Gaussian09 Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 350 [warning: Documents this large are best viewed by clicking the View PDF Link!]
- 官方Gaussian 09文献的引用
- 第1部分 设置和运行Gaussian计算
- 第2部分 Gaussian 09的关键词,LINK0命令和工具
- Gaussian 09关键词
- #
- ADMP
- AM1
- Amber
- B3LYP
- BD
- BOMD
- CacheSize
- CASSCF
- CBS方法
- CBSExtrapolate
- CCD和CCSD
- Charge
- ChkBasis
- CID和CISD
- CIS和CIS(D)
- CNDO
- Complex
- Constants
- Counterpoise
- CPHF
- Density
- DensityFit
- 密度泛函(DFT)方法
- DFTB和DFTBA
- Dreiding
- EOMCCSD
- EPT
- ExtendedHuckel
- External
- ExtraBasis和ExtraDensityBasis
- 冻芯选项
- Field
- FMM
- Force
- Freq
- G1-G4方法
- Gen,GenECP
- GenChk
- Geom
- GFInput
- GFPrint
- Guess
- GVB
- HF
- Huckel
- INDO
- Integral
- IOp
- IRC
- IRCMax
- LSDA
- MaxDisk
- MINDO3
- 分子力学方法
- MNDO
- MP和双杂化DFT方法
- Name
- NMR
- NoDensityFit
- ONIOM
- Opt
- Output
- OVGF
- PBC
- PM3
- PM6
- Polar
- Population
- Pressure
- Prop
- Pseudo
- Punch
- QCISD
- 重新开始计算
- SAC-CI
- Scale
- Scan
- SCF
- SCRF
- 半经验方法
- SP
- Sparse
- Stable
- Symmetry
- TD
- Temperature
- Test
- TestMO
- TrackIO
- Transformation
- UFF
- Units
- Volume
- W1方法
- ZINDO
- 链接0命令总结
- 实用程序
- Gaussian 09关键词
- 附录
- 参考文献


目录
官方Gaussian 09 文献的引用 ..................................................................................................... - 1 -
推荐的附加引用.................................................................................................................. - 1 -
第 1 部分 设置和运行Gaussian计算........................................................................................ - 3 -
Gaussian 09 输入概述 ......................................................................................................... - 5 -
Gaussian 09 输入部分的顺序 ............................................................................................. - 7 -
输入语法.............................................................................................................................. - 9 -
Gaussian 09 任务类型 ....................................................................................................... - 10 -
模型化学............................................................................................................................ - 12 -
基组.................................................................................................................................... - 14 -
添加极化和弥散函数................................................................................................ - 16 -
有关基组的其它关键词............................................................................................- 17 -
纯基函数与笛卡尔基函数产生的问题.................................................................... - 17 -
密度拟合基组............................................................................................................ - 18 -
分子说明概述.................................................................................................................... - 19 -
指定同位素以及其它核参数.................................................................................... - 20 -
指定分子片段............................................................................................................ - 20 -
分子力学原子类型.................................................................................................... - 21 -
PDB文件参数 ........................................................................................................... - 21 -
指定空原子................................................................................................................ - 22 -
指定周期体系............................................................................................................ - 22 -
多步任务............................................................................................................................ - 24 -
运行Gaussian..................................................................................................................... - 25 -
指定擦写文件的操作和位置.................................................................................... - 25 -
跨磁盘分割擦写文件........................................................................................ - 26 -
保存和删除擦写文件........................................................................................ - 26 -
初始化文件................................................................................................................ - 27 -
控制内存的使用........................................................................................................ - 27 -
在UNIX系统运行Gaussian..................................................................................... - 27 -
脚本与Gaussian ................................................................................................. - 28 -
用NQS执行批处理............................................................................................ - 29 -
配置Gaussian.................................................................................................................... - 30 -
系统需求.................................................................................................................... - 30 -
设置Gaussian执行环境........................................................................................... - 30 -
擦写文件的考虑事项........................................................................................ - 30 -
站内定制:Default.Route文件 ............................................................................... - 31 -
默认执行路径.................................................................................................... - 31 -
Default.Route的限制 ......................................................................................... - 32 -
默认内存............................................................................................................ - 32 -
共享内存多处理器的并行执行........................................................................ - 32 -
网络/集群的并行执行....................................................................................... - 32 -
站点名................................................................................................................ - 33 -
主机名................................................................................................................ - 33 -
- 1 -
典型的默认设置................................................................................................ - 33 -
用户默认文件.................................................................................................... - 33 -
用Linda运行集群/LAN并行计算 ............................................................................. - 34 -
Linda并行方法.................................................................................................. - 34 -
安装Linda软件和编译G09/Linda ..................................................................... - 34 -
用Linda运行Gaussian........................................................................................ - 34 -
配置Gaussian 09/Linda...................................................................................... - 35 -
开始并行Gaussian 09 任务 ............................................................................... - 36 -
监测计算............................................................................................................ - 36 -
在基于PPC的Mac机上指定每节点的工作程序.............................................. - 37 -
运行Gaussian测试任务........................................................................................... - 37 -
运行测试文件前,重命名已存在的Default.Route文件.................................. - 37 -
例子.................................................................................................................... - 37 -
Gaussian 09 的链接 ................................................................................................ - 38 -
第 2 部分 Gaussian 09 的关键词,LINK0 命令和工具........................................................ - 41 -
Gaussian 09 关键词........................................................................................................... - 43 -
#................................................................................................................................. - 43 -
ADMP ........................................................................................................................ - 43 -
AM1 ........................................................................................................................... - 47 -
Amber........................................................................................................................ - 47 -
B3LYP ....................................................................................................................... - 47 -
BD.............................................................................................................................. - 47 -
BOMD........................................................................................................................ - 48 -
CacheSize ................................................................................................................ - 53 -
CASSCF ................................................................................................................... - 54 -
CBS方法 ................................................................................................................... - 60 -
CBSExtrapolate ....................................................................................................... - 62 -
CCD和CCSD............................................................................................................ - 63 -
Charge ...................................................................................................................... - 64 -
ChkBasis...................................................................................................................- 64 -
CID和CISD ............................................................................................................... - 65 -
CIS和CIS(D)............................................................................................................. - 66 -
CNDO........................................................................................................................ - 68 -
Complex.................................................................................................................... - 69 -
Constants.................................................................................................................. - 69 -
Counterpoise............................................................................................................ - 70 -
CPHF......................................................................................................................... - 71 -
Density ...................................................................................................................... - 72 -
DensityFit.................................................................................................................. - 74 -
密度泛函(DFT)方法 ............................................................................................ - 75 -
DFTB和DFTBA ........................................................................................................ - 80 -
Dreiding.....................................................................................................................- 81 -
EOMCCSD ............................................................................................................... - 81 -
EPT............................................................................................................................ - 84 -
- 2 -
ExtendedHuckel....................................................................................................... - 85 -
External..................................................................................................................... - 85 -
ExtraBasis和ExtraDensityBasis ............................................................................ - 87 -
冻芯选项.................................................................................................................... - 88 -
Field........................................................................................................................... - 89 -
FMM .......................................................................................................................... - 91 -
Force ......................................................................................................................... - 91 -
Freq ........................................................................................................................... - 92 -
G1-G4 方法............................................................................................................. - 103 -
Gen,GenECP ...................................................................................................... - 105 -
GenChk................................................................................................................... - 110 -
Geom....................................................................................................................... - 110 -
GFInput ................................................................................................................... - 114 -
GFPrint.................................................................................................................... - 115 -
Guess ...................................................................................................................... - 115 -
GVB ......................................................................................................................... - 122 -
HF ............................................................................................................................ - 125 -
Huckel ..................................................................................................................... - 125 -
INDO........................................................................................................................ - 126 -
Integral .................................................................................................................... - 126 -
IOp ........................................................................................................................... - 128 -
IRC........................................................................................................................... - 129 -
IRCMax ................................................................................................................... - 132 -
LSDA ....................................................................................................................... - 135 -
MaxDisk .................................................................................................................. - 135 -
MINDO3 .................................................................................................................. - 135 -
分子力学方法.......................................................................................................... - 136 -
MNDO ..................................................................................................................... - 154 -
MP和双杂化DFT方法............................................................................................ - 155 -
Name....................................................................................................................... - 156 -
NMR ........................................................................................................................ - 156 -
NoDensityFit........................................................................................................... - 158 -
ONIOM .................................................................................................................... - 159 -
Opt ........................................................................................................................... - 164 -
Output...................................................................................................................... - 177 -
OVGF ...................................................................................................................... - 178 -
PBC ......................................................................................................................... - 178 -
PM3 ......................................................................................................................... - 179 -
PM6 ......................................................................................................................... - 179 -
Polar ........................................................................................................................ - 180 -
Population............................................................................................................... - 182 -
Pressure.................................................................................................................. - 186 -
Prop ......................................................................................................................... - 186 -
Pseudo .................................................................................................................... - 188 -
- 3 -
Punch ...................................................................................................................... - 193 -
QCISD..................................................................................................................... - 193 -
重新开始计算.......................................................................................................... - 194 -
SAC-CI .................................................................................................................... - 195 -
Scale........................................................................................................................ - 200 -
Scan ........................................................................................................................ - 200 -
SCF.......................................................................................................................... - 201 -
SCRF....................................................................................................................... - 204 -
PCM计算的附加关键词 ................................................................................. - 211 -
半经验方法.............................................................................................................. - 218 -
SP ............................................................................................................................ - 223 -
Sparse ..................................................................................................................... - 223 -
Stable ...................................................................................................................... - 223 -
Symmetry................................................................................................................ - 225 -
TD ............................................................................................................................ - 225 -
Temperature ........................................................................................................... - 227 -
Tes t .......................................................................................................................... - 227 -
Tes t MO .................................................................................................................... - 227 -
TrackIO.................................................................................................................... - 228 -
Transformation....................................................................................................... - 228 -
UFF.......................................................................................................................... - 228 -
Units......................................................................................................................... - 228 -
Volume .................................................................................................................... - 229 -
W1 方法................................................................................................................... - 229 -
ZINDO ..................................................................................................................... - 231 -
链接 0命令总结 ..................................................................................................... - 232 -
实用程序.......................................................................................................................... - 234 -
GAUSS_MEMDEF环境变量 ................................................................................ - 234 -
c8609....................................................................................................................... - 234 -
chkchk ..................................................................................................................... - 235 -
cubegen .................................................................................................................. - 235 -
cubman ................................................................................................................... - 238 -
formchk ................................................................................................................... - 239 -
freqchk .................................................................................................................... - 240 -
freqmem.................................................................................................................. - 242 -
gauopt ..................................................................................................................... - 243 -
ghelp........................................................................................................................ - 244 -
mm........................................................................................................................... - 244 -
newzmat.................................................................................................................. - 245 -
testrt......................................................................................................................... - 248 -
unfchk...................................................................................................................... - 249 -
附录 ......................................................................................................................................... - 251 -
Gaussian 09 和Gaussian 03 之间的变化......................................................................... - 253 -
新的方法和功能...................................................................................................... - 253 -
- 4 -
能量与导数...................................................................................................... - 253 -
ONIOM ............................................................................................................ - 253 -
溶剂化.............................................................................................................. - 253 -
几何优化和IRC ............................................................................................... - 253 -
分子特性.......................................................................................................... - 254 -
分析和输出...................................................................................................... - 254 -
其它的新功能.................................................................................................. - 254 -
效率的改善.............................................................................................................. - 254 -
Gaussian 09 与Gaussian 03 之间功能的差别 ................................................... - 255 -
工具程序的增强...................................................................................................... - 255 -
效率的考虑...................................................................................................................... - 257 -
估计计算的内存需求.............................................................................................. - 257 -
并行计算的内存需求...................................................................................... - 257 -
SCF流程.................................................................................................................... - 258 -
收敛的问题...................................................................................................... - 258 -
MP2 能量,梯度,和频率....................................................................................... - 259 -
更高的关联方法...................................................................................................... - 259 -
CIS和TD的能量和梯度............................................................................................ - 259 -
积分的存储...................................................................................................... - 259 -
重新开始任务和重复使用波函...................................................................... - 260 -
CIS和TD的激发态密度 .................................................................................. - 260 -
开壳层激发态的缺陷...................................................................................... - 261 -
稳定性计算.............................................................................................................. - 261 -
CASSCF的效率.......................................................................................................... - 261 -
CASSCF频率................................................................................................... - 261 -
程序的限制...................................................................................................................... - 262 -
构造Z矩阵 ....................................................................................................................... - 263 -
使用内坐标.............................................................................................................. - 263 -
混合内坐标和笛卡尔坐标...................................................................................... - 264 -
另一种Z-矩阵格式.................................................................................................. - 265 -
使用虚原子.............................................................................................................. - 265 -
模型构建程序的几何结构定义.............................................................................. - 266 -
格式化检查点文件的结构.............................................................................................. - 268 -
基组数据.................................................................................................................. - 269 -
可用的项目.............................................................................................................. - 270 -
常见问题.................................................................................................................. - 271 -
例子.......................................................................................................................... - 271 -
程序的开发功能.............................................................................................................. - 276 -
程序开发有关的关键词.......................................................................................... - 276 -
一般的任务重新开始...................................................................................... - 276 -
IOP设定关键词............................................................................................... - 276 -
标准关键词的调试选项.................................................................................. - 277 -
改变链接的调用和顺序.................................................................................. - 279 -
指定非标准的执行路径.................................................................................. - 279 -
- 5 -
- 6 -
标准方位的约定...................................................................................................... - 282 -
选择的目的...................................................................................................... - 283 -
一般的考虑...................................................................................................... - 283 -
定位坐标轴的规则.......................................................................................... - 283 -
定位电荷主轴的规则...................................................................................... - 283 -
对称陀螺分子的特殊规则.............................................................................. - 284 -
球陀螺分子的特殊规则.................................................................................. - 284 -
RWF编号................................................................................................................. - 285 -
作废的关键词和过时的功能.................................................................................. - 292 -
作废的工具...................................................................................................... - 293 -
过时的功能...................................................................................................... - 293 -
参考文献.................................................................................................................................. - 297 -

官方Gaussian 09 文献的引用
Gaussian 09代表对以前出版的Gaussian 70,Gaussian 76,Gaussian 80,Gaussian 82,
Gaussian 86,Gaussian 88,Gaussian 90,Gaussian 92,Gaussian 92/DFT,Gaussian 94,
Gaussian 98,和Gaussian 03系统[G70, G76, G80, G82, G86, G88, G90, G92DFT, G94, G98, G03]的进一步
开发。以下给出本项工作在当前需要的引用;注意:需要用实际的修订版程序标识号代替
Revision A.1。描述Gaussian 09科技引用的论文正在准备中。一旦出版,以后应当引用这
篇参考文献(最新的信息参见www.gaussian.com/citation_g09.htm)。
Gaussian 09, Revision A.1, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E.
Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci,
G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F.
Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota,
R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai,
T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J.
J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand,
K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi,
N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo,
J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi,
C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G.
A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas,
J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc.,
Wallingford CT, 2009.
Gaussian 09中首次出现的进展是M. J. Frisch, G. W. Trucks, J. R. Cheeseman, G.
Scalmani, M. Caricato, H. P. Hratchian, X. Li, V. Barone, J. Bloino, G. Zheng, T.
Vreven, J. A. Montgomery, Jr., G. A. Petersson, G. E. Scuseria, H. B. Schlegel, H.
Nakatsuji, A. F. Izmaylov, R. L. Martin, J. L. Sonnenberg, J. E. Peralta, J. J. Heyd,
E. Brothers, F. Ogliaro, M. Bearpark, M. A. Robb, B. Mennucci, K. N. Kudin, V. N.
Staroverov, R. Kobayashi, J. Normand, A. Rendell, R. Gomperts, V. G. Zakrzewski,
M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y.
Honda, O. Kitao,和H. Nakai等人的工作。
推荐的附加引用
在报告Gaussian计算结果时除了给出程序本身的引用之外,通常我们还推荐引用描述计
算所用理论方法的原始参考文献。这些参考文献在本手册有关关键词的详细论述中给出。唯
一的特例是那些建立时间比较长的方法,例如Hartree-Fock理论已经发展到约定俗成的程
度,在这一点上,它基本是自引用的。在某些情况下,Gaussian的输出中会显示与该计算类
型有关的参考文献。
Gaussian还包含作为链接607的程序NBO。若使用该程序,则应另外引用如下:
NBO Version 3.1, E. D. Glendening, A. E. Reed, J. E. Carpenter, and F. Weinhold.
- 1 -
还应引用NBO的原始参考文献[Foster80, Reed83a, Reed85, Reed85a, Carpenter87, Carpenter88, Reed88,
Weinhold88]。
- 2 -
Part
第1部分 设置和运行Gaussian计算
输入概述
任务类型
模型化学
基组
分子说明
多步任务
运行 Gaussian 任务
效率的考虑
在UNIX 系统配置 Gaussian 09
在集群和 LAN 上并运行行计算
Gaussian 测试任务
Gaussian 09 链接的列表
- 3 -
- 4 -

Gaussian 09 输入概述
Gaussian 09输入由写在ASCII文本文件中一系列的行构成。Gaussian输入文件的基本结
构包含几个不同部分:
链接0命令行:定位和命名擦写文件(无结束空行)。
执行路径部分(#行):指定所需的计算类型,模型化学,以及其它选项(有结束
空行)。
标题部分:计算的简要描述(有结束空行)。这一部分在输入中是必须的,但是不
被Gaussian 09程序分析。它出现在输出文件中用于识别和描述计算。典型情况下,
这一部分包含化合物名称,对称性,电子态,以及任何其它有关信息。标题部分不
能超过5行,并且必须有结束空行。以下字符不能出现在标题部分:@ # ! - _
\ 和控制符(特别是Ctrl-G)
分子描述:定义要研究的分子体系(有结束空行)。
可选的附加部分:附加输入用于特定任务类型(通常需要结束空行)。
大多数Gaussian 09任务只包括第二、三、四部分。这里有一个这样的例子,它请求水
分子的单点能计算:
# HF/6-31G(d)
water energy
0 1
O -0.464 0.177 0.0
H -0.464 1.137 0.0
H 0.441 -0.143 0.0
执行路径
标题部分
分子说明
在这个任务中,执行路径和标题部分都只有一行。分子说明部分从分子电荷和自旋多重
度的行开始:本例是0电荷(中性分子),自旋多重度1(单重)。电荷和自旋多重度行之后
是描述分子中每个原子位置的行;本例使用笛卡尔直角坐标。本章后面有关于分子说明更详
细的讨论。
下面是使用链接0命令和附加输入部分的输入示例:
%Chk=heavy
#HF/6-31G(d) Opt=ModRedundant
Opt job
0 1
原子坐标
…
3 8
2 1 3
链接0部分
执行路径部分
标题部分
分子说明部分
几何优化过程中为内坐标加入的键长和键角
- 5 -
这一任务进行几何优化。分子说明之后的输入部分被Opt=ModRedundant关键词使用,
用于在几何优化过程中用内坐标提供附加的键长和键角。该任务还定义了检查点文件名。
为了方便,下面的表列出了Gaussian 09输入文件中所有可能出现的部分,以及相应的
关键词。
- 6 -

Gaussian 09 输入部分的顺序
部分 关键词 结尾空行?
链接 0 命令 %命令
无
执行路径部分(#行) 全部 有
另外的层 ExtraOverlays 有
标题部分 除 Geom=AllCheck 外全部 有
分子说明 除 Geom=AllCheck 外全部 有
原子连接说明 Geom=Connect 或ModConnect 有
修改冻结原子 Geom=ReadFreeze 有
修改坐标 Opt=ModRedundant 有
第二个标题和分子说明 Opt=QST2 或QST3 二者都有
第二套坐标的原子连接说明 Geom=Connect 或ModConnect 以及 Opt=QST2
或QST3 有
第二个修改冻结原子 Geom=ReadFreeze 有
第二套坐标的修改 Opt=QST2 或QST3 有
第三个标题和初始过渡态结构 Opt=QST3 二者都有
第三套坐标的原子连接说明 Geom=Connect 或ModConnect 以及
Opt=(ModRedun, QST3) 有
第三个修改冻结原子 Geom=ReadFreeze 有
第三套坐标的修改 Opt=(ModRedun, QST3) 有
PDB 辅助结构信息 如果分子说明部分有残基信息,是自动的 有
原子质量 ReadIsotopes 选项 有
分子力学参数 HardFirst, SoftFirst, SoftOnly, Modify 有
关心的频率 CPHF=RdFreq 有
背景电荷分布 Charge 有
BOMD/ADMP 输入(一个或多个部分) ADMP 和BOMD 所需的输入,以及 ReadVelocity,
ReadMWVelocity 选项 有
PCM 输入 SCRF=(ExternalIteration,Read) 有
用于 IRC 列表的坐标 IRC=Report 有
谐振束缚 Geom=ReadHarmonic 有
半经验参数(Gaussian 格式) Input 选项,AM1=Both 有
半经验参数(MOPAC 格式) MOPAC, Both 选项 有
基组说明 Gen, GenECP, ExtraBasis 有
基组修改 Massage 有
有限场因子 Field=Read 有
ECP 说明 Pseudo=Cards, GenECP 有
密度拟合基组说明 ExtraDensityBasis 有
PCM 溶剂化模型输入 SCRF=Read 有
DFTB 参数 DFTB 有
初始猜测来源 Guess=Input 有
进行组合的对称类型 Guess=LowSymm 无
轨道说明(分为 α和β部分) Guess=Cards 有
改变轨道(分为 α和β部分) Guess=Alter 有
轨道重新排序(分为 α和β部分) Guess=Permute 无
每个 GVB 对的轨道数 GVB 无
CAS 态平均的权重 CASSCF=StateAverage 无
进行自旋-轨道耦合计算的态 CASSCF=SpinOrbit 无
轨道冻结信息 ReadWindow 选项 有
进行精化的 EPT 轨道 EPT=ReadOrbitals 有
用于自旋-自旋耦合常数的原子列表 NMR=ReadAtoms 有
其它的原子半径 Pop=ReadRadii 或ReadAtRadii 有
静电特性的数据 Prop=Read 或Opt 有
NBO 输入 Pop=NBORead 无
选择简正模式 Freq=SelectNormalModes 有
- 7 -

受阻转子输入 Freq=ReadHindered 有
选择非谐正则模式 Freq=SelectAnharmonicNormalModes 有
FCHT 的简正模式 Freq=SelectFCHTNormalModes 有
Pickett 输出文件名 Output=Pickett 无
PROAIMS 输出文件名 Output=WFN 无
- 8 -
输入语法
一般情况下,Gaussian 输入遵循下面的语法规则:
自由格式输入,且大小写无关。
可以用空格,TAB 键,逗号,正斜杠“/”以任意组合分隔一行内的不同项目。多个空
格作为一个分隔符处理。
关键词的选项可以用以下格式指定:
keyword
=
选项
keyword
(
选项
)
keyword
=(
选项 1, 选项 2, ...
)
keyword
(
选项 1, 选项 2, ...
)
多个选项放在括号中,并用任何有效的分隔符分开(习惯上用逗号,如上所示)。左括
号前的等号可以忽略,这个等号的前后也可以随意加空格。注意有些选项带有数值;本
例中,选项名的后面接等号:如 CBSExtrap(NMin=6)。
在整个 Gaussian 09 系统中,所有的关键词和选项都可以简化为最短的可区分的缩写形
式。因此,SCF 关键词的 Conventional 选项可以缩写成 Conven,但不能写成 Conv(因
为还存在 Convergence 选项)。无论 Conventional 和Convergence 是否都是给定关键
词的有效选项,都要遵守这一规则。
在 Gaussian 09 输入文件中,可以包含外部文件内容,使用的语法为:@
文件名
。这导
致整个文件放到输入命令串的当前位置。命令加上/N 能防止被包含文件的内容在输出
文件的开始部分回显。
注释行以感叹号(!)开始,可以出现在一行中的任何位置。不同的注释行可以出现在输
入文件的任何位置。
- 9 -

Gaussian 09 任务类型
Gaussian 09 输入文件的执行路径部分定义了执行的计算类型。这部分内容有三个关键
部分:
任务类型
计算方法
基组
下面的表列出了 Gaussian 09 中可以使用的任务类型:
关键词 任务类型
SP
Opt
Freq
IRC
IRCMax
Scan
Polar
ADMP 和BOMD
Force
Stable
Volume
Density=Checkpoint Guess=Only
Guess=Only
单点能
几何优化
频率与热化学分析
反应路径跟踪
沿着指定的反应路径寻找能量最大值
势能面扫描
极化率和超极化率
直接动力学轨迹计算
计算核的受力
测试波函稳定性
计算分子体积
只重新计算布居分析
只打印初始猜测;产生基于片断的初始猜测
一般而言,只能指定一种任务类型关键词。例外的情况是:
Polar 和Opt 可以结合 Freq。后一情况下,几何优化之后自动在优化的结构上进行频率
计算。
为了给计算的优化部分指定选项,Opt 可以结合复合方法的关键词,例如,
Opt=(TS,ReadFC) CBS-QB3。
如果在执行路径部分没有指定任务类型关键词,默认的计算类型通常是单点能计算
(SP)。而形式为
方法 2/基组 2 // 方法 1/基组 1
的执行路径,可用于请求在(
方法 1/基
组1
级别的)几何优化计算之后,在优化的结构上(用
方法 2/基组 2
)进行单点能计算。例
如,下面的执行路径用 B3LYP/6-31G(d)进行几何优化,之后用 CCSD/6-31G(d)模型化学进行
单点能计算:
# CCSD/6-31G(d)//B3LYP/6-31G(d) Test
这种情况下,Opt 关键词是可选并且默认的。注意 Opt Freq 计算不能使用此语法。
预测分子特性
下面的表格给出了常用预测量与产生这些量的 Gaussian 09 关键词之间的对应关系:
特性 关键词
反铁磁耦合 Guess=Fragment, Stability
- 10 -

原子电荷
溶剂化 ΔG
偶极矩
电子亲和力
电子密度
电子圆二色性
静电势
静电势导出电荷
电子跃迁带型
极化率/超极化率
高精度能量
超精细耦合常数(各向异性)
超精细光谱张量(包括
g
张量)
电离势
红外和拉曼光谱
预共振拉曼光谱
分子轨道
多极矩
NMR 屏蔽和化学位移
NMR 自旋-自旋耦合常数
旋光性
拉曼光学活性
热化学分析
紫外/可见光谱
振-转耦合
振动圆二色性
Pop
SCRF=SMD
Pop
CBS-QB3, CCSD, EPT
cubegen
CIS, TD, EOM, SAC-CI
cubegen, Prop
Pop=Chelp, CHelpG或MK
Freq=FC, Freq=HT
Freq, Polar [CPHF=RdFreq], Polar=DCSHG
CBS-QB3, G2, G3, G4, W1U, W1BD
Prop
Freq=(VCD,VibRot[,Anharmonic])
CBS-QB3, CCSD, EPT
Freq[=Anharmonic]
Freq CPHF=RdFreq
Pop=Regular
Pop
NMR
NMR=Mixed
Polar=OptRot
Freq=ROA, CPHF=RdFreq
Freq
CIS, ZIndo, TD, EOM, SAC-CI
Freq=VibRot
Freq=VCD
- 11 -

模型化学
方法和基组的组合定义一个Gaussian的模型化学,也就是定义理论级别。每一个
Gaussian任务必须指定方法和基组。这一般通过在输入文件的执行路径部分使用两个独立的
关键词完成,虽然有些方法的关键词已经暗含了基组的选择。有些使用密度泛函方法的任务
还可以包含密度拟合基(更多信息参见基组部分)。
下表列出了Gaussian使用的方法,以及每种方法可用的任务类型。*号表示解析计算,
只能用数值方法的计算用
num
表示(详见特定关键词的讨论)。
Gaussian 09中使用的方法
SP,
Scan
Opt,Force,
BOMD
Freq
IRC
ADMP
Polar
Stable
ONIOM
SCRF
PBC
分子力学方法 * * * * *
AM1,PM3,PM3MM,MP6,PDDG * *
*
* * * * *
HF * * * * * * * * * *
DFT方法 * * * * * * * * * *
CASSCF * * * * * * *
MP2 * * * * * * *
MP3,MP4(SDQ) * *
num
*
num
* *
MP4(SDTQ),MP5 *
num
*
QCISD,CCD,CCSD * *
num
*
num
* *
QCISD(T)或(TQ) *
num
*
BD * * *
EPT * *
CBS,Gn,W1方法 *
CIS * * * * * * *
TD * *
num
*
num
* *
EOM *
num
* *
ZINDO * * *
DFTB * * * * *
CI * * * * * *
GVB * * * *
* = 解析算法可用
num
= 数值差分可用
如果没有指定方法关键词,假定是 HF。大多数方法关键词前可加 R,用于闭壳层限制性
波函,加 U用于非限制性开壳层波函,或者加 RO 用于限制性开壳层波函:例如,ROHF,UMP2,
或RQCISD。RO 仅用于 Hartree-Fock 和密度泛函方法,以及 AM1,PM3,PM3MM,PM6,和 PDDG
的能量和梯度,以及 MP2,MP3,MP4,和 CCSD 能量。
一般情况下,只能指定一种方法的关键词,一种以上的关键词将产生奇怪的结果。然而
也有一些例外:
- CASSCF 可以和 MP2 一起指定,进行包含动态电子关联能的 CASSCF 计算。
- ONIOM 和IRCMax 的任务需要多种方法的说明。但是,这些方法作为相应关键词的选项
给出。
- 12 -
- 前面提到的
模型 2//模型 1
的形式,可用于在几何优化之后,自动在优化的结构上进行
单点能计算。
- 13 -
基组
大多数方法需要定义基组;如果在执行路径部分没有包含基组,则使用 STO-3G 基组。
例外的情况是在一些方法中,基组为该方法不可分割的一部分;这些方法在下面列出:
所有的半经验方法,含用于激发态的 ZINDO。
所有的分子力学方法。
复合的模型化学:所有的 G
n
,CBS 以及 W1 方法。
以下是 Gaussian 09 程序内部存储的基组(完整的说明见引用的参考文献),下面按照
它们对应的 Gaussian 09 关键词列出(有两个例外):
STO-3G [Hehre69, Collins76]
3-21G [Binkley80a, Gordon82, Pietro82, Dobbs86, Dobbs87, Dobbs87a]
6-21G [Binkley80a, Gordon82]
4-31G [Ditchfield71, Hehre72, Hariharan74, Gordon80]
6-31G [Ditchfield71, Hehre72, Hariharan73, Hariharan74, Gordon80, Francl82, Binning90, Blaudeau97,
Rassolov98, Rassolov01]
6-31G†:Gaussian 09 还包含 George Petersson 及合作者的 6-31G†和 6-31G‡基组,定
义为完备基组方法[Petersson88, Petersson91]的一部分。它们可以用关键词 6-31G(d')和
6-31G(d',p')调用,并可以添加单个或两个弥散函数;还可以添加 f 函数:例如,
6-31G(d'f),等。
6-311G:对第一行原子定义 6-311G 基组,对第二行原子定义 MacLean-Chandler 的
(12s,9p)(621111,52111)基组[McLean80, Raghavachari80b] (注意 P,S,和 Cl 的基组是被
MacLean 和 Chandler 称作“负离子”的基组;它们被认为对中性分子能给出更好的结
果),Ca 和 K 是 Blaudeau 等人的基组[Blaudeau97],对第一行过渡元素定义 Wachters-Hay
[Wachters70, Hay77]全电子基组,并使用 Raghavachari 和 Trucks [Raghavachari89]的换算因子,
对第三行其它元素使用 McGrath,Curtiss 等人的 6-311G 基组[Binning90, McGrath91,
Curtiss95]。注意在对第一行过渡元素使用 Wachters-Hay 基组时,Raghavachari 和 Trucks
推荐使用换算因子并包含弥散函数;若包含弥散函数需使用 6-311+G 的形式。MC-311G
是6-311G 的同义词。
D95V:Dunning/Huzinaga 价电子双 zeta 基组[Dunning76]。
D95:Dunning/Huzinaga 完全双 zeta 基组[Dunning76]。
SHC:第一行原子用 D95V,第二行原子用 Goddard/Smedley ECP [ Dunning76, Rappe81]。也就
是SEC。
CEP-4G:Stephens/Basch/Krauss 的 ECP 最小基组[Stevens84, Stevens92, Cundari93]。
CEP-31G:Stephens/Basch/Krauss 的 ECP 分裂价基组[Stevens84, Stevens92, Cundari93]。
CEP-121G:Stephens/Basch/Krauss 的 ECP 三分裂基组[Stevens84, Stevens92, Cundari93]。
注意超过第二行以后,只定义了一种 CEP 基组,所有三个关键词对这些原子都是等价的。
LanL2MB:对第一行原子是 STO-3G [Hehre69, Collins76],对 Na-La,Hf-Bi 是 Los Alamos ECP
加上 MBS [Hay85, Wadt85, Hay85a]。
LanL2DZ:对第一行原子是 D95V [Dunning76],对 Na-La,Hf-Bi 是 Los Alamos ECP 加上
DZ [Hay85, Wadt85, Hay85a]。
SDD:D95V 一直到 Ar 原子[Dunning76],对周期表的其它原子使用 Stuttgart/Dresden ECP
[Fuentealba82, Szentpaly82, Fuentealba83, Stoll84, Fuentealba85, Wedig86, Dolg87, Igel-Mann88, Dolg89,
Schwerdtfeger89, Dolg89a, Andrae90, Dolg91, Kaupp91, Kuechle91, Dolg92, Bergner93, Dolg93, Haeussermann93,
- 14 -

Dolg93a, Kuechle94, Nicklass95, Leininger96, Cao01, Cao02]。在 Gen 的基组输入中,可以用 SDD, SHF,
SDF, MHF, MDF, MWB 等种类指定这些基组/芯势。注意,种类之后必须指定芯电子数(例
如,MDF28 表示替代 28 个芯电子的 MDF 势)。
SDDAll:对 Z>2 的原子选用 Stuttgart 势。
cc-pVDZ, cc-pVTZ, cc-pVQZ, cc-pV5Z, cc-pV6Z:Dunning 的关联一致基组[Dunning89,
Kendall92, Woon93, Peterson94, Wilson96](分别为双-zeta,三-zeta,四-zeta,五-zeta,和
六-zeta)。为了提高计算效率,这些基组删除了冗余函数并进行了旋转[Davidson96]。
这些基组在定义中已包含了极化函数。下面的表列出了各种原子的基组中包含的价极化
函数:
原子 cc-pVDZ cc-pVTZ cc-pVQZ cc-pV5Z cc-pV6Z
H 2s,1p 3s,2p,1d 4s,3p,2d,1f 5s,4p,3d,2f,1g 6s,5p,4d,3f,2g,1h
He 2s,1p 3s,2p,1d 4s,3p,2d,1f 5s,4p,3d,2f,1g
不可用
Li-Be 3s,2p,1d 4s,3p,2d,1f 5s,4p,3d,2f,1g 6s,5p,4d,3f,2g,1h
不可用
B-Ne 3s,2p,1d 4s,3p,2d,1f 5s,4p,3d,2f,1g 6s,5p,4d,3f,2g,1h 7s,6p,5d,4f,3g,2h,1i
Na-Ar 4s,3p,1d 5s,4p,2d,1f 6s,5p,3d,2f,1g 7s,6p,4d,3f,2g,1h
不可用
Ca 5s,4p,2d 6s,5p,3d,1f 7s,6p,4d,2f,1g 8s,7p,5d,3f,2g,1h
不可用
Sc-Zn 6s,5p,3d,1f 7s,6p,4d,2f,1g 8s,7p,5d,3f,2g,1h 9s,8p,6d,4f,3g,2h,1i
不可用
Ga-Kr 5s,4p,1d 6s,5p,3d,1f 7s,6p,4d,2f,1g 8s,7p,5d,3f,2g,1h
不可用
这些基组可以通过给基组关键词添加 AUG-前缀(而不是使用+和++符号——见下),用
弥散函数增大基组。
Ahlrichs 等人的 SV, SVP,TZV,TZVP [Schaefer92, Schaefer94],和 QZVP [Weigend05]基组。
Truhlar 等人的 MIDI!基组[Easton96]。使用这个基组需要 MidiX 关键词。
Epr-II 和EPR-III:Barone [Barone96a]的基组,对 DFT 方法(特别是 B3LYP)的超精细耦
合常数计算进行了优化。EPR-II 是具有一套极化函数的双-zeta 基组,并对 s-部分进
行了加强:对 H 是(6,1)/[4,1],对 B 到 F 是(10,5,1)/[6,2,1]。EPR-III 是三-zeta
基组,包括弥散函数,双 d-极化和一套 f-极化函数。同样,s-部分也进行了改善,以
更好地描述芯区域:对 H 是(6,2)/[4,2],对 B 到 F 是(11,7,2,1)/[7,4,2,1]。
UGBS:de Castro,Jorge 等人[Silver78, Silver78a, Mohallem86, Mohallem87, daCosta87, daSilva89,
Jorge97, Jorge97a, deCastro98]的通用 Gaussian 基组。给关键词加上后缀,可以添加附加的
极化函数:
UGBS
n
P|V|O
其中的
n
是整数,表示是否对常规的 UGBS 基组中的每个函数加上 1 个,2 个或者 3 个
极化函数。第二项是一个字母代码,表示给哪个函数加上极化函数:P表示给所有函数
加上极化函数,V给所有价函数加上极化函数,O使用 Gaussian 03 所用的方案(见下)。
例如,UGBS1P 关键词请求对该基组的所有轨道添加一个附加的极化函数,而 UGBS2V 对
所有价轨道添加两个附加的极化函数。
与 Gaussian 03 的 UGBS
n
P关键词一样,O后缀添加同样的函数。UGBS1O 对每个 s-函数
添加一个 p-函数,对每个 p-函数添加一个 d-函数,等;UGBS2O 对每个 s-函数添加一
个 p-和 d-函数,对每个 p-函数添加一个 d-和 f-函数,等;UGBS3O 对每个 s-函数添加
一个 p-,d-和 f-函数,等。
可以照常用+或++添加弥散函数;第一个可以指定为 2+,表示对重原子加上两个弥散函
数。
Martin 和 de Oliveira 的 MTSmall,定义为 W1 方法的一部分(参见 W1U 关键词)
[Martin99]。
- 15 -

DGauss 中使用的 DGDZVP,DGDZVP2,和 DGTZVP 基组[Godbout92, Sosa92]。
CBSB7:选择 6-311G(2d,d,p)基组,用于 CBS-QB3 高精度能量方法[Montgomery99]。这一名
称对第二行原子指定两个附加的 d 极化函数,对第一行原子指定一个 d 函数,对氢指定
一个 p 函数(注意,Gaussian 09 不支持这种三段的极化函数语法)。
添加极化和弥散函数
添加第一个极化函数也可以使用通常的*或**符号。注意(d,p)与**是相同的——例如
6-31G**和6-31G(d,p)等价——而 3-21G*基组只对第二行原子添加极化函数。有些基组可
以使用弥散函数+和++[Clark83],和使用多个极化函数一样[Frisch84]。最好用例子来解释一下
关键词的语法:6-31+G(3df,2p)表示 6-31G 基组用弥散函数扩充。对重原子添加 3 套 d 函
数和一套 f 函数,对氢原子添加两套 p 函数。
当使用 AUG-前缀为 cc-pV
x
Z基组添加弥散函数时,对于给定的原子,为所用的每种函
数类型都添加一个弥散函数[Kendall92, Woon93]。例如,AUG-cc-pVTZ 基组为氢原子添加一个 s,
一个 p 和一个 d 弥散函数,为 B-Ne 和 Al-Ar 的原子添加一个 s,一个 p,一个 d 和一个 f
弥散函数。
为6-311G 添加一套极化函数(即 6-311G(d))将为第一和第二行原子添加一个 d 函数,
对第一行过渡元素添加一个 f 函数,因为 d 函数已经出现在后者的价电子中了。类似地,给
6-311G 基组添加一个弥散函数,将会对第三行原子产生一个 s,一个 p 和一个 d 弥散函数。
当使用 D95 基组进行冻芯计算的时候,占据的芯轨道和相应的虚轨道都是冻结的。因此
水的 D95**计算有 26 个基函数,而同样的体系用 6-31G**计算,只有 25 个函数,不论用两
个基组中的哪一个,都有 24 个轨道用于冻芯的后-SCF 计算。
下面的表列出了 Gaussian 09 中每种内置基组的极化和弥散函数,以及应用范围:
基组 应用范围 极化函数 弥散函数
3-21G H-Xe +
6-21G H-Cl *
或
**
4-31G H-Ne *
或
**
6-31G H-Kr (3df,3pd) +,++
6-311G H-Kr (3df,3pd) +,++
D95 H-Cl(
除
Mg
和
Na) (3df,3pd) +,++
D95V H-Ne (d)
或
(d,p) +,++
SHC H-Cl *
CEP-4G H-Rn *(
仅用于 Li-Ar
)
CEP-31G H-Rn *(
仅用于 Li-Ar
)
CEP-121G H-Rn *(
仅用于 Li-Ar
)
LANL2MB H-La, Hf-Bi
LANL2DZ H, Li-La, Hf-Bi
SDD, SDDAll
除
Fr
和
Ra
之外
cc-pVDZ H-Ar, Ca-Kr
包含在定义中 加
AUG-
前缀
(H-Ar, Sc-Kr)
cc-pVTZ H-Ar, Ca-Kr
包含在定义中 加
AUG-
前缀
(H-Ar, Sc-Kr)
cc-pVQZ H-Ar, Ca-Kr
包含在定义中 加
AUG-
前缀
(H-Ar, Sc-Kr)
cc-pV5Z H-Ar, Ca-Kr
包含在定义中 加
AUG-
前缀
(H-Na, Al-Ar, Sc-Kr)
cc-pV6Z H, B-Ne
包含在定义中 加
AUG-
前缀
(H, B-O)
SV H-Kr
SVP H-Kr
包含在定义中
TZV 和TZVP H-Kr
包含在定义中
- 16 -

QZVP H-Rn
包含在定义中
MidiX H,C-F,S-Cl,I,Br
包含在定义中
EPR-II, EPR-III H,B,C,N,O,F
包含在定义中
UGBS H-Lr UGBS(1,2,3)P
+,++,2+,2++
MTSmall H-Ar
DGDZVP H-Xe
DGDZVP2 H-F,Al-Ar,Sc-Zn
DGTZVP H,C-F,Al-Ar
CBSB7 H-Kr
包含在定义中
+,++
STO-3G 和3-21G 也接受*后缀,但实际上这不会加上任何极化函数。
有关基组的其它关键词
下面这些有用的关键词与基组关键词一起使用:
5D 和6D:分别使用 5 或 6 个 d 函数(纯 d 函数与笛卡尔 d 函数)。
7F 和10F:分别使用 7 或 10 个 f 函数(纯 f 函数与笛卡尔 f 函数)。它们也用于更高的
函数(g 或更高)。
使用ExtraBasis和Gen关键词,可以为程序输入其它的基组。ChkBasis关键词表示基组
从检查点文件读取(用%Chk命令定义)。详见本章后面关于这些关键词的单独说明。
纯基函数与笛卡尔基函数产生的问题
关于纯基函数与笛卡尔基函数,Gaussian 用户应当知道以下几点:
所有的内置基组使用纯 f 函数。除了 3-21G,6-21G,4-31G,6-31G,6-31G†,6-31G‡,
CEP-31G,D95 以及 D95V 以外,大多数也使用纯 d 函数。上述的几个关键词可以覆盖默
认的纯/笛卡尔设置。注意,如果需要的话,基函数会自动转换到其它类型,例如,当
从检查点文件读取波函,用于使用其它类型函基数进行计算的时候[Schlegel95a]。
在一个 Gaussian 计算中,所有的 d 函数必须都是 5D 或 6D,所有的 f 以及更高的函数
必须都是纯函数或笛卡尔函数。
当使用 ExtraBasis,Gen 和GenECP 关键词时,在执行路径部分直接定义的基组总是决
定基函数的默认类型(对 Gen 是5D 和7F)。例如,如果使用的一般基组中某些函数来
自 3-21G 和 6-31G,将使用纯函数,除非在执行路径部分定义 Gen 之外又特别定义了 6D。
类似地,如果对在执行路径部分定义 6-31G(d)基组的任务,用 ExtraBasis 为过渡金属
添加来自 6-311G(d)基组的基函数,将使用笛卡尔 d 函数。同样,如果对 6-311 基组的
任务,用 ExtraBasis 关键词为 Xe 添加来自 3-21G 基组的基函数,Xe 的基函数将是纯
函数。
- 17 -
密度拟合基组
Gaussian 09对纯DFT计算提供密度拟合近似[Dunlap83, Dunlap00]。这个方法在计算库仑作
用时将密度对一组原子中心的函数进行展开,而不是计算所有的双电子积分。对于因较小而
不能利用线性标度算法的中等大小的分子体系,该方法对于纯DFT计算能够显著提高计算效
率,同时又不会显著牺牲预测的结构、相对能量和分子性质的精度。Gaussian 09能从AO基
自动产生合适的拟合基,用户也可以选择一套内置的拟合基组。
需要的拟合基组可以定义为模型化学的第三部分,例如:
# BLYP/TZVP/TZVPFit
注意,当指定密度拟合基组时,斜杠必须做为方法与基组、基组与拟合基组之间的分隔
符。
在Gaussian 09中可以使用以下的拟合基组关键词:
DGA1和DGA2 [Godbout92, Sosa92]。DGA1可用于H到Xe,DGA2可用于H,He,以及B到Ne。
SVPFit [Eichkorn95, Eichkorn97]和Def2SV [Weigend05],对应于SVP基组。
TZVPFit [Eichkorn95, Eichkorn97]和DefTZV [Weigend05],对应于TZVP基组。
QZVP [Weigend03, Weigend05],对应于QZVP基组。
Ahlrichs及其合作者的W06拟合集[Weigend05, Weigend06]。
Fit:选择对应于指定基组的拟合集。如果没有这样的拟合集,会导致错误。
NoFit:关闭用于该计算的拟合集。该关键词用于将Default.Route文件覆盖DensityFit
关键词。
Auto:自动产生拟合集(见下)。
密度拟合集可以从基组的AO原函数自动产生。这需要使用Auto拟合关键词。程序把拟合
集自动截断到合理的角动量:默认为Max(MaxTyp+1,2*MaxVal),这里的MaxTyp是AO基中最高
角动量,MaxVal是最高的价角动量。用Auto=All,可以请求使用所有产生的函数,或者用
Auto=
N
请求使用直到某一级别的函数,这里的
N
是在拟合函数中保留的最大角动量。最后,
PAuto类型在一个原子中心产生AO函数的所有乘积,而不是AO原函数的平方,但这通常会产
生比需要数量多的函数。
默认不使用拟合集。密度拟合基组可以用 ExtraDensityBasis 关键词扩充,用 Gen 关
键词做完整的定义,也可以选择从检查点文件中读取(使用关键词 ChkBasis)。计算中所用
拟合集的某些属性,可以用 DensityFit 关键词的选项来控制。
- 18 -

分子说明概述
这一输入部分定义核位置和α与β自旋的电子数。有几种定义核构型的方法:可以是Z-
矩阵,可以是笛卡尔直角坐标,或者是二者混合(注意笛卡尔直角坐标是Z-矩阵的一种特殊
形式)。
分子说明部分的第一行指定净电子电荷(一个有正负号的整数)和自旋多重度(一个正
整数)。因此,对于中性分子的单重态,应当输入0 1。对于阴离子自由基,可能是-1 2。对
于有些计算类型,可能或者必须包含多个电荷/自旋输入对。
如果使用了Geom=CheckPoint,那么电荷和自旋行是唯一需要的分子说明输入。如果在
执行路径中包含Geom=AllCheck,那么全部分子说明(以及标题部分)可以忽略。
分子说明余下的部分给出分子中每个原子的元素类型以及核位置。输入行的一般格式如
下:
元素符号[-原子类型[-电荷]] [(参数=数值[, ...])] 原子的位置参数
每一行包含元素类型,可能还有可选的分子力学原子类型和部分电荷。该原子的核参数
在括号内的列表中指定。这一行的余下部分是原子位置信息,可以是用笛卡尔坐标,也可以
是Z-矩阵。我们先考虑第一项和最后一项,然后再讲其它的项。
以下是在分子说明部分指定原子的基本格式(忽略所有其它选项):
元素符号 [固定代码] x, y, z
在这些例子中,虽然用空格分隔一行内的项目,但实际上可以用任何有效的分隔符。原
子位置用笛卡尔直角坐标指定。
固定代码
是可选参数,与ONIOM优化中的冻结原子有关(细
节参见ONIOM)。
元素符号
是包含原子的化学符号或原子序数的字符串。如果使用元素符号,后面可以选
择接其它数字或文字字符,作为区分原子的标识符。一般情况是元素名后加上个用于识别的
整数,如:C1,C2,C3,等;这种技术对于采用传统化学编号方式非常有用。元素符号的最
大长度是4个字符。
在第一种输入方式中,每行其余的项是定义核位置的笛卡尔直角坐标。在第二种输入方
式中1,
原子1
,
原子2
,
原子3
是前面定义的原子符号,它们用于定义当前原子的位置(另一
种方法是,分子说明部分的变量值可以使用其它原子的行号,其中电荷和自旋多重度的行算
作第0行)。
当前原子的位置定义为到
原子1
的给定键长,该键与
原子1
和
原子2
连线形成的键角,以
及由
原子2
和
原子3
连线与当前原子、
原子1
和
原子2
所在平面形成的二面(扭转)角。
这是乙烷的简单分子说明部分,其中对碳原子使用元素标号,对氢原子使用元素类型:
0,1
C1 0.00 0.00 0.00
C2 0.00 0.00 1.52
H 1.02 0.00 -0.39
H -0.51 -0.88 -0.39
H -0.51 0.88 -0.39
H -1.02 0.00 1.92
H 0.51 -0.88 1.92
H 0.51 0.88 1.92
1 在Gaussian 09 的使用手册中,这里忘记给出 Z-矩阵格式,参见附录关于 Z-矩阵的说明——译注。
- 19 -

也可以用Z-矩阵格式的分子说明。细节参见附录。
指定同位素以及其它核参数
同位素和其它核参数可以在原子类型区域内指定,需要使用加上括号的关键词和数值,
例如下面的例子:
C(Iso=13,Spin=3) 0.0 0.0 0.0
这一行指定一个13C原子,核自旋为3/2 (3 * 1/2),位于原点。在参数列表里可以包含
的以下项目:
- Iso=
n
:选择同位素。如果用整数指定原子质量,程序会动使用相应的真实同位素质量(例
如,用18指定18O,Gaussian会使用数值17.99916)。
- Spin=
n
:核自旋,单位是1/2。
- ZEff=
n
:有效电荷。这个参数用在自旋轨道耦合(参见CASSCF=SpinOrbit),ESR
g
张
量以及电子自旋-分子转动超细张量(NMR Output=Pickett)的计算中。
- QMom=
n
:核四极矩。
- GFac=
n
:以核磁子为单位的核磁矩。
指定分子片段
分子体系内的片断可以用Fragment参数定义,它位于原子标记之后的括号中,括号中还
可以加入同位素和/或核参数的值。Fragment的值为一个整数,所有具有相同片段编号的原
子定义为一个片段。片段可用于片段猜测计算,平衡计算,等等。
例如,下面的联苯结构用苯环分为两个片段:
0,1 0,1 0,1
总电荷与自旋多重度,之后是各片段的总电荷与自旋多重度
C(Fragment=1) -3.05015529 -0.24077322 0.00000698
C(Fragment=1) -1.64875545 -0.24070572 0.00067327
C(Fragment=1) -0.94811361 0.97297577 0.00020266
C(Fragment=1) -1.64887160 2.18658975 -0.00093259
C(Fragment=1) -3.05027145 2.18652225 -0.00159819
C(Fragment=1) -3.75091329 0.97284076 -0.00112735
H(Fragment=1) -3.58511088 -1.16744597 0.00036555
H(Fragment=1) -1.11371117 -1.16732692 0.00154256
H(Fragment=1) -1.11391601 3.11326250 -0.00129286
H(Fragment=1) -3.58531573 3.11314346 -0.00246648
H(Fragment=1) -4.82091317 0.97278922 -0.00163655
C(Fragment=2) 0.59188622 0.97304995 0.00093742
C(Fragment=2) 1.29252806 2.18673144 0.00046795
C(Fragment=2) 1.29264421 -0.24056403 0.00207466
C(Fragment=2) 2.69392790 2.18679894 0.00113535
- 20 -

C(Fragment=2) 2.69404405 -0.24049653 0.00274263
C(Fragment=2) 3.39468590 0.97318496 0.00227326
H(Fragment=2) 0.75768862 -1.16723678 0.00243403
H(Fragment=2) 0.75748378 3.11335264 -0.00040118
H(Fragment=2) 3.22888349 3.11347169 0.00077519
H(Fragment=2) 3.22908834 -1.16711773 0.00360969
H(Fragment=2) 4.46468577 0.97323650 0.00278063
这个例子还解释了如何使用片段的电荷和自旋多重度的说明。本例中相应的输入行格式
为:
总电荷,总自旋,片段1的电荷,片段1的自旋,片段2的电荷,片段2的自旋
负的自旋多重度对于Guess=Fragment计算有特殊的含义,表示未成对的片段轨道在指
定的组合片段中为β自旋轨道。在其它任务类型中,负的自旋多重度将导致错误。
对于Guess=Fragment和Counterpoise计算,片段个数必须从1开始并连续递增。对于其
它计算类型,没有这一限制,但违反这一规定会导致输出文件中出现大量的空白数据部分(例
如,全部为零的片段布居分析)。
GaussView提供了用于定义片段的图形工具。
分子力学原子类型
分子力学计算中的分子说明也可以包含原子类型和部分电荷的信息。这里有一些例子:
C-CT
指定一个SP3脂肪族碳原子
C-CT-0.32
指定一个SP3脂肪族碳原子,部分电荷为0.32
O-O--0.5
指定一个羰基氧原子,部分电荷为-0.5
对每一个原子可以指定原子类型以及可选的的部分电荷。也可以指定核参数,如下面的
例子:
C-CT(Iso=13)
C-CT--0.1(Spin=3)
PDB文件参数
有一些附加项可以与核参数和/或片段一起定义。这些项与PDB文件一起使用,用于保留
其中包含的残基以及其它结构信息,这样用户无需再作定义。但是,你可能会在GaussView
用源自PDB文件的结构创建的Gaussian 09输入文件中看到它们。
RESNum指定原子所位于的残基。该值的格式为
n
[X[Y]],其中
n
是一个整数(未必为正),
X是一个可选的单个字符插入代码,是可选的链字母。如果定义了链但没有插入代码,
那么X可以是下划线:ResNum=-17_C用于链C上编号为-17的残基。
RESName指定三个字符的残基名。
PDBName如果不是元素名的话,指定赋值给原子的名称。
- 21 -

指定空原子
具有力学类型 Bq (例如O-Bq)的原子可以设定为相应原子的空原子1[Macbeth],它具有正
常的基函数和数值积分格点,但是没有核电荷或电子。这用于平衡计算。这样的计算和
Gaussian以前版本中Massage关键词的计算略微不同,在DFT的XC积分中,它包含了来自空原
子的格点。新方法与重迭校正更加一致,而且更容易使用。注意,平衡计算也可以用
Counterpoise关键词请求。
指定周期体系
周期体系用通常的分子说明方式指定单位晶胞。唯一需要的额外输入是在分子说明之
后,加上一维、二维或三维平移矢量(中间不需要插入空行),表示体系重复的方向。例如,
下面的输入指定对氯丁橡胶做一维PBC单点能计算:
# PBEPBE/6-31g(d,p)/Auto SCF=Tight
neoprene, -CH2-CH=C(Cl)-CH2- optimized geometry
0 1
C,-1.9267226529, 0.4060180273, 0.0316702826
H,-2.3523143977, 0.9206168644, 0.9131400756
H,-1.8372739404, 1.1548899113,-0.770750797
C,-0.5737182157,-0.1434584477, 0.3762843235
H,-0.5015912465,-0.7653394047, 1.2791284293
C, 0.5790889876, 0.0220081655,-0.3005160849
C, 1.9237098673,-0.5258773194, 0.0966261209
H, 1.772234452, -1.2511397907, 0.915962512
H, 2.3627869487,-1.0792380182,-0.752511583
Cl,0.6209825739, 0.9860944599,-1.7876398696
TV,4.8477468928, 0.1714181332, 0.5112729831
最后一行定义平移矢量。注意TV设定为原子符号。
下面的分子说明用于石墨层的二维PBC计算:
0 1
C 0.000000 0.000000 0.000000
C 0.000000 1.429118 0.000000
TV 2.475315 0.000000 0.000000
TV -1.219952 2.133447 0.000000
这个分子说明用于砷化镓的三维PBC计算:
0 1
Ga 0.000000 0.000000 0.000000
Ga 0.000000 2.825000 2.825000
Ga 2.825000 0.000000 2.825000
Ga 2.825000 2.825000 0.000000
1 空原子(Ghost Atoms),也有文献译为“鬼原子”。它的核电荷数为零,即内部是空的。Ghost 在医学上可以指空细胞,所以
Ghost Atoms 在这里翻译为“空原子”。空原子会影响分子的对称性,这一点和虚原子(Dummy Atoms)不同。空原子作为基函数
的中心,常用来计算基组重叠误差(BSSE,参见 Counterpoise 关键词),以及使用分子核心之外的函数描述分子的孤对电子。
- 22 -
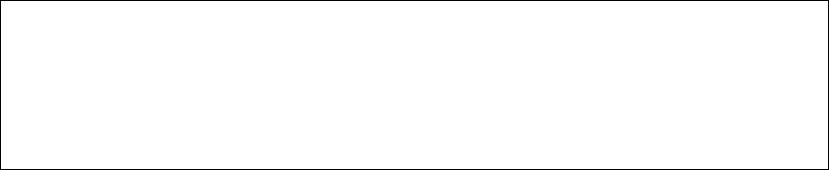
As 1.412500 1.412500 1.412500
As 1.412500 4.237500 4.237500
As 4.237500 1.412500 4.237500
As 4.237500 4.237500 1.412500
TV 5.650000 0.000000 0.000000
TV 0.000000 5.650000 0.000000
TV 0.000000 0.000000 5.650000
- 23 -
多步任务
多个Gaussian任务可以合并为一个输入文件。每次任务的输入可以用下面形式的一行与
前面的任务步骤分开:
--Link1--
这是一个包含两步任务的示例输入文件:
%Chk=freq
# HF/6-31G(d) Freq
Frequencies at STP
(分子说明)
--Link1--
%Chk=freq
%NoSave
# HF/6-31G(d) Geom=Check Guess=Read Freq=(ReadFC,ReadIsotopes)
Frequencies at 300 K
(电荷和自旋)
300.0 2.0
(定义同位素)
这个输入文件在两种不同温度和压强下计算振动频率和进行热化学分析,首先是298.15
K和一个大气压,接下来在300 K和两个大气压。注意--Link1—行之前必须有一个空行。
- 24 -
运行Gaussian
这一部分讲述在基于UNIX的计算机系统运行Gaussian所需的操作系统命令。其它操作系
统的等效信息,见随程序一起的附加说明。这里假定程序已经安装完毕。
运行Gaussian涉及以下步骤:
创建描述所需计算的Gaussian输入文件。
指定各种擦写文件的位置。
指定资源需求
以交互或批处理模式开始执行程序。
在这一部分,我们假定已经创建了基本的Gaussian输入文件,我们的讨论将考察列表中
的其余三项。
指定擦写文件的操作和位置
Gaussian在计算过程中使用数个擦写文件。包括:
检查点文件:
name
.chk
读写文件:
name
.rwf
双电子积分文件:
name
.int(默认为空)
双电子积分的导数文件:
name
.d2e(默认为空)
默认情况下,这些文件由Gaussian进程的进程ID命名,并存储于由(UNIX)环境变量
GAUSS_SCRDIR定义的擦写目录中。在这个目录中还可以看到name.inp形式的文件。它们是
程序使用的内部输入文件。如果没有设置环境变量,则路径默认为Gaussian进程的当前工作
目录。
这些文件在计算成功结束后默认删除。但是,你可能想要保存检查点文件,用于以后其
它的Gaussian计算任务,用于可视化程序,或者重新开始失败的任务,等等。这可以通过在
Gaussian输入文件中使用%Chk命令,对检查点文件直接命名和/或为其提供路径来实现。例
如:
%Chk=water
这一位于输入文件开始的命令(位于执行路径之前),给出检查点文件的名称water.chk,覆
盖通常产生的名称,使这个文件在任务结束后被保存。在这个例子中,这个文件将保留在当
前目录中。但是,下面的命令将指定其它的目录位置和文件名:
%Chk=/chem/scratch2/water
如果擦写文件目录的磁盘空间有限,但是系统有其它的磁盘空间可用,你可能想把擦写
文件分割到几个磁盘位置中。下面的命令允许你定义其它擦写文件的名称和位置:
%RWF=
路径
读写文件
%Int=
路径
积分文件
%D2E=
路径
积分导数文件
一般情况下,读写文件是最大的,所以经常为它指定其它位置。
- 25 -

跨磁盘分割擦写文件
在32位的操作系统如Windows和IA32 Linux,Gaussian 09可对直到16 GB的单个擦写文
件进行寻址,不需要把擦写文件分割成2GB大小的文件。然而,16 GB的总擦写空间限制是32
位整数固有的,无法用分割擦写文件的办法解决1。
还提供了另外一种语法,可以把读写文件,积分文件,和/或积分导数文件分割到两块
或多块磁盘(或文件系统)。这里是%RWF命令的语法:
%RWF=
位置1,大小1,位置2,大小2,
…
其中的每个
位置
是目录位置或文件路径名,每一个
大小
是在该位置下文件片段的最大尺寸。
Gaussian将对任何指定目录的
位置
自动产生唯一的文件名。在UNIX系统中,目录说明(不含
文件名)必须在末尾包含斜线“/”。
默认的尺寸以字为单位;数值后可以接KB,MB或GB(中间不插入空格)分别表示KB,MB
或GB,或接KW,MW或GW,分别表示千字,兆字或吉字。注意1 MB=10242位=1,048,576位(而
不是1,000,000位)。
尺寸参数值-1表示使用所有可用的空间,0值表示使用存在的文件片段的当前大小。-1
仅用于最后指定的文件,这是默认的。
例如,下面的指令把读写文件分割到三块磁盘中:
%RWF=/dalton/s0/,4GB,/scratch/,3GB,/temp/s0/my_job,-1
文件片段的最大尺寸分别是4 GB,3 GB,和无限制。Gaussian将对前两个片段产生文件名,
第三个的名称给定为my_job。注意目录格式的末尾包含斜线。
由于目前UNIX执行的限制,使用-1需要当心,因为它可能把一个文件片段延伸,超过系
统所有剩余的磁盘空间;使用它的副作用是防止包含在列表中的其它文件片段已被用过。
保存和删除擦写文件
未命名的擦写文件在Gaussian计算结束时默认被删除,命名的则被保存。%NoSave命令
可以用来改变这一默认做法。当在输入文件中包含这一指令时,在输入文件中命名擦写文件
的指令若出现在%NoSave以前,那么命名的擦写文件(以及所有未命名的擦写文件)将在计
算结束时被删除。但如果命名文件的%指令出现在%NoSave指令之后,文件将被保存。例如,
下面的命令指定检查点文件名,以及读写文件另外的名称和目录位置,致使在Gaussian任务
结束后只保存检查点文件:
%RWF=/chem/scratch2/water
到这里为止的文件都被删除
%NoSave
%Chk=water
到这里为止的文件都被保存
注意:当任务异常终止时,所有的文件都被保存。
1 64 位机不受此限。
- 26 -
初始化文件
Gaussian系统包含初始化文件,用于设置运行程序的用户环境。这些文件是:
$g09root/g09/bsd/g09.login C shell
$g09root/g09/bsd/g09.profile Bourne shell
注意g09root 环境变量必须由用户设置。因此习惯上在Gaussian用户的.login
或.profile文件中包含类似于下面的命令:
.login文件:
setenv g09root
位置
source $g09root/g09/bsd/g09.login
.profile文件:
g09root=
位置
export g09root
. $g09root/g09/bsd/g09.profile
正确设置后,使用g09命令执行Gaussian 09(见下)。
控制内存的使用
%Mem命令控制Gaussian使用的动态内存总量。默认使用256 MB(32 MW)。这可以变成n
个双精度字,通过定义:
%Mem=n
例如,下面的命令设置使用320兆位内存:
%Mem=40000000
%Mem的值也可接KB,KW,MB,MW,GB或GW(之间无空格)表示其它单位。例如,下面
的命令设置使用1 GB动态内存:
%Mem=1GB
对于非常大的直接SCF计算,需要分配的内存更大——至少3N3字,其中的N是基函数数
目。
警告:如果使用多于实际物理内存的内存总量,会使运行效率极差。
如果Gaussian用在物理内存很有限的计算机上,使得默认的256 MB内存不可用,那么在
安装时应当对默认算法和默认的内存分配进行合理的设置。关于提高Gaussian效率的更多细
节参见
效率的考虑
。
在UNIX系统运行Gaussian
一旦准备好所有的输入和资源说明,就可以准备运行程序了。运行Gaussian 09可以交
互使用两种命令形式之一:
- 27 -

g09
job-name
或
g09 <
input-file
>
output-file
在第一种形式中,程序从job-name.com读取输入,并把输出写到job-name.log中。当没
有指定job-name时,程序从标准的输入端(键盘)读取,并写到标准输出中,这可以使用通
常的UNIX方式重新定位或用管道传递。与任何shell命令方式相同,使用&可以强制在后台运
行两种形式命令中的任何一种。
脚本与Gaussian
运行Gaussian
09的脚本可以用几种方法创建(在这些例子中我们使用C shell)。首先,
像上面的g09命令可以包含在shell脚本中。第二,用<<结构,把Gaussian实际的输入包含在
脚本中:
#!/bin/csh
g09 <<END >water.log
%Chk=water
#RHF/6-31G(d)
water energy
0 1
O
H 1 1.0
H 1 1.0 2 120.0
END
echo "Job done. "
从<<符号之后到字符串前的所有行都看作是g09的输入命令。
最后,可以建立循环,依次运行几个Gaussian任务。例如,下面的脚本运行所有作为命
令行参数指定的Gaussian输入文件,并把活动的日志保存到Status文件中:
#!/bin/csh
echo "Current Job Status:" > Status
echo "Starting file $file at 'date' " >> Status
g09 < $file > $file:r.log
echo "$file Done with status $status" >> Status
end
echo "All Done." >> Status
使用作为脚本命令行参数文件的部分输入,下面更复杂的脚本可以即时创建Gaussian输
入文件。使用的文件缺少完整的执行路径部分;它们的执行路径简单地由#号或#行构成,其
中后者含有用于分子体系的特殊关键词,但没有方法,基组或计算类型。
脚本对每一部分输入文件创建一个两步的任务——Hartree-Fock优化之后是MP2单点能
计算——都由脚本中的文字命令和脚本执行时指定的每个文件内容构成。后者通过使用
Gaussian 09的@包含文件机制来包含:
#!/bin/csh
echo "Current Job Status:" > Status
foreach file ($argv)
- 28 -
echo "Starting file $file at 'date'" >> Status
g09 << END> $file:r.log
%Chk=$file:r
# HF/6-31G(d) FOpt
@$file/N
--Link1—
%Chk=$file:r
%NoSave
# MP2/6-31G(d,p) SP Guess=Read Geom=AllCheck
END
echo "$file Done with status $status" >> Status
end # end of foreach
echo "All Done." >> Status
用NQS执行批处理
Gaussian可以用UNIX系统支持的NQS批处理工具。初始化文件中定义的subg09命令可以
把输入任务提交到批处理队列中。它的语法是:
subg09
队列名 任务名
[-scrdir
dir1
] [-exedir
dir2
] [-p
n
]
两个必需的参数是队列名和文件名。输入来自
job-name.
com,输出为
job-name
.log,与
交互运行一样。NQS日志文件发送到
job-name.
batch-log。可选的参数-scrdir和-exedir分
别用于覆盖默认的擦写和可执行目录。其它参数来自NQS选项。特别是-p
n
可用于设置队列
中优先级为n。它在启动时优先计算(1为最低),并不影响运行时间的优先权。
从交互方式提交NQS任务,需要创建类似于下面的文件(文件名为
name
.job):
# QSUB –r
name
–o
name
.out –eo
# QSUB –lt 2000 –lT 2100
# QSUB –lm 34mw –lM 34mw
g09 <
name
.com
其中的
name
应当用实际计算对应的名称代替。第一行命名运行的任务,输出文件名,并使输
出文件包含出错信息。不同的时间参数允许加入任务控制用于清理(例如,如果任务超过运
行时间限制,就保存检查点文件)。内存参数用于执行任务的初始调度,以及由程序确定动
态内存的使用。
接下来,使用以下命令提交任务:
$ qsub
name
.job
输出的文件将放在当前的工作目录。
- 29 -

配置Gaussian
这一部分讨论在 UNIX 系统安装和配置 Gaussian 的一般流程。一定要检查随程序一起
的说明书和发布说明,以保证附加的或可选的说明书适合于你的计算机系统。
系统需求
根据计算机系统的不同,Gaussian 目录下需要大约 150-750 MB 磁盘空间用于可执
行文件。
Gaussian 09 默认的内存分配是 256 MB。程序中最大的固定维度需要的交换空间大
小为 1-2 GB。当然,如果在任务中用链接 0 命令%Mem 或在 Default.Route 文件使
用-M-命令请求更多的内存,将需要更多的交换空间。这些要求适用于每个同时执
行的任务。
参看随CD一起发布的平台列表。本文档的最新版本总是可以在这里找到:
http://www.gaussian.com/g09_plat.htm
设置Gaussian执行环境
在由几个环境变量定义的目录中,Gaussian 定位可执行文件和创建擦写文件。不过,
用户还需要建立两个环境变量:
g09root:指示 g09 目录所在的位置(即:它之上的目录)。
GAUSS_SCRDIR:指示用于擦写文件的目录。
Gaussian 初始化文件根据需要负责初始化其它别名和环境变量。所有的 Gaussian 用户
需要在 UNIX shell 特定的初始化文件内执行 Gaussian 的初始化文件。详见上一章。
g09.login 和g09.profile 创建的环境变量包括:
GAUSS_EXEDIR:指定存储 Gaussian 映像的目录。默认包含主目录$g09root/g09 和
几个备用目录。
GAUSS_ARCHDIR:指定保存站内主存档文件,以及主存档不可用时放置临时存档文
件的目录。如果未设置,默认为$g09root/g09/arch。
G09BASIS:目录内的文件包含标准的 Gaussian 内置基组,以及一些一般基组输入
格式的外加基组。提供这个环境变量是为了方便使用,专为@包含机制所使用。
使用Linda的网络/集群并行计算还可以用环境变量GAUSS_LFLAGS把选项传递给Linda进
程。细节参见用Linda运行Gaussian任务的讨论。
擦写文件的考虑事项
在UNIX系统中,当用户没有指定文件名时,Gaussian基于进程的ID产生唯一的擦写文件
名。这一机制允许多个Gaussian任务同时运行,并且使用一个共同的擦写目录。
在默认情况下,当一个任务成功完成或彻底死掉后,擦写文件自动被清除。然而,当一
个任务从外部被杀死,或者意外中断时,擦写文件不被删除。因此,余下的文件保留在擦写
- 30 -
目录中。
避免出现大量杂乱的擦写文件的简单方法是所有用户共享同一个擦写文件目录,通过在
系统引导程序脚本(例如,/etc/rc,或者/etc/rc.d/rc3.d下的某个文件)中加入rm命令,
当系统引导时清空擦写目录。如果使用了NQS批处理系统,在开始NQS之前也应清空擦写目录,
并确保在清空时没有计算任务使用该目录。
站内定制:Default.Route文件
根据特定计算机系统的特点,有时由于执行的原因需要覆盖一些程序内置的默认设置。
这可以通过创建站内定制文件来完成。在 Unix 系统中,这个文件名为 Default.Route,位于
$g09root/g09。在 Windows 下,这个文件名为 Default.Rou,位于 Gaussian 09W 的擦写子目
录中(例如,C:\G09W\scratch)。文件的格式在所有的计算机系统中都是相同的。
以下小节讲述默认文件提供的信息类型。
默认执行路径
这些参数由-#-引入,与正常的执行路径命令有同样的格式。例如,这一命令行将把默
认的 SCF 算法设置为传统的(非直接)算法:
-#- SCF=Conventional
文件中可以有一个以上的-#-行。
Default.Route 中列出的命令只改变默认值;这些命令将用输入文件执行路径中定义的
命令覆盖。因此,如果 Default.Route 包含:
-#- MP2=NoDirect
并且执行路径包含 MP2 关键词,那么将使用传统的 MP2 算法。但是,如果执行路径包含
MP2=Direct 关键词,将使用直接算法。
所有的站点想在 Default.Route 文件中用 MaxDisk 关键词指定擦写磁盘空间总量。例如,
下面的行设置 MaxDisk 为800 MB:
-#- MaxDisk=800MB
可用的磁盘空间总量(默认)给定为 8 位的字:这个值后面也可以接上 KB,MB,GB,
KW,MW,或 GW(之间没有空格)来指定千,兆,或吉位或字。擦写磁盘空间默认设为无限
(-1),即,假设执行给定的计算有足够的磁盘空间,没有多余的工作。因此,对于计算执
行的优化,最重要的方法是指定可用的内存和磁盘总量。这样做使得程序在不同的可用算法
之间作出决定,对特定的系统配置选择最优的一种。记住,可用的磁盘空间越多,求解越快,
特别是 MP2。
- 31 -
Default.Route的限制
并不是所有的执行路径关键词都能用于 Default.Route 文件。一般的规则是,只有不影
响计算输出的选项(即,不改变被预测量的数值)才被文件所允许。因此,允许仅改变积分
存储算法的 SCF=Conven,但忽略影响很多计算类型结果的 Int(Grid=3)。
默认内存
不明智地使用大量内存的 Gaussian 任务会导致系统出现严重问题。指令–M–强制默认
的动态内存极限。例如,下面的行设定使用的默认内存为 400 MB:
-M- 50000000
可用的内存总量(默认)给定为 8 位的字:这个值后面也可以接上 KB,MB,GB,KW,
MW,或 GW(之间没有空格)来指定千,兆,或吉位或字。默认的内存大小是 256 MB。注意,
这个限制可由链接 0 命令%Mem 绕开。
共享内存多处理器的并行执行
如果计算机系统有多个处理器/核心,并且 Gaussian 的版本支持并行处理,那么可以在
Default.Route 文件指定默认使用的处理器个数。例如,下面的命令设置默认处理器个数为
4:
-P- 4
通常,程序默认仅用一个处理器执行。对于特定的任务,链接 0 命令%NProcShared 可
用于覆盖默认设置。很明显,请求的处理器个数不能超过可用的处理器个数,否则会使执行
效率大大降低。
还可以用-C-指令指定运行的特定 CPU。例如,下面的指令指定程序运行于六核心系统
的前五个核心上(保留一个核心用于其它用途):
-C- 0,1,2,3,4
网络/集群的并行执行
在Default.Route 文件里通过-W-指令可以指定 Linda 工作程序的列表:
-W- dalton,lavoisier:2,priestley,agassiz:3,curie=20
这个例子使用指定的五个节点用于并行执行,让两个工作进程位于
lavoisier
,三个位于
agassiz
,其它系统每个分配一个工作进程。如果主节点——任务开始的节点——不在这些
系统中,那么还在该系统运行一个工作进程(于是总共有六个节点)。
这个指令对应于链接 0 命令%LindaWorkers,并且可被其覆盖。
- 32 -
关于使用带 Linda 的 Default.Route,如果在集群上使用不同的批处理队列,它们对应
不同的节点集且每一批处理任务运行在自己的临时默认目录中,那么最佳的方法是创建脚
本,把特定队列的标准 Default.Route 复制到当前的默认目录中。
当-W-与-P- n进行组合时,那么
n
路 SMP 并行工作进程从节点列表的每个节点开始(或
一个以上的进程,当在-W-对节点指定了更多的工作进程时)。
早期版本程序使用的-L-指令已经作废,千万不要与-W-组合。
站点名
站点名可由-S-定义,其值定义为 Gaussian 生成存档项的站点名。默认的站点名是 GINC。
例如,下面的行设置站点名为 EXPCONS:
-S- EXPCONS
主机名
主机名可由-H-设定,其值定义为 Gaussian 生成存档项的主机名。默认为当前的主机名。
典型的默认设置
这是一个适于各种机器配置的合理默认设置:
对于 2 GB 内存 200 GB 磁盘的小型工作站,默认的算法和内存分配已经很好。只需
要定义 MaxDisk:
-#- MaxDisk=50GB
对于用于大型任务,有 8 个处理器,8 GB 内存的强大工作站,默认使用全部 8 个处
理器。同样,每个任务需要更多的内存:
-M- 4GB
-P- 8
-#- MaxDisk=100GB
用户默认文件
Gaussian 用户可以通过创建自己的 Default.Route 文件,设置自己的默认值。当一个任
务开始的时候,Gaussian 检查当前工作目录,寻找这个名称的文件。本地文件的设置优先于
站内文件,在任务的执行路径定义的选项优先于前两者。
- 33 -
用Linda运行集群/LAN并行计算
这一部分讲述通过Gaussian公司购买的Linda软件的安装过程,以及创建分布内存并行
版Gaussian的过程。这里假定你已经创建并测试了常规版本的程序。此外还假定你已经阅读
了标准的安装说明,并且有Gaussian 09用户参考手册。
Linda并行方法
分子的HF,CIS=Direct,和DFT计算是Linda并行的,包括能量,结构优化,和频率。TDDFT
的能量和梯度以及MP2的能量和梯度也是Linda并行的。MP2频率和CCSD计算是部分Linda并行
的,其余部分仅为SMP并行,因此使用少数几个节点可以加速,但使用更多的节点则没有进
一步改善。
在节点内最好总是使用SMP并行,Linda仅用于节点之间。例如四节点的集群,每个节点
有两个四核EM64T,那么应当使用
%NProcShared=8
%LindaWorkers=node1,node2,node3,node4
而不能在每个节点使用一个以上的Linda进程。
安装Linda软件和编译G09/Linda
如果购买了 Gaussian 的二进制程序,那么 Linda 发布在与 Gaussian 二进制程序相同的
CD 中,无需另外安装。参照发布 CD 中README 文件的说明。
如果购买了 Gaussian 源代码,那么 Linda 在另外的 CD 中发布。关于安装 Linda 和用
Linda 编译 Gaussian,参照 Gaussian 源代码发布 CD 中README.source 文件的说明。
在两种情况下,必须运行命令 bsd/install,细节参见 README 文件以及安装说明小册
子。
用Linda运行Gaussian
Linda并行程序模型涉及在当前处理器运行的主程序,以及一些可运行于其它网络节点
的工作处理器。因此Gaussian 09/Linda的运行必须指定使用的处理器数目,以及运行任务
的处理器列表,有时还要指定其它任务参数。指定这一信息的最容易办法通常是用环境变量
(下面将会看到)。
这些节点的每一个都需要访问Gaussian 09目录树的权限。建议的配置是在用于并行任
务的每个体系都安装G09。注意,Linda二进制文件需要在每个机器上有相同的路径。如果做
不到,那么G09目录树可通过NFS挂接磁盘访问,它在所有节点下都挂接到相同的位置。
对于MP2计算,每个节点还必须有本地磁盘,用于Gaussian 09放置临时文件。这仍旧通
过GAUSS_SCRDIR环境变量定义,它必须在.cshrc或.profile中设定,这些文件位于每个节点
- 34 -
的帐户下。
配置Gaussian 09/Linda
Gaussian 09获取配置信息有三个主要来源:
在Gaussian输入文件中通过%Link0命令
Default.Route文件
环境变量GAUSS_LFLAGS
关于%Link0和Default.Route文件的细节,可以在Gaussian 09用户参考手册中找到。
Gaussian 09/Linda特定条目的说明见下。
指定工作计算机
%LindaWorkers指令用于指定Linda工作处理器运行的计算机。它的语法如下:
%LindaWorkers=node1[:n1][,node2[:n2]] […]
这里列出了每个启用节点的TCP节点名称。默认情况下,在每个节点开始一个Linda工作程序,
但可以用备选值修改。工作程序总是从开始任务的节点(主节点)开始,不论它是否出现在
节点列表中。%LindaWorkers可以与%NProcShared组合。这种情况下,每个节点上将运行一
个或多个(数量仍旧由%LindaWorkers的值确定)并行工作进程。工作节点列表中每个系统
使用的SMP处理器/核心的个数由%NProcShared的值指定。
不要使用作废的%NProcLinda指令。G09基于%LindaWorkers计算Linda工作程序的总数。
下面的指令指定网络并行任务运行在指定的5个节点上。其中的hamlet和ophelia节点运
行两个工作进程。
%LindaWorkers=hamlet:2,ophelia:2,laertes,horatio,lear
下面的指令指定并行任务运行于norway,italy,和spain主机。其中节点norway和italy
运行一个4路SMP并行工作程序,spain运行两个这样的工作程序。
%NProcShared=4
指定4路SMP并行
%LindaWorkers=norway,italy,spain:2
当norway和italy是4处理器/核心计算机,spain是8处理器/核心计算机时,这些指令才
有意义。
注意,早期版本Gaussian使用的%NProc指令已经作废。
指定并行计算的内存量
就像串行计算一样,内存用%Mem Link0指令指定。
用SSH代替RSH
Linda默认用rsh在节点间通信。通过在GAUSS_LFLAGS环境变量中包含以下选项,可以用
ssh替代:
% setenv GAUSS_LFLAGS '… -opt "Tsnet.Node.lindarsharg: ssh"'
覆盖这一默认设置的另一种方法是在你的主节点根目录下创建配置文件,名
- 35 -

为.tsnet.config,它包含下面的行:
Tsnet.Node.lindarsharg: ssh
这将导致使用ssh。注意必须已经配置了从主节点到所有工作节点的无密码ssh登陆。
指定其它的LINDA选项
有时会用到的Linda选项:
-v
显示详细信息
-vv
显示非常详细的信息
例如,打开非常详细的Linda输出,可以用:
% setenv GAUSS_LFLAGS -vv
还有很多其它Linda选项,但大多数不被Gaussian使用。可以查阅网上的Linda手册:
www.lindaspaces.com/downloads/lindamanual.pdf
GAUSS_LFLAGS中使用的-opt形式可用于调用任何有效的.tsnet.config文件指令。注意,
Gaussian 09/Linda不使用本机的Linda资源
minworker
和
maxworker
。
开始并行Gaussian 09 任务
像往常一样,用g09命令开始分布内存的并行Gaussian 09任务。对于正常执行的Linda
并行任务,必须满足以下条件:
必须已执行合适的Gaussian 09初始化文件($g09root/g09/bsd/g09.login或
$g09root/g09/bsd/g09.profile)。通过在主节点运行一个串行Gaussian 09可作测
试。
在所有节点可以访问$g09root/g09目录。
通过设定环境变量LD_LIBRARY_PATH(参见G09安装说明)来定位Linda共享库文件。
如果需要,每个节点需要有本地的擦写空间(通过GAUSS_SCRDIR)。
程序运行的所有节点被当前主机信任。你应当可以用rlogin或ssh命令远程登录这
些节点,无须提供密码。关于配置网络节点的安全性,请联系系统管理员。
现在可以开始计算,就像串行计算一样:
% g09 input &
Gaussian 09将会开启主进程,以及根据需要的工作进程。
监测计算
对于已实现并行化的链接,你会看到工作节点上开始的进程,例如,它们在G09主目录
中有一个*.exel项目。在工作节点用top或其它命令,你将会看到lxxx.exel,如果它开始的
话。
并行计算性能的有关测试量是逝去时间或时钟时间。最容易的检查方法是使用外部监视
程序,如time,times,或timex。例如,
- 36 -

% times g09 input &
将会报告逝去、CPU、和系统时间。注意,后两个为主节点上的时间,与每个节点上的CPU
和系统时间大致相同。因此加速度为运行串行任务的逝去时间与运行并行任务的逝去时间之
比。
在基于PPC的Mac机上指定每节点的工作程序
在跨越多台基于PowerPC的Mac OS X和其它多处理器的系统上,用Linda运行Gaussian
可以用选项-mp n。它指定每个节点上安排的Linda进程的最大数目。如果所有节点都为双核
心处理器,设定为2。
运行Gaussian测试任务
Gaussian提供了大量的测试任务集,以及相应的输出文件。输入文件可以在
$g09root/g09/tests/com目录下找到。各个测试任务的参考输出(gzip压缩的log文件)位
于$g09root/g09/tests/中的一个或多个由计算机体系类型命名的子目录下(例如,ia64)。
还提供一个脚本文件,用于自动运行各系列的测试任务(说明见下)。
如果在支持的体系和操作系统下,用指定编译器、库和其它软件(如果有的话)从源代
码编译程序,我们推荐运行一些测试任务,以检查程序是否正确创建。但是,全部运行这些
测试集是不必要的。当前支持平台和所需软件的列表,见www.gaussian.com/g09_plat.htm
对于二进制的发布文件,不需要运行任何测试任务。
测试任务的输入文件名形式为test
nnn
.com。测试1,28,94,155,194,296和302包含
了Gaussian的一系列功能。注意有些测试任务需要快速的硬件,而对于较小、较慢的计算机
系统,计算相当昂贵。文件$g09root/g09/tests/tests.idx列出了每个测试任务做什么。
运行测试文件前,重命名已存在的Default.Route文件
如果选择运行某些或全部Gaussian测试任务,需要保证它们使用了程序内置的默认设
置。因此,需要重新命名站内的Default.Route文件(位于$g09root/g09目录)以及在运行
测试任务时任何有优先权的个别版本默认文件。注意这个文件中的某些设置会使一些测试任
务失败。
例子
脚本submit.csh可用于运行测试任务。它使用两个参数:要运行的第一个和最后一个任
务的编号(默认运行全部测试)。注意,应当在另外的目录运行测试任务,以防止它们破坏
参考输出。
下面的命令描述运行测试任务的推荐流程,使用/chem/newtests目录作为任务执行区,
- 37 -

测试任务28作为例子:
$ mkdir /chem/newtests; cd /chem/newtests
$ ln -s $g09root/g09/tests/com .
$ mkdir `gau-machine`
$ $g09root/g09/tests/submit.csh m n &
最后的命令运行测试
m
到
n
。
每个测试任务完成后,检查是否完全正确。接下来用d1脚本比较当前的输出和参考输出。
例如:
$ $g09root/g09/tests/d1 m n
d1脚本会过滤掉
m
到
n
测试输出文件中不重要的差别,并通过more用管道传送余下的输
出。允许出现的差别应当仅限于非实质性的项。
Gaussian 09 的链接
下面的表列出了 Gaussian 09 的构成程序——也就是所说的链接(link)——及其主要功
能:
链接 功能
L0
L1
L101
L102
L103
L105
L106
L107
L108
L109
L110
L111
L113
L114
L115
L116
L117
L118
L120
L121
L122
L123
L124
初始化程序,控制分层
处理执行路径,创建执行链接的列表,并初始化擦写文件
读取标题和分子说明部分
Fletcher-Powell 优化
Berny 优化到最小值和 TS,STQN 过渡态搜索
Murtaugh-Sargent 优化
力或偶极矩的数值差分,用以获得极化率或超极化率
线性同步过渡(LST)的过渡态搜索
非松弛的势能曲面扫描
Newton-Raphson 优化
能量的二次数值差分产生频率
能量的二次数值差分,用以计算极化率和超极化率
使用解析梯度进行 EF 优化
EF 数值优化(仅使用能量)
使用 GS3 算法追踪反应路径
数值的自恰反应场(SCRF)
执行 IPCM 溶剂化计算
BOMD 计算
控制 ONIOM 计算
ADMP 计算
平衡计算
用HPC 算法(及其它)跟踪反应路径
用PCM 及外部迭代 PCM 执行 ONIOM
- 38 -

L202
L301
L302
L303
L308
L310
L311
L314
L316
L319
L401
L402
L405
L502
L503
L506
L508
L510
L601
L602
L604
L607
L608
L609
L610
L701
L702
L703
L716
L801
L802
L804
L811
L901
L902
L903
L904
L905
L906
L908
L913
L914
L915
L916
重新定位坐标,计算对称性,检查变量
产生基组信息
计算重叠,动能,和势能积分
计算多极积分
计算偶极速率和 Rx▽积分
用原函数形式计算 spdf 双电子积分
计算 sp 双电子积分
计算 spdf 双电子积分
打印双电子积分
对近似的自旋-轨道耦合计算单电子积分
形成初始 MO 猜测
进行半经验和分子力学计算
初始化 MCSCF 计算
迭代求解 SCF 方程(用于常规的的 UHF 和ROHF,所有的直接方法,以及 SCRF)
使用直接最小化迭代求解 SCF 方程
进行 ROHF 或GVB-PP 计算
二次收敛 SCF 程序
MC-SCF
布居和有关的分析(包括多极矩)
单电子特性(势,场,和场梯度)
求解 MO,或一个网格点的密度
进行 NBO 分析
非迭代 DFT 能量
分子中的原子特性
数值积分(用于测试积分代码)
单电子积分一阶或二阶导
双电子积分一阶或二阶导(sp)
双电子积分一阶或二阶导数(spdf)
处理优化和频率的信息
双电子积分变换的初始化
进行积分变换(内存算法是 N3标度)
积分变换
变换积分导数,并计算其对 MP2 二阶导的贡献
反对称化双电子积分
确定 Hartree-Fock 波函的稳定性
旧的内存 MP2
Petersson 等人的完全基组外推方法
复数 MP2
半直接 MP2
电子传播子的程序
计算后-SCF 能量和梯度项
CIS,RPA 和Zindo 激发态;SCF 稳定性
计算五阶的量(用于 MP5,QCISD(TQ)和BD(TQ))
旧的 MP4 和CCSD
- 39 -
- 40 -
L918
L923
L1002
L1003
L1014
L1101
L1102
L1110
L1111
L1112
L9999
重新优化波函
SAC-CI 程序
迭代求解 CPHF 方程;计算各种特性(包括 NMR)
迭代求解 CP-MCSCF 方程
计算解析的 CIS 二阶导
计算单电子积分导数
计算偶极导数积分
双电子积分导数对 F(x)的贡献
双电子密度矩阵和后-SCF 导数
MP2 二阶导
完成计算并输出
Part
第2部分 Gaussian 09 的关键词,
LINK0 命令和工具
Gaussian 09 关键词
Gaussian 09 的工具程序
- 41 -
- 42 -
Gaussian 09 关键词
关键词一般按字母顺序排列,除了下面的例外:
- 未出现基组关键词;可用的基组及其关键词的信息见前面的基组部分。但是注意,
ChkBasis,ExtraBasis,Gen和Pseudo关键词将在各自的部分讨论。
- 所有与DFT有关的关键词集中在DFT方法下介绍。
- 链接0命令在ZINDO关键词之后讨论。
- 定义其它执行路径的关键词——ExtraLinks,ExtraOverlays,NonStd,Skip和Use—
—在附录“程序开发功能”的“定义非标准执行路径”部分讨论。有关的信息还出现在
testrt工具的讨论中。
在特定关键词的讨论中,各种选项按照重要性和使用频率排列,而非严格按照字母顺序。
大型的选项列表通常再细分成具有相似功能的几部分。
#
说明
Gaussian 任务的执行路径部分需要以井号(#)开始,作为一行的第一个非空格字符。
这一行的其它部分使用自由格式。对于大部分任务,所有的信息都可以放在这一行,但也可
以延续多行(续行仍可以用#号开始,但不是必须的)。路径部分必须以空行作为结束。
如果在路径部分没有出现关键词,默认的计算为 HF/STO-3G SP。
可选形式
#N 正常打印级别;这是默认的。
#P 产生额外输出。包括每一链接在开始和结束时与计算机有关的信息(包括执行时间
数据),以及 SCF 计算的收敛信息。
#T 精简输出:输出减少为重要的信息和结果。
ADMP
说明
这个关键词请求经典轨迹计算[Bunker71, Raff85, Hase91, Thompson98],使用原子中心密度矩阵
传播的分子动力学模型[Iyengar01, Schlegel01, Schlegel02]。这个方法提供与Born-Oppenheimer分子
动力学(参见BOMD关键词)等价的功能,但大大减少了计算量[Schlegel02]。
ADMP属于分子动力学的扩展Lagrangian方法,使用Gaussian基函数和密度矩阵传播。
这类方法最有名的是Car-Parrinello(CP)分子动力学[Car85],其中选择Kohn-Sham分子轨道ψi
做为动力学变量,代表体系中的电子自由度。执行CP计算通常用平面波基组(虽然有时也
会加上辅助的Gaussian轨道[Martyna91, Lippert97, Lippert99])。和平面波CP不同,ADMP在动力
学计算中不需要对氢原子使用赝势或用氘代替氢。电子自由度的假想质量会自动设定
[Schlegel02],并且数值足够小,因此不需要用于保证能量守衡的恒温方法。
- 43 -
ADMP可以用半经验,HF,纯的和杂化的DFT模型执行计算(细节参见下面的应用)。
它可用于分子,团簇和周期体系。PBC的ADMP计算只使用Γ点(即,没有K空间积分)。
可选的输入
ADMP计算可以使用一些可选的输入,虽然在大部分计算中并不需要:
[原子1的初始速度: x y z
可选的初始笛卡尔速度
原子2的初始速度: x y z
(ReadVelocity和ReadMWVelocity选项)
…]
原子N的初始速度: x y z
…
整个部分重复NTraj次
[原子1,原子2,E0,Len,De,Be
可选的Morse参数,用于每个双原子产物
…]
空行结束部分
首先,如果包含了ReadVelocity或ReadMWVelocity选项,那么先读入每个原子的初始
速度。每一个初始速度要分别定义为原子单位(Bohr/秒)的笛卡尔速度,或者质量加权的
笛卡尔速度(单位:amu1/2*Bohr/秒)。每个需要的轨迹计算都要读入一整套速度。
也可以指定每个双原子产物的Morse参数。Morse参数根据EBK量子化规则,用来确定双
原子片段的振动激发能级。它包括两个原子的原子符号,二者间的键长(
Len
,单位Angstrom),
该键长的能量(E0,单位Hartree),以及Morse势能曲线参数De
(Hartree)和Be
(Angstrom-1)。
这部分输入以空行结束。
选项
MaxPoints=
n
指定每个轨迹采用的最大步数(默认 50)。如果重新开始轨迹任务,那么最
大步数默认为原计算中指定的值。
Lowdin 使用 Löwdin 基做为正交归一集。也可以使用 Cholesky,它使用 Cholesky 基,
并且是默认的。
NKE=
N
设定初始核动能为
N
microHartree。NuclearKineticEnergy 是这个选项的
同义词。默认为 100000(对应 0.1 Hartree)。
DKE=
N
设定初始密度动能为
N
microHartree。DensityKineticEnergy 是这个选项
的同义词。
ElectronMass=
N
设定假想电子质量为|
N
/10000| amu(默认为
N
=1000,对应假想质量 0.1 amu)。
EMass 是这个选项的同义词。如果
N
<0,则所有的基函数作统一的换算。默认
情况下,芯函数比价函数有更大的权重。
FullSCF 在每一点用 SCF 收敛的结果进行动力学计算。
ReadVelocity
从输入串读入初始的笛卡尔速度。注意,速度必须和分子具有相同的对称方
位。这个选项禁止五阶非谐校正。
ReadMWVelocity
从输入串读入初始的质量加权笛卡尔速度。注意,速度必须和分子具有相同
的对称方位。这个选项禁止五阶非谐校正。
StepSize=
n
设定动力学的步长为
n
*0.0001 飞秒。默认为 1000(步长为 0.1 飞秒)。
BandGap 指定在每一步是否把 Fock 矩阵对角化,以获得带隙。默认为 NoBandGap。
Restart 从检查点文件重新开始 ADMP 计算。注意,原任务设定的选项继续有效,不能
- 44 -
修改。
ReadIsotopes
该选项允许指定不同于默认值(分别为 298.15 K,1 大气压,无换算,和最
丰同位素)的温度,压强,频率换算因子,和/或同位素。该选项可用于重新
进行分析,但使用不同于检查点文件数据的参数。
但是需要注意,所有这些参数也可以在执行路径部分(关键词 Temperature,
Pressure,和 Scale)和分子说明部分(Iso=
参数
)指定,如这个例子:
#T
Method
/6-31G(d)
JobType
Temperature=300.0 ...
...
0 1
C(Iso=13)
...
ReadIsotopes 输入具有下面的格式:
temp
pressure
[
scale
]
数值必须为实数。
原子 1 的同位素质量
原子 2 的同位素质量
...
原子 n 的同位素质量
其中的
temp
,
pressure
,和
scale
分别为所需的温度,压强,和当用于热化
学分析时可选的频率数据换算因子(默认不换算)。其余的行是分子中各原
子的同位素质量,按照分子说明部分出现的相同顺序排列。如果原子质量指
定为整数,程序将自动使用相应的真实同位素质量(例如,对 18O 指定为 18,
那么 Gaussian 使用的值为 17.99916)。
适用性
半经验,HF 和 DFT 方法。
有关的关键词
BOMD
例子
下面的ADMP输入文件样本计算H2CO分解为H2 + CO的轨迹,从过渡态开始:
# B3LYP/6-31G(d) ADMP Geom=Crowd
Dissociation of H2CO --> H2 + CO
0 1
C
O 1 r1
H 1 r2 2 a
H 1 r3 3 b 2 180.
r1 1.15275608
r2 1.74415774
- 45 -

r3 1.09413376
a 114.81897892
b 49.08562961
最后的空行
在ADMP计算的一开始,在输出中显示任务所用的参数:
TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ
------------------------------------------------------------------------
INPUT DATA FOR L121
------------------------------------------------------------------------
General parameters:
Maximum Steps = 50
Random Number Generator Seed = 398465
Time Step = 0.10000 femtosec
Ficticious electronic mass = 0.10000 amu
MW individual basis funct. = True
Initial nuclear kin. energy = 0.10000 hartree
Initial electr. kin. energy = 0.00000 hartree
Initial electr. KE scheme = 0
Multitime step - NDtrC = 1
Multitime step - NDtrP = 1
No Thermostats chosen to control nuclear temperature
Integration parameters:
Follow Rxn Path (DVV) = False
Constraint Scheme = 10
Projection of angular mom. = True
Rotate density with nuclei = True
------------------------------------------------------------------------
TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ
分子坐标和速度出现在每个轨迹步骤的开始(为了节省空间,这里截断了一些输出的数
字):
Cartesian coordinates:
I= 1 X= -1.1971360D-01 Y= 0.0000000D+00 Z= -1.0478570D+00
I= 2 X= -1.1971360D-01 Y= 0.0000000D+00 Z= 1.1305362D+00
I= 3 X= 2.8718451D+00 Y= 0.0000000D+00 Z= -2.4313539D+00
I= 4 X= 4.5350603D-01 Y= 0.0000000D+00 Z= -3.0344227D+00
MW Cartesian velocity:
I= 1 X= -4.0368385D+12 Y= 1.4729976D+13 Z= 1.4109897D+14
I= 2 X= 4.4547606D+13 Y= -6.3068948D+12 Z= -2.2951936D+14
I= 3 X= -3.0488505D+13 Y= 6.0922004D+12 Z= 1.8527270D+14
I= 4 X= -1.3305097D+14 Y= -3.1794401D+13 Z= 2.4220839D+14
Cartesian coordinates after ADCart:
I= 1 X= -1.1983609D-01 Y= 4.2521779D-04 Z= -1.0437931D+00
I= 2 X= -1.1859803D-01 Y= -1.5769743D-04 Z= 1.1248052D+00
I= 3 X= 2.8688210D+00 Y= 6.0685035D-04 Z= -2.4129040D+00
I= 4 X= 4.4028377D-01 Y= -3.1670730D-03 Z= -3.0103048D+00
TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ
轨迹计算完成后,轨迹中每一时间步骤的摘要信息会显示在输出中:
Trajectory summary for trajectory 1
Energy/Fock evaluations 51
- 46 -
Gradient evaluations 51
Trajectory summary
Time (fs) Kinetic (au) Potent (au) Delta E (au) Delta A (h-bar)
0.000000 0.1000000 -114.3576722 0.0000000 0.0000000000000000
0.100000 0.0988486 -114.3564837 0.0000371 -0.0000000000000081
0.200000 0.0967812 -114.3543446 0.0001088 -0.0000000000000104
0.300000 0.0948898 -114.3524307 0.0001313 -0.0000000000000115
...
你也可以用GaussView或其它可视化软件通过动画来显示轨迹路径。
AM1
参见半经验方法的说明。
Amber
参见分子力学方法的说明。
B3LYP
参见密度泛函方法的说明。
BD
说明
这个方法关键词请求 Brueckner 双激发计算[Dykstra77, Handy89, Kobayashi91]。BD 具有解析梯
度[Kobayashi91]。
选项
T 请求 Brueckner 双激发计算,并加上三激发贡献[Handy89]。BD-T 是BD(T)的同
义词。
TQ 请求 Brueckner 双激发计算,并加上三、四激发贡献[Raghavachari90]。
FC 这个关键词可以使用所有的冻芯选项。完整的信息参见 FC 选项的说明。
MaxCyc=
n
指定最大的循环次数。
Conver=
N
设定收敛计算对能量为 10-N,对波函为 10-(N-2)。默认对单点能为
N
=4,对梯度
为
N
=6。
SaveAmplitudes
把收敛的波幅保存到检查点文件,用于随后的计算(例如使用更大基组的计
- 47 -

算)。使用这一选项会导致极大的检查点文件,但可以显著加速后面的计算。
ReadAmplitudes
从检查点文件读取收敛的波幅(如果有的话)。注意,新的计算可以使用与原
计算不同的基组,方法(若适用),等。
Read 从检查点文件读取初始轨道,不做 HF 计算。注意,新的计算可以使用与原计
算不同的基组。
适用性
BD 的解析能量和解析梯度,BD(T)的数值梯度,所有方法的数值频率。选项 FC,T,和
TQ 不能用于解析梯度。非限制开壳层计算可用于 BD 能量和梯度。
例子
BD 能量出现在输出文件中标记为 E(CORR),在最后一步关联迭代之后:
DE(Corr)= -0.11126579 E(CORR)= -39.034559769 Delta= 2.76D-08
能量单位是 Hartree。如果使用了三激发(或三、四激发),包含校正的能量出现在上
面的输出之后:
Time for triples= 0.14 seconds.
T4(BD)= -0.17652187D-02
BD(T)= -0.39037034261D+02
三激发校正能量。
BOMD
说明
这个关键词要求使用Born-Oppenheimer分子动力学模型(最初提出在[Helgaker90,
Uggerud92];长篇的评论文章在[Bolton98])做经典轨迹计算[Bunker71, Raff85, Hase91, Thompson98]。在
Gaussian 09的执行[Chen94, Millam99, Li00]中,通过使用非常准确的基于Hessian的算法把通常
的方法作了推广,它在局部二次曲面上结合了预测步骤,接下来是校正步骤。后一步骤在每
一步的开始点和结束点对能量,梯度以及Hessian使用五次多项式或有理函数进行拟合。这
种产生校正步骤的方法能够把步长增加到早期算法的十倍以上。
用准经典固定简正模式取样方法选择初始条件,以及最后产物的分析方式与经典轨迹程
序VENUS [Hase96]相同。也可以选择读入初始笛卡尔坐标和速度。
注意,ADMP方法在Hartree-Fock和DFT级别提供等价的功能,但是计算量更低。
所需的输入
所有的BOMD任务必须指定分解路径的数量;对许多计算工作来说,这个值是0(也可以
用一个空行),不需要其它的BOMD输入。在这种情况下,轨迹会对固定数量的步长进行积分,
程序默认的步长个数是100,也可以用MaxPoints选项的值。
如果
NPath
设置为-1,程序会自动探测分解路径,并使用梯度标准(Hartree/Bohr)替代
一般的分裂路径和终止标准。
当分解路径的数量大于0时,完整BOMD任务的输入必须具有以下的一般结构:
NPath 分解路径的数量(最大=20)
IFrag
1
, …, IFrag
NAtoms
分裂信息
- 48 -
... 重复NPath次
[R1, R2, R3, R4, G5, ITest, IAtom, JAtom, R6 可选的终止标准(ReadStop选项)
...] 重复NPath次
[Estart,DelE,SBeta,Ef,DPert,IFlag] 可选的模拟退火参数(SimAnneal)
[Mode-num, VibEng(Mode-num), ...] 可选的初始简正模式能量(NSample)
[Initial velocity for atom 1: x y z 可选的初始速度
Initial velocity for atom 2: x y z (ReadVelocity或ReadMWVelocity)
...
Initial velocity for atom N: x y z
...] 整个部分重复NTraj次
[Atom1, Atom2, E
0
, Len, D
e
, B
e
对每个双原子产物可选的Morse参数
...]
用空白行结束这一部分
NPath
之后的输入行定义每个路径的分裂信息。每个位置的值指定相应原子所属的特定
片段编号(也即,原子
i
属于片段编号IFragi)。注意,每个路径的片段信息必须从新的一行
开始,但是任一路径内的片段信息根据需要可以写在连续多行内。
当指定了关键词ReadStop,接下来要指定终止标准。每个路径最多可以指定六种终止标
准。当所有指定的标准都满足时,轨迹计算就会终止。但是把任一个参数的值设为0,就会
关闭该终止标准。终止标准的定义如下(括号内是默认参数值):
每对片段质心间的最小距离 >
R1
(18)
不同片段中的原子间的最小距离 >
R2
(20)
任一原子与所在片段质心的最大距离 <
R3
(0)
同一片段内任意两原子间最大距离 <
R4
(0)
片段的梯度 <
G5
(10-6)
如果ITest=1,原子
IAtom
和
JAtom
的距离 >
R6
(0)。否则,原子
IAtom
和
JAtom
的距离 <
R6
(0)
所有的距离定义为Bohr,梯度
G5
的单位是Hartrees/Bohr。
如果定义了选项SimAnneal,输入数据的终止标准之后是模拟退火/分裂的参数:
Estart
是所需的初始动能(Hartree)。
DelE
是获得/损失的能量(Hartree)。
SBeta
是Fermi-Dirac反转温度(1/Hartree)。
Ef
是Fermi能(波数):以波数为单位的频率低于
Ef
的所有振动模式都会被放大,在
Ef
以上的减小。如果
SBeta
为负值,则情况相反。
DPert
是随机微扰的大小。
IFlag
用于决定模拟退火中使用的算法,用于能量微扰 (也即,从本征模式加入或删除
能量)。可能的值有:0(根据频率将每个本征分量加权),1(以随机方式加上
DelE
),2 (0
和1相结合),00 (如果在过渡态附近,把所有能量加到该模式上),10(忽略任何邻近的
过渡态)。
如果使用了NSample选项,接下来的输入部分定义每个简正模式的能量有多大。对每个
模式,
VibEng
是平动能,单位kcal/mol,朝着过渡矢量的方向。如果
VibEng
< 0,则初始速
度方向相反。(可以用Phase选项直接定义前进的方向。)
接下来,如果使用了ReadVelocity或ReadMWVelocity选项,则需要读入每个原子的初
始速度。每个初始速度分别定义为原子单位(Bohr/秒)的笛卡尔速度,或质量加权的笛卡
尔速度(单位 amu1/2*Bohr/秒)。对每个轨迹计算需要读入一组完整的速度。
最后,对每个双原子产物可以指定Morse参数。根据EBK量子化规则,Morse参数数据用
来确定双原子片段的振动激发。它包含两个原子的原子符号,它们之间的键长(
Len
,单位
- 49 -
Angstrom),该键长的能量(E0,单位Hartree),以及Morse曲线参数De(Hartree)和Be
(Angstrom-1)。这一输入部分以空行结束。
选项
MaxPoints=
n
指定每一个轨迹中的最大步数(默认为 100)。如果重新开始计算轨迹任务,
默认的最大步数由原计算指定。
Phase=(
N1,N2
[,
N3
[,
N4
]])
定义过渡矢量的相,使过渡矢量前进的方向对应所指定内坐标的增大方向。
如果提供两个原子编号,那么坐标是这两个原子成键的拉伸方向;如果提供
三个原子编号,则指定键的弯曲,四个原子定义二面角。
ReadVelocity
从输入串读入初始笛卡尔速度。注意,速度必须和分子具有相同的对称方位。
这个选项禁止五阶非谐修正。
ReadMWVelocity
从输入串读入初始的质量加权笛卡尔速度。注意,速度必须和分子具有相同
的对称方位。这个选项禁止五阶非谐修正。
SimAnneal 使用模拟退火(初速度随机产生)。需要读入额外的参数,见下。
ReadVelocity,ReadMWVelocity和SimAnneal只能指定其中的一个。
ReadStop 读入另外的停止标准。
RTemp=
N
指定转动温度。默认是从对称陀螺假设的热分布选择初始转动能(温度默认
为 0 K)。
NSample=
N
读入前
N
个简正模式的初始动能(默认为 0)。其它模式的能量默认由热取样
决定。
NTraj=
N
计算
N
个轨迹。
Update=
n
BOMD 默认在每一点进行二阶求导。使用 Update 关键词会使程序在计算新的
解析 Hessian 之前,在
n
个梯度点上更新 Hessian。GradientOnly 定义只做
梯度计算,并且总是更新 Hessian(不计算完全的二阶微分)。ReCalcFC 是
该选项的同义词。
RandomVelocity
随机产生动力学的初始速度,而不使用任何的二阶导信息。这对
GradientOnly 动力学是默认的。
StepSize=
n
以相应的单位设定动力学的步长为
n
*0.0001。除了 GradientOnly 计算的步
长为 0.025 飞秒之外,默认步长为 0.25 amu1/2*Bohr。
Sample=
type
指定取样类型,其中
type
是这些关键词其中之一:Microcanonical,Fixed
和Local。 默认为 Fixed 简正模式能量,除非指定 RTemp,这种情况下是 Local
模式取样。
Restart 从检查点文件重新开始轨迹计算。注意,原任务设定的选项继续有效,不能
修改。
ReadIsotopes
该选项允许指定不同于默认值(分别为 298.15 K,1 大气压,无换算,和最
丰同位素)的温度,压强,频率换算因子,和/或同位素。该选项可用于重新
进行分析,但使用不同于检查点文件数据的参数。
但是需要注意,所有这些参数也可以在执行路径部分(关键词 Temperature,
- 50 -
Pressure,和 Scale)和分子说明部分(Iso=
参数
)指定,如这个例子:
#T
Method
/6-31G(d)
JobType
Temperature=300.0 ...
...
0 1
C(Iso=13)
...
ReadIsotopes 输入具有下面的格式:
temp
pressure
[
scale
]
数值必须为实数。
原子 1 的同位素质量
原子 2 的同位素质量
...
原子 n 的同位素质量
其中的
temp
,
pressure
,和
scale
分别为所需的温度,压强,和当用于热化
学分析时可选的频率数据换算因子(默认不换算)。其余的行是分子中各原
子的同位素质量,按照分子说明部分出现的相同顺序排列。如果原子质量指
定为整数,程序将自动使用相应的真实同位素质量(例如,对 18O指定为18,
那么 Gaussian 使用的值为 17.99916)。
适用性
所有的半经验,SCF, CASSCF,CIS,MP2 和 DFT 方法。
有关的关键词
ADMP
例子
以下BOMD输入文件的例子演示了多个可用的选项。它将计算H2CO分解为H2 + CO的轨
迹,从过渡态开始。这里是一种片段的路径:C和O属于片段1,两个氢原子属于片段2。
示例的任务也可以指定终止标准。如果:H2和CO的质心距离超过13 Bhor,H2和CO的
最近距离超过11 Bhor,同一片段内所有原子与该片段质心距离小于1.3 Bhor,片段内任何原
子与其它原子间的距离小于2.5 Bhor,使片段分离的梯度小于0.0000005 Hartree/Bohr,原子 1
和3的距离大于12.8 Bohr,轨迹计算都会终止。
沿过渡矢量的初始动能为5.145 kcal/mol,朝着产物的方向(前进的方向是C-H距离增
加的方向)。指定H2和CO的Morse参数,用于确定双原子产物的振动激发;它们由以前的计
算确定。计算在300 K的温度下进行。
# HF/3-21G BOMD(Phase=(1,3),RTemp=300,NSample=1,ReadStop) Geom=Crowd
HF/3-21G dissociation of H2CO --> H2 + CO
0 1
C
O 1 r1
H 1 r2 2 a
H 1 r3 3 b 2 180.
- 51 -

r1 1.15275608
r2 1.74415774
r3 1.09413376
a 114.81897892
b 49.08562961
1
1 1 2 2
13.0 11.0 1.3 2.5 0.0000005 1 1 3 12.8
1 5.145
C O -112.09329898 1.12895435 0.49458169 2.24078955
H H -1.12295984 0.73482237 0.19500473 1.94603924
最后是空行
注意,为了演示,这里使用了所有的六种终止标准。在大多数情况下,一或两个终止标
准就足够了。
在BOMD计算的一开始,输出中先显示计算使用的参数:
TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ-TRJ
-------------------------------------------------------------------
INPUT DATA FOR L118
-------------------------------------------------------------------
General parameters:
Max. points for each Traj. = 100
Total Number of Trajectories = 1
Random Number Generator Seed = 398465
Trajectory Step Size = 0.250 sqrt(amu)*bohr
Sampling parameters:
Vib Energy Sampling Option = Thermal sampling
Vib Sampling Temperature = 300.0 K
Sampling direction = Forward
Rot Energy Sampling Option = Thermal distribution (symmetric top)
Rot Sampling Temperature = 300.0 K
Start point scaling criteria = 1.000D-05 Hartree
…
Reaction Path 1
****************
Fragment 1 center 1 ( C ) 2 ( O )
Fragment 2 center 3 ( H ) 4 ( H )
Termination criteria:
The CM distances are larger than 13.000 bohr
The min atomic distances among fragments are larger than 11.0 bohr
The max atomic and CM distances in frags are shorter than 1.3 bohr
The max atomic distances in fragments are shorter than 2.500 bohr
The change of gradient along CM is less than 5.00D-07 Hartree/bohr
Distance between atom center 1 ( C ) and 3 ( H ) is GE 12.800 bohr
Morse parameters for diatomic fragments:
E0 Re De Be
C O -112.0932990 1.1289544 0.4945817 2.2407896
H H -1.1229598 0.7348224 0.1950047 1.9460392
---------------------------------------------------------------------
在每个轨迹步骤开始的地方是简正模式的初动能:
- 52 -

-------------------------------------------------------
Thermal Sampling of Vibrational Modes
Mode Wavenumber Vib. quant.# Energy (kcal/mol)
-------------------------------------------------------
1 -2212.761 5.14500
2 837.330 0 1.19702
3 1113.182 0 1.59137
4 1392.476 0 1.99064
5 2026.859 0 2.89754
6 3168.689 0 4.52987
-------------------------------------------------------
轨迹计算完成后,输出中显示摘要信息:
Trajectory summary for trajectory 1
Energy/gradient evaluations 76
Hessian evaluations 76
Trajectory summary
Time (fs) Kinetic (au) Potent (au) Delta E (au) Delta A (h-bar)
0.000000 0.0214192 -113.0388912 0.0000000 0.0000000000000000
1.169296 0.0293490 -113.0468302 -0.0000091 0.0000000000053006
2.161873 0.0407383 -113.0582248 -0.0000144 0.0000000000045404
...
轨迹的每个时间步骤会给出这些信息。此外,输出还包括在每个片段中原子的几何参数,
片段间的距离,以及在每个片段内以及片断之间的相对质量加权的速度,在每个事件步骤都
有输出。你还可以用GaussView或其它可视化软件把轨迹显示为动画。
CacheSize
说明
这个关键词指定默认缓存量,用于各种缓存分块的算法。典型情况下,这个关键词用在
Default.Route 文件中。可以用 testrt 工具确定当前系统特定硬件的默认值。在创建 Gaussian
09 二进制文件时,这个值已对当前版本的各种类型 CPU 做了正确设置,但对此后发布的芯
片可能需要进行修改,因为可能使用了不同的缓存大小,或内存之间的缓存共享方案。如果
从源代码创建 Gaussian 09,这个值对应进行编译的系统硬件。
例子
下面 testrt 命令的输出列出了当前系统一个 AMD-64 CPU 的值:
$ testrt hf
Revision: AM64L-GDVRevH.X1 14-Feb-2009 NErtGn=1000 NextEG=818 MaxAtm=250000 CachSize=131072
testrt 的其余输出...
值131,072 对应 128K 工作精度字,或相当于 1 MB,对于有一个处理器 2 MB 缓存的计
算机,这是个合理的值。
在有当前一代处理器的四核 EM64T 计算机上,每对核芯之间有 4MB 共享缓存,在创
建程序时默认用 CacheSize=262144(2 MB),如果一次只运行一个单处理器任务,这是个合
理值。对于占用全部节点的运行,可能要 CacheSize=131072,这对应 4 MB 共享缓存的 1/4
- 53 -
用于每对核芯之间的 Gaussian 数据分块。
在四核芯 Opteron 系统,每个核芯有自己的缓存,CacheSize 值应当是缓存大小的一半
(它随处理器类型变化)。
在IA64 系统,默认在运行时确定,这几乎总是正确的。
注意,在 Linux 系统,可以从/proc/cpuinfo 找到缓存的大小。
CASSCF
说明
这个方法关键词指定进行完全活性空间多组态 SCF(MC-SCF)[Hegarty79, Eade81, Schlegel82a,
Bernardi84, Frisch92, Yamamoto96]计算。MC-SCF 计算是 SCF 计算与用轨道子集进行完全 CI 计算的
组合;轨道子集也就是
活性空间
。必须用下面的关键词指定 CASSCF 活性空间的电子数(
N
)
和轨道数(
M
):CASSCF(
N
,
M
)。注意,其它选项可以任意次序放在
N
和
M
之间或前后。
默认的活性空间定义假设电子来自初始猜测行列式中的最高占据轨道,而其余的活性空
间轨道来自初始猜测的最低虚轨道。因此,一个闭壳层体系的 4 电子,6 轨道 CAS 计算定义
为CASSCF(4,6),其活性空间的构成为:
由初始猜测提供足以容纳 4 个电子的占据轨道。即,包含两个最高的占据 MO。
足够的虚轨道,使得活性空间具有六个分子轨道。由于有了两个占据轨道,因此最低的
四个虚轨道将成为活性空间的一部分。
同理,三重态分子的 4 电子 6 轨道 CAS 计算将包含三个最高的占据轨道(在初始猜测行
列式中,一个双占据轨道,两个单占据轨道)和三个最低的虚轨道。在 Gaussian 09 中,算
法的改进使活性空间可以包含直到 14 个轨道[Siegbahn84, Robb90, Klene00]。
通常需要使用 Guess=Alter 或Guess=Permute,以保证选择的轨道包含感兴趣的电子,
并且对这些电子做正确的关联计算。可以先运行 Guess=Only 计算,快速决定轨道的对称性
(见下面第一个例子)。另一种方法是先进行完整的 Hartree-Fock 单点能计算,接下来的
任务使用 Guess=(Read,Permute)从检查点文件读入经过计算的初始猜测,并进行修改。要
使轨道因子信息包含在输出文件中,需要在预备任务的执行路径中包含 Pop=Regular(如果
要检查除这几个最低虚轨道以外更多的信息,可以使用 Pop=Full)。还 可 以 用 Pop=NBOSave
保存 NBO,它通常是 CAS 初始轨道的最佳选择。也可以用可视化软件查看分子轨道,例如
GaussView。
CASSCF 的同义词是 CAS。
在执行路径部分使用#P 可以在 CASSCF 输出中除了包含能量和单电子密度矩阵以外,还
包含最终的本征值和本征矢。
在《Exploring Chemistry with Electronic Structure Methods》第二版[Foresman96b]的第 9 章
(练习 5 和 6)以及附录 A 中给出了 CASSCF 方法的简要概述。选择活性空间的详细讨论见
参考文献[Bernardi84]。CASSCF 执行效率方面的讨论见附录中效率的考虑。
注意:CASSCF 是一种强大而又先进的方法,需要很多技巧。在进行正式 CASSCF 计算(特
别是对 CASSCF MP2)之前,我们强烈推荐用户研究一下引用的参考文献。应用的例子在参
考文献[Bernardi88, Bernardi88a, Bernardi90, Tonachini90, Bernardi92, Palmer94, Vreven97]中讨论。
变化形式
如果在执行路径除了 CASSCF 之外还指定了 MP2 和关键词,那么在 CASSCF 计算中可以对
CASSSCF 能量计算 MP2 级别的电子关联能修正[McDouall88]。
- 54 -

- 进行分子体系的激发态计算可以使用 NRoot 选项。注意值为 1 表示基态,而非第一激发
态(和 CIS 或TD 关键词的用法不同)。
- 用选项 StateAverage 和NRoot 可以对使用的电子态指定态平均 CASSCF 计算。
- 在 CASSCF 任务的执行路径包含 Opt=Conical,可以计算圆锥交点和避免交叉(见例子)
[Ragazos92, Bearpark94, Bernardi96]。
- 通过包含 SpinOrbit 选项,可以在 CASSCF 计算中计算两自旋态之间的近似自旋轨道耦
合[Walker70, Abegg74, Abegg75, Cimiraglia80, Koseki92, Koseki95, Koseki98]。Gaussian 09 中使用的
方法基于参考文献[Abegg75]。适用于从 H 到 Cl 的元素。
为了计算自旋轨道耦合,积分用包含相对论项的单电子近似进行计算,然后使用有效电
荷,即换算每个原子的 Z 值,经验性地考虑双电子影响。这个值可以在分子说明部分的
核参数列表中对每个原子分别指定。
最后,注意这个计算默认是态平均计算。
- 支持[Klene03]限制活性空间的变体(RASSCF)[Olsen88]。它用选项 RAS 指定。RASSCF 计算
将分子轨道分成五部分:最低的占据轨道(在所有组态中都是双占据的),RAS1 空间
的双占据 MO,RAS2 空间包含问题中最重要的轨道,弱占据 MO 的 RAS3 空间,以及余下
的未占据轨道。因此,CASSCF 计算中的活性空间在 RAS 计算中被分三部分,指定允许
的电子组态需要:RAS1 空间电子的最少数量, RAS3 空间电子的最大数量,以及在这三
个 RAS 空间内的电子总数。设定这些值的方法参见 RAS 选项说明。
选项
NRoot=
j
要求使用 CI 的第
j
个根,因此当
j
>1 时可得到激发态。该选项默认为基态
(
j
=1)。由 NRoot 指定的态称为“感兴趣的态”。
StateAverage
用来指定态平均 CASSCF 计算。直到 NRoot 的所有态被平均。该选项需要输
入各态的权重,格式为
n
F10.8(后面不需空行)。因为 Opt=Conical 或
CASSCF=SpinOrbit 默认进行态平均计算,因此不能和选项 StateAverage 一
起使用。
SpinOrbit 计算两态之间的近似自旋轨道耦合,两个态在另外的输入行指定。这意味着
态平均 CASSCF 计算。
RAS=(
a
,
b
,
c
,
d
)
请求 RASSCF 计算,允许 RAS1 空间的
b
个轨道中最多有
a
个空穴(也就是从
RAS1 到 RAS2 或 RAS3 的激发),RAS3 空间的
d
个轨道中最多有
c
个电子(也
就是从 RAS1 或 RAS2 到 RAS3 的激发)。因此,RAS2 内最少有
2b-a
个电子。
注意,CASSCF 的两个关键词参数指定整个活性空间 RAS1 + RAS2 + RAS3 的
大小(见例子)。
DavidsonDiag
指定使用 Davidson 对角化方法而不是 Lanczos 迭代方法计算 CI 矩阵。当超
过 6 个活性轨道,这是默认的。
LanczosDiag 指定使用Lanczos迭代方法而不是Davidson方法进行CI矩阵对角化。4
FullDiag 对 CI 矩阵使用完全(Jacobi)对角化方法,而不是 Lanczos 或 Davidson 迭
代方法。如果有 6 个或更少的活性轨道,默认使用完全对角化方法,否则用
Davidson 方法。NoFullDiag 禁用完全对角化方法。
4 手册中这里还有一句话:“当有 6个或更少的活性轨道,Lanczos 方法是默认的”,这与 FullDiag 的说明
矛盾。经测试,这种情况下默认为完全对角化方法,而不是 Lanczos 方法。——译注。
- 55 -
在需要二次收敛(见下面的 QC 选项),以及在 CI 本征矢未知的情况下(后
一情况中,指定 FullDiag 进行涉及 6 个以上活性轨道的计算),必须使用
完全 Jacobi 对角化方法。
StateGuess=
k
设定 Lanczos 方法的初始矢量为组态
k
。例如,该选项可用来对所研究的激
发态选定一个具有正确对称性的组态(和基态的组态不同)。这种情况下需
要先进行预备算,以决定所需轨道的对称性。
k
也可设为特殊值 Read,表示由输入数据读入全部的本征矢(格式:
NZ,
(Ind(I), C(Ind(I)), I=1, NZ
)。
当 CI 问题的大小超过 50 时,或者用户通过最初的测试矢量,在计算的开始
就可以认出本征矢的一个或多个主要分量时,默认的对角化方法是最有效
的。默认情况下,初始矢量在第
j+1
个位置初始化,其中的
j
值由 NRoot 选
项给出(或使用其默认值)。这些位置对应于 CI 哈密顿量最低的
j+1
个能
量对角元。该方法对最低的
j
个根通常会得到很好的收敛。
用StateGuess 选项(见下)可以改变默认设置:
CASSCF(...,StatGuess=
k
)指定 C(
k
)为 1.0。
这个矢量的主要要求是在需要的本征矢中不能缺少该矢量。因此,如果 CI
本征矢主要由组态
k
构成,那么把 StateGuess 选项设为
k
将产生很好的初
始矢量(例如,如果 CI 矢量主要是 SCF 波函,StateGuess=1 是合适的)。
但是,如果组态
k
的因子在所需要的根中恰好严格是零(例如,由于对称性),
那么将会缺少该本征矢,计算将收敛到更高的态。
OrbRot OrbRot 包含了来自 CP-MC-SCF 方程的轨道转动导数的贡献,而 NoCPMCSCF 相
反。默认为 OrbRot。
SlaterDet 在 CASSCF 计算中使用 Slater 行列式。该选项用来定位单重态和三重态之间
的圆锥交点/避免交叉点位置。
HWDet 用 Hartree-Waller 行列式而不是 Slater 行列式。对于包含 10 个或更多轨
道的 CAS 计算,这是默认的。它意味着 NoFullDiag。
RFO 请求 RFO 二次步骤。最多只能指定 QC 和RFO 其中的一个。
QC 在 CAS 计算中使用二次收敛算法。使用这个选项要小心:只有对非常好的初
始猜测它才工作得很好。只能指定 QC 和RFO 中的一个。
UNO 请求 CAS 计算的初始轨道为由前一 UHF 计算产生的自然轨道[Hamilton88,
Bofill89]。通常与 Guess=Read 合用。
使用 UNO 猜测必需当心。经常有某些具有中度占据的自然轨道对感兴趣的计
算并不重要。因此,除非对整个价空间做关联计算(通常十分昂贵),通常
用Pop=NaturalOrbital 运行 UHF 计算任务,接下来检查计算的轨道结果。
然后选出属于活性空间的轨道,并进行单点 CASSCF(...,UNO)
Guess=(Read,Alter)计算。接下来检查产生的收敛轨道以验证找到正确的活
性空间,最后用 CASSCF(...,UNO) Guess=Read 进行轨道优化。对于单重态,
整个过程依赖于用户如何将 UHF 波函收敛到合适的破缺自旋对称性(非 RHF)
结果。
NPairs=
n
在 CAS-GVB 计算[Clifford96]中,CAS 活性空间之外的 GVB 对的数量。
适用性和限制
- 56 -

能量,解析梯度,解析的和数值的频率。CASSCF 不能与任何半经验方法合用。解析梯
度和频率只能到 f 函数。
不能用 CASSCF 方法计算解析的极化率。只能使用 CASSCF Polar=Numer。
在执行路径指定SCF=Restart可以重新启动CASSCF计算。若要重新开始CASSCF几何优化
计算,必须在任务的执行路径部分加上关键词CASSCF Opt=Restart Extralinks=L405。
使用PCM溶剂化的CASSCF频率必须用Freq=Numer进行数值计算。
有关的关键词
Opt=Conical,MP2,Guess,Pop,SCF
例子
这一部分,我们看几个 CASSCF 方法最重要的使用。
轨道的初步检查(Guess=Only)。以下执行路径演示的方法用于快速检查轨道,以决
定其对称性以及产生需要的初始态所做的任何更改。我们包含选项 Pop=Reg,以便在布居分
析部分获得分子轨道输出:
# HF/3-21G Guess=Only Pop=Reg Test
研究的分子是 1,3-环丁二烯,对称性为 D2h 的单重态。我们要进行 4x4 CAS 计算,因此
活性空间有四个轨道:两个占据轨道和两个虚轨道。我们需要这四个轨道都是 π轨道。
HOMO 是第 14 个轨道,因此轨道 13 到轨道 16 将构成活性空间。当检查这些分子轨道时,
我们发现只有轨道 14 和轨道 15 的类型是正确的。由于分子位于 YZ 平面,因此 π轨道在 X
方向上会有可观的非零因子。以下是轨道 10 和轨道 13-16 的有关因子:
Molecular Orbital Coefficients
10 13 14 15 16
O O O V V
3 1 C 2PX 0.29536 0.00000 0.34716 0.37752 0.00000
7 3PX 0.16911 0.00000 0.21750 0.24339 0.00000
12 2 C 2PX 0.29536 0.00000 0.34716 -0.37752 0.00000
16 3PX 0.16911 0.00000 0.21750 -0.24339 0.00000
21 3 C 2PX 0.29536 0.00000 -0.34716 -0.37752 0.00000
25 3PX 0.16911 0.00000 -0.21750 -0.24339 0.00000
30 4 C 2PX 0.29536 0.00000 -0.34716 0.37752 0.00000
34 3PX 0.16911 0.00000 -0.21750 0.24339 0.00000
轨道 10 显然是 π轨道。如果观察更高的虚轨道,我们会发现轨道 19 也是 π轨道。我们
已找到四个需要的分子轨道,现在可以使用 Guess=Alter 将这些轨道移入活性空间。以下是
CASSCF 计算的输入文件:
# CASSCF(4,4)/3-21G Guess=Alter Pop=Reg Test
1,3-Cyclobutadiene Singlet, D2H, Pi 4x4 CAS
0 1
分子说明
10,13
交换轨道 10 和 13。
16,19
交换轨道 16 和 19。
CASSCF 能量和单电子密度矩阵。当对环丁二烯进行 CASSCF 计算时,我们将得到预测的
分子能量。它出现在 CASSCF 如下的输出中:
- 57 -

TOTAL -152.836259 …
每一迭代的能量
ITN= 9 MaxIt= 64 E= -152.8402786733 DE=-1.17D-05 Acc= 1.00D-05
ITN= 10 MaxIt= 64 E= -152.8402826495 DE=-3.98D-06 Acc= 1.00D-05
…
DO AN EXTRA-ITERATION FOR FINAL PRINTING
这个例子中,最后一次迭代的 E值是预测的能量:-152.8402826495 Hartree。
检查单电子密度矩阵也很重要,它出现在接下来的输出中:
Final one electron symbolic density matrix:
1 2 3 4
1 0.191842D+01
2 -0.139172D-05 0.182680D+01
3 0.345450D-05 0.130613D-05 0.172679D+00
4 0.327584D-06 0.415187D-05 0.564187D-06 0.820965D-01
MCSCF converged.
对角元表示活性空间内每个轨道依次的近似占据。如果其中的任何一个值是(或基本是)
零,则表示这个轨道在整个计算中几乎都是空的;类似地,如果其中的任何一个值基本是 2,
则表示这个轨道在整个计算中都是双占据的。这两种情况都表示没有从这些轨道或到这些轨
道的电子激发,所以 CASSCF 计算可能有问题。在这个例子中,两个“占据”轨道的值都小
于 2,且活性空间内另两个轨道都有非零占据,这就很好。
CASSCF MP2 能量。当进行包含动态关联的 CASSCF 计算时(执行路径使用 CASSCF MP2),
在 CASSCF 输出中会多出以下几行(第一行与第二行离得很远):
MP2 correction to the MCSCF energy is computed
表明是 CASSCF MP2 任务。
...
E2 = -0.2635549296D+00 EUMP2 = -0.15310383973610D+03
电子关联校正能量。
字符串 EUMP2 表示电子关联校正能量,本例中是-153.1038397361 Hartree。
CAS 组态信息。在 CASSCF 计算输出的开始部分,以下面的格式列出了组态:
Configuration 1 Symmetry 1 1100
Configuration 2 Symmetry 2 1ab0
Configuration 3 Symmetry 1 1010
Configuration 4 Symmetry 1 a1b0
这来自单重参考态的 CAS(4,4)计算,因此每个组态都表示 4 个活性轨道的占据方式。
第一行是参考组态,它有两个最低活性轨道是双占据,用“1”表示。在组态 2 中,第一个
活性轨道仍保持双占据,而一个 β电子从第二个活性轨道激发到第三个活性轨道,这里用“a”
表示 α,用“b”表示 β。在第三个组态中,第一个和第三个活性轨道是双占据,而组态 4
显示一个 β电子从第一个活性轨道激发到第三个活性轨道。默认允许所有的对称类型并报告
每个组态的对称性。关于轨道对称性的分配,参见组态列表之前打印的对称性乘法表。
使用 CASSCF 研究激发态。以下的两步任务演示用 CASSCF 研究激发态的方法。第一步
假设用来检查轨道的初步 Hartree-Fock 单点计算已完成,通过这个任务计算的检查点文件
获得初始猜测:
%chk=CAS1
# CASSCF(2,4) 6-31+G(D) Guess=(Read,Alter) Pop=NaturalOrbital Test
Geom=Check
Alter the guess so that the three LUMOs are all the desired
- 58 -

symmetry, and run the CAS
0,1
轨道交换
--Link1--
%chk=CAS1
%nosave
# CASSCF(2,4,NRoot=2) 6-31+G(D) Guess(Read) Pop(NaturalOrbital) Geom=Check Test
Excited state calculation
0,1
任务的第二步对 CASSCF 使用 NRoot 选项指定第一激发态。计算两个态的能量差作为体
系的第一激发能(关于此项技术的更详细讨论参见《Exploring Chemistry with Electronic
Structure Methods》[Foresman96b]一书第 9 章的练习 5)。
预测圆锥交点。在执行路径使用选项 Opt=Conical,把使用 CASSCF 对指定态进行优化
的计算变成寻找这个态的圆锥交点或避免交叉点的计算。优化的结构将是在圆锥交点或避免
交叉点处的结构。检查 CASSCF 输出文件中最后一步优化计算的本征值(在结构优化之前),
可以区分这两种可能结构:
FINAL EIGENVALUES AND EIGENVECTORS
VECTOR EIGENVALUES CORRESPONDING EIGENVECTOR
态 能量
1 -154.0503161 0.72053292 -0.48879229 ...
-0.16028934E-02 0.31874441E-02 ...
2 -154.0501151 0.45467877 0.77417416 ...
如果这两个本征值(在标有态编号行的第一项)几乎相同,则表示这两个态的能量相同,
为一圆锥交点。否则为避免交叉点。
自旋轨道耦合。这是一个 CASSCF 计算的输出,其中使用 Spin 选项要求进行自旋轨道
耦合计算(由 NRoot 选项指定的态与下一个能量较低态之间的耦合):
****************************
spin-orbit coupling program
****************************
Number of configs= 4
1st state is 1
进行自旋轨道耦合计算的态。
2nd state is 2
Transition Spin Density Matrix
1 2
1 .000000D+00 .141313D+01
2 .553225D-01 .000000D+00
magnitude in x-direction= .0000000 cm-1
magnitude in y-direction= .0000000 cm-1
magnitude in z-direction= 55.2016070 cm-1
total magnitude= 55.2016070 cm-1
自旋轨道耦合。
MCSCF converged.
自旋轨道耦合分解成 X,Y,和 Z 分量,然后是其总值,本例中是 55.2016070 cm-1。
- 59 -

RASSCF的例子。这个例子是RASSCF计算的执行路径:
# CAS(16,18,RASSCF(1,2,3,4)) 6-31G(d)
如果分子是电中性,单重态,那么这个执行路径将定义以下的空间:RAS1 有两个轨道,
在所有的组态中有 3或4个电子;RAS2 有12 个轨道,在参考组态中有 12 个电子;RAS3
有4个轨道,在所有的组态中有 0-3 个电子。因此在所有的组态中,RAS2 空间有 9到13
个电子。从参考行列式得到的活性空间轨道(假设是自旋单重态)是 8个最高占据轨道和
10 个最低虚轨道:也就是说,和正常 CAS(16,18) 的轨道相同。
CBS方法
CBS-4M,CBS-QB3,CBS-APNO
说明
这个方法关键词指定使用由 Petersson 等人开发的各种完备基组(Complete Basis Set;
CBS)方法,用于进行非常精确的能量计算[Nyden81, Petersson88, Petersson91, Petersson91a, Montgomery94,
Ochterski96, Montgomery99, Montgomery00]。这些关键词分别包括改进版的 CBS-4 [Ochterski96,
Montgomery00],CBS-Q//B3 [Montgomery99, Montgomery00],和 CBS-APNO [Ochterski96]方法。使用这些关
键词时不需要指定基组。可以为 CBS-QB3 加上前缀 RO,请求 ROCBS-QB3 方法[Wood06]。
这些方法是复杂的能量计算,对指定的体系包括多个预先定义的计算。当指定了这些关
键词中的一个,即会自动进行多步的计算工作,并将最后计算的能量显示在输出中。
Opt=Maxcyc=n,QCISD=Maxcyc=n,或 CCSD=Maxcyc=n关键词可以和这些关键词中
的任何一个结合使用,分别指定优化和 QCISD 或CCSD 循环的最大数量。
选项
ReadIsotopes
该选项允许指定不同于默认值(分别为 298.15 K,1 大气压,无换算,和最
丰同位素)的温度,压强,频率换算因子,和/或同位素。该选项可用于重新
进行分析,但使用不同于检查点文件数据的参数。
但是需要注意,所有这些参数也可以在执行路径部分(关键词 Temperature,
Pressure,和 Scale)和分子说明部分(Iso=
参数
)指定,如这个例子:
#T
Method
/6-31G(d)
JobType
Temperature=300.0 ...
...
0 1
C(Iso=13)
...
ReadIsotopes 输入具有下面的格式:
temp
pressure
[
scale
]
数值必须为实数。
原子 1 的同位素质量
原子 2 的同位素质量
- 60 -
...
原子 n 的同位素质量
其中的
temp
,
pressure
,和
scale
分别为所需的温度,压强,和当用于热化
学分析时可选的频率数据换算因子(默认不换算)。其余的行是分子中各原
子的同位素质量,按照分子说明部分出现的相同顺序排列。如果原子质量指
定为整数,程序将自动使用相应的真实同位素质量(例如,对 18O 指定为 18,
那么 Gaussian 使用的值为 17.99916)。
Restart 由前一 CBS 计算的检查点文件重新开始计算。新的任务将从前面(未完成)
运行的最后一个成功计算之后开始。
适用性
只能用于能量计算。CBS-4M 和CBS-QB3 只能用于第一和第二行原子;CBS-APNO 只
适用于第一行原子。ROCBS-QB3 [Wood06]用于使用 CBS 方法的限制性开壳层计算。
原来的CBS-4模型化学已经升级,使用了新的局域化流程并改善了经验参数[Montgomery00]。
对于新的研究,建议使用新版本的CBS-4M(M表示使用最小布居局域化)。
RO 可以与CBS-4M和CBS-QB3合用。
例子
CBS 方法每一步骤的计算结果都会包含在输出文件中。文件的最后一部分包括整个计算
结果的总结。
CBS 总结输出。以下输出来自 CH2(三重态)的 CBS-QB3 计算:
Complete Basis Set (CBS) Extrapolation:
M. R. Nyden and G. A. Petersson, JCP 75, 1843 (1981)
G. A. Petersson and M. A. Al-Laham, JCP 94, 6081 (1991)
G. A. Petersson, T. Tensfeldt, and J. A. Montgomery, JCP 94, 6091 (1991)
J. A. Montgomery, J. W. Ochterski, and G. A. Petersson, JCP 101, 5900 (1994)
Temperature= 298.150000 Pressure= 1.000000
E(ZPE)= 0.016991 E(Thermal)= 0.019855
E(SCF)= -38.936447 DE(MP2)= -0.114761
DE(CBS)= -0.011936 DE(MP34)= -0.018720
DE(CCSD)= -0.002759 DE(Int)= 0.004204
DE(Empirical)= -0.006404
CBS-QB3 (0 K)= -39.069832 CBS-QB3 Energy= -39.066969
CBS-QB3 Enthalpy= -39.066025 CBS-QB3 Free Energy= -39.088192
首先给出温度和压强,然后是 CBS-QB3 能量的各个部分。倒数第二行给出 CBS-QB3 的能
量值(通读整行):分别在 0 K 和指定的温度(默认是 298.15 K)下。最后一行是 CBS-Q
的焓(包括在指定温度的热校正)和由 CBS-QB3 方法计算的吉布斯自由能(即含有频率任务
自由能校正的 CBS-QB3 能量)。所有能量的单位都是 Hartree。
能量标记有如下含义(作为例子,使用 CBS-QB3):
CBS-QB3 (0 K) 零点校正电子能量:E0 = Eelec + ZPE
CBS-QB3 Energy 热校正能量:E = E0 + Etrans + Erot + Evib
CBS-QB3 Enthalpy 用CBS-QB3 预测的能量计算的焓:H = E + RT
CBS-QB3 Free Energy 用CBS-QB3 预测的能量计算的 Gibbs 自由能:G = H - TS
- 61 -
在不同温度下重新运行计算。下面的两步任务描述了在第二个(非常快的)CBS 计算中
使用不同温度的方法。这个任务在 298.15 K 和 300 K 的温度下计算 CBS-QB3 能量:
%Chk=cbs
# CBS-QB3 Test
CBS-QB3 on formaldehyde
0 1
分子说明
--Link1--
%Chk=cbs
%NoSave
# CBS-QB3(Restart,ReadIso) Geom=AllCheck Test
300.0 1.0
同位素说明
CBSExtrapolate
说明
这个关键词进行 MP2 能量的一般完全基组外推[Nyden81, Petersson88, Petersson91, Petersson91a]。
这个方法需要两个参数:最小数量的自然轨道对,以及积分格点。前者可以用选项 NMin
指定,它对 6-31G**,6-31G++和6-311G**基组(有无弥散函数均可)默认是 5,对添加了
(2df,p)或(3df,p)极化函数的 6-311G 基组(仍是有无弥散函数均可)默认是 10。在所有的其
它情况下都必需指定 NMin,否则会导致出错。
默认积分格点是(99, 590)网格;其它的格点可用关键词 Int=Grid 指定。因为积分部分
只占整个 CBS 外推计算的一小部分,因此选用了比较大的网格。关于网格的完整讨论,参见
Integral 关键词的说明。
必须的选项
NMin=
N
指定N为自然轨道对的最小个数。
附加的选项
MinPopLocal 使用基于最小基布居的局域化[Montgomery00]。这是默认的。
PopLocal 使用布居局域化,说明在参考文献[Pipek89]中。
BoysLocal 使用 Boys 局域化[Boys60, Foster60, Boys66]。
NoLocal 不使用任何局域化。
NRPopLocal Newton-Raphson 布居局域化。
NRBoysLocal Newton-Raphson Boys 局域化。
NRMinPopLocal
使用二阶最小布居分析。
SaveOrbitals
把局域化的 CBS 轨道保存到读写文件。注意,这些轨道将替换 SCF 轨道。
适用性
- 62 -

只能用于单点能计算,可以用任何电子关联方法。
有关的关键词
Int=Grid,CBS 方法
CCD和CCSD
说明
这个方法关键词请求进行耦合簇[Bartlett78, Pople78]计算,它对 CCD 使用来自
Hartree-Fock 行列式的双激发[Pople78],或者对 CCSD 使用单、双激发[Cizek69, Purvis82, Scuseria88,
Scuseria89]。CC 和QCID 是CCD 的同义词。RO 可以与 CCSD 组合,进行限制性开壳层能量计算
[Watts93]。
选项
FC 这个关键词可以使用所有的冻芯选项。完整的信息参见 FC 选项的说明。
T 包括非迭代三激发[Pople87](只用于 CCSD)。CCSD-T 是CCSD(T)的同义词。
E4T 和选项 T一起使用,对完全 MP4 和CCSD(T)都包含三激发。
T1Diag 计算 T. J. Lee 等人[Lee89]的 T1 诊断流程(只用于 CCSD)。
Conver=
N
设定收敛计算对能量为 10-N,对波函为 10-(N-2)。默认对单点能为
N
=7,对梯度
为
N
=8。
MaxCyc=
n
指定CCSD 计算的最大循环次数。
SaveAmplitudes
把收敛的波幅保存到检查点文件,用于随后的计算(例如使用更大基组的计
算)。使用这一选项会导致极大的检查点文件,但可以显著加速后面的计算。
ReadAmplitudes
从检查点文件读取收敛的波幅(如果有的话)。注意,新的计算可以使用与原
计算不同的基组,方法(若适用),等。
适用性
CCD 和 CCSD 的解析能量和梯度,CCSD(T)的数值梯度,所有方法的数值频率。限制性开
壳层(RO)方法可用于 CCSD 和 CCSD(T)能量计算。
有关的关键词
MP4,Transformation,QCISD
例子
耦合簇能量在输出中显示如下(打印在最后一步关联能迭代计算之后):
DE(CORR)= -.54979226D-01 E(CORR)= -.75019641794D+02
...
CCSD(T)= -.75019717665D+02
CCSD 能量标记为 E(CORR),在最后一行给出包含非迭代三激发贡献的能量。
- 63 -
Charge
说明
Charge 关键词请求在计算中包含背景电荷的分布。电荷分布由点电荷组成[Hall84,
Smith86]。仅对单点能计算有效。
默认从输入数据读入电荷,每行一个电荷,格式如下:
x y z charge
其中 x,y,z 是标准方位的坐标(单位用 Units 关键词指定,默认是埃),
charge
是电荷。
在 Gaussian 09 中,电荷必须以标准方位指定(不是输入方位)。使用链接 0 命令
%KJob=L301 可以快速确定分子的标准方位。
选项
Angstroms 表示输入电荷的位置单位是埃。
Bohrs 表示输入电荷的位置单位是 Bohr。
Check 从检查点文件读入背景电荷分布。
适用性
单点能,优化和频率。不能用于半经验方法或 PBC。
有关的关键词
%Kjob, Units
例子
在有背景电荷存在的情况下进行几何优化计算,必须使用Opt=Z-Matrix NoSymm关键词,
并用传统的Z-矩阵或符号笛卡尔坐标定义输入的结构。这是个例子:
# RHF/STO-3G Opt=Z-Matrix Charge NoSymm
Water, STO-3G, point charges
0,1
O
H 1 R1
H 1 R2 2 A1
Variables:
R1=1.0
R2=1.0
A1=105.
2.0 2.0 2.0 1.2
2.0 -2.0 2.0 1.1
ChkBasis
说明
- 64 -
ChkBasis 关键词指定从检查点文件中读入基组,可用于使用一般基组的组合任务,这
样输入数据中的基组只需输入一次。但是需要注意,ChkBasis 可以用来从检查点文件恢复
任何存在的基组,不论这些基组原先是如何的指定的。同时还恢复基组中指定的 ECP,以
及纯函数或笛卡尔函数的选择。
ChkBasis 默认还会从检查点文件取回任何密度拟合基组。其它的使用参见例子。
当然,使用 ChkBasis 时不用再指定基组关键词。
CheckPointBasis,ReadBasis 和RdBasis 都是 ChkBasis 的同义词。
有关的关键词
Gen, GenECP, Pseudo, ExtraBasis, ExtraDensityBasis
例子
以下执行路径将从检查点文件取回基组和密度拟合基(如果有的话),并把它们用于当
前的任务:
# BLYP/ChkBasis
以下执行路径只从检查点文件取回基组,并使用自动产生的密度拟合基:
# BLYP/ChkBasis/Auto
以下计算路径只从检查点文件取回密度拟合基:
# BLYP/6-31G(d)/ChkBasis
CID和CISD
说明
这个方法关键词指示在 Hartree-Fock 计算之后,从 Hartree-Fock 参考行列式进行全部双
激发(CID )或全部单、双激发(CISD )的组态相互作用计算[Pople77, Raghavachari80a,
Raghavachari81]。CI 是CISD 的同义词。
选项
FC 这个关键词可以使用所有的冻芯选项。完整的信息参见 FC 选项的说明。
Conver=
N
设定收敛计算对能量为 10-N,对波函为 10-(N-2)。默认对单点能为
N
=7,对梯度
为
N
=8。
MaxCyc=
n
指定CISD 计算的最大循环次数。
SaveAmplitudes
把收敛的波幅保存到检查点文件,用于随后的计算(例如使用更大基组的计
算)。使用这一选项会导致极大的检查点文件,但可以显著加速后面的计算。
ReadAmplitudes
从检查点文件读取收敛的波幅(如果有的话)。注意,新的计算可以使用与原
计算不同的基组,方法(若适用),等。
适用性
- 65 -
能量,解析梯度和数值频率。
有关的关键词
Transformation
例子
CI 能量在输出中显示如下:
DE(CI)= -.48299990D-01 E(CI)= -.75009023292D+02
NORM(A) = .10129586D+01
最后一步 CI 迭代的输出之后是预测的总能量。第二个输出行显示 Norm(A)值。波函的
关联校正由 Norm(A)–1 给出;因此 HF 组态的因子就是 1/Norm(A)。注意波函以中间归一化
的形式存储;即:
ijab
ijab
ia
ia
CISD abijTaiT )()(
0
中间归一化波函
其中的 Ψ0是Hartree-Fock 行列式,其因子为 1(也就是中间归一化的含义)。Norm(A)是系
数,它除以上面的波函达到完全归一化。因此:
ia ijab
ijabijabiaia TTTTANorm 1)(
完全归一化波函
在完全对角化波函中,Hartree-Fock 行列式的因子是 1/Norm(A),单激发行列式 Ψi→a的
因子是 Tia/Norm(A),等等。
CIS和CIS(D)
说明
CIS 方法关键词指示用单激发 CI(CI-Singles)方法计算激发态[Foresman92]。这一计算
可用于闭壳层和开壳层体系。
《
Exploring Chemistry with Electronic Structure Methods
》
[Foresman96b]一书的第 9 章有关于该方法及其使用的详细讨论。
CIS(D)用于请求有关的CIS(D)方法(即D选项)
[Head-Gordon94a, Head-Gordon95]。也可以在CIS
任务之后,对附加的态接着进行CIS(D)任务,进行激发能计算(参见例子)。
CIS 任务可以包含 Density 关键词;如果没有指定选项,这个关键词导致布居分析使用
当前的(CIS)密度,而不是默认的 Hartree-Fock 密度。注意,Density 不能与 CIS(D)合用。
选择态的选项
Singlets 只求解单重激发态。该选项只影响闭壳层体系的计算,对这种情况是默认的。
Triplets 只求解三重激发态。该选项只影响闭壳层体系的计算。
50-50 求解一半单重和一半三重激发态。该选项只影响闭壳层体系的计算。
Root=
N
指定感兴趣的态计算广义密度。默认是第一激发态(
N
=1)。
- 66 -
NStates=
M
求解
M
个电子态(默认是 3)。如果使用 50-50,NStates 给出要求解的每种
类型电子态的数量(即,默认计算 3 个单重和 3 个三重激发态)。
Add=
N
从检查点文件中读取收敛的态,并且另外求解
N
个态。这个选项还意味着
Read。NStates 不能和这个选项合用。
与密度有关的选项
AllTransitionDensities
计算任何一对电子态之间的跃迁密度。
与流程和算法有关的选项
FC 这个关键词可以使用所有的冻芯选项。完整的信息参见 FC 选项的说明。
Direct 根据需要使用重新计算的 AO 积分求解 CI-Singles 方程。只有在默认(MO)
算法所需的 4O2N2字磁盘空间不足,或进行非常大的计算(超过 200 个基函
数)时,才需要使用 CIS=Direct。
MO 使用转换过的双电子积分求解 CI-Singles 方程。这是 Gaussian 09 的默认算
法。积分转换受 MaxDisk 关键词的控制,因此进一步降低磁盘需求。
AO 使用 AO 积分求解 CI-Singles 方程,避免积分转换。AO 基很少是最佳选项,
只适用于在小磁盘和内存的系统上计算很小的分子。
Conver=
N
设定收敛计算对能量为 10-N,对波函为 10-(N-2)。默认对单点能为
N
=4,对梯度
为
N
=6。
Read 从检查点文件中读取 CI-Singles 态的初始猜测。注意,和 SCF 计算不同,一
个基组的初始猜测不能用在另一基组的计算中。
Restart 从检查点文件重新开始 CI-Singles 迭代计算。这还意味着 SCF=Restart。
RWFRestart 从读写文件重新开始 CI-Singles 迭代计算。可用于用非标准执行路径进行连
续多个 CI-Singles 计算。
EqSolv 是否进行平衡或非平衡的PCM计算。除了激发态优化和请求激发态密度计算
(例如使用Density=Current或All)之外,默认是NonEqSolv。
NoIVOGuess 指定使用正则单激发做为初始猜测。默认的 IVOGuess 采用改善的虚轨道。
调试选项
ICDiag 对在内存中通过转换积分形成的 CI-Singles 矩阵进行常驻内存式完全对角
化。这主要是调试选项。
MaxDiag=
N
限制 Davidson 流程中的对角化子矩阵维度为
N
。这主要是调试选项。该选项
的同义词为 MaxDavidson。
适用性
CIS 的能量,解析梯度和解析频率(包括开壳层体系),CIS(D)的能量。使用 PCM 溶剂
化的 CAS 频率必须用 Freq=Numer 进行数值计算。
有关的关键词
Zindo, TD, MaxDisk, Transformation, Density
例子
- 67 -

CIS 的输出。CI-Singles 输入部分没有特殊之处或难点。单点 CI-Singles 计算的输出
部分和基态 CI 或 QCI 计算的输出部分相似。SCF 计算之后是积分转换计算,然后求解基态
MP2 能量。接着是迭代求解 CI 问题的信息;注意在第一次迭代计算中还进行额外的初始猜
测,以保证能找出不考虑对称性情况下的指定数目的激发态。在第一次迭代之后,对每一次
迭代中的每个态的解加入一个新的矢量。
每一迭代中,每个态的激发能和波函的改变都打印到输出(用#P 输出的情况):
Iteration 3 Dimension 27
Root 1 not converged, maximum delta is 0.002428737687607
Root 2 not converged, maximum delta is 0.013107675296678
Root 3 not converged, maximum delta is 0.030654755631835
Excitation Energies [eV] at current iteration:
Root 1 : 3.700631883679401 Change is -0.001084398684008
Root 2 : 7.841115226789293 Change is -0.011232152003400
Root 3 : 8.769540624626156 Change is -0.047396173133051
迭代流程可以由两种方式成功结束:仅产生逐渐趋于零的小的展开矢量,或新的波函改
变极小。
当 CI 收敛时,显示的结果以下列标记开始:
*****************************************************************
Excited States From <AA,BB:AA,BB> singles matrix:
*****************************************************************
然后是基态和每一激发态间的跃迁偶极矩表格。接下来总结每个态的结果,包括自旋对
称性和空间对称性,激发能,谐振强度,以及 CI 展开式的最大因子(使用 IOp(9/40=
N
)可以得
到所有大于 10-N 的展开式因子):
Excitation energies and oscillator strengths:
对称性,激发能,谐振强度
Excited State 1: Singlet-A' 3.7006 eV 335.03 nm f=0.0008
8 -> 9 0.69112
每一激发的 CI 展开因子(这里激发是从轨道 8 到轨道 9)
This state for opt. and/or second-order corr.
这是“感兴趣的态”
Total Energy, E(CIS) = -113.696894498
为了方便,这里重复 CIS 能量。
归一化。对于闭壳层的计算,展开因子的平方和归一化到1/2 (因为β电子的因子没有
显示)。对于开壳层计算,归一化的和为1。
寻找附加的态。以下执行路径将从检查点文件中读取CIS结果,然后求解第二个态之上
更高的六个态:
# CIS=(Read,Root=2,NStates=6)
CNDO
说明
这个方法关键词使用 CNDO 哈密顿量[Segal66]进行半经验计算。不需指定基组关键词。
适用性
能量,“解析”梯度,和数值频率。
- 68 -

例子
出现在输出文件中的 CNDO 能量显示如下:
SCF Done: E(UCNDO) = -8.08016620373 A.U. after 11 cycles
能量用 CNDO 模型定义。
Complex
说明
这个关键词允许分子轨道变为复数。它只适用于闭壳层的单重态。
适用性
Hartree-Fock 和MP2 的解析能量,HF 解析梯度和 HF 数值频率。
有关的关键词
SCF
Constants
说明
指定使用哪一套物理常数。注意,只有与旧版Gaussian的结果做比较时才需要使用旧物
理常数。
选项
2006 Gaussian 09中使用的常数,来自[Mohr08]以及其中的参考文献。这是默认的。
1998 Gaussian 03中使用的常数,来自[Mohr00]以及其中的参考文献。
1986 从 Gaussian 88 到 Gaussian 98 使用的常数,来自[CRC80, Cohen86]。
1979 从 Gaussian 80 到 Gaussian 86 使用的常数,大部分来自[Constyear79]。
OldConstants 关键词是 Constants=1979 的同义词。
当前的值
这里汇集了Gaussian 09使用的从标准单位变为原子单位的各种换算因子和物理常数。
Gaussian计算中用到的全部量都是原子单位;换算因子只用于处理输入或者产生打印输出。
没有参考文献的值是从参考文献的量推导而来。
基本常数。直接存储在程序中的常数有:
1 Bohr (a0) = 0.5291772086 Å [Mohr08]
1 原子质量单位(amu, mu) = 1.66053878 x 10-27 千克 [Mohr08]
1 电子电荷(e) = 4.803204 x 10-10 静电单位
- 69 -

= 1.602176487 x 10-19 库仑 [Mohr08]
普朗克常数(h) = 6.62606896 x 10-34 焦耳·秒 [Mohr08]
阿弗伽德罗常数(NA) = 6.02214179 x 1023 [Mohr08]
1 卡= 4.184 焦耳 [Constyear79]
1 Hartree (Eh) = 4.3597439 x 10-18 焦耳 [Mohr08]
光速 (c) = 2.99792458 x 1010 厘米·秒-1 [Mohr08]
波尔兹曼常数 (k) = 1.3806504 x 10-23 焦耳·度-1 [Mohr08]
精细结构常数的倒数 (α-1) = 137.03599968 [Mohr08]
273.15 K理想气体摩尔体积(Vm) = 0.022413996 米3 [Mohr08]
质子静止质量(mp) = 1.67262164 x 10-27 千克 [Mohr08]
电子磁矩(μe) = 9.28476377 x 10-24 焦耳·特斯拉-1 [Mohr08]
自由电子的g-因子(ge) = 2.002319304362(无量纲)[Mohr08]
换算因子。为了方便,下面包含了有用的转换因子:
电子质量(me) = 0.910938 x 10-30 千克 [Mohr08]
质子质量(mp/me) = 1836.1527 电子质量 [Mohr08]
1 原子质量单位(amu) = 1822.8889 电子质量 [Mohr08]
1 电子伏(eV) = 23.06055 千卡·摩尔-1
= 1.60217649 x 10-19焦耳 [Mohr08]
1 Hartree = 627.5095 千卡·摩尔-1
= 27.2114 电子伏
= 219474.63 cm-1(振动频率原子单位)
1 Debye2·Å-2·静电单位-1 = 42.2561 千米·摩尔-1
= 5.82573 x 10-3 厘米-2·大气压-1
(标准温压)
电场:1 au = 5.142206 x 1011 伏·米-1 [Mohr08]
电极化率:1 au = 1.648777 x 10-41 库伦2·米2·焦耳-1 [Mohr08]
1 Bohr-电子(原子单位电偶极矩) = 8.478352 x 10-30 库伦·米 [Mohr08]
= 2.541746 Debye
= 2.541746 x 10-18 静电单位·厘米
Counterpoise
说明
用Counterpoise关键词计算平衡校正[Boys70, Simon96],可用于能量、几何优化或频率计
算,以及BOMD计算。
Counterpoise关键词用一个整数指定分子结构内的片段或单体的数量。我们推荐用新的
语法定义片段(参见第一部分的分子说明概述),这里用的就是这种语法。
选项
NewGhost 使用新型空原子,其中包含了DFT积分的积分格点。 NewBq是NewGhost的同
义词。这是默认的和推荐的方法。
OldGhost 使用旧式空原子。OldBq 是OldGhost 的同义词。只有与以前的结果做比较时
- 70 -

才会用到。
适用性
不能用于 ONIOM。平衡计算不能产生轨道。
例子
平衡计算的输入。下面的输入是平衡计算的例子:
# UHF/6-31G(d) Counterpoise=2
Counterpoise on water dimer
1,2 1,2 0,1
O(Fragment=1) 0.00 0.00 0.00
O(Fragment=2) 0.00 0.00 2.98
H(Fragment=1) 0.49 0.76 -0.29
H(Fragment=1) 0.49 -0.76 -0.29
H(Fragment=2) -0.91 0.00 3.24
H(Fragment=2) -0.01 0.00 2.03
上面的任务还演示了指定片段电荷和片段自旋多重度。第一对电荷和自旋是对整个分子
给出;之后是按照片段编号顺序的每个片段的电荷和自旋。
这是一个用ECP进行平衡优化的例子:
# HF/LANL2DZ Counterpoise=2 NoSymm Opt
HBr + HF, optimization with counterpoise correction using ECP basis
0 1 0 1 0 1
H(Fragment=1) 0.00000000 0.00000000 0.58022808
Br(Fragment=1) 0.00000000 0.00000000 -0.83659185
F(Fragment=2) 0.00000000 0.00000000 2.77788358
H(Fragment=2) 0.00000000 0.00000000 3.69953441
平衡计算的输出。下面是Counterpoise计算的典型输出:
Counterpoise: corrected energy = -2660.083831739527
Counterpoise: BSSE energy = 0.003902746890
这几行分别给出平衡校正后的能量和基组重迭误差。
CPHF
说明
这个关键词选择求解 CPHF 方程的算法[McWeeny60, McWeeny62, Stevens63, Gerratt68, Dodds77,
Dodds77a, Wolinski80, Osamura81, Osamura82, Pulay83, Dykstra84]。
选项
Grid=
grid
定义计算中 CPHF 部分的积分网格。语法与 Int=Grid 选项相同。该选项的自
变量既可以是网格关键词(Fine,UltraFine,等),也可以是特定的网格。
- 71 -
使用的默认网格是 FineGrid。这种情况下,CPHF 的默认网格是 Coarse。当
积分使用 UltraFine 时,CPHF 使用 SG1 网格。如果 SG1 选作网格,那么 CPHF
仍然使用 Coarse 网格。如果对选项 Integral=Grid 指定了特殊的网格,那
么 CPHF 也使用同样的格点。最后注意,对于一些包含 Polar=OptRot,
Freq=Anharmonic,和 Freq=NNROA(这些情况下,CPHF 使用 Coarse)的 DFT
任务,默认使用 SG1 积分网格。
关于指定网格的完整说明,参见 Integral=Grid 的讨论。
RdFreq 执行含频(动态)CPHF,需要读入用于电磁场扰动的入射光频率。所需的频
率必须在输入串中给出。这个值的默认单位是 Hartree。指定其它单位需要加
上cm(cm-1)或 nm(波数)后缀之一。这个选项和 Freq 及 Polar 任务有
关。这是 Freq=ROA 的默认选项。
Static 在进行动态扰动时,自动包含静态扰动。除了 Polar=OptRot 和Freq=ROA
之外,这是默认的。NoStatic 表示在 RdFreq 的动态扰动计算中,不执行静
态扰动。
RecursiveDIIS
用递归 DIIS 求解约化方程。这对以下情况是默认的:方程右侧矩阵个数是约
化矩阵维度的两倍以上,并且约化矩阵维度较大(只会发生在使用电子嵌入
的ONIOM(MO:MM)计算中),或者超过了 MaxInv 的限定设置。否则默认为
NoRecursiveDIIS,表示对约化 A矩阵进行反转。
MaxInv=
N
在联立求解过程中,指定常驻内存反转计算的最大约化空间(直到维度
N
)。
更大的约化问题由二级 DIIS 方法求解。默认是 5000。
EqSolv 使用平衡溶剂。对于静态扰动(即,零频场和核位移)这是默认的。对于动
态扰动(频率非零),默认是 NonEqSolv。
Simultaneous
对所有变量使用同一展开空间。这比使用各自的展开空间快,但精度稍差。
除了用 RdFreq 指定多个频率之外(见上),这是默认的方法。
Separate 在 CPHF 中,每个变量都使用各自的展开空间(与 Simultaneous 相反)。当
用RdFreq 指定多个频率时,这是默认且唯一的选项。
AO 在原子轨道基中求解 CPHF 方程[Stevens63, Osamura81, Osamura82, Pulay83]。这是默认
方法。
MO 在分子轨道基中求解 CPHF 方程。
Conver=
N
设定 CPHF 收敛标准为 10-N。CPHF=Separate 默认为
N
=9,CPHF=Simultaneous
(这是默认选项)默认为
N
=10。
Canonical 使用默认的正则 CPHF。
MOD 对SAC-CI梯度(它使用组态选择)使用MOD轨道导数。
有关的关键词
SCF
Density
说明
- 72 -

布居和其它分析流程默认使用 SCF 密度(也就是说,后-SCF 方法使用 Hartree-Fock 密
度,DFT 任务使用 DFT 密度,CAS 任务使用 CASSCF 密度)。另外也可以使用 MP2,MP3,MP4(SDQ),
QCISD,CCD,CCSD,CID,CISD,BD,和 SAC-CI 方法计算的广义密度。这些广义密度基于
Z-矢量方法[Diercksen81, Diercksen81a, Handy84, Wiberg92]计算,因此产生的多极矩是正确的能量解
析导数。也可以使用二阶(不同于 MP2)的非松弛密度,但不推荐使用。
Density 关键词的选项用来选择分析哪个密度。没有选项的 Density 关键词相当于
Density=Current。
选项
Current 使用当前方法计算的密度矩阵。如果 Density 没有给出选项,这是默认的选
项。
All 使用所有可用的密度。该选项适用于布居分析,但不适用于静电或密度的计
算。注意该选项在 CI-Singles 计算中不会产生所有激发态的密度,只会产生
指定态的密度(实现前者的方法见下面的例子)。
SCF 使用 SCF 密度。HF 是SCF 的同义词。
MP2 使用对应于二阶能量的广义密度。
Transition=
N
或(
N
,
M
)
使用态
M
和态
N
之间的 CIS 跃迁密度。默认的
M
值为 0,对应基态。
AllTransition
使用所有可用的 CIS 跃迁密度。
CI 使用对应 CI 能量的广义密度。
QCI 使用对应 QCI(或耦合簇)能量的广义密度。CC 的同义词是 QCI。
RhoCI 使用由态
N
的 CI 波函计算的单电子密度。这和 CI 密度[Wiberg92]不同,不推
荐使用!在《Exploring Chemistry with Electronic Structure Methods》一书的第
9 章讨论了这个问题[Foresman96b]。
Rho2 在 Møller-Plesset 理论中,使用正确到二阶的密度。这不同于 MP2 密度,不
推荐使用![Wiberg92]
CIS=
N
对态
N
使用总的非松弛 CIS 密度。注意,这不同于 CIS(Root=
N
,...)
Density=Current 计算的密度,后者更好[Wiberg92]。
Checkpoint 从检查点文件恢复密度用于分析。这意味着 Guess=Only CheckBasis:这个
计算不会重算新的积分,SCF,等,并且从检查点文件恢复基组。
有关的关键词
Guess, ChkBasis
例子
下面的执行路径进行 CI-Singles 计算,对要研究的分子预测六个最低激发态。布居和
其它分析计算使用对应最低激发态的 CIS 密度:
%Chk=benzene
# CIS(NStates=6)/6-31+G(d,p) Density=Current Pop=CHelpG
下面的执行路径用来对其它激发态重新进行后 CIS 分析:
%Chk=benzene
# CIS(Read,Root=N) Density=Current Pop=CHelpG
# Guess=Read Geom=AllCheck
- 73 -
这个执行路径从检查点文件中读取收敛的 CIS 和CIS 波函,并进行必要的 CPHF 计算
以产生态 N的松弛密度,接下来用这个密度做布居和其它分析。
DensityFit
说明
对涉及纯(非杂化)DFT泛函计算的库仑问题启用并控制密度拟合。密度拟合基组定义
为任务执行路径中的模型化学的一部分。内置密度拟合基组的列表包含在基组的说明中。
DenFit是这个关键词的同义词。
包含这个关键词会导致使用拟合集,如果对指定的模型化学可能的话。如果没有在执行
路径部分指定特定的拟合集,那么程序将会选择对应给定基组的标准拟合集。如果不存在将
自动生成一个。如果在Default.Route文件中包含DensityFit,那么/Fit意味着对所有的(有
关)计算无需指定拟合集(参见基组部分)。
NoDensityFit关键词在纯DFT计算中关闭密度拟合的使用。由于在程序中默认关闭密度
拟合,这个关键词仅用于覆盖Default.Route中的DensityFit。NoDenFit是NoDensityFit的
同义词。如果在任务的执行路径部分同时指定了拟合集和NoDensityFit,则忽略后者,使用
密度拟合。
选项
Iterative 控制是否建立广义逆或迭代求解拟合方程。除了对 ADMP 和PBC 之外,默认是
NonIterative。
InvToler=
N
对于拟合矩阵广义逆的非平凡特征值,设定阈值为 10-N。
Convergence=
N
指定拟合方程迭代解的收敛标准为 10-N。它意味着 Iterative。对 ADMP 默认
为10-6,对 BOMD 默认为 10-9。
Coulomb Coulomb 指定用于拟合方法的度量。这是默认的。
Overlap Overlap 指定用于拟合方法的度量。默认是 Coulomb。
JNormalization
指定读入的密度基的收缩因子对应于 Coulomb 归一化。这是默认的。
AONormalization
指定读入的密度基的收缩因子对应于 AO(重叠)归一化。默认为
JNormalization。
适用性
仅用于纯泛函(非杂化泛函)的 DFT 计算。
有关的关键词
基组,ExtraDensityBasis, Gen, ChkBasis
- 74 -
密度泛函(DFT)方法
说明
Gaussian 09 提供各种各样的密度泛函理论(DFT)[Hohenberg64, Kohn65, Parr89, Salahub89]模
型(DFT 方法和应用的讨论参见[Salahub89, Labanowski91, Andzelm92, Becke92, Gill92, Perdew92, Scuseria92,
Becke92a, Perdew92a, Perdew93a, Sosa93a, Stephens94, Stephens94a, Ricca95])。所有的 DFT 模型都能计算能
量[Pople92],解析梯度,和真正的解析频率[Johnson93a, Johnson94, Stratmann97]。
自洽反应场(SCRF)可与 DFT 能量,优化和频率计算合用,用于模拟溶液中的体系。
纯 DFT 计算经常需要利用密度拟合。细节参见基组部分的说明。
下一小节简要介绍 DFT 方法。之后给出 Gaussian 09 使用的特定泛函。最后一部分考察
DFT 计算精度的有关考虑。
对于 DFT 频率计算,计算推荐使用由 freqmem 估算的最佳内存大小。
在 DFT 频率计算过程中,默认不计算极化率导数(拉曼强度)和超极化率。用 Freq=Raman
请求这些计算。Polar 计算会包含它们。
注意:
双杂化泛函和MP2关键词一起讨论,因为它们有相似的计算量。
理论背景
在 Hartree-Fock 理论中,能量形式为:
EHF = V + <hP> + 1/2<PJ(P)> - 1/2<PK(P)>
其中的项具有以下意义:
V为核排斥能
P为密度矩阵
<hP>为单电子能量(动能加势能)
1/2<PJ(P)>为电子的经典库仑排斥
-1/2<PK(P)>为来自电子的量子(费米)特性的交换能。
在 Kohn-Sham 公式化的密度泛函理论[Kohn65]中,单行列式的精确交换(HF)被一个更一
般的表达式代替,即交换-关联泛函,该泛函可包含电子交换和电子关联项,其中后者是被
Hartree-Fock 理论忽略的:
EKS = V + <hP> + 1/2<PJ(P)> + EX[P] + EC[P]
其中 EX[P]为交换泛函,EC[P]为关联泛函。
在 Kohn-Sham 公式下,Hartree-Fock 理论可以看作是密度泛函理论的特殊情况,其中
EX[P]由交换积分-1/2<PK(P)>给出,且 EC=0。密度泛函中通常使用的泛函为电子密度以及可
能的密度梯度以某种函数结合的积分:
drrrrrfPEX))(),(),(),((][
其中不同密度泛函的区别在于,对于 EX和EC分别使用哪个函数 f。除了纯 DFT 方法,Gaussian
还支持杂化泛函,其中交换泛函是 Hartree-Fock 交换项和以上形式的泛函积分的线性组合。
建议的泛函所构成的积分无法以封闭的数学形式计算,因此都用数值积分方法求解。
DFT 方法的关键词
- 75 -
各种纯 DFT 模型的名称由交换和关联泛函的名称组合而成。在某些情况下,该领域使用
的标准同义词也可用做关键词。
交换泛函。Gaussian 09 提供以下交换泛函。除非另有说明,否则这些交换泛函必须与
关联泛函组合,才能产生有用的方法。
S Slater 交换,ρ4/3 的理论因子 2/3,也称为局域自旋密度交换[Hohenberg64, Kohn65,
Slater74]。如果关键词单独使用:HFS。
XA XAlpha 交换,ρ4/3 的理论因子 0.7, 通常不加关联泛函,只作为单独的交换泛
函使用[Hohenberg64, Kohn65, Slater74]。如果关键词单独使用:XAlpha。
B Becke 的1988 泛函,其中包括 Slater 交换和含有密度梯度的关联[Becke88b]。如
果关键词单独使用:HFB。
PW91 Perdew 和Wang 的1991 泛函的交换部分[Perdew91, Perdew92, Perdew93a, Perdew96,
Burke98]。
mPW Perdew-Wang 1991 交换泛函,经过 Adamo 和Barone 改进[Adamo98]。
G96 Gill 的1996 交换泛函[Gill96, Adamo98a]。
PBE Perdew,Burke 和Ernzerhof 的1996 泛函[Perdew96a, Perdew97]。
O Becke 的交换泛函,经过 Handy 的OPTX 改进[Handy01, Hoe01]。
TPSS Tao,Perdew,Staroverov,和 Scuseria 的交换泛函[Tao03]。
BRx Becke 的1989 泛函[Becke89a]。
PKZB Perdew,Kurth,Zupan,和 Blaha 泛函的交换部分[Perdew99]。
wPBEh Heyd,Scuseria,和 Ernzerhof 基于屏蔽库伦势的泛函(也被称为 HSE)
[Heyd03,
Izmaylov06, Henderson09]的交换部分。
PBEh PBE 的1998 修订[Ernzerhof98]。
关联泛函。可以使用下面的关联泛函,按其对应的关键词部分排列,所有关联泛函必须
与所需的交换泛函关键词进行组合:
VWN Vo s k o ,Wilk 和Nusair 的1980 关联泛函(III),其中拟合了均匀电子气的 RPA
解,通常称为局域自旋密度(LSD)关联[Vo s k o 8 0 ](论文中的泛函 III)。
VWN5 参考文献[Vo s k o 8 0 ]中的泛函 V,其中拟合了均匀电子气的 Ceperley-Alder 解(这
是[Vo s k o 8 0 ]中推荐的泛函)。
LYP Lee,Yang 和Parr 的关联泛函,其中包括局域和非局域项[Lee88, Miehlich89]。
PL(Perdew Local)
Perdew 的局域(非梯度校正)泛函(1981)[Perdew81]。
P86(Perdew 86)
Perdew 的梯度校正,并加上他的 1981 局域关联泛函[Perdew86]。
PW91(Perdew/Wang 91)
Perdew 和Wang 的1991 梯度校正关联泛函[Perdew91, Perdew92, Perdew93a, Perdew96,
Burke98]。
B95(Becke 95)
Becke 的含 τ梯度校正关联泛函(定义为他的单参数杂化泛函的一部分)
[Becke96]。
PBE Perdew,Burke 和Ernzerhof 的1996 梯度校正关联泛函[Perdew96a, Perdew97]。
TPSS Tao,Perdew,Staroverov,和 Scuseria 的含 τ梯度校正泛函[Tao03]。
KCIS Krieger-Chen-Iafrate-Savin 关联泛函[Rey98, Krieger99, Krieger01, Toulouse02]。
BRC Becke-Roussel 关联泛函[Becke89a]。
- 76 -
PKZB Perdew,Kurth,Zupan,和 Blaha 泛函的关联部分[Perdew99]。
指定实际的泛函。把交换泛函部分的关键词与一个所需的关联泛函进行组合。例如,用
BLYP 关键词请求 Becke 交换泛函(B)与 LYP 关联泛函的组合。类似地,SVWN 请求 Slater
交换泛函(S)与 VWN 关联泛函,它的同义词 LSDA(局域自旋密度近似;Local Spin Density
Approximation)在文献中众所周知。LSDA 是SVWN 的同义词。有些其它具有 DFT 功能的软
件包在请求“LSDA”时,使用相当于 SVWN5 的方法。在进行比较的时候,要仔细阅读所有软
件包的文档。
关联泛函的变体。以下关联泛函结合了来自不同关联泛函的局域和非局域项:
VP86 VWN5 局域和 P86 非局域关联泛函。
V5LYP VWN5 局域和 LYP 非局域关联泛函。
独立的泛函。以下泛函是独自使用的,无需与其它泛函的关键词成分进行组合:
VSXC van Voorhis 和Scuseria 的含 τ梯度校正关联泛函[VanVoorhis98]。
HCTH/* Handy 的泛函系列,包含梯度校正关联[Hamprecht98, Boese00, Boese01]。HCTH 表示
HCTH/407,HCTH93 表示 HCTH/93,HCTH147 表示 HCTH/147,HCTH407
表示 HCTH/407。注意,未执行有关的 HCTH/120 泛函。
tHCTH Handy 泛函系列的含 τ成员[Boese02]。参见下面的 tHCTHhyb。
M06L Truhlar 和Zhao 的纯泛函[Zhao06a]。参见下面的 M06。
B97D Grimme 包含色散的泛函[Grimme06]。
杂化泛函。若干种包含 Hartree-Fock 交换与 DFT 交换-关联混合的杂化泛函,可以通过
关键词调用:
Becke 三参数杂化泛函。这些泛函具有 Becke 于1993 年设计的形式[Becke93a]:
A*ExSlater+(1-A)*ExHF +B*ΔExBecke +EcVWN+C*ΔEcnon-local
其中的 A,B,C是Becke 通过拟合 G1 分子集确定的常数。
这一杂化泛函有数种变体。B3LYP 使用 LYP 表达式提供的非局域关联,局域关联使用
VWN 泛函 III(而不是泛函 V)。注意,由于 LYP 包含局域和非局域项,实际上使用的
关联泛函是:
C*ECLYP+(1-C)*ECVWN。
换句话说,VWN 用来提供过量的局域关联需求,因为 LYP 包含的局域项基本上相当于
VWN。
B3P86 指定同样的泛函,非局域关联由 Perdew 86 提供,而 B3PW91 指定由 Perdew/Wang
91 提供非局域关联的泛函。
Becke 单参数杂化泛函。B1B95 关键词用于指定 Becke 的单参数杂化泛函,在原始文献
[Becke96]中定义。
程序还提供了其它类似的单参数杂化泛函[Becke96],由 Adamo 和Barone 实现[Adamo97]。
在一个变体 B1LYP 中,使用了 LYP 关联泛函(和上面 B3LYP 的说明相同)。另一个版
本mPW1PW91 使用了 Adamo 和Barone 改进的 Perdew-Wang 交换,并与 PW91 关联组
合[Adamo98];还可以使用变体 mPW1LYP,mPW1PBE,mPW3PBE。
Becke 对B97 的1998 修订[Becke97, Schmider98]。关键词是 B98,它执行参考文献[Schmider98]
中的方程 2c。
Handy,Tozer 及合作者对 B97 的修改:B971 [Hamprecht98]。
Wilson,Bradley 和Tozer 对B97 的修改:B972 [Wilson01a]。
- 77 -
Perdew,Burke 和Ernzerhof 的1996 纯泛函[Perdew96a, Perdew97],由 Adamo 做成杂化泛函
[Adamo99a]。关键词是 PBE1PBE。这个泛函使用 25%的交换和 75%的关联的权重,也就
是文献中所说的 PBE0。
HSEh1PBE:完 全 Heyd-Scuseria-Ernzerhof 泛函的推荐版本,文献中称为 HSE06 [Heyd04,
Heyd04a, Heyd05, Heyd06, Izmaylov06, Krukau06, Henderson09]。还可以用两个早期的形式:
HSE2PBE:该泛函的第一种形式,文献中称为 HSE03。
PBEh1PBE:使用 1998 修改形式 PBE 纯泛函(交换与关联)的杂化泛函[Ernzerhof98]。
O3LYP:类似于 B3LYP 的三参数泛函。
A*EXLSD+(1-A)*EXHF+B*ΔEXOPTX+C*ΔECLYP +(1-C)ECVWN
其中 A,B,和 C由Cohen 和Handy 在参考文献[Cohen01]中定义。
TPSSh:使用 TPSS 泛函的杂化泛函[Tao03]。
BMK:Boese 和Martin 的含 τ杂化泛函[Boese04]。
M06:Truhlar 和Zhao 的杂化泛函[Zhao08]。还可以使用变体 M06HF [Zhao06b, Zhao06c]和
M062X [Zhao08],以及早期版本 M05 [Zhao05]和M052X [Zhao06]。
X3LYP:Xu 和Goddard 的泛函[Xu04]。
两者各半泛函:执行下面的泛函。注意它们与 Becke 建议的“两者各半”泛函[Becke93]
不同。包含这些泛函仅用于向下兼容。
BhandH:0.5*ExHF+0.5*ExLSDA+EcLYP
BHandHLYP:0.5*ExHF+0.5*ExLSDA+0.5*ΔExBecke88+EcLYP
长程校正泛函。交换泛函的非库伦部分通常衰减过快,在长距离处变得非常不准确,因
此不适合模拟电子激发到高能轨道一类的过程。于是设计了各种方案处理这类情况。
Gaussian 09 提供以下包含长程校正的泛函:
LC-wPBE 长程校正版的 wPBE [Tawada04, Vydrov06, Vydrov06a, Vydrov07]。
CAM-B3LYP
Handy 及合作者的长程校正版 B3LYP,使用了库伦衰减方法[Yanai04]。
wB97XD 来自 Head-Gordon 及合作者的最新泛函,其中包含了经验色散[Chai08a]。还可
以使用变体 wB97 和wB97X [Chai08]。这些泛函也包含了长程校正。
此外,还可以为任何纯泛函添加前缀 LC-,使用 Hirao 及合作者的长程校正[Iikura01]:例
如,LC-BLYP。
用户定义模型。Gaussian 09 可以使用具有下列一般形式的任何模型:
P2ExHF + P1(P4ExSlater + P3ΔExnon-local) + P6Eclocal + P5ΔEcnon-local
目前可用的局域交换模型只有 Slater(S),当只需要局域交换时必须使用它。也可以
使用任何可进行组合的非局域交换泛函,和可进行组合的关联泛函(见前面的列表)。
可以用多种非标准选项为程序指定公式中的六个参数值:
IOp(3/76=
mmmmmnnnnn
)设定 P1为 mmmmm/10000,P2为 nnnnn/10000。通常 P1设为 0.0
或 1.0,要看是否需要使用交换泛函而定。由 P3和P4完成换算。
IOp(3/77=
mmmmmnnnnn
)指定 P3为 mmmmm/10000,P4为 nnnnn/10000。
-
- IOp(3/78=
mmmmmnnnnn
)指定 P5为 mmmmm/10000,P6为 nnnnn/10000。
例如,IOp(3/76=1000005000)设置 P1为1.0,P2为 0.5。注意所有的值必须用五个数字
表示,并在前面加入必要的零。
这是一个执行路径部分,指定的泛函对应于 B3LYP 关键词:
- 78 -

# BLYP IOp(3/76=1000002000) IOp(3/77=0720008000) IOp(3/78=0810010000)
在输出文件显示了使用的值:
IExCor= 402 DFT=T Ex=B+HF Corr=LYP ExCW=0 ScaHFX= 0.200000
ScaDFX= 0.800000 0.720000 1.000000 0.810000
其中ScaHFX的值是P2,对ScaDFX给出的一系列数值是P4,P3,P6,和P5。
精度的考虑
DFT 计算在 Hartree-Fock 计算的每一主要阶段上添加一个另外的步骤。这一步是泛函
(或各种泛函的导数)的数值积分。因此除了来源于 Hartree-Fock 计算的数值误差(积分
的精度,SCF 收敛,CPHF 收敛)之外,DFT 计算的精度还依赖于数值积分中使用的网格点数。
Gaussian 09 默认为“精密的”积分网格(对应于 Integral=FineGrid)。该网格以最
少的额外耗时最大限度地提高计算精度。不推荐在正式的 DFT 计算中使用更疏的网格。还要
注意,在比较能量时(如计算能量差,生成热,等),所有计算需要使用相同的积分网格。
需要的话可以使用更密的网格(如对某些分子体系进行严格的几何优化计算)。可以在
执行路径中用 Int(Grid=
N
)选取其它的积分网格(详见 Integral 关键词的说明)。
适用性
能量,解析梯度和解析频率;ADMP 计算。
三阶特性,如超极化率和拉曼强度,不能用于未执行三阶导的泛函:交换泛函有 Gill96,
P(Perdew86),BRx,PKZB,TPSS,wPBEh,PBEh;关联泛函有 PKZB 和TPSS;杂化泛函有
HSE1PBE 和HSE2PBE。
有关的关键词
IOp,Int=Grid,Stable,TD, DenFit
例子
DFT 计算得到的能量以类似于 Hartree-Fock 计算的形式打印。以下是 B3LYP 计算的能
量输出:
SCF Done: E(RB+HF-LYP) = -75.3197099428 A.U. after 5 cycles
其中 E 后面括号内的项表示计算能量使用的方法。BLYP 计算的输出采用类似的标记方式:
SCF Done: E(RB-LYP) = -75.2867073414 A.U. after 5 cycles
可用泛函的快速参考
组合形式 独立的泛函
交换 关联 只有交换 纯泛函 杂化泛函
S VWN
HFS VSXC B3LYP
XA VWN5
XAlpha HCTH B3P86
B LYP
HFB HCTH93 B3PW91
PW91 PL
HCTH147 B1B95
mPW P86
HCTH407 mPW1PW91
G96 PW91
tHCTH mPW1LYP
PBE B95
M06L mPW1PBE
- 79 -

O PBE
B97D mPW3PBE
TPSS TPSS
B98
BRx KCIS
B971
PKZB BRC
B972
wPBEh PKZB
PBE1PBE
PBEh VP86
B1LYP
V5LYP
O3LYP
BHandH
长程关联 BHandHLYP
LC- BMK
M06
M06HF
M062X
tHCTHhyb
HSEh1PBE
HSE2PBE
HSEhPBE
PBEh1PBE
wB97XD
wB97
wB97X
TPSSh
X3LYP
LC-wPBE
CAM-B3LYP
DFTB和DFTBA
说明
请求基于密度泛函的紧束缚半经验计算,一种对DFT计算结果参数化的方法。DFTB使用
矩阵元列表,与Elstner及合作者的原执行[Porezag95, Elstner98]相同;DFTBA是对矩阵元使用解
析式的非列表版本[Zheng07]。关于评论文章和校正的研究,见[Frauenheim00, Frauenheim02, Zheng05,
Sattelmeyer06, Otte07]。
选项
这些方法没有内部存储的参数,所以必须指定以下选项之一。
ReadParameters
从输入串读取参数值。
ChkParameters
从检查点文件读取参数值。
适用性
- 80 -

能量,梯度和频率。
DFTBA 已对 H,C,N,和 O之间的所有原子对实现了参数化。它还对金属 Sc,Ti,Fe,
Co,和 Ni 实现了参数化,但仅能结合 H,C,N,O。也就是说,支持 Fe5CO 和Sc5CO,但
不支持 Fe4ScCO。DFTB 参数文件是由 Elstner 教授版权所有,必须从他那里获得。
例子
以下输入文件格式用Gaussian 09提供的参数集运行DFTBA计算:
# DFTBA OPT FREQ
Ala3 DFTB frequencies
0,1
C,0,-4.5929012011,1.0163256276,1.6498020765
O,0,-5.6641782096,0.9622594116,2.2369288649
H,0,-5.788876035,3.2375262156,-2.1703220199
N,0,-4.4446298947,1.4038535552,0.3517633631
分子说明部分继续...
@GAUSS_EXEDIR:dftba.prm
对于DFTB,使用和其它程序相同的参数文件格式:每一对元素一个文件,两个元素的
顺序是有意义的。相应地,H2CO计算将使用类似于下面的参数输入部分:
@cc.prm
@oo.prm
@hh.prm
@co.prm
@oc.prm
@ch.prm
@hc.prm
@oh.prm
@ho.prm
出现在输出中的DFTB能量如下:
SCF Done: E(RDFT-SCTBA) = -33.9465130617 A.U. after 11 cycles
Dreiding
参见分子力学方法的说明。
EOMCCSD
说明
请求用 EOM-CCSD 方法[Koch90, Stanton93, Koch94a, Kallay04]进行激发态计算。EOM-CCSD 是对
CCSD 的扩充,用于模拟激发态。它为激发态计算提供 CCSD 级别的精度,并需要大体相当的
- 81 -
计算量(像 CCSD 一样,标度都是 N6)和附加磁盘空间。这个方法在一开始用 CIS 计算产生
激发态的初始猜测,之后进行 EOM-CCSD 分析。
注意:EOM-CCSD 计算使用阿贝尔群(不能使用更高的点群)。
选项
只能用 NState 和NStPIR 其中的一个指定所需态数。如果指定了两者,则 NState 优先。
如果都没指定,默认为 NStPIR=2。
NStates=
N
在 EOM 中,尝试求解最低的
N
个态。考虑到 CIS 和 EOM 之间可能存在激发态
重新排序,好的办法是把
N
设定为大于所需的态数。
NStPIR=
K
在 EOM 中,对每一对称类型求解的态数。默认是 2。注意,对称类型对应于
最大的阿贝尔子群。
如果
K
小于零,则读取一个由空行终止的输入部分,在其中指定每个对称类
型(不可约表示)的态数。要快速确定对称性顺序,可以用链接 0 命令%KJob
L301 运行一个预备计算。建议同时对 CIS 指定合理的态数 NCISState(见下)。
Singlets 求解单重激发态。该选项只影响闭壳层体系的计算,并且对这种情况是默认
的。
Triplets 求解三重激发态。该选项只影响闭壳层体系的计算。如果同时求解单、三态,
必须同时指定 Singlets。
NCISState=
M
用 CIS 产生猜测的总态数。默认对 NState 是
N*Irr.Reps.
,对是
(K+2)*
Irr.Reps.
。
Root=
N
指定“感兴趣的态”。默认为第一激发态(
N
=1)。
Convergence=
N
设定计算的收敛,对能量是 10
-N
,对波函是 10
-(N-2)
。默认为
N
=7。
CCConvergence=
N
用10
-
N
作为 CCSD 和基态 Z 矢量迭代的收敛。CCSDConvergence 是该选项的同
义词。默认为
N
=8。
LRTransitionDensities
除了 EOM 型(非松弛)跃迁密度外,还请求计算线性响应跃迁密度[Koch90,
Koch94a, Kallay04]。这个算法比默认的 EOM-CCSD 方法更严格,但计算量也更大。
注意,当 CCSD 提供了精确波函时(例如双电子体系),两种算法等价。只能
用于单重的闭壳层和开壳层体系。
EnergyOnly 为了节省时间,只算右本征矢。对于激发能计算足够,但不能算跃迁密度。
读写波幅有关的选项
默认保存波幅,用于随后的计算。可以选择从以前的计算中读入波幅。在随后的计算中
可以增加态数。用于产生猜测的 CIS 也读入矢量,如果需要更多的猜测,它会自动增加态数
(假设基组不变)。
SaveAmplitudes
把收敛的波幅保存到检查点文件,用于随后的计算(例如,使用更大的基组)。
使用该选项会导致极大的检查点文件,但是可以显著加快后面的计算。
ReadAmplitudes
从检查点文件读取收敛的波幅(如果存在的话)。注意新的计算可以使用与
原计算不同的基组,方法(如果适用),等。
- 82 -

ReadGroundStateAmplitudes
只读入基态的(和 Z 矢量的)振幅,不读激发态振幅。当从 EOM 单态计算变
为 EOM 三态计算时,会用到该选项。ReadGSAmplitudes 是该选项的同义词。
NewCIS 当读取 EOM 波幅时,从头开始新的 CIS 计算。该选项用于读入单态,但计算
单态和三态时。如果用 ReadAmplitudes 恢复的前一个波幅计算的基组与当
前基组不同,也需要该选项。
适用性
能量。
例子
使用 EOM-CCSD。经常使用的方法是,先执行初步的、小型的 EOM-CCSD 计算,其中求解
大量的态,接下来对感兴趣的态运行更准确的计算。下面是演示这个方法的执行路径部分:
第一个计算:
%Chk=my_eom
# EOMCCSD(NStates=10,EnergyOnly)/Aug-CC-PVDZ
第二个计算:
%Chk=my_eom
# EOMCCSD(NStates=2,ReadAmplitudes,NewCIS)/Aug-CC-PVQZ
这里是 EOM-CCSD 计算的一些示例输出。下面的输出行引入结果部分
==============================================
EOM-CCSD transition properties
==============================================
接下来是跃迁电偶极矩,分为左右两部分。在每一行的最后给出偶极强度和振荡强度,
前后两部分的结果是相同的,因为偶极强度是跃迁电偶极矩的乘积。
Ground to excited state transition electric dipole moments (Au):
state X Y Z Dip. S. Osc.
1 0.0000 0.0000 -0.3969 0.1601 0.0614
2 0.0000 0.3963 0.0000 0.1638 0.0756
3 0.0000 1.3681 0.0000 1.9183 1.0604
Excited to ground state transition electric dipole moments (Au):
state X Y Z Dip. S. Osc.
1 0.0000 0.0000 -0.4034 0.1601 0.0614
2 0.0000 0.4133 0.0000 0.1638 0.0756
3 0.0000 1.4022 0.0000 1.9183 1.0604
对于每一个态,有一个单独部分列出了激发态的 CI 展开因子,以及相应的轨道阿贝尔
对称类型,分为左右两部分,接下来是激发类型:
Excited State 1: Singlet-A1 15.6603 eV 79.17 nm f=0.0614
Right Eigenvector
Alpha Singles Amplitudes
I SymI A SymA Value
4 1 6 1 0.675597
从轨道 4(占据)到 6(虚)的激发
3 4 7 4 0.122684
Beta Singles Amplitudes
- 83 -

I SymI A SymA Value
4 1 6 1 0.675597
3 4 7 4 0.122684
Alpha-Beta Doubles Amplitudes
类似的信息,但是用于双激发
I SymI J SymJ A SymA B SymB Value
4 1 4 1 6 1 6 1 -0.118378
Left Eigenvector
Alpha Singles Amplitudes
I SymI A SymA Value
4 1 6 1 0.676418
3 4 7 4 0.121856
Beta Singles Amplitudes
I SymI A SymA Value
4 1 6 1 0.676418
3 4 7 4 0.121856
Alpha-Beta Doubles Amplitudes
I SymI J SymJ A SymA B SymB Value
4 1 4 1 6 1 6 1 -0.107806
Total Energy, E(EOM-CCSD) = -74.4340926881
对感兴趣的态打印出总能量
有关的关键词
CCSD, CIS, SAC-CI
EPT
说明
这些方法关键词进行关联电子亲和力和电离势的电子传播子理论计算[Cederbaum75,
Cederbaum77, Ohrn81, vonNiessen84, Ortiz88, Ortiz88a, Ortiz89, Zakrzewski93, Zakrzewski94a, Zakrzewski95, Ortiz96,
Zakrzewski96, Ortiz97, Ferreira01, Linderberg04]。
EPT 计算默认存储<ia||bc>积分,但是可以用 Tran=Full 通过增加磁盘的使用来节省
CPU 时间,或者用 Tran=IJAB 通过增加 CPU 时间来节省磁盘。对于后一种情形,不计算电子
亲和力。
默认只计算 20 eV 以下的电离势。
可以用 ReadOrbitals 选项在输入中指定进行精确计算的起始轨道和终止轨道。默认使
用全部轨道。
这个关键词以前叫 OVGF。
选项
OVGF 使用外部价电子格林函数传播子(Outer Valence Green's Function
propagator)。这是默认的。
P3 使用 P3 传播子。
OVGF+P3 同时使用两种传播子方法。
FC 这个关键词可以使用所有的冻芯选项。完整的信息参见 FC 选项的说明。
ReadOrbitals
指定起始轨道和终止轨道进行精确计算,这一输入部分需要一个空白行作为
结束。对于自旋非限制的计算,需要分别指定 alpha 和 beta 轨道的范围(位
于同一输入行内)。
- 84 -

ForceSort
强制对中间量进行排序,即使这么做没有必要。这个选项出现在一些 G09 测
试任务中,但对正式计算没有用处。
适用性
仅能做单点能计算。
例子
对于 EPT 计算,每个轨道的结果显示如下:
Summary of results for alpha spin-orbital 6 P3:
Koopmans theorem: -0.72022D+00 au -19.598 eV
Converged second order pole: -0.61437D+00 au -16.718 eV 0.840 (PS)
Converged 3rd order P3 pole: -0.63722D+00 au -17.340 eV 0.854 (PS)
第二个输入行对指定轨道给出电离势/电子亲和能(根据轨道是否占据而定)的估算值。
电极强度用于衡量激发的难易程度,最大值是 1.0。注意轨道在输出文件中按照对称性顺序
列出(不一定按照数值大小顺序)。
ExtendedHuckel
参见 Huckel 关键词的说明。
External
说明
指定使用一个外部程序进行计算。这种机制主要是为了便于在ONIOM计算中使用外部程
序做低层计算,但也可以在用Gaussian的优化模块作几何优化计算时,使用外部程序提供函
数值和导数。
Gaussian使用标准化接口运行外部程序,对每个结构产生一个能量(以及可选的偶极矩
或力)。默认产生一个包含当前结构的文本文件,并运行一个名为Gau_External的脚本(关
于指定其他脚本的信息,见下)。用户提供的这个脚本将会:
将文本文件——简称“输入文件”——转换成其它程序的输入文件。
运行这个程序。
把结果转换为标准文本格式,用于Gaussian读取。用于Gaussian的转换文件简称为“输
出文件”。
还可以指定不同的脚本,并把脚本的名称作为External关键词的选项:例如
External=MyScript。
脚本调用
Gau_External 脚本默认传递四个参数:
$ Gau_External layer InputFile OutputFile MsgFile
- 85 -

参数的定义如下:
layer
关键字母,表示执行的计算是对真实体系(R),两层 ONIOM 的模型体系或三
层 ONIOM 的中间体系(M),还是对三层 ONIOM 的模型体系(S)。
InputFile
Gaussian 用作外部程序输入的文件名。
OutputFile
在外部程序完成后,需要读取的文件名。
MsgFile
信息文件名:如果由脚本创建该文件,那么它的内容会复制到 Gaussian 输出
文件中。
一旦获取了结果,所有这些文件会被 Gaussian 删除。
还可以为脚本加入附加参数:
External="RunTink Amber"
在这个例子中,实际命令是:
$ RunTink Amber
layer InputFile OutputFile MsgFile
指定的脚本总是把上面提到的参数作为它的最后四个参数。
输入文件格式
输入文件格式如下:
#atoms derivatives-requested charge spin
atomic# x y z MM-charge
对每个原子重复
第一行指定分子内的原子数,要计算什么导数(0=只算能量,1=一阶导数, 2=二阶导
数),分子的电荷和自旋多重度。其余的行指定每个原子的原子编号,坐标,和分子力学电
荷。
输出文件格式
输出文件是固定格式,包含以下数据(全部是原子单位):
项目 伪代码 行的格式
能量 偶极矩(
xyz
) E, Dip(I), I=1,3 4D20.12
原子的梯度(
xyz
) FX(J,I), J=1,3; I=1,NAtoms 3D20.12
极化率 Polar(I), I=1,6 3D20.12
偶极导数 DDip(I), I=1,9*NAtoms 3D20.12
力常数 FFX(I), I=1,(3*NAtoms*(3*NAtoms+1))/2 3D20.12
只有在做一阶导数或频率计算时才会出现第二部分,只有在做频率计算时才会出现最后
一部分。对于后者,Hessian按照下三角形式给出:αij,i=1到N,j=1到i。偶极矩,极化率,
和偶极导数如果没有值,可以是零。
有关的关键词
外部脚本还可以指定为ONIOM关键词的模型之一(见下面的例子)。
要运行Gaussian的独立MM程序,可以用-external切换,这会导致它以External接口使
用的格式读写数据。
例子
- 86 -

以下执行路径部分指定了外部脚本用于三层ONIOM计算的低层:
# ONIOM(B3LYP/6-31G(d):AM1:External="RunTink Amber") Opt
以下执行路径部分指定了外部脚本用于两层ONIOM任务的高精度层:
# ONIOM(External="RunCC SDT":B3LYP/6-31G(d)) Opt
ExtraBasis和ExtraDensityBasis
说明
这两个关键词表示在路径执行中指定的基组或密度拟合基组上,再(分别)加上额外的
基函数。这些基函数位于输入串的独立部分,可以使用任何有效的格式(格式的详细说明见
Gen 关键词)。
ExtraBasis的最大用处是为标准基组内未定义的元素提供基函数。它不能替代内置基组
中的定义,这样做会导致错误。这个关键词指定的所有基函数会添加到执行路径部分指定的
内置基组上。因此,Gen经常比ExtraBasis更容易使用;使用这个关键词之前先参考说明。
如果在执行路径中没有定义密度拟合基,ExtraDensityBasis会被忽略。
有关的关键词
Gen, Pseudo, GenECP, GFInput, GFPrint
例子
下面的计算对所有的碳原子使用 6-31G(d,p)基组,并添加一个弥散函数:
# HF/6-31G(d,p) ExtraBasis …
标题部分
分子说明部分
C 0
SP 1 1.00
0.4380000000D-01 0.1000000000D+01 0.1000000000D+01
****
以下任务对基组和密度拟合基组都提供附加的函数:
#p rblyp/6-31g*/dga1 extrabasis extradensitybasis 6d
HCl using the internally stored 6-31g* AO basis & DGA1 fitting set,
adding f functions to the AO basis, and f & g fitting functions
0,1
cl
h,1,1.29
! here are some extra AO polarization functions
cl 0
附加的基组函数
F 1 1.00 0.000000000000
0.7500000000D+00 0.1000000000D+01
- 87 -

****
h 0
p 1 1.00 0.000000000000
0.1612777588D+00 0.1000000000D+01
****
! here are some extra fitting functions.
cl 0
附加的密度尼和函数
f 1
1.5
g 1
1.5
****
h 0
spd 1
0.32
****
下面的任务从外部文件读入额外的基组数据:
# B3LYP/6-31++G(d) ExtraBasis
B3LYP/6-31++G(d) with extra diffuse functions
分子说明部分
@tripleplus.gbs
冻芯选项
说明
这些选项指定在后-SCF计算中冻结哪些内层轨道。Gaussian 09对程序中从前已存在的
选项新增了一些附加选项[Austin02]。
选项
FC 指定“冻芯”计算,表示内壳层排除在关联能计算之外。这是默认的计算模
式。注意,FC,Full,RW 和Window 是互相排斥的。除了第三行元素及后面
碱金属及碱土金属原子不冻结最外层
s
和
p
芯轨道之外(与 G2/G3/G4 的规定
一致),在 6-31G 和 6-311G 基组中等价于 FreezeG2,在所有其它基组中等
价于 FreezeNobleGasCore。
FreezeNobleGasCore
在后-SCF 计算中,冻结最大的惰性气体芯轨道。这个选项的同义词是
FrzNGC。
FreezeInnerNobleGasCore
在后-SCF 计算中,冻结惰性气体次大的芯轨道。也就是芯轨道的最外层还保
留在计算中。这个选项的同义词是 FrzINGC 和FC1。
FreezeG2 根据 G2 的规定冻结轨道:冻结主族元素
d
-轨道,但对于第三行元素和以后
的碱金属及碱土金属元素,最外层
sp
芯轨道仍保留在价空间中。
FreezeG3 根据 G3 的规定冻结轨道。
- 88 -
FreezeG4 根据 G4 的规定冻结轨道。
Full 指定在关联能计算中使用全部的电子。
RW “读入窗口”选项,表示保留在后 SCF 计算中的分子轨道特定信息由输入文
件提供。ReadWindow 是RW 的同义词。
这部分的输入只有一行,指定要保留在关联能计算中的起始和终止分子轨道
编号,然后是一空行。根据出现的位置,0 值表示第一个或最后一个分子轨
道。如果第一个轨道编号是负数(-
m
),表示保留能量最高的
m
个轨道;如
果后一个轨道编号为负数(-
n
),表示冻结能量最高的
n
个轨道。如果
m
是
正值且省略
n
,
n
默认是 0。如果
m
是负值且省略
n
,那么保留最高的|m|个占
据轨道和最低的|m|个虚轨道。
以下是计算C4H4的例子:
0,0 等价于 Full。
5,0 冻结 4个芯轨道并保留所有虚轨道(如果基函数有一个单
zeta 芯,这等价于 FC)。
5,-4 冻结 4个芯轨道和能量最高的 4个虚轨道。这适用于使用双
zeta 芯基函数的冻芯。
6,22 在后-SCF 计算中保留 6到22 号分子轨道。例如,C4H4有
28 个电子,如果这是闭壳层计算,则将有 14 个占据轨道,
其中有 5个被冻结,因此后 SCF 计算将使用 9个占据轨道
(6-14)和 8个虚轨道(15-22)。
-6 保留轨道 9到20。
Window=(
m
[,
n
])
执行和 ReadWindow 选项同样的功能,但是它把输入作为执行路径部分的参
数,而不是输入的数据串。
ChkWindow 从检查点文件读取前面计算中的轨道窗口。
ListWindow 从由空白行终止的输入串读入冻结的轨道列表。对于自旋非限制的计算,要
读取两个轨道列表。可以指定轨道范围,例如:2 7-10 14。
Field
说明
Field 关键词指定在计算中加入一个有限场。在 Gaussian 09 中,这个场可以是电多极
场(直到电十六极)或 Fermi 接触项。Field 需要一个参数,以下两个格式均可:
M±N 或 F(M)N
其中
M
表示电多极场,F(
M
)为原子
M
的 Fermi 接触微扰(这部分数据放在输入文件的分子说
明部分之后)。第一种格式中 N*0.0001 指定原子单位的场的大小,第二种格式中指定 Fermi
接触微扰的大小。
因此,Field=X+10 表示在 X 方向加一大小为 0.001 au 的电偶极场,而 Field=XXYZ–20
表示与默认相反的方向(由标准方位决定)加一大小为 0.0020 au 的电十六极场。类似地,
Field=F(3)27 表示在原子 3 处加上 0.0027 乘以自旋密度的微扰。
注意这些因子是笛卡尔算符矩阵的因子;在解释结果时要留意正负号选择的规定。
所有的参数都是输入方位。
- 89 -

场的说明参数根据需要可放在其它选项之间。使用 Field 时禁止存档。
选项
Read 从输入串以自由格式读入 34 个电多极分量的因子。
OldRead 按照旧格式从输入串读入 35 个电多极场分量的因子(包括单极项),格式为
3D20.10 (第一个分量是电荷)。
RWF 从读写文件读取 35 个多极分量。
ERWF 仅从读写文件读取电偶极场的三个分量。
Checkpoint 从检查点文件读取 35 个多极分量。Checkpoint 的同义词是 Chk。当使用
Geom=Check 时,Checkpoint 是默认的。
NoChk 禁止从检查点文件读取场。
EChk 仅从检查点文件读取电偶极场的三个分量。
适用性
单点能,几何优化,Force 和Scan 计算。
限制
注意,如果在 GVB 计算中使用分子对称性,并且选择的场破坏了分子对称性,有限场可
能不会得到正确的数值导数。为安全起见,在同时使用 Field 和GVB 时,应使用
Guess=NoSymm。
例子
在有电场存在的情况下进行几何优化,必须用Opt=Z-Matrix NoSymm关键词,并用传统
的Z矩阵坐标或带符号的笛卡尔坐标定义输入结构。以下是使用Z矩阵的例子:
# RHF/3-21G Field=x+60 Opt=Z-Matrix NoSymm
Z-Matrix optimization
0 1
C
H 1 B1
H 1 B2 2 A1
H 1 B3 2 A2 3 D1
H 1 B4 2 A3 3 D2
B1 1.070000
B2 1.070000
B3 1.070000
B4 1.070000
A1 109.471203
A2 109.471203
A3 109.471231
D1 120.000015
D2 -119.999993
下面这个例子用带符号的笛卡尔坐标:
# HF/6-31G(d) Opt=Z-Matrix Field=z-50 NoSymm
Symbolic Cartesian coordinates optimization
- 90 -
0 1
O 0 x1 y1 z1
H 0 x2 y2 z2
H 0 x3 y3 z3
x1=0.0
y1=0.0
z1=0.12
x2=0.0
y2=0.75
z2=-0.46
x3=0.0
y3=-0.75
z3=-0.46
FMM
说明
如果可能的话,强制使用快速多极方法[Greengard87, Greengard88, Greengard94, Burant96, Strain96,
Burant96a, Millam97, Izmaylov06]。在 Gaussian 09 中自动使用 FMM。即使效率只能得到中等的改善(例
如,1.2 倍),Gaussian 09 一般也会启用 FMM 功能。如果分子没有对称性,对超过 60 个原
子的分子的 Hartree-Fock 和 DFT 计算会使用 FMM。对于高对称性的分子,超过 240 个原子
的 Hartree-Fock 和杂化 DFT 计算,以及超过 360 个原子的纯 DFT 计算,会使用 FMM。具有
较低(但不是没有)对称性的分子,会使用适中的阈值。对于两倍大小尺寸的体系,可以获
得更大的效率改善(两倍或更高)。
当然,结果随实际情况而变(密集体系的速度提高最少,延展开的线性体系提高最大)。
但是当对计算效率有负面的影响时,默认几乎不会使用FMM,而当可以提高到1.5倍或更高的
速度时,默认的选项很少失败。因此,用户几乎不需要对FMM进行人工控制,除非是对某些
特殊情况,例如线形的多肽和很长的碳纳米管。
FMM的选项说明位于附录中程序开发有关的关键词部分。
适用性
HF、纯 DFT 和杂化 DFT 方法的能量,梯度和频率。这个关键词还可用于 ONIOM 层内
的方法说明。
Force
说明
这个计算类型关键词指示在原子核处进行力(即能量梯度)的单个计算。同时也计算偶
极矩(在 MP2,CC,QCI 和CI 方法中偶极矩作为能量的适当解析导数进行计算)
[Raghavachari81,
Wiberg92]。
选项
- 91 -

EnOnly 通过对能量做一次数值差分计算力。对于所有没有解析梯度的计算方法,这
是默认选项。注意,这个流程存在一些数值上的不稳定性,因此对每一情况
必需仔细选用最佳的步长。
Restart 重新开始力的数值求解。
StepSize=
N
指定用于数值差分的步长为 0.0001*N。默认的单位是 Angstrom,除非指定关
键词 Units=Bohr。默认的步长为 0.01 Å。StepSize 只能和 EnOnly 同时使用
才有效。
适用性
解析梯度可用于所有的 SCF 波函,所有 DFT 方法,CIS,MP2,MP3,MP4(SDQ),CID,
CISD,CCD,CCSD,QCISD,BD,CASSF,SAC-CI,以及所有半经验方法。对于其它的
方法,使用数值差分计算力。
例子
原子核上的力在输出文件中显示如下(这个例子来自水的计算):
***** AXES RESTORED TO ORIGINAL SET *****
-------------------------------------------------------------------
Center Atomic Forces (Hartrees/Bohr)
Number Number X Y Z
-------------------------------------------------------------------
1 8 -.049849321 .000000000 -.028780519
2 1 .046711997 .000000000 -.023346514
3 1 .003137324 .000000000 .052127033
-------------------------------------------------------------------
MAX .052127033 RMS .031211490
-------------------------------------------------------------------
Internal Coordinate Forces (Hartree/Bohr or radian)
Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J
-------------------------------------------------------------------
1 O
2 H 1 -.023347( 1)
3 H 1 -.023347( 2) 2 -.088273( 3)
-------------------------------------------------------------------
MAX .088272874 RMS .054412682
在分子标准方位计算力,这些力在打印前转换成在原始坐标(Z 矩阵)轴上的力(如在
输出中所标注的)。如果使用了内坐标,接着是对应内坐标(Z-矩阵上使用的长度和角度)
的导数结果。这两种力之后,是各自的最大值和均方根值。
Freq
说明
这个计算类型关键词用于计算力常数及其振动频率。另外还计算强度。如果可能,默认
用解析方法计算力常数(对 AM1,PM3,PM3MM,PM6,PDDG,RHF,UHF,MP2,CIS,所有的
DFT 方法,以及 CASSCF),对只有一阶导数的方法(MP3,MP4(SDQ),CID,CISD,CCD,CCSD,
BD,QCISD) 默认计算一阶数值差分,对只有能量的方法计算二阶数值差分。
- 92 -
确定能量在笛卡尔核坐标下的二阶导,并变换到质量加权坐标,接下来计算振动频率。
这一变换只对驻点的计算有效!因此,对计算频率所用的方法非驻点的结构,进行频率计
算是毫无意义的。
例如,在 6-31G(d)优化的结构用 6-311G(d)计算频率将会产生无意义的结果。同样,在
用全部电子关联方法优化的结构上,用冻芯关联方法计算频率,或是相反,都是不正确的。
推荐的方法是在前一个几何优化的结构上,使用同样的方法进行频率计算。这通过在任务的
执行路径部分同时指定 Opt 和Freq 自动完成。
还要注意,如果存在一个相同自旋多重度的能量较低波函,用于计算解析频率的 CPHF
(耦合微扰 SCF)方法没有物理意义。可以用 Stable 关键词测试 Hartree-Fock 和 DFT 波函
的稳定性。
频率计算的变化
在频率计算过程中,可能还会计算其它的有关特性,包括:
当计算解析频率时,自动计算极化率;当进行数值差分时(或者使用了 Freq=Numer),
要计算极化率必须用 Polar 关键词定义(例如,CCSD Freq Polar)。
VCD 选项除了在 Hartree-Fock 和 DFT 级别进行简正频率分析之外,还可用于计算振动
圆二色性(VCD)的强度[Cheeseman96a]。
ROA 选项计算解析拉曼光活性强度[Helgaker94, Dukor00, Ruud02a, Barron04, Thorvaldsen08,
Cheeseman09]。
计算预共振拉曼强度可以指定 Raman 的一个选项,在执行路径中加上 CPHF=RdFreq,并
在输入文件中指定所要的频率(关于附加信息参见例子)。
含频极化率和超极化率的计算需要在执行路径内加上 CPHF=RdFreq 选项(受所用方法的
限制)。
可以用 Freq=VibRot 计算振转耦合[Califano76, Miller80, Papousek82, Clabo88, Page88, Adamo90,
Miller90, Page90, Barone03]。如果还指定了 Anharmonic [Adamo90, Barone94, Minichino94, Barone95,
Barone03],还可以沿着振动模式执行数值差分,计算零点能,非谐频率[Califano76, Miller80,
Papousek82, Clabo88, Page88, Miller90, Page90, Barone04, Barone05],和非谐振转耦合。该选项只能
用于具有解析二阶导的方法:Hartree-Fock,DFT,CIS,和 MP2。
有几个选项用于执行 Franck-Condon 和/或 Herzberg-Teller 方法的电子激发分析(见
下)。
Opt=CalcAll 关键词需要在几何优化的每一点进行解析二阶导计算。一旦完成所需的优
化,可以得到频率分析的所有必需信息。于是执行频率分析,并把频率任务的计算结果存档。
选择简正模式的输入
这些部分指定 SelectNormalModes,SelectAnharmonicModes,和
SelectFranckCondonModes 选项输入部分的格式。选择的模式在以空行做为结束的另一输入
部分指定。最初的模式列表总是空的。
不加关键词的整数和整数范围被解释为模式编号,虽然也可以用[not]mode 关键词。关
键词 atoms 和notatoms 可用于定义原子列表,分别表示包含/排除这些原子的模式。通过
[not]layer 关键词,还可以用 ONIOM 层指定原子,它接受以下的值:real 表示真实体系,
model 表示两层 ONIOM 的模型体系,middle 表示三层 ONIOM 的中间体系,small 表示三层
ONIOM 的模型体系。类似地可以用 residue 和notresidue 包含/排除原子,它接受残基名称
或编号的列表。两套关键词的作用都是对原子列表使用简化形式。
这里有一些例子:
- 93 -
2-5
包含模式 2 到 5
atoms=O
包含涉及氧原子的模式
1-20 atoms=Fe
包含模式 1 到 20,以及任何涉及铁原子的模式
layer=real notatoms=H
在低层包含重原子的模式(受默认阈值献之)
请求特定特性/分析的选项
Raman 除 IR 强度之外,还计算拉曼强度。这对 Hartree-Fock 频率计算是默认的。
还可指定用于 DFT 和 MP2 计算。对于 MP2,用偶极导数关于电场的数值差分
方法产生拉曼强度(等价于 NRaman)。
NRaman 通过解析偶极导数对电场的数值差分,计算极化率导数。这对MP2 Freq=Raman
计算是默认的。
NNRaman 用解析极化率对核坐标的数值差分方法计算极化率导数。
NoRaman 在 Hartree-Fock 解析频率计算过程中,跳过计算拉曼强度所需的额外步骤,
可节省 10-30%的 CPU 时间。
VCD 除进行简正频率分析之外,还计算振动圆二色性(VCD)强度。该选项用于
Hartree-Fock 和 DFT 方法。这个选项还计算旋光性(参见 Polar=OptRot)。
ROA 用 GIAO 计算动态解析拉曼光活性强度。这个流程需要在输入中提供一个或多
个入射光频率,用于电磁微扰计算(CPHF=RdFreq 是Freq=ROA 的默认设置)。
该选项对 Hartree-Fock 和 DFT 方法有效。NNROA 表示使用 Gaussian 03 的数
值 ROA 方法;它只是用于重复以前计算的结果。
VibRot 分析振转耦合。
Anharmonic 沿着模式进行数值差分,计算零点能和非谐频率,如果指定了 VibRot,还计
算非谐振转耦合。这个选项只能用于有解析二阶导的方法:Hartree-Fock,
DFT,CIS 和 MP2。
ReadAnharm 从输入部分读取附加参数,(根据 VibRot 或Anharmonic 选项)进行振转耦
合和/或非谐振动分析。可用的输入选项参看下面的例子。
SelectAnharmonicModes
读取输入部分,选择哪些模式用于非谐分析中的差分计算。输入部分格式的
说明见上。SelAnharmonicModes 是该选项的同义词。
Projected 对于质量加权的反应路径(IRC)上的一点,计算振动垂直于路径的投影频率。
在投影中,梯度用来计算路径的切线。注意该计算对结构和路径的精度非常
敏感[Baboul97]。因此,结构至少应定义五位有效数字。该计算在最小值处没
有意义。
HinderedRotor
在谐振分析中,要求识别内转动模式[McClurg97, Ayala98, McClurg99]。如果有模式
被识别为受阻的或自由的内转动,则校正热动力学函数。旋转群的识别可以
通过使用冗余内坐标实现。由于有些结构可能有特殊的成键方式而不能自动
识别,如过渡态,因此冗余内坐标的设置可以用 Geom=Modify 关键词进行修
改。转动涉及的金属需要经过 ReadHinderedRotor 选项(见下)读取附加的
输入部分。
如果前一计算产生的检查点文件中有力常数,可以通过指定 Freq=(ReadFC,
HinderedRotor),进行附加的振动/内转动分析。由于 Opt=CalcAll 自动对
优化的结构执行振动分析,因此也可以使用 Opt=(CalcAll,HinderedRotor)。
ReadHinderedRotor
- 94 -
读入附加的输入部分,其中包含转动能垒的截断高度(单位 kcal/mol)和可
选的周期性,转动模式的对称性编号,和自旋多重度。能垒高度大于截断值
的转动会自动冻结。如果周期值为负,则也冻结相应的转子。必须对金属所
有可转动的键提供周期性,对称性,和自旋多重度。输入部分用空行终止,
具有如下格式:
VMax-value
Atom1 Atom2 periodicity symmetry spin 根据需要重复多次
...
电子激发分析选项
以下选项用相应的方法执行电子激发分析;这些任务使用基态和激发态的振动分析,计
算两个态之间的电子跃迁振幅。基态振动信息取自当前任务(Freq 或Freq=ReadFC),激
发态的振动信息取自检查点文件,文件名由另外的输入部分提供(如果其中包含空格,则需
要把路径放入引号)。激发态计算可以来自 CIS 或 TD-DFT 的 Freq=SaveNormalModes 计算。
可以加上 ReadFCHT 选项,读取用于控制计算的附加输入部分(见下),而 SelFCModes
选项可用于选择涉及的模式。对于后者,激发态的检查点文件在典型情况下用
Freq=(SelectNormalModes,SaveNormalModes)产生,并使用同样的模式选择。
如果 Herzberg-Teller 或 Franck-Condon-Herzberg-Teller 分析中使用 CIS 频率,则必
须用数值方法计算 CIS 频率(Freq=Numer 而不能用 Freq)。这是因为在解析力常数的计算
中,不计算跃迁偶极导数。还需要在基态作相应的 HF 频率计算,可以象通常一样用解析方
法。
FranckCondon
使用 Franck-Condon 方法[Sharp64, Doktorov77, Kupka86, Zhixing89, Berger97, Peluso97,
Berger98, Borrelli03, Weber03, Coutsias04, Dierksen04, Lami04, Dierksen04a, Dierksen05, Liang05,
Jankowiak07, Santoro07, Santoro07a, Barone09](关于执行的描述,见[Santoro07, Santoro07a,
Santoro08, Barone09])。FC 是该选项的同义词。除了激发分析外,还可以做电离
跃迁分析。对于这种情况,分子说明对应于中性分子,而在输入部分命名的
附加检查点文件对应阳离子。
HerzbergTeller
使用 Herzberg-Teller 方法[Herzberg33, Sharp64, Small71, Orlandi73, Lin74, Santoro08]
(关于执行的描述,见[Santoro08])。HT 是该选项的同义词。
FCHT 使用 Franck-Condon-Herzberg-Teller 方法[Santoro08]。
Emission 表示对 Franck-Condon 和/或 Herzberg-Teller 分析模拟发射而不是吸收。这
种情况下,计算中的初态是激发态,末态是基态(但是基态和激发态的频率
数据来源仍然同上:当前任务=基态,第二个检查点文件=激发态)。
ReadFCHT 读取包含参数的输入部分,用于计算。可用的输入选项在示例后面有说明。
如果同时还指定了 ReadAnharmon,那么这个输入部分优先。
SelectFranckCondonModes
读取输入部分,选择哪些模式用于 Franck-Condon 分析中的差分。这一输入
部分格式的说明见上。如果同时还指定了 SelectAnharmonicModes,那么这
个输入部分优先,指定的模式按照通常的 Gaussian 顺序(增序),而不是按
照非谐输出部分显示的顺序。SelFCModes 是该选项的同义词。
简正模式有关的选项
- 95 -
HPModes 在频率输出中除了正常的三个数字输出外,还包括高精度格式(五个数字)
的振动频率本征矢。
InternalModes
把模式打印成冗余内坐标的位移。IntModes 是这个选项的同义词。
SaveNormalModes
把所有模式保存到检查点文件。SaveNM 是该选项的同义词。默认为
NoSaveNormalModes 或NoSaveNM。
ReadNormalModes
从检查点文件读取保存的模式。ReadNM 是该选项的同义词。默认为
NoReadNormalModes 或NoReadNM。
SelectNormalModes
读取输入,选择特定的模式用于显示。SelectNM 是该选项的同义词。默认为
NoSelectNormalModes 或NoSelectNM。AllModes 表示在输出中包含所有模式。
输入部分格式的说明见上。注意,这个选项不影响 SaveNormalModes 的运行,
它总是把所有模式保存到检查点文件。
SortModes 在输出中,按照 ONIOM 层对模式进行排序。
ModelModes 在 ONIOM 计算中,只显示涉及最小模型体系的模式。
MiddleModes
在三层 ONIOM 计算中,只显示涉及两个模型体系的模式。
PrintFrozenAtoms
在模式的输出中,默认不打印冻结原子的零位移。该选项要求列出所有的原
子。
修改分子说明的选项
ModRedundant
读入对冗余内坐标的修改(即,和InternalModes合用)。注意,在Opt Freq
计算中,对优化和模式分析使用相同的坐标,这与Opt=ModRedundant相同。
关于输入数据格式的细节,参见Opt关键词的说明。
ReadIsotopes
该选项允许指定不同于默认值(分别为 298.15 K,1 大气压,无换算,和最
丰同位素)的温度,压强,频率换算因子,和/或同位素。该选项可用于重新
进行分析,但使用不同于检查点文件数据的参数。
但是需要注意,所有这些参数也可以在执行路径部分(关键词 Temperature,
Pressure,和 Scale)和分子说明部分(Iso=
参数
)指定,如这个例子:
#T
Method
/6-31G(d)
JobType
Temperature=300.0 ...
...
0 1
C(Iso=13)
...
ReadIsotopes 输入具有下面的格式:
temp
pressure
[
scale
]
数值必须为实数。
原子 1 的同位素质量
- 96 -
原子 2 的同位素质量
...
原子 n 的同位素质量
其中的
temp
,
pressure
,和
scale
分别为所需的温度,压强,和当用于热化
学分析时可选的频率数据换算因子(默认不换算)。其余的行是分子中各原
子的同位素质量,按照分子说明部分出现的相同顺序排列。如果原子质量指
定为整数,程序将自动使用相应的真实同位素质量(例如,对 18O 指定为 18,
那么 Gaussian 使用的值为 17.99916)。
关于算法和流程的选项
Analytic 指定解析计算能量二阶导。该选项仅能用于 RHF,UHF,CIS,CASSCF,MP2
和全部 DFT 方法,它对这些方法是默认的。
Numerical 请求对能量的一阶解析导进行数值导数计算,得到能量的二阶数值导数。它
可以用于任何具有梯度的方法,并且对那些只能计算解析梯度而不能计算解
析二阶导数的方法是默认的。Freq=Numer 可以和 Polar=Numer 合用于同一个
任务步骤中。
DoubleNumer
请求对能量进行二次数值插分,产生力常数。对于没有解析梯度的方法,这
是默认且唯一的选择。EnOnly 是DoubleNumer 的同义词。
Cubic 进行解析二阶导数的数值差分,产生三阶导数。仅用于有解析频率但没有解
析三阶导的方法。
Step=
N
定义数值差分的步长为 0.0001*
N
(长度单位:埃,除非定义了 Units=Bohr)。
如果同时使用了 Freq=Numer 和Polar=Numer,
N
也用来定义电场的步长。对
于 Hartree-Fock 和关联方法的 Freq=Numer,默认是 0.001 Å,对于 GVB 和
CASSCF 的 Freq=Numer,默认是 0.005 Å,对 Freq=EnOnly 默认是 0.01 Å。
对Freq=Anharmonic 或Freq=VibRot 默认是 0.025 Å。
Restart 该选项用于在最后一个完成的结构计算后,重新开始数值频率计算。失败的
数值频率任务可以从检查点文件重新开始,只需简单地重复原始任务的执行
路径,并对 Freq=Numer 关键词添加 Restart 选项。不需要其它输入。
如果命名并保存了读写文件,也可以用 Restart 关键词从失败的任务重新开
始解析频率计算。
DiagFull 对角化整个(3
N
atoms)2力常数矩阵——包括平移和转动自由度——并报告最低
的振动频率,进行频率计算数值稳定性的测试。这个计算在简正频率分析之
前进行,因为后者将投影掉这些模式。它的输出报告最低的九个模式,其中
较高的三个对应于常规频率分析的三个最低模式。理想情况下,这个分析得
到的最低六个模式具有非常小的数值。如果它们显著非零,则可能表示平移
和转动是该体系的重要模式,需要更好的积分网格,结构未收敛,等。应对
该体系作进一步研究,以获得更准确的频率。参见下面的例子部分关于该选
项的输出。
默认为 DiagFull;NoDiagFull 表示跳过这个分析。
ReadFC 从检查点文件读取前一频率计算中获得的力常数,并使用不同温度、压强或
同位素,用最少的计算花费重新进行模式分析和热化学分析。注意,因为基
组从检查点文件读取,因此不用输入基组。如果前一任务中还指定了 Raman
选项,如果还想使用这个选项也无需再指定它。
- 97 -

TwoPoint 在计算数值导数时,在每个方向做两次位移。这是默认的。FourPoint 将做
四次位移,但只能用于链接 106(Freq=Numer)。对 Freq=DoubleNumer 无效。
NFreq=
N
请求用 Davidson 对角化求解最低的
N
个频率。目前,该选项仅能用于
ONIOM(QM:MM)模型化学。
适用性
解析频率可用于 AM1,PM3,PM3MM,PM6,PDDG,DFTB,DFTBA,HF,DFT,MP2,CIS 和
CASSCF 方法。数值频率用于 MP3,MP4(SDQ),CID,CISD,CCD,CCSD 和 QCISD。Raman 可用
于 HF,DFT,和 MP2 方法。VCD 和ROA 可用于 HF 和 DFT 方法。Anharmonic 可用于 HF,DFT,
MP2,和 CIS 方法。Freq 和NMR 现在可同时位于 HF 和 DFT 的执行路径中。
有关的关键词
Polar,Opt,Stable,NMR
例子
频率输出。频率计算输出的基本内容在《Exploring Chemistry with Electronic Structure
Methods》[Foresman96b]一书的第四章有详细说明。
在频率任务的开始,新的 Gaussian 用户经常会为看到几何优化的输出而感到惊讶:
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Berny optimization.
Initialization pass.
在频率计算的开始和结束时要执行几何优化计算的链接 103。这样做可以使二次优化步
骤用正确的二阶导进行计算。有时候优化使用近似的 Hessian 矩阵,依照通常的标准完成,
而当使用正确的二阶导时,步长实际上大于收敛标准。因为在频率计算的最后打印下一个步
长,所以可以发现这样的问题。一旦发现这个问题,可以在任务的执行路径部分用
Opt=CalcAll 代替 Freq,这将会在结构还未完全收敛时,先完成优化计算(通常,如果在
邻近驻点的位置给出完整的二阶导矩阵,那么仅需要一步附加的优化步骤),并在最终的结
构上自动进行频率分析。
在执行路径部分定义#P 会产生更多的频率计算输出。其中最重要的是极化率和超极化
率张量(它们也可以在正常打印级别任务的存档中找到)。它们分别在标准方位,以下三角
和下四面体的顺序出现(即,αXX,αXY,αYY,αXZ,αYZ,αZZ 和βXXX,βXXY,βXYY,βYYY,
βXXZ,βXYZ,βYYZ,βXZZ,βYZZ,βZZZ):
Dipole = 2.37312183D-16 -6.66133815D-16 -9.39281319D-01
Polarizability= 7.83427191D-01 1.60008472D-15 6.80285860D+00
-3.11369582D-17 2.72397709D-16 3.62729494D+00
HyperPolar = 3.08796953D-16 -6.27350412D-14 4.17080415D-16
5.55019858D-14 -7.26773439D-01 -1.09052038D-14
-2.07727337D+01 4.49920497D-16 -1.40402516D-13
-1.10991697D+01
对个别情况,#P 也可以用来产生模拟光谱的条线图。
在频率和模式数据之后,是热化学分析。
Zero-point correction= .023261 (Hartree/Particle)
Thermal correction to Energy= .026094
Thermal correction to Enthalpy= .027038
Thermal correction to Gibbs Free Energy= .052698
- 98 -

Sum of electronic and zero-point Energies=-527.492585 E0=Eelec+ZPE
Sum of electronic and thermal Energies= -527.489751 E=E0+Evib+Erot+Etrans
Sum of electronic and thermal Enthalpies=-527.488807 H=E+RT
Sum of electronic and thermal Free Energies=-527.463147 G=H-TS
列出了原始零点能校正,以及总能量、焓、吉布斯自由能(全部包含了零点能)的热校
正,之后是相应的校正后的能量。分析使用了正则系综的理想气体标准展开。细节见
McQuarrie[McQuarrie73]以及其它标准的统计力学参考书。在输出中,各种量的标记如下:
E (Thermal)
对热能校正的贡献
CV
恒容摩尔热容
S
熵
Q
配分函数
除了自由转动和平移模式之外,热化学分析处理谐振的所有模式。对于有受阻内转动的
分子,这会对室温下的能量和热容产生微小的误差,并且对熵有显著影响。频率非常低的振
动模式贡献单独列出,因此如果它们是群转动,并且需要高精度计算,它们的谐振贡献可以
从总能量中减去,再加上正确计算的贡献。受阻内转动对这些项产生贡献的表达式见 Benson
[Benson68]。另外还计算了配分函数,以及作为参考的振动势阱最低点和最低(零点)振动态。
预共振拉曼。这个计算类型指定 Raman 的一个选项,并与 CPHF=RdFreq 结合使用。后
者指定的频率应当用以下方式选择:
确定拉曼光谱中感兴趣的峰位和实验入射光的频率差。
用DFT方法进行TD计算,预测同一峰位的位置。
指定CPHF=RdFreq计算所用的频率,这个频率是预测的峰位加上入射光与观测峰之间的
差值。
预共振拉曼的结果出现在简正频率表格中新增加的行中:
Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman
scattering activities (A**4/AMU), depolarization ratios for plane
and unpolarized incident light, reduced masses (AMU), force constants
(mDyne/A), and normal coordinates:
1
B1
Frequencies -- 1315.8011
Red. masses -- 1.3435
Frc consts -- 1.3704
IR Inten -- 7.6649
Raman Activ -- 0.0260
Depolar (P) -- 0.7500
Depolar (U) -- 0.8571
RamAct Fr= 1-- 0.0260
附加输出行从这里开始
Dep-P Fr= 1-- 0.7500
Dep-U Fr= 1-- 0.8571
RamAct Fr= 2-- 0.0023
Dep-P Fr= 2-- 0.7500
Dep-U Fr= 2-- 0.8571
振-转耦合输出。如果指定了 VibRot 选项,则在输出文件的正常热化学分析输出之后,
立即打印简谐振转耦合的分析,开头的标题是:
Vibro-Rotational Analysis at the Harmonic level
如果同时还指定做非谐分析(即,同时指定了VibRot和Anharmonic),则在简谐振转
- 99 -

耦合分析之后,输出非谐振转分析的结果,开头的标题是:
2nd order Perturbative Anharmonic Analysis
非谐频率计算。在正常的频率输出之后,打印 Freq=Anharmonic 计算产生的附加输出。
(如果还指定进行振转耦合计算,则数据打印在振转耦合的输出之后)。这里我们简要讨论
其中最重要的项。
输出中显示平衡结构(即,势能曲面的最低点),接着是0 K时的非谐振动平均结构:
Internal coordinates for the Equilibrium structure (Se)
Interatomic distances:
1 2 3 4
1 C 0.000000
2 O 1.206908 0.000000
3 H 1.083243 2.008999 0.000000
4 H 1.083243 2.008999 1.826598 0.000000
Interatomic angles:
O2-C1-H3=122.5294 O2-C1-H4=122.5294 H3-C1-H4=114.9412
O2-H3-H4= 62.9605
Dihedral angles:
H4-C1-H3-O2= 180.
Internal coordinates for the vibr.aver. structure at 0K (Sz)
Interatomic distances:
1 2 3 4
1 C 0.000000
2 O 1.210431 0.000000
3 H 1.097064 2.024452 0.000000
4 H 1.097064 2.024452 1.849067 0.000000
Interatomic angles:
O2-C1-H3=122.57 O2-C1-H4=122.57 H3-C1-H4=114.8601
O2-H4-H3= 62.8267
Dihedral angles:
H4-C1-H3-O2= 180.
注意,后一结构的键长略长。非谐零点能在其各分量项之后的输出中立即给出:
ZPEharm = 6359.86859 cm-1 = 18.184 Kcal/mol = 76.081 Kj/mol
ZPEfund = 6135.92666 cm-1 = 17.543 Kcal/mol = 73.402 KJ/mol
ZPEaver = 6247.89762 cm-1 = 17.864 Kcal/mol = 74.741 KJ/mol
-1/4sumXii = 22.67024 cm-1 = 0.065 Kcal/mol = 0.271 KJ/mol
x0 = -6.63071 cm-1 = -0.019 Kcal/mol = -0.079 KJ/mol
ZPEtot = 6263.93715 cm-1 = 17.909 Kcal/mol = 74.933 KJ/mol
ZPEtot/ZPEharm = 0.98492 ZPEfund/ZPEharm= 0.96479
非谐频率出现在这个表之后,位于标记为E(anharm)的列:
Vibrational Energies and Rotational Constants (cm-1)
Mode(Quanta) E(harm) E(anharm) Aa(z) Ba(x) Ca(y)
Equilibrium Geometry 10.026637 1.293823 1.145922
Ground State 6359.869 6263.937 9.905085 1.288586 1.136128
Fundamental Bands (DE w.r.t. Ground State)
1(1) 3162.302 2990.777 9.727534 1.287879 1.133639
2(1) 1915.637 1884.683 9.913583 1.284564 1.128397
3(1) 1692.660 1657.100 9.955741 1.294044 1.133257
4(1) 1337.296 1315.965 6.861429 1.277085 1.137163
- 100 -

5(1) 3233.358 3068.112 9.809451 1.286693 1.134405
6(1) 1378.483 1355.216 12.919667 1.290780 1.130316
为了方便,还列出了谐振频率。
检查低频。完整力常数矩阵对角化(默认的 Freq=DiagFull)的输出以如下形式出现,
其中保留了转动和平移自由度:
Low frequencies --- -19.9673 -0.0011 -0.0010 0.0010 14.2959 25.6133
Low frequencies --- 385.4672 988.9028 1083.0692
这个输出来自甲醇的 Opt Freq 计算。注意那些基本为零的模式,最低的模式(忽略符
号)位于大约 14,19,和 25 波数。回到计算中,如果用严格的优化标准和更多的积分网格
(Opt=Tight Int=UltraFine),最低的模式变为:
Low frequencies --- -7.4956 -5.4813 -2.6908 0.0003 0.0007 0.0011
Low frequencies --- 380.1699 988.1436 1081.9083
现在低频模式已经相当小了,而且最低的频率也有所移动。
对于具有小波数频率的分子体系,这个分析特别重要。例如,如果打印出的最低频率位
于大约 30,而在其上大约 25 的位置有一个低频模式,那么前一个数值十分可疑(因为分子
结构甚至可能不在极小点)。
用不同的热化学参数运行频率计算。下面的两步计算任务先进行频率计算,之后使用
不同温度、压强和选择的同位素进行第二个热化学分析:
%Chk=freq
# HF/6-31G(d,p) Freq Test
Frequencies at STP
分子说明
-Link1-
%Chk=freq
%NoSave
# HF/6-31G(d,p) Freq(ReadIso,ReadFC) Geom=Check Test
Repeat at 300 K
0,1
300.0 1.0
16
2
3
...
还要注意,freqchk 工具可以使用存在 Gaussian 检查点文件中的频率数据,重新运行
热化学分析。
FREQ=READANHARM 的附加输入
这个输入从单独部分读入,可以包含以下关键词:
Fermi 还执行各向同性超精细耦合的振动平均。
PrintGeom 打印做振动平均特性计算的几何结构。
- 101 -
TolFre=
x
Fermi 共振的最小频率差(cm-1)(默认值为 10.0)。必须是实数。
DaDeMin=
x
Darling-Dennison 共振的最小频率差(cm-1)(默认值为 10.0)。必须是实
数。
TolCor=
x
Coriolis 耦合的阈值(cm-1)(默认值为 10-3)。必须是实数。
ScHarm=
x
谐振频率做线性换算的换算因子(B3LYP/6-31+G(d)为 1.0 x 10-5)。必须是
实数。默认使用来自 Scale 关键词的值。
FREQ= READFCHT 的附加输入
这个输入从单独部分读入,可以包含以下关键词:
MaxOvr=
N
在计算对应于跃迁到单激发振动态的 Franck-Condon 因子时,设定达到的最
大泛频。默认值为 20。
MaxCMB=
N
设定两个态达到的最大泛频,这两个态组合成最终态。默认值为 13。
MaxInt=
N
设定在每一类跃迁中计算的积分最大个数(单位:百万)。默认值为 100。
NoIntAn 停用 Sharp 和 Rosenstock 分析公式,它用于计算到单个泛频和到双态组合的
跃迁积分。
NoRelI00 给定的光谱范围默认相对于 I00 跃迁的能量。如果由用户给定光谱边界的绝对
能量,必须给出这个关键词。
SpecMin=
x
设定最终光电子谱的下限(单位 cm-1)。必须是实数。默认值为-1000。
SpecMax=
x
设定最终光电子谱的上限(单位 cm-1)。必须是实数。默认值为+8000。
SpecRes=
x
设定离散谱的两点之间间隙(单位 cm-1)。这个值会极大地影响计算时间,
非常低的值会极度减慢计算,特别是当 HWHM 设得很高时。必须是实数。默认
值为 8。
SpecHwHm=
x
设定用高斯函数表示的谱带半高处的半宽度(单位 cm-1)。必须是实数。默
认值为 135。
DeltaSP=
x
设定由于收敛差而终止计算的阈值。这个值应当小于 1.0(对应于非常好的
收敛)。默认为 0.0(不结束计算)。
AllSpectra 在 Gaussian 的输出中,除了最终光谱还打印每一套组合(类型)得到的光谱。
默认停用这个打印。
PrtMat=
N
一串数字,打印不同的矩阵用于积分计算的基:1表示 Duschinsky 矩阵 J,2
表示移动矢量 K,3表示 A,4表示 B,5表示 C,6表示 D,7表示 E,其中的
A,B,C,D,E是 Sharp 和 Rosenstock 的矩阵。数字的顺序不重要。默认值
为。
PrtInt=
x
设定在输出中打印哪些积分。阈值单位是 I00 强度。必须是实数。默认值为
0.01。
DoTemp 在光谱计算中包含温度。默认在 0 K 执行光谱计算。
MinPop=
x
设定做为跃迁起始点的振动态的最小布居。默认值为 0.1。
InFrS0 对于初态,强制程序使用用户提供的频率。这个频率在 Freq=ReadFCHT 选项
行之后的输入中指定。
InFrS1 对于终态,强制程序使用用户提供的频率。这个频率在 Freq=ReadFCHT 选项
行之后的输入中指定。
JDusch, JIdent
强制程序使用标准的 Duschinsky 矩阵(默认为 JDusch),或把单位矩阵做
为 Duschinsky 矩阵(JIdent)。对于后一种情况,不考虑模式的转动。默认
值为 0。
- 102 -
SclVec 进行换算矢量的计算,用初态频率的换算矢量和 Duschinsky 矩阵修改终态的
频率。如果给定这个关键词,会要求用户提供初态的频率,方式同 InFrS0。
EnerInp=
x
使用由用户提供的初态和终态之间的 ΔE,替换计算的值。必须是实数。默认
值为 0。
G1-G4 方法
G1,G2,G2MP2,G3,G3MP2,G3B3,G3MP2B3,G4,G4MP2
说明
这些方法关键词请求用 Gaussian-1(也就是常说的 G1)[Pople89, Curtiss90],Gaussian-2(G2)
[Curtiss91],Gaussian-3(G3)[Curtiss98]和 Gaussian-4(G4)[Curtiss07]方法计算非常精确的能
量。G2MP2 进行修改版的 G2,也就是 G2(MP2)的计算,它对基组展开校正使用 MP2 而不
是MP4 [Curtiss93],不仅具有接近完全 G2 方法的精度,而且节省了计算时间。G3MP3 进行做
了类似修改的 G3(MP2) [Curtiss99]。可以用 G3B3 和G3MP2B3 关键词调用 G3 的变体,其中使用
B3LYP 的结构和频率[Baboul99]。G4 和G4MP2 使用第四代 Gaussian 方法[Curtiss07, Curtiss07a]。
所有这些方法都是复合能量计算,其中对指定的分子体系作多个预定义的计算。在指定
了其中一个关键词后,自动执行所有这些计算步骤,并在输出中显示最终计算的能量值。这
些关键词不需要定义基组关键词。
在选择这些方法之前,用户应当考虑其它的高精度方法。CBS-QB3 同样精确但明显更
快,而 W1U 更准确(但更慢)。
Opt=Maxcyc=n,QCISD=Maxcyc=n或CCSD=Maxcyc=n关键词可以与这些关键词结
合使用,分别用于定义优化的最大步数和 QCISD 或CCSD 循环的最大步数。
选项
ReadIsotopes
该选项允许指定不同于默认值(分别为 298.15 K,1 大气压,无换算,和最
丰同位素)的温度,压强,频率换算因子,和/或同位素。该选项可用于重新
进行分析,但使用不同于检查点文件数据的参数。
但是需要注意,所有这些参数也可以在执行路径部分(关键词 Temperature,
Pressure,和 Scale)和分子说明部分(Iso=
参数
)指定,如这个例子:
#T
Method
/6-31G(d)
JobType
Temperature=300.0 ...
...
0 1
C(Iso=13)
...
ReadIsotopes 输入具有下面的格式:
temp
pressure
[
scale
]
数值必须为实数。
原子 1 的同位素质量
原子 2 的同位素质量
...
- 103 -

原子 n 的同位素质量
其中的
temp
,
pressure
,和
scale
分别为所需的温度,压强,和当用于热化
学分析时可选的频率数据换算因子(默认不换算)。其余的行是分子中各原
子的同位素质量,按照分子说明部分出现的相同顺序排列。如果原子质量指
定为整数,程序将自动使用相应的真实同位素质量(例如,对 18O 指定为 18,
那么 Gaussian 使用的值为 17.99916)。
Restart 从检查点文件恢复部分完成的计算。当与 ReadIso 选项同时使用时,这个选
项允许用不同的热化学参数和/或同位素选择,快速计算能量。
例子
计算的输出总结。在任务的所有组成部分输出之后,Gaussian 为这些方法打印结果的表
格。以下是 G2 计算的输出:
Temperature= 298.150000 Pressure= 1.000000
E(ZPE)= .020511 E(Thermal)= .023346
E(QCISD(T))= -76.276078 E(Empiric)= -.024560
DE(Plus)= -.010827 DE(2DF)= -.037385
G1(0 K)= -76.328339 G1 Energy= -76.325503
G1 Enthalpy= -76.324559 G1 Free Energy= -76.303182
E(Delta-G2)= -.008275 E(G2-Empiric)= .004560
G2(0 K)= -76.332054 G2 Energy= -76.329219
G2 Enthalpy= -76.328274 G2 Free Energy= -76.306897
开始是温度和压强,之后是用于计算 G2 能量的各种成分。输出的最后是在 0 K 和在指
定温度时的 G2 能量(后者包括完整的热校正,而不仅仅是零点能校正),以及(在输出的
最后一行)G2 理论预测的焓和吉布斯自由能(都用热校正的 G2 能量计算)。(注意,这个
总结部分还打印了在 G1 级别预测的同样的量)。
能量标记有如下含义(作为例子,使用 G2):
G2 (0 K) 零点校正电子能量:E0 = Eelec + ZPE
G2 Energy 热校正能量:E = E0 + Etrans + Erot + Evib
G2 Enthalpy 用G2 预测的能量计算的焓:H = E + RT
G2 Free Energy 用G2 预测的能量计算的 Gibbs 自由能:G = H - TS
在不同温度下重新进行计算。下面的两步任务演示了在不同温度下(快速)运行第二个
G2 计算的方法。这个任务先在 298.15 K 计算 G2 能量,接下来在 300 K:
%Chk=formald
# G2 Test
G2 on formaldehyde
0 1
分子说明
--Link1--
%Chk=formald
%NoSave
# G2(Restart,ReadIso) Geom=Check
300.0 1.0
同位素说明
- 104 -
Gen,GenECP
说明
在 Gaussian 内部储存了一套“标准”基组(参见本章前面的基组部分);这些基组可
以在计算的执行路径部分包含相应的关键词来调用。Gen 关键词允许在 Gaussian 计算中使
用用户自定义基组。它用在基组关键词或密度拟合基组关键词的位置。在这种情况下,需要
在输入文件(位于独立的基组输入部分)提供基组的描述。
Gen可用完全类似的方式指定其它的密度拟合基组(见例子)。
变体GenECP可用来同时读入基函数和ECP;它等价于Gen Pseudo=Read。它用在ONIOM计
算中,在ONIOM的一层内使用带有ECP的一般基组。
GFPrint 关键词可用于在输出文件中包含高斯函数表。GFInput 关键词可以把高斯函数
表打印成适合于 Gen 输入的格式。ExtraBasis 关键词可用于给标准基组添加函数。类似地,
ExtraDensityBasis 关键词可用于给标准密度拟合基组添加函数。
在本讨论的最后一部分给出了基函数的简要概述。
基组输入格式
通过在执行路径部分定义 Gen(表示一般基组),把外部基组读入 Gaussian。关键词
5D,6D,7F 和10F 用于定义使用笛卡尔函数或纯 d,f(以及更高的)函数;默认是 5D 和
7F。计算中所有的
d
-壳层必须具有相同的函数数量。类似地,
f
-和更高的壳层也必须全部
是笛卡尔函数或全部是纯函数。
定义壳层。外部基组的输入由链接 301 的 GenBas 例程控制。从基组输入部分读入的基
本信息单元是
壳层定义区
。一 个
壳层定义区
,加上纯函数或笛卡尔函数的全局说明,就包含
了定义一个壳层函数的全部必要信息。它包括一个
壳层描述
行,以及一个或多个原高斯函数
行:
Itype NGauss Sc
壳层说明行:壳层类型,原高斯函数数量,以及换算因子
α1 d1µ
原高斯函数说明:指数和收缩因子
α2 d2µ
...
αN dNµ
总共有
Ngauss
个原高斯函数行
Itype
定义壳层类型和壳层的限制,对
s
-,
p
-,
d
-,
sp
-,
f
-,
g
-等壳层可以是 S,P,
D,SP,F,G, ...。
Ngauss
对描述的壳层定义原高斯壳层的数量(即收缩度)。壳层的换
算因子由
Sc
给出(也就是说,所有原函数的指数都要乘上 Sc2)。
随后的
NGauss
个原高斯函数行定义指数 αk和收缩因子 dkµ。每一行给出一个原函数的
指数,之后是它的收缩因子(对
sp
-壳层是 s 和 p 两个收缩因子)。
还有第二种格式,它把一个壳层定义为一个 Slater 轨道的最小二乘高斯展开。这需要
一个壳层描述行,形式为 STO,
IOrb
,
NGauss
,
Sc
。
IOrb
是1S,2S,2P,2SP,3S,3P,3SP,
3D,4SP 中的一个,并指定需要对哪一个展开。注意 2SP 需要同时进行 S 和 P Slater 轨道
的最小二乘拟合,因此不同于分别指定 S 和 P 的最佳展开。
NGauss
同上。Slater 函数的高
斯展开可以使用 1 到 6 个原函数。
Sc
是换算因子,也就是进行展开的 Slater 函数指数。在
请求 STO 展开的壳层说明行之后,不需要输入原高斯函数行。
- 105 -

为原子或原子类型定义基组。人们习惯将至少一个,但经常是几个壳层,通过
中心定
义区
放在任何给定的核中心(“原子”)上。中心定义区由
中心标识行
,和给定中心上每个
壳层的
壳层定义区
构成。使用从第一到第四列的四个加号或星号作为终止行:
c1 c2
...
0
中心定义行:指定这些壳层的应用范围
IType NGauss Sc 第一个壳层定义区
α
2
d
2µ
...
αN dNµ
... 其余的壳层定义区
IType NGauss Sc 最后一个壳层定义区
α
2
d
2µ
...
αN dNµ
****
分隔符:结束中心定义区
中心标识行定义一组原子中心,把基函数放在这些原子的中心定义区,由 0 作为结束。
它可以包含一个或多个整数,用于表示分子说明部分相应的原子;更常见的做法,是包含一
组所有指定类型原子的元素符号。在一个中心标识行内,中心序号和元素符号可以任意混合。
为了帮助检测输入错误,如果中心定义区指定了分子中未出现的原子,运行就会终止。
如果中心前有一个负号(如-H),且该类型原子没有出现在分子说明部分,则简单地跳过基
组信息(这种情况下终端的 0 也被忽略)。后一语法用于创建包含文件的基组,文件中指定
了多个原子的标准基组;一旦创建这个文件,需要基组时可以在输入串中通过
包含
(@)功
能(见本章前面的说明)包含全部原子的基组。
可以在一个以上的中心定义区指定中心或原子类型。例如,在 Gaussian 09 的基组库目
录——在 UNIX 系统是$g09root/g09/basis——中,有一个指定 6-31G 为一般基组的文件
(631.gbs),另一个文件含有 d 指数,在定义 6-31G*(631s.gbs)时需要和前者同时使用。
在这两个文件中都定义了从 H 到 Cl 的每个原子,实际上它们两个都同时被包含(更常见的
是,对于分子中没有 6-31G 基组的原子,还要为这些原子加上附加的基组说明)。
Integral 关键词的基组转换选项。Integral 关键词有几个选项,用于控制是否以及如
何转换一般收缩基组,以减少原函数个数。Int=BasisTransform=
N
表示转换一般收缩基组
以减少原函数个数,并忽略因子小于等于 10-
N
的原函数。这是默认选项,其中
N
=4。
Int=ExactBasisTransform 表示转换一般收缩基组以减少原函数个数,但使用精确的转换
(不会改变计算的能量)。最后,Int=NoBasisTransform 表示不对一般收缩基组做减少原
函数个数的转换。
在 Gen 输入中使用预定义的基组。Gaussian 对一般基组输入增加了灵活性,允许包含
预定义的基组。在一个原子或原子类型的中心定义区内,整个壳层定义区可以替换成一行标
准基组关键词,表示使用预定义的基组。这种情况下,在对应于指定原子类型的特定基组中,
所有的函数将用于分子中的所有这些原子。
SDD,SHF,SDF,MHF,MDF,MWB 的形式可用于指定 Gen 基组输入中的 Stuttgart/Dresden
基组和赝势。注意,必须还要指定芯电子数。
例子
这是一部分 Gen 输入,对应于 6-31+G(d)基组:
H 0
应用于所有的氢原子
S 3 1.00
0.1873113696D+02 0.3349460434D-01
- 106 -

0.2825394365D+01 0.2347269535D+00
0.6401216923D+00 0.8137573262D+00
S 1 1.00
0.1612777588D+00 0.1000000000D+01
****
C 0
应用于所有的碳原子
S 6 1.00
6-31G 函数
0.3047524880D+04 0.1834737130D-02
0.4573695180D+03 0.1403732280D-01
0.1039486850D+03 0.6884262220D-01
0.2921015530D+02 0.2321844430D+00
0.9286662960D+01 0.4679413480D+00
0.3163926960D+01 0.3623119850D+00
SP 3 1.00
0.7868272350D+01 -0.1193324200D+00 0.6899906660D-01
0.1881288540D+01 -0.1608541520D+00 0.3164239610D+00
0.5442492580D+00 0.1143456440D+01 0.7443082910D+00
SP 1 1.00
0.1687144782D+00 0.1000000000D+01 0.1000000000D+01
D 1 1.00
极化函数
0.8000000000D+00 0.1000000000D+01
****
C 0
应用于所有的碳原子
SP 1 1.00
弥散函数
0.4380000000D-01 0.1000000000D+01 0.1000000000D+01
****
下面的 Gen 输入对分子中的碳和氢原子使用 6-31G(d,p)基组,对氟原子使用 6-31G‡基
组,并对中心编号 1(也就是 1,1-二氟乙烯分子说明部分的第一个碳原子) 添加额外的函数:
C H 0
6-31G(d,p)
****
F 0
6-31G(d',p')
****
1 0
仅对一个碳原子添加弥散函数
SP 1 1.00
0.4380000000D-01 0.1000000000D+01 0.1000000000D+01
****
下面的任务使用 Gaussian 的包含文件机制,为铬原子指定基函数:
# Becke3LYP/Gen Opt Test
HF/6-31G(*) Opt of Cr(CO)6
分子说明
C O 0
6-31G(d)
****
@/home/gwtrucks/basis/chrome.gbs/N
注意:(对于
高斯基组
)基组文件习惯上的扩展名是.gbs。
下面的例子用一般基组输入同时指定基组和密度拟合基组。
# RBLYP/GEN/GEN 6D
- 107 -

HCl: reading in 6-31g* AO basis and DGA1 fitting set.
6D is specified because the default for general basis
input is 5D but the 6-31g* basis is defined to use 6D
0,1
cl
h,1,1.29
! here are the 6-31g* basis sets for Cl and H
cl 0
S 6 1.00
0.2518010000D+05 0.1832959848D-02
0.3780350000D+04 0.1403419883D-01
0.8604740000D+03 0.6909739426D-01
0.2421450000D+03 0.2374519803D+00
0.7733490000D+02 0.4830339599D+00
0.2624700000D+02 0.3398559718D+00
SP 6 1.00
0.4917650000D+03 -0.2297391417D-02 0.3989400879D-02
0.1169840000D+03 -0.3071371894D-01 0.3031770668D-01
0.3741530000D+02 -0.1125280694D+00 0.1298800286D+00
0.1378340000D+02 0.4501632776D-01 0.3279510723D+00
0.5452150000D+01 0.5893533634D+00 0.4535271000D+00
0.2225880000D+01 0.4652062868D+00 0.2521540556D+00
SP 3 1.00
0.3186490000D+01 -0.2518280280D+00 -0.1429931472D-01
0.1144270000D+01 0.6158925141D-01 0.3235723331D+00
0.4203770000D+00 0.1060184328D+01 0.7435077653D+00
SP 1 1.00
0.1426570000D+00 0.1000000000D+01 0.1000000000D+01
D 1 1.00
0.7500000000D+00 0.1000000000D+01
****
h 0
S 3 1.00
0.1873113696D+02 0.3349460434D-01
0.2825394365D+01 0.2347269535D+00
0.6401216923D+00 0.8137573261D+00
S 1 1.00
0.1612777588D+00 0.1000000000D+01
****
! here are the DGA1 fitting sets for Cl and H
cl 0
S 1 1.00
0.2048000000D+05 0.1000000000D+01
S 1 1.00
0.4096000000D+04 0.1000000000D+01
S 1 1.00
0.1024000000D+04 0.1000000000D+01
S 1 1.00
0.2560000000D+03 0.1000000000D+01
S 1 1.00
0.6400000000D+02 0.1000000000D+01
SPD 1 1.00
0.2000000000D+02 0.1000000000D+01 0.1000000000D+01 0.1000000000D+01
SPD 1 1.00
- 108 -

0.4000000000D+01 0.1000000000D+01 0.1000000000D+01 0.1000000000D+01
SPD 1 1.00
0.1000000000D+01 0.1000000000D+01 0.1000000000D+01 0.1000000000D+01
SPD 1 1.00
0.2500000000D+00 0.1000000000D+01 0.1000000000D+01 0.1000000000D+01
000000D+01 0.1000000000D+01
****
h 0
S 1 1.00
0.4500000000D+02 0.1000000000D+01
S 1 1.00
0.7500000000D+01 0.1000000000D+01
S 1 1.00
0.1500000000D+01 0.1000000000D+01
S 1 1.00
0.3000000000D+00 0.1000000000D+01
****
如果用一般基组输入指定密度拟合基组,则需要使用像下面的的执行路径(用实际使用
的基组替换):
# RBLYP/6-31G(d,p)/Gen 6D
有关的关键词
ExtraBasis, ExtraDensityBasis, GFInput, GFPrint, Integral, Pseudo
基函数的概述
单个基函数由一个或多个原始高斯函数构成。例如,
s
-型基函数 φµ(r)是:
22
1
)( rf
N
i
i
i
edr
s
-型基函数
其中 N 是构成基函数的原函数数量,称为基函数的
收缩度
。因子 diµ称作
收缩因子
。数量 aiµ
是指数,fμ是基函数的
换算因子
。Gaussian 允许的最大收缩度是 100。
一个
壳层
是一套有共同指数的基函数φµ。Gaussian支持任意角动量的壳层:
s
,
p
,
d
,
f
,
g
,
h
等5。一个
s
-壳层包含一个单独的
s
-型基函数。一个
p
-壳层包含三个基函数pX,pY和pZ。
sp
-壳层包含四个具有共同高斯指数的基函数:一个
s
-型函数和三个
p
-函数pX,pY和pZ。
d
-壳层可以定义为包含六个二次函数(dX2,dY2,dZ2,dXY,dXZ,dYZ),或者五个“纯
d”基
6-31G 和 6-311G(d)。对于 STO-3G,碳原子有两个壳
层。
函数(d Z2–R2,dX2–Y2,dXY,dXZ,dYZ)中的任何一种。类似地,
f
-壳层可以包含 10
个三次高斯函数或 7 个“纯 f”函数。更高阶壳层的函数类似。注意在给定角动量的全部函
数中,一个壳层内的收缩因子必须相同,但是在
sp
-壳层中,s 和 p 的收缩因子可以不同。
还对每个壳层定义了换算因子。它用于换算壳层中所有原函数的指数。程序具有在两种函数
之间转换的功能[Schlegel95a]。
考虑碳原子的一系列基组 STO-3G,
一个是由三个原高斯函数构成的
s
-壳层(用最小二乘拟合 Slater 1s 轨道得到)。另
一个
sp
-壳层是用三个高斯函数对 Slater 2s 和 2p 轨道进行最小二乘拟合得到的,并加上 s
5 Gaussian 默认支持到 L=13 的角动量。通过修改编译参数重新编译,可以支持更高的角动量。但是注意,
g和g以上的原子轨道基函数使用了未优化的通用积分公式,因此若包含这样的原子轨道基函数,计算非
常慢。——译注。
- 109 -

和 p 函数具有相同指数的限制。这些展开对所有的原子都是相同的。对不同原子,每个壳层
中只有换算因子不同。对于碳原子,
1s
-和
2sp
-壳层的换算因子分别是 5.67 和 1.72。第一
行原子的 6-31G 基组具有三个壳层。一个壳层是收缩的六个原
s
-型高斯函数。第二个壳层
是三个原
sp
-壳层的组合。第三个壳层由一个
sp
-函数构成。这些函数对原子进行了优化。
对于碳的每个壳层,换算因子分别为 1.00,1.00 和 1.04,是由分子计算决定的。正像名称
暗示的那样,6-311G(d)基组有 5 个壳层:6 个原函数构成的一个
s
-壳层,分别由 3 个,1
个和 1 个原函数构成的 3 个
sp
-壳层,以及一个未收缩的
d
-壳层。所有的壳层都是未换算的
(使用单位换算因子)。
GenChk
说明
Opt Freq 中,这个关键词位于自动产生任务步骤的第二步和随后的步
骤。
子
Opt Freq计算的第二个任务步骤,用于演示GenChk关键词:
Link1: Proceeding to internal job step number 2.
---------
在复合计算,如
它用来保证所有的基组,ECP 和拟合集信息(如果有的话)都从检查点文件读取,而
对内部存储的复合计算部分保留关键词(不管它们在例程中是否修改),因此在后面的计算
中,可以在输出和存档项目中打印这些关键词。在由用户创建的执行路径中,这个关键词不
起作用。
例
以下输出来自
----------------------------------------------------
#N Geom=AllCheck Guess=Read SCRF=Check GenChk RHF/STO-3G Freq
-------------------------------------------------------------
Geom
说明
关键词定义分子说明输入的来源,关于坐标定义的选项,以及结构有关的输出。
默认
择项目选项
从检查点文件获取分子说明(包括变量)。只有电荷和多重度从输入串读取。
AllCheck 文件获取分子说明(包括变量),电荷,多重度和标题部分。因此当
Geom
从输入串读入,如前所述。Geom 可以用于定义其它的输入来源,它还可以控制打印哪
些和结构有关的信息,以及检查 Z-矩阵的分子内部连接性。Geom 关键词至少需要一个选项,
否则没有任何意义。
选
Checkpoint
例如,Geom=Checkpoint 可使后续的任务步骤从以前优化步骤的检查点文件
重新获得结构。这种操作很安全,因为如果优化失败,Gaussian 会放弃任务,
后面使用这一优化结构的计算将不再执行。如果想从检查点文件读取分子说
明,并使用冗余内坐标型的坐标修改,可以同时指定该选项和 ModRedundant
选项。
从检查点
- 110 -
使用这一选项,只需要定义执行路径和关键词所需的输入。这个选项对
Modify 无效,但可以和 ModRedundant 一起使用。
从失败的结构优化或部分结构优化恢复第 N步结构(Step=
N
对于成功的优化无效)。
ModRedundant
在执行计算前,使用冗余内坐标修改当前的结构(不考虑其坐标系)。该选项
k或Step 一起使用时,将读入两个输入部分:第一部分包含电
词的 ModRedundant 选项输入具有相同的格
Ac on n
]
Max
]]
N1,N2,N3和N4是原子序号或通配符(编号从 1开始,虚原子不计入)。
的坐标。
关的坐标。
(但不删除有关的坐标)。
前值),
H dv 矩阵中该坐标的对角元改为 dv。
值二阶导。
原 编号位 定
用于指定特殊的坐标类型(坐标类型默认由指定的原子数自动判
直角坐标。这种情况下,Val ue,Min 和Max 每个都有三组数,
B
或四个原子定义的线性弯曲,其中可选的第四个
Step=Original 获得最初的初始结构。这个选项用于从中间的点重新开始几何
优化。它必须与 Checkpoint,AllCheck 或Modify 中的一个结合使用。注意,
并不是所有的步骤都出现在检查点文件中;日志文件的 Hessian updated 信
息表示相应的步骤出现在检查点文件中。
可用于修改输入文件中的分子结构,甚至是那些非几何优化的计算类型。它
可以和 Step,Check 或AllCheck 结合使用,用于读入或修改来自检查点文
件的结构。
当它和 Chec
荷和多重度,第二部分包含读入结构的改变。当与 AllCheck 选项一起使用时,
只需要修改结构的输入部分。
修改冗余坐标的说明与 Opt 关键
式(这里仅对格式做简单介绍;完整介绍见 Opt 关键词的说明):
[
Type
] N1 [N2 [N3 [N4]]] [[+=]
Value
] [
ti
[
Params
]] [[
Mi
Val u e 对指定坐标给出新的值,+=Va l u e 用于把坐标变量增加 Val u e。Action 是
可选的字母代码,表示执行的坐标修改,后面有时需要接参数(忽略此代码
时,默认的操作是添加指定的坐标):
B 添加坐标,并构建所有有关
K 删除坐标,并去掉所有包含该坐标的有
A 在优化中激活坐标,如果这些坐标已被冻结的话。
F 在优化中冻结坐标。
R 从定义列表中删除坐标
S n stp 执行平缓的势能面扫描。设置初值为 Va lu e(或其当
坐标的增幅为 stp,总 共 n步,对每一个获得的初始结构进行
优化。
把初始 Hessian
D 在初始 Hessian 矩阵中,该对坐标的行和列计算数
子 置的星号(*)表示通配符。Min 和Max 为包含通配符的坐标说明
义范围(若 Min 未给出,则为最大值)。只有坐标值在这个范围内才执行
Action。
Type 可以
断):
X
定义 X,Y,Z坐标。
键长。
A 键角。
D 二面角。
L 由三个原子
- 111 -
原子用于确定线性弯曲的两个正交方向(N4 可以是-1)。在
这种情况下,Va lu e,Min 和Max 每个都是两个数,定义两个
正交弯曲分量。
对原子中心(N1)和三
O 个连接原子定义平面外的弯曲坐标。
改坐标选项
指定从检查点文件获取分子结构,并进行修改。它将读取两个输入部分:第
其中 variable 是分子说明中的变量名,new-value 可选,是对它赋予的新值,
Connectivity
在结构说明及其修改部分之后,通过附加的输入部分(需要空行终止)直接
er2
...]
其中的各个 N是和当前原子成键的原子,Order 是相应成键的键级。例如,
8 4 1.0 5 2.0
ModConnectivi
分子说明部分(或从检查点文件获得的结构部分)的原子连接性。该选
.]
其中 当前原子成键的原子,Order 是成键的键
ZMConnectivit
用 Z-矩阵中指定的原子编号(包含虚原子)读入原子连接关系。和虚原子
IHarmonic=
n
在最初结构上加上简谐限制,力常数为
n
/1000 Hartree/Bohr2。该选项的同义
修
Modify
一部分包含电荷和多重度,第二部分包含对读入结构的修改。
Z-矩阵坐标做几何优化计算的结构修改说明具有以下的格式:
variable
[
new-value
] [A|F|D]
最后一项是字母代码,表示变量是激活(也就是进行优化)还是冻结;字母
代码 D对这个变量执行数值差分,并自动激活变量。如果省略字母代码,则
变量的状态保持和分子说明中的原始状态相同。
指定原子成键数据。该选项对每个输入的原子需要一行,顺序和原子说明部
分相同,语法为:
N1 Order1
[
N2 Ord
这个输入指定当前原子与原子 4和5成键,键级分别为 1.0 和2.0:
这个输入部分需要用空行终止。
ty
修改
项在结构说明及其修改部分之后需要附加的输入部分(需要空行终止)。连 接
性的修改使用下面的语法:
M N1 Order1
[
N2 Order2
..
M是原子的序号,各个 N是和
级。键级-1.0 将删除这个键。例如,这个输入指定原子 8与原子 4和5成键,
键级分别为 1.0 和2.0,并删除原子 9的所有键:
8 4 1.0 5 2.0 9 -1.0
这个输入部分需要用空行终止。
y
使
有关的键级将被忽略。
词是 InitialHarmonic。
- 112 -
ChkHarmonic=
检查点文件中储存的初始结构上加上简谐限制,力常数为
n
/1000
ReadHarmonic=
(以输入的方位)添加简谐限制,力常数为
n
/1000
ReadFreeze 。在另外的输入部分(用空
st
]
/或
atoms=3-6,17 notatoms=5
把原子 3,4,6,和 17 加入原子列表
类型
没有关键词的整数表示原子编号:
1,3,5 7
添加原子 1,3,5,和 7
block 和notblock,表示包含/不包含 ONIOM
。
noatoms atoms=1-100 notatoms=H
也可以通过[not]layer 关键词,用 ONIOM 层指定原子,它接受以下的值:real
Micro 标,即是这不是结构优化计算。
出的有关选项
(对 50 个原子以下的分子是默认的)。NoDistance 禁
CAngle 断值确定成键的原子,打印原子间的夹角。默认不打印
CDihedral 印
n
在
Hartree/Bohr2。该选项的同义词是 CHarmonic。
n
从输入串读入的附加结构
Hartree/Bohr2。该选项的同义词是 RHarmonic。
读入输入部分,用于修改那些原子优化或是冻结
行结束)指定原子列表。初始原子列表包含冻结原子(即,不做优化的原子),
这依赖于分子说明部分或检查点文件读入结构的设置
输入部分使用以下格式:
atoms=
list
[notatoms=
li
其中每个
list
是用逗号或空格隔开的原子编号列表,原子编号范围,和
原子类型。可以连续使用关键词,原子列表最初是空的。这里是一些例子:
atoms=3 C 18-30 notatoms=H
加入原子 3,18-30 中所有的碳和非氢原子
atoms=C N notatoms=5
除了原子 5 外,添加所有的 C 和 N 原子
atoms=1-5 notatoms=H atoms=8-10
添加原子 1-5 中的非氢原子,
以及原子 8-10 并且不考虑原子
对于 ONIOM 优化,类似地可以用
分子说明部分定义的刚性区。如果选定的原子和区块内指定的原子有冲突,
例如包含在区块内的原子被原子类型排除,Gaussian 09 会发生错误。
可以把 noatoms 放在输入部分作为第一项,从一个空的冻结原子列表开始
例如,以下输入仅冻结原子 1-100 内的所有非氢原子:
代表真实体系,model 表示 2 层 ONIOM 的模型体系,middle 表示 3 层 ONIOM
的中间层,small 表示 3 层 ONIOM 的模型层。类似地,可以用残基包含或排
除原子,需要使用 residue 和notresidue,它们接受残基名称列表。两对关
键词都具有原子列表缩写形式的功能。
为ONIOM(MO:MM)微迭代设定冗余内坐
输
Distance 要求打印原子距离矩阵
止这一输出。
要求使用距离截
(NoAngle)。Angle 对Opt=Z-matrix 指定打印原子间的夹角(用 Z矩阵确
定哪些原子成键)。CAngle,Angle,和 NoAngle 只能定义其中的一个。
要求使用距离截断值确定原子的连接性,打印二面角。默认不打
- 113 -
(NoDihedral)。Dihedral 对Opt=Z-matrix 指定打印二面角(用 Z矩阵确定
哪些原子成键,得到连接关系)。Dihedral,Cdihedral 和NoDihedral 只能定
义其中的一个。
ent PrintInputOri
格方式给出输入方位的笛卡尔坐标。
构说明和检测选项
eepConstants 保留冻结变量的信息,而 NoKeepConstants 不保留。对 Berny
NewDefinition
生一组新的冗余内坐标,替代检查点文件中的坐标。
wRedundant
从当前笛卡尔坐标重新建立冗余内坐标。如果和 Geom=Modify 合用,在坐
Crowd
Independent ndent 激活检查,NoIndependent
Print 建功能的附加打印。
关的关键词
ead,Opt=ModRedundant
GFInput
说明
GFInput(表示“Gaussian Function Input”)把当前使用的基组以适合
于一
项
alization
Coulomb 归一化打印密度基组。这对拟合集是默认的。
ONormalization
(原子重叠)归一化打印密度拟合基组。基组总是用 AO 归一化打印。
awNormaliza
未归一化的密度拟合集。
关的关键词
以表
结
KeepConstants
K
算法,默认以符号形式保留它们,对旧的优化算法不保留(旧的优化算法不
了解其含义)。
产
Ne
标系统更新前,新的修改信息添加到早先的 Opt=ModRedundant 输入中。
Crowd 激活原子间距的检查,如果在 0.5 Å 以下就终止,而 NoCrowd 关闭
检查。默认对每个读入的结构进行检查。在后面的几何优化点,数值频率等,
如果结构是由任务产生的,默认不检查。
检查 Z-矩阵中变量的线性独立性,Indepe
关闭。该选项只有在用 Berny 算法进行完全优化(Opt=Z-matrix)时才会默
认执行。
打开模型创
有
Guess=R
产生输出的关键词
般基组输入的格式打印出来,
因此可用于添加或修改标准基组。默认打印基组和使用的
拟合集。
选
JNorm
用
A
用AO
R tion
打印
有
- 114 -
Gen,GFPrint
GFPrint
说明
输出的关键词把当前使用的基组和密度拟合基组以表格形式打印出来。变体
GFOl
关的关键词
nput
Guess
说明
Hartree-Fock 波函的初始轨道猜测。Guess 不加选项是没有意义的。
默认
于算法的选项
arris泛函[Harris85]的对角化获得初始猜测。这是对所有HF和DFT计算是
Huckel 这对 CNDO,INDO,MNDO,和 MINDO3 是默认选项。对
RdScale 7.0 倍的
OldHuckel 版 Huckel 初始猜测(Gaussian 03 以前的版本),而非 CNDO 或
INDO 默认猜测:对第一行元素是INDO,第二行元素是CNDO,第
AM1 用于稀疏矩阵代码)。Guess=(AM1,Always)
Core MM,PM6,和PDDG
于轨道的选项
SCF 计算中使用的顺
Alter 通常选择的占
这个产生
dPrint 关键词用 Gaussian 格式打印基组信息。
有
Gen,GFI
这个关键词控制
使用 Harris 猜测(见下)。
关
Harris 通过H
默认选项。
使用 Huckel 初始猜测。
于涉及多个第二行原子的 PM6 计算,应当考虑 Huckel 猜测。
读入原子硬度的换算因子,用于迭代的扩展 Huckel 方法。默认值为
QEq 值。
指示采用旧
升级的 Huckel。
使用Gaussian 98的
三行及以后的元素是Huckel。
对初始猜测做 AM1 计算(目前只适
指定对几何优化后面的几步,在每一点产生新的猜测,并对用旧点和新点的
密度计算的能量进行比较,采用比较好的一个作为猜测。
用芯哈密顿量进行对角化形成初始猜测。这对 AM1,PM3,PM3
是默认的。
关
Permute 读入初始猜测的轨道排列。产生的猜测轨道编号按照在
序给出。可以使用轨道范围(例如,7-12),没有列出的所有轨道则以原有
顺序放在列出的轨道之后。对于开壳层体系必须分别列出 α和β轨道的排列
(两列表用空行隔开)。
表明 Hartree-Fock 波函中选择的占据轨道不是能量最低轨道。
据轨道是根据初始猜测程序使用的单电子哈密顿量,其中具有最低本征值的
- 115 -
轨道。改变的部分包括一系列的轨道交换,表示用一个其它的(虚)轨道代
替一个占据轨道。每个这样的交换位于一个单独的行,由两个整数 N1和N2
构成(自由格式,通常由空格或逗号分开),表示轨道 N1和轨道 N2互换。轨
道交换列表由输入部分末尾的空行终止。
对于 UHF 计算,需要两部分这样的轨道交换,第一部分指定 α轨道交换,第
Mix 以便破坏 α-β和空间对称性。这对于产生单重态的
DensityMix[=
混合占据轨道和虚轨道的贡献,
形成初始猜测的密度。
N
默认值为-3(用
Biorthogonali
非限制性猜测,双正交化 α和β MO,使自旋相反的电子尽可能成对。当
程选项
Guess=Only 具有计算类型关键词的功能,一旦计算并打印了初始猜测,计算
组态。例如,
用于从检查点文件的数据产生布居和其它后期计算
Always 结果用
Fragment=
N
解产生猜测。猜测默认保存到检查点文件。这个选项
Local 。分别局域化占据轨道和虚轨道,(在
Sparse 能的超大 HF 或 DFT
Extra 轨道时(也就是在几何优化的过程中),进行一次额
二部分指定 β轨道交换。这两部分都是必需的。因此,即使只需要 α交换,
也要有 β部分,哪怕只留有一个空行(反之亦然)。必须包含第二个空行,表
明β交换部分是空的。
请求混合 HOMO 和 LUMO,
UHF 波函非常有用。默认只有当产生复数初始猜测时,才会进行轨道混合。
NoMix 表示不做轨道混合。
N
]
是否
Huckel 本征值确定哪些轨道需要混合)。
ze
对于
读入 UHF 波函用作 ROHF 猜测时,这将自动进行。该关键词还可以用在组合
Guess=(BiOrth,Read,Only,Save)中,用 alpha 和 beta 尽可能匹配的轨道替
换正则 UHF 轨道,用于 GaussView 或其它可视化程序包的显示。
流
Only
就终止。注意打印轨道总量的信息由 Pop 关键词控制。Guess=Only 不能用于
基于 MOPAC 的半经验方法(INDO,CNDO,MNDO,MINDO3)。
该选项对于初步运行非常有用,可用来检查是否需要改变电子
Guess=Only 可用于 CASSCF 计算,用来获得 CAS 活性空间中 CI 组态数(以及
初始轨道)的信息。
Guess(Only,Read)也可
的分析。例如,这两个选项将使用检查点文件的波函产生布居分析。
Guess(Only,Read) Prop 将使用检查点文件的波函计算静电特性。
要求在几何优化的每一点产生新的初始猜测。默认是最后一点的 SCF
作下一个点的猜测。
从分子片的猜测或 SCF
通常与 Guess=Only 合用,用于从分子片猜测轨道产生猜测(否则对每个片
段执行完全的 SCF 计算)。关于指定分子片中的原子以及分子片的电荷和多
重度的说明,参见分子说明部分,但负自旋多重度除外,它表示分子片的未
成对轨道在组合时为 β自旋轨道。
使用 Boys 方法[Boys60]进行轨道局域化
使用 LowSymm 或NoSymm 进行可能的合并之后)不可约表示不进行混合。接
下来用第二个任务步骤进行已收敛 SCF 波函的局域化轨道分析,这需要在执
行路径中包含 Guess(Read,Local,Only)和Pop=Full。
对初始猜测做稀疏 SE 计算。这个选项对使用稀疏矩阵功
计算可能会有帮助。
当从 RWF 文件读入分子
- 116 -
外的新初始猜测计算。如果默认的 Harris 猜测是允许的,不需要改变组态,
而且 ILSW 的变量 4 没有标明使用较小步幅进行几何优化的话,这是默认的。
使用 NoExtra 选项可禁止这一功能。
从读写文件或检查点文件读取以前结果Fock 时,重新使用 Fock 矩阵而不是轨道。
取/保存猜测的选项
检查点文件读取初始猜测(Guess=Read 经常和 Geom=Checkpoint 一起
Alpha ss=Read 中对 α和β初始猜测都使用 α轨道。
te 子坐标中。这是默认
Cards 替某些或全部轨道。通
α替换轨道输入部分)由一个指定 Fortran 格式
-1
=按顺序替换所有轨道)。
ec
ra β替换轨道部分的不同仅
Input
通
收敛的 ONIOM 任务,该选项很有用,可以使用像下
=InputOnly 打印各个计算的输入文件。
t SCF=QC 选项进行收
找到正确的态以后,用 Guess=Input 运行 ONIOM 计算,这样能对
如果没有指定 Guess=Alter,这是周期边界条件计算的默认选项。NoFock 禁
止这个功能,这对非周期边界条件计算是默认的。
读
Read 要求从
指定)。这一选项可与 Alter 组合,这时轨道从检查点文件读取,投影到当
前的基组上,接下来进行指定的轨道交换。Checkpoint 是Read 的同义词。
TCheck 选项表明要从检查点文件读入猜测,但如果需要的话会产生新的猜
测。
在Gue
Transla 要求把读入的用于产生猜测的原子坐标平移到当前的原
的。在极个别情况下会失败,例如当波函用于不同化学计量体系的轨道猜测
时,在这种情况下应当定义 Guess=NoTranslate。
在产生初始猜测之后,要求用从输入串读入的轨道代
过替换每个轨道,该选项可用于从输入串读入全部的初始猜测。用于替换的
轨道如果有的话,输入部分位于改变猜测的命令之后。对于 UHF,分别需要 α
和β替换轨道输入部分。
替换轨道输入部分(对 UHF 是
的行开始,用于控制替换轨道输入的读取,外面加上括号。例如:(4E20.8)。
余下的部分包含一个或多个像下面的行:
IVec 用于替换的轨道(
0
表示结束,
(
A
(
I
,
IVec
),
I
=1,
N
)
按照第一行指定格式的新轨道。
Iv
行的格式是 Fort n 格式 I5。对于 UHF 计算,
在于略去了最初的格式定义行。关于替换轨道输入的示例,参见例子部分。
从输入文件读取包含检查点文件名的一行。该选项还接受几个关键词:
generate 表示像往常一样产生猜测,read 和chk 等价于 Guess=Read(像
常一样使用%Chk 指定的检查点文件)。如果检查点文件的路径包含空格,需
要把它放入引号中。
对于 SCF 在初始结构很难
面的流程:
用ONIOM
对于 SCF 有收敛问题的步骤,另外加上 Stable=Op
敛。
在确认
特定的态得到正确的初始猜测。在 ONIOM 输入文件中,每个计算的猜测
需要有一行(即,两层 MO:MO 计算需要三行),其中每一行根据情况可
以是检查点文件名,或 read(对于读取正常检查点文件的 Guess=Read),
或generate(产生新的初始猜测)。例如,如果只有模型体系的高级计
算出问题,那么输入是:
- 117 -

generate
实际体系的低级计算
Save
rbita
Only和Read
Population关键词的说明。
int
的选项
LowSymm 要求分子点群的不可约表示与 SCF 的 N3步骤中的对称性信息相结合,用于允
许降低波函对称性。这可以使轨道(以及可能但未必的总波函)具有比完全
16I2),给出进行组合的不可约表示个数,
0分开;列表本身必须由 9作为结束。这些数值对应链接 301
NoSymm
ForceAbelianS
nSymmetry。
有效的选项组合
只有合理的组合才有效。例如,可以使用 Guess=(Always,Alter)和Guess=(Read,Alter)
等组合(对于前者,先读入一次交换,接着在每一结构做同样的交换)。相反,
Guess=(A
Guess=Only 不能用于基于 MOPAC 的半经验方法:INDO,CNDO,MNDO,和 MINDO3。
有关的关键词
Geom,Pop
例子
用 Guess=Alter 交换两个轨道。这个例子寻找氨基 2A1激发态的 UHF/STO-3G 结构。首
先,运行
HF/STO-3G
guess_for_high.chk
模型体系的高级计算
generate
模型体系的低级计算
在结束 Guess=Only 运行时,把产生的初始猜测保存到检查点文件。这一选
项可用于保存局域化轨道。
ls NaturalO
把自然轨道包含到检查点文件。必须在另外的任务步骤中与选项
同时使用才能完成。细节参见
Pr 打印初始猜测。
对称性有关
分子点群低的对称性。该选项仅用于 GVB 计算,其中对于有对称性的体系的
计算,这经常是必需的(关于使用这个选项的例子,见后面 GVB 关键词的讨
论)。
该选项需要单独一行输入(格式
与新的组之间用
在输出文件中列出的不可约表示的顺序(见下面的例子部分)。
由于这一输入部分总是一行的长度,所以不用空行结束。注意不可约表示在
轨道局域化之前进行组合,因此局域化后的轨道都保持原来的对称性。
Guess=NoSymm 删除所有的轨道对称性限制,不需要读入任何输入。
消除所有的轨道对称性限制。与 SCF=NoSymm 和Symm=NoSCF 是等价的。
ymmetry
强制将初始猜测的轨道根据阿贝尔点群的不可约表示进行转换。默认是
NoForceAbelia
lways,Read)是矛盾的,将会导致不可预知的结果。参见在本章开始部分的输入顺
序表,以决定组合选项在输入部分的顺序,例如 Guess=(Cards,Alter)。
限制
Guess=Only 计算以决定是否必须作轨道交换以获得需要的电子态。默认使用
理论模型:
# Guess=Only Test
- 118 -

Amino radical test of initial guess
nh
hnh
1.03
0 2
n
h 1
h 1 nh 2
nh
hnh 120.0
这里是该任务的轨道对称性部分的摘要输出,它在输出的布居分析之前:
Initial guess orbital symmetries:
A1) (A1) (B2) (A1) (B1)
A1) (A1) (B2) (A1)
ial guess is 2-B1.
<S**2>= 0.7500 S= 0.5000
Alpha Orbitals:
Occupied (
Virtual (A1) (B2)
Beta Orbitals:
Occupied (
Virtual (B1) (A1) (B2)
The electronic state of the init
Initial guess <Sx>= 0.0000 <Sy>= 0.0000 <Sz>= 0.5000
由于涉及二重态,因此分别给出 α和β轨道。通过轨道对称性,初始猜测的电子组态是
a2a
1 1
2b2
2
要改变初始轨道组态:一个 β电子必须从轨
道 4
a1
2b1
1,产生 2B1波函。这实际上是 NH2的基态。打印了非限制初始猜测的期待值
S2。本例中,它对应于纯二重态的值 0.75。
由于我们想要模拟氨基的 2A1激发态,就需
移到轨道 5(电子组态变成 a1
2a1
2b2
2b1
2a1
1
入:
# UHF/6-31G(d) Opt Guess=Alter
no radical: HF/6-31G(d)
nh
hnh
:
空行结束分子说明部分
例为空)
4
)。
这可以用 Guess=Alter 完成。这是几何优化的输
Ami
Structure of 2-A1 state
0 2
n
h 1
h 1 nh 2
Variables
nh 1.03
hnh 120.0
空行结束 alpha 部分(本
4 5
交换 beta 轨道 和 5
结束 beta 轨道交换部分
注意,额外的空行——第 12 行——是必须的,表明这是一个空的 α交换部分。最后的
两行
果,初始猜测程序打印出一列交换轨道列表:
Harris functional with IExCor= 205 diagonalized for initial guess.
pha orbitals switched.
hed:
构成β交换部分。
作为 Alter选项的结
...
No Al
Pairs of Beta orbitals switc
4 5
- 119 -

Initial guess orbital symmetries:
A1) (A1) (B2) (A1) (B1)
A1) (B2) (A1) (A2) (A1)
A1) (A1) (B2) (B1)
A1) (B2) (A1) (B2) (A1) (A2)
is 2-A1.
<S**2>= 0.7500 S= 0.5000
Alpha Orbitals:
Occupied (
Virtual (A1) (B2) (B1) (A1) (B2) (
(B1) (A1) (B2) (A1)
Beta Orbitals:
Occupied (
Virtual (A1) (A1) (B2) (B1) (
(A1) (B1) (A1) (B2) (A1)
The electronic state of the initial guess
Initial guess <Sx>= 0.0000 <Sy>= 0.0000 <Sz>= 0.5000
之后是 SCF 计算,打印了 UHF 波函的能量和 S本征值。如果消除了来自下一可能自旋
多重 的
SCF Done: E(UHF) = -55.4915172451 A.U. after 12 cycles
pin contaminant:
0.7500
2
度(四重态对二重态,五重态对三重态,等) 波函污染,则也打印这个 S2值:
Convg = 0.2693D-08 -V/T = 2.0038
<Sx>= 0.0000 <Sy>= 0.0000 <Sz>= 0.5000 <S**2>= 0.7534 S= 0.5017
<L.S>= 0.000000000000E+00
Annihilation of the first s
S**2 before annihilation 0.7534, after
虽然这个计算结果正确地收敛到了 A1态,但在 SCF 迭代中有时会发生轨道对称性的顺
序发
用Guess=Permute重新排列轨道顺序。这个选项通常是把初始猜测进行复杂修改的最容
易方
# CASSCF/6-31G(d,p) Opt Guess=Permute Pop=Reg
job
说明
65 63 64 66 68 67 61-62 69
定义新顺序
2
生交换。如果最终波函的轨道对称性与在初始猜测中不同(不管 Guess=Alter 使用与
否),我们推荐用 SCF=QC 或SCF=DM 关键词指定直接最小化例程,这通常可以把对称性从一
次迭代保留到下一次迭代。
式,例如这个例子:
CAS
0 1
分子
1-60
这里重新排列轨道61-68。实际上并不需要列出最后的轨道(69),但有助于了解输入。
用 Guess=Cards 读取轨道。在产生了初始猜测后,可以用 Guess=Cards 替换部分或全
部轨
(3E20.8)
809834509E+00 0.4612416518E+00 -0.6437319952E-04
700779642E-13 0.1240395916E-12 -0.3110890228E-12
道。这里是该选项输入的例子,替换轨道 1 和 4(注意,在第一行指定了第三行和以后
各行的格式):
1
0.5
0.1724432549E-02 0.1282235396E-14 0.5417658499E-13
0.1639966912E-02 -0.9146282229E-15 -0.6407549694E-13
-0.4538843604E-03 0.6038992958E-04 -0.1131035485E-03
0.6038992969E-04 -0.1131035471E-03
4
0.7
-0.4479190461E-12 -0.1478805861E-13 0.5807753928E+00
0.6441113412E-12 -0.3119296374E-14 0.1554735923E+00
-0.1190754528E-11 0.2567325943E+00 0.1459733219E+00
- 120 -
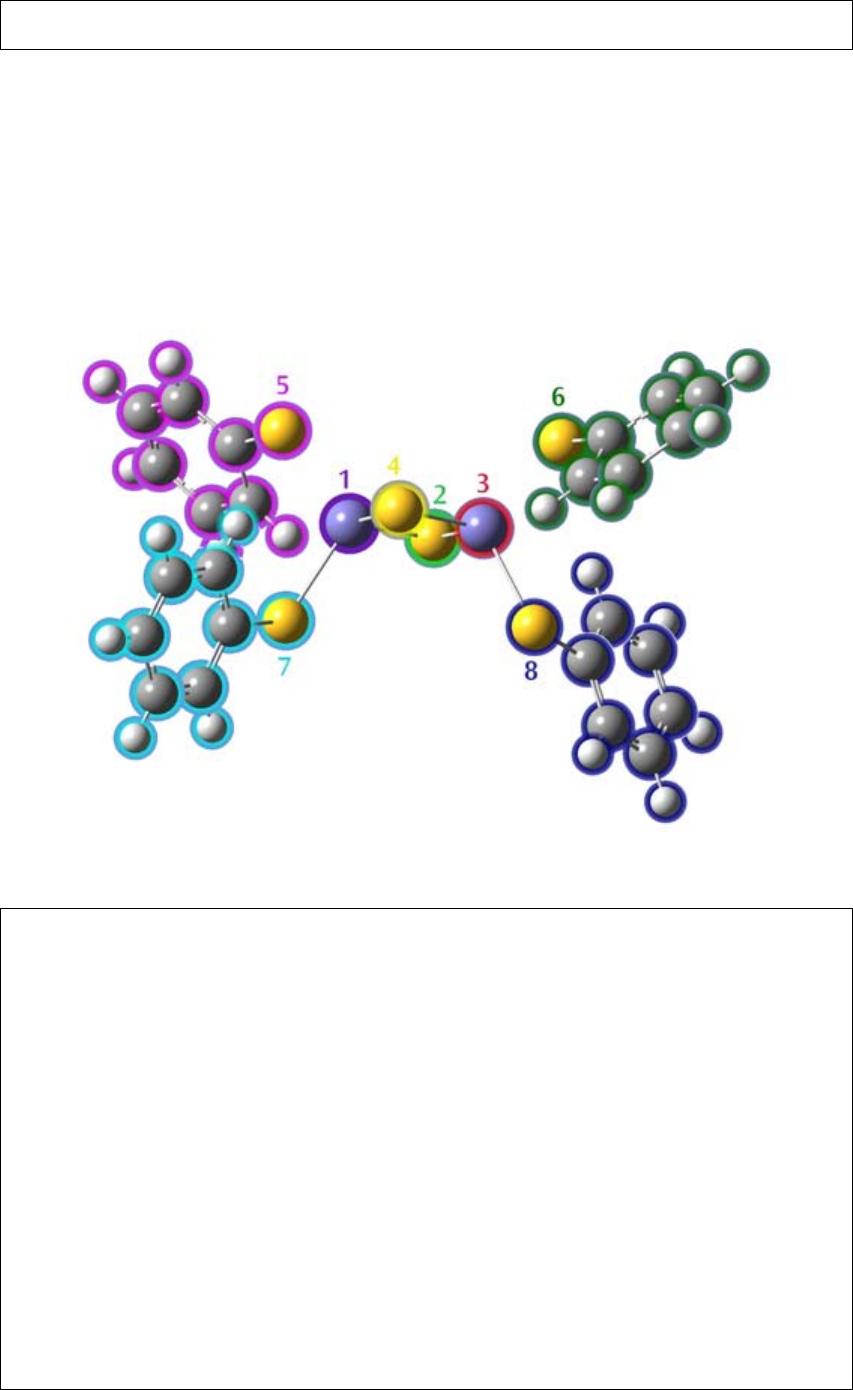
-0.2567325943E+00 -0.1459733219E+00
0
轨道编号 0 结束替换轨道的输入。
反铁磁耦合。这里是一个分子片猜测任务的例子。第一步产生每个分子片的猜测,接下
来把
是S(
它们组合到一起,构成整个分子SCF计算的猜测。第二步读入这个猜测用于随后的计算。
电荷和多重度的行总是整个分子的电荷和多重度在前,之后是每个分子片的电荷和多重度。
本例是Fe2S2 + 4 S-R配体,其中R是苯基。它是单重态,整体电荷为-2。硫原子的猜测
2-)闭壳层(分子片2和4),两个铁为Fe(3+)六重反铁磁耦合(分子片1和3,其中1为alpha
自旋,3为beta自旋),每个S-R分子片(5-8)为闭壳层单重阴离子。
分子片的分配
chk=FragGuess
311G(d) Guess=(Fragment=8,Only) Pop=None
2S2 cluster with phenylthiolates.
,1 3,6 -2,1 3,-6 -2,1 -1,1 -1,1 -1,1 -1,1
%
%mem=64mw
#P UBP86/6-
Fe
Step 1: Generate fragment guess
-2
H(Fragment=7) 23.5010 2.2873 8.5744
S(Fragment=2) 14.8495 1.1490 7.0431
Fe(Fragment=3) 17.0430 1.0091 7.0068
S(Fragment=4) 17.4565 -1.1490 7.0431
S(Fragment=5) 14.3762 -2.1581 8.7983
C(Fragment=5) 12.5993 -2.1848 8.6878
H(Fragment=5) 12.3743 -3.6513 10.1678
C(Fragment=5) 10.4994 -3.1122 9.4309
H(Fragment=5) 9.9929 -3.7579 10.0022
C(Fragment=5) 9.8049 -2.2791 8.5639
H(Fragment=5) 8.8050 -2.2873 8.5744
C(Fragment=5) 10.4833 -1.4146 7.6615
- 121 -

H(Fragment=5) 9.9730 -0.8525 7.0106
S(Fragment=8) 14.3794 -1.8091 5.0446
C(Fragment=5) 11.9048 -1.3675 7.7057
H(Fragment=5) 12.4158 -0.7843 7.0743
C(Fragment=6) 17.2999 3.4265 4.6624
C(Fragment=6) 16.6376 4.1967 5.6090
H(Fragment=6) 16.5022 3.8494 6.5369
C(Fragment=6) 16.1530 5.4856 5.2463
H(Fragment=6) 15.6665 6.0472 5.9155
C(Fragment=6) 16.3468 5.9257 4.0431
H(Fragment=6) 16.0236 6.8408 3.8020
C(Fragment=6) 17.0091 5.1398 3.0522
H(Fragment=6) 17.1330 5.4944 2.1254
C(Fragment=6) 17.4775 3.8823 3.3884
H(Fragment=6) 17.9400 3.3149 2.7071
S(Fragment=7) 17.9298 2.1581 8.7983
C(Fragment=7) 19.7067 2.1848 8.6878
C(Fragment=7) 20.4174 3.0650 9.5194
H(Fragment=7) 19.9317 3.6513 10.1678
C(Fragment=7) 21.8066 3.1122 9.4309
H(Fragment=7) 22.3132 3.7579 10.0022
C(Fragment=7) 22.5011 2.2791 8.5639
C(Fragment=7) 21.8227 1.4146 7.6615
H(Fragment=7) 22.3330 0.8525 7.0106
C(Fragment=7) 20.4012 1.3675 7.7057
H(Fragment=7) 19.8902 0.7843 7.0743
C(Fragment=8) 15.0061 -3.4265 4.6624
Fe(Fragment=1) 15.2630 -1.0091 7.0068
C(Fragment=8) 15.6684 -4.1967 5.6090
H(Fragment=8) 15.8038 -3.8494 6.5369
C(Fragment=8) 16.1530 -5.4856 5.2463
H(Fragment=8) 16.6395 -6.0472 5.9155
C(Fragment=5) 11.8886 -3.0650 9.5194
C(Fragment=8) 15.9592 -5.9257 4.0431
H(Fragment=8) 16.2824 -6.8408 3.8020
C(Fragment=8) 15.2969 -5.1398 3.0522
H(Fragment=8) 15.1730 -5.4944 2.1254
C(Fragment=8) 14.8285 -3.8823 3.3884
H(Fragment=8) 14.3660 -3.3149 2.7071
S(Fragment=6) 17.9266 1.8091 5.0446
--Link1--
ess
6-311G*/Auto Guess=Read Geom=AllCheck ...
%chk=FragGu
%mem=64mw
%nosave
#P UBP86/
GVB
说明
法关键词进行完美配对广义价键理论(perfect-pairing General Valence Bond;
GVB
这个方
-PP)计算。GVB 需要一个参数:进行分离的完美配对电子对数量;例如:GVB(4)。
这个参数也可以用 Npair 选项指定。GVB 电子对的自然轨道来源于初始猜测行列式的占据
- 122 -
轨道和虚轨道(说明见下)。
GVB 计算的输入
计算中,通常大多数输入困难都是指定初始猜测(链接 401)。这经常包含
交换
定的(通常是 1行),
因此
占据的,它们将变成高自旋轨道。
其它占据轨道保持闭壳层。
从2到n1的自然轨道,接下来的的 n2-1 个
的其它(虚)轨道成为 GVB 计算的虚轨道。
测占据轨道和变成相应更高
自然
道数是负值,那么用于 GVB 对及其初始 GVB 因子的 CI 根将以
(I2,5
项
给出完美配对的 GVB 对数目。GVB(N)等价于 GVB(NPair=N)。也接受
InHam=
N
以和完美配对的电
(D15.8)
(I), I =1,NHam)
目 具有相同的 轨道组合到一个“壳层”中,
OSS 态计算。该选项可以和完美配对电子对组
Freeze 第一自然轨道,只允许改变第二
在GVB-PP
轨道,以保证能正确识别高自旋的,完美配对的以及闭壳层的轨道,并尽可能降低 SCF
的对称性以考虑局域化轨道,它们通常代表 GVB-PP 的最低能量解。
GVB 程序读入每个 GVB 对的轨道数(格式 40I2)。读入的行数是固
不需要终止的空行。对于具有自旋多重度 S,N个GVB 对,每对有 n1, ... , nN轨道的分
子,初始猜测的轨道按照下面的方式供 GVB 程序使用:
初始猜测中的 S-1 个最高占据轨道在 ROHF 计算中是单
下面 N个较低的占据轨道在 ROHF 计算中是双占据的,它们将变成 GVB 对的前 N个
自然轨道。
初始猜测中的
最低的 n1-1 个虚轨道变成第一个 GVB 对中
轨道分配给第二个 GVB 电子对,等等。GVB-PP 方案不允许一个轨道被一个以上的 GVB
对共享。
初始猜测中
一般需要指定 Guess=Alter,以保证用作第一自然轨道的猜
轨道的正确猜测虚轨道之间互相匹配。一个有帮助的方法是从 Guess=(Local,Only)开始
计算,检查轨道确定是否需要作交换,接下来进行 Guess=(Local,Alter) 和
GVB(NPair=N,Freeze)计算,以使更高的自然轨道变得更合适。最后,用 Guess=Read 运行
整个计算,在 GVB 中优化全部轨道。如果轨道发生混乱或者关心轨道破缺对称性,应当用
Symm=NoSCF 进行计算,并从 Guess=Local 开始。实际上,一般推荐用这个方法,除非是
那些非常专业的用户。
如果 GVB 对中的轨
D15.8)格式读取。如果 1Σ或1Δ态用 x2 ± y2形式的 GVB 对表示,这是很有用的。
选
NPair
NPair=0,这将会导致闭壳层或自旋限制的 SCF 计算。
读入 N个哈密顿(Fock 算符,耦合因子数集)。该选项可
子对组合。每个哈密顿按照下面的语法读入(格式在括号中):
NO 当前哈密顿中的轨道数(I5)
Fj 占据轨道数(1.0 是闭壳层)
(AJ J因子(5D15.8)
(AK(I), I =1,NHam) K 因子(5D15.8)
前还不支持把几个 AJ 和AK 因子的
因此 NO 总是 1。工具 ham506 可用于在原子计算中对通常的球平均情况产生
平均的哈密顿。在 Bobrowicz 和Goddard 的著作中有哈密顿因子的说明
[Bobrowicz77]。对 GVB 波函的定性解释有一个很好的介绍,见 Goddard 和Harding
的评论文章[Goddard78]。
进行双电子双正交轨道的开壳层单
合。OpenShellSinglet 是OSS 的同义词。
冻结闭壳层和开壳层轨道,以及 GVB 对的
- 123 -

高的和更高的自然轨道。该选项可用于从较困难的波函开始的计算。
用性
,解析梯度,和数值频率。
子
GVB(3/6)计算的例子:
# GVB(3)/6-31G(d) Pop=Full Test
B(3) on CH2
lecule specification
4 0 2 3 9
Guess=LowSym 输入
2 2
GVB 输入
适
能量
例
这是一个对亚甲基单态进行
Guess=(Local,LowSym,Alter)
GV
mo
1
2,3
Guess=Alter 输入
2
三个价电子对的每一个都分裂为一个 GVB 对。执行初步的 Guess=Only 计算以确定局
域化
VB 对的关联的影响,但不包括 GVB 对之间关联的影响。
因此
理
计算使用的对称性信息中进
行组
轨道,以及需要做哪些交换。
完美配对 GVB 方法包含内部 G
,GVB 电子对趋向于局域化。对于单态亚甲基的例子,碳的孤电子对即使是在
Hartree-Fock 论级别也是局域化的。C-H 键的正则 Hartree-Fock 轨道分别离域化到具有 A1
和B2对称性的线性组合(C-H1 + C-H2)和(C-H1 - C-H2)。为了在猜测中进行局域化以产生分开
的成键电子对,这两个不可约表示必须组合。类似地,必须告知 GVB 计算不要对轨道使用
造成离域化的完全分子对称性。A1和B2表示的组合以及 A2和B1表示的组合使计算只能对
各个轨道使用 Cs对称性,才可以使每个键具有各自的 GVB 对。由于对每个键产生的 GVB
对是等价的,因此产生的整个波函和密度仍具有 C2v 对称性。
Guess=LowSym 关键词指定分子点群的不可约表示在 GVB
合。它需要一行输入,由需要组合的不可约表示编号构成,这些序号对应输出文件中列
出的不可约表示顺序(位于标准方位之后)。例如,下面是 C2v 对称性分子的输出:
There are 4 symmetry adapted basis functions of A1 symmetry.
There are 0 symmetry adapted basis functions of A2 symmetry.
There are 1 symmetry adapted basis functions of B1 symmetry.
There are 2 symmetry adapted basis functions of B2 symmetry.
因此对于 C对称性,轨道顺序为 A1, A2, B1, B2,分别对应
2v Guess=LowSym 输入中的
1到
轨道顺序对占据轨道是 C-1s C-H1
C-H
。
4。0用于隔开进行组合的两组不可约表示,9表示结束列表。因此,要把 A1 与B2 组
合,A2 与B1 组合,SCF 对称性降低为 Cs。正确的输入行是:
1 4 0 2 3 9
因为这一信息只需要一行,因此不需要结束空行。
在局域化后,由第一个任务步骤产生的初始猜测的
2 C-2s,对最低虚轨道是 C-2p C-H1* C-H2*。因此,如果没有轨道交换,C-2s 孤对将与
未占据的 p轨道成对,而接下来更低的占据轨道 C-H2将与更高的虚轨道 C-H1*成对。因此,
必须交换两个成键的占据轨道或者两个成键的虚轨道,以匹配出适合的轨道。
最后,GVB 代码的一行输入表示 3个GVB 对中的每一个都有 2个自然轨道
- 124 -
HF
说明
Hartree-Fock 计算。除非明确指定,默认对单重态是 RHF,高多
重度
用性
,解析梯度,RHF 和UHF 的解析频率,ROHF 的数值频率。
例子
能量在输出中显示如下:
SCF Done: E(RHF) = -74.9646569691 A.U. after 4 cycles
这个方法关键词进行
是UHF。对于后者,将分别计算 α和β轨道[Roothaan51, Pople54, McWeeny68]。也可以明确指定
RHF,ROHF 或UHF。
适
能量
Hartree-Fock
Convg = .6164D-03 -V/T = 2.0063
S**2 = .0000
第二行和第三行给出 SCF S2的期待值。
SCF Done: E(UHF) = -38.7068863059 A.U. after 11 cycles
pin contaminant:
2.0001
收敛极限和
对于 UHF 任务,在输出中还打印 S2和有关的值:
Convg = 0.7647D-08 -V/T = 2.0031
<Sx>= 0.0000 <Sy>= 0.0000 <Sz>= 1.0000 <S**2>= 2.0142 S= 1.0047
<L.S>= 0.000000000000E+00
Annihilation of the first s
S**2 before annihilation 2.0142, after
Huckel
说明
个方法关键词指定进行扩展 Huckel 计算[Hoffmann63, Hoffmann64, Hoffmann64a, Hoffmann64b,
Hoffma
项
ann 使用Hueckel研究小组的默认参数进行扩展Hueckel计算。
k86]进行扩展
用性
,“解析”梯度和数值频率。
子
是偶极矩的x,y,z分量):
这
nn66]。ExtendedHuckel 是这个关键词的同义词。不需要指定基组关键词。
选
Hoffm
Muller 使用 Edgar Muller 搜集的参数进行扩展 Hueckel 计算。
Guess 使用 Guess=Huckel 中的修改参数[Pyykko81, Pyykko84, Fitzpatric
Hueckel 计算。
适
能量
例
计算的能量在输出中显示如下(之后
- 125 -

Huc
Energy= -5.968836513622 NIter= 0.
kel eigenvalues -- -1.245 -0.637 -0.558 -0.544 -0.043 0.352
Dipole moment= 0.000000 0.000000 0.000000
能量是用这个半经验模型定义的。注意,半经验方法所得到的能量差的单位是Hartree,
可以直接与其它方法的计算结果进行比较。
有关的关键词
Guess=Huckel
INDO
说明
这个方法关键词使用 INDO 哈密顿量[Pople67]进行半经验计算。不需指定基组关键词。
适用性
能量,“解析”梯度,和数值频率。
例子
出现在输出文件中的 CNDO 能量显示如下:
SCF Done: E(UINDO) = -8.08016620373 A.U. after 11 cycles
能量用 INDO 模型定义。
Integral
说明
Integral 关键词修改计算方法,以及双电子积分及其导数的使用。
选择积
Grid=
grid
指定用于数值积分的积分网格。注意,在比较能量时(例如计算能量差,生
7000
(35,110)
指定(50,194)网格的裁剪版。 比这些网格有更
分网格的选项
成热,等),对所有的计算使用相同的网格非常重要。
“裁剪”的网格已经做了优化,达到给定精度级别所需的格点数最少。如果
可用(目前对 H 到 Kr 做了定义),默认使用裁剪的网格。例如,FineGrid
是裁剪的(75,302)网格,具有 75 个径向壳层,每个壳层具有 302 个角向的点,
使每个原子有大约 个点。Grid=UltraFine 指定(99,590)网格。对于包含
多个四面体中心的分子和计算非常低的频率模式,推荐用这种网格。这种网
格还可用于优化具有多个柔软模式的更大分子,例如甲基的转动,使得优化
更可靠。
这个参数的其它特殊值还有 CoarseGrid,指定 网格的裁剪版,以及
SG1Grid, 但是注意,FineGrid
好的数值精度和转动不变性,因此不推荐用这些网格做正式计算[Krack98]。
- 126 -

Pass0Grid 指定作废的裁剪(35,110)网格,它曾用于严格 SCF 计算的第一步。
默认网格为 FineGrid。这种情况下,CPHF 的默认网格为 Coarse。当积分使
用UltraFine 时,CPHF 使用 SG1;若积分网格选择 SG1,则 CPHF 再次使
用Coarse 网格。如果为 Integral=Grid 选项指定了特殊网格,那么该网格也
用于 CPHF。最后要知道,SG1 是一些 DFT 任务的默认积分网格,包括
Polar=OptRot,Freq=Anharmonic 和Freq=NNROA(在这些情况下,CPHF
使用 Coarse)。
该选项的参数即可以是网格名称关键词,也可以是特定的网格说明。如果选
Grid 的参数。N可以是以下形式之一:
个原子周围径向有
近
对论计算6
请求二阶Douglas-Kroll-Hess标量相对论计算[Douglas74, Hess85, Hess86, Jansen89]
顿量,这是默认的。
DKH0
阶 Douglas-Kroll-Hess
RESC 相对论计算。
择算法的选项
对DFT 计算中的数值积分使用 Scuseria 和Stratman 的加权方案[Stratmann96]。
FMMNAtoms=
N
设定默认打开 FMM 的阈值大小为
N
。默认的
N
是 60 个原子。对称性分子具有
择的是关键词,那么这个选项名称本身可以忽略(即 Integral(Grid=FineGrid)
和Integral(FineGrid)是等价的)。
选择特定网格可以用整数值 N作为
形式为 mmmnnn 的大的正整数,指定的网格为:每个原子周围径向有
mmm 个壳层,每个壳层内分布 nnn 个角度点。每个原子积分点的总数因
此就是 mmm*nnn。例如要指定(99,302)网格,使用 Int(Grid=99302)。角
方向的有效点数是 38,50 [Lebedev75],72 [McLaren63],86,110 [Lebedev75],
146,194,302 [Lebedev76],434 [Lebedev80],590,770 和974 [Lebedev92]。如
果需要大量的角方向点,可以使用球面乘积网格。
形式为-mmmnnn 的大的负整数,指定的网格为:每
mmm 个壳层,每个壳层有一个球面乘积网格,具有 nnn 个θ点和2*nnn
个 φ 点。每个原子上的积分点总数因此是 2*mmm*nnn2。这个形式用于
指定在基准计算中经常引用的(96,32,64)网格:Int(Grid=–96032)。
注意,nnn 允许任何值,即使是无意义的小值(nnn < 15 的值产生大小相
的网格,但和上面第二种格式请求的特殊角向网格相比,执行较差)。大的值
非常昂贵。例如,值 200100 会对每个原子使用 2*200*100*100 也就是 4百万
个点!
相
DKH
(综述参见[Barysz01, deJong01])。这个方法使用Gaussian核模型[Visscher97]。DKH2
和DouglasKrollHess都是这个选项的同义词。
NoDKH 和NonRelativistic 指定非相对论芯哈密
请求零阶 Douglas-Kroll-Hess 标量相对论计算。
DKHSO 请求包含旋轨耦合项(若做 GHF/GKS 计算的话)的四
相对论计算。
请求 RESC 标量
选
SSWeights
这是默认的。
较高的加速交叉点,因此阈值也相应地升高。C2 和 Cs 点群默认是 120 个原
6 这些相对论方法只能用于全电子基组的计算,不能用于 ECP 基组的计算。这些全电子基组要么是用同样的相对论方法重新收
缩的,要么是用非收缩形式(比如 UGBS 系列的基组)。常见的大多数基组都是用非相对论方法进行收缩的,因此不能用于相对
论计算(特别是第二行以上的元素),否则计算结果不可靠。——译注。
- 127 -
子,更高的点群是 240 个原子。
Symm 在求解和存储积分时使用对Symm 称性,而 NoSymm 不使用(默认为 Symm)。
FoFCou 也使用 FoFCou。NoFoFCou 禁止
LTrace 行。主要用于调试。
SP 层。默认是 NoSplitDBFSP。
N
。
AO 基中所有原函数进行非收缩。该选项的同义词是 UncontractAOBasis。
conDBF
NoXCTest 。
般基组有关的选项
一般收缩基组,通过忽略因子小于10-N的原函数来降低原函数个数。这
ExactBasisTra
收缩基组,但只使用精确的转换来降低原函数个数。只删除严格重
NoBasisTransf
一般收缩基组进行转换,不降低原函数个数。
关的关键词
IOp
说明
关键词允许用户设定内部选项(变量位于系统公用区/IOp/)来定义值。语法为:
IOp(1/1 1 2 2 2
iOpi的值为 Ni。由于设置内部选项对计算的影响是不定
,不能传递到第二步和随后自动产生的任务步骤中;这可
能发
由编号的选项控制。每一项分配一个整数值,默认是 0。在
一层
同义词为 Symm=[No]Int,这是推荐用法。
即使在本来不应使用 FoFCou 例程的情况下,
使用 FoFCou。
追踪 Linda 的执
SplitDBF 把密度的 S=P 壳层拆成独立的 S 和 P 壳
ECPAcc=
N
设定 ECP 的精度参数为
N
。
Acc2E=
N
设定双电子积分的精度参数为
UnconAOBasis
将
Un 将密度拟合基中的所有原函数进行非收缩。该选项的同义词是
UncontractDensityBasis。
跳过 XC 积分的数值精度测试
一
BasisTransform=N
转换
是默认的,其中N=4。
nsform
转换一般
复的原函数,能量值不会有变化。
orm
不对
有
SCF
IOp
Ov Op =N,Ov /Op =N, ...)
其中对每一个层 Ov ,设置选项序号
的,因此这一关键词禁止存档。
在执行路径部分设置的 IOp 值
生在关键词组合中,如 Opt Freq,以及原本就是多步的方法中,如 G2 和 CBS 方法。例
如,如果想要对 DFT 几何优化+频率任务指定其它的网格,必须使用关键词 Int=Grid 设定选
项,而不能直接定义 IOp 值。
Gaussian 09 每一层的执行
内所有链接的执行过程中,选项的值自始至终保持不变。因此通过层的传递,一个特殊
- 128 -

选项可以作用于传递过程中所有的组成部分。Gaussian 09 选项的完整列表在《Gaussian 09
IOps Reference》中给出。在我们的网站上也有文档:www.gaussian.com/iops.htm
IRC
说明
这个计算类型关键词要求用内反应坐标积分方法跟踪反应路径[Fukui81, Hratchian05a]。初始
结构(在分子说明部分给出)是过渡态的结构,路径可以从该点的一个或全部两个方向进行
跟踪。当过渡矢量(“相位”)的最大分量为正时,定义为过渡矢量指向的前进方向;它可以
直接用 Phase 选项直接定义。默认对两个反应路径方向都进行跟踪。Gaussian 09 使用新的
算法[Hratchian04a, Hratchian05a, Hratchian05b]计算反应路径上的点,这比早期版本程序使用的方法更有
效率。参见下面 HPC 选项的讨论。
进行 IRC 计算需要初始力常数。必须通过某种方法为计算提供这些信息。通常的方法
是保存前一个频率计算的检查点文件(验证用于 IRC 计算的优化结构是真正的过渡态),接
下来在执行路径中指定 RCFC 选项。另一个方法是在 IRC 计算的开始计算它们(CalcFC)。
注意必须指定 RCFC 和CalcFC 其中的一个(也可以用 CalcAll,但是新的算法在典型情况
下用不到它)。
在Gaussian 09 中已经改变了默认的 IRC 算法。大多数计算默认使用 HPC 算法。
ONIOM(MO:MM)计算使用 Euler 预测-校正积分法。对于有梯度但没有解析二阶导的方法,
计算应当包含 GradientOnly 选项,接下来使用阻尼速率 Verl et 积分程序(DVV),但是如
果需要,也可以指定用 Euler 积分(Euler)。后面详细讨论了可用的算法。
默认仅报告路径上每一点的能量和反应坐标;如果需要沿着路径的结构参数,可以通过
Geom=ModRedundant 定义为冗余内坐标,或者通过 IRC(Report=Read)定义为 IRC 代码的
输入。
可以用 ReadIsotopes 选项为 IRC 任务指定替代的同位素(见下)。
选择路径选项
Phase=(
N1 N2
[
N3
[
N4
]])
定义过渡矢量的相位,使前进运动方向沿着过渡矢量正向,也就是指定的内
坐标增加的方向,这需要最多四个原子序号指定。如果只给出两个原子序号,
坐标为两个原子间的键伸缩;三个原子序号指定键角弯曲;四个原子指定二
面角。
Forward 只跟踪反应路径的正向。
Reverse 只跟踪反应路径的反向。
Downhill 从输入的结构朝着下坡的方向跟踪。
MaxPoints=
N
在反应路径上检查的点数(沿着正反两个方向,如果二者在计算中都考虑的
话)。默认是 10。
StepSize=
N
沿反应路径的步长,单位是 0.01 Bohr。如果 N<0,则步长单位是 0.01
amu1/2-Bohr。默认是 10。
ReadIsotopes
该选项允许指定不同于默认值(分别为 298.15 K,1 大气压,无换算,和最
丰同位素)的温度,压强,频率换算因子,和/或同位素。该选项可用于重新
进行分析,但使用不同于检查点文件数据的参数。
- 129 -
但是需要注意,所有这些参数也可以在执行路径部分(关键词 Temperature,
Pressure,和 Scale)和分子说明部分(Iso=
参数
)指定,如这个例子:
#T
Method
/6-31G(d)
JobType
Temperature=300.0 ...
...
0 1
C(Iso=13)
...
ReadIsotopes 输入具有下面的格式:
temp
pressure
[
scale
]
数值必须为实数。
原子 1 的同位素质量
原子 2 的同位素质量
...
原子 n 的同位素质量
其中的
temp
,
pressure
,和
scale
分别为所需的温度,压强,和当用于热化
学分析时可选的频率数据换算因子(默认不换算)。其余的行是分子中各原
子的同位素质量,按照分子说明部分出现的相同顺序排列。如果原子量指定
为整数,程序将自动使用相应的真实同位素质量(例如,对 18O 指定为 18,
那么 Gaussian 使用的值为 17.99916)。
算法选择选项
HPC 使用基于 Hessian 的预测-校正积分方法[Hratchian04a, Hratchian05a, Hratchian05b]:这是
一种非常准确的算法,它使用基于 Hessian 的局域二次近似作为预测部分,
以及修改的 Bulrisch-Stoer 积分方法用于校正部分。做这个校正积分方法需要
使用距离加权的插值曲面[Collins02],这需要在预测步骤的开始点和结束点对能
量,梯度,和 Hessian 进行拟合才能得到。这对大多数计算是默认的。注意,
这种方法对非常大的分子体系是不实用的。
LQA 对预测步骤使用局域二次近似[Page88, Page90]。
DVV 使用阻尼速率 Verlet 积分方法[Hratchian02]。对于 IRC=GradientOnly 计算是默
认的。
EulerPC 对于预测步骤使用一阶 Euler 积分,同时对校正步骤使用 HPC。这对使用
ONIOM(MO:MM)方法的 IRC 计算是默认算法。这种计算对大分子是现实的
选择。
Euler 对IRC 仅使用一阶 Euler 积分的预测步骤。该选项不建议用于正式计算。
ReCalc=
N
每隔 N个预测步骤或|N|个校正步骤(若 N<0),计算解析 Hessian。必须指定
RCFC 或CalcFC,用于提供初始 Hessian。Update 是ReCalc 的同义词。要
求方法有解析二阶导。
GradientOnly
使用不需要二阶导的算法。注意,对于这类方法必须明确指定该选项(因为
不能自动判断)。可以与 DVV(默认)和 Euler 合用。
坐标系选择选项
MassWeighted
- 130 -
跟踪质量加权笛卡尔坐标的反应路径。MW 是MassWeighted 的同义词。这
是默认选项。
Cartesian 跟踪笛卡尔坐标反应路径,不使用质量加权。
产生初始力常数的选项
RCFC 指定从检查点文件读取频率计算的笛卡尔坐标力常数。ReadCartesianFC 是
RCFC 的同义词。
CalcFC 指定在第一个点计算力常数。
用于和 Gaussian 03 兼容的选项
GS2 选项请求在新的 IRC 执行中使用 Gaussian 03 的 IRC 算法。为了运行 Gaussian 03
的默认代码,需要使用关键词 Use=L115(仅用于重复旧版的结果)。
GS2 使用 Gaussian 03 和早期版本默认的 IRC 算法[Gonzalez89, Gonzalez90]。沿着反应路
径的方向,在每个点进行结构优化,这样,任意两个相邻点之间的一段反应
路径可以用一个圆弧来描述,因此圆弧最后一个点的梯度为路径的切线。默
认的 GS2 IRC 计算使用质量加权内坐标,沿着路径正向 6个点,反向 6个点,
步长为 0.1 amu1/2 Bohr。
CalcAll 指定在每一个点计算力常数。
与进程有关的选项
Restart 重新开始没有完成的 IRC 计算,或对已完成的 IRC 计算在反应路径上加入新
的点继续计算。
Report[=
item
]
控制 IRC 报告哪些几何参数。默认不报告几何参数。不带参数的 Report 将包
含所有产生的内坐标。
item
可能的值有:
Read 读取一列内坐标并输出。内坐标的定义方式同
ModRedundant。
Bonds 从内坐标输出键长(如果出现)。
Angles 从内坐标输出键角(如果出现)。
Dihedrals 从内坐标输出二面角(如果出现)。
Cartesians 输出所有的笛卡尔坐标。
ReCorrect[=when]
对于 HPC 和EulerPC 的IRC 校正步骤,控制测试-重算流程。不带参数的
ReCorrect 和ReCorrect=Yes 表示只要校正大于阈值(降低阈值可以用 Tight
和VTight),就重复校正步骤。参数可以用下面的值:
Never 不重复校正步骤(即,禁止测试阈值)。
Always 不管初始校正多大,总是重新计算校正至少一次。
Test 测试校正步骤的质量并报告结果,但是不进行附加的校正
步骤。计算得到的 IRC 路径与用 ReCorrect=Never 计算
的相同。
对EulerPC 和HPC 默认为 Ye s ,对其它积分方法默认为 Never。
MaxCycle=
N
设定每个几何优化中的最大步数为 N。默认是 20。
Tight 该选项对判断收敛所用的力和步长使用更严格的阈值。对于 DFT 计算,必须
同时指定 Int=UltraFine。
- 131 -

VeryTight 使用极其严格的收敛标准。VTight 是VeryTight 的同义词。对于 DFT 计算,
必须同时指定 Int=UltraFine。
适用性
默认算法可用于 HF,所有的 DFT 方法,CIS,MP2,MP3,MP4(SDQ),CID,CISD,
CCD,CCSD,QCISD,BD,CASSCF,以及所有的半经验方法。
有关的关键词
Opt,Scan,IRCMax
例子
在IRC 完成后,程序打印总结结果的表格:
Reaction path calculation complete.
Energies reported relative to the TS energy of -91.564851
--------------------------------------------------------------------------
Summary of reaction path following
--------------------------------------------------------------------------
Energy Rx Coord
1 -0.00880 -0.54062
2 -0.00567 -0.43250
3 -0.00320 -0.32438
4 -0.00142 -0.21626
5 -0.00035 -0.10815
6 0.00000 0.00000
7 -0.00034 0.10815
8 -0.00131 0.21627
9 -0.00285 0.32439
10 -0.00487 0.43252
11 -0.00725 0.54065
--------------------------------------------------------------------------
初始几何(过渡结构)出现在表格的中间(本例中是第 6个点)。可以通过寻找反应坐
标和能量值为 0.00000 的方法快速识别。
IRCMax
说明
使用 Petersson 及合作者[Eyring35, Truhlar70, Truhlar71, Garrett80, Skodje82, Malick98, Petersson98, Schwartz98,
Petersson98a]的方法进行 IRCMax 计算。这一计算类型用过渡结构做为输入,用 GS2 算法沿着
指定的反应路径寻找最大的能量[Gonzalez89, Gonzalez90](细节参见 IRC=GS2)。
必须用标准方法为 IRCMax 任务指定可选的同位素。
必需的输入
IRCMax 需要两个模型化学作为选项,中间用冒号隔开:IRCMax(
模型
2:
模型
1)。这
里是一个执行路径的例子:
- 132 -
# IRCMax(B3LYP/6-31G(d,p):HF/6-31G(d,p))
这个计算在 HF/6-31G(d,p)反应路径上寻找点,该点使 B3LYP/6-31G(d,p)能量位于最大
值。
选项 Zero 产生用于零曲率变分过渡态理论(ZC-VTST)[Eyring35, Truhlar70, Truhlar71, Garrett80,
Skodje82, Petersson98]的数据。考虑下面的执行路径:
# IRCMax(MP2/6-311G(d):HF/6-31G(d),Zero,Stepsize=15,CalcAll)
这个任务将从 HF/6-31G(d)过渡态开始,沿着 HF/6-31G(d)的IRC 以步长 0.15 amu1/2 Bohr 进
行寻找,直到找到 MP2/6-311G(d)能量的最大值(含 HF/6-31G(d)零点能)为止。对
MP2/6-311G(d)过渡态沿着 HF/6-31G(d)的IRC 进行优化。用 ZC-VTST 版本绝对速率理论进
行反应速率计算所需的全部量都包含输出中:过渡态的转动惯量,所有的实振动频率
(HF/6-31G(d)),隧穿的虚频(拟合到 MP2/6-311G(d), 加 上 ZPE ),以及过渡态的
MP2/6-311G(d)加上 ZPE 的总能量。注意,如果未使用 CalcAll,那么所有这些量(ZPE,频
率,等)只在 HF/6-31G(d)级别进行计算,并且这些量用于 IRCMax 路径上所有的点。
ZC-VTST 选项
Zero 在IRCMax 计算中包含零点能。
选则路径选项
Forward 只沿着反应路径的正向进行跟踪。
Reverse 只沿着反应路径的反向进行跟踪。
ReadVector 读入矢量进行跟踪。格式为 Z-矩阵(FFF(I), I=1,NVAR),以(8F10.6)读入。
MaxPoints=
N
沿反应路径进行检查的点数(沿着正反两个方向,如果计算中都考虑的话)。
默认是 6。
StepSize=
N
反应路径上的步长,单位是 0.01 amu1/2-Bohr。默认是 10。
MaxCyc=
N
设定每个几何优化中的最大步数。默认是 20。
选择坐标系的选项
MassWeighted
跟踪质量加权内坐标(Z-矩阵)的路径(和跟踪质量加权笛卡尔坐标的路径
是等价的)。MW 是MassWeighted 的同义词。这是默认选项。
Internal 跟踪内坐标(Z-矩阵)路径,不使用质量加权。
Cartesian 跟踪笛卡尔坐标路径,不使用质量加权。
收敛有关的选项
VeryTight 在反应路径每一点的优化中,使用严格的收敛标准。该选项对反应路径上很
小的步长是必要的。
产生初始力常数选项
CalcFC 指定在第一个点计算力常数。
CalcAll 指定在每一个点计算力常数。在路径上的每一优化点,对运动计算投影的振
动频率[Baboul97]。注意,这些投影的振动频率仅对质量加权内坐标的反应路径
计算有效。
- 133 -
FCCards 指定从分子说明后面的输入串读入笛卡尔坐标的力和力常数。该选项使用内
部命令 FCList,可以从 Quantum Chemistry Archive 读取力常数。输入的格式
为:
能量
(格式 D24.16)
笛卡尔力
(格式为 6F12.8 的几行)
力常数
(格式为 6F12.8 的几行)
力常数为倒三角形式:((F(J,I),J=1,I),I=1,3Natoms),其中 3Natoms 是笛卡尔坐标
数量。如果同时指定 FCCards 和ReadIsotopes,则在能量,笛卡尔梯度和/
或笛卡尔力常数之前输入原子量。
ReadIsotopes
该选项允许指定不同于默认值(分别为 298.15 K,1 大气压,无换算,和最
丰同位素)的温度,压强,频率换算因子,和/或同位素。该选项可用于重新
进行分析,但使用不同于检查点文件数据的参数。
但是需要注意,所有这些参数也可以在执行路径部分(关键词 Temperature,
Pressure,和 Scale)和分子说明部分(Iso=
参数
)指定,如这个例子:
#T
Method
/6-31G(d)
JobType
Temperature=300.0 ...
...
0 1
C(Iso=13)
...
ReadIsotopes 输入具有下面的格式:
temp
pressure
[
scale
]
数值必须为实数。
原子 1 的同位素质量
原子 2 的同位素质量
...
原子 n 的同位素质量
其中的
temp
,
pressure
,和
scale
分别为所需的温度,压强,和当用于热化学
分析时可选的频率数据换算因子(默认不换算)。其余的行是分子中各原子
的同位素质量,按照分子说明部分出现的相同顺序排列。如果原子量指定为
整数,程序将自动使用相应的真实同位素质量(例如,对18O指定为18,那么
Gaussian使用的值为17.99916)。
重新开始选项
Restart 重新开始没有完成的 IRCMax 计算,或对已完成的 IRCMax 计算在反应路径
上加入新的点继续计算。
适用性
对计算的 IRC 部分(上面的
模型
1)需要解析梯度。对
模型
2可以使用任何非复合的能
量方法和基组。
有关的关键词
IRC,Opt,Freq
- 134 -
LSDA
说明
这个方法关键词进行局域自旋密度近似计算,对 DFT 计算使用 Slater 交换泛函和 VWN
关联泛函。它等价于 SVWN。注意 LSDA 在文献中的定义不是唯一的。实际上,许多不同
的有关方法都使用了这个名称。其它程序提到的 LSDA 方法可能使用了略微不同的泛函。
例如,有些使用了 SVWN5 关键词指定的泛函,而其它则使用了 Perdew 的关联泛函。Gaussian
提供这一关键词是为了方便,根据需要定义准确的泛函可能会更好;关于在 Gaussian 中指
定和使用密度泛函方法的详细说明,参见上面的 DFT 方法。
MaxDisk
说明
MaxDisk 关键词指定临时数据可用的磁盘空间大小,单位是 8 位的字(默认)。这个值
也可以加上单位 KB,MB,GB,KW,MW 或GB(之间无空格),分别表示千,兆,吉位
或字。在工作站中,这通常是在全站范围的 Default.Route 文件内设置。
并非所有计算都可以动态控制磁盘使用量,因此这个关键词的作用依情况而异:
SCF 能量,梯度和频率计算使用固定的磁盘大小。这需要得很少(和分子体系大小的三
次方成正比),通常不够成限制。
MP2 能量和梯度遵从 MaxDisk,必需至少是 2ON2。
MP2 解析频率计算尽可能尝试遵从 MaxDisk,但是有最小磁盘使用量的下限。
用MO 基的 CI-Singles 能量和梯度计算需要 4O2N2字的空间,用于储存有限数目的转
换积分。转换计算中需要额外的临时空间,这受 MaxDisk 指定空间的限制。如果用
CIS=Direct 指定执行直接 CI-Singles 计算,则没有这一磁盘需求的限制。
CID,CISD,CCD,BD 和QCISD 能量计算也需要固定的储存空间,大小和 O2N2成正
比,且比例系数很大,但遵守 MaxDisk 指定的大小以避免更大的储存空间需求。
CCSD,CCSD(T),QCISD(T)和BD(T)的能量计算有固定的磁盘需求,大小和 ON3成正
比,并且无法用 MaxDisk 限制。
CID,CISD,CCD 和QCISD 的密度,以及 BD 和CCSD 的密度及梯度有固定磁盘需求
量,对闭壳层约为 N4/2,对开壳层约为 3N4/4。
EOM-CCSD 在一开始把磁盘需求量设定为需要的最小值。如果这个量比 MaxDisk 大,
则遵从后者,但会牺牲 I/O 和计算时间。
关于在 Gaussian 计算中有效使用磁盘资源的详细讨论,见附录:效率的考虑。
MINDO3
说明
这个方法关键词使用 MINDO3 哈密顿量[Bingham75, Dewar77]进行半经验计算。不需要指定
基组关键词。
- 135 -

适用性
能量,“解析”梯度,和数值频率。限制性开壳层(RO)波函的优化计算只能使用
Fletcher-Powell 和赝 Newton-Raphson 方法(Opt 关键词的选项分别是 FP 和EnOnly)。
例子
出现在输出文件中的 MINDO3 能量显示如下:
SCF Done: E(UMINDO3) = -8.08016620373 A.U. after 11 cycles
能量用 MINDO3 模型定义。
分子力学方法
说明
Gaussian 中可以使用三种分子力学方法。它们用于 ONIOM 计算,但也可以用作独立的
方法。这些方法不需要指定基组关键词。
可以使用以下力场:
Amber:Amber力场在[Cornell95]中描述。这篇文章发表后,实际的参数(parm96.dat)略微
做了更新。我们使用的当前版本来自Amber网站(http://ambermd.org/)。
- Dreiding:Dreiding 力场在[Mayo90]中描述。
- UFF:UFF 力场在[Rappe92]中描述。
分子力学的分子说明
分子力学计算用原子类型确定构成力场的函数和参数。对于单个元素,例如碳,可能有
多种不同的 MM 原子类型。使用哪一种依赖于具体情况,例如杂化,化学环境,等。
对于 UFF 和Dreiding 计算,Gaussian 会尝试自动指定原子类型。但是,Amber 计算需
要在分子说明部分直接指定所有的原子类型,如下面的几个例子:
C-CT
指定
SP3
脂肪族碳原子,用
Amber
的
CT
关键词表示。
C-CT-0.32
指定
SP3
脂肪族碳原子,带有部分电荷
+0.32
。
O-O--0.5
指定羰基氧原子(类型
O
),带有部分电荷
-0.5
。
原子类型关键词在元素符号和一个连字符之后定义。关于原子类型及其有关关键词的定
义,参见 Amber 的论文[Cornell95]。
在示例中的第二行和第三行,还可以在原子说明的第三部分指定部分电荷,前面仍然加
上连字符。部分原子(点)电荷用于计算静电相互作用。Gaussian 用QEq 方法自动分配这
些电荷[Rappe91]。要使用这一方法,可以为 MM 关键词指定 QEq 选项:例如 Dreiding=QEq。
注意,这些电荷依赖于结构,在计算的一开始就进行计算;但是在结构优化和其它结构改变
的过程中,不对电荷进行更新。
如果需要的话,也可以对 UFF 和Dreiding 计算提供原子类型和电荷的信息。要知道,
只有对明确定义的体系,自动分配的 UFF 和Dreiding 原子类型才是可靠的,而直接定义它
们往往会更安全。
当使用 MM 方法时,特别是当修改力场项或定义新的力场项时(说明见下),通常需要
用Geom=Connectivity 直接指定原子之间的连接关系。注意,GaussView 在构造输入文件时,
会自动包含这一输入部分。
- 136 -
电荷分配有关的选项
除非在分子说明部分设定,否则在使用任何分子力学力场时,默认都不会为原子分配电
荷。有些选项可以在初始结构上用QEq算法[Rappe91]估算电荷,可以对任何力学关键词用以下
选项进行控制:
QEq 用 QEq 方法为所有原子分配电荷。
UnTyped 只对在输入文件中没有指定特定类型的原子分配 QEq 电荷。
UnCharged 对所有电荷为零的原子(即,在输入文件中没有设定类型或给定了类型但没
有设定电荷的所有原子)分配 QEq 电荷。
参数优先级选项
术语
: Gaussian对以上所列的内置力场包含内置参数,称为
硬连接
(hard-wired)参数。
软
参数是由用户在当前任务的输入串内指定的参数(或者是从以前任务的检查点文件读取的
参数)。默认不提供有关的选项,只使用硬连接参数。
HardFirst 从输入串读取附加的参数,硬连接参数优先于读入的软参数。因此,只有当
没有相应的硬连接参数时,才会使用读入的参数。注意,和硬连接参数匹配
的通配符结果优先于软参数,即使软参数包含完全匹配的相同项。若要禁止
硬连接参数的匹配,使用 SoftFirst 选项。
SoftFirst 从输入串读入附加的参数,(读入的)软参数优先于硬连接参数值。
SoftOnly 只使用从输入串读入的参数,忽略硬连接参数。
处理多种匹配的参数说明
因为参数可以用通配符指定(见下),符合一个给定结构的参数说明可能有多种。如果
力场存在歧义,默认为中断计算。以下选项指定遇到多种匹配时的其它处理方式:
FirstEquiv 如果所需的参数有等价的匹配,使用最先发现的。
LastEquiv 如果所需的参数有等价的匹配,使用最后发现的。
从检查点文件读入MM参数
当用 Geom=Check 或Geom=AllCheck 从检查点文件读入结构时,默认同时也从检查点
文件提取所有出现的非标准(软)参数。这些读入的参数在使用时比相应的硬连接参数有更
高的优先权,除非同时也指定了 HardFirst。以下选项可用于修改这一默认属性。
ChkParameters
只从检查点文件读入 MM 参数。检查点文件中的非标准(软)参数在使用时比
相应的硬连接参数有更高的优先权,除非同时也指定了 HardFirst。在使用
Geom=Check 或Geom=AllCheck 时无效。
NewParameters
在读入结构时,忽略检查点文件中的所有参数。
Modify 把来自检查点文件的软参数和硬连接参数进行组合后(由上面的选项控制),
读入附加的参数说明并进行修改。该选项的工作方式类似于 Geom=Modify。
打印MM参数
Print 如果指定了#P,默认只打印对能量的贡献(即,来自键伸缩,弯曲,静电,
等)。Print 选项还在第一个结构打印能量贡献和力场参数,以及后面各点
的能量贡献(因为参数不变)。对于 ONIOM(MO:MM)还会打印两套参数,因为
- 137 -

模型体系和真实体系使用了不同的参数(即,H 可能会替代 C 连接原子)。
注意,对体系中的所有原子打印参数,但不是以适合于输入的格式。在
Gaussian 的主目录中,已经提供了输入文件格式的内置参数(amber.prm,
uff.prm,等),对于一些没有内置的力场也提供了文件(amber98.prm 和
oplsaa.prm)。但是打印当前任务中所有原子的参数仍然有用,因为在确定
什么参数实际用于给定的键,二面角,等的时候,会涉及非常复杂的规则。
适用性
解析能量,梯度,和频率。
有关的关键词
ONIOM,Geom=Connectivity
一般分子力学力场的说明
对于想修改或定制MM力场的用户,Gaussian提供了相当大的灵活性。这一部分将讨论
这样做的细节,先从一些理解函数和参数所必须的背景信息开始。
一个基本的MM势函数通常形式为:
22
12 6
1cos
2
ij ij i j
total n
req eq
ij ij ij
A
Bqq
V
EKrr K n rr r
bonds angles dihedrals i<j
伸缩项 弯曲项 扭转项 非键相互作用
分子力学势函数
如上所述,展开的项为(键)伸缩项,(键角)弯曲项,(二面角)扭转项,以及非键
相互作用。在这个方程中,个别函数的特殊形式来自Amber力场,其中对伸缩和弯曲使用简
谐函数,对扭转使用余弦函数,对静电和范德华相互作用使用标准函数。其它的力场使用不
同的函数,其中有些附加类型的项不出现在Amber中,例如偶极相互作用,以及伸缩-弯曲
的耦合。
为了求解MM势函数,Gaussian需要知道体系中有哪些结构——伸缩,弯曲,以及扭转
——以及它们使用的函数和参数。结构可以从分子说明中识别。Gaussian默认会确定哪些原
子成键,以及相应的键级(单键,双键,等)。若输入结构合理,并且所有键级都正确定义,
那么这就是个好计算任务。但是,如果计算从包含更多近似的结构开始,或者键级难以识别,
那么在输入串中指定连接关系会更安全一些,这需要使用Geom=Connectivity关键词(细节
参见Geom的讨论)。
本节余下部分将一般性地讨论MM函数及其参数,接下来的部分讲述所有可用的函数。
显示MM函数。开始这一话题之前,一个有用的方法是检查Gaussian的MM任务的输出。
下面是用Amber计算甲烷的输入文件:
#P Amber Geom=Connectivity IOp(4/33=3)
Methane
0 1
C-CT--0.4 -0.85 0.42 0.00
H-HC-0.1 -0.50 -0.57 0.00
H-HC-0.1 -0.50 0.93 0.87
H-HC-0.1 -0.50 0.93 -0.87
- 138 -

H-HC-0.1 -1.92 0.42 0.00
注意,碳原子的原子类型是CT,氢原子是HC。我们还为每个原子指定了部分电荷。
下面是在任务的执行路径部分加上IOp(4/33=3)后的部分输出:
Atomic parameters:
单个原子的MM函数。
Center VDW
1 1.9080 .1094000
2 1.4870 .0157000
3 1.4870 .0157000
4 1.4870 .0157000
5 1.4870 .0157000
Molecular mechanics terms:
2或多个原子的MM函数。
NBDir 3 1 0 0
非键相互作用主函数。
HrmStr1 1-2 340.00 1.0900
伸缩项。
HrmStr1 1-3 340.00 1.0900
HrmStr1 1-4 340.00 1.0900
HrmStr1 1-5 340.00 1.0900
HrmBnd1 2-1-3 35.00 109.50
弯曲项。
HrmBnd1 2-1-4 35.00 109.50
HrmBnd1 2-1-5 35.00 109.50
HrmBnd1 3-1-4 35.00 109.50
HrmBnd1 3-1-5 35.00 109.50
HrmBnd1 4-1-5 35.00 109.50
NBPair 2-1 3 1 0 0 -1.000 -1.000
临近原子的非键相互作用。
NBPair 3-1 3 1 0 0 -1.000 -1.000
NBPair 3-2 3 1 0 0 -1.000 -1.000
NBPair 4-1 3 1 0 0 -1.000 -1.000
NBPair 4-2 3 1 0 0 -1.000 -1.000
NBPair 4-3 3 1 0 0 -1.000 -1.000
NBPair 5-1 3 1 0 0 -1.000 -1.000
NBPair 5-2 3 1 0 0 -1.000 -1.000
NBPair 5-3 3 1 0 0 -1.000 -1.000
NBPair 5-4 3 1 0 0 -1.000 -1.000
原子参数。有些MM函数只依赖于问题原子的原子类型。在我们的例子中,Amber包含
了每个原子的范德华相互作用,得到的值列于上面输出的Atomic parameters之后。其中的
中心编号对应于分子说明部分的原子位置。DREIDING和UFF力场只包含这一类型的项。
分子力学项。分子力学项部分列出了总势内的项,用于描述多个原子间的相互作用。例
如,涉及每对成键原子(1-2,1-3,和1-4)的伸缩项通过HrmStr1函数计算,使用的力常数
值为340 kcal/(mol-Å2),平衡键长为1.09 Å。确定这些参数值需要根据中心1和2的原子类型:
(分别为)CT和HC。
特殊的函数和参数选择如下:程序可以从连接关系知道在中心1和2之间存在键。Amber
需要伸缩项用于所有成键的原子对,各种函数和参数都存储在程序的内部列表中。在最简单
的情况下,Gaussian用两个原子类型来查找使用的函数和参数。在这种情况下,将使用以下
项目,
HrmStr1 CT HC 340 1.09
因为它对应于甲烷C-H键的两个原子类型。接下来,在计算中使用项目中指定的力常数和平
衡键长。在后面的参考文献部分寻找该函数时,我们看到实际计算的项为ForceC*(R–Req)2,
其中ForceC是力常数,Req是平衡键长,R为键长;在本例中,该项变为340.0*(R–1.09)2。
项的列表中包含所有成键原子对之间的伸缩项,以及所有相应键长的弯曲项。
- 139 -
非键相互作用项。列表中其余的项对应非键相互作用:来自非键原子间相互作用势的贡
献。在进行个别考虑之前,我们需要看一下它们是如何算出来的。
一般情况下,非键项的列表由所有可能的原子对构成,因此项的数目和原子个数是二次
比例关系。但是近距离原子之间的相互作用,基于它们之间的键数,比例关系通常会下降。
典型情况下,由一个键和两个键隔开的相互作用,比例为零(因为这些相互作用由伸缩项和
弯曲项描述)。类似地,由三个键隔开的相互作用,由依赖于力场并且在0与1之间的某个比
例因子换算。很清楚,对于较大体系,非键项的列表非常长。为了管理列表,在大多数MM
程序中通常使用截断。在Gaussian中,我们使用了一种不太常用的方法,可以有效处理非键
相互作用。
体系的总非键能量可以写做如下形式:
11
ji
N
NB vdW vdW Q Q
ij ij ij ij
ij
ji ji
NN
vdW Q vdW vdW Q Q
ij ij ij ij ij ij
ij ij
EsEsE
EE sE sE
所有原子对 多算了的原子对
总非键能量的表达式
其中的EvdW和EQ分别是两中心之间的范德华相互作用和静电相互作用,相应的s值为有关的
换算因子。
第一个方程用通常的形式表示非键相互作用,而第二个方程是等价表达式,其中所有可
能的非键相互作用(不考虑距离)在计算时不做换算(第一项),之后减去多算了的原子对
(第二项)。
这一改写具有相当大的计算优势。第一项可以用有效的方式计算,例如线性标度或其它
技术。此外,第二项的大多数原子对之间的相互作用为零(假如体系不是太小),因此换算
因子sij对大多数原子对为1。程序会简单地保留这些具有非零相互作用的原子对的列表,用
于计算第二项。这是一种不使用截断的有效算法,只需要一个列表,列表长度随体系大小呈
线性比例。
在构成这两种非键相互作用项时,使用了NBDir和NBTerm函数。如前面甲烷任务的输
出所示,单个NBDir函数描述了所有原子对之间相互作用的完备集,即,方程重写后的第一
项。各种NBTerm项目构成了方程中的第二项。这些函数的最后两个参数分别为范德华相互
作用和库仑相互作用的换算因子。在甲烷的例子中,两个换算因子对所有的项都是1,因为
在甲烷中,没有用两个以上的键隔开的原子,最后的非键相互作用相应地为0。
指定缺少的参数。考虑以下分子:2-甲基丙烯。我们对该体系设定了Amber计算,指定
SP2杂化碳的原子类型为CM,SP3杂化碳的原子类型为CT。
- 140 -
2-甲基丙烯
# Amber
2-methylpropene
0 1
C-CM -2.53 0.19 0.00
H-HA -2.00 -0.73 0.00
H-HA -3.60 0.19 0.00
C-CM -1.86 1.37 0.00
C-CT -2.63 2.70 0.00
H-HC -1.93 3.51 0.07
H-HC -3.24 2.76 -0.88
H-HC -3.24 2.76 0.85
C-CT -0.32 1.37 0.00
H-HC 0.03 1.87 -0.83
H-HC 0.03 1.87 0.81
H-HC 0.03 0.36 -0.00
当我们运行此任务时,程序终止并出现以下错误信息:
Angle bend undefined between atoms 2 1 3
Angle bend undefined between atoms 5 4 9
MM function not complete
原子2-1-3对应于类型HA-CM-HA,5-4-9对应于CT-CM-CT。虽然原子类型和次序在化
学上是合理的,但在硬连接列表中没有对应于弯曲项的项目。原因是Amber力场专门为生化
体系开发,其中不会出现原子类型的某些特定序列。因此Amber没有定义这些参数。注意,
Gaussian只对所有的伸缩和弯曲检查是否有函数;而对列表中无对应项目的扭转项及其它
项,仅仅在势函数中简单地忽略它们。
添加和替换参数。当没有硬连接参数时,不论是列表不完整还是包含了新原子类型,都
需要在输入串指定。确定合适的参数很有挑战性。它们经常可以在文献中找到;否则需要从
实验或准确的计算数据获得(这一话题超出了我们的讨论范围)。
指定新的参数需要使用Amber=HardFirst关键词,接着在输入文件另外的部分添加数
据。这个例子中,我们对缺少的原子类型序列定义HrmBnd1函数。于是2-甲基丙烯的任务具
有如下形式:
- 141 -

# Amber=HardFirst
2-methylpropene
0 1
C-CM -2.53 0.19 0.00
分子说明继续...
HrmBnd1 CT CM CT 70.0 120.0
HrmBnd1 HA CM HA 40.0 120.0
本例使用的参数值是基于已有Amber参数的粗糙估计。HardFirst选项表示对函数首先
检查通常的Amber硬连接参数表——简称为硬参数,而输入文件定义的函数仅用于存在不匹
配时。用SoftFirst选项,用户可以替换来自硬连接参数集的参数。注意,只有当一个或多个
函数同时在两个参数列表中定义时,HardFirst和SoftFirst的指定顺序才会有影响。最后,
SoftOnly选项表示在输入文件提供完整的力场定义,并完全忽略硬连接参数集。
使用外部参数文件。可以在外部文件中包含MM函数项目,需要用Gaussian的包含文件
机制替换输入文件中指定的项目:例如,@oplsaa.prm。各种力场的参数文件包含在主
Gaussian目录中(即,$g09root/g09)。如果想用外部文件指定整个力场,需要使用像下面
的输入文件:
# UFF=SoftOnly
Read-in force field example
分子说明
@$g09root/g09/oplsaa.prm
注意,分子力学关键词的选择是任意的,因为整个力场只依赖于读入的函数和参数,而
与内部值无关(换句话说,指定Amber和UFF会产生同样的结果)。
函数说明中的通配符。通配符可用于输入文件给出的函数说明。例如,下面的Amber
弯曲函数指定CM类型中心碳原子的所有弯曲具有相同的参数值:
HrmBnd1 * CM * 70.0 120.0
用通配符还可以包含通用项目和其它更具体的项目:
HrmBnd1 * CM * 70.0 120.0
HrmBnd1 HA CM HA 40.0 120.0
对于特定结构,程序默认使用最具体的项目(这里,键角以CM碳原子为中心)。由项
目内的通配符个数确定相对特殊性。如果恰好有不同的项目具有相同的特定性,程序将用多
匹配项目的出错信息终止计算。在这些情况下,可以用FirstEquiv或LastEquiv选项分别要
求程序使用第一个或最后一个匹配。但是注意,在典型情况下,多匹配项目的信息表示软参
数集存在不一致性,这需要加以研究并解决。
注意,通配符/项目的特定性不会影响使用的项目来自硬连接参数集还是软参数集。因
此,如果指定了SoftFirst,将会使用输入文件中有三个通配符的项目,即使是硬连接参数集
的项目精确地含有所研究的原子类型序列。
子结构:用键级和其它结构特性选择参数。子结构提供了使用附加结构信息的机制,
例如键级和键角的值,用于确定使用哪些参数。考虑丁二烯(图例见下)。碳原子都是CM
- 142 -
类型,但是有两个键形式上为双键,一个为单键。对每种定义的原子类型组合的伸缩项,
Amber默认使用相同的力常数和平衡键长。但是,可以用子结构为不同类型的键分配不同的
值。
子结构编号添加在函数名之后,中间用连字符隔开:例如,HrmStr-1,HrmStr-2,等。
在本例中,数值为键级(参见以下关于子结构的定义)。有时,可以使用多个子结构后缀(例
如,AmbTrs-1-2)。
丁二烯计算的输入文件显示在分子结构之后:
丁二烯分子
# Amber=SoftFirst Geom=Connectivity
Butadiene
0 1
C-CM -2.49 -0.07 0.00
H-HA -1.96 -1.00 0.00
H-HA -3.56 -0.07 0.00
C-CM -1.82 1.09 0.00
H-HA -2.35 2.02 0.00
C-CM -0.28 1.09 0.00
H-HA 0.24 0.16 0.00
C-CM 0.39 2.26 -0.00
H-HA -0.13 3.19 0.00
H-HA 1.46 2.26 -0.00
1 2 1.0 3 1.0 4 2.0
2
3
4 5 1.0 6 1.0
5
6 7 1.0 8 2.0
7
8 9 1.0 10 1.0
9
10
HrmBnd1 HA CM HA 40.0 120.0
参数值仅用于演示。
HrmBnd1 CM CM CM 70.0 120.0
HrmStr1-1 CM CM 350.0 1.51
子结构:特定键级的伸缩参数。
HrmStr1-2 CM CM 500.0 1.37
本例对双键指定了较大的力常数和较小的键长,用于HrmStr1函数。
注意,本例中使用的参数值仅用于演示。事实上,对于正式的计算,我们还需要用相同
的子结构机制,对特定单、双中心的键定义扭转参数。
- 143 -
除了键级,其它子结构的例子还有,项是否位于环状结构,多少氢原子与其相连,等等。
注意,许多函数可以用多个结构后缀:例如,HrmStr1-1-0-4。
在不同函数中,不同子结构位置具有相对一致的含义。第一个子结构后缀定义键级或键
范围,第二个指定与当前原子成键的原子个数,第三个描述环状环境(如果有的话)。任何
子结构位置出现的零值是通配符或占位符,表示对该项目忽略特定的子结构。最后一个子结
构如果不需要,可以类似地设定为0,或直接忽略。
以下子结构用于只依赖于原子类型的函数。
第三个子结构:环状结构(第一和第二个子结构必须为
0
):
-0-0-1 不用于以下的任何环
-0-0-3 原子位于三元环
-0-0-4 原子位于四元环
-0-0-5 原子位于五元环
以下子结构用于键伸缩的函数。
第一个子结构:键级
-0 忽略这个子结构(子结构的“通配符”)
-1 单键:0.00 ≤ 键级 < 1.50
-2 双键:1.50 ≤ 键级 < 2.50
-3 三键:键级 ≥ 2.50
第三个子结构:环状结构(若使用,第二个子结构必须为
0
):
-x-0-1 不用于以下的任何环
-x-0-3 键位于三元环
-x-0-4 键位于四元环
-x-0-5 键位于五元环
以下子结构用于键角的函数(角度单位:度)。
第一个子结构:键角值的范围
-1 0° ≤ θ ≤ 45°
-2 45° < θ ≤ 135°
-3 135° < θ ≤ 180°
第二个子结构:成键原子个数
-x-0 忽略这个子结构(子结构的“通配符”)
-x-n n为与中心原子成键的原子个数
第三个子结构:环状结构
-x-y-0 忽略这个子结构(子结构的“通配符”)
-x-y-1 不用于以下的任何环
-x-y-3 弯曲位于三元环
-x-y-4 弯曲位于四元环
-x-y-5 弯曲位于五元环
第四个子结构:成键的氢原子数
-x-y-z-n 有n+1个氢原子与中心原子成键(不包括仅用来构成键角的氢原子)。
以下子结构用于二面角的函数。
第一个子结构:键级
-0 忽略这个子结构(子结构的“通配符”)
-1 单中心键:0.00 ≤ 键级 < 1.50
-2 双中心键:1.50 ≤ 键级 < 2.50
- 144 -
-3 三中心键:键级 ≥ 2.50
第二个子结构:中心键的键型
-x-0 忽略这个子结构(子结构的“通配符”)
-x-1 共振中心键(1.30 ≤ 键级 ≤ 1.70)
-x-2 氨基中心键(优先于共振)
-x-3 其它中心键型
第三个子结构:环状结构
-x-y-1 不用于以下的任何环
-x-y-4 二面角位于四元环
-x-y-5 二面角位于五元环
例如,HrmStr1-2-0-4指定HrmStr1项仅用于四元环内双键的伸缩(比较HrmStr1-2:它用
于所有的双键)。零值的子结构类似于通配符:HrnStr1-0-0-4指定该项用于任何键级的伸缩,
但必须位于四元环内。
注意,并不总是需要子结构。有些函数的定义中直接包含了键级以及类似的结构信息。
例如,UFF力场的HrmStr3伸缩项在其第三个参数中包含键级。对于每个值设定为-1.0的键,
在UFF计算过程中会把这个参数传递到此函数,用于指定像下面的输入文件(并加上UFF关
键词的合适选项):
HrmStr3 * * -1.0 0.1332
定义新的原子类型。Gaussian从Dreiding,UFF,和 Amber力场知道标准的MM原子类型。
但是,如果力场尚未对所研究的体系参数化,那么可用的原子类型可能都不合适。这种情况
下可以定义新的原子类型,只需在分子说明部分使用新的原子类型标记。当然,由于硬连接
列表中不存在新原子类型,还必须在输入文件中定义所有需要的函数。
非键相互作用主函数。如我们所看到的,指定非键相互作用项可能相当长,需要很多
NBTerm项。如果在输入文件指定非键相互作用(用MM关键词的某个选项),它们可以用
NonBon函数定义。这个函数使用单个项目定义非键相互作用,并由程序自动展开到合适的
NBDir和NBTerm项目中。
这个函数有以下一般形式:
NonBon V-Type C-Type V-Cutoff C-Cutoff VScale1 VScale2 VScale3 CScale1 CScale2 CScale3
V-Type和C-Type为整数,分别表示使用的范德华相互作用和库仑相互作用的类型。其余的项
为每种相互作用类型的截断值和三个换算因子;换算因子用于1,2,3,或更多个键隔开的
原子。若某个换算因子设定为-1.0,则使用标准的Amber换算因子1.0/1.2。
例如,这是一个用于标准Amber力场的函数:
NonBon 3 1 0 0 0.0 0.0 0.5 0.0 0.0 -1.0
这个函数指定用Amber算法的范德华相互作用,1/R库仑相互作用,并且无截断。范德华相
互作用对超过两个键距离的原子用0.5换算,但在更近的原子对中删除。库仑相互作用对超
过两个键距离的原子用1.0/1.2换算,同样也在更近的原子对中删除。
全局参数。有些情况下,势函数所需的参数与特定原子类型无关。一个例子就是介电常
数。对于这类情况,我们使用全局参数:定义在势函数中可用作参数的值。所有定义的全局
参数都在输出文件的MM力场定义部分列出(需要用IOp(4/33=3))。这里是一个定义介电常
数的例子:
- 145 -

Dielc 78.39
全局参数还可以定义成矢量格式或矩阵格式。以下是从MM2力场定义中提取,用于演
示在函数中使用矩阵,它在计算范德华相互作用中指定氢的移动(即,原子间距的换算):
VDWShf1 * 1 0.0
默认对VDWShf2使用索引值1
VDWShf1 MM5 2 -1.0
对原子类型MM5使用索引值2
VDWShf1 MM36 3 -1.0
对原子类型MM36使用索引值3
VDWShf2 1 2 0.915
换算因子:矩阵元(1,2)
VDWShf2 1 3 0.915
换算因子:矩阵元(1,3)
我们将以相反的顺序考虑这两个函数。VDWShf2为原子间距设定换算因子的下三角矩
阵。矩阵元(1,2)和(1,3)都设定为0.915;其它矩阵元——(1,1),(2,2),(2,3),(3,3)——都使用
默认值,表示在计算范德华相互作用时,不对距离进行换算。注意这一用法与原子类型无关。
VDWShf1函数定义哪些矩阵元用于各种原子类型。第一个项目指定默认索引值为1,余
下的两个项目对原子类型MM5和MM36指定索引值为2和3。这个函数的第三个参数指定此
相互作用所涉及的第二个中心。在第一个项目中,零值函数用作通配符,而在其它两个项目
中,-1值通知程序自动输入合适的中心。
这些函数导致只有当一个原子的类型为MM5(H)或MM36(D)而另一原子具有不同
类型时,所涉及的键才会移动。在求解这些函数时,MM5-X型的相互作用使用矩阵元(1,2),
而MM36-X型使用(1,3)。其它类型的相互作用使用(1,1),而MM5-MM5,MM5-MM36,和
MM36-MM36分别使用(2,2),(2,3),和(3,3)——它们都使用未换算的原子间距。
利用这一机制,参数的定义更清楚,更简洁。如果不用它,每对原子就需要一个项目才
能达到同样的目的。
步进(Step-down)表。通配符提供了指定通用参数/默认值的机制,它们不直接依赖于
特定项中所涉及的每种原子类型。步进表具有相同的目的,但是更复杂。考虑下面的例子,
它来自UFF力场。在这些项目中,UseTable和StepDown项目既不是势函数也不包含参数,而
是描述步进机制,用于处理涉及势函数和包含某些参数的项目。
在下面的例子中,第一组UFFBnd3行定义势函数,而UFFBnd2行和第二组UFFBnd3行使
用步进机制:
UseTable UFFBnd2 #3
指定用于特定函数的步进表
UseTable UFFBnd3 #3
StepDown UFFBnd2 0 1 0
特定函数的匹配规则
StepDown UFFBnd2 0 2 0
StepDown UFFBnd3 0 1 0
StepDown UFFBnd3 0 2 0
Table #3 C_R Trig
定义另外的原子类型
Table #3 N_R Trig
Table #3 H_ Lin
Table #3 Li Lin
UFFBnd3 0 C_3 0 109.471 -1.0 -1.0 0.1332
直接用原子类型定义的函数
UFFBnd3 0 N_3 0 106.700 -1.0 -1.0 0.1332
UFFBnd3 0 N_2 0 111.200 -1.0 -1.0 0.1332
UFFBnd3 0 O_3 0 104.510 -1.0 -1.0 0.1332
UFFBnd2-0-2 0 Lin 0 2 -1.0 -1.0 0.1332
直接用步进类型和子结构定义的函数
UFFBnd2-0-3 0 Trig 0 3 -1.0 -1.0 0.1332
UFFBnd3-0-3 0 Lin 0 180.0 -1.0 -1.0 0.1332
UFFBnd3-0-4 0 Lin 0 180.0 -1.0 -1.0 0.1332
UFFBnd3-0-5 0 Lin 0 180.0 -1.0 -1.0 0.1332
UFFBnd3-0-6 0 Lin 0 180.0 -1.0 -1.0 0.1332
UFFBnd3-0-2 0 Trig 0 120.0 -1.0 -1.0 0.1332
- 146 -

UFFBnd3-0-4 0 Trig 0 120.0 -1.0 -1.0 0.1332
UFFBnd3-0-5 0 Trig 0 120.0 -1.0 -1.0 0.1332
UFFBnd3-0-6 0 Trig 0 120.0 -1.0 -1.0 0.1332
在UFF力场中,H_和N_R都是UFF原子类型。如果有键角涉及分子中的这些原子——
H_–N_R–H_——程序需要确定合适的函数和参数。首先它对可用的弯曲函数确定合适的
步进表。在本例中,对可用的弯曲函数UFFBnd2和UFFBnd3使用表#3。
Table项目对指定的原子类型定义替代类型。典型情况下,它们把多个特定原子类型映
射到更通用的伪类型(例如,例子中的Lin和Trig)。这些替代类型允许对函数参数指定范
围更广的默认值,有效地自动估计未定义的参数。
StepDown项目指定选择特定函数项目的规则。在这个例子中,两个弯曲函数使用相同
的两个项目:0 1 0和0 2 0。它们指定程序应当首先寻找能把原始原子类型指定为中心原子的
项目(零值函数是通配符)。若没有出现该项目,那么应当用一个项目把第一个替代原子类
型指定为中心原子。
在例子中,程序会寻找把N_R指定为中心原子的弯曲函数,但是未定义。接下来,程序
寻找中心原子类型为Trig的函数。有四个这样的函数:UFFBnd2-0-3以及第二个子结构为2,
4,5的UFFBnd3-0。使用哪一个依赖于和中心氮原子成键的原子个数。
注意,实际上,步进表可以对各个原子类型指定多个替代类型,并且还能对每个列出的
替代类型包含更多的映射类型。
如果使用步进表,将忽略其它函数说明中的通配符。换句话说,如果使用步进表,那么
就不能在别处使用通配符。
可用的力场项(函数)
一些预备的注意事项:
单位:除非另有说明,否则距离单位都是埃,角度是度,能量是kcal/mol,电荷是原子
单位。
在方程中,R表示距离,θ表示角度。数值和变量下标表示中心,“eq”表示平衡:例
如,R12和Rij分别为原子1,2和原子i,j之间的距离,Ri是原子i的成键半径,Req和θeq是
平衡键长和键角,Req12是原子1,2之间的平衡键长(典型情况下用在角度项中)。
通配符可用在任何函数的定义内。用0或星号表示。
与标准力场函数等效的函数都在说明后用括号表示。
VDW 范德华参数,用于NBDir和NBTerm(MMFF94型的范德华参数见以下的
MMFF94)。
VDW
Atom-type Radius Well-depth
VDW94 MMFF94型范德华参数(用于NBDir和NBTerm)。
VDW94
Atom-type Atomic-pol NE Scale1 Scale2 DFlag
Atomic-pol
原子极化率(Angstrom3)。
NE
价电子的Slater-Kirkwood有效数(无维度)。
Scale1
换算因子(Angstrom1/4)。
Scale2
换算因子(无量纲)。
DFlag
施主型的原子是1.0,受体型的原子是2.0。其它是0.0。
Buf94 MMFF94静电缓冲
Buf94
Atom-type Value
NonBon 非键相互作用主函数。该函数在计算MM能量之前会展开成原子对和一个直
- 147 -

接函数(NBDir和NBTerm)。
NonBon
V-Type C-Type, V-Cutoff C-Cutoff VScale1 VScale2 VScale3 CScale1 CScale2
CScale3
V-Type
是范德华类型:
0 无范德华
1 算数的(同Dreiding)
2 几何的(同UFF)
3 算数的(同Amber)
4 MMFF94型范德华
5 MM2型范德华
6 OPLS型范德华
C-Type
是库仑类型:
0 无库仑作用
1 1/R
2 1/R2
3 1/R缓冲(MMFF94)
10 来自没有截断的偶极-偶极相互作用
V-Cutoff
和
C-Cutoff
分别是范德华和库仑的截断值:
0 无截断
>0 硬截断
<0 软截断
VScale1-3
是间隔为1到3个键的原子对的范德华换算因子。
CScale1-3
是间隔为1到3个键
的原子对的库仑换算因子。如果有换算因子<0.0,则用1.0/1.2换算(同Amber)。
NBDir 库仑和范德华直接(对所有原子对求解)
NBDir
V-Type C-Type V-Cutoff C-Cutoff
V-Type
,
C-Type
,
V-Cutoff
,和
C-Cutoff
的定义同上。
NBTerm 库仑和范德华单项截断值
NBTerm
Atom-type1 Atom-type2 V-Type C-Type V-Cutoff C-Cutoff V-Scale C-Scale
V-Type
,
C-Type
,
V-Cutoff
,
C-Cutoff
,
V-Scale
,和
C-Scale
的定义见上。
AtRad 原子单键半径
AtRad
Atom-type Radius
EffChg 有效电荷(UFF)
EffChg
Atom-type Charge
EleNeg GMP电负性(UFF)
EleNeg
Atom-type Value
Table 步进表
Table
Original-atom-type Stepping-down-type(s)
HrmStr1 简谐伸缩I(Amber 1):ForceC*(R-Req)2
HrmStr1
Atom-type1 Atom-type2 ForceC Req
ForceC
力常数
Req
平衡键长
HrmStr2 简谐伸缩II(Dreiding 4a):ForceC*[R-(Ri+Rj-Delta)]2
HrmStr2
Atom-type1 Atom-type2 ForceC Delta
ForceC 力常数
- 148 -

Delta Delta
Ri
和
Rj为用AtRad设定的原子键半径。
HrmStr3 简谐伸缩III(UFF 1a):k*(R-Rij)2
平衡键长:Rij = (1 - PropC*lnBO)*(Ri + Rj) + Ren
力常数:k = 664.12*Zi*Zj/(Rij3)
电负性校正:Ri*Rj*[Sqrt(Xi) - Sqrt(Xj)]2/(Xi*Ri + Xj*Rj)
HrmStr3
Atom-type1 Atom-type2 BO PropC
BO 键级(小于0时,由即时计算决定)
PropC 比例常数
Ri和Rj是用AtRad定义的原子键半径。Xi和Xj是用EleNeg定义的GMP电负性。Zi和Zj是用
EffChg定义的有效原子电荷。
MrsStr1 Morse伸缩I(Amber):DLim*(e-a(R-Req)-1)2,其中a = Sqrt(
ForceC/DLim
)
MrsStr1
Atom-type1 Atom-type2 ForceC Req DLim
ForceC
力常数
Req
平衡键长
DLim
解离极限
MrsStr2 Morse伸缩II(Dreiding 5a):DLim*(e-a(Ri+Rj-Delta)-1)2,其中a =
Sqrt(
ForceC/DLim
)
MrsStr2
Atom-type1 Atom-type2 ForceC Delta DLim
ForceC
力常数
Delta
Delta
DLim
解离极限
Ri和Rj是用AtRad定义的原子键半径。
MrsStr3 Morse伸缩III(UFF 1b):BO*DLim*(e-a(R-Rij)-1)2,其中a =
Sqrt(
k
/[
BO*PropC
])
平衡键长:Rij = (1 - PropC*lnBO)*(Ri + Rj) + Ren
力常数:k = 664.12*Zi*Zj/Rij3
电负性校正:Ren = Ri*Rj*(Sqrt(Xi) - Sqrt(Xj))2/(Xi*Ri + Xj*Rj)
MrsStr3
Atom-type1 Atom-type2 BO PropC DLim
BO 键级(小于0时,由即时计算决定)
PropC 比例常数
DLim
解离极限
Ri和Rj是用AtRad定义的原子键半径。Xi和Xj是用EleNeg定义的GMP电负性。Zi和Zj是用
EffChg定义的有效原子电荷。
QStr1
四次伸缩I(MMFF94 2):
(Req/2)*(R-ForceC)2*[1+CStr*(R-ForceC+(7/12)*CStr2*(R-ForceC)2]
QStr1
Atom-type1 Atom-type2 ForceC Req CStr
ForceC
力常数(md-Angstrom-1)
Req
平衡键长
CStr
三次伸缩常数(Angstrom-1)
UFFVOx 氧族元素原子扭转能垒(UFF 16)
UFFVOx
Atom-type Barrier
UFFVsp3 原子sp3扭转能垒(UFF 16)
UFFVsp3
Atom-type Barrier
- 149 -

UFFVsp2 原子sp2扭转能垒(UFF 17)
UFFVsp2
Atom-type Barrier
HrmBnd1 简谐弯曲(Amber 1):ForceC*(T-θeq)2
HrmBnd1
Atom-type1 Atom-type2 Atom-type3 ForceC
θ
eq
ForceC
力常数(单位:kcal/(mol*rad2))
θeq 平衡键角
HrmBnd2 简谐弯曲(Dreiding 10a):[ForceC/sin(θeq
2)]*(cos(θ)-cos(θeq))2
HrmBnd2
Atom-type1 Atom-type2 Atom-type3 ForceC
θ
eq
ForceC
力常数
θeq 平衡键角
LinBnd1 Dreiding线性弯曲(Dreiding 10c):ForceC*(1+cos(θ))
LinBnd1
Atom-type1 Atom-type2 Atom-type3 ForceC
ForceC
力常数
UFFBnd3 UFF 3-项弯曲(UFF 11):k*(C0 + C1*cos(θ))+C2*cos(2θ),其中C2=1/(4 *
sin(θeq
2)),C1 = -4*C2*cos(θeq),C0=C2*(2*cos(θeq
2)+1)
力常数:k = 664.12*Zi*Zk*(3*Rij*Rjk*(1-cos(θeq
2))-cos(θeq)*Rik2)/Rik5
UFFBnd3
Atom-type1 Atom-type2 Atom-type3
θ
eq BO12 BO23 PropC
θeq 平衡键角
BO12 原子类型1-原子类型2的键级(小于0时,由即时计算决定)
BO23 原子类型2-原子类型3的键级(小于0时,由即时计算决定)
PropC 比例常数
Ri,Rj和Rk是用AtRad定义的原子键半径。Xi,Xj和Xk是用EleNeg定义的GMP电负性。Zi,
Zj和Zk是用EffChg定义的有效原子电荷。
UFFBnd2 UFF 2-项弯曲(UFF 10):[k/(Per2)]*[1-cos(Per*θ)]
力常数:k = 664.12*Zi*Zk*(3*Rij*Rjk*(1-cos(Per2))-cos(Per)*Rik2)/Rik
5
UFFBnd2
Atom-type1 Atom-type2 Atom-type3 Per BO12 BO23 PropC
Per 周期性:线形是2,三角形是3,平面四方形是4
BO12 原子类型1-原子类型2的键级(小于0时,由即时计算决定)
BO23 原子类型2-原子类型3的键级(小于0时,由即时计算决定)
PropC 比例常数
Ri,Rj和Rk是用AtRad定义的原子键半径。Xi,Xj和Xk是用EleNeg定义的GMP电负性。Zi,
Zj和Zk是用EffChg定义的有效原子电荷。
ZeroBnd 零弯曲项:用于弯曲为零时的罕见情况。使用这一项可以避免由于未定义
角度而产生的程序报错。
ZeroBnd
Atom-type1 Atom-type2 Atom-type3
CubBnd 立方弯曲(MMFF94 3):(ForceC/2)*(1+CBend*(θ-θeq))*(θ-θeq)2
CubBnd
Atom-type1 Atom-type2 Atom-type3 ForceC
θ
eq CBend
ForceC
力常数(单位:md*Angstrom/rad2)
θeq 平衡键角
CBend 三次弯曲常数(单位:deg-1)
LinBnd2 MMFF94线性弯曲(MMFF94 4):ForceC*(1+cos(θ))
LinBnd2
Atom-type1 Atom-type2 Atom-type3 ForceC
ForceC
力常数(单位:md)
AmbTrs Amber扭角(Amber 1):Σi=1,4 (Magi*[1+cos(i*θ-I(i+4))])/NPaths
- 150 -

AmbTrs
Atom-type1 A-type2 A-type3 A-type4 PO1 PO2 PO3 PO4 Mag1 M a g 2 M a g 3 M a g 4 NPaths
PO1-PO4 相偏移
Mag1...Mag4 V/2大小
NPaths 路径数。(小于等于0时,由即时计算决定)
DreiTrs Dreiding扭角(Dreiding 13):V*[1-cos(Period*(θ-PO))]/(2*NPaths)
DreiTrs
Atom-type1 Atom-type2 Atom-type3 Atom-type4 V PO Period NPaths
V 能垒高度V
PO 相偏移
Period 周期性
NPaths 路径数。(小于等于0时,由即时计算决定)
UFFTorC 具有常数能垒高度的UFF 扭角(UFF15):
[V/2]*[1-cos(Period*PO)*cos(V*θ)]/NPaths
UFFTorC
Atom-type1 Atom-type2 Atom-type3 Atom-type4 Period PO V NPaths
Period 周期性
PO 相偏移
V 能垒高度V
NPaths 路径数。小于等于0时,由即时计算决定。
UFFTorB 能垒高度基于键级的UFF扭角(UFF 17):
[V/2]*[1-cos(Period*PO)*cos(Period*θ)]/NPaths,其中 V =
5*Sqrt(Uj*Uk)*[1+4.18*Log(BO12)]
UFFTorB
Atom-type1 Atom-type2 Atom-type3 Atom-type4 Period PO BO12 NPaths
Period 周期性
PO 相偏移
BO12 原子类型1-原子类型2之间的键级(小于0时,由即时计算决定)
NPaths 路径数。小于等于0时,由即时计算决定。
Uj和Uk是UFFVsp2定义的原子常数。
UFFTor1 能垒高度基于原子类型的UFF扭角(UFF 16):
[V/2]*[1-cos(Period*PO)*cos(Period*θ)]/NPaths,其中V=Sqrt(Vj*Vk)
UFFTor1
Atom-type1 Atom-type2 Atom-type3 Atom-type4 Period PO NPaths
Period 周期性
PO 相偏移
NPaths 路径数。小于等于0时,由即时计算决定。
Vj和Vk是UFFVsp3定义的原子常数。
UFFTor2 能垒高度基于原子类型的UFF扭角(UFF 16)(和UFFTor1不同,这里采
用的是原子参数):[V/2]*[1-cos(Period*PO)*cos(Period*θ)]/NPAths,其中
V=Sqrt(Vj*Vk)
UFFTor2
Atom-type1 Atom-type2 Atom-type3 Atom-type4 Period PO NPaths
Period 周期性
PO 相偏移
NPaths 路径数。小于等于0时,由即时计算决定。
Vj和Vk是UFFVOx定义的原子常数。
VDWDreiTRS 与Gaussian 98兼容的Dreiding特殊扭角。处理过程中用DreiTRS代替,使用
以下参数:
如果有三个原子和第三个中心成键,且第四个中心是H,则删除该项。
- 151 -

如果有三个原子和第三个中心成键,且其中至少有一个是H但第四个
中心不是H,则使用以下参数:V=4.0,PO=0.0,Period=3.0,和NPaths=
-1.0。
否则使用以下值:V=1.0,PO=0.0,Period=6.0,和NPaths= -1.0。
OldTor
Atom-type1 Atom-type2 Atom-type3 Atom-type4
ImpTrs 不适当扭角(Amber 1):Mag*[1+cos(Period*(θ-PO))]
ImpTrs
Atom-type1 Atom-type2 Atom-type3 Atom-type4 Mag PO Period
Mag V/2大小
PO 相偏移
Period 周期性
Wilson 三项Wilson角(Dreiding 28c,UFF 19):ForceC*(C1 + C2*cos(θ) +
C3*cos(2θ)),对三个Wilson角θ做平均。
Wilson
Atom-type1 Atom-type2 Atom-type3 Atom-type4 ForceC C1 C2 C3
ForceC 力常数
C1, C2, C3 因子
HrmWil 简谐Wilson角I(MMFF94 6):(ForceC/2)*(θ2),对三个Wilson角θ求和。
HrmWil1
Atom-type1 Atom-type2 Atom-type3 Atom-type4 ForceC
ForceC 力常数
MM2Wil MM2 Wilson六阶弯曲(MM2):Σi=1,2,3(
ForceCi
/2)*(θi
2)*[1+6
Bend
*(θi
4)]
MM2Wil
Atom-type1 Atom-type2 Atom-type3 Atom-type4 ForceC1 ForceC2 ForceC3 6Bend
ForceC1
–
ForceC3
力常数
6Bend
六阶弯曲常数(单位deg-4)
StrBnd 伸缩-弯曲I(MMFF94 5):(ForceC1*(R12-Req12)+ForceC2*(R32-Req23))*(θ-θeq)
StrBnd
Atom-type1 Atom-type2 Atom-type3 ForceC1 ForceC2 Req12 Req23
θ
eq
ForceC1,
ForceC2
力常数
Req12, Req23 平衡键长(如果<0,从合适的伸缩项恢复)
θeq 平衡键角
CubStr1 三次伸缩1(MM2):(
ForceC
/2)*(1–
CStr
*(R–
Req
))*(R–
Req
)2
CubStr1
Atom-type1 Atom-type2 ForceC Req CStr
ForceC 力常数
Req 平衡键长
CStr
三次伸缩常数(Angstrom-1)
SixBnd1 六阶弯曲(MM2):(
ForceC
/2)*(θ–θ
eq
)2(1+6
Bend
*(θ–θ
eq
)4)
SixBnd1
Atom-type1 Atom-type2 Atom-type3 ForceC
θ
eq 6Bend
ForceC 力常数
θ
eq
平衡键角
6Bend
六阶弯曲常数(单位deg-4)
MM2Tors MM2扭角(MM2):
En1
/2(1+cosθ)+
En2
/2(1–cos2θ)+
En3
/2(1+cos3θ)
MM2Tors
Atom-type1 Atom-type2 Atom-type3 Atom-type4 En1 En2 En3
En1
–
En3
能量
MM2VDW MM2范德华函数的参数(MM2)
MM2VDW
Par1 Par1 Par3 Par4 Par5
- 152 -

Par1
–
Par5
参数
VDWX 范德华参数矩阵(MM2)。这些值不依赖原子类型,可用作原子参数的替代,
用于MM2力场(见以上讨论)。
VDWX
Index1 Index2 Radius Well-depth IsDef
IsDef
表示原子对是(1.0)否(0.0)已被定义。
VDWI 原子指数,用于标记VDWXM中的矩阵(MM2)
VDWI
Atom-type Index
VDWShf2 在范德华计算中,用于移动原子的参数矩阵(MM2)。
VDWShf2
Index1 Index2 Shift
VDWShf1 VDWShf2中,原子在矩阵内的索引(MM2)
VDWShf1
Atom-type Index BondTo
BondTo
成键的中心(若为-1.0,由程序确定)
SixBndI 六阶弯曲(MM2):(
ForceC
/2)*(θ–θ
eq
)2(1+6
Bend
*(θ–θ
eq
)4)
SixBndI
Atom-type1 Atom-type2 Atom-type3 ForceC
θ
eq 6Bend S
ForceC 力常数
θ
eq
平衡键角
6Bend
六阶弯曲常数(单位deg-4)
Flag
如果≥ 0.0,禁止投影到平面,即使中心原子为三角形。
对于三角形中心,这个弯曲会投影到平面上。若中心非三角形,则计算常规的弯曲。
Dipole 键偶极
Dipole
Atom-type1 Atom-type2 DMom DPos
DMom
偶极矩(单位:Debye)
DPos
偶极沿着原子1-原子2的位置
DielC 介电常数。它可以通过参数文件指定介电常数;默认是1。
DielC
DielConst
DielConst
介电常数
QStr2 四次伸缩2(MM2/Tinker):
(
ForceC
>/2)*(1+
CStr
*(R–
Req
)+
QStr
*(R–
Req
)2)(R–
Req
)2)
QStr2
Atom-type1 Atom-type2 ForceC Req CStr QStr
ForceC
力常数
Req
平衡键长
CStr
三次伸缩常数(Angstrom-1)
QStr
四次伸缩常数(Angstrom-2)
CubStr2 三次伸缩2(用于兼容旧的MMVB):(
ForceC
>/2)*(1–
CStr
*(R–
Req
))*(R–
Req
)2)
CubStr2
Atom-type1 Atom-type2 ForceC Req CStr
ForceC
力常数
Req
平衡键长
CStr
三次伸缩常数(Angstrom-1);R > 15 Å时设为0
NBonds 键的形式个数,基于该中心的原子类型
NBonds
Atom-type NumBnd
NumBnd
键的形式个数
StrUnit 伸缩函数中的力常数单位
StrUnit
Flag
Flag
0表示单位kcal/(mol*Angstrom2);1表示单位md/Angstrom
- 153 -

BndUnit 弯曲函数中的力常数单位
BndUnit
Flag
Flag
0表示单位kcal/(mol*rad2);1表示单位md*Angstrom/rad2
TorUnit 扭转函数中能垒的单位
TorUnit
Flag
Flag
0表示单位kcal/mol;1表示单位md*Angstrom
OOPUnit 脱离平面弯曲函数的力常数单位
OOPUnit
Flag
Flag
0表示单位kcal/(mol*rad2);1表示单位md*Angstrom/rad2
SBUnit 伸缩-弯曲函数的力常数单位
SBUnit
Flag
Flag
0表示单位kcal/(mol*Angstrom*rad);1表示单位md/rad
StrFact 伸缩函数的因子
StrFact
Value
BndFact 弯曲函数的因子
BndFact
Value
TorFact 扭转函数的因子
TorFact
Value
OOPFact 脱离平面函数的因子
OOPFact
Value
SBFact 伸缩-弯曲函数的因子
SBFact
Value
MNDO
说明
这个方法关键词使用 MNDO 哈密顿量[Dewar77, Dewar78, Dewar78a, Davis81, Dewar83, Dewar83a,
Dewar84, Dewar85a, Dewar86]进行半经验计算。不需要指定基组关键词。
适用性
能量,“解析”梯度,和数值频率。限制性开壳层(RO)波函的优化计算只能使用
Fletcher-Powell 和赝 Newton-Raphson 方法(Opt 关键词的选项分别是 FP 和EnOnly)。
例子
出现在输出文件中的 MNDO 能量显示如下:
SCF Done: E(UMNDO) = -8.08016620373 A.U. after 11 cycles
能量用 MNDO 模型定义。
- 154 -
MP和双杂化DFT方法
MP2,MP3,MP4,MP5,B2PLYP,mPW2PLYP
说明
MPn 方法关键词要求在 Hartree-Fock 计算(默认对单重态用 RHF,高自旋态用 UHF)之
后接着进行 Møller-Plesset 关联能校正的计算[Moller34],对 MP2 截断到二阶[Head-Gordon88a,
Saebo89, Frisch90b, Frisch90c, Head-Gordon94],对 MP3 截断到三阶[Pople76, Pople77],对 MP4 截断到
四阶[Raghavachari78],对 MP5 截断到五阶[Raghavachari90]。解析梯度可用于 MP2 [Pople79, Handy84,
Frisch90b, Frisch90c],MP3 和MP4(SDQ) [Trucks88, Trucks88a],解析频率可用于 MP2 [Head-Gordon94]。
还可以计算 ROMP2,ROMP3,和 ROMP4 能量[Knowles91, Lauderdale91, Lauderdale92]。
Gaussian 09 还包含一些双杂化方法,它把精确 HF 交换和类 MP2 的关联与 DFT 计算组
合。这些方法具有和 MP2(而不是 DFT)相同的计算量。Gaussian 09 还包含 Grimme 的 B2PLYP
[Grimme06a]和 mPW2PLYP [Schwabe06]方法(使用和方法相同的关键词);指定经验色散校正变体需
要为关键词加上后缀 D:例如 B2PLYP 加上经验色散为 B2PLYPD [Schwabe07]。可以计算能量,
梯度,和频率。
MP4 的变体
MP4(DQ)指定进行只使用双激发和四激发的空间,MP4(SDQ)使用单激发,双激发和四
激发,MP4(SDTQ) 使用单激发,双激发,三激发和四激发的完全 MP4 [Raghavachari78,
Raghavachari80]。仅指定 MP4 默认为 MP4(SDTQ)。
MP5 的限制
MP5 代码仅为开壳层情况而写,因此指定 MP5 默认是 UMP5 计算。这个方法需要 O3V3
的磁盘存储空间和 O4V4的 CPU 时间。
冻芯选项
FC 这个关键词可以使用所有的冻芯选项。完整的信息参见 FC 选项的说明。
MP2 和双杂化方法的选择算法选项
注意:将根据%Mem 和MaxDisk 的设置,自动选择 MP2 的合适算法。因此这些选项几乎
用不到(更多信息参见附录:效率的考虑)。
FullDirect 强制使用“完全直接”算法,除了 SCF 所需要的磁盘空间外无额外的磁盘空间
需求。最少需要 2OVN 字的主内存(O为占据轨道数,V为虚轨道数,N为
基函数数量)。这几乎不是一个好的选择,只适用于有很大的主内存而磁盘空
间很有限的计算机。
SemiDirect 强制使用半直接算法。
Direct 使用某种直接方法。根据内存,磁盘空间限制和计算问题的大小,决定选用
常驻内存,完全直接,或是半直接方法。
InCore 强制使用常驻内存算法。这种方法如果适用,会非常快,但需要使用 N4/4 字
的内存。通常和 SCF=InCore 同时使用。NoInCore 禁止使用常驻内存算法。
适用性
- 155 -

MP2,B2PLYP[D],mPW2PLYP[D]:能量,解析梯度和解析频率。
MP3,MP4(SD)和MP4(SDQ):能量,解析梯度和数值频率。
MP4(SDTQ)和MP5:解析能量,数值梯度和数值频率。
RO 可以与 MP2,MP3,和 MP4 组合,仅能用于能量计算。
有关的关键词
HF,SCF,Transformation,MaxDisk
例子
能量。MP2 能量在输出中的形式如下,以 EUMP2 标示:
E2= -.3906492545D-01 EUMP2= -.75003727493390D+02
接着是高阶 Møller-Plesset 方法的能量。以下是 MP4(SDTQ)计算的输出:
Time for triples= .04 seconds.
MP4(T)= -.55601167D-04
E3= -.10847902D-01 EUMP3= -.75014575395D+02
E4(DQ)= -.32068082D-02 UMP4(DQ)= -.75017782203D+02
E4(SDQ)= -.33238377D-02 UMP4(SDQ)= -.75017899233D+02
E4(SDTQ)= -.33794389D-02 UMP4(SDTQ)= -.75017954834D+02
以 EUMP3 标示的是 MP3 能量,然后是多种 MP4 级别的校正能量。最后一行是 MP4(SDTQ)
计算输出(以 UMP4(SDTQ)标示)。
B2PLYP 输出的例子。B2PLYP 能量在输出中的标示为 E(B2PLYP):
E2(B2PLYP) = -0.3262340664D-01 E(B2PLYP) = -0.39113226645200D+02
Name
说明
这个关键词指定把用户名存入到计算的存档项目中。它把所需的用户名作为参数(例如,
Name=RChavez)。在 UNIX 系统中,默认的用户名是操作系统级别的用户在运行该任务时
的登陆名。
有关的关键词
Test
NMR
说明
这个特性关键词指定使用 Hartree-Fock 方法,所有的 DFT 方法和 MP2 方法进行 NMR 屏
蔽张量和磁化率的计算[Gauss92, Gauss93, Gauss95, Cheeseman96]。
NMR 屏蔽张量可以用规范变换连续集(Continuous Set of Gauge Transformations,CSGT)
方法[Keith92, Keith93, Cheeseman96]和规范不变原子轨道(Gauge-Independant Atomic Orbital,
- 156 -
GIAO)方法[London37, McWeeny62, Ditchfield74, Wolinski90, Cheeseman96]进行计算。磁化率也可以用
GIAO [Ruud93]和CSGT 方法计算。Gaussian 还提供了 IGAIM 方法[Keith92, Keith93](略微做了
修改的 CSGT 方法)和单原点(Single Origin)方法,都可以用来计算屏蔽张量和磁化率。
用于 NMR 计算的结构需要先在较高精度的理论级别上优化。注意 CSGT 计算必需使用
大型基组才能得到准确的结果。
在 NMR 任务中使用 SpinSpin 选项,可以计算自旋-自旋耦合常数[Helgaker00, Sychrovsky00,
Barone02, Peralta03, Deng06]。
选项
SpinSpin 除了通常的 NMR 特性以外,还计算自旋-自旋耦合常数。需要知道,这一计
算类型的额外耗时约是振动频率计算的两倍。它只能用于 Hartree-Fock 和
DFT 方法。
Mixed 请求进行两步的自旋-自旋耦合计算[Deng06]。这个选项会导致运行两个任务步
骤。在第一个任务中,通过非收缩基组并对芯添加紧凑的极化函数来修改用
户指定的基组,使其符合 Fermi 接触项。在第二步,用执行路径部分指定的
未修改基组计算自旋-自旋耦合中的其它三项。最终结果出现在第二个任务的
结尾,其中包含了来自第一步的 Fermi 接触贡献。这可以显著改善自旋-自旋
耦合常数的精度,特别是当计算使用了典型的面向价电子的基组时,例如,
6-311G+(d,p),aug-CC-pVDZ,或 aug-CC-pVTZ。与使用修改的基组并加上
紧凑的极化函数计算全部四项相比,这个计算更快。
ReadAtoms 仅对选择的原子读取自旋-自旋耦合常数。原子列表在单独的输入部分指定
(用空行结束)。这个列表在最初是空的。
输入部分使用以下格式:
atoms=
list
[notatoms=
list
]
其中每个 list 是原子序号,原子序号范围,和/或原子类型的列表,用逗号或
空格隔开。可以连续使用关键词,原子列表最初是空的。这里是一些例子:
atoms=3-6,17 notatoms=5
把原子 3,4,6,和 17 加入原子列表。
atoms=3 C 18-30 notatoms=H
加入原子 3,18-30 中的所有 C 和非氢原子。
atoms=C N notatoms=5
加入所有的 C 和 N 原子,原子 5 除外。
atoms=1-5 notatoms=H atoms=8-10
加入原子 1-5 的非氢原子,并假如原子
8-10 但不考虑其对称性。
Bare integers without a keyword are interpreted as atom numbers:
1,3,5 7 Adds atoms 1, 3, 5 and 7.
CSGT 指定只用 CSGT 方法计算 NMR 特性。
GIAO 指定只用 GIAO 方法计算 NMR 特性。这是默认选项。
IGAIM 用原子中心作为规范原点。
SingleOrigin
使用单个规范原点。这个方法只是用于和其它方法作比较,一般并不推荐。
All 使用 SingleOrgin,IGAIM 和CSGT 所有三种方法计算 NMR 特性。
PrintEigenvectors
显示每个原子屏蔽张量的本征矢。
FCOnly 只计算 Fermi 接触自旋-自旋项。
ReadFC 从检查点文件读取 Fermi 接触自旋-自旋项,接下来计算其它的自旋-自旋耦
- 157 -

合项。
Susceptibility
计算磁化率和屏蔽。
适用性
SCF,DFT 和 MP2 方法。NMR 可以跟 SCRF 合用。NMR 和Freq 现在可位于同一个执行路径,
用于 HF 和 DFT。
例子
这是一个 NMR 默认输出的例子:
Magnetic properties (GIAO method)
Magnetic shielding (ppm):
1 C Isotropic = 57.7345 Anisotropy = 194.4092
XX= 48.4143 YX= .0000 ZX= .0000
XY= .0000 YY= -62.5514 ZY= .0000
XZ= .0000 YZ= .0000 ZZ= 187.3406
2 H Isotropic = 23.9397 Anisotropy = 5.2745
XX= 27.3287 YX= .0000 ZX= .0000
XY= .0000 YY= 24.0670 ZY= .0000
XZ= .0000 YZ= .0000 ZZ= 20.4233
对于这个分子体系,所有给定类型原子的值都是相同的,所以前两个原子的输出之后被
截掉。
下面是自旋-自旋耦合计算的附加输出:
Total nuclear spin-spin coupling K (Hz):
1 2
1 0.000000D+00
2 0.147308D+02 0.000000D+00
Total nuclear spin-spin coupling J (Hz):
1 2
1 0.000000D+00
2 0.432614D+03 0.000000D+00
耦合常数的各个分量打印在输出文件的这部分之前。以倒三角形式打印每对原子间各向
同性的自旋-自旋耦合矩阵。K矩阵给出和同位素无关的值,J矩阵给出了在任务中使用特定
同位素的值(不论是另外指定的同位素还是默认同位素)。
NoDensityFit
说明
在纯 DFT 的计算中,默认关闭密度拟合的使用。程序中默认不打开密度拟合,因此这
个关键词主要用于覆盖 Default.Route 文件中由关键词 DensityFit 设置的密度拟合选项。
NoDenFit 是NoDensityFit 的同义词。
适用性
只能用于使用纯泛函(非杂化泛函)的 DFT 计算。
- 158 -
有关的关键词
DensityFit
ONIOM
说明
这个关键词进行两层或三层的 ONIOM [Dapprich99]计算。最近的开发参见[Vreven06, Vreven06b,
Clemente08, Vreven08],早期的工作见[Maseras95, Humbel96, Matsubara96, Svensson96, Svensson96a, Vreven00]。在这个
流程中,要研究的分子体系分成两层或三层,分别用不同的模型化学处理。接下来的结果自
动组合到最终要预测的结果。分层在传统上称为低层,中层,和高层。原子默认放置到高层
(从某种角度来看,常规的计算可以视为单层 ONIOM)。层的分配作为分子说明的一部分
指定(见下)。GaussView 提供了多个图形工具用于简化 ONIOM 计算的设置。
对于MO:MM和MO:MO:MM任务,ONIOM优化流程使用微迭代[Vreven03],还可以使用可选的二
次耦合算法[Vreven06]。后者(需要用Opt=QuadMacro指定)考虑了模型体系内的原子和MM层内
的原子之间的耦合,可以产生比常规微迭代算法更准确的走步[Vreven06a]。
MO:MM 和 MO:MO:MM 计算可以利用电子内嵌方法(ONIOM=EmbedCharge 选项)。电子内
嵌将 MM 区的部分电荷加入到量子力学哈密顿中。这一技术对 QM 区和 MM 区之间的静电相互
作用提供了更好的描述(因为是在 QM 级别进行处理),并且允许极化 QM 波函。
对于 MO:MM 和 MO:MO:MM 优化以及 IRC,有以下几点有关的考虑(注意这几点都不涉及
没有 MM 的 ONIOM):
默认的优化算法对模型体系的内坐标走步使用 RFO 算法,而对真实体系(即,仅用 MM
处理的体系)内的原子用线性微迭代进行优化。对于最小点的计算,这种默认算法通常
是最佳选择。
对于出现收敛问题的情况,主要的替代选项是 Opt=QuadMac,它会对所有原子的坐标进
行二次型走步。这种优化可以对内坐标使用更新的近似 Hessian,或者使用解析计算的
Hessian(见下)。它计算低级(MM)Hessian 与所需矢量的乘积。
如果仍有收敛问题,可以试试 Opt=(QuadMac,CalcFC)或Opt=(QuadMac,CalcAll)。
对于过渡态结构,QuadMac 选项有助于保证在合适的过渡结构方向上移动,因此
Opt=(QuadMac,TS,CalcFC)通常是个很好的选项。
对于最小点和过渡结构,通常最有效的方法是先做力学内嵌优化,接下来用电子内嵌,
对得到的优化结构执行第二次优化(而不是从一开始就用电子内嵌进行优化)。
对于 IRC,首先运行频率计算,确认找到了合适的过渡结构。接下来用任务的力常数,
通过 IRC=RCFC 开始 IRC。对于非常大的体系,需要包含 Freq=SaveNM 选项,这样 IRC=RCFC
就不用重算简正模式了。MO:MM 的默认 IRC 算法为 IRC=EulerPC,它最适合于大体系。
但是对于中小体系,IRC=HPC 可能更好。
使用 ONIOM 的例子,参见[Lundberg09]。
ONIOM 计算也可以对一个或多个理论级别使用外部程序。细节参见 External 关键词。
必需的输入
ONIOM 关键词的选项需要指定两到三个需要的模型化学,顺序为高,中,低(最后一
个可以忽略)。不同模型之间用冒号分开。例如,这是一个三层 ONIOM 计算的执行路径部
- 159 -
分,低层使用 UFF,中层使用 PM6,高层使用 HF:
# ONIOM(HF/6-31G(d):PM6:UFF)
为ONIOM 计算分配原子层是分子说明的一部分,需要在每一行加上一些附加的参数,
语法如下:
atom [freeze-code] coordinate-spec layer [link-atom [bonded-to [scale-fac1 [scale-fac2 [scale-fac3]]]]]
其中 atom 和coordinate-spec 是通常的分子说明中的原子输入。
freeze-code
表示在结构优化过程
中是否冻结原子。如果忽略此值或为
0
,则优化该原子的坐标。若值为
-1
,则冻结该原子。若指定了
其它的负整数,则该原子在优化过程中作为刚性片段的一部分进行处理;所有具有同样负值的原子作
为刚性区域一起移动。刚性区域可以冻结,或者用
Opt=ReadFreeze
激活。
Layer 是对原子分配层的关键词,是 High,Medium 和Low 之一。其它可选的参数指
定如何处理层边界上的原子。可以指定链接原子(可以包含原子类型,部分电荷及其它参数)
替代当前原子。不同层的两个原子之间如果存在共价键,则必须用链接原子使悬键饱和。
注意
:用户必须定义所有链接原子。Gaussian 09不会自动定义链接原子或提供任何默
认设置。GaussView会自动进行设置,但用户仍需做一般考虑并遵循[Clemente08]中的一般规则。
bonded-to
参数指定在高级计算部分,当前原子与哪个原子成键。如果忽略,Gaussian
会尝试自动识别。
通常,Gaussian 09 将原键长(也就是真实体系的键长)乘上一个由程序自动决定的换
算因子,用于确定原子和链接原子间的距离。但是也可以直接指定换算因子。对于两层的计
算,换算因子指定模型系统内链接原子的键长,分别用在低级和高级的计算中。在三层的
ONIOM 计算中,最多可以指定三个换算因子(次序为低,中,高)。所有这些换算因子对应
于文献[Dapprich99]中定义的 g 因子参数,但作了扩展,允许对每一个 ONIOM 计算层使用不同
的值。
如果只指定一个参数,那么在两层的ONIOM计算中,两个换算因子都使用相同的值。三
层ONIOM计算如果只指定一个参数,则三个换算因子都使用相同的值;若只定义了两个参数,
那么第三个换算因子使用第二个值。
如果换算因子直接定义为 0.0,那么程序会用通常的方法确定相应的换算因子。因此,
如果只想改变第二个换算因子(模型体系在中级进行计算),则必须直接设定第一个换算因
子为 0.0。对于三层的 ONIOM 计算,在这种情况下如果不明确指定一个非 0 的值,第三个换
算因子会使用和第二个参数相同的值(也就是说在第二种情况下,忽略这个值和指定为 0.0
具有一样的意义)。
各层的电荷和自旋多重度
可以为ONIOM计算定义多个电荷和自旋多重度。对于两层的ONIOM任务,输入行的格式为:
chrgreal-low spinreal-low [chrgmodel-high spinmodel-high [chrgmodel-low spinmodel-low [chrgreal-high spinreal-high]]]
其中的下标指定这些数值用于哪一层的计算中。第四对数值仅用于ONIOM=SValue计算。如果
只设定一对数值,则所有的层都使用这对数值。如果设定两对数值,则第三对默认使用第二
对的数值。如果在S-value任务中忽略最后一对数值,那么默认为真实体系在低级别的值。
在三层ONIOM的计算中,数值和默认设置的方式类似(在以下的下标中,第一个下标可
以是:
Real
,
Int
=中间体系,
Mod
=模型体系,第二个下标可以是:H,M和L,分别表示高级,
中级,和低级):
cRealL sRealL [cIntM sIntM [cIntL sIntL [cModH sModH [cModM sModM [cModL sModL]]]]]
- 160 -
对于三层的ONIOM=SValue计算,可以额外指定最多三对数值:
... cIntH sIntH [cRealM sRealM [cRealH sRealH]]
如果忽略电荷/自旋多重度的一对值,默认使用下一个较高级计算和/或体系大小的值。
因此,如果指定的数值少于六对或九对,则根据以下方案确定默认的电荷和自旋多重度,其
中每个单元中的数值用于指定用哪一对数值进行相应情况下的计算:
默认的电荷和自旋
指定的数据对个数 (仅用于 SValue)
计算 1 2 3 4 5 6 7 8 9
Real-Low 1 1 1 1 1 1 1 1 1
Int-Med 1 2 2 2 2 2 2 2 2
Int-Low 1 2 3 3 3 3 3 3 3
Model-High 1 2 2 4 4 4 4 4 4
Model-Med 1 2 2 4 5 5 5 5 5
Model-Low 1 2 2 4 5 6 6 6 6
Int-High 1 2 2 2 2 2 7 7 7
Real-Med 1 1 1 1 1 1 1 8 8
Real-High 1 1 1 1 1 1 1 8 9
选项
EmbedCharge 在模型体系的 QM 计算中,使用来自真实体系的 MM 电荷。默认为
NoEmbedCharge。
MK 指定在用电子内嵌的几何优化微迭代过程中,指定使用 Merz-Kollman-Singh
(参见 Population)近似电荷。
MKUFF MKUFF 在用电子内嵌的几何优化微迭代过程中,使用 Merz-Kollman-Singh
近似电荷,但使用对整个周期表做定义的 UFF 半径。这是默认的。
ScaleCharge=
ijklmn
指定在 QM 计算的电子内嵌过程中,MM 电荷的换算参数。这个整数乘以 0.2
得到实际的换算因子。和内层成键的原子使用换算因子 0.2n,两键之外的原
子用 0.2m,等等。但是从 i 到 n 的数值必须单调递减,其中用于所有参数的
最大值位于该参数的最左边。因此,555500,123500 和 500 都等价。默认值
为 500(即,555500),它关闭 QM 区内两键之间的电荷,而其余的电荷不进
行换算。ScaleCharge 意味着 EmbedCharge。
SValue 指定做完全平方进行测试,产生取代的值(S-值)用于 S-值测试[Morokuma01]。
可以为附加的计算指定附加的电荷和自旋多重度数据对(见下)。
Compress 在MO:MM 力学嵌入的二阶导数计算中,进行压缩操作并储存到活性原子中;
这是默认的。NoCompress 进行非压缩的计算。Blank 也进行非压缩的计算,
但去掉非活性原子的贡献(目前仅对核矩微扰不是 0的有:屏蔽和自旋-自旋
耦合张量)。FullMatrix 导致存储整个 Hessian 并用于力学内嵌的
Opt=QuadMac,而不是对真实体系使用算符形式的分子力学 Hessian。这对
中等尺寸的体系更快,但对大体系需要使用更多的磁盘空间。
InputFiles 对每个中间计算打印输入文件,以便独自运行这些计算。OnlyInputFiles 仅
产生文件,但不做任何计算。
- 161 -

适用性
能量,梯度和频率。注意,如果某一指定的模型需要进行数值频率计算,那么将对所有
的模型进行数值频率计算,即使是对这些模型也能进行解析频率计算。
ONIOM也可以对一层或多层进行CIS和TD计算。可以对有关的模型指定Gen,GenECP,和
ChkBas关键词。如果可以的话,也可以用密度拟合基,按照通常的方式指定(参见例子)。
NMR计算也可以使用ONIOM模型。
有关的关键词
Geom=Connect,分子力学关键词,Opt=QuadMacro,External
例子
ONIOM 任务的分子说明。这是一个简单的 ONIOM 输入文件:
# ONIOM(B3LYP/6-31G(d,p):UFF) Opt
2-layer ONIOM optimization
0 1 0 1 0 1
F -1.041506214819 0.000000000000 -2.126109488809 M
F -2.033681935634 -1.142892069126 -0.412218766901 M
F -2.033681935634 1.142892069126 -0.412218766901 M
C -1.299038105677 0.000000000000 -0.750000000000 M H 4
C 0.000000000000 0.000000000000 0.000000000000 H
H 0.000000000000 0.000000000000 1.100000000000 H
O 1.125833024920 0.000000000000 -0.650000000000 H
高层由最后三个原子组成。其它原子位于中层。对第一个碳原子定义了链接原子。
这是一个两层ONIOM计算的输入文件,高层使用DFT方法,低层使用Amber方法。分子说
明部分包含原子类型(对UFF是可选的,而对Amber是必须的)。注意,主原子和链接原子的
说明都要设定原子类型:
# ONIOM(B3LYP/6-31G(d):Amber) Geom=Connectivity
2 layer ONIOM job
0 1 0 1 0 1
整个分子(真实体系),高级模型体系,和低级模型体系的电荷/自旋
C-CA--0.25 0 -4.703834 -1.841116 -0.779093 L
C-CA--0.25 0 -3.331033 -1.841116 -0.779093 L H-HA-0.1 3
C-CA--0.25 0 -2.609095 -0.615995 -0.779093 H
C-CA--0.25 0 -3.326965 0.607871 -0.778723 H
C-CA--0.25 0 -4.748381 0.578498 -0.778569 H
C-CA--0.25 0 -5.419886 -0.619477 -0.778859 L H-HC-0.1 5
H-HA-0.1 0 -0.640022 -1.540960 -0.779336 L
H-HA-0.1 0 -5.264565 -2.787462 -0.779173 L
H-HA-0.1 0 -2.766244 -2.785438 -0.779321 L
C-CA--0.25 0 -1.187368 -0.586452 -0.779356 L H-HA-0.1 3
C-CA--0.25 0 -2.604215 1.832597 -0.778608 H
H-HA-0.1 0 -5.295622 1.532954 -0.778487 L H-HA-0.1 5
H-HA-0.1 0 -6.519523 -0.645844 -0.778757 L
C-CA--0.25 0 -1.231354 1.832665 -0.778881 L H-HC-0.1 11
C-CA--0.25 0 -0.515342 0.610773 -0.779340 L
H-HA-0.1 0 -3.168671 2.777138 -0.778348 L H-HA-0.1 11
H-HA-0.1 0 -0.670662 2.778996 -0.779059 L
H-HA-0.1 0 0.584286 0.637238 -0.779522 L
- 162 -

1 2 1.5 6 1.5 8 1.0
2 3 1.5 9 1.0
3 4 1.5 10 1.5
4 5 1.5 11 1.5
5 6 1.5 12 1.0
6 13 1.0
7 10 1.0
8
9
10 15 1.5
11 14 1.5 16 1.0
12
13
14 15 1.5 17 1.0
15 18 1.0
16
17
18
这个输入文件是由GaussView产生。 注意它用Geom=Connect提供原子连接关系的信息。
这计算任务还演示了在ONOIM任务中使用多个电荷和自旋多重度的值。这个例子只能用于演
示的目的。我们感谢K. Nishimoto教授指出了在运行该计算时的几处科学问题。
复杂的ONIOM计算路径。这是一个复杂ONIOM执行路径的例子:
# ONIOM(BLYP/6-31G(d)/Auto TD=(NStates=8):UFF)
这个例子对DFT高层的含时激发态计算使用密度拟合。
在ONIOM优化计算中冻结原子。ONIOM几何优化可以利用分子说明中可选的第二个位置。
如果忽略的话,这个位置默认是0。如果设为-1,那么在几何优化时相应的原子将被冻结:
C -1 0.0 0.0 0.0
H 0 0.0 0.0 0.9
...
仅在ONIOM计算中,如果这个位置设定为除了-1以外的其它负值,那么在几何优化中会
被作为刚体片段的一部份进行处理:所有具有相同值(< -1)的原子会像刚体一样一起移动。
处理某层内的SCF收敛问题。对初始SCF难于收敛或难于收敛到最低解的情况,以下流程
会有帮助。这是一个初始ONIOM的输入:
%Chk=mychk
# ONIOM(BLYP/3-21G:UFF) Opt Freq
输入文件继续...
首先运行此任务,为高级模型体系的计算创建输入文件,接下来为ONIOM添加选项
OnlyInputFiles用于打印三个不同计算的输入文件:
# ONIOM(BLYP/3-21G:UFF)=OnlyInputFiles Opt Freq
对高级模型使用此输入文件,获得该体系的收敛SCF,并确保把它存入了检查点文件,
例如highmod.chk。使用任何有助于SCF收敛所需的选项(如SCF=QC)。
接下来,用Guess=Input运行ONIOM任务:
- 163 -

%Chk=mychk
# ONIOM(BLYP/3-21G:UFF) Opt Freq Guess=Input
ONIOM Opt Freq
分子说明
highmod.chk
用于 Guess=Input 的检查点文件
当此任务计算初始猜测的时候,它从输入文件读取一行用于判断做什么:generate,
read,或是另一个检查点文件的名称(也就是这里所用的选项)。
对于MO:MO计算,流程是类似的。但是在这种情况下,将执行三次初始猜测(因为所有
的计算都为MO计算),当使用Guess=Input时,对每个猜测都要读入一个输入行。例如,如
果只有模型体系的高级计算需要做另外的收敛计算,输入将是这样:
%chk=mychk
# ONIOM(BLYP/6-31+G*:HF/STO-3G) Opt Freq Guess=Input
2 Layer MO:MO ONIOM Opt Freq
分子说明
generate
对低级真实体系产生初始猜测。
highmod.chk
从该文件读入初始猜测,用于高级模型体系。
generate
对低级模型体系产生初始猜测。
S-值测试。这是ONIOM=SValue选项的部分输出:
S-Values (between gridpoints) and energies:
high 4 -39.322207 7 -39.305712 9
-114.479426 -153.801632 -193.107344
med 2 -39.118688 5 -39.106289 8
-114.041481 -153.160170 -192.266459
low 1 -38.588420 3 -38.577651 6
-112.341899 -150.930320 -189.507971
model mid real
整数是格点号,每个数字之下是能量值。水平方向格点之间是 S-值。这些是用取绝对
能量获得的 s-值。但是在做 S-值测试时需要当心,需要使用相对能量和 S-值(参见文献
[Morokuma01])。
Opt
说明
这个关键词进行分子几何优化计算。计算过程中持续调整分子的几何结构,直到发现势
能面上的稳定点为止。如果使用的方法能计算解析梯度,则利用之。对于 Hartree-Fock,
CIS,MP2,MP3,MP4(SDQ),CID,CISD,CCD,CCSD,QCISD,BD,CASSCF,以及所有的 DFT
和半经验方法,能量最小化计算(优化到局域最小点)和过渡态及高阶鞍点的优化使用的默
认算法是在冗余内坐标下[Pulay79, Fogarasi92, Pulay92, Baker93, Peng93, Peng96]利用 GEDIIS[Li06]的
- 164 -
Berny 方法(对应选项 Redundant)。Berny 算法的简要概述参见本小节的最后一部分。对于
所有无解析梯度的计算方法,默认用本征值跟踪算法(Opt=EF)。
Gaussian 包含用于定位过渡结构的 STQN 方法。该方法由 H. B. Schlegel 等人[Peng93,
Peng96]实现,它用二次同步过渡方法接近过渡态的二次区域,接下来用准牛顿或本征矢跟踪
算法完成优化。和最小化的默认算法类似,它默认用冗余内坐标执行优化。如果提供经验估
算的 Hessian 并有合适的初始结构,这一方法的收敛非常高效。
用QST2 和QST3 选项可以请求使用该方法。QST2 需要在输入中提供两个分子说明,分
别用于反应物和产物,而 QST3 需要三个分子说明:顺序为反应物,产物,和过渡态的初始
结构。在所有分子说明部分,原子的顺序必须相同。关于此方法的输入输出示例,参见例子。
关于几何优化的基本信息,技术以及缺陷,在《Exploring Chemistry with Electronic
Structure Methods》[Foresman96b]一书的第 3 章有详细讨论。对于优化和有关主题的综述文章,
参见[Hratchian05a]。
选择优化的目的
默认的优化搜索局域最小点。
QST2 使用 STQN 方法寻找过渡结构。这个选项需要反应物和产物的结构作为输入,
先后通过两组连续的标题和分子说明部分定义。注意,这两个结构中的原子
顺序必需一致。TS 不能和 QST2 合用。
QST3 使用 STQN 方法寻找过渡结构。这个选项需要反应物,产物和初始的过渡结构
作为输入,先后通过三组连续的标题和分子说明部分定义。注意,这三个结
构中的原子顺序必需一致。TS 不能和 QST3 合用。
TS 请求用 Berny 算法优化过渡态而不是局域最小点。
Saddle=
N
请求用 Berny 算法优化
N
阶鞍点。
Conical 使用态平均 CASSCF 方法搜索圆锥交点或避免交叉点。Conical 的同义词为
Avoided。注意需要使用 CASSCF=SlaterDet 以便找出单重态和三重态之间的
圆锥交点的位置。
修改初始几何的选项
ModRedundant
添加,删除和修改冗余内坐标的定义(包含扫描和坐标限制信息)。这个选项
需要一个单独的输入数据部分,位于分子结构说明部分之后。当与 QST2 或
QST3 合用时,ModRedundant 输入部分必需接在每个几何说明部分之后。
AddRedundant 是ModRedundant 的同义词。
ModRedundant 的输入行部分使用以下语法:
[
Type
]
N
1 [
N
2 [
N
3 [
N
4]]] [[+=]
value
] [A | F] [[
min
]
max
]]
[
Type
]
N
1 [
N
2 [
N
3 [
N
4]]] [[+=]
value
] B [[
min
]
max
]]
[
Type
]
N
1 [
N
2 [
N
3 [
N
4]]] K | R [[
min
]
max
]]
[
Type
]
N
1 [
N
2 [
N
3 [
N
4]]] [[+=]
value
] D [[
min
]
max
]]
[
Type
]
N
1 [
N
2 [
N
3 [
N
4]]] [[+=]
value
] H
diag-elem
[[
min
]
max
]]
[
Type
]
N
1 [
N
2 [
N
3 [
N
4]]] [[+=]
value
] S
nsteps
stepsize
[[
min
]
max
]]
其中的 N1,N2,N3 和N4 为原子编号或通配符(说明见后)。原子编号从
1开始,不包含虚原子。value 对定义的坐标指定新的值,+= value 把坐标增
加value。如果忽略,不改变当前值。
- 165 -
原子编号和坐标值之后是一个字母代码,表示执行的坐标修改;执行代码有
时还要接上面指出的附加参数。如果不包含执行代码,默认执行的操作为增
加坐标。以下是可用的执行代码:
A 激活要优化的坐标,如果它还冻结的话。
F 在优化中冻结坐标。
B 添加坐标,并创建所有有关的坐标。
K 删除坐标,并删除所有包含此坐标的有关坐标。
R 从定义列表中删除坐标(但不包含有关坐标)。
D 从该坐标初始 Hessian 的行和列计算数值二阶导数。
H 把该坐标初始 Hessian 的对角元素改为 diag-elem。
S 执行放松的势能面扫描。设定该坐标的初始值(或它的当前
值)为 value,并把坐标用 stepsize 步长增加 nsteps 次,对得
到的每个初始结构执行优化。
原子编号位置的星号(*)表示通佩符。Min 和Max 用于对包含通配符的坐
标说明部分定义范围(如果没有给出 min 值,则是最大值)。只有在坐标值
位于此范围,才进行执行代码定义的操作。
这里是一些使用通配符的例子:
*
* *
3 *
* * *
* 4 *
* * * *
* 3 4 *
用笛卡尔坐标指定所有原子。
所有定义的键。
原子 3 所有定义的键。
所有定义的键角。
原子 4 周围所有定义的键角。
所有定义的二面角。
在连接原子 3 和 4 的键周围,所有定义的二面角。
默认的坐标类型由定义的原子数决定:一个原子是笛卡尔坐标,两个原子是
键的伸缩,三个原子是键角,四个原子是二面角。此外 type 也可以用来确定
这些坐标类型以及附加的坐标类型:
X 笛卡尔坐标。在这种情况中,value,min 和max 分别解释为 X,
Y,Z坐标。
B 键长。
A 键角。
D 二面角。
L 由三个原子(如果 N4是-1)或四个原子定义的线性弯曲,其
中第四个原子用于确定线性弯曲的两个正交方向。在这种情
况中,value,min 和max 中的每个都是一对数值,用于定义
两个正交的弯曲分量。
O 对一个中心(N1)和三个连接原子,定义平面外的弯曲坐标。
关于使用 ModRedundant 关键词的演示,见本节后面的例子。
ReadFreeze 读取一个输入部分,用于修改原子优化或是冻结的设置。原子列表在一个单
独的输入部分指定(用空行结束)。最初的原子列表包含冻结(即,不做优化)
的原子,它们基于分子说明部分的设定,或从检查点文件读入结构的设定。
输入部分使用以下格式:
atoms=
list
[notatoms=
list
]
其中每个
list
是一列用逗号或空格隔开的原子编号,原子编号范围,或/和
- 166 -
原子类型。依次使用关键词,而原子列表最初是空的。这里有一些例子:
atoms=3-6,17 notatoms=5
把原子 3,4,6 和 17 加入原子列表。
atoms=3 C 18-30 notatoms=H
加入所有的C和3,18-30 中的非氢原子。
atoms=C N notatoms=5
加入除了原子 5 以外的所有 C 和 N 原子。
atoms=1-5 notatoms=H atoms=8-10
加入原子 1-5 内的所有非氢原子,
并加入原子 8-10 不考虑其类型。
没有关键词的单独整数解释为原子编号:
1,3,5 7
加入原子 1,3,5,和 7。
对于 ONIOM 优化,block 和notblock 可同时用于包含/不包含 ONIOM 分子说
明部分定义的刚性区。如果选定的原子和区块内指定的原子有冲突,例如包
含在区块内的原子被原子类型排除,Gaussian 09 会发生错误。
可以把 noatoms 放在输入部分作为第一项,从一个空的冻结原子列表开始。
例如,以下输入仅冻结原子 1-100 内的所有非氢原子:
noatoms atoms=1-100 notatoms=H
也可以通过[not]layer 关键词,用 ONIOM 层指定原子,它接受以下的值:real
代表真实体系,model 表示 2 层 ONIOM 的模型体系,middle 表示 3 层 ONIOM
的中间层,small 表示 3 层 ONIOM 的模型层。类似地,可以用残基包含或排
除原子,需要使用 residue 和notresidue,它们接受残基名称列表。两对关
键词都具有原子列表缩写形式的功能。
对于使用 QST2 或QST3 的过渡态优化,每个结构都要另外读入一部分。要知
道,提供矛盾的输入,例如对反应物和产物使用不同的冻结原子,将产生不
可预知的结果。
RdFreeze 是该选项的同义词。
NoFreeze 将所有变量激活(即解冻,通常与 Geom=Check 合用)。
一般流程选项
MaxCycle=
N
指定优化计算最大迭代次数为
N
。默认是所用冗余内坐标数目的两倍(默认
的流程),或是进行优化的变量个数两倍(其它流程),与 20 之间的最大值。
MaxStep=
N
设定优化走步的最大尺寸(也就是初始可信半径)为 0.01N Bohr 或弧度。N
的默认值是 30。
Restart 从检查点文件重新开始几何优化。在这种情况下,整个执行路径部分由 Opt
关键词以及和原计算任务相同的选项(以及 Restart)构成。不需要其它输入
(见例子)。
InitialHarmonic=
N
对初始结构加上力常数为
N
/1000 Hartree/Bohr2的谐振束缚。该选项的同义词
是IHarmonic。
ChkHarmonic=
N
对检查点件储存的初始结构加上力常数为
N
/1000 Hartree/Bohr2的谐振束缚。
该选项的同义词是 CHarmonic。
ReadHarmonic=
N
对从输入串读入的结构(在输入方位)加上力常数为
N
/1000 Hartree/Bohr2
- 167 -

的谐振束缚。该选项的同义词是 RHarmonic。
初始力常数有关的选项
除非另行指定,否则 Berny 几何优化从二阶导数矩阵(也即 Hessian)的初始猜测开始,
Hessian 矩阵通过从原子半径和简单的价电子力场得到的原子连接关系确定[Schlegel84a,
Peng96]。在每一点使用计算的一阶导数改善这个近似的矩阵。这一方案通常工作得很好,但
是在有些情况下初始猜测可能会非常差,使得优化无法正常开始,或者在优化初期花费较多
的步骤改善 Hessian 而无法接近优化的结构。此外,对于过渡态优化,需要知道鞍点附近的
曲率,因此默认的近似 Hessian 必须在优化过程中总是加以改善。
以下选项选择的方法用于改善力常数:
ReadFC 从检查点文件提取力常数。这通常来自较低理论级别优化计算中的最终
近似力常数,或是在较低理论级别的频率计算正确得到的(更准确的)
力常数(后者远远好于前者)。
CalcFC 指定在第一个点用当前的方法计算力常数(只能用于 HF,MP2,CASSCF,
DFT 和半经验方法)。
CalcHFFC 指定在第一个点进行 HF 解析力常数的计算。CalcHFFC 和 MP2 优化一起使
用,相当于 DFT,AM1,PM3,PM3MM,PM6,和 PDDG 等方法的 CalcFC。
RCFC 指定从检查点文件读入来自于频率计算的笛卡尔坐标力常数。通常情况
下,最好是提取已经转化为内坐标的力常数,说明见上(CalcFC)。但 是 ,
频率计算偶尔会发生分子需要变形为低对称性的情况。这种情况下,用
旧的内坐标计算的力常数无法使用,因此应当用 Opt=RCFC 读取笛卡尔力
常数并进行转换。注意,只有在完成频率计算后,检查点文件中才有笛
卡尔力常数。在优化失败后不能使用该选项,因为近似二阶导数矩阵中
的负本征值个数是错的。在后一种情况下,你可能想要从最近的结构开
始,并用数值方法计算导数(见下)。ReadCartesianFC 是RCFC 的同义
词。
CalcAll 指定在每一个点使用当前的方法计算力常数(只能用于 HF,MP2,CASSCF,
DFT 和半经验方法)。注意在收敛的结构上自动进行振动频率分析,且计
算的结果做为频率任务保存。
VCD 在 Hartree-Fock 或 DFT 的 Opt=CalcAll 优化每一个点,指定计算 VCD 强
度。
NoRaman 在 Hartree-Fock 的 Opt=CalcAll 任务每一点,指定不计算拉曼强度(因
为它包含用最后一个优化点的结果进行的频率分析)。拉曼强度对每一个
中间二阶导数点增加 10-20%的计算量。
还有另外两种提供初始 Hessian 的方法,有时也会用到:
输入新的猜测:可以用默认的近似矩阵,但对 Hessian 矩阵的部分或全部对角元读入新
的猜测。这可以在 ModRedundant 输入或 Z 矩阵的变量定义行中指定。例如:
1 2 3 104.5
1 2 1.05 H 0.55
第一行指定由原子 1,2 和 3 形成的键角从 104.5 开始,第二行设定原子 1 和 2 之间的
初始键长的值为 1.05 埃。第二行中的字母 H表示程序为此坐标指定对角力常数,值为
0.55 Hartree/au2。
- 168 -

用数值方法计算部分或全部 Hessian 矩阵元:可以用优化程序以数值方法计算部分二
阶导数矩阵。在这种情况下,每一个指定的变量都朝一个方向逐渐增加数值,而不是精
确力常数计算中所用的朝上和朝下两个方向的改变。因此计算的二阶导数结果不如从频
率计算得到的准确,但对开始优化计算,这种精度足够了。当然,这需要程序对每个指
定的变量作另外的梯度计算。这一流程可以在变量定义行中用标记(D)指定:
1 2 1.0 D
2 3 1.5
1 2 3 104.5 D
2 3 4 110.0
这个输入要求程序在进行优化计算第一步之前,先做三个单点计算:即原有的第一个点,
原子 1 和 2 之间键长略微增加后的结构,以及原子 1,2 和 3 之间键角略微增加后的结
构。这个程序将对另外两个坐标使用默认的对角力常数,并由这三个点对键(1,2)和角
(1,2,3)估算所有(对角的和非对角的)力常数。这个选项只适用于 Berny 和 EF 算法。
StarOnly 指定对力常数进行数值估算,但不做优化。注意,这与频率计算无关。
NewEstmFC 使用价键力场估算力常数。这是默认选项。
EstmFC 使用旧的对角猜测估算力常数。只适用于 Berny 算法。
收敛有关的选项
这些选项只能用于 Berny 算法。
Tight 这一选项收紧力和步长的截断值,这两个值用来判断收敛。使用 Opt=Tight
的优化计算比用默认截断值的优化计算要多算几步。对于具有很小力常数的
分子体系(低频振动模式),必需使用这个选项以确保合适的收敛和随后任务
步骤计算的频率的可靠性。该选项只能用于 Berny 优化。对于 DFT 计算,还
要同时指定 Int=UltraFine。
VeryTight 使用极严格的优化收敛标准。VeryTight 的同义词是 VTight。对于 DFT 计算,
还要同时指定 Int=UltraFine。
EigenTest EigenTest 在 Berny 优化中测试曲率,用 NoEigenTest 可以禁止。对内坐标
(Z-矩阵)或笛卡尔坐标的过渡态计算,这个测试默认是打开的,并且是推
荐选项。有时虽然没有通过曲率测试,但过渡态结构优化计算仍旧收敛,但
NoEigenTest 仅用于大型计算的预算。
Expert 放松 Berny 程序对最大和最小力常数以及步长的各种限制。该选项可以得到
较快的收敛,但相当危险。通常只适合专家级用户,用于当力和力常数与典
型的分子及 Z-矩阵有很大差异时,有时需要结合 Opt=CalcFC 或Opt=CalcAll
使用。NoExpert 强制用默认的限制,这是默认的。
Loose 设定优化收敛标准的最大步长为 0.01 au,RMS 力为 0.0017 au。 这些值和
Int(Grid=SG1)关键词相符,适用于用 DFT 方法对大分子做初步的优化计算,
通常在该计算之后用默认的(Fine)网格再进行完全收敛优化计算。不推荐
单独使用这个选项。
算法有关的选项
GEDIIS 使用 GEDIIS 优化算法。当有解析梯度时,这对能量最小化计算是默认的。
RFO 在 Berny 优化中使用有理函数优化[Simons83]。它对过渡态优化(Opt=TS)是
默认的。这也是在 Gaussian 03 中有解析梯度时的能量最小化默认算法。
EF 使用本征值跟踪算法[Cerjan81, Simons83, Banerjee85],仅适用于没有解析剃度的方
- 169 -
法(在这种情况下是默认的)。适用于能量最小值和过渡态计算。
EigenvalueFollow 是EF 的同义词。当和 Opt=Z-Matrix 一起使用时,最多
可以优化 50 个变量。
ONIOM 有关的选项
Micro 在ONIOM(MO:MM)优化中使用微迭代。这是默认的,对微迭代选择L120或L103
依赖于电子嵌套是打开还是关闭。NoMicro选项禁止ONIOM(MO:MM)优化中的微
迭代。Mic120指定即使是对力学内嵌,在L120中总对ONIOM(MO:MM)使用微迭
代。这对电子内嵌是默认的。Mic103指定对ONIOM(MO:MM)使用L103进行微迭
代。这对力学内嵌是默认的,但不能用于电子内嵌。
QuadMacro 控制在 ONIOM(MO:MM)的几何优化中是否使用耦合的二次宏走步[Vreven06a]。默
认是 NoQuadMacro。
选择坐标系选项
Redundant 在冗余内坐标内用 Berny 算法进行优化计算。这对具有解析梯度的方法是默
认的。
Z-matrix 用内坐标进行 Berny 算法的优化计算[Schlegel82, Schlegel89, Schlegel95]。在这种
情况下,用关键词 Fopt 而不是 Opt 要求程序确认进行了完全优化计算(即,
含非活性变量在内的变量之间是线性无关的,并且张开的自由度符合分子对
称性)。Popt 形式要求进行内坐标的部分优化。在优化的最后它还禁止频率
分析,即每一点的二阶导数(通过 CalcAll 选项)。关于 Z 矩阵分子说明的
细节和离子,参见附录。
Cartesian 在笛卡尔坐标中使用 Berny 算法进行优化计算。注意初始结构的输入可以用
任何一种坐标系。在使用纯笛卡尔坐标系进行优化计算时,不能做部分优化
或冻结变量的计算;如果需要的话,可以使用 Opt=Z-matrix 选项,并在 Z
矩阵中把所有要优化的原子以直角坐标定义,进行混合坐标的优化计算。如
果分子说明部分的 Z矩阵不含变量,并且指定了 Opt=Z-matrix,那么优化计
算实际上用直角坐标执行。注意,其它各种坐标系,例如距离矩阵坐标,可
以用 ModRedundant 选项构造。
解释数值误差的选项
SG1Error 假定能量和力的数值误差足以满足 SG-1 网格的 DFT 计算(分别是 1.0D-07
和 1.0D-05)。这对使用 Int(Grid=SG1Grid)的 DFT 方法是默认的。
很少使用的选项
Path=
M
与QST2 或QST3 选项组合,用冗余内坐标同时优化过渡态和 M个点的反应
路径[Ayala97]。在这种类型的计算中,没有可以冻结的坐标。
如果指定了 QST2,照常需要反应物和产物的标题和分子说明部分做为输入。
路径上余下的 M-2 个点接下来在反应物和产物输入结构之间用线性内插方法
产生。最高能量的结构做为过渡结构的初始猜测。每一个点在反应路径上进
行优化,而最高点朝着过渡结构优化。
如果指定了 QST3,则在输入文件中还必须包含第三套标题和分子说明部分,
照常用作过渡态的初始猜测。路径上余下的 M-3 个点由接连两次线性内插方
法产生,第一次在反应物和过渡结构之间,第二次在过渡结构和产物之间。
- 170 -
默认的中心点优化到过渡结构,而不管能量顺序。在这种情况下,M必须是
奇数,以保证路径上的点可以均匀地分布在过渡结构的两侧。
在同时优化计算的输出中,对过渡结构预测的优化几何之后是所有 M个收敛
的反应路径结构列表。
由其它选项控制反应物和产物输入的处理:OptReactant,OptProduct,和
BiMolecular。
注意,在反应物能谷结构的 SCF 波函可能完全不同于产物能谷结构处的波
函。Guess=Always 可用于防止类-反应物结构的波函用于类-产物结构的初始
猜测波函。
OptReactant 指定在路径优化(Opt=Path)计算中,反应物的输入结构应当优化到局域最
小值。这是默认的。NoOptReactant 用于保留输入结构,作为反应路径上已
存在的一个点(这一般意味着它在前面的计算中已经优化到了最小值)。
OptReactant 不能和 BiMolecular 一起使用。
BiMolecular 指定反应物或产物是生物分子,输入结构将用作 Opt=Path 优化中的参照点。
这个参照点不会出现在路径的 M个点中。相反,它用来控制过渡态的反应物
一方以何种程度扩散。默认关闭该选项。
OptProduct 指定在路径优化(Opt=Path)计算中,产物的输入结构应当优化到局域最小
值。这是默认的。NoOptProduct 用于保留输入结构,作为反应路径上已存
在的一个点(这一般意味着它在前面的计算中已经优化到了最小值)。
Optproduct 不能和 BiMolecular 一起使用。
Linear 在 Berny 优化计算中使用线性搜索方法,用 NoLinear 禁止。默认是尽可能
使用线性搜索。
TrustUpdate 在 Berny 优化计算中动态更新可信半径,用 NoTrustUpdate 禁止。默认从最
小值更新。
Newton 在 Berny 优化计算中使用 Newton-Raphson 走步而非 RFO 走步。
NRScale 在 Berny 优化计算中若 Newton-Raphson 方法的步长超过最大值时,则用换
算因子减小步长。NoNRScale 在最大步长的球面上进行最小化计算[Golab83]。
默认对过渡状态的优化进行换算,最小化计算默认是在球面上进行最小化。
Steep 在 Berny 优化计算中使用陡降法,而不是 Newton-Raphson 走步。这只和 Berny
局部最小值优化兼容。当从远离最小值的结构开始时,可以用陡降法,但它
通常无法达到完全收敛。
UpdateMethod=
keyword
指定 Hessian 的更新方法。
keyword
可以是以下之一:Powell,BFGS,PDBFGS,
ND2Corr,OD2Corr,D2CorrBFGS,Bofill,D2CMix 和None。
HFError 假定能量和力的数值误差足以满足 HF 和后 SCF 计算(分别是 1.0D-07 和
1.0D-07)。这是使用这两种方法以及半经验方法进行优化计算的默认值。
FineGridError
假定能量和力的数值误差足以满足默认网格的 DFT 计算(分别是 1.0D-07 和
1.0D-06)。这对使用 DFT 方法和默认网格(或指定 Int=FineGrid)进行优
化的计算是默认的。
适用性
解析梯度可用于 HF,所有的 DFT 方法,CIS,MP2,MP3,MP4(SDQ),CID,CISD,CCD,
CCSD,QCISD,BD,CASSCF,以及所有的半经验方法。
- 171 -

选项 Tight,VeryTight,Expert,Eigentest 和EstmFC 只能用于 Berny 算法。
大分子经常有多个低频振动模式,因此用 DFT 对其进行优化时,使用精密的 DFT 积分网
格(Int=UltraFine)往往会更可靠。
有关的关键词
IRC,IRCMax,Scan,Force,Frequency
例子
优化任务的输出。Berny 优化流程的输出以字符串 GradGradGrad...界定。在第一次初
始化通过后,程序对需要优化的变量打印初始值表。对在冗余内坐标系统进行的优化计算,
所有用到的坐标都会显示在表中(不只是分子说明部分出现的坐标和变量):
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Berny optimization.
优化算法由输出头格式和此行标识。
Initialization pass.
----------------------------
! Initial Parameters !
! (Angstroms and Degrees) !
-------------------- ----------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------
! R1 R(2,1) 1. estimate D2E/DX2 !
! R2 R(3,1) 1. estimate D2E/DX2 !
! A1 A(2,1,3) 104.5 estimate D2E/DX2 !
--------------------------------------------------------------------
初始二阶导的类型打印在标题 Derivative Info 以下。本例中的二阶导是估算的。
优化计算的随后步骤用这样的一行加以界定:
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Berny optimization.
Search for a local minimum.
Step number 4 out of a maximum of 20
一旦优化计算完成,程序显示最终结构:
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------- --------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------
! R1 R(2,1) 0.9892 -DE/DX = 0.0002 !
! R2 R(3,1) 0.9892 -DE/DX = 0.0002 !
! A1 A(2,1,3) 100.004 -DE/DX = 0.0001 !
--------------------------------------------------------------------
冗余内坐标的定义在表的第二列给出。括号中的数字指的是分子说明中的原子。例如变
量R1 定义为 R(2,1),表示原子 1 和原子 2 之间的键长。优化结构的能量可以在最后一步优
化的输出中找到,位于输出文件的此表格之前。
- 172 -

复合任务。几何优化之后,通常在优化的结构上进行频率计算。要进行这个流程,可以
在输入文件的执行路径部分把 Opt 关键词与 Freq 组合,这种组合自动产生两步计算任务。
另一个常见做法是在优化计算之后用更高的理论级别进行单点能计算。下面的执行路径
部分自动进行 HF/6-31G(d,p)优化,之后是 MP4/6-31G(d,p)单点能计算:
# MP4/6-31G(d,p)//HF/6-31G(d,p) Test
注意这种情况不需要 Opt 关键词。但是如果需要设定 Opt 需要的选项,则必须包含它。
修改冗余内坐标。下面的输入演示了在输入文件中修改冗余内坐标的方法:
# HF/6-31G(d) Opt=ModRedun Test
Opt job
0,1
C1 0.000 0.000 0.000
C2 0.000 0.000 1.505
O3 1.047 0.000 -0.651
H4 -1.000 -0.006 -0.484
H5 -0.735 0.755 1.898
H6 -0.295 -1.024 1.866
O7 1.242 0.364 2.065
H8 1.938 -0.001 1.499
3 8
添加氢键(而不是键角或二面角)。
2 1 3
添加 C-C-O 键角。
这是用 OH 取代甲基上一个氢的乙醛结构;第一个输入行用于 ModRedundant,创建这个
氢原子和羰基上氧原子之间的氢键。注意这一行只添加这两个原子之间的键。使用执行代码
B,还可以添加相应的键角和二面角:
3 8 B
显示所需坐标的值。对ModRedundant 的第二个输入行定义 C-C=O 键角,可以使它的值
出现在每个优化步骤的结构总结列表中。
在冗余内坐标中使用通配符。距离矩阵坐标系可以用下面的方法激活:
* * B
定义每对原子间所有成键
* * * K
删除所有其它的冗余内坐标
下面的输入定义部分距离矩阵坐标,用于连接原子最近的层:
* * B 1.1
定义距原子 1.1 À 以内的所有原子之间成键
* * * K
删除所有其它的冗余内坐标
下面的输入中设置了冗余内坐标优化,其中从原子 N1 到Nn 被冻结(这样的任务可能需
要NoSymm 关键词)。注意因为只定义了一个原子序号,包含执行代码 B的行将对所有涉及
指定原子的坐标产生直角坐标:
N1 B
产生涉及原子 N1 的直角坐标
...
Nn B
产生涉及原子 Nn 的直角坐标
* F
冻结所有直角坐标
- 173 -

下面的输入通过从冗余内坐标中删除所有的二面角,定义特殊的“球”内坐标,这种坐
标适于描述像 C60 这样的分子:
* * * * R
删除所有二面角
下面的输入沿着 N2-N3 键把体系转动 10 度:
* N2 N3 * +=10.0
把涉及 N2-N3 键的二面角值增加 10.0
其它的例子可以在下面关于平缓势能面扫描的部分找到。
执行部分优化。下面的任务演示了在优化中冻结变量的方法:
# B3LYP/6-31G(d) Opt=ReadFreeze
Partial optimization of Fe2S2
cluster with phenylthiolates.
-2,1
Fe 15.2630 -1.0091 7.0068
S 14.8495 1.1490 7.0431
Fe 17.0430 1.0091 7.0068
S 17.4565 -1.1490 7.0431
S 14.3762 -2.1581 8.7983
C 12.5993 -2.1848 8.6878
…
C 14.8285 -3.8823 3.3884
H 14.3660 -3.3149 2.7071
noatoms atoms=1-4
ReadFreeze 输入。
结构用传统的 Z-矩阵定义,其中变量在单独的部分定义。输入部分最后给出了用于选
项ModRedundant 的值。这个输入固定 O-H 键和到最后一个氢原子的二面角,注意以这种方
法定义的任何值都不能和在前面 Z-矩阵列出的值相同(这里也就是 O-H 键长);通过调整结
构来保证这个限制。限制的值是可选的。例如在这个例子中,第二个修改的冗余内坐标值默
认为 Z-矩阵的值(180.0)。
在优化 phenylthiolate 时,中央的团簇(前四个原子)保持固定。
重新开始优化。失败的优化可以从检查点文件重新开始,只需简单重复原任务的执行路
径部分,并对 Opt 关键词添加 Restart 选项。例如,这个执行路径重新开始二阶鞍点的
B3LYP/6-31G(d) Berny 优化:
%Chk=saddle2
# Opt=(TS,Restart,MaxCyc=50) Test
从检查点文件恢复模型化学和初始几何。在执行路径部分需要指定优化类型和流程的选
项,用于重新开始任务(例如,上面的例子是 TS)。重新开始任务中的某些参数设定选项
可以忽略,这时使用与原任务相同的值,也可以对它们进行修改,例如这个例子中的
MaxCycle。注意,如果在重新开始任务的第一个点计算 Hessian,必须包含 CalcFC。只有
当这个选项出现在重新开始任务的执行路径部分,才会计算二阶导数,而不考虑原任务中是
否指定了该选项。
- 174 -

从检查点文件读入结构。冗余内坐标结构可以从检查点文件恢复,仍是照常使用
Geom=Checkpoint。读入的结构也可以通过指定 Geom=ModRedundant 进行修改;修改结构与
Opt=ModRedundant 的输入具有相同的格式:
[Type] N1 [N2 [N3 [N4]]] [[+=]Value] [Action [Params]] [[Min] Max]]
用 STQN 方法定位过渡结构。QST2 启动过渡结构的搜索,这个过渡结构连接特定的反应
物与产物。该选项输入的一般结构为(忽略了空行):
#HF/6-31G(d,p) Opt=QST2
第一个标题部分
反应物的分子说明
第二个标题部分
产物的分子说明
#HF/6-31G(d) (Opt=QST2,ModRedun)
第一个标题部分
反应物的分子说明
反应物的 ModRedundant 输入
第二个标题部分
产物的分子说明
产物的 ModRedundant 输入(可选)
注意,每个分子说明之前是其标题部分(以及一个空行)。如果指定了 ModRedundant
选项,那么每个分子说明之后是任何对冗余内坐标所作的修改。
Gaussian 将对反应物和产物之间的过渡结构自动产生初始结构,并对一阶鞍点执行优
化。
QST3 选项允许对过渡态指定更好的初始结构。它像 QST2 一样需要对反应物和产物分别
指定两个标题和分子说明部分,另外还对初始过渡态结构指定第三个标题和分子说明部分
(以及分隔用的空行),如果指定了 ModRedundant 选项的话,还有三个对应冗余内坐标的
修改。程序接着将定位连接反应物和产物的过渡结构,并使它最接近指定的初始结构。
由QST2 或QST3 得到的优化结构,它们在输出中出现的格式类似于其它类型的几何优化
输出:
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
--------------------- ----------------------
! Name Definition Value Reactant Product Derivative Info. !
-------------------------------------------------------------------
! R1 R(2,1) 1.0836 1.083 1.084 -DE/DX = 0. !
! R2 R(3,1) 1.4233 1.4047 1.4426 -DE/DX = -0. !
! R3 R(4,1) 1.4154 1.4347 1.3952 -DE/DX = -0. !
! R4 R(5,3) 1.3989 1.3989 1.3984 -DE/DX = 0. !
! R5 R(6,3) 1.1009 1.0985 1.0995 -DE/DX = 0. !
! … !
-------------------------------------------------------------------
除了列出的优化值以外,表中还包含了反应物和产物的值。
进行平缓势能面扫描。Opt=ModRedundant 选项也可以用于进行平缓势能面(PES)的扫
描。类似于 Scan 提供的功能,平缓 PES 扫描步骤使用 PES 上的矩形网格,这涉及所选的内
坐标。它不同于 Scan 关键词之处是,在每一个点都进行限定结构的优化。
平缓 PES 扫描只能用于 Berny 算法。如果有扫描变量在计算中破坏了对称性,那么必须
在任务的执行路径部分包含 NoSymm,否则在报错后失败。
用Opt=ModRedundant 选项指定的冗余内坐标可以用 S字母代码扫描:
N1 N2 [N3 [N4]] [[+=]value]
S
步数 步长
- 175 -

例如,这个输入在原子 2 和 3 之间添加一个键,设定初始值为 1.0 Å,并指定三步扫描,
每步 0.05 Å:
2 3 1.0 S 3 0.05
ModRedundant 输入中的通配符对于设定平缓 PES 扫描可能非常有用。例如,下面的输
入用于包含 N1-N2-N3-N4 二面角的势能面扫描。注意该键周围所有其它二面角都被删除:
* N2 N3 * R
删除所有包含 N2-N3 键的二面角
N1 N2 N3 N4 S 20 2.0
指定 20 步平缓 PES 扫描,步长增量为 2°
笛卡尔坐标也可以包含对扫描的说明:
X atom S x-steps x-size y-steps y-size z-steps z-size
例如,下面的 ModRedundant 输入将执行平缓势能面扫描。它从原子 1 的初始位置开始,
在 X 方向移动 5 次 0.2 埃的步长,在 Z 方向移动 3 次 0.1 埃的步长:
X 1 S 5 0.2 0 0.0 3 0.1
Berny 优化算法
Gaussian 中使用的 Berny 几何优化算法基于 H.B.Schlegel 更早时期写的程序,其中执
行了他所发表的算法[Schlegel82]。这个早期版本使用了其它算法和未发表的技术,因此进行
了相当多的改进,成为这里总结的 Berny 算法。
在 Berny 优化计算的每一步,进行下面的操作:
除了已经计算了解析 Hessian,或者是在计算的第一步(其中的 Hessian 是估算的),
否则都进行 Hessian 更新。在冗余内坐标中,通常用迭代 BFGS 方法对最小值的计算进
行更新,对过渡态使用迭代 Bofill 进行更新,而在内坐标的优化中,使用修改过的
Schlegel 更新流程。默认从价力场得到 Hessian[Schlegel84a],但是作为估算,也可以请
求产生单位矩阵或者对角 Hessian。
如果寻找的是最小值,则使用 Fletcher 的方法[Fletcher80, Bofill94, Bofill95]更新可信半径
(允许的最大 Newton-Raphson 步幅)。
把对应冻结变量的梯度矢量分量设置为零,或投影掉,以消除这些变量在下一个优化步
骤中的直接贡献。
如果寻找最小值,则在上一点和过去的最佳点(已找到的能量最低点)之间进行线性搜
索。如果在每一点能计算二阶导,并且寻找最小值,则先尝试进行五次多项式拟合计算;
如果在这个多项式可接受的范围内(见下)无最小值,或是二阶导不可用,则再尝试束
缚的四次拟合计算。这将在这两点对能量和一阶导数(沿两点连线)进行四次多项式拟
合,并加上多项式的二阶导在其最小值处恰好为零的限制,以保证这个多项式本身只有
一个最小值。如果拟合计算失败,或是计算得到的步长不合理,则再进行简单的三次拟
合计算。
如果上一点是目前的最佳点,则任何一个五次或四次多项式拟合的步幅都认为是可以接
受的,但是如果上一点不是目前的最佳点,则线性搜索必须回到在上一点和已找到的最
佳点之间的某一点上。三次拟合的步幅通常都是不可接受的,除非这些点是在两点之间,
或者步幅不如前一步大。最后,如果所有的拟合都失败,并且上一步找到的步幅是目前
最佳的,则不再进行线性搜索。如果所有的拟合都失败,并且上一步找到的步幅也不是
最佳的,则从上一点和最佳点之间连线的中点上使用线性步幅。
如果上一步找到的点是目前的最佳点,或是寻找过渡态,则使用当前的(可以是近似的)
- 176 -
二阶导确定二次步幅。如果进行线性搜索,则在线性搜索外推得到的点处计算二次步幅,
并且使用该点的力,力的获得需要在线性搜索的两端点处对力进行内插。这一步默认使
用有理函数优化(RFO)方法[Simons83, Banerjee85, Baker86, Baker87]。如果当前位置的曲率不
符合需要,RFO 的步幅比旧版 Gaussian 使用的 Newton-Raphson 方法好。旧的
Newton-Raphson 步幅可以用选项指定。
由二次步幅方法得到的任何步幅矢量分量,如果对应于冻结变量,则设置为零或投影掉。
如果二次步幅超过可信半径,并且寻找最小值,则可信半径的球面上找出二次函数的最
小值,从而把步幅缩短到可信半径的长度以内,见 Jørgensen 的讨论[Golab83]。如果寻
找的是过渡态,或是使用了选项 NRScale,则简单地把二次步幅换算到可信半径。
最后,用最大的力分量、力的均方根、最大步幅分量,和步幅均方根等标准,对收敛进
行测试。步幅是最近一次计算的点和下一个要进行计算的点之间的改变量(线性步幅和
二次步幅之和)。
Output
说明
Output 关键词要求对 Fortran 的非格式化文件产生输出。它的选项控制创建文件的内容。
选项
WFN 写PROAIMS 的波函文件(.wfn)。创建文件的名称从输入串中读取,位于独
立的一行。PSI 是WFN 的同义词。
WfnX 写波函文件,用于新版本的AIMPAC7。
Pickett 把超精细光谱的g张量和其它张量[Curl65, Hirota85, Mills93, Hirota94, Gauss96,
Neese01]写到一个输出文件,使用Pickett的程序[Pickett91]输入格式(参见:
http://spec.jpl.nasa.gov)。以下张量可以用Gaussian 09计算[Barone94,
Minichino94, Barone95, Barone96, Rega96, Barone03]:
核电四极常数:所有的任务
转动常数:Freq=(VibRot[,Anharmonic])
四阶离心畸变项:Freq=(VCD,Anharmonic)
电子自旋转动项:NMR
核自旋转动项:NMR
偶极超精细项:所有的任务
Fermi接触项:所有的任务
SpinRotation
NMR Output=Pickett的同义词。包含所有不用做频率计算就能得到的超精细
张量。
RotationalConstants
Freq=VibRot Output=Pickett的同义词。包含所有只需做谐振频率计算就能
得到的超精细张量。
对于HF和DFT,可以组合以上两个选项。Output=(RotatationalConstants,
7 即AIMPAC作者之一T. A. Keith开发的AILALL程序:http://aim.tkgristmill.com/。它支持spdfgh基函数和
ECP。Gaussian 09.A不能用这个选项。——译注。
- 177 -
SpinRotation)包含所有不超过二阶导就能计算的张量。它等价于
Freq=(VCD,VibRot) Output=Pickett。
QuarticCentrifugal
Freq=(VibRot,Anharm) Output=Pickett的同义词。包含四次振转耦合,但
不包括振转张量,它必须另外计算。
ReadAtoms 读入一组原子,包含在 Pickett 程序的输入中(注意,这个程序只接受八个
核的张量)。原子编号以自由格式给定,这一输入部分用一个空白行结束。
默认为程序自动选择八个“感兴趣的”原子。
有关的关键词
Punch
OVGF
参见 EPT 关键词的讨论。
PBC
说明
这个关键词允许指定周期边界条件任务的选项。注意,通过在输入结构部分包含平移矢
量即可启动 PBC,而这个关键词仅用于控制 PBC 计算的执行。如果不需要以下选项,在执行
PBC 计算时不必使用这个关键词。
选项
GammaOnly 只做 Γ点(k=0)的积分,而非全部的 k-积分。
NKPoint=
N
做近似
N
个 k-点积分。
CellRange=
N
在每个方向上延伸
N
Bohr 长度,用于设定映像晶胞。
NCellMin=
N
至少包含
N
个晶胞。
NCellMax=
N
在计算的任何部分最多包含
N
个晶胞。
NCellDFT=
N
在DFT的XC积分中至少包含
N
个晶胞。NCellXC是该选项的同义词。
NCellK=
N
在精确交换项的计算中至少使用
N
个晶胞。默认如果包含精确交换项,则使
用的晶胞数是用于重叠积分有关的量和 XC 积分的计算中使用的晶胞数的两
倍。
适用性
HF 和DFT 的能量与优化。不能用于 SCRF 或Charge。对任何合理大小的周期体系,
只有用纯 DFT 泛函并结合密度拟合,才能达到可接受的计算性能。
例子
周期体系用通常的分子说明指定单位晶胞。唯一需要的附加输入为接在分子说明之后的
一,二,或三个平移矢量(中间没有空行),表示重复的方向。例如,下面的输入指定氯丁
- 178 -

橡胶一维 PBC 的单点能计算:
# PBEPBE/6-31g(d,p)/Auto SCF=Tight
neoprene, -CH2-CH=C(Cl)-CH2- optimized geometry
0 1
C,-1.9267226529,0.4060180273,0.0316702826
H,-2.3523143977,0.9206168644,0.9131400756
H,-1.8372739404,1.1548899113,-0.770750797
C,-0.5737182157,-0.1434584477,0.3762843235
H,-0.5015912465,-0.7653394047,1.2791284293
C,0.5790889876,0.0220081655,-0.3005160849
C,1.9237098673,-0.5258773194,0.0966261209
H,1.772234452,-1.2511397907,0.915962512
H,2.3627869487,-1.0792380182,-0.752511583
Cl,0.6209825739,0.9860944599,-1.7876398696
TV,4.8477468928,0.1714181332,0.5112729831
最后一行指定平移矢量。注意,原子符号指定为 TV。
下面的分子说明可用于石墨层的二维 PBC 计算:
0 1
C 0.000000 0.000000 0.000000
C 0.000000 1.429118 0.000000
TV 2.475315 0.000000 0.000000
TV -1.219952 2.133447 0.000000
这个分子说明用于砷化镓的三维 PBC 计算:
0 1
Ga 0.000000 0.000000 0.000000
Ga 0.000000 2.825000 2.825000
Ga 2.825000 0.000000 2.825000
Ga 2.825000 2.825000 0.000000
As 1.412500 1.412500 1.412500
As 1.412500 4.237500 4.237500
As 4.237500 1.412500 4.237500
As 4.237500 4.237500 1.412500
TV 5.650000 0.000000 0.000000
TV 0.000000 5.650000 0.000000
TV 0.000000 0.000000 5.650000
PM3
参见半经验方法的说明。
PM6
参见半经验方法的说明。
- 179 -
Polar
说明
这个方法关键词指示进行偶极电场极化率的计算(如果可能的话也计算超极化率)。这
个关键词本身不包括分子结构改变或导数的计算,但是在同一计算任务中,这个关键词可以
与数值差分或力的计算组合,需要在执行路径部分指定 Freq 和Polar。Freq 和Polar 不能
与没有解析梯度的方法组合(如 MP4(SDTQ),QCISD(T),CCSD(T),等)。注意,如果计
算解析二阶导数,默认会进行 Polar 计算。
极化率和超极化率以标准方位出现在输出文件中,分别以下三角和下四面体顺序:αxx,
αxy,αyy,αxz,αyz,αzz 和βxxx,βxxy,βxyy,βyyy,βxxz,βxyz,βyyz,βxzz,βyzz,βzzz。
极化率和超极化率通常用静态频率计算。但是可以在输入文件的执行路径包含关键词
CPHF=RdFreq并指定所用的频率,进行含频极化率和超极化率的计算[Olsen85, Sekino86, Rice90,
Rice91, Rice92]。
用选项 OptRot[Karna91, Helgaker94, Pedersen95, Kondru98, Stephens01, Mennucci02, Ruud02, Stephens02a,
Stephens03]可以预测旋光性[Rosenfeld28, Condon37, Eyring44, Buckingham67, Buckingham68, Atkins69, Barron71,
Charney79, Amos82, Jorgensen88]。应用实例参见[Stephens05, Wilson05, Stephens08]。
选项
OptRot 进行旋光性的计算。用 CPHF=RdFreq 指定所需的频率。仅能用于 HF 和 DFT。
这一选项不能与 NMR 合用。如要计算完全旋光张量,可以在执行路径加上
IOp(10/46=7),用于对偶极-磁偶极极化率包含偶极-四极的贡献[Pedersen95,
Barron04];完全旋光张量在输出中标记为“Optical Rotation G' tensor”。
注意,这么做不会改变旋光性。
DCSHG 对 dc-SHG(直流二次谐波生成)超极化率执行额外的含频 CPHF 计算。该选
项还意味着 CPHF=RdFreq。
Analytic 当有解析三阶导数时,解析计算的极化率和超极化率。该选项对于有解析二
阶导数的方法是默认的:RHF 和 UHF,CASSCF,CIS,MP2,和 DFT。注意在解
析频率的计算中,总是计算极化率。
DoubleNumer
对于有解析梯度(一阶导)的方法,除了极化率以外还计算超极化率。对于
没有解析梯度的方法用能量的二次数值差分计算极化率。EnOnly 是
DoubleNumer 的同义词。
Cubic 对解析极化率做数值差分,计算超极化率。只能用于有解析频率但没有解析
三阶导数的方法。
Numerical 用偶极矩的数值导数计算极化率(偶极矩本身是能量的解析梯度,而非 MP2
或 CI 能量计算中的期待值)。对只有一阶解析导数梯度的方法,这是默认设
置。
Step=
N
指定电场的步长为 0.0001
N
原子单位(用于数值差分)。
Restart 从检查点文件重新开始数值计算。失败的 Polar 计算可以从其检查点文件重
新开始计算,只需简单重复原任务的执行路径部分,并为 Polar 关键词加上
Restart 选项即可。不需要其它输入。
Susceptibility
- 180 -
计算磁化率以及其它特性(参见 NMR)。仅能用于 HF 和 DFT。
TwoPoint 当计算数值导数时,在每一方向做两次位移。这是默认的。FourPoint 将做
四次位移,但只能用于链接 106(Polar=Numer)。对 Polar=DoubleNumer 无
效。
Dipole 计算偶极极化率(这是默认选项)。
适用性
下面的方法表格总结了对可用的方法计算极化率和超极化率所需要的Polar选项。
方法 极化率 超极化率
HF,DFT方法(含有解析三阶导) Polar(默认为Analytic)Polar(默认为Analytic)
其它有解析频率的方法(MP2,CIS,...) Polar(默认为Analytic)Polar=Cubic
有解析梯度的方法(CCSD,BD,...) Polar(默认为Numeric)Polar=DoubleNumer
没有解析梯度的方法(CCSD(T),...) Polar(默认为
DoubleNumer)
无
含频极化率和超极化率(即Polar CPHF=RdFreq)仅能用于HF和DFT方法。
有关的关键词
Freq,CPHF=RdFreq
例子
含频特性。以下任务用ω=0.1 Hartree计算含频极化率和超极化率:
# Polar CPHF=RdFreq B3LYP/6-31G(d)
Frequency-dependent calculation: w=0.1
分子说明部分
0.1
对指定频率执行一个含频Polar计算,计算结果打印在静态极化率计算的结果之后。例
如,这是一个含频(ω=0.1 Hartree)任务的极化率值:
SCF Polarizability for W= 0.000000:
1 2 3
1 0.482729D+01
2 0.000000D+00 0.112001D+02
3 0.000000D+00 0.000000D+00 0.165696D+02
Isotropic polarizability for W= 0.000000 10.87 Bohr**3.
SCF Polarizability for W= 0.100000:
1 2 3
1 0.491893D+01
2 0.000000D+00 0.115663D+02
3 0.000000D+00 0.000000D+00 0.171826D+02
Isotropic polarizability for W= 0.100000 11.22 Bohr**3.
静态极化率计算只有第一部分的输出。之后是超极化率和其它特性的类似输出。
旋光性。以下是旋光性任务(OptRot选项)输出的关键部分。在这个例子中,在执行路
径包含CPHF=RdFreq,并指定频率为589.3 nm进行含频计算:
Dipole-magnetic dipole polarizability for W= 0.077318:
1 2 3
1 -0.428755D+01 -0.175571D+01 0.000000D+00
2 -0.552645D+01 0.987070D+01 0.000000D+00
- 181 -

3 0.000000D+00 0.000000D+00 -0.676292D+00
w= 0.077318 a.u., Optical Rotation Beta= -1.6356 au.
Molar Mass = 172.2694 grams/mole, [Alpha] ( 5893.0 A) = -366.99 deg.
计算的旋光率数值已在示例输出中加粗。
Population
说明
这个特性关键词控制分子轨道,几种布居分析以及原子电荷分布的打印。默认只打印总
原子电荷和轨道能量,但 Guess=Only 任务除外,这种情况默认是 Pop=Full(见下)。 对单
点计算只做一次布居分析,对几何优化在最初和最终的分子结构上各做一次布居分析。注意,
Population 关键词需要选项。
用来做布居分析的密度由关键词 Density 控制。注意在一个任务步骤中只能使用一种密
度和电荷拟合方法。如果需要多种方法的组合,为避免重复耗时的计算,可以在输入中指定
关键词 Guess=Only Density=Check 以添加额外的计算步骤。
布居分析结果以标准分子方位给出。
Pop 关键词的输出控制包括:
分子轨道和轨道能量。默认包含所有轨道,但用 Orbital 选项可以对输出限定特殊的轨
道范围。
原子电荷分布。如果可能,还报告每个片段的总电荷。
多极矩:从偶极到十六极。
在振动频率计算中,默认还计算 APT 电荷[Cioslowski89]。
控制输出文件内容的选项
None 不打印分子轨道,也不做布居分析。这对所有使用 ZIndo 方法的计算是默认
的。
Minimal 打印原子总电荷和轨道能量。除了 Guess=Only 和/或 ZIndo 以外,这对所有
的任务类型和方法是默认的。
Regular 打印五个能量最高的占据轨道,五个能量最低的虚轨道,以及密度矩阵和完
全的(逐个轨道,逐个原子)Mulliken 布居分析。因为输出文件和分子大小
的平方成比,因此大分子的输出文件相当大。
Full 除了它打印所有轨道以外,和 Regular 布居分析相同。这对 Guess=Only 任务
是默认的。
Always 在结构优化计算的每一步,而非第一步和最后一步,执行布居分析。
Orbitals[=N]
对
N
个最高占据轨道和
N
个最低虚轨道执行布居分析(见下面的例子)。默
认的
N
是 10。也可以指定 AllOrbitals 代替 Orbitals=
N
,请求分析所有轨
道。对于开壳层计算,包括 alpha 和 beta 轨道。
ThreshOrbitals=
N
在各个轨道的布居分析中,设定包含轨道贡献的最小百分比。默认是 10。
布居分析选项
Bonding 除了执行标准分析之外,还执行键布居分析。这是 Mulliken 布居分析,计算
- 182 -

中只保留不同原子中心基函数对的密度项。
MBS 执行最小基组 Mulliken 布居分析[Montgomery99, Montgomery00]。NoMBS 通知程序不
做MBS Mulliken 布居分析。
Hirshfeld 执行 Hirshfeld 布居分析[Hirshfeld77, Ritchie85, Ritchie87]。HirshfeldEE 选项还
请求原子间静电相互作用。
Biorthogonalize
双正交化非限制分子轨道,以最大程度地调整电子对。
NaturalTransitionOrbitals
请求执行 CIS 或 TDDFT 激发态的自然跃迁轨道分析[Martin03]。为了指定哪个
跃迁密度用于产生轨道,必须和 Density=(Check,Transition=
N
)一起使用。
要打印多个感兴趣态的轨道,可以在最初的激发态计算之后连续运行多个
Pop=NTO Density=(Check,Transition=
N
) Guess=(Read,Only)任务。NTO 是
该选项的同义词。
SaveNaturalTransitionOrbitals
把产生的轨道保存到检查点文件,若密度是从这里读入的话,可以替换正则
轨道。SaveNTO 是该选项的同义词。如果想显示轨道,需要把它们写入检查
点文件。一个好的办法是对每个态的检查点文件副本做这样的操作。在最初
的激发态计算之后(用%Chk ex.chk),使用类似于下面的技术产生每个态的
显示数据:
$ cp ex.chk staten.chk
复制原检查点文件
$ g09 <<END
运行 Guess=Only 任务产生轨道
%Chk=staten
使用检查点文件的副本
# Geom=AllCheck ChkBas Guess=(Read,Only)
Density=(Check,Transition=n) Pop=SaveNTO
对每个感兴趣的态重复此过程
END
结束 Gaussian 09 输入文件
自然轨道有关的选项
NaturalOrbitals
对总密度进行自然轨道分析。NO 是NaturalOrbitals 的同义词。
NOAB 分别进行 α和β密度的自然轨道分析。NOAB 是NaturalSpinOrbitals 的同
义词。
AlphaNatural
分别进行 α和β密度的自然轨道分析,但只把自旋为 α的密度储存在.wfn
文件中(见 Output=WFN)。NOA 为AlphaNatural 的同义词。
BetaNatural 分别进行 α和β密度的自然轨道分析,但只把自旋为 β的密度储存在.wfn
文件中(见 Output=WFN)。NOB 为BetaNatural 的同义词。
SpinNatural 对自旋密度(以自旋 α为正)产生自然轨道。
自然轨道默认不包含在检查点文件中。使用以下形式的两步任务可以把自然轨道放入检
查点文件:
--Link1--
%Chk=name
# Guess=(Save,Only,NaturalOrbitals) Geom=AllCheck ChkBasis
对产生的检查点文件运行 formchk 工具,可以产生用于显示的轨道。
产生静电势导出电荷的选项
- 183 -

MK 在根据 Merz-Singh-Kollman 方案[Singh84, Besler90]所选的点上,拟合静电势产
生电荷。MK 的同义词是 ESP 和MerzKollman。
CHelp 在根据 CHelp 方案[Chirlian87]所选的点上,拟合静电势产生电荷。
CHelpG 在根据 CHelpG 方案[Breneman90]所选的点上,拟合静电势产生电荷。
Dipole 对势拟合电荷时,限制电荷以便再现偶极矩。ESPDipole 是Dipole 的同义词。
AtomDipole 对势拟合电荷时,同时也在每一个原子中心拟合一个点偶极。
ReadRadii 对每一元素读入不同的半径值(单位:埃),用于拟合势。数据格式是每行
一个元素符号及其半径值,最后用一空行终止。
ReadAtRadii 对每一原子读入不同的半径值(单位:埃),用于拟合势。数据格式是每行
一个原子编号及其半径值,最后用一空行终止。
NBO 有关选项
NBO 使用 NBO 版本 3,进行完整的自然键轨道(Natural Bond Orbital)分析[Foster80,
Reed83a, Reed85, Reed85a, Carpenter87, Carpenter88, Reed88, Weinhold88]。
NCS 用 Bohmann 等人的自然化学屏蔽(Natural Chemical Shielding)分析
[Bohmann97],并基于 NBO 分析方法,把 NMR 屏蔽张量(用 GIAO 计算)划分为来
自键的磁贡献和孤对贡献。默认执行各向同性屏蔽分析。NoNCS 跳过此分析。
NCSDiag 请求对角张量元素的 NCS 分析。
NCSAll 请求所有张量分量的 NCS 分析。
NPA 请求只执行 NBO 的自然布居分析阶段。
NBORead 请求进行完整的 NBO 分析,使用从输入串读入的数据控制分析计算。这个
选项用于指定 NBO 的关键词。关于此输入的细节参见 NBO 文档。
NBODel 删除某些相互作用后,对其影响进行 NBO 分析。仅能用于 SCF 方法。这意
味着需要读入 NBO 输入;细节参见 NBO 文档。注意 NBO 的输入从第二列
开始,因此,UNIX shell 不会解释最前面的$符号。
SaveNBOs 把自然键轨道保存到检查点文件(用于以后的显示)。
SaveNLMOs 把自然局域化分子轨道保存到检查点文件(用于以后的显示)。
SaveMixed 把占据轨道的 NBO 和非占据轨道的 NLMO 保存到检查点文件(用于以后的显
示)。
有关的关键词
Density,Output=WFN
例子
逐个轨道的布居分析。以下执行路径部分请求最低三个虚轨道和最高三个占据轨道的布
居分析:
# UHF/6-311+G(d) Pop=Orbitals=3
以下是FeO+四重态计算的结果输出:
Atomic contributions to Alpha molecular orbitals:
Alpha occ 16 OE=-0.923 is Fe1-d=1.00
Alpha occ 17 OE=-0.699 is O2-p=0.88
Alpha occ 18 OE=-0.690 is O2-p=0.68 Fe1-s=0.21
Alpha vir 19 OE=-0.253 is Fe1-s=0.70 Fe1-p=0.27
Alpha vir 20 OE=-0.188 is Fe1-p=0.71 O2-p=0.29
- 184 -

Alpha vir 21 OE=-0.133 is Fe1-p=1.04
Atomic contributions to Beta molecular orbitals:
Beta occ 13 OE=-0.801 is O2-p=0.79
Beta occ 14 OE=-0.783 is Fe1-d=1.00
Beta occ 15 OE=-0.758 is O2-p=0.89
Beta vir 16 OE=-0.241 is Fe1-s=0.81 Fe1-p=0.17
Beta vir 17 OE=-0.139 is Fe1-p=0.91 Fe1-d=0.14
注意,这里包含了alpha和beta轨道的信息。对于每个轨道,输出中报告了轨道能量,之
后是每个有关原子给定角动量基函数的小数贡献。
这是一个很难确定系统自旋态的例子,因为正则α和β轨道完全不同。如果接下来用
Guess=(Read,Only,BiOrthogonalize) Pop=Orbital计算分析结果,那么程序会(对占据轨道
和虚轨道分别)转换α和β轨道,使之尽可能匹配。这种情况下转换后的轨道不再给出轨道能
量。而Pop=Orbital分析会报告相应的轨道对(即α 19和β 19)之间的重叠,值为1表示100%
地对应。轨道不再标记为占据轨道和虚轨道,而是标记为:
Docc 双占据:alpha和beta都被占据,匹配程度90%或更高。
Asing, Bsing
单占据:alpha与beta占据不匹配,或者较低占据的轨道与任何相反自旋的轨道
都不匹配。
Dvir 具有几乎相同alpha和beta轨道的虚轨道。
AVir, BVir
与任何相反自旋的虚轨道都不匹配的虚轨道。
在输出中,与其它轨道匹配的轨道只被程序列出一次。另外,对每个未成对的额外alpha
自旋占据轨道,总会有一些beta虚轨道与之匹配,后者会被忽略。
这里是FeO+四重态的输出,它的alpha电子比beta电子多3个,实际上是有4个未成对的
alpha占据轨道和1个未成对的beta占据轨道(最低的双占据轨道和最高的未占据轨道已在输
出中截掉):
Atomic contributions to molecular orbitals:
Docc. orb 13 abOv=0.999 is Fe1-d=1.00
Docc. orb 14 abOv=0.990 is O2-p=0.72 Fe1-s=0.14 Fe1-d=0.14
Asing orb 15 abOv=0.316 is Fe1-d=0.99
Asing orb 16 abOv=1.000 is Fe1-d=0.94
Asing orb 17 abOv=1.000 is Fe1-d=0.91
Asing orb 18 abOv=1.000 is Fe1-d=0.91
Dvirt orb 19 abOv=1.000 is O2-s=1.92 Fe1-s=-0.67 O2-p=-0.43 Fe1-p=0.14
Dvirt orb 20 abOv=1.000 is Fe1-s=0.52 Fe1-p=0.37
Bsing orb 15 abOv=0.316 is O2-p=0.92
轨道1-14是双占据的。Alpha轨道15和beta轨道15是不同的单占据轨道;所有的4个未成
对alpha自旋位于铁,1个未成对beta自旋位于氧。在被分析的默认轨道范围内,没有不匹配
的alpha和beta虚轨道。
片段级别的分解。如果有片段信息,输出还会报告片段的布居分析。对于Pop=Orbital
任务,会报告每个轨道用片段所作的分解。
含有片段布居的默认输出:
Mulliken charges with hydrogens summed into heavy atoms:
1
1 Pd -0.265855
2 P 0.346314
3 P 0.346314
- 185 -

4 Cl -0.168156
5 Cl -0.168156
6 C 0.060982
7 C 0.060982
8 C -0.106213
9 C -0.106213
Sum of Mulliken charges with hydrogens summed into heavy atoms = 0.00000
Condensed to fragments (all electrons):
1 -0.265855
2 -0.168156
3 -0.168156
4 0.060982
5 0.060982
6 0.480203
Pop=Orbital 输出:
Alpha occ 60 OE=-0.247 is Cl4-p=0.22 Cl5-p=0.22 P3-p=0.12 P2-p=0.12
Fr6=0.36 Fr2=0.22 Fr3=0.22
NBO 3输入的示例。以下是用NBO 3进行键级分析的输入文件:
# B3LYP/6-31G(d,p) Pop=NBORead
Example of NBO bond orders
0 1
C 0.000000 0.665676 0.000000
H 0.919278 1.237739 0.000000
H -0.919239 1.237787 0.000000
C 0.000000 -0.665676 0.000000
H -0.919278 -1.237739 0.000000
H 0.919239 -1.237787 0.000000
$nbo bndidx $end
Pressure
说明
指定用于热化学分析的压强(单位:大气压)。数值应作为选项定义:
# ... Pressure=1.5
默认是1大气压。
Prop
说明
这个特性关键词指示 Gaussian 计算静电特性[Johnson93, Barone96, Rega96, Barone96a]。默认计
算在每个原子核处的势,电场和电场梯度。用于静电分析的密度由关键词 Density 控制。
- 186 -
选择特性的选项
EFG 指示计算电势,场和场梯度。这是默认的选项。
Potential 指示只计算电势,而不计算场和场梯度。NoPotential 禁止计算电势和更高级
的特性。
Field 指示只计算电势和场,而不计算场梯度。
EPR 指示计算各向异性超精细耦合常数(即,自旋-偶极项)[Barone96, Rega96,
Barone96a]。
输入来源有关的选项
如果同时指定 Read 和Opt,那么输入部分的顺序先是固定点(Read),然后是优化的
点(Opt)。
Read 这个选项通知程序从输入串读入一列额外的原子中心,在这些中心计算特性。
每一中心是自由格式的标准方向直角坐标,单位是埃,一个原子占一行。
Opt 和Prop=Read 一样读入一列额外的中心,但接下来在距每一指定点最近的电
势处寻找最小值。
FitCharge 在范德华曲面上,对静电势拟合原子电荷。
Dipole 对偶极矩限制拟合电荷。
Grid 指定在一个或多个网格点上计算势并写到外部文件(这个选项常用 cubegen
工具代替)。这个选项指定将电势映射到二维网格点上。这些网格点既可以指
定为均匀的矩形网格,也可以是从辅助文件读取的任意点集合(都在下面说
明),或者通过 cubegen 使用的输入格式。
对于均匀的矩形网格,需要以下三个附加的输入行:
KTape,XO,YO,ZO 用于写文件的
Fortran
单元号,以及网格左下角的坐
标。
N1,X1,Y1,Z1 网格的行数以及垂直步长。
N2,X2,Y2,Z2 网格的列数以及水平步长。
如果从辅助文件读入点,则全部所需的输入信息仅有一行:
N,NEFG,LTape,KTape
从LTape 单元读入以埃为单位的 N个点的坐标,格式为 3F20.12。
LTape
默
认值为 52。将会计算势(NEFG=3),势和场(NEFG=2),或者势,场,和
场梯度(NEFG=1),并写到单元 KTape 中。例如以下输入指定从 Fortran 单
元10 读入 19,696 个用于静电势的点(代码 3),并把输出写到 Fortran 单元
11:
19696,3,10,11
适用性
HF,所有的 DFT 方法,CIS,MP2,MP3,MP4(SDQ),CID,CISD,CCD,CCSD 和
QCISD。
有关的关键词
Density,Cube
- 187 -
Pseudo
说明
这一关键词要求用模型势代替芯电子。选项 Cards 是最常用的模式。Gaussian 提供了
一种新的有效芯势(ECP)输入格式(类似于 ExtraBasis 使用的格式),说明见下。当读入
赝势时,不要使用与内置赝势相同的名称:CEP,CHF,LANL1,LANL2,LP-31,SDD 和SHC。
如果与 ONIOM 一起使用,Pseudo 关键词将用于 ONIOM 的所有层。如果仅对一层 ONIOM
读入 ECP,则应当用 GenECP 关键词代替。
没有任何选项的 Pseudo 默认为 Pseudo=Read。
选项
Read 由输入串读入赝势数据。输入说明见下一部分。Cards 是Read 的同义词。
Old 使用旧的格式(用于 Gaussian 92 和更早期版本)读入赝势数据。
CHF 使用无芯的 Hartree-Fock 势。该选项通常用于 LP-31G 基组。
SHC 使用 SHC 势。
LANL1 使用 LANL1 势。
LANL2 使用 LANL2 势。
完整的 ECP 输入格式
有效芯势算符是多项式径向函数,高斯径向函数和角动量投影算符的乘积之和。因此
ECP 输入指定在每一个原子中心使用哪种势,接着对 ECP 的每一角动量中每个势的每一项定
义三项:
(因子,R 的幂,指数)
因为只有最初几个角动量分量有不同的项,因此势可以表示为:(1)一般情况下的项,
典型情况下是 d 或 f,以及更高函数的投影,和(2)每一特殊角动量的额外项。因此对于包
含特殊的 s 和 p 投影项的 LP-31G 势,输入的数据包括一般项(d 及更高角动量),s-d 项(也
就是添加到一般项上构成 s 分量),和 p-d 项。
所有 ECP 输入采用自由格式。每一部分的第一行包括原子中心编号(由分子说明部分定
义),和/或原子符号,用来指定使用的原子和/或原子类型(就像一般的基组输入一样;见
Gen 关键词的说明)。列表以数值 0 作为结束。
接着是这些中心/原子的赝势:
Name,Max,ICore
势的名称,势的最大角动量(也就是特别的 s 和 p 投影为 2,s,p
和 d 投影为 3),以及由势代替的芯电子数。如果
Name
和先前定义的
势函数相同,则使用先前定义的势,除了数据区的终止行(见下)之
外不需要其它输入。
对于当前势的每个分量(
I
=1 到
Max
),读入一组数据,包含以下信息:
Title
数据区的说明,不用于计算。
NTerm
数据区中项的数量。
NPower,Expon,Coef
在 Nterm 个项中,每一个的 R 的幂,指数,和因子。NPower 包括 R2 雅
可比因子。
后面给出一个包括非标准 ECP 及其相应基组的输入文件示例。
简单的 ECP 输入格式
- 188 -

Gaussian 对 ECP 的输入增加了灵活性,可以包含前面定义的基组名。ECP 定义可由一行
含有预定义基组的标准关键词代替。在这种情况下,对应指定原子类型所定义基组的 ECP
将用于该原子(参见例子)。
STUTTGART/DRESDEN 型 ECP 输入的关键词
在Pseudo 输入中,这些 ECP 的关键词形式为 XY
n
,其中
n
是用赝势代替的芯电子数,
X
表示用于产生赝势的参考体系(S表示单个价电子的离子,M表示中性原子)。
Y指定参考数据的理论级别:HF 表示 Hartree-Fock,WB 表示 Wood-Boring 准相对论,
DF 表示 Dirac-Fock 相对论。对于有一个或两个价电子的原子来说,SDF 是很好的选择;否
则推荐使用 MWB 或MDF(虽然对于小原子或为了考虑相对论效应,可能会用到相应的 SHF 和
MHF 赝势)。
适用性
Stuttgart/Dresden ECP 还不能均匀地用于整个周期表。下面的表列出了各种可用的 XY
组合,以及有效的 n 值。在
默认
列中,列出了与 SDD 关键词等价的基组(对于到 Cl 的元素
选择了全电子基组,之后的元素使用 ECP)以及 IOp(3/6)设置为 6 时的等价基组(用于对所
有元素选择 ECP)。
默认 对给定 X 和 Y 值有效的 n 值
原子 IOp(3/6=6) SDD 关键词 MWB SDF SHF MDF MHF
1 H D95 D95
2 He D95 D95
3 Li SDF2 D95
4 Be SDF2 D95 2
5 B MWB2 D95 2 2
6 C MWB2 D95 2 2
7 N MWB2 D95 2 2
8 O MWB2 D95 2 2
9 F MWB2 D95 2 2
10 Ne MWB2 D95 2 2
11 Na SDF10 6-31G 10
12 Mg SDF10 6-31G 10
13 Al MWB10 D95 10 10
14 Si MWB10 D95 10 10
15 P MWB10 D95 10 10
16 S MWB10 D95 10 10
17 Cl MWB10 D95 10 10
18 Ar MWB10 6-31G 10 10
19 K MWB10 MWB10 10 18 18
20 Ca MWB10 MWB10 10 18 18
21 Sc MDF10 MDF10 10 10
22 Ti MDF10 MDF10 10 10
23 V MDF10 MDF10 10 10
24 Cr MDF10 MDF10 10 10
25 Mn MDF10 MDF10 10 10
- 189 -
26 Fe MDF10 MDF10 10 10
27 Co MDF10 MDF10 10 10
28 Ni MDF10 MDF10 10 10
29 Cu MDF10 MDF10 28 10 10
30 Zn MDF10 MDF10 28 28 10 10
31 Ga MWB28 MWB28 28 28
32 Ge MWB28 MWB28 28 28 28
33 As MWB28 MWB28 28 28
34 Se MWB28 MWB28 28 28
35 Br MWB28 MWB28 28 28
36 Kr MWB28 MWB28 28 28
37 Rb MWB28 MWB28 28 36 36
38 Sr MWB28 MWB28 28 36 36
39 Y MWB28 MWB28 28 28
40 Zr MWB28 MWB28 28 28
41 Nb MWB28 MWB28 28 28
42 Mo MWB28 MWB28 28 28
43 Tc MWB28 MWB28 28 28
44 Ru MWB28 MWB28 28 28
45 Rh MWB28 MWB28 28 28
46 Pd MWB28 MWB28 28 28
47 Ag MWB28 MWB28 28 46 28
48 Cd MWB28 MWB28 28 28
49 In MWB46 MWB46 46 46
50 Sn MWB46 MWB46 46 46
51 Sb MWB46 MWB46 46 46
52 Te MWB46 MWB46 46 46
53 I MWB46 MWB46 46 46 46
54 Xe MWB46 MWB46 46 46
55 Cs MWB46 MWB46 46 54 54
56 Ba MWB46 MWB46 46 54
57 La MWB28 MWB28 28,46,47 46,47
58 Ce MWB28 MWB28 28,47,48 47,48
59 Pr MWB28 MWB28 28,48,49 48,49
60 Nd MWB28 MWB28 28,49,50 49,50
61 Pm MWB28 MWB28 28,50,51 50,51
62 Sm MWB28 MWB28 28,51,52 51,52
63 Eu MWB28 MWB28 28,52,53 52,53
64 Gd MWB28 MWB28 28,53,54 53,54
65 Tb MWB28 MWB28 28,54,55 54,55
66 Dy MWB28 MWB28 28,55,56 55,56
67 Ho MWB28 MWB28 28,56,57 56,57
68 Er MWB28 MWB28 28,57,58 57,58
69 Tm MWB28 MWB28 28,58,59 58,59
- 190 -

70 Yb MWB28 MWB28 28,59 59
71 Lu MWB60 MWB60 28,60 60
72 Hf MWB60 MWB60 60 60
73 Ta MWB60 MWB60 60 60
74 W MWB60 MWB60 60 60
75 Re MWB60 MWB60 60 60
76 Os MWB60 MWB60 60 60
77 Ir MWB60 MWB60 60 60
78 Pt MWB60 MWB60 60 60
79 Au MWB60 MWB60 60 78 60 60
80 Hg MWB60 MWB60 60,78 60 60,78
81 Tl MWB78 MWB78 78 78
82 Pb MWB78 MWB78 78 78
83 Bi MWB78 MWB78 78 78
84 Po MWB78 MWB78 78 78
85 At MWB78 MWB78 78 78
86 Rn MWB78 MWB78 78 78
89 Ac MWB60 MWB60 60 60
90 Th MWB60 MWB60 60 60
91 Pa MWB60 MWB60 60 60
92 U MWB60 MWB60 60 60
93 Np MWB60 MWB60 60 60
94 Pu MWB60 MWB60 60 60
95 Am MWB60 MWB60 60 60
96 Cm MWB60 MWB60 60 60
97 Bk MWB60 MWB60 60 60
98 Cf MWB60 MWB60 60 60
99 Es MWB60 MWB60 60 60
100 Fm MWB60 MWB60 60 60
101 Md MWB60 MWB60 60 60
102 No MWB60 MWB60 60 60
103 Lr MWB60 MWB60 60 60
104 Rf 92
注意:这些 ECP 不能用于元素 87(Fr),88(Ra),105,以及原子序数更高的元素。
有关的关键词
ChkBasis,ExtraBasis,Gen,GenECP
例子
指定 ECP。这个输入文件对过氧化氢进行 RHF/LP-31G 计算,并从输入文件中读入基组
和 ECP 数据:
# HF/Gen Pseudo=Read Test
Hydrogen peroxide
- 191 -

0,1
O
H,1,R2
O,1,R3,2,A3
H,3,R2,1,A3,2,180.,0
R2=0.96
R3=1.48
A3=109.47
General basis set input
****
O 0
氧原子的 ECP。
OLP 2 2
ECP 名=OLP,用于 d 和更高的函数,代替 2 个电子。
D component
一般项的说明。
3
接下来的项数。
1 80.0000000 -1.60000000
1 30.0000000 -0.40000000
2 1.0953760 -0.06623814
S-D projection
投影项的校正(最低角动量)。
3
0 0.9212952 0.39552179
0 28.6481971 2.51654843
2 9.3033500 17.04478500
P-D
投影项的校正(最高角动量)。
2
2 52.3427019 27.97790770
2 30.7220233 -16.49630500
空行表示结束 ECP 数据区。
分子说明部分之后是基组数据。ECP 数据的第一行要求对原子 1 和 3(都是氧原子)读
入一个势(类型 7)。原子 2 和 4(都是氢原子)不使用赝势。
ECP 数据的第二行开始需要读入势的原子中心的输入:本例中是氧原子。这些中心的势
名称为 OLP,它是一般项,应用于角动量 2(D)和更高的函数,这个势代替两个电子。接下
来是一般项的标题(D component),该项的分量个数(3),接下来三行是各个分量。然后是
投影项的校正,分为两部分,从最低角动量开始。每一部分仍旧由标题行,项的个数,以及
这些项构成。
用标准基组关键词指定 ECP。下面的输入文件演示简化 ECP 输入格式的使用:
# Becke3LYP/Gen Pseudo=Read Opt Test
HF/6-31G(d) Opt of Cr(CO)6
0 1
Cr 0.0 0.0 0.0
继续分子说明部分
...
C O 0
6-31G(d)
****
Cr 0
LANL2DZ
铬原子用 LANL2DZ 基组。
****
- 192 -

Cr 0
铬原子的 ECP。
LANL2DZ
使用这种基组中的 ECP。
Punch
说明
这一输出说明关键词允许用户在计算各阶段输出有用的信息,用更现代的说法就是发送
个别的输出文件。这个输出通常作为相应操作系统上的 Fortran 备用单元输出的形式进行处
理(例如,在 UNIX 系统中,单元 7发送给文件 fort.7)。选项用于指定输出哪些信息。所有
这些选项可以组合使用,除了 MO 和NaturalOrbitals 只能选用其一。但是注意,合用选项
的输出数据各自独立,不互相影响。例如 Punch(MO, Gamess)把分子轨道和 Gamess 输入信
息发送到文件;它并不把 MO 信息按照 Gamess 输入格式进行格式化。
选项
Archive 指示输出重要计算结果的总结。这一输出具有与浏览量子化学数据库系统
(Browse Quantum Chemistry Database System)相同的格式。
Title 输出标题部分。
Coord 输出原子编号及其直角坐标,使用可由 Gaussian 读取的格式。
Derivatives 输出能量,核直角坐标导数和二阶导数,格式为 6F12.8,可用于随后的
Opt=FCCards 计算。
MO 输出轨道,使用符合 Guess=Cards 输入的格式。
NaturalOrbitals
(对于用关键词 Density 选择的密度)输出自然轨道。
HondoInput 输出 Hondo 某个版本的输入文件,可以很容易地改为其它版本的 Hondo 格式。
GAMESSInput 输出 GAMESS 的输入文件。
All 除了自然轨道之外,输出以上全部。
有关的关键词
Output
QCISD
说明
这个方法关键词进行包括单、双激发的二次 CI[Pople87]计算。注意这个关键词只请求做
QCISD 计算,默认不包括三激发校正 [Gauss88, Salter89] (参见下面的 T)。
选项
T 请求进行二次 CI 计算,包括单、双激发并对能量加入三激发贡献[Pople87]。
E4T 请求进行二次 CI 计算,包括单、双激发并对能量加入三激发贡献,以及求解
MP4 的三激发。必需和选项 T一起指定。
TQ 请求进行二次 CI 计算,包括单、双激发并对能量加入三、四激发贡献
- 193 -

[Raghavachari90]。
SaveAmplitudes
把收敛的波幅保存到检查点文件,用于随后的计算(例如使用更大基组的计
算)。使用这一选项会导致极大的检查点文件,但可以显著加速后面的计算。
ReadAmplitudes
从检查点文件读取收敛的波幅(如果有的话)。注意,新的计算可以使用与原
计算不同的基组,方法(若适用),等。
T1Diag 计算 T. J. Lee 等人[Lee89, Lee90]的 Q1 诊断流程。注意在用 QCISD 而非耦
合簇方法做诊断计算时,Q1 类似于 CCSD 的T1 诊断流程。
FC 这个关键词可以使用所有的冻芯选项。完整的信息参见 FC 选项的说明。
Conver=
N
设定收敛计算对能量为 10-N,对波函为 10-(N-2)。默认对单点能为
N
=7,对梯度
为
N
=8。
MaxCyc=
n
指定最大的循环次数。默认是 50。
适用性
QCISD 的解析能量和梯度,QCISD(T)的数值梯度,所有方法都能计算数值频率。
有关的关键词
CCSD
例子
QCISD 计算的能量出现在最后一步 QCISD 迭代的输出中:
DE(CORR)= -.54999890D-01 E(CORR)= -.7501966245D+02
如果指定了 QCISD(T),则在以上输出之后是包含非迭代三激发贡献的能量:
QCISD(T)= -.75019725718D+02
重新开始计算
说明
这个关键词8重新开始前面失败的任务。这一方法主要用于长时间的任务,其中涉及大
量的中间数据,因此把重新开始数据保存到检查点文件,会使检查点文件大到无法管理,违
背了检查点文件与读写文件相分离的目的。这个流程依赖于读写文件的命名,因此它在任务
异常终止时会保存下来,接下来用它重新开始计算。
例如,在频率计算中需要包含以下的链接 0命令:
%RWF=myrwf
%NoSave
%Chk=mychk
#P Freq ...
输入继续...
8 注意,Restart 并不是可被系统识别的关键词。——译注。
- 194 -

在任务结束时,任何用%行命名的文件默认都会被保留。%RWF 之后%NoSave 的覆盖
着一默认设置,因此如果任务正常结束,读写文件仍被删除(但如果任务提前终止,读写文
件仍被保留)。
需要注意以下几点:
任务结束后,检查点文件经常还会有用,因此典型情况下应把它放在%NoSave 之后。
读写文件可能非常大,应当放置在合适的文件系统中。例如,检查点文件可放在通常的
用户目录,它可能只有适量的自由空间和/或NFS 挂载空间。而足够用于重新开始任务
的读写文件应位于足够大的本地临时文件系统。
可以重新开始的任务是在完成前停止执行的任务。结束时有出错信息的任务,例如收敛
失败或超过优化步数,需要用户干预。
适用性
解析频率计算,包括用 ONIOM 所做的一些特性计算,如 ROA 和VCD;CCSD 和
EOM-CCSD 计算;NMR;Polar=OptRot;CID,CISD,CCD,CISD,和 BD 能量。进行多
个结构的计算,例如结构优化,IRC,和数值频率计算,最好像从前一样在关闭检查点文件
后重新开始。
有关的关键词
Freq,EOM
例子
下面的输入文件重新开始按照以上说明设置的任务:
%RWF=myrwf
%NoSave
%Chk=mychk
#P Restart
在执行路径部分不需要其它关键词,也不需要其它输入。
SAC-CI
说明
这个关键词选择Nakatsuji等人[Nakatsuji78, Nakatsuji79, Nakatsuji79a, Nakatsuji91, Nakatsuji91a,
Nakatsuji93, Nakatsuji96, Nakajima97, Nakatsuji97, Nakajima99, Ishida01, Ishida01a, Ehara02, Toyota02, Toyota03]
的对称匹配簇/组态相互作用(SAC-CI)方法。关于这个方法的详细信息,参见SAC-CI的文
档,位于以下网址:www.sbchem.kyoto-u.ac.jp/nakatsuji-lab
SAC-CI 任务必须指定用于稍后激发态计算的参考态。对于闭壳层体系,SAC-CI 默认使
用 RHF 波函。对于开壳层体系,除了 SAC-CI 关键词以外,还需要在执行路径部分用 ROHF
指定选择 ROHF 基态波函,或者用 AddElectron 或SubElectron 选项指定使用闭壳层态用于
基态计算。更多的信息参见以下的例子。
自旋态选项
Singlet=(
suboptions
)
- 195 -
指定计算单重态。括号内的子选项列表指定要计算的态以及其它计算参数。
其它的自旋态选项有 CationDoublet(同义词是 Doublet),AnionDoublet,
Triplet,Quartet,Quintet,Sextet 和Septet。可指定一个以上的自旋态。
自旋态子选项
在下面的选项中,要用所需要的自旋态替换
SpinState
。
SpinState
=(NState=(i1,i2,...))
对分子点群的各种不可约表示指定计算类型的态数。与分子对称性有关,最
多可以指定八个值(例如,D2h 是8个,C2v 是4个,等等)。简写形式 NState=
N
对每一个不可约表示指定一个
N
值。通过假定最接近的线性对称性来处理简
并(例如,对 Td用D2)。
SpinState
=(Density)
计算非松弛密度矩阵,并对自旋为
SpinState
的所有 SAC-CI 计算的态进行
Mulliken 布居分析。更多信息参见下面的例子。
SpinState
=(SpinDensity)
对自旋为
SpinState
的所有 SAC-CI 计算的态计算自旋密度矩阵。这还意味着
FullActive 选项。
SpinState
=(NoTransitionDensity)
默认情况下,如果
SpinState
是Singlet,则计算 SAC 基态和 SAC-CI 单重激
发态之间的跃迁密度和振荡强度;对于其它的自旋态,则计算最低的 SAC-CI
态和 SAC-CI 激发态之间的跃迁密度和振荡强度。选项 NoTransitionDensity
指定禁止对相应的自旋态进行这些计算。
其它的常用选项
TargetState=(SpinState=
s
, Symmetry=
m
, Root=
n
)
指定用于几何优化或梯度计算的目标态,或者和 Density 关键词合用。
s
是
指定多重度的关键词(即, Singlet,Doublet,等等),
m
是其点群不可约表
示的编号,
n
是所计算自旋态中解的序号(由以前的能量计算决定)。
AddElectron
在开壳层参考 SCF 组态上添加一个电子。这是以下体系的默认选项:
CationDoublet,Doublet,Quartet 和Sextet。
SubElectron 在开壳层参考 SCF 组态上去掉一个电子。这是以下体系的默认选项:
AnionDoublet。
TransitionFrom=(SpinState=
s
, Symmetry=
m
, Root=
n
)
指定用于计算跃迁密度矩阵的初始态。
s
是指定其多重度的关键词(即,
Singlet,Doublet,等),
m
是其点群的不可约表示编号,
n
是所指定的自旋
态中解的序号(同上面的 TargetState)。
AllProperties
计算直到十六极的多极矩,直到四阶的所有力矩,所有静电特性和反磁项(遮
蔽和磁化系数)。这选项适用于用 Density 子选项指定的所有自旋态。
NoProperty 不计算任何分子特性。
SelectCISOnly
在计算了 CIS 初始猜测后就终止计算。可以用这个选项确定感兴趣的特定态
的序号(例如,用于 TargetState)。其它方法参见下面的例子。
- 196 -
SACOnly 只计算参考态,不计算任何激发态。
专业用户的附加选项
附加的自旋态子选项
在下面的选项中,要用所需要的自旋态替换
SpinState
。
SpinState
=(MaxR=
N
)
指定最大激发级别为
N
。
SpinState
=(NonVariational)
对非对称矩阵解 SAC-CI 方程。Variational 通过将对称化矩阵作对角化进行
计算,这是默认的。注意,这个选项只能用于激发态的计算部分(基态计算
总是使用非变分流程)。
SpinState
=(InCoreDiag)
强制使用常驻内存算法。
SpinState
=(Iterative=
item
)
强制使用迭代算法。
item
指定初始猜测的类型:CIS 为 SInitial,CISD 为
SDInitial。
流程的选项
FC 用这个关键词可以指定所有的冻芯选项。完整信息参见 FC 选项的讨论。
一般而言,活性空间的大小对 SAC-CI 计算的精度有很大的影响。因此,推
荐使用全部轨道窗口。对于几何优化和梯度计算,默认为 Full。
LMO=
type
使用指定类型的局域化 MO 做为参考轨道。可用的类型有 PM(Pipek-Mezey)
和Boys。
MacroIteration=
N
在一个几何优化步骤内做
N
次宏迭代。
N
的默认值是0。
InCoreSAC 用常驻内存算法求解 SAC 方程。
MaxItDiag=
N
设定对角化的最大迭代次数。默认 64,最大 999。
MaxItSAC=
N
设定求解 SAC 方程的最大迭代次数。默认 999。
DConvDiag=
M
设定对角化计算的能量收敛标准为10-M。
DConvSAC=
M
设定求解 SAC 方程的能量收敛标准为 10-M。
精度级别选项
SD-R 用单、双连接的激发算符进行计算。这是默认的。
General-R 在计算中使用最高到六阶的连接激发算符。
LevelOne 设定选择双激发算符的阈值为最低的推荐级别。最准确的级别是
LevelThree,这是默认的。LevelTwo的精度介于两者之间。
WithoutDegeneracy
默认进行微扰选择,用以保持简并。该选项禁止这一测试,以降低计算需
求。在正式应用时,不推荐使用该选项。
NoLinkedSelection
禁止连接算符使用微扰选择阈值(也就是包含所有的算符)。
NoUnlinkedSelection
禁止非连接算符使用微扰选择阈值(也就是包含所有的算符)。
- 197 -
FullUnlinked
包含所有类型的非连接项。强制使用常驻内存算法。
如果要包含所有的项,那么三个选项 NoLinkedSelection,NoUnlinkedSelection,
FullUnlinked 都要用到,目前会极大降低计算效率。
WithoutR2S2 忽略R2S2非连接积分。这个选项实现计算精度和计算资源需求之间的折衷。
EgOp 在General-R 方案中使用指数生成算法产生四阶和更高阶的连接算符。这对
单点能计算是默认的。最高阶的激发级别可以用选项 MaxR 指定(最大值为 6)。
微扰选择阈值由 LevelOne,LevelTwo 和LevelThree 选项设定。
FullRGeneration
在General-R 方案中产生所有高阶连接算符,直到 MaxR=4,然后进行上面的
微扰选择。这对梯度计算和几何优化是默认的。
群组求和操作选项
在多点计算类型,如势能曲面扫描中,这些选项用来保证所有点之间的一致性。Scan
计算必须执行三次:在第一点用BeforeGSUM,之后对后面的某些点或所有点用CalcGSUM,
最后对所有的点用AfterGSUM。实际的结果由最后的计算提供。这个流程只适用于单重态,
三重态,电离态和电子亲和态,不能与General-R选项兼容。
BeforeGSUM 初始化一系列的连接计算。在第一点的计算中使用该选项。
CalcGSUM 在指定的点搜集数据,并确定阈值和算符的选择,以构成可用于每一点的一
致的设置。
AfterGSUM 在每一点使用以前用 CalcGSUM 选项收集的 GSUM 数据进行 SAC-CI 计算。
内存使用选项
如果因计算资源不足导致计算失败,可用以下选项增加程序的默认内存。
MaxR2Op=
N
设定 R2 算符在微扰选择之后的最大数量为
N
。默认为 100,000。
MaxEgOp=
N
设定在 General-R 方法中算符的最大数量为
N
。默认为 5,000。
适用性
解析能量,几何优化和数值频率。
几何优化默认使用全部轨道窗口。在几何优化计算中指定不同的冻芯选项会导致用数值
梯度计算,计算效率较差。
有关的关键词
Density
例子
如果要找出能量最低的两个单重激发态,应该使用像下面的执行路径:
# SAC-CI=(Full,Singlet=(NState=8))/6-31G(d) NoSymm ...
计算工作将会寻找8个单重态,忽略对称性。这两个能量最低的激发态将可能出现在计算得
到的电子态之中。
也可以使用以下的执行路径:
# SAC-CI=(Full,Singlet=(NState=4))/6-31G(d) ...
- 198 -
这个计算将会找出每个不可约表示中能量最低的四个单重激发态。
要对具有C2v对称性的分子指定每个不可约表示中单重激发态的数目,可以用这样的执
行路径:
# SAC-CI=(Full,Singlet=(2,2,1,2))/6-31G(d) ...
用便宜的初始计算找出激发态。可用初步的,低精度的计算找出所要的激发态,以节省
计算成本。例如,以下执行路径将对每种对称类型找出4个单重激发态:
# SAC-CI=(Full,Singlet=(NState=4),LevelOne)/6-31G(d) ...
接下来可以对感兴趣的态进行正常的计算(LevelThree)。例如:
# SAC-CI=(Full,Singlet=(1,0,1,0))/6-31G(d) ...
开壳层体系的计算。要预测乙烯基,一种中性的二重态自由基,的激发态,可用以下执
行路径:
# ROHF/6-31G(d) SAC-CI=(Full,Doublet=(NState=3),Quartet=(NState=3)) ...
这里指定在基态使用ROHF波函,对每一个不可约表示计算三个二重和三个四重激发态。对于
亚甲基的三重基态,可以用类似的方法。
几何优化。要优化指定的激发态,可以用TargetState选项:
# Opt SAC-CI=(Singlet=(Nstate=4),
TargetState=(SpinState=Singlet,Symmetry=1,Root=2))/6-31G(d) ...
计算电子密度和分子特性。要对所有预测的激发态计算非松弛密度和进行布居分析,
可以用以下执行路径:
# SAC-CI=(Full,Singlet=(...,Density),Triplet=(...,Density))/6-31G(d) ...
如果只计算三重态的非松弛密度和进行布居分析,可以在Singlet选项中忽略Density子选
项。
如果只对一个特定的态计算松弛密度和进行布居分析,可以用以下执行路径:
# SAC-CI=(Full,Singlet=(NState=4),TargetState=(...)) Density=Current ...
注意,这个任务要求进行完整的梯度计算,因此要比前面的计算要昂贵许多。
SAC-CI 的输出。SAC-CI 计算对每一个要求的自旋态(在这个例子中是单重态)产生如
下的表格:
--------------------------------------------------------------------
Transition dipole moment of singlet state from SAC ground state
--------------------------------------------------------------------
Symmetry Sol Excitation Transition dipole moment (au) Osc.
energy (eV) X Y Z strength
--------------------------------------------------------------------
A1 0 0.0 Excitations are from this state.
A1 1 8.7019 0.0000 0.0000 0.4645 0.0460
A1 2 18.9280 0.0000 0.0000 -0.4502 0.0940
A1 3 18.0422 0.0000 0.0000 -0.8904 0.3505
A1 4 18.5153 0.0000 0.0000 0.0077 0.0000
A2 1 7.1159 0.0000 0.0000 0.0000 0.0000
- 199 -

A2 2 18.2740 0.0000 0.0000 0.0000 0.0000
B1 1 1.0334 -0.2989 0.0000 0.0000 0.0023
B1 2 18.7395 -0.6670 0.0000 0.0000 0.2042
B1 3 22.1915 -0.1500 0.0000 0.0000 0.0122
B1 4 15.8155 0.8252 0.0000 0.0000 0.2639
B2 1 11.0581 0.0000 0.7853 0.0000 0.1671
B2 2 15.6587 0.0000 1.5055 0.0000 0.8696
B2 3 24.6714 0.0000 -0.7764 0.0000 0.3644
B2 4 23.5135 0.0000 -0.1099 0.0000 0.0070
---------------------------------------------------------------------
注意:输出中的各激发态按照对称类型分组,而不是按照能量增加排序。
Scale
说明
指定用于热化学分析的频率换算因子。值以选项方式指定:
# ... Scale=0.95
除了所用计算方法本身已指定了默认值的复合方法以外,默认值为 1.0。
Scan
说明
这个计算类型关键词指示进行势能面(potential energy surface;PES)扫描计算。计算
进行刚性 PES 扫描,它包含所选内坐标系的矩形格点上的一些单点能计算。分子结构必须
用Z-矩阵定义。每一变量的步数和步长在输入文件的变量定义行指定,接在各变量初值之
后。例如:
R1 1.41 3 0.05
A1 104.5 2 1.0
A2 120.0
这个输入表示变量 R1 以步长 0.05 增加三次。因此与其它变量组合的 R1 值一共有四个
(1.41,1.46,1.51 和1.56)。类似地,使用三个 A1 值(104.5,105.5,和 106.5),而 A2
的值固定在 120.0。总共要进行 12 个能量计算。可以扫描的变量数目没有限制。步长的单
位由 Units 关键词控制,默认是埃和度。
松弛 PES 的扫描计算(在每点进行几何优化计算)由关键词 Opt 指定。
如果在计算过程中进行扫描的变量破坏了对称性,那么在任务的执行路径中必须包含
NoSymm,否则任务会因出错而失败。
选项
Restart 重新开始 PES 扫描计算。从检查点文件可重新开始失败的 Scan 计算,只需
简单重复原输入文件的执行路径,并对 Scan 关键词加上 Restart 选项。不需
要其它输入。
- 200 -
有关的关键词
Opt
例子
PES 扫描计算的输出包含一个总结任务结果的表格:
Scan completed.
Summary of the potential surface scan:
N R A HF
---- --------- --------- -----------
1 0.9600 104.5000 -38.39041
2 1.0100 104.5000 -38.41306
3 1.0600 104.5000 -38.42336
4 0.9600 105.5000 -38.39172
5 1.0100 105.5000 -38.41430
6 1.0600 105.5000 -38.42453
7 0.9600 106.5000 -38.39296
8 1.0100 106.5000 -38.41547
9 1.0600 106.5000 -38.42564
10 0.9600 107.5000 -38.39412
11 1.0100 107.5000 -38.41657
12 1.0600 107.5000 -38.42668
---- --------- --------- -----------
在《 Exploring Chemistry with Electronic Structure Methods》一 书 [Foresman96b]的第 8 章提供
了关于势能曲面扫描的详细讨论。
SCF
说明
这个关键词控制 SCF 流程的运作。选项用来指定所需的计算方式,备选的算法,等等。
关于对不同问题最大效率地执行 SCF,更多信息见附录中效率的考虑部分。
默认的 SCF 流程使用 EDIIS
[Kudin02]和 CDIIS 的组合,不加阻尼或 Fermi 展宽。在 Gaussian
09 中,默认是 SCF=Tight。
对于收敛困难的情况,SCF=QC 选项经常会有帮助。对于收敛出现困难的 ROHF 波函,由
于无法使用 QC,这时可以在执行路径部分加上 Use=L506。
关于 SCF 收敛和稳定性的讨论,见参考文献[Schlegel91a]。
选择算法的选项
DIIS 使用 Pulay 的迭代子空间中直接反转(DIIS)外推方法[Pulay82],NoDIIS 禁
止这一选项。
CDIIS 只使用 CDIIS。CDIIS 同时意味着 Damp。
Fermi 指定在初期迭代中使用温度展宽[Rabuck99],并加上 CDIIS 和阻尼。NoFermi
禁止 Fermi 展宽。Fermi 默认还意味着 Damp 并包含能级移动。
Damp 在初期 SCF 迭代中使用动态阻尼。默认是 NoDamp。但是如果指定了 SCF=Fermi
- 201 -
或SCF=CDIIS,则会使用阻尼。注意,阻尼和 EDIIS 不能同时使用。
NDamp=
N
允许对直到
N
次 SCF 迭代使用动态阻尼(默认为 10)。
QC 要求使用二次收敛 SCF 流程[Bacskay81]。默认是当远离收敛时使用线性搜索,
接近收敛时使用 Newton-Raphson 走步方法(除非能量增加)。这个方法较通
常加入 DIIS 外推的 SCF 方法慢,但是更稳定。SCF=QC 不能用于限制性开壳
层(RO)计算。
XQC 对于一阶 SCF 尚未收敛的情况,加入一个额外的 SCF=QC 步骤。
MaxConventionalCycles=
N
在SCF=XQC 计算中,设定传统 SCF 循环的极限为
N
。
PseudoDiagonalization=N
在链接 502 中尽可能使用赝对角化,其中仅在循环的初期,结束,以及中间
的每第
N
个循环进行完全对角化。PDiag 是该选项的同义词。这对半经验方
法是默认选项(默认值为
N
=30)。
FullDiagonalization
在链接 502 中强制使用完全对角化。这对 HF 和 DFT 是默认的。FDiag 是该选
项的同义词。
SD 进行最陡下降 SCF 计算。
SSD 进行缩放的最陡下降 SCF 计算。
SaveKPoint 在 SCF 结束时保存 k 点信息。NoSaveKPoint 不保存这些数据,这是默认的,
但数值频率计算除外,这时默认为 SaveKPoint。
DM 使用直接最小化 SCF 程序[Seeger76]。它通常不如 SCF=QC,主要用于向下兼
容性,不到万不得已不要使用。只适用于闭壳层 RHF 和开壳层 UHF 计算。
VShift[=
N
] 将轨道能量提升
N
*0.001(也就是
N
milliHartree);默认的
N
为100。这
个选项禁止自动存档。
N
=-1 禁止能级移动,等价于 NoVShift。
MaxCycle=
N
改变 SCF 循环的最大数量为
N
;默认的
N
为64(对SCF=DM 和SCF=QC 默认为
512)。注意,当开启 DIIS 后,内存需求会随最大循环次数的增加而增加。
FullLinear 指定在每一次迭代计算中用 L508(SCF=QC,SD 或SSD)进行完全线性搜索。
默认情况下,只有当初始微迭代导致能量上升时才进行完全最小化。
MaxRot=
N
设定SCF=QC 中 Newton-Raphson 走步的最大转动梯度为 10-N。高于此值使用
缩放最陡下降法;高于此值 100 倍以上使用最陡下降法。默认的
N
值是 2。
FinalIteration
在使用 DIIS 的 SCF 或直接 SCF 收敛以后,FinalIteration 进行最后一次非
外推、非增量迭代计算。NoFinalIteration 禁止这一计算。默认是
NoFinalIteration。
IncFock 强制产生增量 Fock 矩阵。这是直接 SCF 方法的默认选项。NoIncFock 禁止产
生增量 Fock 矩阵,这是传统 SCF 方法的默认选项。
Pass 对于常驻内存计算,把积分保存在磁盘上,以避免在链接 1002 中重新计算
这些积分。只适用于加上 SCF=InCore 的频率计算任务。NoPass 强制在每个
常驻内存计算阶段重算积分。
TightLinEq 在整个 SCF=QC 计算的线性方程求解中使用严格的收敛。默认当转动梯度降
低时使用严格的收敛标准。
VeryTightLinEq
在整个 QCSCF 计算的线性方程求解(微迭代)中使用更严格的收敛标准。对
近于线性依赖的情况,有时需要使用该选项。VeryTightLinEq 的同义词是
- 202 -
VTL。
积分存储选项
Direct 进行直接 SCF 计算,其中双电子积分在需要时重新计算。这是 Gaussian 默认
的SCF 计算流程。它能用于所有可用的计算方法,除了 MCSCF 二阶导和使
用复轨道的方法之外。
InCore 要求在进行 SCF 计算时,将完整的积分列表存放于内存。如果有足够的内存,
这在直接 SCF 计算中是自动设置的。使用 SCF=InCore 会强制常驻内存存储,
如果空间不足,计算会终止。NoInCore 用于对 SCF 和 CPHF 禁止使用常驻内
存流程。
Conventional
把双电子积分存储在磁盘上,并在每一 SCF 迭代中读取。NoDirect 是
Conventional 的同义词。
收敛和阈值的有关选项
Conver=
N
设置SCF 的收敛标准为 10-N。 除了 GVB 和CASSCF 之外,这是基于密度的
收敛标准,而 GVB 和CASSCF 的收敛标准分别是轨道的改变和能量的改变。
VarAcc 在直接 SCF 计算的初期使用中等积分精度,稍后再转换成正常精度。这对直
接 SCF 方法是默认的,可以用 NoVarAcc 关闭。VarAcc 的同义词为 VarIn t,
NoVarAcc 的同义词为 NoVarInt。
Tight 在 SCF 中使用正常的严格收敛标准。这是默认的。同义词为 NoSinglePoint,
NoSP,NoSleazy 和TightIntegrals。
Sleazy 要求适用于单点计算的宽松收敛标准;等价于 SCF=(Conv=4,
VarInt,NoFinal,Direct)。这对单点 CASSCF 或直接 SCF 计算是默认的。可
以缩写成 SP。SinglePoint 是Sleazy 的同义词。
VerySleazy 进一步降低阈值;在迭代中使用 Int=CoarseGrid 和单点计算的积分精度,
接着用通常的单点计算格点(MediumGrid)进行一次迭代计算。不推荐用于
正式的计算中。
对称性有关的选项
IDSymm 在第一次迭代中进行密度矩阵的对称化,以和分子的对称性相符(“初始密度
对称化”)。默认是 NoIDSymm。
DSymm 在每一 SCF 迭代中,进行密度矩阵的对称化,以和分子的对称性相符(“密
度对称化”)。默认是 NoDSymm。DSymm 意味着使用 IDSymm。
NoSymm 取消所有轨道对称性的限制。它的同义词为 Guess=NoSymm 和
Symm=NoSCF。
Symm 保留所有对称性限制:使每一对称类型(阿贝尔不可约表示)的占据轨道数
与初始猜测相匹配。使用这一选项可以在整个计算中保持波函特定的态。这
仅对 GVB 计算是默认的。
IntRep 要求在 SCF 流程中利用对称操作,通过复制积分考虑积分的对称性。即使波
函不具有完整的分子对称性,仍可以使用较短的积分列表。适用于 L502(默
认用于 HF,ROHF 和UHF)和 L508(SCF=QC)。
FockSymm 要求 SCF 流程用对称化 Fock 矩阵的方法考虑积分的对称性(使用“小型”积
分列表)。这是默认选项。FockSymm 的同义词是 FSymm。
- 203 -
重新开始计算的有关选项
Save 在每一次迭代中将波函保存在检查点文件中,用来重新进行 SCF 计算。这对
直接 SCF 是默认的。NoSave 禁止保存波函。
Restart 从检查点文件重新开始 SCF 计算。SCF=DM 计算不能重新开始。
SCRF
说明
这一关键词指定在溶剂存在的环境下进行计算,通过把溶质放入溶剂反应场之间的空穴
中。
极化连续模型(Polarizable Continuum Model;PCM)的积分方程形式的变体(IEFPCM)
是 SCRF 方法的默认设置。这一方法通过一系列重叠球创建溶质空穴。它最初是由 Tomasi
及合作者和 Pascual-Ahuir 及合作者[Miertus81, Miertus82, Pascual-Ahuir94, Cossi96, Barone97, Cances97,
Mennucci97, Mennucci97a, Barone98, Cossi98, Barone98a, Cammi99, Cossi99, Tomasi99, Cammi00, Cossi00, Cossi01,
Cossi01a, Cossi02, Cossi03]设计。这一模型对应于 SCRF=PCM。最近的评论参见[Tomasi05]。Chipman
的模型[Chipman00]与此方法密切相关[Cances01]。
其它可用的模型有 IPCM,它对空穴使用静态等密度面[Foresman96],自洽等密度 PCM
(Self-Consistent Isodensity PCM;SCIPCM)模型[Foresman96],以及 Onsager 模型[Kirkwood34,
Onsager36, Wong91, Wong91a, Wong92, Wong92a],它把溶质放在溶剂反应场内的球穴中。
从 Gaussian 03 开始,执行 PCM 的许多方面已发生改变。在 Gaussian 09 中,我们使用
连续表面电荷算法以保证反应场的连续,平滑,和稳定,并且对原子位置和外部扰动场还有
连续的导数[Scalmani09]。它把溶剂-溶剂接触面累积的表面电荷展开为球形高斯函数,这些函
数位于每个曲面单元,从而使空穴表面离散化。曲面导数的不连续性可以通过有效地平滑球
面接触点的区域来消除。
这一算法最初于 1999 年由 Karplus 和 York 对导体屏蔽模型提出[York99],但未得到应有
的重视。在 Gaussian 09 中,我们在 PCM 系列溶剂化方法的框架内对其进行了开发和推广,
现在已成为创建溶剂空穴和计算反应场的默认方法。
Gaussian 09 中的 PCM 方法包括外部迭代流程,程序通过建立溶剂反应场与溶质静电势
的自洽(后者从指定的模型化学算出的电子密度产生),由此计算出溶剂中的能量[Improta06,
Improta07]。与标准方法(基于变分方法或线性响应理论)的差别可用 MP2 来说明。默认的流
程对 SCF 密度计算溶剂效应,接下来应用 MP2 微扰,而外部的迭代方法用 MP2 密度自洽计算
溶剂效应。虽然此项技术主要用于研究如荧光这样的激发态过程,但也可以使用具有梯度的
理论方法,例如后 SCF 方法,用于基态计算。用 ExternalIteration 选项指定这种方法。
溶剂化与激发态
有两种基本方法用于模拟溶液中的激发态:
计算溶剂环境中最低的几个激发态。这种方法早在 Gaussian 03 中即可使用,它把 SCRF
加入到正常的激发态计算如 TD 或CIS 中。这种技术使用线性响应方法,并把必要的项
加入到激发态方法的方程中(因此包含了激发态的溶剂化效应)[Cammi00, Cossi01]。在
Gaussian 09 中,TD 或CIS 计算的溶液中的特定激发态可以做结构优化[Scalmani06]。
单个激发态可以通过特定态方法模拟。在这种情况下,程序通过激发态密度对溶剂反应
场自洽产生的静电势[Improta06, Improta07],并利用上面提到的外部迭代技术来计算溶液中
- 204 -
的能量。
对于溶液中的激发态计算,平衡和非平衡计算之间是有区别的。溶剂对溶质状态变化的
响应有两种不同的方式:它使其电子分布发生极化,这是个非常迅速的过程,以及溶质分子
重新调整其自身(例如,通过转动),这是个较慢的过程。平衡计算描述溶剂(以两种方式)
来得及完全响应溶质的情况,例如,结构优化(这一过程与分子在溶剂中的运动具有相同的
时间标度)。非平衡计算适于溶剂来不及完全响应的非常迅速的过程,例如垂直电子激发。
平衡溶剂化对 CIS 和 TD-DFT 的激发态结构优化是默认的。非平衡溶剂化对使用默认 PCM
流程的 CIS 和 TD-DFT 能量计算是默认的,平衡溶剂化对使用外部迭代方法
(SCRF=ExternalIteration)的计算是默认的。进行非平衡外部迭代的计算方法参见示例。
CASSCF PCM[Cossi99]计算默认对应于溶剂反应场/溶质电子密度极化过程的平衡计算。涉
及两个不同电子态的非平衡溶质-溶剂相互作用计算(例如,垂直跃迁的初态和终态)可以
用NonEq=
type
PCM 关键词,在两个单独的任务步骤中进行(参见下面的 PCM 输入部分)。
必需的和可选的输入:PCM 模型
若同时指定了 Read 选项,则指定 PCM 计算(即默认的 SCRF=PCM 或SCRF=CPCM)
细节的关键词和选项可以在包括一个终止空行的输入部分提供。这一部分的关键词遵循一般
的 Gaussian 输入规则。可以使用的关键词在例子之后的另外部分列出。
必需的输入:其它模型
对于 Onsager 模型(SCRF=Dipole),需要从输入数据的同一行读入两个自由格式的实
数:以埃为单位的溶质分子半径和溶剂的介电常数。合适的溶质半径是由气相分子体积计算
(另外的计算)得来的;见下面 Volume 关键词的说明。
对于 IPCM 和SCI-PCM 模型,输入由一行指定溶剂介电常数和可选的等密度值构成(后
者默认是 0.0004)。
指定溶剂的选项
Solvent=
item
选择用于计算的溶剂。注意对不同的 SCRF 方法,溶剂还可以在输入数据中
用不同的方式指定。如果未指定,默认的溶剂是水。Item 是从这一部分最后
的表格中选出的溶剂名。
选择方法的选项
PCM 使用积分方程形式模型(IEFPCM)进行反应场计算。这是默认选项。形式
的某些细节与执行方式已不同于 Gaussian 03,说明见[Scalmani09]。IEFPCM 是
PCM 的同义词。
如果 PCM 用于各向异性或离子溶剂,那么必须使用 Read 选项和 PCM 输入
部分的项目,从而对这类溶剂选择各向异性或离子介电模型(见下)。这些溶
剂没有连续表面电荷形式,并且不能计算导数。
CPCM 使用 CPCM 极化导体计算模型[Barone98, Cossi03]进行 PCM 计算。
Dipole 进行 Onsager 模型反应场计算。
IPCM 进行 IPCM 模型反应场计算。IPCM 的同义词是 Isodensity。
SCIPCM 进行 SCIPCM 模型反应场计算,即,由等密度面自洽得到空穴,用它进行 SCRF
计算。
- 205 -
PCM 和 CPCM 模型的选项
Read 表示从输入数据的单独部分读取提供计算参数的关键词和选项(说明见上)。
对于各向异性或离子溶剂,必须指定该选项。
Checkpoint 从检查点文件恢复 SCRF 信息。
Modify 从检查点文件恢复 SCRF 信息,并从输入串读入修改。
ONIOMPCM=k
依照代码字母 k选择的方案,执行溶剂中的 ONIOM 计算[Vreven01, Mo03]。可用
的值有:
A 用整合的 ONIOM 密度自洽计算反应场(仅能用于能量,并且不能
用于半经验方法)。
B 对真实体系在低阶方法计算反应场,相应的极化电荷用于模型体系
子计算中的外部电荷(仅用于能量计算)。
C 仅对真实体系在低阶方法计算反应场,而模型体系子计算假设在零
反应场(即,气相)中进行。这种选择可用于能量,优化,和频率
计算。
X 在每个子计算中分别计算反应场,并总是使用真实体系的空穴。若
指定了 ONIOM 和SCRF,这是默认的(可用于能量,优化,和频
率计算)。
ExternalIteration
通过链接 124 的外部迭代,执行自洽 PCM 计算。这一方法通过溶质的静电
势对溶剂反应场自洽来计算溶液中的能量[Improta06, Improta07](见上面的讨论)。
ExternalIteration 仅能用于能量计算。SelfConsistent ,SC ,
StateSpecificPerturbation,和 SSPerturbation 是该选项的同义词。
Restart 从检查点文件重新开始 PCM 外部迭代计算。
SkipVacuum 在溶液的外部迭代 PCM 计算中执行第一次迭代。这可用于加速朝着自洽的
方向收敛,并且是默认设置。在 TD-DFT 计算中,我们推荐使用这一选项以
获得正确的激发态顺序。DoVacuum 在气相执行第一次迭代;在计算溶液的
ΔG时需要这一选项。
SMD 对于 Truhlar 及合作者的 SMD 溶剂化模型[Marenich09]使用半径和非静电项进行
IEFPCM 计算。对于计算溶液的 ΔG,这是推荐的选项,它通过对感兴趣体系
进行气相和 SCRF=SMD 计算,得到能量差来完成。
G03Defaults 修改 PCM 默认设置,以便尽可能地重复 Gaussian 03 PCM 计算的结果。注意
由于程序的改进,不可能实现严格一致。
SCRF=DIPOLE 模型的选项
A0=
val
在执行路径部分(而不是从 SCRF=Dipole 计算的输入字串中读取)设定溶质
分子半径
a0
的值。如果包含该选项,那么也必须使用 Solvent 或Dielectric。
Dielectric=
val
设定溶剂的介电常数值。如果和 Solvent 同时指定,该选项优先。
SCRF=IPCM 模型选项
GradVne 对数值积分使用 Vne 盆。
GradRho 对数值积分使用密度盆。如果出现非核的吸引子,任务将会失败。
- 206 -

SCRF=SCIPCM 模型选项
UseDensity 强制使用密度矩阵以计算电子密度。
UseMOs 强制使用 MO 以计算电子密度。
GasCavity 使用气相等密度面以定义空穴,而不是以自洽方式求解曲面。它主要用作调
试选项。
适用性与限制
下面的表格详细列出了各种 SCRF=PCM 计算类型对理论方法的适用情况:
方法 能量
(外部迭代)
能量 优化 频率 三阶特性 a NMR
MM no yes yes yes no no
AM1,PM3,PM3MM,PM6,PDDG no yes yes yes no no
HF,DFT yes yes yes yes yes yes
MP2 yes yes
b yes
b yes
b yes
b,c yes
b
MP3,MP4(SDQ),CCSD,QCISD,BD yes yes
b no no no no
CASSCF yes yes yes yes e no no
CIS yes yes d yes d yes d no no
TD yes yes d yes d yes d,e no no
ZIndo no yes no no no no
a 例如,Freq=Raman,ROA,或 VCD;b 通过 SCF MO 极化进行计算;c 数值计算拉曼强度(即,如同使
用Freq=NRaman);d 使用线性响应方法;e 仅有数值频率。
使用 PCM 溶剂化的 CIS 和CASSCF 频率必须用 Freq=Numer 做数值计算。
重新开始 SCRF 计算。通过使用 Restart 选项,SCRF=ExternalIteration 和SCRF=IPCM
任务可以从读写文件重新开始。SCF 迭代过程失败的 SCRF=SCIPCM 计算应当通过
SCF=Restart 关键词重新开始。
非默认方法。IPCM 模型只用于 HF,DFT,MP2,MP3,MP4(SDQ),QCISD,CCD,
CCSD,CID 和CISD 能量计算。SCIPCM 模型用于 HF 和DFT 的能量,几何优化和数值频
率计算。Onsager 模型(SCRF=Dipole)适用于 HF,DFT,MP2,MP3,MP4(SDQ),QCISD,
CCD,CCSD,CID 和CISD 能量计算,以及 HF 和DFT 的优化和频率计算。但是,关键词
组合 Opt Freq 不能用于 SCRF=Dipole 计算。
有关的关键词
SCF,Volum e
例子
PCM能量。一般情况下,默认SCRF方法的能量输出以正常的形式显示在输出文件中。例
如,以下是输出文件的一部分,包括从Hartree-Fock和从MP2 PCM计算预测的能量:
Hartree-Fock SCRF计算:
SCF Done: E(RHF) = -99.4687828290 A.U. after 8 cycles
Convg = 0.2586D-08 -V/T = 2.0015
MP2 SCRF计算:
- 207 -

E2 = -0.1192799427D+00 EUMP2 = -0.99584491345297D+02
预测的溶液中的能量包含了所有计算的校正(这与Gaussian 03的输出不同)。
若使用了各种 PCM 选项,还会出现其它的输出行。例如,下面的输出由 HF SCRF(SMD)
计算产生:
SCF Done: E(RHF) = -99.4687828290 A.U. after 8 cycles
Convg = 0.2586D-08 -V/T = 2.0015
SMD-CDS (non-electrostatic) energy (kcal/mol) = 0.54
(included in total energy above)
对于特定态和外部迭代SCRF计算,最后的能量由链接124计算,它用来控制外部迭代,
结果且在另外的输出部分显示,位于非常靠近输出文件结束的位置,如下例所示:
--------------------------------------------------------------------
Self-consistent PCM results
===========================
<psi(f)| H |psi(f)> (a.u.) = -99.577537
(A)
<psi(f)|H+V(f)/2|psi(f)> (a.u.) = -99.584002
(B)
(Polarized solute)-Solvent (kcal/mol) = -4.06
(C)
--------------------------------------------------------------------
Partition over spheres:
Sphere on Atom Surface Charge GEl GCav GDR
1 H1 15.27 -0.157 -2.36 0.00 0.00
2 F2 32.58 0.157 -1.70 0.00 0.00
--------------------------------------------------------------------
外部迭代预测的能量值和特定态的SCRF计算:
After PCM corrections, the energy is -99.5840023899 a.u.
--------------------------------------------------------------------
(A)行给出用极化溶质波函和气相哈密顿计算的能量,(B)行给出用极化溶质波函和溶液
中的哈密顿计算的能量,(C)行给出极化溶质和溶剂之间的相互作用能,对应于积分
<Ψ(f)|V(f)|Ψ(f)>(单位:kcal/mol),最后一行给出包含所有PCM校正的预测能量。
荧光的例子:乙醛从第一激发态的(荧光)发射谱(n→π*)。在这里,我们研究这样
的循环:
发射
乙醛
基态
乙醛
基态形变结构
乙醛
n→
*激发态
平衡结构
乙醛
n→
*激发态
乙醛的激发与发射循环
垂直激发
感兴趣的主要过程是发射,但这个例子演示如何研究包括溶剂化效应在内的整个循环。
第一步:基态的结构优化和频率(平衡溶剂化)。这是对基态包含PCM平衡溶剂化的
- 208 -

标准Opt Freq计算。
%chk=01-ac
# B3LYP/6-31+G(d,p) Opt Freq SCRF=(Solvent=Ethanol)
Acetaldehyde ground state
0 1
C
C,1,RA
X,2,1.,1,A
O,2,RB,3,A,1,180.,0
X,1,1.,2,90.,3,0.,0
H,1,R1,2,A1,5,0.,0
H,1,R23,2,A23,5,B23,0
H,1,R23,2,A23,5,-B23,0
H,2,R4,1,A4,3,180.,0
RA=1.53643
RB=1.21718
R1=1.08516
R23=1.08688
R4=1.10433
A=62.1511
A1=110.51212
A23=109.88119
A4=114.26114
B23=120.56468
第二步:包含线性响应溶剂化的垂直激发。这是垂直激发的TD-DFT计算,因此在基态
平衡结构计算,使用默认溶剂,线性响应,非平衡溶剂化。我们进行单点TD-DFT计算,这
对非平衡溶剂化是默认的。该任务的结果将用于识别感兴趣的态及其顺序。这些结果给出激
发态溶剂化的合理描述,但不如特定态溶剂化计算的结果好。在本例中,我们看到n→π*态
是第一激发态。下一步,我们将使用特定态方法,对垂直激发步骤产生更好的描述。
%chk=02-ac
# B3LYP/6-31+G(d,p) TD=NStates=6 SCRF=(Solvent=Ethanol)
Geom=Check Guess=Read
Acetaldehyde: linear response vertical excited states
0 1
第三步:垂直激发的特定态溶剂化。这需要两个任务步骤:首先做基态计算,为了存
储基态的非平衡溶剂化信息,需要在PCM输入部分指定NonEq=write。接下来做实际的特定
态的计算,需要用NonEq=read读取非平衡溶剂化的必要信息。
%chk=03-ac
# B3LYP/6-31+G(d,p) SCRF=(Solvent=Ethanol,Read)
Geom=Check Guess=Read
Acetaldehyde: prepare for state-specific non-eq solvation
by saving the solvent reaction field from the ground state
0 1
- 209 -

NonEq=write
--link1--
%chk=03-ac
# B3LYP/6-31+G(d,p) TD(NStates=6,Root=1)
SCRF=(Solvent=Ethanol,StateSpecific,Read)
Geom=Check Guess=Read
Acetaldehyde: read non-eq solvation from ground state and
compute energy of the first excited with the state-specific method
0 1
NonEq=read
第四步:放松激发态结构。接下来,我们用平衡的线性响应溶剂化进行TD-DFT几何优
化,以找到激发态势能面上的最小能量点。由于这是TD-DFT优化,程序默认为平衡溶剂化。
作为这类体系的典型情况,基态具有平面对称性,而在激发态对称性被破坏,因此在优化的
开始对基态结构作了略微的扰动以打破对称性。
%chk=04-ac
# B3LYP/6-31+G(d,p) TD=(Read,NStates=6,Root=1) SCRF=(Solvent=Ethanol)
Geom=Modify Guess=Read Opt=RCFC
Acetaldehyde: excited state opt
Modify geometry to break Cs symmetry
since first excited state is A"
0 1
4 1 2 3 10.0
5 1 2 7 -50.0
第五步:激发态结构的振动频率。现在我们通过运行频率计算来确认第四步找到的结
构是能量极小点。如果需要,结果还可用作Franck-Condon计算的一部分(见下)。这是一
个数值频率计算。
%chk=05-ac
# B3LYP/6-31+G(d,p) TD=(Read,NStates=6,Root=1) Freq
SCRF=(Solvent=Ethanol) Geom=Check Guess=Read
Acetaldehyde excited state freq
0 1
第六步:溶液中特定态的发射(第一部分)。这一步在激发态平衡构型计算特定态的
平衡溶剂化,并通过输入中的PCM选项NonEq=write,把溶剂化数据的输出写到文件,用于
下一步计算。
%chk=06-ac
# B3LYP/6-31+G(d,p) TD=(Read,NStates=6,Root=1)
SCRF=(Solvent=Ethanol,StateSpecific,Read)
Geom=Check Guess=Read
Acetaldehyde emission state-specific solvation
at first excited state optimized geometry
- 210 -

0 1
NonEq=write
第七步:到最终基态的发射(第二部分)。最后,我们用非平衡溶剂化和来自激发态
的静态溶剂化,在激发态结构计算基态能量。
%chk=07-ac
# B3LYP/6-31+G(d,p) SCRF=(Solvent=Ethanol,Read) Geom=Check Guess=Read
Acetaldehyde: ground state non-equilibrium
at excited state geometry.
0 1
NonEq=read
对于计算气相的激发和发射能量,第一、二、四步(以及第五步确认驻点的性质)已经
足够。但是当包含溶剂效应时,这是不够的,因为在第四步计算的能量对应于基态溶剂反应
场,而发射发生在对激发态电荷分布进行响应的反应场。在第六和第七步中,这一差别可以
正确计算。
如果计算带形,在气相可以简单地用Freq=(ReadFC,FC,Emission)运行计算,采用第一
步的检查点文件作为该任务的主要检查点文件,并在输入行中提供第五步的检查点文件名,
用于指定其它的电子态。对于溶剂化带形,必须对第一步和第五步使用检查点文件进行
Freq=(ReadFC,FC,Emission,ReadFCHT)计算,同时还要在输入部分提供特定电子态的发射
能量,用于Franck-Condon计算。
PCM计算的附加关键词
PCM SCRF计算可以指定附加的输入关键词。它们位于独立的输入部分,像往常一样用空
行结束,如这个例子:
# B3LYP/6-31G(d) 5D SCRF(SMD,Solvent=Generic,PCM=Read)
Water, solvation by methanol, re-defined as generic solvent.
0 1
O
H,1,0.94
H,1,0.94,2,104.5
stoichiometry=C1H4O1
PCM关键词的输入部分
solventname=methanol
eps=32.63
epsinf=1.758
…
PCM输入的结束空行
可以用以下关键词控制 PCM 计算(有关的项目分组排列):
定义溶剂参数
- 211 -
PCM 计算所用的溶剂通常可以用 SCRF 关键词的常规选项 Solvent 指定。可以用下面
的关键词覆盖一些已知溶剂的默认值:
EPS=
x
指定溶剂的静态(或零频)介电常数。
EPSINF=
x
指定溶剂的动态(或光学)介电常数。
RSOLV=
x
指定溶剂半径(单位:埃)。只用于 AddSph 或Surface=SAS。
未指定的参数默认为Solvent选项所指定溶剂(默认是水)的值。
计算方法变化
NonEq=item 在用 SCRF(Read) 完成 HF ,DFT ,或 CASSCF 计算后,或在任何
SCRF(ExternalIteration,Read)计算的最后,计算并保存非平衡反应场。
NonEq=Write 表示把数据保存到检查点文件。在随后的计算中,可以用
NonEq=Read 从检查点文件恢复数据。
Dis 用Floris 和Tomasi 的模型[Floris89, Floris91],在总能量中计算并包含溶质—溶剂
的色散相互作用能。默认是 NoDis。该选项不能用于 SCRF=SMD 计算。
Rep 用Floris 和Tomasi 的模型[Floris89, Floris91],在总能量中包含溶质—溶剂的排斥
相互作用能。默认是 NoRep。该选项不能用于 SCRF=SMD 计算。
Cav 用Pierotti 的模型[Pierotti76],在总能量中包含溶质空穴生成能。默认是 NoCav。
该选项不能用于 SCRF=SMD 计算。
CavityFieldEffects
用Cammi 及合作者的模型[Cammi00a],在总能量中包含空穴—场相互作用能的
影响(也就是局域场影响)。默认是不包含该影响。
CF=Eps=x 为用于空穴—场能量分布的静态介电常数指定不同的值。这对于调节空穴—
场效应的大小非常有用。
CF=EpsInf=
x
为用于空穴—场能量分布的动态(光学)介电常数指定不同的值。这对于调
节空穴—场效应的大小非常有用。
FITPOT 按照原子或原子团相加贡献的方法分析溶质-溶剂相互作用能。分析涉及用原
子电荷对溶液中的分子静电势进行拟合。
ITERATIVE 通过迭代方法求解 PCM 静电问题,计算极化电荷。
MXITER=
N
在迭代求解静电问题中,指定允许的最大迭代次数。默认为 400。
QCONV=
type
|
N
设定 PCM 极化电荷迭代计算的收敛阈值为 10-N,或为以下几种预定义类型之
一:VeryTight(10-12),Tight(10-9)和 Sleazy(10-6)。默认为 QConv=Tight。
SC=QConv=
x
在外部迭代流程中,指定 PCM 极化电荷的收敛。
MaxExtIt=x 在外部迭代流程中,指定允许的最大迭代次数。
各向异性和离子溶剂
ANISOTROPIC 根据 IEFPCM 方法,对各向异性溶剂进行 PCM 计算。代表介电常数的三秩对
称张量必须用六个附加的关键词指定:EPSX, EPSY, EPSZ, EUPHI, EUTHE,
和EUPSI(它们都带参数,如:EPSX=
value
)。
IONIC 根据 IEFPCM 方法,对各离子溶液进行 PCM 计算。离子强度单位是 mol/dm3,
必须作为关键词 DISM 的值指定。
指定分子空穴
程序默认用 UFF 半径构造空穴,也就是在每个溶质原子周围放置一个球,其半径用因
子1.1 换算。还可以用三种联合原子(UA)模型。
- 212 -
在PCM的输入部分,可以对空穴做大幅修改:可以改变球的参数和空穴的一般拓朴结构,
为默认产生的空穴加入更多的球,等等。用户也可以在输入部分提供整个分子的空穴。
Radii=
model
表明使用拓扑模型和/或原子半径集合。可用的模型和集合有:
UFF 使用UFF力场的半径,并用1.1换算。氢原子使用自己的球
(显式氢原子)。这是默认的。
UA0 对重原子使用联合原子拓朴模型,用于UFF力场的原子半
径。氢原子被与其成键的重原子构成的球所包围。这在
Gaussian 03是默认的。
UAHF 使用联合原子拓朴模型,用于为HF/6-31G(d)理论级别优
化的原子半径。
UAKS 使用联合原子拓朴模型,用于为PBE1PBE/6-31G(d)理论级
别优化的原子半径。
PAULING 使用Pauling(实际上是Merz-Kollman)原子半径(使用
显式氢原子)。
BONDI 使用Bondi的原子半径(使用显式氢原子)。
PDens=
x
为曲面上的积分点设置平均密度,单位:埃-2。默认为0.5。增加此值会导致
更精细的曲面离散化。
Alpha=
scale
指定静电换算因子,用于与球半径相乘。默认值是 1.1。
Surface=
type
指定代表溶剂-溶质边界的分子曲面类型。可用的选项有:
VDW 范德华曲面。使用原子半径(已换算),并跳过产生“增
加的小球”进行曲面平滑的过程。这是默认的。
SES 溶剂占据的曲面。这个球面由原子或原子团的小球,以及
为曲面平滑而自动产生的小球(“增加的小球”)生成。
这在Gaussian 03是默认的。
SAS 溶剂接触曲面。给未换算的原子或原子团的半径加入溶剂
半径。
ModifySph 更改一个或多个球的参数。修改的球在PCM的输入部分提供,格式如下:
ModifySph
atom radius
[
alpha
]
其中
atom
是原子编号或元素类型,
radius
可以用斜杠(“/”)替代,表示
使用内部默认值。当你想提供
alpha
值但不想修改默认半径时,就会用到。
ExtraSph=
N
把用户提供的
N
个球加入到空穴中。球参数用以下的格式指定:
ExtraSph=N
X Y Z radius
[
alpha
]
其中的
X,Y,Z
是标准方位的直角坐标。
NSph=
N
使用用户提供的N个球建立空穴,在按照如下格式的输入行中指定:
atom_number radius
[
alpha
]
X Y Z radius
[
alpha
]
用原子编号指定小球,模拟标准的空穴特性,而指定的直角坐标可以产生不
随结构移动的固定空穴。
其中的
X,Y,Z
是标准方位的直角坐标。
- 213 -

SphereOnH=
N
当使用联合原子拓扑模型时,将一个小球放在原子列表中第
N
个位置的氢原
子上。
SphereOnAcidicHydrogens
当使用联合原子拓扑模型时,将小球放在各个酸性氢原子上(与 N,O,S,P,
Cl 以及 F 原子成键的氢原子)。
OFac=
value
为SES 加入的球指定相嵌球之间的重迭指数[Pascual-Ahuir94]。降低这个指数会
导致加入的球数变少。默认值为 0.89。
RMin=
value
为SES 加入的球指定最小半径,单位是埃。减小这个值会导致加入的球数变
少。默认值为 0.2。
输出选项
GeomView 创建描述空穴的points.off文件。这一文件包含GeomView程序(参见
www.geomview.org)的输入,可用于显示分子空穴。
已定义溶剂的列表
SCRF=Solvent 选项可以接受以下的溶剂关键词。为了方便我们列出了 ε值,但要注意
它仅是定义溶剂的众多内部参数之一。因此,简单改变 ε值并不能正确定义新的溶剂。
Water ε=78.3553
Acetonitrile ε=35.688
Methanol ε=32.613
Ethanol ε=24.852
IsoQuinoline ε=11.00
Quinoline ε=9.16
Chloroform ε=4.7113
DiethylEther ε=4.2400
Dichloromethane ε=8.93
DiChloroEthane ε=10.125
CarbonTetraChloride ε=2.2280
Benzene ε=2.2706
Toluene ε=2.3741
ChloroBenzene ε=5.6968
NitroMethane ε=36.562
Heptane ε=1.9113
CycloHexane ε=2.0165
Aniline ε=6.8882
Acetone ε=20.493
TetraHydroFuran ε=7.4257
DiMethylSulfoxide ε=46.826
Argon ε=1.430
Krypton ε=1.519
Xenon ε=1.706
n-Octanol ε=9.8629
1,1,1-TriChloroEthane ε=7.0826
1,1,2-TriChloroEthane ε=7.1937
- 214 -
1,2,4-TriMethylBenzene ε=2.3653
1,2-DiBromoEthane ε=4.9313
1,2-EthaneDiol ε=40.245
1,4-Dioxane ε=2.2099
1-Bromo-2-MethylPropane ε=7.7792
1-BromoOctane ε=5.0244
1-BromoPentane ε=6.269
1-BromoPropane ε=8.0496
1-Butanol ε=17.332
1-ChloroHexane ε=5.9491
1-ChloroPentane ε=6.5022
1-ChloroPropane ε=8.3548
1-Decanol ε=7.5305
1-FluoroOctane ε=3.89
1-Heptanol ε=11.321
1-Hexanol ε=12.51
1-Hexene ε=2.0717
1-Hexyne ε=2.615
1-IodoButane ε=6.173
1-IodoHexaDecane ε=3.5338
1-IodoPentane ε=5.6973
1-IodoPropane ε=6.9626
1-NitroPropane ε=23.73
1-Nonanol ε=8.5991
1-Pentanol ε=15.13
1-Pentene ε=1.9905
1-Propanol ε=20.524
2,2,2-TriFluoroEthanol ε=26.726
2,2,4-TriMethylPentane ε=1.9358
2,4-DiMethylPentane ε=1.8939
2,4-DiMethylPyridine ε=9.4176
2,6-DiMethylPyridine ε=7.1735
2-BromoPropane ε=9.3610
2-Butanol ε=15.944
2-ChloroButane ε=8.3930
2-Heptanone ε=11.658
2-Hexanone ε=14.136
2-MethoxyEthanol ε=17.2
2-Methyl-1-Propanol ε=16.777
2-Methyl-2-Propanol ε=12.47
2-MethylPentane ε=1.89
2-MethylPyridine ε=9.9533
2-NitroPropane ε=25.654
2-Octanone ε=9.4678
- 215 -
2-Pentanone ε=15.200
2-Propanol ε=19.264
2-Propen-1-ol ε=19.011
3-MethylPyridine ε=11.645
3-Pentanone ε=16.78
4-Heptanone ε=12.257
4-Methyl-2-Pentanone ε=12.887
4-MethylPyridine ε=11.957
5-Nonanone ε=10.6
AceticAcid ε=6.2528
AcetoPhenone ε=17.44
a-ChloroToluene ε=6.7175
Anisole ε=4.2247
Benzaldehyde ε=18.220
BenzoNitrile ε=25.592
BenzylAlcohol ε=12.457
BromoBenzene ε=5.3954
BromoEthane ε=9.01
Bromoform ε=4.2488
Butanal ε=13.45
ButanoicAcid ε=2.9931
Butanone ε=18.246
ButanoNitrile ε=24.291
ButylAmine ε=4.6178
ButylEthanoate ε=4.9941
CarbonDiSulfide ε=2.6105
Cis-1,2-DiMethylCycloHexane ε=2.06
Cis-Decalin ε=2.2139
CycloHexanone ε=15.619
CycloPentane ε=1.9608
CycloPentanol ε=16.989
CycloPentanone ε=13.58
Decalin-mixture ε=2.196
DiBromomEthane ε=7.2273
DiButylEther ε=3.0473
DiEthylAmine ε=3.5766
DiEthylSulfide ε=5.723
DiIodoMethane ε=5.32
DiIsoPropylEther ε=3.38
DiMethylDiSulfide ε=9.6
DiPhenylEther ε=3.73
DiPropylAmine ε=2.9112
e-1,2-DiChloroEthene ε=2.14
e-2-Pentene ε=2.051
- 216 -
EthaneThiol ε=6.667
EthylBenzene ε=2.4339
EthylEthanoate ε=5.9867
EthylMethanoate ε=8.3310
EthylPhenylEther ε=4.1797
FluoroBenzene ε=5.42
Formamide ε=108.94
FormicAcid ε=51.1
HexanoicAcid ε=2.6
IodoBenzene ε=4.5470
IodoEthane ε=7.6177
IodoMethane ε=6.8650
IsoPropylBenzene ε=2.3712
m-Cresol ε=12.44
Mesitylene ε=2.2650
MethylBenzoate ε=6.7367
MethylButanoate ε=5.5607
MethylCycloHexane ε=2.024
MethylEthanoate ε=6.8615
MethylMethanoate ε=8.8377
MethylPropanoate ε=6.0777
m-Xylene ε=2.3478
n-ButylBenzene ε=2.36
n-Decane ε=1.9846
n-Dodecane ε=2.0060
n-Hexadecane ε=2.0402
n-Hexane ε=1.8819
NitroBenzene ε=34.809
NitroEthane ε=28.29
n-MethylAniline ε=5.9600
n-MethylFormamide-mixture ε=181.56
n,n-DiMethylAcetamide ε=37.781
n,n-DiMethylFormamide ε=37.219
n-Nonane ε=1.9605
n-Octane ε=1.9406
n-Pentadecane ε=2.0333
n-Pentane ε=1.8371
n-Undecane ε=1.9910
o-ChloroToluene ε=4.6331
o-Cresol ε=6.76
o-DiChloroBenzene ε=9.9949
o-NitroToluene ε=25.669
o-Xylene ε=2.5454
Pentanal ε=10.0
- 217 -
PentanoicAcid ε=2.6924
PentylAmine ε=4.2010
PentylEthanoate ε=4.7297
PerFluoroBenzene ε=2.029
p-IsoPropylToluene ε=2.2322
Propanal ε=18.5
PropanoicAcid ε=3.44
PropanoNitrile ε=29.324
PropylAmine ε=4.9912
PropylEthanoate ε=5.5205
p-Xylene ε=2.2705
Pyridine ε=12.978
sec-ButylBenzene ε=2.3446
tert-ButylBenzene ε=2.3447
TetraChloroEthene ε=2.268
TetraHydroThiophene-s,s-dioxide ε=43.962
Tetralin ε=2.771
Thiophene ε=2.7270
Thiophenol ε=4.2728
trans-Decalin ε=2.1781
TriButylPhosphate ε=8.1781
TriChloroEthene ε=3.422
TriEthylAmine ε=2.3832
Xylene-mixture ε=2.3879
z-1,2-DiChloroEthene ε=9.2
半经验方法
说明
Gaussian 09中可以使用各种半经验方法。重新实现了AM1和PM3方法[Thiel92, Thiel96,
Frisch09],以便使用标准的积分处理基本结构(而不是使用来自公共领域MOPAC的代码)。除
了效率的提高外,这一变化还提供了解析梯度和解析频率。PM6和PDDG也是以这种方式实
现的。其余的半经验方法使用链接402中的修改版MOPAC,它们将在各自的部分讨论。
AM1:请求用AM1哈密顿进行半经验计算[Dewar77, Dewar78a, Davis81, Dewar85, Dewar86, Dewar88,
Dewar88a, Dewar89, Dewar90, Dewar90a, Anders93]。
PM3:请求用PM3哈密顿进行半经验计算[Stewart89, Stewart89a]。Li的参数已经升级为[Anders93]
中指定的值。PM3MM指定的PM3模型包含了对HCON连接可选的分子力学校正。
PM6:请求用PM6哈密顿进行半经验计算[Stewart07]。还可以使用PDDG变体[Repasky02,
Tubert-Brohman04, Tubert-Brohman05, Sattelmeyer06, Tirado-Rives08]。
任何半经验关键词都不需要指定基组关键词。
支持原子的标准参数自动由程序产生,除非指定了NoGenerate选项。还可以由不同的
方式读入附加的和/或备选的参数(参见选项)。若同时是同两种方式,则读入的参数优先于
内部参数。
- 218 -

选项
Generate 对指定方法产生标准参数。这是默认的。NoGenerate 不产生标准参数;所有
参数必须读入。
Input 从输入串以 Gaussian 格式读取参数。可以指定或覆盖任何参数。输入部分必
须由一空行结束。Cards 是Input 的同义词。
MOPACExternal
从输入串以 MOPAC 外部格式和单位读取参数。可以改变大多数但不是所有
的参数。输入部分必须由一空行结束。
Both 从输入串读取参数,首先以 Gaussian 格式,更多的参数接下来以 MOPAC 格
式读入。两个输入部分必须都由一空行结束。
Checkpoint 从检查点文件读取参数。Chk 和Read 是Checkpoint 的同义词。
TCheckpoint
检查点文件若存在,则从它读取参数;否则由程序产生参数。
RWF 从读写文件读取参数。
Print 在当前任务中以Gaussian格式打印元素的参数。如果从输入文件读取参数,
这是默认的。NoPrint表示不打印参数,若使用标准参数,这是默认的。
PrintAll (以Gaussian格式)打印所有元素的参数,即使是未在分子说明部分出现的
元素。
Zero 打印所有参数,包括值为0的参数。默认为NonZero,表示只打印非零参数。
Old 使用旧的基于MOPAC的代码。二阶导数用数值方法计算。仅能用于AM1和
PM3。默认是New,表示使用以上描述的新执行。
指定半经验参数
半经验参数可以用两种不同的格式指定:Gaussian 和 MOPAC 格式,分别通过 Input 和
MOPACExternal 选项。我们从 G09 原有的半经验参数格式开始,这种格式非常普遍。
这是一个 Gaussian 格式的例子,用于 FeCH:
最开始的部分是全局参数。
Method=40 CoreType=2 PM6R6=0.0000124488 PM6R12=0.0000007621
****
H
氢的参数。
PQN=1 NValence=1 F0ss=0.5309794634 ZetaOverlap=1.2686410000 U=-0.4133181193
Beta=-0.3069665271 CoreKO=0.9416560046 KON=0,0,0,0.9416560046 EISol=-0.4133181193
EHeat=0.0830298228
GCore=0.0016794859,0.8557539899,3.3750716603 DCore=1,3,1.8737858033,2.2435870000
****
C
碳的参数。
PQN=2,2 NValence=4 F0ss=0.4900713271 F0sp=0.4236511476 F0pp=0.3644399975
F2pp=0.1978513243 G1sp=0.0790832988 ZetaOverlap=2.0475580000,1.7028410000
U=-1.8775102825,-1.4676916178 Beta=-0.5653970441,-0.2745883502
DDN=0,1,0.7535642510 DDN=1,1,0.7192361890 CoreKO=1.0202596487
KON=0,0,0,1.0202596487 KON=1,0,1,1.2918442312 KON=0,1,1,1.0202596487
KON=2,1,1,0.7626764584 EISol=-4.2335803497 EHeat=0.2723305520
DipHyp=1.5070417957 GCore=0.0032154961,0.5881175739,2.5208171825
DCore=1,4,0.2878149911,0.2165060000 DCore=2,3,1.6101301385,3.2139710000
DCore=3,3,1.7155258339,16.1800020000 DCore=4,3,2.2293611369,25.0358790000
DCore=5,3,1.5446719761,1.8748590000
DCore=6,5,1.3831173494,0.8135100000,3.1644797074,9.2800000000
****
- 219 -

Fe
铁的参数。
PQN=4,4,3 NValence=8 F0ss=0.2931506917 F0sp=0.2861621092 F0pp=0.2797829041
F0sd=0.3417747898 F0pd=0.3378189937 F0dd=0.5580709105 F2pp=0.1567537881
F2pd=0.1236661383 F2dd=0.2945882511 F4dd=0.1921227725 G1sp=0.2072870321
G1pd=0.1102204721 G2sd=0.0588483485 G3pd=0.0671224585 Rsppd=0.1364112343
Rsdpp=0.1031169651 Rsddd=0.1510228569
ZetaOverlap=1.4791500000,6.0022460000,1.0807470000
Zeta1C=1.4591520000,1.3926140000,2.1619090000
U=-2.5913804076,-2.3138503107,-3.8083983722
Beta=0.2950096563,-0.0413709206,-0.1288993981 DDN=0,1,0.0896587028
DDN=1,1,0.3534210933 DDN=0,2,1.6776352014 DDN=1,2,0.0796789968
DDN=2,2,1.3085519205 CoreKO=1.2720920000 KON=0,0,0,1.7056074374
KON=1,0,1,0.2359511557 KON=0,1,1,1.7056074374 KON=2,1,1,0.4977547907
KON=2,0,2,1.6958541322 KON=1,1,2,0.2947417183 KON=0,2,2,0.8959434914
KON=2,2,2,1.2449263774 EISol=-15.6859079709 EHeat=0.1582446241
DipHyp=0.1793070893 DCore=1,3,0.4521755746,0.0251950000
DCore=6,3,2.1121277473,0.3668350000 DCore=7,3,1.3232002016,0.1553420000
DCore=8,3,0.9135254945,0.1364220000 DCore=9,3,2.2726610620,3.6573500000
DCore=15,3,1.3586804751,0.4312910000 DCore=16,3,0.5233514967,0.0334780000
DCore=17,3,0.6507784269,0.0194730000 DCore=19,3,1.0583544172,6.0000000000
DCore=26,3,1.4397774115,1.8468900000
****
整个输入使用原子单位。各参数部分用四个星号构成的行隔开。
在全局部分指定以下的项目:
Method 对应所需半经验方法的一个整数。作为检查,此值应当对应于执行路径部分
指定的方法。值为 8对应 AM1,9对应 PM3,10 对应 PM3MM,40 对应 PM6,41
对应 PDDG。
CoreType 芯排斥项的类型:1表示 AM1,PM3,或 PDDG,2表示 PM6。
PeptideFC 肽键的力常数。仅用于 PM3MM。
RIJScale AM1 中,O-H 和 N-H 键的 Rij 换算因子。
PM6R6 PM6 芯排斥的 R6 参数。
PM6R12 PM6 芯排斥的 R12 参数。
以下的项目指定每个元素的参数:
PQN 每个壳层(s,p,d)的主量子数。确定哪些基函数用于该元素。
NValence 价电子数。
ZetaOverlap 基函数的 Slater 指数,用于计算重叠对芯哈密顿的贡献。
Zeta1C 基函数的 Slater 指数,用于计算未明确指定的单中心双电子积分。
F0*, G*, Rs*
用于单中心双电子积分的 Slater-Condon 参数。这些项若未指定,则从 Zeta1C
指数计算。当打印内部参数时,将包含所有的值,不管这些值是从 Zeta1C
计算的还是指定的。这些参数的完整列表为:F0ss,F0sp,F0pp,F0sd,F0pd,
F0dd,F2pp,F2pd,F2dd,F4dd,G1sp,G1pd,G2sd,G3pd,Rsppd,Rsdpp,
和Rsddd。
U 芯哈密顿矩阵的对角元,每个角动量有一个值。
Beta 芯哈密顿矩阵的非对角参数,每个角动量有一个值。
DDN 多极近似双中心双电子积分的点电荷距离参数。每一项的形式为
L1
,
L2
,
Value
,用于一个基函数角动量为
L1
,另一个基函数角动量为
L2
的电
荷分布。如果需要的参数未指定,则从 Zeta1C 指数计算获得。
KON 用于双中心双电子积分的 Klopman-Ohno 参数。需要的项若未定义,则通过把
- 220 -

单中心极限匹配到 Slater-Condon 参数和 Zeta1C 给定的单中心积分,并使用
指定的或默认的 DD 值获得。每一项的形式为
LT,L1,L2,Value
,用于角动量
L1
和
L2
函数乘积的 LT 角动量分量。
CoreKO 用于核吸引项的 Klopman-Ohno 参数。若未指定,则使用 0,0,0 (L=0 SS)参
数。
EHeat 孤立原子的生成热。
EISol 孤立原子的能量。若未提供,它由原子的其它参数和标准电子组态计算得到。
DipHyp 偶极距杂化参数。
DCore 芯排斥参数。每一项的形式为
El,IType,Value1,Value2
。每一项指定当前元
素和元素
El
之间的芯排斥。
IType
指定键型:1为通常的 AM1,2为AM1的
N-H 和 O-H,3为通常的 PM6,4为 PM6 的 O-H,5为PM6的CC三键,6为PM6
的 Si-O。根据指定的泛函形式,将有一到两个参数值。
MOPAC 型半经验参数的输入。如果指定 PM6=MOPACExternal 或AM1=MOPACExternal,那
么需要读入一个输入部分,用和 MOPAC 的 External 关键词相同的形式提供参数。这不如
Gaussian 原有的格式普遍,但是包含了最常用的参数。细节参见 MOPAC 文档。这种格式的
单位在 Gaussian 和 MOPAC 中完全相同,都是原子单位与其它单位的混合。
下表给出了 MOPAC 的 External 标记和 Gaussian 原有输入之间的对应关系:
MOPAC Gaussian
USS,UPP,UDD U
ZS,ZP,ZD ZetaOverlap
ZSN,ZPN,ZDN Zeta1C
BetaS,BetaP,BetaD Beta
GSS,GPP,...,FODD,... F0ss, F0pp,等†
DD2 DDN=0,1,
Value
DD3 DDN=1,1,
Value
DD4 DDN=0,2,
Value
DD5 DDN=1,2,
Value
DD6 DDN=2,2,
Value
PO1 KON=0,0,0,
Value
PO2 KON=1,0,1,
Value
PO3 KON=2,1,1,
Value
PO4 KON=2,0,2,
Value
PO5 KON=1,1,2,
Value
PO6 KON=2,2,2,
Value
PO7 KON=0,1,1,
Value
PO8 KON=0,2,2,Value
PO9 CoreKO
EHeat EHeat
EISol EISol
AlpB_NN,XFac_NN DCore=NN,3,Alpha,XFac
† 注意,MOPAC 的 GSP 和 GP2 是 F0sp 和 G1sp 的线性组合。Gaussian 使用标准的 Slater-Condon 命名与参数
定义。
- 221 -

这是一个 MOPAC 对于 Cr 的 External 数据的例子,用 MOPAC 的调试选项打印得到:
PARAMETER VALUES USED IN THE CALCULATION
NI TYPE VALUE UNIT
24 USS -34.86433900 EV ONE-CENTER ENERGY FOR S
24 UPP -26.97861500 EV ONE-CENTER ENERGY FOR P
24 UDD -54.43103600 EV ONE-CENTER ENERGY FOR D
24 ZS 3.28346000 AU ORBITAL EXPONENT FOR S
24 ZP 1.02939400 AU ORBITAL EXPONENT FOR P
24 ZD 1.62311900 AU ORBITAL EXPONENT FOR D
24 BETAS -5.12261500 EV BETA PARAMETER FOR S
24 BETAP 3.92671100 EV BETA PARAMETER FOR P
24 BETAD -4.23055000 EV BETA PARAMETER FOR D
24 GSS 8.85557242 EV ONE-CENTER INTEGRAL (SS,SS)
24 GPP 5.05309383 EV ONE-CENTER INTEGRAL (PP,PP)
24 GSP 5.58863066 EV ONE-CENTER INTEGRAL (SS,PP)
24 GP2 4.42952965 EV ONE-CENTER INTEGRAL (PP*,PP*)
24 HSP 0.64803936 EV ONE-CENTER INTEGRAL (SP,SP)
24 ZSN 1.61985300 AU INTERNAL EXPONENT FOR S - (IJ,KL)
24 ZPN 0.84826600 AU INTERNAL EXPONENT FOR P - (IJ,KL)
24 ZDN 1.40501500 AU INTERNAL EXPONENT FOR D - (IJ,KL)
24 F0DD 9.86923654 EV SLATER-CONDON PARAMETER F0DD
24 F2DD 5.20966257 EV SLATER-CONDON PARAMETER F2DD
24 F4DD 3.39760602 EV SLATER-CONDON PARAMETER F4DD
24 F0SD 6.15013600 EV SLATER-CONDON PARAMETER F0SD
24 G2SD 2.00030000 EV SLATER-CONDON PARAMETER G2SD
24 F0PD 5.63536196 EV SLATER-CONDON PARAMETER F0PD
24 F2PD 1.91648791 EV SLATER-CONDON PARAMETER F2PD
24 G1PD 1.58022558 EV SLATER-CONDON PARAMETER G1PD
24 G3PD 0.96233144 EV SLATER-CONDON PARAMETER G3PD
24 DD2 0.28669123 BOHR CHARGE SEPARATION, SP, L=1
24 DD3 2.91433601 BOHR CHARGE SEPARATION, PP, L=2
24 DD4 1.12394737 BOHR CHARGE SEPARATION, SD, L=2
24 DD5 0.81804068 BOHR CHARGE SEPARATION, PD, L=1
24 DD6 1.23219554 BOHR CHARGE SEPARATION, DD, L=2
24 PO1 1.53639890 BOHR KLOPMAN-OHNO TERM, SS, L=0
24 PO2 0.72875078 BOHR KLOPMAN-OHNO TERM, SP, L=1
24 PO3 1.96024483 BOHR KLOPMAN-OHNO TERM, PP, L=2
24 PO4 0.94950312 BOHR KLOPMAN-OHNO TERM, SD, L=2
24 PO5 1.66105265 BOHR KLOPMAN-OHNO TERM, PD, L=1
24 PO6 1.08400979 BOHR KLOPMAN-OHNO TERM, DD, L=2
24 PO7 1.53639890 BOHR KLOPMAN-OHNO TERM, PP, L=0
24 PO8 1.37859617 BOHR KLOPMAN-OHNO TERM, DD, L=0
24 PO9 1.53639890 BOHR KLOPMAN-OHNO TERM, CORE
24 CORE 6.00000000 E CORE CHARGE
24 EHEAT 95.00000000 KCAL/MOL HEAT OF FORMATION OF THE ATOM (EXP)
24 EISOL -185.72482255 EV TOTAL ENERGY OF THE ATOM (CALC)
24 ALPB_24 4.65541900 ALPB factor
24 XFAC_24 10.31860700 XFAC factor
适用性
能量,优化,和频率。
程序对以下元素存储了参数:
AM1:H,Li-F,Mg-Cl,Cr,Zn,Ge,Br,Sn,I,和 Hg
PM3:H,Li-F,Na-Cl,K,Ca,Cr,Zn-Br,Rb,Sr,Cd-I,Cs,Ba,和 Hg-Bi。
- 222 -

PM6:H-Ba 和 Lu-Bi。
有关的关键词
CNDO,INDO,MINDO,MINDO3。
例子
这些计算出现在输出文件中的能量如下:
SCF Done: E(RAM1) = -0.185015348024 A.U. after 14 cycles AM1
SCF Done: E(RPM3) = -0.185015348024 A.U. after 14 cycles PM3
SCF Done: E(RPM3MM) = -0.185015348024 A.U. after 14 cycles PM3MM
SCF Done: E(RPM6) = -0.185015348024 A.U. after 14 cycles PM6
SCF Done: E(RPDDG) = -0.185015348024 A.U. after 14 cycles PDDG
打印的能量是各模型计算的生成热。
SP
说明
这个计算类型关键词指定进行单点能计算。如果没有指定计算类型关键词,这是默认的
计算类型。
适用性
用于所有方法。
例子
参见其它方法关键词的例子中关于能量输出格式的说明。
Sparse
说明
使用稀疏矩阵存储,用于改进大计算(400 个原子以上)的执行效率[Daniels97]。这个关
键词选项允许定义截断值,低于这个值的矩阵元素认为是 0。
参见附录中程序开发有关的关键词关于此关键词的选项。
Stable
说明
这一计算类型方法指定对 Hartree-Fock 或DFT 波函的稳定性进行测试。Gaussian 可以
测试单行列式波函在放松各种限制时的稳定性[Seeger77, Bauernschmitt96](另外参见
[Schlegel91a])。这包括:
- 223 -
允许 RHF 行列式变成 UHF 行列式。
允许轨道变成复值。
降低轨道对称性。
Stable 关键词导致程序象通常一样计算波函,接下来用指定需要考虑的自由度确定得
到的行列式是否为局域最小。默认是测试所有的不稳定性,但不重新优化波函。如果指定
Stable=Opt,必要的话波函默认可以变为自旋非限制的。
注意,只有当波函无内部不稳定性时,解析频率计算才有效。在做频率计算之前先检查
计算结果,对自旋限制的波函检查单态是否存在不稳定性,对自旋非限制的波函检查(单态
或三态)是否存在不稳定性。Møller-Plesset 计算只对波函在给定对称约束下无内部不稳定
性时才有效。在进行 Møller-Plesset 计算之前先检查计算结果,只有混合的一对分子轨道属
于相同空间对称性时,内部的不稳定性才会影响结果的有效性。如果波函在变成 UHF 时不
稳定,那么根据这个波函进行的限制性 Møller-Plesset 能量的正确性值得怀疑[Carsky91]。
默认只考虑实型轨道(而不是复型轨道)的不稳定性。检查复型轨道不稳定性的代码(链
接 902)较老,而且不太可靠,只有研究复型轨道的稳定性时才能使用。
一般选项
RExt 测试外部实型不稳定性和内部不稳定性(默认)。
Int 只测试内部不稳定性(具有相同约束的较低行列式)。
RRHF 指定波函测试或重优化是实型自旋限制的。同义词是 Singlet。
RUHF 指定波函测试或重优化是实型自旋非限制的。同义词是 Triplet。
CRHF 允许对自旋限制波函测试从实型变为复型的不稳定性。
CUHF 允许对自旋非限制波函测试从实型变为复型的不稳定性。
波函再优化选项
Opt 如果发现不稳定性,适当降低限制重新优化波函,反复进行稳定性测试和波
函重优化,直到找到稳定的波函为止。Opt 的同义词是 RepOpt。默认的 NoOpt
禁止重新优化。
1Opt 一旦发现不稳定性,重新进行 SCF 计算。
算法有关的选项
Direct 强制使用直接算法。这是默认的。
MO 强制使用转换的双电子积分(即,以分子轨道基)进行稳定性计算。
AO 强制使用原子轨道积分(写在磁盘上)进行稳定性计算,避免积分转换。AO
基通常不是最佳选择,除非是在有限磁盘空间的系统上计算小分子。在指定
SCF=Conven 时,这是默认选项。
InCore 使用常驻内存算法。
ICDiag 强制把存储器内由转换积分构成的矩阵进行常驻内存的完全对角化。它意味
着使用 MO 积分。
QCOnly 在Stable=Opt 迭代后期的 SCF 计算中,禁止使用常规的 SCF 流程(即,不
用二次收敛;用链接 502)。
XQC 在Stable=Opt 迭代后期的 SCF 计算中,在二次收敛 SCF(链接 508)之前尝
试使用常规的 SCF 流程(链接 502)。
Restart 从检查点文件重新开始计算。这还意味着 SCF=Restart。
适用性
HF 和 DFT 方法。
- 224 -
有关的关键词
SCF
Symmetry
说明
这个关键词指定在计算中使用分子对称性。如果使用了对称性,分子可能先被旋转到一
个不同的坐标系中,称为
标准方位
,然后才进行计算。计算的导数再转回原来的(输入)方
位。分子轨道以标准方位打印。性质和背景电荷分布的输入数据必需使用标准方位。
NoSymmetry 关键词可避免重新定位的运算,并导致所有计算以输入方位进行。默认是
只要有可能尽量使用分子对称性,以降低 CPU,磁盘储存空间和 I/O 需求。使用 NoSymm
可以完全禁止对称性,也可以用 Symm 关键词及下列选项修改对称性。
选项
Int Int 使用积分对称性(使用“小型列表”),NoInt 不使用积分对称性。等同于
Int=[No]Symm。
Grad 在积分的导数计算中使用对称性。NoGrad 禁止使用对称性。
SCF 在 SCF 计算中使用 N3对称性,NoSCF 禁止使用对称性。在 GVB 计算中默认
使用对称性。Symm=NoSCF 等价于 Guess=LowSym,并把所有的不可约表
示合并到一起。
Loose 在第一个点的计算中,指示程序在确定对称性时使用较松的截断值。它用于
不满足理想对称性的输入结构。Tight 表示在第一步计算使用常规标准,这
是默认的。
Follow 在优化过程中,尝试保持点群和方位。
PG=
group
使用的对称性不能高于指定点群。
Axis=[X|Y|Z]
指定轴以帮助确定子群。
On 在某些关闭对称性的情况中启用对称性,例如在massage中。这可能会导致错
误的结果,因此,只有在你知道自己所做的一切时才能使用!
CenterOfCharge
在确定标准方位时使用原子电荷中心。这是默认的。COC是该选项的同义词。
CenterOfMass
在确定标准方位时使用原子质心。COM是该选项的同义词。
有关的关键词
Int,SCF
TD
说明
- 225 -
这个方法关键词使用含时 Hartree-Fock 或 DFT 方法进行激发态计算[Bauernschmitt96a,
Casida98, Stratmann98, VanCaillie99, VanCaillie00, Furche02, Scalmani06];Gaussian 09 可以使用解析梯
度[Furche02, Scalmani06]。
注意,使用的归一化标准是<X+Y|X-Y>=1。
在这些计算中也可以进行电子圆二色性(ECD)分析[Helgaker91, Bak93, Bak95, Olsen95, Hansen99,
Autschbach02]。
选项
Singlets 只求解单重激发态。仅用于闭壳层体系,这是默认选项。
Triplets 只求解三重激发态。仅用于闭壳层体系。
50-50 求解一半单重激发态和一半三重态。仅用于闭壳层体系。
Root=
N
指定感兴趣的态。默认是第一激发态(
N
=1)。
NStates=
M
求解
M
个电子态(默认是 3)。如果使用 50-50,则 NStates 给出每种要求解
的电子态类型的数量(即,默认计算 3 个单重和 3 个三重态)。
Add=
N
从检查点文件中读取收敛的态,并求解另外
N
个态。该选项还意味着 Read。
Read 从检查点文件中读取态的初始猜测。注意和 SCF 计算不同,一个基组的初始
猜测不能用在另一基组的计算中。
EqSolv 指定是否进行平衡或非平衡的PCM溶剂计算。除了激发态优化和进行激发态
密度计算(例如,使用Density=Current或All)外,默认是NonEqSolv。
IVOGuess 强制使用 IVO 猜测。这对 TD Hartree-Fock 是默认的。NoIVOGuess 强制使用
正则单激发作为初始猜测,这对 TD-DFT 是默认的。HFIVOGuess 选项强制使
用 Hartree-Fock IVO 作为初始猜测,即使对 TD-DFT 也是如此。
SOS 进行电子态极化率的求和计算。默认对所有求解的激发态。需要读入一列进
行求和的频率。零频率总是要被计算,因此不必列出。
Conver=
N
设定收敛计算对能量为 10-N,对波函为 10-(N-2)。对单点默认
N
=4,对梯度
N
=6。
适用性
使用 Hartree-Fock 或 DFT 方法的能量和梯度计算。
有关的关键词
CIS,Zindo,OutPut
例子
这里是 TD 激发态计算输出的关键部分:
Excitation energies and oscillator strengths:
Excited State 1: Singlet-A2 4.0147 eV 308.83 nm f=0.0000 <S**2>=0.000
8 -> 9 0.70701
This state for optimization and/or second-order correction.
Copying the excited state density for this state as the 1-particle RhoCI density.
Excited State 2: Singlet-B1 9.1612 eV 135.34 nm f=0.0017 <S**2>=0.000
6 -> 9 0.70617
Excited State 3: Singlet-B2 9.5662 eV 129.61 nm f=0.1563 <S**2>=0.000
8 -> 10 0.70616
- 226 -
其中总结了每个态的结果,包括自旋和空间对称性,激发能,振荡强度,S2,以及(在
每个态的第二行)CI 展开的最大因子。
ECD 结果出现在输出的稍前位置,如下:
1/2[<0|r|b>*<b|rxdel|0> + (<0|rxdel|b>*<b|r|0>)*]
Rotatory Strengths (R) in cgs (10**-40 erg-esu-cm/Gauss)
state XX YY ZZ R(length) R(au)
1 0.0000 0.0000 0.0000 0.0000 0.0000
2 0.0000 0.0000 0.0000 0.0000 0.0000
3 0.0000 0.0000 0.0000 0.0000 0.0000
1/2[<0|del|b>*<b|r|0> + (<0|r|b>*<b|del|0>)*] (Au)
state X Y Z Dip. S. Osc.(frdel)
1 0.0000 0.0000 0.0000 0.0000 0.0000
2 -0.0050 0.0000 0.0000 0.0050 0.0033
3 0.0000 -0.2099 0.0000 0.2099 0.1399
Temperature
说明
指定用于热化学分析的温度(单位:开)。其数值应当作为选项指定:
# ... Temperature=300
默认是 298.15 K。
Test
说明
这个关键词禁止自动产生存档数据(以前用于 Browse Quantum Chemistry Database
System)。反义字是默认的 Archive。注意,存档数据可以通过 pluck 工具,从 Gaussian 的
日志文件提取。
TestMO
说明
计算和存储积分时使用的截断值,以及SCF和CPHF计算中使用的收敛标准,它们适用
于大部分的分子和基组。但是如果使用近于线性依赖的基组时,可能会出现极大的MO因子,
当和有限精度的其它项组合时,会产生显著的数值误差。在CPHF和后-SCF计算中,如果任
何一个MO因子大于1000,默认将终止计算。(注意,这对应于一个AO积分对涉及四个虚轨
道的MO积分的贡献因子达到1012。)关键词NoTestMO可禁止这一检查。它必需在仔细考虑
后才能使用。TestMO是默认的。
- 227 -
TrackIO
说明
这个关键词请求每个进程的 I/O 以及 CPU 使用的统计数据。
有关的关键词
#P
Transformation
说明
这个关键词控制用于积分转换的算法,以及产生的转换积分的类型。
积分转换算法选项
Direct 指定使用直接转换进程。等价于 L804。链接 804 将会在常驻内存(in-core),
完全直接和半直接方法之间自动选择。这是默认选项。
InCore 指定在链接 804 内使用常驻内存算法。
FullDirect 指定在链接 804 内使用完全直接(MO 积分常驻内存)方法。
SemiDirect 指定在链接 804 内使用半直接算法。
积分选择选项
Full 强制对所有轨道(包括转换积分涉及的所有虚轨道)进行转换。Full 的同义
词是 ABCD。
IJAB 只产生<IJ||AB>积分。
IAJB 产生<IJ||AB>和<IA||JB>积分。
IJKL 产生<IJ||AB>,<IA||JB>和<IJ||KL>积分。
IJKA 产生<IJ||AB>,<IA||JB>,<IJ||KL>和<IJ||KA>积分
IABC 产生<IJ||AB>,<IA||JB>,<IJ||KL>,<IJ||KA>和<IA||BC>积分
UFF
参见分子力学方法的说明。
Units
说明
Units 关键词控制 Z-矩阵内使用的单位,用于距离,角度以及有关的值,如数值差分的
步长单位。默认为埃和度。
选项
- 228 -
Ang 距离以埃为单位(这是默认的)。
AU 距离为原子单位(Bohr)。
Deg 角度以度为单位(这是默认的)。
Rad 角度以弧度为单位。
限制
Charge,Cube 和Massage 关键词不受 Units 关键词设置的影响,它们的输入所用的单
位总是解释为埃和度。
Volume
说明
这个关键词指定计算分子体积,定义为密度在 0.001 电子/Bohr3轮廓之内的空间。关键
词Density 可用来指定所使用的电子密度。由于用 Monte-Carlo 方法做积分计算,计算的体
积只能精确到小数点后面两位有效数字,但足以用来估计 Onsager 溶剂反应场模型中使用的
半径。输出中将打印这个建议的半径(比计算体积对应的半径大 0.5Å)。
由于 Gaussian 09 还提供其它更精确的溶剂模型,因此这个关键词只适用于使用
SCRF=Dipole 作频率计算的准备工作。
选项
Tight 指定增加点的密度以提高积分的精度。默认计算的体积精度在 10%左右。如
果计算更高精度的分子体积,推荐使用该选项。
适用性
Hartree-Fock,所有的 DFT 方法,CIS,MP2,MP3,MP4(SDQ),CID,CISD,CCD,
CCSD,以及 QCISD。
有关关键词
SCRF=Dipole
W1 方法
W1U,W1BD,W1RO
说明
这些方法关键词指定进行Martin的W1方法[Martin99, Parthiban01]。W1U关键词指定修改的W1
方法,对开壳层体系使用UCCSD而不是ROCCSD [Barnes09];ROCCSD方法由Handy,Pople,及合
作者提出[Knowles91];W1BD使用有关的方法,但用BD代替耦合簇[Barnes09]。这种方法比CBS-QB3
和G3更昂贵,但也更准确(所有W1方法都是如此);当需要非常准确的能量时,我们推荐使
用W1BD。W1RO是[Martin99]中描述的W1方法,其中使用了略微改善的标量相对论校正,描述见
- 229 -

[Barnes09]。
选项
ReadAmplitudes 选项对所有 W1 方法都是默认的。SaveAmplitudes 对W1BD 也是默认的。
ReadIsotopes
该选项允许指定不同于默认值(分别为 298.15 K,1 大气压,无换算,和最
丰同位素)的温度,压强,频率换算因子,和/或同位素。该选项可用于重新
进行分析,但使用不同于检查点文件数据的参数。
但是需要注意,所有这些参数也可以在执行路径部分(关键词 Temperature,
Pressure,和 Scale)和分子说明部分(Iso=
参数
)指定,如这个例子:
#T
Method
/6-31G(d)
JobType
Temperature=300.0 ...
...
0 1
C(Iso=13)
...
ReadIsotopes 输入具有下面的格式:
temp
pressure
[
scale
]
数值必须为实数。
原子 1 的同位素质量
原子 2 的同位素质量
...
原子 n 的同位素质量
其中的
temp
,
pressure
,和
scale
分别为所需的温度,压强,和当用于热化学
分析时可选的频率数据换算因子(默认不换算)。其余的行是分子中各原子
的同位素质量,按照分子说明部分出现的相同顺序排列。如果原子质量指定
为整数,程序将自动使用相应的真实同位素质量(例如,对18O指定为18,那
么Gaussian使用的值为17.99916)。
ReadAmplitudes
从检查点文件(如果存在)读取收敛的幅度。注意,新的计算可以使用不同
于原计算的基组,方法(如果适用),等。
SaveAmplitudes
把收敛的幅度保存到检查点文件,用于随后的计算(例如,使用更大的基组)。
使用该选项会产生非常大的检查点文件,但可以显著加速后面的计算。
Restart 重新开始一个未完成的 W1 计算。
例子
计算的总结输出。在任务子步骤的所有输出之后,Gaussian对这些方法打印结果的表格。
这里是W1BD计算输出的关键部分:
自旋校正前的结果。
Temperature= 298.150000 Pressure= 1.000000
E(ZPE)= 0.016919 E(Thermal)= 0.019783
W1BD (0 K)= -39.139927 W1BD Energy= -39.137063
W1BD Enthalpy= -39.136119 W1BD Free Energy= -39.158277
- 230 -
W1U spin correction:
G.P.F. Wood, L. Radom, G.A. Petersson, E.C. Barnes, M.J. Frisch
and J.A. Montgomery, Jr., JCP 125, 94106 (2006).
DE(Spin)= -0.000051
W1Bsc Electronic Energy -39.156897
预测的能量。
自旋校正的结果。
Temperature= 298.150000 Pressure= 1.000000
E(ZPE)= 0.016919 E(Thermal)= 0.019783
W1Bsc(0 K)= -39.139978 W1Bsc Energy= -39.137114
W1Bsc Enthalpy= -39.136170 W1Bsc Free Energy= -39.158328
预测的能量在常规的和自旋校正的热化学分析数据之间给出。
能量标记具有如下含义(以自旋校正 W1BD 为例):
W1Bsc (0 K) 零点校正的电子能量:E0 = Eelec + ZPE
W1Bsc Energy 热校正能量:E = E0 + Etrans + Erot + Evib
W1Bsc Enthalpy 用自旋校正 W1BD 能量计算的焓:H = E + RT
W1Bsc Free Energy 用自旋校正 W1BD 能量计算的 Gibbs 自由能:G = H - TS
ZINDO
说明
这个方法关键词使用 ZINDO-S 方法[Ridley73, Ridley76, Bacon79, Zerner80, Zerner82, Anderson86,
Hanson87, Thompson91, Zerner91]进行激发态能量的计算。注意 ZINDO 计算不能指定基组关键词。
选项
Singlets 只求解单重激发态。仅用于闭壳层体系。对闭壳层体系这是默认选项。
Triplets 只求解三重激发态。仅用于闭壳层体系。
50-50 求解一半单重激发态和一半三重态。仅用于闭壳层体系。
Root=
N
指定感兴趣的态。默认是第一激发态(
N
=1)。
NStates=
M
求解
M
个电子态(默认是 3)。如果使用 50-50,则 NStates 给出每种要求解
的电子态类型的数量(即,默认计算 3 个单重和 3 个三重态)。
Add=
N
从检查点文件中读取收敛的态,并求解另外
N
个态。
Window=
(m[,n])
这两个值定义使用的起始和终止轨道。默认使用全部轨道。根据使用位置的
不同,零值表示第一个或最后一个轨道。如果第一个轨道的值为负(-
m
),
则保留最高的
m
个轨道;如果最后一个轨道的值为负(-
n
),则冻结最高的
n
个轨道。如果
m
为正并忽略
n
,那么
n
默认是 0。如果
m
为负并忽略
n
,则
保留最高的|
m
|个占据轨道和最低的|
m
|个虚轨道。
适用性
仅能量计算。ZINDO 计算忽略 Density 关键词。
- 231 -
有关的关键词
CIS,TD
链接 0命令总结
这一部分列出了所有的链接0命令,它们都是可选的,并且位于执行路径部分之前。详
细的使用说明见后。
%Mem=
N
指定使用
N
个8位字(默认单位)的动态内存;
N
值后也可以接KB,MB,GB,KW,
MB或GW(中间没有空格)分别指定单位为千,兆,吉位或字。默认内存大小
是256 MB。
%Chk=
file
定位和命名检查点文件。
%RWF=
file
定位和命名一个单独的、统一的读写文件(旧式语法)。
%RWF=
位置1,尺寸1,位置2,尺寸2,...
用于在两块或更多的磁盘(或文件系统)中分割读写文件的另一种语法。每
个位置之后接该位置存储的文件片段的最大尺寸。每个尺寸的单位是字(默
认);数值后也可以接KB,MB,GB,KW,MB或GW(中间没有空格)分别指定
单位为千,兆,吉位或字。如果任何一个尺寸参数为-1,表示可以使用任何
可用的空间,如果值为0,表示已存在的片断应保持其现有大小。位置可以是
目录位置,也可以是完整的路径名。注意在UNIX系统中,目录说明必须包含
结尾的正斜杠(/)。
%Int=
spec
定位和命名双电子积分文件。
spec
可采用用于读写文件的两种格式之一。
%D2E=
spec
定位和命名双电子积分导数文件。
spec
可采用用于读写文件的两种格式之一。
%KJob L
N
[M]
指示程序在第
M
次执行链接
N
之后停止。例如,%KJob L502 2将在执行链接502
两次后停止。
M
值可以省略,默认值为1。
%Save 指定链接0在运行结束时保存临时文件。在运行成功结束时,默认删除所有未
指定的临时文件,只保留所有命名的临时文件。
%NoSave 指定链接0在运行结束时删除临时文件,包括在此命令之前命名的任何文件。
换句话说,如果在%NoSave之前命名了一个文件,它将不被保存。但是,如果
在%NoSave指令之后用%指令命名了文件,文件将会保存。
如果指定了%Save和%NoSave,那么在输入文件中最后出现的优先。
%Subst L
N
dir
通知链接0从另一目录dir取得链接的可执行文件(.exe文件)。例如:%SUBST
L913 /user/chem表示使用/usr/chem/l913.exe而非默认的可执行文件(在
$g09root中)。目录的说明应当是相应计算机的通用格式。只能定义目录;文
件名必须是l
nnnn
.exe的标准形式,其中
nnnn
是链接编号。
并行任务指令
%NProcShared=
N
在SMP多处理器计算机上,指定在共享内存并行执行中使用直到
N
个处理器。
这个功能只能在某些计算机系统中使用。在并行机中,通常在Default.Route
文件中设定正式计算使用的处理器数量,而%NProcShared 链接0命令用来覆
盖着一本地默认值(例如,在单处理器上运行调试任务,即使是默认使用4
- 232 -

个处理器)。如果没有使用%NProcShared,并且在Default.Roulte文件中也
没有提供默认值的话,则只使用一个处理器。
早期版本使用的%NProc已作废。
%LindaWorkers=
node1[:n1] [,node2[:n2]]
…
它对每个要使用的节点列出了TCP节点名。默认在每个节点上开始一个Linda
工作程序,但可选的值允许对此作出改变。任务开始的节点(主节点)上总
是开始一个工作程序,不论它是否出现在节点的列表中。%LindaWorkers可
以与%NProcShared组合。在这种情况下,在每个节点上将运行一个或多个并
行工作进程(个数仍旧由%LindaWorkers中的值确定)。在工作节点列表中,
每个系统使用的SMP处理器/核心的个数由%NProcShared的值指定。
不要使用作废的%NProcLinda指令。基于%LindaWorkers输入,G09将会计算
Linda工作程序的总数。
例子
这些例子指定检查点文件名称,以及读写文件的替代名称和目录位置,将导致在
Gaussian任务结束后只保存检查点文件:
%RWF=/chem/scratch2/water
到这里为止,文件都被删除
%NoSave
%Chk=water
到这里为止,文件都被保存
下面的指令导致网络并行任务在 5个指定的节点上运行。注意,hamlet 和ophelia 每个
都运行两个工作进程。
%LindaWorkers=hamlet:2,ophelia:2,laertes,horatio,lear
下面的指令指定并行任务将在主机 norway,italy,和 spain 上执行。节点 norway 和italy
每个都运行一个四路 SMP 并行工作程序,而 spain 将运行两个这样的工作程序:
%NProcShared=4
指定四路 SMP 并行。
%LindaWorkers=norway,italy,spain:2
只有当 norway 和italy 为4处理器/核心的计算机,spain 是8处理器/核心的计算机时,
这些指令才有意义。
- 233 -
实用程序
这一部分讨论Gaussian 09中包含的各种实用程序。这些实用程序按照字母顺序讨论。
大多数实用程序既可以在UNIX版也可以在Windows版的Gaussian中使用。但是,必须考
虑到和程序一起发布的说明信息只适用于特定的操作系统。
下面的表列出了所有的实用程序及其功能(加*号的项包含在Gaussian 09W的Utilities菜
单中)。
工具 功能
c8609 将旧版 Gaussian 程序产生的检查点文件转换成 Gaussian 09 格式。
chkchk* 显示检查点文件的执行路径和标题部分。
cubegen* 独立的立方体生成工具
cubman* 处理 Gaussian 产生的电子密度和静电势的立方体(允许它们进行加、减,等
等)。
formchk* 将二进制的检查点文件转换成 ASCII 码形式,以便可视化程序使用,以及在
各种不同的计算机系统之间移动。
freqchk* 由检查点文件打印频率和热化学数据,还可以指定用于热化学分析的同位素、
温度、压强以及换算因子。
freqmem 确定频率计算需要的内存。
gauopt 进行变量优化,而不是分子坐标的优化。
ghelp Gaussian 的在线帮助。
mm 独立的分子力学程序。
newzmat* 不同分子结构说明格式之间相互转换。
testrt* 对输入文件的执行路径部分进行语法检查,并产生非标准的执行路径。
unfchk* 把格式化的检查点文件转换回到二进制格式(例如,在各种不同的计算机系
统之间移动以后)。
GAUSS_MEMDEF环境变量
GAUSS_MEMDEF环境变量可以给实用程序增加使用的内存,因为实用程序不提供这类选项
的。它的值应设定为以字为单位的所需内存总量。
c8609
c8609程序从Gaussian 86经由Gaussian 03,到Gaussian 09之间转换检查点文件格式。它
使用检查点文件名作为参数,并在同一目录下进行转换,使重新格式化的文件与原文件有相
同的文件名。例如,下面的命令把Gaussian临时目录下的检察点文件taxol.chk转化到
Gaussian 09格式:
$ c8609 $GAUSS_SCRDIR/taxol.chk
- 234 -
chkchk
chkchk实用程序显示检查点文件的执行路径和标题部分,并标出文件中出现的其它信
息。对于那些忘记其内容,且文件名是非描述性的随机检查点文件,该程序可以用来确定其
内容。它使用检查点文件名作为参数。这是一个使用的例子:
$ chkchk important
Checkpoint file important.chk:
Title: Optimization and frequencies for pentaprismane
Route: #T BECKE3LYP/6-31+G(D,P) OPT FREQ POP=FULL
Atomic coordinates present.
SCF restart data present.
This file appears to be from the middle of a restartable job.
Internal force constants may be present.
-p选项可用于打印检查点文件的附加信息,包括分子坐标,Gen格式的基组,为
Guess=Cards打印正确格式的分子轨道。
cubegen
Gaussian提供一个独立的实用程序,用于从格式化检查点文件的数据产生立方体(等价
于以前的Cube关键词)。这个工具名为cubegen,语法如下:
cubegen
memory kind fchkfile cubefile npts format
所有的参数都是可选的;如果需要的话,cubegen将提示
fchkfile
。默认的命令为:
cubegen 0 density=scf test.cube 0 h
不分大小写的参数含义如下:
memory
以前该参数用于指定分配的内存总量。现在被忽略,使用GAUSS_MEMDEF环境
变量。
kind
指定产生立方体类型的关键词:
MO=
n
分子轨道
n
。关键词Homo,Lumo,All,OccA(全部是占据
的alpha),OccB(全部是占据的beta),Valence(所有
价轨道)和Virtuals(所有虚轨道)也可以用来代替特定
的轨道序号。没有默认的
n
,如果忽略将会导致错误。类
似地,AMO和BMO可(分别)用于选择alpha或beta轨道。
对于开壳层体系,Homo同时选择alpha和beta轨道。
Density=
type
指定类型的总密度。关键词
type
是Density关键词的一个
密度选项:SCF,MP2,CI,QCI等 (注意,不支持Current)。
Fdensity=的各种形式要求使用全部电子密度而不是冻芯
密度。默认的关键词
type
是SCF。
Spin=
type
指定类型的自旋密度(α和β密度之差)。
Alpha=
type
指定类型的alpha自旋密度。Fdensity=要求使用全部电子
- 235 -
密度而不是冻芯密度。
Beta=
type
指定类型的beta自旋密度。Fdensity=要求使用全部电子
密度而不是冻芯密度。
Potential=
type
使用指定类型密度的静电势。
Gradient 计算密度和梯度。
Laplacian 计算密度的拉普拉斯量(∇2ρ)。
NormGradient 计算每个点密度梯度的模。
CurrentDensity=I
磁致(GIAO)流密度的大小,其中I是应用的磁场方向(X,
Y或Z)。
ShieldingDensity=IJN
磁屏蔽密度。其中I是应用的磁场方向,J是诱导场方向(X,
Y或Z),N是计算屏蔽密度(GIAO)的核子数量。
fchkfile
格式化的检查点文件名。如果没有出现,cubegen将会提示。
cubefile
输出的立方体文件名;如果没有明确指定,默认是test.cube(也就是说,指
定检查点文件名不会改变默认的立方体文件名)。
npts
立方体每个边的点数。选择 0使用默认值,由程序自动产生 803个点,均匀
分布在长方体网格(不一定是立方网格)内。类似地,正
npts
值指定每个边
上的点数:例如,100 指定 1,000,000(1003)个网格点。
值-2,-3 和-4 分别对应于关键词 Coarse,Medium 和Fine,对应值为 3 个点
/ Bohr,6 个点/Bohr,和 12 个点/Bohr。npts≤-5 的负值指定网格内的点间
隔为
npts
*10-3 Angstrom。
值-1 表示从输入字符串中读取立方体的说明,格式为:
IFlag, X0, Y0, Z0
输出单元编号和初始点。
N1, X1, Y1, Z1
X-方向的点数和点间隔。
N2, X2, Y2, Z2
Y-方向的点数和点间隔。
N3, X3, Y3, Z3
Z-方向的点数和点间隔。
IFlag
是输出单元编号。如果
IFlag
小于零,那么将产生格式化文件;否则
写非格式化文件。
如果 N1<0,假定输入的立方体坐标单位是 Bohr,反之则为 Angstrom。无论
何种情况,都使用|N1|作为 X 轴向的点数;N2和N
3分别指定 Y 轴向和 Z 轴向的
点数。注意这三个轴必须严格按照定义使用;它们不必互相垂直,所以网格
不一定是长方体。
值-5 表示从标准的输入读取任意的一列点。如果是用手输入的,结束输入时
应当用文件结束命令(在 Unix 下,也就是 Ctrl-D)。还可以把标准输入改向
到一个包含点列表的文件中(在文件结束不要加空行或 Ctrl-D)。
format
格式化的输出文件格式:h表示包含头信息(默认);n表示不包含头。当产
生非格式化的立方体文件时,将忽略这个参数。
输出文件格式
立方体文件的全部值都是原子单位,与输入单位无关。
对于密度和势的格点,未格式化文件的每个记录有一行(即,N1*N2个记录,每个长度
N3)。对于格式化文件,每行的书写格式为(6E13.5)。这种情况下,N3不再是 6的倍数,于
是在某些行内可能有空格。
- 236 -

密度梯度的模和拉普拉斯量都是标量(即,每点一个值),并以同样的方式书写。密度
+梯度格点是类似的,但每行有两组值(长度为 N3和3*N3)。密 度 +梯度+拉普拉斯量格点每
行有三组值(长度为 N3,3*N3,和 N3)。
例如,对于密度立方体,输出文件形式为:
NAtoms, X-Origin, Y-Origin, Z-Origin
N1, X1, Y1, Z1
最外层指数方向的步数
N2, X2, Y2, Z2
N3, X3, Y3, Z3
最底层指数方向的步数
IA1, Chg1, X1, Y1, Z1
第一个原子的原子序号,电荷,和原子数
…
IAn, Chgn, Xn, Yn, Zn
最后一个原子的原子序号,电荷,和原子数
(N1*N2)
个记录,每个长度 N3
网格中每个点的密度值
注意,每个记录单独书写。
对于分子轨道输出,NAtoms 小于 0,在最后的原子数据后接上附加的记录(若是格式
化文件,格式为 10I5):
NMO, (MO(I),I=1,NMO)
MO 数及其编号
如果求解 NMO 个轨道,则每个记录长度为 NMO*N3,每个点所有轨道的值放在一起。
用FORTRAN 程序读取立方体文件
如果想把密度,拉普拉斯量,或势读入到维度为 X(N3,N2,N1)的数组中,代码可以使用
如下的 Fortran 循环:
Do 10 I1 = 1, N1
Do 10 I2 = 1, N2
Read(n,'(6E13.5)') (X(I3,I2,I1),I3=1,N3)
10 Continue
其中的 n为立方体文件的单元号。
如果原点为(X0,Y0,Z0),步长为(X1,Y1,Z1),那么点(I1,I2,I3)的坐标为:
X 坐标:X0+(I1-1)*X1+(I2-1)*X2+(I3-1)*X3
Y 坐标:Y0+(I1-1)*Y1+(I2-1)*Y2+(I3-1)*Y3
Z 坐标:Z0+(I1-1)*Z1+(I2-1)*Z2+(I3-1)*Z3
如果还需要电荷密度的梯度或梯度和拉普拉斯量,除了每对 I1,I2值有两个或三个记录
外,输出是类似的。因此,如果要把密度和梯度读入到数组 D(N3,N2,N1),G(3,N3,N2,N1),
RL(N3,N2,N1),正确的 Fortran 循环是:
Do 10 I1 = 1, N1
Do 10 I2 = 1, N2
Read(n,'(6F13.5)') (D(I3,I2,I1),I3=1,N3)
Read(n,'(6F13.5)') ((G(IXYZ,I3,I2,I1),IXYZ=1,3), I3=1,N3)
10 Continue
其中的 n仍为立方体文件的单元号。
- 237 -

cubman
cubman 程序对 Gaussian 产生的电子密度值和静电势值的立方体进行操作。该程序会对
进行的操作和接下来需要的文件名产生提示。可进行的操作及其相应的子命令有:
add 将两立方体内相加,产生一个新的立方体。
copy 复制一个立方体,可能会在格式化和非格式化之间相互转换。
diff 计算两个立方体之差的特性,但不产生新的立方体。
prop 计算一个立方体的特性。
subtract 计算两个立方体的差,并产生一个新的立方体。
scale 将一立方体乘以一个常数因子,并产生一个新的立方体。
square 将一立方体乘以自身,并产生一个新的立方体。
所有操作的子命令都可以缩写为能够相互区分开的最短形式。
这里有一些运行 cubman 的例子,加了注释(用户输入内容是黑体字,由于空间的考虑,
输出略微做了压缩):
第一个例子
$ cubman
Action [Add, Copy, Difference, Properties, SUbtract, SCale]? p
Input file? b.cube
Is it formatted [no,yes,old]? y
Opened special file b.cube.
Input file titles:
First excited state of propellane
任务的标题行
CI Total Density
立方体文件的内容
SumAP= 13.39263 SumAN= .00000 SumA= 13.39263
立方体内容的统计
CAMax= 3.35320 XYZ= .18898 -1.32280 .000004
CAMin= .00000 XYZ= -9999.00000 -9999.00000 -9999.00000
DipAE= -.8245357658 .7624198057 .1127178115
DipAN= -.0000060000 -.0000060000 .0000000000
DipA= -.8245417658 .7624138057 .1127178115
第二个例子
$ cubman
Action [Add, Copy, Difference, Properties, SUbtract, SCale]? su
First input? b.cube
Is it formatted [no,yes,old]? y
Opened special file b.cube.
Second input? a.cube
Is it formatted [no,yes,old]? y
Opened special file a.cube.
Output file? c.cube
保存新立方体的文件
Should it be formatted [no,yes,old]? y
Opened special file c.cube.
Input file titles:
First excited state of propellane
第一个文件的标题
CI Total Density
第一个立方体的内容
Input file titles:
Propellane HF/6-31G*
第二个文件的标题
- 238 -

SCF Total Density
第二个立方体的内容
Output file titles:
合成后的新文件标题
First excited state of propellane || Propellane HF/6-31G*
CI Total Density - SCF Total Density
需要计算的差
SumAP= 13.39263 SumAN= .00000 SumA= 13.39263
第一个立方体文件统计
CAMax= 3.35320 XYZ= .18898 -1.32280 .000004
CAMin= .00000 XYZ= -9999.00000 -9999.00000 -9999.00000
SumBP= 13.38168 SumBN= .00000 SumB= 13.38168
第二个立方体文件统计
CBMax= 3.39683 CBMin= .00000
SumOP= .63453 SumON=-.62358 SumO= .01094
输出立方体文件的统计
COMax= .49089 COMin=-.39885
DipAE= -.8245357658 .7624198057 .1127178115
DipAN= -.0000060000 -.0000060000 .0000000000
DipA= -.8245417658 .7624138057 .1127178115
DipBE= -.8306292172 .5490287046 .1243830393
DipBN= -.0000060000 -.0000060000 .0000000000
DipB= -.8306352172 .5490227046 .1243830393
DipOE= .0060934514 .2133911011 -.0116652278
DipON= -.0000060000 -.0000060000 .0000000000
DipO= .0060874514 .2133851011 -.0116652278
在输出文件中,输入的立方体标记为A和B,输出的立方文件定义为O。其它字母代码为:
N依赖于上下文,表示“负值”或“原子核”,P表示“正值”,E表示“电子”,C表示“电荷”,
Dip表示“偶极”,Sum表示“求和”,Max表示“最大值”,Min表示“最小值”。因此SumAN是
第一个输入立方体的负值的总和, 而DipON是输出立方体中原子核对偶极矩的贡献。类似地,
CBMax是第二个输入的立方体的最大电荷,SumO是输出的立方体内包含正、负值在内的数值
总和。
formchk
formchk 把 Gaussian 检查点文件中的数据转换到格式化的形式,用作各种可视化软件
的输入。
formchk 的语法是:
formchk [
选项
]
chkpt-file formatted-file
其中的
chkpt-file
是需要格式化的二进制的检查点文件名,
formatted-file
是输出结果的
文件名。例如,下面的命令将从检查点文件 propell.chk 产生格式化的检查点文件
propell.fchk:
$ formchk propel.chk propel.fchk
在 UNIX 系统和其它支持可变长度扩展名的计算机中,格式化检查点文件通常的扩展名
是.fchk,而其它扩展名限制在三个字符的操作系统如 Windows 中,扩展名是.fck。
- 239 -
注意,格式化检查点文件可用作不同计算机平台间的数据交换格式。在原计算机使用
formchk,在目标计算机使用 unfchk 创建二进制检查点文件。当在不同类型计算机之间移动
数据时,总要使用-3 选项(见下)。
选项
formchk 默认产生的格式化检查点文件是对所有以前版本 Gaussian 向下兼容的。
-3 产生版本 3 的格式化检查点文件,包括 Gaussian 09 支持的全部功能(例如,
用户定义的 MM 类型,冗余坐标的定义,数值,力,和 Hessian)。
-2 产生版本 2 的格式化检查点文件。这是 Gaussian 03 使用的版本。
-c 导致出现在格式化检查点文件的分子力学原子类型为字符串形式,而非整数
形式。
freqchk
freqchk 实用程序用于从检查点文件获取频率和热化学数据,以及可选的其它温度,气
压,换算因子,和/或同位素替代等定义。
freqchk 命令的完整语法为:
$ freqchk
检查点文件
[
选项
] [
对提示的回答
]
若检查点文件名未包含扩展名,假定是.fchk;格式化和非格式化检查点文件都可接受。
选项
以下是支持的选项:
-o
文件名
把输出发送到指定文件。输出默认显示在屏幕上。
-nfd 跳过完全的(3Natoms)2矩阵对角化(同 Freq=NoDiagFull)。
-sel 选择包含的简正模式。实用工具像往常一样提示所需的信息,接着停下来等
待用户输入所需的模式(没有提示)。你可以指定模式编号,或者像
Freq=ReadNormalModes 那样指定原子列表。见下面的例子。
-read 从检查点文件读取简正模式,不做计算。
-save 保存简正模式;仅用于二进制检查点文件。当与-sel 组合时,仅有选择的模
式保存在检查点文件中,用于随后的-read 操作。
-np=
N
用
N
个处理器计算频率分析。
示例
freqchk 会对其它需要的信息给出提示。以下带有注释的记录说明了它在这种模式下的
使用(用户输入设定为黑体):
$ freqchk
Checkpoint file? solvent.chk
Write Hyperchem files? n
Temperature (K)? [0=>298.15] 0
必须输入 0;回车无效
Pressure (Atm)? [0=>1 atm] 0
Scale factor for frequencies during thermochemistry? [0=>1/1.12] 0
Do you want the principal isotope masses? [Y]:
回车接受默认值
- 240 -

打印每个原子的同位素
Full mass-weighted force constant matrix:
Low frequencies --- -948.3077 .0008 .0020 .0026
...
下面是 Gaussian 的简正频率输出
...
1 2
?A ?A
Frequencies -- 1885.3939 3853.5773
Red. masses -- 1.0920 1.0366
Frc consts -- 2.2871 9.0697
IR Inten -- 17.3416 21.5997
Raman Activ -- 7.8442 67.0384
Depolar -- .7428 .2248
Atom AN X Y Z X Y Z
简正模式
1 8 .06 .00 .04 .04 .00 .02
2 1 -.70 .00 .03 .01 .00 -.71
...
-------------------
- Thermochemistry -
-------------------
Temperature 298.150 Kelvin. Pressure 1.00000 Atm.
Thermochemistry will use frequencies scaled by .8929.
...
Zero-point vibrational energy 53494.5 (Joules/Mol)
12.78550 (Kcal/Mol)
VIBRATIONAL TEMPERATURES: 2422.01 4950.36 5495.38 (KELVIN)
零点能和热校正:
Zero-point correction= .020375 (Hartree/Particle)
Thermal corr to Energy= .023210
Thermal corr to Enthalpy= .024154
Thermal corr to Gibbs Free Energy= .045589
E=热能;CV=定容摩尔热容;S=熵
E CV S
KCAL/MOL CAL/MOL-KELVIN CAL/MOL-KELVIN
TOTAL 14.564 6.001 45.114
ELECTRONIC .000 .000 .000
TRANSLATIONAL .889 2.981 34.609
ROTATIONAL .889 2.981 10.500
VIBRATIONAL 12.787 .039 .005
配分函数
Q LOG10(Q) LN(Q)
TOTAL BOT .561443D-01 -1.250695 -2.880127
TOTAL V=0 .132155D+09 8.121085 18.699192
VIB (BOT) .424961D-09 -9.371650 -21.579023
VIB (V=0) .100030D+01 .000129 .000297
ELECTRONIC .100000D+01 .000000 .000000
TRANSLATIONAL .300436D+07 6.477751 14.915574
ROTATIONAL .439749D+02 1.643204 3.783618
$ freqchk solvent.chk
检查点文件名可位于命令行
Write Hyperchem files? n
Temperature (K)? [0=>298.15] 300
改变温度
Pressure (Atm)? [0=>1 atm] 1.5
改变气压
Scale factor for freqs during thermochem? [0=>1/1.12] 1
不换算
Do you want to use the principal isotope masses? [Y]: n
For each atom, give the integer mass number.
- 241 -

In each case, the default is the principal isotope.
Atom number 1, atomic number 8: [16]
回车,接受默认值
Atom number 2, atomic number 1: [1] 2
用整数指定同位素质量
后面是频率输出...
接下来是频率输出,反映了上面指定的值。注意如果定义了换算因子,只会反映在热化
学数据中,频率本身不做换算。
在freqchk记录中有时也会出现其它提示:
Project out gradient direction? [N]
当力显著不为零时,就会出现这一提示。力非零的可能原因有,频率是在IRC上的点做
的计算,因此可以投影掉梯度的方向。如果在其它情况下力非零,那么结构不是稳定点,为
了做有意义的频率分析必须对结构作优化。但是如果你想检查频率,例如看优化的初始点是
否有正确的曲率,那么梯度方向不能被投影掉。
在交互模式下,你还可以在一个命令行中指定所有的freqchk输入。例如下面的例子,
执行与上面最后一个交互记录相同的操作:
$ freqchk solvent.chk N 300 1.5 1 N N
如果倒数第二个参数是N的话,程序将会提示同位素。最后的参数是回答是否出现梯度方向
提示;若忽略此参数并且遇到了该提示,那么实用程序就会提示你。
选择模式。
下面的命令和输入选择了模式 1-5,以及所有涉及氮原子的其它模式,并把输出发送到
ala_freq.out 文件:
$ freqchk ala.chk -sel -o ala_freq.out N 0 0 0 N N
1-5 atoms=N
输入以空行结束
$ more ala_freq.out
准备查看频率数据
…
freqmem
freqmem实用程序获取频率计算的参数,并确定用单通道完成所有计算步骤所需的内存
总量,以达到最大的计算效率。所有的参数必须在命令行提供,使用下面的语法:
freqmem
natoms nbasis
r|u c|d
functions
其中的参量为:
natoms 分子中的原子数。
nbasis 体系所用基组的基函数总数。
r|u 一个字母代码,分别表示 RHF(闭壳层)或 UHF(开壳层)计算。
c|d 一个字母代码,表示计算使用传统算法(c)还是直接算法(d)。
functions 一个字符串,表示所选基组的基函数类型:sp,spd,spdf 等。
下面是一个使用的例子,估算紫杉酚(113 个原子)RHF/STO-3G 频率计算的内存资源需
求量:
$ freqmem 113 361 r d sp
- 242 -

RHF direct frequencies with sp functions:
One pass requires 44.80 megawords.
输出显示:单通道完成频率计算,程序需要大约 360 MB 内存。
如果 freqmem 定义的内存需要量不足,用多通道仍能完成频率计算。在链接 0 命令部分
使用%Mem 定义可用的内存量。通常最佳的选择是设定这个参数为 freqmem 推荐内存量的二
分之一或三分之一。
Gaussain 计算中使用的基函数个数打印在输出文件的较靠前的位置。还可以通过设置
计算任务的输入文件,包含链接 0 命令%KJob=301 来计算。它告诉程序,一旦执行到模块 301
程序就(几乎是立即)停止。可以从 log 文件得到分子特定基组使用的基函数数量,命令为:
$ grep "basis func"
name.
log
361 basis functions 1083 primitive gaussians
gauopt
gauopt 实用程序用来反复执行 Gaussian 进行优化。通过这种方式,它可以优化输入串
中的任何参数,包括一般的和修改的基函数。它通过反复创建运行 Gaussian 的子进程来操
作。典型情况下,gauopt 用来优化基组的参数,因为在 Gaussian 中没有标准的优化方法。
使用时,用 gauopt 命令调用程序,并由键盘输入所需数据。
gauopt 的输入由模板文件构成,其中某些内容用优化的变量值替换。模板文件用于构
造 Gaussian 实际使用的输入文件,它包含每个能量计算中的变量当前值。接下来在计算的
每一步,通过运行 Gaussian 单点能计算,自动计算能量。模板第一行的格式为:
NVar, MaxIt, SaveFlag, Conv, ConvV
使用的格式为 2I3, L2, D9.2。含义定义如下:
NVar
变量数目。
MaxIt
优化计算进行循环的最大次数。
T|F 逻辑变量标记,决定是否保存 Gaussian 产生的中间输出文件。这些中间文件
的名称为 fork.com,fork.log,fork.rwf,等。默认是删除,但是可以保存,
用来帮助对模板输入进行调试。
Conv
指定变量的 RMS 变化作为收敛条件。如果参数设置为 0.0,则使用相当严格
的默认值。
ConvV
能量的收敛条件。如果参数设置为 0.0,则使用默认值 1 milliHartree。
模板文件的下一行有一对或多对值,使用下面的语法:
Value
C|V
重复 n 次(不要插入空格)
其中的
Value
是变量的值,第二个值是一个字符标记,可以设置为 C约束变量(即在当前的
运行中不进行优化), 或设置为 V对变量进行优化。这一行的每一对变量使用格式 F14.9,
A1。
模板文件余下的部分包含 Gaussian 输入文件模板。在输入文件中,每个需要插入先前
定义变量的部分应当包含<
n x.y>
,表示在该点以格式 F
x.y
插入第 n 个变量,或<
-n x.y
>,
表示在这里插入-1 倍的变量
n
。
- 243 -

一个例子将有助于弄清这些概念。下面的 gauopt 模板文件对水分子最小基组 STO-2G
展开式中的换算因子进行优化:
3 10 T 0.00 0.00
7.660000000V 2.250000000V 1.240000000V
# RHF/Gen Test
Water RHF/STO-2G basis with optimized scale factors
0,1
O
H,1,r
H,1,r,2,a
r 0.96
a 104.5
1 0
sto 1s 2 <1 12.10>
sto 2sp 2 <2 12.10>
****
2 0
sto 1s 2 <3 12.10>
****
3 0
sto 1s 2 <3 12.10>
****
在多处使用相同的 gauopt 变量,可以使两个氢原子的换算因子保持相等。当然,指定
每个氢原子使用相同的基组,也可以达到同样的效果。
ghelp
ghelp 是 Gaussian 的分层帮助工具。输入 ghelp 将显示一般的信息,以及可能用到帮
助的主题列表。命令 ghelp topics 只显示主题列表。
显示 Gaussian 关键词和选项的信息,可以使用下面的格式:
ghelp route
keyword [option]
关于 Gaussian 实用程序的信息,可以用实用程序名作为主题来获取,或者用主题
utilities。
关于层
n
中的内部选项
m
(IOp(
n/m
))的信息,在 Unix 系统下使用下面的命令获得:
$ ghelp "ov
n
iop
(m)
"
mm
独立的分子力学程序。这个程序从标准键盘输入读取Gaussian输入文件,并把(可能优
- 244 -
化的)分子结构作为标准输出,写到一个新的输入文件中。需要的力场必须用-Dreiding,
-UFF,-Amber,或-Param选项选取(最后一个见下)。运行的任务类型用-Force,-Freq,
-Opt,和-Micro命令行选项指定;默认为能量计算。为了预优化分子的MM部分,-Micro
只优化ONIOM(即,真实体系)的低层原子。
附加的命令行选项
-Param N 使用力场
N
(与 Gaussian 中的 IOp(1/64)=N相同)。
-ReadParam 读入附加的参数。内部存储的参数优先于读入的参数。
-ReplaceParam
读入附加的参数。读入的参数优先于内部存储的参数。
-OptCyc N 定义优化循环的最大数量为
N
。
-ReadCon 从输入文件读取分子连接的信息(也就是说,在输入文件中使用
Geom=Connect)。
-Test N 设定调试标记为
N
(数值越大,调试输出越多)。
-TRScale
num
对刚性平移/转动使用换算方案
num
:0=无换算(默认);1表示在刚性区用
1/N 换算 N 个原子;2表示在刚性区用 1/SQRT(N)换算 N 个原子,负值用
|N|/1000 换算。
-External 读取输入,并按照 External 关键词使用的格式写输出。
newzmat
这个实用程序把不同标准格式的分子结构说明相互转化。它还能值型多种有关的功能,
例如从 Gaussian 检查点文件提取分子结构说明。它的全部功能如下:
newzmat 可以将分子结构说明部分在各种数据文件格式之间进行转换。这包括由其它
程序产生的文件转换成 Z-矩阵(因此可用于 Gaussian 输入),以及在这些程序的格式
之间进行转换。因此 newzmat 可用于从几个常见分子图形和力学程序的数据文件产生
Gaussian 输入文件,允许它们作为 Gaussian 的图形输入前端。这样产生的数据文件
将具有正确的对称性限制(若可能的话),以提高计算效率。
newzmat 可以从其它数据文件产生 Gaussian 09 检查点文件,并且(更重要的是)可
以从检查点文件产生数据文件。这个功能可用来提取数据供可视化程序显示。
newzmat 可以从来自几何优化(或优化过程中)的检查点文件恢复中间结构,用于重
新计算或绘图。
命令语法
newzmat具有下面的一般语法:
newzmat
option(s) input-file output-file
其中的
option(s)
可以是一个或多个选项,指定需要的操作。
input-file
是包含要被转换(或
恢复)的分子结构的文件,
output-file
用来保存新的分子说明(或 Gaussian 输入)的文
件。
如果省略输出文件名,则输出文件名与输入文件名具有相同的基本名,并加上常规的扩
展名表示其类型。一般来说,如果遵守扩展名命名习惯的话,扩展名可以从文件说明中忽略。
默认的扩展名在下面的表中列出:
- 245 -
扩展名 描述 选项形式
.bgf Biograf 内部数据文件 bgf
.cac CaChe 分子文件 cache
.chk Gaussian 09 检查点文件 chk
.com Gaussian 输入文件(Z-矩阵分子说明) zmat
.com Gaussian 输入文件(笛卡尔坐标分子说明) cart
.con QUIPU 系统数据文件 con
.dat Model/XModel/MM2 数据文件 model
.dat MacroModel 数据文件(可以是格式化的和非格式化的) mmodel,ummodel
.ent Brookhaven 数据文件(即 PDB) ent
.com 晶体结构的分数坐标(需要三个平移操作) fract
.inp MOPAC 输入文件 mopac
.pdb 蛋白质数据库格式文件(即 Brookhaven) pdb
.ppp 某个 PPP 程序(仅输出文件) ppp
.xyz 未修改的笛卡尔坐标 xyz
.zin 老版本 ZINDO zindo
输入和输出选项
指定输入和输出分子说明格式的选项分别由字符串-i 和-o 开始,其后紧接上表中所列
的适当字符串(不留空格),对应所需的分子说明格式。例如-ipdb 表示以 PDB 格式输入分
子结构说明,如果没有指定扩展名的话,.pdb 用作输入文件名的扩展名。同理,-oxyz 表示
以笛卡尔坐标格式输出分子结构说明,且输出文件名以.xyz 作为默认的扩展名。默认的输
入和输出选项分别是-izmat 和-ozmat。注 意 -izmat 和-icart 是同义词,两者都可读入一个
Gaussian 输入文件,不论其中的分子说明格式是 Z-矩阵,笛卡尔坐标,或是内坐标与笛卡
尔坐标的混合。
其它关于输入和输出的选项
下面的选项进一步定义 newzmat 的输入:
-step
N
读取Gaussian 09检查点文件中第N步几何优化的分子结构(必须和输入选项
-ichk合用)。
这个选项不能用于冗余内坐标(默认的坐标系)的优化。相反,在以后的任
务中,从检查点文件读入结构需要在执行路径部分包含
Geom=(Check,Step=
N
)。
-ubhor 输入文件内的长度单位为Bohr(默认为Angstrom)。
-urad 输入文件内的角度单位为弧度(默认为度)。
下面的选项进一步定义输出文件格式:
-mof1 使用macromodel第一种格式(仅用于-ommodel)。
-mof2 使用macromodel第二种格式(如果定义了-ommodel,这是默认的)。
-optprompt 提示优化哪些参数;用于设定几何优化使用的分子结构说明,并且需要指定
-ozmat(或者不指定输出选项)。默认对所有不受对称性限制而固定的参数都
进行优化。
-prompt 当使用了-ozmat时(或不指定输出选项),将对执行路径,标题行,以及电
荷与多重度部分进行提示。newzmat 产生的默认Gaussian输入文件是
HF/6-31G(d)单点能计算。
- 246 -

例子
下面的命令从 PDB 文件 water.pdb 读取分子说明,并把含有等价 Z-矩阵的 Gaussian 输
入文件写到文件 h2o.com:
$ newzmat -ipdb water h2o
-ozmat 是默认的,所以可以忽略。
Charge and multiplicity [0,1]?
回车接受显示的默认值。
由于电荷和多重度不能从 PDB 文件决定,newzmat 将会对 Z-矩阵提示这些信息。
下面的命令从 Gaussian 09 的检查点文件 job-11234.chk 读取分子说明,并写出 PDB
文件 propell.pdb:
$ newzmat -ichk -opdb job-11234 propell
下面的命令从检查点文件 newopt.chk 读取几何优化第五步的分子说明,并产生 Mopac
文件 step5.inp:
$ newzmat -ichk -omopac -step 5 newopt step5
下面的命令从 Mopac 文件 newsalt.inp 读取分子说明,写到包含等价 Z-矩阵的 Gaussian
输入文件 newsalt.com,并对执行路径,标题部分,以及分子的电荷与多重度部分给出提示:
$ newzmat -imopac -prompt newsalt
Percent or Route card? # Becke3LYP/6-31G(d,p) Opt
Route card?
用一空行结束执行路径部分。
Titles? Optimization of caffeine at Becke3LYP/6-31G**
Titles?
用一空行结束标题部分
Charge and Multiplicity? 0,1
选择输出格式
为了与不支持的可视化系统进行通信,格式的首选是尝试 PDB 文件。这种格式包括原子
相互连接的信息,并得到广泛支持。注意有些软件包使用.ent 扩展名,而不是.pdb;-ient
和-oent 选项选择前者,而-ipdb 和-opdb 选择后者。另一个经常使用的选择是 Mopac 文件
格式。
其它 newzmat 选项
newzmat 其它选项与产生原子连接的信息有关,其中使用标准几何参数,并且确定和使
用分子对称性。完整的原子连接表格可用来产生适合于包含对称限制的 Z-矩阵。这个表格
对于分子力学程序数据文件的输出,也是必需的。如果使用了包括全部原子连接关系的输入
文件格式(如 MacroModel 数据文件),则使用提供的原子连接关系。
但是,当输入文件是 Z-矩阵或 MOPAC 格式,则只能使用内坐标包含的原子连接信息。
因此,如果要产生包括分子对称性的新 Z-矩阵,则必需产生其它原子的连接信息。因为笛
卡尔坐标文件不含原子连接信息,当读入笛卡尔坐标时,程序默认利用内部存储的原子半径
数据产生原子连接信息。此外,当用分子力学程序生成输入结构时,可能不会产生正好满足
实际对称性的键长和坐标。可以用以下选项控制些情况。
-allbonded 当建立新的原子连接信息时,假设所有原子都成键。
-bmodel 在决定键长时,使用内部数值和标准模型 B 键长数据。
-density N 产生密度编号
N
的自然轨道。该选项仅用来产生 CAChe 文件。对于 HF 计算,
- 247 -
N
值设为 0,MP2 计算
N
值设为 2,CI 计算
N
值设为 6,QCISD 或 CCD 计算
N
值设为 7。
-fudge 利用内部存储的数值猜测合理的键长,对模型输入是默认的,不要在其它地
方用。
-gencon 利用内部存储的原子半径数据产生原子连接信息。
-getfile 要求所有输入参数必需以文件名定义,而不是用标准的输入和输出终端。
-lsymm 确定分子对称性时使用较松的截断值,该选项意味着-symav。
-mdensity M 由选项-density 指定的密度减去广义密度
M
以产生密度差,然后再转换成自
然轨道。
-nofudge 不猜测键长。除了对模型输入以外,这是默认且唯一的选项。
-nogetfile 取消-getfile。
-noround 不做 Z-矩阵参数的四舍五入处理。
-nosymav 不做输入坐标的平均。
-nosymm 不使用对称性。
-order 尽可能保持和输入文件相同的原子顺序。
-round 将 Z-矩阵参数四舍五入到 0.01 Å和 1 度。
-symav 用近似的对称性操作把输入坐标值平均,以达到准确的对称性。
-symm 指定分子的对称性。
-tsymm 在确定分子对称性时使用较严格的截断值。该选项是默认的。
-rebuildzmat 建立新的 Z-矩阵而不是用读入的 Z-矩阵(对于 Z-矩阵或 MOPAC 输入是默认
的)。该选项意味着-gencon,并可缩写成-redoz。
使用 newzmat 时的已知困难
对称性平均程序通过给定的坐标猜测指定的对称性,但这只是近似的对称性,并不总
是指定的对称性。它需要从 Gaussian 输出文件中读取六位数字的原子坐标来重建分
子对称性,如果分子力学程序的优化计算能够在合理的步数内收敛,其结果可用来创
建坐标。以 MacroModel 产生的坐标为例,在正常的能量最小化计算后,有时需要再
做最终的完全 Newton-Raphson 步骤。
newzmat 对最初读入结构和最后产生的结构分别计算核排斥能,用于一致性检查。如
果二者不一致则会打印警告信息。如果二者相差很大,则表示程序的计算失败。
testrt
testrt 是把标准的 Gaussian 计算执行路径作为输入,并产生等价的非标准执行路径的
实用程序。执行路径通常在命令行定义(外面加引号):
$ testrt "# rhf/sto-3g"
如果执行路径没有包含在命令行中,testrt 将提示要测试的执行路径。
如果指定的执行路径有效,testrt 将打印出相应的非标准路经。如果出现语法错误则
显示出错信息。因此,即使是在用户对非标准计算路径一无所知的情况下,也可使用 testrt
检查计算执行路径部分的语法正确性。
下面是一些运行 testrt 的例子:
- 248 -

$ testrt "# ccsd(modredun)/6-31G* scf=driect"
-----------------------------------
# ccsd(modredun)/6-31G* scf=driect
-----------------------------------
QPErr ---- A syntax error was detected in the input line.
# ccsd(modredundant)
ModRedundant 是一个有效的 Gaussian 选项,但对 CCSD 无效。
'
$ testrt "mp4 nmr"
NMR 不能用于 MP4。
-------
mp4 nmr
-------
NMR only for HF, DFT, and MP2.
$ testrt
Please type in the route specification, terminated with a blank line:
# HF/6-31G(d) Opt=QST2
End of testrt input.
----------------------
# HF/6-31G(d) Opt=QST2
----------------------
1/5=1,18=20,27=202,38=1/1,3;
2/9=110,12=2,17=6,18=5,40=1/2;
...
第一个例子表明,执行路径部分只有第一个错误被标记。第二个例子表示,错误信息来
自无效的关键词组合。最后一个例子来自正确的路径测试输出。
注意 testrt 不能判断关键词使用正确与否。它仅检查给定的执行路径部分的语法。因
此,它不会警告在计算执行路径部分包含两次 MP2 关键词会导致不可预期的结果(运行一个
MP4 任务)。
Testrt 的输出可以用标准的 UNIX 输出转向到一个文件中:
$ testrt " rhf/sto-3g"
> output file
unfchk
这个实用程序与 formChk 相反。它以适于本地计算机的格式,把格式化的检查点文件转
化为二进制检查点文件:
$ unfchk
Formatted Checkpoint file? water
Read formatted file water.fchk
Write checkpoint file water.chk
在 Windows 操作系统中,这个实用程序对指定的文件名使用扩展名.fch,在其它的计算
机系统中,扩展名是.fchk。
注意,格式化的检查点文件可以用作不同计算机平台间的数据交换格式。在原计算机使
用formchk,在目标计算机用 unfchk 创建二进制检查点文件。
- 249 -
- 250 -
Appendix
附录
Gaussian 09 和Gaussian 03 之间的变化
程序的限制
构造 Z矩阵
格式化检查点文件的结构
程序的开发功能
作废的关键词和过时的功能
- 251 -
- 252 -

Gaussian 09 和Gaussian 03 之间的变化
这一部分简要总结Gaussian 09和Gaussian 03之间的主要差别。这些功能(及参考文献)
的全部细节见手册前面的部分。Gaussian各个版本功能的回顾性总结,见我们的网站
www.gaussian.com/g_tech/gdiffs.pdf
新的方法和功能
能量与导数
最近的半经验模型的最新执行,包括 AM1,PM3,PM3MM,PDDG,和 PM6,它们包括解析
的一阶和二阶导数,用户指定参数,以及与 PCM 溶剂化模型相结合。
TD-DFT 梯度和数值频率。
使用 EOM-CCSD 计算电子激发能。
许多新的 DFT 泛函,包括 HSE,wB97,m05/m06,LC 类泛函,以及双杂化 B2PLYP。
包含经验色散模型和相应的泛函。
ROMP3,ROMP4,ROCCSD,ROCCSD(T)能量。
W1RO,W1BD,G4 复合方法计算能量。
DFTB 半经验模型,以及使用解析矩阵元的 DFTBA 版本。
ONIOM
ONIOM可以与PCM组合。有数种ONIOM+PCM模型。
现在可以用ONIOM计算IRC,即使分子包含上千个原子,效率也很高。
溶剂化
新的PCM溶剂化算法,能使能量成为核坐标的连续函数,它包含了所有SCF特性的有效溶
剂化。现在,使用PCM的几何优化与相应的气相优化有相同的收敛速率。
可以使用特定态的自洽溶剂化,用于模拟荧光和其它发射过程。
可以使用SMD溶剂化模型。它的参数化可以对上百种溶剂给出非常好的总溶剂化自由能。
几何优化和IRC
能量最小化默认使用GEDIIS几何优化算法,这对大的柔软分子特别有帮助。
对力学部分或电子嵌入部分,以及对能量极小点和过渡结构使用二次收敛ONIOM(MO:MM)
- 253 -
优化。
读取一个输入部分,用于控制优化中哪些原子冻结或非冻结。可以用原子、元素、残基、
或ONIOM层来指定原子。
分子特性
解析的含频ROA强度。
解析的DFT超极化率。
对两个态使用谐振动模式,通过Franck-Condon原理计算电子激发、发射、光电离的谱
带带型。
用Herzberg-Teller或Franck-Condon-Herzberg-Teller理论计算电子激发的谱带带型。
选择简正模式用于显示,非谐校正,和FC/HT/FCHT分析。可通过原子、元素、ONIOM层、
或残基来选择。
分析和输出
蛋白质二级结构的信息可以包含在分子说明输入部分,或者.fchk文件中。
逐个轨道执行轨道布居分析,给出原子或角动量对一个轨道的贡献。
正则UHF/UDFT进行二次正交化,用于显示或用于ROHF计算的初始猜测。
CIS和TD激发的自然跃迁轨道分析。
把占据轨道投影到最小基之后,可以进行Mulliken布居分析。当基组增大时,这能给出
稳定的布居。
其它的新功能
SCF的初始猜测可以用片段计算组合产生,需要指定每个片段的电荷和自旋。
用四点差分而不是默认的两点差分计算数值频率,具有更好的精度和数值稳定性。
效率的改善
HF和DFT对大分子的频率计算更快,特别是当并行时。
FMM以及线性标度的库仑和交换对集群实现了并行。
大体系的ONIOM(MO:MM)频率计算更快,特别是对电子嵌入。可以计算100-200个量子力
学原子和6000个分子力学原子的频率。
在大型频率计算中保存简正模式,用于显示或打印模式,以及开始IRC=RCFC任务。
CC,BD,和EOM-CCSD幅度可以保存在检查点文件中,在以后的计算中读入,包括使用不
同基组的情况。还保存了BD轨道并在以后读入。
半经验,HF,和DFT的频率计算可以在中间计算阶段重新开始。重新开始数值频率参见
- 254 -
Freq=Restart,Restart关键词用于重新开始解析频率。
CC和EOM-CC计算可以在中间计算阶段重新开始。
在ONIOM计算中,个别步骤的初始猜测可以来自另外的检查点文件。
ONIOM=OnlyInputFiles选项会打印ONIOM计算每一部分的输入文件,便于分别产生波
函。
加入了密度拟合基,对应于SVP,TZVP,和QZV基组。/Fit关键词请求与特定AO基匹配的
拟合基,没有特定的拟合基时需要用/Auto关键词。因此BVP86/SVP/Fit使用的密度拟
合定义为伴随SVP基的数集,而BVP86/6-31G*/Fit等同于BVP86/6-31G*/Auto。
可以在Default.Route文件中包含DensityFit关键词,只要执行纯密度泛函,默认就使
用密度拟合。
读入的密度基组可以是非归一化原函数的因子,密度归一化原函数,或按照AO基组归一
化的原函数。每种形式都有一些程序在用,因此如果用G09读入这些发表的基组时,需
要使用相应的选项。
Gaussian 09 与Gaussian 03 之间功能的差别
单点SCF计算现在默认为完全精度(SCF=Tight)。
Freq=ROA默认为CPHF=RdFreq,因为含频ROA强度是解析的,而在静态极限是数值的,
精度较低。
后-SCF方法,如MP,BD,和CC,默认为Tran=IABC,在大多数机器上比完全转换的效率
更高。
IRC默认为新的链接L123。除非明确指定,否则旧的IRC链接(L115)仅用于IRCMax任务。
除了使用默认算法IRC=EulerPC 的ONIOM(MO:MM)计算之外,L123中的默认算法是
IRC=HPC。L123可以使用IRC=GS2算法(L115所用的算法),但这通常比默认设置更昂贵。
默认的IRC结果报告仅为路径上每点的能量和反应坐标。使用IRC=Report指定还把内坐
标的值列表。
常规的量子力学频率计算和ONIOM(MO:MM)频率默认为CPHF=Simultaneous,与G03相同。
但是半经验频率用CPHF=Separate执行的效率更高,这种情况下为默认设置。
在平衡和Guess=Fragment计算中,或者做片断的布居分析时,需要把原子归属到片断,
这位于分子说明的核特性部分而非行尾,因为在行尾会与ONIOM输入冲突。例如,
C(Fragment=3) 0.0 1.0 2.0
而非旧的格式:
C 0.0 1.0 2.0 3
同位素通常在原子说明行的核性质部分定义。如果另外读入,它们需要在分子说明之后
一次读入,而不是由程序的不同部分(IRC,Freq,等)分别读入。
工具程序的增强
formchk工具可把其它信息加入到.fchk文件,包括用户指定的分子力学类型和其它字符
- 255 -
串。当调用formchk时使用–3选项,就打印这一信息。
freqchk工具可以从.chk或.fchk文件提取简正模式,并把提取的简正模式保存到.chk
文件(但不能存于.fchk文件)。
- 256 -
效率的考虑
Gaussian设计为在各种给定的计算机配置中能够有效地工作。在给定的内存和磁盘限制
下,程序尝试选择最有效的算法。由于Gaussian提供多种算法供选择,使用者需对各种算法
的优缺点有足够了解,才能实现最佳的运行效果。
在开始前,我们强调非常重要的两点:
除了非常大的任务之外,程序选择的默认算法对所有的任务可以得到相当理想的计算效
率。注意,为了反映目前典型问题的尺寸,Gaussian 09改变了一些默认的设置。程序
早期版本使用的默认值对小任务设置为100个基函数以下。Gaussian使用的默认算法一
般为更长时间的任务而设计。
对于经常进行大型任务的用户或站点而言,以下位于Default.Route文件的默认值一般
产生很好的执行效率:
-M-
可用内存
# MaxDisk=
可用磁盘大小
其中定义了使用内存和磁盘的总量。二者给定值的默认单位是8位的字,也可以是数值
后加上KB,MB,GB,KW,MW或GW(不要插入空格),来定义千-、兆-、或吉-位或字的单
位。默认内存大小为256 MB,擦写磁盘空间默认设定为无限制(-1),即,假定有足够
的磁盘空间执行给定的计算,无须多余的工作。对大多数站点来说,一旦设置了
Default.Route文件,不需要其它的特殊操作就可以完全高效地使用程序了。
估计计算的内存需求
使用Gaussian 09和当前的计算机,256 MB的默认内存大小足够用于大多数任务类型和
方法,使用的计函数可以直到g函数。如果基组包含h或更高角动量的函数(例如cc-PVQZ),
则可能需要为任务增加内存。下面的公式可以用来估计Gaussian各种类型任务的内存需求
(单位是8位的字):
M + 2(NB)2
其中NB是计算中使用的基函数数量,M是一个最小值,通常默认内存量足以大于这个值。注
意1 MW = 1,048,576字(= 8,388,608位)。对于收缩的高角动量函数(f或更高)的求导,
需要更大的内存。
后面的部分适用于那些希望了解更多关于权衡各种选项的用户,以对个别的而非所有的
任务得到最佳的运行效果。技术还涵盖了非常大的和小型的任务。此外,有关信息可以在参
考文献[Schlegel91]中找到。
并行计算的内存需求
当使用有共享内存的多个处理器时,
内存需求的较好估计是单处理器任务的内存量。对
- 257 -

于分布式内存计算(即,通过Linda执行的计算),那么在%Mem指定的内存量应当大于或等于
单处理器任务的值。
在Gaussian 09中,两种并行方法可以组合。例如,为了在有8个CPU位于4台双接头共享
内存的多处理器平台上运行任务,应当使用以下指令(假定上面表格给出的内存值为10 MW):
%Mem=128MW
每个多处理器需要的内存
%LindaWorkers=sysa,sysb,sysc,sysd
使用4个Linda工作计算机:全部是多处理器系统
%NProcShared=2
在每个多处理器计算机上使用两个共享内存处理器
SCF流程
为了加快直接 HF 和 DFT 计算的速度,迭代计算分两步进行:
用准确到六个数字的积分,在 DFT 计算中还要使用中等的积分格点,将电子密度收敛到
10-5。这一步在 21 次迭代后结束。不论是否完全收敛。如果出现了过渡金属原子,默
认忽略这一步。
用准确到十个数字的积分将电子密度收敛到 10-8,两步总共可进行 128 次循环。
在迄今所进行的大部分测试中,这种方法比始终使用完全积分精度快很多,并且不会减
慢收敛。在遇到困难的情况下可指定 SCF=NoVarAcc,要求始终使用完全精度的积分。更多
细节参见 SCF 关键词的讨论。
收敛的问题
默认的 SCF 算法使用两种迭代子空间直接反转(DIIS)方法的组合:EDIIS 和 CDIIS。
EDIIS [Kudin02]使用能量外推,用在 SCF 收敛过程的初期迭代。CDIIS 对 Fock 矩阵和密度矩
阵交替进行外推,用于 SCF 收敛的较后阶段。这种算法非常可靠,以前棘手的 SCF 收敛问题,
现在用默认算法几乎总能实现收敛。对于个别的病态收敛情况,Gaussian 09 提供了与 CDIIS
结合使用的 Fermi 展宽和阻尼(包含自动的能级移动)。
如果默认的方法收敛失败,可以尝试其它方法(用对应的关键词标明):
SCF=Fermi 在初期迭代中要求温度展宽[Rabuck99],需要与 SCF 初期迭代的 CDIIS 和动态
阻尼结合使用。
SCF=QC 基于 Bacskay 的方法[Bacskay81],是二次收敛 SCF。这种方法结合了 Bacskay
建议的 Newton-Raphson 算法和线性最小化,因此保证最后一定会收敛到一
稳定点。典型情况下,SCF=QC 的计算量是传统 SCF 计算量的两倍。由于
SCF=QC 可靠,并可用于直接 SCF,因此通常是遇到计算不收敛情况时的第
二种选择。它可用于 RHF 和UHF,但不适用于复数 HF 或ROHF。
Guess=Alter 有时,收敛困难是初始猜测占据了错误轨道的一种警告。应检查猜测,特别
是占据轨道的对称性。Guess=Alter 可用来修改占据轨道的选择。
SCF(MaxCyc=
N
)
将SCF 计算迭代总数增加到 N。注意,对默认算法只增加 SCF 循环数通常没
有太大的帮助。
这些方法都强制收敛到轨道空间内最接近的一个稳定点上,对轨道转动而言这个稳定点
可能不是最小值。要验证是否已得到合适的SCF解,可用稳定性计算来验证(见Stable关键
- 258 -
词)。需要注意,应检验最后的波函数是否对应所要的电子态,特别当使用Guess=Alter时。
工具程序freqmem对频率计算的各种参数返回最优的内存大小(即,一次计算完成主要
步骤所需的内存量)。
MP2 能量,梯度,和频率
MP2 有四种算法可用,但大部分情况下由程序自动作决定。对决定起重要作用的是
MaxDisk 的值,这个值应按照特定的计算机配置来设定。MaxDisk 以字为单位,指定可用的
磁盘空间最大量。若不在执行路径部分或者 Default.Route 文件中指定 MaxDisk 的值,
Gaussian 假设有足够的磁盘空间来执行计算,因此不会做多余的工作,这对大型计算可能
会造成问题。因此,要想最有效地进行 MP2 计算,最重要的方式是指定可用的内存和磁盘空
间的大小。这使得程序能够在各种可用算法中确定最适于计算机配置的算法。这最好由
Default.Route 文件中的-M-指令和 MaxDisk 关键词指定(尽管也可以在输入文件中包含
MaxDisk 和%Mem)。
更高的关联方法
MP2 之上的关联方法(MP3,MP4,CCSD,CISD,QCISD,等)都需要将一些转换的 MO 积
分储存在磁盘上(这与 MP2 能量和梯度计算不同),因此磁盘空间的需求以分子大小四次方
的比例增加。这些转换的积分的产生,储存,和其余项的计算有几种方式。
Gaussian 默认为半直接方法。AO 积分可以写到磁盘上,在计算的 SCF 阶段用到时再读
回来,或是用直接或内存方式进行 SCF 计算。转换重新计算需要的 AO 积分,使磁盘上存放
的 MO 积分数量最少(见下)。其余的项用重算的 AO 积分计算。
以下几点总结了 MaxDisk 对后-SCF 计算的影响:
CID,CISD,CCD,BD,和QCISD能量也有固定的磁盘存储需求量,比例为O2N2,还要乘
上一个很大的因子,但是遵守MaxDisk的定义以避免更大的存储需求。
CCSD,CCSD(T),QCISD(T),和BD(T)能量有固定的磁盘存储需求量,比例为ON3,不受
MaxDisk的限制。
CID,CISD,CCD,QCISD密度和CCSD梯度有固定的磁盘存储需求量,对闭壳层是N4/2,
开壳层是3N4/4。
CIS和TD的能量和梯度
除了积分存储的选择以外,恰当使用重新开始功能可以改善CIS和TD计算的效率。
积分的存储
用单激发 CI 进行激发态计算可以使用五种方法(用 CIS 关键词中相应的选项标记)。注
- 259 -
意,只有前两个选项可用于 TD 方法:
Direct 由实时计算的双电子积分建立乘积向量,利用迭代对角化求解指定数目的电
子态。这种算法把内存和磁盘的需求降至 O(N2)。这对 TD 是默认的。
InCore 指定在内存中储存 AO 的 Raffenetti 组合。存于内存是相当高效的方法,但
只适用于小分子体系或具有大内存的计算机,因为需要 N4/4 字内存。如果有
足够的内存,程序会自动使用这个方法。
MO 由 MO 积分建立乘积向量,利用迭代(Davidson)对角化对指定数目的电子态
求解。这是最快也是默认的方法。对于直到 150 个基函数,依占据轨道数而
定,这种算法是高效率的选择。占据轨道越多,越应使用直接方法。因为计
算只需要用到涉及两个虚轨道的积分(即使是梯度计算也是如此),程 序 会 设
法使用由 MaxDisk 指定的磁盘空间。最小磁盘需求约为 4O2N2(开壳层是
6O2N2)。这对 CIS 是默认的。
重新开始任务和重复使用波函
CIS 和 TD 任务可以从 Gaussian 的检查点文件重新开始。这限定为用于小型计算,因为
可在 MO 基中进行,必需计算新的积分和进行转换,但对直接 CIS 计算却无价值。如果直接
CIS 任务在 CIS 阶段因故终止,则除了 CIS=Restart 或TD=Restart 之外,应同时指定
SCF=Restart,因为最后的 SCF 波函只有在整个任务步骤(或优化步骤)完成后才会写到永
久存放的位置(适合于 Guess=Read)。
CIS和TD的激发态密度
若只进行密度分析,并且已找到激发态,那么 CIS 密度可以从检查点文件中恢复,使用
选项 Density=(Check,Current) Guess=Only。这将恢复当前方法(假设是 CIS)所储存的
任何广义密度,并重复布居分析计算。注意可以检查单电子(非松弛的)密度和广义的(松
弛)密度,但如果使用单电子密度(也就是忽略轨道的松弛项),在 CIS 级别计算的偶极矩
和其它特性的精度会很差[Foresman92, Wiberg92]。因此应避免使用 CIS 单电子密度,除非要与正
确的密度或与不能提供广义密度的其它程序结果比较时才可使用。
产生多个电子态的广义密度需分别计算,因为必需对每个电子态分别进行 CPHF 计算。
要做这样的计算,首先要解出所有的电子态,以及第一激发态的密度。如果对
N
个电子态感
兴趣:
# CIS=(Root=1,NStates=N) Density=Current
接下来对电子态 M=2 到 N 做 N-1 个另外的计算,使用的执行路径部分形式为:
# CIS=(Read,Root=M,NStates=N) Density=Current
- 260 -
开壳层激发态的缺陷
因为UHF参考态不是S2的本征函数,因此CIS或TD产生的激发态也不是S2的本征函数
[Foresman93]。
稳定性计算
对于RHF和UHF以及限制和非限制DFT波函的三重态和单重态,可用关键词Stable要求进
行稳定性测试。稳定性计算可以重新开始,参见上面CIS的说明。
CASSCF的效率
CASSCF 计算工作的主要困难在于选择合适的活性空间轨道。有以下几个技巧:
用Pop=SaveMixed 把占据轨道的 NBO 和未占据轨道的 NLMO 保存到检查点文件。接下来
用 GaussView 布居和重排所需的轨道,构成活性空间。这是目前最容易也最有效的方法。
使用标准的离域化初始猜测轨道。有时候用这种方法就够了,例如,如果活性空间全由
p
电子构成。使用 Guess=Only 选项检查各轨道,并在实际计算之前决定是否需要更改。
使用局域化初始猜测轨道。这种方法适用于要将特定的成键电子对包含在活性空间的情
况,因为局域化会使电子对分离。
使用由 UHF 计算的总密度构成的自然轨道(CAS-UNO)
[Hamilton88, Bofill89]。 对于单重态,
这需要先将 UHF 的计算收敛到对称性破缺的波函数(通常使用 Guess =Mix)。它适用
于复杂体系,其中不清楚哪些电子最不适合用双占据轨道描述。
在所有的情况下,任何优化计算之前应先做单点计算,以便检查收敛的活性空间,以保
证在继续计算之前指定的电子已包含关联。对激发态求解 CASSCF 波函还有另外的考虑(详
细情况在 CASSCF 关键词中讨论)。
CASSCF频率
CASSCF 频率计算需要使用大量的内存。因此增加可用的内存总可以改善 CASSCF 频率任
务的执行效率(而其它类型的频率计算不会因可用内存增加而使效率显著改善)。这些计算
同样需要 O2N2磁盘空间。
- 261 -
程序的限制
这一部分列出了Gaussian 09存在的各种尺寸的限制。
积分程序有以下限制:
原子最大个数为250,000。
原壳层的最大总数为750,000。
原始d和更高的壳层的最大个数为250,000。
收缩壳层的最大个数是250,000。
允许的最大收缩度是100。
Opt=(EF,EnOnly)优化——仅对没有解析梯度的方法有用——限制为50个变量。
GVB程序限制为100个成对轨道(在实用中算不上是限制)。
NBO的维度限制为250,000个原子和10,000个基函数。
- 262 -

构造Z矩阵
这一部分简要介绍分子体系的传统 Z-矩阵描述。对 Z矩阵的大小存在限制:计算中的
最多变量数和最多原子数。它们设定为最多 250,000 个真实原子(包括空原子但不包括虚原
子),以及最多 250,000 个Z矩阵中心(原子,空原子,和虚原子)。
使用内坐标
每一行 Z 矩阵给出分子中每一原子的内坐标。最常用的 Z 矩阵格式使用如下语法:
元素标记,原子 1,键长,原子 2,键角,原子 3,二面角[,格式代码]
虽然例子中用逗号分隔一行内的项目,但是可以用有效的分隔符。
元素标记
是由原子的
化学符号或原子序数表示的字符串。如果使用元素符号,则可在其后接其它的字母或数字9,
创建该原子的可识别标记。常见做法在元素名后接辅助的识别整数,例如C1,C2,等等。
原子 1,原子 2,原子 3
是在先前定义的原子符号,用于定义当前原子的位置。也可以
使用其它原子在分子说明部分的行号表示变量值,其中电荷和自旋多重度的行是第 0 行。
当前原子的位置由三个量定义:连接
原子
1的键长,由该键长和
原子
1,
原子
2之间键
长形成的角度,包含
原子
1,
原子
2,
原子
3的平面和包含当前原子,原子 1,原子 2的平
面之间的二面角。注意键角范围必须是 0º<键角<180º。二面角可以用任何值。
可选的格式代码参数定义 Z 矩阵输入的格式。在这里描述的语法,代码总是 0。这个代
码仅用于当正常的 Z 矩阵说明数据之后接附加参数时,就像在 ONIOM 计算中一样。
作为第一个例子,考虑过氧化氢。该结构的 Z 矩阵为:
H
O 1 0.9
O 2 1.4 1 105.0
H 3 0.9 2 105.0 1 120.0
Z矩阵第一行简单地指定氢原子。下一行列出氧原子,并指定它与氢原子的核间距是
0.9 埃。第三行定义另一个氧原子,其中 O-O 键长 1.4 埃(从原子 2开始,也即另一个氧原
子),O-O-H 键角(使用原子 2和1)为 105 度。第四行也就是最后一行是唯一需要给出三
个内坐标的。它定义另一个氢到第二个氧的 H-O 距离为 0.9 埃,H-O-O 角度 105 度,以及
H-O-O-H 二面角 120 度。
也可以用变量定义 Z矩阵中的某些或所有的值。以下是前一个 Z矩阵的另一版本:
H
O 1 R1
O 2 R2 1 A
H 3 R1 2 A 1 D
Variables:
R1 0.9
R2 1.4
A 105.0
9 除了数字标记外(例如 H1,H-1),从 Gaussian 09 开始可以使用字母标记。例如 Ha,H-a 也都表示 H原
子。但不能用 Hg,Hf,等,因为与现有元素符号冲突。这种情况下只能用 H-g 和H-f。——译注。
- 263 -

D 120.0
分子的对称性约束表现在内坐标上。两个H-O距离由同一个变量定义,与两个H-O-O键
角类似。当这样的Z矩阵用于内坐标几何优化时(Opt=Z-matrix),
将会优化这些变量值来定
位最低能量的结构。当进行完全优化(FOpt)时,这些变量必需是线性独立的,并且包含分
子的所有自由度。在进行部分优化(POpt)时,第二部分变量(常标明为Constants:)的数
值必需保持固定,只对第一部分变量的数值进行优化:
Variables:
R1 0.9
R2 1.4
A 105.0
Constants:
D 120.0
关于内坐标优化的更多信息,参见Opt关键词讨论中的例子。
混合内坐标和笛卡尔坐标
笛卡尔坐标实际上是 Z矩阵的特例,例如这个例子:
C 0.00 0.00 0.00
C 0.00 0.00 1.52
H 1.02 0.00 -0.39
H -0.51 -0.88 -0.39
H -0.51 0.88 -0.39
H -1.02 0.00 1.92
H 0.51 -0.88 1.92
H 0.51 0.88 1.92
在同一 Z矩阵中,还可以同时使用内坐标和笛卡尔坐标,如这个例子:
O 0 xo 0. zo
C 0 0. yc 0.
C 0 0. -yc 0.
N 0 xn 0. 0.
H 2 r1 3 a1 1 b1
H 2 r2 3 a2 1 b2
H 3 r1 2 a1 1 -b1
H 3 r2 2 a2 1 -b2
H 4 r3 2 a3 3 d3
Variables:
xo -1.
zo 0.
yc 1.
xn 1.
r1 1.08
r2 1.08
r3 1.02
a1 125.
a2 125.
d3 160.
b1 90.
b2 -90.
- 264 -

这个 Z-矩阵有几点值得注意:
笛卡尔坐标的变量名以符号给出,这与内坐标变量的方式相同。
原子符号后的 0表示接下来是有符号的笛卡尔坐标。
就像二面角一样,笛卡尔坐标与正负号的改变有关系。
另一种Z-矩阵格式
另一种 Z-矩阵格式允许用两个键角,而非一个键角和一个二面角,来定义核的位置。
这在第二个角之后的附加字段中用 1表示(这一字段默认是 0,表示第三部分是二面角):
C4 O1 0.9 C2 120.3 O2 180.0 0
C5 O1 1.0 C2 110.4 C4 105.4 1
C6 O1 R C2 A1 C3 A2 1
第一行使用二面角,而后面两行都使用两个键角。
使用虚原子
这一部分讲解在 Z 矩阵中使用虚原子,它由赝原子符号 X表示。下面的例子描述使用虚
原子固定 C3v 群氨分子的三重轴:
N
X 1 1.
H 1 nh 2 hnx
H 1 nh 2 hnx 3 120.0
H 1 nh 2 hnx 3 -120.0
nh 1.0
hnx 70.0
虚原子在轴上的位置并不重要,使用的键长 1.0 可以用任何其它的正数代替。hnx 是
N-H 键和三重轴间的夹角。
这是一个环氧乙烷的 Z 矩阵:
X
C1 X halfcc
O X ox C1 90.
C2 X halfcc O 90. C1 180.0
H1 C1 ch X hcc O hcco
H2 C1 ch X hcc O -hcco
H3 C2 ch X hcc O hcco
H4 C2 ch X hcc O -hcco
halfcc 0.75
ox 1.0
ch 1.08
hcc 130.0
hcco 130.0
- 265 -

这个例子说明两点。第一,虚原子位于 C-C 键的中心以帮助约束 CCO 为等边三角形。
ox 是从 O到C-C 键的垂直距离,OXC 角度保持在 90 度。第二,Z 矩阵的某些项可以用负
的二面角变量 hcco 表示。
下面的例子演示用虚原子指定线形键。使用内坐标的几何优化无法处理线形分子片断中
键角为 180 度的状况,例如乙炔,或丁三烯中的 C4链。问题还可能发生在接近线形的情况,
例如不对称分子中的乙炔基。在键角平分线上引入虚原子,并用半角作为变量或常数,可以
避免这些问题:
N
C 1 cn
X 2 1. 1 90.
H 2 ch 3 90. 1 180.
cn 1.20
ch 1.06
类似地,在这个用于几何优化的 Z 矩阵中,half 表示接近直线的 NCO 角度的一半。注
意,小于 90 度的 half 值表示顺式构型:
N
C 1 cn
X 2 1. 1 half
O 2 co 3 half 1 180.0
H 4 oh 2 coh 3 0.0
cn 1.20
co 1.3
oh 1.0
half 80.0
coh 105.
模型构建程序的几何结构定义
模型构建程序是 Gaussian 中的另一个工具,用来快速指定某类的分子系统[Pople67a]。
它可以由 Geom 关键词的 ModelA 或ModelB 选项来调用,并且需要在输入文件另外的部分提
供额外输入。
模型构建程序的基本输入称为
短公式矩阵
,由几行组成,每一行(用原子符号)定义一
个原子及其连接关系,最多可以指定六项。每一项既可以用与当前原子成键的原子行数表示,
也可以用端键与当前原子相连的原子符号(例如,H,F)表示,或者用与当前原子成键的终
端官能团符号表示。目前可用的官能团有 OH,NH2,Me,Et,Npr,IPr,NBu,IBu 和TBu。
短公式矩阵还按以下方式隐含定义每一键的旋转几何关系。假设原子 X 和原子 Y 已经定
义。那么原子 X 出现在 Y 行,Y 原子出现在 X 行。令 I 为在 Y 行中原子 X 右边的原子,J 为
在 X 行中原子 Y 右边的原子。那么原子 I 和 J 被放置在 X-Y 键的反式位置。短公式矩阵之
后可加入可选的几行,用于修改产生的结构。下面每个这样的行中,都有零个或更多的这样
的项,它们必需以下列次序成组出现:
AtomGeom,
I
,
Geom
一个原子的局部结构通常可由该原子成键的数目和类型定义(例如甲烷分子
中的碳原子是四面体,乙烯分子中的碳原子是三角形,等等)。中心原子处的
- 266 -
所有键角必须都相等。在 AtomGeom 行更改在中心原子
I
处的键值。
Geom
可
以是浮点数或用字符串 Tetr,Pyra,Trig,Bent,或 Line 之一表示的角度。
BondRot,
I,J,K,L,Geom
改变 I-J 和 K-L 键相对于 J-K 键的方向。Geom 可以是二面角或字符串 Cis
(≥0),Trans(≥180),Gaup(≥+60)或 Gaum(≥-60)之一。
BondLen,
I,J,NewLen
设置 I-J 键长为 NewLen(一个浮点数)。
模型构建程序只能建立原子具有正常价态的结构。如果建立自由基分子,则可以用虚原
子有效地对价态加以限制,这可以通过在原子符号前加一负号(如-H)来指定。只有键端
的原子可以是虚原子。
两种可选的模型(A和B)不同之处在于,模型A中的键型(单键,双键,三键等)是以
键长来区分,而模型B中的键长仅依赖于有关原子的类型。除了He和Ne以外,从H到Cl的所有
原子都可以用模型B。如果使用模型A,但键长在模型A中没有定义时,程序会从模型B中选用
适当的键长做替换。
- 267 -
格式化检查点文件的结构
这个文件用于计算机无关的结构,易于后处理器提取所需的数据并忽略其余部分。后者
实际上对可扩展性很重要,因为将来增加的数据不应影响以前版本的应用。通常,运行任务
会指定一个.chk文件,它是二进制的,包含一个计算中可能会对以后计算或后期处理有用的
结果,接下来在Gaussian 09计算完成后,运行formchk工具从二进制.chk文件产生文本
的.fchk文件。还有一个工具unfchk,用于逆过程。为了向下兼容,运行formchk时不加任何
选项会产生完整信息的子集。本文当描述了运行formchk -3
chkfile fchkfile
的结果,它
会产生版本3(当前的和功能最完成的版本)的格式化检查点文件。
这里是一个Fortran格式的数据说明,虽然并没有特别理由使用Fortran而非其它语言来
读取数据。
文件中的前两行包含描述任务的字符串:
标题部分的前72个字符 完整的标题和路径出现在后面
类型,方法,基 格式:A10,A30,A30
类型
是以下关键词之一:
SP 单点
FOPT 完全优化到最小
POPT 部分优化到最小
FTS 完全优化到过渡态
PTS 部分优化到过渡态
FSADDLE 完全优化到 2 阶或高阶鞍点
PSADDLE 部分优化到 2 阶或高阶鞍点
FORCE 能量+梯度计算
FREQ 振动频率(二阶导)计算
SCAN 势能面扫描
GUESS=ONLY 只产生分子轨道,还可用于产生局域轨道
LST 线性同步过渡
STABILITY 测试 SCF/KS 稳定性
REARCHIVE/MS-RESTART 从检查点文件产生存档信息
MIXED 混合方法的模型化学(CBS-x,G1,G2,等),其中的方法和基
组包含在模型中
方法
是能量的计算方法(AM1,RHF,CASSCF,MP4,等),
基
是基组。
文件中包含的所有其它数据位于由以下形式之一标记的行或部分:
标量值与其数据标记出现在同一行。这一行由描述数据项的一个字串,一个数据类型标
签,以及数值构成:
整型标量:
Name,
I
,IValue
,格式为A40,3X,A1,5X,I12
实型标量:
Name,
R
,Value
,格式为A40,3X,A1,5X,E22.15
字符串型标量:
Name,
C
,Value
,格式为A40,3X,A1,5X,A12
逻辑型标量:
Name,
L
,Value
,格式为A40,3X,A1,5X,L1
矢量和数组数据部分的第一行命名数据,给出类型和数据的个数,接下来的多行(行数
根据需要)给出数据:
- 268 -
整型数组:
Name,
I
,Num
,格式为A40,3X,A1,3X,'N=',I12。N=表明这是一个数组,
字串之后接数值的个数。从接下来的行开始是数组元素,格式为6I12。
实型数组:
Name,
R
,Num
,格式为A40,3X,A1,3X,'N=',I12,其中N=字串再次表明这
是一个数组,之后接数组元素的个数。从接下来的行开始是数组元素,格式为
5E16.8。注意,所选的实型格式保证了数组元素之间至少有一个空格,以便用C语
言程序读取数据。
字符串型数组(第一类):
Name,
C
,Num
,格式为A40,3X,A1,3X,'N=',I12,其中N=
字串表明这是一个数组,之后接数组元素的个数。从接下来的行开始是数组元素,
格式为5A12。
字符串型数组(第二类):
Name,
H
,Num
,格式为A40,3X,A1,3X,'N=',I12,其中N=
字串表明这是一个数组,之后接数组元素的个数。从接下来的行开始是数组元素,
格式为9A8。
逻辑型数组:
Name,
L
,Num
,格式为A40,3X,A1,3X,'N=',I12,其中N=字串表明这是
一个数组,之后接数组元素的个数。从接下来的行开始是数组元素,格式为72L1。
所有的量如果来自Gaussian计算,那么都是原子单位,位于标准方位。标准方位很少是
一个有趣的视觉角度,但它却是矢量场的自然方位。字段名称应当是详细信息,使之提供充
分信息,但也不必视为累赘,因为只有接口程序需要用到它们。随Gaussian一起发布的示例
程序demofc演示了如何提取命名的字段。
基组数据
基组信息以合理的一般方式提供,而非采用特殊的 Gaussian 的 Common /B/结构,因为
后者难于理解,之所以用它更多是历史原因而非为了清晰。基组数据包括标量,给出壳层个
数(
NShell
),最大的收缩度,出现的最高角动量,以及原始壳层个数(
NPrim
)。接下来的
数组包含:
壳层类型(NShell个数值):0=s,1=p,-1=sp,2=6d,-2=5d,3=10f,-3=7f
每个壳层的原函数个数(NShell个值)
壳层到原子的映射(NShell个值):每个壳层所属原子的编号
原函数指数(NPrim个值)
收缩因子(NPrim个值):每个归一化原始壳层的收缩因子。对任何S=P壳层包含S因子。
P(S=P)收缩因子(NPrim个值):S=P的p部分的收缩因子。如果没有S=P壳层则不出现。
对不属于S=P壳层部分的原函数,值为0
每个壳层的坐标:(3,NShell)数组,每个壳层的XYZ坐标
其它的数据,例如基函数索引数组,可以很容易地从上面得到。一个壳层内的基函数顺
序采用通常的 Gaussian 顺序:
S,X,Y,Z,XX,YY,ZZ,XY,XZ,YZ,XXX,YYY,ZZZ,XYY,XXY,XXZ,XZZ,YZZ,YYZ,XYZ
或
3ZZ-RR,XZ,YZ,XX-YY,XY,ZZZ-ZRR,XZZ-XRR,YZZ-YRR,XXZ-YYZ,XYZ,XXX-XYY,XXY-YYY
- 269 -
可用的项目
以下是当前定义的项目:
执行路径
完整标题
原子数
电荷
多重度
电子数
alpha 电子数
beta 电子数
基函数个数
收缩壳层个数
最高角动量
最大收缩度
原始壳层个数
维里比例
原子序号
核电荷
当前的笛卡尔坐标
Alpha 轨道能量
Beta 轨道能量
Alpha MO 因子
Beta MO 因子
壳层类型
每个壳层的原始函数个数
壳层到原子的映射
原函数指数
收缩因子
P(S=P) 收缩因子
每个壳层的坐标
总 SCF 密度
自旋 SCF 密度
总 MP2 密度
自旋 MP2 密度
总 CI 密度
自旋 CI 密度
总 CC 密度
自旋 CC 密度
笛卡尔力
笛卡尔力常数
偶极矩
偶极导数
- 270 -
极化率
偶极二阶导数
极化率导数
超极化率
常见问题
默认应当使用哪个能量?
Total Energy 字段具有用户请求的任何理论级别的能量。由于这个原因,其它程序不
必再判断
方法
字串的能量在哪。特别是我们可以加入新的方法,但不必修改逻辑判断去
寻找通常想要的能量。
为什么字段描述符包含数据类型信息?
对每一字段包含数据类型的目的是,如果对字段不感兴趣,可以很容易跳过,如下面演
示程序所示。
ECP 原子电荷是如何操作的?
若使用 ECP,“核电荷”将不同于原子序号。
会出现哪些密度字段?
总是出现总密度;仅对开壳层体系存储自旋密度。默认这是 SCF 密度。如果需要后-SCF
密度,需要在 Gaussian 输入中包含 Density 关键词。
何时会出现力常数?
出现的力常数可能为零,例如实际只计算一阶导的情况,或者仅在结构优化的第一点计
算力常数,此后不再计算的情况。如果任务类型为 Freq,它们将用于振动分析。
为什么没有壳层与原函数之间的映射关系数组?
需要指出,壳层到原函数之间的映射关系未明确给出,因此需要对每个原子分别存储原
函数数据,即使是某些原子具有相同的基组。原子具有相同基组的信息在 Gaussian 的
早期被丢弃,并且写文件时的时间不允许书写代码重新生成这一信息。只有当还使用了
轨道或密度时,才会对基组感兴趣。由于后者对分子大小呈二次方,因此对于大分子来
说,删除多余原函数的潜在省时似乎并不大。
例子
这个例子是三重态亚甲基 MP2/STO-3G 计算产生的格式化检查点文件:
Triplet Methylene
SP UMP2-FC STO-3G
Number of atoms I 3
IOpCl I 1
IROHF I 0
Charge I 0
Multiplicity I 3
Number of electrons I 8
Number of alpha electrons I 5
Number of beta electrons I 3
Number of basis functions I 7
- 271 -

Number of independent functions I 7
Number of point charges in /Mol/ I 0
Number of translation vectors I 0
Number of residues I 0
Number of secondary structures I 0
Number of symbols in /Mol/ I 0
Info1-9 I N= 9
8 8 0 0 0 111
1 2 1
Num ILSW I 100
ILSW I N= 100
1 0 0 0 2 0
0 0 0 0 0 -1
0 0 0 0 0 0
0 0 2 0 0 0
1 0 0 0 0 0
0 0 100000 0 -1 0
0 0 0 0 0 0
0 0 0 1 0 0
0 0 1 0 0 0
0 0 4 40 0 0
0 0 0 0 0 0
0 0 0 0 0 0
0 0 0 0 0 0
0 0 0 0 0 0
0 0 0 0 0 0
0 0 0 0 0 0
0 0 0 0
Num RLSW I 40
RLSW R N= 40
1.00000000E+00 1.00000000E+00 1.00000000E+00 1.00000000E+00 1.00000000E+00
0.00000000E+00 0.00000000E+00 1.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 1.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 1.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 1.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 1.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00 1.00000000E+00 1.00000000E+00
Number of contracted shells I 4
Highest angular momentum I 1
Largest degree of contraction I 3
Number of primitive shells I 12
Pure/Cartesian d shells I 0
Pure/Cartesian f shells I 0
Virial Ratio R 1.995814616819217E+00
SCF Energy R -3.843551207731927E+01
MP2 Energy R -3.845916855965901E+01
Total Energy R -3.845916855965901E+01
PUHF Energy R -3.843794045556613E+01
PMP2-0 Energy R -3.846088420969759E+01
Post-SCF wavefunction norm R 1.004516257004766E+00
S**2 R 2.014461674556861E+00
S**2 after annihilation R 2.000077930794892E+00
S**2 corrected to first order R 2.005942183926543E+00
RMS Density R 1.581648813313776E-09
Atomic numbers I N= 3
6 1 1
Nuclear charges R N= 3
- 272 -

6.00000000E+00 1.00000000E+00 1.00000000E+00
Current cartesian coordinates R N= 9
-6.16297582E-33 0.00000000E+00 2.55113028E-01 0.00000000E+00 1.76747490E+00
-7.65339084E-01 -2.16453248E-16 -1.76747490E+00 -7.65339084E-01
Atom Types C N= 3
Int Atom Types I N= 3
0 0 0
Force Field I 0
MM charges R N= 3
0.00000000E+00 0.00000000E+00 0.00000000E+00
Integer atomic weights I N= 3
12 1 1
Real atomic weights R N= 3
1.20000000E+01 1.00782504E+00 1.00782504E+00
Atom fragment info I N= 3
0 0 0
Atom residue num I N= 3
0 0 0
Nuclear spins I N= 3
0 1 1
Nuclear ZEff R N= 3
-3.60000000E+00 -1.00000000E+00 -1.00000000E+00
Nuclear QMom R N= 3
0.00000000E+00 0.00000000E+00 0.00000000E+00
Nuclear GFac R N= 3
0.00000000E+00 2.79284600E+00 2.79284600E+00
MicOpt I N= 3
-1 -1 -1
Constraint Structure R N= 9
-2.35838313E-33 0.00000000E+00 2.55113028E-01 -3.41380879E-32 1.76747490E+00
-7.65339084E-01 -2.16453248E-16 -1.76747490E+00 -7.65339084E-01
ONIOM Charges I N= 16
0 0 0 0 0 0
0 0 0 0 0 0
0 0 0 0
ONIOM Multiplicities I N= 16
3 0 0 0 0 0
0 0 0 0 0 0
0 0 0 0
Atom Layers I N= 3
1 1 1
Atom Modifiers I N= 3
0 0 0
Atom Modified Types C N= 3
Int Atom Modified Types I N= 3
0 0 0
Link Atoms I N= 3
0 0 0
Atom Modified MM Charges R N= 3
0.00000000E+00 0.00000000E+00 0.00000000E+00
Link Distances R N= 12
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00
MxBond I 2
NBond I N= 3
- 273 -

2 1 1
IBond I N= 6
2 3 1 0 1 0
RBond R N= 6
1.00000000E+00 1.00000000E+00 1.00000000E+00 0.00000000E+00 1.00000000E+00
0.00000000E+00
Shell types I N= 4
0 -1 0 0
Number of primitives per shell I N= 4
3 3 3 3
Shell to atom map I N= 4
1 1 2 3
Primitive exponents R N= 12
7.16168373E+01 1.30450963E+01 3.53051216E+00 2.94124936E+00 6.83483096E-01
2.22289916E-01 3.42525091E+00 6.23913730E-01 1.68855404E-01 3.42525091E+00
6.23913730E-01 1.68855404E-01
Contraction coefficients R N= 12
1.54328967E-01 5.35328142E-01 4.44634542E-01 -9.99672292E-02 3.99512826E-01
7.00115469E-01 1.54328967E-01 5.35328142E-01 4.44634542E-01 1.54328967E-01
5.35328142E-01 4.44634542E-01
P(S=P) Contraction coefficients R N= 12
0.00000000E+00 0.00000000E+00 0.00000000E+00 1.55916275E-01 6.07683719E-01
3.91957393E-01 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00
Coordinates of each shell R N= 12
-2.35838313E-33 0.00000000E+00 2.55113028E-01 -2.35838313E-33 0.00000000E+00
2.55113028E-01 -3.41380879E-32 1.76747490E+00 -7.65339084E-01 -2.16453248E-16
-1.76747490E+00 -7.65339084E-01
Alpha Orbital Energies R N= 7
-1.09934649E+01 -8.99280967E-01 -5.45391261E-01 -4.22615172E-01 -3.66596942E-01
6.71551044E-01 7.55382975E-01
Beta Orbital Energies R N= 7
-1.09350319E+01 -7.15686644E-01 -5.03082985E-01 3.05387126E-01 3.76749183E-01
7.45159737E-01 8.42123674E-01
Alpha MO coefficients R N= 49
9.92143921E-01 3.41561086E-02 0.00000000E+00 0.00000000E+00 -3.96834245E-03
-7.70608078E-03 -7.70608078E-03 -2.45353746E-01 7.88800679E-01 0.00000000E+00
0.00000000E+00 -1.51998297E-02 2.07368129E-01 2.07368129E-01 0.00000000E+00
0.00000000E+00 0.00000000E+00 5.81713695E-01 0.00000000E+00 4.03972000E-01
-4.03972000E-01 -7.91331978E-02 4.18822356E-01 0.00000000E+00 0.00000000E+00
8.40280330E-01 -2.41428132E-01 -2.41428132E-01 0.00000000E+00 0.00000000E+00
1.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
-1.73156512E-01 1.11677115E+00 0.00000000E+00 0.00000000E+00 -7.07495939E-01
-9.12363520E-01 -9.12363520E-01 0.00000000E+00 0.00000000E+00 0.00000000E+00
1.14003960E+00 0.00000000E+00 -8.92668310E-01 8.92668310E-01
Beta MO coefficients R N= 49
9.93625814E-01 2.80174965E-02 0.00000000E+00 0.00000000E+00 -3.90152527E-03
-6.44780850E-03 -6.44780850E-03 -1.95727052E-01 5.46887218E-01 0.00000000E+00
0.00000000E+00 -1.74242092E-01 3.44389565E-01 3.44389565E-01 0.00000000E+00
0.00000000E+00 0.00000000E+00 4.87826885E-01 0.00000000E+00 4.74684290E-01
-4.74684290E-01 -1.54812376E-01 7.12820567E-01 0.00000000E+00 0.00000000E+00
7.39978976E-01 -2.53618910E-01 -2.53618910E-01 0.00000000E+00 0.00000000E+00
1.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
-1.76706118E-01 1.11264520E+00 0.00000000E+00 0.00000000E+00 -7.93047228E-01
-8.66479868E-01 -8.66479868E-01 0.00000000E+00 0.00000000E+00 0.00000000E+00
1.18326077E+00 0.00000000E+00 -8.57160962E-01 8.57160962E-01
Total SCF Density R N= 28
2.07641142E+00 -2.71991893E-01 1.09865593E+00 6.74377463E-34 -4.51874795E-33
- 274 -

1.00000000E+00 0.00000000E+00 0.00000000E+00 -4.00593428E-32 5.76365893E-01
-3.64746665E-02 2.44402925E-01 -1.67021577E-32 3.77373431E-49 7.36693344E-01
-1.13232171E-01 2.50355011E-01 -3.08148791E-33 4.66559803E-01 -2.65970693E-01
6.08512766E-01 -1.13232171E-01 2.50355011E-01 1.23259516E-32 -4.66559803E-01
-2.65970693E-01 -1.68524337E-01 6.08512766E-01
Spin SCF Density R N= 28
2.52087462E-02 -1.13588463E-01 4.98914708E-01 1.79211577E-33 -8.04643841E-33
1.00000000E+00 0.00000000E+00 0.00000000E+00 -3.38963670E-32 1.00415753E-01
-9.69291361E-02 4.35203093E-01 -1.55789358E-32 3.77373431E-49 6.75942287E-01
3.43939549E-02 -1.25968187E-01 3.08148791E-33 3.43228637E-03 -1.46006689E-01
-7.94290766E-02 3.43939549E-02 -1.25968187E-01 6.16297582E-33 -3.43228637E-03
-1.46006689E-01 4.48345193E-02 -7.94290766E-02
Cartesian Gradient R N= 9
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
Grdnt Energy R 0.000000000000000E+00
Grdnt NVar I 2
Grdnt IGetFC I 0
Internal Forces R N= 2
0.00000000E+00 0.00000000E+00
Internal Force Constants R N= 3
0.00000000E+00 0.00000000E+00 0.00000000E+00
Mulliken Charges R N= 3
-9.27503627E-02 4.63751814E-02 4.63751814E-02
Full Title C N= 2
Triplet Methylene
Route C N= 3
#t mp2/sto-3g test geom=modela
External E-field R N= 35
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00
- 275 -
程序的开发功能
这一部分讲解的关键词和选项,对那些打算扩展或接口到Gaussian 09的开发人员有用。
它还讨论了非标准路径和确定标准方位。
程序开发有关的关键词
这里讲述的关键词和选项用于开发新的方法和其它调试目的,但不推荐用于产品级的计
算。
一般的任务重新开始
Restart 我们这里讨论 Restart 用于调试时的一般使用。产品级使用见 Restart 部分。
这个关键词通过重新使用读写文件来重新开始计算。Restart L1 的形式重
新使用读写文件,但产生新的执行路径。
使用原来执行路径的重新开始可以指定特定链接的发生次数,以及是否清除
或保留层和链接的临时文件,使用以下语法:
#P Restart [Ln[(
m
)]] [Clean|KeepOverlay|KeepAll]
如果指定了所有参数,在第
m
次调用链接
n
时重新开始任务。Clean 请求用
Link1 清除所有的执行路径和层的临时文件;KeepOverlay 请求保留层的临
时文件,但清除执行路径的临时文件;KeepAll 保留所有的临时文件。如果
对内部连接设置了读写文件,默认为 KeepAll,否则默认是 Clean。
IOP设定关键词
IOp1=
关键词
这个关键词控制操作系统接口的各种细节。这些选项是标准选项,但并非所
有选项在每一版本里都执行(或与之有关)。
选项
FileIODump 在每一链接的最后转存 FileIO 表。FDump 是这个关键词的同义词。
TimeStamp 打开时间印记。TStamp 是这个关键词的同义词。
FileIOPrint 在 FileIO 中打开附加的调试打印。
Synch 目前无操作(在一些测试任务中出现)。
NoDFTJ 对于非杂化 DFT,关闭使用纯库仑项(在正式任务中很少用到)。
AbelianOnly 强制使用阿贝尔对称性(在正式任务中很少用到)。
NoPackSort 在排序时关闭对地址的 32 位压缩,即使是排序的地址空间小于 231
(在正式任务中很少用到)。
- 276 -

IOp2 该选项设定动态分配的最大内存量。MDV 和Core 是IOp2 的同义词。
IOp33 它设定指定的标准调试打印选项。下面在层 2 的所有调用中设定 IOp(33)为3,
层 7 的所有调用中设定 IOp(33)为1:
IOp33(2=3,7=1)
《
Gaussian 09 IOp
参考》提供了所有内部选项(IOP)的说明。它们在我们的网站上也
有说明:www.gaussian.com/iops.htm
标准关键词的调试选项
CPHF 有以下选项用于调试:
选项
KeepMicro 在 CPHF 中保留所有的 EE 中心,即使是 Opt=CalcFC 或Opt=CalcAll
使用了非二次的微迭代,其中未用于内坐标的原子不必包含在
CPHF 中。
NoReuse 在频率计算的过程中,不重复使用二阶(核的)CPHF 中的电场 CPHF
解。默认是 ReUse。
XY 把实微扰和虚微扰一起处理。反义词是 NoXY,把它们分开处理。
默认是如果还做核微扰的话就分别处理,但若仅做电磁微扰就一起
处理。
ZVector 对后-SCF 梯度使用 Z 矢量方法[Diercksen81, Diercksen81a, Handy84]。如
果不需要 Hartree-Fock 二阶导的话,该选项是允许的并且是默认
的。NoZVector 关键词对后-SCF 梯度使用完整的 3×NAtoms CPHF。
FMM 有以下选项用于调试:
选项
LMax=
N
指定多极的最高阶。默认是 25。
Levels=
N
指定 FMM 使用的层数。默认对分子是 8,对 PBC 动态调节。
Tolerance=
N
指定精度级别为 10-N。
N
的默认值为 11,例外是 SCF 的第 0 步,默
认为 7。
JBoxLen=
N
在进行 J 时,设定最小箱长(尺寸)为 N/1000 Bohr。默认
N
为2.5。
如果同时进行 J 和 K,则使用 JBoxLen 和KBoxLen 的最大值。BoxLen
是JBoxLen 的同义词。
KBoxLen=
N
在进行 K 时,设定最小箱长(尺寸)为 N/1000 Bohr。默认
N
为 0.75。
如果同时进行 J 和 K,则使用 JBoxLen 和KBoxLen 的最大值。
AllNearField 在 FMM 中打开所有的近场。
NoParallelCPHF CPHF 阶段的 FMM 中禁止并行执行。NoParCPHF 是该选项的同义词。
Integral 有以下选项用于调试:
选项
CNDO 在主代码中用 CNDO/2 积分做计算。
INDO 在主代码中用 INDO/2 积分做计算。
ZIndo1 在主代码中用 ZIndo/1 积分做计算。
ZIndoS 在主代码中用 ZIndo/S 积分做计算。
DPRISM 对 spdf 积分的求导使用 PRISM 算法[Gill94]。这是默认的。
- 277 -
Rys1E 用 Rys 方法而非默认方法求解单电子积分[Dupuis76, King76, Rys83]。这
对内存很小的计算机是必要的。
Rys2E 若写双电子积分,则使用 Rys 方法(L314)[Dupuis76, King76, Rys83,
Schlegel84]。这比默认方法慢,但内存较小的计算机可能会需要,如
果请求常规(非 Raffenetti)积分会(由 NoRaff 选项)默认选择。
DSRys 使用标量 Rys 积分求导代码。可以与 Berny 合用,对 df 仅使用 Rys。
Berny 使用 Berny sp 积分求导和二阶导代码(L702)。
Pass Pass 指定积分通过磁盘存储到内存,NoPass 禁止这一功能。与
SCF=[No]Pass 同义,推荐使用。
NoJEngine 禁止使用特殊的库仑代码。
NoSP 在把积分写到磁盘时,不使用特殊的 sp 积分程序(L311)。
RevDagSam 在 Prism 中把对角取样的选择反向。
NoSchwartz 不使用 Schwartz 积分估算(即,仅使用试探集)。Schwartz 除了
使用试探集外,还使用 Schwartz 积分估算。默认为使用二者。
RevRepFock 反向 Scat20 与重复 Fock 矩阵的选择。
NoDFTCut 关闭额外的 DFT 截断。
SplitSP 把 AO S=P 壳层分裂到分别的 S 和 P 壳层。默认为 NoSplitSP。
SplitSPDF 把 AO S=P=D 和 S=P=D=F 壳层分裂到分别的 S=P,D,和 F。默认为
NoSplitSPDF。
SplitDBFSPDF 把密度 S=P=D 和 S=P=D=F 分裂到 S=P,D,和 F。默认为
NoSplitDBFSPDF。
NoGather 禁止使用聚散消解,即使是处理少数的密度矩阵。Splatter 是该
选项的同义词。
ForceNuc 用电子-电子做核-电子库仑计算。
SepJK 在 HF 和杂化 DFT 中分别作 J 和 K 用于测试。
Seq2E 设定并行双电子积分求解,但接下来并不以并行方式运行(用于调
试)。
SeqXC 设置并行 XC 积分求解,但接下来并不以并行方式运行(用于调试)。
SeqLinda 导致串行运行 Linda 工作程序。目前仅使 Linda 工作程序而非主程
序同时在主程序之前运行。
BigAtoms 在 XC 积分中使所有原子尺寸变大。
BigShells 在 XC 积分中使所有壳层尺寸变大。
NoSymAtGrid 不使用能够减少对称唯一原子的格点数的(阿贝尔)对称性。
LinMIO 在FoFCou 中转为线存储,用于测试。
RevDistanceMatrix
反向选择是否在数值积分中预计算距离矩阵。默认对分子预计算,
但对 PBC 不计算。
NoDynParallel 关闭动态工作分配。
Sparse 有以下选项用于调试:
选项
Loose 设定截断为 5 * 10-5。
Medium 设定截断为 5 * 10-7。这对半经验方法是默认的。
Tight 设定截断为 1 * 10-10。这对 DFT 方法是默认的。
N
设定截断为 1 * 10-N。
- 278 -
改变链接的调用和顺序
ExtraLinks 请求执行附加的链接。这些链接添加到常规链接后面所有层的项目中。
例如 ExtraLink=L9997 将导致 99 层的每一项目以这样的顺序包含链接:9999
(默认)和 9997。
ExtraOverlays
这一命令请求以非标准的执行路径格式读入额外的层卡,并立即插入到最后
一个卡(层 99)之前的标准执行路径。
Skip 在执行路径中跳过最初的几个层卡。
Skip=Ov
NNN
跳过所有层,直到首次出现层
NNN
。
Skip=
M
跳过前 M 个卡。
Use=L
nnn
通过程序指定另外的执行路径。
选项
L123 对 IRC 使用 L123 而不是 L115。除了 IRCMax 任务之外,这对 IRC
是默认的。
L402 对半经验使用旧链接 402 代码。
L503 对 SCF 使用链接 503。
L506 对 ROHF 使用链接 506。
指定非标准的执行路径
当所需的选项或链接的组合与标准执行路径有明显差异时,可读入一系列完整的层和具
有相应选项的链接。在任务类型输入部分,应以下面的指令行开始:
# NonStd
在这之后,按照执行的顺序每个需要的层占据一行,需要给出层编号,一个斜线/,需要的
选项,另一个斜线/,执行链接的列表,最后是一个分号:
层/选项=值,选项=值,.../链接,链接,...;
例如:
7/5=3,7=4/2,3,16;
表示以链接702,703,和716的次序进行计算,每一个链接中,选项5设置为3,选项7设置为
4。如果所有选项都使用默认值,则这一行应写成:
7//2,3,16;
执行路径描述的另一特点是
跳转行数
。它在链接列表行尾分号前的括号内给出。它表示
在完成当前层后,执行哪个层的行。如果省略,默认值是+0,表示程序进入列表的下一行(不
跳行)。另一方面,如果跳转行数设置为-4,例如
7//2,3,16(-4);
- 279 -
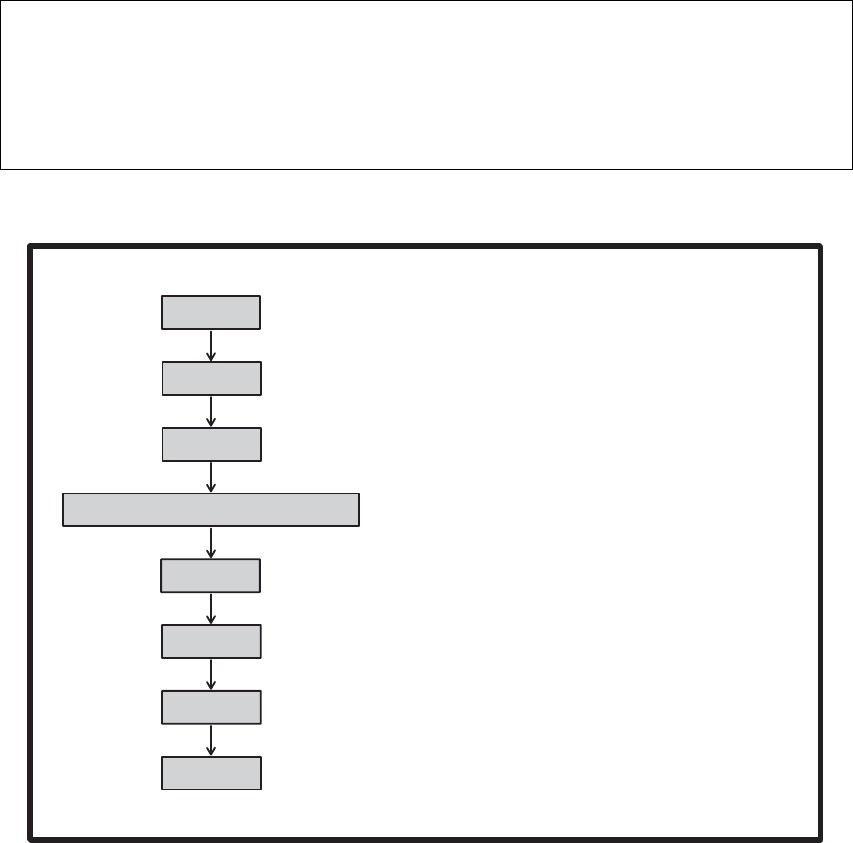
则表示完成本层后,将执行往前数到第四个路径行的层(本行不计)。
这个功能可以在执行路径部分建立循环结构,用于结构优化计算。连接路径的程序有一
个参数可以禁止跳跃。它在几何优化计算中对一系列层的行执行循环,直到优化完成为止,
这时再执行循环后的第一行计算。
注意,非标准执行路径通常并非从头创建,而是从最相似的标准计算产生的打印输出创
建,并修改执行的次序。这可以用工具程序testrt很容易地完成。
一个简单的执行路径示例。标准路径为:
# RHF/STO-3G
产生以下非标准执行路径:
1/38=1/1;
2/12=2,17=6,18=5,40=1/2;
3/6=3,11=1,16=1,25=1,30=1,116=1/1,2,3;
4//1;
5/5=2,38=5/2;
6/7=2,8=2,9=2,10=2,28=1/1;
99/5=1,9=1/99;
程序执行的顺序如下图所示。
L1
L101
L202
L301,L302,L303
L401
L502
L601
L9999
߃ຉे࠶డનى࠶
߃ຉ࠶Ᏻಿ
๕ު࠶Ᏻߖڮᅢ
ᎅᐉ˨ৼྛ݉ދᏳߢৡे࠶
ڄ༖ईݤ
੶
SCF
ࠝڴ
ᎅईݤेزऩݨߖڮᅢ˨ܸईݤ˨
جઌᅞ
Mulliken
نૅ࠶
֟བྷܬݡ
checkpoint
႖˨جੵདྷৼ
ྛ
除了链接1(用于读取并解释执行路径)先于实际的计算以外,程序执行的基本顺序与
任何其它从头计算程序相同,接下来是链接9999(写入检查点文件)。MP4单点计算类似,在
布居分析(链接601)和链接9999之前插入积分转换(链接801和804)和MP计算(链接913)。
链接9999在计算完成后自动终止任务步骤。
一个含有循环的执行路径例子。标准执行路径为:
- 280 -
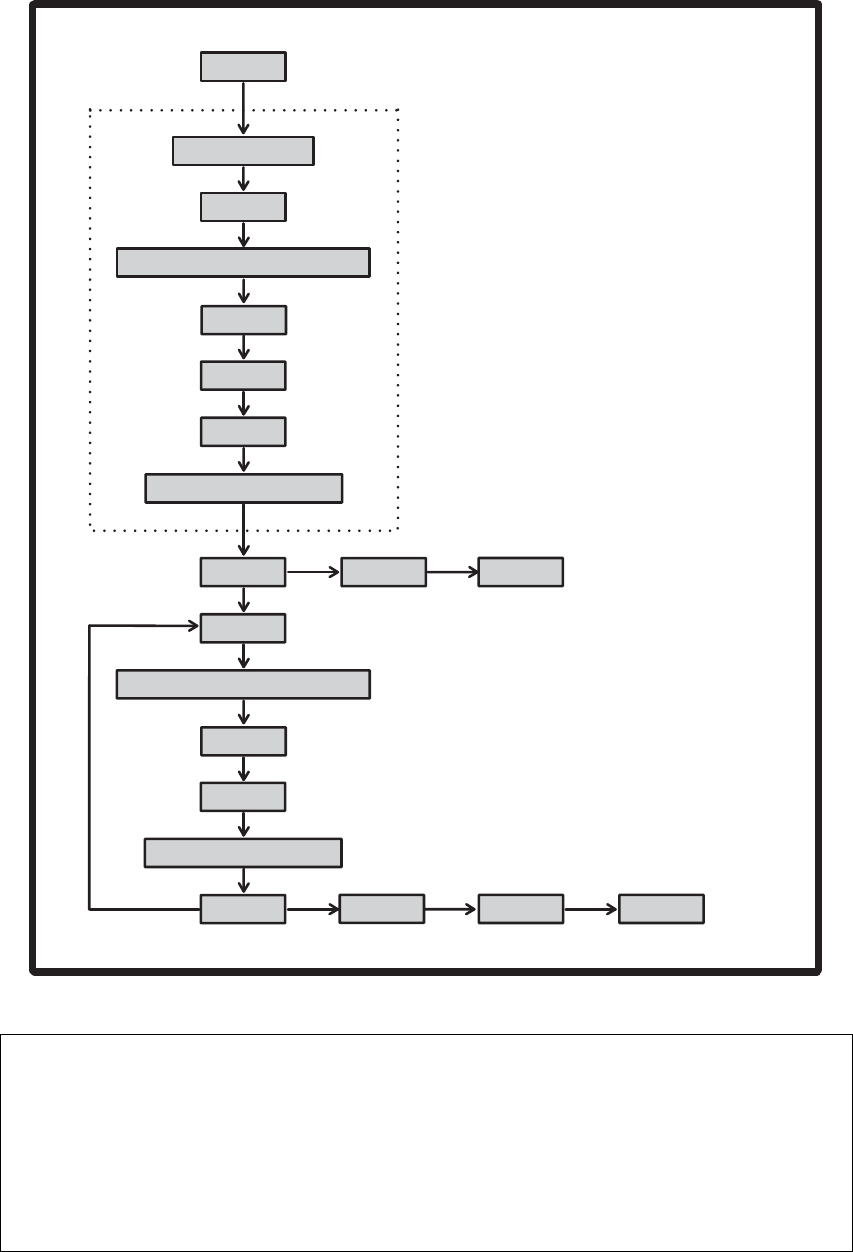
# RHF/STO-3G Opt
产生下面的非标准执行路径:
L1
L101, L103
L202
L301,L302,L303
L401
L502
L601
L701,L702,L703,L716
L103
L202
L301,L302,L303
L401
L502
L701,L702,L703,L716
L103
L202
L9999L601L202
ፆህى࠶ዠ
Opt=Restart
་ဎङ
L9999
1
1/18=20,19=15,38=1/1,3;
2
2/9=110,12=2,17=6,18=5,40=1/2;
3
3/6=3,11=1,16=1,25=1,30=1,71=1,116=1/1,2,3;
4
4//1;
5
5/5=2,38=5/2;
6
6/7=2,8=2,9=2,10=2,28=1/1;
7
7//1,2,3,16;
8
1/18=20,19=15/3(2);
9
2/9=110/2;
- 281 -
10
99//99;
11
2/9=110/2;
12
3/6=3,11=1,16=1,25=1,30=1,71=1,116=1/1,2,3;
13
4/5=5,16=3/1;
14
5/5=2,38=5/2;
15
7//1,2,3,16;
16
1/18=20,19=15/3(-5);
17
2/9=110/2;
18
6/7=2,8=2,9=2,10=2,19=2,28=1/1;
19
99/9=1/99;
程序的执行顺序如图所示。
使这一路径复杂化的几点考虑:
优化计算的第一点必须和以后的步骤分开处理,因为有几个操作只需做一次。这包括读
入初始的分子说明和产生初始轨道。
必需有一个分子构型的循环,用优化程序(本例中使用Berny优化程序链接103)决定是
否需要另一个结构,或者结构已被优化。
如果提供了收敛的结构,则程序计算一次能量梯度,在确认结构已优化后,退出。
默认只在第一个和最后一个分子结构进行布居分析并打印轨道,不要在不感兴趣的中间
结构进行这些操作。
第一个点的处理分成两个基本序列,由执行路径中一系列的积分、猜测、SCF、以及积
分求导组成。第一序列包括链接 101(读入初始结构),链接 103(进行自身的初始化),并
有选项集指示链接 401 产生初始猜测。第二序列在优化过程中使用由链接 103 产生的结构,
并且有选项集指示链接 401 取回前一结构的波函,用作下一步的初始猜测。
第八行指示向前跳行,表示如果链接 103 正常退出(没有任何特殊的操作),则跳过下
面的行(调用链接 202 和 9999)。通常在第二次调用链接 103 时,会检查最初的能量梯度,
然后选出新的结构。下一个要执行的是链接 202,用来处理新的结构,接着是第二个能量和
梯度序列中其余的计算,它们构成主要的优化循环。如果在链接 103 的第二次调用时发现结
构已收敛,则链接退出并产生禁止向前跳行的信号,然后调用下一行的链接 202,601,和
9999,完成整个计算。
第 10-16 行构成主要的优化循环。该循环计算积分、波函,以及优化计算中第二个点与
后面各点的梯度。它以链接 103 结束。如果结构还未收敛,链接 103 会选择新的结构并正常
结束计算,这会使执行次序在执行到第 16 行时发生跳行,下一步进行到第 11 行,开始新的
循环。如果链接 103 发现结构已收敛,将会退出并禁止跳行,执行结束行(17-19)。
最后一次调用的链接 601 打印最终的多极矩,如果需要的话还有轨道和布居分析。最后,
链接 9999 产生存档项目并结束任务步骤。
MP 和 CI 的优化计算需要在层 7 之前插入转换层和关联层(8 和 9),以及后-SCF 梯度层(按
照顺序 11 和 10)。用数值差分产生频率或极化率的计算也使用相同的两段路径结构。
Opt=Restart 的执行路径基本上就是原始优化计算的主循环部分,省略了第一步中的几
行。保留了对链接 103 的第二次调用,用于重新开始计算。
标准方位的约定
在执行计算前,分子可能被重定位到使用分子对称性的不同坐标系,称为标准方位。
这一部分说明分子标准方位的目的,考虑的因素,以及定位坐标轴的各种规则。
- 282 -
选择的目的
标准方位选择习惯的目的是:
通过重定位分子,简化 3×3转换矩阵。
两个 Z矩阵的内坐标值不同但整数的量相同(例如发生在结构优化随后的点上),应当
产生同样的标准方位。
同一分子的两个不同 Z矩阵应当产生同样的坐标,除了原子可能会重新编号。
利用对称性,使分子轨道因子为零的个数最大。
一般的考虑
标准方位的考虑因素有:
整个计算使用右手坐标系。
平移分子使之电荷中心位于原点。
相对于输入顺序,原子不做重新排序。
直角坐标轴按优先级增加的顺序是 X < Y < Z。
定位坐标轴的规则
转动和对齐坐标轴的标准列在下面。如果需要转动满足这些标准之一,那么它必须是绕
X,Y,或 Z轴的 180 度转动,定义如下:
X 绕Y旋转
Y 绕Z旋转
Z 绕X旋转
电荷的转轴或主轴可以按照两种方式之一与直角坐标轴对齐——平行或反平行,选择的
方式通过连续应用以下测试直到满足确定的结果为止:
原子序数最高原子的坐标在该轴的总和必须为正。
电荷的三阶矩必须为正。
原子坐标到参考轴投影的总和必须为正。
到参考轴具有非零投影的第一个原子在该轴必须有正投影。
定位电荷主轴的规则
在没有其它规则时,对应于电荷最大主矩的主轴必须与可用的优先级最高的直角坐标轴
对齐。不同点群有特殊的考虑:
Cs 分子平面必须与 XY 平面重合。注意,虽然这一约定与 Mulliken 的建议冲突,
但与 Cotton 和Herzberg 的特征标表一致。于是根据下面给出的 Cn分子规则
- 283 -
绕Z轴转动分子。
C2v 按照 Mulliken 对平面 C2v 分子的建议,分子平面位于 XY 平面。以下测试连
续应用于非平面分子:(1) 含原子最多的镜面位于 YZ 平面;(2) 含非氢原子
最多的镜面位于 YZ 平面;(3) 含最低编号原子的镜面与 YZ 重合;最后,应
用电荷的轴规则(说明见上)。
平面,D2h 按照 Mulliken 的建议,分子平面位于 YZ 平面。分子绕 X轴转动,因此 Z轴
要么穿过最多数量的原子,若不起决定作用,要么穿过穿过最多数量的键。
Cn 遵照一般的对称陀螺分子规则。
Ci 平移但不重定位。
C1 平移但不重定位。
对称陀螺分子的特殊规则
利用三个转动惯量中的两个相等,
可以识别对称陀螺分子。因此第三个转动惯量可唯一
地确定参考轴,通过考虑原子的循环集,可以分析点群。
以下规则用于对称陀螺分子:
对称唯一轴与 Z轴对齐。
原子循环集由位于平面具有相同原子序数的原子构成,并且到垂直平面的参考轴等距。
参考轴上的原子不包含在任何循环集中。原子的循环集由合适的转轴产生。
对称陀螺分子的关键原子是在关键循环集中具有最低编号的原子。通过连续应用以下测
试来寻找关键循环集:
哪个集合最接近 XY 平面?
哪个集合在 Z轴有正投影?
哪个集合最接近 Z轴?
哪个原子集合有最低的原子编号?
接下来对特定的点群选择方位:
Cn,Cnh,Sn:分子绕 Z轴转动,使平行于 Y轴的成对重原子数最大。若不满足这
种排列,则关键原子位于具有正 Z坐标的 YZ 平面。
Dn,Dnh:其中一个 C2轴与直角坐标 Y轴重合。下面的测试用于确定哪个 C2轴满
足这样的定位。
Dnd,Cnv:其中一个垂直平面与直角坐标 YZ 平面重合。下面的测试用于确定哪个
平面满足这样的定位。
以下是对 Dn,Dnh,Dnd,和 Cnv 分子选择轴的测试:
使关键原子在 Y轴的投影最大。
若两种方位都在 Y轴给出最大的投影,则选择在 X轴投影最大的一个。
若分子位于 XY 平面,则把应用标准轴方位规则(见上)用于 X轴,完成方位的
指定。
球陀螺分子的特殊规则
利用三个转动惯量都相等,可以确定球陀螺分子,它可以通过识别原子的球型集来标记。
原子的球型集由到原点等距,
具有相同原子序数的原子构成。球型集应当按照距原点距
- 284 -
离增加,同一距离原子序数增加的顺序排列。关键原子是第一个球型集内原子编号最低的一
个。
虽然不是一般情况,但在合适的结构束缚下,球陀螺还可能是 D2d,D2h,或 D2分子。
这样的分子具有三个相互垂直的二重轴,按照上述规则分别与 X,Y,和 Z轴对齐。
RWF编号
以下是读写文件的列表。永久保存在检查点文件中的文件用字母 P标记,临时保存在
检查点文件中的文件用字母 T标记。保存的 T文件用于重新开始优化或数值频率计算,但
当任务步骤成功完成后会被删除。
类型 读写文件 说明
P 501 Gen 数组。
P 502 /LABEL/——标题和原子轨道标记。
503 原子连接信息(MxBond,0),NBond(NAtoms),IBond(MxBond,NAtoms),
RBond(MxBond,NAtoms),其中的数组大小调整到 IntPWP 的倍数。
504 偶极导数矩阵(NTT,3,NAt3)。
P 505 来自势能面扫描的/Gen/数组拷贝。
P 506 在调整、非收缩等之前保存的基组信息。
P 507 /ZMAT/和/ZSUBST/。
P 508 /IBF/积分缓冲区格式。
509 不完全的积分缓冲区。
T 510 /FPINFO/ Fletcher-Powell 优化程序数据。
P 511 /GRDNT/ 能量对 NVAR 个变量的一阶和二阶导数。
P 512 赝势信息。
P 513 /DIBF/ 积分导数缓冲格式。
514 重叠矩阵,之后可以选择接绝对重叠和原函数的绝对重叠。
515 芯哈密顿量。这里有四个矩阵:H(α),α芯哈密顿量;H(β),β芯哈密
顿量;G'(α),α G'对Fock 矩阵的贡献;G'(β),β G'对Fock 矩阵的贡献。
H(α)和H(β)的不同之处仅仅在于是否加入了 Fermi 接触积分。G'矩阵用
于微扰,它在密度中是真正二次的(因此与真正的单电子项相比,其
对能量的贡献有因子 1/2),但在外部对 SCF 进行计算。
516 动能和对 α及β芯哈密顿量的修改。这包括 ECP 项,Douglas-Kroll-Hess
校正,多极微扰,以及 Fermi 接触微扰。后者用于核与电子库仑项一起
计算的情况,例如 Harris 泛函和 PBC 计算。对于半经验方法,在初始
猜测中使用没有核吸引项的芯哈密顿量。
517 Fermi 接触积分。
518 多极积分,顺序为:X,Y,Z,XX,YY,ZZ,XY,XZ,YZ,XXX,
YYY,ZZZ,XYY,XXY,XXZ,XZZ,YZZ,YYZ,XYZ,XXXX,
YYYY,ZZZZ,XXY,XXXZ,YYYX,YYYZ,ZZZX,ZZZY,XXYY,
XXZZ,YYZZ,XXYZ,YYXZ,ZZXY。
T 519 公共块/OptEn/——链接 109 的优化控制。
T 520 电子态:计数和压缩的字符串(1+9 个整数)。
- 285 -
P 521 电子态:计数和压缩的字符串(1+9 个整数)。
P 522 alpha 特征值,必要的话还有 beta。
523 对称性分配。
P 524 MO 因子:实 alpha。
P 525 (不再使用)
P 526 MO 因子:实 beta。
P 527 (不再使用)
T 528 SCF 密度矩阵:实 alpha。
T 529 (不再使用)
T 530 SCF 密度矩阵:实 beta。
T 531 (不再使用)
T 532 SCF 密度矩阵:实总和。
T 533 (不再使用)
T 534 SCF 密度矩阵:实自旋。
535 (不再使用)
536 Fock 矩阵:实 alpha。
537 Fock 矩阵:虚 alpha。
538 Fock 矩阵:实 beta。
539 Fock 矩阵:虚 beta。
540 分子的 alpha-beta 重叠(U),实。
541 分子的 alpha-beta 重叠(U),虚。
T 542 赝势信息。
T 543 赝势信息。
T 544 赝势信息。
P 545 /ORB/——窗口信息。
546 存储段条目的点。
547 特征值(双精度,带有窗口:总是 alpha 和beta,即使是在 RHF 的情
况下)。
P 548 MO 因子(双精度,带有窗口: alpha,必要的话还有 beta)。必要的话
为复值。
549 分子轨道的 alpha-beta 重叠,双精度,带有窗口。
T 550 势能面扫描公共块。
T 551 对称操作信息(排列,变换矩阵,等)。
P 552 包含化学式和框架群名称的字符串。
T 553 在FP 优化中临时存储公共块/gen/。
T 554 轮流的初始 MO 因子,从 L918 到L503,实 alpha。对 L509 还有以 S-1/2
为基的 MO 因子,和从 L914 到L508 的转动角度。
555 轮流的初始 MO 因子,从 L918 到L503,虚 alpha。
T 556 轮流的初始 MO 因子,从 L918 到L503,实 beta。对 L509 还有以 S-1/2
为基的 MO 因子,和从 L914 到L508 的转动角度。
557 轮流的初始 MO 因子,从 L918 到L503,虚 beta。
558 对G1,G2,等保存 HF 二阶导信息。
559 公共块/MAP/。
560 芯哈密顿量(AO 基)并加入了删除轨道(即,冻芯)的 2 j - k 部分。
- 286 -
P 561 外部点电荷或 SCIPCM 信息。
P 562 完全点群的对称操作和特征标表。
T 563 整数的对称性归属(α)。
T 564 整数的对称性归属(β)。
T 565 对称等价壳层和基函数的列表。
T 566 未在 G09 中使用。
T 567 GVB 对信息(目前仅有 100 个成对轨道尺寸)。
P 568 保存从 L504 到L506 的哈密顿量信息。
P 569 保存读入的窗口。
P 570 保存的幅度(IAS1,IAS2,IAD1,IAD2,IAD3;闭壳层仅有 IAS1 和IAD2)。
571 能量加权密度矩阵。
572 偶极-速率积分<Phi|Del|Phi'>,X,Y,Z分量,之后是 R x Del 积分(R
x X,R x Y,R x Z)。
573 更多 SCIPCM 信息。
T 574 /MSINFO/ Murtaugh-Sargent 程序数据。
T 575 /OPTGRD/ L103,L115,和 L509 的梯度优化程序数据。
T 576 /TESTS/ 控制 L105 中的常数。
T 577 对称匹配基函数数据。
T 578 逻辑矢量,表明哪些 MO 被占据。
T 579 NEQATM (NATOMS*NOP2)用于对称性。
T 580 NEQBAS (NBASIS*NOP2+NBas6D*NOp2)用于对称性。
T 581 NSABF (NBASIS*NOP2)用于对称性。之后是匹配整数的特征标表,总
是(8,8)。
T 582 MAPROT (3*NBASIS)用于对称性。
T 583 MAPPER (NATOMS)用于对称性。
P 584 FXYZ (3*NATOMS)笛卡尔力。在 PSCF 梯度运行中,这里有两个数组:
首先是 PSCF 梯度,接下来是仅有 HF 的分量(对于使用 HF 二阶导的
PSCF 是必须的)。
P 585 FFXYZ (NAT3TT)笛卡尔力常数(下三角)。
T 586 L106,L110,和 L111 的信息。
T 587 L107 (LST)数据。
588 AO 基的笛卡尔 Sx。
589 AO 基的笛卡尔 Hx。
590 AO 基的笛卡尔 Fx(都是 α,之后对 UHF 都是 β;没有 CPHF 项)。
591 U1(A,I)——MO 因子对电场和核坐标的导数。
592 电场和核 P1(AO 基)。
593 电场和核 W1(AO 基)。
594 电场和核 S1(MO 基)。
595 磁场 U1(A,I) -- Del(X,Y,Z),接下来是 R x (X,Y,Z),6 α之后是 6 β。
596 MO 基的完全 MO Fock 导数,包含 CPHF 项。
P 597 Guess=Alter 的组态变化。
598 用户名。
599 密度基组信息:NDBFn,NVar,U0,DenBfn(4,NDBfn),ITypDB(NDBfn),
Var(NVar),IJAnDB(NDBfn),IVar(4,NDBfn)。
- 287 -
600 保存数据,用于内部链接的重新开始。
P 601 保存沿反应路径的结构,以及可能的力和力常数。全部的结构,接下
来是全部的力和全部力常数。
602 后SCF 双粒子密度矩阵。
P 603 各种理论级别的密度矩阵。
T 604 来自 drt 程序的公共块/drt1/。杂项:整数 ci 项之后是可变维度的 drt 数
组。
P 605 来自 Mulliken 布居的原子电荷,ESP 拟合,等。数位映射之后是 0个
或更多的 NAtoms 数组。Bits 0/1/2/3/4 Mulliken/ESP-fit/Bader/NPA/APT。
606 阿贝尔点群的 SCF 轨道对称性。Alpha 以及可能的 beta 完全集,之后
是定义窗口的轨道集。
607 窗口轨道的对称性,类似于读写文件 606(总是分 alpha 和beta)。
608 IBF 用于排序的积分(通常位于 SAO 单元)。
609 用于排序积分的数位映射(通常位于 SAO 单元)。
610 排序的 AO 积分(通常位于 SAO 单元)。
611 用于排序积分的 NTT 映射(通常位于 SAO 单元)。
612 某些 1E 生成元,用于产生直接 CI 矩阵元。
613 更多的 1E 生成元,用于产生直接 CI 矩阵元。
614 CAS-MP2 的组态信息。
615-616 用于 CAS-MP2。
617 自旋-轨道积分。
P 618 核坐标的三阶导。
P 619 电场导数:1 WP 字的数位映射,偶极,偶极导数,极化率,偶极二阶
导,极化率导数,超极化率。
620 GIAO 的磁场导数。
621 极化率和化学位移张量。
622 部分重叠导数(<Mu|dNu/da>, NBasis*NBasis*NAt3)。
P 623 Born-Oppenheimer 波函导数(<Phi|d2Phi/dadb>对电子 Phi 和a,b 核,
NAt3TT)。
624 未在 G09 中使用。
625 展开来自 CPHF 的矢量和 AY 乘积,顺序为 Y α,AY α,Y β,AY β。
626 MCSCF MO 1PDM (NTT)。
627 MCSCF MO 拉格朗日量(NTT)
628 MCSCF MO 2PDM (NTT,NTT)或NVTTTT。
629 AO 2PDM(壳层顺序)。
T 630 MCSCF 信息。
631 后SCF 拉格朗日量(TA,若 UHF 接下来是 TB)。
632 O*V*3*NAtoms,之后是 O*V*NVar d2E/d(V,O)d(XYZ,Atom)。
P 633 激发态 CI 密度。
T 634 SCF 重新开始信息(alpha MO,接下来可能有 beta MO)。
P 635 CIS 和CASSCF 的CI 因子和重新开始信息。
636 NBO 分析信息。
637 链接 601 产生的自然轨道。
640 MCSCF 数据或 CIS AO Tx,用于二阶导。
- 288 -
641 MCSCF 数据用于二阶导。
642 MCSCF 数据用于二阶导。
643 MCSCF 数据用于二阶导。
644 MCSCF 数据用于二阶导。
645 MCSCF 数据用于二阶导。
646 MCSCF 数据用于二阶导。
647 MCSCF 数据用于二阶导。
648 MCSCF 数据用于二阶导。
649 特征值导数(即使是正则计算,也使用非正则形式)。
650 2PDM 导数,顺序为 LenTQ,NDeriv,ShellQuartet。
651 完全的 U,根据需要可以是正则或非正则。
652 产生当前方法的密度矩阵(NTT,NDeriv,IOpCl+1)。
653 当前方法的拉格朗日导数(NTT,NDeriv,IOpCl+1)。
654 Gx(Gamma)。
655 G(Gamma)。
656 MP2 或CIS 二阶导拉格朗日量的非对称 S1 和S2 部分。
657 来自 L811 或L1112 的t*Ix 和t*Ix/D。
658 来自 L1111 的L(x)。
659 MO 有关的 W,用于有关的频率。
660 二阶 CPHF 结果:Pia,xy, Sxy, Fxy,全部以 MO 基,PSF α,如果是 UHF
接下来是 PSF β。
661 从L602 计算的电场。
662 用于静电求解的点。
T 663 为L117 和L124 保存信息。
664 自旋投影数据。
P 665 冗余坐标信息。
666 (不再使用)
667 CIS AO Fock 矩阵
668 CIS Gx(T)矩阵。
669 在冗余优化过程中保存的/ZMat/和/ZSubst/。
P 670 新格式的基组数据(压缩的/B/)。
P 671 新的优化(L103/L104)数据。
P 672 未在 G09 中使用。
673 全局优化数据。
674 ONIOM 内部数据。
675 保存的文件用于 ONIOM 过程中的 LS。
676 保存的文件用于 ONIOM 过程中的 MS。
677 保存的文件用于 ONIOM 过程中的 LM。
678 保存的文件用于 ONIOM 过程中的 HS。
679 保存的文件用于 ONIOM 过程中的 MM。
680 保存的文件用于 ONIOM 过程中的 LL。
681 保存的文件用于 ONIOM 过程中的 HM。
682 保存的文件用于 ONIOM 过程中的 ML。
683 保存的文件用于 ONIOM 过程中的 HL。
- 289 -
684 用于 DBFS 的SABF 信息:对于 AO 等价于文件 577 和581。
685 Cholesky U,或者到残存基函数的转换。
686 Cholesky U-1。
687 分子力学参数。
688 正交基(α自旋)的密度用于 ADMP 或稀疏 SCF。
691 保存 ONIOM 过程中的初始文件(格点 17,因此 674+17=691)。
694 排列应用于 MO,用于后 SCF 对称性。
695 分子特性。
696 保存磁场密度导数。
698 保存结构优化的初始结构,并且为标准方位,它还用于跟踪结构的力
常数的束缚优化。
699 正交基(β自旋)的密度用于 ADMP 或稀疏 SCF。
700 保存的/Mol/用于 ONIOM。
701 保存轨迹/ IRC/优化的历史。
702 用于库仑的拟合密度。
703 用于库仑的拟合密度。
704 把XC 贡献保存到电场 F(xa)用于极化导数。
P 710 基本 PCM 信息。
P 711 其它 PCM 数据。
P 712 用于 PCM 的非平衡数据。
713 保存的信息用于含有 ONIOM 微迭代的 RFO。
714 保存的模型体系信息用于 ONIOM 微迭代。
715 保存的刚性片段信息用于 ONIOM 微迭代。
T 716 保存基组数据的拷贝用于基组平衡计算。
T 717 保存 ECP 数据的拷贝用于基组平衡计算。
T 718 保存拟合基的拷贝用于基组平衡计算。
719 保存的 DiNa 信息。
P 720 保存的 DiNa 信息。
721 含频特性。
722 含频特性的导数。
723 密度拟合矩阵(度量)。
724 密度拟合基(格式同/B/)。
725 DBF 对称性信息(NEqDBF(NDBF,NOp2),NEqDB6(NDBF6D,NOp2))。
726 DBF 壳层对称性信息(NEqDBS(NDBShl,NOpAll))。
727 F(x)(P-Pfit)用于密度拟合二阶导。
728 PBC 晶胞的重复信息。
729 优化过程中另一新的猜测。
730 基组平衡的输入规格。
731 基组平衡计算的中间数据。
732 有限核的基组。
733 PBC 晶胞的标量,以及整数晶胞指数。
734 SAC-CI 的特定态的输入参数。
735 SAC 和SAC-CI 的激发标记。
736 SAC 和SAC-CI 的特征值和特征矢。
- 290 -
737 H 矩阵及其非零元的指数,用于 SAC/SAC-CI。
738 保存的原子参数用于 DFTB/EHTSC。
739 临时存储,用于芯哈密顿量微扰的虚部。
740 用于 SAC/SAC-CI 梯度和 GSUM 计算 PES 的轨道信息。
741 用于 SAC 梯度的 MOD 轨道信息。
742 保存的积分格点。
743 正交基的 alpha Fock 矩阵用于 ADMP,以及非 HF 后SCF 方法的 alpha
HF Fock 矩阵。
744 正交基的 beta Fock 矩阵用于 ADMP,以及非 HF 后SCF 方法的 beta HF
Fock 矩阵。
745 K 积分网格信息。
746 全部 k点上的特征值和轨道。
747 在ONIOM 方法中,外部低级别计算的信息。
748 ONIOM TS 优化的 TS 矢量信息。
749 ONIOM 的圆锥交点信息。
750 未在 G09 中使用。
751 SO ECP 积分的临时存储。
752 赝正则 MO Fock 矩阵,用于 ROMP 和ROCC。
753 FD 极化导数的数据。
754 保存的 PCM 电荷导数。
755 PCM 逆矩阵。
756 ONIOM 的电荷信息。
757 用于 L924 的MO:MO 嵌入电荷数据。
758 当直接计算时,嵌入电荷的导数。
759 密度嵌入的基组信息。
P 760 一整套赝势轨道用于 RO。
761 来自外部 PCM 迭代的电荷(L117 和L124)。
762 保存非对称 Mulliken 分析的权重。
763 用于 FC/HT 积分的文件。
764 用于 FC/HT 积分的文件。
P 765 保存简正模式。
766 保存 QuadMac 矢量(临时的)。
767 按照对称性重新排序的 CIS 因子。
768 半经验参数。
769 保存数值差分过程中的 MO。
P 770 保存基态到激发态的能量和跃迁矩。
771 EOM 迭代信息。
772 阿贝尔群的对称操作和特征标表。
989 多步任务信息(1000 个实数,2000 个整数)。
990 在某些执行中的 KJob 信息。
991 保存文件名,ID,并保存标记。
992 在某些执行中用于链接替代信息。
993 公共块/INFO/。
994 公共块/PHYCON/。
- 291 -

995 公共块/MUNIT/。
996 公共块/IOP/。
P 997 公共块/MOL/。
P 998 公共块/ILSW/。
999 层的数据。
作废的关键词和过时的功能
下表列出了从前Gaussian版本中使用过的作废关键词。虽然Gaussian 09仍支持全部这
些关键词,但我们强烈建议使用表中列出的最新的等价关键词。
作废的关键词 替换的关键词和选项
Alter
BD-T
BeckeHalfandHalf
Camp-King
CCSD-T
CubeDensity
GridDensity
Cube
DIIS
Direct
Formchk
Guess=Restart
MP2=Stingy和VeryStingy
NoDIIS
NoExtrap
NoRaff
OldConstants
Opt=AddRedundant
OptCyc=
n
PlotDensity
Prop=Grid
QCID
QCISD-T
QCSCF
Raff
Save
SCFCon=
n
SCFCyc=
n
SCFDM
SCFQC
SCRF=Checkpoint
Guess=Alter
BD(T)
BHandH
SCF=Camp-King
CCSD(T)
cubegen工具
cubegen工具
cubegen工具
SCF=Fermi
SCF=Direct(默认)
formchk工具
SCF=Restart
无(选项无操作)
SCF=NoDIIS
SCF=NoExtrap
Int=NoRaff
Constants=1979
Opt=ModRedundant
Opt(MaxCycle=
n
)
cubegen工具
cubegen工具
CCD
QCISD(T)
SCF=QC
Int=NoRaff
无
(Save
无操作
)
SCF(Conver=
n
)
SCF(MaxCyc=
n
)
SCF=DM
SCF=QC
Field=EChk
- 292 -
VShift[=
n
]
%NProcLinda
-L-
%NProc
chkmove工具
SCF(VShift[=
n
])
%LindaWorkers
-W-
%NProcShared
formchk和unfchk
作废的工具
chkmove 工具用于不同计算机架构之间的检查点文件在二进制和文本格式之间相互转
化,现在不再提供。它的功能由 formchk 和unfchk 控制。
过时的功能
NoFMM 这一关键词禁止使用FMM的功能,即使是它能改善执行。当在集群或装有Linda
的LAN上并行运行Gaussian 03时,在某些情况下需要该选项。由于已经修复
了有关的问题,故不再需要此关键词。
CCD+STCCD 指定耦合簇计算使用双取代,并使用CCD波函通过四激发求解单、三激发的贡
献。它被CCSD(T)代替。
CBS-Q, CBS-Lq
请求CBS-Q [Ochterski96]和CBS-q [Petersson91a]方法(即,Lq表示“小q”)。它
们被CBS-QB3代替。
CBS-QB3O 使用CBS-QB3的原始参数化[Montgomery99]。它已作废,仅用于向下兼容。
CBS-4O 请求CBS-4的原始参数化[Ochterski96]。它已作废,仅用于向下兼容。
Geom=Coord 表明结构说明为笛卡尔坐标。笛卡尔坐标可以包含在分子说明部分,无需任
何特殊的选项。
Geom=OldRedundant
使用Gaussian 94的冗余内坐标生成程序。
Geom=ModLargeRedundant
对Opt=Big使用最小设置。它不能用于周期边界计算。
Int=Raff 仅用于SCF=Conventional (不推荐)。Raff 对使用的双电子积分需要
Raffenetti格式[Raffenetti73]。NoRaff要求使用常规的积分格式。在直接CPHF
过程中它还禁止使用Raffenetti积分。这会影响传统的SCF以及传统的和直接
的频率计算。
Int=BWeights
对于数值积分使用Becke的加权方案。
ReUse 使用存在的积分文件。积分文件和检查点文件必须已在前一计算中保存。仅
允许单点计算和Polar=Restart。
WriteD2E 在HF频率计算中,强制写入内部导数文件。仅用于调试新的导数代码。
LST和LSTCyc
对要产生的过渡结构使用线性同步过渡[Halgren77]获得初始猜测。LST程序沿着
连接两个结构的路径定位最大值,并对连接它们的过渡结构产生猜测。
注意:
- 293 -
LST计算实际上并不能定位合适的过渡态。
LST方法已经被Opt=QST2代替。
Massage Massage关键词用于在分子说明部分和基组数据部分产生后,再进行修改。
不赞成使用,推荐用ExtraBasis,Charge,Counterpoise和其它的关键词。
Opt=EnOnly 使用固定Hessian和能量数值差分产生梯度的赝Newton-Raphson方法进行优
化。该选项要求Hessian用ReadFC或RCFC读入。它可以用来定位过渡结构和
较高的鞍点。要求用Z矩阵指定分子。对仅有能量的方法默认为
Opt=(EnOnly,EF)。
Opt=FP 使用Fletcher-Powell优化算法[Fletcher63],它不需要解析梯度。
Fletcher-Powell优化允许的最大变量个数是30。要求用Z矩阵指定分子。
Opt=Grad 使用默认方法进行梯度优化,除非定义了其它选项。只要可以使用解析梯度,
这就是默认的,其它情况无效。
Opt=MNDOFC 进行MNDO (或AM1,如果可能的话)力常数计算,并用于开始(可能是从头计
算的)几何优化。我们推荐执行PM6 Freq计算,之后接Opt=RCFC而非此选项。
Opt=MS 指定Murtaugh-Sargent优化算法[Murtaugh70]。Murtaugh-Sargent优化算法是作
废的选项,在Gaussian 09中保留仅为向下兼容。Murtaugh-Sargent优化允许
的最大变量个数是50。要求用Z矩阵指定分子。
Opt=UnitFC 要求对Hessian使用单位矩阵而非通常的价电子力场猜测。
Opt=GDIIS 指定使用修改的GDIIS算法[Csaszar84, Farkas95, Farkas99]。默认的GEDIIS算法总
是更好。
Opt=Big 请求对坐标变换用快速方程求解方法[Farkas98]以及Newton-Raphson或RFO走
步进行优化。这一方法避免了矩阵对角化。因此,特征矢跟踪方法(Opt=TS)
不能与之结合使用。该选项不可靠,不推荐。
Output=PolyAtom
请求单变量积分文件输出,使用PolyAtom积分程序的格式。默认产生Caltech
MQM程序的格式,但是可以很容易地通过修改链接9999的代码来产生同样内容
的其它类型格式。
Output=Trans
用Caltech (Tran2P5)格式写MO因子文件。只对Caltech程序的用户有用。
SCRF=DPCM 使用极化电介质模型[Miertus81, Miertus82, Cossi96],除了某些小的执行细节不同
以外,它对应于Gaussian 98的SCRF=PCM选项[Cossi02]。对一般的使用,不推
荐这一模型。默认的SCRF方法是IEFPCM。
SCRF=Numer 进行数值SCRF计算而不是解析计算。这个关键词用于Dipole以上的多极计算。
该选项意味着使用球形空穴,不推荐使用。这个选项没有梯度。
SCRF=Dipole Dipole,Quadrupole,Octopole和Hexadecapole选项指定在SCRF计算中使用
的多极的级别。除了Dipole之外的所有选项都需要同时指定Numer选项。
SCRF=Cards 在指定介电常数和半径(三个自由格式的实数)的行之后,马上从输入资料
读入以前计算的反应场,开始SCRF=Numer计算。
%SCR 用于指定.SCR临时文件的位置。
Stable=Symm 维持对称性限制。默认的NoSymm放松对称性限制。
Transformation=Old2PDM
在后SCF梯度中(在L1111中排序,接下来在L702和L703中进行处理),强 制 按
旧的方式处理2PDM。这非常慢,但会减少内存需求。该选项不能用于冻芯计
算。
Transformation=New2PDM
- 294 -
导致产生并使用2PDM,并在后SCF梯度计算中用L1111丢弃2PDM。
Transformation=Conventional
请求使用原始转换方法,它基于使用外部存储的积分。
- 295 -
- 296 -
参考文献
Abegg74 P. W. Abegg and T.-K. Ha, “Ab initio calculation of spin-orbit-coupling constant from
Gaussian lobe SCF molecular wavefunctions,” Mol. Phys., 27 (1974) 763-67.
Abegg75 P. W. Abegg, “Ab initio calculation of spin-orbit-coupling constants for Gaussian lo
b
e and
Gaussian-type wave-functions,” Mol. Phys., 30 (1975) 579-96.
Abraham93 M. H. Abraham, “Scales of solute hydrogen-
b
onding: their construction and application to
physicochemical and biochemical processes,” Chem. Soc. Reviews, 22 (1993) 73.
Adamo90 C. Adamo, M. Cossi, N. Rega, and V. Barone, in Theoretical Biochemistry: Processes and
Properties of Biological Systems, Theoretical and Computational Chemistry, Vol. 9
(Elsevier, New York, 1990).
Adamo97 C. Adamo and V. Barone, “Toward reliable adiabatic connection models free from
adjustable parameters,” Chem. Phys. Lett., 274 (1997) 242-50.
Adamo98 C. Adamo and V. Barone, “Exchange functionals with improved long-range behavior and
adiabatic connection methods without adjustable parameters: The mPW and mPW1PW
models,” J. Chem. Phys., 108 (1998) 664-75.
Adamo98a C. Adamo and V. Barone, “Implementation and validation of the Lacks-Gordon exchange
functional in conventional density functional and adiabatic connection methods,”
J
. Comp.
Chem., 19 (1998) 418-29.
Adamo99a C. Adamo and V. Barone, “Toward reliable density functional methods without adjustable
parameters: The PBE0 model,” J. Chem. Phys., 110 (1999) 6158-69.
Almlof82 J. Almlöf, K. Korsell, and K. Fægri Jr., “Principles for a direct SCF approach to LCAO-MO
ab-initio calculations,” J. Comp. Chem., 3 (1982) 385-99.
Amos82 R. D. Amos, “Electric and Magnetic Properties of CO, HF, HCl and CH3F,” Chem. Phys.
Lett., 87 (1982) 23-26.
Amos84 R. D. Amos, “Dipole-moment derivatives of H2O and H2S,” Chem. Phys. Lett., 108 (1984)
185-90.
Anders93 E. Anders, R. Koch, and P. Freunscht, “Optimization and application of lithium parameters
for PM3,” J. Comp. Chem., 14 (1993) 1301-12.
Anderson86 W. P. Anderson, W. D. Edwards, and M. C. Zerner, “Calculated Spectra of Hydrated Ions o
f
the First Transition-Metal Series,” Inorganic Chem., 25 (1986) 2728-32.
Andrae90 D. Andrae, U. Haeussermann, M. Dolg, H. Stoll, and H. Preuss, “Energy-adjusted ab initio
pseudopotentials for the 2nd and 3rd row transition-elements,” Theor. Chem. Acc., 77 (1990)
123-41.
Andzelm92 J. Andzelm and E. Wimmer, “Density functional Gaussian-type-orbital approach to
molecular geometries, vibrations, and reaction energies,” J. Chem. Phys., 96 (1992)
1280-303.
Atkins69 P. W. Atkins and L. D. Barron, “Rayleigh Scattering of Polarized Photons by Molecules,”
Mol. Phys., 16 (1969) 453.
Austin02 A. J. Austin, M. J. Frisch, J. A. Montgomery Jr., and G. A. Petersson, “An overlap criterion
for selection of core orbitals,” Theor. Chem. Acc., 107 (2002) 180-86.
- 297 -
Autschbach02 J.
Autschbach, T. Ziegler, S. J. A. van Gisbergen, and E. J. Baerends, “Chiroptical properties
from time-dependent density functional theory. I. Circular dichroism spectra of organic
molecules,” J. Chem. Phys., 116 (2002) 6930-40.
Ayala97 P. Y. Ayala and H. B. Schlegel, “A combined method for determining reaction paths, minima
and transition state geometries,” J. Chem. Phys., 107 (1997) 375-84.
Ayala98 P. Y. Ayala and H. B. Schlegel, “Identification and treatment of internal rotation in normal
mode vibrational analysis,” J. Chem. Phys., 108 (1998) 2314-25.
Baboul97 A. G. Baboul and H. B. Schlegel, “Improved Method for Calculating Projected Frequencies
along a Reaction Path,” J. Chem. Phys., 107 (1997) 9413-17.
Baboul99 A. G. Baboul, L. A. Curtiss, P. C. Redfern, and K. Raghavachari, “Gaussian-3 theory using
density functional geometries and zero-point energies,” J. Chem. Phys., 110 (1999) 7650-57.
Bacon79 A. D. Bacon and M. C. Zerner, “An Intermediate Neglect of Differential Overlap Theory for
Transition Metal Complexes: Fe, Co, and Cu Chlorides,” Theor. Chem. Acc., 53 (1979)
21-54.
Bacskay81 G. B. Bacskay, “A Quadratically Convergent Hartree-Fock (QC-SCF) Method. Application
to Closed Systems,” Chem. Phys., 61 (1981) 385-404.
Bak93 K. L. Bak, P. Jørgensen, T. Helgaker, K. Ruud, and H. J. A. Jensen, “Gauge-Origin
Independent Multiconfigurational Self-Consistent-Field Theory for Vibrational
Circular-Dichroism,” J. Chem. Phys., 98 (1993) 8873-87.
Bak94 K. L. Bak, P. Jørgensen, T. Helgaker, and K. Ruud, “Basis Set Convergence and Correlation
Effects in Vibrational Circular Dichroism Calculations Using London Atomic Orbitals,”
Faraday Discuss., 99 (1994) 121.
Bak95 K. L. Bak, A. E. Hansen, K. Ruud, T. Helgaker, J. Olsen, and P. Jørgensen, “Ab Initio
Calculation of Electronic Circular-Dichroism for trans-Cyclooctene Using London Atomic
Orbitals,” Theor. Chem. Acc., 90 (1995) 441-58.
Baker86 J. Baker, “An algorithm for the location of transition-states,” J. Comp. Chem., 7 (1986)
385-95.
Baker87 J. Baker, “An algorithm for geometry optimization without analytical gradients,”
J
. Comp.
Chem., 8 (1987) 563-74.
Baker93 J. Baker, “Techniques for geometry optimization - a comparison of cartesian and natural
internal coordinates,” J. Comp. Chem., 14 (1993) 1085-100.
Bakken99 V. Bakken, J. M. Millam, and H. B. Schlegel, “Ab initio classical trajectories on the
Born-Oppenheimer Surface: Updating methods for Hessian-Based Integrators,”
J
. Chem.
Phys., 111 (1999) 8773-77.
Banerjee85 A. Banerjee, N. Adams, J. Simons, and R. Shepard, “Search for Stationary Points on
Surfaces,” J. Phys. Chem., 89 (1985) 52-57.
Barnes09 E. C. F. Barnes, M.J., G. A. Petersson, and J. M. L. Martin, (2009) in prep.
Barone02 V. Barone, J. E. Peralta, R. H. Contreras, and J. P. Snyder, “DFT Calculation of NMR JFF
Spin-Spin Coupling Constants in Fluorinated Pyridines,” J. Phys. Chem. A, 106 (2002)
5607-12.
Barone03 V. Barone, M. Cossi, N. Rega, and G. Scalmani, “Energies, structures, and electronic
properties of molecules in solution with the C-PCM solvation model,” J. Comp. Chem., 24
(2003) 669-81.
- 298 -
Barone04 V. Barone, “Vibrational zero-
p
oint energies and thermodynamic functions beyond the
harmonic approximation,” J. Chem. Phys., 120 (2004) 3059-65.
Barone05 V. Barone, “Anharmonic vibrational properties by a fully automated second-order
perturbative approach,” J. Chem. Phys., 122 (2005) 014108: 1-10.
Barone09 V. Barone, J. Bloino, M. Biczysko, and F. Santoro, “Fully integrated approach to compute
vibrationally resolved optical spectra: From small molecules to macrosystems,”
J
. Chem.
Theory and Comput., 5 (2009) 540-54.
Barone94 V. Barone, “Characterization of the potential energy surface of the HO2 molecular system
by a density functional approach,” J. Chem. Phys., 101 (1994) 10666-76.
Barone95 V. Barone and C. Minichino, “From concepts to algorithms for the treatment of
large-amplitude internal motions and unimolecular reactions,” J. Mol. Struct. (Theochem),
330 (1995) 365-76.
Barone96 V. Barone, “Electronic, vibrational and environmental effects on the hyperfine coupling
constants of nitroside radicals. H2NO as a case study,” Chem. Phys. Lett., 262 (1996)
201-06.
Barone96a V. Barone, in Recent Advances in Density Functional Methods, Part I, Ed. D. P. Chong
(World Scientific Publ. Co., Singapore, 1996).
Barone97 V. Barone, M. Cossi, and J. Tomasi, “A new definition of cavities for the computation of
solvation free energies by the polarizable continuum model,” J. Chem. Phys., 107 (1997)
3210-21.
Barone98 V. Barone and M. Cossi, “Quantum calculation of molecular energies and energy gradients
in solution by a conductor solvent model,” J. Phys. Chem. A, 102 (1998) 1995-2001.
Barone98a V. Barone, M. Cossi, and J. Tomasi, “Geometry optimization of molecular structures in
solution by the polarizable continuum model,” J. Comp. Chem., 19 (1998) 404-17.
Barron04 L. D. Barron, Molecular Light Scattering and Optical Activity, 2nd ed. (Cambridge
University Press, Cambridge, UK, 2004).
Barron71 L. D. Barron and A. D. Buckingham, “Rayleigh and Raman Scattering from Optically
Active Molecules,” Mol. Phys., 20 (1971) 1111.
Bartlett78 R. J. Bartlett and G. D. Purvis III, “Many-body perturbation-theory, coupled-
p
air
many-electron theory, and importance of quadruple excitations for correlation problem,” Int.
J. Quantum Chem., 14 (1978) 561-81.
Barysz01 M. Barysz and A. J. Sadlej, “Two-component methods of relativistic quantum chemistry:
From the Douglas-Kroll approximation to the exact two-component formalism,”
J
. Mol.
Struct. (Theochem), 573 (2001) 181-200.
Bauernschmitt96 R. Bauernschmitt and R. Ahlrichs, “Stability analysis for solutions of the closed shell
Kohn-Sham equation,” J. Chem. Phys., 104 (1996) 9047-52.
Bauernschmitt96a R. Bauernschmitt and R. Ahlrichs, “Treatment of electronic excitations within the adiabatic
approximation of time dependent density functional theory,” Chem. Phys. Lett., 256 (1996)
454-64.
Bearpark94 M. J. Bearpark, M. A. Robb, and H. B. Schlegel, “A Direct Method for the Location of the
Lowest Energy Point on a Potential Surface Crossing,” Chem. Phys. Lett., 223 (1994)
269-74.
- 299 -
Becke88b A. D. Becke, “Density-functional exchange-energy approximation with correct
asymptotic-behavior,” Phys. Rev. A, 38 (1988) 3098-100.
Becke89a A. D. Becke and M. R. Roussel, “Exchange holes in inhomogeneous systems: A
coordinate-space model,” Phys. Rev. A, 39 (1989) 3761-67.
Becke92 A. D. Becke, “Density-functional thermochemistry. I. The effect of the exchange-only
gradient correction,” J. Chem. Phys., 96 (1992) 2155-60.
Becke92a A. D. Becke, “Density-functional thermochemistry. II. The effect of the Perdew-Wang
generalized-gradient correlation correction,” J. Chem. Phys., 97 (1992) 9173-77.
Becke93 A. D. Becke, “A new mixing of Hartree-Fock and local density-functional theories,”
J
.
Chem. Phys., 98 (1993) 1372-77.
Becke93a A. D. Becke, “Density-functional thermochemistry. III. The role of exact exchange,”
J
.
Chem. Phys., 98 (1993) 5648-52.
Becke96 A. D. Becke, “Density-functional thermochemistry. IV. A new dynamical correlation
functional and implications for exact-exchange mixing,” J. Chem. Phys., 104 (1996)
1040-46.
Becke97 A. D. Becke, “Density-functional thermochemistry. V. Systematic optimization of
exchange-correlation functionals,” J. Chem. Phys., 107 (1997) 8554-60.
Benson68 S. W. Benson, Thermochemical Kinetics (Wiley and Sons, New York, 1968).
Berger97 R. Berger and M. Klessinger, “Algorithms for exact counting of energy levels of
spectroscopic transitions at different temperatures,” J. Comp. Chem., 18 (1997) 1312-19.
Berger98 R. Berger, C. Fischer, and M. Klessinger, “Calculation of the vibronic fine structure in
electronic spectra at higher temperatures. 1. Benzene and pyrazine,” J. Phys. Chem. A, 102
(1998) 7157-67.
Bergner93 A. Bergner, M. Dolg, W. Kuechle, H. Stoll, and H. Preuss, “Ab-initio energy-adjusted
pseudopotentials for elements of groups 13-17,” Mol. Phys., 80 (1993) 1431-41.
Bernardi84 F. Bernardi, A. Bottini, J. J. W. McDougall, M. A. Robb, and H. B. Schlegel, “MCSCF
gradient calculation of transition structures in organic reactions,” Far. Symp. Chem. Soc., 19
(1984) 137-47.
Bernardi88 F. Bernardi, A. Bottoni, M. J. Field, M. F. Guest, I. H. Hillier, M. A. Robb, and A. Venturini,
“MC-SCF Study of the Diels-Alder Reaction Between Ethylene and Butadiene,”
J
. Am.
Chem. Soc., 110 (1988) 3050-55.
Bernardi88a F. Bernardi, A. Bottoni, M. Olivucci, M. A. Robb, H. B. Schlegel, and G. Tonachini, “Do
Supra-Antara Paths Really Exist for 2+2 Cycloaddition Reactions? Analytical Computation
of the MC-SCF Hessians for Transition States of C2H4 with C2H4, Singlet O2, and
Ketene,” J. Am. Chem. Soc., 110 (1988) 5993-95.
Bernardi90 F. Bernardi, A. Bottoni, M. A. Robb, and A. Venturini, “MC-SCF study of the cycloaddition
reaction between ketene and ethylene,” J. Am. Chem. Soc., 112 (1990) 2106-14.
Bernardi92 F. Bernardi, M. Olivucci, I. Palmer, and M. A. Robb, “An MC-SCF study of the thermal and
photochemical cycloaddition of Dewar benzene,” J. Organic Chem., 57 (1992) 5081-87.
Bernardi96 F. Bernardi, M. A. Robb, and M. Olivucci, “Potential energy surface crossings in organic
photochemistry,” Chem. Soc. Reviews, 25 (1996) 321.
Besler90 B. H. Besler, K. M. Merz Jr., and P. A. Kollman, “Atomic charges derived from
semiempirical methods,” J. Comp. Chem., 11 (1990) 431-39.
- 300 -
Bingham75 R. C. Bingham, M. J. S. Dewar, and D. H. Lo, “Ground-states of molecules. 25. MINDO-3 -
Improved version of MINDO semiempirical SCF-MO method,” J. Am. Chem. Soc., 97
(1975) 1285-93.
Binkley80a J. S. Binkley, J. A. Pople, and W. J. Hehre, “Self-Consistent Molecular Orbital Methods. 21.
Small Split-Valence Basis Sets for First-Row Elements,” J. Am. Chem. Soc., 102 (1980)
939-47.
Binning90 R. C. Binning Jr. and L. A. Curtiss, “Compact contracted basis-sets for 3rd-row atoms -
GA-KR,” J. Comp. Chem., 11 (1990) 1206-16.
Blaudeau97 J.-P. Blaudeau, M. P. McGrath, L. A. Curtiss, and L. Radom, “Extension of Gaussian-2 (G2)
theory to molecules containing third-row atoms K and Ca,” J. Chem. Phys., 107 (1997)
5016-21.
Bobrowicz77 F. W. Bobrowicz and W. A. Goddard III, in Methods of Electronic Structure Theory, Ed. H.
F. Schaefer III, Modern Theoretical Chemistry, Vol. 3 (Plenum, New York, 1977) 79-126.
Boese00 A. D. Boese, N. L. Doltsinis, N. C. Handy, and M. Sprik, “New generalized gradient
approximation functionals,” J. Chem. Phys., 112 (2000) 1670-78.
Boese01 A. D. Boese and N. C. Handy, “A new parametrization of exchange-correlation generalized
gradient approximation functionals,” J. Chem. Phys., 114 (2001) 5497-503.
Boese02 A. D. Boese and N. C. Handy, “New exchange-correlation density functionals: The role o
f
the kinetic-energy density,” J. Chem. Phys., 116 (2002) 9559-69.
Boese04 A. D. Boese and J. M. L. Martin, “Development of Density Functionals for Thermochemical
Kinetics,” J. Chem. Phys., 121 (2004) 3405-16.
Bofill89 J. M. Bofill and P. Pulay, “The unrestricted natural orbital-complete active space
(UNO-CAS) method: An inexpensive alternative to the complete active
space-self-consistent-field (CAS-SCF) method,” J. Chem. Phys., 90 (1989) 3637-46.
Bofill94 J. M. Bofill, “Updated Hessian matrix and the restricted step method for locating transition
structures,” J. Comp. Chem., 15 (1994) 1-11.
Bofill95 J. M. Bofill and M. Comajuan, “Analysis of the updated Hessian matrices for locating
transition structures,” J. Comp. Chem., 16 (1995) 1326-38.
Bohmann97 J. A. Bohmann, F. Weinhold, and T. C. Farrar, “Natural Chemical Shielding Analysis of
Nuclear Magnetic Resonance Shielding Tensors from Gauge-Including Atomic Orbital
Calculations,” J. Chem. Phys., 107 (1997) 1173-84.
Bolton98 K. Bolton, W. L. Hase, and G. H. Peslherbe, in
M
odern Methods for Multidimensional
Dynamics Computation in Chemistry, Ed. D. L. Thompson (World Scientific, Singapore,
1998) 143.
Borrelli03 R. Borrelli and A. Peluso, “Dynamics of radiationless transitions in large molecular systems:
A Franck-Condon-
b
ased method accounting for displacements and rotations of all the
normal coordinates,” J. Chem. Phys., 119 (2003) 8437-48.
Boys60 S. F. Boys, “Construction of Molecular Orbitals to be Approximately Invariant for Changes
from One Molecule to Another,” Rev. Mod. Phys., 32 (1960) 296-99.
Boys66 S. F. Boys, in Quantum Theory of Atoms, Molecules and the Solid State, Ed. P.-O. Löwdin
(Academic Press, New York, 1966) 253.
Boys70 S. F. Boys and F. Bernardi, “Calculation of Small Molecular Interactions by Differences of
Separate Total Energies - Some Procedures with Reduced Errors,” Mol. Phys., 19 (1970)
- 301 -
553.
Breneman90 C. M. Breneman and K. B. Wiberg, “Determining atom-centered mono
p
oles from molecular
electrostatic potentials - the need for high sampling density in formamide
conformational-analysis,” J. Comp. Chem., 11 (1990) 361-73.
Buckingham67 A. D. Buckingham, in Advances in Chemical Physics, Ed. I. Prigogine, Vol. 12 (Wiley
Interscience, New York, 1967) 107.
Buckingham68 A. D. Buckingham and G. C. Longuet-Higgins, “Quadrupole Moments of Dipolar
Molecules,” Mol. Phys., 14 (1968) 63.
Bunker71 D. L. Bunker, “Classical Trajectory Methods,” Meth. Comp. Phys., 10 (1971) 287.
Burant96 J. C. Burant, M. C. Strain, G. E. Scuseria, and M. J. Frisch, “Kohn-Sham Analytic Energy
Second Derivatives with the Gaussian Very Fast Multipole Method (GvFMM),” Chem.
Phys. Lett., 258 (1996) 45-52.
Burant96a J. C. Burant, M. C. Strain, G. E. Scuseria, and M. J. Frisch, “Analytic Energy Gradients for
the Gaussian Very Fast Multipole Method (GvFMM),” Chem. Phys. Lett., 248 (1996) 43-49.
Burant96b J. C. Burant, G. E. Scuseria, and M. J. Frisch, “Linear scaling method for Hartree-Fock
exchange calculations of large molecules,” J. Chem. Phys., 105 (1996) 8969-72.
Burke98 K. Burke, J. P. Perdew, and Y. Wang, in Electronic Density Functional Theory: Recent
Progress and New Directions, Ed. J. F. Dobson, G. Vignale, and M. P. Das (Plenum, 1998).
Califano76 S. Califano, Vibrational States (Wiley, London, 1976).
Cammi00 R. Cammi, B. Mennucci, and J. Tomasi, “Fast evaluation of geometries and properties of
excited molecules in solution: A Tamm-Dancoff model with application to
4-dimethylaminobenzonitrile,” J. Phys. Chem. A, 104 (2000) 5631-37.
Cammi00a R. Cammi, C. Cappelli, S. Corni, and J. Tomasi, “On the calculation of infrared intensities in
solution within the polarizable continuum model,” J. Phys. Chem. A, 104 (2000) 9874-79.
Cammi99 R. Cammi, B. Mennucci, and J. Tomasi, “Second-order Møller-Plesset analytical derivatives
for the polarizable continuum model using the relaxed density approach,” J. Phys. Chem. A,
103 (1999) 9100-08.
Cances01 E. Cancès and B. Mennucci, “Comment on‘Reaction field treatment of charge penetration‘,”
J. Chem. Phys., 114 (2001) 4744-45.
Cances97 E. Cancès, B. Mennucci, and J. Tomasi, “A new integral equation formalism for the
p
olarizable continuum model: Theoretical background and applications to isotropic and
anistropic dielectrics,” J. Chem. Phys., 107 (1997) 3032-41.
Cao01 X. Y. Cao and M. Dolg, “Valence basis sets for relativistic energy-consistent small-core
lanthanide pseudopotentials,” J. Chem. Phys., 115 (2001) 7348-55.
Cao02 X. Y. Cao and M. Dolg, “Segmented contraction scheme for small-core lanthanide
pseudopotential basis sets,” J. Mol. Struct. (Theochem), 581 (2002) 139-47.
Cappelli02 C. Cappelli, S. Corni, B. Mennucci, R. Cammi, and J. Tomasi, “Vibrational Circular
Dichroism within the Polarizable Continuum Model: A Theoretical Evidence o
f
Conformation Effects and Hydrogen Bonding for (S)-(-)-3-Butyn-2-ol in CCl4 Solution,”
J
.
Phys. Chem. A, 106 (2002) 12331-39.
Car85 R. Car and M. Parrinello, “Unified Approach for Molecular-Dynamics and
Density-Functional Theory,” Phys. Rev. Lett., 55 (1985) 2471-74.
- 302 -
Caricato06 M. Caricato, B. Mennucci, J. Tomasi, F. Ingrosso, R. Cammi, S. Corni, and G. Scalmani,
“Formation and relaxation of excited states in solution: A new time dependent polarizable
continuum model based on time dependent density functional theory,” J. Chem. Phys., 124
(2006) 124520.
Carpenter87 J. E. Carpenter, Extension of Lewis structure concepts to open-shell and excited-
s
tate
molecular species, Ph. D. thesis, University of Wisconsin, Madison, WI, 1987.
Carpenter88 J. E. Carpenter and F. Weinhold, “Analysis of the geometry of the hydroxymethyl radical by
the different hybrids for different spins natural bond orbital procedure,”
J
. Mol. Struct.
(Theochem), 46 (1988) 41-62.
Carsky91 P. Cársky and E. Hubak, “Restricted Hartree-Fock and Unrestricted Hartree-Fock as
reference states in many-body perturbation-theory: A critical comparison of the two
approaches,” Theor. Chem. Acc., 80 (1991) 407-25.
Casida98 M. E. Casida, C. Jamorski, K. C. Casida, and D. R. Salahub, “Molecular excitation energies
to high-lying bound states from time-dependent density-functional response theory:
Characterization and correction of the time-dependent local density approximation
ionization threshold,” J. Chem. Phys., 108 (1998) 4439-49.
Cederbaum75 L. S. Cederbaum, “One-
b
ody Green's function for atoms and molecules: theory and
application,” J. Phys. B, 8 (1975) 290-303.
Cederbaum77 L. S. Cederbaum and W. Domcke, in Advances in Chemical Physics, Ed. I. Prigogine and S.
A. Rice, Vol. 36 (Wiley & Sons, New York, 1977) 205.
Cerjan81 C. J. Cerjan and W. H. Miller, “On Finding Transition States,” J. Chem. Phys., 75 (1981)
2800-06.
Chai08 J.-D. Chai and M. Head-Gordon, “Systematic optimization of long-range corrected hybrid
density functionals,” J. Chem. Phys., 128 (2008) 084106.
Chai08a J.-D. Chai and M. Head-Gordon, “Long-range corrected hybrid density functionals with
damped atom-atom dispersion corrections,” Phys. Chem. Chem. Phys., 10 (2008) 6615-20.
Charney79 E. Charney, The Molecular Basis of Optical Activity (Wiley, New York, 1979).
Cheeseman09 J. R. Cheeseman, G. S. Scalmani, and M. J. Frisch, “Analytic Vibrational Raman Optical
Activity Intensities,” J. Chem. Theory and Comput., (2009) to be submitted.
Cheeseman96 J. R. Cheeseman, G. W. Trucks, T. A. Keith, and M. J. Frisch, “A Comparison of Models for
Calculating Nuclear Magnetic Resonance Shielding Tensors,” J. Chem. Phys., 104 (1996)
5497-509.
Cheeseman96a J. R. Cheeseman, M. J. Frisch, F. J. Devlin, and P. J. Stephens, “Ab Initio Calculation of
Atomic Axial Tensors and Vibrational Rotational Strengths Using Density Functional
Theory,” Chem. Phys. Lett., 252 (1996) 211-20.
Chen94 W. Chen, W. L. Hase, and H. B. Schlegel, “Ab initio classical trajectory study of H2C -->
H2 + CO dissociation,” Chem. Phys. Lett., 228 (1994) 436-42.
Chipman00 D. M. Chipman, “Reaction field treatment of charge penetration,” J. Chem. Phys., 112
(2000) 5558-65.
Chirlian87 L. E. Chirlian and M. M. Francl, “Atomic charges derived from electrostatic potentials - a
detailed study,” J. Comp. Chem., 8 (1987) 894-905.
Cimiraglia80 R. Cimiraglia, M. Persico, and J. Tomasi, “Roto-electronic and spin-orbit couplings in the
predissociation of HNO - a theoretical calculation,” Chem. Phys. Lett., 76 (1980) 169-71.
- 303 -
Cioslowski89 J. Cioslowski, “A New Population Analysis Based on Atomic Polar Tensors,”
J
. Am. Chem.
Soc., 111 (1989) 8333-36.
Cizek69 J. Cizek, in Advances in Chemical Physics, Ed. P. C. Hariharan, Vol. 14 (Wiley Interscience,
New York, 1969) 35.
Clabo88 D. A. Clabo, W. D. Allen, R. B. Remington, Y. Yamaguchi, and H. F. Schaefer III, “A
systematic study of molecular vibrational anharmonicity and vibration-rotation interaction
by self-consistent-field higher-derivative methods - asymmetric-top molecules,” Chem.
Phys., 123 (1988) 187-239.
Clark83 T. Clark, J. Chandrasekhar, G. W. Spitznagel, and P. v. R. Schleyer, “Efficient diffuse
function-augmented basis-sets for anion calculations. 3. The 3-21+G basis set for 1st-row
elements, Li-F,” J. Comp. Chem., 4 (1983) 294-301.
Clemente08 F. Clemente, T. Vreven, and M. J. Frisch, in Quantum Biochemistry, Ed. C. Matta (Wiley
VCH, 2008).
Clifford96 S. Clifford, M. J. Bearpark, and M. A. Robb, “A hybrid MC-SCF method: Generalized
valence bond (GVB) with complete active space SCF (CASSCF),” Chem. Phys. Lett., 255
(1996) 320-26.
Cohen01 A. J. Cohen and N. C. Handy, “Dynamic correlation,” Mol. Phys., 99 (2001) 607-15.
Cohen86 E. R. Cohen and B. N. Taylor, The 1986 Adjustment of the Fundamental Physical Constants,
CODATA Bulletin (Pergamon, Elmsford, NY, 1986).
Collins76 J. B. Collins, P. v. R. Schleyer, J. S. Binkley, and J. A. Pople, “Self-Consistent Molecular
Orbital Methods. 17. Geometries and binding energies of second-row molecules. A
comparison of three basis sets,” J. Chem. Phys., 64 (1976) 5142-51.
Collins02 M. A. Collins, “Molecular potential-energy surfaces for chemical reaction dynamics,” Theor.
Chem. Acc., 108 (2002) 313-24.
Condon37 E. U. Condon, “Theories of optical rotatory power,” Rev. Mod. Phys., 9 (1937) 432-57.
Constyear73 Pure and Applied Chemistry, 2 (1973) 717.
Constyear79 Pure and Applied Chemistry, 51 (1979) 1.
Cornell95 W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz Jr., D. M. Ferguson, D. C.
Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman, “A second generation force-field for
the simulation of proteins, nucleic-acids, and organic-molecules,” J. Am. Chem. Soc., 117
(1995) 5179-97.
Cossi00 M. Cossi and V. Barone, “Solvent effect on vertical electronic transitions by the polarizable
continuum model,” J. Chem. Phys., 112 (2000) 2427-35.
Cossi01 M. Cossi and V. Barone, “Time-dependent density functional theory for molecules in liquid
solutions,” J. Chem. Phys., 115 (2001) 4708-17.
Cossi01a M. Cossi, N. Rega, G. Scalmani, and V. Barone, “Polarizable dielectric model of solvation
with inclusion of charge penetration effects,” J. Chem. Phys., 114 (2001) 5691-701.
Cossi02 M. Cossi, G. Scalmani, N. Rega, and V. Barone, “New developments in the polarizable
continuum model for quantum mechanical and classical calculations on molecules in
solution,” J. Chem. Phys., 117 (2002) 43-54.
Cossi03 M. Cossi, N. Rega, G. Scalmani, and V. Barone, “Energies, structures, and electronic
properties of molecules in solution with the C-PCM solvation model,” J. Comp. Chem., 24
(2003) 669-81.
- 304 -
Cossi96 M. Cossi, V. Barone, R. Cammi, and J. Tomasi, “Ab initio study of solvated molecules: A
new implementation of the polarizable continuum model,” Chem. Phys. Lett., 255 (1996)
327-35.
Cossi98 M. Cossi, V. Barone, B. Mennucci, and J. Tomasi, “Ab initio study of ionic solutions by a
polarizable continuum dielectric model,” Chem. Phys. Lett., 286 (1998) 253-60.
Cossi99 M. Cossi, V. Barone, and M. A. Robb, “A direct procedure for the evaluation of solvent
effects in MC-SCF calculations,” J. Chem. Phys., 111 (1999) 5295-302.
Coutsias04 E. A. Coutsias, C. Seok, and K. A. Dill, “Using quaternions to calculate RMSD,”
J
. Comp.
Chem., 25 (2004) 1849-57.
CRC80 CRC Handbook of Chemistry and Physics, 60th ed., Ed. D. R. Lide (CRC Press, Boca
Raton, FL, 1980).
Csaszar84 P. Csaszar and P. Pulay, “Geometry optimization by direct inversion in the iterative
subspace,” J. Mol. Struct. (Theochem), 114 (1984) 31-34.
Cundari93 T. R. Cundari and W. J. Stevens, “Effective core potential methods for the lanthanides,”
J
.
Chem. Phys., 98 (1993) 5555-65.
Curl65 R. F. Curl Jr., “Relationship between Electron Spin Rotation Coupling Constants and
G-Tensor Components,” Mol. Phys., 9 (1965) 585.
Curtiss07 L. A. Curtiss, P. C. Redfern, and K. Raghavachari, “Gaussian-4 theory,” J. Chem. Phys., 126
(2007) 084108.
Curtiss07a L. A. Curtiss, P. C. Redfern, and K. Raghavachari, “Gaussian-4 theory using reduced order
perturbation theory,” J. Chem. Phys., 127 (2007) 124105.
Curtiss90 L. A. Curtiss, C. Jones, G. W. Trucks, K. Raghavachari, and J. A. Pople, “Gaussian-1 theory
of molecular energies for second-row compounds,” J. Chem. Phys., 93 (1990) 2537-45.
Curtiss91 L. A. Curtiss, K. Raghavachari, G. W. Trucks, and J. A. Pople, “Gaussian-2 theory for
molecular energies of first- and second-row compounds,” J. Chem. Phys., 94 (1991)
7221-30.
Curtiss93 L. A. Curtiss, K. Raghavachari, and J. A. Pople, “Gaussian-2 theory using reduced
Møller-Plesset orders,” J. Chem. Phys., 98 (1993) 1293-98.
Curtiss95 L. A. Curtiss, M. P. McGrath, J.-P. Blaudeau, N. E. Davis, R. C. Binning Jr., and L. Radom,
“Extension of Gaussian-2 theory to molecules containing third-row atoms Ga-Kr,”
J
. Chem.
Phys., 103 (1995) 6104-13.
Curtiss98 L. A. Curtiss, K. Raghavachari, P. C. Redfern, V. Rassolov, and J. A. Pople, “Gaussian-3
(G3) theory for molecules containing first and second-row atoms,” J. Chem. Phys., 109
(1998) 7764-76.
Curtiss99 L. A. Curtiss, P. C. Redfern, K. Raghavachari, V. Rassolov, and J. A. Pople, “Gaussian-3
theory using reduced Møller-Plesset order,” J. Chem. Phys., 110 (1999) 4703-09.
daCosta87 H. F. M. da Costa, M. Trsic, and J. R. Mohallem, “Universal Gaussian and Slater-type
basis-sets for atoms He to Ar based on an integral version of the Hartree-Fock equations,”
Mol. Phys., 62 (1987) 91-95.
Daniels97 A. D. Daniels, J. M. Millam, and G. E. Scuseria, “Semiempirical methods with conjugate
gradient density matrix search to replace diagonalization for molecular systems containing
thousands of atoms,” J. Chem. Phys., 107 (1997) 425-31.
- 305 -
Dapprich99 S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma, and M. J. Frisch, “A New ONIOM
Implementation in Gaussian 98. 1. The Calculation of Energies, Gradients and Vibrational
Frequencies and Electric Field Derivatives,” J. Mol. Struct. (Theochem), 462 (1999) 1-21.
daSilva89 A. B. F. da Silva, H. F. M. da Costa, and M. Trsic, “Universal Gaussian and Slater-type
bases for atoms H to Xe based on the generator-coordinate Hartree-Fock method .1. Ground
and certain low-lying excited-states of the neutral atoms,” Mol. Phys., 68 (1989) 433-45.
Davidson96 E. R. Davidson, “Comment on "Comment on Dunning's correlation-consistent basis sets",”
Chem. Phys. Lett., 260 (1996) 514-18.
Davis81 L. P. Davis, et. al., “MNDO calculations for compounds containing aluminum and boron,”
J
.
Comp. Chem., 2 (1981) 433-45.
deCastro98 E. V. R. de Castro and F. E. Jorge, “Accurate universal gaussian basis set for all atoms of the
periodic table,” J. Chem. Phys., 108 (1998) 5225-29.
deJong01 W. A. deJong, R. J. Harrison, and D. A. Dixon, “Parallel Douglas-Kroll energy and gradients
in NWChem: Estimating scalar relativistic effects using Douglas-Kroll contracted basis
sets,” J. Chem. Phys., 114 (2001) 48-53.
Deng06 W. Deng, J. R. Cheeseman, and M. J. Frisch, “Calculation of Nuclear Spin-Spin Coupling
Constants of Molecules with First and Second Row Atoms in Study of Basis Set
Dependence,” J. Chem. Theory and Comput., 2 (2006) 1028-37.
Dewar77 M. J. S. Dewar and W. Thiel, “Ground-States of Molecules. 38. The MNDO Method:
Approximations and Parameters,” J. Am. Chem. Soc., 99 (1977) 4899-907.
Dewar78 M. J. S. Dewar and H. S. Rzepa, “Ground-states of molecules. 45. MNDO results for
molecules containing beryllium,” J. Am. Chem. Soc., 100 (1978) 777-84.
Dewar78a M. J. S. Dewar, M. L. McKee, and H. S. Rzepa, “MNDO parameters for 3rd period
elements,” J. Am. Chem. Soc., 100 (1978) 3607-07.
Dewar83 M. J. S. Dewar and M. L. McKee, “Ground-states of molecules. 56. MNDO calculations for
molecules containing sulfur,” J. Comp. Chem., 4 (1983) 84-103.
Dewar83a M. J. S. Dewar and E. F. Healy, “Ground-states of molecules. 64. MNDO calculations for
compounds containing bromine,” J. Comp. Chem., 4 (1983) 542-51.
Dewar84 M. J. S. Dewar, G. L. Grady, and J. J. P. Stewart, “Ground-states of molecules. 68. MNDO
calculations for compounds containing tin,” J. Am. Chem. Soc., 106 (1984) 6771-73.
Dewar85 M. J. S. Dewar, E. G. Zoebisch, and E. F. Healy, “AM1: A New General Purpose Quantum
Mechanical Molecular Model,” J. Am. Chem. Soc., 107 (1985) 3902-09.
Dewar85a M. J. S. Dewar, et. al., “Ground-states of molecules. 74. MNDO calculations for compounds
containing mercury,” Organometallics, 4 (1985) 1964-66.
Dewar86 M. J. S. Dewar and C. H. Reynolds, “An improved set of MNDO parameters for sulfur,”
J
.
Comp. Chem., 7 (1986) 140-43.
Dewar88 M. J. S. Dewar, C. Jie, and E. G. Zoebisch, “AM1 calculations for compounds containing
boron,” Organometallics, 7 (1988) 513-21.
Dewar88a M. J. S. Dewar and K. M. Merz Jr., “AM1 parameters for zinc,” Organometallics, 7 (1988)
522-24.
Dewar89 M. J. S. Dewar and C. Jie, “AM1 parameters for phosphorus,” J. Mol. Struct. (Theochem),
187 (1989) 1.
Dewar90 M. J. S. Dewar and Y.-C. Yuan, “AM1 parameters for sulfur,” Inorganic Chem., 29 (1990)
- 306 -
3881-90.
Dewar90a M. J. S. Dewar and A. J. Holder, “AM1 parameters for aluminum,” Organometallics, 9
(1990) 508-11.
Diercksen81 G. H. F. Diercksen, B. O. Roos, and A. J. Sadlej, “Legitimate calculation of 1st-order
molecular-properties in the case of limited CI functions - dipole-moments,” Chem. Phys., 59
(1981) 29-39.
Diercksen81a G. H. F. Diercksen and A. J. Sadlej, “Perturbation-theory of the electron correlation-effects
for atomic and molecular-properties - 2nd-order and 3rd-order correlation corrections to
molecular dipole-moments and polarizabilities,” J. Chem. Phys., 75 (1981) 1253-66.
Dierksen04 M. Dierksen and S. Grimme, “Density functional calculations of the vibronic structure o
f
electronic absorption spectra,” J. Chem. Phys., 120 (2004) 3544-54.
Dierksen04a M. Dierksen and S. Grimme, “The vibronic structure of electronic absorption spectra o
f
large molecules: A time-dependent density functional study on the influence of
‘Exact‘ Hartree-Fock exchange,” J. Phys. Chem. A, 108 (2004) 10225-37.
Dierksen05 M. Dierksen and S. Grimme, “An efficient approach for the calculation of Franck-Condon
integrals of large molecules,” J. Chem. Phys., 122 (2005) 244101.
Ditchfield71 R. Ditchfield, W. J. Hehre, and J. A. Pople, “Self-Consistent Molecular Orbital Methods. 9.
Extended Gaussian-type basis for molecular-orbital studies of organic molecules,”
J
. Chem.
Phys., 54 (1971) 724.
Ditchfield74 R. Ditchfield, “Self-consistent perturbation theory of diamagnetism. 1. Gauge-invariant
LCAO method for N.M.R. chemical shifts,” Mol. Phys., 27 (1974) 789-807.
Dobbs86 K. D. Dobbs and W. J. Hehre, “Molecular-orbital theory of the properties of inorganic and
organometallic compounds. 4. Extended basis-sets for 3rd row and 4th row, main-group
elements,” J. Comp. Chem., 7 (1986) 359-78.
Dobbs87 K. D. Dobbs and W. J. Hehre, “Molecular-orbital theory of the properties of inorganic and
organometallic compounds. 5. Extended basis-sets for 1st-row transition-metals,”
J
. Comp.
Chem., 8 (1987) 861-79.
Dobbs87a K. D. Dobbs and W. J. Hehre, “Molecular-orbital theory of the properties of inorganic and
organometallic compounds. 6. Extended basis-sets for 2nd-row transition-metals,”
J
. Comp.
Chem., 8 (1987) 880-93.
Dodds77 J. L. Dodds, R. McWeeny, W. T. Raynes, and J. P. Riley, “SCF theory for multiple
perturbations,” Mol. Phys., 33 (1977) 611-17.
Dodds77a J. L. Dodds, R. McWeeny, and A. J. Sadlej, “Self-consistent perturbation theory:
Generalization for perturbation-dependent non-orthogonal basis set,” Mol. Phys., 34 (1977)
1779-91.
Doktorov77 E. V. Doktorov, I. A. Malkin, and V. I. Manko, “Dynamical symmetry of vibronic transitions
in polyatomic-molecules and Franck-Condon principle. 2. ,” J. Mol. Spec., 64 (1977)
302-26.
Dolg87 M. Dolg, U. Wedig, H. Stoll, and H. Preuss, “Energy-adjusted ab initio pseudopotentials for
the first row transition elements,” J. Chem. Phys., 86 (1987) 866-72.
Dolg89 M. Dolg, H. Stoll, and H. Preuss, “Energy-adjusted ab initio pseudopotentials for the rare
earth elements,” J. Chem. Phys., 90 (1989) 1730-34.
- 307 -
Dolg89a M. Dolg, H. Stoll, A. Savin, and H. Preuss, “Energy-adjusted pseudopotentials for the
rare-earth elements,” Theor. Chem. Acc., 75 (1989) 173-94.
Dolg91 M. Dolg, P. Fulde, W. Kuechle, C.-S. Neumann, and H. Stoll, “Ground state calculations o
f
di-pi-cyclooctatetraene cerium,” J. Chem. Phys., 94 (1991) 3011-17.
Dolg92 M. Dolg, H. Stoll, H.-J. Flad, and H. Preuss, “Ab initio pseudopotential study of Yb and
YbO,” J. Chem. Phys., 97 (1992) 1162-73.
Dolg93 M. Dolg, H. Stoll, and H. Preuss, “A combination of quasi-relativistic pseudopotential and
ligand-field calculations for lanthanoid compounds,” Theor. Chem. Acc., 85 (1993) 441-50.
Dolg93a M. Dolg, H. Stoll, H. Preuss, and R. M. Pitzer, “Relativistic and correlation-effects for
element 105 (Hahnium, Ha) - a comparative-study of M and MO (M = NB, TA, HA) using
energy-adjusted ab initio pseudopotentials,” J. Phys. Chem., 97 (1993) 5852-59.
Douglas74 M. Douglas and N. M. Kroll, “Quantum electrodynamical corrections to fine-structure of
helium,” Ann. Phys. (NY), 82 (1974) 89-155.
Dukor00 R. K. Dukor and L. A. Nafie, in Encyclopedia of Analytical Chemistry: Instrumentation and
Applications, Ed. R. A. Meyers (Wiley & Sons, Chichester, 2000) 662-76.
Dunlap00 B. I. Dunlap, “Robust and variational fitting: Removing the four-center integrals from center
stage in quantum chemistry,” J. Mol. Struct. (Theochem), 529 (2000) 37-40.
Dunlap83 B. I. Dunlap, “Fitting the Coulomb Potential Variationally in X-Alpha Molecular
Calculations,” J. Chem. Phys., 78 (1983) 3140-42.
Dunning76 T. H. Dunning Jr. and P. J. Hay, in Modern Theoretical Chemistry, Ed. H. F. Schaefer III,
Vol. 3 (Plenum, New York, 1976) 1-28.
Dunning89 T. H. Dunning Jr., “Gaussian basis sets for use in correlated molecular calculations. I. The
atoms boron through neon and hydrogen,” J. Chem. Phys., 90 (1989) 1007-23.
Dupuis76 M. Dupuis, J. Rys, and H. F. King, “Evaluation of molecular integrals over Gaussian basis
functions,” J. Chem. Phys., 65 (1976) 111-16.
Dykstra77 C. E. Dykstra, “Examination of Brueckner condition for selection of molecular-orbitals in
correlated wavefunctions,” Chem. Phys. Lett., 45 (1977) 466-69.
Dykstra84 C. E. Dykstra and P. G. Jasien, “Derivative Hartree-Fock theory to all orders,” Chem. Phys.
Lett., 109 (1984) 388-93.
Eade81 R. H. A. Eade and M. A. Robb, “Direct minimization in MC SCF theory - the Quasi-Newton
method,” Chem. Phys. Lett., 83 (1981) 362-68.
Easton96 R. E. Easton, D. J. Giesen, A. Welch, C. J. Cramer, and D. G. Truhlar, “The MIDI! basis set
for quantum mechanical calculations of molecular geometries and partial charges,” Theor.
Chem. Acc., 93 (1996) 281-301.
Ehara02 M. Ehara, M. Ishida, K. Toyota, and H. Nakatsuji, in
R
eviews in Modern Quantum
Chemistry, Ed. K. D. Sen (World Scientific, Singapore, 2002) 293.
Eichkorn95 K. Eichkorn, O. Treutler, H. Ohm, M. Haser, and R. Ahlrichs, “Auxiliary basis-sets to
approximate Coulomb potentials,” Chem. Phys. Lett., 240 (1995) 283-89.
Eichkorn97 K. Eichkorn, F. Weigend, O. Treutler, and R. Ahlrichs, “Auxiliary basis sets for main row
atoms and transition metals and their use to approximate Coulomb potentials,” Theor. Chem.
Acc., 97 (1997) 119-24.
- 308 -
Elstner98 M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai, and G.
Seifert, “Self-consistent-charge density-functional tight-
b
inding method for simulations of
complex materials properties,” Phys. Rev. B, 58 (1998) 7260-68.
Ernzerhof98 M. Ernzerhof and J. P. Perdew, “Generalized gradient approximation to the angle- and
system-averaged exchange hole,” J. Chem. Phys., 109 (1998)
Eyring35 H. Eyring, “The activated complex in chemical reactions,” J. Chem. Phys., 3 (1935) 107-15.
Eyring44 H. Eyring, J. Walter, and G. E. Kimball, Quantum Chemistry (Wiley, New York, 1944).
Farkas95 Ö. Farkas, PhD (CsC) thesis, Eötvös Loránd University and Hungarian Academy o
f
Sciences, Budapest, 1995 (in Hungarian).
Farkas98 Ö. Farkas and H. B. Schlegel, “Methods for geometry optimization of large molecules. I. An
O(N2) algorithm for solving systems of linear equations for the transformation of
coordinates and forces,” J. Chem. Phys., 109 (1998) 7100-04.
Farkas99 Ö. Farkas and H. B. Schlegel, “Methods for optimizing large molecules. II. Quadratic
search,” J. Chem. Phys., 111 (1999) 10806-14.
Ferreira01 A. M. Ferreira, G. Seabra, O. Dolgounitcheva, V. G. Zakrzewski, and J. V. Ortiz, in
Quantum-Mechanical Prediction of Thermochemical Data, Ed. J. Cioslowski,
Understanding Chemical Reactivity, Vol. 22 (Kluwer Academic, Dordrecht, 2001) 131-60.
Fitzpatrick86 N. J. Fitzpatrick and G. H. Murphy, “Double Zeta-D Radial Wave-Functions for
Transition-Elements,” Inorg. Chim. Acta, 111 (1986) 139-40.
Fletcher63 R. Fletcher and M. J. D. Powell, “A Rapidly Convergent Descent Method for
Minimization,” Comput. J., 6 (1963) 163-68.
Fletcher80 R. Fletcher, Practical Methods of Optimization (Wiley, New York, 1980).
Floris89 F. Florsi and J. Tomasi, “Evaluation of the dispersion contribution to the solvation energy. A
simple computational model in the continuum approximation,” J. Comp. Chem., 10 (1989)
616.
Floris91 F. Florsi, J. Tomasi, and J. L. Pascual-Ahuir, “Dispersion and repulsion contributions to the
solvation energy: Refinements to a simple computational model in the continuum
approximation,” J. Comp. Chem., 12 (1991) 784.
Fogarasi92 G. Fogarasi, X. Zhou, P. Taylor, and P. Pulay, “The calculation of ab initio molecular
geometries: Efficient optimization by natural internal coordinates and empirical correction
by offset forces,” J. Am. Chem. Soc., 114 (1992) 8191-201.
Foresman92 J. B. Foresman, M. Head-Gordon, J. A. Pople, and M. J. Frisch, “Toward a Systematic
Molecular Orbital Theory for Excited States,” J. Phys. Chem., 96 (1992) 135-49.
Foresman93 J. B. Foresman and H. B. Schlegel, in Recent experimental and computational advances in
molecular spectroscopy, Ed. R. Fausto and J. M. Hollas, NATO-ASI Series C, vol. 406
(Kluwer Academic, The Netherlands, 1993) 11-26.
Foresman96 J. B. Foresman, T. A. Keith, K. B. Wiberg, J. Snoonian, and M. J. Frisch, “Solvent Effects 5.
The Influence of Cavity Shape, Truncation of Electrostatics, and Electron Correlation on ab
initio Reaction Field Calculations,” J. Phys. Chem., 100 (1996) 16098-104.
Foresman96b J. B. Foresman and Æ. Frisch, Exploring Chemistry with Electronic Structure Methods, 2nd
ed. (Gaussian, Inc., Pittsburgh, PA, 1996).
Foster60 J. M. Foster and S. F. Boys, “Canonical configurational interaction procedure,”
R
ev. Mod.
Phys., 32 (1960) 300-02.
- 309 -
Foster80 J. P. Foster and F. Weinhold, “Natural hybrid orbitals,” J. Am. Chem. Soc., 102 (1980)
7211-18.
Francl82 M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. DeFrees, J. A. Pople, and M. S.
Gordon, “Self-Consistent Molecular Orbital Methods. 23. A polarization-type basis set for
2nd-row elements,” J. Chem. Phys., 77 (1982) 3654-65.
Frauenheim00 T. Frauenheim, G. Seifert, M. Elstner, Z. Hajnal, G. Jungnickel, D. Porezag, S. Suhai, and R.
Scholz, “A self-consistent charge density-functional based tight-
b
inding method for
predictive materials simulations in physics, chemistry and biology,” Physica Stat. Sol. B,
217 (2000) 41-62.
Frauenheim02 T. Frauenheim, G. Seifert, M. Elstner, T. Niehaus, C. Kohler, M. Amkreutz, M. Sternberg, Z.
Hajnal, A. D. Carlo, and S. Suhai, “Atomistic simulations of complex materials:
ground-state and excited-state properties,” J. Phys.: Condens. Matter, 14 (2002) 3015-47.
Frisch09 M. J. Frisch, G. Scalmani, T. Vreven, and G. Zheng, “Analytic second derivatives for
semiempirical models based on MNDO,” (for Mol. Phys.), (2009)
Frisch84 M. J. Frisch, J. A. Pople, and J. S. Binkley, “Self-Consistent Molecular Orbital Methods. 25.
Supplementary Functions for Gaussian Basis Sets,” J. Chem. Phys., 80 (1984) 3265-69.
Frisch86a M. J. Frisch, Y. Yamaguchi, J. Gaw, H. F. Schaefer III, and J. S. Binkley, “Analytic Raman
intensities from molecular electronic wave functions,” J. Chem. Phys., 84 (1986) 531-32.
Frisch90a M. J. Frisch, M. Head-Gordon, and J. A. Pople, “Direct analytic SCF second derivatives and
electric field properties,” Chem. Phys., 141 (1990) 189-96.
Frisch90b M. J. Frisch, M. Head-Gordon, and J. A. Pople, “Direct MP2 gradient method,” Chem. Phys.
Lett., 166 (1990) 275-80.
Frisch90c M. J. Frisch, M. Head-Gordon, and J. A. Pople, “Semi-direct algorithms for the MP2 energy
and gradient,” Chem. Phys. Lett., 166 (1990) 281-89.
Frisch92 M. J. Frisch, I. N. Ragazos, M. A. Robb, and H. B. Schlegel, “An Evaluation of 3 Direct
MC-SCF Procedures,” Chem. Phys. Lett., 189 (1992) 524-28.
Fuentealba82 P. Fuentealba, H. Preuss, H. Stoll, and L. v. Szentpaly, “A Proper Account of
Core-polarization with Pseudopotentials - Single Valence-Electron Alkali Compounds,”
Chem. Phys. Lett., 89 (1982) 418-22.
Fuentealba83 P. Fuentealba, H. Stoll, L. v. Szentpaly, P. Schwerdtfeger, and H. Preuss, “On the reliability
of semi-empirical pseudopotentials - simulation of Hartree-Fock and Dirac-Fock results,”
J
.
Phys. B, 16 (1983) L323-L28.
Fuentealba85 P. Fuentealba, L. v. Szentpaly, H. Preuss, and H. Stoll, “Pseudopotential calculations for
alkaline-earth atoms,” J. Phys. B, 18 (1985) 1287-96.
Fukui81 K. Fukui, “The path of chemical-reactions - The IRC approach,” Acc. Chem. Res., 14 (1981)
363-68.
Furche02 F. Furche and R. Ahlrichs, “Adiabatic time-dependent density functional methods for
excited state properties,” J. Chem. Phys., 117 (2002) 7433-47.
- 310 -
G03 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Rob, J. R. Cheeseman, J. A.
Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J.
Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H.
Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima,
Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V.
Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R.
Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J.
Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K.
Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul,
S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.
L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M.
Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A.
Pople, Gaussian 03 (Gaussian, Inc., Wallingford, CT, 2003).
G70 W. J. Hehre, W. A. Lathan, R. Ditchfield, M. D. Newton, and J. A. Pople, Gaussian 70
(Quantum Chemistry Program Exchange, Program No. 237, 1970).
G76 J. S. Binkley, R. A. Whiteside, P. C. Hariharan, R. Seeger, J. A. Pople, W. J. Hehre, and M.
D. Newton, Gaussian 76 (Carnegie-Mellon University, Pittsburgh, PA, 1976).
G80 J. S. Binkley, R. A. Whiteside, R. Krishnan, R. Seeger, D. J. Defrees, H. B. Schlegel, S.
Topiol, L. R. Kahn, and J. A. Pople, Gaussian 80 (Carnegie-Mellon Quantum Chemistry
Publishing Unit, Pittsburgh, PA, 1980).
G82 J. S. Binkley, M. J. Frisch, D. J. Defrees, R. Krishnan, R. A. Whiteside, H. B. Schlegel, E.
M. Fluder, and J. A. Pople, Gaussian 82 (Carnegie-Mellon Quantum Chemistry Publishing
Unit, Pittsburgh, PA, 1982).
G86 M. J. Frisch, J. S. Binkley, H. B. Schlegel, K. Raghavachari, C. F. Melius, R. L. Martin, J. J.
P. Stewart, F. W. Bobrowicz, C. M. Rohlfing, L. R. Kahn, D. J. Defrees, R. Seeger, R. A.
Whiteside, D. J. Fox, E. M. Fluder, and J. A. Pople, Gaussian 86 (Gaussian, Inc., Pittsburgh,
PA, 1986).
G88 M. J. Frisch, M. Head-Gordon, H. B. Schlegel, K. Raghavachari, J. S. Binkley, C. Gonzalez,
D. J. Defrees, D. J. Fox, R. A. Whiteside, R. Seeger, C. F. Melius, J. Baker, L. R. Kahn, J. J.
P. Stewart, E. M. Fluder, S. Topiol, and J. A. Pople, Gaussian 88 (Gaussian, Inc., Pittsburgh,
PA, 1988).
G90 M. J. Frisch, M. Head-Gordon, G. W. Trucks, J. B. Foresman, K. Raghavachari, H. B.
Schlegel, M. Robb, J. S. Binkley, C. Gonzalez, D. J. Defrees, D. J. Fox, R. A. Whiteside, R.
Seeger, C. F. Melius, J. Baker, L. R. Kahn, J. J. P. Stewart, E. M. Fluder, S. Topiol, and J. A.
Pople, Gaussian 90 (Gaussian, Inc., Pittsburgh, PA, 1990).
G92 M. J. Frisch, G. W. Trucks, M. Head-Gordon, P. M. W. Gill, M. W. Wong, J. B. Foresman, B.
G. Johnson, H. B. Schlegel, M. A. Robb, E. S. Replogle, R. Gomperts, J. L. Andres, K.
Raghavachari, J. S. Binkley, C. Gonzalez, R. L. Martin, D. J. Fox, D. J. Defrees, J. Baker, J.
J. P. Stewart, and J. A. Pople, Gaussian 92 (Gaussian, Inc., Pittsburgh, PA, 1992).
G92DFT M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. W. Wong, J. B.
Foresman, M. A. Robb, M. Head-Gordon, E. S. Replogle, R. Gomperts, J. L. Andres, K.
Raghavachari, J. S. Binkley, C. Gonzalez, R. L. Martin, D. J. Fox, D. J. Defrees, J. Baker, J.
J. P. Stewart, and J. A. Pople, Gaussian 92/DFT (Gaussian, Inc., Pittsburgh, PA, 1993).
- 311 -
G94 M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. A. Robb, J. R.
Cheeseman, T. A. Keith, G. A. Petersson, J. A. Montgomery Jr., K. Raghavachari, M. A.
Al-Laham, V. G. Zakrzewski, J. V. Ortiz, J. B. Foresman, J. Cioslowski, B. B. Stefanov, A.
Nanayakkara, M. Challacombe, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres,
E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker,
J. P. Stewart, M. Head-Gordon, C. Gonzalez, and J. A. Pople, Gaussian 94 (Gaussian, Inc.,
Pittsburgh, PA, 1995).
G98 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V.
G. Zakrzewski, J. A. Montgomery Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M.
Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M.
Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A.
Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, P. Salvador, J. J. Dannenberg, D. K. Malick,
A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B.
B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J.
Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W.
Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S.
Replogle, and J. A. Pople, Gaussian 98 (Gaussian, Inc., Pittsburgh, PA, 1998).
Garrett80 B. C. Garrett, D. G. Truhlar, R. S. Grev, and A. W. Magnusson, “Improved treatment o
f
threshold contributions in variational transition-state theory,” J. Phys. Chem., 84 (1980)
1730-48.
Gauss88 J. Gauss and D. Cremer, “Analytical evaluation of energy gradients in quadratic
configuration-interaction theory,” Chem. Phys. Lett., 150 (1988) 280-86.
Gauss92 J. Gauss, “Calculation of NMR chemical shifts at second-order many-
b
ody perturbation
theory using gauge-including atomic orbitals,” Chem. Phys. Lett., 191 (1992) 614-20.
Gauss93 J. Gauss, “Effects of Electron Correlation in the Calculation of Nuclear-
Magnetic-Resonance Chemical-Shifts,” J. Chem. Phys., 99 (1993) 3629-43.
Gauss95 J. Gauss, “Accurate Calculation of NMR Chemical-Shifts,” Phys. Chem. Chem. Phys., 99
(1995) 1001-08.
Gauss96 J. Gauss, K. Ruud, and T. Helgaker, “Perturbation-dependent atomic orbitals for the
calculation of spin-rotation constants and rotational g tensors,” J. Chem. Phys., 105 (1996)
2804-12.
Gerratt68 J. Gerratt and I. M. Mills, “Force constants and dipole-moment derivatives of molecules
from perturbed Hartree-Fock calculations. I.,” J. Chem. Phys., 49 (1968) 1719.
Gill92 P. M. W. Gill, B. G. Johnson, J. A. Pople, and M. J. Frisch, “The performance of the
Becke-Lee-Yang-Parr (B-LYP) density functional theory with various basis sets,” Chem.
Phys. Lett., 197 (1992) 499-505.
Gill94 P. M. W. Gill, in Advances in Quantum Chemistry, Vol. 25 (Academic Press, San Diego,
CA, 1994) 141-205.
Gill96 P. M. W. Gill, “A new gradient-corrected exchange functional,” Mol. Phys., 89 (1996)
433-45.
Godbout92 N. Godbout, D. R. Salahub, J. Andzelm, and E. Wimmer, “Optimization of Gaussian-type
b
asis sets for local spin density functional calculations. Part I. Boron through neon,
optimization technique and validation,” Can. J. Chem., 70 (1992) 560-71.
- 312 -
Goddard78 W. A. Goddard III and L. B. Harding, in Annual Review of Physical Chemistry, Ed. B. S.
Rabinovitch, Vol. 29 (Annual Reviews, Inc., Palo Alto, CA, 1978) 363-96.
Golab83 J. T. Golab, D. L. Yeager, and P. Jørgensen, “Proper characterization of MC SCF
stationary-points,” Chem. Phys., 78 (1983) 175-99.
Gonzalez89 C. Gonzalez and H. B. Schlegel, “An Improved Algorithm for Reaction Path Following,”
J
.
Chem. Phys., 90 (1989) 2154-61.
Gonzalez90 C. Gonzalez and H. B. Schlegel, “Reaction Path Following in Mass-Weighted Internal
Coordinates,” J. Phys. Chem., 94 (1990) 5523-27.
Gordon80 M. S. Gordon, “The isomers of silacyclopropane,” Chem. Phys. Lett., 76 (1980) 163-68.
Gordon82 M. S. Gordon, J. S. Binkley, J. A. Pople, W. J. Pietro, and W. J. Hehre, “Self-Consistent
Molecular Orbital Methods. 22. Small Split-Valence Basis Sets for Second-Row Elements,”
J. Am. Chem. Soc., 104 (1982) 2797-803.
Gready77 J. E. Gready, G. B. Bacskay, and N. S. Hush, “Finite-field Method Calculations. III. Dipole
moment gradients, polarisability gradients and field-induced shifts in bond lengths,
vibrational levels, spectroscopic constants and dipole functions -- Application to LiH,”
Chem. Phys., 24 (1977) 333-41.
Greengard87 L. Greengard and V. Rokhlin, “A fast algorithm for particle simulations,” J. Comp. Phys., 73
(1987) 325-48.
Greengard88 L. Greengard, The Rapid Evaluation of Potential Fields in Particle Systems (MIT Press,
Cambridge, MA, 1988).
Greengard94 L. Greengard, “Fast algorithms for classical physics,” Science, 265 (1994) 909-14.
Grimme06 S. Grimme, “Semiempirical GGA-type density functional constructed with a long-range
dispersion correction,” J. Comp. Chem., 27 (2006) 1787-99.
Grimme06a S. Grimme, “Semiempirical hybrid density functional with perturbative second-order
correlation,” J. Chem. Phys., 124 (2006) 034108.
Haeussermann93 U. Haeussermann, M. Dolg, H. Stoll, and H. Preuss, “Accuracy of energy-adjusted
quasi-relativistic ab initio pseudopotentials - all-electron and pseudopotential benchmark
calculations for HG, HGH and their cations,” Mol. Phys., 78 (1993) 1211-24.
Halgren77 T. A. Halgren and W. N. Lipscomb, “The Synchronous Transit Method for Determining
Reaction Pathways and Locating Transition States,” Chem. Phys. Lett., 49 (1977) 225-32.
Hall84 G. G. Hall and C. M. Smith, “Fitting electron-densities of molecules,” Int. J. Quantum
Chem., 25 (1984) 881-90.
Hamilton88 T. P. Hamilton and P. Pulay, “UHF natural orbitals for defining and starting MC-SCF
calculations,” J. Chem. Phys., 88 (1988) 4926-33.
Hamprecht98 F. A. Hamprecht, A. Cohen, D. J. Tozer, and N. C. Handy, “Development and assessment of
new exchange-correlation functionals,” J. Chem. Phys., 109 (1998) 6264-71.
Handy01 N. C. Handy and A. J. Cohen, “Left-right correlation energy,” Mol. Phys., 99 (2001) 403-12.
Handy84 N. C. Handy and H. F. Schaefer III, “On the evaluation of analytic energy derivatives for
correlated wave-functions,” J. Chem. Phys., 81 (1984) 5031-33.
Handy89 N. C. Handy, J. A. Pople, M. Head-Gordon, K. Raghavachari, and G. W. Trucks,
“Size-consistent Brueckner theory limited to double substitutions,” Chem. Phys. Lett., 164
(1989) 185-92.
- 313 -
Hansen99 A. E. Hansen and K. L. Bak, “Ab initio calculations of electronic circular dichroism,”
ENANTIOMER, 4 (1999) 455-76.
Hanson87 L. K. Hanson, J. Fajer, M. A. Thompson, and M. C. Zerner, “Electrochromic Effects o
f
Charge Separation in Bacterial Photosynthesis - Theoretical Models,” J. Am. Chem. Soc.,
109 (1987) 4728-30.
Hariharan73 P. C. Hariharan and J. A. Pople, “Influence of polarization functions on molecular-orbital
hydrogenation energies,” Theor. Chem. Acc., 28 (1973) 213-22.
Hariharan74 P. C. Hariharan and J. A. Pople, “Accuracy of AH equilibrium geometries by single
determinant molecular-orbital theory,” Mol. Phys., 27 (1974) 209-14.
Harris85 J. Harris, “Simplified method for calculating the energy of weakly interacting fragments,”
Phys. Rev. B, 31 (1985) 1770-79.
Hase91 Advances in Classical Trajectory Methods, Vol. 1-3, Ed. W. L. Hase (JAI, Stamford, CT,
1991).
Hase96 W. L. Hase, R. J. Duchovic, X. Hu, A. Komornicki, K. F. Lim, D.-H. Lu, G. H. Peslherbe, K.
N. Swamy, S. R. V. Linde, A. Varandas, H. Wang, and R. J. Wolfe, “VENUS96: A General
Chemical Dynamics Computer Program,” QCPE, 16 (1996) 671.
Hay77 P. J. Hay, “Gaussian basis sets for molecular calculations - representation of 3D orbitals in
transition-metal atoms,” J. Chem. Phys., 66 (1977) 4377-84.
Hay85 P. J. Hay and W. R. Wadt, “Ab initio effective core potentials for molecular calculations -
potentials for the transition-metal atoms Sc to Hg,” J. Chem. Phys., 82 (1985) 270-83.
Hay85a P. J. Hay and W. R. Wadt, “Ab initio effective core potentials for molecular calculations -
potentials for K to Au including the outermost core orbitals,” J. Chem. Phys., 82 (1985)
299-310.
Head-Gordon88 M. Head-Gordon and J. A. Pople, “A Method for Two-Electron Gaussian Integral and
Integral Derivative Evaluation Using Recurrence Relations,” J. Chem. Phys., 89 (1988)
5777-86.
Head-Gordon88a M. Head-Gordon, J. A. Pople, and M. J. Frisch, “MP2 energy evaluation by direct methods,”
Chem. Phys. Lett., 153 (1988) 503-06.
Head-Gordon94 M. Head-Gordon and T. Head-Gordon, “Analytic MP2 Frequencies Without Fifth Order
Storage: Theory and Application to Bifurcated Hydrogen Bonds in the Water Hexamer,”
Chem. Phys. Lett., 220 (1994) 122-28.
Head-Gordon94a M. Head-Gordon, R. J. Rico, M. Oumi, and T. J. Lee, “A Doubles Correction to Electronic
Excited-States from Configuration-Interaction in the Space of Single Substitutions,” Chem.
Phys. Lett., 219 (1994) 21-29.
Head-Gordon95 M. Head-Gordon, D. Maurice, and M. Oumi, “A Perturbative Correction to Restricted
Open-Shell Configuration-Interaction with Single Substitutions for Excited-States o
f
Radicals,” Chem. Phys. Lett., 246 (1995) 114-21.
Hegarty79 D. Hegarty and M. A. Robb, “Application of unitary group-methods to
configuration-interaction calculations,” Mol. Phys., 38 (1979) 1795-812.
Hehre69 W. J. Hehre, R. F. Stewart, and J. A. Pople, “Self-Consistent Molecular Orbital Methods. 1.
Use of Gaussian expansions of Slater-type atomic orbitals,” J. Chem. Phys., 51 (1969)
2657-64.
- 314 -
Hehre72 W. J. Hehre, R. Ditchfield, and J. A. Pople, “Self-Consistent Molecular Orbital Methods. 12.
Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of
organic-molecules,” J. Chem. Phys., 56 (1972) 2257.
Helgaker00 T. Helgaker, M. Watson, and N. C. Handy, “Analytical calculation of nuclear magnetic
resonance indirect spin-spin coupling constants at the generalized gradient approximation
and hybrid levels of density-functional theory,” J. Chem. Phys., 113 (2000) 9402-09.
Helgaker90 T. Helgaker, E. Uggerud, and H. J. A. Jensen, “Integration of the Classical Equations o
f
Motion on ab initio Molecular-Potential Energy Surfaces Using Gradients and Hessians -
Application to Translational Energy-Release Upon Fragmentation,” Chem. Phys. Lett., 173
(1990) 145-50.
Helgaker91 T. Helgaker and P. Jørgensen, “An Electronic Hamiltonian for Origin Independent
Calculations of Magnetic-Properties,” J. Chem. Phys., 95 (1991) 2595-601.
Helgaker94 T. Helgaker, K. Ruud, K. L. Bak, P. Jørgensen, and J. Olsen, “Vibrational Raman
Optical-Activity Calculations Using London Atomic Orbitals,” Faraday Discuss., 99 (1994)
165-80.
Henderson09 T. M. Henderson, A. F. Izmaylov, G. Scalmani, and G. E. Scuseria, “Can short-range hybrids
describe long-range-dependent properties?,” J. Chem. Phys., 131 (2009) 044108.
Herzberg33 G. Herzberg and E. Teller, “Fluctuation structure of electron transfer in multiatomic
molecules,” Z. Phys. Chemie, 21 (1933) 410.
Hess85 B. A. Hess, “Applicability of the no-pair equation with free-
p
article projection operators to
atomic and molecular-structure calculations,” Phys. Rev. A, 32 (1985) 756-63.
Hess86 B. A. Hess, “Relativistic electronic-structure calculations employing a 2-component no-
p
air
formalism with external-field projection operators,” Phys. Rev. A, 33 (1986) 3742-48.
Heyd03 J. Heyd, G. Scuseria, and M. Ernzerhof, “Hybrid functionals based on a screened Coulomb
potential,” J. Chem. Phys., 118 (2003) 8207-15.
Heyd04 J. Heyd and G. Scuseria, “Efficient hybrid density functional calculations in solids: The
HS-Ernzerhof screened Coulomb hybrid functional,” J. Chem. Phys., 121 (2004) 1187-92.
Heyd04a J. Heyd and G. E. Scuseria, “Assessment and validation of a screened Coulomb hybrid
density functional,” J. Chem. Phys., 120 (2004) 7274.
Heyd05 J. Heyd, J. E. Peralta, G. E. Scuseria, and R. L. Martin, “Energy band gaps and lattice
parameters evaluated with the Heyd-Scuseria-Ernzerhof screened hybrid functional,”
J
.
Chem. Phys., 123 (2005) 174101: 1-8.
Heyd06 J. Heyd, G. E. Scuseria, and M. Ernzerhof, “Erratum: "Hybrid functionals based on a
screened Coulomb potential",” J. Chem. Phys., 124 (2006) 219906.
Hirota85 E. Hirota, High-Resolution Spectroscopy of Transient Molecules, Springer Series in
Chemical Physics, Vol. 40 (Springer-Verlag, Berlin, 1985).
Hirota94 E. Hirota, J. M. Brown, J. T. Hougen, T. Shida, and N. Hirota, “Symbols for fine and
hyperfine-structure parameters,” Pure & Appl. Chem., 66 (1994) 571-76.
Hirshfeld77 F. L. Hirshfeld, “Bonded-atom fragments for describing molecular charge densities,” Theor.
Chem. Acc., 44 (1977) 129-38.
Hoe01 W.-M. Hoe, A. Cohen, and N. C. Handy, “Assessment of a new local exchange functional
OPTX,” Chem. Phys. Lett., 341 (2001) 319-28.
Hoffmann63 R. Hoffmann, “An Extended Huckel Theory. I. Hydrocarbons,” J. Chem. Phys., 39 (1963)
- 315 -
1397.
Hoffmann64 R. Hoffmann, “An Extended Huckel Theory. II. Sigma Orbitals in the Azines,”
J
. Chem.
Phys., 40 (1964) 2745.
Hoffmann64a R. Hoffmann, “An Extended Huckel Theory. III. Compounds of Boron and Nitrogen,”
J
.
Chem. Phys., 40 (1964) 2474.
Hoffmann64b R. Hoffmann, “An Extended Huckel Theory. IV. Carbonium Ions,” J. Chem. Phys., 40
(1964) 2480.
Hoffmann66 R. Hoffmann, “Extended Huckel Theory. V. Cumulenes, Polyenes, Polyacetylenes and Cn,”
Tetrahedron, 22 (1966) 521.
Hohenberg64 P. Hohenberg and W. Kohn, “Inhomogeneous Electron Gas,” Phys. Rev., 136 (1964)
B864-B71.
Hratchian02 H. P. Hratchian and H. B. Schlegel, “Following reaction pathways using a damped classical
trajectory algorithm,” J. Phys. Chem. A, 106 (2002) 165-69.
Hratchian04a H. P. Hratchian and H. B. Schlegel, “Accurate reaction paths using a Hessian based
predictor-corrector integrator,” J. Chem. Phys., 120 (2004) 9918-24.
Hratchian05a H. P. Hratchian and H. B. Schlegel, in Theory and Applications of Computational
Chemistry: The First 40 Years, Ed. C. E. Dykstra, G. Frenking, K. S. Kim, and G. Scuseria
(Elsevier, Amsterdam, 2005) 195-249.
Hratchian05b H. P. Hratchian and H. B. Schlegel, “Using Hessian updating to increase the efficiency of a
Hessian based predictor-corrector reaction path following method,”
J
. Chem. Theory and
Comput., 1 (2005) 61-69.
Humbel96 S. Humbel, S. Sieber, and K. Morokuma, “The IMOMO method: Integration of different
levels of molecular orbital approximations for geometry optimization of large systems: Test
for n-butane conformation and SN2 reaction: RCI+Cl-,” J. Chem. Phys., 105 (1996)
1959-67.
Igel-Mann88 G. Igel-Mann, H. Stoll, and H. Preuss, “Pseudopotentials for main group elements (IIIA
through VIIA),” Mol. Phys., 65 (1988) 1321-28.
Iikura01 H. Iikura, T. Tsuneda, T. Yanai, and K. Hirao, “Long-range correction scheme for
generalized-gradient-approximation exchange functionals,” J. Chem. Phys., 115 (2001)
3540-44.
Improta06 R. Improta, V. Barone, G. Scalmani, and M. J. Frisch, “A state-specific polarizable
continuum model time dependent density functional method for excited state calculations in
solution,” J. Chem. Phys., 125 (2006) 054103: 1-9.
Improta07 R. Improta, G. Scalmani, M. J. Frisch, and V. Barone, “Toward effective and reliable
fluorescence energies in solution by a new State Specific Polarizable Continuum Model
Time Dependent Density Functional Theory Approach,” J. Chem. Phys., 127 (2007)
074504: 1-9.
Ishida01 M. Ishida, K. Toyota, M. Ehara, and H. Nakatsuji, “Analytical energy gradients of the
excited, ionized and electron-attached states calculated by the SAC-CI general-R method,”
Chem. Phys. Lett., 347 (2001) 493-98.
Ishida01a M. Ishida, K. Toyota, M. Ehara, and H. Nakatsuji, “Analytical energy gradient of high-spin
multiplet state calculated by the SAC-CI method,” Chem. Phys. Lett., 350 (2001) 351-58.
- 316 -
Iyengar01 S. S. Iyengar, H. B. Schlegel, J. M. Millam, G. A. Voth, G. E. Scuseria, and M. J. Frisch, “Ab
initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. II.
Generalizations based on mass-weighting, idempotency, energy conservation and choice o
f
initial conditions,” J. Chem. Phys., 115 (2001) 10291-302.
Izmaylov06 A. F. Izmaylov, G. Scuseria, and M. J. Frisch, “Efficient evaluation of short-range
Hartree-Fock exchange in large molecules and periodic systems,” J. Chem. Phys., 125
(2006) 104103: 1-8.
Jankowiak07 H.-C. Jankowiak, J. L. Stuber, and R. Berger, “Vibronic transitions in large molecular
systems: rigorous prescreening conditions for Franck-Condon factors,” J. Chem. Phys., 127
(2007) 234101.
Jansen89 G. Jansen and B. A. Hess, “Revision of the Douglas-Kroll transformation,” Phys. Rev. A, 39
(1989) 6016-17.
Johnson93 B. G. Johnson, P. M. W. Gill, and J. A. Pople, “Computing Molecular Electrostatic Potentials
with the PRISM Algorithm,” Chem. Phys. Lett., 206 (1993) 239-46.
Johnson93a B. G. Johnson and M. J. Frisch, “Analytic second derivatives of the gradient-corrected
density functional energy: Effect of quadrature weight derivatives,” Chem. Phys. Lett., 216
(1993) 133-40.
Johnson94 B. G. Johnson and M. J. Frisch, “An implementation of analytic second derivatives of the
gradient-corrected density functional energy,” J. Chem. Phys., 100 (1994) 7429-42.
Jorge97 F. E. Jorge, E. V. R. de Castro, and A. B. F. da Silva, “A universal Gaussian basis set for
atoms Cerium through Lawrencium generated with the generator coordinate Hartree-Fock
method,” J. Comp. Chem., 18 (1997) 1565-69.
Jorge97a F. E. Jorge, E. V. R. de Castro, and A. B. F. da Silva, “Accurate universal Gaussian basis set
for hydrogen through lanthanum generated with the generator coordinate Hartree-Fock
method,” Chem. Phys., 216 (1997) 317-21.
Jorgensen88 P. Jørgensen, H. J. A. Jensen, and J. Olsen, “Linear Response Calculations for Large-Scale
Multiconfiguration Self-Consistent Field Wave-Functions,” J. Chem. Phys., 89 (1988)
3654-61.
Kallay04 M. Kállay and J. Gauss, “Calculation of excited-state properties using general
coupled-cluster and configuration-interaction models,” J. Chem. Phys., 121 (2004) 9257.
Karna91 S. P. Karna and M. Dupuis, “Frequency-Dependent Nonlinear Optical-Properties of
Molecules - Formulation and Implementation in the Hondo Program,” J. Comp. Chem., 12
(1991) 487-504.
Kaupp91 M. Kaupp, P. v. R. Schleyer, H. Stoll, and H. Preuss, “Pseudopotential approaches to CA,
SR, and BA hydrides. Why are some alkaline-earth MX2 compounds bent?,”
J
. Chem.
Phys., 94 (1991) 1360-66.
Keith92 T. A. Keith and R. F. W. Bader, “Calculation of magnetic response properties using atoms in
molecules,” Chem. Phys. Lett., 194 (1992) 1-8.
Keith93 T. A. Keith and R. F. W. Bader, “Calculation of magnetic response properties using a
continuous set of gauge transformations,” Chem. Phys. Lett., 210 (1993) 223-31.
Kendall92 R. A. Kendall, T. H. Dunning Jr., and R. J. Harrison, “Electron affinities of the first-row
atoms revisited. Systematic basis sets and wave functions,” J. Chem. Phys., 96 (1992)
6796-806.
- 317 -
King76 H. F. King and M. Dupuis, “Numerical Integration Using Rys Polynomials,” J. Comp. Phys.,
21 (1976) 144-65.
Kirkwood34 J. G. Kirkwood, “Theory of Solutions of Molecules Containing Widely Separated Charges
with Special Application to Zwitterions,” J. Chem. Phys., 2 (1934) 351.
Klene00 M. Klene, M. A. Robb, M. J. Frisch, and P. Celani, “Parallel implementation of the
CI-vector evaluation in full CI/CAS-SCF,” J. Chem. Phys., 113 (2000) 5653-65.
Klene03 M. Klene, M. A. Robb, L. Blancafort, and M. J. Frisch, “A New Efficient Approach to the
Direct RASSCF Method,” J. Chem. Phys., 119 (2003) 713-28.
Knowles91 P. J. Knowles, J. S. Andrews, R. D. Amos, N. C. Handy, and J. A. Pople, “Restricted
Møller-Plesset theory for open shell molecules,” Chem. Phys. Lett., 186 (1991) 130-36.
Kobayashi91 R. Kobayashi, N. C. Handy, R. D. Amos, G. W. Trucks, M. J. Frisch, and J. A. Pople,
“Gradient theory applied to the Brueckner doubles method,” J. Chem. Phys., 95 (1991)
6723-33.
Koch90 H. Koch and P. Jørgensen, “Coupled cluster response functions,” J. Chem. Phys., 93 (1990)
3333-44.
Koch94a H. Koch, R. Kobayashi, A. Sánchez de Merás, and P. Jørgensen, “Calculation o
f
size-intensive transition moments from the coupled cluster singles and doubles linear
response function,” J. Chem. Phys., 100 (1994) 4393.
Kohn65 W. Kohn and L. J. Sham, “Self-Consistent Equations Including Exchange and Correlation
Effects,” Phys. Rev., 140 (1965) A1133-A38.
Komornicki79 A. Komornicki and R. L. Jaffe, “Ab initio investigation of the structure, vibrational
frequencies, and intensities of HO2 and HOCl,” J. Chem. Phys., 71 (1979) 2150-55.
Kondru98 R. K. Kondru, P. Wipf, and D. N. Beratan, “Theory-assisted determination of absolute
stereochemistry for complex natural products via computation of molar rotation angles,”
J
.
Am. Chem. Soc., 120 (1998) 2204-05.
Koseki92 S. Koseki, M. W. Schmidt, and M. S. Gordon, “MCSCF/6-31G(d,p) calculations o
f
one-electron spin-orbit-coupling constants in diatomic-molecules,” J. Phys. Chem., 96
(1992) 10768-72.
Koseki95 S. Koseki, M. S. Gordon, M. W. Schmidt, and N. Matsunaga, “Main-group effective nuclear
charges for spin-orbit calculations,” J. Phys. Chem., 99 (1995) 12764-72.
Koseki98 S. Koseki, M. W. Schmidt, and M. S. Gordon, “Effective nuclear charges for the first-
through third-row transition metal elements in spin-orbit calculations,” J. Phys. Chem. A,
102 (1998) 10430-35.
Krack98 M. Krack and A. M. Köster, “An adaptive numerical integrator for molecular integrals,”
J
.
Chem. Phys., 108 (1998) 3226-34.
Krieger01 J. B. Krieger, J. Q. Chen, and S. Kurth, in Density Functional Theory and its Application to
Materials, Ed. V. VanDoren, C. VanAlsenoy, and P. Geerlings,
A
.I.P. Conference
Proceedings, Vol. 577 (A.I.P., New York, 2001) 48-69.
Krieger99 J. B. Krieger, J. Q. Chen, G. J. Iafrate, and A. Savin, in Electron Correlations and Materials
Properties, Ed. A. Gonis, N. Kioussis, and M. Ciftan (Kluwer Academic, New York, 1999)
463-77.
- 318 -
Krukau06 A. V. Krukau, O. A. Vydrov, A. F. Izmaylov, and G. E. Scuseria, “Influence of the exchange
screening parameter on the performance of screened hybrid functionals,” J. Chem. Phys.,
125 (2006) 224106.
Kudin02 K. N. Kudin, G. E. Scuseria, and E. Cancès, “A black-box self-consistent field convergence
algorithm: One step closer,” J. Chem. Phys., 116 (2002) 8255-61.
Kuechle91 W. Kuechle, M. Dolg, H. Stoll, and H. Preuss, “Ab initio pseudopotentials for HG through
RN. 1. Parameter sets and atomic calculations,” Mol. Phys., 74 (1991) 1245-63.
Kuechle94 W. Kuechle, M. Dolg, H. Stoll, and H. Preuss, “Energy-adjusted pseudopotentials for the
actinides. Parameter sets and test calculations for thorium and thorium molecules,”
J
. Chem.
Phys., 100 (1994) 7535-42.
Kupka86 H. Kupka and P. H. Cribb, “Multidimensional Franck-Condon integrals and Duschinsky
mixing effects,” J. Chem. Phys., 85 (1986) 1303-15.
Labanowski91 Density Functional Methods in Chemistry, Ed. J. K. Labanowski and J. W. Andzelm
(Springer-Verlag, New York, 1991).
Lami04 A. Lami, C. Petrongolo, and F. Santoro, in Conical Intersections: Electronic Structure,
Dynamics & Spectroscopy, Ed. W. Domcke, D. R. Yarkony, and H. Koppel (World
Scientific, Singapore, 2004).
Lauderdale91 W. J. Lauderdale, J. F. Stanton, J. Gauss, J. D. Watts, and R. J. Bartlett, “Many-
b
ody
perturbation theory with a restricted open-shell Hartree-Fock reference,” Chem. Phys. Lett.,
187 (1991)
Lauderdale92 W. J. Lauderdale, J. F. Stanton, J. Gauss, J. D. Watts, and R. J. Bartlett, “Restricted
open-shell Hartree-Fock based many-
b
ody perturbation theory: Theory and application of
energy and gradient calculations,” J. Chem. Phys., 97 (1992)
Lebedev75 V. I. Lebedev, “Weights and Nodes of Gauss-Markov Quadrature Formulas of Orders 9 to
17 for the Sphere that are Invariant under the Octahedron Group with Inversion,”
Z
h.
Vychisl. Mat. Mat. Fiz., 15 (1975) 48-54.
Lebedev76 V. I. Lebedev, “Quadratures on a Sphere,” Zh. Vychisl. Mat. Mat. Fiz., 16 (1976) 293-306.
Lebedev80 V. I. Lebedev, in Theory of Cubature Formulas and Computational Mathematics, Ed. S. L.
Sobolev (Nauka, Novosibirsk, 1980) 75-82 [in Russian].
Lebedev92 V. I. Lebedev and L. Skorokhodov, “Quadrature formulas of orders 41,47 and 53 for the
sphere,” Russian Acad. Sci. Dokl. Math., 45 (1992) 587-92.
Lee88 C. Lee, W. Yang, and R. G. Parr, “Development of the Colle-Salvetti correlation-energy
formula into a functional of the electron density,” Phys. Rev. B, 37 (1988) 785-89.
Lee89 T. J. Lee and P. R. Taylor, “A diagnostic for determining the quality of single-reference
electron correlation methods,” Int. J. Quantum Chem., Quant. Chem. Symp., S23 (1989)
199-207.
Lee90 T. J. Lee, A. P. Rendell, and P. R. Taylor, “Comparison of the Quadratic Configuration
Interaction and Coupled-Cluster Approaches to Electron Correlation Including the Effect o
f
Triple Excitations,” J. Phys. Chem., 94 (1990) 5463-68.
Leininger96 T. Leininger, A. Nicklass, H. Stoll, M. Dolg, and P. Schwerdtfeger, “The accuracy of the
pseudopotential approximation. II. A comparison of various core sizes for indium
pseudopotentials in calculations for spectroscopic constants of InH, InF, and InCI,”
J
. Chem.
Phys., 105 (1996) 1052-59.
- 319 -
Li00 X. Li, J. M. Millam, and H. B. Schlegel, “Ab initio molecular dynamics studies of the
photodissociation of formaldehyde, H2CO --> H2+CO: Direct classical trajectory
calculations by MP2 and density functional theory,” J. Chem. Phys., 113 (2000) 10062-67.
Li06 X. Li and M. J. Frisch, “Energy-represented DIIS within a hybrid geometry optimization
method,” J. Chem. Theory and Comput., 2 (2006) 835-39.
Liang05 J. Liang and H. Y. Li, “Calculation of the multimode Franck-Condon factors based on the
coherent state method,” Mol. Phys., 103 (2005) 3337-42.
Lin74 S. H. Lin and H. Eyring, “Study of Franck-Condon and Herzberg-Teller approximations,”
Proceedings of the National Acad. of Sciences, 71 (1974) 3802-04.
Linderberg04 J. Linderberg and Y. Öhrn, Propagators in Quantum Chemistry, 2nd ed. (Wiley & Sons,
Hoboken, NJ, 2004).
Lippert97 G. Lippert, J. Hutter, and M. Parrinello, “A hybrid Gaussian and plane wave density
functional scheme,” Mol. Phys., 92 (1997) 477-87.
Lippert99 G. Lippert, J. Hutter, and M. Parrinello, “The Gaussian and augmented-plane-wave density
functional method for ab initio molecular dynamics simulations,” Theor. Chem. Acc., 103
(1999) 124-40.
London37 F. London, “The quantic theory of inter-atomic currents in aromatic combinations,”
J
. Phys.
Radium, 8 (1937) 397-409.
Lundberg09 M. Lundberg, T. Kawatsu, T. Vreven, M. J. Frisch, and K. Morokuma, “Transition States in
the Protein Environment -- ONIOM QM:MM Modeling of Isopenicillin N Synthesis,”
J
.
Chem. Theory and Comput., 5 (2009) 222-34.
Macbeth W. Shakespeare, Macbeth, III.iv.40-107 (London, c.1606-1611).
Malick98 D. K. Malick, G. A. Petersson, and J. A. Montgomery Jr., “Transition states for chemical
reactions. I. Geometry and classical barrier height,” J. Chem. Phys., 108 (1998) 5704-13.
Marenich09 A. V. Marenich, C. J. Cramer, and D. G. Truhlar, “Universal solvation model based on solute
electron density and a continuum model of the solvent defined by the bulk dielectric
constant and atomic surface tensions,” J. Phys. Chem. B, 113 (2009) 6378-96.
Martin03 R. L. Martin, “Natural transition orbitals,” J. Chem. Phys., 118 (2003) 4775-77.
Martin99 J. M. L. Martin and G. de Oliveira, “Towards standard methods for benchmark quality ab
initio thermochemistry - W1 and W2 theory,” J. Chem. Phys., 111 (1999) 1843-56.
Martyna91 G. Martyna, C. Cheng, and M. L. Klein, “Electronic States and Dynamic Behavior of Lixen
and Csxen Clusters,” J. Chem. Phys., 95 (1991) 1318-36.
Maseras95 F. Maseras and K. Morokuma, “IMOMM - A new integrated ab-initio plus molecular
mechanics geometry optimization scheme of equilibrium structures and transition-states,”
J
.
Comp. Chem., 16 (1995) 1170-79.
Matsubara96 T. Matsubara, S. Sieber, and K. Morokuma, “A Test of the New "Integrated MO + MM"
(IMOMM) Method for the Conformational Energy of Ethane and n-Butane,” Int. J.
Quantum Chem., 60 (1996) 1101-09.
Mayo90 S. L. Mayo, B. D. Olafson, and W. A. Goddard III, “Dreiding - A generic force-field for
molecular simulations,” J. Phys. Chem., 94 (1990) 8897-909.
McClurg97 R. B. McClurg, R. C. Flagan, and W. A. Goddard III, “The hindered rotor density-of-states
interpolation function,” J. Chem. Phys., 106 (1997) 6675.
- 320 -
McClurg99 R. B. McClurg, “Comment on: 'The hindered rotor density-of-states interpolation function'
[J. Chem. Phys. 106, 6675 (1997)] and 'The hindered rotor density-of-states' [J. Chem. Phys.
108, 2314 (1998)],” J. Chem. Phys., 111 (1999) 7163.
McDouall88 J. J. McDouall, K. Peasley, and M. A. Robb, “A Simple MC-SCF Perturbation Theory:
Orthogonal Valence Bond Møller-Plesset 2 (OVB-MP2),” Chem. Phys. Lett., 148 (1988)
183-89.
McGrath91 M. P. McGrath and L. Radom, “Extension of Gaussian-1 (G1) theory to bromine-containing
molecules,” J. Chem. Phys., 94 (1991) 511-16.
McLaren63 A. D. McLaren, “Optimal Numerical Integration on a Sphere,” Math. Comp., 17 (1963)
361-83.
McLean80 A. D. McLean and G. S. Chandler, “Contracted Gaussian-
b
asis sets for molecular
calculations. 1. 2nd row atoms, Z=11-18,” J. Chem. Phys., 72 (1980) 5639-48.
McQuarrie73 D. A. McQuarrie, Statistical Thermodynamics (Harper and Row, New York, 1973).
McWeeny60 R. McWeeny, “Some recent advances in density matrix theory,” Rev. Mod. Phys., 32 (1960)
335-69.
McWeeny62 R. McWeeny, “Perturbation Theory for Fock-Dirac Density Matrix,” Phys. Rev., 126 (1962)
1028.
McWeeny68 R. McWeeny and G. Dierksen, “Self-consistent perturbation theory. 2. Extension to open
shells,” J. Chem. Phys., 49 (1968) 4852.
Mennucci02 B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel,
and P. J. Stephens, “Polarizable continuum model (PCM) calculations of solvent effects on
optical rotations of chiral molecules,” J. Phys. Chem. A, 106 (2002) 6102-13.
Mennucci97 B. Mennucci and J. Tomasi, “Continuum solvation models: A new approach to the problem
of solute's charge distribution and cavity boundaries,” J. Chem. Phys., 106 (1997) 5151-58.
Mennucci97a B. Mennucci, E. Cancès, and J. Tomasi, “Evaluation of Solvent Effects in Isotropic and
Anisotropic Dielectrics, and in Ionic Solutions with a Unified Integral Equation Method:
Theoretical Bases, Computational Implementation and Numerical Applications,”
J
. Phys.
Chem. B, 101 (1997) 10506-17.
Miehlich89 B. Miehlich, A. Savin, H. Stoll, and H. Preuss, “Results obtained with the correlation-energy
density functionals of Becke and Lee, Yang and Parr,” Chem. Phys. Lett., 157 (1989)
200-06.
Miertus81 S. Miertuš, E. Scrocco, and J. Tomasi, “Electrostatic Interaction of a Solute with a
Continuum. A Direct Utilization of ab initio Molecular Potentials for the Prevision o
f
Solvent Effects,” Chem. Phys., 55 (1981) 117-29.
Miertus82 S. Miertuš and J. Tomasi, “Approximate Evaluations of the Electrostatic Free Energy and
Internal Energy Changes in Solution Processes,” Chem. Phys., 65 (1982) 239-45.
Millam97 J. M. Millam and G. E. Scuseria, “Linear scaling conjugate gradient density matrix search as
an alternative to diagonalization for first principles electronic structure calculations,”
J
.
Chem. Phys., 106 (1997) 5569-77.
Millam99 J. M. Millam, V. Bakken, W. Chen, W. L. Hase, and H. B. Schlegel, “Ab initio classical
trajectories on the Born-Oppenheimer Surface: Hessian-Based Integrators using Fifth Order
Polynomial and Rational Function Fits,” J. Chem. Phys., 111 (1999) 3800-05.
- 321 -
Miller80 W. H. Miller, N. C. Handy, and J. E. Adams, “Reaction-
p
ath Hamiltonian for
polyatomic-molecules,” J. Chem. Phys., 72 (1980) 99-112.
Miller81 W. H. Miller, in Potential Energy Surfaces and Dynamical Calculations, Ed. D. G. Truhlar
(Plenum, New York, 1981) 265.
Miller88 W. H. Miller, B. A. Ruf, and Y. T. Chang, “A diabatic reaction path Hamiltonian,”
J
. Chem.
Phys., 89 (1988) 6298-304.
Miller90 W. H. Miller, R. Hernandez, N. C. Handy, D. Jayatilaka, and A. Willets, “Ab initio
calculation of anharmonic constants for a transition-state, with application to semiclassical
transition-state tunneling probabilities,” Chem. Phys. Lett., 172 (1990) 62-68.
Mills93 Quantities, Units and Symbols in Physical Chemistry, 2nd ed., Ed. I. Mills, T. Cvitaš, K.
Homann, N. Kállay, and K. Kuchitsu (Blackwell, Oxford; dist. CRC Press, Boca Raton,
1993).
Minichino94 C. Minichino and V. Barone, “From concepts to algorithms for the characterization of
reaction mechanisms. H2CS as a case study,” J. Chem. Phys., 100 (1994) 3717-41.
Mo03 S. J. Mo, T. Vreven, B. Mennucci, K. Morokuma, and J. Tomasi, “Theoretical study of the
SN2 reaction of Cl-(H2O) + CH3Cl using our own N-layered integrated molecular orbital
and molecular mechanics polarizable continuum model method (ONIOM-PCM),” Theor.
Chem. Acc., 111 (2003) 154-61.
Mohallem86 J. R. Mohallem, R. M. Dreizler, and M. Trsic, “A Griffin-Hill-Wheeler version of the
Hartree-Fock equations,” Int. J. Quantum Chem., Quant. Chem. Symp., 30 (S20) (1986)
45-55.
Mohallem87 J. R. Mohallem and M. Trsic, “A universal Gaussian basis set for atoms Li through Ne based
on a generator coordinate version of the Hartree-Fock equations,” J. Chem. Phys., 86 (1987)
5043-44.
Mohr00 P. J. Mohr and B. N. Taylor, “CODATA Recommended Values of the Fundamental Physical
Constants: 1998,” Rev. Mod. Phys., 72 (2000) 351-495.
Mohr08 P. J. Mohr, B. N. Taylor, and D. B. Newell, “CODATA Recommended Values of the
Fundamental Physical Constants: 2006,” Rev. Mod. Phys., 80 (2008) 633-730.
Moller34 C. Møller and M. S. Plesset, “Note on an approximation treatment for many-electron
systems,” Phys. Rev., 46 (1934) 0618-22.
Montgomery00 J. A. Montgomery Jr., M. J. Frisch, J. W. Ochterski, and G. A. Petersson, “A complete basis
set model chemistry. VII. Use of the minimum population localization method,”
J
. Chem.
Phys., 112 (2000) 6532-42.
Montgomery94 J. A. Montgomery Jr., J. W. Ochterski, and G. A. Petersson, “A complete basis set model
chemistry. IV. An improved atomic pair natural orbital method,” J. Chem. Phys., 101 (1994)
5900-09.
Montgomery99 J. A. Montgomery Jr., M. J. Frisch, J. W. Ochterski, and G. A. Petersson, “A complete basis
set model chemistry. VI. Use of density functional geometries and frequencies,”
J
. Chem.
Phys., 110 (1999) 2822-27.
Morokuma01 K. Morokuma, D. G. Musaev, T. Vreven, H. Basch, M. Torrent, and D. V. Khoroshun,
“Model Studies of the Structures, Reactivities, and Reaction Mechanisms of
Metalloenzymes,” IBM J. Res. & Dev., 45 (2001) 367-95.
- 322 -
Mulliken55 R. S. Mulliken, “Electronic Population Analysis on LCAO-MO Molecular Wave
Functions,” J. Chem. Phys., 23 (1955) 1833-40.
Murtaugh70 B. A. Murtaugh and R. W. H. Sargent, “Computational Experience with Quadratically
Convergent Minimization Methods,” Comput. J., 13 (1970) 185-94.
Nakajima97 T. Nakajima and H. Nakatsuji, “Analytical energy gradient of the ground, excited, ionized
and electron-attached states calculated by the SAC/SAC-CI method,” Chem. Phys. Lett., 280
(1997) 79-84.
Nakajima99 T. Nakajima and H. Nakatsuji, “Energy gradient method for the ground, excited, ionized,
and electron-attached states calculated by the SAC (symmetry-adapted cluster)/SAC-CI
(configuration interaction) method,” Chem. Phys., 242 (1999) 177-93.
Nakatsuji78 H. Nakatsuji and K. Hirao, “Cluster expansion of the wavefunction:
Symmetry-adapted-cluster expansion, its variational determination, and extension of
open-shell orbital theory,” J. Chem. Phys., 68 (1978) 2053-65.
Nakatsuji79 H. Nakatsuji, “Cluster expansion of the wavefunction: Calculation of electron correlations
in ground and excited states by SAC and SAC CI theories,” Chem. Phys. Lett., 67 (1979)
334-42.
Nakatsuji79a H. Nakatsuji, “Cluster expansion of the wavefunction: Electron correlations in ground and
excited states by SAC (Symmetry-Adapted-Cluster) and SAC CI theories,” Chem. Phys.
Lett., 67 (1979) 329-33.
Nakatsuji91 H. Nakatsuji, “Description of 2-electron and many-electron processes by the SAC-CI
method,” Chem. Phys. Lett., 177 (1991) 331-37.
Nakatsuji91a H. Nakatsuji, “Exponentially generated configuration interaction theory. Descriptions of
excited, ionized and electron attached states,” J. Chem. Phys., 94 (1991) 6716-27.
Nakatsuji93 H. Nakatsuji and M. Ehara, “Symmetry-adapted cluster-configuration interaction method
applied to high-spin multiplicity,” J. Chem. Phys., 98 (1993) 7179-84.
Nakatsuji96 H. Nakatsuji, “Dipped adcluster model for chemisorption and catalytic reactions,” Prog.
Surf. Sci., 54 (1996) 1.
Nakatsuji97 H. Nakatsuji, in Computational Chemistry: Reviews of Current Trends, Ed. J. Leszczynski,
Vol. 2 (World Scientific, Singapore, 1997) 62-124.
Neese01 F. Neese, “Prediction of electron paramagnetic resonance g values using coupled perturbed
Hartree-Fock and Kohn-Sham theory,” J. Chem. Phys., 115 (2001) 11080-96.
Nicklass95 A. Nicklass, M. Dolg, H. Stoll, and H. Preuss, “Ab initio energy-adjusted pseudopotentials
for the noble gases Ne through Xe: Calculation of atomic dipole and quadrupole
polarizabilities,” J. Chem. Phys., 102 (1995) 8942-52.
Nyden81 M. R. Nyden and G. A. Petersson, “Complete basis set correlation energies. I. The
asymptotic convergence of pair natural orbital expansions,” J. Chem. Phys., 75 (1981)
1843-62.
Ochterski96 J. W. Ochterski, G. A. Petersson, and J. A. Montgomery Jr., “A complete basis set model
chemistry. V. Extensions to six or more heavy atoms,” J. Chem. Phys., 104 (1996)
2598-619.
Ohrn81 Y. Öhrn and G. Born, in Advances in Quantum Chemistry, Ed. P.-O. Löwdin, Vol. 13
(Academic Press, San Diego, CA, 1981) 1-88.
- 323 -
Olsen85 J. Olsen and P. Jørgensen, “Linear and Nonlinear Response Functions for an Exact State and
for an MCSCF State,” J. Chem. Phys., 82 (1985) 3235-64.
Olsen88 J. Olsen, B. O. Roos, P. Jørgensen, and H. J. A. Jensen, “Determinant Based
Configuration-Interaction Algorithms for Complete and Restricted Configuration-Interaction
Spaces,” J. Chem. Phys., 89 (1988) 2185-92.
Olsen95 J. Olsen, K. L. Bak, K. Ruud, T. Helgaker, and P. Jørgensen, “Orbital Connections for
Perturbation-Dependent Basis-Sets,” Theor. Chem. Acc., 90 (1995) 421-39.
Onsager36 L. Onsager, “Electric Moments of Molecules in Liquids,” J. Am. Chem. Soc., 58 (1936)
1486-93.
Orlandi73 G. Orlandi and W. Siebrand, “Theory of vibronic intensity borrowing - Comparison o
f
Herzberg-Teller and Born-Oppenheimer coupling,” J. Chem. Phys., 58 (1973) 4513-23.
Ortiz88 J. V. Ortiz, “Electron binding energies of anionic alkali metal atoms from partial fourth
order electron propagator theory calculations,” J. Chem. Phys., 89 (1988) 6348-52.
Ortiz88a J. V. Ortiz, “Partial fourth order electron propagator theory,” Int. J. Quantum Chem., Quant.
Chem. Symp., 34 (S22) (1988) 431-36.
Ortiz89 J. V. Ortiz, “Electron propagator calculations with nondiagonal partial 4th-order
self-energies and unrestricted Hartree-Fock reference states,” Int. J. Quantum Chem., Quant.
Chem. Symp., S23 (1989) 321-32.
Ortiz96 J. V. Ortiz, “Partial third-order quasiparticle theory: Comparisons for closed-shell ionization
energies and an application to the Borazine photoelectron spectrum,” J. Chem. Phys., 104
(1996) 7599-605.
Ortiz97 J. V. Ortiz, V. G. Zakrzewski, and O. Dolgounircheva, in Conceptual Perspectives in
Quantum Chemistry, Ed. J.-L. Calais and E. Kryachko (Kluwer Academic, Dordrecht, 1997)
465-518.
Osamura81 Y. Osamura, Y. Yamaguchi, and H. F. Schaefer III, “Analytic configuration-interaction (CI)
gradient techniques for potential-energy hypersurfaces - a method for openshell molecular
wave-functions,” J. Chem. Phys., 75 (1981) 2919-22.
Osamura82 Y. Osamura, Y. Yamaguchi, and H. F. Schaefer III, “Generalization of analytic
configuration-interaction (CI) gradient techniques for potential-energy hypersurfaces,
including a solution to the coupled perturbed Hartree-Fock equations for multiconfiguration
SCF molecular wave-functions,” J. Chem. Phys., 77 (1982) 383-90.
Otte07 N. Otte, M. Scholten, and W. Thiel, “Looking at self-consistent-charge density functional
tight binding from a semiempirical perspective,” J. Phys. Chem. A, 111 (2007) 5751-55.
Page88 M. Page and J. W. McIver Jr., “On evaluating the reaction path Hamiltonian,”
J
. Chem.
Phys., 88 (1988) 922-35.
Page90 M. Page, C. Doubleday Jr., and J. W. McIver Jr., “Following steepest descent reaction paths
- the use of higher energy derivatives with ab initio electronic-structure methods,”
J
. Chem.
Phys., 93 (1990) 5634-42.
Palmer94 I. J. Palmer, F. Bernardi, M. Olivucci, I. N. Ragazos, and M. A. Robb, “An MC-SCF study
of the (photochemical) Paterno-Buchi reaction,” J. Am. Chem. Soc., 116 (1994) 2121-32.
Papousek82 D. Papousek and M. R. Aliev, in Molecular Vibrational Spectra, Ed. J. R. Durig (Elsevier,
New York, 1982).
- 324 -
Parr89 R. G. Parr and W. Yang, Density-functional theory of atoms and molecules (Oxford Univ.
Press, Oxford, 1989).
Parthiban01 S. Parthiban and J. M. L. Martin, “Assessment of W1 and W2 theories for the computation
of electron affinities, ionization potentials, heats of formation, and proton affinities,”
J
.
Chem. Phys., 114 (2001) 6014-29.
Pascual-Ahuir94 J. L. Pascual-Ahuir, E. Silla, and I. Tuñón, “GEPOL: An improved description of
molecular-surfaces. 3. A new algorithm for the computation of a solvent-excluding surface,”
J. Comp. Chem., 15 (1994) 1127-38.
Pedersen95 T. B. Pedersen and A. E. Hansen, “Ab initio calculation and display of the rotatory strength
tensor in the random phase approximation. Method and model studies,” Chem. Phys. Lett.,
246 (1995) 1-8.
Peluso97 A. Peluso, F. Santoro, and G. del Re, “Vibronic coupling in electronic transitions with
significant Duschinsky effect,” Int. J. Quantum Chem., 63 (1997) 233-44.
Peng93 C. Peng and H. B. Schlegel, “Combining Synchronous Transit and Quasi-Newton Methods
for Finding Transition States,” Israel J. Chem., 33 (1993) 449-54.
Peng96 C. Peng, P. Y. Ayala, H. B. Schlegel, and M. J. Frisch, “Using redundant internal coordinates
to optimize equilibrium geometries and transition states,” J. Comp. Chem., 17 (1996) 49-56.
Peralta03 J. E. Peralta, G. E. Scuseria, J. R. Cheeseman, and M. J. Frisch, “Basis set dependence of
NMR Spin-Spin Couplings in Density Functional Theory Calculations: First row and
hydrogen atoms,” Chem. Phys. Lett., 375 (2003) 452-58.
Perdew81 J. P. Perdew and A. Zunger, “Self-interaction correction to density-functional
approximations for many-electron systems,” Phys. Rev. B, 23 (1981) 5048-79.
Perdew86 J. P. Perdew, “Density-functional approximation for the correlation energy of the
inhomogeneous electron gas,” Phys. Rev. B, 33 (1986) 8822-24.
Perdew91 J. P. Perdew, in Electronic Structure of Solids ‘91, Ed. P. Ziesche and H. Eschrig (Akademie
Verlag, Berlin, 1991) 11.
Perdew92 J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh, and C.
Fiolhais, “Atoms, molecules, solids, and surfaces: Applications of the generalized gradient
approximation for exchange and correlation,” Phys. Rev. B, 46 (1992) 6671-87.
Perdew92a J. P. Perdew and Y. Wang, “Accurate and Simple Analytic Representation of the Electron
Gas Correlation Energy,” Phys. Rev. B, 45 (1992) 13244-49.
Perdew93a J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh, and C.
Fiolhais, “Erratum: Atoms, molecules, solids, and surfaces - Applications of the generalized
gradient approximation for exchange and correlation,” Phys. Rev. B, 48 (1993) 4978.
Perdew96 J. P. Perdew, K. Burke, and Y. Wang, “Generalized gradient approximation for the
exchange-correlation hole of a many-electron system,” Phys. Rev. B, 54 (1996) 16533-39.
Perdew96a J. P. Perdew, K. Burke, and M. Ernzerhof, “Generalized gradient approximation made
simple,” Phys. Rev. Lett., 77 (1996) 3865-68.
Perdew97 J. P. Perdew, K. Burke, and M. Ernzerhof, “Errata: Generalized gradient approximation
made simple,” Phys. Rev. Lett., 78 (1997) 1396.
Perdew99 J. P. Perdew, S. Kurth, A. Zupan, and P. Blaha, “Accurate density functional with correct
formal properties: A step beyond the generalized gradient approximation,” Phys. Rev. Lett.,
82 (1999) 2544-47.
- 325 -

Peterson94 K. A. Peterson, D. E. Woon, and T. H. Dunning Jr., “Benchmark calculations with correlated
molecular wave functions. IV. The classical barrier height of the H+H2 ® H2+H reaction,”
J. Chem. Phys., 100 (1994) 7410-15.
Petersson88 G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley, and J.
Mantzaris, “A complete basis set model chemistry. I. The total energies of closed-shell
atoms and hydrides of the first-row atoms,” J. Chem. Phys., 89 (1988) 2193-218.
Petersson91 G. A. Petersson and M. A. Al-Laham, “A complete basis set model chemistry. II. Open-shell
systems and the total energies of the first-row atoms,” J. Chem. Phys., 94 (1991) 6081-90.
Petersson91a G. A. Petersson, T. G. Tensfeldt, and J. A. Montgomery Jr., “A complete basis set model
chemistry. III. The complete basis set-quadratic configuration interaction family o
f
methods,” J. Chem. Phys., 94 (1991) 6091-101.
Petersson98 G. A. Petersson, in Computational Thermochemistry: Prediction and Estimation o
f
Molecular Thermodynamics, Ed. K. K. Irikura and D. J. Frurip, ACS Symposium Series, Vol.
677 (ACS, Washington, D.C., 1998) 237.
Petersson98a G. A. Petersson, D. K. Malick, W. G. Wilson, J. W. Ochterski, J. A. Montgomery Jr., and M.
J. Frisch, “Calibration and comparison of the Gaussian-2, complete basis set, and density
functional methods for computational thermochemistry,” J. Chem. Phys., 109 (1998)
10570-79.
Pickett91 H. M. Pickett, “The Fitting and Prediction of Vibration-Rotation Spectra with Spin
Interactions,” J. Mol. Spec., 148 (1991) 371-77. See spec.jpl.nasa.gov
Pierotti76 R. A. Pierotti, “A scaled particle theory of aqueous and nonaqueous solutions,” Chem. Rev.,
76 (1976) 717.
Pietro82 W. J. Pietro, M. M. Francl, W. J. Hehre, D. J. Defrees, J. A. Pople, and J. S. Binkley,
“Self-Consistent Molecular Orbital Methods. 24. Supplemented small split-valence
basis-sets for 2nd-row elements,” J. Am. Chem. Soc., 104 (1982) 5039-48.
Pipek89 J. Pipek and P. G. Mezey, “A fast intrinsic localization procedure applicable for ab initio and
semiempirical linear combination of atomic orbital wave functions,” J. Chem. Phys., 90
(1989) 4916-26.
Pople54 J. A. Pople and R. K. Nesbet, “Self-Consistent Orbitals for Radicals,” J. Chem. Phys., 22
(1954) 571-72.
Pople67 J. A. Pople, D. Beveridge, and P. Dobosh, “Approximate self-consistent molecular-orbital
theory. 5. Intermediate neglect of differential overlap,” J. Chem. Phys., 47 (1967) 2026-33.
Pople67a J. A. Pople and M. S. Gordon, “Molecular orbital theory of electronic structure of organic
compounds. 1. Substituent effects and dipole methods,” J. Am. Chem. Soc., 89 (1967) 4253.
Pople76 J. A. Pople, J. S. Binkley, and R. Seeger, “Theoretical Models Incorporating Electron
Correlation,” Int. J. Quantum Chem., Suppl. Y-10 (1976) 1-19.
Pople77 J. A. Pople, R. Seeger, and R. Krishnan, “Variational Configuration Interaction Methods and
Comparison with Perturbation Theory,” Int. J. Quantum Chem., Suppl. Y-11 (1977) 149-63.
Pople78 J. A. Pople, R. Krishnan, H. B. Schlegel, and J. S. Binkley, “Electron Correlation Theories
and Their Application to the Study of Simple Reaction Potential Surfaces,” Int. J. Quantum
Chem., 14 (1978) 545-60.
Pople79 J. A. Pople, K. Raghavachari, H. B. Schlegel, and J. S. Binkley, “Derivative Studies in
Hartree-Fock and Møller-Plesset Theories,” Int. J. Quantum Chem., Quant. Chem. Symp.,
- 326 -
S13 (1979) 225-41.
Pople87 J. A. Pople, M. Head-Gordon, and K. Raghavachari, “Quadratic configuration interaction - a
general technique for determining electron correlation energies,” J. Chem. Phys., 87 (1987)
5968-75.
Pople89 J. A. Pople, M. Head-Gordon, D. J. Fox, K. Raghavachari, and L. A. Curtiss, “Gaussian-1
theory: A general procedure for prediction of molecular energies,” J. Chem. Phys., 90 (1989)
5622-29.
Pople92 J. A. Pople, P. M. W. Gill, and B. G. Johnson, “Kohn-Sham density-functional theory within
a finite basis set,” Chem. Phys. Lett., 199 (1992) 557-60.
Porezag95 D. Porezag, T. Frauenheim, T. Köhler, G. Seifert, and R. Kaschner, “Construction of
tight-binding-like potentials on the basis of density-functional theory: Application to
carbon,” Phys. Rev. B, 51 (1995) 12947-57.
Pulay72 P. Pulay and W. Meyer, “Force constants and dipole-moment derivatives of ammonia from
Hartree-Fock calculations,” J. Chem. Phys., 57 (1972) 3337.
Pulay79 P. Pulay, G. Fogarasi, F. Pang, and J. E. Boggs, “Systematic ab initio gradient calculation o
f
molecular geometries, force constants, and dipole-moment derivatives,” J. Am. Chem. Soc.,
101 (1979) 2550-60.
Pulay82 P. Pulay, “Improved SCF convergence acceleration,” J. Comp. Chem., 3 (1982) 556-60.
Pulay83 P. Pulay, “2nd and 3rd derivatives of variational energy expressions - application to
multi-configurational self-consistent field wave-functions,” J. Chem. Phys., 78 (1983)
5043-51.
Pulay92 P. Pulay and G. Fogarasi, “Geometry optimization in redundant internal coordinates,”
J
.
Chem. Phys., 96 (1992) 2856-60.
Purvis82 G. D. Purvis III and R. J. Bartlett, “A full coupled-cluster singles and doubles model - the
inclusion of disconnected triples,” J. Chem. Phys., 76 (1982) 1910-18.
Pyykko81 P. Pyykko and L. L. Lohr, “Relativistically Parameterized Extended Huckel Calculations. 3.
Structure and Bonding for Some Compounds of Uranium and Other Heavy-Elements,”
Inorganic Chem., 20 (1981) 1950-59.
Pyykko84 P. Pyykko and L. Laaksonen, “Relativistically Parameterized Extended Huckel Calculations.
8. Double-Zeta Parameters for the Actinoids Th, Pa, U, Np, Pu, and Am and an Application
on Uranyl,” J. Phys. Chem., 88 (1984) 4892-95.
Rabuck99 A. Rabuck and G. E. Scuseria, “Improving self-consistent field convergence by varying
occupation numbers,” J. Chem. Phys., 110 (1999) 695-700.
Raff85 L. M. Raff and D. L. Thompson, in Theory of Chemical Reaction Dynamics, Ed. M. Baer
(CRC, Boca Raton, FL, 1985).
Raffenetti73 R. C. Raffenetti, “Pre-processing Two-Electron Integrals for Efficient Utilization in
Many-Electron Self-Consistent Field Calculations,” Chem. Phys. Lett., 20 (1973) 335-38.
Ragazos92 I. N. Ragazos, M. A. Robb, F. Bernardi, and M. Olivucci, “Optimization and
Characterization of the Lowest Energy Point on a Conical Intersection using an MC-SCF
Lagrangian,” Chem. Phys. Lett., 197 (1992) 217-23.
Raghavachari78 K. Raghavachari and J. A. Pople, “Approximate 4th-order perturbation-theory of electron
correlation energy,” Int. J. Quantum Chem., 14 (1978) 91-100.
- 327 -
Raghavachari80 K. Raghavachari, M. J. Frisch, and J. A. Pople, “Contribution of triple substitutions to the
electron correlation energy in fourth-order perturbation theory,” Chem. Phys. Lett., 72
(1980) 4244-45.
Raghavachari80a K. Raghavachari, H. B. Schlegel, and J. A. Pople, “Derivative studies in
configuration-interaction theory,” J. Chem. Phys., 72 (1980) 4654-55.
Raghavachari80b K. Raghavachari, J. S. Binkley, R. Seeger, and J. A. Pople, “Self-Consistent Molecular
Orbital Methods. 20. Basis set for correlated wave-functions,” J. Chem. Phys., 72 (1980)
650-54.
Raghavachari81 K. Raghavachari and J. A. Pople, “Calculation of one-electron properties using limited
configuration-interaction techniques,” Int. J. Quantum Chem., 20 (1981) 1067-71.
Raghavachari89 K. Raghavachari and G. W. Trucks, “Highly correlated systems: Excitation energies of first
row transition metals Sc-Cu,” J. Chem. Phys., 91 (1989) 1062-65.
Raghavachari90 K. Raghavachari, J. A. Pople, E. S. Replogle, and M. Head-Gordon, “Fifth Order
Møller-Plesset Perturbation Theory: Comparison of Existing Correlation Methods and
Implementation of New Methods Correct to Fifth Order,” J. Phys. Chem., 94 (1990)
5579-86.
Rappe81 A. K. Rappé, T. Smedly, and W. A. Goddard III, “The Shape and Hamiltonian Consistent
(SHC) Effective Potentials,” J. Phys. Chem., 85 (1981) 1662-66.
Rappe91 A. K. Rappé and W. A. Goddard III, “Charge equilibration for molecular-dynamics
simulations,” J. Phys. Chem., 95 (1991) 3358-63.
Rappe92 A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. G. III, and W. M. Skiff, “UFF, a full
periodic-table force-field for molecular mechanics and molecular-dynamics simulations,”
J
.
Am. Chem. Soc., 114 (1992) 10024-35.
Rassolov01 V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern, and L. A. Curtiss, “6-31G* Basis
Set for Third-Row Atoms,” J. Comp. Chem., 22 (2001) 976-84.
Rassolov98 V. A. Rassolov, J. A. Pople, M. A. Ratner, and T. L. Windus, “6-31G* basis set for atoms K
through Zn,” J. Chem. Phys., 109 (1998) 1223-29.
Reed83a A. E. Reed and F. Weinhold, “Natural bond orbital analysis of near-Hartree-Fock water
dimer,” J. Chem. Phys., 78 (1983) 4066-73.
Reed85 A. E. Reed, R. B. Weinstock, and F. Weinhold, “Natural-population analysis,”
J
. Chem.
Phys., 83 (1985) 735-46.
Reed85a A. E. Reed and F. Weinhold, “Natural Localized Molecular Orbitals,” J. Chem. Phys., 83
(1985) 1736-40.
Reed88 A. E. Reed, L. A. Curtiss, and F. Weinhold, “Intermolecular interactions from a natural bond
orbital, donor-acceptor viewpoint,” Chem. Rev., 88 (1988) 899-926.
Rega96 N. Rega, M. Cossi, and V. Barone, “Development and validation of reliable quantum
mechanical approaches for the study of free radicals in solution,” J. Chem. Phys., 105
(1996) 11060-67.
Repasky02 M. P. Repasky, J. Chandrasekhar, and W. L. Jorgensen, “PDDG/PM3 and PDDG/MNDO:
Improved semiempirical methods,” J. Comp. Chem., 23 (2002) 1601-22.
Rey98 J. Rey and A. Savin, “Virtual space level shifting and correlation energies,” Int. J. Quantum
Chem., 69 (1998) 581-90.
- 328 -
Ricca95 A. Ricca and C. W. Bauschlicher Jr., “Successive H2O binding energies for Fe(H2O)N+,”
J
.
Phys. Chem., 99 (1995) 9003-07.
Rice90 J. E. Rice, R. D. Amos, S. M. Colwell, N. C. Handy, and J. Sanz, “Frequency-Dependent
Hyperpolarizabilities with Application to Formaldehyde and Methyl-Fluoride,”
J
. Chem.
Phys., 93 (1990) 8828-39.
Rice91 J. E. Rice and N. C. Handy, “The Calculation of Frequency-Dependent Polarizabilities as
Pseudo-Energy Derivatives,” J. Chem. Phys., 94 (1991) 4959-71.
Rice92 J. E. Rice and N. C. Handy, “The Calculation of Frequency-Dependent Hyperpolarizabilities
Including Electron Correlation-Effects,” Int. J. Quantum Chem., 43 (1992) 91-118.
Ridley73 J. E. Ridley and M. C. Zerner, “An Intermediate Neglect of Differential Overlap Technique
for Spectroscopy: Pyrrole and the Azines,” Theor. Chem. Acc., 32 (1973) 111-34.
Ridley76 J. E. Ridley and M. C. Zerner, “Triplet states via Intermediate Neglect of Differential
Overlap: Benzene, Pyridine, and Diazines,” Theor. Chem. Acc., 42 (1976) 223-36.
Ritchie85 J. P. Ritchie, “Electron density distribution analysis for nitromethane, nitromethide, and
nitramide,” J. Am. Chem. Soc., 107 (1985) 1829-37.
Ritchie87 J. P. Ritchie and S. M. Bachrach, “Some methods and applications of electron density
distribution analysis,” J. Comp. Chem., 8 (1987) 499-509.
Robb90 M. A. Robb and U. Niazi, Reports in Molecular Theory, 1 (1990) 23.
Roothaan51 C. C. J. Roothaan, “New Developments in Molecular Orbital Theory,” Rev. Mod. Phys., 23
(1951) 69.
Rosenfeld28 L. Z. Rosenfeld, Physik, 52 (1928) 161.
Ruud02 K. Ruud and T. Helgaker, “Optical rotation studied by density-functional and
coupled-cluster methods,” Chem. Phys. Lett., 352 (2002) 533-39.
Ruud02a K. Ruud, T. Helgaker, and P. Bour, “Gauge-origin independent density-functional theory
calculations of vibrational Raman optical activity,” J. Phys. Chem. A, 106 (2002) 7448-55.
Ruud93 K. Ruud, T. Helgaker, K. L. Bak, P. Jørgensen, and H. J. A. Jensen, “Hartree-Fock Limit
Magnetizabilities from London Orbitals,” J. Chem. Phys., 99 (1993) 3847-59.
Rys83 J. Rys, M. Dupuis, and H. F. King, “Computation of electron repulsion integrals using the
Rys quadrature method,” J. Comp. Chem., 4 (1983) 154-57.
Saebo89 S. Saebø and J. Almlöf, “Avoiding the integral storage bottleneck in LCAO calculations o
f
electron correlation,” Chem. Phys. Lett., 154 (1989) 83-89.
Salahub89 The Challenge of d and f Electrons, Ed. D. R. Salahub and M. C. Zerner (ACS, Washington,
D.C., 1989).
Salter89 E. A. Salter, G. W. Trucks, and R. J. Bartlett, “Analytic Energy Derivatives in Many-Body
Methods. I. First Derivatives,” J. Chem. Phys., 90 (1989) 1752-66.
Santoro07 F. Santoro, R. Improta, A. Lami, J. Bloino, and V. Barone, “Effective method to compute
Franck-Condon integrals for optical spectra of large molecules in solution,” J. Chem. Phys.,
126 (2007) 084509 1-13.
Santoro07a F. Santoro, A. Lami, R. Improta, and V. Barone, “Effective method to compute vibrationally
resolved optical spectra of large molecules at finite temperature in the gas phase and in
solution,” J. Chem. Phys., 126 (2007) 184102.
- 329 -
Santoro08 F. Santoro, A. Lami, R. Improta, J. Bloino, and V. Barone, “Effective method for the
computation of optical spectra of large molecules at finite temperature including the
Duschinsky and Herzberg-Teller effect: The Qx band of porphyrin as a case study,”
J
. Chem.
Phys., 128 (2008) 224311.
Sattelmeyer06 K. W. Sattelmeyer, J. Tirado-Rives, and W. L. Jorgensen, “Comparison of SCC-DFTB and
NDDO-based semiempirical molecular orbital methods for organic molecules,”
J
. Phys.
Chem. A, 110 (2006) 13551-59.
Scalmani06 G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi, and V. Barone, “Geometries
and properties of excited states in the gas phase and in solution: Theory and application of a
time-dependent density functional theory polarizable continuum model,” J. Chem. Phys.,
124 (2006) 094107: 1-15.
Scalmani09 G. Scalmani and M. J. Frisch, “A continuous surface charge formalism for the polarizable
continuum model of solvation,” (2009) in prep.
Schaefer92 A. Schaefer, H. Horn, and R. Ahlrichs, “Fully optimized contracted Gaussian-
b
asis sets for
atoms Li to Kr,” J. Chem. Phys., 97 (1992) 2571-77.
Schaefer94 A. Schaefer, C. Huber, and R. Ahlrichs, “Fully optimized contracted Gaussian-
b
asis sets o
f
triple zeta valence quality for atoms Li to Kr,” J. Chem. Phys., 100 (1994) 5829-35.
Schlegel01 H. B. Schlegel, J. M. Millam, S. S. Iyengar, G. A. Voth, G. E. Scuseria, A. D. Daniels, and
M. J. Frisch, “Ab initio molecular dynamics: Propagating the density matrix with Gaussian
orbitals,” J. Chem. Phys., 114 (2001) 9758-63.
Schlegel02 H. B. Schlegel, S. S. Iyengar, X. Li, J. M. Millam, G. A. Voth, G. E. Scuseria, and M. J.
Frisch, “Ab initio molecular dynamics: Propagating the density matrix with Gaussian
orbitals. III. Comparison with Born-Oppenheimer dynamics,” J. Chem. Phys., 117 (2002)
8694-704.
Schlegel82 H. B. Schlegel, “Optimization of Equilibrium Geometries and Transition Structures,”
J
.
Comp. Chem., 3 (1982) 214-18.
Schlegel82a H. B. Schlegel and M. A. Robb, “MC SCF gradient optimization of the H2CO --> H2 + CO
transition structure,” Chem. Phys. Lett., 93 (1982) 43-46.
Schlegel84 H. B. Schlegel, J. S. Binkley, and J. A. Pople, “First and Second Derivatives of Two
Electron Integrals over Cartesian Gaussians using Rys Polynomials,” J. Chem. Phys., 80
(1984) 1976-81.
Schlegel84a H. B. Schlegel, “Estimating the Hessian for gradient-type geometry optimizations,” Theor.
Chem. Acc., 66 (1984) 333-40.
Schlegel89 H. B. Schlegel, in New Theoretical Concepts for Understanding Organic Reactions, Ed. J.
Bertran and I. G. Csizmadia, NATO-ASI series C, vol. 267 (Kluwer Academic, The
Netherlands, 1989) 33-53.
Schlegel91 H. B. Schlegel and M. J. Frisch, in Theoretical and Computational Models for Organic
Chemistry, Ed. J. S. Formosinho, I. G. Csizmadia, and L. G. Arnaut, NATO-
A
SI Series C,
vol. 339 (Kluwer Academic, The Netherlands, 1991) 5-33.
Schlegel91a H. B. Schlegel and J. J. McDouall, in Computational Advances in Organic Chemistry, Ed.
C. Ögretir and I. G. Csizmadia (Kluwer Academic, The Netherlands, 1991) 167-85.
Schlegel95 H. B. Schlegel, in Modern Electronic Structure Theory, Ed. D. R. Yarkony (World Scientific
Publishing, Singapore, 1995) 459-500.
- 330 -
Schlegel95a H. B. Schlegel and M. J. Frisch, “Transformation between Cartesian and Pure Spherical
Harmonic Gaussians,” Int. J. Quantum Chem., 54 (1995) 83-87.
Schmider98 H. L. Schmider and A. D. Becke, “Optimized density functionals from the extended G2 test
set,” J. Chem. Phys., 108 (1998) 9624-31.
Schwabe06 T. Schwabe and S. Grimme, “Towards chemical accuracy for the thermodynamics of large
molecules: new hybrid density functionals including non-local correlation effects,” Phys.
Chem. Chem. Phys., 8 (2006) 4398.
Schwabe07 T. Schwabe and S. Grimme, “Double-hybrid density functionals with long-range dispersion
corrections: higher accuracy and extended applicability,” Phys. Chem. Chem. Phys., 9
(2007) 3397.
Schwartz98 M. Schwartz, P. Marshall, R. J. Berry, C. J. Ehlers, and G. A. Petersson, “Computational
study of the kinetics of hydrogen abstraction from fluoromethanes by the hydroxyl radical,”
J. Phys. Chem. A, 102 (1998) 10074-81.
Schwerdtfeger89 P. Schwerdtfeger, M. Dolg, W. H. E. Schwarz, G. A. Bowmaker, and P. D. W. Boyd,
“Relativistic effects in gold chemistry. 1. Diatomic gold compounds,” J. Chem. Phys., 91
(1989) 1762-74.
Scuseria88 G. E. Scuseria, C. L. Janssen, and H. F. Schaefer III, “An efficient reformulation of the
closed-shell coupled cluster single and double excitation (CCSD) equations,”
J
. Chem.
Phys., 89 (1988) 7382-87.
Scuseria89 G. E. Scuseria and H. F. Schaefer III, “Is coupled cluster singles and doubles (CCSD) more
computationally intensive than quadratic configuration-interaction (QCISD)?,”
J
. Chem.
Phys., 90 (1989) 3700-03.
Scuseria92 G. E. Scuseria, “Comparison of coupled-cluster results with a hybrid of Hartree-Fock and
density functional theory,” J. Chem. Phys., 97 (1992) 7528-30.
Seeger76 R. Seeger and J. A. Pople, “Self-Consistent Molecular Orbital Methods. 16. Numerically
stable direct energy minimization procedures for solution of Hartree-Fock equations,”
J
.
Chem. Phys., 65 (1976) 265-71.
Seeger77 R. Seeger and J. A. Pople, “Self-Consistent Molecular Orbital Methods. 28. Constraints and
Stability in Hartree-Fock Theory,” J. Chem. Phys., 66 (1977) 3045-50.
Segal66 G. Segal and J. Pople, “Approximate self-consistent molecular orbital theory. 3. CNDO
results for AB2 and AB3 systems,” J. Chem. Phys., 44 (1966) 3289-96.
Sekino86 H. Sekino and R. J. Bartlett, “Frequency-Dependent Nonlinear Optical-Properties of
Molecules,” J. Chem. Phys., 85 (1986) 976-89.
Sharp64 T. E. Sharp and H. M. Rosenstock, “Franck-Condon factors for polyatomic molecules,”
J
.
Chem. Phys., 41 (1964) 3453.
Siegbahn84 P. E. M. Siegbahn, “A new direct CI method for large CI expansions in a small orbital
space,” Chem. Phys. Lett., 109 (1984) 417-23.
Silver78 D. M. Silver, S. Wilson, and W. C. Nieuwpoort, “Universal basis sets and transferability o
f
integrals,” Int. J. Quantum Chem., 14 (1978) 635-39.
Silver78a D. M. Silver and W. C. Nieuwpoort, “Universal atomic basis sets,” Chem. Phys. Lett., 57
(1978) 421-22.
Simon96 S. Simon, M. Duran, and J. J. Dannenberg, “How does basis set superposition error change
the potential surfaces for hydrogen bonded dimers?,” J. Chem. Phys., 105 (1996) 11024-31.
- 331 -
Simons83 J. Simons, P. Jørgensen, H. Taylor, and J. Ozment, “Walking on Potential Energy Surfaces,”
J. Phys. Chem., 87 (1983) 2745-53.
Singh84 U. C. Singh and P. A. Kollman, “An approach to computing electrostatic charges for
molecules,” J. Comp. Chem., 5 (1984) 129-45.
Skodje82 R. T. Skodje, D. G. Truhlar, and B. C. Garrett, “Vibrationally adiabatic models for reactive
tunneling,” J. Chem. Phys., 77 (1982) 5955-76.
Slater74 J. C. Slater, The Self-Consistent Field for Molecular and Solids, Quantum Theory o
f
Molecular and Solids, Vol. 4 (McGraw-Hill, New York, 1974).
Small71 G. J. Small, “Herzberg-Teller vibronic coupling and Duschinsky effect,” J. Chem. Phys., 54
(1971) 3300.
Smith86 C. M. Smith and G. G. Hall, “Approximation of electron-densities,” Theor. Chem. Acc., 69
(1986) 63-69.
Sosa92 C. Sosa, J. Andzelm, B. C. Elkin, E. Wimmer, K. D. Dobbs, and D. A. Dixon, “A Local
Density Functional Study of the Structure and Vibrational Frequencies of Molecular
Transition-Metal Compounds,” J. Phys. Chem., 96 (1992) 6630-36.
Sosa93a C. Sosa and C. Lee, “Density-functional description of transition structures using nonlocal
corrections: Silylene insertion reactions into the hydrogen molecule,” J. Chem. Phys., 98
(1993) 8004-11.
Stanton93 J. F. Stanton and R. J. Bartlett, “Equation of motion coupled-cluster method: A systematic
biorthogonal approach to molecular excitation energies, transition probabilities, and excited
state properties,” J. Chem. Phys., 98 (1993) 7029-39.
Stephens01 P. J. Stephens, F. J. Devlin, J. R. Cheeseman, and M. J. Frisch, “Calculation of optical
rotation using density functional theory,” J. Phys. Chem. A, 105 (2001) 5356-71.
Stephens02a P. J. Stephens, F. J. Devlin, J. R. Cheeseman, M. J. Frisch, and C. Rosini, “Determination o
f
Absolute Configuration Using Optical Rotation Calculated Using Density Functional
Theory,” Org. Lett., 4 (2002) 4595-98.
Stephens03 P. J. Stephens, F. J. Devlin, J. R. Cheeseman, M. J. Frisch, O. Bortolini, and P. Besse,
“Determination of Absolute Configuration Using Ab Initio Calculation of Optical Rotation,”
Chirality, 15 (2003) S57-S64.
Stephens05 P. J. Stephens, D. M. McCann, J. R. Cheeseman, and M. J. Frisch, “Determination of
absolute configurations of chiral molecules using ab initio time-dependent density
functional theory calculations of optical rotation: How reliable are absolute configurations
obtained for molecules with small rotations?,” Chirality, 17 (2005) S52-S64.
Stephens08 J. P. Stephens, J. J. Pan, F. J. Devlin, and J. R. Cheeseman, “Determination of the Absolute
Configurations of Natural Products Using TDDFT Optical Rotation Calculations: The
Iridoid Oruwacin,” J. Natural Prod., 71 (2008) 285-88.
Stephens94 P. J. Stephens, F. J. Devlin, M. J. Frisch, and C. F. Chabalowski, “Ab initio Calculation of
Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force
Fields,” J. Phys. Chem., 98 (1994) 11623-27.
Stephens94a P. J. Stephens, F. J. Devlin, C. S. Ashvar, C. F. Chabalowski, and M. J. Frisch, “Theoretical
Calculation of Vibrational Circular Dichroism Spectra,” Faraday Discuss., 99 (1994)
103-19.
- 332 -
Stevens63 R. M. Stevens, R. M. Pitzer, and W. N. Lipscomb, “Perturbed Hartree-Fock calculations. 1.
Magnetic susceptibility and shielding in LiH molecule,” J. Chem. Phys., 38 (1963) 550.
Stevens84 W. J. Stevens, H. Basch, and M. Krauss, “Compact effective potentials and efficient
shared-exponent basis-sets for the 1st-row and 2nd-row atoms,” J. Chem. Phys., 81 (1984)
6026-33.
Stevens92 W. J. Stevens, M. Krauss, H. Basch, and P. G. Jasien, “Relativistic compact effective
potentials and efficient, shared-exponent basis-sets for the 3rd-row, 4th-row, and 5th-row
atoms,” Can. J. Chem., 70 (1992) 612-30.
Stewart07 J. J. P. Stewart, “Optimization of parameters for semiempirical methods. V. Modification of
NDDO approximations and application to 70 elements,” J. Mol. Model., 13 (2007)
1173-213.
Stewart89 J. J. P. Stewart, “Optimization of parameters for semiempirical methods. I. Method,”
J
.
Comp. Chem., 10 (1989) 209-20.
Stewart89a J. J. P. Stewart, “Optimization of parameters for semiempirical methods. II. Applications,”
J
.
Comp. Chem., 10 (1989) 221-64.
Stoll84 H. Stoll, P. Fuentealba, P. Schwerdtfeger, J. Flad, L. v. Szentpaly, and H. Preuss, “Cu and Ag
as one-valence-electron atoms - CI results and quadrupole corrections of Cu2, Ag2, CuH,
and AgH,” J. Chem. Phys., 81 (1984) 2732-36.
Strain96 M. C. Strain, G. E. Scuseria, and M. J. Frisch, “Achieving Linear Scaling for the Electronic
Quantum Coulomb Problem,” Science, 271 (1996) 51-53.
Stratmann96 R. E. Stratmann, G. E. Scuseria, and M. J. Frisch, “Achieving linear scaling in
exchange-correlation density functional quadratures,” Chem. Phys. Lett., 257 (1996) 213-23.
Stratmann97 R. E. Stratmann, J. C. Burant, G. E. Scuseria, and M. J. Frisch, “Improving harmonic
vibrational frequencies calculations in density functional theory,” J. Chem. Phys., 106
(1997) 10175-83.
Stratmann98 R. E. Stratmann, G. E. Scuseria, and M. J. Frisch, “An efficient implementation of
time-dependent density-functional theory for the calculation of excitation energies of large
molecules,” J. Chem. Phys., 109 (1998) 8218-24.
Svensson96 M. Svensson, S. Humbel, R. D. J. Froese, T. Matsubara, S. Sieber, and K. Morokuma,
“ONIOM: A multi-layered integrated MO+MM method for geometry optimizations and
single point energy predictions. A test for Diels-Alder reactions and Pt(P(t-Bu)3)2+H2
oxidative addition,” J. Phys. Chem., 100 (1996) 19357-63.
Svensson96a M. Svensson, S. Humbel, and K. Morokuma, “Energetics using the single point IMOMO
(integrated molecular orbital plus molecular orbital) calculations: Choices of computational
levels and model system,” J. Chem. Phys., 105 (1996) 3654-61.
Sychrovsky00 V. Sychrovsky, J. Gräfenstein, and D. Cremer, “Nuclear magnetic resonance spin-spin
coupling constants from coupled perturbed density functional theory,” J. Chem. Phys., 113
(2000) 3530-47.
Szentpaly82 L. v. Szentpaly, P. Fuentealba, H. Preuss, and H. Stoll, “Pseudopotential calculations on
Rb+2, Cs+2, RbH+, CsH+ and the mixed alkali dimer ions ” Chem. Phys. Lett., 93 (1982)
555-59.
- 333 -
Tao03 J. M. Tao, J. P. Perdew, V. N. Staroverov, and G. E. Scuseria, “Climbing the density
functional ladder: Nonempirical meta-generalized gradient approximation designed for
molecules and solids,” Phys. Rev. Lett., 91 (2003) 146401.
Tawada04 Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai, and K. Hirao, “A long-range-corrected
time-dependent density functional theory,” J. Chem. Phys., 120 (2004) 8425.
Taylor87 P. R. Taylor, “Integral processing in beyond-Hartree-Fock calculations,” Int. J. Quantum
Chem., 31 (1987) 521-34.
Thiel92 W. Thiel and A. A. Voityuk, “Extension of the MNDO formalism to d orbitals: Integral
approximations and preliminary numerical results,” Theor. Chem. Acc., 81 (1992) 391-404.
Thiel96 W. Thiel and A. A. Voityuk, “Extension of MNDO to d orbitals: Parameters and results for
the second-row elements and for the zinc group,” J. Phys. Chem., 100 (1996) 616-26.
Thompson91 M. A. Thompson and M. C. Zerner, “A Theoretical Examination of the Electronic Structure
and Spectroscopy of the Photosynthetic Reaction Center from Rhodopseudomonas viridis,”
J. Am. Chem. Soc., 113 (1991) 8210-15.
Thompson98 D. L. Thompson, in Encyclopedia of Computational Chemistry, Ed. P. v. R. Schleyer, N. L.
Allinger, P. A. Kollman, T. Clark, H. F. Schaefer III, J. Gasteiger, and P. R. Schreiner (Wiley,
Chichester, 1998) 3056-73.
Thorvaldsen08 A. J. Thorvaldsen, K. Ruud, K. Kristensen, P. Jørgensen, and S. Coriani, “A density
matrix-based quasienergy formulation of the Kohn-Sham density functional response theory
using perturbation- and time-dependent basis sets,” J. Chem. Phys., 129 (2008) 214108.
Tirado-Rives08 J. Tirado-Rives and W. L. Jorgensen, “Performance of B3LYP density functional methods
for a large set of organic molecules,” J. Chem. Theory and Comput., 4 (2008) 297-306.
Tomasi02 J. Tomasi, R. Cammi, B. Mennucci, C. Cappelli, and S. Corni, “Molecular properties in
solution described with a continuum solvation model,” Phys. Chem. Chem. Phys., 4 (2002)
5697-712.
Tomasi05 J. Tomasi, B. Mennucci, and R. Cammi, “Quantum mechanical continuum solvation
models,” Chem. Rev., 105 (2005) 2999-3093.
Tomasi99 J. Tomasi, B. Mennucci, and E. Cancès, “The IEF version of the PCM solvation method: An
overview of a new method addressed to study molecular solutes at the QM ab initio level,”
J. Mol. Struct. (Theochem), 464 (1999) 211-26.
Tonachini90 G. Tonachini, H. B. Schlegel, F. Bernardi, and M. A. Robb, “MC-SCF Study of the Addition
Reaction of the 1Dg Oxygen Molecule to Ethene,” J. Am. Chem. Soc., 112 (1990) 483-91.
Torrent02 M. Torrent, T. Vreven, D. G. Musaev, K. Morokuma, Ö. Farkas, and H. B. Schlegel, “Effects
of the protein environment on the structure and energetics of active sites of metalloenzymes:
ONIOM study of methane monooxygenase and ribonucleotide reductase,”
J
. Am. Chem.
Soc., 124 (2002) 192-93.
Toulouse02 J. Toulouse, A. Savin, and C. Adamo, “Validation and assessment of an accurate approach to
the correlation problem in density functional theory: The Krieger-Chen-Iafrate-Savin
model,” J. Chem. Phys., 117 (2002) 10465-73.
Toyota02 K. Toyota, M. Ehara, and H. Nakatsuji, “Elimination of singularities in molecular orbital
derivatives: Minimum orbital-deformation (MOD) method,” Chem. Phys. Lett., 356 (2002)
1-6.
- 334 -
Toyota03 K. Toyota, I. Mayumi, M. Ehara, M. J. Frisch, and H. Nakatsuji, “Singularity-free analytical
energy gradients for the SAC/SAC-CI method: Coupled perturbed minimum
orbital-deformation (CPMOD) approach,” Chem. Phys. Lett., 367 (2003) 730-36.
Trucks88 G. W. Trucks, J. D. Watts, E. A. Salter, and R. J. Bartlett, “Analytical MBPT(4) Gradients,”
Chem. Phys. Lett., 153 (1988) 490-95.
Trucks88a G. W. Trucks, E. A. Salter, C. Sosa, and R. J. Bartlett, “Theory and Implementation of the
MBPT Density Matrix: An Application to One-Electron Properties,” Chem. Phys. Lett., 147
(1988) 359-66.
Truhlar70 D. G. Truhlar, “Adiabatic theory of chemical reactions,” J. Chem. Phys., 53 (1970) 2041.
Truhlar71 D. G. Truhlar and A. Kuppermann, “Exact tunneling calculations,” J. Am. Chem. Soc., 93
(1971) 1840.
Tubert-Brohman04 I. Tubert-Brohman, C. R. W. Guimaraes, M. P. Repasky, and W. L. Jorgensen, “Extension o
f
the PDDG/PM3 and PDDG/MNDO Semiempirical Molecular Orbital Methods to the
Halogens,” J. Comp. Chem., 25 (2004) 138-50.
Tubert-Brohman05 I. Tubert-Brohman, C. R. W. Guimaraes, and W. L. Jorgensen, “Extension of the
PDDG/PM3 Semiempirical Molecular Orbit Method to Sulfur, Silicon, and Phosphorus,”
J
.
Chem. Theory and Comput., 1 (2005) 817-23.
Uggerud92 E. Uggerud and T. Helgaker, “Dynamics of the Reaction CH2OH+ --> CHO+ + H2.
Translational Energy-Release from ab initio Trajectory Calculations,” J. Am. Chem. Soc.,
114 (1992) 4265-68.
VanCaillie00 C. Van Caillie and R. D. Amos, “Geometric derivatives of density functional theory
excitation energies using gradient-corrected functionals,” Chem. Phys. Lett., 317 (2000)
159-64.
VanCaillie99 C. Van Caillie and R. D. Amos, “Geometric derivatives of excitation energies using SCF and
DFT,” Chem. Phys. Lett., 308 (1999) 249-55.
VanVoorhis98 T. Van Voorhis and G. E. Scuseria, “A never form for the exchange-correlation energy
functional,” J. Chem. Phys., 109 (1998) 400-10.
Visscher97 L. Visscher and K. G. Dyall, “Dirac-Fock atomic electronic structure calculations using
different nuclear charge distributions,” Atomic Data and Nuclear Data Tables, 67 (1997)
207-24.
vonNiessen84 W. von Niessen, J. Schirmer, and L. S. Cederbaum, “Computational methods for the
one-particle Green's function,” Comp. Phys. Rep., 1 (1984) 57-125.
Vosko80 S. H. Vosko, L. Wilk, and M. Nusair, “Accurate spin-dependent electron liquid correlation
energies for local spin density calculations: A critical analysis,” Can. J. Phys., 58 (1980)
1200-11.
Vreven00 T. Vreven and K. Morokuma, “On the application of the IMOMO (Integrated Molecular
Orbital + Molecular Orbital) method,” J. Comp. Chem., 21 (2000) 1419-32.
Vreven01 T. Vreven, B. Mennucci, C. O. da Silva, K. Morokuma, and J. Tomasi, “The ONIOM-PCM
method: Combining the hybrid molecular orbital method and the polarizable continuum
model for solvation. Application to the geometry and properties of a merocyanine in
solution,” J. Chem. Phys., 115 (2001) 62-72.
- 335 -
Vreven03 T. Vreven, K. Morokuma, Ö. Farkas, H. B. Schlegel, and M. J. Frisch, “Geometry
optimization with QM/MM, ONIOM and other combined methods. I. Microiterations and
constraints,” J. Comp. Chem., 24 (2003) 760-69.
Vreven06 T. Vreven, K. S. Byun, I. Komáromi, S. Dapprich, J. A. Montgomery Jr., K. Morokuma, and
M. J. Frisch, “Combining quantum mechanics methods with molecular mechanics methods
in ONIOM,” J. Chem. Theory and Comput., 2 (2006) 815-26.
Vreven06a T. Vreven, M. J. Frisch, K. N. Kudin, H. B. Schlegel, and K. Morokuma, “Geometry
optimization with QM/MM Methods. II. Explicit Quadratic Coupling,” Mol. Phys., 104
(2006) 701-14.
Vreven06b T. Vreven and K. Morokuma, in Annual Reports in Computational Chemistry, Ed. D. C.
Spellmeyer, Vol. 2 (Elsevier, 2006) 35 - 51.
Vreven08 T. Vreven and K. Morokuma, in Continuum Solvation Models in Chemical Physics: From
Theory to Applications , Ed. B. Mennucci and R. Cammi (Wiley, 2008).
Vreven97 T. Vreven, F. Bernardi, M. Garavelli, M. Olivucci, M. A. Robb, and H. B. Schlegel, “Ab
initio photoisomerization dynamics of a simple retinal chromophore model,”
J
. Am. Chem.
Soc., 119 (1997) 12687-88.
Vydrov06 O. A. Vydrov and G. E. Scuseria, “Assessment of a long range corrected hybrid functional,”
J. Chem. Phys., 125 (2006) 234109.
Vydrov06a O. A. Vydrov, J. Heyd, A. Krukau, and G. E. Scuseria, “Importance of short-range versus
long-range Hartree-Fock exchange for the performance of hybrid density functionals,”
J
.
Chem. Phys., 125 (2006) 074106.
Vydrov07 O. A. Vydrov, G. E. Scuseria, and J. P. Perdew, “Tests of functionals for systems with
fractional electron number,” J. Chem. Phys., 126 (2007) 154109.
Wachters70 A. J. H. Wachters, “Gaussian basis set for molecular wavefunctions containing third-row
atoms,” J. Chem. Phys., 52 (1970) 1033.
Wadt85 W. R. Wadt and P. J. Hay, “Ab initio effective core potentials for molecular calculations -
potentials for main group elements Na to Bi,” J. Chem. Phys., 82 (1985) 284-98.
Walker70 T. E. H. Walker, “Molecular spin-orbit coupling constants. Role of core polarization,”
J
.
Chem. Phys., 52 (1970) 1311.
Watts93 J. D. Watts, J. Gauss, and R. J. Bartlett, “Coupled-cluster methods with noniterative triple
excitations for restricted open-shell Hartree-Fock and other general single determinant
reference functions. Energies and analytical gradients,” J. Chem. Phys., 98 (1993)
Weber03 J. Weber and G. Hohlneicher, “Franck-Condon factors for polyatomic molecules,”
M
ol.
Phys., 101 (2003) 2125-44.
Wedig86 U. Wedig, M. Dolg, H. Stoll, and H. Preuss, in Quantum Chemistry: The Challenge o
f
Transition Metals and Coordination Chemistry, Ed. A. Veillard, Reidel, and Dordrecht
(1986) 79.
Weigend03 F. Weigend, F. Furche, and R. Ahlrichs, “Gaussian basis sets of quadruple zeta valence
quality for atoms H-Kr,” J. Chem. Phys., 119 (2003) 12753-62.
Weigend05 F. Weigend and R. Ahlrichs, “Balanced basis sets of split valence, triple zeta valence and
quadruple zeta valence quality for H to Rn: Design and assessment of accuracy,” Phys.
Chem. Chem. Phys., 7 (2005) 3297-305.
- 336 -
Weigend06 F. Weigend, “Accurate Coulomb-fitting basis sets for H to Rn,” Phys. Chem. Chem. Phys., 8
(2006) 1057-65.
Weinhold88 F. Weinhold and J. E. Carpenter, in The Structure of Small Molecules and Ions, Ed. R.
Naaman and Z. Vager (Plenum, 1988) 227-36.
Wiberg92 K. B. Wiberg, C. M. Hadad, T. J. LePage, C. M. Breneman, and M. J. Frisch, “An Analysis
of the Effect of Electron Correlation on Charge Density Distributions,” J. Phys. Chem., 96
(1992) 671-79.
Wilson01a P. J. Wilson, T. J. Bradley, and D. J. Tozer, “Hybrid exchange-correlation functional
determined from thermochemical data and ab initio potentials,” J. Chem. Phys., 115 (2001)
9233-42.
Wilson05 S. M. Wilson, K. B. Wiberg, J. R. Cheeseman, M. J. Frisch, and P. H. Vaccaro,
“Nonresonant optical activity of isolated organic molecules,” J. Phys. Chem. A, 109 (2005)
11752-64.
Wilson96 A. K. Wilson, T. van Mourik, and T. H. Dunning Jr., “Gaussian Basis Sets for use in
Correlated Molecular Calculations. VI. Sextuple zeta correlation consistent basis sets for
boron through neon,” J. Mol. Struct. (Theochem), 388 (1996) 339-49.
Wolinski80 K. Wolinski and A. Sadlej, “Self-consistent perturbation theory: Open-shell states in
perturbation-dependent non-orthogonal basis sets,” Mol. Phys., 41 (1980) 1419-30.
Wolinski90 K. Wolinski, J. F. Hilton, and P. Pulay, “Efficient Implementation of the Gauge-Independent
Atomic Orbital Method for NMR Chemical Shift Calculations,” J. Am. Chem. Soc., 112
(1990) 8251-60.
Wong91 M. W. Wong, M. J. Frisch, and K. B. Wiberg, “Solvent Effects 1. The Mediation of
Electrostatic Effects by Solvents,” J. Am. Chem. Soc., 113 (1991) 4776-82.
Wong91a M. W. Wong, K. B. Wiberg, and M. J. Frisch, “Hartree-Fock Second Derivatives and
Electric Field Properties in a Solvent Reaction Field - Theory and Application,”
J
. Chem.
Phys., 95 (1991) 8991-98.
Wong92 M. W. Wong, K. B. Wiberg, and M. J. Frisch, “Solvent Effects 2. Medium Effect on the
Structure, Energy, Charge Density, and Vibrational Frequencies of Sulfamic Acid,”
J
. Am.
Chem. Soc., 114 (1992) 523-29.
Wong92a M. W. Wong, K. B. Wiberg, and M. J. Frisch, “Solvent Effects 3. Tautomeric Equilibria of
Formamide and 2-Pyridone in the Gas Phase and Solution: An ab initio SCRF Study,”
J
.
Am. Chem. Soc., 114 (1992) 1645-52.
Wood06 G. P. F. Wood, L. Radom, G. A. Petersson, E. C. Barnes, M. J. Frisch, and J. A. Montgomery
Jr. , “A restricted-open-shell complete-basis-set model chemistry,” J. Chem. Phys., 125
(2006) 094106: 1-16.
Woon93 D. E. Woon and T. H. Dunning Jr., “Gaussian-
b
asis sets for use in correlated molecular
calculations. 3. The atoms aluminum through argon,” J. Chem. Phys., 98 (1993) 1358-71.
Xu04 X. Xu and W. A. Goddard III, “The X3LYP extended density functional for accurate
descriptions of nonbond interactions, spin states, and thermochemical properties,” Proc.
Natl. Acad. Sci. USA, 101 (2004) 2673-77.
Yamaguchi86 Y. Yamaguchi, M. J. Frisch, J. Gaw, H. F. Schaefer III, and J. S. Binkley, “Analytic
computation and basis set dependence of Intensities of Infrared Spectra,” J. Chem. Phys., 84
(1986) 2262-78.
- 337 -
Yamamoto96 N. Yamamoto, T. Vreven, M. A. Robb, M. J. Frisch, and H. B. Schlegel, “A Direct
Derivative MC-SCF Procedure,” Chem. Phys. Lett., 250 (1996) 373-78.
Yanai04 T. Yanai, D. Tew, and N. Handy, “A new hybrid exchange-correlation functional using the
Coulomb-attenuating method (CAM-B3LYP),” Chem. Phys. Lett., 393 (2004) 51-57.
York99 D. M. York and M. Karplus, “Smooth solvation potential based on the conductor-like
screening model,” J. Phys. Chem. A, 103 (1999) 11060-79.
Zakrzewski93 V. G. Zakrzewski and W. von Niessen, “Vectorizable algorithm for Green function and
many-body perturbation methods,” J. Comp. Chem., 14 (1993) 13-18.
Zakrzewski94a V. G. Zakrzewski and J. V. Ortiz, “Semidirect algorithms in electron propagator
calculations,” Int. J. Quantum Chem., Quant. Chem. Symp., S28 (1994) 23-27.
Zakrzewski95 V. G. Zakrzewski and J. V. Ortiz, “Semidirect algorithms for third-order electron propagator
calculations,” Int. J. Quantum Chem., 53 (1995) 583-90.
Zakrzewski96 V. G. Zakrzewski, J. V. Ortiz, J. A. Nichols, D. Heryadi, D. L. Yeager, and J. T. Golab,
“Comparison of perturbative and multiconfigurational electron propagator methods,” Int. J.
Quant. Chem., 60 (1996) 29-36.
Zerner80 M. C. Zerner, G. H. Lowe, R. F. Kirchner, and U. T. Mueller-Westerhoff, “An Intermediate
Neglect of Differential Overlap Technique for Spectroscopy of Transition-Metal Complexes.
Ferrocene,” J. Am. Chem. Soc., 102 (1980) 589-99.
Zerner82 M. C. Zerner, P. Correa de Mello, and M. Hehenberger, “Converging SCF Calculations on
Excited States,” Int. J. Quantum Chem., 21 (1982) 251-59.
Zerner91 M. C. Zerner, in Reviews of Computational Chemistry, Ed. K. B. Lipkowitz and D. B. Boyd,
Vol. 2 (VCH Publishing, New York, 1991) 313-66.
Zhao05 Y. Zhao, N. E. Schultz, and D. G. Truhlar, “Exchange-correlation functional with broad
accuracy for metallic and nonmetallic compounds, kinetics, and noncovalent interactions,”
J. Chem. Phys., 123 (2005)
Zhao06 Y. Zhao, N. E. Schultz, and D. G. Truhlar, “Design of density functionals by combining the
method of constraint satisfaction with parametrization for thermochemistry, thermochemical
kinetics, and noncovalent interactions,” J. Chem. Theory and Comput., 2 (2006) 364-82.
Zhao06a Y. Zhao and D. G. Truhlar, “A new local density functional for main-group thermochemistry,
transition metal bonding, thermochemical kinetics, and noncovalent interactions,”
J
. Chem.
Phys., 125 (2006), 194101: 1-18.
Zhao06b Y. Zhao and D. G. Truhlar, “Comparative DFT study of van der Waals complexes: Rare-gas
dimers, alkaline-earth dimers, zinc dimer, and zinc-rare-gas dimers,” J. Phys. Chem., 110
(2006) 5121-29.
Zhao06c Y. Zhao and D. G. Truhlar, “Density Functional for Spectroscopy: No Long-Range
Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and
Better Performance on Average than B3LYP for Ground States,” J. Phys. Chem. A, 110
(2006) 13126-30.
Zhao08 Y. Zhao and D. G. Truhlar, “The M06 suite of density functionals for main group
thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and
transition elements: two new functionals and systematic testing of four M06-class
functionals and 12 other functionals,” Theor. Chem. Acc., 120 (2008) 215-41.
- 338 -
Zheng05 G. Zheng, S. Irle, and K. Morokuma, “Performance of the DFTB method in comparison to
DFT and semiempirical methods for geometries and energies of C20-C86 fullerene
isomers,” Chem. Phys. Lett., 412 (2005) 210-16.
Zheng07 G. Zheng, H. Witek, P. Bobadova-Parvanova, S. Irle, D. G. Musaev, R. Prabhakar, K.
Morokuma, M. Lundberg, M. Elstner, C. Kohler, and T. Frauenheim, “Parameter calibration
of transition-metal elements for the spin-polarized self-consistent-charge density-functional
tight-binding (DFTB) method: Sc, Ti, Fe, Co and Ni,” J. Chem. Theory and Comput., 3
(2007) 1349-67.
Zhixing89 C. Zhixing, “Rotation procedure in intrinsic reaction coordinate calculations,” Theor. Chim.
Acta., 75 (1989) 481-84.
- 339 -
- 340 -
