,
double ∗psource )
Source function SX(k, τ)at a given conformal time tau.
Evaluate source functions at given conformal time tau by reading the pre-computed table and interpolating.
Generated by Doxygen

5.13 perturbations.c File Reference 95
Parameters
ppt Input: pointer to perturbation structure containing interpolation tables
index_md Input: index of requested mode
index_ic Input: index of requested initial condition
index_type Input: index of requested source function type
tau Input: any value of conformal time
psource Output: vector (already allocated) of source function as a function of k
Returns
the error status
Summary:
• interpolate in pre-computed table contained in ppt
5.13.2.2 perturb_init()
int perturb_init (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt )
Initialize the perturbs structure, and in particular the table of source functions.
Main steps:
• given the values of the flags describing which kind of perturbations should be considered (modes←-
: scalar/vector/tensor, initial conditions, type of source functions needed...), initialize indices and wavenumber
list
• define the time sampling for the output source functions
• for each mode (scalar/vector/tensor): initialize the indices of relevant perturbations, integrate the differential
system, compute and store the source functions.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
ppt Output: Initialized perturbation structure
Generated by Doxygen
96 File Documentation
Returns
the error status
Summary:
• define local variables
• perform preliminary checks
• initialize all indices and lists in perturbs structure using perturb_indices_of_perturbs()
• define the common time sampling for all sources using perturb_timesampling_for_sources()
• if we want to store perturbations, write titles and allocate storage
• create an array of workspaces in multi-thread case
• loop over modes (scalar, tensors, etc). For each mode:
• –>(a) create a workspace (one per thread in multi-thread case)
• –>(b) initialize indices of vectors of perturbations with perturb_indices_of_current_vectors()
• –>(c) loop over initial conditions and wavenumbers; for each of them, evolve perturbations and compute
source functions with perturb_solve()
5.13.2.3 perturb_free()
int perturb_free (
struct perturbs ∗ppt )
Free all memory space allocated by perturb_init().
To be called at the end of each run, only when no further calls to perturb_sources_at_tau() are needed.
Parameters
ppt Input: perturbation structure to be freed
Returns
the error status
Stuff related to perturbations output:
• Free non-NULL pointers
Generated by Doxygen

5.13 perturbations.c File Reference 97
5.13.2.4 perturb_indices_of_perturbs()
int perturb_indices_of_perturbs (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt )
Initialize all indices and allocate most arrays in perturbs structure.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
ppt Input/Output: Initialized perturbation structure
Returns
the error status
Summary:
• define local variables
• count modes (scalar, vector, tensor) and assign corresponding indices
• allocate array of number of types for each mode, ppt->tp_size[index_md]
• allocate array of number of initial conditions for each mode, ppt->ic_size[index_md]
• allocate array of arrays of source functions for each mode, ppt->source[index_md]
• initialization of all flags to false (will eventually be set to true later)
• source flags and indices, for sources that all modes have in common (temperature, polarization, ...). For
temperature, the term t2 is always non-zero, while other terms are non-zero only for scalars and vectors. For
polarization, the term e is always non-zero, while the term b is only for vectors and tensors.
• define k values with perturb_get_k_list()
• loop over modes. Initialize flags and indices which are specific to each mode.
• (a) scalars
• –>source flags and indices, for sources that are specific to scalars
• –>count scalar initial conditions (for scalars: ad, cdi, nid, niv; for tensors: only one) and assign corresponding
indices
• (b) vectors
• –>source flags and indices, for sources that are specific to vectors
• –>initial conditions for vectors
• (c) tensors
• –>source flags and indices, for sources that are specific to tensors
• –>only one initial condition for tensors
• (d) for each mode, allocate array of arrays of source functions for each initial conditions and wavenumber,
(ppt->source[index_md])[index_ic][index_type]
Generated by Doxygen

98 File Documentation
5.13.2.5 perturb_timesampling_for_sources()
int perturb_timesampling_for_sources (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt )
Define time sampling for source functions.
For each type, compute the list of values of tau at which sources will be sampled. Knowing the number of tau values,
allocate all arrays of source functions.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
ppt Input/Output: Initialized perturbation structure
Returns
the error status
Summary:
• define local variables
• allocate background/thermodynamics vectors
• first, just count the number of sampling points in order to allocate the array containing all values
• (a) if CMB requested, first sampling point = when the universe stops being opaque; otherwise, start sampling
gravitational potential at recombination [however, if perturbed recombination is requested, we also need to
start the system before recombination. Otherwise, the initial conditions for gas temperature and ionization
fraction perturbations (delta_T = 1/3 delta_b, delta_x_e) are not valid].
• (b) next sampling point = previous + ppr->perturb_sampling_stepsize ∗timescale_source, where:
• –>if CMB requested: timescale_source1 = |g/ ˙g|=|˙κ−¨κ/ ˙κ|−1; timescale_source2 = |2¨a/a −( ˙a/a)2|−1/2
(to sample correctly the late ISW effect; and timescale_source=1/(1/timescale_source1+1/timescale_←-
source2); repeat till today.
• –>if CMB not requested: timescale_source = 1/aH; repeat till today.
• –>infer total number of time steps, ppt->tau_size
• –>allocate array of time steps, ppt->tau_sampling[index_tau]
• –>repeat the same steps, now filling the array with each tau value:
• –>(b.1.) first sampling point = when the universe stops being opaque
• –>(b.2.) next sampling point = previous + ppr->perturb_sampling_stepsize ∗timescale_source, where
timescale_source1 = |g/ ˙g|=|˙κ−¨κ/ ˙κ|−1; timescale_source2 = |2¨a/a −( ˙a/a)2|−1/2(to sample correctly
the late ISW effect; and timescale_source=1/(1/timescale_source1+1/timescale_source2); repeat till today. If
CMB not requested: timescale_source = 1/aH; repeat till today.
• last sampling point = exactly today
• loop over modes, initial conditions and types. For each of them, allocate array of source functions.
Generated by Doxygen

5.13 perturbations.c File Reference 99
5.13.2.6 perturb_get_k_list()
int perturb_get_k_list (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt )
Define the number of comoving wavenumbers using the information passed in the precision structure.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
ppt Input: pointer to perturbation structure
Returns
the error status
Summary:
• allocate arrays related to k list for each mode
• scalar modes
• –>find k_max (as well as k_max_cmb[ppt->index_md_scalars], k_max_cl[ppt->index_md_scalars])
• –>test that result for k_min, k_max make sense
• vector modes
• –>find k_max (as well as k_max_cmb[ppt->index_md_vectors], k_max_cl[ppt->index_md_vectors])
• –>test that result for k_min, k_max make sense
• tensor modes
• –>find k_max (as well as k_max_cmb[ppt->index_md_tensors], k_max_cl[ppt->index_md_tensors])
• –>test that result for k_min, k_max make sense
• If user asked for k_output_values, add those to all k lists:
• –>Find indices in ppt->k[index_md] corresponding to 'k_output_values'. We are assuming that ppt->k is
sorted and growing, and we have made sure that ppt->k_output_values is also sorted and growing.
• –>Decide if we should add k_output_value now. This has to be this complicated, since we can only compare
the k-values when both indices are in range.
• –>The two MIN statements are here because in a normal run, the cl and cmb arrays contain a single k value
larger than their respective k_max. We are mimicking this behavior.
• finally, find the global k_min and k_max for the ensemble of all modes 9scalars, vectors, tensors)
Generated by Doxygen
100 File Documentation
5.13.2.7 perturb_workspace_init()
int perturb_workspace_init (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
int index_md,
struct perturb_workspace ∗ppw )
Initialize a perturb_workspace structure. All fields are allocated here, with the exception of the perturb_vector '–>pv'
field, which is allocated separately in perturb_vector_init. We allocate one such perturb_workspace structure per
thread and per mode (scalar/../tensor). Then, for each thread, all initial conditions and wavenumbers will use the
same workspace.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to the thermodynamics structure
ppt Input: pointer to the perturbation structure
index_md Input: index of mode under consideration (scalar/.../tensor)
ppw Input/Output: pointer to perturb_workspace structure which fields are allocated or filled here
Returns
the error status
Summary:
• define local variables
• Compute maximum l_max for any multipole
• Allocate sl[ ] array for freestreaming of multipoles (see arXiv:1305.3261) and initialize to 1.0, which is the K=0
value.
• define indices of metric perturbations obeying constraint equations (this can be done once and for all, be-
cause the vector of metric perturbations is the same whatever the approximation scheme, unlike the vector of
quantities to be integrated, which is allocated separately in perturb_vector_init)
• allocate some workspace in which we will store temporarily the values of background, thermodynamics, metric
and source quantities at a given time
• count number of approximations, initialize their indices, and allocate their flags
• For definiteness, initialize approximation flags to arbitrary values (correct values are overwritten in pertub_←-
find_approximation_switches)
• allocate fields where some of the perturbations are stored
Generated by Doxygen

5.13 perturbations.c File Reference 101
5.13.2.8 perturb_workspace_free()
int perturb_workspace_free (
struct perturbs ∗ppt,
int index_md,
struct perturb_workspace ∗ppw )
Free the perturb_workspace structure (with the exception of the perturb_vector '–>pv' field, which is freed sepa-
rately in perturb_vector_free).
Parameters
ppt Input: pointer to the perturbation structure
index_md Input: index of mode under consideration (scalar/.../tensor)
ppw Input: pointer to perturb_workspace structure to be freed
Returns
the error status
5.13.2.9 perturb_solve()
int perturb_solve (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
int index_md,
int index_ic,
int index_k,
struct perturb_workspace ∗ppw )
Solve the perturbation evolution for a given mode, initial condition and wavenumber, and compute the corresponding
source functions.
For a given mode, initial condition and wavenumber, this function finds the time ranges over which the perturbations
can be described within a given approximation. For each such range, it initializes (or redistributes) perturbations
using perturb_vector_init(), and integrates over time. Whenever a "source sampling time" is passed, the source
terms are computed and stored in the source table using perturb_sources().
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to the thermodynamics structure
ppt Input/Output: pointer to the perturbation structure (output source functions S(k,tau) written here)
index_md Input: index of mode under consideration (scalar/.../tensor)
index_ic Input: index of initial condition under consideration (ad, iso...)
index_k Input: index of wavenumber
ppw Input: pointer to perturb_workspace structure containing index values and workspaces
Generated by Doxygen

102 File Documentation
Returns
the error status
Summary:
• define local variables
• initialize indices relevant for back/thermo tables search
• get wavenumber value
• If non-zero curvature, update array of free-streaming coefficients ppw->s_l
• maximum value of tau for which sources are calculated for this wavenumber
• using bisection, compute minimum value of tau for which this wavenumber is integrated
• find the number of intervals over which approximation scheme is constant
• fill the structure containing all fixed parameters, indices and workspaces needed by perturb_derivs
• check whether we need to print perturbations to a file for this wavenumber
• loop over intervals over which approximation scheme is uniform. For each interval:
• –>(a) fix the approximation scheme
• –>(b) get the previous approximation scheme. If the current interval starts from the initial time tau_ini,
the previous approximation is set to be a NULL pointer, so that the function perturb_vector_init() knows that
perturbations must be initialized
• –>(c) define the vector of perturbations to be integrated over. If the current interval starts from the initial time
tau_ini, fill the vector with initial conditions for each mode. If it starts from an approximation switching point,
redistribute correctly the perturbations from the previous to the new vector of perturbations.
• –>(d) integrate the perturbations over the current interval.
• if perturbations were printed in a file, close the file
• fill the source terms array with zeros for all times between the last integrated time tau_max and tau_today.
• free quantities allocated at the beginning of the routine
5.13.2.10 perturb_prepare_output()
int perturb_prepare_output (
struct background ∗pba,
struct perturbs ∗ppt )
Write titles for all perturbations that we would like to print/store.
Generated by Doxygen
5.13 perturbations.c File Reference 103
5.13.2.11 perturb_find_approximation_number()
int perturb_find_approximation_number (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
int index_md,
double k,
struct perturb_workspace ∗ppw,
double tau_ini,
double tau_end,
int ∗interval_number,
int ∗interval_number_of )
For a given mode and wavenumber, find the number of intervals of time between tau_ini and tau_end such that the
approximation scheme (and the number of perturbation equations) is uniform.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to the thermodynamics structure
ppt Input: pointer to the perturbation structure
index_md Input: index of mode under consideration (scalar/.../tensor)
kInput: index of wavenumber
ppw Input: pointer to perturb_workspace structure containing index values and workspaces
tau_ini Input: initial time of the perturbation integration
tau_end Input: final time of the perturbation integration
interval_number Output: total number of intervals
interval_number←-
_of
Output: number of intervals with respect to each particular approximation
Returns
the error status
Summary:
• fix default number of intervals to one (if no approximation switch)
• loop over each approximation and add the number of approximation switching times
5.13.2.12 perturb_find_approximation_switches()
int perturb_find_approximation_switches (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
Generated by Doxygen
104 File Documentation
struct perturbs ∗ppt,
int index_md,
double k,
struct perturb_workspace ∗ppw,
double tau_ini,
double tau_end,
double precision,
int interval_number,
int ∗interval_number_of,
double ∗interval_limit,
int ∗∗ interval_approx )
For a given mode and wavenumber, find the values of time at which the approximation changes.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to the thermodynamics structure
ppt Input: pointer to the perturbation structure
index_md Input: index of mode under consideration (scalar/.../tensor)
kInput: index of wavenumber
ppw Input: pointer to perturb_workspace structure containing index values and workspaces
tau_ini Input: initial time of the perturbation integration
tau_end Input: final time of the perturbation integration
precision Input: tolerance on output values
interval_number Input: total number of intervals
interval_number←-
_of
Input: number of intervals with respect to each particular approximation
interval_limit Output: value of time at the boundary of the intervals: tau_ini, tau_switch1, ..., tau_end
interval_approx Output: value of approximations in each interval
Returns
the error status
Summary:
• write in output arrays the initial time and approximation
• if there are no approximation switches, just write final time and return
• if there are switches, consider approximations one after each other. Find switching time by bisection. Store
all switches in arbitrary order in array unsorted_tau_switch[ ]
• now sort interval limits in correct order
• store each approximation in chronological order
Generated by Doxygen

5.13 perturbations.c File Reference 105
5.13.2.13 perturb_vector_init()
int perturb_vector_init (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
int index_md,
int index_ic,
double k,
double tau,
struct perturb_workspace ∗ppw,
int ∗pa_old )
Initialize the field '–>pv' of a perturb_workspace structure, which is a perturb_vector structure. This structure con-
tains indices and values of all quantities which need to be integrated with respect to time (and only them: quantities
fixed analytically or obeying constraint equations are NOT included in this vector). This routine distinguishes be-
tween two cases:
–>the input pa_old is set to the NULL pointer:
This happens when we start integrating over a new wavenumber and we want to set initial conditions for the per-
turbations. Then, it is assumed that ppw–>pv is not yet allocated. This routine allocates it, defines all indices, and
then fills the vector ppw–>pv–>y with the initial conditions defined in perturb_initial_conditions.
–>the input pa_old is not set to the NULL pointer and describes some set of approximations:
This happens when we need to change approximation scheme while integrating over a given wavenumber. The
new approximation described by ppw–>pa is then different from pa_old. Then, this routine allocates a new vector
with a new size and new index values; it fills this vector with initial conditions taken from the previous vector passed
as an input in ppw–>pv, and eventually with some analytic approximations for the new variables appearing at this
time; then the new vector comes in replacement of the old one, which is freed.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to the thermodynamics structure
ppt Input: pointer to the perturbation structure
index_md Input: index of mode under consideration (scalar/.../tensor)
index_ic Input: index of initial condition under consideration (ad, iso...)
kInput: wavenumber
tau Input: conformal time
ppw Input/Output: workspace containing in input the approximation scheme, the
background/thermodynamics/metric quantities, and eventually the previous vector y; and in output
the new vector y.
pa_old Input: NULL is we need to set y to initial conditions for a new wavenumber; points towards a
perturb_approximations if we want to switch of approximation.
Returns
the error status
Summary:
Generated by Doxygen

106 File Documentation
• define local variables
• allocate a new perturb_vector structure to which ppw–>pv will point at the end of the routine
• initialize pointers to NULL (they will be allocated later if needed), relevant for perturb_vector_free()
• define all indices in this new vector (depends on approximation scheme, described by the input structure
ppw–>pa)
• (a) metric perturbations V or hvdepending on gauge
• (b) metric perturbation h is a propagating degree of freedom, so h and hdot are included in the vector of
ordinary perturbations, no in that of metric perturbations
• allocate vectors for storing the values of all these quantities and their time-derivatives at a given time
• specify which perturbations are needed in the evaluation of source terms
• case of setting initial conditions for a new wavenumber
• –>(a) check that current approximation scheme is consistent with initial conditions
• –>(b) let ppw–>pv points towards the perturb_vector structure that we just created
• –>(c) fill the vector ppw–>pv–>y with appropriate initial conditions
• case of switching approximation while a wavenumber is being integrated
• –>(a) for the scalar mode:
• —>(a.1.) check that the change of approximation scheme makes sense (note: before calling this routine
there is already a check that we wish to change only one approximation flag at a time)
• —>(a.2.) some variables (b, cdm, fld, ...) are not affected by any approximation. They need to be recon-
ducted whatever the approximation switching is. We treat them here. Below we will treat other variables case
by case.
• –>(b) for the vector mode
• —>(b.1.) check that the change of approximation scheme makes sense (note: before calling this routine
there is already a check that we wish to change only one approximation flag at a time)
• —>(b.2.) some variables (gw, gwdot, ...) are not affected by any approximation. They need to be recon-
ducted whatever the approximation switching is. We treat them here. Below we will treat other variables case
by case.
• –>(c) for the tensor mode
• —>(c.1.) check that the change of approximation scheme makes sense (note: before calling this routine
there is already a check that we wish to change only one approximation flag at a time)
• —>(c.2.) some variables (gw, gwdot, ...) are not affected by any approximation. They need to be recon-
ducted whatever the approximation switching is. We treat them here. Below we will treat other variables case
by case.
• –>(d) free the previous vector of perturbations
• –>(e) let ppw–>pv points towards the perturb_vector structure that we just created
5.13.2.14 perturb_vector_free()
int perturb_vector_free (
struct perturb_vector ∗pv )
Free the perturb_vector structure.
Generated by Doxygen
5.13 perturbations.c File Reference 107
Parameters
pv Input: pointer to perturb_vector structure to be freed
Returns
the error status
5.13.2.15 perturb_initial_conditions()
int perturb_initial_conditions (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
int index_md,
int index_ic,
double k,
double tau,
struct perturb_workspace ∗ppw )
For each mode, wavenumber and initial condition, this function initializes in the vector all values of perturbed vari-
ables (in a given gauge). It is assumed here that all values have previously been set to zero, only non-zero values
are set here.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
ppt Input: pointer to the perturbation structure
index_md Input: index of mode under consideration (scalar/.../tensor)
index_ic Input: index of initial condition under consideration (ad, iso...)
kInput: wavenumber
tau Input: conformal time
ppw Input/Output: workspace containing in input the approximation scheme, the
background/thermodynamics/metric quantities, and eventually the previous vector y; and in output
the new vector y.
Returns
the error status
Summary:
–>Declare local variables
–>For scalars
• (a) compute relevant background quantities: compute rho_r, rho_m, rho_nu (= all relativistic except photons),
and their ratio.
Generated by Doxygen
108 File Documentation
• (b) starts by setting everything in synchronous gauge. If another gauge is needed, we will perform a gauge
transformation below.
• –>(b.1.) adiabatic
• —>Canonical field (solving for the perturbations): initial perturbations set to zero, they should reach the
attractor soon enough.
• —>TODO: Incorporate the attractor IC from 1004.5509. delta_phi =−(a/k)2/φ0(ρ+p)θ, delta_phi_prime
=a2/φ0(delta_rho_phi + V'delta_phi), and assume theta, delta_rho as for perfect fluid with c2
s= 1 and w =
1/3 (ASSUMES radiation TRACKING)
• –>(b.2.) Cold dark matter Isocurvature
• –>(b.3.) Baryon Isocurvature
• –>(b.4.) Neutrino density Isocurvature
• –>(b.5.) Neutrino velocity Isocurvature
• (c) If the needed gauge is really the synchronous gauge, we need to affect the previously computed value of
eta to the actual variable eta
• (d) If the needed gauge is the newtonian gauge, we must compute alpha and then perform a gauge transfor-
mation for each variable
• (e) In any gauge, we should now implement the relativistic initial conditions in ur and ncdm variables
–>For tensors
tensor initial conditions take into account the fact that scalar (resp. tensor) Cl's are related to the real space power
spectrum of curvature (resp. of the tensor part of metric perturbations)
< R(x)R(x)>X
ij
< hij (x)hij (x)>
In momentum space it is conventional to use the modes R(k) and h(k) where the quantity h obeying to the equation
of propagation:
h00 +2a0
ah+ [k2+2K]h= 12πGa2(ρ+p)σ= 8πGa2pπ
and the power spectra in real space and momentum space are related through:
< R(x)R(x)>=Zdk
kk3
2π2< R(k)R(k)∗>=Zdk
kPR(k)
X
ij
< hij (x)hij (x)>=dk
kk3
2π2Fk2
K< h(k)h(k)∗>=Zdk
kFk2
KPh(k)
where PRand Phare the dimensionless spectrum of curvature R, and F is a function of k2/K, where K is the curva-
ture parameter. F is equal to one in flat space (K=0), and coming from the contraction of the laplacian eigentensor
Qij with itself. We will give F explicitly below.
Similarly the scalar (S) and tensor (T) Cl's are given by
Generated by Doxygen

5.13 perturbations.c File Reference 109
CS
l= 4πZdk
k[∆S
l(q)]2PR(k)
CT
l= 4πZdk
k[∆T
l(q)]2Fk2
KPh(k)
The usual convention for the tensor-to-scalar ratio r=At/Asat pivot scale = 16 epsilon in single-field inflation is
such that for constant PR(k)and Ph(k),
r= 6 Ph(k)
PR(k)
so
Ph(k) = PR(k)r
6=Asr
6=At
6
A priori it would make sense to say that for a power-law primordial spectrum there is an extra factor (k/kpivot)nt
(and eventually running and so on and so forth...)
However it has been shown that the minimal models of inflation in a negatively curved bubble lead to Ph(k) =
tanh(π∗ν/2). In open models it is customary to define the tensor tilt in a non-flat universe as a deviation from this
behavior rather than from true scale-invariance in the above sense.
Hence we should have
Ph(k) = At
6[tanh(π∗ν
2)](k/kpivot)(nt+...)
where the brackets
[...]
mean "if K<0"
Then
CT
l= 4πZdk
k[∆T
l(q)]2Fk2
KAt
6[tanh(π∗ν
2)](k/kpivot)(nt+...)
In the code, it is then a matter of choice to write:
• In the primordial module: Ph(k) = At
6tanh (π∗ν
2)(k/k∗)nT
• In the perturbation initial conditions: h= 1
• In the spectra module: CT
l=4
πRdk
k[∆T
l(q)]2Fk2
KPh(k)
or:
• In the primordial module: Ph(k) = At(k/k∗)nT
Generated by Doxygen
110 File Documentation
• In the perturbation initial conditions: h=q[Fk2
K/6] tanh (π∗ν
2)
• In the spectra module: CT
l=4
πRdk
k[∆T
l(q)]2Ph(k)
We choose this last option, such that the primordial and spectra module differ minimally in flat and non-flat space.
Then we must impose
h=sF
6tanh (π∗ν
2)
The factor F is found to be given by:
X
ij
< hij (x)hij (x)>=Zdk
k
k2(k2−K)
(k2+3K)(k2+2K)Ph(k)
Introducing as usual q2 = k2−3Kand using qdq = kdk this gives
X
ij
< hij (x)hij (x)>=Zdk
k
(q2−3K)(q2−4K)
q2(q2−K)Ph(k)
Using qdq = kdk this is equivalent to
X
ij
< hij (x)hij (x)>=Zdq
q
q2−4K
q2−KPh(k(q))
Finally, introducing ν=q/p|K|and sgnK=SIGN(k) =±1, this could also be written
X
ij
< hij (x)hij (x)>=Zdν
ν
(ν2−4sgnK)
(ν2−sgnK)Ph(k(ν))
Equation (43,44) of Hu, Seljak, White, Zaldarriaga is equivalent to absorbing the above factor (ν2−4sgnK)/(ν2−
sgnK)in the definition of the primordial spectrum. Since the initial condition should be written in terms of k rather
than nu, they should read
h=r[k2(k2−K)]/[(k2+3K)(k2+2K)]/6∗tanh (π∗ν
2)
We leave the freedom to multiply by an arbitrary number ppr->gw_ini. The standard convention corresponding to
standard definitions of r, AT,nTis however ppr->gw_ini=1.
Generated by Doxygen

5.13 perturbations.c File Reference 111
5.13.2.16 perturb_approximations()
int perturb_approximations (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
int index_md,
double k,
double tau,
struct perturb_workspace ∗ppw )
Evaluate background/thermodynamics at τ, infer useful flags / time scales for integrating perturbations.
Evaluate background quantities at τ, as well as thermodynamics for scalar mode; infer useful flags and time scales
for integrating the perturbations:
• check whether tight-coupling approximation is needed.
• check whether radiation (photons, massless neutrinos...) perturbations are needed.
• choose step of integration: step = ppr->perturb_integration_stepsize ∗min_time_scale, where min_time_←-
scale = smallest time scale involved in the equations. There are three time scales to compare:
1. that of recombination, τc= 1/κ0
2. Hubble time scale, τh=a/a0
3. Fourier mode, τk= 1/k
So, in general, min_time_scale = min(τc, τb, τh, τk).
However, if τcτhand τcτk, we can use the tight-coupling regime for photons and write equations in such
way that the time scale τcbecomes irrelevant (no effective mass term in 1/τc). Then, the smallest scale in the
equations is only min(τh, τk). In practise, it is sufficient to use only the condition τcτh.
Also, if ρmatter ρradiation and kaH, we can switch off radiation perturbations (i.e. switch on the free-
streaming approximation) and then the smallest scale is simply τh.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
ppt Input: pointer to the perturbation structure
index_md Input: index of mode under consideration (scalar/.../tensor)
kInput: wavenumber
tau Input: conformal time
ppw Input/Output: in output contains the approximation to be used at this time
Returns
the error status
Summary:
Generated by Doxygen

112 File Documentation
• define local variables
• compute Fourier mode time scale = τk= 1/k
• evaluate background quantities with background_at_tau() and Hubble time scale τh=a/a0
• for scalar modes:
• –>(a) evaluate thermodynamical quantities with thermodynamics_at_z()
• —>(b.1.) if κ0= 0, recombination is finished; tight-coupling approximation must be off
• —>(b.2.) if κ06= 0, recombination is not finished: check tight-coupling approximation
• -—>(b.2.a) compute recombination time scale for photons, τγ= 1/κ0
• -—>(b.2.b) check whether tight-coupling approximation should be on
• –>(c) free-streaming approximations
• for tensor modes:
• –>(a) evaluate thermodynamical quantities with thermodynamics_at_z()
• —>(b.1.) if κ0= 0, recombination is finished; tight-coupling approximation must be off
• —>(b.2.) if κ06= 0, recombination is not finished: check tight-coupling approximation
• -—>(b.2.a) compute recombination time scale for photons, τγ= 1/κ0
• -—>(b.2.b) check whether tight-coupling approximation should be on
5.13.2.17 perturb_timescale()
int perturb_timescale (
double tau,
void ∗parameters_and_workspace,
double ∗timescale,
ErrorMsg error_message )
Compute typical timescale over which the perturbation equations vary. Some integrators (e.g. Runge-Kunta) benefit
from calling this routine at each step in order to adapt the next step.
This is one of the few functions in the code which is passed to the generic_integrator() routine. Since generic_←-
integrator() should work with functions passed from various modules, the format of the arguments is a bit special:
• fixed parameters and workspaces are passed through a generic pointer. generic_integrator() doesn't know
the content of this pointer.
• the error management is a bit special: errors are not written as usual to pth->error_message, but to a generic
error_message passed in the list of arguments.
Parameters
tau Input: conformal time
parameters_and_workspace Input: fixed parameters (e.g. indices), workspace, approximation used, etc.
timescale Output: perturbation variation timescale (given the approximation used)
error_message Output: error message
Generated by Doxygen
5.13 perturbations.c File Reference 113
Summary:
• define local variables
• extract the fields of the parameter_and_workspace input structure
• compute Fourier mode time scale = τk= 1/k
• evaluate background quantities with background_at_tau() and Hubble time scale τh=a/a0
• for scalars modes:
• –>compute recombination time scale for photons, τγ= 1/κ0
• for vector modes:
• –>compute recombination time scale for photons, τγ= 1/κ0
• for tensor modes:
• –>compute recombination time scale for photons, τγ= 1/κ0
5.13.2.18 perturb_einstein()
int perturb_einstein (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
int index_md,
double k,
double tau,
double ∗y,
struct perturb_workspace ∗ppw )
Compute metric perturbations (those not integrated over time) using Einstein equations
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
ppt Input: pointer to the perturbation structure
index_md Input: index of mode under consideration (scalar/.../tensor)
kInput: wavenumber
tau Input: conformal time
yInput: vector of perturbations (those integrated over time) (already allocated)
ppw Input/Output: in output contains the updated metric perturbations
Returns
the error status
Generated by Doxygen

114 File Documentation
Summary:
• define local variables
• define wavenumber and scale factor related quantities
• sum up perturbations from all species
• for scalar modes:
• –>infer metric perturbations from Einstein equations
• for vector modes
• for tensor modes
5.13.2.19 perturb_total_stress_energy()
int perturb_total_stress_energy (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
int index_md,
double k,
double ∗y,
struct perturb_workspace ∗ppw )
Summary:
• define local variables
• wavenumber and scale factor related quantities
• for scalar modes
• –>(a) deal with approximation schemes
• —>(a.1.) photons
• -—>(a.1.1.) no approximation
• -—>(a.1.2.) radiation streaming approximation
• -—>(a.1.3.) tight coupling approximation
• —>(a.2.) ur
• –>(b) compute the total density, velocity and shear perturbations
• for vector modes
• –>photon contribution to vector sources:
• –>baryons
• for tensor modes
• –>photon contribution to gravitational wave source:
• –>ur contribution to gravitational wave source:
• –>ncdm contribution to gravitational wave source:
Generated by Doxygen
5.13 perturbations.c File Reference 115
5.13.2.20 perturb_sources()
int perturb_sources (
double tau,
double ∗y,
double ∗dy,
int index_tau,
void ∗parameters_and_workspace,
ErrorMsg error_message )
Compute the source functions (three terms for temperature, one for E or B modes, etc.)
This is one of the few functions in the code which is passed to the generic_integrator() routine. Since generic_←-
integrator() should work with functions passed from various modules, the format of the arguments is a bit special:
• fixed parameters and workspaces are passed through a generic pointer. generic_integrator() doesn't know
the content of this pointer.
• the error management is a bit special: errors are not written as usual to pth->error_message, but to a generic
error_message passed in the list of arguments.
Parameters
tau Input: conformal time
yInput: vector of perturbations
dy Input: vector of time derivative of perturbations
index_tau Input: index in the array tau_sampling
parameters_and_workspace Input/Output: in input, all parameters needed by perturb_derivs, in output,
source terms
error_message Output: error message
Returns
the error status
Summary:
• define local variables
• rename structure fields (just to avoid heavy notations)
• get background/thermo quantities in this point
• for scalars
• –>compute metric perturbations
• –>compute quantities depending on approximation schemes
• –>for each type, compute source terms
• for tensors
• –>compute quantities depending on approximation schemes
Generated by Doxygen

116 File Documentation
5.13.2.21 perturb_print_variables()
int perturb_print_variables (
double tau,
double ∗y,
double ∗dy,
void ∗parameters_and_workspace,
ErrorMsg error_message )
When testing the code or a cosmological model, it can be useful to output perturbations at each step of integration
(and not just the delta's at each source sampling point, which is achieved simply by asking for matter transfer
functions). Then this function can be passed to the generic_evolver routine.
By default, instead of passing this function to generic_evolver, one passes a null pointer. Then this function is just
not used.
Parameters
tau Input: conformal time
yInput: vector of perturbations
dy Input: vector of its derivatives (already allocated)
parameters_and_workspace Input: fixed parameters (e.g. indices)
error_message Output: error message
Summary:
• define local variables
• ncdm sector begins
• ncdm sector ends
• rename structure fields (just to avoid heavy notations)
• calculate perturbed recombination
• for scalar modes
• –>Get delta, deltaP/rho, theta, shear and store in array
• –>Do gauge transformation of delta, deltaP/rho (?) and theta using -= 3aH(1+w_ncdm) alpha for delta.
• –>Handle (re-)allocation
• for tensor modes:
• –>Handle (re-)allocation
Generated by Doxygen

5.13 perturbations.c File Reference 117
5.13.2.22 perturb_derivs()
int perturb_derivs (
double tau,
double ∗y,
double ∗dy,
void ∗parameters_and_workspace,
ErrorMsg error_message )
Compute derivative of all perturbations to be integrated
For each mode (scalar/vector/tensor) and each wavenumber k, this function computes the derivative of all values in
the vector of perturbed variables to be integrated.
This is one of the few functions in the code which is passed to the generic_integrator() routine. Since generic_←-
integrator() should work with functions passed from various modules, the format of the arguments is a bit special:
• fixed parameters and workspaces are passed through a generic pointer. generic_integrator() doesn't know
what the content of this pointer is.
• errors are not written as usual in pth->error_message, but in a generic error_message passed in the list of
arguments.
Parameters
tau Input: conformal time
yInput: vector of perturbations
dy Output: vector of its derivatives (already allocated)
parameters_and_workspace Input/Output: in input, fixed parameters (e.g. indices); in output, background
and thermo quantities evaluated at tau.
error_message Output: error message
Summary:
• define local variables
• rename the fields of the input structure (just to avoid heavy notations)
• get background/thermo quantities in this point
• get metric perturbations with perturb_einstein()
• compute related background quantities
• Compute 'generalised cotK function of argument p|K| ∗ τ, for closing hierarchy. (see equation 2.34 in
arXiv:1305.3261):
• for scalar modes:
• –>(a) define short-cut notations for the scalar perturbations
• –>(b) perturbed recombination
• –>(c) compute metric-related quantities (depending on gauge; additional gauges can be coded below)
–Each continuity equation contains a term in (theta+metric_continuity) with metric_continuity = (h_←-
prime/2) in synchronous gauge, (-3 phi_prime) in newtonian gauge
Generated by Doxygen

118 File Documentation
–Each Euler equation contains a source term metric_euler with metric_euler = 0 in synchronous gauge,
(k2 psi) in newtonian gauge
–Each shear derivative equation contains a source term metric_shear equal to metric_shear = (h_←-
prime+6eta_prime)/2 in synchronous gauge, 0 in newtonian gauge
–metric_shear_prime is the derivative of metric_shear
–In the ufa_class approximation, the leading-order source term is (h_prime/2) in synchronous gauge, (-3
(phi_prime+psi_prime)) in newtonian gauge: we approximate the later by (-6 phi_prime)
• –>(d) if some approximation schemes are turned on, enforce a few y[] values computed in perturb_einstein
• –>(e) BEGINNING OF ACTUAL SYSTEM OF EQUATIONS OF EVOLUTION
• —>photon temperature density
• —>baryon density
• —>baryon velocity (depends on tight-coupling approximation=tca)
• -—>perturbed recombination has an impact
• —>photon temperature higher momenta and photon polarization (depend on tight-coupling approximation)
• -—>if photon tight-coupling is off
• —–>define Π = Gγ0+Gγ2+Fγ2
• —–>photon temperature velocity
• —–>photon temperature shear
• —–>photon temperature l=3
• —–>photon temperature l>3
• —–>photon temperature lmax
• —–>photon polarization l=0
• —–>photon polarization l=1
• —–>photon polarization l=2
• —–>photon polarization l>2
• —–>photon polarization lmax_pol
• -—>if photon tight-coupling is on:
• —–>in that case, only need photon velocity
• —>cdm
• -—>newtonian gauge: cdm density and velocity
• -—>synchronous gauge: cdm density only (velocity set to zero by definition of the gauge)
• —>dcdm and dr
• -—>dcdm
• —>dr
• -—>dr F0
• -—>dr F1
• -—>exact dr F2
• -—>exact dr l=3
Generated by Doxygen

5.13 perturbations.c File Reference 119
• -—>exact dr l>3
• -—>exact dr lmax_dr
• —>fluid (fld)
• -—>factors w, w_prime, adiabatic sound speed ca2 (all three background-related), plus actual sound speed
in the fluid rest frame cs2
• -—>fluid density
• -—>fluid velocity
• —>scalar field (scf)
• -—>field value
• -—>Klein Gordon equation
• —>ultra-relativistic neutrino/relics (ur)
• -—>if radiation streaming approximation is off
• —–>ur density
• —–>ur velocity
• —–>exact ur shear
• —–>exact ur l=3
• —–>exact ur l>3
• —–>exact ur lmax_ur
• —–>in fluid approximation (ufa): only ur shear needed
• —>non-cold dark matter (ncdm): massive neutrinos, WDM, etc.
• -—>first case: use a fluid approximation (ncdmfa)
• —–>loop over species
• —–>define intermediate quantitites
• —–>exact continuity equation
• —–>exact euler equation
• —–>different ansatz for approximate shear derivative
• —–>jump to next species
• -—>second case: use exact equation (Boltzmann hierarchy on momentum grid)
• —–>loop over species
• —–>loop over momentum
• —–>define intermediate quantities
• —–>ncdm density for given momentum bin
• —–>ncdm velocity for given momentum bin
• —–>ncdm shear for given momentum bin
• —–>ncdm l>3 for given momentum bin
• —–>ncdm lmax for given momentum bin (truncation as in Ma and Bertschinger) but with curvature taken
into account a la arXiv:1305.3261
Generated by Doxygen

120 File Documentation
• —–>jump to next momentum bin or species
• —>metric
• —>eta of synchronous gauge
• vector mode
• –>baryon velocity
• tensor modes:
• –>non-cold dark matter (ncdm): massive neutrinos, WDM, etc.
• —>loop over species
• -—>loop over momentum
• -—>define intermediate quantities
• -—>ncdm density for given momentum bin
• -—>ncdm l>0 for given momentum bin
• -—>ncdm lmax for given momentum bin (truncation as in Ma and Bertschinger) but with curvature taken into
account a la arXiv:1305.3261
• -—>jump to next momentum bin or species
• –>tensor metric perturbation h (gravitational waves)
• –>its time-derivative
5.13.2.23 perturb_tca_slip_and_shear()
int perturb_tca_slip_and_shear (
double ∗y,
void ∗parameters_and_workspace,
ErrorMsg error_message )
Summary:
• define local variables
• rename the fields of the input structure (just to avoid heavy notations)
• compute related background quantities
• –>(a) define short-cut notations for the scalar perturbations
• –>(b) define short-cut notations used only in tight-coupling approximation
• –>(c) compute metric-related quantities (depending on gauge; additional gauges can be coded below)
–Each continuity equation contains a term in (theta+metric_continuity) with metric_continuity = (h_←-
prime/2) in synchronous gauge, (-3 phi_prime) in newtonian gauge
–Each Euler equation contains a source term metric_euler with metric_euler = 0 in synchronous gauge,
(k2 psi) in newtonian gauge
–Each shear derivative equation contains a source term metric_shear equal to metric_shear = (h_←-
prime+6eta_prime)/2 in synchronous gauge, 0 in newtonian gauge
Generated by Doxygen
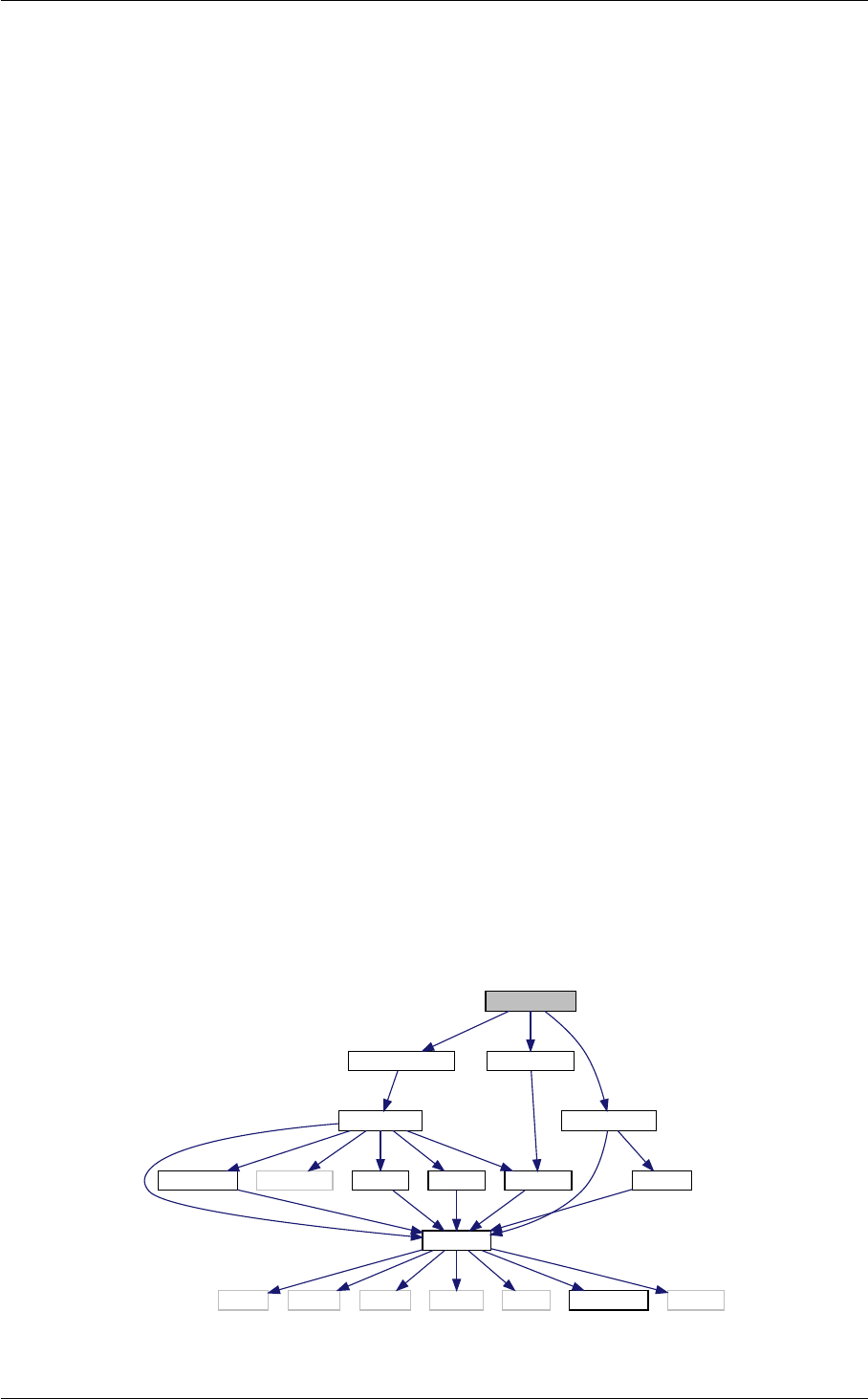
5.14 perturbations.h File Reference 121
–metric_shear_prime is the derivative of metric_shear
–In the ufa_class approximation, the leading-order source term is (h_prime/2) in synchronous gauge, (-3
(phi_prime+psi_prime)) in newtonian gauge: we approximate the later by (-6 phi_prime)
• –>(d) if some approximation schemes are turned on, enforce a few y[ ] values computed in perturb_einstein
• —>like Ma & Bertschinger
• —>relax assumption dkappa∼a−2(like in CAMB)
• —>also relax assumption cb2∼a−1
• —>intermediate quantities for 2nd order tca: shear_g at first order in tight-coupling
• —>intermediate quantities for 2nd order tca: zero order for theta_b' = theta_g'
• -—>perturbed recombination has an impact
• —>intermediate quantities for 2nd order tca: shear_g_prime at first order in tight-coupling
• —>2nd order as in CRS
• —>2nd order like in CLASS paper
• —>add only the most important 2nd order terms
• —>store tight-coupling values of photon shear and its derivative
5.14 perturbations.h File Reference
#include "thermodynamics.h"
#include "evolver_ndf15.h"
#include "evolver_rkck.h"
Include dependency graph for perturbations.h:
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
Generated by Doxygen
122 File Documentation
This graph shows which files directly or indirectly include this file:
perturbations.h
input.h
class.h
primordial.h perturbations.c
input.c
class.c
nonlinear.h
nonlinear_conflict
-20170920-132422.h
nonlinear_conflict
-20170920-150212.h nonlinear_exp.h nonlinear_test.h primordial.c
transfer.h nonlinear.c
spectra.h transfer.c
lensing.hspectra.c
output.h lensing.c
output.c
Data Structures
• struct perturbs
• struct perturb_vector
• struct perturb_workspace
• struct perturb_parameters_and_workspace
Macros
• #define _MAX_NUMBER_OF_K_FILES_ 30
Enumerations
• enum tca_flags
• enum tca_method
• enum possible_gauges {newtonian,synchronous }
• #define _SELECTION_NUM_MAX_ 100
Generated by Doxygen
5.14 perturbations.h File Reference 123
5.14.1 Detailed Description
Documented includes for perturbation module
5.14.2 Data Structure Documentation
5.14.2.1 struct perturbs
Structure containing everything about perturbations that other modules need to know, in particular tabled values of
the source functions S(k, τ)for all requested modes (scalar/vector/tensor), initial conditions, types (temperature,
E-polarization, B-polarization, lensing potential, etc), multipole l and wavenumber k.
Data Fields
short has_perturbations do we need to compute perturbations at
all ?
short has_cls do we need any harmonic space
spectrum Cl(and hence Bessel
functions, transfer functions, ...)?
short has_scalars do we need scalars?
short has_vectors do we need vectors?
short has_tensors do we need tensors?
short has_ad do we need adiabatic mode?
short has_bi do we need isocurvature bi mode?
short has_cdi do we need isocurvature cdi mode?
short has_nid do we need isocurvature nid mode?
short has_niv do we need isocurvature niv mode?
short has_perturbed_recombination Do we want to consider perturbed
temperature and ionization fraction?
enum tensor_methods tensor_method Neutrino contribution to tensors way to
treat neutrinos in tensor
perturbations(neglect, approximate as
massless, take exact equations)
short evolve_tensor_ur will we evolve ur tensor perturbations
(either because we have ur species, or
we have ncdm species with massless
approximation) ?
short evolve_tensor_ncdm will we evolve ncdm tensor perturbations
(if we have ncdm species and we use
the exact method) ?
short has_cl_cmb_temperature do we need Cl's for CMB temperature?
short has_cl_cmb_polarization do we need Cl's for CMB polarization?
short has_cl_cmb_lensing_potential do we need Cl's for CMB lensing
potential?
short has_cl_lensing_potential do we need Cl's for galaxy lensing
potential?
short has_cl_number_count do we need Cl's for density number
count?
short has_pk_matter do we need matter Fourier spectrum?
short has_density_transfers do we need to output individual matter
density transfer functions?
Generated by Doxygen

124 File Documentation
Data Fields
short has_velocity_transfers do we need to output individual matter
velocity transfer functions?
short has_metricpotential_transfers do we need to output individual transfer
functions for scalar metric
perturbations?
short has_nl_corrections_based_on_delta_m do we want to compute non-linear
corrections with an algorithm relying on
delta_m (like halofit)?
short has_nc_density in dCl, do we want density terms ?
short has_nc_rsd in dCl, do we want redshift space
distortion terms ?
short has_nc_lens in dCl, do we want lensing terms ?
short has_nc_gr in dCl, do we want gravity terms ?
int l_scalar_max maximum l value for CMB scalars Cl's
int l_vector_max maximum l value for CMB vectors Cl's
int l_tensor_max maximum l value for CMB tensors Cl's
int l_lss_max maximum l value for LSS Cl's (density
and lensing potential in bins)
double k_max_for_pk maximum value of k in 1/Mpc in P(k) (if
Cl's also requested, overseeded by
value kmax inferred from l_scalar_max if
it is bigger)
int selection_num number of selection functions (i.e. bins)
for matter density Cl's
enum selection_type selection type of selection functions
double selection_mean[_SELECTION_NUM_MAX_]centers of selection functions
double selection_width[_SELECTION_NUM_MAX_]widths of selection functions
int switch_sw in temperature calculation, do we want
to include the intrinsic temperature +
Sachs Wolfe term?
int switch_eisw in temperature calculation, do we want
to include the early integrated Sachs
Wolfe term?
int switch_lisw in temperature calculation, do we want
to include the late integrated Sachs
Wolfe term?
int switch_dop in temperature calculation, do we want
to include the Doppler term?
int switch_pol in temperature calculation, do we want
to include the polarization-related term?
double eisw_lisw_split_z at which redshift do we define the cut
between eisw and lisw ?
int store_perturbations Do we want to store perturbations?
int k_output_values_num Number of perturbation outputs
(default=0)
double k_output_values[_MAX_NUMBER_OF_K_FILES_]List of k values where perturbation
output is requested.
int ∗index_k_output_values List of indices corresponding to k-values
close to k_output_values for each mode.
[index_md∗k_output_values_num+ik]
char scalar_titles[_MAXTITLESTRINGLENGTH_]DELIMITER separated string of titles for
scalar perturbation output files.
Generated by Doxygen

5.14 perturbations.h File Reference 125
Data Fields
char vector_titles[_MAXTITLESTRINGLENGTH_]DELIMITER separated string of titles for
vector perturbation output files.
char tensor_titles[_MAXTITLESTRINGLENGTH_]DELIMITER separated string of titles for
tensor perturbation output files.
int number_of_scalar_titles number of titles/columns in scalar
perturbation output files
int number_of_vector_titles number of titles/columns in vector
perturbation output files
int number_of_tensor_titles number of titles/columns in tensor
perturbation output files
double ∗scalar_perturbations_data[_MAX_NUMBER_OF_K_FILES_]Array of double pointers to perturbation
output for scalars
double ∗vector_perturbations_data[_MAX_NUMBER_OF_K_FILES_]Array of double pointers to perturbation
output for vectors
double ∗tensor_perturbations_data[_MAX_NUMBER_OF_K_FILES_]Array of double pointers to perturbation
output for tensors
int size_scalar_perturbation_data[_MAX_NUMBER_OF_K_FILES_]Array of sizes of scalar double pointers
int size_vector_perturbation_data[_MAX_NUMBER_OF_K_FILES_]Array of sizes of vector double pointers
int size_tensor_perturbation_data[_MAX_NUMBER_OF_K_FILES_]Array of sizes of tensor double pointers
double three_ceff2_ur 3 x effective squared sound speed for
the ultrarelativistic perturbations
double three_cvis2_ur 3 x effective viscosity parameter for the
ultrarelativistic perturbations
double z_max_pk when we compute only the matter
spectrum / transfer functions, but not the
CMB, we are sometimes interested to
sample source functions at very high
redshift, way before recombination. This
z_max_pk will then fix the initial
sampling time of the sources.
short has_cmb do we need CMB-related sources
(temperature, polarization) ?
short has_lss do we need LSS-related sources
(lensing potential, ...) ?
enum possible_gauges gauge gauge in which to perform this
calculation
int index_md_scalars index value for scalars
int index_md_tensors index value for tensors
int index_md_vectors index value for vectors
int md_size number of modes included in
computation
int index_ic_ad index value for adiabatic
int index_ic_cdi index value for CDM isocurvature
int index_ic_bi index value for baryon isocurvature
int index_ic_nid index value for neutrino density
isocurvature
int index_ic_niv index value for neutrino velocity
isocurvature
int index_ic_ten index value for unique possibility for
tensors
Generated by Doxygen

126 File Documentation
Data Fields
int ∗ic_size for a given mode, ic_size[index_md] =
number of initial conditions included in
computation
short has_source_t do we need source for CMB
temperature?
short has_source_p do we need source for CMB
polarization?
short has_source_delta_m do we need source for delta of total
matter?
short has_source_delta_cb do we ALSO need source for delta of
ONLY cdm and baryon?
short has_source_delta_g do we need source for delta of gammas?
short has_source_delta_b do we need source for delta of baryons?
short has_source_delta_cdm do we need source for delta of cold dark
matter?
short has_source_delta_dcdm do we need source for delta of DCDM?
short has_source_delta_fld do we need source for delta of dark
energy?
short has_source_delta_scf do we need source for delta from scalar
field?
short has_source_delta_dr do we need source for delta of decay
radiation?
short has_source_delta_ur do we need source for delta of
ultra-relativistic neutrinos/relics?
short has_source_delta_ncdm do we need source for delta of all
non-cold dark matter species (e.g.
massive neutrinos)?
short has_source_theta_m do we need source for theta of total
matter?
short has_source_theta_cb do we ALSO need source for theta of
ONLY cdm and baryon?
short has_source_theta_g do we need source for theta of
gammas?
short has_source_theta_b do we need source for theta of baryons?
short has_source_theta_cdm do we need source for theta of cold dark
matter?
short has_source_theta_dcdm do we need source for theta of DCDM?
short has_source_theta_fld do we need source for theta of dark
energy?
short has_source_theta_scf do we need source for theta of scalar
field?
short has_source_theta_dr do we need source for theta of
ultra-relativistic neutrinos/relics?
short has_source_theta_ur do we need source for theta of
ultra-relativistic neutrinos/relics?
short has_source_theta_ncdm do we need source for theta of all
non-cold dark matter species (e.g.
massive neutrinos)?
short has_source_phi do we need source for metric fluctuation
phi?
short has_source_phi_prime do we need source for metric fluctuation
phi'?
Generated by Doxygen
5.14 perturbations.h File Reference 127
Data Fields
short has_source_phi_plus_psi do we need source for metric fluctuation
(phi+psi)?
short has_source_psi do we need source for metric fluctuation
psi?
short has_source_h do we need source for metric fluctuation
h?
short has_source_h_prime do we need source for metric fluctuation
h'?
short has_source_eta do we need source for metric fluctuation
eta?
short has_source_eta_prime do we need source for metric fluctuation
eta'?
int index_tp_t0 index value for temperature (j=0 term)
int index_tp_t1 index value for temperature (j=1 term)
int index_tp_t2 index value for temperature (j=2 term)
int index_tp_p index value for polarization
int index_tp_delta_m index value for delta tot
int index_tp_delta_cb index value for delta cb
int index_tp_delta_g index value for delta of gammas
int index_tp_delta_b index value for delta of baryons
int index_tp_delta_cdm index value for delta of cold dark matter
int index_tp_delta_dcdm index value for delta of DCDM
int index_tp_delta_fld index value for delta of dark energy
int index_tp_delta_scf index value for delta of scalar field
int index_tp_delta_dr index value for delta of decay radiation
int index_tp_delta_ur index value for delta of ultra-relativistic
neutrinos/relics
int index_tp_delta_ncdm1 index value for delta of first non-cold
dark matter species (e.g. massive
neutrinos)
int index_tp_perturbed_recombination_delta_tempGas temperature perturbation
int index_tp_perturbed_recombination_delta_chiInionization fraction perturbation
int index_tp_theta_m index value for theta tot
int index_tp_theta_cb index value for theta cb
int index_tp_theta_g index value for theta of gammas
int index_tp_theta_b index value for theta of baryons
int index_tp_theta_cdm index value for theta of cold dark matter
int index_tp_theta_dcdm index value for theta of DCDM
int index_tp_theta_fld index value for theta of dark energy
int index_tp_theta_scf index value for theta of scalar field
int index_tp_theta_ur index value for theta of ultra-relativistic
neutrinos/relics
int index_tp_theta_dr index value for F1 of decay radiation
int index_tp_theta_ncdm1 index value for theta of first non-cold
dark matter species (e.g. massive
neutrinos)
int index_tp_phi index value for metric fluctuation phi
int index_tp_phi_prime index value for metric fluctuation phi'
int index_tp_phi_plus_psi index value for metric fluctuation phi+psi
Generated by Doxygen
128 File Documentation
Data Fields
int index_tp_psi index value for metric fluctuation psi
int index_tp_h index value for metric fluctuation h
int index_tp_h_prime index value for metric fluctuation h'
int index_tp_eta index value for metric fluctuation eta
int index_tp_eta_prime index value for metric fluctuation eta'
int ∗tp_size number of types tp_size[index_md]
included in computation for each mode
int ∗k_size_cmb k_size_cmb[index_md] number of k
values used for CMB calculations,
requiring a fine sampling in k-space
int ∗k_size_cl k_size_cl[index_md] number of k values
used for non-CMB Clcalculations,
requiring a coarse sampling in k-space.
int ∗k_size k_size[index_md] = total number of k
values, including those needed for P(k)
but not for Cl's
double ∗∗ k k[index_md][index_k] = list of values
double k_min minimum value (over all modes)
double k_max maximum value (over all modes)
int tau_size tau_size = number of values
double ∗tau_sampling tau_sampling[index_tau] = list of tau
values
double selection_min_of_tau_min used in presence of selection functions
(for matter density, cosmic shear...)
double selection_max_of_tau_max used in presence of selection functions
(for matter density, cosmic shear...)
double selection_delta_tau used in presence of selection functions
(for matter density, cosmic shear...)
double ∗selection_tau_min value of conformal time below which
W(tau) is considered to vanish for each
bin
double ∗selection_tau_max value of conformal time above which
W(tau) is considered to vanish for each
bin
double ∗selection_tau value of conformal time at the center of
each bin
double ∗selection_function selection function W(tau), normalized to
RW(tau)dtau = 1, stored in
selection_function[bin∗ppt->tau_←-
size+index_tau]
double ∗∗∗ sources Pointer towards the source interpolation
table sources[index_md] [index_ic ∗
ppt->tp_size[index_md] + index_type]
[index_tau ∗ppt->k_size + index_k]
short perturbations_verbose flag regulating the amount of information
sent to standard output (none if set to
zero)
ErrorMsg error_message zone for writing error messages
Generated by Doxygen

5.14 perturbations.h File Reference 129
5.14.2.2 struct perturb_vector
Structure containing the indices and the values of the perturbation variables which are integrated over time (as
well as their time-derivatives). For a given wavenumber, the size of these vectors changes when the approximation
scheme changes.
Data Fields
int index_pt_delta_g photon density
int index_pt_theta_g photon velocity
int index_pt_shear_g photon shear
int index_pt_l3_g photon l=3
int l_max_g max momentum in Boltzmann hierarchy (at
least 3)
int index_pt_pol0_g photon polarization, l=0
int index_pt_pol1_g photon polarization, l=1
int index_pt_pol2_g photon polarization, l=2
int index_pt_pol3_g photon polarization, l=3
int l_max_pol_g max momentum in Boltzmann hierarchy (at
least 3)
int index_pt_delta_b baryon density
int index_pt_theta_b baryon velocity
int index_pt_delta_cdm cdm density
int index_pt_theta_cdm cdm velocity
int index_pt_delta_dcdm dcdm density
int index_pt_theta_dcdm dcdm velocity
int index_pt_delta_fld dark energy density in true fluid case
int index_pt_theta_fld dark energy velocity in true fluid case
int index_pt_Gamma_fld unique dark energy dynamical variable in PPF
case
int index_pt_phi_scf scalar field density
int index_pt_phi_prime_scf scalar field velocity
int index_pt_delta_ur density of ultra-relativistic neutrinos/relics
int index_pt_theta_ur velocity of ultra-relativistic neutrinos/relics
int index_pt_shear_ur shear of ultra-relativistic neutrinos/relics
int index_pt_l3_ur l=3 of ultra-relativistic neutrinos/relics
int l_max_ur max momentum in Boltzmann hierarchy (at
least 3)
int index_pt_perturbed_recombination_delta_temp Gas temperature perturbation
int index_pt_perturbed_recombination_delta_chi Inionization fraction perturbation
int index_pt_F0_dr The index to the first Legendre multipole of the
DR expansion. Not that this is not exactly the
usual delta, see Kaplinghat et al.,
astro-ph/9907388.
int l_max_dr max momentum in Boltzmann hierarchy for dr)
int index_pt_psi0_ncdm1 first multipole of perturbation of first ncdm
species, Psi_0
int N_ncdm number of distinct non-cold-dark-matter (ncdm)
species
int ∗l_max_ncdm mutipole l at which Boltzmann hierarchy is
truncated (for each ncdm species)
Generated by Doxygen

130 File Documentation
Data Fields
int ∗q_size_ncdm number of discrete momenta (for each ncdm
species)
int index_pt_eta synchronous gauge metric perturbation eta
int index_pt_phi newtonian gauge metric perturbation phi
int index_pt_hv_prime vector metric perturbation h_v' in synchronous
gauge
int index_pt_V vector metric perturbation V in Newtonian
gauge
int index_pt_gw tensor metric perturbation h (gravitational
waves)
int index_pt_gwdot its time-derivative
int pt_size size of perturbation vector
double ∗y vector of perturbations to be integrated
double ∗dy time-derivative of the same vector
int ∗used_in_sources boolean array specifying which perturbations
enter in the calculation of source functions
5.14.2.3 struct perturb_workspace
Workspace containing, among other things, the value at a given time of all background/perturbed quantities, as well
as their indices. There will be one such structure created for each mode (scalar/.../tensor) and each thread (in case
of parallel computing)
Data Fields
int index_mt_psi psi in longitudinal gauge
int index_mt_phi_prime (d phi/d conf.time) in longitudinal gauge
int index_mt_h_prime h' (wrt conf. time) in synchronous gauge
int index_mt_h_prime_prime h'' (wrt conf. time) in synchronous gauge
int index_mt_eta_prime eta' (wrt conf. time) in synchronous gauge
int index_mt_alpha α= (h0+ 6η0)/(2k2)in synchronous gauge
int index_mt_alpha_prime α0wrt conf. time) in synchronous gauge
int index_mt_gw_prime_prime second derivative wrt conformal time of gravitational
wave field, often called h
int index_mt_V_prime derivative of Newtonian gauge vector metric
perturbation V
int index_mt_hv_prime_prime Second derivative of Synchronous gauge vector
metric perturbation hv
int mt_size size of metric perturbation vector
double ∗pvecback background quantities
double ∗pvecthermo thermodynamics quantities
double ∗pvecmetric metric quantities
struct perturb_vector ∗pv pointer to vector of integrated perturbations and their
time-derivatives
double delta_rho total density perturbation (gives delta Too)
double rho_plus_p_theta total (rho+p)∗theta perturbation (gives delta Toi)
double rho_plus_p_shear total (rho+p)∗shear (gives delta Tij)
double delta_p total pressure perturbation (gives Tii)
Generated by Doxygen

5.14 perturbations.h File Reference 131
Data Fields
double gw_source stress-energy source term in Einstein's tensor
equations (gives Tij[tensor])
double vector_source_pi first stress-energy source term in Einstein's vector
equations
double vector_source_v second stress-energy source term in Einstein's vector
equations
double tca_shear_g photon shear in tight-coupling approximation
double tca_slip photon-baryon slip in tight-coupling approximation
double rsa_delta_g photon density in radiation streaming approximation
double rsa_theta_g photon velocity in radiation streaming approximation
double rsa_delta_ur photon density in radiation streaming approximation
double rsa_theta_ur photon velocity in radiation streaming approximation
double ∗delta_ncdm relative density perturbation of each ncdm species
double ∗theta_ncdm velocity divergence theta of each ncdm species
double ∗shear_ncdm shear for each ncdm species
double delta_m relative density perturbation of all non-relativistic
species
double theta_m velocity divergence theta of all non-relativistic species
double delta_cb relative density perturbation of only cdm and baryon
double theta_cb velocity divergence theta of only cdm and baryon
double delta_rho_fld density perturbation of fluid, not so trivial in PPF
scheme
double rho_plus_p_theta_fld velocity divergence of fluid, not so trivial in PPF
scheme
double S_fld S quantity sourcing Gamma_prime evolution in PPF
scheme (equivalent to eq. 15 in 0808.3125)
double Gamma_prime_fld Gamma_prime in PPF scheme (equivalent to eq. 14 in
0808.3125)
FILE ∗perturb_output_file filepointer to output file
int index_ikout index for output k value (when k_output_values is set)
short inter_mode flag defining the method used for interpolation
background/thermo quantities tables
int last_index_back the background interpolation function
background_at_tau() keeps memory of the last point
called through this index
int last_index_thermo the thermodynamics interpolation function
thermodynamics_at_z() keeps memory of the last
point called through this index
int index_ap_tca index for tight-coupling approximation
int index_ap_rsa index for radiation streaming approximation
int index_ap_ufa index for ur fluid approximation
int index_ap_ncdmfa index for ncdm fluid approximation
int ap_size number of relevant approximations for a given mode
int ∗approx array of approximation flags holding at a given time:
approx[index_ap]
int max_l_max maximum l_max for any multipole
double ∗s_l array of freestreaming coefficients
sl=p1−K∗(l2−1)/k2
Generated by Doxygen
132 File Documentation
5.14.2.4 struct perturb_parameters_and_workspace
Structure pointing towards all what the function that perturb_derivs needs to know: fixed input parameters and
indices contained in the various structures, workspace, etc.
Data Fields
struct precision ∗ppr pointer to the precision structure
struct background ∗pba pointer to the background structure
struct thermo ∗pth pointer to the thermodynamics structure
struct perturbs ∗ppt pointer to the precision structure
int index_md index of mode (scalar/.../vector/tensor)
int index_ic index of initial condition (adiabatic/isocurvature(s)/...)
int index_k index of wavenumber
double k current value of wavenumber in 1/Mpc
struct perturb_workspace ∗ppw workspace defined above
5.14.3 Macro Definition Documentation
5.14.3.1 _SELECTION_NUM_MAX_
#define _SELECTION_NUM_MAX_ 100
maximum number and types of selection function (for bins of matter density or cosmic shear)
5.14.3.2 _MAX_NUMBER_OF_K_FILES_
#define _MAX_NUMBER_OF_K_FILES_ 30
maximum number of k-values for perturbation output
5.14.4 Enumeration Type Documentation
5.14.4.1 tca_flags
enum tca_flags
flags for various approximation schemes (tca = tight-coupling approximation, rsa = radiation streaming approxima-
tion, ufa = massless neutrinos / ultra-relativistic relics fluid approximation)
CAUTION: must be listed below in chronological order, and cannot be reversible. When integrating equations for a
given mode, it is only possible to switch from left to right in the lists below.
Generated by Doxygen
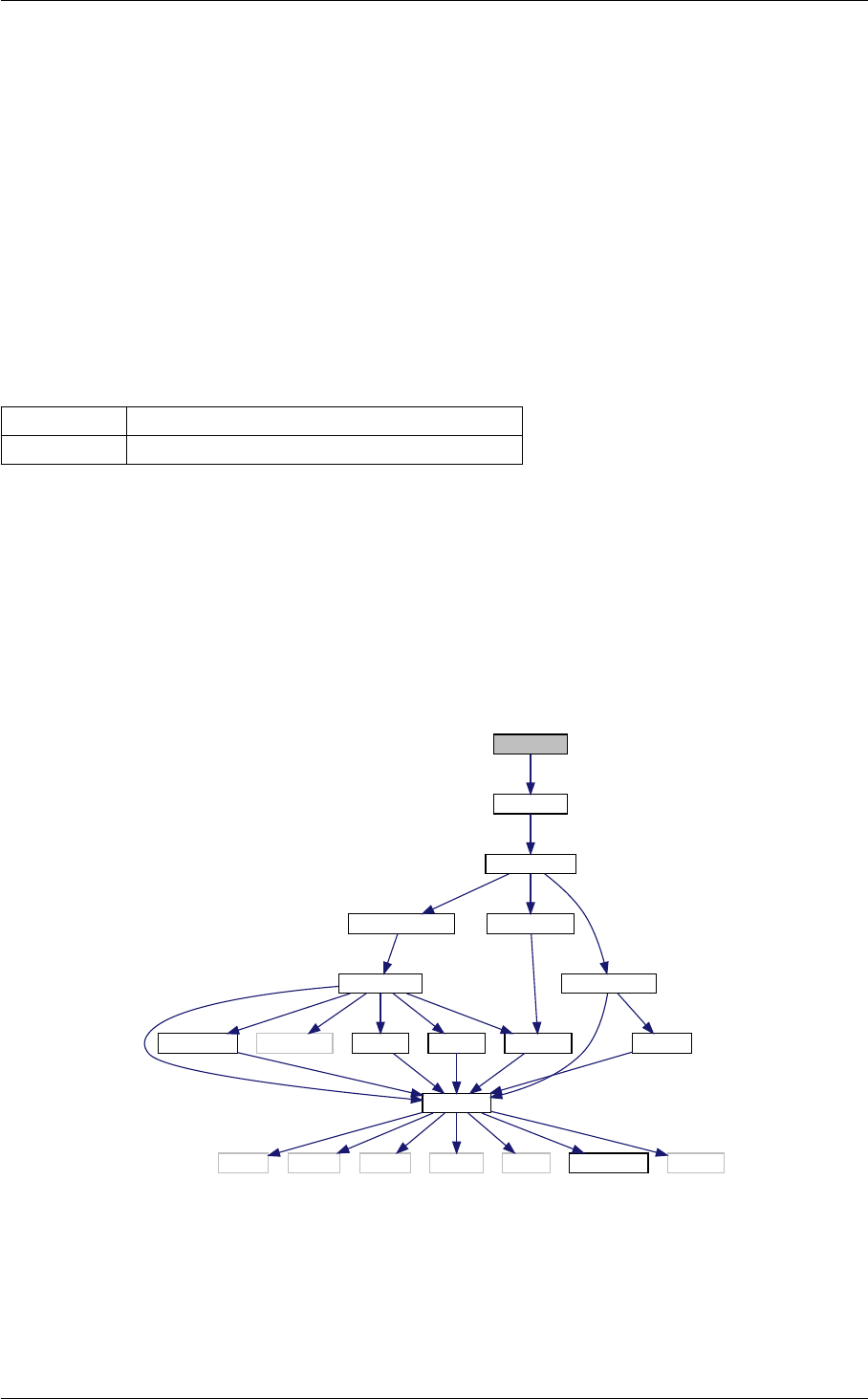
5.15 primordial.c File Reference 133
5.14.4.2 tca_method
enum tca_method
labels for the way in which each approximation scheme is implemented
5.14.4.3 possible_gauges
enum possible_gauges
List of coded gauges. More gauges can in principle be defined.
Enumerator
newtonian newtonian (or longitudinal) gauge
synchronous synchronous gauge with θcdm = 0 by convention
5.15 primordial.c File Reference
#include "primordial.h"
Include dependency graph for primordial.c:
primordial.c
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
Functions
• int primordial_spectrum_at_k (struct primordial ∗ppm, int index_md, enum linear_or_logarithmic mode, dou-
ble input, double ∗output)
Generated by Doxygen

134 File Documentation
• int primordial_init (struct precision ∗ppr, struct perturbs ∗ppt, struct primordial ∗ppm)
• int primordial_free (struct primordial ∗ppm)
• int primordial_indices (struct perturbs ∗ppt, struct primordial ∗ppm)
• int primordial_get_lnk_list (struct primordial ∗ppm, double kmin, double kmax, double k_per_decade)
• int primordial_analytic_spectrum_init (struct perturbs ∗ppt, struct primordial ∗ppm)
• int primordial_analytic_spectrum (struct primordial ∗ppm, int index_md, int index_ic1_ic2, double k, double
∗pk)
• int primordial_inflation_potential (struct primordial ∗ppm, double phi, double ∗V, double ∗dV, double ∗ddV)
• int primordial_inflation_hubble (struct primordial ∗ppm, double phi, double ∗H, double ∗dH, double ∗ddH,
double ∗dddH)
• int primordial_inflation_indices (struct primordial ∗ppm)
• int primordial_inflation_solve_inflation (struct perturbs ∗ppt, struct primordial ∗ppm, struct precision ∗ppr)
• int primordial_inflation_analytic_spectra (struct perturbs ∗ppt, struct primordial ∗ppm, struct precision ∗ppr,
double ∗y_ini)
• int primordial_inflation_spectra (struct perturbs ∗ppt, struct primordial ∗ppm, struct precision ∗ppr, double
∗y_ini)
• int primordial_inflation_one_wavenumber (struct perturbs ∗ppt, struct primordial ∗ppm, struct precision ∗ppr,
double ∗y_ini, int index_k)
• int primordial_inflation_one_k (struct primordial ∗ppm, struct precision ∗ppr, double k, double ∗y, double ∗dy,
double ∗curvature, double ∗tensor)
• int primordial_inflation_find_attractor (struct primordial ∗ppm, struct precision ∗ppr, double phi_0, double pre-
cision, double ∗y, double ∗dy, double ∗H_0, double ∗dphidt_0)
• int primordial_inflation_evolve_background (struct primordial ∗ppm, struct precision ∗ppr, double ∗y, double
∗dy, enum target_quantity target, double stop, short check_epsilon, enum integration_direction direction,
enum time_definition time)
• int primordial_inflation_check_potential (struct primordial ∗ppm, double phi, double ∗V, double ∗dV, double
∗ddV)
• int primordial_inflation_check_hubble (struct primordial ∗ppm, double phi, double ∗H, double ∗dH, double
∗ddH, double ∗dddH)
• int primordial_inflation_get_epsilon (struct primordial ∗ppm, double phi, double ∗epsilon)
• int primordial_inflation_find_phi_pivot (struct primordial ∗ppm, struct precision ∗ppr, double ∗y, double ∗dy)
• int primordial_inflation_derivs (double tau, double ∗y, double ∗dy, void ∗parameters_and_workspace, Error←-
Msg error_message)
• int primordial_external_spectrum_init (struct perturbs ∗ppt, struct primordial ∗ppm)
5.15.1 Detailed Description
Documented primordial module.
Julien Lesgourgues, 24.08.2010
This module computes the primordial spectra. It can be used in different modes: simple parametric form, evolving
inflaton perturbations, etc. So far only the mode corresponding to a simple analytic form in terms of amplitudes, tilts
and runnings has been developed.
The following functions can be called from other modules:
1. primordial_init() at the beginning (anytime after perturb_init() and before spectra_init())
2. primordial_spectrum_at_k() at any time for computing P(k) at any k
3. primordial_free() at the end
Generated by Doxygen
5.15 primordial.c File Reference 135
5.15.2 Function Documentation
5.15.2.1 primordial_spectrum_at_k()
int primordial_spectrum_at_k (
struct primordial ∗ppm,
int index_md,
enum linear_or_logarithmic mode,
double input,
double ∗output )
Primordial spectra for arbitrary argument and for all initial conditions.
This routine evaluates the primordial spectrum at a given value of k by interpolating in the pre-computed table.
When k is not in the pre-computed range but the spectrum can be found analytically, it finds it. Otherwise returns
an error.
Can be called in two modes; linear or logarithmic:
• linear: takes k, returns P(k)
• logarithmic: takes ln(k), return ln(P(k))
One little subtlety: in case of several correlated initial conditions, the cross-correlation spectrum can be negative.
Then, in logarithmic mode, the non-diagonal elements contain the cross-correlation angle P12/√P11P22 (from -1
to 1) instead of ln P12
This function can be called from whatever module at whatever time, provided that primordial_init() has been called
before, and primordial_free() has not been called yet.
Parameters
ppm Input: pointer to primordial structure containing tabulated primordial spectrum
index_md Input: index of mode (scalar, tensor, ...)
mode Input: linear or logarithmic
input Input: wavenumber in 1/Mpc (linear mode) or its logarithm (logarithmic mode)
output Output: for each pair of initial conditions, primordial spectra P(k) in Mpc3(linear mode), or their
logarithms and cross-correlation angles (logarithmic mode)
Returns
the error status
Summary:
• define local variables
• infer ln(k) from input. In linear mode, reject negative value of input k value.
Generated by Doxygen

136 File Documentation
• if ln(k) is not in the interpolation range, return an error, unless we are in the case of a analytic spectrum, for
which a direct computation is possible
• otherwise, interpolate in the pre-computed table
5.15.2.2 primordial_init()
int primordial_init (
struct precision ∗ppr,
struct perturbs ∗ppt,
struct primordial ∗ppm )
This routine initializes the primordial structure (in particular, it computes table of primordial spectrum values)
Parameters
ppr Input: pointer to precision structure (defines method and precision for all computations)
ppt Input: pointer to perturbation structure (useful for knowing k_min, k_max, etc.)
ppm Output: pointer to initialized primordial structure
Returns
the error status
Summary:
• define local variables
• check that we really need to compute the primordial spectra
• get kmin and kmax from perturbation structure. Test that they make sense.
• allocate and fill values of ln k's
• define indices and allocate tables in primordial structure
• deal with case of analytic primordial spectra (with amplitudes, tilts, runnings, etc.)
• deal with case of inflation with given V(φ)or H(φ)
• deal with the case of external calculation of Pk
• compute second derivative of each ln Pkversus lnk with spline, in view of interpolation
• derive spectral parameters from numerically computed spectra (not used by the rest of the code, but useful
to keep in memory for several types of investigation)
• expression for alpha_s comes from:
ns_2 = (lnpk_plus-lnpk_pivot)/(dlnk)+1
ns_1 = (lnpk_pivot-lnpk_minus)/(dlnk)+1
alpha_s = dns/dlnk = (ns_2-ns_1)/dlnk = (lnpk_plus-lnpk_pivot-lnpk_←-
pivot+lnpk_minus)/(dlnk)/(dlnk)
• expression for beta_s:
ppm->beta_s = (alpha_plus-alpha_minus)/dlnk = (lnpk_plusplus-2.∗lnpk_←-
plus+lnpk_pivot - (lnpk_pivot-2.∗lnpk_minus+lnpk_minusminus)/pow(dlnk,3)
Generated by Doxygen

5.15 primordial.c File Reference 137
5.15.2.3 primordial_free()
int primordial_free (
struct primordial ∗ppm )
This routine frees all the memory space allocated by primordial_init().
To be called at the end of each run.
Parameters
ppm Input: pointer to primordial structure (which fields must be freed)
Returns
the error status
5.15.2.4 primordial_indices()
int primordial_indices (
struct perturbs ∗ppt,
struct primordial ∗ppm )
This routine defines indices and allocates tables in the primordial structure
Parameters
ppt Input: pointer to perturbation structure
ppm Input/output: pointer to primordial structure
Returns
the error status
5.15.2.5 primordial_get_lnk_list()
int primordial_get_lnk_list (
struct primordial ∗ppm,
double kmin,
double kmax,
double k_per_decade )
This routine allocates and fills the list of wavenumbers k
Generated by Doxygen

138 File Documentation
Parameters
ppm Input/output: pointer to primordial structure
kmin Input: first value
kmax Input: last value that we should encompass
k_per_decade Input: number of k per decade
Returns
the error status
5.15.2.6 primordial_analytic_spectrum_init()
int primordial_analytic_spectrum_init (
struct perturbs ∗ppt,
struct primordial ∗ppm )
This routine interprets and stores in a condensed form the input parameters in the case of a simple analytic spectra
with amplitudes, tilts, runnings, in such way that later on, the spectrum can be obtained by a quick call to the routine
primordial_analytic_spectrum(()
Parameters
ppt Input: pointer to perturbation structure
ppm Input/output: pointer to primordial structure
Returns
the error status
5.15.2.7 primordial_analytic_spectrum()
int primordial_analytic_spectrum (
struct primordial ∗ppm,
int index_md,
int index_ic1_ic2,
double k,
double ∗pk )
This routine returns the primordial spectrum in the simple analytic case with amplitudes, tilts, runnings, for each
mode (scalar/tensor...), pair of initial conditions, and wavenumber.
Parameters
ppm Input/output: pointer to primordial structure
index_md Input: index of mode (scalar, tensor, ...)
index_ic1_ic2 Input: pair of initial conditions (ic1, ic2)
kInput: wavenumber in same units as pivot scale, i.e. in 1/Mpc
pk Output: primordial power spectrum A (k/k_pivot)∧(n+...)
Generated by Doxygen

5.15 primordial.c File Reference 139
Returns
the error status
5.15.2.8 primordial_inflation_potential()
int primordial_inflation_potential (
struct primordial ∗ppm,
double phi,
double ∗V,
double ∗dV,
double ∗ddV )
This routine encodes the inflaton scalar potential
Parameters
ppm Input: pointer to primordial structure
phi Input: background inflaton field value in units of Mp
VOutput: inflaton potential in units of Mp4
dV Output: first derivative of inflaton potential wrt the field
ddV Output: second derivative of inflaton potential wrt the field
Returns
the error status
5.15.2.9 primordial_inflation_hubble()
int primordial_inflation_hubble (
struct primordial ∗ppm,
double phi,
double ∗H,
double ∗dH,
double ∗ddH,
double ∗dddH )
This routine encodes the function H(φ)
Parameters
ppm Input: pointer to primordial structure
phi Input: background inflaton field value in units of Mp
HOutput: Hubble parameters in units of Mp
dH Output: dH/dφ
ddH Output: d2H/dφ2
dddH Output: d3H/dφ3
Generated by Doxygen
140 File Documentation
Returns
the error status
5.15.2.10 primordial_inflation_indices()
int primordial_inflation_indices (
struct primordial ∗ppm )
This routine defines indices used by the inflation simulator
Parameters
ppm Input/output: pointer to primordial structure
Returns
the error status
5.15.2.11 primordial_inflation_solve_inflation()
int primordial_inflation_solve_inflation (
struct perturbs ∗ppt,
struct primordial ∗ppm,
struct precision ∗ppr )
Main routine of inflation simulator. Its goal is to check the background evolution before and after the pivot value
phi=phi_pivot, and then, if this evolution is suitable, to call the routine primordial_inflation_spectra().
Parameters
ppt Input: pointer to perturbation structure
ppm Input/output: pointer to primordial structure
ppr Input: pointer to precision structure
Returns
the error status
Summary:
• define local variables
• allocate vectors for background/perturbed quantities
• eventually, needs first to find phi_pivot
Generated by Doxygen
5.15 primordial.c File Reference 141
• compute H_pivot at phi_pivot
• check positivity and negative slope of potential in field pivot value, and find value of phi_dot and H for field's
pivot value, assuming slow-roll attractor solution has been reached. If no solution, code will stop there.
• check positivity and negative slope of H(φ)in field pivot value, and get H_pivot
• find a_pivot, value of scale factor when k_pivot crosses horizon while phi=phi_pivot
• integrate background solution starting from phi_pivot and until k_max>>aH. This ensures that the inflationary
model considered here is valid and that the primordial spectrum can be computed. Otherwise, if slow-roll
brakes too early, model is not suitable and run stops.
• starting from this time, i.e. from y_ini[ ], we run the routine which takes care of computing the primordial
spectrum.
• before ending, we want to compute and store the values of φcorresponding to k=aH for k_min and k_max
• finally, we can de-allocate
5.15.2.12 primordial_inflation_analytic_spectra()
int primordial_inflation_analytic_spectra (
struct perturbs ∗ppt,
struct primordial ∗ppm,
struct precision ∗ppr,
double ∗y_ini )
Routine for the computation of an analytic apporoximation to the the primordial spectrum. In general, should be
used only for comparing with exact numerical computation performed by primordial_inflation_spectra().
Parameters
ppt Input: pointer to perturbation structure
ppm Input/output: pointer to primordial structure
ppr Input: pointer to precision structure
y_ini Input: initial conditions for the vector of background/perturbations, already allocated and filled
Returns
the error status
Summary
• allocate vectors for background/perturbed quantities
• initialize the background part of the running vector
• loop over Fourier wavenumbers
• read value of phi at time when k=aH
• get potential (and its derivatives) at this value
• calculate the analytic slow-roll formula for the spectra
• store the obtained result for curvature and tensor perturbations
Generated by Doxygen
142 File Documentation
5.15.2.13 primordial_inflation_spectra()
int primordial_inflation_spectra (
struct perturbs ∗ppt,
struct primordial ∗ppm,
struct precision ∗ppr,
double ∗y_ini )
Routine with a loop over wavenumbers for the computation of the primordial spectrum. For each wavenumber it
calls primordial_inflation_one_wavenumber()
Parameters
ppt Input: pointer to perturbation structure
ppm Input/output: pointer to primordial structure
ppr Input: pointer to precision structure
y_ini Input: initial conditions for the vector of background/perturbations, already allocated and filled
Returns
the error status
5.15.2.14 primordial_inflation_one_wavenumber()
int primordial_inflation_one_wavenumber (
struct perturbs ∗ppt,
struct primordial ∗ppm,
struct precision ∗ppr,
double ∗y_ini,
int index_k )
Routine coordinating the computation of the primordial spectrum for one wavenumber. It calls primordial_inflation←-
_one_k() to integrate the perturbation equations, and then it stores the result for the scalar/tensor spectra.
Parameters
ppt Input: pointer to perturbation structure
ppm Input/output: pointer to primordial structure
ppr Input: pointer to precision structure
y_ini Input: initial conditions for the vector of background/perturbations, already allocated and filled
index←-
_k
Input: index of wavenumber to be considered
Returns
the error status
Summary
Generated by Doxygen
5.15 primordial.c File Reference 143
• allocate vectors for background/perturbed quantities
• initialize the background part of the running vector
–evolve the background until the relevant initial time for integrating perturbations
–evolve the background/perturbation equations from this time and until some time after Horizon crossing
• store the obtained result for curvature and tensor perturbations
5.15.2.15 primordial_inflation_one_k()
int primordial_inflation_one_k (
struct primordial ∗ppm,
struct precision ∗ppr,
double k,
double ∗y,
double ∗dy,
double ∗curvature,
double ∗tensor )
Routine integrating the background plus perturbation equations for each wavenumber, and returning the scalar and
tensor spectrum.
Parameters
ppm Input: pointer to primordial structure
ppr Input: pointer to precision structure
kInput: Fourier wavenumber
yInput: running vector of background/perturbations, already allocated and initialized
dy Input: running vector of background/perturbation derivatives, already allocated
curvature Output: curvature perturbation
tensor Output: tensor perturbation
Returns
the error status
Summary:
• define local variables
• initialize the generic integrator (same integrator already used in background, thermodynamics and perturba-
tion modules)
• initialize variable used for deciding when to stop the calculation (= when the curvature remains stable)
• initialize conformal time to arbitrary value (here, only variations of tau matter: the equations that we integrate
do not depend explicitly on time)
• compute derivative of initial vector and infer first value of adaptive time-step
• loop over time
Generated by Doxygen

144 File Documentation
• clean the generic integrator
• store final value of curvature for this wavenumber
• store final value of tensor perturbation for this wavenumber
5.15.2.16 primordial_inflation_find_attractor()
int primordial_inflation_find_attractor (
struct primordial ∗ppm,
struct precision ∗ppr,
double phi_0,
double precision,
double ∗y,
double ∗dy,
double ∗H_0,
double ∗dphidt_0 )
Routine searching for the inflationary attractor solution at a given phi_0, by iterations, with a given tolerance. If no
solution found within tolerance, returns error message. The principle is the following. The code starts integrating
the background equations from various values of phi, corresponding to earlier and earlier value before phi_0, and
separated by a small arbitrary step size, corresponding roughly to 1 e-fold of inflation. Each time, the integration
starts with the initial condition φ=−V0/3H(slow-roll prediction). If the found value of φ0in phi_0 is stable (up to
the parameter "precision"), the code considers that there is an attractor, and stops iterating. If this process does not
converge, it returns an error message.
Parameters
ppm Input: pointer to primordial structure
ppr Input: pointer to precision structure
phi_0 Input: field value at which we wish to find the solution
precision Input: tolerance on output values (if too large, an attractor will always considered to be found)
yInput: running vector of background variables, already allocated and initialized
dy Input: running vector of background derivatives, already allocated
H_0 Output: Hubble value at phi_0 for attractor solution
dphidt←-
_0
Output: field derivative value at phi_0 for attractor solution
Returns
the error status
5.15.2.17 primordial_inflation_evolve_background()
int primordial_inflation_evolve_background (
struct primordial ∗ppm,
struct precision ∗ppr,
Generated by Doxygen
5.15 primordial.c File Reference 145
double ∗y,
double ∗dy,
enum target_quantity target,
double stop,
short check_epsilon,
enum integration_direction direction,
enum time_definition time )
Routine integrating background equations only, from initial values stored in y, to a final value (if target = aH, until aH
= aH_stop; if target = phi, till phi = phi_stop; if target = end_inflation, until d2a/dt2= 0 (here t = proper time)). In
output, y contains the final background values. In addition, if check_epsilon is true, the routine controls at each step
that the expansion is accelerated and that inflation holds (wepsilon>1), otherwise it returns an error. Thanks to the
last argument, it is also possible to specify whether the integration should be carried forward or backward in time.
For the inflation_H case, only a 1st order differential equation is involved, so the forward and backward case can
be done exactly without problems. For the inflation_V case, the equation of motion is 2nd order. What the module
will do in the backward case is to search for an approximate solution, corresponding to the (first-order) attractor
inflationary solution. This approximate backward solution is used in order to estimate some initial times, but the
approximation made here will never impact the final result: the module is written in such a way that after using this
approximation, the code always computes (and relies on) the exact forward solution.
Parameters
ppm Input: pointer to primordial structure
ppr Input: pointer to precision structure
yInput/output: running vector of background variables, already allocated and initialized
dy Input: running vector of background derivatives, already allocated
target Input: whether the goal is to reach a given aH or φ
stop Input: the target value of either aH or φ
check_epsilon Input: whether we should impose inflation (epsilon>1) at each step
direction Input: whether we should integrate forward or backward in time
time Input: definition of time (proper or conformal)
Returns
the error status
5.15.2.18 primordial_inflation_check_potential()
int primordial_inflation_check_potential (
struct primordial ∗ppm,
double phi,
double ∗V,
double ∗dV,
double ∗ddV )
Routine checking positivity and negative slope of potential. The negative slope is an arbitrary choice. Currently the
code can only deal with monotonic variations of the inflaton during inflation. So the slope had to be always negative
or always positive... we took the first option.
Generated by Doxygen
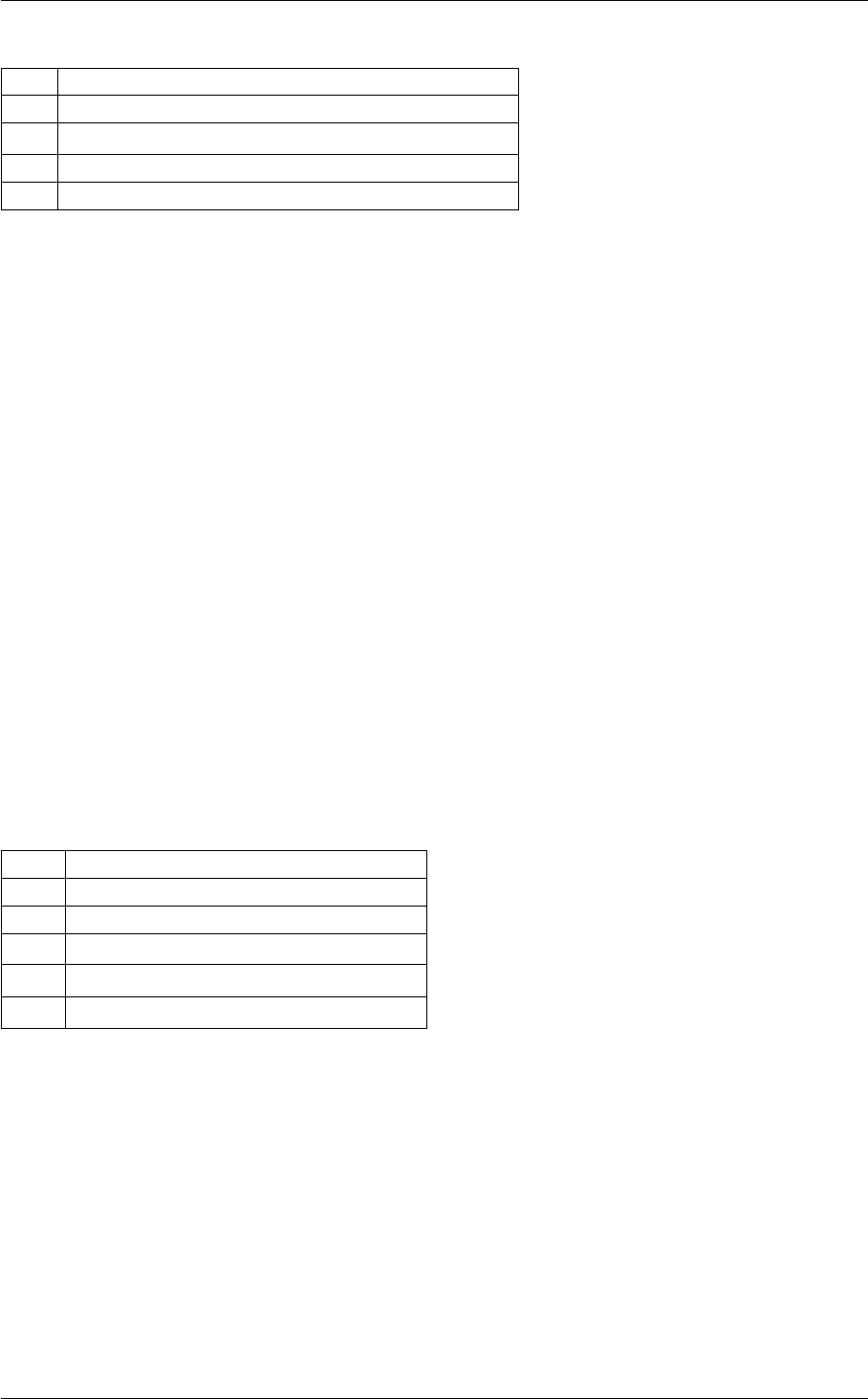
146 File Documentation
Parameters
ppm Input: pointer to primordial structure
phi Input: field value where to perform the check
VOutput: inflaton potential in units of Mp4
dV Output: first derivative of inflaton potential wrt the field
ddV Output: second derivative of inflaton potential wrt the field
Returns
the error status
5.15.2.19 primordial_inflation_check_hubble()
int primordial_inflation_check_hubble (
struct primordial ∗ppm,
double phi,
double ∗H,
double ∗dH,
double ∗ddH,
double ∗dddH )
Routine checking positivity and negative slope of H(φ). The negative slope is an arbitrary choice. Currently the
code can only deal with monotonic variations of the inflaton during inflation. And H can only decrease with time.
So the slope dH/dφ has to be always negative or always positive... we took the first option: phi increases, H
decreases.
Parameters
ppm Input: pointer to primordial structure
phi Input: field value where to perform the check
HOutput: Hubble parameters in units of Mp
dH Output: dH/dφ
ddH Output: d2H/dφ2
dddH Output: d3H/dφ3
Returns
the error status
5.15.2.20 primordial_inflation_get_epsilon()
int primordial_inflation_get_epsilon (
struct primordial ∗ppm,
Generated by Doxygen

5.15 primordial.c File Reference 147
double phi,
double ∗epsilon )
Routine computing the first slow-roll parameter epsilon
Generated by Doxygen
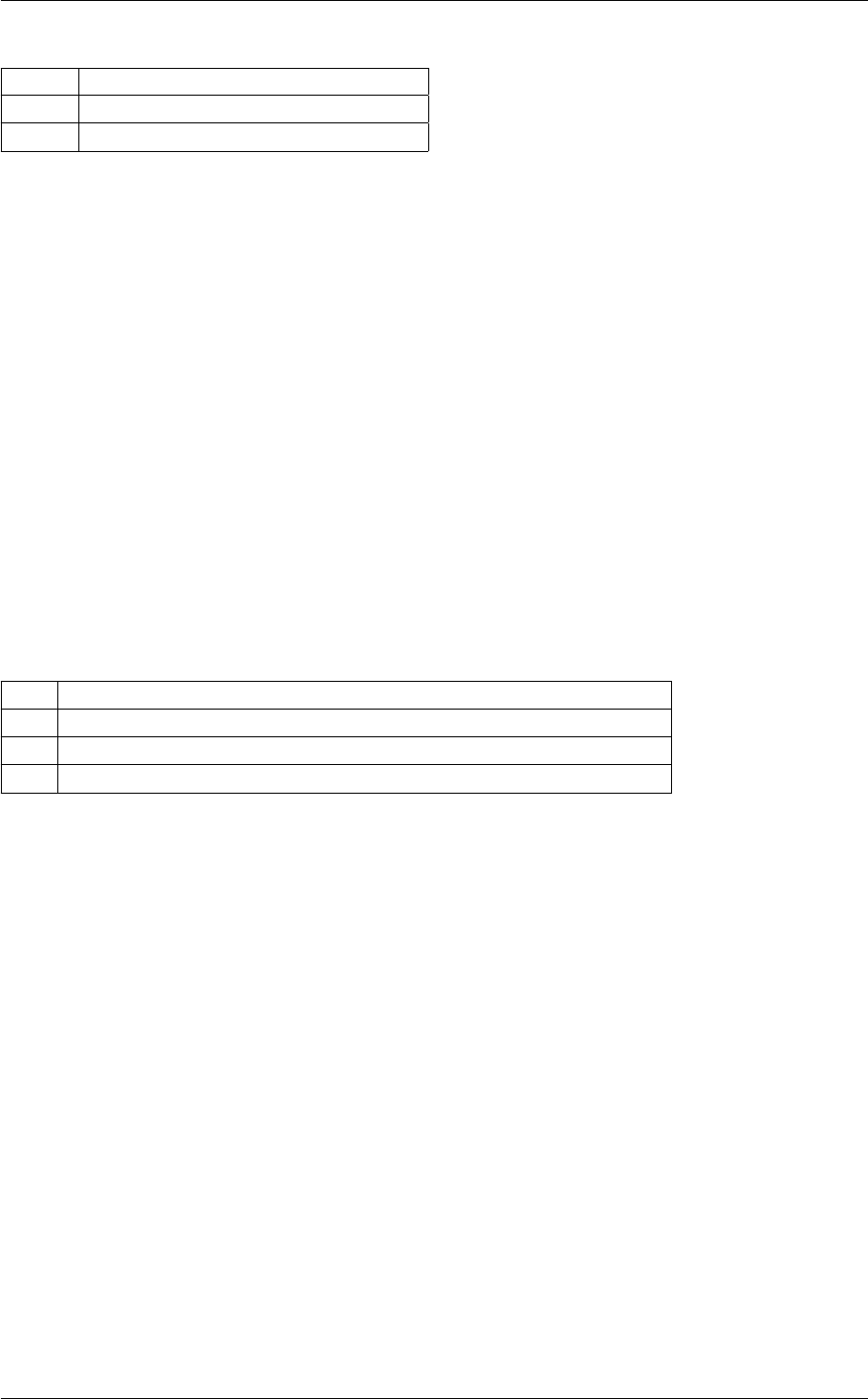
148 File Documentation
Parameters
ppm Input: pointer to primordial structure
phi Input: field value where to compute epsilon
epsilon Output: result
Returns
the error status
5.15.2.21 primordial_inflation_find_phi_pivot()
int primordial_inflation_find_phi_pivot (
struct primordial ∗ppm,
struct precision ∗ppr,
double ∗y,
double ∗dy )
Routine searching phi_pivot when a given amount of inflation is requested.
Parameters
ppm Input/output: pointer to primordial structure
ppr Input: pointer to precision structure
yInput: running vector of background variables, already allocated and initialized
dy Input: running vector of background derivatives, already allocated
Returns
the error status
Summary:
• define local variables
• check whether in vicinity of phi_end, inflation is still ongoing
• case in which epsilon>1: hence we must find the value phi_stop <phi_end where inflation ends up naturally
• –>find latest value of the field such that epsilon = primordial_inflation_small_epsilon (default: 0.1)
• –>bracketing right-hand value is phi_end (but the potential will not be evaluated exactly there, only closeby
• –>bracketing left-hand value is found by iterating with logarithmic step until epsilon <primordial_inflation←-
_small_epsilon
• –>find value such that epsilon = primordial_inflation_small_epsilon by bisection
• –>value found and stored as phi_small_epsilon
• –>find inflationary attractor in phi_small_epsilon (should exist since epsilon<<1 there)
Generated by Doxygen

5.15 primordial.c File Reference 149
• -->compute amount of inflation between this phi_small_epsilon and the end of inflation
• –>by starting from phi_small_epsilon and integrating an approximate solution backward in time, try to esti-
mate roughly a value close to phi_pivot but a bit smaller. This is done by trying to reach an amount of inflation
equal to the requested one, minus the amount after phi_small_epsilon, and plus primordial_inflation_extra←-
_efolds efolds (default: two). Note that it is not aggressive to require two extra e-folds of inflation before the
pivot, since the calculation of the spectrum in the observable range will require even more.
• –>find attractor in phi_try
• –>check the total amount of inflation between phi_try and the end of inflation
• –>go back to phi_try, and now find phi_pivot such that the amount of inflation between phi_pivot and the end
of inflation is exactly the one requested.
• case in which epsilon<1:
• –>find inflationary attractor in phi_small_epsilon (should exist since epsilon<1 there)
• -->by starting from phi_end and integrating an approximate solution backward in time, try to estimate roughly
a value close to phi_pivot but a bit smaller. This is done by trying to reach an amount of inflation equal to the
requested one, minus the amount after phi_small_epsilon, and plus primordial_inflation_extra_efolds efolds
(default: two). Note that it is not aggressive to require two extra e-folds of inflation before the pivot, since the
calculation of the spectrum in the observable range will require even more.
• –>we now have a value phi_try believed to be close to and slightly smaller than phi_pivot
• –>find attractor in phi_try
• –>check the total amount of inflation between phi_try and the end of inflation
• –>go back to phi_try, and now find phi_pivot such that the amount of inflation between phi_pivot and the end
of inflation is exactly the one requested.
• –>In verbose mode, check that phi_pivot is correct. Done by restarting from phi_pivot and going again till
the end of inflation.
5.15.2.22 primordial_inflation_derivs()
int primordial_inflation_derivs (
double tau,
double ∗y,
double ∗dy,
void ∗parameters_and_workspace,
ErrorMsg error_message )
Routine returning derivative of system of background/perturbation variables. Like other routines used by the generic
integrator (background_derivs, thermodynamics_derivs, perturb_derivs), this routine has a generic list of argu-
ments, and a slightly different error management, with the error message returned directly in an ErrMsg field.
Parameters
tau Input: time (not used explicitly inside the routine, but requested by the generic
integrator)
yInput/output: running vector of background variables, already allocated and
initialized
dy Input: running vector of background derivatives, already allocated
parameters_and_workspace Input: all necessary input variables apart from y
error_message Output: error message
Generated by Doxygen

150 File Documentation
Returns
the error status
5.15.2.23 primordial_external_spectrum_init()
int primordial_external_spectrum_init (
struct perturbs ∗ppt,
struct primordial ∗ppm )
This routine reads the primordial spectrum from an external command, and stores the tabulated values. The sam-
pling of the k's given by the external command is preserved.
Author: Jesus Torrado (torradocacho@lorentz.leidenuniv.nl) Date: 2013-12-20
Parameters
ppt Input/output: pointer to perturbation structure
ppm Input/output: pointer to primordial structure
Returns
the error status
Summary:
• Initialization
• Launch the command and retrieve the output
• Store the read results into CLASS structures
• Make room
• Store values
• Release the memory used locally
• Tell CLASS that there are scalar (and tensor) modes
5.16 primordial.h File Reference
#include "perturbations.h"
Generated by Doxygen
5.16 primordial.h File Reference 151
Include dependency graph for primordial.h:
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
This graph shows which files directly or indirectly include this file:
primordial.h
nonlinear.h
input.h
class.h
nonlinear_conflict
-20170920-132422.h
nonlinear_conflict
-20170920-150212.h nonlinear_exp.h nonlinear_test.h primordial.c
transfer.h nonlinear.c
spectra.htransfer.c
input.c
class.c
lensing.hspectra.c
output.h lensing.c
output.c
Data Structures
• struct primordial
Enumerations
• enum primordial_spectrum_type
• enum linear_or_logarithmic
• enum potential_shape
• enum target_quantity
• enum integration_direction
• enum time_definition
• enum phi_pivot_methods
• enum inflation_module_behavior
Generated by Doxygen
152 File Documentation
5.16.1 Detailed Description
Documented includes for primordial module.
5.16.2 Data Structure Documentation
5.16.2.1 struct primordial
Structure containing everything about primordial spectra that other modules need to know.
Once initialized by primordial_init(), contains a table of all primordial spectra as a function of wavenumber, mode,
and pair of initial conditions.
Data Fields
double k_pivot pivot scale in Mpc−1
enum primordial_spectrum_type primordial_spec_type type of primordial spectrum (simple analytic from,
integration of inflationary perturbations, etc.)
double A_s usual scalar amplitude = curvature power
spectrum at pivot scale
double n_s usual scalar tilt = [curvature power spectrum tilt at
pivot scale -1]
double alpha_s usual scalar running
double beta_s running of running
double r usual tensor to scalar ratio of power spectra,
r=AT/AS=Ph/PR
double n_t usual tensor tilt = [GW power spectrum tilt at pivot
scale]
double alpha_t usual tensor running
double f_bi baryon isocurvature (BI) entropy-to-curvature ratio
Sbi/R
double n_bi BI tilt
double alpha_bi BI running
double f_cdi CDM isocurvature (CDI) entropy-to-curvature ratio
Scdi/R
double n_cdi CDI tilt
double alpha_cdi CDI running
double f_nid neutrino density isocurvature (NID)
entropy-to-curvature ratio Snid/R
double n_nid NID tilt
double alpha_nid NID running
double f_niv neutrino velocity isocurvature (NIV)
entropy-to-curvature ratio Sniv/R
double n_niv NIV tilt
double alpha_niv NIV running
double c_ad_bi ADxBI cross-correlation at pivot scale, from -1 to 1
double n_ad_bi ADxBI cross-correlation tilt
double alpha_ad_bi ADxBI cross-correlation running
double c_ad_cdi ADxCDI cross-correlation at pivot scale, from -1 to
1
Generated by Doxygen
5.16 primordial.h File Reference 153
Data Fields
double n_ad_cdi ADxCDI cross-correlation tilt
double alpha_ad_cdi ADxCDI cross-correlation running
double c_ad_nid ADxNID cross-correlation at pivot scale, from -1 to
1
double n_ad_nid ADxNID cross-correlation tilt
double alpha_ad_nid ADxNID cross-correlation running
double c_ad_niv ADxNIV cross-correlation at pivot scale, from -1 to
1
double n_ad_niv ADxNIV cross-correlation tilt
double alpha_ad_niv ADxNIV cross-correlation running
double c_bi_cdi BIxCDI cross-correlation at pivot scale, from -1 to
1
double n_bi_cdi BIxCDI cross-correlation tilt
double alpha_bi_cdi BIxCDI cross-correlation running
double c_bi_nid BIxNIV cross-correlation at pivot scale, from -1 to
1
double n_bi_nid BIxNIV cross-correlation tilt
double alpha_bi_nid BIxNIV cross-correlation running
double c_bi_niv BIxNIV cross-correlation at pivot scale, from -1 to
1
double n_bi_niv BIxNIV cross-correlation tilt
double alpha_bi_niv BIxNIV cross-correlation running
double c_cdi_nid CDIxNID cross-correlation at pivot scale, from -1
to 1
double n_cdi_nid CDIxNID cross-correlation tilt
double alpha_cdi_nid CDIxNID cross-correlation running
double c_cdi_niv CDIxNIV cross-correlation at pivot scale, from -1
to 1
double n_cdi_niv CDIxNIV cross-correlation tilt
double alpha_cdi_niv CDIxNIV cross-correlation running
double c_nid_niv NIDxNIV cross-correlation at pivot scale, from -1
to 1
double n_nid_niv NIDxNIV cross-correlation tilt
double alpha_nid_niv NIDxNIV cross-correlation running
enum potential_shape potential parameters describing the case
primordial_spec_type = inflation_V
double V0 one parameter of the function V(phi)
double V1 one parameter of the function V(phi)
double V2 one parameter of the function V(phi)
double V3 one parameter of the function V(phi)
double V4 one parameter of the function V(phi)
double H0 one parameter of the function H(phi)
double H1 one parameter of the function H(phi)
double H2 one parameter of the function H(phi)
double H3 one parameter of the function H(phi)
double H4 one parameter of the function H(phi)
double phi_end value of inflaton at the end of inflation
enum phi_pivot_methods phi_pivot_method flag for method used to define and find the pivot
scale
Generated by Doxygen
154 File Documentation
Data Fields
double phi_pivot_target For each of the above methods, critical value to
be reached between pivot and end of inflation
(N_star, [aH]ratio, etc.)
enum inflation_module_behavior behavior Specifies if the inflation module computes the
primordial spectrum numerically (default) or
analytically
char ∗command 'external_Pk' mode: command generating the
table of Pk and custom parameters to be passed
to it string with the command for calling
'external_Pk'
double custom1 one parameter of the primordial computed in
'external_Pk'
double custom2 one parameter of the primordial computed in
'external_Pk'
double custom3 one parameter of the primordial computed in
'external_Pk'
double custom4 one parameter of the primordial computed in
'external_Pk'
double custom5 one parameter of the primordial computed in
'external_Pk'
double custom6 one parameter of the primordial computed in
'external_Pk'
double custom7 one parameter of the primordial computed in
'external_Pk'
double custom8 one parameter of the primordial computed in
'external_Pk'
double custom9 one parameter of the primordial computed in
'external_Pk'
double custom10 one parameter of the primordial computed in
'external_Pk'
int md_size number of modes included in computation
int ∗ic_size for a given mode, ic_size[index_md] = number of
initial conditions included in computation
int ∗ic_ic_size number of ordered pairs of (index_ic1, index_ic2);
this number is just N(N+1)/2 where N =
ic_size[index_md]
int lnk_size number of ln(k) values
double ∗lnk list of ln(k) values lnk[index_k]
Generated by Doxygen
5.16 primordial.h File Reference 155
Data Fields
double ∗∗ lnpk depends on indices index_md, index_ic1,
index_ic2, index_k as: lnpk[index_md][index_←-
k∗ppm->ic_ic_size[index_md]+index_ic1_ic2]
where index_ic1_ic2 labels ordered pairs
(index_ic1, index_ic2) (since the primordial
spectrum is symmetric in (index_ic1, index_ic2)).
• for diagonal elements (index_ic1 =
index_ic2) this arrays contains ln[P(k)]
where P(k) is positive by construction.
• for non-diagonal elements this arrays
contains the k-dependent cosine of the
correlation angle, namely P(k )_(index_ic1,
index_ic2)/sqrt[P(k)_index_ic1
P(k)_index_ic2] This choice is convenient
since the sign of the non-diagonal
cross-correlation is arbitrary. For fully
correlated or anti-correlated initial
conditions, this non -diagonal element is
independent on k, and equal to +1 or -1.
double ∗∗ ddlnpk second derivative of above array, for spline
interpolation. So:
• for index_ic1 = index_ic, we spline ln[P(k)]
vs. ln(k), which is good since this function is
usually smooth.
• for non-diagonal coefficients, we spline
P(k)_(index_ic1,
index_ic2)/sqrt[P(k)_index_ic1
P(k)_index_ic2] vs. ln(k), which is fine since
this quantity is often assumed to be
constant (e.g for fully
correlated/anticorrelated initial conditions)
or nearly constant, and with arbitrary sign.
short ∗∗ is_non_zero is_non_zero[index_md][index_ic1_ic2] set to false
if pair (index_ic1, index_ic2) is uncorrelated
(ensures more precision and saves time with
respect to the option of simply setting
P(k)_(index_ic1, index_ic2) to zero)
double ∗∗ amplitude all amplitudes in matrix form:
amplitude[index_md][index_ic1_ic2]
double ∗∗ tilt all tilts in matrix form: tilt[index_md][index_ic1_ic2]
double ∗∗ running all runnings in matrix form:
running[index_md][index_ic1_ic2]
int index_in_a scale factor
int index_in_phi inflaton vev
int index_in_dphi its time derivative
int index_in_ksi_re Mukhanov variable (real part)
int index_in_ksi_im Mukhanov variable (imaginary part)
int index_in_dksi_re Mukhanov variable (real part, time derivative)
int index_in_dksi_im Mukhanov variable (imaginary part, time
derivative)
Generated by Doxygen
156 File Documentation
Data Fields
int index_in_ah_re tensor perturbation (real part)
int index_in_ah_im tensor perturbation (imaginary part)
int index_in_dah_re tensor perturbation (real part, time derivative)
int index_in_dah_im tensor perturbation (imaginary part, time
derivative)
int in_bg_size size of vector of background quantities only
int in_size full size of vector
double phi_pivot in inflationary module, value of phi_pivot (set to 0
for inflation_V, inflation_H; found by code for
inflation_V_end)
double phi_min in inflationary module, value of phi when
kmin =aH
double phi_max in inflationary module, value of phi when
kmax =aH
double phi_stop in inflationary module, value of phi at the end of
inflation
short primordial_verbose flag regulating the amount of information sent to
standard output (none if set to zero)
ErrorMsg error_message zone for writing error messages
5.16.3 Enumeration Type Documentation
5.16.3.1 primordial_spectrum_type
enum primordial_spectrum_type
enum defining how the primordial spectrum should be computed
5.16.3.2 linear_or_logarithmic
enum linear_or_logarithmic
enum defining whether the spectrum routine works with linear or logarithmic input/output
5.16.3.3 potential_shape
enum potential_shape
enum defining the type of inflation potential function V(phi)
Generated by Doxygen

5.16 primordial.h File Reference 157
5.16.3.4 target_quantity
enum target_quantity
enum defining which quantity plays the role of a target for evolving inflationary equations
5.16.3.5 integration_direction
enum integration_direction
enum specifying if we want to integrate equations forward or backward in time
5.16.3.6 time_definition
enum time_definition
enum specifying if we want to evolve quantities with conformal or proper time
5.16.3.7 phi_pivot_methods
enum phi_pivot_methods
enum specifying how, in the inflation_V_end case, the value of phi_pivot should calculated
5.16.3.8 inflation_module_behavior
enum inflation_module_behavior
enum specifying how the inflation module computes the primordial spectrum (default: numerical)
Generated by Doxygen
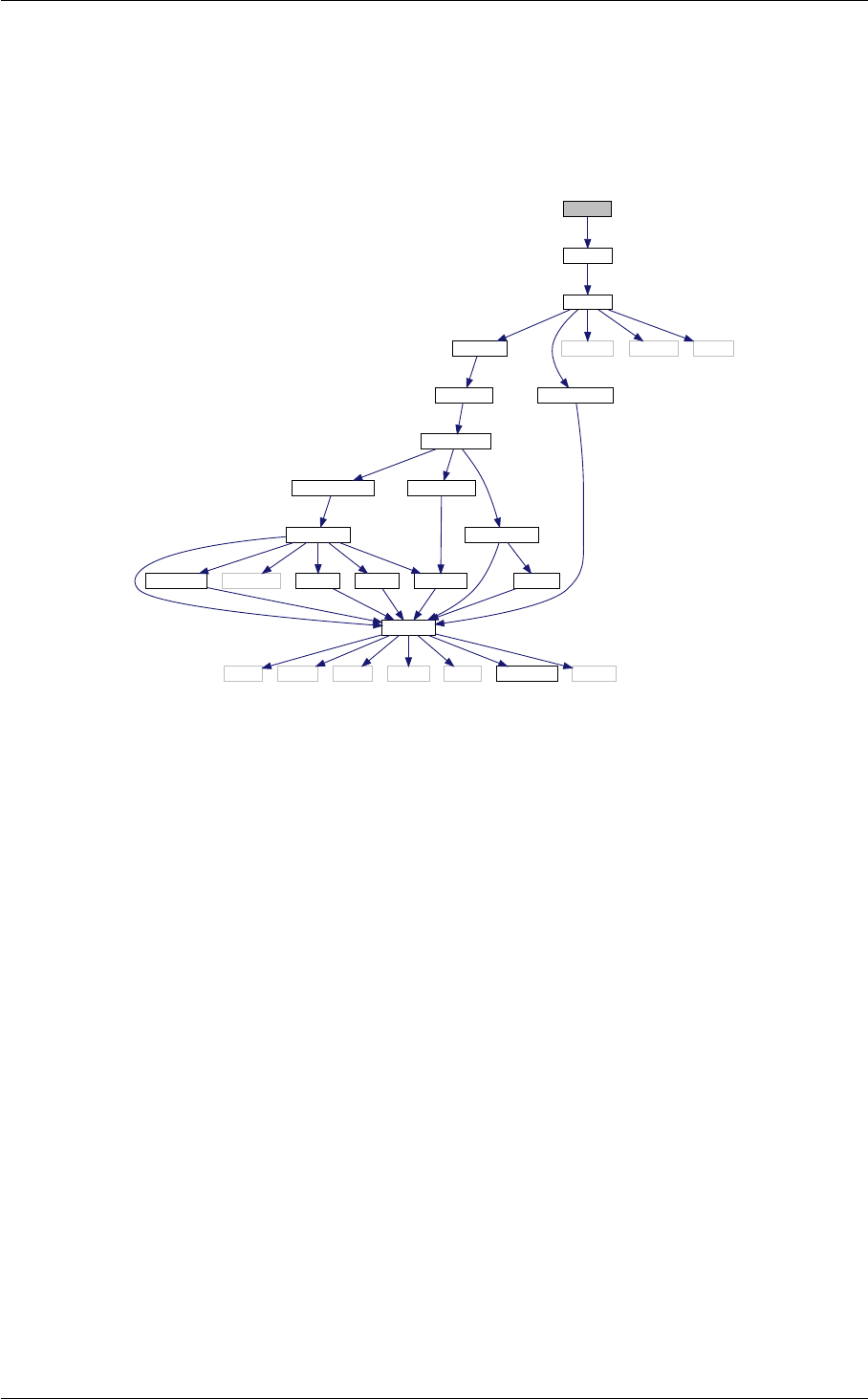
158 File Documentation
5.17 spectra.c File Reference
#include "spectra.h"
Include dependency graph for spectra.c:
spectra.c
spectra.h
transfer.h
nonlinear.h
hyperspherical.h
sys/shm.h sys/stat.h errno.h
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
Functions
• int spectra_cl_at_l (struct spectra ∗psp, double l, double ∗cl_tot, double ∗∗cl_md, double ∗∗cl_md_ic)
• int spectra_pk_at_z (struct background ∗pba, struct spectra ∗psp, enum linear_or_logarithmic mode, double
z, double ∗output_tot, double ∗output_ic, double ∗output_cb_tot, double ∗output_cb_ic)
• int spectra_pk_at_k_and_z (struct background ∗pba, struct primordial ∗ppm, struct spectra ∗psp, double k,
double z, double ∗pk_tot, double ∗pk_ic, double ∗pk_cb_tot, double ∗pk_cb_ic)
• int spectra_pk_nl_at_z (struct background ∗pba, struct spectra ∗psp, enum linear_or_logarithmic mode, dou-
ble z, double ∗output_tot, double ∗output_cb_tot)
• int spectra_pk_nl_at_k_and_z (struct background ∗pba, struct primordial ∗ppm, struct spectra ∗psp, double
k, double z, double ∗pk_tot, double ∗pk_cb_tot)
• int spectra_tk_at_z (struct background ∗pba, struct spectra ∗psp, double z, double ∗output)
• int spectra_tk_at_k_and_z (struct background ∗pba, struct spectra ∗psp, double k, double z, double ∗output)
• int spectra_init (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, struct primordial ∗ppm,
struct nonlinear ∗pnl, struct transfers ∗ptr, struct spectra ∗psp)
• int spectra_free (struct spectra ∗psp)
• int spectra_indices (struct background ∗pba, struct perturbs ∗ppt, struct transfers ∗ptr, struct primordial ∗ppm,
struct spectra ∗psp)
• int spectra_cls (struct background ∗pba, struct perturbs ∗ppt, struct transfers ∗ptr, struct primordial ∗ppm,
struct spectra ∗psp)
• int spectra_compute_cl (struct background ∗pba, struct perturbs ∗ppt, struct transfers ∗ptr, struct primordial
∗ppm, struct spectra ∗psp, int index_md, int index_ic1, int index_ic2, int index_l, int cl_integrand_num_←-
columns, double ∗cl_integrand, double ∗primordial_pk, double ∗transfer_ic1, double ∗transfer_ic2)
• int spectra_k_and_tau (struct background ∗pba, struct perturbs ∗ppt, struct nonlinear ∗pnl, struct spectra
∗psp)
Generated by Doxygen

5.17 spectra.c File Reference 159
• int spectra_pk (struct background ∗pba, struct perturbs ∗ppt, struct primordial ∗ppm, struct nonlinear ∗pnl,
struct spectra ∗psp)
• int spectra_sigma (struct background ∗pba, struct primordial ∗ppm, struct spectra ∗psp, double R, double z,
double ∗sigma)
• int spectra_matter_transfers (struct background ∗pba, struct perturbs ∗ppt, struct spectra ∗psp)
• int spectra_output_tk_data (struct background ∗pba, struct perturbs ∗ppt, struct spectra ∗psp, enum file_←-
format output_format, double z, int number_of_titles, double ∗data)
• int spectra_fast_pk_at_kvec_and_zvec (struct background ∗pba, struct spectra ∗psp, double ∗kvec, int kvec←-
_size, double ∗zvec, int zvec_size, double ∗pk_tot_out, double ∗pk_cb_tot_out, int nonlinear)
5.17.1 Detailed Description
Documented spectra module
Julien Lesgourgues, 25.08.2010
This module computes the anisotropy and Fourier power spectra CX
l, P (k), ...'s given the transfer and Bessel
functions (for anisotropy spectra), the source functions (for Fourier spectra) and the primordial spectra.
The following functions can be called from other modules:
1. spectra_init() at the beginning (but after transfer_init())
2. spectra_cl_at_l() at any time for computing Clat any l
3. spectra_spectrum_at_z() at any time for computing P(k) at any z
4. spectra_spectrum_at_k_and z() at any time for computing P at any k and z
5. spectra_free() at the end
5.17.2 Function Documentation
5.17.2.1 spectra_cl_at_l()
int spectra_cl_at_l (
struct spectra ∗psp,
double l,
double ∗cl_tot,
double ∗∗ cl_md,
double ∗∗ cl_md_ic )
Anisotropy power spectra Cl's for all types, modes and initial conditions.
This routine evaluates all the Cl's at a given value of l by interpolating in the pre-computed table. When relevant, it
also sums over all initial conditions for each mode, and over all modes.
This function can be called from whatever module at whatever time, provided that spectra_init() has been called
before, and spectra_free() has not been called yet.
Generated by Doxygen

160 File Documentation
Parameters
psp Input: pointer to spectra structure (containing pre-computed table)
lInput: multipole number
cl_tot Output: total Cl's for all types (TT, TE, EE, etc..)
cl_md Output: Cl's for all types (TT, TE, EE, etc..) decomposed mode by mode (scalar, tensor, ...) when
relevant
cl_md←-
_ic
Output: Cl's for all types (TT, TE, EE, etc..) decomposed by pairs of initial conditions (adiabatic,
isocurvatures) for each mode (usually, only for the scalar mode) when relevant
Returns
the error status
Summary:
• define local variables
• (a) treat case in which there is only one mode and one initial condition. Then, only cl_tot needs to be filled.
• (b) treat case in which there is only one mode with several initial condition. Fill cl_md_ic[index_md=0] and
sum it to get cl_tot.
• (c) loop over modes
• –>(c.1.) treat case in which the mode under consideration has only one initial condition. Fill cl_md[index_←-
md].
• –>(c.2.) treat case in which the mode under consideration has several initial conditions. Fill cl_md_ic[index←-
_md] and sum it to get cl_md[index_md]
• –>(c.3.) add contribution of cl_md[index_md] to cl_tot
5.17.2.2 spectra_pk_at_z()
int spectra_pk_at_z (
struct background ∗pba,
struct spectra ∗psp,
enum linear_or_logarithmic mode,
double z,
double ∗output_tot,
double ∗output_ic,
double ∗output_cb_tot,
double ∗output_cb_ic )
Matter power spectrum for arbitrary redshift and for all initial conditions.
This routine evaluates the matter power spectrum at a given value of z by interpolating in the pre-computed table (if
several values of z have been stored) or by directly reading it (if it only contains values at z=0 and we want P(k,z=0))
Can be called in two modes: linear or logarithmic.
Generated by Doxygen

5.17 spectra.c File Reference 161
• linear: returns P(k) (units: Mpc3)
• logarithmic: returns ln P(k)
One little subtlety: in case of several correlated initial conditions, the cross-correlation spectrum can be negative.
Then, in logarithmic mode, the non-diagonal elements contain the cross-correlation angle P12/√P11P22 (from -1
to 1) instead of ln P12
This function can be called from whatever module at whatever time, provided that spectra_init() has been called
before, and spectra_free() has not been called yet.
Generated by Doxygen

162 File Documentation
Parameters
pba Input: pointer to background structure (used for converting z into tau)
psp Input: pointer to spectra structure (containing pre-computed table)
mode Input: linear or logarithmic
zInput: redshift
output_tot Output: total matter power spectrum P(k) in M pc3(linear mode), or its logarithms (logarithmic
mode)
output_ic Output: for each pair of initial conditions, matter power spectra P(k) in Mpc3(linear mode), or
their logarithms and cross-correlation angles (logarithmic mode)
output_cb_tot Output: b+CDM power spectrum P_cb(k) in M pc3(linear mode), or its logarithms (logarithmic
mode)
output_cb_ic Output: for each pair of initial conditions, b+CDM power spectra P_cb(k) in Mpc3(linear
mode), or their logarithms and cross-correlation angles (logarithmic mode)
Returns
the error status
Summary:
• define local variables
• first step: convert z into ln τ
• second step: for both modes (linear or logarithmic), store the spectrum in logarithmic format in the output
array(s)
• –>(a) if only values at tau=tau_today are stored and we want P(k, z = 0), no need to interpolate
• –>(b) if several values of tau have been stored, use interpolation routine to get spectra at correct redshift
• third step: if there are several initial conditions, compute the total P(k) and set back all uncorrelated coeffi-
cients to exactly zero. Check positivity of total P(k).
• fourth step: depending on requested mode (linear or logarithmic), apply necessary transformation to the
output arrays
• –>(a) linear mode: if only one initial condition, convert output_tot to linear format; if several initial conditions,
convert output_ic to linear format, output_tot is already in this format
• –>(b) logarithmic mode: if only one initial condition, nothing to be done; if several initial conditions, convert
output_tot to logarithmic format, output_ic is already in this format
5.17.2.3 spectra_pk_at_k_and_z()
int spectra_pk_at_k_and_z (
struct background ∗pba,
struct primordial ∗ppm,
struct spectra ∗psp,
double k,
double z,
Generated by Doxygen
5.17 spectra.c File Reference 163
double ∗pk_tot,
double ∗pk_ic,
double ∗pk_cb_tot,
double ∗pk_cb_ic )
Matter power spectrum for arbitrary wavenumber, redshift and initial condition.
This routine evaluates the matter power spectrum at a given value of k and z by interpolating in a table of all P(k)'s
computed at this z by spectra_pk_at_z() (when kmin <= k <= kmax), or eventually by using directly the primordial
spectrum (when 0 <= k <kmin): the latter case is an approximation, valid when kmin << comoving Hubble scale
today. Returns zero when k=0. Returns an error when k<0 or k >kmax.
This function can be called from whatever module at whatever time, provided that spectra_init() has been called
before, and spectra_free() has not been called yet.
Parameters
pba Input: pointer to background structure (used for converting z into tau)
ppm Input: pointer to primordial structure (used only in the case 0 <k<kmin)
psp Input: pointer to spectra structure (containing pre-computed table)
kInput: wavenumber in 1/Mpc
zInput: redshift
pk_tot Output: total matter power spectrum P(k) in M pc3
pk_ic Output: for each pair of initial conditions, matter power spectra P(k) in Mpc3
pk_cb_tot Output: b+CDM power spectrum P(k) in M pc3
pk_cb_ic Output: for each pair of initial conditions, b+CDM power spectra P(k) in Mpc3
Returns
the error status
Summary:
• define local variables
• first step: check that k is in valid range [0:kmax] (the test for z will be done when calling spectra_pk_at_z())
• deal with case 0 <= k <kmin
• –>(a) subcase k=0: then P(k)=0
• –>(b) subcase 0<k<kmin: in this case we know that on super-Hubble scales: P(k) = [some number] ∗k
∗P_primordial(k) so P(k) = P(kmin) ∗(k P_primordial(k)) / (kmin P_primordial(kmin)) (note that the result is
accurate only if kmin is such that [a0 kmin] << H0)
• deal with case kmin <= k <= kmax
• last step: if more than one condition, sum over pk_ic to get pk_tot, and set back coefficients of non-correlated
pairs to exactly zero.
Generated by Doxygen
164 File Documentation
5.17.2.4 spectra_pk_nl_at_z()
int spectra_pk_nl_at_z (
struct background ∗pba,
struct spectra ∗psp,
enum linear_or_logarithmic mode,
double z,
double ∗output_tot,
double ∗output_cb_tot )
Non-linear total matter power spectrum for arbitrary redshift.
This routine evaluates the non-linear matter power spectrum at a given value of z by interpolating in the pre-
computed table (if several values of z have been stored) or by directly reading it (if it only contains values at z=0 and
we want P(k,z=0))
Can be called in two modes: linear or logarithmic.
• linear: returns P(k) (units: Mpc∧3)
• logarithmic: returns ln(P(k))
This function can be called from whatever module at whatever time, provided that spectra_init() has been called
before, and spectra_free() has not been called yet.
Parameters
pba Input: pointer to background structure (used for converting z into tau)
psp Input: pointer to spectra structure (containing pre-computed table)
mode Input: linear or logarithmic
zInput: redshift
output_tot Output: total matter power spectrum P(k) in M pc3(linear mode), or its logarithms (logarithmic
mode)
output_cb_tot Output: b+CDM power spectrum P(k) in M pc3(linear mode), or its logarithms (logarithmic
mode)
Returns
the error status
Summary:
• define local variables
• first step: convert z into ln(tau)
• second step: for both modes (linear or logarithmic), store the spectrum in logarithmic format in the output
array(s)
• –>(a) if only values at tau=tau_today are stored and we want P(k,z=0), no need to interpolate
• –>(b) if several values of tau have been stored, use interpolation routine to get spectra at correct redshift
• fourth step: eventually convert to linear format
Generated by Doxygen
5.17 spectra.c File Reference 165
5.17.2.5 spectra_pk_nl_at_k_and_z()
int spectra_pk_nl_at_k_and_z (
struct background ∗pba,
struct primordial ∗ppm,
struct spectra ∗psp,
double k,
double z,
double ∗pk_tot,
double ∗pk_cb_tot )
Non-linear total matter power spectrum for arbitrary wavenumber and redshift.
This routine evaluates the matter power spectrum at a given value of k and z by interpolating in a table of all P(k)'s
computed at this z by spectra_pk_nl_at_z() (when kmin <= k <= kmax), or eventually by using directly the primordial
spectrum (when 0 <= k <kmin): the latter case is an approximation, valid when kmin << comoving Hubble scale
today. Returns zero when k=0. Returns an error when k<0 or k >kmax.
This function can be called from whatever module at whatever time, provided that spectra_init() has been called
before, and spectra_free() has not been called yet.
Parameters
pba Input: pointer to background structure (used for converting z into tau)
ppm Input: pointer to primordial structure (used only in the case 0 <k<kmin)
psp Input: pointer to spectra structure (containing pre-computed table)
kInput: wavenumber in 1/Mpc
zInput: redshift
pk_tot Output: total matter power spectrum P(k) in M pc3
pk_cb_tot Output: b+CDM power spectrum P(k) in M pc3
Returns
the error status
Summary:
• define local variables
• check that k is in valid range [0:kmax] (the test for z will be done when calling spectra_pk_at_z())
• compute P(k,z) (in logarithmic format for more accurate interpolation)
• get its second derivatives with spline, then interpolate, then convert to linear format
Generated by Doxygen

166 File Documentation
5.17.2.6 spectra_tk_at_z()
int spectra_tk_at_z (
struct background ∗pba,
struct spectra ∗psp,
double z,
double ∗output )
Matter transfer functions Ti(k)for arbitrary redshift and for all initial conditions.
This routine evaluates the matter transfer functions at a given value of z by interpolating in the pre-computed table
(if several values of z have been stored) or by directly reading it (if it only contains values at z=0 and we want
Ti(k, z = 0))
This function can be called from whatever module at whatever time, provided that spectra_init() has been called
before, and spectra_free() has not been called yet.
Parameters
pba Input: pointer to background structure (used for converting z into tau)
psp Input: pointer to spectra structure (containing pre-computed table)
zInput: redshift
output Output: matter transfer functions
Returns
the error status
Summary:
• define local variables
• first step: convert z into ln(tau)
• second step: store the matter transfer functions in the output array
• –>(a) if only values at tau=tau_today are stored and we want Ti(k, z = 0), no need to interpolate
• –>(b) if several values of tau have been stored, use interpolation routine to get spectra at correct redshift
5.17.2.7 spectra_tk_at_k_and_z()
int spectra_tk_at_k_and_z (
struct background ∗pba,
struct spectra ∗psp,
double k,
double z,
double ∗output )
Matter transfer functions Ti(k)for arbitrary wavenumber, redshift and initial condition.
This routine evaluates the matter transfer functions at a given value of k and z by interpolating in a table of all
Ti(k, z)'s computed at this z by spectra_tk_at_z() (when kmin <= k <= kmax). Returns an error when k<kmin or
k>kmax.
This function can be called from whatever module at whatever time, provided that spectra_init() has been called
before, and spectra_free() has not been called yet.
Generated by Doxygen

5.17 spectra.c File Reference 167
Parameters
pba Input: pointer to background structure (used for converting z into tau)
psp Input: pointer to spectra structure (containing pre-computed table)
kInput: wavenumber in 1/Mpc
zInput: redshift
output Output: matter transfer functions
Returns
the error status
Summary:
• define local variables
• check that k is in valid range [0:kmax] (the test for z will be done when calling spectra_tk_at_z())
• compute T_i(k,z)
• get its second derivatives w.r.t. k with spline, then interpolate
5.17.2.8 spectra_init()
int spectra_init (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
struct primordial ∗ppm,
struct nonlinear ∗pnl,
struct transfers ∗ptr,
struct spectra ∗psp )
This routine initializes the spectra structure (in particular, computes table of anisotropy and Fourier spectra
CX
l, P (k), ...)
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure (will provide H, Omega_m at redshift of interest)
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfer structure
ppm Input: pointer to primordial structure
pnl Input: pointer to nonlinear structure
psp Output: pointer to initialized spectra structure
Generated by Doxygen
168 File Documentation
Returns
the error status
Summary:
• check that we really want to compute at least one spectrum
• initialize indices and allocate some of the arrays in the spectra structure
• deal with Cl's, if any
• deal with P(k, τ )and Ti(k, τ )
5.17.2.9 spectra_free()
int spectra_free (
struct spectra ∗psp )
This routine frees all the memory space allocated by spectra_init().
To be called at the end of each run, only when no further calls to spectra_cls_at_l(), spectra_pk_at_z(),spectra_←-
pk_at_k_and_z() are needed.
Parameters
psp Input: pointer to spectra structure (which fields must be freed)
Returns
the error status
5.17.2.10 spectra_indices()
int spectra_indices (
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
struct primordial ∗ppm,
struct spectra ∗psp )
This routine defines indices and allocates tables in the spectra structure
Parameters
pba Input: pointer to background structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
ppm Input: pointer to primordial structure
psp Input/output: pointer to spectra structure
Generated by Doxygen
5.17 spectra.c File Reference 169
Returns
the error status
5.17.2.11 spectra_cls()
int spectra_cls (
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
struct primordial ∗ppm,
struct spectra ∗psp )
This routine computes a table of values for all harmonic spectra Cl's, given the transfer functions and primordial
spectra.
Parameters
pba Input: pointer to background structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
ppm Input: pointer to primordial structure
psp Input/Output: pointer to spectra structure
Returns
the error status
Summary:
• define local variables
• allocate pointers to arrays where results will be stored
• store values of l
• loop over modes (scalar, tensors, etc). For each mode:
• –>(a) store number of l values for this mode
• –>(b) allocate arrays where results will be stored
• –>(c) loop over initial conditions
• —>loop over l values defined in the transfer module. For each l, compute the Cl's for all types (TT, TE, ...) by
convolving primordial spectra with transfer functions. This elementary task is assigned to spectra_compute←-
_cl()
• –>(d) now that for a given mode, all possible Cl's have been computed, compute second derivative of the
array in which they are stored, in view of spline interpolation.
Generated by Doxygen

170 File Documentation
5.17.2.12 spectra_compute_cl()
int spectra_compute_cl (
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
struct primordial ∗ppm,
struct spectra ∗psp,
int index_md,
int index_ic1,
int index_ic2,
int index_l,
int cl_integrand_num_columns,
double ∗cl_integrand,
double ∗primordial_pk,
double ∗transfer_ic1,
double ∗transfer_ic2 )
This routine computes the Cl's for a given mode, pair of initial conditions and multipole, but for all types (TT, TE...),
by convolving the transfer functions with the primordial spectra.
Parameters
pba Input: pointer to background structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
ppm Input: pointer to primordial structure
psp Input/Output: pointer to spectra structure (result stored here)
index_md Input: index of mode under consideration
index_ic1 Input: index of first initial condition in the correlator
index_ic2 Input: index of second initial condition in the correlator
index_l Input: index of multipole under consideration
cl_integrand_num_columns Input: number of columns in cl_integrand
cl_integrand Input: an allocated workspace
primordial_pk Input: table of primordial spectrum values
transfer_ic1 Input: table of transfer function values for first initial condition
transfer_ic2 Input: table of transfer function values for second initial condition
Returns
the error status
5.17.2.13 spectra_k_and_tau()
int spectra_k_and_tau (
struct background ∗pba,
struct perturbs ∗ppt,
struct nonlinear ∗pnl,
struct spectra ∗psp )
This routine computes the values of k and tau at which the matter power spectra P(k, τ)and the matter transfer
functions Ti(k, τ)will be stored.
Generated by Doxygen

5.17 spectra.c File Reference 171
Parameters
pba Input: pointer to background structure (for z to tau conversion)
ppt Input: pointer to perturbation structure (contain source functions)
pnl Input: pointer to nonlinear structure (contain nonlinear corrections)
psp Input/Output: pointer to spectra structure
Returns
the error status
Summary:
• define local variables
• check the presence of scalar modes
• check the maximum redshift z_max_pk at which P(k, z)and Ti(k, z)should be computable by interpolation.
If it is equal to zero, only P(k, z = 0) needs to be computed. If it is higher, we will store in a table various
P(k,tau) at several values of tau generously encompassing the range 0<z<z_max_pk
• allocate and fill table of tau values at which P(k, τ)and Ti(k, τ )are stored
• allocate and fill table of k values at which P(k, τ)is stored
• if the non-linear power spectrum is requested, we should store it only at values of tau where non-linear
corrections were really computed and not brutally set to one. Hence we must find here ln_tau_nl_size which
might be smaller than ln_tau_size. But the same table ln_tau will be used for both.
5.17.2.14 spectra_pk()
int spectra_pk (
struct background ∗pba,
struct perturbs ∗ppt,
struct primordial ∗ppm,
struct nonlinear ∗pnl,
struct spectra ∗psp )
This routine computes a table of values for all matter power spectra P(k), given the source functions and primordial
spectra.
Parameters
pba Input: pointer to background structure (will provide H, Omega_m at redshift of interest)
ppt Input: pointer to perturbation structure (contain source functions)
ppm Input: pointer to primordial structure
pnl Input: pointer to nonlinear structure
psp Input/Output: pointer to spectra structure
Generated by Doxygen

172 File Documentation
Returns
the error status
Summary:
• define local variables
• check the presence of scalar modes
• allocate temporary vectors where the primordial spectrum and the background quantities will be stored
• allocate and fill array of P(k, τ)values
• if interpolation of P(k, τ )will be needed (as a function of tau), compute array of second derivatives in view of
spline interpolation
• if interpolation of PNL(k, τ)will be needed (as a function of tau), compute array of second derivatives in view
of spline interpolation
5.17.2.15 spectra_sigma()
int spectra_sigma (
struct background ∗pba,
struct primordial ∗ppm,
struct spectra ∗psp,
double R,
double z,
double ∗sigma )
This routine computes sigma(R) given P(k) (does not check that k_max is large enough)
Parameters
pba Input: pointer to background structure
ppm Input: pointer to primordial structure
psp Input: pointer to spectra structure
zInput: redshift
RInput: radius in Mpc
sigma Output: variance in a sphere of radius R (dimensionless)
5.17.2.16 spectra_matter_transfers()
int spectra_matter_transfers (
struct background ∗pba,
struct perturbs ∗ppt,
struct spectra ∗psp )
This routine computes a table of values for all matter power spectra P(k), given the source functions and primordial
spectra.
Generated by Doxygen

5.17 spectra.c File Reference 173
Parameters
pba Input: pointer to background structure (will provide density of each species)
ppt Input: pointer to perturbation structure (contain source functions)
psp Input/Output: pointer to spectra structure
Returns
the error status
Summary:
• define local variables
• check the presence of scalar modes
• allocate and fill array of Ti(k, τ)values
• allocate temporary vectors where the background quantities will be stored
• if interpolation of P(k, τ )will be needed (as a function of tau), compute array of second derivatives in view of
spline interpolation
5.17.2.17 spectra_output_tk_data()
int spectra_output_tk_data (
struct background ∗pba,
struct perturbs ∗ppt,
struct spectra ∗psp,
enum file_format output_format,
double z,
int number_of_titles,
double ∗data )
• compute Ti(k)for each k (if several ic's, compute it for each ic; if z_pk = 0, this is done by directly reading
inside the pre-computed table; if not, this is done by interpolating the table at the correct value of tau.
• store data
Generated by Doxygen

174 File Documentation
5.17.2.18 spectra_fast_pk_at_kvec_and_zvec()
int spectra_fast_pk_at_kvec_and_zvec (
struct background ∗pba,
struct spectra ∗psp,
double ∗kvec,
int kvec_size,
double ∗zvec,
int zvec_size,
double ∗pk_tot_out,
double ∗pk_cb_tot_out,
int nonlinear )
Summary:
• define local variables
Compute spline over ln(k)
Construct table of log(pk) on the computed k nodes but requested redshifts:
Construct ln(kvec):
I will assume that the k vector is sorted in ascending order. Case k<kmin:
If needed, add some extrapolation here
Implement some extrapolation perhaps
Case kmin<=k<=kmax. Do not loop through kvec, but loop through the interpolation nodes.
Loop through k's that fall in this interval
Perform interpolation
case k>kmax
If needed, add some extrapolation here
Generated by Doxygen

5.18 spectra.h File Reference 175
5.18 spectra.h File Reference
#include "transfer.h"
Include dependency graph for spectra.h:
spectra.h
transfer.h
nonlinear.h
hyperspherical.h
sys/shm.h sys/stat.h errno.h
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
Generated by Doxygen
176 File Documentation
This graph shows which files directly or indirectly include this file:
spectra.h
input.h
class.h
lensing.h spectra.c
input.c
class.c
output.h lensing.c
output.c
Data Structures
• struct spectra
5.18.1 Detailed Description
Documented includes for spectra module
5.18.2 Data Structure Documentation
5.18.2.1 struct spectra
Structure containing everything about anisotropy and Fourier power spectra that other modules need to know.
Once initialized by spectra_init(), contains a table of all Cl's and P(k) as a function of multipole/wavenumber, mode
(scalar/tensor...), type (for Cl's: TT, TE...), and pairs of initial conditions (adiabatic, isocurvatures...).
Generated by Doxygen
5.18 spectra.h File Reference 177
Data Fields
double z_max_pk maximum value of z at which matter spectrum P(k,z) will be evaluated;
keep fixed to zero if P(k) only needed today
int non_diag sets the number of cross-correlation spectra that you want to calculate:
0 means only auto-correlation, 1 means only adjacent bins, and number
of bins minus one means all correlations
int md_size number of modes (scalar, tensor, ...) included in computation
int index_md_scalars index for scalar modes
int ∗ic_size for a given mode, ic_size[index_md] = number of initial conditions
included in computation
int ∗ic_ic_size for a given mode, ic_ic_size[index_md] = number of pairs of (index_ic1,
index_ic2) with index_ic2 >= index_ic1; this number is just N(N+1)/2
where N = ic_size[index_md]
short ∗∗ is_non_zero for a given mode, is_non_zero[index_md][index_ic1_ic2] is set to true if
the pair of initial conditions (index_ic1, index_ic2) are statistically
correlated, or to false if they are uncorrelated
int has_tt do we want CT T
l? (T = temperature)
int has_ee do we want CEE
l? (E = E-polarization)
int has_te do we want CT E
l?
int has_bb do we want CBB
l? (B = B-polarization)
int has_pp do we want Cφφ
l? ( φ= CMB lensing potential)
int has_tp do we want CT φ
l?
int has_ep do we want CEφ
l?
int has_dd do we want Cdd
l? (d = density)
int has_td do we want CT d
l?
int has_pd do we want Cφd
l?
int has_ll do we want Cll
l? (l = galaxy lensing potential)
int has_tl do we want CT l
l?
int has_dl do we want Cdl
l?
int index_ct_tt index for type CT T
l
int index_ct_ee index for type CEE
l
int index_ct_te index for type CT E
l
int index_ct_bb index for type CBB
l
int index_ct_pp index for type Cφφ
l
int index_ct_tp index for type CT φ
l
int index_ct_ep index for type CEφ
l
int index_ct_dd first index for type
Cdd
l((d_size∗d_size-(d_size-non_diag)∗(d_size-non_diag-1)/2) values)
int index_ct_td first index for type CT d
l(d_size values)
int index_ct_pd first index for type Cpd
l(d_size values)
int index_ct_ll first index for type
Cll
l((d_size∗d_size-(d_size-non_diag)∗(d_size-non_diag-1)/2) values)
int index_ct_tl first index for type CT l
l(d_size values)
int index_ct_dl first index for type Cdl
l(d_size values)
Generated by Doxygen

178 File Documentation
Data Fields
int d_size number of bins for which density Cl's are computed
int ct_size number of Cltypes requested
int ∗l_size number of multipole values for each requested mode, l_size[index_md]
int l_size_max greatest of all l_size[index_md]
double ∗l list of multipole values l[index_l]
int ∗∗ l_max_ct last multipole (given as an input) at which we want to output Cl's for a
given mode and type; l[index_md][l_size[index_md]-1] can be larger
than l_max[index_md], in order to ensure a better interpolation with no
boundary effects
int ∗l_max last multipole (given as an input) at which we want to output Cl's for a
given mode (maximized over types); l[index_md][l_size[index_md]-1]
can be larger than l_max[index_md], in order to ensure a better
interpolation with no boundary effects
int l_max_tot last multipole (given as an input) at which we want to output Cl's
(maximized over modes and types); l[index_md][l_size[index_md]-1] can
be larger than l_max[index_md], in order to ensure a better interpolation
with no boundary effects
double ∗∗ cl table of anisotropy spectra for each mode, multipole, pair of initial
conditions and types, cl[index_md][(index_l ∗
psp->ic_ic_size[index_md] + index_ic1_ic2) ∗psp->ct_size + index_ct]
double ∗∗ ddcl second derivatives of previous table with respect to l, in view of spline
interpolation
double alpha_II_2_20 parameter describing adiabatic versus isocurvature contribution in
mutipole range [2,20] (see Planck parameter papers)
double alpha_RI_2_20 parameter describing adiabatic versus isocurvature contribution in
mutipole range [2,20] (see Planck parameter papers)
double alpha_RR_2_20 parameter describing adiabatic versus isocurvature contribution in
mutipole range [2,20] (see Planck parameter papers)
double alpha_II_21_200 parameter describing adiabatic versus isocurvature contribution in
mutipole range [21,200] (see Planck parameter papers)
double alpha_RI_21_200 parameter describing adiabatic versus isocurvature contribution in
mutipole range [21,200] (see Planck parameter papers)
double alpha_RR_21_200 parameter describing adiabatic versus isocurvature contribution in
mutipole range [21,200] (see Planck parameter papers)
double alpha_II_201_2500 parameter describing adiabatic versus isocurvature contribution in
mutipole range [201,2500] (see Planck parameter papers)
double alpha_RI_201_2500 parameter describing adiabatic versus isocurvature contribution in
mutipole range [201,2500] (see Planck parameter papers)
double alpha_RR_201_2500 parameter describing adiabatic versus isocurvature contribution in
mutipole range [201,2500] (see Planck parameter papers)
double alpha_II_2_2500 parameter describing adiabatic versus isocurvature contribution in
mutipole range [2,2500] (see Planck parameter papers)
double alpha_RI_2_2500 parameter describing adiabatic versus isocurvature contribution in
mutipole range [2,2500] (see Planck parameter papers)
double alpha_RR_2_2500 parameter describing adiabatic versus isocurvature contribution in
mutipole range [2,2500] (see Planck parameter papers)
double alpha_kp parameter describing adiabatic versus isocurvature contribution at pivot
scale (see Planck parameter papers)
double alpha_k1 parameter describing adiabatic versus isocurvature contribution at scale
k1 (see Planck parameter papers)
Generated by Doxygen

5.18 spectra.h File Reference 179
Data Fields
double alpha_k2 parameter describing adiabatic versus isocurvature contribution at scale
k2 (see Planck parameter papers)
int ln_k_size number ln(k) values
double ∗ln_k list of ln(k) values ln_k[index_k]
int ln_tau_size number of ln(tau) values, for the matter power spectrum and the matter
transfer functions, (only one if z_max_pk = 0)
double ∗ln_tau list of ln(tau) values ln_tau[index_tau], for the matter power spectrum
and the matter transfer functions, in growing order. So exp(ln_tau[0]) is
the earliest time (i.e. highest redshift), while exp(ln_tau[ln_tau_size-1])
is today (i.e z=0).
double ∗ln_pk Matter power spectrum. depends on indices index_ic1_ic2, index_k,
index_tau as: ln_pk[(index_tau ∗psp->k_size + index_k)∗
psp->ic_ic_size[index_md] + index_ic1_ic2] where index_ic1_ic2 labels
ordered pairs (index_ic1, index_ic2) (since the primordial spectrum is
symmetric in (index_ic1, index_ic2)).
• for diagonal elements (index_ic1 = index_ic2) this arrays contains
ln[P(k)] where P(k) is positive by construction.
• for non-diagonal elements this arrays contains the k-dependent
cosine of the correlation angle, namely P(k)_(index_ic1,
index_ic2)/sqrt[P(k)_index_ic1 P(k)_index_ic2] This choice is
convenient since the sign of the non-diagonal cross-correlation is
arbitrary. For fully correlated or anti-correlated initial conditions,
this non-diagonal element is independent on k, and equal to +1 or
-1.
double ∗ddln_pk second derivative of above array with respect to log(tau), for spline
interpolation. So:
• for index_ic1 = index_ic, we spline ln[P(k)] vs. ln(k), which is good
since this function is usually smooth.
• for non-diagonal coefficients, we spline P(k)_(index_ic1,
index_ic2)/sqrt[P(k)_index_ic1 P(k)_index_ic2] vs. ln(k), which is
fine since this quantity is often assumed to be constant (e.g for
fully correlated/anticorrelated initial conditions) or nearly constant,
and with arbitrary sign.
double sigma8 sigma8 parameter
double sigma8_cb if ncdm present: contribution to sigma8 from only baryons and cdm
double ∗ln_pk_l
double ∗ddln_pk_l q<Total linear matter power spectrum, just depending on indices
index_k, index_tau as: ln_pk[index_tau ∗psp->k_size + index_k]
Range of k and tau value identical to ln_pk array. second derivative of
above array with respect to log(tau), for spline interpolation.
int ln_tau_nl_size number of ln(tau) values for non-linear spectrum (possibly smaller than
ln_tau_size, because the non-linear spectrum is stored only in the
time/redhsift range where the non-linear corrections were really
computed, to avoid dealing with discontinuities in the spline
interpolation)
double ∗ln_tau_nl list of ln(tau) values ln_tau_nl[index_tau], for the non-linear power
spectrum, in growing order. So exp(ln_tau_nl[0]) is the earliest time (i.e.
highest redshift), while exp(ln_tau_nl[ln_tau_nl_size-1]) is today (i.e
z=0).
Generated by Doxygen

180 File Documentation
Data Fields
double ∗ln_pk_nl Non-linear matter power spectrum. depends on indices index_k,
index_tau as: ln_pk_nl[index_tau ∗psp->k_size + index_k]
double ∗ddln_pk_nl second derivative of above array with respect to log(tau), for spline
interpolation.
double ∗ln_pk_cb same as ln_pk_l for baryon+cdm component only
double ∗ddln_pk_cb same as ddln_pk_cb for baryon+cdm component only
double ∗ln_pk_cb_l same as ln_pk_cb_l for baryon+cdm component only
double ∗ddln_pk_cb_l same as ddln_pk_cb_l for baryon+cdm component only
double ∗ln_pk_cb_nl same as ln_pk_cb_nl for baryon+cdm component only
double ∗ddln_pk_cb_nl same as ddln_pk_cb_nl for baryon+cdm component only
int index_tr_delta_g index of gamma density transfer function
int index_tr_delta_b index of baryon density transfer function
int index_tr_delta_cdm index of cold dark matter density transfer function
int index_tr_delta_dcdm index of decaying cold dark matter density transfer function
int index_tr_delta_scf index of scalar field phi transfer function
int index_tr_delta_fld index of dark energy fluid density transfer function
int index_tr_delta_ur index of ultra-relativistic neutrinos/relics density transfer function
int index_tr_delta_dr index of decay radiation density transfer function
int index_tr_delta_ncdm1 index of first species of non-cold dark matter (massive neutrinos, ...)
density transfer function
int index_tr_delta_tot index of total matter density transfer function
int index_tr_theta_g index of gamma velocity transfer function
int index_tr_theta_b index of baryon velocity transfer function
int index_tr_theta_cdm index of cold dark matter velocity transfer function
int index_tr_theta_dcdm index of decaying cold dark matter velocity transfer function
int index_tr_theta_scf index of derivative of scalar field phi transfer function
int index_tr_theta_fld index of dark energy fluid velocity transfer function
int index_tr_theta_ur index of ultra-relativistic neutrinos/relics velocity transfer function
int index_tr_theta_dr index of decay radiation velocity transfer function
int index_tr_theta_ncdm1 index of first species of non-cold dark matter (massive neutrinos, ...)
velocity transfer function
int index_tr_theta_tot index of total matter velocity transfer function
int index_tr_phi index of Bardeen potential phi
int index_tr_psi index of Bardeen potential psi
int index_tr_phi_prime index of derivative of Bardeen potential phi
int index_tr_h index of synchronous gauge metric perturbation h
int index_tr_h_prime index of synchronous gauge metric perturbation h'
int index_tr_eta index of synchronous gauge metric perturbation eta
int index_tr_eta_prime index of synchronous gauge metric perturbation eta'
int tr_size total number of species in transfer functions
double ∗matter_transfer Matter transfer functions. Depends on indices
index_md,index_tau,index_ic,index_k, index_tr as:
matter_transfer[((index_tau∗psp->ln_k_size + index_k) ∗
psp->ic_size[index_md] + index_ic) ∗psp->tr_size + index_tr]
double ∗ddmatter_transfer second derivative of above array with respect to log(tau), for spline
interpolation.
short spectra_verbose flag regulating the amount of information sent to standard output (none if
set to zero)
Generated by Doxygen
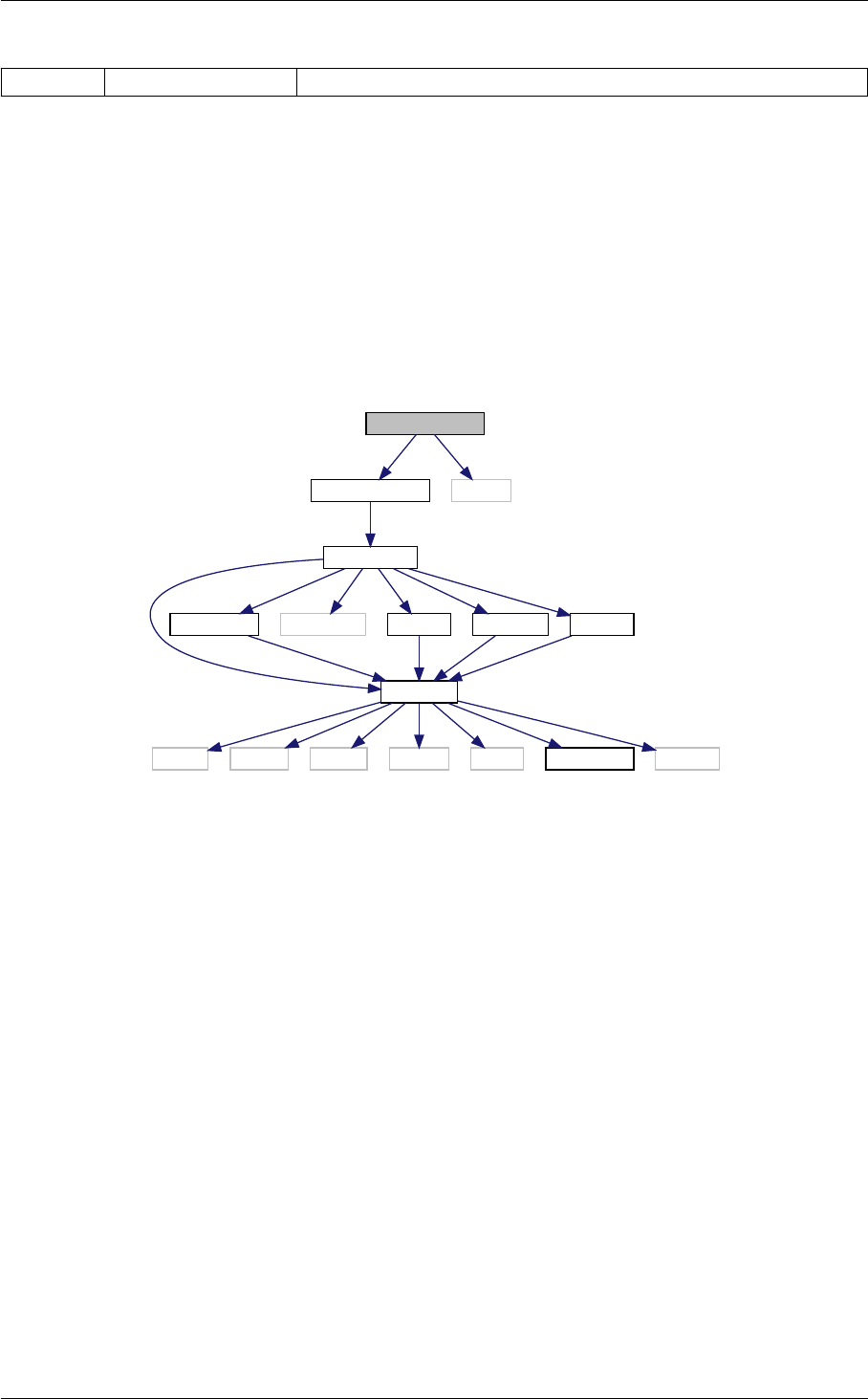
5.19 thermodynamics.c File Reference 181
Data Fields
ErrorMsg error_message zone for writing error messages
5.19 thermodynamics.c File Reference
#include "thermodynamics.h"
#include "hyrec.h"
Include dependency graph for thermodynamics.c:
thermodynamics.c
thermodynamics.h hyrec.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.h parser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
Functions
• int thermodynamics_at_z (struct background ∗pba, struct thermo ∗pth, double z, short inter_mode, int ∗last←-
_index, double ∗pvecback, double ∗pvecthermo)
• int thermodynamics_init (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth)
• int thermodynamics_free (struct thermo ∗pth)
• int thermodynamics_indices (struct thermo ∗pth, struct recombination ∗preco, struct reionization ∗preio)
• int thermodynamics_helium_from_bbn (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth)
• int thermodynamics_onthespot_energy_injection (struct precision ∗ppr, struct background ∗pba, struct re-
combination ∗preco, double z, double ∗energy_rate, ErrorMsg error_message)
• int thermodynamics_energy_injection (struct precision ∗ppr, struct background ∗pba, struct recombination
∗preco, double z, double ∗energy_rate, ErrorMsg error_message)
• int thermodynamics_reionization_function (double z, struct thermo ∗pth, struct reionization ∗preio, double
∗xe)
• int thermodynamics_get_xe_before_reionization (struct precision ∗ppr, struct thermo ∗pth, struct recombina-
tion ∗preco, double z, double ∗xe)
• int thermodynamics_reionization (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct
recombination ∗preco, struct reionization ∗preio, double ∗pvecback)
• int thermodynamics_reionization_sample (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth,
struct recombination ∗preco, struct reionization ∗preio, double ∗pvecback)
Generated by Doxygen

182 File Documentation
• int thermodynamics_recombination (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct
recombination ∗preco, double ∗pvecback)
• int thermodynamics_recombination_with_hyrec (struct precision ∗ppr, struct background ∗pba, struct thermo
∗pth, struct recombination ∗preco, double ∗pvecback)
• int thermodynamics_recombination_with_recfast (struct precision ∗ppr, struct background ∗pba, struct
thermo ∗pth, struct recombination ∗preco, double ∗pvecback)
• int thermodynamics_derivs_with_recfast (double z, double ∗y, double ∗dy, void ∗parameters_and_workspace,
ErrorMsg error_message)
• int thermodynamics_merge_reco_and_reio (struct precision ∗ppr, struct thermo ∗pth, struct recombination
∗preco, struct reionization ∗preio)
• int thermodynamics_output_titles (struct background ∗pba, struct thermo ∗pth, char titles[_MAXTITLESTR←-
INGLENGTH_])
5.19.1 Detailed Description
Documented thermodynamics module
Julien Lesgourgues, 6.09.2010
Deals with the thermodynamical evolution. This module has two purposes:
• at the beginning, to initialize the thermodynamics, i.e. to integrate the thermodynamical equations, and store
all thermodynamical quantities as a function of redshift inside an interpolation table. The current version of
recombination is based on RECFAST v1.5. The current version of reionization is based on exactly the same
reionization function as in CAMB, in order to make allow for comparison. It should be easy to generalize the
module to more complicated reionization histories.
• to provide a routine which allow other modules to evaluate any thermodynamical quantities at a given redshift
value (by interpolating within the interpolation table).
The logic is the following:
• in a first step, the code assumes that there is no reionization, and computes the ionization fraction, Thom-
son scattering rate, baryon temperature, etc., using RECFAST. The result is stored in a temporary table
'recombination_table' (within a temporary structure of type 'recombination') for each redshift in a range 0 <z
<z_initial. The sampling in z space is done with a simple linear step size.
• in a second step, the code adds the reionization history, starting from a redshift z_reio_start. The ionization
fraction at this redshift is read in the previous recombination table in order to ensure a perfect matching.
The code computes the ionization fraction, Thomson scattering rate, baryon temperature, etc., using a given
parametrization of the reionization history. The result is stored in a temporary table 'reionization_table' (within
a temporary structure of type 'reionization') for each redshift in the range 0 <z<z_reio_start. The sampling
in z space is found automatically, given the precision parameter 'reionization_sampling'.
• in a third step, the code merges the two tables 'recombination_table' and 'reionization_table' inside the ta-
ble 'thermodynamics_table', and the temporary structures 'recombination' and 'reionization' are freed. In
'thermodynamics_table', the sampling in z space is the one defined in the recombination algorithm for z_←-
reio_start <z<z_initial, and the one defined in the reionization algorithm for 0 <z<z_reio_start.
• at this stage, only a few columns in the table 'thermodynamics_table' have been filled. In a fourth step,
the remaining columns are filled, using some numerical integration/derivation routines from the 'array.c' tools
module.
Generated by Doxygen

5.19 thermodynamics.c File Reference 183
• small detail: one of the columns contains the maximum variation rate of a few relevant thermodynamical
quantities. This rate will be used for defining automatically the sampling step size in the perturbation module.
Hence, the exact value of this rate is unimportant, but its order of magnitude at a given z defines the sampling
precision of the perturbation module. Hence, it is harmless to use a smoothing routine in order to make this
rate look nicer, although this will not affect the final result significantly. The last step in the thermodynamics←-
_init module is to perform this smoothing.
In summary, the following functions can be called from other modules:
1. thermodynamics_init() at the beginning (but after background_init())
2. thermodynamics_at_z() at any later time
3. thermodynamics_free() at the end, when no more calls to thermodynamics_at_z() are needed
5.19.2 Function Documentation
5.19.2.1 thermodynamics_at_z()
int thermodynamics_at_z (
struct background ∗pba,
struct thermo ∗pth,
double z,
short inter_mode,
int ∗last_index,
double ∗pvecback,
double ∗pvecthermo )
Thermodynamics quantities at given redshift z.
Evaluates all thermodynamics quantities at a given value of the redshift by reading the pre-computed table and
interpolating.
Parameters
pba Input: pointer to background structure
pth Input: pointer to the thermodynamics structure (containing pre-computed table)
zInput: redshift
inter_mode Input: interpolation mode (normal or growing_closeby)
last_index Input/Output: index of the previous/current point in the interpolation array (input only for closeby
mode, output for both)
pvecback Input: vector of background quantities (used only in case z>z_initial for getting ddkappa and
dddkappa; in that case, should be already allocated and filled, with format short_info or larger; in
other cases, will be ignored)
pvecthermo Output: vector of thermodynamics quantities (assumed to be already allocated)
Generated by Doxygen
184 File Documentation
Returns
the error status
Summary:
• define local variables
• interpolate in table with array_interpolate_spline() (normal mode) or array_interpolate_spline_growing_←-
closeby() (closeby mode)
5.19.2.2 thermodynamics_init()
int thermodynamics_init (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth )
Initialize the thermo structure, and in particular the thermodynamics interpolation table.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input/Output: pointer to initialized thermo structure
Returns
the error status
Summary:
• define local variables
• initialize pointers, allocate background vector
• compute and check primordial Helium fraction
• check energy injection parameters
• assign values to all indices in the structures with thermodynamics_indices()
• solve recombination and store values of z, xe, dκ/dτ, Tb, c2
bwith thermodynamics_recombination()
• if there is reionization, solve reionization and store values of z, xe, dκ/dτ, Tb, c2
bwith thermodynamics_←-
reionization()
• merge tables in recombination and reionization structures into a single table in thermo structure
• compute table of corresponding conformal times
• store initial value of conformal time in the structure
Generated by Doxygen

5.19 thermodynamics.c File Reference 185
• fill missing columns (quantities not computed previously but related)
• –>baryon drag interaction rate time minus one, -[1/R ∗kappa'], with R = 3 rho_b / 4 rho_gamma, stored
temporarily in column ddkappa
• –>second derivative of this rate, -[1/R ∗kappa']'', stored temporarily in column dddkappa
• –>compute tau_d = [int_{tau_today}∧{tau} dtau -dkappa_d/dtau]
• –>compute r_d = 2pi/k_d = 2pi ∗[int_{tau_ini}∧{tau} dtau (1/kappa') (R∧2+4/5(1+R))/(1+R∧2)/6 ]∧1/2 (see
e.g. Wayne Hu's thesis eq. (5.59)
• –>second derivative with respect to tau of dkappa (in view of spline interpolation)
• –>first derivative with respect to tau of dkappa (using spline interpolation)
• –>compute -kappa = [int_{tau_today}∧{tau} dtau dkappa/dtau], store temporarily in column "g"
• –>derivatives of baryon sound speed (only computed if some non-minimal tight-coupling schemes is re-
quested)
• —>second derivative with respect to tau of cb2
• —>first derivative with respect to tau of cb2 (using spline interpolation)
• –>compute visibility: g= (dκ/dτ)e−κ
• —>compute g
• —>compute exp(-kappa)
• —>compute g' (the plus sign of the second term is correct, see def of -kappa in thermodynamics module!)
• —>compute g''
• —>store g
• —>compute variation rate
• smooth the rate (details of smoothing unimportant: only the order of magnitude of the rate matters)
• fill tables of second derivatives with respect to z (in view of spline interpolation)
• find maximum of g
• find conformal recombination time using background_tau_of_z()
• find damping scale at recombination (using linear interpolation)
• find time (always after recombination) at which tau_c/tau falls below some threshold, defining tau_free_←-
streaming
• find baryon drag time (when tau_d crosses one, using linear interpolation) and sound horizon at that time
• find time above which visibility falls below a given fraction of its maximum
• if verbose flag set to next-to-minimum value, print the main results
5.19.2.3 thermodynamics_free()
int thermodynamics_free (
struct thermo ∗pth )
Free all memory space allocated by thermodynamics_init().
Generated by Doxygen
186 File Documentation
Parameters
pth Input/Output: pointer to thermo structure (to be freed)
Returns
the error status
5.19.2.4 thermodynamics_indices()
int thermodynamics_indices (
struct thermo ∗pth,
struct recombination ∗preco,
struct reionization ∗preio )
Assign value to each relevant index in vectors of thermodynamical quantities, as well as in vector containing reion-
ization parameters.
Parameters
pth Input/Output: pointer to thermo structure
preco Input/Output: pointer to recombination structure
preio Input/Output: pointer to reionization structure
Returns
the error status
Summary:
• define local variables
• initialization of all indices and flags in thermo structure
• initialization of all indices and flags in recombination structure
• initialization of all indices and flags in reionization structure
• same with parameters of the function Xe(z)
5.19.2.5 thermodynamics_helium_from_bbn()
int thermodynamics_helium_from_bbn (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth )
Infer the primordial helium fraction from standard BBN, as a function of the baryon density and expansion rate during
BBN.
This module is simpler then the one used in arXiv:0712.2826 because it neglects the impact of a possible significant
chemical potentials for electron neutrinos. The full code with xi_nu_e could be introduced here later.
Generated by Doxygen

5.19 thermodynamics.c File Reference 187
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input/Output: pointer to initialized thermo structure
Returns
the error status
Summary:
• Infer effective number of neutrinos at the time of BBN
• 8.6173e-11 converts from Kelvin to MeV. We randomly choose 0.1 MeV to be the temperature of BBN
• compute Delta N_eff as defined in bbn file, i.e. ∆Neff = 0 means Neff = 3.046
• spline in one dimension (along deltaN)
• interpolate in one dimension (along deltaN)
• spline in remaining dimension (along omegab)
• interpolate in remaining dimension (along omegab)
• deallocate arrays
5.19.2.6 thermodynamics_onthespot_energy_injection()
int thermodynamics_onthespot_energy_injection (
struct precision ∗ppr,
struct background ∗pba,
struct recombination ∗preco,
double z,
double ∗energy_rate,
ErrorMsg error_message )
In case of non-minimal cosmology, this function determines the energy rate injected in the IGM at a given redshift z
(= on-the-spot annihilation). This energy injection may come e.g. from dark matter annihilation or decay.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
preco Input: pointer to recombination structure
zInput: redshift
energy_rate Output: energy density injection rate
error_message Output: error message
Generated by Doxygen
188 File Documentation
Returns
the error status
5.19.2.7 thermodynamics_energy_injection()
int thermodynamics_energy_injection (
struct precision ∗ppr,
struct background ∗pba,
struct recombination ∗preco,
double z,
double ∗energy_rate,
ErrorMsg error_message )
In case of non-minimal cosmology, this function determines the effective energy rate absorbed by the IGM at a given
redshift (beyond the on-the-spot annihilation). This energy injection may come e.g. from dark matter annihilation or
decay.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
preco Input: pointer to recombination structure
zInput: redshift
energy_rate Output: energy density injection rate
error_message Output: error message
Returns
the error status
5.19.2.8 thermodynamics_reionization_function()
int thermodynamics_reionization_function (
double z,
struct thermo ∗pth,
struct reionization ∗preio,
double ∗xe )
This subroutine contains the reionization function Xe(z)(one for each scheme; so far, only the function correspond-
ing to the reio_camb scheme is coded)
Parameters
zInput: redshift
pth Input: pointer to thermo structure, to know which scheme is used
preio Input: pointer to reionization structure, containing the parameters of the function Xe(z)
xe Output: Xe(z)
Generated by Doxygen
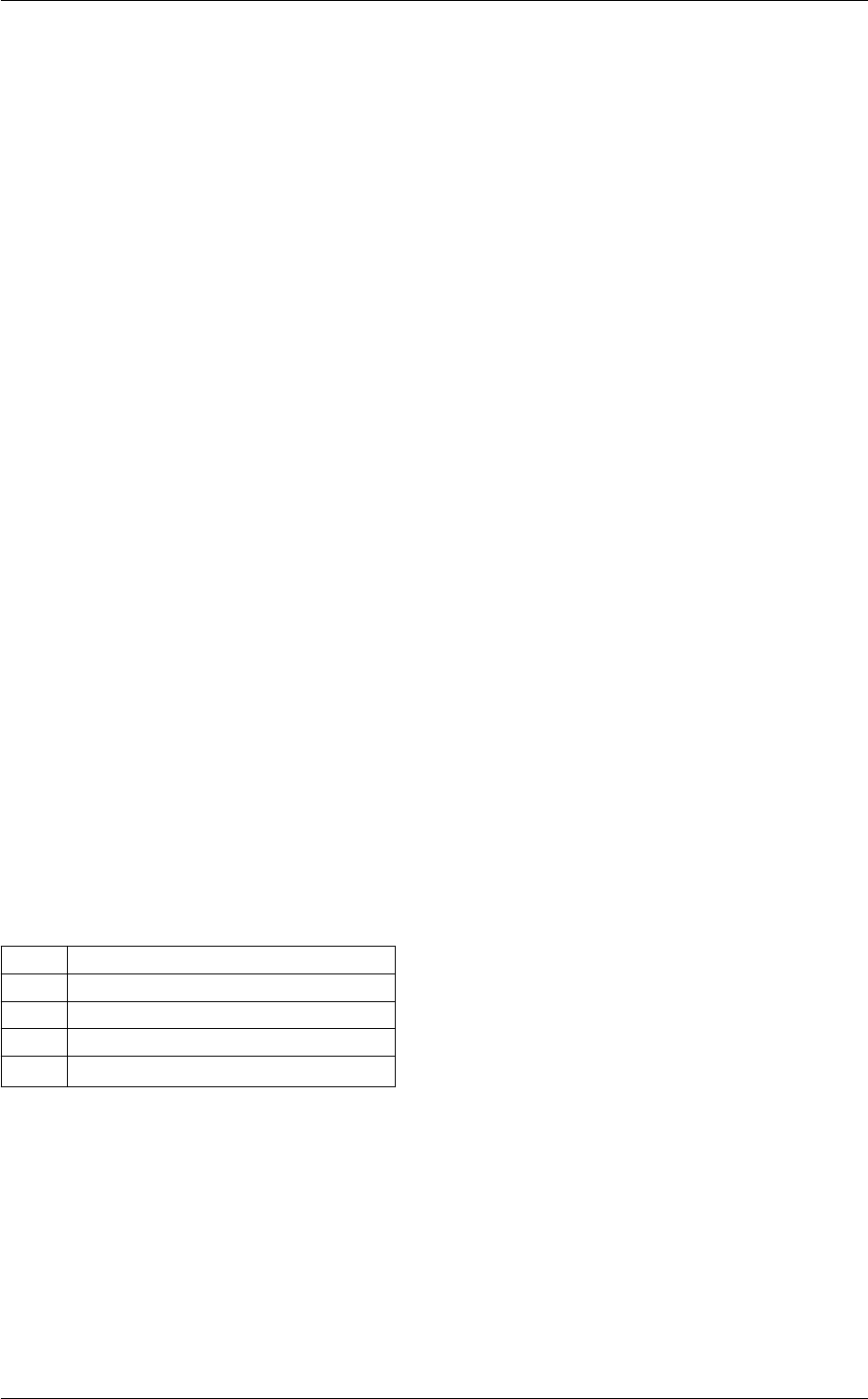
5.19 thermodynamics.c File Reference 189
Summary:
• define local variables
• implementation of ionization function similar to the one in CAMB
• –>case z >z_reio_start
• –>case z <z_reio_start: hydrogen contribution (tanh of complicated argument)
• –>case z <z_reio_start: helium contribution (tanh of simpler argument)
• implementation of binned ionization function similar to astro-ph/0606552
• –>case z >z_reio_start
• implementation of many tanh jumps
• –>case z >z_reio_start
• implementation of reio_inter
• –>case z >z_reio_start
5.19.2.9 thermodynamics_get_xe_before_reionization()
int thermodynamics_get_xe_before_reionization (
struct precision ∗ppr,
struct thermo ∗pth,
struct recombination ∗preco,
double z,
double ∗xe )
This subroutine reads Xe(z)in the recombination table at the time at which reionization starts. Hence it provides
correct initial conditions for the reionization function.
Parameters
ppr Input: pointer to precision structure
pth Input: pointer to thermo structure
preco Input: pointer to recombination structure
zInput: redshift z_reio_start
xe Output: Xe(z)at z
5.19.2.10 thermodynamics_reionization()
int thermodynamics_reionization (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
Generated by Doxygen

190 File Documentation
struct recombination ∗preco,
struct reionization ∗preio,
double ∗pvecback )
This routine computes the reionization history. In the reio_camb scheme, this is straightforward if the input parameter
is the reionization redshift. If the input is the optical depth, need to find z_reio by dichotomy (trying several z_reio
until the correct tau_reio is approached).
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermo structure
preco Input: pointer to filled recombination structure
preio Input/Output: pointer to reionization structure (to be filled)
pvecback Input: vector of background quantities (used as workspace: must be already allocated, with format
short_info or larger, but does not need to be filled)
Returns
the error status
Summary:
• define local variables
• allocate the vector of parameters defining the function Xe(z)
• (a) if reionization implemented like in CAMB
• –>set values of these parameters, excepted those depending on the reionization redshift
• –>if reionization redshift given as an input, initialize the remaining values and fill reionization table
• –>if reionization optical depth given as an input, find reionization redshift by dichotomy and initialize the
remaining values
• (b) if reionization implemented with reio_bins_tanh scheme
• (c) if reionization implemented with reio_many_tanh scheme
• (d) if reionization implemented with reio_inter scheme
5.19.2.11 thermodynamics_reionization_sample()
int thermodynamics_reionization_sample (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct recombination ∗preco,
struct reionization ∗preio,
double ∗pvecback )
For fixed input reionization parameters, this routine computes the reionization history and fills the reionization table.
Generated by Doxygen

5.19 thermodynamics.c File Reference 191
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermo structure
preco Input: pointer to filled recombination structure
preio Input/Output: pointer to reionization structure (to be filled)
pvecback Input: vector of background quantities (used as workspace: must be already allocated, with format
short_info or larger, but does not need to be filled)
Returns
the error status
Summary:
• define local variables
• (a) allocate vector of values related to reionization
• (b) create a growTable with gt_init()
• (c) first line is taken from thermodynamics table, just before reionization starts
• –>look where to start in current thermodynamics table
• –>get redshift
• –>get Xe
• –>get dκ/dz = (dκ/dτ)∗(dτ/dz) = −(dκ/dτ )/H
• –>get baryon temperature
• –>after recombination, Tb scales like (1+z)∗∗2. Compute constant factor Tb/(1+z)∗∗2.
• –>get baryon sound speed
• –>store these values in growing table
• (d) set the maximum step value (equal to the step in thermodynamics table)
• (e) loop over redshift values in order to find values of z, x_e, kappa' (Tb and cb2 found later by integration).
The sampling in z space is found here.
• (f) allocate reionization_table with correct size
• (g) retrieve data stored in the growTable with gt_getPtr()
• (h) copy growTable to reionization_temporary_table (invert order of lines, so that redshift is growing, like in
recombination table)
• (i) free the growTable with gt_free() , free vector of reionization variables
• (j) another loop on z, to integrate equation for Tb and to compute cb2
• –>derivative of baryon temperature
• –>increment baryon temperature
• –>get baryon sound speed
• –>spline dτ/dz with respect to z in view of integrating for optical depth
• –>integrate for optical depth
Generated by Doxygen

192 File Documentation
5.19.2.12 thermodynamics_recombination()
int thermodynamics_recombination (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct recombination ∗preco,
double ∗pvecback )
Integrate thermodynamics with your favorite recombination code.
5.19.2.13 thermodynamics_recombination_with_hyrec()
int thermodynamics_recombination_with_hyrec (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct recombination ∗preco,
double ∗pvecback )
Integrate thermodynamics with HyRec.
Integrate thermodynamics with HyRec, allocate and fill the part of the thermodynamics interpolation table (the rest
is filled in thermodynamics_init()). Called once by thermodynamics_recombination(), from thermodynamics_init().
HYREC: Hydrogen and Helium Recombination Code
Written by Yacine Ali-Haimoud and Chris Hirata (Caltech)
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
preco Output: pointer to recombination structure
pvecback Input: pointer to an allocated (but empty) vector of background variables
Summary:
• Fill hyrec parameter structure
• Build effective rate tables
• distribute addresses for each table
• Normalize 2s–1s differential decay rate to L2s1s (can be set by user in hydrogen.h)
• Compute the recombination history by calling a function in hyrec (no CLASS-like error management here)
• fill a few parameters in preco and pth
• allocate memory for thermodynamics interpolation tables (size known in advance) and fill it
• –>get redshift, corresponding results from hyrec, and background quantities
• –>store the results in the table
Generated by Doxygen

5.19 thermodynamics.c File Reference 193
5.19.2.14 thermodynamics_recombination_with_recfast()
int thermodynamics_recombination_with_recfast (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct recombination ∗preco,
double ∗pvecback )
Integrate thermodynamics with RECFAST.
Integrate thermodynamics with RECFAST, allocate and fill the part of the thermodynamics interpolation table (the
rest is filled in thermodynamics_init()). Called once by thermodynamics_recombination, from thermodynamics_←-
init().
RECFAST is an integrator for Cosmic Recombination of Hydrogen and Helium, developed by Douglas Scott
(dscott@astro.ubc.ca) based on calculations in the paper Seager, Sasselov & Scott (ApJ, 523, L1, 1999).
and "fudge" updates in Wong, Moss & Scott (2008).
Permission to use, copy, modify and distribute without fee or royalty at any tier, this software and its documentation,
for any purpose and without fee or royalty is hereby granted, provided that you agree to comply with the following
copyright notice and statements, including the disclaimer, and that the same appear on ALL copies of the software
and documentation, including modifications that you make for internal use or for distribution:
Copyright 1999-2010 by University of British Columbia. All rights reserved.
THIS SOFTWARE IS PROVIDED "AS IS", AND U.B.C. MAKES NO REPRESENTATIONS OR WARRANTIES,
EXPRESS OR IMPLIED. BY WAY OF EXAMPLE, BUT NOT LIMITATION, U.B.C. MAKES NO REPRESENTAT←-
IONS OR WARRANTIES OF MERCHANTABILITY OR FITNESS FOR ANY PARTICULAR PURPOSE OR THAT
THE USE OF THE LICENSED SOFTWARE OR DOCUMENTATION WILL NOT INFRINGE ANY THIRD PARTY
PATENTS, COPYRIGHTS, TRADEMARKS OR OTHER RIGHTS.
Version 1.5: includes extra fitting function from Rubino-Martin et al. arXiv:0910.4383v1 [astro-ph.CO]
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
preco Output: pointer to recombination structure
pvecback Input: pointer to an allocated (but empty) vector of background variables
Returns
the error status
Summary:
• define local variables
• allocate memory for thermodynamics interpolation tables (size known in advance)
• initialize generic integrator with initialize_generic_integrator()
• read a few precision/cosmological parameters
Generated by Doxygen

194 File Documentation
• define the fields of the 'thermodynamics parameter and workspace' structure
• impose initial conditions at early times
• loop over redshift steps Nz; integrate over each step with generic_integrator(), store the results in the table
using thermodynamics_derivs_with_recfast()
• –>first approximation: H and Helium fully ionized
• –>second approximation: first Helium recombination (analytic approximation)
• –>third approximation: first Helium recombination completed
• –>fourth approximation: second Helium recombination starts (analytic approximation)
• –>fifth approximation: second Helium recombination (full evolution for Helium), H recombination starts (an-
alytic approximation)
• –>last case: full evolution for H and Helium
• –>store the results in the table
• cleanup generic integrator with cleanup_generic_integrator()
5.19.2.15 thermodynamics_derivs_with_recfast()
int thermodynamics_derivs_with_recfast (
double z,
double ∗y,
double ∗dy,
void ∗parameters_and_workspace,
ErrorMsg error_message )
Subroutine evaluating the derivative with respect to redshift of thermodynamical quantities (from RECFAST version
1.4).
Computes derivatives of the three variables to integrate: dxH/dz, dxHe/dz, dTmat/dz.
This is one of the few functions in the code which are passed to the generic_integrator() routine. Since generic_←-
integrator() should work with functions passed from various modules, the format of the arguments is a bit special:
• fixed parameters and workspaces are passed through a generic pointer. Here, this pointer contains the pre-
cision, background and recombination structures, plus a background vector, but generic_integrator() doesn't
know its fine structure.
• the error management is a bit special: errors are not written as usual to pth->error_message, but to a generic
error_message passed in the list of arguments.
Parameters
zInput: redshift
yInput: vector of variable to integrate
dy Output: its derivative (already allocated)
parameters_and_workspace Input: pointer to fixed parameters (e.g. indices) and workspace (already allocated)
error_message Output: error message
Generated by Doxygen
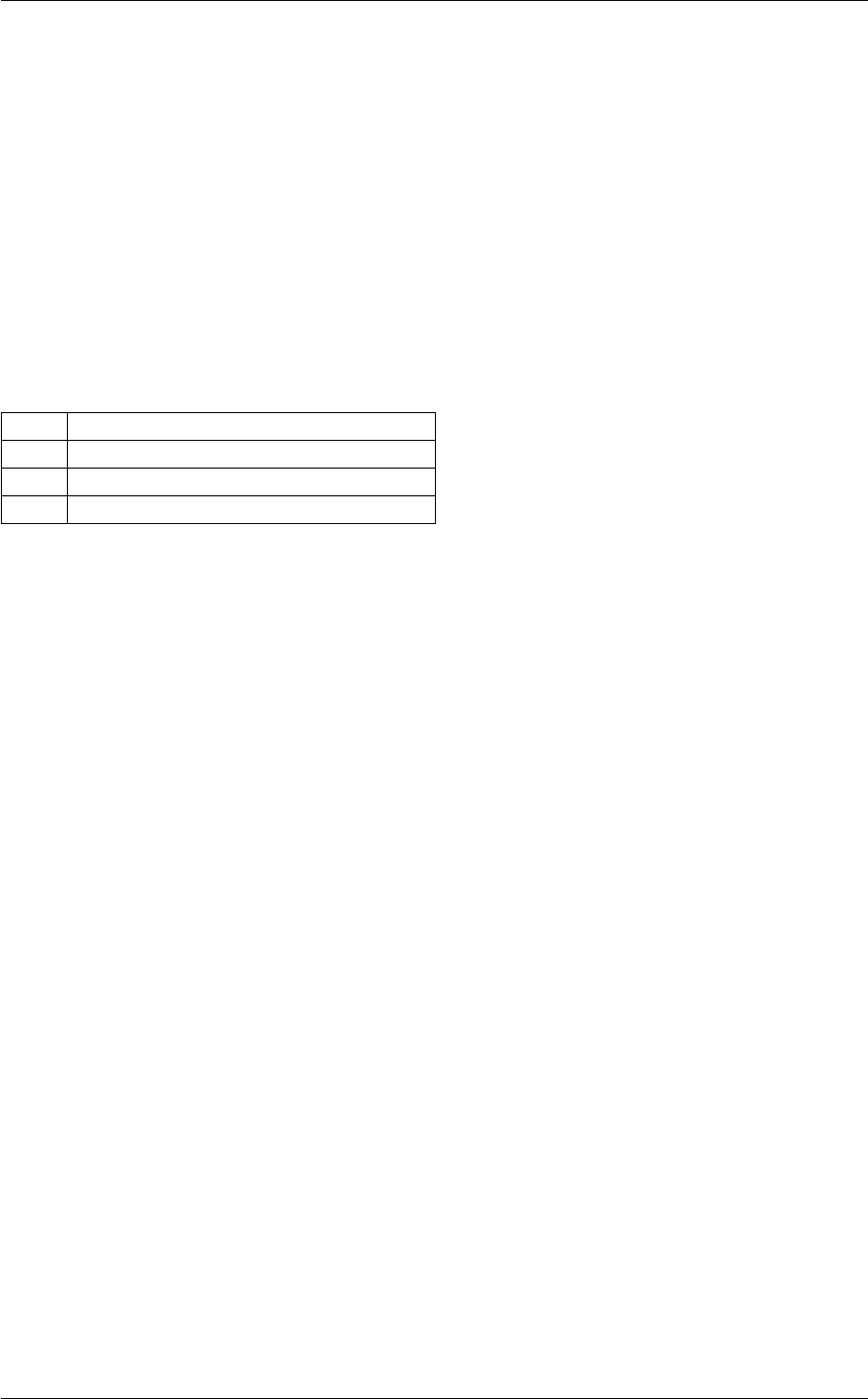
5.19 thermodynamics.c File Reference 195
5.19.2.16 thermodynamics_merge_reco_and_reio()
int thermodynamics_merge_reco_and_reio (
struct precision ∗ppr,
struct thermo ∗pth,
struct recombination ∗preco,
struct reionization ∗preio )
This routine merges the two tables 'recombination_table' and 'reionization_table' inside the table 'thermodynamics←-
_table', and frees the temporary structures 'recombination' and 'reionization'.
Parameters
ppr Input: pointer to precision structure
pth Input/Output: pointer to thermo structure
preco Input: pointer to filled recombination structure
preio Input: pointer to reionization structure
Returns
the error status
Summary:
• define local variables
• first, a little check that the two tables match each other and can be merged
• find number of redshift in full table = number in reco + number in reio - overlap
• allocate arrays in thermo structure
• fill these arrays
• free the temporary structures
5.19.2.17 thermodynamics_output_titles()
int thermodynamics_output_titles (
struct background ∗pba,
struct thermo ∗pth,
char titles[_MAXTITLESTRINGLENGTH_] )
Subroutine for formatting thermodynamics output
Generated by Doxygen
196 File Documentation
5.20 thermodynamics.h File Reference
#include "background.h"
Include dependency graph for thermodynamics.h:
thermodynamics.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.h parser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
This graph shows which files directly or indirectly include this file:
thermodynamics.h
input.h
class.h
perturbations.h thermodynamics.c
input.c
class.c
primordial.h perturbations.c
nonlinear.h
nonlinear_conflict
-20170920-132422.h
nonlinear_conflict
-20170920-150212.h nonlinear_exp.h nonlinear_test.h primordial.c
transfer.h nonlinear.c
spectra.h transfer.c
lensing.hspectra.c
output.hlensing.c
output.c
Data Structures
• struct thermo
• struct recombination
• struct reionization
• struct thermodynamics_parameters_and_workspace
Generated by Doxygen

5.20 thermodynamics.h File Reference 197
Macros
• #define f1(x) (-0.75∗x∗(x∗x/3.-1.)+0.5)
• #define f2(x) (x∗x∗(0.5-x/3.)∗6.)
• #define _YHE_BIG_ 0.5
• #define _YHE_SMALL_ 0.01
Enumerations
• enum recombination_algorithm
• enum reionization_parametrization {
reio_none,reio_camb,reio_bins_tanh,reio_half_tanh,
reio_many_tanh,reio_inter }
• enum reionization_z_or_tau {reio_z,reio_tau }
5.20.1 Detailed Description
Documented includes for thermodynamics module
5.20.2 Data Structure Documentation
5.20.2.1 struct thermo
All thermodynamics parameters and evolution that other modules need to know.
Once initialized by thermodynamics_init(), contains all the necessary information on the thermodynamics, and in
particular, a table of thermodynamical quantities as a function of the redshift, used for interpolation in other modules.
Data Fields
double YHe YHe: primordial helium fraction
enum recombination_algorithm recombination recombination code
enum reionization_parametrization reio_parametrization reionization scheme
enum reionization_z_or_tau reio_z_or_tau is the input parameter the
reionization redshift or optical depth?
double tau_reio if above set to tau, input value of
reionization optical depth
double z_reio if above set to z, input value of
reionization redshift
short compute_cb2_derivatives do we want to include in computation
derivatives of baryon sound speed?
short compute_damping_scale do we want to compute the simplest
analytic approximation to the photon
damping (or diffusion) scale?
double reionization_width parameters for reio_camb width of H
reionization
Generated by Doxygen
198 File Documentation
Data Fields
double reionization_exponent shape of H reionization
double helium_fullreio_redshift redshift for of helium reionization
double helium_fullreio_width width of helium reionization
int binned_reio_num parameters for reio_bins_tanh with
how many bins do we want to
describe reionization?
double ∗binned_reio_z central z value for each bin
double ∗binned_reio_xe imposed Xe(z)value at center of
each bin
double binned_reio_step_sharpness sharpness of tanh() step interpolating
between binned values
int many_tanh_num parameters for reio_many_tanh with
how many jumps do we want to
describe reionization?
double ∗many_tanh_z central z value for each tanh jump
double ∗many_tanh_xe imposed Xe(z)value at the end of
each jump (ie at later times)
double many_tanh_width sharpness of tanh() steps
int reio_inter_num parameters for reio_inter with how
many jumps do we want to describe
reionization?
double ∗reio_inter_z discrete z values
double ∗reio_inter_xe discrete Xe(z)values
double annihilation parameters for energy injection
short has_on_the_spot parameter describing CDM
annihilation (f <sigma∗v>/ m_cdm,
see e.g. 0905.0003)
double decay flag to specify if we want to use the
on-the-spot approximation
double annihilation_variation parameter describing CDM decay
(f/tau, see e.g. 1109.6322)
double annihilation_z if this parameter is non-zero, the
function F(z)=(f <sigma∗v>/
m_cdm)(z) will be a parabola in
log-log scale between zmin and
zmax, with a curvature given by
annihlation_variation (must be
negative), and with a maximum in
zmax; it will be constant outside this
range
double annihilation_zmax if annihilation_variation is non-zero,
this is the value of z at which the
parameter annihilation is defined, i.e.
F(annihilation_z)=annihilation
double annihilation_zmin if annihilation_variation is non-zero,
redshift above which annihilation rate
is maximal
double annihilation_f_halo if annihilation_variation is non-zero,
redshift below which annihilation rate
is constant
double annihilation_z_halo takes the contribution of DM
annihilation in halos into account
int index_th_xe ionization fraction xe
Generated by Doxygen

5.20 thermodynamics.h File Reference 199
Data Fields
int index_th_dkappa Thomson scattering rate dκ/dτ
(units 1/Mpc)
int index_th_tau_d Baryon drag optical depth
int index_th_ddkappa scattering rate derivative d2κ/dτ 2
int index_th_dddkappa scattering rate second derivative
d3κ/dτ3
int index_th_exp_m_kappa exp−κ
int index_th_g visibility function
g= (dκ/dτ)∗exp−κ
int index_th_dg visibility function derivative (dg/dτ)
int index_th_ddg visibility function second derivative
(d2g/dτ2)
int index_th_Tb baryon temperature Tb
int index_th_cb2 squared baryon sound speed c2
b
int index_th_dcb2 derivative wrt conformal time of
squared baryon sound speed
d[c2
b]/dτ (only computed if some
non-minimal tight-coupling schemes
is requested)
int index_th_ddcb2 second derivative wrt conformal time
of squared baryon sound speed
d2[c2
b]/dτ2(only computed if some
non0-minimal tight-coupling schemes
is requested)
int index_th_rate maximum variation rate of exp−κ, g
and (dg/dτ), used for computing
integration step in perturbation
module
int index_th_r_d simple analytic approximation to the
photon comoving damping scale
int th_size size of thermodynamics vector
int tt_size number of lines (redshift steps) in the
tables
double ∗z_table vector z_table[index_z] with values of
redshift (vector of size tt_size)
double ∗thermodynamics_table table thermodynamics_table[index←-
_z∗pth->tt_size+pba->index_th]
with all other quantities (array of size
th_size∗tt_size)
double ∗d2thermodynamics_dz2_table table d2thermodynamics_dz2_←-
table[index_z∗pth->tt_size+pba-
>index_th] with values of d2ti/dz2
(array of size th_size∗tt_size)
double z_rec z at which the visibility reaches its
maximum (= recombination redshift)
double tau_rec conformal time at which the visibility
reaches its maximum (=
recombination time)
double rs_rec comoving sound horizon at
recombination
Generated by Doxygen
200 File Documentation
Data Fields
double ds_rec physical sound horizon at
recombination
double ra_rec conformal angular diameter distance
to recombination
double da_rec physical angular diameter distance to
recombination
double rd_rec comoving photon damping scale at
recombination
double z_d baryon drag redshift
double tau_d baryon drag time
double ds_d physical sound horizon at baryon
drag
double rs_d comoving sound horizon at baryon
drag
double tau_cut at at which the visibility goes below a
fixed fraction of the maximum
visibility, used for an approximation in
perturbation module
double angular_rescaling [ratio ra_rec / (tau0-tau_rec)]: gives
CMB rescaling in angular space
relative to flat model (=1 for curvature
K=0)
double tau_free_streaming minimum value of tau at which
sfree-streaming approximation can
be switched on
double tau_ini initial conformal time at which
thermodynamical variables have
been be integrated
double n_e total number density of electrons
today (free or not)
short inter_normal flag for calling thermodynamics_at_z
and find position in interpolation table
normally
short inter_closeby flag for calling thermodynamics_at_z
and find position in interpolation table
starting from previous position in
previous call
short thermodynamics_verbose flag regulating the amount of
information sent to standard output
(none if set to zero)
ErrorMsg error_message zone for writing error messages
5.20.2.2 struct recombination
Temporary structure where all the recombination history is defined and stored.
This structure is used internally by the thermodynamics module, but never passed to other modules.
Data Fields
int index_re_z redshift z
Generated by Doxygen

5.20 thermodynamics.h File Reference 201
Data Fields
int index_re_xe ionization fraction xe
int index_re_Tb baryon temperature Tb
int index_re_cb2 squared baryon sound speed c2
b
int index_re_dkappadtau Thomson scattering rate dκ/dτ (units 1/Mpc)
int re_size size of this vector
int rt_size number of lines (redshift steps) in the table
double ∗recombination_table table recombination_table[index_z∗preco->re_size+index_re] with all
other quantities (array of size preco->rt_size∗preco->re_size)
double CDB defined as in RECFAST
double CR defined as in RECFAST
double CK defined as in RECFAST
double CL defined as in RECFAST
double CT defined as in RECFAST
double fHe defined as in RECFAST
double CDB_He defined as in RECFAST
double CK_He defined as in RECFAST
double CL_He defined as in RECFAST
double fu defined as in RECFAST
double H_frac defined as in RECFAST
double Tnow defined as in RECFAST
double Nnow defined as in RECFAST
double Bfact defined as in RECFAST
double CB1 defined as in RECFAST
double CB1_He1 defined as in RECFAST
double CB1_He2 defined as in RECFAST
double H0 defined as in RECFAST
double YHe defined as in RECFAST
double annihilation parameter describing CDM annihilation (f <sigma∗v>/ m_cdm, see e.g.
0905.0003)
short has_on_the_spot flag to specify if we want to use the on-the-spot approximation
double decay parameter describing CDM decay (f/tau, see e.g. 1109.6322)
double annihilation_variation if this parameter is non-zero, the function F(z)=(f <sigma∗v>/ m_cdm)(z)
will be a parabola in log-log scale between zmin and zmax, with a
curvature given by annihlation_variation (must be negative), and with a
maximum in zmax; it will be constant outside this range
double annihilation_z if annihilation_variation is non-zero, this is the value of z at which the
parameter annihilation is defined, i.e. F(annihilation_z)=annihilation
double annihilation_zmax if annihilation_variation is non-zero, redshift above which annihilation rate
is maximal
double annihilation_zmin if annihilation_variation is non-zero, redshift below which annihilation rate
is constant
double annihilation_f_halo takes the contribution of DM annihilation in halos into account
double annihilation_z_halo characteristic redshift for DM annihilation in halos
5.20.2.3 struct reionization
Temporary structure where all the reionization history is defined and stored.
This structure is used internally by the thermodynamics module, but never passed to other modules.
Generated by Doxygen

202 File Documentation
Data Fields
int index_re_z redshift z
int index_re_xe ionization fraction xe
int index_re_Tb baryon temperature Tb
int index_re_cb2 squared baryon sound speed c2
b
int index_re_dkappadtau Thomson scattering rate dκ/dτ (units 1/Mpc)
int index_re_dkappadz Thomson scattering rate with respect to redshift dκ/dz (units
1/Mpc)
int index_re_d3kappadz3 second derivative of previous quantity with respect to redshift
int re_size size of this vector
int rt_size number of lines (redshift steps) in the table
double ∗reionization_table table reionization_table[index_z∗preio->re_size+index_re] with all
other quantities (array of size preio->rt_size∗preio->re_size)
double reionization_optical_depth reionization optical depth inferred from reionization history
int index_reio_redshift hydrogen reionization redshift
int index_reio_exponent an exponent used in the function x_e(z) in the reio_camb scheme
int index_reio_width a width defining the duration of hydrogen reionization in the
reio_camb scheme
int index_reio_xe_before ionization fraction at redshift 'reio_start'
int index_reio_xe_after ionization fraction after full reionization
int index_helium_fullreio_fraction helium full reionization fraction inferred from primordial helium
fraction
int index_helium_fullreio_redshift helium full reionization redshift
int index_helium_fullreio_width a width defining the duration of helium full reionization in the
reio_camb scheme
int reio_num_z number of reionization jumps
int index_reio_first_z redshift at which we start to impose reionization function
int index_reio_first_xe ionization fraction at redshift first_z (inferred from recombination
code)
int index_reio_step_sharpness sharpness of tanh jump
int index_reio_start redshift above which hydrogen reionization neglected
double ∗reionization_parameters vector containing all reionization parameters necessary to
compute xe(z)
int reio_num_params length of vector reionization_parameters
int index_reco_when_reio_start index of line in recombination table corresponding to first line of
reionization table
5.20.2.4 struct thermodynamics_parameters_and_workspace
temporary parameters and workspace passed to the thermodynamics_derivs function
5.20.3 Macro Definition Documentation
Generated by Doxygen

5.20 thermodynamics.h File Reference 203
5.20.3.1 f1
#define f1(
x) (-0.75∗x∗(x∗x/3.-1.)+0.5)
Two useful smooth step functions, for smoothing transitions in recfast.goes from 0 to 1 when x goes from -1 to 1
5.20.3.2 f2
#define f2(
x) (x∗x∗(0.5-x/3.)∗6.)
goes from 0 to 1 when x goes from 0 to 1
5.20.3.3 _YHE_BIG_
#define _YHE_BIG_ 0.5
maximal YHe
5.20.3.4 _YHE_SMALL_
#define _YHE_SMALL_ 0.01
minimal YHe
5.20.4 Enumeration Type Documentation
5.20.4.1 recombination_algorithm
enum recombination_algorithm
List of possible recombination algorithms.
5.20.4.2 reionization_parametrization
enum reionization_parametrization
List of possible reionization schemes.
Enumerator
reio_none no reionization
reio_camb reionization parameterized like in CAMB
reio_bins_tanh binned reionization history with tanh inteprolation between bins
reio_half_tanh half a tanh, instead of the full tanh
reio_many_tanh similar to reio_camb but with more than one tanh
reio_inter linear interpolation between specified points
Generated by Doxygen
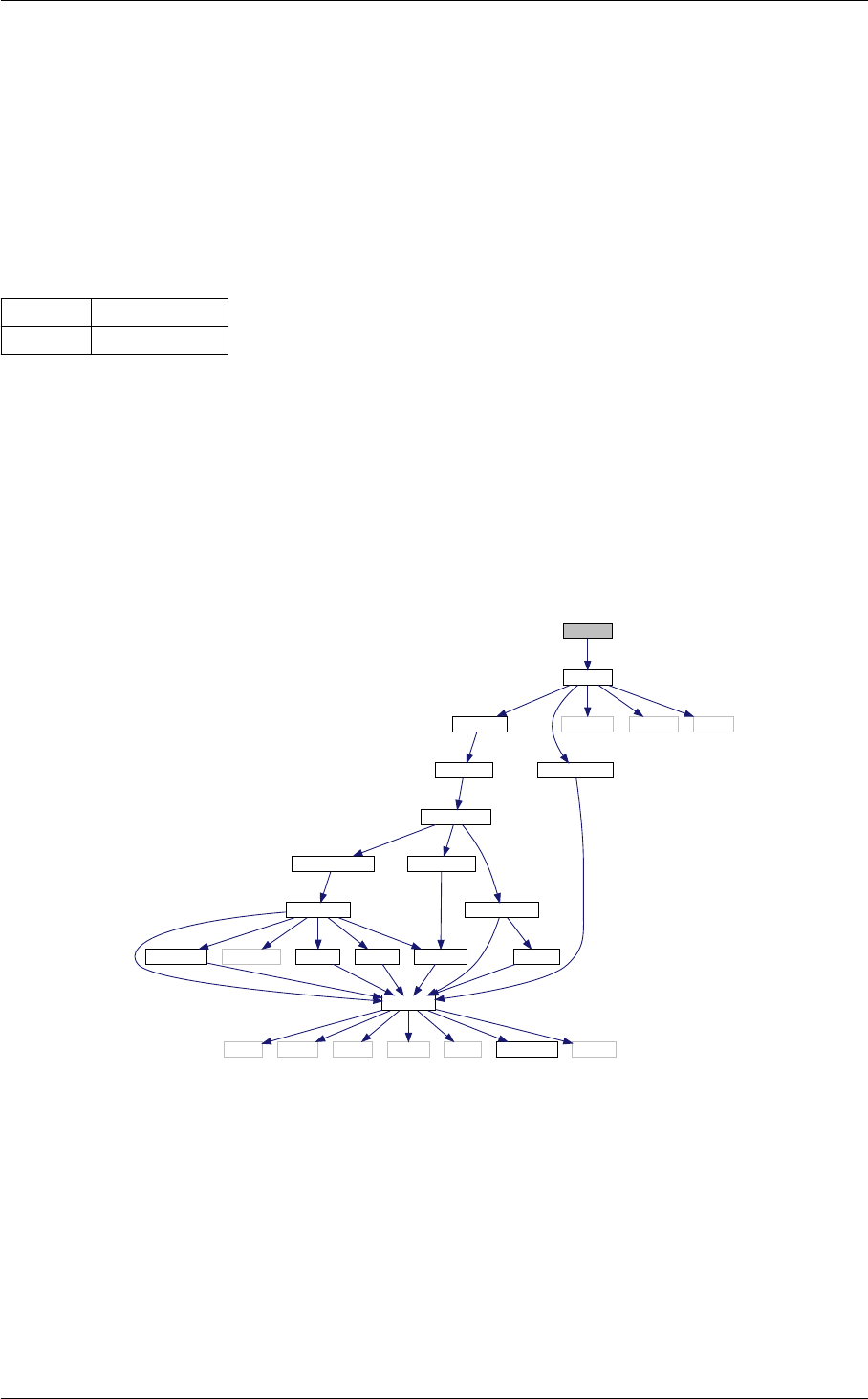
204 File Documentation
5.20.4.3 reionization_z_or_tau
enum reionization_z_or_tau
Is the input parameter the reionization redshift or optical depth?
Enumerator
reio_z input = redshift
reio_tau input = tau
5.21 transfer.c File Reference
#include "transfer.h"
Include dependency graph for transfer.c:
transfer.c
transfer.h
nonlinear.h
hyperspherical.h
sys/shm.h sys/stat.h errno.h
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
Functions
• int transfer_functions_at_q (struct transfers ∗ptr, int index_md, int index_ic, int index_tt, int index_l, double q,
double ∗transfer_function)
• int transfer_init (struct precision ∗ppr, struct background ∗pba, struct thermo ∗pth, struct perturbs ∗ppt, struct
nonlinear ∗pnl, struct transfers ∗ptr)
• int transfer_free (struct transfers ∗ptr)
• int transfer_indices_of_transfers (struct precision ∗ppr, struct perturbs ∗ppt, struct transfers ∗ptr, double q_←-
period, double K, int sgnK)
Generated by Doxygen

5.21 transfer.c File Reference 205
• int transfer_get_l_list (struct precision ∗ppr, struct perturbs ∗ppt, struct transfers ∗ptr)
• int transfer_get_q_list (struct precision ∗ppr, struct perturbs ∗ppt, struct transfers ∗ptr, double q_period, dou-
ble K, int sgnK)
• int transfer_get_k_list (struct perturbs ∗ppt, struct transfers ∗ptr, double K)
• int transfer_get_source_correspondence (struct perturbs ∗ppt, struct transfers ∗ptr, int ∗∗tp_of_tt)
• int transfer_source_tau_size (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, struct trans-
fers ∗ptr, double tau_rec, double tau0, int index_md, int index_tt, int ∗tau_size)
• int transfer_compute_for_each_q (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, struct
transfers ∗ptr, int ∗∗tp_of_tt, int index_q, int tau_size_max, double tau_rec, double ∗∗∗pert_sources, double
∗∗∗pert_sources_spline, struct transfer_workspace ∗ptw)
• int transfer_interpolate_sources (struct perturbs ∗ppt, struct transfers ∗ptr, int index_q, int index_md, int
index_ic, int index_type, double ∗pert_source, double ∗pert_source_spline, double ∗interpolated_sources)
• int transfer_sources (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, struct transfers ∗ptr,
double ∗interpolated_sources, double tau_rec, int index_q, int index_md, int index_tt, double ∗sources, dou-
ble ∗tau0_minus_tau, double ∗w_trapz, int ∗tau_size_out)
• int transfer_selection_function (struct precision ∗ppr, struct perturbs ∗ppt, struct transfers ∗ptr, int bin, double
z, double ∗selection)
• int transfer_dNdz_analytic (struct transfers ∗ptr, double z, double ∗dNdz, double ∗dln_dNdz_dz)
• int transfer_selection_sampling (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, struct
transfers ∗ptr, int bin, double ∗tau0_minus_tau, int tau_size)
• int transfer_lensing_sampling (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, struct
transfers ∗ptr, int bin, double tau0, double ∗tau0_minus_tau, int tau_size)
• int transfer_source_resample (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, struct trans-
fers ∗ptr, int bin, double ∗tau0_minus_tau, int tau_size, int index_md, double tau0, double ∗interpolated_←-
sources, double ∗sources)
• int transfer_selection_times (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, struct trans-
fers ∗ptr, int bin, double ∗tau_min, double ∗tau_mean, double ∗tau_max)
• int transfer_selection_compute (struct precision ∗ppr, struct background ∗pba, struct perturbs ∗ppt, struct
transfers ∗ptr, double ∗selection, double ∗tau0_minus_tau, double ∗w_trapz, int tau_size, double ∗pvecback,
double tau0, int bin)
• int transfer_compute_for_each_l (struct transfer_workspace ∗ptw, struct precision ∗ppr, struct perturbs ∗ppt,
struct transfers ∗ptr, int index_q, int index_md, int index_ic, int index_tt, int index_l, double l, double q_max←-
_bessel, radial_function_type radial_type)
• int transfer_integrate (struct perturbs ∗ppt, struct transfers ∗ptr, struct transfer_workspace ∗ptw, int index_q,
int index_md, int index_tt, double l, int index_l, double k, radial_function_type radial_type, double ∗trsf)
• int transfer_limber (struct transfers ∗ptr, struct transfer_workspace ∗ptw, int index_md, int index_q, double l,
double q, radial_function_type radial_type, double ∗trsf)
• int transfer_limber_interpolate (struct transfers ∗ptr, double ∗tau0_minus_tau, double ∗sources, int tau_size,
double tau0_minus_tau_limber, double ∗S)
• int transfer_limber2 (int tau_size, struct transfers ∗ptr, int index_md, int index_k, double l, double k, double
∗tau0_minus_tau, double ∗sources, radial_function_type radial_type, double ∗trsf)
5.21.1 Detailed Description
Documented transfer module.
Julien Lesgourgues, 28.07.2013
This module has two purposes:
• at the beginning, to compute the transfer functions ∆X
l(q), and store them in tables used for interpolation in
other modules.
• at any time in the code, to evaluate the transfer functions (for a given mode, initial condition, type and multipole
l) at any wavenumber q (by interpolating within the interpolation table).
Generated by Doxygen

206 File Documentation
Hence the following functions can be called from other modules:
1. transfer_init() at the beginning (but after perturb_init() and bessel_init())
2. transfer_functions_at_q() at any later time
3. transfer_free() at the end, when no more calls to transfer_functions_at_q() are needed
Note that in the standard implementation of CLASS, only the pre-computed values of the transfer functions are
used, no interpolation is necessary; hence the routine transfer_functions_at_q() is actually never called.
5.21.2 Function Documentation
5.21.2.1 transfer_functions_at_q()
int transfer_functions_at_q (
struct transfers ∗ptr,
int index_md,
int index_ic,
int index_tt,
int index_l,
double q,
double ∗transfer_function )
Transfer function ∆X
l(q)at a given wavenumber q.
For a given mode (scalar, vector, tensor), initial condition, type (temperature, polarization, lensing, etc) and multipole,
computes the transfer function for an arbitrary value of q by interpolating between pre-computed values of q. This
function can be called from whatever module at whatever time, provided that transfer_init() has been called before,
and transfer_free() has not been called yet.
Wavenumbers are called q in this module and k in the perturbation module. In flat universes k=q. In non-flat
universes q and k differ through q2 = k2 + K(1 + m), where m=0,1,2 for scalar, vector, tensor. q should be
used throughout the transfer module, excepted when interpolating or manipulating the source functions S(k,tau)
calculated in the perturbation module: for a given value of q, this should be done at the corresponding k(q).
Parameters
ptr Input: pointer to transfer structure
index_md Input: index of requested mode
index_ic Input: index of requested initial condition
index_tt Input: index of requested type
index_l Input: index of requested multipole
qInput: any wavenumber
transfer_function Output: transfer function
Generated by Doxygen

5.21 transfer.c File Reference 207
Returns
the error status
Summary:
• interpolate in pre-computed table using array_interpolate_two()
5.21.2.2 transfer_init()
int transfer_init (
struct precision ∗ppr,
struct background ∗pba,
struct thermo ∗pth,
struct perturbs ∗ppt,
struct nonlinear ∗pnl,
struct transfers ∗ptr )
This routine initializes the transfers structure, (in particular, computes table of transfer functions ∆X
l(q))
Main steps:
• initialize all indices in the transfers structure and allocate all its arrays using transfer_indices_of_transfers().
• for each thread (in case of parallel run), initialize the fields of a memory zone called the transfer_workspace
with transfer_workspace_init()
• loop over q values. For each q, compute the Bessel functions if needed with transfer_update_HIS(), and defer
the calculation of all transfer functions to transfer_compute_for_each_q()
• for each thread, free the the workspace with transfer_workspace_free()
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
pth Input: pointer to thermodynamics structure
ppt Input: pointer to perturbation structure
pnl Input: pointer to nonlinear structure
ptr Output: pointer to initialized transfers structure
Returns
the error status
Summary:
• define local variables
Generated by Doxygen
208 File Documentation
• array with the correspondence between the index of sources in the perturbation module and in the transfer
module, tp_of_tt[index_md][index_tt]
• check whether any spectrum in harmonic space (i.e., any Cl's) is actually requested
• get number of modes (scalars, tensors...)
• get conformal age / recombination time from background / thermodynamics structures (only place where
these structures are used in this module)
• correspondence between k and l depend on angular diameter distance, i.e. on curvature.
• order of magnitude of the oscillation period of transfer functions
• initialize all indices in the transfers structure and allocate all its arrays using transfer_indices_of_transfers()
• copy sources to a local array sources (in fact, only the pointers are copied, not the data), and eventually apply
non-linear corrections to the sources
• spline all the sources passed by the perturbation module with respect to k (in order to interpolate later at a
given value of k)
• allocate and fill array describing the correspondence between perturbation types and transfer types
• evaluate maximum number of sampled times in the transfer sources: needs to be known here, in order to
allocate a large enough workspace
• compute flat spherical bessel functions
• eventually read the selection and evolution functions
• loop over all wavenumbers (parallelized).
• finally, free arrays allocated outside parallel zone
5.21.2.3 transfer_free()
int transfer_free (
struct transfers ∗ptr )
This routine frees all the memory space allocated by transfer_init().
To be called at the end of each run, only when no further calls to transfer_functions_at_k() are needed.
Parameters
ptr Input: pointer to transfers structure (which fields must be freed)
Returns
the error status
Generated by Doxygen
5.21 transfer.c File Reference 209
5.21.2.4 transfer_indices_of_transfers()
int transfer_indices_of_transfers (
struct precision ∗ppr,
struct perturbs ∗ppt,
struct transfers ∗ptr,
double q_period,
double K,
int sgnK )
This routine defines all indices and allocates all tables in the transfers structure
Compute list of (k, l) values, allocate and fill corresponding arrays in the transfers structure. Allocate the array of
transfer function tables.
Parameters
ppr Input: pointer to precision structure
ppt Input: pointer to perturbation structure
ptr Input/Output: pointer to transfer structure
q_period Input: order of magnitude of the oscillation period of transfer functions
KInput: spatial curvature (in absolute value)
sgnK Input: spatial curvature sign (open/closed/flat)
Returns
the error status
Summary:
• define local variables
• define indices for transfer types
• type indices common to scalars and tensors
• type indices for scalars
• type indices for vectors
• type indices for tensors
• allocate arrays of (k, l) values and transfer functions
• get q values using transfer_get_q_list()
• get k values using transfer_get_k_list()
• get l values using transfer_get_l_list()
• loop over modes (scalar, etc). For each mode:
• allocate arrays of transfer functions, (ptr->transfer[index_md])[index_ic][index_tt][index_l][index_k]
Generated by Doxygen

210 File Documentation
5.21.2.5 transfer_get_l_list()
int transfer_get_l_list (
struct precision ∗ppr,
struct perturbs ∗ppt,
struct transfers ∗ptr )
This routine defines the number and values of multipoles l for all modes.
Parameters
ppr Input: pointer to precision structure
ppt Input: pointer to perturbation structure
ptr Input/Output: pointer to transfers structure containing l's
Returns
the error status
Summary:
• allocate and fill l array
• start from l = 2 and increase with logarithmic step
• when the logarithmic step becomes larger than some linear step, stick to this linear step till l_max
• last value set to exactly l_max
• so far we just counted the number of values. Now repeat the whole thing but fill array with values.
5.21.2.6 transfer_get_q_list()
int transfer_get_q_list (
struct precision ∗ppr,
struct perturbs ∗ppt,
struct transfers ∗ptr,
double q_period,
double K,
int sgnK )
This routine defines the number and values of wavenumbers q for each mode (goes smoothly from logarithmic step
for small q's to linear step for large q's).
Parameters
ppr Input: pointer to precision structure
ppt Input: pointer to perturbation structure
ptr Input/Output: pointer to transfers structure containing q's
q_period Input: order of magnitude of the oscillation period of transfer functions
KInput: spatial curvature (in absolute value)
sgnK Input: spatial curvature sign (open/closed/flat) Generated by Doxygen
5.21 transfer.c File Reference 211
Returns
the error status
5.21.2.7 transfer_get_k_list()
int transfer_get_k_list (
struct perturbs ∗ppt,
struct transfers ∗ptr,
double K)
This routine infers from the q values a list of corresponding k values for each mode.
Parameters
ppt Input: pointer to perturbation structure
ptr Input/Output: pointer to transfers structure containing q's
KInput: spatial curvature
Returns
the error status
5.21.2.8 transfer_get_source_correspondence()
int transfer_get_source_correspondence (
struct perturbs ∗ppt,
struct transfers ∗ptr,
int ∗∗ tp_of_tt )
This routine defines the correspondence between the sources in the perturbation and transfer module.
Parameters
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure containing l's
tp_of←-
_tt
Input/Output: array with the correspondence (allocated before, filled here)
Returns
the error status
Summary:
Generated by Doxygen
212 File Documentation
• running index on modes
• running index on transfer types
• which source are we considering? Define correspondence between transfer types and source types
5.21.2.9 transfer_source_tau_size()
int transfer_source_tau_size (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
double tau_rec,
double tau0,
int index_md,
int index_tt,
int ∗tau_size )
the code makes a distinction between "perturbation sources" (e.g. gravitational potential) and "transfer sources"
(e.g. total density fluctuations, obtained through the Poisson equation, and observed with a given selection function).
This routine computes the number of sampled time values for each type of transfer sources.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
tau_rec Input: recombination time
tau0 Input: time today
index_md Input: index of the mode (scalar, tensor)
index_tt Input: index of transfer type
tau_size Output: pointer to number of sampled times
Returns
the error status
5.21.2.10 transfer_compute_for_each_q()
int transfer_compute_for_each_q (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
Generated by Doxygen

5.21 transfer.c File Reference 213
int ∗∗ tp_of_tt,
int index_q,
int tau_size_max,
double tau_rec,
double ∗∗∗ pert_sources,
double ∗∗∗ pert_sources_spline,
struct transfer_workspace ∗ptw )
Summary:
• define local variables
–we deal with workspaces, i.e. with contiguous memory zones (one per thread) containing various fields
used by the integration routine
• for a given l, maximum value of k such that we can convolve the source with Bessel functions j_l(x) without
reaching x_max
• store the sources in the workspace and define all fields in this workspace
• loop over all modes. For each mode
• loop over initial conditions.
• check if we must now deal with a new source with a new index ppt->index_type. If yes, interpolate it at the
right values of k.
• Select radial function type
5.21.2.11 transfer_interpolate_sources()
int transfer_interpolate_sources (
struct perturbs ∗ppt,
struct transfers ∗ptr,
int index_q,
int index_md,
int index_ic,
int index_type,
double ∗pert_source,
double ∗pert_source_spline,
double ∗interpolated_sources )
This routine interpolates sources S(k, τ)for each mode, initial condition and type (of perturbation module), to get
them at the right values of k, using the spline interpolation method.
Parameters
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
index_q Input: index of wavenumber
index_md Input: index of mode
index_ic Input: index of initial condition
index_type Input: index of type of source (in perturbation module)
pert_source Input: array of sources
pert_source_spline Input: array of second derivative of sources
interpolated_sources Output: array of interpolated sources (filled here but allocated in transfer_init() to avoid
numerous reallocation)
Generated by Doxygen
214 File Documentation
Returns
the error status
Summary:
• define local variables
• interpolate at each k value using the usual spline interpolation algorithm.
5.21.2.12 transfer_sources()
int transfer_sources (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
double ∗interpolated_sources,
double tau_rec,
int index_q,
int index_md,
int index_tt,
double ∗sources,
double ∗tau0_minus_tau,
double ∗w_trapz,
int ∗tau_size_out )
The code makes a distinction between "perturbation sources" (e.g. gravitational potential) and "transfer sources"
(e.g. total density fluctuations, obtained through the Poisson equation, and observed with a given selection function).
This routine computes the transfer source given the interpolated perturbation source, and copies it in the workspace.
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
interpolated_sources Input: interpolated perturbation source
tau_rec Input: recombination time
index_q Input: index of wavenumber
index_md Input: index of mode
index_tt Input: index of type of (transfer) source
sources Output: transfer source
tau0_minus_tau Output: values of (tau0-tau) at which source are sample
w_trapz Output: trapezoidal weights for integration over tau
tau_size_out Output: pointer to size of previous two arrays, converted to double
Generated by Doxygen

5.21 transfer.c File Reference 215
Returns
the error status
Summary:
• define local variables
–in which cases are perturbation and transfer sources are different? I.e., in which case do we need
to multiply the sources by some background and/or window function, and eventually to resample it, or
redefine its time limits?
–case where we need to redefine by a window function (or any function of the background and of k)
• case where we do not need to redefine
–return tau_size value that will be stored in the workspace (the workspace wants a double)
5.21.2.13 transfer_selection_function()
int transfer_selection_function (
struct precision ∗ppr,
struct perturbs ∗ppt,
struct transfers ∗ptr,
int bin,
double z,
double ∗selection )
Arbitrarily normalized selection function dN/dz(z,bin)
Parameters
ppr Input: pointer to precision structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
bin Input: redshift bin number
zInput: one value of redshift
selection Output: pointer to selection function
Returns
the error status
5.21.2.14 transfer_dNdz_analytic()
int transfer_dNdz_analytic (
struct transfers ∗ptr,
Generated by Doxygen

216 File Documentation
double z,
double ∗dNdz,
double ∗dln_dNdz_dz )
Analytic form for dNdz distribution, from arXiv:1004.4640
Parameters
ptr Input: pointer to transfer structure
zInput: redshift
dNdz Output: density per redshift, dN/dZ
dln_dNdz_dz Output: dln(dN/dz)/dz, used optionally for the source evolution
Returns
the error status
5.21.2.15 transfer_selection_sampling()
int transfer_selection_sampling (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
int bin,
double ∗tau0_minus_tau,
int tau_size )
For sources that need to be multiplied by a selection function, redefine a finer time sampling in a small range
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
bin Input: redshift bin number
tau0_minus_tau Output: values of (tau0-tau) at which source are sample
tau_size Output: pointer to size of previous array
Returns
the error status
Generated by Doxygen
5.21 transfer.c File Reference 217
5.21.2.16 transfer_lensing_sampling()
int transfer_lensing_sampling (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
int bin,
double tau0,
double ∗tau0_minus_tau,
int tau_size )
For lensing sources that need to be convolved with a selection function, redefine the sampling within the range
extending from the tau_min of the selection function up to tau0
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
bin Input: redshift bin number
tau0 Input: time today
tau0_minus_tau Output: values of (tau0-tau) at which source are sample
tau_size Output: pointer to size of previous array
Returns
the error status
5.21.2.17 transfer_source_resample()
int transfer_source_resample (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
int bin,
double ∗tau0_minus_tau,
int tau_size,
int index_md,
double tau0,
double ∗interpolated_sources,
double ∗sources )
For sources that need to be multiplied by a selection function, redefine a finer time sampling in a small range, and
resample the perturbation sources at the new value by linear interpolation
Parameters
ppr Input: pointer to precision structure
Generated by Doxygen
218 File Documentation
Parameters
pba Input: pointer to background structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
bin Input: redshift bin number
tau0_minus_tau Output: values of (tau0-tau) at which source are sample
tau_size Output: pointer to size of previous array
index_md Input: index of mode
tau0 Input: time today
interpolated_sources Input: interpolated perturbation source
sources Output: resampled transfer source
Returns
the error status
5.21.2.18 transfer_selection_times()
int transfer_selection_times (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
int bin,
double ∗tau_min,
double ∗tau_mean,
double ∗tau_max )
For each selection function, compute the min, mean and max values of conformal time (associated to the min, mean
and max values of redshift specified by the user)
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
bin Input: redshift bin number
tau_min Output: smallest time in the selection interval
tau_mean Output: time corresponding to z_mean
tau_max Output: largest time in the selection interval
Returns
the error status
Generated by Doxygen
5.21 transfer.c File Reference 219
5.21.2.19 transfer_selection_compute()
int transfer_selection_compute (
struct precision ∗ppr,
struct background ∗pba,
struct perturbs ∗ppt,
struct transfers ∗ptr,
double ∗selection,
double ∗tau0_minus_tau,
double ∗w_trapz,
int tau_size,
double ∗pvecback,
double tau0,
int bin )
Compute and normalize selection function for a set of time values
Parameters
ppr Input: pointer to precision structure
pba Input: pointer to background structure
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
selection Output: normalized selection function
tau0_minus_tau Input: values of (tau0-tau) at which source are sample
w_trapz Input: trapezoidal weights for integration over tau
tau_size Input: size of previous two arrays
pvecback Input: allocated array of background values
tau0 Input: time today
bin Input: redshift bin number
Returns
the error status
5.21.2.20 transfer_compute_for_each_l()
int transfer_compute_for_each_l (
struct transfer_workspace ∗ptw,
struct precision ∗ppr,
struct perturbs ∗ppt,
struct transfers ∗ptr,
int index_q,
int index_md,
int index_ic,
int index_tt,
int index_l,
double l,
double q_max_bessel,
radial_function_type radial_type )
Generated by Doxygen

220 File Documentation
This routine computes the transfer functions ∆X
l(k)) as a function of wavenumber k for a given mode, initial condi-
tion, type and multipole l passed in input.
For a given value of k, the transfer function is inferred from the source function (passed in input in the array
interpolated_sources) and from Bessel functions (passed in input in the bessels structure), either by convolving
them along tau, or by a Limber approximation. This elementary task is distributed either to transfer_integrate() or to
transfer_limber(). The task of this routine is mainly to loop over k values, and to decide at which k_max the calcula-
tion can be stopped, according to some approximation scheme designed to find a compromise between execution
time and precision. The approximation scheme is defined by parameters in the precision structure.
Parameters
ptw Input: pointer to transfer_workspace structure (allocated in transfer_init() to avoid numerous
reallocation)
ppr Input: pointer to precision structure
ppt Input: pointer to perturbation structure
ptr Input/output: pointer to transfers structure (result stored there)
index_q Input: index of wavenumber
index_md Input: index of mode
index_ic Input: index of initial condition
index_tt Input: index of type of transfer
index_l Input: index of multipole
lInput: multipole
q_max_bessel Input: maximum value of argument q at which Bessel functions are computed
radial_type Input: type of radial (Bessel) functions to convolve with
Returns
the error status
Summary:
• define local variables
• return zero transfer function if l is above l_max
• store transfer function in transfer structure
5.21.2.21 transfer_integrate()
int transfer_integrate (
struct perturbs ∗ppt,
struct transfers ∗ptr,
struct transfer_workspace ∗ptw,
int index_q,
int index_md,
int index_tt,
double l,
int index_l,
Generated by Doxygen

5.21 transfer.c File Reference 221
double k,
radial_function_type radial_type,
double ∗trsf )
This routine computes the transfer functions ∆X
l(k)) for each mode, initial condition, type, multipole l and wavenum-
ber k, by convolving the source function (passed in input in the array interpolated_sources) with Bessel functions
(passed in input in the bessels structure).
Generated by Doxygen

222 File Documentation
Parameters
ppt Input: pointer to perturbation structure
ptr Input: pointer to transfers structure
ptw Input: pointer to transfer_workspace structure (allocated in transfer_init() to avoid numerous
reallocation)
index_q Input: index of wavenumber
index_md Input: index of mode
index_tt Input: index of type
lInput: multipole
index_l Input: index of multipole
kInput: wavenumber
radial_type Input: type of radial (Bessel) functions to convolve with
trsf Output: transfer function ∆l(k)
Returns
the error status
Summary:
• define local variables
• find minimum value of (tau0-tau) at which jl(k[τ0−τ]) is known, given that jl(x)is sampled above some
finite value xmin (below which it can be approximated by zero)
• if there is no overlap between the region in which bessels and sources are non-zero, return zero
• if there is an overlap:
• –>trivial case: the source is a Dirac function and is sampled in only one point
• –>other cases
• —>(a) find index in the source's tau list corresponding to the last point in the overlapping region. After this
step, index_tau_max can be as small as zero, but not negative.
• —>(b) the source function can vanish at large τ. Check if further points can be eliminated. After this step
and if we did not return a null transfer function, index_tau_max can be as small as zero, but not negative.
• Compute the radial function:
• Now we do most of the convolution integral:
• This integral is correct for the case where no truncation has occurred. If it has been truncated at some
index_tau_max because f[index_tau_max+1]==0, it is still correct. The 'mistake' in using the wrong weight
w_trapz[index_tau_max] is exactly compensated by the triangle we miss. However, for the Bessel cut off, we
must subtract the wrong triangle and add the correct triangle.
Generated by Doxygen
5.21 transfer.c File Reference 223
5.21.2.22 transfer_limber()
int transfer_limber (
struct transfers ∗ptr,
struct transfer_workspace ∗ptw,
int index_md,
int index_q,
double l,
double q,
radial_function_type radial_type,
double ∗trsf )
This routine computes the transfer functions ∆X
l(k)) for each mode, initial condition, type, multipole l and wavenum-
ber k, by using the Limber approximation, i.e by evaluating the source function (passed in input in the array
interpolated_sources) at a single value of tau (the Bessel function being approximated as a Dirac distribution).
Parameters
ptr Input: pointer to transfers structure
ptw Input: pointer to transfer workspace structure
index_md Input: index of mode
index_q Input: index of wavenumber
lInput: multipole
qInput: wavenumber
radial_type Input: type of radial (Bessel) functions to convolve with
trsf Output: transfer function ∆l(k)
Returns
the error status
Summary:
• define local variables
• get k, l and infer tau such that k(tau0-tau)=l+1/2; check that tau is in appropriate range
• get transfer = source ∗pπ/(2l+ 1)/q = source∗[tau0-tau] ∗pπ/(2l+ 1)/(l+ 1/2)
5.21.2.23 transfer_limber_interpolate()
int transfer_limber_interpolate (
struct transfers ∗ptr,
double ∗tau0_minus_tau,
double ∗sources,
int tau_size,
double tau0_minus_tau_limber,
double ∗S)
Generated by Doxygen
224 File Documentation
• find bracketing indices. index_tau must be at least 1 (so that index_tau-1 is at least 0) and at most tau_size-2
(so that index_tau+1 is at most tau_size-1).
• interpolate by fitting a polynomial of order two; get source and its first two derivatives. Note that we are
not interpolating S, but the product S∗(tau0-tau). Indeed this product is regular in tau=tau0, while S alone
diverges for lensing.
5.21.2.24 transfer_limber2()
int transfer_limber2 (
int tau_size,
struct transfers ∗ptr,
int index_md,
int index_k,
double l,
double k,
double ∗tau0_minus_tau,
double ∗sources,
radial_function_type radial_type,
double ∗trsf )
This routine computes the transfer functions ∆X
l(k)) for each mode, initial condition, type, multipole l and wavenum-
ber k, by using the Limber approximation at order two, i.e as a function of the source function and its first two
derivatives at a single value of tau
Parameters
tau_size Input: size of conformal time array
ptr Input: pointer to transfers structure
index_md Input: index of mode
index_k Input: index of wavenumber
lInput: multipole
kInput: wavenumber
tau0_minus_tau Input: array of values of (tau_today - tau)
sources Input: source functions
radial_type Input: type of radial (Bessel) functions to convolve with
trsf Output: transfer function ∆l(k)
Returns
the error status
Summary:
• define local variables
• get k, l and infer tau such that k(tau0-tau)=l+1/2; check that tau is in appropriate range
• find bracketing indices
• interpolate by fitting a polynomial of order two; get source and its first two derivatives
• get transfer from 2nd order Limber approx (inferred from 0809.5112 [astro-ph])
Generated by Doxygen

5.22 transfer.h File Reference 225
5.22 transfer.h File Reference
#include "nonlinear.h"
#include "hyperspherical.h"
#include <sys/shm.h>
#include <sys/stat.h>
#include "errno.h"
Include dependency graph for transfer.h:
transfer.h
nonlinear.h
hyperspherical.h
sys/shm.h sys/stat.h errno.h
primordial.h
perturbations.h
thermodynamics.h
evolver_ndf15.h
evolver_rkck.h
background.h
common.h
quadrature.h growTable.h arrays.h dei_rkck.hparser.h
stdio.h stdlib.h math.h string.h float.h svnversion.h stdarg.h
sparse.h
Generated by Doxygen
226 File Documentation
This graph shows which files directly or indirectly include this file:
transfer.h
input.h
class.h
spectra.h transfer.c
input.c
class.c
lensing.h spectra.c
output.hlensing.c
output.c
Data Structures
• struct transfers
• struct transfer_workspace
Enumerations
• enum radial_function_type
5.22.1 Detailed Description
Documented includes for transfer module.
5.22.2 Data Structure Documentation
5.22.2.1 struct transfers
Structure containing everything about transfer functions in harmonic space ∆X
l(q)that other modules need to know.
Once initialized by transfer_init(), contains all tables of transfer functions used for interpolation in other modules, for
all requested modes (scalar/vector/tensor), initial conditions, types (temperature, polarization, etc), multipoles l, and
wavenumbers q.
Generated by Doxygen

5.22 transfer.h File Reference 227
Wavenumbers are called q in this module and k in the perturbation module. In flat universes k=q. In non-flat
universes q and k differ through q2 = k2 + K(1+m), where m=0,1,2 for scalar, vector, tensor. q should be used
throughout the transfer module, except when interpolating or manipulating the source functions S(k,tau) calculated
in the perturbation module: for a given value of q, this should be done at the corresponding k(q).
The content of this structure is entirely computed in this module, given the content of the 'precision', 'bessels',
'background', 'thermodynamics' and 'perturbation' structures.
Generated by Doxygen

228 File Documentation
Data Fields
double lcmb_rescale normally set to one, can be used exceptionally
to rescale by hand the CMB lensing potential
double lcmb_tilt normally set to zero, can be used exceptionally
to tilt by hand the CMB lensing potential
double lcmb_pivot if lcmb_tilt non-zero, corresponding pivot scale
double selection_bias[_SELECTION_NUM_MAX_] light-to-mass bias in the transfer function of
density number count
double selection_magnification_bias[_SELECTION_NUM_MAX_]magnification bias in the transfer function of
density number count
short has_nz_file Has dN/dz (selection function) input file?
short has_nz_analytic Use analytic form for dN/dz (selection function)
distribution?
FileName nz_file_name dN/dz (selection function) input file name
int nz_size number of redshift values in input tabulated
selection function
double ∗nz_z redshift values in input tabulated selection
function
double ∗nz_nz input tabulated values of selection function
double ∗nz_ddnz second derivatives in splined selection function
short has_nz_evo_file Has dN/dz (evolution function) input file?
short has_nz_evo_analytic Use analytic form for dN/dz (evolution function)
distribution?
FileName nz_evo_file_name dN/dz (evolution function) input file name
int nz_evo_size number of redshift values in input tabulated
evolution function
double ∗nz_evo_z redshift values in input tabulated evolution
function
double ∗nz_evo_nz input tabulated values of evolution function
double ∗nz_evo_dlog_nz log of tabulated values of evolution function
double ∗nz_evo_dd_dlog_nz second derivatives in splined log of evolution
function
short has_cls copy of same flag in perturbation structure
int md_size number of modes included in computation
int index_tt_t0 index for transfer type = temperature (j=0 term)
int index_tt_t1 index for transfer type = temperature (j=1 term)
int index_tt_t2 index for transfer type = temperature (j=2 term)
int index_tt_e index for transfer type = E-polarization
int index_tt_b index for transfer type = B-polarization
int index_tt_lcmb index for transfer type = CMB lensing
int index_tt_density index for first bin of transfer type = matter
density
int index_tt_lensing index for first bin of transfer type = galaxy
lensing
int index_tt_rsd index for first bin of transfer type = redshift
space distortion of number count
int index_tt_d0 index for first bin of transfer type = doppler
effect for of number count (j=0 term)
int index_tt_d1 index for first bin of transfer type = doppler
effect for of number count (j=1 term)
Generated by Doxygen

5.22 transfer.h File Reference 229
Data Fields
int index_tt_nc_lens index for first bin of transfer type = lensing for
of number count
int index_tt_nc_g1 index for first bin of transfer type = gravity term
G1 for of number count
int index_tt_nc_g2 index for first bin of transfer type = gravity term
G2 for of number count
int index_tt_nc_g3 index for first bin of transfer type = gravity term
G3 for of number count
int index_tt_nc_g4 index for first bin of transfer type = gravity term
G3 for of number count
int index_tt_nc_g5 index for first bin of transfer type = gravity term
G3 for of number count
int ∗tt_size number of requested transfer types
tt_size[index_md] for each mode
int ∗∗ l_size_tt number of multipole values for which we
effectively compute the transfer
function,l_size_tt[index_md][index_tt]
int ∗l_size number of multipole values for each requested
mode, l_size[index_md]
int l_size_max greatest of all l_size[index_md]
int ∗l list of multipole values l[index_l]
double angular_rescaling correction between l and k space due to
curvature (= comoving angular diameter
distance to recombination / comoving radius to
recombination)
size_t q_size number of wavenumber values
double ∗q list of wavenumber values, q[index_q]
double ∗∗ k list of wavenumber values for each requested
mode, k[index_md][index_q]. In flat universes
k=q. In non-flat universes q and k differ through
q2 = k2 + K(1+m), where m=0,1,2 for scalar,
vector, tensor. q should be used throughout the
transfer module, excepted when interpolating
or manipulating the source functions S(k,tau):
for a given value of q this should be done in
k(q).
int index_q_flat_approximation index of the first q value using the flat rescaling
approximation
double ∗∗ transfer table of transfer functions for each mode, initial
condition, type, multipole and wavenumber,
with argument transfer[index_md][((index_ic ∗
ptr->tt_size[index_md] + index_tt) ∗
ptr->l_size[index_md] + index_l) ∗ptr->q_size
+ index_q]
short initialise_HIS_cache only true if we are using CLASS for setting up a
cache of HIS structures
short transfer_verbose flag regulating the amount of information sent
to standard output (none if set to zero)
ErrorMsg error_message zone for writing error messages
Generated by Doxygen

230 File Documentation
5.22.2.2 struct transfer_workspace
Structure containing all the quantities that each thread needs to know for computing transfer functions (but that can
be forgotten once the transfer functions are known, otherwise they would be stored in the transfer module)
Data Fields
HyperInterpStruct HIS structure containing all hyperspherical bessel functions (flat
case) or all hyperspherical bessel functions for a given value of
beta=q/sqrt(|K|) (non-flat case). HIS = Hyperspherical
Interpolation Structure.
int HIS_allocated flag specifying whether the previous structure has been
allocated
HyperInterpStruct ∗pBIS pointer to structure containing all the spherical bessel functions
of the flat case (used even in the non-flat case, for
approximation schemes). pBIS = pointer to Bessel Interpolation
Structure.
int l_size number of l values
int tau_size number of discrete time values for a given type
int tau_size_max maximum number of discrete time values for all types
double ∗interpolated_sources interpolated_sources[index_tau]: sources interpolated from the
perturbation module at the right value of k
double ∗sources sources[index_tau]: sources used in transfer module, possibly
differing from those in the perturbation module by some
resampling or rescaling
double ∗tau0_minus_tau tau0_minus_tau[index_tau]: values of (tau0 - tau)
double ∗w_trapz w_trapz[index_tau]: values of weights in trapezoidal integration
(related to time steps)
double ∗chi chi[index_tau]: value of argument of bessel function:
k(tau0-tau) (flat case) or sqrt(|K|)(tau0-tau) (non-flat case)
double ∗cscKgen cscKgen[index_tau]: useful trigonometric function
double ∗cotKgen cotKgen[index_tau]: useful trigonometric function
double K curvature parameter (see background module for details)
int sgnK 0 (flat), 1 (positive curvature, spherical, closed), -1 (negative
curvature, hyperbolic, open)
double tau0_minus_tau_cut critical value of (tau0-tau) in time cut approximation for the
wavenumber at hand
short neglect_late_source flag stating whether we use the time cut approximation for the
wavenumber at hand
5.22.3 Enumeration Type Documentation
5.22.3.1 radial_function_type
enum radial_function_type
enumeration of possible source types. This looks redundant with respect to the definition of indices index_tt_... This
definition is however convenient and time-saving: it allows to use a "case" statement in transfer_radial_function()
Generated by Doxygen
Chapter 6
The ‘external_Pk‘ mode
• Author: Jesus Torrado (torradocacho [@] lorentz.leidenuniv.nl)
• Date: 2013-12-20
Introduction
This mode allows for an arbitrary primordial spectrum P(k) to be calculated by an external command and passed
to CLASS. That external command may be anything that can be run in the shell: a python script, some compiled
C or Fortran code... This command is executed from within CLASS, and CLASS is able to pass it a number of
parameters defining the spectrum (an amplitude, a tilt...). Those parameters can be used in a Markov chain search
performed by MontePython.
This mode includes the simple case of a precomputed primordial spectrum stored in a text file. In that case, the
cat shell command will do the trick (see below).
Currently, scalar and tensor spectra of perturbations of adiabatic modes are supported.
Use case #1: reading the spectrum from a table
In this case, say the file with the table is called spectrum.txt, located under /path/to, simply include in the
.ini file
command = cat path/to/spectrum.txt
It is necessary that 1st 4 characters are exactly cat.

232 The ‘external_Pk‘ mode
Use case #2: getting the spectrum from an external command
Here an external command is called to generate the spectrum; it may be some compiled C or Fortran code, a python
script... This command may be passed up to 10 floating point arguments, named custom1 to custom10, which
are assigned values inside the .ini file of CLASS. The command parameter would look like
command = /path/to/example.py
if it starts with #/usr/bin/python, otherwise
command = python /path/to/example.py
As an example of the 1st use case, one may use the included script generate_Pk_example.py, which imple-
ments a single-field slow-roll spectrum without running, and takes 3 arguments:
•custom1 – the pivot scale (k_0 = 0.05 1/Mpc for Planck).
•custom2 – the amplitude of the scalar power spectrum.
•custom3 – the scalar spectral index.
In order to use it, the following lines must be present in the parameter file:
P_k_ini type = external_Pk
command = /path/to/CLASS/external_Pk/generate_Pk_example.py
custom1 = 0.05
custom2 = 2.2e-9
custom3 = 1.
Defined or not (in that case, 0-valued), parameters from custom4 to custom10 will be passed to the example
script, which should ignore them. In this case, CLASS will run in the shell the command
/path/to/CLASS/external_Pk/generate_Pk_example.py 0.05 2.2e-9 1. 0 0 0 0 0 0 0
If CLASS fails to run the command, try to do it directly yourself by hand, using exactly the same string that was
given in command.
Output of the command / format of the table
The command must generate an output separated into lines, each containing a tuple (k,P(k)). The following
requirements must be fulfilled:
• Each line must contain 2 (3, if tensors) floating point numbers: k(in 1/Mpc units) and P_s(k) (and P←-
_t(k), if tensors), separated by any number of spaces or tabs. The numbers can be in scientific notation,
e.g. 1.4e-3.
• The lines must be sorted in increasing values of k.
• There must be at least two points (k, P(k)) before and after the interval of krequested by CLASS, in
order not to introduce unnecessary interpolation error. Otherwise, an error will be raised. In most of the
cases, generating the spectrum between 1e-6 and 1 1/Mpc should be more than enough.
Generated by Doxygen

233
Precision
This implementation properly handles double-precision floating point numbers (i.e. about 17 significant figures),
both for the input parameters of the command and for the output of the command (or the table).
The sampling of kgiven by the command (or table) is preserved to be used internally by CLASS. It must be fine
enough a sampling to clearly show the features of the spectrum. The best way to test this is to plot the output/table
and check it with the naked eye.
Another thing to have in mind arises at the time of convolving with the transfer functions. Two precision parameters
are implied: the sampling of kin the integral, given by k_step_trans, and the sampling of the transfer functions
in l, given by l_logstep and l_linstep. In general, it will be enough to reduce the values of the first and
the third parameters. A good start is to give them rather small values, say k_step_trans=0.01 and l_←-
linstep=1, and to increase them slowly until the point at which the effect of increasing them gets noticeable.
Parameter fit with MontePython
(MontePython)[http://montepython.net/] is able to interact with the external_Pk mode transparently,
using the custom parameters in an MCMC fit. One must just add the appropriate lines to the input file of Monte←-
Python. For our example, if we wanted to fit the amplitude and spectral index of the primordial spectrum, it would
be:
data.cosmo_arguments[’P_k_ini type’] = ’external_Pk’
data.cosmo_arguments[’command’] = ’/path/to/CLASS/external_Pk/generate_Pk_example.py’
data.cosmo_arguments[’custom1’] = 0.05 # k_pivot
data.parameters[’custom2’] = [ 2.2, 0, -1, 0.055, 1.e-9, ’cosmo’] # A_s
data.parameters[’custom3’] = [ 1., 0, -1, 0.0074, 1, ’cosmo’] # n_s
Notice that since in our case custom1 represents the pivot scale, it is passed as a (non-varying) argument, instead
of as a (varying) parameter.
In this case, one would not include the corresponding lines for the primordial parameters of CLASS: k_pivot,
A_s,n_s,alpha_s, etc. They would simply be ignored.
Limitations
• So far, this mode cannot handle vector perturbations, nor isocurvature initial conditions.
• The external script knows nothing about the rest of the CLASS parameters, so if it needs, e.g., k_pivot, it
should be either hard coded, or its value passed as one of the custom parameters.
Generated by Doxygen

234 The ‘external_Pk‘ mode
Generated by Doxygen
Chapter 7
Updating the manual
Author: D. C. Hooper (hooper@physik.rwth-aachen.de)
This pdf manual and accompanying web version have been generated using the doxygen software (http←-
://www.doxygen.org). This software directly reads the code and extracts the necessary comments to form
the manual, meaning it is very easy to generate newer versions of the manual as desired.
For CLASS developpers:
To maintain the usefulness of the manual, a new version should be generated after any major upgrade to CLASS.
To keep track of how up-to-date the manual is the title page also displays the last modification date. The manual is
generated automatically from the code, excepted a few chapters written manually in the files
README.md
doc/input/chap2.md
doc/input/chap3.md
doc/input/mod.md
external_Pk/README.md
You can update these files, or add new ones that should be declared in the INPUT= field of doc/input/doxyconf.
Generating a new version of this manual is straightforward. First, you need to install the doxygen software, which
can be done by following the instructions on the software's webpage. The location where you install this software
is irrelevant; it doesn't need to be in the same folder as CLASS. For Mac OSX, homebrew users can install the
software with brew install doxygen --with-graphviz.
Once installed, navigate to the class/doc/input directory and run the first script
. make1.sh
This will generate a new version of the html manual and the necessary files to make the pdf version. Unfortunately,
doxygen does not yet offer the option to automatically order the output chapters in the pdf version of the manual.
Hence, before compiling the pdf, this must be done manually. To do this you need to find the refman.tex file in
class/doc/manual/latex. With this file you can modify the title page, headers, footers, and chapter ordering for the
final pdf. Usually we just make two things: add manually the line
\vspace*{1cm}
{\large Last updated \today}\\
after

236 Updating the manual
{\Large C\+L\+A\+SS M\+A\+N\+U\+AL }\\
and move manually the chapters "The external Pk mode" and "Updating the manual" to the end,
after the automatically generated part. Once you have this file with your desired configuration, navigate back to the
class/doc/input directory, and run the second script
. make2.sh
You should now be able to find the finished pdf in class/doc/manual/CLASS_MANUAL.pdf. Finally you
can commit the changes to git, but not all the content of doc/ is necessary: only doc/README,doc/input/,
doc/manual/CLASS_MANUAL.pdf,doc/manual/html/. This means that before committing you will have
to do a: git add doc/manual/html/, but NOT a: git add doc/manual/latex/!
As a final comment, doxygen uses two main configuration files: doxyconf and doxygen.sty, both located in
class/doc/input. Changes to these files can dramatically impact the outcome, so any modifications to these files
should be done with great care.
Generated by Doxygen
Index
_MAX_NUMBER_OF_K_FILES_
perturbations.h, 132
_M_EV_TOO_BIG_FOR_HALOFIT_
nonlinear.h, 83
_SELECTION_NUM_MAX_
perturbations.h, 132
_YHE_BIG_
thermodynamics.h, 203
_YHE_SMALL_
thermodynamics.h, 203
_Z_PK_NUM_MAX_
output.h, 92
background, 38
background.c, 19
background_at_tau, 21
background_derivs, 34
background_find_equality, 33
background_free, 26
background_free_input, 27
background_free_noinput, 26
background_functions, 22
background_indices, 27
background_init, 25
background_initial_conditions, 32
background_ncdm_M_from_Omega, 30
background_ncdm_distribution, 28
background_ncdm_init, 29
background_ncdm_momenta, 29
background_ncdm_test_function, 28
background_output_data, 33
background_output_titles, 33
background_solve, 31
background_tau_of_z, 22
background_w_fld, 23
V_e_scf, 35
V_p_scf, 35
V_scf, 36
background.h, 37
equation_of_state, 42
spatial_curvature, 42
background_at_tau
background.c, 21
background_derivs
background.c, 34
background_find_equality
background.c, 33
background_free
background.c, 26
background_free_input
background.c, 27
background_free_noinput
background.c, 26
background_functions
background.c, 22
background_indices
background.c, 27
background_init
background.c, 25
background_initial_conditions
background.c, 32
background_ncdm_M_from_Omega
background.c, 30
background_ncdm_distribution
background.c, 28
background_ncdm_init
background.c, 29
background_ncdm_momenta
background.c, 29
background_ncdm_test_function
background.c, 28
background_output_data
background.c, 33
background_output_titles
background.c, 33
background_parameters_and_workspace, 42
background_parameters_for_distributions, 42
background_solve
background.c, 31
background_tau_of_z
background.c, 22
background_w_fld
background.c, 23
class.c, 43
class_fzero_ridder
input.c, 61
common.h, 43
evolver_type, 53
file_format, 54
pk_def, 53
equation_of_state
background.h, 42
error_message
nonlinear, 18
evolver_type
common.h, 53
f1

238 INDEX
thermodynamics.h, 202
f2
thermodynamics.h, 203
file_format
common.h, 54
get_machine_precision
input.c, 61
has_pk_eq
nonlinear, 17
index_pk_eq_Omega_m
nonlinear, 17
index_pk_eq_w
nonlinear, 17
index_tau_min_nl
nonlinear, 17
inflation_module_behavior
primordial.h, 157
input.c, 54
class_fzero_ridder, 61
get_machine_precision, 61
input_default_params, 59
input_default_precision, 60
input_find_root, 62
input_get_guess, 62
input_init, 56
input_init_from_arguments, 55
input_prepare_pk_eq, 62
input_read_parameters, 57
input_try_unknown_parameters, 61
input.h, 63
target_names, 65
input_default_params
input.c, 59
input_default_precision
input.c, 60
input_find_root
input.c, 62
input_get_guess
input.c, 62
input_init
input.c, 56
input_init_from_arguments
input.c, 55
input_prepare_pk_eq
input.c, 62
input_read_parameters
input.c, 57
input_try_unknown_parameters
input.c, 61
integration_direction
primordial.h, 157
k
nonlinear, 16
k_nl
nonlinear, 17,18
k_size
nonlinear, 16
lensing, 79
lensing.c, 66
lensing_addback_cl_ee_bb, 72
lensing_addback_cl_te, 71
lensing_addback_cl_tt, 70
lensing_cl_at_l, 67
lensing_d00, 72
lensing_d11, 73
lensing_d1m1, 73
lensing_d20, 74
lensing_d22, 74
lensing_d2m2, 74
lensing_d31, 75
lensing_d3m1, 75
lensing_d3m3, 76
lensing_d40, 76
lensing_d4m2, 76
lensing_d4m4, 77
lensing_free, 69
lensing_indices, 69
lensing_init, 68
lensing_lensed_cl_ee_bb, 71
lensing_lensed_cl_te, 70
lensing_lensed_cl_tt, 70
lensing.h, 77
lensing_addback_cl_ee_bb
lensing.c, 72
lensing_addback_cl_te
lensing.c, 71
lensing_addback_cl_tt
lensing.c, 70
lensing_cl_at_l
lensing.c, 67
lensing_d00
lensing.c, 72
lensing_d11
lensing.c, 73
lensing_d1m1
lensing.c, 73
lensing_d20
lensing.c, 74
lensing_d22
lensing.c, 74
lensing_d2m2
lensing.c, 74
lensing_d31
lensing.c, 75
lensing_d3m1
lensing.c, 75
lensing_d3m3
lensing.c, 76
lensing_d40
lensing.c, 76
lensing_d4m2
lensing.c, 76
lensing_d4m4
Generated by Doxygen

INDEX 239
lensing.c, 77
lensing_free
lensing.c, 69
lensing_indices
lensing.c, 69
lensing_init
lensing.c, 68
lensing_lensed_cl_ee_bb
lensing.c, 71
lensing_lensed_cl_te
lensing.c, 70
lensing_lensed_cl_tt
lensing.c, 70
linear_or_logarithmic
primordial.h, 156
method
nonlinear, 16
nl_corr_density
nonlinear, 17,18
nonlinear, 15
error_message, 18
has_pk_eq, 17
index_pk_eq_Omega_m, 17
index_pk_eq_w, 17
index_tau_min_nl, 17
k, 16
k_nl, 17,18
k_size, 16
method, 16
nl_corr_density, 17,18
nonlinear_verbose, 18
pk_eq_ddw_and_ddOmega, 18
pk_eq_size, 17
pk_eq_tau, 18
pk_eq_tau_size, 18
pk_eq_w_and_Omega, 18
pk_size, 16
tau, 16
tau_size, 16
nonlinear.c, 80
nonlinear_halofit, 81
nonlinear_init, 81
nonlinear.h, 82
_M_EV_TOO_BIG_FOR_HALOFIT_, 83
nonlinear_halofit
nonlinear.c, 81
nonlinear_init
nonlinear.c, 81
nonlinear_verbose
nonlinear, 18
output, 92
output.c, 83
output_cl, 85
output_init, 84
output_one_line_of_cl, 88
output_one_line_of_pk, 90
output_open_cl_file, 88
output_open_pk_file, 89
output_pk, 86
output_pk_nl, 86
output_print_data, 87
output_tk, 87
output.h, 90
_Z_PK_NUM_MAX_, 92
output_cl
output.c, 85
output_init
output.c, 84
output_one_line_of_cl
output.c, 88
output_one_line_of_pk
output.c, 90
output_open_cl_file
output.c, 88
output_open_pk_file
output.c, 89
output_pk
output.c, 86
output_pk_nl
output.c, 86
output_print_data
output.c, 87
output_tk
output.c, 87
perturb_approximations
perturbations.c, 110
perturb_derivs
perturbations.c, 116
perturb_einstein
perturbations.c, 113
perturb_find_approximation_number
perturbations.c, 102
perturb_find_approximation_switches
perturbations.c, 103
perturb_free
perturbations.c, 96
perturb_get_k_list
perturbations.c, 98
perturb_indices_of_perturbs
perturbations.c, 96
perturb_init
perturbations.c, 95
perturb_initial_conditions
perturbations.c, 107
perturb_parameters_and_workspace, 131
perturb_prepare_output
perturbations.c, 102
perturb_print_variables
perturbations.c, 115
perturb_solve
perturbations.c, 101
perturb_sources
perturbations.c, 114
perturb_sources_at_tau
Generated by Doxygen

240 INDEX
perturbations.c, 94
perturb_tca_slip_and_shear
perturbations.c, 120
perturb_timesampling_for_sources
perturbations.c, 97
perturb_timescale
perturbations.c, 112
perturb_total_stress_energy
perturbations.c, 114
perturb_vector, 128
perturb_vector_free
perturbations.c, 106
perturb_vector_init
perturbations.c, 104
perturb_workspace, 130
perturb_workspace_free
perturbations.c, 100
perturb_workspace_init
perturbations.c, 99
perturbations.c, 93
perturb_approximations, 110
perturb_derivs, 116
perturb_einstein, 113
perturb_find_approximation_number, 102
perturb_find_approximation_switches, 103
perturb_free, 96
perturb_get_k_list, 98
perturb_indices_of_perturbs, 96
perturb_init, 95
perturb_initial_conditions, 107
perturb_prepare_output, 102
perturb_print_variables, 115
perturb_solve, 101
perturb_sources, 114
perturb_sources_at_tau, 94
perturb_tca_slip_and_shear, 120
perturb_timesampling_for_sources, 97
perturb_timescale, 112
perturb_total_stress_energy, 114
perturb_vector_free, 106
perturb_vector_init, 104
perturb_workspace_free, 100
perturb_workspace_init, 99
perturbations.h, 121
_MAX_NUMBER_OF_K_FILES_, 132
_SELECTION_NUM_MAX_, 132
possible_gauges, 133
tca_flags, 132
tca_method, 132
perturbs, 123
phi_pivot_methods
primordial.h, 157
pk_def
common.h, 53
pk_eq_ddw_and_ddOmega
nonlinear, 18
pk_eq_size
nonlinear, 17
pk_eq_tau
nonlinear, 18
pk_eq_tau_size
nonlinear, 18
pk_eq_w_and_Omega
nonlinear, 18
pk_size
nonlinear, 16
possible_gauges
perturbations.h, 133
potential_shape
primordial.h, 156
precision, 44
primordial, 152
primordial.c, 133
primordial_analytic_spectrum, 138
primordial_analytic_spectrum_init, 138
primordial_external_spectrum_init, 150
primordial_free, 136
primordial_get_lnk_list, 137
primordial_indices, 137
primordial_inflation_analytic_spectra, 141
primordial_inflation_check_hubble, 146
primordial_inflation_check_potential, 145
primordial_inflation_derivs, 149
primordial_inflation_evolve_background, 144
primordial_inflation_find_attractor, 144
primordial_inflation_find_phi_pivot, 148
primordial_inflation_get_epsilon, 146
primordial_inflation_hubble, 139
primordial_inflation_indices, 140
primordial_inflation_one_k, 143
primordial_inflation_one_wavenumber, 142
primordial_inflation_potential, 139
primordial_inflation_solve_inflation, 140
primordial_inflation_spectra, 141
primordial_init, 136
primordial_spectrum_at_k, 135
primordial.h, 150
inflation_module_behavior, 157
integration_direction, 157
linear_or_logarithmic, 156
phi_pivot_methods, 157
potential_shape, 156
primordial_spectrum_type, 156
target_quantity, 156
time_definition, 157
primordial_analytic_spectrum
primordial.c, 138
primordial_analytic_spectrum_init
primordial.c, 138
primordial_external_spectrum_init
primordial.c, 150
primordial_free
primordial.c, 136
primordial_get_lnk_list
primordial.c, 137
primordial_indices
Generated by Doxygen

INDEX 241
primordial.c, 137
primordial_inflation_analytic_spectra
primordial.c, 141
primordial_inflation_check_hubble
primordial.c, 146
primordial_inflation_check_potential
primordial.c, 145
primordial_inflation_derivs
primordial.c, 149
primordial_inflation_evolve_background
primordial.c, 144
primordial_inflation_find_attractor
primordial.c, 144
primordial_inflation_find_phi_pivot
primordial.c, 148
primordial_inflation_get_epsilon
primordial.c, 146
primordial_inflation_hubble
primordial.c, 139
primordial_inflation_indices
primordial.c, 140
primordial_inflation_one_k
primordial.c, 143
primordial_inflation_one_wavenumber
primordial.c, 142
primordial_inflation_potential
primordial.c, 139
primordial_inflation_solve_inflation
primordial.c, 140
primordial_inflation_spectra
primordial.c, 141
primordial_init
primordial.c, 136
primordial_spectrum_at_k
primordial.c, 135
primordial_spectrum_type
primordial.h, 156
radial_function_type
transfer.h, 230
recombination, 200
recombination_algorithm
thermodynamics.h, 203
reionization, 201
reionization_parametrization
thermodynamics.h, 203
reionization_z_or_tau
thermodynamics.h, 204
spatial_curvature
background.h, 42
spectra, 176
spectra.c, 158
spectra_cl_at_l, 159
spectra_cls, 169
spectra_compute_cl, 169
spectra_fast_pk_at_kvec_and_zvec, 173
spectra_free, 168
spectra_indices, 168
spectra_init, 167
spectra_k_and_tau, 170
spectra_matter_transfers, 172
spectra_output_tk_data, 173
spectra_pk, 171
spectra_pk_at_k_and_z, 162
spectra_pk_at_z, 160
spectra_pk_nl_at_k_and_z, 164
spectra_pk_nl_at_z, 163
spectra_sigma, 172
spectra_tk_at_k_and_z, 166
spectra_tk_at_z, 165
spectra.h, 175
spectra_cl_at_l
spectra.c, 159
spectra_cls
spectra.c, 169
spectra_compute_cl
spectra.c, 169
spectra_fast_pk_at_kvec_and_zvec
spectra.c, 173
spectra_free
spectra.c, 168
spectra_indices
spectra.c, 168
spectra_init
spectra.c, 167
spectra_k_and_tau
spectra.c, 170
spectra_matter_transfers
spectra.c, 172
spectra_output_tk_data
spectra.c, 173
spectra_pk
spectra.c, 171
spectra_pk_at_k_and_z
spectra.c, 162
spectra_pk_at_z
spectra.c, 160
spectra_pk_nl_at_k_and_z
spectra.c, 164
spectra_pk_nl_at_z
spectra.c, 163
spectra_sigma
spectra.c, 172
spectra_tk_at_k_and_z
spectra.c, 166
spectra_tk_at_z
spectra.c, 165
target_names
input.h, 65
target_quantity
primordial.h, 156
tau
nonlinear, 16
tau_size
nonlinear, 16
tca_flags
Generated by Doxygen

242 INDEX
perturbations.h, 132
tca_method
perturbations.h, 132
thermo, 197
thermodynamics.c, 181
thermodynamics_at_z, 183
thermodynamics_derivs_with_recfast, 194
thermodynamics_energy_injection, 188
thermodynamics_free, 185
thermodynamics_get_xe_before_reionization, 189
thermodynamics_helium_from_bbn, 186
thermodynamics_indices, 186
thermodynamics_init, 184
thermodynamics_merge_reco_and_reio, 195
thermodynamics_onthespot_energy_injection, 187
thermodynamics_output_titles, 195
thermodynamics_recombination, 191
thermodynamics_recombination_with_hyrec, 192
thermodynamics_recombination_with_recfast, 192
thermodynamics_reionization, 189
thermodynamics_reionization_function, 188
thermodynamics_reionization_sample, 190
thermodynamics.h, 196
_YHE_BIG_, 203
_YHE_SMALL_, 203
f1, 202
f2, 203
recombination_algorithm, 203
reionization_parametrization, 203
reionization_z_or_tau, 204
thermodynamics_at_z
thermodynamics.c, 183
thermodynamics_derivs_with_recfast
thermodynamics.c, 194
thermodynamics_energy_injection
thermodynamics.c, 188
thermodynamics_free
thermodynamics.c, 185
thermodynamics_get_xe_before_reionization
thermodynamics.c, 189
thermodynamics_helium_from_bbn
thermodynamics.c, 186
thermodynamics_indices
thermodynamics.c, 186
thermodynamics_init
thermodynamics.c, 184
thermodynamics_merge_reco_and_reio
thermodynamics.c, 195
thermodynamics_onthespot_energy_injection
thermodynamics.c, 187
thermodynamics_output_titles
thermodynamics.c, 195
thermodynamics_parameters_and_workspace, 202
thermodynamics_recombination
thermodynamics.c, 191
thermodynamics_recombination_with_hyrec
thermodynamics.c, 192
thermodynamics_recombination_with_recfast
thermodynamics.c, 192
thermodynamics_reionization
thermodynamics.c, 189
thermodynamics_reionization_function
thermodynamics.c, 188
thermodynamics_reionization_sample
thermodynamics.c, 190
time_definition
primordial.h, 157
transfer.c, 204
transfer_compute_for_each_l, 219
transfer_compute_for_each_q, 212
transfer_dNdz_analytic, 215
transfer_free, 208
transfer_functions_at_q, 206
transfer_get_k_list, 211
transfer_get_l_list, 209
transfer_get_q_list, 210
transfer_get_source_correspondence, 211
transfer_indices_of_transfers, 208
transfer_init, 207
transfer_integrate, 220
transfer_interpolate_sources, 213
transfer_lensing_sampling, 216
transfer_limber, 222
transfer_limber2, 224
transfer_limber_interpolate, 223
transfer_selection_compute, 218
transfer_selection_function, 215
transfer_selection_sampling, 216
transfer_selection_times, 218
transfer_source_resample, 217
transfer_source_tau_size, 212
transfer_sources, 214
transfer.h, 225
radial_function_type, 230
transfer_compute_for_each_l
transfer.c, 219
transfer_compute_for_each_q
transfer.c, 212
transfer_dNdz_analytic
transfer.c, 215
transfer_free
transfer.c, 208
transfer_functions_at_q
transfer.c, 206
transfer_get_k_list
transfer.c, 211
transfer_get_l_list
transfer.c, 209
transfer_get_q_list
transfer.c, 210
transfer_get_source_correspondence
transfer.c, 211
transfer_indices_of_transfers
transfer.c, 208
transfer_init
transfer.c, 207
Generated by Doxygen

INDEX 243
transfer_integrate
transfer.c, 220
transfer_interpolate_sources
transfer.c, 213
transfer_lensing_sampling
transfer.c, 216
transfer_limber
transfer.c, 222
transfer_limber2
transfer.c, 224
transfer_limber_interpolate
transfer.c, 223
transfer_selection_compute
transfer.c, 218
transfer_selection_function
transfer.c, 215
transfer_selection_sampling
transfer.c, 216
transfer_selection_times
transfer.c, 218
transfer_source_resample
transfer.c, 217
transfer_source_tau_size
transfer.c, 212
transfer_sources
transfer.c, 214
transfer_workspace, 229
transfers, 226
V_e_scf
background.c, 35
V_p_scf
background.c, 35
V_scf
background.c, 36
Generated by Doxygen
