FASTAptamer Users Guide
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 25

A Bioinformatic Toolkit for Combinatorial Selections
User’s Guide
Version 1.0
Khalid K Alam1, Jonathan L Chang2 & Donald H Burke1,2
1Department of Biochemistry
2Department of Molecular Microbiology and Immunology
University of Missouri, Columbia, Missouri, USA
© 2014. The FASTAptamer software package, including the source code and users guide, is distributed under a GNU
General Public License v.3.0. For a copy of the full text, see the LICENSE document included with the software. Adnan S.
Hussaini (Saint Louis University School of Medicine) designed the FASTAptamer logo.

If you use FASTAptamer, please cite the paper:
Khalid K. Alam, Jonathan L. Chang, Donald H. Burke. “FASTAptamer: A Bioinformatic
Toolkit for High-Throughput Sequence Analysis of Combinatorial Selections.” Molecular
Therapy – Nucleic Acids. 2015; 4:e1 DOI: 10.1038/mtna.2015.4
For feedback, suggestions, technical support, etc., please email us at
burkelab@missouri.edu or tweet us @BurkeLabRNA.
Table of Contents
1. Introduction
a. Overview of Features
b. Sample Pipeline
2. Installation & Use
a. User Requirements
b. System Requirements
c. Installing as Executable
d. Use Without Installation
3. Tutorials
a. Data Requirements & Pre-Processing
b. Sample Data
c. FASTAptamer-Count
d. FASTAptamer-Compare
e. FASTAptamer-Cluster
f. FASTAptamer-Enrich
g. FASTAptamer-Search
4. Miscellaneous
a. Quick Reference Table – FASTAptamer Input/Output
b. Quick Reference Table – Command Line Options
c. Resources and Links

FASTAptamer is an open source toolkit designed to address the primary sequence
analysis needs from high-throughput sequencing of combinatorial selection
populations. FASTAptamer performs the simple tasks of counting, normalizing,
ranking and sorting the abundance of each unique sequence in a population,
comparing sequence distributions for two populations, clustering sequences into
sequence families based on Levenshtein edit distance, calculating fold-enrichment for
all of the sequences present in 2 or 3 populations, and searching degenerately for
nucleotide sequence motifs. While FASTAptamer was originally developed for
analysis of high-throughput sequencing data from aptamer selections, it offers broad
utility for those working on ribozyme or DNAzyme selections, surface display (phage
display, mRNA display, etc.) selections, in vivo SELEX, protein mutagenesis
selection, or any biocombinatorial selection that results in a DNA-encoded library for
sequencing.
What FASTAptamer cannot do is merge paired-end reads, trim constant regions from
FASTQ files, calculate secondary structure, perform sequence alignments, or any
other function for which software already exists (see section 4c for more on these
resources). Rather than re-inventing the wheel, we decided to simply address the
needs of the selections field and ensure that the output from FASTAptamer remains
compatible for downstream analysis. Doing so allows users the flexibility of plugging
into the FASTAptamer toolkit for rapid identification of candidate biomolecules and
performing additional analysis on a smaller subset of their high-throughput
sequencing data, all while preserving the sequence metrics important in combinatorial
selections.
FASTAptamer makes extensive use of the FASTA file format, the widely used and de
facto format for representing nucleotide or amino acid sequence information. This
format contains two main features - a description line and a sequence line.
FASTAptamer exploits the format by utilizing the description line to preserve
sequence metrics, such as the abundance of the sequence or it’s degree of
relatedness to other sequences. By assigning each sequence in a population a
unique description line, FASTAptamer is able to use this information throughout the
toolkit to perform a variety of primary sequence analysis tasks.
1. Introduction
>3420-14-7.04-83-1-0
TGAAAATGCAGACCAAGAAA…
Sequence Line
Description Line

FASTAptamer-Count
is the gateway to the FASTAptamer toolkit and will rapidly
parse through a FASTQ file to perform the following tasks:
!Count the occurrence of each unique sequence (often referred to as
abundance, read counts, copy number, multiplicity or frequency).
!Normalize the sequence abundance to reads per million (RPM).
!Rank the abundance of each sequence in the population.
!Sort the population by decreasing abundance.
Output from FASTAptamer-Count is provided in FASTA format and is required for all
subsequent FASTAptamer scripts to function properly.
FASTAptamer-Compare
is a tool to compare the sequence distribution of two
populations. Using two input files from FASTAptamer-Count, the script will:
!List the RPM for each sequence present in both input populations, along with
the sequence information itself, to allow rapid generation of XY-scatter plots.
!Calculate the binary logarithm (Log2) of the ratio of RPM in each sequence in
both populations.
!Generate “bin buckets” of the binary log values for effortless generation of a
sequence distribution histogram.
Output from FASTAptamer-Compare is a tab-separated (or “tab-delimited”) plain text
file.
FASTAptamer-Cluster
is a tool that can generate families, or “clusters”, of
closely-related sequences based on a user-defined Levenshtein edit distance. Using
an input file processed with FASTAptamer-Count, the script will:
!Identify “seed” sequences for cluster generation based on abundance.
!Calculate the Levenshtein edit distance (the number of insertions, deletions or
substitutions necessary to transform a sequence into the seed sequence) for
each unclustered sequence.
!Cluster sequences together if the edit distance from the seed sequence is less
than or equal to the edit distance specified.
!Assign each sequence within a cluster a rank based on abundance.
Output from FASTAptamer-Cluster remains in the FASTA format, allowing for
downstream analysis within the toolkit or with other software.
1a. Overview of Features

FASTAptamer-Enrich will accept up to 3 input files from FASTAptamer-Count or
FASTAptamer-Cluster and rapidly:
!Calculate the fold-enrichment ratio for each sequence present in more than
one population.
!List each sequence along with its length, rank, reads, RPM and cluster
information (if provided) for each population, in a sortable format for facile
candidate identification.
!Filter output to include only those sequences present across all input
populations greater than a user-defined RPM.
Output from FASTAptamer-Enrich is provided as a tab-separated plain text file.
FASTAptamer-Search
accepts multiple input files from FASTAptamer-Count or
FASTAptamer-Cluster and will:
!Search for multiple sequence motifs at a time, using degenerate IUPAC-
IUBMB single letter nomenclature for nucleotides.
!Highlight sequence motif matches by enclosing each match in parentheses.
!Generate a new file containing only matched sequences for downstream
analysis.
Output from FASTAptamer-Search preserves the FASTA format of each input file.
For more detailed information on FASTAptamer, please see our publication:
Khalid K. Alam, Jonathan L. Chang, Donald H. Burke. “FASTAptamer: A
Bioinformatic Toolkit for High-Throughput Sequence Analysis of Combinatorial
Selections.” Molecular Therapy – Nucleic Acids. 2015; 4:e22X DOI:
10.1038/mtna.2015.4
1a. Overview of Features
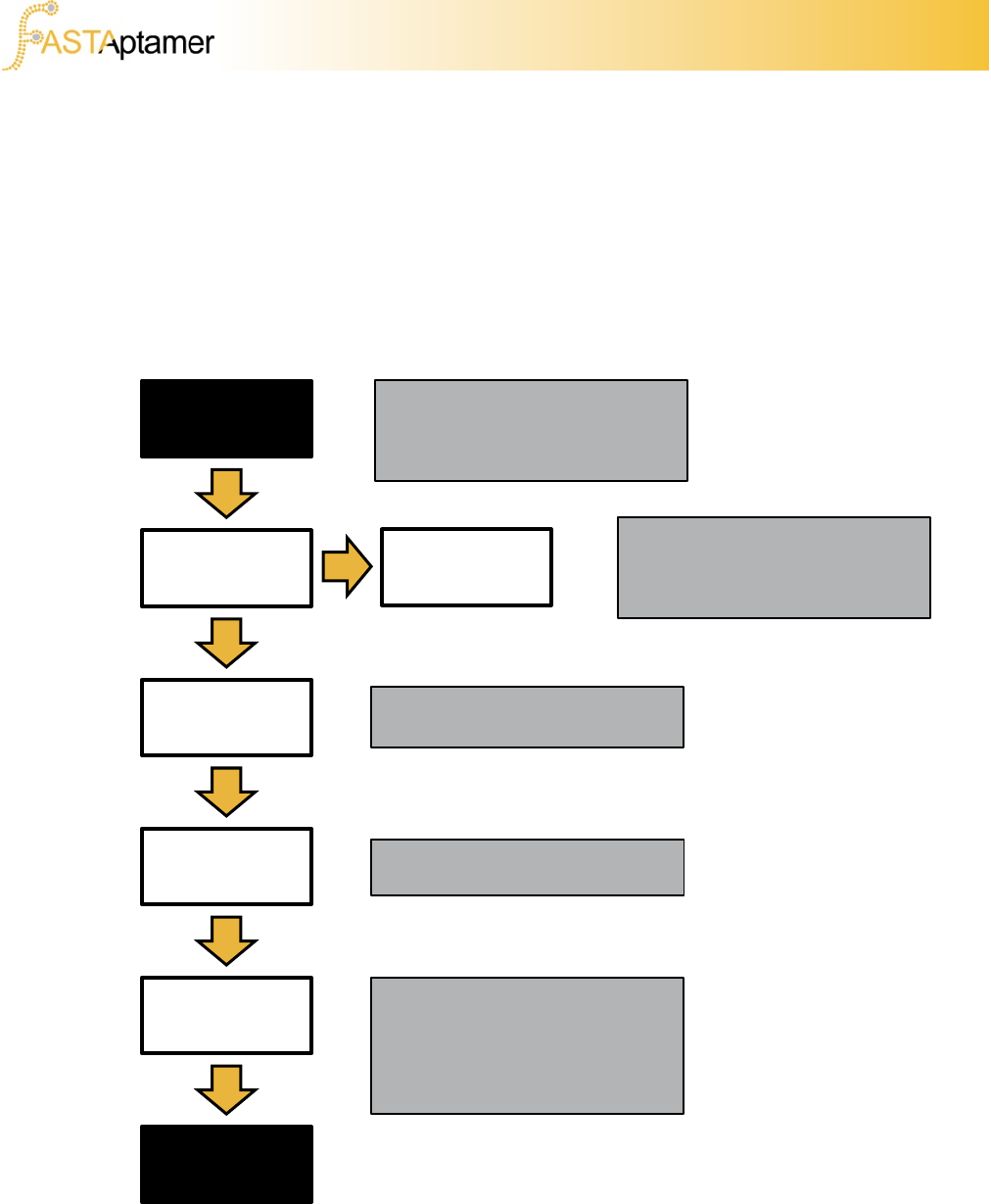
FASTAptamer is provided as a modular collection of scripts that can be configured in
several ways to extract the information from the dataset that you deem important.
Below is a sample pipeline that we use in our research, but be aware that several
more configurations exist. For a complete list of input and output compatibilities for
each script refer to section 4a.
1b. Sample Pipeline
Data must pass through
FASTAptamer-Count for the scripts
to extract sequence abundance
information and function properly.
FASTAptamer
-Search
Other
software
FASTAptamer
-Compare
FASTQ input
file
FASTAptamer
-Count
FASTAptamer
-Cluster
FASTAptamer
-Enrich
Population comparison can facilitate
the analysis of the degree to which
a population has evolved relative to
others.
Optional clustering step generates
sequence families.
Identify candidate molecules based
on fold-enrichment and other data.
Search for interesting sequence
motifs and generate an FASTA
formatted output file for downstream
sequence alignment, comparative
sequence analysis and secondary
structure prediction.

FASTAptamer is provided as a compressed folder containing:
!The 5 FASTAptamer scripts.
!LICENSE.txt – a plain text file containing the GNU GPL v3 software licensing
information.
!README.txt – a plain text file with the essential information.
!This PDF user’s guide.
After “unzipping” the folder, the FASTAptamer scripts can be installed as executable
programs or simply used without installation. Refer to sections 2c and 2d for more
information.
2. Installation & Use
2a. User Requirements
FASTAptamer is designed to be EASY to use. Installation and use of the
FASTAptamer toolkit assumes a basic working knowledge of command line
operation. If you can navigate around your computer’s directories (“cd”), copy (“cp”)
and move (“mv
”) files, and can tolerate the inability of using a mouse, then you should
be able to start using FASTAptamer immediately. If you can’t – don’t fear. Several
resources are provided in section 4c that should get you up to speed quickly. The
installation instructions and tutorials are designed for the inexperienced user. If you
continue to experience problems don’t hesitate to tweet us (@BurkeLabRNA
) or email
us (burkelab@missouri.edu) for support.
2b. System Requirements
FASTAptamer is written in the Perl programming language with no external
dependencies. What this means for you is that virtually every modern computer can
run it. Linux and Mac users rejoice, as nearly every instance of Linux and Mac OS X
can run Perl out of the box. If you’re running Windows you’ll need to download a Perl
interpreter such as Strawberry Perl (open source - always free) or ActiveState’s
ActivePerl (they provide a free “community distribution”). We’ve personally tested the
toolkit, without issue, on CentOS Linux 5.4, Mac OS X 10.6+, Debian GNU/Linux 7.0
and Strawberry Perl 5.20.1.
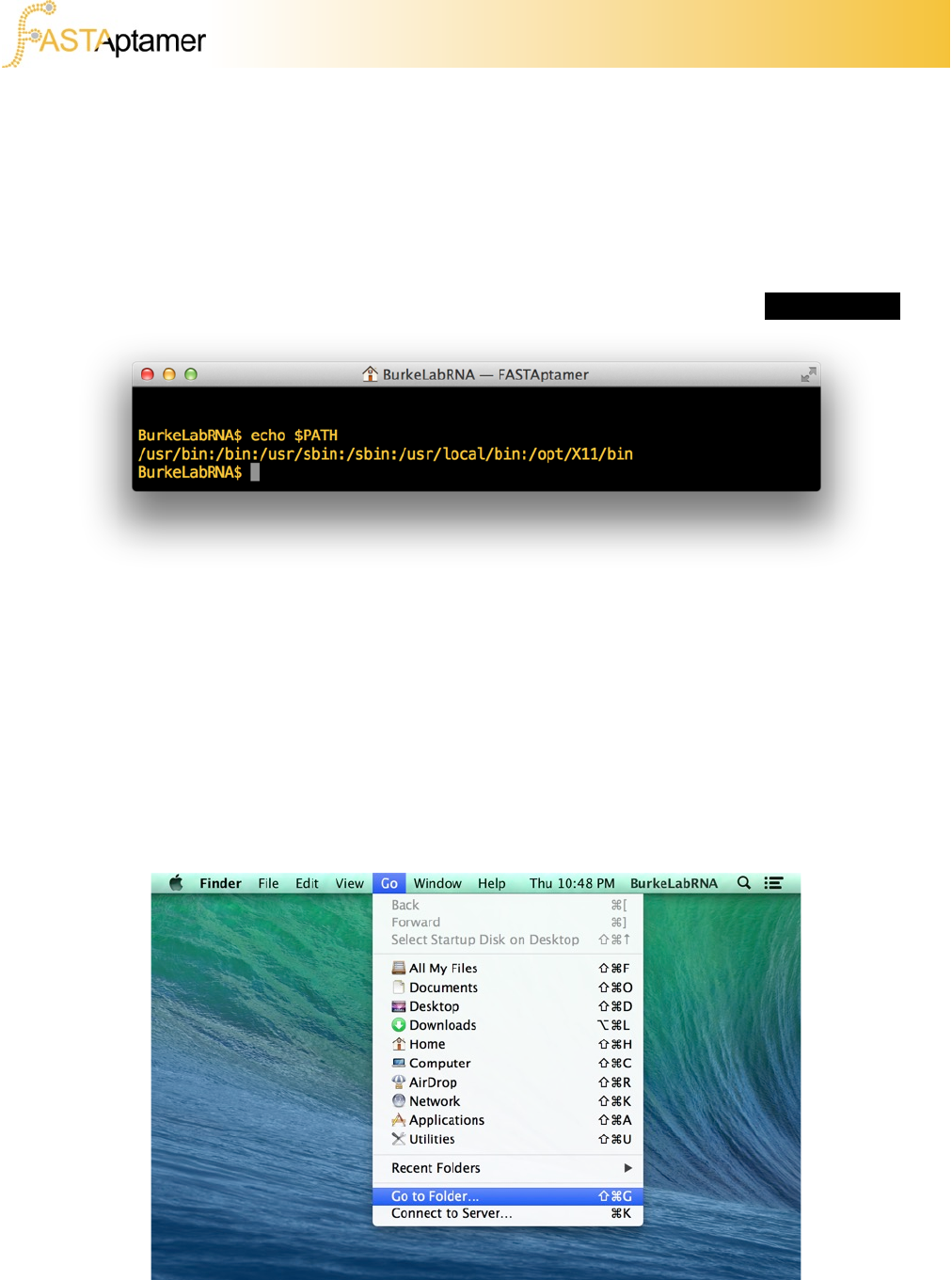
The PATH variable in a UNIX-like system is where “executable” programs are called
upon by the operating system. Having the FASTAptamer scripts saved in one of
these directories will ensure that no matter where you are in the file system, you’ll be
able to call upon the scripts to execute from the prompt. To find these directories,
open up a terminal emulator (the “Terminal” app on all Macs) and enter echo $PATH
2c. Installing as Executable
Each directory in the PATH variable is then displayed and separated by a colon.
When you enter a command in the command line, the system searches from the left-
most directories first for the program to execute. Be aware that some of these
directories require administrator privileges and will require your system password to
access. In Mac OS X, the /usr/local/bin directory is listed in the PATH but the folder
doesn’t usually exist until you create it.
Copy the scripts to the directory you have access to. Depending on your system and
your comfort level, this can be performed using the command line or by “dragging-
and-dropping” using the graphical user interface. On a Mac, we’ll do this by returning
to our desktop and clicking on “Go” in the menu bar.
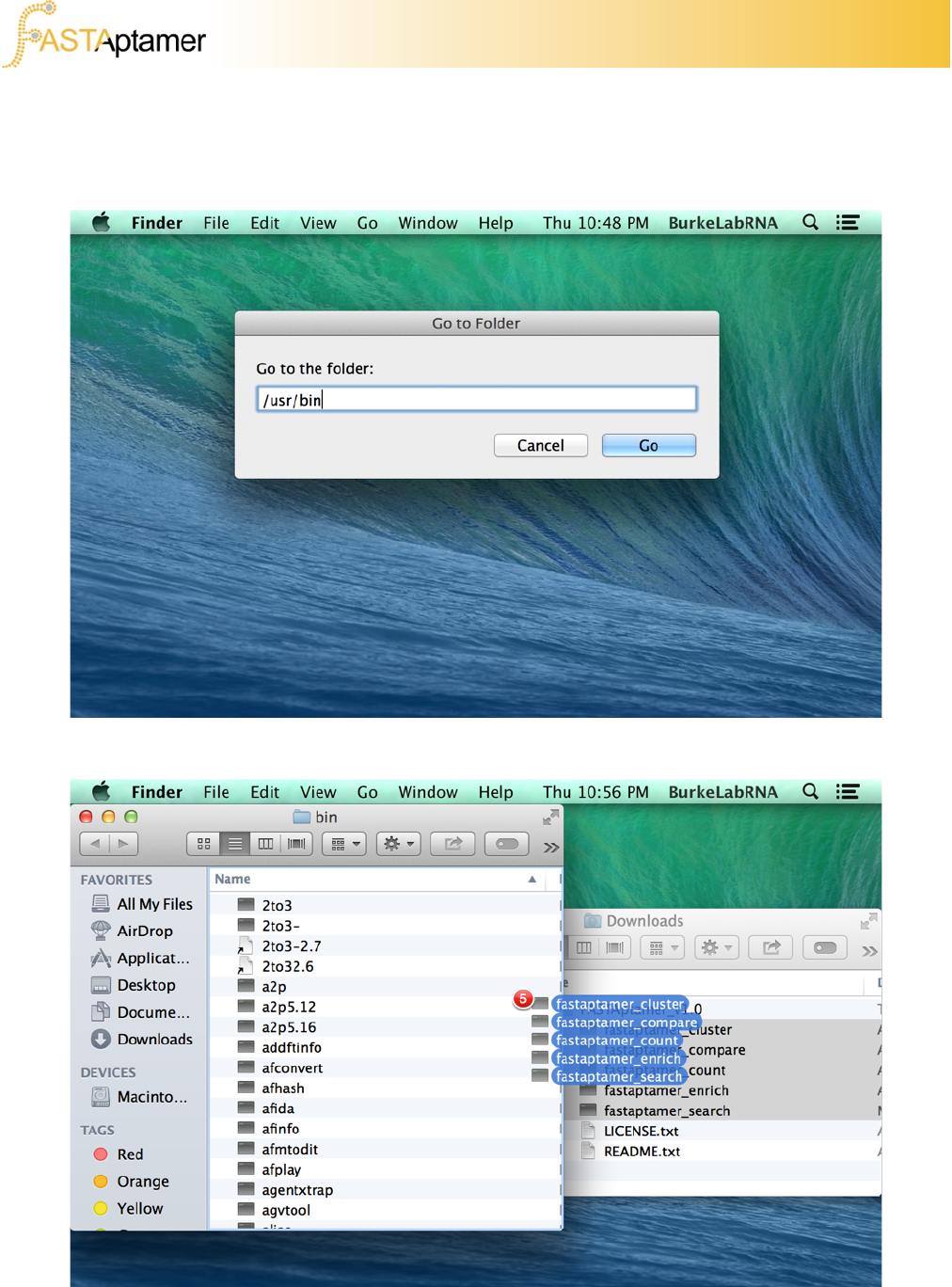
Enter the directory you wish to copy or save the scripts to.
2c. Installing as Executable
Copy or move the scripts into the executable directory.
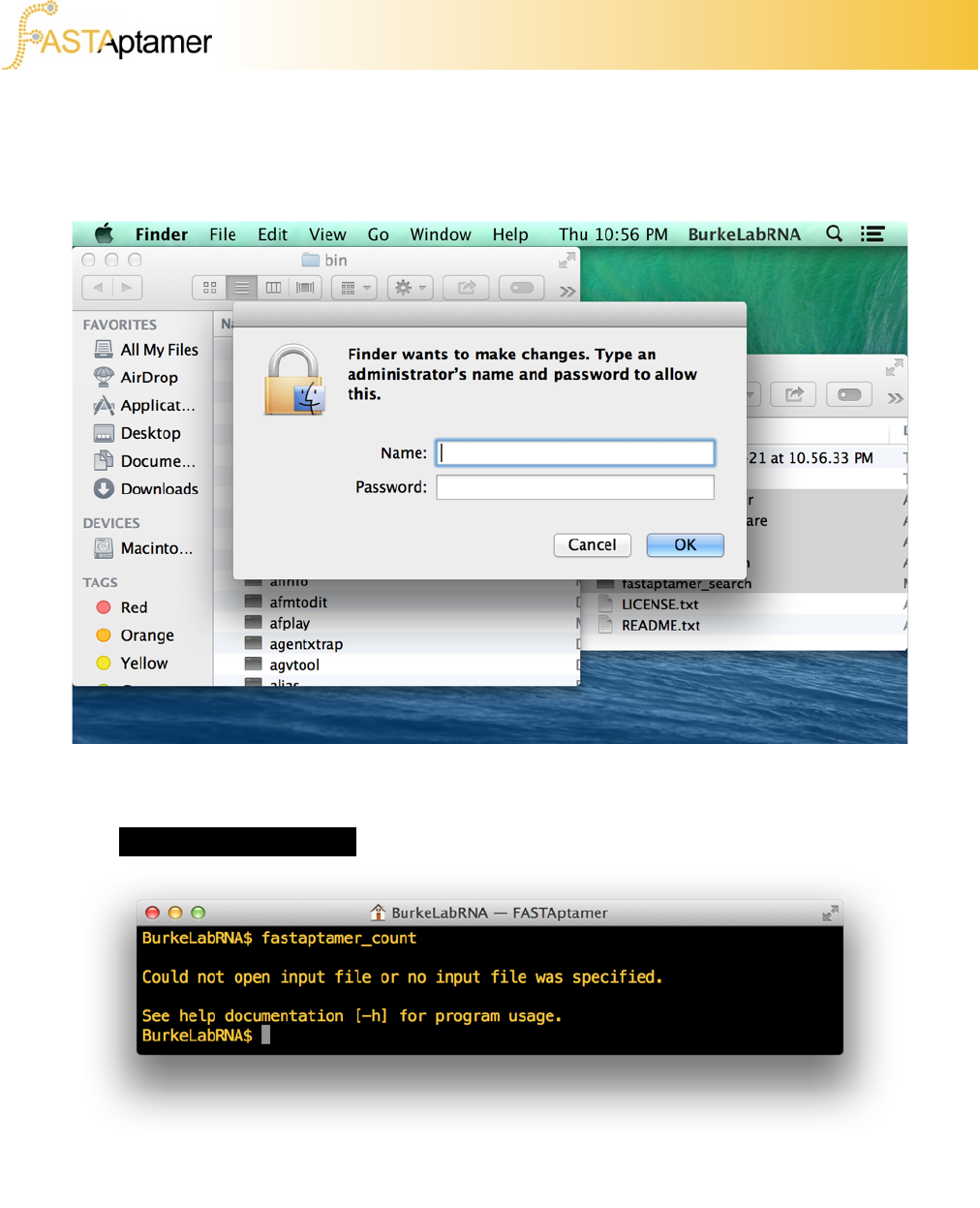
If necessary, enter an administrator password to complete the installation.
2c. Installing as Executable
At this point, the toolkit should be ready to use and executable from anywhere in your
directory. To test the installation, try to call up FASTAptamer-Count in the terminal by
typing
fastaptamer_count
at the prompt and hitting enter.
If you see an error message similar to the one above then you’re ready to proceed to
the tutorial in section 3. If you’re having issues with the installation, try using
FASTAptamer without installation.
An easier, yet inelegant, solution to using FASTAptamer is by creating a folder where
you’ll be doing your data analysis and copying the scripts in that folder. Rather than
entering the name of the script in the command line prompt, you’ll have to first enter
perl, followed by the script you wish to use.
2d. Use Without Installation
If you navigate out of the directory containing the scripts you’ll have to enter the
relative path of the script so that Perl can find it. For example, if you’re within a
subdirectory of where the scripts are located you may have to enter something like
the following:
Alternatively, if you’re in a parent directory you’ll have to enter something like this:
You should now be ready to use the FASTAptamer toolkit. If you’re still having issues
installing or using the software, feel free to contact us with a thorough description of
what you’ve tried and what’s happening.
The tutorials that follow are intended to get you familiar with FASTAptamer and the
various options provided by the scripts. As a convenience, we’ve provided two quick
reference tables (section 4) to refer to. We’ve also included a help screen for each
script that can be invoked by using the –h command.
3. Tutorial
3a. Data Requirements & Pre-Processing
FASTAptamer-Count, the gateway to the toolkit, requires input files in the FASTQ
format, the de facto standard for high-throughput DNA sequence files. SAM, BAM, or
other file formats are not currently supported. If your sequencing information is, or will
be, in one of these other formats, contact your sequencing provider and request
FASTQ files, or use one of the several utilities listed in the resources (section 4c) to
convert your files to FASTQ.
Although FASTAptamer-Count will accept any raw FASTQ file, it is prudent to ensure
that the input file itself undergoes some level of pre-processing. Typically this
involves the removal of 5’ and/or 3’ constant regions (“trimming”) and quality filtering
for only those reads whose bases have been called with high confidence. Several
pre-processing tools are listed in the resources (section 4c).
3b. Sample Data
Sample data for FASTAptamer can be downloaded as compressed FASTQ files from
the Burke Lab website at http://burkelab.missouri.edu/fastapamer.html and through
our GitHub site http://github.com/FASTAptamer.
The two population files (70HRT14.fastq.zip and 70HRT15.fastq.zip) are already pre-
processed (trimmed and filtered) and can be used directly in FASTAptamer after
decompression (“unzipping”).

FASTAptamer-Count determines the abundance of each sequence in a population
file. It also normalizes the reads for each sequence to RPM (reads per million), sorts
by decreasing abundance, and rank sequences before sending the information to
output. The FASTA formatted output file exploits the FASTA format by populating the
description line with the rank, reads, and RPM of each unique sequence.
3c. FASTAptamer-Count
>3420-14-7.04
TGAAAATGCAGACCAAGAAA…
Sequence Line
Description Line
Rank
Reads
Reads Per Million
All data must first be processed through FASTAptamer-Count to generate a non-
redundant FASTA file for use throughout the toolkit. Assuming you have already
decompressed the sample data file and that the files are located in our current
directory, let’s process both rounds using FASTAptamer-Count.
The command for FASTAptamer-Count is fastaptamer_count.
Before we begin, recall that for all FASTAptamer scripts we can always call up a help
screen using –help (or –h) to review requirements and options.
From the help screen we should be able to deduce that FASTAptamer-Count requires
an input file in FASTQ format.
We’ll begin with the 70HRT14.fastq file (which should already be “unzipped”). We can
specify this file by using the –i flag. Our command should read:
fastaptamer_count –i 70HRT14.fastq
We’ll also need to specify an output file using the –o flag. For simplicity sake we’ll
append _count to the current file name. Remember that the output file will now be in
FASTA format.
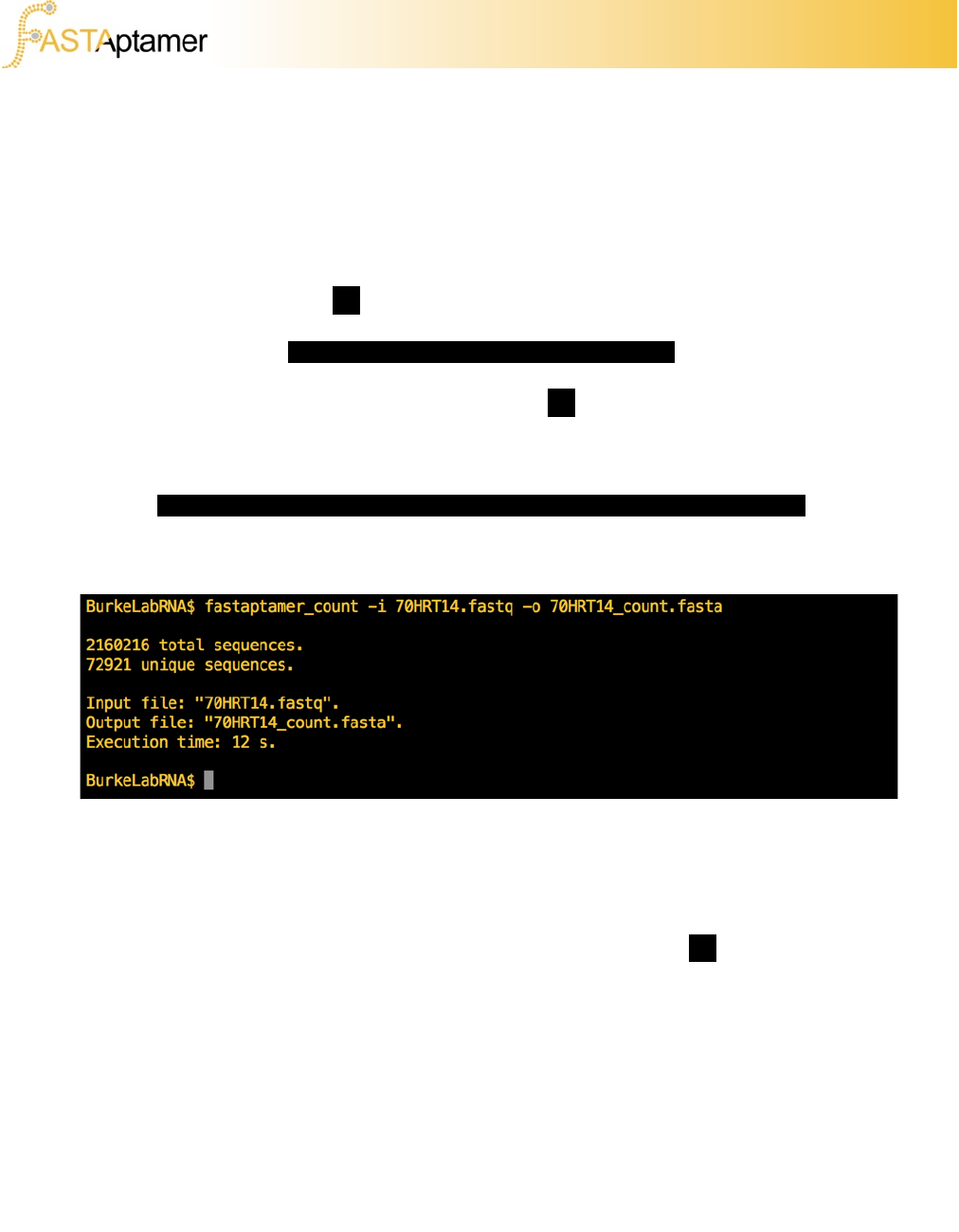
fastaptamer_count –i 70HRT14.fastq –o 70HRT14_count.fasta
Execute the command. Your output should look like this:
3c. FASTAptamer-Count
You’ll notice that FASTAptamer-Count provides a summary report showing the
number of sequence entries in the FASTQ file, as well as the number of non-
redundant entries created by FASTAptamer-Count.
This summary report can be suppressed by invoking the option –q on the command
line prior to execution. Just like the help screen, the ability to suppress summary
reports using the same command remains true for all FASTAptamer scripts.
For the next tutorials we’ll need a FASTAptamer-Count file for the 70HRT15
population. We’ll be able to use these two population files to compare the sequence
distribution, cluster into sequence families, calculate fold-enrichment across the
populations and search for the presence of sequence motifs.
Repeat these steps to generate a FASTAptamer-Count file called
70HRT15_count.fasta.

FASTAptamer-Compare compares the sequence distribution between two
populations files by generating a plain-text file with tab-separated values (.TSV) of
each sequence present in both populations, along with their respective reads per
million (RPM) and the calculated binary logarithm for the RPM (for each sequence,
Log2 RPMy/RPMx). FASTAptamer-Compare also facilitates the creation of a
histogram by taking those binary logarithm values and calculating the number of
times each value falls within one of its 102 bin buckets.
The command for FASTAptamer-Compare is fastaptamer_compare.
Pull up the help screen for FASTAptamer-Compare and review the usage and options
provided by the script.
fastaptamer_compare –h
We’ll need to specify two input files, called x and y, using the –i flag. We’ll use our
counted files generated using FASTAptamer-Count.
fastaptamer_compare –x 70HRT14_count.fasta –y 70HRT15_count.fasta
Before we execute the command, we’ll have to specify where we want our output file
to go and what we want to call it (using –o). For this example, let’s call it
70HRT14_vs_15_compare.tsv.
fastaptamer_compare –x 70HRT14_count.fasta –y 70HRT15_count.fasta –o
70HRT14_vs_15_compare.tsv
Execute the command. Your output should look like this:
3d. FASTAptamer-Compare
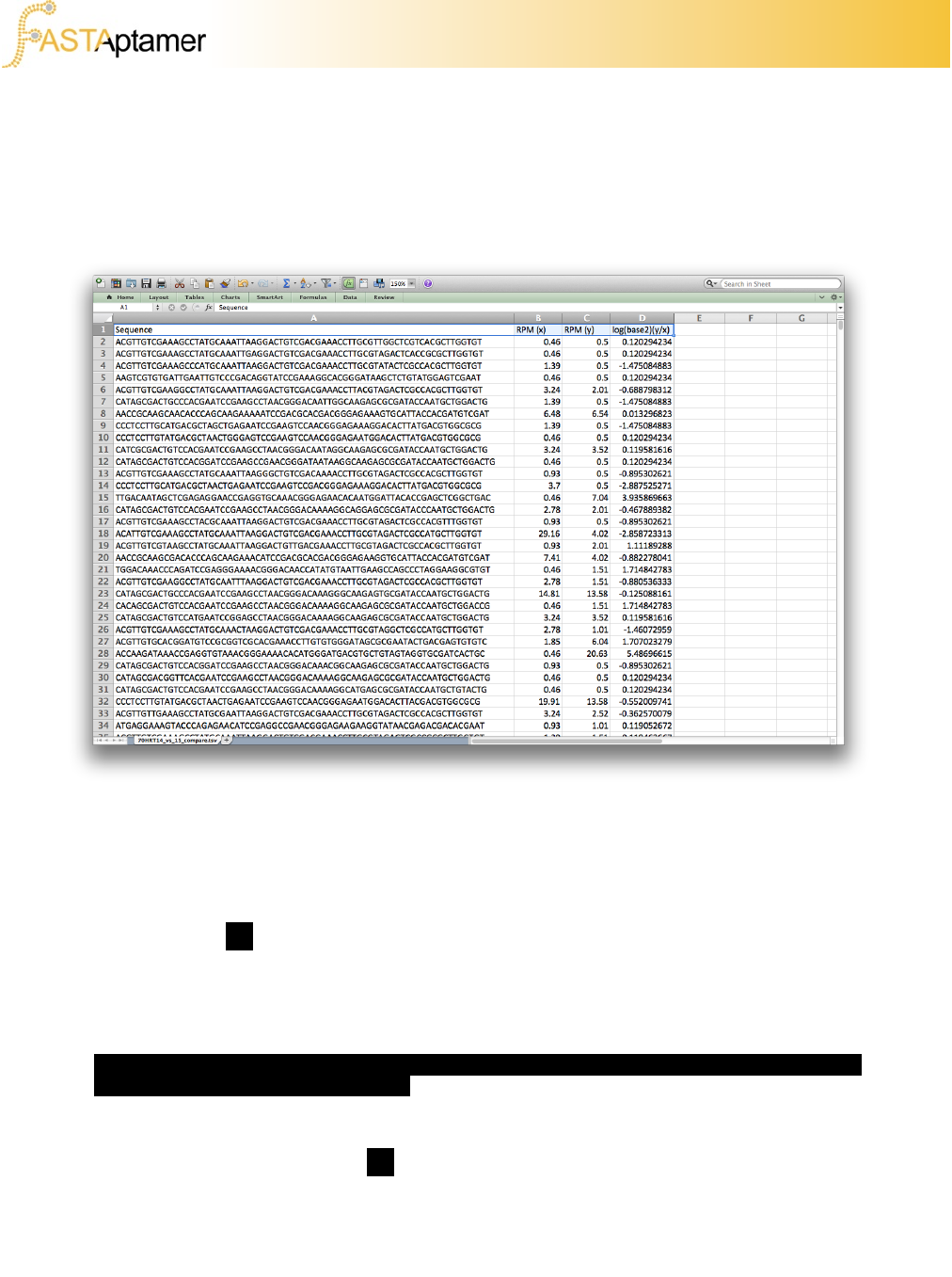
FASTAptamer-Compare created a .TSV file that can be opened using any standard
text editor or spreadsheet software.
3d. FASTAptamer-Compare
The histogram data is the last output generated and is available in the bottom-most
rows of the document.
Recall that the default of FASTAptamer-Compare is to only output sequences that
were present in BOTH population files. To output all the data, regardless of a match,
invoke the option –a on the command line prior to execution. For these “unmatched”
sequences, the output will leave out the RPM value for the population in which the
sequence was not found. It will also not calculate the binary logarithm or send any
additional values to the histogram bins for these sequences.
fastaptamer_compare –x 70HRT14_count.fasta –y 70HRT15_count.fasta –o
70HRT14_vs_15_compare.tsv -a
Lastly, the summary report listing the input and output files and execution time can be
suppressed with the addition of –q to the command line.
FASTAptamer-Cluster can generate clusters of closely-related sequences using
Levenshtein edit distance. The script preserves FASTA formatting and appends
cluster identity information to the description line information provided by
FASTAptamer-Count, including the cluster in which the sequence was grouped, the
rank within that cluster (as determined by reads), and the edit distance from the “seed
sequence” of the cluster.
3e. FASTAptamer-Cluster
The command for FASTAptamer-Cluster is fastaptamer_cluster.
If you call up the help screen (–h
) you’ll see that, similar to FASTAptamer-Count, we’ll
have to specify and input file (–i) and an output file (–o
). The output file will remain in
FASTA format, and for consistency’s sake, we’ll call the file 70HRT15_cluster.fasta
(for the 70HRT15 population).
fastaptamer_cluster –i 70HRT15_count.fasta –o 70HRT15_cluster.fasta
We’ll also have to specify the Levenshtein edit distance using the flag –d. Edit
distance is the number of insertions, deletions, or substitutions required to transform
one sequence string into another, for this reason, only integers can be used. For the
70HRT15 population we’ll use an edit distance of 7.
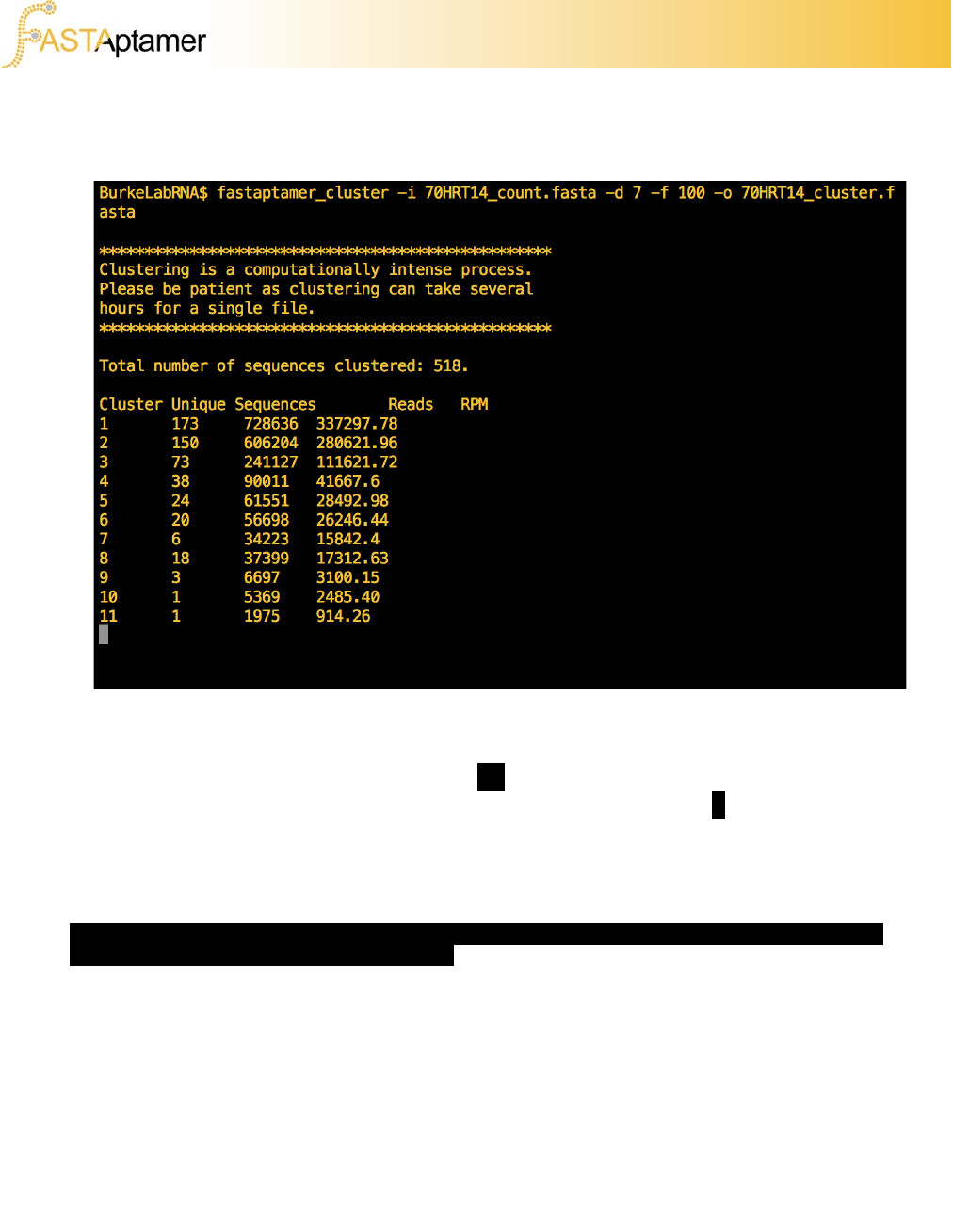
fastaptamer_cluster –i 70HRT15_count.fasta –o 70HRT15_cluster.fasta –d 7
Clustering is a slow process and can take tens of hours for diverse populations. For
our 70HRT14 population, if we were only interested in clustering sequences with an
RPM ≥ 100, we can apply the filtering option using the command line option –f.
fastaptamer_cluster –i 70HRT15_count.fasta –o 70HRT15_cluster.fasta –d 7
-f 100
Execute the command.
>56-2494-1254.61-2-10-1
ACCAAGGTAAACCGAGGTGTAAA…
Sequence Line
Description Line
Cluster
Rank
Edit Distance
3e. FASTAptamer-Cluster
You’ll notice that FASTAptamer-Cluster provides additional information on the cluster
size (in terms of unique sequences and total reads and RPM) as it finishes each
cluster. This can be suppressed using the (–q) flag, or you may elect to ‘redirect’ the
output to a new file using the standard Unix redirection command (>). This redirected
output file will contain the cluster statistics in a tab-delimited format and can be used
to create graphs of cluster sizes, useful for making informed decisions on which
clusters to analyze further.
fastaptamer_cluster –i 70HRT15_count.fasta –o 70HRT15_cluster.fasta –d 7
-f 100 > 70HRT15_cluster_sizes.tsv

3f. FASTAptamer-Enrich
FASTAptamer-Enrich calculates fold-enrichment for individual sequences across
populations. The script generates a plain-text file with tab-separated values for use in
any standard spreadsheet software. Output information contains each sequence, the
sequence length, the rank, reads and RPM for each sequence and population the
sequence was detected in and cluster information (if available). Lastly, the output file
contains the fold-enrichment ratio for each possible pairwise comparison (y/x, z/y and
z/x).
FASTAptamer-Enrich requires two files, but can process up to 3 files simultaneously.
Each file must come from either FASTAptamer-Count or FASTAptamer-Cluster.
FASTAptamer-Enrich will adjust output accordingly to accommodate for populations
with cluster information.
The command for FASTAptamer-Enrich is fastaptamer_enrich.
If you call up the help screen (-h) you’ll notice that the input files will be designated
using the flags (-x,-y, and an optional third file -z).
Let’s provide FASTAptamer-Enrich with a 70HRT14 population file from
FASTAptamer-Count and a FASTAptamer-Cluster file for the 70HRT15 population.
fastaptamer_enrich –x 70HRT14_count.fasta –y 70HRT15_cluster.fasta
We’ll also have to specify a name and location for the output file (-o). We’ll keep this
file in the current directory and call it 70HRT14_vs_15_enrich.tsv.
3f. FASTAptamer-Enrich
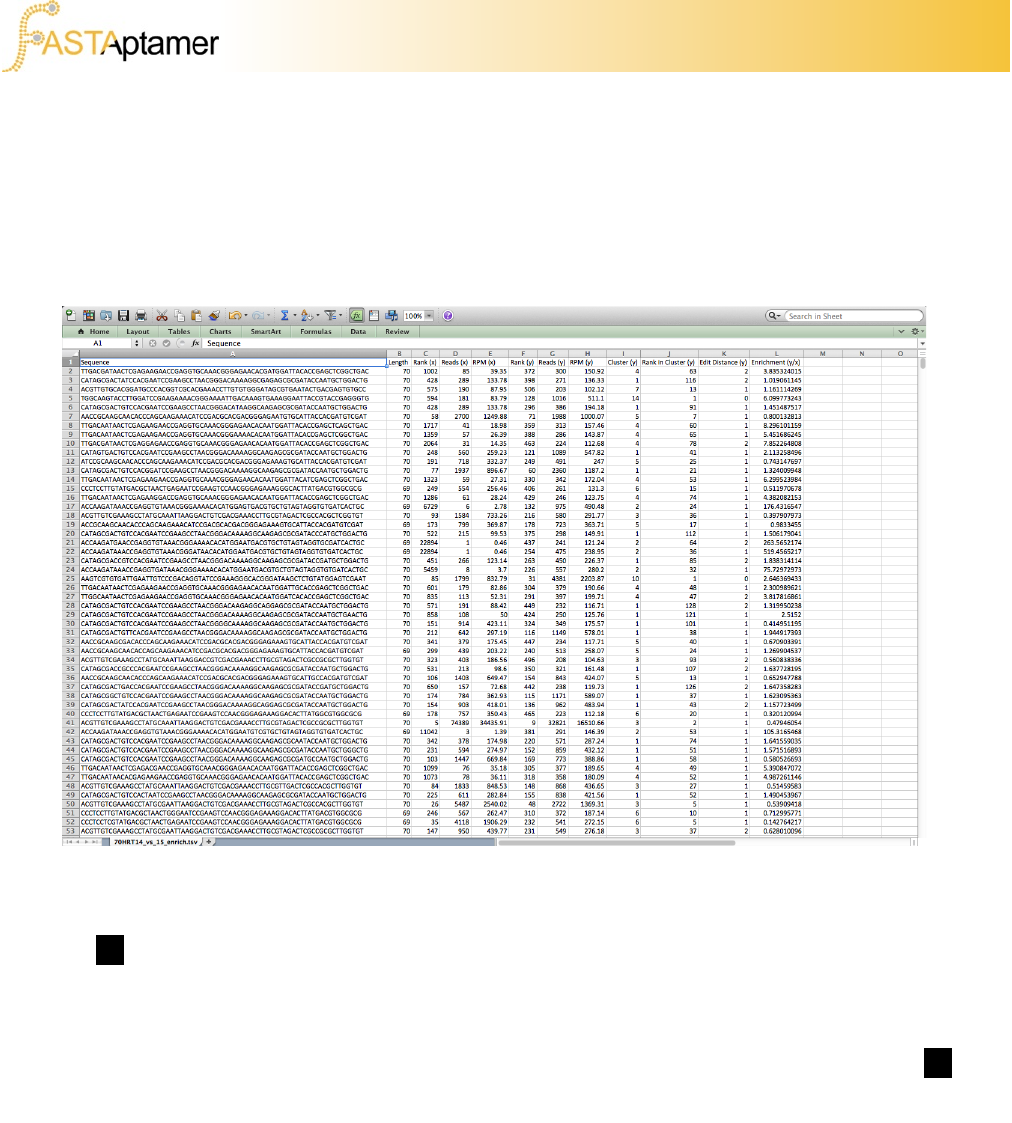
We can open up this file in our favorite spreadsheet software and sort by metrics such
as the rank in the final population or by overall enrichment. We have found that data
from this step is useful for identifying candidate molecules for further investigation.
FASTAptamer-Enrich also provides a tool to filter output to only a subset of
sequences that were highly sampled. To invoke this command use the command line
flag –f and specify a RPM value that sequences must meet or exceed to get sent to
output. The script will tally the RPM values for each sequence across all populations it
was present in to determine whether the criterion is being met.
Like all FASTAptamer scripts, the summary report can be suppressed by invoking -q
at the command line prior to execution.

3g. FASTAptamer-Search
FASTAptamer-Search is a script that allows for degenerate motif searches using
IUPAC-IUBMB single nucleotide codes. Keep in mind that the use of T and U are
interchangeable.
A/T/G/C/U single bases
R puRines (A/G)
Y pYrimidines (C/T)
W Weak (A/T)
S Strong (G/C)
M aMino (A/C)
K Keto (G/T)
B not A
D not C
H not G
V not T
N aNy base (not a gap)
With FASTAptamer-Search you can search for the co-occurrence of more than one
motif and across multiple files. FASTAptamer-Search will generate an output file
containing only those sequences that matched the patterns.
The command for FASTAptamer-Search is fastaptamer_search.
A notable difference between this script and the others in the toolkit is that the help
screen requires the use of the full command line flag –help, to avoid ambiguity with
the –highlight flag which allows us to place parentheses around matched patterns
for easy visualization.
For the both the 70HRT14 and 70HRT15 clustered population, we’ll search for the
presence of the dominant family 1 pseudoknot motif. This motif contains two patterns,
UCCG and CGGGANAA. For each input file we’ll have to use an input flag (-i), and
for each pattern we’ll have to use a pattern flag (-p).
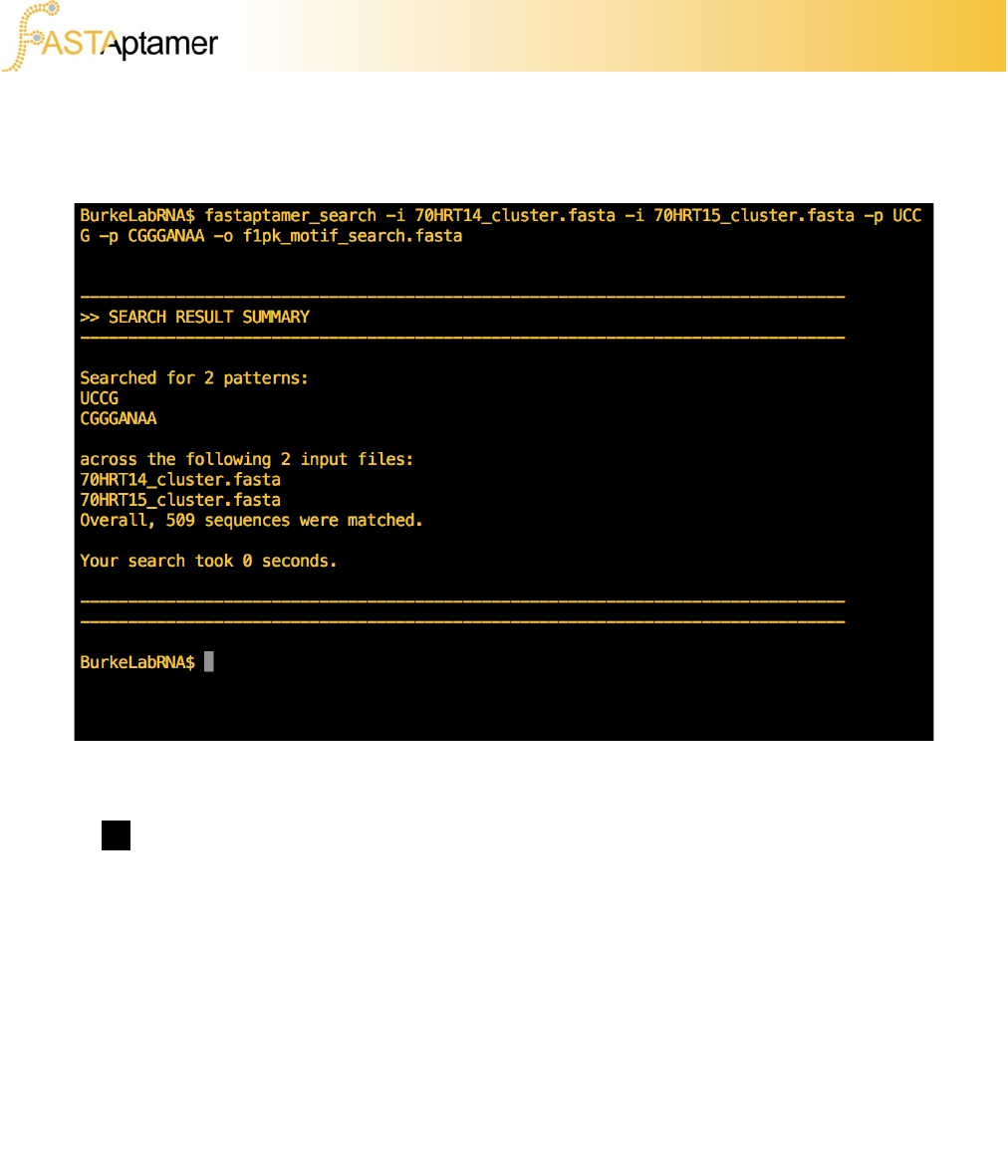
fastaptamer_search –i 70HRT14_cluster.fasta –i 70HRT15_cluster.fasta
-p UCCG –p CGGANAA –o f1pk_motif_search.fasta
Execute the search. If you find that the output is being displayed on screen, you’ll be
aware that the script can be used to quickly find a motif, or can create an output file of
matches using the -o flag.
3g. FASTAptamer-Search
Summary reports listing the number of matched sequences can be suppressed using
the -q flag.
You may notice that the number of matched sequences seems rather low. If we revist
the process that we took to get to these steps you’ll notice that we’re using clustered
files, and that during the clustering process for this tutorial, we restricted our output to
only those sequences that were sampled with 100 RPM or more.

4a. FASTAptamer Input/Output
Script Input Output
fastaptamer_count FASTQ FASTA
fastaptamer_compare 2 FASTA files
(from FASTAptamer-Count)
Tab separated values plain text
fastaptamer_cluster FASTA
(from FASTAptamer-Count)
FASTA
fastaptamer_enrich
2 or 3 FASTA files
(from FASTAptamer-Count or
FASTAptamer-Cluster)
Tab separated values plain text
fastaptamer_search FASTA FASTA
FASTA

Script Function
Command Line
Flags
Interpretation
fastaptamer_count
Determines abundance of each
sequence, normalizes value to
total reads per million, ranks and
sorts by decreasing abundance.
-i
Input file (.FASTQ)*
-o
Output file (.FASTA)*
-h
Help screen
-q
Quiet mode - suppresses summary report
-v
Display version
fastaptamer_compare
Calculates log2 values of RPM
y/x, generates table containing
RPM for each sequence in both
files, generates and fills values for
histogram of sequence
distribution.
-x
Input file 1 (.FASTA from FASTAptamer-
Count or FASTAptamer-Cluster)*
-y
Input file 2 (.FASTA from FASTAptamer-
Count or FASTAptamer-Cluster)*
-o
Output file (.TSV)*
-h
Help screen
-a
Output all sequences
-q
Quiet mode - suppresses summary report
-v
Display version
fastaptamer_cluster
Generates sequence clusters
based on a user-defined
Levenshtein edit distance.
-i
Input file (.FASTA from FASTAptamer-
Count)*
-o
Output file (.FASTA)*
-h
Help screen
-d
Edit Distance*
-f
Read filter
-q
Quiet mode - suppresses summary report
-v
Display version
fastaptamer_enrich
Calculates fold-enrichment values
for each sequence in 2 or 3
populations.
-x
Input file 1 (.FASTA from FASTAptamer-
Count or FASTAptamer-Cluster)*
-y
Input file 2 (.FASTA from FASTAptamer-
Count or FASTAptamer-Cluster)*
-z
Input file 3 (optional - .FASTA from
FASTAptamer-Count or FASTAptamer-
Cluster)
-o
Output file (.TSV)*
-h
Help screen
-f
RPM threshold filter
-q
Quiet mode - suppresses summary report
-v
Display version
fastaptamer_search
Degenerately searches for
multiple sequence patterns
across several files.
-i
Input files(s) (.FASTA from FASTAptamer-
Count, FASTAptamer-Cluster or other)*
-o
Output file (.FASTA)
-p
Pattern(s) *
-help
Help screen
-highlight
Highlight matched motifs
-q
Quiet mode - suppresses summary report
-v
Display version
*Required
4b. FASTAptamer Command Line Options

!
Learning Command Line:
•
Web Resources
oLinuxCommand.Org (http://linuxcommand.org) is a highly
recommended resource and includes a free downloadable PDF.
oA pair of frequently recommended online tutorials can be found at
http://ryanstutorials.net/linuxtutorial/ and http://cli.learncodethehardway.org.
•
Books
oUNIX and Perl to the Rescue!: A Field Guide for the Life Sciences
(and Other Data-Rich Pursuits) by Keith Bradnam and Ian Korf.
oPractical Computing for Biologists by Steven Haddock and Casey
Dunn
Data pre-processing (trimming, filtering for quality, etc.):
•cutadapt (http://code.google.com/p/cutadapt/) - what we use to trim
constant regions from our sequencing data.
•FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) - our go to set
of tools for quality analysis and filtering.
OMICtools (http://omictools.com/common-tools-c1219-p1.html) provides a
long list of common software for high-throughput sequence analysis. Some
other tools we keep coming across are listed below.
•PRINSEQ (http://prinseq.sourceforge.net)
•FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
•SolexaQA (http://solexaqa.sourceforge.net)
•ea-utils (http://code.google.com/p/ea-utils/)
•Trimmomatic (http://www.usadellab.org/cms/?page=trimmomatic)
4c. Resources and Links
