41406_Egrifta_DocDiscussionGuide_CVR_SPANISH_M01 EGRIFTA Doctor Discussion Guide Spanish
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 56

Una transformación desde su interior
®
Una transformación desde su interior
EGRIFTA.com
Regreso del exceso de VAT
Las personas que usaron EGRIFTA®
de forma continua por 1 año
mantuvieron sus resultados durante
este período de tiempo.
UNA REDUCCIÓN
DEL
18%
EN
Promedio del Ensayo 1
VAT
UNA REDUCCIÓN
DEL
14%
EN
Promedio del Ensayo 2
VAT
Cómo ayudar a reducir el exceso de
tejido adiposo visceral (VAT) abdominal
(y controlarlo)
EGRIFTA® (tesamorelin inyectable) es el único tratamiento aprobado por
la Administración de Alimentos y Medicamentos de los Estados Unidos
(US Food and Drug Administration, FDA) para tratar el exceso de VAT
abdominal en personas con VIH que tienen lipodistrofia. EGRIFTA® es
un medicamento recetado inyectable via subcutánea (debajo de la piel),
una vez al día en la zona inferior de su abdomen.
Se han realizado estudios de EGRIFTA® en 2 ensayos clínicos de
personas con VIH que desarrollaron lipodistrofia.
• Cada ensayo duró 6 meses, con una fase de extensión de
6 meses adicionales
• Los pacientes de estos ensayos presentaron reducciones
significativas del tejido adiposo visceral (VAT)
EGRIFTA® no está indicado para el manejo de la pérdida de peso.
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
Informe a su proveedor de atención médica sobre todos los
medicamentos que toma, incluidos los que se venden con receta
y los de venta libre, vitaminas y suplementos a base de hierbas.
EGRIFTA® puede afectar el funcionamiento de otros medicamentos
y estos mismos pueden afectar el funcionamiento de EGRIFTA ®.
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
Nuestro personal de enfermería de EGRIFTA ASSIST ®
está listo para ayudar a:
• Brindarle capacitación sobre las técnicas
apropiadas de inyección y reconstitución
• Asesorarlo sobre cómo comenzar
y permanecer bajo tratamiento
• Contestar preguntas frecuentes relacionadas
con el tratamiento través de su tratamiento
Asistencia de expertos con tan solo una llamada
Los asesores están listos para comunicarse con usted
y ayudar a contestar sus preguntas.
1-844-EGRIFTA (1-844-347-4382)
De lunes a viernes, de 8a.m. a 8p.m. hora del Este
Los mensajes de correos de voz se responderán dentro
del próximo día laborable.
Los efectos secundarios más frecuentes de EGRIFTA
®
incluyen:
• dolor de las articulaciones
• dolor en los brazos y en las piernas
• hinchazón en las piernas
• dolor muscular
• hormigueo, adormecimiento y pinchazos
• náuseas
• vómitos
• sarpullido
• picazón
Informe a su proveedor de atención médica si tiene cualquier efecto
secundario que le cause molestias o que persista.
Estos no son todos los efectos secundarios posibles de EGRIFTA ®.
Para recibir más información, consulte con su proveedor de atención
médica o a su farmacéutico.
EGRIFTA.com
No pierda tiempo. Hable con su HCP hoy.
Pregunte a su proveedor de atención médica (HCP, en inglés)
sobre
EGRIFTA
®
(tesamorelin inyectable)
.
Para obtener más información, vaya a EGRIFTA.com.
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
Llame a su médico para que lo asesore acerca de los efectos
secundarios. Para informar sobre sospechas de reacciones adversas,
comuníquese con EGRIFTA ASSIST ® al número de teléfono gratuito
1-844-EGRIFTA (1-844-347-4382), o bien con la FDA al
1-800-FDA-1088 o en www.fda.gov/medwatch.
¿Cómo debo almacenar EGRIFTA ®?
• EGRIFTA® viene en dos cajas dispensadas por la farmacia:
Almacene los frascos de la caja del medicamento EGRIFTA ®
en el refrigerador entre 2 °C y 8 °C (36 °F y 46 °F)
Almacene la caja de agua estéril para inyección, las jeringas
y las agujas a temperatura ambiente entre 20 °C y 25 °C
(68 °F y 77 °F).
• Guarde los frascos de EGRIFTA® en la caja del medicamento
alejados de la luz.
• No se debe congelar.
• No use EGRIFTA® después de la fecha de vencimiento impresa en
la caja y en las etiquetas del frasco.
• Después de mezclar, use EGRIFTA® de inmediato y descarte todo
el EGRIFTA ® restante. No almacene la mezcla de EGRIFTA ®.
Además, deseche el frasco usado de agua estéril para inyección.
Mantenga EGRIFTA ® y todos los medicamentos fuera del
alcance de los niños.
Información general sobre el uso eficaz y seguro de EGRIFTA ®
Los medicamentos a veces se recetan con fines distintos de los que se
indican en la Información para el paciente. No use EGRIFTA® para una
afección para la cual no fue recetado. No administre EGRIFTA® a otras
personas, aunque tengan los mismos síntomas que usted. Hacer esto
puede causarles daño.
No comparta su jeringa o agujas de EGRIFTA ® con otras personas.
Usted puede provocarles una infección o infectarse por ellos.
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
EGRIFTA ® es una marca registrada de Theratechnologies Inc.
© 2016 Theratechnologies Inc. Todos los derechos reservados. 210-01-04/16
Si es VIH positivo, es posible que se
enfrente con otro desafío médico
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
¿Qué es EGRIFTA ®?
• EGRIFTA ® es un medicamento recetado inyectable para reducir
el exceso de grasa abdominal en pacientes infectados con el VIH que
tienen lipodistrofia. EGRIFTA® contiene un factor de liberación de la
hormona de crecimiento (growth hormone-releasing factor, GRF).
• No se han realizado estudios sobre el impacto y seguridad de EGRIFTA ®
en la salud cardiovascular.
• EGRIFTA® no está indicado para el manejo de la pérdida de peso.
• No se sabe si EGRIFTA® ayuda a mejorar la adhesión al tratamiento del VIH.
• No se sabe si EGRIFTA® es seguro y eficaz en niños.
No se recomienda el uso de EGRIFTA ® en niños.
Por favor, lea el prospecto,
junto con la información para
el paciente y las instrucciones
de uso adjuntas.
Aprenda más sobre EGRIFTA ® (tesamorelin inyectable)
Es el único tratamiento, aprobado por la FDA, para tratar
el exceso de tejido adiposo visceral en personas
VIH positivas que tienen lipodistrofia
Obtenga más información
en EGRIFTA.com
Ayude a reducir el exceso de tejido
adiposo visceral en su abdomen
(VAT, en inglés)
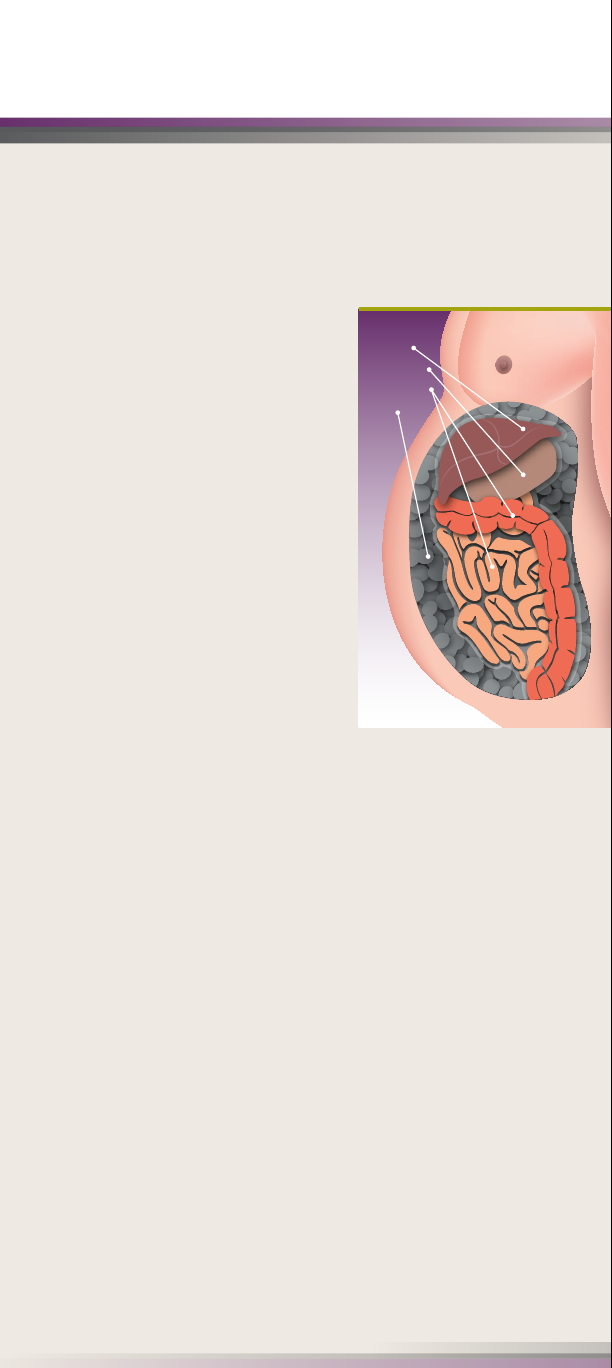
Hígado
Estómago
Intestinos
VAT
Una transformación desde su interior
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
Entender qué es VAT
(y por qué es distinta a la grasa regular)
Cuando el VIH está controlado, usted puede enfocarse más en su
salud con el correr de los años. La lipodistrofia es una afeccion que
puede provocar la formación de tejido adiposo visceral (VAT) y puede
convertirse en un reto para su salud.
El VAT es un tipo de grasa que se
encuentra, principalmente, dentro del área
abdominal. La grasa normal, también
conocida como tejido adiposo subcutáneo
(SAT, en inglés), se encuentra justo debajo
de la piel.
El VAT se encuentra en un área más
profunda y:
• Puede rodear los órganos
• Puede estar asociada con la
acumulación de grasa en los órganos
• Se siente más dura que la grasa
regular (SAT)
• Puede ser difícil de diagnosticar
durante un examen físico de rutina
en el consultorio
• Puede ser difícil de reducir solamente
con dieta y ejercicio
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
¿Quiénes no deben usar EGRIFTA ® (tesamorelin inyectable)?
No use EGRIFTA ® si usted:
• Tiene un tumor de la glándula pituitaria, cirugía de la glándula
pituitaria u otros problemas relacionados con su glándula pituitaria.
• Tiene cáncer activo (ya sea recientemente diagnosticado
o recurrente) o está recibiendo tratamiento para el cáncer.
• Es alérgico a tesamorelin o a cualquiera de los ingredientes
de EGRIFTA®. Lea la última página de este folleto para ver una lista
completa de los ingredientes de EGRIFTA ®.
• Está embarazada o planea quedar embarazada. Si queda
embarazada, deje de usar EGRIFTA® y hable con su proveedor
de atención médica. Consulte la sección “¿Qué debo decirle
a mi proveedor de atención médica antes de usar EGRIFTA®?”
Cómo saber si existe exceso de VAT abdominal
(y cómo ayudar a mantenerlo bajo control)
El exceso de VAT abdominal puede estar asociado con:
• Graves problemas de salud
• Una disminución en las habilidades físicas
No se han realizado estudios sobre la seguridad y eficacia
de EGRIFTA ® en otros problemas de salud que pueden estar
relacionados con VAT.
Su proveedor de atención médica (HCP) puede verificar
si hay exceso de VAT de una forma simple. Esto incluye:
Si no puede controlar la presencia de VAT con ejercicio fisico
y una dieta, pregunte a su HCP sobre EGRIFTA ®, el cual se ha
demostrado que reduce el exceso de VAT abdominal en las personas
con VIH que tienen lipodistrofia.
¿Qué debo decirle a mi proveedor de atención médica antes
de tomar EGRIFTA ®?
Antes de tomar EGRIFTA ®, informe a su proveedor de atención médica
si usted:
• tiene o ha tenido cáncer
• tiene diabetes
• está amamantando o planea hacerlo. No se sabe si EGRIFTA® pasa
a la leche materna. Los Centros para el Control y la Prevención de
Enfermedades (Centers for Disease Control and Prevention, CDC)
recomiendan que las madres infectadas por el virus del VIH no
amamanten para evitar el riesgo de pasar la infección del VIH a su
bebés. Hable con su proveedor de atención médica sobre la mejor
forma de alimentar a su bebé si está tomando EGRIFTA ®
• tiene problemas renales o hepáticos
• tiene otras afecciones médicas.
EGRIFTA.com
Medir la firmeza de su abdomen
Medir la circumferencia
de su cintura y sus caderas

Hígado
Estómago
Intestinos
VAT
Una transformación desde su interior
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
Entender qué es VAT
(y por qué es distinta a la grasa regular)
Cuando el VIH está controlado, usted puede enfocarse más en su
salud con el correr de los años. La lipodistrofia es una afeccion que
puede provocar la formación de tejido adiposo visceral (VAT) y puede
convertirse en un reto para su salud.
El VAT es un tipo de grasa que se
encuentra, principalmente, dentro del área
abdominal. La grasa normal, también
conocida como tejido adiposo subcutáneo
(SAT, en inglés), se encuentra justo debajo
de la piel.
El VAT se encuentra en un área más
profunda y:
• Puede rodear los órganos
• Puede estar asociada con la
acumulación de grasa en los órganos
• Se siente más dura que la grasa
regular (SAT)
• Puede ser difícil de diagnosticar
durante un examen físico de rutina
en el consultorio
• Puede ser difícil de reducir solamente
con dieta y ejercicio
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
¿Quiénes no deben usar EGRIFTA ® (tesamorelin inyectable)?
No use EGRIFTA ® si usted:
• Tiene un tumor de la glándula pituitaria, cirugía de la glándula
pituitaria u otros problemas relacionados con su glándula pituitaria.
• Tiene cáncer activo (ya sea recientemente diagnosticado
o recurrente) o está recibiendo tratamiento para el cáncer.
• Es alérgico a tesamorelin o a cualquiera de los ingredientes
de EGRIFTA®. Lea la última página de este folleto para ver una lista
completa de los ingredientes de EGRIFTA ®.
• Está embarazada o planea quedar embarazada. Si queda
embarazada, deje de usar EGRIFTA® y hable con su proveedor
de atención médica. Consulte la sección “¿Qué debo decirle
a mi proveedor de atención médica antes de usar EGRIFTA®?”
Cómo saber si existe exceso de VAT abdominal
(y cómo ayudar a mantenerlo bajo control)
El exceso de VAT abdominal puede estar asociado con:
• Graves problemas de salud
• Una disminución en las habilidades físicas
No se han realizado estudios sobre la seguridad y eficacia
de EGRIFTA ® en otros problemas de salud que pueden estar
relacionados con VAT.
Su proveedor de atención médica (HCP) puede verificar
si hay exceso de VAT de una forma simple. Esto incluye:
Si no puede controlar la presencia de VAT con ejercicio fisico
y una dieta, pregunte a su HCP sobre EGRIFTA ®, el cual se ha
demostrado que reduce el exceso de VAT abdominal en las personas
con VIH que tienen lipodistrofia.
¿Qué debo decirle a mi proveedor de atención médica antes
de tomar EGRIFTA ®?
Antes de tomar EGRIFTA ®, informe a su proveedor de atención médica
si usted:
• tiene o ha tenido cáncer
• tiene diabetes
• está amamantando o planea hacerlo. No se sabe si EGRIFTA® pasa
a la leche materna. Los Centros para el Control y la Prevención de
Enfermedades (Centers for Disease Control and Prevention, CDC)
recomiendan que las madres infectadas por el virus del VIH no
amamanten para evitar el riesgo de pasar la infección del VIH a su
bebés. Hable con su proveedor de atención médica sobre la mejor
forma de alimentar a su bebé si está tomando EGRIFTA ®
• tiene problemas renales o hepáticos
• tiene otras afecciones médicas.
EGRIFTA.com
Medir la firmeza de su abdomen
Medir la circumferencia
de su cintura y sus caderas
Una transformación desde su interior
®
Una transformación desde su interior
EGRIFTA.com
Regreso del exceso de VAT
Las personas que usaron EGRIFTA®
de forma continua por 1 año
mantuvieron sus resultados durante
este período de tiempo.
UNA REDUCCIÓN
DEL
18%
EN
Promedio del Ensayo 1
VAT
UNA REDUCCIÓN
DEL
14%
EN
Promedio del Ensayo 2
VAT
Cómo ayudar a reducir el exceso de
tejido adiposo visceral (VAT) abdominal
(y controlarlo)
EGRIFTA® (tesamorelin inyectable) es el único tratamiento aprobado por
la Administración de Alimentos y Medicamentos de los Estados Unidos
(US Food and Drug Administration, FDA) para tratar el exceso de VAT
abdominal en personas con VIH que tienen lipodistrofia. EGRIFTA® es
un medicamento recetado inyectable via subcutánea (debajo de la piel),
una vez al día en la zona inferior de su abdomen.
Se han realizado estudios de EGRIFTA® en 2 ensayos clínicos de
personas con VIH que desarrollaron lipodistrofia.
• Cada ensayo duró 6 meses, con una fase de extensión de
6 meses adicionales
• Los pacientes de estos ensayos presentaron reducciones
significativas del tejido adiposo visceral (VAT)
EGRIFTA® no está indicado para el manejo de la pérdida de peso.
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
Informe a su proveedor de atención médica sobre todos los
medicamentos que toma, incluidos los que se venden con receta
y los de venta libre, vitaminas y suplementos a base de hierbas.
EGRIFTA® puede afectar el funcionamiento de otros medicamentos
y estos mismos pueden afectar el funcionamiento de EGRIFTA ®.
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
Nuestro personal de enfermería de EGRIFTA ASSIST ®
está listo para ayudar a:
• Brindarle capacitación sobre las técnicas
apropiadas de inyección y reconstitución
• Asesorarlo sobre cómo comenzar
y permanecer bajo tratamiento
• Contestar preguntas frecuentes relacionadas
con el tratamiento través de su tratamiento
Asistencia de expertos con tan solo una llamada
Los asesores están listos para comunicarse con usted
y ayudar a contestar sus preguntas.
1-844-EGRIFTA (1-844-347-4382)
De lunes a viernes, de 8a.m. a 8p.m. hora del Este
Los mensajes de correos de voz se responderán dentro
del próximo día laborable.
Los efectos secundarios más frecuentes de EGRIFTA
®
incluyen:
• dolor de las articulaciones
• dolor en los brazos y en las piernas
• hinchazón en las piernas
• dolor muscular
• hormigueo, adormecimiento y pinchazos
• náuseas
• vómitos
• sarpullido
• picazón
Informe a su proveedor de atención médica si tiene cualquier efecto
secundario que le cause molestias o que persista.
Estos no son todos los efectos secundarios posibles de EGRIFTA ®.
Para recibir más información, consulte con su proveedor de atención
médica o a su farmacéutico.
EGRIFTA.com
No pierda tiempo. Hable con su HCP hoy.
Pregunte a su proveedor de atención médica (HCP, en inglés)
sobre
EGRIFTA
®
(tesamorelin inyectable)
.
Para obtener más información, vaya a EGRIFTA.com.
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
Llame a su médico para que lo asesore acerca de los efectos
secundarios. Para informar sobre sospechas de reacciones adversas,
comuníquese con EGRIFTA ASSIST ® al número de teléfono gratuito
1-844-EGRIFTA (1-844-347-4382), o bien con la FDA al
1-800-FDA-1088 o en www.fda.gov/medwatch.
¿Cómo debo almacenar EGRIFTA ®?
• EGRIFTA® viene en dos cajas dispensadas por la farmacia:
Almacene los frascos de la caja del medicamento EGRIFTA ®
en el refrigerador entre 2 °C y 8 °C (36 °F y 46 °F)
Almacene la caja de agua estéril para inyección, las jeringas
y las agujas a temperatura ambiente entre 20 °C y 25 °C
(68 °F y 77 °F).
• Guarde los frascos de EGRIFTA® en la caja del medicamento
alejados de la luz.
• No se debe congelar.
• No use EGRIFTA® después de la fecha de vencimiento impresa en
la caja y en las etiquetas del frasco.
• Después de mezclar, use EGRIFTA® de inmediato y descarte todo
el EGRIFTA ® restante. No almacene la mezcla de EGRIFTA ®.
Además, deseche el frasco usado de agua estéril para inyección.
Mantenga EGRIFTA ® y todos los medicamentos fuera del
alcance de los niños.
Información general sobre el uso eficaz y seguro de EGRIFTA ®
Los medicamentos a veces se recetan con fines distintos de los que se
indican en la Información para el paciente. No use EGRIFTA® para una
afección para la cual no fue recetado. No administre EGRIFTA® a otras
personas, aunque tengan los mismos síntomas que usted. Hacer esto
puede causarles daño.
No comparta su jeringa o agujas de EGRIFTA ® con otras personas.
Usted puede provocarles una infección o infectarse por ellos.
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
EGRIFTA ® es una marca registrada de Theratechnologies Inc.
© 2016 Theratechnologies Inc. Todos los derechos reservados. 210-01-04/16
Si es VIH positivo, es posible que se
enfrente con otro desafío médico
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
¿Qué es EGRIFTA ®?
• EGRIFTA ® es un medicamento recetado inyectable para reducir
el exceso de grasa abdominal en pacientes infectados con el VIH que
tienen lipodistrofia. EGRIFTA® contiene un factor de liberación de la
hormona de crecimiento (growth hormone-releasing factor, GRF).
• No se han realizado estudios sobre el impacto y seguridad de EGRIFTA ®
en la salud cardiovascular.
• EGRIFTA® no está indicado para el manejo de la pérdida de peso.
• No se sabe si EGRIFTA® ayuda a mejorar la adhesión al tratamiento del VIH.
• No se sabe si EGRIFTA® es seguro y eficaz en niños.
No se recomienda el uso de EGRIFTA ® en niños.
Por favor, lea el prospecto,
junto con la información para
el paciente y las instrucciones
de uso adjuntas.
Aprenda más sobre EGRIFTA ® (tesamorelin inyectable)
Es el único tratamiento, aprobado por la FDA, para tratar
el exceso de tejido adiposo visceral en personas
VIH positivas que tienen lipodistrofia
Obtenga más información
en EGRIFTA.com
Ayude a reducir el exceso de tejido
adiposo visceral en su abdomen
(VAT, en inglés)

Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
EGRIFTA.com Una transformación desde su interior
EGRIFTA
®
puede ayudar a reducir el exceso de tejido adiposo visceral
(VAT) en el área abdominal, al cambiar la forma en que se liberan ciertas
hormonas (sustancias químicas que su cuerpo produce naturalmente) se
liberan. Sin embargo, para pacientes con VIH, el exceso de VAT es una
afección crónica. Para mantener una reducción en el exceso de VAT, es
posible que su proveedor de atención médica (health care provider, HCP)
recomiende que continúe el tratmiento con EGRIFTA
®
.
En ensayos clínicos:
¿Cómo debo usar EGRIFTA
®
?
• Lea las “Instrucciones de uso” detalladas que vienen con
EGRIFTA®
antes de comenzar a usar
EGRIFTA®
. Su proveedor de atención
médica le indicará cómo debe inyectarse
EGRIFTA®
.
• Use
EGRIFTA®
exactamente como lo indicó su proveedor de atención
médica.
•
EGRIFTA®
debe inyectarse via subcutánea (debajo de la piel)
en el area abdominal.
• Rote diariamente el lugar de la inyección. No inyecte
EGRIFTA®
en tejido cicatrizal, en moretones o en el ombligo.
• No comparta las agujas o jeringas con otras personas. Compartir
agujas puede causar la transmisión de enfermedades infecciosas,
como el VIH.
Las personas que dejaron de tomar
EGRIFTA® después de los 6 meses de
tratamiento, desarrollaron la
formación de nuevo VAT relacionado
con la infección del VIH
Para mantener la reducción en VAT
Regreso del exceso de VAT
Las personas que usaron EGRIFTA®
de forma continua por 1 año
mantuvieron sus resultados durante
este período de tiempo.

Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
Cómo hablar con su proveedor de atención
médica (HCP)
(y cómo prepararse para la conversación)
Antes de visitar a su HCP, usted debe hacerse algunas preguntas.
Esto puede servirle de guía en su conversación:
1. ¿Alguna de las situaciones descritas aquí corresponden a su
caso? Marque todas las casillas que correspondan.
__ Mi abdomen se siente duro o incómodo
__ Mi cintura se ha ensanchado con el tiempo
__ El cambio en el tamaño y la forma de mi abdomen
está afectando la manera que calzo la ropa
__ No puedo deshacerme del exceso de grasa abdominal
solamente con dieta y ejercicio
2. ¿Alguno de estos factores de riesgo de exceso de VAT
abdominal relacionados con el VIH corresponden a su caso?
__ Tiene más de 40 años de edad
__ Hace más de 3 años que vive con el VIH
__ Recibe terapia antirretroviral (ART)
__ Tiene un alto índice de masa corporal (BMI, en inglés)
3. ¿Cuánto hace que recibe tratamiento para el VIH?
________ años ________ meses
¿Cuánto tiempo después de haber comenzado el tratamiento
usted cree que desarrolló exceso de VAT?
________ años ________ meses
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
¿Cuáles son los posibles efectos secundarios de EGRIFTA ®
(tesamorelin inyectable)?
EGRIFTA® puede causar efectos secundarios graves, incluidos:
• Reacción alérgica grave. Algunas personas que reciben tratamiento
con EGRIFTA® pueden tener una reacción alérgica.
Deje de usar EGRIFTA ® y obtenga ayuda de emergencia
inmediatamente si tiene alguno de los siguientes síntomas:
sarpullido en su cuerpo
urticaria
hinchazón de la cara o garganta
falta de aire o dificultad para respirar
pulso acelerado
sensación de que va a desmayarse o desmayos
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
EGRIFTA.com Una transformación desde su interior
4. Verifique qué medidas ha tomado para tratar de reducir
el exceso de VAT abdominal.
__ Cambié mi dieta
__ Comencé o mejoré una rutina de ejercicios
__ Cambié las terapias de VIH (con el permiso de mi HCP)
5.
El exceso de VAT puede estar asociado con otros problemas
de salud. Si ha sido diagnosticado con otras afecciones médicas,
enumérelas más abajo y asegúrese de mencionárselas a su HCP:
_______________________________________
_______________________________________
_______________________________________
_______________________________________
_______________________________________
No se han realizado estudios sobre la seguridad y eficacia de
EGRIFTA ® en otros problemas de salud que pueden estar
relacionados con el VAT.
Si usted o su HCP consideran que tiene un exceso de VAT
abdominal, consulte sobre el tratamiento con EGRIFTA®.
¿Cuáles son los posibles efectos secundarios de EGRIFTA ®?
EGRIFTA® puede causar efectos secundarios graves, incluidos los
siguientes:
• Hinchazón (retención de líquidos). EGRIFTA® puede causar
hinchazón en algunas partes de su cuerpo. Llame a su proveedor
de atención médica si tiene un aumento del dolor en las
articulaciones, o dolor o entumecimiento en sus manos o muñecas
(síndrome del túnel carpiano).
• Aumento de intolerancia a la glucosa (azúcar en sangre) y diabetes.
Su proveedor de atención médica medirá su azúcar en sangre
periódicamente.

Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
Servicio personal para ayudarle a conseguir
el mayor beneficio de su tratamiento
Junto con la consulta con su HCP, EGRIFTA ASSIST
®
es el programa
que debe usar cuando necesita asesoría y apoyo adicional mientras
recibe tratamiento con EGRIFTA
®
(tesamorelin inyectable).
Los consejeros profesionales están listos para ayudar a:
• Identificar las mejores opciones de seguro
de salud y cobertura
• Hacer un seguimiento de sus beneficios
del seguro de salud
• Prepararlo para el tratamiento con materiales
educativos y recipientes para objetos
cortantes y punzantes (si fuera necesario)
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
¿Cuáles son los posibles efectos secundarios de EGRIFTA
®
?
EGRIFTA
®
puede causar efectos secundarios graves, incluidos
los siguientes:
• Reacciones en el lugar de la inyección. Rote el área de la inyección
para disminuir su riesgo de reacciones en el lugar de la inyección.
Llame a su proveedor de atención médica para obtener asesoría
médica si tiene los siguientes síntomas alrededor del sitio de la
inyección:
enrojecimiento
picazón
dolor
irritación
sangrado
sarpullido
hinchazón

Una transformación desde su interior
®
Una transformación desde su interior
EGRIFTA.com
Regreso del exceso de VAT
Las personas que usaron EGRIFTA®
de forma continua por 1 año
mantuvieron sus resultados durante
este período de tiempo.
UNA REDUCCIÓN
DEL
18%
EN
Promedio del Ensayo 1
VAT
UNA REDUCCIÓN
DEL
14%
EN
Promedio del Ensayo 2
VAT
Cómo ayudar a reducir el exceso de
tejido adiposo visceral (VAT) abdominal
(y controlarlo)
EGRIFTA® (tesamorelin inyectable) es el único tratamiento aprobado por
la Administración de Alimentos y Medicamentos de los Estados Unidos
(US Food and Drug Administration, FDA) para tratar el exceso de VAT
abdominal en personas con VIH que tienen lipodistrofia. EGRIFTA® es
un medicamento recetado inyectable via subcutánea (debajo de la piel),
una vez al día en la zona inferior de su abdomen.
Se han realizado estudios de EGRIFTA® en 2 ensayos clínicos de
personas con VIH que desarrollaron lipodistrofia.
• Cada ensayo duró 6 meses, con una fase de extensión de
6 meses adicionales
• Los pacientes de estos ensayos presentaron reducciones
significativas del tejido adiposo visceral (VAT)
EGRIFTA® no está indicado para el manejo de la pérdida de peso.
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
Informe a su proveedor de atención médica sobre todos los
medicamentos que toma, incluidos los que se venden con receta
y los de venta libre, vitaminas y suplementos a base de hierbas.
EGRIFTA® puede afectar el funcionamiento de otros medicamentos
y estos mismos pueden afectar el funcionamiento de EGRIFTA ®.
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
Nuestro personal de enfermería de EGRIFTA ASSIST ®
está listo para ayudar a:
• Brindarle capacitación sobre las técnicas
apropiadas de inyección y reconstitución
• Asesorarlo sobre cómo comenzar
y permanecer bajo tratamiento
• Contestar preguntas frecuentes relacionadas
con el tratamiento través de su tratamiento
Asistencia de expertos con tan solo una llamada
Los asesores están listos para comunicarse con usted
y ayudar a contestar sus preguntas.
1-844-EGRIFTA (1-844-347-4382)
De lunes a viernes, de 8a.m. a 8p.m. hora del Este
Los mensajes de correos de voz se responderán dentro
del próximo día laborable.
Los efectos secundarios más frecuentes de EGRIFTA
®
incluyen:
• dolor de las articulaciones
• dolor en los brazos y en las piernas
• hinchazón en las piernas
• dolor muscular
• hormigueo, adormecimiento y pinchazos
• náuseas
• vómitos
• sarpullido
• picazón
Informe a su proveedor de atención médica si tiene cualquier efecto
secundario que le cause molestias o que persista.
Estos no son todos los efectos secundarios posibles de EGRIFTA ®.
Para recibir más información, consulte con su proveedor de atención
médica o a su farmacéutico.
EGRIFTA.com
No pierda tiempo. Hable con su HCP hoy.
Pregunte a su proveedor de atención médica (HCP, en inglés)
sobre
EGRIFTA
®
(tesamorelin inyectable)
.
Para obtener más información, vaya a EGRIFTA.com.
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
Llame a su médico para que lo asesore acerca de los efectos
secundarios. Para informar sobre sospechas de reacciones adversas,
comuníquese con EGRIFTA ASSIST ® al número de teléfono gratuito
1-844-EGRIFTA (1-844-347-4382), o bien con la FDA al
1-800-FDA-1088 o en www.fda.gov/medwatch.
¿Cómo debo almacenar EGRIFTA ®?
• EGRIFTA® viene en dos cajas dispensadas por la farmacia:
Almacene los frascos de la caja del medicamento EGRIFTA ®
en el refrigerador entre 2 °C y 8 °C (36 °F y 46 °F)
Almacene la caja de agua estéril para inyección, las jeringas
y las agujas a temperatura ambiente entre 20 °C y 25 °C
(68 °F y 77 °F).
• Guarde los frascos de EGRIFTA® en la caja del medicamento
alejados de la luz.
• No se debe congelar.
• No use EGRIFTA® después de la fecha de vencimiento impresa en
la caja y en las etiquetas del frasco.
• Después de mezclar, use EGRIFTA® de inmediato y descarte todo
el EGRIFTA ® restante. No almacene la mezcla de EGRIFTA ®.
Además, deseche el frasco usado de agua estéril para inyección.
Mantenga EGRIFTA ® y todos los medicamentos fuera del
alcance de los niños.
Información general sobre el uso eficaz y seguro de EGRIFTA ®
Los medicamentos a veces se recetan con fines distintos de los que se
indican en la Información para el paciente. No use EGRIFTA® para una
afección para la cual no fue recetado. No administre EGRIFTA® a otras
personas, aunque tengan los mismos síntomas que usted. Hacer esto
puede causarles daño.
No comparta su jeringa o agujas de EGRIFTA ® con otras personas.
Usted puede provocarles una infección o infectarse por ellos.
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
EGRIFTA ® es una marca registrada de Theratechnologies Inc.
© 2016 Theratechnologies Inc. Todos los derechos reservados. 210-01-04/16
Si es VIH positivo, es posible que se
enfrente con otro desafío médico
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
¿Qué es EGRIFTA ®?
• EGRIFTA ® es un medicamento recetado inyectable para reducir
el exceso de grasa abdominal en pacientes infectados con el VIH que
tienen lipodistrofia. EGRIFTA® contiene un factor de liberación de la
hormona de crecimiento (growth hormone-releasing factor, GRF).
• No se han realizado estudios sobre el impacto y seguridad de EGRIFTA ®
en la salud cardiovascular.
• EGRIFTA® no está indicado para el manejo de la pérdida de peso.
• No se sabe si EGRIFTA® ayuda a mejorar la adhesión al tratamiento del VIH.
• No se sabe si EGRIFTA® es seguro y eficaz en niños.
No se recomienda el uso de EGRIFTA ® en niños.
Por favor, lea el prospecto,
junto con la información para
el paciente y las instrucciones
de uso adjuntas.
Aprenda más sobre EGRIFTA ® (tesamorelin inyectable)
Es el único tratamiento, aprobado por la FDA, para tratar
el exceso de tejido adiposo visceral en personas
VIH positivas que tienen lipodistrofia
Obtenga más información
en EGRIFTA.com
Ayude a reducir el exceso de tejido
adiposo visceral en su abdomen
(VAT, en inglés)

Una transformación desde su interior
®
Una transformación desde su interior
EGRIFTA.com
Regreso del exceso de VAT
Las personas que usaron EGRIFTA®
de forma continua por 1 año
mantuvieron sus resultados durante
este período de tiempo.
UNA REDUCCIÓN
DEL
18%
EN
Promedio del Ensayo 1
VAT
UNA REDUCCIÓN
DEL
14%
EN
Promedio del Ensayo 2
VAT
Cómo ayudar a reducir el exceso de
tejido adiposo visceral (VAT) abdominal
(y controlarlo)
EGRIFTA® (tesamorelin inyectable) es el único tratamiento aprobado por
la Administración de Alimentos y Medicamentos de los Estados Unidos
(US Food and Drug Administration, FDA) para tratar el exceso de VAT
abdominal en personas con VIH que tienen lipodistrofia. EGRIFTA® es
un medicamento recetado inyectable via subcutánea (debajo de la piel),
una vez al día en la zona inferior de su abdomen.
Se han realizado estudios de EGRIFTA® en 2 ensayos clínicos de
personas con VIH que desarrollaron lipodistrofia.
• Cada ensayo duró 6 meses, con una fase de extensión de
6 meses adicionales
• Los pacientes de estos ensayos presentaron reducciones
significativas del tejido adiposo visceral (VAT)
EGRIFTA® no está indicado para el manejo de la pérdida de peso.
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
Informe a su proveedor de atención médica sobre todos los
medicamentos que toma, incluidos los que se venden con receta
y los de venta libre, vitaminas y suplementos a base de hierbas.
EGRIFTA® puede afectar el funcionamiento de otros medicamentos
y estos mismos pueden afectar el funcionamiento de EGRIFTA ®.
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
Nuestro personal de enfermería de EGRIFTA ASSIST ®
está listo para ayudar a:
• Brindarle capacitación sobre las técnicas
apropiadas de inyección y reconstitución
• Asesorarlo sobre cómo comenzar
y permanecer bajo tratamiento
• Contestar preguntas frecuentes relacionadas
con el tratamiento través de su tratamiento
Asistencia de expertos con tan solo una llamada
Los asesores están listos para comunicarse con usted
y ayudar a contestar sus preguntas.
1-844-EGRIFTA (1-844-347-4382)
De lunes a viernes, de 8a.m. a 8p.m. hora del Este
Los mensajes de correos de voz se responderán dentro
del próximo día laborable.
Los efectos secundarios más frecuentes de EGRIFTA
®
incluyen:
• dolor de las articulaciones
• dolor en los brazos y en las piernas
• hinchazón en las piernas
• dolor muscular
• hormigueo, adormecimiento y pinchazos
• náuseas
• vómitos
• sarpullido
• picazón
Informe a su proveedor de atención médica si tiene cualquier efecto
secundario que le cause molestias o que persista.
Estos no son todos los efectos secundarios posibles de EGRIFTA ®.
Para recibir más información, consulte con su proveedor de atención
médica o a su farmacéutico.
EGRIFTA.com
No pierda tiempo. Hable con su HCP hoy.
Pregunte a su proveedor de atención médica (HCP, en inglés)
sobre
EGRIFTA
®
(tesamorelin inyectable)
.
Para obtener más información, vaya a EGRIFTA.com.
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
Llame a su médico para que lo asesore acerca de los efectos
secundarios. Para informar sobre sospechas de reacciones adversas,
comuníquese con EGRIFTA ASSIST ® al número de teléfono gratuito
1-844-EGRIFTA (1-844-347-4382), o bien con la FDA al
1-800-FDA-1088 o en www.fda.gov/medwatch.
¿Cómo debo almacenar EGRIFTA ®?
• EGRIFTA® viene en dos cajas dispensadas por la farmacia:
Almacene los frascos de la caja del medicamento EGRIFTA ®
en el refrigerador entre 2 °C y 8 °C (36 °F y 46 °F)
Almacene la caja de agua estéril para inyección, las jeringas
y las agujas a temperatura ambiente entre 20 °C y 25 °C
(68 °F y 77 °F).
• Guarde los frascos de EGRIFTA® en la caja del medicamento
alejados de la luz.
• No se debe congelar.
• No use EGRIFTA® después de la fecha de vencimiento impresa en
la caja y en las etiquetas del frasco.
• Después de mezclar, use EGRIFTA® de inmediato y descarte todo
el EGRIFTA ® restante. No almacene la mezcla de EGRIFTA ®.
Además, deseche el frasco usado de agua estéril para inyección.
Mantenga EGRIFTA ® y todos los medicamentos fuera del
alcance de los niños.
Información general sobre el uso eficaz y seguro de EGRIFTA ®
Los medicamentos a veces se recetan con fines distintos de los que se
indican en la Información para el paciente. No use EGRIFTA® para una
afección para la cual no fue recetado. No administre EGRIFTA® a otras
personas, aunque tengan los mismos síntomas que usted. Hacer esto
puede causarles daño.
No comparta su jeringa o agujas de EGRIFTA ® con otras personas.
Usted puede provocarles una infección o infectarse por ellos.
Por favor, lea el prospecto, junto con la información
para el paciente y las instrucciones de uso adjuntas.
EGRIFTA ® es una marca registrada de Theratechnologies Inc.
© 2016 Theratechnologies Inc. Todos los derechos reservados. 210-01-04/16
Si es VIH positivo, es posible que se
enfrente con otro desafío médico
INFORMACIÓN IMPORTANTE SOBRE RIESGOS
¿Qué es EGRIFTA ®?
• EGRIFTA ® es un medicamento recetado inyectable para reducir
el exceso de grasa abdominal en pacientes infectados con el VIH que
tienen lipodistrofia. EGRIFTA® contiene un factor de liberación de la
hormona de crecimiento (growth hormone-releasing factor, GRF).
• No se han realizado estudios sobre el impacto y seguridad de EGRIFTA ®
en la salud cardiovascular.
• EGRIFTA® no está indicado para el manejo de la pérdida de peso.
• No se sabe si EGRIFTA® ayuda a mejorar la adhesión al tratamiento del VIH.
• No se sabe si EGRIFTA® es seguro y eficaz en niños.
No se recomienda el uso de EGRIFTA ® en niños.
Por favor, lea el prospecto,
junto con la información para
el paciente y las instrucciones
de uso adjuntas.
Aprenda más sobre EGRIFTA ® (tesamorelin inyectable)
Es el único tratamiento, aprobado por la FDA, para tratar
el exceso de tejido adiposo visceral en personas
VIH positivas que tienen lipodistrofia
Obtenga más información
en EGRIFTA.com
Ayude a reducir el exceso de tejido
adiposo visceral en su abdomen
(VAT, en inglés)

DATOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN
Estos datos destacados no incluyen toda la información necesaria para usar
EGRIFTA® de manera segura y eficaz. Consulte la información de
prescripción completa para EGRIFTA®.
EGRIFTA® (tesamorelin inyectable) para uso subcutáneo
Aprobación inicial en EE. UU.: 2010
____________________ INDICACIONES Y USO ___________________
EGRIFTA® es un análogo del factor liberador de la hormona de crecimiento
(hormone growth releasing factor, GRF) indicado para la reducción del exceso
de grasa abdominal en pacientes infectados por VIH con lipodistrofia. (1)
Limitaciones de uso:
No se ha estudiado la seguridad y el beneficio cardiovascular a largo plazo
de EGRIFTA®. (1)
No está indicado para el manejo de la pérdida de peso (efecto neutro en el
peso). (1)
No existen datos que respalden una mejora en el cumplimiento de las terapias
antirretrovirales en pacientes VIH positivo que toman EGRIFTA®. (1)
_____________ DOSIFICACIÓN Y ADMINISTRACIÓN ____________
La dosis recomendada de EGRIFTA® es de 2 mg inyectados por vía
subcutánea una vez al día. (2.1)
Reconstituir con el diluyente suministrado según las recomendaciones. (2.2)
Administrar por vía subcutánea en la piel abdominal, alternando los lugares
de aplicación. (2.3)
___________ FORMAS Y CONCENTRACIÓN DE DOSIS ___________
Cada frasco de EGRIFTA® contiene 1 mg de tesamorelin (3). Otro frasco
contiene el diluyente para la reconstitución, agua estéril para inyección,
Farmacopea de Estados Unidos (United States Pharmacopea, USP). (3)
___________________ CONTRAINDICACIONES __________________
Inhibición del eje hipotálamico-hipofisiario debido a hipofisectomía,
hipopituitarismo o tumor/cirugía de hipófisis, radioterapia en la cabeza o
traumatismo de cráneo (4.1)
Neoplasia maligna activa (4.2)
Hipersensibilidad conocida a tesamorelin y/o manitol (4.3)
Embarazo (4.4)
_____________ ADVERTENCIAS Y PRECAUCIONES ______________
Neoplasias: La neoplasia maligna preexistente debe estar inactiva y se debe
haber completado su tratamiento antes de comenzar la terapia con
EGRIFTA®. (5.1)
Aumento del IGF-1: Controlar en forma regular en todos los pacientes.
Considerar la discontinuación en pacientes con aumentos persistentes. (5.2)
Retención de líquidos: Puede incluir edema, artralgia y síndrome del túnel
carpiano. (5.3)
Intolerancia a la glucosa: Puede desarrollarse con el uso de EGRIFTA®.
Evaluar el estado de la glucosa antes de la terapia con EGRIFTA® y durante
esta. (5.4)
Reacciones de hipersensibilidad (p. ej., erupción, urticaria): Aconsejar a los
pacientes solicitar atención médica inmediata ante una sospecha. (5.5)
Reacciones en el lugar de la inyección: Aconsejar a los pacientes alternar
los lugares de aplicación. (5.6)
Enfermedad crítica aguda: Considerar la discontinuación. (5.7)
__________________ REACCIONES ADVERSAS __________________
Las reacciones adversas reportadas con más frecuencia (>5 % y más frecuentes
que con placebo): Artralgia, eritema en el lugar de la inyección, prurito en el lugar
de la inyección, dolor en las extremidades, edema periférico y mialgia. (6.1)
Para reportar acerca de SOSPECHAS DE REACCIONES ADVERSAS,
comuníquese con ® al número de teléfono gratuito 1-
844-EGRIFTA (1-844-347-4382), o bien con la FDA al 1-800-FDA-1088 o
en www.fda.gov/medwatch
____________ INTERACCIONES MEDICAMENTOSAS ____________
Fármacos metabolizados por el citocromo P450: Controlar atentamente si
se usan con EGRIFTA®. (7.1)
____________ USO EN POBLACIONES ESPECÍFICAS _____________
Madres en período de lactancia: Las madres infectadas por VIH-1 no deben
amamantar para evitar la posibilidad de contagio posnatal del VIH-1. (8.3)
Uso pediátrico: No se ha establecido la seguridad y la eficacia. (8.4)
Consulte la INFORMACIÓN DE ASESORAMIENTO PARA EL
PACIENTE y la ficha técnica con información para el paciente aprobada
por la FDA en la Sección 17.
Revisado: 06/2015
INFORMACIÓN DE PRESCRIPCIÓN COMPLETA:
CONTENIDO*
1 INDICACIONES Y USO
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Información general de dosificación
2.2 Procedimiento de reconstitución
2.3 Administración
3 FORMAS Y CONCENTRACIÓN DE DOSIS
4 CONTRAINDICACIONES
4.1 Inhibición del eje hipotálamico-hipofisiario
4.2 Neoplasia maligna activa
4.3 Hipersensibilidad
4.4 Embarazo
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Neoplasias
5.2 Aumento del IGF-1
5.3 Retención de líquidos
5.4 Intolerancia a la glucosa
5.5 Reacciones de hipersensibilidad
5.6 Reacciones en el lugar de la inyección
5.7 Enfermedad crítica aguda
6 REACCIONES ADVERSAS
6.1 Experiencia en ensayos clínicos
6.2 Inmunogenicidad
7 INTERACCIONES MEDICAMENTOSAS
7.1 Fármacos metabolizados por el citocromo P450
7.2 11β-hidroxiesteroide deshidrogenasa tipo 1
(11βHSD-1)
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
8.3 Madres en período de lactancia
8.4 Uso pediátrico
8.5 Uso geriátrico
8.6 Insuficiencia renal y hepática
10 SOBREDOSIS
11 DESCRIPCIÓN
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
12.2 Farmacodinámica
12.3 Farmacocinética
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la
fertilidad
14 ESTUDIOS CLÍNICOS
16 PRESENTACIÓN/ALMACENAMIENTO Y
MANIPULACIÓN
17 INFORMACIÓN DE ORIENTACIÓN PARA EL
PACIENTE
*Las secciones o subsecciones omitidas en el prospecto, no están
descriptas en la lista.
INFORMACIÓN DE PRESCRIPCIÓN COMPLETA
1 INDICACIONES Y USO
EGRIFTA® (tesamorelin inyectable) está indicado para la reducción del exceso de grasa abdominal en
pacientes infectados por VIH con lipodistrofia (consulte Estudios clínicos [14]).
Limitaciones de uso:
Debido a que no se ha estudiado y se desconoce la seguridad cardiovascular a largo plazo y el
posible beneficio cardiovascular a largo plazo del tratamiento con EGRIFTA®, se debe
considerar cuidadosamente si se continuará el tratamiento con EGRIFTA® en pacientes que no
demuestran una respuesta de eficacia clara, de acuerdo con el grado de reducción en el tejido
adiposo visceral medido por la circunferencia de la cintura o mediante exploración por
tomografía computarizada (computerized tomography, CT).
EGRIFTA® no está indicado para el manejo de la pérdida de peso (efecto neutro en el peso).
No existen datos que respalden una mejora en el cumplimiento de las terapias antirretrovirales
en pacientes VIH positivo que toman EGRIFTA®.
2 DOSIFICACIÓN Y ADMINISTRACIÓN
2.1 Información general de dosificación
La dosis recomendada de EGRIFTA® es de 2 mg inyectados por vía subcutánea una vez al día.
El lugar de inyección recomendado es el abdomen. Los lugares de inyección deben alternarse en
diferentes áreas del abdomen. No se debe inyectar en tejido cicatricial, en moretones o en el ombligo.
2.2 Procedimiento de reconstitución
En el folleto de INSTRUCCIONES DE USO incluido en las cajas que contienen EGRIFTA® y el
diluyente se proveen instrucciones detalladas para reconstituir EGRIFTA®.
Se deben reconstituir dos frascos de 1 mg de EGRIFTA® con el diluyente suministrado con el
producto.
Reconstituir el primer frasco de 1 mg de EGRIFTA® con 2.2 ml de diluyente. Mezclar haciendo rodar el
frasco entre las manos con cuidado durante 30 segundos. No agitar. Reconstituir el segundo frasco de
1 mg de EGRIFTA® con toda la solución del primer frasco. Mezclar haciendo rodar el frasco entre las
manos con cuidado durante 30 segundos. No agitar.
Administrar EGRIFTA® inmediatamente después de la reconstitución y desechar toda la solución de
EGRIFTA® no utilizada. Si no se usa inmediatamente, la solución de EGRIFTA® reconstituida debe
desecharse. No congelar ni refrigerar la solución de EGRIFTA® reconstituida.
2.3 Administración
Siempre se debe inspeccionar visualmente la solución de EGRIFTA®reconstituida para detectar
partículas y alteración del color antes de la administración, si la solución y el recipiente lo permiten.
Solo se debe inyectar EGRIFTA® si la solución es transparente, incolora y no contiene partículas.
EGRIFTA® debe inyectarse por vía subcutánea en la piel del abdomen. Los lugares de inyección deben
alternarse en diferentes áreas del abdomen. No se debe inyectar en tejido cicatricial, en moretones o en
el ombligo.
3 FORMAS Y CONCENTRACIÓN DE DOSIS
EGRIFTA® (tesamorelin inyectable) se suministra en un frasco que contiene 1 mg de tesamorelin en
forma de polvo liofilizado. El diluyente (agua estéril para inyección, USP 10 ml) se provee en un frasco
separado.
4 CONTRAINDICACIONES
4.1 Inhibición del eje hipotálamico-hipofisiario
EGRIFTA® está contraindicado en pacientes con inhibición del eje hipotálamico-hipofisiario debido a
hipofisectomía, hipopituitarismo o tumor/cirugía de hipófisis, radioterapia en la cabeza o traumatismo de
cráneo.
4.2 Neoplasia maligna activa
EGRIFTA® está contraindicado en pacientes con neoplasias malignas activas (recientemente
diagnosticadas o recurrentes). Cualquier neoplasia maligna preexistente debe estar inactiva y se debe
haber completado su tratamiento antes de iniciar la terapia con EGRIFTA®.
4.3 Hipersensibilidad
EGRIFTA® está contraindicado en pacientes con hipersensibilidad conocida a tesamorelin y/o manitol
(un excipiente)(consulte Advertencias y precauciones [5.5]).
4.4 Embarazo
EGRIFTA® está contraindicado en mujeres embarazadas. Durante el embarazo, el tejido adiposo
visceral aumenta debido a los cambios metabólicos y hormonales normales. Modificar este cambio
fisiológico del embarazo con EGRIFTA® no ofrece ningún beneficio conocido y podría provocar daño
fetal. La administración de acetato de tesamorelin a ratas durante la organogénesis y la lactancia provocó
hidrocefalia en las crías a una dosis aproximadamente dos y cuatro veces mayor que la dosis clínica,
respectivamente, en función de la exposición al fármaco medida (área bajo la curva [area under the
curve, AUC]). Si ocurre un embarazo, discontinuar la terapia con EGRIFTA®. Si este fármaco se usa
durante el embarazo, o si la paciente queda embarazada mientras toma este fármaco, se debe informar a
la paciente sobre el riesgo potencial para el feto (consulte Uso en poblaciones específicas [8.1]).
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Neoplasias
EGRIFTA® induce la liberación de la hormona de crecimiento (growth hormone, GH) endógena, un
factor de crecimiento conocido. Por lo tanto, los pacientes con una neoplasia maligna activa no deben
ser tratados con EGRIFTA® (consulte Contraindicaciones [4.2]).
En pacientes con antecedentes de neoplasias no malignas, la terapia con EGRIFTA® debe iniciarse luego
de una evaluación minuciosa del posible beneficio del tratamiento. En pacientes con antecedentes de
neoplasias malignas tratadas y estables, la terapia con EGRIFTA® debe iniciarse solo luego de una
evaluación minuciosa del posible beneficio del tratamiento en relación con el riesgo de reactivación de
la neoplasia maligna subyacente.
Asimismo, la decisión de iniciar el tratamiento con EGRIFTA® se debe considerar minuciosamente
debido al riesgo elevado de desarrollo de neoplasias malignas en pacientes VIH positivos.
5.2 Aumento del IGF-1
EGRIFTA® estimula la producción de GH y aumenta el IGF-1 sérico. Debido a que el IGF-1 es un
factor de crecimiento y se desconoce el efecto de los aumentos prolongados en los niveles de IGF-1 en
el desarrollo o la progresión de neoplasias malignas, deben controlarse atentamente los niveles de IGF-1
durante la terapia con EGRIFTA®. Se debe considerar minuciosamente la discontinuación de
EGRIFTA® en pacientes con aumentos persistentes de los niveles de IGF-1 (p. ej., puntaje de desviación
estándar [standard deviation score, SDS] >3 ), especialmente si la respuesta de eficacia no es sólida
(p. ej., en función de los cambios en el tejido adiposo visceral medidos por la circunferencia de la
cintura o mediante exploración por CT).
Durante los ensayos clínicos se controló a los pacientes cada tres meses. Entre los pacientes que
recibieron EGRIFTA® durante 26 semanas, el 47.4 % tuvo niveles de IGF-1 mayores que 2 en el puntaje
de desviación estándar (SDS) y el 35.6 % tuvo SDS >3, y este efecto se observó con tan solo 13 semanas
de tratamiento. Entre los pacientes que continuaron bajo tratamiento con EGRIFTA® por un total de
52 semanas, al final del tratamiento, el 33.7 % tenía IGF-1 SDS >2 y el 22.6 % tenía IGF-1 SDS >3.
5.3 Retención de líquidos
Puede ocurrir retención de líquidos durante la terapia con EGRIFTA®, lo cual se cree que está
relacionado con la inducción de la secreción de GH. Se manifiesta como un aumento de la turgencia del
tejido y molestia musculoesquelética que provoca una variedad de reacciones adversas (p. ej., edema,
artralgia, síndrome del túnel carpiano) que son pasajeras o se resuelven con la discontinuación
del tratamiento.
5.4 Intolerancia a la glucosa
El tratamiento con EGRIFTA® puede provocar intolerancia a la glucosa. Durante los ensayos clínicos de
fase 3, los porcentajes de pacientes con HbA1c elevada (≥6.5 %) desde el inicio a la semana 26 fueron
del 4.5 % y el 1.3 % en los grupos de EGRIFTA® y placebo, respectivamente. Se observó un mayor
riesgo de desarrollar diabetes con EGRIFTA® (nivel de HbA1c ≥6.5 %) en relación con el placebo (razón
de riesgo y probabilidades por intención de tratar de 3.3 [CI 1.4, 9.6]). Por lo tanto, el estado de la
glucosa se debe evaluar minuciosamente antes de iniciar el tratamiento con EGRIFTA®. Además, se
debe controlar periódicamente a todos los pacientes tratados con EGRIFTA® para detectar cambios en el
metabolismo de la glucosa a fin de diagnosticar a quienes desarrollen una deficiencia de tolerancia a la
glucosa o diabetes. La diabetes es un factor de riesgo cardiovascular conocido y los pacientes que
desarrollan intolerancia a la glucosa tienen un mayor riesgo de desarrollar diabetes. Se debe ejercer
precaución al tratar con EGRIFTA® a pacientes VIH positivos con lipodistrofia si desarrollan
intolerancia a la glucosa o diabetes, y se debe considerar minuciosamente la discontinuación del
tratamiento con EGRIFTA® en pacientes que no demuestran una respuesta de eficacia clara de acuerdo
con el grado de reducción en el tejido adiposo visceral medido por la circunferencia de la cintura o
mediante exploración por CT.
Dado que EGRIFTA® aumenta el IGF-1, se debe controlar, a intervalos regulares, a los pacientes con
diabetes que están recibiendo tratamiento constante con EGRIFTA® para detectar el posible desarrollo o
empeoramiento de retinopatía.
5.5 Reacciones de hipersensibilidad
Pueden ocurrir reacciones de hipersensibilidad en pacientes tratados con EGRIFTA®. Ocurrieron
reacciones de hipersensibilidad en el 3.6 % de los pacientes con lipodistrofia asociada a VIH tratados
con EGRIFTA® en los ensayos clínicos de fase 3. Estas reacciones incluyeron prurito, eritema, sofocos,
urticaria y otras erupciones. En casos de sospecha de reacciones de hipersensibilidad se debe aconsejar a
los pacientes que soliciten atención médica sin demora, y el tratamiento con EGRIFTA® se debe
discontinuar de inmediato.
5.6 Reacciones en el lugar de la inyección
El tratamiento con EGRIFTA® puede causar reacciones en el lugar de la inyección, que incluyen
eritema, prurito, dolor, irritación y moretones en el lugar de la inyección. La incidencia de reacciones en
el lugar de la inyección fue del 24.5 % en pacientes tratados con EGRIFTA® y del 14.4 % en pacientes
tratados con placebo durante las primeras 26 semanas de tratamiento en los ensayos clínicos de fase 3.
En el caso de los pacientes que continuaron EGRIFTA® por otras 26 semanas, la incidencia de
reacciones en el lugar de la inyección fue del 6.1 %. Para reducir la incidencia de reacciones en el lugar
de la inyección, se recomienda alternar el lugar de la inyección a diferentes áreas del abdomen.
5.7 Enfermedad crítica aguda
Después del tratamiento con cantidades farmacológicas de la hormona de crecimiento, se ha informado
aumento de la mortalidad en pacientes con enfermedad crítica aguda debido a complicaciones
posteriores a una cirugía a corazón abierto, cirugía abdominal o politraumatismo accidental, o en
quienes padecen insuficiencia respiratoria aguda. No se ha estudiado a EGRIFTA® en pacientes con
enfermedad crítica aguda. Dado que EGRIFTA® estimula la producción de la hormona de crecimiento,
se debe considerar minuciosamente la discontinuación de EGRIFTA® en pacientes con enfermedad
crítica.
6 REACCIONES ADVERSAS
Las reacciones adversas informadas con más frecuencia son reacciones de hipersensibilidad (p. ej.,
erupción, urticaria) debido al efecto de la GH (p. ej., artralgia, dolor en las extremidades, edema
periférico, hiperglucemia, síndrome del túnel carpiano), reacciones en el lugar de la inyección (eritema,
prurito, dolor, urticaria, irritación, hinchazón, hemorragia en el lugar de la inyección).
Durante las primeras 26 semanas de tratamiento (fase principal) ocurrieron interrupciones como
consecuencia de reacciones adversas en el 9.6 % de los pacientes que recibían EGRIFTA® y en el 6.8 %
de los pacientes que recibían placebo. Aparte de los pacientes con reacciones de hipersensibilidad
identificados durante los estudios y que discontinuaron la participación en el estudio según el protocolo
(2.2 %), los motivos más frecuentes de discontinuación del tratamiento con EGRIFTA® fueron
reacciones adversas debido al efecto de la GH (4.2 %) y reacciones en el lugar de la inyección (4.6 %).
Durante las siguientes 26 semanas de tratamiento (fase de extensión) ocurrieron discontinuaciones como
consecuencia de eventos adversos en el 2.4 % de los pacientes en el grupo de T-T (pacientes tratados
con tesamorelin de la semana 0 a la 26 y con tesamorelin de la semana 26 a la 52) y el 5.2 % de los
pacientes en el grupo de T-P (pacientes tratados con tesamorelin de la semana 0 a la 26 y con placebo de
la semana 26 a la 52).
6.1 Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones
adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las
tasas en los ensayos clínicos de otro fármaco, y posiblemente no reflejen las tasas observadas en la
práctica.
Setecientos cuarenta pacientes VIH positivos, con lipodistrofia y exceso de grasa abdominal se
expusieron a EGRIFTA® en los ensayos clínicos de fase 3; de estos, 543 recibieron EGRIFTA® durante
la fase inicial de 26 semanas, controlada con placebo (consulte Estudios clínicos [14]).
En la Tabla 1 se presentan las reacciones adversas que ocurrieron con más frecuencia con EGRIFTA® en
relación con el placebo y tuvieron una incidencia de ≥1 % durante las primeras 26 semanas entre todos
los estudios.

Tabla 1. Reacciones adversas reportadas en ≥1 % y más frecuentes en pacientes tratados con
EGRIFTA® que en pacientes tratados con placebo durante la fase principal de 26 semanas
(estudios combinados)
Incidencia de pacientes (%) con reacciones adversas
al fármaco
Clasificación de sistema de órganos
Término preferido
EGRIFTA®
(N=543)
Placebo
(N=263)
Trastornos musculoesqueléticos y del
tejido conjuntivo
Artralgia
Dolor en las extremidades
Mialgia
Dolor musculoesquelético
Rigidez musculoesquelética
Rigidez de las articulaciones
Espasmos musculares
Hinchazón de las articulaciones
13.3
6.1
5.5
1.8
1.7
1.5
1.1
1.1
11.0
4.6
1.9
0.8
0.4
0.8
0.8
0.0
Trastornos generales y afecciones en
el sitio de administración
Eritema en el lugar de la inyección
Prurito en el lugar de la inyección
Edema periférico
Dolor en el lugar de la inyección
Irritación en el lugar de la inyección
Dolor
Hemorragia en el lugar de la
inyección
Urticaria en el lugar de la inyección
Hinchazón en el lugar de la
inyección
Reacción en el lugar de la inyección
Dolor en el pecho
Erupción en el lugar de la inyección
8.5
7.6
6.1
4.1
2.9
1.7
1.7
1.7
1.5
1.3
1.1
1.1
2.7
0.8
2.3
3.0
1.1
1.1
0.4
0.4
0.4
0.8
0.8
0.0
Trastornos del sistema nervioso
Parestesia
Hipoestesia
Síndrome del túnel carpiano
4.8
4.2
1.5
2.3
1.5
0.0
Trastornos gastrointestinales
Náuseas
Vómitos
Dispepsia
Dolor abdominal superior
4.4
2.6
1.7
1.1
3.8
0.0
0.8
0.8
Trastornos cardíacos
Palpitaciones
1.1
0.4
Trastornos psiquiátricos
Depresión
2.0
1.5
Trastornos de la piel y el tejido
subcutáneo

Incidencia de pacientes (%) con reacciones adversas
al fármaco
Clasificación de sistema de órganos
Término preferido
EGRIFTA®
(N=543)
Placebo
(N=263)
Erupción
Prurito
Sudoración nocturna
3.7
2.4
1.1
1.5
1.1
0.4
Trastornos vasculares
Hipertensión
1.3
0.8
Lesión, intoxicación y complicaciones
del procedimiento
Distensión muscular
1.1
0.0
Investigaciones
Aumento de la creatina
fosfoquinasa en sangre
1.5
0.4
Los niveles medios de glucosa sanguínea en ayunas e insulina en ayunas no fueron significativamente
diferentes entre pacientes tratados con EGRIFTA® y pacientes tratados con placebo después de
26 semanas de tratamiento.
En los ensayos clínicos de fase 3 de EGRIFTA®, la HbA1c inicial (semana 0) media fue de 5.26 % entre
los pacientes del grupo de EGRIFTA® y de 5.28 % entre los pacientes del grupo del placebo. En la
semana 26, la HbA1c media fue más alta entre los pacientes tratados con EGRIFTA® en comparación
con el placebo (5.39 % frente a 5.28 % para los grupos de EGRIFTA® y placebo, respectivamente, con
una diferencia media del tratamiento de 0.12 %, p=0.0004). Los pacientes que recibían EGRIFTA®
tuvieron un mayor riesgo de desarrollar diabetes (nivel de HbA1c ≥6.5 %) en comparación con el placebo
(4.5 % frente a 1.3 %), con una razón de riesgo de 3.3 (intervalo de confianza [confidence interval, CI]
de 1.4, 9.6).
En la Tabla 2 se presentan las reacciones adversas observadas durante la semana 26 a 52 de los ensayos
clínicos de fase 3, que tuvieron una incidencia de ≥1 % y se observaron con más frecuencia con
EGRIFTA® en relación con el placebo:

Tabla 2. Reacciones adversas reportadas en ≥1 % y más frecuentes en pacientes tratados con
EGRIFTA® que en pacientes tratados con placebo durante la fase de extensión de 26 semanas de
los estudios combinados (semana 26 a semana 52 de los estudios)
Incidencia de pacientes (%) con reacciones adversas al fármaco
Clasificación de sistema de
órganos
Término preferido
T-T1 (semana 26 a 52)
(N=246)
T-P2(semana 26 a 52)
(N=135)
Trastornos
musculoesqueléticos y del
tejido conjuntivo
Dolor en las extremidades
Mialgia
3.3
1.2
0.7
0.0
Trastornos generales y
afecciones en el sitio de
administración
Prurito en el lugar de la
inyección
Edema periférico
Eritema en el lugar de la
inyección
2.0
2.0
1.2
0.0
0.0
0.0
Trastornos del sistema
nervioso
Parestesia
Hipoestesia
Neuropatía periférica
1.6
1.6
1.6
1.5
0.7
1.5
Trastornos gastrointestinales
Vómitos
2.0
0.7
Trastornos psiquiátricos
Depresión
Insomnio
1.6
1.2
0.7
0.0
Trastornos de la piel y el
tejido subcutáneo
Prurito
Urticaria
Sudoración nocturna
1.2
1.2
1.2
0.7
0.0
0.0
Trastornos vasculares
Hipertensión
Sofocos
1.6
1.2
1.5
0.7
1T-T = tesamorelin para la semana 0 a 26 y tesamorelin para la semana 26 a 52
2T-P = tesamorelin para la semana 0 a 26 y placebo para la semana 26 a 52
Para los pacientes que continuaron desde la semana 26 a la 52, los niveles medios de glucosa sanguínea
en ayunas, insulina en ayunas y HbA1c no fueron diferentes entre los grupos de T-T y T-P.
6.2 Inmunogenicidad
Como ocurre con todas las proteínas y péptidos terapéuticos, existe la posibilidad de que se desarrollen
anticuerpos contra EGRIFTA® in vivo. En ensayos clínicos de fase 3 combinados se detectaron
anticuerpos IgG contra tesamorelin en el 49.5 % de los pacientes tratados con EGRIFTA® durante
26 semanas y en el 47.4 % de los pacientes que recibieron EGRIFTA® durante 52 semanas. En el
subconjunto de pacientes con reacciones de hipersensibilidad se detectaron anticuerpos IgG contra
tesamorelin en el 85.2 %. Se observó reactividad cruzada a la hormona liberadora de la hormona de
crecimiento (growth hormone-releasing hormone, GHRH) endógena en aproximadamente el 60 % de los
pacientes que desarrollaron anticuerpos contra tesamorelin. Los pacientes con y sin anticuerpos IgG
contra tesamorelin tuvieron reducciones medias similares en el tejido adiposo visceral (visceral adipose
tissue, VAT) y la respuesta del factor de crecimiento insulínico tipo 1 (insulin-like growth factor-1, IGF-
1), que sugiere la presencia de anticuerpos, no alteró la eficacia de EGRIFTA®. En un grupo de
pacientes que tenían anticuerpos contra tesamorelin después de 26 semanas de tratamiento (56 %) y que
fueron reevaluados 6 meses más tarde, después de interrumpir el tratamiento con EGRIFTA®, el 18 %
aún arrojaba resultados positivos de anticuerpos.
Se detectaron anticuerpos neutralizantes contra tesamorelin y hGHRH in vitro en la semana 52 en el
10 % y el 5 % de los pacientes tratados con EGRIFTA®, respectivamente. No parecieron influir en la
eficacia, según se demuestra en los cambios comparables en el VAT y el nivel de IGF-1 en pacientes
con y sin anticuerpos neutralizantes in vitro.
La incidencia de positividad de anticuerpos observada en un ensayo depende en gran medida de varios
factores, como la sensibilidad y especificidad del ensayo, la metodología, la manipulación de la muestra,
el momento de obtención de la muestra, los medicamentos concomitantes y la enfermedad subyacente.
Por estos motivos, la comparación de la incidencia de anticuerpos contra EGRIFTA® y de la incidencia
de anticuerpos contra otros productos puede llevar a conclusiones erradas.
7 INTERACCIONES MEDICAMENTOSAS
7.1 Fármacos metabolizados por el citocromo P450
La administración concomitante de EGRIFTA® con simvastatina, un sustrato que se metaboliza a través
de CYP3A, demostró que EGRIFTA® no afectó significativamente los perfiles farmacocinéticos de la
simvastatina en sujetos sanos. Este resultado sugiere que EGRIFTA® podría no afectar
significativamente la actividad de CYP3A. No se han evaluado otras isoenzimas de CYP450 con
EGRIFTA®. Sin embargo, los datos publicados indican que la GH puede modular la depuración de
antipirina mediada por el citocromo P450 (CYP450) en el hombre. Estos datos sugieren que la GH
puede alterar la depuración de compuestos conocidos por ser metabolizados por las enzimas hepáticas
CYP450 (p. ej., corticosteroides, esteroides sexuales, anticonvulsivos, ciclosporina). Debido a que
tesamorelin estimula la producción de GH, se recomienda un control minucioso cuando EGRIFTA® se
administra en combinación con otros fármacos conocidos por ser metabolizados por las enzimas
hepáticas CYP450 (consulte Farmacología clínica [12.3]).
7.2 11β-hidroxiesteroide deshidrogenasa tipo 1 (11βHSD-1)
Se sabe que la GH inhibe a la 11β-hidroxiesteroide deshidrogenasa tipo 1 (11βHSD-1), una enzima
microsomal requerida para la conversión de la cortisona a su metabolito activo, cortisol, en el tejido
hepático y adiposo. Debido a que tesamorelin estimula la producción de la GH, los pacientes que reciben
reemplazo de glucocorticoides para el hipoadrenalismo diagnosticado previamente pueden requerir un
aumento de las dosis de mantenimiento o dosis de estrés luego de iniciar EGRIFTA®, especialmente en
pacientes tratados con acetato de cortisona y prednisona, ya que la conversión de estos fármacos a sus
metabolitos biológicamente activos depende de la actividad de 11βHSD-1.
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Categoría X del embarazo (consulte Contraindicaciones [4.4]).
EGRIFTA® está contraindicado en mujeres embarazadas. Durante el embarazo, el tejido adiposo
visceral aumenta debido a los cambios metabólicos y hormonales normales. Modificar este cambio
fisiológico del embarazo con EGRIFTA® no ofrece ningún beneficio conocido y podría provocar daño
fetal. La administración de acetato de tesamorelin a ratas durante la organogénesis y la lactancia provocó
hidrocefalia en las crías a una dosis aproximadamente dos y cuatro veces mayor que la dosis clínica,
respectivamente, en función de la exposición al fármaco medida (AUC). Si ocurre un embarazo,
discontinuar la terapia con EGRIFTA®. Si se usa el fármaco durante el embarazo, o si la paciente queda
embarazada mientras toma este fármaco, se debe informar a la paciente sobre el riesgo potencial para
el feto.
La administración de acetato de tesamorelin a ratas durante la organogénesis y la lactancia provocó
hidrocefalia en las crías a una dosis aproximadamente dos y cuatro veces mayor que la dosis clínica,
respectivamente, en función de la exposición al fármaco medida (AUC). La dosis real en animales fue
de 1.2 mg/kg. Durante la organogénesis, dosis más bajas, aproximadamente 0.1 a 1 vez mayores que la
dosis clínica, causaron un retraso en la osificación craneal en ratas. Las dosis reales en animales fueron
de 0.1 a 0.6 mg/kg. No ocurrieron efectos adversos en el desarrollo en conejos que recibieron dosis
aproximadamente hasta 500 veces mayores que la dosis clínica.
8.3 Madres en período de lactancia
Los Centros para el Control y la Prevención de Enfermedades recomiendan que las madres infectadas
por VIH en los Estados Unidos no amamanten a sus bebés para evitar el riesgo de contagio posnatal de
la infección por VIH-1. Debido tanto al potencial de contagio de la infección con VIH-1 como a las
reacciones adversas graves en los lactantes, se debe indicar a las madres que reciben EGRIFTA® que no
amamanten.
Se desconoce si EGRIFTA® se excreta en la leche humana. La administración de acetato de tesamorelin
a ratas durante la organogénesis y la lactancia provocó hidrocefalia en las crías a una dosis
aproximadamente dos y cuatro veces mayor que la dosis clínica, respectivamente, en función de la
exposición al fármaco medida (AUC). La dosis real en animales fue de 1.2 mg/kg.
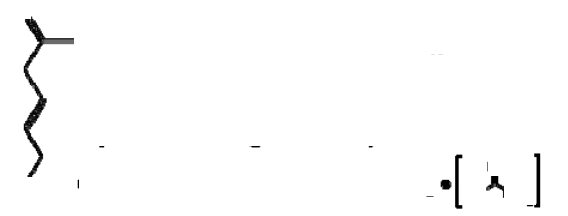
8.4 Uso pediátrico
No se ha establecido la seguridad ni la eficacia en pacientes pediátricos. EGRIFTA® no se debe usar en
niños con epífisis abiertas, entre quienes el exceso de GH y IGF-1 puede provocar aceleración del
crecimiento lineal y crecimiento excesivo.
8.5 Uso geriátrico
No existe información sobre el uso de EGRIFTA® en pacientes mayores de 65 años de edad con VIH y
lipodistrofia.
8.6 Insuficiencia renal y hepática
No se ha establecido la seguridad, la eficacia y la farmacocinética de EGRIFTA® en pacientes con
disfunción renal o hepática.
10 SOBREDOSIS
No se dispone de datos de sobredosis.
11 DESCRIPCIÓN
EGRIFTA® contiene tesamorelin (como sal de acetato), un análogo del factor de liberación de la
hormona de crecimiento (GRF) humana. El precursor peptídico del acetato de tesamorelin se produce en
forma sintética y está compuesto por la secuencia de aminoácidos 44 del GRF humano. El acetato de
tesamorelin se produce adhiriendo una porción de hexenoil, una cadena C6 con una doble unión en la
posición 3, al residuo de tirosina en la parte N-terminal de la molécula. La fórmula molecular del acetato
de tesamorelin es C221H366N72O67S • x C2H4O2 (x ≈ 7) y su peso molecular (base libre) es de
5135.9 daltones. La fórmula estructural del acetato de tesamorelin es:
EGRIFTA® es un polvo liofilizado estéril, de color blanco a blanquecino, sin conservantes para
inyección subcutánea. Después de la reconstitución con el diluyente suministrado (agua estéril para
inyección, USP), una solución de EGRIFTA® es trasparente e incolora. Cada frasco de dosis única de
EGRIFTA® contiene 1 mg de tesamorelin como base libre (1.1 mg de acetato de tesamorelin, anhidro) y
el siguiente ingrediente inactivo: 50 mg de manitol, USP.
O
H3C
O
HO CH3X
Tyr-Ala-Asp-Ala-Ile-Phe-Thr-Asn-Ser-Tyr-Arg-Lys-
10
Val-Leu-Gly-Gln-Leu-Ser-Ala-Arg-Lys-Leu-Leu-Gln-
20
Asp-Ile-Met-Ser-Arg-Gln-Gln-Gly-Glu-Ser-Asn-Gln-
30
Glu-Arg-Gly-Ala-Arg-Ala-Arg-Leu-NH2
40

12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
In vitro, tesamorelin se une y estimula a los receptores de GRF humanos con potencia similar al GRF
endógeno (consulte Farmacología clínica [12.2]).
El factor de liberación de la hormona de crecimiento (GRF), también conocido como hormona
liberadora de la hormona de crecimiento (GHRH), es un péptido hipotálamico que actúa en las células
somatotropas pituitarias para estimular la síntesis y la liberación pulsátil de la hormona de crecimiento
endógena (GH), que es anabólica y lipolítica. La GH ejerce sus efectos interactuando con receptores
específicos en una variedad de células diana, incluidos condrocitos, osteoblastos, miocitos, hepatocitos y
adipocitos, lo cual provoca una gran cantidad de efectos farmacodinámicos. Algunos, pero no todos
estos efectos, son mediados principalmente por el IGF-1 producido en el hígado y en los tejidos
periféricos.
12.2 Farmacodinámica
Efectos en los niveles de IGF-1 e IGFBP-3
Tesamorelin estimula la secreción de la hormona de crecimiento y posteriormente aumenta los niveles
de IGF-1 y de la proteína transportadora 3 del factor de crecimiento similar a la insulina (insulin-like
growth factor binding protein, IGFBP-3) (consulte Estudios clínicos [14]).
Otras hormonas pituitarias
No se observaron cambios clínicamente significativos en los niveles de otras hormonas pituitarias,
incluidas la hormona estimuladora de la tiroides (thyroid-stimulating hormone, TSH), la hormona
luteinizante (luteinizing hormone, LH), la hormona adrenocorticotrópica (adrenocorticotropic hormone,
ACTH) y la prolactina, en sujetos que recibieron EGRIFTA® en ensayos clínicos de fase 3.
12.3 Farmacocinética
Absorción
Se determinó que la biodisponibilidad absoluta de EGRIFTA® luego de la administración subcutánea de
una dosis de 2 mg es inferior al 4 % en sujetos adultos sanos. La farmacocinética de dosis únicas y
múltiples de EGRIFTA® se ha caracterizado en sujetos sanos y pacientes infectados por VIH sin
lipodistrofia luego de la administración subcutánea de 2 mg.
Los valores medios [coeficiente de variación [coefficient of variation, CV]) de la medida de absorción
(AUC) para tesamorelin fueron 634.6 (72.4) y 852.8 (91.9) pg.h/ml en sujetos sanos y pacientes
infectados por VIH, respectivamente, luego de la administración subcutánea de una dosis única de
EGRIFTA® de 2 mg. Los valores de concentración máxima media de tesamorelin (CV) (Cmáx) fueron
2874.6 (43.9) pg/ml en sujetos sanos y 2822.3 (48.9) pg/ml en pacientes infectados por VIH. La
mediana de la concentración plasmática máxima de tesamorelin (Tmáx) fue de 0.15 h en ambas
poblaciones.
Distribución
El volumen de distribución medio (± desviación estándar [standard deviation, SD]) de tesamorelin luego
de la administración subcutánea de una dosis única fue de 9.4±3.1 l/kg en sujetos sanos y 10.5±6.1 l/kg
en pacientes infectados por VIH.
Metabolismo
No se han realizado estudios de metabolismo formales en seres humanos.
Eliminación
La media de la vida media de eliminación (T1/2) de tesamorelin fue de 26 y 38 minutos en sujetos sanos
y pacientes infectados por VIH, respectivamente, luego de la administración subcutánea durante 14 días
consecutivos.
Interacciones medicamentosas
Simvastatina
El efecto de la administración de dosis múltiples de EGRIFTA® (2 mg) en la farmacocinética de
simvastatina y ácido de simvastatina se evaluó en sujetos sanos. La administración concomitante de
EGRIFTA® y simvastatina (un sustrato de CYP3A sensible) provocó una disminución del 8 % en la
medida de absorción (AUCinf) y un aumento del 5 % en la tasa de absorción (Cmáx) de simvastatina. Para
el ácido de simvastatina, hubo una disminución del 15 % en el AUCinf y una disminución del 1 % en la
Cmáx (consulte Interacciones medicamentosas [7.1]).
Ritonavir
El efecto de la administración de dosis múltiples de EGRIFTA® (2 mg) en la farmacocinética de
ritonavir se evaluó en sujetos sanos. La administración concomitante de EGRIFTA® con ritonavir
provocó una disminución del 9 % en el AUCinf y una disminución del 11 % en la Cmáx de ritonavir
(consulte Interacciones medicamentosas).
Poblaciones específicas
No se ha establecido la farmacocinética de tesamorelin en pacientes con disfunción renal o hepática, en
pacientes pediátricos o en pacientes de edad avanzada.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
No se han realizado estudios de carcinogenicidad de por vida en roedores con acetato de tesamorelin. No
se reveló potencial de mutagenicidad del acetato de tesamorelin en una batería de pruebas que
incluyeron la inducción de mutaciones genéticas en bacterias (prueba Ames), mutaciones genéticas en
células de mamíferos cultivadas in vitro (células de hámster CHOK1) y daño cromosómico en animales
intactos (células de médula ósea en ratones). No se observaron efectos en la fertilidad en ratas macho y
hembra luego de la administración de acetato de tesamorelin en dosis de hasta 0.6 mg/kg
(aproximadamente equivalentes a la exposición clínica) durante 28 días en machos y 14 días en
hembras. En el estudio de toxicidad de 26 semanas, las hembras que recibieron dosis aproximadamente
entre 16 y 25 veces mayores que la dosis clínica tuvieron más probabilidades de estar en diestro.
14 ESTUDIOS CLÍNICOS
Se realizaron dos estudios multicéntricos, aleatorizados, doble ciego, controlados con placebo en
pacientes infectados por VIH con lipodistrofia y exceso de grasa abdominal (lipohipertrofia abdominal).
Ambos estudios (Estudio 1 y 2) consistieron en una fase principal de 26 semanas y una fase de extensión
de 26 semanas. Los principales criterios de inclusión fueron la edad de 18 a 65 años, una circunferencia
de cintura de ≥95 cm (37.4 pulgadas), una relación de cintura a cadera de ≥0.94 para los hombres y
≥94 cm (37.0 pulgadas) y ≥0.88 para las mujeres, respectivamente, y una glucosa sanguínea en ayunas
(fasting blood glucose, FBG) <150 mg/dl (8.33 mmol/l). Los criterios de exclusión principales
incluyeron el IMC ≤ 20 kg/m2, diabetes tipo 1, diabetes tipo 2, si el paciente recibía tratamiento previo
con insulina o con agentes hipoglucémicos o sensibilizadores de la insulina orales, antecedentes de
neoplasia maligna e hipopituitarismo. Los pacientes recibían un régimen antirretroviral estable durante
al menos 8 semanas antes de la aleatorización. Los pacientes que cumplían los criterios de
inclusión/exclusión fueron aleatorizados en una proporción 2:1 para recibir 2 mg de EGRIFTA® o
placebo por vía subcutánea todos los días durante 26 semanas. La evaluación de eficacia principal para
cada uno de estos estudios fue el cambio porcentual desde el inicio a la semana 26 (fase principal) en el
tejido adiposo visceral (VAT), según lo evaluado mediante exploración por tomografía computarizada
(CT) al nivel vertebral L4-L5. Los criterios de valoración secundarios incluyeron cambios desde el
inicio en los resultados informados por el paciente en relación con imagen corporal, triglicéridos,
relación de colesterol total con colesterol HDL, niveles de IGF-1 y parámetros de seguridad. Otros
criterios de valoración incluyeron cambios desde el inicio en la circunferencia de la cintura, el tejido
subcutáneo abdominal (abdominal subcutaneous tissue, SAT), grasa del tronco y masa corporal magra.
En ambos estudios, los pacientes tratados con EGRIFTA® que completaron el período de tratamiento de
26 semanas fueron aleatorizados nuevamente a la terapia a ciego con placebo o 2 mg de EGRIFTA®
todos los días por un período de tratamiento adicional de 26 semanas (fase de extensión) para evaluar el
mantenimiento de la reducción del VAT y recopilar datos de seguridad a largo plazo. Para la inclusión
en los estudios de fase de extensión, los sujetos debían haber completado la fase principal con FBG
≤150 mg/dl.

Fase principal (inicio a la semana 26):
Estudio 1
Este estudio aleatorizó a 412 pacientes infectados por VIH con lipodistrofia y exceso de grasa
abdominal para recibir EGRIFTA® (N=273) o placebo (N=137). Al inicio para los dos grupos
combinados, la edad media era de 48 años; el 86 % eran hombres; el 75 % eran blancos, el 14 % eran
negros/afroamericanos y el 8 % eran hispanos; el peso medio era de 90 kg; el IMC medio era de
29 kg/m2; la circunferencia media de la cintura era de 104 cm; la circunferencia media de la cadera era
de 100 cm; el VAT medio era de 176 cm2; el recuento de células CD4 medio era de
606 células/mm3; el 69 % tenía carga viral no detectable (<50 copias/ml); y el 33.7 % aleatorizado a
EGRIFTA® y el 36.6 % aleatorizado a placebo tenía deficiencia de tolerancia a la glucosa, mientras que
el 5.6 % aleatorizado a EGRIFTA® y el 6.7 % aleatorizado a placebo tenía diabetes mellitus controlada
por la dieta. La tasa de cumplimiento de la semana 26 en el Estudio 1 fue del 80 %.
Estudio 2
Este estudio aleatorizó a 404 pacientes infectados por VIH con lipodistrofia y exceso de grasa
abdominal para recibir EGRIFTA® (N=270) o placebo (N=126). Al inicio para los dos grupos
combinados, la edad media era de 48 años; el 84 % eran hombres; el 77 % eran blancos, el 12 % eran
negros/afroamericanos y el 9 % eran hispanos; el peso medio era de 88 kg; el IMC medio era de
29 kg/m2; la circunferencia media de la cintura era de 105 cm; la circunferencia media de la cadera era
de 100 cm; el VAT medio era de 189 cm2; el recuento de células CD4 medio era de
592 células/mm3; el 83 % tenía carga viral no detectable (<50 copias/ml); y el 44.1 % aleatorizado a
EGRIFTA® y el 39.7 % aleatorizado a placebo tenía deficiencia de tolerancia a la glucosa, mientras que
el 9.3 % aleatorizado a EGRIFTA® y el 9.5 % aleatorizado a placebo tenía diabetes mellitus controlada
por la dieta. La tasa de cumplimiento de la semana 26 en el Estudio 2 fue del 74 %.
En las Tablas 3 y 4 se presentan los resultados de las fases principales de los Estudios 1 y 2.

Tabla 3: Cambios desde el inicio a la semana 26 en el tejido adiposo visceral (cm2) por grupo de
tratamiento (población con intención de tratar con extrapolación de la última observación
realizada)
FASE PRINCIPAL (inicio a la semana 26)
Estudio 1 Estudio 2
EGRIFTA®
(N=273)
Placebo
(N=137)
EGRIFTA®
(N=270)
Placebo
(N=126)
Inicio (cm2) 178 (77) 171 (77) 186 (87) 195 (95)
Cambio (cm2) -27 4 -21 -0
Media de la
diferencia en el
tratamiento (CI del
95 %)
-31 (-39,-24) -21 (-29,-12)
Media del cambio
(%)1 -18 2 -14 -2
Media de la
diferencia en el
tratamiento (CI del
95 %)1
-20 (-24, -15) -12 (-16, -7)
Los datos iniciales se expresan como media (SD); el cambio se refiere a las medias de mínimos
cuadrados (least-squares mean, LSM); CI: intervalo de confianza.
1 Resultados derivados del modelo estadístico: Ln(VAT semana 26/VAT inicio) = Ln(VAT inicio) +
grupo de tratamiento
Tabla 4: Cambios desde el inicio a la semana 26 en IGF-1, IGFBP-3, peso y circunferencia de la
cintura por grupo de tratamiento (población con intención de tratar con extrapolación de la
última observación realizada)
FASE PRINCIPAL (inicio a la semana 26)
Estudio 1 Estudio 2
EGRIFTA®
(N=273)
Placebo
(N=137)
EGRIFTA®
(N=270)
Placebo
(N=126)
IGF-1
(ng/ml)
Inicio 161 (59) 168 (75) 146 (66) 149 (59)
Cambio 107 -15 108 3
Media de la
diferencia en el
tratamiento (CI del
95 %)
122 (101, 141) 105 (85, 126)
IGFBP-3
(mg/l)
Inicio 3 (1) 3 (1) 3 (1) 3 (1)
Cambio 0.4 -0.2 0.8 -0.0
Media de la
diferencia en el
tratamiento (CI del
95 %)
0.6 (0.5, 0.8) 0.8 (0.5, 1.0)
Peso (kg)
Inicio 90 (14) 90 (14) 89 (14) 87 (16)
Cambio -0.4 0.0 0.5 0.3
Media de la
diferencia en el
tratamiento (CI del
95 %)
-0.4 (-1.3, 0.5) 0.2 (-0.7, 1.3)
Circunferenci
a de la cintura
(cm)
Inicio 104 (10) 105 (9) 105 (9) 105 (9)
Cambio -3 (5) -1 (4) -2 (5) -1 (5)
Media de la
diferencia en el
tratamiento (CI del
95 %)
-2 (-2.8, -0.9) -1 (-2.5, -0.3)
Los datos iniciales se expresan como media (SD); el cambio se refiere a las medias de mínimos
cuadrados (LSM); CI: intervalo de confianza.
Un análisis de subgrupos por sexo demostró que no hubo diferencias significativas en el cambio
porcentual desde el inicio en el tejido adiposo visceral (VAT) y las respuestas del IFG-1,
respectivamente, entre hombres y mujeres.
En la semana 26, el tratamiento con EGRIFTA® provocó una reducción desde el inicio en la grasa del
tronco media de 1.0 kg en el Estudio 1 y 0.8 kg en el Estudio 2, respectivamente (comparada con un
aumento de 0.4 kg en el Estudio 1 y de 0.2 kg en el Estudio 2, respectivamente, en pacientes que
recibían placebo). El tratamiento con EGRIFTA® provocó un aumento desde el inicio en la masa
corporal magra media de 1.3 kg en el Estudio 1 y de 1.2 kg en el Estudio 2, respectivamente (comparado
con una disminución de 0.2 kg en el Estudio 1 y 0.03 kg en el Estudio 2, respectivamente, en pacientes
que recibían placebo).
Treatment:
Placebo
tesamorelin
-100 -50 0 50 100
Belly Appearance Distress change
-100 -50 0 50 100
Belly Appearance Distress change
100%
50%
0%
Study 2Study 2Study 1Study 1
En promedio, no hubo efectos adversos de EGRIFTA® en los lípidos o el tejido adiposo subcutáneo
(SAT). EGRIFTA® no afectó en forma adversa la efectividad antirretroviral, como niveles circulantes
medios de recuentos de CD4 o ARN del VIH-1 (carga viral).
Resultados informados por el paciente
Los pacientes calificaron el grado de angustia asociado con el aspecto del vientre en una escala de
calificación de 9 puntos que luego se transformó a un puntaje de 0 (extremadamente penoso y
angustiante) a 100 (extremadamente alentador). Un puntaje de 50 indicaba un sentimiento neutro (de
ninguna de las dos formas). Un cambio positivo en el puntaje del inicio indicaba una mejora, es decir,
menos angustia.
La distribución de respuesta acumulativa (cambio desde el inicio a la semana 26) se muestra en la Figura
1 para ambos grupos de tratamiento. Una curva desviada a la derecha en esta escala indica un mayor
porcentaje de pacientes que informaron mejora.
Figura 1. Distribución de respuesta acumulativa de angustia por el aspecto del vientre
Pacientes acumulados (%)
→me
j
o
r
peor ← peor ← → me
j
o
r
Estudio 1 Estudio 2
100 %
50 %
0 % -100 -50 0 50 100
-100 -50 0 50 100
Cambio en la angustia por el
aspecto del vientre Cambio en la angustia por el aspecto
del vientre
Tratamiento
Placebo
Tesamorelin

Fase de extensión (semanas 26 a 52):
En la fase de extensión doble ciego, los pacientes que recibían EGRIFTA® y completaron la fase
principal de 26 semanas fueron aleatorizados nuevamente para recibir 2 mg de EGRIFTA® o placebo.
Estudio 1
Este estudio aleatorizó nuevamente a 207 pacientes infectados por VIH con lipodistrofia que
completaron el tratamiento con EGRIFTA® en la fase principal para recibir EGRIFTA® (N=154) o
placebo (N=50) por otras 26 semanas (relación de aleatorización de 3:1). Al inicio (semana 26) para los
dos grupos combinados, la edad media era de 48 años; el 88 % eran hombres; el 78 % eran blancos,
el 12 % eran negros/afroamericanos y el 8 % eran hispanos; el peso medio era de 90 kg; el IMC medio
era de 29 kg/m2; la circunferencia media de la cintura era de 102 cm; la circunferencia media de la
cadera era de 100 cm; el VAT medio era de 145 cm2; el recuento de células CD4 medio era de
639 células/mm3; el 68 % tenía carga viral no detectable (<50 copias/ml); y para los pacientes tratados
con EGRIFTA® que completaron el período de tratamiento de 26 semanas que fueron aleatorizados
nuevamente a EGRIFTA® (grupo T-T) o aleatorizados nuevamente a placebo, el 36.6 % y el 32.0 %,
respectivamente, tenía deficiencia de tolerancia a la glucosa, mientras que el 2.0 % aleatorizado
nuevamente a EGRIFTA® y el 6.0 % aleatorizado nuevamente a placebo tenía diabetes mellitus
controlada por la dieta. La tasa de cumplimiento para pacientes aleatorizados en la fase de extensión del
Estudio 1 fue del 83 %.
Estudio 2
Este estudio aleatorizó nuevamente a 177 pacientes infectados por VIH con lipodistrofia que
completaron el tratamiento con EGRIFTA® en la fase principal para recibir EGRIFTA® (N=92) o
placebo (N=85) por otras 26 semanas (relación de aleatorización de 1:1). Al inicio (semana 26) para los
dos grupos combinados, la edad media era de 48 años; el 90 % eran hombres; el 84 % eran blancos, el
9 % eran negros/afroamericanos y el 7 % eran hispanos; el peso medio era de 89 kg; el IMC medio era
de 28 kg/m2; la circunferencia media de la cintura era de 105 cm; la circunferencia media de la cadera
era de 100 cm; el VAT medio era de 172 cm2; el recuento de células CD4 medio era de
579 células/mm3; el 82 % tenía carga viral no detectable (<50 copias/ml); y para los pacientes tratados
con EGRIFTA® que completaron el período de tratamiento de 26 semanas que fueron aleatorizados
nuevamente a EGRIFTA® (grupo T-T) o aleatorizados nuevamente a placebo, el 48.9 % y el 50.6 %,
respectivamente, tenía deficiencia de tolerancia a la glucosa, mientras que el 4.3 % aleatorizado
nuevamente a EGRIFTA® y el 12.9 % aleatorizado nuevamente a placebo tenía diabetes mellitus
controlada por la dieta. La tasa de cumplimiento para pacientes aleatorizados en la fase de extensión del
Estudio 2 fue del 81 %.
En las Tablas 5 y 6 se presentan los resultados de las fases de extensión de los Estudios 1 y 2.
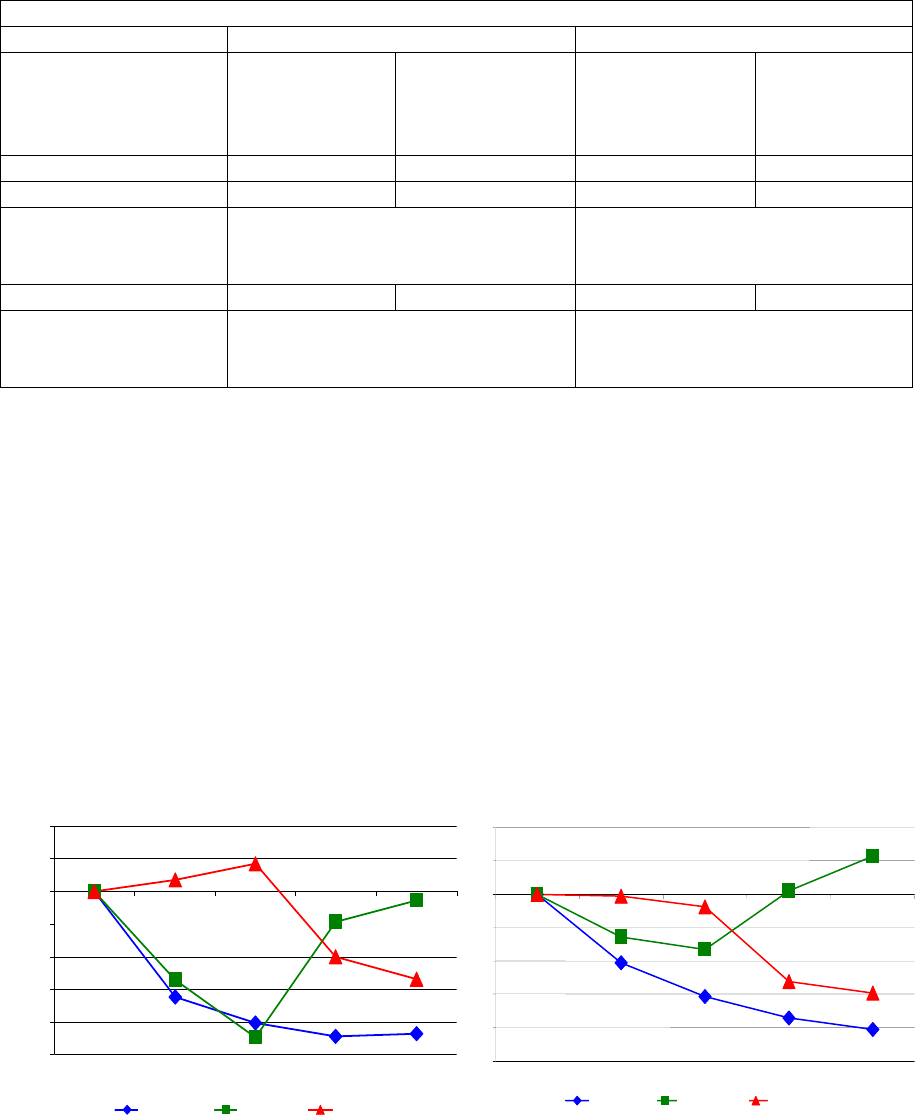
Tabla 5: Cambios desde el inicio de la semana 26 a la semana 52 en el tejido adiposo visceral (cm2)
por grupo de tratamiento (población con intención de tratar con extrapolación de la última
observación realizada)
Los datos iniciales de la semana 26 se expresan como media (SD). El cambio se refiere a la media de
mínimos cuadrados (LSM); CI: intervalo de confianza.
1T-T = tesamorelin para las semanas 0 a 26 y tesamorelin para las semanas 26 a 52
2T-P = tesamorelin para las semanas 0 a 26 y placebo para las semanas 26 a 52
3 Resultados derivados del modelo estadístico: Ln(VAT semana 52/semana 26) = Ln(VAT semana 26) +
grupo de tratamiento
La Figura 2 muestra el cambio porcentual en el VAT desde el inicio (semana 0) a través del tiempo
hasta las 52 semanas en pacientes que completaron el estudio.
Figura 2. Cambio porcentual en el VAT desde el inicio con el paso del tiempo
FASE DE EXTENSIÓN (semanas 26 a 52)
Estudio 1 Estudio 2
T-T1
(Semana 26 a
52)
(N=154)
T-P2
(Semana 26 a 52)
(N=50)
T-T1
(Semana 26 a 52)
(N=92)
T-P2
(Semana 26 a
52)
(N=85)
Semana 26 (cm2) 145 (72) 144 (72) 166 (89) 177 (88)
Cambio (cm2) 3 25 -11 24
Media de la diferencia
en el tratamiento (CI
del 95 %)
-22 (-34, -10) -35 (-48, -22)
Media del cambio (%)3 0 22 -5 16
Media de la diferencia
en el tratamiento (CI
del 95 %)3
-17 (-24, -10) -18 (-24, -11)
-25
-20
-15
-10
-5
0
5
10
Baseline Week 13 Week 26 Week 39 Week 52
T-T (n=108) T-P (n=28) P-T (n=70)
-25
-20
-15
-10
-5
0
5
10
Baseline Week 13 Week 26 Week 39 Week 52
T-T (n=55) T-P (n=40) P-T (n=48)
ESTUDIO 1 ESTUDIO 2
Inicio Semana 13 Semana 26 Semana 39 Semana 52
T-T (n=108) T-P (n=28) P-T (n=70)
10
5
0
-5
-10
-15
-20
-25
Inicio Semana 52Semana 39
Semana 26
Semana 13
T-T (n=55) T-P (n=40) P-T (n=48)
10
5
0
-5
-10
-15
-20
-25
Cambio % en el VAT
desde el inicio
Los datos de la Figura 2 se expresan como valores medios. T-T (tesamorelin a tesamorelin) se refiere al
grupo de pacientes que recibió tesamorelin para las semanas 0 a 26 y fueron aleatorizados nuevamente a
tesamorelin para las semanas 26 a 52. T-P (tesamorelin a placebo) se refiere al grupo de pacientes que
recibió tesamorelin para las semanas 0 a 26 y fueron aleatorizados nuevamente a placebo para las
semanas 26 a 52. P-T (placebo a tesamorelin) se refiere al grupo de pacientes que recibió placebo para
las semanas 0 a 26 y cambiaron a tesamorelin (tratados a etiqueta abierta) para las semanas 26 a 52.
Tabla 6: Cambios desde el inicio a la semana 26 a la semana 52 en IGF-1, IGFBP-3, peso y
circunferencia de la cintura por grupo de tratamiento (población con intención de tratar con
extrapolación de la última observación realizada)
FASE DE EXTENSIÓN (semanas 26 a 52)
Estudio 1 Estudio 2
T-T1
(Semana 26 a
52)
(N=154)
T-P2
(Semana 26 a
52)
(N=50)
T-T1
(Semana 26 a 52)
(N=92)
T-P2
(Semana 26
a 52)
(N=85)
IGF-1
(ng/ml)
Semana 26 291 (124) 281 (105) 280 (134) 269 (110)
Cambio -59 -137 -25 -135
Media de la
diferencia en el
tratamiento (CI del
95 %)
78 (50, 106) 110 (87, 134)
IGFBP-3
(mg/l)
Semana 26 3 (1) 3 (1) 3 (1) 3 (1)
Cambio -0.2 -0.5 -0.3 -0.9
Media de la
diferencia en el
tratamiento (CI del
95 %)
0.3 (-0.0, 0.6) 0.6 (0.3, 0.9)
Peso (kg)
Semana 26 89 (14) 92 (17) 89 (13) 90 (14)
Cambio 0.2% 0.6 -0.5 0.1
Media de la
diferencia en el
tratamiento (CI del
95 %)
-0.4 (-2, 1) -0.6 (-2, 1)
Circunferencia
de la cintura
(cm)
Semana 26 101 (10) 102 (12) 101 (9) 103 (11)
Cambio -0.2 2.4 -1.1 0.2%
Media de la
diferencia en el
tratamiento (CI del
95 %)
-2.6 (-4, -1) -1.3 (-2, 0)
Los datos iniciales de la semana 26 se expresan como media (SD); el cambio se refiere a la media de
mínimos cuadrados (LSM); CI: intervalo de confianza.
1T-T = tesamorelin para la semana 0 a 26 y tesamorelin para la semana 26 a 52
2T-P = tesamorelin para la semana 0 a 26 y placebo para la semana 26 a 52
Los pacientes tratados con EGRIFTA® durante 52 semanas (grupo T-T) no demostraron cambios
entre las semanas 26 y 52 en la grasa del tronco media (aumento de 0.1 kg en el Estudio 1 y disminución
de 0.5 kg en el Estudio 2, respectivamente, en comparación con un aumento de 1.4 kg en pacientes en el
grupo de T-P en el Estudio 1 y un aumento de 1.09 kg en el Estudio 2, respectivamente), ni hubo un
cambio desde el inicio de la semana 26 en la masa corporal magra media (disminución de 0.1 kg en el
Estudio 1 y aumento de 0.1 kg en el Estudio 2, respectivamente, en comparación con una disminución
de 1.8 kg en pacientes en el grupo de T-P en el Estudio 1 y una disminución de 1.7 kg en el Estudio 2,
respectivamente).
No hubo efectos adversos de EGRIFTA® en los lípidos o el tejido adiposo subcutáneo (SAT).
EGRIFTA® no afectó en forma adversa la efectividad antirretroviral, como niveles circulantes medios de
recuentos de CD4 o ARN del VIH-1 (carga viral).
16 PRESENTACIÓN/ALMACENAMIENTO Y MANIPULACIÓN
EGRIFTA® (tesamorelin inyectable) se suministra como polvo liofilizado estéril, de color blanco a
blanquecino. Cada frasco de dosis única de EGRIFTA® contiene 1 mg de tesamorelin como base libre
(1.1 mg de acetato de tesamorelin, anhidro) y el siguiente ingrediente inactivo: 50 mg de manitol, USP.
EGRIFTA® está disponible en un paquete que incluye dos cajas. Una caja contiene 60 frascos de 1 mg
cada uno de EGRIFTA® y una segunda caja contiene 30 frascos de 10 ml de dosis única de diluyente
para la reconstitución (agua estéril para inyección, USP), jeringas desechables y agujas suficientes para
un suministro para 30 días.
Después de la reconstitución con agua estéril para inyección, USP, la concentración de la solución
reconstituida es de 1 mg/ml y debe inyectarse de inmediato.
Los frascos de EGRIFTA® deben protegerse de la luz y conservarse en la caja original hasta el momento
de usarlos. EGRIFTA® no reconstituido debe almacenarse a temperatura refrigerada, entre 2 °C y 8 °C
(entre 36 °F y 46 °F). El diluyente para la reconstitución (agua estéril para inyección, USP), las jeringas
y agujas deben almacenarse a una temperatura ambiente controlada entre 20 °C y 25 °C (entre 68 °F y
77 °F).
Las jeringas y las agujas son para un solo uso por parte de un solo paciente y nunca se deben compartir
entre los pacientes.
NDC 62064-011-60

17 INFORMACIÓN DE ORIENTACIÓN PARA EL PACIENTE
Consulte la ficha técnica con información para el paciente aprobada por la FDA (Información
para el paciente e Instrucciones de uso).
Retención de líquidos (5.3): advertir a los pacientes que el tratamiento con EGRIFTA® puede
causar síntomas que coinciden con la retención de líquidos, entre ellos, edema, artralgia y
síndrome del túnel carpiano. Estas reacciones son transitorias o se resuelven con la
discontinuación del tratamiento.
Reacciones de hipersensibilidad (5.5): advertir a los pacientes que pueden ocurrir reacciones de
hipersensibilidad (p. ej., erupción, urticaria) durante el tratamiento con EGRIFTA®. Aconsejar a
los pacientes solicitar atención médica sin demora y discontinuar el tratamiento con EGRIFTA®
de inmediato.
Reacciones en el lugar de la inyección (5.6): advertir a los pacientes de la posibilidad de que
ocurran reacciones en el lugar de la inyección, entre ellas, eritema, prurito, dolor, irritación y
moretones en el lugar de la inyección. Para reducir la incidencia de reacciones en el lugar de la
inyección, aconsejar a los pacientes alternar el lugar de la inyección.
Aconsejar a los pacientes que nunca deben compartir una jeringa de EGRIFTA® con otra
persona, aunque cambien la aguja. Compartir jeringas o agujas entre pacientes puede implicar
un riesgo de contagio de la infección.
Embarazo
Asesorar a las mujeres que discontinúen EGRIFTA® si ocurre un embarazo, ya que el fármaco no ofrece
ningún beneficio conocido a mujeres embarazadas y podría provocar daño fetal (consulte
Contraindicaciones [4.4] y Uso en poblaciones específicas [8.1]).
Madres en período de lactancia
Debido tanto al potencial de contagio de la infección por VIH-1 como a las reacciones adversas graves
en los lactantes, se debe indicar a las madres que reciben EGRIFTA® que no amamanten (consulte Uso
en poblaciones específicas [8.3]).
EGRIFTA® es una marca registrada de Theratechnologies Inc.
Distribuido por: Theratechnologies Inc., Montreal, Québec, Canadá H3A 1T8
Revisado: 06/2015
Información para el paciente
EGRIFTA®
(tesamorelin inyectable)
para uso subcutáneo
Lea la Información para el paciente que viene con EGRIFTA® antes de
comenzar a tomar el medicamento y cada vez que resurta su receta. Puede
haber nueva información. Este folleto no sustituye una conversación con su
proveedor de atención médica acerca de su afección médica o su
tratamiento.
¿Qué es EGRIFTA®?
EGRIFTA® es un medicamento recetado inyectable para reducir el exceso
de grasa abdominal en pacientes infectados con el VIH que tienen
lipodistrofia. EGRIFTA® contiene un factor de liberación de la hormona de
crecimiento (GRF).
No se han realizado estudios sobre el impacto y seguridad de EGRIFTA®
en la salud cardiovascular.
EGRIFTA® no está indicado para el manejo de la pérdida de peso.
Se desconoce si el hecho de tomar EGRIFTA® ayuda a mejorar el
cumplimiento con los medicamentos antirretrovirales.
No se sabe si EGRIFTA® es seguro y efectivo en niños. No se recomienda
el uso de EGRIFTA® en niños.
¿Quién no debe usar EGRIFTA®?
No use EGRIFTA® si usted:
Tiene un tumor de la glándula pituitaria, cirugía de la glándula pituitaria u
otros problemas relacionados con su glándula pituitaria.
Tiene cáncer activo (ya sea recientemente diagnosticado o recurrente) o
está recibiendo tratamiento para el cáncer.
Es alérgico al tesamorelin o a cualquiera de los ingredientes de EGRIFTA®.
Consulte el final de este folleto para ver una lista completa de los
ingredientes de EGRIFTA®
Está embarazada o planea quedar embarazada. Si queda embarazada,
deje de usar EGRIFTA® y hable con su proveedor de atención médica.
Consulte la sección “¿Qué debo decirle a mi proveedor de atención médica
antes de usar EGRIFTA®?”
¿Qué debo decirle a mi proveedor de atención médica antes de usar
EGRIFTA®?
Antes de tomar EGRIFTA®, informe a su proveedor de atención médica si
usted:
tiene o ha tenido cáncer
tiene diabetes
está amamantando o planea amamantar. No se sabe si EGRIFTA® pasa a
la leche materna. Los Centros para el Control y la Prevención de
Enfermedades (Centers for Disease Control and Prevention, CDC)
recomiendan que las madres infectadas por el virus del VIH no
amamanten para evitar el riesgo de pasar la infección del VIH a su bebés.
Hable con su proveedor de atención médica sobre la mejor forma de
alimentar a su bebé si está tomando EGRIFTA®
tiene problemas renales o hepáticos
tiene cualquier otra afección médica.
Informe a su proveedor de atención médica sobre todos los medicamentos
que toma, incluidos los que se venden con receta y los de venta libre,
vitaminas y suplementos a base de hierbas. EGRIFTA® afectar la forma en
que otras medicinas funcionan, y otras medicinas pueden afectar el
funcionamiento de EGRIFTA®.
Conozca los medicamentos que toma. Tenga una lista y muéstresela a su
proveedor de atención médica y farmacéutico cuando adquiera un
medicamento nuevo.
¿Cómo debo usar EGRIFTA®?
Lea las “Instrucciones de uso” detalladas que vienen con EGRIFTA®
antes de comenzar a usar EGRIFTA®. Su proveedor de atención médica
le indicará cómo debe inyectarse EGRIFTA®.
Use EGRIFTA® exactamente como lo indicó su proveedor de atención
médica.
Inyecte EGRIFTA® por debajo de la piel (subcutáneamente) del área de su
estómago (abdomen).
Cambie (alterne) el lugar de inyección en su área del estómago
(abdomen) con cada dosis. No inyecte EGRIFTA® en tejido cicatricial, en
moretones o en el ombligo.
No comparta las agujas o jeringas con otras personas. Compartir agujas
puede provocar el contagio de enfermedades infecciosas, como el VIH.
¿Cuáles son los posibles efectos secundarios de EGRIFTA®?
EGRIFTA® puede causar efectos secundarios graves, incluidos los
siguientes:
Reacción alérgica grave. Algunas personas que toman EGRIFTA®
pueden tener una reacción alérgica.
Deje de usar EGRIFTA® y obtenga asistencia de emergencia de
inmediato si tiene uno de los siguientes síntomas:
- erupción en el cuerpo
- urticaria
- hinchazón de la cara o garganta
- falta de aire o dificultad para respirar
- latidos cardíacos rápidos
- sensación de que va a desmayarse o desmayos
hinchazón (retención de líquidos). EGRIFTA® puede causar hinchazón
en algunas partes de su cuerpo. Llame a su proveedor de atención médica
si tiene un aumento del dolor en las articulaciones, o dolor o
entumecimiento en sus manos o muñecas (síndrome del túnel carpiano).
Aumento de intolerancia a la glucosa (azúcar en sangre) y
diabetes. Su proveedor de atención médica medirá su azúcar en sangre
periódicamente.
Reacciones en el lugar de la inyección. Cambie (alterne) el lugar de la
inyección para disminuir su riesgo de reacciones en el lugar de la
inyección. Llame a su proveedor de atención médica para obtener
asesoría médica si tiene los siguientes síntomas alrededor del lugar de la
inyección:
- enrojecimiento
- picazón
- dolor
- irritación
- sangrado
- erupción
- hinchazón

Los efectos secundarios más frecuentes de EGRIFTA® incluyen:
- dolor de las articulaciones
- dolor en las piernas y en los brazos
- hinchazón en las piernas
- dolor muscular
- hormigueo, adormecimiento y pinchazos
- náuseas
- vómitos
- erupción
- picazón
Informe a su proveedor de atención médica si tiene cualquier efecto
secundario que le cause molestias o que persista.
Estos no son todos los efectos secundarios posibles de EGRIFTA®. Para recibir
más información, consulte a su proveedor de atención médica o a su
farmacéutico.
Llame a su médico para que le asesore acerca de los efectos secundarios.
Puede informar los efectos secundarios a la FDA llamando al 1-800-FDA-
1088.
También puede informar los efectos secundarios llamando al número de
teléfono gratuito de ® 1-844-EGRIFTA (1-844-347-4382).
¿Cómo debo almacenar EGRIFTA®?
EGRIFTA® viene en dos cajas dispensadas por la farmacia:
- Almacene los frascos de la caja del medicamento EGRIFTA® en
el refrigerador entre 2 °C y 8 °C (36 °F y 46 °F).
- Almacene la caja de agua estéril para inyección, las jeringas y
las agujas a temperatura ambiente entre 20 °C y 25 °C (68 °F y
77 °F).
Guarde los frascos de EGRIFTA® en la caja del medicamento alejados de
la luz.
No congelar.
No use EGRIFTA® después de la fecha de vencimiento impresa en la caja
y en las etiquetas del frasco.
Después de mezclar, use EGRIFTA® de inmediato y descarte todo el
EGRIFTA® restante. No almacene la mezcla de EGRIFTA®. Además,
deseche el frasco usado de agua estéril para inyección.

Mantenga EGRIFTA® y todos los medicamentos fuera del alcance de
los niños.
Información general sobre el uso eficaz y seguro de EGRIFTA®
Los medicamentos a veces se recetan con fines distintos de los que se
indican en el folleto de Información para el paciente. No use EGRIFTA® para
una afección para la cual no fue recetado. No administre EGRIFTA® a otras
personas, aunque tengan los mismos síntomas que usted. Hacer esto puede
causarles daño.
No comparta su jeringa o agujas de EGRIFTA® con otras personas.
Puede contagiarles una infección o contagiarse de ellos.
Este folleto de Información para el paciente resume la información más
importante sobre EGRIFTA®. Si desea obtener más información, consulte a su
proveedor de atención médica. Puede solicitar a su proveedor de atención
médica o farmacéutico información acerca de EGRIFTA® redactada para
profesionales de la salud.
Para obtener más información sobre EGRIFTA®, visite www.EGRIFTA.com o
comuníquese con ® al número de teléfono gratuito 1-844-
EGRIFTA (1-844-347-4382).
¿Cuáles son los ingredientes de EGRIFTA®?
Ingrediente activo: tesamorelin
Ingredientes inactivos: manitol y agua estéril para inyección
Esta información para pacientes ha sido aprobada por la Administración de
Alimentos y Medicamentos de los EE. UU.
Fabricado por: Jubilant HollisterStier General Partnership, 16751 Trans-
Canada Highway, Montreal, Québec, Canadá H9H 4J4
Revisado: 06/2015

Instrucciones de uso
EGRIFTA®
(tesamorelin inyectable)
para uso subcutáneo
Asegúrese de leer, comprender y seguir estas “Instrucciones de uso” antes
de usar EGRIFTA®. Su proveedor de atención médica debería mostrarle cómo
mezclar e inyectarse EGRIFTA® antes de inyectárselo por primera vez. Si
tiene alguna pregunta, consulte a su proveedor de atención médica.
Conserve este folleto en caso de que necesite volver a leerlo más adelante.
Información importante para el uso de EGRIFTA®
Después de mezclar EGRIFTA® con agua estéril para inyección, debe
tener un aspecto trasparente e incoloro, sin partículas visibles. No use
EGRIFTA® si tiene aspecto turbio, cambios de coloración o partículas
visibles. Si tiene alguna pregunta, consulte a su proveedor de atención
médica.
No use EGRIFTA® después de la fecha indicada en la caja del
medicamento y el frasco de EGRIFTA®.
No use una jeringa o aguja más de 1 vez.
No comparta sus agujas de EGRIFTA® con otras personas.
Puede contagiarles una infección o contagiarse de ellos.
Compartir agujas puede provocar el contagio de enfermedades
infecciosas, como el VIH. No comparta su jeringa de EGRIFTA® con
otras personas, aunque cambie la aguja.
Si falta algún insumo de la caja del medicamento o la caja de la
inyección, o si algo parece estar dañado, llame a su farmacéutico o
comuníquese con ® al número de teléfono gratuito 1-
844-EGRIFTA (1-844-347-4382) de inmediato.
Cómo preparar la inyección de EGRIFTA®
Paso 1: Encuentre una superficie bien iluminada, limpia y plana, como
una mesa.
Paso 2: Reúna todos los insumos:
La caja del medicamento que contiene 60 frascos de EGRIFTA® en
polvo
La caja de la inyección con el siguiente contenido:
a) 30 frascos de 10 ml de agua estéril para inyección, que se
usan para la mezcla
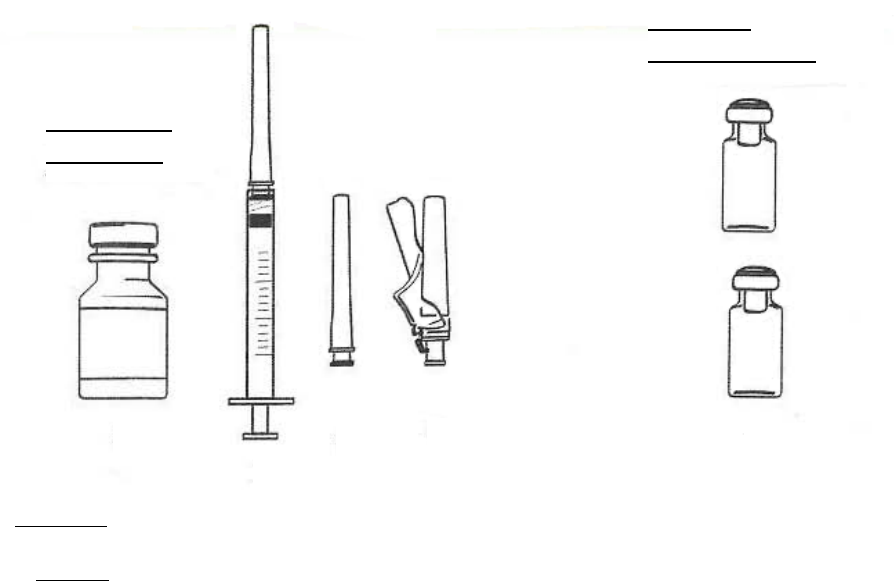
b) 30 jeringas estériles de 3 ml con aguja estéril ya conectada
c) 30 agujas estériles individuales, calibre 18, de 1½”, que se
usan para la mezcla
d) 30 agujas para inyección, calibre 27, de ½”
Otros insumos necesarios
Toallitas con alcohol
Gasa estéril
Un “recipiente para objetos punzantes” o un recipiente
resistente a punciones para desechar las agujas y las
jeringas usadas cuando haya terminado.
Material incluido en la caja de la inyección (a, b, c, d) y la caja del
medicamento (e):
Figura A
Paso 3: Retire los siguientes insumos de la caja de la inyección:
Un frasco de agua estéril para inyección (Figura A, a)
Una jeringa con la aguja ya conectada (Figura A, b)
Una aguja calibre 18, de 1½” (Figura A, c)
Una aguja para inyección calibre 27, de ½” (Figura A, d)
Caja de la
inyección
Caja del
medicamento
a
b
c d e
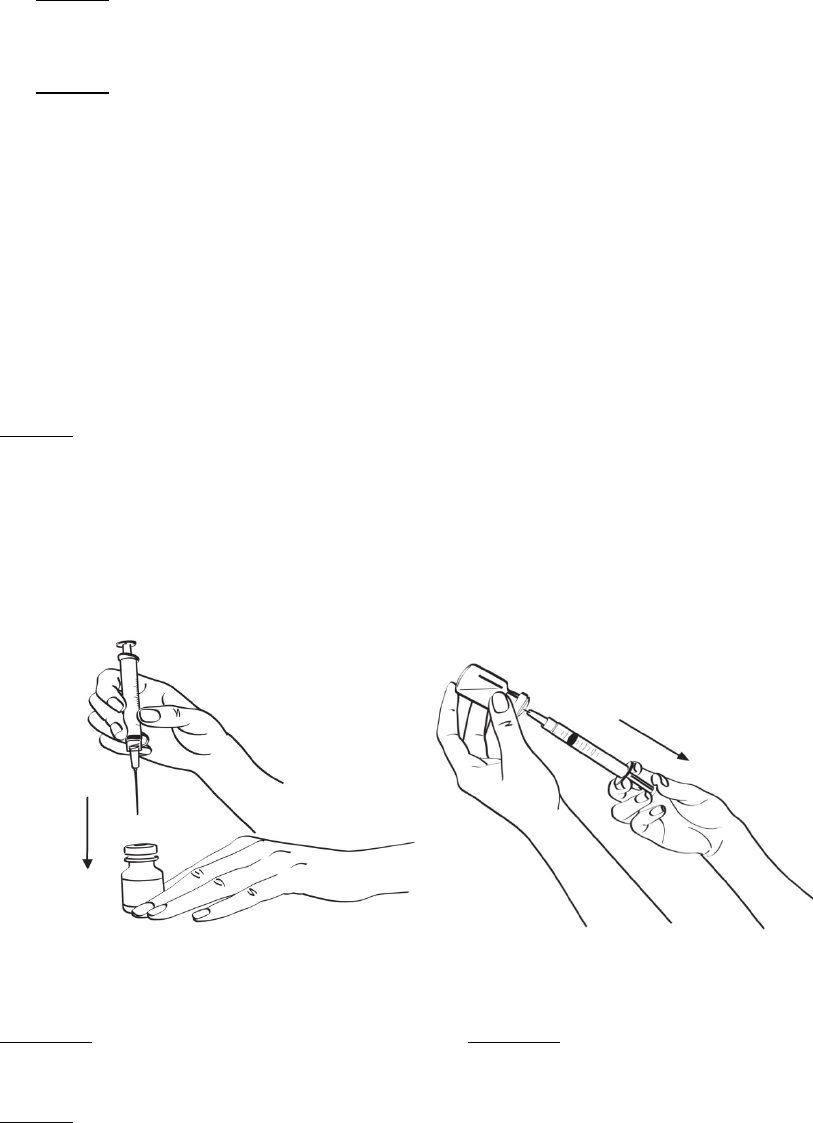
Paso 4: Tome 2 frascos de EGRIFTA® (Figura A, e) de la caja del
medicamento. Vuelva a guardar la caja con el resto de los
frascos en el refrigerador de inmediato.
Paso 5: Prepárese para usar los insumos:
Lávese las manos con agua y jabón. Séquese las manos
con una toalla limpia.
Retire las tapas plásticas de los frascos de EGRIFTA® y el
frasco de agua estéril.
Limpie los tapones de caucho sobre los 2 frascos de
EGRIFTA® y el frasco de agua estéril con un algodón
impregnado en alcohol.
Cómo mezclar EGRIFTA®
Paso 1: Tome la jeringa con la aguja conectada (Figura A, b), retire la tapa
protectora e inserte la aguja a través del tapón de caucho del frasco de agua
estéril (Figura A, a; consulte la Figura B a modo de ilustración). Invierta la
posición de ambos y tire del émbolo hasta que el líquido alcance la marca de
2.2 ml en la jeringa. (Consulte la Figura C)
Figura B Figura C
Paso 2: Saque la jeringa con la aguja conectada del frasco de agua estéril e
inserte la aguja en uno de los frascos de EGRIFTA®. Presione el émbolo hacia
adentro lentamente en un ligero ángulo de manera que el agua descienda
por la pared interior del frasco de EGRIFTA® en lugar de hacerlo
directamente hacia el polvo para evitar que se forme espuma. (Consulte la
Figura D)

Figura D
Paso 3: Mientras mantiene la jeringa con la aguja conectada en el frasco y el
frasco en posición vertical, haga rodar el frasco con cuidado en sus manos
durante 30 segundos, hasta que el agua estéril y el polvo de EGRIFTA® se
mezclen bien. No agite el frasco. (Consulte la Figura E)
Figura E
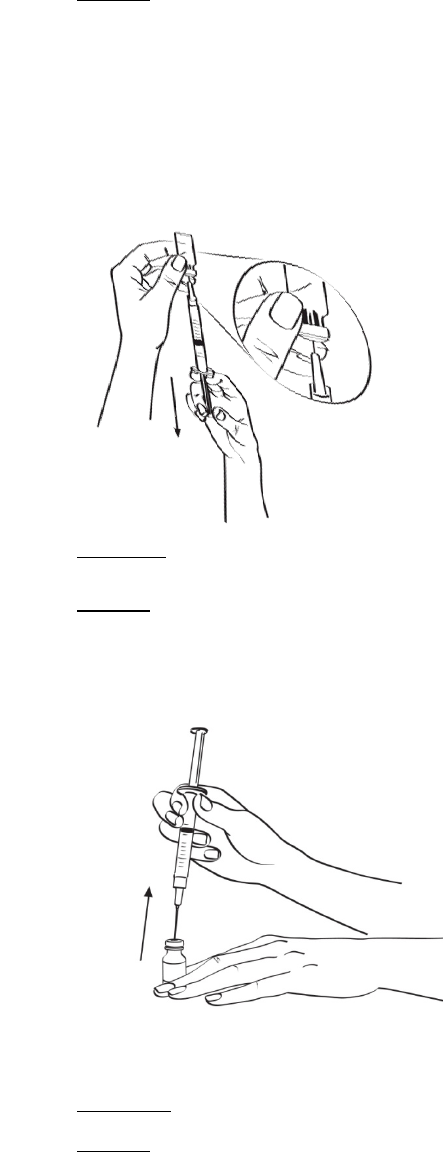
Paso 4: Aún manteniendo la jeringa con la aguja conectada en el frasco,
inviértalos a ambos hasta enderezar la jeringa. Presione la jeringa hacia
abajo hasta ver solo la punta de la aguja que atraviesa el tapón de caucho;
luego, tire del émbolo hasta que todo el líquido en el interior del frasco pase
a la jeringa. El nivel de medicamento en la jeringa debe estar
aproximadamente en la marca de 2.2 ml de la jeringa. (Consulte la Figura F)
Figura F
Paso 5: Retire la aguja del frasco. (Consulte la Figura G)
Figura G
Paso 6: Coloque la tapa de la aguja a un costado sobre una superficie limpia
y plana. Sin tocar la aguja, sostenga la jeringa y deslice la aguja
cuidadosamente dentro de la tapa protectora (Consulte la Figura H). Empuje
la tapa hasta el fondo o hasta que se cierre a presión (Consulte la Figura I).
No toque la tapa hasta que no cubra la aguja por completo.
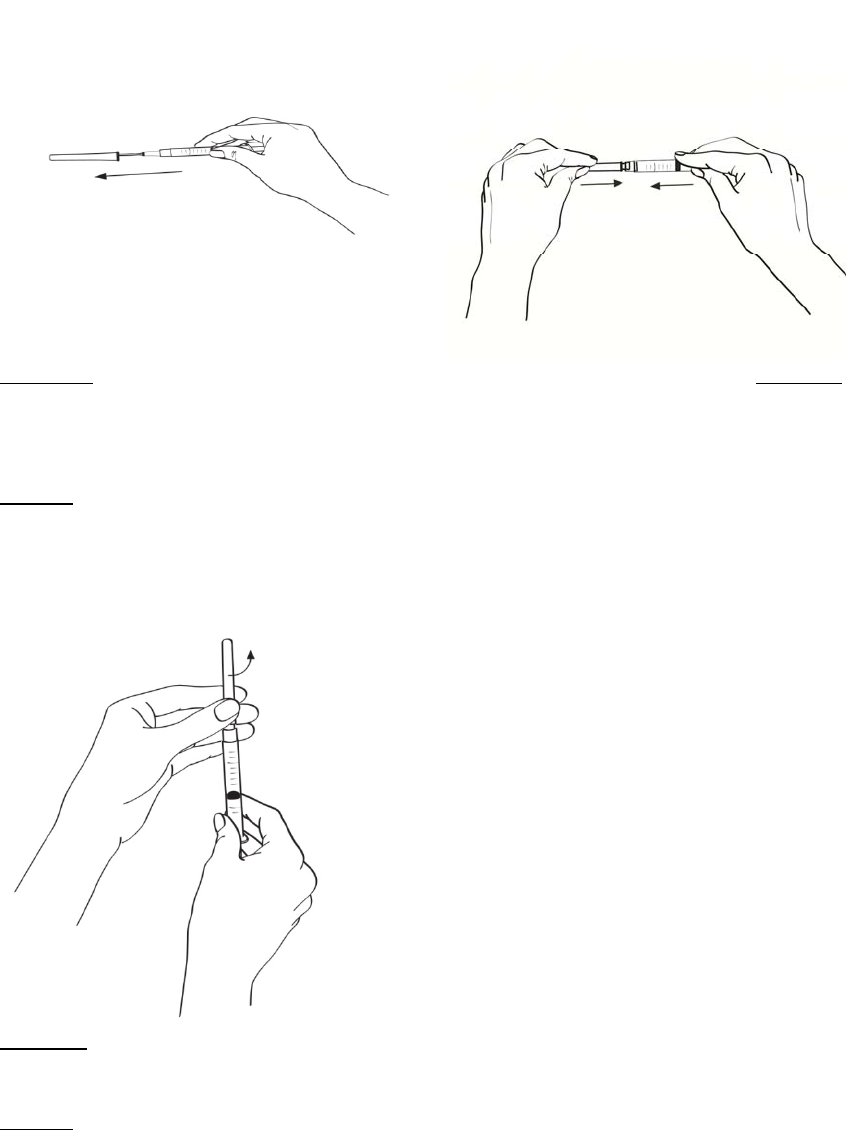
Figura H Figura I
Paso 7: Con la tapa en la aguja, retire la aguja sosteniendo la jeringa con
firmeza y haciendo girar la tapa en sentido antihorario (hacia la izquierda).
(Consulte la Figura J)
Figura J
Paso 8: Coloque la aguja para mezclar, calibre 18, de 1½” (Figura A, c), con
la tapa protectora colocada, en la jeringa. Gire la tapa en sentido horario
(hacia la derecha) hasta que quede ajustada. (Consulte la Figura K)

Figura K
Paso 9: Retire la tapa protectora e inserte la aguja en el segundo frasco de
EGRIFTA® (Figura A, e). Presione el émbolo hacia adentro lentamente en un
ligero ángulo de manera que la mezcla descienda por la pared interior del
frasco de EGRIFTA® en lugar de hacerlo directamente hacia el polvo para
evitar que se forme espuma (consulte la Figura L).
Figura L
Paso 10: Mientras mantiene la jeringa en el frasco y el frasco en posición
vertical, haga rodar el frasco con cuidado en sus manos durante
30 segundos, hasta que el agua y el polvo se mezclen bien. (No agite el
frasco). La solución debe tener un aspecto trasparente e incoloro, sin
partículas visibles. (Consulte la Figura M)

Figura M
Paso 11: Manteniendo aún la jeringa en el frasco, inviértalos a ambos hasta
enderezar la jeringa. Presione la jeringa hacia abajo hasta ver solo la punta
de la aguja que atraviesa el tapón de caucho; luego, tire del émbolo hasta
que todo el líquido en el interior del frasco pase a la jeringa. El nivel de
medicamento en la jeringa debe estar aproximadamente en la marca de
2.2 ml de la jeringa. (Consulte la Figura N)
Figura N
Paso 12: Retire la aguja del frasco. (Consulte la Figura O)
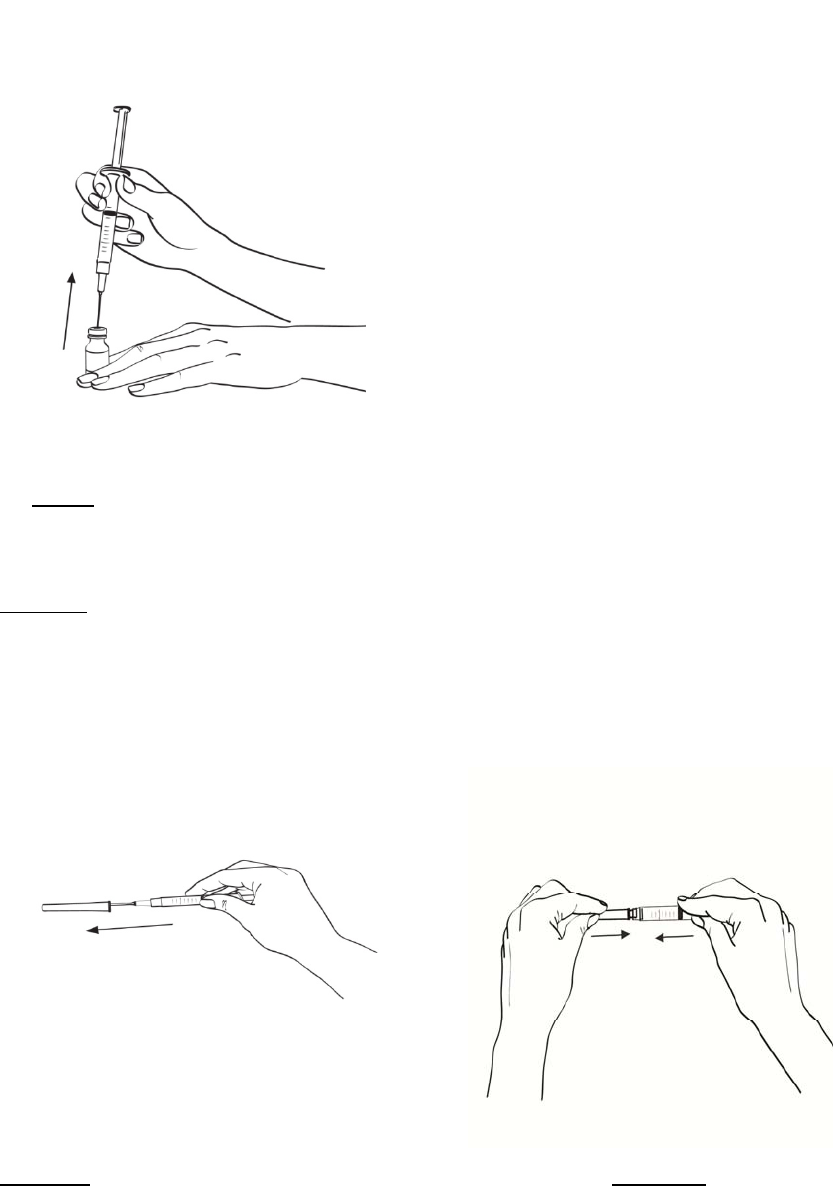
Figura O
Paso 13: Coloque la tapa de la aguja a un costado sobre una superficie limpia
y plana. Sin tocar la aguja, sostenga la jeringa y deslice la aguja
cuidadosamente dentro de la tapa protectora (consulte la Figura P). Empuje
la tapa hasta el fondo y hasta que se cierre a presión (consulte la Figura Q).
No toque la tapa hasta que no cubra la aguja por completo.
Figura P Figura Q
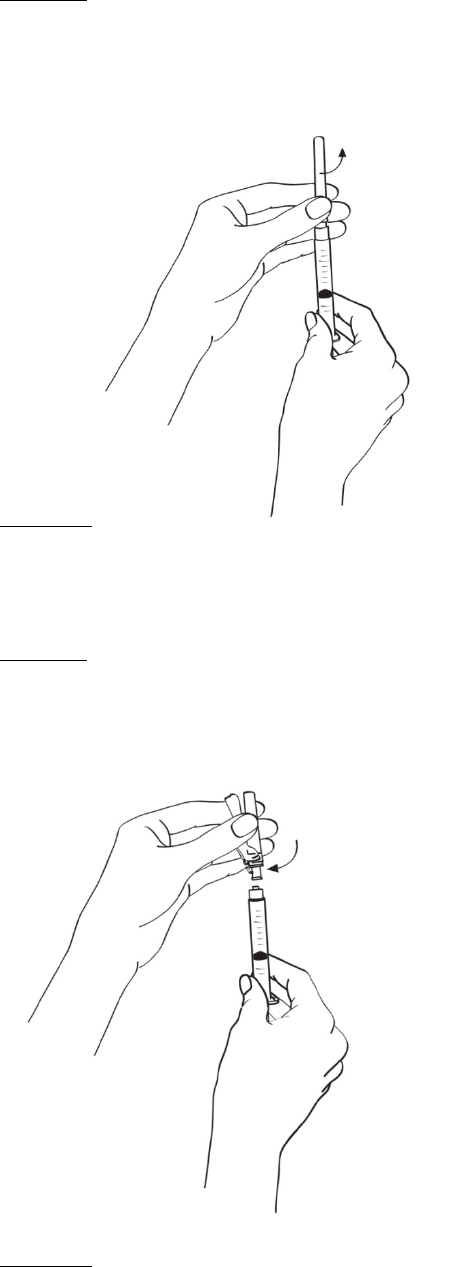
Paso 14: Con la tapa colocada en la aguja, retire la aguja para mezclar
sosteniendo la jeringa con firmeza y haciendo girar la tapa en sentido
antihorario (hacia la izquierda). (Consulte la Figura R)
Figura R
Paso 15: Coloque la aguja para inyección (Figura A, d), con la tapa protectora
colocada, en la jeringa. Sostenga la jeringa con firmeza y gire la tapa en
sentido horario (hacia la derecha) hasta que se cierre bien. (Consulte la
Figura S)
Figura S
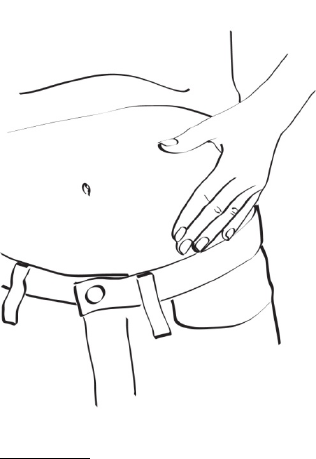
¿Dónde me inyecto EGRIFTA®?
Debe inyectarse EGRIFTA® en la piel del estómago (abdomen). (Consulte la
Figura T)
Elija un lugar de inyección cerca del ombligo, hacia la izquierda o la
derecha.
No elija ningún área donde haya tejido cicatricial, moretones,
enrojecimiento, infección o irritación.
Evite las zonas en las que haya protuberancias duras a causa de
inyecciones previas.
Cambie el lugar de aplicación de la inyección de un día al otro. De
esta forma puede evitarse la formación de hematomas o irritación.
Es conveniente anotar la fecha y el lugar de cada inyección diaria
como ayuda para recordar.
Figura T
Cómo inyectarse EGRIFTA®
Tome la jeringa y retire la tapa de la aguja de la inyección. No la gire.
(Consulte la Figura U)
Figura U
Dé golpecitos ligeros y suaves con el dedo para hacer que las burbujas
de aire suban. Presione el émbolo para que salgan las burbujas.
(Consulte la Figura V)
Figura V
Limpie el lugar de la inyección que ha elegido con un algodón
impregnado en alcohol y déjelo secar. Sostenga la jeringa con una
mano. Con la otra mano, sostenga un pliegue de piel limpio para la

inyección. Sostenga la piel entre el pulgar y los dedos. (Consulte la
Figura W)
Figura W
Sostenga la jeringa en un ángulo recto respecto de la piel, como un
dardo. Empuje la aguja de la inyección insertándola en la piel con un
movimiento rápido. Casi toda la aguja debe quedar debajo de la
superficie de la piel. (Consulte la Figura X)
Figura X
Retire la mano del área de piel pellizcada después de introducir la
aguja. Asegúrese de que la aguja quede dentro de la piel. (Consulte la
Figura Y)
Figura Y
Presione lentamente el émbolo hasta el fondo, hasta que todo el
medicamento en la jeringa se haya inyectado debajo de la piel.
Retire la aguja de la inyección de la piel cuando la jeringa esté vacía:
Tenga cuidado de retirarla en el mismo ángulo que la introdujo.
Vuelva a colocar la protección de la aguja de color rosa hasta que
cierre a presión, cubriendo la aguja de la inyección por completo.
Siga presionando hasta escuchar un clic; eso significa que la aguja
de la inyección está protegida. (Consulte la Figura Z)
Figura Z

Use un trozo de gasa estéril para limpiar el lugar de la inyección. Si
sangra, aplique presión en el lugar de la inyección con una gasa durante
30 segundos. Si el sangrado continúa, aplique un vendaje en el lugar.
¿Cómo debo desechar las jeringas, agujas, frascos y frascos usados?
No vuelva a tapar la aguja ni retire la aguja de la jeringa después de
inyectarse EGRIFTA®.
Coloque las agujas y jeringas de EGRIFTA® usadas en un recipiente
para el descarte de objetos punzantes aprobado por la FDA
inmediatamente después del uso. No tire (deseche) agujas y
jeringas sueltas en la basura de su hogar.
Si no tiene un recipiente para el descarte de objetos punzantes
aprobado por la FDA, puede usar un recipiente doméstico que:
o esté hecho de plástico duro,
o pueda cerrarse bien fuerte, con tapa resistente a punciones, de
manera que las objetos punzantes no puedan salirse,
o esté vertical y estable durante el uso,
o sea resistente a filtraciones, y
o este adecuadamente etiquetado para advertir que contiene
desechos peligrosos en su interior.
Cuando el recipiente para desechar objetos punzantes esté casi lleno,
deberá seguir las pautas de su comunidad para el desecho adecuado
de este tipo de recipiente. Pueden existir leyes estatales o locales
acerca de cómo se deben desechar las agujas y los bolígrafos para
inyección usados. Para obtener más información sobre el desecho
seguro de objetos punzantes e información específica sobre el desecho
de objetos punzantes en el estado donde usted vive, visite el sitio web
de la FDA en: http://www.fda.gov/safesharpsdisposal.
No deseche el recipiente para desechar objetos punzantes en la
basura de su hogar a menos que las pautas de su comunidad lo
permitan. No recicle el recipiente para desechar objetos punzantes.
Si por accidente pincha a otra persona con una aguja usada, se debe
informar a esa persona que se comunique con un proveedor de
atención médica de inmediato para consultar sobre el accidente.
Mantenga el recipiente de objetos punzantes lejos de niños y mascotas.
Si tiene alguna pregunta, llame a su proveedor de atención médica. Puede
llamar a ® al número de teléfono gratuito 1-844-EGRIFTA (1-
844-347-4382) o visitar el sitio web de EGRIFTA® en www.EGRIFTA.com
para obtener más información.
¿Cómo debo almacenar EGRIFTA®?
EGRIFTA® viene en dos cajas dispensadas por la farmacia:
o Almacene los frascos de la caja del medicamento EGRIFTA® en el
refrigerador entre 2 °C y 8 °C (36 °F y 46 °F).
Almacene la caja de agua estéril para inyección, las jeringas y las
agujas a temperatura ambiente entre 20 °C y 25 °C (68 °F y 77 °F).
Guarde los frascos de EGRIFTA® alejados de la luz.
No congelar.
Después de mezclar, use EGRIFTA® de inmediato y descarte todo el
EGRIFTA® restante. No almacene la mezcla de EGRIFTA®. Además,
deseche el frasco usado de agua estéril para inyección.
No use EGRIFTA® después de la fecha de vencimiento impresa en la caja
del medicamento y en las etiquetas del frasco.

Información general sobre el uso eficaz y seguro de EGRIFTA®.
Mantenga EGRIFTA® y todos los medicamentos fuera del alcance de los
niños.
Estas Instrucciones de uso han sido aprobadas por la Administración de
Alimentos y Medicamentos de los EE. UU.
Fabricado por: Jubilant HollisterStier General Partnership, 16751 Trans-
Canada Highway, Montreal, Québec, Canadá H9H 4J4
EGRIFTA® es una marca registrada de Theratechnologies Inc.
Distribuido por: Theratechnologies Inc., Montreal, Québec, Canadá H3A 1T8
Revisado: 06/2015
