IP4M.manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 75
Integrated Platform for Metabolomics Data
Mining (IP4M, V1.8)
User Manual
Feb 25 2019
Contents
1. Introduction ................................................................................................................................. 1
1.1 Aim and main features ........................................................................................................ 1
1.2 Running environments ........................................................................................................ 2
2. Usage rules ................................................................................................................................... 3
2.1 Interface structure and workflow ........................................................................................ 3
2.2 Inputs and outputs: .............................................................................................................. 7
3. Functional modules ..................................................................................................................... 8
3.1 Raw data Preprocessing ...................................................................................................... 8
3.1.1 LC-MS preprocessing .............................................................................................. 8
3.1.2 GC-MS preprocessing ............................................................................................ 10
3.2 Annotation by Public and Custom Libraries ..................................................................... 14
3.2.1 GC-MS peak annotation ......................................................................................... 14
3.2.2 LC-MS peak annotation ......................................................................................... 15
3.3 Peak Table Operations ....................................................................................................... 18
3.3.1 Pretreatment ........................................................................................................... 18
3.3.3 Other operations ..................................................................................................... 21
3.3.4 Transformation ....................................................................................................... 23
3.3.5 Merge tables ........................................................................................................... 24
3.4 Statistical Analysis ............................................................................................................ 26
3.4.1 Univariate statistical analysis ................................................................................. 26
3.4.2 Multivariate statistical analysis .............................................................................. 29
3.5 Pathway Analysis .............................................................................................................. 43
Tool: Compounds ID mapping ........................................................................................ 43
Tool: Pathway analysis on compounds ID mapping results ............................................ 44
Tool: Enrichment analysis on compounds ID mapping results ....................................... 45
3.6 Workflows ......................................................................................................................... 47
3.6.1 GC-MS data preprocessing workflow: from raw data to peak table ...................... 47
3.6.2 LC-MS data preprocessing workflow: from raw data to peak table ....................... 48
3.6.3 Statistical analysis based on peak table .................................................................. 49
3.6.4 Pathway and enrichment analysis .......................................................................... 51
3.7 Other Tools ........................................................................................................................ 53
3.7.1 Merge LECO CSV files ......................................................................................... 53
3.7.2 GLM on two groups ............................................................................................... 53
3.7.3 ROC analysis .......................................................................................................... 54
3.7.4 Hierarchical cluster analysis .................................................................................. 55
3.7.5 Plot tree .................................................................................................................. 57
3.7.6 Plot heatmap with tree ............................................................................................ 58
3.7.7 Sub-cluster expression analysis .............................................................................. 58
3.7.8 Correlation and distance analysis ........................................................................... 61
3.7.9 Plotting tools .......................................................................................................... 66
3.7.10 Sample size and power analysis ........................................................................... 69

IP4M V1.0
1
1. Introduction
1.1 Aim and main features
Metabolomics depends more and more on bioinformatics tools, along with its rapid evolution
and broad application. Currently, a number of free or commercial, desktop or web based, separate
or comprehensive tools have been developed but there is still an unmet demand for a green and
user-friendly desktop platform to cover all the steps of computational metabolomics. Here, an
all-in-one platform for mass spectrometry-based untargeted metabolomics data mining (IP4M)
was developed to provide an alternative tool for beginners and advanced users.
The main features of IP4M include the following: 1) IP4M developed using Java, Perl, and R
is a freely available, green, and instrument-independent tool. 2) IP4M covers all the representative
steps and functions of computational metabolomics, including peak identification and annotation,
raw data and peak table preprocessing, univariate and multivariate difference analysis, correlation
analysis, cluster and sub-cluster analysis, linear regression analysis, ROC analysis, pathway
analysis, venn analysis, and sample size and power analysis. The integrated functions and
packages are selected from numerous popular and representative ones. 3) IP4M is suitable for
beginners and advanced users, as it provides workflows for a quick and reproducible analysis and
offers sufficient basic/advanced parameters for a more refined analysis.
Compared with other multi-function platforms, the strengths of IP4M are the GC-MS peak
identification, many simple but useful tools, and rich knowledgebase. However, it is limited in
integration with other omics data. IP4M can be further extended to an online platform and NMR
data preprocessing module is could be incorporated. Nevertheless, it is still an attractive
alternative to existing platforms.
IP4M V1.0
2
1.2 Running environments
Software: Windows 7 and above
Hardware: CPU > 3.0 GHz; Memory > 8 Gb
Programming language: Java, Perl, and R
Administrator privileges are required.
This is a green desktop software. No registration is required.
IP4M V1.0
3
2. Usage rules
2.1 Interface structure and workflow
The software interface includes four parts: tools window, main window, task window and file
window.
Workflow (Fig. 1): select a tool in tools window–> Set parameters and execute in main
window –> View running status in task window –> View results in main and file window.
Fig.1 Workflow of usage
Specific steps:
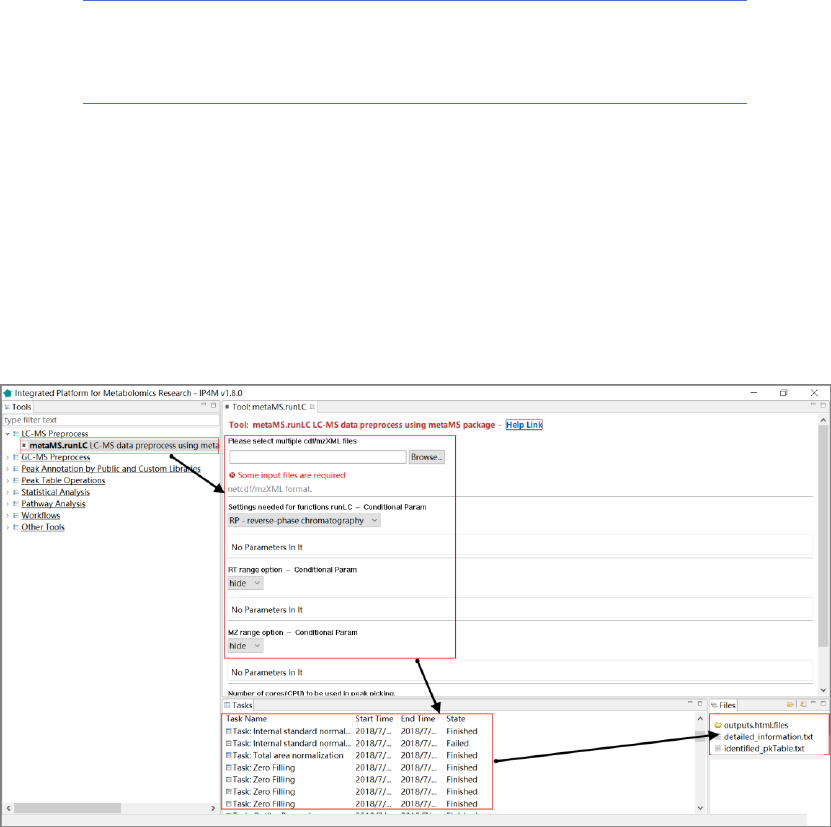
1) In the tools window, double-click the tool you want to use and the parameters setting panel
will pop up automatically (fig. 2).
IP4M V1.0
4
Fig.2 Select a tool
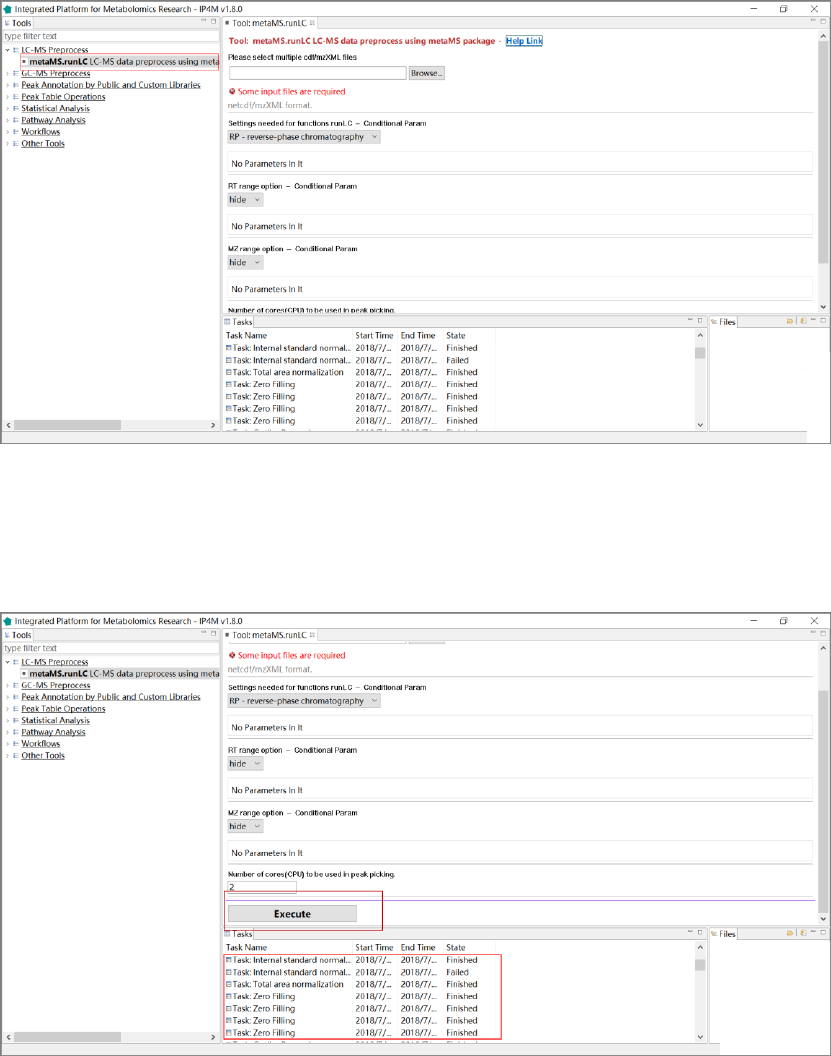
2) Use the default parameters or edit them as you want. Click the “Execute” button to run and
the corresponding task information will appear in the task window (fig. 3).
Fig.3 Execute the task
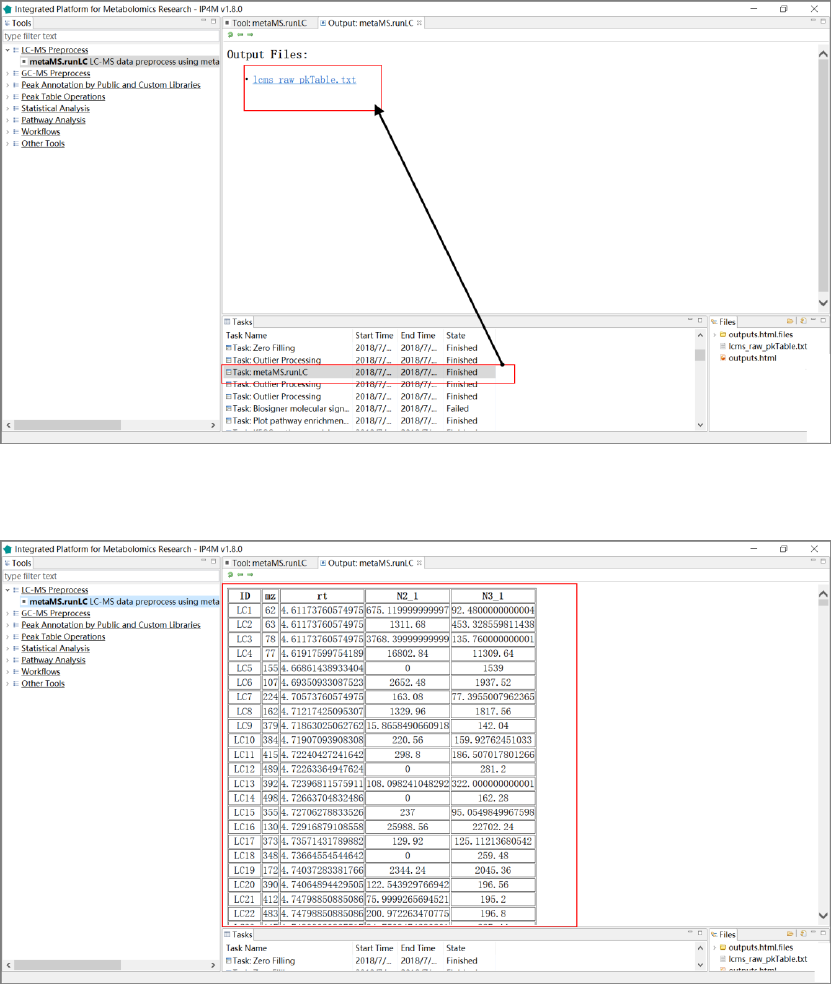
3) When the task is finished, double-click the task to view the list of result files in main window
and file window. Click the files to view the specific results (Fig. 4-5).
IP4M V1.0
5
Fig.4 View the results 1
Fig.5 View the results 2
IP4M V1.0
6
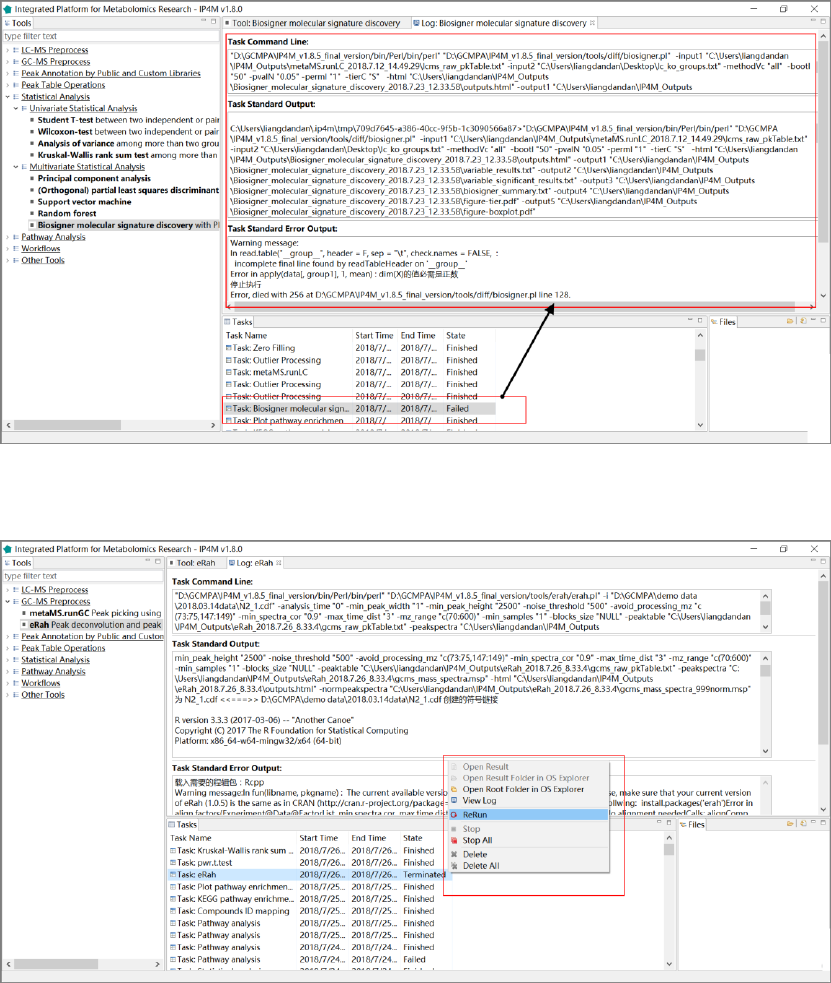
4) If the task has failed, you can double-click the task item to view the log information (fig. 6).
5) Right-click on the task item and select „Rerun‟ to edit the inputs and/or parameters as the error
messages and then re-run the task (fig. 7).
Fig.6 View the log information
Fig.7 Rerun the task
IP4M V1.0
7
2.2 Inputs and outputs:
Raw data of mzXML and NetCDF formats and other files (peak table, sample information,
compound list etc.) of tab-delimited text format are supported inputs. The free software
ProteoWizard (http://proteowizard.sourceforge.net/) is recommended for converting raw data files
from various instrument vendors to mzXML format. All the intermediate and final results are
exported as .txt files (data) or .pdf (figures) files.

IP4M V1.0
8
3. Functional modules
3.1 Raw data Preprocessing
3.1.1 LC-MS preprocessing
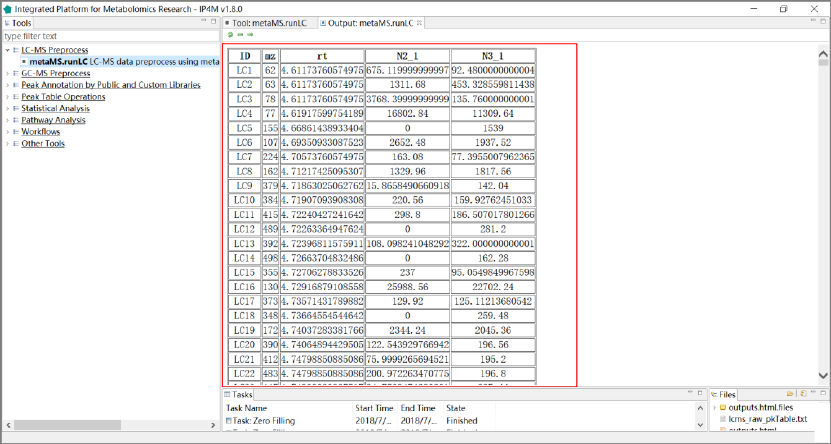
Tool: metaMS.runLC LC-MS data preprocessing using metaMS
package
This tool is a wrapper for the function 'runLC()' in the R 'metaMS' package. It is designed to
process a series of LC-MS data files and to produce a peak table with mz, rt, and intensities of
peaks in all samples. The popular package xcms is used to perform the peak picking, grouping and
retention correction, and peak filling operations.
Parameter:
1. RP - reverse-phase chromatography: This particular setting is fine-tuned for the analysis of
LC-MS runs.
2. NP - normal-phase chromatography: This particular setting is fine-tuned for the analysis of
LC-MS runs.
3. RT range: RT range to process in minutes, for example, 5,25.
4. MZ range option: MZ range retained for the analysis, for example, 50,500.
5. matchedFilter: Method to use for peak detection. This function identifies peaks in the
chromatographic time domain. The intensity values are binned by cutting the LC/MS data
into slices (bins) of a mass unit (binSize m/z) wide. Within each bin, the maximal intensity is
selected. The peak detection is then performed in each bin by extending it based on the steps
parameter to generate slices comprising bins current _bin - steps +1 to current _bin + steps -
1. Each of these slices is then filtered with matched filtration using a second-derivative
Gaussian as the model peak shape. After filtration peaks are detected using a signal-to-ration
cut-off.
6. step size: The peak detection algorithm creates extracted base peak chromatograms (EIBPC)
on a fixed step size.
7. FWHM: Full width at half maximum of matched filtration gaussian model peak. Can only be
used to calculate the actual sigma.
8. max: Maximum number of peak per extracted ion chromatogram.
9. snthresh: Signal to noise ratio cutoff.
IP4M V1.0
9
10. min. class. Fraction: Minimum fraction of sample necessary in at least one of the sample
groups for it to be a valid group.
11. min. class. Size: Minimum number of sample necessary in at least one of the sample groups
for it to be a valid group.
12. mzwid: Width of overlapping m/z slices to use for creating peak density chromatograms and
grouping peaks across samples.
13. bws: The two bandwidths used for grouping before and after retention time alignment.
14. missing ratio: Ratio of missing samples to allow in retention time correction groups.
15. extra ratio: Ratio of extra peaks to allow in retention time correction groups.
16. centWave: Method to use for peak detection. The centWave algorithm performs peak density
and wavelet-based chromatographic peak detection. It is most suitable for high-resolution
LC/{TOF,OrbiTrap,FTICR}-MS data in centroid mode. In the first phase the method
identifies regions of interest (ROIs) representing mass traces that are characterized as regions
with less than ppm m/z deviation in consecutive scans in the LC/MS map. These ROIs are
then subsequently analyzed using continuous wavelet transform (CWT) to locate
chromatographic peaks on different scales. The first analysis step is skipped, if regions of
interest are passed via the param parameter.
17. ppm: Numeric defining the maximal tolerated m/z deviation in consecutive scans in parts per
million (ppm) for the initial ROI definition
18. peakwidth: numeric with the expected approximate peak width in chromatographic space.
Given as a range (min, max) in seconds.
19. prefilter: numeric: c (k, I) specifying the prefilter step for the first analysis step (ROI
detection). Mass traces are only retained if they contain at least k peaks with intensity >= I.
Reference:
[1] R. Wehrens, G. Weingart and F. Mattivi, metaMS: An open-source pipeline for GC-MS-based
untargeted metabolomics J. Chrom. B (2014), v966, 109-116.
[2] Colin A. Smith, Elizabeth J. Want, Grace O‟Maille, Ruben Abagyan and Gary Siuzdak.
"XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak
Alignment, Matching, and Identification" Anal. Chem. 2006, 78:779-787.
[3] Ralf Tautenhahn, Christoph B\"ottcher, and Steffen Neumann "Highly sensitive feature
detection for high resolution LC/MS" BMC Bioinformatics 2008, 9:504
IP4M V1.0
10
Results and visualization:
Fig. 1 the outputted Peak table with two samples of metaMS.runLC tool
3.1.2 GC-MS preprocessing
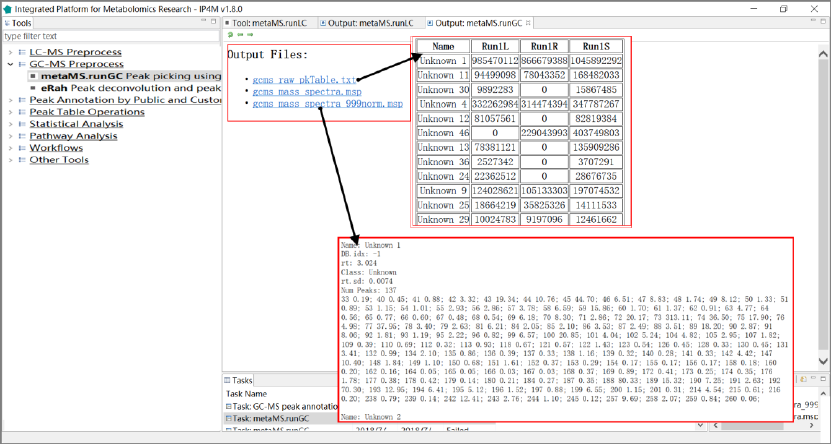
Tool: metaMS.runGC peak picking using metaMS package
This tool is a wrapper for the function 'runGC()' in the R 'metaMS' package which is designed
to process a series of GC-MS data files and to produce a peak table. It performs a
pseudospectrum-based analysis, where the basic entity is a collection of (mz, I) pairs at specific
retention times. The standard workflow of metaMS for GC-MS data is the following:
1. peak picking;
2. definition of pseudospectra;
3. identification and elimination of artefacts;
4. annotation by comparison to a database of standards;
5. definition of unknowns;
6. output.
Parameter:
1. RT range: part of the chromatograms that is to be analyzed. If given, it should be a vector of
two numbers indicating minimal and maximal retention time (in minutes). For example 5, 25.
2. FWHM: numeric specifying the full width at half maximum of matched filtration gaussian
model peak. Can only be used to calculate the actual sigma.
3. RT_ Diff: the allowed RT shift in minutes between different samples.
IP4M V1.0
11
4. Min_ Features: the minimum number of ion in a mass spectrum.
5. similarity_ threshold: the minimum similarity allowed between mass spectra considered as
the same compound.
6. min. class. fract: the fraction of samples in which a pseudospectrum is present before it is
regarded as an unknown.
7. min. class. size: the absolute number of samples in which a pseudospectrum is present before
it is regarded as an unknown.
Reference:
[1] R. Wehrens, G. Weingart and F. Mattivi, metaMS: An open-source pipeline for GC-MS-based
untargeted metabolomics J. Chrom. B (2014), v966, 109-116.
Results and visualization:
Fig.2 The outputted total results files, peak table, and normalized mass spectra information of
metaMS.runGC.
IP4M V1.0
12
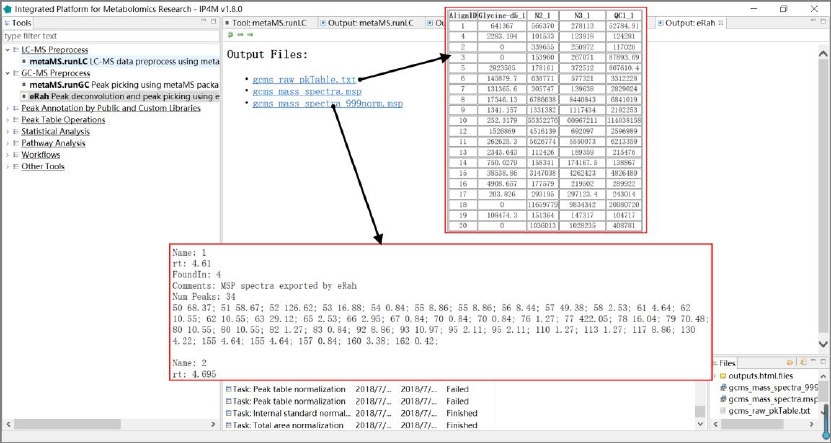
Tool: eRah Peak deconvolution and peak picking using eRah package
This tool is a wrapper of the R 'eRah' package for GC-MS data processing. 'eRah' is an R
package that allows for an innovative deconvolution of GC-MS chromatograms using multivariate
techniques based on blind source separation (BSS). It automatically detects and deconvolves the
spectra of the compounds appearing in GC-MS chromatograms. Then, compounds are aligned by
spectral similarity and retention time distance. It computes the Euclidean distance between
retention time distance and spectral similarity for all compounds in the chromatograms, resulting
in compounds appearing across the maximum number of samples and with the least retention time
and spectral distance. After that, a missing compound recovery step can be applied to recover
those compounds that are missing in some samples. Missing compounds appear as a result of an
incorrect deconvolution or alignment - due to a low compound concentration in a sample - , or
because it is not present in the sample. This forces the final data table with compound names and
compounds area, to not have any missing (zero) values. Please see the references for detailed
descriptions.
Parameter:
1. RT window: The chromatographic retention time window to process. If 0 all the
chromatogram is processed.
2. Minimum peak width: This is a critical parameter that conditions the efficiency of eRah.
Typically, it should be the half of the mean compound width.
3. noise. threshold: Data above this threshold will be considered as noise
4. avoid.processing.mz: The masses that do not want to be considered for processing. Typically,
in GCMS those masses are 73,74,75,147,148 and 149, since they are ubiquitous mass
fragments typically generated from compounds carrying a trimethylsilyl moiety.
5. Minimum spectral correlation value: From 0 (no similar) to 1 (very similar). This value sets
how similar two or more compounds have to be considered for alignment between them.
6. Maximum retention time distance: This value (in seconds) sets how far two or more
compounds can be considered for alignment between them.
7. Minimum. sample: The minimum number of samples in which a compound has to appear to
be considered for searching into the rest of the samples where this compound is missing.
8. blocks. size: For experiments containing more than 100 (Windows) or 1000 (Mac or Linux)
samples (numbers depending on the computer resources and sample type). In those cases,
alignment can be conducted by block segmentation. For an experiment of e.g. 1000 samples,
the block.size can be set to 100, so the alignment will perform as multiple (ten) 100-samples
experiments, to later align them into a single experiment.
This parameter is designed to solve the typical problem that appears when aligning under the
IP4M V1.0
13
Windows operating system: "Error: cannot allocate vector of size XX Gb". Such a problem
will not appear with Mac or Linux, but several hours of computation are expected when
aligning a large number of samples. Using block segmentation provides a greatly improved
run-time performance.
Reference:
[1] X. Domingo-Almenara, et al., eRah: a computational tool integrating spectral deconvolution
and alignment with quantification and identification of metabolites in GC{MS-based
metabolomics. Analytical Chemistry. 88 (2016) 9821{9829. DOI:
10.1021/acs.analchem.6v02927
[2] X. Domingo-Almenara, et al., Compound identification in gas chromatography/mass
spectrometry-based metabolomics by blind source separation. Journal of Chromatography A
1409 (2015) 226{233. DOI: 10.1016/j.chroma.2015.07.044
Results visualization:
Fig.3 The outputted results files, peak table, and normalized mass spectra of eRah package.
IP4M V1.0
14
3.2 Annotation by Public and Custom Libraries
3.2.1 GC-MS peak annotation
Tool: GC-MS peak annotation on msp database files
This tool intends to annotate compounds from the GC-MS peak table by matching mass
spectra and/or retention times of public/custom library and detected peaks. If you want to use the
custom library for annotation, a standard MSP format file is required.
Parameter:
1. normalized dot product: Matching factor function for mass spectrum. The function applies
weights to an input to get weighted outputs.
2. normalized Euclidean distance: Matching factor function for mass spectrum.
3. mass spectrum similarity cutoff: 0-1, more similar larger matching factor.
4. RT window: The retention time difference that can be allowed.
5. NSEN: An integrated library derived from NIST/EPA/NIH. It is the default public library.
6. GMD_ALK: A public database from the Golm Metabolome Database (GMD). ALK - based
on 9 n-alkanes (C10–C36).
7. GMD_FAME: A public database from the Golm Metabolome Database (GMD). FAME -
based on 13 fatty acid methyl esters (C8 ME–C30 ME).
8. GMD_MSIR: The 'Q_MSRI_ID' GC-Quadrupole-MS MSRI Database of Golm Metabolome
library.
9. MoNA-HMDB: It is derived from MassBank of North America, with 4620
spectra(http://mona.fiehnlab.ucdavis.edu/downloads).
10. MoNA-MetaboBASE: It is derived from MassBank of North America, with 1254 spectra
(http://mona.fiehnlab.ucdavis.edu/downloads).
11. MoNA-ReSpect: It is derived from MassBank of North America, with 6290
spectra(http://mona.fiehnlab.ucdavis.edu/downloads).
Note:
There is no retention time field in the public library and only mass spectrum information is
used for annotation. For a custom library, this tool supports the joint annotation by mass spectrum
and retention time. Users can provide an in-house library file in MSP format containing the field
'rt'. This is an optional field. A compound can be repeated in the database with the same 'Name '
IP4M V1.0
15
but a different mass spectrum. In this case, the best hit will be outputted.
Reference:
[1] Schauer N, Steinhauser D, Strelkov S, Schomburg D, Allison G, Moritz T, Lundgren K,
Roessner-Tunali U, Forbes MG, Willmitzer L, Fernie AR, Kopka J: GC-MS libraries for the
rapid identification of metabolites in complex biological samples. FEBS Lett 2005,
579(6):1332–1337. 10.1016/j.febslet.2005.01.029
[2] Kopka, J., Schauer, N., Krueger, S., Birkemeyer, C., Usadel, B., Bergmuller, E., Dormann, P.,
Weckwerth, W., Gibon, Y., Stitt, M., Willmitzer, L., Fernie, A.R. and Steinhauser, D. (2005)
GMD@CSB.DB: the Golm Metabolome Database, Bioinformatics, 21, 1635-1638.
Results and visualization:
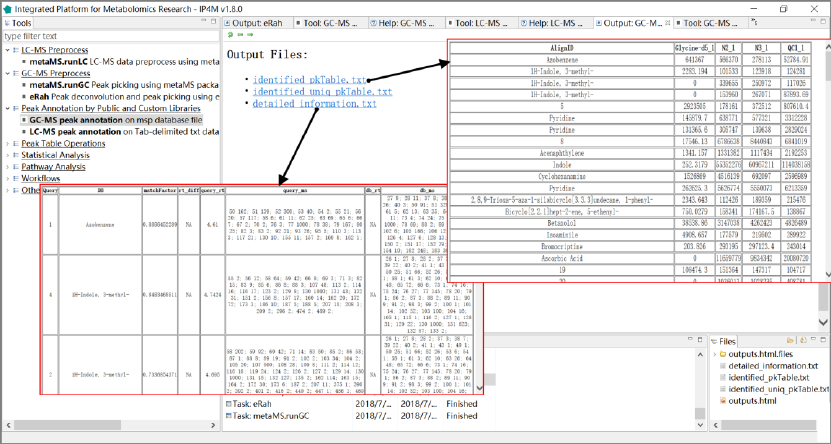
Fig.4 The outputted total files, identified peak table, and compounds detailed information of
GC-MS peak table annotation.
3.2.2 LC-MS peak annotation
Tool: LC-MS peak annotation on xls database files
This tool is used to annotate compounds from the LC-MS peak table by comparing m/z and
RT with the public/custom library. The best top five hits will be shown in the results. If you want
to use the custom library for annotation, a two-column Tab-delimited text file is required with the
first column as compound name and the second column as precise MZ.
IP4M V1.0
16
Parameter:
1. mz cutoff: A hit that difference of mz must be <=mz_cutoff.
2. RT window: The acceptable retention time difference.
3. ppm: Parts per million. numeric the relative error for matching peaks that is a window of user
specified error (or the default 10) in ppm for each fragment mass.
(|M- M0|÷m)×106 (ppm). „M‟ is the measured value of the ion mass; „M0‟ is the theoretical
value of the ion mass; „m‟, an integer, is the mass of the ion.
For example, the molecular ion measured value of a compound is 364.2504, the theoretical
value is 364.2509, and the mass measurement accuracy is:
|364.2504-364.2509|÷364×106=1.4ppm
4. adducts type: There are several possible adducts and the recommended type that most
commonly occurs is “M+H” or “M-H”.
Note:
1. There is no retention time field in the public library and only mass spectrum information is
used for annotation. For a custom library, this tool supports the joint annotation by precise
MZ and retention time. Users can provide an in-house two-column Tab-delimited text file
with the first column as compound name and the second column as precise MZ.
2. The tool will identify all possible matching compounds based on all adduct types selected,
sort them according to the matching score, and output them all as 'detailed_information.txt'.
Also, the compound with the smallest MZ and RT deviation will be outputted as the final
identified compound in 'identified_pkTable.txt' file.
Results and visualization:
IP4M V1.0
17
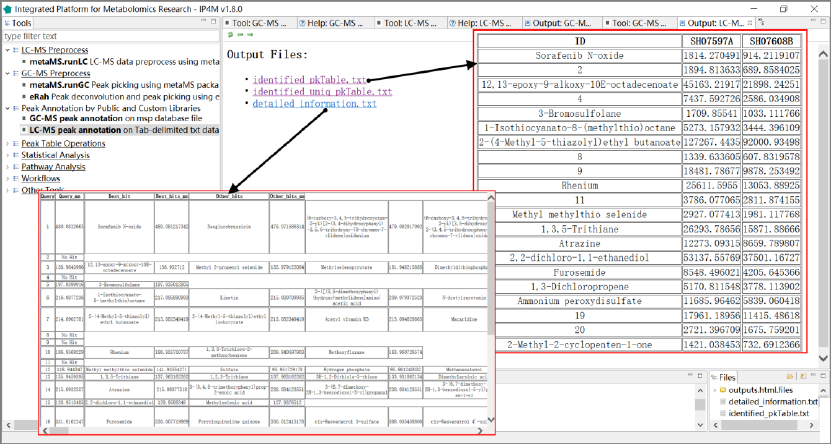
Fig.5 The outputted results files, identified peak table, and detailed information of compounds of
LC-MS peak annotation.
IP4M V1.0
18
3.3 Peak Table Operations
3.3.1 Pretreatment
Tool: Outlier processing on peak table
The tool takes a peak table file as input and processes the outliers using the capping method.
Default boundary is [0, Q3+1.5*IQR]. If the value > (Q3+1.5*IQR), it is identified as an outlier
and replaced by the maximum value within the normal range.
Parameter:
1. Q3: The third quartile (Q3), also known as the "larger quartile", equals to the value ranked at
75% of all values in ascending order.
2. IQR: InterQuartile Range, equals to |Q3 minus Q1|.
Tool: Zero filling on peak table
This tool takes a peak table file as input and fills the missing values (zero , null value or „NA‟,
or negative values) with 1) the a*min value, where 'a' is a user-defined coefficient; 2) 'min' which
is the minimum non-negative value in the peak table; 3) user-specified value; 4) values computed
by 'KNN'; and 5) values computed by 'qirlc'. The „qirlc‟ algorithm is especially suitable for
left-censored data.
Reference:
If you use 'KNN' method, references:
[1] Hastie, T., Tibshirani, R., Sherlock, G., Eisen, M., Brown, P. and Botstein, D., Imputing
Missing Data for Gene Expression Arrays, Stanford University Statistics Department
Technical report (1999).
[2] Olga Troyanskaya, Michael Cantor, Gavin Sherlock, Pat Brown, Trevor Hastie, Robert
Tibshirani, David Botstein and Russ B. Altman, Missing value estimation methods for DNA
microarrays BIOINFORMATICS Vol. 17 no. 6, 2001 Pages 520-525
If you use 'qirlc' method, references:
[3] QRILC: a quantile regression approach for the imputation of left-censored missing data in
quantitative proteomics, Cosmin Lazar et al.
[4] Wei R, Wang J, Su M, et al. Missing Value Imputation Approach for Mass Spectrometry-based

IP4M V1.0
19
Metabolomics Data: [J]. Scientific Reports, 2018, 8(1).
[5] Wei R, Wang J, Jia E, et al. GSimp: A Gibbs sampler based left-censored missing value
imputation approach for metabolomics studies: [J]. Plos Computational Biology, 2018,
14(1):e1005973.
3.3.2 Normalization
Tool: Total area normalization on peak table
This tool takes a peak table file as input and performs total intensity normalization within
samples. The formula is (x/sum of total intensity within the corresponding sample) *1000.
Tool: Internal standard normalization
This tool takes a peak table file as input and performs internal standard (IS) normalization
within samples. The normalization formula is (x/internal standard) *10000.
Parameters:
Set the standard compound: for example Chlorophenylalanine
Note:
1. The IS compound must exist in the inputted peak table.
2. Experimental preparation: The internal standard must be prepared in advance and added
quantitatively to each sample.
Tool: Peak table normalization based on QC (pooled samples)
This tool takes a peak table file as input and performs normalization based on quality control
samples (QCs).
QCs are pooled samples. They contain the same compounds as the subject samples and are
supposed to reflect the average metabolite concentrations within a study. QCs are pretreated
according to the same protocols as the subject samples and are evenly injected throughout the
analyses. The performances of the pretreatment and the analytical platform can be assessed using
the QCs. The normalization formula is (x metabolite/QC metabolite) *10000.
Input files:

IP4M V1.0
20
1. Peak table file in Tab-delimited text format, with the first column as the compound identifier
and others as samples.
For example:
Table.1 Peak table with QC
AlignID
STDmix_GC_01
STDmix_GC_02
QC1
STDmix_GC_03
QC2
Unknown 1
1486892478
561322777
3448620272
3448620272
561322777
Nitrogen dioxide
5492977592
684434115
3265669981
3265669981
3265669981
Ethanol, 2-fluoro-
2265686433
4182838129
4365291513
4365291513
4182838129
3-Pentanone, 2,2,4,4-
tetramethyl-
13390154
12612932
21155307
21155307
21155322
Hydrazine
14588107
8510918
7224351
7224351
7224380
2. Sample-to-QC design file, a Tab-delimited text file with two columns, "sample" and "QC".
For example:
Table.2 Sample-to-QC group file
STDmix_GC_01
QC1
STDmix_GC_02
QC1
STDmix_GC_03
QC2
Output files:
'QC_norm_pkTable.txt', normalized peak table.
For example:
Table.3 Normalized peak table based on QC
AlignID
STDmix_GC_01
STDmix_GC_02
QC1
STDmix_GC_03
QC2
1
4311.558
1627.673
10000
61437.38
10000
Nitrogen dioxide
16820.37
2095.846
10000
10000
10000
Ethanol, 2-fluoro-
5190.229
9582.036
10000
10436.2
10000
3-Pentanone, 2,2,4,4-tetramethyl-
6329.454
5962.065
10000
9999.993
10000
Hydrazine
20192.97
11780.88
10000
9999.96
10000
IP4M V1.0
21
3.3.3 Other operations
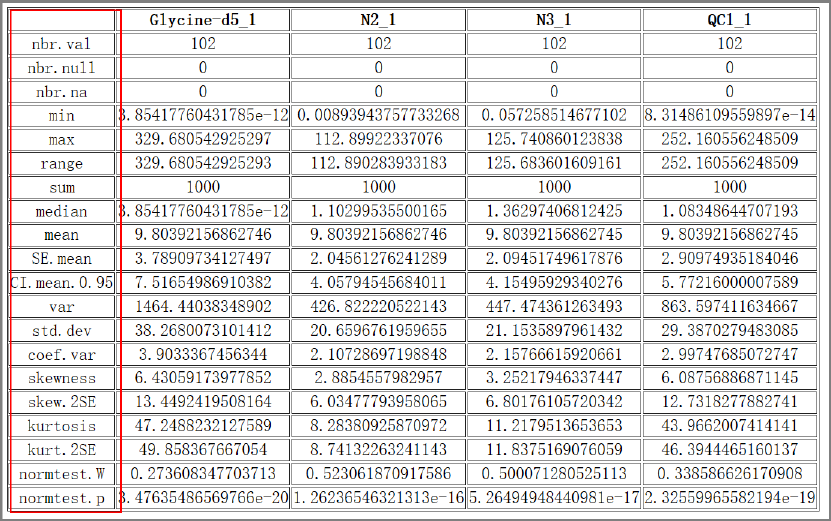
Tool: Basic statistics summary
This tool takes a peak table file as input and outputs the basic statistics, including 'nbr.val',
'nbr.null', 'nbr.na', 'min', 'max', 'range', 'sum', 'median', 'mean', 'SE.mean', 'CI.mean.0.95' ,'var',
'std.dev', 'coef.var', 'skewness', 'skew.2SE', 'kurtosis', 'kurt.2SE', 'normtest.W', and 'normtest.p'. .
Results and visualization:
Fig.6 The basic statistics summary of four samples
Tool: retrieve rows from peak table
The tool takes a peak table file and a one-column compounds list file as inputs and outputs a
sub-peak table file which rows correspond to the compounds list.
Input files:
1. Peak table file in Tab-delimited text format, with the first column as the compound identifier
and others as samples.
For example:
Table.4 Peak table

IP4M V1.0
22
HU_011
HU_014
HU_015
HU_017
HU_018
HU_019
(2-methoxyethoxy)propanoic
acid isomer
3.019766
3.814339
3.519691
2.562183
3.781922
4.161074
(gamma)Glu-Leu/Ile
3.888479
4.277149
4.195649
4.32376
4.629329
4.412266
1-Methyluric acid
3.869006
3.837704
4.102254
4.53852
4.178829
4.516805
1-Methylxanthine
3.717259
3.776851
4.291665
4.432216
4.11736
4.562052
1,3-Dimethyluric acid
3.535461
3.932581
3.955376
4.228491
4.005545
4.320582
2. A one-column compound list file in text format.
For example:
Table.5 Compound list file
1-methyluric acid
1-Methylxanthine
1,3-Dimethyluric acid
1,7-Dimethyluric acid
Output files:
A sub-peak table file in Tab-delimited text format, with the retrieved information according
to the compounds list.
For example:
Table.6 Sub-peak table
HU_011
HU_014
HU_015
HU_017
HU_018
HU_019
1-Methyluric acid
3.869006
3.837704
4.102254
4.53852
4.178829
4.516805
1-Methylxanthine
3.717259
3.776851
4.291665
4.432216
4.11736
4.562052
1,3-Dimethyluric acid
3.535461
3.932581
3.955376
4.228491
4.005545
4.320582
1,7-Dimethyluric acid
3.325199
4.025125
3.972904
4.109927
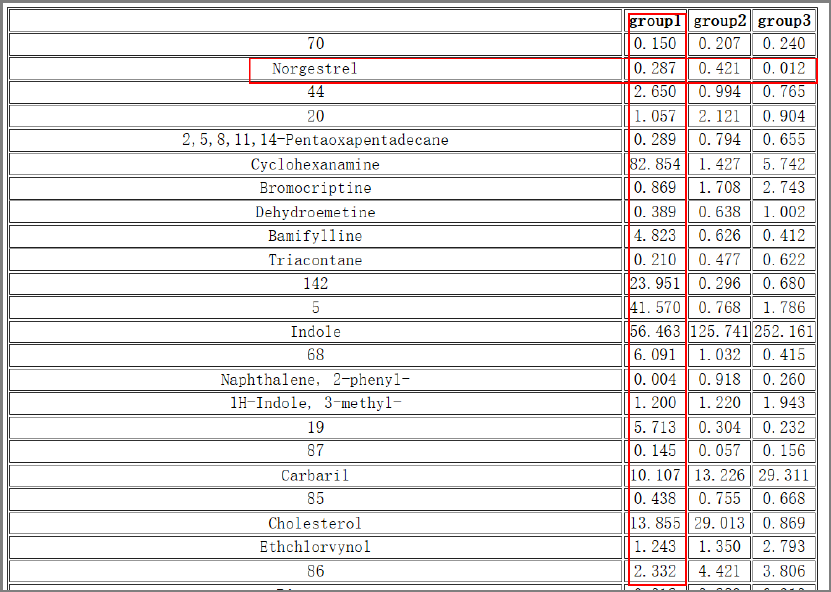
Tool: Row average by groups
This tool takes a peak table file and a samples-to-group design file as inputs, and outputs the
averaged intensity of every compound in every group.
Results and visualization:
IP4M V1.0
23
Fig.7 The outputted averaged intensity of every compound in 3 groups.
3.3.4 Transformation
Tool: Log2 transformation
This tool takes a peak table file as input and performs log transformation (base 2) or median
centered log2 transformation on the peaks.
Note:
The transformation formula is: log2 (value+1)
The median center is performed on the row (compound data).
Tool: Z-score transformation
This tool takes a peak table file as input and performs z-score transformation on the peaks.
This method standardizes the data by mean and standard deviation of the original data. It is
applicable to the cases where the maximum and minimum values are unknown, or there is outlier
data beyond the range of values. Formula is new data = (original data - mean)/standard deviation.

IP4M V1.0
24
Tool: Transpose
This tool takes a matrix data as input and performs transpose operation.
3.3.5 Merge tables
Tool: Merge tables by compound name
This tool takes multiple peak tables as input and merges them together. The outputted peak
table will have more samples and compounds. If a compound exists in some but not all tables, it
will be filled as NA in missing position in the final merged table. If same sample names exist in
different tables, their common compounds will be averaged and outputted in the final table.
Input files:
Multiple peak table files in Tab-delimited text format, with the first column as the compound
identifier and the others as samples.
For example:
Peak table 1:
Table.7 The inputted table1
AlignID
STDmix_GC_01
STDmix_GC_02
1
1486892478
451322711
Nitrogen dioxide
5492977400
684433223
Ethanol, 2-fluoro-
2265686433
4182838129
Peak table2:
Table.8 The inputted table2
AlignID
STDmix_GC_02
STDmix_GC_03
1
0
3448620100
Nitrogen dioxide
3265968000
3265668000
Norgestrel
789.33
5315.224
Output files:
'merged_matrix.txt', merged peak table file in Tab-delimited text format.
Table.9 The merged table
AlignID
STDmix_GC_01
STDmix_GC_02
STDmix_GC_03
1
1486892478
451322711
3448620272

IP4M V1.0
25
Nitrogen dioxide
5492977592
1975200611.5
3265669981
Ethanol, 2-fluoro-
2265686433
4182838129
NA
Norgestrel
NA
789.33
5315.224
IP4M V1.0
26
3.4 Statistical Analysis
3.4.1 Univariate statistical analysis
Tool: Student t test between two independent or paired groups
This tool performs the Student t-test and multiple comparison correction on the peaks of the
inputted table. Group information is given by a group design file (Tab-delimited text file). The
number of groups should be 2. For paired t-test, pairs are defined according to the order in each of
the two groups and the number of samples must be equal in the two groups.
Note:
Groups number must be 2 in the sample group file.
Group names of characters or string are preferred. Numbers are also supported but not
recommended.
Input files:
Group design file. For paired t-test, pairs are according to the order in each of the two groups.
For example:
Table.10 The group file for paired t-test, with 3 pairs (_p1, _p2, and _p3) in different color blocks
HU_01_p1
M
HU_02_p1
F
HU_03_p2
M
HU_04_p3
M
HU_05_p2
F
HU_06_p3
F
Output files:
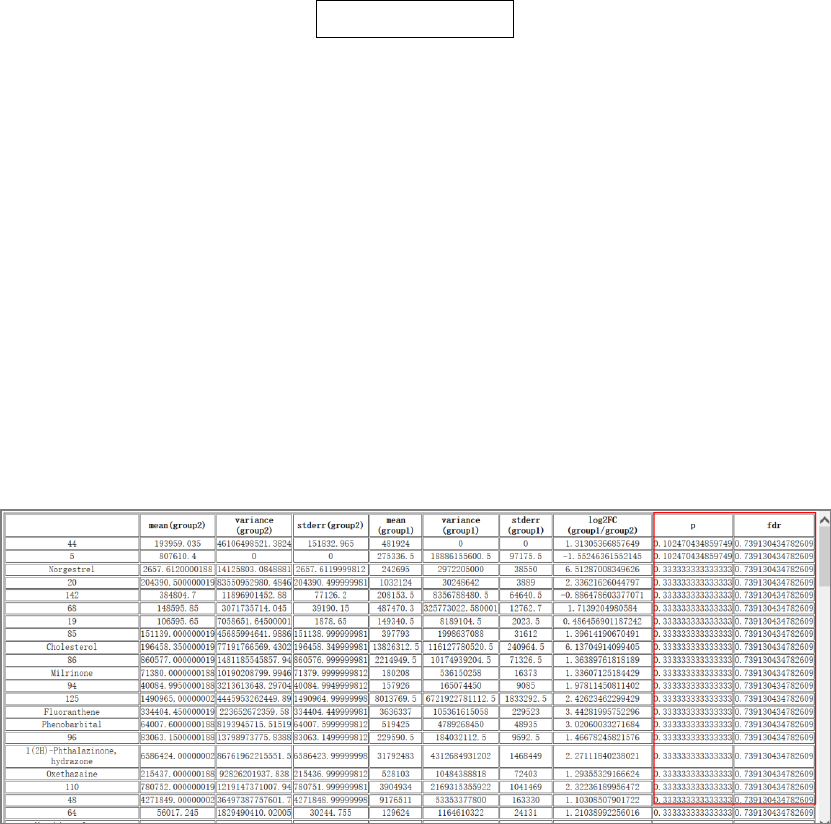
1. 't_test_results.txt', t-test results with p value, log2FC, and q value.
2. 't_test_significant_results.txt', significant t-test results.
Note:
Groups number must be 2 in the sample group file.
Group names of characters or string are preferred. Numbers are also supported but not

IP4M V1.0
27
recommended.
Results and visualization:
Fig.8 The outputted files with the full results and significant results of t-test method.
Tool: Wilcoxon-signed-rank-test between two paired groups
This tool performs the Wilcoxon-test and multiple comparison corrections to find the
significant peaks on the peak table data. Group information is given by a group design file
(Tab-delimited text file). The number of groups should be 2. For a paired test, pairs are according
to the order in each of the two groups. For example, A-group-first-sample and
B-group-first-sample are a pair. For a paired test, the number of samples must be equal in the two
groups. For a paired-test, a Wilcoxon rank sum test (equivalent to the Mann-Whitney test) is
carried out, otherwise, a Wilcoxon signed rank test is performed.
Input files:
Group design file. For a paired t-test, pairs are according to the order in each two groups.
For example:
Table.11 The group file for paired test, with 3 pairs in different color blocks
HU_01_p1
M
HU_02_p1
F
HU_03_p2
M
HU_04_p3
M
HU_05_p2
F
IP4M V1.0
28
HU_06_p3
F
Output files:
1. 'wilcox_test_results.txt', Wilcoxon-test results with p value, log2FC, and q value.
2. 'wilcox _test_significant_results.txt', significant Wilcoxon-test results.
Note:
Group number must be 2 in the sample group file.
Group names of characters or string are preferred. Numbers are also supported but not
recommended.
Results and visualization:
Fig.10 The results of Wilcoxon-signed-rank-test between two paired groups
Tool: Analysis of variance among more than two groups
This tool fits an analysis of variance model to find the significant peaks on the inputted peak
table. Group information is given by a group design file (Tab-delimited text file).
Note:
Group names of characters or string are preferred. Numbers are also supported but not
recommended.
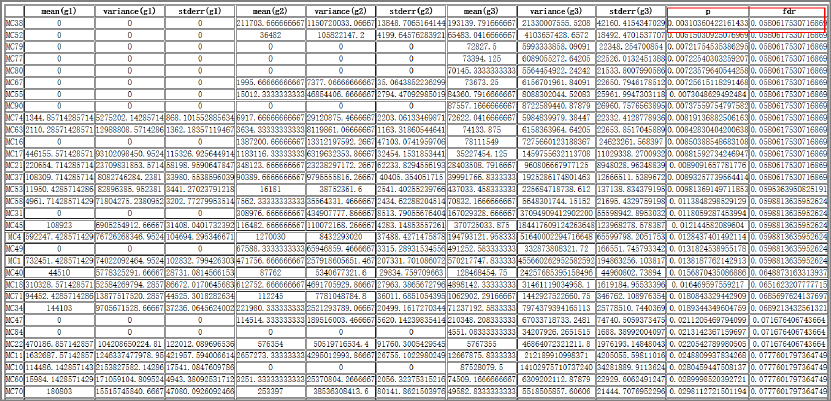
Results and visualization:
IP4M V1.0
29
Fig.11 The result of analysis of variance among 3 groups
Tool: Kruskal-Wallis rank test among more than two groups
This tool performs a Kruskal-Wallis rank sum test and multiple comparison corrections to
find the significant peaks on the inputted peak table. Group information is given by a group design
file (Tab-delimited text file).
Note:
Group names of characters or string are preferred. Numbers are also supported but not
recommended.
3.4.2 Multivariate statistical analysis
Tool: Principal component analysis
This tool performs a principal components analysis on the inputted peak table data. If the
group design file (a Tab-delimited text file) is provided, samples in the same group will be plotted
as the same color.
Input files:
1. Peak table file in Tab-delimited text format, with the first column as compound identifier, the
others as samples.
For example:

IP4M V1.0
30
Table.12 The inputted peak table file
HU_01
1
HU_01
4
HU_01
5
HU_01
7
HU_01
8
HU_01
9
(2-methoxyethoxy)propanic
acid isomer
3.0197
66
3.8143
39
3.5196
91
2.5621
83
3.7819
22
4.1610
74
(gamma)Glu-Leu/Ile
3.8884
79
4.2771
49
4.1956
49
4.3237
6
4.6293
29
4.4122
66
1-Methyluric acid
3.8690
06
3.8377
04
4.1022
54
4.5385
2
4.1788
29
4.5168
05
1-Methylxanthine
3.7172
59
3.7768
51
4.2916
65
4.4322
16
4.1173
6
4.5620
52
1,3-Dimethyluric acid
3.5354
61
3.9325
81
3.9553
76
4.2284
91
4.0055
45
4.3205
82
1,7-Dimethyluric acid
3.3251
99
4.0251
25
3.9729
04
4.1099
27
4.0240
92
4.3268
56
2-acetamido-4-methylphenyl
acetate
4.2047
54
5.1818
58
3.8856
8
4.2379
15
1.8529
94
4.0806
81
2-Aminoadipic acid
4.0802
04
4.3592
46
4.2491
11
4.2314
04
4.3236
79
4.2444
85
2.(Optional), Group design file in Tab-delimited text file.
For example:
Table.13 The inputted group design file
HU_011
M
HU 014
F
HU_015
M
HU_017
M
HU_018
M
HU_019
M
Output files:
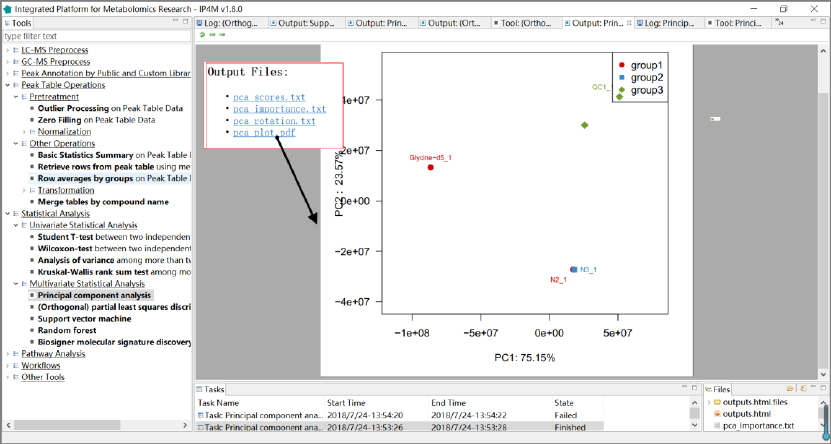
1. 'pca_scores.txt', PCs (scores) matrix.
2. 'pca_importance.txt', importance of PCs.
3. 'pca_rotation.txt', the matrix of variable loadings (i.e., a matrix whose columns contain the
eigenvectors).
4. 'pca_plot.pdf', PCA plot using PCs score values, the default is PC1 and PC2.
Results and visualization:
IP4M V1.0
31
Fig.12 The resulting files and the PCA scores plot
Tool: (Orthogonal) partial least squares discriminant analysis
This tool performs the OPLS-DA algorithm to rank peaks on the inputted table by variable
importance in projection (VIP). Group information is given by a group design file (Tab-delimited
text file). OPLS-DA is only available for binary classification and the number of groups should be
2.
The orthogonal Partial Least-Squares (OPLS) algorithm was introduced by J. Trygg and
Wold (2002) in order to model separately the variations of the predictors correlated and orthogonal
to the response. It has a similar predictive capacity compared to PLS and improves the
interpretation of the predictive components and of the systematic variation (Pinto, Trygg, and
Gottfries 2012). In particular, OPLS modeling of single responses only requires one predictive
component. Diagnostics such as the Q2Y metrics and permutation testing are of high importance
to avoid overfitting and assess the statistical significance of the model. The VIP, which reflects
both the loading weights for each component and the variability of the response explained by this
component (Pinto, Trygg, and Gottfries 2012; Mehmood et al. 2012), can be used for feature
ranking and selection (J. Trygg and Wold 2002; Pinto, Trygg, and Gottfries 2012).
Input files:
1. Peak table file in Tab-delimited text format, with the first column as the compound identifier
and others as samples.
For example:
Table.13 The inputted peak table file

IP4M V1.0
32
HU_01
1
HU_01
4
HU_01
5
HU_01
7
HU_01
8
HU_01
9
(2-methoxyethoxy)propanoic
acid isomer
3.0197
66
3.8143
39
3.5196
91
2.5621
83
3.7819
22
4.1610
74
(gamma)Glu-Leu/Ile
3.8884
79
4.2771
49
4.1956
49
4.3237
6
4.6293
29
4.4122
66
1-Methyluric acid
3.8690
06
3.8377
04
4.1022
54
4.5385
2
4.1788
29
4.5168
05
1-Methylxanthine
3.7172
59
3.7768
51
4.2916
65
4.4322
16
4.1173
6
4.5620
52
1,3-Dimethyluric acid
3.5354
61
3.9325
81
3.9553
76
4.2284
91
4.0055
45
4.3205
82
1,7-Dimethyluric acid
3.3251
99
4.0251
25
3.9729
04
4.1099
27
4.0240
92
4.3268
56
2-acetamido-4-methylphenyl
acetate
4.2047
54
5.1818
58
3.8856
8
4.2379
15
1.8529
94
4.0806
81
2-Aminoadipic acid
4.0802
04
4.3592
46
4.2491
11
4.2314
04
4.3236
79
4.2444
85
2. Group design file in Tab-delimited text file with two columns (samplename groupname).
For example:
Table.14 The inputted group design file
HU_011
M
HU 014
F
HU_015
M
HU_017
M
HU_018
M
HU_019
M
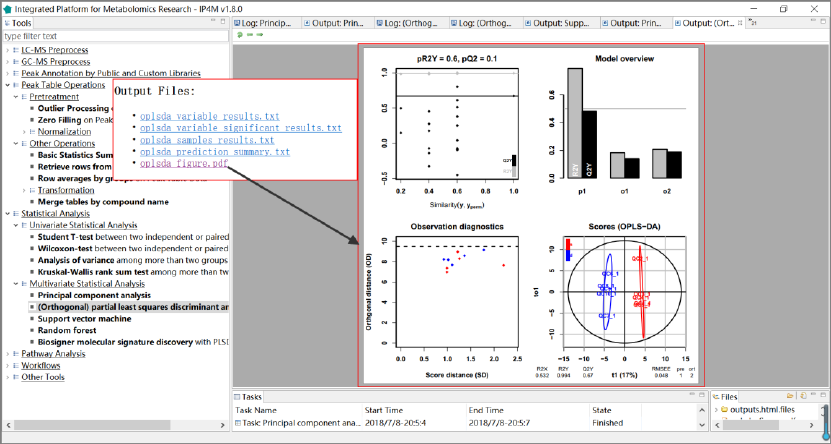
Output files:
1. 'oplsda_variable_results.txt', feature ranked results that are sorted by VIP.
2. 'oplsda_variable_significant_results.txt', significant feature results.
3. 'oplsda_samples_results.txt', OPLS-DA model sample prediction results using inputted
data.
4. 'oplsda_prediction_summary.txt', prediction summary.
5. 'oplsda_figure.pdf', OPLS-DA plot.
Parameter:
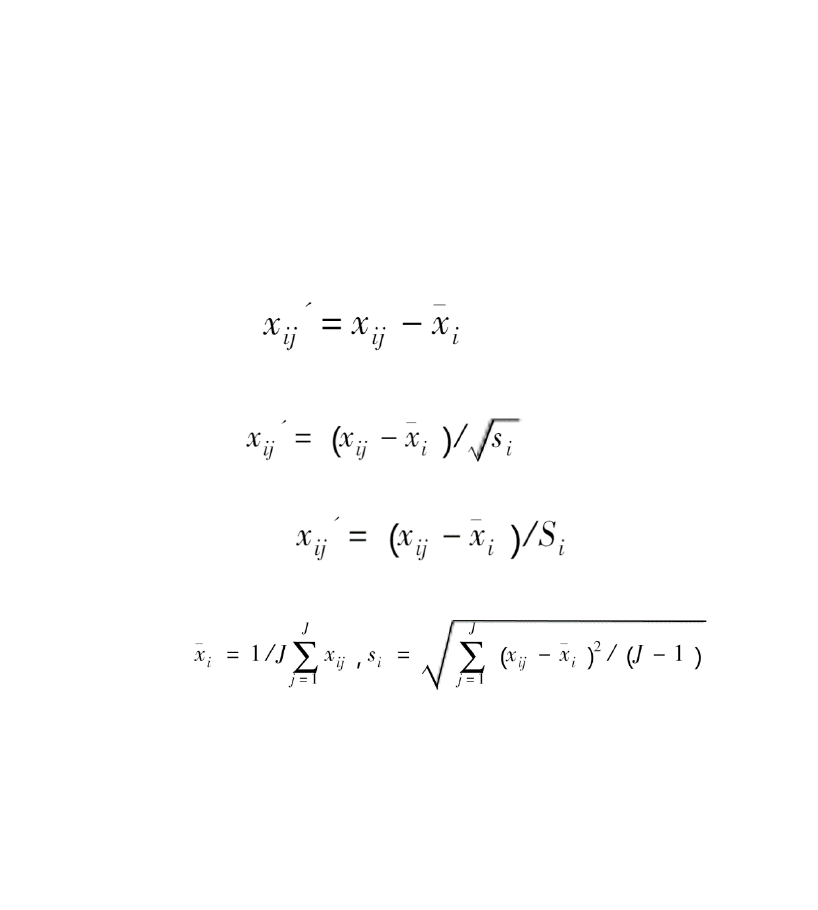
IP4M V1.0
33
1. VIP-value: A numerical variable indicating the Variable Importance in Projection.
2. orthogonal components: The number of orthogonal components (for OPLS only); when
set to 0 [default], PLS will be performed; otherwise OPLS will be performed; when set
to NA, OPLS is performed and the number of orthogonal components is automatically
computed by using the cross-validation (with a maximum of 9 orthogonal components).
3. scaling methods: Either no centering nor scaling (‟none‟), mean-centering only (‟center‟),
mean-centering and Pareto scaling (‟Pareto‟), or mean-centering and unit variance
scaling (‟standard‟) [default].
Mean-centering:
Pareto scaling:
unit variance scaling:
Comments:
4. crossvalI: Number of cross-validation segments (default is 7); The number of samples
(rows of ‟x‟) must be at least >= crossvalI
5. permutation: Number of random permutations of response labels to estimate R2Y and
Q2Y significance by permutation testing [default is 20 for single response models
(without train/test partition), and 0 otherwise]
6. graphical parameters: This tool provides ten graphic parameters for ten different graphic
types. They are displayed in 'oplsda_figure.pdf' file.
Note:
Group number must be 2 in the sample group file.
Group names of characters or string are preferred. Numbers are also supported but not
recommended.
Reference:
[1] Thevenot, E.A., Roux, A., Xu, Y., Ezan, E., Junot, C. 2015. Analysis of the human adult
urinary metabolome variations with age, body mass index and gender by implementing a
comprehensive workflow for univariate and OPLS statistical analyses. Journal of Proteome
IP4M V1.0
34
Research. 14: 3322-3335.
[2] Trygg J, Wold S. Orthogonal projections to latent structures (O-PLS) [J]. Journal of
Chemometrics 2002,16:119 –128.
[3] Rui C P, Trygg J, Gottfries J. Advantages of orthogonal inspection in chemometrics[J]. Journal
of Chemometrics, 2012, 26(6):231–235.
[4] Mehmood, T., KH. Liland, L. Snipen, and S. Saebo. 2012. “A Review of Variable Selection
Methods in Partial Least Squares Regression.” Chemometrics and Intelligent Laboratory
Systems 118 (0): 62–69.
[5] Galindo-Prieto B., Eriksson L. and Trygg J. (2014). Variable influence on projection (VIP) for
orthogonal projections to latent structures (OPLS). Journal of Chemometrics 28, 623-632.
Results and visualization:
Fig.13 The resulting files and the summary plot of OPLS-DA
Tool: Support vector machine
This tool performs support vector machines to rank peaks in the inputted table by SVM-RFE.
Group information is given by a group design file (Tab-delimited text file).
The SVM-RFE algorithm proposed by Guyon returns a ranking of the features of a
classification problem by training an SVM with a linear kernel and removing the feature with the
smallest ranking criterion. This criterion is the w value of the decision hyperplane given by the
SVM. For more detailed information, please review the original paper.
Input files:

IP4M V1.0
35
1. Peak table file in Tab-delimited text format, with the first column as the compound identifier
and the others as samples.
For example:
Table.15 The inputted peak table file
HU_01
1
HU_01
4
HU_01
5
HU_01
7
HU_01
8
HU_01
9
(2-methoxyethoxy)propanoic
acid isomer
3.0197
66
3.8143
39
3.5196
91
2.5621
83
3.7819
22
4.1610
74
(gamma)Glu-Leu/Ile
3.8884
79
4.2771
49
4.1956
49
4.3237
6
4.6293
29
4.4122
66
1-Methyluric acid
3.8690
06
3.8377
04
4.1022
54
4.5385
2
4.1788
29
4.5168
05
1-Methylxanthine
3.7172
59
3.7768
51
4.2916
65
4.4322
16
4.1173
6
4.5620
52
1,3-Dimethyluric acid
3.5354
61
3.9325
81
3.9553
76
4.2284
91
4.0055
45
4.3205
82
1,7-Dimethyluric acid
3.3251
99
4.0251
25
3.9729
04
4.1099
27
4.0240
92
4.3268
56
2-acetamido-4-methylphenyl
acetate
4.2047
54
5.1818
58
3.8856
8
4.2379
15
1.8529
94
4.0806
81
2-Aminoadipic acid
4.0802
04
4.3592
46
4.2491
11
4.2314
04
4.3236
79
4.2444
85
2. Group design file in Tab-delimited text file with two columns (samplename groupname).
For example:
Table.16 The inputted group design file
HU_011
M
HU 014
F
HU_015
M
HU_017
M
HU_018
M
HU_019
M
Output files:
1. 'svm_summary.txt', summary information about SVM.
2. 'svm_variable_results.txt', feature ranked results that are sorted by SVM-RFE.
3. ' svm_samples_results.txt', SVM model sample prediction results using inputted data.
4. 'svm_prediction_summary.txt', prediction summary.
5. 'support_vectors.txt', support vectors in the model.

IP4M V1.0
36
6. 'svm_plot.pdf', SVM plot.
Parameter:
kernel function: The kernel function reflects the similarity between the inputted data. The
correct choice of kernel parameters is crucial for obtaining good results, which practically
means that an extensive search must be conducted on the parameter space before results can
be trusted.
1. Linear kernel: Simple and safe, try it first. The model is interpretative. It indicates which
features or data points in the model are important. But it is not available if the data is not
linearly separable.
2. Polynomial kernel: Less restrictive than linear applications, it can solve non-linear separable
data. But it is more complicated with three parameters.
3. Radial basis function (RBF): Usually defined as a monotonic function of the Euclidean
distance between any points in space to a certain center. It maps primitive features to infinite
dimensions. It is able to achieve nonlinear mapping and also has less numerical difficulties.
4. Sigmoid: Squashes numbers to the range [0, 1]. Historically popular since they have a nice
interpretation as a saturating “firing rate” of a neuron. But there are some fatal disadvantages.
For instance, saturated neurons “kill” the gradients, sigmoid outputs are not zero-centered,
and exp () is a bit computationally expensive.
Note:
Group names of characters or string are preferred. Numbers are also supported but not
recommended.
Reference:
[1] Marchiori E, Sebag M. Bayesian Learning with Local Support Vector Machines for Cancer
Classification with Gene Expression Data[M]// Applications of Evolutionary Computing.
Springer Berlin Heidelberg, 2005:74-83.[2] Gene Selection for Cancer Classification using
Support Vector Machines (2002) Isabelle Guyon, Jason Weston, Stephen Barnhill, Vladimir
Vapnik.
Results and visualization:
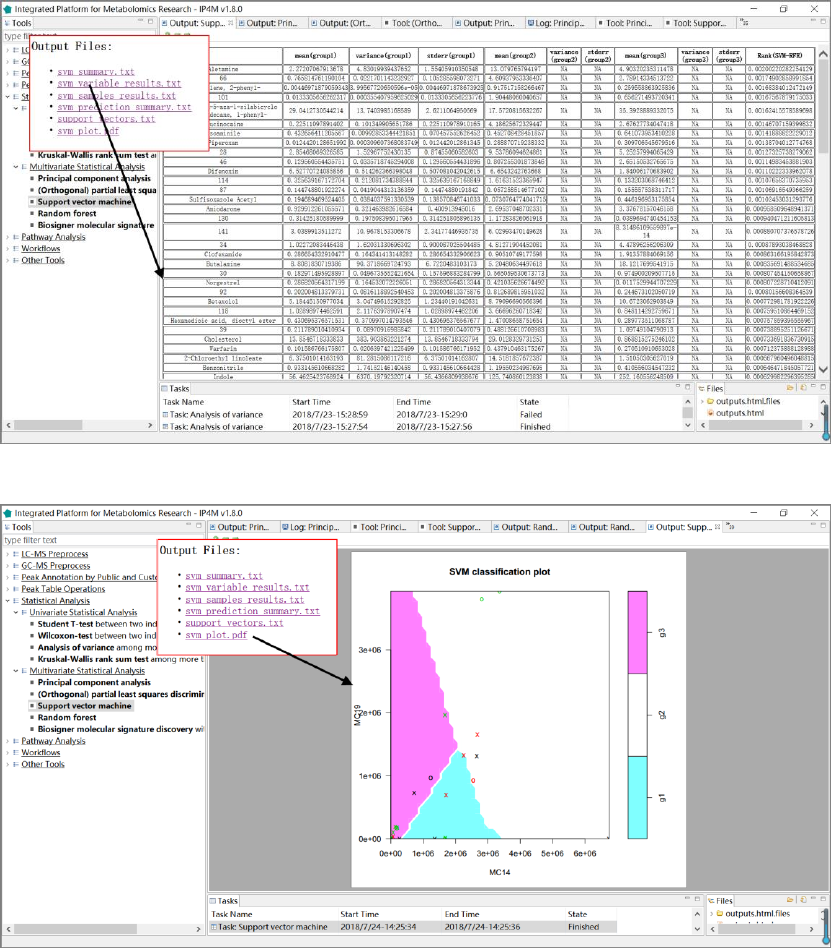
IP4M V1.0
37
Fig.14 The resulting files and the variable ranks of SVM (3 groups)
Fig.15 The resulting files and the classification plot of SVM (3 groups)
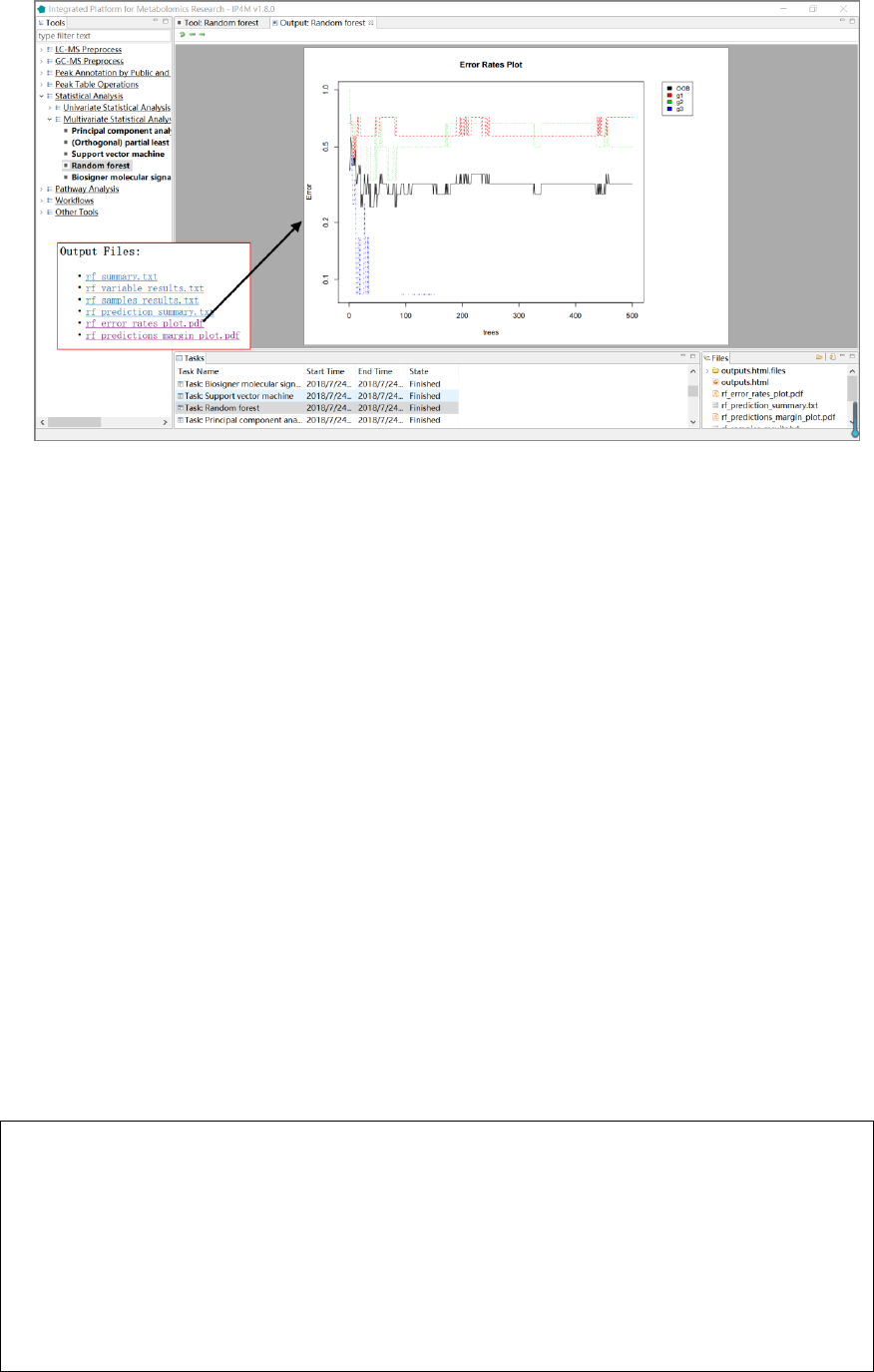
Tool: Random forest
This tool implements Breiman's random forest algorithm (R randomforest package) for
classification and peak ranking based on the inputted table. The peaks are ranked by the mean
decrease in Gini index. Group information is given by a group design file (Tab-delimited text file).
Input files:
1. Peak table file in Tab-delimited text format, with the first column as the compound identifier

IP4M V1.0
38
and the others as samples.
For example:
Table.17 The inputted peak table file
HU_01
1
HU_01
4
HU_01
5
HU_01
7
HU_01
8
HU_01
9
(2-methoxyethoxy)propanoic
acid isomer
3.0197
66
3.8143
39
3.5196
91
2.5621
83
3.7819
22
4.1610
74
(gamma)Glu-Leu/Ile
3.8884
79
4.2771
49
4.1956
49
4.3237
6
4.6293
29
4.4122
66
1-Methyluric acid
3.8690
06
3.8377
04
4.1022
54
4.5385
2
4.1788
29
4.5168
05
1-Methylxanthine
3.7172
59
3.7768
51
4.2916
65
4.4322
16
4.1173
6
4.5620
52
1,3-Dimethyluric acid
3.5354
61
3.9325
81
3.9553
76
4.2284
91
4.0055
45
4.3205
82
1,7-Dimethyluric acid
3.3251
99
4.0251
25
3.9729
04
4.1099
27
4.0240
92
4.3268
56
2-acetamido-4-methylphenyl
acetate
4.2047
54
5.1818
58
3.8856
8
4.2379
15
1.8529
94
4.0806
81
2-Aminoadipic acid
4.0802
04
4.3592
46
4.2491
11
4.2314
04
4.3236
79
4.2444
85
2. Group design file in Tab-delimited text format with two columns (samplename
groupname).
For example:
Table.18 The inputted group design file
HU_011
M
HU 014
F
HU_015
M
HU_017
M
HU_018
M
HU_019
M
Output files:
1. 'rf_summary.txt ', summary information about random forest model.
2. 'rf_variable_results.txt ', feature rank results that sorted by mean decrease in Gini index.
3. 'rf_prediction_summary.txt ', random forest model sample prediction results using inputted
data.
4. 'rf_prediction_summary.txt', prediction summary.
5. 'rf_error_rates_plot.pdf ', error rates plot in the model.
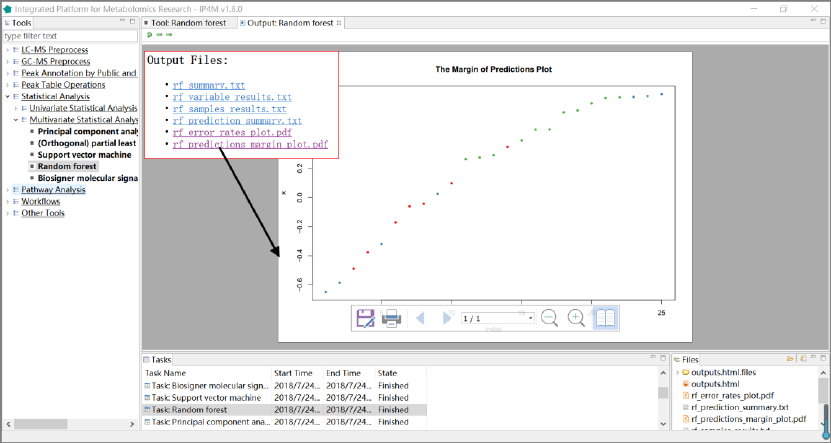
IP4M V1.0
39
6. 'rf_predictions_margin_plot.pdf ', predictions_margin plot.
Parameter:
1. number of trees: It specifies the number of decision trees included in the random forest. The
default is 500.
2. mtry: Mtry specifies the number of variables used in the node for the binary tree. The default
is the quadratic root of the data set variable (classification model) or one- third (predictive
model). Generally, it is necessary to carry out artificial selection step by step to determine the
optimal m value.
3. replacement: Specify the way to randomly sample Bootstrap. The default is resampling.
4. nodesize: The minimum number of decision tree nodes. By default, the discriminant model is
1 and the regression model is 5.
5. maxnodes: The maximum number of decision tree nodes
Results and visualization:
Fig.16 The resulting files and the margin of prediction plot of RF
IP4M V1.0
40
Fig.17 The resulting files and error rate plot of RF
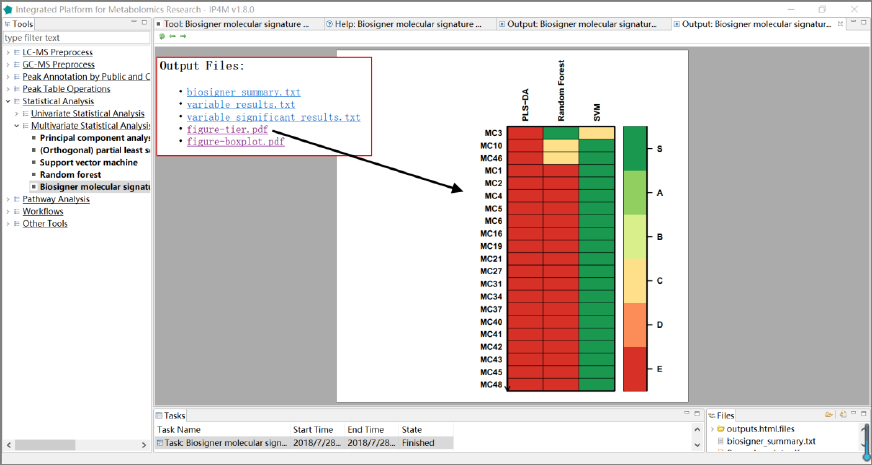
Tool: Biosigner molecular signature discovery with PLSDA, RF, and
SVM
This tool is the wrapper of the R package 'biosigner' and aims to find the significant peaks in
the inputted table. Three binary classifiers have been jointly used in biosigner, namely Partial
Least Square Discriminant Analysis (PLS-DA), Random Forest (RF) and Support Vector
Machines (SVM), to achieve high levels of prediction accuracy. Group information is given by a
group design file (Tab-delimited text file).
Input files:
1. Peak table file in Tab-delimited text format, with the first column as the compound identifier
and the others as samples.
For example:
Table.19 The inputted peak table file
HU_01
1
HU_01
4
HU_01
5
HU_01
7
HU_01
8
HU_01
9
(2-methoxyethoxy)propanoic
acid isomer
3.0197
66
3.8143
39
3.5196
91
2.5621
83
3.7819
22
4.1610
74
(gamma)Glu-Leu/Ile
3.8884
79
4.2771
49
4.1956
49
4.3237
6
4.6293
29
4.4122
66
1-Methyluric acid
3.8690
06
3.8377
04
4.1022
54
4.5385
2
4.1788
29
4.5168
05

IP4M V1.0
41
1-Methylxanthine
3.7172
59
3.7768
51
4.2916
65
4.4322
16
4.1173
6
4.5620
52
1,3-Dimethyluric acid
3.5354
61
3.9325
81
3.9553
76
4.2284
91
4.0055
45
4.3205
82
1,7-Dimethyluric acid
3.3251
99
4.0251
25
3.9729
04
4.1099
27
4.0240
92
4.3268
56
2-acetamido-4-methylphenyl
acetate
4.2047
54
5.1818
58
3.8856
8
4.2379
15
1.8529
94
4.0806
81
2-Aminoadipic acid
4.0802
04
4.3592
46
4.2491
11
4.2314
04
4.3236
79
4.2444
85
2. Group design file in Tab-delimited text format with two columns (samplename
groupname).
For example:
Table.20 The inputted group design file
HU_011
M
HU 014
F
HU_015
M
HU_017
M
HU_018
M
HU_019
M
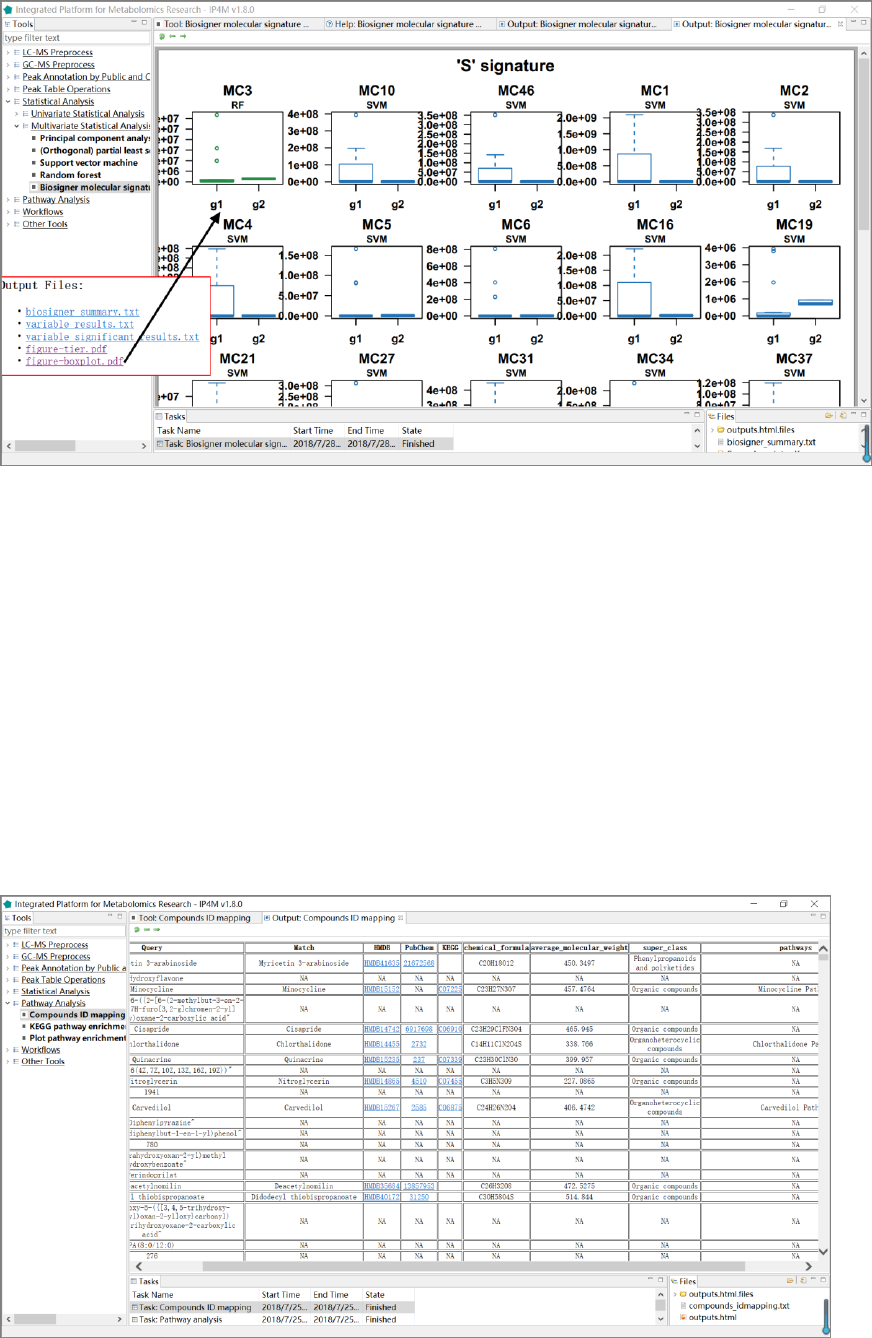
Output files:
1. 'biosigner_summary.txt', summary information about biosigner algorithm.
2. 'biosigner_variable_results.txt', ranked feature results by biosigner algorithm.
3. 'biosigner_variable_significant_results.txt', significant feature results.
4. ' biosigner_figure-tier.pdf ', displays classifier tiers from selected features.
5. ' biosigner_figure-boxplot.pdf ', individual boxplots from selected features.
Parameter:
1. bootstraps for resampling: The number of bootstraps is set to 5 to speed up computations
when generating this vignette; we however recommend to keep the default 50 value for
analyzing (otherwise signatures may be less stable).
2. pvalN: To speed up the selection, only variables which significantly improve the model up to
two times this threshold (to take into account potential fluctuations) are computed.
3. Selection tiers: Tiers from S, A, up to E by decreasing relevance. The (S) tier corresponds to
the final signature, i.e. features which passed through all the backward selection steps. In
contrast, features from the other tiers were discarded during the last (A) or previous (B to E)
selection rounds. Note that tierMaxC = „A‟ argument in the print and plot methods can be
used to view the features from the larger S+A signatures (especially when no S features have
IP4M V1.0
42
been found, or when the performance of the S model is much lower than the S+A model).
Note:
1. Group number must be 2 in the sample group file.
2. Group names of characters or string are preferred. Numbers are also supported but not
recommended.
3. The algorithm returns the tier of each feature for the selected classifier (s): tier S corresponds
to the final signature, i.e., features which have been found significant in all the selection steps;
features with tier A have been found significant in all but the last selection, and so on for tier
B to D. Tier E regroup all previous round of selection.
Reference:
[1] Rinaudo P, Boudah S, Junot C, et al. biosigner: A New Method for the Discovery of Significant
Molecular Signatures from Omics Data[J]. Frontiers in Molecular Biosciences, 2016, 3.
Results and visualization:
Fig.18 The resulting files and the potential biomarker (signatures) plot of biosigner
IP4M V1.0
43
Fig.19 The resulting files and the boxplot of „S‟ signatures by biosigner.
3.5 Pathway Analysis
Tool: Compounds ID mapping
The tool takes a one-column compound list file as input and performs libraries (HMDB,
PubChem, KEGG, etc.) IDs and basic information searching. This is a wrapper of the popular R
package metaboAnalystR (https://github.com/xialab/MetaboAnalystR).
Results and visualization:
Fig.20 The resulting file of compound IDs annotation.
IP4M V1.0
44
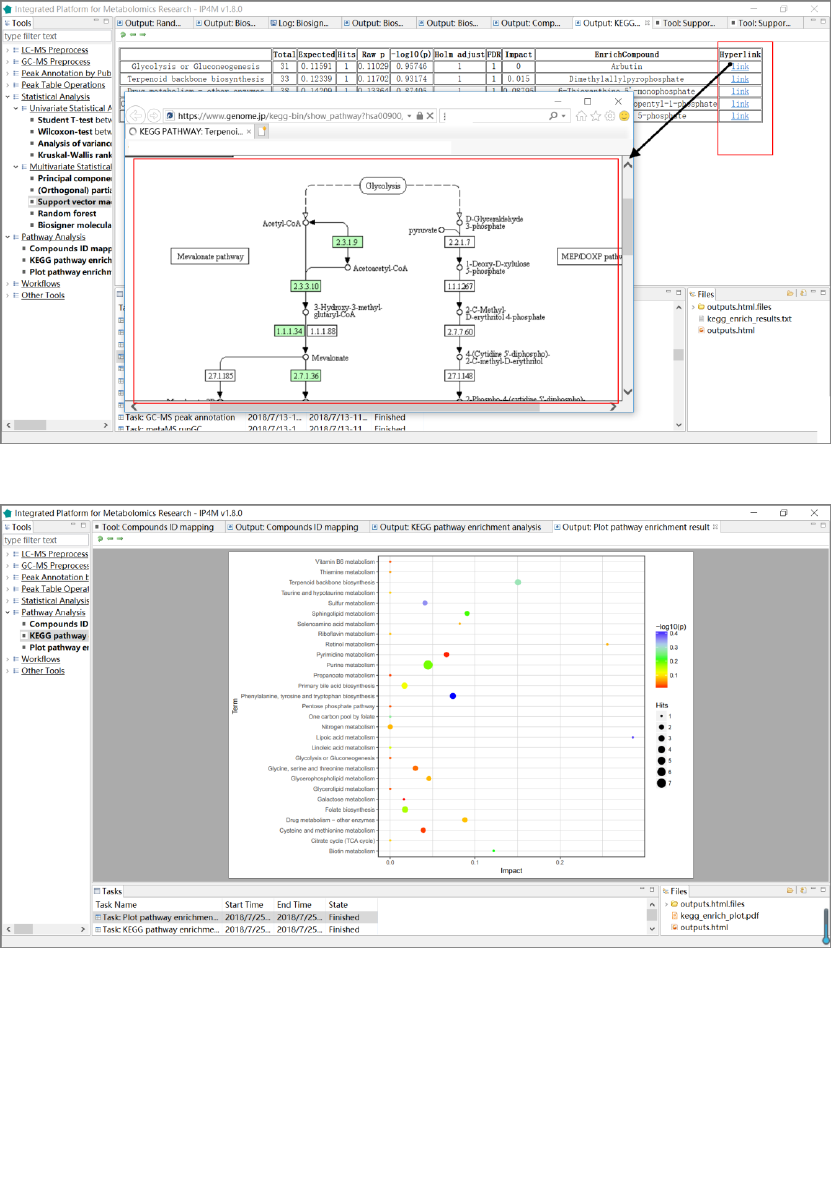
Tool: Pathway analysis on compounds ID mapping results
KEGG (Kyoto Encyclopedia of Genes and Genomes) is a database resource that integrates
genomic, chemical and systemic functional information. Gene catalogs from completely
sequenced genomes are linked to higher-level systemic functions of the cell, the organism, and the
ecosystem. This tool is a wrapper of the „metabolic pathway analysis‟ modules of the popular
MetaboAnalyst platform.
The tool takes a compounds annotation file as input and performs pathway analysis based on
information from KEGG.
Parameter:
1. pathway library: 21 different species libraries have been provided, including Human, Mouse,
Rat, Cow, Chicken, Zebrafish, Arabidopsis thaliana, Rice, Drosophila, Malaria, Budding
yeast, E.coli., etc., with a total of 1600 pathways.
2. representation analysis algorithm:
hypergeometric test: In statistics, the hypergeometric test uses the hypergeometric
distribution to calculate the statistical significance of having drawn a specific {\displaystyle k}
k successes (out of {\displaystyle n} n total draws) from the aforementioned population. The
test is often used to identify which subpopulations are over- or under-represented in a sample.
Fisher's exact test: It is a statistical significance test used in the analysis of contingency tables.
The test is useful for categorical data that result from classifying objects in two different
ways; it is used to examine the significance of the association (contingency) between the two
kinds of classification.
3. Specify pathway topology analysis algorithm:
The module provides two popular topological measures found on the left-panel to provide
users greater insight into their networks.
Out-degree centrality: It refers to the number of links a node has to other nodes.
Relative betweenness centrality: It represents the degree of centrality a node has in a network
by measuring the number of shortest paths that pass through that node.
Nodes with high scores in both measures are more likely to be important hubs.
Reference:
[1] Xia J, Wishart D S. Web-based inference of biological patterns, functions and pathways from
metabolomic data using MetaboAnalyst[J]. Nature Protocols, 2011, 6(6):743-760.
[2] Chong, J., et al. (2018) MetaboAnalyst 4.0: towards more transparent and integrative
metabolomics analysis. Nucleic acids research, 46, W486-w494.
http://www.metaboanalyst.ca.
IP4M V1.0
45
Results and visualization:
Fig.21 The results of pathway analysis with detailed information and hyperlink of the pathways.
Fig.22 The pathway analysis plot of the first 30 compounds
Tool: Enrichment analysis on compounds ID mapping
results
The tool takes a one-column compound list file and performs metabolite set enrichment
analysis for human and mammalian species. The analysis is based on eight metabolite set libraries
containing ~7000 groups of biologically meaningful metabolite sets collected primarily from
human studies. This tool is a wrapper of the „enrichment analysis‟ modules of the popular
IP4M V1.0
46
MetaboAnalyst platform.
Parameter:
metabolite set library: Eight different metabolite set libraries have been provided, containing
~6300 groups of biologically meaningful metabolite sets collected primarily from human
studies. Pathway-associated metabolite set library contains 99 metabolite sets based on
normal metabolic pathways. Diseased-associated metabolite set library contains 344
metabolite sets reported in human blood. Disease-associated metabolite set library contains
384 metabolite sets reported in human urine. Disease-associated metabolite set (CSF) library
contains 166 metabolite sets reported in human cerebral spinal fluid (CSF). SNP-associated
metabolite set library contains 4598 metabolite sets based on their associations with detected
single nucleotide polymorphisms (SNPs) loci. Predicted metabolite set library contains 912
metabolic sets that are predicted to be changed in the case of dysfunctional enzymes using
genome-scale network model of human metabolism. Location-based metabolite set library
contains 73 metabolite sets based on organ, tissue and subcellular localizations.
Drug-pathway-associated metabolite set library contains 461 metabolite sets based on drug
pathway.
Reference:
[1] Xia J, Wishart D S. Web-based inference of biological patterns, functions and pathways from
metabolomic data using MetaboAnalyst[J]. Nature Protocols, 2011, 6(6):743-760.
[2] Chong, J., et al. (2018) MetaboAnalyst 4.0: towards more transparent and integrative
metabolomics analysis. Nucleic acids research, 46, W486-w494.
http://www.metaboanalyst.ca.
IP4M V1.0
47
3.6 Workflows
3.6.1 GC-MS data preprocessing workflow: from raw data to
peak table
This workflow takes multiple GC-MS raw data files in netCDF or mzXML format as inputs
and outputs a peak table. It performs GC-MS data preprocessing and peak table operation,
including mainly peak detection, spectrum aligning, metabolites annotation and peak table
pretreatment.
Input files:
Multiple GC-MS raw data files in netCDF or mzXML format.
Output files:
'gcms_raw_pkTable.txt', raw peak table is generated with one line per "compound" and one
column per sample.
'gcms_mass_spectra.msp', Corresponding pseudospectrum(compound) mass spectrum
information in MSP format, the identifier is same in peak table file.
'gcms_mass_spectra_999norm.msp', intensities normalized mass spectrum information in
MSP format, intensities sum=999.
'identified_pkTable.txt', identified peak table file in Tab-delimited text format.
'identified_uniq_pkTable.txt', identified unique peak table file in Tab-delimited text format.
When row names are duplication, the row with the maximum intensity will be retained.
'detailed_information.txt', detailed information about query and database relationship in
library searching.
'zero_filled_pkTable.txt', zero filled peak table file in Tab-delimited text format.
'total_area_norm_pkTable.txt', total area normalized peak table file in Tab-delimited text
format.
'log2_transformed_pkTable.txt', log2 transformed peak table file in Tab-delimited text format.
Results and visualization:
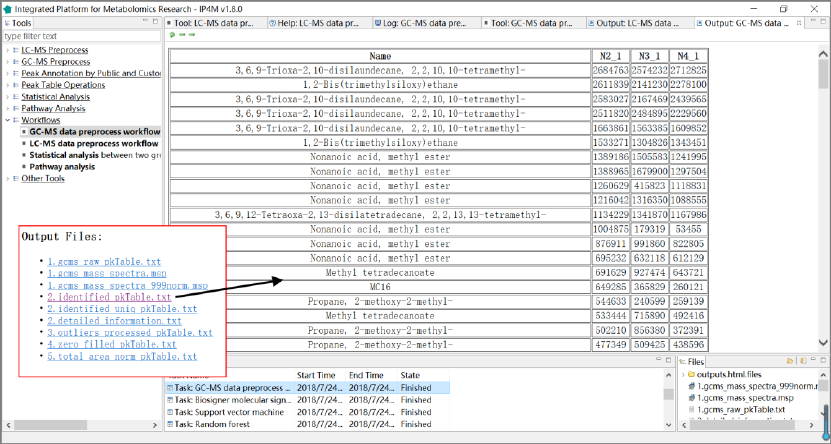
IP4M V1.0
48
Fig.23 The outputted files and the identified peak table of GC-MS data processing workflow
3.6.2 LC-MS data preprocessing workflow: from raw data to
peak table
This workflow takes multiple LC-MS raw data files in netCDF or mzXML format as inputs
and outputs peak table. It performs LC-MS data preprocessing and peak table operation, including
peak detection, spectrum aligning, metabolites annotation and peak table pretreatment.
Input files:
Multiple GC-MS raw data files in netCDF or mzXML format.
Output files:
'lcms_raw_pkTable.txt', a peak table is generated with one line per "compound" and one
column per sample.
'identified_pkTable.txt', identified peak table file in Tab-delimited text format.
'identified_uniq_pkTable.txt', identified unique peak table file in Tab-delimited text format.
When row names are duplication, the row with the maximum intensity will be retained.
'detailed_information.txt', detailed information about query and database relationship in
library searching.
'zero_filled_pkTable.txt', zero filled peak table file in Tab-delimited text format.
'total_area_norm_pkTable.txt', total area normalized peak table file in Tab-delimited text
format.
'log2_transformed_pkTable.txt', log2 transformed peak table file in Tab-delimited text format.
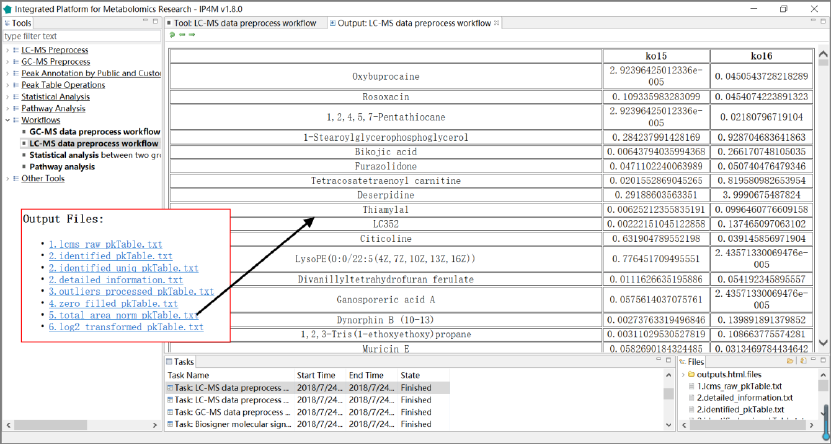
IP4M V1.0
49
Results and visualization:
Fig.24 The outputted files and the total area normalized peak table of LC-MS data
processing workflow
3.6.3 Statistical analysis based on peak table
This workflow takes a peak table file and a group design file as inputs. It performs all
univariate and multivariate statistical analysis as user selected (between two groups).
Input files:
1. Peak table file in Tab-delimited text format, with the first column as the compound identifier
and the others as samples.
2. Group design file in Tab-delimited text format with two columns (samplename
groupname).
Output files:
'pkTable_summary.txt', basic statistics summary information on columns (sample data).
't_test_results.txt', t-test results with p value, log2FC, and q value.
't_test_significant_results.txt', significant t-test results.
'wilcox_test_results.txt', Wilcoxon-test results with p value, log2FC, and q value.
'wilcox _test_significant_results.txt', significant Wilcoxon-test results.
'aov_results.txt', analysis of variance model results with p-value and q value.
'aov_significant_results.txt', significant analysis of variance model results.
IP4M V1.0
50
'kw_test_results.txt ', Kruskal-Wallis rank sum test results with p-value and q value.
'kw_test_significant_results.txt ', significant Kruskal-Wallis rank sum test results.
'pca_scores.txt', PCs (scores) matrix.
'pca_importance.txt', the importance of PCs.
'pca_rotation.txt', the matrix of variable loadings (i.e., a matrix whose columns contain the
eigenvectors).
'pca_plot.pdf', PCA plot using PCs score values, the default is PC1 and PC2.
'oplsda_variable_results.txt', feature ranked results that are sorted by VIP.
'oplsda_variable_significant_results.txt', significant feature results.
'oplsda_samples_results.txt', OPLS-DA model sample prediction results using inputted data.
'oplsda_prediction_summary.txt', prediction summary.
'oplsda_figure.pdf', OPLS-DA Plot.
'svm_summary.txt', summary information about SVM.
'svm_variable_results.txt', feature ranked results that are sorted by SVM-RFE.
' svm_samples_results.txt', SVM model sample prediction results using inputted data.
'svm_prediction_summary.txt', prediction summary of SVM.
'support_vectors.txt', support vectors in the model of SVM.
'svm_plot.pdf', SVM plot.
'rf_summary.txt ', summary information about random forest model.
'rf_variable_results.txt ', feature ranked results that are sorted by mean decrease in Gini index
using RF.
'rf_samples_results.txt ', random forest model sample prediction results using inputted data.
'rf_prediction_summary.txt', prediction summary of RF.
'rf_error_rates_plot.pdf ', error rates plot in the RF model.
'rf_predictions_margin_plot.pdf ', predictions _margin plot of RF.
'biosigner_summary.txt', summary information about biosigner algorithm.
'biosigner_variable_results.txt', feature ranked results by biosigner algorithm.
'biosigner_variable_significant_results.txt', significant feature results by biosigner algorithm.
' biosigner_figure-tier.pdf ', displaying classifier tiers from selected features by biosigner
algorithm.
' biosigner_figure-boxplot.pdf ', individual boxplots from selected features by biosigner
algorithm.
Results and visualization:
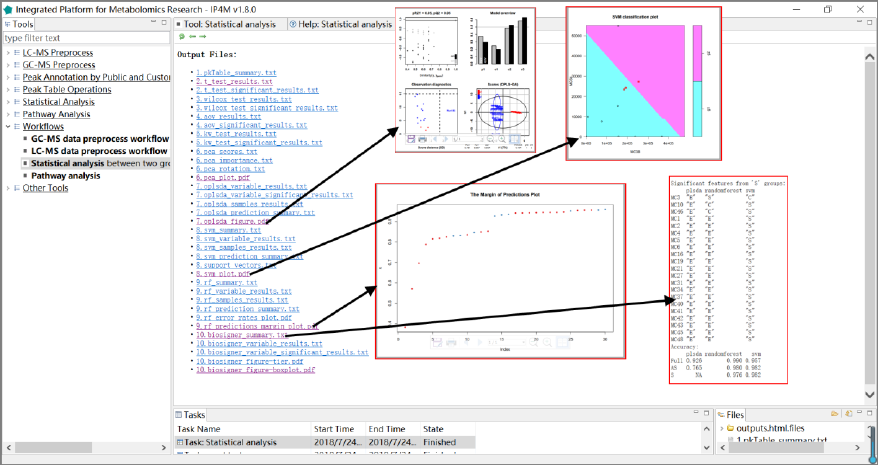
IP4M V1.0
51
Fig.25 The outputted files and demos (opls-da plot, SVM plot, the margin of prediction plot by
RF, and the biosigner summary) of statistical analysis workflow
3.6.4 Pathway and enrichment analysis
The tool takes a one-column compound list file as input and performs pathway analysis and
enrichment analysis, including compounds ID mapping, KEGG pathway, and enrichment analysis.
KEGG pathway libraries with ~1600 pathways are the knowledgebase for this tool which covers
21 species (human, mouse, rat, cow, chicken, zebrafish, arabidopsis thaliana, drosophila, malaria,
etc.). The enrichment analysis performs metabolite set enrichment analysis for human and
mammalian species. The analysis is based on 8 libraries containing ~6300 groups of biologically
meaningful metabolite sets collected primarily from human studies.
Input files:
A one-column compound list file in text format.
Output files:
'compounds_idmapping.txt ', compounds annotation result.
' pathway_results.txt ', KEGG pathway enrichment analysis result.
'pathway_results_plot.txt ', KEGG pathway enrichment result visualization diagram.
' enrichment_results.txt ', enrichment analysis result.
' enrichment_plot.pdf', enrichment result visualization diagram.
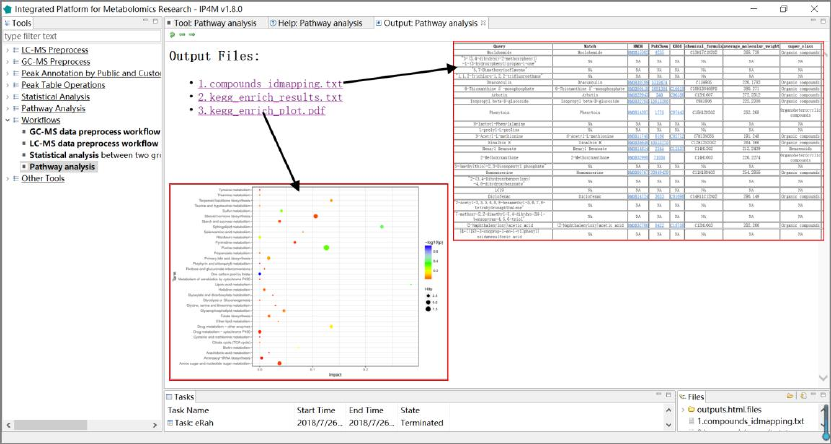
IP4M V1.0
52
Results and visualization:
Fig.26 The outputted files and visualization of pathway and enrichment analysis workflow
IP4M V1.0
53
3.7 Other Tools
3.7.1 Merge LECO CSV files
Tool: Merge LECO CSV files on peak table
The tool takes multiple .CSV files as inputs (outputted from the Chromatof software of
LECO., USA, reference mode) and merges them to generate a combined peak table file, according
to „R.T.‟, „Quant mass‟, and „Area‟. The .CSV files from BT, 4D, and HRT GC-TOF/MS
instruments (LECO, USA) are supported.
Parameter:
Merge method:
Same mass and RT difference within the cutoff: if same quant mass and rt difference within
the cutoff are met, the corresponding compounds of multiple samples is considered as the same
one. The “area” values of the same compound will be merged and the name of the compound
outputted is the one with the highest frequency of occurrence. For same frequency names, the one
with the maximum average strength is taken. If more than one variable (in the same sample) meet
the criteria, the largest “area” will be taken.
Same mass and one by one according to RT order: all the inputted files will be sorted by
“Quant mass” and “R.T.”, and then merged directly one by one. This option is simple but
effective.
Note:
This tool is strict to file format. Make sure these columns exist: the retention time column
with the name starts with „R.T.‟, the peak area column named „Area‟, the quant mass column
named „Quant Masses‟, and the compound name column named „Name‟. Other columns are also
permitted but will not be involved in process. The row number of all the inputted files should be
the same.
3.7.2 GLM on two groups
Tool: GLM on two groups
This tool is the wrapper of the R „glm‟ function and aims at peak ranking by coefficients of
linear regression. Group information is given by a group design file (Tab-delimited text file). The
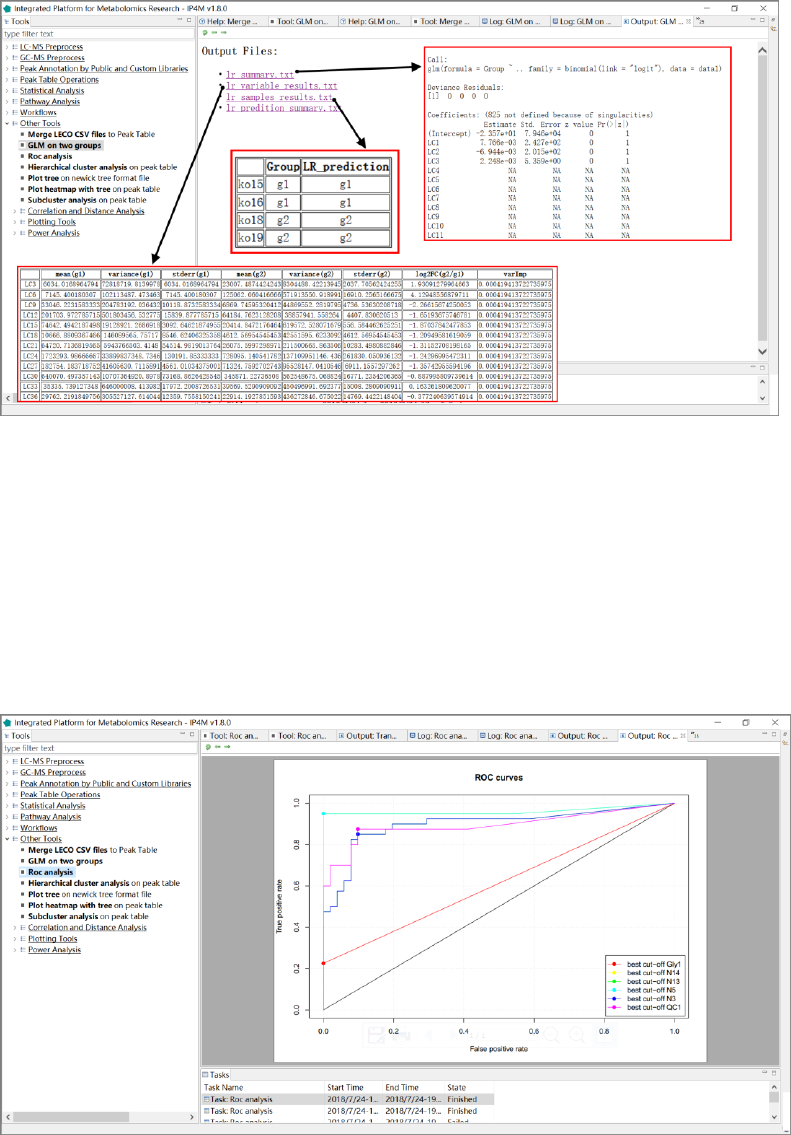
IP4M V1.0
54
tool is only available for binary classification and the number of groups should be 2.
Results and visualization:
Fig.28 The outputted files and corresponding information of GLM analysis
3.7.3 ROC analysis
Tool: ROC analysis
This tool takes a peak table file as input and computes ROC curves for every peak.
Fig.29 ROC curves of six peaks

IP4M V1.0
55
3.7.4 Hierarchical cluster analysis
Tool: Hierarchical cluster analysis on peak table
This tool takes a peak table file as input and performs hierarchical cluster analysis on it.
Parameter:
Distance calculate method:
1. Euclidean:
The Euclidean distance between points p and q is the length of the line segment connecting
them.
2. Correlation distance:
Correlation coefficient:
3. Canberra distance:sum (|p_i - q_i| / |p_i + q_i|). Terms with zero numerator and
denominator are omitted from the sum and treated as if the values were missing. This is
intended for non-negative values (e.g., counts): take the absolute value of the denominator.
4. Binary distance: The vectors are regarded as binary bits, so non-zero elements are „on‟ and
zero elements are „off‟. The distance is the proportion of bits in which the only one is on
amongst those in which at least one is on.
5. Minkowski distance: The p norm, the pth root of the sum of the pth powers of the differences
between the components.
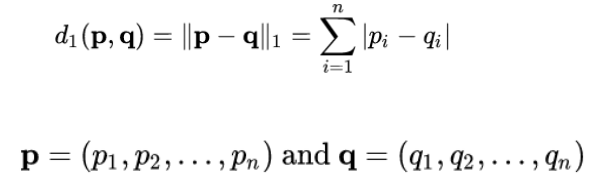
IP4M V1.0
56
6. Manhattan: Absolute distance between the two vectors.
where (p, q) are vectors.
7. maximum distance:Maximum distance between two components of x and y (supremum
norm).
Cluster methods:
1. ward: Ward's minimum variance method aims at finding compact, spherical clusters.
2. complete: The complete linkage method finds similar clusters.
3. single: The single linkage method (which is closely related to the minimal spanning tree)
adopts a „friends of friends‟ clustering strategy.
The other methods can be regarded as aiming for clusters with characteristics somewhere
between the single and complete link methods.
4. centroid: Method "centroid" is typically meant to be used with squared Euclidean distances.
5. average: The average distance method measures the average distance between each pair of
observations
6. mcquitty: It finds the similar cluster.
7. median: Median distance method.
Results and visualization:
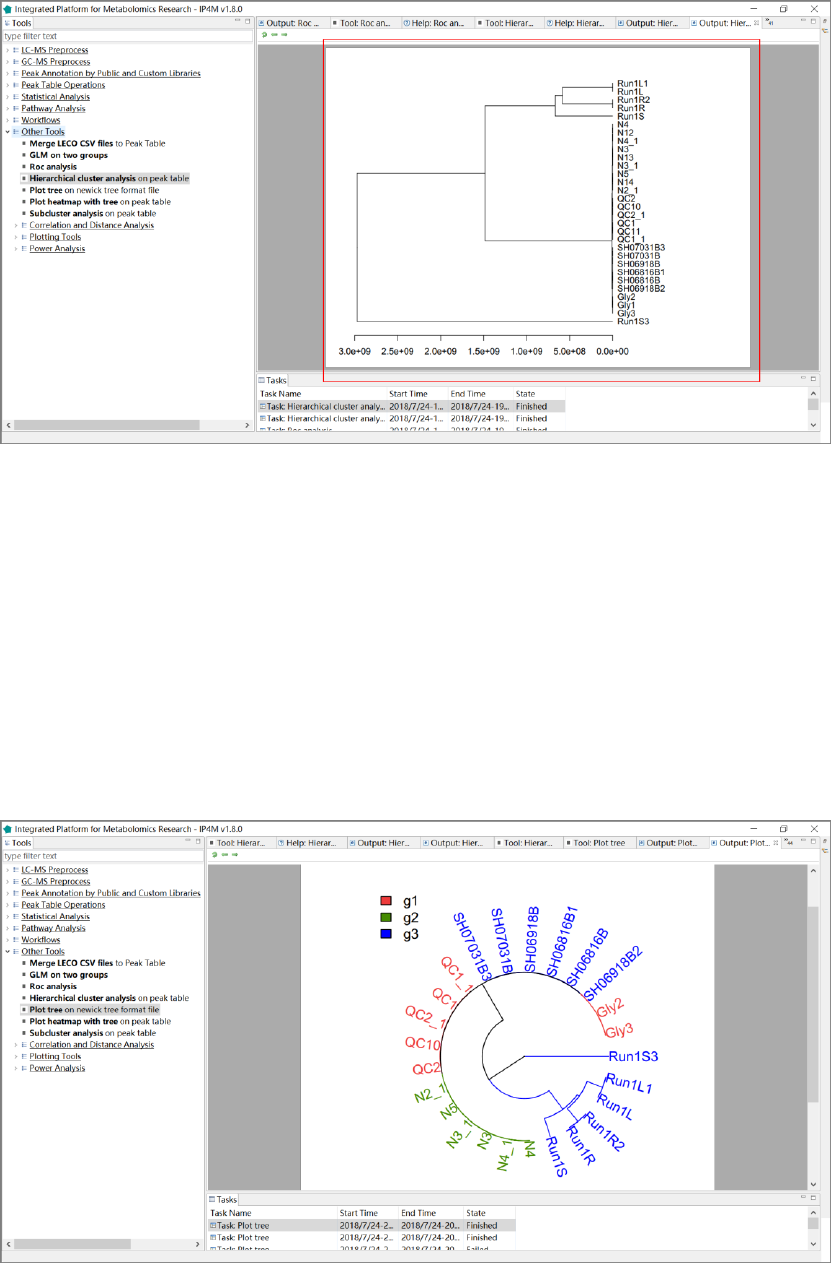
IP4M V1.0
57
Fig.30 The hierarchical cluster tree plot.
3.7.5 Plot tree
Tool: Plot tree on Newick tree format file
This tool takes a standard Newick format tree file as input and plots hierarchical tree by
phylogram, fan, or cladogram mode.
Fig.31 The hierarchical tree plot by fan mode.
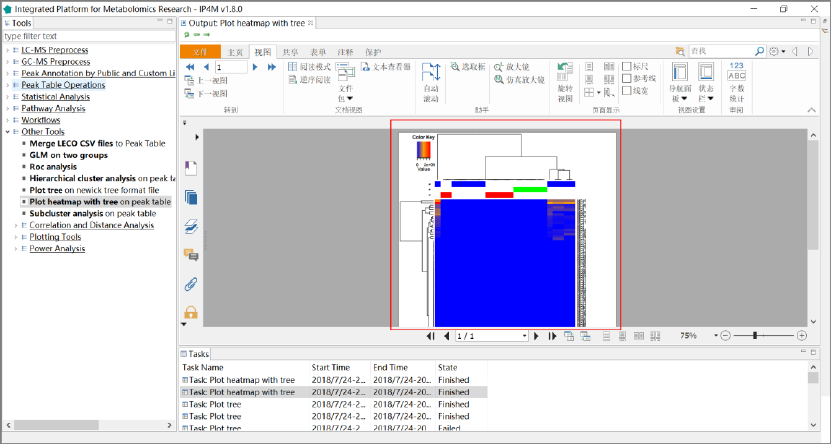
IP4M V1.0
58
3.7.6 Plot heatmap with tree
Tool: Plot heatmap with tree on peak table
This tool takes a peak table file as input and plots heatmap with clusters on it.
Results and visualization:
Fig.32 The heatmap with tree in pdf edit box.
3.7.7 Sub-cluster expression analysis
Tool: Sub-cluster expression analysis on peak table
This tool takes a peak table file as input and performs cluster analysis on it. The metabolites
are classified into several groups (clusters) according to their distance or variation similarity.
Parameter:
Distance calculate method:
1. Euclidean:
The Euclidean distance between points p and q is the length of the line segment connecting
them.
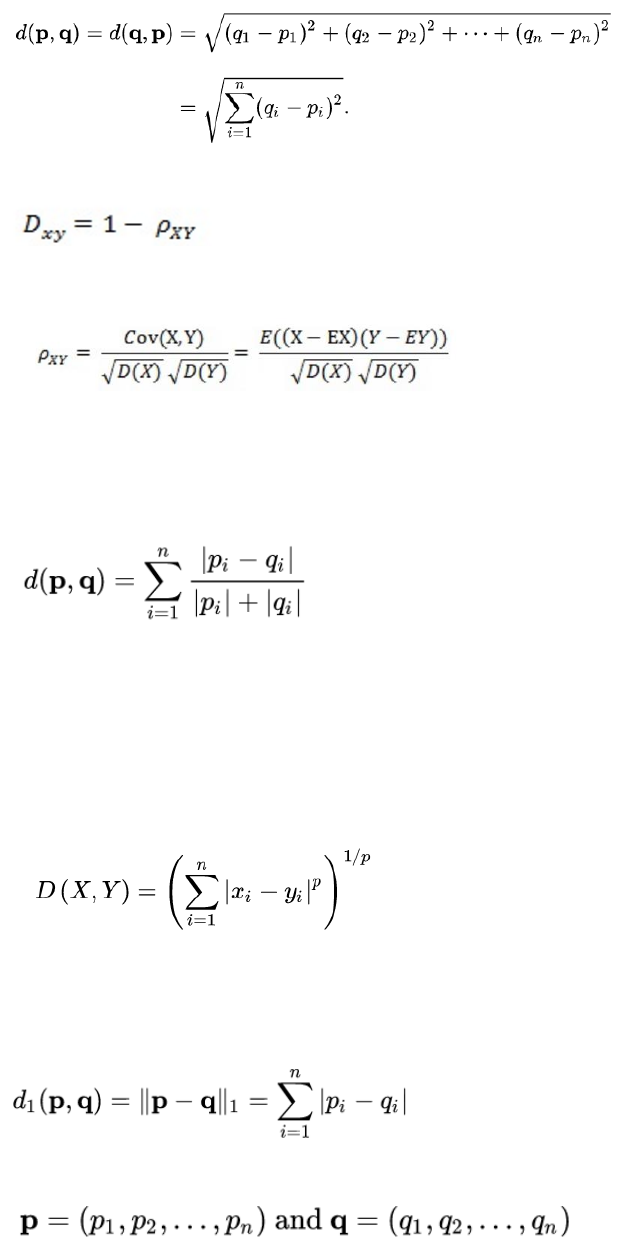
IP4M V1.0
59
2. Correlation distance:
Correlation coefficient:
3. Canberra distance: sum (|p_i - q_i| / |p_i + q_i|). Terms with zero numerator and
denominator are omitted from the sum and treated as if the values were missing. This is
intended for non-negative values (e.g., counts): take the absolute value of the denominator.
4. Binary distance: The vectors are regarded as binary bits, so non-zero elements are „on‟ and
zero elements are „off‟. The distance is the proportion of bits in which the only one is on
amongst those in which at least one is on.
5. Minkowski distance: The p norm, the pth root of the sum of the pth powers of the differences
between the components.
6. Manhattan: Absolute distance between the two vectors.
where (p, q) are vectors.
7. maximum distance:Maximum distance between two components of x and y (supreme norm).
Cluster methods:
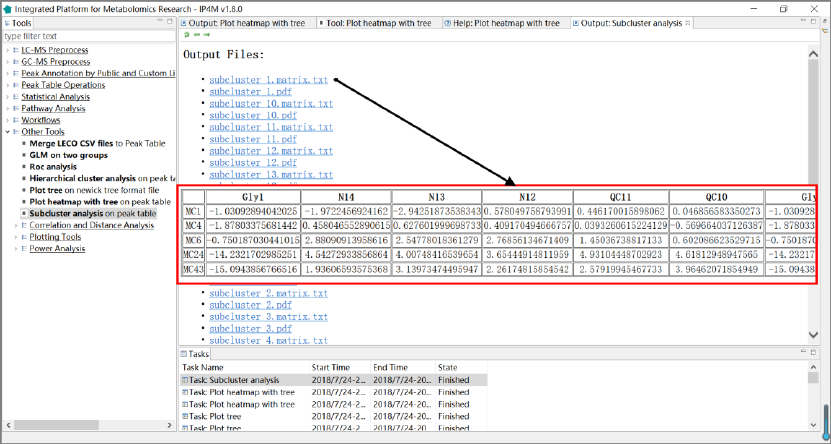
IP4M V1.0
60
1. ward: Ward's minimum variance method aims at finding compact, spherical clusters.
2. complete: The complete linkage method finds similar clusters.
3. single: The single linkage method (which is closely related to the minimal spanning tree)
adopts a „friends of friends‟ clustering strategy.
The other methods can be regarded as aiming for clusters with characteristics somewhere
between the single and complete link methods.
4. centroid: Method "centroid" is typically meant to be used with squared Euclidean distances.
5. average: The average distance method measures the average distance between each pair of
observations
6. mcquitty: It finds the similar cluster.
7. median: Median distance method.
Results and visualization:
Fig.33 Th outputted files and one matrix of sub-cluster analysis.
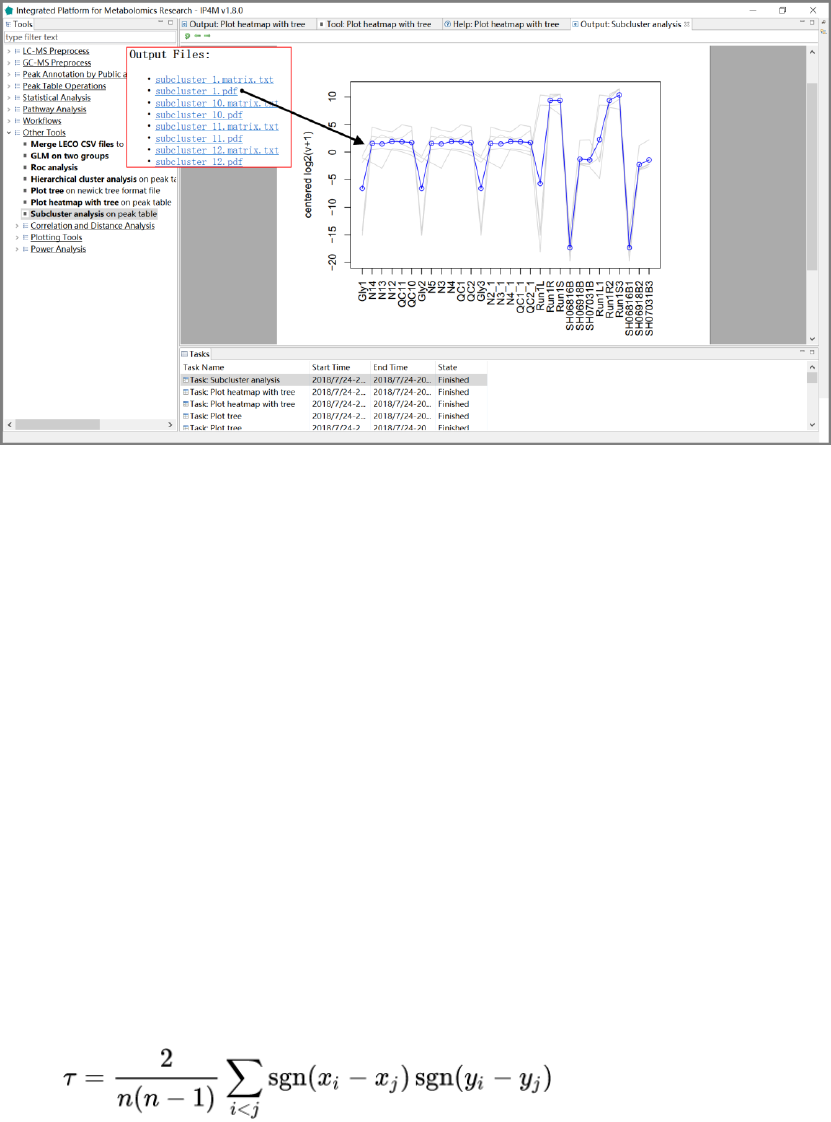
IP4M V1.0
61
Fig.34 The line chart of sub-cluster1.
3.7.8 Correlation and distance analysis
Tool: Create sample correlation matrix and make heatmap plot
This tool takes a peak table file as input and performs correlation analysis on it.
Parameter:
Correlation methods:
1. Kendall rank correlation:
The Kendall rank correlation coefficient, commonly referred to as Kendall's tau coefficient
(after the Greek letter τ), is a statistic used to measure the ordinal association between two
measured quantities. A tau test is a non-parametric hypothesis test for statistical dependence
based on the tau coefficient.
2. Pearson correlation:
the Pearson correlation coefficient, also referred to as Pearson's r is a measure of the linear
correlation between two variables X and Y. It has a value between +1 and −1, where 1 is a
total positive linear correlation, 0 is no linear correlation, and −1 is a total negative linear
correlation.
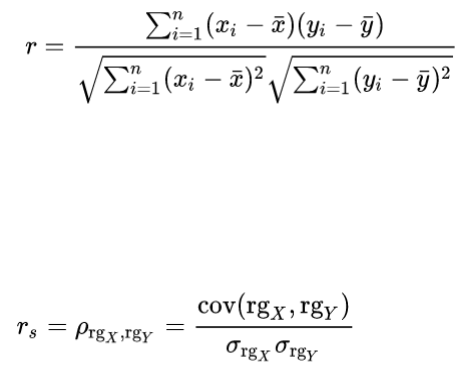
IP4M V1.0
62
3. Spearman correlation:
Spearman's rank correlation coefficient or Spearman's rho is a nonparametric measure of rank
correlation. It assesses how well the relationship between two variables can be described
using a monotonic function.
Cluster methods:
1. ward: Ward's minimum variance method aims at finding compact, spherical clusters.
2. complete: The complete linkage method finds similar clusters.
3. single: The single linkage method (which is closely related to the minimal spanning tree)
adopts a „friends of friends‟ clustering strategy.
The other methods can be regarded as aiming for clusters with characteristics somewhere
between the single and complete link methods.
4. centroid: Method "centroid" is typically meant to be used with squared Euclidean distances.
5. average: The average distance method measures the average distance between each pair of
observations
6. mcquitty: It finds the similar cluster.
7. median: Median distance method.
Results and visualization:

IP4M V1.0
63
Fig.35 The heatmap of correlation analysis
Tool: Generate distance matrix on peak table
This tool takes a peak table file as input and generates the distance matrix.
Parameter:
Distance calculate method:
1. Euclidean:
The Euclidean distance between points p and q is the length of the line segment connecting
them.
2. Correlation distance:
Correlation coefficient:
3. Canberra distance: sum (|p_i - q_i| / |p_i + q_i|). Terms with zero numerator and

IP4M V1.0
64
denominator are omitted from the sum and treated as if the values were missing. This is
intended for non-negative values (e.g., counts): take the absolute value of the denominator.
4. Binary distance: The vectors are regarded as binary bits, so non-zero elements are „on‟ and
zero elements are „off‟. The distance is the proportion of bits in which the only one is on
amongst those in which at least one is on.
5. Minkowski distance: The p norm, the pth root of the sum of the pth powers of the differences
between the components.
6. Manhattan: Absolute distance between the two vectors.
where (p, q) are vectors.
7. maximum distance:Maximum distance between two components of x and y (supremum
norm).
Results and visualization:
Fig.36 The distance matrix.
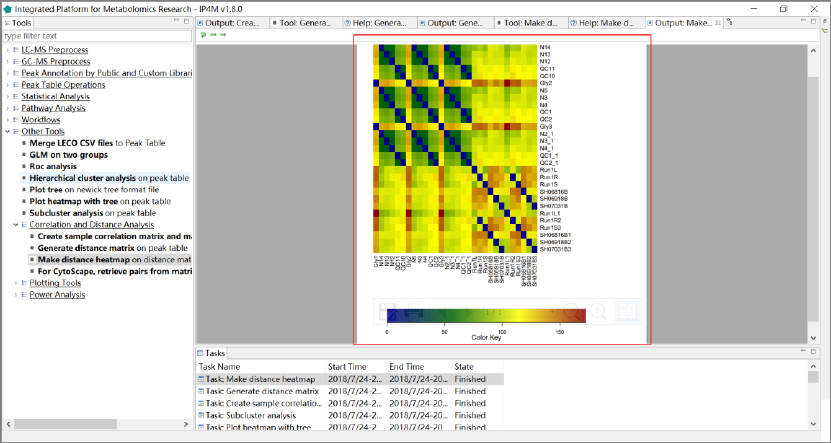
IP4M V1.0
65
Tool: Make distance heatmap on distance matrix
This tool takes a distance matrix as input and makes a heat map plot based on it.
Results and visualization:
Fig.37 The distance heatmap
Tool: For Cytoscape: retrieve pairs from matrix according to specific
criterion
This tool retrieves pairs from matrix according to a specific criterion. The result can be
imported directly into Cytoscape for network construction.
Results and visualization:
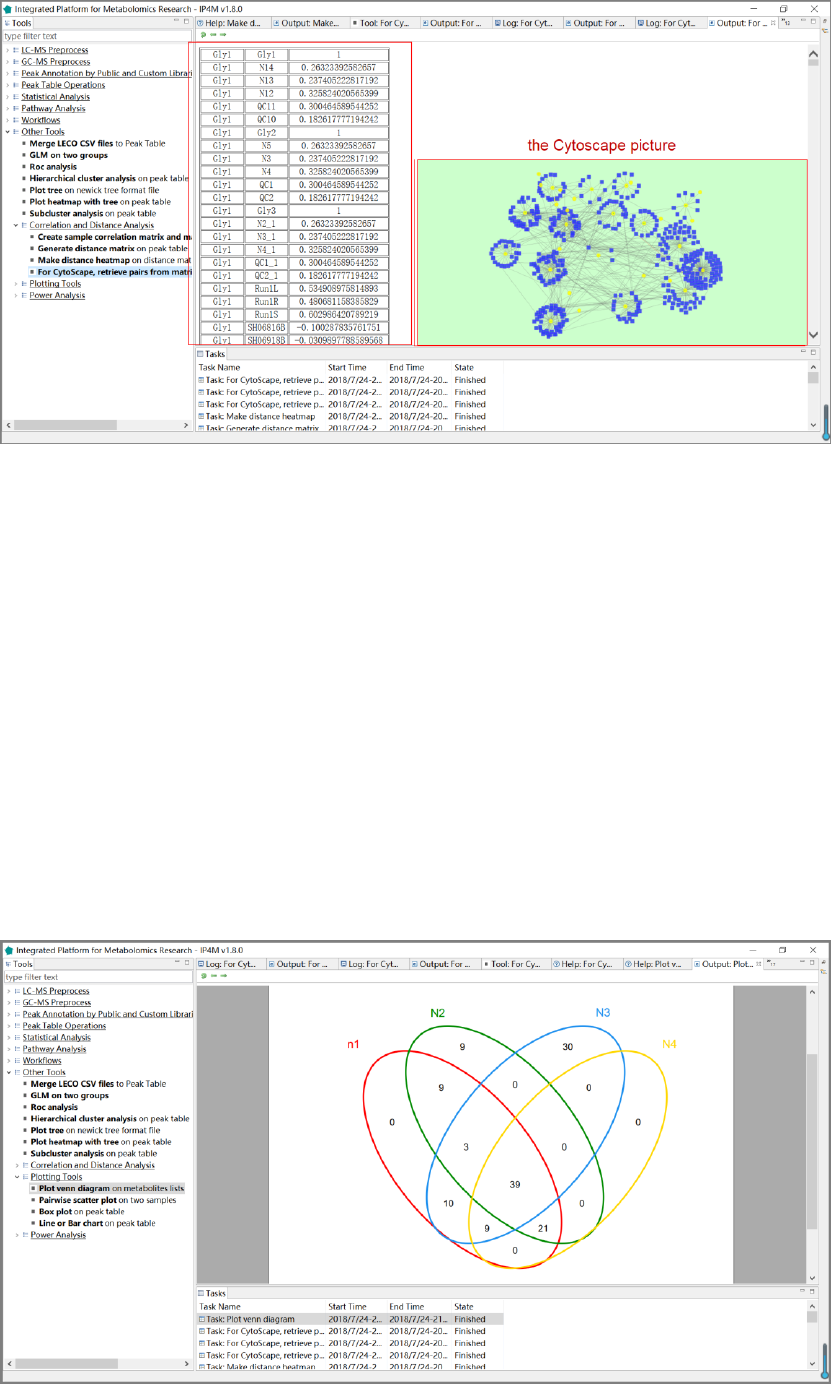
IP4M V1.0
66
Fig.38 The retrieved pairs for Cytoscape, and the outputted network of Cytoscape.
3.7.9 Plotting tools
Tool: Plot Venn diagram on metabolites lists
With this tool, you can calculate the intersection(s) of the list of elements. It will generate a
Venn plot and textual output indicating which elements are exist in each intersection or are unique
to a certain list/group.
Results and visualization:
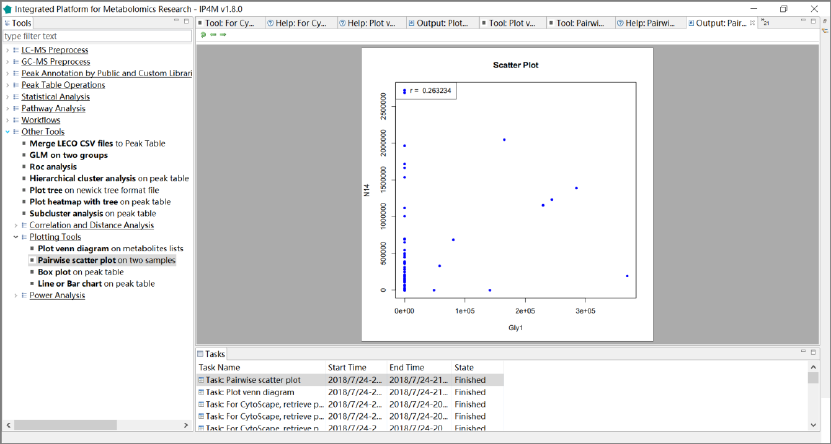
IP4M V1.0
67
Fig.39 The Venn plot of four groups.
Tool: Pairwise scatter plot on two samples
This tool is used for plotting pairwise scatter figures in batch mode. All the scatter plots will
be saved in one pdf file.
Results and visualization:
Fig.40 The pairwise scatter plot on two samples.
Tool: Box plot on peak table
This tool is used for plotting box figures in batch mode. All the box plots will be saved in one
pdf file.
Results and visualization:
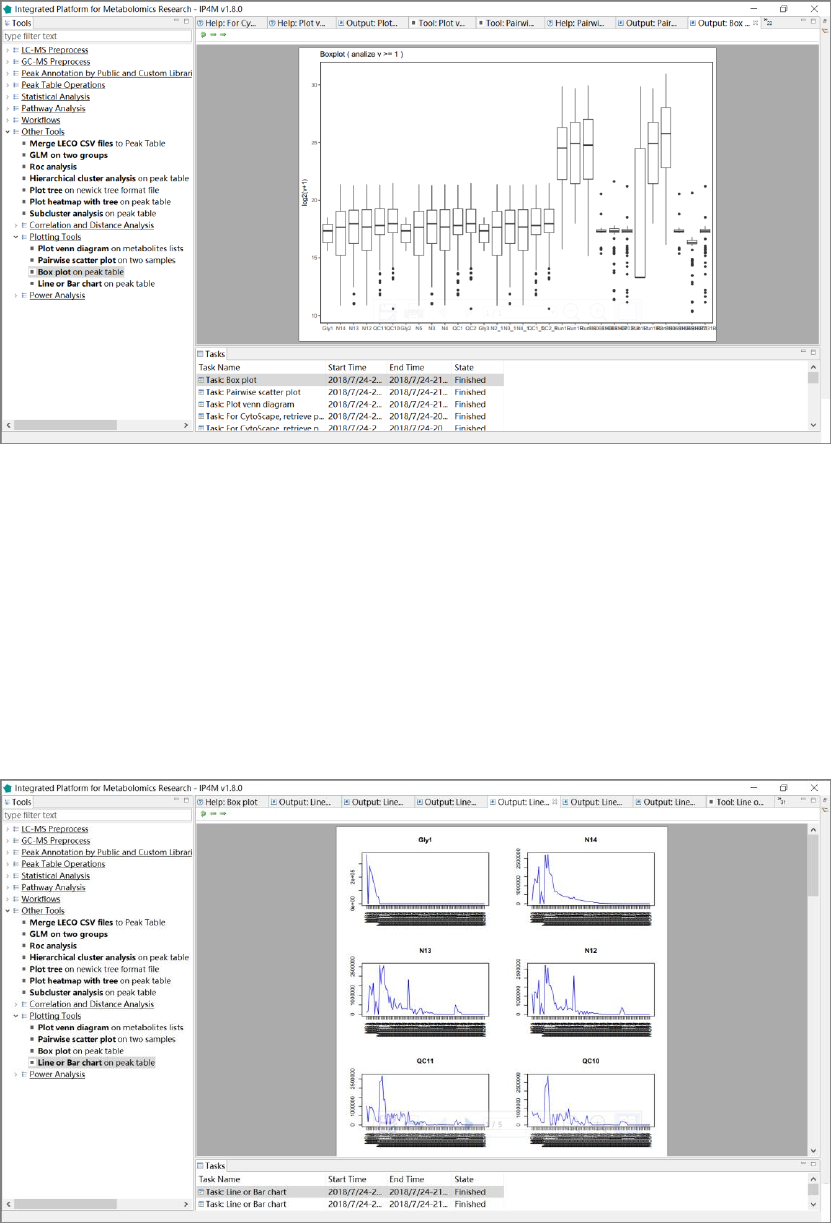
IP4M V1.0
68
Fig.41 The box plot of samples (one box per sample)
Tool: Line chart on peak table
This tool is used for plotting line or bar charts in batch mode. All the plots will be saved in
one pdf file.
Results and visualization:
Fig.42 The line charts of samples.

IP4M V1.0
69
Fig.43 The bar charts of samples.
3.7.10 Sample size and power analysis
Sample size and power analysis is helpful to estimate a reasonable sample size before
experiments or to evaluate the power of analysis results after experiments.
1) Tool: Pwr.t.test power calculation for t-test (one, two, and paired
samples)
For t-tests, the following functions are used:
pwr.t.test(n = , d = , sig.level = , power = , type = c("two.sample", "one.sample", "paired"))
where n is the sample size, d is the effect size, and type indicates a two-sample t-test, one-sample
t-test or paired t-test. If you have unequal sample sizes, use
pwr.t2n.test(n1 =, n2=, d =, sig.level =, power = )
where n1 and n2 are the sample sizes.
For t-tests, the effect size is assessed as
μ1: mean of group1
μ2: mean of group1
σ2: common error variance
Cohen suggests that d values of 0.2, 0.5, and 0.8 represent small, medium, and large effect sizes
respectively.
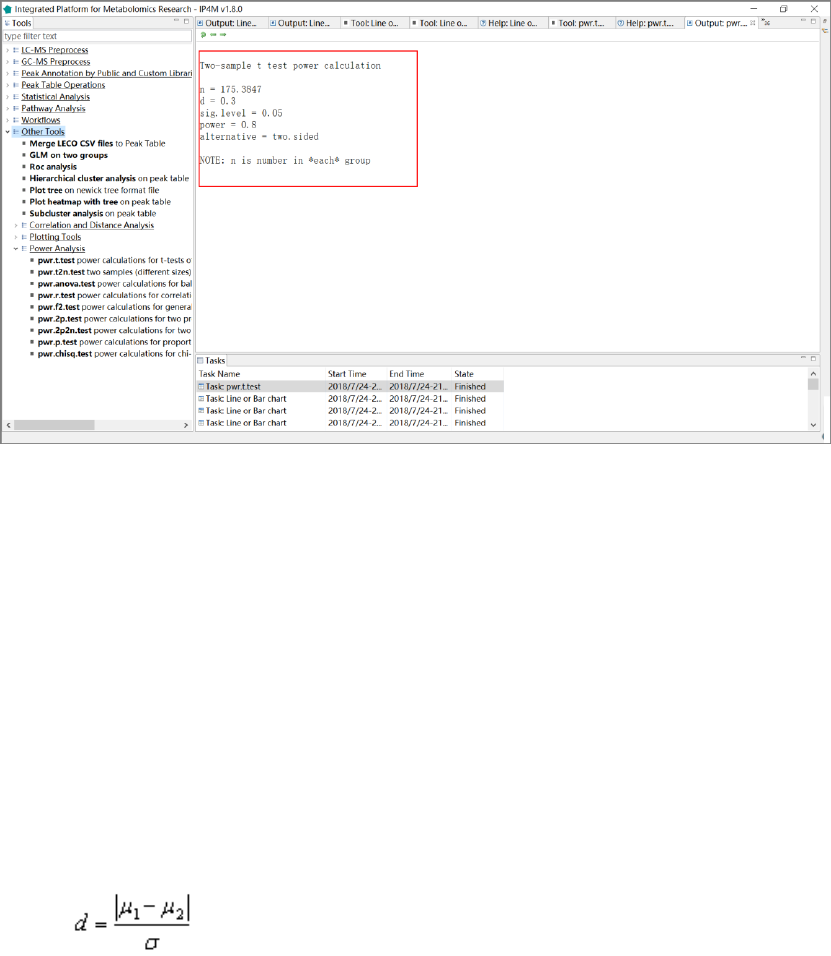
IP4M V1.0
70
You can specify alternative="two.sided", "less", or "greater" to indicate a two-tailed, or one-tailed
test. A two-tailed test is the default.
Results and visualization:
Fig.44 The results of power analysis when two samples t- test is used.
2) Tool: Pwr.t2n.test power calculation for t-test (different sizes)
For t-tests, the following functions are used:
pwr.t.test(n = , d = , sig.level = , power = , type = c("two.sample", "one.sample", "paired"))
where n is the sample size, d is the effect size, and type indicates a two-sample t-test, one-sample
t-test or paired t-test. If you have unequal sample sizes, use
pwr.t2n.test(n1 = , n2= , d = , sig.level =, power = )
where n1 and n2 are the sample sizes.
For t-tests, the effect size is assessed as
μ1: mean of group1
μ2: mean of group1
σ2: common error variance

IP4M V1.0
71
3) Tool: Pwr.anova.test power calculation for balanced one way
ANOVA
For a one-way analysis of variance, the following functions are used:pwr.anova.test(k = , n = , f
= , sig.level = , power = )
where k is the number of groups and n is the common sample size in each group.
For a one-way ANOVA, effect size is measured by f where
pi=ni / N
ni=number of observations in group i
N=total number of observations
μi= mean of group i
μ=grand mean
σ2= error variance within groups
Cohen suggests that f values of 0.1, 0.25, and 0.4 represent small, medium, and large
effect sizes respectively.
4) Tool: Pwr.r.test power calculation for correlation test
For correlation coefficients, the following functions are used:
pwr.r.test(n = , r = , sig.level = , power = )
where n is the sample size and r is the correlation. We use the population correlation coefficient as
the effect size measure. Cohen suggests that r values of 0.1, 0.3, and 0.5 represent small, medium,
and large effect sizes respectively.
5) Tool: Pwr.f2.test power calculation for general linear model
For linear models (e.g., multiple regression), the following functions are used:
pwr.f2.test(u =, v = , f2 = , sig.level = , power = )
where u and v are the numerator and denominator degrees of freedom. We use f2 as the effect size
measure.

IP4M V1.0
72
R2 = population squared multiple correlation
R2A = variance accounted for in the population by variable set A
R2AB = variance accounted for in the population by variable set A and B together
The first formula is appropriate when we are evaluating the impact of a set of predictors on an
outcome. The second formula is appropriate when we are evaluating the impact of one set of
predictors above and beyond the second set of predictors (or covariates). Cohen suggests f2 values
of 0.02, 0.15, and 0.35 represent small, medium, and large effect sizes.
6) Tool: Pwr.2p.test power calculation for two proportions (equal n)
When comparing two proportions, the following functions are used:
pwr.2p.test(h = , n = , sig.level =, power = )
where h is the effect size and n is the common sample size in each group.
Cohen suggests that h values of 0.2, 0.5, and 0.8 represent small, medium, and large effect sizes
respectively.
7) Pwr.2p2n.test power calculation for two proportions (unequal n)
When comparing two proportions, the following functions are used:
pwr.2p.test(h = , n = , sig.level =, power = )
For unequal n's
pwr.2p2n.test(h = , n1 = , n2 = , sig.level = , power = )
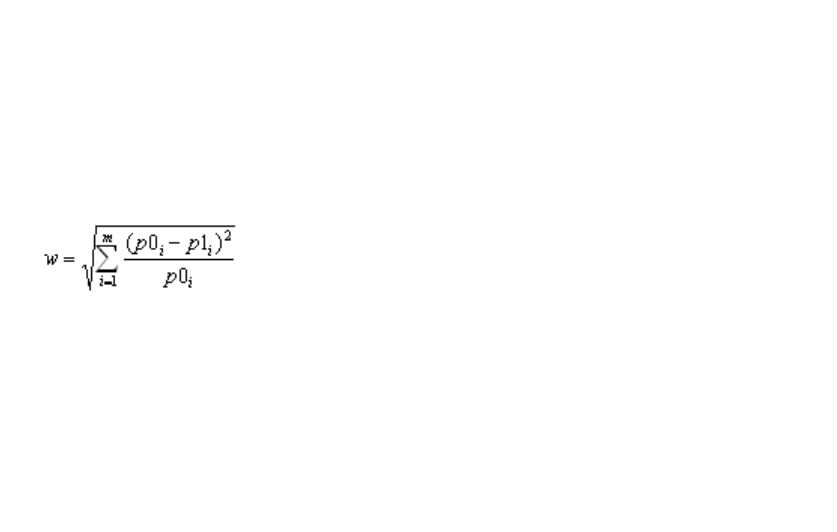
IP4M V1.0
73
8) Pwr.p.test power calculation for proportions (one sample)
To test a single proportion, the following functions are used:
pwr.p.test (h = , n = , sig.level = power = )
For both two sample and one sample proportion tests, you can specify alternative="two. sided",
"less", or "greater" to indicate a two-tailed, or one-tailed test. A two-tailed test is the default.
9) Pwr.chisq.test power calculation for the chi-square test
For chi-square tests, the following functions are used:pwr.chisq.test(w =, N = , df = , sig.level =,
power = )
where w is the effect size, N is the total sample size, and df is the degrees of freedom. The effect
size w is defined as
p0i = cell probability in an ith cell under H0
p1i = cell probability in an ith cell under H1
Cohen suggests that w values of 0.1, 0.3, and 0.5 represent small, medium, and large effect sizes
respectively.
