DIA NN GUI Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 6

DIA-NN GUI manual
1
version 13/02/2019
Contents
Introduction ....................................................................................................................................................... 2
GUI window ...................................................................................................................................................... 2
Files ................................................................................................................................................................... 3
Main Settings ..................................................................................................................................................... 3
Spectral library .............................................................................................................................................. 3
FASTA .......................................................................................................................................................... 4
Library-free search ............................................................................................................................................ 4
Use library-free search / generate spectral library......................................................................................... 4
Training library ............................................................................................................................................. 4
Protease ......................................................................................................................................................... 5
Modifications ................................................................................................................................................ 5
Precursor ion mass range ............................................................................................................................... 5
Fragment ion mass range ............................................................................................................................... 5
Optimise for training ..................................................................................................................................... 5
Algorithm .......................................................................................................................................................... 5
Output ................................................................................................................................................................ 6
Protein.Group ................................................................................................................................................ 6
Protein.Names and Genes ............................................................................................................................. 6
Protein.Q.Value ............................................................................................................................................. 6
Quantities ...................................................................................................................................................... 6
Extra information .......................................................................................................................................... 6
Quality control ............................................................................................................................................... 6
Pipelines ............................................................................................................................................................ 6
1
Please email any comments, suggestions, questions or feedback to Vadim Demichev (vadim.demichev[аt]gmail.com) or create a
new issue on GitHub (github.com/vdemichev/diann).
Introduction
DIA-NN is a fast and easy to use tool for processing data-independent acquisition (DIA or SWATH) proteomics
data. DIA-NN is designed to do as much as possible automatically, eliminating the need to optimise the
processing parameters for each experiment.
DIA-NN distribution contains DiaNN.exe, which is a command line tool, and DIA-NN.exe, a GUI wrapper for
this tool. The GUI works by launching DiaNN.exe and displaying its output. A report (with precursor ion and
protein identification and quantification information) is generated by the command line tool. Optionally, a
spectral library can be generated. Any analysis that can be performed with the GUI can also be performed with
the command line tool and vice versa. Using the GUI is recommended for any workflow that does not involve
automated processing of several experiments. Here we provide detailed information on data formats supported
by DIA-NN and consider the ways to fine-tune its performance.
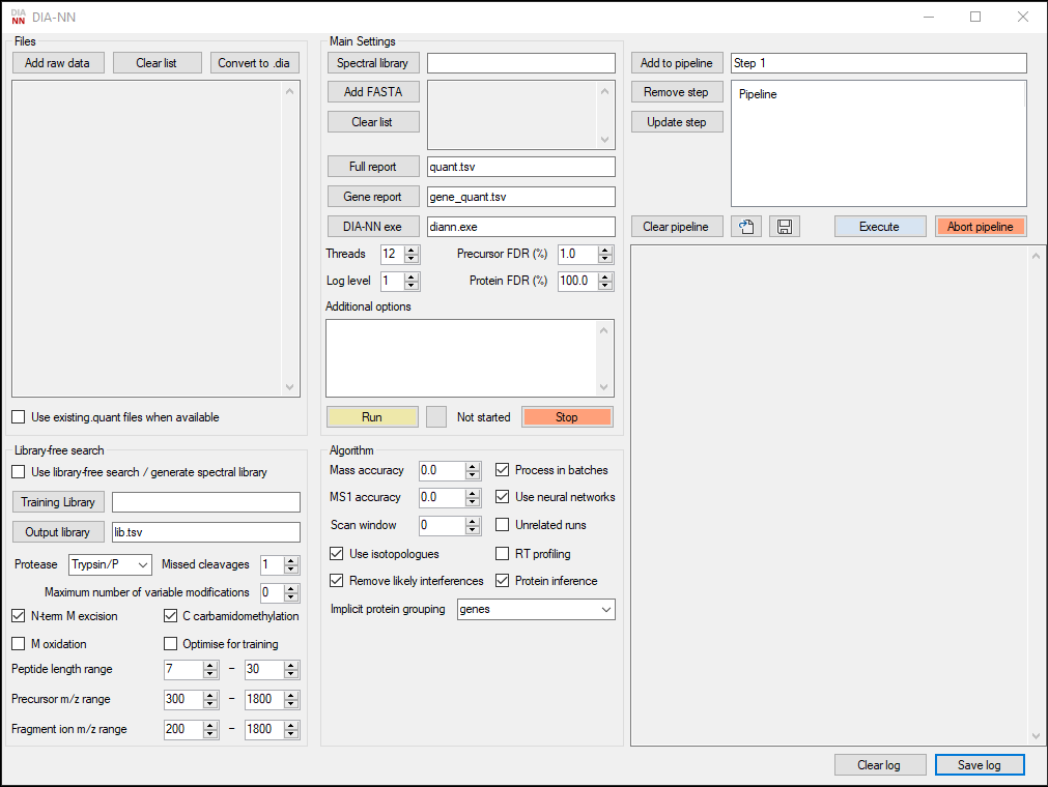
GUI window
The GUI window features four panels used to specify the data and processing settings: Files, Main Settings,
Library-free search and Algorithm. To the right, there is a pipeline editor at the top and a panel used to display
the output of the command-line tool at the bottom. Extra commands entered in the Additional options text box
will also be passed to the command line tool; quotes can be used when passing options but are not required unless
file names featuring multiple adjacent spaces need to be specified. See the project description on the GitHub for
the information on DIA-NN’s command line syntax and some useful commands. Most GUI elements feature
tooltips with a short description of their function, activated by hovering a mouse cursor over the element.
Files
Sciex .wiff, Thermo .raw, .mzML and .dia (native DIA-NN format) files are supported. Reading Sciex .wiff files
requires the DIA-NN.Wiff.dll file (included in the DIA-NN distribution) as well as the Sciex DLL libraries to be
present in the same folder as DiaNN.exe. For example, one can first install ProteoWizard (which includes the
Sciex DLLs) and then install DIA-NN in the same folder using the installer provided (DIA-NN-Setup.msi).
Reading Thermo .raw files requires Thermo MS File Reader to be installed. It is highly recommended that .mzML
files are centroided, preferably using the vendor centroiding algorithm for both MS1 and MS2 spectra. DIA-NN
has been tested on .mzML files produced from Sciex .wiff and Thermo .raw by MSConvert GUI (part of the
ProteoWizard project) with 32-bit binary precision, vendor centroiding enabled and all other options disabled.
DIA-NN can convert raw files to its own .dia format. This format allows very quick loading, essentially limited
only by the hard drive speed. Converted files are placed in the same location as the raw files.
During the analysis, DIA-NN generates a .quant file containing identification and quantification information for
each run analysed. These .quant files are placed in the same location as the raw files. Thus, it is not recommended
to analyse the same raw data files simultaneously with multiple instances of DIA-NN (unless .quant files are
stored in memory – see below), although multiple instances of DIA-NN can be used to analyse different
experiments. The size of the .quant files is roughly proportional to the number of precursors identified at the
given FDR setting and is negligible unless a library-free search without any FDR filtering is used (not
recommended). The .quant files can be reused to speed up reanalysis of the data, but only if the spectral library
and FASTA files specified as well as the FDR filtering settings are exactly the same as the settings that were
used to generate these .quant files.
An alternative location for the .quant files can be specified using the --temp command, e.g. --temp C:/Temp.
The --no-quant-files command instructs DIA-NN to store .quant files in memory, without saving to disk.
Main Settings
Spectral library
Used to specify the spectral library. The library is required unless a library-free search against a FASTA database
is to be performed. In that case, if specified, the spectral library will be used to enhance and speed up library-free
search (experimental). DIA-NN accepts spectral libraries in a plain text format (tab-separated or comma-
separated, i.e. .tsv, .csv, .xls or .txt).
Libraries produced by TargetedFileConverter (part of OpenMS), exported from Spectronaut (Biognosys) in the
.xls format or generated by DIA-NN itself are supported “as is”.
For libraries generated by other means, DIA-NN needs to be told the header names (separated by commas) (for
the columns it requires) using the --library-headers command. The headers should be specified in the
following order:
- Modified peptide sequences. Outside parentheses, all symbols apart from letters are ignored.
Modifications are specified within parentheses (“()” or “[]”) after the modified amino acid or (e.g.
“[Acetyl (Protein N-term)]”) before the first amino acid. Recognition of some common modifications is
built in. Custom modifications can be specified using the --mod command, e.g. “--mod
UniMod:5,43.005814” will add carbamylation (KR) to the list of modifications recognized by DIA-
NN. DIA-NN does not need to recognize modifications if decoys are provided in the library (see below).
- Precursor charge.
- Precursor m/z.
- Reference retention time. There are no restrictions on the retention time scale used here.
- Fragment ion m/z.
- Relative intensity of the fragment ion. There are no restrictions on the scale used here.
The following optional headers can also be specified:
- UniProt identifiers of the proteins matched to the peptide (separated by semicolon). These will be used
for peptide annotation as well as protein quantification. If a FASTA file is specified, UniProt identifiers
will be searched against this file and annotated with protein and gene names. If the library contains no
column specifying which peptides are proteotypic (see below), DIA-NN will consider any peptide
matched to a single UniProt identifier proteotypic. DIA-NN does not check the validity of the UniProt
identifiers provided, i.e. arbitrary text can be used instead.
- Protein names. These will be used for annotation if a FASTA file is not provided.
- Gene names. These will be used for annotation if a FASTA file is not provided.
- Proteotypicity. The value should be 1 if the peptide is to be considered proteotypic. DIA-NN calculates
protein q-values using proteotypic peptides only. If the protein FDR filter setting is below 100%,
proteotypicity definition also affects protein inference and grouping.
- Decoy. Indicates whether the peptide is a decoy. If there are decoy peptides in the library, DIA-NN uses
these and does not generate its own decoys.
- Fragment ion charge. If this column is not provided, DIA-NN infers the charges itself, although this
inference relies on recognition of all peptide modifications and is not guaranteed to be correct in all the
cases. However, DIA-NN needs to know fragment ion charges only if it is told to consider isotopologues
of the peptides and fragments in the library (this is enabled by default).
FASTA
One or several FASTA files can optionally be specified. These will be used for protein annotation (if in UniProt
format). If library-free search is used, the FASTA files will be digested in silico to generate a spectral library.
Optionally, peptides considered can be filtered using the --fasta-filter command, e.g. “--fasta-filter
C:/human_peptides.fasta”. Peptides can be provided as a fasta file (e.g. DIA-NN recognises the format of
peptide lists from PeptideAtlas builds) or as a simple list of stripped sequences (one per line); all lines starting
with ‘>’ are ignored. Filtering allows to dramatically reduce the search space, speeding up the analysis and
improving identification performance.
Library-free search
Use library-free search / generate spectral library
This will instruct DIA-NN to in silico generate a spectral library from the FASTA file provided and use it to
analyse the data files. In addition to the analysis report, a new spectral library will be generated. If a spectral
library is provided in addition to the FASTA file, it will be used (experimental) to enhance and speed up the
library-free search. In this mode, q-values for precursor ions that belong to the library will be calculated
separately. If a spectral library is provided, but there is no FASTA file, this library will be used to analyse the
data files and a new spectral library will be generated using only identified precursor ions (at the specified
precursor and protein FDR cutoff rates) and featuring refined spectra and retention times; the retention time scale
used in the spectral library will be retained. A new spectral library is saved in an OpenMS-compatible format.
Training library
Specifying a training library allows DIA-NN to in silico predict peptide fragmentation patterns (only charge one
y-series fragments without neutral losses are considered) using an algorithm similar to that of MS Simulator. In
addition, a linear retention time predictor is trained. It is essential that if a spectral library is also specified (in the
Main Settings panel), its retention time scale should be the same (e.g. both the training and the spectral library
may use the iRT scale). It is desirable but not essential that the training library is generated on the same LC-MS
setup. If no suitable training library is available, one can first run DIA-NN without it (identification performance
will be somewhat worse) and then run it again using the newly generated library as the training library. For
efficient training of the predictors at least 10’000 precursors should be present in the training library.
Protease
Several built-in options are available for the in silico digest. Alternatively, one can specify custom cleavage
specificity, e.g. “--cut-after KR --no-cut-before P” corresponds to a tryptic digest. Multiple proteases
are not supported, however DIA-NN can be provided with an in silico digest in the FASTA format instead of the
FASTA database.
Modifications
In general, it is not recommended to enable variable modifications, as this slows down analysis considerably and
expands the search space, potentially negatively affecting identification performance. It is essential that the
removal of likely interferences is enabled (see the Algorithm settings below), if e.g. deamidation is enabled and
the runs have been acquired on an instrument that does not allow to effectively distinguish between deamidated
ions and heavy isotopologues.
Arbitrary user-defined modifications can be added using the --fixed-mod and --var-mod commands, e.g. -
-var-mod UniMod:5,43.005814,KR will enable lysine and arginine carbamylation as a variable
modification. Use lower-case letters for the amino acids to restrict the modification to N-terminus.
Precursor ion mass range
This allows to restrict the range of precursors being considered, e.g. to the range covered by the SWATH cycle
of the instrument. Allows to slightly reduce memory consumption.
Fragment ion mass range
This allows to restrict the range of fragment ion masses being considered.
Optimise for training
This option instructs DIA-NN to strictly filter the fragment ions that are being saved to the spectral library. Use
of this option is recommended only for generating libraries that will be used to train the peptide fragmentation
predictor.
Algorithm
Mass accuracies can be determined automatically (when set to the default 0.0) or specified by the user. Initially,
DIA-NN performs a preliminary search with mass accuracy set to 100ppm, followed by mass correction. If the
masses reported by the instrument are guaranteed not to be that much off (e.g. the instrument is regularly
calibrated), this setting can be reduced, e.g. to 30ppm with “--mass-acc-cal 30”. This can produce a
noticeable speed up for library-free searches.
The implicit protein grouping option allows to specify the proteotypicity definition that will be used by DIA-NN.
For example, if set to the default “genes”, DIA-NN will consider a peptide proteotypic, if all of its associated
proteins correspond (according to the FASTA files provided) to a single gene. Proteotypicity definition affects
protein q-value calculation, as the latter relies on proteotypic peptides only, and, if the protein FDR filter setting
is below 100%, protein inference and grouping.
DIA-NN can use an ensemble of neural networks to calculate q-values. By default, these are trained for a single
epoch, minimizing the chance of overfitting. It is not recommended to disable the removal of likely interferences
when using neural networks. The number of epochs can be increased by setting--nn-epochs, and the number
of neural networks in the ensemble – by setting --nn-bagging (more = better and slower; default = 12). To
almost completely eliminate any potential overfitting and allow for an increased number of epochs, neural
networks can be used in a “cross-validation mode”, when each network is only used to score peptide-spectrum
matches that have not been used for its training. However, in this case it is desirable to also increase the number
of networks, slowing down the analysis. For example, “--nn-cross-val --nn-bagging 96 --nn-epochs
5”.
Output
Default output header names can be replaced (in the full report only) with those specified with the --output-
headers command, analogous to --library-headers (see above).
Protein.Group
DIA-NN aims to reduce the number of proteins matched to each precursor ion. Maximal parsimony approach is
implemented using a greedy set cover algorithm. Furthermore, TrEMBL proteins can be omitted, if the precursor
in question is also matched to at least one SwissProt protein (this can be disabled with --no-swissprot).
Protein.Names and Genes
Protein names and genes corresponding to proteins listed in the Protein.Group column.
Protein.Q.Value
The best q-value across all proteins matched to the precursor ion. Protein q-values are calculated separately for
all protein isoform names, protein names or genes (not protein groups or gene groups) – depending on the implicit
protein grouping setting. Only proteotypic peptides are used to calculate q-values. Thus, when DIA-NN reports
the number of proteins identified at 1% FDR (for each run separately, once it is analysed), it underestimates the
number of protein groups that are actually detected. Importantly, proteins with no proteotypic peptides found are
omitted from the report if the protein FDR threshold is set below 100%.
Quantities
PG.Quantity – protein group (i.e. the set of proteins listed in Protein.Group), Gene.Group.Quantity – gene group
(i.e. the set of genes listed in Genes), Gene.Quantity.Unique – the gene quantified using only peptides that are
specific to it. Quantification is performed using the top 3 method, i.e. the intensities of the top 3 precursor ions
identified at 1% FDR are summed for each run separately. Normalised – the same quantities with cross-run
normalisation applied (on the precursor level). Normalisation is performed using the top 40% least variable
(across runs) precursors identified at 1% FDR. These figures can be adjusted, e.g. --norm-fraction 0.3 -
-norm-qvalue 0.001.
Extra information
CScore and Decoy.CScore columns provide information on the scores for each precursor ion and the respective
decoy, which were used to calculate q-values. Fragment.Quant.Raw, Fragment.Quant.Corrected and
Fragment.Correlations columns list raw and interference-corrected fragment intensities and correlation scores
(higher = better) (the fragments being ordered by their library intensities from highest to lowest). This information
allows to easily direct DIA-NN output to scripts that perform custom protein inference, quantification, etc.
Quality control
Along with the main report, DIA-NN produces a file [main report name].stats.tsv with a quality control
information for all the samples in the experiment. For example, it contains such information as the precursor and
protein identification numbers, total quantity (sum of all precursor quantities), average full width at half
maximum (FWHM) for chromatographic peaks, expressed both as the number of SWATH scans and as time in
minutes, mass accuracy correction data as well as retention time prediction accuracy, average precursor length
and charge. The generation of this file can be turned off with the --no-stats command.
Pipelines
Pipeline support allows to set up automatic analysis of a series of experiments, using a separate set of settings for
each. The “Add to pipeline” button adds the current set of settings (including all input and output file names) to
the pipeline. Selecting one of the pipeline steps (with a mouse) loads the respective set of settings. The “Update
step” button overwrites the selected pipeline step with the current set of settings. Buttons with pictograms to the
right from the “Clear pipeline” button can be used top open and save .pipeline files. Note that the .pipeline format
is not guaranteed to retain compatibility across different versions of DIA-NN GUI.
