Manual_1.3x EStok TP Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 42
1
EStokTP
From Electronic Structure to
Temperature and Pressure
Dependent Rate Constants
Carlo Cavallotti, Matteo Pelucchi
Dipartimento di Chimica, Materiali e Ingegneria Chimica "Giulio
Natta", Politecnico di Milano, 20131 Milano, Italy
Stephen J. Klippenstein
Chemical Sciences and Engineering Division, Argonne National
Laboratory, Argonne, IL, 60439, USA
2
Table of Contents
1.Overview of EStokTP................................................................................................................4
1.1.Purpose......................................................................................................................................4
1.2.Capabilities...............................................................................................................................5
1.2.1.Current....................................................................................................................................5
1.2.2.Planned...................................................................................................................................6
1.3.EStokTP Jobs: theory and sequential module calls...........................................................6
1.3.1.Well.........................................................................................................................................6
1.3.2.Abstraction............................................................................................................................7
1.3.3.Addition...............................................................................................................................11
1.3.4.BetaScission.........................................................................................................................14
1.3.5.Isomerization.......................................................................................................................15
1.4.Input/Output Overview...........................................................................................................16
2.Installation.................................................................................................................................18
2.1.Distribution.............................................................................................................................18
2.2.Compiling................................................................................................................................18
2.3.Execution Environment.........................................................................................................18
2.4.Running...................................................................................................................................19
3.Input files...................................................................................................................................20
3.1.estoktp.dat: job selection and modules..............................................................................20
3.1.1.Reaction Type/Global Keywords.....................................................................................20
3.1.2.List of Modules...................................................................................................................22
3.1.3.Computational environment.............................................................................................26
3.2.Species Data Files..................................................................................................................26
3.2.1.reac1.dat, reac2.dat, prod1.dat, prod2.dat.....................................................................26
3.2.2.wellr.dat, wellp.dat.............................................................................................................29
3.2.3.ts.dat.....................................................................................................................................30
3.3.Electronic Structure and Master Equation Inputs..........................................................32
3.3.1.Theory files..........................................................................................................................32
3.3.1.1.Theory.dat........................................................................................................................32
3
3.3.1.2.Molpro theory files..........................................................................................................35
3.3.2.Master Equation files.........................................................................................................37
3.3.2.1.me_head.dat.....................................................................................................................37
4.Output........................................................................................................................................38
4.1.EStokTP Outputs..................................................................................................................38
4.1.1.Geometries...........................................................................................................................38
4.1.2.Frequencies..........................................................................................................................39
4.1.3.Hindered Rotor...................................................................................................................39
4.1.4.IRC.......................................................................................................................................39
4.1.5.Energies................................................................................................................................39
4.1.6.Multi Dimensional Tunneling...........................................................................................40
4.2.Rate Output............................................................................................................................40
5.Examples....................................................................................................................................41
6.Advices/Suggestions/Troubleshooting...................................................................................41
7.Index...........................................................................................................................................42
4
1. Overview of EStokTP
1.1. Purpose
In recent years, theoretical chemical kinetics has transformed from an empirical to a
predictive science, with achievable accuracies approaching or even exceeding that of
experiments. This transformation arises from the dramatic progress in the accuracy of electronic
structure calculations, as well as improved procedures for incorporating such predictions within
treatments of chemical dynamics and kinetics. The ab initio transition state theory based master
equation (AITSTME) approach, which provides the basis for much of the work in this area, is
finding widespread utility as a tool for chemical kinetic modeling. This utility could be greatly
enhanced by reducing the human effort involved in the implementation of such calculations.
Typically, such calculations are performed in three distinct steps: First the molecular
properties of the stationary points on the potential energy surface are predicted with ab initio
(AI) electronic structure codes. Microcanonical rates for chemical transfer are then predicted
with transition state theory (TST) codes that incorporate these stationary point properties. Finally,
thermal rate estimates are obtained with master equation (ME) solver codes, which rely on the
microcanonical and energy transfer rates. While, the transition state theory and master equation
calculations are often performed within the same code, the electronic structure calculations are
almost always performed independently. This decoupling of the electronic structure evaluations
leads to considerable human effort wasted on the transfer of data from one set of codes to the
other.
The EStokTP computational environment is designed to directly couple the electronic
structure, TST, and ME evaluations in automatically obtaining a priori predictions of the
temperature and pressure dependent rate constants. The code requires as input the reference
molecular structures, the transition states to be searched, and the electronic structure and
transition state theory methodologies to be employed. Given this set of input files, the code
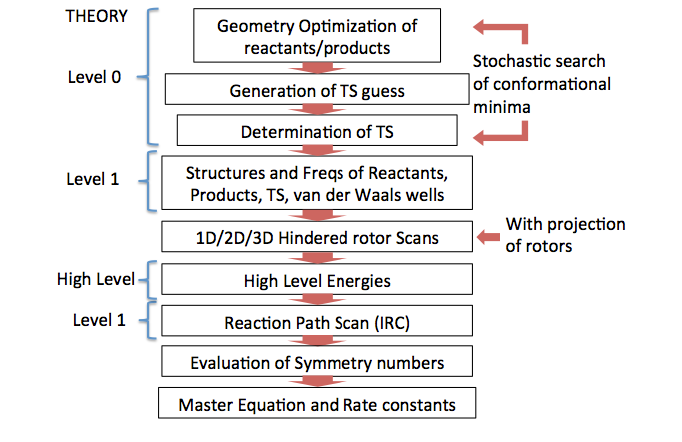
performs sequentially the series of calculations listed in Figure 1.
Figure1:EStokTPprogramstructure.
5
EStokTP relies on calls to external codes to perform electronic structure and master equation
calculations. Internal torsions can be treated as hindered rotors, in which case they are projected
out from the Hessian so that no identification and removal from the frequency list is necessary.
Tunneling can be accounted for using both Eckart and multidimensional models, such as small
curvature theory. The code has been written following two principles. The first is that it must be
able to reliably and automatically determine a large number of rate constants for classes of
reactions of a target molecule (e.g. H-abstractions). This requirement is motivated by the
perspective of the upcoming availability of exascale computational resources. The code is thus
robust, with several automatic fallback options implemented so that human intervention can be
limited. The second is that it must be capable of determining highly accurate rate constants,
differing by no more than a factor of two from experiments. At present the following reaction
types are implemented: abstraction, addition, isomerization, and beta-decomposition.
EStokTP can be used also to investigate complicated potential energy surfaces. For this
purpose all reaction steps are studied separately applying repeatedly EStokTP. There is no need
of repeating calculations for reactants or products more than once as scripts allow copying all
reactants and products data necessary for a calculation from one directory to the other. After all
the desired reaction steps have been performed, a multiple well master equation input can be
written using a wrapper script.
1.2. Capabilities
1.2.1. Current
- determination of minimum energy structures for wells and saddle points performing
Monte Carlo conformational analysis over specified list of internal coordinates;
- 1D, 2D or 3D rotational scan over list of dihedral coordinates;
- project out torsional motions from Hessian (implies no need to identify torsional
vibrational frequencies manually);
- possibility to project out of the Hessian the motion along the reaction path
- automatic multilevel determination of structures (level0 followed by level1
calculations);
- search for high quality guesses for abstraction, addition, beta-scission and
isomerization reactions;
- determination of internal and external rotational symmetry numbers, including optical
symmetry numbers;
- search for structures of van der Waals wells on reactant and product side;
- IRC calculations with PES evaluation and projection from the Hessian of hindered
rotors along the IRC path at selected points with interpolation for intermediate points;
rescaling of energy at a high level quality along the IRC path is also supported;
- Small Curvature Tunneling calculations, implemented using reaction path
Hamiltonian frequencies;
- preparation of input for the Mess master equation solver, which is at present used to
compute thermodynamic data, multi dimensional partition functions for hindered
rotors, and pressure dependent rate constants;
- two electronic structure calculation codes are currently supported: G09 and molpro
(2008-2015 are supported). All calculation steps can be done with any of the two
6
codes made exception for the internal reaction coordinate calculations, which are
supported only for G09.
1.2.2. Planned
- automatic determination of rate constants for barrierless reactions
- anharmonic analysis
1.3. EStokTP Jobs: theory and sequential module calls
EStokTP is made of a series of modules that can be called separately. Each module requires a set
of data, which are reported in the ./data subdirectory for the initial calculations (e.g. level 0
geometry optimizations), while geometries and Hessian are stored in the ./output and ./geoms
subdirectories for later stages of the calculations (such as for saddle point determination or high
level calculations).
EStokTP modules can be used to perform five types of calculations (jobs): well, abstraction,
addition, beta-scission, or isomerization. For each job we suggest a set of modules to call, as
listed below. Changes to the proposed call list are possible, though in order to obtain the
production of the master equation blocks and, for the reactions, of the master equation input
for the Mess solver the level0, level1 and high-level calculations must always be performed (see
section 1.4 for a description of what is stored in a block or master equation input file). The
hindered rotor analysis, IRC, and symmetry calculations can be omitted, if thought unnecessary.
1.3.1. Well
1) Opt_Reac1
2) Opt_Reac1_1
3) 1dTau_Reac1
4) mdtau_reac1
5) HL_Reac1
6) Symm_reac1
7) kTP
In step 1, the minimum energy structure for reactant 1 is searched at level 0 of theory. In step 2
geometry and Hessian are determined at level 1. In step 3 1D hindered rotor scans are performed.
In step 4 multidimensional hindered rotor scans are performed. If 1d and multi dimensional HR
scans are not requested, step 3 and 4 can be omitted. In step 5 energies are determined at the
specified high level of theory on level1 geometries. In step 6 symmetry numbers are determined.
In step 7 the output are written in the Mess block format (briefly, me block), which can be used
to generate the input necessary to compute thermodynamic parameters or for multi well master
question calculations. The calculation output, which comprises level 1 geometry, level 1
frequencies from which hindered rotors have been projected, list of 1d hindered rotors, with
rotating groups, axis and rotational pes, and symmetry is summarized in one file that is written
by the kTP module in the ./me_block subdirectory in the reac1.me file. The high level energy
expressed in Hartrees is stored in the same subdirectory in the reac_en.me file. If multi
dimensional hindered rotors calculations have been requested, the PES files are copied as well in
7
the ./me_block subdirectory and are named reac1_2dhr_01.dat and reac1_2dhr_nofr01.dat, with
the first file containing the PES and the frequencies calculated and projected along the hindered
rotor path and the second one only the PES.
1.3.2. Abstraction
Rate constant calculations for abstraction reactions can be performed using three different PESs,
which differ depending on whether van der Waals wells are considered both on the reactant and
product sides (3TS calculation), only on the reactant side (2TS calculation) or not considered
(1TS calculations). The approach is chosen with the ‘reactiontype’ keyword (see section 3.1.1).
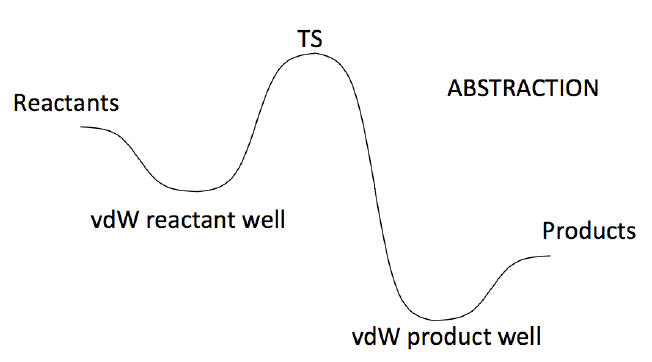
A scheme of the 3TS PES is sketched in Figure 2.
Figure2:PESusedtocalculatedtheabstractionreactionusingathreetransitionstates(3TS)model.
In the 3TS model, the abstraction rate constant is computed solving a master equation consisting
of three channels: entrance, exit and reaction. The rates of the entrance and exit channels are
computed using phase space theory, while that of the reaction channel is determined using
transition state theory. Phase space theory calculations are set up and performed automatically by
the Mess solver, which requires only information on the wells it connects. In this case they are
the bimolecular reactants and the vdW reactant well for the entrance channel and the vdW
product well and the bimolecular products for the exit channel. Usually the rates of entrance and
exit channels largely exceed those of the reaction channel, so that the latter becomes the
controlling step. However at sufficiently low temperatures the van der Waals well may live long
enough time to get collisionally stabilized, in which case they would appear as a possible product
channel in the master equation results. The depth of the van der Waals well may have a
significant impact, especially at low temperatures and when the reaction barrier height is small
with respect to reactants, on the quantum tunneling contribution to the rate constant. It is thus
adviced to compute the wells energy and structure at the same level of theory as that of reactants
and products, though it is it is not necessary, unless the rate of formation of the van der Waals
well is desired, to perform a hindered rotor study of the van der Waals wells. In case of need, the
hindered rotor study of the van der Waals wells should be carefully checked as it may easily lead
to the finding of other van der Waals wells, which may not be those connected to the reaction
transition state. The 2TS and 1TS abstraction PESs are similar to that reported in Figure 2, from
8
which they differ for the absence of the van der Waals product well and for the absence of both
van der Waals wells, respectively.
The search for the transition state for the reaction channel is performed through a set of
restrained geometry optimizations where the energy of the structure consisting of the two
reactants is minimized while keeping fixed a coordinate, named RTS, which describes the
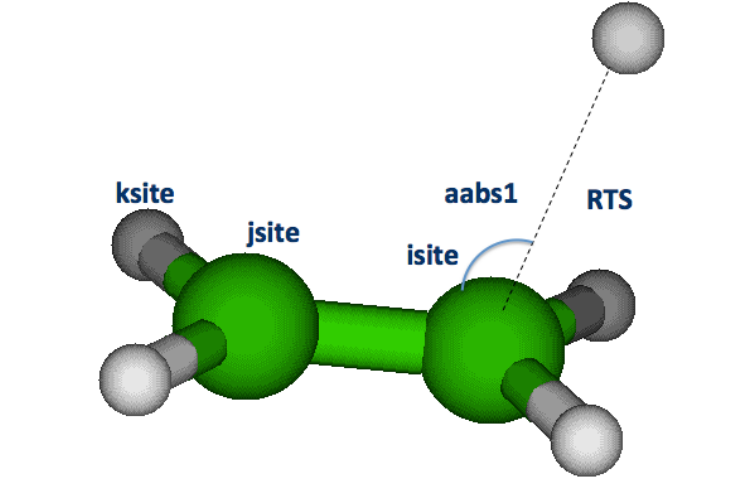
approach of reactant 2 to the reaction site (isite) of reactant 1. In particular the z-matrix
containing the two reactants is automatically constructed using as input the z-matrixes specified
for reactant 1 and reactant 2 in the reac1.dat and reac2.dat files. The relative position of reactant
2 with respect to reactant 1 is defined through the definition of 6 degrees of freedom (3 if
reactant 2 is an atom, 5 if it is a biatomic species). Since the abstracting atom is often co-linear
with the bond of the abstracted atom, a dummy atom is positioned at 2.0 Å from the abstraction
site (same as abstracted atom) and perpendicularly to the bond that is being broken in reactant 1
through the abstraction reaction. To construct the transition state z-matrix the following
information is needed: a list of three atoms of reactant 1 used to define the relative position of
reactant 2 and 5 angle coordinates: aabs1, babs1, aabs2, babs2, and babs3. The three atoms,
defined as isite, jsite and ksite are the abstraction site (isite), the atom to which the abstracted
atom is bound (jsite), and an atom, usually connected to jsite, which is used to define the dihedral
angle for the dummy atom. The dihedral angle is placed at 2.0 Å from isite, at 90 degrees with
respect to isite and jsite and on a dummy atom-isite-jsite plane that forms a 180 angle with the
isite-jsite-ksite plane. The first atom of reactant 2 is assumed to be the extracting atom (that is the
atom that will bind the extracted atom).
The definition of the 5 angle coordinates (transitional degrees of freedom) is the following:
aabs1: angle between first atom of reactant2, isite and dummy atom (usually 90°).
babs1: dihedral angle between first atom of reactant2, isite, dummy atom, and jsite (usually
180°).
aabs2: angle between second atom of reactant 2, fisrt atom of reactant2, and isite. It depends on
the reactant, often a good choice is 90°, if it is about 180° then it may give problems in
the optimization.
babs2: dihedral angle between second atom of reactant 2, first atom of reactant2, isite, and the
dummy atom. Its value depends on the system. It is likely that it is a torsional degree of
freedom, and should thus be considered as a hindered rotor and randomly scanned in the
tauo_ts module.
babs3: dihedral angle between third atom of reactant 2, the two atoms to which it is connected in
the reactant2 z-matrix structure, and isite. Its value depends on the system. It is likely that
it is a torsional degree of freedom, and should thus be considered as a hindered rotor and
randomly scanned in the tauo_ts module.
An example of a possible definition of isite, jsite, ksite, dummy atom, scanned coordinate as well
as of aabs1 and aabs2 is shown in Figure 3. Once the transition state z-matrix has been
constructed, the RTS distance is scanned in the interval and for the number of points specified in
the ts.dat input file (see section 3.2.3 for further details).
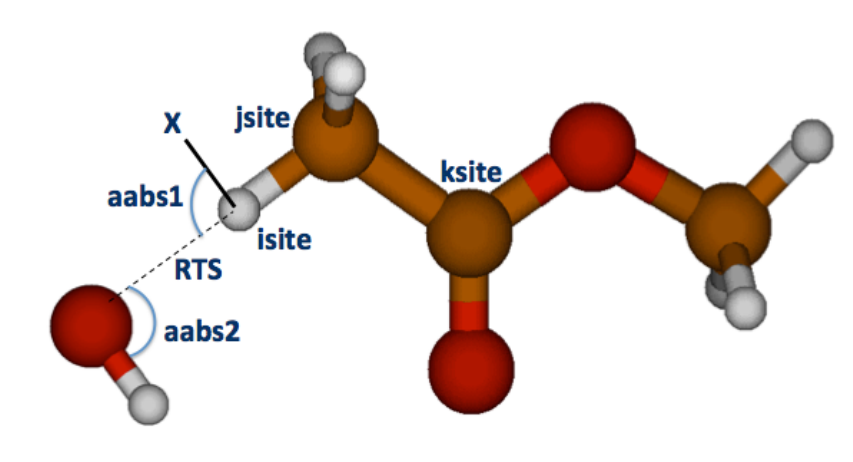
9
Figure3:TransitionstategeometryconstructedautomaticallyforHabstractionintheOH+methylactetatereaction.
Theisite,jsite,andksiteatomsspecifiedforthecalculationandtheresultingpositionofthedummyatom(X)areexplicitly
shown,togetherwithsomeofthegeometricalvariablesusedinthecalculations(thegridscanneddistanceRTS,andtwoof
theanglesdefiningthepositionoftheOHresiduerelativelytomethylacetate.
For a 3 TS abstraction reaction calculation the sequential list of modules that should be called is
the following:
First the geometries of reactants and products is determined at level 0 of theory.
Opt_Reac1
Opt_Reac2
Opt_Prod1
Opt_Prod2
Then the RTS coordinate is scanned in the interval and for the number of points requested in the
ts.dat file.
Grid_Opt_TS
The structure with the highest energy is used as guess for a saddle point search, which is
performed at level 0 of theory.
Opt_TS_0
Once the saddle point has been found, a random search is performed on the dihedral angles
specified in the ts.dat input for the requested number of times.
Tauo_Ts
Once the TS minimum energy structure has been determined, the structure of reactants, products
and transition state are determined at level 1 of theory.
Opt_Reac1_1
Opt_Reac2_1
Opt_Prod1_1
10
Opt_Prod2_1
Opt_TS_1
If the ‘wellr findgeom level1’ and ‘wellp findgeom level1’ keywords have been used in the
estoktp.dat file, than the structures of wellr and wellp are searched at level 1 of theory. A
minimum input is requested for the well input files (see section 3.2.2).
Opt_wellr_1
Opt_wellp_1
Hindered rotor scans are then performed for all the considered stationary points and wells.
1dTau_Reac1
1dTau_Reac2
1dTau_Prod1
1dTau_Prod2
1dTau_ts
1dTau_wellr
1dTau_wellp
High level energies are determined for all the considered stationary points and wells.
HL_Reac1
HL_Reac2
HL_Prod1
HL_Prod2
HL_WellR
HL_WellP
HL_Ts
Symmetry numbers are determined for all the considered stationary points and wells.
Symm_reac1
Symm_reac2
Symm_prod1
Symm_prod2
Symm_wellr
Symm_wellp
Symm_ts
An IRC calculation is performed for the number of points and using the algorithm specified in
the theory.dat file. It can be used to determine variationally the rate constant if the ‘variational’
keyword is present in the estoktp.dat file.
Irc
Having performed all the above calculation, we are now ready to compute the rate constant
calling the ktp module.
kTP
11
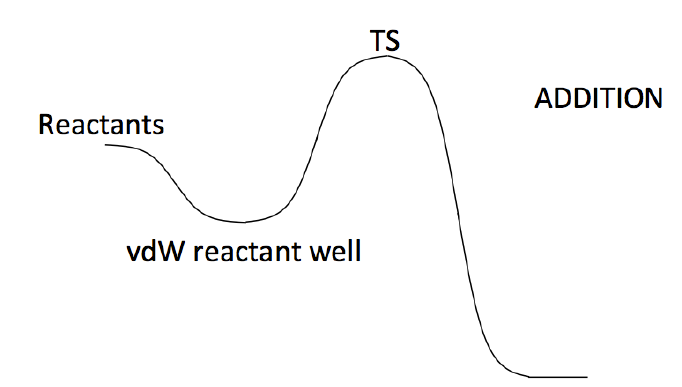
1.3.3. Addition
Rate constant calculations for addition reactions can be performed using two different PESs,
which differ depending on whether a van der Waals wells is considered on the reactant side (2TS
calculation) or not (1TS calculations). The selection is made with the ‘reactiontype’ keyword
(see section 3.1.1). A scheme of the 2TS PES is sketched in Figure 4.
Figure4:PESusedtocalculatedanadditionreactionusingatwotransitionstates(2TS)model.
Similarly to what seen for abstraction reactions, also in this case in the 2TS model the abstraction
rate constant is computed solving a master equation consisting of two channels: entrance, and
reaction. The rate of the entrance channel is computed using phase space theory, while that of the
reaction channel is determined using transition state theory. We refer to section 1.3.2 for a
discussion of what implies including a van der Waals well in a master equation and to set up a
calculation for the well.
The algorithm used to search for the reaction transition state is similar to the one used for
abstraction reactions. The main difference is that no dummy atom is used in this case to construct
the z-matrix of the transition state. Also in this case, however, a list of three atoms (isite, jsite,
and ksite) must be given in order to define the orientation of reactant 2 with respect to reactant1
in the z-matrix. In this case however isite, jsite and ksite are used to define the position of the
first atom of reactant 2, which is assumed to be the atom with which a bond between reactant 1
and 2 will be formed. The parameters aabs1, babs1, aabs2, babs2, and babs3 have the following
meaning:
aabs1: angle between first atom of reactant2, isite and jsite.
babs1: dihedral angle between first atom of reactant2, isite, jsite, and ksite.
aabs2: angle between second atom of reactant 2, first atom of reactant2, and isite.
babs2: dihedral angle between second atom of reactant 2, first atom of reactant2, isite and jsite.
Its value depends on the system. It is likely that it is a torsional degree of freedom, and
should thus be considered as a hindered rotor and randomly scanned in the tauo_ts
module.
12
babs3: dihedral angle between third atom of reactant 2, the two atoms to which it is connected in
the reactant2 z-matrix structure, and isite. Its value depends on the system. It is likely that
it is a torsional degree of freedom, and should thus be considered as a hindered rotor and
randomly scanned in the tauo_ts module.
An example of a possible definition of isite, jsite, ksite, dummy atom, scanned coordinate as well
as of aabs1 and aabs2 is shown in Figure 5. Once the transition state z-matrix has been
constructed, the RTS distance is scanned in the interval and for the number of points specified in
the ts.dat input file (see section 3.2.3 for further details).
Figure5:TransitionstategeometryconstructedautomaticallyforHadditiontoC2H4.Theisite,jsite,andksiteatoms
specifiedforthecalculationareexplicitlyshown,togetherwithsomeofthegeometricalvariablesusedinthecalculations
(thegridscanneddistanceRTS,andtwooftheanglesdefiningthepositionoftheHatomrelativelytoethylene.
For a 2 TS addition reaction calculation the sequential list of modules that should be called is the
following:
First the geometries of the two reactants are determined at level 0 of theory. Note that no product
needs to be specified here as it will be determined by the code as wellp using the findgeom
keyword.
Opt_Reac1
Opt_Reac2
Then the RTS coordinate is scanned in the interval and for the number of points requested in the
ts.dat file.
Grid_Opt_TS
The structure with the highest energy is used as guess for a saddle point search, which is
13
performed at level 0 of theory.
Opt_TS_0
Once the saddle point has been found, a random search is performed on the dihedral angles
specified in the ts.dat input for the requested number of times.
Tauo_Ts
Level 1 structures and vibrational frequencies are determined for all the considered stationary
points and wells. The structure of the van der Waals reactant well is determined using the
findgeom algorithm. The same algorithm is used to locate the product well, wellp, which in this
case will coincide with the reaction product.
Opt_Reac1_1
Opt_Reac2_1
Opt_TS_1
Opt_wellr_1
Opt_wellp_1
Hindered rotor scans are then performed for all the considered stationary points and wells.
1dTau_Reac1
1dTau_Reac2
1dTau_ts
1dTau_wellr
1dTau_wellp
High level energies are determined for all the considered stationary points and wells.
HL_Reac1
HL_Reac2
HL_WellR
HL_WellP
HL_Ts
Symmetry numbers are determined for all the considered stationary points and wells.
Symm_reac1
Symm_reac2
Symm_wellr
Symm_wellp
Symm_ts
An IRC calculation is performed for the number of points and using the algorithm specified in
the theory.dat file. It can be used to determine variationally the rate constant if the ‘variational’
keyword is present in the estoktp.dat file.
Irc
Having performed all the above calculation, we are now ready to compute the rate constant
calling the ktp module.
kTP
14
1.3.4. BetaScission
Beta-scission reactions are the inverse of addition, so that their rate constant may be determined
through detailed balance from the addition rate constant. Alternatively, given a reactant, a rate
constant for a beta-scission reaction can be determined using the betascission algorithm. This is a
relatively simple bond pulling algorithm that stretches the breaking bond for a given number of
steps, specified in the ts.dat input file. To use this algorithm the ‘reactiontype’ keyword (see
section 3.1.1) of the estoktp.dat file must be set to ‘betascission’. The rate of the bet-
decomposition reaction is determined solving the master equation calling the Mess code, so that
pressure dependence is properly accounted for. The parameters of the master equation
calculation are set in the me_head.dat file, located in the ./data subdirectory. No van der Waals
well is used for this type of calculation. The breaking bond is determined through the data
specified in the ts.dat input file. The number of the first of the two atoms of the breaking bond is
identified as isite. The jsite and ksite atom number should be specified as well in the input file
but are at present not used in the calculation, so that they can assume any value. The second atom
is the last number specified in the ts.dat input identified by the rmax1 keyword, namely ireact.
The other numbers are distance1, step1, distance2, and step2. The beta-scission algorithm is a
pushing algorithm working so that the distance between two atoms, isite and ireact, is
progressively increased from that of the minimum energy structure to distance1 with step step1
and then to distance2 with step step2. See section 3.2.3 for further details and an example.
The sequential list of modules that should be called for a standard beta-scission calculation is the
following:
First the geometries of the reactant and two products are determined at level 0 of theory.
Opt_Reac1
Opt_Prod1
Opt_Prod2
Then the breaking bond coordinate is scanned in the intervals requested in the ts.dat file using
the specified steps.
Grid_Opt_TS
The structure with the highest energy is used as guess for a saddle point search, which is
performed at level 0 of theory.
Opt_TS_0
Once the saddle point has been found, a random search is performed on the dihedral angles
specified in the ts.dat input for the requested number of times.
Tauo_Ts
Level 1 structures and vibrational frequencies are determined for all the considered stationary
points and wells.
Opt_Reac1_1
Opt_Prod1_1
Opt_Prod2_1
Opt_TS_1
Hindered rotor scans are then performed for all the considered stationary points and wells.
1dTau_Reac1
15
1dTau_Prod1
1dTau_Prod2
1dTau_ts
High level energies are determined for all the considered stationary points and wells.
HL_Reac1
HL_Prod1
HL_Prod2
HL_Ts
Symmetry numbers are determined for all the considered stationary points and wells.
Symm_reac1
Symm_prod1
Symm_prod2
Symm_ts
An IRC calculation is performed for the number of points and using the algorithm specified in
the theory.dat file. It can be used to determine variationally the rate constant if the ‘variational’
keyword is present in the estoktp.dat file.
Irc
Having performed all the above calculation, we are now ready to compute the rate constant
calling the ktp module.
kTP
1.3.5. Isomerization
Isomerization reactions are pressure independent unimolecular reactions that involve one
reactant and one product. Transition states for isomerizations are searched constructing a product
like z-matrix where the bond that is to be formed as a result of the reaction is explicitly expressed
as a z-matrix parameter. This is performed by substituting the z-matrix line that contains the
reacting atom (ireact) with a line where the forming bond appears explicitly. To use this
algorithm the ‘reactiontype’ keyword (see section 3.1.1) of the estoktp.dat file must be set to
‘isomerization’. The breaking bond is determined through the data specified in the ts.dat input
file. The number of the first of the two atoms of the breaking bond is the last number specified in
the ts.dat input identified by the rmin1 keyword, namely ireact. The other numbers are distance1,
step1, distance2, and step2. The second atom is identified as isite. The jsite and ksite atom
numbers are used to define the connectivity (angle and dihedral) of the ireact and isite atoms.
The isomerization algorithm is a pulling algorithm working so that the distance between two
atoms, isite and ireact, is progressively decreased from that of the starting structure to distance1
with step step1 and then to distance2 with step step2. The starting structure is not the minimum
energy structure, but the conformer found in the Monte Carlo search for the minimum energy
structure of reactant 1 that exhibits the minimum distance between ireact and isite. See section
3.2.3 for further details and an example.
The sequential list of modules that should be called for a standard isomerization calculation is
the following:
First the geometry of the reactant and is optimized at level 0 of theory.
16
Opt_Reac1
Then the forming bond coordinate is scanned in the specified intervals (distance1 and distance 2)
using the step specified in the ts.dat file.
Grid_Opt_TS
The structure with the highest energy is used as guess for a saddle point search, which is
performed at level 0 of theory.
Opt_TS_0
Once the saddle point has been found, a random search is performed on the dihedral angles
specified in the ts.dat input for the requested number of times.
Tauo_Ts
Level 1 structures and vibrational frequencies are determined for all the considered stationary
points and wells. The findgeom wellp algorithm is in this case used to find the product structure.
Opt_Reac1_1
Opt_TS_1
Opt_wellp_1
Hindered rotor scans are then performed for all the considered stationary points and wells.
1dTau_Reac1
1dTau_wellp
1dTau_ts
High level energies are determined for all the considered stationary points and wells.
HL_Reac1
HL_wellp
HL_Ts
Symmetry numbers are determined for all the considered stationary points and wells.
Symm_reac1
Symm_wellp
Symm_ts
Having performed all the above calculations, we are now ready to compute the rate constant
calling the ktp module.
kTP
1.4. Input/Output Overview
In order to run EStokTP it is suggested to create a dedicated directory for each calculation type,
may it be just a well or a full reaction calculation. The directory must contain the ./data
subdirectory, which must contain all input files necessary to run EStokTP.
The primary input is contained in the estoktp.dat file, where the type of calculation and list of
modules to call, as well as the computational environment (number of nodes and memory to use)
are defined. See section 3.1 for details on what should go in estoktp.dat.
17
A dedicated file must be used for each species considered in the calculations, so that it may be
necessary to compile up to seven species input files: reac1.dat, reac2.dat, wellr.dat, ts.dat,
wellp.dat, prod1.dat, prod2.dat. See section 3.2 for details.
The level of theory for the Electronic Structure calculations is specified in the theory.dat file.
This may be sufficient if only g09 is used to perform the calculations. If molpro is used as well
or alternatively, module specific theory files will be needed. These are level0_molpro.dat,
level1_molpro.dat, hl_molpro.dat, and onedtau_molpro.dat. Additionally, specific inputs for
saddle point calculations can be requested, in which case we should add level0_ts_molpro.dat,
level1_ts_molpro.dat, hl_ts_molpro.dat, and onedtau_ts_molpro.dat. See section 3.3 for
details.
The input necessary to run master equation simulations using Mess is specified in the
me_head.dat file, while if only thermochemical data are requested, the thermp.dat file must be
compiled.
The calculation outputs are stored into eight subdirectories that are created contextually to the
first call of EStokTP: level0 and level1 geometries are stored in the ./geoms subdirectory;
modules output and z-matrix parameters of geometries are stored in the ./output subdirectory;
files used in irc calculations are stored in the ./irc_files subdirectory; input and output files for
multi dimensional tunneling calculations are stored in the ./md_tunn subdirectory; the log files
of all high level calculations are stored in the ./hl_logs subdirectory; geometries of hindered
rotor structures computed while performing the PES scan of 1D rotors are stored in
the ./hr_geoms subdirectory; data necessary to build the input for the master question
calculations as well as for the me block files are stored in the ./me_files subdirectory; the me
blocks and energies (multi dimensional rotational PESs and high level energies) are stored in
the ./me_blocks subdirectory.
The main results of an EStokTP job are contained in two different sets of files. The first is the
input for the Mess solver, namely the me_ktp.inp file. The second is the me_block file produced
for each species (reactant1, reactant2, product1, product2, reactant van derWaals well, product
van der Waals well, or transition state). Me block files are named after the species whose data
they contain as reac1.me, reac2.me, prod1.me, prod2.me, wellr.me, wellp.me, or ts.me and
contain essentially the same information as the me_ktp.inp file, that is geometry, hindered rotor
data, frequencies, symmetry, and electronic energy levels. It is not difficult to assemble an input
for the Mess solver starting from the me_block files.
18
2. Installation
2.1. Distribution
EStokTP is distributed as a tar file. Once opened it will create three subdirectories: example, exe,
and sources. Commands:
2.2. Compiling
EStokTP comprises two separate codes: the EStokTP source and the source code used to
performe the projection of the Hessian and to apply multidimensional tunneling theory. The two
codes are reported in two subdirectories in the sources subdirectory: estoktp_1.6 and projRot_1.0.
Both must be compiled. Makefiles are already present in the subdirectory and should be edited to
choose the preferred compilers and options. The pre-selected compilers and gfortran and gcc.
The code has also been compiled with intel fortran without problems. To compile the codes, it is
sufficient to hit ‘make’ in each subdirectory. One executable file should be created in the ./exe
subdirectory for each code: estoktp.x and RPHt.exe. Commands:
tar –xvf estoktp_dis_1.0.tar
cd estoktp_dis_1.0
cd sources
cd estoktp_1.6
make
cd ../projRot_1.0
make
2.3. Execution Environment
EStokTP uses the executables and the scripts that are present in the ./exe subdirectory. It is thus
sufficient to initialize the execution environment to put the ./exe subdirectory in your path. If the
code is run on a cluster the same applies.
To run EStokTP up to the production of a rate constants it is necessary to have installed at least
one electronic structure code (at present Gaussian 09 and molpro 2008-2015 are both supported)
and the Mess master equation solver (see http://tcg.cse.anl.gov/papr/codes/mess.html for
distribution and installation instructions).
EStokTP calls Gaussian with the ‘g09’ command, so that if Gaussian is properly installed no
further action is needed. Molpro is called through the molprop script reported in the exe directory.
The molprop script takes as input the number of processors to use. The script should be edited to
point to the proper molpro executable. Also, if compiling with gfortran, it is suggested to set the
gfortran_unbuffered_all variable to Y, to increase the frequency at which output is written.
Commands:
Bash:
export PATH=$estoktpdir/estoktp_dis_1.0/exe:$PATH
export GFORTRAN_UNBUFFERED_ALL=Y
csh/tcsh
set PATH = ($estoktpdir/estoktp_dis_1.0/exe $PATH)
19
setenv GFORTRAN_UNBUFFERED_ALL Y
2.4. Running
A script that launches EStokTP on a node of a cluster is the run_estoktp.com script reported in
the ./exe subdirectory. It should probably be edited to reflect your needs. It is used to launch
EStokTP as: ‘run_estoktp.com nodename &’. Alternatively EStokTP can be launched directly
from a node through the command ‘estoktp.x’.
20
3. Input files
All data files necessary to perform an EStokTP job are stored in the ./data subdirectory of the
main job directory. Section 1.4 reports an overview of the input files needed. In the following a
detailed description of each file is reported. It should be noted that the input is not case sensitive.
3.1. estoktp.dat: job selection and modules
Estoktp.dat is made of three sequential parts. The first contains a list of keywords, the second the
list of modules to use, and the third a concise description of the computational environment.
3.1.1. Reaction Type/Global Keywords
These keywords define the type of EStokTP job that should be performed. Each one should stay
on a separate line. The order is not important.
- ReactionType reaction tsnumber : this keyword defines which type of reaction is studied. The
reaction field can be either Addition, Abstraction, Isomerization, or Betascission. In the absence
of this keyword the code assumes that the job is dedicated to studying a single well, with data in
the reac1.dat file. The tsnumber, which must be specified, can be either 1TS (active for all
reaction types), 2TS (active only for addition and abstraction) or 3TS (active only for
abstraction). See section 1.3 for how the choice of tsnumber determines the model used for the
calculations. Example: ‘ReactionType abstraction 3TS’.
- Wellr algorithm leveloftheory: it means that a van der Waals well must be searched on the
reactant side. If the algorithm and leveloftheory keywords are left blank than the search is
performed starting from the z-matrix structure that must be specified in the wellr.dat file. If the
algorithm keyword is ‘findgeom’ then a guess structure is generated from the TS geometry. No
z-matrix specification is in this case needed in the wellr.dat file. This is the recommended option
when a van der Waals well is searched. The leveloftheory keyword, which can be either ‘level1’
or ‘level0’, specifies whether the search should be performed starting from level0 of theory or
directly from level1. Example: ‘wellr findgeom level1’.
- Wellp algorithm leveloftheory: it means that a van der Waals well must be searched on the
product side. If the algorithm and leveloftheory keywords are left blank than the search is
performed starting from the z-matrix structure that must be specified in the wellp.dat file. If the
algorithm keyword is ‘findgeom’ then a guess structure is generated from the TS geometry. No
z-matrix specification is in this case needed in the wellp.dat file. This is the recommended option
when a van der Waals well is searched. The leveloftheory keyword, which can be either ‘level1’
or ‘level0’, specifies whether the search should be performed starting from level0 of theory or
directly from level1. Example: ‘wellp findgeom level1’
- Prods: it tells the code that the formation of products should be explicitly considered in writing
the Mess input.
21
- Variational : it specifies that an IRC scan will be performed (and thus the IRC module will be
called and should be activated in the requested series of calculations) and variational transition
state theory will be used to determine the rate constant.
- FrozRTS distance: it means calculations are performed keeping the RTS coordinate frozen. If
activated, the code will be able to run up to the determination of the rate constant determining
geometries and frequencies and performing hindered rotor scans performing all geometry
optimizations constraining the RTS coordinate. This option can be used to perform several types
of calculations. One possibility for example is to use it to perform variational TST calculations as
a function of the RTS coordinate, so that the minimum energy path can be determined for a
barrierless reaction. The distance parameter is used by the Opt_TS_0 module to set the RTS
value. It should be noted (see sections 1.3.2 and 1.3.3) that RTS is the name of the distance
coordinate used by EStokTP to define the distance between react1 and react2 in abstraction and
addition reaction. Example: ‘FrozRTS 2.5’.
- ResIRC: this keyword requests a linear rescaling of the potential energy profile calculated in
the IRC module to high level quality energies. For this purpose two additional high level
calculations are performed for the first and last geometries computed along the IRC path.
- ResIRCAll: same as ResIRC, except that high level energies are computed for all points
determined along the IRC path. It is, obviously, quite expansive computationally.
- MdTunnel: it requests to compute tunneling contributions using multi dimensional tunneling
theory, in particular the present implementation uses small curvature theory with reaction path
Hamiltonian frequencies. It should be noted that this calculation requires that a large number of
points is taken along the reaction path (on the average 200 points with a stepsize of 0.3 Bohr). It
is suggested to use this keyword in conjunction with the ResIRC keyword. By default, in the
absence of this keyword, the Eckart tunneling model is used.
- NoTunnel: this keyword disables the use of tunneling in rate constant calculations.
- Recover: Recovers the output from a calculation that was interrupted after generating output
prior to processing it (e.g., a high level MOLPRO calculation). It can be used only if a single
module is active (that is, the one from which we want to recover the data).
- Debug debuglevel: it is a keyword that defines the density of writing in the output. At present,
as this is the first release of the code, use 2 (that is dense output), as it will help with debugging
the code. Example: “debug 2”
- Stoichiometry atomstypenumber : this keyword specifies the number of atoms of each type
present in the molecule. It is an input for an external call to a code that is to be used to determine
Lennard Jones parameters, which are needed for the master equation calculation. It is not used at
present. Example: “stoichiometry C4H8”
22
3.1.2. List of Modules
An EStokTP job typically consists of a set of electronic structure calculations, each used to
produce certain data (geometries, frequencies, or energies most typically) that will then be
necessary either to perform successive higher level calculations or, eventually, to write a master
equation input file or a master equation block. These calculations are grouped in modules, which
can be called independently, provided that the necessary input is available. Each job type (be it a
reaction or a well job) requires that a minimal set of modules is called, either sequentially or one
by one. See sections 1.3.1-1.3.5 for a list of the modules requested by each job type. The full list
of all the available modules is reported in the following, with a brief description. The Module
section must be terminated with a line containing the ‘End’ word.
Opt_Reac1
Opt_Reac2
Opt_Prod1
Opt_Prod2
Opt_WellR
Opt_WellP
Modules that perform the search for the minimum energy structure of reactants, products or
wells at level 0 of theory. Structural parameters are saved in the ./output subdirectory as
reac1_opt.out, reac2_opt.out, prod1_opt.out, prod2_opt.out, wellr_opt.out, wellp_opt.out (briefly,
$species_opt.out). Structural data for all minimum energy structures found in the stochastic scan
are saved as well in the order in which they are found as reac1_opt_1.out, reac1_opt_2.out, and
so on. Geometries in xyz format are saved in the ./geoms subdirectory as reac1_01.xyz,
reac1_02.xyz, etc. Level 0 energies are reported in the title line of the xyz geometries and as last
line of the $species_opt.out geometries. A log of the calculations is reported in the reac1.out,
reac2.out and so on files in the ./output subdirectory. It can be used to check the progress of the
calculations. If the ‘wellr findgeom level0’ or ‘wellp findgeom level0’ keywords are used then
the guess geometry for the van der Waals well optimization will be taken from
the ./output/ts_opt.out file and modified by changing the bond breaking/bond forming length.
Grid_Opt_TS
Module that performs a grid scan along a distance coordinate (usually RTS) to determine a
suitable guess for a TS search. The structure (z-matrix parameters) with the highest energy found
along the scan is saved in the ./output/ts_opt.out file and used for successive calculations. A log
of the grid scan is saved in the ./output/grid_opt.out file and can be used to check the status of
the calculations.
Opt_TS_0
Module used to determine the transition state structure at level 0 of theory. The structure so
determined is saved in the ./output/ts_opt.out file.
TauO_TS
Module that performs a MonteCarlo search along the selected dihedral coordinates to search for
rotational conformers of the transition state. The one with the minimum energy is saved in
the ./output/ts_opt.out file and used for successive calculations.
23
Opt_Reac1_1
Opt_Reac2_1
Opt_Prod1_1
Opt_Prod2_1
Opt_TS_1
Opt_WellR_1
Opt_WellP_1
These modules are used to determine the minimum energy structure of reactants, products or
wells at level 1 of theory. Structural parameters are saved in the ./output subdirectory as
reac1_opt.out, reac2_opt.out, prod1_opt.out, prod2_opt.out, wellr_opt.out, wellp_opt.out (briefly,
$species_opt.out). Thus overwriting the files written by level0 calculations. Geometries in xyz
format are saved in the ./geoms subdirectory as reac1_l1.xyz, reac2_l1.xyz, etc. Level 1 energies
are reported in the title line of the xyz geometries and as last line of the $species_opt.out
geometries. A log of the calculations is reported in the level1.out file in the ./output
subdirectory. Structures determined at level1 of theory are then used for me calculations and are
saved in the ./me_files subdirectory as reac1_ge.me, reac2_ge.me and so on. Frequencies are
calculated in these modules from the Hessian matrix if no hindered rotor calculation is requested
and are saved in the ./me_files subdirectory as reac1_fr.me, reac2_fr.me and so on. If hindered
rotors are present, then the Hessian matrix (reac1_fcmat.log, reac2_fcmat.log and so on) is saved
in the ./output subdirectory, and will then be used by the hindered rotor module (see the
1dTau_xxx module description) to compute a set of frequencies from which hindered rotors have
been projected. Zero point energies are computed by these modules and saved in the ./me_files
subdirectory as reac1_zpe.me, reac2_zpe.me, and so on. If the ‘wellr findgeom level1’ or ‘wellp
findgeom level1’ keywords are used then the guess geometry for the van der Waals well
optimization will be taken form the ./output/ts_opt.out file and modified by changing the bond
breaking/bond forming length. It means that no preliminary call to the Opt_WellR or Opt_WellP
module is necessary. The global symmetry number (ratio of external rotational and external
optical symmetry numbers, which should account for optical isomers found in hindered rotor
scans) is here set to 1 and should then be edited or corrected calling the symmetry modules (see
below).
1dTau_Reac1
1dTau_Reac2
1dTau_Prod1
1dTau_Prod2
1dTau_WellR
1dTau_WellP
1dTau_TS
The 1dTau modules perform one dimensional hindered rotor scans along the dihedral coordinates
indicated in the input data files for reactants, products, wells or transition state (nhind keyword).
These modules should also be called when a multidimensional hindered rotor scan is requested
for the dihedral coordinates treated multidimensionally. Log files of the calculations are saved in
the ./output subdirectory as reac1_hr.out, reac2_hr.out, and so on. The calculated 1D hindered
rotor PES, the list of atoms in the rotating top and that belong to the rotation axis are saved in
the ./me_files subdirectory as reac1_hr.me, reac2_hr.me, and so on. The Hessian matrix is then
24
projected and projected frequencies are saved in the ./me_files subdirectory as reac1_fr.me,
reac2_fr.me and so on. Hindered rotor structures for each point are saved in the ./hr_geoms
subdirectory.
MdTau_Reac1
MdTau_Reac2
MdTau_Prod1
MdTau_Prod2
MdTau_WellR
MdTau_WellP
MdTau_TS
The mdTau modules perform a multidimensional hindered rotor scan along the dihedral
coordinates indicated in the input data files for reactants, products, wells or transition state
(nhind2D or nhind3D keywords). At the present level of theory, only one multidimensional
hindered rotor scan is allowed for molecule in order to preserve the correct coupling between
internal and external rotational motions as implemented in the Mess master equation solver. The
calculated potential energy surface is saved in the ./me_files subdirectory in the
reac1_2dhr_nofr01.dat (or reac2_.. or reac1_3dhr_.. and so on) if no frequencies were estimated
at each point of the PES and in the reac1_2dhr_01.dat (or reac2_.. or reac1_3dhr_.. and so on)
file otherwise (if the mhr_freqs keyword has been used in the species input data). The
frequencies, if calculated along the multi dimensional PES, are determined from a Hessian from
which the corresponding hindered rotors have been projected out. Details on rotating groups
necessary to solve the multi dimensional hindered rotor equation as implemented in Mess are
saved in the reac1_mdhr.me file (or reac2, prod1, and so on).
Symm_Reac1
Symm_Reac2
Symm_Prod1
Symm_Prod2
Symm_WellR
Symm_WellP
Symm_TS
The symmetry keywords are used to determine the rotational and optical symmetry numbers for
external and internal rotations. The implemented algorithm works as follows. The external
rotational and optical symmetry numbers are determined through the symmetry_number code
that is part of the Mess suite of codes. A tolerance of 0.07 is used as input to symmetry_number,
as it was found to give better predictions than obtained using the default 0.001 parameter.
Successively the hindered rotor PES is scanned to check if optical isomers are found along the
rotational scan. For this purpose structures saved in the ./hr_geoms subdirectory are used to find
local minima along the rotational PES for each hindered rotor. If the computed energy differs by
less than 0.00001 Hartrees from the absolute minimum than it is considered as an optical isomer.
The species external optical isomer number is then divided by the number of rotational optical
isomers. It should be mentioned that this algorithm is not yet extended to treat optical symmetry
in multidimensional hindered rotors. For this reason if a multidimensional rotor treatment has
been requested all optical symmetry numbers (internal and external) for the considered species
are set to 1, as it is supposed than an eventual external optical symmetry would be captured by
25
the multi dimensional hindered rotor analysis. The log files for symmetry calculations is saved in
the ./output subdirectory as reac1_sym.out (or reac2_sym.out, and so on). The reac1_ge.me (or
reac2_ge.me and so on) file, which contains the global symmetry number for each species (ratio
of external rotational and external optical symmetry numbers), which is set to 1 when written for
the first time in the level1 calculations (opt_reac1_1 and so on), is then updated on the basis of
what calculated in the symmetry module.
HL_Reac1
HL_Reac2
HL_Prod1
HL_Prod2
HL_WellR
HL_WellP
HL_TS
These modules perform high level calculations for all species. The geometry adopted for this
purpose is the one determine at level 1 of theory and saved in the ./output/reac1_opt.out and so
on files. The output of all high-level calculations are saved in the ./hl_logs subdirectory as
reac1_molpro.out or reac1_g09.out and so on.
IRC
The internal reaction coordinate module is at present set to process files produced through g09
IRC calculations. The required information are Hessians and energies calculated along the
reaction path, which is started from the transition state structure located using level 1
calculations whose geometry is stored in the ./output/ts_opt.out. It is important that the level of
theory at which IRC calculations are performed (specified in the theory.dat file) is the same as
that used to find the transition state. All files used in IRC calculations are stored in the ./irc_files
subdirectory. When the IRC module is called the ‘variational’ keyword should also be present in
the estoktp.dat input, and vice-versa. The output of the IRC module is saved in the variational.me
file, which is stored in the ./me_files subdirectory. Additionally, the IRC module should be
called whenever multi dimensional tunneling calculations are requested. As MD tunneling
calculations are fast compared to electronic structure calculations, they are performed always
when the IRC module is called. The input and output files for such calculations are saved in
the ./md_tunn subdirectory. Notably, the trajec.xyz contains all the geometries of each point of
the IRC scan saved in xyz format and thus easily readable using molecular structure visualization
softwares (such as molden or vmd), while the mdtunn.out files contains the log file of the multi
dimensional tunneling calculation. The tunneling coefficient is reported as kSCT at the end of
the file. The file created and used by the Mess solver for multi dimensional tunneling
calculations is the imactint.dat file, which contains the natural logarithm of the imaginary action
integral as a function of the energy (in cm-1), measured from the energy of the saddle point.
Please look at the mdTunnel keyword description for further information on how to run
multidimensional tunneling calculations.
kTP
The kTP module generates, when the ‘reaction’ keyword is present in the estoktp.dat file, the
me_ktp.inp file, which is the input for the Mess solver. The data used for this purpose are those
saved by previous calls to all modules necessary for the desired calculation in the ./me_files
26
subdirectory. Also, the me_blocks for all species participating to the reaction are created and
saved in the ./me_blocks subdirectory. The output of the Mess solver is saved in the rate.out file,
which can be found in the job directory and is also copied to the ./output subdirectory.
If the reaction keyword is not present in the estoktp.dat file then it is assumed that this is a well
job and thus only the me_block files for the reac1 species are generated.
ModArr
This module is not yet active. Its purpose is to fit all rate constants reported in the rate.out file
produced by the Mess solver to the modified Arrhenius form.
3.1.3. Computational environment
The computational environment is defined by the last 4 lines of the estoktp.dat file, which must
be preceded by the ‘End’ line that marks the end of the module section. The first line contains
the number of cores used for the low and high level calculations, separated by a comma, the
second is a comment line, the third line contains the memory requested for low and high level
calculations (expressed in Mega words, with the MW letters attached to the number), and the
fourth line is a comment line. In practice, the calculations for each module are performed using
the low level parameters except for high level modules, which use the high level parameters.
Example:
12,4
numprocll, numprochl
200MW 300MW
gmemll gmemhl
3.2. Species Data Files
An EStokTP job can include up to seven species, each one requiring a dedicated input file: two
reactants (reac1.dat and reac2.dat), two products (prod1.dat and prod2.dat), two van der Waals
wells (wellr.dat and wellp.dat) and one transition state (ts.dat). Each file should terminate with a
line containing the ‘end’ word. EStokTP retrieves data from the input files searching for
keywords that correspond to a specific set of input data. For example the geometry is reported
after the charge keyword, while the hindered rotor section is reported after the nhind keyword.
The format of each file is described in detail in the following.
3.2.1. reac1.dat, reac2.dat, prod1.dat, prod2.dat
Each of these files should contain the following data blocks. The order in which the blocks are
reported in the file is not relevant.
- nosmp: it gives the number of sampling points to be used in the Monte Carlo sampling of the
configurational space defined by the coordinates listed in the ntau block, the geometric (in
degrees) and the energy (in Hartree) threshold to check if the new geometry found in the
27
stochastic search differs from geometries found in previous scans. The whole block is formed
by two lines (keyword and data).
Example (requests 5 sampling points):
nosmp
5 1.0 0.00001
- ntau: lists the number and names of the coordinates that are stochastically sampled to search
for the absolute minimum energy structure. The whole block is formed by three lines
(keyword, number of sampled coordinates and comment line) plus a list of the names of the
sampled coordinates, each followed by its sampling interval. It is assumed that the sampled
coordinates are dihedral angles whose names and z-matrix coordinates are defined in the
‘charge’ block.
Example (scans b1 and b5 coordinates randomly sampling their values in the 0-360 interval
for b1 and in the 0-120 interval for b2):
ntau
2
-- name and sampling interval
b1 0 360.
b2 0 120.
- nhind: lists the number and names of the dihedral coordinates that are to be treated as
hindered rotors. The whole block is formed by three lines (keyword, number of dihedral
coordinates and a comment line) plus a list of the names of the dihedral coordinates, each
followed by its scan interval, the number of points to take on the PES, and periodicity. The
scan step is obtained dividing the scan interval by the number of PES points. It is assumed
that the first and last point of the scan interval correspond to the same molecular structure,
thus the calculation is performed only once for the first point, so that the total number of
electronic structure calculations is equal to the number of PES points indicated. Though the
ntau and nhind coordinates are often the same this may not always be necessarily the case and
is thus not enforced.
Example (treats b1 and b5 coordinates as hindered rotors, using a symmetry number of 1 for
b1 and of 3 for b2, that is in fact scanned between 0 and 120. The calculations at 360. and 120.
degress for b1 and b2 are not performed, as they are assumed to give the same energy as those
performed at 0 degrees).
nhind
2
-- name hindmin hindmax points periodicity
b1 0 360. 12 1
b2 0 120. 4 3
- natom: it must be followed by three numbers: the total number atoms in the molecule
excluding dummy atoms, the total number of atoms including dummy atoms and an index,
which can be either 0 and 1, that indicates whether the molecule is linear or not (not necessary
for diatomic molecules).
Example:
28
natom natomt ilin
6 6 0
- charge: this block contains the charge and spin multiplicity and is followed by the z-matrix of
the molecule. In specifying the z-matrix each coordinate name must differ from the others for
the same species, and for the reac1 and reac2 species in abstraction and addition reactions.
Example (ethylene charge, spin and z-matrix):
charge spin atomlabel
0 1
c1
c2 c1 rcc1
h3 c1 rch1 c2 ahcc1
h4 c1 rch2 c2 ahcc2 h3 b1
h5 c2 rch3 c1 ahcc3 h3 b2
h6 c2 rch4 c1 ahcc4 h5 b3
- intcoor: this blocks contains first guess values for all coordinates reported in the z-matrix,
made exception for the coordinates listed in the ntau block, whose value is determined
through stochastic sampling. Coordinate names and guess values must be separated by a space.
Example (coordinates for ethylene):
intcoor
rcc1 1.5
rch1 1.08
rch2 1.08
rch3 1.08
rch4 1.08
ahcc1 110.
ahcc2 110.
ahcc3 110.
ahcc4 110.
b1 178.
b2 5.
b3 177.
- symmetryfactor: it is the global symmetry factor for the molecule, defined as the ratio
between external rotational symmetry number and external optical symmetry number. It
should also account for optical isomers found in the hindered rotor scan. Though it has to be
specified in this file, it is recalculated when the Symmetry modules are called.
Example (symmetry for ethylene):
SymmetryFactor
4.
- nelec: it lists the number of electronic states for the considered species and their multiplicity
and energy relative to the ground state, including the ground state. The nelec keyword must be
29
followed by one line giving the number Ne of electronic states and by Ne lines giving energy
(in cm-1) and multiplicity of each electronic state.
Example (electronic states for OH):
nelec
2
0. 2
140. 2
In addition to these data blocks, which should always be included in the data file, even in the
absence of hindered rotors or coordinates to sample stochastically (in which case the nhind and
ntau parameters should be set to 0), the following data blocks can be optionally added:
- nhind2D (same rules for nhind1D and nhind3D): number and name of rotors for which a
multidimensional scan is requested. The block is structured similarly to the nhind block, thus
it is formed by three lines (keyword, number of multi dimensional rotors and a comment line)
plus nlines containing the list of the names of the dihedral coordinates for which a
multidimensional treatment is requested (nlines = rotor dimensionality x number of
multidimensional rotors). At present, this keyword is designed to generate inputs for the Mess
solver, and thus for the multidimensional hindered rotor model it implements. This is a
sophisticated model that accounts for the coupling between internal and external rotations.
Because of this it does not make sense (and it is thus not supported at present) to use this
treatment for more than 1 multi dimensional rotor at a time, as the correct theoretical
treatment would be to increase the dimensionality of the rotor, rather than have several rotors
of lower dimensionality (which also means that nhind1d, nhind2d, and nhind3d calculations
are alternative ones with respect to the other). This block is read by the mdTau modules,
which must therefore be present in the estoktp.dat input file.
Example (treats b1 and b6 as 2D hindered rotors, using a symmetry number of 3 for each, so
that 4 points are used to scan the coordinates between 0 and 120 for 4 times, for a total
number of 16 points taken along the PES).
nhind2D
1
-->namehind,hindmn,hindmx,hindstep period
b1 0 120 4 3
b6 0 120 4 3
3.2.2. wellr.dat, wellp.dat
The wellr.dat and wellp.dat data files should always be filled in when the wellp and wellr
keywords are present in the estoktp.dat file. They should be filled in as described in section 3.2.1
unless the findgeom option is associated to the wellr and wellp keywords (which is
recommended). In that case, the only blocks that should be included are nosmp, ntau, nhind,
charge, Symmetryfactor, and nelec. If the findgeom option is used, it is suggested to set the
number of sampling points to 1 and that of sampling coordinates to 0, as the findgeom algorithm
should help to find the van der Waals wells connected to the TS, which may not be that of
30
absolute minimum energy which may be found using the stochastic sampling strategy. It is also
suggested to set the nhind keyword to 0 (that is, do not consider hindered rotors), as what is
necessary for rate calculations is the energy of the well, rather than an accurate evaluation of its
density of states so that the RRHO approximation should be sufficient.
3.2.3. ts.dat
The data file for the transition state contains several blocks that are the same as those of the
species data described in section 3.2.1. In particular it must contain the: nosmp; ntau, nhind,
Symmetryfactor, and nelec blocks, organized as described in section 3.2.1. The natom and
intcoor blocks should not be present, as the geometry of the TS is constructed from EStokTP
following rules that depend on the reaction type starting from the data reported in the reac1.dat
and reac2.dat data files. In addition, the following blocks must be present:
- charge: requires two integers: charge and spin multiplicity.
Example:
charge spin
0 2
- isite: requires three integers: the atom numbers for isite, jsite, and ksite. The meaning of these
three numbers depends on the reaction type. They are used to construct the z-matrix of the
transition state and to define the reacting atoms. For example isite is the abstracted atom in an
abstraction reaction, the site to which an atom or molecule is added in an addition reaction;
the site to which an atom is transferred in an isomerization reaction, and one of the two atoms
whose bond is being broken in a beta-scission reaction. See sections 1.3.2 – 1.3.5 and
keywords rmin, rmin1 and rmax1 for the meaning.
Example:
isite jsite ksite
11 3 2
- rmin: this block must be present in abstraction and addition reactions. It is not read in
isomerization and beta-scission reactions. It reports data for constructing the TS z-matrix and
defines the TS search grid. It is a four lines block, with the first line being the keyword, the
second three numbers defining the interval of the grid search. The grid search is a restrained
geometry optimization with the RTS coordinate kept frozen. The first number is the lowest
value of RTS, the second the highest and the last the number of steps to progressively move
from the lowest to the highest value of RTS. The third line is a comment line while the fourth
contains 5 numbers, separated by commas, which defines coordinates aabs1, babs1, aabs2,
babs2, and babs3 that are used to construct the transition state z-matrix. The 5 coordinates are
two angles (aabs1 and aabs2) and three dihedrals (babs1, babs2, and babs3) that are used to
define the initial relative orientation of fragments reac1 and reac2. See sections 1.3.2 – 1.3.5
for a precise definition of these coordinates.
Example (scans RTS between 1.4 and 2.2 in 5 steps):
31
rmin rmax nr
1.4 2.2 5
-->aabs1,babs1,aabs2,babs2,babs3
90., 90., 90., 175., 90.
- rmin1: this block is used in isomerization reactions. It consists of the keyword line and a line
containing 5 numbers: distance1, step1, distance2, step2, and ireact. The isomerization
algorithm is a pulling algorithm working so that the distance between two atoms, the isite and
ireact is progressively diminished from the minimum value found among all the react1
conformers located in the stochastic conformational analysis to distance1 with step step1 and
then decreased to distance2 with step step2.
Example (atom 4 is moved toward atom isite in steps of 0.2 Å down to 1.7 Å and then down
to distance 1.2 Å in steps of 0.1 Å):
rmin1 rstp1 rmin2 rstp2 ireact
1.7 0.2 1.2 0.1 4
- rmax1: this block is used in beta-scission reactions. It consists of the keyword line and a line
containing 5 numbers: distance1, step1, distance2, step2, and ireact. The beta-scission
algorithm is a pushing algorithm working so that the distance between two atoms, the isite
and ireact is progressively increased from that of the minimum energy structure to distance1
with step step1 and then to distance2 with step step2.
Example (the distance between atom 8 and isite is increased in steps of 0.2 Å up to 1.8 Å and
then up to distance 2.4 Å in steps of 0.1 Å):
rmax1 rstp1 rmax2 rstp2 ireact
1.8 0.2 2.4 0.1 8
Additional blocks for the ts.dat file are the nhind1D, nhind2D, and nhind3D blocks, organized
as described in section 3.2.1 and the ts_geom block:
- ts_geom: this block, read by module opt_TS_1, is optionally used to request for level1
calculations the use of the geometry of a transition state different from that of the minimum
energy conformer found in the stochastic search performed at level 0 of theory by the
1dTau_TS module. It is made by the keyword line followed by an integer number iTS
indicating the structure to use for level1, and thus successive, TS calculations. When invoked,
the geometry of conformer iTs saved in the ./output subdirectory as ts_opt_iTS.out is copied
to file ./output/ts_opt.out, which is thus overwritten.
Example (uses geometry, in particular z-matrix parameters, saved in ./output/ts_opt_4.out to
perform level1 and successive calculations, as the ./output/ts_opt.out file is overwritten)
ts_geom
4
32
3.3. Electronic Structure and Master Equation Inputs
EStokTP relies on external codes to perform electronic structure and master equation
calculations. At present the supported electronic structure codes are Gaussian 09 and molpro
(versions from 2008 to 2015). Extension to NWchem and Gamess is planned within the next year.
The supported master equation code is Mess, which is part of the Papr distribution
(http://tcg.cse.anl.gov/papr/). Sections 3.3.1 and 3.3.2 describe how to select the level of theory
to use for electronic structure calculations and the input for master equation calculations
performed with Mess.
3.3.1. Theory files
The electronic structure code to use for each calculation module is defined in the theory.dat file.
The level of theory to be used for each module is also defined in theory.dat when Gaussian 09 is
used and in a separate set of files for molpro.
3.3.1.1. Theory.dat
The theory.dat file is organized in blocks, each one identified by a keyword. Each block defines
the electronic structure code to use for a set of modules through a string reported after the
keyword. If the code of choice is g09, the level of theory to use in the calculations is reported in
the lines that follow the keyword, which are then used as the route section for Gaussian
calculations. All optimizations should be performed using internal coordinates, thus any opt g09
call should include the internal option. Also it is suggested to suppress the use of symmetry, and
thus include the ‘nosym’ option in the g09 route section. If the code is molpro one or more
separate files must be prepared, as described in section 3.3.1.2.
- level0: level of theory for modules Opt_Reac1, Opt_Reac2, Opt_Prod1, Opt_Prod2,
Opt_WellR, Opt_WellP, Grid_Opt_TS. If the g09 code is used the g09 route section is
reported in two separate lines after the keyword.
Example (performs optimization using g09 at the b3lyp/6-31+g(d,p) level of theory):
level0 g09
b3lyp/6-31+g(d,p) opt=internal
int=ultrafine nosym
- level0_ts: level of theory for modules Opt_TS_0 and TauO_TS. If the g09 code is used the
g09 route section is reported in two separate lines after the keyword.
Example (performs ts search at using g09 at the b3lyp/6-31+g(d,p) level of theory):
level0_ts g09
b3lyp/6-31+g(d,p) opt=(ts,calcfc,noeig,intern,maxcyc=50)
int=ultrafine nosym
33
- level1: level of theory for modules Opt_Reac1_1, Opt_Reac2_1, Opt_Prod1_1,
Opt_Prod2_1, Opt_WellR_1, Opt_WellP_1. If the g09 code is used the g09 route section is
reported in two separate lines after the keyword.
Example (performs optimization using g09 at the m062x/6-311+g(d,p) level of theory):
level1 g09
m062x/6-311+g(d,p) opt=internal
int=ultrafine nosym freq
- level1_ts: level of theory for module Opt_TS_1. If the g09 code is used the g09 route section
is reported in two separate lines after the keyword.
Example (performs transition state search using g09 at the m062x/6-311+g(d,p) level of
theory):
level1_ts g09
m062x/6-311+g(d,p) opt=(ts,calcfc,noeig,intern,maxcyc=50)
int=ultrafine nosym freq
- hind_rotor: level of theory for modules 1dTau_Reac1, 1dTau_Reac2, 1dTau_Prod1,
1dTau_Prod2, 1dTau_WellR, 1dTau_WellP, MdTau_Reac1, MdTau_Reac2,
MdTau_Prod1, MdTau_Prod2, MdTau_WellR, MdTau_WellP. If the g09 code is used
the g09 route section is reported in two separate lines after the keyword. If the mdtau module
is called, it searches for an additional line for the ‘mhr_freqs’ keyword. If present, frequencies
are computed for each point along the hindered rotor path.
Example (performs hindered rotor scans using g09 at the b3lyp/6-31+g(d,p) level of theory,
when called from an mdtau module, performs a frequency analysis for each point of the
hindered rotor PES).
hind_rotor g09
b3lyp/6-31+g(d,p) opt=internal
int=ultrafine nosym
mhr_freqs
- hind_rotor_ts: level of theory for modules 1dTau_TS and MdTau_TS. If the g09 code is
used two different g09 route sections are reported in four separate lines after the keyword.
The first two lines are used for the first attempt at determining the torsional PES, while the
other two are used as fallback option if the first attempt fails. It is assumed that in the first
attempt the search is performed for a constrained TS, that is a saddle point where one (or
more) dihedral coordinates is kept fixed. If this fails, the hindered rotor analysis is performed
fixing the length of the bond that is being formed/broken to the TS value. If the mdtau_ts
module is called, it searches for an additional line for the ‘mhr_freqs’ keyword. If present,
frequencies are computed for each point along the hindered rotor path.
Example (performs hindered rotor scans using g09 at the b3lyp/6-31+g(d,p) level of theory
first searching for a constrained TS and then performing a constrained optimization if this
fails).
hind_rotor_ts g09
34
b3lyp/6-31+g(d,p) opt=(ts,calcfc,noeig,intern,maxcyc=50)
int=ultrafine nosym
b3lyp/6-31+g(d,p) opt=internal
int=ultrafine nosym
- symmetry: level of theory for modules Symm_Reac1, Symm_Reac2, Symm_Prod1,
Symm_Prod2, Symm_WellR, Symm_WellP. If the g09 code is used the g09 route section is
reported in two separate lines after the keyword.
Example (if requested by the symmetry algorithm, performs optimization using g09 at the
b3lyp/6-31+g(d,p) level of theory):
symmetry g09
b3lyp/6-31+g(d,p) opt=internal
int=ultrafine nosym
- symmetry_ts: level of theory for module Symm_TS. If the g09 code is used the g09 route
section is reported in two separate lines after the keyword.
Example (if needed, performs optimization using g09 at the b3lyp/6-31+g(d,p) level of
theory):
symmetry_ts g09
b3lyp/6-31+g(d,p) opt=(ts,calcfc,noeig,intern,maxcyc=50)
int=ultrafine nosym
- hlevel: level of theory for modules HL_Reac1, HL_Reac2, HL_Prod1, HL_Prod2,
HL_WellR, HL_WellP. If the g09 code is used the g09 route section is reported in two
separate lines after the keyword.
Example (searches for a molpro input file):
hlevel molpro
- hlevel_ts: level of theory for module HL_TS. If the g09 code is used the g09 route section is
reported in two separate lines after the keyword.
Example (searches for a molpro input file):
hlevel_ts molpro
- irc: level of theory for module IRC. At present only g09 is supported. It requires that four
lines are reported after the keyword. The first two contain the g09 route section for the
forward IRC scan, while the second two contain the route section for the backward scan. An
additional line is also searched by the IRC module to check if the HRcc option is active. It is
the HRcc keyword followed two numbers, ifor and iback. This option is used to rescale the
hindered rotor potentials along the IRC path by performing ifor +1 and iback+1 times the
hindered rotor analysis at equally spaced points along the IRC path. To produce consistent
results, the level of theory should be the same used to determine the level1 geometry and
frequencies. This is not enforced at present and it is left to the user to check that this condition
is respected.
35
Example (performs an IRC scan at the m062x/6-311+g(d,p) level taking 10 points in the
forward direction and 10 in the backward using a step of 0.03 Bohr for a total number of 21
points. The hindered rotor potential is re-evaluated 4 times at points 1, 5, 16, and 21. Point 11
is the transition state and it is assumed that its potential is already available):
irc g09
m062x/6-311+g(d,p) irc(forward,calcall,stepsize=3,maxpoints=10)
int=ultrafine nosym iop(7/33=1)
m062x/6-311+g(d,p) irc(reverse,calcall,stepsize=3,maxpoints=10)
int=ultrafine nosym iop(7/33=1)
HRcc 1 1
All keywords may have additional options, some of which are keyword specific. In particular if
the word after the electronic structure code is ‘restart’ it means that in case of failure the
electronic structure calculation is restarted for the number of times reported after the ‘restart’
keyword.
Example (restarts 3 times the calculation from the last available geometry if it fails):
level0 g09 restart 3
It is possible to specify module specific theory levels by introducing keyword whose first part is
the general module keyword and the second part, separated by an underscore from the first part,
is an integer number that identifies each species (1 for reac1, 2 for reac2, 3 for prod1, 4 for prod2,
5 for wellr, 51 for wellr searched using the findgeom algorithm, 6 for wellr, 61 for wellr searched
using the findgeom algorithm).
Example (level 0 of theory used only for product 2, supersedes what was written in the level0
keyword):
level0_4 g09
b3lyp/6-31+g(d,p) opt(internal,calcall)
int=ultrafine nosym
3.3.1.2. Molpro theory files
If the molpro option is specified as the code to use for a specific module, a dedicated file must be
written. The file is formatted so that a molpro input file can be easily reconstructed. The file
should contain three lines: ‘End1’, ‘End2’, and ‘End3’. All that is reported above line End1 will
be inserted in the molpro input above the z-matrix, which will be inserted by EStokTP, all that is
reported between lines ‘End1’ and ‘End2’ will be inserted after the z-matrix for closed shell
species, while all that is reported between lines ‘End2’ and ‘End3’ will be inserted after the z-
matrix for open shell species. Additionally, two lines must be present both in the open shell and
in the closed shell species. One line that saves the molpro output in molden format:
‘put,molden,molpro.molden’, and one line where the ‘CBSen’ variable is initialized to the
desired energy value. The CBSen variable defines the high level energy that will be employed in
the master equation analysis.
The correspondence between molpro file names and theory.dat keywords is the following:
36
Level0 molpro à level0_molpro.dat
Level0_ts molpro à level0_ts_molpro.dat
Level1 molpro à level1_molpro.dat
Level1_ts molpro à level1_ts_molpro.dat
hindrotor molpro à onedtau_molpro.dat
hindrotor_ts molpro à onedtau_ts_molpro.dat
symmetry molpro à symm_molpro.dat
symmetry_ts molpro à symm_ts_molpro.dat
hlevel molpro à hl_molpro.dat
hlevel_ts molpro à hl_ts_molpro.dat
Additionally, if a specific theory file is requested for a particular species, this is searched by
appending the species number to the molpro file as defined above (see the bottom of section
3.3.1.1 for details about the correspondence between number and species):
Level0_1 molpro à level0_reac1_molpro.dat
Level0_2 molpro à level0_reac2_molpro.dat
Level0_3 molpro à level0_prod1_molpro.dat
Level0_4 molpro à level0_prod2_molpro.dat
Level0_5 molpro à level0_wellr_molpro.dat
Level0_51 molpro à level0_wellr_molpro.dat
Level0_6 molpro à level0_wellp_molpro.dat
Level0_61 molpro à level0_wellp_molpro.dat
and so on
Example (file to read when the ‘hlevel molpro’ keyword is present in the theory.dat file. It
performs CCSD(T) calculations followed by MP2 corrections for basis set size for the reac1,
reac2, prod1, prod2, wellr, and wellp species if called from the hl_reac1, hl_reac2 and so on
modules):
hl_molpro.dat
nosym
End1
!closed shell input
basis=aug-cc-pvtz
hf
ccsd(t)
en_cc=energy
gdirect
basis=aug-cc-pvtz
hf
df-mp2
en_mp2_tz=energy
basis=aug-cc-pvqz
hf
df-mp2
37
en_mp2_qz=energy
! these lines must be always included in molpro input
! CBSen should be defined as desired
! the molden line should be left as it is
put,molden,molpro.molden
CBSen = en_cc+en_mp2_qz-en_mp2_tz
---
End2
!open shell input
basis=cc-pvdz
uhf
basis=cc-pvtz
uhf
rhf
basis=aug-cc-pvtz
rhf
uccsd(t)
en_cc = energy
gdirect
basis=aug-cc-pvtz
rhf
df-rmp2
en_mp2_tz=energy
basis=aug-cc-pvqz
rhf
df-rmp2
en_mp2_qz=energy
!these lines must be always included in molpro input
! CBSen should be defined as desired
! the molden line should be left as it is
put,molden,molpro.molden
CBSen = en_cc+en_mp2_qz-en_mp2_tz
---
End3
3.3.2. Master Equation files
3.3.2.1. me_head.dat
The input for master equation simulations is reported in the me_head.dat file, which should
always be in the ./data subdirectory. Please look at EStokTP examples for examples on how it
should be formatted and at the Mess manual for an explanation of its syntax
(http://tcg.cse.anl.gov/papr/codes/mess.html). It is in many ways self – explanatory, so it is
suggested to copy it from the EStokTP example subdirectory and modify it according to needs.
38
4. Output
The output of EStokTP calculations is stored in several files and subdirectories. The log files of
an EStokTP calculation (the ones to control to check for error or warning messages) are
the ./output/estoktp.out file and several standard output messages (screen). It is suggested to re-
direct the standard output to a log file (we usually call this file estoktp.log) when running
EStokTP, which should therefore be called as ‘estoktp.x > estoktp.log). Estoktp.log reports all
the unix shell commands called by EStokTP together with some error messages. The
output/estoktp.out reports several calculation log messages, which may be useful to track the
progression of the code and should be checked in case of an unexpected termination.
4.1. EStokTP Outputs
EStokTP saves as much as possible of the data produced from the electronic structure
calculations. Briefly, calculation logs of each module are reported in the ./output subdirectory,
geometries in the ./geoms subdirectory, files used to create the master equation input in
the ./me_files subdirectory, and logs of high level calculations in the ./hl_logs subdirectory.
More details are reported in dedicated subsections.
4.1.1. Geometries
Optimized geometries are reported in the ./geoms, ./ouput, and ./me_files subdirectories. Level 0
geometries are reported in the ./geoms subdirectory, where they are named $species_$numb.xyz,
where $species may be reac1, reac2, prod1, prod2, wellr, wellp, or ts, and $numb refers to the
progressive number of the structure optimized using the Monte Carlo protocol. The optimized
geometries are numbered following the order on which they are found, not the energy. The
structures are reported in xyz format, with the level0 energy of the structure reported in the
second line. The EStokTP log file of a level0 geometry optimization is reported in the ./output
subdirectory as $species.out, which describes the progress of the Monte Carlo sampling
procedure and reports which structure has been selected for successive level1 calculations. The
z-matrix parameters of each structure are reported in the ./output subdirectory as
$species_opt_$numb.out. The minimum energy structure z-matrix parameters are reported in
the ./output subdirectory as $species_opt.out. This file is the reference file for all geometry
calculations, so that it is overwritten by level1 calculations and used in high level calculations.
Level1 optimized structures are reported in the ./geoms subdirectory as $species_l1.xyz, where
$species may be reac1, reac2, prod1, prod2, wellr, wellp, or tsgta (for the transition state
structure). The level1 module generates also the geometry structures that will be used by the
master equation solver to compute inertia moments. These structures are saved in the ./me_files
subdirectory as $species_ge.me files ($species may be reac1, reac2, prod1, prod2, wellr, wellp,
or ts).
Log files produced by the electronic structure code for level1 and level0 calculations are saved as
well, with the same name as that of the optimized xyz structure, but with the .log extension
($species_$numb.log and $species_l1.log). In case molpro is used, the molden file generated is
also saved in the ./geoms subdirectory using the molden extension ($species_$numb.molden and
$species_l1.molden).
39
4.1.2. Frequencies
Frequencies are computed in the level 1 module and saved in the ./me_files subdirectory as
$species_fr.me. In addition, level1 calculations by default generate a log file with the Cartesian
Hessian, which is stored in the ./output subdirectory as $species_fcmat.log (where $species may
be reac1, reac2, prod1, prod2, wellr, wellp, or ts). The list of frequencies to be used for master
equation calculations is updated after a call to the hindered rotor module if hindered rotor
treatment has been requested in the species input file. This is done by projecting out the torsional
motions from the Hessian, orthonormalizing and diagonalizing it as suggested by Green et al.
(Sharma et al., J. Phys. Chem. A 2010, 114, 5689–5701). After projection, the updated list of
frequencies is stored in the ./me_files subdirectory as $species_fr.me, while the unprojected
frequencies are saved as $species_unpfr.me. In case of need, an animation of the frequencies is
possible post-processing the electronic structure calculation log file (the .molden file if molpro
was used) reported in the ./geoms subdirectory, using a visualization program such as, for
example, molden.
4.1.3. Hindered Rotor
The log file of hindered rotor calculations is reported in the ./output subdirectory as
$species_hr.out. It describes the progress of the calculation and the adopted algorithm. The
hindered rotor potential together with details on the rotating top is reported in the ./me_files
subdirectory as $species_hr.me. If a multi dimensional hindered rotor treatment is requested, the
multi dimensional hindered rotor PES and the frequencies at each point along the PES (if
requested) are stored in the ./me_files subdirectory in the $species_$dimension_$number.dat file,
with $dimension being 1dhr, 2dhr or 3dhr and $number being a progressive number starting
from 01 (at present only one multidimensional hindered rotor is supported). For 1D hindered
rotors geometries of point taken on the PES are stored in the ./hr_geoms subdirectory. Structures
are saved in xyz format as geom_isp$numb1_hr$numb2_hpt$numb3.xyz, where $numb1 refers
to the species (01 for reac1, 02 for reac2, 03 for prod1, 04 for prod2, 05 for wellr, 06 for wellp,
00 for transition state), $numb2 refers to the hindered rotor progressive number (as numbered in
the specie input file) and $numb3 is the number of the point taken along the hindered rotor PES.
These files are used by the symmetry module to determine the optical symmetry number for
internal rotations.
4.1.4. IRC
Files used in internal reaction coordinate calculations (for now supported only for Gaussian) are
stored in the ./irc_files subdirectory. In particular irc_g09f.log and irc_g09b.log are the log files
of the Gaussian forward and backward IRC scan. Input files for projection of hindered rotor
frequencies are stored as well in this subdirectory as input$numb. The main output of IRC
calculations is reported in the variational.me file, which is stored in the ./me_files subdirectory.
The log file of irc calculations is irc.out and is reported in the ./ouptut subdirectory.
4.1.5. Energies
The electronic structure log files of high level calculations are stored in the ./hl_logs
subdirectory as $species_$code.out, where $code could be either molpro or g09. The energy (In
Hartree) determined through high level calculations is saved in the ./me_files subdirectory as
40
$species_en.me. The EStokTP log file of high level calculations is very terse, as the electronic
structure log files contain most of the information necessary to check the progress and success of
these calculations.
4.1.6. Multi Dimensional Tunneling
File used to perform multidimensional tunneling calculations are stored in the ./md_tunn
subdirectory. They are produced whenever the IRC module is called. The trajec.xyz file reports
all the structures along the irc in xyz format and can be viewed using a structure visualization
software, such as molden or vmd. The reduced mass computed through the SCT formalism is
stored in the mueff.txt file (last column) in atomic units, while the vibrationally adiabatic energy
is reported in the VaG.txt file. The SCT tunneling coefficients are reported in the last rows of the
mdtunn.out file, in the kSCT column. The imaginary action integral file that is used by the
master equation solver (Mess) to determine tunneling contributions is stored in the imactint.dat
file, which is saved in the main directory of the EStokTP calculation and in the ./me_files
subdirectory.
4.2. Rate Output
The master equation input file is created by the kTP module, which then runs the master equation
calculations calling the Mess program. The Mess output is composed of two parts, the rate.out
file, which contains all the temperature and pressure dependent rate constants, and the
me_ktp.log file, which is the log file of the Mess code. A detailed description of the Mess output
is reported in the Mess manual at http://tcg.cse.anl.gov/papr/codes/mess.html.
41
5. Examples
Several examples of uses of EStokTP are included in the distribution in the ./examples
subdirectory. Only the input data files are reported in the distribution, while the log files for all
the examples are distributed as a separate tar file. With reference to the subdirectory path, the
examples are:
- Wells/C3H8/c3h8_1dhr: minimum energy search for propane with 1D hindered rotor
treatment up to the production of the Mess block.
- Wells/C3H8/c3h8_2dhr: minimum energy search for propane with 2D hindered rotor
treatment up to the production of the Mess block.
- Wells/C3H8/c3h8_2dhr_fr: minimum energy search for propane with 2D hindered rotor
treatment and evaluation of frequencies at each point of the hindered rotor PES up to the
production of the Mess block.
- Abstraction/ c3h8_h_n3h7_h2_1TS: rate constant evaluation using the 1TS model for H
abstraction from propane (see section 1.3.2).
- Abstraction/ c3h8_h_n3h7_h2_3TS: rate constant evaluation using the 3TS model for H
abstraction from propane (see section 1.3.2).
- Addition/c2h4_h_1TS: rate constant for addition of H to ethylene using the 1TS model (see
section 1.3.3).
- Addition/c2h4_h_2TS: rate constant for addition of H to ethylene using the 2TS model (see
section 1.3.3).
- Betascission/nc3h7_c3h6_h; rate constant calculation for betascission of n-propyl to propene
and H (see section 1.3.4).
- Isomerization/nc3h7_nc3h7: rate constant calculation for n-propyl isomerization (see section
1.3.5). The product well (nc3h7) is determined using the findgeom keyword, so that no z-
matrix has to be specified in the input.
- Isomerization/nc3h7_nc3h7_well5: rate constant calculation for n-propyl isomerization (see
section 1.3.5). The product well (nc3h7) is specified explicitly in the input as wellp.
6. Advices/Suggestions/Troubleshooting
- Highly endothermic reactions must be studied from the endothermic side.
- Search of van der Waals wells with H atoms may be problematic, it may be skipped
without loosing much accuracy.
42
7. Index
1
1dTau,14,24
C
Capabilities,6
D
Debug,12
G
Grid_Opt,13,23
H
HL,16,25
HL_Prod,16,25
HL_Reac,16,25
HL_TS,16
HL_Well,16,25
I
IRC,16
K
kTP,16
M
MdTau,15,24
MdTunnel,12
ModArr,17
N
NoTunnel,12
O
Opt,13,14,23
Overview,5
R
ResIRC,12
ResIRCAll,12
S
Stoichiometry,12
Symm,15,25
