Episo User Guide
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 6

Episo-User Guide-V1.0
1) Quick Reference
Episo needs a working version of Perl and it is run from the command line. Meanwhile,
Bowtie, Tophat and Cufflinks need to be installed on your computer. First you need to
download a transcript annotation file from the Ensembl or NCBI websites. Episo supports the
reference trancriptom sequence files in FastA format, allowed file extensions are either .fa
or .fasta. The following examples will use the paired-ends files
‘example_1.fastq&example_2.fastq’ (it contains 2,500,876 reads in FastQ format, 101 bp
long reads, simulated by Fluxsimulator) and the transcript annotation file.
(1) Compiling the program
When you use the UNIX (linux, Mac OSX) you should compile some programs. You can use
gcc or any ANSI C-compatible compiler. The source codes are from my Episo package. The
commands are as follow.
gcc -o contrans contrans.c
gcc -o compare-paired compare-paired.c -lm
gcc -o anti-bisulfite anti-bisulfite.c
gcc -o selsam selsam.c
gcc -o methylation_ratio methylation_ratio.c –lm
gcc -o isofrom_filter isoform_filter.c
(2) Generating reference transcriptome
The first step is to generate the transcript file according to annotation transcript by using
the program Cufflinks. The command is as follows.
Usage: cufflinks -G annotation.gtf convert.sam
Note. The file annotation.gtf is from the Ensembl or NCBI websites and the file convert.sam
is from the Episo package.
This will produce one output file: transcripts.gtf.
The second step is to generate the reference transcriptome by using the program contrans.
The command is as follows.
Usage: contrans contrans.ctl
Note. The format of control file contrans.ctl is as follows. The genome file genome.fa is from
the Ensembl or NCBI websites.
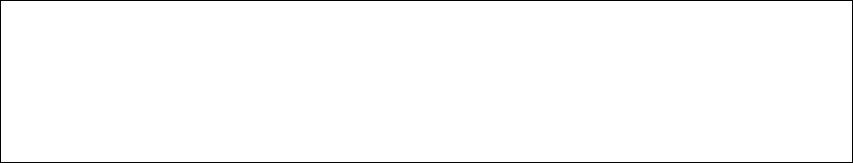
outfile = out * recording the running information
gtffile = transcripts.gtf * gtf file generated by cufflinks
fafile = genome.fa * genome file
transfile = out_trans * recording the transcript
seqfile = out_seq.fa * recording the sequence of each trascript
seqlength = 50 * the length of sequence in the seqfile
This will produce two output files whose names are from the parameter “seqfile” and
“transfile” in the control file contrans.ctl.
(3) Generating transcriptome indexing
Usage: bismark_genome_preparation [options] <path_to_trancriptome_folder>
Note. The output seqfile generated by the program contrans or reference trancriptome
sequence file downloaded from the Ensembl or NCBI websites should be put in the
<path_to_transcriptome_folder>.
A typical trancriptome indexing could be like this:
bismark_genome_preparation --path_to_bowtie /usr/local/bowtie --verbose
/data/transcriptome/
(4) Calling the methylation site
Usage: bismark-liu [options] <transcriptiome_folder> -1 <mates> -2 <mates>
A typical calling example could be like this:
bismark-liu --path_to_bowtie /usr/local/bowtie --vanilla --sam -n 2 /data/transcriptome/ -1
example_1.fastq -2 example_2.fastq
This will produce three output files:
(a) example_1.fastq_bismark_pe.txt (contains all alignments and methylation call strings)
(b) example_1.fastq_bismark_pe_mul.txt (contains the transcript information, where the
alignment belongs)
(c) example_1.fastq_bismark_PE_report.txt (contains alignment and methylation summary)
Note. The options “vanilla” and “sam” are necessary and the bowtie version must be
bowtie1. The program compare-paired and the transfile generated by contrans must be in
the same directory in which bismark-liu is.
(5) Generating the anti-bisulfite RNA-Seq data
Usage: anti-bisulfite anti-bisulfite.ctl
Note. The format of control file anti-bisulfite.ctl is as follows.
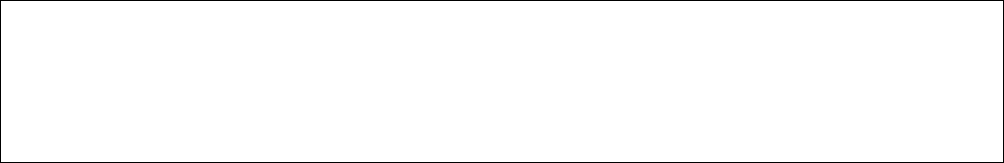
outfile = out * recording the running information
intxtfile = example_1.fastq_bismark_pe.txt * bismark_pe.txt file generated by bismark-liu
outreadfile = anti-example * the index of anti-bisulfite RNA-Seq fastq file
flag = p * p means paired-ends; s means singled-end
skipped_number = 1 * the number of the rows which will be skipped
inmultxtfile = example_1.fastq_bismark_pe_mul.txt * bismark_pe_mul.txt file generated by bismark-liu
This will produce three output files according to the control file anti-bisulfite.ctl:
(a) anti-example_1.fastq and anti-example_2.fastq (an anti-bisulfite RAN-Seq paired-end file)
(b) methylation_summary (contains the transcript information, where the methylation
alignment belongs)
(6) Estimating the methylation level of each isoform
The first step is to analysis the anti-example_1.fastq and anti-example_2.fastq by using the
program TopHat. The options “--bowtie1” and “--no-convert-bam” must be chosen. The
second step is to generate the file which contains the methylation alignments only according
to the sam file generated by TopHat. The command is as follows.
Usage: selsam <.sam> <skipped_number>
Note. The .sam file was generated by TopHat. The skipped_number is the number of rows
which include the sign “@” in the .sam file. The output file name is
accepted_hits_methylation.sam.
The third step is to use the program Cufflinks to analysis the sam file which was generated
by TopHat and the file accepted_hits_methylation.sam generated by selsam respectively.
The option “-G” must be chosen and the gtf file comes from the contrans.ctl file.
The last step is to estimate the methylation level of each transcript. The command is as
follows.
Usage: methylation_ratio <isoform_filter> <isoform_methylation_filter> <total_number>
<methylation_number>
Note. The files in <isoform_filter> and <isoform_methylation_filter> are got by using the
program isoform_filter according to the files in <isoform> and <isoform_methylation>. The
file in <isoform> is the output file isoforms.fpkm_tracking when using the Cufflinks to
analysis the sam file which was generated by TopHat. The file in <isoform_methylation> is
the output file isoforms.fpkm_tracking when using the Cufflinks to analysis the file
accepted_hits_sam generated by selsam. The number in <total_number> is the number of
rows of the file ***_bismark_pe_mul.txt generated by bismark-liu. The number in
<methylation_number> is the number of rows of the file methylation_summary generated
by anti-bisulfite.
The filtering command is as follows.
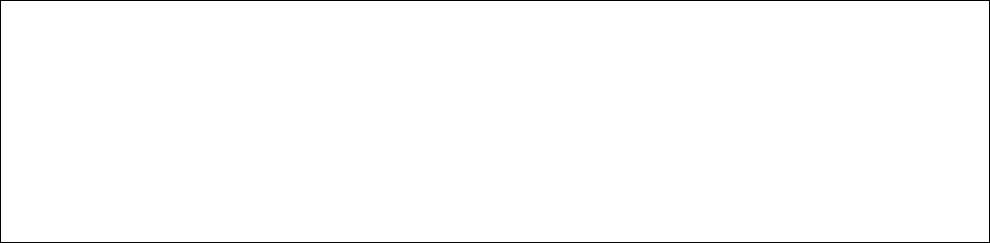
Usage: isoform_filter <isoform> <isoform_methylation> <filter_number>
Note. The number in <filter_number> is FPKM value under which the records in the files
<isoform> and <isoform_methylation> are deleted.
2) Estimating the methylation level of each transcript at a single site
After generating the anti-bisulfite RNA-Seq data, we can estimate the methylation level of
each transcript at a single site. In order to estimate the methylation level of each transcript
at a single site, we need the output files generated by TopHat&selsam according to the
output files generated by anti-bisulfite. The pipeline is as follows.
(1) Compiling the program
gcc -o selreads selreads.c -lm
gcc -o selsam-single-parallel selsam-single-parallel.c
(2) Outputting the alignments which include the assigned single site
Usage: selreads selreads.ctl
Note. The format of control file selreads.ctl is as follows. The value of parameter “length” is
the length of read which in the fastq file generated by anti-bisulfite.
outfile = out * recording the running information
intxtfile = example_1.fastq_bismark_pe.txt * bismark_pe.txt file generated by bismark-liu
intransfile = out_trans * trans file generated by contrans
outreadfile = sel_reads * the index of reads file in which the reads include assigned methylated site
location = 59 * the methylated site location
flag = p * p means paired-ends; s means singled-end
length = 75 * the length of read
chrom_name = test_chromosome * the name for chromosome
skipped_number = 1 * the number of the rows which will be skipped
This will produce one output file: methylation_summary_sam (contains the names of
alignments which include the assigned single site in the control file selreads.ctl)
(3) Generating the sam files which are used to be analysed by Cufflinks
Usage: selsam-single-parallel <sam file> <methylation_summary_sam> <skipped_number>
<tag> <the number of rows in methylation_summary_sam> 1 <output file name>
Note. The value in <tag> is 1 or 0. When the value in <tag> is 1, the file in <sam file> is the
output file generated by TopHat according to the output files generated by anti-bisulfite.
When the value in <tag> is 0, the file in <sam file> is the output file generated by selsam. The
value in <skipped_number> is the number of rows which include the sign “@” in the .sam
file.
(4) Using the program Cufflinks to analysis the output files generated by selsam-single-
parallel when the value in <tag> is 1 and 0 respectively. The option “-G” must be chosen
and the gtf file comes from the contrans.ctl file.
(5) Estimating the methylation level of each isoform at a single site
Usage: methylation_ratio <isoform_filter> <isoform_methylation_filter> <total_number>
<methylation_number>
Note. The files in <isoform_filter> and <isoform_methylation_filter> are got by using the
program isoform_filter according to the files in <isoform> and <isoform_methylation>. The
file in <isoform> is the output file isoforms.fpkm_tracking when using the Cufflinks to
analysis the output file generated by selsam-single-parallel when the value in <tag> is 1. The
file in <isoform_methylation> is the output file isoforms.fpkm_tracking when using the
Cufflinks to analysis the output file generated by selsam-single-parallel when the value in
<tag> is 0. The number in <total_number> is the number of rows of the file
methylation_summary_sam generated by selreads. The number in <methylation_number> is
the number of rows which include the character “methylated_liu” in the file
methylation_summary_sam generated by selreads.
3) Computing the methylation rate of each site
The methylation rate of each site is the ratio of methylated reads in all reads which include
the site. The methylated read is the read in which the assigned site is methylated. The
pipeline is as follows.
(1) Compiling the program
gcc -o trans2genom-bismark trans2genom-bismark.c -lm
gcc -o trans2genom-bismark-methy trans2genom-bismark-methy.c -lm
(2) Extracting the methylation call for every single C analysed
Usage: bismark_methylation_extractor-liu [options] <filenames>
A typical command to extract context-dependent (CpG/CHG/CHH) methylation could like
this:
Bismark_methylation_extractor-liu -p --vanilla --no_overlap --comprehensive
example_1.fastq_bismark_pe.txt
Note. The file in <filenames> is generated by bismark-liu.
This will produce six output files:
(a) CHG_context_ example_1.fastq_bismark_pe.txt
(b) CHH_context_ example_1.fastq_bismark_pe.txt
(c) CpG_context_ example_1.fastq_bismark_pe.txt
(3) Computing the methylation rate of each site
In order to compute the methylation rate of each site, a series of shell script commands
should be executed. These commands are as follows.

#!/bin/sh
sort -k 2 <out_trans> >> out_trans-sort
sed '1d' <CHG_context_*_pe.txt> | sort -k 3 >> CHG_CTOB-sort.txt
nice ./trans2genom-bismark CHG_CTOB-sort.txt out_trans-sort
cat methylation_genom >> methylation_genom-all
rm methylation_genom -f
sed '1d' <CHH_context_*_pe.txt> | sort -k 3 >> CHH_CTOB-sort.txt
nice ./trans2genom-bismark CHH_CTOB-sort.txt out_trans-sort
cat methylation_genom >> methylation_genom-all
rm methylation_genom -f
sed '1d' <CpG_context_*_pe.txt> | sort -k 3 >> CpG_CTOB-sort.txt
nice ./trans2genom-bismark CpG_CTOB-sort.txt out_trans-sort
cat methylation_genom >> methylation_genom-all
rm methylation_genom -f
sort -k 4 methylation_genom-all >> methylation_genom-all-sort
nice ./trans2genom-bismark-methy methylation_genom-all-sort
Note. The file in <out_trans> is generated by contrans. The files in <CHG_context_*_pe.txt>,
<CHH_context_*_pe.txt>, and <CpG_context_*_pe.txt> are the output files generated by
bismark_methylation_extractor-liu.
