LAMMPS Users Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 1950 [warning: Documents this large are best viewed by clicking the View PDF Link!]
- Table of Contents
- LAMMPS Documentation
- 31 Mar 2017 version
- 1. Introduction
- 2. Getting Started
- 3. Commands
- 4. Packages
- 5. Accelerating LAMMPS performance
- 6. How-to discussions
- 6.1 Restarting a simulation
- 6.2 2d simulations
- 6.3 CHARMM, AMBER, and DREIDING force fields
- 6.4 Running multiple simulations from one input script
- 6.5 Multi-replica simulations
- 6.6 Granular models
- 6.7 TIP3P water model
- 6.8 TIP4P water model
- 6.9 SPC water model
- 6.10 Coupling LAMMPS to other codes
- 6.11 Visualizing LAMMPS snapshots
- 6.12 Triclinic (non-orthogonal) simulation boxes
- 6.13 NEMD simulations
- 6.14 Finite-size spherical and aspherical particles
- 6.15 Output from LAMMPS (thermo, dumps, computes, fixes, variables)
- 6.16 Thermostatting, barostatting, and computing temperature
- 6.17 Walls
- 6.18 Elastic constants
- 6.19 Library interface to LAMMPS
- 6.20 Calculating thermal conductivity
- 6.21 Calculating viscosity
- 6.22 Calculating a diffusion coefficient
- 6.23 Using chunks to calculate system properties
- 6.24 Setting parameters for the kspace_style pppm/disp command
- 6.25 Polarizable models
- 6.26 Adiabatic core/shell model
- 6.27 Drude induced dipoles
- 7. Example problems
- 8. Performance & scalability
- 9. Additional tools
- amber2lmp tool
- binary2txt tool
- ch2lmp tool
- chain tool
- colvars tools
- createatoms tool
- drude tool
- eam database tool
- eam generate tool
- eff tool
- emacs tool
- fep tool
- i-pi tool
- ipp tool
- kate tool
- lmp2arc tool
- lmp2cfg tool
- matlab tool
- micelle2d tool
- moltemplate tool
- msi2lmp tool
- phonon tool
- polybond tool
- pymol_asphere tool
- python tool
- reax tool
- smd tool
- vim tool
- xmgrace tool
- 10. Modifying & extending LAMMPS
- 10.1 Atom styles
- 10.2 Bond, angle, dihedral, improper potentials
- 10.3 Compute styles
- 10.4 Dump styles
- 10.5 Dump custom output options
- 10.6 Fix styles
- 10.7 Input script commands
- 10.8 Kspace computations
- 10.9 Minimization styles
- 10.10 Pairwise potentials
- 10.11 Region styles
- 10.12 Body styles
- 10.13 Thermodynamic output options
- 10.14 Variable options
- 10.15 Submitting new features for inclusion in LAMMPS
- 11. Python interface to LAMMPS
- 11.1 Overview of running LAMMPS from Python
- 11.2 Overview of using Python from a LAMMPS script
- 11.3 Building LAMMPS as a shared library
- 11.4 Installing the Python wrapper into Python
- 11.5 Extending Python with MPI to run in parallel
- 11.6 Testing the Python-LAMMPS interface
- 11.7 Using LAMMPS from Python
- 11.8 Example Python scripts that use LAMMPS
- 11.9 PyLammps interface
- 12. Errors
- 13. Future and history
- Tutorial for Thermalized Drude oscillators in LAMMPS
- LAMMPS GitHub tutorial
- LAMMPS Documentation
- PyLammps Tutorial
- Body particles
- Manifolds (surfaces)
- angle_coeff command
- angle_style command
- atom_modify command
- atom_style command
- balance command
- bond_coeff command
- bond_style command
- bond_write command
- boundary command
- box command
- change_box command
- clear command
- comm_modify command
- comm_style command
- compute command
- compute_modify command
- create_atoms command
- create_bonds command
- create_box command
- delete_atoms command
- delete_bonds command
- dielectric command
- dihedral_coeff command
- dihedral_style command
- dimension command
- displace_atoms command
- dump command
- dump custom/vtk command
- dump h5md command
- dump image command
- dump movie command
- dump molfile command
- dump nc command
- dump custom/vtk command
- dump h5md command
- dump image command
- dump movie command
- dump_modify command
- dump molfile command
- dump nc command
- dump nc/mpiio command
- echo command
- fix command
- fix_modify command
- group command
- group2ndx command
- ndx2group command
- if command
- improper_coeff command
- improper_style command
- include command
- info command
- jump command
- kspace_modify command
- kspace_style command
- label command
- lattice command
- log command
- mass command
- min_modify command
- min_style command
- minimize command
- molecule command
- neb command
- neigh_modify command
- neighbor command
- newton command
- next command
- package command
- pair_coeff command
- pair_modify command
- pair_style command
- pair_write command
- partition command
- prd command
- print command
- processors command
- python command
- quit command
- read_data command
- read_dump command
- read_restart command
- region command
- replicate command
- rerun command
- reset_timestep command
- restart command
- run command
- run_style command
- set command
- shell command
- special_bonds command
- suffix command
- tad command
- temper command
- temper/grem command
- thermo command
- thermo_modify command
- thermo_style command
- timer command
- timestep command
- uncompute command
- undump command
- unfix command
- units command
- variable command
- velocity command
- write_coeff command
- write_data command
- write_dump command
- write_restart command
- fix adapt command
- fix adapt/fep command
- fix addforce command
- fix addtorque command
- fix append/atoms command
- fix atc command
- fix atom/swap command
- fix ave/atom command
- fix ave/chunk command
- fix ave/correlate command
- fix ave/correlate/long command
- fix ave/histo command
- fix ave/histo/weight command
- fix ave/time command
- fix aveforce command
- fix balance command
- fix bond/break command
- fix bond/create command
- fix bond/swap command
- fix box/relax command
- fix cmap command
- fix colvars command
- fix controller command
- fix deform command
- fix deform/kk command
- fix deposit command
- fix dpd/energy command
- fix drag command
- fix drude command
- fix drude/transform/direct command
- fix drude/transform/inverse command
- fix dt/reset command
- fix efield command
- fix ehex command
- fix enforce2d command
- fix eos/cv command
- fix eos/table command
- fix eos/table/rx command
- fix evaporate command
- fix external command
- fix filter/corotate command
- fix flow/gauss command
- fix freeze command
- fix gcmc command
- fix gld command
- fix gle command
- fix gravity command
- fix gravity/omp command
- fix grem command
- fix halt command
- fix heat command
- fix imd command
- fix indent command
- fix ipi command
- fix langevin command
- fix langevin/kk command
- fix langevin/drude command
- fix langevin/eff command
- fix lb/fluid command
- fix lb/momentum command
- fix lb/pc command
- fix lb/rigid/pc/sphere command
- fix lb/viscous command
- fix lineforce command
- fix manifoldforce command
- fix meso command
- fix meso/stationary command
- fix momentum command
- fix momentum/kk command
- fix move command
- fix mscg command
- fix msst command
- fix neb command
- fix nvt command
- fix nvt/intel command
- fix nvt/kk command
- fix nvt/omp command
- fix npt command
- fix npt/intel command
- fix npt/kk command
- fix npt/omp command
- fix nph command
- fix nph/kk command
- fix nph/omp command
- fix nvt/eff command
- fix npt/eff command
- fix nph/eff command
- fix nph/asphere command
- fix nph/asphere/omp command
- fix nph/body command
- fix nph/sphere command
- fix nph/sphere/omp command
- fix nphug command
- fix nphug/omp command
- fix npt/asphere command
- fix npt/asphere/omp command
- fix npt/body command
- fix npt/sphere command
- fix npt/sphere/omp command
- fix nve command
- fix nve/intel command
- fix nve/kk command
- fix nve/omp command
- fix nve/asphere command
- fix nve/asphere/intel command
- fix nve/asphere/noforce command
- fix nve/body command
- fix nve/dot command
- fix nve/dotc/langevin command
- fix nve/eff command
- fix nve/limit command
- fix nve/line command
- fix nve/manifold/rattle command
- fix nve/noforce command
- fix nve/sphere command
- fix nve/sphere/omp command
- fix nve/tri command
- fix nvk command
- fix nvt/asphere command
- fix nvt/asphere/omp command
- fix nvt/body command
- fix nvt/manifold/rattle command
- fix nvt/sllod command
- fix nvt/sllod/intel command
- fix nvt/sllod/omp command
- fix nvt/sllod/eff command
- fix nvt/sphere command
- fix nvt/sphere/omp command
- fix oneway command
- fix orient/fcc command
- fix orient/bcc command
- fix phonon command
- fix pimd command
- fix planeforce command
- fix poems
- fix pour command
- fix press/berendsen command
- fix print command
- fix property/atom command
- fix qbmsst command
- fix qeq/point command
- fix qeq/shielded command
- fix qeq/slater command
- fix qeq/dynamic command
- fix qeq/fire command
- fix qeq/comb command
- fix qeq/comb/omp command
- fix qeq/reax command
- fix qeq/reax/kk command
- fix qmmm command
- fix qtb command
- fix reax/bonds command
- fix reax/c/bonds command
- fix reax/c/bonds/kk command
- fix reax/c/species command
- fix reax/c/species/kk command
- fix recenter command
- fix restrain command
- fix rigid command
- fix rigid/nve command
- fix rigid/nvt command
- fix rigid/npt command
- fix rigid/nph command
- fix rigid/small command
- fix rigid/nve/small command
- fix rigid/nvt/small command
- fix rigid/npt/small command
- fix rigid/nph/small command
- fix rx command
- fix saed/vtk command
- fix setforce command
- fix setforce/kk command
- fix shake command
- fix rattle command
- fix shardlow command
- fix smd command
- fix smd/adjust_dt command
- fix smd/integrate_tlsph command
- fix smd/integrate_ulsph command
- fix smd/move_tri_surf command
- fix smd/setvel command
- fix smd/wall_surface command
- fix spring command
- fix spring/chunk command
- fix spring/rg command
- fix spring/self command
- fix srd command
- fix store/force command
- fix store/state command
- fix temp/berendsen command
- fix temp/csvr command
- fix temp/csld command
- fix temp/rescale command
- fix temp/rescale/eff command
- fix tfmc command
- fix thermal/conductivity command
- fix ti/spring command
- fix tmd command
- fix ttm command
- fix ttm/mod command
- fix tune/kspace command
- fix vector command
- fix viscosity command
- fix viscous command
- fix wall/lj93 command
- fix wall/lj126 command
- fix wall/lj1043 command
- fix wall/colloid command
- fix wall/harmonic command
- fix wall/gran command
- fix wall/gran/region command
- fix wall/piston command
- fix wall/reflect command
- fix wall/reflect/kk command
- fix wall/region command
- fix wall/srd command
- compute ackland/atom command
- compute angle command
- compute angle/local command
- compute angmom/chunk command
- compute basal/atom command
- compute body/local command
- compute bond command
- compute bond/local command
- compute centro/atom command
- compute chunk/atom command
- compute cluster/atom command
- compute cna/atom command
- compute com command
- compute com/chunk command
- compute contact/atom command
- compute coord/atom command
- compute damage/atom command
- compute dihedral command
- compute dihedral/local command
- compute dilatation/atom command
- compute dipole/chunk command
- compute displace/atom command
- compute dpd command
- compute dpd/atom command
- compute erotate/asphere command
- compute erotate/rigid command
- compute erotate/sphere command
- compute erotate/sphere/atom command
- compute event/displace command
- compute fep command
- compute global/atom command
- compute group/group command
- compute gyration command
- compute gyration/chunk command
- compute heat/flux command
- compute hexorder/atom command
- compute improper command
- compute improper/local command
- compute inertia/chunk command
- compute ke command
- compute ke/atom command
- compute ke/atom/eff command
- compute ke/eff command
- compute ke/rigid command
- compute meso/e/atom command
- compute meso/rho/atom command
- compute meso/t/atom command
- compute msd command
- compute msd/chunk command
- compute msd/nongauss command
- compute omega/chunk command
- compute orientorder/atom command
- compute pair command
- compute pair/local command
- compute pe command
- compute pe/atom command
- compute plasticity/atom command
- compute pressure command
- compute property/atom command
- compute property/chunk command
- compute property/local command
- compute rdf command
- compute reduce command
- compute reduce/region command
- compute rigid/local command
- compute saed command
- compute slice command
- compute smd/contact/radius command
- compute smd/damage command
- compute smd/hourglass/error command
- compute smd/internal/energy command
- compute smd/plastic/strain command
- compute smd/plastic/strain/rate command
- compute smd/rho command
- compute smd/tlsph/defgrad command
- compute smd/tlsph/dt command
- compute smd/tlsph/num/neighs command
- compute smd/tlsph/shape command
- compute smd/tlsph/strain command
- compute smd/tlsph/strain/rate command
- compute smd/tlsph/stress command
- compute smd/triangle/mesh/vertices
- compute smd/ulsph/num/neighs command
- compute smd/ulsph/strain command
- compute smd/ulsph/strain/rate command
- compute smd/ulsph/stress command
- compute smd/vol command
- compute sna/atom command
- compute snad/atom command
- compute snav/atom command
- compute stress/atom command
- compute force/tally command
- compute heat/flux/tally command
- compute pe/tally command
- compute pe/mol/tally command
- compute stress/tally command
- compute temp command
- compute temp/kk command
- compute temp/asphere command
- compute temp/body command
- compute temp/chunk command
- compute temp/com command
- compute temp/cs command
- compute temp/deform command
- compute temp/deform/eff command
- compute temp/drude command
- compute temp/eff command
- compute temp/partial command
- compute temp/profile command
- compute temp/ramp command
- compute temp/region command
- compute temp/region/eff command
- compute temp/rotate command
- compute temp/sphere command
- compute ti command
- compute torque/chunk command
- compute vacf command
- compute vcm/chunk command
- compute voronoi/atom command
- compute xrd command
- pair_style adp command
- pair_style adp/omp command
- pair_style agni command
- pair_style agni/omp command
- pair_style airebo command
- pair_style airebo/omp command
- pair_style airebo/morse command
- pair_style airebo/morse/omp command
- pair_style rebo command
- pair_style rebo/omp command
- pair_style awpmd/cut command
- pair_style beck command
- pair_style beck/gpu command
- pair_style beck/omp command
- pair_style body command
- pair_style bop command
- pair_style born command
- pair_style born/omp command
- pair_style born/gpu command
- pair_style born/coul/long command
- pair_style born/coul/long/cs command
- pair_style born/coul/long/gpu command
- pair_style born/coul/long/omp command
- pair_style born/coul/msm command
- pair_style born/coul/msm/omp command
- pair_style born/coul/wolf command
- pair_style born/coul/wolf/gpu command
- pair_style born/coul/wolf/omp command
- pair_style born/coul/dsf command
- pair_style born/coul/dsf/cs command
- pair_style brownian command
- pair_style brownian/omp command
- pair_style brownian/poly command
- pair_style brownian/poly/omp command
- pair_style buck command
- pair_style buck/gpu command
- pair_style buck/intel command
- pair_style buck/kk command
- pair_style buck/omp command
- pair_style buck/coul/cut command
- pair_style buck/coul/cut/gpu command
- pair_style buck/coul/cut/intel command
- pair_style buck/coul/cut/kk command
- pair_style buck/coul/cut/omp command
- pair_style buck/coul/long command
- pair_style buck/coul/long/cs command
- pair_style buck/coul/long/gpu command
- pair_style buck/coul/long/intel command
- pair_style buck/coul/long/kk command
- pair_style buck/coul/long/omp command
- pair_style buck/coul/msm command
- pair_style buck/coul/msm/omp command
- pair_style buck/long/coul/long command
- pair_style buck/long/coul/long/omp command
- pair_style lj/charmm/coul/charmm command
- pair_style lj/charmm/coul/charmm/omp command
- pair_style lj/charmm/coul/charmm/implicit command
- pair_style lj/charmm/coul/charmm/implicit/omp command
- pair_style lj/charmm/coul/long command
- pair_style lj/charmm/coul/long/gpu command
- pair_style lj/charmm/coul/long/intel command
- pair_style lj/charmm/coul/long/opt command
- pair_style lj/charmm/coul/long/omp command
- pair_style lj/charmm/coul/msm command
- pair_style lj/charmm/coul/msm/omp command
- pair_style lj/charmmfsw/coul/charmmfsh command
- pair_style lj/charmmfsw/coul/long command
- pair_style lj/class2 command
- pair_style lj/class2/gpu command
- pair_style lj/class2/kk command
- pair_style lj/class2/omp command
- pair_style lj/class2/coul/cut command
- pair_style lj/class2/coul/cut/kk command
- pair_style lj/class2/coul/cut/omp command
- pair_style lj/class2/coul/long command
- pair_style lj/class2/coul/long/gpu command
- pair_style lj/class2/coul/long/kk command
- pair_style lj/class2/coul/long/omp command
- pair_style colloid command
- pair_style colloid/gpu command
- pair_style colloid/omp command
- pair_style comb command
- pair_style comb/omp command
- pair_style comb3 command
- pair_style coul/cut command
- pair_style coul/cut/gpu command
- pair_style coul/cut/kk command
- pair_style coul/cut/omp command
- pair_style coul/debye command
- pair_style coul/debye/gpu command
- pair_style coul/debye/kk command
- pair_style coul/debye/omp command
- pair_style coul/dsf command
- pair_style coul/dsf/gpu command
- pair_style coul/dsf/kk command
- pair_style coul/dsf/omp command
- pair_style coul/long command
- pair_style coul/long/cs command
- pair_style coul/long/omp command
- pair_style coul/long/gpu command
- pair_style coul/long/kk command
- pair_style coul/msm command
- pair_style coul/msm/omp command
- pair_style coul/streitz command
- pair_style coul/wolf command
- pair_style coul/wolf/kk command
- pair_style coul/wolf/omp command
- pair_style tip4p/cut command
- pair_style tip4p/long command
- pair_style tip4p/cut/omp command
- pair_style tip4p/long/omp command
- pair_style coul/diel command
- pair_style coul/diel/omp command
- pair_style born/coul/long/cs command
- pair_style buck/coul/long/cs command
- pair_style born/coul/dsf/cs command
- pair_style lj/cut/dipole/cut command
- pair_style lj/cut/dipole/cut/gpu command
- pair_style lj/cut/dipole/cut/omp command
- pair_style lj/sf/dipole/sf command
- pair_style lj/sf/dipole/sf/gpu command
- pair_style lj/sf/dipole/sf/omp command
- pair_style lj/cut/dipole/long command
- pair_style lj/long/dipole/long command
- pair_style dpd command
- pair_style dpd/gpu command
- pair_style dpd/omp command
- pair_style dpd/tstat command
- pair_style dpd/tstat/gpu command
- pair_style dpd/tstat/omp command
- pair_style dpd/fdt command
- pair_style dpd/fdt/energy command
- pair_style dsmc command
- pair_style eam command
- pair_style eam/gpu command
- pair_style eam/intel command
- pair_style eam/kk command
- pair_style eam/omp command
- pair_style eam/opt command
- pair_style eam/alloy command
- pair_style eam/alloy/gpu command
- pair_style eam/alloy/kk command
- pair_style eam/alloy/omp command
- pair_style eam/alloy/opt command
- pair_style eam/cd command
- pair_style eam/cd/omp command
- pair_style eam/fs command
- pair_style eam/fs/gpu command
- pair_style eam/fs/kk command
- pair_style eam/fs/omp command
- pair_style eam/fs/opt command
- pair_style edip command
- pair_style eff/cut command
- pair_style eim command
- pair_style eim/omp command
- pair_style exp6/rx command
- pair_style gauss command
- pair_style gauss/gpu command
- pair_style gauss/omp command
- pair_style gauss/cut command
- pair_style gauss/cut/omp command
- pair_style gayberne command
- pair_style gayberne/gpu command
- pair_style gayberne/intel command
- pair_style gayberne/omp command
- pair_style gran/hooke command
- pair_style gran/omp command
- pair_style gran/hooke/history command
- pair_style gran/hooke/history/omp command
- pair_style gran/hertz/history command
- pair_style gran/hertz/history/omp command
- pair_style lj/gromacs command
- pair_style lj/gromacs/gpu command
- pair_style lj/gromacs/omp command
- pair_style lj/gromacs/coul/gromacs command
- pair_style lj/gromacs/coul/gromacs/omp command
- pair_style hbond/dreiding/lj command
- pair_style hbond/dreiding/lj/omp command
- pair_style hbond/dreiding/morse command
- pair_style hbond/dreiding/morse/omp command
- pair_style hybrid command
- pair_style hybrid/omp command
- pair_style hybrid/overlay command
- pair_style hybrid/overlay/omp command
- pair_style kim command
- pair_style kolmogorov/crespi/z command
- pair_style lcbop command
- pair_style line/lj command
- pair_style list command
- pair_style lj/cut command
- pair_style lj/cut/gpu command
- pair_style lj/cut/intel command
- pair_style lj/cut/kk command
- pair_style lj/cut/opt command
- pair_style lj/cut/omp command
- pair_style lj/cut/coul/cut command
- pair_style lj/cut/coul/cut/gpu command
- pair_style lj/cut/coul/cut/omp command
- pair_style lj/cut/coul/debye command
- pair_style lj/cut/coul/debye/gpu command
- pair_style lj/cut/coul/debye/kk command
- pair_style lj/cut/coul/debye/omp command
- pair_style lj/cut/coul/dsf command
- pair_style lj/cut/coul/dsf/gpu command
- pair_style lj/cut/coul/dsf/kk command
- pair_style lj/cut/coul/dsf/omp command
- pair_style lj/cut/coul/long command
- pair_style lj/cut/coul/long/cs command
- pair_style lj/cut/coul/long/gpu command
- pair_style lj/cut/coul/long/intel command
- pair_style lj/cut/coul/long/opt command
- pair_style lj/cut/coul/long/omp command
- pair_style lj/cut/coul/msm command
- pair_style lj/cut/coul/msm/gpu command
- pair_style lj/cut/coul/msm/omp command
- pair_style lj/cut/tip4p/cut command
- pair_style lj/cut/tip4p/cut/omp command
- pair_style lj/cut/tip4p/long command
- pair_style lj/cut/tip4p/long/omp command
- pair_style lj/cut/tip4p/long/opt command
- pair_style lj96/cut command
- pair_style lj96/cut/gpu command
- pair_style lj96/cut/omp command
- pair_style lj/cubic command
- pair_style lj/cubic/gpu command
- pair_style lj/cubic/omp command
- pair_style lj/expand command
- pair_style lj/expand/gpu command
- pair_style lj/expand/omp command
- pair_style lj/long/coul/long command
- pair_style lj/long/coul/long/omp command
- pair_style lj/long/coul/long/opt command
- pair_style lj/long/tip4p/long command
- pair_style lj/sf command
- pair_style lj/sf/omp command
- pair_style lj/smooth command
- pair_style lj/smooth/omp command
- pair_style lj/smooth/linear command
- pair_style lj/smooth/linear/omp command
- pair_style lj/cut/soft command
- pair_style lj/cut/soft/omp command
- pair_style lj/cut/coul/cut/soft command
- pair_style lj/cut/coul/cut/soft/omp command
- pair_style lj/cut/coul/long/soft command
- pair_style lj/cut/coul/long/soft/omp command
- pair_style lj/cut/tip4p/long/soft command
- pair_style lj/cut/tip4p/long/soft/omp command
- pair_style lj/charmm/coul/long/soft command
- pair_style lj/charmm/coul/long/soft/omp command
- pair_style coul/cut/soft command
- pair_style coul/cut/soft/omp command
- pair_style coul/long/soft command
- pair_style coul/long/soft/omp command
- pair_style tip4p/long/soft command
- pair_style tip4p/long/soft/omp command
- pair_style lubricate command
- pair_style lubricate/omp command
- pair_style lubricate/poly command
- pair_style lubricate/poly/omp command
- pair_style lubricateU command
- pair_style lubricateU/poly command
- pair_style lj/mdf command
- pair_style buck/mdf command
- pair_style lennard/mdf command
- pair_style meam command
- pair_style meam/spline
- pair_style meam/spline/omp
- pair_style meam/sw/spline
- pair_style meam/sw/spline/omp
- pair_style mgpt command
- pair_style mie/cut command
- pair_style mie/cut/gpu command
- pair_style momb command
- pair_style morse command
- pair_style morse/gpu command
- pair_style morse/omp command
- pair_style morse/opt command
- pair_style morse/smooth/linear command
- pair_style morse/smooth/linear/omp command
- pair_style morse/soft command
- pair_style morse/kk command
- pair_style multi/lucy command
- pair_style multi/lucy/rx command
- pair_style nb3b/harmonic command
- pair_style nb3b/harmonic/omp command
- pair_style nm/cut command
- pair_style nm/cut/coul/cut command
- pair_style nm/cut/coul/long command
- pair_style nm/cut/omp command
- pair_style nm/cut/coul/cut/omp command
- pair_style nm/cut/coul/long/omp command
- pair_style none command
- pair_style oxdna/excv command
- pair_style oxdna/stk command
- pair_style oxdna/hbond command
- pair_style oxdna/xstk command
- pair_style oxdna/coaxstk command
- pair_style oxdna2/excv command
- pair_style oxdna2/stk command
- pair_style oxdna2/hbond command
- pair_style oxdna2/xstk command
- pair_style oxdna2/coaxstk command
- pair_style oxdna2/dh command
- pair_style peri/pmb command
- pair_style peri/pmb/omp command
- pair_style peri/lps command
- pair_style peri/lps/omp command
- pair_style peri/ves command
- pair_style peri/eps command
- pair_style polymorphic command
- pair_style quip command
- pair_style reax command
- pair_style reax/c command
- pair_style reax/c/kk command
- pair_style resquared command
- pair_style resquared/gpu command
- pair_style resquared/omp command
- pair_style lj/sdk command
- pair_style lj/sdk/gpu command
- pair_style lj/sdk/kk command
- pair_style lj/sdk/omp command
- pair_style lj/sdk/coul/long command
- pair_style lj/sdk/coul/long/gpu command
- pair_style lj/sdk/coul/long/omp command
- pair_style smd/hertz command
- pair_style smd/tlsph command
- pair_style smd/tri_surface command
- pair_style smd/ulsph command
- pair_style smtbq command
- pair_style snap command
- pair_style soft command
- pair_style soft/gpu command
- pair_style soft/omp command
- pair_style sph/heatconduction command
- pair_style sph/idealgas command
- pair_style sph/lj command
- pair_style sph/rhosum command
- pair_style sph/taitwater command
- pair_style sph/taitwater/morris command
- pair_style srp command
- pair_style sw command
- pair_style sw/gpu command
- pair_style sw/intel command
- pair_style sw/kk command
- pair_style sw/omp command
- pair_style table command
- pair_style table/gpu command
- pair_style table/kk command
- pair_style table/omp command
- pair_style table/rx command
- pair_style tersoff command
- pair_style tersoff/table command
- pair_style tersoff/gpu
- pair_style tersoff/intel
- pair_style tersoff/kk
- pair_style tersoff/omp
- pair_style tersoff/table/omp command
- pair_style tersoff/mod command
- pair_style tersoff/mod/c command
- pair_style tersoff/mod/gpu command
- pair_style tersoff/mod/kk command
- pair_style tersoff/mod/omp command
- pair_style tersoff/mod/c/omp command
- pair_style tersoff/zbl command
- pair_style tersoff/zbl/gpu command
- pair_style tersoff/zbl/kk command
- pair_style tersoff/zbl/omp command
- pair_style thole command
- pair_style lj/cut/thole/long command
- pair_style lj/cut/thole/long/omp command
- pair_style tri/lj command
- pair_style vashishta command
- pair_style vashishta/omp command
- pair_style vashishta/kk command
- pair_style vashishta/table command
- pair_style vashishta/table/omp command
- pair_style yukawa command
- pair_style yukawa/gpu command
- pair_style yukawa/omp command
- pair_style yukawa/colloid command
- pair_style yukawa/colloid/gpu command
- pair_style yukawa/colloid/omp command
- pair_style zbl command
- pair_style zbl/gpu command
- pair_style zbl/omp command
- pair_style zero command
- bond_style class2 command
- bond_style class2/omp command
- bond_style class2/kk command
- bond_style fene command
- bond_style fene/intel command
- bond_style fene/kk command
- bond_style fene/omp command
- bond_style fene/expand command
- bond_style fene/expand/omp command
- bond_style oxdna/fene command
- bond_style oxdna2/fene command
- bond_style harmonic command
- bond_style harmonic/intel command
- bond_style harmonic/kk command
- bond_style harmonic/omp command
- bond_style harmonic/shift command
- bond_style harmonic/shift/omp command
- bond_style harmonic/shift/cut command
- bond_style harmonic/shift/cut/omp command
- bond_style hybrid command
- bond_style morse command
- bond_style morse/omp command
- bond_style none command
- bond_style nonlinear command
- bond_style nonlinear/omp command
- bond_style quartic command
- bond_style quartic/omp command
- bond_style table command
- bond_style table/omp command
- bond_style zero command
- angle_style charmm command
- angle_style charmm/intel command
- angle_style charmm/kk command
- angle_style charmm/omp command
- angle_style class2 command
- angle_style class2/omp command
- angle_style class2/kk command
- angle_style cosine command
- angle_style cosine/omp command
- angle_style cosine/delta command
- angle_style cosine/delta/omp command
- angle_style cosine/periodic command
- angle_style cosine/periodic/omp command
- angle_style cosine/shift command
- angle_style cosine/shift/omp command
- angle_style cosine/shift/exp command
- angle_style cosine/shift/exp/omp command
- angle_style cosine/squared command
- angle_style cosine/squared/omp command
- angle_style dipole command
- angle_style dipole/omp command
- angle_style fourier command
- angle_style fourier/omp command
- angle_style fourier/simple command
- angle_style fourier/simple/omp command
- angle_style harmonic command
- angle_style harmonic/intel command
- angle_style harmonic/kk command
- angle_style harmonic/omp command
- angle_style hybrid command
- angle_style none command
- angle_style quartic command
- angle_style quartic/omp command
- angle_style sdk command
- angle_style table command
- angle_style table/omp command
- angle_style zero command
- dihedral_style charmm command
- dihedral_style charmm/intel command
- dihedral_style charmm/kk command
- dihedral_style charmm/omp command
- dihedral_style charmmfsh command
- dihedral_style class2 command
- dihedral_style class2/omp command
- dihedral_style class2/kk command
- dihedral_style cosine/shift/exp command
- dihedral_style cosine/shift/exp/omp command
- dihedral_style fourier command
- dihedral_style fourier/omp command
- dihedral_style harmonic command
- dihedral_style harmonic/intel command
- dihedral_style harmonic/omp command
- dihedral_style helix command
- dihedral_style helix/omp command
- dihedral_style hybrid command
- dihedral_style multi/harmonic command
- dihedral_style multi/harmonic/omp command
- dihedral_style nharmonic command
- dihedral_style nharmonic/omp command
- dihedral_style none command
- dihedral_style opls command
- dihedral_style opls/intel command
- dihedral_style opls/kk command
- dihedral_style opls/omp command
- dihedral_style quadratic command
- dihedral_style quadratic/omp command
- dihedral_style spherical command
- dihedral_style table command
- dihedral_style table/omp command
- dihedral_style zero command
- improper_style class2 command
- improper_style class2/omp command
- improper_style class2/kk command
- improper_style cossq command
- improper_style cossq/omp command
- improper_style cvff command
- improper_style cvff/intel command
- improper_style cvff/omp command
- improper_style distance command
- improper_style fourier command
- improper_style fourier/omp command
- improper_style harmonic command
- improper_style harmonic/intel command
- improper_style harmonic/kk command
- improper_style harmonic/omp command
- improper_style hybrid command
- improper_style none command
- improper_style ring command
- improper_style ring/omp command
- improper_style umbrella command
- improper_style umbrella/omp command
- improper_style zero command
- Body particles
- fix_modify AtC add_molecule
- fix_modify AtC add_species
- fix_modify AtC atom_element_map
- fix_modify AtC atom_weight
- fix_modify AtC atomic_charge
- fix_modify AtC boundary
- fix_modify AtC boundary_dynamics
- fix_modify AtC boundary_faceset
- fix_modify AtC output boundary_integral
- fix_modify AtC consistent_fe_initialization
- fix_modify AtC output contour_integral
- fix_modify AtC control
- fix_modify AtC control momentum
- fix_modify AtC control thermal
- fix_modify AtC control thermal correction_max_iterations
- fix_modify AtC decomposition
- fix_modify AtC extrinsic electron_integration
- fix_modify AtC equilibrium_start
- fix_modify AtC extrinsic exchange
- fix_modify AtC fe_md_boundary
- fix_modify AtC fem create mesh
- fix_modify AtC filter scale
- fix_modify AtC filter type
- fix atc command
- fix_modify AtC fix_flux
- fix_modify AtC fix
- fix_modify AtC computes
- fix_modify AtC fields
- fix_modify AtC gradients
- fix_modify AtC kernel
- fix_modify AtC on_the_fly
- fix_modify AtC rates
- fix_modify AtC initial
- fix_modify AtC internal_atom_integrate
- fix_modify AtC internal_element_set
- fix_modify AtC internal_quadrature
- fix_modify AtC kernel
- fix_modify AtC control localized_lambda
- fix_modify AtC control lumped_lambda_solve
- fix_modify AtC control mask_direction
- fix_modify AtC mass_matrix
- fix_modify AtC material
- fix_modify AtC mesh add_to_nodeset
- fix_modify AtC mesh create
- fix_modify AtC mesh create_elementset
- fix_modify AtC mesh create_faceset box
- fix_modify AtC mesh create_faceset plane
- fix_modify AtC mesh create_nodeset
- fix_modify AtC mesh delete_elements
- fix_modify AtC mesh nodeset_to_elementset
- fix_modify AtC mesh output
- fix_modify AtC mesh quadrature
- fix_modify AtC mesh read
- fix_modify AtC mesh write
- fix_modify AtC time_integration (momentum)
- fix_modify AtC output
- fix_modify AtC output elementset
- fix_modify AtC output nodeset
- fix_modify AtC pair_interactions/bond_interactions
- fix_modify AtC poisson_solver
- fix_modify AtC read_restart
- fix_modify AtC remove_molecule
- fix_modify AtC remove_source
- fix_modify AtC remove_species
- fix_modify AtC reset_atomic_reference_positions
- fix_modify AtC reset_time
- fix_modify AtC sample_frequency
- fix_modify AtC set
- fix_modify AtC source
- fix_modify AtC source_integration
- fix_modify AtC temperature_definition
- fix_modify AtC time_integration (thermal)
- fix_modify AtC filter
- fix_modify AtC track_displacement
- fix_modify AtC unfix_flux
- fix_modify AtC unfix
- fix_modify AtC write_atom_weights
- fix_modify AtC write_restart
LAMMPS Users Manual
31 Mar 2017 version
http://lammps.sandia.gov - Sandia National Laboratories
Copyright (2003) Sandia Corporation. This software and manual is distributed under the GNU General Public License.
Table of Contents
LAMMPS Documentation.......................................................................................................................1
31 Mar 2017 version..........................................................................................................................1
Version info:...............................................................................................................................1
1. Introduction...................................................................................................................................4
1.1 What is LAMMPS................................................................................................................4
1.2 LAMMPS features................................................................................................................5
1.3 LAMMPS non-features.........................................................................................................8
1.4 Open source distribution.....................................................................................................10
1.5 Acknowledgments and citations.........................................................................................11
2. Getting Started.............................................................................................................................13
2.1 What's in the LAMMPS distribution..................................................................................13
2.2 Making LAMMPS..............................................................................................................14
2.3 Making LAMMPS with optional packages........................................................................22
2.4 Building LAMMPS via the Make.py tool..........................................................................26
2.5 Building LAMMPS as a library..........................................................................................28
2.6 Running LAMMPS.............................................................................................................30
2.7 Command-line options........................................................................................................32
2.8 LAMMPS screen output.....................................................................................................38
2.9 Tips for users of previous LAMMPS versions...................................................................40
3. Commands...................................................................................................................................42
3.1 LAMMPS input script........................................................................................................42
3.2 Parsing rules........................................................................................................................43
3.3 Input script structure...........................................................................................................44
3.4 Commands listed by category.............................................................................................46
3.5 Individual commands..........................................................................................................47
Fix styles...................................................................................................................................47
Compute styles..........................................................................................................................48
Pair_style potentials..................................................................................................................49
Bond_style potentials................................................................................................................50
Angle_style potentials...............................................................................................................51
Dihedral_style potentials..........................................................................................................51
Improper_style potentials.........................................................................................................51
Kspace solvers..........................................................................................................................52
4. Packages......................................................................................................................................53
4.1 Standard packages...............................................................................................................53
4.2 User packages.....................................................................................................................70
5. Accelerating LAMMPS performance.........................................................................................84
5.1 Measuring performance......................................................................................................84
5.2 General strategies................................................................................................................85
5.3 Packages with optimized styles..........................................................................................86
5.4 Comparison of various accelerator packages......................................................................89
6. How-to discussions....................................................................................................................115
6.1 Restarting a simulation.....................................................................................................115
6.2 2d simulations...................................................................................................................117
6.3 CHARMM, AMBER, and DREIDING force fields.........................................................117
6.4 Running multiple simulations from one input script........................................................119
6.5 Multi-replica simulations..................................................................................................120
6.6 Granular models................................................................................................................121
LAMMPS Users Manual
i
Table of Contents
6.7 TIP3P water model...........................................................................................................122
6.8 TIP4P water model...........................................................................................................123
6.9 SPC water model..............................................................................................................124
6.10 Coupling LAMMPS to other codes................................................................................125
6.11 Visualizing LAMMPS snapshots....................................................................................126
6.12 Triclinic (non-orthogonal) simulation boxes..................................................................127
6.13 NEMD simulations.........................................................................................................131
6.14 Finite-size spherical and aspherical particles..................................................................132
6.15 Output from LAMMPS (thermo, dumps, computes, fixes, variables)...........................135
6.16 Thermostatting, barostatting, and computing temperature.............................................140
6.17 Walls...............................................................................................................................142
6.18 Elastic constants..............................................................................................................143
6.19 Library interface to LAMMPS.......................................................................................143
6.20 Calculating thermal conductivity....................................................................................146
6.21 Calculating viscosity.......................................................................................................147
6.22 Calculating a diffusion coefficient..................................................................................149
6.23 Using chunks to calculate system properties..................................................................149
6.24 Setting parameters for the kspace_style pppm/disp command.......................................152
6.25 Polarizable models..........................................................................................................153
6.26 Adiabatic core/shell model.............................................................................................154
6.27 Drude induced dipoles....................................................................................................157
7. Example problems.....................................................................................................................160
Lowercase directories.............................................................................................................160
Uppercase directories..............................................................................................................162
8. Performance & scalability.........................................................................................................163
9. Additional tools.........................................................................................................................165
amber2lmp tool.......................................................................................................................165
binary2txt tool.........................................................................................................................165
ch2lmp tool.............................................................................................................................166
chain tool.................................................................................................................................166
colvars tools............................................................................................................................166
createatoms tool......................................................................................................................166
drude tool................................................................................................................................167
eam database tool....................................................................................................................167
eam generate tool....................................................................................................................167
eff tool.....................................................................................................................................167
emacs tool...............................................................................................................................167
fep tool....................................................................................................................................167
i-pi tool....................................................................................................................................168
ipp tool....................................................................................................................................168
kate tool...................................................................................................................................168
lmp2arc tool............................................................................................................................168
lmp2cfg tool............................................................................................................................168
matlab tool..............................................................................................................................169
micelle2d tool.........................................................................................................................169
moltemplate tool.....................................................................................................................169
msi2lmp tool...........................................................................................................................169
phonon tool.............................................................................................................................169
LAMMPS Users Manual
ii
Table of Contents
polybond tool..........................................................................................................................170
pymol_asphere tool.................................................................................................................170
python tool..............................................................................................................................170
reax tool..................................................................................................................................170
smd tool...................................................................................................................................170
vim tool...................................................................................................................................171
xmgrace tool...........................................................................................................................171
10. Modifying & extending LAMMPS.........................................................................................172
10.1 Atom styles.....................................................................................................................173
10.2 Bond, angle, dihedral, improper potentials.....................................................................175
10.3 Compute styles................................................................................................................175
10.4 Dump styles....................................................................................................................176
10.5 Dump custom output options..........................................................................................176
10.6 Fix styles.........................................................................................................................177
10.7 Input script commands....................................................................................................179
10.8 Kspace computations......................................................................................................179
10.9 Minimization styles.........................................................................................................179
10.10 Pairwise potentials........................................................................................................180
10.11 Region styles.................................................................................................................180
10.12 Body styles....................................................................................................................180
10.13 Thermodynamic output options....................................................................................181
10.14 Variable options............................................................................................................181
10.15 Submitting new features for inclusion in LAMMPS....................................................182
11. Python interface to LAMMPS.................................................................................................186
11.1 Overview of running LAMMPS from Python................................................................187
11.2 Overview of using Python from a LAMMPS script.......................................................187
11.3 Building LAMMPS as a shared library..........................................................................188
11.4 Installing the Python wrapper into Python.....................................................................189
11.5 Extending Python with MPI to run in parallel................................................................190
11.6 Testing the Python-LAMMPS interface.........................................................................192
11.7 Using LAMMPS from Python........................................................................................194
11.8 Example Python scripts that use LAMMPS...................................................................197
11.9 PyLammps interface.......................................................................................................199
12. Errors.......................................................................................................................................200
12.1 Common problems..........................................................................................................200
12.2 Reporting bugs................................................................................................................201
12.3 Error & warning messages..............................................................................................202
Errors:.....................................................................................................................................202
Warnings:................................................................................................................................307
13. Future and history....................................................................................................................317
13.1 Coming attractions..........................................................................................................317
13.2 Past versions...................................................................................................................317
Tutorial for Thermalized Drude oscillators in LAMMPS.............................................................320
LAMMPS GitHub tutorial.............................................................................................................327
PyLammps Tutorial........................................................................................................................................342
Body particles......................................................................................................................................343
Manifolds (surfaces)......................................................................................................................347
LAMMPS Users Manual
iii
Table of Contents
PyLammps Tutorial
angle_coeff command...................................................................................................................348
angle_style command....................................................................................................................350
atom_modify command.................................................................................................................352
atom_style command.....................................................................................................................355
balance command..........................................................................................................................360
bond_coeff command....................................................................................................................368
bond_style command.....................................................................................................................370
bond_write command....................................................................................................................372
boundary command.......................................................................................................................373
box command................................................................................................................................375
change_box command...................................................................................................................376
clear command..............................................................................................................................381
comm_modify command...............................................................................................................382
comm_style command...................................................................................................................385
compute command........................................................................................................................386
compute_modify command...........................................................................................................390
create_atoms command.................................................................................................................392
create_bonds command.................................................................................................................397
create_box command.....................................................................................................................399
delete_atoms command.................................................................................................................402
delete_bonds command.................................................................................................................405
dielectric command.......................................................................................................................408
dihedral_coeff command...............................................................................................................409
dihedral_style command................................................................................................................411
dimension command......................................................................................................................413
displace_atoms command..............................................................................................................414
dump command.............................................................................................................................416
dump custom/vtk command..........................................................................................................416
dump h5md command...................................................................................................................416
dump image command..................................................................................................................416
dump movie command..................................................................................................................416
dump molfile command.................................................................................................................416
dump nc command........................................................................................................................416
dump custom/vtk command..........................................................................................................426
dump h5md command...................................................................................................................431
dump image command..................................................................................................................433
dump movie command..................................................................................................................433
dump_modify command................................................................................................................443
dump molfile command.................................................................................................................456
dump nc command........................................................................................................................458
dump nc/mpiio command..............................................................................................................458
echo command...............................................................................................................................459
fix command..................................................................................................................................460
fix_modify command....................................................................................................................465
group command.............................................................................................................................467
group2ndx command.....................................................................................................................472
ndx2group command.....................................................................................................................472
LAMMPS Users Manual
iv
Table of Contents
PyLammps Tutorial
if command....................................................................................................................................473
improper_coeff command.............................................................................................................476
improper_style command..............................................................................................................478
include command..........................................................................................................................480
info command................................................................................................................................481
jump command..............................................................................................................................483
kspace_modify command..............................................................................................................486
kspace_style command..................................................................................................................491
label command..............................................................................................................................497
lattice command............................................................................................................................498
log command.................................................................................................................................502
mass command..............................................................................................................................503
min_modify command..................................................................................................................505
min_style command......................................................................................................................507
minimize command.......................................................................................................................509
molecule command........................................................................................................................513
neb command................................................................................................................................520
neigh_modify command................................................................................................................526
neighbor command........................................................................................................................530
newton command..........................................................................................................................532
next command...............................................................................................................................533
package command.........................................................................................................................536
pair_coeff command......................................................................................................................545
pair_modify command..................................................................................................................548
pair_style command......................................................................................................................552
pair_write command......................................................................................................................556
partition command.........................................................................................................................558
prd command.................................................................................................................................560
print command...............................................................................................................................565
processors command.....................................................................................................................567
python command...........................................................................................................................572
quit command................................................................................................................................580
read_data command.......................................................................................................................581
read_dump command....................................................................................................................598
read_restart command...................................................................................................................603
region command............................................................................................................................607
replicate command........................................................................................................................613
rerun command..............................................................................................................................615
reset_timestep command...............................................................................................................618
restart command............................................................................................................................619
run command.................................................................................................................................622
run_style command.......................................................................................................................626
set command..................................................................................................................................631
shell command...............................................................................................................................638
special_bonds command................................................................................................................640
suffix command.............................................................................................................................644
tad command.................................................................................................................................646
LAMMPS Users Manual
v
Table of Contents
PyLammps Tutorial
temper command...........................................................................................................................651
temper/grem command..................................................................................................................654
thermo command...........................................................................................................................656
thermo_modify command.............................................................................................................657
thermo_style command.................................................................................................................660
timer command..............................................................................................................................666
timestep command.........................................................................................................................668
uncompute command....................................................................................................................669
undump command.........................................................................................................................670
unfix command..............................................................................................................................671
units command..............................................................................................................................672
variable command.........................................................................................................................676
Numbers, constants, and thermo keywords............................................................................683
Math Operators.......................................................................................................................683
Math Functions.......................................................................................................................684
Group and Region Functions..................................................................................................686
Special Functions....................................................................................................................686
Feature Functions....................................................................................................................687
Atom Values and Vectors.......................................................................................................688
Compute References...............................................................................................................689
Fix References........................................................................................................................690
Variable References................................................................................................................690
velocity command.........................................................................................................................695
write_coeff command....................................................................................................................699
write_data command.....................................................................................................................700
write_dump command...................................................................................................................702
write_restart command..................................................................................................................704
fix adapt command........................................................................................................................706
fix adapt/fep command..................................................................................................................710
fix addforce command...................................................................................................................714
fix addtorque command.................................................................................................................717
fix append/atoms command...........................................................................................................719
fix atc command............................................................................................................................721
fix atom/swap command...............................................................................................................727
fix ave/atom command..................................................................................................................730
fix ave/chunk command................................................................................................................733
fix ave/correlate command............................................................................................................740
fix ave/correlate/long command....................................................................................................745
fix ave/histo command..................................................................................................................748
fix ave/histo/weight command......................................................................................................748
fix ave/time command...................................................................................................................754
fix aveforce command...................................................................................................................759
fix balance command.....................................................................................................................761
fix bond/break command...............................................................................................................767
fix bond/create command..............................................................................................................770
fix bond/swap command...............................................................................................................774
fix box/relax command..................................................................................................................777
LAMMPS Users Manual
vi
Table of Contents
PyLammps Tutorial
fix cmap command........................................................................................................................783
fix colvars command.....................................................................................................................785
fix controller command.................................................................................................................787
fix deform command.....................................................................................................................791
fix deform/kk command................................................................................................................791
fix deposit command.....................................................................................................................800
fix dpd/energy command...............................................................................................................805
fix drag command..........................................................................................................................807
fix drude command........................................................................................................................808
fix drude/transform/direct command.............................................................................................809
fix drude/transform/inverse command..........................................................................................809
fix dt/reset command.....................................................................................................................812
fix efield command........................................................................................................................814
fix ehex command.........................................................................................................................817
fix enforce2d command.................................................................................................................820
fix eos/cv command.......................................................................................................................821
fix eos/table command...................................................................................................................822
fix eos/table/rx command..............................................................................................................824
fix evaporate command.................................................................................................................827
fix external command....................................................................................................................829
fix filter/corotate command...........................................................................................................832
fix flow/gauss command...............................................................................................................834
fix freeze command.......................................................................................................................837
fix gcmc command........................................................................................................................839
fix gld command............................................................................................................................846
fix gle command............................................................................................................................849
fix gravity command.....................................................................................................................852
fix gravity/omp command.............................................................................................................852
fix grem command.........................................................................................................................855
fix halt command...........................................................................................................................857
fix heat command..........................................................................................................................860
fix imd command...........................................................................................................................862
fix indent command.......................................................................................................................865
fix ipi command.............................................................................................................................868
fix langevin command...................................................................................................................870
fix langevin/kk command..............................................................................................................870
fix langevin/drude command.........................................................................................................875
fix langevin/eff command.............................................................................................................879
fix lb/fluid command.....................................................................................................................881
fix lb/momentum command..........................................................................................................888
fix lb/pc command.........................................................................................................................890
fix lb/rigid/pc/sphere command.....................................................................................................891
fix lb/viscous command.................................................................................................................894
fix lineforce command...................................................................................................................896
fix manifoldforce command..........................................................................................................897
fix meso command........................................................................................................................898
fix meso/stationary command.......................................................................................................899
LAMMPS Users Manual
vii
Table of Contents
PyLammps Tutorial
fix momentum command...............................................................................................................900
fix momentum/kk command.........................................................................................................900
fix move command........................................................................................................................902
fix mscg command........................................................................................................................906
fix msst command........................................................................................................................908
fix neb command...........................................................................................................................911
fix nvt command............................................................................................................................913
fix nvt/intel command...................................................................................................................913
fix nvt/kk command.......................................................................................................................913
fix nvt/omp command...................................................................................................................913
fix npt command............................................................................................................................913
fix npt/intel command...................................................................................................................913
fix npt/kk command.......................................................................................................................913
fix npt/omp command...................................................................................................................913
fix nph command...........................................................................................................................913
fix nph/kk command......................................................................................................................913
fix nph/omp command...................................................................................................................913
fix nvt/eff command......................................................................................................................923
fix npt/eff command......................................................................................................................923
fix nph/eff command.....................................................................................................................923
fix nph/asphere command.............................................................................................................926
fix nph/asphere/omp command.....................................................................................................926
fix nph/body command..................................................................................................................929
fix nph/sphere command...............................................................................................................932
fix nph/sphere/omp command.......................................................................................................932
fix nphug command.......................................................................................................................935
fix nphug/omp command...............................................................................................................935
fix npt/asphere command..............................................................................................................939
fix npt/asphere/omp command......................................................................................................939
fix npt/body command...................................................................................................................942
fix npt/sphere command................................................................................................................945
fix npt/sphere/omp command........................................................................................................945
fix nve command...........................................................................................................................948
fix nve/intel command...................................................................................................................948
fix nve/kk command......................................................................................................................948
fix nve/omp command...................................................................................................................948
fix nve/asphere command..............................................................................................................950
fix nve/asphere/intel command.....................................................................................................950
fix nve/asphere/noforce command................................................................................................952
fix nve/body command..................................................................................................................953
fix nve/dot command.....................................................................................................................954
fix nve/dotc/langevin command....................................................................................................955
fix nve/eff command.....................................................................................................................957
fix nve/limit command..................................................................................................................958
fix nve/line command....................................................................................................................960
fix nve/manifold/rattle command..................................................................................................961
fix nve/noforce command..............................................................................................................963
LAMMPS Users Manual
viii
Table of Contents
PyLammps Tutorial
fix nve/sphere command...............................................................................................................964
fix nve/sphere/omp command.......................................................................................................964
fix nve/tri command......................................................................................................................966
fix nvk command...........................................................................................................................967
fix nvt/asphere command..............................................................................................................969
fix nvt/asphere/omp command......................................................................................................969
fix nvt/body command...................................................................................................................972
fix nvt/manifold/rattle command...................................................................................................975
fix nvt/sllod command...................................................................................................................977
fix nvt/sllod/intel command...........................................................................................................977
fix nvt/sllod/omp command...........................................................................................................977
fix nvt/sllod/eff command.............................................................................................................980
fix nvt/sphere command................................................................................................................982
fix nvt/sphere/omp command........................................................................................................982
fix oneway command....................................................................................................................985
fix orient/fcc command.................................................................................................................986
fix orient/bcc command.................................................................................................................986
fix phonon command.....................................................................................................................990
fix pimd command.........................................................................................................................994
fix planeforce command................................................................................................................998
fix poems.......................................................................................................................................999
fix pour command.......................................................................................................................1001
fix press/berendsen command.....................................................................................................1005
fix print command.......................................................................................................................1009
fix property/atom command........................................................................................................1011
fix qbmsst command...................................................................................................................1015
fix qeq/point command................................................................................................................1019
fix qeq/shielded command...........................................................................................................1019
fix qeq/slater command...............................................................................................................1019
fix qeq/dynamic command..........................................................................................................1019
fix qeq/fire command..................................................................................................................1019
fix qeq/comb command...............................................................................................................1023
fix qeq/comb/omp command.......................................................................................................1023
fix qeq/reax command.................................................................................................................1025
fix qeq/reax/kk command............................................................................................................1025
fix qmmm command...................................................................................................................1027
fix qtb command..........................................................................................................................1028
fix reax/bonds command.............................................................................................................1031
fix reax/c/bonds command..........................................................................................................1031
fix reax/c/bonds/kk command.....................................................................................................1031
fix reax/c/species command........................................................................................................1033
fix reax/c/species/kk command...................................................................................................1033
fix recenter command..................................................................................................................1036
fix restrain command...................................................................................................................1038
fix rigid command.......................................................................................................................1042
fix rigid/nve command................................................................................................................1042
fix rigid/nvt command.................................................................................................................1042
LAMMPS Users Manual
ix
Table of Contents
PyLammps Tutorial
fix rigid/npt command.................................................................................................................1042
fix rigid/nph command................................................................................................................1042
fix rigid/small command.............................................................................................................1042
fix rigid/nve/small command.......................................................................................................1042
fix rigid/nvt/small command.......................................................................................................1042
fix rigid/npt/small command.......................................................................................................1042
fix rigid/nph/small command......................................................................................................1042
fix rx command...........................................................................................................................1053
fix saed/vtk command.................................................................................................................1057
fix setforce command..................................................................................................................1060
fix setforce/kk command.............................................................................................................1060
fix shake command......................................................................................................................1062
fix rattle command.......................................................................................................................1062
fix shardlow command................................................................................................................1066
fix smd command........................................................................................................................1068
fix smd/adjust_dt command........................................................................................................1071
fix smd/integrate_tlsph command...............................................................................................1072
fix smd/integrate_ulsph command..............................................................................................1073
fix smd/move_tri_surf command................................................................................................1074
fix smd/setvel command..............................................................................................................1076
fix smd/wall_surface command...................................................................................................1078
fix spring command.....................................................................................................................1080
fix spring/chunk command..........................................................................................................1083
fix spring/rg command................................................................................................................1085
fix spring/self command..............................................................................................................1087
fix srd command..........................................................................................................................1089
fix store/force command..............................................................................................................1095
fix store/state command...............................................................................................................1097
fix temp/berendsen command.....................................................................................................1099
fix temp/csvr command...............................................................................................................1102
fix temp/csld command...............................................................................................................1102
fix temp/rescale command...........................................................................................................1105
fix temp/rescale/eff command.....................................................................................................1108
fix tfmc command.......................................................................................................................1110
fix thermal/conductivity command.............................................................................................1113
fix ti/spring command.................................................................................................................1116
fix tmd command.........................................................................................................................1119
fix ttm command.........................................................................................................................1121
fix ttm/mod command.................................................................................................................1121
fix tune/kspace command............................................................................................................1127
fix vector command.....................................................................................................................1129
fix viscosity command.................................................................................................................1132
fix viscous command...................................................................................................................1135
fix wall/lj93 command.................................................................................................................1137
fix wall/lj126 command...............................................................................................................1137
fix wall/lj1043 command.............................................................................................................1137
fix wall/colloid command............................................................................................................1137
LAMMPS Users Manual
x
Table of Contents
PyLammps Tutorial
fix wall/harmonic command........................................................................................................1137
fix wall/gran command................................................................................................................1142
fix wall/gran/region command....................................................................................................1145
fix wall/piston command.............................................................................................................1149
fix wall/reflect command.............................................................................................................1151
fix wall/reflect/kk command.......................................................................................................1151
fix wall/region command.............................................................................................................1154
fix wall/srd command..................................................................................................................1158
compute ackland/atom command................................................................................................1161
compute angle command.............................................................................................................1163
compute angle/local command....................................................................................................1164
compute angmom/chunk command.............................................................................................1166
compute basal/atom command....................................................................................................1168
compute body/local command.....................................................................................................1170
compute bond command.............................................................................................................1172
compute bond/local command.....................................................................................................1173
compute centro/atom command..................................................................................................1176
compute chunk/atom command...................................................................................................1179
compute cluster/atom command..................................................................................................1188
compute cna/atom command.......................................................................................................1190
compute com command...............................................................................................................1192
compute com/chunk command....................................................................................................1193
compute contact/atom command.................................................................................................1195
compute coord/atom command...................................................................................................1196
compute damage/atom command................................................................................................1198
compute dihedral command........................................................................................................1199
compute dihedral/local command...............................................................................................1200
compute dilatation/atom command.............................................................................................1202
compute dipole/chunk command.................................................................................................1203
compute displace/atom command...............................................................................................1205
compute dpd command...............................................................................................................1206
compute dpd/atom command......................................................................................................1208
compute erotate/asphere command.............................................................................................1209
compute erotate/rigid command..................................................................................................1211
compute erotate/sphere command...............................................................................................1212
compute erotate/sphere/atom command......................................................................................1213
compute event/displace command...............................................................................................1214
compute fep command................................................................................................................1215
compute global/atom command..................................................................................................1220
compute group/group command..................................................................................................1223
compute gyration command........................................................................................................1226
compute gyration/chunk command.............................................................................................1228
compute heat/flux command.......................................................................................................1230
compute hexorder/atom command..............................................................................................1234
compute improper command.......................................................................................................1236
compute improper/local command..............................................................................................1237
compute inertia/chunk command................................................................................................1239
LAMMPS Users Manual
xi
Table of Contents
PyLammps Tutorial
compute ke command..................................................................................................................1241
compute ke/atom command.........................................................................................................1242
compute ke/atom/eff command...................................................................................................1243
compute ke/eff command............................................................................................................1245
compute ke/rigid command.........................................................................................................1247
compute meso/e/atom command.................................................................................................1248
compute meso/rho/atom command.............................................................................................1249
compute meso/t/atom command..................................................................................................1250
compute msd command...............................................................................................................1251
compute msd/chunk command....................................................................................................1253
compute msd/nongauss command...............................................................................................1255
compute omega/chunk command................................................................................................1257
compute orientorder/atom command...........................................................................................1259
compute pair command...............................................................................................................1262
compute pair/local command......................................................................................................1264
compute pe command..................................................................................................................1266
compute pe/atom command.........................................................................................................1268
compute plasticity/atom command..............................................................................................1270
compute pressure command........................................................................................................1271
compute property/atom command...............................................................................................1274
compute property/chunk command.............................................................................................1277
compute property/local command...............................................................................................1279
compute rdf command.................................................................................................................1282
compute reduce command...........................................................................................................1285
compute reduce/region command...............................................................................................1285
compute rigid/local command.....................................................................................................1288
compute saed command..............................................................................................................1291
compute slice command..............................................................................................................1295
compute smd/contact/radius command.......................................................................................1297
compute smd/damage command.................................................................................................1298
compute smd/hourglass/error command.....................................................................................1299
compute smd/internal/energy command.....................................................................................1300
compute smd/plastic/strain command.........................................................................................1301
compute smd/plastic/strain/rate command..................................................................................1302
compute smd/rho command........................................................................................................1303
compute smd/tlsph/defgrad command.........................................................................................1304
compute smd/tlsph/dt command..................................................................................................1305
compute smd/tlsph/num/neighs command..................................................................................1306
compute smd/tlsph/shape command............................................................................................1307
compute smd/tlsph/strain command............................................................................................1308
compute smd/tlsph/strain/rate command.....................................................................................1309
compute smd/tlsph/stress command............................................................................................1310
compute smd/triangle/mesh/vertices...........................................................................................1311
compute smd/ulsph/num/neighs command.................................................................................1312
compute smd/ulsph/strain command...........................................................................................1313
compute smd/ulsph/strain/rate command....................................................................................1314
compute smd/ulsph/stress command...........................................................................................1315
LAMMPS Users Manual
xii
Table of Contents
PyLammps Tutorial
compute smd/vol command.........................................................................................................1316
compute sna/atom command.......................................................................................................1317
compute snad/atom command.....................................................................................................1317
compute snav/atom command.....................................................................................................1317
compute stress/atom command...................................................................................................1322
compute force/tally command.....................................................................................................1325
compute heat/flux/tally command...............................................................................................1325
compute pe/tally command.........................................................................................................1325
compute pe/mol/tally command..................................................................................................1325
compute stress/tally command....................................................................................................1325
compute temp command.............................................................................................................1327
compute temp/kk command........................................................................................................1327
compute temp/asphere command................................................................................................1329
compute temp/body command....................................................................................................1332
compute temp/chunk command...................................................................................................1334
compute temp/com command.....................................................................................................1338
compute temp/cs command.........................................................................................................1340
compute temp/deform command.................................................................................................1342
compute temp/deform/eff command...........................................................................................1344
compute temp/drude command...................................................................................................1345
compute temp/eff command........................................................................................................1347
compute temp/partial command..................................................................................................1349
compute temp/profile command..................................................................................................1351
compute temp/ramp command....................................................................................................1354
compute temp/region command..................................................................................................1356
compute temp/region/eff command.............................................................................................1358
compute temp/rotate command...................................................................................................1359
compute temp/sphere command..................................................................................................1361
compute ti command...................................................................................................................1363
compute torque/chunk command................................................................................................1365
compute vacf command...............................................................................................................1367
compute vcm/chunk command....................................................................................................1369
compute voronoi/atom command................................................................................................1371
compute xrd command................................................................................................................1375
pair_style adp command..............................................................................................................1380
pair_style adp/omp command.....................................................................................................1380
pair_style agni command.............................................................................................................1383
pair_style agni/omp command....................................................................................................1383
pair_style airebo command.........................................................................................................1386
pair_style airebo/omp command.................................................................................................1386
pair_style airebo/morse command...............................................................................................1386
pair_style airebo/morse/omp command......................................................................................1386
pair_style rebo command............................................................................................................1386
pair_style rebo/omp command....................................................................................................1386
pair_style awpmd/cut command..................................................................................................1390
pair_style beck command............................................................................................................1392
pair_style beck/gpu command.....................................................................................................1392
LAMMPS Users Manual
xiii
Table of Contents
PyLammps Tutorial
pair_style beck/omp command....................................................................................................1392
pair_style body command...........................................................................................................1394
pair_style bop command.............................................................................................................1396
pair_style born command............................................................................................................1403
pair_style born/omp command....................................................................................................1403
pair_style born/gpu command.....................................................................................................1403
pair_style born/coul/long command............................................................................................1403
pair_style born/coul/long/cs command.......................................................................................1403
pair_style born/coul/long/gpu command.....................................................................................1403
pair_style born/coul/long/omp command....................................................................................1403
pair_style born/coul/msm command...........................................................................................1403
pair_style born/coul/msm/omp command...................................................................................1403
pair_style born/coul/wolf command............................................................................................1403
pair_style born/coul/wolf/gpu command.....................................................................................1403
pair_style born/coul/wolf/omp command...................................................................................1403
pair_style born/coul/dsf command..............................................................................................1403
pair_style born/coul/dsf/cs command..........................................................................................1403
pair_style brownian command....................................................................................................1407
pair_style brownian/omp command............................................................................................1407
pair_style brownian/poly command............................................................................................1407
pair_style brownian/poly/omp command....................................................................................1407
pair_style buck command............................................................................................................1410
pair_style buck/gpu command.....................................................................................................1410
pair_style buck/intel command...................................................................................................1410
pair_style buck/kk command.......................................................................................................1410
pair_style buck/omp command...................................................................................................1410
pair_style buck/coul/cut command..............................................................................................1410
pair_style buck/coul/cut/gpu command.......................................................................................1410
pair_style buck/coul/cut/intel command.....................................................................................1410
pair_style buck/coul/cut/kk command.........................................................................................1410
pair_style buck/coul/cut/omp command.....................................................................................1410
pair_style buck/coul/long command...........................................................................................1410
pair_style buck/coul/long/cs command.......................................................................................1410
pair_style buck/coul/long/gpu command....................................................................................1410
pair_style buck/coul/long/intel command...................................................................................1410
pair_style buck/coul/long/kk command......................................................................................1410
pair_style buck/coul/long/omp command...................................................................................1410
pair_style buck/coul/msm command...........................................................................................1410
pair_style buck/coul/msm/omp command...................................................................................1410
pair_style buck/long/coul/long command...................................................................................1414
pair_style buck/long/coul/long/omp command...........................................................................1414
pair_style lj/charmm/coul/charmm command.............................................................................1417
pair_style lj/charmm/coul/charmm/omp command.....................................................................1417
pair_style lj/charmm/coul/charmm/implicit command...............................................................1417
pair_style lj/charmm/coul/charmm/implicit/omp command.......................................................1417
pair_style lj/charmm/coul/long command...................................................................................1417
pair_style lj/charmm/coul/long/gpu command............................................................................1417
LAMMPS Users Manual
xiv
Table of Contents
PyLammps Tutorial
pair_style lj/charmm/coul/long/intel command...........................................................................1417
pair_style lj/charmm/coul/long/opt command.............................................................................1417
pair_style lj/charmm/coul/long/omp command...........................................................................1417
pair_style lj/charmm/coul/msm command..................................................................................1417
pair_style lj/charmm/coul/msm/omp command..........................................................................1417
pair_style lj/charmmfsw/coul/charmmfsh command..................................................................1417
pair_style lj/charmmfsw/coul/long command.............................................................................1417
pair_style lj/class2 command......................................................................................................1422
pair_style lj/class2/gpu command...............................................................................................1422
pair_style lj/class2/kk command.................................................................................................1422
pair_style lj/class2/omp command..............................................................................................1422
pair_style lj/class2/coul/cut command........................................................................................1422
pair_style lj/class2/coul/cut/kk command...................................................................................1422
pair_style lj/class2/coul/cut/omp command................................................................................1422
pair_style lj/class2/coul/long command......................................................................................1422
pair_style lj/class2/coul/long/gpu command...............................................................................1422
pair_style lj/class2/coul/long/kk command.................................................................................1422
pair_style lj/class2/coul/long/omp command..............................................................................1422
pair_style colloid command........................................................................................................1425
pair_style colloid/gpu command.................................................................................................1425
pair_style colloid/omp command................................................................................................1425
pair_style comb command...........................................................................................................1430
pair_style comb/omp command..................................................................................................1430
pair_style comb3 command.........................................................................................................1430
pair_style coul/cut command.......................................................................................................1434
pair_style coul/cut/gpu command...............................................................................................1434
pair_style coul/cut/kk command.................................................................................................1434
pair_style coul/cut/omp command..............................................................................................1434
pair_style coul/debye command..................................................................................................1434
pair_style coul/debye/gpu command...........................................................................................1434
pair_style coul/debye/kk command.............................................................................................1434
pair_style coul/debye/omp command..........................................................................................1434
pair_style coul/dsf command.......................................................................................................1434
pair_style coul/dsf/gpu command...............................................................................................1434
pair_style coul/dsf/kk command.................................................................................................1434
pair_style coul/dsf/omp command..............................................................................................1434
pair_style coul/long command....................................................................................................1434
pair_style coul/long/cs command................................................................................................1434
pair_style coul/long/omp command............................................................................................1434
pair_style coul/long/gpu command.............................................................................................1434
pair_style coul/long/kk command...............................................................................................1434
pair_style coul/msm command....................................................................................................1434
pair_style coul/msm/omp command...........................................................................................1434
pair_style coul/streitz command..................................................................................................1434
pair_style coul/wolf command....................................................................................................1434
pair_style coul/wolf/kk command...............................................................................................1435
pair_style coul/wolf/omp command............................................................................................1435
LAMMPS Users Manual
xv
Table of Contents
PyLammps Tutorial
pair_style tip4p/cut command.....................................................................................................1435
pair_style tip4p/long command...................................................................................................1435
pair_style tip4p/cut/omp command.............................................................................................1435
pair_style tip4p/long/omp command...........................................................................................1435
pair_style coul/diel command.....................................................................................................1441
pair_style coul/diel/omp command.............................................................................................1441
pair_style born/coul/long/cs command.......................................................................................1443
pair_style buck/coul/long/cs command.......................................................................................1443
pair_style born/coul/dsf/cs command..........................................................................................1443
pair_style lj/cut/dipole/cut command..........................................................................................1445
pair_style lj/cut/dipole/cut/gpu command...................................................................................1445
pair_style lj/cut/dipole/cut/omp command..................................................................................1445
pair_style lj/sf/dipole/sf command..............................................................................................1445
pair_style lj/sf/dipole/sf/gpu command.......................................................................................1445
pair_style lj/sf/dipole/sf/omp command......................................................................................1445
pair_style lj/cut/dipole/long command........................................................................................1445
pair_style lj/long/dipole/long command......................................................................................1445
pair_style dpd command.............................................................................................................1453
pair_style dpd/gpu command......................................................................................................1453
pair_style dpd/omp command.....................................................................................................1453
pair_style dpd/tstat command......................................................................................................1453
pair_style dpd/tstat/gpu command...............................................................................................1453
pair_style dpd/tstat/omp command.............................................................................................1453
pair_style dpd/fdt command........................................................................................................1457
pair_style dpd/fdt/energy command............................................................................................1457
pair_style dsmc command...........................................................................................................1460
pair_style eam command.............................................................................................................1463
pair_style eam/gpu command......................................................................................................1463
pair_style eam/intel command.....................................................................................................1463
pair_style eam/kk command........................................................................................................1463
pair_style eam/omp command.....................................................................................................1463
pair_style eam/opt command.......................................................................................................1463
pair_style eam/alloy command....................................................................................................1463
pair_style eam/alloy/gpu command.............................................................................................1463
pair_style eam/alloy/kk command...............................................................................................1463
pair_style eam/alloy/omp command...........................................................................................1463
pair_style eam/alloy/opt command.............................................................................................1463
pair_style eam/cd command........................................................................................................1463
pair_style eam/cd/omp command................................................................................................1463
pair_style eam/fs command.........................................................................................................1463
pair_style eam/fs/gpu command..................................................................................................1463
pair_style eam/fs/kk command....................................................................................................1463
pair_style eam/fs/omp command.................................................................................................1463
pair_style eam/fs/opt command...................................................................................................1463
pair_style edip command.............................................................................................................1471
pair_style eff/cut command.........................................................................................................1474
pair_style eim command.............................................................................................................1480
LAMMPS Users Manual
xvi
Table of Contents
PyLammps Tutorial
pair_style eim/omp command.....................................................................................................1480
pair_style exp6/rx command.......................................................................................................1484
pair_style gauss command...........................................................................................................1487
pair_style gauss/gpu command...................................................................................................1487
pair_style gauss/omp command..................................................................................................1487
pair_style gauss/cut command.....................................................................................................1487
pair_style gauss/cut/omp command............................................................................................1487
pair_style gayberne command.....................................................................................................1490
pair_style gayberne/gpu command..............................................................................................1490
pair_style gayberne/intel command.............................................................................................1490
pair_style gayberne/omp command.............................................................................................1490
pair_style gran/hooke command.................................................................................................1494
pair_style gran/omp command....................................................................................................1494
pair_style gran/hooke/history command.....................................................................................1494
pair_style gran/hooke/history/omp command.............................................................................1494
pair_style gran/hertz/history command.......................................................................................1494
pair_style gran/hertz/history/omp command...............................................................................1494
pair_style lj/gromacs command...................................................................................................1499
pair_style lj/gromacs/gpu command...........................................................................................1499
pair_style lj/gromacs/omp command..........................................................................................1499
pair_style lj/gromacs/coul/gromacs command............................................................................1499
pair_style lj/gromacs/coul/gromacs/omp command....................................................................1499
pair_style hbond/dreiding/lj command........................................................................................1502
pair_style hbond/dreiding/lj/omp command...............................................................................1502
pair_style hbond/dreiding/morse command................................................................................1502
pair_style hbond/dreiding/morse/omp command........................................................................1502
pair_style hybrid command.........................................................................................................1507
pair_style hybrid/omp command.................................................................................................1507
pair_style hybrid/overlay command............................................................................................1507
pair_style hybrid/overlay/omp command....................................................................................1507
pair_style kim command.............................................................................................................1513
pair_style kolmogorov/crespi/z command..................................................................................1515
pair_style lcbop command...........................................................................................................1517
pair_style line/lj command..........................................................................................................1519
pair_style list command...............................................................................................................1522
pair_style lj/cut command...........................................................................................................1525
pair_style lj/cut/gpu command....................................................................................................1525
pair_style lj/cut/intel command...................................................................................................1525
pair_style lj/cut/kk command......................................................................................................1525
pair_style lj/cut/opt command.....................................................................................................1525
pair_style lj/cut/omp command...................................................................................................1525
pair_style lj/cut/coul/cut command.............................................................................................1525
pair_style lj/cut/coul/cut/gpu command......................................................................................1525
pair_style lj/cut/coul/cut/omp command.....................................................................................1525
pair_style lj/cut/coul/debye command.........................................................................................1525
pair_style lj/cut/coul/debye/gpu command.................................................................................1525
pair_style lj/cut/coul/debye/kk command...................................................................................1525
LAMMPS Users Manual
xvii
Table of Contents
PyLammps Tutorial
pair_style lj/cut/coul/debye/omp command................................................................................1525
pair_style lj/cut/coul/dsf command.............................................................................................1525
pair_style lj/cut/coul/dsf/gpu command......................................................................................1525
pair_style lj/cut/coul/dsf/kk command........................................................................................1525
pair_style lj/cut/coul/dsf/omp command.....................................................................................1525
pair_style lj/cut/coul/long command...........................................................................................1525
pair_style lj/cut/coul/long/cs command.......................................................................................1525
pair_style lj/cut/coul/long/gpu command....................................................................................1525
pair_style lj/cut/coul/long/intel command...................................................................................1525
pair_style lj/cut/coul/long/opt command.....................................................................................1526
pair_style lj/cut/coul/long/omp command...................................................................................1526
pair_style lj/cut/coul/msm command..........................................................................................1526
pair_style lj/cut/coul/msm/gpu command...................................................................................1526
pair_style lj/cut/coul/msm/omp command..................................................................................1526
pair_style lj/cut/tip4p/cut command............................................................................................1526
pair_style lj/cut/tip4p/cut/omp command....................................................................................1526
pair_style lj/cut/tip4p/long command..........................................................................................1526
pair_style lj/cut/tip4p/long/omp command.................................................................................1526
pair_style lj/cut/tip4p/long/opt command...................................................................................1526
pair_style lj96/cut command.......................................................................................................1532
pair_style lj96/cut/gpu command................................................................................................1532
pair_style lj96/cut/omp command...............................................................................................1532
pair_style lj/cubic command.......................................................................................................1534
pair_style lj/cubic/gpu command................................................................................................1534
pair_style lj/cubic/omp command...............................................................................................1534
pair_style lj/expand command.....................................................................................................1536
pair_style lj/expand/gpu command.............................................................................................1536
pair_style lj/expand/omp command............................................................................................1536
pair_style lj/long/coul/long command.........................................................................................1538
pair_style lj/long/coul/long/omp command.................................................................................1538
pair_style lj/long/coul/long/opt command...................................................................................1538
pair_style lj/long/tip4p/long command.......................................................................................1538
pair_style lj/sf command.............................................................................................................1542
pair_style lj/sf/omp command.....................................................................................................1542
pair_style lj/smooth command....................................................................................................1544
pair_style lj/smooth/omp command............................................................................................1544
pair_style lj/smooth/linear command..........................................................................................1546
pair_style lj/smooth/linear/omp command..................................................................................1546
pair_style lj/cut/soft command....................................................................................................1548
pair_style lj/cut/soft/omp command............................................................................................1548
pair_style lj/cut/coul/cut/soft command......................................................................................1548
pair_style lj/cut/coul/cut/soft/omp command..............................................................................1548
pair_style lj/cut/coul/long/soft command....................................................................................1548
pair_style lj/cut/coul/long/soft/omp command............................................................................1548
pair_style lj/cut/tip4p/long/soft command...................................................................................1548
pair_style lj/cut/tip4p/long/soft/omp command..........................................................................1548
pair_style lj/charmm/coul/long/soft command............................................................................1548
LAMMPS Users Manual
xviii
Table of Contents
PyLammps Tutorial
pair_style lj/charmm/coul/long/soft/omp command...................................................................1548
pair_style coul/cut/soft command...............................................................................................1548
pair_style coul/cut/soft/omp command.......................................................................................1548
pair_style coul/long/soft command.............................................................................................1548
pair_style coul/long/soft/omp command.....................................................................................1548
pair_style tip4p/long/soft command............................................................................................1548
pair_style tip4p/long/soft/omp command....................................................................................1548
pair_style lubricate command.....................................................................................................1553
pair_style lubricate/omp command.............................................................................................1553
pair_style lubricate/poly command.............................................................................................1553
pair_style lubricate/poly/omp command.....................................................................................1553
pair_style lubricateU command...................................................................................................1557
pair_style lubricateU/poly command..........................................................................................1557
pair_style lj/mdf command..........................................................................................................1561
pair_style buck/mdf command....................................................................................................1561
pair_style lennard/mdf command................................................................................................1561
pair_style meam command..........................................................................................................1564
pair_style meam/spline................................................................................................................1570
pair_style meam/spline/omp........................................................................................................1570
pair_style meam/sw/spline..........................................................................................................1573
pair_style meam/sw/spline/omp..................................................................................................1573
pair_style mgpt command...........................................................................................................1576
pair_style mie/cut command.......................................................................................................1580
pair_style mie/cut/gpu command................................................................................................1580
pair_style momb command.........................................................................................................1582
pair_style morse command..........................................................................................................1584
pair_style morse/gpu command...................................................................................................1584
pair_style morse/omp command.................................................................................................1584
pair_style morse/opt command...................................................................................................1584
pair_style morse/smooth/linear command...................................................................................1584
pair_style morse/smooth/linear/omp command..........................................................................1584
pair_style morse/soft command...................................................................................................1584
pair_style morse/kk command.....................................................................................................1584
pair_style multi/lucy command...................................................................................................1587
pair_style multi/lucy/rx command...............................................................................................1591
pair_style nb3b/harmonic command...........................................................................................1595
pair_style nb3b/harmonic/omp command...................................................................................1595
pair_style nm/cut command........................................................................................................1597
pair_style nm/cut/coul/cut command..........................................................................................1597
pair_style nm/cut/coul/long command........................................................................................1597
pair_style nm/cut/omp command................................................................................................1597
pair_style nm/cut/coul/cut/omp command..................................................................................1597
pair_style nm/cut/coul/long/omp command................................................................................1597
pair_style none command............................................................................................................1600
pair_style oxdna/excv command.................................................................................................1601
pair_style oxdna/stk command....................................................................................................1601
pair_style oxdna/hbond command...............................................................................................1601
LAMMPS Users Manual
xix
Table of Contents
PyLammps Tutorial
pair_style oxdna/xstk command..................................................................................................1601
pair_style oxdna/coaxstk command............................................................................................1601
pair_style oxdna2/excv command...............................................................................................1603
pair_style oxdna2/stk command..................................................................................................1603
pair_style oxdna2/hbond command.............................................................................................1603
pair_style oxdna2/xstk command................................................................................................1603
pair_style oxdna2/coaxstk command..........................................................................................1603
pair_style oxdna2/dh command...................................................................................................1603
pair_style peri/pmb command.....................................................................................................1605
pair_style peri/pmb/omp command.............................................................................................1605
pair_style peri/lps command.......................................................................................................1605
pair_style peri/lps/omp command...............................................................................................1605
pair_style peri/ves command.......................................................................................................1605
pair_style peri/eps command.......................................................................................................1605
pair_style polymorphic command...............................................................................................1609
pair_style quip command............................................................................................................1615
pair_style reax command.............................................................................................................1617
pair_style reax/c command..........................................................................................................1621
pair_style reax/c/kk command.....................................................................................................1621
pair_style resquared command....................................................................................................1627
pair_style resquared/gpu command.............................................................................................1627
pair_style resquared/omp command............................................................................................1627
pair_style lj/sdk command...........................................................................................................1631
pair_style lj/sdk/gpu command...................................................................................................1631
pair_style lj/sdk/kk command.....................................................................................................1631
pair_style lj/sdk/omp command..................................................................................................1631
pair_style lj/sdk/coul/long command..........................................................................................1631
pair_style lj/sdk/coul/long/gpu command...................................................................................1631
pair_style lj/sdk/coul/long/omp command..................................................................................1631
pair_style smd/hertz command....................................................................................................1634
pair_style smd/tlsph command....................................................................................................1635
pair_style smd/tri_surface command...........................................................................................1636
pair_style smd/ulsph command...................................................................................................1637
pair_style smtbq command..........................................................................................................1639
pair_style snap command............................................................................................................1645
pair_style soft command.............................................................................................................1648
pair_style soft/gpu command......................................................................................................1648
pair_style soft/omp command.....................................................................................................1648
pair_style sph/heatconduction command....................................................................................1651
pair_style sph/idealgas command................................................................................................1652
pair_style sph/lj command...........................................................................................................1654
pair_style sph/rhosum command.................................................................................................1656
pair_style sph/taitwater command...............................................................................................1657
pair_style sph/taitwater/morris command...................................................................................1659
pair_style srp command...............................................................................................................1661
pair_style sw command...............................................................................................................1664
pair_style sw/gpu command........................................................................................................1664
LAMMPS Users Manual
xx
Table of Contents
PyLammps Tutorial
pair_style sw/intel command.......................................................................................................1664
pair_style sw/kk command..........................................................................................................1664
pair_style sw/omp command.......................................................................................................1664
pair_style table command............................................................................................................1668
pair_style table/gpu command.....................................................................................................1668
pair_style table/kk command.......................................................................................................1668
pair_style table/omp command...................................................................................................1668
pair_style table/rx command.......................................................................................................1672
pair_style tersoff command.........................................................................................................1676
pair_style tersoff/table command................................................................................................1676
pair_style tersoff/gpu...................................................................................................................1676
pair_style tersoff/intel..................................................................................................................1676
pair_style tersoff/kk.....................................................................................................................1676
pair_style tersoff/omp..................................................................................................................1676
pair_style tersoff/table/omp command........................................................................................1676
pair_style tersoff/mod command.................................................................................................1681
pair_style tersoff/mod/c command..............................................................................................1681
pair_style tersoff/mod/gpu command..........................................................................................1681
pair_style tersoff/mod/kk command............................................................................................1681
pair_style tersoff/mod/omp command.........................................................................................1681
pair_style tersoff/mod/c/omp command......................................................................................1681
pair_style tersoff/zbl command...................................................................................................1685
pair_style tersoff/zbl/gpu command............................................................................................1685
pair_style tersoff/zbl/kk command..............................................................................................1685
pair_style tersoff/zbl/omp command...........................................................................................1685
pair_style thole command...........................................................................................................1691
pair_style lj/cut/thole/long command..........................................................................................1691
pair_style lj/cut/thole/long/omp command..................................................................................1691
pair_style tri/lj command.............................................................................................................1694
pair_style vashishta command.....................................................................................................1696
pair_style vashishta/omp command............................................................................................1696
pair_style vashishta/kk command...............................................................................................1696
pair_style vashishta/table command............................................................................................1696
pair_style vashishta/table/omp command...................................................................................1696
pair_style yukawa command.......................................................................................................1701
pair_style yukawa/gpu command................................................................................................1701
pair_style yukawa/omp command...............................................................................................1701
pair_style yukawa/colloid command...........................................................................................1703
pair_style yukawa/colloid/gpu command....................................................................................1703
pair_style yukawa/colloid/omp command...................................................................................1703
pair_style zbl command...............................................................................................................1706
pair_style zbl/gpu command.......................................................................................................1706
pair_style zbl/omp command......................................................................................................1706
pair_style zero command.............................................................................................................1709
bond_style class2 command........................................................................................................1711
bond_style class2/omp command................................................................................................1711
bond_style class2/kk command...................................................................................................1711
LAMMPS Users Manual
xxi
Table of Contents
PyLammps Tutorial
bond_style fene command...........................................................................................................1713
bond_style fene/intel command...................................................................................................1713
bond_style fene/kk command......................................................................................................1713
bond_style fene/omp command...................................................................................................1713
bond_style fene/expand command..............................................................................................1715
bond_style fene/expand/omp command......................................................................................1715
bond_style oxdna/fene command................................................................................................1717
bond_style oxdna2/fene command..............................................................................................1717
bond_style harmonic command...................................................................................................1719
bond_style harmonic/intel command..........................................................................................1719
bond_style harmonic/kk command.............................................................................................1719
bond_style harmonic/omp command..........................................................................................1719
bond_style harmonic/shift command..........................................................................................1721
bond_style harmonic/shift/omp command..................................................................................1721
bond_style harmonic/shift/cut command....................................................................................1723
bond_style harmonic/shift/cut/omp command............................................................................1723
bond_style hybrid command.......................................................................................................1725
bond_style morse command........................................................................................................1727
bond_style morse/omp command................................................................................................1727
bond_style none command..........................................................................................................1729
bond_style nonlinear command...................................................................................................1730
bond_style nonlinear/omp command..........................................................................................1730
bond_style quartic command.......................................................................................................1732
bond_style quartic/omp command..............................................................................................1732
bond_style table command..........................................................................................................1734
bond_style table/omp command..................................................................................................1734
bond_style zero command...........................................................................................................1737
angle_style charmm command....................................................................................................1738
angle_style charmm/intel command............................................................................................1738
angle_style charmm/kk command...............................................................................................1738
angle_style charmm/omp command............................................................................................1738
angle_style class2 command.......................................................................................................1740
angle_style class2/omp command...............................................................................................1740
angle_style class2/kk command..................................................................................................1740
angle_style cosine command.......................................................................................................1743
angle_style cosine/omp command...............................................................................................1743
angle_style cosine/delta command..............................................................................................1745
angle_style cosine/delta/omp command......................................................................................1745
angle_style cosine/periodic command.........................................................................................1747
angle_style cosine/periodic/omp command................................................................................1747
angle_style cosine/shift command...............................................................................................1749
angle_style cosine/shift/omp command......................................................................................1749
angle_style cosine/shift/exp command........................................................................................1751
angle_style cosine/shift/exp/omp command...............................................................................1751
angle_style cosine/squared command.........................................................................................1753
angle_style cosine/squared/omp command.................................................................................1753
angle_style dipole command.......................................................................................................1755
LAMMPS Users Manual
xxii
Table of Contents
PyLammps Tutorial
angle_style dipole/omp command...............................................................................................1755
angle_style fourier command......................................................................................................1758
angle_style fourier/omp command..............................................................................................1758
angle_style fourier/simple command..........................................................................................1760
angle_style fourier/simple/omp command..................................................................................1760
angle_style harmonic command..................................................................................................1762
angle_style harmonic/intel command..........................................................................................1762
angle_style harmonic/kk command.............................................................................................1762
angle_style harmonic/omp command..........................................................................................1762
angle_style hybrid command.......................................................................................................1764
angle_style none command.........................................................................................................1766
angle_style quartic command......................................................................................................1767
angle_style quartic/omp command..............................................................................................1767
angle_style sdk command...........................................................................................................1769
angle_style table command.........................................................................................................1770
angle_style table/omp command.................................................................................................1770
angle_style zero command..........................................................................................................1773
dihedral_style charmm command...............................................................................................1774
dihedral_style charmm/intel command.......................................................................................1774
dihedral_style charmm/kk command..........................................................................................1774
dihedral_style charmm/omp command.......................................................................................1774
dihedral_style charmmfsh command...........................................................................................1774
dihedral_style class2 command...................................................................................................1777
dihedral_style class2/omp command...........................................................................................1777
dihedral_style class2/kk command..............................................................................................1777
dihedral_style cosine/shift/exp command...................................................................................1781
dihedral_style cosine/shift/exp/omp command...........................................................................1781
dihedral_style fourier command..................................................................................................1783
dihedral_style fourier/omp command.........................................................................................1783
dihedral_style harmonic command.............................................................................................1785
dihedral_style harmonic/intel command.....................................................................................1785
dihedral_style harmonic/omp command.....................................................................................1785
dihedral_style helix command.....................................................................................................1787
dihedral_style helix/omp command............................................................................................1787
dihedral_style hybrid command..................................................................................................1789
dihedral_style multi/harmonic command....................................................................................1791
dihedral_style multi/harmonic/omp command............................................................................1791
dihedral_style nharmonic command...........................................................................................1793
dihedral_style nharmonic/omp command...................................................................................1793
dihedral_style none command.....................................................................................................1795
dihedral_style opls command......................................................................................................1796
dihedral_style opls/intel command..............................................................................................1796
dihedral_style opls/kk command.................................................................................................1796
dihedral_style opls/omp command..............................................................................................1796
dihedral_style quadratic command..............................................................................................1798
dihedral_style quadratic/omp command.....................................................................................1798
dihedral_style spherical command..............................................................................................1800
LAMMPS Users Manual
xxiii
Table of Contents
PyLammps Tutorial
dihedral_style table command.....................................................................................................1802
dihedral_style table/omp command.............................................................................................1802
dihedral_style zero command......................................................................................................1805
improper_style class2 command.................................................................................................1806
improper_style class2/omp command.........................................................................................1806
improper_style class2/kk command............................................................................................1806
improper_style cossq command..................................................................................................1809
improper_style cossq/omp command..........................................................................................1809
improper_style cvff command.....................................................................................................1811
improper_style cvff/intel command............................................................................................1811
improper_style cvff/omp command............................................................................................1811
improper_style distance command..............................................................................................1813
improper_style fourier command................................................................................................1815
improper_style fourier/omp command........................................................................................1815
improper_style harmonic command............................................................................................1817
improper_style harmonic/intel command....................................................................................1817
improper_style harmonic/kk command.......................................................................................1817
improper_style harmonic/omp command....................................................................................1817
improper_style hybrid command.................................................................................................1819
improper_style none command...................................................................................................1820
improper_style ring command.....................................................................................................1821
improper_style ring/omp command............................................................................................1821
improper_style umbrella command.............................................................................................1823
improper_style umbrella/omp command.....................................................................................1823
improper_style zero command....................................................................................................1825
fix_modify AtC add_molecule.....................................................................................................................1827
syntax................................................................................................................................................1827
examples...........................................................................................................................................1827
description.........................................................................................................................................1827
restrictions.........................................................................................................................................1827
related................................................................................................................................................1827
default...............................................................................................................................................1827
fix_modify AtC add_species.........................................................................................................................1828
syntax................................................................................................................................................1828
examples...........................................................................................................................................1828
description.........................................................................................................................................1828
restrictions.........................................................................................................................................1828
related................................................................................................................................................1828
default...............................................................................................................................................1828
fix_modify AtC atom_element_map...........................................................................................................1829
syntax................................................................................................................................................1829
examples...........................................................................................................................................1829
description.........................................................................................................................................1829
restrictions.........................................................................................................................................1829
LAMMPS Users Manual
xxiv
Table of Contents
fix_modify AtC atom_element_map
related................................................................................................................................................1829
default...............................................................................................................................................1829
fix_modify AtC atom_weight.......................................................................................................................1830
syntax................................................................................................................................................1830
examples...........................................................................................................................................1830
description.........................................................................................................................................1830
restrictions.........................................................................................................................................1830
related................................................................................................................................................1830
default...............................................................................................................................................1830
fix_modify AtC atomic_charge....................................................................................................................1831
syntax................................................................................................................................................1831
examples...........................................................................................................................................1831
description.........................................................................................................................................1831
restrictions.........................................................................................................................................1831
related................................................................................................................................................1831
default...............................................................................................................................................1831
fix_modify AtC boundary............................................................................................................................1832
syntax................................................................................................................................................1832
examples...........................................................................................................................................1832
description.........................................................................................................................................1832
restrictions.........................................................................................................................................1832
default...............................................................................................................................................1832
fix_modify AtC boundary_dynamics..........................................................................................................1833
syntax................................................................................................................................................1833
description.........................................................................................................................................1833
restrictions.........................................................................................................................................1833
related................................................................................................................................................1833
default...............................................................................................................................................1833
fix_modify AtC boundary_faceset...............................................................................................................1834
syntax................................................................................................................................................1834
examples...........................................................................................................................................1834
description.........................................................................................................................................1834
restrictions.........................................................................................................................................1834
related................................................................................................................................................1834
default...............................................................................................................................................1834
fix_modify AtC output boundary_integral.................................................................................................1835
syntax................................................................................................................................................1835
examples...........................................................................................................................................1835
description.........................................................................................................................................1835
restrictions.........................................................................................................................................1835
related................................................................................................................................................1835
LAMMPS Users Manual
xxv
Table of Contents
fix_modify AtC output boundary_integral
default...............................................................................................................................................1835
fix_modify AtC consistent_fe_initialization...............................................................................................1836
syntax................................................................................................................................................1836
examples...........................................................................................................................................1836
description.........................................................................................................................................1836
restrictions.........................................................................................................................................1836
related................................................................................................................................................1836
default...............................................................................................................................................1836
fix_modify AtC output contour_integral....................................................................................................1837
syntax................................................................................................................................................1837
examples...........................................................................................................................................1837
description.........................................................................................................................................1837
restrictions.........................................................................................................................................1837
related................................................................................................................................................1837
default...............................................................................................................................................1837
fix_modify AtC control.................................................................................................................................1838
syntax................................................................................................................................................1838
examples...........................................................................................................................................1838
description.........................................................................................................................................1838
restrictions.........................................................................................................................................1838
related................................................................................................................................................1838
default...............................................................................................................................................1838
fix_modify AtC control momentum............................................................................................................1840
syntax................................................................................................................................................1840
examples...........................................................................................................................................1840
description.........................................................................................................................................1840
restrictions.........................................................................................................................................1840
related................................................................................................................................................1840
default...............................................................................................................................................1840
fix_modify AtC control thermal..................................................................................................................1841
syntax................................................................................................................................................1841
examples...........................................................................................................................................1841
description.........................................................................................................................................1841
restrictions.........................................................................................................................................1841
related................................................................................................................................................1841
default...............................................................................................................................................1842
fix_modify AtC control thermal correction_max_iterations....................................................................1843
syntax................................................................................................................................................1843
examples...........................................................................................................................................1843
description.........................................................................................................................................1843
restrictions.........................................................................................................................................1843
LAMMPS Users Manual
xxvi
Table of Contents
fix_modify AtC control thermal correction_max_iterations
related................................................................................................................................................1843
default...............................................................................................................................................1843
fix_modify AtC decomposition....................................................................................................................1844
syntax................................................................................................................................................1844
examples...........................................................................................................................................1844
description.........................................................................................................................................1844
restrictions.........................................................................................................................................1844
related................................................................................................................................................1844
default...............................................................................................................................................1844
fix_modify AtC extrinsic electron_integration...........................................................................................1845
syntax................................................................................................................................................1845
examples...........................................................................................................................................1845
description.........................................................................................................................................1845
restrictions.........................................................................................................................................1845
default...............................................................................................................................................1845
fix_modify AtC equilibrium_start...............................................................................................................1846
syntax................................................................................................................................................1846
examples...........................................................................................................................................1846
description.........................................................................................................................................1846
restrictions.........................................................................................................................................1846
related................................................................................................................................................1846
default...............................................................................................................................................1846
fix_modify AtC extrinsic exchange..............................................................................................................1847
syntax................................................................................................................................................1847
examples...........................................................................................................................................1847
description.........................................................................................................................................1847
restrictions.........................................................................................................................................1847
related................................................................................................................................................1847
default...............................................................................................................................................1847
fix_modify AtC fe_md_boundary................................................................................................................1848
syntax................................................................................................................................................1848
examples...........................................................................................................................................1848
description.........................................................................................................................................1848
restrictions.........................................................................................................................................1848
related................................................................................................................................................1848
default...............................................................................................................................................1848
fix_modify AtC fem create mesh.................................................................................................................1849
syntax................................................................................................................................................1849
examples...........................................................................................................................................1849
description.........................................................................................................................................1849
restrictions.........................................................................................................................................1849
LAMMPS Users Manual
xxvii
Table of Contents
fix_modify AtC fem create mesh
related................................................................................................................................................1849
default...............................................................................................................................................1849
fix_modify AtC filter scale...........................................................................................................................1850
syntax................................................................................................................................................1850
examples...........................................................................................................................................1850
description.........................................................................................................................................1850
restrictions.........................................................................................................................................1850
related................................................................................................................................................1850
default...............................................................................................................................................1850
fix_modify AtC filter type............................................................................................................................1851
syntax................................................................................................................................................1851
examples...........................................................................................................................................1851
description.........................................................................................................................................1851
restrictions.........................................................................................................................................1851
related................................................................................................................................................1851
default...............................................................................................................................................1851
fix atc command............................................................................................................................................1852
syntax................................................................................................................................................1852
examples...........................................................................................................................................1852
description.........................................................................................................................................1852
restrictions.........................................................................................................................................1854
related................................................................................................................................................1854
default...............................................................................................................................................1856
fix_modify AtC fix_flux................................................................................................................................1857
syntax................................................................................................................................................1857
examples...........................................................................................................................................1857
description.........................................................................................................................................1857
restrictions.........................................................................................................................................1857
related................................................................................................................................................1857
default...............................................................................................................................................1857
fix_modify AtC fix.........................................................................................................................................1858
syntax................................................................................................................................................1858
examples...........................................................................................................................................1858
description.........................................................................................................................................1858
restrictions.........................................................................................................................................1858
related................................................................................................................................................1858
default...............................................................................................................................................1858
fix_modify AtC computes.............................................................................................................................1859
syntax................................................................................................................................................1859
examples...........................................................................................................................................1859
description.........................................................................................................................................1859
LAMMPS Users Manual
xxviii
Table of Contents
fix_modify AtC computes
restrictions.........................................................................................................................................1859
related................................................................................................................................................1859
default...............................................................................................................................................1859
fix_modify AtC fields....................................................................................................................................1860
syntax................................................................................................................................................1860
examples...........................................................................................................................................1860
description.........................................................................................................................................1860
restrictions.........................................................................................................................................1861
related................................................................................................................................................1861
default...............................................................................................................................................1861
fix_modify AtC gradients.............................................................................................................................1862
syntax................................................................................................................................................1862
examples...........................................................................................................................................1862
description.........................................................................................................................................1862
restrictions.........................................................................................................................................1862
related................................................................................................................................................1862
default...............................................................................................................................................1862
fix_modify AtC kernel..................................................................................................................................1863
syntax................................................................................................................................................1863
examples...........................................................................................................................................1863
description.........................................................................................................................................1863
restrictions.........................................................................................................................................1863
related................................................................................................................................................1863
default...............................................................................................................................................1863
fix_modify AtC on_the_fly...........................................................................................................................1864
syntax................................................................................................................................................1864
examples...........................................................................................................................................1864
description.........................................................................................................................................1864
restrictions.........................................................................................................................................1864
related................................................................................................................................................1864
default...............................................................................................................................................1864
fix_modify AtC rates.....................................................................................................................................1865
syntax................................................................................................................................................1865
examples...........................................................................................................................................1865
description.........................................................................................................................................1865
restrictions.........................................................................................................................................1865
related................................................................................................................................................1865
default...............................................................................................................................................1865
fix_modify AtC initial...................................................................................................................................1866
syntax................................................................................................................................................1866
examples...........................................................................................................................................1866
LAMMPS Users Manual
xxix
Table of Contents
fix_modify AtC initial
description.........................................................................................................................................1866
restrictions.........................................................................................................................................1866
default...............................................................................................................................................1866
fix_modify AtC internal_atom_integrate...................................................................................................1867
syntax................................................................................................................................................1867
description.........................................................................................................................................1867
default...............................................................................................................................................1867
fix_modify AtC internal_element_set.........................................................................................................1868
syntax................................................................................................................................................1868
examples...........................................................................................................................................1868
description.........................................................................................................................................1868
restrictions.........................................................................................................................................1868
related................................................................................................................................................1868
default...............................................................................................................................................1868
fix_modify AtC internal_quadrature..........................................................................................................1869
syntax................................................................................................................................................1869
examples...........................................................................................................................................1869
description.........................................................................................................................................1869
optional.............................................................................................................................................1869
restrictions.........................................................................................................................................1869
related................................................................................................................................................1869
default...............................................................................................................................................1869
fix_modify AtC kernel..................................................................................................................................1870
syntax................................................................................................................................................1870
examples...........................................................................................................................................1870
description.........................................................................................................................................1870
restrictions.........................................................................................................................................1870
related................................................................................................................................................1870
default...............................................................................................................................................1870
fix_modify AtC control localized_lambda..................................................................................................1871
syntax................................................................................................................................................1871
examples...........................................................................................................................................1871
description.........................................................................................................................................1871
restrictions.........................................................................................................................................1871
related................................................................................................................................................1871
default...............................................................................................................................................1871
fix_modify AtC control lumped_lambda_solve..........................................................................................1872
syntax................................................................................................................................................1872
examples...........................................................................................................................................1872
description.........................................................................................................................................1872
restrictions.........................................................................................................................................1872
LAMMPS Users Manual
xxx
Table of Contents
fix_modify AtC control lumped_lambda_solve
related................................................................................................................................................1872
default...............................................................................................................................................1872
fix_modify AtC control mask_direction.....................................................................................................1873
syntax................................................................................................................................................1873
examples...........................................................................................................................................1873
description.........................................................................................................................................1873
restrictions.........................................................................................................................................1873
related................................................................................................................................................1873
default...............................................................................................................................................1873
fix_modify AtC mass_matrix.......................................................................................................................1874
syntax................................................................................................................................................1874
examples...........................................................................................................................................1874
description.........................................................................................................................................1874
restrictions.........................................................................................................................................1874
related................................................................................................................................................1874
default...............................................................................................................................................1874
fix_modify AtC material...............................................................................................................................1875
syntax................................................................................................................................................1875
examples...........................................................................................................................................1875
description.........................................................................................................................................1875
restrictions.........................................................................................................................................1875
related................................................................................................................................................1875
default...............................................................................................................................................1875
fix_modify AtC mesh add_to_nodeset........................................................................................................1876
syntax................................................................................................................................................1876
examples...........................................................................................................................................1876
description.........................................................................................................................................1876
restrictions.........................................................................................................................................1876
related................................................................................................................................................1876
default...............................................................................................................................................1876
fix_modify AtC mesh create.........................................................................................................................1877
syntax................................................................................................................................................1877
examples...........................................................................................................................................1877
description.........................................................................................................................................1877
restrictions.........................................................................................................................................1877
related................................................................................................................................................1877
default...............................................................................................................................................1877
fix_modify AtC mesh create_elementset.....................................................................................................1878
syntax................................................................................................................................................1878
examples...........................................................................................................................................1878
description.........................................................................................................................................1878
LAMMPS Users Manual
xxxi
Table of Contents
fix_modify AtC mesh create_elementset
restrictions.........................................................................................................................................1878
related................................................................................................................................................1878
default...............................................................................................................................................1878
fix_modify AtC mesh create_faceset box....................................................................................................1879
syntax................................................................................................................................................1879
examples...........................................................................................................................................1879
description.........................................................................................................................................1879
restrictions.........................................................................................................................................1879
related................................................................................................................................................1879
default...............................................................................................................................................1879
fix_modify AtC mesh create_faceset plane.................................................................................................1880
syntax................................................................................................................................................1880
examples...........................................................................................................................................1880
description.........................................................................................................................................1880
restrictions.........................................................................................................................................1880
related................................................................................................................................................1880
default...............................................................................................................................................1880
fix_modify AtC mesh create_nodeset..........................................................................................................1881
syntax................................................................................................................................................1881
examples...........................................................................................................................................1881
description.........................................................................................................................................1881
restrictions.........................................................................................................................................1881
related................................................................................................................................................1881
default...............................................................................................................................................1881
fix_modify AtC mesh delete_elements........................................................................................................1882
syntax................................................................................................................................................1882
examples...........................................................................................................................................1882
description.........................................................................................................................................1882
restrictions.........................................................................................................................................1882
related................................................................................................................................................1882
default...............................................................................................................................................1882
fix_modify AtC mesh nodeset_to_elementset.............................................................................................1883
syntax................................................................................................................................................1883
examples...........................................................................................................................................1883
description.........................................................................................................................................1883
restrictions.........................................................................................................................................1883
related................................................................................................................................................1883
default...............................................................................................................................................1883
fix_modify AtC mesh output........................................................................................................................1884
syntax................................................................................................................................................1884
examples...........................................................................................................................................1884
LAMMPS Users Manual
xxxii
Table of Contents
fix_modify AtC mesh output
description.........................................................................................................................................1884
restrictions.........................................................................................................................................1884
related................................................................................................................................................1884
default...............................................................................................................................................1884
fix_modify AtC mesh quadrature................................................................................................................1885
syntax................................................................................................................................................1885
examples...........................................................................................................................................1885
description.........................................................................................................................................1885
restrictions.........................................................................................................................................1885
related................................................................................................................................................1885
default...............................................................................................................................................1885
fix_modify AtC mesh read...........................................................................................................................1886
syntax................................................................................................................................................1886
examples...........................................................................................................................................1886
description.........................................................................................................................................1886
restrictions.........................................................................................................................................1886
related................................................................................................................................................1886
default...............................................................................................................................................1886
fix_modify AtC mesh write..........................................................................................................................1887
syntax................................................................................................................................................1887
examples...........................................................................................................................................1887
description.........................................................................................................................................1887
restrictions.........................................................................................................................................1887
related................................................................................................................................................1887
default...............................................................................................................................................1887
fix_modify AtC time_integration (momentum).........................................................................................1888
syntax................................................................................................................................................1888
description.........................................................................................................................................1888
examples...........................................................................................................................................1888
description.........................................................................................................................................1888
related................................................................................................................................................1888
default...............................................................................................................................................1888
fix_modify AtC output..................................................................................................................................1889
syntax................................................................................................................................................1889
examples...........................................................................................................................................1889
description.........................................................................................................................................1889
restrictions.........................................................................................................................................1889
related................................................................................................................................................1889
default...............................................................................................................................................1889
LAMMPS Users Manual
xxxiii
Table of Contents
fix_modify AtC output elementset...............................................................................................................1890
syntax................................................................................................................................................1890
examples...........................................................................................................................................1890
description.........................................................................................................................................1890
restrictions.........................................................................................................................................1890
related................................................................................................................................................1890
default...............................................................................................................................................1890
fix_modify AtC output nodeset....................................................................................................................1891
syntax................................................................................................................................................1891
examples...........................................................................................................................................1891
description.........................................................................................................................................1891
restrictions.........................................................................................................................................1891
related................................................................................................................................................1891
default...............................................................................................................................................1891
fix_modify AtC pair_interactions/bond_interactions................................................................................1892
syntax................................................................................................................................................1892
examples...........................................................................................................................................1892
description.........................................................................................................................................1892
restrictions.........................................................................................................................................1892
related................................................................................................................................................1892
default...............................................................................................................................................1892
fix_modify AtC poisson_solver....................................................................................................................1893
syntax................................................................................................................................................1893
examples...........................................................................................................................................1893
description.........................................................................................................................................1893
restrictions.........................................................................................................................................1893
related................................................................................................................................................1893
default...............................................................................................................................................1893
fix_modify AtC read_restart........................................................................................................................1894
syntax................................................................................................................................................1894
examples...........................................................................................................................................1894
description.........................................................................................................................................1894
restrictions.........................................................................................................................................1894
related................................................................................................................................................1894
default...............................................................................................................................................1894
fix_modify AtC remove_molecule...............................................................................................................1895
syntax................................................................................................................................................1895
examples...........................................................................................................................................1895
description.........................................................................................................................................1895
restrictions.........................................................................................................................................1895
related................................................................................................................................................1895
default...............................................................................................................................................1895
LAMMPS Users Manual
xxxiv
Table of Contents
fix_modify AtC remove_source...................................................................................................................1896
syntax................................................................................................................................................1896
examples...........................................................................................................................................1896
description.........................................................................................................................................1896
restrictions.........................................................................................................................................1896
related................................................................................................................................................1896
default...............................................................................................................................................1896
fix_modify AtC remove_species...................................................................................................................1897
syntax................................................................................................................................................1897
examples...........................................................................................................................................1897
description.........................................................................................................................................1897
restrictions.........................................................................................................................................1897
related................................................................................................................................................1897
default...............................................................................................................................................1897
fix_modify AtC reset_atomic_reference_positions....................................................................................1898
syntax................................................................................................................................................1898
examples...........................................................................................................................................1898
description.........................................................................................................................................1898
restrictions.........................................................................................................................................1898
related................................................................................................................................................1898
default...............................................................................................................................................1898
fix_modify AtC reset_time...........................................................................................................................1899
syntax................................................................................................................................................1899
examples...........................................................................................................................................1899
description.........................................................................................................................................1899
restrictions.........................................................................................................................................1899
related................................................................................................................................................1899
default...............................................................................................................................................1899
syntax................................................................................................................................................1899
examples...........................................................................................................................................1899
description.........................................................................................................................................1899
restrictions.........................................................................................................................................1899
related................................................................................................................................................1899
default...............................................................................................................................................1899
fix_modify AtC sample_frequency..............................................................................................................1901
syntax................................................................................................................................................1901
examples...........................................................................................................................................1901
description.........................................................................................................................................1901
restrictions.........................................................................................................................................1901
related................................................................................................................................................1901
default...............................................................................................................................................1901
LAMMPS Users Manual
xxxv
Table of Contents
fix_modify AtC set........................................................................................................................................1902
syntax................................................................................................................................................1902
examples...........................................................................................................................................1902
description.........................................................................................................................................1902
restrictions.........................................................................................................................................1902
related................................................................................................................................................1902
default...............................................................................................................................................1902
fix_modify AtC source..................................................................................................................................1903
syntax................................................................................................................................................1903
examples...........................................................................................................................................1903
description.........................................................................................................................................1903
restrictions.........................................................................................................................................1903
related................................................................................................................................................1903
default...............................................................................................................................................1903
fix_modify AtC source_integration.............................................................................................................1904
syntax................................................................................................................................................1904
examples...........................................................................................................................................1904
description.........................................................................................................................................1904
restrictions.........................................................................................................................................1904
related................................................................................................................................................1904
default...............................................................................................................................................1904
fix_modify AtC temperature_definition.....................................................................................................1905
syntax................................................................................................................................................1905
examples...........................................................................................................................................1905
description.........................................................................................................................................1905
restrictions.........................................................................................................................................1905
default...............................................................................................................................................1905
fix_modify AtC time_integration (thermal)...............................................................................................1906
syntax................................................................................................................................................1906
description.........................................................................................................................................1906
examples...........................................................................................................................................1906
description.........................................................................................................................................1906
related................................................................................................................................................1906
default...............................................................................................................................................1906
fix_modify AtC filter.....................................................................................................................................1907
syntax................................................................................................................................................1907
examples...........................................................................................................................................1907
description.........................................................................................................................................1907
restrictions.........................................................................................................................................1907
related................................................................................................................................................1907
default...............................................................................................................................................1907
LAMMPS Users Manual
xxxvi
Table of Contents
fix_modify AtC track_displacement...........................................................................................................1908
syntax................................................................................................................................................1908
examples...........................................................................................................................................1908
description.........................................................................................................................................1908
restrictions.........................................................................................................................................1908
default...............................................................................................................................................1908
fix_modify AtC unfix_flux...........................................................................................................................1909
syntax................................................................................................................................................1909
examples...........................................................................................................................................1909
description.........................................................................................................................................1909
restrictions.........................................................................................................................................1909
related................................................................................................................................................1909
default...............................................................................................................................................1909
fix_modify AtC unfix....................................................................................................................................1910
syntax................................................................................................................................................1910
examples...........................................................................................................................................1910
description.........................................................................................................................................1910
restrictions.........................................................................................................................................1910
related................................................................................................................................................1910
default...............................................................................................................................................1910
fix_modify AtC write_atom_weights...........................................................................................................1911
syntax................................................................................................................................................1911
examples...........................................................................................................................................1911
description.........................................................................................................................................1911
restrictions.........................................................................................................................................1911
related................................................................................................................................................1911
default...............................................................................................................................................1911
fix_modify AtC write_restart.......................................................................................................................1912
syntax................................................................................................................................................1912
examples...........................................................................................................................................1912
description.........................................................................................................................................1912
restrictions.........................................................................................................................................1912
related................................................................................................................................................1912
default...............................................................................................................................................1912
LAMMPS Users Manual
xxxvii

LAMMPS Documentation
31 Mar 2017 version
Version info:
The LAMMPS "version" is the date when it was released, such as 1 May 2010. LAMMPS is updated
continuously. Whenever we fix a bug or add a feature, we release it immediately, and post a notice on this
page of the WWW site. Every 2-4 months one of the incremental releases is subjected to more thorough
testing and labeled as a stable version.
Each dated copy of LAMMPS contains all the features and bug-fixes up to and including that version date.
The version date is printed to the screen and logfile every time you run LAMMPS. It is also in the file
src/version.h and in the LAMMPS directory name created when you unpack a tarball, and at the top of the
first page of the manual (this page).
If you browse the HTML doc pages on the LAMMPS WWW site, they always describe the most
current development version of LAMMPS.
•
If you browse the HTML doc pages included in your tarball, they describe the version you have.• The PDF file on the WWW site or in the tarball is updated about once per month. This is because it is
large, and we don't want it to be part of every patch.
•
There is also a Developer.pdf file in the doc directory, which describes the internal structure and
algorithms of LAMMPS.
•
LAMMPS stands for Large-scale Atomic/Molecular Massively Parallel Simulator.
LAMMPS is a classical molecular dynamics simulation code designed to run efficiently on parallel
computers. It was developed at Sandia National Laboratories, a US Department of Energy facility, with
funding from the DOE. It is an open-source code, distributed freely under the terms of the GNU Public
License (GPL).
The current core group of LAMMPS developers is at Sandia National Labs and Temple University:
Steve Plimpton, sjplimp at sandia.gov• Aidan Thompson, athomps at sandia.gov• Stan Moore, stamoor at sandia.gov• Axel Kohlmeyer, akohlmey at gmail.com•
Past core developers include Paul Crozier, Ray Shan and Mark Stevens, all at Sandia. The LAMMPS home
page at http://lammps.sandia.gov has more information about the code and its uses. Interaction with external
LAMMPS developers, bug reports and feature requests are mainly coordinated through the LAMMPS project
on GitHub. The lammps.org domain, currently hosting public continuous integration testing and precompiled
Linux RPM and Windows installer packages is located at Temple University and managed by Richard Berger,
richard.berger at temple.edu.
The LAMMPS documentation is organized into the following sections. If you find errors or omissions in this
manual or have suggestions for useful information to add, please send an email to the developers so we can
improve the LAMMPS documentation.
Once you are familiar with LAMMPS, you may want to bookmark this page at
1
Section_commands.html#comm since it gives quick access to documentation for all LAMMPS commands.
PDF file of the entire manual, generated by htmldoc
Introduction
1.1 What is LAMMPS
1.2 LAMMPS features
1.3 LAMMPS non-features
1.4 Open source distribution
1.5 Acknowledgments and citations
1.
Getting started
2.1 What's in the LAMMPS distribution
2.2 Making LAMMPS
2.3 Making LAMMPS with optional packages
2.4 Building LAMMPS via the Make.py script
2.5 Building LAMMPS as a library
2.6 Running LAMMPS
2.7 Command-line options
2.8 Screen output
2.9 Tips for users of previous versions
2.
Commands
3.1 LAMMPS input script
3.2 Parsing rules
3.3 Input script structure
3.4 Commands listed by category
3.5 Commands listed alphabetically
3.
Packages
4.1 Standard packages
4.2 User packages
4.
Accelerating LAMMPS performance
5.1 Measuring performance
5.2 Algorithms and code options to boost performace
5.3 Accelerator packages with optimized styles
5.3.1 GPU package
5.3.2 USER-INTEL package
5.3.3 KOKKOS package
5.3.4 USER-OMP package
5.3.5 OPT package
5.4 Comparison of various accelerator packages
5.
How-to discussions
6.1 Restarting a simulation
6.2 2d simulations
6.3 CHARMM and AMBER force fields
6.4 Running multiple simulations from one input script
6.5 Multi-replica simulations
6.6 Granular models
6.7 TIP3P water model
6.8 TIP4P water model
6.9 SPC water model
6.10 Coupling LAMMPS to other codes
6.11 Visualizing LAMMPS snapshots
6.
LAMMPS Users Manual
2
6.12 Triclinic (non-orthogonal) simulation boxes
6.13 NEMD simulations
6.14 Finite-size spherical and aspherical particles
6.15 Output from LAMMPS (thermo, dumps, computes, fixes, variables)
6.16 Thermostatting, barostatting, and compute temperature
6.17 Walls
6.18 Elastic constants
6.19 Library interface to LAMMPS
6.20 Calculating thermal conductivity
6.21 Calculating viscosity
6.22 Calculating a diffusion coefficient
6.23 Using chunks to calculate system properties
6.24 Setting parameters for pppm/disp
6.25 Polarizable models
6.26 Adiabatic core/shell model
6.27 Drude induced dipoles
Example problems7. Performance & scalability8. Additional tools9. Modifying & extending LAMMPS
10.1 Atom styles
10.2 Bond, angle, dihedral, improper potentials
10.3 Compute styles
10.4 Dump styles
10.5 Dump custom output options
10.6 Fix styles
10.7 Input script commands
10.8 Kspace computations
10.9 Minimization styles
10.10 Pairwise potentials
10.11 Region styles
10.12 Body styles
10.13 Thermodynamic output options
10.14 Variable options
10.15 Submitting new features for inclusion in LAMMPS
10.
Python interface
11.1 Overview of running LAMMPS from Python
11.2 Overview of using Python from a LAMMPS script
11.3 Building LAMMPS as a shared library
11.4 Installing the Python wrapper into Python
11.5 Extending Python with MPI to run in parallel
11.6 Testing the Python-LAMMPS interface
11.7 Using LAMMPS from Python
11.8 Example Python scripts that use LAMMPS
11.
Errors
12.1 Common problems
12.2 Reporting bugs
12.3 Error & warning messages
12.
Future and history
13.1 Coming attractions
13.2 Past versions
13.
LAMMPS Users Manual
3

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
1. Introduction
This section provides an overview of what LAMMPS can and can't do, describes what it means for LAMMPS
to be an open-source code, and acknowledges the funding and people who have contributed to LAMMPS over
the years.
1.1 What is LAMMPS
1.2 LAMMPS features
1.3 LAMMPS non-features
1.4 Open source distribution
1.5 Acknowledgments and citations
1.1 What is LAMMPS
LAMMPS is a classical molecular dynamics code that models an ensemble of particles in a liquid, solid, or
gaseous state. It can model atomic, polymeric, biological, metallic, granular, and coarse-grained systems using
a variety of force fields and boundary conditions.
For examples of LAMMPS simulations, see the Publications page of the LAMMPS WWW Site.
LAMMPS runs efficiently on single-processor desktop or laptop machines, but is designed for parallel
computers. It will run on any parallel machine that compiles C++ and supports the MPI message-passing
library. This includes distributed- or shared-memory parallel machines and Beowulf-style clusters.
LAMMPS can model systems with only a few particles up to millions or billions. See Section 8 for
information on LAMMPS performance and scalability, or the Benchmarks section of the LAMMPS WWW
Site.
LAMMPS is a freely-available open-source code, distributed under the terms of the GNU Public License,
which means you can use or modify the code however you wish. See this section for a brief discussion of the
open-source philosophy.
LAMMPS is designed to be easy to modify or extend with new capabilities, such as new force fields, atom
types, boundary conditions, or diagnostics. See Section 10 for more details.
The current version of LAMMPS is written in C++. Earlier versions were written in F77 and F90. See Section
13 for more information on different versions. All versions can be downloaded from the LAMMPS WWW
Site.
LAMMPS was originally developed under a US Department of Energy CRADA (Cooperative Research and
Development Agreement) between two DOE labs and 3 companies. It is distributed by Sandia National Labs.
See this section for more information on LAMMPS funding and individuals who have contributed to
LAMMPS.
In the most general sense, LAMMPS integrates Newton's equations of motion for collections of atoms,
molecules, or macroscopic particles that interact via short- or long-range forces with a variety of initial and/or
LAMMPS Users Manual
4

boundary conditions. For computational efficiency LAMMPS uses neighbor lists to keep track of nearby
particles. The lists are optimized for systems with particles that are repulsive at short distances, so that the
local density of particles never becomes too large. On parallel machines, LAMMPS uses
spatial-decomposition techniques to partition the simulation domain into small 3d sub-domains, one of which
is assigned to each processor. Processors communicate and store "ghost" atom information for atoms that
border their sub-domain. LAMMPS is most efficient (in a parallel sense) for systems whose particles fill a 3d
rectangular box with roughly uniform density. Papers with technical details of the algorithms used in
LAMMPS are listed in this section.
1.2 LAMMPS features
This section highlights LAMMPS features, with pointers to specific commands which give more details. If
LAMMPS doesn't have your favorite interatomic potential, boundary condition, or atom type, see Section 10,
which describes how you can add it to LAMMPS.
General features
runs on a single processor or in parallel• distributed-memory message-passing parallelism (MPI)• spatial-decomposition of simulation domain for parallelism• open-source distribution• highly portable C++• optional libraries used: MPI and single-processor FFT• GPU (CUDA and OpenCL), Intel(R) Xeon Phi(TM) coprocessors, and OpenMP support for many
code features
•
easy to extend with new features and functionality• runs from an input script• syntax for defining and using variables and formulas• syntax for looping over runs and breaking out of loops• run one or multiple simulations simultaneously (in parallel) from one script• build as library, invoke LAMMPS thru library interface or provided Python wrapper• couple with other codes: LAMMPS calls other code, other code calls LAMMPS, umbrella code calls
both
•
Particle and model types
(atom style command)
atoms• coarse-grained particles (e.g. bead-spring polymers)• united-atom polymers or organic molecules• all-atom polymers, organic molecules, proteins, DNA• metals• granular materials• coarse-grained mesoscale models• finite-size spherical and ellipsoidal particles• finite-size line segment (2d) and triangle (3d) particles• point dipole particles• rigid collections of particles• hybrid combinations of these•
LAMMPS Users Manual
5
Force fields
(pair style, bond style, angle style, dihedral style, improper style, kspace style commands)
pairwise potentials: Lennard-Jones, Buckingham, Morse, Born-Mayer-Huggins, Yukawa, soft, class 2
(COMPASS), hydrogen bond, tabulated
•
charged pairwise potentials: Coulombic, point-dipole• manybody potentials: EAM, Finnis/Sinclair EAM, modified EAM (MEAM), embedded ion method
(EIM), EDIP, ADP, Stillinger-Weber, Tersoff, REBO, AIREBO, ReaxFF, COMB, SNAP,
Streitz-Mintmire, 3-body polymorphic
•
long-range interactions for charge, point-dipoles, and LJ dispersion: Ewald, Wolf, PPPM (similar to
particle-mesh Ewald)
•
polarization models: QEq, core/shell model, Drude dipole model• charge equilibration (QEq via dynamic, point, shielded, Slater methods)• coarse-grained potentials: DPD, GayBerne, REsquared, colloidal, DLVO• mesoscopic potentials: granular, Peridynamics, SPH• electron force field (eFF, AWPMD)• bond potentials: harmonic, FENE, Morse, nonlinear, class 2, quartic (breakable)• angle potentials: harmonic, CHARMM, cosine, cosine/squared, cosine/periodic, class 2 (COMPASS)• dihedral potentials: harmonic, CHARMM, multi-harmonic, helix, class 2 (COMPASS), OPLS• improper potentials: harmonic, cvff, umbrella, class 2 (COMPASS)• polymer potentials: all-atom, united-atom, bead-spring, breakable• water potentials: TIP3P, TIP4P, SPC• implicit solvent potentials: hydrodynamic lubrication, Debye• force-field compatibility with common CHARMM, AMBER, DREIDING, OPLS, GROMACS,
COMPASS options
•
access to KIM archive of potentials via pair kim• hybrid potentials: multiple pair, bond, angle, dihedral, improper potentials can be used in one
simulation
•
overlaid potentials: superposition of multiple pair potentials•
Atom creation
(read_data, lattice, create_atoms, delete_atoms, displace_atoms, replicate commands)
read in atom coords from files• create atoms on one or more lattices (e.g. grain boundaries)• delete geometric or logical groups of atoms (e.g. voids)• replicate existing atoms multiple times• displace atoms•
Ensembles, constraints, and boundary conditions
(fix command)
2d or 3d systems• orthogonal or non-orthogonal (triclinic symmetry) simulation domains• constant NVE, NVT, NPT, NPH, Parinello/Rahman integrators• thermostatting options for groups and geometric regions of atoms• pressure control via Nose/Hoover or Berendsen barostatting in 1 to 3 dimensions• simulation box deformation (tensile and shear)•
LAMMPS Users Manual
6
harmonic (umbrella) constraint forces• rigid body constraints• SHAKE bond and angle constraints• Monte Carlo bond breaking, formation, swapping• atom/molecule insertion and deletion• walls of various kinds• non-equilibrium molecular dynamics (NEMD)• variety of additional boundary conditions and constraints•
Integrators
(run, run_style, minimize commands)
velocity-Verlet integrator• Brownian dynamics• rigid body integration• energy minimization via conjugate gradient or steepest descent relaxation• rRESPA hierarchical timestepping• rerun command for post-processing of dump files•
Diagnostics
see the various flavors of the fix and compute commands•
Output
(dump, restart commands)
log file of thermodynamic info• text dump files of atom coords, velocities, other per-atom quantities• binary restart files• parallel I/O of dump and restart files• per-atom quantities (energy, stress, centro-symmetry parameter, CNA, etc)• user-defined system-wide (log file) or per-atom (dump file) calculations• spatial and time averaging of per-atom quantities• time averaging of system-wide quantities• atom snapshots in native, XYZ, XTC, DCD, CFG formats•
Multi-replica models
nudged elastic band parallel replica dynamics temperature accelerated dynamics parallel tempering
Pre- and post-processing
Various pre- and post-processing serial tools are packaged with LAMMPS; see these doc pages.• Our group has also written and released a separate toolkit called Pizza.py which provides tools for
doing setup, analysis, plotting, and visualization for LAMMPS simulations. Pizza.py is written in
Python and is available for download from the Pizza.py WWW site.
•
LAMMPS Users Manual
7

Specialized features
These are LAMMPS capabilities which you may not think of as typical molecular dynamics options:
static and dynamic load-balancing• generalized aspherical particles• stochastic rotation dynamics (SRD)• real-time visualization and interactive MD• calculate virtual diffraction patterns• atom-to-continuum coupling with finite elements• coupled rigid body integration via the POEMS library• QM/MM coupling• path-integral molecular dynamics (PIMD) and this as well• Monte Carlo via GCMC and tfMC atom swapping and bond swapping• Direct Simulation Monte Carlo for low-density fluids• Peridynamics mesoscale modeling• Lattice Boltzmann fluid• targeted and steered molecular dynamics• two-temperature electron model•
1.3 LAMMPS non-features
LAMMPS is designed to efficiently compute Newton's equations of motion for a system of interacting
particles. Many of the tools needed to pre- and post-process the data for such simulations are not included in
the LAMMPS kernel for several reasons:
the desire to keep LAMMPS simple• they are not parallel operations• other codes already do them• limited development resources•
Specifically, LAMMPS itself does not:
run thru a GUI• build molecular systems• assign force-field coefficients automagically• perform sophisticated analyses of your MD simulation• visualize your MD simulation• plot your output data•
A few tools for pre- and post-processing tasks are provided as part of the LAMMPS package; they are
described in this section. However, many people use other codes or write their own tools for these tasks.
As noted above, our group has also written and released a separate toolkit called Pizza.py which addresses
some of the listed bullets. It provides tools for doing setup, analysis, plotting, and visualization for LAMMPS
simulations. Pizza.py is written in Python and is available for download from the Pizza.py WWW site.
LAMMPS requires as input a list of initial atom coordinates and types, molecular topology information, and
force-field coefficients assigned to all atoms and bonds. LAMMPS will not build molecular systems and
assign force-field parameters for you.
LAMMPS Users Manual
8

For atomic systems LAMMPS provides a create_atoms command which places atoms on solid-state lattices
(fcc, bcc, user-defined, etc). Assigning small numbers of force field coefficients can be done via the pair
coeff, bond coeff, angle coeff, etc commands. For molecular systems or more complicated simulation
geometries, users typically use another code as a builder and convert its output to LAMMPS input format, or
write their own code to generate atom coordinate and molecular topology for LAMMPS to read in.
For complicated molecular systems (e.g. a protein), a multitude of topology information and hundreds of
force-field coefficients must typically be specified. We suggest you use a program like CHARMM or
AMBER or other molecular builders to setup such problems and dump its information to a file. You can then
reformat the file as LAMMPS input. Some of the tools in this section can assist in this process.
Similarly, LAMMPS creates output files in a simple format. Most users post-process these files with their
own analysis tools or re-format them for input into other programs, including visualization packages. If you
are convinced you need to compute something on-the-fly as LAMMPS runs, see Section 10 for a discussion of
how you can use the dump and compute and fix commands to print out data of your choosing. Keep in mind
that complicated computations can slow down the molecular dynamics timestepping, particularly if the
computations are not parallel, so it is often better to leave such analysis to post-processing codes.
For high-quality visualization we recommend the following packages:
VMD• AtomEye• OVITO• ParaView• PyMol• Raster3d• RasMol•
Other features that LAMMPS does not yet (and may never) support are discussed in Section 13.
Finally, these are freely-available molecular dynamics codes, most of them parallel, which may be well-suited
to the problems you want to model. They can also be used in conjunction with LAMMPS to perform
complementary modeling tasks.
CHARMM• AMBER• NAMD• NWCHEM• DL_POLY• Tinker•
CHARMM, AMBER, NAMD, NWCHEM, and Tinker are designed primarily for modeling biological
molecules. CHARMM and AMBER use atom-decomposition (replicated-data) strategies for parallelism;
NAMD and NWCHEM use spatial-decomposition approaches, similar to LAMMPS. Tinker is a serial code.
DL_POLY includes potentials for a variety of biological and non-biological materials; both a replicated-data
and spatial-decomposition version exist.
LAMMPS Users Manual
9

1.4 Open source distribution
LAMMPS comes with no warranty of any kind. As each source file states in its header, it is a copyrighted
code that is distributed free-of- charge, under the terms of the GNU Public License (GPL). This is often
referred to as open-source distribution - see www.gnu.org or www.opensource.org for more details. The legal
text of the GPL is in the LICENSE file that is included in the LAMMPS distribution.
Here is a summary of what the GPL means for LAMMPS users:
(1) Anyone is free to use, modify, or extend LAMMPS in any way they choose, including for commercial
purposes.
(2) If you distribute a modified version of LAMMPS, it must remain open-source, meaning you distribute it
under the terms of the GPL. You should clearly annotate such a code as a derivative version of LAMMPS.
(3) If you release any code that includes LAMMPS source code, then it must also be open-sourced, meaning
you distribute it under the terms of the GPL.
(4) If you give LAMMPS files to someone else, the GPL LICENSE file and source file headers (including the
copyright and GPL notices) should remain part of the code.
In the spirit of an open-source code, these are various ways you can contribute to making LAMMPS better.
You can send email to the developers on any of these items.
Point prospective users to the LAMMPS WWW Site. Mention it in talks or link to it from your
WWW site.
•
If you find an error or omission in this manual or on the LAMMPS WWW Site, or have a suggestion
for something to clarify or include, send an email to the developers.
•
If you find a bug, Section 12.2 describes how to report it.• If you publish a paper using LAMMPS results, send the citation (and any cool pictures or movies if
you like) to add to the Publications, Pictures, and Movies pages of the LAMMPS WWW Site, with
links and attributions back to you.
•
Create a new Makefile.machine that can be added to the src/MAKE directory.• The tools sub-directory of the LAMMPS distribution has various stand-alone codes for pre- and
post-processing of LAMMPS data. More details are given in Section 9. If you write a new tool that
users will find useful, it can be added to the LAMMPS distribution.
•
LAMMPS is designed to be easy to extend with new code for features like potentials, boundary
conditions, diagnostic computations, etc. This section gives details. If you add a feature of general
interest, it can be added to the LAMMPS distribution.
•
The Benchmark page of the LAMMPS WWW Site lists LAMMPS performance on various platforms.
The files needed to run the benchmarks are part of the LAMMPS distribution. If your machine is
sufficiently different from those listed, your timing data can be added to the page.
•
You can send feedback for the User Comments page of the LAMMPS WWW Site. It might be added
to the page. No promises.
•
Cash. Small denominations, unmarked bills preferred. Paper sack OK. Leave on desk. VISA also
accepted. Chocolate chip cookies encouraged.
•
LAMMPS Users Manual
10
1.5 Acknowledgments and citations
LAMMPS development has been funded by the US Department of Energy (DOE), through its CRADA,
LDRD, ASCI, and Genomes-to-Life programs and its OASCR and OBER offices.
Specifically, work on the latest version was funded in part by the US Department of Energy's Genomics:GTL
program (www.doegenomestolife.org) under the project, "Carbon Sequestration in Synechococcus Sp.: From
Molecular Machines to Hierarchical Modeling".
The following paper describe the basic parallel algorithms used in LAMMPS. If you use LAMMPS results in
your published work, please cite this paper and include a pointer to the LAMMPS WWW Site
(http://lammps.sandia.gov):
S. Plimpton, Fast Parallel Algorithms for Short-Range Molecular Dynamics, J Comp Phys, 117, 1-19
(1995).
Other papers describing specific algorithms used in LAMMPS are listed under the Citing LAMMPS link of
the LAMMPS WWW page.
The Publications link on the LAMMPS WWW page lists papers that have cited LAMMPS. If your paper is
not listed there for some reason, feel free to send us the info. If the simulations in your paper produced cool
pictures or animations, we'll be pleased to add them to the Pictures or Movies pages of the LAMMPS WWW
site.
The core group of LAMMPS developers is at Sandia National Labs:
Steve Plimpton, sjplimp at sandia.gov• Aidan Thompson, athomps at sandia.gov• Paul Crozier, pscrozi at sandia.gov•
The following folks are responsible for significant contributions to the code, or other aspects of the LAMMPS
development effort. Many of the packages they have written are somewhat unique to LAMMPS and the code
would not be as general-purpose as it is without their expertise and efforts.
Axel Kohlmeyer (Temple U), akohlmey at gmail.com, SVN and Git repositories, indefatigable mail
list responder, USER-CG-CMM and USER-OMP packages
•
Roy Pollock (LLNL), Ewald and PPPM solvers• Mike Brown (ORNL), brownw at ornl.gov, GPU package• Greg Wagner (Sandia), gjwagne at sandia.gov, MEAM package for MEAM potential• Mike Parks (Sandia), mlparks at sandia.gov, PERI package for Peridynamics• Rudra Mukherjee (JPL), Rudranarayan.M.Mukherjee at jpl.nasa.gov, POEMS package for articulated
rigid body motion
•
Reese Jones (Sandia) and collaborators, rjones at sandia.gov, USER-ATC package for
atom/continuum coupling
•
Ilya Valuev (JIHT), valuev at physik.hu-berlin.de, USER-AWPMD package for wave-packet MD• Christian Trott (U Tech Ilmenau), christian.trott at tu-ilmenau.de, USER-CUDA package• Andres Jaramillo-Botero (Caltech), ajaramil at wag.caltech.edu, USER-EFF package for electron
force field
•
Christoph Kloss (JKU), Christoph.Kloss at jku.at, USER-LIGGGHTS package for granular models
and granular/fluid coupling
•
Metin Aktulga (LBL), hmaktulga at lbl.gov, USER-REAXC package for C version of ReaxFF•
LAMMPS Users Manual
11
Georg Gunzenmuller (EMI), georg.ganzenmueller at emi.fhg.de, USER-SPH package•
As discussed in Section 13, LAMMPS originated as a cooperative project between DOE labs and industrial
partners. Folks involved in the design and testing of the original version of LAMMPS were the following:
John Carpenter (Mayo Clinic, formerly at Cray Research)• Terry Stouch (Lexicon Pharmaceuticals, formerly at Bristol Myers Squibb)• Steve Lustig (Dupont)• Jim Belak (LLNL)•
LAMMPS Users Manual
12
Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
2. Getting Started
This section describes how to build and run LAMMPS, for both new and experienced users.
2.1 What's in the LAMMPS distribution
2.2 Making LAMMPS
2.3 Making LAMMPS with optional packages
2.4 Building LAMMPS via the Make.py script
2.5 Building LAMMPS as a library
2.6 Running LAMMPS
2.7 Command-line options
2.8 Screen output
2.9 Tips for users of previous versions
2.1 What's in the LAMMPS distribution
When you download a LAMMPS tarball you will need to unzip and untar the downloaded file with the
following commands, after placing the tarball in an appropriate directory.
tar -xzvf lammps*.tar.gz
This will create a LAMMPS directory containing two files and several sub-directories:
README text file
LICENSE the GNU General Public License (GPL)
bench benchmark problems
doc documentation
examples simple test problems
potentials embedded atom method (EAM) potential files
src source files
tools pre- and post-processing tools
Note that the download page also has links to download pre-build Windows installers, as well as pre-built
packages for several widely used Linux distributions. It also has instructions for how to download/install
LAMMPS for Macs (via Homebrew), and to download and update LAMMPS from SVN and Git repositories,
which gives you access to the up-to-date sources that are used by the LAMMPS core developers.
The Windows and Linux packages for serial or parallel include only selected packages and bug-fixes/upgrades
listed on this page up to a certain date, as stated on the download page. If you want an executable with
non-included packages or that is more current, then you'll need to build LAMMPS yourself, as discussed in
the next section.
Skip to the Running LAMMPS sections for info on how to launch a LAMMPS Windows executable on a
Windows box.
LAMMPS Users Manual
13

2.2 Making LAMMPS
This section has the following sub-sections:
2.2.1 Read this first
2.2.1 Steps to build a LAMMPS executable
2.2.3 Common errors that can occur when making LAMMPS
2.2.4 Additional build tips
2.2.5 Building for a Mac
2.2.6 Building for Windows
Read this first
If you want to avoid building LAMMPS yourself, read the preceding section about options available for
downloading and installing executables. Details are discussed on the download page.
Building LAMMPS can be simple or not-so-simple. If all you need are the default packages installed in
LAMMPS, and MPI is already installed on your machine, or you just want to run LAMMPS in serial, then
you can typically use the Makefile.mpi or Makefile.serial files in src/MAKE by typing one of these lines
(from the src dir):
make mpi
make serial
Note that on a facility supercomputer, there are often "modules" loaded in your environment that provide the
compilers and MPI you should use. In this case, the "mpicxx" compile/link command in Makefile.mpi should
just work by accessing those modules.
It may be the case that one of the other Makefile.machine files in the src/MAKE sub-directories is a better
match to your system (type "make" to see a list), you can use it as-is by typing (for example):
make stampede
If any of these builds (with an existing Makefile.machine) works on your system, then you're done!
If you want to do one of the following:
use optional LAMMPS features that require additional libraries• use optional packages that require additional libraries• use optional accelerator packages that require special compiler/linker settings• run on a specialized platform that has its own compilers, settings, or other libs to use•
then building LAMMPS is more complicated. You may need to find where auxiliary libraries exist on your
machine or install them if they don't. You may need to build additional libraries that are part of the LAMMPS
package, before building LAMMPS. You may need to edit a Makefile.machine file to make it compatible with
your system.
Note that there is a Make.py tool in the src directory that automates several of these steps, but you still have to
know what you are doing. Section 2.4 below describes the tool. It is a convenient way to work with
installing/un-installing various packages, the Makefile.machine changes required by some packages, and the
auxiliary libraries some of them use.
LAMMPS Users Manual
14

Please read the following sections carefully. If you are not comfortable with makefiles, or building codes on a
Unix platform, or running an MPI job on your machine, please find a local expert to help you. Many
compilation, linking, and run problems that users have are often not really LAMMPS issues - they are peculiar
to the user's system, compilers, libraries, etc. Such questions are better answered by a local expert.
If you have a build problem that you are convinced is a LAMMPS issue (e.g. the compiler complains about a
line of LAMMPS source code), then please post the issue to the LAMMPS mail list.
If you succeed in building LAMMPS on a new kind of machine, for which there isn't a similar machine
Makefile included in the src/MAKE/MACHINES directory, then send it to the developers and we can include
it in the LAMMPS distribution.
Steps to build a LAMMPS executable
Step 0
The src directory contains the C++ source and header files for LAMMPS. It also contains a top-level Makefile
and a MAKE sub-directory with low-level Makefile.* files for many systems and machines. See the
src/MAKE/README file for a quick overview of what files are available and what sub-directories they are
in.
The src/MAKE dir has a few files that should work as-is on many platforms. The src/MAKE/OPTIONS dir
has more that invoke additional compiler, MPI, and other setting options commonly used by LAMMPS, to
illustrate their syntax. The src/MAKE/MACHINES dir has many more that have been tweaked or optimized
for specific machines. These files are all good starting points if you find you need to change them for your
machine. Put any file you edit into the src/MAKE/MINE directory and it will be never be touched by any
LAMMPS updates.
>From within the src directory, type "make" or "gmake". You should see a list of available choices from
src/MAKE and all of its sub-directories. If one of those has the options you want or is the machine you want,
you can type a command like:
make mpi
or
make serial
or
gmake mac
Note that the corresponding Makefile.machine can exist in src/MAKE or any of its sub-directories. If a file
with the same name appears in multiple places (not a good idea), the order they are used is as follows:
src/MAKE/MINE, src/MAKE, src/MAKE/OPTIONS, src/MAKE/MACHINES. This gives preference to a
file you have created/edited and put in src/MAKE/MINE.
Note that on a multi-processor or multi-core platform you can launch a parallel make, by using the "-j" switch
with the make command, which will build LAMMPS more quickly.
If you get no errors and an executable like lmp_mpi or lmp_serial or lmp_mac is produced, then you're
done; it's your lucky day.
LAMMPS Users Manual
15
Note that by default only a few of LAMMPS optional packages are installed. To build LAMMPS with
optional packages, see this section below.
Step 1
If Step 0 did not work, you will need to create a low-level Makefile for your machine, like Makefile.foo. You
should make a copy of an existing Makefile.* in src/MAKE or one of its sub-directories as a starting point.
The only portions of the file you need to edit are the first line, the "compiler/linker settings" section, and the
"LAMMPS-specific settings" section. When it works, put the edited file in src/MAKE/MINE and it will not
be altered by any future LAMMPS updates.
Step 2
Change the first line of Makefile.foo to list the word "foo" after the "#", and whatever other options it will set.
This is the line you will see if you just type "make".
Step 3
The "compiler/linker settings" section lists compiler and linker settings for your C++ compiler, including
optimization flags. You can use g++, the open-source GNU compiler, which is available on all Unix systems.
You can also use mpicxx which will typically be available if MPI is installed on your system, though you
should check which actual compiler it wraps. Vendor compilers often produce faster code. On boxes with
Intel CPUs, we suggest using the Intel icc compiler, which can be downloaded from Intel's compiler site.
If building a C++ code on your machine requires additional libraries, then you should list them as part of the
LIB variable. You should not need to do this if you use mpicxx.
The DEPFLAGS setting is what triggers the C++ compiler to create a dependency list for a source file. This
speeds re-compilation when source (*.cpp) or header (*.h) files are edited. Some compilers do not support
dependency file creation, or may use a different switch than -D. GNU g++ and Intel icc works with -D. If your
compiler can't create dependency files, then you'll need to create a Makefile.foo patterned after
Makefile.storm, which uses different rules that do not involve dependency files. Note that when you build
LAMMPS for the first time on a new platform, a long list of *.d files will be printed out rapidly. This is not an
error; it is the Makefile doing its normal creation of dependencies.
Step 4
The "system-specific settings" section has several parts. Note that if you change any -D setting in this section,
you should do a full re-compile, after typing "make clean" (which will describe different clean options).
The LMP_INC variable is used to include options that turn on ifdefs within the LAMMPS code. The options
that are currently recognized are:
-DLAMMPS_GZIP• -DLAMMPS_JPEG• -DLAMMPS_PNG• -DLAMMPS_FFMPEG• -DLAMMPS_MEMALIGN• -DLAMMPS_XDR• -DLAMMPS_SMALLBIG• -DLAMMPS_BIGBIG• -DLAMMPS_SMALLSMALL•
LAMMPS Users Manual
16
-DLAMMPS_LONGLONG_TO_LONG• -DLAMMPS_EXCEPTIONS• -DPACK_ARRAY• -DPACK_POINTER• -DPACK_MEMCPY•
The read_data and dump commands will read/write gzipped files if you compile with -DLAMMPS_GZIP. It
requires that your machine supports the "popen()" function in the standard runtime library and that a gzip
executable can be found by LAMMPS during a run.
NOTE: on some clusters with high-speed networks, using the fork() library calls (required by popen()) can
interfere with the fast communication library and lead to simulations using compressed output or input to
hang or crash. For selected operations, compressed file I/O is also available using a compression library
instead, which are provided in the COMPRESS package. From more details about compiling LAMMPS with
packages, please see below.
If you use -DLAMMPS_JPEG, the dump image command will be able to write out JPEG image files. For
JPEG files, you must also link LAMMPS with a JPEG library, as described below. If you use
-DLAMMPS_PNG, the dump image command will be able to write out PNG image files. For PNG files, you
must also link LAMMPS with a PNG library, as described below. If neither of those two defines are used,
LAMMPS will only be able to write out uncompressed PPM image files.
If you use -DLAMMPS_FFMPEG, the dump movie command will be available to support on-the-fly
generation of rendered movies the need to store intermediate image files. It requires that your machines
supports the "popen" function in the standard runtime library and that an FFmpeg executable can be found by
LAMMPS during the run.
NOTE: Similar to the note above, this option can conflict with high-speed networks, because it uses popen().
Using -DLAMMPS_MEMALIGN= enables the use of the posix_memalign() call instead of malloc() when
large chunks or memory are allocated by LAMMPS. This can help to make more efficient use of vector
instructions of modern CPUS, since dynamically allocated memory has to be aligned on larger than default
byte boundaries (e.g. 16 bytes instead of 8 bytes on x86 type platforms) for optimal performance.
If you use -DLAMMPS_XDR, the build will include XDR compatibility files for doing particle dumps in
XTC format. This is only necessary if your platform does have its own XDR files available. See the
Restrictions section of the dump command for details.
Use at most one of the -DLAMMPS_SMALLBIG, -DLAMMPS_BIGBIG, -DLAMMPS_SMALLSMALL
settings. The default is -DLAMMPS_SMALLBIG. These settings refer to use of 4-byte (small) vs 8-byte (big)
integers within LAMMPS, as specified in src/lmptype.h. The only reason to use the BIGBIG setting is to
enable simulation of huge molecular systems (which store bond topology info) with more than 2 billion
atoms, or to track the image flags of moving atoms that wrap around a periodic box more than 512 times.
Normally, the only reason to use SMALLSMALL is if your machine does not support 64-bit integers, though
you can use SMALLSMALL setting if you are running in serial or on a desktop machine or small cluster
where you will never run large systems or for long time (more than 2 billion atoms, more than 2 billion
timesteps). See the Additional build tips section below for more details on these settings.
Note that the USER-ATC package is not currently compatible with -DLAMMPS_BIGBIG. Also the GPU
package requires the lib/gpu library to be compiled with the same setting, or the link will fail.
LAMMPS Users Manual
17
The -DLAMMPS_LONGLONG_TO_LONG setting may be needed if your system or MPI version does not
recognize "long long" data types. In this case a "long" data type is likely already 64-bits, in which case this
setting will convert to that data type.
The -DLAMMPS_EXCEPTIONS setting can be used to activate alternative versions of error handling inside
of LAMMPS. This is useful when external codes drive LAMMPS as a library. Using this option, LAMMPS
errors do not kill the caller. Instead, the call stack is unwound and control returns to the caller. The library
interface provides the lammps_has_error() and lammps_get_last_error_message() functions to detect and find
out more about a LAMMPS error.
Using one of the -DPACK_ARRAY, -DPACK_POINTER, and -DPACK_MEMCPY options can make for
faster parallel FFTs (in the PPPM solver) on some platforms. The -DPACK_ARRAY setting is the default.
See the kspace_style command for info about PPPM. See Step 6 below for info about building LAMMPS
with an FFT library.
Step 5
The 3 MPI variables are used to specify an MPI library to build LAMMPS with. Note that you do not need to
set these if you use the MPI compiler mpicxx for your CC and LINK setting in the section above. The MPI
wrapper knows where to find the needed files.
If you want LAMMPS to run in parallel, you must have an MPI library installed on your platform. If MPI is
installed on your system in the usual place (under /usr/local), you also may not need to specify these 3
variables, assuming /usr/local is in your path. On some large parallel machines which use "modules" for their
compile/link environments, you may simply need to include the correct module in your build environment,
before building LAMMPS. Or the parallel machine may have a vendor-provided MPI which the compiler has
no trouble finding.
Failing this, these 3 variables can be used to specify where the mpi.h file (MPI_INC) and the MPI library file
(MPI_PATH) are found and the name of the library file (MPI_LIB).
If you are installing MPI yourself, we recommend Argonne's MPICH2 or OpenMPI. MPICH can be
downloaded from the Argonne MPI site. OpenMPI can be downloaded from the OpenMPI site. Other MPI
packages should also work. If you are running on a big parallel platform, your system people or the vendor
should have already installed a version of MPI, which is likely to be faster than a self-installed MPICH or
OpenMPI, so find out how to build and link with it. If you use MPICH or OpenMPI, you will have to
configure and build it for your platform. The MPI configure script should have compiler options to enable you
to use the same compiler you are using for the LAMMPS build, which can avoid problems that can arise when
linking LAMMPS to the MPI library.
If you just want to run LAMMPS on a single processor, you can use the dummy MPI library provided in
src/STUBS, since you don't need a true MPI library installed on your system. See src/MAKE/Makefile.serial
for how to specify the 3 MPI variables in this case. You will also need to build the STUBS library for your
platform before making LAMMPS itself. Note that if you are building with src/MAKE/Makefile.serial, e.g. by
typing "make serial", then the STUBS library is built for you.
To build the STUBS library from the src directory, type "make mpi-stubs", or from the src/STUBS dir, type
"make". This should create a libmpi_stubs.a file suitable for linking to LAMMPS. If the build fails, you will
need to edit the STUBS/Makefile for your platform.
LAMMPS Users Manual
18
The file STUBS/mpi.c provides a CPU timer function called MPI_Wtime() that calls gettimeofday() . If your
system doesn't support gettimeofday() , you'll need to insert code to call another timer. Note that the
ANSI-standard function clock() rolls over after an hour or so, and is therefore insufficient for timing long
LAMMPS simulations.
Step 6
The 3 FFT variables allow you to specify an FFT library which LAMMPS uses (for performing 1d FFTs)
when running the particle-particle particle-mesh (PPPM) option for long-range Coulombics via the
kspace_style command.
LAMMPS supports common open-source or vendor-supplied FFT libraries for this purpose. If you leave these
3 variables blank, LAMMPS will use the open-source KISS FFT library, which is included in the LAMMPS
distribution. This library is portable to all platforms and for typical LAMMPS simulations is almost as fast as
FFTW or vendor optimized libraries. If you are not including the KSPACE package in your build, you can
also leave the 3 variables blank.
Otherwise, select which kinds of FFTs to use as part of the FFT_INC setting by a switch of the form
-DFFT_XXX. Recommended values for XXX are: MKL or FFTW3. FFTW2 and NONE are supported as
legacy options. Selecting -DFFT_FFTW will use the FFTW3 library and -DFFT_NONE will use the KISS
library described above.
You may also need to set the FFT_INC, FFT_PATH, and FFT_LIB variables, so the compiler and linker can
find the needed FFT header and library files. Note that on some large parallel machines which use "modules"
for their compile/link environments, you may simply need to include the correct module in your build
environment. Or the parallel machine may have a vendor-provided FFT library which the compiler has no
trouble finding.
FFTW is a fast, portable library that should also work on any platform. You can download it from
www.fftw.org. Both the legacy version 2.1.X and the newer 3.X versions are supported as -DFFT_FFTW2 or
-DFFT_FFTW3. Building FFTW for your box should be as simple as ./configure; make. Note that on some
platforms FFTW2 has been pre-installed, and uses renamed files indicating the precision it was compiled
with, e.g. sfftw.h, or dfftw.h instead of fftw.h. In this case, you can specify an additional define variable for
FFT_INC called -DFFTW_SIZE, which will select the correct include file. In this case, for FFT_LIB you
must also manually specify the correct library, namely -lsfftw or -ldfftw.
The FFT_INC variable also allows for a -DFFT_SINGLE setting that will use single-precision FFTs with
PPPM, which can speed-up long-range calculations, particularly in parallel or on GPUs. Fourier transform and
related PPPM operations are somewhat insensitive to floating point truncation errors and thus do not always
need to be performed in double precision. Using the -DFFT_SINGLE setting trades off a little accuracy for
reduced memory use and parallel communication costs for transposing 3d FFT data. Note that single precision
FFTs have only been tested with the FFTW3, FFTW2, MKL, and KISS FFT options.
Step 7
The 3 JPG variables allow you to specify a JPEG and/or PNG library which LAMMPS uses when writing out
JPEG or PNG files via the dump image command. These can be left blank if you do not use the
-DLAMMPS_JPEG or -DLAMMPS_PNG switches discussed above in Step 4, since in that case JPEG/PNG
output will be disabled.
LAMMPS Users Manual
19

A standard JPEG library usually goes by the name libjpeg.a or libjpeg.so and has an associated header file
jpeglib.h. Whichever JPEG library you have on your platform, you'll need to set the appropriate JPG_INC,
JPG_PATH, and JPG_LIB variables, so that the compiler and linker can find it.
A standard PNG library usually goes by the name libpng.a or libpng.so and has an associated header file
png.h. Whichever PNG library you have on your platform, you'll need to set the appropriate JPG_INC,
JPG_PATH, and JPG_LIB variables, so that the compiler and linker can find it.
As before, if these header and library files are in the usual place on your machine, you may not need to set
these variables.
Step 8
Note that by default only a few of LAMMPS optional packages are installed. To build LAMMPS with
optional packages, see this section below, before proceeding to Step 9.
Step 9
That's it. Once you have a correct Makefile.foo, and you have pre-built any other needed libraries (e.g. MPI,
FFT, etc) all you need to do from the src directory is type something like this:
make foo
make -j N foo
gmake foo
gmake -j N foo
The -j or -j N switches perform a parallel build which can be much faster, depending on how many cores your
compilation machine has. N is the number of cores the build runs on.
You should get the executable lmp_foo when the build is complete.
Errors that can occur when making LAMMPS: h5
NOTE: If an error occurs when building LAMMPS, the compiler or linker will state very explicitly what the
problem is. The error message should give you a hint as to which of the steps above has failed, and what you
need to do in order to fix it. Building a code with a Makefile is a very logical process. The compiler and linker
need to find the appropriate files and those files need to be compatible with LAMMPS source files. When a
make fails, there is usually a very simple reason, which you or a local expert will need to fix.
Here are two non-obvious errors that can occur:
(1) If the make command breaks immediately with errors that indicate it can't find files with a "*" in their
names, this can be because your machine's native make doesn't support wildcard expansion in a makefile. Try
gmake instead of make. If that doesn't work, try using a -f switch with your make command to use a
pre-generated Makefile.list which explicitly lists all the needed files, e.g.
make makelist
make -f Makefile.list linux
gmake -f Makefile.list mac
The first "make" command will create a current Makefile.list with all the file names in your src dir. The 2nd
"make" command (make or gmake) will use it to build LAMMPS. Note that you should include/exclude any
LAMMPS Users Manual
20

desired optional packages before using the "make makelist" command.
(2) If you get an error that says something like 'identifier "atoll" is undefined', then your machine does not
support "long long" integers. Try using the -DLAMMPS_LONGLONG_TO_LONG setting described above
in Step 4.
Additional build tips
Building LAMMPS for multiple platforms.
You can make LAMMPS for multiple platforms from the same src directory. Each target creates its own
object sub-directory called Obj_target where it stores the system-specific *.o files.
Cleaning up.
Typing "make clean-all" or "make clean-machine" will delete *.o object files created when LAMMPS is built,
for either all builds or for a particular machine.
Changing the LAMMPS size limits via -DLAMMPS_SMALLBIG or -DLAMMPS_BIGBIG or -DLAMMPS_SMALLSMALL
As explained above, any of these 3 settings can be specified on the LMP_INC line in your low-level
src/MAKE/Makefile.foo.
The default is -DLAMMPS_SMALLBIG which allows for systems with up to 2^63 atoms and 2^63 timesteps
(about 9e18). The atom limit is for atomic systems which do not store bond topology info and thus do not
require atom IDs. If you use atom IDs for atomic systems (which is the default) or if you use a molecular
model, which stores bond topology info and thus requires atom IDs, the limit is 2^31 atoms (about 2 billion).
This is because the IDs are stored in 32-bit integers.
Likewise, with this setting, the 3 image flags for each atom (see the dump doc page for a discussion) are
stored in a 32-bit integer, which means the atoms can only wrap around a periodic box (in each dimension) at
most 512 times. If atoms move through the periodic box more than this many times, the image flags will "roll
over", e.g. from 511 to -512, which can cause diagnostics like the mean-squared displacement, as calculated
by the compute msd command, to be faulty.
To allow for larger atomic systems with atom IDs or larger molecular systems or larger image flags, compile
with -DLAMMPS_BIGBIG. This stores atom IDs and image flags in 64-bit integers. This enables atomic or
molecular systems with atom IDS of up to 2^63 atoms (about 9e18). And image flags will not "roll over" until
they reach 2^20 = 1048576.
If your system does not support 8-byte integers, you will need to compile with the
-DLAMMPS_SMALLSMALL setting. This will restrict the total number of atoms (for atomic or molecular
systems) and timesteps to 2^31 (about 2 billion). Image flags will roll over at 2^9 = 512.
Note that in src/lmptype.h there are definitions of all these data types as well as the MPI data types associated
with them. The MPI types need to be consistent with the associated C data types, or else LAMMPS will
generate a run-time error. As far as we know, the settings defined in src/lmptype.h are portable and work on
every current system.
In all cases, the size of problem that can be run on a per-processor basis is limited by 4-byte integer storage to
2^31 atoms per processor (about 2 billion). This should not normally be a limitation since such a problem
would have a huge per-processor memory footprint due to neighbor lists and would run very slowly in terms
LAMMPS Users Manual
21

of CPU secs/timestep.
Building for a Mac
OS X is a derivative of BSD Unix, so it should just work. See the src/MAKE/MACHINES/Makefile.mac and
Makefile.mac_mpi files.
Building for Windows
If you want to build a Windows version of LAMMPS, you can build it yourself, but it may require some
effort. LAMMPS expects a Unix-like build environment for the default build procedure. This can be done
using either Cygwin or MinGW; the latter also exists as a ready-to-use Linux-to-Windows cross-compiler in
several Linux distributions. In these cases, you can do the installation after installing several unix-style
commands like make, grep, sed and bash with some shell utilities.
For Cygwin and the MinGW cross-compilers, suitable makefiles are provided in src/MAKE/MACHINES.
When using other compilers, like Visual C++ or Intel compilers for Windows, you may have to implement
your own build system. Since none of the current LAMMPS core developers has significant experience
building executables on Windows, we are happy to distribute contributed instructions and modifications, but
we cannot provide support for those.
With the so-called "Anniversary Update" to Windows 10, there is a Ubuntu Linux subsystem available for
Windows, that can be installed and then used to compile/install LAMMPS as if you are running on a Ubuntu
Linux system instead of Windows.
As an alternative, you can download "daily builds" (and some older versions) of the installer packages from
rpm.lammps.org/windows.html. These executables are built with most optional packages and the download
includes documentation, potential files, some tools and many examples, but no source code.
2.3 Making LAMMPS with optional packages
This section has the following sub-sections:
2.3.1 Package basics
2.3.2 Including/excluding packages
2.3.3 Packages that require extra libraries
2.3.4 Packages that require Makefile.machine settings
Note that the following Section 2.4 describes the Make.py tool which can be used to install/un-install
packages and build the auxiliary libraries which some of them use. It can also auto-edit a Makefile.machine to
add settings needed by some packages.
Package basics:
The source code for LAMMPS is structured as a set of core files which are always included, plus optional
packages. Packages are groups of files that enable a specific set of features. For example, force fields for
molecular systems or granular systems are in packages.
Section 4 in the manual has details about all the packages, including specific instructions for building
LAMMPS with each package, which are covered in a more general manner below.
LAMMPS Users Manual
22

You can see the list of all packages by typing "make package" from within the src directory of the LAMMPS
distribution. This also lists various make commands that can be used to manipulate packages.
If you use a command in a LAMMPS input script that is part of a package, you must have built LAMMPS
with that package, else you will get an error that the style is invalid or the command is unknown. Every
command's doc page specifies if it is part of a package. You can also type
lmp_machine -h
to run your executable with the optional -h command-line switch for "help", which will simply list the styles
and commands known to your executable, and immediately exit.
There are two kinds of packages in LAMMPS, standard and user packages. More information about the
contents of standard and user packages is given in Section 4 of the manual. The difference between standard
and user packages is as follows:
Standard packages, such as molecule or kspace, are supported by the LAMMPS developers and are written in
a syntax and style consistent with the rest of LAMMPS. This means we will answer questions about them,
debug and fix them if necessary, and keep them compatible with future changes to LAMMPS.
User packages, such as user-atc or user-omp, have been contributed by users, and always begin with the user
prefix. If they are a single command (single file), they are typically in the user-misc package. Otherwise, they
are a set of files grouped together which add a specific functionality to the code.
User packages don't necessarily meet the requirements of the standard packages. If you have problems using a
feature provided in a user package, you may need to contact the contributor directly to get help. Information
on how to submit additions you make to LAMMPS as single files or either a standard or user-contributed
package are given in this section of the documentation.
Including/excluding packages
To use (or not use) a package you must include it (or exclude it) before building LAMMPS. From the src
directory, this is typically as simple as:
make yes-colloid
make mpi
or
make no-manybody
make mpi
NOTE: You should NOT include/exclude packages and build LAMMPS in a single make command using
multiple targets, e.g. make yes-colloid mpi. This is because the make procedure creates a list of source files
that will be out-of-date for the build if the package configuration changes within the same command.
Some packages have individual files that depend on other packages being included. LAMMPS checks for this
and does the right thing. I.e. individual files are only included if their dependencies are already included.
Likewise, if a package is excluded, other files dependent on that package are also excluded.
If you will never run simulations that use the features in a particular packages, there is no reason to include it
in your build. For some packages, this will keep you from having to build auxiliary libraries (see below), and
LAMMPS Users Manual
23

will also produce a smaller executable which may run a bit faster.
When you download a LAMMPS tarball, these packages are pre-installed in the src directory: KSPACE,
MANYBODY,MOLECULE, because they are so commonly used. When you download LAMMPS source
files from the SVN or Git repositories, no packages are pre-installed.
Packages are included or excluded by typing "make yes-name" or "make no-name", where "name" is the name
of the package in lower-case, e.g. name = kspace for the KSPACE package or name = user-atc for the
USER-ATC package. You can also type "make yes-standard", "make no-standard", "make yes-std", "make
no-std", "make yes-user", "make no-user", "make yes-lib", "make no-lib", "make yes-all", or "make no-all" to
include/exclude various sets of packages. Type "make package" to see all of the package-related make
options.
NOTE: Inclusion/exclusion of a package works by simply moving files back and forth between the main src
directory and sub-directories with the package name (e.g. src/KSPACE, src/USER-ATC), so that the files are
seen or not seen when LAMMPS is built. After you have included or excluded a package, you must re-build
LAMMPS.
Additional package-related make options exist to help manage LAMMPS files that exist in both the src
directory and in package sub-directories. You do not normally need to use these commands unless you are
editing LAMMPS files or have downloaded a patch from the LAMMPS WWW site.
Typing "make package-update" or "make pu" will overwrite src files with files from the package
sub-directories if the package has been included. It should be used after a patch is installed, since patches only
update the files in the package sub-directory, but not the src files. Typing "make package-overwrite" will
overwrite files in the package sub-directories with src files.
Typing "make package-status" or "make ps" will show which packages are currently included. For those that
are included, it will list any files that are different in the src directory and package sub-directory. Typing
"make package-diff" lists all differences between these files. Again, type "make package" to see all of the
package-related make options.
Packages that require extra libraries
A few of the standard and user packages require additional auxiliary libraries. Many of them are provided
with LAMMPS, in which case they must be compiled first, before LAMMPS is built, if you wish to include
that package. If you get a LAMMPS build error about a missing library, this is likely the reason. See the
Section 4 doc page for a list of packages that have these kinds of auxiliary libraries.
The lib directory in the distribution has sub-directories with package names that correspond to the needed
auxiliary libs, e.g. lib/gpu. Each sub-directory has a README file that gives more details. Code for most of
the auxiliary libraries is included in that directory. Examples are the USER-ATC and MEAM packages.
A few of the lib sub-directories do not include code, but do include instructions (and sometimes scripts) that
automate the process of downloading the auxiliary library and installing it so LAMMPS can link to it.
Examples are the KIM, VORONOI, USER-MOLFILE, and USER-SMD packages.
The lib/python directory (for the PYTHON package) contains only a choice of Makefile.lammps.* files. This
is because no auxiliary code or libraries are needed, only the Python library and other system libs that should
already available on your system. However, the Makefile.lammps file is needed to tell LAMMPS which libs
to use and where to find them.
LAMMPS Users Manual
24

For libraries with provided code, the sub-directory README file (e.g. lib/atc/README) has instructions on
how to build that library. This information is also summarized in Section 4. Typically this is done by typing
something like:
make -f Makefile.g++
If one of the provided Makefiles is not appropriate for your system you will need to edit or add one. Note that
all the Makefiles have a setting for EXTRAMAKE at the top that specifies a Makefile.lammps.* file.
If the library build is successful, it will produce 2 files in the lib directory:
libpackage.a
Makefile.lammps
The Makefile.lammps file will typically be a copy of one of the Makefile.lammps.* files in the library
directory.
Note that you must insure that the settings in Makefile.lammps are appropriate for your system. If they are
not, the LAMMPS build may fail. To fix this, you can edit or create a new Makefile.lammps.* file for your
system, and copy it to Makefile.lammps.
As explained in the lib/package/README files, the settings in Makefile.lammps are used to specify
additional system libraries and their locations so that LAMMPS can build with the auxiliary library. For
example, if the MEAM package is used, the auxiliary library consists of F90 code, built with a Fortran
complier. To link that library with LAMMPS (a C++ code) via whatever C++ compiler LAMMPS is built
with, typically requires additional Fortran-to-C libraries be included in the link. Another example are the
BLAS and LAPACK libraries needed to use the USER-ATC or USER-AWPMD packages.
For libraries without provided code, the sub-directory README file has information on where to download
the library and how to build it, e.g. lib/voronoi/README and lib/smd/README. The README files also
describe how you must either (a) create soft links, via the "ln" command, in those directories to point to where
you built or installed the packages, or (b) check or edit the Makefile.lammps file in the same directory to
provide that information.
Some of the sub-directories, e.g. lib/voronoi, also have an install.py script which can be used to automate the
process of downloading/building/installing the auxiliary library, and setting the needed soft links. Type
"python install.py" for further instructions.
As with the sub-directories containing library code, if the soft links or settings in the
lib/package/Makefile.lammps files are not correct, the LAMMPS build will typically fail.
Packages that require Makefile.machine settings
A few packages require specific settings in Makefile.machine, to either build or use the package effectively.
These are the USER-INTEL, KOKKOS, USER-OMP, and OPT packages, used for accelerating code
performance on CPUs or other hardware, as discussed in Section 5.3.
A summary of what Makefile.machine changes are needed for each of these packages is given in Section 4.
The details are given on the doc pages that describe each of these accelerator packages in detail:
LAMMPS Users Manual
25

5.3.1 USER-INTEL package
5.3.3 KOKKOS package
5.3.4 USER-OMP package
5.3.5 OPT package
You can also look at the following machine Makefiles in src/MAKE/OPTIONS, which include the changes.
Note that the USER-INTEL and KOKKOS packages allow for settings that build LAMMPS for different
hardware. The USER-INTEL package builds for CPU and the Xeon Phi, the KOKKOS package builds for
OpenMP, GPUs (Cuda), and the Xeon Phi.
Makefile.intel_cpu• Makefile.intel_phi• Makefile.kokkos_omp• Makefile.kokkos_cuda• Makefile.kokkos_phi• Makefile.omp• Makefile.opt•
Also note that the Make.py tool, described in the next Section 2.4 can automatically add the needed info to an
existing machine Makefile, using simple command-line arguments.
2.4 Building LAMMPS via the Make.py tool
The src directory includes a Make.py script, written in Python, which can be used to automate various steps of
the build process. It is particularly useful for working with the accelerator packages, as well as other packages
which require auxiliary libraries to be built.
The goal of the Make.py tool is to allow any complex multi-step LAMMPS build to be performed as a single
Make.py command. And you can archive the commands, so they can be re-invoked later via the -r (redo)
switch. If you find some LAMMPS build procedure that can't be done in a single Make.py command, let the
developers know, and we'll see if we can augment the tool.
You can run Make.py from the src directory by typing either:
Make.py -h
python Make.py -h
which will give you help info about the tool. For the former to work, you may need to edit the first line of
Make.py to point to your local Python. And you may need to insure the script is executable:
chmod +x Make.py
Here are examples of build tasks you can perform with Make.py:
Install/uninstall packages Make.py -p no-lib kokkos omp intel
Build specific auxiliary libs Make.py -a lib-atc lib-meam
Build libs for all installed packages Make.py -p cuda gpu -gpu mode=double
arch=31 -a lib-all
Create a Makefile from scratch with compiler and MPI
settings Make.py -m none -cc g++ -mpi mpich -a file
LAMMPS Users Manual
26

Augment Makefile.serial with settings for installed packages Make.py -p intel -intel cpu -m serial -a file
Add JPG and FFTW support to Makefile.mpi Make.py -m mpi -jpg -fft fftw -a file
Build LAMMPS with a parallel make using Makefile.mpi Make.py -j 16 -m mpi -a exe
Build LAMMPS and libs it needs using Makefile.serial with
accelerator settings Make.py -p gpu intel -intel cpu -a lib-all file
serial
The bench and examples directories give Make.py commands that can be used to build LAMMPS with the
various packages and options needed to run all the benchmark and example input scripts. See these files for
more details:
bench/README• bench/FERMI/README• bench/KEPLER/README• bench/PHI/README• examples/README• examples/accelerate/README• examples/accelerate/make.list•
All of the Make.py options and syntax help can be accessed by using the "-h" switch.
E.g. typing "Make.py -h" gives
Syntax: Make.py switch args ...
switches can be listed in any order
help switch:
-h prints help and syntax for all other specified switches
switch for actions:
-a lib-all, lib-dir, clean, file, exe or machine
list one or more actions, in any order
machine is a Makefile.machine suffix, must be last if used
one-letter switches:
-d (dir), -j (jmake), -m (makefile), -o (output),
-p (packages), -r (redo), -s (settings), -v (verbose)
switches for libs:
-atc, -awpmd, -colvars, -cuda
-gpu, -meam, -poems, -qmmm, -reax
switches for build and makefile options:
-intel, -kokkos, -cc, -mpi, -fft, -jpg, -png
Using the "-h" switch with other switches and actions gives additional info on all the other specified switches
or actions. The "-h" can be anywhere in the command-line and the other switches do not need their arguments.
E.g. type "Make.py -h -d -atc -intel" will print:
-d dir
dir = LAMMPS home dir
if -d not specified, working dir must be lammps/src
-atc make=suffix lammps=suffix2
all args are optional and can be in any order
make = use Makefile.suffix (def = g++)
lammps = use Makefile.lammps.suffix2 (def = EXTRAMAKE in makefile)
-intel mode
mode = cpu or phi (def = cpu)
build Intel package for CPU or Xeon Phi
LAMMPS Users Manual
27

Note that Make.py never overwrites an existing Makefile.machine. Instead, it creates
src/MAKE/MINE/Makefile.auto, which you can save or rename if desired. Likewise it creates an executable
named src/lmp_auto, which you can rename using the -o switch if desired.
The most recently executed Make.py command is saved in src/Make.py.last. You can use the "-r" switch (for
redo) to re-invoke the last command, or you can save a sequence of one or more Make.py commands to a file
and invoke the file of commands using "-r". You can also label the commands in the file and invoke one or
more of them by name.
A typical use of Make.py is to start with a valid Makefile.machine for your system, that works for a vanilla
LAMMPS build, i.e. when optional packages are not installed. You can then use Make.py to add various
settings (FFT, JPG, PNG) to the Makefile.machine as well as change its compiler and MPI options. You can
also add additional packages to the build, as well as build the needed supporting libraries.
You can also use Make.py to create a new Makefile.machine from scratch, using the "-m none" switch, if you
also specify what compiler and MPI options to use, via the "-cc" and "-mpi" switches.
2.5 Building LAMMPS as a library
LAMMPS can be built as either a static or shared library, which can then be called from another application or
a scripting language. See this section for more info on coupling LAMMPS to other codes. See this section for
more info on wrapping and running LAMMPS from Python.
Static library
To build LAMMPS as a static library (*.a file on Linux), type
make foo mode=lib
where foo is the machine name. This kind of library is typically used to statically link a driver application to
LAMMPS, so that you can insure all dependencies are satisfied at compile time. This will use the ARCHIVE
and ARFLAGS settings in src/MAKE/Makefile.foo. The build will create the file liblammps_foo.a which
another application can link to. It will also create a soft link liblammps.a, which will point to the most recently
built static library.
Shared library
To build LAMMPS as a shared library (*.so file on Linux), which can be dynamically loaded, e.g. from
Python, type
make foo mode=shlib
where foo is the machine name. This kind of library is required when wrapping LAMMPS with Python; see
Section 11 for details. This will use the SHFLAGS and SHLIBFLAGS settings in src/MAKE/Makefile.foo
and perform the build in the directory Obj_shared_foo. This is so that each file can be compiled with the -fPIC
flag which is required for inclusion in a shared library. The build will create the file liblammps_foo.so which
another application can link to dynamically. It will also create a soft link liblammps.so, which will point to the
most recently built shared library. This is the file the Python wrapper loads by default.
Note that for a shared library to be usable by a calling program, all the auxiliary libraries it depends on must
also exist as shared libraries. This will be the case for libraries included with LAMMPS, such as the dummy
LAMMPS Users Manual
28
MPI library in src/STUBS or any package libraries in lib/packages, since they are always built as shared
libraries using the -fPIC switch. However, if a library like MPI or FFTW does not exist as a shared library, the
shared library build will generate an error. This means you will need to install a shared library version of the
auxiliary library. The build instructions for the library should tell you how to do this.
Here is an example of such errors when the system FFTW or provided lib/colvars library have not been built
as shared libraries:
/usr/bin/ld: /usr/local/lib/libfftw3.a(mapflags.o): relocation
R_X86_64_32 against '.rodata' can not be used when making a shared
object; recompile with -fPIC
/usr/local/lib/libfftw3.a: could not read symbols: Bad value
/usr/bin/ld: ../../lib/colvars/libcolvars.a(colvarmodule.o):
relocation R_X86_64_32 against '__pthread_key_create' can not be used
when making a shared object; recompile with -fPIC
../../lib/colvars/libcolvars.a: error adding symbols: Bad value
As an example, here is how to build and install the MPICH library, a popular open-source version of MPI,
distributed by Argonne National Labs, as a shared library in the default /usr/local/lib location:
./configure --enable-shared
make
make install
You may need to use "sudo make install" in place of the last line if you do not have write privileges for
/usr/local/lib. The end result should be the file /usr/local/lib/libmpich.so.
Additional requirement for using a shared library:
The operating system finds shared libraries to load at run-time using the environment variable
LD_LIBRARY_PATH. So you may wish to copy the file src/liblammps.so or src/liblammps_g++.so (for
example) to a place the system can find it by default, such as /usr/local/lib, or you may wish to add the
LAMMPS src directory to LD_LIBRARY_PATH, so that the current version of the shared library is always
available to programs that use it.
For the csh or tcsh shells, you would add something like this to your ~/.cshrc file:
setenv LD_LIBRARY_PATH ${LD_LIBRARY_PATH}:/home/sjplimp/lammps/src
Calling the LAMMPS library
Either flavor of library (static or shared) allows one or more LAMMPS objects to be instantiated from the
calling program.
When used from a C++ program, all of LAMMPS is wrapped in a LAMMPS_NS namespace; you can safely
use any of its classes and methods from within the calling code, as needed.
When used from a C or Fortran program or a scripting language like Python, the library has a simple
function-style interface, provided in src/library.cpp and src/library.h.
See the sample codes in examples/COUPLE/simple for examples of C++ and C and Fortran codes that invoke
LAMMPS thru its library interface. There are other examples as well in the COUPLE directory which are
LAMMPS Users Manual
29
discussed in Section 6.10 of the manual. See Section 11 of the manual for a description of the Python wrapper
provided with LAMMPS that operates through the LAMMPS library interface.
The files src/library.cpp and library.h define the C-style API for using LAMMPS as a library. See Section
6.19 of the manual for a description of the interface and how to extend it for your needs.
2.6 Running LAMMPS
By default, LAMMPS runs by reading commands from standard input. Thus if you run the LAMMPS
executable by itself, e.g.
lmp_linux
it will simply wait, expecting commands from the keyboard. Typically you should put commands in an input
script and use I/O redirection, e.g.
lmp_linux <in.file
For parallel environments this should also work. If it does not, use the '-in' command-line switch, e.g.
lmp_linux -in in.file
This section describes how input scripts are structured and what commands they contain.
You can test LAMMPS on any of the sample inputs provided in the examples or bench directory. Input scripts
are named in.* and sample outputs are named log.*.name.P where name is a machine and P is the number of
processors it was run on.
Here is how you might run a standard Lennard-Jones benchmark on a Linux box, using mpirun to launch a
parallel job:
cd src
make linux
cp lmp_linux ../bench
cd ../bench
mpirun -np 4 lmp_linux -in in.lj
See this page for timings for this and the other benchmarks on various platforms. Note that some of the
example scripts require LAMMPS to be built with one or more of its optional packages.
On a Windows box, you can skip making LAMMPS and simply download an installer package from here
For running the non-MPI executable, follow these steps:
Get a command prompt by going to Start->Run... , then typing "cmd".• Move to the directory where you have your input, e.g. a copy of the in.lj input from the bench folder.
(e.g. by typing: cd "Documents").
•
At the command prompt, type "lmp_serial -in in.lj", replacing in.lj with the name of your LAMMPS
input script.
•
For the MPI version, which allows you to run LAMMPS under Windows on multiple processors, follow these
steps:
LAMMPS Users Manual
30

Download and install MPICH2 for Windows.
The LAMMPS Windows installer packages will automatically adjust your path for the default
location of this MPI package. After the installation of the MPICH software, it needs to be integrated
into the system. For this you need to start a Command Prompt in Administrator Mode (right click on
the icon and select it). Change into the MPICH2 installation directory, then into the subdirectory bin
and execute smpd.exe -install. Exit the command window.
•
Get a new, regular command prompt by going to Start->Run... , then typing "cmd".• Move to the directory where you have your input file (e.g. by typing: cd "Documents").
Then type something like this:
mpiexec -localonly 4 lmp_mpi -in in.lj
or
mpiexec -np 4 lmp_mpi -in in.lj
•
replacing in.lj with the name of your LAMMPS input script. For the latter case, you may be prompted
to enter your password.
•
In this mode, output may not immediately show up on the screen, so if your input script takes a long
time to execute, you may need to be patient before the output shows up.
The parallel executable can also run on a single processor by typing something like:
lmp_mpi -in in.lj
•
The screen output from LAMMPS is described in a section below. As it runs, LAMMPS also writes a
log.lammps file with the same information.
Note that this sequence of commands copies the LAMMPS executable (lmp_linux) to the directory with the
input files. This may not be necessary, but some versions of MPI reset the working directory to where the
executable is, rather than leave it as the directory where you launch mpirun from (if you launch lmp_linux on
its own and not under mpirun). If that happens, LAMMPS will look for additional input files and write its
output files to the executable directory, rather than your working directory, which is probably not what you
want.
If LAMMPS encounters errors in the input script or while running a simulation it will print an ERROR
message and stop or a WARNING message and continue. See Section 12 for a discussion of the various kinds
of errors LAMMPS can or can't detect, a list of all ERROR and WARNING messages, and what to do about
them.
LAMMPS can run a problem on any number of processors, including a single processor. In theory you should
get identical answers on any number of processors and on any machine. In practice, numerical round-off can
cause slight differences and eventual divergence of molecular dynamics phase space trajectories.
LAMMPS can run as large a problem as will fit in the physical memory of one or more processors. If you run
out of memory, you must run on more processors or setup a smaller problem.
LAMMPS Users Manual
31
2.7 Command-line options
At run time, LAMMPS recognizes several optional command-line switches which may be used in any order.
Either the full word or a one-or-two letter abbreviation can be used:
-e or -echo• -h or -help• -i or -in• -k or -kokkos• -l or -log• -nc or -nocite• -pk or -package• -p or -partition• -pl or -plog• -ps or -pscreen• -r or -restart• -ro or -reorder• -sc or -screen• -sf or -suffix• -v or -var•
For example, lmp_ibm might be launched as follows:
mpirun -np 16 lmp_ibm -v f tmp.out -l my.log -sc none -in in.alloy
mpirun -np 16 lmp_ibm -var f tmp.out -log my.log -screen none -in in.alloy
Here are the details on the options:
-echo style
Set the style of command echoing. The style can be none or screen or log or both. Depending on the style,
each command read from the input script will be echoed to the screen and/or logfile. This can be useful to
figure out which line of your script is causing an input error. The default value is log. The echo style can also
be set by using the echo command in the input script itself.
-help
Print a brief help summary and a list of options compiled into this executable for each LAMMPS style
(atom_style, fix, compute, pair_style, bond_style, etc). This can tell you if the command you want to use was
included via the appropriate package at compile time. LAMMPS will print the info and immediately exit if
this switch is used.
-in file
Specify a file to use as an input script. This is an optional switch when running LAMMPS in one-partition
mode. If it is not specified, LAMMPS reads its script from standard input, typically from a script via I/O
redirection; e.g. lmp_linux < in.run. I/O redirection should also work in parallel, but if it does not (in the
unlikely case that an MPI implementation does not support it), then use the -in flag. Note that this is a
required switch when running LAMMPS in multi-partition mode, since multiple processors cannot all read
from stdin.
-kokkos on/off keyword/value ...
LAMMPS Users Manual
32
Explicitly enable or disable KOKKOS support, as provided by the KOKKOS package. Even if LAMMPS is
built with this package, as described above in Section 2.3, this switch must be set to enable running with the
KOKKOS-enabled styles the package provides. If the switch is not set (the default), LAMMPS will operate as
if the KOKKOS package were not installed; i.e. you can run standard LAMMPS or with the GPU or
USER-OMP packages, for testing or benchmarking purposes.
Additional optional keyword/value pairs can be specified which determine how Kokkos will use the
underlying hardware on your platform. These settings apply to each MPI task you launch via the "mpirun" or
"mpiexec" command. You may choose to run one or more MPI tasks per physical node. Note that if you are
running on a desktop machine, you typically have one physical node. On a cluster or supercomputer there may
be dozens or 1000s of physical nodes.
Either the full word or an abbreviation can be used for the keywords. Note that the keywords do not use a
leading minus sign. I.e. the keyword is "t", not "-t". Also note that each of the keywords has a default setting.
Example of when to use these options and what settings to use on different platforms is given in Section 5.3.
d or device• g or gpus• t or threads• n or numa•
device Nd
This option is only relevant if you built LAMMPS with CUDA=yes, you have more than one GPU per node,
and if you are running with only one MPI task per node. The Nd setting is the ID of the GPU on the node to
run on. By default Nd = 0. If you have multiple GPUs per node, they have consecutive IDs numbered as
0,1,2,etc. This setting allows you to launch multiple independent jobs on the node, each with a single MPI
task per node, and assign each job to run on a different GPU.
gpus Ng Ns
This option is only relevant if you built LAMMPS with CUDA=yes, you have more than one GPU per node,
and you are running with multiple MPI tasks per node (up to one per GPU). The Ng setting is how many
GPUs you will use. The Ns setting is optional. If set, it is the ID of a GPU to skip when assigning MPI tasks
to GPUs. This may be useful if your desktop system reserves one GPU to drive the screen and the rest are
intended for computational work like running LAMMPS. By default Ng = 1 and Ns is not set.
Depending on which flavor of MPI you are running, LAMMPS will look for one of these 3 environment
variables
SLURM_LOCALID (various MPI variants compiled with SLURM support)
MV2_COMM_WORLD_LOCAL_RANK (Mvapich)
OMPI_COMM_WORLD_LOCAL_RANK (OpenMPI)
which are initialized by the "srun", "mpirun" or "mpiexec" commands. The environment variable setting for
each MPI rank is used to assign a unique GPU ID to the MPI task.
threads Nt
This option assigns Nt number of threads to each MPI task for performing work when Kokkos is executing in
OpenMP or pthreads mode. The default is Nt = 1, which essentially runs in MPI-only mode. If there are Np
MPI tasks per physical node, you generally want Np*Nt = the number of physical cores per node, to use your
LAMMPS Users Manual
33
available hardware optimally. This also sets the number of threads used by the host when LAMMPS is
compiled with CUDA=yes.
numa Nm
This option is only relevant when using pthreads with hwloc support. In this case Nm defines the number of
NUMA regions (typically sockets) on a node which will be utilized by a single MPI rank. By default Nm = 1.
If this option is used the total number of worker-threads per MPI rank is threads*numa. Currently it is always
almost better to assign at least one MPI rank per NUMA region, and leave numa set to its default value of 1.
This is because letting a single process span multiple NUMA regions induces a significant amount of cross
NUMA data traffic which is slow.
-log file
Specify a log file for LAMMPS to write status information to. In one-partition mode, if the switch is not used,
LAMMPS writes to the file log.lammps. If this switch is used, LAMMPS writes to the specified file. In
multi-partition mode, if the switch is not used, a log.lammps file is created with hi-level status information.
Each partition also writes to a log.lammps.N file where N is the partition ID. If the switch is specified in
multi-partition mode, the hi-level logfile is named "file" and each partition also logs information to a file.N.
For both one-partition and multi-partition mode, if the specified file is "none", then no log files are created.
Using a log command in the input script will override this setting. Option -plog will override the name of the
partition log files file.N.
-nocite
Disable writing the log.cite file which is normally written to list references for specific cite-able features used
during a LAMMPS run. See the citation page for more details.
-package style args ....
Invoke the package command with style and args. The syntax is the same as if the command appeared at the
top of the input script. For example "-package gpu 2" or "-pk gpu 2" is the same as package gpu 2 in the input
script. The possible styles and args are documented on the package doc page. This switch can be used multiple
times, e.g. to set options for the USER-INTEL and USER-OMP packages which can be used together.
Along with the "-suffix" command-line switch, this is a convenient mechanism for invoking accelerator
packages and their options without having to edit an input script.
-partition 8x2 4 5 ...
Invoke LAMMPS in multi-partition mode. When LAMMPS is run on P processors and this switch is not used,
LAMMPS runs in one partition, i.e. all P processors run a single simulation. If this switch is used, the P
processors are split into separate partitions and each partition runs its own simulation. The arguments to the
switch specify the number of processors in each partition. Arguments of the form MxN mean M partitions,
each with N processors. Arguments of the form N mean a single partition with N processors. The sum of
processors in all partitions must equal P. Thus the command "-partition 8x2 4 5" has 10 partitions and runs on
a total of 25 processors.
Running with multiple partitions can e useful for running multi-replica simulations, where each replica runs
on on one or a few processors. Note that with MPI installed on a machine (e.g. your desktop), you can run on
more (virtual) processors than you have physical processors.
LAMMPS Users Manual
34
To run multiple independent simulations from one input script, using multiple partitions, see Section 6.4 of
the manual. World- and universe-style variables are useful in this context.
-plog file
Specify the base name for the partition log files, so partition N writes log information to file.N. If file is none,
then no partition log files are created. This overrides the filename specified in the -log command-line option.
This option is useful when working with large numbers of partitions, allowing the partition log files to be
suppressed (-plog none) or placed in a sub-directory (-plog replica_files/log.lammps) If this option is not used
the log file for partition N is log.lammps.N or whatever is specified by the -log command-line option.
-pscreen file
Specify the base name for the partition screen file, so partition N writes screen information to file.N. If file is
none, then no partition screen files are created. This overrides the filename specified in the -screen
command-line option. This option is useful when working with large numbers of partitions, allowing the
partition screen files to be suppressed (-pscreen none) or placed in a sub-directory (-pscreen
replica_files/screen). If this option is not used the screen file for partition N is screen.N or whatever is
specified by the -screen command-line option.
-restart restartfile remap datafile keyword value ...
Convert the restart file into a data file and immediately exit. This is the same operation as if the following
2-line input script were run:
read_restart restartfile remap
write_data datafile keyword value ...
Note that the specified restartfile and datafile can have wild-card characters ("*",%") as described by the
read_restart and write_data commands. But a filename such as file.* will need to be enclosed in quotes to
avoid shell expansion of the "*" character.
Note that following restartfile, the optional flag remap can be used. This has the same effect as adding it to the
read_restart command, as explained on its doc page. This is only useful if the reading of the restart file
triggers an error that atoms have been lost. In that case, use of the remap flag should allow the data file to still
be produced.
Also note that following datafile, the same optional keyword/value pairs can be listed as used by the
write_data command.
-reorder nth N
-reorder custom filename
Reorder the processors in the MPI communicator used to instantiate LAMMPS, in one of several ways. The
original MPI communicator ranks all P processors from 0 to P-1. The mapping of these ranks to physical
processors is done by MPI before LAMMPS begins. It may be useful in some cases to alter the rank order.
E.g. to insure that cores within each node are ranked in a desired order. Or when using the run_style
verlet/split command with 2 partitions to insure that a specific Kspace processor (in the 2nd partition) is
matched up with a specific set of processors in the 1st partition. See the Section 5 doc pages for more details.
If the keyword nth is used with a setting N, then it means every Nth processor will be moved to the end of the
ranking. This is useful when using the run_style verlet/split command with 2 partitions via the -partition
LAMMPS Users Manual
35
command-line switch. The first set of processors will be in the first partition, the 2nd set in the 2nd partition.
The -reorder command-line switch can alter this so that the 1st N procs in the 1st partition and one proc in the
2nd partition will be ordered consecutively, e.g. as the cores on one physical node. This can boost
performance. For example, if you use "-reorder nth 4" and "-partition 9 3" and you are running on 12
processors, the processors will be reordered from
0 1 2 3 4 5 6 7 8 9 10 11
to
0 1 2 4 5 6 8 9 10 3 7 11
so that the processors in each partition will be
0 1 2 4 5 6 8 9 10
3 7 11
See the "processors" command for how to insure processors from each partition could then be grouped
optimally for quad-core nodes.
If the keyword is custom, then a file that specifies a permutation of the processor ranks is also specified. The
format of the reorder file is as follows. Any number of initial blank or comment lines (starting with a "#"
character) can be present. These should be followed by P lines of the form:
I J
where P is the number of processors LAMMPS was launched with. Note that if running in multi-partition
mode (see the -partition switch above) P is the total number of processors in all partitions. The I and J values
describe a permutation of the P processors. Every I and J should be values from 0 to P-1 inclusive. In the set
of P I values, every proc ID should appear exactly once. Ditto for the set of P J values. A single I,J pairing
means that the physical processor with rank I in the original MPI communicator will have rank J in the
reordered communicator.
Note that rank ordering can also be specified by many MPI implementations, either by environment variables
that specify how to order physical processors, or by config files that specify what physical processors to
assign to each MPI rank. The -reorder switch simply gives you a portable way to do this without relying on
MPI itself. See the processors out command for how to output info on the final assignment of physical
processors to the LAMMPS simulation domain.
-screen file
Specify a file for LAMMPS to write its screen information to. In one-partition mode, if the switch is not used,
LAMMPS writes to the screen. If this switch is used, LAMMPS writes to the specified file instead and you
will see no screen output. In multi-partition mode, if the switch is not used, hi-level status information is
written to the screen. Each partition also writes to a screen.N file where N is the partition ID. If the switch is
specified in multi-partition mode, the hi-level screen dump is named "file" and each partition also writes
screen information to a file.N. For both one-partition and multi-partition mode, if the specified file is "none",
then no screen output is performed. Option -pscreen will override the name of the partition screen files file.N.
-suffix style args
LAMMPS Users Manual
36
Use variants of various styles if they exist. The specified style can be cuda, gpu, intel, kk, omp, opt, or hybrid.
These refer to optional packages that LAMMPS can be built with, as described above in Section 2.3. The
"gpu" style corresponds to the GPU package, the "intel" style to the USER-INTEL package, the "kk" style to
the KOKKOS package, the "opt" style to the OPT package, and the "omp" style to the USER-OMP package.
The hybrid style is the only style that accepts arguments. It allows for two packages to be specified. The first
package specified is the default and will be used if it is available. If no style is available for the first package,
the style for the second package will be used if available. For example, "-suffix hybrid intel omp" will use
styles from the USER-INTEL package if they are installed and available, but styles for the USER-OMP
package otherwise.
Along with the "-package" command-line switch, this is a convenient mechanism for invoking accelerator
packages and their options without having to edit an input script.
As an example, all of the packages provide a pair_style lj/cut variant, with style names lj/cut/gpu, lj/cut/intel,
lj/cut/kk, lj/cut/omp, and lj/cut/opt. A variant style can be specified explicitly in your input script, e.g.
pair_style lj/cut/gpu. If the -suffix switch is used the specified suffix (gpu,intel,kk,omp,opt) is automatically
appended whenever your input script command creates a new atom, pair, fix, compute, or run style. If the
variant version does not exist, the standard version is created.
For the GPU package, using this command-line switch also invokes the default GPU settings, as if the
command "package gpu 1" were used at the top of your input script. These settings can be changed by using
the "-package gpu" command-line switch or the package gpu command in your script.
For the USER-INTEL package, using this command-line switch also invokes the default USER-INTEL
settings, as if the command "package intel 1" were used at the top of your input script. These settings can be
changed by using the "-package intel" command-line switch or the package intel command in your script. If
the USER-OMP package is also installed, the hybrid style with "intel omp" arguments can be used to make
the omp suffix a second choice, if a requested style is not available in the USER-INTEL package. It will also
invoke the default USER-OMP settings, as if the command "package omp 0" were used at the top of your
input script. These settings can be changed by using the "-package omp" command-line switch or the package
omp command in your script.
For the KOKKOS package, using this command-line switch also invokes the default KOKKOS settings, as if
the command "package kokkos" were used at the top of your input script. These settings can be changed by
using the "-package kokkos" command-line switch or the package kokkos command in your script.
For the OMP package, using this command-line switch also invokes the default OMP settings, as if the
command "package omp 0" were used at the top of your input script. These settings can be changed by using
the "-package omp" command-line switch or the package omp command in your script.
The suffix command can also be used within an input script to set a suffix, or to turn off or back on any suffix
setting made via the command line.
-var name value1 value2 ...
Specify a variable that will be defined for substitution purposes when the input script is read. This switch can
be used multiple times to define multiple variables. "Name" is the variable name which can be a single
character (referenced as $x in the input script) or a full string (referenced as ${abc}). An index-style variable
will be created and populated with the subsequent values, e.g. a set of filenames. Using this command-line
option is equivalent to putting the line "variable name index value1 value2 ..." at the beginning of the input
script. Defining an index variable as a command-line argument overrides any setting for the same index
LAMMPS Users Manual
37

variable in the input script, since index variables cannot be re-defined. See the variable command for more
info on defining index and other kinds of variables and this section for more info on using variables in input
scripts.
NOTE: Currently, the command-line parser looks for arguments that start with "-" to indicate new switches.
Thus you cannot specify multiple variable values if any of they start with a "-", e.g. a negative numeric value.
It is OK if the first value1 starts with a "-", since it is automatically skipped.
2.8 LAMMPS screen output
As LAMMPS reads an input script, it prints information to both the screen and a log file about significant
actions it takes to setup a simulation. When the simulation is ready to begin, LAMMPS performs various
initializations and prints the amount of memory (in MBytes per processor) that the simulation requires. It also
prints details of the initial thermodynamic state of the system. During the run itself, thermodynamic
information is printed periodically, every few timesteps. When the run concludes, LAMMPS prints the final
thermodynamic state and a total run time for the simulation. It then appends statistics about the CPU time and
storage requirements for the simulation. An example set of statistics is shown here:
Loop time of 2.81192 on 4 procs for 300 steps with 2004 atoms
Performance: 18.436 ns/day 1.302 hours/ns 106.689 timesteps/s
97.0% CPU use with 4 MPI tasks x no OpenMP threads
MPI task timings breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 1.9808 | 2.0134 | 2.0318 | 1.4 | 71.60
Bond | 0.0021894 | 0.0060319 | 0.010058 | 4.7 | 0.21
Kspace | 0.3207 | 0.3366 | 0.36616 | 3.1 | 11.97
Neigh | 0.28411 | 0.28464 | 0.28516 | 0.1 | 10.12
Comm | 0.075732 | 0.077018 | 0.07883 | 0.4 | 2.74
Output | 0.00030518 | 0.00042665 | 0.00078821 | 1.0 | 0.02
Modify | 0.086606 | 0.086631 | 0.086668 | 0.0 | 3.08
Other | | 0.007178 | | | 0.26
Nlocal: 501 ave 508 max 490 min
Histogram: 1 0 0 0 0 0 1 1 0 1
Nghost: 6586.25 ave 6628 max 6548 min
Histogram: 1 0 1 0 0 0 1 0 0 1
Neighs: 177007 ave 180562 max 170212 min
Histogram: 1 0 0 0 0 0 0 1 1 1
Total # of neighbors = 708028
Ave neighs/atom = 353.307
Ave special neighs/atom = 2.34032
Neighbor list builds = 26
Dangerous builds = 0
The first section provides a global loop timing summary. The loop time is the total wall time for the section.
The Performance line is provided for convenience to help predicting the number of loop continuations
required and for comparing performance with other, similar MD codes. The CPU use line provides the CPU
utilization per MPI task; it should be close to 100% times the number of OpenMP threads (or 1 of no
OpenMP). Lower numbers correspond to delays due to file I/O or insufficient thread utilization.
The MPI task section gives the breakdown of the CPU run time (in seconds) into major categories:
LAMMPS Users Manual
38
Pair stands for all non-bonded force computation• Bond stands for bonded interactions: bonds, angles, dihedrals, impropers• Kspace stands for reciprocal space interactions: Ewald, PPPM, MSM• Neigh stands for neighbor list construction• Comm stands for communicating atoms and their properties• Output stands for writing dumps and thermo output• Modify stands for fixes and computes called by them• Other is the remaining time•
For each category, there is a breakdown of the least, average and most amount of wall time a processor spent
on this section. Also you have the variation from the average time. Together these numbers allow to gauge the
amount of load imbalance in this segment of the calculation. Ideally the difference between minimum,
maximum and average is small and thus the variation from the average close to zero. The final column shows
the percentage of the total loop time is spent in this section.
When using the timer full setting, an additional column is present that also prints the CPU utilization in
percent. In addition, when using timer full and the package omp command are active, a similar timing
summary of time spent in threaded regions to monitor thread utilization and load balance is provided. A new
entry is the Reduce section, which lists the time spent in reducing the per-thread data elements to the storage
for non-threaded computation. These thread timings are taking from the first MPI rank only and and thus, as
the breakdown for MPI tasks can change from MPI rank to MPI rank, this breakdown can be very different for
individual ranks. Here is an example output for this section:
Thread timings breakdown (MPI rank 0):
Total threaded time 0.6846 / 90.6%
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 0.5127 | 0.5147 | 0.5167 | 0.3 | 75.18
Bond | 0.0043139 | 0.0046779 | 0.0050418 | 0.5 | 0.68
Kspace | 0.070572 | 0.074541 | 0.07851 | 1.5 | 10.89
Neigh | 0.084778 | 0.086969 | 0.089161 | 0.7 | 12.70
Reduce | 0.0036485 | 0.003737 | 0.0038254 | 0.1 | 0.55
The third section lists the number of owned atoms (Nlocal), ghost atoms (Nghost), and pair-wise neighbors
stored per processor. The max and min values give the spread of these values across processors with a 10-bin
histogram showing the distribution. The total number of histogram counts is equal to the number of
processors.
The last section gives aggregate statistics for pair-wise neighbors and special neighbors that LAMMPS keeps
track of (see the special_bonds command). The number of times neighbor lists were rebuilt during the run is
given as well as the number of potentially "dangerous" rebuilds. If atom movement triggered neighbor list
rebuilding (see the neigh_modify command), then dangerous reneighborings are those that were triggered on
the first timestep atom movement was checked for. If this count is non-zero you may wish to reduce the delay
factor to insure no force interactions are missed by atoms moving beyond the neighbor skin distance before a
rebuild takes place.
If an energy minimization was performed via the minimize command, additional information is printed, e.g.
Minimization stats:
Stopping criterion = linesearch alpha is zero
Energy initial, next-to-last, final =
-6372.3765206 -8328.46998942 -8328.46998942
Force two-norm initial, final = 1059.36 5.36874
LAMMPS Users Manual
39

Force max component initial, final = 58.6026 1.46872
Final line search alpha, max atom move = 2.7842e-10 4.0892e-10
Iterations, force evaluations = 701 1516
The first line prints the criterion that determined the minimization to be completed. The third line lists the
initial and final energy, as well as the energy on the next-to-last iteration. The next 2 lines give a measure of
the gradient of the energy (force on all atoms). The 2-norm is the "length" of this force vector; the inf-norm is
the largest component. Then some information about the line search and statistics on how many iterations and
force-evaluations the minimizer required. Multiple force evaluations are typically done at each iteration to
perform a 1d line minimization in the search direction.
If a kspace_style long-range Coulombics solve was performed during the run (PPPM, Ewald), then additional
information is printed, e.g.
FFT time (% of Kspce) = 0.200313 (8.34477)
FFT Gflps 3d 1d-only = 2.31074 9.19989
The first line gives the time spent doing 3d FFTs (4 per timestep) and the fraction it represents of the total
KSpace time (listed above). Each 3d FFT requires computation (3 sets of 1d FFTs) and communication
(transposes). The total flops performed is 5Nlog_2(N), where N is the number of points in the 3d grid. The
FFTs are timed with and without the communication and a Gflop rate is computed. The 3d rate is with
communication; the 1d rate is without (just the 1d FFTs). Thus you can estimate what fraction of your FFT
time was spent in communication, roughly 75% in the example above.
2.9 Tips for users of previous LAMMPS versions
The current C++ began with a complete rewrite of LAMMPS 2001, which was written in F90. Features of
earlier versions of LAMMPS are listed in Section 13. The F90 and F77 versions (2001 and 99) are also freely
distributed as open-source codes; check the LAMMPS WWW Site for distribution information if you prefer
those versions. The 99 and 2001 versions are no longer under active development; they do not have all the
features of C++ LAMMPS.
If you are a previous user of LAMMPS 2001, these are the most significant changes you will notice in C++
LAMMPS:
(1) The names and arguments of many input script commands have changed. All commands are now a single
word (e.g. read_data instead of read data).
(2) All the functionality of LAMMPS 2001 is included in C++ LAMMPS, but you may need to specify the
relevant commands in different ways.
(3) The format of the data file can be streamlined for some problems. See the read_data command for details.
The data file section "Nonbond Coeff" has been renamed to "Pair Coeff" in C++ LAMMPS.
(4) Binary restart files written by LAMMPS 2001 cannot be read by C++ LAMMPS with a read_restart
command. This is because they were output by F90 which writes in a different binary format than C or C++
writes or reads. Use the restart2data tool provided with LAMMPS 2001 to convert the 2001 restart file to a
text data file. Then edit the data file as necessary before using the C++ LAMMPS read_data command to read
it in.
(5) There are numerous small numerical changes in C++ LAMMPS that mean you will not get identical
LAMMPS Users Manual
40
answers when comparing to a 2001 run. However, your initial thermodynamic energy and MD trajectory
should be close if you have setup the problem for both codes the same.
LAMMPS Users Manual
41

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
3. Commands
This section describes how a LAMMPS input script is formatted and the input script commands used to define
a LAMMPS simulation.
3.1 LAMMPS input script
3.2 Parsing rules
3.3 Input script structure
3.4 Commands listed by category
3.5 Commands listed alphabetically
3.1 LAMMPS input script
LAMMPS executes by reading commands from a input script (text file), one line at a time. When the input
script ends, LAMMPS exits. Each command causes LAMMPS to take some action. It may set an internal
variable, read in a file, or run a simulation. Most commands have default settings, which means you only need
to use the command if you wish to change the default.
In many cases, the ordering of commands in an input script is not important. However the following rules
apply:
(1) LAMMPS does not read your entire input script and then perform a simulation with all the settings.
Rather, the input script is read one line at a time and each command takes effect when it is read. Thus this
sequence of commands:
timestep 0.5
run 100
run 100
does something different than this sequence:
run 100
timestep 0.5
run 100
In the first case, the specified timestep (0.5 fmsec) is used for two simulations of 100 timesteps each. In the
2nd case, the default timestep (1.0 fmsec) is used for the 1st 100 step simulation and a 0.5 fmsec timestep is
used for the 2nd one.
(2) Some commands are only valid when they follow other commands. For example you cannot set the
temperature of a group of atoms until atoms have been defined and a group command is used to define which
atoms belong to the group.
(3) Sometimes command B will use values that can be set by command A. This means command A must
precede command B in the input script if it is to have the desired effect. For example, the read_data command
initializes the system by setting up the simulation box and assigning atoms to processors. If default values are
not desired, the processors and boundary commands need to be used before read_data to tell LAMMPS how
LAMMPS Users Manual
42

to map processors to the simulation box.
Many input script errors are detected by LAMMPS and an ERROR or WARNING message is printed. This
section gives more information on what errors mean. The documentation for each command lists restrictions
on how the command can be used.
3.2 Parsing rules
Each non-blank line in the input script is treated as a command. LAMMPS commands are case sensitive.
Command names are lower-case, as are specified command arguments. Upper case letters may be used in file
names or user-chosen ID strings.
Here is how each line in the input script is parsed by LAMMPS:
(1) If the last printable character on the line is a "&" character, the command is assumed to continue on the
next line. The next line is concatenated to the previous line by removing the "&" character and line break.
This allows long commands to be continued across two or more lines. See the discussion of triple quotes in
(6) for how to continue a command across multiple line without using "&" characters.
(2) All characters from the first "#" character onward are treated as comment and discarded. See an exception
in (6). Note that a comment after a trailing "&" character will prevent the command from continuing on the
next line. Also note that for multi-line commands a single leading "#" will comment out the entire command.
(3) The line is searched repeatedly for $ characters, which indicate variables that are replaced with a text
string. See an exception in (6).
If the $ is followed by curly brackets, then the variable name is the text inside the curly brackets. If no curly
brackets follow the $, then the variable name is the single character immediately following the $. Thus
${myTemp} and $x refer to variable names "myTemp" and "x".
How the variable is converted to a text string depends on what style of variable it is; see the variable doc page
for details. It can be a variable that stores multiple text strings, and return one of them. The returned text string
can be multiple "words" (space separated) which will then be interpreted as multiple arguments in the input
command. The variable can also store a numeric formula which will be evaluated and its numeric result
returned as a string.
As a special case, if the $ is followed by parenthesis, then the text inside the parenthesis is treated as an
"immediate" variable and evaluated as an equal-style variable. This is a way to use numeric formulas in an
input script without having to assign them to variable names. For example, these 3 input script lines:
variable X equal (xlo+xhi)/2+sqrt(v_area)
region 1 block $X 2 INF INF EDGE EDGE
variable X delete
can be replaced by
region 1 block $((xlo+xhi)/2+sqrt(v_area)) 2 INF INF EDGE EDGE
so that you do not have to define (or discard) a temporary variable X.
LAMMPS Users Manual
43

Note that neither the curly-bracket or immediate form of variables can contain nested $ characters for other
variables to substitute for. Thus you cannot do this:
variable a equal 2
variable b2 equal 4
print "B2 = ${b$a}"
Nor can you specify this $($x-1.0) for an immediate variable, but you could use $(v_x-1.0), since the latter is
valid syntax for an equal-style variable.
See the variable command for more details of how strings are assigned to variables and evaluated, and how
they can be used in input script commands.
(4) The line is broken into "words" separated by whitespace (tabs, spaces). Note that words can thus contain
letters, digits, underscores, or punctuation characters.
(5) The first word is the command name. All successive words in the line are arguments.
(6) If you want text with spaces to be treated as a single argument, it can be enclosed in either single or double
or triple quotes. A long single argument enclosed in single or double quotes can span multiple lines if the "&"
character is used, as described above. When the lines are concatenated together (and the "&" characters and
line breaks removed), the text will become a single line. If you want multiple lines of an argument to retain
their line breaks, the text can be enclosed in triple quotes, in which case "&" characters are not needed. For
example:
print "Volume = $v"
print 'Volume = $v'
if "${steps} > 1000" then quit
variable a string "red green blue &
purple orange cyan"
print """
System volume = $v
System temperature = $t
"""
In each case, the single, double, or triple quotes are removed when the single argument they enclose is stored
internally.
See the dump modify format, print, if, and python commands for examples.
A "#" or "$" character that is between quotes will not be treated as a comment indicator in (2) or substituted
for as a variable in (3).
NOTE: If the argument is itself a command that requires a quoted argument (e.g. using a print command as
part of an if or run every command), then single, double, or triple quotes can be nested in the usual manner.
See the doc pages for those commands for examples. Only one of level of nesting is allowed, but that should
be sufficient for most use cases.
3.3 Input script structure
This section describes the structure of a typical LAMMPS input script. The "examples" directory in the
LAMMPS distribution contains many sample input scripts; the corresponding problems are discussed in
Section 7, and animated on the LAMMPS WWW Site.
LAMMPS Users Manual
44
A LAMMPS input script typically has 4 parts:
Initialization1. Atom definition2. Settings3. Run a simulation4.
The last 2 parts can be repeated as many times as desired. I.e. run a simulation, change some settings, run
some more, etc. Each of the 4 parts is now described in more detail. Remember that almost all the commands
need only be used if a non-default value is desired.
(1) Initialization
Set parameters that need to be defined before atoms are created or read-in from a file.
The relevant commands are units, dimension, newton, processors, boundary, atom_style, atom_modify.
If force-field parameters appear in the files that will be read, these commands tell LAMMPS what kinds of
force fields are being used: pair_style, bond_style, angle_style, dihedral_style, improper_style.
(2) Atom definition
There are 3 ways to define atoms in LAMMPS. Read them in from a data or restart file via the read_data or
read_restart commands. These files can contain molecular topology information. Or create atoms on a lattice
(with no molecular topology), using these commands: lattice, region, create_box, create_atoms. The entire set
of atoms can be duplicated to make a larger simulation using the replicate command.
(3) Settings
Once atoms and molecular topology are defined, a variety of settings can be specified: force field coefficients,
simulation parameters, output options, etc.
Force field coefficients are set by these commands (they can also be set in the read-in files): pair_coeff,
bond_coeff, angle_coeff, dihedral_coeff, improper_coeff, kspace_style, dielectric, special_bonds.
Various simulation parameters are set by these commands: neighbor, neigh_modify, group, timestep,
reset_timestep, run_style, min_style, min_modify.
Fixes impose a variety of boundary conditions, time integration, and diagnostic options. The fix command
comes in many flavors.
Various computations can be specified for execution during a simulation using the compute, compute_modify,
and variable commands.
Output options are set by the thermo, dump, and restart commands.
(4) Run a simulation
A molecular dynamics simulation is run using the run command. Energy minimization (molecular statics) is
performed using the minimize command. A parallel tempering (replica-exchange) simulation can be run using
the temper command.
LAMMPS Users Manual
45

3.4 Commands listed by category
This section lists core LAMMPS commands, grouped by category. The next section lists all commands
alphabetically. The next section also includes (long) lists of style options for entries that appear in the
following categories as a single command (fix, compute, pair, etc). Commands that are added by user
packages are not included in the categories here, but they are in the next section.
Initialization:
newton, package, processors, suffix, units
Setup simulation box:
boundary, box, change_box, create_box, dimension, lattice, region
Setup atoms:
atom_modify, atom_style, balance, create_atoms, create_bonds, delete_atoms, delete_bonds, displace_atoms,
group, mass, molecule, read_data, read_dump, read_restart, replicate, set, velocity
Force fields:
angle_coeff, angle_style, bond_coeff, bond_style, bond_write, dielectric, dihedral_coeff, dihedral_style,
improper_coeff, improper_style, kspace_modify, kspace_style, pair_coeff, pair_modify, pair_style,
pair_write, special_bonds
Settings:
comm_modify, comm_style, info, min_modify, min_style, neigh_modify, neighbor, partition, reset_timestep,
run_style, timer, timestep
Operations within timestepping (fixes) and diagnostics (computes):
compute, compute_modify, fix, fix_modify, uncompute, unfix
Output:
dump image, dump movie, dump, dump_modify, restart, thermo, thermo_modify, thermo_style, undump,
write_coeff, write_data, write_dump, write_restart
Actions:
minimize, neb, prd, rerun, run, tad, temper
Input script control:
clear, echo, if, include, jump, label, log, next, print, python, quit, shell, variable
LAMMPS Users Manual
46
3.5 Individual commands
This section lists all LAMMPS commands alphabetically, with a separate listing below of styles within certain
commands. The previous section lists the same commands, grouped by category. Note that some style options
for some commands are part of specific LAMMPS packages, which means they cannot be used unless the
package was included when LAMMPS was built. Not all packages are included in a default LAMMPS build.
These dependencies are listed as Restrictions in the command's documentation.
angle_coeff angle_style atom_modify atom_style balance bond_coeff
bond_style bond_write boundary box change_box clear
comm_modify comm_style compute compute_modify create_atoms create_bonds
create_box delete_atoms delete_bonds dielectric dihedral_coeff dihedral_style
dimension displace_atoms dump dump image dump_modify dump movie
echo fix fix_modify group if info
improper_coeff improper_style include jump kspace_modify kspace_style
label lattice log mass minimize min_modify
min_style molecule neb neigh_modify neighbor newton
next package pair_coeff pair_modify pair_style pair_write
partition prd print processors python quit
read_data read_dump read_restart region replicate rerun
reset_timestep restart run run_style set shell
special_bonds suffix tad temper thermo thermo_modify
thermo_style timer timestep uncompute undump unfix
units variable velocity write_coeff write_data write_dump
write_restart
These are additional commands in USER packages, which can be used if LAMMPS is built with the
appropriate package.
dump custom/vtk dump nc dump nc/mpiio
group2ndx ndx2group temper/grem
Fix styles
See the fix command for one-line descriptions of each style or click on the style itself for a full description.
Some of the styles have accelerated versions, which can be used if LAMMPS is built with the appropriate
accelerated package. This is indicated by additional letters in parenthesis: g = GPU, i = USER-INTEL, k =
KOKKOS, o = USER-OMP, t = OPT.
adapt addforce append/atoms atom/swap aveforce ave/atom ave/chunk ave/correlate
ave/histo ave/histo/weight ave/time balance bond/break bond/create bond/swap box/relax
cmap controller deform (k) deposit drag dt/reset efield ehex
enforce2d evaporate external freeze gcmc gld gravity (o) halt
heat indent langevin (k) lineforce momentum (k) move mscg msst
neb nph (ko) nphug (o) nph/asphere (o) nph/body nph/sphere (o) npt (kio) npt/asphere
(o)
LAMMPS Users Manual
47
npt/body npt/sphere (o) nve (kio) nve/asphere (i) nve/asphere/noforce nve/body nve/limit nve/line
nve/noforce nve/sphere (o) nve/tri nvt (iko) nvt/asphere (o) nvt/body nvt/sllod (io) nvt/sphere
(o)
oneway orient/bcc orient/fcc planeforce poems pour press/berendsen print
property/atom qeq/comb (o) qeq/dynamic qeq/fire qeq/point qeq/shielded qeq/slater rattle
reax/bonds recenter restrain rigid (o) rigid/nph (o) rigid/npt (o) rigid/nve (o) rigid/nvt (o)
rigid/small (o) rigid/small/nph
(o) rigid/small/npt
(o) rigid/small/nve (o) rigid/small/nvt (o) setforce (k) shake spring
spring/chunk spring/rg spring/self srd store/force store/state temp/berendsen temp/csld
temp/csvr temp/rescale tfmc thermal/conductivity tmd ttm tune/kspace vector
viscosity viscous wall/colloid wall/gran wall/gran/region wall/harmonic wall/lj1043 wall/lj126
wall/lj93 wall/piston wall/reflect
(k) wall/region wall/srd
These are additional fix styles in USER packages, which can be used if LAMMPS is built with the appropriate
package.
adapt/fep addtorque atc ave/correlate/long colvars dpd/energy
drude drude/transform/direct drude/transform/reverse eos/cv eos/table eos/table/rx
filter/corotate flow/gauss gle grem imd ipi
langevin/drude langevin/eff lb/fluid lb/momentum lb/pc lb/rigid/pc/sphere
lb/viscous meso manifoldforce meso/stationary nve/dot nve/dotc/langevin
nve/manifold/rattle nvk nvt/manifold/rattle nph/eff npt/eff nve/eff
nvt/eff nvt/sllod/eff phonon pimd qbmsst qeq/reax
qmmm qtb reax/c/bonds reax/c/species rx saed/vtk
shardlow smd smd/adjust/dt smd/integrate/tlsph smd/integrate/ulsph smd/move/triangulated/surface
smd/setvel smd/wall/surface temp/rescale/eff ti/spring ttm/mod
Compute styles
See the compute command for one-line descriptions of each style or click on the style itself for a full
description. Some of the styles have accelerated versions, which can be used if LAMMPS is built with the
appropriate accelerated package. This is indicated by additional letters in parenthesis: g = GPU, i =
USER-INTEL, k = KOKKOS, o = USER-OMP, t = OPT.
angle angle/local angmom/chunk body/local bond bond/local
centro/atom chunk/atom cluster/atom cna/atom com com/chunk
contact/atom coord/atom damage/atom dihedral dihedral/local dilatation/atom
dipole/chunk displace/atom erotate/asphere erotate/rigid erotate/sphere erotate/sphere/atom
event/displace global/atom group/group gyration gyration/chunk heat/flux
hexorder/atom improper improper/local inertia/chunk ke ke/atom
ke/rigid msd msd/chunk msd/nongauss omega/chunk orientorder/atom
pair pair/local pe pe/atom plasticity/atom pressure
property/atom property/local property/chunk rdf reduce reduce/region
rigid/local slice sna/atom snad/atom snav/atom stress/atom
LAMMPS Users Manual
48

temp (k) temp/asphere temp/body temp/chunk temp/com temp/deform
temp/partial temp/profile temp/ramp temp/region temp/sphere ti
torque/chunk vacf vcm/chunk voronoi/atom
These are additional compute styles in USER packages, which can be used if LAMMPS is built with the
appropriate package.
ackland/atom basal/atom dpd dpd/atom fep force/tally
heat/flux/tally ke/eff ke/atom/eff meso/e/atom meso/rho/atom meso/t/atom
pe/tally pe/mol/tally saed smd/contact/radius smd/damage smd/hourglass/error
smd/internal/energy smd/plastic/strain smd/plastic/strain/rate smd/rho smd/tlsph/defgrad smd/tlsph/dt
smd/tlsph/num/neighs smd/tlsph/shape smd/tlsph/strain smd/tlsph/strain/rate smd/tlsph/stress smd/triangle/mesh/vertices
smd/ulsph/num/neighs smd/ulsph/strain smd/ulsph/strain/rate smd/ulsph/stress smd/vol stress/tally
temp/drude temp/eff temp/deform/eff temp/region/eff temp/rotate xrd
Pair_style potentials
See the pair_style command for an overview of pair potentials. Click on the style itself for a full description.
Many of the styles have accelerated versions, which can be used if LAMMPS is built with the appropriate
accelerated package. This is indicated by additional letters in parenthesis: g = GPU, i = USER-INTEL, k =
KOKKOS, o = USER-OMP, t = OPT.
none zero hybrid hybrid/overlay
adp (o) airebo (o) airebo/morse (o) beck (go)
body bop born (go) born/coul/dsf
born/coul/dsf/cs born/coul/long (go) born/coul/long/cs born/coul/msm (o)
born/coul/wolf (go) brownian (o) brownian/poly (o) buck (gkio)
buck/coul/cut (gkio) buck/coul/long (gkio) buck/coul/long/cs buck/coul/msm (o)
buck/long/coul/long (o) colloid (go) comb (o) comb3
coul/cut (gko) coul/debye (gko) coul/dsf (gko) coul/long (gko)
coul/long/cs coul/msm coul/streitz coul/wolf (ko)
dpd (go) dpd/tstat (go) dsmc eam (gkiot)
eam/alloy (gkot) eam/fs (gkot) eim (o) gauss (go)
gayberne (gio) gran/hertz/history (o) gran/hooke (o) gran/hooke/history
(o)
hbond/dreiding/lj (o) hbond/dreiding/morse (o) kim lcbop
line/lj lj/charmm/coul/charmm (ko) lj/charmm/coul/charmm/implicit
(ko) lj/charmm/coul/long
(giko)
lj/charmm/coul/msm lj/charmmfsw/coul/charmmfsh lj/charmmfsw/coul/long lj/class2 (gko)
lj/class2/coul/cut (ko) lj/class2/coul/long (gko) lj/cubic (go) lj/cut (gikot)
lj/cut/coul/cut (gko) lj/cut/coul/debye (gko) lj/cut/coul/dsf (gko) lj/cut/coul/long
(gikot)
lj/cut/coul/long/cs lj/cut/coul/msm (go) lj/cut/dipole/cut (go) lj/cut/dipole/long
lj/cut/tip4p/cut (o) lj/cut/tip4p/long (ot) lj/expand (gko) lj/gromacs (gko)
lj/long/coul/long (o) lj/long/dipole/long lj/long/tip4p/long
LAMMPS Users Manual
49
lj/gromacs/coul/gromacs
(ko)
lj/smooth (o) lj/smooth/linear (o) lj96/cut (go) lubricate (o)
lubricate/poly (o) lubricateU lubricateU/poly meam
mie/cut (o) morse (gkot) nb3b/harmonic (o) nm/cut (o)
nm/cut/coul/cut (o) nm/cut/coul/long (o) peri/eps peri/lps (o)
peri/pmb (o) peri/ves polymorphic reax
rebo (o) resquared (go) snap soft (go)
sw (gkio) table (gko) tersoff (gkio) tersoff/mod (gko)
tersoff/mod/c (o) tersoff/zbl (gko) tip4p/cut (o) tip4p/long (o)
tri/lj vashishta (ko) vashishta/table (o) yukawa (go)
yukawa/colloid (go) zbl (go)
These are additional pair styles in USER packages, which can be used if LAMMPS is built with the
appropriate package.
agni (o) awpmd/cut buck/mdf coul/cut/soft (o)
coul/diel (o) coul/long/soft (o) dpd/fdt dpd/fdt/energy
eam/cd (o) edip (o) eff/cut exp6/rx
gauss/cut kolmogorov/crespi/z lennard/mdf list
lj/charmm/coul/long/soft (o) lj/cut/coul/cut/soft (o) lj/cut/coul/long/soft (o) lj/cut/dipole/sf (go)
lj/cut/soft (o) lj/cut/thole/long (o) lj/cut/tip4p/long/soft (o) lj/mdf
lj/sdk (gko) lj/sdk/coul/long (go) lj/sdk/coul/msm (o) lj/sf (o)
meam/spline (o) meam/sw/spline mgpt momb
morse/smooth/linear morse/soft multi/lucy multi/lucy/rx
oxdna/coaxstk oxdna/excv oxdna/hbond oxdna/stk
oxdna/xstk oxdna2/coaxstk oxdna2/dh oxdna2/excv
oxdna2/stk quip reax/c (k) smd/hertz
smd/tlsph smd/triangulated/surface smd/ulsph smtbq
sph/heatconduction sph/idealgas sph/lj sph/rhosum
sph/taitwater sph/taitwater/morris srp table/rx
tersoff/table (o) thole tip4p/long/soft (o)
Bond_style potentials
See the bond_style command for an overview of bond potentials. Click on the style itself for a full
description. Some of the styles have accelerated versions, which can be used if LAMMPS is built with the
appropriate accelerated package. This is indicated by additional letters in parenthesis: g = GPU, i =
USER-INTEL, k = KOKKOS, o = USER-OMP, t = OPT.
none zero hybrid class2 (ko)
fene (iko) fene/expand (o) harmonic (ko) morse (o)
nonlinear (o) quartic (o) table (o)
These are additional bond styles in USER packages, which can be used if LAMMPS is built with the
appropriate package.
LAMMPS Users Manual
50
harmonic/shift (o) harmonic/shift/cut (o) oxdna/fene oxdna2/fene
Angle_style potentials
See the angle_style command for an overview of angle potentials. Click on the style itself for a full
description. Some of the styles have accelerated versions, which can be used if LAMMPS is built with the
appropriate accelerated package. This is indicated by additional letters in parenthesis: g = GPU, i =
USER-INTEL, k = KOKKOS, o = USER-OMP, t = OPT.
none zero hybrid charmm (ko)
class2 (ko) cosine (o) cosine/delta (o) cosine/periodic (o)
cosine/squared (o) harmonic (iko) table (o)
These are additional angle styles in USER packages, which can be used if LAMMPS is built with the
appropriate package.
cosine/shift (o) cosine/shift/exp (o) dipole (o) fourier (o)
fourier/simple (o) quartic (o) sdk
Dihedral_style potentials
See the dihedral_style command for an overview of dihedral potentials. Click on the style itself for a full
description. Some of the styles have accelerated versions, which can be used if LAMMPS is built with the
appropriate accelerated package. This is indicated by additional letters in parenthesis: g = GPU, i =
USER-INTEL, k = KOKKOS, o = USER-OMP, t = OPT.
none zero hybrid charmm (ko)
charmmfsh class2 (ko) harmonic (io) helix (o)
multi/harmonic (o) opls (iko)
These are additional dihedral styles in USER packages, which can be used if LAMMPS is built with the
appropriate package.
cosine/shift/exp (o) fourier (o) nharmonic (o) quadratic (o)
spherical (o) table (o)
Improper_style potentials
See the improper_style command for an overview of improper potentials. Click on the style itself for a full
description. Some of the styles have accelerated versions, which can be used if LAMMPS is built with the
appropriate accelerated package. This is indicated by additional letters in parenthesis: g = GPU, i =
USER-INTEL, k = KOKKOS, o = USER-OMP, t = OPT.
none zero hybrid class2
(ko)
cvff (io) harmonic (ko) umbrella (o)
These are additional improper styles in USER packages, which can be used if LAMMPS is built with the
appropriate package.
LAMMPS Users Manual
51
cossq (o) distance fourier (o) ring (o)
Kspace solvers
See the kspace_style command for an overview of Kspace solvers. Click on the style itself for a full
description. Some of the styles have accelerated versions, which can be used if LAMMPS is built with the
appropriate accelerated package. This is indicated by additional letters in parenthesis: g = GPU, i =
USER-INTEL, k = KOKKOS, o = USER-OMP, t = OPT.
ewald (o) ewald/disp msm (o) msm/cg (o)
pppm (go) pppm/cg (o) pppm/disp pppm/disp/tip4p
pppm/stagger pppm/tip4p (o)
LAMMPS Users Manual
52

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
4. Packages
This section gives an overview of the add-on optional packages that extend LAMMPS functionality. Packages
are groups of files that enable a specific set of features. For example, force fields for molecular systems or
granular systems are in packages. You can see the list of all packages by typing "make package" from within
the src directory of the LAMMPS distribution.
Here are links for two tables below, which list standard and user packages.
4.1 Standard packages
4.2 User packages
Section 2.3 of the manual describes the difference between standard packages and user packages. It also has
general details on how to include/exclude specific packages as part of the LAMMPS build process, and on
how to build auxiliary libraries or modify a machine Makefile if a package requires it.
Following the two tables below, is a sub-section for each package. It has a summary of what the package
contains. It has specific instructions on how to install it, build or obtain any auxiliary library it requires, and
any Makefile.machine changes it requires. It also lists pointers to examples of its use or documentation
provided in the LAMMPS distribution. If you want to know the complete list of commands that a package
adds to LAMMPS, simply list the files in its directory, e.g. "ls src/GRANULAR". Source files with names
that start with compute, fix, pair, bond, etc correspond to command styles with the same names.
NOTE: The USER package sub-sections below are still being filled in, as of March 2016.
Unless otherwise noted below, every package is independent of all the others. I.e. any package can be
included or excluded in a LAMMPS build, independent of all other packages. However, note that some
packages include commands derived from commands in other packages. If the other package is not installed,
the derived command from the new package will also not be installed when you include the new one. E.g. the
pair lj/cut/coul/long/omp command from the USER-OMP package will not be installed as part of the
USER-OMP package if the KSPACE package is not also installed, since it contains the pair lj/cut/coul/long
command. If you later install the KSPACE package and the USER-OMP package is already installed, both the
pair lj/cut/coul/long and lj/cut/coul/long/omp commands will be installed.
4.1 Standard packages
The current list of standard packages is as follows. Each package name links to a sub-section below with more
details.
Package Description Author(s) Doc page Example Library
ASPHERE aspherical particles - Section 6.6.14 ellipse -
BODY body-style particles - body body -
CLASS2 class 2 force fields - pair_style
lj/class2 - -
COLLOID colloidal particles Kumar (1) atom_style
colloid colloid -
COMPRESS I/O compression dump */gz - -
LAMMPS Users Manual
53
Axel Kohlmeyer (Temple
U)
CORESHELL adiabatic core/shell model Hendrik Heenen
(Technical U of Munich) Section 6.6.25 coreshell -
DIPOLE point dipole particles - pair_style
dipole/cut dipole -
GPU GPU-enabled styles Mike Brown (ORNL) Section 5.3.1 gpu lib/gpu
GRANULAR granular systems - Section 6.6.6 pour -
KIM openKIM potentials Smirichinski & Elliot &
Tadmor (3) pair_style kim kim KIM
KOKKOS Kokkos-enabled styles Trott & Moore (4) Section 5.3.3 kokkos lib/kokkos
KSPACE long-range Coulombic
solvers -kspace_style peptide -
MANYBODY many-body potentials - pair_style tersoff shear -
MEAM modified EAM potential Greg Wagner (Sandia) pair_style meam meam lib/meam
MC Monte Carlo options - fix gcmc - -
MOLECULE molecular system force
fields -Section 6.6.3 peptide -
OPT optimized pair styles Fischer & Richie &
Natoli (2) Section 5.3.5 - -
PERI Peridynamics models Mike Parks (Sandia) pair_style peri peri -
POEMS coupled rigid body
motion Rudra Mukherjee (JPL) fix poems rigid lib/poems
PYTHON embed Python code in an
input script -python python lib/python
REAX ReaxFF potential Aidan Thompson
(Sandia) pair_style reax reax lib/reax
REPLICA multi-replica methods - Section 6.6.5 tad -
RIGID rigid bodies - fix rigid rigid -
SHOCK shock loading methods - fix msst - -
SNAP quantum-fit potential Aidan Thompson
(Sandia) pair snap snap -
SRD stochastic rotation
dynamics -fix srd srd -
VORONOI Voronoi tesselations Daniel Schwen (LANL) compute
voronoi/atom - Voro++
The "Authors" column lists a name(s) if a specific person is responsible for creating and maintaining the
package.
(1) The COLLOID package includes Fast Lubrication Dynamics pair styles which were created by Amit
Kumar and Michael Bybee from Jonathan Higdon's group at UIUC.
(2) The OPT package was created by James Fischer (High Performance Technologies), David Richie, and
Vincent Natoli (Stone Ridge Technolgy).
(3) The KIM package was created by Valeriu Smirichinski, Ryan Elliott, and Ellad Tadmor (U Minn).
LAMMPS Users Manual
54

(4) The KOKKOS package was created primarily by Christian Trott and Stan Moore (Sandia). It uses the
Kokkos library which was developed by Carter Edwards, Christian Trott, and others at Sandia.
The "Doc page" column links to either a sub-section of the Section 6 of the manual, or an input script
command implemented as part of the package, or to additional documentation provided within the package.
The "Example" column is a sub-directory in the examples directory of the distribution which has an input
script that uses the package. E.g. "peptide" refers to the examples/peptide directory.
The "Library" column lists an external library which must be built first and which LAMMPS links to when it
is built. If it is listed as lib/package, then the code for the library is under the lib directory of the LAMMPS
distribution. See the lib/package/README file for info on how to build the library. If it is not listed as
lib/package, then it is a third-party library not included in the LAMMPS distribution. See details on all of this
below for individual packages.
ASPHERE package
Contents: Several computes, time-integration fixes, and pair styles for aspherical particle models: ellipsoids,
2d lines, 3d triangles.
To install via make or Make.py:
make yes-asphere
make machine
Make.py -p asphere -a machine
To un-install via make or Make.py:
make no-asphere
make machine
Make.py -p ^asphere -a machine
Supporting info: Section 6.14, pair_style gayberne, pair_style resquared, doc/PDF/pair_gayberne_extra.pdf,
doc/PDF/pair_resquared_extra.pdf, examples/ASPHERE, examples/ellipse
BODY package
Contents: Support for body-style particles. Computes, time-integration fixes, pair styles, as well as the body
styles themselves. See the body doc page for an overview.
To install via make or Make.py:
make yes-body
make machine
Make.py -p body -a machine
To un-install via make or Make.py:
LAMMPS Users Manual
55

make no-body
make machine
Make.py -p ^body -a machine
Supporting info: atom_style body, body, pair_style body, examples/body
CLASS2 package
Contents: Bond, angle, dihedral, improper, and pair styles for the COMPASS CLASS2 molecular force field.
To install via make or Make.py:
make yes-class2
make machine
Make.py -p class2 -a machine
To un-install via make or Make.py:
make no-class2
make machine
Make.py -p ^class2 -a machine
Supporting info: bond_style class2, angle_style class2, dihedral_style class2, improper_style class2, pair_style
lj/class2
COLLOID package
Contents: Support for coarse-grained colloidal particles. Wall fix and pair styles that implement colloidal
interaction models for finite-size particles. This includes the Fast Lubrication Dynamics method for
hydrodynamic interactions, which is a simplified approximation to Stokesian dynamics.
To install via make or Make.py:
make yes-colloid
make machine
Make.py -p colloid -a machine
To un-install via make or Make.py:
make no-colloid
make machine
Make.py -p ^colloid -a machine
Supporting info: fix wall/colloid, pair_style colloid, pair_style yukawa/colloid, pair_style brownian, pair_style
lubricate, pair_style lubricateU, examples/colloid, examples/srd
LAMMPS Users Manual
56
COMPRESS package
Contents: Support for compressed output of dump files via the zlib compression library, using dump styles
with a "gz" in their style name.
Building with the COMPRESS package assumes you have the zlib compression library available on your
system. The build uses the lib/compress/Makefile.lammps file in the compile/link process. You should only
need to edit this file if the LAMMPS build cannot find the zlib info it specifies.
To install via make or Make.py:
make yes-compress
make machine
Make.py -p compress -a machine
To un-install via make or Make.py:
make no-compress
make machine
Make.py -p ^compress -a machine
Supporting info: src/COMPRESS/README, lib/compress/README, dump atom/gz, dump cfg/gz, dump
custom/gz, dump xyz/gz
CORESHELL package
Contents: Compute and pair styles that implement the adiabatic core/shell model for polarizability. The
compute temp/cs command measures the temperature of a system with core/shell particles. The pair styles
augment Born, Buckingham, and Lennard-Jones styles with core/shell capabilities. See Section 6.26 for an
overview of how to use the package.
To install via make or Make.py:
make yes-coreshell
make machine
Make.py -p coreshell -a machine
To un-install via make or Make.py:
make no-coreshell
make machine
Make.py -p ^coreshell -a machine
Supporting info: Section 6.26, compute temp/cs, pair_style born/coul/long/cs, >pair_style buck/coul/long/cs,
pair_style lj/cut/coul/long/cs, examples/coreshell
LAMMPS Users Manual
57

DIPOLE package
Contents: An atom style and several pair styles to support point dipole models with short-range or long-range
interactions.
To install via make or Make.py:
make yes-dipole
make machine
Make.py -p dipole -a machine
To un-install via make or Make.py:
make no-dipole
make machine
Make.py -p ^dipole -a machine
Supporting info: atom_style dipole, pair_style lj/cut/dipole/cut, pair_style lj/cut/dipole/long, pair_style
lj/long/dipole/long, examples/dipole
GPU package
Contents: Dozens of pair styles and a version of the PPPM long-range Coulombic solver for NVIDIA GPUs.
All of them have a "gpu" in their style name. Section 5.3.1 gives details of what hardware and Cuda software
is required on your system, and how to build and use this package. See the KOKKOS package, which also has
GPU-enabled styles.
Building LAMMPS with the GPU package requires first building the GPU library itself, which is a set of C
and Cuda files in lib/gpu. Details of how to do this are in lib/gpu/README. As illustrated below, perform a
"make" using one of the Makefile.machine files in lib/gpu which should create a lib/reax/libgpu.a file.
Makefile.linux.* and Makefile.xk7 are examples for different platforms. There are 3 important settings in the
Makefile.machine you use:
CUDA_HOME = where NVIDIA Cuda software is installed on your system• CUDA_ARCH = appropriate to your GPU hardware• CUDA_PREC = precision (double, mixed, single) you desire•
See example Makefile.machine files in lib/gpu for the syntax of these settings. See
lib/gpu/Makefile.linux.double for ARCH settings for various NVIDIA GPUs. The "make" also creates a
lib/gpu/Makefile.lammps file. This file has settings that enable LAMMPS to link with Cuda libraries. If the
settings in Makefile.lammps for your machine are not correct, the LAMMPS link will fail. Note that the
Make.py script has a "-gpu" option to allow the GPU library (with several of its options) and LAMMPS to be
built in one step, with Type "python src/Make.py -h -gpu" to see the details.
To install via make or Make.py:
cd ~/lammps/lib/gpu
make -f Makefile.linux.mixed # for example
cd ~/lammps/src
make yes-gpu
LAMMPS Users Manual
58

make machine
Make.py -p gpu -gpu mode=mixed arch=35 -a machine
To un-install via make or Make.py:
make no-gpu
make machine
Make.py -p ^gpu -a machine
Supporting info: src/GPU/README, lib/gpu/README, Section 5.3, Section 5.3.1, Pair Styles section of
Section 3.5 for any pair style listed with a (g), kspace_style, package gpu, examples/accelerate, bench/FERMI,
bench/KEPLER
GRANULAR package
Contents: Fixes and pair styles that support models of finite-size granular particles, which interact with each
other and boundaries via frictional and dissipative potentials.
To install via make or Make.py:
make yes-granular
make machine
Make.py -p granular -a machine
To un-install via make or Make.py:
make no-granular
make machine
Make.py -p ^granular -a machine
Supporting info: Section 6.6, fix pour, fix wall/gran, pair_style gran/hooke, pair_style gran/hertz/history,
examples/pour, bench/in.chute
KIM package
Contents: A pair style that interfaces to the Knowledge Base for Interatomic Models (KIM) repository of
interatomic potentials, so that KIM potentials can be used in a LAMMPS simulation.
To build LAMMPS with the KIM package you must have previously installed the KIM API (library) on your
system. The lib/kim/README file explains how to download and install KIM. Building with the KIM
package also uses the lib/kim/Makefile.lammps file in the compile/link process. You should not need to edit
this file.
To install via make or Make.py:
make yes-kim
make machine
LAMMPS Users Manual
59

Make.py -p kim -a machine
To un-install via make or Make.py:
make no-kim
make machine
Make.py -p ^kim -a machine
Supporting info: src/KIM/README, lib/kim/README, pair_style kim, examples/kim
KOKKOS package
Contents: Dozens of atom, pair, bond, angle, dihedral, improper styles which run with the Kokkos library to
provide optimization for multicore CPUs (via OpenMP), NVIDIA GPUs, or the Intel Xeon Phi (in native
mode). All of them have a "kk" in their style name. Section 5.3.3 gives details of what hardware and software
is required on your system, and how to build and use this package. See the GPU, OPT, USER-INTEL,
USER-OMP packages, which also provide optimizations for the same range of hardware.
Building with the KOKKOS package requires choosing which of 3 hardware options you are optimizing for:
CPU acceleration via OpenMP, GPU acceleration, or Intel Xeon Phi. (You can build multiple times to create
LAMMPS executables for different hardware.) It also requires a C++11 compatible compiler. For GPUs, the
NVIDIA "nvcc" compiler is used, and an appropriate KOKKOS_ARCH setting should be made in your
Makefile.machine for your GPU hardware and NVIDIA software.
The simplest way to do this is to use Makefile.kokkos_cuda or Makefile.kokkos_omp or Makefile.kokkos_phi
in src/MAKE/OPTIONS, via "make kokkos_cuda" or "make kokkos_omp" or "make kokkos_phi". (Check the
KOKKOS_ARCH setting in Makefile.kokkos_cuda), Or, as illustrated below, you can use the Make.py script
with its "-kokkos" option to choose which hardware to build for. Type "python src/Make.py -h -kokkos" to see
the details. If these methods do not work on your system, you will need to read the Section 5.3.3 doc page for
details of what Makefile.machine settings are needed.
To install via make or Make.py for each of 3 hardware options:
make yes-kokkos
make kokkos_omp # for CPUs with OpenMP
make kokkos_cuda # for GPUs, check the KOKKOS_ARCH setting in Makefile.kokkos_cuda
make kokkos_phi # for Xeon Phis
Make.py -p kokkos -kokkos omp -a machine # for CPUs with OpenMP Make.py -p kokkos -kokkos cuda
arch=35 -a machine # for GPUs of style arch Make.py -p kokkos -kokkos phi -a machine # for Xeon Phis
To un-install via make or Make.py:
make no-kokkos
make machine
Make.py -p ^kokkos -a machine
Supporting info: src/KOKKOS/README, lib/kokkos/README, Section 5.3, Section 5.3.3, Pair Styles
section of Section 3.5 for any pair style listed with a (k), package kokkos, examples/accelerate, bench/FERMI,
bench/KEPLER
LAMMPS Users Manual
60
KSPACE package
Contents: A variety of long-range Coulombic solvers, and pair styles which compute the corresponding
short-range portion of the pairwise Coulombic interactions. These include Ewald, particle-particle
particle-mesh (PPPM), and multilevel summation method (MSM) solvers.
Building with the KSPACE package requires a 1d FFT library be present on your system for use by the PPPM
solvers. This can be the KISS FFT library provided with LAMMPS, or 3rd party libraries like FFTW or a
vendor-supplied FFT library. See step 6 of Section 2.2.2 of the manual for details of how to select different
FFT options in your machine Makefile. The Make.py tool has an "-fft" option which can insert these settings
into your machine Makefile automatically. Type "python src/Make.py -h -fft" to see the details.
To install via make or Make.py:
make yes-kspace
make machine
Make.py -p kspace -a machine
To un-install via make or Make.py:
make no-kspace
make machine
Make.py -p ^kspace -a machine
Supporting info: kspace_style, doc/PDF/kspace.pdf, Section 6.7, Section 6.8, Section 6.9, pair_style coul,
other pair style command doc pages which have "long" or "msm" in their style name, examples/peptide,
bench/in.rhodo
MANYBODY package
Contents: A variety of many-body and bond-order potentials. These include (AI)REBO, EAM, EIM, BOP,
Stillinger-Weber, and Tersoff potentials. Do a directory listing, "ls src/MANYBODY", to see the full list.
To install via make or Make.py:
make yes-manybody
make machine
Make.py -p manybody -a machine
To un-install via make or Make.py:
make no-manybody
make machine
Make.py -p ^manybody -a machine
Supporting info:
LAMMPS Users Manual
61

Examples: Pair Styles section of Section 3.5, examples/comb, examples/eim, examples/nb3d,
examples/vashishta
MC package
Contents: Several fixes and a pair style that have Monte Carlo (MC) or MC-like attributes. These include fixes
for creating, breaking, and swapping bonds, and for performing atomic swaps and grand-canonical MC in
conjuction with dynamics.
To install via make or Make.py:
make yes-mc
make machine
Make.py -p mc -a machine
To un-install via make or Make.py:
make no-mc
make machine
Make.py -p ^mc -a machine
Supporting info: fix atom/swap, fix bond/break, fix bond/create, fix bond/swap, fix gcmc, pair_style dsmc
MEAM package
Contents: A pair style for the modified embedded atom (MEAM) potential.
Building LAMMPS with the MEAM package requires first building the MEAM library itself, which is a set
of Fortran 95 files in lib/meam. Details of how to do this are in lib/meam/README. As illustrated below,
perform a "make" using one of the Makefile.machine files in lib/meam which should create a
lib/meam/libmeam.a file. Makefile.gfortran and Makefile.ifort are examples for the GNU Fortran and Intel
Fortran compilers. The "make" also copies a lib/meam/Makefile.lammps.machine file to
lib/meam/Makefile.lammps. This file has settings that enable the C++ compiler used to build LAMMPS to
link with a Fortran library (typically the 2 compilers to be consistent e.g. both Intel compilers, or both GNU
compilers). If the settings in Makefile.lammps for your compilers and machine are not correct, the LAMMPS
link will fail. Note that the Make.py script has a "-meam" option to allow the MEAM library and LAMMPS to
be built in one step. Type "python src/Make.py -h -meam" to see the details.
NOTE: The MEAM potential can run dramatically faster if built with the Intel Fortran compiler, rather than
the GNU Fortran compiler.
To install via make or Make.py:
cd ~/lammps/lib/meam
make -f Makefile.gfortran # for example
cd ~/lammps/src
make yes-meam
make machine
LAMMPS Users Manual
62

Make.py -p meam -meam make=gfortran -a machine
To un-install via make or Make.py:
make no-meam
make machine
Make.py -p ^meam -a machine
Supporting info: lib/meam/README, pair_style meam, examples/meam
MISC package
Contents: A variety of computes, fixes, and pair styles that are not commonly used, but don't align with other
packages. Do a directory listing, "ls src/MISC", to see the list of commands.
To install via make or Make.py:
make yes-misc
make machine
Make.py -p misc -a machine
To un-install via make or Make.py:
make no-misc
make machine
Make.py -p ^misc -a machine
Supporting info: compute ti, fix evaporate, fix tmm, fix viscosity, examples/misc
MOLECULE package
Contents: A large number of atom, pair, bond, angle, dihedral, improper styles that are used to model
molecular systems with fixed covalent bonds. The pair styles include terms for the Dreiding
(hydrogen-bonding) and CHARMM force fields, and TIP4P water model.
To install via make or Make.py:
make yes-molecule
make machine
Make.py -p molecule -a machine
To un-install via make or Make.py:
make no-molecule
make machine
Make.py -p ^molecule -a machine
LAMMPS Users Manual
63

Supporting info:atom_style, bond_style, angle_style, dihedral_style, improper_style, pair_style
hbond/dreiding/lj, pair_style lj/charmm/coul/charmm, Section 6.3, examples/micelle, examples/peptide,
bench/in.chain, bench/in.rhodo
MPIIO package
Contents: Support for parallel output/input of dump and restart files via the MPIIO library, which is part of the
standard message-passing interface (MPI) library. It adds dump styles with a "mpiio" in their style name.
Restart files with an ".mpiio" suffix are also written and read in parallel.
To install via make or Make.py:
make yes-mpiio
make machine
Make.py -p mpiio -a machine
To un-install via make or Make.py:
make no-mpiio
make machine
Make.py -p ^mpiio -a machine
Supporting info: dump, restart, write_restart, read_restart
OPT package
Contents: A handful of pair styles with an "opt" in their style name which are optimized for improved CPU
performance on single or multiple cores. These include EAM, LJ, CHARMM, and Morse potentials. Section
5.3.5 gives details of how to build and use this package. See the KOKKOS, USER-INTEL, and USER-OMP
packages, which also have styles optimized for CPU performance.
Some C++ compilers, like the Intel compiler, require the compile flag "-restrict" to build LAMMPS with the
OPT package. It should be added to the CCFLAGS line of your Makefile.machine. Or use Makefile.opt in
src/MAKE/OPTIONS, via "make opt". For compilers that use the flag, the Make.py command adds it
automatically to the Makefile.auto file it creates and uses.
To install via make or Make.py:
make yes-opt
make machine
Make.py -p opt -a machine
To un-install via make or Make.py:
make no-opt
make machine
Make.py -p ^opt -a machine
LAMMPS Users Manual
64
Supporting info: Section 5.3, Section 5.3.5, Pair Styles section of Section 3.5 for any pair style listed with an
(t), examples/accelerate, bench/KEPLER
PERI package
Contents: Support for the Peridynamics method, a particle-based meshless continuum model. The package
includes an atom style, several computes which calculate diagnostics, and several Peridynamic pair styles
which implement different materials models.
To install via make or Make.py:
make yes-peri
make machine
Make.py -p peri -a machine
To un-install via make or Make.py:
make no-peri
make machine
Make.py -p ^peri -a machine
Supporting info: doc/PDF/PDLammps_overview.pdf, doc/PDF/PDLammps_EPS.pdf,
doc/PDF/PDLammps_VES.pdf, atom_style peri, compute damage/atom, pair_style peri/pmb, examples/peri
POEMS package
Contents: A fix that wraps the Parallelizable Open source Efficient Multibody Software (POEMS) librar,
which is able to simulate the dynamics of articulated body systems. These are systems with multiple rigid
bodies (collections of atoms or particles) whose motion is coupled by connections at hinge points.
Building LAMMPS with the POEMS package requires first building the POEMS library itself, which is a set
of C++ files in lib/poems. Details of how to do this are in lib/poems/README. As illustrated below, perform
a "make" using one of the Makefile.machine files in lib/poems which should create a lib/meam/libpoems.a
file. Makefile.g++ and Makefile.icc are examples for the GNU and Intel C++ compilers. The "make" also
creates a lib/poems/Makefile.lammps file which you should not need to change. Note the Make.py script has a
"-poems" option to allow the POEMS library and LAMMPS to be built in one step. Type "python
src/Make.py -h -poems" to see the details.
To install via make or Make.py:
cd ~/lammps/lib/poems
make -f Makefile.g++ # for example
cd ~/lammps/src
make yes-poems
make machine
Make.py -p poems -poems make=g++ -a machine
To un-install via make or Make.py:
LAMMPS Users Manual
65
make no-meam
make machine
Make.py -p ^meam -a machine
Supporting info: src/POEMS/README, lib/poems/README, fix poems, examples/rigid
PYTHON package
Contents: A python command which allow you to execute Python code from a LAMMPS input script. The
code can be in a separate file or embedded in the input script itself. See Section 11.2 for an overview of using
Python from LAMMPS and for other ways to use LAMMPS and Python together.
Building with the PYTHON package assumes you have a Python shared library available on your system,
which needs to be a Python 2 version, 2.6 or later. Python 3 is not yet supported. The build uses the contents
of the lib/python/Makefile.lammps file to find all the Python files required in the build/link process. See the
lib/python/README file if the settings in that file do not work on your system. Note that the Make.py script
has a "-python" option to allow an alternate lib/python/Makefile.lammps file to be specified and LAMMPS to
be built in one step. Type "python src/Make.py -h -python" to see the details.
To install via make or Make.py:
make yes-python
make machine
Make.py -p python -a machine
To un-install via make or Make.py:
make no-python
make machine
Make.py -p ^python -a machine
Supporting info: examples/python
QEQ package
Contents: Several fixes for performing charge equilibration (QEq) via severeal different algorithms. These can
be used with pair styles that use QEq as part of their formulation.
To install via make or Make.py:
make yes-qeq
make machine
Make.py -p qeq -a machine
To un-install via make or Make.py:
make no-qeq
LAMMPS Users Manual
66
make machine
Make.py -p ^qeq -a machine
Supporting info: fix qeq/*, examples/qeq
REAX package
Contents: A pair style for the ReaxFF potential, a universal reactive force field, as well as a fix reax/bonds
command for monitoring molecules as bonds are created and destroyed.
Building LAMMPS with the REAX package requires first building the REAX library itself, which is a set of
Fortran 95 files in lib/reax. Details of how to do this are in lib/reax/README. As illustrated below, perform a
"make" using one of the Makefile.machine files in lib/reax which should create a lib/reax/libreax.a file.
Makefile.gfortran and Makefile.ifort are examples for the GNU Fortran and Intel Fortran compilers. The
"make" also copies a lib/reax/Makefile.lammps.machine file to lib/reax/Makefile.lammps. This file has
settings that enable the C++ compiler used to build LAMMPS to link with a Fortran library (typically the 2
compilers to be consistent e.g. both Intel compilers, or both GNU compilers). If the settings in
Makefile.lammps for your compilers and machine are not correct, the LAMMPS link will fail. Note that the
Make.py script has a "-reax" option to allow the REAX library and LAMMPS to be built in one step. Type
"python src/Make.py -h -reax" to see the details.
To install via make or Make.py:
cd ~/lammps/lib/reax
make -f Makefile.gfortran # for example
cd ~/lammps/src
make yes-reax
make machine
Make.py -p reax -reax make=gfortran -a machine
To un-install via make or Make.py:
make no-reax
make machine
Make.py -p ^reax -a machine
Supporting info: lib/reax/README, pair_style reax, fix reax/bonds, examples/reax
REPLICA package
Contents: A collection of multi-replica methods that are used by invoking multiple instances (replicas) of
LAMMPS simulations. Communication between individual replicas is performed in different ways by the
different methods. See Section 6.5 for an overview of how to run multi-replica simulations in LAMMPS.
Multi-replica methods included in the package are nudged elastic band (NEB), parallel replica dynamics
(PRD), temperature accelerated dynamics (TAD), parallel tempering, and a verlet/split algorithm for
performing long-range Coulombics on one set of processors, and the remainder of the force field calculation
on another set.
LAMMPS Users Manual
67

To install via make or Make.py:
make yes-replica
make machine
Make.py -p replica -a machine
To un-install via make or Make.py:
make no-replica
make machine
Make.py -p ^replica -a machine
Supporting info: Section 6.5, neb, prd, tad, temper, run_style verlet/split, examples/neb, examples/prd,
examples/tad
RIGID package
Contents: A collection of computes and fixes which enforce rigid constraints on collections of atoms or
particles. This includes SHAKE and RATTLE, as well as variants of rigid-body time integrators for a few
large bodies or many small bodies.
To install via make or Make.py:
make yes-rigid
make machine
Make.py -p rigid -a machine
To un-install via make or Make.py:
make no-rigid
make machine
Make.py -p ^rigid -a machine
Supporting info: compute erotate/rigid, fix shake, fix rattle, fix rigid/*, examples/ASPHERE, examples/rigid
SHOCK package
Contents: A small number of fixes useful for running impact simulations where a shock-wave passes through
a material.
To install via make or Make.py:
make yes-shock
make machine
Make.py -p shock -a machine
LAMMPS Users Manual
68

To un-install via make or Make.py:
make no-shock
make machine
Make.py -p ^shock -a machine
Supporting info: fix append/atoms, fix msst, fix nphug, fix wall/piston, examples/hugoniostat, examples/msst
SNAP package
Contents: A pair style for the spectral neighbor analysis potential (SNAP), which is an empirical potential
which can be quantum accurate when fit to an archive of DFT data. Computes useful for analyzing properties
of the potential are also included.
To install via make or Make.py:
make yes-snap
make machine
Make.py -p snap -a machine
To un-install via make or Make.py:
make no-snap
make machine
Make.py -p ^snap -a machine
Supporting info: pair snap, compute sna/atom, compute snad/atom, compute snav/atom, examples/snap
SRD package
Contents: Two fixes which implement the Stochastic Rotation Dynamics (SRD) method for coarse-graining of
a solvent, typically around large colloidal-scale particles.
To install via make or Make.py:
make yes-srd
make machine
Make.py -p srd -a machine
To un-install via make or Make.py:
make no-srd
make machine
Make.py -p ^srd -a machine
Supporting info: fix srd, fix wall/srd, examples/srd, examples/ASPHERE
LAMMPS Users Manual
69

VORONOI package
Contents: A compute voronoi/atom command which computes the Voronoi tesselation of a collection of
atoms or particles by wrapping the Voro++ lib
To build LAMMPS with the KIM package you must have previously installed the KIM API (library) on your
system. The lib/kim/README file explains how to download and install KIM. Building with the KIM
package also uses the lib/kim/Makefile.lammps file in the compile/link process. You should not need to edit
this file.
To build LAMMPS with the VORONOI package you must have previously installed the Voro++ library on
your system. The lib/voronoi/README file explains how to download and install Voro++. There is a
lib/voronoi/install.py script which automates the process. Type "python install.py" to see instructions. The
final step is to create soft links in the lib/voronoi directory for "includelink" and "liblink" which point to
installed Voro++ directories. Building with the VORONOI package uses the contents of the
lib/voronoi/Makefile.lammps file in the compile/link process. You should not need to edit this file. Note that
the Make.py script has a "-voronoi" option to allow the Voro++ library to be downloaded and/or installed and
LAMMPS to be built in one step. Type "python src/Make.py -h -voronoi" to see the details.
To install via make or Make.py:
cd ~/lammps/lib/voronoi
python install.py -g -b -l # download Voro++, build in lib/voronoi, create links
cd ~/lammps/src
make yes-voronoi
make machine
Make.py -p voronoi -voronoi install="-g -b -l" -a machine
To un-install via make or Make.py:
make no-voronoi
make machine
Make.py -p ^voronoi -a machine
Supporting info: src/VORONOI/README, lib/voronoi/README, compute voronoi/atom, examples/voronoi
4.2 User packages
The current list of user-contributed packages is as follows:
Package Description Author(s) Doc page Example Pic/movie Library
USER-ATC atom-to-continuum
coupling Jones & Templeton &
Zimmerman (1) fix atc USER/atc atc lib/atc
USER-AWPMD wave-packet MD Ilya Valuev (JIHT) pair_style awpmd/cut USER/awpmd - lib/awpmd
USER-CG-CMM coarse-graining
model Axel Kohlmeyer
(Temple U) pair_style lj/sdk USER/cg-cmm cg -
USER-CGDNA src/USER-CGDNA/README USER/cgdna - -
LAMMPS Users Manual
70

coarse-grained
DNA force fields Oliver Henrich (U
Strathclyde Glasgow)
USER-COLVARS collective variables Fiorin & Henin &
Kohlmeyer (2) fix colvars USER/colvars colvars lib/colvars
USER-DIFFRACTION virutal x-ray and
electron diffraction Shawn Coleman (ARL) compute xrd USER/diffraction - -
USER-DPD reactive dissipative
particle dynamics
(DPD)
Larentzos & Mattox &
Brennan (5) src/USER-DPD/README USER/dpd - -
USER-DRUDE Drude oscillators Dequidt & Devemy &
Padua (3) tutorial USER/drude - -
USER-EFF electron force field Andres Jaramillo-Botero
(Caltech) pair_style eff/cut USER/eff eff -
USER-FEP free energy
perturbation
Agilio Padua (U Blaise
Pascal
Clermont-Ferrand) compute fep USER/fep - -
USER-H5MD dump output via
HDF5 Pierre de Buyl (KU
Leuven) dump h5md - - lib/h5md
USER-INTEL Vectorized CPU
and Intel(R)
coprocessor styles
W. Michael Brown
(Intel) Section 5.3.2 examples/intel - -
USER-LB Lattice Boltzmann
fluid Colin Denniston (U
Western Ontario) fix lb/fluid USER/lb - -
USER-MGPT fast MGPT
multi-ion potentials Tomas Oppelstrup &
John Moriarty (LLNL) pair_style mgpt USER/mgpt - -
USER-MISC single-file
contributions USER-MISC/README USER-MISC/README - - -
USER-MANIFOLD motion on 2d
surface
Stefan Paquay
(Eindhoven U of
Technology) fix manifoldforce USER/manifold manifold -
USER-MOLFILE VMD molfile
plug-ins Axel Kohlmeyer
(Temple U) dump molfile - - VMD-MOLFILE
USER-NC-DUMP dump output via
NetCDF
Lars Pastewka
(Karlsruhe Institute of
Technology KIT) dump nc / dump
nc/mpiio - - lib/netcdf
USER-OMP OpenMP threaded
styles Axel Kohlmeyer
(Temple U) Section 5.3.4 - - -
USER-PHONON phonon dynamical
matrix Ling-Ti Kong (Shanghai
Jiao Tong U) fix phonon USER/phonon - -
USER-QMMM QM/MM coupling Axel Kohlmeyer
(Temple U) fix qmmm USER/qmmm - lib/qmmm
USER-QTB quantum nuclear
effects Yuan Shen (Stanford) fix qtb fix qbmsst qtb - -
USER-QUIP QUIP/libatoms
interface Albert Bartok-Partay (U
Cambridge) pair_style quip USER/quip - lib/quip
USER-REAXC Metin Aktulga (LBNL) pair_style reaxc reax - -
LAMMPS Users Manual
71

C version of
ReaxFF
USER-SMD smoothed Mach
dynamics Georg Ganzenmuller
(EMI) SMD User Guide USER/smd - -
USER-SMTBQ Second Moment
Tight Binding -
QEq potential
Salles & Maras &
Politano & Tetot (4) pair_style smtbq USER/smtbq - -
USER-SPH smoothed particle
hydrodynamics Georg Ganzenmuller
(EMI) SPH User Guide USER/sph sph -
USER-TALLY Pairwise tallied
computes Axel Kohlmeyer
(Temple U) compute XXX/tally USER/tally - -
USER-VTK VTK-style dumps Berger and Queteschiner
(6) compute custom/vtk - - lib/vtk
The "Authors" column lists a name(s) if a specific person is responsible for creating and maintaining the
package.
(1) The ATC package was created by Reese Jones, Jeremy Templeton, and Jon Zimmerman (Sandia).
(2) The COLVARS package was created by Axel Kohlmeyer (Temple U) using the colvars module library
written by Giacomo Fiorin (Temple U) and Jerome Henin (LISM, Marseille, France).
(3) The DRUDE package was created by Alain Dequidt (U Blaise Pascal Clermont-Ferrand) and co-authors
Julien Devemy (CNRS) and Agilio Padua (U Blaise Pascal).
(4) The SMTBQ package was created by Nicolas Salles, Emile Maras, Olivier Politano, and Robert Tetot
(LAAS-CNRS, France).
(5) The USER-DPD package was created by James Larentzos (ARL), Timothy Mattox (Engility), and John
Brennan (ARL).
(6) The USER-VTK package was created by Richard Berger (JKU) and Daniel Queteschiner (DCS
Computing).
The "Doc page" column links to either a sub-section of the Section 6 of the manual, or an input script
command implemented as part of the package, or to additional documentation provided within the package.
The "Example" column is a sub-directory in the examples directory of the distribution which has an input
script that uses the package. E.g. "peptide" refers to the examples/peptide directory.
The "Library" column lists an external library which must be built first and which LAMMPS links to when it
is built. If it is listed as lib/package, then the code for the library is under the lib directory of the LAMMPS
distribution. See the lib/package/README file for info on how to build the library. If it is not listed as
lib/package, then it is a third-party library not included in the LAMMPS distribution. See details on all of this
below for individual packages.
LAMMPS Users Manual
72

USER-ATC package
Contents: ATC stands for atoms-to-continuum. This package implements a fix atc command to either couple
MD with continuum finite element equations or perform on-the-fly post-processing of atomic information to
continuum fields. See src/USER-ATC/README for more details.
To build LAMMPS with this package ...
To install via make or Make.py:
make yes-user-atc
make machine
Make.py -p atc -a machine
To un-install via make or Make.py:
make no-user-atc
make machine
Make.py -p ^atc -a machine
Supporting info:src/USER-ATC/README, fix atc, examples/USER/atc
Authors: Reese Jones (rjones at sandia.gov), Jeremy Templeton (jatempl at sandia.gov) and Jon Zimmerman
(jzimmer at sandia.gov) at Sandia. Contact them directly if you have questions.
USER-AWPMD package
Contents: AWPMD stands for Antisymmetrized Wave Packet Molecular Dynamics. This package implements
an atom, pair, and fix style which allows electrons to be treated as explicit particles in an MD calculation. See
src/USER-AWPMD/README for more details.
To build LAMMPS with this package ...
Supporting info: src/USER-AWPMD/README, fix awpmd/cut, examples/USER/awpmd
Author: Ilya Valuev at the JIHT in Russia (valuev at physik.hu-berlin.de). Contact him directly if you have
questions.
USER-CG-CMM package
Contents: CG-CMM stands for coarse-grained ??. This package implements several pair styles and an angle
style using the coarse grained parametrization of Shinoda, DeVane, Klein, Mol Sim, 33, 27 (2007) (SDK),
with extensions to simulate ionic liquids, electrolytes, lipids and charged amino acids. See
src/USER-CG-CMM/README for more details.
Supporting info: src/USER-CG-CMM/README, pair lj/sdk, pair lj/sdk/coul/long, angle sdk,
examples/USER/cg-cmm
LAMMPS Users Manual
73
Author: Axel Kohlmeyer at Temple U (akohlmey at gmail.com). Contact him directly if you have questions.
USER-CGDNA package
Contents: The CGDNA package implements coarse-grained force fields for single- and double-stranded DNA.
These are at the moment mainly the oxDNA and oxDNA2 models, developed by Doye, Louis and Ouldridge
at the University of Oxford. The package also contains Langevin-type rigid-body integrators with improved
stability.
See these doc pages to get started:
bond_style oxdna/fene• bond_style oxdna2/fene• pair_style oxdna/...• pair_style oxdna2/...• fix nve/dotc/langevin•
Supporting info: /src/USER-CGDNA/README, bond_style oxdna/fene, bond_style oxdna2/fene, pair_style
oxdna/..., pair_style oxdna2/..., fix nve/dotc/langevin
Author: Oliver Henrich at the University of Strathclyde, Glasgow, UK and University of Edinburgh
(ohenrich@ph.ed.ac.uk). Contact him directly if you have any questions.
USER-COLVARS package
Contents: COLVARS stands for collective variables which can be used to implement Adaptive Biasing Force,
Metadynamics, Steered MD, Umbrella Sampling and Restraints. This package implements a fix colvars
command which wraps a COLVARS library which can perform those kinds of simulations. See
src/USER-COLVARS/README for more details.
Supporting info: doc/PDF/colvars-refman-lammps.pdf, src/USER-COLVARS/README,
lib/colvars/README, fix colvars, examples/USER/colvars
Authors: Axel Kohlmeyer at Temple U (akohlmey at gmail.com) wrote the fix. The COLVARS library itself
is written and maintained by Giacomo Fiorin (ICMS, Temple University, Philadelphia, PA, USA) and Jerome
Henin (LISM, CNRS, Marseille, France). Contact them directly if you have questions.
USER-DIFFRACTION package
Contents: This packages implements two computes and a fix for calculating x-ray and electron diffraction
intensities based on kinematic diffraction theory. See src/USER-DIFFRACTION/README for more details.
Supporting info: compute saed, compute xrd, fix saed/vtk, examples/USER/diffraction
Author: Shawn P. Coleman (shawn.p.coleman8.ctr at mail.mil) while at the University of Arkansas. Contact
him directly if you have questions.
LAMMPS Users Manual
74

USER-DPD package
Contents: DPD stands for dissipative particle dynamics, This package implements DPD for isothermal,
isoenergetic, isobaric and isenthalpic conditions. It also has extensions for performing reactive DPD, where
each particle has internal state for multiple species and a coupled set of chemical reaction ODEs are integrated
each timestep. The DPD equations of motion are integrated efficiently through the Shardlow splitting
algorithm. See src/USER-DPD/README for more details.
Supporting info: /src/USER-DPD/README, compute dpd compute dpd/atom fix eos/cv fix eos/table fix
eos/table/rx fix shardlow fix rx pair table/rx pair dpd/fdt pair dpd/fdt/energy pair exp6/rx pair multi/lucy pair
multi/lucy/rx, examples/USER/dpd
Authors: James Larentzos (ARL) (james.p.larentzos.civ at mail.mil), Timothy Mattox (Engility Corp)
(Timothy.Mattox at engilitycorp.com) and John Brennan (ARL) (john.k.brennan.civ at mail.mil). Contact
them directly if you have questions.
USER-DRUDE package
Contents: This package contains methods for simulating polarizable systems using thermalized Drude
oscillators. It has computes, fixes, and pair styles for this purpose. See Section 6.27 for an overview of how to
use the package. See src/USER-DRUDE/README for additional details. There are auxiliary tools for using
this package in tools/drude.
Supporting info: Section 6.27, src/USER-DRUDE/README, fix drude, fix drude/transform/*, compute
temp/drude, pair thole, pair lj/cut/thole/long, examples/USER/drude, tools/drude
Authors: Alain Dequidt at Universite Blaise Pascal Clermont-Ferrand (alain.dequidt at univ-bpclermont.fr);
co-authors: Julien Devemy, Agilio Padua. Contact them directly if you have questions.
USER-EFF package
Contents: EFF stands for electron force field. This package contains atom, pair, fix and compute styles which
implement the eFF as described in A. Jaramillo-Botero, J. Su, Q. An, and W.A. Goddard III, JCC, 2010. The
eFF potential was first introduced by Su and Goddard, in 2007. See src/USER-EFF/README for more
details. There are auxiliary tools for using this package in tools/eff; see its README file.
Supporting info:
Author: Andres Jaramillo-Botero at CalTech (ajaramil at wag.caltech.edu). Contact him directly if you have
questions.
USER-FEP package
Contents: FEP stands for free energy perturbation. This package provides methods for performing FEP
simulations by using a fix adapt/fep command with soft-core pair potentials, which have a "soft" in their style
name. See src/USER-FEP/README for more details. There are auxiliary tools for using this package in
tools/fep; see its README file.
LAMMPS Users Manual
75

Supporting info: src/USER-FEP/README, fix adapt/fep, compute fep, pair_style */soft, examples/USER/fep
Author: Agilio Padua at Universite Blaise Pascal Clermont-Ferrand (agilio.padua at univ-bpclermont.fr).
Contact him directly if you have questions.
USER-H5MD package
Contents: H5MD stands for HDF5 for MD. HDF5 is a binary, portable, self-describing file format, used by
many scientific simulations. H5MD is a format for molecular simulations, built on top of HDF5. This package
implements a dump h5md command to output LAMMPS snapshots in this format. See
src/USER-H5MD/README for more details.
Supporting info: src/USER-H5MD/README, lib/h5md/README, dump h5md
Author: Pierre de Buyl at KU Leuven (see http://pdebuyl.be) created this package as well as the H5MD format
and library. Contact him directly if you have questions.
USER-INTEL package
Contents: Dozens of pair, bond, angle, dihedral, and improper styles that are optimized for Intel CPUs and the
Intel Xeon Phi (in offload mode). All of them have an "intel" in their style name. Section 5.3.2 gives details of
what hardware and compilers are required on your system, and how to build and use this package. Also see
src/USER-INTEL/README for more details. See the KOKKOS, OPT, and USER-OMP packages, which
also have CPU and Phi-enabled styles.
Supporting info: examples/accelerate, src/USER-INTEL/TEST
Section 5.3
Author: Mike Brown at Intel (michael.w.brown at intel.com). Contact him directly if you have questions.
For the USER-INTEL package, you have 2 choices when building. You can build with CPU or Phi support.
The latter uses Xeon Phi chips in "offload" mode. Each of these modes requires additional settings in your
Makefile.machine for CCFLAGS and LINKFLAGS.
For CPU mode (if using an Intel compiler):
CCFLAGS: add -fopenmp, -DLAMMPS_MEMALIGN=64, -restrict, -xHost, -fno-alias, -ansi-alias,
-override-limits
•
LINKFLAGS: add -fopenmp•
For Phi mode add the following in addition to the CPU mode flags:
CCFLAGS: add -DLMP_INTEL_OFFLOAD and• LINKFLAGS: add -offload•
And also add this to CCFLAGS:
-offload-option,mic,compiler,"-fp-model fast=2 -mGLOB_default_function_attrs=\"gather_scatter_loop_unroll=4\""
LAMMPS Users Manual
76

Examples:
USER-LB package
Supporting info:
This package contains a LAMMPS implementation of a background Lattice-Boltzmann fluid, which can be
used to model MD particles influenced by hydrodynamic forces.
See this doc page and its related commands to get started:
fix lb/fluid
The people who created this package are Frances Mackay (fmackay at uwo.ca) and Colin (cdennist at uwo.ca)
Denniston, University of Western Ontario. Contact them directly if you have questions.
Examples: examples/USER/lb
USER-MGPT package
Supporting info:
This package contains a fast implementation for LAMMPS of quantum-based MGPT multi-ion potentials.
The MGPT or model GPT method derives from first-principles DFT-based generalized pseudopotential theory
(GPT) through a series of systematic approximations valid for mid-period transition metals with nearly
half-filled d bands. The MGPT method was originally developed by John Moriarty at Lawrence Livermore
National Lab (LLNL).
In the general matrix representation of MGPT, which can also be applied to f-band actinide metals, the
multi-ion potentials are evaluated on the fly during a simulation through d- or f-state matrix multiplication,
and the forces that move the ions are determined analytically. The mgpt pair style in this package calculates
forces and energies using an optimized matrix-MGPT algorithm due to Tomas Oppelstrup at LLNL.
See this doc page to get started:
pair_style mgpt
The persons who created the USER-MGPT package are Tomas Oppelstrup (oppelstrup2@llnl.gov) and John
Moriarty (moriarty2@llnl.gov) Contact them directly if you have any questions.
Examples: examples/USER/mgpt
USER-MISC package
Supporting info:
The files in this package are a potpourri of (mostly) unrelated features contributed to LAMMPS by users.
Each feature is a single pair of files (*.cpp and *.h).
LAMMPS Users Manual
77

More information about each feature can be found by reading its doc page in the LAMMPS doc directory. The
doc page which lists all LAMMPS input script commands is as follows:
Section 3.5
User-contributed features are listed at the bottom of the fix, compute, pair, etc sections.
The list of features and author of each is given in the src/USER-MISC/README file.
You should contact the author directly if you have specific questions about the feature or its coding.
Examples: examples/USER/misc
USER-MANIFOLD package
Supporting info:
This package contains a dump molfile command which uses molfile plugins that are bundled with the VMD
molecular visualization and analysis program, to enable LAMMPS to dump its information in formats
compatible with various molecular simulation tools.
This package allows LAMMPS to perform MD simulations of particles constrained on a manifold (i.e., a 2D
subspace of the 3D simulation box). It achieves this using the RATTLE constraint algorithm applied to
single-particle constraint functions g(xi,yi,zi) = 0 and their derivative (i.e. the normal of the manifold) n =
grad(g).
See this doc page to get started:
fix manifoldforce
The person who created this package is Stefan Paquay, at the Eindhoven University of Technology (TU/e),
The Netherlands (s.paquay at tue.nl). Contact him directly if you have questions.
USER-MOLFILE package
Supporting info:
This package contains a dump molfile command which uses molfile plugins that are bundled with the VMD
molecular visualization and analysis program, to enable LAMMPS to dump its information in formats
compatible with various molecular simulation tools.
The package only provides the interface code, not the plugins. These can be obtained from a VMD installation
which has to match the platform that you are using to compile LAMMPS for. By adding plugins to VMD,
support for new file formats can be added to LAMMPS (or VMD or other programs that use them) without
having to recompile the application itself.
See this doc page to get started:
dump molfile
LAMMPS Users Manual
78
The person who created this package is Axel Kohlmeyer at Temple U (akohlmey at gmail.com). Contact him
directly if you have questions.
USER-NC-DUMP package
Contents: Dump styles for writing NetCDF format files. NetCDF is a binary, portable, self-describing file
format on top of HDF5. The file format contents follow the AMBER NetCDF trajectory conventions
(http://ambermd.org/netcdf/nctraj.xhtml), but include extensions to this convention. This package implements
a dump nc command and a dump nc/mpiio command to output LAMMPS snapshots in this format. See
src/USER-NC-DUMP/README for more details.
NetCDF files can be directly visualized with the following tools:
Ovito (http://www.ovito.org/). Ovito supports the AMBER convention and all of the above
extensions.
•
VMD (http://www.ks.uiuc.edu/Research/vmd/)• AtomEye (http://www.libatoms.org/). The libAtoms version of AtomEye contains a NetCDF reader
that is not present in the standard distribution of AtomEye
•
The person who created these files is Lars Pastewka at Karlsruhe Institute of Technology (lars.pastewka at
kit.edu). Contact him directly if you have questions.
USER-OMP package
Supporting info:
This package provides OpenMP multi-threading support and other optimizations of various LAMMPS pair
styles, dihedral styles, and fix styles.
See this section of the manual to get started:
Section 5.3
The person who created this package is Axel Kohlmeyer at Temple U (akohlmey at gmail.com). Contact him
directly if you have questions.
For the USER-OMP package, your Makefile.machine needs additional settings for CCFLAGS and
LINKFLAGS.
CCFLAGS: add -fopenmp and -restrict• LINKFLAGS: add -fopenmp•
Examples: examples/accelerate, bench/KEPLER
USER-PHONON package
This package contains a fix phonon command that calculates dynamical matrices, which can then be used to
compute phonon dispersion relations, directly from molecular dynamics simulations.
LAMMPS Users Manual
79

See this doc page to get started:
fix phonon
The person who created this package is Ling-Ti Kong (konglt at sjtu.edu.cn) at Shanghai Jiao Tong
University. Contact him directly if you have questions.
Examples: examples/USER/phonon
USER-QMMM package
Supporting info:
This package provides a fix qmmm command which allows LAMMPS to be used in a QM/MM simulation,
currently only in combination with pw.x code from the Quantum ESPRESSO package.
The current implementation only supports an ONIOM style mechanical coupling to the Quantum ESPRESSO
plane wave DFT package. Electrostatic coupling is in preparation and the interface has been written in a
manner that coupling to other QM codes should be possible without changes to LAMMPS itself.
See this doc page to get started:
fix qmmm
as well as the lib/qmmm/README file.
The person who created this package is Axel Kohlmeyer at Temple U (akohlmey at gmail.com). Contact him
directly if you have questions.
USER-QTB package
Supporting info:
This package provides a self-consistent quantum treatment of the vibrational modes in a classical molecular
dynamics simulation. By coupling the MD simulation to a colored thermostat, it introduces zero point energy
into the system, alter the energy power spectrum and the heat capacity towards their quantum nature. This
package could be of interest if one wants to model systems at temperatures lower than their classical limits or
when temperatures ramp up across the classical limits in the simulation.
See these two doc pages to get started:
fix qtb provides quantum nulcear correction through a colored thermostat and can be used with other time
integration schemes like fix nve or fix nph.
fix qbmsst enables quantum nuclear correction of a multi-scale shock technique simulation by coupling the
quantum thermal bath with the shocked system.
The person who created this package is Yuan Shen (sy0302 at stanford.edu) at Stanford University. Contact
him directly if you have questions.
LAMMPS Users Manual
80

Examples: examples/USER/qtb
USER-QUIP package
Supporting info:
Examples: examples/USER/quip
USER-REAXC package
Supporting info:
This package contains a implementation for LAMMPS of the ReaxFF force field. ReaxFF uses
distance-dependent bond-order functions to represent the contributions of chemical bonding to the potential
energy. It was originally developed by Adri van Duin and the Goddard group at CalTech.
The USER-REAXC version of ReaxFF (pair_style reax/c), implemented in C, should give identical or very
similar results to pair_style reax, which is a ReaxFF implementation on top of a Fortran library, a version of
which library was originally authored by Adri van Duin.
The reax/c version should be somewhat faster and more scalable, particularly with respect to the charge
equilibration calculation. It should also be easier to build and use since there are no complicating issues with
Fortran memory allocation or linking to a Fortran library.
For technical details about this implementation of ReaxFF, see this paper:
Parallel and Scalable Reactive Molecular Dynamics: Numerical Methods and Algorithmic Techniques, H. M.
Aktulga, J. C. Fogarty, S. A. Pandit, A. Y. Grama, Parallel Computing, in press (2011).
See the doc page for the pair_style reax/c command for details of how to use it in LAMMPS.
The person who created this package is Hasan Metin Aktulga (hmaktulga at lbl.gov), while at Purdue
University. Contact him directly, or Aidan Thompson at Sandia (athomps at sandia.gov), if you have
questions.
Examples: examples/reax
USER-SMD package
Supporting info:
This package implements smoothed Mach dynamics (SMD) in LAMMPS. Currently, the package has the
following features:
* Does liquids via traditional Smooth Particle Hydrodynamics (SPH)
* Also solves solids mechanics problems via a state of the art stabilized meshless method with hourglass
control.
LAMMPS Users Manual
81

* Can specify hydrostatic interactions independently from material strength models, i.e. pressure and
deviatoric stresses are separated.
* Many material models available (Johnson-Cook, plasticity with hardening, Mie-Grueneisen, Polynomial
EOS). Easy to add new material models.
* Rigid boundary conditions (walls) can be loaded as surface geometries from *.STL files.
See the file doc/PDF/SMD_LAMMPS_userguide.pdf to get started.
There are example scripts for using this package in examples/USER/smd.
The person who created this package is Georg Ganzenmuller at the Fraunhofer-Institute for High-Speed
Dynamics, Ernst Mach Institute in Germany (georg.ganzenmueller at emi.fhg.de). Contact him directly if you
have questions.
Examples: examples/USER/smd
USER-SMTBQ package
Supporting info:
This package implements the Second Moment Tight Binding - QEq (SMTB-Q) potential for the description of
ionocovalent bonds in oxides.
There are example scripts for using this package in examples/USER/smtbq.
See this doc page to get started:
pair_style smtbq
The persons who created the USER-SMTBQ package are Nicolas Salles, Emile Maras, Olivier Politano,
Robert Tetot, who can be contacted at these email addresses: lammps@u-bourgogne.fr, nsalles@laas.fr.
Contact them directly if you have any questions.
Examples: examples/USER/smtbq
USER-SPH package
Supporting info:
This package implements smoothed particle hydrodynamics (SPH) in LAMMPS. Currently, the package has
the following features:
* Tait, ideal gas, Lennard-Jones equation of states, full support for complete (i.e. internal-energy dependent)
equations of state
* Plain or Monaghans XSPH integration of the equations of motion
LAMMPS Users Manual
82

* Density continuity or density summation to propagate the density field
* Commands to set internal energy and density of particles from the input script
* Output commands to access internal energy and density for dumping and thermo output
See the file doc/PDF/SPH_LAMMPS_userguide.pdf to get started.
There are example scripts for using this package in examples/USER/sph.
The person who created this package is Georg Ganzenmuller at the Fraunhofer-Institute for High-Speed
Dynamics, Ernst Mach Institute in Germany (georg.ganzenmueller at emi.fhg.de). Contact him directly if you
have questions.
Examples: examples/USER/sph
USER-TALLY package
Supporting info:
Examples: examples/USER/tally
USER-VTK package
LAMMPS Users Manual
83

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
5. Accelerating LAMMPS performance
This section describes various methods for improving LAMMPS performance for different classes of
problems running on different kinds of machines.
There are two thrusts to the discussion that follows. The first is using code options that implement alternate
algorithms that can speed-up a simulation. The second is to use one of the several accelerator packages
provided with LAMMPS that contain code optimized for certain kinds of hardware, including multi-core
CPUs, GPUs, and Intel Xeon Phi coprocessors.
5.1 Measuring performance• 5.2 Algorithms and code options to boost performace• 5.3 Accelerator packages with optimized styles• 5.3.1 GPU package• 5.3.2 USER-INTEL package• 5.3.3 KOKKOS package• 5.3.4 USER-OMP package• 5.3.5 OPT package• 5.4 Comparison of various accelerator packages•
The Benchmark page of the LAMMPS web site gives performance results for the various accelerator
packages discussed in Section 5.2, for several of the standard LAMMPS benchmark problems, as a function
of problem size and number of compute nodes, on different hardware platforms.
5.1 Measuring performance
Before trying to make your simulation run faster, you should understand how it currently performs and where
the bottlenecks are.
The best way to do this is run the your system (actual number of atoms) for a modest number of timesteps
(say 100 steps) on several different processor counts, including a single processor if possible. Do this for an
equilibrium version of your system, so that the 100-step timings are representative of a much longer run.
There is typically no need to run for 1000s of timesteps to get accurate timings; you can simply extrapolate
from short runs.
For the set of runs, look at the timing data printed to the screen and log file at the end of each LAMMPS run.
This section of the manual has an overview.
Running on one (or a few processors) should give a good estimate of the serial performance and what portions
of the timestep are taking the most time. Running the same problem on a few different processor counts
should give an estimate of parallel scalability. I.e. if the simulation runs 16x faster on 16 processors, its 100%
parallel efficient; if it runs 8x faster on 16 processors, it's 50% efficient.
The most important data to look at in the timing info is the timing breakdown and relative percentages. For
example, trying different options for speeding up the long-range solvers will have little impact if they only
consume 10% of the run time. If the pairwise time is dominating, you may want to look at GPU or OMP
versions of the pair style, as discussed below. Comparing how the percentages change as you increase the
LAMMPS Users Manual
84

processor count gives you a sense of how different operations within the timestep are scaling. Note that if you
are running with a Kspace solver, there is additional output on the breakdown of the Kspace time. For PPPM,
this includes the fraction spent on FFTs, which can be communication intensive.
Another important detail in the timing info are the histograms of atoms counts and neighbor counts. If these
vary widely across processors, you have a load-imbalance issue. This often results in inaccurate relative
timing data, because processors have to wait when communication occurs for other processors to catch up.
Thus the reported times for "Communication" or "Other" may be higher than they really are, due to
load-imbalance. If this is an issue, you can uncomment the MPI_Barrier() lines in src/timer.cpp, and
recompile LAMMPS, to obtain synchronized timings.
5.2 General strategies
NOTE: this section 5.2 is still a work in progress
Here is a list of general ideas for improving simulation performance. Most of them are only applicable to
certain models and certain bottlenecks in the current performance, so let the timing data you generate be your
guide. It is hard, if not impossible, to predict how much difference these options will make, since it is a
function of problem size, number of processors used, and your machine. There is no substitute for identifying
performance bottlenecks, and trying out various options.
rRESPA• 2-FFT PPPM• Staggered PPPM• single vs double PPPM• partial charge PPPM• verlet/split run style• processor command for proc layout and numa layout• load-balancing: balance and fix balance•
2-FFT PPPM, also called analytic differentiation or ad PPPM, uses 2 FFTs instead of the 4 FFTs used by the
default ik differentiation PPPM. However, 2-FFT PPPM also requires a slightly larger mesh size to achieve
the same accuracy as 4-FFT PPPM. For problems where the FFT cost is the performance bottleneck (typically
large problems running on many processors), 2-FFT PPPM may be faster than 4-FFT PPPM.
Staggered PPPM performs calculations using two different meshes, one shifted slightly with respect to the
other. This can reduce force aliasing errors and increase the accuracy of the method, but also doubles the
amount of work required. For high relative accuracy, using staggered PPPM allows one to half the mesh size
in each dimension as compared to regular PPPM, which can give around a 4x speedup in the kspace time.
However, for low relative accuracy, using staggered PPPM gives little benefit and can be up to 2x slower in
the kspace time. For example, the rhodopsin benchmark was run on a single processor, and results for kspace
time vs. relative accuracy for the different methods are shown in the figure below. For this system, staggered
PPPM (using ik differentiation) becomes useful when using a relative accuracy of slightly greater than 1e-5
and above.
LAMMPS Users Manual
85

NOTE: Using staggered PPPM may not give the same increase in accuracy of energy and pressure as it does
in forces, so some caution must be used if energy and/or pressure are quantities of interest, such as when using
a barostat.
5.3 Packages with optimized styles
Accelerated versions of various pair_style, fixes, computes, and other commands have been added to
LAMMPS, which will typically run faster than the standard non-accelerated versions. Some require
appropriate hardware to be present on your system, e.g. GPUs or Intel Xeon Phi coprocessors.
All of these commands are in packages provided with LAMMPS. An overview of packages is give in Section
packages.
These are the accelerator packages currently in LAMMPS, either as standard or user packages:
GPU Package for NVIDIA GPUs as well as OpenCL support
USER-INTEL Package for Intel CPUs and Intel Xeon Phi
KOKKOS Package for Nvidia GPUs, Intel Xeon Phi, and OpenMP threading
USER-OMP Package for OpenMP threading and generic CPU optimizations
OPT Package generic CPU optimizations
Inverting this list, LAMMPS currently has acceleration support for three kinds of hardware, via the listed
packages:
Many-core CPUs USER-INTEL, KOKKOS, USER-OMP, OPT packages
NVIDIA GPUs GPU, KOKKOS packages
Intel Phi USER-INTEL, KOKKOS packages
Which package is fastest for your hardware may depend on the size problem you are running and what
commands (accelerated and non-accelerated) are invoked by your input script. While these doc pages include
LAMMPS Users Manual
86
performance guidelines, there is no substitute for trying out the different packages appropriate to your
hardware.
Any accelerated style has the same name as the corresponding standard style, except that a suffix is appended.
Otherwise, the syntax for the command that uses the style is identical, their functionality is the same, and the
numerical results it produces should also be the same, except for precision and round-off effects.
For example, all of these styles are accelerated variants of the Lennard-Jones pair_style lj/cut:
pair_style lj/cut/gpu• pair_style lj/cut/intel• pair_style lj/cut/kk• pair_style lj/cut/omp• pair_style lj/cut/opt•
To see what accelerate styles are currently available, see Section 3.5 of the manual. The doc pages for
individual commands (e.g. pair lj/cut or fix nve) also list any accelerated variants available for that style.
To use an accelerator package in LAMMPS, and one or more of the styles it provides, follow these general
steps. Details vary from package to package and are explained in the individual accelerator doc pages, listed
above:
build the accelerator library only for GPU package
install the accelerator package make yes-opt, make yes-user-intel, etc
add compile/link flags to Makefile.machine in src/MAKE only for USER-INTEL, KOKKOS,
USER-OMP, OPT packages
re-build LAMMPS make machine
prepare and test a regular LAMMPS simulation lmp_machine -in in.script; mpirun -np
32 lmp_machine -in in.script
enable specific accelerator support via '-k on' command-line switch, only needed for KOKKOS package
set any needed options for the package via "-pk" command-line switch
or package command, only if defaults need to be changed
use accelerated styles in your input via "-sf" command-line switch or
suffix command lmp_machine -in in.script -sf gpu
Note that the first 4 steps can be done as a single command, using the src/Make.py tool. This tool is discussed
in Section 2.4 of the manual, and its use is illustrated in the individual accelerator sections. Typically these
steps only need to be done once, to create an executable that uses one or more accelerator packages.
The last 4 steps can all be done from the command-line when LAMMPS is launched, without changing your
input script, as illustrated in the individual accelerator sections. Or you can add package and suffix commands
to your input script.
NOTE: With a few exceptions, you can build a single LAMMPS executable with all its accelerator packages
installed. Note however that the USER-INTEL and KOKKOS packages require you to choose one of their
hardware options when building for a specific platform. I.e. CPU or Phi option for the USER-INTEL package.
Or the OpenMP, Cuda, or Phi option for the KOKKOS package.
These are the exceptions. You cannot build a single executable with:
LAMMPS Users Manual
87
both the USER-INTEL Phi and KOKKOS Phi options• the USER-INTEL Phi or Kokkos Phi option, and the GPU package•
See the examples/accelerate/README and make.list files for sample Make.py commands that build
LAMMPS with any or all of the accelerator packages. As an example, here is a command that builds with all
the GPU related packages installed (GPU, KOKKOS with Cuda), including settings to build the needed
auxiliary GPU libraries for Kepler GPUs:
Make.py -j 16 -p omp gpu kokkos -cc nvcc wrap=mpi -gpu mode=double arch=35 -kokkos cuda arch=35 lib-all file mpi
The examples/accelerate directory also has input scripts that can be used with all of the accelerator packages.
See its README file for details.
Likewise, the bench directory has FERMI and KEPLER and PHI sub-directories with Make.py commands
and input scripts for using all the accelerator packages on various machines. See the README files in those
dirs.
As mentioned above, the Benchmark page of the LAMMPS web site gives performance results for the various
accelerator packages for several of the standard LAMMPS benchmark problems, as a function of problem size
and number of compute nodes, on different hardware platforms.
Here is a brief summary of what the various packages provide. Details are in the individual accelerator
sections.
Styles with a "gpu" suffix are part of the GPU package, and can be run on NVIDIA GPUs. The
speed-up on a GPU depends on a variety of factors, discussed in the accelerator sections.
•
Styles with an "intel" suffix are part of the USER-INTEL package. These styles support vectorized
single and mixed precision calculations, in addition to full double precision. In extreme cases, this can
provide speedups over 3.5x on CPUs. The package also supports acceleration in "offload" mode to
Intel(R) Xeon Phi(TM) coprocessors. This can result in additional speedup over 2x depending on the
hardware configuration.
•
Styles with a "kk" suffix are part of the KOKKOS package, and can be run using OpenMP on
multicore CPUs, on an NVIDIA GPU, or on an Intel Xeon Phi in "native" mode. The speed-up
depends on a variety of factors, as discussed on the KOKKOS accelerator page.
•
Styles with an "omp" suffix are part of the USER-OMP package and allow a pair-style to be run in
multi-threaded mode using OpenMP. This can be useful on nodes with high-core counts when using
less MPI processes than cores is advantageous, e.g. when running with PPPM so that FFTs are run on
fewer MPI processors or when the many MPI tasks would overload the available bandwidth for
communication.
•
Styles with an "opt" suffix are part of the OPT package and typically speed-up the pairwise
calculations of your simulation by 5-25% on a CPU.
•
The individual accelerator package doc pages explain:
what hardware and software the accelerated package requires• how to build LAMMPS with the accelerated package• how to run with the accelerated package either via command-line switches or modifying the input
script
•
speed-ups to expect• guidelines for best performance• restrictions•
LAMMPS Users Manual
88

5.4 Comparison of various accelerator packages
NOTE: this section still needs to be re-worked with additional KOKKOS and USER-INTEL information.
The next section compares and contrasts the various accelerator options, since there are multiple ways to
perform OpenMP threading, run on GPUs, and run on Intel Xeon Phi coprocessors.
All 3 of these packages accelerate a LAMMPS calculation using NVIDIA hardware, but they do it in different
ways.
As a consequence, for a particular simulation on specific hardware, one package may be faster than the other.
We give guidelines below, but the best way to determine which package is faster for your input script is to try
both of them on your machine. See the benchmarking section below for examples where this has been done.
Guidelines for using each package optimally:
The GPU package allows you to assign multiple CPUs (cores) to a single GPU (a common
configuration for "hybrid" nodes that contain multicore CPU(s) and GPU(s)) and works effectively in
this mode.
•
The GPU package moves per-atom data (coordinates, forces) back-and-forth between the CPU and
GPU every timestep. The KOKKOS/CUDA package only does this on timesteps when a CPU
calculation is required (e.g. to invoke a fix or compute that is non-GPU-ized). Hence, if you can
formulate your input script to only use GPU-ized fixes and computes, and avoid doing I/O too often
(thermo output, dump file snapshots, restart files), then the data transfer cost of the KOKKOS/CUDA
package can be very low, causing it to run faster than the GPU package.
•
The GPU package is often faster than the KOKKOS/CUDA package, if the number of atoms per GPU
is smaller. The crossover point, in terms of atoms/GPU at which the KOKKOS/CUDA package
becomes faster depends strongly on the pair style. For example, for a simple Lennard Jones system
the crossover (in single precision) is often about 50K-100K atoms per GPU. When performing double
precision calculations the crossover point can be significantly smaller.
•
Both packages compute bonded interactions (bonds, angles, etc) on the CPU. If the GPU package is
running with several MPI processes assigned to one GPU, the cost of computing the bonded
interactions is spread across more CPUs and hence the GPU package can run faster.
•
When using the GPU package with multiple CPUs assigned to one GPU, its performance depends to
some extent on high bandwidth between the CPUs and the GPU. Hence its performance is affected if
full 16 PCIe lanes are not available for each GPU. In HPC environments this can be the case if
S2050/70 servers are used, where two devices generally share one PCIe 2.0 16x slot. Also many
multi-GPU mainboards do not provide full 16 lanes to each of the PCIe 2.0 16x slots.
•
Differences between the two packages:
The GPU package accelerates only pair force, neighbor list, and PPPM calculations.• The GPU package requires neighbor lists to be built on the CPU when using exclusion lists, hybrid
pair styles, or a triclinic simulation box.
•
LAMMPS Users Manual
89

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
Return to Section accelerate overview
5.3.1 GPU package
The GPU package was developed by Mike Brown at ORNL and his collaborators, particularly Trung Nguyen
(ORNL). It provides GPU versions of many pair styles, including the 3-body Stillinger-Weber pair style, and
for kspace_style pppm for long-range Coulombics. It has the following general features:
It is designed to exploit common GPU hardware configurations where one or more GPUs are coupled
to many cores of one or more multi-core CPUs, e.g. within a node of a parallel machine.
•
Atom-based data (e.g. coordinates, forces) moves back-and-forth between the CPU(s) and GPU every
timestep.
•
Neighbor lists can be built on the CPU or on the GPU• The charge assignment and force interpolation portions of PPPM can be run on the GPU. The FFT
portion, which requires MPI communication between processors, runs on the CPU.
•
Asynchronous force computations can be performed simultaneously on the CPU(s) and GPU.• It allows for GPU computations to be performed in single or double precision, or in mixed-mode
precision, where pairwise forces are computed in single precision, but accumulated into
double-precision force vectors.
•
LAMMPS-specific code is in the GPU package. It makes calls to a generic GPU library in the lib/gpu
directory. This library provides NVIDIA support as well as more general OpenCL support, so that the
same functionality can eventually be supported on a variety of GPU hardware.
•
Here is a quick overview of how to enable and use the GPU package:
build the library in lib/gpu for your GPU hardware with the desired precision settings• install the GPU package and build LAMMPS as usual• use the mpirun command to set the number of MPI tasks/node which determines the number of MPI
tasks/GPU
•
specify the # of GPUs per node• use GPU styles in your input script•
The latter two steps can be done using the "-pk gpu" and "-sf gpu" command-line switches respectively. Or
the effect of the "-pk" or "-sf" switches can be duplicated by adding the package gpu or suffix gpu commands
respectively to your input script.
Required hardware/software:
To use this package, you currently need to have an NVIDIA GPU and install the NVIDIA Cuda software on
your system:
Check if you have an NVIDIA GPU: cat /proc/driver/nvidia/gpus/0/information• Go to http://www.nvidia.com/object/cuda_get.html• Install a driver and toolkit appropriate for your system (SDK is not necessary)• Run lammps/lib/gpu/nvc_get_devices (after building the GPU library, see below) to list supported
devices and properties
•
Building LAMMPS with the GPU package:
LAMMPS Users Manual
90
This requires two steps (a,b): build the GPU library, then build LAMMPS with the GPU package.
You can do both these steps in one line, using the src/Make.py script, described in Section 2.4 of the manual.
Type "Make.py -h" for help. If run from the src directory, this command will create src/lmp_gpu using
src/MAKE/Makefile.mpi as the starting Makefile.machine:
Make.py -p gpu -gpu mode=single arch=31 -o gpu -a lib-gpu file mpi
Or you can follow these two (a,b) steps:
(a) Build the GPU library
The GPU library is in lammps/lib/gpu. Select a Makefile.machine (in lib/gpu) appropriate for your system.
You should pay special attention to 3 settings in this makefile.
CUDA_HOME = needs to be where NVIDIA Cuda software is installed on your system• CUDA_ARCH = needs to be appropriate to your GPUs• CUDA_PREC = precision (double, mixed, single) you desire•
See lib/gpu/Makefile.linux.double for examples of the ARCH settings for different GPU choices, e.g. Fermi
vs Kepler. It also lists the possible precision settings:
CUDA_PREC = -D_SINGLE_SINGLE # single precision for all calculations
CUDA_PREC = -D_DOUBLE_DOUBLE # double precision for all calculations
CUDA_PREC = -D_SINGLE_DOUBLE # accumulation of forces, etc, in double
The last setting is the mixed mode referred to above. Note that your GPU must support double precision to
use either the 2nd or 3rd of these settings.
To build the library, type:
make -f Makefile.machine
If successful, it will produce the files libgpu.a and Makefile.lammps.
The latter file has 3 settings that need to be appropriate for the paths and settings for the CUDA system
software on your machine. Makefile.lammps is a copy of the file specified by the EXTRAMAKE setting in
Makefile.machine. You can change EXTRAMAKE or create your own Makefile.lammps.machine if needed.
Note that to change the precision of the GPU library, you need to re-build the entire library. Do a "clean" first,
e.g. "make -f Makefile.linux clean", followed by the make command above.
(b) Build LAMMPS with the GPU package
cd lammps/src
make yes-gpu
make machine
No additional compile/link flags are needed in Makefile.machine.
Note that if you change the GPU library precision (discussed above) and rebuild the GPU library, then you
also need to re-install the GPU package and re-build LAMMPS, so that all affected files are re-compiled and
LAMMPS Users Manual
91
linked to the new GPU library.
Run with the GPU package from the command line:
The mpirun or mpiexec command sets the total number of MPI tasks used by LAMMPS (one or multiple per
compute node) and the number of MPI tasks used per node. E.g. the mpirun command in MPICH does this via
its -np and -ppn switches. Ditto for OpenMPI via -np and -npernode.
When using the GPU package, you cannot assign more than one GPU to a single MPI task. However multiple
MPI tasks can share the same GPU, and in many cases it will be more efficient to run this way. Likewise it
may be more efficient to use less MPI tasks/node than the available # of CPU cores. Assignment of multiple
MPI tasks to a GPU will happen automatically if you create more MPI tasks/node than there are GPUs/mode.
E.g. with 8 MPI tasks/node and 2 GPUs, each GPU will be shared by 4 MPI tasks.
Use the "-sf gpu" command-line switch, which will automatically append "gpu" to styles that support it. Use
the "-pk gpu Ng" command-line switch to set Ng = # of GPUs/node to use.
lmp_machine -sf gpu -pk gpu 1 -in in.script # 1 MPI task uses 1 GPU
mpirun -np 12 lmp_machine -sf gpu -pk gpu 2 -in in.script # 12 MPI tasks share 2 GPUs on a single 16-core (or whatever) node
mpirun -np 48 -ppn 12 lmp_machine -sf gpu -pk gpu 2 -in in.script # ditto on 4 16-core nodes
Note that if the "-sf gpu" switch is used, it also issues a default package gpu 1 command, which sets the
number of GPUs/node to 1.
Using the "-pk" switch explicitly allows for setting of the number of GPUs/node to use and additional options.
Its syntax is the same as same as the "package gpu" command. See the package command doc page for
details, including the default values used for all its options if it is not specified.
Note that the default for the package gpu command is to set the Newton flag to "off" pairwise interactions. It
does not affect the setting for bonded interactions (LAMMPS default is "on"). The "off" setting for pairwise
interaction is currently required for GPU package pair styles.
Or run with the GPU package by editing an input script:
The discussion above for the mpirun/mpiexec command, MPI tasks/node, and use of multiple MPI tasks/GPU
is the same.
Use the suffix gpu command, or you can explicitly add an "gpu" suffix to individual styles in your input
script, e.g.
pair_style lj/cut/gpu 2.5
You must also use the package gpu command to enable the GPU package, unless the "-sf gpu" or "-pk gpu"
command-line switches were used. It specifies the number of GPUs/node to use, as well as other options.
Speed-ups to expect:
The performance of a GPU versus a multi-core CPU is a function of your hardware, which pair style is used,
the number of atoms/GPU, and the precision used on the GPU (double, single, mixed).
See the Benchmark page of the LAMMPS web site for performance of the GPU package on various hardware,
LAMMPS Users Manual
92
including the Titan HPC platform at ORNL.
You should also experiment with how many MPI tasks per GPU to use to give the best performance for your
problem and machine. This is also a function of the problem size and the pair style being using. Likewise, you
should experiment with the precision setting for the GPU library to see if single or mixed precision will give
accurate results, since they will typically be faster.
Guidelines for best performance:
Using multiple MPI tasks per GPU will often give the best performance, as allowed my most
multi-core CPU/GPU configurations.
•
If the number of particles per MPI task is small (e.g. 100s of particles), it can be more efficient to run
with fewer MPI tasks per GPU, even if you do not use all the cores on the compute node.
•
The package gpu command has several options for tuning performance. Neighbor lists can be built on
the GPU or CPU. Force calculations can be dynamically balanced across the CPU cores and GPUs.
GPU-specific settings can be made which can be optimized for different hardware. See the packakge
command doc page for details.
•
As described by the package gpu command, GPU accelerated pair styles can perform computations
asynchronously with CPU computations. The "Pair" time reported by LAMMPS will be the maximum
of the time required to complete the CPU pair style computations and the time required to complete
the GPU pair style computations. Any time spent for GPU-enabled pair styles for computations that
run simultaneously with bond, angle, dihedral, improper, and long-range calculations will not be
included in the "Pair" time.
•
When the mode setting for the package gpu command is force/neigh, the time for neighbor list
calculations on the GPU will be added into the "Pair" time, not the "Neigh" time. An additional
breakdown of the times required for various tasks on the GPU (data copy, neighbor calculations, force
computations, etc) are output only with the LAMMPS screen output (not in the log file) at the end of
each run. These timings represent total time spent on the GPU for each routine, regardless of
asynchronous CPU calculations.
•
The output section "GPU Time Info (average)" reports "Max Mem / Proc". This is the maximum
memory used at one time on the GPU for data storage by a single MPI process.
•
Restrictions:
None.
LAMMPS Users Manual
93

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
Return to Section accelerate overview
5.3.2 USER-INTEL package
The USER-INTEL package is maintained by Mike Brown at Intel Corporation. It provides two methods for
accelerating simulations, depending on the hardware you have. The first is acceleration on Intel CPUs by
running in single, mixed, or double precision with vectorization. The second is acceleration on Intel Xeon Phi
coprocessors via offloading neighbor list and non-bonded force calculations to the Phi. The same C++ code is
used in both cases. When offloading to a coprocessor from a CPU, the same routine is run twice, once on the
CPU and once with an offload flag. This allows LAMMPS to run on the CPU cores and coprocessor cores
simultaneously.
Currently Available USER-INTEL Styles:
Angle Styles: charmm, harmonic• Bond Styles: fene, harmonic• Dihedral Styles: charmm, harmonic, opls• Fixes: nve, npt, nvt, nvt/sllod• Improper Styles: cvff, harmonic• Pair Styles: buck/coul/cut, buck/coul/long, buck, eam, gayberne, charmm/coul/long, lj/cut,
lj/cut/coul/long, sw, tersoff
•
K-Space Styles: pppm•
Speed-ups to expect:
The speedups will depend on your simulation, the hardware, which styles are used, the number of atoms, and
the floating-point precision mode. Performance improvements are shown compared to LAMMPS without
using other acceleration packages as these are under active development (and subject to performance
changes). The measurements were performed using the input files available in the src/USER-INTEL/TEST
directory. These are scalable in size; the results given are with 512K particles (524K for Liquid Crystal). Most
of the simulations are standard LAMMPS benchmarks (indicated by the filename extension in parenthesis)
with modifications to the run length and to add a warmup run (for use with offload benchmarks).
LAMMPS Users Manual
94
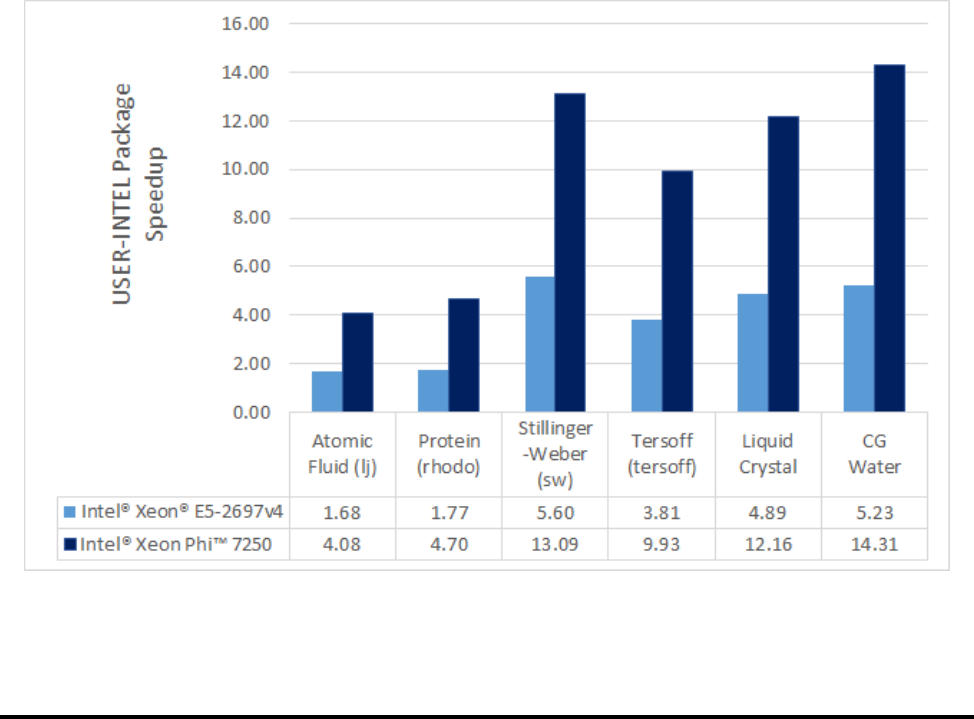
Results are speedups obtained on Intel Xeon E5-2697v4 processors (code-named Broadwell) and Intel Xeon
Phi 7250 processors (code-named Knights Landing) with "18 Jun 2016" LAMMPS built with Intel Parallel
Studio 2016 update 3. Results are with 1 MPI task per physical core. See src/USER-INTEL/TEST/README
for the raw simulation rates and instructions to reproduce.
Quick Start for Experienced Users:
LAMMPS should be built with the USER-INTEL package installed. Simulations should be run with 1 MPI
task per physical core, not hardware thread.
For Intel Xeon CPUs:
Edit src/MAKE/OPTIONS/Makefile.intel_cpu_intelmpi as necessary.• If using kspace_style pppm in the input script, add "neigh_modify binsize 3" and "kspace_modify diff
ad" to the input script for better performance.
•
"-pk intel 0 omp 2 -sf intel" added to LAMMPS command-line•
For Intel Xeon Phi CPUs for simulations without kspace_style pppm in the input script
Edit src/MAKE/OPTIONS/Makefile.knl as necessary.• Runs should be performed using MCDRAM.• "-pk intel 0 omp 2 -sf intel" or "-pk intel 0 omp 4 -sf intel" should be added to the LAMMPS
command-line. Choice for best performance will depend on the simulation.
•
For Intel Xeon Phi CPUs for simulations with kspace_style pppm in the input script:
Edit src/MAKE/OPTIONS/Makefile.knl as necessary.• Runs should be performed using MCDRAM.•
LAMMPS Users Manual
95

Add "neigh_modify binsize 3" to the input script for better performance.• Add "kspace_modify diff ad" to the input script for better performance.• export KMP_AFFINITY=none• "-pk intel 0 omp 3 lrt yes -sf intel" or "-pk intel 0 omp 1 lrt yes -sf intel" added to LAMMPS
command-line. Choice for best performance will depend on the simulation.
•
For Intel Xeon Phi coprocessors (Offload):
Edit src/MAKE/OPTIONS/Makefile.intel_coprocessor as necessary• "-pk intel N omp 1" added to command-line where N is the number of coprocessors per node.•
Required hardware/software:
In order to use offload to coprocessors, an Intel Xeon Phi coprocessor and an Intel compiler are required. For
this, the recommended version of the Intel compiler is 14.0.1.106 or versions 15.0.2.044 and higher.
Although any compiler can be used with the USER-INTEL package, currently, vectorization directives are
disabled by default when not using Intel compilers due to lack of standard support and observations of
decreased performance. The OpenMP standard now supports directives for vectorization and we plan to
transition the code to this standard once it is available in most compilers. We expect this to allow improved
performance and support with other compilers.
For Intel Xeon Phi x200 series processors (code-named Knights Landing), there are multiple configuration
options for the hardware. For best performance, we recommend that the MCDRAM is configured in "Flat"
mode and with the cluster mode set to "Quadrant" or "SNC4". "Cache" mode can also be used, although the
performance might be slightly lower.
Notes about Simultaneous Multithreading:
Modern CPUs often support Simultaneous Multithreading (SMT). On Intel processors, this is called
Hyper-Threading (HT) technology. SMT is hardware support for running multiple threads efficiently on a
single core. Hardware threads or logical cores are often used to refer to the number of threads that are
supported in hardware. For example, the Intel Xeon E5-2697v4 processor is described as having 36 cores and
72 threads. This means that 36 MPI processes or OpenMP threads can run simultaneously on separate cores,
but that up to 72 MPI processes or OpenMP threads can be running on the CPU without costly operating
system context switches.
Molecular dynamics simulations will often run faster when making use of SMT. If a thread becomes stalled,
for example because it is waiting on data that has not yet arrived from memory, another thread can start
running so that the CPU pipeline is still being used efficiently. Although benefits can be seen by launching a
MPI task for every hardware thread, for multinode simulations, we recommend that OpenMP threads are used
for SMT instead, either with the USER-INTEL package, USER-OMP package, or KOKKOS package. In the
example above, up to 36X speedups can be observed by using all 36 physical cores with LAMMPS. By using
all 72 hardware threads, an additional 10-30% performance gain can be achieved.
The BIOS on many platforms allows SMT to be disabled, however, we do not recommend this on modern
processors as there is little to no benefit for any software package in most cases. The operating system will
report every hardware thread as a separate core allowing one to determine the number of hardware threads
available. On Linux systems, this information can normally be obtained with:
cat /proc/cpuinfo
LAMMPS Users Manual
96
Building LAMMPS with the USER-INTEL package:
The USER-INTEL package must be installed into the source directory:
make yes-user-intel
Several example Makefiles for building with the Intel compiler are included with LAMMPS in the
src/MAKE/OPTIONS/ directory:
Makefile.intel_cpu_intelmpi # Intel Compiler, Intel MPI, No Offload
Makefile.knl # Intel Compiler, Intel MPI, No Offload
Makefile.intel_cpu_mpich # Intel Compiler, MPICH, No Offload
Makefile.intel_cpu_openpmi # Intel Compiler, OpenMPI, No Offload
Makefile.intel_coprocessor # Intel Compiler, Intel MPI, Offload
Makefile.knl is identical to Makefile.intel_cpu_intelmpi except that it explicitly specifies that vectorization
should be for Intel Xeon Phi x200 processors making it easier to cross-compile. For users with recent
installations of Intel Parallel Studio, the process can be as simple as:
make yes-user-intel
source /opt/intel/parallel_studio_xe_2016.3.067/psxevars.sh
# or psxevars.csh for C-shell
make intel_cpu_intelmpi
Alternatively, the build can be accomplished with the src/Make.py script, described in Section 2.4 of the
manual. Type "Make.py -h" for help. For an example:
Make.py -v -p intel omp -intel cpu -a file intel_cpu_intelmpi
Note that if you build with support for a Phi coprocessor, the same binary can be used on nodes with or
without coprocessors installed. However, if you do not have coprocessors on your system, building without
offload support will produce a smaller binary.
The general requirements for Makefiles with the USER-INTEL package are as follows.
"-DLAMMPS_MEMALIGN=64" is required for CCFLAGS. When using Intel compilers, "-restrict" is
required and "-qopenmp" is highly recommended for CCFLAGS and LINKFLAGS. LIB should include
"-ltbbmalloc". For builds supporting offload, "-DLMP_INTEL_OFFLOAD" is required for CCFLAGS and
"-qoffload" is required for LINKFLAGS. Other recommended CCFLAG options for best performance are
"-O2 -fno-alias -ansi-alias -qoverride-limits fp-model fast=2 -no-prec-div". The Make.py command will add
all of these automatically.
NOTE: The vectorization and math capabilities can differ depending on the CPU. For Intel compilers, the "-x"
flag specifies the type of processor for which to optimize. "-xHost" specifies that the compiler should build for
the processor used for compiling. For Intel Xeon Phi x200 series processors, this option is "-xMIC-AVX512".
For fourth generation Intel Xeon (v4/Broadwell) processors, "-xCORE-AVX2" should be used. For older Intel
Xeon processors, "-xAVX" will perform best in general for the different simulations in LAMMPS. The
default in most of the example Makefiles is to use "-xHost", however this should not be used when
cross-compiling.
Running LAMMPS with the USER-INTEL package:
Running LAMMPS with the USER-INTEL package is similar to normal use with the exceptions that one
should 1) specify that LAMMPS should use the USER-INTEL package, 2) specify the number of OpenMP
LAMMPS Users Manual
97
threads, and 3) optionally specify the specific LAMMPS styles that should use the USER-INTEL package. 1)
and 2) can be performed from the command-line or by editing the input script. 3) requires editing the input
script. Advanced performance tuning options are also described below to get the best performance.
When running on a single node (including runs using offload to a coprocessor), best performance is normally
obtained by using 1 MPI task per physical core and additional OpenMP threads with SMT. For Intel Xeon
processors, 2 OpenMP threads should be used for SMT. For Intel Xeon Phi CPUs, 2 or 4 OpenMP threads
should be used (best choice depends on the simulation). In cases where the user specifies that LRT mode is
used (described below), 1 or 3 OpenMP threads should be used. For multi-node runs, using 1 MPI task per
physical core will often perform best, however, depending on the machine and scale, users might get better
performance by decreasing the number of MPI tasks and using more OpenMP threads. For performance, the
product of the number of MPI tasks and OpenMP threads should not exceed the number of available hardware
threads in almost all cases.
NOTE: Setting core affinity is often used to pin MPI tasks and OpenMP threads to a core or group of cores so
that memory access can be uniform. Unless disabled at build time, affinity for MPI tasks and OpenMP threads
on the host (CPU) will be set by default on the host when using offload to a coprocessor. In this case, it is
unnecessary to use other methods to control affinity (e.g. taskset, numactl, I_MPI_PIN_DOMAIN, etc.). This
can be disabled with the no_affinity option to the package intel command or by disabling the option at build
time (by adding -DINTEL_OFFLOAD_NOAFFINITY to the CCFLAGS line of your Makefile). Disabling
this option is not recommended, especially when running on a machine with Intel Hyper-Threading
technology disabled.
Run with the USER-INTEL package from the command line:
To enable USER-INTEL optimizations for all available styles used in the input script, the "-sf intel"
command-line switch can be used without any requirement for editing the input script. This switch will
automatically append "intel" to styles that support it. It also invokes a default command: package intel 1. This
package command is used to set options for the USER-INTEL package. The default package command will
specify that USER-INTEL calculations are performed in mixed precision, that the number of OpenMP threads
is specified by the OMP_NUM_THREADS environment variable, and that if coprocessors are present and the
binary was built with offload support, that 1 coprocessor per node will be used with automatic balancing of
work between the CPU and the coprocessor.
You can specify different options for the USER-INTEL package by using the "-pk intel Nphi" command-line
switch with keyword/value pairs as specified in the documentation. Here, Nphi = # of Xeon Phi
coprocessors/node (ignored without offload support). Common options to the USER-INTEL package include
omp to override any OMP_NUM_THREADS setting and specify the number of OpenMP threads, mode to set
the floating-point precision mode, and lrt to enable Long-Range Thread mode as described below. See the
package intel command for details, including the default values used for all its options if not specified, and
how to set the number of OpenMP threads via the OMP_NUM_THREADS environment variable if desired.
Examples (see documentation for your MPI/Machine for differences in launching MPI applications):
mpirun -np 72 -ppn 36 lmp_machine -sf intel -in in.script # 2 nodes, 36 MPI tasks/node, $OMP_NUM_THREADS OpenMP Threads
mpirun -np 72 -ppn 36 lmp_machine -sf intel -in in.script -pk intel 0 omp 2 mode double # Don't use any coprocessors that might be available, use 2 OpenMP threads for each task, use double precision
Or run with the USER-INTEL package by editing an input script:
As an alternative to adding command-line arguments, the input script can be edited to enable the
USER-INTEL package. This requires adding the package intel command to the top of the input script. For the
LAMMPS Users Manual
98
second example above, this would be:
package intel 0 omp 2 mode double
To enable the USER-INTEL package only for individual styles, you can add an "intel" suffix to the individual
style, e.g.:
pair_style lj/cut/intel 2.5
Alternatively, the suffix intel command can be added to the input script to enable USER-INTEL styles for the
commands that follow in the input script.
Tuning for Performance:
NOTE: The USER-INTEL package will perform better with modifications to the input script when PPPM is
used: kspace_modify diff ad and neigh_modify binsize 3 should be added to the input script.
Long-Range Thread (LRT) mode is an option to the package intel command that can improve performance
when using PPPM for long-range electrostatics on processors with SMT. It generates an extra pthread for each
MPI task. The thread is dedicated to performing some of the PPPM calculations and MPI communications.
On Intel Xeon Phi x200 series CPUs, this will likely always improve performance, even on a single node. On
Intel Xeon processors, using this mode might result in better performance when using multiple nodes,
depending on the machine. To use this mode, specify that the number of OpenMP threads is one less than
would normally be used for the run and add the "lrt yes" option to the "-pk" command-line suffix or "package
intel" command. For example, if a run would normally perform best with "-pk intel 0 omp 4", instead use "-pk
intel 0 omp 3 lrt yes". When using LRT, you should set the environment variable "KMP_AFFINITY=none".
LRT mode is not supported when using offload.
Not all styles are supported in the USER-INTEL package. You can mix the USER-INTEL package with styles
from the OPT package or the USER-OMP package. Of course, this requires that these packages were installed
at build time. This can performed automatically by using "-sf hybrid intel opt" or "-sf hybrid intel omp"
command-line options. Alternatively, the "opt" and "omp" suffixes can be appended manually in the input
script. For the latter, the package omp command must be in the input script or the "-pk omp Nt"
command-line switch must be used where Nt is the number of OpenMP threads. The number of OpenMP
threads should not be set differently for the different packages. Note that the suffix hybrid intel omp command
can also be used within the input script to automatically append the "omp" suffix to styles when
USER-INTEL styles are not available.
When running on many nodes, performance might be better when using fewer OpenMP threads and more
MPI tasks. This will depend on the simulation and the machine. Using the verlet/split run style might also
give better performance for simulations with PPPM electrostatics. Note that this is an alternative to LRT mode
and the two cannot be used together.
Currently, when using Intel MPI with Intel Xeon Phi x200 series CPUs, better performance might be obtained
by setting the environment variable "I_MPI_SHM_LMT=shm" for Linux kernels that do not yet have full
support for AVX-512. Runs on Intel Xeon Phi x200 series processors will always perform better using
MCDRAM. Please consult your system documentation for the best approach to specify that MPI runs are
performed in MCDRAM.
Tuning for Offload Performance:
LAMMPS Users Manual
99
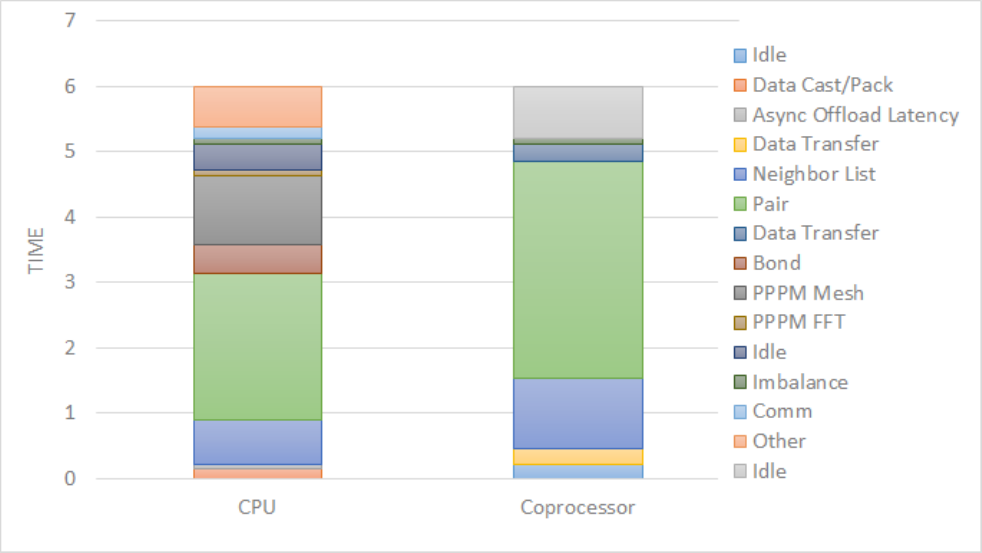
The default settings for offload should give good performance.
When using LAMMPS with offload to Intel coprocessors, best performance will typically be achieved with
concurrent calculations performed on both the CPU and the coprocessor. This is achieved by offloading only a
fraction of the neighbor and pair computations to the coprocessor or using hybrid pair styles where only one
style uses the "intel" suffix. For simulations with long-range electrostatics or bond, angle, dihedral, improper
calculations, computation and data transfer to the coprocessor will run concurrently with computations and
MPI communications for these calculations on the host CPU. This is illustrated in the figure below for the
rhodopsin protein benchmark running on E5-2697v2 processors with a Intel Xeon Phi 7120p coprocessor. In
this plot, the vertical access is time and routines running at the same time are running concurrently on both the
host and the coprocessor.
The fraction of the offloaded work is controlled by the balance keyword in the package intel command. A
balance of 0 runs all calculations on the CPU. A balance of 1 runs all supported calculations on the
coprocessor. A balance of 0.5 runs half of the calculations on the coprocessor. Setting the balance to -1 (the
default) will enable dynamic load balancing that continously adjusts the fraction of offloaded work throughout
the simulation. Because data transfer cannot be timed, this option typically produces results within 5 to 10
percent of the optimal fixed balance.
If running short benchmark runs with dynamic load balancing, adding a short warm-up run (10-20 steps) will
allow the load-balancer to find a near-optimal setting that will carry over to additional runs.
The default for the package intel command is to have all the MPI tasks on a given compute node use a single
Xeon Phi coprocessor. In general, running with a large number of MPI tasks on each node will perform best
with offload. Each MPI task will automatically get affinity to a subset of the hardware threads available on the
coprocessor. For example, if your card has 61 cores, with 60 cores available for offload and 4 hardware
threads per core (240 total threads), running with 24 MPI tasks per node will cause each MPI task to use a
subset of 10 threads on the coprocessor. Fine tuning of the number of threads to use per MPI task or the
number of threads to use per core can be accomplished with keyword settings of the package intel command.
LAMMPS Users Manual
100
The USER-INTEL package has two modes for deciding which atoms will be handled by the coprocessor. This
choice is controlled with the ghost keyword of the package intel command. When set to 0, ghost atoms (atoms
at the borders between MPI tasks) are not offloaded to the card. This allows for overlap of MPI
communication of forces with computation on the coprocessor when the newton setting is "on". The default is
dependent on the style being used, however, better performance may be achieved by setting this option
explicitly.
When using offload with CPU Hyper-Threading disabled, it may help performance to use fewer MPI tasks
and OpenMP threads than available cores. This is due to the fact that additional threads are generated
internally to handle the asynchronous offload tasks.
If pair computations are being offloaded to an Intel Xeon Phi coprocessor, a diagnostic line is printed to the
screen (not to the log file), during the setup phase of a run, indicating that offload mode is being used and
indicating the number of coprocessor threads per MPI task. Additionally, an offload timing summary is
printed at the end of each run. When offloading, the frequency for atom sorting is changed to 1 so that the
per-atom data is effectively sorted at every rebuild of the neighbor lists. All the available coprocessor threads
on each Phi will be divided among MPI tasks, unless the tptask option of the "-pk intel" command-line switch
is used to limit the coprocessor threads per MPI task.
Restrictions:
When offloading to a coprocessor, hybrid styles that require skip lists for neighbor builds cannot be offloaded.
Using hybrid/overlay is allowed. Only one intel accelerated style may be used with hybrid styles.
Special_bonds exclusion lists are not currently supported with offload, however, the same effect can often be
accomplished by setting cutoffs for excluded atom types to 0. None of the pair styles in the USER-INTEL
package currently support the "inner", "middle", "outer" options for rRESPA integration via the run_style
respa command; only the "pair" option is supported.
References:
Brown, W.M., Carrillo, J.-M.Y., Mishra, B., Gavhane, N., Thakker, F.M., De Kraker, A.R., Yamada,
M., Ang, J.A., Plimpton, S.J., "Optimizing Classical Molecular Dynamics in LAMMPS," in Intel
Xeon Phi Processor High Performance Programming: Knights Landing Edition, J. Jeffers, J. Reinders,
A. Sodani, Eds. Morgan Kaufmann.
•
Brown, W. M., Semin, A., Hebenstreit, M., Khvostov, S., Raman, K., Plimpton, S.J. Increasing
Molecular Dynamics Simulation Rates with an 8-Fold Increase in Electrical Power Efficiency. 2016
International Conference for High Performance Computing. In press.
•
Brown, W.M., Carrillo, J.-M.Y., Gavhane, N., Thakkar, F.M., Plimpton, S.J. Optimizing Legacy
Molecular Dynamics Software with Directive-Based Offload. Computer Physics Communications.
2015. 195: p. 95-101.
•
LAMMPS Users Manual
101

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
Return to Section accelerate overview
5.3.3 KOKKOS package
The KOKKOS package was developed primarily by Christian Trott (Sandia) with contributions of various
styles by others, including Sikandar Mashayak (UIUC), Stan Moore (Sandia), and Ray Shan (Sandia). The
underlying Kokkos library was written primarily by Carter Edwards, Christian Trott, and Dan Sunderland (all
Sandia).
The KOKKOS package contains versions of pair, fix, and atom styles that use data structures and macros
provided by the Kokkos library, which is included with LAMMPS in lib/kokkos.
The Kokkos library is part of Trilinos and can also be downloaded from Github. Kokkos is a templated C++
library that provides two key abstractions for an application like LAMMPS. First, it allows a single
implementation of an application kernel (e.g. a pair style) to run efficiently on different kinds of hardware,
such as a GPU, Intel Phi, or many-core CPU.
The Kokkos library also provides data abstractions to adjust (at compile time) the memory layout of basic data
structures like 2d and 3d arrays and allow the transparent utilization of special hardware load and store
operations. Such data structures are used in LAMMPS to store atom coordinates or forces or neighbor lists.
The layout is chosen to optimize performance on different platforms. Again this functionality is hidden from
the developer, and does not affect how the kernel is coded.
These abstractions are set at build time, when LAMMPS is compiled with the KOKKOS package installed.
All Kokkos operations occur within the context of an individual MPI task running on a single node of the
machine. The total number of MPI tasks used by LAMMPS (one or multiple per compute node) is set in the
usual manner via the mpirun or mpiexec commands, and is independent of Kokkos.
Kokkos currently provides support for 3 modes of execution (per MPI task). These are OpenMP (for
many-core CPUs), Cuda (for NVIDIA GPUs), and OpenMP (for Intel Phi). Note that the KOKKOS package
supports running on the Phi in native mode, not offload mode like the USER-INTEL package supports. You
choose the mode at build time to produce an executable compatible with specific hardware.
Here is a quick overview of how to use the KOKKOS package for CPU acceleration, assuming one or more
16-core nodes. More details follow.
use a C++11 compatible compiler
make yes-kokkos
make mpi KOKKOS_DEVICES=OpenMP # build with the KOKKOS package
make kokkos_omp # or Makefile.kokkos_omp already has variable set
Make.py -v -p kokkos -kokkos omp -o mpi -a file mpi # or one-line build via Make.py
mpirun -np 16 lmp_mpi -k on -sf kk -in in.lj # 1 node, 16 MPI tasks/node, no threads
mpirun -np 2 -ppn 1 lmp_mpi -k on t 16 -sf kk -in in.lj # 2 nodes, 1 MPI task/node, 16 threads/task
mpirun -np 2 lmp_mpi -k on t 8 -sf kk -in in.lj # 1 node, 2 MPI tasks/node, 8 threads/task
mpirun -np 32 -ppn 4 lmp_mpi -k on t 4 -sf kk -in in.lj # 8 nodes, 4 MPI tasks/node, 4 threads/task
specify variables and settings in your Makefile.machine that enable OpenMP, GPU, or Phi support• include the KOKKOS package and build LAMMPS•
LAMMPS Users Manual
102
enable the KOKKOS package and its hardware options via the "-k on" command-line switch use
KOKKOS styles in your input script
•
Here is a quick overview of how to use the KOKKOS package for GPUs, assuming one or more nodes, each
with 16 cores and a GPU. More details follow.
discuss use of NVCC, which Makefiles to examine
use a C++11 compatible compiler
KOKKOS_DEVICES = Cuda, OpenMP
KOKKOS_ARCH = Kepler35
make yes-kokkos
make machine
Make.py -p kokkos -kokkos cuda arch=31 -o kokkos_cuda -a file kokkos_cuda
mpirun -np 1 lmp_cuda -k on t 6 -sf kk -in in.lj # one MPI task, 6 threads on CPU
mpirun -np 4 -ppn 1 lmp_cuda -k on t 6 -sf kk -in in.lj # ditto on 4 nodes
mpirun -np 2 lmp_cuda -k on t 8 g 2 -sf kk -in in.lj # two MPI tasks, 8 threads per CPU
mpirun -np 32 -ppn 2 lmp_cuda -k on t 8 g 2 -sf kk -in in.lj # ditto on 16 nodes
Here is a quick overview of how to use the KOKKOS package for the Intel Phi:
use a C++11 compatible compiler
KOKKOS_DEVICES = OpenMP
KOKKOS_ARCH = KNC
make yes-kokkos
make machine
Make.py -p kokkos -kokkos phi -o kokkos_phi -a file mpi
host=MIC, Intel Phi with 61 cores (240 threads/phi via 4x hardware threading):
mpirun -np 1 lmp_g++ -k on t 240 -sf kk -in in.lj # 1 MPI task on 1 Phi, 1*240 = 240
mpirun -np 30 lmp_g++ -k on t 8 -sf kk -in in.lj # 30 MPI tasks on 1 Phi, 30*8 = 240
mpirun -np 12 lmp_g++ -k on t 20 -sf kk -in in.lj # 12 MPI tasks on 1 Phi, 12*20 = 240
mpirun -np 96 -ppn 12 lmp_g++ -k on t 20 -sf kk -in in.lj # ditto on 8 Phis
Required hardware/software:
Kokkos support within LAMMPS must be built with a C++11 compatible compiler. If using gcc, version
4.7.2 or later is required.
To build with Kokkos support for CPUs, your compiler must support the OpenMP interface. You should have
one or more multi-core CPUs so that multiple threads can be launched by each MPI task running on a CPU.
To build with Kokkos support for NVIDIA GPUs, NVIDIA Cuda software version 7.5 or later must be
installed on your system. See the discussion for the GPU package for details of how to check and do this.
NOTE: For good performance of the KOKKOS package on GPUs, you must have Kepler generation GPUs
(or later). The Kokkos library exploits texture cache options not supported by Telsa generation GPUs (or
older).
To build with Kokkos support for Intel Xeon Phi coprocessors, your sysmte must be configured to use them in
"native" mode, not "offload" mode like the USER-INTEL package supports.
LAMMPS Users Manual
103
Building LAMMPS with the KOKKOS package:
You must choose at build time whether to build for CPUs (OpenMP), GPUs, or Phi.
You can do any of these in one line, using the src/Make.py script, described in Section 2.4 of the manual.
Type "Make.py -h" for help. If run from the src directory, these commands will create src/lmp_kokkos_omp,
lmp_kokkos_cuda, and lmp_kokkos_phi. Note that the OMP and PHI options use src/MAKE/Makefile.mpi as
the starting Makefile.machine. The CUDA option uses src/MAKE/OPTIONS/Makefile.kokkos_cuda.
The latter two steps can be done using the "-k on", "-pk kokkos" and "-sf kk" command-line switches
respectively. Or the effect of the "-pk" or "-sf" switches can be duplicated by adding the package kokkos or
suffix kk commands respectively to your input script.
Or you can follow these steps:
CPU-only (run all-MPI or with OpenMP threading):
cd lammps/src
make yes-kokkos
make kokkos_omp
CPU-only (only MPI, no threading):
cd lammps/src
make yes-kokkos
make kokkos_mpi
Intel Xeon Phi (Intel Compiler, Intel MPI):
cd lammps/src
make yes-kokkos
make kokkos_phi
CPUs and GPUs (with MPICH):
cd lammps/src
make yes-kokkos
make kokkos_cuda_mpich
These examples set the KOKKOS-specific OMP, MIC, CUDA variables on the make command line which
requires a GNU-compatible make command. Try "gmake" if your system's standard make complains.
NOTE: If you build using make line variables and re-build LAMMPS twice with different KOKKOS options
and the *same* target, e.g. g++ in the first two examples above, then you *must* perform a "make clean-all"
or "make clean-machine" before each build. This is to force all the KOKKOS-dependent files to be
re-compiled with the new options.
NOTE: Currently, there are no precision options with the KOKKOS package. All compilation and
computation is performed in double precision.
There are other allowed options when building with the KOKKOS package. As above, they can be set either
as variables on the make command line or in Makefile.machine. This is the full list of options, including those
discussed above, Each takes a value shown below. The default value is listed, which is set in the
LAMMPS Users Manual
104
lib/kokkos/Makefile.kokkos file.
#Default settings specific options #Options: force_uvm,use_ldg,rdc
KOKKOS_DEVICES, values = OpenMP, Serial, Pthreads, Cuda, default = OpenMP• KOKKOS_ARCH, values = KNC, SNB, HSW, Kepler, Kepler30, Kepler32, Kepler35, Kepler37,
Maxwell, Maxwell50, Maxwell52, Maxwell53, ARMv8, BGQ, Power7, Power8, default = none
•
KOKKOS_DEBUG, values = yes, no, default = no• KOKKOS_USE_TPLS, values = hwloc, librt, default = none• KOKKOS_CUDA_OPTIONS, values = force_uvm, use_ldg, rdc•
KOKKOS_DEVICE sets the parallelization method used for Kokkos code (within LAMMPS).
KOKKOS_DEVICES=OpenMP means that OpenMP will be used. KOKKOS_DEVICES=Pthreads means
that pthreads will be used. KOKKOS_DEVICES=Cuda means an NVIDIA GPU running CUDA will be used.
If KOKKOS_DEVICES=Cuda, then the lo-level Makefile in the src/MAKE directory must use "nvcc" as its
compiler, via its CC setting. For best performance its CCFLAGS setting should use -O3 and have a
KOKKOS_ARCH setting that matches the compute capability of your NVIDIA hardware and software
installation, e.g. KOKKOS_ARCH=Kepler30. Note the minimal required compute capability is 2.0, but this
will give significantly reduced performance compared to Kepler generation GPUs with compute capability
3.x. For the LINK setting, "nvcc" should not be used; instead use g++ or another compiler suitable for linking
C++ applications. Often you will want to use your MPI compiler wrapper for this setting (i.e. mpicxx).
Finally, the lo-level Makefile must also have a "Compilation rule" for creating *.o files from *.cu files. See
src/Makefile.cuda for an example of a lo-level Makefile with all of these settings.
KOKKOS_USE_TPLS=hwloc binds threads to hardware cores, so they do not migrate during a simulation.
KOKKOS_USE_TPLS=hwloc should always be used if running with KOKKOS_DEVICES=Pthreads for
pthreads. It is not necessary for KOKKOS_DEVICES=OpenMP for OpenMP, because OpenMP provides
alternative methods via environment variables for binding threads to hardware cores. More info on binding
threads to cores is given in Section 5.3.
KOKKOS_ARCH=KNC enables compiler switches needed when compiling for an Intel Phi processor.
KOKKOS_USE_TPLS=librt enables use of a more accurate timer mechanism on most Unix platforms. This
library is not available on all platforms.
KOKKOS_DEBUG is only useful when developing a Kokkos-enabled style within LAMMPS.
KOKKOS_DEBUG=yes enables printing of run-time debugging information that can be useful. It also
enables runtime bounds checking on Kokkos data structures.
KOKKOS_CUDA_OPTIONS are additional options for CUDA.
For more information on Kokkos see the Kokkos programmers' guide here: /lib/kokkos/doc/Kokkos_PG.pdf.
Run with the KOKKOS package from the command line:
The mpirun or mpiexec command sets the total number of MPI tasks used by LAMMPS (one or multiple per
compute node) and the number of MPI tasks used per node. E.g. the mpirun command in MPICH does this via
its -np and -ppn switches. Ditto for OpenMPI via -np and -npernode.
LAMMPS Users Manual
105
When using KOKKOS built with host=OMP, you need to choose how many OpenMP threads per MPI task
will be used (via the "-k" command-line switch discussed below). Note that the product of MPI tasks *
OpenMP threads/task should not exceed the physical number of cores (on a node), otherwise performance will
suffer.
When using the KOKKOS package built with device=CUDA, you must use exactly one MPI task per physical
GPU.
When using the KOKKOS package built with host=MIC for Intel Xeon Phi coprocessor support you need to
insure there are one or more MPI tasks per coprocessor, and choose the number of coprocessor threads to use
per MPI task (via the "-k" command-line switch discussed below). The product of MPI tasks * coprocessor
threads/task should not exceed the maximum number of threads the coprocessor is designed to run, otherwise
performance will suffer. This value is 240 for current generation Xeon Phi(TM) chips, which is 60 physical
cores * 4 threads/core. Note that with the KOKKOS package you do not need to specify how many Phi
coprocessors there are per node; each coprocessors is simply treated as running some number of MPI tasks.
You must use the "-k on" command-line switch to enable the KOKKOS package. It takes additional
arguments for hardware settings appropriate to your system. Those arguments are documented here. The two
most commonly used options are:
-k on t Nt g Ng
The "t Nt" option applies to host=OMP (even if device=CUDA) and host=MIC. For host=OMP, it specifies
how many OpenMP threads per MPI task to use with a node. For host=MIC, it specifies how many Xeon Phi
threads per MPI task to use within a node. The default is Nt = 1. Note that for host=OMP this is effectively
MPI-only mode which may be fine. But for host=MIC you will typically end up using far less than all the 240
available threads, which could give very poor performance.
The "g Ng" option applies to device=CUDA. It specifies how many GPUs per compute node to use. The
default is 1, so this only needs to be specified is you have 2 or more GPUs per compute node.
The "-k on" switch also issues a "package kokkos" command (with no additional arguments) which sets
various KOKKOS options to default values, as discussed on the package command doc page.
Use the "-sf kk" command-line switch, which will automatically append "kk" to styles that support it. Use the
"-pk kokkos" command-line switch if you wish to change any of the default package kokkos optionns set by
the "-k on" command-line switch.
Note that the default for the package kokkos command is to use "full" neighbor lists and set the Newton flag
to "off" for both pairwise and bonded interactions. This typically gives fastest performance. If the newton
command is used in the input script, it can override the Newton flag defaults.
However, when running in MPI-only mode with 1 thread per MPI task, it will typically be faster to use "half"
neighbor lists and set the Newton flag to "on", just as is the case for non-accelerated pair styles. You can do
this with the "-pk" command-line switch.
Or run with the KOKKOS package by editing an input script:
The discussion above for the mpirun/mpiexec command and setting appropriate thread and GPU values for
host=OMP or host=MIC or device=CUDA are the same.
LAMMPS Users Manual
106
You must still use the "-k on" command-line switch to enable the KOKKOS package, and specify its
additional arguments for hardware options appropriate to your system, as documented above.
Use the suffix kk command, or you can explicitly add a "kk" suffix to individual styles in your input script,
e.g.
pair_style lj/cut/kk 2.5
You only need to use the package kokkos command if you wish to change any of its option defaults, as set by
the "-k on" command-line switch.
Speed-ups to expect:
The performance of KOKKOS running in different modes is a function of your hardware, which
KOKKOS-enable styles are used, and the problem size.
Generally speaking, the following rules of thumb apply:
When running on CPUs only, with a single thread per MPI task, performance of a KOKKOS style is
somewhere between the standard (un-accelerated) styles (MPI-only mode), and those provided by the
USER-OMP package. However the difference between all 3 is small (less than 20%).
•
When running on CPUs only, with multiple threads per MPI task, performance of a KOKKOS style is
a bit slower than the USER-OMP package.
•
When running large number of atoms per GPU, KOKKOS is typically faster than the GPU package.• When running on Intel Xeon Phi, KOKKOS is not as fast as the USER-INTEL package, which is
optimized for that hardware.
•
See the Benchmark page of the LAMMPS web site for performance of the KOKKOS package on different
hardware.
Guidelines for best performance:
Here are guidline for using the KOKKOS package on the different hardware configurations listed above.
Many of the guidelines use the package kokkos command See its doc page for details and default settings.
Experimenting with its options can provide a speed-up for specific calculations.
Running on a multi-core CPU:
If N is the number of physical cores/node, then the number of MPI tasks/node * number of threads/task should
not exceed N, and should typically equal N. Note that the default threads/task is 1, as set by the "t" keyword of
the "-k" command-line switch. If you do not change this, no additional parallelism (beyond MPI) will be
invoked on the host CPU(s).
You can compare the performance running in different modes:
run with 1 MPI task/node and N threads/task• run with N MPI tasks/node and 1 thread/task• run with settings in between these extremes•
Examples of mpirun commands in these modes are shown above.
LAMMPS Users Manual
107
When using KOKKOS to perform multi-threading, it is important for performance to bind both MPI tasks to
physical cores, and threads to physical cores, so they do not migrate during a simulation.
If you are not certain MPI tasks are being bound (check the defaults for your MPI installation), binding can be
forced with these flags:
OpenMPI 1.8: mpirun -np 2 -bind-to socket -map-by socket ./lmp_openmpi ...
Mvapich2 2.0: mpiexec -np 2 -bind-to socket -map-by socket ./lmp_mvapich ...
For binding threads with the KOKKOS OMP option, use thread affinity environment variables to force
binding. With OpenMP 3.1 (gcc 4.7 or later, intel 12 or later) setting the environment variable
OMP_PROC_BIND=true should be sufficient. For binding threads with the KOKKOS pthreads option,
compile LAMMPS the KOKKOS HWLOC=yes option, as discussed in Section 2.3.4 of the manual.
Running on GPUs:
Insure the -arch setting in the machine makefile you are using, e.g. src/MAKE/Makefile.cuda, is correct for
your GPU hardware/software (see this section of the manual for details).
The -np setting of the mpirun command should set the number of MPI tasks/node to be equal to the # of
physical GPUs on the node.
Use the "-k" command-line switch to specify the number of GPUs per node, and the number of threads per
MPI task. As above for multi-core CPUs (and no GPU), if N is the number of physical cores/node, then the
number of MPI tasks/node * number of threads/task should not exceed N. With one GPU (and one MPI task)
it may be faster to use less than all the available cores, by setting threads/task to a smaller value. This is
because using all the cores on a dual-socket node will incur extra cost to copy memory from the 2nd socket to
the GPU.
Examples of mpirun commands that follow these rules are shown above.
NOTE: When using a GPU, you will achieve the best performance if your input script does not use any fix or
compute styles which are not yet Kokkos-enabled. This allows data to stay on the GPU for multiple timesteps,
without being copied back to the host CPU. Invoking a non-Kokkos fix or compute, or performing I/O for
thermo or dump output will cause data to be copied back to the CPU.
You cannot yet assign multiple MPI tasks to the same GPU with the KOKKOS package. We plan to support
this in the future, similar to the GPU package in LAMMPS.
You cannot yet use both the host (multi-threaded) and device (GPU) together to compute pairwise interactions
with the KOKKOS package. We hope to support this in the future, similar to the GPU package in LAMMPS.
Running on an Intel Phi:
Kokkos only uses Intel Phi processors in their "native" mode, i.e. not hosted by a CPU.
As illustrated above, build LAMMPS with OMP=yes (the default) and MIC=yes. The latter insures code is
correctly compiled for the Intel Phi. The OMP setting means OpenMP will be used for parallelization on the
Phi, which is currently the best option within Kokkos. In the future, other options may be added.
LAMMPS Users Manual
108
Current-generation Intel Phi chips have either 61 or 57 cores. One core should be excluded for running the
OS, leaving 60 or 56 cores. Each core is hyperthreaded, so there are effectively N = 240 (4*60) or N = 224
(4*56) cores to run on.
The -np setting of the mpirun command sets the number of MPI tasks/node. The "-k on t Nt" command-line
switch sets the number of threads/task as Nt. The product of these 2 values should be N, i.e. 240 or 224. Also,
the number of threads/task should be a multiple of 4 so that logical threads from more than one MPI task do
not run on the same physical core.
Examples of mpirun commands that follow these rules are shown above.
Restrictions:
As noted above, if using GPUs, the number of MPI tasks per compute node should equal to the number of
GPUs per compute node. In the future Kokkos will support assigning multiple MPI tasks to a single GPU.
Currently Kokkos does not support AMD GPUs due to limits in the available backend programming models.
Specifically, Kokkos requires extensive C++ support from the Kernel language. This is expected to change in
the future.
LAMMPS Users Manual
109

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
Return to Section 5 overview
5.3.4 USER-OMP package
The USER-OMP package was developed by Axel Kohlmeyer at Temple University. It provides
multi-threaded versions of most pair styles, nearly all bonded styles (bond, angle, dihedral, improper), several
Kspace styles, and a few fix styles. The package currently uses the OpenMP interface for multi-threading.
Here is a quick overview of how to use the USER-OMP package, assuming one or more 16-core nodes. More
details follow.
use -fopenmp with CCFLAGS and LINKFLAGS in Makefile.machine
make yes-user-omp
make mpi # build with USER-OMP package, if settings added to Makefile.mpi
make omp # or Makefile.omp already has settings
Make.py -v -p omp -o mpi -a file mpi # or one-line build via Make.py
lmp_mpi -sf omp -pk omp 16 <in.script # 1 MPI task, 16 threads
mpirun -np 4 lmp_mpi -sf omp -pk omp 4 -in in.script # 4 MPI tasks, 4 threads/task
mpirun -np 32 -ppn 4 lmp_mpi -sf omp -pk omp 4 -in in.script # 8 nodes, 4 MPI tasks/node, 4 threads/task
Required hardware/software:
Your compiler must support the OpenMP interface. You should have one or more multi-core CPUs so that
multiple threads can be launched by each MPI task running on a CPU.
Building LAMMPS with the USER-OMP package:
The lines above illustrate how to include/build with the USER-OMP package in two steps, using the "make"
command. Or how to do it with one command via the src/Make.py script, described in Section 2.4 of the
manual. Type "Make.py -h" for help.
Note that the CCFLAGS and LINKFLAGS settings in Makefile.machine must include "-fopenmp". Likewise,
if you use an Intel compiler, the CCFLAGS setting must include "-restrict". The Make.py command will add
these automatically.
Run with the USER-OMP package from the command line:
The mpirun or mpiexec command sets the total number of MPI tasks used by LAMMPS (one or multiple per
compute node) and the number of MPI tasks used per node. E.g. the mpirun command in MPICH does this via
its -np and -ppn switches. Ditto for OpenMPI via -np and -npernode.
You need to choose how many OpenMP threads per MPI task will be used by the USER-OMP package. Note
that the product of MPI tasks * threads/task should not exceed the physical number of cores (on a node),
otherwise performance will suffer.
As in the lines above, use the "-sf omp" command-line switch, which will automatically append "omp" to
styles that support it. The "-sf omp" switch also issues a default package omp 0 command, which will set the
number of threads per MPI task via the OMP_NUM_THREADS environment variable.
LAMMPS Users Manual
110
You can also use the "-pk omp Nt" command-line switch, to explicitly set Nt = # of OpenMP threads per MPI
task to use, as well as additional options. Its syntax is the same as the package omp command whose doc page
gives details, including the default values used if it is not specified. It also gives more details on how to set the
number of threads via the OMP_NUM_THREADS environment variable.
Or run with the USER-OMP package by editing an input script:
The discussion above for the mpirun/mpiexec command, MPI tasks/node, and threads/MPI task is the same.
Use the suffix omp command, or you can explicitly add an "omp" suffix to individual styles in your input
script, e.g.
pair_style lj/cut/omp 2.5
You must also use the package omp command to enable the USER-OMP package. When you do this you also
specify how many threads per MPI task to use. The command doc page explains other options and how to set
the number of threads via the OMP_NUM_THREADS environment variable.
Speed-ups to expect:
Depending on which styles are accelerated, you should look for a reduction in the "Pair time", "Bond time",
"KSpace time", and "Loop time" values printed at the end of a run.
You may see a small performance advantage (5 to 20%) when running a USER-OMP style (in serial or
parallel) with a single thread per MPI task, versus running standard LAMMPS with its standard
un-accelerated styles (in serial or all-MPI parallelization with 1 task/core). This is because many of the
USER-OMP styles contain similar optimizations to those used in the OPT package, described in Section 5.3.5.
With multiple threads/task, the optimal choice of number of MPI tasks/node and OpenMP threads/task can
vary a lot and should always be tested via benchmark runs for a specific simulation running on a specific
machine, paying attention to guidelines discussed in the next sub-section.
A description of the multi-threading strategy used in the USER-OMP package and some performance
examples are presented here
Guidelines for best performance:
For many problems on current generation CPUs, running the USER-OMP package with a single thread/task is
faster than running with multiple threads/task. This is because the MPI parallelization in LAMMPS is often
more efficient than multi-threading as implemented in the USER-OMP package. The parallel efficiency (in a
threaded sense) also varies for different USER-OMP styles.
Using multiple threads/task can be more effective under the following circumstances:
Individual compute nodes have a significant number of CPU cores but the CPU itself has limited
memory bandwidth, e.g. for Intel Xeon 53xx (Clovertown) and 54xx (Harpertown) quad-core
processors. Running one MPI task per CPU core will result in significant performance degradation, so
that running with 4 or even only 2 MPI tasks per node is faster. Running in hybrid MPI+OpenMP
mode will reduce the inter-node communication bandwidth contention in the same way, but offers an
additional speedup by utilizing the otherwise idle CPU cores.
•
LAMMPS Users Manual
111
The interconnect used for MPI communication does not provide sufficient bandwidth for a large
number of MPI tasks per node. For example, this applies to running over gigabit ethernet or on Cray
XT4 or XT5 series supercomputers. As in the aforementioned case, this effect worsens when using an
increasing number of nodes.
•
The system has a spatially inhomogeneous particle density which does not map well to the domain
decomposition scheme or load-balancing options that LAMMPS provides. This is because
multi-threading achives parallelism over the number of particles, not via their distribution in space.
•
A machine is being used in "capability mode", i.e. near the point where MPI parallelism is maxed out.
For example, this can happen when using the PPPM solver for long-range electrostatics on large
numbers of nodes. The scaling of the KSpace calculation (see the kspace_style command) becomes
the performance-limiting factor. Using multi-threading allows less MPI tasks to be invoked and can
speed-up the long-range solver, while increasing overall performance by parallelizing the pairwise
and bonded calculations via OpenMP. Likewise additional speedup can be sometimes be achived by
increasing the length of the Coulombic cutoff and thus reducing the work done by the long-range
solver. Using the run_style verlet/split command, which is compatible with the USER-OMP package,
is an alternative way to reduce the number of MPI tasks assigned to the KSpace calculation.
•
Additional performance tips are as follows:
The best parallel efficiency from omp styles is typically achieved when there is at least one MPI task
per physical CPU chip, i.e. socket or die.
•
It is usually most efficient to restrict threading to a single socket, i.e. use one or more MPI task per
socket.
•
NOTE: By default, several current MPI implementations use a processor affinity setting that restricts
each MPI task to a single CPU core. Using multi-threading in this mode will force all threads to share
the one core and thus is likely to be counterproductive. Instead, binding MPI tasks to a (multi-core)
socket, should solve this issue.
•
Restrictions:
None.
LAMMPS Users Manual
112

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
Return to Section accelerate overview
5.3.5 OPT package
The OPT package was developed by James Fischer (High Performance Technologies), David Richie, and
Vincent Natoli (Stone Ridge Technologies). It contains a handful of pair styles whose compute() methods
were rewritten in C++ templated form to reduce the overhead due to if tests and other conditional code.
Here is a quick overview of how to use the OPT package. More details follow.
make yes-opt
make mpi # build with the OPT package
Make.py -v -p opt -o mpi -a file mpi # or one-line build via Make.py
lmp_mpi -sf opt -in in.script # run in serial
mpirun -np 4 lmp_mpi -sf opt -in in.script # run in parallel
Required hardware/software:
None.
Building LAMMPS with the OPT package:
The lines above illustrate how to build LAMMPS with the OPT package in two steps, using the "make"
command. Or how to do it with one command via the src/Make.py script, described in Section 2.4 of the
manual. Type "Make.py -h" for help.
Note that if you use an Intel compiler to build with the OPT package, the CCFLAGS setting in your
Makefile.machine must include "-restrict". The Make.py command will add this automatically.
Run with the OPT package from the command line:
As in the lines above, use the "-sf opt" command-line switch, which will automatically append "opt" to styles
that support it.
Or run with the OPT package by editing an input script:
Use the suffix opt command, or you can explicitly add an "opt" suffix to individual styles in your input script,
e.g.
pair_style lj/cut/opt 2.5
Speed-ups to expect:
You should see a reduction in the "Pair time" value printed at the end of a run. On most machines for
reasonable problem sizes, it will be a 5 to 20% savings.
Guidelines for best performance:
Just try out an OPT pair style to see how it performs.
LAMMPS Users Manual
113
Restrictions:
None.
LAMMPS Users Manual
114

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
6. How-to discussions
This section describes how to perform common tasks using LAMMPS.
6.1 Restarting a simulation
6.2 2d simulations
6.3 CHARMM, AMBER, and DREIDING force fields
6.4 Running multiple simulations from one input script
6.5 Multi-replica simulations
6.6 Granular models
6.7 TIP3P water model
6.8 TIP4P water model
6.9 SPC water model
6.10 Coupling LAMMPS to other codes
6.11 Visualizing LAMMPS snapshots
6.12 Triclinic (non-orthogonal) simulation boxes
6.13 NEMD simulations
6.14 Finite-size spherical and aspherical particles
6.15 Output from LAMMPS (thermo, dumps, computes, fixes, variables)
6.16 Thermostatting, barostatting and computing temperature
6.17 Walls
6.18 Elastic constants
6.19 Library interface to LAMMPS
6.20 Calculating thermal conductivity
6.21 Calculating viscosity
6.22 Calculating a diffusion coefficient
6.23 Using chunks to calculate system properties
6.24 Setting parameters for the kspace_style pppm/disp command
6.25 Polarizable models
6.26 Adiabatic core/shell model
6.27 Drude induced dipoles
The example input scripts included in the LAMMPS distribution and highlighted in Section 7 also show how
to setup and run various kinds of simulations.
6.1 Restarting a simulation
There are 3 ways to continue a long LAMMPS simulation. Multiple run commands can be used in the same
input script. Each run will continue from where the previous run left off. Or binary restart files can be saved to
disk using the restart command. At a later time, these binary files can be read via a read_restart command in a
new script. Or they can be converted to text data files using the -r command-line switch and read by a
read_data command in a new script.
Here we give examples of 2 scripts that read either a binary restart file or a converted data file and then issue a
new run command to continue where the previous run left off. They illustrate what settings must be made in
the new script. Details are discussed in the documentation for the read_restart and read_data commands.
LAMMPS Users Manual
115
Look at the in.chain input script provided in the bench directory of the LAMMPS distribution to see the
original script that these 2 scripts are based on. If that script had the line
restart 50 tmp.restart
added to it, it would produce 2 binary restart files (tmp.restart.50 and tmp.restart.100) as it ran.
This script could be used to read the 1st restart file and re-run the last 50 timesteps:
read_restart tmp.restart.50
neighbor 0.4 bin
neigh_modify every 1 delay 1
fix 1 all nve
fix 2 all langevin 1.0 1.0 10.0 904297
timestep 0.012
run 50
Note that the following commands do not need to be repeated because their settings are included in the restart
file: units, atom_style, special_bonds, pair_style, bond_style. However these commands do need to be used,
since their settings are not in the restart file: neighbor, fix, timestep.
If you actually use this script to perform a restarted run, you will notice that the thermodynamic data match at
step 50 (if you also put a "thermo 50" command in the original script), but do not match at step 100. This is
because the fix langevin command uses random numbers in a way that does not allow for perfect restarts.
As an alternate approach, the restart file could be converted to a data file as follows:
lmp_g++ -r tmp.restart.50 tmp.restart.data
Then, this script could be used to re-run the last 50 steps:
units lj
atom_style bond
pair_style lj/cut 1.12
pair_modify shift yes
bond_style fene
special_bonds 0.0 1.0 1.0
read_data tmp.restart.data
neighbor 0.4 bin
neigh_modify every 1 delay 1
fix 1 all nve
fix 2 all langevin 1.0 1.0 10.0 904297
timestep 0.012
reset_timestep 50
run 50
LAMMPS Users Manual
116
Note that nearly all the settings specified in the original in.chain script must be repeated, except the pair_coeff
and bond_coeff commands since the new data file lists the force field coefficients. Also, the reset_timestep
command is used to tell LAMMPS the current timestep. This value is stored in restart files, but not in data
files.
6.2 2d simulations
Use the dimension command to specify a 2d simulation.
Make the simulation box periodic in z via the boundary command. This is the default.
If using the create box command to define a simulation box, set the z dimensions narrow, but finite, so that the
create_atoms command will tile the 3d simulation box with a single z plane of atoms - e.g.
create box 1 -10 10 -10 10 -0.25 0.25
If using the read data command to read in a file of atom coordinates, set the "zlo zhi" values to be finite but
narrow, similar to the create_box command settings just described. For each atom in the file, assign a z
coordinate so it falls inside the z-boundaries of the box - e.g. 0.0.
Use the fix enforce2d command as the last defined fix to insure that the z-components of velocities and forces
are zeroed out every timestep. The reason to make it the last fix is so that any forces induced by other fixes
will be zeroed out.
Many of the example input scripts included in the LAMMPS distribution are for 2d models.
NOTE: Some models in LAMMPS treat particles as finite-size spheres, as opposed to point particles. See the
atom_style sphere and fix nve/sphere commands for details. By default, for 2d simulations, such particles will
still be modeled as 3d spheres, not 2d discs (circles), meaning their moment of inertia will be that of a sphere.
If you wish to model them as 2d discs, see the set density/disc command and the disc option for the fix
nve/sphere, fix nvt/sphere, fix nph/sphere, fix npt/sphere commands.
6.3 CHARMM, AMBER, and DREIDING force fields
A force field has 2 parts: the formulas that define it and the coefficients used for a particular system. Here we
only discuss formulas implemented in LAMMPS that correspond to formulas commonly used in the
CHARMM, AMBER, and DREIDING force fields. Setting coefficients is done in the input data file via the
read_data command or in the input script with commands like pair_coeff or bond_coeff. See Section 9 for
additional tools that can use CHARMM or AMBER to assign force field coefficients and convert their output
into LAMMPS input.
See (MacKerell) for a description of the CHARMM force field. See (Cornell) for a description of the AMBER
force field.
These style choices compute force field formulas that are consistent with common options in CHARMM or
AMBER. See each command's documentation for the formula it computes.
bond_style harmonic• angle_style charmm•
LAMMPS Users Manual
117

dihedral_style charmmfsh• dihedral_style charmm• pair_style lj/charmmfsw/coul/charmmfsh• pair_style lj/charmmfsw/coul/long• pair_style lj/charmm/coul/charmm• pair_style lj/charmm/coul/charmm/implicit• pair_style lj/charmm/coul/long•
special_bonds charmm• special_bonds amber•
NOTE: For CHARMM, the newer charmmfsw or charmmfsh styles were released in March 2017. We
recommend they be used instead of the older charmm styles. See discussion of the differences on the pair
charmm and dihedral charmm doc pages.
DREIDING is a generic force field developed by the Goddard group at Caltech and is useful for predicting
structures and dynamics of organic, biological and main-group inorganic molecules. The philosophy in
DREIDING is to use general force constants and geometry parameters based on simple hybridization
considerations, rather than individual force constants and geometric parameters that depend on the particular
combinations of atoms involved in the bond, angle, or torsion terms. DREIDING has an explicit hydrogen
bond term to describe interactions involving a hydrogen atom on very electronegative atoms (N, O, F).
See (Mayo) for a description of the DREIDING force field
These style choices compute force field formulas that are consistent with the DREIDING force field. See each
command's documentation for the formula it computes.
bond_style harmonic• bond_style morse•
angle_style harmonic• angle_style cosine• angle_style cosine/periodic•
dihedral_style charmm• improper_style umbrella•
pair_style buck• pair_style buck/coul/cut• pair_style buck/coul/long• pair_style lj/cut• pair_style lj/cut/coul/cut• pair_style lj/cut/coul/long•
pair_style hbond/dreiding/lj• pair_style hbond/dreiding/morse•
special_bonds dreiding•
LAMMPS Users Manual
118
6.4 Running multiple simulations from one input script
This can be done in several ways. See the documentation for individual commands for more details on how
these examples work.
If "multiple simulations" means continue a previous simulation for more timesteps, then you simply use the
run command multiple times. For example, this script
units lj
atom_style atomic
read_data data.lj
run 10000
run 10000
run 10000
run 10000
run 10000
would run 5 successive simulations of the same system for a total of 50,000 timesteps.
If you wish to run totally different simulations, one after the other, the clear command can be used in between
them to re-initialize LAMMPS. For example, this script
units lj
atom_style atomic
read_data data.lj
run 10000
clear
units lj
atom_style atomic
read_data data.lj.new
run 10000
would run 2 independent simulations, one after the other.
For large numbers of independent simulations, you can use variables and the next and jump commands to
loop over the same input script multiple times with different settings. For example, this script, named
in.polymer
variable d index run1 run2 run3 run4 run5 run6 run7 run8
shell cd $d
read_data data.polymer
run 10000
shell cd ..
clear
next d
jump in.polymer
would run 8 simulations in different directories, using a data.polymer file in each directory. The same concept
could be used to run the same system at 8 different temperatures, using a temperature variable and storing the
output in different log and dump files, for example
variable a loop 8
variable t index 0.8 0.85 0.9 0.95 1.0 1.05 1.1 1.15
log log.$a
read data.polymer
velocity all create $t 352839
LAMMPS Users Manual
119

fix 1 all nvt $t $t 100.0
dump 1 all atom 1000 dump.$a
run 100000
clear
next t
next a
jump in.polymer
All of the above examples work whether you are running on 1 or multiple processors, but assumed you are
running LAMMPS on a single partition of processors. LAMMPS can be run on multiple partitions via the
"-partition" command-line switch as described in this section of the manual.
In the last 2 examples, if LAMMPS were run on 3 partitions, the same scripts could be used if the "index" and
"loop" variables were replaced with universe-style variables, as described in the variable command. Also, the
"next t" and "next a" commands would need to be replaced with a single "next a t" command. With these
modifications, the 8 simulations of each script would run on the 3 partitions one after the other until all were
finished. Initially, 3 simulations would be started simultaneously, one on each partition. When one finished,
that partition would then start the 4th simulation, and so forth, until all 8 were completed.
6.5 Multi-replica simulations
Several commands in LAMMPS run mutli-replica simulations, meaning that multiple instances (replicas) of
your simulation are run simultaneously, with small amounts of data exchanged between replicas periodically.
These are the relevant commands:
neb for nudged elastic band calculations• prd for parallel replica dynamics• tad for temperature accelerated dynamics• temper for parallel tempering• fix pimd for path-integral molecular dynamics (PIMD)•
NEB is a method for finding transition states and barrier energies. PRD and TAD are methods for performing
accelerated dynamics to find and perform infrequent events. Parallel tempering or replica exchange runs
different replicas at a series of temperature to facilitate rare-event sampling.
These commands can only be used if LAMMPS was built with the REPLICA package. See the Making
LAMMPS section for more info on packages.
PIMD runs different replicas whose individual particles are coupled together by springs to model a system or
ring-polymers.
This commands can only be used if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info on packages.
In all these cases, you must run with one or more processors per replica. The processors assigned to each
replica are determined at run-time by using the -partition command-line switch to launch LAMMPS on
multiple partitions, which in this context are the same as replicas. E.g. these commands:
mpirun -np 16 lmp_linux -partition 8x2 -in in.temper
mpirun -np 8 lmp_linux -partition 8x1 -in in.neb
LAMMPS Users Manual
120

would each run 8 replicas, on either 16 or 8 processors. Note the use of the -in command-line switch to
specify the input script which is required when running in multi-replica mode.
Also note that with MPI installed on a machine (e.g. your desktop), you can run on more (virtual) processors
than you have physical processors. Thus the above commands could be run on a single-processor (or
few-processor) desktop so that you can run a multi-replica simulation on more replicas than you have physical
processors.
6.6 Granular models
Granular system are composed of spherical particles with a diameter, as opposed to point particles. This
means they have an angular velocity and torque can be imparted to them to cause them to rotate.
To run a simulation of a granular model, you will want to use the following commands:
atom_style sphere• fix nve/sphere• fix gravity•
This compute
compute erotate/sphere•
calculates rotational kinetic energy which can be output with thermodynamic info.
Use one of these 3 pair potentials, which compute forces and torques between interacting pairs of particles:
pair_style gran/history• pair_style gran/no_history• pair_style gran/hertzian•
These commands implement fix options specific to granular systems:
fix freeze• fix pour• fix viscous• fix wall/gran•
The fix style freeze zeroes both the force and torque of frozen atoms, and should be used for granular system
instead of the fix style setforce.
For computational efficiency, you can eliminate needless pairwise computations between frozen atoms by
using this command:
neigh_modify exclude•
NOTE: By default, for 2d systems, granular particles are still modeled as 3d spheres, not 2d discs (circles),
meaning their moment of inertia will be the same as in 3d. If you wish to model granular particles in 2d as 2d
discs, see the note on this topic in Section 6.2, where 2d simulations are disussed.
LAMMPS Users Manual
121

6.7 TIP3P water model
The TIP3P water model as implemented in CHARMM (MacKerell) specifies a 3-site rigid water molecule
with charges and Lennard-Jones parameters assigned to each of the 3 atoms. In LAMMPS the fix shake
command can be used to hold the two O-H bonds and the H-O-H angle rigid. A bond style of harmonic and an
angle style of harmonic or charmm should also be used.
These are the additional parameters (in real units) to set for O and H atoms and the water molecule to run a
rigid TIP3P-CHARMM model with a cutoff. The K values can be used if a flexible TIP3P model (without fix
shake) is desired. If the LJ epsilon and sigma for HH and OH are set to 0.0, it corresponds to the original 1983
TIP3P model (Jorgensen).
O mass = 15.9994
H mass = 1.008
O charge = -0.834
H charge = 0.417
LJ epsilon of OO = 0.1521
LJ sigma of OO = 3.1507
LJ epsilon of HH = 0.0460
LJ sigma of HH = 0.4000
LJ epsilon of OH = 0.0836
LJ sigma of OH = 1.7753
K of OH bond = 450
r0 of OH bond = 0.9572
K of HOH angle = 55
theta of HOH angle = 104.52
These are the parameters to use for TIP3P with a long-range Coulombic solver (e.g. Ewald or PPPM in
LAMMPS), see (Price) for details:
O mass = 15.9994
H mass = 1.008
O charge = -0.830
H charge = 0.415
LJ epsilon of OO = 0.102
LJ sigma of OO = 3.188
LJ epsilon, sigma of OH, HH = 0.0
K of OH bond = 450
r0 of OH bond = 0.9572
K of HOH angle = 55
theta of HOH angle = 104.52
Wikipedia also has a nice article on water models.
LAMMPS Users Manual
122
6.8 TIP4P water model
The four-point TIP4P rigid water model extends the traditional three-point TIP3P model by adding an
additional site, usually massless, where the charge associated with the oxygen atom is placed. This site M is
located at a fixed distance away from the oxygen along the bisector of the HOH bond angle. A bond style of
harmonic and an angle style of harmonic or charmm should also be used.
A TIP4P model is run with LAMMPS using either this command for a cutoff model:
pair_style lj/cut/tip4p/cut
or these two commands for a long-range model:
pair_style lj/cut/tip4p/long• kspace_style pppm/tip4p•
For both models, the bond lengths and bond angles should be held fixed using the fix shake command.
These are the additional parameters (in real units) to set for O and H atoms and the water molecule to run a
rigid TIP4P model with a cutoff (Jorgensen). Note that the OM distance is specified in the pair_style
command, not as part of the pair coefficients.
O mass = 15.9994
H mass = 1.008
O charge = -1.040
H charge = 0.520
r0 of OH bond = 0.9572
theta of HOH angle = 104.52
OM distance = 0.15
LJ epsilon of O-O = 0.1550
LJ sigma of O-O = 3.1536
LJ epsilon, sigma of OH, HH = 0.0
Coulombic cutoff = 8.5
For the TIP4/Ice model (J Chem Phys, 122, 234511 (2005); http://dx.doi.org/10.1063/1.1931662) these values
can be used:
O mass = 15.9994
H mass = 1.008
O charge = -1.1794
H charge = 0.5897
r0 of OH bond = 0.9572
theta of HOH angle = 104.52
OM distance = 0.1577
LJ epsilon of O-O = 0.21084
LJ sigma of O-O = 3.1668
LJ epsilon, sigma of OH, HH = 0.0
Coulombic cutoff = 8.5
For the TIP4P/2005 model (J Chem Phys, 123, 234505 (2005); http://dx.doi.org/10.1063/1.2121687), these
values can be used:
LAMMPS Users Manual
123

O mass = 15.9994
H mass = 1.008
O charge = -1.1128
H charge = 0.5564
r0 of OH bond = 0.9572
theta of HOH angle = 104.52
OM distance = 0.1546
LJ epsilon of O-O = 0.1852
LJ sigma of O-O = 3.1589
LJ epsilon, sigma of OH, HH = 0.0
Coulombic cutoff = 8.5
These are the parameters to use for TIP4P with a long-range Coulombic solver (e.g. Ewald or PPPM in
LAMMPS):
O mass = 15.9994
H mass = 1.008
O charge = -1.0484
H charge = 0.5242
r0 of OH bond = 0.9572
theta of HOH angle = 104.52
OM distance = 0.1250
LJ epsilon of O-O = 0.16275
LJ sigma of O-O = 3.16435
LJ epsilon, sigma of OH, HH = 0.0
Note that the when using the TIP4P pair style, the neighbor list cutoff for Coulomb interactions is effectively
extended by a distance 2 * (OM distance), to account for the offset distance of the fictitious charges on O
atoms in water molecules. Thus it is typically best in an efficiency sense to use a LJ cutoff >= Coulomb cutoff
+ 2*(OM distance), to shrink the size of the neighbor list. This leads to slightly larger cost for the long-range
calculation, so you can test the trade-off for your model. The OM distance and the LJ and Coulombic cutoffs
are set in the pair_style lj/cut/tip4p/long command.
Wikipedia also has a nice article on water models.
6.9 SPC water model
The SPC water model specifies a 3-site rigid water molecule with charges and Lennard-Jones parameters
assigned to each of the 3 atoms. In LAMMPS the fix shake command can be used to hold the two O-H bonds
and the H-O-H angle rigid. A bond style of harmonic and an angle style of harmonic or charmm should also
be used.
These are the additional parameters (in real units) to set for O and H atoms and the water molecule to run a
rigid SPC model.
O mass = 15.9994
H mass = 1.008
O charge = -0.820
H charge = 0.410
LJ epsilon of OO = 0.1553
LAMMPS Users Manual
124

LJ sigma of OO = 3.166
LJ epsilon, sigma of OH, HH = 0.0
r0 of OH bond = 1.0
theta of HOH angle = 109.47
Note that as originally proposed, the SPC model was run with a 9 Angstrom cutoff for both LJ and
Coulommbic terms. It can also be used with long-range Coulombics (Ewald or PPPM in LAMMPS), without
changing any of the parameters above, though it becomes a different model in that mode of usage.
The SPC/E (extended) water model is the same, except the partial charge assignments change:
O charge = -0.8476
H charge = 0.4238
See the (Berendsen) reference for more details on both the SPC and SPC/E models.
Wikipedia also has a nice article on water models.
6.10 Coupling LAMMPS to other codes
LAMMPS is designed to allow it to be coupled to other codes. For example, a quantum mechanics code might
compute forces on a subset of atoms and pass those forces to LAMMPS. Or a continuum finite element (FE)
simulation might use atom positions as boundary conditions on FE nodal points, compute a FE solution, and
return interpolated forces on MD atoms.
LAMMPS can be coupled to other codes in at least 3 ways. Each has advantages and disadvantages, which
you'll have to think about in the context of your application.
(1) Define a new fix command that calls the other code. In this scenario, LAMMPS is the driver code. During
its timestepping, the fix is invoked, and can make library calls to the other code, which has been linked to
LAMMPS as a library. This is the way the POEMS package that performs constrained rigid-body motion on
groups of atoms is hooked to LAMMPS. See the fix poems command for more details. See this section of the
documentation for info on how to add a new fix to LAMMPS.
(2) Define a new LAMMPS command that calls the other code. This is conceptually similar to method (1), but
in this case LAMMPS and the other code are on a more equal footing. Note that now the other code is not
called during the timestepping of a LAMMPS run, but between runs. The LAMMPS input script can be used
to alternate LAMMPS runs with calls to the other code, invoked via the new command. The run command
facilitates this with its every option, which makes it easy to run a few steps, invoke the command, run a few
steps, invoke the command, etc.
In this scenario, the other code can be called as a library, as in (1), or it could be a stand-alone code, invoked
by a system() call made by the command (assuming your parallel machine allows one or more processors to
start up another program). In the latter case the stand-alone code could communicate with LAMMPS thru files
that the command writes and reads.
See Section 10 of the documentation for how to add a new command to LAMMPS.
(3) Use LAMMPS as a library called by another code. In this case the other code is the driver and calls
LAMMPS as needed. Or a wrapper code could link and call both LAMMPS and another code as libraries.
LAMMPS Users Manual
125

Again, the run command has options that allow it to be invoked with minimal overhead (no setup or clean-up)
if you wish to do multiple short runs, driven by another program.
Examples of driver codes that call LAMMPS as a library are included in the examples/COUPLE directory of
the LAMMPS distribution; see examples/COUPLE/README for more details:
simple: simple driver programs in C++ and C which invoke LAMMPS as a library• lammps_quest: coupling of LAMMPS and Quest, to run classical MD with quantum forces calculated
by a density functional code
•
lammps_spparks: coupling of LAMMPS and SPPARKS, to couple a kinetic Monte Carlo model for
grain growth using MD to calculate strain induced across grain boundaries
•
This section of the documentation describes how to build LAMMPS as a library. Once this is done, you can
interface with LAMMPS either via C++, C, Fortran, or Python (or any other language that supports a vanilla
C-like interface). For example, from C++ you could create one (or more) "instances" of LAMMPS, pass it an
input script to process, or execute individual commands, all by invoking the correct class methods in
LAMMPS. From C or Fortran you can make function calls to do the same things. See Section 11 of the
manual for a description of the Python wrapper provided with LAMMPS that operates through the LAMMPS
library interface.
The files src/library.cpp and library.h contain the C-style interface to LAMMPS. See Section 6.19 of the
manual for a description of the interface and how to extend it for your needs.
Note that the lammps_open() function that creates an instance of LAMMPS takes an MPI communicator as an
argument. This means that instance of LAMMPS will run on the set of processors in the communicator. Thus
the calling code can run LAMMPS on all or a subset of processors. For example, a wrapper script might
decide to alternate between LAMMPS and another code, allowing them both to run on all the processors. Or it
might allocate half the processors to LAMMPS and half to the other code and run both codes simultaneously
before syncing them up periodically. Or it might instantiate multiple instances of LAMMPS to perform
different calculations.
6.11 Visualizing LAMMPS snapshots
LAMMPS itself does not do visualization, but snapshots from LAMMPS simulations can be visualized (and
analyzed) in a variety of ways.
LAMMPS snapshots are created by the dump command which can create files in several formats. The native
LAMMPS dump format is a text file (see "dump atom" or "dump custom") which can be visualized by several
popular visualization tools. The dump image and dump movie styles can output internally rendered images
and convert a sequence of them to a movie during the MD run. Several programs included with LAMMPS as
auxiliary tools can convert between LAMMPS format files and other formats. See the Section 9 doc page for
details.
A Python-based toolkit distributed by our group can read native LAMMPS dump files, including custom
dump files with additional columns of user-specified atom information, and convert them to various formats
or pipe them into visualization software directly. See the Pizza.py WWW site for details. Specifically,
Pizza.py can convert LAMMPS dump files into PDB, XYZ, Ensight, and VTK formats. Pizza.py can pipe
LAMMPS dump files directly into the Raster3d and RasMol visualization programs. Pizza.py has tools that
do interactive 3d OpenGL visualization and one that creates SVG images of dump file snapshots.
LAMMPS Users Manual
126

6.12 Triclinic (non-orthogonal) simulation boxes
By default, LAMMPS uses an orthogonal simulation box to encompass the particles. The boundary command
sets the boundary conditions of the box (periodic, non-periodic, etc). The orthogonal box has its "origin" at
(xlo,ylo,zlo) and is defined by 3 edge vectors starting from the origin given by a = (xhi-xlo,0,0); b =
(0,yhi-ylo,0); c = (0,0,zhi-zlo). The 6 parameters (xlo,xhi,ylo,yhi,zlo,zhi) are defined at the time the
simulation box is created, e.g. by the create_box or read_data or read_restart commands. Additionally,
LAMMPS defines box size parameters lx,ly,lz where lx = xhi-xlo, and similarly in the y and z dimensions.
The 6 parameters, as well as lx,ly,lz, can be output via the thermo_style custom command.
LAMMPS also allows simulations to be performed in triclinic (non-orthogonal) simulation boxes shaped as a
parallelepiped with triclinic symmetry. The parallelepiped has its "origin" at (xlo,ylo,zlo) and is defined by 3
edge vectors starting from the origin given by a = (xhi-xlo,0,0); b = (xy,yhi-ylo,0); c = (xz,yz,zhi-zlo).
xy,xz,yz can be 0.0 or positive or negative values and are called "tilt factors" because they are the amount of
displacement applied to faces of an originally orthogonal box to transform it into the parallelepiped. In
LAMMPS the triclinic simulation box edge vectors a, b, and c cannot be arbitrary vectors. As indicated, a
must lie on the positive x axis. b must lie in the xy plane, with strictly positive y component. c may have any
orientation with strictly positive z component. The requirement that a, b, and c have strictly positive x, y, and
z components, respectively, ensures that a, b, and c form a complete right-handed basis. These restrictions
impose no loss of generality, since it is possible to rotate/invert any set of 3 crystal basis vectors so that they
conform to the restrictions.
For example, assume that the 3 vectors A,B,C are the edge vectors of a general parallelepiped, where there is
no restriction on A,B,C other than they form a complete right-handed basis i.e. A x B . C > 0. The equivalent
LAMMPS a,b,c are a linear rotation of A, B, and C and can be computed as follows:
LAMMPS Users Manual
127
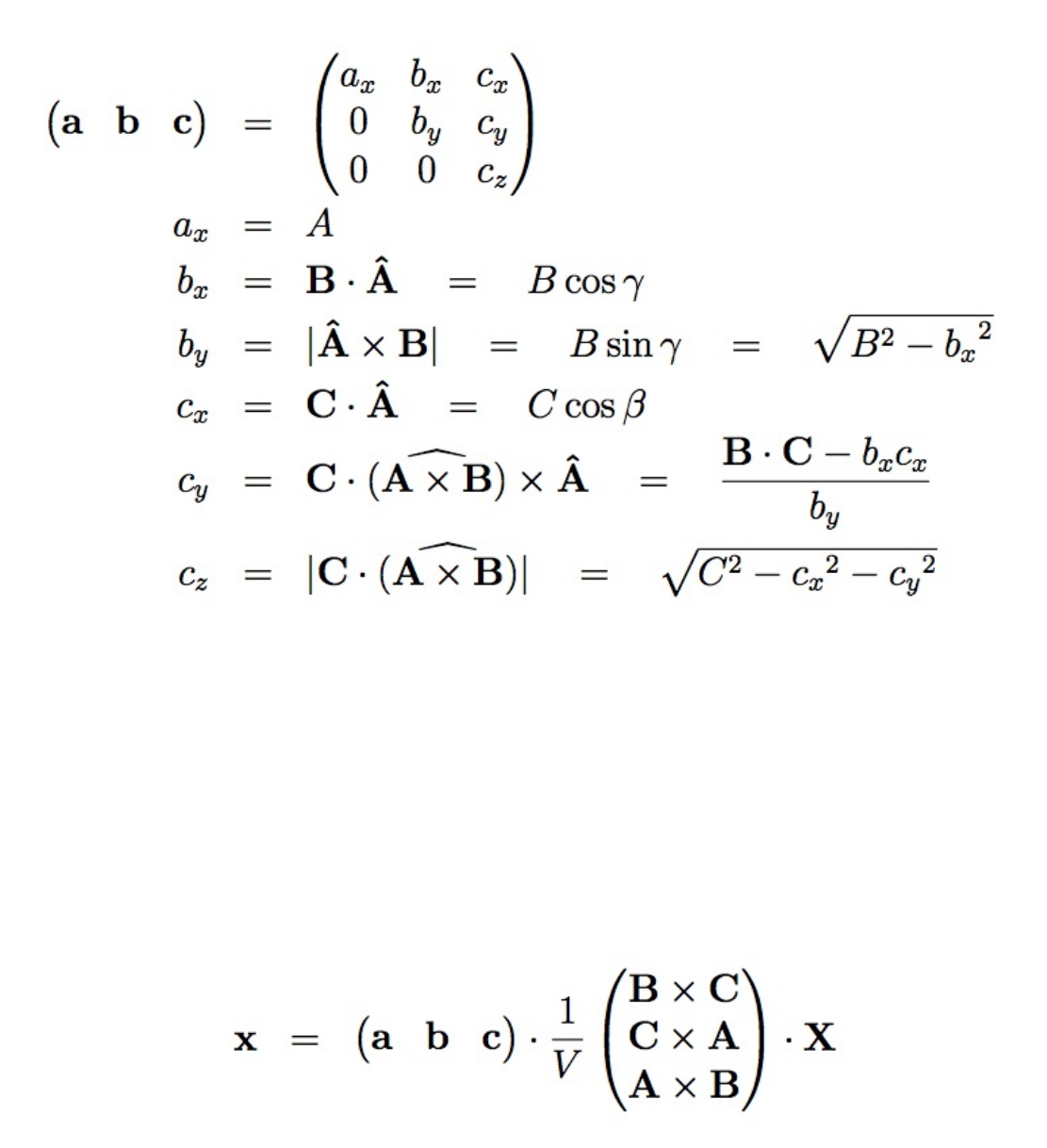
where A = | A | indicates the scalar length of A. The hat symbol (^) indicates the corresponding unit vector.
beta and gamma are angles between the vectors described below. Note that by construction, a, b, and c have
strictly positive x, y, and z components, respectively. If it should happen that A, B, and C form a left-handed
basis, then the above equations are not valid for c. In this case, it is necessary to first apply an inversion. This
can be achieved by interchanging two basis vectors or by changing the sign of one of them.
For consistency, the same rotation/inversion applied to the basis vectors must also be applied to atom
positions, velocities, and any other vector quantities. This can be conveniently achieved by first converting to
fractional coordinates in the old basis and then converting to distance coordinates in the new basis. The
transformation is given by the following equation:
where V is the volume of the box, X is the original vector quantity and x is the vector in the LAMMPS basis.
There is no requirement that a triclinic box be periodic in any dimension, though it typically should be in at
least the 2nd dimension of the tilt (y in xy) if you want to enforce a shift in periodic boundary conditions
LAMMPS Users Manual
128
across that boundary. Some commands that work with triclinic boxes, e.g. the fix deform and fix npt
commands, require periodicity or non-shrink-wrap boundary conditions in specific dimensions. See the
command doc pages for details.
The 9 parameters (xlo,xhi,ylo,yhi,zlo,zhi,xy,xz,yz) are defined at the time the simulation box is created. This
happens in one of 3 ways. If the create_box command is used with a region of style prism, then a triclinic box
is setup. See the region command for details. If the read_data command is used to define the simulation box,
and the header of the data file contains a line with the "xy xz yz" keyword, then a triclinic box is setup. See
the read_data command for details. Finally, if the read_restart command reads a restart file which was written
from a simulation using a triclinic box, then a triclinic box will be setup for the restarted simulation.
Note that you can define a triclinic box with all 3 tilt factors = 0.0, so that it is initially orthogonal. This is
necessary if the box will become non-orthogonal, e.g. due to the fix npt or fix deform commands.
Alternatively, you can use the change_box command to convert a simulation box from orthogonal to triclinic
and vice versa.
As with orthogonal boxes, LAMMPS defines triclinic box size parameters lx,ly,lz where lx = xhi-xlo, and
similarly in the y and z dimensions. The 9 parameters, as well as lx,ly,lz, can be output via the thermo_style
custom command.
To avoid extremely tilted boxes (which would be computationally inefficient), LAMMPS normally requires
that no tilt factor can skew the box more than half the distance of the parallel box length, which is the 1st
dimension in the tilt factor (x for xz). This is required both when the simulation box is created, e.g. via the
create_box or read_data commands, as well as when the box shape changes dynamically during a simulation,
e.g. via the fix deform or fix npt commands.
For example, if xlo = 2 and xhi = 12, then the x box length is 10 and the xy tilt factor must be between -5 and
5. Similarly, both xz and yz must be between -(xhi-xlo)/2 and +(yhi-ylo)/2. Note that this is not a limitation,
since if the maximum tilt factor is 5 (as in this example), then configurations with tilt = ..., -15, -5, 5, 15, 25,
... are geometrically all equivalent. If the box tilt exceeds this limit during a dynamics run (e.g. via the fix
deform command), then the box is "flipped" to an equivalent shape with a tilt factor within the bounds, so the
run can continue. See the fix deform doc page for further details.
One exception to this rule is if the 1st dimension in the tilt factor (x for xy) is non-periodic. In that case, the
limits on the tilt factor are not enforced, since flipping the box in that dimension does not change the atom
positions due to non-periodicity. In this mode, if you tilt the system to extreme angles, the simulation will
simply become inefficient, due to the highly skewed simulation box.
The limitation on not creating a simulation box with a tilt factor skewing the box more than half the distance
of the parallel box length can be overridden via the box command. Setting the tilt keyword to large allows any
tilt factors to be specified.
Box flips that may occur using the fix deform or fix npt commands can be turned off using the flip no option
with either of the commands.
Note that if a simulation box has a large tilt factor, LAMMPS will run less efficiently, due to the large volume
of communication needed to acquire ghost atoms around a processor's irregular-shaped sub-domain. For
extreme values of tilt, LAMMPS may also lose atoms and generate an error.
Triclinic crystal structures are often defined using three lattice constants a, b, and c, and three angles alpha,
beta and gamma. Note that in this nomenclature, the a, b, and c lattice constants are the scalar lengths of the
LAMMPS Users Manual
129
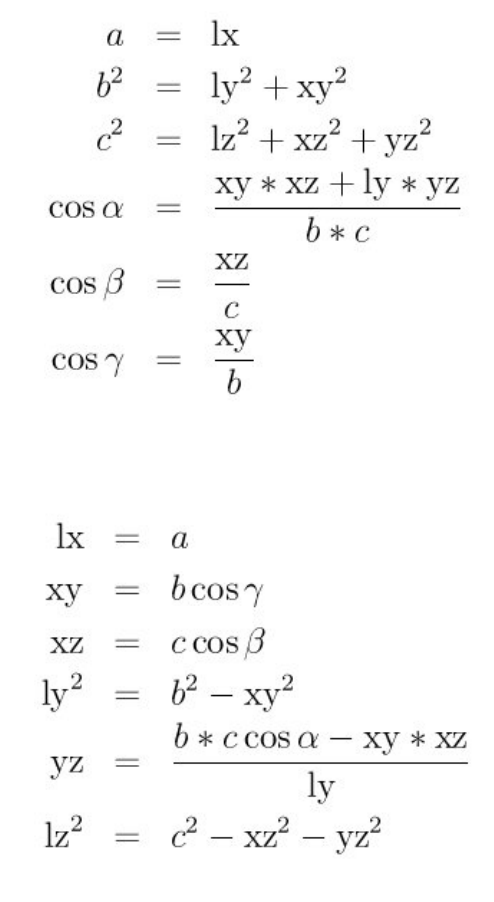
edge vectors a, b, and c defined above. The relationship between these 6 quantities (a,b,c,alpha,beta,gamma)
and the LAMMPS box sizes (lx,ly,lz) = (xhi-xlo,yhi-ylo,zhi-zlo) and tilt factors (xy,xz,yz) is as follows:
The inverse relationship can be written as follows:
The values of a, b, c , alpha, beta , and gamma can be printed out or accessed by computes using the
thermo_style custom keywords cella, cellb, cellc, cellalpha, cellbeta, cellgamma, respectively.
As discussed on the dump command doc page, when the BOX BOUNDS for a snapshot is written to a dump
file for a triclinic box, an orthogonal bounding box which encloses the triclinic simulation box is output, along
with the 3 tilt factors (xy, xz, yz) of the triclinic box, formatted as follows:
ITEM: BOX BOUNDS xy xz yz
xlo_bound xhi_bound xy
ylo_bound yhi_bound xz
zlo_bound zhi_bound yz
LAMMPS Users Manual
130

This bounding box is convenient for many visualization programs and is calculated from the 9 triclinic box
parameters (xlo,xhi,ylo,yhi,zlo,zhi,xy,xz,yz) as follows:
xlo_bound = xlo + MIN(0.0,xy,xz,xy+xz)
xhi_bound = xhi + MAX(0.0,xy,xz,xy+xz)
ylo_bound = ylo + MIN(0.0,yz)
yhi_bound = yhi + MAX(0.0,yz)
zlo_bound = zlo
zhi_bound = zhi
These formulas can be inverted if you need to convert the bounding box back into the triclinic box parameters,
e.g. xlo = xlo_bound - MIN(0.0,xy,xz,xy+xz).
One use of triclinic simulation boxes is to model solid-state crystals with triclinic symmetry. The lattice
command can be used with non-orthogonal basis vectors to define a lattice that will tile a triclinic simulation
box via the create_atoms command.
A second use is to run Parinello-Rahman dynamics via the fix npt command, which will adjust the xy, xz, yz
tilt factors to compensate for off-diagonal components of the pressure tensor. The analog for an energy
minimization is the fix box/relax command.
A third use is to shear a bulk solid to study the response of the material. The fix deform command can be used
for this purpose. It allows dynamic control of the xy, xz, yz tilt factors as a simulation runs. This is discussed
in the next section on non-equilibrium MD (NEMD) simulations.
6.13 NEMD simulations
Non-equilibrium molecular dynamics or NEMD simulations are typically used to measure a fluid's rheological
properties such as viscosity. In LAMMPS, such simulations can be performed by first setting up a
non-orthogonal simulation box (see the preceding Howto section).
A shear strain can be applied to the simulation box at a desired strain rate by using the fix deform command.
The fix nvt/sllod command can be used to thermostat the sheared fluid and integrate the SLLOD equations of
motion for the system. Fix nvt/sllod uses compute temp/deform to compute a thermal temperature by
subtracting out the streaming velocity of the shearing atoms. The velocity profile or other properties of the
fluid can be monitored via the fix ave/chunk command.
As discussed in the previous section on non-orthogonal simulation boxes, the amount of tilt or skew that can
be applied is limited by LAMMPS for computational efficiency to be 1/2 of the parallel box length. However,
fix deform can continuously strain a box by an arbitrary amount. As discussed in the fix deform command,
when the tilt value reaches a limit, the box is flipped to the opposite limit which is an equivalent tiling of
periodic space. The strain rate can then continue to change as before. In a long NEMD simulation these box
re-shaping events may occur many times.
In a NEMD simulation, the "remap" option of fix deform should be set to "remap v", since that is what fix
nvt/sllod assumes to generate a velocity profile consistent with the applied shear strain rate.
An alternative method for calculating viscosities is provided via the fix viscosity command.
NEMD simulations can also be used to measure transport properties of a fluid through a pore or channel.
Simulations of steady-state flow can be performed using the fix flow/gauss command.
LAMMPS Users Manual
131

6.14 Finite-size spherical and aspherical particles
Typical MD models treat atoms or particles as point masses. Sometimes it is desirable to have a model with
finite-size particles such as spheroids or ellipsoids or generalized aspherical bodies. The difference is that such
particles have a moment of inertia, rotational energy, and angular momentum. Rotation is induced by torque
coming from interactions with other particles.
LAMMPS has several options for running simulations with these kinds of particles. The following aspects are
discussed in turn:
atom styles• pair potentials• time integration• computes, thermodynamics, and dump output• rigid bodies composed of finite-size particles•
Example input scripts for these kinds of models are in the body, colloid, dipole, ellipse, line, peri, pour, and tri
directories of the examples directory in the LAMMPS distribution.
Atom styles
There are several atom styles that allow for definition of finite-size particles: sphere, dipole, ellipsoid, line, tri,
peri, and body.
The sphere style defines particles that are spheriods and each particle can have a unique diameter and mass (or
density). These particles store an angular velocity (omega) and can be acted upon by torque. The "set"
command can be used to modify the diameter and mass of individual particles, after then are created.
The dipole style does not actually define finite-size particles, but is often used in conjunction with spherical
particles, via a command like
atom_style hybrid sphere dipole
This is because when dipoles interact with each other, they induce torques, and a particle must be finite-size
(i.e. have a moment of inertia) in order to respond and rotate. See the atom_style dipole command for details.
The "set" command can be used to modify the orientation and length of the dipole moment of individual
particles, after then are created.
The ellipsoid style defines particles that are ellipsoids and thus can be aspherical. Each particle has a shape,
specified by 3 diameters, and mass (or density). These particles store an angular momentum and their
orientation (quaternion), and can be acted upon by torque. They do not store an angular velocity (omega),
which can be in a different direction than angular momentum, rather they compute it as needed. The "set"
command can be used to modify the diameter, orientation, and mass of individual particles, after then are
created. It also has a brief explanation of what quaternions are.
The line style defines line segment particles with two end points and a mass (or density). They can be used in
2d simulations, and they can be joined together to form rigid bodies which represent arbitrary polygons.
LAMMPS Users Manual
132
The tri style defines triangular particles with three corner points and a mass (or density). They can be used in
3d simulations, and they can be joined together to form rigid bodies which represent arbitrary particles with a
triangulated surface.
The peri style is used with Peridynamic models and defines particles as having a volume, that is used
internally in the pair_style peri potentials.
The body style allows for definition of particles which can represent complex entities, such as surface meshes
of discrete points, collections of sub-particles, deformable objects, etc. The body style is discussed in more
detail on the body doc page.
Note that if one of these atom styles is used (or multiple styles via the atom_style hybrid command), not all
particles in the system are required to be finite-size or aspherical.
For example, in the ellipsoid style, if the 3 shape parameters are set to the same value, the particle will be a
sphere rather than an ellipsoid. If the 3 shape parameters are all set to 0.0 or if the diameter is set to 0.0, it will
be a point particle. In the line or tri style, if the lineflag or triflag is specified as 0, then it will be a point
particle.
Some of the pair styles used to compute pairwise interactions between finite-size particles also compute the
correct interaction with point particles as well, e.g. the interaction between a point particle and a finite-size
particle or between two point particles. If necessary, pair_style hybrid can be used to insure the correct
interactions are computed for the appropriate style of interactions. Likewise, using groups to partition particles
(ellipsoids versus spheres versus point particles) will allow you to use the appropriate time integrators and
temperature computations for each class of particles. See the doc pages for various commands for details.
Also note that for 2d simulations, atom styles sphere and ellipsoid still use 3d particles, rather than as circular
disks or ellipses. This means they have the same moment of inertia as the 3d object. When temperature is
computed, the correct degrees of freedom are used for rotation in a 2d versus 3d system.
Pair potentials
When a system with finite-size particles is defined, the particles will only rotate and experience torque if the
force field computes such interactions. These are the various pair styles that generate torque:
pair_style gran/history• pair_style gran/hertzian• pair_style gran/no_history• pair_style dipole/cut• pair_style gayberne• pair_style resquared• pair_style brownian• pair_style lubricate• pair_style line/lj• pair_style tri/lj• pair_style body•
The granular pair styles are used with spherical particles. The dipole pair style is used with the dipole atom
style, which could be applied to spherical or ellipsoidal particles. The GayBerne and REsquared potentials
require ellipsoidal particles, though they will also work if the 3 shape parameters are the same (a sphere). The
Brownian and lubrication potentials are used with spherical particles. The line, tri, and body potentials are
LAMMPS Users Manual
133
used with line segment, triangular, and body particles respectively.
Time integration
There are several fixes that perform time integration on finite-size spherical particles, meaning the integrators
update the rotational orientation and angular velocity or angular momentum of the particles:
fix nve/sphere• fix nvt/sphere• fix npt/sphere•
Likewise, there are 3 fixes that perform time integration on ellipsoidal particles:
fix nve/asphere• fix nvt/asphere• fix npt/asphere•
The advantage of these fixes is that those which thermostat the particles include the rotational degrees of
freedom in the temperature calculation and thermostatting. The fix langevin command can also be used with
its omgea or angmom options to thermostat the rotational degrees of freedom for spherical or ellipsoidal
particles. Other thermostatting fixes only operate on the translational kinetic energy of finite-size particles.
These fixes perform constant NVE time integration on line segment, triangular, and body particles:
fix nve/line• fix nve/tri• fix nve/body•
Note that for mixtures of point and finite-size particles, these integration fixes can only be used with groups
which contain finite-size particles.
Computes, thermodynamics, and dump output
There are several computes that calculate the temperature or rotational energy of spherical or ellipsoidal
particles:
compute temp/sphere• compute temp/asphere• compute erotate/sphere• compute erotate/asphere•
These include rotational degrees of freedom in their computation. If you wish the thermodynamic output of
temperature or pressure to use one of these computes (e.g. for a system entirely composed of finite-size
particles), then the compute can be defined and the thermo_modify command used. Note that by default
thermodynamic quantities will be calculated with a temperature that only includes translational degrees of
freedom. See the thermo_style command for details.
These commands can be used to output various attributes of finite-size particles:
dump custom• compute property/atom•
LAMMPS Users Manual
134

dump local• compute body/local•
Attributes include the dipole moment, the angular velocity, the angular momentum, the quaternion, the torque,
the end-point and corner-point coordinates (for line and tri particles), and sub-particle attributes of body
particles.
Rigid bodies composed of finite-size particles
The fix rigid command treats a collection of particles as a rigid body, computes its inertia tensor, sums the
total force and torque on the rigid body each timestep due to forces on its constituent particles, and integrates
the motion of the rigid body.
If any of the constituent particles of a rigid body are finite-size particles (spheres or ellipsoids or line segments
or triangles), then their contribution to the inertia tensor of the body is different than if they were point
particles. This means the rotational dynamics of the rigid body will be different. Thus a model of a dimer is
different if the dimer consists of two point masses versus two spheroids, even if the two particles have the
same mass. Finite-size particles that experience torque due to their interaction with other particles will also
impart that torque to a rigid body they are part of.
See the "fix rigid" command for example of complex rigid-body models it is possible to define in LAMMPS.
Note that the fix shake command can also be used to treat 2, 3, or 4 particles as a rigid body, but it always
assumes the particles are point masses.
Also note that body particles cannot be modeled with the fix rigid command. Body particles are treated by
LAMMPS as single particles, though they can store internal state, such as a list of sub-particles. Individual
body partices are typically treated as rigid bodies, and their motion integrated with a command like fix
nve/body. Interactions between pairs of body particles are computed via a command like pair_style body.
6.15 Output from LAMMPS (thermo, dumps, computes, fixes, variables)
There are four basic kinds of LAMMPS output:
Thermodynamic output, which is a list of quantities printed every few timesteps to the screen and
logfile.
•
Dump files, which contain snapshots of atoms and various per-atom values and are written at a
specified frequency.
•
Certain fixes can output user-specified quantities to files: fix ave/time for time averaging, fix
ave/chunk for spatial or other averaging, and fix print for single-line output of variables. Fix print can
also output to the screen.
•
Restart files.•
A simulation prints one set of thermodynamic output and (optionally) restart files. It can generate any number
of dump files and fix output files, depending on what dump and fix commands you specify.
As discussed below, LAMMPS gives you a variety of ways to determine what quantities are computed and
printed when the thermodynamics, dump, or fix commands listed above perform output. Throughout this
discussion, note that users can also add their own computes and fixes to LAMMPS which can then generate
values that can then be output with these commands.
LAMMPS Users Manual
135

The following sub-sections discuss different LAMMPS command related to output and the kind of data they
operate on and produce:
Global/per-atom/local data• Scalar/vector/array data• Thermodynamic output• Dump file output• Fixes that write output files• Computes that process output quantities• Fixes that process output quantities• Computes that generate values to output• Fixes that generate values to output• Variables that generate values to output• Summary table of output options and data flow between commands•
Global/per-atom/local data
Various output-related commands work with three different styles of data: global, per-atom, or local. A global
datum is one or more system-wide values, e.g. the temperature of the system. A per-atom datum is one or
more values per atom, e.g. the kinetic energy of each atom. Local datums are calculated by each processor
based on the atoms it owns, but there may be zero or more per atom, e.g. a list of bond distances.
Scalar/vector/array data
Global, per-atom, and local datums can each come in three kinds: a single scalar value, a vector of values, or a
2d array of values. The doc page for a "compute" or "fix" or "variable" that generates data will specify both
the style and kind of data it produces, e.g. a per-atom vector.
When a quantity is accessed, as in many of the output commands discussed below, it can be referenced via the
following bracket notation, where ID in this case is the ID of a compute. The leading "c_" would be replaced
by "f_" for a fix, or "v_" for a variable:
c_ID entire scalar, vector, or array
c_ID[I] one element of vector, one column of array
c_ID[I][J] one element of array
In other words, using one bracket reduces the dimension of the data once (vector -> scalar, array -> vector).
Using two brackets reduces the dimension twice (array -> scalar). Thus a command that uses scalar values as
input can typically also process elements of a vector or array.
Thermodynamic output
The frequency and format of thermodynamic output is set by the thermo, thermo_style, and thermo_modify
commands. The thermo_style command also specifies what values are calculated and written out. Pre-defined
keywords can be specified (e.g. press, etotal, etc). Three additional kinds of keywords can also be specified
(c_ID, f_ID, v_name), where a compute or fix or variable provides the value to be output. In each case, the
compute, fix, or variable must generate global values for input to the thermo_style custom command.
Note that thermodynamic output values can be "extensive" or "intensive". The former scale with the number
of atoms in the system (e.g. total energy), the latter do not (e.g. temperature). The setting for thermo_modify
norm determines whether extensive quantities are normalized or not. Computes and fixes produce either
extensive or intensive values; see their individual doc pages for details. Equal-style variables produce only
LAMMPS Users Manual
136
intensive values; you can include a division by "natoms" in the formula if desired, to make an extensive
calculation produce an intensive result.
Dump file output
Dump file output is specified by the dump and dump_modify commands. There are several pre-defined
formats (dump atom, dump xtc, etc).
There is also a dump custom format where the user specifies what values are output with each atom.
Pre-defined atom attributes can be specified (id, x, fx, etc). Three additional kinds of keywords can also be
specified (c_ID, f_ID, v_name), where a compute or fix or variable provides the values to be output. In each
case, the compute, fix, or variable must generate per-atom values for input to the dump custom command.
There is also a dump local format where the user specifies what local values to output. A pre-defined index
keyword can be specified to enumerate the local values. Two additional kinds of keywords can also be
specified (c_ID, f_ID), where a compute or fix or variable provides the values to be output. In each case, the
compute or fix must generate local values for input to the dump local command.
Fixes that write output files
Several fixes take various quantities as input and can write output files: fix ave/time, fix ave/chunk, fix
ave/histo, fix ave/correlate, and fix print.
The fix ave/time command enables direct output to a file and/or time-averaging of global scalars or vectors.
The user specifies one or more quantities as input. These can be global compute values, global fix values, or
variables of any style except the atom style which produces per-atom values. Since a variable can refer to
keywords used by the thermo_style custom command (like temp or press) and individual per-atom values, a
wide variety of quantities can be time averaged and/or output in this way. If the inputs are one or more scalar
values, then the fix generate a global scalar or vector of output. If the inputs are one or more vector values,
then the fix generates a global vector or array of output. The time-averaged output of this fix can also be used
as input to other output commands.
The fix ave/chunk command enables direct output to a file of chunk-averaged per-atom quantities like those
output in dump files. Chunks can represent spatial bins or other collections of atoms, e.g. individual
molecules. The per-atom quantities can be atom density (mass or number) or atom attributes such as position,
velocity, force. They can also be per-atom quantities calculated by a compute, by a fix, or by an atom-style
variable. The chunk-averaged output of this fix can also be used as input to other output commands.
The fix ave/histo command enables direct output to a file of histogrammed quantities, which can be global or
per-atom or local quantities. The histogram output of this fix can also be used as input to other output
commands.
The fix ave/correlate command enables direct output to a file of time-correlated quantities, which can be
global values. The correlation matrix output of this fix can also be used as input to other output commands.
The fix print command can generate a line of output written to the screen and log file or to a separate file,
periodically during a running simulation. The line can contain one or more variable values for any style
variable except the vector or atom styles). As explained above, variables themselves can contain references to
global values generated by thermodynamic keywords, computes, fixes, or other variables, or to per-atom
values for a specific atom. Thus the fix print command is a means to output a wide variety of quantities
separate from normal thermodynamic or dump file output.
LAMMPS Users Manual
137
Computes that process output quantities
The compute reduce and compute reduce/region commands take one or more per-atom or local vector
quantities as inputs and "reduce" them (sum, min, max, ave) to scalar quantities. These are produced as output
values which can be used as input to other output commands.
The compute slice command take one or more global vector or array quantities as inputs and extracts a subset
of their values to create a new vector or array. These are produced as output values which can be used as input
to other output commands.
The compute property/atom command takes a list of one or more pre-defined atom attributes (id, x, fx, etc)
and stores the values in a per-atom vector or array. These are produced as output values which can be used as
input to other output commands. The list of atom attributes is the same as for the dump custom command.
The compute property/local command takes a list of one or more pre-defined local attributes (bond info, angle
info, etc) and stores the values in a local vector or array. These are produced as output values which can be
used as input to other output commands.
Fixes that process output quantities
The fix vector command can create global vectors as output from global scalars as input, accumulating them
one element at a time.
The fix ave/atom command performs time-averaging of per-atom vectors. The per-atom quantities can be
atom attributes such as position, velocity, force. They can also be per-atom quantities calculated by a
compute, by a fix, or by an atom-style variable. The time-averaged per-atom output of this fix can be used as
input to other output commands.
The fix store/state command can archive one or more per-atom attributes at a particular time, so that the old
values can be used in a future calculation or output. The list of atom attributes is the same as for the dump
custom command, including per-atom quantities calculated by a compute, by a fix, or by an atom-style
variable. The output of this fix can be used as input to other output commands.
Computes that generate values to output
Every compute in LAMMPS produces either global or per-atom or local values. The values can be scalars or
vectors or arrays of data. These values can be output using the other commands described in this section. The
doc page for each compute command describes what it produces. Computes that produce per-atom or local
values have the word "atom" or "local" in their style name. Computes without the word "atom" or "local"
produce global values.
Fixes that generate values to output
Some fixes in LAMMPS produces either global or per-atom or local values which can be accessed by other
commands. The values can be scalars or vectors or arrays of data. These values can be output using the other
commands described in this section. The doc page for each fix command tells whether it produces any output
quantities and describes them.
LAMMPS Users Manual
138
Variables that generate values to output
Variables defined in an input script can store one or more strings. But equal-style, vector-style, and atom-style
or atomfile-style variables generate a global scalar value, global vector or values, or a per-atom vector,
respectively, when accessed. The formulas used to define these variables can contain references to the
thermodynamic keywords and to global and per-atom data generated by computes, fixes, and other variables.
The values generated by variables can be used as input to and thus output by the other commands described in
this section.
Summary table of output options and data flow between commands
This table summarizes the various commands that can be used for generating output from LAMMPS. Each
command produces output data of some kind and/or writes data to a file. Most of the commands can take data
from other commands as input. Thus you can link many of these commands together in pipeline form, where
data produced by one command is used as input to another command and eventually written to the screen or to
a file. Note that to hook two commands together the output and input data types must match, e.g.
global/per-atom/local data and scalar/vector/array data.
Also note that, as described above, when a command takes a scalar as input, that could be an element of a
vector or array. Likewise a vector input could be a column of an array.
Command Input Output
thermo_style custom global scalars screen, log file
dump custom per-atom vectors dump file
dump local local vectors dump file
fix print global scalar from variable screen, file
print global scalar from variable screen
computes N/A global/per-atom/local scalar/vector/array
fixes N/A global/per-atom/local scalar/vector/array
variables global scalars and vectors, per-atom vectors global scalar and vector, per-atom vector
compute reduce per-atom/local vectors global scalar/vector
compute slice global vectors/arrays global vector/array
compute
property/atom per-atom vectors per-atom vector/array
compute
property/local local vectors local vector/array
fix vector global scalars global vector
fix ave/atom per-atom vectors per-atom vector/array
fix ave/time global scalars/vectors global scalar/vector/array, file
fix ave/chunk per-atom vectors global array, file
fix ave/histo global/per-atom/local scalars and vectors global array, file
fix ave/correlate global scalars global array, file
fix store/state per-atom vectors per-atom vector/array
LAMMPS Users Manual
139
6.16 Thermostatting, barostatting, and computing temperature
Thermostatting means controlling the temperature of particles in an MD simulation. Barostatting means
controlling the pressure. Since the pressure includes a kinetic component due to particle velocities, both these
operations require calculation of the temperature. Typically a target temperature (T) and/or pressure (P) is
specified by the user, and the thermostat or barostat attempts to equilibrate the system to the requested T
and/or P.
Temperature is computed as kinetic energy divided by some number of degrees of freedom (and the
Boltzmann constant). Since kinetic energy is a function of particle velocity, there is often a need to distinguish
between a particle's advection velocity (due to some aggregate motion of particles) and its thermal velocity.
The sum of the two is the particle's total velocity, but the latter is often what is wanted to compute a
temperature.
LAMMPS has several options for computing temperatures, any of which can be used in thermostatting and
barostatting. These compute commands calculate temperature, and the compute pressure command calculates
pressure.
compute temp• compute temp/sphere• compute temp/asphere• compute temp/com• compute temp/deform• compute temp/partial• compute temp/profile• compute temp/ramp• compute temp/region•
All but the first 3 calculate velocity biases directly (e.g. advection velocities) that are removed when
computing the thermal temperature. Compute temp/sphere and compute temp/asphere compute kinetic energy
for finite-size particles that includes rotational degrees of freedom. They both allow for velocity biases
indirectly, via an optional extra argument, another temperature compute that subtracts a velocity bias. This
allows the translational velocity of spherical or aspherical particles to be adjusted in prescribed ways.
Thermostatting in LAMMPS is performed by fixes, or in one case by a pair style. Several thermostatting fixes
are available: Nose-Hoover (nvt), Berendsen, CSVR, Langevin, and direct rescaling (temp/rescale).
Dissipative particle dynamics (DPD) thermostatting can be invoked via the dpd/tstat pair style:
fix nvt• fix nvt/sphere• fix nvt/asphere• fix nvt/sllod• fix temp/berendsen• fix temp/csvr• fix langevin• fix temp/rescale• pair_style dpd/tstat•
Fix nvt only thermostats the translational velocity of particles. Fix nvt/sllod also does this, except that it
subtracts out a velocity bias due to a deforming box and integrates the SLLOD equations of motion. See the
NEMD simulations section of this page for further details. Fix nvt/sphere and fix nvt/asphere thermostat not
LAMMPS Users Manual
140
only translation velocities but also rotational velocities for spherical and aspherical particles.
DPD thermostatting alters pairwise interactions in a manner analogous to the per-particle thermostatting of fix
langevin.
Any of the thermostatting fixes can use temperature computes that remove bias which has two effects. First,
the current calculated temperature, which is compared to the requested target temperature, is calculated with
the velocity bias removed. Second, the thermostat adjusts only the thermal temperature component of the
particle's velocities, which are the velocities with the bias removed. The removed bias is then added back to
the adjusted velocities. See the doc pages for the individual fixes and for the fix_modify command for
instructions on how to assign a temperature compute to a thermostatting fix. For example, you can apply a
thermostat to only the x and z components of velocity by using it in conjunction with compute temp/partial.
Of you could thermostat only the thermal temperature of a streaming flow of particles without affecting the
streaming velocity, by using compute temp/profile.
NOTE: Only the nvt fixes perform time integration, meaning they update the velocities and positions of
particles due to forces and velocities respectively. The other thermostat fixes only adjust velocities; they do
NOT perform time integration updates. Thus they should be used in conjunction with a constant NVE
integration fix such as these:
fix nve• fix nve/sphere• fix nve/asphere•
Barostatting in LAMMPS is also performed by fixes. Two barosttating methods are currently available:
Nose-Hoover (npt and nph) and Berendsen:
fix npt• fix npt/sphere• fix npt/asphere• fix nph• fix press/berendsen•
The fix npt commands include a Nose-Hoover thermostat and barostat. Fix nph is just a Nose/Hoover
barostat; it does no thermostatting. Both fix nph and fix press/berendsen can be used in conjunction with any
of the thermostatting fixes.
As with the thermostats, fix npt and fix nph only use translational motion of the particles in computing T and
P and performing thermo/barostatting. Fix npt/sphere and fix npt/asphere thermo/barostat using not only
translation velocities but also rotational velocities for spherical and aspherical particles.
All of the barostatting fixes use the compute pressure compute to calculate a current pressure. By default, this
compute is created with a simple compute temp (see the last argument of the compute pressure command),
which is used to calculated the kinetic component of the pressure. The barostatting fixes can also use
temperature computes that remove bias for the purpose of computing the kinetic component which contributes
to the current pressure. See the doc pages for the individual fixes and for the fix_modify command for
instructions on how to assign a temperature or pressure compute to a barostatting fix.
NOTE: As with the thermostats, the Nose/Hoover methods (fix npt and fix nph) perform time integration. Fix
press/berendsen does NOT, so it should be used with one of the constant NVE fixes or with one of the NVT
fixes.
LAMMPS Users Manual
141

Finally, thermodynamic output, which can be setup via the thermo_style command, often includes
temperature and pressure values. As explained on the doc page for the thermo_style command, the default T
and P are setup by the thermo command itself. They are NOT the ones associated with any thermostatting or
barostatting fix you have defined or with any compute that calculates a temperature or pressure. Thus if you
want to view these values of T and P, you need to specify them explicitly via a thermo_style custom
command. Or you can use the thermo_modify command to re-define what temperature or pressure compute is
used for default thermodynamic output.
6.17 Walls
Walls in an MD simulation are typically used to bound particle motion, i.e. to serve as a boundary condition.
Walls in LAMMPS can be of rough (made of particles) or idealized surfaces. Ideal walls can be smooth,
generating forces only in the normal direction, or frictional, generating forces also in the tangential direction.
Rough walls, built of particles, can be created in various ways. The particles themselves can be generated like
any other particle, via the lattice and create_atoms commands, or read in via the read_data command.
Their motion can be constrained by many different commands, so that they do not move at all, move together
as a group at constant velocity or in response to a net force acting on them, move in a prescribed fashion (e.g.
rotate around a point), etc. Note that if a time integration fix like fix nve or fix nvt is not used with the group
that contains wall particles, their positions and velocities will not be updated.
fix aveforce - set force on particles to average value, so they move together• fix setforce - set force on particles to a value, e.g. 0.0• fix freeze - freeze particles for use as granular walls• fix nve/noforce - advect particles by their velocity, but without force• fix move - prescribe motion of particles by a linear velocity, oscillation, rotation, variable•
The fix move command offers the most generality, since the motion of individual particles can be specified
with variable formula which depends on time and/or the particle position.
For rough walls, it may be useful to turn off pairwise interactions between wall particles via the neigh_modify
exclude command.
Rough walls can also be created by specifying frozen particles that do not move and do not interact with
mobile particles, and then tethering other particles to the fixed particles, via a bond. The bonded particles do
interact with other mobile particles.
Idealized walls can be specified via several fix commands. Fix wall/gran creates frictional walls for use with
granular particles; all the other commands create smooth walls.
fix wall/reflect - reflective flat walls• fix wall/lj93 - flat walls, with Lennard-Jones 9/3 potential• fix wall/lj126 - flat walls, with Lennard-Jones 12/6 potential• fix wall/colloid - flat walls, with pair_style colloid potential• fix wall/harmonic - flat walls, with repulsive harmonic spring potential• fix wall/region - use region surface as wall• fix wall/gran - flat or curved walls with pair_style granular potential•
LAMMPS Users Manual
142
The lj93, lj126, colloid, and harmonic styles all allow the flat walls to move with a constant velocity, or
oscillate in time. The fix wall/region command offers the most generality, since the region surface is treated as
a wall, and the geometry of the region can be a simple primitive volume (e.g. a sphere, or cube, or plane), or a
complex volume made from the union and intersection of primitive volumes. Regions can also specify a
volume "interior" or "exterior" to the specified primitive shape or union or intersection. Regions can also be
"dynamic" meaning they move with constant velocity, oscillate, or rotate.
The only frictional idealized walls currently in LAMMPS are flat or curved surfaces specified by the fix
wall/gran command. At some point we plan to allow regoin surfaces to be used as frictional walls, as well as
triangulated surfaces.
6.18 Elastic constants
Elastic constants characterize the stiffness of a material. The formal definition is provided by the linear
relation that holds between the stress and strain tensors in the limit of infinitesimal deformation. In tensor
notation, this is expressed as s_ij = C_ijkl * e_kl, where the repeated indices imply summation. s_ij are the
elements of the symmetric stress tensor. e_kl are the elements of the symmetric strain tensor. C_ijkl are the
elements of the fourth rank tensor of elastic constants. In three dimensions, this tensor has 3^4=81 elements.
Using Voigt notation, the tensor can be written as a 6x6 matrix, where C_ij is now the derivative of s_i w.r.t.
e_j. Because s_i is itself a derivative w.r.t. e_i, it follows that C_ij is also symmetric, with at most 7*6/2 = 21
distinct elements.
At zero temperature, it is easy to estimate these derivatives by deforming the simulation box in one of the six
directions using the change_box command and measuring the change in the stress tensor. A general-purpose
script that does this is given in the examples/elastic directory described in this section.
Calculating elastic constants at finite temperature is more challenging, because it is necessary to run a
simulation that perfoms time averages of differential properties. One way to do this is to measure the change
in average stress tensor in an NVT simulations when the cell volume undergoes a finite deformation. In order
to balance the systematic and statistical errors in this method, the magnitude of the deformation must be
chosen judiciously, and care must be taken to fully equilibrate the deformed cell before sampling the stress
tensor. Another approach is to sample the triclinic cell fluctuations that occur in an NPT simulation. This
method can also be slow to converge and requires careful post-processing (Shinoda)
6.19 Library interface to LAMMPS
As described in Section 2.5, LAMMPS can be built as a library, so that it can be called by another code, used
in a coupled manner with other codes, or driven through a Python interface.
All of these methodologies use a C-style interface to LAMMPS that is provided in the files src/library.cpp and
src/library.h. The functions therein have a C-style argument list, but contain C++ code you could write
yourself in a C++ application that was invoking LAMMPS directly. The C++ code in the functions illustrates
how to invoke internal LAMMPS operations. Note that LAMMPS classes are defined within a LAMMPS
namespace (LAMMPS_NS) if you use them from another C++ application.
Library.cpp contains these functions for creating and destroying an instance of LAMMPS and sending it
commands to execute. See the documentation in the src/library.cpp file for details:
void lammps_open(int, char **, MPI_Comm, void **)
LAMMPS Users Manual
143
void lammps_open_no_mpi(int, char **, void **)
void lammps_close(void *)
int lammps_version(void *)
void lammps_file(void *, char *)
char *lammps_command(void *, char *)
void lammps_commands_list(void *, int, char **)
void lammps_commands_string(void *, char *)
void lammps_free(void *)
The lammps_open() function is used to initialize LAMMPS, passing in a list of strings as if they were
command-line arguments when LAMMPS is run in stand-alone mode from the command line, and a MPI
communicator for LAMMPS to run under. It returns a ptr to the LAMMPS object that is created, and which is
used in subsequent library calls. The lammps_open() function can be called multiple times, to create multiple
instances of LAMMPS.
LAMMPS will run on the set of processors in the communicator. This means the calling code can run
LAMMPS on all or a subset of processors. For example, a wrapper script might decide to alternate between
LAMMPS and another code, allowing them both to run on all the processors. Or it might allocate half the
processors to LAMMPS and half to the other code and run both codes simultaneously before syncing them up
periodically. Or it might instantiate multiple instances of LAMMPS to perform different calculations.
The lammps_open_no_mpi() function is similar except that no MPI communicator is passed from the caller.
Instead, MPI_COMM_WORLD is used to instantiate LAMMPS, and MPI is initialized if necessary.
The lammps_close() function is used to shut down an instance of LAMMPS and free all its memory.
The lammps_version() function can be used to determined the specific version of the underlying LAMMPS
code. This is particularly useful when loading LAMMPS as a shared library via dlopen(). The code using the
library interface can than use this information to adapt to changes to the LAMMPS command syntax between
versions. The returned LAMMPS version code is an integer (e.g. 2 Sep 2015 results in 20150902) that grows
with every new LAMMPS version.
The lammps_file(), lammps_command(), lammps_commands_list(), and lammps_commands_string()
functions are used to pass one or more commands to LAMMPS to execute, the same as if they were coming
from an input script.
Via these functions, the calling code can read or generate a series of LAMMPS commands one or multiple at a
time and pass it thru the library interface to setup a problem and then run it in stages. The caller can interleave
the command function calls with operations it performs, calls to extract information from or set information
within LAMMPS, or calls to another code's library.
The lammps_file() function passes the filename of an input script. The lammps_command() function passes a
single command as a string. The lammps_commands_list() function passes multiple commands in a char**
list. In both lammps_command() and lammps_commands_list(), individual commands may or may not have a
trailing newline. The lammps_commands_string() function passes multiple commands concatenated into one
long string, separated by newline characters. In both lammps_commands_list() and
lammps_commands_string(), a single command can be spread across multiple lines, if the last printable
character of all but the last line is "&", the same as if the lines appeared in an input script.
The lammps_free() function is a clean-up function to free memory that the library allocated previously via
other function calls. See comments in src/library.cpp file for which other functions need this clean-up.
LAMMPS Users Manual
144
Library.cpp also contains these functions for extracting information from LAMMPS and setting value within
LAMMPS. Again, see the documentation in the src/library.cpp file for details, including which quantities can
be queried by name:
void *lammps_extract_global(void *, char *)
void lammps_extract_box(void *, double *, double *,
double *, double *, double *, int *, int *)
void *lammps_extract_atom(void *, char *)
void *lammps_extract_compute(void *, char *, int, int)
void *lammps_extract_fix(void *, char *, int, int, int, int)
void *lammps_extract_variable(void *, char *, char *)
void lammps_reset_box(void *, double *, double *, double, double, double)
int lammps_set_variable(void *, char *, char *)
double lammps_get_thermo(void *, char *)
int lammps_get_natoms(void *)
void lammps_gather_atoms(void *, double *)
void lammps_scatter_atoms(void *, double *)
void lammps_create_atoms(void *, int, tagint *, int *, double *, double *,
imageint *, int)
The extract functions return a pointer to various global or per-atom quantities stored in LAMMPS or to values
calculated by a compute, fix, or variable. The pointer returned by the extract_global() function can be used as
a permanent reference to a value which may change. For the extract_atom() method, see the extract() method
in the src/atom.cpp file for a list of valid per-atom properties. New names could easily be added if the
property you want is not listed. For the other extract functions, the underlying storage may be reallocated as
LAMMPS runs, so you need to re-call the function to assure a current pointer or returned value(s).
The lammps_reset_box() function resets the size and shape of the simulation box, e.g. as part of restoring a
previously extracted and saved state of a simulation.
The lammps_set_variable() function can set an existing string-style variable to a new string value, so that
subsequent LAMMPS commands can access the variable.
The lammps_get_thermo() function returns the current value of a thermo keyword as a double precision value.
The lammps_get_natoms() function returns the total number of atoms in the system and can be used by the
caller to allocate space for the lammps_gather_atoms() and lammps_scatter_atoms() functions. The gather
function collects peratom info of the requested type (atom coords, types, forces, etc) from all processors,
orders them by atom ID, and returns a full list to each calling processor. The scatter function does the inverse.
It distributes the same peratom values, passed by the caller, to each atom owned by individual processors.
Both methods are thus a means to extract or assign (overwrite) any peratom quantities within LAMMPS. See
the extract() method in the src/atom.cpp file for a list of valid per-atom properties. New names could easily be
added if the property you want is not listed. A special treatment is applied for accessing image flags via the
"image" property. Image flags are stored in a packed format with all three image flags stored in a single
integer. When signaling to access the image flags as 3 individual values per atom instead of 1, the data is
transparently packed or unpacked by the library interface.
The lammps_create_atoms() function takes a list of N atoms as input with atom types and coords (required),
an optionally atom IDs and velocities and image flags. It uses the coords of each atom to assign it as a new
atom to the processor that owns it. This function is useful to add atoms to a simulation or (in tandem with
LAMMPS Users Manual
145

lammps_reset_box()) to restore a previously extracted and saved state of a simulation. Additional properties
for the new atoms can then be assigned via the lammps_scatter_atoms() or lammps_extract_atom() functions.
The examples/COUPLE and python directories have example C++ and C and Python codes which show how
a driver code can link to LAMMPS as a library, run LAMMPS on a subset of processors, grab data from
LAMMPS, change it, and put it back into LAMMPS.
NOTE: You can write code for additional functions as needed to define how your code talks to LAMMPS and
add them to src/library.cpp and src/library.h, as well as to the Python interface. The added functions can
access or change any LAMMPS data you wish.
6.20 Calculating thermal conductivity
The thermal conductivity kappa of a material can be measured in at least 4 ways using various options in
LAMMPS. See the examples/KAPPA directory for scripts that implement the 4 methods discussed here for a
simple Lennard-Jones fluid model. Also, see this section of the manual for an analogous discussion for
viscosity.
The thermal conductivity tensor kappa is a measure of the propensity of a material to transmit heat energy in a
diffusive manner as given by Fourier's law
J = -kappa grad(T)
where J is the heat flux in units of energy per area per time and grad(T) is the spatial gradient of temperature.
The thermal conductivity thus has units of energy per distance per time per degree K and is often
approximated as an isotropic quantity, i.e. as a scalar.
The first method is to setup two thermostatted regions at opposite ends of a simulation box, or one in the
middle and one at the end of a periodic box. By holding the two regions at different temperatures with a
thermostatting fix, the energy added to the hot region should equal the energy subtracted from the cold region
and be proportional to the heat flux moving between the regions. See the papers by Ikeshoji and Hafskjold and
Wirnsberger et al for details of this idea. Note that thermostatting fixes such as fix nvt, fix langevin, and fix
temp/rescale store the cumulative energy they add/subtract.
Alternatively, as a second method, the fix heat or fix ehex commands can be used in place of thermostats on
each of two regions to add/subtract specified amounts of energy to both regions. In both cases, the resulting
temperatures of the two regions can be monitored with the "compute temp/region" command and the
temperature profile of the intermediate region can be monitored with the fix ave/chunk and compute ke/atom
commands.
The third method is to perform a reverse non-equilibrium MD simulation using the fix thermal/conductivity
command which implements the rNEMD algorithm of Muller-Plathe. Kinetic energy is swapped between
atoms in two different layers of the simulation box. This induces a temperature gradient between the two
layers which can be monitored with the fix ave/chunk and compute ke/atom commands. The fix tallies the
cumulative energy transfer that it performs. See the fix thermal/conductivity command for details.
The fourth method is based on the Green-Kubo (GK) formula which relates the ensemble average of the
auto-correlation of the heat flux to kappa. The heat flux can be calculated from the fluctuations of per-atom
potential and kinetic energies and per-atom stress tensor in a steady-state equilibrated simulation. This is in
contrast to the two preceding non-equilibrium methods, where energy flows continuously between hot and
LAMMPS Users Manual
146

cold regions of the simulation box.
The compute heat/flux command can calculate the needed heat flux and describes how to implement the
Green_Kubo formalism using additional LAMMPS commands, such as the fix ave/correlate command to
calculate the needed auto-correlation. See the doc page for the compute heat/flux command for an example
input script that calculates the thermal conductivity of solid Ar via the GK formalism.
6.21 Calculating viscosity
The shear viscosity eta of a fluid can be measured in at least 5 ways using various options in LAMMPS. See
the examples/VISCOSITY directory for scripts that implement the 5 methods discussed here for a simple
Lennard-Jones fluid model. Also, see this section of the manual for an analogous discussion for thermal
conductivity.
Eta is a measure of the propensity of a fluid to transmit momentum in a direction perpendicular to the
direction of velocity or momentum flow. Alternatively it is the resistance the fluid has to being sheared. It is
given by
J = -eta grad(Vstream)
where J is the momentum flux in units of momentum per area per time. and grad(Vstream) is the spatial
gradient of the velocity of the fluid moving in another direction, normal to the area through which the
momentum flows. Viscosity thus has units of pressure-time.
The first method is to perform a non-equilibrium MD (NEMD) simulation by shearing the simulation box via
the fix deform command, and using the fix nvt/sllod command to thermostat the fluid via the SLLOD
equations of motion. Alternatively, as a second method, one or more moving walls can be used to shear the
fluid in between them, again with some kind of thermostat that modifies only the thermal (non-shearing)
components of velocity to prevent the fluid from heating up.
In both cases, the velocity profile setup in the fluid by this procedure can be monitored by the fix ave/chunk
command, which determines grad(Vstream) in the equation above. E.g. the derivative in the y-direction of the
Vx component of fluid motion or grad(Vstream) = dVx/dy. The Pxy off-diagonal component of the pressure
or stress tensor, as calculated by the compute pressure command, can also be monitored, which is the J term in
the equation above. See this section of the manual for details on NEMD simulations.
The third method is to perform a reverse non-equilibrium MD simulation using the fix viscosity command
which implements the rNEMD algorithm of Muller-Plathe. Momentum in one dimension is swapped between
atoms in two different layers of the simulation box in a different dimension. This induces a velocity gradient
which can be monitored with the fix ave/chunk command. The fix tallies the cumulative momentum transfer
that it performs. See the fix viscosity command for details.
The fourth method is based on the Green-Kubo (GK) formula which relates the ensemble average of the
auto-correlation of the stress/pressure tensor to eta. This can be done in a fully equilibrated simulation which
is in contrast to the two preceding non-equilibrium methods, where momentum flows continuously through
the simulation box.
Here is an example input script that calculates the viscosity of liquid Ar via the GK formalism:
# Sample LAMMPS input script for viscosity of liquid Ar
LAMMPS Users Manual
147
units real
variable T equal 86.4956
variable V equal vol
variable dt equal 4.0
variable p equal 400 # correlation length
variable s equal 5 # sample interval
variable d equal $p*$s # dump interval
# convert from LAMMPS real units to SI
variable kB equal 1.3806504e-23 # [J/K/ Boltzmann
variable atm2Pa equal 101325.0
variable A2m equal 1.0e-10
variable fs2s equal 1.0e-15
variable convert equal ${atm2Pa}*${atm2Pa}*${fs2s}*${A2m}*${A2m}*${A2m}
# setup problem
dimension 3
boundary p p p
lattice fcc 5.376 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
region box block 0 4 0 4 0 4
create_box 1 box
create_atoms 1 box
mass 1 39.948
pair_style lj/cut 13.0
pair_coeff * * 0.2381 3.405
timestep ${dt}
thermo $d
# equilibration and thermalization
velocity all create $T 102486 mom yes rot yes dist gaussian
fix NVT all nvt temp $T $T 10 drag 0.2
run 8000
# viscosity calculation, switch to NVE if desired
#unfix NVT
#fix NVE all nve
reset_timestep 0
variable pxy equal pxy
variable pxz equal pxz
variable pyz equal pyz
fix SS all ave/correlate $s $p $d &
v_pxy v_pxz v_pyz type auto file S0St.dat ave running
variable scale equal ${convert}/(${kB}*$T)*$V*$s*${dt}
variable v11 equal trap(f_SS[3])*${scale}
variable v22 equal trap(f_SS[4])*${scale}
variable v33 equal trap(f_SS[5])*${scale}
thermo_style custom step temp press v_pxy v_pxz v_pyz v_v11 v_v22 v_v33
run 100000
variable v equal (v_v11+v_v22+v_v33)/3.0
variable ndens equal count(all)/vol
print "average viscosity: $v [Pa.s] @ $T K, ${ndens} /A^3"
The fifth method is related to the above Green-Kubo method, but uses the Einstein formulation, analogous to
the Einstein mean-square-displacement formulation for self-diffusivity. The time-integrated momentum fluxes
LAMMPS Users Manual
148

play the role of Cartesian coordinates, whose mean-square displacement increases linearly with time at
sufficiently long times.
6.22 Calculating a diffusion coefficient
The diffusion coefficient D of a material can be measured in at least 2 ways using various options in
LAMMPS. See the examples/DIFFUSE directory for scripts that implement the 2 methods discussed here for
a simple Lennard-Jones fluid model.
The first method is to measure the mean-squared displacement (MSD) of the system, via the compute msd
command. The slope of the MSD versus time is proportional to the diffusion coefficient. The instantaneous
MSD values can be accumulated in a vector via the fix vector command, and a line fit to the vector to
compute its slope via the variable slope function, and thus extract D.
The second method is to measure the velocity auto-correlation function (VACF) of the system, via the
compute vacf command. The time-integral of the VACF is proportional to the diffusion coefficient. The
instantaneous VACF values can be accumulated in a vector via the fix vector command, and time integrated
via the variable trap function, and thus extract D.
6.23 Using chunks to calculate system properties
In LAMMS, "chunks" are collections of atoms, as defined by the compute chunk/atom command, which
assigns each atom to a chunk ID (or to no chunk at all). The number of chunks and the assignment of chunk
IDs to atoms can be static or change over time. Examples of "chunks" are molecules or spatial bins or atoms
with similar values (e.g. coordination number or potential energy).
The per-atom chunk IDs can be used as input to two other kinds of commands, to calculate various properties
of a system:
fix ave/chunk• any of the compute */chunk commands•
Here, each of the 3 kinds of chunk-related commands is briefly overviewed. Then some examples are given of
how to compute different properties with chunk commands.
Compute chunk/atom command:
This compute can assign atoms to chunks of various styles. Only atoms in the specified group and optional
specified region are assigned to a chunk. Here are some possible chunk definitions:
atoms in same molecule chunk ID = molecule ID
atoms of same atom type chunk ID = atom type
all atoms with same atom property (charge,
radius, etc) chunk ID = output of compute property/atom
atoms in same cluster chunk ID = output of compute cluster/atom command
atoms in same spatial bin chunk ID = bin ID
atoms in same rigid body chunk ID = molecule ID used to define rigid bodies
LAMMPS Users Manual
149

atoms with similar potential energy chunk ID = output of compute pe/atom
atoms with same local defect structure chunk ID = output of compute centro/atom or compute
coord/atom command
Note that chunk IDs are integer values, so for atom properties or computes that produce a floating point value,
they will be truncated to an integer. You could also use the compute in a variable that scales the floating point
value to spread it across multiple integers.
Spatial bins can be of various kinds, e.g. 1d bins = slabs, 2d bins = pencils, 3d bins = boxes, spherical bins,
cylindrical bins.
This compute also calculates the number of chunks Nchunk, which is used by other commands to tally
per-chunk data. Nchunk can be a static value or change over time (e.g. the number of clusters). The chunk ID
for an individual atom can also be static (e.g. a molecule ID), or dynamic (e.g. what spatial bin an atom is in
as it moves).
Note that this compute allows the per-atom output of other computes, fixes, and variables to be used to define
chunk IDs for each atom. This means you can write your own compute or fix to output a per-atom quantity to
use as chunk ID. See Section 10 of the documentation for how to do this. You can also define a per-atom
variable in the input script that uses a formula to generate a chunk ID for each atom.
Fix ave/chunk command:
This fix takes the ID of a compute chunk/atom command as input. For each chunk, it then sums one or more
specified per-atom values over the atoms in each chunk. The per-atom values can be any atom property, such
as velocity, force, charge, potential energy, kinetic energy, stress, etc. Additional keywords are defined for
per-chunk properties like density and temperature. More generally any per-atom value generated by other
computes, fixes, and per-atom variables, can be summed over atoms in each chunk.
Similar to other averaging fixes, this fix allows the summed per-chunk values to be time-averaged in various
ways, and output to a file. The fix produces a global array as output with one row of values per chunk.
Compute */chunk commands:
Currently the following computes operate on chunks of atoms to produce per-chunk values.
compute com/chunk• compute gyration/chunk• compute inertia/chunk• compute msd/chunk• compute property/chunk• compute temp/chunk• compute torque/chunk• compute vcm/chunk•
They each take the ID of a compute chunk/atom command as input. As their names indicate, they calculate the
center-of-mass, radius of gyration, moments of inertia, mean-squared displacement, temperature, torque, and
velocity of center-of-mass for each chunk of atoms. The compute property/chunk command can tally the
count of atoms in each chunk and extract other per-chunk properties.
The reason these various calculations are not part of the fix ave/chunk command, is that each requires a more
complicated operation than simply summing and averaging over per-atom values in each chunk. For example,
LAMMPS Users Manual
150

many of them require calculation of a center of mass, which requires summing mass*position over the atoms
and then dividing by summed mass.
All of these computes produce a global vector or global array as output, wih one or more values per chunk.
They can be used in various ways:
As input to the fix ave/time command, which can write the values to a file and optionally time
average them.
•
As input to the fix ave/histo command to histogram values across chunks. E.g. a histogram of cluster
sizes or molecule diffusion rates.
•
As input to special functions of equal-style variables, like sum() and max(). E.g. to find the largest
cluster or fastest diffusing molecule.
•
Example calculations with chunks
Here are examples using chunk commands to calculate various properties:
(1) Average velocity in each of 1000 2d spatial bins:
compute cc1 all chunk/atom bin/2d x 0.0 0.1 y lower 0.01 units reduced
fix 1 all ave/chunk 100 10 1000 cc1 vx vy file tmp.out
(2) Temperature in each spatial bin, after subtracting a flow velocity:
compute cc1 all chunk/atom bin/2d x 0.0 0.1 y lower 0.1 units reduced
compute vbias all temp/profile 1 0 0 y 10
fix 1 all ave/chunk 100 10 1000 cc1 temp bias vbias file tmp.out
(3) Center of mass of each molecule:
compute cc1 all chunk/atom molecule
compute myChunk all com/chunk cc1
fix 1 all ave/time 100 1 100 c_myChunk[*] file tmp.out mode vector
(4) Total force on each molecule and ave/max across all molecules:
compute cc1 all chunk/atom molecule
fix 1 all ave/chunk 1000 1 1000 cc1 fx fy fz file tmp.out
variable xave equal ave(f_1[2])
variable xmax equal max(f_1[2])
thermo 1000
thermo_style custom step temp v_xave v_xmax
(5) Histogram of cluster sizes:
compute cluster all cluster/atom 1.0
compute cc1 all chunk/atom c_cluster compress yes
compute size all property/chunk cc1 count
fix 1 all ave/histo 100 1 100 0 20 20 c_size mode vector ave running beyond ignore file tmp.histo
LAMMPS Users Manual
151
6.24 Setting parameters for the kspace_style pppm/disp command
The PPPM method computes interactions by splitting the pair potential into two parts, one of which is
computed in a normal pairwise fashion, the so-called real-space part, and one of which is computed using the
Fourier transform, the so called reciprocal-space or kspace part. For both parts, the potential is not computed
exactly but is approximated. Thus, there is an error in both parts of the computation, the real-space and the
kspace error. The just mentioned facts are true both for the PPPM for Coulomb as well as dispersion
interactions. The deciding difference - and also the reason why the parameters for pppm/disp have to be
selected with more care - is the impact of the errors on the results: The kspace error of the PPPM for Coulomb
and dispersion interaction and the real-space error of the PPPM for Coulomb interaction have the character of
noise. In contrast, the real-space error of the PPPM for dispersion has a clear physical interpretation: the
underprediction of cohesion. As a consequence, the real-space error has a much stronger effect than the
kspace error on simulation results for pppm/disp. Parameters must thus be chosen in a way that this error is
much smaller than the kspace error.
When using pppm/disp and not making any specifications on the PPPM parameters via the kspace modify
command, parameters will be tuned such that the real-space error and the kspace error are equal. This will
result in simulations that are either inaccurate or slow, both of which is not desirable. For selecting parameters
for the pppm/disp that provide fast and accurate simulations, there are two approaches, which both have their
up- and downsides.
The first approach is to set desired real-space an kspace accuracies via the kspace_modify force/disp/real and
kspace_modify force/disp/kspace commands. Note that the accuracies have to be specified in force units and
are thus dependent on the chosen unit settings. For real units, 0.0001 and 0.002 seem to provide reasonable
accurate and efficient computations for the real-space and kspace accuracies. 0.002 and 0.05 work well for
most systems using lj units. PPPM parameters will be generated based on the desired accuracies. The upside
of this approach is that it usually provides a good set of parameters and will work for both the kspace_modify
diff ad and kspace_modify diff ik options. The downside of the method is that setting the PPPM parameters
will take some time during the initialization of the simulation.
The second approach is to set the parameters for the pppm/disp explicitly using the kspace_modify mesh/disp,
kspace_modify order/disp, and kspace_modify gewald/disp commands. This approach requires a more
experienced user who understands well the impact of the choice of parameters on the simulation accuracy and
performance. This approach provides a fast initialization of the simulation. However, it is sensitive to errors:
A combination of parameters that will perform well for one system might result in far-from-optimal
conditions for other simulations. For example, parameters that provide accurate and fast computations for
all-atomistic force fields can provide insufficient accuracy or united-atomistic force fields (which is related to
that the latter typically have larger dispersion coefficients).
To avoid inaccurate or inefficient simulations, the pppm/disp stops simulations with an error message if no
action is taken to control the PPPM parameters. If the automatic parameter generation is desired and
real-space and kspace accuracies are desired to be equal, this error message can be suppressed using the
kspace_modify disp/auto yes command.
A reasonable approach that combines the upsides of both methods is to make the first run using the
kspace_modify force/disp/real and kspace_modify force/disp/kspace commands, write down the PPPM
parameters from the outut, and specify these parameters using the second approach in subsequent runs (which
have the same composition, force field, and approximately the same volume).
Concerning the performance of the pppm/disp there are two more things to consider. The first is that when
using the pppm/disp, the cutoff parameter does no longer affect the accuracy of the simulation (subject to that
LAMMPS Users Manual
152

gewald/disp is adjusted when changing the cutoff). The performance can thus be increased by examining
different values for the cutoff parameter. A lower bound for the cutoff is only set by the truncation error of the
repulsive term of pair potentials.
The second is that the mixing rule of the pair style has an impact on the computation time when using the
pppm/disp. Fastest computations are achieved when using the geometric mixing rule. Using the arithmetic
mixing rule substantially increases the computational cost. The computational overhead can be reduced using
the kspace_modify mix/disp geom and kspace_modify splittol commands. The first command simply enforces
geometric mixing of the dispersion coefficients in kspace computations. This introduces some error in the
computations but will also significantly speed-up the simulations. The second keyword sets the accuracy with
which the dispersion coefficients are approximated using a matrix factorization approach. This may result in
better accuracy then using the first command, but will usually also not provide an equally good increase of
efficiency.
Finally, pppm/disp can also be used when no mixing rules apply. This can be achieved using the
kspace_modify mix/disp none command. Note that the code does not check automatically whether any mixing
rule is fulfilled. If mixing rules do not apply, the user will have to specify this command explicitly.
6.25 Polarizable models
In polarizable force fields the charge distributions in molecules and materials respond to their electrostatic
environments. Polarizable systems can be simulated in LAMMPS using three methods:
the fluctuating charge method, implemented in the QEQ package,• the adiabatic core-shell method, implemented in the CORESHELL package,• the thermalized Drude dipole method, implemented in the USER-DRUDE package.•
The fluctuating charge method calculates instantaneous charges on interacting atoms based on the
electronegativity equalization principle. It is implemented in the fix qeq which is available in several variants.
It is a relatively efficient technique since no additional particles are introduced. This method allows for charge
transfer between molecules or atom groups. However, because the charges are located at the interaction sites,
off-plane components of polarization cannot be represented in planar molecules or atom groups.
The two other methods share the same basic idea: polarizable atoms are split into one core atom and one
satellite particle (called shell or Drude particle) attached to it by a harmonic spring. Both atoms bear a charge
and they represent collectively an induced electric dipole. These techniques are computationally more
expensive than the QEq method because of additional particles and bonds. These two charge-on-spring
methods differ in certain features, with the core-shell model being normally used for ionic/crystalline
materials, whereas the so-called Drude model is normally used for molecular systems and fluid states.
The core-shell model is applicable to crystalline materials where the high symmetry around each site leads to
stable trajectories of the core-shell pairs. However, bonded atoms in molecules can be so close that a core
would interact too strongly or even capture the Drude particle of a neighbor. The Drude dipole model is
relatively more complex in order to remediate this and other issues. Specifically, the Drude model includes
specific thermostating of the core-Drude pairs and short-range damping of the induced dipoles.
The three polarization methods can be implemented through a self-consistent calculation of charges or
induced dipoles at each timestep. In the fluctuating charge scheme this is done by the matrix inversion method
in fix qeq/point, but for core-shell or Drude-dipoles the relaxed-dipoles technique would require an slow
iterative procedure. These self-consistent solutions yield accurate trajectories since the additional degrees of
LAMMPS Users Manual
153

freedom representing polarization are massless. An alternative is to attribute a mass to the additional degrees
of freedom and perform time integration using an extended Lagrangian technique. For the fluctuating charge
scheme this is done by fix qeq/dynamic, and for the charge-on-spring models by the methods outlined in the
next two sections. The assignment of masses to the additional degrees of freedom can lead to unphysical
trajectories if care is not exerted in choosing the parameters of the polarizable models and the simulation
conditions.
In the core-shell model the vibration of the shells is kept faster than the ionic vibrations to mimic the fast
response of the polarizable electrons. But in molecular systems thermalizing the core-Drude pairs at
temperatures comparable to the rest of the simulation leads to several problems (kinetic energy transfer, too
short a timestep, etc.) In order to avoid these problems the relative motion of the Drude particles with respect
to their cores is kept "cold" so the vibration of the core-Drude pairs is very slow, approaching the
self-consistent regime. In both models the temperature is regulated using the velocities of the center of mass
of core+shell (or Drude) pairs, but in the Drude model the actual relative core-Drude particle motion is
thermostated separately as well.
6.26 Adiabatic core/shell model
The adiabatic core-shell model by Mitchell and Fincham is a simple method for adding polarizability to a
system. In order to mimic the electron shell of an ion, a satellite particle is attached to it. This way the ions are
split into a core and a shell where the latter is meant to react to the electrostatic environment inducing
polarizability.
Technically, shells are attached to the cores by a spring force f = k*r where k is a parametrized spring constant
and r is the distance between the core and the shell. The charges of the core and the shell add up to the ion
charge, thus q(ion) = q(core) + q(shell). This setup introduces the ion polarizability (alpha) given by alpha =
q(shell)^2 / k. In a similar fashion the mass of the ion is distributed on the core and the shell with the core
having the larger mass.
To run this model in LAMMPS, atom_style full can be used since atom charge and bonds are needed. Each
kind of core/shell pair requires two atom types and a bond type. The core and shell of a core/shell pair should
be bonded to each other with a harmonic bond that provides the spring force. For example, a data file for
NaCl, as found in examples/coreshell, has this format:
432 atoms # core and shell atoms
216 bonds # number of core/shell springs
4 atom types # 2 cores and 2 shells for Na and Cl
2 bond types
0.0 24.09597 xlo xhi
0.0 24.09597 ylo yhi
0.0 24.09597 zlo zhi
Masses # core/shell mass ratio = 0.1
1 20.690784 # Na core
2 31.90500 # Cl core
3 2.298976 # Na shell
4 3.54500 # Cl shell
Atoms
LAMMPS Users Manual
154
1 1 2 1.5005 0.00000000 0.00000000 0.00000000 # core of core/shell pair 1
2 1 4 -2.5005 0.00000000 0.00000000 0.00000000 # shell of core/shell pair 1
3 2 1 1.5056 4.01599500 4.01599500 4.01599500 # core of core/shell pair 2
4 2 3 -0.5056 4.01599500 4.01599500 4.01599500 # shell of core/shell pair 2
(...)
Bonds # Bond topology for spring forces
1 2 1 2 # spring for core/shell pair 1
2 2 3 4 # spring for core/shell pair 2
(...)
Non-Coulombic (e.g. Lennard-Jones) pairwise interactions are only defined between the shells. Coulombic
interactions are defined between all cores and shells. If desired, additional bonds can be specified between
cores.
The special_bonds command should be used to turn-off the Coulombic interaction within core/shell pairs,
since that interaction is set by the bond spring. This is done using the special_bonds command with a 1-2
weight = 0.0, which is the default value. It needs to be considered whether one has to adjust the special_bonds
weighting according to the molecular topology since the interactions of the shells are bypassed over an extra
bond.
Note that this core/shell implementation does not require all ions to be polarized. One can mix core/shell pairs
and ions without a satellite particle if desired.
Since the core/shell model permits distances of r = 0.0 between the core and shell, a pair style with a "cs"
suffix needs to be used to implement a valid long-range Coulombic correction. Several such pair styles are
provided in the CORESHELL package. See this doc page for details. All of the core/shell enabled pair styles
require the use of a long-range Coulombic solver, as specified by the kspace_style command. Either the
PPPM or Ewald solvers can be used.
For the NaCL example problem, these pair style and bond style settings are used:
pair_style born/coul/long/cs 20.0 20.0
pair_coeff * * 0.0 1.000 0.00 0.00 0.00
pair_coeff 3 3 487.0 0.23768 0.00 1.05 0.50 #Na-Na
pair_coeff 3 4 145134.0 0.23768 0.00 6.99 8.70 #Na-Cl
pair_coeff 4 4 405774.0 0.23768 0.00 72.40 145.40 #Cl-Cl
bond_style harmonic
bond_coeff 1 63.014 0.0
bond_coeff 2 25.724 0.0
When running dynamics with the adiabatic core/shell model, the following issues should be considered. The
relative motion of the core and shell particles corresponds to the polarization, hereby an instantaneous
relaxation of the shells is approximated and a fast core/shell spring frequency ensures a nearly constant
internal kinetic energy during the simulation. Thermostats can alter this polarization behaviour, by scaling the
internal kinetic energy, meaning the shell will not react freely to its electrostatic environment. Therefore it is
typically desirable to decouple the relative motion of the core/shell pair, which is an imaginary degree of
freedom, from the real physical system. To do that, the compute temp/cs command can be used, in
conjunction with any of the thermostat fixes, such as fix nvt or fix langevin. This compute uses the
center-of-mass velocity of the core/shell pairs to calculate a temperature, and insures that velocity is what is
rescaled for thermostatting purposes. This compute also works for a system with both core/shell pairs and
non-polarized ions (ions without an attached satellite particle). The compute temp/cs command requires input
LAMMPS Users Manual
155
of two groups, one for the core atoms, another for the shell atoms. Non-polarized ions which might also be
included in the treated system should not be included into either of these groups, they are taken into account
by the group-ID (2nd argument) of the compute. The groups can be defined using the group type command.
Note that to perform thermostatting using this definition of temperature, the fix modify temp command should
be used to assign the compute to the thermostat fix. Likewise the thermo_modify temp command can be used
to make this temperature be output for the overall system.
For the NaCl example, this can be done as follows:
group cores type 1 2
group shells type 3 4
compute CSequ all temp/cs cores shells
fix thermoberendsen all temp/berendsen 1427 1427 0.4 # thermostat for the true physical system
fix thermostatequ all nve # integrator as needed for the berendsen thermostat
fix_modify thermoberendsen temp CSequ
thermo_modify temp CSequ # output of center-of-mass derived temperature
The pressure for the core/shell system is computed via the regular LAMMPS convention by treating the cores
and shells as individual particles. For the thermo output of the pressure as well as for the application of a
barostat, it is necessary to use an additional pressure compute based on the default temperature and specifying
it as a second argument in fix modify and thermo_modify resulting in:
(...)
compute CSequ all temp/cs cores shells
compute thermo_press_lmp all pressure thermo_temp # pressure for individual particles
thermo_modify temp CSequ press thermo_press_lmp # modify thermo to regular pressure
fix press_bar all npt temp 300 300 0.04 iso 0 0 0.4
fix_modify press_bar temp CSequ press thermo_press_lmp # pressure modification for correct kinetic scalar
If compute temp/cs is used, the decoupled relative motion of the core and the shell should in theory be stable.
However numerical fluctuation can introduce a small momentum to the system, which is noticable over long
trajectories. Therefore it is recommendable to use the fix momentum command in combination with compute
temp/cs when equilibrating the system to prevent any drift.
When initializing the velocities of a system with core/shell pairs, it is also desirable to not introduce energy
into the relative motion of the core/shell particles, but only assign a center-of-mass velocity to the pairs. This
can be done by using the bias keyword of the velocity create command and assigning the compute temp/cs
command to the temp keyword of the velocity command, e.g.
velocity all create 1427 134 bias yes temp CSequ
velocity all scale 1427 temp CSequ
To maintain the correct polarizability of the core/shell pairs, the kinetic energy of the internal motion shall
remain nearly constant. Therefore the choice of spring force and mass ratio need to ensure much faster relative
motion of the 2 atoms within the core/shell pair than their center-of-mass velocity. This allows the shells to
effectively react instantaneously to the electrostatic environment and limits energy transfer to or from the
core/shell oscillators. This fast movement also dictates the timestep that can be used.
The primary literature of the adiabatic core/shell model suggests that the fast relative motion of the core/shell
pairs only allows negligible energy transfer to the environment. The mentioned energy transfer will typically
lead to a small drift in total energy over time. This internal energy can be monitored using the compute
chunk/atom and compute temp/chunk commands. The internal kinetic energies of each core/shell pair can
then be summed using the sum() special function of the variable command. Or they can be time/averaged and
LAMMPS Users Manual
156

output using the fix ave/time command. To use these commands, each core/shell pair must be defined as a
"chunk". If each core/shell pair is defined as its own molecule, the molecule ID can be used to define the
chunks. If cores are bonded to each other to form larger molecules, the chunks can be identified by the fix
property/atom via assigning a core/shell ID to each atom using a special field in the data file read by the
read_data command. This field can then be accessed by the compute property/atom command, to use as input
to the compute chunk/atom command to define the core/shell pairs as chunks.
For example if core/shell pairs are the only molecules:
read_data NaCl_CS_x0.1_prop.data
compute prop all property/atom molecule
compute cs_chunk all chunk/atom c_prop
compute cstherm all temp/chunk cs_chunk temp internal com yes cdof 3.0 # note the chosen degrees of freedom for the core/shell pairs
fix ave_chunk all ave/time 10 1 10 c_cstherm file chunk.dump mode vector
For example if core/shell pairs and other molecules are present:
fix csinfo all property/atom i_CSID # property/atom command
read_data NaCl_CS_x0.1_prop.data fix csinfo NULL CS-Info # atom property added in the data-file
compute prop all property/atom i_CSID
(...)
The additional section in the date file would be formatted like this:
CS-Info # header of additional section
1 1 # column 1 = atom ID, column 2 = core/shell ID
2 1
3 2
4 2
5 3
6 3
7 4
8 4
(...)
6.27 Drude induced dipoles
The thermalized Drude model, similarly to the core-shell model, represents induced dipoles by a pair of
charges (the core atom and the Drude particle) connected by a harmonic spring. The Drude model has a
number of features aimed at its use in molecular systems (Lamoureux and Roux):
Thermostating of the additional degrees of freedom associated with the induced dipoles at very low
temperature, in terms of the reduced coordinates of the Drude particles with respect to their cores.
This makes the trajectory close to that of relaxed induced dipoles.
•
Consistent definition of 1-2 to 1-4 neighbors. A core-Drude particle pair represents a single
(polarizable) atom, so the special screening factors in a covalent structure should be the same for the
core and the Drude particle. Drude particles have to inherit the 1-2, 1-3, 1-4 special neighbor relations
from their respective cores.
•
Stabilization of the interactions between induced dipoles. Drude dipoles on covalently bonded atoms
interact too strongly due to the short distances, so an atom may capture the Drude particle of a
neighbor, or the induced dipoles within the same molecule may align too much. To avoid this,
damping at short range can be done by Thole functions (for which there are physical grounds). This
•
LAMMPS Users Manual
157

Thole damping is applied to the point charges composing the induced dipole (the charge of the Drude
particle and the opposite charge on the core, not to the total charge of the core atom).
A detailed tutorial covering the usage of Drude induced dipoles in LAMMPS is available here.
As with the core-shell model, the cores and Drude particles should appear in the data file as standard atoms.
The same holds for the springs between them, which are described by standard harmonic bonds. The nature of
the atoms (core, Drude particle or non-polarizable) is specified via the fix drude command. The special list of
neighbors is automatically refactored to account for the equivalence of core and Drude particles as regards
special 1-2 to 1-4 screening. It may be necessary to use the extra keyword of the special_bonds command. If
using fix shake, make sure no Drude particle is in this fix group.
There are two ways to thermostat the Drude particles at a low temperature: use either fix langevin/drude for a
Langevin thermostat, or fix drude/transform/* for a Nose-Hoover thermostat. The former requires use of the
command comm_modify vel yes. The latter requires two separate integration fixes like nvt or npt. The correct
temperatures of the reduced degrees of freedom can be calculated using the compute temp/drude. This
requires also to use the command comm_modify vel yes.
Short-range damping of the induced dipole interactions can be achieved using Thole functions through the
pair style thole in pair_style hybrid/overlay with a Coulomb pair style. It may be useful to use coul/long/cs or
similar from the CORESHELL package if the core and Drude particle come too close, which can cause
numerical issues.
(Berendsen) Berendsen, Grigera, Straatsma, J Phys Chem, 91, 6269-6271 (1987).
(Cornell) Cornell, Cieplak, Bayly, Gould, Merz, Ferguson, Spellmeyer, Fox, Caldwell, Kollman, JACS 117,
5179-5197 (1995).
(Horn) Horn, Swope, Pitera, Madura, Dick, Hura, and Head-Gordon, J Chem Phys, 120, 9665 (2004).
(Ikeshoji) Ikeshoji and Hafskjold, Molecular Physics, 81, 251-261 (1994).
(Wirnsberger) Wirnsberger, Frenkel, and Dellago, J Chem Phys, 143, 124104 (2015).
(MacKerell) MacKerell, Bashford, Bellott, Dunbrack, Evanseck, Field, Fischer, Gao, Guo, Ha, et al, J Phys
Chem, 102, 3586 (1998).
(Mayo) Mayo, Olfason, Goddard III, J Phys Chem, 94, 8897-8909 (1990).
(Jorgensen) Jorgensen, Chandrasekhar, Madura, Impey, Klein, J Chem Phys, 79, 926 (1983).
LAMMPS Users Manual
158
(Price) Price and Brooks, J Chem Phys, 121, 10096 (2004).
(Shinoda) Shinoda, Shiga, and Mikami, Phys Rev B, 69, 134103 (2004).
(Mitchell and Fincham) Mitchell, Fincham, J Phys Condensed Matter, 5, 1031-1038 (1993).
(Fincham) Fincham, Mackrodt and Mitchell, J Phys Condensed Matter, 6, 393-404 (1994).
(Lamoureux and Roux) G. Lamoureux, B. Roux, J. Chem. Phys 119, 3025 (2003)
LAMMPS Users Manual
159

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
7. Example problems
The LAMMPS distribution includes an examples sub-directory with many sample problems. Many are 2d
models that run quickly are are straightforward to visualize, requiring at most a couple of minutes to run on a
desktop machine. Each problem has an input script (in.*) and produces a log file (log.*) when it runs. Some
use a data file (data.*) of initial coordinates as additional input. A few sample log file run on different
machines and different numbers of processors are included in the directories to compare your answers to. E.g.
a log file like log.date.crack.foo.P means the "crack" example was run on P processors of machine "foo" on
that date (i.e. with that version of LAMMPS).
Many of the input files have commented-out lines for creating dump files and image files.
If you uncomment the dump command in the input script, a text dump file will be produced, which can be
animated by various visualization programs.
If you uncomment the dump image command in the input script, and assuming you have built LAMMPS with
a JPG library, JPG snapshot images will be produced when the simulation runs. They can be quickly
post-processed into a movie using commands described on the dump image doc page.
Animations of many of the examples can be viewed on the Movies section of the LAMMPS web site.
There are two kinds of sub-directories in the examples dir. Lowercase dirs contain one or a few simple,
quick-to-run problems. Uppercase dirs contain up to several complex scripts that illustrate a particular kind of
simulation method or model. Some of these run for longer times, e.g. to measure a particular quantity.
Lists of both kinds of directories are given below.
Lowercase directories
accelerate run with various acceleration options (OpenMP, GPU, Phi)
balance dynamic load balancing, 2d system
body body particles, 2d system
cmap CMAP 5-body contributions to CHARMM force field
colloid big colloid particles in a small particle solvent, 2d system
comb models using the COMB potential
coreshell core/shell model using CORESHELL package
controller use of fix controller as a thermostat
crack crack propagation in a 2d solid
deposit deposit atoms and molecules on a surface
dipole point dipolar particles, 2d system
dreiding methanol via Dreiding FF
eim NaCl using the EIM potential
ellipse ellipsoidal particles in spherical solvent, 2d system
flow Couette and Poiseuille flow in a 2d channel
friction frictional contact of spherical asperities between 2d surfaces
LAMMPS Users Manual
160
gcmc Grand Canonical Monte Carlo (GCMC) via the fix gcmc command
granregion use of fix wall/region/gran as boundary on granular particles
hugoniostat Hugoniostat shock dynamics
indent spherical indenter into a 2d solid
kim use of potentials in Knowledge Base for Interatomic Models (KIM)
meam MEAM test for SiC and shear (same as shear examples)
melt rapid melt of 3d LJ system
micelle self-assembly of small lipid-like molecules into 2d bilayers
min energy minimization of 2d LJ melt
mscg parameterize a multi-scale coarse-graining (MSCG) model
msst MSST shock dynamics
nb3b use of nonbonded 3-body harmonic pair style
neb nudged elastic band (NEB) calculation for barrier finding
nemd non-equilibrium MD of 2d sheared system
obstacle flow around two voids in a 2d channel
peptide dynamics of a small solvated peptide chain (5-mer)
peri Peridynamic model of cylinder impacted by indenter
pour pouring of granular particles into a 3d box, then chute flow
prd parallel replica dynamics of vacancy diffusion in bulk Si
python using embedded Python in a LAMMPS input script
qeq use of the QEQ package for charge equilibration
reax RDX and TATB models using the ReaxFF
rigid rigid bodies modeled as independent or coupled
shear sideways shear applied to 2d solid, with and without a void
snap NVE dynamics for BCC tantalum crystal using SNAP potential
srd stochastic rotation dynamics (SRD) particles as solvent
streitz use of Streitz/Mintmire potential with charge equilibration
tad temperature-accelerated dynamics of vacancy diffusion in bulk Si
vashishta use of the Vashishta potential
voronoi Voronoi tesselation via compute voronoi/atom command
Here is how you can run and visualize one of the sample problems:
cd indent
cp ../../src/lmp_linux . # copy LAMMPS executable to this dir
lmp_linux -in in.indent # run the problem
Running the simulation produces the files dump.indent and log.lammps. You can visualize the dump file of
snapshots with a variety of 3rd-party tools highlighted on the Visualization page of the LAMMPS web site.
If you uncomment the dump image line(s) in the input script a series of JPG images will be produced by the
run (assuming you built LAMMPS with JPG support; see Section 2.2 for details). These can be viewed
individually or turned into a movie or animated by tools like ImageMagick or QuickTime or various
Windows-based tools. See the dump image doc page for more details. E.g. this Imagemagick command would
create a GIF file suitable for viewing in a browser.
LAMMPS Users Manual
161

% convert -loop 1 *.jpg foo.gif
Uppercase directories
ASPHERE various aspherical particle models, using ellipsoids, rigid bodies, line/triangle particles, etc
COUPLE examples of how to use LAMMPS as a library
DIFFUSE compute diffusion coefficients via several methods
ELASTIC compute elastic constants at zero temperature
ELASTIC_T compute elastic constants at finite temperature
KAPPA compute thermal conductivity via several methods
MC using LAMMPS in a Monte Carlo mode to relax the energy of a system
USER examples for USER packages and USER-contributed commands
VISCOSITY compute viscosity via several methods
Nearly all of these directories have README files which give more details on how to understand and use
their contents.
The USER directory has a large number of sub-directories which correspond by name to a USER package.
They contain scripts that illustrate how to use the command(s) provided in that package. Many of the
sub-directories have their own README files which give further instructions. See the Section 4 doc page for
more info on specific USER packages.
LAMMPS Users Manual
162

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
8. Performance & scalability
Current LAMMPS performance is discussed on the Benchmarks page of the LAMMPS WWW Site where
CPU timings and parallel efficiencies are listed. The page has several sections, which are briefly described
below:
CPU performance on 5 standard problems, strong and weak scaling• GPU and Xeon Phi performance on same and related problems• Comparison of cost of interatomic potentials• Performance of huge, billion-atom problems•
The 5 standard problems are as follow:
LJ = atomic fluid, Lennard-Jones potential with 2.5 sigma cutoff (55 neighbors per atom), NVE
integration
1.
Chain = bead-spring polymer melt of 100-mer chains, FENE bonds and LJ pairwise interactions with
a 2^(1/6) sigma cutoff (5 neighbors per atom), NVE integration
2.
EAM = metallic solid, Cu EAM potential with 4.95 Angstrom cutoff (45 neighbors per atom), NVE
integration
3.
Chute = granular chute flow, frictional history potential with 1.1 sigma cutoff (7 neighbors per atom),
NVE integration
4.
Rhodo = rhodopsin protein in solvated lipid bilayer, CHARMM force field with a 10 Angstrom LJ
cutoff (440 neighbors per atom), particle-particle particle-mesh (PPPM) for long-range Coulombics,
NPT integration
5.
Input files for these 5 problems are provided in the bench directory of the LAMMPS distribution. Each has
32,000 atoms and runs for 100 timesteps. The size of the problem (number of atoms) can be varied using
command-line switches as described in the bench/README file. This is an easy way to test performance and
either strong or weak scalability on your machine.
The bench directory includes a few log.* files that show performance of these 5 problems on 1 or 4 cores of
Linux desktop. The bench/FERMI and bench/KEPLER dirs have input files and scripts and instructions for
running the same (or similar) problems using OpenMP or GPU or Xeon Phi acceleration options. See the
README files in those dirs and the Section 5.3 doc pages for instructions on how to build LAMMPS and run
on that kind of hardware.
The bench/POTENTIALS directory has input files which correspond to the table of results on the Potentials
section of the Benchmarks web page. So you can also run those test problems on your machine.
The billion-atom section of the Benchmarks web page has performance data for very large benchmark runs of
simple Lennard-Jones (LJ) models, which use the bench/in.lj input script.
For all the benchmarks, a useful metric is the CPU cost per atom per timestep. Since performance scales
roughly linearly with problem size and timesteps for all LAMMPS models (i.e. interatomic or coarse-grained
potentials), the run time of any problem using the same model (atom style, force field, cutoff, etc) can then be
estimated.
LAMMPS Users Manual
163
Performance on a parallel machine can also be predicted from one-core or one-node timings if the parallel
efficiency can be estimated. The communication bandwidth and latency of a particular parallel machine
affects the efficiency. On most machines LAMMPS will give parallel efficiencies on these benchmarks above
50% so long as the number of atoms/core is a few 100 or greater, and closer to 100% for large numbers of
atoms/core. This is for all-MPI mode with one MPI task per core. For nodes with accelerator options or
hardware (OpenMP, GPU, Phi), you should first measure single node performance. Then you can estimate
parallel performance for multi-node runs using the same logic as for all-MPI mode, except that now you will
typically need many more atoms/node to achieve good scalability.
LAMMPS Users Manual
164

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
9. Additional tools
LAMMPS is designed to be a computational kernel for performing molecular dynamics computations.
Additional pre- and post-processing steps are often necessary to setup and analyze a simulation. A list of such
tools can be found on the LAMMPS home page at http://lammps.sandia.gov/prepost.html
A few additional tools are provided with the LAMMPS distribution and are described in this section.
Our group has also written and released a separate toolkit called Pizza.py which provides tools for doing
setup, analysis, plotting, and visualization for LAMMPS simulations. Pizza.py is written in Python and is
available for download from the Pizza.py WWW site.
Note that many users write their own setup or analysis tools or use other existing codes and convert their
output to a LAMMPS input format or vice versa. The tools listed here are included in the LAMMPS
distribution as examples of auxiliary tools. Some of them are not actively supported by Sandia, as they were
contributed by LAMMPS users. If you have problems using them, we can direct you to the authors.
The source code for each of these codes is in the tools sub-directory of the LAMMPS distribution. There is a
Makefile (which you may need to edit for your platform) which will build several of the tools which reside in
that directory. Most of them are larger packages in their own sub-directories with their own Makefiles and/or
README files.
amber2lmp binary2txt ch2lmp chain colvars createatoms drude eam database eam generate eff emacs fep i-pi
ipp kate lmp2arc lmp2cfg matlab micelle2d moltemplate msi2lmp phonon polybond pymol_asphere python
reax smd vim xmgrace
amber2lmp tool
The amber2lmp sub-directory contains two Python scripts for converting files back-and-forth between the
AMBER MD code and LAMMPS. See the README file in amber2lmp for more information.
These tools were written by Keir Novik while he was at Queen Mary University of London. Keir is no longer
there and cannot support these tools which are out-of-date with respect to the current LAMMPS version (and
maybe with respect to AMBER as well). Since we don't use these tools at Sandia, you'll need to experiment
with them and make necessary modifications yourself.
binary2txt tool
The file binary2txt.cpp converts one or more binary LAMMPS dump file into ASCII text files. The syntax for
running the tool is
binary2txt file1 file2 ...
which creates file1.txt, file2.txt, etc. This tool must be compiled on a platform that can read the binary file
created by a LAMMPS run, since binary files are not compatible across all platforms.
LAMMPS Users Manual
165
ch2lmp tool
The ch2lmp sub-directory contains tools for converting files back-and-forth between the CHARMM MD code
and LAMMPS.
They are intended to make it easy to use CHARMM as a builder and as a post-processor for LAMMPS. Using
charmm2lammps.pl, you can convert a PDB file with associated CHARMM info, including CHARMM force
field data, into its LAMMPS equivalent. Using lammps2pdb.pl you can convert LAMMPS atom dumps into
PDB files.
See the README file in the ch2lmp sub-directory for more information.
These tools were created by Pieter in't Veld (pjintve at sandia.gov) and Paul Crozier (pscrozi at sandia.gov) at
Sandia.
chain tool
The file chain.f creates a LAMMPS data file containing bead-spring polymer chains and/or monomer solvent
atoms. It uses a text file containing chain definition parameters as an input. The created chains and solvent
atoms can strongly overlap, so LAMMPS needs to run the system initially with a "soft" pair potential to
un-overlap it. The syntax for running the tool is
chain <def.chain > data.file
See the def.chain or def.chain.ab files in the tools directory for examples of definition files. This tool was used
to create the system for the chain benchmark.
colvars tools
The colvars directory contains a collection of tools for postprocessing data produced by the colvars collective
variable library. To compile the tools, edit the makefile for your system and run "make".
Please report problems and issues the colvars library and its tools at: https://github.com/colvars/colvars/issues
abf_integrate:
MC-based integration of multidimensional free energy gradient Version 20110511
Syntax: ./abf_integrate <filename > [-n <nsteps >] [-t <temp >] [-m [0|1] (metadynamics)] [-h <hill_height >] [-f <variable_hill_factor >]
The LAMMPS interface to the colvars collective variable library, as well as these tools, were created by Axel
Kohlmeyer (akohlmey at gmail.com) at ICTP, Italy.
createatoms tool
The tools/createatoms directory contains a Fortran program called createAtoms.f which can generate a variety
of interesting crystal structures and geometries and output the resulting list of atom coordinates in LAMMPS
or other formats.
See the included Manual.pdf for details.
LAMMPS Users Manual
166

The tool is authored by Xiaowang Zhou (Sandia), xzhou at sandia.gov.
drude tool
The tools/drude directory contains a Python script called polarizer.py which can add Drude oscillators to a
LAMMPS data file in the required format.
See the header of the polarizer.py file for details.
The tool is authored by Agilio Padua and Alain Dequidt: agilio.padua at univ-bpclermont.fr, alain.dequidt at
univ-bpclermont.fr
eam database tool
The tools/eam_database directory contains a Fortran program that will generate EAM alloy setfl potential files
for any combination of 16 elements: Cu, Ag, Au, Ni, Pd, Pt, Al, Pb, Fe, Mo, Ta, W, Mg, Co, Ti, Zr. The files
can then be used with the pair_style eam/alloy command.
The tool is authored by Xiaowang Zhou (Sandia), xzhou at sandia.gov, and is based on his paper:
X. W. Zhou, R. A. Johnson, and H. N. G. Wadley, Phys. Rev. B, 69, 144113 (2004).
eam generate tool
The tools/eam_generate directory contains several one-file C programs that convert an analytic formula into a
tabulated embedded atom method (EAM) setfl potential file. The potentials they produce are in the potentials
directory, and can be used with the pair_style eam/alloy command.
The source files and potentials were provided by Gerolf Ziegenhain (gerolf at ziegenhain.com).
eff tool
The tools/eff directory contains various scripts for generating structures and post-processing output for
simulations using the electron force field (eFF).
These tools were provided by Andres Jaramillo-Botero at CalTech (ajaramil at wag.caltech.edu).
emacs tool
The tools/emacs directory contains a Lips add-on file for Emacs that enables a lammps-mode for editing of
input scripts when using Emacs, with various highlighting options setup.
These tools were provided by Aidan Thompson at Sandia (athomps at sandia.gov).
fep tool
The tools/fep directory contains Python scripts useful for post-processing results from performing free-energy
perturbation simulations using the USER-FEP package.
LAMMPS Users Manual
167
The scripts were contributed by Agilio Padua (Universite Blaise Pascal Clermont-Ferrand), agilio.padua at
univ-bpclermont.fr.
See README file in the tools/fep directory.
i-pi tool
The tools/i-pi directory contains a version of the i-PI package, with all the LAMMPS-unrelated files removed.
It is provided so that it can be used with the fix ipi command to perform path-integral molecular dynamics
(PIMD).
The i-PI package was created and is maintained by Michele Ceriotti, michele.ceriotti at gmail.com, to
interface to a variety of molecular dynamics codes.
See the tools/i-pi/manual.pdf file for an overview of i-PI, and the fix ipi doc page for further details on
running PIMD calculations with LAMMPS.
ipp tool
The tools/ipp directory contains a Perl script ipp which can be used to facilitate the creation of a complicated
file (say, a lammps input script or tools/createatoms input file) using a template file.
ipp was created and is maintained by Reese Jones (Sandia), rjones at sandia.gov.
See two examples in the tools/ipp directory. One of them is for the tools/createatoms tool's input file.
kate tool
The file in the tools/kate directory is an add-on to the Kate editor in the KDE suite that allow syntax
highlighting of LAMMPS input scripts. See the README.txt file for details.
The file was provided by Alessandro Luigi Sellerio (alessandro.sellerio at ieni.cnr.it).
lmp2arc tool
The lmp2arc sub-directory contains a tool for converting LAMMPS output files to the format for Accelrys'
Insight MD code (formerly MSI/Biosym and its Discover MD code). See the README file for more
information.
This tool was written by John Carpenter (Cray), Michael Peachey (Cray), and Steve Lustig (Dupont). John is
now at the Mayo Clinic (jec at mayo.edu), but still fields questions about the tool.
This tool was updated for the current LAMMPS C++ version by Jeff Greathouse at Sandia (jagreat at
sandia.gov).
lmp2cfg tool
The lmp2cfg sub-directory contains a tool for converting LAMMPS output files into a series of *.cfg files
which can be read into the AtomEye visualizer. See the README file for more information.
LAMMPS Users Manual
168

This tool was written by Ara Kooser at Sandia (askoose at sandia.gov).
matlab tool
The matlab sub-directory contains several MATLAB scripts for post-processing LAMMPS output. The scripts
include readers for log and dump files, a reader for EAM potential files, and a converter that reads LAMMPS
dump files and produces CFG files that can be visualized with the AtomEye visualizer.
See the README.pdf file for more information.
These scripts were written by Arun Subramaniyan at Purdue Univ (asubrama at purdue.edu).
micelle2d tool
The file micelle2d.f creates a LAMMPS data file containing short lipid chains in a monomer solution. It uses
a text file containing lipid definition parameters as an input. The created molecules and solvent atoms can
strongly overlap, so LAMMPS needs to run the system initially with a "soft" pair potential to un-overlap it.
The syntax for running the tool is
micelle2d <def.micelle2d > data.file
See the def.micelle2d file in the tools directory for an example of a definition file. This tool was used to create
the system for the micelle example.
moltemplate tool
The moltemplate sub-directory contains a Python-based tool for building molecular systems based on a
text-file description, and creating LAMMPS data files that encode their molecular topology as lists of bonds,
angles, dihedrals, etc. See the README.TXT file for more information.
This tool was written by Andrew Jewett (jewett.aij at gmail.com), who supports it. It has its own WWW page
at http://moltemplate.org.
msi2lmp tool
The msi2lmp sub-directory contains a tool for creating LAMMPS input data files from BIOVIA's Materias
Studio files (formerly Accelrys' Insight MD code, formerly MSI/Biosym and its Discover MD code).
This tool was written by John Carpenter (Cray), Michael Peachey (Cray), and Steve Lustig (Dupont). Several
people contributed changes to remove bugs and adapt its output to changes in LAMMPS.
See the README file for more information.
phonon tool
The phonon sub-directory contains a post-processing tool useful for analyzing the output of the fix phonon
command in the USER-PHONON package.
See the README file for instruction on building the tool and what library it needs. And see the
examples/USER/phonon directory for example problems that can be post-processed with this tool.
LAMMPS Users Manual
169

This tool was written by Ling-Ti Kong at Shanghai Jiao Tong University.
polybond tool
The polybond sub-directory contains a Python-based tool useful for performing "programmable polymer
bonding". The Python file lmpsdata.py provides a "Lmpsdata" class with various methods which can be
invoked by a user-written Python script to create data files with complex bonding topologies.
See the Manual.pdf for details and example scripts.
This tool was written by Zachary Kraus at Georgia Tech.
pymol_asphere tool
The pymol_asphere sub-directory contains a tool for converting a LAMMPS dump file that contains
orientation info for ellipsoidal particles into an input file for the PyMol visualization package or its open
source variant.
Specifically, the tool triangulates the ellipsoids so they can be viewed as true ellipsoidal particles within
PyMol. See the README and examples directory within pymol_asphere for more information.
This tool was written by Mike Brown at Sandia.
python tool
The python sub-directory contains several Python scripts that perform common LAMMPS post-processing
tasks, such as:
extract thermodynamic info from a log file as columns of numbers• plot two columns of thermodynamic info from a log file using GnuPlot• sort the snapshots in a dump file by atom ID• convert multiple NEB dump files into one dump file for viz• convert dump files into XYZ, CFG, or PDB format for viz by other packages•
These are simple scripts built on Pizza.py modules. See the README for more info on Pizza.py and how to
use these scripts.
reax tool
The reax sub-directory contains stand-alond codes that can post-process the output of the fix reax/bonds
command from a LAMMPS simulation using ReaxFF. See the README.txt file for more info.
These tools were written by Aidan Thompson at Sandia.
smd tool
The smd sub-directory contains a C++ file dump2vtk_tris.cpp and Makefile which can be compiled and used
to convert triangle output files created by the Smooth-Mach Dynamics (USER-SMD) package into a
VTK-compatible unstructured grid file. It could then be read in and visualized by VTK.
LAMMPS Users Manual
170

See the header of dump2vtk.cpp for more details.
This tool was written by the USER-SMD package author, Georg Ganzenmuller at the Fraunhofer-Institute for
High-Speed Dynamics, Ernst Mach Institute in Germany (georg.ganzenmueller at emi.fhg.de).
vim tool
The files in the tools/vim directory are add-ons to the VIM editor that allow easier editing of LAMMPS input
scripts. See the README.txt file for details.
These files were provided by Gerolf Ziegenhain (gerolf at ziegenhain.com)
xmgrace tool
The files in the tools/xmgrace directory can be used to plot the thermodynamic data in LAMMPS log files via
the xmgrace plotting package. There are several tools in the directory that can be used in post-processing
mode. The lammpsplot.cpp file can be compiled and used to create plots from the current state of a running
LAMMPS simulation.
See the README file for details.
These files were provided by Vikas Varshney (vv0210 at gmail.com)
LAMMPS Users Manual
171

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
10. Modifying & extending LAMMPS
This section describes how to customize LAMMPS by modifying and extending its source code.
10.1 Atom styles
10.2 Bond, angle, dihedral, improper potentials
10.3 Compute styles
10.4 Dump styles
10.5 Dump custom output options
10.6 Fix styles which include integrators, temperature and pressure control, force constraints, boundary
conditions, diagnostic output, etc
10.7 Input script commands
10.8 Kspace computations
10.9 Minimization styles
10.10 Pairwise potentials
10.11 Region styles
10.12 Body styles
10.13 Thermodynamic output options
10.14 Variable options
10.15 Submitting new features for inclusion in LAMMPS
LAMMPS is designed in a modular fashion so as to be easy to modify and extend with new functionality. In
fact, about 75% of its source code is files added in this fashion.
In this section, changes and additions users can make are listed along with minimal instructions. If you add a
new feature to LAMMPS and think it will be of interest to general users, we encourage you to submit it to the
developers for inclusion in the released version of LAMMPS. Information about how to do this is provided
below.
The best way to add a new feature is to find a similar feature in LAMMPS and look at the corresponding
source and header files to figure out what it does. You will need some knowledge of C++ to be able to
understand the hi-level structure of LAMMPS and its class organization, but functions (class methods) that do
actual computations are written in vanilla C-style code and operate on simple C-style data structures (vectors
and arrays).
Most of the new features described in this section require you to write a new C++ derived class (except for
exceptions described below, where you can make small edits to existing files). Creating a new class requires 2
files, a source code file (*.cpp) and a header file (*.h). The derived class must provide certain methods to
work as a new option. Depending on how different your new feature is compared to existing features, you can
either derive from the base class itself, or from a derived class that already exists. Enabling LAMMPS to
invoke the new class is as simple as putting the two source files in the src dir and re-building LAMMPS.
The advantage of C++ and its object-orientation is that all the code and variables needed to define the new
feature are in the 2 files you write, and thus shouldn't make the rest of LAMMPS more complex or cause
side-effect bugs.
Here is a concrete example. Suppose you write 2 files pair_foo.cpp and pair_foo.h that define a new class
PairFoo that computes pairwise potentials described in the classic 1997 paper by Foo, et al. If you wish to
LAMMPS Users Manual
172

invoke those potentials in a LAMMPS input script with a command like
pair_style foo 0.1 3.5
then your pair_foo.h file should be structured as follows:
#ifdef PAIR_CLASS
PairStyle(foo,PairFoo)
#else
...
(class definition for PairFoo)
...
#endif
where "foo" is the style keyword in the pair_style command, and PairFoo is the class name defined in your
pair_foo.cpp and pair_foo.h files.
When you re-build LAMMPS, your new pairwise potential becomes part of the executable and can be
invoked with a pair_style command like the example above. Arguments like 0.1 and 3.5 can be defined and
processed by your new class.
As illustrated by this pairwise example, many kinds of options are referred to in the LAMMPS documentation
as the "style" of a particular command.
The instructions below give the header file for the base class that these styles are derived from. Public
variables in that file are ones used and set by the derived classes which are also used by the base class.
Sometimes they are also used by the rest of LAMMPS. Virtual functions in the base class header file which
are set = 0 are ones you must define in your new derived class to give it the functionality LAMMPS expects.
Virtual functions that are not set to 0 are functions you can optionally define.
Additionally, new output options can be added directly to the thermo.cpp, dump_custom.cpp, and variable.cpp
files as explained below.
Here are additional guidelines for modifying LAMMPS and adding new functionality:
Think about whether what you want to do would be better as a pre- or post-processing step. Many
computations are more easily and more quickly done that way.
•
Don't do anything within the timestepping of a run that isn't parallel. E.g. don't accumulate a bunch of
data on a single processor and analyze it. You run the risk of seriously degrading the parallel
efficiency.
•
If your new feature reads arguments or writes output, make sure you follow the unit conventions
discussed by the units command.
•
If you add something you think is truly useful and doesn't impact LAMMPS performance when it isn't
used, send an email to the developers. We might be interested in adding it to the LAMMPS
distribution. See further details on this at the bottom of this page.
•
10.1 Atom styles
Classes that define an atom style are derived from the AtomVec class and managed by the Atom class. The
atom style determines what attributes are associated with an atom. A new atom style can be created if one of
LAMMPS Users Manual
173

the existing atom styles does not define all the attributes you need to store and communicate with atoms.
Atom_vec_atomic.cpp is a simple example of an atom style.
Here is a brief description of methods you define in your new derived class. See atom_vec.h for details.
init one time setup (optional)
grow re-allocate atom arrays to longer lengths (required)
grow_reset make array pointers in Atom and AtomVec classes consistent (required)
copy copy info for one atom to another atom's array locations (required)
pack_comm store an atom's info in a buffer communicated every timestep (required)
pack_comm_vel add velocity info to communication buffer (required)
pack_comm_hybrid store extra info unique to this atom style (optional)
unpack_comm retrieve an atom's info from the buffer (required)
unpack_comm_vel also retrieve velocity info (required)
unpack_comm_hybrid retrieve extra info unique to this atom style (optional)
pack_reverse store an atom's info in a buffer communicating partial forces (required)
pack_reverse_hybrid store extra info unique to this atom style (optional)
unpack_reverse retrieve an atom's info from the buffer (required)
unpack_reverse_hybrid retrieve extra info unique to this atom style (optional)
pack_border store an atom's info in a buffer communicated on neighbor re-builds (required)
pack_border_vel add velocity info to buffer (required)
pack_border_hybrid store extra info unique to this atom style (optional)
unpack_border retrieve an atom's info from the buffer (required)
unpack_border_vel also retrieve velocity info (required)
unpack_border_hybrid retrieve extra info unique to this atom style (optional)
pack_exchange store all an atom's info to migrate to another processor (required)
unpack_exchange retrieve an atom's info from the buffer (required)
size_restart number of restart quantities associated with proc's atoms (required)
pack_restart pack atom quantities into a buffer (required)
unpack_restart unpack atom quantities from a buffer (required)
create_atom create an individual atom of this style (required)
data_atom parse an atom line from the data file (required)
data_atom_hybrid parse additional atom info unique to this atom style (optional)
data_vel parse one line of velocity information from data file (optional)
data_vel_hybrid parse additional velocity data unique to this atom style (optional)
memory_usage tally memory allocated by atom arrays (required)
The constructor of the derived class sets values for several variables that you must set when defining a new
atom style, which are documented in atom_vec.h. New atom arrays are defined in atom.cpp. Search for the
word "customize" and you will find locations you will need to modify.
NOTE: It is possible to add some attributes, such as a molecule ID, to atom styles that do not have them via
the fix property/atom command. This command also allows new custom attributes consisting of extra integer
or floating-point values to be added to atoms. See the fix property/atom doc page for examples of cases where
LAMMPS Users Manual
174

this is useful and details on how to initialize, access, and output the custom values.
New pair styles, fixes, or computes can be added to LAMMPS, as discussed below. The code for these classes
can use the per-atom properties defined by fix property/atom. The Atom class has a find_custom() method
that is useful in this context:
int index = atom->find_custom(char *name, int &flag);
The "name" of a custom attribute, as specified in the fix property/atom command, is checked to verify that it
exists and its index is returned. The method also sets flag = 0/1 depending on whether it is an integer or
floating-point attribute. The vector of values associated with the attribute can then be accessed using the
returned index as
int *ivector = atom->ivector[index];
double *dvector = atom->dvector[index];
Ivector or dvector are vectors of length Nlocal = # of owned atoms, which store the attributes of individual
atoms.
10.2 Bond, angle, dihedral, improper potentials
Classes that compute molecular interactions are derived from the Bond, Angle, Dihedral, and Improper
classes. New styles can be created to add new potentials to LAMMPS.
Bond_harmonic.cpp is the simplest example of a bond style. Ditto for the harmonic forms of the angle,
dihedral, and improper style commands.
Here is a brief description of common methods you define in your new derived class. See bond.h, angle.h,
dihedral.h, and improper.h for details and specific additional methods.
init check if all coefficients are set, calls init_style (optional)
init_style check if style specific conditions are met (optional)
compute compute the molecular interactions (required)
settings apply global settings for all types (optional)
coeff set coefficients for one type (required)
equilibrium_distance length of bond, used by SHAKE (required, bond only)
equilibrium_angle opening of angle, used by SHAKE (required, angle only)
write & read_restart writes/reads coeffs to restart files (required)
single force and energy of a single bond or angle (required, bond or angle only)
memory_usage tally memory allocated by the style (optional)
10.3 Compute styles
Classes that compute scalar and vector quantities like temperature and the pressure tensor, as well as classes
that compute per-atom quantities like kinetic energy and the centro-symmetry parameter are derived from the
Compute class. New styles can be created to add new calculations to LAMMPS.
LAMMPS Users Manual
175

Compute_temp.cpp is a simple example of computing a scalar temperature. Compute_ke_atom.cpp is a
simple example of computing per-atom kinetic energy.
Here is a brief description of methods you define in your new derived class. See compute.h for details.
init perform one time setup (required)
init_list neighbor list setup, if needed (optional)
compute_scalar compute a scalar quantity (optional)
compute_vector compute a vector of quantities (optional)
compute_peratom compute one or more quantities per atom (optional)
compute_local compute one or more quantities per processor (optional)
pack_comm pack a buffer with items to communicate (optional)
unpack_comm unpack the buffer (optional)
pack_reverse pack a buffer with items to reverse communicate (optional)
unpack_reverse unpack the buffer (optional)
remove_bias remove velocity bias from one atom (optional)
remove_bias_all remove velocity bias from all atoms in group (optional)
restore_bias restore velocity bias for one atom after remove_bias (optional)
restore_bias_all same as before, but for all atoms in group (optional)
pair_tally_callback callback function for tally-style computes (optional).
memory_usage tally memory usage (optional)
Tally-style computes are a special case, as their computation is done in two stages: the callback function is
registered with the pair style and then called from the Pair::ev_tally() function, which is called for each pair
after force and energy has been computed for this pair. Then the tallied values are retrieved with the standard
compute_scalar or compute_vector or compute_peratom methods. The USER-TALLY package provides
examples_compute_tally.html for utilizing this mechanism.
10.4 Dump styles
10.5 Dump custom output options
Classes that dump per-atom info to files are derived from the Dump class. To dump new quantities or in a new
format, a new derived dump class can be added, but it is typically simpler to modify the DumpCustom class
contained in the dump_custom.cpp file.
Dump_atom.cpp is a simple example of a derived dump class.
Here is a brief description of methods you define in your new derived class. See dump.h for details.
write_header write the header section of a snapshot of atoms
count count the number of lines a processor will output
pack pack a proc's output data into a buffer
write_data write a proc's data to a file
LAMMPS Users Manual
176

See the dump command and its custom style for a list of keywords for atom information that can already be
dumped by DumpCustom. It includes options to dump per-atom info from Compute classes, so adding a new
derived Compute class is one way to calculate new quantities to dump.
Alternatively, you can add new keywords to the dump custom command. Search for the word "customize" in
dump_custom.cpp to see the half-dozen or so locations where code will need to be added.
10.6 Fix styles
In LAMMPS, a "fix" is any operation that is computed during timestepping that alters some property of the
system. Essentially everything that happens during a simulation besides force computation, neighbor list
construction, and output, is a "fix". This includes time integration (update of coordinates and velocities), force
constraints or boundary conditions (SHAKE or walls), and diagnostics (compute a diffusion coefficient). New
styles can be created to add new options to LAMMPS.
Fix_setforce.cpp is a simple example of setting forces on atoms to prescribed values. There are dozens of fix
options already in LAMMPS; choose one as a template that is similar to what you want to implement.
Here is a brief description of methods you can define in your new derived class. See fix.h for details.
setmask determines when the fix is called during the timestep (required)
init initialization before a run (optional)
setup_pre_exchange called before atom exchange in setup (optional)
setup_pre_force called before force computation in setup (optional)
setup called immediately before the 1st timestep and after forces are computed (optional)
min_setup_pre_force like setup_pre_force, but for minimizations instead of MD runs (optional)
min_setup like setup, but for minimizations instead of MD runs (optional)
initial_integrate called at very beginning of each timestep (optional)
pre_exchange called before atom exchange on re-neighboring steps (optional)
pre_neighbor called before neighbor list build (optional)
pre_force called before pair & molecular forces are computed (optional)
post_force called after pair & molecular forces are computed and communicated (optional)
final_integrate called at end of each timestep (optional)
end_of_step called at very end of timestep (optional)
write_restart dumps fix info to restart file (optional)
restart uses info from restart file to re-initialize the fix (optional)
grow_arrays allocate memory for atom-based arrays used by fix (optional)
copy_arrays copy atom info when an atom migrates to a new processor (optional)
pack_exchange store atom's data in a buffer (optional)
unpack_exchange retrieve atom's data from a buffer (optional)
pack_restart store atom's data for writing to restart file (optional)
unpack_restart retrieve atom's data from a restart file buffer (optional)
size_restart size of atom's data (optional)
maxsize_restart max size of atom's data (optional)
LAMMPS Users Manual
177
setup_pre_force_respa same as setup_pre_force, but for rRESPA (optional)
initial_integrate_respa same as initial_integrate, but for rRESPA (optional)
post_integrate_respa called after the first half integration step is done in rRESPA (optional)
pre_force_respa same as pre_force, but for rRESPA (optional)
post_force_respa same as post_force, but for rRESPA (optional)
final_integrate_respa same as final_integrate, but for rRESPA (optional)
min_pre_force called after pair & molecular forces are computed in minimizer (optional)
min_post_force called after pair & molecular forces are computed and communicated in minimizer
(optional)
min_store store extra data for linesearch based minimization on a LIFO stack (optional)
min_pushstore push the minimization LIFO stack one element down (optional)
min_popstore pop the minimization LIFO stack one element up (optional)
min_clearstore clear minimization LIFO stack (optional)
min_step reset or move forward on line search minimization (optional)
min_dof report number of degrees of freedom added by this fix in minimization (optional)
max_alpha report maximum allowed step size during linesearch minimization (optional)
pack_comm pack a buffer to communicate a per-atom quantity (optional)
unpack_comm unpack a buffer to communicate a per-atom quantity (optional)
pack_reverse_comm pack a buffer to reverse communicate a per-atom quantity (optional)
unpack_reverse_comm unpack a buffer to reverse communicate a per-atom quantity (optional)
dof report number of degrees of freedom removed by this fix during MD (optional)
compute_scalar return a global scalar property that the fix computes (optional)
compute_vector return a component of a vector property that the fix computes (optional)
compute_array return a component of an array property that the fix computes (optional)
deform called when the box size is changed (optional)
reset_target called when a change of the target temperature is requested during a run (optional)
reset_dt is called when a change of the time step is requested during a run (optional)
modify_param called when a fix_modify request is executed (optional)
memory_usage report memory used by fix (optional)
thermo compute quantities for thermodynamic output (optional)
Typically, only a small fraction of these methods are defined for a particular fix. Setmask is mandatory, as it
determines when the fix will be invoked during the timestep. Fixes that perform time integration (nve, nvt,
npt) implement initial_integrate() and final_integrate() to perform velocity Verlet updates. Fixes that constrain
forces implement post_force().
Fixes that perform diagnostics typically implement end_of_step(). For an end_of_step fix, one of your fix
arguments must be the variable "nevery" which is used to determine when to call the fix and you must set this
variable in the constructor of your fix. By convention, this is the first argument the fix defines (after the ID,
group-ID, style).
If the fix needs to store information for each atom that persists from timestep to timestep, it can manage that
memory and migrate the info with the atoms as they move from processors to processor by implementing the
grow_arrays, copy_arrays, pack_exchange, and unpack_exchange methods. Similarly, the pack_restart and
unpack_restart methods can be implemented to store information about the fix in restart files. If you wish an
LAMMPS Users Manual
178
integrator or force constraint fix to work with rRESPA (see the run_style command), the initial_integrate,
post_force_integrate, and final_integrate_respa methods can be implemented. The thermo method enables a
fix to contribute values to thermodynamic output, as printed quantities and/or to be summed to the potential
energy of the system.
10.7 Input script commands
New commands can be added to LAMMPS input scripts by adding new classes that have a "command"
method. For example, the create_atoms, read_data, velocity, and run commands are all implemented in this
fashion. When such a command is encountered in the LAMMPS input script, LAMMPS simply creates a class
with the corresponding name, invokes the "command" method of the class, and passes it the arguments from
the input script. The command method can perform whatever operations it wishes on LAMMPS data
structures.
The single method your new class must define is as follows:
command operations performed by the new command
Of course, the new class can define other methods and variables as needed.
10.8 Kspace computations
Classes that compute long-range Coulombic interactions via K-space representations (Ewald, PPPM) are
derived from the KSpace class. New styles can be created to add new K-space options to LAMMPS.
Ewald.cpp is an example of computing K-space interactions.
Here is a brief description of methods you define in your new derived class. See kspace.h for details.
init initialize the calculation before a run
setup computation before the 1st timestep of a run
compute every-timestep computation
memory_usage tally of memory usage
10.9 Minimization styles
Classes that perform energy minimization derived from the Min class. New styles can be created to add new
minimization algorithms to LAMMPS.
Min_cg.cpp is an example of conjugate gradient minimization.
Here is a brief description of methods you define in your new derived class. See min.h for details.
init initialize the minimization before a run
run perform the minimization
memory_usage tally of memory usage
LAMMPS Users Manual
179
10.10 Pairwise potentials
Classes that compute pairwise interactions are derived from the Pair class. In LAMMPS, pairwise calculation
include manybody potentials such as EAM or Tersoff where particles interact without a static bond topology.
New styles can be created to add new pair potentials to LAMMPS.
Pair_lj_cut.cpp is a simple example of a Pair class, though it includes some optional methods to enable its use
with rRESPA.
Here is a brief description of the class methods in pair.h:
compute workhorse routine that computes pairwise interactions
settings reads the input script line with arguments you define
coeff set coefficients for one i,j type pair
init_one perform initialization for one i,j type pair
init_style initialization specific to this pair style
write & read_restart write/read i,j pair coeffs to restart files
write & read_restart_settings write/read global settings to restart files
single force and energy of a single pairwise interaction between 2 atoms
compute_inner/middle/outer versions of compute used by rRESPA
The inner/middle/outer routines are optional.
10.11 Region styles
Classes that define geometric regions are derived from the Region class. Regions are used elsewhere in
LAMMPS to group atoms, delete atoms to create a void, insert atoms in a specified region, etc. New styles
can be created to add new region shapes to LAMMPS.
Region_sphere.cpp is an example of a spherical region.
Here is a brief description of methods you define in your new derived class. See region.h for details.
inside determine whether a point is in the region
surface_interior determine if a point is within a cutoff distance inside of surc
surface_exterior determine if a point is within a cutoff distance outside of surf
shape_update change region shape if set by time-dependent variable
10.12 Body styles
Classes that define body particles are derived from the Body class. Body particles can represent complex
entities, such as surface meshes of discrete points, collections of sub-particles, deformable objects, etc.
See Section 6.14 of the manual for an overview of using body particles and the body doc page for details on
LAMMPS Users Manual
180

the various body styles LAMMPS supports. New styles can be created to add new kinds of body particles to
LAMMPS.
Body_nparticle.cpp is an example of a body particle that is treated as a rigid body containing N sub-particles.
Here is a brief description of methods you define in your new derived class. See body.h for details.
data_body process a line from the Bodies section of a data file
noutrow number of sub-particles output is generated for
noutcol number of values per-sub-particle output is generated for
output output values for the Mth sub-particle
pack_comm_body body attributes to communicate every timestep
unpack_comm_body unpacking of those attributes
pack_border_body body attributes to communicate when reneighboring is done
unpack_border_body unpacking of those attributes
10.13 Thermodynamic output options
There is one class that computes and prints thermodynamic information to the screen and log file; see the file
thermo.cpp.
There are two styles defined in thermo.cpp: "one" and "multi". There is also a flexible "custom" style which
allows the user to explicitly list keywords for quantities to print when thermodynamic info is output. See the
thermo_style command for a list of defined quantities.
The thermo styles (one, multi, etc) are simply lists of keywords. Adding a new style thus only requires
defining a new list of keywords. Search for the word "customize" with references to "thermo style" in
thermo.cpp to see the two locations where code will need to be added.
New keywords can also be added to thermo.cpp to compute new quantities for output. Search for the word
"customize" with references to "keyword" in thermo.cpp to see the several locations where code will need to
be added.
Note that the thermo_style custom command already allows for thermo output of quantities calculated by
fixes, computes, and variables. Thus, it may be simpler to compute what you wish via one of those constructs,
than by adding a new keyword to the thermo command.
10.14 Variable options
There is one class that computes and stores variable information in LAMMPS; see the file variable.cpp. The
value associated with a variable can be periodically printed to the screen via the print, fix print, or
thermo_style custom commands. Variables of style "equal" can compute complex equations that involve the
following types of arguments:
thermo keywords = ke, vol, atoms, ...
other variables = v_a, v_myvar, ...
math functions = div(x,y), mult(x,y), add(x,y), ...
LAMMPS Users Manual
181

group functions = mass(group), xcm(group,x), ...
atom values = x[123], y[3], vx[34], ...
compute values = c_mytemp[0], c_thermo_press[3], ...
Adding keywords for the thermo_style custom command (which can then be accessed by variables) was
discussed here on this page.
Adding a new math function of one or two arguments can be done by editing one section of the
Variable::evaluate() method. Search for the word "customize" to find the appropriate location.
Adding a new group function can be done by editing one section of the Variable::evaluate() method. Search
for the word "customize" to find the appropriate location. You may need to add a new method to the Group
class as well (see the group.cpp file).
Accessing a new atom-based vector can be done by editing one section of the Variable::evaluate() method.
Search for the word "customize" to find the appropriate location.
Adding new compute styles (whose calculated values can then be accessed by variables) was discussed here
on this page.
10.15 Submitting new features for inclusion in LAMMPS
We encourage users to submit new features or modifications for LAMMPS to the core developers so they can
be added to the LAMMPS distribution. The preferred way to manage and coordinate this is as of Fall 2016 via
the LAMMPS project on GitHub. An alternative is to contact the LAMMPS developers or the indicated
developer of a package or feature directly and send in your contribution via e-mail.
For any larger modifications or programming project, you are encouraged to contact the LAMMPS developers
ahead of time, in order to discuss implementation strategies and coding guidelines, that will make it easier to
integrate your contribution and result in less work for everybody involved. You are also encouraged to search
through the list of open issues on GitHub and submit a new issue for a planned feature, so you would not
duplicate the work of others (and possibly get scooped by them) or have your work duplicated by others.
How quickly your contribution will be integrated depends largely on how much effort it will cause to integrate
and test it, how much it requires changes to the core codebase, and of how much interest it is to the larger
LAMMPS community. Please see below for a checklist of typical requirements. Once you have prepared
everything, see this tutorial for instructions on how to submit your changes or new files through a GitHub pull
request. If you prefer to submit patches or full files, you should first make certain, that your code works
correctly with the latest patch-level version of LAMMPS and contains all bugfixes from it. Then create a
gzipped tar file of all changed or added files or a corresponding patch file using 'diff -u' or 'diff -c' and
compress it with gzip. Please only use gzip compression, as this works well on all platforms.
If the new features/files are broadly useful we may add them as core files to LAMMPS or as part of a standard
package. Else we will add them as a user-contributed file or package. Examples of user packages are in src
sub-directories that start with USER. The USER-MISC package is simply a collection of (mostly) unrelated
single files, which is the simplest way to have your contribution quickly added to the LAMMPS distribution.
You can see a list of the both standard and user packages by typing "make package" in the LAMMPS src
directory.
LAMMPS Users Manual
182
Note that by providing us files to release, you are agreeing to make them open-source, i.e. we can release
them under the terms of the GPL, used as a license for the rest of LAMMPS. See Section 1.4 for details.
With user packages and files, all we are really providing (aside from the fame and fortune that accompanies
having your name in the source code and on the Authors page of the LAMMPS WWW site), is a means for
you to distribute your work to the LAMMPS user community, and a mechanism for others to easily try out
your new feature. This may help you find bugs or make contact with new collaborators. Note that you're also
implicitly agreeing to support your code which means answer questions, fix bugs, and maintain it if
LAMMPS changes in some way that breaks it (an unusual event).
NOTE: If you prefer to actively develop and support your add-on feature yourself, then you may wish to make
it available for download from your own website, as a user package that LAMMPS users can add to their copy
of LAMMPS. See the Offsite LAMMPS packages and tools page of the LAMMPS web site for examples of
groups that do this. We are happy to advertise your package and web site from that page. Simply email the
developers with info about your package and we will post it there.
The previous sections of this doc page describe how to add new "style" files of various kinds to LAMMPS.
Packages are simply collections of one or more new class files which are invoked as a new style within a
LAMMPS input script. If designed correctly, these additions typically do not require changes to the main core
of LAMMPS; they are simply add-on files. If you think your new feature requires non-trivial changes in core
LAMMPS files, you'll need to communicate with the developers, since we may or may not want to make
those changes. An example of a trivial change is making a parent-class method "virtual" when you derive a
new child class from it.
Here is a checklist of steps you need to follow to submit a single file or user package for our consideration.
Following these steps will save both you and us time. See existing files in packages in the src dir for
examples. If you are uncertain, please ask.
All source files you provide must compile with the most current version of LAMMPS with multiple
configurations. In particular you need to test compiling LAMMPS from scratch with
-DLAMMPS_BIGBIG set in addition to the default -DLAMMPS_SMALLBIG setting. Your code
will need to work correctly in serial and in parallel using MPI.
•
For consistency with the rest of LAMMPS and especially, if you want your contribution(s) to be
added to main LAMMPS code or one of its standard packages, it needs to be written in a style
compatible with other LAMMPS source files. This means: 2-character indentation per level, no tabs,
no lines over 80 characters. I/O is done via the C-style stdio library, class header files should not
import any system headers outside , STL containers should be avoided in headers, and forward
declarations used where possible or needed. All added code should be placed into the LAMMPS_NS
namespace or a sub-namespace; global or static variables should be avoided, as they conflict with the
modular nature of LAMMPS and the C++ class structure. Header files must not import namespaces
with using. This all is so the developers can more easily understand, integrate, and maintain your
contribution and reduce conflicts with other parts of LAMMPS. This basically means that the code
accesses data structures, performs its operations, and is formatted similar to other LAMMPS source
files, including the use of the error class for error and warning messages.
•
If you want your contribution to be added as a user-contributed feature, and it's a single file (actually a
*.cpp and *.h file) it can rapidly be added to the USER-MISC directory. Send us the one-line entry to
add to the USER-MISC/README file in that dir, along with the 2 source files. You can do this
multiple times if you wish to contribute several individual features.
•
If you want your contribution to be added as a user-contribution and it is several related features, it is
probably best to make it a user package directory with a name like USER-FOO. In addition to your
new files, the directory should contain a README text file. The README should contain your name
•
LAMMPS Users Manual
183

and contact information and a brief description of what your new package does. If your files depend
on other LAMMPS style files also being installed (e.g. because your file is a derived class from the
other LAMMPS class), then an Install.sh file is also needed to check for those dependencies. See
other README and Install.sh files in other USER directories as examples. Send us a tarball of this
USER-FOO directory.
Your new source files need to have the LAMMPS copyright, GPL notice, and your name and email
address at the top, like other user-contributed LAMMPS source files. They need to create a class that
is inside the LAMMPS namespace. If the file is for one of the USER packages, including
USER-MISC, then we are not as picky about the coding style (see above). I.e. the files do not need to
be in the same stylistic format and syntax as other LAMMPS files, though that would be nice for
developers as well as users who try to read your code.
•
You must also create a documentation file for each new command or style you are adding to
LAMMPS. For simplicity and convenience, the documentation of groups of closely related commands
or styles may be combined into a single file. This will be one file for a single-file feature. For a
package, it might be several files. These are simple text files with a specific markup language, that are
then auto-converted to HTML and PDF. The tools for this conversion are included in the source
distribution, and the translation can be as simple as doing "make html pdf" in the doc folder. Thus the
documentation source files must be in the same format and style as other *.txt files in the
lammps/doc/src directory for similar commands and styles; use one or more of them as a starting
point. A description of the markup can also be found in lammps/doc/utils/txt2html/README.html As
appropriate, the text files can include links to equations (see doc/Eqs/*.tex for examples, we
auto-create the associated JPG files), or figures (see doc/JPG for examples), or even additional PDF
files with further details (see doc/PDF for examples). The doc page should also include literature
citations as appropriate; see the bottom of doc/fix_nh.txt for examples and the earlier part of the same
file for how to format the cite itself. The "Restrictions" section of the doc page should indicate that
your command is only available if LAMMPS is built with the appropriate USER-MISC or
USER-FOO package. See other user package doc files for examples of how to do this. The
prerequisite for building the HTML format files are Python 3.x and virtualenv, the requirement for
generating the PDF format manual is the htmldoc software. Please run at least "make html" and
carefully inspect and proofread the resulting HTML format doc page before submitting your code.
•
For a new package (or even a single command) you should include one or more example scripts
demonstrating its use. These should run in no more than a couple minutes, even on a single processor,
and not require large data files as input. See directories under examples/USER for examples of input
scripts other users provided for their packages. These example inputs are also required for validating
memory accesses and testing for memory leaks with valgrind
•
If there is a paper of yours describing your feature (either the algorithm/science behind the feature
itself, or its initial usage, or its implementation in LAMMPS), you can add the citation to the *.cpp
source file. See src/USER-EFF/atom_vec_electron.cpp for an example. A LaTeX citation is stored in
a variable at the top of the file and a single line of code that references the variable is added to the
constructor of the class. Whenever a user invokes your feature from their input script, this will cause
LAMMPS to output the citation to a log.cite file and prompt the user to examine the file. Note that
you should only use this for a paper you or your group authored. E.g. adding a cite in the code for a
paper by Nose and Hoover if you write a fix that implements their integrator is not the intended usage.
That kind of citation should just be in the doc page you provide.
•
Finally, as a general rule-of-thumb, the more clear and self-explanatory you make your documentation and
README files, and the easier you make it for people to get started, e.g. by providing example scripts, the
more likely it is that users will try out your new feature.
LAMMPS Users Manual
184
(Foo) Foo, Morefoo, and Maxfoo, J of Classic Potentials, 75, 345 (1997).
LAMMPS Users Manual
185

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
11. Python interface to LAMMPS
LAMMPS can work together with Python in three ways. First, Python can wrap LAMMPS through the
LAMMPS library interface, so that a Python script can create one or more instances of LAMMPS and launch
one or more simulations. In Python lingo, this is "extending" Python with LAMMPS.
Second, the low-level Python interface can be used indirectly through the PyLammps and IPyLammps
wrapper classes in Python. These wrappers try to simplify the usage of LAMMPS in Python by providing an
object-based interface to common LAMMPS functionality. It also reduces the amount of code necessary to
parameterize LAMMPS scripts through Python and makes variables and computes directly accessible. See
PyLammps interface for more details.
Third, LAMMPS can use the Python interpreter, so that a LAMMPS input script can invoke Python code, and
pass information back-and-forth between the input script and Python functions you write. The Python code
can also callback to LAMMPS to query or change its attributes. In Python lingo, this is "embedding" Python
in LAMMPS.
This section describes how to use these three approaches.
11.1 Overview of running LAMMPS from Python• 11.2 Overview of using Python from a LAMMPS script• 11.3 Building LAMMPS as a shared library• 11.4 Installing the Python wrapper into Python• 11.5 Extending Python with MPI to run in parallel• 11.6 Testing the Python-LAMMPS interface• 11.7 Using LAMMPS from Python• 11.8 Example Python scripts that use LAMMPS• 11.9 PyLammps interface•
If you are not familiar with it, Python is a powerful scripting and programming language which can essentially
do anything that faster, lower-level languages like C or C++ can do, but typically with much fewer lines of
code. When used in embedded mode, Python can perform operations that the simplistic LAMMPS input script
syntax cannot. Python can be also be used as a "glue" language to drive a program through its library
interface, or to hook multiple pieces of software together, such as a simulation package plus a visualization
package, or to run a coupled multiscale or multiphysics model.
See Section 6.10 of the manual and the couple directory of the distribution for more ideas about coupling
LAMMPS to other codes. See Section 6.19 for a description of the LAMMPS library interface provided in
src/library.cpp and src/library.h, and how to extend it for your needs. As described below, that interface is
what is exposed to Python either when calling LAMMPS from Python or when calling Python from a
LAMMPS input script and then calling back to LAMMPS from Python code. The library interface is designed
to be easy to add functions to. Thus the Python interface to LAMMPS is also easy to extend as well.
If you create interesting Python scripts that run LAMMPS or interesting Python functions that can be called
from a LAMMPS input script, that you think would be useful to other users, please email them to the
developers. We can include them in the LAMMPS distribution.
LAMMPS Users Manual
186

11.1 Overview of running LAMMPS from Python
The LAMMPS distribution includes a python directory with all you need to run LAMMPS from Python. The
python/lammps.py file wraps the LAMMPS library interface, with one wrapper function per LAMMPS
library function. This file makes it is possible to do the following either from a Python script, or interactively
from a Python prompt: create one or more instances of LAMMPS, invoke LAMMPS commands or give it an
input script, run LAMMPS incrementally, extract LAMMPS results, an modify internal LAMMPS variables.
From a Python script you can do this in serial or parallel. Running Python interactively in parallel does not
generally work, unless you have a version of Python that extends standard Python to enable multiple instances
of Python to read what you type.
To do all of this, you must first build LAMMPS as a shared library, then insure that your Python can find the
python/lammps.py file and the shared library. These steps are explained in subsequent sections 11.3 and 11.4.
Sections 11.5 and 11.6 discuss using MPI from a parallel Python program and how to test that you are ready
to use LAMMPS from Python. Section 11.7 lists all the functions in the current LAMMPS library interface
and how to call them from Python.
Section 11.8 gives some examples of coupling LAMMPS to other tools via Python. For example, LAMMPS
can easily be coupled to a GUI or other visualization tools that display graphs or animations in real time as
LAMMPS runs. Examples of such scripts are included in the python directory.
Two advantages of using Python to run LAMMPS are how concise the language is, and that it can be run
interactively, enabling rapid development and debugging of programs. If you use it to mostly invoke costly
operations within LAMMPS, such as running a simulation for a reasonable number of timesteps, then the
overhead cost of invoking LAMMPS thru Python will be negligible.
The Python wrapper for LAMMPS uses the amazing and magical (to me) "ctypes" package in Python, which
auto-generates the interface code needed between Python and a set of C interface routines for a library. Ctypes
is part of standard Python for versions 2.5 and later. You can check which version of Python you have
installed, by simply typing "python" at a shell prompt.
11.2 Overview of using Python from a LAMMPS script
NOTE: It is not currently possible to use the python command described in this section with Python 3, only
with Python 2. The C API changed from Python 2 to 3 and the LAMMPS code is not compatible with both.
LAMMPS has a python command which can be used in an input script to define and execute a Python
function that you write the code for. The Python function can also be assigned to a LAMMPS python-style
variable via the variable command. Each time the variable is evaluated, either in the LAMMPS input script
itself, or by another LAMMPS command that uses the variable, this will trigger the Python function to be
invoked.
The Python code for the function can be included directly in the input script or in an auxiliary file. The
function can have arguments which are mapped to LAMMPS variables (also defined in the input script) and it
can return a value to a LAMMPS variable. This is thus a mechanism for your input script to pass information
to a piece of Python code, ask Python to execute the code, and return information to your input script.
Note that a Python function can be arbitrarily complex. It can import other Python modules, instantiate Python
classes, call other Python functions, etc. The Python code that you provide can contain more code than the
single function. It can contain other functions or Python classes, as well as global variables or other
LAMMPS Users Manual
187

mechanisms for storing state between calls from LAMMPS to the function.
The Python function you provide can consist of "pure" Python code that only performs operations provided by
standard Python. However, the Python function can also "call back" to LAMMPS through its Python-wrapped
library interface, in the manner described in the previous section 11.1. This means it can issue LAMMPS
input script commands or query and set internal LAMMPS state. As an example, this can be useful in an input
script to create a more complex loop with branching logic, than can be created using the simple looping and
branching logic enabled by the next and if commands.
See the python doc page and the variable doc page for its python-style variables for more info, including
examples of Python code you can write for both pure Python operations and callbacks to LAMMPS.
To run pure Python code from LAMMPS, you only need to build LAMMPS with the PYTHON package
installed:
make yes-python
make machine
Note that this will link LAMMPS with the Python library on your system, which typically requires several
auxiliary system libraries to also be linked. The list of these libraries and the paths to find them are specified
in the lib/python/Makefile.lammps file. You need to insure that file contains the correct information for your
version of Python and your machine to successfully build LAMMPS. See the lib/python/README file for
more info.
If you want to write Python code with callbacks to LAMMPS, then you must also follow the steps overviewed
in the preceding section (11.1) for running LAMMPS from Python. I.e. you must build LAMMPS as a shared
library and insure that Python can find the python/lammps.py file and the shared library.
11.3 Building LAMMPS as a shared library
Instructions on how to build LAMMPS as a shared library are given in Section 2.5. A shared library is one
that is dynamically loadable, which is what Python requires to wrap LAMMPS. On Linux this is a library file
that ends in ".so", not ".a".
From the src directory, type
make foo mode=shlib
where foo is the machine target name, such as linux or g++ or serial. This should create the file
liblammps_foo.so in the src directory, as well as a soft link liblammps.so, which is what the Python wrapper
will load by default. Note that if you are building multiple machine versions of the shared library, the soft link
is always set to the most recently built version.
NOTE: If you are building LAMMPS with an MPI or FFT library or other auxiliary libraries (used by various
packages), then all of these extra libraries must also be shared libraries. If the LAMMPS shared-library build
fails with an error complaining about this, see Section 2.5 for more details.
LAMMPS Users Manual
188
11.4 Installing the Python wrapper into Python
For Python to invoke LAMMPS, there are 2 files it needs to know about:
python/lammps.py• src/liblammps.so•
Lammps.py is the Python wrapper on the LAMMPS library interface. Liblammps.so is the shared LAMMPS
library that Python loads, as described above.
You can insure Python can find these files in one of two ways:
set two environment variables• run the python/install.py script•
If you set the paths to these files as environment variables, you only have to do it once. For the csh or tcsh
shells, add something like this to your ~/.cshrc file, one line for each of the two files:
setenv PYTHONPATH ${PYTHONPATH}:/home/sjplimp/lammps/python
setenv LD_LIBRARY_PATH ${LD_LIBRARY_PATH}:/home/sjplimp/lammps/src
If you use the python/install.py script, you need to invoke it every time you rebuild LAMMPS (as a shared
library) or make changes to the python/lammps.py file.
You can invoke install.py from the python directory as
% python install.py [libdir] [pydir]
The optional libdir is where to copy the LAMMPS shared library to; the default is /usr/local/lib. The optional
pydir is where to copy the lammps.py file to; the default is the site-packages directory of the version of Python
that is running the install script.
Note that libdir must be a location that is in your default LD_LIBRARY_PATH, like /usr/local/lib or /usr/lib.
And pydir must be a location that Python looks in by default for imported modules, like its site-packages dir.
If you want to copy these files to non-standard locations, such as within your own user space, you will need to
set your PYTHONPATH and LD_LIBRARY_PATH environment variables accordingly, as above.
If the install.py script does not allow you to copy files into system directories, prefix the python command
with "sudo". If you do this, make sure that the Python that root runs is the same as the Python you run. E.g.
you may need to do something like
% sudo /usr/local/bin/python install.py [libdir] [pydir]
You can also invoke install.py from the make command in the src directory as
% make install-python
In this mode you cannot append optional arguments. Again, you may need to prefix this with "sudo". In this
mode you cannot control which Python is invoked by root.
Note that if you want Python to be able to load different versions of the LAMMPS shared library (see this
section below), you will need to manually copy files like liblammps_g++.so into the appropriate system
LAMMPS Users Manual
189

directory. This is not needed if you set the LD_LIBRARY_PATH environment variable as described above.
11.5 Extending Python with MPI to run in parallel
If you wish to run LAMMPS in parallel from Python, you need to extend your Python with an interface to
MPI. This also allows you to make MPI calls directly from Python in your script, if you desire.
There are several Python packages available that purport to wrap MPI as a library and allow MPI functions to
be called from Python. However, development on most of them seems to be halted except on:
mpi4py• PyPar•
Both packages, PyPar and mpi4py have been successfully tested with LAMMPS. PyPar is simpler and easy to
set up and use, but supports only a subset of MPI. Mpi4py is more MPI-feature complete, but also a bit more
complex to use. As of version 2.0.0, mpi4py is the only python MPI wrapper that allows passing a custom
MPI communicator to the LAMMPS constructor, which means one can easily run one or more LAMMPS
instances on subsets of the total MPI ranks.
PyPar requires the ubiquitous Numpy package be installed in your Python. After launching Python, type
import numpy
to see if it is installed. If not, here is how to install it (version 1.3.0b1 as of April 2009). Unpack the numpy
tarball and from its top-level directory, type
python setup.py build
sudo python setup.py install
The "sudo" is only needed if required to copy Numpy files into your Python distribution's site-packages
directory.
To install PyPar (version pypar-2.1.4_94 as of Aug 2012), unpack it and from its "source" directory, type
python setup.py build
sudo python setup.py install
Again, the "sudo" is only needed if required to copy PyPar files into your Python distribution's site-packages
directory.
If you have successfully installed PyPar, you should be able to run Python and type
import pypar
without error. You should also be able to run python in parallel on a simple test script
% mpirun -np 4 python test.py
where test.py contains the lines
import pypar
LAMMPS Users Manual
190
print "Proc %d out of %d procs" % (pypar.rank(),pypar.size())
and see one line of output for each processor you run on.
NOTE: To use PyPar and LAMMPS in parallel from Python, you must insure both are using the same version
of MPI. If you only have one MPI installed on your system, this is not an issue, but it can be if you have
multiple MPIs. Your LAMMPS build is explicit about which MPI it is using, since you specify the details in
your lo-level src/MAKE/Makefile.foo file. PyPar uses the "mpicc" command to find information about the
MPI it uses to build against. And it tries to load "libmpi.so" from the LD_LIBRARY_PATH. This may or
may not find the MPI library that LAMMPS is using. If you have problems running both PyPar and
LAMMPS together, this is an issue you may need to address, e.g. by moving other MPI installations so that
PyPar finds the right one.
To install mpi4py (version mpi4py-2.0.0 as of Oct 2015), unpack it and from its main directory, type
python setup.py build
sudo python setup.py install
Again, the "sudo" is only needed if required to copy mpi4py files into your Python distribution's site-packages
directory. To install with user privilege into the user local directory type
python setup.py install --user
If you have successfully installed mpi4py, you should be able to run Python and type
from mpi4py import MPI
without error. You should also be able to run python in parallel on a simple test script
% mpirun -np 4 python test.py
where test.py contains the lines
from mpi4py import MPI
comm = MPI.COMM_WORLD
print "Proc %d out of %d procs" % (comm.Get_rank(),comm.Get_size())
and see one line of output for each processor you run on.
NOTE: To use mpi4py and LAMMPS in parallel from Python, you must insure both are using the same
version of MPI. If you only have one MPI installed on your system, this is not an issue, but it can be if you
have multiple MPIs. Your LAMMPS build is explicit about which MPI it is using, since you specify the
details in your lo-level src/MAKE/Makefile.foo file. Mpi4py uses the "mpicc" command to find information
about the MPI it uses to build against. And it tries to load "libmpi.so" from the LD_LIBRARY_PATH. This
may or may not find the MPI library that LAMMPS is using. If you have problems running both mpi4py and
LAMMPS together, this is an issue you may need to address, e.g. by moving other MPI installations so that
mpi4py finds the right one.
LAMMPS Users Manual
191
11.6 Testing the Python-LAMMPS interface
To test if LAMMPS is callable from Python, launch Python interactively and type:
>>> from lammps import lammps
>>> lmp = lammps()
If you get no errors, you're ready to use LAMMPS from Python. If the 2nd command fails, the most common
error to see is
OSError: Could not load LAMMPS dynamic library
which means Python was unable to load the LAMMPS shared library. This typically occurs if the system can't
find the LAMMPS shared library or one of the auxiliary shared libraries it depends on, or if something about
the library is incompatible with your Python. The error message should give you an indication of what went
wrong.
You can also test the load directly in Python as follows, without first importing from the lammps.py file:
>>> from ctypes import CDLL
>>> CDLL("liblammps.so")
If an error occurs, carefully go thru the steps in Section 2.5 and above about building a shared library and
about insuring Python can find the necessary two files it needs.
Test LAMMPS and Python in serial:
To run a LAMMPS test in serial, type these lines into Python interactively from the bench directory:
>>> from lammps import lammps
>>> lmp = lammps()
>>> lmp.file("in.lj")
Or put the same lines in the file test.py and run it as
% python test.py
Either way, you should see the results of running the in.lj benchmark on a single processor appear on the
screen, the same as if you had typed something like:
lmp_g++ -in in.lj
Test LAMMPS and Python in parallel:
To run LAMMPS in parallel, assuming you have installed the PyPar package as discussed above, create a
test.py file containing these lines:
import pypar
from lammps import lammps
lmp = lammps()
lmp.file("in.lj")
print "Proc %d out of %d procs has" % (pypar.rank(),pypar.size()),lmp
pypar.finalize()
LAMMPS Users Manual
192

To run LAMMPS in parallel, assuming you have installed the mpi4py package as discussed above, create a
test.py file containing these lines:
from mpi4py import MPI
from lammps import lammps
lmp = lammps()
lmp.file("in.lj")
me = MPI.COMM_WORLD.Get_rank()
nprocs = MPI.COMM_WORLD.Get_size()
print "Proc %d out of %d procs has" % (me,nprocs),lmp
MPI.Finalize()
You can either script in parallel as:
% mpirun -np 4 python test.py
and you should see the same output as if you had typed
% mpirun -np 4 lmp_g++ -in in.lj
Note that if you leave out the 3 lines from test.py that specify PyPar commands you will instantiate and run
LAMMPS independently on each of the P processors specified in the mpirun command. In this case you
should get 4 sets of output, each showing that a LAMMPS run was made on a single processor, instead of one
set of output showing that LAMMPS ran on 4 processors. If the 1-processor outputs occur, it means that
PyPar is not working correctly.
Also note that once you import the PyPar module, PyPar initializes MPI for you, and you can use MPI calls
directly in your Python script, as described in the PyPar documentation. The last line of your Python script
should be pypar.finalize(), to insure MPI is shut down correctly.
Running Python scripts:
Note that any Python script (not just for LAMMPS) can be invoked in one of several ways:
% python foo.script
% python -i foo.script
% foo.script
The last command requires that the first line of the script be something like this:
#!/usr/local/bin/python
#!/usr/local/bin/python -i
where the path points to where you have Python installed, and that you have made the script file executable:
% chmod +x foo.script
Without the "-i" flag, Python will exit when the script finishes. With the "-i" flag, you will be left in the
Python interpreter when the script finishes, so you can type subsequent commands. As mentioned above, you
can only run Python interactively when running Python on a single processor, not in parallel.
LAMMPS Users Manual
193
11.7 Using LAMMPS from Python
As described above, the Python interface to LAMMPS consists of a Python "lammps" module, the source
code for which is in python/lammps.py, which creates a "lammps" object, with a set of methods that can be
invoked on that object. The sample Python code below assumes you have first imported the "lammps" module
in your Python script, as follows:
from lammps import lammps
These are the methods defined by the lammps module. If you look at the files src/library.cpp and src/library.h
you will see that they correspond one-to-one with calls you can make to the LAMMPS library from a C++ or
C or Fortran program, and which are described in Section 6.19 of the manual.
lmp = lammps() # create a LAMMPS object using the default liblammps.so library
# 4 optional args are allowed: name, cmdargs, ptr, comm
lmp = lammps(ptr=lmpptr) # use lmpptr as previously created LAMMPS object
lmp = lammps(comm=split) # create a LAMMPS object with a custom communicator, requires mpi4py 2.0.0 or later
lmp = lammps(name="g++") # create a LAMMPS object using the liblammps_g++.so library
lmp = lammps(name="g++",cmdargs=list) # add LAMMPS command-line args, e.g. list = ["-echo","screen"]
lmp.close() # destroy a LAMMPS object
version = lmp.version() # return the numerical version id, e.g. LAMMPS 2 Sep 2015 -> 20150902
lmp.file(file) # run an entire input script, file = "in.lj"
lmp.command(cmd) # invoke a single LAMMPS command, cmd = "run 100"
lmp.commands_list(cmdlist) # invoke commands in cmdlist = "run 10", "run 20"
lmp.commands_string(multicmd) # invoke commands in multicmd = "run 10\nrun 20"
xlo = lmp.extract_global(name,type) # extract a global quantity
# name = "boxxlo", "nlocal", etc
# type = 0 = int
# 1 = double
coords = lmp.extract_atom(name,type) # extract a per-atom quantity
# name = "x", "type", etc
# type = 0 = vector of ints
# 1 = array of ints
# 2 = vector of doubles
# 3 = array of doubles
eng = lmp.extract_compute(id,style,type) # extract value(s) from a compute
v3 = lmp.extract_fix(id,style,type,i,j) # extract value(s) from a fix
# id = ID of compute or fix
# style = 0 = global data
# 1 = per-atom data
# 2 = local data
# type = 0 = scalar
# 1 = vector
# 2 = array
# i,j = indices of value in global vector or array
var = lmp.extract_variable(name,group,flag) # extract value(s) from a variable
# name = name of variable
# group = group ID (ignored for equal-style variables)
# flag = 0 = equal-style variable
LAMMPS Users Manual
194

# 1 = atom-style variable
flag = lmp.set_variable(name,value) # set existing named string-style variable to value, flag = 0 if successful
value = lmp.get_thermo(name) # return current value of a thermo keyword
natoms = lmp.get_natoms() # total # of atoms as int
data = lmp.gather_atoms(name,type,count) # return per-atom property of all atoms gathered into data, ordered by atom ID
# name = "x", "charge", "type", etc
# count = # of per-atom values, 1 or 3, etc
lmp.scatter_atoms(name,type,count,data) # scatter per-atom property to all atoms from data, ordered by atom ID
# name = "x", "charge", "type", etc
# count = # of per-atom values, 1 or 3, etc
The lines
from lammps import lammps
lmp = lammps()
create an instance of LAMMPS, wrapped in a Python class by the lammps Python module, and return an
instance of the Python class as lmp. It is used to make all subsequent calls to the LAMMPS library.
Additional arguments to lammps() can be used to tell Python the name of the shared library to load or to pass
arguments to the LAMMPS instance, the same as if LAMMPS were launched from a command-line prompt.
If the ptr argument is set like this:
lmp = lammps(ptr=lmpptr)
then lmpptr must be an argument passed to Python via the LAMMPS python command, when it is used to
define a Python function that is invoked by the LAMMPS input script. This mode of using Python with
LAMMPS is described above in 11.2. The variable lmpptr refers to the instance of LAMMPS that called the
embedded Python interpreter. Using it as an argument to lammps() allows the returned Python class instance
"lmp" to make calls to that instance of LAMMPS. See the python command doc page for examples using this
syntax.
Note that you can create multiple LAMMPS objects in your Python script, and coordinate and run multiple
simulations, e.g.
from lammps import lammps
lmp1 = lammps()
lmp2 = lammps()
lmp1.file("in.file1")
lmp2.file("in.file2")
The file(), command(), commands_list(), commands_string() methods allow an input script, a single
command, or multiple commands to be invoked.
The extract_global(), extract_atom(), extract_compute(), extract_fix(), and extract_variable() methods return
values or pointers to data structures internal to LAMMPS.
For extract_global() see the src/library.cpp file for the list of valid names. New names could easily be added.
A double or integer is returned. You need to specify the appropriate data type via the type argument.
LAMMPS Users Manual
195
For extract_atom(), a pointer to internal LAMMPS atom-based data is returned, which you can use via normal
Python subscripting. See the extract() method in the src/atom.cpp file for a list of valid names. Again, new
names could easily be added if the property you want is not listed. A pointer to a vector of doubles or integers,
or a pointer to an array of doubles (double **) or integers (int **) is returned. You need to specify the
appropriate data type via the type argument.
For extract_compute() and extract_fix(), the global, per-atom, or local data calculated by the compute or fix
can be accessed. What is returned depends on whether the compute or fix calculates a scalar or vector or
array. For a scalar, a single double value is returned. If the compute or fix calculates a vector or array, a
pointer to the internal LAMMPS data is returned, which you can use via normal Python subscripting. The one
exception is that for a fix that calculates a global vector or array, a single double value from the vector or
array is returned, indexed by I (vector) or I and J (array). I,J are zero-based indices. The I,J arguments can be
left out if not needed. See Section 6.15 of the manual for a discussion of global, per-atom, and local data, and
of scalar, vector, and array data types. See the doc pages for individual computes and fixes for a description of
what they calculate and store.
For extract_variable(), an equal-style or atom-style variable is evaluated and its result returned.
For equal-style variables a single double value is returned and the group argument is ignored. For atom-style
variables, a vector of doubles is returned, one value per atom, which you can use via normal Python
subscripting. The values will be zero for atoms not in the specified group.
The get_natoms() method returns the total number of atoms in the simulation, as an int.
The gather_atoms() method allows any per-atom property (coordinates, velocities, etc) to be extracted from
LAMMPS. It returns a ctypes vector of ints or doubles as specified by type, of length count*natoms, for the
named property for all atoms in the simulation. The data is ordered by count and then by atom ID. See the
extract() method in the src/atom.cpp file for a list of valid names. Again, new names could easily be added if
the property you want is missing. The vector can be used via normal Python subscripting. If atom IDs are not
consecutively ordered within LAMMPS, a None is returned as indication of an error. A special treatment is
applied for image flags stored in the "image" property. All three image flags are stored in a packed format in a
single integer, so count would be 1 to retrieve that integer, however also a count value of 3 can be used and
then the image flags will be unpacked into 3 individual integers, ordered in a similar fashion as coordinates.
Note that the data structure gather_atoms("x") returns is different from the data structure returned by
extract_atom("x") in four ways. (1) Gather_atoms() returns a vector which you index as x[i]; extract_atom()
returns an array which you index as x[i][j]. (2) Gather_atoms() orders the atoms by atom ID while
extract_atom() does not. (3) Gathert_atoms() returns a list of all atoms in the simulation; extract_atoms()
returns just the atoms local to each processor. (4) Finally, the gather_atoms() data structure is a copy of the
atom coords stored internally in LAMMPS, whereas extract_atom() returns an array that effectively points
directly to the internal data. This means you can change values inside LAMMPS from Python by assigning a
new values to the extract_atom() array. To do this with the gather_atoms() vector, you need to change values
in the vector, then invoke the scatter_atoms() method.
The scatter_atoms() method allows any per-atom property (coordinates, velocities, etc) to be inserted into
LAMMPS, overwriting the current property. It takes a vector of ints or doubles as specified by type, of length
count*natoms, for the named property for all atoms in the simulation. The data should be ordered by count
and then by atom ID. See the extract() method in the src/atom.cpp file for a list of valid names. Again, new
names could easily be added if the property you want is missing. It uses the vector of data to overwrite the
corresponding properties for each atom inside LAMMPS. This requires LAMMPS to have its "map" option
enabled; see the atom_modify command for details. If it is not, or if atom IDs are not consecutively ordered,
LAMMPS Users Manual
196
no coordinates are reset. Similar as for gather_atoms() a special treatment is applied for image flags, which
can be provided in packed (count = 1) or unpacked (count = 3) format and in the latter case, they will be
packed before applied to atoms.
The array of coordinates passed to scatter_atoms() must be a ctypes vector of ints or doubles, allocated and
initialized something like this:
from ctypes import *
natoms = lmp.get_natoms()
n3 = 3*natoms
x = (n3*c_double)()
x[0] = x coord of atom with ID 1
x[1] = y coord of atom with ID 1
x[2] = z coord of atom with ID 1
x[3] = x coord of atom with ID 2
...
x[n3-1] = z coord of atom with ID natoms
lmp.scatter_atoms("x",1,3,x)
Alternatively, you can just change values in the vector returned by gather_atoms("x",1,3), since it is a ctypes
vector of doubles.
As noted above, these Python class methods correspond one-to-one with the functions in the LAMMPS
library interface in src/library.cpp and library.h. This means you can extend the Python wrapper via the
following steps:
Add a new interface function to src/library.cpp and src/library.h.• Rebuild LAMMPS as a shared library.• Add a wrapper method to python/lammps.py for this interface function.• You should now be able to invoke the new interface function from a Python script. Isn't ctypes
amazing?
•
11.8 Example Python scripts that use LAMMPS
These are the Python scripts included as demos in the python/examples directory of the LAMMPS
distribution, to illustrate the kinds of things that are possible when Python wraps LAMMPS. If you create
your own scripts, send them to us and we can include them in the LAMMPS distribution.
trivial.py read/run a LAMMPS input script thru Python
demo.py invoke various LAMMPS library interface
routines
simple.py run in parallel
similar to examples/COUPLE/simple/simple.cpp split.py
same as simple.py but running in parallel on a subset of
procs gui.py
GUI go/stop/temperature-slider to control LAMMPS plot.py
real-time temperature plot with GnuPlot via Pizza.py viz_tool.py
real-time viz via some viz package vizplotgui_tool.py
LAMMPS Users Manual
197

combination of viz_tool.py and plot.py and gui.py
For the viz_tool.py and vizplotgui_tool.py commands, replace "tool" with "gl" or "atomeye" or "pymol" or
"vmd", depending on what visualization package you have installed.
Note that for GL, you need to be able to run the Pizza.py GL tool, which is included in the pizza
sub-directory. See the Pizza.py doc pages for more info:
Note that for AtomEye, you need version 3, and there is a line in the scripts that specifies the path and name
of the executable. See the AtomEye WWW pages here or here for more details:
http://mt.seas.upenn.edu/Archive/Graphics/A
http://mt.seas.upenn.edu/Archive/Graphics/A3/A3.html
The latter link is to AtomEye 3 which has the scriping capability needed by these Python scripts.
Note that for PyMol, you need to have built and installed the open-source version of PyMol in your Python, so
that you can import it from a Python script. See the PyMol WWW pages here or here for more details:
http://www.pymol.org
http://sourceforge.net/scm/?type=svn&group_id=4546
The latter link is to the open-source version.
Note that for VMD, you need a fairly current version (1.8.7 works for me) and there are some lines in the
pizza/vmd.py script for 4 PIZZA variables that have to match the VMD installation on your system.
See the python/README file for instructions on how to run them and the source code for individual scripts
for comments about what they do.
Here are screenshots of the vizplotgui_tool.py script in action for different visualization package options.
Click to see larger images:
LAMMPS Users Manual
198

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
12. Errors
This section describes the errors you can encounter when using LAMMPS, either conceptually, or as printed
out by the program.
12.1 Common problems
12.2 Reporting bugs
12.3 Error & warning messages
12.1 Common problems
If two LAMMPS runs do not produce the exact same answer on different machines or different numbers of
processors, this is typically not a bug. In theory you should get identical answers on any number of processors
and on any machine. In practice, numerical round-off can cause slight differences and eventual divergence of
molecular dynamics phase space trajectories within a few 100s or few 1000s of timesteps. However, the
statistical properties of the two runs (e.g. average energy or temperature) should still be the same.
If the velocity command is used to set initial atom velocities, a particular atom can be assigned a different
velocity when the problem is run on a different number of processors or on different machines. If this
happens, the phase space trajectories of the two simulations will rapidly diverge. See the discussion of the
loop option in the velocity command for details and options that avoid this issue.
Similarly, the create_atoms command generates a lattice of atoms. For the same physical system, the ordering
and numbering of atoms by atom ID may be different depending on the number of processors.
Some commands use random number generators which may be setup to produce different random number
streams on each processor and hence will produce different effects when run on different numbers of
processors. A commonly-used example is the fix langevin command for thermostatting.
A LAMMPS simulation typically has two stages, setup and run. Most LAMMPS errors are detected at setup
time; others like a bond stretching too far may not occur until the middle of a run.
LAMMPS tries to flag errors and print informative error messages so you can fix the problem. For most errors
it will also print the last input script command that it was processing. Of course, LAMMPS cannot figure out
your physics or numerical mistakes, like choosing too big a timestep, specifying erroneous force field
coefficients, or putting 2 atoms on top of each other! If you run into errors that LAMMPS doesn't catch that
you think it should flag, please send an email to the developers.
If you get an error message about an invalid command in your input script, you can determine what command
is causing the problem by looking in the log.lammps file or using the echo command to see it on the screen. If
you get an error like "Invalid ... style", with ... being fix, compute, pair, etc, it means that you mistyped the
style name or that the command is part of an optional package which was not compiled into your executable.
The list of available styles in your executable can be listed by using the -h command-line argument. The
installation and compilation of optional packages is explained in the installation instructions.
LAMMPS Users Manual
200

For a given command, LAMMPS expects certain arguments in a specified order. If you mess this up,
LAMMPS will often flag the error, but it may also simply read a bogus argument and assign a value that is
valid, but not what you wanted. E.g. trying to read the string "abc" as an integer value of 0. Careful reading of
the associated doc page for the command should allow you to fix these problems. In most cases, where
LAMMPS expects to read a number, either integer or floating point, it performs a stringent test on whether the
provided input actually is an integer or floating-point number, respectively, and reject the input with an error
message (for instance, when an integer is required, but a floating-point number 1.0 is provided):
ERROR: Expected integer parameter in input script or data file
Some commands allow for using variable references in place of numeric constants so that the value can be
evaluated and may change over the course of a run. This is typically done with the syntax v_name for a
parameter, where name is the name of the variable. On the other hand, immediate variable expansion with the
syntax $name is performed while reading the input and before parsing commands,
NOTE: Using a variable reference (i.e. v_name) is only allowed if the documentation of the corresponding
command explicitly says it is.
Generally, LAMMPS will print a message to the screen and logfile and exit gracefully when it encounters a
fatal error. Sometimes it will print a WARNING to the screen and logfile and continue on; you can decide if
the WARNING is important or not. A WARNING message that is generated in the middle of a run is only
printed to the screen, not to the logfile, to avoid cluttering up thermodynamic output. If LAMMPS crashes or
hangs without spitting out an error message first then it could be a bug (see this section) or one of the
following cases:
LAMMPS runs in the available memory a processor allows to be allocated. Most reasonable MD runs are
compute limited, not memory limited, so this shouldn't be a bottleneck on most platforms. Almost all large
memory allocations in the code are done via C-style malloc's which will generate an error message if you run
out of memory. Smaller chunks of memory are allocated via C++ "new" statements. If you are unlucky you
could run out of memory just when one of these small requests is made, in which case the code will crash or
hang (in parallel), since LAMMPS doesn't trap on those errors.
Illegal arithmetic can cause LAMMPS to run slow or crash. This is typically due to invalid physics and
numerics that your simulation is computing. If you see wild thermodynamic values or NaN values in your
LAMMPS output, something is wrong with your simulation. If you suspect this is happening, it is a good idea
to print out thermodynamic info frequently (e.g. every timestep) via the thermo so you can monitor what is
happening. Visualizing the atom movement is also a good idea to insure your model is behaving as you
expect.
In parallel, one way LAMMPS can hang is due to how different MPI implementations handle buffering of
messages. If the code hangs without an error message, it may be that you need to specify an MPI setting or
two (usually via an environment variable) to enable buffering or boost the sizes of messages that can be
buffered.
12.2 Reporting bugs
If you are confident that you have found a bug in LAMMPS, follow these steps.
Check the New features and bug fixes section of the LAMMPS WWW site to see if the bug has already been
reported or fixed or the Unfixed bug to see if a fix is pending.
LAMMPS Users Manual
201

Check the mailing list to see if it has been discussed before.
If not, send an email to the mailing list describing the problem with any ideas you have as to what is causing it
or where in the code the problem might be. The developers will ask for more info if needed, such as an input
script or data files.
The most useful thing you can do to help us fix the bug is to isolate the problem. Run it on the smallest
number of atoms and fewest number of processors and with the simplest input script that reproduces the bug
and try to identify what command or combination of commands is causing the problem.
As a last resort, you can send an email directly to the developers.
12.3 Error & warning messages
These are two alphabetic lists of the ERROR and WARNING messages LAMMPS prints out and the reason
why. If the explanation here is not sufficient, the documentation for the offending command may help. Error
and warning messages also list the source file and line number where the error was generated. For example,
this message
ERROR: Illegal velocity command (velocity.cpp:78)
means that line #78 in the file src/velocity.cpp generated the error. Looking in the source code may help you
figure out what went wrong.
Note that error messages from user-contributed packages are not listed here. If such an error occurs and is not
self-explanatory, you'll need to look in the source code or contact the author of the package.
Errors:
1-3 bond count is inconsistent
An inconsistency was detected when computing the number of 1-3 neighbors for each atom. This
likely means something is wrong with the bond topologies you have defined.
1-4 bond count is inconsistent
An inconsistency was detected when computing the number of 1-4 neighbors for each atom. This
likely means something is wrong with the bond topologies you have defined.
Accelerator sharing is not currently supported on system
Multiple MPI processes cannot share the accelerator on your system. For NVIDIA GPUs, see the
nvidia-smi command to change this setting.
All angle coeffs are not set
All angle coefficients must be set in the data file or by the angle_coeff command before running a
simulation.
All atom IDs = 0 but atom_modify id = yes
Self-explanatory.
All atoms of a swapped type must have same charge.
Self-explanatory.
All atoms of a swapped type must have the same charge.
Self-explanatory.
All bond coeffs are not set
All bond coefficients must be set in the data file or by the bond_coeff command before running a
simulation.
All dihedral coeffs are not set
LAMMPS Users Manual
202
All dihedral coefficients must be set in the data file or by the dihedral_coeff command before running
a simulation.
All improper coeffs are not set
All improper coefficients must be set in the data file or by the improper_coeff command before
running a simulation.
All masses are not set
For atom styles that define masses for each atom type, all masses must be set in the data file or by the
mass command before running a simulation. They must also be set before using the velocity
command.
All mol IDs should be set for fix gcmc group atoms
The molecule flag is on, yet not all molecule ids in the fix group have been set to non-zero positive
values by the user. This is an error since all atoms in the fix gcmc group are eligible for deletion,
rotation, and translation and therefore must have valid molecule ids.
All pair coeffs are not set
All pair coefficients must be set in the data file or by the pair_coeff command before running a
simulation.
All read_dump x,y,z fields must be specified for scaled, triclinic coords
For triclinic boxes and scaled coordinates you must specify all 3 of the x,y,z fields, else LAMMPS
cannot reconstruct the unscaled coordinates.
All universe/uloop variables must have same # of values
Self-explanatory.
All variables in next command must be same style
Self-explanatory.
Angle atom missing in delete_bonds
The delete_bonds command cannot find one or more atoms in a particular angle on a particular
processor. The pairwise cutoff is too short or the atoms are too far apart to make a valid angle.
Angle atom missing in set command
The set command cannot find one or more atoms in a particular angle on a particular processor. The
pairwise cutoff is too short or the atoms are too far apart to make a valid angle.
Angle atoms %d %d %d missing on proc %d at step %ld
One or more of 3 atoms needed to compute a particular angle are missing on this processor. Typically
this is because the pairwise cutoff is set too short or the angle has blown apart and an atom is too far
away.
Angle atoms missing on proc %d at step %ld
One or more of 3 atoms needed to compute a particular angle are missing on this processor. Typically
this is because the pairwise cutoff is set too short or the angle has blown apart and an atom is too far
away.
Angle coeff for hybrid has invalid style
Angle style hybrid uses another angle style as one of its coefficients. The angle style used in the
angle_coeff command or read from a restart file is not recognized.
Angle coeffs are not set
No angle coefficients have been assigned in the data file or via the angle_coeff command.
Angle extent > half of periodic box length
This error was detected by the neigh_modify check yes setting. It is an error because the angle atoms
are so far apart it is ambiguous how it should be defined.
Angle potential must be defined for SHAKE
When shaking angles, an angle_style potential must be used.
Angle style hybrid cannot have hybrid as an argument
Self-explanatory.
Angle style hybrid cannot have none as an argument
Self-explanatory.
LAMMPS Users Manual
203
Angle style hybrid cannot use same angle style twice
Self-explanatory.
Angle table must range from 0 to 180 degrees
Self-explanatory.
Angle table parameters did not set N
List of angle table parameters must include N setting.
Angle_coeff command before angle_style is defined
Coefficients cannot be set in the data file or via the angle_coeff command until an angle_style has
been assigned.
Angle_coeff command before simulation box is defined
The angle_coeff command cannot be used before a read_data, read_restart, or create_box command.
Angle_coeff command when no angles allowed
The chosen atom style does not allow for angles to be defined.
Angle_style command when no angles allowed
The chosen atom style does not allow for angles to be defined.
Angles assigned incorrectly
Angles read in from the data file were not assigned correctly to atoms. This means there is something
invalid about the topology definitions.
Angles defined but no angle types
The data file header lists angles but no angle types.
Append boundary must be shrink/minimum
The boundary style of the face where atoms are added must be of type m (shrink/minimum).
Arccos of invalid value in variable formula
Argument of arccos() must be between -1 and 1.
Arcsin of invalid value in variable formula
Argument of arcsin() must be between -1 and 1.
Assigning body parameters to non-body atom
Self-explanatory.
Assigning ellipsoid parameters to non-ellipsoid atom
Self-explanatory.
Assigning line parameters to non-line atom
Self-explanatory.
Assigning quat to non-body atom
Self-explanatory.
Assigning tri parameters to non-tri atom
Self-explanatory.
At least one atom of each swapped type must be present to define charges.
Self-explanatory.
Atom IDs must be consecutive for velocity create loop all
Self-explanatory.
Atom IDs must be used for molecular systems
Atom IDs are used to identify and find partner atoms in bonds.
Atom count changed in fix neb
This is not allowed in a NEB calculation.
Atom count is inconsistent, cannot write data file
The sum of atoms across processors does not equal the global number of atoms. Probably some atoms
have been lost.
Atom count is inconsistent, cannot write restart file
Sum of atoms across processors does not equal initial total count. This is probably because you have
lost some atoms.
Atom in too many rigid bodies - boost MAXBODY
LAMMPS Users Manual
204
Fix poems has a parameter MAXBODY (in fix_poems.cpp) which determines the maximum number
of rigid bodies a single atom can belong to (i.e. a multibody joint). The bodies you have defined
exceed this limit.
Atom sort did not operate correctly
This is an internal LAMMPS error. Please report it to the developers.
Atom sorting has bin size = 0.0
The neighbor cutoff is being used as the bin size, but it is zero. Thus you must explicitly list a bin size
in the atom_modify sort command or turn off sorting.
Atom style hybrid cannot have hybrid as an argument
Self-explanatory.
Atom style hybrid cannot use same atom style twice
Self-explanatory.
Atom style template molecule must have atom types
The defined molecule(s) does not specify atom types.
Atom style was redefined after using fix property/atom
This is not allowed.
Atom type must be zero in fix gcmc mol command
Self-explanatory.
Atom vector in equal-style variable formula
Atom vectors generate one value per atom which is not allowed in an equal-style variable.
Atom-style variable in equal-style variable formula
Atom-style variables generate one value per atom which is not allowed in an equal-style variable.
Atom_modify id command after simulation box is defined
The atom_modify id command cannot be used after a read_data, read_restart, or create_box
command.
Atom_modify map command after simulation box is defined
The atom_modify map command cannot be used after a read_data, read_restart, or create_box
command.
Atom_modify sort and first options cannot be used together
Self-explanatory.
Atom_style command after simulation box is defined
The atom_style command cannot be used after a read_data, read_restart, or create_box command.
Atom_style line can only be used in 2d simulations
Self-explanatory.
Atom_style tri can only be used in 3d simulations
Self-explanatory.
Atomfile variable could not read values
Check the file assigned to the variable.
Atomfile variable in equal-style variable formula
Self-explanatory.
Atomfile-style variable in equal-style variable formula
Self-explanatory.
Attempt to pop empty stack in fix box/relax
Internal LAMMPS error. Please report it to the developers.
Attempt to push beyond stack limit in fix box/relax
Internal LAMMPS error. Please report it to the developers.
Attempting to rescale a 0.0 temperature
Cannot rescale a temperature that is already 0.0.
Bad FENE bond
Two atoms in a FENE bond have become so far apart that the bond cannot be computed.
Bad TIP4P angle type for PPPM/TIP4P
LAMMPS Users Manual
205
Specified angle type is not valid.
Bad TIP4P angle type for PPPMDisp/TIP4P
Specified angle type is not valid.
Bad TIP4P bond type for PPPM/TIP4P
Specified bond type is not valid.
Bad TIP4P bond type for PPPMDisp/TIP4P
Specified bond type is not valid.
Bad fix ID in fix append/atoms command
The value of the fix_id for keyword spatial must start with 'f_'.
Bad grid of processors
The 3d grid of processors defined by the processors command does not match the number of
processors LAMMPS is being run on.
Bad kspace_modify kmax/ewald parameter
Kspace_modify values for the kmax/ewald keyword must be integers > 0
Bad kspace_modify slab parameter
Kspace_modify value for the slab/volume keyword must be >= 2.0.
Bad matrix inversion in mldivide3
This error should not occur unless the matrix is badly formed.
Bad principal moments
Fix rigid did not compute the principal moments of inertia of a rigid group of atoms correctly.
Bad quadratic solve for particle/line collision
This is an internal error. It should normally not occur.
Bad quadratic solve for particle/tri collision
This is an internal error. It should normally not occur.
Bad real space Coulomb cutoff in fix tune/kspace
Fix tune/kspace tried to find the optimal real space Coulomb cutoff using the Newton-Rhaphson
method, but found a non-positive or NaN cutoff
Balance command before simulation box is defined
The balance command cannot be used before a read_data, read_restart, or create_box command.
Balance produced bad splits
This should not occur. It means two or more cutting plane locations are on top of each other or out of
order. Report the problem to the developers.
Balance rcb cannot be used with comm_style brick
Comm_style tiled must be used instead.
Balance shift string is invalid
The string can only contain the characters "x", "y", or "z".
Bias compute does not calculate a velocity bias
The specified compute must compute a bias for temperature.
Bias compute does not calculate temperature
The specified compute must compute temperature.
Bias compute group does not match compute group
The specified compute must operate on the same group as the parent compute.
Big particle in fix srd cannot be point particle
Big particles must be extended spheriods or ellipsoids.
Bigint setting in lmptype.h is invalid
Size of bigint is less than size of tagint.
Bigint setting in lmptype.h is not compatible
Format of bigint stored in restart file is not consistent with LAMMPS version you are running. See the
settings in src/lmptype.h
Bitmapped lookup tables require int/float be same size
LAMMPS Users Manual
206
Cannot use pair tables on this machine, because of word sizes. Use the pair_modify command with
table 0 instead.
Bitmapped table in file does not match requested table
Setting for bitmapped table in pair_coeff command must match table in file exactly.
Bitmapped table is incorrect length in table file
Number of table entries is not a correct power of 2.
Bond and angle potentials must be defined for TIP4P
Cannot use TIP4P pair potential unless bond and angle potentials are defined.
Bond atom missing in box size check
The 2nd atoms needed to compute a particular bond is missing on this processor. Typically this is
because the pairwise cutoff is set too short or the bond has blown apart and an atom is too far away.
Bond atom missing in delete_bonds
The delete_bonds command cannot find one or more atoms in a particular bond on a particular
processor. The pairwise cutoff is too short or the atoms are too far apart to make a valid bond.
Bond atom missing in image check
The 2nd atom in a particular bond is missing on this processor. Typically this is because the pairwise
cutoff is set too short or the bond has blown apart and an atom is too far away.
Bond atom missing in set command
The set command cannot find one or more atoms in a particular bond on a particular processor. The
pairwise cutoff is too short or the atoms are too far apart to make a valid bond.
Bond atoms %d %d missing on proc %d at step %ld
The 2nd atom needed to compute a particular bond is missing on this processor. Typically this is
because the pairwise cutoff is set too short or the bond has blown apart and an atom is too far away.
Bond atoms missing on proc %d at step %ld
The 2nd atom needed to compute a particular bond is missing on this processor. Typically this is
because the pairwise cutoff is set too short or the bond has blown apart and an atom is too far away.
Bond coeff for hybrid has invalid style
Bond style hybrid uses another bond style as one of its coefficients. The bond style used in the
bond_coeff command or read from a restart file is not recognized.
Bond coeffs are not set
No bond coefficients have been assigned in the data file or via the bond_coeff command.
Bond extent > half of periodic box length
This error was detected by the neigh_modify check yes setting. It is an error because the bond atoms
are so far apart it is ambiguous how it should be defined.
Bond potential must be defined for SHAKE
Cannot use fix shake unless bond potential is defined.
Bond style hybrid cannot have hybrid as an argument
Self-explanatory.
Bond style hybrid cannot have none as an argument
Self-explanatory.
Bond style hybrid cannot use same bond style twice
Self-explanatory.
Bond style quartic cannot be used with 3,4-body interactions
No angle, dihedral, or improper styles can be defined when using bond style quartic.
Bond style quartic cannot be used with atom style template
This bond style can change the bond topology which is not allowed with this atom style.
Bond style quartic requires special_bonds = 1,1,1
This is a restriction of the current bond quartic implementation.
Bond table parameters did not set N
List of bond table parameters must include N setting.
Bond table values are not increasing
LAMMPS Users Manual
207
The values in the tabulated file must be monotonically increasing.
BondAngle coeff for hybrid angle has invalid format
No "ba" field should appear in data file entry.
BondBond coeff for hybrid angle has invalid format
No "bb" field should appear in data file entry.
Bond_coeff command before bond_style is defined
Coefficients cannot be set in the data file or via the bond_coeff command until an bond_style has
been assigned.
Bond_coeff command before simulation box is defined
The bond_coeff command cannot be used before a read_data, read_restart, or create_box command.
Bond_coeff command when no bonds allowed
The chosen atom style does not allow for bonds to be defined.
Bond_style command when no bonds allowed
The chosen atom style does not allow for bonds to be defined.
Bonds assigned incorrectly
Bonds read in from the data file were not assigned correctly to atoms. This means there is something
invalid about the topology definitions.
Bonds defined but no bond types
The data file header lists bonds but no bond types.
Both restart files must use % or neither
Self-explanatory.
Both restart files must use MPI-IO or neither
Self-explanatory.
Both sides of boundary must be periodic
Cannot specify a boundary as periodic only on the lo or hi side. Must be periodic on both sides.
Boundary command after simulation box is defined
The boundary command cannot be used after a read_data, read_restart, or create_box command.
Box bounds are invalid
The box boundaries specified in the read_data file are invalid. The lo value must be less than the hi
value for all 3 dimensions.
Box command after simulation box is defined
The box command cannot be used after a read_data, read_restart, or create_box command.
CPU neighbor lists must be used for ellipsoid/sphere mix.
When using Gay-Berne or RE-squared pair styles with both ellipsoidal and spherical particles, the
neighbor list must be built on the CPU
Can not specify Pxy/Pxz/Pyz in fix box/relax with non-triclinic box
Only triclinic boxes can be used with off-diagonal pressure components. See the region prism
command for details.
Can not specify Pxy/Pxz/Pyz in fix nvt/npt/nph with non-triclinic box
Only triclinic boxes can be used with off-diagonal pressure components. See the region prism
command for details.
Can only use -plog with multiple partitions
Self-explanatory. See doc page discussion of command-line switches.
Can only use -pscreen with multiple partitions
Self-explanatory. See doc page discussion of command-line switches.
Can only use Kokkos supported regions with Kokkos package
Self-explanatory.
Can only use NEB with 1-processor replicas
This is current restriction for NEB as implemented in LAMMPS.
Can only use TAD with 1-processor replicas for NEB
This is current restriction for NEB as implemented in LAMMPS.
LAMMPS Users Manual
208
Cannot (yet) do analytic differentiation with pppm/gpu
This is a current restriction of this command.
Cannot (yet) request ghost atoms with Kokkos half neighbor list
This feature is not yet supported.
Cannot (yet) use 'electron' units with dipoles
This feature is not yet supported.
Cannot (yet) use Ewald with triclinic box and slab correction
This feature is not yet supported.
Cannot (yet) use K-space slab correction with compute group/group for triclinic systems
This option is not yet supported.
Cannot (yet) use MSM with 2d simulation
This feature is not yet supported.
Cannot (yet) use PPPM with triclinic box and TIP4P
This feature is not yet supported.
Cannot (yet) use PPPM with triclinic box and kspace_modify diff ad
This feature is not yet supported.
Cannot (yet) use PPPM with triclinic box and slab correction
This feature is not yet supported.
Cannot (yet) use kspace slab correction with long-range dipoles and non-neutral systems or per-atom energy
This feature is not yet supported.
Cannot (yet) use kspace_modify diff ad with compute group/group
This option is not yet supported.
Cannot (yet) use kspace_style pppm/stagger with triclinic systems
This feature is not yet supported.
Cannot (yet) use molecular templates with Kokkos
Self-explanatory.
Cannot (yet) use respa with Kokkos
Self-explanatory.
Cannot (yet) use rigid bodies with fix deform and Kokkos
Self-explanatory.
Cannot (yet) use rigid bodies with fix nh and Kokkos
Self-explanatory.
Cannot (yet) use single precision with MSM (remove -DFFT_SINGLE from Makefile and recompile)
Single precision cannot be used with MSM.
Cannot add atoms to fix move variable
Atoms can not be added afterwards to this fix option.
Cannot append atoms to a triclinic box
The simulation box must be defined with edges aligned with the Cartesian axes.
Cannot balance in z dimension for 2d simulation
Self-explanatory.
Cannot change box ortho/triclinic with certain fixes defined
This is because those fixes store the shape of the box. You need to use unfix to discard the fix, change
the box, then redefine a new fix.
Cannot change box ortho/triclinic with dumps defined
This is because some dumps store the shape of the box. You need to use undump to discard the dump,
change the box, then redefine a new dump.
Cannot change box tilt factors for orthogonal box
Cannot use tilt factors unless the simulation box is non-orthogonal.
Cannot change box to orthogonal when tilt is non-zero
Self-explanatory.
Cannot change box z boundary to nonperiodic for a 2d simulation
LAMMPS Users Manual
209
Self-explanatory.
Cannot change dump_modify every for dump dcd
The frequency of writing dump dcd snapshots cannot be changed.
Cannot change dump_modify every for dump xtc
The frequency of writing dump xtc snapshots cannot be changed.
Cannot change timestep once fix srd is setup
This is because various SRD properties depend on the timestep size.
Cannot change timestep with fix pour
This is because fix pour pre-computes the time delay for particles to fall out of the insertion volume
due to gravity.
Cannot change to comm_style brick from tiled layout
Self-explanatory.
Cannot change_box after reading restart file with per-atom info
This is because the restart file info cannot be migrated with the atoms. You can get around this by
performing a 0-timestep run which will assign the restart file info to actual atoms.
Cannot change_box in xz or yz for 2d simulation
Self-explanatory.
Cannot change_box in z dimension for 2d simulation
Self-explanatory.
Cannot clear group all
This operation is not allowed.
Cannot close restart file - MPI error: %s
This error was generated by MPI when reading/writing an MPI-IO restart file.
Cannot compute initial g_ewald_disp
LAMMPS failed to compute an initial guess for the PPPM_disp g_ewald_6 factor that partitions the
computation between real space and k-space for Dispersion interactions.
Cannot create an atom map unless atoms have IDs
The simulation requires a mapping from global atom IDs to local atoms, but the atoms that have been
defined have no IDs.
Cannot create atoms with undefined lattice
Must use the lattice command before using the create_atoms command.
Cannot create/grow a vector/array of pointers for %s
LAMMPS code is making an illegal call to the templated memory allocaters, to create a vector or
array of pointers.
Cannot create_atoms after reading restart file with per-atom info
The per-atom info was stored to be used when by a fix that you may re-define. If you add atoms
before re-defining the fix, then there will not be a correct amount of per-atom info.
Cannot create_box after simulation box is defined
A simulation box can only be defined once.
Cannot currently use pair reax with pair hybrid
This is not yet supported.
Cannot currently use pppm/gpu with fix balance.
Self-explanatory.
Cannot delete group all
Self-explanatory.
Cannot delete group currently used by a compute
Self-explanatory.
Cannot delete group currently used by a dump
Self-explanatory.
Cannot delete group currently used by a fix
Self-explanatory.
LAMMPS Users Manual
210
Cannot delete group currently used by atom_modify first
Self-explanatory.
Cannot delete_atoms bond yes for non-molecular systems
Self-explanatory.
Cannot displace_atoms after reading restart file with per-atom info
This is because the restart file info cannot be migrated with the atoms. You can get around this by
performing a 0-timestep run which will assign the restart file info to actual atoms.
Cannot do GCMC on atoms in atom_modify first group
This is a restriction due to the way atoms are organized in a list to enable the atom_modify first
command.
Cannot do atom/swap on atoms in atom_modify first group
This is a restriction due to the way atoms are organized in a list to enable the atom_modify first
command.
Cannot dump sort on atom IDs with no atom IDs defined
Self-explanatory.
Cannot dump sort when multiple dump files are written
In this mode, each processor dumps its atoms to a file, so no sorting is allowed.
Cannot embed Python when also extending Python with LAMMPS
When running LAMMPS via Python through the LAMMPS library interface you cannot also user the
input script python command.
Cannot evaporate atoms in atom_modify first group
This is a restriction due to the way atoms are organized in a list to enable the atom_modify first
command.
Cannot find create_bonds group ID
Self-explanatory.
Cannot find delete_bonds group ID
Group ID used in the delete_bonds command does not exist.
Cannot find specified group ID for core particles
Self-explanatory.
Cannot find specified group ID for shell particles
Self-explanatory.
Cannot have both pair_modify shift and tail set to yes
These 2 options are contradictory.
Cannot intersect groups using a dynamic group
This operation is not allowed.
Cannot mix molecular and molecule template atom styles
Self-explanatory.
Cannot open -reorder file
Self-explanatory.
Cannot open ADP potential file %s
The specified ADP potential file cannot be opened. Check that the path and name are correct.
Cannot open AIREBO potential file %s
The specified AIREBO potential file cannot be opened. Check that the path and name are correct.
Cannot open BOP potential file %s
The specified BOP potential file cannot be opened. Check that the path and name are correct.
Cannot open COMB potential file %s
The specified COMB potential file cannot be opened. Check that the path and name are correct.
Cannot open COMB3 lib.comb3 file
The COMB3 library file cannot be opened. Check that the path and name are correct.
Cannot open COMB3 potential file %s
The specified COMB3 potential file cannot be opened. Check that the path and name are correct.
LAMMPS Users Manual
211
Cannot open EAM potential file %s
The specified EAM potential file cannot be opened. Check that the path and name are correct.
Cannot open EIM potential file %s
The specified EIM potential file cannot be opened. Check that the path and name are correct.
Cannot open LCBOP potential file %s
The specified LCBOP potential file cannot be opened. Check that the path and name are correct.
Cannot open MEAM potential file %s
The specified MEAM potential file cannot be opened. Check that the path and name are correct.
Cannot open SNAP coefficient file %s
The specified SNAP coefficient file cannot be opened. Check that the path and name are correct.
Cannot open SNAP parameter file %s
The specified SNAP parameter file cannot be opened. Check that the path and name are correct.
Cannot open Stillinger-Weber potential file %s
The specified SW potential file cannot be opened. Check that the path and name are correct.
Cannot open Tersoff potential file %s
The specified potential file cannot be opened. Check that the path and name are correct.
Cannot open Vashishta potential file %s
The specified Vashishta potential file cannot be opened. Check that the path and name are correct.
Cannot open balance output file
Self-explanatory.
Cannot open coul/streitz potential file %s
The specified coul/streitz potential file cannot be opened. Check that the path and name are correct.
Cannot open custom file
Self-explanatory.
Cannot open data file %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open dir to search for restart file
Using a "*" in the name of the restart file will open the current directory to search for matching file
names.
Cannot open dump file
Self-explanatory.
Cannot open dump file %s
The output file for the dump command cannot be opened. Check that the path and name are correct.
Cannot open file %s
The specified file cannot be opened. Check that the path and name are correct. If the file is a
compressed file, also check that the gzip executable can be found and run.
Cannot open file variable file %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix ave/chunk file %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix ave/correlate file %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix ave/histo file %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix ave/spatial file %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix ave/time file %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix balance output file
Self-explanatory.
Cannot open fix poems file %s
LAMMPS Users Manual
212
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix print file %s
The output file generated by the fix print command cannot be opened
Cannot open fix qeq parameter file %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix qeq/comb file %s
The output file for the fix qeq/combs command cannot be opened. Check that the path and name are
correct.
Cannot open fix reax/bonds file %s
The output file for the fix reax/bonds command cannot be opened. Check that the path and name are
correct.
Cannot open fix rigid infile %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix rigid restart file %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix rigid/small infile %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open fix tmd file %s
The output file for the fix tmd command cannot be opened. Check that the path and name are correct.
Cannot open fix ttm file %s
The output file for the fix ttm command cannot be opened. Check that the path and name are correct.
Cannot open gzipped file
LAMMPS was compiled without support for reading and writing gzipped files through a pipeline to
the gzip program with -DLAMMPS_GZIP.
Cannot open input script %s
Self-explanatory.
Cannot open log.cite file
This file is created when you use some LAMMPS features, to indicate what paper you should cite on
behalf of those who implemented the feature. Check that you have write privileges into the directory
you are running in.
Cannot open log.lammps for writing
The default LAMMPS log file cannot be opened. Check that the directory you are running in allows
for files to be created.
Cannot open logfile
The LAMMPS log file named in a command-line argument cannot be opened. Check that the path
and name are correct.
Cannot open logfile %s
The LAMMPS log file specified in the input script cannot be opened. Check that the path and name
are correct.
Cannot open molecule file %s
The specified file cannot be opened. Check that the path and name are correct.
Cannot open nb3b/harmonic potential file %s
The specified potential file cannot be opened. Check that the path and name are correct.
Cannot open pair_write file
The specified output file for pair energies and forces cannot be opened. Check that the path and name
are correct.
Cannot open polymorphic potential file %s
The specified polymorphic potential file cannot be opened. Check that the path and name are correct.
Cannot open print file %s
Self-explanatory.
Cannot open processors output file
LAMMPS Users Manual
213
Self-explanatory.
Cannot open restart file %s
Self-explanatory.
Cannot open restart file for reading - MPI error: %s
This error was generated by MPI when reading/writing an MPI-IO restart file.
Cannot open restart file for writing - MPI error: %s
This error was generated by MPI when reading/writing an MPI-IO restart file.
Cannot open screen file
The screen file specified as a command-line argument cannot be opened. Check that the directory you
are running in allows for files to be created.
Cannot open temporary file for world counter.
Self-explanatory.
Cannot open universe log file
For a multi-partition run, the master log file cannot be opened. Check that the directory you are
running in allows for files to be created.
Cannot open universe screen file
For a multi-partition run, the master screen file cannot be opened. Check that the directory you are
running in allows for files to be created.
Cannot read from restart file - MPI error: %s
This error was generated by MPI when reading/writing an MPI-IO restart file.
Cannot read_data without add keyword after simulation box is defined
Self-explanatory.
Cannot read_restart after simulation box is defined
The read_restart command cannot be used after a read_data, read_restart, or create_box command.
Cannot redefine variable as a different style
An equal-style variable can be re-defined but only if it was originally an equal-style variable.
Cannot replicate 2d simulation in z dimension
The replicate command cannot replicate a 2d simulation in the z dimension.
Cannot replicate with fixes that store atom quantities
Either fixes are defined that create and store atom-based vectors or a restart file was read which
included atom-based vectors for fixes. The replicate command cannot duplicate that information for
new atoms. You should use the replicate command before fixes are applied to the system.
Cannot reset timestep with a dynamic region defined
Dynamic regions (see the region command) have a time dependence. Thus you cannot change the
timestep when one or more of these are defined.
Cannot reset timestep with a time-dependent fix defined
You cannot reset the timestep when a fix that keeps track of elapsed time is in place.
Cannot run 2d simulation with nonperiodic Z dimension
Use the boundary command to make the z dimension periodic in order to run a 2d simulation.
Cannot set bond topology types for atom style template
The bond, angle, etc types cannot be changed for this atom style since they are static settings in the
molecule template files.
Cannot set both respa pair and inner/middle/outer
In the rRESPA integrator, you must compute pairwise potentials either all together (pair), or in pieces
(inner/middle/outer). You can't do both.
Cannot set cutoff/multi before simulation box is defined
Self-explanatory.
Cannot set dpd/theta for this atom style
Self-explanatory.
Cannot set dump_modify flush for dump xtc
Self-explanatory.
LAMMPS Users Manual
214
Cannot set mass for this atom style
This atom style does not support mass settings for each atom type. Instead they are defined on a
per-atom basis in the data file.
Cannot set meso/cv for this atom style
Self-explanatory.
Cannot set meso/e for this atom style
Self-explanatory.
Cannot set meso/rho for this atom style
Self-explanatory.
Cannot set non-zero image flag for non-periodic dimension
Self-explanatory.
Cannot set non-zero z velocity for 2d simulation
Self-explanatory.
Cannot set quaternion for atom that has none
Self-explanatory.
Cannot set quaternion with xy components for 2d system
Self-explanatory.
Cannot set respa hybrid and any of pair/inner/middle/outer
In the rRESPA integrator, you must compute pairwise potentials either all together (pair), with
different cutoff regions (inner/middle/outer), or per hybrid sub-style (hybrid). You cannot mix those.
Cannot set respa middle without inner/outer
In the rRESPA integrator, you must define both a inner and outer setting in order to use a middle
setting.
Cannot set restart file size - MPI error: %s
This error was generated by MPI when reading/writing an MPI-IO restart file.
Cannot set smd/contact/radius for this atom style
Self-explanatory.
Cannot set smd/mass/density for this atom style
Self-explanatory.
Cannot set temperature for fix rigid/nph
The temp keyword cannot be specified.
Cannot set theta for atom that is not a line
Self-explanatory.
Cannot set this attribute for this atom style
The attribute being set does not exist for the defined atom style.
Cannot set variable z velocity for 2d simulation
Self-explanatory.
Cannot skew triclinic box in z for 2d simulation
Self-explanatory.
Cannot subtract groups using a dynamic group
This operation is not allowed.
Cannot union groups using a dynamic group
This operation is not allowed.
Cannot use -cuda on and -kokkos on together
This is not allowed since both packages can use GPUs.
Cannot use -cuda on without USER-CUDA installed
The USER-CUDA package must be installed via "make yes-user-cuda" before LAMMPS is built.
Cannot use -kokkos on without KOKKOS installed
Self-explanatory.
Cannot use -reorder after -partition
Self-explanatory. See doc page discussion of command-line switches.
LAMMPS Users Manual
215
Cannot use Ewald with 2d simulation
The kspace style ewald cannot be used in 2d simulations. You can use 2d Ewald in a 3d simulation;
see the kspace_modify command.
Cannot use Ewald/disp solver on system with no charge, dipole, or LJ particles
No atoms in system have a non-zero charge or dipole, or are LJ particles. Change charges/dipoles or
change options of the kspace solver/pair style.
Cannot use EwaldDisp with 2d simulation
This is a current restriction of this command.
Cannot use GPU package with USER-CUDA package enabled
You cannot use both the GPU and USER-CUDA packages together. Use one or the other.
Cannot use Kokkos pair style with rRESPA inner/middle
Self-explanatory.
Cannot use NEB unless atom map exists
Use the atom_modify command to create an atom map.
Cannot use NEB with a single replica
Self-explanatory.
Cannot use NEB with atom_modify sort enabled
This is current restriction for NEB implemented in LAMMPS.
Cannot use PPPM with 2d simulation
The kspace style pppm cannot be used in 2d simulations. You can use 2d PPPM in a 3d simulation;
see the kspace_modify command.
Cannot use PPPMDisp with 2d simulation
The kspace style pppm/disp cannot be used in 2d simulations. You can use 2d pppm/disp in a 3d
simulation; see the kspace_modify command.
Cannot use PRD with a changing box
The current box dimensions are not copied between replicas
Cannot use PRD with a time-dependent fix defined
PRD alters the timestep in ways that will mess up these fixes.
Cannot use PRD with a time-dependent region defined
PRD alters the timestep in ways that will mess up these regions.
Cannot use PRD with atom_modify sort enabled
This is a current restriction of PRD. You must turn off sorting, which is enabled by default, via the
atom_modify command.
Cannot use PRD with multi-processor replicas unless atom map exists
Use the atom_modify command to create an atom map.
Cannot use TAD unless atom map exists for NEB
See atom_modify map command to set this.
Cannot use TAD with a single replica for NEB
NEB requires multiple replicas.
Cannot use TAD with atom_modify sort enabled for NEB
This is a current restriction of NEB.
Cannot use a damped dynamics min style with fix box/relax
This is a current restriction in LAMMPS. Use another minimizer style.
Cannot use a damped dynamics min style with per-atom DOF
This is a current restriction in LAMMPS. Use another minimizer style.
Cannot use append/atoms in periodic dimension
The boundary style of the face where atoms are added can not be of type p (periodic).
Cannot use atomfile-style variable unless atom map exists
Self-explanatory. See the atom_modify command to create a map.
Cannot use both com and bias with compute temp/chunk
Self-explanatory.
LAMMPS Users Manual
216
Cannot use chosen neighbor list style with buck/coul/cut/kk
Self-explanatory.
Cannot use chosen neighbor list style with buck/coul/long/kk
Self-explanatory.
Cannot use chosen neighbor list style with buck/kk
That style is not supported by Kokkos.
Cannot use chosen neighbor list style with coul/cut/kk
That style is not supported by Kokkos.
Cannot use chosen neighbor list style with coul/debye/kk
Self-explanatory.
Cannot use chosen neighbor list style with coul/dsf/kk
That style is not supported by Kokkos.
Cannot use chosen neighbor list style with coul/wolf/kk
That style is not supported by Kokkos.
Cannot use chosen neighbor list style with lj/charmm/coul/charmm/implicit/kk
Self-explanatory.
Cannot use chosen neighbor list style with lj/charmm/coul/charmm/kk
Self-explanatory.
Cannot use chosen neighbor list style with lj/charmm/coul/long/kk
Self-explanatory.
Cannot use chosen neighbor list style with lj/class2/coul/cut/kk
Self-explanatory.
Cannot use chosen neighbor list style with lj/class2/coul/long/kk
Self-explanatory.
Cannot use chosen neighbor list style with lj/class2/kk
Self-explanatory.
Cannot use chosen neighbor list style with lj/cut/coul/cut/kk
That style is not supported by Kokkos.
Cannot use chosen neighbor list style with lj/cut/coul/debye/kk
Self-explanatory.
Cannot use chosen neighbor list style with lj/cut/coul/long/kk
That style is not supported by Kokkos.
Cannot use chosen neighbor list style with lj/cut/kk
That style is not supported by Kokkos.
Cannot use chosen neighbor list style with lj/expand/kk
Self-explanatory.
Cannot use chosen neighbor list style with lj/gromacs/coul/gromacs/kk
Self-explanatory.
Cannot use chosen neighbor list style with lj/gromacs/kk
Self-explanatory.
Cannot use chosen neighbor list style with lj/sdk/kk
That style is not supported by Kokkos.
Cannot use chosen neighbor list style with pair eam/kk
That style is not supported by Kokkos.
Cannot use chosen neighbor list style with pair eam/kk/alloy
Self-explanatory.
Cannot use chosen neighbor list style with pair eam/kk/fs
Self-explanatory.
Cannot use chosen neighbor list style with pair sw/kk
Self-explanatory.
Cannot use chosen neighbor list style with tersoff/kk
LAMMPS Users Manual
217
Self-explanatory.
Cannot use chosen neighbor list style with tersoff/zbl/kk
Self-explanatory.
Cannot use compute chunk/atom bin z for 2d model
Self-explanatory.
Cannot use compute cluster/atom unless atoms have IDs
Atom IDs are used to identify clusters.
Cannot use create_atoms rotate unless single style
Self-explanatory.
Cannot use create_bonds unless atoms have IDs
This command requires a mapping from global atom IDs to local atoms, but the atoms that have been
defined have no IDs.
Cannot use create_bonds with non-molecular system
Self-explanatory.
Cannot use cwiggle in variable formula between runs
This is a function of elapsed time.
Cannot use delete_atoms bond yes with atom_style template
This is because the bonds for that atom style are hardwired in the molecule template.
Cannot use delete_atoms unless atoms have IDs
Your atoms do not have IDs, so the delete_atoms command cannot be used.
Cannot use delete_bonds with non-molecular system
Your choice of atom style does not have bonds.
Cannot use dump_modify fileper without % in dump file name
Self-explanatory.
Cannot use dump_modify nfile without % in dump file name
Self-explanatory.
Cannot use dynamic group with fix adapt atom
This is not yet supported.
Cannot use fix TMD unless atom map exists
Using this fix requires the ability to lookup an atom index, which is provided by an atom map. An
atom map does not exist (by default) for non-molecular problems. Using the atom_modify map
command will force an atom map to be created.
Cannot use fix ave/spatial z for 2 dimensional model
Self-explanatory.
Cannot use fix bond/break with non-molecular systems
Only systems with bonds that can be changed can be used. Atom_style template does not qualify.
Cannot use fix bond/create with non-molecular systems
Only systems with bonds that can be changed can be used. Atom_style template does not qualify.
Cannot use fix bond/swap with non-molecular systems
Only systems with bonds that can be changed can be used. Atom_style template does not qualify.
Cannot use fix box/relax on a 2nd non-periodic dimension
When specifying an off-diagonal pressure component, the 2nd of the two dimensions must be
periodic. E.g. if the xy component is specified, then the y dimension must be periodic.
Cannot use fix box/relax on a non-periodic dimension
When specifying a diagonal pressure component, the dimension must be periodic.
Cannot use fix box/relax with both relaxation and scaling on a tilt factor
When specifying scaling on a tilt factor component, that component can not also be controlled by the
barostat. E.g. if scalexy yes is specified and also keyword tri or xy, this is wrong.
Cannot use fix box/relax with tilt factor scaling on a 2nd non-periodic dimension
When specifying scaling on a tilt factor component, the 2nd of the two dimensions must be periodic.
E.g. if the xy component is specified, then the y dimension must be periodic.
LAMMPS Users Manual
218
Cannot use fix deform on a shrink-wrapped boundary
The x, y, z options cannot be applied to shrink-wrapped dimensions.
Cannot use fix deform tilt on a shrink-wrapped 2nd dim
This is because the shrink-wrapping will change the value of the strain implied by the tilt factor.
Cannot use fix deform trate on a box with zero tilt
The trate style alters the current strain.
Cannot use fix deposit rigid and not molecule
Self-explanatory.
Cannot use fix deposit rigid and shake
These two attributes are conflicting.
Cannot use fix deposit shake and not molecule
Self-explanatory.
Cannot use fix enforce2d with 3d simulation
Self-explanatory.
Cannot use fix gcmc in a 2d simulation
Fix gcmc is set up to run in 3d only. No 2d simulations with fix gcmc are allowed.
Cannot use fix gcmc shake and not molecule
Self-explanatory.
Cannot use fix msst without per-type mass defined
Self-explanatory.
Cannot use fix npt and fix deform on same component of stress tensor
This would be changing the same box dimension twice.
Cannot use fix nvt/npt/nph on a 2nd non-periodic dimension
When specifying an off-diagonal pressure component, the 2nd of the two dimensions must be
periodic. E.g. if the xy component is specified, then the y dimension must be periodic.
Cannot use fix nvt/npt/nph on a non-periodic dimension
When specifying a diagonal pressure component, the dimension must be periodic.
Cannot use fix nvt/npt/nph with both xy dynamics and xy scaling
Self-explanatory.
Cannot use fix nvt/npt/nph with both xz dynamics and xz scaling
Self-explanatory.
Cannot use fix nvt/npt/nph with both yz dynamics and yz scaling
Self-explanatory.
Cannot use fix nvt/npt/nph with xy scaling when y is non-periodic dimension
The 2nd dimension in the barostatted tilt factor must be periodic.
Cannot use fix nvt/npt/nph with xz scaling when z is non-periodic dimension
The 2nd dimension in the barostatted tilt factor must be periodic.
Cannot use fix nvt/npt/nph with yz scaling when z is non-periodic dimension
The 2nd dimension in the barostatted tilt factor must be periodic.
Cannot use fix pour rigid and not molecule
Self-explanatory.
Cannot use fix pour rigid and shake
These two attributes are conflicting.
Cannot use fix pour shake and not molecule
Self-explanatory.
Cannot use fix pour with triclinic box
This option is not yet supported.
Cannot use fix press/berendsen and fix deform on same component of stress tensor
These commands both change the box size/shape, so you cannot use both together.
Cannot use fix press/berendsen on a non-periodic dimension
Self-explanatory.
LAMMPS Users Manual
219
Cannot use fix press/berendsen with triclinic box
Self-explanatory.
Cannot use fix reax/bonds without pair_style reax
Self-explanatory.
Cannot use fix rigid npt/nph and fix deform on same component of stress tensor
This would be changing the same box dimension twice.
Cannot use fix rigid npt/nph on a non-periodic dimension
When specifying a diagonal pressure component, the dimension must be periodic.
Cannot use fix rigid/small npt/nph on a non-periodic dimension
When specifying a diagonal pressure component, the dimension must be periodic.
Cannot use fix shake with non-molecular system
Your choice of atom style does not have bonds.
Cannot use fix ttm with 2d simulation
This is a current restriction of this fix due to the grid it creates.
Cannot use fix ttm with triclinic box
This is a current restriction of this fix due to the grid it creates.
Cannot use fix tune/kspace without a kspace style
Self-explanatory.
Cannot use fix tune/kspace without a pair style
This fix (tune/kspace) can only be used when a pair style has been specified.
Cannot use fix wall in periodic dimension
Self-explanatory.
Cannot use fix wall zlo/zhi for a 2d simulation
Self-explanatory.
Cannot use fix wall/reflect in periodic dimension
Self-explanatory.
Cannot use fix wall/reflect zlo/zhi for a 2d simulation
Self-explanatory.
Cannot use fix wall/srd in periodic dimension
Self-explanatory.
Cannot use fix wall/srd more than once
Nor is their a need to since multiple walls can be specified in one command.
Cannot use fix wall/srd without fix srd
Self-explanatory.
Cannot use fix wall/srd zlo/zhi for a 2d simulation
Self-explanatory.
Cannot use fix_deposit unless atoms have IDs
Self-explanatory.
Cannot use fix_pour unless atoms have IDs
Self-explanatory.
Cannot use include command within an if command
Self-explanatory.
Cannot use lines with fix srd unless overlap is set
This is because line segments are connected to each other.
Cannot use multiple fix wall commands with pair brownian
Self-explanatory.
Cannot use multiple fix wall commands with pair lubricate
Self-explanatory.
Cannot use multiple fix wall commands with pair lubricate/poly
Self-explanatory.
Cannot use multiple fix wall commands with pair lubricateU
LAMMPS Users Manual
220
Self-explanatory.
Cannot use neigh_modify exclude with GPU neighbor builds
This is a current limitation of the GPU implementation in LAMMPS.
Cannot use neighbor bins - box size << cutoff
Too many neighbor bins will be created. This typically happens when the simulation box is very small
in some dimension, compared to the neighbor cutoff. Use the "nsq" style instead of "bin" style.
Cannot use newton pair with beck/gpu pair style
Self-explanatory.
Cannot use newton pair with born/coul/long/gpu pair style
Self-explanatory.
Cannot use newton pair with born/coul/wolf/gpu pair style
Self-explanatory.
Cannot use newton pair with born/gpu pair style
Self-explanatory.
Cannot use newton pair with buck/coul/cut/gpu pair style
Self-explanatory.
Cannot use newton pair with buck/coul/long/gpu pair style
Self-explanatory.
Cannot use newton pair with buck/gpu pair style
Self-explanatory.
Cannot use newton pair with colloid/gpu pair style
Self-explanatory.
Cannot use newton pair with coul/cut/gpu pair style
Self-explanatory.
Cannot use newton pair with coul/debye/gpu pair style
Self-explanatory.
Cannot use newton pair with coul/dsf/gpu pair style
Self-explanatory.
Cannot use newton pair with coul/long/gpu pair style
Self-explanatory.
Cannot use newton pair with dipole/cut/gpu pair style
Self-explanatory.
Cannot use newton pair with dipole/sf/gpu pair style
Self-explanatory.
Cannot use newton pair with dpd/gpu pair style
Self-explanatory.
Cannot use newton pair with dpd/tstat/gpu pair style
Self-explanatory.
Cannot use newton pair with eam/alloy/gpu pair style
Self-explanatory.
Cannot use newton pair with eam/fs/gpu pair style
Self-explanatory.
Cannot use newton pair with eam/gpu pair style
Self-explanatory.
Cannot use newton pair with gauss/gpu pair style
Self-explanatory.
Cannot use newton pair with gayberne/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/charmm/coul/long/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/class2/coul/long/gpu pair style
LAMMPS Users Manual
221
Self-explanatory.
Cannot use newton pair with lj/class2/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/cubic/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/cut/coul/cut/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/cut/coul/debye/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/cut/coul/dsf/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/cut/coul/long/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/cut/coul/msm/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/cut/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/expand/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/gromacs/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/sdk/coul/long/gpu pair style
Self-explanatory.
Cannot use newton pair with lj/sdk/gpu pair style
Self-explanatory.
Cannot use newton pair with lj96/cut/gpu pair style
Self-explanatory.
Cannot use newton pair with mie/cut/gpu pair style
Self-explanatory.
Cannot use newton pair with morse/gpu pair style
Self-explanatory.
Cannot use newton pair with resquared/gpu pair style
Self-explanatory.
Cannot use newton pair with soft/gpu pair style
Self-explanatory.
Cannot use newton pair with table/gpu pair style
Self-explanatory.
Cannot use newton pair with yukawa/colloid/gpu pair style
Self-explanatory.
Cannot use newton pair with yukawa/gpu pair style
Self-explanatory.
Cannot use newton pair with zbl/gpu pair style
Self-explanatory.
Cannot use non-zero forces in an energy minimization
Fix setforce cannot be used in this manner. Use fix addforce instead.
Cannot use nonperiodic boundares with fix ttm
This fix requires a fully periodic simulation box.
Cannot use nonperiodic boundaries with Ewald
For kspace style ewald, all 3 dimensions must have periodic boundaries unless you use the
kspace_modify command to define a 2d slab with a non-periodic z dimension.
Cannot use nonperiodic boundaries with EwaldDisp
LAMMPS Users Manual
222
For kspace style ewald/disp, all 3 dimensions must have periodic boundaries unless you use the
kspace_modify command to define a 2d slab with a non-periodic z dimension.
Cannot use nonperiodic boundaries with PPPM
For kspace style pppm, all 3 dimensions must have periodic boundaries unless you use the
kspace_modify command to define a 2d slab with a non-periodic z dimension.
Cannot use nonperiodic boundaries with PPPMDisp
For kspace style pppm/disp, all 3 dimensions must have periodic boundaries unless you use the
kspace_modify command to define a 2d slab with a non-periodic z dimension.
Cannot use order greater than 8 with pppm/gpu.
Self-explanatory.
Cannot use package gpu neigh yes with triclinic box
This is a current restriction in LAMMPS.
Cannot use pair hybrid with GPU neighbor list builds
Neighbor list builds must be done on the CPU for this pair style.
Cannot use pair tail corrections with 2d simulations
The correction factors are only currently defined for 3d systems.
Cannot use processors part command without using partitions
See the command-line -partition switch.
Cannot use ramp in variable formula between runs
This is because the ramp() function is time dependent.
Cannot use read_data add before simulation box is defined
Self-explanatory.
Cannot use read_data extra with add flag
Self-explanatory.
Cannot use read_data offset without add flag
Self-explanatory.
Cannot use read_data shift without add flag
Self-explanatory.
Cannot use region INF or EDGE when box does not exist
Regions that extend to the box boundaries can only be used after the create_box command has been
used.
Cannot use set atom with no atom IDs defined
Atom IDs are not defined, so they cannot be used to identify an atom.
Cannot use set mol with no molecule IDs defined
Self-explanatory.
Cannot use swiggle in variable formula between runs
This is a function of elapsed time.
Cannot use tris with fix srd unless overlap is set
This is because triangles are connected to each other.
Cannot use variable energy with constant efield in fix efield
LAMMPS computes the energy itself when the E-field is constant.
Cannot use variable energy with constant force in fix addforce
This is because for constant force, LAMMPS can compute the change in energy directly.
Cannot use variable every setting for dump dcd
The format of DCD dump files requires snapshots be output at a constant frequency.
Cannot use variable every setting for dump xtc
The format of this file requires snapshots at regular intervals.
Cannot use vdisplace in variable formula between runs
This is a function of elapsed time.
Cannot use velocity bias command without temp keyword
Self-explanatory.
LAMMPS Users Manual
223
Cannot use velocity create loop all unless atoms have IDs
Atoms in the simulation to do not have IDs, so this style of velocity creation cannot be performed.
Cannot use wall in periodic dimension
Self-explanatory.
Cannot use write_restart fileper without % in restart file name
Self-explanatory.
Cannot use write_restart nfile without % in restart file name
Self-explanatory.
Cannot wiggle and shear fix wall/gran
Cannot specify both options at the same time.
Cannot write to restart file - MPI error: %s
This error was generated by MPI when reading/writing an MPI-IO restart file.
Cannot yet use KSpace solver with grid with comm style tiled
This is current restriction in LAMMPS.
Cannot yet use comm_style tiled with multi-mode comm
Self-explanatory.
Cannot yet use comm_style tiled with triclinic box
Self-explanatory.
Cannot yet use compute tally with Kokkos
This feature is not yet supported.
Cannot yet use fix bond/break with this improper style
This is a current restriction in LAMMPS.
Cannot yet use fix bond/create with this improper style
This is a current restriction in LAMMPS.
Cannot yet use minimize with Kokkos
This feature is not yet supported.
Cannot yet use pair hybrid with Kokkos
This feature is not yet supported.
Cannot zero Langevin force of 0 atoms
The group has zero atoms, so you cannot request its force be zeroed.
Cannot zero gld force for zero atoms
There are no atoms currently in the group.
Cannot zero momentum of no atoms
Self-explanatory.
Change_box command before simulation box is defined
Self-explanatory.
Change_box volume used incorrectly
The "dim volume" option must be used immediately following one or two settings for "dim1 ..." (and
optionally "dim2 ...") and must be for a different dimension, i.e. dim != dim1 and dim != dim2.
Chunk/atom compute does not exist for compute angmom/chunk
Self-explanatory.
Chunk/atom compute does not exist for compute com/chunk
Self-explanatory.
Chunk/atom compute does not exist for compute gyration/chunk
Self-explanatory.
Chunk/atom compute does not exist for compute inertia/chunk
Self-explanatory.
Chunk/atom compute does not exist for compute msd/chunk
Self-explanatory.
Chunk/atom compute does not exist for compute omega/chunk
Self-explanatory.
LAMMPS Users Manual
224
Chunk/atom compute does not exist for compute property/chunk
Self-explanatory.
Chunk/atom compute does not exist for compute temp/chunk
Self-explanatory.
Chunk/atom compute does not exist for compute torque/chunk
Self-explanatory.
Chunk/atom compute does not exist for compute vcm/chunk
Self-explanatory.
Chunk/atom compute does not exist for fix ave/chunk
Self-explanatory.
Comm tiled invalid index in box drop brick
Internal error check in comm_style tiled which should not occur. Contact the developers.
Comm tiled mis-match in box drop brick
Internal error check in comm_style tiled which should not occur. Contact the developers.
Comm_modify group != atom_modify first group
Self-explanatory.
Communication cutoff for comm_style tiled cannot exceed periodic box length
Self-explanatory.
Communication cutoff too small for SNAP micro load balancing
This can happen if you change the neighbor skin after your pair_style command or if your box
dimensions grow during a run. You can set the cutoff explicitly via the comm_modify cutoff
command.
Compute %s does not allow use of dynamic group
Dynamic groups have not yet been enabled for this compute.
Compute ID for compute chunk /atom does not exist
Self-explanatory.
Compute ID for compute chunk/atom does not exist
Self-explanatory.
Compute ID for compute reduce does not exist
Self-explanatory.
Compute ID for compute slice does not exist
Self-explanatory.
Compute ID for fix ave/atom does not exist
Self-explanatory.
Compute ID for fix ave/chunk does not exist
Self-explanatory.
Compute ID for fix ave/correlate does not exist
Self-explanatory.
Compute ID for fix ave/histo does not exist
Self-explanatory.
Compute ID for fix ave/spatial does not exist
Self-explanatory.
Compute ID for fix ave/time does not exist
Self-explanatory.
Compute ID for fix store/state does not exist
Self-explanatory.
Compute ID for fix vector does not exist
Self-explanatory.
Compute ID must be alphanumeric or underscore characters
Self-explanatory.
Compute angle/local used when angles are not allowed
LAMMPS Users Manual
225
The atom style does not support angles.
Compute angmom/chunk does not use chunk/atom compute
The style of the specified compute is not chunk/atom.
Compute body/local requires atom style body
Self-explanatory.
Compute bond/local used when bonds are not allowed
The atom style does not support bonds.
Compute centro/atom requires a pair style be defined
This is because the computation of the centro-symmetry values uses a pairwise neighbor list.
Compute chunk/atom bin/cylinder radius is too large for periodic box
Radius cannot be bigger than 1/2 of a non-axis periodic dimension.
Compute chunk/atom bin/sphere radius is too large for periodic box
Radius cannot be bigger than 1/2 of any periodic dimension.
Compute chunk/atom compute array is accessed out-of-range
The index for the array is out of bounds.
Compute chunk/atom compute does not calculate a per-atom array
Self-explanatory.
Compute chunk/atom compute does not calculate a per-atom vector
Self-explanatory.
Compute chunk/atom compute does not calculate per-atom values
Self-explanatory.
Compute chunk/atom cylinder axis must be z for 2d
Self-explanatory.
Compute chunk/atom fix array is accessed out-of-range
the index for the array is out of bounds.
Compute chunk/atom fix does not calculate a per-atom array
Self-explanatory.
Compute chunk/atom fix does not calculate a per-atom vector
Self-explanatory.
Compute chunk/atom fix does not calculate per-atom values
Self-explanatory.
Compute chunk/atom for triclinic boxes requires units reduced
Self-explanatory.
Compute chunk/atom ids once but nchunk is not once
You cannot assign chunks IDs to atom permanently if the number of chunks may change.
Compute chunk/atom molecule for non-molecular system
Self-explanatory.
Compute chunk/atom sphere z origin must be 0.0 for 2d
Self-explanatory.
Compute chunk/atom stores no IDs for compute property/chunk
It will only store IDs if its compress option is enabled.
Compute chunk/atom stores no coord1 for compute property/chunk
Only certain binning options for compute chunk/atom store coordinates.
Compute chunk/atom stores no coord2 for compute property/chunk
Only certain binning options for compute chunk/atom store coordinates.
Compute chunk/atom stores no coord3 for compute property/chunk
Only certain binning options for compute chunk/atom store coordinates.
Compute chunk/atom variable is not atom-style variable
Self-explanatory.
Compute chunk/atom without bins cannot use discard mixed
That discard option only applies to the binning styles.
LAMMPS Users Manual
226
Compute cluster/atom cutoff is longer than pairwise cutoff
Cannot identify clusters beyond cutoff.
Compute cluster/atom requires a pair style be defined
This is so that the pair style defines a cutoff distance which is used to find clusters.
Compute cna/atom cutoff is longer than pairwise cutoff
Self-explanatory.
Compute cna/atom requires a pair style be defined
Self-explanatory.
Compute com/chunk does not use chunk/atom compute
The style of the specified compute is not chunk/atom.
Compute contact/atom requires a pair style be defined
Self-explanatory.
Compute contact/atom requires atom style sphere
Self-explanatory.
Compute coord/atom cutoff is longer than pairwise cutoff
Cannot compute coordination at distances longer than the pair cutoff, since those atoms are not in the
neighbor list.
Compute coord/atom requires a pair style be defined
Self-explanatory.
Compute damage/atom requires peridynamic potential
Damage is a Peridynamic-specific metric. It requires you to be running a Peridynamics simulation.
Compute dihedral/local used when dihedrals are not allowed
The atom style does not support dihedrals.
Compute dilatation/atom cannot be used with this pair style
Self-explanatory.
Compute dilatation/atom requires Peridynamic pair style
Self-explanatory.
Compute does not allow an extra compute or fix to be reset
This is an internal LAMMPS error. Please report it to the developers.
Compute erotate/asphere requires atom style ellipsoid or line or tri
Self-explanatory.
Compute erotate/asphere requires extended particles
This compute cannot be used with point particles.
Compute erotate/rigid with non-rigid fix-ID
Self-explanatory.
Compute erotate/sphere requires atom style sphere
Self-explanatory.
Compute erotate/sphere/atom requires atom style sphere
Self-explanatory.
Compute event/displace has invalid fix event assigned
This is an internal LAMMPS error. Please report it to the developers.
Compute group/group group ID does not exist
Self-explanatory.
Compute gyration/chunk does not use chunk/atom compute
The style of the specified compute is not chunk/atom.
Compute heat/flux compute ID does not compute ke/atom
Self-explanatory.
Compute heat/flux compute ID does not compute pe/atom
Self-explanatory.
Compute heat/flux compute ID does not compute stress/atom
Self-explanatory.
LAMMPS Users Manual
227
Compute hexorder/atom cutoff is longer than pairwise cutoff
Cannot compute order parameter beyond cutoff.
Compute hexorder/atom requires a pair style be defined
Self-explanatory.
Compute improper/local used when impropers are not allowed
The atom style does not support impropers.
Compute inertia/chunk does not use chunk/atom compute
The style of the specified compute is not chunk/atom.
Compute ke/rigid with non-rigid fix-ID
Self-explanatory.
Compute msd/chunk does not use chunk/atom compute
The style of the specified compute is not chunk/atom.
Compute msd/chunk nchunk is not static
This is required because the MSD cannot be computed consistently if the number of chunks is
changing. Compute chunk/atom allows setting nchunk to be static.
Compute nve/asphere requires atom style ellipsoid
Self-explanatory.
Compute nvt/nph/npt asphere requires atom style ellipsoid
Self-explanatory.
Compute nvt/nph/npt body requires atom style body
Self-explanatory.
Compute omega/chunk does not use chunk/atom compute
The style of the specified compute is not chunk/atom.
Compute orientorder/atom cutoff is longer than pairwise cutoff
Cannot compute order parameter beyond cutoff.
Compute orientorder/atom requires a pair style be defined
Self-explanatory.
Compute pair must use group all
Pair styles accumulate energy on all atoms.
Compute pe must use group all
Energies computed by potentials (pair, bond, etc) are computed on all atoms.
Compute plasticity/atom cannot be used with this pair style
Self-explanatory.
Compute plasticity/atom requires Peridynamic pair style
Self-explanatory.
Compute pressure must use group all
Virial contributions computed by potentials (pair, bond, etc) are computed on all atoms.
Compute pressure requires temperature ID to include kinetic energy
The keflag cannot be used unless a temperature compute is provided.
Compute pressure temperature ID does not compute temperature
The compute ID assigned to a pressure computation must compute temperature.
Compute property/atom floating point vector does not exist
The command is accessing a vector added by the fix property/atom command, that does not exist.
Compute property/atom for atom property that isn't allocated
Self-explanatory.
Compute property/atom integer vector does not exist
The command is accessing a vector added by the fix property/atom command, that does not exist.
Compute property/chunk does not use chunk/atom compute
The style of the specified compute is not chunk/atom.
Compute property/local cannot use these inputs together
LAMMPS Users Manual
228
Only inputs that generate the same number of datums can be used together. E.g. bond and angle
quantities cannot be mixed.
Compute property/local does not (yet) work with atom_style template
Self-explanatory.
Compute property/local for property that isn't allocated
Self-explanatory.
Compute rdf requires a pair style be defined
Self-explanatory.
Compute reduce compute array is accessed out-of-range
An index for the array is out of bounds.
Compute reduce compute calculates global values
A compute that calculates peratom or local values is required.
Compute reduce compute does not calculate a local array
Self-explanatory.
Compute reduce compute does not calculate a local vector
Self-explanatory.
Compute reduce compute does not calculate a per-atom array
Self-explanatory.
Compute reduce compute does not calculate a per-atom vector
Self-explanatory.
Compute reduce fix array is accessed out-of-range
An index for the array is out of bounds.
Compute reduce fix calculates global values
A fix that calculates peratom or local values is required.
Compute reduce fix does not calculate a local array
Self-explanatory.
Compute reduce fix does not calculate a local vector
Self-explanatory.
Compute reduce fix does not calculate a per-atom array
Self-explanatory.
Compute reduce fix does not calculate a per-atom vector
Self-explanatory.
Compute reduce replace requires min or max mode
Self-explanatory.
Compute reduce variable is not atom-style variable
Self-explanatory.
Compute slice compute array is accessed out-of-range
An index for the array is out of bounds.
Compute slice compute does not calculate a global array
Self-explanatory.
Compute slice compute does not calculate a global vector
Self-explanatory.
Compute slice compute does not calculate global vector or array
Self-explanatory.
Compute slice compute vector is accessed out-of-range
The index for the vector is out of bounds.
Compute slice fix array is accessed out-of-range
An index for the array is out of bounds.
Compute slice fix does not calculate a global array
Self-explanatory.
Compute slice fix does not calculate a global vector
LAMMPS Users Manual
229
Self-explanatory.
Compute slice fix does not calculate global vector or array
Self-explanatory.
Compute slice fix vector is accessed out-of-range
The index for the vector is out of bounds.
Compute sna/atom cutoff is longer than pairwise cutoff
Self-explanatory.
Compute sna/atom requires a pair style be defined
Self-explanatory.
Compute snad/atom cutoff is longer than pairwise cutoff
Self-explanatory.
Compute snad/atom requires a pair style be defined
Self-explanatory.
Compute snav/atom cutoff is longer than pairwise cutoff
Self-explanatory.
Compute snav/atom requires a pair style be defined
Self-explanatory.
Compute stress/atom temperature ID does not compute temperature
The specified compute must compute temperature.
Compute temp/asphere requires atom style ellipsoid
Self-explanatory.
Compute temp/asphere requires extended particles
This compute cannot be used with point particles.
Compute temp/body requires atom style body
Self-explanatory.
Compute temp/body requires bodies
This compute can only be applied to body particles.
Compute temp/chunk does not use chunk/atom compute
The style of the specified compute is not chunk/atom.
Compute temp/cs requires ghost atoms store velocity
Use the comm_modify vel yes command to enable this.
Compute temp/cs used when bonds are not allowed
This compute only works on pairs of bonded particles.
Compute temp/partial cannot use vz for 2d systemx
Self-explanatory.
Compute temp/profile cannot bin z for 2d systems
Self-explanatory.
Compute temp/profile cannot use vz for 2d systemx
Self-explanatory.
Compute temp/sphere requires atom style sphere
Self-explanatory.
Compute ti kspace style does not exist
Self-explanatory.
Compute ti pair style does not exist
Self-explanatory.
Compute ti tail when pair style does not compute tail corrections
Self-explanatory.
Compute torque/chunk does not use chunk/atom compute
The style of the specified compute is not chunk/atom.
Compute used in dump between runs is not current
LAMMPS Users Manual
230
The compute was not invoked on the current timestep, therefore it cannot be used in a dump between
runs.
Compute used in variable between runs is not current
Computes cannot be invoked by a variable in between runs. Thus they must have been evaluated on
the last timestep of the previous run in order for their value(s) to be accessed. See the doc page for the
variable command for more info.
Compute used in variable thermo keyword between runs is not current
Some thermo keywords rely on a compute to calculate their value(s). Computes cannot be invoked by
a variable in between runs. Thus they must have been evaluated on the last timestep of the previous
run in order for their value(s) to be accessed. See the doc page for the variable command for more
info.
Compute vcm/chunk does not use chunk/atom compute
The style of the specified compute is not chunk/atom.
Computed temperature for fix temp/berendsen cannot be 0.0
Self-explanatory.
Computed temperature for fix temp/rescale cannot be 0.0
Cannot rescale the temperature to a new value if the current temperature is 0.0.
Core/shell partner atom not found
Could not find one of the atoms in the bond pair.
Core/shell partners were not all found
Could not find or more atoms in the bond pairs.
Could not adjust g_ewald_6
The Newton-Raphson solver failed to converge to a good value for g_ewald. This error should not
occur for typical problems. Please send an email to the developers.
Could not compute g_ewald
The Newton-Raphson solver failed to converge to a good value for g_ewald. This error should not
occur for typical problems. Please send an email to the developers.
Could not compute grid size
The code is unable to compute a grid size consistent with the desired accuracy. This error should not
occur for typical problems. Please send an email to the developers.
Could not compute grid size for Coulomb interaction
The code is unable to compute a grid size consistent with the desired accuracy. This error should not
occur for typical problems. Please send an email to the developers.
Could not compute grid size for Dispersion
The code is unable to compute a grid size consistent with the desired accuracy. This error should not
occur for typical problems. Please send an email to the developers.
Could not create 3d FFT plan
The FFT setup for the PPPM solver failed, typically due to lack of memory. This is an unusual error.
Check the size of the FFT grid you are requesting.
Could not create 3d grid of processors
The specified constraints did not allow a Px by Py by Pz grid to be created where Px * Py * Pz = P =
total number of processors.
Could not create 3d remap plan
The FFT setup in pppm failed.
Could not create Python function arguments
This is an internal Python error, possibly because the number of inputs to the function is too large.
Could not create numa grid of processors
The specified constraints did not allow this style of grid to be created. Usually this is because the total
processor count is not a multiple of the cores/node or the user specified processor count is > 1 in one
of the dimensions.
Could not create twolevel 3d grid of processors
LAMMPS Users Manual
231
The specified constraints did not allow this style of grid to be created.
Could not evaluate Python function input variable
Self-explanatory.
Could not find Python function
The provided Python code was run successfully, but it not define a callable function with the required
name.
Could not find atom_modify first group ID
Self-explanatory.
Could not find change_box group ID
Group ID used in the change_box command does not exist.
Could not find compute ID for PRD
Self-explanatory.
Could not find compute ID for TAD
Self-explanatory.
Could not find compute ID for temperature bias
Self-explanatory.
Could not find compute ID to delete
Self-explanatory.
Could not find compute displace/atom fix ID
Self-explanatory.
Could not find compute event/displace fix ID
Self-explanatory.
Could not find compute group ID
Self-explanatory.
Could not find compute heat/flux compute ID
Self-explanatory.
Could not find compute msd fix ID
Self-explanatory.
Could not find compute msd/chunk fix ID
The compute creates an internal fix, which has been deleted.
Could not find compute pressure temperature ID
The compute ID for calculating temperature does not exist.
Could not find compute stress/atom temperature ID
Self-explanatory.
Could not find compute vacf fix ID
Self-explanatory.
Could not find compute/voronoi surface group ID
Self-explanatory.
Could not find compute_modify ID
Self-explanatory.
Could not find custom per-atom property ID
Self-explanatory.
Could not find delete_atoms group ID
Group ID used in the delete_atoms command does not exist.
Could not find delete_atoms region ID
Region ID used in the delete_atoms command does not exist.
Could not find displace_atoms group ID
Group ID used in the displace_atoms command does not exist.
Could not find dump custom compute ID
Self-explanatory.
Could not find dump custom fix ID
LAMMPS Users Manual
232
Self-explanatory.
Could not find dump custom variable name
Self-explanatory.
Could not find dump group ID
A group ID used in the dump command does not exist.
Could not find dump local compute ID
Self-explanatory.
Could not find dump local fix ID
Self-explanatory.
Could not find dump modify compute ID
Self-explanatory.
Could not find dump modify custom atom floating point property ID
Self-explanatory.
Could not find dump modify custom atom integer property ID
Self-explanatory.
Could not find dump modify fix ID
Self-explanatory.
Could not find dump modify variable name
Self-explanatory.
Could not find fix ID to delete
Self-explanatory.
Could not find fix adapt storage fix ID
This should not happen unless you explicitly deleted a secondary fix that fix adapt created internally.
Could not find fix gcmc exclusion group ID
Self-explanatory.
Could not find fix gcmc rotation group ID
Self-explanatory.
Could not find fix group ID
A group ID used in the fix command does not exist.
Could not find fix msst compute ID
Self-explanatory.
Could not find fix poems group ID
A group ID used in the fix poems command does not exist.
Could not find fix recenter group ID
A group ID used in the fix recenter command does not exist.
Could not find fix rigid group ID
A group ID used in the fix rigid command does not exist.
Could not find fix srd group ID
Self-explanatory.
Could not find fix_modify ID
A fix ID used in the fix_modify command does not exist.
Could not find fix_modify pressure ID
The compute ID for computing pressure does not exist.
Could not find fix_modify temperature ID
The compute ID for computing temperature does not exist.
Could not find group clear group ID
Self-explanatory.
Could not find group delete group ID
Self-explanatory.
Could not find pair fix ID
A fix is created internally by the pair style to store shear history information. You cannot delete it.
LAMMPS Users Manual
233
Could not find set group ID
Group ID specified in set command does not exist.
Could not find specified fix gcmc group ID
Self-explanatory.
Could not find thermo compute ID
Compute ID specified in thermo_style command does not exist.
Could not find thermo custom compute ID
The compute ID needed by thermo style custom to compute a requested quantity does not exist.
Could not find thermo custom fix ID
The fix ID needed by thermo style custom to compute a requested quantity does not exist.
Could not find thermo custom variable name
Self-explanatory.
Could not find thermo fix ID
Fix ID specified in thermo_style command does not exist.
Could not find thermo variable name
Self-explanatory.
Could not find thermo_modify pressure ID
The compute ID needed by thermo style custom to compute pressure does not exist.
Could not find thermo_modify temperature ID
The compute ID needed by thermo style custom to compute temperature does not exist.
Could not find undump ID
A dump ID used in the undump command does not exist.
Could not find velocity group ID
A group ID used in the velocity command does not exist.
Could not find velocity temperature ID
The compute ID needed by the velocity command to compute temperature does not exist.
Could not find/initialize a specified accelerator device
Could not initialize at least one of the devices specified for the gpu package
Could not grab element entry from EIM potential file
Self-explanatory
Could not grab global entry from EIM potential file
Self-explanatory.
Could not grab pair entry from EIM potential file
Self-explanatory.
Could not initialize embedded Python
The main module in Python was not accessible.
Could not open Python file
The specified file of Python code cannot be opened. Check that the path and name are correct.
Could not process Python file
The Python code in the specified file was not run successfully by Python, probably due to errors in the
Python code.
Could not process Python string
The Python code in the here string was not run successfully by Python, probably due to errors in the
Python code.
Coulomb PPPMDisp order has been reduced below minorder
The default minimum order is 2. This can be reset by the kspace_modify minorder command.
Coulomb cut not supported in pair_style buck/long/coul/coul
Must use long-range Coulombic interactions.
Coulomb cut not supported in pair_style lj/long/coul/long
Must use long-range Coulombic interactions.
Coulomb cut not supported in pair_style lj/long/tip4p/long
LAMMPS Users Manual
234
Must use long-range Coulombic interactions.
Coulomb cutoffs of pair hybrid sub-styles do not match
If using a Kspace solver, all Coulomb cutoffs of long pair styles must be the same.
Coulombic cut not supported in pair_style lj/long/dipole/long
Must use long-range Coulombic interactions.
Cound not find dump_modify ID
Self-explanatory.
Create_atoms command before simulation box is defined
The create_atoms command cannot be used before a read_data, read_restart, or create_box command.
Create_atoms molecule has atom IDs, but system does not
The atom_style id command can be used to force atom IDs to be stored.
Create_atoms molecule must have atom types
The defined molecule does not specify atom types.
Create_atoms molecule must have coordinates
The defined molecule does not specify coordinates.
Create_atoms region ID does not exist
A region ID used in the create_atoms command does not exist.
Create_bonds command before simulation box is defined
Self-explanatory.
Create_bonds command requires no kspace_style be defined
This is so that atom pairs that are already bonded to not appear in the neighbor list.
Create_bonds command requires special_bonds 1-2 weights be 0.0
This is so that atom pairs that are already bonded to not appear in the neighbor list.
Create_bonds max distance > neighbor cutoff
Can only create bonds for atom pairs that will be in neighbor list.
Create_bonds requires a pair style be defined
Self-explanatory.
Create_box region ID does not exist
Self-explanatory.
Create_box region does not support a bounding box
Not all regions represent bounded volumes. You cannot use such a region with the create_box
command.
Custom floating point vector for fix store/state does not exist
The command is accessing a vector added by the fix property/atom command, that does not exist.
Custom integer vector for fix store/state does not exist
The command is accessing a vector added by the fix property/atom command, that does not exist.
Custom per-atom property ID is not floating point
Self-explanatory.
Custom per-atom property ID is not integer
Self-explanatory.
Cut-offs missing in pair_style lj/long/dipole/long
Self-explanatory.
Cutoffs missing in pair_style buck/long/coul/long
Self-explanatory.
Cutoffs missing in pair_style lj/long/coul/long
Self-explanatory.
Cyclic loop in joint connections
Fix poems cannot (yet) work with coupled bodies whose joints connect the bodies in a ring (or cycle).
Degenerate lattice primitive vectors
Invalid set of 3 lattice vectors for lattice command.
Delete region ID does not exist
LAMMPS Users Manual
235
Self-explanatory.
Delete_atoms command before simulation box is defined
The delete_atoms command cannot be used before a read_data, read_restart, or create_box command.
Delete_atoms cutoff > max neighbor cutoff
Can only delete atoms in atom pairs that will be in neighbor list.
Delete_atoms mol yes requires atom attribute molecule
Cannot use this option with a non-molecular system.
Delete_atoms requires a pair style be defined
This is because atom deletion within a cutoff uses a pairwise neighbor list.
Delete_bonds command before simulation box is defined
The delete_bonds command cannot be used before a read_data, read_restart, or create_box command.
Delete_bonds command with no atoms existing
No atoms are yet defined so the delete_bonds command cannot be used.
Deposition region extends outside simulation box
Self-explanatory.
Did not assign all atoms correctly
Atoms read in from a data file were not assigned correctly to processors. This is likely due to some
atom coordinates being outside a non-periodic simulation box.
Did not assign all restart atoms correctly
Atoms read in from the restart file were not assigned correctly to processors. This is likely due to
some atom coordinates being outside a non-periodic simulation box. Normally this should not happen.
You may wish to use the "remap" option on the read_restart command to see if this helps.
Did not find all elements in MEAM library file
The requested elements were not found in the MEAM file.
Did not find fix shake partner info
Could not find bond partners implied by fix shake command. This error can be triggered if the
delete_bonds command was used before fix shake, and it removed bonds without resetting the 1-2,
1-3, 1-4 weighting list via the special keyword.
Did not find keyword in table file
Keyword used in pair_coeff command was not found in table file.
Did not set pressure for fix rigid/nph
The press keyword must be specified.
Did not set temp for fix rigid/nvt/small
Self-explanatory.
Did not set temp or press for fix rigid/npt/small
Self-explanatory.
Did not set temperature for fix rigid/nvt
The temp keyword must be specified.
Did not set temperature or pressure for fix rigid/npt
The temp and press keywords must be specified.
Dihedral atom missing in delete_bonds
The delete_bonds command cannot find one or more atoms in a particular dihedral on a particular
processor. The pairwise cutoff is too short or the atoms are too far apart to make a valid dihedral.
Dihedral atom missing in set command
The set command cannot find one or more atoms in a particular dihedral on a particular processor.
The pairwise cutoff is too short or the atoms are too far apart to make a valid dihedral.
Dihedral atoms %d %d %d %d missing on proc %d at step %ld
One or more of 4 atoms needed to compute a particular dihedral are missing on this processor.
Typically this is because the pairwise cutoff is set too short or the dihedral has blown apart and an
atom is too far away.
Dihedral atoms missing on proc %d at step %ld
LAMMPS Users Manual
236
One or more of 4 atoms needed to compute a particular dihedral are missing on this processor.
Typically this is because the pairwise cutoff is set too short or the dihedral has blown apart and an
atom is too far away.
Dihedral charmm is incompatible with Pair style
Dihedral style charmm must be used with a pair style charmm in order for the 1-4 epsilon/sigma
parameters to be defined.
Dihedral coeff for hybrid has invalid style
Dihedral style hybrid uses another dihedral style as one of its coefficients. The dihedral style used in
the dihedral_coeff command or read from a restart file is not recognized.
Dihedral coeffs are not set
No dihedral coefficients have been assigned in the data file or via the dihedral_coeff command.
Dihedral style hybrid cannot have hybrid as an argument
Self-explanatory.
Dihedral style hybrid cannot have none as an argument
Self-explanatory.
Dihedral style hybrid cannot use same dihedral style twice
Self-explanatory.
Dihedral/improper extent > half of periodic box length
This error was detected by the neigh_modify check yes setting. It is an error because the dihedral
atoms are so far apart it is ambiguous how it should be defined.
Dihedral_coeff command before dihedral_style is defined
Coefficients cannot be set in the data file or via the dihedral_coeff command until an dihedral_style
has been assigned.
Dihedral_coeff command before simulation box is defined
The dihedral_coeff command cannot be used before a read_data, read_restart, or create_box
command.
Dihedral_coeff command when no dihedrals allowed
The chosen atom style does not allow for dihedrals to be defined.
Dihedral_style command when no dihedrals allowed
The chosen atom style does not allow for dihedrals to be defined.
Dihedrals assigned incorrectly
Dihedrals read in from the data file were not assigned correctly to atoms. This means there is
something invalid about the topology definitions.
Dihedrals defined but no dihedral types
The data file header lists dihedrals but no dihedral types.
Dimension command after simulation box is defined
The dimension command cannot be used after a read_data, read_restart, or create_box command.
Dispersion PPPMDisp order has been reduced below minorder
The default minimum order is 2. This can be reset by the kspace_modify minorder command.
Displace_atoms command before simulation box is defined
The displace_atoms command cannot be used before a read_data, read_restart, or create_box
command.
Distance must be > 0 for compute event/displace
Self-explanatory.
Divide by 0 in influence function
This should not normally occur. It is likely a problem with your model.
Divide by 0 in influence function of pair peri/lps
This should not normally occur. It is likely a problem with your model.
Divide by 0 in variable formula
Self-explanatory.
Domain too large for neighbor bins
LAMMPS Users Manual
237
The domain has become extremely large so that neighbor bins cannot be used. Most likely, one or
more atoms have been blown out of the simulation box to a great distance.
Double precision is not supported on this accelerator
Self-explanatory
Dump atom/gz only writes compressed files
The dump atom/gz output file name must have a .gz suffix.
Dump cfg arguments can not mix xs|ys|zs with xsu|ysu|zsu
Self-explanatory.
Dump cfg arguments must start with 'mass type xs ys zs' or 'mass type xsu ysu zsu'
This is a requirement of the CFG output format. See the dump cfg doc page for more details.
Dump cfg requires one snapshot per file
Use the wildcard "*" character in the filename.
Dump cfg/gz only writes compressed files
The dump cfg/gz output file name must have a .gz suffix.
Dump custom and fix not computed at compatible times
The fix must produce per-atom quantities on timesteps that dump custom needs them.
Dump custom compute does not calculate per-atom array
Self-explanatory.
Dump custom compute does not calculate per-atom vector
Self-explanatory.
Dump custom compute does not compute per-atom info
Self-explanatory.
Dump custom compute vector is accessed out-of-range
Self-explanatory.
Dump custom fix does not compute per-atom array
Self-explanatory.
Dump custom fix does not compute per-atom info
Self-explanatory.
Dump custom fix does not compute per-atom vector
Self-explanatory.
Dump custom fix vector is accessed out-of-range
Self-explanatory.
Dump custom variable is not atom-style variable
Only atom-style variables generate per-atom quantities, needed for dump output.
Dump custom/gz only writes compressed files
The dump custom/gz output file name must have a .gz suffix.
Dump dcd of non-matching # of atoms
Every snapshot written by dump dcd must contain the same # of atoms.
Dump dcd requires sorting by atom ID
Use the dump_modify sort command to enable this.
Dump every variable returned a bad timestep
The variable must return a timestep greater than the current timestep.
Dump file MPI-IO output not allowed with % in filename
This is because a % signifies one file per processor and MPI-IO creates one large file for all
processors.
Dump file does not contain requested snapshot
Self-explanatory.
Dump file is incorrectly formatted
Self-explanatory.
Dump image body yes requires atom style body
Self-explanatory.
LAMMPS Users Manual
238
Dump image bond not allowed with no bond types
Self-explanatory.
Dump image cannot perform sorting
Self-explanatory.
Dump image line requires atom style line
Self-explanatory.
Dump image persp option is not yet supported
Self-explanatory.
Dump image requires one snapshot per file
Use a "*" in the filename.
Dump image tri requires atom style tri
Self-explanatory.
Dump local and fix not computed at compatible times
The fix must produce per-atom quantities on timesteps that dump local needs them.
Dump local attributes contain no compute or fix
Self-explanatory.
Dump local cannot sort by atom ID
This is because dump local does not really dump per-atom info.
Dump local compute does not calculate local array
Self-explanatory.
Dump local compute does not calculate local vector
Self-explanatory.
Dump local compute does not compute local info
Self-explanatory.
Dump local compute vector is accessed out-of-range
Self-explanatory.
Dump local count is not consistent across input fields
Every column of output must be the same length.
Dump local fix does not compute local array
Self-explanatory.
Dump local fix does not compute local info
Self-explanatory.
Dump local fix does not compute local vector
Self-explanatory.
Dump local fix vector is accessed out-of-range
Self-explanatory.
Dump modify bcolor not allowed with no bond types
Self-explanatory.
Dump modify bdiam not allowed with no bond types
Self-explanatory.
Dump modify compute ID does not compute per-atom array
Self-explanatory.
Dump modify compute ID does not compute per-atom info
Self-explanatory.
Dump modify compute ID does not compute per-atom vector
Self-explanatory.
Dump modify compute ID vector is not large enough
Self-explanatory.
Dump modify element names do not match atom types
Number of element names must equal number of atom types.
Dump modify fix ID does not compute per-atom array
LAMMPS Users Manual
239
Self-explanatory.
Dump modify fix ID does not compute per-atom info
Self-explanatory.
Dump modify fix ID does not compute per-atom vector
Self-explanatory.
Dump modify fix ID vector is not large enough
Self-explanatory.
Dump modify variable is not atom-style variable
Self-explanatory.
Dump sort column is invalid
Self-explanatory.
Dump xtc requires sorting by atom ID
Use the dump_modify sort command to enable this.
Dump xyz/gz only writes compressed files
The dump xyz/gz output file name must have a .gz suffix.
Dump_modify buffer yes not allowed for this style
Self-explanatory.
Dump_modify format string is too short
There are more fields to be dumped in a line of output than your format string specifies.
Dump_modify region ID does not exist
Self-explanatory.
Dumping an atom property that isn't allocated
The chosen atom style does not define the per-atom quantity being dumped.
Duplicate atom IDs exist
Self-explanatory.
Duplicate fields in read_dump command
Self-explanatory.
Duplicate particle in PeriDynamic bond - simulation box is too small
This is likely because your box length is shorter than 2 times the bond length.
Electronic temperature dropped below zero
Something has gone wrong with the fix ttm electron temperature model.
Element not defined in potential file
The specified element is not in the potential file.
Empty brackets in variable
There is no variable syntax that uses empty brackets. Check the variable doc page.
Energy was not tallied on needed timestep
You are using a thermo keyword that requires potentials to have tallied energy, but they didn't on this
timestep. See the variable doc page for ideas on how to make this work.
Epsilon or sigma reference not set by pair style in PPPMDisp
Self-explanatory.
Epsilon or sigma reference not set by pair style in ewald/n
The pair style is not providing the needed epsilon or sigma values.
Error in vdw spline: inner radius > outer radius
A pre-tabulated spline is invalid. Likely a problem with the potential parameters.
Error writing averaged chunk data
Something in the output to the file triggered an error.
Error writing file header
Something in the output to the file triggered an error.
Error writing out correlation data
Something in the output to the file triggered an error.
Error writing out histogram data
LAMMPS Users Manual
240
Something in the output to the file triggered an error.
Error writing out time averaged data
Something in the output to the file triggered an error.
Failed to allocate %ld bytes for array %s
Your LAMMPS simulation has run out of memory. You need to run a smaller simulation or on more
processors.
Failed to open FFmpeg pipeline to file %s
The specified file cannot be opened. Check that the path and name are correct and writable and that
the FFmpeg executable can be found and run.
Failed to reallocate %ld bytes for array %s
Your LAMMPS simulation has run out of memory. You need to run a smaller simulation or on more
processors.
Fewer SRD bins than processors in some dimension
This is not allowed. Make your SRD bin size smaller.
File variable could not read value
Check the file assigned to the variable.
Final box dimension due to fix deform is < 0.0
Self-explanatory.
Fix %s does not allow use of dynamic group
Dynamic groups have not yet been enabled for this fix.
Fix ID for compute chunk/atom does not exist
Self-explanatory.
Fix ID for compute erotate/rigid does not exist
Self-explanatory.
Fix ID for compute ke/rigid does not exist
Self-explanatory.
Fix ID for compute reduce does not exist
Self-explanatory.
Fix ID for compute slice does not exist
Self-explanatory.
Fix ID for fix ave/atom does not exist
Self-explanatory.
Fix ID for fix ave/chunk does not exist
Self-explanatory.
Fix ID for fix ave/correlate does not exist
Self-explanatory.
Fix ID for fix ave/histo does not exist
Self-explanatory.
Fix ID for fix ave/spatial does not exist
Self-explanatory.
Fix ID for fix ave/time does not exist
Self-explanatory.
Fix ID for fix store/state does not exist
Self-explanatory
Fix ID for fix vector does not exist
Self-explanatory.
Fix ID for read_data does not exist
Self-explanatory.
Fix ID for velocity does not exist
Self-explanatory.
Fix ID must be alphanumeric or underscore characters
LAMMPS Users Manual
241
Self-explanatory.
Fix SRD: bad bin assignment for SRD advection
Something has gone wrong in your SRD model; try using more conservative settings.
Fix SRD: bad search bin assignment
Something has gone wrong in your SRD model; try using more conservative settings.
Fix SRD: bad stencil bin for big particle
Something has gone wrong in your SRD model; try using more conservative settings.
Fix SRD: too many big particles in bin
Reset the ATOMPERBIN parameter at the top of fix_srd.cpp to a larger value, and re-compile the
code.
Fix SRD: too many walls in bin
This should not happen unless your system has been setup incorrectly.
Fix adapt interface to this pair style not supported
New coding for the pair style would need to be done.
Fix adapt kspace style does not exist
Self-explanatory.
Fix adapt pair style does not exist
Self-explanatory
Fix adapt pair style param not supported
The pair style does not know about the parameter you specified.
Fix adapt requires atom attribute charge
The atom style being used does not specify an atom charge.
Fix adapt requires atom attribute diameter
The atom style being used does not specify an atom diameter.
Fix adapt type pair range is not valid for pair hybrid sub-style
Self-explanatory.
Fix append/atoms requires a lattice be defined
Use the lattice command for this purpose.
Fix ave/atom compute array is accessed out-of-range
Self-explanatory.
Fix ave/atom compute does not calculate a per-atom array
Self-explanatory.
Fix ave/atom compute does not calculate a per-atom vector
A compute used by fix ave/atom must generate per-atom values.
Fix ave/atom compute does not calculate per-atom values
A compute used by fix ave/atom must generate per-atom values.
Fix ave/atom fix array is accessed out-of-range
Self-explanatory.
Fix ave/atom fix does not calculate a per-atom array
Self-explanatory.
Fix ave/atom fix does not calculate a per-atom vector
A fix used by fix ave/atom must generate per-atom values.
Fix ave/atom fix does not calculate per-atom values
A fix used by fix ave/atom must generate per-atom values.
Fix ave/atom variable is not atom-style variable
A variable used by fix ave/atom must generate per-atom values.
Fix ave/chunk compute does not calculate a per-atom array
Self-explanatory.
Fix ave/chunk compute does not calculate a per-atom vector
Self-explanatory.
Fix ave/chunk compute does not calculate per-atom values
LAMMPS Users Manual
242
Self-explanatory.
Fix ave/chunk compute vector is accessed out-of-range
Self-explanatory.
Fix ave/chunk does not use chunk/atom compute
The specified compute is not for a compute chunk/atom command.
Fix ave/chunk fix does not calculate a per-atom array
Self-explanatory.
Fix ave/chunk fix does not calculate a per-atom vector
Self-explanatory.
Fix ave/chunk fix does not calculate per-atom values
Self-explanatory.
Fix ave/chunk fix vector is accessed out-of-range
Self-explanatory.
Fix ave/chunk variable is not atom-style variable
Self-explanatory.
Fix ave/correlate compute does not calculate a scalar
Self-explanatory.
Fix ave/correlate compute does not calculate a vector
Self-explanatory.
Fix ave/correlate compute vector is accessed out-of-range
The index for the vector is out of bounds.
Fix ave/correlate fix does not calculate a scalar
Self-explanatory.
Fix ave/correlate fix does not calculate a vector
Self-explanatory.
Fix ave/correlate fix vector is accessed out-of-range
The index for the vector is out of bounds.
Fix ave/correlate variable is not equal-style variable
Self-explanatory.
Fix ave/histo cannot input local values in scalar mode
Self-explanatory.
Fix ave/histo cannot input per-atom values in scalar mode
Self-explanatory.
Fix ave/histo compute array is accessed out-of-range
Self-explanatory.
Fix ave/histo compute does not calculate a global array
Self-explanatory.
Fix ave/histo compute does not calculate a global scalar
Self-explanatory.
Fix ave/histo compute does not calculate a global vector
Self-explanatory.
Fix ave/histo compute does not calculate a local array
Self-explanatory.
Fix ave/histo compute does not calculate a local vector
Self-explanatory.
Fix ave/histo compute does not calculate a per-atom array
Self-explanatory.
Fix ave/histo compute does not calculate a per-atom vector
Self-explanatory.
Fix ave/histo compute does not calculate local values
Self-explanatory.
LAMMPS Users Manual
243
Fix ave/histo compute does not calculate per-atom values
Self-explanatory.
Fix ave/histo compute vector is accessed out-of-range
Self-explanatory.
Fix ave/histo fix array is accessed out-of-range
Self-explanatory.
Fix ave/histo fix does not calculate a global array
Self-explanatory.
Fix ave/histo fix does not calculate a global scalar
Self-explanatory.
Fix ave/histo fix does not calculate a global vector
Self-explanatory.
Fix ave/histo fix does not calculate a local array
Self-explanatory.
Fix ave/histo fix does not calculate a local vector
Self-explanatory.
Fix ave/histo fix does not calculate a per-atom array
Self-explanatory.
Fix ave/histo fix does not calculate a per-atom vector
Self-explanatory.
Fix ave/histo fix does not calculate local values
Self-explanatory.
Fix ave/histo fix does not calculate per-atom values
Self-explanatory.
Fix ave/histo fix vector is accessed out-of-range
Self-explanatory.
Fix ave/histo input is invalid compute
Self-explanatory.
Fix ave/histo input is invalid fix
Self-explanatory.
Fix ave/histo input is invalid variable
Self-explanatory.
Fix ave/histo inputs are not all global, peratom, or local
All inputs in a single fix ave/histo command must be of the same style.
Fix ave/histo/weight value and weight vector lengths do not match
Self-explanatory.
Fix ave/spatial compute does not calculate a per-atom array
Self-explanatory.
Fix ave/spatial compute does not calculate a per-atom vector
A compute used by fix ave/spatial must generate per-atom values.
Fix ave/spatial compute does not calculate per-atom values
A compute used by fix ave/spatial must generate per-atom values.
Fix ave/spatial compute vector is accessed out-of-range
The index for the vector is out of bounds.
Fix ave/spatial fix does not calculate a per-atom array
Self-explanatory.
Fix ave/spatial fix does not calculate a per-atom vector
A fix used by fix ave/spatial must generate per-atom values.
Fix ave/spatial fix does not calculate per-atom values
A fix used by fix ave/spatial must generate per-atom values.
Fix ave/spatial fix vector is accessed out-of-range
LAMMPS Users Manual
244
The index for the vector is out of bounds.
Fix ave/spatial for triclinic boxes requires units reduced
Self-explanatory.
Fix ave/spatial settings invalid with changing box size
If the box size changes, only the units reduced option can be used.
Fix ave/spatial variable is not atom-style variable
A variable used by fix ave/spatial must generate per-atom values.
Fix ave/time cannot set output array intensive/extensive from these inputs
One of more of the vector inputs has individual elements which are flagged as intensive or extensive.
Such an input cannot be flagged as all intensive/extensive when turned into an array by fix ave/time.
Fix ave/time cannot use variable with vector mode
Variables produce scalar values.
Fix ave/time columns are inconsistent lengths
Self-explanatory.
Fix ave/time compute array is accessed out-of-range
An index for the array is out of bounds.
Fix ave/time compute does not calculate a scalar
Self-explanatory.
Fix ave/time compute does not calculate a vector
Self-explanatory.
Fix ave/time compute does not calculate an array
Self-explanatory.
Fix ave/time compute vector is accessed out-of-range
The index for the vector is out of bounds.
Fix ave/time fix array cannot be variable length
Self-explanatory.
Fix ave/time fix array is accessed out-of-range
An index for the array is out of bounds.
Fix ave/time fix does not calculate a scalar
Self-explanatory.
Fix ave/time fix does not calculate a vector
Self-explanatory.
Fix ave/time fix does not calculate an array
Self-explanatory.
Fix ave/time fix vector cannot be variable length
Self-explanatory.
Fix ave/time fix vector is accessed out-of-range
The index for the vector is out of bounds.
Fix ave/time variable is not equal-style variable
Self-explanatory.
Fix balance rcb cannot be used with comm_style brick
Comm_style tiled must be used instead.
Fix balance shift string is invalid
The string can only contain the characters "x", "y", or "z".
Fix bond/break needs ghost atoms from further away
This is because the fix needs to walk bonds to a certain distance to acquire needed info, The
comm_modify cutoff command can be used to extend the communication range.
Fix bond/create angle type is invalid
Self-explanatory.
Fix bond/create cutoff is longer than pairwise cutoff
This is not allowed because bond creation is done using the pairwise neighbor list.
LAMMPS Users Manual
245
Fix bond/create dihedral type is invalid
Self-explanatory.
Fix bond/create improper type is invalid
Self-explanatory.
Fix bond/create induced too many angles/dihedrals/impropers per atom
See the read_data command for info on setting the "extra angle per atom", etc header values to allow
for additional angles, etc to be formed.
Fix bond/create needs ghost atoms from further away
This is because the fix needs to walk bonds to a certain distance to acquire needed info, The
comm_modify cutoff command can be used to extend the communication range.
Fix bond/swap cannot use dihedral or improper styles
These styles cannot be defined when using this fix.
Fix bond/swap requires pair and bond styles
Self-explanatory.
Fix bond/swap requires special_bonds = 0,1,1
Self-explanatory.
Fix box/relax generated negative box length
The pressure being applied is likely too large. Try applying it incrementally, to build to the high
pressure.
Fix command before simulation box is defined
The fix command cannot be used before a read_data, read_restart, or create_box command.
Fix deform cannot use yz variable with xy
The yz setting cannot be a variable if xy deformation is also specified. This is because LAMMPS
cannot determine if the yz setting will induce a box flip which would be invalid if xy is also changing.
Fix deform is changing yz too much with xy
When both yz and xy are changing, it induces changes in xz if the box must flip from one tilt extreme
to another. Thus it is not allowed for yz to grow so much that a flip is induced.
Fix deform tilt factors require triclinic box
Cannot deform the tilt factors of a simulation box unless it is a triclinic (non-orthogonal) box.
Fix deform volume setting is invalid
Cannot use volume style unless other dimensions are being controlled.
Fix deposit and fix rigid/small not using same molecule template ID
Self-explanatory.
Fix deposit and fix shake not using same molecule template ID
Self-explanatory.
Fix deposit molecule must have atom types
The defined molecule does not specify atom types.
Fix deposit molecule must have coordinates
The defined molecule does not specify coordinates.
Fix deposit molecule template ID must be same as atom_style template ID
When using atom_style template, you cannot deposit molecules that are not in that template.
Fix deposit region cannot be dynamic
Only static regions can be used with fix deposit.
Fix deposit region does not support a bounding box
Not all regions represent bounded volumes. You cannot use such a region with the fix deposit
command.
Fix deposit shake fix does not exist
Self-explanatory.
Fix efield requires atom attribute q or mu
The atom style defined does not have this attribute.
Fix efield with dipoles cannot use atom-style variables
LAMMPS Users Manual
246
This option is not supported.
Fix evaporate molecule requires atom attribute molecule
The atom style being used does not define a molecule ID.
Fix external callback function not set
This must be done by an external program in order to use this fix.
Fix for fix ave/atom not computed at compatible time
Fixes generate their values on specific timesteps. Fix ave/atom is requesting a value on a non-allowed
timestep.
Fix for fix ave/chunk not computed at compatible time
Fixes generate their values on specific timesteps. Fix ave/chunk is requesting a value on a
non-allowed timestep.
Fix for fix ave/correlate not computed at compatible time
Fixes generate their values on specific timesteps. Fix ave/correlate is requesting a value on a
non-allowed timestep.
Fix for fix ave/histo not computed at compatible time
Fixes generate their values on specific timesteps. Fix ave/histo is requesting a value on a non-allowed
timestep.
Fix for fix ave/spatial not computed at compatible time
Fixes generate their values on specific timesteps. Fix ave/spatial is requesting a value on a
non-allowed timestep.
Fix for fix ave/time not computed at compatible time
Fixes generate their values on specific timesteps. Fix ave/time is requesting a value on a non-allowed
timestep.
Fix for fix store/state not computed at compatible time
Fixes generate their values on specific timesteps. Fix store/state is requesting a value on a
non-allowed timestep.
Fix for fix vector not computed at compatible time
Fixes generate their values on specific timesteps. Fix vector is requesting a value on a non-allowed
timestep.
Fix freeze requires atom attribute torque
The atom style defined does not have this attribute.
Fix gcmc and fix shake not using same molecule template ID
Self-explanatory.
Fix gcmc atom has charge, but atom style does not
Self-explanatory.
Fix gcmc cannot exchange individual atoms belonging to a molecule
This is an error since you should not delete only one atom of a molecule. The user has specified
atomic (non-molecular) gas exchanges, but an atom belonging to a molecule could be deleted.
Fix gcmc does not (yet) work with atom_style template
Self-explanatory.
Fix gcmc molecule command requires that atoms have molecule attributes
Should not choose the gcmc molecule feature if no molecules are being simulated. The general
molecule flag is off, but gcmc's molecule flag is on.
Fix gcmc molecule has charges, but atom style does not
Self-explanatory.
Fix gcmc molecule must have atom types
The defined molecule does not specify atom types.
Fix gcmc molecule must have coordinates
The defined molecule does not specify coordinates.
Fix gcmc molecule template ID must be same as atom_style template ID
When using atom_style template, you cannot insert molecules that are not in that template.
LAMMPS Users Manual
247
Fix gcmc put atom outside box
This should not normally happen. Contact the developers.
Fix gcmc ran out of available atom IDs
See the setting for tagint in the src/lmptype.h file.
Fix gcmc ran out of available molecule IDs
See the setting for tagint in the src/lmptype.h file.
Fix gcmc region cannot be dynamic
Only static regions can be used with fix gcmc.
Fix gcmc region does not support a bounding box
Not all regions represent bounded volumes. You cannot use such a region with the fix gcmc
command.
Fix gcmc region extends outside simulation box
Self-explanatory.
Fix gcmc shake fix does not exist
Self-explanatory.
Fix gld c coefficients must be >= 0
Self-explanatory.
Fix gld needs more prony series coefficients
Self-explanatory.
Fix gld prony terms must be > 0
Self-explanatory.
Fix gld series type must be pprony for now
Self-explanatory.
Fix gld start temperature must be >= 0
Self-explanatory.
Fix gld stop temperature must be >= 0
Self-explanatory.
Fix gld tau coefficients must be > 0
Self-explanatory.
Fix heat group has no atoms
Self-explanatory.
Fix heat kinetic energy of an atom went negative
This will cause the velocity rescaling about to be performed by fix heat to be invalid.
Fix heat kinetic energy went negative
This will cause the velocity rescaling about to be performed by fix heat to be invalid.
Fix in variable not computed at compatible time
Fixes generate their values on specific timesteps. The variable is requesting the values on a
non-allowed timestep.
Fix langevin angmom is not yet implemented with kokkos
This option is not yet available.
Fix langevin angmom requires atom style ellipsoid
Self-explanatory.
Fix langevin angmom requires extended particles
This fix option cannot be used with point particles.
Fix langevin omega is not yet implemented with kokkos
This option is not yet available.
Fix langevin omega requires atom style sphere
Self-explanatory.
Fix langevin omega requires extended particles
One of the particles has radius 0.0.
Fix langevin period must be > 0.0
LAMMPS Users Manual
248
The time window for temperature relaxation must be > 0
Fix langevin variable returned negative temperature
Self-explanatory.
Fix momentum group has no atoms
Self-explanatory.
Fix move cannot define z or vz variable for 2d problem
Self-explanatory.
Fix move cannot rotate aroung non z-axis for 2d problem
Self-explanatory.
Fix move cannot set linear z motion for 2d problem
Self-explanatory.
Fix move cannot set wiggle z motion for 2d problem
Self-explanatory.
Fix msst compute ID does not compute potential energy
Self-explanatory.
Fix msst compute ID does not compute pressure
Self-explanatory.
Fix msst compute ID does not compute temperature
Self-explanatory.
Fix msst requires a periodic box
Self-explanatory.
Fix msst tscale must satisfy 0 <= tscale < 1
Self-explanatory.
Fix npt/nph has tilted box too far in one step - periodic cell is too far from equilibrium state
Self-explanatory. The change in the box tilt is too extreme on a short timescale.
Fix nve/asphere requires extended particles
This fix can only be used for particles with a shape setting.
Fix nve/asphere/noforce requires atom style ellipsoid
Self-explanatory.
Fix nve/asphere/noforce requires extended particles
One of the particles is not an ellipsoid.
Fix nve/body requires atom style body
Self-explanatory.
Fix nve/body requires bodies
This fix can only be used for particles that are bodies.
Fix nve/line can only be used for 2d simulations
Self-explanatory.
Fix nve/line requires atom style line
Self-explanatory.
Fix nve/line requires line particles
Self-explanatory.
Fix nve/sphere dipole requires atom attribute mu
An atom style with this attribute is needed.
Fix nve/sphere requires atom style sphere
Self-explanatory.
Fix nve/sphere requires extended particles
This fix can only be used for particles of a finite size.
Fix nve/tri can only be used for 3d simulations
Self-explanatory.
Fix nve/tri requires atom style tri
Self-explanatory.
LAMMPS Users Manual
249
Fix nve/tri requires tri particles
Self-explanatory.
Fix nvt/nph/npt asphere requires extended particles
The shape setting for a particle in the fix group has shape = 0.0, which means it is a point particle.
Fix nvt/nph/npt body requires bodies
Self-explanatory.
Fix nvt/nph/npt sphere requires atom style sphere
Self-explanatory.
Fix nvt/npt/nph damping parameters must be > 0.0
Self-explanatory.
Fix nvt/npt/nph dilate group ID does not exist
Self-explanatory.
Fix nvt/sphere requires extended particles
This fix can only be used for particles of a finite size.
Fix orient/fcc file open failed
The fix orient/fcc command could not open a specified file.
Fix orient/fcc file read failed
The fix orient/fcc command could not read the needed parameters from a specified file.
Fix orient/fcc found self twice
The neighbor lists used by fix orient/fcc are messed up. If this error occurs, it is likely a bug, so send
an email to the developers.
Fix peri neigh does not exist
Somehow a fix that the pair style defines has been deleted.
Fix pour and fix rigid/small not using same molecule template ID
Self-explanatory.
Fix pour and fix shake not using same molecule template ID
Self-explanatory.
Fix pour insertion count per timestep is 0
Self-explanatory.
Fix pour molecule must have atom types
The defined molecule does not specify atom types.
Fix pour molecule must have coordinates
The defined molecule does not specify coordinates.
Fix pour molecule template ID must be same as atom style template ID
When using atom_style template, you cannot pour molecules that are not in that template.
Fix pour polydisperse fractions do not sum to 1.0
Self-explanatory.
Fix pour region ID does not exist
Self-explanatory.
Fix pour region cannot be dynamic
Only static regions can be used with fix pour.
Fix pour region does not support a bounding box
Not all regions represent bounded volumes. You cannot use such a region with the fix pour command.
Fix pour requires atom attributes radius, rmass
The atom style defined does not have these attributes.
Fix pour rigid fix does not exist
Self-explanatory.
Fix pour shake fix does not exist
Self-explanatory.
Fix press/berendsen damping parameters must be > 0.0
Self-explanatory.
LAMMPS Users Manual
250
Fix property/atom cannot specify mol twice
Self-explanatory.
Fix property/atom cannot specify q twice
Self-explanatory.
Fix property/atom mol when atom_style already has molecule attribute
Self-explanatory.
Fix property/atom q when atom_style already has charge attribute
Self-explanatory.
Fix property/atom vector name already exists
The name for an integer or floating-point vector must be unique.
Fix qeq has negative upper Taper radius cutoff
Self-explanatory.
Fix qeq/comb group has no atoms
Self-explanatory.
Fix qeq/comb requires atom attribute q
An atom style with charge must be used to perform charge equilibration.
Fix qeq/dynamic group has no atoms
Self-explanatory.
Fix qeq/dynamic requires atom attribute q
Self-explanatory.
Fix qeq/fire group has no atoms
Self-explanatory.
Fix qeq/fire requires atom attribute q
Self-explanatory.
Fix qeq/point group has no atoms
Self-explanatory.
Fix qeq/point has insufficient QEq matrix size
Occurs when number of neighbor atoms for an atom increased too much during a run. Increase
SAFE_ZONE and MIN_CAP in fix_qeq.h and recompile.
Fix qeq/point requires atom attribute q
Self-explanatory.
Fix qeq/shielded group has no atoms
Self-explanatory.
Fix qeq/shielded has insufficient QEq matrix size
Occurs when number of neighbor atoms for an atom increased too much during a run. Increase
SAFE_ZONE and MIN_CAP in fix_qeq.h and recompile.
Fix qeq/shielded requires atom attribute q
Self-explanatory.
Fix qeq/slater could not extract params from pair coul/streitz
This should not happen unless pair coul/streitz has been altered.
Fix qeq/slater group has no atoms
Self-explanatory.
Fix qeq/slater has insufficient QEq matrix size
Occurs when number of neighbor atoms for an atom increased too much during a run. Increase
SAFE_ZONE and MIN_CAP in fix_qeq.h and recompile.
Fix qeq/slater requires atom attribute q
Self-explanatory.
Fix reax/bonds numbonds > nsbmax_most
The limit of the number of bonds expected by the ReaxFF force field was exceeded.
Fix recenter group has no atoms
Self-explanatory.
LAMMPS Users Manual
251
Fix restrain requires an atom map, see atom_modify
Self-explanatory.
Fix rigid atom has non-zero image flag in a non-periodic dimension
Image flags for non-periodic dimensions should not be set.
Fix rigid file has no lines
Self-explanatory.
Fix rigid langevin period must be > 0.0
Self-explanatory.
Fix rigid molecule requires atom attribute molecule
Self-explanatory.
Fix rigid npt/nph dilate group ID does not exist
Self-explanatory.
Fix rigid npt/nph does not yet allow triclinic box
This is a current restriction in LAMMPS.
Fix rigid npt/nph period must be > 0.0
Self-explanatory.
Fix rigid npt/small t_chain should not be less than 1
Self-explanatory.
Fix rigid npt/small t_order must be 3 or 5
Self-explanatory.
Fix rigid nvt/npt/nph damping parameters must be > 0.0
Self-explanatory.
Fix rigid nvt/small t_chain should not be less than 1
Self-explanatory.
Fix rigid nvt/small t_iter should not be less than 1
Self-explanatory.
Fix rigid nvt/small t_order must be 3 or 5
Self-explanatory.
Fix rigid xy torque cannot be on for 2d simulation
Self-explanatory.
Fix rigid z force cannot be on for 2d simulation
Self-explanatory.
Fix rigid/npt period must be > 0.0
Self-explanatory.
Fix rigid/npt temperature order must be 3 or 5
Self-explanatory.
Fix rigid/npt/small period must be > 0.0
Self-explanatory.
Fix rigid/nvt period must be > 0.0
Self-explanatory.
Fix rigid/nvt temperature order must be 3 or 5
Self-explanatory.
Fix rigid/nvt/small period must be > 0.0
Self-explanatory.
Fix rigid/small atom has non-zero image flag in a non-periodic dimension
Image flags for non-periodic dimensions should not be set.
Fix rigid/small langevin period must be > 0.0
Self-explanatory.
Fix rigid/small molecule must have atom types
The defined molecule does not specify atom types.
Fix rigid/small molecule must have coordinates
LAMMPS Users Manual
252
The defined molecule does not specify coordinates.
Fix rigid/small npt/nph period must be > 0.0
Self-explanatory.
Fix rigid/small nvt/npt/nph damping parameters must be > 0.0
Self-explanatory.
Fix rigid/small nvt/npt/nph dilate group ID does not exist
Self-explanatory.
Fix rigid/small requires an atom map, see atom_modify
Self-explanatory.
Fix rigid/small requires atom attribute molecule
Self-explanatory.
Fix rigid: Bad principal moments
The principal moments of inertia computed for a rigid body are not within the required tolerances.
Fix shake cannot be used with minimization
Cannot use fix shake while doing an energy minimization since it turns off bonds that should
contribute to the energy.
Fix shake molecule template must have shake info
The defined molecule does not specify SHAKE information.
Fix spring couple group ID does not exist
Self-explanatory.
Fix srd can only currently be used with comm_style brick
This is a current restriction in LAMMPS.
Fix srd lamda must be >= 0.6 of SRD grid size
This is a requirement for accuracy reasons.
Fix srd no-slip requires atom attribute torque
This is because the SRD collisions will impart torque to the solute particles.
Fix srd requires SRD particles all have same mass
Self-explanatory.
Fix srd requires ghost atoms store velocity
Use the comm_modify vel yes command to enable this.
Fix srd requires newton pair on
Self-explanatory.
Fix store/state compute array is accessed out-of-range
Self-explanatory.
Fix store/state compute does not calculate a per-atom array
The compute calculates a per-atom vector.
Fix store/state compute does not calculate a per-atom vector
The compute calculates a per-atom vector.
Fix store/state compute does not calculate per-atom values
Computes that calculate global or local quantities cannot be used with fix store/state.
Fix store/state fix array is accessed out-of-range
Self-explanatory.
Fix store/state fix does not calculate a per-atom array
The fix calculates a per-atom vector.
Fix store/state fix does not calculate a per-atom vector
The fix calculates a per-atom array.
Fix store/state fix does not calculate per-atom values
Fixes that calculate global or local quantities cannot be used with fix store/state.
Fix store/state for atom property that isn't allocated
Self-explanatory.
Fix store/state variable is not atom-style variable
LAMMPS Users Manual
253
Only atom-style variables calculate per-atom quantities.
Fix temp/berendsen period must be > 0.0
Self-explanatory.
Fix temp/berendsen variable returned negative temperature
Self-explanatory.
Fix temp/csld is not compatible with fix rattle or fix shake
These two commands cannot currently be used together with fix temp/csld.
Fix temp/csld variable returned negative temperature
Self-explanatory.
Fix temp/csvr variable returned negative temperature
Self-explanatory.
Fix temp/rescale variable returned negative temperature
Self-explanatory.
Fix tfmc displacement length must be > 0
Self-explanatory.
Fix tfmc is not compatible with fix shake
These two commands cannot currently be used together.
Fix tfmc temperature must be > 0
Self-explanatory.
Fix thermal/conductivity swap value must be positive
Self-explanatory.
Fix tmd must come after integration fixes
Any fix tmd command must appear in the input script after all time integration fixes (nve, nvt, npt).
See the fix tmd documentation for details.
Fix ttm electron temperatures must be > 0.0
Self-explanatory.
Fix ttm electronic_density must be > 0.0
Self-explanatory.
Fix ttm electronic_specific_heat must be > 0.0
Self-explanatory.
Fix ttm electronic_thermal_conductivity must be >= 0.0
Self-explanatory.
Fix ttm gamma_p must be > 0.0
Self-explanatory.
Fix ttm gamma_s must be >= 0.0
Self-explanatory.
Fix ttm number of nodes must be > 0
Self-explanatory.
Fix ttm v_0 must be >= 0.0
Self-explanatory.
Fix used in compute chunk/atom not computed at compatible time
The chunk/atom compute cannot query the output of the fix on a timestep it is needed.
Fix used in compute reduce not computed at compatible time
Fixes generate their values on specific timesteps. Compute reduce is requesting a value on a
non-allowed timestep.
Fix used in compute slice not computed at compatible time
Fixes generate their values on specific timesteps. Compute slice is requesting a value on a
non-allowed timestep.
Fix vector cannot set output array intensive/extensive from these inputs
The inputs to the command have conflicting intensive/extensive attributes. You need to use more than
one fix vector command.
LAMMPS Users Manual
254
Fix vector compute does not calculate a scalar
Self-explanatory.
Fix vector compute does not calculate a vector
Self-explanatory.
Fix vector compute vector is accessed out-of-range
Self-explanatory.
Fix vector fix does not calculate a scalar
Self-explanatory.
Fix vector fix does not calculate a vector
Self-explanatory.
Fix vector fix vector is accessed out-of-range
Self-explanatory.
Fix vector variable is not equal-style variable
Self-explanatory.
Fix viscosity swap value must be positive
Self-explanatory.
Fix viscosity vtarget value must be positive
Self-explanatory.
Fix wall cutoff <= 0.0
Self-explanatory.
Fix wall/colloid requires atom style sphere
Self-explanatory.
Fix wall/colloid requires extended particles
One of the particles has radius 0.0.
Fix wall/gran is incompatible with Pair style
Must use a granular pair style to define the parameters needed for this fix.
Fix wall/gran requires atom style sphere
Self-explanatory.
Fix wall/piston command only available at zlo
The face keyword must be zlo.
Fix wall/region colloid requires atom style sphere
Self-explanatory.
Fix wall/region colloid requires extended particles
One of the particles has radius 0.0.
Fix wall/region cutoff <= 0.0
Self-explanatory.
Fix_modify pressure ID does not compute pressure
The compute ID assigned to the fix must compute pressure.
Fix_modify temperature ID does not compute temperature
The compute ID assigned to the fix must compute temperature.
For triclinic deformation, specified target stress must be hydrostatic
Triclinic pressure control is allowed using the tri keyword, but non-hydrostatic pressure control can
not be used in this case.
Found no restart file matching pattern
When using a "*" in the restart file name, no matching file was found.
GPU library not compiled for this accelerator
Self-explanatory.
GPU package does not (yet) work with atom_style template
Self-explanatory.
GPU particle split must be set to 1 for this pair style.
For this pair style, you cannot run part of the force calculation on the host. See the package command.
LAMMPS Users Manual
255
GPU split param must be positive for hybrid pair styles
See the package gpu command.
GPUs are requested but Kokkos has not been compiled for CUDA
Recompile Kokkos with CUDA support to use GPUs.
Ghost velocity forward comm not yet implemented with Kokkos
This is a current restriction.
Gmask function in equal-style variable formula
Gmask is per-atom operation.
Gravity changed since fix pour was created
The gravity vector defined by fix gravity must be static.
Gravity must point in -y to use with fix pour in 2d
Self-explanatory.
Gravity must point in -z to use with fix pour in 3d
Self-explanatory.
Grmask function in equal-style variable formula
Grmask is per-atom operation.
Group ID does not exist
A group ID used in the group command does not exist.
Group ID in variable formula does not exist
Self-explanatory.
Group all cannot be made dynamic
This operation is not allowed.
Group command before simulation box is defined
The group command cannot be used before a read_data, read_restart, or create_box command.
Group dynamic cannot reference itself
Self-explanatory.
Group dynamic parent group cannot be dynamic
Self-explanatory.
Group dynamic parent group does not exist
Self-explanatory.
Group region ID does not exist
A region ID used in the group command does not exist.
If read_dump purges it cannot replace or trim
These operations are not compatible. See the read_dump doc page for details.
Illegal ... command
Self-explanatory. Check the input script syntax and compare to the documentation for the command.
You can use -echo screen as a command-line option when running LAMMPS to see the offending
line.
Illegal COMB parameter
One or more of the coefficients defined in the potential file is invalid.
Illegal COMB3 parameter
One or more of the coefficients defined in the potential file is invalid.
Illegal Stillinger-Weber parameter
One or more of the coefficients defined in the potential file is invalid.
Illegal Tersoff parameter
One or more of the coefficients defined in the potential file is invalid.
Illegal Vashishta parameter
One or more of the coefficients defined in the potential file is invalid.
Illegal compute voronoi/atom command (occupation and (surface or edges))
Self-explanatory.
Illegal coul/streitz parameter
LAMMPS Users Manual
256
One or more of the coefficients defined in the potential file is invalid.
Illegal dump_modify sfactor value (must be > 0.0)
Self-explanatory.
Illegal dump_modify tfactor value (must be > 0.0)
Self-explanatory.
Illegal fix gcmc gas mass <= 0
The computed mass of the designated gas molecule or atom type was less than or equal to zero.
Illegal fix tfmc random seed
Seeds can only be nonzero positive integers.
Illegal fix wall/piston velocity
The piston velocity must be positive.
Illegal integrate style
Self-explanatory.
Illegal nb3b/harmonic parameter
One or more of the coefficients defined in the potential file is invalid.
Illegal number of angle table entries
There must be at least 2 table entries.
Illegal number of bond table entries
There must be at least 2 table entries.
Illegal number of pair table entries
There must be at least 2 table entries.
Illegal or unset periodicity in restart
This error should not normally occur unless the restart file is invalid.
Illegal range increment value
The increment must be >= 1.
Illegal simulation box
The lower bound of the simulation box is greater than the upper bound.
Illegal size double vector read requested
This error should not normally occur unless the restart file is invalid.
Illegal size integer vector read requested
This error should not normally occur unless the restart file is invalid.
Illegal size string or corrupt restart
This error should not normally occur unless the restart file is invalid.
Imageint setting in lmptype.h is invalid
Imageint must be as large or larger than smallint.
Imageint setting in lmptype.h is not compatible
Format of imageint stored in restart file is not consistent with LAMMPS version you are running. See
the settings in src/lmptype.h
Improper atom missing in delete_bonds
The delete_bonds command cannot find one or more atoms in a particular improper on a particular
processor. The pairwise cutoff is too short or the atoms are too far apart to make a valid improper.
Improper atom missing in set command
The set command cannot find one or more atoms in a particular improper on a particular processor.
The pairwise cutoff is too short or the atoms are too far apart to make a valid improper.
Improper atoms %d %d %d %d missing on proc %d at step %ld
One or more of 4 atoms needed to compute a particular improper are missing on this processor.
Typically this is because the pairwise cutoff is set too short or the improper has blown apart and an
atom is too far away.
Improper atoms missing on proc %d at step %ld
One or more of 4 atoms needed to compute a particular improper are missing on this processor.
Typically this is because the pairwise cutoff is set too short or the improper has blown apart and an
LAMMPS Users Manual
257
atom is too far away.
Improper coeff for hybrid has invalid style
Improper style hybrid uses another improper style as one of its coefficients. The improper style used
in the improper_coeff command or read from a restart file is not recognized.
Improper coeffs are not set
No improper coefficients have been assigned in the data file or via the improper_coeff command.
Improper style hybrid cannot have hybrid as an argument
Self-explanatory.
Improper style hybrid cannot have none as an argument
Self-explanatory.
Improper style hybrid cannot use same improper style twice
Self-explanatory.
Improper_coeff command before improper_style is defined
Coefficients cannot be set in the data file or via the improper_coeff command until an improper_style
has been assigned.
Improper_coeff command before simulation box is defined
The improper_coeff command cannot be used before a read_data, read_restart, or create_box
command.
Improper_coeff command when no impropers allowed
The chosen atom style does not allow for impropers to be defined.
Improper_style command when no impropers allowed
The chosen atom style does not allow for impropers to be defined.
Impropers assigned incorrectly
Impropers read in from the data file were not assigned correctly to atoms. This means there is
something invalid about the topology definitions.
Impropers defined but no improper types
The data file header lists improper but no improper types.
Incomplete use of variables in create_atoms command
The var and set options must be used together.
Inconsistent iparam/jparam values in fix bond/create command
If itype and jtype are the same, then their maxbond and newtype settings must also be the same.
Inconsistent line segment in data file
The end points of the line segment are not equal distances from the center point which is the atom
coordinate.
Inconsistent triangle in data file
The centroid of the triangle as defined by the corner points is not the atom coordinate.
Inconsistent use of finite-size particles by molecule template molecules
Not all of the molecules define a radius for their constituent particles.
Incorrect # of floating-point values in Bodies section of data file
See doc page for body style.
Incorrect # of integer values in Bodies section of data file
See doc page for body style.
Incorrect %s format in data file
A section of the data file being read by fix property/atom does not have the correct number of values
per line.
Incorrect SNAP parameter file
The file cannot be parsed correctly, check its internal syntax.
Incorrect args for angle coefficients
Self-explanatory. Check the input script or data file.
Incorrect args for bond coefficients
Self-explanatory. Check the input script or data file.
LAMMPS Users Manual
258
Incorrect args for dihedral coefficients
Self-explanatory. Check the input script or data file.
Incorrect args for improper coefficients
Self-explanatory. Check the input script or data file.
Incorrect args for pair coefficients
Self-explanatory. Check the input script or data file.
Incorrect args in pair_style command
Self-explanatory.
Incorrect atom format in data file
Number of values per atom line in the data file is not consistent with the atom style.
Incorrect atom format in neb file
The number of fields per line is not what expected.
Incorrect bonus data format in data file
See the read_data doc page for a description of how various kinds of bonus data must be formatted for
certain atom styles.
Incorrect boundaries with slab Ewald
Must have periodic x,y dimensions and non-periodic z dimension to use 2d slab option with Ewald.
Incorrect boundaries with slab EwaldDisp
Must have periodic x,y dimensions and non-periodic z dimension to use 2d slab option with Ewald.
Incorrect boundaries with slab PPPM
Must have periodic x,y dimensions and non-periodic z dimension to use 2d slab option with PPPM.
Incorrect boundaries with slab PPPMDisp
Must have periodic x,y dimensions and non-periodic z dimension to use 2d slab option with
pppm/disp.
Incorrect element names in ADP potential file
The element names in the ADP file do not match those requested.
Incorrect element names in EAM potential file
The element names in the EAM file do not match those requested.
Incorrect format in COMB potential file
Incorrect number of words per line in the potential file.
Incorrect format in COMB3 potential file
Incorrect number of words per line in the potential file.
Incorrect format in MEAM potential file
Incorrect number of words per line in the potential file.
Incorrect format in SNAP coefficient file
Incorrect number of words per line in the coefficient file.
Incorrect format in SNAP parameter file
Incorrect number of words per line in the parameter file.
Incorrect format in Stillinger-Weber potential file
Incorrect number of words per line in the potential file.
Incorrect format in TMD target file
Format of file read by fix tmd command is incorrect.
Incorrect format in Tersoff potential file
Incorrect number of words per line in the potential file.
Incorrect format in Vashishta potential file
Incorrect number of words per line in the potential file.
Incorrect format in coul/streitz potential file
Incorrect number of words per line in the potential file.
Incorrect format in nb3b/harmonic potential file
Incorrect number of words per line in the potential file.
Incorrect integer value in Bodies section of data file
LAMMPS Users Manual
259
See doc page for body style.
Incorrect multiplicity arg for dihedral coefficients
Self-explanatory. Check the input script or data file.
Incorrect number of elements in potential file
Self-explanatory.
Incorrect rigid body format in fix rigid file
The number of fields per line is not what expected.
Incorrect rigid body format in fix rigid/small file
The number of fields per line is not what expected.
Incorrect sign arg for dihedral coefficients
Self-explanatory. Check the input script or data file.
Incorrect table format check for element types
Self-explanatory.
Incorrect velocity format in data file
Each atom style defines a format for the Velocity section of the data file. The read-in lines do not
match.
Incorrect weight arg for dihedral coefficients
Self-explanatory. Check the input script or data file.
Index between variable brackets must be positive
Self-explanatory.
Indexed per-atom vector in variable formula without atom map
Accessing a value from an atom vector requires the ability to lookup an atom index, which is provided
by an atom map. An atom map does not exist (by default) for non-molecular problems. Using the
atom_modify map command will force an atom map to be created.
Initial temperatures not all set in fix ttm
Self-explanatory.
Input line quote not followed by whitespace
An end quote must be followed by whitespace.
Insertion region extends outside simulation box
Self-explanatory.
Insufficient Jacobi rotations for POEMS body
Eigensolve for rigid body was not sufficiently accurate.
Insufficient Jacobi rotations for body nparticle
Eigensolve for rigid body was not sufficiently accurate.
Insufficient Jacobi rotations for rigid body
Eigensolve for rigid body was not sufficiently accurate.
Insufficient Jacobi rotations for rigid molecule
Eigensolve for rigid body was not sufficiently accurate.
Insufficient Jacobi rotations for triangle
The calculation of the inertia tensor of the triangle failed. This should not happen if it is a reasonably
shaped triangle.
Insufficient memory on accelerator
There is insufficient memory on one of the devices specified for the gpu package
Internal error in atom_style body
This error should not occur. Contact the developers.
Invalid -reorder N value
Self-explanatory.
Invalid Angles section in molecule file
Self-explanatory.
Invalid Bonds section in molecule file
Self-explanatory.
LAMMPS Users Manual
260
Invalid Boolean syntax in if command
Self-explanatory.
Invalid Charges section in molecule file
Self-explanatory.
Invalid Coords section in molecule file
Self-explanatory.
Invalid Diameters section in molecule file
Self-explanatory.
Invalid Dihedrals section in molecule file
Self-explanatory.
Invalid Impropers section in molecule file
Self-explanatory.
Invalid Kokkos command-line args
Self-explanatory. See Section 2.7 of the manual for details.
Invalid LAMMPS restart file
The file does not appear to be a LAMMPS restart file since it doesn't contain the correct magic string
at the beginning.
Invalid Masses section in molecule file
Self-explanatory.
Invalid REAX atom type
There is a mis-match between LAMMPS atom types and the elements listed in the ReaxFF force field
file.
Invalid Special Bond Counts section in molecule file
Self-explanatory.
Invalid Types section in molecule file
Self-explanatory.
Invalid angle count in molecule file
Self-explanatory.
Invalid angle table length
Length must be 2 or greater.
Invalid angle type in Angles section of data file
Angle type must be positive integer and within range of specified angle types.
Invalid angle type in Angles section of molecule file
Self-explanatory.
Invalid angle type index for fix shake
Self-explanatory.
Invalid args for non-hybrid pair coefficients
"NULL" is only supported in pair_coeff calls when using pair hybrid
Invalid argument to factorial %d
N must be >= 0 and <= 167, otherwise the factorial result is too large.
Invalid atom ID in %s section of data file
An atom in a section of the data file being read by fix property/atom has an invalid atom ID that is <=
0 or > the maximum existing atom ID.
Invalid atom ID in Angles section of data file
Atom IDs must be positive integers and within range of defined atoms.
Invalid atom ID in Angles section of molecule file
Self-explanatory.
Invalid atom ID in Atoms section of data file
Atom IDs must be positive integers.
Invalid atom ID in Bodies section of data file
Atom IDs must be positive integers and within range of defined atoms.
LAMMPS Users Manual
261
Invalid atom ID in Bonds section of data file
Atom IDs must be positive integers and within range of defined atoms.
Invalid atom ID in Bonds section of molecule file
Self-explanatory.
Invalid atom ID in Bonus section of data file
Atom IDs must be positive integers and within range of defined atoms.
Invalid atom ID in Dihedrals section of data file
Atom IDs must be positive integers and within range of defined atoms.
Invalid atom ID in Impropers section of data file
Atom IDs must be positive integers and within range of defined atoms.
Invalid atom ID in Velocities section of data file
Atom IDs must be positive integers and within range of defined atoms.
Invalid atom ID in dihedrals section of molecule file
Self-explanatory.
Invalid atom ID in impropers section of molecule file
Self-explanatory.
Invalid atom ID in variable file
Self-explanatory.
Invalid atom IDs in neb file
An ID in the file was not found in the system.
Invalid atom diameter in molecule file
Diameters must be >= 0.0.
Invalid atom mass for fix shake
Mass specified in fix shake command must be > 0.0.
Invalid atom mass in molecule file
Masses must be > 0.0.
Invalid atom type in Atoms section of data file
Atom types must range from 1 to specified # of types.
Invalid atom type in create_atoms command
The create_box command specified the range of valid atom types. An invalid type is being requested.
Invalid atom type in create_atoms mol command
The atom types in the defined molecule are added to the value specified in the create_atoms
command, as an offset. The final value for each atom must be between 1 to N, where N is the number
of atom types.
Invalid atom type in fix atom/swap command
The atom type specified in the atom/swap command does not exist.
Invalid atom type in fix bond/create command
Self-explanatory.
Invalid atom type in fix deposit command
Self-explanatory.
Invalid atom type in fix deposit mol command
The atom types in the defined molecule are added to the value specified in the create_atoms
command, as an offset. The final value for each atom must be between 1 to N, where N is the number
of atom types.
Invalid atom type in fix gcmc command
The atom type specified in the gcmc command does not exist.
Invalid atom type in fix pour command
Self-explanatory.
Invalid atom type in fix pour mol command
The atom types in the defined molecule are added to the value specified in the create_atoms
command, as an offset. The final value for each atom must be between 1 to N, where N is the number
LAMMPS Users Manual
262
of atom types.
Invalid atom type in molecule file
Atom types must range from 1 to specified # of types.
Invalid atom type in neighbor exclusion list
Atom types must range from 1 to Ntypes inclusive.
Invalid atom type index for fix shake
Atom types must range from 1 to Ntypes inclusive.
Invalid atom types in pair_write command
Atom types must range from 1 to Ntypes inclusive.
Invalid atom vector in variable formula
The atom vector is not recognized.
Invalid atom_style body command
No body style argument was provided.
Invalid atom_style command
Self-explanatory.
Invalid attribute in dump custom command
Self-explanatory.
Invalid attribute in dump local command
Self-explanatory.
Invalid attribute in dump modify command
Self-explanatory.
Invalid basis setting in create_atoms command
The basis index must be between 1 to N where N is the number of basis atoms in the lattice. The type
index must be between 1 to N where N is the number of atom types.
Invalid basis setting in fix append/atoms command
The basis index must be between 1 to N where N is the number of basis atoms in the lattice. The type
index must be between 1 to N where N is the number of atom types.
Invalid bin bounds in compute chunk/atom
The lo/hi values are inconsistent.
Invalid bin bounds in fix ave/spatial
The lo/hi values are inconsistent.
Invalid body nparticle command
Arguments in atom-style command are not correct.
Invalid bond count in molecule file
Self-explanatory.
Invalid bond table length
Length must be 2 or greater.
Invalid bond type in Bonds section of data file
Bond type must be positive integer and within range of specified bond types.
Invalid bond type in Bonds section of molecule file
Self-explanatory.
Invalid bond type in create_bonds command
Self-explanatory.
Invalid bond type in fix bond/break command
Self-explanatory.
Invalid bond type in fix bond/create command
Self-explanatory.
Invalid bond type index for fix shake
Self-explanatory. Check the fix shake command in the input script.
Invalid coeffs for this dihedral style
Cannot set class 2 coeffs in data file for this dihedral style.
LAMMPS Users Manual
263
Invalid color in dump_modify command
The specified color name was not in the list of recognized colors. See the dump_modify doc page.
Invalid color map min/max values
The min/max values are not consistent with either each other or with values in the color map.
Invalid command-line argument
One or more command-line arguments is invalid. Check the syntax of the command you are using to
launch LAMMPS.
Invalid compute ID in variable formula
The compute is not recognized.
Invalid create_atoms rotation vector for 2d model
The rotation vector can only have a z component.
Invalid custom OpenCL parameter string.
There are not enough or too many parameters in the custom string for package GPU.
Invalid cutoff in comm_modify command
Specified cutoff must be >= 0.0.
Invalid cutoffs in pair_write command
Inner cutoff must be larger than 0.0 and less than outer cutoff.
Invalid d1 or d2 value for pair colloid coeff
Neither d1 or d2 can be < 0.
Invalid data file section: Angle Coeffs
Atom style does not allow angles.
Invalid data file section: AngleAngle Coeffs
Atom style does not allow impropers.
Invalid data file section: AngleAngleTorsion Coeffs
Atom style does not allow dihedrals.
Invalid data file section: AngleTorsion Coeffs
Atom style does not allow dihedrals.
Invalid data file section: Angles
Atom style does not allow angles.
Invalid data file section: Bodies
Atom style does not allow bodies.
Invalid data file section: Bond Coeffs
Atom style does not allow bonds.
Invalid data file section: BondAngle Coeffs
Atom style does not allow angles.
Invalid data file section: BondBond Coeffs
Atom style does not allow angles.
Invalid data file section: BondBond13 Coeffs
Atom style does not allow dihedrals.
Invalid data file section: Bonds
Atom style does not allow bonds.
Invalid data file section: Dihedral Coeffs
Atom style does not allow dihedrals.
Invalid data file section: Dihedrals
Atom style does not allow dihedrals.
Invalid data file section: Ellipsoids
Atom style does not allow ellipsoids.
Invalid data file section: EndBondTorsion Coeffs
Atom style does not allow dihedrals.
Invalid data file section: Improper Coeffs
Atom style does not allow impropers.
LAMMPS Users Manual
264
Invalid data file section: Impropers
Atom style does not allow impropers.
Invalid data file section: Lines
Atom style does not allow lines.
Invalid data file section: MiddleBondTorsion Coeffs
Atom style does not allow dihedrals.
Invalid data file section: Triangles
Atom style does not allow triangles.
Invalid delta_conf in tad command
The value must be between 0 and 1 inclusive.
Invalid density in Atoms section of data file
Density value cannot be <= 0.0.
Invalid density in set command
Density must be > 0.0.
Invalid diameter in set command
Self-explanatory.
Invalid dihedral count in molecule file
Self-explanatory.
Invalid dihedral type in Dihedrals section of data file
Dihedral type must be positive integer and within range of specified dihedral types.
Invalid dihedral type in dihedrals section of molecule file
Self-explanatory.
Invalid dipole length in set command
Self-explanatory.
Invalid displace_atoms rotate axis for 2d
Axis must be in z direction.
Invalid dump dcd filename
Filenames used with the dump dcd style cannot be binary or compressed or cause multiple files to be
written.
Invalid dump frequency
Dump frequency must be 1 or greater.
Invalid dump image element name
The specified element name was not in the standard list of elements. See the dump_modify doc page.
Invalid dump image filename
The file produced by dump image cannot be binary and must be for a single processor.
Invalid dump image persp value
Persp value must be >= 0.0.
Invalid dump image theta value
Theta must be between 0.0 and 180.0 inclusive.
Invalid dump image zoom value
Zoom value must be > 0.0.
Invalid dump movie filename
The file produced by dump movie cannot be binary or compressed and must be a single file for a
single processor.
Invalid dump xtc filename
Filenames used with the dump xtc style cannot be binary or compressed or cause multiple files to be
written.
Invalid dump xyz filename
Filenames used with the dump xyz style cannot be binary or cause files to be written by each
processor.
Invalid dump_modify threshold operator
LAMMPS Users Manual
265
Operator keyword used for threshold specification in not recognized.
Invalid entry in -reorder file
Self-explanatory.
Invalid fix ID in variable formula
The fix is not recognized.
Invalid fix ave/time off column
Self-explanatory.
Invalid fix box/relax command for a 2d simulation
Fix box/relax styles involving the z dimension cannot be used in a 2d simulation.
Invalid fix box/relax command pressure settings
If multiple dimensions are coupled, those dimensions must be specified.
Invalid fix box/relax pressure settings
Settings for coupled dimensions must be the same.
Invalid fix nvt/npt/nph command for a 2d simulation
Cannot control z dimension in a 2d model.
Invalid fix nvt/npt/nph command pressure settings
If multiple dimensions are coupled, those dimensions must be specified.
Invalid fix nvt/npt/nph pressure settings
Settings for coupled dimensions must be the same.
Invalid fix press/berendsen for a 2d simulation
The z component of pressure cannot be controlled for a 2d model.
Invalid fix press/berendsen pressure settings
Settings for coupled dimensions must be the same.
Invalid fix qeq parameter file
Element index > number of atom types.
Invalid fix rigid npt/nph command for a 2d simulation
Cannot control z dimension in a 2d model.
Invalid fix rigid npt/nph command pressure settings
If multiple dimensions are coupled, those dimensions must be specified.
Invalid fix rigid/small npt/nph command for a 2d simulation
Cannot control z dimension in a 2d model.
Invalid fix rigid/small npt/nph command pressure settings
If multiple dimensions are coupled, those dimensions must be specified.
Invalid flag in force field section of restart file
Unrecognized entry in restart file.
Invalid flag in header section of restart file
Unrecognized entry in restart file.
Invalid flag in peratom section of restart file
The format of this section of the file is not correct.
Invalid flag in type arrays section of restart file
Unrecognized entry in restart file.
Invalid frequency in temper command
Nevery must be > 0.
Invalid group ID in neigh_modify command
A group ID used in the neigh_modify command does not exist.
Invalid group function in variable formula
Group function is not recognized.
Invalid group in comm_modify command
Self-explanatory.
Invalid image up vector
Up vector cannot be (0,0,0).
LAMMPS Users Manual
266
Invalid immediate variable
Syntax of immediate value is incorrect.
Invalid improper count in molecule file
Self-explanatory.
Invalid improper type in Impropers section of data file
Improper type must be positive integer and within range of specified improper types.
Invalid improper type in impropers section of molecule file
Self-explanatory.
Invalid index for non-body particles in compute body/local command
Only indices 1,2,3 can be used for non-body particles.
Invalid index in compute body/local command
Self-explanatory.
Invalid is_active() function in variable formula
Self-explanatory.
Invalid is_available() function in variable formula
Self-explanatory.
Invalid is_defined() function in variable formula
Self-explanatory.
Invalid keyword in angle table parameters
Self-explanatory.
Invalid keyword in bond table parameters
Self-explanatory.
Invalid keyword in compute angle/local command
Self-explanatory.
Invalid keyword in compute bond/local command
Self-explanatory.
Invalid keyword in compute dihedral/local command
Self-explanatory.
Invalid keyword in compute improper/local command
Self-explanatory.
Invalid keyword in compute pair/local command
Self-explanatory.
Invalid keyword in compute property/atom command
Self-explanatory.
Invalid keyword in compute property/chunk command
Self-explanatory.
Invalid keyword in compute property/local command
Self-explanatory.
Invalid keyword in dump cfg command
Self-explanatory.
Invalid keyword in pair table parameters
Keyword used in list of table parameters is not recognized.
Invalid length in set command
Self-explanatory.
Invalid mass in set command
Self-explanatory.
Invalid mass line in data file
Self-explanatory.
Invalid mass value
Self-explanatory.
Invalid math function in variable formula
LAMMPS Users Manual
267
Self-explanatory.
Invalid math/group/special function in variable formula
Self-explanatory.
Invalid option in lattice command for non-custom style
Certain lattice keywords are not supported unless the lattice style is "custom".
Invalid order of forces within respa levels
For respa, ordering of force computations within respa levels must obey certain rules. E.g. bonds
cannot be compute less frequently than angles, pairwise forces cannot be computed less frequently
than kspace, etc.
Invalid pair table cutoff
Cutoffs in pair_coeff command are not valid with read-in pair table.
Invalid pair table length
Length of read-in pair table is invalid
Invalid param file for fix qeq/shielded
Invalid value of gamma.
Invalid param file for fix qeq/slater
Zeta value is 0.0.
Invalid partitions in processors part command
Valid partitions are numbered 1 to N and the sender and receiver cannot be the same partition.
Invalid python command
Self-explanatory. Check the input script syntax and compare to the documentation for the command.
You can use -echo screen as a command-line option when running LAMMPS to see the offending
line.
Invalid radius in Atoms section of data file
Radius must be >= 0.0.
Invalid random number seed in fix ttm command
Random number seed must be > 0.
Invalid random number seed in set command
Random number seed must be > 0.
Invalid replace values in compute reduce
Self-explanatory.
Invalid rigid body ID in fix rigid file
The ID does not match the number of an existing ID of rigid bodies that are defined by the fix rigid
command.
Invalid rigid body ID in fix rigid/small file
The ID does not match the number of an existing ID of rigid bodies that are defined by the fix
rigid/small command.
Invalid run command N value
The number of timesteps must fit in a 32-bit integer. If you want to run for more steps than this,
perform multiple shorter runs.
Invalid run command start/stop value
Self-explanatory.
Invalid run command upto value
Self-explanatory.
Invalid seed for Marsaglia random # generator
The initial seed for this random number generator must be a positive integer less than or equal to 900
million.
Invalid seed for Park random # generator
The initial seed for this random number generator must be a positive integer.
Invalid shake angle type in molecule file
Self-explanatory.
LAMMPS Users Manual
268
Invalid shake atom in molecule file
Self-explanatory.
Invalid shake bond type in molecule file
Self-explanatory.
Invalid shake flag in molecule file
Self-explanatory.
Invalid shape in Ellipsoids section of data file
Self-explanatory.
Invalid shape in Triangles section of data file
Two or more of the triangle corners are duplicate points.
Invalid shape in set command
Self-explanatory.
Invalid shear direction for fix wall/gran
Self-explanatory.
Invalid special atom index in molecule file
Self-explanatory.
Invalid special function in variable formula
Self-explanatory.
Invalid style in pair_write command
Self-explanatory. Check the input script.
Invalid syntax in variable formula
Self-explanatory.
Invalid t_event in prd command
Self-explanatory.
Invalid t_event in tad command
The value must be greater than 0.
Invalid template atom in Atoms section of data file
The atom indices must be between 1 to N, where N is the number of atoms in the template molecule
the atom belongs to.
Invalid template index in Atoms section of data file
The template indices must be between 1 to N, where N is the number of molecules in the template.
Invalid thermo keyword in variable formula
The keyword is not recognized.
Invalid threads_per_atom specified.
For 3-body potentials on the GPU, the threads_per_atom setting cannot be greater than 4 for NVIDIA
GPUs.
Invalid timestep reset for fix ave/atom
Resetting the timestep has invalidated the sequence of timesteps this fix needs to process.
Invalid timestep reset for fix ave/chunk
Resetting the timestep has invalidated the sequence of timesteps this fix needs to process.
Invalid timestep reset for fix ave/correlate
Resetting the timestep has invalidated the sequence of timesteps this fix needs to process.
Invalid timestep reset for fix ave/histo
Resetting the timestep has invalidated the sequence of timesteps this fix needs to process.
Invalid timestep reset for fix ave/spatial
Resetting the timestep has invalidated the sequence of timesteps this fix needs to process.
Invalid timestep reset for fix ave/time
Resetting the timestep has invalidated the sequence of timesteps this fix needs to process.
Invalid tmax in tad command
The value must be greater than 0.0.
Invalid type for mass set
LAMMPS Users Manual
269
Mass command must set a type from 1-N where N is the number of atom types.
Invalid use of library file() function
This function is called thru the library interface. This error should not occur. Contact the developers if
it does.
Invalid value in set command
The value specified for the setting is invalid, likely because it is too small or too large.
Invalid variable evaluation in variable formula
A variable used in a formula could not be evaluated.
Invalid variable in next command
Self-explanatory.
Invalid variable name
Variable name used in an input script line is invalid.
Invalid variable name in variable formula
Variable name is not recognized.
Invalid variable style in special function next
Only file-style or atomfile-style variables can be used with next().
Invalid variable style with next command
Variable styles equal and world cannot be used in a next command.
Invalid volume in set command
Volume must be > 0.0.
Invalid wiggle direction for fix wall/gran
Self-explanatory.
Invoked angle equil angle on angle style none
Self-explanatory.
Invoked angle single on angle style none
Self-explanatory.
Invoked bond equil distance on bond style none
Self-explanatory.
Invoked bond single on bond style none
Self-explanatory.
Invoked pair single on pair style none
A command (e.g. a dump) attempted to invoke the single() function on a pair style none, which is
illegal. You are probably attempting to compute per-atom quantities with an undefined pair style.
Invoking coulombic in pair style lj/coul requires atom attribute q
The atom style defined does not have this attribute.
Invoking coulombic in pair style lj/long/dipole/long requires atom attribute q
The atom style defined does not have these attributes.
KIM neighbor iterator exceeded range
This should not happen. It likely indicates a bug in the KIM implementation of the interatomic
potential where it is requesting neighbors incorrectly.
KOKKOS package does not yet support comm_style tiled
Self-explanatory.
KOKKOS package requires a kokkos enabled atom_style
Self-explanatory.
KSpace accuracy must be > 0
The kspace accuracy designated in the input must be greater than zero.
KSpace accuracy too large to estimate G vector
Reduce the accuracy request or specify gwald explicitly via the kspace_modify command.
KSpace accuracy too low
Requested accuracy must be less than 1.0.
KSpace solver requires a pair style
LAMMPS Users Manual
270
No pair style is defined.
KSpace style does not yet support triclinic geometries
The specified kspace style does not allow for non-orthogonal simulation boxes.
KSpace style has not yet been set
Cannot use kspace_modify command until a kspace style is set.
KSpace style is incompatible with Pair style
Setting a kspace style requires that a pair style with matching long-range Coulombic or dispersion
components be used.
Keyword %s in MEAM parameter file not recognized
Self-explanatory.
Kokkos has been compiled for CUDA but no GPUs are requested
One or more GPUs must be used when Kokkos is compiled for CUDA.
Kspace style does not support compute group/group
Self-explanatory.
Kspace style pppm/disp/tip4p requires newton on
Self-explanatory.
Kspace style pppm/tip4p requires newton on
Self-explanatory.
Kspace style requires atom attribute q
The atom style defined does not have these attributes.
Kspace_modify eigtol must be smaller than one
Self-explanatory.
LAMMPS is not built with Python embedded
This is done by including the PYTHON package before LAMMPS is built. This is required to use
python-style variables.
LAMMPS unit_style lj not supported by KIM models
Self-explanatory. Check the input script or data file.
LJ6 off not supported in pair_style buck/long/coul/long
Self-explanatory.
Label wasn't found in input script
Self-explanatory.
Lattice orient vectors are not orthogonal
The three specified lattice orientation vectors must be mutually orthogonal.
Lattice orient vectors are not right-handed
The three specified lattice orientation vectors must create a right-handed coordinate system such that
a1 cross a2 = a3.
Lattice primitive vectors are collinear
The specified lattice primitive vectors do not for a unit cell with non-zero volume.
Lattice settings are not compatible with 2d simulation
One or more of the specified lattice vectors has a non-zero z component.
Lattice spacings are invalid
Each x,y,z spacing must be > 0.
Lattice style incompatible with simulation dimension
2d simulation can use sq, sq2, or hex lattice. 3d simulation can use sc, bcc, or fcc lattice.
Log of zero/negative value in variable formula
Self-explanatory.
Lost atoms via balance: original %ld current %ld
This should not occur. Report the problem to the developers.
Lost atoms: original %ld current %ld
Lost atoms are checked for each time thermo output is done. See the thermo_modify lost command
for options. Lost atoms usually indicate bad dynamics, e.g. atoms have been blown far out of the
LAMMPS Users Manual
271
simulation box, or moved further than one processor's sub-domain away before reneighboring.
MEAM library error %d
A call to the MEAM Fortran library returned an error.
MPI_LMP_BIGINT and bigint in lmptype.h are not compatible
The size of the MPI datatype does not match the size of a bigint.
MPI_LMP_TAGINT and tagint in lmptype.h are not compatible
The size of the MPI datatype does not match the size of a tagint.
MSM can only currently be used with comm_style brick
This is a current restriction in LAMMPS.
MSM grid is too large
The global MSM grid is larger than OFFSET in one or more dimensions. OFFSET is currently set to
16384. You likely need to decrease the requested accuracy.
MSM order must be 4, 6, 8, or 10
This is a limitation of the MSM implementation in LAMMPS: the MSM order can only be 4, 6, 8, or
10.
Mass command before simulation box is defined
The mass command cannot be used before a read_data, read_restart, or create_box command.
Matrix factorization to split dispersion coefficients failed
This should not normally happen. Contact the developers.
Min_style command before simulation box is defined
The min_style command cannot be used before a read_data, read_restart, or create_box command.
Minimization could not find thermo_pe compute
This compute is created by the thermo command. It must have been explicitly deleted by a uncompute
command.
Minimize command before simulation box is defined
The minimize command cannot be used before a read_data, read_restart, or create_box command.
Mismatched brackets in variable
Self-explanatory.
Mismatched compute in variable formula
A compute is referenced incorrectly or a compute that produces per-atom values is used in an
equal-style variable formula.
Mismatched fix in variable formula
A fix is referenced incorrectly or a fix that produces per-atom values is used in an equal-style variable
formula.
Mismatched variable in variable formula
A variable is referenced incorrectly or an atom-style variable that produces per-atom values is used in
an equal-style variable formula.
Modulo 0 in variable formula
Self-explanatory.
Molecule IDs too large for compute chunk/atom
The IDs must not be larger than can be stored in a 32-bit integer since chunk IDs are 32-bit integers.
Molecule auto special bond generation overflow
Counts exceed maxspecial setting for other atoms in system.
Molecule file has angles but no nangles setting
Self-explanatory.
Molecule file has body params but no setting for them
Self-explanatory.
Molecule file has bonds but no nbonds setting
Self-explanatory.
Molecule file has dihedrals but no ndihedrals setting
Self-explanatory.
LAMMPS Users Manual
272
Molecule file has impropers but no nimpropers setting
Self-explanatory.
Molecule file has no Body Doubles section
Self-explanatory.
Molecule file has no Body Integers section
Self-explanatory.
Molecule file has special flags but no bonds
Self-explanatory.
Molecule file needs both Special Bond sections
Self-explanatory.
Molecule file requires atom style body
Self-explanatory.
Molecule file shake flags not before shake atoms
The order of the two sections is important.
Molecule file shake flags not before shake bonds
The order of the two sections is important.
Molecule file shake info is incomplete
All 3 SHAKE sections are needed.
Molecule file special list does not match special count
The number of values in an atom's special list does not match count.
Molecule file z center-of-mass must be 0.0 for 2d
Self-explanatory.
Molecule file z coord must be 0.0 for 2d
Self-explanatory.
Molecule natoms must be 1 for body particle
Self-explanatory.
Molecule sizescale must be 1.0 for body particle
Self-explanatory.
Molecule template ID for atom_style template does not exist
Self-explanatory.
Molecule template ID for create_atoms does not exist
Self-explanatory.
Molecule template ID for fix deposit does not exist
Self-explanatory.
Molecule template ID for fix gcmc does not exist
Self-explanatory.
Molecule template ID for fix pour does not exist
Self-explanatory.
Molecule template ID for fix rigid/small does not exist
Self-explanatory.
Molecule template ID for fix shake does not exist
Self-explanatory.
Molecule template ID must be alphanumeric or underscore characters
Self-explanatory.
Molecule topology/atom exceeds system topology/atom
The number of bonds, angles, etc per-atom in the molecule exceeds the system setting. See the
create_box command for how to specify these values.
Molecule topology type exceeds system topology type
The number of bond, angle, etc types in the molecule exceeds the system setting. See the create_box
command for how to specify these values.
More than one fix deform
LAMMPS Users Manual
273
Only one fix deform can be defined at a time.
More than one fix freeze
Only one of these fixes can be defined, since the granular pair potentials access it.
More than one fix shake
Only one fix shake can be defined.
Mu not allowed when not using semi-grand in fix atom/swap command
Self-explanatory.
Must define angle_style before Angle Coeffs
Must use an angle_style command before reading a data file that defines Angle Coeffs.
Must define angle_style before BondAngle Coeffs
Must use an angle_style command before reading a data file that defines Angle Coeffs.
Must define angle_style before BondBond Coeffs
Must use an angle_style command before reading a data file that defines Angle Coeffs.
Must define bond_style before Bond Coeffs
Must use a bond_style command before reading a data file that defines Bond Coeffs.
Must define dihedral_style before AngleAngleTorsion Coeffs
Must use a dihedral_style command before reading a data file that defines AngleAngleTorsion Coeffs.
Must define dihedral_style before AngleTorsion Coeffs
Must use a dihedral_style command before reading a data file that defines AngleTorsion Coeffs.
Must define dihedral_style before BondBond13 Coeffs
Must use a dihedral_style command before reading a data file that defines BondBond13 Coeffs.
Must define dihedral_style before Dihedral Coeffs
Must use a dihedral_style command before reading a data file that defines Dihedral Coeffs.
Must define dihedral_style before EndBondTorsion Coeffs
Must use a dihedral_style command before reading a data file that defines EndBondTorsion Coeffs.
Must define dihedral_style before MiddleBondTorsion Coeffs
Must use a dihedral_style command before reading a data file that defines MiddleBondTorsion
Coeffs.
Must define improper_style before AngleAngle Coeffs
Must use an improper_style command before reading a data file that defines AngleAngle Coeffs.
Must define improper_style before Improper Coeffs
Must use an improper_style command before reading a data file that defines Improper Coeffs.
Must define pair_style before Pair Coeffs
Must use a pair_style command before reading a data file that defines Pair Coeffs.
Must define pair_style before PairIJ Coeffs
Must use a pair_style command before reading a data file that defines PairIJ Coeffs.
Must have more than one processor partition to temper
Cannot use the temper command with only one processor partition. Use the -partition command-line
option.
Must read Atoms before Angles
The Atoms section of a data file must come before an Angles section.
Must read Atoms before Bodies
The Atoms section of a data file must come before a Bodies section.
Must read Atoms before Bonds
The Atoms section of a data file must come before a Bonds section.
Must read Atoms before Dihedrals
The Atoms section of a data file must come before a Dihedrals section.
Must read Atoms before Ellipsoids
The Atoms section of a data file must come before a Ellipsoids section.
Must read Atoms before Impropers
The Atoms section of a data file must come before an Impropers section.
LAMMPS Users Manual
274
Must read Atoms before Lines
The Atoms section of a data file must come before a Lines section.
Must read Atoms before Triangles
The Atoms section of a data file must come before a Triangles section.
Must read Atoms before Velocities
The Atoms section of a data file must come before a Velocities section.
Must set both respa inner and outer
Cannot use just the inner or outer option with respa without using the other.
Must set number of threads via package omp command
Because you are using the USER-OMP package, set the number of threads via its settings, not by the
pair_style snap nthreads setting.
Must shrink-wrap piston boundary
The boundary style of the face where the piston is applied must be of type s (shrink-wrapped).
Must specify a region in fix deposit
The region keyword must be specified with this fix.
Must specify a region in fix pour
Self-explanatory.
Must specify at least 2 types in fix atom/swap command
Self-explanatory.
Must use 'kspace_modify pressure/scalar no' for rRESPA with kspace_style MSM
The kspace scalar pressure option cannot (yet) be used with rRESPA.
Must use 'kspace_modify pressure/scalar no' for tensor components with kspace_style msm
Otherwise MSM will compute only a scalar pressure. See the kspace_modify command for details on
this setting.
Must use 'kspace_modify pressure/scalar no' to obtain per-atom virial with kspace_style MSM
The kspace scalar pressure option cannot be used to obtain per-atom virial.
Must use 'kspace_modify pressure/scalar no' with GPU MSM Pair styles
The kspace scalar pressure option is not (yet) compatible with GPU MSM Pair styles.
Must use 'kspace_modify pressure/scalar no' with kspace_style msm/cg
The kspace scalar pressure option is not compatible with kspace_style msm/cg.
Must use -in switch with multiple partitions
A multi-partition simulation cannot read the input script from stdin. The -in command-line option
must be used to specify a file.
Must use Kokkos half/thread or full neighbor list with threads or GPUs
Using Kokkos half-neighbor lists with threading is not allowed.
Must use a block or cylinder region with fix pour
Self-explanatory.
Must use a block region with fix pour for 2d simulations
Self-explanatory.
Must use a bond style with TIP4P potential
TIP4P potentials assume bond lengths in water are constrained by a fix shake command.
Must use a molecular atom style with fix poems molecule
Self-explanatory.
Must use a z-axis cylinder region with fix pour
Self-explanatory.
Must use an angle style with TIP4P potential
TIP4P potentials assume angles in water are constrained by a fix shake command.
Must use atom map style array with Kokkos
See the atom_modify map command.
Must use atom style with molecule IDs with fix bond/swap
Self-explanatory.
LAMMPS Users Manual
275
Must use pair_style comb or comb3 with fix qeq/comb
Self-explanatory.
Must use variable energy with fix addforce
Must define an energy variable when applying a dynamic force during minimization.
Must use variable energy with fix efield
You must define an energy when performing a minimization with a variable E-field.
NEB command before simulation box is defined
Self-explanatory.
NEB requires damped dynamics minimizer
Use a different minimization style.
NEB requires use of fix neb
Self-explanatory.
NL ramp in wall/piston only implemented in zlo for now
The ramp keyword can only be used for piston applied to face zlo.
Need nswaptypes mu values in fix atom/swap command
Self-explanatory.
Needed bonus data not in data file
Some atom styles require bonus data. See the read_data doc page for details.
Needed molecular topology not in data file
The header of the data file indicated bonds, angles, etc would be included, but they are not present.
Neigh_modify exclude molecule requires atom attribute molecule
Self-explanatory.
Neigh_modify include group != atom_modify first group
Self-explanatory.
Neighbor delay must be 0 or multiple of every setting
The delay and every parameters set via the neigh_modify command are inconsistent. If the delay
setting is non-zero, then it must be a multiple of the every setting.
Neighbor include group not allowed with ghost neighbors
This is a current restriction within LAMMPS.
Neighbor list overflow, boost neigh_modify one
There are too many neighbors of a single atom. Use the neigh_modify command to increase the max
number of neighbors allowed for one atom. You may also want to boost the page size.
Neighbor multi not yet enabled for ghost neighbors
This is a current restriction within LAMMPS.
Neighbor multi not yet enabled for granular
Self-explanatory.
Neighbor multi not yet enabled for rRESPA
Self-explanatory.
Neighbor page size must be >= 10x the one atom setting
This is required to prevent wasting too much memory.
New atom IDs exceed maximum allowed ID
See the setting for tagint in the src/lmptype.h file.
New bond exceeded bonds per atom in create_bonds
See the read_data command for info on setting the "extra bond per atom" header value to allow for
additional bonds to be formed.
New bond exceeded bonds per atom in fix bond/create
See the read_data command for info on setting the "extra bond per atom" header value to allow for
additional bonds to be formed.
New bond exceeded special list size in fix bond/create
See the special_bonds extra command for info on how to leave space in the special bonds list to allow
for additional bonds to be formed.
LAMMPS Users Manual
276
Newton bond change after simulation box is defined
The newton command cannot be used to change the newton bond value after a read_data, read_restart,
or create_box command.
Next command must list all universe and uloop variables
This is to insure they stay in sync.
No Kspace style defined for compute group/group
Self-explanatory.
No OpenMP support compiled in
An OpenMP flag is set, but LAMMPS was not built with OpenMP support.
No angle style is defined for compute angle/local
Self-explanatory.
No angles allowed with this atom style
Self-explanatory.
No atoms in data file
The header of the data file indicated that atoms would be included, but they are not present.
No basis atoms in lattice
Basis atoms must be defined for lattice style user.
No bodies allowed with this atom style
Self-explanatory. Check data file.
No bond style is defined for compute bond/local
Self-explanatory.
No bonds allowed with this atom style
Self-explanatory.
No box information in dump. You have to use 'box no'
Self-explanatory.
No count or invalid atom count in molecule file
The number of atoms must be specified.
No dihedral style is defined for compute dihedral/local
Self-explanatory.
No dihedrals allowed with this atom style
Self-explanatory.
No dump custom arguments specified
The dump custom command requires that atom quantities be specified to output to dump file.
No dump local arguments specified
Self-explanatory.
No ellipsoids allowed with this atom style
Self-explanatory. Check data file.
No fix gravity defined for fix pour
Gravity is required to use fix pour.
No improper style is defined for compute improper/local
Self-explanatory.
No impropers allowed with this atom style
Self-explanatory.
No input values for fix ave/spatial
Self-explanatory.
No lines allowed with this atom style
Self-explanatory. Check data file.
No matching element in ADP potential file
The ADP potential file does not contain elements that match the requested elements.
No matching element in EAM potential file
The EAM potential file does not contain elements that match the requested elements.
LAMMPS Users Manual
277
No molecule topology allowed with atom style template
The data file cannot specify the number of bonds, angles, etc, because this info if inferred from the
molecule templates.
No overlap of box and region for create_atoms
Self-explanatory.
No pair coul/streitz for fix qeq/slater
These commands must be used together.
No pair hbond/dreiding coefficients set
Self-explanatory.
No pair style defined for compute group/group
Cannot calculate group interactions without a pair style defined.
No pair style is defined for compute pair/local
Self-explanatory.
No pair style is defined for compute property/local
Self-explanatory.
No rigid bodies defined
The fix specification did not end up defining any rigid bodies.
No triangles allowed with this atom style
Self-explanatory. Check data file.
No values in fix ave/chunk command
Self-explanatory.
No values in fix ave/time command
Self-explanatory.
Non digit character between brackets in variable
Self-explanatory.
Non integer # of swaps in temper command
Swap frequency in temper command must evenly divide the total # of timesteps.
Non-numeric box dimensions - simulation unstable
The box size has apparently blown up.
Non-zero atom IDs with atom_modify id = no
Self-explanatory.
Non-zero read_data shift z value for 2d simulation
Self-explanatory.
Nprocs not a multiple of N for -reorder
Self-explanatory.
Number of core atoms != number of shell atoms
There must be a one-to-one pairing of core and shell atoms.
Numeric index is out of bounds
A command with an argument that specifies an integer or range of integers is using a value that is less
than 1 or greater than the maximum allowed limit.
One or more Atom IDs is negative
Atom IDs must be positive integers.
One or more atom IDs is too big
The limit on atom IDs is set by the SMALLBIG, BIGBIG, SMALLSMALL setting in your Makefile.
See Section_start 2.2 of the manual for more details.
One or more atom IDs is zero
Either all atoms IDs must be zero or none of them.
One or more atoms belong to multiple rigid bodies
Two or more rigid bodies defined by the fix rigid command cannot contain the same atom.
One or more rigid bodies are a single particle
Self-explanatory.
LAMMPS Users Manual
278
One or zero atoms in rigid body
Any rigid body defined by the fix rigid command must contain 2 or more atoms.
Only 2 types allowed when not using semi-grand in fix atom/swap command
Self-explanatory.
Only one cut-off allowed when requesting all long
Self-explanatory.
Only one cutoff allowed when requesting all long
Self-explanatory.
Only zhi currently implemented for fix append/atoms
Self-explanatory.
Out of range atoms - cannot compute MSM
One or more atoms are attempting to map their charge to a MSM grid point that is not owned by a
processor. This is likely for one of two reasons, both of them bad. First, it may mean that an atom near
the boundary of a processor's sub-domain has moved more than 1/2 the neighbor skin distance
without neighbor lists being rebuilt and atoms being migrated to new processors. This also means you
may be missing pairwise interactions that need to be computed. The solution is to change the
re-neighboring criteria via the neigh_modify command. The safest settings are "delay 0 every 1 check
yes". Second, it may mean that an atom has moved far outside a processor's sub-domain or even the
entire simulation box. This indicates bad physics, e.g. due to highly overlapping atoms, too large a
timestep, etc.
Out of range atoms - cannot compute PPPM
One or more atoms are attempting to map their charge to a PPPM grid point that is not owned by a
processor. This is likely for one of two reasons, both of them bad. First, it may mean that an atom near
the boundary of a processor's sub-domain has moved more than 1/2 the neighbor skin distance
without neighbor lists being rebuilt and atoms being migrated to new processors. This also means you
may be missing pairwise interactions that need to be computed. The solution is to change the
re-neighboring criteria via the neigh_modify command. The safest settings are "delay 0 every 1 check
yes". Second, it may mean that an atom has moved far outside a processor's sub-domain or even the
entire simulation box. This indicates bad physics, e.g. due to highly overlapping atoms, too large a
timestep, etc.
Out of range atoms - cannot compute PPPMDisp
One or more atoms are attempting to map their charge to a PPPM grid point that is not owned by a
processor. This is likely for one of two reasons, both of them bad. First, it may mean that an atom near
the boundary of a processor's sub-domain has moved more than 1/2 the neighbor skin distance
without neighbor lists being rebuilt and atoms being migrated to new processors. This also means you
may be missing pairwise interactions that need to be computed. The solution is to change the
re-neighboring criteria via the neigh_modify command. The safest settings are "delay 0 every 1 check
yes". Second, it may mean that an atom has moved far outside a processor's sub-domain or even the
entire simulation box. This indicates bad physics, e.g. due to highly overlapping atoms, too large a
timestep, etc.
Overflow of allocated fix vector storage
This should not normally happen if the fix correctly calculated how long the vector will grow to.
Contact the developers.
Overlapping large/large in pair colloid
This potential is infinite when there is an overlap.
Overlapping small/large in pair colloid
This potential is infinite when there is an overlap.
POEMS fix must come before NPT/NPH fix
NPT/NPH fix must be defined in input script after all poems fixes, else the fix contribution to the
pressure virial is incorrect.
PPPM can only currently be used with comm_style brick
LAMMPS Users Manual
279
This is a current restriction in LAMMPS.
PPPM grid is too large
The global PPPM grid is larger than OFFSET in one or more dimensions. OFFSET is currently set to
4096. You likely need to decrease the requested accuracy.
PPPM grid stencil extends beyond nearest neighbor processor
This is not allowed if the kspace_modify overlap setting is no.
PPPM order < minimum allowed order
The default minimum order is 2. This can be reset by the kspace_modify minorder command.
PPPM order cannot be < 2 or > than %d
This is a limitation of the PPPM implementation in LAMMPS.
PPPMDisp Coulomb grid is too large
The global PPPM grid is larger than OFFSET in one or more dimensions. OFFSET is currently set to
4096. You likely need to decrease the requested accuracy.
PPPMDisp Dispersion grid is too large
The global PPPM grid is larger than OFFSET in one or more dimensions. OFFSET is currently set to
4096. You likely need to decrease the requested accuracy.
PPPMDisp can only currently be used with comm_style brick
This is a current restriction in LAMMPS.
PPPMDisp coulomb order cannot be greater than %d
This is a limitation of the PPPM implementation in LAMMPS.
PPPMDisp used but no parameters set, for further information please see the pppm/disp documentation
An efficient and accurate usage of the pppm/disp requires settings via the kspace_modify command.
Please see the pppm/disp documentation for further instructions.
PRD command before simulation box is defined
The prd command cannot be used before a read_data, read_restart, or create_box command.
PRD nsteps must be multiple of t_event
Self-explanatory.
PRD t_corr must be multiple of t_event
Self-explanatory.
Package command after simulation box is defined
The package command cannot be used afer a read_data, read_restart, or create_box command.
Package cuda command without USER-CUDA package enabled
The USER-CUDA package must be installed via "make yes-user-cuda" before LAMMPS is built, and
the "-c on" must be used to enable the package.
Package gpu command without GPU package installed
The GPU package must be installed via "make yes-gpu" before LAMMPS is built.
Package intel command without USER-INTEL package installed
The USER-INTEL package must be installed via "make yes-user-intel" before LAMMPS is built.
Package kokkos command without KOKKOS package enabled
The KOKKOS package must be installed via "make yes-kokkos" before LAMMPS is built, and the
"-k on" must be used to enable the package.
Package omp command without USER-OMP package installed
The USER-OMP package must be installed via "make yes-user-omp" before LAMMPS is built.
Pair body requires atom style body
Self-explanatory.
Pair body requires body style nparticle
This pair style is specific to the nparticle body style.
Pair brownian requires atom style sphere
Self-explanatory.
Pair brownian requires extended particles
One of the particles has radius 0.0.
LAMMPS Users Manual
280
Pair brownian requires monodisperse particles
All particles must be the same finite size.
Pair brownian/poly requires atom style sphere
Self-explanatory.
Pair brownian/poly requires extended particles
One of the particles has radius 0.0.
Pair brownian/poly requires newton pair off
Self-explanatory.
Pair coeff for hybrid has invalid style
Style in pair coeff must have been listed in pair_style command.
Pair coul/wolf requires atom attribute q
The atom style defined does not have this attribute.
Pair cutoff < Respa interior cutoff
One or more pairwise cutoffs are too short to use with the specified rRESPA cutoffs.
Pair dipole/cut requires atom attributes q, mu, torque
The atom style defined does not have these attributes.
Pair dipole/cut/gpu requires atom attributes q, mu, torque
The atom style defined does not have this attribute.
Pair dipole/long requires atom attributes q, mu, torque
The atom style defined does not have these attributes.
Pair dipole/sf/gpu requires atom attributes q, mu, torque
The atom style defined does not one or more of these attributes.
Pair distance < table inner cutoff
Two atoms are closer together than the pairwise table allows.
Pair distance > table outer cutoff
Two atoms are further apart than the pairwise table allows.
Pair dpd requires ghost atoms store velocity
Use the comm_modify vel yes command to enable this.
Pair gayberne epsilon a,b,c coeffs are not all set
Each atom type involved in pair_style gayberne must have these 3 coefficients set at least once.
Pair gayberne requires atom style ellipsoid
Self-explanatory.
Pair gayberne requires atoms with same type have same shape
Self-explanatory.
Pair gayberne/gpu requires atom style ellipsoid
Self-explanatory.
Pair gayberne/gpu requires atoms with same type have same shape
Self-explanatory.
Pair granular requires atom attributes radius, rmass
The atom style defined does not have these attributes.
Pair granular requires ghost atoms store velocity
Use the comm_modify vel yes command to enable this.
Pair granular with shear history requires newton pair off
This is a current restriction of the implementation of pair granular styles with history.
Pair hybrid single calls do not support per sub-style special bond values
Self-explanatory.
Pair hybrid sub-style does not support single call
You are attempting to invoke a single() call on a pair style that doesn't support it.
Pair hybrid sub-style is not used
No pair_coeff command used a sub-style specified in the pair_style command.
Pair inner cutoff < Respa interior cutoff
LAMMPS Users Manual
281
One or more pairwise cutoffs are too short to use with the specified rRESPA cutoffs.
Pair inner cutoff >= Pair outer cutoff
The specified cutoffs for the pair style are inconsistent.
Pair line/lj requires atom style line
Self-explanatory.
Pair lj/long/dipole/long requires atom attributes mu, torque
The atom style defined does not have these attributes.
Pair lubricate requires atom style sphere
Self-explanatory.
Pair lubricate requires ghost atoms store velocity
Use the comm_modify vel yes command to enable this.
Pair lubricate requires monodisperse particles
All particles must be the same finite size.
Pair lubricate/poly requires atom style sphere
Self-explanatory.
Pair lubricate/poly requires extended particles
One of the particles has radius 0.0.
Pair lubricate/poly requires ghost atoms store velocity
Use the comm_modify vel yes command to enable this.
Pair lubricate/poly requires newton pair off
Self-explanatory.
Pair lubricateU requires atom style sphere
Self-explanatory.
Pair lubricateU requires ghost atoms store velocity
Use the comm_modify vel yes command to enable this.
Pair lubricateU requires monodisperse particles
All particles must be the same finite size.
Pair lubricateU/poly requires ghost atoms store velocity
Use the comm_modify vel yes command to enable this.
Pair lubricateU/poly requires newton pair off
Self-explanatory.
Pair peri lattice is not identical in x, y, and z
The lattice defined by the lattice command must be cubic.
Pair peri requires a lattice be defined
Use the lattice command for this purpose.
Pair peri requires an atom map, see atom_modify
Even for atomic systems, an atom map is required to find Peridynamic bonds. Use the atom_modify
command to define one.
Pair resquared epsilon a,b,c coeffs are not all set
Self-explanatory.
Pair resquared epsilon and sigma coeffs are not all set
Self-explanatory.
Pair resquared requires atom style ellipsoid
Self-explanatory.
Pair resquared requires atoms with same type have same shape
Self-explanatory.
Pair resquared/gpu requires atom style ellipsoid
Self-explanatory.
Pair resquared/gpu requires atoms with same type have same shape
Self-explanatory.
Pair style AIREBO requires atom IDs
LAMMPS Users Manual
282
This is a requirement to use the AIREBO potential.
Pair style AIREBO requires newton pair on
See the newton command. This is a restriction to use the AIREBO potential.
Pair style BOP requires atom IDs
This is a requirement to use the BOP potential.
Pair style BOP requires newton pair on
See the newton command. This is a restriction to use the BOP potential.
Pair style COMB requires atom IDs
This is a requirement to use the AIREBO potential.
Pair style COMB requires atom attribute q
Self-explanatory.
Pair style COMB requires newton pair on
See the newton command. This is a restriction to use the COMB potential.
Pair style COMB3 requires atom IDs
This is a requirement to use the COMB3 potential.
Pair style COMB3 requires atom attribute q
Self-explanatory.
Pair style COMB3 requires newton pair on
See the newton command. This is a restriction to use the COMB3 potential.
Pair style LCBOP requires atom IDs
This is a requirement to use the LCBOP potential.
Pair style LCBOP requires newton pair on
See the newton command. This is a restriction to use the Tersoff potential.
Pair style MEAM requires newton pair on
See the newton command. This is a restriction to use the MEAM potential.
Pair style SNAP requires newton pair on
See the newton command. This is a restriction to use the SNAP potential.
Pair style Stillinger-Weber requires atom IDs
This is a requirement to use the SW potential.
Pair style Stillinger-Weber requires newton pair on
See the newton command. This is a restriction to use the SW potential.
Pair style Tersoff requires atom IDs
This is a requirement to use the Tersoff potential.
Pair style Tersoff requires newton pair on
See the newton command. This is a restriction to use the Tersoff potential.
Pair style Vashishta requires atom IDs
This is a requirement to use the Vashishta potential.
Pair style Vashishta requires newton pair on
See the newton command. This is a restriction to use the Vashishta potential.
Pair style bop requires comm ghost cutoff at least 3x larger than %g
Use the communicate ghost command to set this. See the pair bop doc page for more details.
Pair style born/coul/long requires atom attribute q
An atom style that defines this attribute must be used.
Pair style born/coul/long/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style born/coul/wolf requires atom attribute q
The atom style defined does not have this attribute.
Pair style buck/coul/cut requires atom attribute q
The atom style defined does not have this attribute.
Pair style buck/coul/long requires atom attribute q
The atom style defined does not have these attributes.
LAMMPS Users Manual
283
Pair style buck/coul/long/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style buck/long/coul/long requires atom attribute q
The atom style defined does not have this attribute.
Pair style coul/cut requires atom attribute q
The atom style defined does not have these attributes.
Pair style coul/cut/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style coul/debye/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style coul/dsf requires atom attribute q
The atom style defined does not have this attribute.
Pair style coul/dsf/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style coul/long/gpu requires atom attribute q
The atom style defined does not have these attributes.
Pair style coul/streitz requires atom attribute q
Self-explanatory.
Pair style does not have extra field requested by compute pair/local
The pair style does not support the pN value requested by the compute pair/local command.
Pair style does not support bond_style quartic
The pair style does not have a single() function, so it can not be invoked by bond_style quartic.
Pair style does not support compute group/group
The pair_style does not have a single() function, so it cannot be invoked by the compute group/group
command.
Pair style does not support compute pair/local
The pair style does not have a single() function, so it can not be invoked by compute pair/local.
Pair style does not support compute property/local
The pair style does not have a single() function, so it can not be invoked by fix bond/swap.
Pair style does not support fix bond/swap
The pair style does not have a single() function, so it can not be invoked by fix bond/swap.
Pair style does not support pair_write
The pair style does not have a single() function, so it can not be invoked by pair write.
Pair style does not support rRESPA inner/middle/outer
You are attempting to use rRESPA options with a pair style that does not support them.
Pair style granular with history requires atoms have IDs
Atoms in the simulation do not have IDs, so history effects cannot be tracked by the granular pair
potential.
Pair style hbond/dreiding requires an atom map, see atom_modify
Self-explanatory.
Pair style hbond/dreiding requires atom IDs
Self-explanatory.
Pair style hbond/dreiding requires molecular system
Self-explanatory.
Pair style hbond/dreiding requires newton pair on
See the newton command for details.
Pair style hybrid cannot have hybrid as an argument
Self-explanatory.
Pair style hybrid cannot have none as an argument
Self-explanatory.
Pair style is incompatible with KSpace style
LAMMPS Users Manual
284
If a pair style with a long-range Coulombic component is selected, then a kspace style must also be
used.
Pair style is incompatible with TIP4P KSpace style
The pair style does not have the requires TIP4P settings.
Pair style lj/charmm/coul/charmm requires atom attribute q
The atom style defined does not have these attributes.
Pair style lj/charmm/coul/long requires atom attribute q
The atom style defined does not have these attributes.
Pair style lj/charmm/coul/long/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/class2/coul/cut requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/class2/coul/long requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/class2/coul/long/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/cut/coul/cut requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/cut/coul/cut/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/cut/coul/debye/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/cut/coul/dsf requires atom attribute q
The atom style defined does not have these attributes.
Pair style lj/cut/coul/dsf/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/cut/coul/long requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/cut/coul/long/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/cut/tip4p/cut requires atom IDs
This is a requirement to use this potential.
Pair style lj/cut/tip4p/cut requires atom attribute q
The atom style defined does not have this attribute.
Pair style lj/cut/tip4p/cut requires newton pair on
See the newton command. This is a restriction to use this potential.
Pair style lj/cut/tip4p/long requires atom IDs
There are no atom IDs defined in the system and the TIP4P potential requires them to find O,H atoms
with a water molecule.
Pair style lj/cut/tip4p/long requires atom attribute q
The atom style defined does not have these attributes.
Pair style lj/cut/tip4p/long requires newton pair on
This is because the computation of constraint forces within a water molecule adds forces to atoms
owned by other processors.
Pair style lj/gromacs/coul/gromacs requires atom attribute q
An atom_style with this attribute is needed.
Pair style lj/long/dipole/long does not currently support respa
This feature is not yet supported.
Pair style lj/long/tip4p/long requires atom IDs
There are no atom IDs defined in the system and the TIP4P potential requires them to find O,H atoms
with a water molecule.
LAMMPS Users Manual
285
Pair style lj/long/tip4p/long requires atom attribute q
The atom style defined does not have these attributes.
Pair style lj/long/tip4p/long requires newton pair on
This is because the computation of constraint forces within a water molecule adds forces to atoms
owned by other processors.
Pair style lj/sdk/coul/long/gpu requires atom attribute q
The atom style defined does not have this attribute.
Pair style nb3b/harmonic requires atom IDs
This is a requirement to use this potential.
Pair style nb3b/harmonic requires newton pair on
See the newton command. This is a restriction to use this potential.
Pair style nm/cut/coul/cut requires atom attribute q
The atom style defined does not have this attribute.
Pair style nm/cut/coul/long requires atom attribute q
The atom style defined does not have this attribute.
Pair style peri requires atom style peri
Self-explanatory.
Pair style polymorphic requires atom IDs
This is a requirement to use the polymorphic potential.
Pair style polymorphic requires newton pair on
See the newton command. This is a restriction to use the polymorphic potential.
Pair style reax requires atom IDs
This is a requirement to use the ReaxFF potential.
Pair style reax requires atom attribute q
The atom style defined does not have this attribute.
Pair style reax requires newton pair on
This is a requirement to use the ReaxFF potential.
Pair style requires a KSpace style
No kspace style is defined.
Pair style requires use of kspace_style ewald/disp
Self-explanatory.
Pair style sw/gpu requires atom IDs
This is a requirement to use this potential.
Pair style sw/gpu requires newton pair off
See the newton command. This is a restriction to use this potential.
Pair style tersoff/gpu requires atom IDs
This is a requirement to use the tersoff/gpu potential.
Pair style tersoff/gpu requires newton pair off
See the newton command. This is a restriction to use this pair style.
Pair style tip4p/cut requires atom IDs
This is a requirement to use this potential.
Pair style tip4p/cut requires atom attribute q
The atom style defined does not have this attribute.
Pair style tip4p/cut requires newton pair on
See the newton command. This is a restriction to use this potential.
Pair style tip4p/long requires atom IDs
There are no atom IDs defined in the system and the TIP4P potential requires them to find O,H atoms
with a water molecule.
Pair style tip4p/long requires atom attribute q
The atom style defined does not have these attributes.
Pair style tip4p/long requires newton pair on
LAMMPS Users Manual
286
This is because the computation of constraint forces within a water molecule adds forces to atoms
owned by other processors.
Pair table cutoffs must all be equal to use with KSpace
When using pair style table with a long-range KSpace solver, the cutoffs for all atom type pairs must
all be the same, since the long-range solver starts at that cutoff.
Pair table parameters did not set N
List of pair table parameters must include N setting.
Pair tersoff/zbl requires metal or real units
This is a current restriction of this pair potential.
Pair tersoff/zbl/kk requires metal or real units
This is a current restriction of this pair potential.
Pair tri/lj requires atom style tri
Self-explanatory.
Pair yukawa/colloid requires atom style sphere
Self-explanatory.
Pair yukawa/colloid requires atoms with same type have same radius
Self-explanatory.
Pair yukawa/colloid/gpu requires atom style sphere
Self-explanatory.
PairKIM only works with 3D problems
This is a current limitation.
Pair_coeff command before pair_style is defined
Self-explanatory.
Pair_coeff command before simulation box is defined
The pair_coeff command cannot be used before a read_data, read_restart, or create_box command.
Pair_modify command before pair_style is defined
Self-explanatory.
Pair_modify special setting for pair hybrid incompatible with global special_bonds setting
Cannot override a setting of 0.0 or 1.0 or change a setting between 0.0 and 1.0.
Pair_write command before pair_style is defined
Self-explanatory.
Particle on or inside fix wall surface
Particles must be "exterior" to the wall in order for energy/force to be calculated.
Particle outside surface of region used in fix wall/region
Particles must be inside the region for energy/force to be calculated. A particle outside the region
generates an error.
Per-atom compute in equal-style variable formula
Equal-style variables cannot use per-atom quantities.
Per-atom energy was not tallied on needed timestep
You are using a thermo keyword that requires potentials to have tallied energy, but they didn't on this
timestep. See the variable doc page for ideas on how to make this work.
Per-atom fix in equal-style variable formula
Equal-style variables cannot use per-atom quantities.
Per-atom virial was not tallied on needed timestep
You are using a thermo keyword that requires potentials to have tallied the virial, but they didn't on
this timestep. See the variable doc page for ideas on how to make this work.
Per-processor system is too big
The number of owned atoms plus ghost atoms on a single processor must fit in 32-bit integer.
Potential energy ID for fix neb does not exist
Self-explanatory.
Potential energy ID for fix nvt/nph/npt does not exist
LAMMPS Users Manual
287
A compute for potential energy must be defined.
Potential file has duplicate entry
The potential file has more than one entry for the same element.
Potential file is missing an entry
The potential file does not have a needed entry.
Power by 0 in variable formula
Self-explanatory.
Pressure ID for fix box/relax does not exist
The compute ID needed to compute pressure for the fix does not exist.
Pressure ID for fix modify does not exist
Self-explanatory.
Pressure ID for fix npt/nph does not exist
Self-explanatory.
Pressure ID for fix press/berendsen does not exist
The compute ID needed to compute pressure for the fix does not exist.
Pressure ID for fix rigid npt/nph does not exist
Self-explanatory.
Pressure ID for thermo does not exist
The compute ID needed to compute pressure for thermodynamics does not exist.
Pressure control can not be used with fix nvt
Self-explanatory.
Pressure control can not be used with fix nvt/asphere
Self-explanatory.
Pressure control can not be used with fix nvt/body
Self-explanatory.
Pressure control can not be used with fix nvt/sllod
Self-explanatory.
Pressure control can not be used with fix nvt/sphere
Self-explanatory.
Pressure control must be used with fix nph
Self-explanatory.
Pressure control must be used with fix nph/asphere
Self-explanatory.
Pressure control must be used with fix nph/body
Self-explanatory.
Pressure control must be used with fix nph/small
Self-explanatory.
Pressure control must be used with fix nph/sphere
Self-explanatory.
Pressure control must be used with fix nphug
A pressure control keyword (iso, aniso, tri, x, y, or z) must be provided.
Pressure control must be used with fix npt
Self-explanatory.
Pressure control must be used with fix npt/asphere
Self-explanatory.
Pressure control must be used with fix npt/body
Self-explanatory.
Pressure control must be used with fix npt/sphere
Self-explanatory.
Processor count in z must be 1 for 2d simulation
Self-explanatory.
LAMMPS Users Manual
288
Processor partitions do not match number of allocated processors
The total number of processors in all partitions must match the number of processors LAMMPS is
running on.
Processors command after simulation box is defined
The processors command cannot be used after a read_data, read_restart, or create_box command.
Processors custom grid file is inconsistent
The vales in the custom file are not consistent with the number of processors you are running on or
the Px,Py,Pz settings of the processors command. Or there was not a setting for every processor.
Processors grid numa and map style are incompatible
Using numa for gstyle in the processors command requires using cart for the map option.
Processors part option and grid style are incompatible
Cannot use gstyle numa or custom with the part option.
Processors twogrid requires proc count be a multiple of core count
Self-explanatory.
Pstart and Pstop must have the same value
Self-explanatory.
Python function evaluation failed
The Python function did not run successfully and/or did not return a value (if it is supposed to return a
value). This is probably due to some error condition in the function.
Python function is not callable
The provided Python code was run successfully, but it not define a callable function with the required
name.
Python invoke of undefined function
Cannot invoke a function that has not been previously defined.
Python variable does not match Python function
This matching is defined by the python-style variable and the python command.
Python variable has no function
No python command was used to define the function associated with the python-style variable.
QEQ with 'newton pair off' not supported
See the newton command. This is a restriction to use the QEQ fixes.
R0 < 0 for fix spring command
Equilibrium spring length is invalid.
RATTLE coordinate constraints are not satisfied up to desired tolerance
Self-explanatory.
RATTLE determinant = 0.0
The determinant of the matrix being solved for a single cluster specified by the fix rattle command is
numerically invalid.
RATTLE failed
Certain constraints were not satisfied.
RATTLE velocity constraints are not satisfied up to desired tolerance
Self-explanatory.
Read data add offset is too big
It cannot be larger than the size of atom IDs, e.g. the maximum 32-bit integer.
Read dump of atom property that isn't allocated
Self-explanatory.
Read rerun dump file timestep > specified stop
Self-explanatory.
Read restart MPI-IO input not allowed with % in filename
This is because a % signifies one file per processor and MPI-IO creates one large file for all
processors.
Read_data shrink wrap did not assign all atoms correctly
LAMMPS Users Manual
289
This is typically because the box-size specified in the data file is large compared to the actual extent
of atoms in a shrink-wrapped dimension. When LAMMPS shrink-wraps the box atoms will be lost if
the processor they are re-assigned to is too far away. Choose a box size closer to the actual extent of
the atoms.
Read_dump command before simulation box is defined
The read_dump command cannot be used before a read_data, read_restart, or create_box command.
Read_dump field not found in dump file
Self-explanatory.
Read_dump triclinic status does not match simulation
Both the dump snapshot and the current LAMMPS simulation must be using either an orthogonal or
triclinic box.
Read_dump xyz fields do not have consistent scaling/wrapping
Self-explanatory.
Reading from MPI-IO filename when MPIIO package is not installed
Self-explanatory.
Reax_defs.h setting for NATDEF is too small
Edit the setting in the ReaxFF library and re-compile the library and re-build LAMMPS.
Reax_defs.h setting for NNEIGHMAXDEF is too small
Edit the setting in the ReaxFF library and re-compile the library and re-build LAMMPS.
Receiving partition in processors part command is already a receiver
Cannot specify a partition to be a receiver twice.
Region ID for compute chunk/atom does not exist
Self-explanatory.
Region ID for compute reduce/region does not exist
Self-explanatory.
Region ID for compute temp/region does not exist
Self-explanatory.
Region ID for dump custom does not exist
Self-explanatory.
Region ID for fix addforce does not exist
Self-explanatory.
Region ID for fix atom/swap does not exist
Self-explanatory.
Region ID for fix ave/spatial does not exist
Self-explanatory.
Region ID for fix aveforce does not exist
Self-explanatory.
Region ID for fix deposit does not exist
Self-explanatory.
Region ID for fix efield does not exist
Self-explanatory.
Region ID for fix evaporate does not exist
Self-explanatory.
Region ID for fix gcmc does not exist
Self-explanatory.
Region ID for fix heat does not exist
Self-explanatory.
Region ID for fix setforce does not exist
Self-explanatory.
Region ID for fix wall/region does not exist
Self-explanatory.
LAMMPS Users Manual
290
Region ID for group dynamic does not exist
Self-explanatory.
Region ID in variable formula does not exist
Self-explanatory.
Region cannot have 0 length rotation vector
Self-explanatory.
Region for fix oneway does not exist
Self-explanatory.
Region intersect region ID does not exist
Self-explanatory.
Region union or intersect cannot be dynamic
The sub-regions can be dynamic, but not the combined region.
Region union region ID does not exist
One or more of the region IDs specified by the region union command does not exist.
Replacing a fix, but new style != old style
A fix ID can be used a 2nd time, but only if the style matches the previous fix. In this case it is
assumed you with to reset a fix's parameters. This error may mean you are mistakenly re-using a fix
ID when you do not intend to.
Replicate command before simulation box is defined
The replicate command cannot be used before a read_data, read_restart, or create_box command.
Replicate did not assign all atoms correctly
Atoms replicated by the replicate command were not assigned correctly to processors. This is likely
due to some atom coordinates being outside a non-periodic simulation box.
Replicated system atom IDs are too big
See the setting for tagint in the src/lmptype.h file.
Replicated system is too big
See the setting for bigint in the src/lmptype.h file.
Required border comm not yet implemented with Kokkos
There are various limitations in the communication options supported by Kokkos.
Rerun command before simulation box is defined
The rerun command cannot be used before a read_data, read_restart, or create_box command.
Rerun dump file does not contain requested snapshot
Self-explanatory.
Resetting timestep size is not allowed with fix move
This is because fix move is moving atoms based on elapsed time.
Respa inner cutoffs are invalid
The first cutoff must be <= the second cutoff.
Respa levels must be >= 1
Self-explanatory.
Respa middle cutoffs are invalid
The first cutoff must be <= the second cutoff.
Restart file MPI-IO output not allowed with % in filename
This is because a % signifies one file per processor and MPI-IO creates one large file for all
processors.
Restart file byte ordering is not recognized
The file does not appear to be a LAMMPS restart file since it doesn't contain a recognized
byte-orderomg flag at the beginning.
Restart file byte ordering is swapped
The file was written on a machine with different byte-ordering than the machine you are reading it on.
Convert it to a text data file instead, on the machine you wrote it on.
Restart file incompatible with current version
LAMMPS Users Manual
291
This is probably because you are trying to read a file created with a version of LAMMPS that is too
old compared to the current version. Use your older version of LAMMPS and convert the restart file
to a data file.
Restart file is a MPI-IO file
The file is inconsistent with the filename you specified for it.
Restart file is a multi-proc file
The file is inconsistent with the filename you specified for it.
Restart file is not a MPI-IO file
The file is inconsistent with the filename you specified for it.
Restart file is not a multi-proc file
The file is inconsistent with the filename you specified for it.
Restart variable returned a bad timestep
The variable must return a timestep greater than the current timestep.
Restrain atoms %d %d %d %d missing on proc %d at step %ld
The 4 atoms in a restrain dihedral specified by the fix restrain command are not all accessible to a
processor. This probably means an atom has moved too far.
Restrain atoms %d %d %d missing on proc %d at step %ld
The 3 atoms in a restrain angle specified by the fix restrain command are not all accessible to a
processor. This probably means an atom has moved too far.
Restrain atoms %d %d missing on proc %d at step %ld
The 2 atoms in a restrain bond specified by the fix restrain command are not all accessible to a
processor. This probably means an atom has moved too far.
Reuse of compute ID
A compute ID cannot be used twice.
Reuse of dump ID
A dump ID cannot be used twice.
Reuse of molecule template ID
The template IDs must be unique.
Reuse of region ID
A region ID cannot be used twice.
Rigid body atoms %d %d missing on proc %d at step %ld
This means that an atom cannot find the atom that owns the rigid body it is part of, or vice versa. The
solution is to use the communicate cutoff command to insure ghost atoms are acquired from far
enough away to encompass the max distance printed when the fix rigid/small command was invoked.
Rigid body has degenerate moment of inertia
Fix poems will only work with bodies (collections of atoms) that have non-zero principal moments of
inertia. This means they must be 3 or more non-collinear atoms, even with joint atoms removed.
Rigid fix must come before NPT/NPH fix
NPT/NPH fix must be defined in input script after all rigid fixes, else the rigid fix contribution to the
pressure virial is incorrect.
Rmask function in equal-style variable formula
Rmask is per-atom operation.
Run command before simulation box is defined
The run command cannot be used before a read_data, read_restart, or create_box command.
Run command start value is after start of run
Self-explanatory.
Run command stop value is before end of run
Self-explanatory.
Run_style command before simulation box is defined
The run_style command cannot be used before a read_data, read_restart, or create_box command.
SRD bin size for fix srd differs from user request
LAMMPS Users Manual
292
Fix SRD had to adjust the bin size to fit the simulation box. See the cubic keyword if you want this
message to be an error vs warning.
SRD bins for fix srd are not cubic enough
The bin shape is not within tolerance of cubic. See the cubic keyword if you want this message to be
an error vs warning.
SRD particle %d started inside big particle %d on step %ld bounce %d
See the inside keyword if you want this message to be an error vs warning.
SRD particle %d started inside wall %d on step %ld bounce %d
See the inside keyword if you want this message to be an error vs warning.
Same dimension twice in fix ave/spatial
Self-explanatory.
Sending partition in processors part command is already a sender
Cannot specify a partition to be a sender twice.
Set command before simulation box is defined
The set command cannot be used before a read_data, read_restart, or create_box command.
Set command floating point vector does not exist
Self-explanatory.
Set command integer vector does not exist
Self-explanatory.
Set command with no atoms existing
No atoms are yet defined so the set command cannot be used.
Set region ID does not exist
Region ID specified in set command does not exist.
Shake angles have different bond types
All 3-atom angle-constrained SHAKE clusters specified by the fix shake command that are the same
angle type, must also have the same bond types for the 2 bonds in the angle.
Shake atoms %d %d %d %d missing on proc %d at step %ld
The 4 atoms in a single shake cluster specified by the fix shake command are not all accessible to a
processor. This probably means an atom has moved too far.
Shake atoms %d %d %d missing on proc %d at step %ld
The 3 atoms in a single shake cluster specified by the fix shake command are not all accessible to a
processor. This probably means an atom has moved too far.
Shake atoms %d %d missing on proc %d at step %ld
The 2 atoms in a single shake cluster specified by the fix shake command are not all accessible to a
processor. This probably means an atom has moved too far.
Shake cluster of more than 4 atoms
A single cluster specified by the fix shake command can have no more than 4 atoms.
Shake clusters are connected
A single cluster specified by the fix shake command must have a single central atom with up to 3
other atoms bonded to it.
Shake determinant = 0.0
The determinant of the matrix being solved for a single cluster specified by the fix shake command is
numerically invalid.
Shake fix must come before NPT/NPH fix
NPT fix must be defined in input script after SHAKE fix, else the SHAKE fix contribution to the
pressure virial is incorrect.
Shear history overflow, boost neigh_modify one
There are too many neighbors of a single atom. Use the neigh_modify command to increase the max
number of neighbors allowed for one atom. You may also want to boost the page size.
Small to big integers are not sized correctly
LAMMPS Users Manual
293
This error occurs whenthe sizes of smallint, imageint, tagint, bigint, as defined in src/lmptype.h are
not what is expected. Contact the developers if this occurs.
Smallint setting in lmptype.h is invalid
It has to be the size of an integer.
Smallint setting in lmptype.h is not compatible
Smallint stored in restart file is not consistent with LAMMPS version you are running.
Special list size exceeded in fix bond/create
See the read_data command for info on setting the "extra special per atom" header value to allow for
additional special values to be stored.
Specified processors != physical processors
The 3d grid of processors defined by the processors command does not match the number of
processors LAMMPS is being run on.
Specified target stress must be uniaxial or hydrostatic
Self-explanatory.
Sqrt of negative value in variable formula
Self-explanatory.
Subsequent read data induced too many angles per atom
See the create_box extra/angle/per/atom or read_data "extra angle per atom" header value to set this
limit larger.
Subsequent read data induced too many bonds per atom
See the create_box extra/bond/per/atom or read_data "extra bond per atom" header value to set this
limit larger.
Subsequent read data induced too many dihedrals per atom
See the create_box extra/dihedral/per/atom or read_data "extra dihedral per atom" header value to set
this limit larger.
Subsequent read data induced too many impropers per atom
See the create_box extra/improper/per/atom or read_data "extra improper per atom" header value to
set this limit larger.
Substitution for illegal variable
Input script line contained a variable that could not be substituted for.
Support for writing images in JPEG format not included
LAMMPS was not built with the -DLAMMPS_JPEG switch in the Makefile.
Support for writing images in PNG format not included
LAMMPS was not built with the -DLAMMPS_PNG switch in the Makefile.
Support for writing movies not included
LAMMPS was not built with the -DLAMMPS_FFMPEG switch in the Makefile
System in data file is too big
See the setting for bigint in the src/lmptype.h file.
System is not charge neutral, net charge = %g
The total charge on all atoms on the system is not 0.0. For some KSpace solvers this is an error.
TAD nsteps must be multiple of t_event
Self-explanatory.
TIP4P hydrogen has incorrect atom type
The TIP4P pairwise computation found an H atom whose type does not agree with the specified H
type.
TIP4P hydrogen is missing
The TIP4P pairwise computation failed to find the correct H atom within a water molecule.
TMD target file did not list all group atoms
The target file for the fix tmd command did not list all atoms in the fix group.
Tad command before simulation box is defined
Self-explanatory.
LAMMPS Users Manual
294
Tagint setting in lmptype.h is invalid
Tagint must be as large or larger than smallint.
Tagint setting in lmptype.h is not compatible
Format of tagint stored in restart file is not consistent with LAMMPS version you are running. See the
settings in src/lmptype.h
Target pressure for fix rigid/nph cannot be < 0.0
Self-explanatory.
Target pressure for fix rigid/npt/small cannot be < 0.0
Self-explanatory.
Target temperature for fix nvt/npt/nph cannot be 0.0
Self-explanatory.
Target temperature for fix rigid/npt cannot be 0.0
Self-explanatory.
Target temperature for fix rigid/npt/small cannot be 0.0
Self-explanatory.
Target temperature for fix rigid/nvt cannot be 0.0
Self-explanatory.
Target temperature for fix rigid/nvt/small cannot be 0.0
Self-explanatory.
Temper command before simulation box is defined
The temper command cannot be used before a read_data, read_restart, or create_box command.
Temperature ID for fix bond/swap does not exist
Self-explanatory.
Temperature ID for fix box/relax does not exist
Self-explanatory.
Temperature ID for fix nvt/npt does not exist
Self-explanatory.
Temperature ID for fix press/berendsen does not exist
Self-explanatory.
Temperature ID for fix rigid nvt/npt/nph does not exist
Self-explanatory.
Temperature ID for fix temp/berendsen does not exist
Self-explanatory.
Temperature ID for fix temp/csld does not exist
Self-explanatory.
Temperature ID for fix temp/csvr does not exist
Self-explanatory.
Temperature ID for fix temp/rescale does not exist
Self-explanatory.
Temperature compute degrees of freedom < 0
This should not happen if you are calculating the temperature on a valid set of atoms.
Temperature control can not be used with fix nph
Self-explanatory.
Temperature control can not be used with fix nph/asphere
Self-explanatory.
Temperature control can not be used with fix nph/body
Self-explanatory.
Temperature control can not be used with fix nph/sphere
Self-explanatory.
Temperature control must be used with fix nphug
The temp keyword must be provided.
LAMMPS Users Manual
295
Temperature control must be used with fix npt
Self-explanatory.
Temperature control must be used with fix npt/asphere
Self-explanatory.
Temperature control must be used with fix npt/body
Self-explanatory.
Temperature control must be used with fix npt/sphere
Self-explanatory.
Temperature control must be used with fix nvt
Self-explanatory.
Temperature control must be used with fix nvt/asphere
Self-explanatory.
Temperature control must be used with fix nvt/body
Self-explanatory.
Temperature control must be used with fix nvt/sllod
Self-explanatory.
Temperature control must be used with fix nvt/sphere
Self-explanatory.
Temperature control must not be used with fix nph/small
Self-explanatory.
Temperature for fix nvt/sllod does not have a bias
The specified compute must compute temperature with a bias.
Tempering could not find thermo_pe compute
This compute is created by the thermo command. It must have been explicitly deleted by a uncompute
command.
Tempering fix ID is not defined
The fix ID specified by the temper command does not exist.
Tempering temperature fix is not valid
The fix specified by the temper command is not one that controls temperature (nvt or langevin).
Test_descriptor_string already allocated
This is an internal error. Contact the developers.
The package gpu command is required for gpu styles
Self-explanatory.
Thermo and fix not computed at compatible times
Fixes generate values on specific timesteps. The thermo output does not match these timesteps.
Thermo compute array is accessed out-of-range
Self-explanatory.
Thermo compute does not compute array
Self-explanatory.
Thermo compute does not compute scalar
Self-explanatory.
Thermo compute does not compute vector
Self-explanatory.
Thermo compute vector is accessed out-of-range
Self-explanatory.
Thermo custom variable cannot be indexed
Self-explanatory.
Thermo custom variable is not equal-style variable
Only equal-style variables can be output with thermodynamics, not atom-style variables.
Thermo every variable returned a bad timestep
The variable must return a timestep greater than the current timestep.
LAMMPS Users Manual
296
Thermo fix array is accessed out-of-range
Self-explanatory.
Thermo fix does not compute array
Self-explanatory.
Thermo fix does not compute scalar
Self-explanatory.
Thermo fix does not compute vector
Self-explanatory.
Thermo fix vector is accessed out-of-range
Self-explanatory.
Thermo keyword in variable requires thermo to use/init pe
You are using a thermo keyword in a variable that requires potential energy to be calculated, but your
thermo output does not use it. Add it to your thermo output.
Thermo keyword in variable requires thermo to use/init press
You are using a thermo keyword in a variable that requires pressure to be calculated, but your thermo
output does not use it. Add it to your thermo output.
Thermo keyword in variable requires thermo to use/init temp
You are using a thermo keyword in a variable that requires temperature to be calculated, but your
thermo output does not use it. Add it to your thermo output.
Thermo style does not use press
Cannot use thermo_modify to set this parameter since the thermo_style is not computing this quantity.
Thermo style does not use temp
Cannot use thermo_modify to set this parameter since the thermo_style is not computing this quantity.
Thermo_modify every variable returned a bad timestep
The returned timestep is less than or equal to the current timestep.
Thermo_modify int format does not contain d character
Self-explanatory.
Thermo_modify pressure ID does not compute pressure
The specified compute ID does not compute pressure.
Thermo_modify temperature ID does not compute temperature
The specified compute ID does not compute temperature.
Thermo_style command before simulation box is defined
The thermo_style command cannot be used before a read_data, read_restart, or create_box command.
This variable thermo keyword cannot be used between runs
Keywords that refer to time (such as cpu, elapsed) do not make sense in between runs.
Threshhold for an atom property that isn't allocated
A dump threshold has been requested on a quantity that is not defined by the atom style used in this
simulation.
Timestep must be >= 0
Specified timestep is invalid.
Too big a problem to use velocity create loop all
The system size must fit in a 32-bit integer to use this option.
Too big a timestep for dump dcd
The timestep must fit in a 32-bit integer to use this dump style.
Too big a timestep for dump xtc
The timestep must fit in a 32-bit integer to use this dump style.
Too few bits for lookup table
Table size specified via pair_modify command does not work with your machine's floating point
representation.
Too few lines in %s section of data file
Self-explanatory.
LAMMPS Users Manual
297
Too few values in body lines in data file
Self-explanatory.
Too few values in body section of molecule file
Self-explanatory.
Too many -pk arguments in command line
The string formed by concatenating the arguments is too long. Use a package command in the input
script instead.
Too many MSM grid levels
The max number of MSM grid levels is hardwired to 10.
Too many args in variable function
More args are used than any variable function allows.
Too many atom pairs for pair bop
The number of atomic pairs exceeds the expected number. Check your atomic structure to ensure that
it is realistic.
Too many atom sorting bins
This is likely due to an immense simulation box that has blown up to a large size.
Too many atom triplets for pair bop
The number of three atom groups for angle determinations exceeds the expected number. Check your
atomic structure to ensure that it is realistic.
Too many atoms for dump dcd
The system size must fit in a 32-bit integer to use this dump style.
Too many atoms for dump xtc
The system size must fit in a 32-bit integer to use this dump style.
Too many atoms to dump sort
Cannot sort when running with more than 2^31 atoms.
Too many exponent bits for lookup table
Table size specified via pair_modify command does not work with your machine's floating point
representation.
Too many groups
The maximum number of atom groups (including the "all" group) is given by MAX_GROUP in
group.cpp and is 32.
Too many iterations
You must use a number of iterations that fit in a 32-bit integer for minimization.
Too many lines in one body in data file - boost MAXBODY
MAXBODY is a setting at the top of the src/read_data.cpp file. Set it larger and re-compile the code.
Too many local+ghost atoms for neighbor list
The number of nlocal + nghost atoms on a processor is limited by the size of a 32-bit integer with 2
bits removed for masking 1-2, 1-3, 1-4 neighbors.
Too many mantissa bits for lookup table
Table size specified via pair_modify command does not work with your machine's floating point
representation.
Too many masses for fix shake
The fix shake command cannot list more masses than there are atom types.
Too many molecules for fix poems
The limit is 2^31 = ~2 billion molecules.
Too many molecules for fix rigid
The limit is 2^31 = ~2 billion molecules.
Too many neighbor bins
This is likely due to an immense simulation box that has blown up to a large size.
Too many timesteps
The cumulative timesteps must fit in a 64-bit integer.
LAMMPS Users Manual
298
Too many timesteps for NEB
You must use a number of timesteps that fit in a 32-bit integer for NEB.
Too many total atoms
See the setting for bigint in the src/lmptype.h file.
Too many total bits for bitmapped lookup table
Table size specified via pair_modify command is too large. Note that a value of N generates a 2^N
size table.
Too many values in body lines in data file
Self-explanatory.
Too many values in body section of molecule file
Self-explanatory.
Too much buffered per-proc info for dump
The size of the buffered string must fit in a 32-bit integer for a dump.
Too much per-proc info for dump
Number of local atoms times number of columns must fit in a 32-bit integer for dump.
Tree structure in joint connections
Fix poems cannot (yet) work with coupled bodies whose joints connect the bodies in a tree structure.
Triclinic box skew is too large
The displacement in a skewed direction must be less than half the box length in that dimension. E.g.
the xy tilt must be between -half and +half of the x box length. This constraint can be relaxed by using
the box tilt command.
Tried to convert a double to int, but input_double > INT_MAX
Self-explanatory.
Trying to build an occasional neighbor list before initialization completed
This is not allowed. Source code caller needs to be modified.
Two fix ave commands using same compute chunk/atom command in incompatible ways
They are both attempting to "lock" the chunk/atom command so that the chunk assignments persist
for some number of timesteps, but are doing it in different ways.
Two groups cannot be the same in fix spring couple
Self-explanatory.
USER-CUDA mode requires CUDA variant of min style
CUDA mode is enabled, so the min style must include a cuda suffix.
USER-CUDA mode requires CUDA variant of run style
CUDA mode is enabled, so the run style must include a cuda suffix.
USER-CUDA package does not yet support comm_style tiled
Self-explanatory.
USER-CUDA package requires a cuda enabled atom_style
Self-explanatory.
Unable to initialize accelerator for use
There was a problem initializing an accelerator for the gpu package
Unbalanced quotes in input line
No matching end double quote was found following a leading double quote.
Unexpected end of -reorder file
Self-explanatory.
Unexpected end of AngleCoeffs section
Read a blank line.
Unexpected end of BondCoeffs section
Read a blank line.
Unexpected end of DihedralCoeffs section
Read a blank line.
Unexpected end of ImproperCoeffs section
LAMMPS Users Manual
299
Read a blank line.
Unexpected end of PairCoeffs section
Read a blank line.
Unexpected end of custom file
Self-explanatory.
Unexpected end of data file
LAMMPS hit the end of the data file while attempting to read a section. Something is wrong with the
format of the data file.
Unexpected end of dump file
A read operation from the file failed.
Unexpected end of fix rigid file
A read operation from the file failed.
Unexpected end of fix rigid/small file
A read operation from the file failed.
Unexpected end of molecule file
Self-explanatory.
Unexpected end of neb file
A read operation from the file failed.
Units command after simulation box is defined
The units command cannot be used after a read_data, read_restart, or create_box command.
Universe/uloop variable count < # of partitions
A universe or uloop style variable must specify a number of values >= to the number of processor
partitions.
Unknown angle style
The choice of angle style is unknown.
Unknown atom style
The choice of atom style is unknown.
Unknown body style
The choice of body style is unknown.
Unknown bond style
The choice of bond style is unknown.
Unknown category for info is_active()
Self-explanatory.
Unknown category for info is_available()
Self-explanatory.
Unknown category for info is_defined()
Self-explanatory.
Unknown command: %s
The command is not known to LAMMPS. Check the input script.
Unknown compute style
The choice of compute style is unknown.
Unknown dihedral style
The choice of dihedral style is unknown.
Unknown dump reader style
The choice of dump reader style via the format keyword is unknown.
Unknown dump style
The choice of dump style is unknown.
Unknown error in GPU library
Self-explanatory.
Unknown fix style
The choice of fix style is unknown.
LAMMPS Users Manual
300
Unknown identifier in data file: %s
A section of the data file cannot be read by LAMMPS.
Unknown improper style
The choice of improper style is unknown.
Unknown keyword in thermo_style custom command
One or more specified keywords are not recognized.
Unknown kspace style
The choice of kspace style is unknown.
Unknown name for info newton category
Self-explanatory.
Unknown name for info package category
Self-explanatory.
Unknown name for info pair category
Self-explanatory.
Unknown pair style
The choice of pair style is unknown.
Unknown pair_modify hybrid sub-style
The choice of sub-style is unknown.
Unknown region style
The choice of region style is unknown.
Unknown section in molecule file
Self-explanatory.
Unknown table style in angle style table
Self-explanatory.
Unknown table style in bond style table
Self-explanatory.
Unknown table style in pair_style command
Style of table is invalid for use with pair_style table command.
Unknown unit_style
Self-explanatory. Check the input script or data file.
Unrecognized lattice type in MEAM file 1
The lattice type in an entry of the MEAM library file is not valid.
Unrecognized lattice type in MEAM file 2
The lattice type in an entry of the MEAM parameter file is not valid.
Unrecognized pair style in compute pair command
Self-explanatory.
Unrecognized virial argument in pair_style command
Only two options are supported: LAMMPSvirial and KIMvirial
Unsupported mixing rule in kspace_style ewald/disp
Only geometric mixing is supported.
Unsupported order in kspace_style ewald/disp
Only 1/r^6 dispersion or dipole terms are supported.
Unsupported order in kspace_style pppm/disp, pair_style %s
Only pair styles with 1/r and 1/r^6 dependence are currently supported.
Use cutoff keyword to set cutoff in single mode
Mode is single so cutoff/multi keyword cannot be used.
Use cutoff/multi keyword to set cutoff in multi mode
Mode is multi so cutoff keyword cannot be used.
Using fix nvt/sllod with inconsistent fix deform remap option
Fix nvt/sllod requires that deforming atoms have a velocity profile provided by "remap v" as a fix
deform option.
LAMMPS Users Manual
301
Using fix nvt/sllod with no fix deform defined
Self-explanatory.
Using fix srd with inconsistent fix deform remap option
When shearing the box in an SRD simulation, the remap v option for fix deform needs to be used.
Using pair lubricate with inconsistent fix deform remap option
Must use remap v option with fix deform with this pair style.
Using pair lubricate/poly with inconsistent fix deform remap option
If fix deform is used, the remap v option is required.
Using suffix cuda without USER-CUDA package enabled
Self-explanatory.
Using suffix gpu without GPU package installed
Self-explanatory.
Using suffix intel without USER-INTEL package installed
Self-explanatory.
Using suffix kk without KOKKOS package enabled
Self-explanatory.
Using suffix omp without USER-OMP package installed
Self-explanatory.
Using update dipole flag requires atom attribute mu
Self-explanatory.
Using update dipole flag requires atom style sphere
Self-explanatory.
Variable ID in variable formula does not exist
Self-explanatory.
Variable atom ID is too large
Specified ID is larger than the maximum allowed atom ID.
Variable evaluation before simulation box is defined
Cannot evaluate a compute or fix or atom-based value in a variable before the simulation has been
setup.
Variable evaluation in fix wall gave bad value
The returned value for epsilon or sigma < 0.0.
Variable evaluation in region gave bad value
Variable returned a radius < 0.0.
Variable for compute ti is invalid style
Self-explanatory.
Variable for create_atoms is invalid style
The variables must be equal-style variables.
Variable for displace_atoms is invalid style
It must be an equal-style or atom-style variable.
Variable for dump every is invalid style
Only equal-style variables can be used.
Variable for dump image center is invalid style
Must be an equal-style variable.
Variable for dump image persp is invalid style
Must be an equal-style variable.
Variable for dump image phi is invalid style
Must be an equal-style variable.
Variable for dump image theta is invalid style
Must be an equal-style variable.
Variable for dump image zoom is invalid style
Must be an equal-style variable.
LAMMPS Users Manual
302
Variable for fix adapt is invalid style
Only equal-style variables can be used.
Variable for fix addforce is invalid style
Self-explanatory.
Variable for fix aveforce is invalid style
Only equal-style variables can be used.
Variable for fix deform is invalid style
The variable must be an equal-style variable.
Variable for fix efield is invalid style
The variable must be an equal- or atom-style variable.
Variable for fix gravity is invalid style
Only equal-style variables can be used.
Variable for fix heat is invalid style
Only equal-style or atom-style variables can be used.
Variable for fix indent is invalid style
Only equal-style variables can be used.
Variable for fix indent is not equal style
Only equal-style variables can be used.
Variable for fix langevin is invalid style
It must be an equal-style variable.
Variable for fix move is invalid style
Only equal-style variables can be used.
Variable for fix setforce is invalid style
Only equal-style variables can be used.
Variable for fix temp/berendsen is invalid style
Only equal-style variables can be used.
Variable for fix temp/csld is invalid style
Only equal-style variables can be used.
Variable for fix temp/csvr is invalid style
Only equal-style variables can be used.
Variable for fix temp/rescale is invalid style
Only equal-style variables can be used.
Variable for fix wall is invalid style
Only equal-style variables can be used.
Variable for fix wall/reflect is invalid style
Only equal-style variables can be used.
Variable for fix wall/srd is invalid style
Only equal-style variables can be used.
Variable for group dynamic is invalid style
The variable must be an atom-style variable.
Variable for group is invalid style
Only atom-style variables can be used.
Variable for region cylinder is invalid style
Only equal-style variables are allowed.
Variable for region is invalid style
Only equal-style variables can be used.
Variable for region is not equal style
Self-explanatory.
Variable for region sphere is invalid style
Only equal-style variables are allowed.
Variable for restart is invalid style
LAMMPS Users Manual
303
Only equal-style variables can be used.
Variable for set command is invalid style
Only atom-style variables can be used.
Variable for thermo every is invalid style
Only equal-style variables can be used.
Variable for velocity set is invalid style
Only atom-style variables can be used.
Variable for voronoi radius is not atom style
Self-explanatory.
Variable formula compute array is accessed out-of-range
Self-explanatory.
Variable formula compute vector is accessed out-of-range
Self-explanatory.
Variable formula fix array is accessed out-of-range
Self-explanatory.
Variable formula fix vector is accessed out-of-range
Self-explanatory.
Variable has circular dependency
A circular dependency is when variable "a" in used by variable "b" and variable "b" is also used by
variable "a". Circular dependencies with longer chains of dependence are also not allowed.
Variable name between brackets must be alphanumeric or underscore characters
Self-explanatory.
Variable name for compute chunk/atom does not exist
Self-explanatory.
Variable name for compute reduce does not exist
Self-explanatory.
Variable name for compute ti does not exist
Self-explanatory.
Variable name for create_atoms does not exist
Self-explanatory.
Variable name for displace_atoms does not exist
Self-explanatory.
Variable name for dump every does not exist
Self-explanatory.
Variable name for dump image center does not exist
Self-explanatory.
Variable name for dump image persp does not exist
Self-explanatory.
Variable name for dump image phi does not exist
Self-explanatory.
Variable name for dump image theta does not exist
Self-explanatory.
Variable name for dump image zoom does not exist
Self-explanatory.
Variable name for fix adapt does not exist
Self-explanatory.
Variable name for fix addforce does not exist
Self-explanatory.
Variable name for fix ave/atom does not exist
Self-explanatory.
Variable name for fix ave/chunk does not exist
LAMMPS Users Manual
304
Self-explanatory.
Variable name for fix ave/correlate does not exist
Self-explanatory.
Variable name for fix ave/histo does not exist
Self-explanatory.
Variable name for fix ave/spatial does not exist
Self-explanatory.
Variable name for fix ave/time does not exist
Self-explanatory.
Variable name for fix aveforce does not exist
Self-explanatory.
Variable name for fix deform does not exist
Self-explanatory.
Variable name for fix efield does not exist
Self-explanatory.
Variable name for fix gravity does not exist
Self-explanatory.
Variable name for fix heat does not exist
Self-explanatory.
Variable name for fix indent does not exist
Self-explanatory.
Variable name for fix langevin does not exist
Self-explanatory.
Variable name for fix move does not exist
Self-explanatory.
Variable name for fix setforce does not exist
Self-explanatory.
Variable name for fix store/state does not exist
Self-explanatory.
Variable name for fix temp/berendsen does not exist
Self-explanatory.
Variable name for fix temp/csld does not exist
Self-explanatory.
Variable name for fix temp/csvr does not exist
Self-explanatory.
Variable name for fix temp/rescale does not exist
Self-explanatory.
Variable name for fix vector does not exist
Self-explanatory.
Variable name for fix wall does not exist
Self-explanatory.
Variable name for fix wall/reflect does not exist
Self-explanatory.
Variable name for fix wall/srd does not exist
Self-explanatory.
Variable name for group does not exist
Self-explanatory.
Variable name for group dynamic does not exist
Self-explanatory.
Variable name for region cylinder does not exist
Self-explanatory.
LAMMPS Users Manual
305
Variable name for region does not exist
Self-explanatory.
Variable name for region sphere does not exist
Self-explanatory.
Variable name for restart does not exist
Self-explanatory.
Variable name for set command does not exist
Self-explanatory.
Variable name for thermo every does not exist
Self-explanatory.
Variable name for velocity set does not exist
Self-explanatory.
Variable name for voronoi radius does not exist
Self-explanatory.
Variable name must be alphanumeric or underscore characters
Self-explanatory.
Variable uses atom property that isn't allocated
Self-explanatory.
Velocity command before simulation box is defined
The velocity command cannot be used before a read_data, read_restart, or create_box command.
Velocity command with no atoms existing
A velocity command has been used, but no atoms yet exist.
Velocity ramp in z for a 2d problem
Self-explanatory.
Velocity rigid used with non-rigid fix-ID
Self-explanatory.
Velocity temperature ID does calculate a velocity bias
The specified compute must compute a bias for temperature.
Velocity temperature ID does not compute temperature
The compute ID given to the velocity command must compute temperature.
Verlet/split can only currently be used with comm_style brick
This is a current restriction in LAMMPS.
Verlet/split does not yet support TIP4P
This is a current limitation.
Verlet/split requires 2 partitions
See the -partition command-line switch.
Verlet/split requires Rspace partition layout be multiple of Kspace partition layout in each dim
This is controlled by the processors command.
Verlet/split requires Rspace partition size be multiple of Kspace partition size
This is so there is an equal number of Rspace processors for every Kspace processor.
Virial was not tallied on needed timestep
You are using a thermo keyword that requires potentials to have tallied the virial, but they didn't on
this timestep. See the variable doc page for ideas on how to make this work.
Voro++ error: narea and neigh have a different size
This error is returned by the Voro++ library.
Wall defined twice in fix wall command
Self-explanatory.
Wall defined twice in fix wall/reflect command
Self-explanatory.
Wall defined twice in fix wall/srd command
Self-explanatory.
LAMMPS Users Manual
306
Water H epsilon must be 0.0 for pair style lj/cut/tip4p/cut
This is because LAMMPS does not compute the Lennard-Jones interactions with these particles for
efficiency reasons.
Water H epsilon must be 0.0 for pair style lj/cut/tip4p/long
This is because LAMMPS does not compute the Lennard-Jones interactions with these particles for
efficiency reasons.
Water H epsilon must be 0.0 for pair style lj/long/tip4p/long
This is because LAMMPS does not compute the Lennard-Jones interactions with these particles for
efficiency reasons.
World variable count doesn't match # of partitions
A world-style variable must specify a number of values equal to the number of processor partitions.
Write_data command before simulation box is defined
Self-explanatory.
Write_restart command before simulation box is defined
The write_restart command cannot be used before a read_data, read_restart, or create_box command.
Writing to MPI-IO filename when MPIIO package is not installed
Self-explanatory.
Zero length rotation vector with displace_atoms
Self-explanatory.
Zero length rotation vector with fix move
Self-explanatory.
Zero-length lattice orient vector
Self-explanatory.
Warnings:
Adjusting Coulombic cutoff for MSM, new cutoff = %g
The adjust/cutoff command is turned on and the Coulombic cutoff has been adjusted to match the
user-specified accuracy.
Angle atoms missing at step %ld
One or more of 3 atoms needed to compute a particular angle are missing on this processor. Typically
this is because the pairwise cutoff is set too short or the angle has blown apart and an atom is too far
away.
Angle style in data file differs from currently defined angle style
Self-explanatory.
Atom style in data file differs from currently defined atom style
Self-explanatory.
Bond atom missing in box size check
The 2nd atoms needed to compute a particular bond is missing on this processor. Typically this is
because the pairwise cutoff is set too short or the bond has blown apart and an atom is too far away.
Bond atom missing in image check
The 2nd atom in a particular bond is missing on this processor. Typically this is because the pairwise
cutoff is set too short or the bond has blown apart and an atom is too far away.
Bond atoms missing at step %ld
The 2nd atom needed to compute a particular bond is missing on this processor. Typically this is
because the pairwise cutoff is set too short or the bond has blown apart and an atom is too far away.
Bond style in data file differs from currently defined bond style
Self-explanatory.
Bond/angle/dihedral extent > half of periodic box length
This is a restriction because LAMMPS can be confused about which image of an atom in the bonded
interaction is the correct one to use. "Extent" in this context means the maximum end-to-end length of
LAMMPS Users Manual
307
the bond/angle/dihedral. LAMMPS computes this by taking the maximum bond length, multiplying
by the number of bonds in the interaction (e.g. 3 for a dihedral) and adding a small amount of stretch.
Both groups in compute group/group have a net charge; the Kspace boundary correction to energy will be
non-zero
Self-explanatory.
Calling write_dump before a full system init.
The write_dump command is used before the system has been fully initialized as part of a 'run' or
'minimize' command. Not all dump styles and features are fully supported at this point and thus the
command may fail or produce incomplete or incorrect output. Insert a "run 0" command, if a full
system init is required.
Cannot count rigid body degrees-of-freedom before bodies are fully initialized
This means the temperature associated with the rigid bodies may be incorrect on this timestep.
Cannot count rigid body degrees-of-freedom before bodies are initialized
This means the temperature associated with the rigid bodies may be incorrect on this timestep.
Cannot include log terms without 1/r terms; setting flagHI to 1
Self-explanatory.
Cannot include log terms without 1/r terms; setting flagHI to 1.
Self-explanatory.
Charges are set, but coulombic solver is not used
Self-explanatory.
Charges did not converge at step %ld: %lg
Self-explanatory.
Communication cutoff is too small for SNAP micro load balancing, increased to %lf
Self-explanatory.
Compute cna/atom cutoff may be too large to find ghost atom neighbors
The neighbor cutoff used may not encompass enough ghost atoms to perform this operation correctly.
Computing temperature of portions of rigid bodies
The group defined by the temperature compute does not encompass all the atoms in one or more rigid
bodies, so the change in degrees-of-freedom for the atoms in those partial rigid bodies will not be
accounted for.
Create_bonds max distance > minimum neighbor cutoff
This means atom pairs for some atom types may not be in the neighbor list and thus no bond can be
created between them.
Delete_atoms cutoff > minimum neighbor cutoff
This means atom pairs for some atom types may not be in the neighbor list and thus an atom in that
pair cannot be deleted.
Dihedral atoms missing at step %ld
One or more of 4 atoms needed to compute a particular dihedral are missing on this processor.
Typically this is because the pairwise cutoff is set too short or the dihedral has blown apart and an
atom is too far away.
Dihedral problem
Conformation of the 4 listed dihedral atoms is extreme; you may want to check your simulation
geometry.
Dihedral problem: %d %ld %d %d %d %d
Conformation of the 4 listed dihedral atoms is extreme; you may want to check your simulation
geometry.
Dihedral style in data file differs from currently defined dihedral style
Self-explanatory.
Dump dcd/xtc timestamp may be wrong with fix dt/reset
If the fix changes the timestep, the dump dcd file will not reflect the change.
Energy tally does not account for 'zero yes'
LAMMPS Users Manual
308
The energy removed by using the 'zero yes' flag is not accounted for in the energy tally and thus
energy conservation cannot be monitored in this case.
Estimated error in splitting of dispersion coeffs is %g
Error is greater than 0.0001 percent.
Ewald/disp Newton solver failed, using old method to estimate g_ewald
Self-explanatory. Choosing a different cutoff value may help.
FENE bond too long
A FENE bond has stretched dangerously far. It's interaction strength will be truncated to attempt to
prevent the bond from blowing up.
FENE bond too long: %ld %d %d %g
A FENE bond has stretched dangerously far. It's interaction strength will be truncated to attempt to
prevent the bond from blowing up.
FENE bond too long: %ld %g
A FENE bond has stretched dangerously far. It's interaction strength will be truncated to attempt to
prevent the bond from blowing up.
Fix SRD walls overlap but fix srd overlap not set
You likely want to set this in your input script.
Fix bond/swap will ignore defined angles
See the doc page for fix bond/swap for more info on this restriction.
Fix deposit near setting < possible overlap separation %g
This test is performed for finite size particles with a diameter, not for point particles. The near setting
is smaller than the particle diameter which can lead to overlaps.
Fix evaporate may delete atom with non-zero molecule ID
This is probably an error, since you should not delete only one atom of a molecule.
Fix gcmc using full_energy option
Fix gcmc has automatically turned on the full_energy option since it is required for systems like the
one specified by the user. User input included one or more of the following: kspace, triclinic, a hybrid
pair style, an eam pair style, or no "single" function for the pair style.
Fix property/atom mol or charge w/out ghost communication
A model typically needs these properties defined for ghost atoms.
Fix qeq CG convergence failed (%g) after %d iterations at %ld step
Self-explanatory.
Fix qeq has non-zero lower Taper radius cutoff
Absolute value must be <= 0.01.
Fix qeq has very low Taper radius cutoff
Value should typically be >= 5.0.
Fix qeq/dynamic tolerance may be too small for damped dynamics
Self-explanatory.
Fix qeq/fire tolerance may be too small for damped fires
Self-explanatory.
Fix rattle should come after all other integration fixes
This fix is designed to work after all other integration fixes change atom positions. Thus it should be
the last integration fix specified. If not, it will not satisfy the desired constraints as well as it otherwise
would.
Fix recenter should come after all other integration fixes
Other fixes may change the position of the center-of-mass, so fix recenter should come last.
Fix srd SRD moves may trigger frequent reneighboring
This is because the SRD particles may move long distances.
Fix srd grid size > 1/4 of big particle diameter
This may cause accuracy problems.
Fix srd particle moved outside valid domain
LAMMPS Users Manual
309
This may indicate a problem with your simulation parameters.
Fix srd particles may move > big particle diameter
This may cause accuracy problems.
Fix srd viscosity < 0.0 due to low SRD density
This may cause accuracy problems.
Fix thermal/conductivity comes before fix ave/spatial
The order of these 2 fixes in your input script is such that fix thermal/conductivity comes first. If you
are using fix ave/spatial to measure the temperature profile induced by fix viscosity, then this may
cause a glitch in the profile since you are averaging immediately after swaps have occurred. Flipping
the order of the 2 fixes typically helps.
Fix viscosity comes before fix ave/spatial
The order of these 2 fixes in your input script is such that fix viscosity comes first. If you are using fix
ave/spatial to measure the velocity profile induced by fix viscosity, then this may cause a glitch in the
profile since you are averaging immediately after swaps have occurred. Flipping the order of the 2
fixes typically helps.
Fixes cannot send data in Kokkos communication, switching to classic communication
This is current restriction with Kokkos.
For better accuracy use 'pair_modify table 0'
The user-specified force accuracy cannot be achieved unless the table feature is disabled by using
'pair_modify table 0'.
Geometric mixing assumed for 1/r^6 coefficients
Self-explanatory.
Group for fix_modify temp != fix group
The fix_modify command is specifying a temperature computation that computes a temperature on a
different group of atoms than the fix itself operates on. This is probably not what you want to do.
H matrix size has been exceeded: m_fill=%d H.m=%d\n
This is the size of the matrix.
Ignoring unknown or incorrect info command flag
Self-explanatory. An unknown argument was given to the info command. Compare your input with
the documentation.
Improper atoms missing at step %ld
One or more of 4 atoms needed to compute a particular improper are missing on this processor.
Typically this is because the pairwise cutoff is set too short or the improper has blown apart and an
atom is too far away.
Improper problem: %d %ld %d %d %d %d
Conformation of the 4 listed improper atoms is extreme; you may want to check your simulation
geometry.
Improper style in data file differs from currently defined improper style
Self-explanatory.
Inconsistent image flags
The image flags for a pair on bonded atoms appear to be inconsistent. Inconsistent means that when
the coordinates of the two atoms are unwrapped using the image flags, the two atoms are far apart.
Specifically they are further apart than half a periodic box length. Or they are more than a box length
apart in a non-periodic dimension. This is usually due to the initial data file not having correct image
flags for the 2 atoms in a bond that straddles a periodic boundary. They should be different by 1 in
that case. This is a warning because inconsistent image flags will not cause problems for dynamics or
most LAMMPS simulations. However they can cause problems when such atoms are used with the
fix rigid or replicate commands. Note that if you have an infinite periodic crystal with bonds then it is
impossible to have fully consistent image flags, since some bonds will cross periodic boundaries and
connect two atoms with the same image flag.
KIM Model does not provide 'energy'; Potential energy will be zero
LAMMPS Users Manual
310
Self-explanatory.
KIM Model does not provide 'forces'; Forces will be zero
Self-explanatory.
KIM Model does not provide 'particleEnergy'; energy per atom will be zero
Self-explanatory.
KIM Model does not provide 'particleVirial'; virial per atom will be zero
Self-explanatory.
Kspace_modify slab param < 2.0 may cause unphysical behavior
The kspace_modify slab parameter should be larger to insure periodic grids padded with empty space
do not overlap.
Less insertions than requested
The fix pour command was unsuccessful at finding open space for as many particles as it tried to
insert.
Library error in lammps_gather_atoms
This library function cannot be used if atom IDs are not defined or are not consecutively numbered.
Library error in lammps_scatter_atoms
This library function cannot be used if atom IDs are not defined or are not consecutively numbered, or
if no atom map is defined. See the atom_modify command for details about atom maps.
Lost atoms via change_box: original %ld current %ld
The command options you have used caused atoms to be lost.
Lost atoms via displace_atoms: original %ld current %ld
The command options you have used caused atoms to be lost.
Lost atoms: original %ld current %ld
Lost atoms are checked for each time thermo output is done. See the thermo_modify lost command
for options. Lost atoms usually indicate bad dynamics, e.g. atoms have been blown far out of the
simulation box, or moved further than one processor's sub-domain away before reneighboring.
MSM mesh too small, increasing to 2 points in each direction
Self-explanatory.
Mismatch between velocity and compute groups
The temperature computation used by the velocity command will not be on the same group of atoms
that velocities are being set for.
Mixing forced for lj coefficients
Self-explanatory.
Molecule attributes do not match system attributes
An attribute is specified (e.g. diameter, charge) that is not defined for the specified atom style.
Molecule has bond topology but no special bond settings
This means the bonded atoms will not be excluded in pair-wise interactions.
Molecule template for create_atoms has multiple molecules
The create_atoms command will only create molecules of a single type, i.e. the first molecule in the
template.
Molecule template for fix gcmc has multiple molecules
The fix gcmc command will only create molecules of a single type, i.e. the first molecule in the
template.
Molecule template for fix shake has multiple molecules
The fix shake command will only recognize molecules of a single type, i.e. the first molecule in the
template.
More than one compute centro/atom
It is not efficient to use compute centro/atom more than once.
More than one compute cluster/atom
It is not efficient to use compute cluster/atom more than once.
More than one compute cna/atom defined
LAMMPS Users Manual
311
It is not efficient to use compute cna/atom more than once.
More than one compute contact/atom
It is not efficient to use compute contact/atom more than once.
More than one compute coord/atom
It is not efficient to use compute coord/atom more than once.
More than one compute damage/atom
It is not efficient to use compute ke/atom more than once.
More than one compute dilatation/atom
Self-explanatory.
More than one compute erotate/sphere/atom
It is not efficient to use compute erorate/sphere/atom more than once.
More than one compute hexorder/atom
It is not efficient to use compute hexorder/atom more than once.
More than one compute ke/atom
It is not efficient to use compute ke/atom more than once.
More than one compute orientorder/atom
It is not efficient to use compute orientorder/atom more than once.
More than one compute plasticity/atom
Self-explanatory.
More than one compute sna/atom
Self-explanatory.
More than one compute snad/atom
Self-explanatory.
More than one compute snav/atom
Self-explanatory.
More than one fix poems
It is not efficient to use fix poems more than once.
More than one fix rigid
It is not efficient to use fix rigid more than once.
Neighbor exclusions used with KSpace solver may give inconsistent Coulombic energies
This is because excluding specific pair interactions also excludes them from long-range interactions
which may not be the desired effect. The special_bonds command handles this consistently by
insuring excluded (or weighted) 1-2, 1-3, 1-4 interactions are treated consistently by both the
short-range pair style and the long-range solver. This is not done for exclusions of charged atom pairs
via the neigh_modify exclude command.
New thermo_style command, previous thermo_modify settings will be lost
If a thermo_style command is used after a thermo_modify command, the settings changed by the
thermo_modify command will be reset to their default values. This is because the thermo_modify
command acts on the currently defined thermo style, and a thermo_style command creates a new
style.
No Kspace calculation with verlet/split
The 2nd partition performs a kspace calculation so the kspace_style command must be used.
No automatic unit conversion to XTC file format conventions possible for units lj
This means no scaling will be performed.
No fixes defined, atoms won't move
If you are not using a fix like nve, nvt, npt then atom velocities and coordinates will not be updated
during timestepping.
No joints between rigid bodies, use fix rigid instead
The bodies defined by fix poems are not connected by joints. POEMS will integrate the body motion,
but it would be more efficient to use fix rigid.
Not using real units with pair reax
LAMMPS Users Manual
312
This is most likely an error, unless you have created your own ReaxFF parameter file in a different set
of units.
Number of MSM mesh points changed to be a multiple of 2
MSM requires that the number of grid points in each direction be a multiple of two and the number of
grid points in one or more directions have been adjusted to meet this requirement.
OMP_NUM_THREADS environment is not set.
This environment variable must be set appropriately to use the USER-OMP package.
One or more atoms are time integrated more than once
This is probably an error since you typically do not want to advance the positions or velocities of an
atom more than once per timestep.
One or more chunks do not contain all atoms in molecule
This may not be what you intended.
One or more dynamic groups may not be updated at correct point in timestep
If there are other fixes that act immediately after the initial stage of time integration within a timestep
(i.e. after atoms move), then the command that sets up the dynamic group should appear after those
fixes. This will insure that dynamic group assignments are made after all atoms have moved.
One or more respa levels compute no forces
This is computationally inefficient.
Pair COMB charge %.10f with force %.10f hit max barrier
Something is possibly wrong with your model.
Pair COMB charge %.10f with force %.10f hit min barrier
Something is possibly wrong with your model.
Pair brownian needs newton pair on for momentum conservation
Self-explanatory.
Pair dpd needs newton pair on for momentum conservation
Self-explanatory.
Pair dsmc: num_of_collisions > number_of_A
Collision model in DSMC is breaking down.
Pair dsmc: num_of_collisions > number_of_B
Collision model in DSMC is breaking down.
Pair style in data file differs from currently defined pair style
Self-explanatory.
Particle deposition was unsuccessful
The fix deposit command was not able to insert as many atoms as needed. The requested volume
fraction may be too high, or other atoms may be in the insertion region.
Proc sub-domain size < neighbor skin, could lead to lost atoms
The decomposition of the physical domain (likely due to load balancing) has led to a processor's
sub-domain being smaller than the neighbor skin in one or more dimensions. Since reneighboring is
triggered by atoms moving the skin distance, this may lead to lost atoms, if an atom moves all the way
across a neighboring processor's sub-domain before reneighboring is triggered.
Reducing PPPM order b/c stencil extends beyond nearest neighbor processor
This may lead to a larger grid than desired. See the kspace_modify overlap command to prevent
changing of the PPPM order.
Reducing PPPMDisp Coulomb order b/c stencil extends beyond neighbor processor
This may lead to a larger grid than desired. See the kspace_modify overlap command to prevent
changing of the PPPM order.
Reducing PPPMDisp dispersion order b/c stencil extends beyond neighbor processor
This may lead to a larger grid than desired. See the kspace_modify overlap command to prevent
changing of the PPPM order.
Replacing a fix, but new group != old group
LAMMPS Users Manual
313
The ID and style of a fix match for a fix you are changing with a fix command, but the new group you
are specifying does not match the old group.
Replicating in a non-periodic dimension
The parameters for a replicate command will cause a non-periodic dimension to be replicated; this
may cause unwanted behavior.
Resetting reneighboring criteria during PRD
A PRD simulation requires that neigh_modify settings be delay = 0, every = 1, check = yes. Since
these settings were not in place, LAMMPS changed them and will restore them to their original
values after the PRD simulation.
Resetting reneighboring criteria during TAD
A TAD simulation requires that neigh_modify settings be delay = 0, every = 1, check = yes. Since
these settings were not in place, LAMMPS changed them and will restore them to their original
values after the PRD simulation.
Resetting reneighboring criteria during minimization
Minimization requires that neigh_modify settings be delay = 0, every = 1, check = yes. Since these
settings were not in place, LAMMPS changed them and will restore them to their original values after
the minimization.
Restart file used different # of processors
The restart file was written out by a LAMMPS simulation running on a different number of
processors. Due to round-off, the trajectories of your restarted simulation may diverge a little more
quickly than if you ran on the same # of processors.
Restart file used different 3d processor grid
The restart file was written out by a LAMMPS simulation running on a different 3d grid of
processors. Due to round-off, the trajectories of your restarted simulation may diverge a little more
quickly than if you ran on the same # of processors.
Restart file used different boundary settings, using restart file values
Your input script cannot change these restart file settings.
Restart file used different newton bond setting, using restart file value
The restart file value will override the setting in the input script.
Restart file used different newton pair setting, using input script value
The input script value will override the setting in the restart file.
Restrain problem: %d %ld %d %d %d %d
Conformation of the 4 listed dihedral atoms is extreme; you may want to check your simulation
geometry.
Running PRD with only one replica
This is allowed, but you will get no parallel speed-up.
SRD bin shifting turned on due to small lamda
This is done to try to preserve accuracy.
SRD bin size for fix srd differs from user request
Fix SRD had to adjust the bin size to fit the simulation box. See the cubic keyword if you want this
message to be an error vs warning.
SRD bins for fix srd are not cubic enough
The bin shape is not within tolerance of cubic. See the cubic keyword if you want this message to be
an error vs warning.
SRD particle %d started inside big particle %d on step %ld bounce %d
See the inside keyword if you want this message to be an error vs warning.
SRD particle %d started inside wall %d on step %ld bounce %d
See the inside keyword if you want this message to be an error vs warning.
Shake determinant < 0.0
The determinant of the quadratic equation being solved for a single cluster specified by the fix shake
command is numerically suspect. LAMMPS will set it to 0.0 and continue.
LAMMPS Users Manual
314
Shell command '%s' failed with error '%s'
Self-explanatory.
Shell command returned with non-zero status
This may indicate the shell command did not operate as expected.
Should not allow rigid bodies to bounce off relecting walls
LAMMPS allows this, but their dynamics are not computed correctly.
Should not use fix nve/limit with fix shake or fix rattle
This will lead to invalid constraint forces in the SHAKE/RATTLE computation.
Simulations might be very slow because of large number of structure factors
Self-explanatory.
Slab correction not needed for MSM
Slab correction is intended to be used with Ewald or PPPM and is not needed by MSM.
System is not charge neutral, net charge = %g
The total charge on all atoms on the system is not 0.0. For some KSpace solvers this is only a
warning.
Table inner cutoff >= outer cutoff
You specified an inner cutoff for a Coulombic table that is longer than the global cutoff. Probably not
what you wanted.
Temperature for MSST is not for group all
User-assigned temperature to MSST fix does not compute temperature for all atoms. Since MSST
computes a global pressure, the kinetic energy contribution from the temperature is assumed to also
be for all atoms. Thus the pressure used by MSST could be inaccurate.
Temperature for NPT is not for group all
User-assigned temperature to NPT fix does not compute temperature for all atoms. Since NPT
computes a global pressure, the kinetic energy contribution from the temperature is assumed to also
be for all atoms. Thus the pressure used by NPT could be inaccurate.
Temperature for fix modify is not for group all
The temperature compute is being used with a pressure calculation which does operate on group all,
so this may be inconsistent.
Temperature for thermo pressure is not for group all
User-assigned temperature to thermo via the thermo_modify command does not compute temperature
for all atoms. Since thermo computes a global pressure, the kinetic energy contribution from the
temperature is assumed to also be for all atoms. Thus the pressure printed by thermo could be
inaccurate.
The fix ave/spatial command has been replaced by the more flexible fix ave/chunk and compute chunk/atom
commands -- fix ave/spatial will be removed in the summer of 2015
Self-explanatory.
The minimizer does not re-orient dipoles when using fix efield
This means that only the atom coordinates will be minimized, not the orientation of the dipoles.
Too many common neighbors in CNA %d times
More than the maximum # of neighbors was found multiple times. This was unexpected.
Too many inner timesteps in fix ttm
Self-explanatory.
Too many neighbors in CNA for %d atoms
More than the maximum # of neighbors was found multiple times. This was unexpected.
Triclinic box skew is large
The displacement in a skewed direction is normally required to be less than half the box length in that
dimension. E.g. the xy tilt must be between -half and +half of the x box length. You have relaxed the
constraint using the box tilt command, but the warning means that a LAMMPS simulation may be
inefficient as a result.
Use special bonds = 0,1,1 with bond style fene
LAMMPS Users Manual
315
Most FENE models need this setting for the special_bonds command.
Use special bonds = 0,1,1 with bond style fene/expand
Most FENE models need this setting for the special_bonds command.
Using a manybody potential with bonds/angles/dihedrals and special_bond exclusions
This is likely not what you want to do. The exclusion settings will eliminate neighbors in the neighbor
list, which the manybody potential needs to calculated its terms correctly.
Using compute temp/deform with inconsistent fix deform remap option
Fix nvt/sllod assumes deforming atoms have a velocity profile provided by "remap v" or "remap
none" as a fix deform option.
Using compute temp/deform with no fix deform defined
This is probably an error, since it makes little sense to use compute temp/deform in this case.
Using fix srd with box deformation but no SRD thermostat
The deformation will heat the SRD particles so this can be dangerous.
Using kspace solver on system with no charge
Self-explanatory.
Using largest cut-off for lj/long/dipole/long long long
Self-explanatory.
Using largest cutoff for buck/long/coul/long
Self-explanatory.
Using largest cutoff for lj/long/coul/long
Self-explanatory.
Using largest cutoff for pair_style lj/long/tip4p/long
Self-explanatory.
Using package gpu without any pair style defined
Self-explanatory.
Using pair potential shift with pair_modify compute no
The shift effects will thus not be computed.
Using pair tail corrections with nonperiodic system
This is probably a bogus thing to do, since tail corrections are computed by integrating the density of
a periodic system out to infinity.
Using pair tail corrections with pair_modify compute no
The tail corrections will thus not be computed.
pair style reax is now deprecated and will soon be retired. Users should switch to pair_style reax/c
Self-explanatory.
LAMMPS Users Manual
316

Previous Section - LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands - Next Section
13. Future and history
This section lists features we plan to add to LAMMPS, features of previous versions of LAMMPS, and
features of other parallel molecular dynamics codes our group has distributed.
13.1 Coming attractions
13.2 Past versions
13.1 Coming attractions
As of summer 2016 we are using the LAMMPS project issue tracker on GitHub for keeping track of
suggested, planned or pending new features. This includes discussions of how to best implement them, or why
they would be useful. Especially if a planned or proposed feature is non-trivial to add, e.g. because it requires
changes to some of the core classes of LAMMPS, people planning to contribute a new feature to LAMMS are
encouraged to submit an issue about their planned implementation this way in order to receive feedback from
the LAMMPS core developers. They will provide suggestions about the validity of the proposed approach and
possible improvements, pitfalls or alternatives.
Please see some of the closed issues for examples of how to suggest code enhancements, submit proposed
changes, or report possible bugs and how they are resolved.
As an alternative to using GitHub, you may e-mail the core developers or send an e-mail to the LAMMPS
Mail list if you want to have your suggestion added to the list.
13.2 Past versions
LAMMPS development began in the mid 1990s under a cooperative research & development agreement
(CRADA) between two DOE labs (Sandia and LLNL) and 3 companies (Cray, Bristol Myers Squibb, and
Dupont). The goal was to develop a large-scale parallel classical MD code; the coding effort was led by Steve
Plimpton at Sandia.
After the CRADA ended, a final F77 version, LAMMPS 99, was released. As development of LAMMPS
continued at Sandia, its memory management was converted to F90; a final F90 version was released as
LAMMPS 2001.
The current LAMMPS is a rewrite in C++ and was first publicly released as an open source code in 2004. It
includes many new features beyond those in LAMMPS 99 or 2001. It also includes features from older
parallel MD codes written at Sandia, namely ParaDyn, Warp, and GranFlow (see below).
In late 2006 we began merging new capabilities into LAMMPS that were developed by Aidan Thompson at
Sandia for his MD code GRASP, which has a parallel framework similar to LAMMPS. Most notably, these
have included many-body potentials - Stillinger-Weber, Tersoff, ReaxFF - and the associated
charge-equilibration routines needed for ReaxFF.
The History link on the LAMMPS WWW page gives a timeline of features added to the C++ open-source
version of LAMMPS over the last several years.
LAMMPS Users Manual
317
These older codes are available for download from the LAMMPS WWW site, except for Warp & GranFlow
which were primarily used internally. A brief listing of their features is given here.
LAMMPS 2001
F90 + MPI• dynamic memory• spatial-decomposition parallelism• NVE, NVT, NPT, NPH, rRESPA integrators• LJ and Coulombic pairwise force fields• all-atom, united-atom, bead-spring polymer force fields• CHARMM-compatible force fields• class 2 force fields• 3d/2d Ewald & PPPM• various force and temperature constraints• SHAKE• Hessian-free truncated-Newton minimizer• user-defined diagnostics•
LAMMPS 99
F77 + MPI• static memory allocation• spatial-decomposition parallelism• most of the LAMMPS 2001 features with a few exceptions• no 2d Ewald & PPPM• molecular force fields are missing a few CHARMM terms• no SHAKE•
Warp
F90 + MPI• spatial-decomposition parallelism• embedded atom method (EAM) metal potentials + LJ• lattice and grain-boundary atom creation• NVE, NVT integrators• boundary conditions for applying shear stresses• temperature controls for actively sheared systems• per-atom energy and centro-symmetry computation and output•
ParaDyn
F77 + MPI• atom- and force-decomposition parallelism• embedded atom method (EAM) metal potentials• lattice atom creation• NVE, NVT, NPT integrators• all serial DYNAMO features for controls and constraints•
GranFlow
LAMMPS Users Manual
318
F90 + MPI• spatial-decomposition parallelism• frictional granular potentials• NVE integrator• boundary conditions for granular flow and packing and walls• particle insertion•
LAMMPS Users Manual
319

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
Tutorial for Thermalized Drude oscillators in LAMMPS
This tutorial explains how to use Drude oscillators in LAMMPS to simulate polarizable systems using the
USER-DRUDE package. As an illustration, the input files for a simulation of 250 phenol molecules are
documented. First of all, LAMMPS has to be compiled with the USER-DRUDE package activated. Then, the
data file and input scripts have to be modified to include the Drude dipoles and how to handle them.
Overview of Drude induced dipoles
Polarizable atoms acquire an induced electric dipole moment under the action of an external electric field, for
example the electric field created by the surrounding particles. Drude oscillators represent these dipoles by
two fixed charges: the core (DC) and the Drude particle (DP) bound by a harmonic potential. The Drude
particle can be thought of as the electron cloud whose center can be displaced from the position of the
corresponding nucleus.
The sum of the masses of a core-Drude pair should be the mass of the initial (unsplit) atom, \(m_C + m_D =
m\). The sum of their charges should be the charge of the initial (unsplit) atom, \(q_C + q_D = q\). A
harmonic potential between the core and Drude partners should be present, with force constant \(k_D\) and an
equilibrium distance of zero. The (half-)stiffness of the harmonic bond \(K_D = k_D/2\) and the Drude charge
\(q_D\) are related to the atom polarizability \(\alpha\) by
\begin{equation} K_D = \frac 1 2\, \frac {q_D^2} \alpha \end{equation}
Ideally, the mass of the Drude particle should be small, and the stiffness of the harmonic bond should be large,
so that the Drude particle remains close ot the core. The values of Drude mass, Drude charge, and force
constant can be chosen following different strategies, as in the following examples of polarizable force fields:
Lamoureux and Roux suggest adopting a global half-stiffness, \(K_D\) = 500 kcal/(mol Ang \({}^2\))
- which corresponds to a force constant \(k_D\) = 4184 kJ/(mol Ang \({}^2\)) - for all types of
core-Drude bond, a global mass \(m_D\) = 0.4 g/mol (or u) for all types of Drude particles, and to
calculate the Drude charges for individual atom types from the atom polarizabilities using equation
(1). This choice is followed in the polarizable CHARMM force field.
•
Alternately Schroeder and Steinhauser suggest adopting a global charge \(q_D\) = -1.0e and a global
mass \(m_D\) = 0.1 g/mol (or u) for all Drude particles, and to calculate the force constant for each
type of core-Drude bond from equation (1). The timesteps used by these authors are between 0.5 and
2 fs, with the degrees of freedom of the Drude oscillators kept cold at 1 K.
•
In both these force fields hydrogen atoms are treated as non-polarizable.•
The motion of of the Drude particles can be calculated by minimizing the energy of the induced dipoles at
each timestep, by an iterative, self-consistent procedure. The Drude particles can be massless and therefore do
not contribute to the kinetic energy. However, the relaxed method is computational slow. An
extended-lagrangian method can be used to calculate the positions of the Drude particles, but this requires
them to have mass. It is important in this case to decouple the degrees of freedom associated with the Drude
oscillators from those of the normal atoms. Thermalizing the Drude dipoles at temperatures comparable to the
rest of the simulation leads to several problems (kinetic energy transfer, very short timestep, etc.), which can
be remediate by the "cold Drude" technique (Lamoureux and Roux).
LAMMPS Users Manual
320
Two closely related models are used to represent polarization through "charges on a spring": the core-shell
model and the Drude model. Although the basic idea is the same, the core-shell model is normally used for
ionic/crystalline materials, whereas the Drude model is normally used for molecular systems and fluid states.
In ionic crystals the symmetry around each ion and the distance between them are such that the core-shell
model is sufficiently stable. But to be applicable to molecular/covalent systems the Drude model includes two
important features:
The possibility to thermostat the additional degrees of freedom associated with the induced dipoles at
very low temperature, in terms of the reduced coordinates of the Drude particles with respect to their
cores. This makes the trajectory close to that of relaxed induced dipoles.
1.
The Drude dipoles on covalently bonded atoms interact too strongly due to the short distances, so an
atom may capture the Drude particle (shell) of a neighbor, or the induced dipoles within the same
molecule may align too much. To avoid this, damping at short of the interactions between the point
charges composing the induced dipole can be done by Thole functions.
2.
Preparation of the data file
The data file is similar to a standard LAMMPS data file for atom_style full. The DPs and the harmonic bonds
connecting them to their DC should appear in the data file as normal atoms and bonds.
You can use the polarizer tool (Python script distributed with the USER-DRUDE package) to convert a
non-polarizable data file (here data.102494.lmp) to a polarizable data file (data-p.lmp)
polarizer -q -f phenol.dff data.102494.lmp data-p.lmp
This will automatically insert the new atoms and bonds. The masses and charges of DCs and DPs are
computed from phenol.dff, as well as the DC-DP bond constants. The file phenol.dff contains the
polarizabilities of the atom types and the mass of the Drude particles, for instance:
# units: kJ/mol, A, deg
# kforce is in the form k/2 r_D^2
# type m_D/u q_D/e k_D alpha/A3 thole
OH 0.4 -1.0 4184.0 0.63 0.67
CA 0.4 -1.0 4184.0 1.36 2.51
CAI 0.4 -1.0 4184.0 1.09 2.51
The hydrogen atoms are absent from this file, so they will be treated as non-polarizable atoms. In the
non-polarizable data file data.102494.lmp, atom names corresponding to the atom type numbers have to be
specified as comments at the end of lines of the Masses section. You probably need to edit it to add these
names. It should look like
Masses
1 12.011 # CAI
2 12.011 # CA
3 15.999 # OH
4 1.008 # HA
5 1.008 # HO
Basic input file
The atom style should be set to (or derive from) full, so that you can define atomic charges and molecular
bonds, angles, dihedrals...
LAMMPS Users Manual
321
The polarizer tool also outputs certain lines related to the input script (the use of these lines will be explained
below). In order for LAMMPS to recognize that you are using Drude oscillators, you should use the fix drude.
The command is
fix DRUDE all drude C C C N N D D D
The N, C, D following the drude keyword have the following meaning: There is one tag for each atom type.
This tag is C for DCs, D for DPs and N for non-polarizable atoms. Here the atom types 1 to 3 (C and O atoms)
are DC, atom types 4 and 5 (H atoms) are non-polarizable and the atom types 6 to 8 are the newly created
DPs.
By recognizing the fix drude, LAMMPS will find and store matching DC-DP pairs and will treat DP as
equivalent to their DC in the special bonds relations. It may be necessary to extend the space for storing such
special relations. In this case extra space should be reserved by using the extra keyword of the special_bonds
command. With our phenol, there is 1 more special neighbor for which space is required. Otherwise
LAMMPS crashes and gives the required value.
special_bonds lj/coul 0.0 0.0 0.5 extra 1
Let us assume we want to run a simple NVT simulation at 300 K. Note that Drude oscillators need to be
thermalized at a low temperature in order to approximate a self-consistent field (SCF), therefore it is not
possible to simulate an NVE ensemble with this package. Since dipoles are approximated by a charged
DC-DP pair, the pair_style must include Coulomb interactions, for instance lj/cut/coul/long with kspace_style
pppm. For example, with a cutoff of 10. and a precision 1.e-4:
pair_style lj/cut/coul/long 10.0
kspace_style pppm 1.0e-4
As compared to the non-polarizable input file, pair_coeff lines need to be added for the DPs. Since the DPs
have no Lennard-Jones interactions, their epsilon is 0. so the only pair_coeff line that needs to be added is
pair_coeff * 6* 0.0 0.0 # All-DPs
Now for the thermalization, the simplest choice is to use the fix langevin/drude.
fix LANG all langevin/drude 300. 100 12435 1. 20 13977
This applies a Langevin thermostat at temperature 300. to the centers of mass of the DC-DP pairs, with
relaxation time 100 and with random seed 12345. This fix applies also a Langevin thermostat at temperature
1. to the relative motion of the DPs around their DCs, with relaxation time 20 and random seed 13977. Only
the DCs and non-polarizable atoms need to be in this fix's group. LAMMPS will thermostate the DPs together
with their DC. For this, ghost atoms need to know their velocities. Thus you need to add the following
command:
comm_modify vel yes
In order to avoid that the center of mass of the whole system drifts due to the random forces of the Langevin
thermostat on DCs, you can add the zero yes option at the end of the fix line.
If the fix shake is used to constrain the C-H bonds, it should be invoked after the fix langevin/drude for more
accuracy.
LAMMPS Users Manual
322

fix SHAKE ATOMS shake 0.0001 20 0 t 4 5
NOTE: The group of the fix shake must not include the DPs. If the group ATOMS is defined by non-DPs atom
types, you could use
Since the fix langevin/drude does not perform time integration (just modification of forces but no
position/velocity updates), the fix nve should be used in conjunction.
fix NVE all nve
Finally, do not forget to update the atom type elements if you use them in a dump_modify ... element ...
command, by adding the element type of the DPs. Here for instance
dump DUMP all custom 10 dump.lammpstrj id mol type element x y z ix iy iz
dump_modify DUMP element C C O H H D D D
The input file should now be ready for use!
You will notice that the global temperature thermo_temp computed by LAMMPS is not 300. K as wanted.
This is because LAMMPS treats DPs as standard atoms in his default compute. If you want to output the
temperatures of the DC-DP pair centers of mass and of the DPs relative to their DCs, you should use the
compute temp_drude
compute TDRUDE all temp/drude
And then output the correct temperatures of the Drude oscillators using thermo_style custom with respectively
c_TDRUDE[1] and c_TDRUDE[2]. These should be close to 300.0 and 1.0 on average.
thermo_style custom step temp c_TDRUDE[1] c_TDRUDE[2]
Thole screening
Dipolar interactions represented by point charges on springs may not be stable, for example if the atomic
polarizability is too high for instance, a DP can escape from its DC and be captured by another DC, which
makes the force and energy diverge and the simulation crash. Even without reaching this extreme case, the
correlation between nearby dipoles on the same molecule may be exaggerated. Often, special bond relations
prevent bonded neighboring atoms to see the charge of each other's DP, so that the problem does not always
appear. It is possible to use screened dipole dipole interactions by using the pair_style thole. This is
implemented as a correction to the Coulomb pair_styles, which dampens at short distance the interactions
between the charges representing the induced dipoles. It is to be used as hybrid/overlay with any standard coul
pair style. In our example, we would use
pair_style hybrid/overlay lj/cut/coul/long 10.0 thole 2.6 10.0
This tells LAMMPS that we are using two pair_styles. The first one is as above (lj/cut/coul/long 10.0). The
second one is a thole pair_style with default screening factor 2.6 (Noskov) and cutoff 10.0.
Since hybrid/overlay does not support mixing rules, the interaction coefficients of all the pairs of atom types
with i < j should be explicitly defined. The output of the polarizer script can be used to complete the
pair_coeff section of the input file. In our example, this will look like:
pair_coeff 1 1 lj/cut/coul/long 0.0700 3.550
LAMMPS Users Manual
323

pair_coeff 1 2 lj/cut/coul/long 0.0700 3.550
pair_coeff 1 3 lj/cut/coul/long 0.1091 3.310
pair_coeff 1 4 lj/cut/coul/long 0.0458 2.985
pair_coeff 2 2 lj/cut/coul/long 0.0700 3.550
pair_coeff 2 3 lj/cut/coul/long 0.1091 3.310
pair_coeff 2 4 lj/cut/coul/long 0.0458 2.985
pair_coeff 3 3 lj/cut/coul/long 0.1700 3.070
pair_coeff 3 4 lj/cut/coul/long 0.0714 2.745
pair_coeff 4 4 lj/cut/coul/long 0.0300 2.420
pair_coeff * 5 lj/cut/coul/long 0.0000 0.000
pair_coeff * 6* lj/cut/coul/long 0.0000 0.000
pair_coeff 1 1 thole 1.090 2.510
pair_coeff 1 2 thole 1.218 2.510
pair_coeff 1 3 thole 0.829 1.590
pair_coeff 1 6 thole 1.090 2.510
pair_coeff 1 7 thole 1.218 2.510
pair_coeff 1 8 thole 0.829 1.590
pair_coeff 2 2 thole 1.360 2.510
pair_coeff 2 3 thole 0.926 1.590
pair_coeff 2 6 thole 1.218 2.510
pair_coeff 2 7 thole 1.360 2.510
pair_coeff 2 8 thole 0.926 1.590
pair_coeff 3 3 thole 0.630 0.670
pair_coeff 3 6 thole 0.829 1.590
pair_coeff 3 7 thole 0.926 1.590
pair_coeff 3 8 thole 0.630 0.670
pair_coeff 6 6 thole 1.090 2.510
pair_coeff 6 7 thole 1.218 2.510
pair_coeff 6 8 thole 0.829 1.590
pair_coeff 7 7 thole 1.360 2.510
pair_coeff 7 8 thole 0.926 1.590
pair_coeff 8 8 thole 0.630 0.670
For the thole pair style the coefficients are
the atom polarizability in units of cubic length1. the screening factor of the Thole function (optional, default value specified by the pair_style
command)
2.
the cutoff (optional, default value defined by the pair_style command)3.
The special neighbors have charge-charge and charge-dipole interactions screened by the coul factors of the
special_bonds command (0.0, 0.0, and 0.5 in the example above). Without using the pair_style thole,
dipole-dipole interactions are screened by the same factor. By using the pair_style thole, dipole-dipole
interactions are screened by Thole's function, whatever their special relationship (except within each DC-DP
pair of course). Consider for example 1-2 neighbors: using the pair_style thole, their dipoles will see each
other (despite the coul factor being 0.) and the interactions between these dipoles will be damped by Thole's
function.
Thermostats and barostats
Using a Nose-Hoover barostat with the langevin/drude thermostat is straightforward using fix nph instead of
nve. For example:
fix NPH all nph iso 1. 1. 500
LAMMPS Users Manual
324

It is also possible to use a Nose-Hoover instead of a Langevin thermostat. This requires to use fix
drude/transform just before and after the time intergation fixes. The fix drude/transform/direct converts the
atomic masses, positions, velocities and forces into a reduced representation, where the DCs transform into
the centers of mass of the DC-DP pairs and the DPs transform into their relative position with respect to their
DC. The fix drude/transform/inverse performs the reverse transformation. For a NVT simulation, with the
DCs and atoms at 300 K and the DPs at 1 K relative to their DC one would use
fix DIRECT all drude/transform/direct
fix NVT1 ATOMS nvt temp 300. 300. 100
fix NVT2 DRUDES nvt temp 1. 1. 20
fix INVERSE all drude/transform/inverse
For our phenol example, the groups would be defined as
group ATOMS type 1 2 3 4 5 # DCs and non-polarizable atoms
group CORES type 1 2 3 # DCs
group DRUDES type 6 7 8 # DPs
Note that with the fixes drude/transform, it is not required to specify comm_modify vel yes because the fixes
do it anyway (several times and for the forces also). To avoid the flying ice cube artifact (Lamoureux), where
the atoms progressively freeze and the center of mass of the whole system drifts faster and faster, the fix
momentum can be used. For instance:
fix MOMENTUM all momentum 100 linear 1 1 1
It is a bit more tricky to run a NPT simulation with Nose-Hoover barostat and thermostat. First, the volume
should be integrated only once. So the fix for DCs and atoms should be npt while the fix for DPs should be
nvt (or vice versa). Second, the fix npt computes a global pressure and thus a global temperature whatever the
fix group. We do want the pressure to correspond to the whole system, but we want the temperature to
correspond to the fix group only. We must then use the fix_modify command for this. In the end, the block of
instructions for thermostating and barostating will look like
compute TATOMS ATOMS temp
fix DIRECT all drude/transform/direct
fix NPT ATOMS npt temp 300. 300. 100 iso 1. 1. 500
fix_modify NPT temp TATOMS press thermo_press
fix NVT DRUDES nvt temp 1. 1. 20
fix INVERSE all drude/transform/inverse
Rigid bodies
You may want to simulate molecules as rigid bodies (but polarizable). Common cases are water models such
as SWM4-NDP, which is a kind of polarizable TIP4P water. The rigid bodies and the DPs should be
integrated separately, even with the Langevin thermostat. Let us review the different thermostats and
ensemble combinations.
NVT ensemble using Langevin thermostat:
comm_modify vel yes
fix LANG all langevin/drude 300. 100 12435 1. 20 13977
fix RIGID ATOMS rigid/nve/small molecule
fix NVE DRUDES nve
NVT ensemble using Nose-Hoover thermostat:
LAMMPS Users Manual
325

fix DIRECT all drude/transform/direct
fix RIGID ATOMS rigid/nvt/small molecule temp 300. 300. 100
fix NVT DRUDES nvt temp 1. 1. 20
fix INVERSE all drude/transform/inverse
NPT ensemble with Langevin thermostat:
comm_modify vel yes
fix LANG all langevin/drude 300. 100 12435 1. 20 13977
fix RIGID ATOMS rigid/nph/small molecule iso 1. 1. 500
fix NVE DRUDES nve
NPT ensemble using Nose-Hoover thermostat:
compute TATOM ATOMS temp
fix DIRECT all drude/transform/direct
fix RIGID ATOMS rigid/npt/small molecule temp 300. 300. 100 iso 1. 1. 500
fix_modify RIGID temp TATOM press thermo_press
fix NVT DRUDES nvt temp 1. 1. 20
fix INVERSE all drude/transform/inverse
(Lamoureux) Lamoureux and Roux, J Chem Phys, 119, 3025-3039 (2003)
(Schroeder) Schroeder and Steinhauser, J Chem Phys, 133, 154511 (2010).
(Jiang) Jiang, Hardy, Phillips, MacKerell, Schulten, and Roux, J Phys Chem Lett, 2, 87-92 (2011).
(Thole) Chem Phys, 59, 341 (1981).
(Noskov) Noskov, Lamoureux and Roux, J Phys Chem B, 109, 6705 (2005).
(SWM4-NDP) Lamoureux, Harder, Vorobyov, Roux, MacKerell, Chem Phys Let, 418, 245-249 (2006)
LAMMPS Users Manual
326
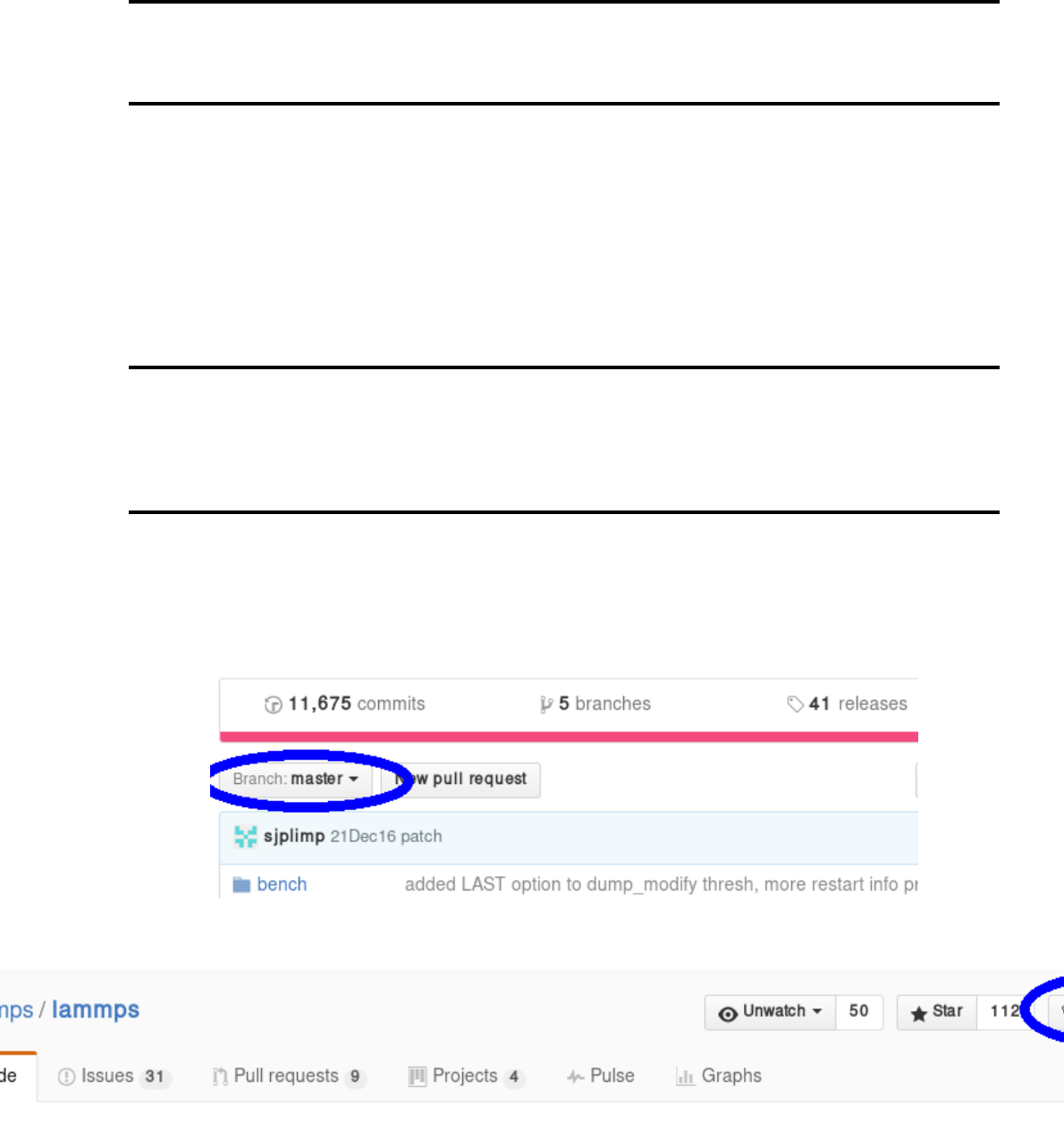
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
LAMMPS GitHub tutorial
written by Stefan Paquay
This document describes the process of how to use GitHub to integrate changes or additions you have made to
LAMMPS into the official LAMMPS distribution. It uses the process of updating this very tutorial as an
example to describe the individual steps and options. You need to be familiar with git and you may want to
have a look at the Git book to reacquaint yourself with some of the more advanced git features used below.
As of fall 2016, submitting contributions to LAMMPS via pull requests on GitHub is the preferred option for
integrating contributed features or improvements to LAMMPS, as it significantly reduces the amount of work
required by the LAMMPS developers. Consequently, creating a pull request will increase your chances to
have your contribution included and will reduce the time until the integration is complete. For more
information on the requirements to have your code included into LAMMPS please see Section 10.15
Making an account
First of all, you need a GitHub account. This is fairly simple, just go to GitHub and create an account by
clicking the "Sign up for GitHub" button. Once your account is created, you can sign in by clicking the button
in the top left and filling in your username or e-mail address and password.
Forking the repository
To get changes into LAMMPS, you need to first fork the `lammps/lammps` repository on GitHub. At the time
of writing, master is the preferred target branch. Thus go to LAMMPS on GitHub and make sure branch is set
to "master", as shown in the figure below.
If it is not, use the button to change it to master. Once it is, use the fork button to create a fork.
LAMMPS Users Manual
327
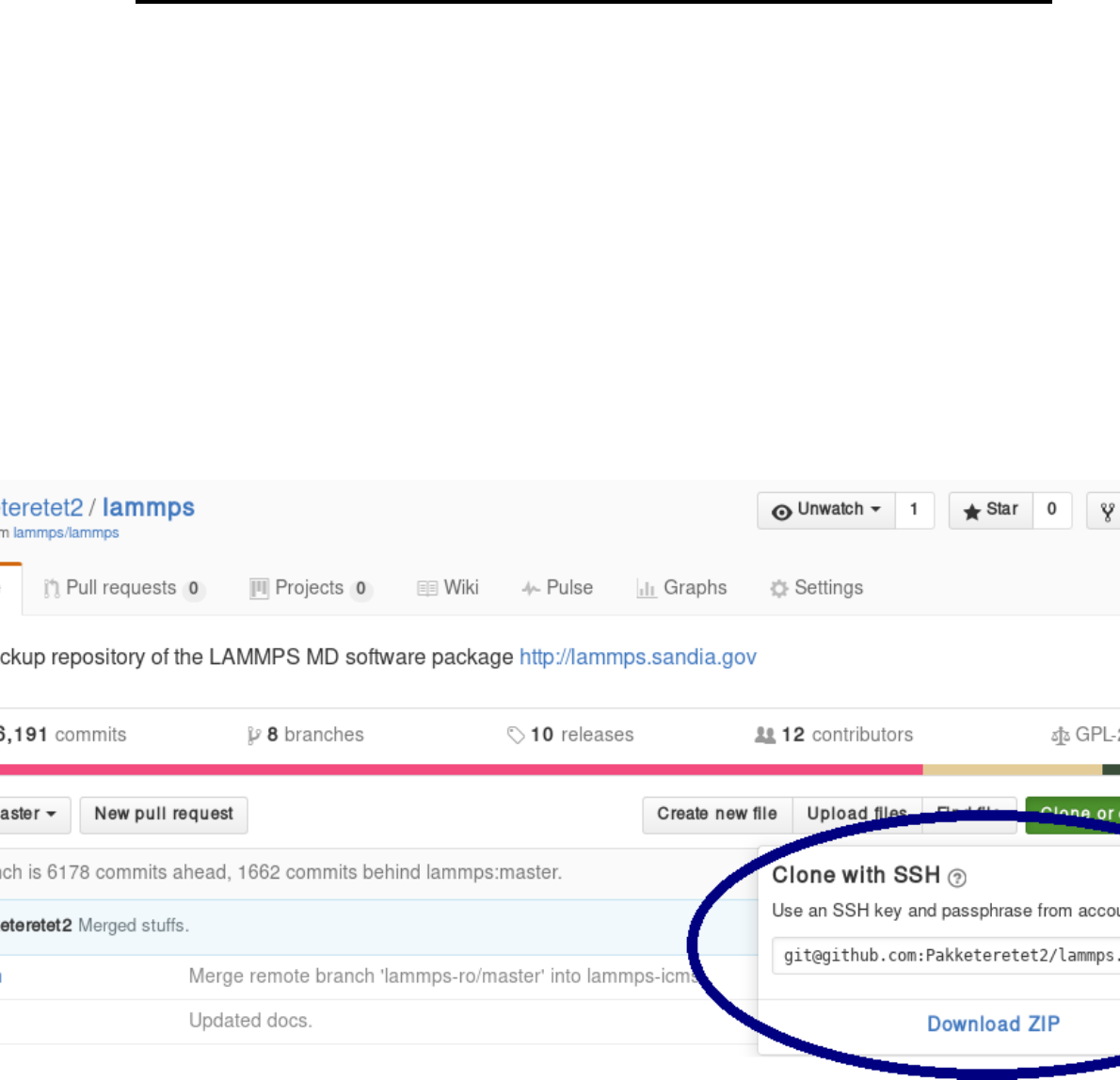
This will create a fork (which is essentially a copy, but uses less resources) of the LAMMPS repository under
your own GitHub account. You can make changes in this fork and later file pull requests to allow the
upstream repository to merge changes from your own fork into the one we just forked from (or others that
were forked from the same repository). At the same time, you can set things up, so you can include changes
from upstream into your repository and thus keep it in sync with the ongoing LAMMPS development.
Adding changes to your own fork
Additions to the upstream version of LAMMPS are handled using feature branches. For every new feature, a
so-called feature branch is created, which contains only those modification relevant to one specific feature.
For example, adding a single fix would consist of creating a branch with only the fix header and source file
and nothing else. It is explained in more detail here: feature branch workflow.
Feature branches
First of all, create a clone of your version on github on your local machine via HTTPS:
$ git clone https://github.com//lammps.git
or, if you have set up your GitHub account for using SSH keys, via SSH:
$ git clone git@github.com:/lammps.git
You can find the proper URL by clicking the "Clone or download"-button:
LAMMPS Users Manual
328
The above command copies ("clones") the git repository to your local machine to a directory with the name
you chose. If none is given, it will default to "lammps". Typical names are "mylammps" or something similar.
You can use this local clone to make changes and test them without interfering with the repository on Github.
To pull changes from upstream into this copy, you can go to the directory and use git pull:
$ cd mylammps
$ git checkout master
$ git pull https://github.com/lammps/lammps
You can also add this URL as a remote:
$ git remote add lammps_upstream https://www.github.com/lammps/lammps
At this point, you typically make a feature branch from the updated master branch for the feature you want to
work on. This tutorial contains the workflow that updated this tutorial, and hence we will call the branch
"github-tutorial-update":
$ git checkout -b github-tutorial-update master
Now that we have changed branches, we can make our changes to our local repository. Just remember that if
you want to start working on another, unrelated feature, you should switch branches!
After changes are made
After everything is done, add the files to the branch and commit them:
$ git add doc/src/tutorial_github.txt
$ git add doc/src/JPG/tutorial*.png
IMPORTANT NOTE: Do not use git commit -a (or git add -A). The -a flag (or -A flag) will automatically
include _all_ modified or new files and that is rarely the behavior you want. It can easily lead to accidentally
adding unrelated and unwanted changes into the repository. Instead it is preferable to explicitly use git add, git
rm, git mv for adding, removing, renaming individual files, respectively, and then git commit to finalize the
commit. Carefully check all pending changes with git status before committing them. If you find doing this on
the command line too tedious, consider using a GUI, for example the one included in git distributions written
in Tk, i.e. use git gui (on some Linux distributions it may be required to install an additional package to use
it).
After adding all files, the change set can be committed with some useful message that explains the change.
$ git commit -m 'Finally updated the github tutorial'
After the commit, the changes can be pushed to the same branch on GitHub:
$ git push
Git will ask you for your user name and password on GitHub if you have not configured anything. If your
local branch is not present on Github yet, it will ask you to add it by running
$ git push --set-upstream origin github-tutorial-update
LAMMPS Users Manual
329
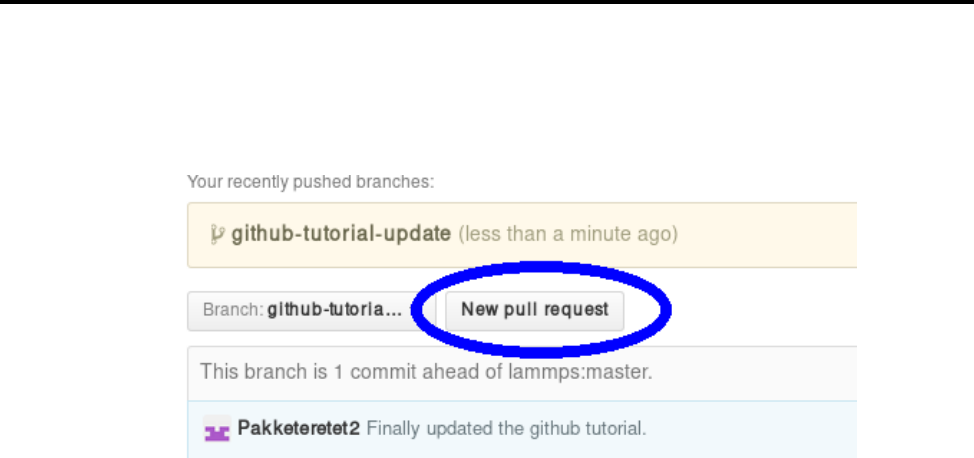
If you correctly type your user name and password, the feature branch should be added to your fork on
GitHub.
If you want to make really sure you push to the right repository (which is good practice), you can provide it
explicitly:
$ git push origin
or using an explicit URL:
$ git push git@github.com:Pakketeretet2/lammps.git
Filing a pull request
Up to this point in the tutorial, all changes were to your clones of LAMMPS. Eventually, however, you want
this feature to be included into the official LAMMPS version. To do this, you will want to file a pull request
by clicking on the "New pull request" button:
Make sure that the current branch is set to the correct one, which, in this case, is "github-tutorial-update". If
done correctly, the only changes you will see are those that were made on this branch.
This will open up a new window that lists changes made to the repository. If you are just adding new files,
there is not much to do, but I suppose merge conflicts are to be resolved here if there are changes in existing
files. If all changes can automatically be merged, green text at the top will say so and you can click the
"Create pull request" button, see image.
LAMMPS Users Manual
330
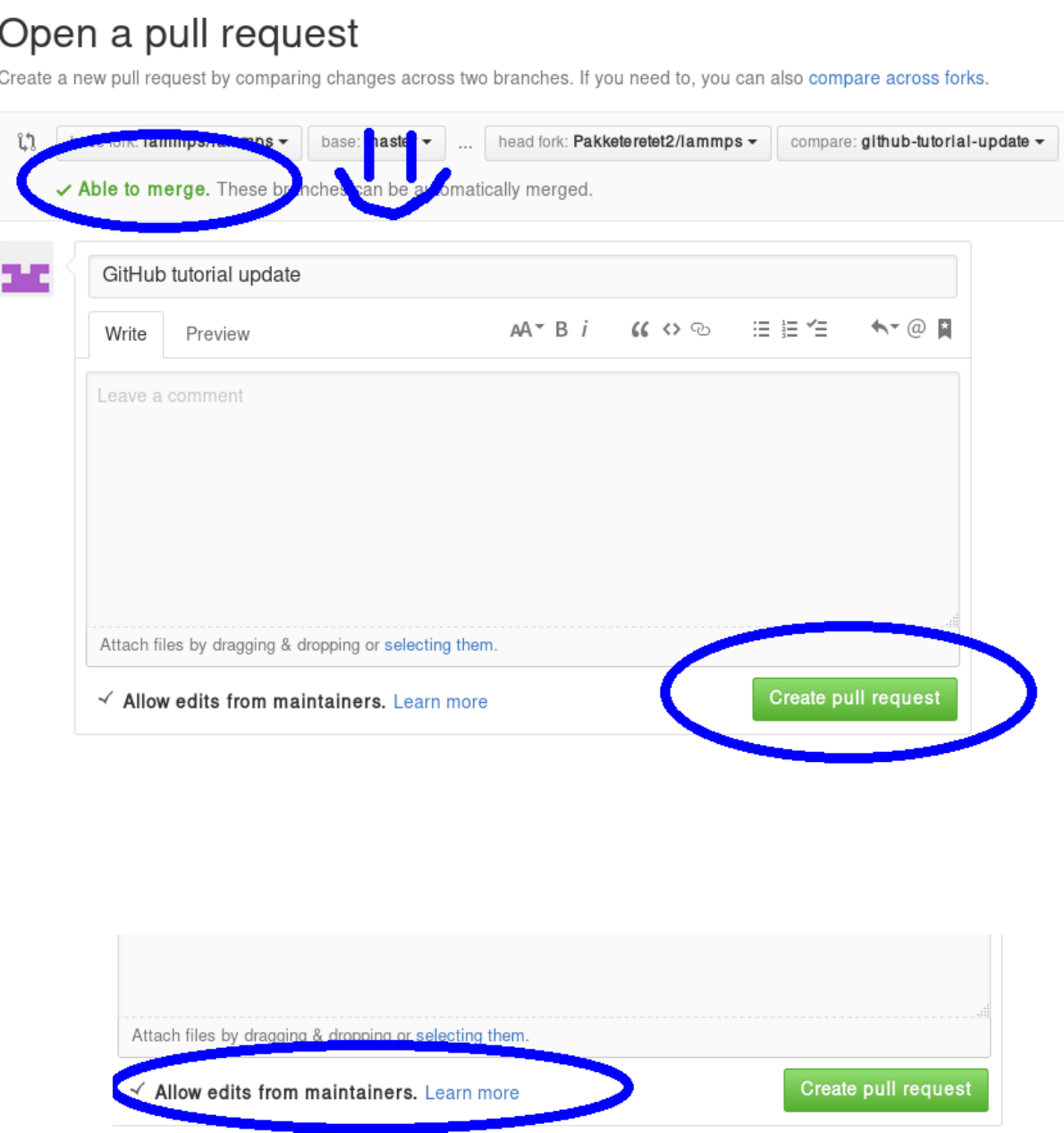
Before creating the pull request, make sure the short title is accurate and add a comment with details about
your pull request. Here you write what your modifications do and why they should be incorporated upstream.
Note the checkbox that says "Allow edits from maintainers". This is checked by default checkbox (although in
my version of Firefox, only the checkmark is visible):
If it is checked, maintainers can immediately add their own edits to the pull request. This helps the inclusion
of your branch significantly, as simple/trivial changes can be added directly to your pull request branch by the
LAMMPS Users Manual
331
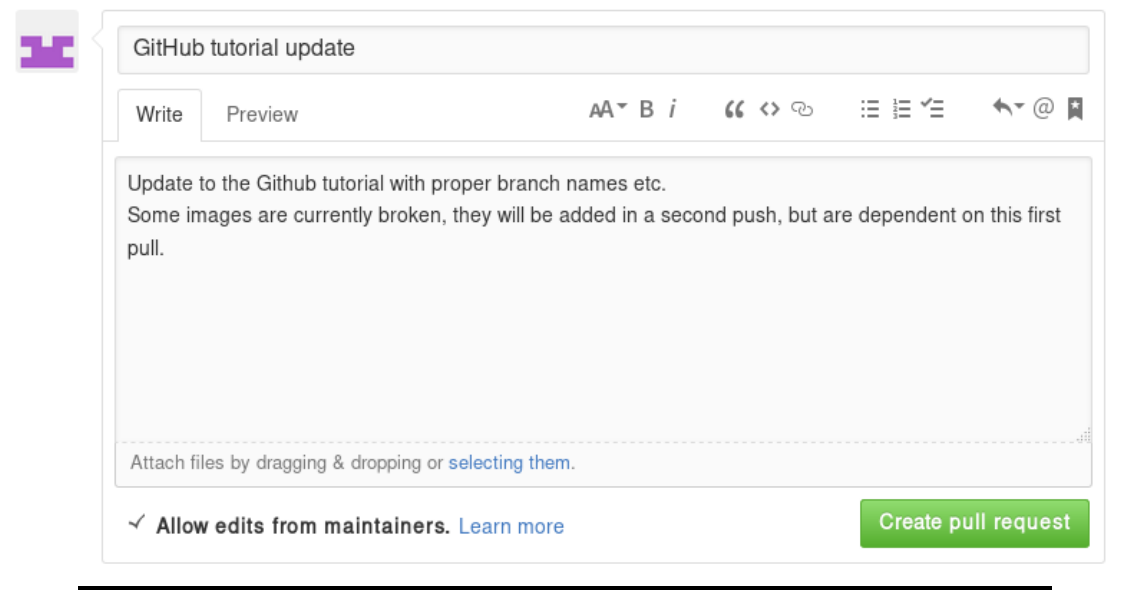
LAMMPS maintainers. The alternative would be that they make changes on their own version of the branch
and file a reverse pull request to you. Just leave this box checked unless you have a very good reason not to.
Now just write some nice comments and click on "Create pull request".
After filing a pull request
NOTE: When you submit a pull request (or ask for a pull request) for the first time, you will receive an
invitation to become a LAMMPS project collaborator. Please accept this invite as being a collaborator will
simplify certain administrative tasks and will probably speed up the merging of your feature, too.
You will notice that after filing the pull request, some checks are performed automatically:
LAMMPS Users Manual
332
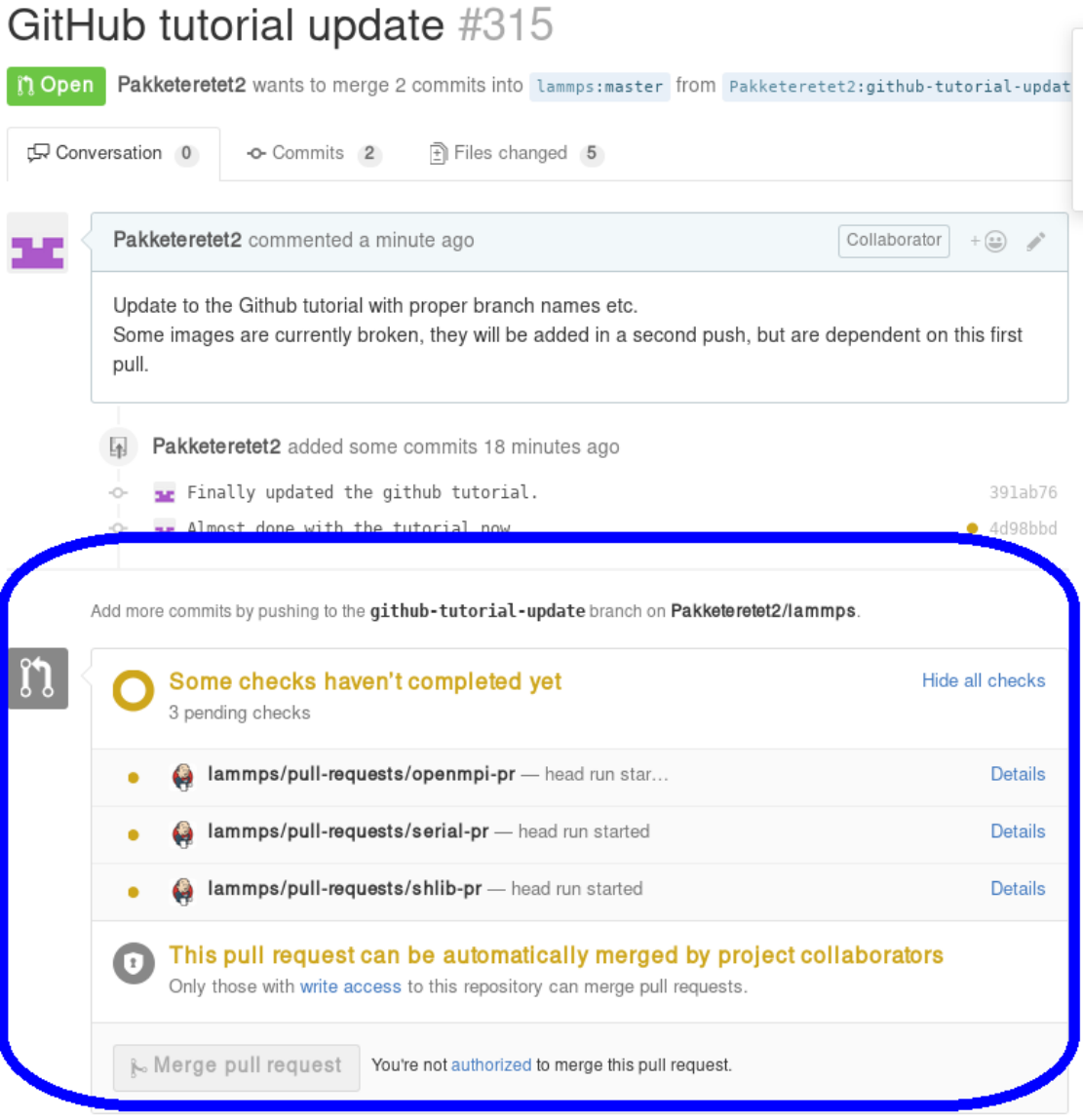
If all is fine, you will see this:
LAMMPS Users Manual
333
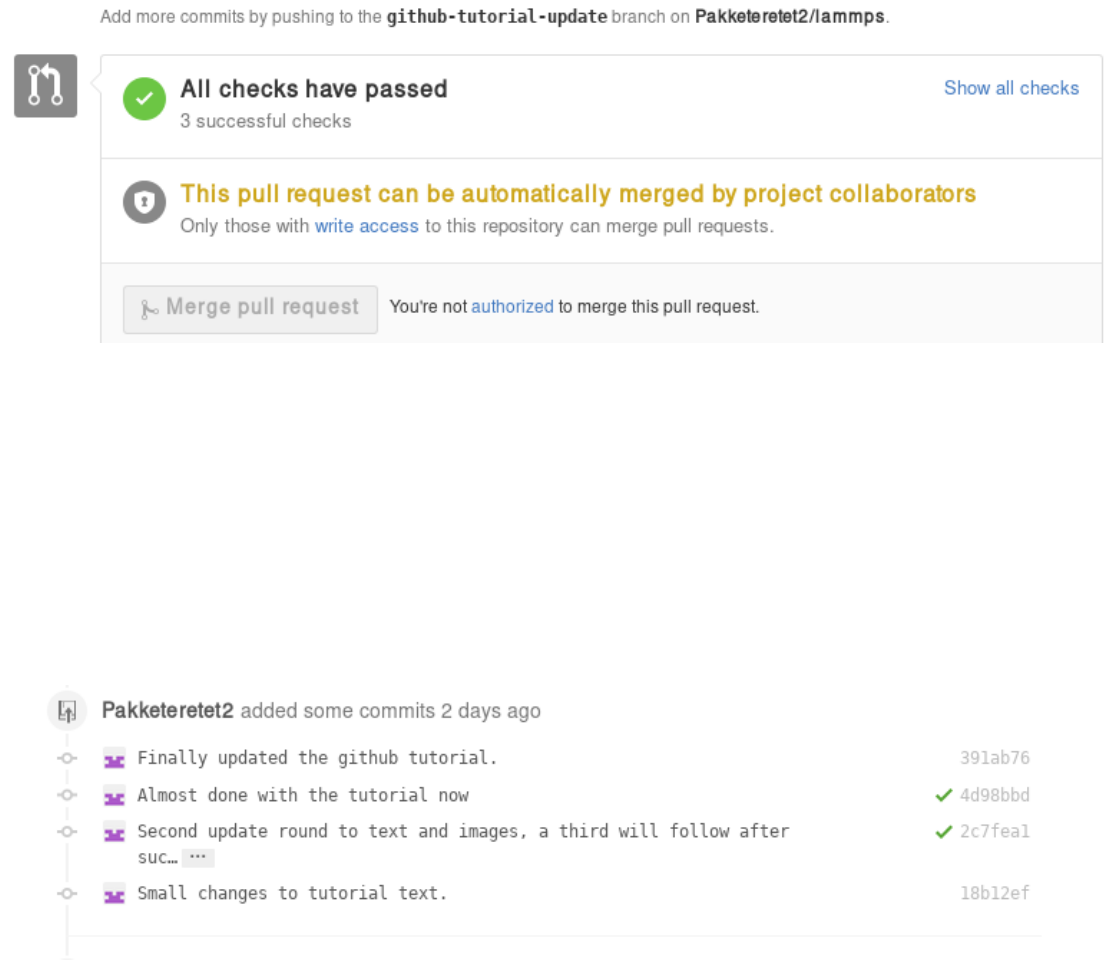
If any of the checks are failing, your pull request will not be processed, as your changes may break
compilation for certain configurations or may not merge cleanly. It is your responsibility to remove the
reason(s) for the failed test(s). If you need help with this, please contact the LAMMPS developers by adding a
comment explaining your problems with resolving the failed tests.
A few further interesting things (can) happen to pull requests before they are included.
Additional changes
First of all, any additional changes you push into your branch in your repository will automatically become
part of the pull request:
This means you can add changes that should be part of the feature after filing the pull request, which is useful
in case you have forgotten them, or if a developer has requested that something needs to be changed before
the feature can be accepted into the official LAMMPS version. After each push, the automated checks are run
again.
Assignees
There is an assignee label for pull requests. If the request has not been reviewed by any developer yet, it is not
assigned to anyone. After revision, a developer can choose to assign it to either a) you, b) a LAMMPS
developer (including him/herself) or c) Steve Plimpton (sjplimp).
Case a) happens if changes are required on your part• Case b) means that at the moment, it is being tested and reviewed by a LAMMPS developer with the•
LAMMPS Users Manual
334

expectation that some changes would be required. After the review, the developer can choose to
implement changes directly or suggest them to you.
Case c) means that the pull request has been assigned to the lead developer Steve Plimpton and means
it is considered ready for merging.
•
In this case, Axel assigned the tutorial to Steve:
Edits from LAMMPS maintainers
If you allowed edits from maintainers (the default), any LAMMPS maintainer can add changes to your pull
request. In this case, both Axel and Richard made changes to the tutorial:
Reverse pull requests
Sometimes, however, you might not feel comfortable having other people push changes into your own branch,
or maybe the maintainers are not sure their idea was the right one. In such a case, they can make changes,
reassign you as the assignee, and file a "reverse pull request", i.e. file a pull request in your GitHub repository
to include changes in the branch, that you have submitted as a pull request yourself. In that case, you can
choose to merge their changes back into your branch, possibly make additional changes or corrections and
proceed from there. It looks something like this:
LAMMPS Users Manual
335
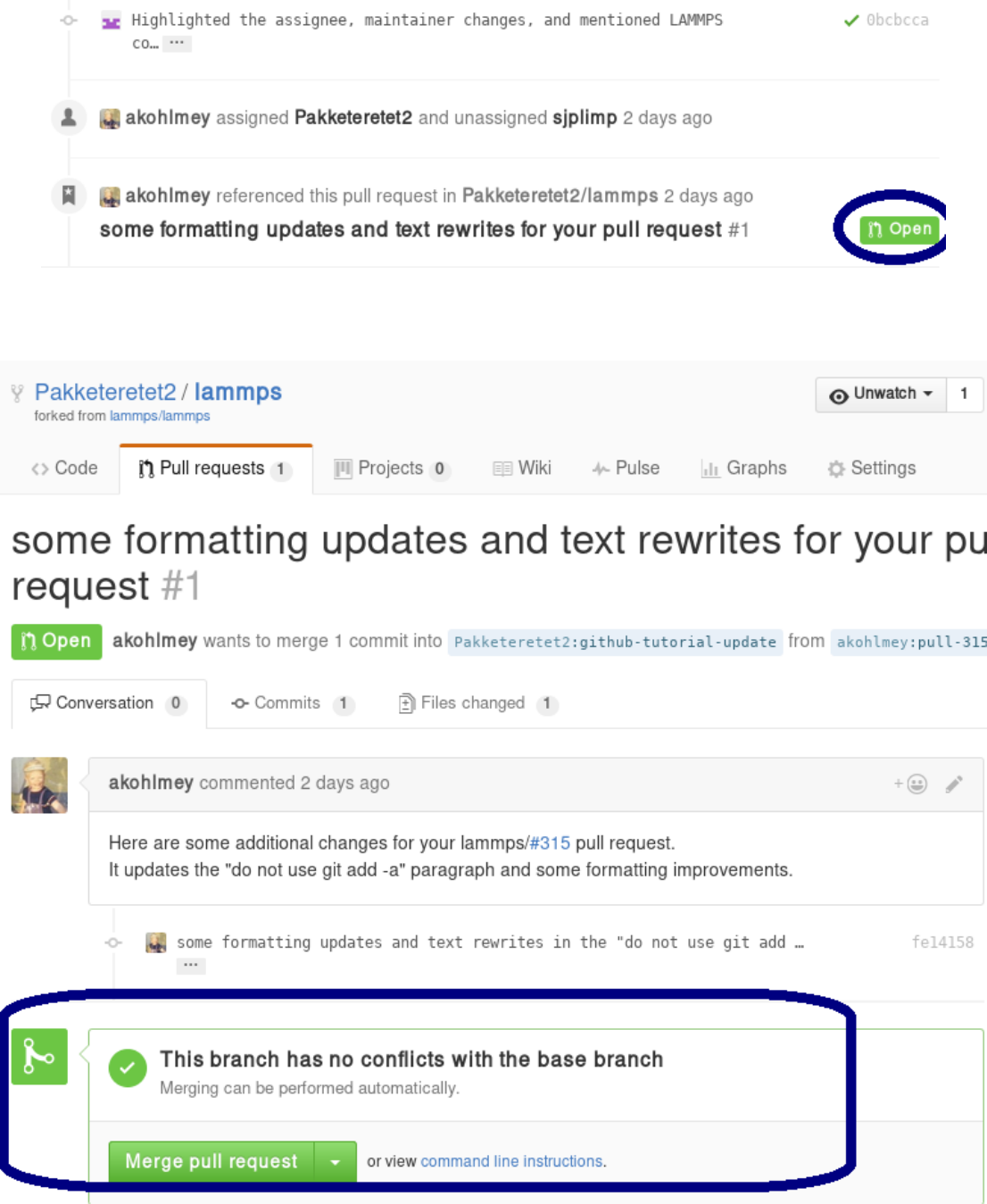
For some reason, the highlighted button didn't work in my case, but I can go to my own repository and merge
the pull request from there:
LAMMPS Users Manual
336
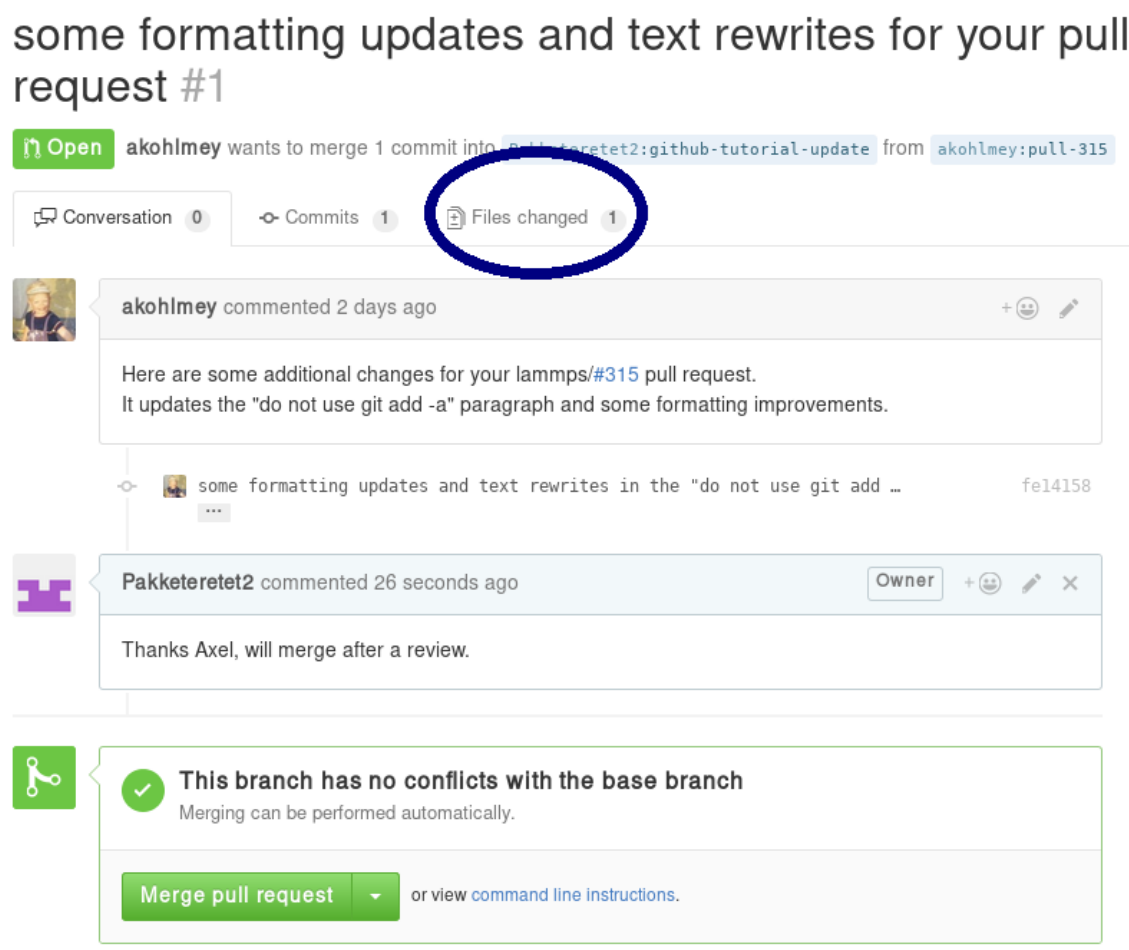
Be sure to check the changes to see if you agree with them by clicking on the tab button:
In this case, most of it is changes in the markup and a short rewrite of Axel's explanation of the "git gui" and
"git add" commands.
LAMMPS Users Manual
337
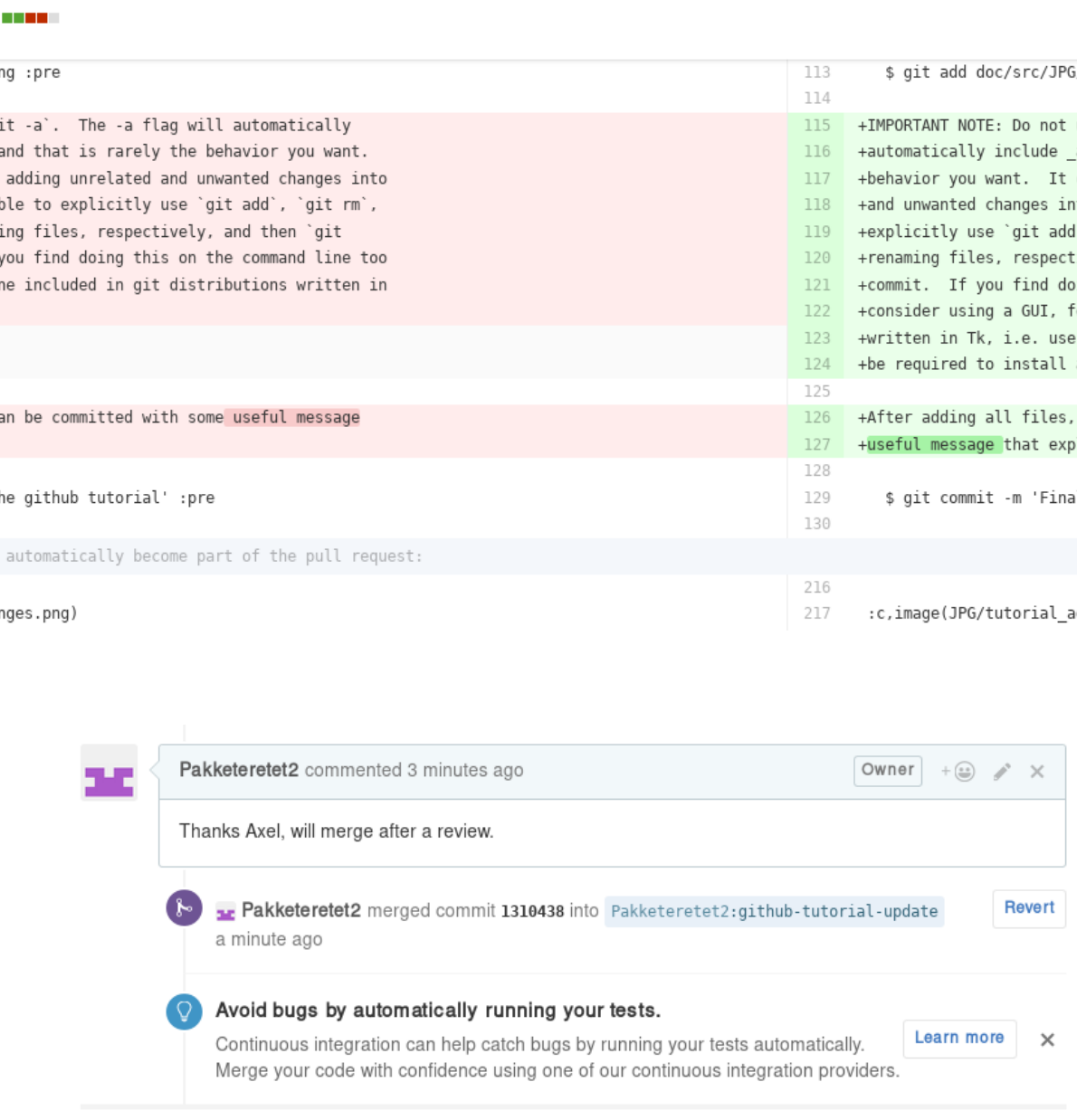
Because the changes are OK with us, we are going to merge by clicking on "Merge pull request". After a
merge it looks like this:
Now, since in the meantime our local text for the tutorial also changed, we need to pull Axel's change back
into our branch, and merge them:
LAMMPS Users Manual
338
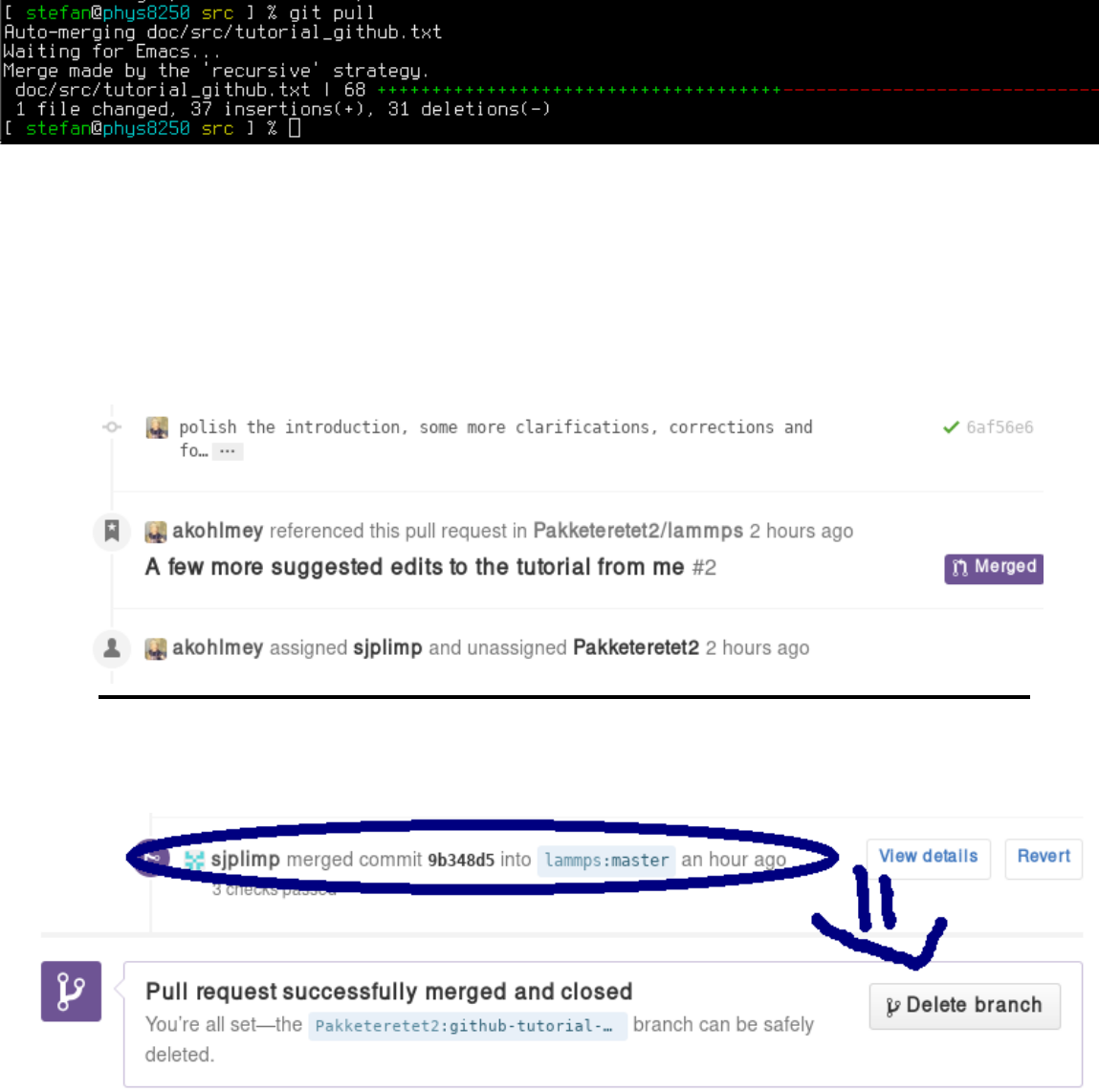
$ git add tutorial_github.txt
$ git add JPG/tutorial_reverse_pull_request*.png
$ git commit -m "Updated text and images on reverse pull requests"
$ git pull
In this case, the merge was painless because git could auto-merge:
With Axel's changes merged in and some final text updates, our feature branch is now perfect as far as we are
concerned, so we are going to commit and push again:
$ git add tutorial_github.txt
$ git add JPG/tutorial_reverse_pull_request6.png
$ git commit -m "Merged Axel's suggestions and updated text"
$ git push git@github.com:Pakketeretet2/lammps
This merge also shows up on the lammps Github page:
After a merge
When everything is fine, the feature branch is merged into the master branch:
LAMMPS Users Manual
339
Now one question remains: What to do with the feature branch that got merged into upstream?
It is in principle safe to delete them from your own fork. This helps keep it a bit more tidy. Note that you first
have to switch to another branch!
$ git checkout master
$ git pull master
$ git branch -d github-tutorial-update
If you do not pull first, it is not really a problem but git will warn you at the next statement that you are
deleting a local branch that was not yet fully merged into HEAD. This is because git does not yet know your
branch just got merged into LAMMPS upstream. If you first delete and then pull, everything should still be
fine.
Finally, if you delete the branch locally, you might want to push this to your remote(s) as well:
$ git push origin :github-tutorial-update
Recent changes in the workflow
Some changes to the workflow are not captured in this tutorial. For example, in addition to the master branch,
to which all new features should be submitted, there is now also an "unstable" and a "stable" branch; these
have the same content as "master", but are only updated after a patch release or stable release was made.
Furthermore, the naming of the patches now follow the pattern "patch_" to simplify comparisons between
releases. Finally, all patches and submissions are subject to automatic testing and code checks to make sure
they at the very least compile.
LAMMPS Users Manual
340

PyLammps Tutorial
342
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
Body particles
Overview:
This doc page is not about a LAMMPS input script command, but about body particles, which are generalized
finite-size particles. Individual body particles can represent complex entities, such as surface meshes of
discrete points, collections of sub-particles, deformable objects, etc. Note that other kinds of finite-size
spherical and aspherical particles are also supported by LAMMPS, such as spheres, ellipsoids, line segments,
and triangles, but they are simpler entities that body particles. See Section 6.14 for a general overview of all
these particle types.
Body particles are used via the atom_style body command. It takes a body style as an argument. The current
body styles supported by LAMMPS are as follows. The name in the first column is used as the bstyle
argument for the atom_style body command.
nparticle rigid body with N sub-particles
rounded/polygon 2d convex polygon with N vertices
The body style determines what attributes are stored for each body and thus how they can be used to compute
pairwise body/body or bond/non-body (point particle) interactions. More details of each style are described
below.
NOTE: The rounded/polygon style listed in the table above and described below has not yet been relesed in
LAMMPS. It will be soon.
We hope to add more styles in the future. See Section 10.12 for details on how to add a new body style to the
code.
When to use body particles:
You should not use body particles to model a rigid body made of simpler particles (e.g. point, sphere,
ellipsoid, line segment, triangular particles), if the interaction between pairs of rigid bodies is just the
summation of pairwise interactions between the simpler particles. LAMMPS already supports this kind of
model via the fix rigid command. Any of the numerous pair styles that compute interactions between simpler
particles can be used. The fix rigid command time integrates the motion of the rigid bodies. All of the
standard LAMMPS commands for thermostatting, adding constraints, performing output, etc will operate as
expected on the simple particles.
By contrast, when body particles are used, LAMMPS treats an entire body as a single particle for purposes of
computing pairwise interactions, building neighbor lists, migrating particles between processors, outputting
particles to a dump file, etc. This means that interactions between pairs of bodies or between a body and
non-body (point) particle need to be encoded in an appropriate pair style. If such a pair style were to mimic
the fix rigid model, it would need to loop over the entire collection of interactions between pairs of simple
particles within the two bodies, each time a single body/body interaction was computed.
Thus it only makes sense to use body particles and develop such a pair style, when particle/particle
interactions are more complex than what the fix rigid command can already calculate. For example, if
particles have one or more of the following attributes:
LAMMPS Users Manual
343

represented by a surface mesh• represented by a collection of geometric entities (e.g. planes + spheres)• deformable• internal stress that induces fragmentation•
then the interaction between pairs of particles is likely to be more complex than the summation of simple
sub-particle interactions. An example is contact or frictional forces between particles with planar surfaces that
inter-penetrate.
These are additional LAMMPS commands that can be used with body particles of different styles
fix nve/body integrate motion of a body particle in NVE ensemble
fix nvt/body ditto for NVT ensemble
fix npt/body ditto for NPT ensemble
fix nph/body ditto for NPH ensemble
compute body/local store sub-particle attributes of a body particle
compute temp/body compute temperature of body particles
dump local output sub-particle attributes of a body particle
dump image output body particle attributes as an image
The pair styles defined for use with specific body styles are listed in the sections below.
Specifics of body style nparticle:
The nparticle body style represents body particles as a rigid body with a variable number N of sub-particles. It
is provided as a vanilla, prototypical example of a body particle, although as mentioned above, the fix rigid
command already duplicates its functionality.
The atom_style body command for this body style takes two additional arguments:
atom_style body nparticle Nmin Nmax
Nmin = minimum # of sub-particles in any body in the system
Nmax = maximum # of sub-particles in any body in the system
The Nmin and Nmax arguments are used to bound the size of data structures used internally by each particle.
When the read_data command reads a data file for this body style, the following information must be provided
for each entry in the Bodies section of the data file:
atom-ID 1 M
N
ixx iyy izz ixy ixz iyz
x1 y1 z1
...
xN yN zN
N is the number of sub-particles in the body particle. M = 6 + 3*N. The integer line has a single value N. The
floating point line(s) list 6 moments of inertia followed by the coordinates of the N sub-particles (x1 to zN) as
3N values. These values can be listed on as many lines as you wish; see the read_data command for more
details.
LAMMPS Users Manual
344

The 6 moments of inertia (ixx,iyy,izz,ixy,ixz,iyz) should be the values consistent with the current orientation
of the rigid body around its center of mass. The values are with respect to the simulation box XYZ axes, not
with respect to the principal axes of the rigid body itself. LAMMPS performs the latter calculation internally.
The coordinates of each sub-particle are specified as its x,y,z displacement from the center-of-mass of the
body particle. The center-of-mass position of the particle is specified by the x,y,z values in the Atoms section
of the data file, as is the total mass of the body particle.
The pair_style body command can be used with this body style to compute body/body and body/non-body
interactions.
For output purposes via the compute body/local and dump local commands, this body style produces one
datum for each of the N sub-particles in a body particle. The datum has 3 values:
1 = x position of sub-particle
2 = y position of sub-particle
3 = z position of sub-particle
These values are the current position of the sub-particle within the simulation domain, not a displacement
from the center-of-mass (COM) of the body particle itself. These values are calculated using the current COM
and orientation of the body particle.
For images created by the dump image command, if the body keyword is set, then each body particle is drawn
as a collection of spheres, one for each sub-particle. The size of each sphere is determined by the bflag1
parameter for the body keyword. The bflag2 argument is ignored.
Specifics of body style rounded/polygon:
NOTE: Aug 2016 - This body style has not yet been added to LAMMPS. The info below is a placeholder.
The rounded/polygon body style represents body particles as a convex polygon with a variable number N > 2
of vertices, which can only be used for 2d models. One example use of this body style is for 2d discrete
element models, as described in Fraige. Similar to body style nparticle, the atom_style body command for this
body style takes two additional arguments:
atom_style body rounded/polygon Nmin Nmax
Nmin = minimum # of vertices in any body in the system
Nmax = maximum # of vertices in any body in the system
The Nmin and Nmax arguments are used to bound the size of data structures used internally by each particle.
When the read_data command reads a data file for this body style, the following information must be provided
for each entry in the Bodies section of the data file:
atom-ID 1 M
N
ixx iyy izz ixy ixz iyz
x1 y1 z1
...
xN yN zN
i j j k k ...
radius
LAMMPS Users Manual
345

N is the number of vertices in the body particle. M = 6 + 3*N + 2*N + 1. The integer line has a single value
N. The floating point line(s) list 6 moments of inertia followed by the coordinates of the N vertices (x1 to zN)
as 3N values, followed by 2N vertex indices corresponding to the end points of the N edges, followed by a
single radius value = the smallest circle encompassing the polygon. That last value is used to facilitate the
body/body contact detection. These floating-point values can be listed on as many lines as you wish; see the
read_data command for more details.
The 6 moments of inertia (ixx,iyy,izz,ixy,ixz,iyz) should be the values consistent with the current orientation
of the rigid body around its center of mass. The values are with respect to the simulation box XYZ axes, not
with respect to the principal axes of the rigid body itself. LAMMPS performs the latter calculation internally.
The coordinates of each vertex are specified as its x,y,z displacement from the center-of-mass of the body
particle. The center-of-mass position of the particle is specified by the x,y,z values in the Atoms section of the
data file.
For example, the following information would specify a square particles whose edge length is sqrt(2):
3 1 27
4
1 1 4 0 0 0
-0.7071 -0.7071 0
-0.7071 0.7071 0
0.7071 0.7071 0
0.7071 -0.7071 0
0 1 1 2 2 3 3 0
1.0
The pair_style body/rounded/polygon command can be used with this body style to compute body/body
interactions.
For output purposes via the compute body/local and dump local commands, this body style produces one
datum for each of the N sub-particles in a body particle. The datum has 3 values:
1 = x position of vertex
2 = y position of vertex
3 = z position of vertex
These values are the current position of the vertex within the simulation domain, not a displacement from the
center-of-mass (COM) of the body particle itself. These values are calculated using the current COM and
orientation of the body particle.
For images created by the dump image command, if the body keyword is set, then each body particle is drawn
as a convex polygon consisting of N line segments. Note that the line segments are drawn between the N
vertices, which does not correspond exactly to the physical extent of the body (because the pair_style
rounded/polygon defines finite-size spheres at those point and the line segments between the spheres are
tangent to the spheres). The drawn diameter of each line segment is determined by the bflag1 parameter for
the body keyword. The bflag2 argument is ignored.
(Fraige) F. Y. Fraige, P. A. Langston, A. J. Matchett, J. Dodds, Particuology, 6, 455 (2008).
LAMMPS Users Manual
346

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
Manifolds (surfaces)
Overview:
This doc page is not about a LAMMPS input script command, but about manifolds, which are generalized
surfaces, as defined and used by the USER-MANIFOLD package, to track particle motion on the manifolds.
See the src/USER-MANIFOLD/README file for more details about the package and its commands.
Below is a list of currently supported manifolds by the USER-MANIFOLD package, their parameters and a
short description of them. The parameters listed here are in the same order as they should be passed to the
relevant fixes.
manifold parameters equation description
cylinder R x^2 + y^2 - R^2 = 0 Cylinder along z-axis, axis going
through (0,0,0)
cylinder_dent R l a x^2 + y^2 - r(z)^2 = 0, r(x) = R if | z | > l,
r(z) = R - a*(1 + cos(z/l))/2 otherwise A cylinder with a dent around z = 0
dumbbell a A B c -( x^2 + y^2 ) * (a^2 - z^2/c^2) * ( 1 +
(A*sin(B*z^2))^4) = 0 A dumbbell
ellipsoid a b c (x/a)^2 + (y/b)^2 + (z/c)^2 = 0 An ellipsoid
plane a b c x0 y0
z0 a*(x-x0) + b*(y-y0) + c*(z-z0) = 0 A plane with normal (a,b,c) going
through point (x0,y0,z0)
plane_wiggle a w z - a*sin(w*x) = 0 A plane with a sinusoidal modulation
on z along x.
sphere R x^2 + y^2 + z^2 - R^2 = 0 A sphere of radius R
supersphere R q | x |^q + | y |^q + | z |^q - R^q = 0 A supersphere of hyperradius R
spine a, A, B, B2,
c
-(x^2 + y^2)*(a^2 - z^2/f(z)^2)*(1 +
(A*sin(g(z)*z^2))^4), f(z) = c if z > 0, 1
otherwise; g(z) = B if z > 0, B2 otherwise
An approximation to a dendtritic
spine
spine_two a, A, B, B2,
c
-(x^2 + y^2)*(a^2 - z^2/f(z)^2)*(1 +
(A*sin(g(z)*z^2))^2), f(z) = c if z > 0, 1
otherwise; g(z) = B if z > 0, B2 otherwise
Another approximation to a dendtritic
spine
thylakoid wB LB lB Various, see (Paquay)
A model grana thylakoid consisting of
two block-like compartments
connected by a bridge of width wB,
length LB and taper length lB
torus R r (R - sqrt( x^2 + y^2 ) )^2 + z^2 - r^2 A torus with large radius R and small
radius r, centered on (0,0,0)
(Paquay) Paquay and Kusters, Biophys. J., 110, 6, (2016). preprint available at arXiv:1411.3019.
LAMMPS Users Manual
347

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_coeff command
Syntax:
angle_coeff N args
N = angle type (see asterisk form below)• args = coefficients for one or more angle types•
Examples:
angle_coeff 1 300.0 107.0
angle_coeff * 5.0
angle_coeff 2*10 5.0
Description:
Specify the angle force field coefficients for one or more angle types. The number and meaning of the
coefficients depends on the angle style. Angle coefficients can also be set in the data file read by the read_data
command or in a restart file.
N can be specified in one of two ways. An explicit numeric value can be used, as in the 1st example above. Or
a wild-card asterisk can be used to set the coefficients for multiple angle types. This takes the form "*" or "*n"
or "n*" or "m*n". If N = the number of angle types, then an asterisk with no numeric values means all types
from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A trailing asterisk means all types
from n to N (inclusive). A middle asterisk means all types from m to n (inclusive).
Note that using an angle_coeff command can override a previous setting for the same angle type. For
example, these commands set the coeffs for all angle types, then overwrite the coeffs for just angle type 2:
angle_coeff * 200.0 107.0 1.2
angle_coeff 2 50.0 107.0
A line in a data file that specifies angle coefficients uses the exact same format as the arguments of the
angle_coeff command in an input script, except that wild-card asterisks should not be used since coefficients
for all N types must be listed in the file. For example, under the "Angle Coeffs" section of a data file, the line
that corresponds to the 1st example above would be listed as
1 300.0 107.0
The angle_style class2 is an exception to this rule, in that an additional argument is used in the input script to
allow specification of the cross-term coefficients. See its doc page for details.
Here is an alphabetic list of angle styles defined in LAMMPS. Click on the style to display the formula it
computes and coefficients specified by the associated angle_coeff command.
Note that there are also additional angle styles submitted by users which are included in the LAMMPS
distribution. The list of these with links to the individual styles are given in the angle section of this page.
LAMMPS Users Manual
348

angle_style none - turn off angle interactions• angle_style hybrid - define multiple styles of angle interactions•
angle_style charmm - CHARMM angle• angle_style class2 - COMPASS (class 2) angle• angle_style cosine - cosine angle potential• angle_style cosine/delta - difference of cosines angle potential• angle_style cosine/periodic - DREIDING angle• angle_style cosine/squared - cosine squared angle potential• angle_style harmonic - harmonic angle• angle_style table - tabulated by angle•
Restrictions:
This command must come after the simulation box is defined by a read_data, read_restart, or create_box
command.
An angle style must be defined before any angle coefficients are set, either in the input script or in a data file.
Related commands:
angle_style
Default: none
LAMMPS Users Manual
349
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style command
Syntax:
angle_style style
style = none or hybrid or charmm or class2 or cosine or cosine/squared or harmonic•
Examples:
angle_style harmonic
angle_style charmm
angle_style hybrid harmonic cosine
Description:
Set the formula(s) LAMMPS uses to compute angle interactions between triplets of atoms, which remain in
force for the duration of the simulation. The list of angle triplets is read in by a read_data or read_restart
command from a data or restart file.
Hybrid models where angles are computed using different angle potentials can be setup using the hybrid angle
style.
The coefficients associated with a angle style can be specified in a data or restart file or via the angle_coeff
command.
All angle potentials store their coefficient data in binary restart files which means angle_style and angle_coeff
commands do not need to be re-specified in an input script that restarts a simulation. See the read_restart
command for details on how to do this. The one exception is that angle_style hybrid only stores the list of
sub-styles in the restart file; angle coefficients need to be re-specified.
NOTE: When both an angle and pair style is defined, the special_bonds command often needs to be used to
turn off (or weight) the pairwise interaction that would otherwise exist between 3 bonded atoms.
In the formulas listed for each angle style, theta is the angle between the 3 atoms in the angle.
Here is an alphabetic list of angle styles defined in LAMMPS. Click on the style to display the formula it
computes and coefficients specified by the associated angle_coeff command.
Note that there are also additional angle styles submitted by users which are included in the LAMMPS
distribution. The list of these with links to the individual styles are given in the angle section of this page.
angle_style none - turn off angle interactions• angle_style zero - topology but no interactions• angle_style hybrid - define multiple styles of angle interactions•
angle_style charmm - CHARMM angle• angle_style class2 - COMPASS (class 2) angle• angle_style cosine - cosine angle potential•
LAMMPS Users Manual
350

angle_style cosine/delta - difference of cosines angle potential• angle_style cosine/periodic - DREIDING angle• angle_style cosine/squared - cosine squared angle potential• angle_style harmonic - harmonic angle• angle_style table - tabulated by angle•
Restrictions:
Angle styles can only be set for atom_styles that allow angles to be defined.
Most angle styles are part of the MOLECULE package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info on packages. The doc pages for individual
bond potentials tell if it is part of a package.
Related commands:
angle_coeff
Default:
angle_style none
LAMMPS Users Manual
351

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
atom_modify command
Syntax:
atom_modify keyword values ...
one or more keyword/value pairs may be appended• keyword = id or map or first or sort
id value = yes or no
map value = array or hash
first value = group-ID = group whose atoms will appear first in internal atom lists
sort values = Nfreq binsize
Nfreq = sort atoms spatially every this many time steps
binsize = bin size for spatial sorting (distance units)
•
Examples:
atom_modify map hash
atom_modify map array sort 10000 2.0
atom_modify first colloid
Description:
Modify certain attributes of atoms defined and stored within LAMMPS, in addition to what is specified by the
atom_style command. The id and map keywords must be specified before a simulation box is defined; other
keywords can be specified any time.
The id keyword determines whether non-zero atom IDs can be assigned to each atom. If the value is yes,
which is the default, IDs are assigned, whether you use the create atoms or read_data or read_restart
commands to initialize atoms. If the value is no the IDs for all atoms are assumed to be 0.
If atom IDs are used, they must all be positive integers. They should also be unique, though LAMMPS does
not check for this. Typically they should also be consecutively numbered (from 1 to Natoms), though this is
not required. Molecular atom styles are those that store bond topology information (styles bond, angle,
molecular, full). These styles require atom IDs since the IDs are used to encode the topology. Some other
LAMMPS commands also require the use of atom IDs. E.g. some many-body pair styles use them to avoid
double computation of the I-J interaction between two atoms.
The only reason not to use atom IDs is if you are running an atomic simulation so large that IDs cannot be
uniquely assigned. For a default LAMMPS build this limit is 2^31 or about 2 billion atoms. However, even in
this case, you can use 64-bit atom IDs, allowing 2^63 or about 9e18 atoms, if you build LAMMPS with the -
DLAMMPS_BIGBIG switch. This is described in Section 2.2 of the manual. If atom IDs are not used, they
must be specified as 0 for all atoms, e.g. in a data or restart file.
The map keyword determines how atom ID lookup is done for molecular atom styles. Lookups are performed
by bond (angle, etc) routines in LAMMPS to find the local atom index associated with a global atom ID.
When the array value is used, each processor stores a lookup table of length N, where N is the largest atom ID
in the system. This is a fast, simple method for many simulations, but requires too much memory for large
LAMMPS Users Manual
352
simulations. The hash value uses a hash table to perform the lookups. This can be slightly slower than the
array method, but its memory cost is proportional to the number of atoms owned by a processor, i.e. N/P
when N is the total number of atoms in the system and P is the number of processors.
When this setting is not specified in your input script, LAMMPS creates a map, if one is needed, as an array
or hash. See the discussion of default values below for how LAMMPS chooses which kind of map to build.
Note that atomic systems do not normally need to create a map. However, even in this case some LAMMPS
commands will create a map to find atoms (and then destroy it), or require a permanent map. An example of
the former is the velocity loop all command, which uses a map when looping over all atoms and insuring the
same velocity values are assigned to an atom ID, no matter which processor owns it.
The first keyword allows a group to be specified whose atoms will be maintained as the first atoms in each
processor's list of owned atoms. This in only useful when the specified group is a small fraction of all the
atoms, and there are other operations LAMMPS is performing that will be sped-up significantly by being able
to loop over the smaller set of atoms. Otherwise the reordering required by this option will be a net
slow-down. The neigh_modify include and comm_modify group commands are two examples of commands
that require this setting to work efficiently. Several fixes, most notably time integration fixes like fix nve, also
take advantage of this setting if the group they operate on is the group specified by this command. Note that
specifying "all" as the group-ID effectively turns off the first option.
It is OK to use the first keyword with a group that has not yet been defined, e.g. to use the atom_modify first
command at the beginning of your input script. LAMMPS does not use the group until a simulation is run.
The sort keyword turns on a spatial sorting or reordering of atoms within each processor's sub-domain every
Nfreq timesteps. If Nfreq is set to 0, then sorting is turned off. Sorting can improve cache performance and
thus speed-up a LAMMPS simulation, as discussed in a paper by (Meloni). Its efficacy depends on the
problem size (atoms/processor), how quickly the system becomes disordered, and various other factors. As a
general rule, sorting is typically more effective at speeding up simulations of liquids as opposed to solids. In
tests we have done, the speed-up can range from zero to 3-4x.
Reordering is performed every Nfreq timesteps during a dynamics run or iterations during a minimization.
More precisely, reordering occurs at the first reneighboring that occurs after the target timestep. The
reordering is performed locally by each processor, using bins of the specified binsize. If binsize is set to 0.0,
then a binsize equal to half the neighbor cutoff distance (force cutoff plus skin distance) is used, which is a
reasonable value. After the atoms have been binned, they are reordered so that atoms in the same bin are
adjacent to each other in the processor's 1d list of atoms.
The goal of this procedure is for atoms to put atoms close to each other in the processor's one-dimensional list
of atoms that are also near to each other spatially. This can improve cache performance when pairwise
interactions and neighbor lists are computed. Note that if bins are too small, there will be few atoms/bin.
Likewise if bins are too large, there will be many atoms/bin. In both cases, the goal of cache locality will be
undermined.
NOTE: Running a simulation with sorting on versus off should not change the simulation results in a
statistical sense. However, a different ordering will induce round-off differences, which will lead to diverging
trajectories over time when comparing two simulations. Various commands, particularly those which use
random numbers (e.g. velocity create, and fix langevin), may generate (statistically identical) results which
depend on the order in which atoms are processed. The order of atoms in a dump file will also typically
change if sorting is enabled.
Restrictions:
LAMMPS Users Manual
353

The first and sort options cannot be used together. Since sorting is on by default, it will be turned off if the
first keyword is used with a group-ID that is not "all".
Related commands: none
Default:
By default, id is yes. By default, atomic systems (no bond topology info) do not use a map. For molecular
systems (with bond topology info), a map is used. The default map style is array if no atom ID is larger than 1
million, otherwise the default is hash. By default, a "first" group is not defined. By default, sorting is enabled
with a frequency of 1000 and a binsize of 0.0, which means the neighbor cutoff will be used to set the bin
size.
(Meloni) Meloni, Rosati and Colombo, J Chem Phys, 126, 121102 (2007).
LAMMPS Users Manual
354

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
atom_style command
Syntax:
atom_style style args
style = angle or atomic or body or bond or charge or dipole or dpd or electron or ellipsoid or full or
line or meso or molecular or peri or smd or sphere or tri or template or hybrid
args = none for any style except the following
body args = bstyle bstyle-args
bstyle = style of body particles
bstyle-args = additional arguments specific to the bstyle
see the body doc page for details
template args = template-ID
template-ID = ID of molecule template specified in a separate molecule command
hybrid args = list of one or more sub-styles, each with their args
•
accelerated styles (with same args) = angle/kk or atomic/kk or bond/kk or charge/kk or full/kk or
molecular/kk
•
Examples:
atom_style atomic
atom_style bond
atom_style full
atom_style body nparticle 2 10
atom_style hybrid charge bond
atom_style hybrid charge body nparticle 2 5
atom_style template myMols
Description:
Define what style of atoms to use in a simulation. This determines what attributes are associated with the
atoms. This command must be used before a simulation is setup via a read_data, read_restart, or create_box
command.
NOTE: Many of the atom styles discussed here are only enabled if LAMMPS was built with a specific
package, as listed below in the Restrictions section.
Once a style is assigned, it cannot be changed, so use a style general enough to encompass all attributes. E.g.
with style bond, angular terms cannot be used or added later to the model. It is OK to use a style more general
than needed, though it may be slightly inefficient.
The choice of style affects what quantities are stored by each atom, what quantities are communicated
between processors to enable forces to be computed, and what quantities are listed in the data file read by the
read_data command.
These are the additional attributes of each style and the typical kinds of physical systems they are used to
model. All styles store coordinates, velocities, atom IDs and types. See the read_data, create_atoms, and set
commands for info on how to set these various quantities.
LAMMPS Users Manual
355

angle bonds and angles bead-spring polymers with stiffness
atomic only the default values coarse-grain liquids, solids, metals
body mass, inertia moments, quaternion, angular momentum arbitrary bodies
bond bonds bead-spring polymers
charge charge atomic system with charges
dipole charge and dipole moment system with dipolar particles
dpd internal temperature and internal energies DPD particles
electron charge and spin and eradius electronic force field
ellipsoid shape, quaternion, angular momentum aspherical particles
full molecular + charge bio-molecules
line end points, angular velocity rigid bodies
meso rho, e, cv SPH particles
molecular bonds, angles, dihedrals, impropers uncharged molecules
peri mass, volume mesocopic Peridynamic models
smd volume, kernel diameter, contact radius, mass solid and fluid SPH particles
sphere diameter, mass, angular velocity granular models
template template index, template atom small molecules with fixed topology
tri corner points, angular momentum rigid bodies
wavepacket charge, spin, eradius, etag, cs_re, cs_im AWPMD
NOTE: It is possible to add some attributes, such as a molecule ID, to atom styles that do not have them via
the fix property/atom command. This command also allows new custom attributes consisting of extra integer
or floating-point values to be added to atoms. See the fix property/atom doc page for examples of cases where
this is useful and details on how to initialize, access, and output the custom values.
All of the above styles define point particles, except the sphere, ellipsoid, electron, peri, wavepacket, line, tri,
and body styles, which define finite-size particles. See Section 6.14 for an overview of using finite-size
particle models with LAMMPS.
All of the point-particle styles assign mass to particles on a per-type basis, using the mass command, The
finite-size particle styles assign mass to individual particles on a per-particle basis.
For the sphere style, the particles are spheres and each stores a per-particle diameter and mass. If the diameter
> 0.0, the particle is a finite-size sphere. If the diameter = 0.0, it is a point particle. Note that by use of the disc
keyword with the fix nve/sphere, fix nvt/sphere, fix nph/sphere, fix npt/sphere commands, spheres can be
effectively treated as 2d discs for a 2d simulation if desired. See also the set density/disc command.
For the ellipsoid style, the particles are ellipsoids and each stores a flag which indicates whether it is a
finite-size ellipsoid or a point particle. If it is an ellipsoid, it also stores a shape vector with the 3 diameters of
the ellipsoid and a quaternion 4-vector with its orientation.
For the dipole style, a point dipole is defined for each point particle. Note that if you wish the particles to be
finite-size spheres as in a Stockmayer potential for a dipolar fluid, so that the particles can rotate due to
dipole-dipole interactions, then you need to use atom_style hybrid sphere dipole, which will assign both a
diameter and dipole moment to each particle.
For the electron style, the particles representing electrons are 3d Gaussians with a specified position and
bandwidth or uncertainty in position, which is represented by the eradius = electron size.
LAMMPS Users Manual
356
For the peri style, the particles are spherical and each stores a per-particle mass and volume.
The dpd style is for dissipative particle dynamics (DPD) particles. Note that it is part of the USER-DPD
package, and is not for use with the pair_style dpd or dpd/stat commands, which can simply use atom_style
atomic. Atom_style dpd extends DPD particle properties with internal temperature (dpdTheta), internal
conductive energy (uCond), internal mechanical energy (uMech), and internal chemical energy (uChem).
The meso style is for smoothed particle hydrodynamics (SPH) particles which store a density (rho), energy
(e), and heat capacity (cv).
The smd style is for a general formulation of Smooth Particle Hydrodynamics. Both fluids and solids can be
modeled. Particles store the mass and volume of an integration point, a kernel diameter used for calculating
the field variables (e.g. stress and deformation) and a contact radius for calculating repulsive forces which
prevent individual physical bodies from penetrating each other.
The wavepacket style is similar to electron, but the electrons may consist of several Gaussian wave packets,
summed up with coefficients cs= (cs_re,cs_im). Each of the wave packets is treated as a separate particle in
LAMMPS, wave packets belonging to the same electron must have identical etag values.
For the line style, the particles are idealized line segments and each stores a per-particle mass and length and
orientation (i.e. the end points of the line segment).
For the tri style, the particles are planar triangles and each stores a per-particle mass and size and orientation
(i.e. the corner points of the triangle).
The template style allows molecular topology (bonds,angles,etc) to be defined via a molecule template using
the molecule command. The template stores one or more molecules with a single copy of the topology info
(bonds,angles,etc) of each. Individual atoms only store a template index and template atom to identify which
molecule and which atom-within-the-molecule they represent. Using the template style instead of the bond,
angle, molecular styles can save memory for systems comprised of a large number of small molecules, all of a
single type (or small number of types). See the paper by Grime and Voth, in (Grime), for examples of how
this can be advantageous for large-scale coarse-grained systems.
NOTE: When using the template style with a molecule template that contains multiple molecules, you should
insure the atom types, bond types, angle_types, etc in all the molecules are consistent. E.g. if one molecule
represents H2O and another CO2, then you probably do not want each molecule file to define 2 atom types
and a single bond type, because they will conflict with each other when a mixture system of H2O and CO2
molecules is defined, e.g. by the read_data command. Rather the H2O molecule should define atom types 1
and 2, and bond type 1. And the CO2 molecule should define atom types 3 and 4 (or atom types 3 and 2 if a
single oxygen type is desired), and bond type 2.
For the body style, the particles are arbitrary bodies with internal attributes defined by the "style" of the
bodies, which is specified by the bstyle argument. Body particles can represent complex entities, such as
surface meshes of discrete points, collections of sub-particles, deformable objects, etc.
The body doc page describes the body styles LAMMPS currently supports, and provides more details as to the
kind of body particles they represent. For all styles, each body particle stores moments of inertia and a
quaternion 4-vector, so that its orientation and position can be time integrated due to forces and torques.
Note that there may be additional arguments required along with the bstyle specification, in the atom_style
body command. These arguments are described in the body doc page.
LAMMPS Users Manual
357

Typically, simulations require only a single (non-hybrid) atom style. If some atoms in the simulation do not
have all the properties defined by a particular style, use the simplest style that defines all the needed properties
by any atom. For example, if some atoms in a simulation are charged, but others are not, use the charge style.
If some atoms have bonds, but others do not, use the bond style.
The only scenario where the hybrid style is needed is if there is no single style which defines all needed
properties of all atoms. For example, as mentioned above, if you want dipolar particles which will rotate due
to torque, you need to use "atom_style hybrid sphere dipole". When a hybrid style is used, atoms store and
communicate the union of all quantities implied by the individual styles.
When using the hybrid style, you cannot combine the template style with another molecular style that stores
bond,angle,etc info on a per-atom basis.
LAMMPS can be extended with new atom styles as well as new body styles; see this section.
Styles with a kk suffix are functionally the same as the corresponding style without the suffix. They have been
optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The
accelerated styles take the same arguments and should produce the same results, except for round-off and
precision issues.
Note that other acceleration packages in LAMMPS, specifically the GPU, USER-INTEL, USER-OMP, and
OPT packages do not use accelerated atom styles.
The accelerated styles are part of the KOKKOS package. They are only enabled if LAMMPS was built with
those packages. See the Making LAMMPS section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This command cannot be used after the simulation box is defined by a read_data or create_box command.
Many of the styles listed above are only enabled if LAMMPS was built with a specific package, as listed
below. See the Making LAMMPS section for more info.
The angle, bond, full, molecular, and template styles are part of the MOLECULE package.
The line and tri styles are part of the ASPHERE package.
The body style is part of the BODY package.
The dipole style is part of the DIPOLE package.
The peri style is part of the PERI package for Peridynamics.
LAMMPS Users Manual
358

The electron style is part of the USER-EFF package for electronic force fields.
The dpd style is part of the USER-DPD package for dissipative particle dynamics (DPD).
The meso style is part of the USER-SPH package for smoothed particle hydrodynamics (SPH). See this PDF
guide to using SPH in LAMMPS.
The wavepacket style is part of the USER-AWPMD package for the antisymmetrized wave packet MD
method.
Related commands:
read_data, pair_style
Default:
atom_style atomic
(Grime) Grime and Voth, to appear in J Chem Theory & Computation (2014).
LAMMPS Users Manual
359

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
balance command
Syntax:
balance thresh style args ... keyword args ...
thresh = imbalance threshold that must be exceeded to perform a re-balance• one style/arg pair can be used (or multiple for x,y,z)• style = x or y or z or shift or rcb
x args = uniform or Px-1 numbers between 0 and 1
uniform = evenly spaced cuts between processors in x dimension
numbers = Px-1 ascending values between 0 and 1, Px - # of processors in x dimension
x can be specified together with y or z
y args = uniform or Py-1 numbers between 0 and 1
uniform = evenly spaced cuts between processors in y dimension
numbers = Py-1 ascending values between 0 and 1, Py - # of processors in y dimension
y can be specified together with x or z
z args = uniform or Pz-1 numbers between 0 and 1
uniform = evenly spaced cuts between processors in z dimension
numbers = Pz-1 ascending values between 0 and 1, Pz - # of processors in z dimension
z can be specified together with x or y
shift args = dimstr Niter stopthresh
dimstr = sequence of letters containing "x" or "y" or "z", each not more than once
Niter = # of times to iterate within each dimension of dimstr sequence
stopthresh = stop balancing when this imbalance threshold is reached
rcb args = none
•
zero or more keyword/arg pairs may be appended• keyword = weight or out
weight style args = use weighted particle counts for the balancing
style = group or neigh or time or var or store
group args = Ngroup group1 weight1 group2 weight2 ...
Ngroup = number of groups with assigned weights
group1, group2, ... = group IDs
weight1, weight2, ... = corresponding weight factors
neigh factor = compute weight based on number of neighbors
factor = scaling factor (> 0)
time factor = compute weight based on time spend computing
factor = scaling factor (> 0)
var name = take weight from atom-style variable
name = name of the atom-style variable
store name = store weight in custom atom property defined by fix property/atom command
name = atom property name (without d_ prefix)
out arg = filename
filename = write each processor's sub-domain to a file
•
Examples:
balance 0.9 x uniform y 0.4 0.5 0.6
balance 1.2 shift xz 5 1.1
balance 1.0 shift xz 5 1.1
balance 1.1 rcb
balance 1.0 shift x 10 1.1 weight group 2 fast 0.5 slow 2.0
balance 1.0 shift x 10 1.1 weight time 0.8 weight neigh 0.5 weight store balance
LAMMPS Users Manual
360
balance 1.0 shift x 20 1.0 out tmp.balance
Description:
This command adjusts the size and shape of processor sub-domains within the simulation box, to attempt to
balance the number of atoms or particles and thus indirectly the computational cost (load) more evenly across
processors. The load balancing is "static" in the sense that this command performs the balancing once, before
or between simulations. The processor sub-domains will then remain static during the subsequent run. To
perform "dynamic" balancing, see the fix balance command, which can adjust processor sub-domain sizes and
shapes on-the-fly during a run.
Load-balancing is typically most useful if the particles in the simulation box have a spatially-varying density
distribution or when the computational cost varies significantly between different particles. E.g. a model of a
vapor/liquid interface, or a solid with an irregular-shaped geometry containing void regions, or hybrid pair
style simulations which combine pair styles with different computational cost. In these cases, the LAMMPS
default of dividing the simulation box volume into a regular-spaced grid of 3d bricks, with one equal-volume
sub-domain per processor, may assign numbers of particles per processor in a way that the computational
effort varies significantly. This can lead to poor performance when the simulation is run in parallel.
The balancing can be performed with or without per-particle weighting. With no weighting, the balancing
attempts to assign an equal number of particles to each processor. With weighting, the balancing attempts to
assign an equal aggregate computational weight to each processor, which typically induces a different number
of atoms assigned to each processor. Details on the various weighting options and examples for how they can
be used are given below.
Note that the processors command allows some control over how the box volume is split across processors.
Specifically, for a Px by Py by Pz grid of processors, it allows choice of Px, Py, and Pz, subject to the
constraint that Px * Py * Pz = P, the total number of processors. This is sufficient to achieve good
load-balance for some problems on some processor counts. However, all the processor sub-domains will still
have the same shape and same volume.
The requested load-balancing operation is only performed if the current "imbalance factor" in particles owned
by each processor exceeds the specified thresh parameter. The imbalance factor is defined as the maximum
number of particles (or weight) owned by any processor, divided by the average number of particles (or
weight) per processor. Thus an imbalance factor of 1.0 is perfect balance.
As an example, for 10000 particles running on 10 processors, if the most heavily loaded processor has 1200
particles, then the factor is 1.2, meaning there is a 20% imbalance. Note that a re-balance can be forced even if
the current balance is perfect (1.0) be specifying a thresh < 1.0.
NOTE: Balancing is performed even if the imbalance factor does not exceed the thresh parameter if a "grid"
style is specified when the current partitioning is "tiled". The meaning of "grid" vs "tiled" is explained below.
This is to allow forcing of the partitioning to "grid" so that the comm_style brick command can then be used
to replace a current comm_style tiled setting.
When the balance command completes, it prints statistics about the result, including the change in the
imbalance factor and the change in the maximum number of particles on any processor. For "grid" methods
(defined below) that create a logical 3d grid of processors, the positions of all cutting planes in each of the 3
dimensions (as fractions of the box length) are also printed.
LAMMPS Users Manual
361
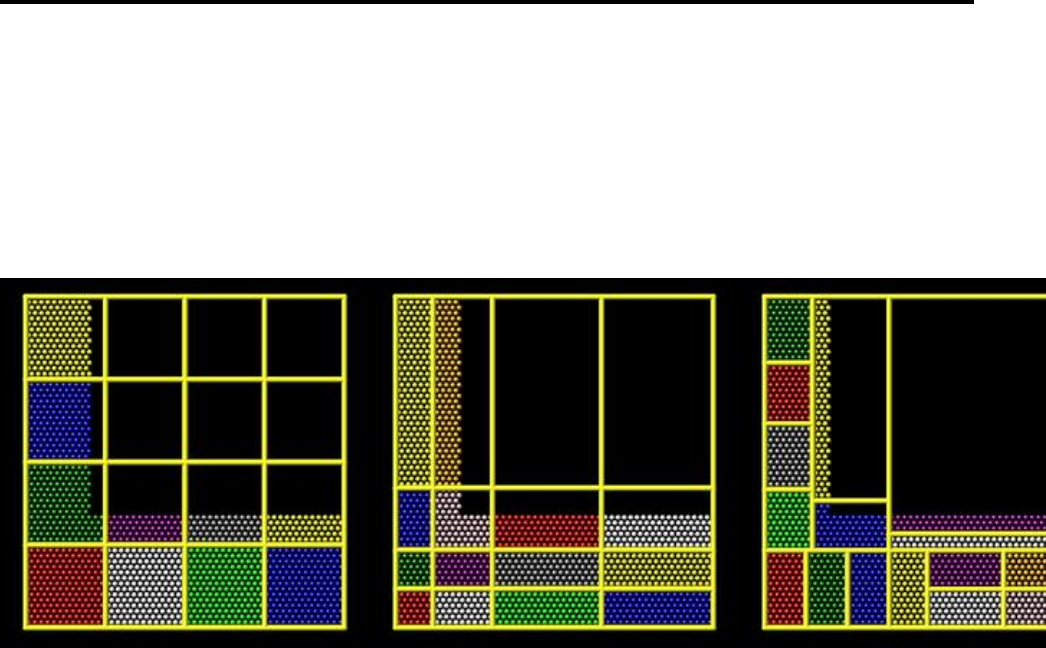
NOTE: This command attempts to minimize the imbalance factor, as defined above. But depending on the
method a perfect balance (1.0) may not be achieved. For example, "grid" methods (defined below) that create
a logical 3d grid cannot achieve perfect balance for many irregular distributions of particles. Likewise, if a
portion of the system is a perfect lattice, e.g. the initial system is generated by the create_atoms command,
then "grid" methods may be unable to achieve exact balance. This is because entire lattice planes will be
owned or not owned by a single processor.
NOTE: The imbalance factor is also an estimate of the maximum speed-up you can hope to achieve by
running a perfectly balanced simulation versus an imbalanced one. In the example above, the 10000 particle
simulation could run up to 20% faster if it were perfectly balanced, versus when imbalanced. However,
computational cost is not strictly proportional to particle count, and changing the relative size and shape of
processor sub-domains may lead to additional computational and communication overheads, e.g. in the PPPM
solver used via the kspace_style command. Thus you should benchmark the run times of a simulation before
and after balancing.
The method used to perform a load balance is specified by one of the listed styles (or more in the case of
x,y,z), which are described in detail below. There are 2 kinds of styles.
The x, y, z, and shift styles are "grid" methods which produce a logical 3d grid of processors. They operate by
changing the cutting planes (or lines) between processors in 3d (or 2d), to adjust the volume (area in 2d)
assigned to each processor, as in the following 2d diagram where processor sub-domains are shown and
particles are colored by the processor that owns them. The leftmost diagram is the default partitioning of the
simulation box across processors (one sub-box for each of 16 processors); the middle diagram is after a "grid"
method has been applied.
The rcb style is a "tiling" method which does not produce a logical 3d grid of processors. Rather it tiles the
simulation domain with rectangular sub-boxes of varying size and shape in an irregular fashion so as to have
equal numbers of particles (or weight) in each sub-box, as in the rightmost diagram above.
The "grid" methods can be used with either of the comm_style command options, brick or tiled. The "tiling"
methods can only be used with comm_style tiled. Note that it can be useful to use a "grid" method with
comm_style tiled to return the domain partitioning to a logical 3d grid of processors so that "comm_style
brick" can afterwords be specified for subsequent run commands.
When a "grid" method is specified, the current domain partitioning can be either a logical 3d grid or a tiled
partitioning. In the former case, the current logical 3d grid is used as a starting point and changes are made to
LAMMPS Users Manual
362

improve the imbalance factor. In the latter case, the tiled partitioning is discarded and a logical 3d grid is
created with uniform spacing in all dimensions. This becomes the starting point for the balancing operation.
When a "tiling" method is specified, the current domain partitioning ("grid" or "tiled") is ignored, and a new
partitioning is computed from scratch.
The x, y, and z styles invoke a "grid" method for balancing, as described above. Note that any or all of these 3
styles can be specified together, one after the other, but they cannot be used with any other style. This style
adjusts the position of cutting planes between processor sub-domains in specific dimensions. Only the
specified dimensions are altered.
The uniform argument spaces the planes evenly, as in the left diagrams above. The numeric argument requires
listing Ps-1 numbers that specify the position of the cutting planes. This requires knowing Ps = Px or Py or Pz
= the number of processors assigned by LAMMPS to the relevant dimension. This assignment is made (and
the Px, Py, Pz values printed out) when the simulation box is created by the "create_box" or "read_data" or
"read_restart" command and is influenced by the settings of the processors command.
Each of the numeric values must be between 0 and 1, and they must be listed in ascending order. They
represent the fractional position of the cutting place. The left (or lower) edge of the box is 0.0, and the right
(or upper) edge is 1.0. Neither of these values is specified. Only the interior Ps-1 positions are specified. Thus
is there are 2 processors in the x dimension, you specify a single value such as 0.75, which would make the
left processor's sub-domain 3x larger than the right processor's sub-domain.
The shift style invokes a "grid" method for balancing, as described above. It changes the positions of cutting
planes between processors in an iterative fashion, seeking to reduce the imbalance factor, similar to how the
fix balance shift command operates.
The dimstr argument is a string of characters, each of which must be an "x" or "y" or "z". Eacn character can
appear zero or one time, since there is no advantage to balancing on a dimension more than once. You should
normally only list dimensions where you expect there to be a density variation in the particles.
Balancing proceeds by adjusting the cutting planes in each of the dimensions listed in dimstr, one dimension
at a time. For a single dimension, the balancing operation (described below) is iterated on up to Niter times.
After each dimension finishes, the imbalance factor is re-computed, and the balancing operation halts if the
stopthresh criterion is met.
A rebalance operation in a single dimension is performed using a recursive multisectioning algorithm, where
the position of each cutting plane (line in 2d) in the dimension is adjusted independently. This is similar to a
recursive bisectioning for a single value, except that the bounds used for each bisectioning take advantage of
information from neighboring cuts if possible. At each iteration, the count of particles on either side of each
plane is tallied. If the counts do not match the target value for the plane, the position of the cut is adjusted to
be halfway between a low and high bound. The low and high bounds are adjusted on each iteration, using new
count information, so that they become closer together over time. Thus as the recursion progresses, the count
of particles on either side of the plane gets closer to the target value.
Once the rebalancing is complete and final processor sub-domains assigned, particles are migrated to their
new owning processor, and the balance procedure ends.
NOTE: At each rebalance operation, the bisectioning for each cutting plane (line in 2d) typically starts with
low and high bounds separated by the extent of a processor's sub-domain in one dimension. The size of this
LAMMPS Users Manual
363

bracketing region shrinks by 1/2 every iteration. Thus if Niter is specified as 10, the cutting plane will
typically be positioned to 1 part in 1000 accuracy (relative to the perfect target position). For Niter = 20, it
will be accurate to 1 part in a million. Thus there is no need ot set Niter to a large value. LAMMPS will check
if the threshold accuracy is reached (in a dimension) is less iterations than Niter and exit early. However, Niter
should also not be set too small, since it will take roughly the same number of iterations to converge even if
the cutting plane is initially close to the target value.
The rcb style invokes a "tiled" method for balancing, as described above. It performs a recursive coordinate
bisectioning (RCB) of the simulation domain. The basic idea is as follows.
The simulation domain is cut into 2 boxes by an axis-aligned cut in one of the dimensions, leaving one new
sub-box on either side of the cut. Which dimension is chosen for the cut depends on the particle (weight)
distribution within the parent box. Normally the longest dimension of the box is cut, but if all (or most) of the
particles are at one end of the box, a cut may be performed in another dimension to induce sub-boxes that are
more cube-ish (3d) or square-ish (2d) in shape.
After the cut is made, all the processors are also partitioned into 2 groups, half assigned to the box on the
lower side of the cut, and half to the box on the upper side. (If the processor count is odd, one side gets an
extra processor.) The cut is positioned so that the number of (weighted) particles in the lower box is exactly
the number that the processors assigned to that box should own for load balance to be perfect. This also makes
load balance for the upper box perfect. The positioning of the cut is done iteratively, by a bisectioning method
(median search). Note that counting particles on either side of the cut requires communication between all
processors at each iteration.
That is the procedure for the first cut. Subsequent cuts are made recursively, in exactly the same manner. The
subset of processors assigned to each box make a new cut in one dimension of that box, splitting the box, the
subset of processors, and the particles in the box in two. The recursion continues until every processor is
assigned a sub-box of the entire simulation domain, and owns the (weighted) particles in that sub-box.
This sub-section describes how to perform weighted load balancing using the weight keyword.
By default, all particles have a weight of 1.0, which means each particle is assumed to require the same
amount of computation during a timestep. There are, however, scenarios where this is not a good assumption.
Measuring the computational cost for each particle accurately would be impractical and slow down the
computation. Instead the weight keyword implements several ways to influence the per-particle weights
empirically by properties readily available or using the user's knowledge of the system. Note that the absolute
value of the weights are not important; only their relative ratios affect which particle is assigned to which
processor. A particle with a weight of 2.5 is assumed to require 5x more computational than a particle with a
weight of 0.5. For all the options below the weight assigned to a particle must be a positive value; an error
will be be generated if a weight is <= 0.0.
Below is a list of possible weight options with a short description of their usage and some example scenarios
where they might be applicable. It is possible to apply multiple weight flags and the weightings they induce
will be combined through multiplication. Most of the time, however, it is sufficient to use just one method.
The group weight style assigns weight factors to specified groups of particles. The group style keyword is
followed by the number of groups, then pairs of group IDs and the corresponding weight factor. If a particle
belongs to none of the specified groups, its weight is not changed. If it belongs to multiple groups, its weight
is the product of the weight factors.
LAMMPS Users Manual
364
This weight style is useful in combination with pair style hybrid, e.g. when combining a more costly
manybody potential with a fast pair-wise potential. It is also useful when using run_style respa where some
portions of the system have many bonded interactions and others none. It assumes that the computational cost
for each group remains constant over time. This is a purely empirical weighting, so a series test runs to tune
the assigned weight factors for optimal performance is recommended.
The neigh weight style assigns the same weight to each particle owned by a processor based on the total count
of neighbors in the neighbor list owned by that processor. The motivation is that more neighbors means a
higher computational cost. The style does not use neighbors per atom to assign a unique weight to each atom,
because that value can vary depending on how the neighbor list is built.
The factor setting is applied as an overall scale factor to the neigh weights which allows adjustment of their
impact on the balancing operation. The specified factor value must be positive. A value > 1.0 will increase the
weights so that the ratio of max weight to min weight increases by factor. A value < 1.0 will decrease the
weights so that the ratio of max weight to min weight decreases by factor. In both cases the intermediate
weight values increase/decrease proportionally as well. A value = 1.0 has no effect on the neigh weights. As a
rule of thumb, we have found a factor of about 0.8 often results in the best performance, since the number of
neighbors is likely to overestimate the ideal weight.
This weight style is useful for systems where there are different cutoffs used for different pairs of interactions,
or the density fluctuates, or a large number of particles are in the vicinity of a wall, or a combination of these
effects. If a simulation uses multiple neighbor lists, this weight style will use the first suitable neighbor list it
finds. It will not request or compute a new list. A warning will be issued if there is no suitable neighbor list
available or if it is not current, e.g. if the balance command is used before a run or minimize command is
used, in which case the neighbor list may not yet have been built. In this case no weights are computed.
Inserting a run 0 post no command before issuing the balance command, may be a workaround for this case,
as it will induce the neighbor list to be built.
The time weight style uses timer data to estimate weights. It assigns the same weight to each particle owned
by a processor based on the total computational time spent by that processor. See details below on what time
window is used. It uses the same timing information as is used for the MPI task timing breakdown, namely,
for sections Pair, Bond, Kspace, and Neigh. The time spent in those portions of the timestep are measured for
each MPI rank, summed, then divided by the number of particles owned by that processor. I.e. the weight is
an effective CPU time/particle averaged over the particles on that processor.
The factor setting is applied as an overall scale factor to the time weights which allows adjustment of their
impact on the balancing operation. The specified factor value must be positive. A value > 1.0 will increase the
weights so that the ratio of max weight to min weight increases by factor. A value < 1.0 will decrease the
weights so that the ratio of max weight to min weight decreases by factor. In both cases the intermediate
weight values increase/decrease proportionally as well. A value = 1.0 has no effect on the time weights. As a
rule of thumb, effective values to use are typically between 0.5 and 1.2. Note that the timer quantities
mentioned above can be affected by communication which occurs in the middle of the operations, e.g. pair
styles with intermediate exchange of data witin the force computation, and likewise for KSpace solves.
When using the time weight style with the balance command, the timing data is taken from the preceding run
command, i.e. the timings are for the entire previous run. For the fix balance command the timing data is for
only the timesteps since the last balancing operation was performed. If timing information for the required
sections is not available, e.g. at the beginning of a run, or when the timer command is set to either loop or off,
a warning is issued. In this case no weights are computed.
LAMMPS Users Manual
365

NOTE: The time weight style is the most generic option, and should be tried first, unless the group style is
easily applicable. However, since the computed cost function is averaged over all particles on a processor, the
weights may not be highly accurate. This style can also be effective as a secondary weight in combination
with either group or neigh to offset some of inaccuracies in either of those heuristics.
The var weight style assigns per-particle weights by evaluating an atom-style variable specified by name. This
is provided as a more flexible alternative to the group weight style, allowing definition of a more complex
heuristics based on information (global and per atom) available inside of LAMMPS. For example, atom-style
variables can reference the position of a particle, its velocity, the volume of its Voronoi cell, etc.
The store weight style does not compute a weight factor. Instead it stores the current accumulated weights in a
custom per-atom property specified by name. This must be a property defined as d_name via the fix
property/atom command. Note that these custom per-atom properties can be output in a dump file, so this is a
way to examine, debug, or visualize the per-particle weights computed during the load-balancing operation.
The out keyword writes a text file to the specified filename with the results of the balancing operation. The
file contains the bounds of the sub-domain for each processor after the balancing operation completes. The
format of the file is compatible with the Pizza.py mdump tool which has support for manipulating and
visualizing mesh files. An example is shown here for a balancing by 4 processors for a 2d problem:
ITEM: TIMESTEP
0
ITEM: NUMBER OF NODES
16
ITEM: BOX BOUNDS
0 10
0 10
0 10
ITEM: NODES
1 1 0 0 0
2 1 5 0 0
3 1 5 5 0
4 1 0 5 0
5 1 5 0 0
6 1 10 0 0
7 1 10 5 0
8 1 5 5 0
9 1 0 5 0
10 1 5 5 0
11 1 5 10 0
12 1 10 5 0
13 1 5 5 0
14 1 10 5 0
15 1 10 10 0
16 1 5 10 0
ITEM: TIMESTEP
0
ITEM: NUMBER OF SQUARES
4
ITEM: SQUARES
1 1 1 2 3 4
2 1 5 6 7 8
3 1 9 10 11 12
4 1 13 14 15 16
LAMMPS Users Manual
366

The coordinates of all the vertices are listed in the NODES section, 5 per processor. Note that the 4
sub-domains share vertices, so there will be duplicate nodes in the list.
The "SQUARES" section lists the node IDs of the 4 vertices in a rectangle for each processor (1 to 4).
For a 3d problem, the syntax is similar with 8 vertices listed for each processor, instead of 4, and
"SQUARES" replaced by "CUBES".
Restrictions:
For 2d simulations, the z style cannot be used. Nor can a "z" appear in dimstr for the shift style.
Related commands:
group, processors, fix balance
Default: none
LAMMPS Users Manual
367

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_coeff command
Syntax:
bond_coeff N args
N = bond type (see asterisk form below)• args = coefficients for one or more bond types•
Examples:
bond_coeff 5 80.0 1.2
bond_coeff * 30.0 1.5 1.0 1.0
bond_coeff 1*4 30.0 1.5 1.0 1.0
bond_coeff 1 harmonic 200.0 1.0
Description:
Specify the bond force field coefficients for one or more bond types. The number and meaning of the
coefficients depends on the bond style. Bond coefficients can also be set in the data file read by the read_data
command or in a restart file.
N can be specified in one of two ways. An explicit numeric value can be used, as in the 1st example above. Or
a wild-card asterisk can be used to set the coefficients for multiple bond types. This takes the form "*" or "*n"
or "n*" or "m*n". If N = the number of bond types, then an asterisk with no numeric values means all types
from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A trailing asterisk means all types
from n to N (inclusive). A middle asterisk means all types from m to n (inclusive).
Note that using a bond_coeff command can override a previous setting for the same bond type. For example,
these commands set the coeffs for all bond types, then overwrite the coeffs for just bond type 2:
bond_coeff * 100.0 1.2
bond_coeff 2 200.0 1.2
A line in a data file that specifies bond coefficients uses the exact same format as the arguments of the
bond_coeff command in an input script, except that wild-card asterisks should not be used since coefficients
for all N types must be listed in the file. For example, under the "Bond Coeffs" section of a data file, the line
that corresponds to the 1st example above would be listed as
5 80.0 1.2
Here is an alphabetic list of bond styles defined in LAMMPS. Click on the style to display the formula it
computes and coefficients specified by the associated bond_coeff command.
Note that here are also additional bond styles submitted by users which are included in the LAMMPS
distribution. The list of these with links to the individual styles are given in the bond section of this page.
bond_style none - turn off bonded interactions• bond_style hybrid - define multiple styles of bond interactions•
LAMMPS Users Manual
368

bond_style class2 - COMPASS (class 2) bond• bond_style fene - FENE (finite-extensible non-linear elastic) bond• bond_style fene/expand - FENE bonds with variable size particles• bond_style harmonic - harmonic bond• bond_style morse - Morse bond• bond_style nonlinear - nonlinear bond• bond_style quartic - breakable quartic bond• bond_style table - tabulated by bond length•
Restrictions:
This command must come after the simulation box is defined by a read_data, read_restart, or create_box
command.
A bond style must be defined before any bond coefficients are set, either in the input script or in a data file.
Related commands:
bond_style
Default: none
LAMMPS Users Manual
369

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style command
Syntax:
bond_style style args
style = none or hybrid or class2 or fene or fene/expand or harmonic or morse or nonlinear or quartic•
args = none for any style except hybrid
hybrid args = list of one or more styles
Examples:
bond_style harmonic
bond_style fene
bond_style hybrid harmonic fene
Description:
Set the formula(s) LAMMPS uses to compute bond interactions between pairs of atoms. In LAMMPS, a bond
differs from a pairwise interaction, which are set via the pair_style command. Bonds are defined between
specified pairs of atoms and remain in force for the duration of the simulation (unless the bond breaks which
is possible in some bond potentials). The list of bonded atoms is read in by a read_data or read_restart
command from a data or restart file. By contrast, pair potentials are typically defined between all pairs of
atoms within a cutoff distance and the set of active interactions changes over time.
Hybrid models where bonds are computed using different bond potentials can be setup using the hybrid bond
style.
The coefficients associated with a bond style can be specified in a data or restart file or via the bond_coeff
command.
All bond potentials store their coefficient data in binary restart files which means bond_style and bond_coeff
commands do not need to be re-specified in an input script that restarts a simulation. See the read_restart
command for details on how to do this. The one exception is that bond_style hybrid only stores the list of
sub-styles in the restart file; bond coefficients need to be re-specified.
NOTE: When both a bond and pair style is defined, the special_bonds command often needs to be used to turn
off (or weight) the pairwise interaction that would otherwise exist between 2 bonded atoms.
In the formulas listed for each bond style, r is the distance between the 2 atoms in the bond.
Here is an alphabetic list of bond styles defined in LAMMPS. Click on the style to display the formula it
computes and coefficients specified by the associated bond_coeff command.
Note that there are also additional bond styles submitted by users which are included in the LAMMPS
distribution. The list of these with links to the individual styles are given in the bond section of this page.
bond_style none - turn off bonded interactions•
LAMMPS Users Manual
370

bond_style zero - topology but no interactions• bond_style hybrid - define multiple styles of bond interactions•
bond_style class2 - COMPASS (class 2) bond• bond_style fene - FENE (finite-extensible non-linear elastic) bond• bond_style fene/expand - FENE bonds with variable size particles• bond_style harmonic - harmonic bond• bond_style morse - Morse bond• bond_style nonlinear - nonlinear bond• bond_style quartic - breakable quartic bond• bond_style table - tabulated by bond length•
Restrictions:
Bond styles can only be set for atom styles that allow bonds to be defined.
Most bond styles are part of the MOLECULE package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info on packages. The doc pages for individual
bond potentials tell if it is part of a package.
Related commands:
bond_coeff, delete_bonds
Default:
bond_style none
LAMMPS Users Manual
371

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_write command
Syntax:
bond_write btype N inner outer file keyword itype jtype
btype = bond types• N = # of values• inner,outer = inner and outer bond length (distance units)• file = name of file to write values to• keyword = section name in file for this set of tabulated values• itype,jtype = 2 atom types (optional)•
•
Examples:
bond_write 1 500 0.5 3.5 table.txt Harmonic_1
bond_write 3 1000 0.1 6.0 table.txt Morse
Description:
Write energy and force values to a file as a function of distance for the currently defined bond potential. This
is useful for plotting the potential function or otherwise debugging its values. If the file already exists, the
table of values is appended to the end of the file to allow multiple tables of energy and force to be included in
one file.
The energy and force values are computed at distances from inner to outer for 2 interacting atoms forming a
bond of type btype, using the appropriate bond_coeff coefficients. N evenly spaced distances are used.
For example, for N = 7, inner = 1.0, and outer = 4.0, values are computed at r = 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0.
The file is written in the format used as input for the bond_style table option with keyword as the section
name. Each line written to the file lists an index number (1-N), a distance (in distance units), an energy (in
energy units), and a force (in force units).
Restrictions:
All force field coefficients for bond and other kinds of interactions must be set before this command can be
invoked.
Due to how the bond force is computed, an inner value > 0.0 must be specified even if the potential has a
finite value at r = 0.0.
Related commands:
bond_style table, bond_style, bond_coeff
Default: none
LAMMPS Users Manual
372

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
boundary command
Syntax:
boundary x y z
x,y,z = p or s or f or m, one or two letters
p is periodic
f is non-periodic and fixed
s is non-periodic and shrink-wrapped
m is non-periodic and shrink-wrapped with a minimum value
•
Examples:
boundary p p f
boundary p fs p
boundary s f fm
Description:
Set the style of boundaries for the global simulation box in each dimension. A single letter assigns the same
style to both the lower and upper face of the box. Two letters assigns the first style to the lower face and the
second style to the upper face. The initial size of the simulation box is set by the read_data, read_restart, or
create_box commands.
The style p means the box is periodic, so that particles interact across the boundary, and they can exit one end
of the box and re-enter the other end. A periodic dimension can change in size due to constant pressure
boundary conditions or box deformation (see the fix npt and fix deform commands). The p style must be
applied to both faces of a dimension.
The styles f, s, and m mean the box is non-periodic, so that particles do not interact across the boundary and
do not move from one side of the box to the other.
For style f, the position of the face is fixed. If an atom moves outside the face it will be deleted on the next
timestep that reneighboring occurs. This will typically generate an error unless you have set the
thermo_modify lost option to allow for lost atoms.
For style s, the position of the face is set so as to encompass the atoms in that dimension (shrink-wrapping),
no matter how far they move. Note that when the difference between the current box dimensions and the
shrink-wrap box dimensions is large, this can lead to lost atoms at the beginning of a run when running in
parallel. This is due to the large change in the (global) box dimensions also causing significant changes in the
individual sub-domain sizes. If these changes are farther than the communication cutoff, atoms will be lost.
This is best addressed by setting initial box dimensions to match the shrink-wrapped dimensions more closely,
by using m style boundaries (see below).
For style m, shrink-wrapping occurs, but is bounded by the value specified in the data or restart file or set by
the create_box command. For example, if the upper z face has a value of 50.0 in the data file, the face will
always be positioned at 50.0 or above, even if the maximum z-extent of all the atoms becomes less than 50.0.
This can be useful if you start a simulation with an empty box or if you wish to leave room on one side of the
LAMMPS Users Manual
373
box, e.g. for atoms to evaporate from a surface.
For triclinic (non-orthogonal) simulation boxes, if the 2nd dimension of a tilt factor (e.g. y for xy) is periodic,
then the periodicity is enforced with the tilt factor offset. If the 1st dimension is shrink-wrapped, then the
shrink wrapping is applied to the tilted box face, to encompass the atoms. E.g. for a positive xy tilt, the xlo
and xhi faces of the box are planes tilting in the +y direction as y increases. These tilted planes are
shrink-wrapped around the atoms to determine the x extent of the box.
See Section 6.12 of the doc pages for a geometric description of triclinic boxes, as defined by LAMMPS, and
how to transform these parameters to and from other commonly used triclinic representations.
Restrictions:
This command cannot be used after the simulation box is defined by a read_data or create_box command or
read_restart command. See the change_box command for how to change the simulation box boundaries after
it has been defined.
For 2d simulations, the z dimension must be periodic.
Related commands:
See the thermo_modify command for a discussion of lost atoms.
Default:
boundary p p p
LAMMPS Users Manual
374

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
box command
Syntax:
box keyword value ...
one or more keyword/value pairs may be appended• keyword = tilt
tilt value = small or large
•
Examples:
box tilt large
box tilt small
Description:
Set attributes of the simulation box.
For triclinic (non-orthogonal) simulation boxes, the tilt keyword allows simulation domains to be created with
arbitrary tilt factors, e.g. via the create_box or read_data commands. Tilt factors determine how skewed the
triclinic box is; see this section of the manual for a discussion of triclinic boxes in LAMMPS.
LAMMPS normally requires that no tilt factor can skew the box more than half the distance of the parallel
box length, which is the 1st dimension in the tilt factor (x for xz). If tilt is set to small, which is the default,
then an error will be generated if a box is created which exceeds this limit. If tilt is set to large, then no limit
is enforced. You can create a box with any tilt factors you wish.
Note that if a simulation box has a large tilt factor, LAMMPS will run less efficiently, due to the large volume
of communication needed to acquire ghost atoms around a processor's irregular-shaped sub-domain. For
extreme values of tilt, LAMMPS may also lose atoms and generate an error.
Restrictions:
This command cannot be used after the simulation box is defined by a read_data or create_box command or
read_restart command.
Related commands: none
Default:
The default value is tilt = small.
LAMMPS Users Manual
375

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
change_box command
Syntax:
change_box group-ID parameter args ... keyword args ...
group-ID = ID of group of atoms to (optionally) displace• one or more parameter/arg pairs may be appended
parameter = x or y or z or xy or xz or yz or boundary or ortho or triclinic or set or remap
x, y, z args = style value(s)
style = final or delta or scale or volume
final values = lo hi
lo hi = box boundaries after displacement (distance units)
delta values = dlo dhi
dlo dhi = change in box boundaries after displacement (distance units)
scale values = factor
factor = multiplicative factor for change in box length after displacement
volume value = none = adjust this dim to preserve volume of system
xy, xz, yz args = style value
style = final or delta
final value = tilt
tilt = tilt factor after displacement (distance units)
delta value = dtilt
dtilt = change in tilt factor after displacement (distance units)
boundary args = x y z
x,y,z = p or s or f or m, one or two letters
p is periodic
f is non-periodic and fixed
s is non-periodic and shrink-wrapped
m is non-periodic and shrink-wrapped with a minimum value
ortho args = none = change box to orthogonal
triclinic args = none = change box to triclinic
set args = none = store state of current box
remap args = none = remap atom coords from last saved state to current box
•
zero or more keyword/value pairs may be appended• keyword = units
units value = lattice or box
lattice = distances are defined in lattice units
box = distances are defined in simulation box units
•
Examples:
change_box all xy final -2.0 z final 0.0 5.0 boundary p p f remap units box
change_box all x scale 1.1 y volume z volume remap
Description:
Change the volume and/or shape and/or boundary conditions for the simulation box. Orthogonal simulation
boxes have 3 adjustable size parameters (x,y,z). Triclinic (non-orthogonal) simulation boxes have 6 adjustable
size/shape parameters (x,y,z,xy,xz,yz). Any or all of them can be adjusted independently by this command.
Thus it can be used to expand or contract a box, or to apply a shear strain to a non-orthogonal box. It can also
be used to change the boundary conditions for the simulation box, similar to the boundary command.
LAMMPS Users Manual
376
The size and shape of the initial simulation box are specified by the create_box or read_data or read_restart
command used to setup the simulation. The size and shape may be altered by subsequent runs, e.g. by use of
the fix npt or fix deform commands. The create_box, read data, and read_restart commands also determine
whether the simulation box is orthogonal or triclinic and their doc pages explain the meaning of the xy,xz,yz
tilt factors.
See Section 6.12 of the doc pages for a geometric description of triclinic boxes, as defined by LAMMPS, and
how to transform these parameters to and from other commonly used triclinic representations.
The keywords used in this command are applied sequentially to the simulation box and the atoms in it, in the
order specified.
Before the sequence of keywords are invoked, the current box size/shape is stored, in case a remap keyword is
used to map the atom coordinates from a previously stored box size/shape to the current one.
After all the keywords have been processed, any shrink-wrap boundary conditions are invoked (see the
boundary command) which may change simulation box boundaries, and atoms are migrated to new owning
processors.
NOTE: This means that you cannot use the change_box command to enlarge a shrink-wrapped box, e.g. to
make room to insert more atoms via the create_atoms command, because the simulation box will be
re-shrink-wrapped before the change_box command completes. Instead you could do something like this,
assuming the simulation box is non-periodic and atoms extend from 0 to 20 in all dimensions:
change_box all x final -10 20
create_atoms 1 single -5 5 5 # this will fail to insert an atom
change_box all x final -10 20 boundary f s s
create_atoms 1 single -5 5 5
change_box all boundary s s s # this will work
NOTE: Unlike the earlier "displace_box" version of this command, atom remapping is NOT performed by
default. This command allows remapping to be done in a more general way, exactly when you specify it (zero
or more times) in the sequence of transformations. Thus if you do not use the remap keyword, atom
coordinates will not be changed even if the box size/shape changes. If a uniformly strained state is desired, the
remap keyword should be specified.
NOTE: It is possible to lose atoms with this command. E.g. by changing the box without remapping the
atoms, and having atoms end up outside of non-periodic boundaries. It is also possible to alter bonds between
atoms straddling a boundary in bad ways. E.g. by converting a boundary from periodic to non-periodic. It is
also possible when remapping atoms to put them (nearly) on top of each other. E.g. by converting a boundary
from non-periodic to periodic. All of these will typically lead to bad dynamics and/or generate error messages.
NOTE: The simulation box size/shape can be changed by arbitrarily large amounts by this command. This is
not a problem, except that the mapping of processors to the simulation box is not changed from its initial 3d
configuration; see the processors command. Thus, if the box size/shape changes dramatically, the mapping of
processors to the simulation box may not end up as optimal as the initial mapping attempted to be.
NOTE: Because the keywords used in this command are applied one at a time to the simulation box and the
atoms in it, care must be taken with triclinic cells to avoid exceeding the limits on skew after each
transformation in the sequence. If skew is exceeded before the final transformation this can be avoided by
changing the order of the sequence, or breaking the transformation into two or more smaller transformations.
LAMMPS Users Manual
377

For more information on the allowed limits for box skew see the discussion on triclinic boxes on this page.
For the x, y, and z parameters, this is the meaning of their styles and values.
For style final, the final lo and hi box boundaries of a dimension are specified. The values can be in lattice or
box distance units. See the discussion of the units keyword below.
For style delta, plus or minus changes in the lo/hi box boundaries of a dimension are specified. The values can
be in lattice or box distance units. See the discussion of the units keyword below.
For style scale, a multiplicative factor to apply to the box length of a dimension is specified. For example, if
the initial box length is 10, and the factor is 1.1, then the final box length will be 11. A factor less than 1.0
means compression.
The volume style changes the specified dimension in such a way that the overall box volume remains constant
with respect to the operation performed by the preceding keyword. The volume style can only be used
following a keyword that changed the volume, which is any of the x, y, z keywords. If the preceding keyword
"key" had a volume style, then both it and the current keyword apply to the keyword preceding "key". I.e. this
sequence of keywords is allowed:
change_box all x scale 1.1 y volume z volume
The volume style changes the associated dimension so that the overall box volume is unchanged relative to its
value before the preceding keyword was invoked.
If the following command is used, then the z box length will shrink by the same 1.1 factor the x box length
was increased by:
change_box all x scale 1.1 z volume
If the following command is used, then the y,z box lengths will each shrink by sqrt(1.1) to keep the volume
constant. In this case, the y,z box lengths shrink so as to keep their relative aspect ratio constant:
change_box all"x scale 1.1 y volume z volume
If the following command is used, then the final box will be a factor of 10% larger in x and y, and a factor of
21% smaller in z, so as to keep the volume constant:
change_box all x scale 1.1 z volume y scale 1.1 z volume
NOTE: For solids or liquids, when one dimension of the box is expanded, it may be physically undesirable to
hold the other 2 box lengths constant since that implies a density change. For solids, adjusting the other
dimensions via the volume style may make physical sense (just as for a liquid), but may not be correct for
materials and potentials whose Poisson ratio is not 0.5.
For the scale and volume styles, the box length is expanded or compressed around its mid point.
For the xy, xz, and yz parameters, this is the meaning of their styles and values. Note that changing the tilt
factors of a triclinic box does not change its volume.
LAMMPS Users Manual
378

For style final, the final tilt factor is specified. The value can be in lattice or box distance units. See the
discussion of the units keyword below.
For style delta, a plus or minus change in the tilt factor is specified. The value can be in lattice or box distance
units. See the discussion of the units keyword below.
All of these styles change the xy, xz, yz tilt factors. In LAMMPS, tilt factors (xy,xz,yz) for triclinic boxes are
required to be no more than half the distance of the parallel box length. For example, if xlo = 2 and xhi = 12,
then the x box length is 10 and the xy tilt factor must be between -5 and 5. Similarly, both xz and yz must be
between -(xhi-xlo)/2 and +(yhi-ylo)/2. Note that this is not a limitation, since if the maximum tilt factor is 5
(as in this example), then configurations with tilt = ..., -15, -5, 5, 15, 25, ... are all equivalent. Any tilt factor
specified by this command must be within these limits.
The boundary keyword takes arguments that have exactly the same meaning as they do for the boundary
command. In each dimension, a single letter assigns the same style to both the lower and upper face of the
box. Two letters assigns the first style to the lower face and the second style to the upper face.
The style p means the box is periodic; the other styles mean non-periodic. For style f, the position of the face
is fixed. For style s, the position of the face is set so as to encompass the atoms in that dimension
(shrink-wrapping), no matter how far they move. For style m, shrink-wrapping occurs, but is bounded by the
current box edge in that dimension, so that the box will become no smaller. See the boundary command for
more explanation of these style options.
Note that the "boundary" command itself can only be used before the simulation box is defined via a
read_data or create_box or read_restart command. This command allows the boundary conditions to be
changed later in your input script. Also note that the read_restart will change boundary conditions to match
what is stored in the restart file. So if you wish to change them, you should use the change_box command
after the read_restart command.
The ortho and triclinic keywords convert the simulation box to be orthogonal or triclinic (non-orthogonal).
See this section for a discussion of how non-orthogonal boxes are represented in LAMMPS.
The simulation box is defined as either orthogonal or triclinic when it is created via the create_box, read_data,
or read_restart commands.
These keywords allow you to toggle the existing simulation box from orthogonal to triclinic and vice versa.
For example, an initial equilibration simulation can be run in an orthogonal box, the box can be toggled to
triclinic, and then a non-equilibrium MD (NEMD) simulation can be run with deformation via the fix deform
command.
If the simulation box is currently triclinic and has non-zero tilt in xy, yz, or xz, then it cannot be converted to
an orthogonal box.
The set keyword saves the current box size/shape. This can be useful if you wish to use the remap keyword
more than once or if you wish it to be applied to an intermediate box size/shape in a sequence of keyword
operations. Note that the box size/shape is saved before any of the keywords are processed, i.e. the box
size/shape at the time the create_box command is encountered in the input script.
The remap keyword remaps atom coordinates from the last saved box size/shape to the current box state. For
example, if you stretch the box in the x dimension or tilt it in the xy plane via the x and xy keywords, then the
LAMMPS Users Manual
379

remap command will dilate or tilt the atoms to conform to the new box size/shape, as if the atoms moved with
the box as it deformed.
Note that this operation is performed without regard to periodic boundaries. Also, any shrink-wrapping of
non-periodic boundaries (see the boundary command) occurs after all keywords, including this one, have been
processed.
Only atoms in the specified group are remapped.
The units keyword determines the meaning of the distance units used to define various arguments. A box
value selects standard distance units as defined by the units command, e.g. Angstroms for units = real or
metal. A lattice value means the distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacing.
Restrictions:
If you use the ortho or triclinic keywords, then at the point in the input script when this command is issued, no
dumps can be active, nor can a fix deform be active. This is because these commands test whether the
simulation box is orthogonal when they are first issued. Note that these commands can be used in your script
before a change_box command is issued, so long as an undump or unfix command is also used to turn them
off.
Related commands:
fix deform, boundary
Default:
The option default is units = lattice.
LAMMPS Users Manual
380

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
clear command
Syntax:
clear
Examples:
(commands for 1st simulation)
clear
(commands for 2nd simulation)
Description:
This command deletes all atoms, restores all settings to their default values, and frees all memory allocated by
LAMMPS. Once a clear command has been executed, it is almost as if LAMMPS were starting over, with
only the exceptions noted below. This command enables multiple jobs to be run sequentially from one input
script.
These settings are not affected by a clear command: the working directory (shell command), log file status
(log command), echo status (echo command), and input script variables (variable command).
Restrictions: none
Related commands: none
Default: none
LAMMPS Users Manual
381

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
comm_modify command
Syntax:
comm_modify keyword value ...
zero or more keyword/value pairs may be appended• keyword = mode or cutoff or cutoff/multi or group or vel
mode value = single or multi = communicate atoms within a single or multiple distances
cutoff value = Rcut (distance units) = communicate atoms from this far away
cutoff/multi type value
type = atom type or type range (supports asterisk notation)
value = Rcut (distance units) = communicate atoms for selected types from this far away
group value = group-ID = only communicate atoms in the group
vel value = yes or no = do or do not communicate velocity info with ghost atoms
•
Examples:
comm_modify mode multi
comm_modify mode multi group solvent
comm_modift mode multi cutoff/multi 1 10.0 cutoff/multi 2*4 15.0
comm_modify vel yes
comm_modify mode single cutoff 5.0 vel yes
comm_modify cutoff/multi * 0.0
Description:
This command sets parameters that affect the inter-processor communication of atom information that occurs
each timestep as coordinates and other properties are exchanged between neighboring processors and stored as
properties of ghost atoms.
NOTE: These options apply to the currently defined comm style. When you specify a comm_style command,
all communication settings are restored to their default values, including those previously reset by a
comm_modify command. Thus if your input script specifies a comm_style command, you should use the
comm_modify command after it.
The mode keyword determines whether a single or multiple cutoff distances are used to determine which
atoms to communicate.
The default mode is single which means each processor acquires information for ghost atoms that are within a
single distance from its sub-domain. The distance is by default the maximum of the neighbor cutoff across all
atom type pairs.
For many systems this is an efficient algorithm, but for systems with widely varying cutoffs for different type
pairs, the multi mode can be faster. In this case, each atom type is assigned its own distance cutoff for
communication purposes, and fewer atoms will be communicated. See the neighbor multi command for a
neighbor list construction option that may also be beneficial for simulations of this kind.
The cutoff keyword allows you to extend the ghost cutoff distance for communication mode single, which is
the distance from the borders of a processor's sub-domain at which ghost atoms are acquired from other
LAMMPS Users Manual
382
processors. By default the ghost cutoff = neighbor cutoff = pairwise force cutoff + neighbor skin. See the
neighbor command for more information about the skin distance. If the specified Rcut is greater than the
neighbor cutoff, then extra ghost atoms will be acquired. If the provided cutoff is smaller, the provided value
will be ignored and the ghost cutoff is set to the neighbor cutoff. Specifying a cutoff value of 0.0 will reset
any previous value to the default.
The cutoff/multi option is equivalent to cutoff, but applies to communication mode multi instead. Since in this
case the communication cutoffs are determined per atom type, a type specifier is needed and cutoff for one or
multiple types can be extended. Also ranges of types using the usual asterisk notation can be given.
These are simulation scenarios in which it may be useful or even necessary to set a ghost cutoff > neighbor
cutoff:
a single polymer chain with bond interactions, but no pairwise interactions• bonded interactions (e.g. dihedrals) extend further than the pairwise cutoff• ghost atoms beyond the pairwise cutoff are needed for some computation•
In the first scenario, a pairwise potential is not defined. Thus the pairwise neighbor cutoff will be 0.0. But
ghost atoms are still needed for computing bond, angle, etc interactions between atoms on different
processors, or when the interaction straddles a periodic boundary.
The appropriate ghost cutoff depends on the newton bond setting. For newton bond off, the distance needs to
be the furthest distance between any two atoms in the bond, angle, etc. E.g. the distance between 1-4 atoms in
a dihedral. For newton bond on, the distance between the central atom in the bond, angle, etc and any other
atom is sufficient. E.g. the distance between 2-4 atoms in a dihedral.
In the second scenario, a pairwise potential is defined, but its neighbor cutoff is not sufficiently long enough
to enable bond, angle, etc terms to be computed. As in the previous scenario, an appropriate ghost cutoff
should be set.
In the last scenario, a fix or compute or pairwise potential needs to calculate with ghost atoms beyond the
normal pairwise cutoff for some computation it performs (e.g. locate neighbors of ghost atoms in a multibody
pair potential). Setting the ghost cutoff appropriately can insure it will find the needed atoms.
NOTE: In these scenarios, if you do not set the ghost cutoff long enough, and if there is only one processor in
a periodic dimension (e.g. you are running in serial), then LAMMPS may "find" the atom it is looking for
(e.g. the partner atom in a bond), that is on the far side of the simulation box, across a periodic boundary. This
will typically lead to bad dynamics (i.e. the bond length is now the simulation box length). To detect if this is
happening, see the neigh_modify cluster command.
The group keyword will limit communication to atoms in the specified group. This can be useful for models
where no ghost atoms are needed for some kinds of particles. All atoms (not just those in the specified group)
will still migrate to new processors as they move. The group specified with this option must also be specified
via the atom_modify first command.
The vel keyword enables velocity information to be communicated with ghost particles. Depending on the
atom_style, velocity info includes the translational velocity, angular velocity, and angular momentum of a
particle. If the vel option is set to yes, then ghost atoms store these quantities; if no then they do not. The yes
setting is needed by some pair styles which require the velocity state of both the I and J particles to compute a
pairwise I,J interaction, as well as by some compute and fix commands.
LAMMPS Users Manual
383
Note that if the fix deform command is being used with its "remap v" option enabled, then the velocities for
ghost atoms (in the fix deform group) mirrored across a periodic boundary will also include components due
to any velocity shift that occurs across that boundary (e.g. due to dilation or shear).
Restrictions:
Communication mode multi is currently only available for comm_style brick.
Related commands:
comm_style, neighbor
Default:
The option defauls are mode = single, group = all, cutoff = 0.0, vel = no. The cutoff default of 0.0 means that
ghost cutoff = neighbor cutoff = pairwise force cutoff + neighbor skin.
LAMMPS Users Manual
384

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
comm_style command
Syntax:
comm_style style
style = brick or tiled•
Examples:
comm_style brick
comm_style tiled
Description:
This command sets the style of inter-processor communication of atom information that occurs each timestep
as coordinates and other properties are exchanged between neighboring processors and stored as properties of
ghost atoms.
For the default brick style, the domain decomposition used by LAMMPS to partition the simulation box must
be a regular 3d grid of bricks, one per processor. Each processor communicates with its 6 Cartesian neighbors
in the grid to acquire information for nearby atoms.
For the tiled style, a more general domain decomposition can be used, as triggered by the balance or fix
balance commands. The simulation box can be partitioned into non-overlapping rectangular-shaped "tiles" or
varying sizes and shapes. Again there is one tile per processor. To acquire information for nearby atoms,
communication must now be done with a more complex pattern of neighboring processors.
Note that this command does not actually define a partitioning of the simulation box (a domain
decomposition), rather it determines what kinds of decompositions are allowed and the pattern of
communication used to enable the decomposition. A decomposition is created when the simulation box is first
created, via the create_box or read_data or read_restart commands. For both the brick and tiled styles, the
initial decomposition will be the same, as described by create_box and processors commands. The
decomposition can be changed via the balance or fix balance commands.
Restrictions: none
Related commands:
comm_modify, processors, balance, fix balance
Default:
The default style is brick.
LAMMPS Users Manual
385

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute command
Syntax:
compute ID group-ID style args
ID = user-assigned name for the computation• group-ID = ID of the group of atoms to perform the computation on• style = one of a list of possible style names (see below)• args = arguments used by a particular style•
Examples:
compute 1 all temp
compute newtemp flow temp/partial 1 1 0
compute 3 all ke/atom
Description:
Define a computation that will be performed on a group of atoms. Quantities calculated by a compute are
instantaneous values, meaning they are calculated from information about atoms on the current timestep or
iteration, though a compute may internally store some information about a previous state of the system.
Defining a compute does not perform a computation. Instead computes are invoked by other LAMMPS
commands as needed, e.g. to calculate a temperature needed for a thermostat fix or to generate
thermodynamic or dump file output. See this howto section for a summary of various LAMMPS output
options, many of which involve computes.
The ID of a compute can only contain alphanumeric characters and underscores.
Computes calculate one of three styles of quantities: global, per-atom, or local. A global quantity is one or
more system-wide values, e.g. the temperature of the system. A per-atom quantity is one or more values per
atom, e.g. the kinetic energy of each atom. Per-atom values are set to 0.0 for atoms not in the specified
compute group. Local quantities are calculated by each processor based on the atoms it owns, but there may
be zero or more per atom, e.g. a list of bond distances. Computes that produce per-atom quantities have the
word "atom" in their style, e.g. ke/atom. Computes that produce local quantities have the word "local" in their
style, e.g. bond/local. Styles with neither "atom" or "local" in their style produce global quantities.
Note that a single compute produces either global or per-atom or local quantities, but never more than one of
these (with only a few exceptions, as documented by individual compute commands).
Global, per-atom, and local quantities each come in three kinds: a single scalar value, a vector of values, or a
2d array of values. The doc page for each compute describes the style and kind of values it produces, e.g. a
per-atom vector. Some computes produce more than one kind of a single style, e.g. a global scalar and a
global vector.
When a compute quantity is accessed, as in many of the output commands discussed below, it can be
referenced via the following bracket notation, where ID is the ID of the compute:
c_ID entire scalar, vector, or array
LAMMPS Users Manual
386

c_ID[I] one element of vector, one column of array
c_ID[I][J] one element of array
In other words, using one bracket reduces the dimension of the quantity once (vector -> scalar, array ->
vector). Using two brackets reduces the dimension twice (array -> scalar). Thus a command that uses scalar
compute values as input can also process elements of a vector or array.
Note that commands and variables which use compute quantities typically do not allow for all kinds, e.g. a
command may require a vector of values, not a scalar. This means there is no ambiguity about referring to a
compute quantity as c_ID even if it produces, for example, both a scalar and vector. The doc pages for various
commands explain the details.
In LAMMPS, the values generated by a compute can be used in several ways:
The results of computes that calculate a global temperature or pressure can be used by fixes that do
thermostatting or barostatting or when atom velocities are created.
•
Global values can be output via the thermo_style custom or fix ave/time command. Or the values can
be referenced in a variable equal or variable atom command.
•
Per-atom values can be output via the dump custom command. Or they can be time-averaged via the
fix ave/atom command or reduced by the compute reduce command. Or the per-atom values can be
referenced in an atom-style variable.
•
Local values can be reduced by the compute reduce command, or histogrammed by the fix ave/histo
command, or output by the dump local command.
•
The results of computes that calculate global quantities can be either "intensive" or "extensive" values.
Intensive means the value is independent of the number of atoms in the simulation, e.g. temperature.
Extensive means the value scales with the number of atoms in the simulation, e.g. total rotational kinetic
energy. Thermodynamic output will normalize extensive values by the number of atoms in the system,
depending on the "thermo_modify norm" setting. It will not normalize intensive values. If a compute value is
accessed in another way, e.g. by a variable, you may want to know whether it is an intensive or extensive
value. See the doc page for individual computes for further info.
LAMMPS creates its own computes internally for thermodynamic output. Three computes are always created,
named "thermo_temp", "thermo_press", and "thermo_pe", as if these commands had been invoked in the input
script:
compute thermo_temp all temp
compute thermo_press all pressure thermo_temp
compute thermo_pe all pe
Additional computes for other quantities are created if the thermo style requires it. See the documentation for
the thermo_style command.
Fixes that calculate temperature or pressure, i.e. for thermostatting or barostatting, may also create computes.
These are discussed in the documentation for specific fix commands.
In all these cases, the default computes LAMMPS creates can be replaced by computes defined by the user in
the input script, as described by the thermo_modify and fix modify commands.
Properties of either a default or user-defined compute can be modified via the compute_modify command.
LAMMPS Users Manual
387

Computes can be deleted with the uncompute command.
Code for new computes can be added to LAMMPS (see this section of the manual) and the results of their
calculations accessed in the various ways described above.
Each compute style has its own doc page which describes its arguments and what it does. Here is an
alphabetic list of compute styles available in LAMMPS. They are also given in more compact form in the
Compute section of this page.
There are also additional compute styles (not listed here) submitted by users which are included in the
LAMMPS distribution. The list of these with links to the individual styles are given in the compute section of
this page.
angle/local - theta and energy of each angle• angmom/chunk - angular momentum for each chunk• body/local - attributes of body sub-particles• bond - values computed by a bond style• bond/local - distance and energy of each bond• centro/atom - centro-symmetry parameter for each atom• chunk/atom - assign chunk IDs to each atom• cluster/atom - cluster ID for each atom• cna/atom - common neighbor analysis (CNA) for each atom• com - center-of-mass of group of atoms• com/chunk - center-of-mass for each chunk• contact/atom - contact count for each spherical particle• coord/atom - coordination number for each atom• damage/atom - Peridynamic damage for each atom• dihedral/local - angle of each dihedral• dilatation/atom - Peridynamic dilatation for each atom• displace/atom - displacement of each atom• erotate/asphere - rotational energy of aspherical particles• erotate/rigid - rotational energy of rigid bodies• erotate/sphere - rotational energy of spherical particles• erotate/sphere/atom - rotational energy for each spherical particle• event/displace - detect event on atom displacement• group/group - energy/force between two groups of atoms• gyration - radius of gyration of group of atoms• gyration/chunk - radius of gyration for each chunk• heat/flux - heat flux through a group of atoms• hexorder/atom - bond orientational order parameter q6• improper/local - angle of each improper• inertia/chunk - inertia tensor for each chunk• ke - translational kinetic energy• ke/atom - kinetic energy for each atom• ke/rigid - translational kinetic energy of rigid bodies• msd - mean-squared displacement of group of atoms• msd/chunk - mean-squared displacement for each chunk• msd/nongauss - MSD and non-Gaussian parameter of group of atoms• omega/chunk - angular velocity for each chunk• orientorder/atom - Steinhardt bond orientational order parameters Ql• pair - values computed by a pair style•
LAMMPS Users Manual
388
pair/local - distance/energy/force of each pairwise interaction• pe - potential energy• pe/atom - potential energy for each atom• plasticity/atom - Peridynamic plasticity for each atom• pressure - total pressure and pressure tensor• property/atom - convert atom attributes to per-atom vectors/arrays• property/local - convert local attributes to localvectors/arrays• property/chunk - extract various per-chunk attributes• rdf - radial distribution function g(r) histogram of group of atoms• reduce - combine per-atom quantities into a single global value• reduce/region - same as compute reduce, within a region• rigid/local - extract rigid body attributes• slice - extract values from global vector or array• sna/atom - calculate bispectrum coefficients for each atom• snad/atom - derivative of bispectrum coefficients for each atom• snav/atom - virial contribution from bispectrum coefficients for each atom• stress/atom - stress tensor for each atom• temp - temperature of group of atoms• temp/asphere - temperature of aspherical particles• temp/body - temperature of body particles• temp/chunk - temperature of each chunk• temp/com - temperature after subtracting center-of-mass velocity• temp/deform - temperature excluding box deformation velocity• temp/partial - temperature excluding one or more dimensions of velocity• temp/profile - temperature excluding a binned velocity profile• temp/ramp - temperature excluding ramped velocity component• temp/region - temperature of a region of atoms• temp/sphere - temperature of spherical particles• ti - thermodynamic integration free energy values• torque/chunk - torque applied on each chunk• vacf - velocity-autocorrelation function of group of atoms• vcm/chunk - velocity of center-of-mass for each chunk• voronoi/atom - Voronoi volume and neighbors for each atom•
There are also additional compute styles submitted by users which are included in the LAMMPS distribution.
The list of these with links to the individual styles are given in the compute section of this page.
There are also additional accelerated compute styles included in the LAMMPS distribution for faster
performance on CPUs and GPUs. The list of these with links to the individual styles are given in the pair
section of this page.
Restrictions: none
Related commands:
uncompute, compute_modify, fix ave/atom, fix ave/time, fix ave/histo
Default: none
LAMMPS Users Manual
389

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute_modify command
Syntax:
compute_modify compute-ID keyword value ...
compute-ID = ID of the compute to modify• one or more keyword/value pairs may be listed• keyword = extra/dof or extra or dynamic/dof or dynamic
extra/dof value = N
N = # of extra degrees of freedom to subtract
extra syntax is identical to extra/dof, will be disabled at some point
dynamic/dof value = yes or no
yes/no = do or do not recompute the number of degrees of freedom (DOF) contributing to the temperature
dynamic syntax is identical to dynamic/dof, will be disabled at some point
•
Examples:
compute_modify myTemp extra/dof 0
compute_modify newtemp dynamic/dof yes extra/dof 600
Description:
Modify one or more parameters of a previously defined compute. Not all compute styles support all
parameters.
The extra/dof or extra keyword refers to how many degrees-of-freedom are subtracted (typically from 3N) as
a normalizing factor in a temperature computation. Only computes that compute a temperature use this option.
The default is 2 or 3 for 2d or 3d systems which is a correction factor for an ensemble of velocities with zero
total linear momentum. For compute temp/partial, if one or more velocity components are excluded, the value
used for extra is scaled accordingly. You can use a negative number for the extra parameter if you need to add
degrees-of-freedom. See the compute temp/asphere command for an example.
The dynamic/dof or dynamic keyword determines whether the number of atoms N in the compute group and
their associated degrees of freedom are re-computed each time a temperature is computed. Only compute
styles that calculate a temperature use this option. By default, N and their DOF are assumed to be constant. If
you are adding atoms or molecules to the system (see the fix pour, fix deposit, and fix gcmc commands) or
expect atoms or molecules to be lost (e.g. due to exiting the simulation box or via fix evaporate), then this
option should be used to insure the temperature is correctly normalized.
NOTE: The extra and dynamic keywords should not be used as they are deprecated (March 2017) and will
eventually be disabled. Instead, use the equivalent extra/dof and dynamic/dof keywords.
Restrictions: none
Related commands:
compute
LAMMPS Users Manual
390
Default:
The option defaults are extra/dof = 2 or 3 for 2d or 3d systems and dynamic/dof = no.
LAMMPS Users Manual
391

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
create_atoms command
Syntax:
create_atoms type style args keyword values ...
type = atom type (1-Ntypes) of atoms to create (offset for molecule creation)• style = box or region or single or random
box args = none
region args = region-ID
region-ID = particles will only be created if contained in the region
single args = x y z
x,y,z = coordinates of a single particle (distance units)
random args = N seed region-ID
N = number of particles to create
seed = random # seed (positive integer)
region-ID = create atoms within this region, use NULL for entire simulation box
•
zero or more keyword/value pairs may be appended• keyword = mol or basis or remap or var or set or units
mol value = template-ID seed
template-ID = ID of molecule template specified in a separate molecule command
seed = random # seed (positive integer)
basis values = M itype
M = which basis atom
itype = atom type (1-N) to assign to this basis atom
remap value = yes or no
var value = name = variable name to evaluate for test of atom creation
set values = dim name
dim = x or y or z
name = name of variable to set with x, y, or z atom position
rotate values = Rx Ry Rz theta
Rx,Ry,Rz = rotation vector for single molecule
theta = rotation angle for single molecule (degrees)
units value = lattice or box
lattice = the geometry is defined in lattice units
box = the geometry is defined in simulation box units
•
Examples:
create_atoms 1 box
create_atoms 3 region regsphere basis 2 3
create_atoms 3 single 0 0 5
create_atoms 1 box var v set x xpos set y ypos
Description:
This command creates atoms (or molecules) on a lattice, or a single atom (or molecule), or a random
collection of atoms (or molecules), as an alternative to reading in their coordinates explicitly via a read_data
or read_restart command. A simulation box must already exist, which is typically created via the create_box
command. Before using this command, a lattice must also be defined using the lattice command, unless you
specify the single style with units = box or the random style. For the remainder of this doc page, a created
atom or molecule is referred to as a "particle".
LAMMPS Users Manual
392
If created particles are individual atoms, they are assigned the specified atom type, though this can be altered
via the basis keyword as discussed below. If molecules are being created, the type of each atom in the created
molecule is specified in the file read by the molecule command, and those values are added to the specified
atom type. E.g. if type = 2, and the file specifies atom types 1,2,3, then each created molecule will have atom
types 3,4,5.
For the box style, the create_atoms command fills the entire simulation box with particles on the lattice. If
your simulation box is periodic, you should insure its size is a multiple of the lattice spacings, to avoid
unwanted atom overlaps at the box boundaries. If your box is periodic and a multiple of the lattice spacing in
a particular dimension, LAMMPS is careful to put exactly one particle at the boundary (on either side of the
box), not zero or two.
For the region style, a geometric volume is filled with particles on the lattice. This volume what is inside the
simulation box and is also consistent with the region volume. See the region command for details. Note that a
region can be specified so that its "volume" is either inside or outside a geometric boundary. Also note that if
your region is the same size as a periodic simulation box (in some dimension), LAMMPS does not implement
the same logic described above as for the box style, to insure exactly one particle at periodic boundaries. if
this is what you desire, you should either use the box style, or tweak the region size to get precisely the
particles you want.
For the single style, a single particle is added to the system at the specified coordinates. This can be useful for
debugging purposes or to create a tiny system with a handful of particles at specified positions.
For the random style, N particles are added to the system at randomly generated coordinates, which can be
useful for generating an amorphous system. The particles are created one by one using the specified random
number seed, resulting in the same set of particles coordinates, independent of how many processors are being
used in the simulation. If the region-ID argument is specified as NULL, then the created particles will be
anywhere in the simulation box. If a region-ID is specified, a geometric volume is filled which is both inside
the simulation box and is also consistent with the region volume. See the region command for details. Note
that a region can be specified so that its "volume" is either inside or outside a geometric boundary.
NOTE: Particles generated by the random style will typically be highly overlapped which will cause many
interatomic potentials to compute large energies and forces. Thus you should either perform an energy
minimization or run dynamics with fix nve/limit to equilibrate such a system, before running normal
dynamics.
Note that this command adds particles to those that already exist. This means it can be used to add particles to
a system previously read in from a data or restart file. Or the create_atoms command can be used multiple
times, to add multiple sets of particles to the simulation. For example, grain boundaries can be created, by
interleaving create_atoms with lattice commands specifying different orientations. By using the create_atoms
command in conjunction with the delete_atoms command, reasonably complex geometries can be created, or
a protein can be solvated with a surrounding box of water molecules.
In all these cases, care should be taken to insure that new atoms do not overlap existing atoms inappropriately,
especially if molecules are being added. The delete_atoms command can be used to remove overlapping
atoms or molecules.
NOTE: You cannot use any of the styles explained above to create atoms that are outside the simulation box;
they will just be ignored by LAMMPS. This is true even if you are using shrink-wrapped box boundaries, as
specified by the boundary command. However, you can first use the change_box command to temporarily
expand the box, then add atoms via create_atoms, then finally use change_box command again if needed to
LAMMPS Users Manual
393
re-shrink-wrap the new atoms. See the change_box doc page for an example of how to do this, using the
create_atoms single style to insert a new atom outside the current simulation box.
Individual atoms are inserted by this command, unless the mol keyword is used. It specifies a template-ID
previously defined using the molecule command, which reads a file that defines the molecule. The
coordinates, atom types, charges, etc, as well as any bond/angle/etc and special neighbor information for the
molecule can be specified in the molecule file. See the molecule command for details. The only settings
required to be in this file are the coordinates and types of atoms in the molecule.
Using a lattice to add molecules, e.g. via the box or region or single styles, is exactly the same as adding
atoms on lattice points, except that entire molecules are added at each point, i.e. on the point defined by each
basis atom in the unit cell as it tiles the simulation box or region. This is done by placing the geometric center
of the molecule at the lattice point, and giving the molecule a random orientation about the point. The random
seed specified with the mol keyword is used for this operation, and the random numbers generated by each
processor are different. This means the coordinates of individual atoms (in the molecules) will be different
when running on different numbers of processors, unlike when atoms are being created in parallel.
Also note that because of the random rotations, it may be important to use a lattice with a large enough
spacing that adjacent molecules will not overlap, regardless of their relative orientations.
NOTE: If the create_box command is used to create the simulation box, followed by the create_atoms
command with its mol option for adding molecules, then you typically need to use the optional keywords
allowed by the create_box command for extra bonds (angles,etc) or extra special neighbors. This is because
by default, the create_box command sets up a non-molecular system which doesn't allow molecules to be
added.
This is the meaning of the other allowed keywords.
The basis keyword is only used when atoms (not molecules) are being created. It specifies an atom type that
will be assigned to specific basis atoms as they are created. See the lattice command for specifics on how
basis atoms are defined for the unit cell of the lattice. By default, all created atoms are assigned the argument
type as their atom type.
The remap keyword only applies to the single style. If it is set to yes, then if the specified position is outside
the simulation box, it will mapped back into the box, assuming the relevant dimensions are periodic. If it is set
to no, no remapping is done and no particle is created if its position is outside the box.
The var and set keywords can be used together to provide a criterion for accepting or rejecting the addition of
an individual atom, based on its coordinates. The name specified for the var keyword is the name of an
equal-style variable which should evaluate to a zero or non-zero value based on one or two or three variables
which will store the x, y, or z coordinates of an atom (one variable per coordinate). If used, these other
variables must be internal-style variables defined in the input script; their initial numeric value can be
anything. They must be internal-style variables, because this command resets their values directly. The set
keyword is used to identify the names of these other variables, one variable for the x-coordinate of a created
atom, one for y, and one for z.
When an atom is created, its x,y,z coordinates become the values for any set variable that is defined. The var
variable is then evaluated. If the returned value is 0.0, the atom is not created. If it is non-zero, the atom is
created.
LAMMPS Users Manual
394

As an example, these commands can be used in a 2d simulation, to create a sinusoidal surface. Note that the
surface is "rough" due to individual lattice points being "above" or "below" the mathematical expression for
the sinusoidal curve. If a finer lattice were used, the sinusoid would appear to be "smoother". Also note the
use of the "xlat" and "ylat" thermo_style keywords which converts lattice spacings to distance. Click on the
image for a larger version.
variable x equal 100
variable y equal 25
lattice hex 0.8442
region box block 0 $x 0 $y -0.5 0.5
create_box 1 box
variable xx equal 0.0
variable yy equal 0.0
variable v equal "(0.2*v_y*ylat * cos(v_xx/xlat * 2.0*PI*4.0/v_x) + 0.5*v_y*ylat - v_yy) > 0.0"
create_atoms 1 box var v set x xx set y yy
The rotate keyword can be used with the single style, when adding a single molecule to specify the orientation
at which the molecule is inserted. The axis of rotation is determined by the rotation vector (Rx,Ry,Rz) that
goes through the insertion point. The specified theta determines the angle of rotation around that axis. Note
that the direction of rotation for the atoms around the rotation axis is consistent with the right-hand rule: if
your right-hand's thumb points along R, then your fingers wrap around the axis in the direction of rotation.
The units keyword determines the meaning of the distance units used to specify the coordinates of the one
particle created by the single style. A box value selects standard distance units as defined by the units
command, e.g. Angstroms for units = real or metal. A lattice value means the distance units are in lattice
spacings.
Atom IDs are assigned to created atoms in the following way. The collection of created atoms are assigned
consecutive IDs that start immediately following the largest atom ID existing before the create_atoms
command was invoked. When a simulation is performed on different numbers of processors, there is no
guarantee a particular created atom will be assigned the same ID. If molecules are being created, molecule IDs
are assigned to created molecules in a similar fashion.
Aside from their ID, atom type, and xyz position, other properties of created atoms are set to default values,
depending on which quantities are defined by the chosen atom style. See the atom style command for more
details. See the set and velocity commands for info on how to change these values.
charge = 0.0•
LAMMPS Users Manual
395

dipole moment magnitude = 0.0• diameter = 1.0• shape = 0.0 0.0 0.0• density = 1.0• volume = 1.0• velocity = 0.0 0.0 0.0• angular velocity = 0.0 0.0 0.0• angular momentum = 0.0 0.0 0.0• quaternion = (1,0,0,0)• bonds, angles, dihedrals, impropers = none•
If molecules are being created, these defaults can be overridden by values specified in the file read by the
molecule command. E.g. the file typically defines bonds (angles,etc) between atoms in the molecule, and can
optionally define charges on each atom.
Note that the sphere atom style sets the default particle diameter to 1.0 as well as the density. This means the
mass for the particle is not 1.0, but is PI/6 * diameter^3 = 0.5236.
Note that the ellipsoid atom style sets the default particle shape to (0.0 0.0 0.0) and the density to 1.0 which
means it is a point particle, not an ellipsoid, and has a mass of 1.0.
Note that the peri style sets the default volume and density to 1.0 and thus also set the mass for the particle to
1.0.
The set command can be used to override many of these default settings.
Restrictions:
An atom_style must be previously defined to use this command.
A rotation vector specified for a single molecule must be in the z-direction for a 2d model.
Related commands:
lattice, region, create_box, read_data, read_restart
Default:
The default for the basis keyword is that all created atoms are assigned the argument type as their atom type
(when single atoms are being created). The other defaults are remap = no, rotate = random, and units = lattice.
LAMMPS Users Manual
396

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
create_bonds command
Syntax:
create_bonds group-ID group2-ID btype rmin rmax
group-ID = ID of first group• group2-ID = ID of second group, bonds will be between atoms in the 2 groups• btype = bond type of created bonds• rmin = minimum distance between pair of atoms to bond together• rmax = minimum distance between pair of atoms to bond together•
Examples:
create_bonds all all 1 1.0 1.2
create_bonds surf solvent 3 2.0 2.4
Description:
Create bonds between pairs of atoms that meet specified distance criteria. The bond interactions can then be
computed during a simulation by the bond potential defined by the bond_style and bond_coeff commands.
This command is useful for adding bonds to a system, e.g. between nearest neighbors in a lattice of atoms,
without having to enumerate all the bonds in the data file read by the read_data command.
Note that the flexibility of this command is limited. It can be used several times to create different types of
bond at different distances. But it cannot typically create all the bonds that would normally be defined in a
complex system of molecules. Also note that this command does not add any 3-body or 4-body interactions
which, depending on your model, may be induced by added bonds, e.g. angle, dihedral, or improper
interactions.
All created bonds will be between pairs of atoms I,J where I is in one of the two specified groups, and J is in
the other. The two groups can be the same, e.g. group "all". The created bonds will be of bond type btype,
where btype must be a value between 1 and the number of bond types defined. This maximum value is set by
the "bond types" field in the header of the data file read by the read_data command, or via the optional
"bond/types" argument of the create_box command.
For a bond to be created, an I,J pair of atoms must be a distance D apart such that rmin <= D <= rmax.
The following settings must have been made in an input script before this command is used:
special_bonds weight for 1-2 interactions must be 0.0• a pair_style must be defined• no kspace_style defined• minimum pair_style cutoff + neighbor skin >= rmax•
These settings are required so that a neighbor list can be created to search for nearby atoms. Pairs of atoms
that are already bonded cannot appear in the neighbor list, to avoid creation of duplicate bonds. The neighbor
list for all atom type pairs must also extend to a distance that encompasses the rmax for new bonds to create.
LAMMPS Users Manual
397
An additional requirement is that your system must be ready to perform a simulation. This means, for
example, that all pair_style coefficients be set via the pair_coeff command. A bond_style command and all
bond coefficients must also be set, even if no bonds exist before this command is invoked. This is because the
building of neighbor list requires initialization and setup of a simulation, similar to what a run command
would require.
Note that you can change any of these settings after this command executes, e.g. if you wish to use long-range
Coulombic interactions via the kspace_style command for your subsequent simulation.
NOTE: If the system has no bonds to begin with, or if more bonds per atom are being added than currently
exist, then you must insure that the number of bond types and the maximum number of bonds per atom are set
to large enough values. Otherwise an error may occur when too many bonds are added to an atom. If the
read_data command is used to define the system, these 2 parameters can be set via the "bond types" and "extra
bond per atom" fields in the header section of the data file. If the create_box command is used to define the
system, these 2 parameters can be set via its optional "bond/types" and "extra/bond/per/atom" arguments. See
the doc pages for the 2 commands for details.
Restrictions:
This command cannot be used with molecular systems defined using molecule template files via the molecule
and atom_style template commands.
Related commands:
create_atoms, delete_bonds
Default: none
LAMMPS Users Manual
398

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
create_box command
Syntax:
create_box N region-ID keyword value ...
N = # of atom types to use in this simulation• region-ID = ID of region to use as simulation domain• zero or more keyword/value pairs may be appended• keyword = bond/types or angle/types or dihedral/types or improper/types or extra/bond/per/atom or
extra/angle/per/atom or extra/dihedral/per/atom or extra/improper/per/atom
bond/types value = # of bond types
angle/types value = # of angle types
dihedral/types value = # of dihedral types
improper/types value = # of improper types
extra/bond/per/atom value = # of bonds per atom
extra/angle/per/atom value = # of angles per atom
extra/dihedral/per/atom value = # of dihedrals per atom
extra/improper/per/atom value = # of impropers per atom
extra/special/per/atom value = # of special neighbors per atom
•
Examples:
create_box 2 mybox
create_box 2 mybox bond/types 2 extra/bond/per/atom 1
Description:
This command creates a simulation box based on the specified region. Thus a region command must first be
used to define a geometric domain. It also partitions the simulation box into a regular 3d grid of rectangular
bricks, one per processor, based on the number of processors being used and the settings of the processors
command. The partitioning can later be changed by the balance or fix balance commands.
The argument N is the number of atom types that will be used in the simulation.
If the region is not of style prism, then LAMMPS encloses the region (block, sphere, etc) with an axis-aligned
orthogonal bounding box which becomes the simulation domain.
If the region is of style prism, LAMMPS creates a non-orthogonal simulation domain shaped as a
parallelepiped with triclinic symmetry. As defined by the region prism command, the parallelepiped has its
"origin" at (xlo,ylo,zlo) and is defined by 3 edge vectors starting from the origin given by A = (xhi-xlo,0,0); B
= (xy,yhi-ylo,0); C = (xz,yz,zhi-zlo). Xy,xz,yz can be 0.0 or positive or negative values and are called "tilt
factors" because they are the amount of displacement applied to faces of an originally orthogonal box to
transform it into the parallelipiped.
By default, a prism region used with the create_box command must have tilt factors (xy,xz,yz) that do not
skew the box more than half the distance of the parallel box length. For example, if xlo = 2 and xhi = 12, then
the x box length is 10 and the xy tilt factor must be between -5 and 5. Similarly, both xz and yz must be
between -(xhi-xlo)/2 and +(yhi-ylo)/2. Note that this is not a limitation, since if the maximum tilt factor is 5
LAMMPS Users Manual
399

(as in this example), then configurations with tilt = ..., -15, -5, 5, 15, 25, ... are all geometrically equivalent. If
you wish to define a box with tilt factors that exceed these limits, you can use the box tilt command, with a
setting of large; a setting of small is the default.
See Section 6.12 of the doc pages for a geometric description of triclinic boxes, as defined by LAMMPS, and
how to transform these parameters to and from other commonly used triclinic representations.
When a prism region is used, the simulation domain should normally be periodic in the dimension that the tilt
is applied to, which is given by the second dimension of the tilt factor (e.g. y for xy tilt). This is so that pairs
of atoms interacting across that boundary will have one of them shifted by the tilt factor. Periodicity is set by
the boundary command. For example, if the xy tilt factor is non-zero, then the y dimension should be periodic.
Similarly, the z dimension should be periodic if xz or yz is non-zero. LAMMPS does not require this
periodicity, but you may lose atoms if this is not the case.
Also note that if your simulation will tilt the box, e.g. via the fix deform command, the simulation box must
be setup to be triclinic, even if the tilt factors are initially 0.0. You can also change an orthogonal box to a
triclinic box or vice versa by using the change box command with its ortho and triclinic options.
NOTE: If the system is non-periodic (in a dimension), then you should not make the lo/hi box dimensions (as
defined in your region command) radically smaller/larger than the extent of the atoms you eventually plan to
create, e.g. via the create_atoms command. For example, if your atoms extend from 0 to 50, you should not
specify the box bounds as -10000 and 10000. This is because as described above, LAMMPS uses the
specified box size to layout the 3d grid of processors. A huge (mostly empty) box will be sub-optimal for
performance when using "fixed" boundary conditions (see the boundary command). When using
"shrink-wrap" boundary conditions (see the boundary command), a huge (mostly empty) box may cause a
parallel simulation to lose atoms the first time that LAMMPS shrink-wraps the box around the atoms.
The optional keywords can be used to create a system that allows for bond (angle, dihedral, improper)
interactions, or for molecules with special 1-2,1-3,1-4 neighbors to be added later. These optional keywords
serve the same purpose as the analogous keywords that can be used in a data file which are recognized by the
read_data command when it sets up a system.
Note that if these keywords are not used, then the create_box command creates an atomic (non-molecular)
simulation that does not allow bonds between pairs of atoms to be defined, or a bond potential to be specified,
or for molecules with special neighbors to be added to the system by commands such as create_atoms mol, fix
deposit or fix pour.
As an example, see the examples/deposit/in.deposit.molecule script, which deposits molecules onto a
substrate. Initially there are no molecules in the system, but they are added later by the fix deposit command.
The create_box command in the script uses the bond/types and extra/bond/per/atom keywords to allow this. If
the added molecule contained more than 1 special bond (allowed by default), an extra/special/per/atom
keyword would also need to be specified.
Restrictions:
An atom_style and region must have been previously defined to use this command.
Related commands:
read_data, create_atoms, region
LAMMPS Users Manual
400
Default: none
LAMMPS Users Manual
401

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
delete_atoms command
Syntax:
delete_atoms style args keyword value ...
style = group or region or overlap or porosity
group args = group-ID
region args = region-ID
overlap args = cutoff group1-ID group2-ID
cutoff = delete one atom from pairs of atoms within the cutoff (distance units)
group1-ID = one atom in pair must be in this group
group2-ID = other atom in pair must be in this group
porosity args = region-ID fraction seed
region-ID = region within which to perform deletions
fraction = delete this fraction of atoms
seed = random number seed (positive integer)
•
zero or more keyword/value pairs may be appended• keyword = compress or bond or mol
compress value = no or yes
bond value = no or yes
mol value = no or yes
•
Examples:
delete_atoms group edge
delete_atoms region sphere compress no
delete_atoms overlap 0.3 all all
delete_atoms overlap 0.5 solvent colloid
delete_atoms porosity cube 0.1 482793 bond yes
Description:
Delete the specified atoms. This command can be used to carve out voids from a block of material or to delete
created atoms that are too close to each other (e.g. at a grain boundary).
For style group, all atoms belonging to the group are deleted.
For style region, all atoms in the region volume are deleted. Additional atoms can be deleted if they are in a
molecule for which one or more atoms were deleted within the region; see the mol keyword discussion below.
For style overlap pairs of atoms whose distance of separation is within the specified cutoff distance are
searched for, and one of the 2 atoms is deleted. Only pairs where one of the two atoms is in the first group
specified and the other atom is in the second group are considered. The atom that is in the first group is the
one that is deleted.
Note that it is OK for the two group IDs to be the same (e.g. group all), or for some atoms to be members of
both groups. In these cases, either atom in the pair may be deleted. Also note that if there are atoms which are
members of both groups, the only guarantee is that at the end of the deletion operation, enough deletions will
have occurred that no atom pairs within the cutoff will remain (subject to the group restriction). There is no
LAMMPS Users Manual
402
guarantee that the minimum number of atoms will be deleted, or that the same atoms will be deleted when
running on different numbers of processors.
For style porosity a specified fraction of atoms are deleted within the specified region. For example, if
fraction is 0.1, then 10% of the atoms will be deleted. The atoms to delete are chosen randomly. There is no
guarantee that the exact fraction of atoms will be deleted, or that the same atoms will be deleted when running
on different numbers of processors.
If the compress keyword is set to yes, then after atoms are deleted, then atom IDs are re-assigned so that they
run from 1 to the number of atoms in the system. Note that this is not done for molecular systems (see the
atom_style command), regardless of the compress setting, since it would foul up the bond connectivity that
has already been assigned.
A molecular system with fixed bonds, angles, dihedrals, or improper interactions, is one where the topology of
the interactions is typically defined in the data file read by the read_data command, and where the interactions
themselves are defined with the bond_style, angle_style, etc commands. If you delete atoms from such a
system, you must be careful not to end up with bonded interactions that are stored by remaining atoms but
which include deleted atoms. This will cause LAMMPS to generate a "missing atoms" error when the bonded
interaction is computed. The bond and mol keywords offer two ways to do that.
It the bond keyword is set to yes then any bond or angle or dihedral or improper interaction that includes a
deleted atom is also removed from the lists of such interactions stored by non-deleted atoms. Note that simply
deleting interactions due to dangling bonds (e.g. at a surface) may result in a inaccurate or invalid model for
the remaining atoms.
It the mol keyword is set to yes, then for every atom that is deleted, all other atoms in the same molecule (with
the same molecule ID) will also be deleted. This is not done for atoms with molecule ID = 0, since such an ID
is assumed to flag isolated atoms that are not part of molecules.
NOTE: The molecule deletion operation in invoked after all individual atoms have been deleted using the
rules described above for each style. This means additional atoms may be deleted that are not in the group or
region, that are not required by the overlap cutoff criterion, or that will create a higher fraction of porosity
than was requested.
Restrictions:
The overlap styles requires inter-processor communication to acquire ghost atoms and build a neighbor list.
This means that your system must be ready to perform a simulation before using this command (force fields
setup, atom masses set, etc). Since a neighbor list is used to find overlapping atom pairs, it also means that
you must define a pair style with the minimum force cutoff distance between any pair of atoms types (plus the
neighbor skin) >= the specified overlap cutoff.
If the special_bonds command is used with a setting of 0, then a pair of bonded atoms (1-2, 1-3, or 1-4) will
not appear in the neighbor list, and thus will not be considered for deletion by the overlap styles. You
probably don't want to be deleting one atom in a bonded pair anyway.
The bond yes option cannot be used with molecular systems defined using molecule template files via the
molecule and atom_style template commands.
Related commands:
LAMMPS Users Manual
403

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
delete_bonds command
Syntax:
delete_bonds group-ID style arg keyword ...
group-ID = group ID• style = multi or atom or bond or angle or dihedral or improper or stats
multi arg = none
atom arg = an atom type or range of types (see below)
bond arg = a bond type or range of types (see below)
angle arg = an angle type or range of types (see below)
dihedral arg = a dihedral type or range of types (see below)
improper arg = an improper type or range of types (see below)
stats arg = none
•
zero or more keywords may be appended• keyword = any or undo or remove or special•
Examples:
delete_bonds frozen multi remove
delete_bonds all atom 4 special
delete_bonds all bond 0*3 special
delete_bonds all stats
Description:
Turn off (or on) molecular topology interactions, i.e. bonds, angles, dihedrals, impropers. This command is
useful for deleting interactions that have been previously turned off by bond-breaking potentials. It is also
useful for turning off topology interactions between frozen or rigid atoms. Pairwise interactions can be turned
off via the neigh_modify exclude command. The fix shake command also effectively turns off certain bond
and angle interactions.
For all styles, by default, an interaction is only turned off (or on) if all the atoms involved are in the specified
group. See the any keyword to change the behavior.
Several of the styles (atom, bond, angle, dihedral, improper) take a type as an argument. The specified type
should be an integer from 0 to N, where N is the number of relevant types (atom types, bond types, etc). A
value of 0 is only relevant for style bond; see details below. In all cases, a wildcard asterisk can be used in
place of or in conjunction with the type argument to specify a range of types. This takes the form "*" or "*n"
or "n*" or "m*n". If N = the number of types, then an asterisk with no numeric values means all types from 0
to N. A leading asterisk means all types from 0 to n (inclusive). A trailing asterisk means all types from n to N
(inclusive). A middle asterisk means all types from m to n (inclusive). Note that it is fine to include a type of 0
for non-bond styles; it will simply be ignored.
For style multi all bond, angle, dihedral, and improper interactions of any type, involving atoms in the group,
are turned off.
Style atom is the same as style multi except that in addition, one or more of the atoms involved in the bond,
LAMMPS Users Manual
405
angle, dihedral, or improper interaction must also be of the specified atom type.
For style bond, only bonds are candidates for turn-off, and the bond must also be of the specified type. Styles
angle, dihedral, and improper are treated similarly.
For style bond, you can set the type to 0 to delete bonds that have been previously broken by a bond-breaking
potential (which sets the bond type to 0 when a bond is broken); e.g. see the bond_style quartic command.
For style stats no interactions are turned off (or on); the status of all interactions in the specified group is
simply reported. This is useful for diagnostic purposes if bonds have been turned off by a bond-breaking
potential during a previous run.
The default behavior of the delete_bonds command is to turn off interactions by toggling their type to a
negative value, but not to permanently remove the interaction. E.g. a bond_type of 2 is set to -2. The neighbor
list creation routines will not include such an interaction in their interaction lists. The default is also to not
alter the list of 1-2, 1-3, 1-4 neighbors computed by the special_bonds command and used to weight pairwise
force and energy calculations. This means that pairwise computations will proceed as if the bond (or angle,
etc) were still turned on.
Several keywords can be appended to the argument list to alter the default behaviors.
The any keyword changes the requirement that all atoms in the bond (angle, etc) must be in the specified
group in order to turn-off the interaction. Instead, if any of the atoms in the interaction are in the specified
group, it will be turned off (or on if the undo keyword is used).
The undo keyword inverts the delete_bonds command so that the specified bonds, angles, etc are turned on if
they are currently turned off. This means a negative value is toggled to positive. For example, for style angle,
if type is specified as 2, then all angles with current type = -2, are reset to type = 2. Note that the fix shake
command also sets bond and angle types negative, so this option should not be used on those interactions.
The remove keyword is invoked at the end of the delete_bonds operation. It causes turned-off bonds (angles,
etc) to be removed from each atom's data structure and then adjusts the global bond (angle, etc) counts
accordingly. Removal is a permanent change; removed bonds cannot be turned back on via the undo keyword.
Removal does not alter the pairwise 1-2, 1-3, 1-4 weighting list.
The special keyword is invoked at the end of the delete_bonds operation, after (optional) removal. It
re-computes the pairwise 1-2, 1-3, 1-4 weighting list. The weighting list computation treats turned-off bonds
the same as turned-on. Thus, turned-off bonds must be removed if you wish to change the weighting list.
Note that the choice of remove and special options affects how 1-2, 1-3, 1-4 pairwise interactions will be
computed across bonds that have been modified by the delete_bonds command.
Restrictions:
This command requires inter-processor communication to acquire ghost atoms, to coordinate the deleting of
bonds, angles, etc between atoms shared by multiple processors. This means that your system must be ready
to perform a simulation before using this command (force fields setup, atom masses set, etc). Just as would be
needed to run dynamics, the force field you define should define a cutoff (e.g. through a pair_style command)
which is long enough for a processor to acquire the ghost atoms its needs to compute bond, angle, etc
interactions.
LAMMPS Users Manual
406
If deleted bonds (angles, etc) are removed but the 1-2, 1-3, 1-4 weighting list is not recomputed, this can cause
a later fix shake command to fail due to an atom's bonds being inconsistent with the weighting list. This
should only happen if the group used in the fix command includes both atoms in the bond, in which case you
probably should be recomputing the weighting list.
Related commands:
neigh_modify exclude, special_bonds, fix shake
Default: none
LAMMPS Users Manual
407

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dielectric command
Syntax:
dielectric value
value = dielectric constant•
Examples:
dielectric 2.0
Description:
Set the dielectric constant for Coulombic interactions (pairwise and long-range) to this value. The constant is
unitless, since it is used to reduce the strength of the interactions. The value is used in the denominator of the
formulas for Coulombic interactions - e.g. a value of 4.0 reduces the Coulombic interactions to 25% of their
default strength. See the pair_style command for more details.
Restrictions: none
Related commands:
pair_style
Default:
dielectric 1.0
LAMMPS Users Manual
408

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_coeff command
Syntax:
dihedral_coeff N args
N = dihedral type (see asterisk form below)• args = coefficients for one or more dihedral types•
Examples:
dihedral_coeff 1 80.0 1 3
dihedral_coeff * 80.0 1 3 0.5
dihedral_coeff 2* 80.0 1 3 0.5
Description:
Specify the dihedral force field coefficients for one or more dihedral types. The number and meaning of the
coefficients depends on the dihedral style. Dihedral coefficients can also be set in the data file read by the
read_data command or in a restart file.
N can be specified in one of two ways. An explicit numeric value can be used, as in the 1st example above. Or
a wild-card asterisk can be used to set the coefficients for multiple dihedral types. This takes the form "*" or
"*n" or "n*" or "m*n". If N = the number of dihedral types, then an asterisk with no numeric values means all
types from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A trailing asterisk means all
types from n to N (inclusive). A middle asterisk means all types from m to n (inclusive).
Note that using a dihedral_coeff command can override a previous setting for the same dihedral type. For
example, these commands set the coeffs for all dihedral types, then overwrite the coeffs for just dihedral type
2:
dihedral_coeff * 80.0 1 3
dihedral_coeff 2 200.0 1 3
A line in a data file that specifies dihedral coefficients uses the exact same format as the arguments of the
dihedral_coeff command in an input script, except that wild-card asterisks should not be used since
coefficients for all N types must be listed in the file. For example, under the "Dihedral Coeffs" section of a
data file, the line that corresponds to the 1st example above would be listed as
1 80.0 1 3
The dihedral_style class2 is an exception to this rule, in that an additional argument is used in the input script
to allow specification of the cross-term coefficients. See its doc page for details.
NOTE: When comparing the formulas and coefficients for various LAMMPS dihedral styles with dihedral
equations defined by other force fields, note that some force field implementations divide/multiply the energy
prefactor K by the multiple number of torsions that contain the J-K bond in an I-J-K-L torsion. LAMMPS
does not do this, i.e. the listed dihedral equation applies to each individual dihedral. Thus you need to define K
appropriately to account for this difference if necessary.
LAMMPS Users Manual
409

Here is an alphabetic list of dihedral styles defined in LAMMPS. Click on the style to display the formula it
computes and coefficients specified by the associated dihedral_coeff command.
Note that there are also additional dihedral styles submitted by users which are included in the LAMMPS
distribution. The list of these with links to the individual styles are given in the dihedral section of this page.
dihedral_style none - turn off dihedral interactions• dihedral_style hybrid - define multiple styles of dihedral interactions•
dihedral_style charmm - CHARMM dihedral• dihedral_style class2 - COMPASS (class 2) dihedral• dihedral_style harmonic - harmonic dihedral• dihedral_style helix - helix dihedral• dihedral_style multi/harmonic - multi-harmonic dihedral• dihedral_style opls - OPLS dihedral•
Restrictions:
This command must come after the simulation box is defined by a read_data, read_restart, or create_box
command.
A dihedral style must be defined before any dihedral coefficients are set, either in the input script or in a data
file.
Related commands:
dihedral_style
Default: none
LAMMPS Users Manual
410

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style command
Syntax:
dihedral_style style
style = none or hybrid or charmm or class2 or harmonic or helix or multi/harmonic or opls•
Examples:
dihedral_style harmonic
dihedral_style multi/harmonic
dihedral_style hybrid harmonic charmm
Description:
Set the formula(s) LAMMPS uses to compute dihedral interactions between quadruplets of atoms, which
remain in force for the duration of the simulation. The list of dihedral quadruplets is read in by a read_data or
read_restart command from a data or restart file.
Hybrid models where dihedrals are computed using different dihedral potentials can be setup using the hybrid
dihedral style.
The coefficients associated with a dihedral style can be specified in a data or restart file or via the
dihedral_coeff command.
All dihedral potentials store their coefficient data in binary restart files which means dihedral_style and
dihedral_coeff commands do not need to be re-specified in an input script that restarts a simulation. See the
read_restart command for details on how to do this. The one exception is that dihedral_style hybrid only
stores the list of sub-styles in the restart file; dihedral coefficients need to be re-specified.
NOTE: When both a dihedral and pair style is defined, the special_bonds command often needs to be used to
turn off (or weight) the pairwise interaction that would otherwise exist between 4 bonded atoms.
In the formulas listed for each dihedral style, phi is the torsional angle defined by the quadruplet of atoms.
This angle has a sign convention as shown in this diagram:
where the I,J,K,L ordering of the 4 atoms that define the dihedral is from left to right.
LAMMPS Users Manual
411

This sign convention effects several of the dihedral styles listed below (e.g. charmm, helix) in the sense that
the energy formula depends on the sign of phi, which may be reflected in the value of the coefficients you
specify.
NOTE: When comparing the formulas and coefficients for various LAMMPS dihedral styles with dihedral
equations defined by other force fields, note that some force field implementations divide/multiply the energy
prefactor K by the multiple number of torsions that contain the J-K bond in an I-J-K-L torsion. LAMMPS
does not do this, i.e. the listed dihedral equation applies to each individual dihedral. Thus you need to define K
appropriately via the dihedral_coeff command to account for this difference if necessary.
Here is an alphabetic list of dihedral styles defined in LAMMPS. Click on the style to display the formula it
computes and coefficients specified by the associated dihedral_coeff command.
Note that there are also additional dihedral styles submitted by users which are included in the LAMMPS
distribution. The list of these with links to the individual styles are given in the dihedral section of this page.
dihedral_style none - turn off dihedral interactions• dihedral_style zero - topology but no interactions• dihedral_style hybrid - define multiple styles of dihedral interactions•
dihedral_style charmm - CHARMM dihedral• dihedral_style class2 - COMPASS (class 2) dihedral• dihedral_style harmonic - harmonic dihedral• dihedral_style helix - helix dihedral• dihedral_style multi/harmonic - multi-harmonic dihedral• dihedral_style opls - OPLS dihedral•
Restrictions:
Dihedral styles can only be set for atom styles that allow dihedrals to be defined.
Most dihedral styles are part of the MOLECULE package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info on packages. The doc pages for individual
dihedral potentials tell if it is part of a package.
Related commands:
dihedral_coeff
Default:
dihedral_style none
LAMMPS Users Manual
412

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dimension command
Syntax:
dimension N
N = 2 or 3•
Examples:
dimension 2
Description:
Set the dimensionality of the simulation. By default LAMMPS runs 3d simulations. To run a 2d simulation,
this command should be used prior to setting up a simulation box via the create_box or read_data commands.
Restart files also store this setting.
See the discussion in Section 6 for additional instructions on how to run 2d simulations.
NOTE: Some models in LAMMPS treat particles as finite-size spheres or ellipsoids, as opposed to point
particles. In 2d, the particles will still be spheres or ellipsoids, not circular disks or ellipses, meaning their
moment of inertia will be the same as in 3d.
Restrictions:
This command must be used before the simulation box is defined by a read_data or create_box command.
Related commands:
fix enforce2d
Default:
dimension 3
LAMMPS Users Manual
413

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
displace_atoms command
Syntax:
displace_atoms group-ID style args keyword value ...
group-ID = ID of group of atoms to displace• style = move or ramp or random or rotate
move args = delx dely delz
delx,dely,delz = distance to displace in each dimension (distance units)
any of delx,dely,delz can be a variable (see below)
ramp args = ddim dlo dhi dim clo chi
ddim = x or y or z
dlo,dhi = displacement distance between dlo and dhi (distance units)
dim = x or y or z
clo,chi = lower and upper bound of domain to displace (distance units)
random args = dx dy dz seed
dx,dy,dz = random displacement magnitude in each dimension (distance units)
seed = random # seed (positive integer)
rotate args = Px Py Pz Rx Ry Rz theta
Px,Py,Pz = origin point of axis of rotation (distance units)
Rx,Ry,Rz = axis of rotation vector
theta = angle of rotation (degrees)
•
zero or more keyword/value pairs may be appended
keyword = units
value = box or lattice
•
Examples:
displace_atoms top move 0 -5 0 units box
displace_atoms flow ramp x 0.0 5.0 y 2.0 20.5
Description:
Displace a group of atoms. This can be used to move atoms a large distance before beginning a simulation or
to randomize atoms initially on a lattice. For example, in a shear simulation, an initial strain can be imposed
on the system. Or two groups of atoms can be brought into closer proximity.
The move style displaces the group of atoms by the specified 3d displacement vector. Any of the 3 quantities
defining the vector components can be specified as an equal-style or atom-style variable. If the value is a
variable, it should be specified as v_name, where name is the variable name. In this case, the variable will be
evaluated, and its value(s) used for the displacement(s). The scale factor implied by the units keyword will
also be applied to the variable result.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Atom-style variables
can specify the same formulas as equal-style variables but can also include per-atom values, such as atom
coordinates or per-atom values read from a file. Note that if the variable references other compute or fix
commands, those values must be up-to-date for the current timestep. See the "Variable Accuracy" section of
the variable doc page for more details.
LAMMPS Users Manual
414

The ramp style displaces atoms a variable amount in one dimension depending on the atom's coordinate in a
(possibly) different dimension. For example, the second example command displaces atoms in the x-direction
an amount between 0.0 and 5.0 distance units. Each atom's displacement depends on the fractional distance its
y coordinate is between 2.0 and 20.5. Atoms with y-coordinates outside those bounds will be moved the
minimum (0.0) or maximum (5.0) amount.
The random style independently moves each atom in the group by a random displacement, uniformly sampled
from a value between -dx and +dx in the x dimension, and similarly for y and z. Random numbers are used in
such a way that the displacement of a particular atom is the same, regardless of how many processors are
being used.
The rotate style rotates each atom in the group by the angle theta around a rotation axis R = (Rx,Ry,Rz) that
goes thru a point P = (Px,Py,Pz). The direction of rotation for the atoms around the rotation axis is consistent
with the right-hand rule: if your right-hand thumb points along R, then your fingers wrap around the axis in
the direction of positive theta.
If the defined atom_style assigns an orientation to each atom (atom styles ellipsoid, line, tri, body), then that
property is also updated appropriately to correspond to the atom's rotation.
Distance units for displacements and the origin point of the rotate style are determined by the setting of box or
lattice for the units keyword. Box means distance units as defined by the units command - e.g. Angstroms for
real units. Lattice means distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacing.
NOTE: Care should be taken not to move atoms on top of other atoms. After the move, atoms are remapped
into the periodic simulation box if needed, and any shrink-wrap boundary conditions (see the boundary
command) are enforced which may change the box size. Other than this effect, this command does not change
the size or shape of the simulation box. See the change_box command if that effect is desired.
NOTE: Atoms can be moved arbitrarily long distances by this command. If the simulation box is non-periodic
and shrink-wrapped (see the boundary command), this can change its size or shape. This is not a problem,
except that the mapping of processors to the simulation box is not changed by this command from its initial 3d
configuration; see the processors command. Thus, if the box size/shape changes dramatically, the mapping of
processors to the simulation box may not end up as optimal as the initial mapping attempted to be.
Restrictions:
For a 2d simulation, only rotations around the a vector parallel to the z-axis are allowed.
Related commands:
lattice, change_box, fix move
Default:
The option defaults are units = lattice.
LAMMPS Users Manual
415

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dump command
dump custom/vtk command
dump h5md command
dump image command
dump movie command
dump molfile command
dump nc command
Syntax:
dump ID group-ID style N file args
ID = user-assigned name for the dump• group-ID = ID of the group of atoms to be dumped• style = atom or atom/gz or atom/mpiio or cfg or cfg/gz or cfg/mpiio or dcd or xtc or xyz or xyz/gz or
xyz/mpiio or h5md or image or movie or molfile or local or custom or custom/gz or custom/mpiio
•
N = dump every this many timesteps• file = name of file to write dump info to• args = list of arguments for a particular style
atom args = none
atom/gz args = none
atom/mpiio args = none
cfg args = same as custom args, see below
cfg/gz args = same as custom args, see below
cfg/mpiio args = same as custom args, see below
dcd args = none
xtc args = none
xyz args = none
xyz/gz args = none
xyz/mpiio args = none
custom/vtk args = similar to custom args below, discussed on dump custom/vtk doc page
h5md args = discussed on dump h5md doc page
image args = discussed on dump image doc page
movie args = discussed on dump image doc page
molfile args = discussed on dump molfile doc page
•
LAMMPS Users Manual
416
nc args = discussed on dump nc doc page
local args = list of local attributes
possible attributes = index, c_ID, c_ID[I], f_ID, f_ID[I]
index = enumeration of local values
c_ID = local vector calculated by a compute with ID
c_ID[I] = Ith column of local array calculated by a compute with ID, I can include wildcard (see below)
f_ID = local vector calculated by a fix with ID
f_ID[I] = Ith column of local array calculated by a fix with ID, I can include wildcard (see below)
custom or custom/gz or custom/mpiio args = list of atom attributes
possible attributes = id, mol, proc, procp1, type, element, mass,
x, y, z, xs, ys, zs, xu, yu, zu,
xsu, ysu, zsu, ix, iy, iz,
vx, vy, vz, fx, fy, fz,
q, mux, muy, muz, mu,
radius, diameter, omegax, omegay, omegaz,
angmomx, angmomy, angmomz, tqx, tqy, tqz,
c_ID, c_ID[N], f_ID, f_ID[N], v_name
id = atom ID
mol = molecule ID
proc = ID of processor that owns atom
procp1 = ID+1 of processor that owns atom
type = atom type
element = name of atom element, as defined by dump_modify command
mass = atom mass
x,y,z = unscaled atom coordinates
xs,ys,zs = scaled atom coordinates
xu,yu,zu = unwrapped atom coordinates
xsu,ysu,zsu = scaled unwrapped atom coordinates
ix,iy,iz = box image that the atom is in
vx,vy,vz = atom velocities
fx,fy,fz = forces on atoms
q = atom charge
mux,muy,muz = orientation of dipole moment of atom
mu = magnitude of dipole moment of atom
radius,diameter = radius,diameter of spherical particle
omegax,omegay,omegaz = angular velocity of spherical particle
angmomx,angmomy,angmomz = angular momentum of aspherical particle
tqx,tqy,tqz = torque on finite-size particles
c_ID = per-atom vector calculated by a compute with ID
c_ID[I] = Ith column of per-atom array calculated by a compute with ID, I can include wildcard (see below)
f_ID = per-atom vector calculated by a fix with ID
f_ID[I] = Ith column of per-atom array calculated by a fix with ID, I can include wildcard (see below)
v_name = per-atom vector calculated by an atom-style variable with name
d_name = per-atom floating point vector with name, managed by fix property/atom
i_name = per-atom integer vector with name, managed by fix property/atom
Examples:
dump myDump all atom 100 dump.atom
dump myDump all atom/mpiio 100 dump.atom.mpiio
dump myDump all atom/gz 100 dump.atom.gz
dump 2 subgroup atom 50 dump.run.bin
dump 2 subgroup atom 50 dump.run.mpiio.bin
dump 4a all custom 100 dump.myforce.* id type x y vx fx
dump 4b flow custom 100 dump.%.myforce id type c_myF[3] v_ke
dump 4b flow custom 100 dump.%.myforce id type c_myF[*] v_ke
dump 2 inner cfg 10 dump.snap.*.cfg mass type xs ys zs vx vy vz
LAMMPS Users Manual
417

dump snap all cfg 100 dump.config.*.cfg mass type xs ys zs id type c_Stress[2]
dump 1 all xtc 1000 file.xtc
Description:
Dump a snapshot of atom quantities to one or more files every N timesteps in one of several styles. The image
and movie styles are the exception: the image style renders a JPG, PNG, or PPM image file of the atom
configuration every N timesteps while the movie style combines and compresses them into a movie file; both
are discussed in detail on the dump image doc page. The timesteps on which dump output is written can also
be controlled by a variable. See the dump_modify every command.
Only information for atoms in the specified group is dumped. The dump_modify thresh and region commands
can also alter what atoms are included. Not all styles support all these options; see details below.
As described below, the filename determines the kind of output (text or binary or gzipped, one big file or one
per timestep, one big file or multiple smaller files).
NOTE: Because periodic boundary conditions are enforced only on timesteps when neighbor lists are rebuilt,
the coordinates of an atom written to a dump file may be slightly outside the simulation box. Re-neighbor
timesteps will not typically coincide with the timesteps dump snapshots are written. See the dump_modify
pbc command if you with to force coordinates to be strictly inside the simulation box.
NOTE: Unless the dump_modify sort option is invoked, the lines of atom information written to dump files
(typically one line per atom) will be in an indeterminate order for each snapshot. This is even true when
running on a single processor, if the atom_modify sort option is on, which it is by default. In this case atoms
are re-ordered periodically during a simulation, due to spatial sorting. It is also true when running in parallel,
because data for a single snapshot is collected from multiple processors, each of which owns a subset of the
atoms.
For the atom, custom, cfg, and local styles, sorting is off by default. For the dcd, xtc, xyz, and molfile styles,
sorting by atom ID is on by default. See the dump_modify doc page for details.
The atom/gz, cfg/gz, custom/gz, and xyz/gz styles are identical in command syntax to the corresponding styles
without "gz", however, they generate compressed files using the zlib library. Thus the filename suffix ".gz" is
mandatory. This is an alternative approach to writing compressed files via a pipe, as done by the regular dump
styles, which may be required on clusters where the interface to the high-speed network disallows using the
fork() library call (which is needed for a pipe). For the remainder of this doc page, you should thus consider
the atom and atom/gz styles (etc) to be inter-changeable, with the exception of the required filename suffix.
As explained below, the atom/mpiio, cfg/mpiio, custom/mpiio, and xyz/mpiio styles are identical in command
syntax and in the format of the dump files they create, to the corresponding styles without "mpiio", except the
single dump file they produce is written in parallel via the MPI-IO library. For the remainder of this doc page,
you should thus consider the atom and atom/mpiio styles (etc) to be inter-changeable. The one exception is
how the filename is specified for the MPI-IO styles, as explained below.
The precision of values output to text-based dump files can be controlled by the dump_modify format
command and its options.
The style keyword determines what atom quantities are written to the file and in what format. Settings made
via the dump_modify command can also alter the format of individual values and the file itself.
LAMMPS Users Manual
418
The atom, local, and custom styles create files in a simple text format that is self-explanatory when viewing a
dump file. Many of the LAMMPS post-processing tools, including Pizza.py, work with this format, as does
the rerun command.
For post-processing purposes the atom, local, and custom text files are self-describing in the following sense.
The dimensions of the simulation box are included in each snapshot. For an orthogonal simulation box this
information is is formatted as:
ITEM: BOX BOUNDS xx yy zz
xlo xhi
ylo yhi
zlo zhi
where xlo,xhi are the maximum extents of the simulation box in the x-dimension, and similarly for y and z.
The "xx yy zz" represent 6 characters that encode the style of boundary for each of the 6 simulation box
boundaries (xlo,xhi and ylo,yhi and zlo,zhi). Each of the 6 characters is either p = periodic, f = fixed, s =
shrink wrap, or m = shrink wrapped with a minimum value. See the boundary command for details.
For triclinic simulation boxes (non-orthogonal), an orthogonal bounding box which encloses the triclinic
simulation box is output, along with the 3 tilt factors (xy, xz, yz) of the triclinic box, formatted as follows:
ITEM: BOX BOUNDS xy xz yz xx yy zz
xlo_bound xhi_bound xy
ylo_bound yhi_bound xz
zlo_bound zhi_bound yz
The presence of the text "xy xz yz" in the ITEM line indicates that the 3 tilt factors will be included on each of
the 3 following lines. This bounding box is convenient for many visualization programs. The meaning of the 6
character flags for "xx yy zz" is the same as above.
Note that the first two numbers on each line are now xlo_bound instead of xlo, etc, since they represent a
bounding box. See this section of the doc pages for a geometric description of triclinic boxes, as defined by
LAMMPS, simple formulas for how the 6 bounding box extents (xlo_bound,xhi_bound,etc) are calculated
from the triclinic parameters, and how to transform those parameters to and from other commonly used
triclinic representations.
The "ITEM: ATOMS" line in each snapshot lists column descriptors for the per-atom lines that follow. For
example, the descriptors would be "id type xs ys zs" for the default atom style, and would be the atom
attributes you specify in the dump command for the custom style.
For style atom, atom coordinates are written to the file, along with the atom ID and atom type. By default,
atom coords are written in a scaled format (from 0 to 1). I.e. an x value of 0.25 means the atom is at a location
1/4 of the distance from xlo to xhi of the box boundaries. The format can be changed to unscaled coords via
the dump_modify settings. Image flags can also be added for each atom via dump_modify.
Style custom allows you to specify a list of atom attributes to be written to the dump file for each atom.
Possible attributes are listed above and will appear in the order specified. You cannot specify a quantity that is
not defined for a particular simulation - such as q for atom style bond, since that atom style doesn't assign
charges. Dumps occur at the very end of a timestep, so atom attributes will include effects due to fixes that are
applied during the timestep. An explanation of the possible dump custom attributes is given below.
LAMMPS Users Manual
419

For style local, local output generated by computes and fixes is used to generate lines of output that is written
to the dump file. This local data is typically calculated by each processor based on the atoms it owns, but there
may be zero or more entities per atom, e.g. a list of bond distances. An explanation of the possible dump local
attributes is given below. Note that by using input from the compute property/local command with dump
local, it is possible to generate information on bonds, angles, etc that can be cut and pasted directly into a data
file read by the read_data command.
Style cfg has the same command syntax as style custom and writes extended CFG format files, as used by the
AtomEye visualization package. Since the extended CFG format uses a single snapshot of the system per file,
a wildcard "*" must be included in the filename, as discussed below. The list of atom attributes for style cfg
must begin with either "mass type xs ys zs" or "mass type xsu ysu zsu" since these quantities are needed to
write the CFG files in the appropriate format (though the "mass" and "type" fields do not appear explicitly in
the file). Any remaining attributes will be stored as "auxiliary properties" in the CFG files. Note that you will
typically want to use the dump_modify element command with CFG-formatted files, to associate element
names with atom types, so that AtomEye can render atoms appropriately. When unwrapped coordinates xsu,
ysu, and zsu are requested, the nominal AtomEye periodic cell dimensions are expanded by a large factor
UNWRAPEXPAND = 10.0, which ensures atoms that are displayed correctly for up to UNWRAPEXPAND/2
periodic boundary crossings in any direction. Beyond this, AtomEye will rewrap the unwrapped coordinates.
The expansion causes the atoms to be drawn farther away from the viewer, but it is easy to zoom the atoms
closer, and the interatomic distances are unaffected.
The dcd style writes DCD files, a standard atomic trajectory format used by the CHARMM, NAMD, and
XPlor molecular dynamics packages. DCD files are binary and thus may not be portable to different
machines. The number of atoms per snapshot cannot change with the dcd style. The unwrap option of the
dump_modify command allows DCD coordinates to be written "unwrapped" by the image flags for each
atom. Unwrapped means that if the atom has passed through a periodic boundary one or more times, the value
is printed for what the coordinate would be if it had not been wrapped back into the periodic box. Note that
these coordinates may thus be far outside the box size stored with the snapshot.
The xtc style writes XTC files, a compressed trajectory format used by the GROMACS molecular dynamics
package, and described here. The precision used in XTC files can be adjusted via the dump_modify
command. The default value of 1000 means that coordinates are stored to 1/1000 nanometer accuracy. XTC
files are portable binary files written in the NFS XDR data format, so that any machine which supports XDR
should be able to read them. The number of atoms per snapshot cannot change with the xtc style. The unwrap
option of the dump_modify command allows XTC coordinates to be written "unwrapped" by the image flags
for each atom. Unwrapped means that if the atom has passed thru a periodic boundary one or more times, the
value is printed for what the coordinate would be if it had not been wrapped back into the periodic box. Note
that these coordinates may thus be far outside the box size stored with the snapshot.
The xyz style writes XYZ files, which is a simple text-based coordinate format that many codes can read.
Specifically it has a line with the number of atoms, then a comment line that is usually ignored followed by
one line per atom with the atom type and the x-, y-, and z-coordinate of that atom. You can use the
dump_modify element option to change the output from using the (numerical) atom type to an element name
(or some other label). This will help many visualization programs to guess bonds and colors.
Note that atom, custom, dcd, xtc, and xyz style dump files can be read directly by VMD, a popular molecular
viewing program.
Dumps are performed on timesteps that are a multiple of N (including timestep 0) and on the last timestep of a
minimization if the minimization converges. Note that this means a dump will not be performed on the initial
timestep after the dump command is invoked, if the current timestep is not a multiple of N. This behavior can
LAMMPS Users Manual
420
be changed via the dump_modify first command, which can also be useful if the dump command is invoked
after a minimization ended on an arbitrary timestep. N can be changed between runs by using the
dump_modify every command (not allowed for dcd style). The dump_modify every command also allows a
variable to be used to determine the sequence of timesteps on which dump files are written. In this mode a
dump on the first timestep of a run will also not be written unless the dump_modify first command is used.
The specified filename determines how the dump file(s) is written. The default is to write one large text file,
which is opened when the dump command is invoked and closed when an undump command is used or when
LAMMPS exits. For the dcd and xtc styles, this is a single large binary file.
Dump filenames can contain two wildcard characters. If a "*" character appears in the filename, then one file
per snapshot is written and the "*" character is replaced with the timestep value. For example, tmp.dump.*
becomes tmp.dump.0, tmp.dump.10000, tmp.dump.20000, etc. This option is not available for the dcd and xtc
styles. Note that the dump_modify pad command can be used to insure all timestep numbers are the same
length (e.g. 00010), which can make it easier to read a series of dump files in order with some post-processing
tools.
If a "%" character appears in the filename, then each of P processors writes a portion of the dump file, and the
"%" character is replaced with the processor ID from 0 to P-1. For example, tmp.dump.% becomes
tmp.dump.0, tmp.dump.1, ... tmp.dump.P-1, etc. This creates smaller files and can be a fast mode of output on
parallel machines that support parallel I/O for output. This option is not available for the dcd, xtc, and xyz
styles.
By default, P = the number of processors meaning one file per processor, but P can be set to a smaller value
via the nfile or fileper keywords of the dump_modify command. These options can be the most efficient way
of writing out dump files when running on large numbers of processors.
Note that using the "*" and "%" characters together can produce a large number of small dump files!
For the atom/mpiio, cfg/mpiio, custom/mpiio, and xyz/mpiio styles, a single dump file is written in parallel via
the MPI-IO library, which is part of the MPI standard for versions 2.0 and above. Using MPI-IO requires two
steps. First, build LAMMPS with its MPIIO package installed, e.g.
make yes-mpiio # installs the MPIIO package
make mpi # build LAMMPS for your platform
Second, use a dump filename which contains ".mpiio". Note that it does not have to end in ".mpiio", just
contain those characters. Unlike MPI-IO restart files, which must be both written and read using MPI-IO, the
dump files produced by these MPI-IO styles are identical in format to the files produced by their non-MPI-IO
style counterparts. This means you can write a dump file using MPI-IO and use the read_dump command or
perform other post-processing, just as if the dump file was not written using MPI-IO.
Note that MPI-IO dump files are one large file which all processors write to. You thus cannot use the "%"
wildcard character described above in the filename since that specifies generation of multiple files. You can
use the ".bin" suffix described below in an MPI-IO dump file; again this file will be written in parallel and
have the same binary format as if it were written without MPI-IO.
If the filename ends with ".bin", the dump file (or files, if "*" or "%" is also used) is written in binary format.
A binary dump file will be about the same size as a text version, but will typically write out much faster. Of
course, when post-processing, you will need to convert it back to text format (see the binary2txt tool) or write
your own code to read the binary file. The format of the binary file can be understood by looking at the
LAMMPS Users Manual
421

tools/binary2txt.cpp file. This option is only available for the atom and custom styles.
If the filename ends with ".gz", the dump file (or files, if "*" or "%" is also used) is written in gzipped format.
A gzipped dump file will be about 3x smaller than the text version, but will also take longer to write. This
option is not available for the dcd and xtc styles.
Note that in the discussion which follows, for styles which can reference values from a compute or fix, like
the custom, cfg, or local styles, the bracketed index I can be specified using a wildcard asterisk with the index
to effectively specify multiple values. This takes the form "*" or "*n" or "n*" or "m*n". If N = the size of the
vector (for mode = scalar) or the number of columns in the array (for mode = vector), then an asterisk with no
numeric values means all indices from 1 to N. A leading asterisk means all indices from 1 to n (inclusive). A
trailing asterisk means all indices from n to N (inclusive). A middle asterisk means all indices from m to n
(inclusive).
Using a wildcard is the same as if the individual columns of the array had been listed one by one. E.g. these 2
dump commands are equivalent, since the compute stress/atom command creates a per-atom array with 6
columns:
compute myPress all stress/atom NULL
dump 2 all custom 100 tmp.dump id myPress[*]
dump 2 all custom 100 tmp.dump id myPress[1] myPress[2] myPress[3] &
myPress[4] myPress[5] myPress[6]
This section explains the local attributes that can be specified as part of the local style.
The index attribute can be used to generate an index number from 1 to N for each line written into the dump
file, where N is the total number of local datums from all processors, or lines of output that will appear in the
snapshot. Note that because data from different processors depend on what atoms they currently own, and
atoms migrate between processor, there is no guarantee that the same index will be used for the same info
(e.g. a particular bond) in successive snapshots.
The c_ID and c_ID[I] attributes allow local vectors or arrays calculated by a compute to be output. The ID in
the attribute should be replaced by the actual ID of the compute that has been defined previously in the input
script. See the compute command for details. There are computes for calculating local information such as
indices, types, and energies for bonds and angles.
Note that computes which calculate global or per-atom quantities, as opposed to local quantities, cannot be
output in a dump local command. Instead, global quantities can be output by the thermo_style custom
command, and per-atom quantities can be output by the dump custom command.
If c_ID is used as a attribute, then the local vector calculated by the compute is printed. If c_ID[I] is used,
then I must be in the range from 1-M, which will print the Ith column of the local array with M columns
calculated by the compute. See the discussion above for how I can be specified with a wildcard asterisk to
effectively specify multiple values.
The f_ID and f_ID[I] attributes allow local vectors or arrays calculated by a fix to be output. The ID in the
attribute should be replaced by the actual ID of the fix that has been defined previously in the input script.
If f_ID is used as a attribute, then the local vector calculated by the fix is printed. If f_ID[I] is used, then I
must be in the range from 1-M, which will print the Ith column of the local with M columns calculated by the
fix. See the discussion above for how I can be specified with a wildcard asterisk to effectively specify
LAMMPS Users Manual
422

multiple values.
Here is an example of how to dump bond info for a system, including the distance and energy of each bond:
compute 1 all property/local batom1 batom2 btype
compute 2 all bond/local dist eng
dump 1 all local 1000 tmp.dump index c_1[1] c_1[2] c_1[3] c_2[1] c_2[2]
This section explains the atom attributes that can be specified as part of the custom and cfg styles.
The id, mol, proc, procp1, type, element, mass, vx, vy, vz, fx, fy, fz, q attributes are self-explanatory.
Id is the atom ID. Mol is the molecule ID, included in the data file for molecular systems. Proc is the ID of the
processor (0 to Nprocs-1) that currently owns the atom. Procp1 is the proc ID+1, which can be convenient in
place of a type attribute (1 to Ntypes) for coloring atoms in a visualization program. Type is the atom type (1
to Ntypes). Element is typically the chemical name of an element, which you must assign to each type via the
dump_modify element command. More generally, it can be any string you wish to associated with an atom
type. Mass is the atom mass. Vx, vy, vz, fx, fy, fz, and q are components of atom velocity and force and atomic
charge.
There are several options for outputting atom coordinates. The x, y, z attributes write atom coordinates
"unscaled", in the appropriate distance units (Angstroms, sigma, etc). Use xs, ys, zs if you want the
coordinates "scaled" to the box size, so that each value is 0.0 to 1.0. If the simulation box is triclinic (tilted),
then all atom coords will still be between 0.0 and 1.0. I.e. actual unscaled (x,y,z) = xs*A + ys*B + zs*C,
where (A,B,C) are the non-orthogonal vectors of the simulation box edges, as discussed in Section 6.12.
Use xu, yu, zu if you want the coordinates "unwrapped" by the image flags for each atom. Unwrapped means
that if the atom has passed thru a periodic boundary one or more times, the value is printed for what the
coordinate would be if it had not been wrapped back into the periodic box. Note that using xu, yu, zu means
that the coordinate values may be far outside the box bounds printed with the snapshot. Using xsu, ysu, zsu is
similar to using xu, yu, zu, except that the unwrapped coordinates are scaled by the box size. Atoms that have
passed through a periodic boundary will have the corresponding coordinate increased or decreased by 1.0.
The image flags can be printed directly using the ix, iy, iz attributes. For periodic dimensions, they specify
which image of the simulation box the atom is considered to be in. An image of 0 means it is inside the box as
defined. A value of 2 means add 2 box lengths to get the true value. A value of -1 means subtract 1 box length
to get the true value. LAMMPS updates these flags as atoms cross periodic boundaries during the simulation.
The mux, muy, muz attributes are specific to dipolar systems defined with an atom style of dipole. They give
the orientation of the atom's point dipole moment. The mu attribute gives the magnitude of the atom's dipole
moment.
The radius and diameter attributes are specific to spherical particles that have a finite size, such as those
defined with an atom style of sphere.
The omegax, omegay, and omegaz attributes are specific to finite-size spherical particles that have an angular
velocity. Only certain atom styles, such as sphere define this quantity.
The angmomx, angmomy, and angmomz attributes are specific to finite-size aspherical particles that have an
angular momentum. Only the ellipsoid atom style defines this quantity.
LAMMPS Users Manual
423

The tqx, tqy, tqz attributes are for finite-size particles that can sustain a rotational torque due to interactions
with other particles.
The c_ID and c_ID[I] attributes allow per-atom vectors or arrays calculated by a compute to be output. The
ID in the attribute should be replaced by the actual ID of the compute that has been defined previously in the
input script. See the compute command for details. There are computes for calculating the per-atom energy,
stress, centro-symmetry parameter, and coordination number of individual atoms.
Note that computes which calculate global or local quantities, as opposed to per-atom quantities, cannot be
output in a dump custom command. Instead, global quantities can be output by the thermo_style custom
command, and local quantities can be output by the dump local command.
If c_ID is used as a attribute, then the per-atom vector calculated by the compute is printed. If c_ID[I] is used,
then I must be in the range from 1-M, which will print the Ith column of the per-atom array with M columns
calculated by the compute. See the discussion above for how I can be specified with a wildcard asterisk to
effectively specify multiple values.
The f_ID and f_ID[I] attributes allow vector or array per-atom quantities calculated by a fix to be output. The
ID in the attribute should be replaced by the actual ID of the fix that has been defined previously in the input
script. The fix ave/atom command is one that calculates per-atom quantities. Since it can time-average
per-atom quantities produced by any compute, fix, or atom-style variable, this allows those time-averaged
results to be written to a dump file.
If f_ID is used as a attribute, then the per-atom vector calculated by the fix is printed. If f_ID[I] is used, then I
must be in the range from 1-M, which will print the Ith column of the per-atom array with M columns
calculated by the fix. See the discussion above for how I can be specified with a wildcard asterisk to
effectively specify multiple values.
The v_name attribute allows per-atom vectors calculated by a variable to be output. The name in the attribute
should be replaced by the actual name of the variable that has been defined previously in the input script. Only
an atom-style variable can be referenced, since it is the only style that generates per-atom values. Variables of
style atom can reference individual atom attributes, per-atom atom attributes, thermodynamic keywords, or
invoke other computes, fixes, or variables when they are evaluated, so this is a very general means of creating
quantities to output to a dump file.
The d_name and i_name attributes allow to output custom per atom floating point or integer properties that
are managed by fix property/atom.
See Section 10 of the manual for information on how to add new compute and fix styles to LAMMPS to
calculate per-atom quantities which could then be output into dump files.
Restrictions:
To write gzipped dump files, you must either compile LAMMPS with the -DLAMMPS_GZIP option or use
the styles from the COMPRESS package - see the Making LAMMPS section of the documentation.
The atom/gz, cfg/gz, custom/gz, and xyz/gz styles are part of the COMPRESS package. They are only enabled
if LAMMPS was built with that package. See the Making LAMMPS section for more info.
The atom/mpiio, cfg/mpiio, custom/mpiio, and xyz/mpiio styles are part of the MPIIO package. They are only
enabled if LAMMPS was built with that package. See the Making LAMMPS section for more info.
LAMMPS Users Manual
424
The xtc style is part of the MISC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info. This is because some machines may not support the low-level XDR
data format that XTC files are written with, which will result in a compile-time error when a low-level include
file is not found. Putting this style in a package makes it easy to exclude from a LAMMPS build for those
machines. However, the MISC package also includes two compatibility header files and associated functions,
which should be a suitable substitute on machines that do not have the appropriate native header files. This
option can be invoked at build time by adding -DLAMMPS_XDR to the CCFLAGS variable in the
appropriate low-level Makefile, e.g. src/MAKE/Makefile.foo. This compatibility mode has been tested
successfully on Cray XT3/XT4/XT5 and IBM BlueGene/L machines and should also work on IBM BG/P, and
Windows XP/Vista/7 machines.
Related commands:
dump h5md, dump image, dump molfile, dump_modify, undump
Default:
The defaults for the image and movie styles are listed on the dump image doc page.
LAMMPS Users Manual
425

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dump custom/vtk command
Syntax:
dump ID group-ID style N file args
ID = user-assigned name for the dump• group-ID = ID of the group of atoms to be dumped• style = custom/vtk• N = dump every this many timesteps• file = name of file to write dump info to• args = list of arguments for a particular style
custom/vtk args = list of atom attributes
possible attributes = id, mol, proc, procp1, type, element, mass,
x, y, z, xs, ys, zs, xu, yu, zu,
xsu, ysu, zsu, ix, iy, iz,
vx, vy, vz, fx, fy, fz,
q, mux, muy, muz, mu,
radius, diameter, omegax, omegay, omegaz,
angmomx, angmomy, angmomz, tqx, tqy, tqz,
c_ID, c_ID[N], f_ID, f_ID[N], v_name
id = atom ID
mol = molecule ID
proc = ID of processor that owns atom
procp1 = ID+1 of processor that owns atom
type = atom type
element = name of atom element, as defined by dump_modify command
mass = atom mass
x,y,z = unscaled atom coordinates
xs,ys,zs = scaled atom coordinates
xu,yu,zu = unwrapped atom coordinates
xsu,ysu,zsu = scaled unwrapped atom coordinates
ix,iy,iz = box image that the atom is in
vx,vy,vz = atom velocities
fx,fy,fz = forces on atoms
q = atom charge
mux,muy,muz = orientation of dipole moment of atom
mu = magnitude of dipole moment of atom
radius,diameter = radius,diameter of spherical particle
omegax,omegay,omegaz = angular velocity of spherical particle
angmomx,angmomy,angmomz = angular momentum of aspherical particle
tqx,tqy,tqz = torque on finite-size particles
c_ID = per-atom vector calculated by a compute with ID
c_ID[I] = Ith column of per-atom array calculated by a compute with ID, I can include wildcard (see below)
f_ID = per-atom vector calculated by a fix with ID
f_ID[I] = Ith column of per-atom array calculated by a fix with ID, I can include wildcard (see below)
v_name = per-atom vector calculated by an atom-style variable with name
d_name = per-atom floating point vector with name, managed by fix property/atom
i_name = per-atom integer vector with name, managed by fix property/atom
•
Examples:
dump dmpvtk all custom/vtk 100 dump*.myforce.vtk id type vx fx
LAMMPS Users Manual
426

dump dmpvtp flow custom/vtk 100 dump*.%.displace.vtp id type c_myD[1] c_myD[2] c_myD[3] v_ke
The style custom/vtk is similar to the custom style but uses the VTK library to write data to VTK simple
legacy or XML format depending on the filename extension specified. This can be either *.vtk for the legacy
format or *.vtp and *.vtu, respectively, for the XML format; see the VTK homepage for a detailed description
of these formats. Since this naming convention conflicts with the way binary output is usually specified (see
below), dump_modify binary allows to set the binary flag for this dump style explicitly.
Description:
Dump a snapshot of atom quantities to one or more files every N timesteps in a format readable by the VTK
visualization toolkit or other visualization tools that use it, e.g. ParaView. The timesteps on which dump
output is written can also be controlled by a variable; see the dump_modify every command for details.
Only information for atoms in the specified group is dumped. The dump_modify thresh and region commands
can also alter what atoms are included; see details below.
As described below, special characters ("*", "%") in the filename determine the kind of output.
IMPORTANT NOTE: Because periodic boundary conditions are enforced only on timesteps when neighbor
lists are rebuilt, the coordinates of an atom written to a dump file may be slightly outside the simulation box.
IMPORTANT NOTE: Unless the dump_modify sort option is invoked, the lines of atom information written
to dump files will be in an indeterminate order for each snapshot. This is even true when running on a single
processor, if the atom_modify sort option is on, which it is by default. In this case atoms are re-ordered
periodically during a simulation, due to spatial sorting. It is also true when running in parallel, because data
for a single snapshot is collected from multiple processors, each of which owns a subset of the atoms.
For the custom/vtk style, sorting is off by default. See the dump_modify doc page for details.
The dimensions of the simulation box are written to a separate file for each snapshot (either in legacy VTK or
XML format depending on the format of the main dump file) with the suffix _boundingBox appended to the
given dump filename.
For an orthogonal simulation box this information is saved as a rectilinear grid (legacy .vtk or .vtr XML
format).
Triclinic simulation boxes (non-orthogonal) are saved as hexahedrons in either legacy .vtk or .vtu XML
format.
Style custom/vtk allows you to specify a list of atom attributes to be written to the dump file for each atom.
Possible attributes are listed above. In contrast to the custom style, the attributes are rearranged to ensure
correct ordering of vector components (except for computes and fixes - these have to be given in the right
order) and duplicate entries are removed.
You cannot specify a quantity that is not defined for a particular simulation - such as q for atom style bond,
since that atom style doesn't assign charges. Dumps occur at the very end of a timestep, so atom attributes will
include effects due to fixes that are applied during the timestep. An explanation of the possible dump
custom/vtk attributes is given below. Since position data is required to write VTK files "x y z" do not have to
be specified explicitly.
LAMMPS Users Manual
427

The VTK format uses a single snapshot of the system per file, thus a wildcard "*" must be included in the
filename, as discussed below. Otherwise the dump files will get overwritten with the new snapshot each time.
Dumps are performed on timesteps that are a multiple of N (including timestep 0) and on the last timestep of a
minimization if the minimization converges. Note that this means a dump will not be performed on the initial
timestep after the dump command is invoked, if the current timestep is not a multiple of N. This behavior can
be changed via the dump_modify first command, which can also be useful if the dump command is invoked
after a minimization ended on an arbitrary timestep. N can be changed between runs by using the
dump_modify every command. The dump_modify every command also allows a variable to be used to
determine the sequence of timesteps on which dump files are written. In this mode a dump on the first
timestep of a run will also not be written unless the dump_modify first command is used.
Dump filenames can contain two wildcard characters. If a "*" character appears in the filename, then one file
per snapshot is written and the "*" character is replaced with the timestep value. For example, tmp.dump*.vtk
becomes tmp.dump0.vtk, tmp.dump10000.vtk, tmp.dump20000.vtk, etc. Note that the dump_modify pad
command can be used to insure all timestep numbers are the same length (e.g. 00010), which can make it
easier to read a series of dump files in order with some post-processing tools.
If a "%" character appears in the filename, then each of P processors writes a portion of the dump file, and the
"%" character is replaced with the processor ID from 0 to P-1 preceded by an underscore character. For
example, tmp.dump%.vtp becomes tmp.dump_0.vtp, tmp.dump_1.vtp, ... tmp.dump_P-1.vtp, etc. This creates
smaller files and can be a fast mode of output on parallel machines that support parallel I/O for output.
By default, P = the number of processors meaning one file per processor, but P can be set to a smaller value
via the nfile or fileper keywords of the dump_modify command. These options can be the most efficient way
of writing out dump files when running on large numbers of processors.
For the legacy VTK format "%" is ignored and P = 1, i.e., only processor 0 does write files.
Note that using the "*" and "%" characters together can produce a large number of small dump files!
If dump_modify binary is used, the dump file (or files, if "*" or "%" is also used) is written in binary format. A
binary dump file will be about the same size as a text version, but will typically write out much faster.
This section explains the atom attributes that can be specified as part of the custom/vtk style.
The id, mol, proc, procp1, type, element, mass, vx, vy, vz, fx, fy, fz, q attributes are self-explanatory.
Id is the atom ID. Mol is the molecule ID, included in the data file for molecular systems. Proc is the ID of the
processor (0 to Nprocs-1) that currently owns the atom. Procp1 is the proc ID+1, which can be convenient in
place of a type attribute (1 to Ntypes) for coloring atoms in a visualization program. Type is the atom type (1
to Ntypes). Element is typically the chemical name of an element, which you must assign to each type via the
dump_modify element command. More generally, it can be any string you wish to associated with an atom
type. Mass is the atom mass. Vx, vy, vz, fx, fy, fz, and q are components of atom velocity and force and atomic
charge.
There are several options for outputting atom coordinates. The x, y, z attributes write atom coordinates
"unscaled", in the appropriate distance units (Angstroms, sigma, etc). Use xs, ys, zs if you want the
coordinates "scaled" to the box size, so that each value is 0.0 to 1.0. If the simulation box is triclinic (tilted),
then all atom coords will still be between 0.0 and 1.0. I.e. actual unscaled (x,y,z) = xs*A + ys*B + zs*C,
LAMMPS Users Manual
428
where (A,B,C) are the non-orthogonal vectors of the simulation box edges, as discussed in Section 6.12.
Use xu, yu, zu if you want the coordinates "unwrapped" by the image flags for each atom. Unwrapped means
that if the atom has passed thru a periodic boundary one or more times, the value is printed for what the
coordinate would be if it had not been wrapped back into the periodic box. Note that using xu, yu, zu means
that the coordinate values may be far outside the box bounds printed with the snapshot. Using xsu, ysu, zsu is
similar to using xu, yu, zu, except that the unwrapped coordinates are scaled by the box size. Atoms that have
passed through a periodic boundary will have the corresponding coordinate increased or decreased by 1.0.
The image flags can be printed directly using the ix, iy, iz attributes. For periodic dimensions, they specify
which image of the simulation box the atom is considered to be in. An image of 0 means it is inside the box as
defined. A value of 2 means add 2 box lengths to get the true value. A value of -1 means subtract 1 box length
to get the true value. LAMMPS updates these flags as atoms cross periodic boundaries during the simulation.
The mux, muy, muz attributes are specific to dipolar systems defined with an atom style of dipole. They give
the orientation of the atom's point dipole moment. The mu attribute gives the magnitude of the atom's dipole
moment.
The radius and diameter attributes are specific to spherical particles that have a finite size, such as those
defined with an atom style of sphere.
The omegax, omegay, and omegaz attributes are specific to finite-size spherical particles that have an angular
velocity. Only certain atom styles, such as sphere define this quantity.
The angmomx, angmomy, and angmomz attributes are specific to finite-size aspherical particles that have an
angular momentum. Only the ellipsoid atom style defines this quantity.
The tqx, tqy, tqz attributes are for finite-size particles that can sustain a rotational torque due to interactions
with other particles.
The c_ID and c_ID[I] attributes allow per-atom vectors or arrays calculated by a compute to be output. The
ID in the attribute should be replaced by the actual ID of the compute that has been defined previously in the
input script. See the compute command for details. There are computes for calculating the per-atom energy,
stress, centro-symmetry parameter, and coordination number of individual atoms.
Note that computes which calculate global or local quantities, as opposed to per-atom quantities, cannot be
output in a dump custom/vtk command. Instead, global quantities can be output by the thermo_style custom
command, and local quantities can be output by the dump local command.
If c_ID is used as a attribute, then the per-atom vector calculated by the compute is printed. If c_ID[I] is used,
then I must be in the range from 1-M, which will print the Ith column of the per-atom array with M columns
calculated by the compute. See the discussion above for how I can be specified with a wildcard asterisk to
effectively specify multiple values.
The f_ID and f_ID[I] attributes allow vector or array per-atom quantities calculated by a fix to be output. The
ID in the attribute should be replaced by the actual ID of the fix that has been defined previously in the input
script. The fix ave/atom command is one that calculates per-atom quantities. Since it can time-average
per-atom quantities produced by any compute, fix, or atom-style variable, this allows those time-averaged
results to be written to a dump file.
LAMMPS Users Manual
429

If f_ID is used as a attribute, then the per-atom vector calculated by the fix is printed. If f_ID[I] is used, then I
must be in the range from 1-M, which will print the Ith column of the per-atom array with M columns
calculated by the fix. See the discussion above for how I can be specified with a wildcard asterisk to
effectively specify multiple values.
The v_name attribute allows per-atom vectors calculated by a variable to be output. The name in the attribute
should be replaced by the actual name of the variable that has been defined previously in the input script. Only
an atom-style variable can be referenced, since it is the only style that generates per-atom values. Variables of
style atom can reference individual atom attributes, per-atom atom attributes, thermodynamic keywords, or
invoke other computes, fixes, or variables when they are evaluated, so this is a very general means of creating
quantities to output to a dump file.
The d_name and i_name attributes allow to output custom per atom floating point or integer properties that
are managed by fix property/atom.
See Section 10 of the manual for information on how to add new compute and fix styles to LAMMPS to
calculate per-atom quantities which could then be output into dump files.
Restrictions:
The custom/vtk style does not support writing of gzipped dump files.
The custom/vtk dump style is part of the USER-VTK package. It is only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info.
To use this dump style, you also must link to the VTK library. See the info in lib/vtk/README and insure the
Makefile.lammps file in that directory is appropriate for your machine.
The custom/vtk dump style neither supports buffering nor custom format strings.
Related commands:
dump, dump image, dump_modify, undump
Default:
By default, files are written in ASCII format. If the file extension is not one of .vtk, .vtp or .vtu, the legacy
VTK file format is used.
LAMMPS Users Manual
430

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dump h5md command
Syntax:
dump ID group-ID h5md N file.h5 args
ID = user-assigned name for the dump• group-ID = ID of the group of atoms to be imaged• h5md = style of dump command (other styles atom or cfg or dcd or xtc or xyz or local or custom are
discussed on the dump doc page)
•
N = dump every this many timesteps• file.h5 = name of file to write to• args = list of data elements to dump, with their dump "subintervals". At least one element must be
given and image may only be present if position is specified first.
position options
image
velocity options
force options
species options
file_from ID: do not open a new file, re-use the already opened file from dump ID
box value = yes or no
create_group value = yes or no
author value = quoted string
For the elements position, velocity, force and species, one may specify a sub-interval to write the data
only every N_element iterations of the dump (i.e. every N*N_element time steps). This is specified by
the option
every N_element
that follows directly the element declaration.
•
Examples:
dump h5md1 all h5md 100 dump_h5md.h5 position image
dump h5md1 all h5md 100 dump_h5md.h5 position velocity every 10
dump h5md1 all h5md 100 dump_h5md.h5 velocity author "John Doe"
Description:
Dump a snapshot of atom coordinates every N timesteps in the HDF5 based H5MD file format (de Buyl).
HDF5 files are binary, portable and self-describing. This dump style will write only one file, on the root node.
Several dumps may write to the same file, by using file_from and referring to a previously defined dump.
Several groups may also be stored within the same file by defining several dumps. A dump that refers (via
file_from) to an already open dump ID and that concerns another particle group must specify create_group
yes.
Each data element is written every N*N_element steps. For image, no subinterval is needed as it must be
present at the same interval as position. image must be given after position in any case. The box information
LAMMPS Users Manual
431

(edges in each dimension) is stored at the same interval than the position element, if present. Else it is stored
every N steps.
NOTE: Because periodic boundary conditions are enforced only on timesteps when neighbor lists are rebuilt,
the coordinates of an atom written to a dump file may be slightly outside the simulation box.
Use from write_dump:
It is possible to use this dump style with the write_dump command. In this case, the subintervals must not be
set at all. The write_dump command can be used either to create a new file or to add current data to an
existing dump file by using the file_from keyword.
Typically, the species data is fixed. The following two commands store the position data every 100 timesteps,
with the image data, and store once the species data in the same file.
dump h5md1 all h5md 100 dump.h5 position image
write_dump all h5md dump.h5 file_from h5md1 species
Restrictions:
The number of atoms per snapshot cannot change with the h5md style. The position data is stored wrapped
(box boundaries not enforced, see note above). Only orthogonal domains are currently supported. This is a
limitation of the present dump h5md command and not of H5MD itself.
The h5md dump style is part of the USER-H5MD package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info. It also requires (i) building the ch5md library
provided with LAMMPS (See the Making LAMMPS section for more info.) and (ii) having the HDF5 library
installed (C bindings are sufficient) on your system. The library ch5md is compiled with the h5cc wrapper
provided by the HDF5 library.
Related commands:
dump, dump_modify, undump
(de Buyl) de Buyl, Colberg and Hofling, H5MD: A structured, efficient, and portable file format for
molecular data, Comp. Phys. Comm. 185(6), 1546-1553 (2014) - [arXiv:1308.6382].
LAMMPS Users Manual
432

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dump image command
dump movie command
Syntax:
dump ID group-ID style N file color diameter keyword value ...
ID = user-assigned name for the dump• group-ID = ID of the group of atoms to be imaged• style = image or movie = style of dump command (other styles atom or cfg or dcd or xtc or xyz or
local or custom are discussed on the dump doc page)
•
N = dump every this many timesteps• file = name of file to write image to• color = atom attribute that determines color of each atom• diameter = atom attribute that determines size of each atom• zero or more keyword/value pairs may be appended• keyword = atom or adiam or bond or line or tri or body or fix or size or view or center or up or zoom
or persp or box or axes or subbox or shiny or ssao
atom = yes/no = do or do not draw atoms
adiam size = numeric value for atom diameter (distance units)
bond values = color width = color and width of bonds
color = atom or type or none
width = number or atom or type or none
number = numeric value for bond width (distance units)
line = color width
color = type
width = numeric value for line width (distance units)
tri = color tflag width
color = type
tflag = 1 for just triangle, 2 for just tri edges, 3 for both
width = numeric value for tringle edge width (distance units)
body = color bflag1 bflag2
color = type
bflag1,bflag2 = 2 numeric flags to affect how bodies are drawn
fix = fixID color fflag1 fflag2
fixID = ID of fix that generates objects to dray
color = type
fflag1,fflag2 = 2 numeric flags to affect how fix objects are drawn
size values = width height = size of images
width = width of image in # of pixels
height = height of image in # of pixels
view values = theta phi = view of simulation box
theta = view angle from +z axis (degrees)
phi = azimuthal view angle (degrees)
theta or phi can be a variable (see below)
center values = flag Cx Cy Cz = center point of image
flag = "s" for static, "d" for dynamic
Cx,Cy,Cz = center point of image as fraction of box dimension (0.5 = center of box)
Cx,Cy,Cz can be variables (see below)
up values = Ux Uy Uz = direction that is "up" in image
Ux,Uy,Uz = components of up vector
Ux,Uy,Uz can be variables (see below)
•
LAMMPS Users Manual
433
zoom value = zfactor = size that simulation box appears in image
zfactor = scale image size by factor > 1 to enlarge, factor <1 to shrink
zfactor can be a variable (see below)
persp value = pfactor = amount of "perspective" in image
pfactor = amount of perspective (0 = none, <1 = some, > 1 = highly skewed)
pfactor can be a variable (see below)
box values = yes/no diam = draw outline of simulation box
yes/no = do or do not draw simulation box lines
diam = diameter of box lines as fraction of shortest box length
axes values = yes/no length diam = draw xyz axes
yes/no = do or do not draw xyz axes lines next to simulation box
length = length of axes lines as fraction of respective box lengths
diam = diameter of axes lines as fraction of shortest box length
subbox values = yes/no diam = draw outline of processor sub-domains
yes/no = do or do not draw sub-domain lines
diam = diameter of sub-domain lines as fraction of shortest box length
shiny value = sfactor = shinyness of spheres and cylinders
sfactor = shinyness of spheres and cylinders from 0.0 to 1.0
ssao value = yes/no seed dfactor = SSAO depth shading
yes/no = turn depth shading on/off
seed = random # seed (positive integer)
dfactor = strength of shading from 0.0 to 1.0
Examples:
dump d0 all image 100 dump.*.jpg type type
dump d1 mobile image 500 snap.*.png element element ssao yes 4539 0.6
dump d2 all image 200 img-*.ppm type type zoom 2.5 adiam 1.5 size 1280 720
dump m0 all movie 1000 movie.mpg type type size 640 480
dump m1 all movie 1000 movie.avi type type size 640 480
dump m2 all movie 100 movie.m4v type type zoom 1.8 adiam v_value size 1280 720
Description:
Dump a high-quality rendered image of the atom configuration every N timesteps and save the images either
as a sequence of JPEG or PNG or PPM files, or as a single movie file. The options for this command as well
as the dump_modify command control what is included in the image or movie and how it appears. A series of
such images can easily be manually converted into an animated movie of your simulation or the process can
be automated without writing the intermediate files using the dump movie style; see further details below.
Other dump styles store snapshots of numerical data associated with atoms in various formats, as discussed on
the dump doc page.
Note that a set of images or a movie can be made after a simulation has been run, using the rerun command to
read snapshots from an existing dump file, and using these dump commands in the rerun script to generate the
images/movie.
Here are two sample images, rendered as 1024x1024 JPEG files. Click to see the full-size images:
LAMMPS Users Manual
434
Only atoms in the specified group are rendered in the image. The dump_modify region and thresh commands
can also alter what atoms are included in the image.
The filename suffix determines whether a JPEG, PNG, or PPM file is created with the image dump style. If
the suffix is ".jpg" or ".jpeg", then a JPEG format file is created, if the suffix is ".png", then a PNG format is
created, else a PPM (aka NETPBM) format file is created. The JPEG and PNG files are binary; PPM has a
text mode header followed by binary data. JPEG images have lossy compression; PNG has lossless
compression; and PPM files are uncompressed but can be compressed with gzip, if LAMMPS has been
compiled with -DLAMMPS_GZIP and a ".gz" suffix is used.
Similarly, the format of the resulting movie is chosen with the movie dump style. This is handled by the
underlying FFmpeg converter and thus details have to be looked up in the FFmpeg documentation. Typical
examples are: .avi, .mpg, .m4v, .mp4, .mkv, .flv, .mov, .gif Additional settings of the movie compression like
bitrate and framerate can be set using the dump_modify command.
To write out JPEG and PNG format files, you must build LAMMPS with support for the corresponding JPEG
or PNG library. To convert images into movies, LAMMPS has to be compiled with the
-DLAMMPS_FFMPEG flag. See this section of the manual for instructions on how to do this.
NOTE: Because periodic boundary conditions are enforced only on timesteps when neighbor lists are rebuilt,
the coordinates of an atom in the image may be slightly outside the simulation box.
Dumps are performed on timesteps that are a multiple of N (including timestep 0) and on the last timestep of a
minimization if the minimization converges. Note that this means a dump will not be performed on the initial
timestep after the dump command is invoked, if the current timestep is not a multiple of N. This behavior can
be changed via the dump_modify first command, which can be useful if the dump command is invoked after a
minimization ended on an arbitrary timestep. N can be changed between runs by using the dump_modify
every command.
Dump image filenames must contain a wildcard character "*", so that one image file per snapshot is written.
The "*" character is replaced with the timestep value. For example, tmp.dump.*.jpg becomes tmp.dump.0.jpg,
tmp.dump.10000.jpg, tmp.dump.20000.jpg, etc. Note that the dump_modify pad command can be used to
insure all timestep numbers are the same length (e.g. 00010), which can make it easier to convert a series of
images into a movie in the correct ordering.
Dump movie filenames on the other hand, must not have any wildcard character since only one file combining
all images into a single movie will be written by the movie encoder.
LAMMPS Users Manual
435

The color and diameter settings determine the color and size of atoms rendered in the image. They can be any
atom attribute defined for the dump custom command, including type and element. This includes per-atom
quantities calculated by a compute, fix, or variable, which are prefixed by "c_", "f_", or "v_" respectively.
Note that the diameter setting can be overridden with a numeric value applied to all atoms by the optional
adiam keyword.
If type is specified for the color setting, then the color of each atom is determined by its atom type. By default
the mapping of types to colors is as follows:
type 1 = red• type 2 = green• type 3 = blue• type 4 = yellow• type 5 = aqua• type 6 = cyan•
and repeats itself for types > 6. This mapping can be changed by the dump_modify acolor command.
If type is specified for the diameter setting then the diameter of each atom is determined by its atom type. By
default all types have diameter 1.0. This mapping can be changed by the dump_modify adiam command.
If element is specified for the color and/or diameter setting, then the color and/or diameter of each atom is
determined by which element it is, which in turn is specified by the element-to-type mapping specified by the
"dump_modify element" command. By default every atom type is C (carbon). Every element has a color and
diameter associated with it, which is the same as the colors and sizes used by the AtomEye visualization
package.
If other atom attributes are used for the color or diameter settings, they are interpreted in the following way.
If "vx", for example, is used as the color setting, then the color of the atom will depend on the x-component of
its velocity. The association of a per-atom value with a specific color is determined by a "color map", which
can be specified via the dump_modify command. The basic idea is that the atom-attribute will be within a
range of values, and every value within the range is mapped to a specific color. Depending on how the color
map is defined, that mapping can take place via interpolation so that a value of -3.2 is halfway between "red"
and "blue", or discretely so that the value of -3.2 is "orange".
If "vx", for example, is used as the diameter setting, then the atom will be rendered using the x-component of
its velocity as the diameter. If the per-atom value <= 0.0, them the atom will not be drawn. Note that
finite-size spherical particles, as defined by atom_style sphere define a per-particle radius or diameter, which
can be used as the diameter setting.
The various keywords listed above control how the image is rendered. As listed below, all of the keywords
have defaults, most of which you will likely not need to change. The dump modify also has options specific to
the dump image style, particularly for assigning colors to atoms, bonds, and other image features.
The atom keyword allow you to turn off the drawing of all atoms, if the specified value is no. Note that this
will not turn off the drawing of particles that are represented as lines, triangles, or bodies, as discussed below.
These particles can be drawn separately if the line, tri, or body keywords are used.
LAMMPS Users Manual
436
The adiam keyword allows you to override the diameter setting to set a single numeric size. All atoms will be
drawn with that diameter, e.g. 1.5, which is in whatever distance units the input script defines, e.g. Angstroms.
The bond keyword allows to you to alter how bonds are drawn. A bond is only drawn if both atoms in the
bond are being drawn due to being in the specified group and due to other selection criteria (e.g. region,
threshold settings of the dump_modify command). By default, bonds are drawn if they are defined in the input
data file as read by the read_data command. Using none for both the bond color and width value will turn off
the drawing of all bonds.
If atom is specified for the bond color value, then each bond is drawn in 2 halves, with the color of each half
being the color of the atom at that end of the bond.
If type is specified for the color value, then the color of each bond is determined by its bond type. By default
the mapping of bond types to colors is as follows:
type 1 = red• type 2 = green• type 3 = blue• type 4 = yellow• type 5 = aqua• type 6 = cyan•
and repeats itself for bond types > 6. This mapping can be changed by the dump_modify bcolor command.
The bond width value can be a numeric value or atom or type (or none as indicated above).
If a numeric value is specified, then all bonds will be drawn as cylinders with that diameter, e.g. 1.0, which is
in whatever distance units the input script defines, e.g. Angstroms.
If atom is specified for the width value, then each bond will be drawn with a width corresponding to the
minimum diameter of the 2 atoms in the bond.
If type is specified for the width value then the diameter of each bond is determined by its bond type. By
default all types have diameter 0.5. This mapping can be changed by the dump_modify bdiam command.
The line keyword can be used when atom_style line is used to define particles as line segments, and will draw
them as lines. If this keyword is not used, such particles will be drawn as spheres, the same as if they were
regular atoms. The only setting currently allowed for the color value is type, which will color the lines
according to the atom type of the particle. By default the mapping of types to colors is as follows:
type 1 = red• type 2 = green• type 3 = blue• type 4 = yellow• type 5 = aqua• type 6 = cyan•
and repeats itself for types > 6. There is not yet an option to change this via the dump_modify command.
The line width can only be a numeric value, which specifies that all lines will be drawn as cylinders with that
LAMMPS Users Manual
437

diameter, e.g. 1.0, which is in whatever distance units the input script defines, e.g. Angstroms.
The tri keyword can be used when atom_style tri is used to define particles as triangles, and will draw them as
triangles or edges (3 lines) or both, depending on the setting for tflag. If edges are drawn, the width setting
determines the diameters of the line segments. If this keyword is not used, triangle particles will be drawn as
spheres, the same as if they were regular atoms. The only setting currently allowed for the color value is type,
which will color the triangles according to the atom type of the particle. By default the mapping of types to
colors is as follows:
type 1 = red• type 2 = green• type 3 = blue• type 4 = yellow• type 5 = aqua• type 6 = cyan•
and repeats itself for types > 6. There is not yet an option to change this via the dump_modify command.
The body keyword can be used when atom_style body is used to define body particles with internal state (e.g.
sub-particles), and will drawn them in a manner specific to the body style. If this keyword is not used, such
particles will be drawn as spheres, the same as if they were regular atoms.
The body doc page describes the body styles LAMMPS currently supports, and provides more details as to the
kind of body particles they represent and how they are drawn by this dump image command. For all the body
styles, individual atoms can be either a body particle or a usual point (non-body) particle. Non-body particles
will be drawn the same way they would be as a regular atom. The bflag1 and bflag2 settings are numerical
values which are passed to the body style to affect how the drawing of a body particle is done. See the body
doc page for a description of what these parameters mean for each body style.
The only setting currently allowed for the color value is type, which will color the body particles according to
the atom type of the particle. By default the mapping of types to colors is as follows:
type 1 = red• type 2 = green• type 3 = blue• type 4 = yellow• type 5 = aqua• type 6 = cyan•
and repeats itself for types > 6. There is not yet an option to change this via the dump_modify command.
The fix keyword can be used with a fix that produces objects to be drawn. An example is the fix surface/global
command which can draw lines or triangles for 2d/3d simulations.
NOTE: Aug 2016 - The fix surface/global command is not yet added to LAMMPS.
The fflag1 and fflag2 settings are numerical values which are passed to the fix to affect how the drawing of its
objects is done. See the individual fix doc page for a description of what these parameters mean for a
particular fix.
LAMMPS Users Manual
438

The only setting currently allowed for the color value is type, which will color the fix objects according to
their type. By default the mapping of types to colors is as follows:
type 1 = red• type 2 = green• type 3 = blue• type 4 = yellow• type 5 = aqua• type 6 = cyan•
and repeats itself for types > 6. There is not yet an option to change this via the dump_modify command.
The size keyword sets the width and height of the created images, i.e. the number of pixels in each direction.
The view, center, up, zoom, and persp values determine how 3d simulation space is mapped to the 2d plane of
the image. Basically they control how the simulation box appears in the image.
All of the view, center, up, zoom, and persp values can be specified as numeric quantities, whose meaning is
explained below. Any of them can also be specified as an equal-style variable, by using v_name as the value,
where "name" is the variable name. In this case the variable will be evaluated on the timestep each image is
created to create a new value. If the equal-style variable is time-dependent, this is a means of changing the
way the simulation box appears from image to image, effectively doing a pan or fly-by view of your
simulation.
The view keyword determines the viewpoint from which the simulation box is viewed, looking towards the
center point. The theta value is the vertical angle from the +z axis, and must be an angle from 0 to 180
degrees. The phi value is an azimuthal angle around the z axis and can be positive or negative. A value of 0.0
is a view along the +x axis, towards the center point. If theta or phi are specified via variables, then the
variable values should be in degrees.
The center keyword determines the point in simulation space that will be at the center of the image. Cx, Cy,
and Cz are specified as fractions of the box dimensions, so that (0.5,0.5,0.5) is the center of the simulation
box. These values do not have to be between 0.0 and 1.0, if you want the simulation box to be offset from the
center of the image. Note, however, that if you choose strange values for Cx, Cy, or Cz you may get a blank
image. Internally, Cx, Cy, and Cz are converted into a point in simulation space. If flag is set to "s" for static,
then this conversion is done once, at the time the dump command is issued. If flag is set to "d" for dynamic
then the conversion is performed every time a new image is created. If the box size or shape is changing, this
will adjust the center point in simulation space.
The up keyword determines what direction in simulation space will be "up" in the image. Internally it is stored
as a vector that is in the plane perpendicular to the view vector implied by the theta and pni values, and which
is also in the plane defined by the view vector and user-specified up vector. Thus this internal vector is
computed from the user-specified up vector as
up_internal = view cross (up cross view)
This means the only restriction on the specified up vector is that it cannot be parallel to the view vector,
implied by the theta and phi values.
The zoom keyword scales the size of the simulation box as it appears in the image. The default zfactor value
of 1 should display an image mostly filled by the atoms in the simulation box. A zfactor > 1 will make the
LAMMPS Users Manual
439

simulation box larger; a zfactor < 1 will make it smaller. Zfactor must be a value > 0.0.
The persp keyword determines how much depth perspective is present in the image. Depth perspective makes
lines that are parallel in simulation space appear non-parallel in the image. A pfactor value of 0.0 means that
parallel lines will meet at infinity (1.0/pfactor), which is an orthographic rendering with no perspective. A
pfactor value between 0.0 and 1.0 will introduce more perspective. A pfactor value > 1 will create a highly
skewed image with a large amount of perspective.
NOTE: The persp keyword is not yet supported as an option.
The box keyword determines if and how the simulation box boundaries are rendered as thin cylinders in the
image. If no is set, then the box boundaries are not drawn and the diam setting is ignored. If yes is set, the 12
edges of the box are drawn, with a diameter that is a fraction of the shortest box length in x,y,z (for 3d) or x,y
(for 2d). The color of the box boundaries can be set with the dump_modify boxcolor command.
The axes keyword determines if and how the coordinate axes are rendered as thin cylinders in the image. If no
is set, then the axes are not drawn and the length and diam settings are ignored. If yes is set, 3 thin cylinders
are drawn to represent the x,y,z axes in colors red,green,blue. The origin of these cylinders will be offset from
the lower left corner of the box by 10%. The length setting determines how long the cylinders will be as a
fraction of the respective box lengths. The diam setting determines their thickness as a fraction of the shortest
box length in x,y,z (for 3d) or x,y (for 2d).
The subbox keyword determines if and how processor sub-domain boundaries are rendered as thin cylinders
in the image. If no is set (default), then the sub-domain boundaries are not drawn and the diam setting is
ignored. If yes is set, the 12 edges of each processor sub-domain are drawn, with a diameter that is a fraction
of the shortest box length in x,y,z (for 3d) or x,y (for 2d). The color of the sub-domain boundaries can be set
with the dump_modify boxcolor command.
The shiny keyword determines how shiny the objects rendered in the image will appear. The sfactor value
must be a value 0.0 <= sfactor <= 1.0, where sfactor = 1 is a highly reflective surface and sfactor = 0 is a
rough non-shiny surface.
The ssao keyword turns on/off a screen space ambient occlusion (SSAO) model for depth shading. If yes is
set, then atoms further away from the viewer are darkened via a randomized process, which is perceived as
depth. The calculation of this effect can increase the cost of computing the image by roughly 2x. The strength
of the effect can be scaled by the dfactor parameter. If no is set, no depth shading is performed.
A series of JPEG, PNG, or PPM images can be converted into a movie file and then played as a movie using
commonly available tools. Using dump style movie automates this step and avoids the intermediate step of
writing (many) image snapshot file. But LAMMPS has to be compiled with -DLAMMPS_FFMPEG and an
FFmpeg executable have to be installed.
To manually convert JPEG, PNG or PPM files into an animated GIF or MPEG or other movie file you can
use:
a) Use the ImageMagick convert program.
% convert *.jpg foo.gif
% convert -loop 1 *.ppm foo.mpg
•
LAMMPS Users Manual
440

Animated GIF files from ImageMagick are unoptimized. You can use a program like gifsicle to
optimize and massively shrink them. MPEG files created by ImageMagick are in MPEG-1 format
with rather inefficient compression and low quality.
b) Use QuickTime.
Select "Open Image Sequence" under the File menu Load the images into QuickTime to animate them
Select "Export" under the File menu Save the movie as a QuickTime movie (*.mov) or in another
format. QuickTime can generate very high quality and efficiently compressed movie files. Some of
the supported formats require to buy a license and some are not readable on all platforms until
specific runtime libraries are installed.
•
c) Use FFmpeg
FFmpeg is a command line tool that is available on many platforms and allows extremely flexible
encoding and decoding of movies.
cat snap.*.jpg | ffmpeg -y -f image2pipe -c:v mjpeg -i - -b:v 2000k movie.m4v
cat snap.*.ppm | ffmpeg -y -f image2pipe -c:v ppm -i - -b:v 2400k movie.avi
Frontends for FFmpeg exist for multiple platforms. For more information see the FFmpeg homepage
•
Play the movie:
a) Use your browser to view an animated GIF movie.
Select "Open File" under the File menu Load the animated GIF file
•
b) Use the freely available mplayer or ffplay tool to view a movie. Both are available for multiple
OSes and support a large variety of file formats and decoders.
% mplayer foo.mpg
% ffplay bar.avi
•
c) Use the Pizza.py animate tool, which works directly on a series of image files.
a = animate("foo*.jpg")
•
d) QuickTime and other Windows- or MacOS-based media players can obviously play movie files
directly. Similarly for corresponding tools bundled with Linux desktop environments. However, due
to licensing issues with some file formats, the formats may require installing additional libraries,
purchasing a license, or may not be supported.
•
See Section 10 of the manual for information on how to add new compute and fix styles to LAMMPS to
calculate per-atom quantities which could then be output into dump files.
Restrictions:
To write JPEG images, you must use the -DLAMMPS_JPEG switch when building LAMMPS and link with a
JPEG library. To write PNG images, you must use the -DLAMMPS_PNG switch when building LAMMPS
and link with a PNG library.
To write movie dumps, you must use the -DLAMMPS_FFMPEG switch when building LAMMPS and have
the FFmpeg executable available on the machine where LAMMPS is being run. Typically it's name is
lowercase, i.e. ffmpeg.
LAMMPS Users Manual
441
See the Making LAMMPS section of the documentation for details on how to compile with optional switches.
Note that since FFmpeg is run as an external program via a pipe, LAMMPS has limited control over its
execution and no knowledge about errors and warnings printed by it. Those warnings and error messages will
be printed to the screen only. Due to the way image data is communicated to FFmpeg, it will often print the
message
pipe:: Input/output error
which can be safely ignored. Other warnings and errors have to be addressed according to the FFmpeg
documentation. One known issue is that certain movie file formats (e.g. MPEG level 1 and 2 format streams)
have video bandwidth limits that can be crossed when rendering too large of image sizes. Typical warnings
look like this:
[mpeg @ 0x98b5e0] packet too large, ignoring buffer limits to mux it
[mpeg @ 0x98b5e0] buffer underflow st=0 bufi=281407 size=285018
[mpeg @ 0x98b5e0] buffer underflow st=0 bufi=283448 size=285018
In this case it is recommended to either reduce the size of the image or encode in a different format that is also
supported by your copy of FFmpeg, and which does not have this limitation (e.g. .avi, .mkv, mp4).
Related commands:
dump, dump_modify, undump
Default:
The defaults for the keywords are as follows:
adiam = not specified (use diameter setting)• atom = yes• bond = none none (if no bonds in system)• bond = atom 0.5 (if bonds in system)• size = 512 512• view = 60 30 (for 3d)• view = 0 0 (for 2d)• center = s 0.5 0.5 0.5• up = 0 0 1 (for 3d)• up = 0 1 0 (for 2d)• zoom = 1.0• persp = 0.0• box = yes 0.02• axes = no 0.0 0.0• subbox no 0.0• shiny = 1.0• ssao = no•
LAMMPS Users Manual
442

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dump_modify command
Syntax:
dump_modify dump-ID keyword values ...
dump-ID = ID of dump to modify• one or more keyword/value pairs may be appended• these keywords apply to various dump styles• keyword = append or buffer or element or every or fileper or first or flush or format or image or label
or nfile or pad or precision or region or scale or sort or thresh or unwrap
append arg = yes or no
buffer arg = yes or no
element args = E1 E2 ... EN, where N = # of atom types
E1,...,EN = element name, e.g. C or Fe or Ga
every arg = N
N = dump every this many timesteps
N can be a variable (see below)
fileper arg = Np
Np = write one file for every this many processors
first arg = yes or no
format args = line string, int string, float string, M string, or none
string = C-style format string
M = integer from 1 to N, where N = # of per-atom quantities being output
flush arg = yes or no
image arg = yes or no
label arg = string
string = character string (e.g. BONDS) to use in header of dump local file
nfile arg = Nf
Nf = write this many files, one from each of Nf processors
pad arg = Nchar = # of characters to convert timestep to
pbc arg = yes or no = remap atoms via periodic boundary conditions
precision arg = power-of-10 value from 10 to 1000000
region arg = region-ID or "none"
scale arg = yes or no
sfactor arg = coordinate scaling factor (> 0.0)
tfactor arg = time scaling factor (> 0.0)
sort arg = off or id or N or -N
off = no sorting of per-atom lines within a snapshot
id = sort per-atom lines by atom ID
N = sort per-atom lines in ascending order by the Nth column
-N = sort per-atom lines in descending order by the Nth column
thresh args = attribute operator value
attribute = same attributes (x,fy,etotal,sxx,etc) used by dump custom style
operator = "" or ">=" or "==" or "!=" or "|^"
value = numeric value to compare to, or LAST
these 3 args can be replaced by the word "none" to turn off thresholding
unwrap arg = yes or no
•
these keywords apply only to the image and movie styles• keyword = acolor or adiam or amap or backcolor or bcolor or bdiam or boxcolor or color or bitrate
or framerate
acolor args = type color
type = atom type or range of types (see below)
•
LAMMPS Users Manual
443
color = name of color or color1/color2/...
adiam args = type diam
type = atom type or range of types (see below)
diam = diameter of atoms of that type (distance units)
amap args = lo hi style delta N entry1 entry2 ... entryN
lo = number or min = lower bound of range of color map
hi = number or max = upper bound of range of color map
style = 2 letters = "c" or "d" or "s" plus "a" or "f"
"c" for continuous
"d" for discrete
"s" for sequential
"a" for absolute
"f" for fractional
delta = binsize (only used for style "s", otherwise ignored)
binsize = range is divided into bins of this width
N = # of subsequent entries
entry = value color (for continuous style)
value = number or min or max = single value within range
color = name of color used for that value
entry = lo hi color (for discrete style)
lo/hi = number or min or max = lower/upper bound of subset of range
color = name of color used for that subset of values
entry = color (for sequential style)
color = name of color used for a bin of values
backcolor arg = color
color = name of color for background
bcolor args = type color
type = bond type or range of types (see below)
color = name of color or color1/color2/...
bdiam args = type diam
type = bond type or range of types (see below)
diam = diameter of bonds of that type (distance units)
boxcolor arg = color
color = name of color for simulation box lines and processor sub-domain lines
color args = name R G B
name = name of color
R,G,B = red/green/blue numeric values from 0.0 to 1.0
bitrate arg = rate
rate = target bitrate for movie in kbps
framerate arg = fps
fps = frames per second for movie
Examples:
dump_modify 1 format line "%d %d %20.15g %g %g" scale yes
dump_modify 1 format float %20.15g scale yes
dump_modify myDump image yes scale no flush yes
dump_modify 1 region mySphere thresh x <0.0 thresh epair >= 3.2
dump_modify xtcdump precision 10000 sfactor 0.1
dump_modify 1 every 1000 nfile 20
dump_modify 1 every v_myVar
dump_modify 1 amap min max cf 0.0 3 min green 0.5 yellow max blue boxcolor red
Description:
Modify the parameters of a previously defined dump command. Not all parameters are relevant to all dump
styles.
LAMMPS Users Manual
444

As explained on the dump doc page, the atom/mpiio, custom/mpiio, and xyz/mpiio dump styles are identical in
command syntax and in the format of the dump files they create, to the corresponding styles without "mpiio",
except the single dump file they produce is written in parallel via the MPI-IO library. Thus if a dump_modify
option below is valid for the atom style, it is also valid for the atom/mpiio style, and similarly for the other
styles which allow for use of MPI-IO.
These keywords apply to various dump styles, including the dump image and dump movie styles. The
description gives details.
The append keyword applies to all dump styles except cfg and xtc and dcd. It also applies only to text output
files, not to binary or gzipped or image/movie files. If specified as yes, then dump snapshots are appended to
the end of an existing dump file. If specified as no, then a new dump file will be created which will overwrite
an existing file with the same name. This keyword can only take effect if the dump_modify command is used
after the dump command, but before the first command that causes dump snapshots to be output, e.g. a run or
minimize command. Once the dump file has been opened, this keyword has no further effect.
The buffer keyword applies only to dump styles atom, cfg, custom, local, and xyz. It also applies only to text
output files, not to binary or gzipped files. If specified as yes, which is the default, then each processor writes
its output into an internal text buffer, which is then sent to the processor(s) which perform file writes, and
written by those processors(s) as one large chunk of text. If specified as no, each processor sends its per-atom
data in binary format to the processor(s) which perform file wirtes, and those processor(s) format and write it
line by line into the output file.
The buffering mode is typically faster since each processor does the relatively expensive task of formatting
the output for its own atoms. However it requires about twice the memory (per processor) for the extra
buffering.
The element keyword applies only to the dump cfg, xyz, and image styles. It associates element names (e.g. H,
C, Fe) with LAMMPS atom types. See the list of element names at the bottom of this page.
In the case of dump cfg, this allows the AtomEye visualization package to read the dump file and render
atoms with the appropriate size and color.
In the case of dump image, the output images will follow the same AtomEye convention. An element name is
specified for each atom type (1 to Ntype) in the simulation. The same element name can be given to multiple
atom types.
In the case of xyz format dumps, there are no restrictions to what label can be used as an element name. Any
whitespace separated text will be accepted.
The every keyword changes the dump frequency originally specified by the dump command to a new value.
The every keyword can be specified in one of two ways. It can be a numeric value in which case it must be >
0. Or it can be an equal-style variable, which should be specified as v_name, where name is the variable
name.
In this case, the variable is evaluated at the beginning of a run to determine the next timestep at which a dump
snapshot will be written out. On that timestep the variable will be evaluated again to determine the next
timestep, etc. Thus the variable should return timestep values. See the stagger() and logfreq() and stride()
math functions for equal-style variables, as examples of useful functions to use in this context. Other similar
LAMMPS Users Manual
445

math functions could easily be added as options for equal-style variables. Also see the next() function, which
allows use of a file-style variable which reads successive values from a file, each time the variable is
evaluated. Used with the every keyword, if the file contains a list of ascending timesteps, you can output
snapshots whenever you wish.
Note that when using the variable option with the every keyword, you need to use the first option if you want
an initial snapshot written to the dump file. The every keyword cannot be used with the dump dcd style.
For example, the following commands will write snapshots at timesteps
0,10,20,30,100,200,300,1000,2000,etc:
variable s equal logfreq(10,3,10)
dump 1 all atom 100 tmp.dump
dump_modify 1 every v_s first yes
The following commands would write snapshots at the timesteps listed in file tmp.times:
variable f file tmp.times
variable s equal next(f)
dump 1 all atom 100 tmp.dump
dump_modify 1 every v_s
NOTE: When using a file-style variable with the every keyword, the file of timesteps must list a first timestep
that is beyond the current timestep (e.g. it cannot be 0). And it must list one or more timesteps beyond the
length of the run you perform. This is because the dump command will generate an error if the next timestep it
reads from the file is not a value greater than the current timestep. Thus if you wanted output on steps
0,15,100 of a 100-timestep run, the file should contain the values 15,100,101 and you should also use the
dump_modify first command. Any final value > 100 could be used in place of 101.
The first keyword determines whether a dump snapshot is written on the very first timestep after the dump
command is invoked. This will always occur if the current timestep is a multiple of N, the frequency specified
in the dump command, including timestep 0. But if this is not the case, a dump snapshot will only be written if
the setting of this keyword is yes. If it is no, which is the default, then it will not be written.
The flush keyword determines whether a flush operation is invoked after a dump snapshot is written to the
dump file. A flush insures the output in that file is current (no buffering by the OS), even if LAMMPS halts
before the simulation completes. Flushes cannot be performed with dump style xtc.
The format keyword can be used to change the default numeric format output by the text-based dump styles:
atom, custom, cfg, and xyz styles, and their MPIIO variants. Only the line or none options can be used with the
atom and xyz styles.
All the specified format strings are C-style formats, e.g. as used by the C/C++ printf() command. The line
keyword takes a single argument which is the format string for an entire line of output for each atom (do not
include a trailing "\n"), with N fields, which you must enclose in quotes if it is more than one field. The int
and float keywords take a single format argument and are applied to all integer or floating-point quantities
output. The setting for M string also takes a single format argument which is used for the Mth value output in
each line, e.g. the 5th column is output in high precision for "format 5 %20.15g".
NOTE: When using the line keyword for the cfg style, the first two fields (atom ID and type) are not actually
written into the CFG file, however you must include formats for them in the format string.
LAMMPS Users Manual
446

The format keyword can be used multiple times. The precedence is that for each value in a line of output, the
M format (if specified) is used, else the int or float setting (if specified) is used, else the line setting (if
specified) for that value is used, else the default setting is used. A setting of none clears all previous settings,
reverting all values to their default format.
NOTE: Atom and molecule IDs are stored internally as 4-byte or 8-byte signed integers, depending on how
LAMMPS was compiled. When specifying the format int option you can use a "%d"-style format identifier in
the format string and LAMMPS will convert this to the corresponding 8-byte form it it is needed when
outputting those values. However, when specifying the line option or format M string option for those values,
you should specify a format string appropriate for an 8-byte signed integer, e.g. one with "%ld", if LAMMPS
was compiled with the -DLAMMPS_BIGBIG option for 8-byte IDs.
NOTE: Any value written to a text-based dump file that is a per-atom quantity calculated by a compute or fix
is stored internally as a floating-point value. If the value is actually an integer and you wish it to appear in the
text dump file as a (large) integer, then you need to use an appropriate format. For example, these commands:
compute 1 all property/local batom1 batom2
dump 1 all local 100 tmp.bonds index c_1[1] c_1[2]
dump_modify 1 format "%d %0.0f %0.0f"
will output the two atom IDs for atoms in each bond as integers. If the dump_modify command were omitted,
they would appear as floating-point values, assuming they were large integers (more than 6 digits). The
"index" keyword should use the "%d" format since it is not generated by a compute or fix, and is stored
internally as an integer.
The fileper keyword is documented below with the nfile keyword.
The image keyword applies only to the dump atom style. If the image value is yes, 3 flags are appended to
each atom's coords which are the absolute box image of the atom in each dimension. For example, an x image
flag of -2 with a normalized coord of 0.5 means the atom is in the center of the box, but has passed thru the
box boundary 2 times and is really 2 box lengths to the left of its current coordinate. Note that for dump style
custom these various values can be printed in the dump file by using the appropriate atom attributes in the
dump command itself.
The label keyword applies only to the dump local style. When it writes local information, such as bond or
angle topology to a dump file, it will use the specified label to format the header. By default this includes 2
lines:
ITEM: NUMBER OF ENTRIES
ITEM: ENTRIES ...
The word "ENTRIES" will be replaced with the string specified, e.g. BONDS or ANGLES.
The nfile or fileper keywords can be used in conjunction with the "%" wildcard character in the specified
dump file name, for all dump styles except the dcd, image, movie, xtc, and xyz styles (for which "%" is not
allowed). As explained on the dump command doc page, the "%" character causes the dump file to be written
in pieces, one piece for each of P processors. By default P = the number of processors the simulation is
running on. The nfile or fileper keyword can be used to set P to a smaller value, which can be more efficient
when running on a large number of processors.
LAMMPS Users Manual
447

The nfile keyword sets P to the specified Nf value. For example, if Nf = 4, and the simulation is running on
100 processors, 4 files will be written, by processors 0,25,50,75. Each will collect information from itself and
the next 24 processors and write it to a dump file.
For the fileper keyword, the specified value of Np means write one file for every Np processors. For example,
if Np = 4, every 4th processor (0,4,8,12,etc) will collect information from itself and the next 3 processors and
write it to a dump file.
The pad keyword only applies when the dump filename is specified with a wildcard "*" character which
becomes the timestep. If pad is 0, which is the default, the timestep is converted into a string of unpadded
length, e.g. 100 or 12000 or 2000000. When pad is specified with Nchar > 0, the string is padded with leading
zeroes so they are all the same length = Nchar. For example, pad 7 would yield 0000100, 0012000, 2000000.
This can be useful so that post-processing programs can easily read the files in ascending timestep order.
The pbc keyword applies to all the dump styles. As explained on the dump doc page, atom coordinates in a
dump file may be slightly outside the simulation box. This is because periodic boundary conditions are
enforced only on timesteps when neighbor lists are rebuilt, which will not typically coincide with the
timesteps dump snapshots are written. If the setting of this keyword is set to yes, then all atoms will be
remapped to the periodic box before the snapshot is written, then restored to their original position. If it is set
to no they will not be. The no setting is the default because it requires no extra computation.
The precision keyword only applies to the dump xtc style. A specified value of N means that coordinates are
stored to 1/N nanometer accuracy, e.g. for N = 1000, the coordinates are written to 1/1000 nanometer
accuracy.
The sfactor and tfactor keywords only apply to the dump xtc style. They allow customization of the unit
conversion factors used when writing to XTC files. By default they are initialized for whatever units style is
being used, to write out coordinates in nanometers and time in picoseconds. I.e. for real units, LAMMPS
defines sfactor = 0.1 and tfactor = 0.001, since the Angstroms and fmsec used by real units are 0.1 nm and
0.001 psec respectively. If you are using a units system with distance and time units far from nm and psec,
you may wish to write XTC files with different units, since the compression algorithm used in XTC files is
most effective when the typical magnitude of position data is between 10.0 and 0.1.
The region keyword only applies to the dump custom, cfg, image, and movie styles. If specified, only atoms in
the region will be written to the dump file or included in the image/movie. Only one region can be applied as a
filter (the last one specified). See the region command for more details. Note that a region can be defined as
the "inside" or "outside" of a geometric shape, and it can be the "union" or "intersection" of a series of simpler
regions.
The scale keyword applies only to the dump atom style. A scale value of yes means atom coords are written in
normalized units from 0.0 to 1.0 in each box dimension. If the simulation box is triclinic (tilted), then all atom
coords will still be between 0.0 and 1.0. A value of no means they are written in absolute distance units (e.g.
Angstroms or sigma).
The sort keyword determines whether lines of per-atom output in a snapshot are sorted or not. A sort value of
off means they will typically be written in indeterminate order, either in serial or parallel. This is the case even
in serial if the atom_modify sort option is turned on, which it is by default, to improve performance. A sort
value of id means sort the output by atom ID. A sort value of N or -N means sort the output by the value in the
Nth column of per-atom info in either ascending or descending order.
LAMMPS Users Manual
448

The dump local style cannot be sorted by atom ID, since there are typically multiple lines of output per atom.
Some dump styles, such as dcd and xtc, require sorting by atom ID to format the output file correctly. If
multiple processors are writing the dump file, via the "%" wildcard in the dump filename, then sorting cannot
be performed.
NOTE: Unless it is required by the dump style, sorting dump file output requires extra overhead in terms of
CPU and communication cost, as well as memory, versus unsorted output.
The thresh keyword only applies to the dump custom, cfg, image, and movie styles. Multiple thresholds can be
specified. Specifying none turns off all threshold criteria. If thresholds are specified, only atoms whose
attributes meet all the threshold criteria are written to the dump file or included in the image. The possible
attributes that can be tested for are the same as those that can be specified in the dump custom command, with
the exception of the element attribute, since it is not a numeric value. Note that a different attributes can be
used than those output by the dump custom command. E.g. you can output the coordinates and stress of atoms
whose energy is above some threshold.
If an atom-style variable is used as the attribute, then it can produce continuous numeric values or effective
Boolean 0/1 values which may be useful for the comparison operator. Boolean values can be generated by
variable formulas that use comparison or Boolean math operators or special functions like gmask() and
rmask() and grmask(). See the variable command doc page for details.
The specified value must be a simple numeric value or the word LAST. If LAST is used, it refers to the value
of the attribute the last time the dump command was invoked to produce a snapshot. This is a way to only
dump atoms whose attribute has changed (or not changed). Three examples follow.
dump_modify ... thresh ix != LAST
This will dump atoms which have crossed the periodic x boundary of the simulation box since the last dump.
(Note that atoms that crossed once and then crossed back between the two dump timesteps would not be
included.)
region foo sphere 10 20 10 15 variable inregion atom rmask(foo) dump_modify ... thresh v_inregion |^ LAST
This will dump atoms which crossed the boundary of the spherical region since the last dump.
variable charge atom "(q > 0.5) || (q < -0.5)" dump_modify ... thresh v_charge |^ LAST
This will dump atoms whose charge has changed from an absolute value less than 1/2 to greater than 1/2 (or
vice versa) since the last dump. E.g. due to reactions and subsequent charge equilibration in a reactive force
field.
The choice of operators listed above are the usual comparison operators. The XOR operation (exclusive or) is
also included as "|^". In this context, XOR means that if either the attribute or value is 0.0 and the other is
non-zero, then the result is "true" and the threshold criterion is met. Otherwise it is not met.
The unwrap keyword only applies to the dump dcd and xtc styles. If set to yes, coordinates will be written
"unwrapped" by the image flags for each atom. Unwrapped means that if the atom has passed thru a periodic
boundary one or more times, the value is printed for what the coordinate would be if it had not been wrapped
back into the periodic box. Note that these coordinates may thus be far outside the box size stored with the
snapshot.
LAMMPS Users Manual
449

These keywords apply only to the dump image and dump movie styles. Any keyword that affects an image,
also affects a movie, since the movie is simply a collection of images. Some of the keywords only affect the
dump movie style. The descriptions give details.
The acolor keyword can be used with the dump image command, when its atom color setting is type, to set
the color that atoms of each type will be drawn in the image.
The specified type should be an integer from 1 to Ntypes = the number of atom types. A wildcard asterisk can
be used in place of or in conjunction with the type argument to specify a range of atom types. This takes the
form "*" or "*n" or "n*" or "m*n". If N = the number of atom types, then an asterisk with no numeric values
means all types from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A trailing asterisk
means all types from n to N (inclusive). A middle asterisk means all types from m to n (inclusive).
The specified color can be a single color which is any of the 140 pre-defined colors (see below) or a color
name defined by the dump_modify color option. Or it can be two or more colors separated by a "/" character,
e.g. red/green/blue. In the former case, that color is assigned to all the specified atom types. In the latter case,
the list of colors are assigned in a round-robin fashion to each of the specified atom types.
The adiam keyword can be used with the dump image command, when its atom diameter setting is type, to set
the size that atoms of each type will be drawn in the image. The specified type should be an integer from 1 to
Ntypes. As with the acolor keyword, a wildcard asterisk can be used as part of the type argument to specify a
range of atomt types. The specified diam is the size in whatever distance units the input script is using, e.g.
Angstroms.
The amap keyword can be used with the dump image command, with its atom keyword, when its atom setting
is an atom-attribute, to setup a color map. The color map is used to assign a specific RGB (red/green/blue)
color value to an individual atom when it is drawn, based on the atom's attribute, which is a numeric value,
e.g. its x-component of velocity if the atom-attribute "vx" was specified.
The basic idea of a color map is that the atom-attribute will be within a range of values, and that range is
associated with a series of colors (e.g. red, blue, green). An atom's specific value (vx = -3.2) can then mapped
to the series of colors (e.g. halfway between red and blue), and a specific color is determined via an
interpolation procedure.
There are many possible options for the color map, enabled by the amap keyword. Here are the details.
The lo and hi settings determine the range of values allowed for the atom attribute. If numeric values are used
for lo and/or hi, then values that are lower/higher than that value are set to the value. I.e. the range is static. If
lo is specified as min or hi as max then the range is dynamic, and the lower and/or upper bound will be
calculated each time an image is drawn, based on the set of atoms being visualized.
The style setting is two letters, such as "ca". The first letter is either "c" for continuous, "d" for discrete, or "s"
for sequential. The second letter is either "a" for absolute, or "f" for fractional.
A continuous color map is one in which the color changes continuously from value to value within the range.
A discrete color map is one in which discrete colors are assigned to sub-ranges of values within the range. A
sequential color map is one in which discrete colors are assigned to a sequence of sub-ranges of values
covering the entire range.
LAMMPS Users Manual
450
An absolute color map is one in which the values to which colors are assigned are specified explicitly as
values within the range. A fractional color map is one in which the values to which colors are assigned are
specified as a fractional portion of the range. For example if the range is from -10.0 to 10.0, and the color red
is to be assigned to atoms with a value of 5.0, then for an absolute color map the number 5.0 would be used.
But for a fractional map, the number 0.75 would be used since 5.0 is 3/4 of the way from -10.0 to 10.0.
The delta setting must be specified for all styles, but is only used for the sequential style; otherwise the value
is ignored. It specifies the bin size to use within the range for assigning consecutive colors to. For example, if
the range is from -10.0 to 10.0 and a delta of 1.0 is used, then 20 colors will be assigned to the range. The first
will be from -10.0 <= color1 < -9.0, then 2nd from -9.0 <= color2 < -8.0, etc.
The N setting is how many entries follow. The format of the entries depends on whether the color map style is
continuous, discrete or sequential. In all cases the color setting can be any of the 140 pre-defined colors (see
below) or a color name defined by the dump_modify color option.
For continuous color maps, each entry has a value and a color. The value is either a number within the range
of values or min or max. The value of the first entry must be min and the value of the last entry must be max.
Any entries in between must have increasing values. Note that numeric values can be specified either as
absolute numbers or as fractions (0.0 to 1.0) of the range, depending on the "a" or "f" in the style setting for
the color map.
Here is how the entries are used to determine the color of an individual atom, given the value X of its atom
attribute. X will fall between 2 of the entry values. The color of the atom is linearly interpolated (in each of
the RGB values) between the 2 colors associated with those entries. For example, if X = -5.0 and the 2
surrounding entries are "red" at -10.0 and "blue" at 0.0, then the atom's color will be halfway between "red"
and "blue", which happens to be "purple".
For discrete color maps, each entry has a lo and hi value and a color. The lo and hi settings are either numbers
within the range of values or lo can be min or hi can be max. The lo and hi settings of the last entry must be
min and max. Other entries can have any lo and hi values and the sub-ranges of different values can overlap.
Note that numeric lo and hi values can be specified either as absolute numbers or as fractions (0.0 to 1.0) of
the range, depending on the "a" or "f" in the style setting for the color map.
Here is how the entries are used to determine the color of an individual atom, given the value X of its atom
attribute. The entries are scanned from first to last. The first time that lo <= X <= hi, X is assigned the color
associated with that entry. You can think of the last entry as assigning a default color (since it will always be
matched by X), and the earlier entries as colors that override the default. Also note that no interpolation of a
color RGB is done. All atoms will be drawn with one of the colors in the list of entries.
For sequential color maps, each entry has only a color. Here is how the entries are used to determine the color
of an individual atom, given the value X of its atom attribute. The range is partitioned into N bins of width
binsize. Thus X will fall in a specific bin from 1 to N, say the Mth bin. If it falls on a boundary between 2
bins, it is considered to be in the higher of the 2 bins. Each bin is assigned a color from the E entries. If E < N,
then the colors are repeated. For example if 2 entries with colors red and green are specified, then the odd
numbered bins will be red and the even bins green. The color of the atom is the color of its bin. Note that the
sequential color map is really a shorthand way of defining a discrete color map without having to specify
where all the bin boundaries are.
Here is an example of using a sequential color map to color all the atoms in individual molecules with a
different color. See the examples/pour/in.pour.2d.molecule input script for an example of how this is used.
LAMMPS Users Manual
451

variable colors string &
"red green blue yellow white &
purple pink orange lime gray"
variable mol atom mol%10
dump 1 all image 250 image.*.jpg v_mol type &
zoom 1.6 adiam 1.5
dump_modify 1 pad 5 amap 0 10 sa 1 10 ${colors}
In this case, 10 colors are defined, and molecule IDs are mapped to one of the colors, even if there are 1000s
of molecules.
The backcolor sets the background color of the images. The color name can be any of the 140 pre-defined
colors (see below) or a color name defined by the dump_modify color option.
The bcolor keyword can be used with the dump image command, with its bond keyword, when its color
setting is type, to set the color that bonds of each type will be drawn in the image.
The specified type should be an integer from 1 to Nbondtypes = the number of bond types. A wildcard
asterisk can be used in place of or in conjunction with the type argument to specify a range of bond types. This
takes the form "*" or "*n" or "n*" or "m*n". If N = the number of bond types, then an asterisk with no
numeric values means all types from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A
trailing asterisk means all types from n to N (inclusive). A middle asterisk means all types from m to n
(inclusive).
The specified color can be a single color which is any of the 140 pre-defined colors (see below) or a color
name defined by the dump_modify color option. Or it can be two or more colors separated by a "/" character,
e.g. red/green/blue. In the former case, that color is assigned to all the specified bond types. In the latter case,
the list of colors are assigned in a round-robin fashion to each of the specified bond types.
The bdiam keyword can be used with the dump image command, with its bond keyword, when its diam
setting is type, to set the diameter that bonds of each type will be drawn in the image. The specified type
should be an integer from 1 to Nbondtypes. As with the bcolor keyword, a wildcard asterisk can be used as
part of the type argument to specify a range of bond types. The specified diam is the size in whatever distance
units you are using, e.g. Angstroms.
The bitrate keyword can be used with the dump movie command to define the size of the resulting movie file
and its quality via setting how many kbits per second are to be used for the movie file. Higher bitrates require
less compression and will result in higher quality movies. The quality is also determined by the compression
format and encoder. The default setting is 2000 kbit/s, which will result in average quality with older
compression formats.
NOTE: Not all movie file formats supported by dump movie allow the bitrate to be set. If not, the setting is
silently ignored.
The boxcolor keyword sets the color of the simulation box drawn around the atoms in each image as well as
the color of processor sub-domain boundaries. See the "dump image box" command for how to specify that a
box be drawn via the box keyword, and the sub-domain boundaries via the subbox keyword. The color name
can be any of the 140 pre-defined colors (see below) or a color name defined by the dump_modify color
option.
LAMMPS Users Manual
452

The color keyword allows definition of a new color name, in addition to the 140-predefined colors (see
below), and associates 3 red/green/blue RGB values with that color name. The color name can then be used
with any other dump_modify keyword that takes a color name as a value. The RGB values should each be
floating point values between 0.0 and 1.0 inclusive.
When a color name is converted to RGB values, the user-defined color names are searched first, then the 140
pre-defined color names. This means you can also use the color keyword to overwrite one of the pre-defined
color names with new RBG values.
The framerate keyword can be used with the dump movie command to define the duration of the resulting
movie file. Movie files written by the dump movie command have a default frame rate of 24 frames per
second and the images generated will be converted at that rate. Thus a sequence of 1000 dump images will
result in a movie of about 42 seconds. To make a movie run longer you can either generate images more
frequently or lower the frame rate. To speed a movie up, you can do the inverse. Using a frame rate higher
than 24 is not recommended, as it will result in simply dropping the rendered images. It is more efficient to
dump images less frequently.
Restrictions: none
Related commands:
dump, dump image, undump
Default:
The option defaults are
append = no• buffer = yes for dump styles atom, custom, loca, and xyz• element = "C" for every atom type• every = whatever it was set to via the dump command• fileper = # of processors• first = no• flush = yes• format = %d and %g for each integer or floating point value• image = no• label = ENTRIES• nfile = 1• pad = 0• pbc = no• precision = 1000• region = none• scale = yes• sort = off for dump styles atom, custom, cfg, and local• sort = id for dump styles dcd, xtc, and xyz• thresh = none• unwrap = no•
acolor = * red/green/blue/yellow/aqua/cyan•
LAMMPS Users Manual
453
adiam = * 1.0• amap = min max cf 0.0 2 min blue max red• backcolor = black• bcolor = * red/green/blue/yellow/aqua/cyan• bdiam = * 0.5• bitrate = 2000• boxcolor = yellow• color = 140 color names are pre-defined as listed below• framerate = 24•
These are the standard 109 element names that LAMMPS pre-defines for use with the dump image and
dump_modify commands.
1-10 = "H", "He", "Li", "Be", "B", "C", "N", "O", "F", "Ne"• 11-20 = "Na", "Mg", "Al", "Si", "P", "S", "Cl", "Ar", "K", "Ca"• 21-30 = "Sc", "Ti", "V", "Cr", "Mn", "Fe", "Co", "Ni", "Cu", "Zn"• 31-40 = "Ga", "Ge", "As", "Se", "Br", "Kr", "Rb", "Sr", "Y", "Zr"• 41-50 = "Nb", "Mo", "Tc", "Ru", "Rh", "Pd", "Ag", "Cd", "In", "Sn"• 51-60 = "Sb", "Te", "I", "Xe", "Cs", "Ba", "La", "Ce", "Pr", "Nd"• 61-70 = "Pm", "Sm", "Eu", "Gd", "Tb", "Dy", "Ho", "Er", "Tm", "Yb"• 71-80 = "Lu", "Hf", "Ta", "W", "Re", "Os", "Ir", "Pt", "Au", "Hg"• 81-90 = "Tl", "Pb", "Bi", "Po", "At", "Rn", "Fr", "Ra", "Ac", "Th"• 91-100 = "Pa", "U", "Np", "Pu", "Am", "Cm", "Bk", "Cf", "Es", "Fm"• 101-109 = "Md", "No", "Lr", "Rf", "Db", "Sg", "Bh", "Hs", "Mt"•
These are the 140 colors that LAMMPS pre-defines for use with the dump image and dump_modify
commands. Additional colors can be defined with the dump_modify color command. The 3 numbers listed for
each name are the RGB (red/green/blue) values. Divide each value by 255 to get the equivalent 0.0 to 1.0
value.
aliceblue = 240,
248, 255 antiquewhite = 250, 235,
215 aqua = 0, 255, 255 aquamarine = 127,
255, 212 azure = 240, 255,
255
beige = 245, 245,
220 bisque = 255, 228, 196 black = 0, 0, 0 blanchedalmond =
255, 255, 205 blue = 0, 0, 255
blueviolet = 138,
43, 226 brown = 165, 42, 42 burlywood = 222, 184,
135 cadetblue = 95, 158,
160 chartreuse = 127,
255, 0
chocolate = 210,
105, 30 coral = 255, 127, 80 cornflowerblue = 100,
149, 237 cornsilk = 255, 248,
220 crimson = 220,
20, 60
cyan = 0, 255, 255 darkblue = 0, 0, 139 darkcyan = 0, 139, 139 darkgoldenrod =
184, 134, 11 darkgray = 169,
169, 169
darkgreen = 0, 100,
0darkkhaki = 189, 183,
107 darkmagenta = 139, 0,
139 darkolivegreen = 85,
107, 47 darkorange =
255, 140, 0
darkorchid = 153,
50, 204 darkred = 139, 0, 0 darksalmon = 233,
150, 122 darkseagreen = 143,
188, 143 darkslateblue =
72, 61, 139
darkslategray = 47,
79, 79 darkturquoise = 0, 206,
209 darkviolet = 148, 0,
211 deeppink = 255, 20,
147 deepskyblue = 0,
191, 255
dimgray = 105, 105,
105 dodgerblue = 30, 144,
255 firebrick = 178, 34, 34 floralwhite = 255,
250, 240 forestgreen = 34,
139, 34
LAMMPS Users Manual
454
fuchsia = 255, 0,
255 gainsboro = 220, 220,
220 ghostwhite = 248, 248,
255 gold = 255, 215, 0 goldenrod = 218,
165, 32
gray = 128, 128,
128 green = 0, 128, 0 greenyellow = 173,
255, 47 honeydew = 240,
255, 240 hotpink = 255,
105, 180
indianred = 205, 92,
92 indigo = 75, 0, 130 ivory = 255, 240, 240 khaki = 240, 230,
140 lavender = 230,
230, 250
lavenderblush =
255, 240, 245 lawngreen = 124, 252, 0 lemonchiffon = 255,
250, 205 lightblue = 173, 216,
230 lightcoral = 240,
128, 128
lightcyan = 224,
255, 255 lightgoldenrodyellow =
250, 250, 210 lightgreen = 144, 238,
144 lightgrey = 211,
211, 211 lightpink = 255,
182, 193
lightsalmon = 255,
160, 122 lightseagreen = 32, 178,
170 lightskyblue = 135,
206, 250 lightslategray = 119,
136, 153 lightsteelblue =
176, 196, 222
lightyellow = 255,
255, 224 lime = 0, 255, 0 limegreen = 50, 205,
50 linen = 250, 240,
230 magenta = 255,
0, 255
maroon = 128, 0, 0 mediumaquamarine =
102, 205, 170 mediumblue = 0, 0,
205 mediumorchid =
186, 85, 211 mediumpurple =
147, 112, 219
mediumseagreen =
60, 179, 113 mediumslateblue = 123,
104, 238 mediumspringgreen =
0, 250, 154 mediumturquoise =
72, 209, 204 mediumvioletred
= 199, 21, 133
midnightblue = 25,
25, 112 mintcream = 245, 255,
250 mistyrose = 255, 228,
225 moccasin = 255,
228, 181 navajowhite =
255, 222, 173
navy = 0, 0, 128 oldlace = 253, 245, 230 olive = 128, 128, 0 olivedrab = 107,
142, 35 orange = 255,
165, 0
orangered = 255,
69, 0 orchid = 218, 112, 214 palegoldenrod = 238,
232, 170 palegreen = 152,
251, 152 paleturquoise =
175, 238, 238
palevioletred = 219,
112, 147 papayawhip = 255, 239,
213 peachpuff = 255, 239,
213 peru = 205, 133, 63 pink = 255, 192,
203
plum = 221, 160,
221 powderblue = 176, 224,
230 purple = 128, 0, 128 red = 255, 0, 0 rosybrown =
188, 143, 143
royalblue = 65, 105,
225 saddlebrown = 139, 69,
19 salmon = 250, 128,
114 sandybrown = 244,
164, 96 seagreen = 46,
139, 87
seashell = 255, 245,
238 sienna = 160, 82, 45 silver = 192, 192, 192 skyblue = 135, 206,
235 slateblue = 106,
90, 205
slategray = 112,
128, 144 snow = 255, 250, 250 springgreen = 0, 255,
127 steelblue = 70, 130,
180 tan = 210, 180,
140
teal = 0, 128, 128 thistle = 216, 191, 216 tomato = 253, 99, 71 turquoise = 64, 224,
208 violet = 238,
130, 238
wheat = 245, 222,
179 white = 255, 255, 255 whitesmoke = 245,
245, 245 yellow = 255, 255, 0 yellowgreen =
154, 205, 50
LAMMPS Users Manual
455

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dump molfile command
Syntax:
dump ID group-ID molfile N file format path
ID = user-assigned name for the dump• group-ID = ID of the group of atoms to be imaged• molfile = style of dump command (other styles atom or cfg or dcd or xtc or xyz or local or custom are
discussed on the dump doc page)
•
N = dump every this many timesteps• file = name of file to write to• format = file format to be used• path = file path with plugins (optional)•
Examples:
dump mf1 all molfile 10 melt1.xml hoomd
dump mf2 all molfile 10 melt2-*.pdb pdb .
dump mf3 all molfile 50 melt3.xyz xyz .:/home/akohlmey/vmd/plugins/LINUX/molfile
Description:
Dump a snapshot of atom coordinates and selected additional quantities to one or more files every N timesteps
in one of several formats. Only information for atoms in the specified group is dumped. This specific dump
style uses molfile plugins that are bundled with the VMD molecular visualization and analysis program.
Unless the filename contains a * character, the output will be written to one single file with the specified
format. Otherwise there will be one file per snapshot and the * will be replaced by the time step number when
the snapshot is written.
NOTE: Because periodic boundary conditions are enforced only on timesteps when neighbor lists are rebuilt,
the coordinates of an atom written to a dump file may be slightly outside the simulation box.
The molfile plugin API has a few restrictions that have to be honored by this dump style: the number of atoms
must not change, the atoms must be sorted, outside of the coordinates no change in atom properties (like type,
mass, charge) will be recorded.
The format keyword determines what format is used to write out the dump. For this to work, LAMMPS must
be able to find and load a compatible molfile plugin that supports this format. Settings made via the
dump_modify command can alter per atom properties like element names.
The path keyword determines which in directories. This is a "path" like other search paths, i.e. it can contain
multiple directories separated by a colon (or semi-colon on windows). This keyword is optional and default to
".", the current directory.
The unwrap option of the dump_modify command allows coordinates to be written "unwrapped" by the
image flags for each atom. Unwrapped means that if the atom has passed through a periodic boundary one or
more times, the value is printed for what the coordinate would be if it had not been wrapped back into the
LAMMPS Users Manual
456

periodic box. Note that these coordinates may thus be far outside the box size stored with the snapshot.
Dumps are performed on timesteps that are a multiple of N (including timestep 0) and on the last timestep of a
minimization if the minimization converges. Note that this means a dump will not be performed on the initial
timestep after the dump command is invoked, if the current timestep is not a multiple of N. This behavior can
be changed via the dump_modify first command, which can be useful if the dump command is invoked after a
minimization ended on an arbitrary timestep. N can be changed between runs by using the dump_modify
every command. The dump_modify every command also allows a variable to be used to determine the
sequence of timesteps on which dump files are written.
Restrictions:
The molfile dump style is part of the USER-MOLFILE package. It is only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info.
Molfile plugins provide a consistent programming interface to read and write file formats commonly used in
molecular simulations. The USER-MOLFILE package only provides the interface code, not the plugins.
These can be obtained from a VMD installation which has to match the platform that you are using to compile
LAMMPS for. By adding plugins to VMD, support for new file formats can be added to LAMMPS (or VMD
or other programs that use them) without having to recompile the application itself. The plugins are installed
in the directory: /plugins//molfile
NOTE: while the programming interface (API) to the plugins is backward compatible, the binary interface
(ABI) has been changing over time, so it is necessary to compile this package with the plugin header files
from VMD that match the binary plugins. These header files in the directory: /plugins/include For
convenience, the package ships with a set of header files that are compatible with VMD 1.9 and 1.9.1 (June
2012)
Related commands:
dump, dump_modify, undump
Default:
The default path is ".". All other properties have to be specified.
LAMMPS Users Manual
457

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dump nc command
dump nc/mpiio command
Syntax:
dump ID group-ID nc N file.nc args
dump ID group-ID nc/mpiio N file.nc args
ID = user-assigned name for the dump• group-ID = ID of the group of atoms to be imaged• nc or nc/mpiio = style of dump command (other styles atom or cfg or dcd or xtc or xyz or local or
custom are discussed on the dump doc page)
•
N = dump every this many timesteps• file.nc = name of file to write to• args = list of per atom data elements to dump, same as for the 'custom' dump style.•
Examples:
dump 1 all nc 100 traj.nc type x y z vx vy vz
dump_modify 1 append yes at -1 global c_thermo_pe c_thermo_temp c_thermo_press
dump 1 all nc/mpiio 1000 traj.nc id type x y z
Description:
Dump a snapshot of atom coordinates every N timesteps in Amber-style NetCDF file format. NetCDF files
are binary, portable and self-describing. This dump style will write only one file on the root node. The dump
style nc uses the standard NetCDF library all data is collected on one processor and then written to the dump
file. Dump style nc/mpiio used the parallel NetCDF library and MPI-IO; it has better performance on a larger
number of processors. Note that 'nc' outputs all atoms sorted by atom tag while 'nc/mpiio' outputs in order of
the MPI rank.
In addition to per-atom data, also global (i.e. not per atom, but per frame) quantities can be included in the
dump file. This can be variables, output from computes or fixes data prefixed with v_, c_ and f_, respectively.
These properties are included via dump_modify global.
Restrictions:
The nc and nc/mpiio dump styles are part of the USER-NC-DUMP package. It is only enabled if LAMMPS
was built with that package. See the Making LAMMPS section for more info.
Related commands:
dump, dump_modify, undump
LAMMPS Users Manual
458

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
echo command
Syntax:
echo style
style = none or screen or log or both•
Examples:
echo both
echo log
Description:
This command determines whether LAMMPS echoes each input script command to the screen and/or log file
as it is read and processed. If an input script has errors, it can be useful to look at echoed output to see the last
command processed.
The command-line switch -echo can be used in place of this command.
Restrictions: none
Related commands: none
Default:
echo log
LAMMPS Users Manual
459

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix command
Syntax:
fix ID group-ID style args
ID = user-assigned name for the fix• group-ID = ID of the group of atoms to apply the fix to• style = one of a long list of possible style names (see below)• args = arguments used by a particular style•
Examples:
fix 1 all nve
fix 3 all nvt temp 300.0 300.0 0.01
fix mine top setforce 0.0 NULL 0.0
Description:
Set a fix that will be applied to a group of atoms. In LAMMPS, a "fix" is any operation that is applied to the
system during timestepping or minimization. Examples include updating of atom positions and velocities due
to time integration, controlling temperature, applying constraint forces to atoms, enforcing boundary
conditions, computing diagnostics, etc. There are dozens of fixes defined in LAMMPS and new ones can be
added; see this section for a discussion.
Fixes perform their operations at different stages of the timestep. If 2 or more fixes operate at the same stage
of the timestep, they are invoked in the order they were specified in the input script.
The ID of a fix can only contain alphanumeric characters and underscores.
Fixes can be deleted with the unfix command.
NOTE: The unfix command is the only way to turn off a fix; simply specifying a new fix with a similar style
will not turn off the first one. This is especially important to realize for integration fixes. For example, using a
fix nve command for a second run after using a fix nvt command for the first run, will not cancel out the NVT
time integration invoked by the "fix nvt" command. Thus two time integrators would be in place!
If you specify a new fix with the same ID and style as an existing fix, the old fix is deleted and the new one is
created (presumably with new settings). This is the same as if an "unfix" command were first performed on
the old fix, except that the new fix is kept in the same order relative to the existing fixes as the old one
originally was. Note that this operation also wipes out any additional changes made to the old fix via the
fix_modify command.
The fix modify command allows settings for some fixes to be reset. See the doc page for individual fixes for
details.
Some fixes store an internal "state" which is written to binary restart files via the restart or write_restart
commands. This allows the fix to continue on with its calculations in a restarted simulation. See the
read_restart command for info on how to re-specify a fix in an input script that reads a restart file. See the doc
LAMMPS Users Manual
460

pages for individual fixes for info on which ones can be restarted.
Some fixes calculate one of three styles of quantities: global, per-atom, or local, which can be used by other
commands or output as described below. A global quantity is one or more system-wide values, e.g. the energy
of a wall interacting with particles. A per-atom quantity is one or more values per atom, e.g. the displacement
vector for each atom since time 0. Per-atom values are set to 0.0 for atoms not in the specified fix group.
Local quantities are calculated by each processor based on the atoms it owns, but there may be zero or more
per atoms.
Note that a single fix may produces either global or per-atom or local quantities (or none at all), but never
more than one of these.
Global, per-atom, and local quantities each come in three kinds: a single scalar value, a vector of values, or a
2d array of values. The doc page for each fix describes the style and kind of values it produces, e.g. a
per-atom vector. Some fixes produce more than one kind of a single style, e.g. a global scalar and a global
vector.
When a fix quantity is accessed, as in many of the output commands discussed below, it can be referenced via
the following bracket notation, where ID is the ID of the fix:
f_ID entire scalar, vector, or array
f_ID[I] one element of vector, one column of array
f_ID[I][J] one element of array
In other words, using one bracket reduces the dimension of the quantity once (vector -> scalar, array ->
vector). Using two brackets reduces the dimension twice (array -> scalar). Thus a command that uses scalar
fix values as input can also process elements of a vector or array.
Note that commands and variables which use fix quantities typically do not allow for all kinds, e.g. a
command may require a vector of values, not a scalar. This means there is no ambiguity about referring to a
fix quantity as f_ID even if it produces, for example, both a scalar and vector. The doc pages for various
commands explain the details.
In LAMMPS, the values generated by a fix can be used in several ways:
Global values can be output via the thermo_style custom or fix ave/time command. Or the values can
be referenced in a variable equal or variable atom command.
•
Per-atom values can be output via the dump custom command. Or they can be time-averaged via the
fix ave/atom command or reduced by the compute reduce command. Or the per-atom values can be
referenced in an atom-style variable.
•
Local values can be reduced by the compute reduce command, or histogrammed by the fix ave/histo
command.
•
See this howto section for a summary of various LAMMPS output options, many of which involve fixes.
The results of fixes that calculate global quantities can be either "intensive" or "extensive" values. Intensive
means the value is independent of the number of atoms in the simulation, e.g. temperature. Extensive means
the value scales with the number of atoms in the simulation, e.g. total rotational kinetic energy.
Thermodynamic output will normalize extensive values by the number of atoms in the system, depending on
the "thermo_modify norm" setting. It will not normalize intensive values. If a fix value is accessed in another
way, e.g. by a variable, you may want to know whether it is an intensive or extensive value. See the doc page
LAMMPS Users Manual
461

for individual fixes for further info.
Each fix style has its own documentation page which describes its arguments and what it does, as listed
below. Here is an alphabetic list of fix styles available in LAMMPS. They are also given in more compact
form in the Fix section of this page.
There are also additional fix styles (not listed here) submitted by users which are included in the LAMMPS
distribution. The list of these with links to the individual styles are given in the fix section of this page.
adapt - change a simulation parameter over time• addforce - add a force to each atom• append/atoms - append atoms to a running simulation• atom/swap - Monte Carlo atom type swapping• aveforce - add an averaged force to each atom• ave/atom - compute per-atom time-averaged quantities• ave/chunk - compute per-chunk time-averaged quantities• ave/correlate - compute/output time correlations• ave/histo - compute/output time-averaged histograms• ave/time - compute/output global time-averaged quantities• balance - perform dynamic load-balancing• bond/break - break bonds on the fly• bond/create - create bonds on the fly• bond/swap - Monte Carlo bond swapping• box/relax - relax box size during energy minimization• deform - change the simulation box size/shape• deposit - add new atoms above a surface• drag - drag atoms towards a defined coordinate• dt/reset - reset the timestep based on velocity, forces• efield - impose electric field on system• ehex - ehanced heat exchange algorithm• enforce2d - zero out z-dimension velocity and force• evaporate - remove atoms from simulation periodically• external - callback to an external driver program• freeze - freeze atoms in a granular simulation• gcmc - grand canonical insertions/deletions• gld - generalized Langevin dynamics integrator• gravity - add gravity to atoms in a granular simulation• halt - terminate a dynamics run or minimization• heat - add/subtract momentum-conserving heat• indent - impose force due to an indenter• langevin - Langevin temperature control• lineforce - constrain atoms to move in a line• momentum - zero the linear and/or angular momentum of a group of atoms• move - move atoms in a prescribed fashion• msst - multi-scale shock technique (MSST) integration• neb - nudged elastic band (NEB) spring forces• nph - constant NPH time integration via Nose/Hoover• nphug - constant-stress Hugoniostat integration• nph/asphere - NPH for aspherical particles• nph/body - NPH for body particles• nph/sphere - NPH for spherical particles•
LAMMPS Users Manual
462
npt - constant NPT time integration via Nose/Hoover• npt/asphere - NPT for aspherical particles• npt/body - NPT for body particles• npt/sphere - NPT for spherical particles• nve - constant NVE time integration• nve/asphere - NVE for aspherical particles• nve/asphere/noforce - NVE for aspherical particles without forces"• nve/body - NVE for body particles• nve/limit - NVE with limited step length• nve/line - NVE for line segments• nve/noforce - NVE without forces (v only)• nve/sphere - NVE for spherical particles• nve/tri - NVE for triangles• nvt - constant NVT time integration via Nose/Hoover• nvt/asphere - NVT for aspherical particles• nvt/body - NVT for body particles• nvt/sllod - NVT for NEMD with SLLOD equations• nvt/sphere - NVT for spherical particles• oneway - constrain particles on move in one direction• orient/bcc - add grain boundary migration force for BCC• orient/fcc - add grain boundary migration force for FCC• planeforce - constrain atoms to move in a plane• poems - constrain clusters of atoms to move as coupled rigid bodies• pour - pour new atoms/molecules into a granular simulation domain• press/berendsen - pressure control by Berendsen barostat• print - print text and variables during a simulation• property/atom - add customized per-atom values• qeq/comb - charge equilibration for COMB potential qeq/dynamic - charge equilibration via dynamic
method qeq/fire - charge equilibration via FIRE minimizer qeq/point - charge equilibration via point
method qeq/shielded - charge equilibration via shielded method qeq/slater - charge equilibration via
Slater method rattle - RATTLE constraints on bonds and/or angles
•
reax/bonds - write out ReaxFF bond information recenter - constrain the center-of-mass position of a
group of atoms
•
restrain - constrain a bond, angle, dihedral• rigid - constrain one or more clusters of atoms to move as a rigid body with NVE integration• rigid/nph - constrain one or more clusters of atoms to move as a rigid body with NPH integration• rigid/npt - constrain one or more clusters of atoms to move as a rigid body with NPT integration• rigid/nve - constrain one or more clusters of atoms to move as a rigid body with alternate NVE
integration
•
rigid/nvt - constrain one or more clusters of atoms to move as a rigid body with NVT integration• rigid/small - constrain many small clusters of atoms to move as a rigid body with NVE integration• rigid/small/nph - constrain many small clusters of atoms to move as a rigid body with NPH
integration
•
rigid/small/npt - constrain many small clusters of atoms to move as a rigid body with NPT integration• rigid/small/nve - constrain many small clusters of atoms to move as a rigid body with alternate NVE
integration
•
rigid/small/nvt - constrain many small clusters of atoms to move as a rigid body with NVT integration• setforce - set the force on each atom• shake - SHAKE constraints on bonds and/or angles• spring - apply harmonic spring force to group of atoms• spring/chunk - apply harmonic spring force to each chunk of atoms•
LAMMPS Users Manual
463
spring/rg - spring on radius of gyration of group of atoms• spring/self - spring from each atom to its origin• srd - stochastic rotation dynamics (SRD)• store/force - store force on each atom• store/state - store attributes for each atom• temp/berendsen - temperature control by Berendsen thermostat• temp/csld - canonical sampling thermostat with Langevin dynamics• temp/csvr - canonical sampling thermostat with Hamiltonian dynamics• temp/rescale - temperature control by velocity rescaling• tfmc - perform force-bias Monte Carlo with time-stamped method• thermal/conductivity - Muller-Plathe kinetic energy exchange for thermal conductivity calculation• tmd - guide a group of atoms to a new configuration• ttm - two-temperature model for electronic/atomic coupling• tune/kspace - auto-tune KSpace parameters• vector - accumulate a global vector every N timesteps• viscosity - Muller-Plathe momentum exchange for viscosity calculation• viscous - viscous damping for granular simulations• wall/colloid - Lennard-Jones wall interacting with finite-size particles• wall/gran - frictional wall(s) for granular simulations• wall/harmonic - harmonic spring wall• wall/lj1043 - Lennard-Jones 10-4-3 wall• wall/lj126 - Lennard-Jones 12-6 wall• wall/lj93 - Lennard-Jones 9-3 wall• wall/piston - moving reflective piston wall• wall/reflect - reflecting wall(s)• wall/region - use region surface as wall• wall/srd - slip/no-slip wall for SRD particles•
Restrictions:
Some fix styles are part of specific packages. They are only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info on packages. The doc pages for individual fixes tell if it is
part of a package.
Related commands:
unfix, fix_modify
Default: none
LAMMPS Users Manual
464

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix_modify command
Syntax:
fix_modify fix-ID keyword value ...
fix-ID = ID of the fix to modify• one or more keyword/value pairs may be appended• keyword = temp or press or energy or respa or dynamic/dof
temp value = compute ID that calculates a temperature
press value = compute ID that calculates a pressure
energy value = yes or no
respa value = 1 to max respa level or 0 (for outermost level)
dynamic/dof value = yes or no
yes/no = do or do not recompute the number of degrees of freedom (DOF) contributing to the temperature
•
Examples:
fix_modify 3 temp myTemp press myPress
fix_modify 1 energy yes
fix_modify tether respa 2
Description:
Modify one or more parameters of a previously defined fix. Only specific fix styles support specific
parameters. See the doc pages for individual fix commands for info on which ones support which fix_modify
parameters.
The temp keyword is used to determine how a fix computes temperature. The specified compute ID must have
been previously defined by the user via the compute command and it must be a style of compute that
calculates a temperature. All fixes that compute temperatures define their own compute by default, as
described in their documentation. Thus this option allows the user to override the default method for
computing T.
The press keyword is used to determine how a fix computes pressure. The specified compute ID must have
been previously defined by the user via the compute command and it must be a style of compute that
calculates a pressure. All fixes that compute pressures define their own compute by default, as described in
their documentation. Thus this option allows the user to override the default method for computing P.
For fixes that calculate a contribution to the potential energy of the system, the energy keyword will include
that contribution in thermodynamic output of potential energy. This is because the energy yes setting must be
specified to include the fix's global or per-atom energy in the calculation performed by the compute pe or
compute pe/atom commands. See the thermo_style command for info on how potential energy is output. For
fixes that tally a global energy, it can be printed by using the keyword f_ID in the thermo_style custom
command, where ID is the fix-ID of the appropriate fix.
NOTE: You must also specify the energy yes setting for a fix if you are using it when performing an energy
minimization and if you want the energy and forces it produces to be part of the optimization criteria.
LAMMPS Users Manual
465
For fixes that set or modify forces, it may be possible to select at which r-RESPA level the fix operates via the
respa keyword. The RESPA level at which the fix is active can be selected. This is a number ranging from 1
to the number of levels. If the RESPA level is larger than the current maximum, the outermost level will be
used, which is also the default setting. This default can be restored using a value of 0 for the RESPA level.
The affected fix has to be enabled to support this feature; if not, fix_modify will report an error. Active fixes
with a custom RESPA level setting are reported with their specified level at the beginning of a r-RESPA run.
The dynamic/dof keyword determines whether the number of atoms N in the fix group and their associated
degrees of freedom are re-computed each time a temperature is computed. Only fix styles that calculate their
own internal temperature use this option. Currently this is only the fix rigid/nvt/small and fix rigid/npt/small
commands for the purpose of thermostatting rigid body translation and rotation. By default, N and their DOF
are assumed to be constant. If you are adding atoms or molecules to the system (see the fix pour, fix deposit,
and fix gcmc commands) or expect atoms or molecules to be lost (e.g. due to exiting the simulation box or via
fix evaporate), then this option should be used to insure the temperature is correctly normalized.
NOTE: Other thermostatting fixes, such as fix nvt, do not use the dynamic/dof keyword because they use a
temperature compute to calculate temperature. See the compute_modify dynamic/dof command for a similar
way to insure correct temperature normalization for those thermostats.
Restrictions: none
Related commands:
fix, compute temp, compute pressure, thermo_style
Default:
The option defaults are temp = ID defined by fix, press = ID defined by fix, energy = no, respa = 0.
LAMMPS Users Manual
466

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
group command
Syntax:
group ID style args
ID = user-defined name of the group• style = delete or region or type or id or molecule or variable or include or subtract or union or
intersect or dynamic or static
delete = no args
clear = no args
region args = region-ID
type or id or molecule
args = list of one or more atom types, atom IDs, or molecule IDs
any entry in list can be a sequence formatted as A:B or A:B:C where
A = starting index, B = ending index,
C = increment between indices, 1 if not specified
args = logical value
logical = "" or ">=" or "==" or "!="
value = an atom type or atom ID or molecule ID (depending on style)
args = logical value1 value2
logical = ""
value1,value2 = atom types or atom IDs or molecule IDs (depending on style)
variable args = variable-name
include args = molecule
molecule = add atoms to group with same molecule ID as atoms already in group
subtract args = two or more group IDs
union args = one or more group IDs
intersect args = two or more group IDs
dynamic args = parent-ID keyword value ...
one or more keyword/value pairs may be appended
keyword = region or var or every
region value = region-ID
var value = name of variable
every value = N = update group every this many timesteps
static = no args
•
Examples:
group edge region regstrip
group water type 3 4
group sub id 10 25 50
group sub id 10 25 50 500:1000
group sub id 100:10000:10
group sub id <= 150
group polyA molecule 50 250
group hienergy variable eng
group hienergy include molecule
group boundary subtract all a2 a3
group boundary union lower upper
group boundary intersect upper flow
group boundary delete
group mine dynamic all region myRegion every 100
LAMMPS Users Manual
467
Description:
Identify a collection of atoms as belonging to a group. The group ID can then be used in other commands such
as fix, compute, dump, or velocity to act on those atoms together.
If the group ID already exists, the group command adds the specified atoms to the group.
NOTE: By default groups are static, meaning the atoms are permanently assigned to the group. For example,
if the region style is used to assign atoms to a group, the atoms will remain in the group even if they later
move out of the region. As explained below, the dynamic style can be used to make a group dynamic so that a
periodic determination is made as to which atoms are in the group. Since many LAMMPS commands operate
on groups of atoms, you should think carefully about whether making a group dynamic makes sense for your
model.
A group with the ID all is predefined. All atoms belong to this group. This group cannot be deleted, or made
dynamic.
The delete style removes the named group and un-assigns all atoms that were assigned to that group. Since
there is a restriction (see below) that no more than 32 groups can be defined at any time, the delete style
allows you to remove groups that are no longer needed, so that more can be specified. You cannot delete a
group if it has been used to define a current fix or compute or dump.
The clear style un-assigns all atoms that were assigned to that group. This may be dangerous to do during a
simulation run, e.g. using the run every command if a fix or compute or other operation expects the atoms in
the group to remain constant, but LAMMPS does not check for this.
The region style puts all atoms in the region volume into the group. Note that this is a static one-time
assignment. The atoms remain assigned (or not assigned) to the group even in they later move out of the
region volume.
The type, id, and molecule styles put all atoms with the specified atom types, atom IDs, or molecule IDs into
the group. These 3 styles can use arguments specified in one of two formats.
The first format is a list of values (types or IDs). For example, the 2nd command in the examples above puts
all atoms of type 3 or 4 into the group named water. Each entry in the list can be a colon-separated sequence
A:B or A:B:C, as in two of the examples above. A "sequence" generates a sequence of values (types or IDs),
with an optional increment. The first example with 500:1000 has the default increment of 1 and would add all
atom IDs from 500 to 1000 (inclusive) to the group sub, along with 10,25,50 since they also appear in the list
of values. The second example with 100:10000:10 uses an increment of 10 and would thus would add atoms
IDs 100,110,120, ... 9990,10000 to the group sub.
The second format is a logical followed by one or two values (type or ID). The 7 valid logicals are listed
above. All the logicals except take a single argument. The 3rd example above adds all atoms with IDs from 1
to 150 to the group named sub. The logical means "between" and takes 2 arguments. The 4th example above
adds all atoms belonging to molecules with IDs from 50 to 250 (inclusive) to the group named polyA.
The variable style evaluates a variable to determine which atoms to add to the group. It must be an atom-style
variable previously defined in the input script. If the variable evaluates to a non-zero value for a particular
atom, then that atom is added to the specified group.
LAMMPS Users Manual
468
Atom-style variables can specify formulas that include thermodynamic quantities, per-atom values such as
atom coordinates, or per-atom quantities calculated by computes, fixes, or other variables. They can also
include Boolean logic where 2 numeric values are compared to yield a 1 or 0 (effectively a true or false). Thus
using the variable style, is a general way to flag specific atoms to include or exclude from a group.
For example, these lines define a variable "eatom" that calculates the potential energy of each atom and
includes it in the group if its potential energy is above the threshold value -3.0.
compute 1 all pe/atom
compute 2 all reduce sum c_1
thermo_style custom step temp pe c_2
run 0
variable eatom atom "c_1 > -3.0"
group hienergy variable eatom
Note that these lines
compute 2 all reduce sum c_1
thermo_style custom step temp pe c_2
run 0
are necessary to insure that the "eatom" variable is current when the group command invokes it. Because the
eatom variable computes the per-atom energy via the pe/atom compute, it will only be current if a run has
been performed which evaluated pairwise energies, and the pe/atom compute was actually invoked during the
run. Printing the thermodynamic info for compute 2 insures that this is the case, since it sums the pe/atom
compute values (in the reduce compute) to output them to the screen. See the "Variable Accuracy" section of
the variable doc page for more details on insuring that variables are current when they are evaluated between
runs.
The include style with its arg molecule adds atoms to a group that have the same molecule ID as atoms
already in the group. The molecule ID = 0 is ignored in this operation, since it is assumed to flag isolated
atoms that are not part of molecules. An example of where this operation is useful is if the region style has
been used previously to add atoms to a group that are within a geometric region. If molecules straddle the
region boundary, then atoms outside the region that are part of molecules with atoms inside the region will not
be in the group. Using the group command a 2nd time with include molecule will add those atoms that are
outside the region to the group.
NOTE: The include molecule operation is relatively expensive in a parallel sense. This is because it requires
communication of relevant molecule IDs between all the processors and each processor to loop over its atoms
once per processor, to compare its atoms to the list of molecule IDs from every other processor. Hence it
scales as N, rather than N/P as most of the group operations do, where N is the number of atoms, and P is the
number of processors.
The subtract style takes a list of two or more existing group names as arguments. All atoms that belong to the
1st group, but not to any of the other groups are added to the specified group.
The union style takes a list of one or more existing group names as arguments. All atoms that belong to any of
the listed groups are added to the specified group.
The intersect style takes a list of two or more existing group names as arguments. Atoms that belong to every
one of the listed groups are added to the specified group.
LAMMPS Users Manual
469

The dynamic style flags an existing or new group as dynamic. This means atoms will be (re)assigned to the
group periodically as a simulation runs. This is in contrast to static groups where atoms are permanently
assigned to the group. The way the assignment occurs is as follows. Only atoms in the group specified as the
parent group via the parent-ID are assigned to the dynamic group before the following conditions are applied.
If the region keyword is used, atoms not in the specified region are removed from the dynamic group. If the
var keyword is used, the variable name must be an atom-style or atomfile-style variable. The variable is
evaluated and atoms whose per-atom values are 0.0, are removed from the dynamic group.
The assignment of atoms to a dynamic group is done at the beginning of each run and on every timestep that
is a multiple of N, which is the argument for the every keyword (N = 1 is the default). For an energy
minimization, via the minimize command, an assignment is made at the beginning of the minimization, but
not during the iterations of the minimizer.
The point in the timestep at which atoms are assigned to a dynamic group is after the initial stage of velocity
Verlet time integration has been performed, and before neighbor lists or forces are computed. This is the point
in the timestep where atom positions have just changed due to the time integration, so the region criterion
should be accurate, if applied.
NOTE: If the region keyword is used to determine what atoms are in the dynamic group, atoms can move
outside of the simulation box between reneighboring events. Thus if you want to include all atoms on the left
side of the simulation box, you probably want to set the left boundary of the region to be outside the
simulation box by some reasonable amount (e.g. up to the cutoff of the potential), else they may be excluded
from the dynamic region.
Here is an example of using a dynamic group to shrink the set of atoms being integrated by using a spherical
region with a variable radius (shrinking from 18 to 5 over the course of the run). This could be used to model
a quench of the system, freezing atoms outside the shrinking sphere, then converting the remaining atoms to a
static group and running further.
variable nsteps equal 5000
variable rad equal 18-(step/v_nsteps)*(18-5)
region ss sphere 20 20 0 v_rad
group mobile dynamic all region ss
fix 1 mobile nve
run ${nsteps}
group mobile static
run ${nsteps}
NOTE: All fixes and computes take a group ID as an argument, but they do not all allow for use of a dynamic
group. If you get an error message that this is not allowed, but feel that it should be for the fix or compute in
question, then please post your reasoning to the LAMMPS mail list and we can change it.
The static style removes the setting for a dynamic group, converting it to a static group (the default). The
atoms in the static group are those currently in the dynamic group.
Restrictions:
There can be no more than 32 groups defined at one time, including "all".
The parent group of a dynamic group cannot itself be a dynamic group.
LAMMPS Users Manual
470
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
group2ndx command
ndx2group command
Syntax:
group2ndx file group-ID ...
ndx2group file group-ID ...
file = name of index file to write out or read in• zero or more group IDs may be appended•
Examples:
group2ndx allindex.ndx
group2ndx someindex.ndx upper lower mobile
ndx2group someindex.ndx
ndx2group someindex.ndx mobile
Description:
Write or read a Gromacs style index file in text format that associates atom IDs with the corresponding group
definitions. This index file can be used with in combination with Gromacs analysis tools or to import group
definitions into the fix colvars input file. It can also be used to save and restore group definitions for static
groups.
The group2ndx command will write group definitions to an index file. Without specifying any group IDs, all
groups will be written to the index file. When specifying group IDs, only those groups will be written to the
index file. In order to follow the Gromacs conventions, the group all will be renamed to System in the index
file.
The ndx2group command will create of update group definitions from those stored in an index file. Without
specifying any group IDs, all groups except System will be read from the index file and the corresponding
groups recreated. If a group of the same name already exists, it will be completely reset. When specifying
group IDs, those groups, if present, will be read from the index file and restored.
Restrictions:
This command requires that atoms have atom IDs, since this is the information that is written to the index file.
These commands are part of the USER-COLVARS package. They are only enabled if LAMMPS was built
with that package. See the Making LAMMPS section for more info.
Related commands:
group, dump, fix colvars
Default: none
LAMMPS Users Manual
472

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
if command
Syntax:
if boolean then t1 t2 ... elif boolean f1 f2 ... elif boolean f1 f2 ... else e1 e2 ...
boolean = a Boolean expression evaluated as TRUE or FALSE (see below)• then = required word• t1,t2,...,tN = one or more LAMMPS commands to execute if condition is met, each enclosed in quotes• elif = optional word, can appear multiple times• f1,f2,...,fN = one or more LAMMPS commands to execute if elif condition is met, each enclosed in
quotes (optional arguments)
•
else = optional argument• e1,e2,...,eN = one or more LAMMPS commands to execute if no condition is met, each enclosed in
quotes (optional arguments)
•
Examples:
if "${steps} > 1000" then quit
if "${myString} == a10" then quit
if "$x <= $y" then "print X is smaller = $x" else "print Y is smaller = $y"
if "(${eng} > 0.0) || ($n <1000)" then &
"timestep 0.005" &
elif $n ${eng_previous}" then "jump file1" else "jump file2"
Description:
This command provides an if-then-else capability within an input script. A Boolean expression is evaluated
and the result is TRUE or FALSE. Note that as in the examples above, the expression can contain variables, as
defined by the variable command, which will be evaluated as part of the expression. Thus a user-defined
formula that reflects the current state of the simulation can be used to issue one or more new commands.
If the result of the Boolean expression is TRUE, then one or more commands (t1, t2, ..., tN) are executed. If it
is FALSE, then Boolean expressions associated with successive elif keywords are evaluated until one is found
to be true, in which case its commands (f1, f2, ..., fN) are executed. If no Boolean expression is TRUE, then
the commands associated with the else keyword, namely (e1, e2, ..., eN), are executed. The elif and else
keywords and their associated commands are optional. If they aren't specified and the initial Boolean
expression is FALSE, then no commands are executed.
The syntax for Boolean expressions is described below.
Each command (t1, f1, e1, etc) can be any valid LAMMPS input script command, except an include
command, which is not allowed. If the command is more than one word, it must enclosed in quotes, so it will
be treated as a single argument, as in the examples above.
NOTE: If a command itself requires a quoted argument (e.g. a print command), then double and single quotes
can be used and nested in the usual manner, as in the examples above and below. See Section 3.2 of the
manual for more details on using quotes in arguments. Only one of level of nesting is allowed, but that should
be sufficient for most use cases.
LAMMPS Users Manual
473

Note that by using the line continuation character "&", the if command can be spread across many lines,
though it is still a single command:
if "$a <$b" then &
"print 'Minimum value = $a'" &
"run 1000" &
else &
'print "Minimum value = $b"' &
"minimize 0.001 0.001 1000 10000"
Note that if one of the commands to execute is quit, as in the first example above, then executing the
command will cause LAMMPS to halt.
Note that by jumping to a label in the same input script, the if command can be used to break out of a loop.
See the variable delete command for info on how to delete the associated loop variable, so that it can be
re-used later in the input script.
Here is an example of a loop which checks every 1000 steps if the system temperature has reached a certain
value, and if so, breaks out of the loop to finish the run. Note that any variable could be checked, so long as it
is current on the timestep when the run completes. As explained on the variable doc page, this can be insured
by including the variable in thermodynamic output.
variable myTemp equal temp
label loop
variable a loop 1000
run 1000
if "${myTemp} <300.0" then "jump SELF break"
next a
jump SELF loop
label break
print "ALL DONE"
Here is an example of a double loop which uses the if and jump commands to break out of the inner loop
when a condition is met, then continues iterating thru the outer loop.
label loopa
variable a loop 5
label loopb
variable b loop 5
print "A,B = $a,$b"
run 10000
if "$b > 2" then "jump SELF break"
next b
jump in.script loopb
label break
variable b delete
next a
jump SELF loopa
The Boolean expressions for the if and elif keywords have a C-like syntax. Note that each expression is a
single argument within the if command. Thus if you want to include spaces in the expression for clarity, you
must enclose the entire expression in quotes.
An expression is built out of numbers (which start with a digit or period or minus sign) or strings (which start
with a letter and can contain alphanumeric characters or underscores):
LAMMPS Users Manual
474

0.2, 100, 1.0e20, -15.4, etc
InP, myString, a123, ab_23_cd, etc
and Boolean operators:
A == B, A != B, A <B, A <= B, A > B, A >= B, A && B, A || B, A |^ B, !A
Each A and B is a number or string or a variable reference like $a or ${abc}, or A or B can be another
Boolean expression.
If a variable is used it can produce a number when evaluated, like an equal-style variable. Or it can produce a
string, like an index-style variable. For an individual Boolean operator, A and B must both be numbers or
must both be strings. You cannot compare a number to a string.
Expressions are evaluated left to right and have the usual C-style precedence: the unary logical NOT operator
"!" has the highest precedence, the 4 relational operators "", and ">=" are next; the two remaining relational
operators "==" and "!=" are next; then the logical AND operator "&&"; and finally the logical OR operator
"||" and logical XOR (exclusive or) operator "|^" have the lowest precedence. Parenthesis can be used to group
one or more portions of an expression and/or enforce a different order of evaluation than what would occur
with the default precedence.
When the 6 relational operators (first 6 in list above) compare 2 numbers, they return either a 1.0 or 0.0
depending on whether the relationship between A and B is TRUE or FALSE. When the 6 relational operators
compare 2 strings, they also return a 1.0 or 0.0 for TRUE or FALSE, but the comparison is done by the C
function strcmp().
When the 3 logical operators (last 3 in list above) compare 2 numbers, they also return either a 1.0 or 0.0
depending on whether the relationship between A and B is TRUE or FALSE (or just A). The logical AND
operator will return 1.0 if both its arguments are non-zero, else it returns 0.0. The logical OR operator will
return 1.0 if either of its arguments is non-zero, else it returns 0.0. The logical XOR operator will return 1.0 if
one of its arguments is zero and the other non-zero, else it returns 0.0. The logical NOT operator returns 1.0 if
its argument is 0.0, else it returns 0.0. The 3 logical operators can only be used to operate on numbers, not on
strings.
The overall Boolean expression produces a TRUE result if the result is non-zero. If the result is zero, the
expression result is FALSE.
Restrictions: none
Related commands:
variable, print
Default: none
LAMMPS Users Manual
475

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_coeff command
Syntax:
improper_coeff N args
N = improper type (see asterisk form below)• args = coefficients for one or more improper types•
Examples:
improper_coeff 1 300.0 0.0
improper_coeff * 80.2 -1 2
improper_coeff *4 80.2 -1 2
Description:
Specify the improper force field coefficients for one or more improper types. The number and meaning of the
coefficients depends on the improper style. Improper coefficients can also be set in the data file read by the
read_data command or in a restart file.
N can be specified in one of two ways. An explicit numeric value can be used, as in the 1st example above. Or
a wild-card asterisk can be used to set the coefficients for multiple improper types. This takes the form "*" or
"*n" or "n*" or "m*n". If N = the number of improper types, then an asterisk with no numeric values means
all types from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A trailing asterisk means all
types from n to N (inclusive). A middle asterisk means all types from m to n (inclusive).
Note that using an improper_coeff command can override a previous setting for the same improper type. For
example, these commands set the coeffs for all improper types, then overwrite the coeffs for just improper
type 2:
improper_coeff * 300.0 0.0
improper_coeff 2 50.0 0.0
A line in a data file that specifies improper coefficients uses the exact same format as the arguments of the
improper_coeff command in an input script, except that wild-card asterisks should not be used since
coefficients for all N types must be listed in the file. For example, under the "Improper Coeffs" section of a
data file, the line that corresponds to the 1st example above would be listed as
1 300.0 0.0
The improper_style class2 is an exception to this rule, in that an additional argument is used in the input script
to allow specification of the cross-term coefficients. See its doc page for details.
Here is an alphabetic list of improper styles defined in LAMMPS. Click on the style to display the formula it
computes and coefficients specified by the associated improper_coeff command.
Note that there are also additional improper styles submitted by users which are included in the LAMMPS
distribution. The list of these with links to the individual styles are given in the improper section of this page.
LAMMPS Users Manual
476

improper_style none - turn off improper interactions• improper_style hybrid - define multiple styles of improper interactions•
improper_style class2 - COMPASS (class 2) improper• improper_style cvff - CVFF improper• improper_style harmonic - harmonic improper• improper_style umbrella - DREIDING improper•
Restrictions:
This command must come after the simulation box is defined by a read_data, read_restart, or create_box
command.
An improper style must be defined before any improper coefficients are set, either in the input script or in a
data file.
Related commands:
improper_style
Default: none
LAMMPS Users Manual
477
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style command
Syntax:
improper_style style
style = none or hybrid or class2 or cvff or harmonic•
Examples:
improper_style harmonic
improper_style cvff
improper_style hybrid cvff harmonic
Description:
Set the formula(s) LAMMPS uses to compute improper interactions between quadruplets of atoms, which
remain in force for the duration of the simulation. The list of improper quadruplets is read in by a read_data or
read_restart command from a data or restart file. Note that the ordering of the 4 atoms in an improper
quadruplet determines the definition of the improper angle used in the formula for each style. See the doc
pages of individual styles for details.
Hybrid models where impropers are computed using different improper potentials can be setup using the
hybrid improper style.
The coefficients associated with an improper style can be specified in a data or restart file or via the
improper_coeff command.
All improper potentials store their coefficient data in binary restart files which means improper_style and
improper_coeff commands do not need to be re-specified in an input script that restarts a simulation. See the
read_restart command for details on how to do this. The one exception is that improper_style hybrid only
stores the list of sub-styles in the restart file; improper coefficients need to be re-specified.
NOTE: When both an improper and pair style is defined, the special_bonds command often needs to be used
to turn off (or weight) the pairwise interaction that would otherwise exist between a group of 4 bonded atoms.
Here is an alphabetic list of improper styles defined in LAMMPS. Click on the style to display the formula it
computes and coefficients specified by the associated improper_coeff command.
Note that there are also additional improper styles submitted by users which are included in the LAMMPS
distribution. The list of these with links to the individual styles are given in the improper section of this page.
improper_style none - turn off improper interactions• improper_style zero - topology but no interactions• improper_style hybrid - define multiple styles of improper interactions•
improper_style class2 - COMPASS (class 2) improper• improper_style cvff - CVFF improper• improper_style harmonic - harmonic improper•
LAMMPS Users Manual
478

improper_style umbrella - DREIDING improper•
Restrictions:
Improper styles can only be set for atom_style choices that allow impropers to be defined.
Most improper styles are part of the MOLECULE package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info on packages. The doc pages for individual
improper potentials tell if it is part of a package.
Related commands:
improper_coeff
Default:
improper_style none
LAMMPS Users Manual
479

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
include command
Syntax:
include file
file = filename of new input script to switch to•
Examples:
include newfile
include in.run2
Description:
This command opens a new input script file and begins reading LAMMPS commands from that file. When
the new file is finished, the original file is returned to. Include files can be nested as deeply as desired. If input
script A includes script B, and B includes A, then LAMMPS could run for a long time.
If the filename is a variable (see the variable command), different processor partitions can run different input
scripts.
Restrictions: none
Related commands:
variable, jump
Default: none
LAMMPS Users Manual
480

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
info command
Syntax:
info args
args = one or more of the following keywords: out, all, system, memory, communication, computes,
dumps, fixes, groups, regions, variables, styles, time, or configuration
•
out values = screen, log, append filename, overwrite filename• styles values = all, angle, atom, bond, compute, command, dump, dihedral, fix, improper, integrate,
kspace, minimize, pair, region
•
Examples:
info system
info groups computes variables
info all out log
info all out append info.txt
info styles all
info styles atom
Description:
Print out information about the current internal state of the running LAMMPS process. This can be helpful
when debugging or validating complex input scripts. Several output categories are available and one or more
output category may be requested.
The out flag controls where the output is sent. It can only be sent to one target. By default this is the screen, if
it is active. The log argument selects the log file instead. With the append and overwrite option, followed by a
filename, the output is written to that file, which is either appended to or overwritten, respectively.
The all flag activates printing all categories listed below.
The configuration category prints some information about the LAMMPS version as well as architecture and
OS it is run on.
The memory category prints some information about the current memory allocation of MPI rank 0 (this the
amount of dynamically allocated memory reported by LAMMPS classes). Where supported, also some OS
specific information about the size of the reserved memory pool size (this is where malloc() and the new
operator request memory from) and the maximum resident set size is reported (this is the maximum amount of
physical memory occupied so far).
The system category prints a general system overview listing. This includes the unit style, atom style, number
of atoms, bonds, angles, dihedrals, and impropers and the number of the respective types, box dimensions and
properties, force computing styles and more.
The communication category prints a variety of information about communication and parallelization: the
MPI library version level, the number of MPI ranks and OpenMP threads, the communication style and
layout, the processor grid dimensions, ghost atom communication mode, cutoff, and related settings.
LAMMPS Users Manual
481
The computes category prints a list of all currently defined computes, their IDs and styles and groups they
operate on.
The dumps category prints a list of all currently active dumps, their IDs, styles, filenames, groups, and dump
frequencies.
The fixes category prints a list of all currently defined fixes, their IDs and styles and groups they operate on.
The groups category prints a list of all currently defined groups.
The regions category prints a list of all currently defined regions, their IDs and styles and whether "inside" or
"outside" atoms are selected.
The variables category prints a list of all currently defined variables, their names, styles, definition and last
computed value, if available.
The styles category prints the list of styles available in the current LAMMPS binary. It supports one of the
following options to control which category of styles is printed out:
all• angle• atom• bond• compute• command• dump• dihedral• fix• improper• integrate• kspace• minimize• pair• region•
The time category prints the accumulated CPU and wall time for the process that writes output (usually MPI
rank 0).
Restrictions: none
Related commands:
print
Default:
The out option has the default screen.
The styles option has the default all.
LAMMPS Users Manual
482

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
jump command
Syntax:
jump file label
file = filename of new input script to switch to• label = optional label within file to jump to•
Examples:
jump newfile
jump in.run2 runloop
jump SELF runloop
Description:
This command closes the current input script file, opens the file with the specified name, and begins reading
LAMMPS commands from that file. Unlike the include command, the original file is not returned to, although
by using multiple jump commands it is possible to chain from file to file or back to the original file.
If the word "SELF" is used for the filename, then the current input script is re-opened and read again.
NOTE: The SELF option is not guaranteed to work when the current input script is being read through stdin
(standard input), e.g.
lmp_g++ <in.script
since the SELF option invokes the C-library rewind() call, which may not be supported for stdin on some
systems or by some MPI implementations. This can be worked around by using the -in command-line
argument, e.g.
lmp_g++ -in in.script
or by using the -var command-line argument to pass the script name as a variable to the input script. In the
latter case, a variable called "fname" could be used in place of SELF, e.g.
lmp_g++ -var fname in.script <in.script
The 2nd argument to the jump command is optional. If specified, it is treated as a label and the new file is
scanned (without executing commands) until the label is found, and commands are executed from that point
forward. This can be used to loop over a portion of the input script, as in this example. These commands
perform 10 runs, each of 10000 steps, and create 10 dump files named file.1, file.2, etc. The next command is
used to exit the loop after 10 iterations. When the "a" variable has been incremented for the tenth time, it will
cause the next jump command to be skipped.
variable a loop 10
label loop
dump 1 all atom 100 file.$a
run 10000
LAMMPS Users Manual
483
undump 1
next a
jump in.lj loop
If the jump file argument is a variable, the jump command can be used to cause different processor partitions
to run different input scripts. In this example, LAMMPS is run on 40 processors, with 4 partitions of 10 procs
each. An in.file containing the example variable and jump command will cause each partition to run a
different simulation.
mpirun -np 40 lmp_ibm -partition 4x10 -in in.file
variable f world script.1 script.2 script.3 script.4
jump $f
Here is an example of a loop which checks every 1000 steps if the system temperature has reached a certain
value, and if so, breaks out of the loop to finish the run. Note that any variable could be checked, so long as it
is current on the timestep when the run completes. As explained on the variable doc page, this can be insured
by including the variable in thermodynamic output.
variable myTemp equal temp
label loop
variable a loop 1000
run 1000
if "${myTemp} <300.0" then "jump SELF break"
next a
jump SELF loop
label break
print "ALL DONE"
Here is an example of a double loop which uses the if and jump commands to break out of the inner loop
when a condition is met, then continues iterating thru the outer loop.
label loopa
variable a loop 5
label loopb
variable b loop 5
print "A,B = $a,$b"
run 10000
if "$b > 2" then "jump SELF break"
next b
jump in.script loopb
label break
variable b delete
next a
jump SELF loopa
Restrictions:
If you jump to a file and it does not contain the specified label, LAMMPS will come to the end of the file and
exit.
Related commands:
variable, include, label, next
LAMMPS Users Manual
484
Default: none
LAMMPS Users Manual
485

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
kspace_modify command
Syntax:
kspace_modify keyword value ...
one or more keyword/value pairs may be listed
keyword = mesh or order or order/disp or mix/disp or overlap or minorder or force or gewald or gewald/disp or slab or (nozforce or compute or cutoff/adjust or fftbench or collective or diff or kmax/ewald or force/disp/real or force/disp/kspace or splittol or disp/auto:l
mesh value = x y z
x,y,z = grid size in each dimension for long-range Coulombics
mesh/disp value = x y z
x,y,z = grid size in each dimension for 1/r^6 dispersion
order value = N
N = extent of Gaussian for PPPM or MSM mapping of charge to grid
order/disp value = N
N = extent of Gaussian for PPPM mapping of dispersion term to grid
mix/disp value = pair or geom or none
overlap = yes or no = whether the grid stencil for PPPM is allowed to overlap into more than the nearest-neighbor processor
minorder value = M
M = min allowed extent of Gaussian when auto-adjusting to minimize grid communication
force value = accuracy (force units)
gewald value = rinv (1/distance units)
rinv = G-ewald parameter for Coulombics
gewald/disp value = rinv (1/distance units)
rinv = G-ewald parameter for dispersion
slab value = volfactor or nozforce
volfactor = ratio of the total extended volume used in the
2d approximation compared with the volume of the simulation domain
nozforce turns off kspace forces in the z direction
compute value = yes or no
cutoff/adjust value = yes or no
pressure/scalar value = yes or no
fftbench value = yes or no
collective value = yes or no
diff value = ad or ik = 2 or 4 FFTs for PPPM in smoothed or non-smoothed mode
kmax/ewald value = kx ky kz
kx,ky,kz = number of Ewald sum kspace vectors in each dimension
force/disp/real value = accuracy (force units)
force/disp/kspace value = accuracy (force units)
splittol value = tol
tol = relative size of two eigenvalues (see discussion below)
disp/auto value = yes or no
•
Examples:
kspace_modify mesh 24 24 30 order 6
kspace_modify slab 3.0
Description:
Set parameters used by the kspace solvers defined by the kspace_style command. Not all parameters are
relevant to all kspace styles.
LAMMPS Users Manual
486
The mesh keyword sets the grid size for kspace style pppm or msm. In the case of PPPM, this is the FFT mesh,
and each dimension must be factorizable into powers of 2, 3, and 5. In the case of MSM, this is the finest scale
real-space mesh, and each dimension must be factorizable into powers of 2. When this option is not set, the
PPPM or MSM solver chooses its own grid size, consistent with the user-specified accuracy and pairwise
cutoff. Values for x,y,z of 0,0,0 unset the option.
The mesh/disp keyword sets the grid size for kspace style pppm/disp. This is the FFT mesh for long-range
dispersion and ach dimension must be factorizable into powers of 2, 3, and 5. When this option is not set, the
PPPM solver chooses its own grid size, consistent with the user-specified accuracy and pairwise cutoff.
Values for x,y,z of 0,0,0 unset the option.
The order keyword determines how many grid spacings an atom's charge extends when it is mapped to the
grid in kspace style pppm or msm. The default for this parameter is 5 for PPPM and 8 for MSM, which means
each charge spans 5 or 8 grid cells in each dimension, respectively. For the LAMMPS implementation of
MSM, the order can range from 4 to 10 and must be even. For PPPM, the minimum allowed setting is 2 and
the maximum allowed setting is 7. The larger the value of this parameter, the smaller that LAMMPS will set
the grid size, to achieve the requested accuracy. Conversely, the smaller the order value, the larger the grid
size will be. Note that there is an inherent trade-off involved: a small grid will lower the cost of FFTs or MSM
direct sum, but a larger order parameter will increase the cost of interpolating charge/fields to/from the grid.
The order/disp keyword determines how many grid spacings an atom's dispersion term extends when it is
mapped to the grid in kspace style pppm/disp. It has the same meaning as the order setting for Coulombics.
The overlap keyword can be used in conjunction with the minorder keyword with the PPPM styles to adjust
the amount of communication that occurs when values on the FFT grid are exchanged between processors.
This communication is distinct from the communication inherent in the parallel FFTs themselves, and is
required because processors interpolate charge and field values using grid point values owned by neighboring
processors (i.e. ghost point communication). If the overlap keyword is set to yes then this communication is
allowed to extend beyond nearest-neighbor processors, e.g. when using lots of processors on a small problem.
If it is set to no then the communication will be limited to nearest-neighbor processors and the order setting
will be reduced if necessary, as explained by the minorder keyword discussion. The overlap keyword is
always set to yes in MSM.
The minorder keyword allows LAMMPS to reduce the order setting if necessary to keep the communication
of ghost grid point limited to exchanges between nearest-neighbor processors. See the discussion of the
overlap keyword for details. If the overlap keyword is set to yes, which is the default, this is never needed. If
it set to no and overlap occurs, then LAMMPS will reduce the order setting, one step at a time, until the ghost
grid overlap only extends to nearest neighbor processors. The minorder keyword limits how small the order
setting can become. The minimum allowed value for PPPM is 2, which is the default. If minorder is set to the
same value as order then no reduction is allowed, and LAMMPS will generate an error if the grid
communication is non-nearest-neighbor and overlap is set to no. The minorder keyword is not currently
supported in MSM.
The PPPM order parameter may be reset by LAMMPS when it sets up the FFT grid if the implied grid stencil
extends beyond the grid cells owned by neighboring processors. Typically this will only occur when small
problems are run on large numbers of processors. A warning will be generated indicating the order parameter
is being reduced to allow LAMMPS to run the problem. Automatic adjustment of the order parameter is not
supported in MSM.
The force keyword overrides the relative accuracy parameter set by the kspace_style command with an
absolute accuracy. The accuracy determines the RMS error in per-atom forces calculated by the long-range
LAMMPS Users Manual
487
solver and is thus specified in force units. A negative value for the accuracy setting means to use the relative
accuracy parameter. The accuracy setting is used in conjunction with the pairwise cutoff to determine the
number of K-space vectors for style ewald, the FFT grid size for style pppm, or the real space grid size for
style msm.
The gewald keyword sets the value of the Ewald or PPPM G-ewald parameter for charge as rinv in reciprocal
distance units. Without this setting, LAMMPS chooses the parameter automatically as a function of cutoff,
precision, grid spacing, etc. This means it can vary from one simulation to the next which may not be
desirable for matching a KSpace solver to a pre-tabulated pairwise potential. This setting can also be useful if
Ewald or PPPM fails to choose a good grid spacing and G-ewald parameter automatically. If the value is set to
0.0, LAMMPS will choose the G-ewald parameter automatically. MSM does not use the gewald parameter.
The gewald/disp keyword sets the value of the Ewald or PPPM G-ewald parameter for dispersion as rinv in
reciprocal distance units. It has the same meaning as the gewald setting for Coulombics.
The slab keyword allows an Ewald or PPPM solver to be used for a systems that are periodic in x,y but
non-periodic in z - a boundary setting of "boundary p p f". This is done by treating the system as if it were
periodic in z, but inserting empty volume between atom slabs and removing dipole inter-slab interactions so
that slab-slab interactions are effectively turned off. The volfactor value sets the ratio of the extended
dimension in z divided by the actual dimension in z. The recommended value is 3.0. A larger value is
inefficient; a smaller value introduces unwanted slab-slab interactions. The use of fixed boundaries in z means
that the user must prevent particle migration beyond the initial z-bounds, typically by providing a wall-style
fix. The methodology behind the slab option is explained in the paper by (Yeh). The slab option is also
extended to non-neutral systems (Ballenegger).
An alternative slab option can be invoked with the nozforce keyword in lieu of the volfactor. This turns off all
kspace forces in the z direction. The nozforce option is not supported by MSM. For MSM, any combination of
periodic, non-periodic, or shrink-wrapped boundaries can be set using boundary (the slab approximation in
not needed). The slab keyword is not currently supported by Ewald or PPPM when using a triclinic simulation
cell. The slab correction has also been extended to point dipole interactions (Klapp) in kspace_style
ewald/disp.
NOTE: If you wish to apply an electric field in the Z-direction, in conjunction with the slab keyword, you
should do it by adding explicit charged particles to the +/- Z surfaces. If you do it via the fix efield command,
it will not give the correct dielectric constant due to the Yeh/Berkowitz (Yeh) correction not being compatible
with how fix efield works.
The compute keyword allows Kspace computations to be turned off, even though a kspace_style is defined.
This is not useful for running a real simulation, but can be useful for debugging purposes or for computing
only partial forces that do not include the Kspace contribution. You can also do this by simply not defining a
kspace_style, but a Kspace-compatible pair_style requires a kspace style to be defined. This keyword gives
you that option.
The cutoff/adjust keyword applies only to MSM. If this option is turned on, the Coulombic cutoff will be
automatically adjusted at the beginning of the run to give the desired estimated error. Other cutoffs such as LJ
will not be affected. If the grid is not set using the mesh command, this command will also attempt to use the
optimal grid that minimizes cost using an estimate given by (Hardy). Note that this cost estimate is not exact,
somewhat experimental, and still may not yield the optimal parameters.
The pressure/scalar keyword applies only to MSM. If this option is turned on, only the scalar pressure (i.e.
(Pxx + Pyy + Pzz)/3.0) will be computed, which can be used, for example, to run an isotropic barostat.
LAMMPS Users Manual
488
Computing the full pressure tensor with MSM is expensive, and this option provides a faster alternative. The
scalar pressure is computed using a relationship between the Coulombic energy and pressure (Hummer)
instead of using the virial equation. This option cannot be used to access individual components of the
pressure tensor, to compute per-atom virial, or with suffix kspace/pair styles of MSM, like OMP or GPU.
The fftbench keyword applies only to PPPM. It is on by default. If this option is turned off, LAMMPS will not
take the time at the end of a run to give FFT benchmark timings, and will finish a few seconds faster than it
would if this option were on.
The collective keyword applies only to PPPM. It is set to no by default, except on IBM BlueGene machines. If
this option is set to yes, LAMMPS will use MPI collective operations to remap data for 3d-FFT operations
instead of the default point-to-point communication. This is faster on IBM BlueGene machines, and may also
be faster on other machines if they have an efficient implementation of MPI collective operations and
adequate hardware.
The diff keyword specifies the differentiation scheme used by the PPPM method to compute forces on
particles given electrostatic potentials on the PPPM mesh. The ik approach is the default for PPPM and is the
original formulation used in (Hockney). It performs differentiation in Kspace, and uses 3 FFTs to transfer
each component of the computed fields back to real space for total of 4 FFTs per timestep.
The analytic differentiation ad approach uses only 1 FFT to transfer information back to real space for a total
of 2 FFTs per timestep. It then performs analytic differentiation on the single quantity to generate the 3
components of the electric field at each grid point. This is sometimes referred to as "smoothed" PPPM. This
approach requires a somewhat larger PPPM mesh to achieve the same accuracy as the ik method. Currently,
only the ik method (default) can be used for a triclinic simulation cell with PPPM. The ad method is always
used for MSM.
NOTE: Currently, not all PPPM styles support the ad option. Support for those PPPM variants will be added
later.
The kmax/ewald keyword sets the number of kspace vectors in each dimension for kspace style ewald. The
three values must be positive integers, or else (0,0,0), which unsets the option. When this option is not set, the
Ewald sum scheme chooses its own kspace vectors, consistent with the user-specified accuracy and pairwise
cutoff. In any case, if kspace style ewald is invoked, the values used are printed to the screen and the log file
at the start of the run.
With the mix/disp keyword one can select the mixing rule for the dispersion coefficients. With pair, the
dispersion coefficients of unlike types are computed as indicated with pair_modify. With geom, geometric
mixing is enforced on the dispersion coefficients in the kspace coefficients. When using the arithmetic mixing
rule, this will speed-up the simulations but introduces some error in the force computations, as shown in
(Wennberg). With none, it is assumed that no mixing rule is applicable. Splitting of the dispersion coefficients
will be performed as described in (Isele-Holder). This splitting can be influenced with the splittol keywords.
Only the eigenvalues that are larger than tol compared to the largest eigenvalues are included. Using this
keywords the original matrix of dispersion coefficients is approximated. This leads to faster computations, but
the accuracy in the reciprocal space computations of the dispersion part is decreased.
The force/disp/real and force/disp/kspace keywords set the force accuracy for the real and space computations
for the dispersion part of pppm/disp. As shown in (Isele-Holder), optimal performance and accuracy in the
results is obtained when these values are different.
LAMMPS Users Manual
489

The disp/auto option controls whether the pppm/disp is allowed to generate PPPM parameters automatically.
If set to no, parameters have to be specified using the gewald/disp, mesh/disp, force/disp/real or
force/disp/kspace keywords, or the code will stop with an error message. When this option is set to yes, the
error message will not appear and the simulation will start. For a typical application, using the automatic
parameter generation will provide simulations that are either inaccurate or slow. Using this option is thus not
recommended. For guidelines on how to obtain good parameters, see the How-To discussion.
Restrictions: none
Related commands:
kspace_style, boundary
Default:
The option defaults are mesh = mesh/disp = 0 0 0, order = order/disp = 5 (PPPM), order = 10 (MSM),
minorder = 2, overlap = yes, force = -1.0, gewald = gewald/disp = 0.0, slab = 1.0, compute = yes, cutoff/adjust
= yes (MSM), pressure/scalar = yes (MSM), fftbench = yes (PPPM), diff = ik (PPPM), mix/disp = pair,
force/disp/real = -1.0, force/disp/kspace = -1.0, split = 0, tol = 1.0e-6, and disp/auto = no.
(Hockney) Hockney and Eastwood, Computer Simulation Using Particles, Adam Hilger, NY (1989).
(Yeh) Yeh and Berkowitz, J Chem Phys, 111, 3155 (1999).
(Ballenegger) Ballenegger, Arnold, Cerda, J Chem Phys, 131, 094107 (2009).
(Klapp) Klapp, Schoen, J Chem Phys, 117, 8050 (2002).
(Hardy) David Hardy thesis: Multilevel Summation for the Fast Evaluation of Forces for the Simulation of
Biomolecules, University of Illinois at Urbana-Champaign, (2006).
(Hummer) Hummer, Gronbech-Jensen, Neumann, J Chem Phys, 109, 2791 (1998)
(Isele-Holder) Isele-Holder, Mitchell, Hammond, Kohlmeyer, Ismail, J Chem Theory Comput, 9, 5412
(2013).
(Wennberg) Wennberg, Murtola, Hess, Lindahl, J Chem Theory Comput, 9, 3527 (2013).
LAMMPS Users Manual
490

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
kspace_style command
Syntax:
kspace_style style value
style = none or ewald or ewald/disp or ewald/omp or pppm or pppm/cg or pppm/disp or pppm/tip4p or
pppm/stagger or pppm/disp/tip4p or pppm/gpu or pppm/kk or pppm/omp or pppm/cg/omp or
pppm/tip4p/omp or msm or msm/cg or msm/omp or msm/cg/omp
none value = none
ewald value = accuracy
accuracy = desired relative error in forces
ewald/disp value = accuracy
accuracy = desired relative error in forces
ewald/omp value = accuracy
accuracy = desired relative error in forces
pppm value = accuracy
accuracy = desired relative error in forces
pppm/cg value = accuracy (smallq)
accuracy = desired relative error in forces
smallq = cutoff for charges to be considered (optional) (charge units)
pppm/disp value = accuracy
accuracy = desired relative error in forces
pppm/tip4p value = accuracy
accuracy = desired relative error in forces
pppm/disp/tip4p value = accuracy
accuracy = desired relative error in forces
pppm/gpu value = accuracy
accuracy = desired relative error in forces
pppm/kk value = accuracy
accuracy = desired relative error in forces
pppm/omp value = accuracy
accuracy = desired relative error in forces
pppm/cg/omp value = accuracy
accuracy = desired relative error in forces
pppm/tip4p/omp value = accuracy
accuracy = desired relative error in forces
pppm/stagger value = accuracy
accuracy = desired relative error in forces
msm value = accuracy
accuracy = desired relative error in forces
msm/cg value = accuracy (smallq)
accuracy = desired relative error in forces
smallq = cutoff for charges to be considered (optional) (charge units)
msm/omp value = accuracy
accuracy = desired relative error in forces
msm/cg/omp value = accuracy (smallq)
accuracy = desired relative error in forces
smallq = cutoff for charges to be considered (optional) (charge units)
•
Examples:
kspace_style pppm 1.0e-4
kspace_style pppm/cg 1.0e-5 1.0e-6
kspace style msm 1.0e-4
LAMMPS Users Manual
491

kspace_style none
Description:
Define a long-range solver for LAMMPS to use each timestep to compute long-range Coulombic interactions
or long-range 1/r^6 interactions. Most of the long-range solvers perform their computation in K-space, hence
the name of this command.
When such a solver is used in conjunction with an appropriate pair style, the cutoff for Coulombic or 1/r^N
interactions is effectively infinite. If the Coulombic case, this means each charge in the system interacts with
charges in an infinite array of periodic images of the simulation domain.
Note that using a long-range solver requires use of a matching pair style to perform consistent short-range
pairwise calculations. This means that the name of the pair style contains a matching keyword to the name of
the KSpace style, as in this table:
Pair style KSpace style
coul/long ewald or pppm
coul/msm msm
lj/long or buck/long disp (for dispersion)
tip4p/long tip4p
The ewald style performs a standard Ewald summation as described in any solid-state physics text.
The ewald/disp style adds a long-range dispersion sum option for 1/r^6 potentials and is useful for simulation
of interfaces (Veld). It also performs standard Coulombic Ewald summations, but in a more efficient manner
than the ewald style. The 1/r^6 capability means that Lennard-Jones or Buckingham potentials can be used
without a cutoff, i.e. they become full long-range potentials. The ewald/disp style can also be used with
point-dipoles (Toukmaji) and is currently the only kspace solver in LAMMPS with this capability.
The pppm style invokes a particle-particle particle-mesh solver (Hockney) which maps atom charge to a 3d
mesh, uses 3d FFTs to solve Poisson's equation on the mesh, then interpolates electric fields on the mesh
points back to the atoms. It is closely related to the particle-mesh Ewald technique (PME) (Darden) used in
AMBER and CHARMM. The cost of traditional Ewald summation scales as N^(3/2) where N is the number
of atoms in the system. The PPPM solver scales as Nlog(N) due to the FFTs, so it is almost always a faster
choice (Pollock).
The pppm/cg style is identical to the pppm style except that it has an optimization for systems where most
particles are uncharged. Similarly the msm/cg style implements the same optimization for msm. The optional
smallq argument defines the cutoff for the absolute charge value which determines whether a particle is
considered charged or not. Its default value is 1.0e-5.
The pppm/tip4p style is identical to the pppm style except that it adds a charge at the massless 4th site in each
TIP4P water molecule. It should be used with pair styles with a tip4p/long in their style name.
The pppm/stagger style performs calculations using two different meshes, one shifted slightly with respect to
the other. This can reduce force aliasing errors and increase the accuracy of the method for a given mesh size.
Or a coarser mesh can be used for the same target accuracy, which saves CPU time. However, there is a
trade-off since FFTs on two meshes are now performed which increases the computation required. See
LAMMPS Users Manual
492
(Cerutti), (Neelov), and (Hockney) for details of the method.
For high relative accuracy, using staggered PPPM allows the mesh size to be reduced by a factor of 2 in each
dimension as compared to regular PPPM (for the same target accuracy). This can give up to a 4x speedup in
the KSpace time (8x less mesh points, 2x more expensive). However, for low relative accuracy, the staggered
PPPM mesh size may be essentially the same as for regular PPPM, which means the method will be up to 2x
slower in the KSpace time (simply 2x more expensive). For more details and timings, see Section 5.
NOTE: Using pppm/stagger may not give the same increase in the accuracy of energy and pressure as it does
in forces, so some caution must be used if energy and/or pressure are quantities of interest, such as when using
a barostat.
The pppm/disp and pppm/disp/tip4p styles add a mesh-based long-range dispersion sum option for 1/r^6
potentials (Isele-Holder), similar to the ewald/disp style. The 1/r^6 capability means that Lennard-Jones or
Buckingham potentials can be used without a cutoff, i.e. they become full long-range potentials.
For these styles, you will possibly want to adjust the default choice of parameters by using the kspace_modify
command. This can be done by either choosing the Ewald and grid parameters, or by specifying separate
accuracies for the real and kspace calculations. When not making any settings, the simulation will stop with
an error message. Further information on the influence of the parameters and how to choose them is described
in (Isele-Holder), (Isele-Holder2) and the How-To discussion.
NOTE: All of the PPPM styles can be used with single-precision FFTs by using the compiler switch
-DFFT_SINGLE for the FFT_INC setting in your lo-level Makefile. This setting also changes some of the
PPPM operations (e.g. mapping charge to mesh and interpolating electric fields to particles) to be performed
in single precision. This option can speed-up long-range calculations, particularly in parallel or on GPUs. The
use of the -DFFT_SINGLE flag is discussed in this section of the manual. MSM does not currently support
the -DFFT_SINGLE compiler switch.
The msm style invokes a multi-level summation method MSM solver, (Hardy) or (Hardy2), which maps atom
charge to a 3d mesh, and uses a multi-level hierarchy of coarser and coarser meshes on which direct coulomb
solves are done. This method does not use FFTs and scales as N. It may therefore be faster than the other
K-space solvers for relatively large problems when running on large core counts. MSM can also be used for
non-periodic boundary conditions and for mixed periodic and non-periodic boundaries.
MSM is most competitive versus Ewald and PPPM when only relatively low accuracy forces, about 1e-4
relative error or less accurate, are needed. Note that use of a larger coulomb cutoff (i.e. 15 angstroms instead
of 10 angstroms) provides better MSM accuracy for both the real space and grid computed forces.
Currently calculation of the full pressure tensor in MSM is expensive. Using the kspace_modify
pressure/scalar yes command provides a less expensive way to compute the scalar pressure (Pxx + Pyy +
Pzz)/3.0. The scalar pressure can be used, for example, to run an isotropic barostat. If the full pressure tensor
is needed, then calculating the pressure at every timestep or using a fixed pressure simulation with MSM will
cause the code to run slower.
The specified accuracy determines the relative RMS error in per-atom forces calculated by the long-range
solver. It is set as a dimensionless number, relative to the force that two unit point charges (e.g. 2 monovalent
ions) exert on each other at a distance of 1 Angstrom. This reference value was chosen as representative of the
magnitude of electrostatic forces in atomic systems. Thus an accuracy value of 1.0e-4 means that the RMS
error will be a factor of 10000 smaller than the reference force.
LAMMPS Users Manual
493

The accuracy setting is used in conjunction with the pairwise cutoff to determine the number of K-space
vectors for style ewald or the grid size for style pppm or msm.
Note that style pppm only computes the grid size at the beginning of a simulation, so if the length or triclinic
tilt of the simulation cell increases dramatically during the course of the simulation, the accuracy of the
simulation may degrade. Likewise, if the kspace_modify slab option is used with shrink-wrap boundaries in
the z-dimension, and the box size changes dramatically in z. For example, for a triclinic system with all three
tilt factors set to the maximum limit, the PPPM grid should be increased roughly by a factor of 1.5 in the y
direction and 2.0 in the z direction as compared to the same system using a cubic orthogonal simulation cell.
One way to handle this issue if you have a long simulation where the box size changes dramatically, is to
break it into shorter simulations (multiple run commands). This works because the grid size is re-computed at
the beginning of each run. Another way to ensure the described accuracy requirement is met is to run a short
simulation at the maximum expected tilt or length, note the required grid size, and then use the kspace_modify
mesh command to manually set the PPPM grid size to this value for the long run. The simulation then will be
"too accurate" for some portion of the run.
RMS force errors in real space for ewald and pppm are estimated using equation 18 of (Kolafa), which is also
referenced as equation 9 of (Petersen). RMS force errors in K-space for ewald are estimated using equation 11
of (Petersen), which is similar to equation 32 of (Kolafa). RMS force errors in K-space for pppm are estimated
using equation 38 of (Deserno). RMS force errors for msm are estimated using ideas from chapter 3 of
(Hardy), with equation 3.197 of particular note. When using msm with non-periodic boundary conditions, it is
expected that the error estimation will be too pessimistic. RMS force errors for dipoles when using ewald/disp
are estimated using equations 33 and 46 of (Wang).
See the kspace_modify command for additional options of the K-space solvers that can be set, including a
force option for setting an absolute RMS error in forces, as opposed to a relative RMS error.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
More specifically, the pppm/gpu style performs charge assignment and force interpolation calculations on the
GPU. These processes are performed either in single or double precision, depending on whether the
-DFFT_SINGLE setting was specified in your lo-level Makefile, as discussed above. The FFTs themselves
are still calculated on the CPU. If pppm/gpu is used with a GPU-enabled pair style, part of the PPPM
calculation can be performed concurrently on the GPU while other calculations for non-bonded and bonded
force calculation are performed on the CPU.
The pppm/kk style also performs charge assignment and force interpolation calculations on the GPU while the
FFTs themselves are calculated on the CPU in non-threaded mode.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP, and OPT packages
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
494

Note that the long-range electrostatic solvers in LAMMPS assume conducting metal (tinfoil) boundary
conditions for both charge and dipole interactions. Vacuum boundary conditions are not currently supported.
The ewald/disp, ewald, pppm, and msm styles support non-orthogonal (triclinic symmetry) simulation boxes.
However, triclinic simulation cells may not yet be supported by suffix versions of these styles.
All of the kspace styles are part of the KSPACE package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info. Note that the KSPACE package is installed by
default.
For MSM, a simulation must be 3d and one can use any combination of periodic, non-periodic, or
shrink-wrapped boundaries (specified using the boundary command).
For Ewald and PPPM, a simulation must be 3d and periodic in all dimensions. The only exception is if the
slab option is set with kspace_modify, in which case the xy dimensions must be periodic and the z dimension
must be non-periodic.
Related commands:
kspace_modify, pair_style lj/cut/coul/long, pair_style lj/charmm/coul/long, pair_style lj/long/coul/long,
pair_style buck/coul/long
Default:
kspace_style none
(Darden) Darden, York, Pedersen, J Chem Phys, 98, 10089 (1993).
(Deserno) Deserno and Holm, J Chem Phys, 109, 7694 (1998).
(Hockney) Hockney and Eastwood, Computer Simulation Using Particles, Adam Hilger, NY (1989).
(Kolafa) Kolafa and Perram, Molecular Simulation, 9, 351 (1992).
(Petersen) Petersen, J Chem Phys, 103, 3668 (1995).
(Wang) Wang and Holm, J Chem Phys, 115, 6277 (2001).
(Pollock) Pollock and Glosli, Comp Phys Comm, 95, 93 (1996).
(Cerutti) Cerutti, Duke, Darden, Lybrand, Journal of Chemical Theory and Computation 5, 2322 (2009)
LAMMPS Users Manual
495
(Neelov) Neelov, Holm, J Chem Phys 132, 234103 (2010)
(Veld) In 't Veld, Ismail, Grest, J Chem Phys, 127, 144711 (2007).
(Toukmaji) Toukmaji, Sagui, Board, and Darden, J Chem Phys, 113, 10913 (2000).
(Isele-Holder) Isele-Holder, Mitchell, Ismail, J Chem Phys, 137, 174107 (2012).
(Isele-Holder2) Isele-Holder, Mitchell, Hammond, Kohlmeyer, Ismail, J Chem Theory Comput 9, 5412
(2013).
(Hardy) David Hardy thesis: Multilevel Summation for the Fast Evaluation of Forces for the Simulation of
Biomolecules, University of Illinois at Urbana-Champaign, (2006).
(Hardy2) Hardy, Stone, Schulten, Parallel Computing 35 (2009) 164-177.
LAMMPS Users Manual
496

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
label command
Syntax:
label ID
ID = string used as label name•
Examples:
label xyz
label loop
Description:
Label this line of the input script with the chosen ID. Unless a jump command was used previously, this does
nothing. But if a jump command was used with a label argument to begin invoking this script file, then all
command lines in the script prior to this line will be ignored. I.e. execution of the script will begin at this line.
This is useful for looping over a section of the input script as discussed in the jump command.
Restrictions: none
Related commands: none
Default: none
LAMMPS Users Manual
497

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
lattice command
Syntax:
lattice style scale keyword values ...
style = none or sc or bcc or fcc or hcp or diamond or sq or sq2 or hex or custom• scale = scale factor between lattice and simulation box
scale = reduced density rho* (for LJ units)
scale = lattice constant in distance units (for all other units)
•
zero or more keyword/value pairs may be appended• keyword = origin or orient or spacing or a1 or a2 or a3 or basis
origin values = x y z
x,y,z = fractions of a unit cell (0 <= x,y,z <1)
orient values = dim i j k
dim = x or y or z
i,j,k = integer lattice directions
spacing values = dx dy dz
dx,dy,dz = lattice spacings in the x,y,z box directions
a1,a2,a3 values = x y z
x,y,z = primitive vector components that define unit cell
basis values = x y z
x,y,z = fractional coords of a basis atom (0 <= x,y,z <1)
•
Examples:
lattice fcc 3.52
lattice hex 0.85
lattice sq 0.8 origin 0.0 0.5 0.0 orient x 1 1 0 orient y -1 1 0
lattice custom 3.52 a1 1.0 0.0 0.0 a2 0.5 1.0 0.0 a3 0.0 0.0 0.5 &
basis 0.0 0.0 0.0 basis 0.5 0.5 0.5
lattice none 2.0
Description:
Define a lattice for use by other commands. In LAMMPS, a lattice is simply a set of points in space,
determined by a unit cell with basis atoms, that is replicated infinitely in all dimensions. The arguments of the
lattice command can be used to define a wide variety of crystallographic lattices.
A lattice is used by LAMMPS in two ways. First, the create_atoms command creates atoms on the lattice
points inside the simulation box. Note that the create_atoms command allows different atom types to be
assigned to different basis atoms of the lattice. Second, the lattice spacing in the x,y,z dimensions implied by
the lattice, can be used by other commands as distance units (e.g. create_box, region and velocity), which are
often convenient to use when the underlying problem geometry is atoms on a lattice.
The lattice style must be consistent with the dimension of the simulation - see the dimension command. Styles
sc or bcc or fcc or hcp or diamond are for 3d problems. Styles sq or sq2 or hex are for 2d problems. Style
custom can be used for either 2d or 3d problems.
LAMMPS Users Manual
498
A lattice consists of a unit cell, a set of basis atoms within that cell, and a set of transformation parameters
(scale, origin, orient) that map the unit cell into the simulation box. The vectors a1,a2,a3 are the edge vectors
of the unit cell. This is the nomenclature for "primitive" vectors in solid-state crystallography, but in
LAMMPS the unit cell they determine does not have to be a "primitive cell" of minimum volume.
Note that the lattice command can be used multiple times in an input script. Each time it is invoked, the lattice
attributes are re-defined and are used for all subsequent commands (that use lattice attributes). For example, a
sequence of lattice, region, and create_atoms commands can be repeated multiple times to build a
poly-crystalline model with different geometric regions populated with atoms in different lattice orientations.
A lattice of style none does not define a unit cell and basis set, so it cannot be used with the create_atoms
command. However it does define a lattice spacing via the specified scale parameter. As explained above the
lattice spacings in x,y,z can be used by other commands as distance units. No additional keyword/value pairs
can be specified for the none style. By default, a "lattice none 1.0" is defined, which means the lattice spacing
is the same as one distance unit, as defined by the units command.
Lattices of style sc, fcc, bcc, and diamond are 3d lattices that define a cubic unit cell with edge length = 1.0.
This means a1 = 1 0 0, a2 = 0 1 0, and a3 = 0 0 1. Style hcp has a1 = 1 0 0, a2 = 0 sqrt(3) 0, and a3 = 0 0
sqrt(8/3). The placement of the basis atoms within the unit cell are described in any solid-state physics text. A
sc lattice has 1 basis atom at the lower-left-bottom corner of the cube. A bcc lattice has 2 basis atoms, one at
the corner and one at the center of the cube. A fcc lattice has 4 basis atoms, one at the corner and 3 at the cube
face centers. A hcp lattice has 4 basis atoms, two in the z = 0 plane and 2 in the z = 0.5 plane. A diamond
lattice has 8 basis atoms.
Lattices of style sq and sq2 are 2d lattices that define a square unit cell with edge length = 1.0. This means a1
= 1 0 0 and a2 = 0 1 0. A sq lattice has 1 basis atom at the lower-left corner of the square. A sq2 lattice has 2
basis atoms, one at the corner and one at the center of the square. A hex style is also a 2d lattice, but the unit
cell is rectangular, with a1 = 1 0 0 and a2 = 0 sqrt(3) 0. It has 2 basis atoms, one at the corner and one at the
center of the rectangle.
A lattice of style custom allows you to specify a1, a2, a3, and a list of basis atoms to put in the unit cell. By
default, a1 and a2 and a3 are 3 orthogonal unit vectors (edges of a unit cube). But you can specify them to be
of any length and non-orthogonal to each other, so that they describe a tilted parallelepiped. Via the basis
keyword you add atoms, one at a time, to the unit cell. Its arguments are fractional coordinates (0.0 <= x,y,z <
1.0). The position vector x of a basis atom within the unit cell is thus a linear combination of the the unit cell's
3 edge vectors, i.e. x = bx a1 + by a2 + bz a3, where bx,by,bz are the 3 values specified for the basis keyword.
This sub-section discusses the arguments that determine how the idealized unit cell is transformed into a
lattice of points within the simulation box.
The scale argument determines how the size of the unit cell will be scaled when mapping it into the
simulation box. I.e. it determines a multiplicative factor to apply to the unit cell, to convert it to a lattice of the
desired size and distance units in the simulation box. The meaning of the scale argument depends on the units
being used in your simulation.
For all unit styles except lj, the scale argument is specified in the distance units defined by the unit style. For
example, in real or metal units, if the unit cell is a unit cube with edge length 1.0, specifying scale = 3.52
would create a cubic lattice with a spacing of 3.52 Angstroms. In cgs units, the spacing would be 3.52 cm.
For unit style lj, the scale argument is the Lennard-Jones reduced density, typically written as rho*. LAMMPS
LAMMPS Users Manual
499

converts this value into the multiplicative factor via the formula "factor^dim = rho/rho*", where rho = N/V
with V = the volume of the lattice unit cell and N = the number of basis atoms in the unit cell (described
below), and dim = 2 or 3 for the dimensionality of the simulation. Effectively, this means that if LJ particles
of size sigma = 1.0 are used in the simulation, the lattice of particles will be at the desired reduced density.
The origin option specifies how the unit cell will be shifted or translated when mapping it into the simulation
box. The x,y,z values are fractional values (0.0 <= x,y,z < 1.0) meaning shift the lattice by a fraction of the
lattice spacing in each dimension. The meaning of "lattice spacing" is discussed below.
The orient option specifies how the unit cell will be rotated when mapping it into the simulation box. The dim
argument is one of the 3 coordinate axes in the simulation box. The other 3 arguments are the crystallographic
direction in the lattice that you want to orient along that axis, specified as integers. E.g. "orient x 2 1 0" means
the x-axis in the simulation box will be the [210] lattice direction, and similarly for y and z. The 3 lattice
directions you specify do not have to be unit vectors, but they must be mutually orthogonal and obey the
right-hand rule, i.e. (X cross Y) points in the Z direction.
NOTE: The preceding paragraph describing lattice directions is only valid for orthogonal cubic unit cells (or
square in 2d). If you are using a hcp or hex lattice or the more general lattice style custom with non-orthogonal
a1,a2,a3 vectors, then you should think of the 3 orient vectors as creating a 3x3 rotation matrix which is
applied to a1,a2,a3 to rotate the original unit cell to a new orientation in the simulation box.
Several LAMMPS commands have the option to use distance units that are inferred from "lattice spacings" in
the x,y,z box directions. E.g. the region command can create a block of size 10x20x20, where 10 means 10
lattice spacings in the x direction.
NOTE: Though they are called lattice spacings, all the commands that have a "units lattice" option, simply use
the 3 values as scale factors on the distance units defined by the units command. Thus if you do not like the
lattice spacings computed by LAMMPS (e.g. for a non-orthogonal or rotated unit cell), you can define the 3
values to be whatever you wish, via the spacing option.
If the spacing option is not specified, the lattice spacings are computed by LAMMPS in the following way. A
unit cell of the lattice is mapped into the simulation box (scaled and rotated), so that it now has (perhaps) a
modified size and orientation. The lattice spacing in X is defined as the difference between the min/max
extent of the x coordinates of the 8 corner points of the modified unit cell (4 in 2d). Similarly, the Y and Z
lattice spacings are defined as the difference in the min/max of the y and z coordinates.
Note that if the unit cell is orthogonal with axis-aligned edges (no rotation via the orient keyword), then the
lattice spacings in each dimension are simply the scale factor (described above) multiplied by the length of
a1,a2,a3. Thus a hex style lattice with a scale factor of 3.0 Angstroms, would have a lattice spacing of 3.0 in x
and 3*sqrt(3.0) in y.
NOTE: For non-orthogonal unit cells and/or when a rotation is applied via the orient keyword, then the lattice
spacings computed by LAMMPS are typically less intuitive. In particular, in these cases, there is no guarantee
that a particular lattice spacing is an integer multiple of the periodicity of the lattice in that direction. Thus, if
you create an orthogonal periodic simulation box whose size in a dimension is a multiple of the lattice
spacing, and then fill it with atoms via the create_atoms command, you will NOT necessarily create a periodic
system. I.e. atoms may overlap incorrectly at the faces of the simulation box.
The spacing option sets the 3 lattice spacings directly. All must be non-zero (use 1.0 for dz in a 2d
simulation). The specified values are multiplied by the multiplicative factor described above that is associated
with the scale factor. Thus a spacing of 1.0 means one unit cell edge length independent of the scale factor. As
LAMMPS Users Manual
500
mentioned above, this option can be useful if the spacings LAMMPS computes are inconvenient to use in
subsequent commands, which can be the case for non-orthogonal or rotated lattices.
Note that whenever the lattice command is used, the values of the lattice spacings LAMMPS calculates are
printed out. Thus their effect in commands that use the spacings should be decipherable.
Example commands for generating a Wurtzite crystal (courtesy of Aidan Thompson), with its 8 atom unit cell.
variable a equal 4.340330
variable b equal $a*sqrt(3.0)
variable c equal $a*sqrt(8.0/3.0)
variable 1_3 equal 1.0/3.0
variable 2_3 equal 2.0/3.0
variable 1_6 equal 1.0/6.0
variable 5_6 equal 5.0/6.0
variable 1_12 equal 1.0/12.0
variable 5_12 equal 5.0/12.0
lattice custom 1.0 &
a1 $a 0.0 0.0 &
a2 0.0 $b 0.0 &
a3 0.0 0.0 $c &
basis 0.0 0.0 0.0 &
basis 0.5 0.5 0.0 &
basis ${1_3} 0.0 0.5 &
basis ${5_6} 0.5 0.5 &
basis 0.0 0.0 0.625 &
basis 0.5 0.5 0.625 &
basis ${1_3} 0.0 0.125 &
basis ${5_6} 0.5 0.125
region myreg block 0 1 0 1 0 1
create_box 2 myreg
create_atoms 1 box &
basis 5 2 &
basis 6 2 &
basis 7 2 &
basis 8 2
Restrictions:
The a1,a2,a3,basis keywords can only be used with style custom.
Related commands:
dimension, create_atoms, region
Default:
lattice none 1.0
For other lattice styles, the option defaults are origin = 0.0 0.0 0.0, orient = x 1 0 0, orient = y 0 1 0, orient = z
0 0 1, a1 = 1 0 0, a2 = 0 1 0, and a3 = 0 0 1.
LAMMPS Users Manual
501

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
log command
Syntax:
log file keyword
file = name of new logfile• keyword = append if output should be appended to logfile (optional)•
Examples:
log log.equil
log log.equil append
Description:
This command closes the current LAMMPS log file, opens a new file with the specified name, and begins
logging information to it. If the specified file name is none, then no new log file is opened. If the optional
keyword append is specified, then output will be appended to an existing log file, instead of overwriting it.
If multiple processor partitions are being used, the file name should be a variable, so that different processors
do not attempt to write to the same log file.
The file "log.lammps" is the default log file for a LAMMPS run. The name of the initial log file can also be
set by the command-line switch -log. See Section 2.7 for details.
Restrictions: none
Related commands: none
Default:
The default LAMMPS log file is named log.lammps
LAMMPS Users Manual
502

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
mass command
Syntax:
mass I value
I = atom type (see asterisk form below)• value = mass•
Examples:
mass 1 1.0
mass * 62.5
mass 2* 62.5
Description:
Set the mass for all atoms of one or more atom types. Per-type mass values can also be set in the read_data
data file using the "Masses" keyword. See the units command for what mass units to use.
The I index can be specified in one of two ways. An explicit numeric value can be used, as in the 1st example
above. Or a wild-card asterisk can be used to set the mass for multiple atom types. This takes the form "*" or
"*n" or "n*" or "m*n". If N = the number of atom types, then an asterisk with no numeric values means all
types from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A trailing asterisk means all
types from n to N (inclusive). A middle asterisk means all types from m to n (inclusive).
A line in a data file that follows the "Masses" keyword specifies mass using the same format as the arguments
of the mass command in an input script, except that no wild-card asterisk can be used. For example, under the
"Masses" section of a data file, the line that corresponds to the 1st example above would be listed as
1 1.0
Note that the mass command can only be used if the atom style requires per-type atom mass to be set.
Currently, all but the sphere and ellipsoid and peri styles do. They require mass to be set for individual
particles, not types. Per-atom masses are defined in the data file read by the read_data command, or set to
default values by the create_atoms command. Per-atom masses can also be set to new values by the set mass
or set density commands.
Also note that pair_style eam and pair_style bop commands define the masses of atom types in their
respective potential files, in which case the mass command is normally not used.
If you define a hybrid atom style which includes one (or more) sub-styles which require per-type mass and
one (or more) sub-styles which require per-atom mass, then you must define both. However, in this case the
per-type mass will be ignored; only the per-atom mass will be used by LAMMPS.
Restrictions:
This command must come after the simulation box is defined by a read_data, read_restart, or create_box
command.
LAMMPS Users Manual
503

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
min_modify command
Syntax:
min_modify keyword values ...
one or more keyword/value pairs may be listed
keyword = dmax or line
dmax value = max
max = maximum distance for line search to move (distance units)
line value = backtrack or quadratic or forcezero
backtrack,quadratic,forcezero = style of linesearch to use
•
Examples:
min_modify dmax 0.2
Description:
This command sets parameters that affect the energy minimization algorithms selected by the min_style
command. The various settings may affect the convergence rate and overall number of force evaluations
required by a minimization, so users can experiment with these parameters to tune their minimizations.
The cg and sd minimization styles have an outer iteration and an inner iteration which is steps along a
one-dimensional line search in a particular search direction. The dmax parameter is how far any atom can
move in a single line search in any dimension (x, y, or z). For the quickmin and fire minimization styles, the
dmax setting is how far any atom can move in a single iteration (timestep). Thus a value of 0.1 in real units
means no atom will move further than 0.1 Angstroms in a single outer iteration. This prevents highly
overlapped atoms from being moved long distances (e.g. through another atom) due to large forces.
The choice of line search algorithm for the cg and sd minimization styles can be selected via the line keyword.
The default quadratic line search algorithm starts out using the robust backtracking method described below.
However, once the system gets close to a local minimum and the linesearch steps get small, so that the energy
is approximately quadratic in the step length, it uses the estimated location of zero gradient as the linesearch
step, provided the energy change is downhill. This becomes more efficient than backtracking for
highly-converged relaxations. The forcezero line search algorithm is similar to quadratic. It may be more
efficient than quadratic on some systems.
The backtracking search is robust and should always find a local energy minimum. However, it will
"converge" when it can no longer reduce the energy of the system. Individual atom forces may still be larger
than desired at this point, because the energy change is measured as the difference of two large values (energy
before and energy after) and that difference may be smaller than machine epsilon even if atoms could move in
the gradient direction to reduce forces further.
Restrictions: none
Related commands:
min_style, minimize
LAMMPS Users Manual
505
Default:
The option defaults are dmax = 0.1 and line = quadratic.
LAMMPS Users Manual
506

LAMMPS WWW Page - LAMMPS Documentation - LAMMPS Commands
min_style command
Syntax:
min_style style
style = cg or hftn or sd or quickmin or fire•
Examples:
min_style cg
min_style fire
Description:
Choose a minimization algorithm to use when a minimize command is performed.
Style cg is the Polak-Ribiere version of the conjugate gradient (CG) algorithm. At each iteration the force
gradient is combined with the previous iteration information to compute a new search direction perpendicular
(conjugate) to the previous search direction. The PR variant affects how the direction is chosen and how the
CG method is restarted when it ceases to make progress. The PR variant is thought to be the most effective
CG choice for most problems.
Style hftn is a Hessian-free truncated Newton algorithm. At each iteration a quadratic model of the energy
potential is solved by a conjugate gradient inner iteration. The Hessian (second derivatives) of the energy is
not formed directly, but approximated in each conjugate search direction by a finite difference directional
derivative. When close to an energy minimum, the algorithm behaves like a Newton method and exhibits a
quadratic convergence rate to high accuracy. In most cases the behavior of hftn is similar to cg, but it offers an
alternative if cg seems to perform poorly. This style is not affected by the min_modify command.
Style sd is a steepest descent algorithm. At each iteration, the search direction is set to the downhill direction
corresponding to the force vector (negative gradient of energy). Typically, steepest descent will not converge
as quickly as CG, but may be more robust in some situations.
Style quickmin is a damped dynamics method described in (Sheppard), where the damping parameter is
related to the projection of the velocity vector along the current force vector for each atom. The velocity of
each atom is initialized to 0.0 by this style, at the beginning of a minimization.
Style fire is a damped dynamics method described in (Bitzek), which is similar to quickmin but adds a
variable timestep and alters the projection operation to maintain components of the velocity non-parallel to the
current force vector. The velocity of each atom is initialized to 0.0 by this style, at the beginning of a
minimization.
Either the quickmin and fire styles are useful in the context of nudged elastic band (NEB) calculations via the
neb command.
NOTE: The damped dynamic minimizers use whatever timestep you have defined via the timestep command.
Often they will converge more quickly if you use a timestep about 10x larger than you would normally use for
dynamics simulations.
LAMMPS Users Manual
507

NOTE: The quickmin, fire, and hftn styles do not yet support the use of the fix box/relax command or
minimizations involving the electron radius in eFF models.
Restrictions: none
Related commands:
min_modify, minimize, neb
Default:
min_style cg
(Sheppard) Sheppard, Terrell, Henkelman, J Chem Phys, 128, 134106 (2008). See ref 1 in this paper for
original reference to Qmin in Jonsson, Mills, Jacobsen.
(Bitzek) Bitzek, Koskinen, Gahler, Moseler, Gumbsch, Phys Rev Lett, 97, 170201 (2006).
LAMMPS Users Manual
508

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
minimize command
Syntax:
minimize etol ftol maxiter maxeval
etol = stopping tolerance for energy (unitless)• ftol = stopping tolerance for force (force units)• maxiter = max iterations of minimizer• maxeval = max number of force/energy evaluations•
Examples:
minimize 1.0e-4 1.0e-6 100 1000
minimize 0.0 1.0e-8 1000 100000
Description:
Perform an energy minimization of the system, by iteratively adjusting atom coordinates. Iterations are
terminated when one of the stopping criteria is satisfied. At that point the configuration will hopefully be in
local potential energy minimum. More precisely, the configuration should approximate a critical point for the
objective function (see below), which may or may not be a local minimum.
The minimization algorithm used is set by the min_style command. Other options are set by the min_modify
command. Minimize commands can be interspersed with run commands to alternate between relaxation and
dynamics. The minimizers bound the distance atoms move in one iteration, so that you can relax systems with
highly overlapped atoms (large energies and forces) by pushing the atoms off of each other.
Alternate means of relaxing a system are to run dynamics with a small or limited timestep. Or dynamics can
be run using fix viscous to impose a damping force that slowly drains all kinetic energy from the system. The
pair_style soft potential can be used to un-overlap atoms while running dynamics.
Note that you can minimize some atoms in the system while holding the coordinates of other atoms fixed by
applying fix setforce to the other atoms. See a fuller discussion of using fixes while minimizing below.
The minimization styles cg, sd, and hftn involves an outer iteration loop which sets the search direction along
which atom coordinates are changed. An inner iteration is then performed using a line search algorithm. The
line search typically evaluates forces and energies several times to set new coordinates. Currently, a
backtracking algorithm is used which may not be optimal in terms of the number of force evaluations
performed, but appears to be more robust than previous line searches we've tried. The backtracking method is
described in Nocedal and Wright's Numerical Optimization (Procedure 3.1 on p 41).
The minimization styles quickmin and fire perform damped dynamics using an Euler integration step. Thus
they require a timestep be defined.
NOTE: The damped dynamic minimizers use whatever timestep you have defined via the timestep command.
Often they will converge more quickly if you use a timestep about 10x larger than you would normally use for
dynamics simulations.
LAMMPS Users Manual
509
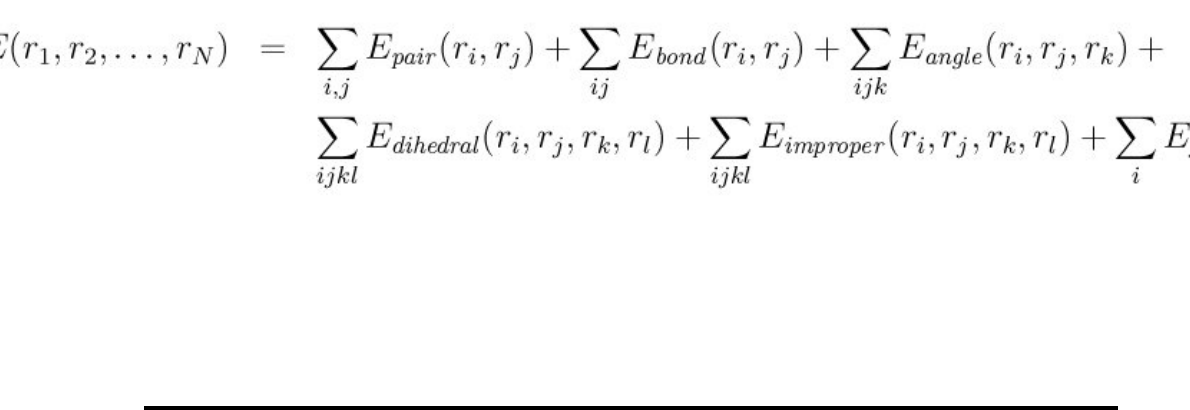
In all cases, the objective function being minimized is the total potential energy of the system as a function of
the N atom coordinates:
where the first term is the sum of all non-bonded pairwise interactions including long-range Coulombic
interactions, the 2nd thru 5th terms are bond, angle, dihedral, and improper interactions respectively, and the
last term is energy due to fixes which can act as constraints or apply force to atoms, such as thru interaction
with a wall. See the discussion below about how fix commands affect minimization.
The starting point for the minimization is the current configuration of the atoms.
The minimization procedure stops if any of several criteria are met:
the change in energy between outer iterations is less than etol• the 2-norm (length) of the global force vector is less than the ftol• the line search fails because the step distance backtracks to 0.0• the number of outer iterations or timesteps exceeds maxiter• the number of total force evaluations exceeds maxeval•
NOTE: You can also use the fix halt command to specify a general criterion for exiting a minimization, that is
a calculation performed on the state of the current system, as defined by an equal-style variable.
For the first criterion, the specified energy tolerance etol is unitless; it is met when the energy change between
successive iterations divided by the energy magnitude is less than or equal to the tolerance. For example, a
setting of 1.0e-4 for etol means an energy tolerance of one part in 10^4. For the damped dynamics minimizers
this check is not performed for a few steps after velocities are reset to 0, otherwise the minimizer would
prematurely converge.
For the second criterion, the specified force tolerance ftol is in force units, since it is the length of the global
force vector for all atoms, e.g. a vector of size 3N for N atoms. Since many of the components will be near
zero after minimization, you can think of ftol as an upper bound on the final force on any component of any
atom. For example, a setting of 1.0e-4 for ftol means no x, y, or z component of force on any atom will be
larger than 1.0e-4 (in force units) after minimization.
Either or both of the etol and ftol values can be set to 0.0, in which case some other criterion will terminate the
minimization.
During a minimization, the outer iteration count is treated as a timestep. Output is triggered by this timestep,
e.g. thermodynamic output or dump and restart files.
Using the thermo_style custom command with the fmax or fnorm keywords can be useful for monitoring the
progress of the minimization. Note that these outputs will be calculated only from forces on the atoms, and
will not include any extra degrees of freedom, such as from the fix box/relax command.
LAMMPS Users Manual
510

Following minimization, a statistical summary is printed that lists which convergence criterion caused the
minimizer to stop, as well as information about the energy, force, final line search, and iteration counts. An
example is as follows:
Minimization stats:
Stopping criterion = max iterations
Energy initial, next-to-last, final =
-0.626828169302 -2.82642039062 -2.82643549739
Force two-norm initial, final = 2052.1 91.9642
Force max component initial, final = 346.048 9.78056
Final line search alpha, max atom move = 2.23899e-06 2.18986e-05
Iterations, force evaluations = 2000 12724
The 3 energy values are for before and after the minimization and on the next-to-last iteration. This is what the
etol parameter checks.
The two-norm force values are the length of the global force vector before and after minimization. This is
what the ftol parameter checks.
The max-component force values are the absolute value of the largest component (x,y,z) in the global force
vector, i.e. the infinity-norm of the force vector.
The alpha parameter for the line-search, when multiplied by the max force component (on the last iteration),
gives the max distance any atom moved during the last iteration. Alpha will be 0.0 if the line search could not
reduce the energy. Even if alpha is non-zero, if the "max atom move" distance is tiny compared to typical
atom coordinates, then it is possible the last iteration effectively caused no atom movement and thus the
evaluated energy did not change and the minimizer terminated. Said another way, even with non-zero forces,
it's possible the effect of those forces is to move atoms a distance less than machine precision, so that the
energy cannot be further reduced.
The iterations and force evaluation values are what is checked by the maxiter and maxeval parameters.
NOTE: There are several force fields in LAMMPS which have discontinuities or other approximations which
may prevent you from performing an energy minimization to high tolerances. For example, you should use a
pair style that goes to 0.0 at the cutoff distance when performing minimization (even if you later change it
when running dynamics). If you do not do this, the total energy of the system will have discontinuities when
the relative distance between any pair of atoms changes from cutoff+epsilon to cutoff-epsilon and the
minimizer may behave poorly. Some of the manybody potentials use splines and other internal cutoffs that
inherently have this problem. The long-range Coulombic styles (PPPM, Ewald) are approximate to within the
user-specified tolerance, which means their energy and forces may not agree to a higher precision than the
Kspace-specified tolerance. In all these cases, the minimizer may give up and stop before finding a minimum
to the specified energy or force tolerance.
Note that a cutoff Lennard-Jones potential (and others) can be shifted so that its energy is 0.0 at the cutoff via
the pair_modify command. See the doc pages for individual pair styles for details. Note that Coulombic
potentials always have a cutoff, unless versions with a long-range component are used (e.g. pair_style
lj/cut/coul/long). The CHARMM potentials go to 0.0 at the cutoff (e.g. pair_style lj/charmm/coul/charmm), as
do the GROMACS potentials (e.g. pair_style lj/gromacs).
If a soft potential (pair_style soft) is used the Astop value is used for the prefactor (no time dependence).
LAMMPS Users Manual
511

The fix box/relax command can be used to apply an external pressure to the simulation box and allow it to
shrink/expand during the minimization.
Only a few other fixes (typically those that apply force constraints) are invoked during minimization. See the
doc pages for individual fix commands to see which ones are relevant. Current examples of fixes that can be
used include:
fix addforce• fix addtorque• fix efield• fix enforce2d• fix indent• fix lineforce• fix planeforce• fix setforce• fix spring• fix spring/self• fix viscous• fix wall• fix wall/region•
NOTE: Some fixes which are invoked during minimization have an associated potential energy. For that
energy to be included in the total potential energy of the system (the quantity being minimized), you MUST
enable the fix_modify energy option for that fix. The doc pages for individual fix commands specify if this
should be done.
Restrictions:
Features that are not yet implemented are listed here, in case someone knows how they could be coded:
It is an error to use fix shake with minimization because it turns off bonds that should be included in the
potential energy of the system. The effect of a fix shake can be approximated during a minimization by using
stiff spring constants for the bonds and/or angles that would normally be constrained by the SHAKE
algorithm.
Fix rigid is also not supported by minimization. It is not an error to have it defined, but the energy
minimization will not keep the defined body(s) rigid during the minimization. Note that if bonds, angles, etc
internal to a rigid body have been turned off (e.g. via neigh_modify exclude), they will not contribute to the
potential energy which is probably not what is desired.
Pair potentials that produce torque on a particle (e.g. granular potentials or the GayBerne potential for
ellipsoidal particles) are not relaxed by a minimization. More specifically, radial relaxations are induced, but
no rotations are induced by a minimization, so such a system will not fully relax.
Related commands:
min_modify, min_style, run_style
Default: none
LAMMPS Users Manual
512

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
molecule command
Syntax:
molecule ID file1 keyword values ... file2 keyword values ... fileN ...
ID = user-assigned name for the molecule template• file1,file2,... = names of files containing molecule descriptions• zero or more keyword/value pairs may be appended after each file• keyword = offset or toff or boff or aoff or doff or ioff or scale
offset values = Toff Boff Aoff Doff Ioff
Toff = offset to add to atom types
Boff = offset to add to bond types
Aoff = offset to add to angle types
Doff = offset to add to dihedral types
Ioff = offset to add to improper types
toff value = Toff
Toff = offset to add to atom types
boff value = Boff
Boff = offset to add to bond types
aoff value = Aoff
Aoff = offset to add to angle types
doff value = Doff
Doff = offset to add to dihedral types
ioff value = Ioff
Ioff = offset to add to improper types
scale value = sfactor
sfactor = scale factor to apply to the size and mass of the molecule
•
Examples:
molecule 1 mymol.txt
molecule 1 co2.txt h2o.txt
molecule CO2 co2.txt boff 3 aoff 2
molecule 1 mymol.txt offset 6 9 18 23 14
molecule objects file.1 scale 1.5 file.1 scale 2.0 file.2 scale 1.3
Description:
Define a molecule template that can be used as part of other LAMMPS commands, typically to define a
collection of particles as a bonded molecule or a rigid body. Commands that currently use molecule templates
include:
fix deposit• fix pour• fix rigid/small• fix shake• fix gcmc• create_atoms• atom_style template•
LAMMPS Users Manual
513
The ID of a molecule template can only contain alphanumeric characters and underscores.
A single template can contain multiple molecules, listed one per file. Some of the commands listed above
currently use only the first molecule in the template, and will issue a warning if the template contains multiple
molecules. The atom_style template command allows multiple-molecule templates to define a system with
more than one templated molecule.
Each filename can be followed by optional keywords which are applied only to the molecule in the file as
used in this template. This is to make it easy to use the same molecule file in different molecule templates or
in different simulations. You can specify the same file multiple times with different optional keywords.
The offset, toff, aoff, doff, ioff keywords add the specified offset values to the atom types, bond types, angle
types, dihedral types, and/or improper types as they are read from the molecule file. E.g. if toff = 2, and the
file uses atom types 1,2,3, then each created molecule will have atom types 3,4,5. For the offset keyword, all
five offset values must be specified, but individual values will be ignored if the molecule template does not
use that attribute (e.g. no bonds).
The scale keyword scales the size of the molecule. This can be useful for modeling polydisperse granular rigid
bodies. The scale factor is applied to each of these properties in the molecule file, if they are defined: the
individual particle coordinates (Coords section), the individual mass of each particle (Masses section), the
individual diameters of each particle (Diameters section), the total mass of the molecule (header keyword =
mass), the center-of-mass of the molecule (header keyword = com), and the moments of inertia of the
molecule (header keyword = inertia).
NOTE: The molecule command can be used to define molecules with bonds, angles, dihedrals, imporopers, or
special bond lists of neighbors within a molecular topology, so that you can later add the molecules to your
simulation, via one or more of the commands listed above. If such molecules do not already exist when
LAMMPS creates the simulation box, via the create_box or read_data command, when you later add them
you may overflow the pre-allocated data structures which store molecular topology information with each
atom, and an error will be generated. Both the create_box command and the data files read by the read_data
command have "extra" options which insure space is allocated for storing topology info for molecules that are
added later.
The format of an individual molecule file is similar to the data file read by the read_data commands, and is as
follows.
A molecule file has a header and a body. The header appears first. The first line of the header is always
skipped; it typically contains a description of the file. Then lines are read one at a time. Lines can have a
trailing comment starting with '#' that is ignored. If the line is blank (only whitespace after comment is
deleted), it is skipped. If the line contains a header keyword, the corresponding value(s) is read from the line.
If it doesn't contain a header keyword, the line begins the body of the file.
The body of the file contains zero or more sections. The first line of a section has only a keyword. The next
line is skipped. The remaining lines of the section contain values. The number of lines depends on the section
keyword as described below. Zero or more blank lines can be used between sections. Sections can appear in
any order, with a few exceptions as noted below.
These are the recognized header keywords. Header lines can come in any order. The numeric value(s) are read
from the beginning of the line. The keyword should appear at the end of the line. All these settings have
default values, as explained below. A line need only appear if the value(s) are different than the default.
LAMMPS Users Manual
514

N atoms = # of atoms N in molecule, default = 0• Nb bonds = # of bonds Nb in molecule, default = 0• Na angles = # of angles Na in molecule, default = 0• Nd dihedrals = # of dihedrals Nd in molecule, default = 0• Ni impropers = # of impropers Ni in molecule, default = 0• Mtotal mass = total mass of molecule• Xc Yc Zc com = coordinates of center-of-mass of molecule• Ixx Iyy Izz Ixy Ixz Iyz inertia = 6 components of inertia tensor of molecule•
For mass, com, and inertia, the default is for LAMMPS to calculate this quantity itself if needed, assuming the
molecules consists of a set of point particles or finite-size particles (with a non-zero diameter) that do not
overlap. If finite-size particles in the molecule do overlap, LAMMPS will not account for the overlap effects
when calculating any of these 3 quantities, so you should pre-compute them yourself and list the values in the
file.
The mass and center-of-mass coordinates (Xc,Yc,Zc) are self-explanatory. The 6 moments of inertia
(ixx,iyy,izz,ixy,ixz,iyz) should be the values consistent with the current orientation of the rigid body around
its center of mass. The values are with respect to the simulation box XYZ axes, not with respect to the
principal axes of the rigid body itself. LAMMPS performs the latter calculation internally.
These are the allowed section keywords for the body of the file.
Coords, Types, Charges, Diameters, Masses = atom-property sections• Bonds, Angles, Dihedrals, Impropers = molecular topology sections• Special Bond Counts, Special Bonds = special neighbor info• Shake Flags, Shake Atoms, Shake Bond Types = SHAKE info•
If a Bonds section is specified then the Special Bond Counts and Special Bonds sections can also be used, if
desired, to explicitly list the 1-2, 1-3, 1-4 neighbors within the molecule topology (see details below). This is
optional since if these sections are not included, LAMMPS will auto-generate this information. Note that
LAMMPS uses this info to properly exclude or weight bonded pairwise interactions between bonded atoms.
See the special_bonds command for more details. One reason to list the special bond info explicitly is for the
thermalized Drude oscillator model which treats the bonds between nuclear cores and Drude electrons in a
different manner.
NOTE: Whether a section is required depends on how the molecule template is used by other LAMMPS
commands. For example, to add a molecule via the fix deposit command, the Coords and Types sections are
required. To add a rigid body via the fix pour command, the Bonds (Angles, etc) sections are not required,
since the molecule will be treated as a rigid body. Some sections are optional. For example, the fix pour
command can be used to add "molecules" which are clusters of finite-size granular particles. If the Diameters
section is not specified, each particle in the molecule will have a default diameter of 1.0. See the doc pages for
LAMMPS commands that use molecule templates for more details.
Each section is listed below in alphabetic order. The format of each section is described including the number
of lines it must contain and rules (if any) for whether it can appear in the data file. In each case the ID is
ignored; it is simply included for readability, and should be a number from 1 to Nlines for the section,
indicating which atom (or bond, etc) the entry applies to. The lines are assumed to be listed in order from 1 to
Nlines, but LAMMPS does not check for this.
Coords section:
LAMMPS Users Manual
515

one line per atom• line syntax: ID x y z• x,y,z = coordinate of atom•
Types section:
one line per atom• line syntax: ID type• type = atom type of atom•
Charges section:
one line per atom• line syntax: ID q• q = charge on atom•
This section is only allowed for atom styles that support charge. If this section is not included, the default
charge on each atom in the molecule is 0.0.
Diameters section:
one line per atom• line syntax: ID diam• diam = diameter of atom•
This section is only allowed for atom styles that support finite-size spherical particles, e.g. atom_style sphere.
If not listed, the default diameter of each atom in the molecule is 1.0.
Masses section:
one line per atom• line syntax: ID mass• mass = mass of atom•
This section is only allowed for atom styles that support per-atom mass, as opposed to per-type mass. See the
mass command for details. If this section is not included, the default mass for each atom is derived from its
volume (see Diameters section) and a default density of 1.0, in units of mass/volume.
Bonds section:
one line per bond• line syntax: ID type atom1 atom2• type = bond type (1-Nbondtype)• atom1,atom2 = IDs of atoms in bond•
The IDs for the two atoms in each bond should be values from 1 to Natoms, where Natoms = # of atoms in the
molecule.
Angles section:
LAMMPS Users Manual
516

one line per angle• line syntax: ID type atom1 atom2 atom3• type = angle type (1-Nangletype)• atom1,atom2,atom3 = IDs of atoms in angle•
The IDs for the three atoms in each angle should be values from 1 to Natoms, where Natoms = # of atoms in
the molecule. The 3 atoms are ordered linearly within the angle. Thus the central atom (around which the
angle is computed) is the atom2 in the list.
Dihedrals section:
one line per dihedral• line syntax: ID type atom1 atom2 atom3 atom4• type = dihedral type (1-Ndihedraltype)• atom1,atom2,atom3,atom4 = IDs of atoms in dihedral•
The IDs for the four atoms in each dihedral should be values from 1 to Natoms, where Natoms = # of atoms in
the molecule. The 4 atoms are ordered linearly within the dihedral.
Impropers section:
one line per improper• line syntax: ID type atom1 atom2 atom3 atom4• type = improper type (1-Nimpropertype)• atom1,atom2,atom3,atom4 = IDs of atoms in improper•
The IDs for the four atoms in each improper should be values from 1 to Natoms, where Natoms = # of atoms
in the molecule. The ordering of the 4 atoms determines the definition of the improper angle used in the
formula for the defined improper style. See the doc pages for individual styles for details.
Special Bond Counts section:
one line per atom• line syntax: ID N1 N2 N3• N1 = # of 1-2 bonds• N2 = # of 1-3 bonds• N3 = # of 1-4 bonds•
N1, N2, N3 are the number of 1-2, 1-3, 1-4 neighbors respectively of this atom within the topology of the
molecule. See the special_bonds doc page for more discussion of 1-2, 1-3, 1-4 neighbors. If this section
appears, the Special Bonds section must also appear.
As explained above, LAMMPS will auto-generate this information if this section is not specified. If specified,
this section will override what would be auto-generated.
Special Bonds section:
one line per atom• line syntax: ID a b c d ...• a,b,c,d,... = IDs of atoms in N1+N2+N3 special bonds•
LAMMPS Users Manual
517

A, b, c, d, etc are the IDs of the n1+n2+n3 atoms that are 1-2, 1-3, 1-4 neighbors of this atom. The IDs should
be values from 1 to Natoms, where Natoms = # of atoms in the molecule. The first N1 values should be the
1-2 neighbors, the next N2 should be the 1-3 neighbors, the last N3 should be the 1-4 neighbors. No atom ID
should appear more than once. See the special_bonds doc page for more discussion of 1-2, 1-3, 1-4 neighbors.
If this section appears, the Special Bond Counts section must also appear.
As explained above, LAMMPS will auto-generate this information if this section is not specified. If specified,
this section will override what would be auto-generated.
Shake Flags section:
one line per atom• line syntax: ID flag• flag = 0,1,2,3,4•
This section is only needed when molecules created using the template will be constrained by SHAKE via the
"fix shake" command. The other two Shake sections must also appear in the file, following this one.
The meaning of the flag for each atom is as follows. See the fix shake doc page for a further description of
SHAKE clusters.
0 = not part of a SHAKE cluster• 1 = part of a SHAKE angle cluster (two bonds and the angle they form)• 2 = part of a 2-atom SHAKE cluster with a single bond• 3 = part of a 3-atom SHAKE cluster with two bonds• 4 = part of a 4-atom SHAKE cluster with three bonds•
Shake Atoms section:
one line per atom• line syntax: ID a b c d• a,b,c,d = IDs of atoms in cluster•
This section is only needed when molecules created using the template will be constrained by SHAKE via the
"fix shake" command. The other two Shake sections must also appear in the file.
The a,b,c,d values are atom IDs (from 1 to Natoms) for all the atoms in the SHAKE cluster that this atom
belongs to. The number of values that must appear is determined by the shake flag for the atom (see the Shake
Flags section above). All atoms in a particular cluster should list their a,b,c,d values identically.
If flag = 0, no a,b,c,d values are listed on the line, just the (ignored) ID.
If flag = 1, a,b,c are listed, where a = ID of central atom in the angle, and b,c the other two atoms in the angle.
If flag = 2, a,b are listed, where a = ID of atom in bond with the the lowest ID, and b = ID of atom in bond
with the highest ID.
If flag = 3, a,b,c are listed, where a = ID of central atom, and b,c = IDs of other two atoms bonded to the
central atom.
LAMMPS Users Manual
518
If flag = 4, a,b,c,d are listed, where a = ID of central atom, and b,c,d = IDs of other three atoms bonded to the
central atom.
See the fix shake doc page for a further description of SHAKE clusters.
Shake Bond Types section:
one line per atom• line syntax: ID a b c• a,b,c = bond types (or angle type) of bonds (or angle) in cluster•
This section is only needed when molecules created using the template will be constrained by SHAKE via the
"fix shake" command. The other two Shake sections must also appear in the file.
The a,b,c values are bond types (from 1 to Nbondtypes) for all bonds in the SHAKE cluster that this atom
belongs to. The number of values that must appear is determined by the shake flag for the atom (see the Shake
Flags section above). All atoms in a particular cluster should list their a,b,c values identically.
If flag = 0, no a,b,c values are listed on the line, just the (ignored) ID.
If flag = 1, a,b,c are listed, where a = bondtype of the bond between the central atom and the first non-central
atom (value b in the Shake Atoms section), b = bondtype of the bond between the central atom and the 2nd
non-central atom (value c in the Shake Atoms section), and c = the angle type (1 to Nangletypes) of the angle
between the 3 atoms.
If flag = 2, only a is listed, where a = bondtype of the bond between the 2 atoms in the cluster.
If flag = 3, a,b are listed, where a = bondtype of the bond between the central atom and the first non-central
atom (value b in the Shake Atoms section), and b = bondtype of the bond between the central atom and the
2nd non-central atom (value c in the Shake Atoms section).
If flag = 4, a,b,c are listed, where a = bondtype of the bond between the central atom and the first non-central
atom (value b in the Shake Atoms section), b = bondtype of the bond between the central atom and the 2nd
non-central atom (value c in the Shake Atoms section), and c = bondtype of the bond between the central
atom and the 3rd non-central atom (value d in the Shake Atoms section).
See the fix shake doc page for a further description of SHAKE clusters.
Restrictions: none
Related commands:
fix deposit, fix pour, fix gcmc
Default:
The default keywords values are offset 0 0 0 0 0 and scale = 1.0.
LAMMPS Users Manual
519

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
neb command
Syntax:
neb etol ftol N1 N2 Nevery file-style arg
etol = stopping tolerance for energy (energy units)• ftol = stopping tolerance for force (force units)• N1 = max # of iterations (timesteps) to run initial NEB• N2 = max # of iterations (timesteps) to run barrier-climbing NEB• Nevery = print replica energies and reaction coordinates every this many timesteps• file-style= final or each or none
final arg = filename
filename = file with initial coords for final replica
coords for intermediate replicas are linearly interpolated between first and last replica
each arg = filename
filename = unique filename for each replica (except first) with its initial coords
none arg = no argument
all replicas assumed to already have their initial coords
•
Examples:
neb 0.1 0.0 1000 500 50 final coords.final
neb 0.0 0.001 1000 500 50 each coords.initial.$i
neb 0.0 0.001 1000 500 50 none
Description:
Perform a nudged elastic band (NEB) calculation using multiple replicas of a system. Two or more replicas
must be used; the first and last are the end points of the transition path.
NEB is a method for finding both the atomic configurations and height of the energy barrier associated with a
transition state, e.g. for an atom to perform a diffusive hop from one energy basin to another in a coordinated
fashion with its neighbors. The implementation in LAMMPS follows the discussion in these 3 papers:
(HenkelmanA), (HenkelmanB), and (Nakano).
Each replica runs on a partition of one or more processors. Processor partitions are defined at run-time using
the -partition command-line switch; see Section 2.7 of the manual. Note that if you have MPI installed, you
can run a multi-replica simulation with more replicas (partitions) than you have physical processors, e.g you
can run a 10-replica simulation on just one or two processors. You will simply not get the performance
speed-up you would see with one or more physical processors per replica. See Section 6.5 of the manual for
further discussion.
NOTE: As explained below, a NEB calculation perfoms a damped dynamics minimization across all the
replicas. The minimizer uses whatever timestep you have defined in your input script, via the timestep
command. Often NEB will converge more quickly if you use a timestep about 10x larger than you would
normally use for dynamics simulations.
LAMMPS Users Manual
520

When a NEB calculation is performed, it is assumed that each replica is running the same system, though
LAMMPS does not check for this. I.e. the simulation domain, the number of atoms, the interaction potentials,
and the starting configuration when the neb command is issued should be the same for every replica.
In a NEB calculation each atom in a replica is connected to the same atom in adjacent replicas by springs,
which induce inter-replica forces. These forces are imposed by the fix neb command, which must be used in
conjunction with the neb command. The group used to define the fix neb command defines the NEB atoms
which are the only ones that inter-replica springs are applied to. If the group does not include all atoms, then
non-NEB atoms have no inter-replica springs and the forces they feel and their motion is computed in the
usual way due only to other atoms within their replica. Conceptually, the non-NEB atoms provide a
background force field for the NEB atoms. They can be allowed to move during the NEB minimization
procedure (which will typically induce different coordinates for non-NEB atoms in different replicas), or held
fixed using other LAMMPS commands such as fix setforce. Note that the partition command can be used to
invoke a command on a subset of the replicas, e.g. if you wish to hold NEB or non-NEB atoms fixed in only
the end-point replicas.
The initial atomic configuration for each of the replicas can be specified in different manners via the file-style
setting, as discussed below. Only atoms whose initial coordinates should differ from the current configuration
need be specified.
Conceptually, the initial configuration for the first replica should be a state with all the atoms (NEB and
non-NEB) having coordinates on one side of the energy barrier. A perfect energy minimum is not required,
since atoms in the first replica experience no spring forces from the 2nd replica. Thus the damped dynamics
minimization will drive the first replica to an energy minimum if it is not already there. However, you will
typically get better convergence if the initial state is already at a minimum. For example, for a system with a
free surface, the surface should be fully relaxed before attempting a NEB calculation.
Likewise, the initial configuration of the final replica should be a state with all the atoms (NEB and non-NEB)
on the other side of the energy barrier. Again, a perfect energy minimum is not required, since the atoms in
the last replica also experience no spring forces from the next-to-last replica, and thus the damped dynamics
minimization will drive it to an energy minimum.
As explained below, the initial configurations of intermediate replicas can be atomic coordinates interpolated
in a linear fashion between the first and last replicas. This is often adequate state for simple transitions. For
more complex transitions, it may lead to slow convergence or even bad results if the minimum energy path
(MEP, see below) of states over the barrier cannot be correctly converged to from such an initial
configuration. In this case, you will want to generate initial states for the intermediate replicas that are
geometrically closer to the MEP and read them in.
For a file-style setting of final, a filename is specified which contains atomic coordinates for zero or more
atoms, in the format described below. For each atom that appears in the file, the new coordinates are assigned
to that atom in the final replica. Each intermediate replica also assigns a new position to that atom in an
interpolated manner. This is done by using the current position of the atom as the starting point and the
read-in position as the final point. The distance between them is calculated, and the new position is assigned
to be a fraction of the distance. E.g. if there are 10 replicas, the 2nd replica will assign a position that is 10%
of the distance along a line between the starting and final point, and the 9th replica will assign a position that
is 90% of the distance along the line. Note that this procedure to produce consistent coordinates across all the
replicas, the current coordinates need to be the same in all replicas. LAMMPS does not check for this, but
invalid initial configurations will likely result if it is not the case.
LAMMPS Users Manual
521

NOTE: The "distance" between the starting and final point is calculated in a minimum-image sense for a
periodic simulation box. This means that if the two positions are on opposite sides of a box (periodic in that
dimension), the distance between them will be small, because the periodic image of one of the atoms is close
to the other. Similarly, even if the assigned position resulting from the interpolation is outside the periodic
box, the atom will be wrapped back into the box when the NEB calculation begins.
For a file-style setting of each, a filename is specified which is assumed to be unique to each replica. This can
be done by using a variable in the filename, e.g.
variable i equal part
neb 0.0 0.001 1000 500 50 each coords.initial.$i
which in this case will substitute the partition ID (0 to N-1) for the variable I, which is also effectively the
replica ID. See the variable command for other options, such as using world-, universe-, or uloop-style
variables.
Each replica (except the first replica) will read its file, formatted as described below, and for any atom that
appears in the file, assign the specified coordinates to its atom. The various files do not need to contain the
same set of atoms.
For a file-style setting of none, no filename is specified. Each replica is assumed to already be in its initial
configuration at the time the neb command is issued. This allows each replica to define its own configuration
by reading a replica-specific data or restart or dump file, via the read_data, read_restart, or read_dump
commands. The replica-specific names of these files can be specified as in the discussion above for the each
file-style. Also see the section below for how a NEB calculation can produce restart files, so that a long
calculation can be restarted if needed.
NOTE: None of the file-style settings change the initial configuration of any atom in the first replica. The first
replica must thus be in the correct initial configuration at the time the neb command is issued.
A NEB calculation proceeds in two stages, each of which is a minimization procedure, performed via damped
dynamics. To enable this, you must first define a damped dynamics min_style, such as quickmin or fire. The
cg, sd, and hftn styles cannot be used, since they perform iterative line searches in their inner loop, which
cannot be easily synchronized across multiple replicas.
The minimizer tolerances for energy and force are set by etol and ftol, the same as for the minimize command.
A non-zero etol means that the NEB calculation will terminate if the energy criterion is met by every replica.
The energies being compared to etol do not include any contribution from the inter-replica forces, since these
are non-conservative. A non-zero ftol means that the NEB calculation will terminate if the force criterion is
met by every replica. The forces being compared to ftol include the inter-replica forces between an atom and
its images in adjacent replicas.
The maximum number of iterations in each stage is set by N1 and N2. These are effectively timestep counts
since each iteration of damped dynamics is like a single timestep in a dynamics run. During both stages, the
potential energy of each replica and its normalized distance along the reaction path (reaction coordinate RD)
will be printed to the screen and log file every Nevery timesteps. The RD is 0 and 1 for the first and last
replica. For intermediate replicas, it is the cumulative distance (normalized by the total cumulative distance)
between adjacent replicas, where "distance" is defined as the length of the 3N-vector of differences in atomic
coordinates, where N is the number of NEB atoms involved in the transition. These outputs allow you to
monitor NEB's progress in finding a good energy barrier. N1 and N2 must both be multiples of Nevery.
LAMMPS Users Manual
522

In the first stage of NEB, the set of replicas should converge toward the minimum energy path (MEP) of
conformational states that transition over the barrier. The MEP for a barrier is defined as a sequence of
3N-dimensional states that cross the barrier at its saddle point, each of which has a potential energy gradient
parallel to the MEP itself. The replica states will also be roughly equally spaced along the MEP due to the
inter-replica spring force added by the fix neb command.
In the second stage of NEB, the replica with the highest energy is selected and the inter-replica forces on it are
converted to a force that drives its atom coordinates to the top or saddle point of the barrier, via the
barrier-climbing calculation described in (HenkelmanB). As before, the other replicas rearrange themselves
along the MEP so as to be roughly equally spaced.
When both stages are complete, if the NEB calculation was successful, one of the replicas should be an atomic
configuration at the top or saddle point of the barrier, the potential energies for the set of replicas should
represent the energy profile of the barrier along the MEP, and the configurations of the replicas should be a
sequence of configurations along the MEP.
A few other settings in your input script are required or advised to perform a NEB calculation. See the NOTE
about the choice of timestep at the beginning of this doc page.
An atom map must be defined which it is not by default for atom_style atomic problems. The atom_modify
map command can be used to do this.
The minimizers in LAMMPS operate on all atoms in your system, even non-NEB atoms, as defined above. To
prevent non-NEB atoms from moving during the minimization, you should use the fix setforce command to
set the force on each of those atoms to 0.0. This is not required, and may not even be desired in some cases,
but if those atoms move too far (e.g. because the initial state of your system was not well-minimized), it can
cause problems for the NEB procedure.
The damped dynamics minimizers, such as quickmin and fire), adjust the position and velocity of the atoms
via an Euler integration step. Thus you must define an appropriate timestep to use with NEB. As mentioned
above, NEB will often converge more quickly if you use a timestep about 10x larger than you would normally
use for dynamics simulations.
Each file read by the neb command containing atomic coordinates used to initialize one or more replicas must
be formatted as follows.
The file can be ASCII text or a gzipped text file (detected by a .gz suffix). The file can contain initial blank
lines or comment lines starting with "#" which are ignored. The first non-blank, non-comment line should list
N = the number of lines to follow. The N successive lines contain the following information:
ID1 x1 y1 z1
ID2 x2 y2 z2
...
IDN xN yN zN
The fields are the atom ID, followed by the x,y,z coordinates. The lines can be listed in any order. Additional
trailing information on the line is OK, such as a comment.
Note that for a typical NEB calculation you do not need to specify initial coordinates for very many atoms to
produce differing starting and final replicas whose intermediate replicas will converge to the energy barrier.
Typically only new coordinates for atoms geometrically near the barrier need be specified.
LAMMPS Users Manual
523

Also note there is no requirement that the atoms in the file correspond to the NEB atoms in the group defined
by the fix neb command. Not every NEB atom need be in the file, and non-NEB atoms can be listed in the
file.
Four kinds of output can be generated during a NEB calculation: energy barrier statistics, thermodynamic
output by each replica, dump files, and restart files.
When running with multiple partitions (each of which is a replica in this case), the print-out to the screen and
master log.lammps file contains a line of output, printed once every Nevery timesteps. It contains the timestep,
the maximum force per replica, the maximum force per atom (in any replica), potential gradients in the initial,
final, and climbing replicas, the forward and backward energy barriers, the total reaction coordinate (RDT),
and the normalized reaction coordinate and potential energy of each replica.
The "maximum force per replica" is the two-norm of the 3N-length force vector for the atoms in each replica,
maximized across replicas, which is what the ftol setting is checking against. In this case, N is all the atoms in
each replica. The "maximum force per atom" is the maximum force component of any atom in any replica.
The potential gradients are the two-norm of the 3N-length force vector solely due to the interaction potential
i.e. without adding in inter-replica forces. Note that inter-replica forces are zero in the initial and final
replicas, and only affect the direction in the climbing replica. For this reason, the "maximum force per replica"
is often equal to the potential gradient in the climbing replica. In the first stage of NEB, there is no climbing
replica, and so the potential gradient in the highest energy replica is reported, since this replica will become
the climbing replica in the second stage of NEB.
The "reaction coordinate" (RD) for each replica is the two-norm of the 3N-length vector of distances between
its atoms and the preceding replica's atoms, added to the RD of the preceding replica. The RD of the first
replica RD1 = 0.0; the RD of the final replica RDN = RDT, the total reaction coordinate. The normalized RDs
are divided by RDT, so that they form a monotonically increasing sequence from zero to one. When
computing RD, N only includes the atoms being operated on by the fix neb command.
The forward (reverse) energy barrier is the potential energy of the highest replica minus the energy of the first
(last) replica.
When running on multiple partitions, LAMMPS produces additional log files for each partition, e.g.
log.lammps.0, log.lammps.1, etc. For a NEB calculation, these contain the thermodynamic output for each
replica.
If dump commands in the input script define a filename that includes a universe or uloop style variable, then
one dump file (per dump command) will be created for each replica. At the end of the NEB calculation, the
final snapshot in each file will contain the sequence of snapshots that transition the system over the energy
barrier. Earlier snapshots will show the convergence of the replicas to the MEP.
Likewise, restart filenames can be specified with a universe or uloop style variable, to generate restart files for
each replica. These may be useful if the NEB calculation fails to converge properly to the MEP, and you wish
to restart the calculation from an intermediate point with altered parameters.
There are 2 Python scripts provided in the tools/python directory, neb_combine.py and neb_final.py, which
are useful in analyzing output from a NEB calculation. Assume a NEB simulation with M replicas, and the
NEB atoms labeled with a specific atom type.
The neb_combine.py script extracts atom coords for the NEB atoms from all M dump files and creates a
single dump file where each snapshot contains the NEB atoms from all the replicas and one copy of non-NEB
LAMMPS Users Manual
524

atoms from the first replica (presumed to be identical in other replicas). This can be visualized/animated to see
how the NEB atoms relax as the NEB calculation proceeds.
The neb_final.py script extracts the final snapshot from each of the M dump files to create a single dump file
with M snapshots. This can be visualized to watch the system make its transition over the energy barrier.
To illustrate, here are images from the final snapshot produced by the neb_combine.py script run on the dump
files produced by the two example input scripts in examples/neb. Click on them to see a larger image.
Restrictions:
This command can only be used if LAMMPS was built with the REPLICA package. See the Making
LAMMPS section for more info on packages.
Related commands:
prd, temper, fix langevin, fix viscous
Default: none
(HenkelmanA) Henkelman and Jonsson, J Chem Phys, 113, 9978-9985 (2000).
(HenkelmanB) Henkelman, Uberuaga, Jonsson, J Chem Phys, 113, 9901-9904 (2000).
(Nakano) Nakano, Comp Phys Comm, 178, 280-289 (2008).
LAMMPS Users Manual
525

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
neigh_modify command
Syntax:
neigh_modify keyword values ...
one or more keyword/value pairs may be listed
keyword = delay or every or check or once or cluster or include or exclude or page or one or binsize
delay value = N
N = delay building until this many steps since last build
every value = M
M = build neighbor list every this many steps
check value = yes or no
yes = only build if some atom has moved half the skin distance or more
no = always build on 1st step that every and delay are satisfied
once
yes = only build neighbor list once at start of run and never rebuild
no = rebuild neighbor list according to other settings
cluster
yes = check bond,angle,etc neighbor list for nearby clusters
no = do not check bond,angle,etc neighbor list for nearby clusters
include value = group-ID
group-ID = only build pair neighbor lists for atoms in this group
exclude values:
type M N
M,N = exclude if one atom in pair is type M, other is type N
group group1-ID group2-ID
group1-ID,group2-ID = exclude if one atom is in 1st group, other in 2nd
molecule/intra group-ID
group-ID = exclude if both atoms are in the same molecule and in group
molecule/inter group-ID
group-ID = exclude if both atoms are in different molecules and in group
none
delete all exclude settings
page value = N
N = number of pairs stored in a single neighbor page
one value = N
N = max number of neighbors of one atom
binsize value = size
size = bin size for neighbor list construction (distance units)
•
Examples:
neigh_modify every 2 delay 10 check yes page 100000
neigh_modify exclude type 2 3
neigh_modify exclude group frozen frozen check no
neigh_modify exclude group residue1 chain3
neigh_modify exclude molecule/intra rigid
Description:
This command sets parameters that affect the building and use of pairwise neighbor lists. Depending on what
pair interactions and other commands are defined, a simulation may require one or more neighbor lists.
LAMMPS Users Manual
526
The every, delay, check, and once options affect how often lists are built as a simulation runs. The delay
setting means never build new lists until at least N steps after the previous build. The every setting means
build lists every M steps (after the delay has passed). If the check setting is no, the lists are built on the first
step that satisfies the delay and every settings. If the check setting is yes, then the every and delay settings
determine when a build may possibly be performed, but an actual build only occurs if some atom has moved
more than half the skin distance (specified in the neighbor command) since the last build.
If the once setting is yes, then the neighbor list is only built once at the beginning of each run, and never
rebuilt, except on steps when a restart file is written, or steps when a fix forces a rebuild to occur (e.g. fixes
that create or delete atoms, such as fix deposit or fix evaporate). This setting should only be made if you are
certain atoms will not move far enough that the neighbor list should be rebuilt, e.g. running a simulation of a
cold crystal. Note that it is not that expensive to check if neighbor lists should be rebuilt.
When the rRESPA integrator is used (see the run_style command), the every and delay parameters refer to the
longest (outermost) timestep.
The cluster option does a sanity test every time neighbor lists are built for bond, angle, dihedral, and improper
interactions, to check that each set of 2, 3, or 4 atoms is a cluster of nearby atoms. It does this by computing
the distance between pairs of atoms in the interaction and insuring they are not further apart than half the
periodic box length. If they are, an error is generated, since the interaction would be computed between
far-away atoms instead of their nearby periodic images. The only way this should happen is if the pairwise
cutoff is so short that atoms that are part of the same interaction are not communicated as ghost atoms. This is
an unusual model (e.g. no pair interactions at all) and the problem can be fixed by use of the comm_modify
cutoff command. Note that to save time, the default cluster setting is no, so that this check is not performed.
The include option limits the building of pairwise neighbor lists to atoms in the specified group. This can be
useful for models where a large portion of the simulation is particles that do not interact with other particles or
with each other via pairwise interactions. The group specified with this option must also be specified via the
atom_modify first command.
The exclude option turns off pairwise interactions between certain pairs of atoms, by not including them in the
neighbor list. These are sample scenarios where this is useful:
In crack simulations, pairwise interactions can be shut off between 2 slabs of atoms to effectively
create a crack.
•
When a large collection of atoms is treated as frozen, interactions between those atoms can be turned
off to save needless computation. E.g. Using the fix setforce command to freeze a wall or portion of a
bio-molecule.
•
When one or more rigid bodies are specified, interactions within each body can be turned off to save
needless computation. See the fix rigid command for more details.
•
The exclude type option turns off the pairwise interaction if one atom is of type M and the other of type N. M
can equal N. The exclude group option turns off the interaction if one atom is in the first group and the other
is the second. Group1-ID can equal group2-ID. The exclude molecule/intra option turns off the interaction if
both atoms are in the specified group and in the same molecule, as determined by their molecule ID. The
exclude molecule/inter turns off the interaction between pairs of atoms that have different molecule IDs and
are both in the specified group.
Each of the exclude options can be specified multiple times. The exclude type option is the most efficient
option to use; it requires only a single check, no matter how many times it has been specified. The other
exclude options are more expensive if specified multiple times; they require one check for each time they
LAMMPS Users Manual
527
have been specified.
Note that the exclude options only affect pairwise interactions; see the delete_bonds command for information
on turning off bond interactions.
NOTE: Excluding pairwise interactions will not work correctly when also using a long-range solver via the
kspace_style command. LAMMPS will give a warning to this effect. This is because the short-range pairwise
interaction needs to subtract off a term from the total energy for pairs whose short-range interaction is
excluded, to compensate for how the long-range solver treats the interaction. This is done correctly for
pairwise interactions that are excluded (or weighted) via the special_bonds command. But it is not done for
interactions that are excluded via these neigh_modify exclude options.
The page and one options affect how memory is allocated for the neighbor lists. For most simulations the
default settings for these options are fine, but if a very large problem is being run or a very long cutoff is
being used, these parameters can be tuned. The indices of neighboring atoms are stored in "pages", which are
allocated one after another as they fill up. The size of each page is set by the page value. A new page is
allocated when the next atom's neighbors could potentially overflow the list. This threshold is set by the one
value which tells LAMMPS the maximum number of neighbor's one atom can have.
NOTE: LAMMPS can crash without an error message if the number of neighbors for a single particle is larger
than the page setting, which means it is much, much larger than the one setting. This is because LAMMPS
doesn't error check these limits for every pairwise interaction (too costly), but only after all the particle's
neighbors have been found. This problem usually means something is very wrong with the way you've setup
your problem (particle spacing, cutoff length, neighbor skin distance, etc). If you really expect that many
neighbors per particle, then boost the one and page settings accordingly.
The binsize option allows you to specify what size of bins will be used in neighbor list construction to sort and
find neighboring atoms. By default, for neighbor style bin, LAMMPS uses bins that are 1/2 the size of the
maximum pair cutoff. For neighbor style multi, the bins are 1/2 the size of the minimum pair cutoff. Typically
these are good values values for minimizing the time for neighbor list construction. This setting overrides the
default. If you make it too big, there is little overhead due to looping over bins, but more atoms are checked. If
you make it too small, the optimal number of atoms is checked, but bin overhead goes up. If you set the
binsize to 0.0, LAMMPS will use the default binsize of 1/2 the cutoff.
Restrictions:
If the "delay" setting is non-zero, then it must be a multiple of the "every" setting.
The molecule/intra and molecule/inter exclude options can only be used with atom styles that define molecule
IDs.
The value of the page setting must be at least 10x larger than the one setting. This insures neighbor pages are
not mostly empty space.
Related commands:
neighbor, delete_bonds
Default:
LAMMPS Users Manual
528
The option defaults are delay = 10, every = 1, check = yes, once = no, cluster = no, include = all, exclude =
none, page = 100000, one = 2000, and binsize = 0.0.
LAMMPS Users Manual
529

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
neighbor command
Syntax:
neighbor skin style
skin = extra distance beyond force cutoff (distance units)• style = bin or nsq or multi•
Examples:
neighbor 0.3 bin
neighbor 2.0 nsq
Description:
This command sets parameters that affect the building of pairwise neighbor lists. All atom pairs within a
neighbor cutoff distance equal to the their force cutoff plus the skin distance are stored in the list. Typically,
the larger the skin distance, the less often neighbor lists need to be built, but more pairs must be checked for
possible force interactions every timestep. The default value for skin depends on the choice of units for the
simulation; see the default values below.
The skin distance is also used to determine how often atoms migrate to new processors if the check option of
the neigh_modify command is set to yes. Atoms are migrated (communicated) to new processors on the same
timestep that neighbor lists are re-built.
The style value selects what algorithm is used to build the list. The bin style creates the list by binning which
is an operation that scales linearly with N/P, the number of atoms per processor where N = total number of
atoms and P = number of processors. It is almost always faster than the nsq style which scales as (N/P)^2. For
unsolvated small molecules in a non-periodic box, the nsq choice can sometimes be faster. Either style should
give the same answers.
The multi style is a modified binning algorithm that is useful for systems with a wide range of cutoff
distances, e.g. due to different size particles. For the bin style, the bin size is set to 1/2 of the largest cutoff
distance between any pair of atom types and a single set of bins is defined to search over for all atom types.
This can be inefficient if one pair of types has a very long cutoff, but other type pairs have a much shorter
cutoff. For style multi the bin size is set to 1/2 of the shortest cutoff distance and multiple sets of bins are
defined to search over for different atom types. This imposes some extra setup overhead, but the searches
themselves may be much faster for the short-cutoff cases. See the comm_modify mode multi command for a
communication option option that may also be beneficial for simulations of this kind.
The neigh_modify command has additional options that control how often neighbor lists are built and which
pairs are stored in the list.
When a run is finished, counts of the number of neighbors stored in the pairwise list and the number of times
neighbor lists were built are printed to the screen and log file. See this section for details.
Restrictions: none
LAMMPS Users Manual
530

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
newton command
Syntax:
newton flag
newton flag1 flag2
flag = on or off for both pairwise and bonded interactions• flag1 = on or off for pairwise interactions• flag2 = on or off for bonded interactions•
Examples:
newton off
newton on off
Description:
This command turns Newton's 3rd law on or off for pairwise and bonded interactions. For most problems,
setting Newton's 3rd law to on means a modest savings in computation at the cost of two times more
communication. Whether this is faster depends on problem size, force cutoff lengths, a machine's
compute/communication ratio, and how many processors are being used.
Setting the pairwise newton flag to off means that if two interacting atoms are on different processors, both
processors compute their interaction and the resulting force information is not communicated. Similarly, for
bonded interactions, newton off means that if a bond, angle, dihedral, or improper interaction contains atoms
on 2 or more processors, the interaction is computed by each processor.
LAMMPS should produce the same answers for any newton flag settings, except for round-off issues.
With run_style respa and only bonded interactions (bond, angle, etc) computed in the innermost timestep, it
may be faster to turn newton off for bonded interactions, to avoid extra communication in the innermost loop.
Restrictions:
The newton bond setting cannot be changed after the simulation box is defined by a read_data or create_box
command.
Related commands:
run_style respa
Default:
newton on
LAMMPS Users Manual
532

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
next command
Syntax:
next variables
variables = one or more variable names•
Examples:
next x
next a t x myTemp
Description:
This command is used with variables defined by the variable command. It assigns the next value to the
variable from the list of values defined for that variable by the variable command. Thus when that variable is
subsequently substituted for in an input script command, the new value is used.
See the variable command for info on how to define and use different kinds of variables in LAMMPS input
scripts. If a variable name is a single lower-case character from "a" to "z", it can be used in an input script
command as $a or $z. If it is multiple letters, it can be used as ${myTemp}.
If multiple variables are used as arguments to the next command, then all must be of the same variable style:
index, loop, file, universe, or uloop. An exception is that universe- and uloop-style variables can be mixed in
the same next command.
All the variables specified with the next command are incremented by one value from their respective list of
values. A file-style variable reads the next line from its associated file. An atomfile-style variable reads the
next set of lines (one per atom) from its associated file. String- or atom- or equal- or world-style variables
cannot be used with the next command, since they only store a single value.
When any of the variables in the next command has no more values, a flag is set that causes the input script to
skip the next jump command encountered. This enables a loop containing a next command to exit. As
explained in the variable command, the variable that has exhausted its values is also deleted. This allows it to
be used and re-defined later in the input script. File-style and atomfile-style variables are exhausted when the
end-of-file is reached.
When the next command is used with index- or loop-style variables, the next value is assigned to the variable
for all processors. When the next command is used with file-style variables, the next line is read from its file
and the string assigned to the variable. When the next command is used with atomfile-style variables, the next
set of per-atom values is read from its file and assigned to the variable.
When the next command is used with universe- or uloop-style variables, all universe- or uloop-style variables
must be listed in the next command. This is because of the manner in which the incrementing is done, using a
single lock file for all variables. The next value (for each variable) is assigned to whichever processor
partition executes the command first. All processors in the partition are assigned the same value(s). Running
LAMMPS on multiple partitions of processors via the "-partition" command-line switch is described in this
section of the manual. Universe- and uloop-style variables are incremented using the files
LAMMPS Users Manual
533
"tmp.lammps.variable" and "tmp.lammps.variable.lock" which you will see in your directory during and after
such a LAMMPS run.
Here is an example of running a series of simulations using the next command with an index-style variable. If
this input script is named in.polymer, 8 simulations would be run using data files from directories run1 thru
run8.
variable d index run1 run2 run3 run4 run5 run6 run7 run8
shell cd $d
read_data data.polymer
run 10000
shell cd ..
clear
next d
jump in.polymer
If the variable "d" were of style universe, and the same in.polymer input script were run on 3 partitions of
processors, then the first 3 simulations would begin, one on each set of processors. Whichever partition
finished first, it would assign variable "d" the 4th value and run another simulation, and so forth until all 8
simulations were finished.
Jump and next commands can also be nested to enable multi-level loops. For example, this script will run 15
simulations in a double loop.
variable i loop 3
variable j loop 5
clear
...
read_data data.polymer.$i$j
print Running simulation $i.$j
run 10000
next j
jump in.script
next i
jump in.script
Here is an example of a double loop which uses the if and jump commands to break out of the inner loop
when a condition is met, then continues iterating thru the outer loop.
label loopa
variable a loop 5
label loopb
variable b loop 5
print "A,B = $a,$b"
run 10000
if $b > 2 then "jump in.script break"
next b
jump in.script loopb
label break
variable b delete
next a
jump in.script loopa
Restrictions:
LAMMPS Users Manual
534

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
package command
Syntax:
package style args
style = gpu or intel or kokkos or omp• args = arguments specific to the style
gpu args = Ngpu keyword value ...
Ngpu = # of GPUs per node
zero or more keyword/value pairs may be appended
keywords = neigh or newton or binsize or split or gpuID or tpa or device or blocksize
neigh value = yes or no
yes = neighbor list build on GPU (default)
no = neighbor list build on CPU
newton = off or on
off = set Newton pairwise flag off (default and required)
on = set Newton pairwise flag on (currently not allowed)
binsize value = size
size = bin size for neighbor list construction (distance units)
split = fraction
fraction = fraction of atoms assigned to GPU (default = 1.0)
gpuID values = first last
first = ID of first GPU to be used on each node
last = ID of last GPU to be used on each node
tpa value = Nthreads
Nthreads = # of GPU threads used per atom
device value = device_type
device_type = kepler or fermi or cypress or generic
blocksize value = size
size = thread block size for pair force computation
intel args = NPhi keyword value ...
Nphi = # of coprocessors per node
zero or more keyword/value pairs may be appended
keywords = mode or omp or lrt or balance or ghost or tpc or tptask or no_affinity
mode value = single or mixed or double
single = perform force calculations in single precision
mixed = perform force calculations in mixed precision
double = perform force calculations in double precision
omp value = Nthreads
Nthreads = number of OpenMP threads to use on CPU (default = 0)
lrt value = yes or no
yes = use additional thread dedicated for some PPPM calculations
no = do not dedicate an extra thread for some PPPM calculations
balance value = split
split = fraction of work to offload to coprocessor, -1 for dynamic
ghost value = yes or no
yes = include ghost atoms for offload
no = do not include ghost atoms for offload
tpc value = Ntpc
Ntpc = max number of coprocessor threads per coprocessor core (default = 4)
tptask value = Ntptask
Ntptask = max number of coprocessor threads per MPI task (default = 240)
no_affinity values = none
kokkos args = keyword value ...
zero or more keyword/value pairs may be appended
•
LAMMPS Users Manual
536
keywords = neigh or neigh/qeq or newton or binsize or comm or comm/exchange or comm/forward
neigh value = full or half
full = full neighbor list
half = half neighbor list built in thread-safe manner
neigh/qeq value = full or half
full = full neighbor list
half = half neighbor list built in thread-safe manner
newton = off or on
off = set Newton pairwise and bonded flags off (default)
on = set Newton pairwise and bonded flags on
binsize value = size
size = bin size for neighbor list construction (distance units)
comm value = no or host or device
use value for both comm/exchange and comm/forward
comm/exchange value = no or host or device
comm/forward value = no or host or device
no = perform communication pack/unpack in non-KOKKOS mode
host = perform pack/unpack on host (e.g. with OpenMP threading)
device = perform pack/unpack on device (e.g. on GPU)
omp args = Nthreads keyword value ...
Nthread = # of OpenMP threads to associate with each MPI process
zero or more keyword/value pairs may be appended
keywords = neigh
neigh value = yes or no
yes = threaded neighbor list build (default)
no = non-threaded neighbor list build
Examples:
package gpu 1
package gpu 1 split 0.75
package gpu 2 split -1.0
package kokkos neigh half comm device
package omp 0 neigh no
package omp 4
package intel 1
package intel 2 omp 4 mode mixed balance 0.5
Description:
This command invokes package-specific settings for the various accelerator packages available in LAMMPS.
Currently the following packages use settings from this command: GPU, USER-INTEL, KOKKOS, and
USER-OMP.
If this command is specified in an input script, it must be near the top of the script, before the simulation box
has been defined. This is because it specifies settings that the accelerator packages use in their initialization,
before a simulation is defined.
This command can also be specified from the command-line when launching LAMMPS, using the "-pk"
command-line switch. The syntax is exactly the same as when used in an input script.
Note that all of the accelerator packages require the package command to be specified (except the OPT
package), if the package is to be used in a simulation (LAMMPS can be built with an accelerator package
without using it in a particular simulation). However, in all cases, a default version of the command is
typically invoked by other accelerator settings.
LAMMPS Users Manual
537

The KOKKOS package requires a "-k on" command-line switch respectively, which invokes a "package
kokkos" command with default settings.
For the GPU, USER-INTEL, and USER-OMP packages, if a "-sf gpu" or "-sf intel" or "-sf omp"
command-line switch is used to auto-append accelerator suffixes to various styles in the input script, then
those switches also invoke a "package gpu", "package intel", or "package omp" command with default
settings.
NOTE: A package command for a particular style can be invoked multiple times when a simulation is setup,
e.g. by the "-c on", "-k on", "-sf", and "-pk" command-line switches, and by using this command in an input
script. Each time it is used all of the style options are set, either to default values or to specified settings. I.e.
settings from previous invocations do not persist across multiple invocations.
See the Section 5.3 section of the manual for more details about using the various accelerator packages for
speeding up LAMMPS simulations.
The gpu style invokes settings associated with the use of the GPU package.
The Ngpu argument sets the number of GPUs per node. There must be at least as many MPI tasks per node as
GPUs, as set by the mpirun or mpiexec command. If there are more MPI tasks (per node) than GPUs, multiple
MPI tasks will share each GPU.
Optional keyword/value pairs can also be specified. Each has a default value as listed below.
The neigh keyword specifies where neighbor lists for pair style computation will be built. If neigh is yes,
which is the default, neighbor list building is performed on the GPU. If neigh is no, neighbor list building is
performed on the CPU. GPU neighbor list building currently cannot be used with a triclinic box. GPU
neighbor list calculation currently cannot be used with hybrid pair styles. GPU neighbor lists are not
compatible with commands that are not GPU-enabled. When a non-GPU enabled command requires a
neighbor list, it will also be built on the CPU. In these cases, it will typically be more efficient to only use
CPU neighbor list builds.
The newton keyword sets the Newton flags for pairwise (not bonded) interactions to off or on, the same as the
newton command allows. Currently, only an off value is allowed, since all the GPU package pair styles
require this setting. This means more computation is done, but less communication. In the future a value of on
may be allowed, so the newton keyword is included as an option for compatibility with the package command
for other accelerator styles. Note that the newton setting for bonded interactions is not affected by this
keyword.
The binsize keyword sets the size of bins used to bin atoms in neighbor list builds performed on the GPU, if
neigh = yes is set. If binsize is set to 0.0 (the default), then bins = the size of the pairwise cutoff + neighbor
skin distance. This is 2x larger than the LAMMPS default used for neighbor list building on the CPU. This
will be close to optimal for the GPU, so you do not normally need to use this keyword. Note that if you use a
longer-than-usual pairwise cutoff, e.g. to allow for a smaller fraction of KSpace work with a long-range
Coulombic solver because the GPU is faster at performing pairwise interactions, then it may be optimal to
make the binsize smaller than the default. For example, with a cutoff of 20*sigma in LJ units and a neighbor
skin distance of sigma, a binsize = 5.25*sigma can be more efficient than the default.
The split keyword can be used for load balancing force calculations between CPU and GPU cores in
GPU-enabled pair styles. If 0 < split < 1.0, a fixed fraction of particles is offloaded to the GPU while force
calculation for the other particles occurs simultaneously on the CPU. If split < 0.0, the optimal fraction (based
LAMMPS Users Manual
538

on CPU and GPU timings) is calculated every 25 timesteps, i.e. dynamic load-balancing across the CPU and
GPU is performed. If split = 1.0, all force calculations for GPU accelerated pair styles are performed on the
GPU. In this case, other hybrid pair interactions, bond, angle, dihedral, improper, and long-range calculations
can be performed on the CPU while the GPU is performing force calculations for the GPU-enabled pair style.
If all CPU force computations complete before the GPU completes, LAMMPS will block until the GPU has
finished before continuing the timestep.
As an example, if you have two GPUs per node and 8 CPU cores per node, and would like to run on 4 nodes
(32 cores) with dynamic balancing of force calculation across CPU and GPU cores, you could specify
mpirun -np 32 -sf gpu -in in.script # launch command
package gpu 2 split -1 # input script command
In this case, all CPU cores and GPU devices on the nodes would be utilized. Each GPU device would be
shared by 4 CPU cores. The CPU cores would perform force calculations for some fraction of the particles at
the same time the GPUs performed force calculation for the other particles.
The gpuID keyword allows selection of which GPUs on each node will be used for a simulation. The first and
last values specify the GPU IDs to use (from 0 to Ngpu-1). By default, first = 0 and last = Ngpu-1, so that all
GPUs are used, assuming Ngpu is set to the number of physical GPUs. If you only wish to use a subset, set
Ngpu to a smaller number and first/last to a sub-range of the available GPUs.
The tpa keyword sets the number of GPU thread per atom used to perform force calculations. With a default
value of 1, the number of threads will be chosen based on the pair style, however, the value can be set
explicitly with this keyword to fine-tune performance. For large cutoffs or with a small number of particles
per GPU, increasing the value can improve performance. The number of threads per atom must be a power of
2 and currently cannot be greater than 32.
The device keyword can be used to tune parameters optimized for a specific accelerator, when using OpenCL.
For CUDA, the device keyword is ignored. Currently, the device type is limited to NVIDIA Kepler, NVIDIA
Fermi, AMD Cypress, or a generic device. More devices may be added later. The default device type can be
specified when building LAMMPS with the GPU library, via settings in the lib/gpu/Makefile that is used.
The blocksize keyword allows you to tweak the number of threads used per thread block. This number should
be a multiple of 32 (for GPUs) and its maximum depends on the specific GPU hardware. Typical choices are
64, 128, or 256. A larger blocksize increases occupancy of individual GPU cores, but reduces the total number
of thread blocks, thus may lead to load imbalance.
The intel style invokes settings associated with the use of the USER-INTEL package. All of its settings,
except the omp and mode keywords, are ignored if LAMMPS was not built with Xeon Phi coprocessor
support. All of its settings, including the omp and mode keyword are applicable if LAMMPS was built with
coprocessor support.
The Nphi argument sets the number of coprocessors per node. This can be set to any value, including 0, if
LAMMPS was not built with coprocessor support.
Optional keyword/value pairs can also be specified. Each has a default value as listed below.
The omp keyword determines the number of OpenMP threads allocated for each MPI task when any portion
of the interactions computed by a USER-INTEL pair style are run on the CPU. This can be the case even if
LAMMPS was built with coprocessor support; see the balance keyword discussion below. If you are running
LAMMPS Users Manual
539
with less MPI tasks/node than there are CPUs, it can be advantageous to use OpenMP threading on the CPUs.
NOTE: The omp keyword has nothing to do with coprocessor threads on the Xeon Phi; see the tpc and tptask
keywords below for a discussion of coprocessor threads.
The Nthread value for the omp keyword sets the number of OpenMP threads allocated for each MPI task.
Setting Nthread = 0 (the default) instructs LAMMPS to use whatever value is the default for the given
OpenMP environment. This is usually determined via the OMP_NUM_THREADS environment variable or the
compiler runtime, which is usually a value of 1.
For more details, including examples of how to set the OMP_NUM_THREADS environment variable, see the
discussion of the Nthreads setting on this doc page for the "package omp" command. Nthreads is a required
argument for the USER-OMP package. Its meaning is exactly the same for the USER-INTEL package.
NOTE: If you build LAMMPS with both the USER-INTEL and USER-OMP packages, be aware that both
packages allow setting of the Nthreads value via their package commands, but there is only a single global
Nthreads value used by OpenMP. Thus if both package commands are invoked, you should insure the two
values are consistent. If they are not, the last one invoked will take precedence, for both packages. Also note
that if the "-sf hybrid intel omp" command-line"> switch is used, it invokes a "package intel" command,
followed by a "package omp" command, both with a setting of Nthreads = 0.
The mode keyword determines the precision mode to use for computing pair style forces, either on the CPU or
on the coprocessor, when using a USER-INTEL supported pair style. It can take a value of single, mixed
which is the default, or double. Single means single precision is used for the entire force calculation. Mixed
means forces between a pair of atoms are computed in single precision, but accumulated and stored in double
precision, including storage of forces, torques, energies, and virial quantities. Double means double precision
is used for the entire force calculation.
The lrt keyword can be used to enable "Long Range Thread (LRT)" mode. It can take a value of yes to enable
and no to disable. LRT mode generates an extra thread (in addition to any OpenMP threads specified with the
OMP_NUM_THREADS environment variable or the omp keyword). The extra thread is dedicated for
performing part of the PPPM solver computations and communications. This can improve parallel
performance on processors supporting Simultaneous Multithreading (SMT) such as Hyperthreading on Intel
processors. In this mode, one additional thread is generated per MPI process. LAMMPS will generate a
warning in the case that more threads are used than available in SMT hardware on a node. If the PPPM solver
from the USER-INTEL package is not used, then the LRT setting is ignored and no extra threads are
generated. Enabling LRT will replace the run_style with the verlet/lrt/intel style that is identical to the default
verlet style aside from supporting the LRT feature.
The balance keyword sets the fraction of pair style work offloaded to the coprocessor for split values between
0.0 and 1.0 inclusive. While this fraction of work is running on the coprocessor, other calculations will run on
the host, including neighbor and pair calculations that are not offloaded, as well as angle, bond, dihedral,
kspace, and some MPI communications. If split is set to -1, the fraction of work is dynamically adjusted
automatically throughout the run. This typically give performance within 5 to 10 percent of the optimal fixed
fraction.
The ghost keyword determines whether or not ghost atoms, i.e. atoms at the boundaries of processor
sub-domains, are offloaded for neighbor and force calculations. When the value = "no", ghost atoms are not
offloaded. This option can reduce the amount of data transfer with the coprocessor and can also overlap MPI
communication of forces with computation on the coprocessor when the newton pair setting is "on". When the
value = "yes", ghost atoms are offloaded. In some cases this can provide better performance, especially if the
LAMMPS Users Manual
540

balance fraction is high.
The tpc keyword sets the max # of coprocessor threads Ntpc that will run on each core of the coprocessor. The
default value = 4, which is the number of hardware threads per core supported by the current generation Xeon
Phi chips.
The tptask keyword sets the max # of coprocessor threads (Ntptask assigned to each MPI task. The default
value = 240, which is the total # of threads an entire current generation Xeon Phi chip can run (240 = 60 cores
* 4 threads/core). This means each MPI task assigned to the Phi will enough threads for the chip to run the
max allowed, even if only 1 MPI task is assigned. If 8 MPI tasks are assigned to the Phi, each will run with 30
threads. If you wish to limit the number of threads per MPI task, set tptask to a smaller value. E.g. for tptask =
16, if 8 MPI tasks are assigned, each will run with 16 threads, for a total of 128.
Note that the default settings for tpc and tptask are fine for most problems, regardless of how many MPI tasks
you assign to a Phi.
The no_affinity keyword will turn off automatic setting of core affinity for MPI tasks and OpenMP threads on
the host when using offload to a coprocessor. Affinity settings are used when possible to prevent MPI tasks
and OpenMP threads from being on separate NUMA domains and to prevent offload threads from interfering
with other processes/threads used for LAMMPS.
The kokkos style invokes settings associated with the use of the KOKKOS package.
All of the settings are optional keyword/value pairs. Each has a default value as listed below.
The neigh keyword determines how neighbor lists are built. A value of half uses a thread-safe variant of
half-neighbor lists, the same as used by most pair styles in LAMMPS.
A value of full uses a full neighbor lists and is the default. This performs twice as much computation as the
half option, however that is often a win because it is thread-safe and doesn't require atomic operations in the
calculation of pair forces. For that reason, full is the default setting. However, when running in MPI-only
mode with 1 thread per MPI task, half neighbor lists will typically be faster, just as it is for non-accelerated
pair styles. Similarly, the neigh/qeq keyword determines how neighbor lists are built for fix qeq/reax/kk. If not
explicitly set, the value of neigh/qeq will match neigh.
The newton keyword sets the Newton flags for pairwise and bonded interactions to off or on, the same as the
newton command allows. The default is off because this will almost always give better performance for the
KOKKOS package. This means more computation is done, but less communication. However, when running
in MPI-only mode with 1 thread per MPI task, a value of on will typically be faster, just as it is for
non-accelerated pair styles.
The binsize keyword sets the size of bins used to bin atoms in neighbor list builds. The same value can be set
by the neigh_modify binsize command. Making it an option in the package kokkos command allows it to be
set from the command line. The default value is 0.0, which means the LAMMPS default will be used, which
is bins = 1/2 the size of the pairwise cutoff + neighbor skin distance. This is fine when neighbor lists are built
on the CPU. For GPU builds, a 2x larger binsize equal to the pairwise cutoff + neighbor skin, is often faster,
which can be set by this keyword. Note that if you use a longer-than-usual pairwise cutoff, e.g. to allow for a
smaller fraction of KSpace work with a long-range Coulombic solver because the GPU is faster at performing
pairwise interactions, then this rule of thumb may give too large a binsize.
LAMMPS Users Manual
541

The comm and comm/exchange and comm/forward keywords determine whether the host or device performs
the packing and unpacking of data when communicating per-atom data between processors. "Exchange"
communication happens only on timesteps that neighbor lists are rebuilt. The data is only for atoms that
migrate to new processors. "Forward" communication happens every timestep. The data is for atom
coordinates and any other atom properties that needs to be updated for ghost atoms owned by each processor.
The comm keyword is simply a short-cut to set the same value for both the comm/exchange and
comm/forward keywords.
The value options for all 3 keywords are no or host or device. A value of no means to use the standard
non-KOKKOS method of packing/unpacking data for the communication. A value of host means to use the
host, typically a multi-core CPU, and perform the packing/unpacking in parallel with threads. A value of
device means to use the device, typically a GPU, to perform the packing/unpacking operation.
The optimal choice for these keywords depends on the input script and the hardware used. The no value is
useful for verifying that the Kokkos-based host and device values are working correctly. It may also be the
fastest choice when using Kokkos styles in MPI-only mode (i.e. with a thread count of 1).
When running on CPUs or Xeon Phi, the host and device values work identically. When using GPUs, the
device value will typically be optimal if all of your styles used in your input script are supported by the
KOKKOS package. In this case data can stay on the GPU for many timesteps without being moved between
the host and GPU, if you use the device value. This requires that your MPI is able to access GPU memory
directly. Currently that is true for OpenMPI 1.8 (or later versions), Mvapich2 1.9 (or later), and CrayMPI. If
your script uses styles (e.g. fixes) which are not yet supported by the KOKKOS package, then data has to be
move between the host and device anyway, so it is typically faster to let the host handle communication, by
using the host value. Using host instead of no will enable use of multiple threads to pack/unpack
communicated data.
The omp style invokes settings associated with the use of the USER-OMP package.
The Nthread argument sets the number of OpenMP threads allocated for each MPI task. For example, if your
system has nodes with dual quad-core processors, it has a total of 8 cores per node. You could use two MPI
tasks per node (e.g. using the -ppn option of the mpirun command in MPICH or -npernode in OpenMPI), and
set Nthreads = 4. This would use all 8 cores on each node. Note that the product of MPI tasks * threads/task
should not exceed the physical number of cores (on a node), otherwise performance will suffer.
Setting Nthread = 0 instructs LAMMPS to use whatever value is the default for the given OpenMP
environment. This is usually determined via the OMP_NUM_THREADS environment variable or the compiler
runtime. Note that in most cases the default for OpenMP capable compilers is to use one thread for each
available CPU core when OMP_NUM_THREADS is not explicitly set, which can lead to poor performance.
Here are examples of how to set the environment variable when launching LAMMPS:
env OMP_NUM_THREADS=4 lmp_machine -sf omp -in in.script
env OMP_NUM_THREADS=2 mpirun -np 2 lmp_machine -sf omp -in in.script
mpirun -x OMP_NUM_THREADS=2 -np 2 lmp_machine -sf omp -in in.script
or you can set it permanently in your shell's start-up script. All three of these examples use a total of 4 CPU
cores.
LAMMPS Users Manual
542

Note that different MPI implementations have different ways of passing the OMP_NUM_THREADS
environment variable to all MPI processes. The 2nd example line above is for MPICH; the 3rd example line
with -x is for OpenMPI. Check your MPI documentation for additional details.
What combination of threads and MPI tasks gives the best performance is difficult to predict and can depend
on many components of your input. Not all features of LAMMPS support OpenMP threading via the
USER-OMP package and the parallel efficiency can be very different, too.
Optional keyword/value pairs can also be specified. Each has a default value as listed below.
The neigh keyword specifies whether neighbor list building will be multi-threaded in addition to force
calculations. If neigh is set to no then neighbor list calculation is performed only by MPI tasks with no
OpenMP threading. If mode is yes (the default), a multi-threaded neighbor list build is used. Using neigh = yes
is almost always faster and should produce identical neighbor lists at the expense of using more memory.
Specifically, neighbor list pages are allocated for all threads at the same time and each thread works within its
own pages.
Restrictions:
This command cannot be used after the simulation box is defined by a read_data or create_box command.
The gpu style of this command can only be invoked if LAMMPS was built with the GPU package. See the
Making LAMMPS section for more info.
The intel style of this command can only be invoked if LAMMPS was built with the USER-INTEL package.
See the Making LAMMPS section for more info.
The kk style of this command can only be invoked if LAMMPS was built with the KOKKOS package. See
the Making LAMMPS section for more info.
The omp style of this command can only be invoked if LAMMPS was built with the USER-OMP package.
See the Making LAMMPS section for more info.
Related commands:
suffix, "-pk" command-line setting
Default:
For the GPU package, the default is Ngpu = 1 and the option defaults are neigh = yes, newton = off, binsize =
0.0, split = 1.0, gpuID = 0 to Ngpu-1, tpa = 1, and device = not used. These settings are made automatically if
the "-sf gpu" command-line switch is used. If it is not used, you must invoke the package gpu command in
your input script or via the "-pk gpu" command-line switch.
For the USER-INTEL package, the default is Nphi = 1 and the option defaults are omp = 0, mode = mixed, lrt
= no, balance = -1, tpc = 4, tptask = 240. The default ghost option is determined by the pair style being used.
This value is output to the screen in the offload report at the end of each run. Note that all of these settings,
except "omp" and "mode", are ignored if LAMMPS was not built with Xeon Phi coprocessor support. These
settings are made automatically if the "-sf intel" command-line switch is used. If it is not used, you must
invoke the package intel command in your input script or or via the "-pk intel" command-line switch.
LAMMPS Users Manual
543
For the KOKKOS package, the option defaults neigh = full, neigh/qeq = full, newton = off, binsize = 0.0, and
comm = device. These settings are made automatically by the required "-k on" command-line switch. You can
change them bu using the package kokkos command in your input script or via the "-pk kokkos"
command-line switch.
For the OMP package, the default is Nthreads = 0 and the option defaults are neigh = yes. These settings are
made automatically if the "-sf omp" command-line switch is used. If it is not used, you must invoke the
package omp command in your input script or via the "-pk omp" command-line switch.
LAMMPS Users Manual
544

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_coeff command
Syntax:
pair_coeff I J args
I,J = atom types (see asterisk form below)• args = coefficients for one or more pairs of atom types•
Examples:
pair_coeff 1 2 1.0 1.0 2.5
pair_coeff 2 * 1.0 1.0
pair_coeff 3* 1*2 1.0 1.0 2.5
pair_coeff * * 1.0 1.0
pair_coeff * * nialhjea 1 1 2
pair_coeff * 3 morse.table ENTRY1
pair_coeff 1 2 lj/cut 1.0 1.0 2.5 (for pair_style hybrid)
Description:
Specify the pairwise force field coefficients for one or more pairs of atom types. The number and meaning of
the coefficients depends on the pair style. Pair coefficients can also be set in the data file read by the read_data
command or in a restart file.
I and J can be specified in one of two ways. Explicit numeric values can be used for each, as in the 1st
example above. I <= J is required. LAMMPS sets the coefficients for the symmetric J,I interaction to the same
values.
A wildcard asterisk can be used in place of or in conjunction with the I,J arguments to set the coefficients for
multiple pairs of atom types. This takes the form "*" or "*n" or "n*" or "m*n". If N = the number of atom
types, then an asterisk with no numeric values means all types from 1 to N. A leading asterisk means all types
from 1 to n (inclusive). A trailing asterisk means all types from n to N (inclusive). A middle asterisk means all
types from m to n (inclusive). Note that only type pairs with I <= J are considered; if asterisks imply type
pairs where J < I, they are ignored.
Note that a pair_coeff command can override a previous setting for the same I,J pair. For example, these
commands set the coeffs for all I,J pairs, then overwrite the coeffs for just the I,J = 2,3 pair:
pair_coeff * * 1.0 1.0 2.5
pair_coeff 2 3 2.0 1.0 1.12
A line in a data file that specifies pair coefficients uses the exact same format as the arguments of the
pair_coeff command in an input script, with the exception of the I,J type arguments. In each line of the "Pair
Coeffs" section of a data file, only a single type I is specified, which sets the coefficients for type I interacting
with type I. This is because the section has exactly N lines, where N = the number of atom types. For this
reason, the wild-card asterisk should also not be used as part of the I argument. Thus in a data file, the line
corresponding to the 1st example above would be listed as
2 1.0 1.0 2.5
LAMMPS Users Manual
545

For many potentials, if coefficients for type pairs with I != J are not set explicitly by a pair_coeff command,
the values are inferred from the I,I and J,J settings by mixing rules; see the pair_modify command for a
discussion. Details on this option as it pertains to individual potentials are described on the doc page for the
potential.
Many pair styles, typically for many-body potentials, use tabulated potential files as input, when specifying
the pair_coeff command. Potential files provided with LAMMPS are in the potentials directory of the
distribution. For some potentials, such as EAM, other archives of suitable files can be found on the Web. They
can be used with LAMMPS so long as they are in the format LAMMPS expects, as discussed on the
individual doc pages.
When a pair_coeff command using a potential file is specified, LAMMPS looks for the potential file in 2
places. First it looks in the location specified. E.g. if the file is specified as "niu3.eam", it is looked for in the
current working directory. If it is specified as "../potentials/niu3.eam", then it is looked for in the potentials
directory, assuming it is a sister directory of the current working directory. If the file is not found, it is then
looked for in the directory specified by the LAMMPS_POTENTIALS environment variable. Thus if this is set
to the potentials directory in the LAMMPS distro, then you can use those files from anywhere on your system,
without copying them into your working directory. Environment variables are set in different ways for
different shells. Here are example settings for
csh, tcsh:
% setenv LAMMPS_POTENTIALS /path/to/lammps/potentials
bash:
% export LAMMPS_POTENTIALS=/path/to/lammps/potentials
Windows:
% set LAMMPS_POTENTIALS="C:\\Path to LAMMPS\\Potentials"
The alphabetic list of pair styles defined in LAMMPS is given on the pair_style doc page. They are also given
in more compact form in the pair section of this page.
Click on the style to display the formula it computes, arguments specified in the pair_style command, and
coefficients specified by the associated pair_coeff command.
Note that there are also additional pair styles (not listed on the pair_style doc page) submitted by users which
are included in the LAMMPS distribution. The list of these with links to the individual styles are given in the
pair section of this page.
There are also additional accelerated pair styles (not listed on the pair_style doc page) included in the
LAMMPS distribution for faster performance on CPUs and GPUs. The list of these with links to the
individual styles are given in the pair section of this page.
Restrictions:
This command must come after the simulation box is defined by a read_data, read_restart, or create_box
command.
LAMMPS Users Manual
546

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_modify command
Syntax:
pair_modify keyword values ...
one or more keyword/value pairs may be listed• keyword = pair or shift or mix or table or table/disp or tabinner or tabinner/disp or tail or compute
pair values = sub-style N special which wt1 wt2 wt3
sub-style = sub-style of pair hybrid
N = which instance of sub-style (only if sub-style is used multiple times)
special which wt1 wt2 wt3 = override special_bonds settings (optional)
which = lj/coul or lj or coul
w1,w2,w3 = 1-2, 1-3, and 1-4 weights from 0.0 to 1.0 inclusive
mix value = geometric or arithmetic or sixthpower
shift value = yes or no
table value = N
2^N = # of values in table
table/disp value = N
2^N = # of values in table
tabinner value = cutoff
cutoff = inner cutoff at which to begin table (distance units)
tabinner/disp value = cutoff
cutoff = inner cutoff at which to begin table (distance units)
tail value = yes or no
compute value = yes or no
•
Examples:
pair_modify shift yes mix geometric
pair_modify tail yes
pair_modify table 12
pair_modify pair lj/cut compute no
pair_modify pair lj/cut/coul/long 1 special lj/coul 0.0 0.0 0.0
Description:
Modify the parameters of the currently defined pair style. Not all parameters are relevant to all pair styles.
If used, the pair keyword must appear first in the list of keywords. It can only be used with the hybrid and
hybrid/overlay pair styles. It means that all the following parameters will only be modified for the specified
sub-style. If the sub-style is defined multiple times, then an additional numeric argument N must also be
specified, which is a number from 1 to M where M is the number of times the sub-style was listed in the
pair_style hybrid command. The extra number indicates which instance of the sub-style the remaining
keywords will be applied to. Note that if the pair keyword is not used, and the pair style is hybrid or
hybrid/overlay, then all the specified keywords will be applied to all sub-styles.
The special keyword can only be used in conjunction with the pair keyword and must directly follow it. It
allows to override the special_bonds settings for the specified sub-style. More details are given below.
The mix keyword affects pair coefficients for interactions between atoms of type I and J, when I != J and the
LAMMPS Users Manual
548
coefficients are not explicitly set in the input script. Note that coefficients for I = J must be set explicitly,
either in the input script via the "pair_coeff" command or in the "Pair Coeffs" section of the data file. For
some pair styles it is not necessary to specify coefficients when I != J, since a "mixing" rule will create them
from the I,I and J,J settings. The pair_modify mix value determines what formulas are used to compute the
mixed coefficients. In each case, the cutoff distance is mixed the same way as sigma.
Note that not all pair styles support mixing. Also, some mix options are not available for certain pair styles.
See the doc page for individual pair styles for those restrictions. Note also that the pair_coeff command also
can be to directly set coefficients for a specific I != J pairing, in which case no mixing is performed.
mix geometric
epsilon_ij = sqrt(epsilon_i * epsilon_j)
sigma_ij = sqrt(sigma_i * sigma_j)
mix arithmetic
epsilon_ij = sqrt(epsilon_i * epsilon_j)
sigma_ij = (sigma_i + sigma_j) / 2
mix sixthpower
epsilon_ij = (2 * sqrt(epsilon_i*epsilon_j) * sigma_i^3 * sigma_j^3) /
(sigma_i^6 + sigma_j^6)
sigma_ij = ((sigma_i**6 + sigma_j**6) / 2) ^ (1/6)
The shift keyword determines whether a Lennard-Jones potential is shifted at its cutoff to 0.0. If so, this adds
an energy term to each pairwise interaction which will be included in the thermodynamic output, but does not
affect pair forces or atom trajectories. See the doc page for individual pair styles to see which ones support
this option.
The table and table/disp keywords apply to pair styles with a long-range Coulombic term or long-range
dispersion term respectively; see the doc page for individual styles to see which potentials support these
options. If N is non-zero, a table of length 2^N is pre-computed for forces and energies, which can shrink their
computational cost by up to a factor of 2. The table is indexed via a bit-mapping technique (Wolff) and a
linear interpolation is performed between adjacent table values. In our experiments with different table styles
(lookup, linear, spline), this method typically gave the best performance in terms of speed and accuracy.
The choice of table length is a tradeoff in accuracy versus speed. A larger N yields more accurate force
computations, but requires more memory which can slow down the computation due to cache misses. A
reasonable value of N is between 8 and 16. The default value of 12 (table of length 4096) gives approximately
the same accuracy as the no-table (N = 0) option. For N = 0, forces and energies are computed directly, using
a polynomial fit for the needed erfc() function evaluation, which is what earlier versions of LAMMPS did.
Values greater than 16 typically slow down the simulation and will not improve accuracy; values from 1 to 8
give unreliable results.
The tabinner and tabinner/disp keywords set an inner cutoff above which the pairwise computation is done by
table lookup (if tables are invoked), for the corresponding Coulombic and dispersion tables discussed with the
table and table/disp keywords. The smaller the cutoff is set, the less accurate the table becomes (for a given
number of table values), which can require use of larger tables. The default cutoff value is sqrt(2.0) distance
units which means nearly all pairwise interactions are computed via table lookup for simulations with "real"
units, but some close pairs may be computed directly (non-table) for simulations with "lj" units.
LAMMPS Users Manual
549

When the tail keyword is set to yes, certain pair styles will add a long-range VanderWaals tail "correction" to
the energy and pressure. These corrections are bookkeeping terms which do not affect dynamics, unless a
constant-pressure simulation is being performed. See the doc page for individual styles to see which support
this option. These corrections are included in the calculation and printing of thermodynamic quantities (see
the thermo_style command). Their effect will also be included in constant NPT or NPH simulations where the
pressure influences the simulation box dimensions (e.g. the fix npt and fix nph commands). The formulas used
for the long-range corrections come from equation 5 of (Sun).
NOTE: The tail correction terms are computed at the beginning of each run, using the current atom counts of
each atom type. If atoms are deleted (or lost) or created during a simulation, e.g. via the fix gcmc command,
the correction factors are not re-computed. If you expect the counts to change dramatically, you can break a
run into a series of shorter runs so that the correction factors are re-computed more frequently.
Several additional assumptions are inherent in using tail corrections, including the following:
The simulated system is a 3d bulk homogeneous liquid. This option should not be used for systems
that are non-liquid, 2d, have a slab geometry (only 2d periodic), or inhomogeneous.
•
G(r), the radial distribution function (rdf), is unity beyond the cutoff, so a fairly large cutoff should be
used (i.e. 2.5 sigma for an LJ fluid), and it is probably a good idea to verify this assumption by
checking the rdf. The rdf is not exactly unity beyond the cutoff for each pair of interaction types, so
the tail correction is necessarily an approximation.
The tail corrections are computed at the beginning of each simulation run. If the number of atoms
changes during the run, e.g. due to atoms leaving the simulation domain, or use of the fix gcmc
command, then the corrections are not updates to relect the changed atom count. If this is a large
effect in your simulation, you should break the long run into several short runs, so that the correction
factors are re-computed multiple times.
•
Thermophysical properties obtained from calculations with this option enabled will not be
thermodynamically consistent with the truncated force-field that was used. In other words, atoms do
not feel any LJ pair interactions beyond the cutoff, but the energy and pressure reported by the
simulation include an estimated contribution from those interactions.
•
The compute keyword allows pairwise computations to be turned off, even though a pair_style is defined. This
is not useful for running a real simulation, but can be useful for debugging purposes or for performing a rerun
simulation, when you only wish to compute partial forces that do not include the pairwise contribution.
Two examples are as follows. First, this option allows you to perform a simulation with pair_style hybrid with
only a subset of the hybrid sub-styles enabled. Second, this option allows you to perform a simulation with
only long-range interactions but no short-range pairwise interactions. Doing this by simply not defining a pair
style will not work, because the kspace_style command requires a Kspace-compatible pair style be defined.
The special keyword allows to override the 1-2, 1-3, and 1-4 exclusion settings for individual sub-styles of a
hybrid pair style. It requires 4 arguments similar to the special_bonds command, which and wt1,wt2,wt3. The
which argument can be lj to change the Lennard-Jones settings, coul to change the Coulombic settings, or
lj/coul to change both to the same set of 3 values. The wt1,wt2,wt3 values are numeric weights from 0.0 to 1.0
inclusive, for the 1-2, 1-3, and 1-4 bond topology neighbors, respectively. The special keyword can only be
used in conjunction with the pair keyword and has to directly follow it.
NOTE: The global settings specified by the special_bonds command affect the construction of neighbor lists.
Weights of 0.0 (for 1-2, 1-3, or 1-4 neighbors) exclude those pairs from the neighbor list entirely. Weights of
1.0 store the neighbor with no weighting applied. Thus only global values different from exactly 0.0 or 1.0 can
LAMMPS Users Manual
550

be overridden and an error is generated if the requested setting is not compatible with the global setting.
Substituting 1.0e-10 for 0.0 and 0.9999999999 for 1.0 is usually a sufficient workaround in this case without
causing a significant error.
Restrictions: none
You cannot use shift yes with tail yes, since those are conflicting options. You cannot use tail yes with 2d
simulations.
Related commands:
pair_style, pair_coeff, thermo_style
Default:
The option defaults are mix = geometric, shift = no, table = 12, tabinner = sqrt(2.0), tail = no, and compute =
yes.
Note that some pair styles perform mixing, but only a certain style of mixing. See the doc pages for individual
pair styles for details.
(Wolff) Wolff and Rudd, Comp Phys Comm, 120, 200-32 (1999).
(Sun) Sun, J Phys Chem B, 102, 7338-7364 (1998).
LAMMPS Users Manual
551

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style command
Syntax:
pair_style style args
style = one of the styles from the list below• args = arguments used by a particular style•
Examples:
pair_style lj/cut 2.5
pair_style eam/alloy
pair_style hybrid lj/charmm/coul/long 10.0 eam
pair_style table linear 1000
pair_style none
Description:
Set the formula(s) LAMMPS uses to compute pairwise interactions. In LAMMPS, pair potentials are defined
between pairs of atoms that are within a cutoff distance and the set of active interactions typically changes
over time. See the bond_style command to define potentials between pairs of bonded atoms, which typically
remain in place for the duration of a simulation.
In LAMMPS, pairwise force fields encompass a variety of interactions, some of which include many-body
effects, e.g. EAM, Stillinger-Weber, Tersoff, REBO potentials. They are still classified as "pairwise"
potentials because the set of interacting atoms changes with time (unlike molecular bonds) and thus a
neighbor list is used to find nearby interacting atoms.
Hybrid models where specified pairs of atom types interact via different pair potentials can be setup using the
hybrid pair style.
The coefficients associated with a pair style are typically set for each pair of atom types, and are specified by
the pair_coeff command or read from a file by the read_data or read_restart commands.
The pair_modify command sets options for mixing of type I-J interaction coefficients and adding energy
offsets or tail corrections to Lennard-Jones potentials. Details on these options as they pertain to individual
potentials are described on the doc page for the potential. Likewise, info on whether the potential information
is stored in a restart file is listed on the potential doc page.
In the formulas listed for each pair style, E is the energy of a pairwise interaction between two atoms
separated by a distance r. The force between the atoms is the negative derivative of this expression.
If the pair_style command has a cutoff argument, it sets global cutoffs for all pairs of atom types. The
distance(s) can be smaller or larger than the dimensions of the simulation box.
Typically, the global cutoff value can be overridden for a specific pair of atom types by the pair_coeff
command. The pair style settings (including global cutoffs) can be changed by a subsequent pair_style
command using the same style. This will reset the cutoffs for all atom type pairs, including those previously
LAMMPS Users Manual
552

set explicitly by a pair_coeff command. The exceptions to this are that pair_style table and hybrid settings
cannot be reset. A new pair_style command for these styles will wipe out all previously specified pair_coeff
values.
Here is an alphabetic list of pair styles defined in LAMMPS. They are also given in more compact form in the
pair section of this page.
Click on the style to display the formula it computes, arguments specified in the pair_style command, and
coefficients specified by the associated pair_coeff command.
There are also additional pair styles (not listed here) submitted by users which are included in the LAMMPS
distribution. The list of these with links to the individual styles are given in the pair section of this page.
There are also additional accelerated pair styles (not listed here) included in the LAMMPS distribution for
faster performance on CPUs and GPUs. The list of these with links to the individual styles are given in the
pair section of this page.
pair_style none - turn off pairwise interactions• pair_style hybrid - multiple styles of pairwise interactions• pair_style hybrid/overlay - multiple styles of superposed pairwise interactions• pair_style zero - neighbor list but no interactions•
pair_style adp - angular dependent potential (ADP) of Mishin• pair_style airebo - AIREBO potential of Stuart• pair_style airebo/morse - AIREBO with Morse instead of LJ• pair_style beck - Beck potential• pair_style body - interactions between body particles• pair_style bop - BOP potential of Pettifor• pair_style born - Born-Mayer-Huggins potential• pair_style born/coul/long - Born-Mayer-Huggins with long-range Coulombics• pair_style born/coul/long/cs - Born-Mayer-Huggins with long-range Coulombics and core/shell• pair_style born/coul/msm - Born-Mayer-Huggins with long-range MSM Coulombics• pair_style born/coul/wolf - Born-Mayer-Huggins with Coulombics via Wolf potential• pair_style brownian - Brownian potential for Fast Lubrication Dynamics• pair_style brownian/poly - Brownian potential for Fast Lubrication Dynamics with polydispersity• pair_style buck - Buckingham potential• pair_style buck/coul/cut - Buckingham with cutoff Coulomb• pair_style buck/coul/long - Buckingham with long-range Coulombics• pair_style buck/coul/long/cs - Buckingham with long-range Coulombics and core/shell• pair_style buck/coul/msm - Buckingham long-range MSM Coulombics• pair_style buck/long/coul/long - long-range Buckingham with long-range Coulombics• pair_style colloid - integrated colloidal potential• pair_style comb - charge-optimized many-body (COMB) potential• pair_style comb3 - charge-optimized many-body (COMB3) potential• pair_style coul/cut - cutoff Coulombic potential• pair_style coul/debye - cutoff Coulombic potential with Debye screening• pair_style coul/dsf - Coulombics via damped shifted forces• pair_style coul/long - long-range Coulombic potential• pair_style coul/long/cs - long-range Coulombic potential and core/shell• pair_style coul/msm - long-range MSM Coulombics• pair_style coul/streitz - Coulombics via Streitz/Mintmire Slater orbitals•
LAMMPS Users Manual
553
pair_style coul/wolf - Coulombics via Wolf potential• pair_style dpd - dissipative particle dynamics (DPD)• pair_style dpd/tstat - DPD thermostatting• pair_style dsmc - Direct Simulation Monte Carlo (DSMC)• pair_style eam - embedded atom method (EAM)• pair_style eam/alloy - alloy EAM• pair_style eam/fs - Finnis-Sinclair EAM• pair_style eim - embedded ion method (EIM)• pair_style gauss - Gaussian potential• pair_style gayberne - Gay-Berne ellipsoidal potential• pair_style gran/hertz/history - granular potential with Hertzian interactions• pair_style gran/hooke - granular potential with history effects• pair_style gran/hooke/history - granular potential without history effects• pair_style hbond/dreiding/lj - DREIDING hydrogen bonding LJ potential• pair_style hbond/dreiding/morse - DREIDING hydrogen bonding Morse potential• pair_style kim - interface to potentials provided by KIM project• pair_style lcbop - long-range bond-order potential (LCBOP)• pair_style line/lj - LJ potential between line segments• pair_style lj/charmm/coul/charmm - CHARMM potential with cutoff Coulomb• pair_style lj/charmm/coul/charmm/implicit - CHARMM for implicit solvent• pair_style lj/charmm/coul/long - CHARMM with long-range Coulomb• pair_style lj/charmm/coul/msm - CHARMM with long-range MSM Coulombics• pair_style lj/class2 - COMPASS (class 2) force field with no Coulomb• pair_style lj/class2/coul/cut - COMPASS with cutoff Coulomb• pair_style lj/class2/coul/long - COMPASS with long-range Coulomb• pair_style lj/cubic - LJ with cubic after inflection point• pair_style lj/cut - cutoff Lennard-Jones potential with no Coulomb• pair_style lj/cut/coul/cut - LJ with cutoff Coulomb• pair_style lj/cut/coul/debye - LJ with Debye screening added to Coulomb• pair_style lj/cut/coul/dsf - LJ with Coulombics via damped shifted forces• pair_style lj/cut/coul/long - LJ with long-range Coulombics• pair_style lj/cut/coul/long/cs - LJ with long-range Coulombics and core/shell• pair_style lj/cut/coul/msm - LJ with long-range MSM Coulombics• pair_style lj/cut/dipole/cut - point dipoles with cutoff• pair_style lj/cut/dipole/long - point dipoles with long-range Ewald• pair_style lj/cut/tip4p/cut - LJ with cutoff Coulomb for TIP4P water• pair_style lj/cut/tip4p/long - LJ with long-range Coulomb for TIP4P water• pair_style lj/expand - Lennard-Jones for variable size particles• pair_style lj/gromacs - GROMACS-style Lennard-Jones potential• pair_style lj/gromacs/coul/gromacs - GROMACS-style LJ and Coulombic potential• pair_style lj/long/coul/long - long-range LJ and long-range Coulombics• pair_style lj/long/dipole/long - long-range LJ and long-range point dipoles• pair_style lj/long/tip4p/long - long-range LJ and long-range Coulomb for TIP4P water• pair_style lj/smooth - smoothed Lennard-Jones potential• pair_style lj/smooth/linear - linear smoothed Lennard-Jones potential• pair_style lj96/cut - Lennard-Jones 9/6 potential• pair_style lubricate - hydrodynamic lubrication forces• pair_style lubricate/poly - hydrodynamic lubrication forces with polydispersity• pair_style lubricateU - hydrodynamic lubrication forces for Fast Lubrication Dynamics• pair_style lubricateU/poly - hydrodynamic lubrication forces for Fast Lubrication with polydispersity• pair_style meam - modified embedded atom method (MEAM)•
LAMMPS Users Manual
554

pair_style mie/cut - Mie potential• pair_style morse - Morse potential• pair_style nb3b/harmonic - nonbonded 3-body harmonic potential• pair_style nm/cut - N-M potential• pair_style nm/cut/coul/cut - N-M potential with cutoff Coulomb• pair_style nm/cut/coul/long - N-M potential with long-range Coulombics• pair_style peri/eps - peridynamic EPS potential• pair_style peri/lps - peridynamic LPS potential• pair_style peri/pmb - peridynamic PMB potential• pair_style peri/ves - peridynamic VES potential• pair_style polymorphic - polymorphic 3-body potential• pair_style reax - ReaxFF potential• pair_style rebo - 2nd generation REBO potential of Brenner• pair_style resquared - Everaers RE-Squared ellipsoidal potential• pair_style snap - SNAP quantum-accurate potential• pair_style soft - Soft (cosine) potential• pair_style sw - Stillinger-Weber 3-body potential• pair_style table - tabulated pair potential• pair_style tersoff - Tersoff 3-body potential• pair_style tersoff/mod - modified Tersoff 3-body potential• pair_style tersoff/zbl - Tersoff/ZBL 3-body potential• pair_style tip4p/cut - Coulomb for TIP4P water w/out LJ• pair_style tip4p/long - long-range Coulombics for TIP4P water w/out LJ• pair_style tri/lj - LJ potential between triangles• pair_style vashishta - Vashishta 2-body and 3-body potential• pair_style yukawa - Yukawa potential• pair_style yukawa/colloid - screened Yukawa potential for finite-size particles• pair_style zbl - Ziegler-Biersack-Littmark potential•
Restrictions:
This command must be used before any coefficients are set by the pair_coeff, read_data, or read_restart
commands.
Some pair styles are part of specific packages. They are only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info on packages. The doc pages for individual pair
potentials tell if it is part of a package.
Related commands:
pair_coeff, read_data, pair_modify, kspace_style, dielectric, pair_write
Default:
pair_style none
LAMMPS Users Manual
555

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_write command
Syntax:
pair_write itype jtype N style inner outer file keyword Qi Qj
itype,jtype = 2 atom types• N = # of values• style = r or rsq or bitmap• inner,outer = inner and outer cutoff (distance units)• file = name of file to write values to• keyword = section name in file for this set of tabulated values• Qi,Qj = 2 atom charges (charge units) (optional)•
Examples:
pair_write 1 3 500 r 1.0 10.0 table.txt LJ
pair_write 1 1 1000 rsq 2.0 8.0 table.txt Yukawa_1_1 -0.5 0.5
Description:
Write energy and force values to a file as a function of distance for the currently defined pair potential. This is
useful for plotting the potential function or otherwise debugging its values. If the file already exists, the table
of values is appended to the end of the file to allow multiple tables of energy and force to be included in one
file.
The energy and force values are computed at distances from inner to outer for 2 interacting atoms of type
itype and jtype, using the appropriate pair_coeff coefficients. If the style is r, then N distances are used,
evenly spaced in r; if the style is rsq, N distances are used, evenly spaced in r^2.
For example, for N = 7, style = r, inner = 1.0, and outer = 4.0, values are computed at r = 1.0, 1.5, 2.0, 2.5,
3.0, 3.5, 4.0.
If the style is bitmap, then 2^N values are written to the file in a format and order consistent with how they are
read in by the pair_coeff command for pair style table. For reasonable accuracy in a bitmapped table, choose
N >= 12, an inner value that is smaller than the distance of closest approach of 2 atoms, and an outer value <=
cutoff of the potential.
If the pair potential is computed between charged atoms, the charges of the pair of interacting atoms can
optionally be specified. If not specified, values of Qi = Qj = 1.0 are used.
The file is written in the format used as input for the pair_style table option with keyword as the section name.
Each line written to the file lists an index number (1-N), a distance (in distance units), an energy (in energy
units), and a force (in force units).
Restrictions:
All force field coefficients for pair and other kinds of interactions must be set before this command can be
invoked.
LAMMPS Users Manual
556
Due to how the pairwise force is computed, an inner value > 0.0 must be specified even if the potential has a
finite value at r = 0.0.
For EAM potentials, the pair_write command only tabulates the pairwise portion of the potential, not the
embedding portion.
Related commands:
pair_style table, pair_style, pair_coeff
Default: none
LAMMPS Users Manual
557

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
partition command
Syntax:
partition style N command ...
style = yes or no• N = partition number (see asterisk form below)• command = any LAMMPS command•
Examples:
partition yes 1 processors 4 10 6
partition no 5 print "Active partition"
partition yes *5 fix all nve
partition yes 6* fix all nvt temp 1.0 1.0 0.1
Description:
This command invokes the specified command on a subset of the partitions of processors you have defined
via the -partition command-line switch. See Section 2.6 for an explanation of the switch.
Normally, every input script command in your script is invoked by every partition. This behavior can be
modified by defining world- or universe-style variables that have different values for each partition. This
mechanism can be used to cause your script to jump to different input script files on different partitions, if
such a variable is used in a jump command.
The "partition" command is another mechanism for having as input script operate differently on different
partitions. It is basically a prefix on any LAMMPS command. The command will only be invoked on the
partition(s) specified by the style and N arguments.
If the style is yes, the command will be invoked on any partition which matches the N argument. If the style is
no the command will be invoked on all the partitions which do not match the Np argument.
Partitions are numbered from 1 to Np, where Np is the number of partitions specified by the -partition
command-line switch.
N can be specified in one of two ways. An explicit numeric value can be used, as in the 1st example above. Or
a wild-card asterisk can be used to span a range of partition numbers. This takes the form "*" or "*n" or "n*"
or "m*n". An asterisk with no numeric values means all partitions from 1 to Np. A leading asterisk means all
partitions from 1 to n (inclusive). A trailing asterisk means all partitions from n to Np (inclusive). A middle
asterisk means all partitions from m to n (inclusive).
This command can be useful for the "run_style verlet/split" command which imposed requirements on how
the processors command lays out a 3d grid of processors in each of 2 partitions.
Restrictions: none
Related commands:
LAMMPS Users Manual
558

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
prd command
Syntax:
prd N t_event n_dephase t_dephase t_correlate compute-ID seed keyword value ...
N = # of timesteps to run (not including dephasing/quenching)• t_event = timestep interval between event checks• n_dephase = number of velocity randomizations to perform in each dephase run• t_dephase = number of timesteps to run dynamics after each velocity randomization during dephase• t_correlate = number of timesteps within which 2 consecutive events are considered to be correlated• compute-ID = ID of the compute used for event detection• random_seed = random # seed (positive integer)• zero or more keyword/value pairs may be appended• keyword = min or temp or vel
min values = etol ftol maxiter maxeval
etol = stopping tolerance for energy, used in quenching
ftol = stopping tolerance for force, used in quenching
maxiter = max iterations of minimize, used in quenching
maxeval = max number of force/energy evaluations, used in quenching
temp value = Tdephase
Tdephase = target temperature for velocity randomization, used in dephasing
vel values = loop dist
loop = all or local or geom, used in dephasing
dist = uniform or gaussian, used in dephasing
time value = steps or clock
steps = simulation runs for N timesteps on each replica (default)
clock = simulation runs for N timesteps across all replicas
•
Examples:
prd 5000 100 10 10 100 1 54982
prd 5000 100 10 10 100 1 54982 min 0.1 0.1 100 200
Description:
Run a parallel replica dynamics (PRD) simulation using multiple replicas of a system. One or more replicas
can be used. The total number of steps N to run can be interpreted in one of two ways; see discussion of the
time keyword below.
PRD is described in this paper by Art Voter. It is a method for performing accelerated dynamics that is
suitable for infrequent-event systems that obey first-order kinetics. A good overview of accelerated dynamics
methods for such systems in given in this review paper from the same group. To quote from the paper: "The
dynamical evolution is characterized by vibrational excursions within a potential basin, punctuated by
occasional transitions between basins." The transition probability is characterized by p(t) = k*exp(-kt) where k
is the rate constant. Running multiple replicas gives an effective enhancement in the timescale spanned by the
multiple simulations, while waiting for an event to occur.
Each replica runs on a partition of one or more processors. Processor partitions are defined at run-time using
the -partition command-line switch; see Section 2.7 of the manual. Note that if you have MPI installed, you
LAMMPS Users Manual
560
can run a multi-replica simulation with more replicas (partitions) than you have physical processors, e.g you
can run a 10-replica simulation on one or two processors. However for PRD, this makes little sense, since
running a replica on virtual instead of physical processors,offers no effective parallel speed-up in searching
for infrequent events. See Section 6.5 of the manual for further discussion.
When a PRD simulation is performed, it is assumed that each replica is running the same model, though
LAMMPS does not check for this. I.e. the simulation domain, the number of atoms, the interaction potentials,
etc should be the same for every replica.
A PRD run has several stages, which are repeated each time an "event" occurs in one of the replicas, as
defined below. The logic for a PRD run is as follows:
while (time remains):
dephase for n_dephase*t_dephase steps
until (event occurs on some replica):
run dynamics for t_event steps
quench
check for uncorrelated event on any replica
until (no correlated event occurs):
run dynamics for t_correlate steps
quench
check for correlated event on this replica
event replica shares state with all replicas
Before this loop begins, the state of the system on replica 0 is shared with all replicas, so that all replicas
begin from the same initial state. The first potential energy basin is identified by quenching (an energy
minimization, see below) the initial state and storing the resulting coordinates for reference.
In the first stage, dephasing is performed by each replica independently to eliminate correlations between
replicas. This is done by choosing a random set of velocities, based on the random_seed that is specified, and
running t_dephase timesteps of dynamics. This is repeated n_dephase times. At each of the n_dephase stages,
if an event occurs during the t_dephase steps of dynamics for a particular replica, the replica repeats the stage
until no event occurs.
If the temp keyword is not specified, the target temperature for velocity randomization for each replica is the
current temperature of that replica. Otherwise, it is the specified Tdephase temperature. The style of velocity
randomization is controlled using the keyword vel with arguments that have the same meaning as their
counterparts in the velocity command.
In the second stage, each replica runs dynamics continuously, stopping every t_event steps to check if a
transition event has occurred. This check is performed by quenching the system and comparing the resulting
atom coordinates to the coordinates from the previous basin. The first time through the PRD loop, the
"previous basin" is the set of quenched coordinates from the initial state of the system.
A quench is an energy minimization and is performed by whichever algorithm has been defined by the
min_style command. Minimization parameters may be set via the min_modify command and by the min
keyword of the PRD command. The latter are the settings that would be used with the minimize command.
Note that typically, you do not need to perform a highly-converged minimization to detect a transition event.
The event check is performed by a compute with the specified compute-ID. Currently there is only one
compute that works with the PRD command, which is the compute event/displace command. Other
event-checking computes may be added. Compute event/displace checks whether any atom in the compute
group has moved further than a specified threshold distance. If so, an "event" has occurred.
LAMMPS Users Manual
561

In the third stage, the replica on which the event occurred (event replica) continues to run dynamics to search
for correlated events. This is done by running dynamics for t_correlate steps, quenching every t_event steps,
and checking if another event has occurred.
The first time no correlated event occurs, the final state of the event replica is shared with all replicas, the new
basin reference coordinates are updated with the quenched state, and the outer loop begins again. While the
replica event is searching for correlated events, all the other replicas also run dynamics and event checking
with the same schedule, but the final states are always overwritten by the state of the event replica.
The outer loop of the pseudo-code above continues until N steps of dynamics have been performed. Note that
N only includes the dynamics of stages 2 and 3, not the steps taken during dephasing or the minimization
iterations of quenching. The specified N is interpreted in one of two ways, depending on the time keyword. If
the time value is steps, which is the default, then each replica runs for N timesteps. If the time value is clock,
then the simulation runs until N aggregate timesteps across all replicas have elapsed. This aggregate time is
the "clock" time defined below, which typically advances nearly M times faster than the timestepping on a
single replica, where M is the number of replicas.
Four kinds of output can be generated during a PRD run: event statistics, thermodynamic output by each
replica, dump files, and restart files.
When running with multiple partitions (each of which is a replica in this case), the print-out to the screen and
master log.lammps file is limited to event statistics. Note that if a PRD run is performed on only a single
replica then the event statistics will be intermixed with the usual thermodynamic output discussed below.
The quantities printed each time an event occurs are the timestep, CPU time, clock, event number, a
correlation flag, the number of coincident events, and the replica number of the chosen event.
The timestep is the usual LAMMPS timestep, except that time does not advance during dephasing or
quenches, but only during dynamics. Note that are two kinds of dynamics in the PRD loop listed above that
contribute to this timestepping. The first is when all replicas are performing independent dynamics, waiting
for an event to occur. The second is when correlated events are being searched for, but only one replica is
running dynamics.
The CPU time is the total elapsed time on each processor, since the start of the PRD run.
The clock is the same as the timestep except that it advances by M steps per timestep during the first kind of
dynamics when the M replicas are running independently. The clock advances by only 1 step per timestep
during the second kind of dynamics, when only a single replica is checking for a correlated event. Thus
"clock" time represents the aggregate time (in steps) that has effectively elapsed during a PRD simulation on
M replicas. If most of the PRD run is spent in the second stage of the loop above, searching for infrequent
events, then the clock will advance nearly M times faster than it would if a single replica was running. Note
the clock time between successive events should be drawn from p(t).
The event number is a counter that increments with each event, whether it is uncorrelated or correlated.
The correlation flag will be 0 when an uncorrelated event occurs during the second stage of the loop listed
above, i.e. when all replicas are running independently. The correlation flag will be 1 when a correlated event
occurs during the third stage of the loop listed above, i.e. when only one replica is running dynamics.
When more than one replica detects an event at the end of the same event check (every t_event steps) during
the second stage, then one of them is chosen at random. The number of coincident events is the number of
LAMMPS Users Manual
562

replicas that detected an event. Normally, this value should be 1. If it is often greater than 1, then either the
number of replicas is too large, or t_event is too large.
The replica number is the ID of the replica (from 0 to M-1) in which the event occurred.
When running on multiple partitions, LAMMPS produces additional log files for each partition, e.g.
log.lammps.0, log.lammps.1, etc. For the PRD command, these contain the thermodynamic output for each
replica. You will see short runs and minimizations corresponding to the dynamics and quench operations of
the loop listed above. The timestep will be reset appropriately depending on whether the operation advances
time or not.
After the PRD command completes, timing statistics for the PRD run are printed in each replica's log file,
giving a breakdown of how much CPU time was spent in each stage (dephasing, dynamics, quenching, etc).
Any dump files defined in the input script, will be written to during a PRD run at timesteps corresponding to
both uncorrelated and correlated events. This means the requested dump frequency in the dump command is
ignored. There will be one dump file (per dump command) created for all partitions.
The atom coordinates of the dump snapshot are those of the minimum energy configuration resulting from
quenching following a transition event. The timesteps written into the dump files correspond to the timestep at
which the event occurred and NOT the clock. A dump snapshot corresponding to the initial minimum state
used for event detection is written to the dump file at the beginning of each PRD run.
If the restart command is used, a single restart file for all the partitions is generated, which allows a PRD run
to be continued by a new input script in the usual manner.
The restart file is generated at the end of the loop listed above. If no correlated events are found, this means it
contains a snapshot of the system at time T + t_correlate, where T is the time at which the uncorrelated event
occurred. If correlated events were found, then it contains a snapshot of the system at time T + t_correlate,
where T is the time of the last correlated event.
The restart frequency specified in the restart command is interpreted differently when performing a PRD run.
It does not mean the timestep interval between restart files. Instead it means an event interval for uncorrelated
events. Thus a frequency of 1 means write a restart file every time an uncorrelated event occurs. A frequency
of 10 means write a restart file every 10th uncorrelated event.
When an input script reads a restart file from a previous PRD run, the new script can be run on a different
number of replicas or processors. However, it is assumed that t_correlate in the new PRD command is the
same as it was previously. If not, the calculation of the "clock" value for the first event in the new run will be
slightly off.
Restrictions:
This command can only be used if LAMMPS was built with the REPLICA package. See the Making
LAMMPS section for more info on packages.
The N and t_correlate settings must be integer multiples of t_event.
Runs restarted from restart file written during a PRD run will not produce identical results due to changes in
the random numbers used for dephasing.
LAMMPS Users Manual
563

This command cannot be used when any fixes are defined that keep track of elapsed time to perform
time-dependent operations. Examples include the "ave" fixes such as fix ave/chunk. Also fix dt/reset and fix
deposit.
Related commands:
compute event/displace, min_modify, min_style, run_style, minimize, velocity, temper, neb, tad
Default:
The option defaults are min = 0.1 0.1 40 50, no temp setting, vel = geom gaussian, and time = steps.
(Voter1998) Voter, Phys Rev B, 57, 13985 (1998).
(Voter2002) Voter, Montalenti, Germann, Annual Review of Materials Research 32, 321 (2002).
LAMMPS Users Manual
564

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
print command
Syntax:
print string keyword value
string = text string to print, which may contain variables• zero or more keyword/value pairs may be appended• keyword = file or append or screen
file value = filename
append value = filename
screen value = yes or no
•
Examples:
print "Done with equilibration" file info.dat
print Vol=$v append info.dat screen no
print "The system volume is now $v"
print 'The system volume is now $v'
print """
System volume = $v
System temperature = $t
"""
Description:
Print a text string to the screen and logfile. The text string must be a single argument, so if it is one line but
more than one word, it should be enclosed in single or double quotes. To generate multiple lines of output, the
string can be enclosed in triple quotes, as in the last example above. If the text string contains variables, they
will be evaluated and their current values printed.
If the file or append keyword is used, a filename is specified to which the output will be written. If file is used,
then the filename is overwritten if it already exists. If append is used, then the filename is appended to if it
already exists, or created if it does not exist.
If the screen keyword is used, output to the screen and logfile can be turned on or off as desired.
If you want the print command to be executed multiple times (with changing variable values), there are 3
options. First, consider using the fix print command, which will print a string periodically during a simulation.
Second, the print command can be used as an argument to the every option of the run command. Third, the
print command could appear in a section of the input script that is looped over (see the jump and next
commands).
See the variable command for a description of equal style variables which are typically the most useful ones
to use with the print command. Equal-style variables can calculate formulas involving mathematical
operations, atom properties, group properties, thermodynamic properties, global values calculated by a
compute or fix, or references to other variables.
Restrictions: none
LAMMPS Users Manual
565

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
processors command
Syntax:
processors Px Py Pz keyword args ...
Px,Py,Pz = # of processors in each dimension of 3d grid overlaying the simulation domain• zero or more keyword/arg pairs may be appended• keyword = grid or map or part or file
grid arg = gstyle params ...
gstyle = onelevel or twolevel or numa or custom
onelevel params = none
twolevel params = Nc Cx Cy Cz
Nc = number of cores per node
Cx,Cy,Cz = # of cores in each dimension of 3d sub-grid assigned to each node
numa params = none
custom params = infile
infile = file containing grid layout
map arg = cart or cart/reorder or xyz or xzy or yxz or yzx or zxy or zyx
cart = use MPI_Cart() methods to map processors to 3d grid with reorder = 0
cart/reorder = use MPI_Cart() methods to map processors to 3d grid with reorder = 1
xyz,xzy,yxz,yzx,zxy,zyx = map processors to 3d grid in IJK ordering
numa arg = none
part args = Psend Precv cstyle
Psend = partition # (1 to Np) which will send its processor layout
Precv = partition # (1 to Np) which will recv the processor layout
cstyle = multiple
multiple = Psend grid will be multiple of Precv grid in each dimension
file arg = outfile
outfile = name of file to write 3d grid of processors to
•
Examples:
processors * * 5
processors 2 4 4
processors * * 8 map xyz
processors * * * grid numa
processors * * * grid twolevel 4 * * 1
processors 4 8 16 grid custom myfile
processors * * * part 1 2 multiple
Description:
Specify how processors are mapped as a regular 3d grid to the global simulation box. The mapping involves 2
steps. First if there are P processors it means choosing a factorization P = Px by Py by Pz so that there are Px
processors in the x dimension, and similarly for the y and z dimensions. Second, the P processors are mapped
to the regular 3d grid. The arguments to this command control each of these 2 steps.
The Px, Py, Pz parameters affect the factorization. Any of the 3 parameters can be specified with an asterisk
"*", which means LAMMPS will choose the number of processors in that dimension of the grid. It will do this
based on the size and shape of the global simulation box so as to minimize the surface-to-volume ratio of each
processor's sub-domain.
LAMMPS Users Manual
567

Choosing explicit values for Px or Py or Pz can be used to override the default manner in which LAMMPS
will create the regular 3d grid of processors, if it is known to be sub-optimal for a particular problem. E.g. a
problem where the extent of atoms will change dramatically in a particular dimension over the course of the
simulation.
The product of Px, Py, Pz must equal P, the total # of processors LAMMPS is running on. For a 2d
simulation, Pz must equal 1.
Note that if you run on a prime number of processors P, then a grid such as 1 x P x 1 will be required, which
may incur extra communication costs due to the high surface area of each processor's sub-domain.
Also note that if multiple partitions are being used then P is the number of processors in this partition; see this
section for an explanation of the -partition command-line switch. Also note that you can prefix the processors
command with the partition command to easily specify different Px,Py,Pz values for different partitions.
You can use the partition command to specify different processor grids for different partitions, e.g.
partition yes 1 processors 4 4 4
partition yes 2 processors 2 3 2
NOTE: This command only affects the initial regular 3d grid created when the simulation box is first specified
via a create_box or read_data or read_restart command. Or if the simulation box is re-created via the replicate
command. The same regular grid is initially created, regardless of which comm_style command is in effect.
If load-balancing is never invoked via the balance or fix balance commands, then the initial regular grid will
persist for all simulations. If balancing is performed, some of the methods invoked by those commands retain
the logical topology of the initial 3d grid, and the mapping of processors to the grid specified by the
processors command. However the grid spacings in different dimensions may change, so that processors own
sub-domains of different sizes. If the comm_style tiled command is used, methods invoked by the balancing
commands may discard the 3d grid of processors and tile the simulation domain with sub-domains of different
sizes and shapes which no longer have a logical 3d connectivity. If that occurs, all the information specified
by the processors command is ignored.
The grid keyword affects the factorization of P into Px,Py,Pz and it can also affect how the P processor IDs
are mapped to the 3d grid of processors.
The onelevel style creates a 3d grid that is compatible with the Px,Py,Pz settings, and which minimizes the
surface-to-volume ratio of each processor's sub-domain, as described above. The mapping of processors to the
grid is determined by the map keyword setting.
The twolevel style can be used on machines with multicore nodes to minimize off-node communication. It
insures that contiguous sub-sections of the 3d grid are assigned to all the cores of a node. For example if Nc is
4, then 2x2x1 or 2x1x2 or 1x2x2 sub-sections of the 3d grid will correspond to the cores of each node. This
affects both the factorization and mapping steps.
The Cx, Cy, Cz settings are similar to the Px, Py, Pz settings, only their product should equal Nc. Any of the 3
parameters can be specified with an asterisk "*", which means LAMMPS will choose the number of cores in
that dimension of the node's sub-grid. As with Px,Py,Pz, it will do this based on the size and shape of the
global simulation box so as to minimize the surface-to-volume ratio of each processor's sub-domain.
LAMMPS Users Manual
568

NOTE: For the twolevel style to work correctly, it assumes the MPI ranks of processors LAMMPS is running
on are ordered by core and then by node. E.g. if you are running on 2 quad-core nodes, for a total of 8
processors, then it assumes processors 0,1,2,3 are on node 1, and processors 4,5,6,7 are on node 2. This is the
default rank ordering for most MPI implementations, but some MPIs provide options for this ordering, e.g. via
environment variable settings.
The numa style operates similar to the twolevel keyword except that it auto-detects which cores are running on
which nodes. Currently, it does this in only 2 levels, but it may be extended in the future to account for socket
topology and other non-uniform memory access (NUMA) costs. It also uses a different algorithm than the
twolevel keyword for doing the two-level factorization of the simulation box into a 3d processor grid to
minimize off-node communication, and it does its own MPI-based mapping of nodes and cores to the regular
3d grid. Thus it may produce a different layout of the processors than the twolevel options.
The numa style will give an error if the number of MPI processes is not divisible by the number of cores used
per node, or any of the Px or Py of Pz values is greater than 1.
NOTE: Unlike the twolevel style, the numa style does not require any particular ordering of MPI ranks i
norder to work correctly. This is because it auto-detects which processes are running on which nodes.
The custom style uses the file infile to define both the 3d factorization and the mapping of processors to the
grid.
The file should have the following format. Any number of initial blank or comment lines (starting with a "#"
character) can be present. The first non-blank, non-comment line should have 3 values:
Px Py Py
These must be compatible with the total number of processors and the Px, Py, Pz settings of the processors
command.
This line should be immediately followed by P = Px*Py*Pz lines of the form:
ID I J K
where ID is a processor ID (from 0 to P-1) and I,J,K are the processors location in the 3d grid. I must be a
number from 1 to Px (inclusive) and similarly for J and K. The P lines can be listed in any order, but no
processor ID should appear more than once.
The map keyword affects how the P processor IDs (from 0 to P-1) are mapped to the 3d grid of processors. It
is only used by the onelevel and twolevel grid settings.
The cart style uses the family of MPI Cartesian functions to perform the mapping, namely MPI_Cart_create(),
MPI_Cart_get(), MPI_Cart_shift(), and MPI_Cart_rank(). It invokes the MPI_Cart_create() function with its
reorder flag = 0, so that MPI is not free to reorder the processors.
The cart/reorder style does the same thing as the cart style except it sets the reorder flag to 1, so that MPI can
reorder processors if it desires.
The xyz, xzy, yxz, yzx, zxy, and zyx styles are all similar. If the style is IJK, then it maps the P processors to the
grid so that the processor ID in the I direction varies fastest, the processor ID in the J direction varies next
fastest, and the processor ID in the K direction varies slowest. For example, if you select style xyz and you
LAMMPS Users Manual
569
have a 2x2x2 grid of 8 processors, the assignments of the 8 octants of the simulation domain will be:
proc 0 = lo x, lo y, lo z octant
proc 1 = hi x, lo y, lo z octant
proc 2 = lo x, hi y, lo z octant
proc 3 = hi x, hi y, lo z octant
proc 4 = lo x, lo y, hi z octant
proc 5 = hi x, lo y, hi z octant
proc 6 = lo x, hi y, hi z octant
proc 7 = hi x, hi y, hi z octant
Note that, in principle, an MPI implementation on a particular machine should be aware of both the machine's
network topology and the specific subset of processors and nodes that were assigned to your simulation. Thus
its MPI_Cart calls can optimize the assignment of MPI processes to the 3d grid to minimize communication
costs. In practice, however, few if any MPI implementations actually do this. So it is likely that the cart and
cart/reorder styles simply give the same result as one of the IJK styles.
Also note, that for the twolevel grid style, the map setting is used to first map the nodes to the 3d grid, then
again to the cores within each node. For the latter step, the cart and cart/reorder styles are not supported, so
an xyz style is used in their place.
The part keyword affects the factorization of P into Px,Py,Pz.
It can be useful when running in multi-partition mode, e.g. with the run_style verlet/split command. It
specifies a dependency between a sending partition Psend and a receiving partition Precv which is enforced
when each is setting up their own mapping of their processors to the simulation box. Each of Psend and Precv
must be integers from 1 to Np, where Np is the number of partitions you have defined via the -partition
command-line switch.
A "dependency" means that the sending partition will create its regular 3d grid as Px by Py by Pz and after it
has done this, it will send the Px,Py,Pz values to the receiving partition. The receiving partition will wait to
receive these values before creating its own regular 3d grid and will use the sender's Px,Py,Pz values as a
constraint. The nature of the constraint is determined by the cstyle argument.
For a cstyle of multiple, each dimension of the sender's processor grid is required to be an integer multiple of
the corresponding dimension in the receiver's processor grid. This is a requirement of the run_style verlet/split
command.
For example, assume the sending partition creates a 4x6x10 grid = 240 processor grid. If the receiving
partition is running on 80 processors, it could create a 4x2x10 grid, but it will not create a 2x4x10 grid, since
in the y-dimension, 6 is not an integer multiple of 4.
NOTE: If you use the partition command to invoke different "processors" commands on different partitions,
and you also use the part keyword, then you must insure that both the sending and receiving partitions invoke
the "processors" command that connects the 2 partitions via the part keyword. LAMMPS cannot easily check
for this, but your simulation will likely hang in its setup phase if this error has been made.
The file keyword writes the mapping of the factorization of P processors and their mapping to the 3d grid to
the specified file outfile. This is useful to check that you assigned physical processors in the manner you
desired, which can be tricky to figure out, especially when running on multiple partitions or on, a multicore
machine or when the processor ranks were reordered by use of the -reorder command-line switch or due to use
of MPI-specific launch options such as a config file.
LAMMPS Users Manual
570

If you have multiple partitions you should insure that each one writes to a different file, e.g. using a
world-style variable for the filename. The file has a self-explanatory header, followed by one-line per
processor in this format:
world-ID universe-ID original-ID: I J K: name
The IDs are the processor's rank in this simulation (the world), the universe (of multiple simulations), and the
original MPI communicator used to instantiate LAMMPS, respectively. The world and universe IDs will only
be different if you are running on more than one partition; see the -partition command-line switch. The
universe and original IDs will only be different if you used the -reorder command-line switch to reorder the
processors differently than their rank in the original communicator LAMMPS was instantiated with.
I,J,K are the indices of the processor in the regular 3d grid, each from 1 to Nd, where Nd is the number of
processors in that dimension of the grid.
The name is what is returned by a call to MPI_Get_processor_name() and should represent an identifier
relevant to the physical processors in your machine. Note that depending on the MPI implementation, multiple
cores can have the same name.
Restrictions:
This command cannot be used after the simulation box is defined by a read_data or create_box command. It
can be used before a restart file is read to change the 3d processor grid from what is specified in the restart
file.
The grid numa keyword only currently works with the map cart option.
The part keyword (for the receiving partition) only works with the grid onelevel or grid twolevel options.
Related commands:
partition, -reorder command-line switch
Default:
The option defaults are Px Py Pz = * * *, grid = onelevel, and map = cart.
LAMMPS Users Manual
571

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
python command
Syntax:
python func keyword args ...
func = name of Python function• one or more keyword/args pairs must be appended
keyword = invoke or input or return or format or length or file or here or exists
invoke arg = none = invoke the previously defined Python function
input args = N i1 i2 ... iN
N = # of inputs to function
i1,...,iN = value, SELF, or LAMMPS variable name
value = integer number, floating point number, or string
SELF = reference to LAMMPS itself which can be accessed by Python function
variable = v_name, where name = name of LAMMPS variable, e.g. v_abc
return arg = varReturn
varReturn = v_name = LAMMPS variable name which return value of function will be assigned to
format arg = fstring with M characters
M = N if no return value, where N = # of inputs
M = N+1 if there is a return value
fstring = each character (i,f,s,p) corresponds in order to an input or return value
'i' = integer, 'f' = floating point, 's' = string, 'p' = SELF
length arg = Nlen
Nlen = max length of string returned from Python function
file arg = filename
filename = file of Python code, which defines func
here arg = inline
inline = one or more lines of Python code which defines func
must be a single argument, typically enclosed between triple quotes
exists arg = none = Python code has been loaded by previous python command
•
Examples:
python pForce input 2 v_x 20.0 return v_f format fff file force.py
python pForce invoke
python factorial input 1 myN return v_fac format ii here """
def factorial(n):
if n == 1: return n
return n * factorial(n-1)
"""
python loop input 1 SELF return v_value format -f here """
def loop(lmpptr,N,cut0):
from lammps import lammps
lmp = lammps(ptr=lmpptr)
# loop N times, increasing cutoff each time
for i in range(N):
cut = cut0 + i*0.1
lmp.set_variable("cut",cut) # set a variable in LAMMPS
lmp.command("pair_style lj/cut ${cut}") # LAMMPS commands
LAMMPS Users Manual
572

lmp.command("pair_coeff * * 1.0 1.0")
lmp.command("run 100")
"""
Description:
NOTE: It is not currently possible to use the python command described in this section with Python 3, only
with Python 2. The C API changed from Python 2 to 3 and the LAMMPS code is not compatible with both.
Define a Python function or execute a previously defined function. Arguments, including LAMMPS variables,
can be passed to the function from the LAMMPS input script and a value returned by the Python function to a
LAMMPS variable. The Python code for the function can be included directly in the input script or in a
separate Python file. The function can be standard Python code or it can make "callbacks" to LAMMPS
through its library interface to query or set internal values within LAMMPS. This is a powerful mechanism
for performing complex operations in a LAMMPS input script that are not possible with the simple input
script and variable syntax which LAMMPS defines. Thus your input script can operate more like a true
programming language.
Use of this command requires building LAMMPS with the PYTHON package which links to the Python
library so that the Python interpreter is embedded in LAMMPS. More details about this process are given
below.
There are two ways to invoke a Python function once it has been defined. One is using the invoke keyword.
The other is to assign the function to a python-style variable defined in your input script. Whenever the
variable is evaluated, it will execute the Python function to assign a value to the variable. Note that variables
can be evaluated in many different ways within LAMMPS. They can be substituted for directly in an input
script. Or they can be passed to various commands as arguments, so that the variable is evaluated during a
simulation run.
A broader overview of how Python can be used with LAMMPS is given in Section 11. There is an
examples/python directory which illustrates use of the python command.
The func setting specifies the name of the Python function. The code for the function is defined using the file
or here keywords as explained below.
If the invoke keyword is used, no other keywords can be used, and a previous python command must have
defined the Python function referenced by this command. This invokes the Python function with the
previously defined arguments and return value processed as explained below. You can invoke the function as
many times as you wish in your input script.
The input keyword defines how many arguments N the Python function expects. If it takes no arguments, then
the input keyword should not be used. Each argument can be specified directly as a value, e.g. 6 or 3.14159 or
abc (a string of characters). The type of each argument is specified by the format keyword as explained below,
so that Python will know how to interpret the value. If the word SELF is used for an argument it has a special
meaning. A pointer is passed to the Python function which it converts into a reference to LAMMPS itself.
This enables the function to call back to LAMMPS through its library interface as explained below. This
allows the Python function to query or set values internal to LAMMPS which can affect the subsequent
execution of the input script. A LAMMPS variable can also be used as an argument, specified as v_name,
where "name" is the name of the variable. Any style of LAMMPS variable can be used, as defined by the
variable command. Each time the Python function is invoked, the LAMMPS variable is evaluated and its
value is passed to the Python function.
LAMMPS Users Manual
573

The return keyword is only needed if the Python function returns a value. The specified varReturn must be of
the form v_name, where "name" is the name of a python-style LAMMPS variable, defined by the variable
command. The Python function can return a numeric or string value, as specified by the format keyword.
As explained on the variable doc page, the definition of a python-style variable associates a Python function
name with the variable. This must match the func setting for this command. For example these two commands
would be self-consistent:
variable foo python myMultiply
python myMultiply return v_foo format f file funcs.py
The two commands can appear in either order in the input script so long as both are specified before the
Python function is invoked for the first time.
The format keyword must be used if the input or return keyword is used. It defines an fstring with M
characters, where M = sum of number of inputs and outputs. The order of characters corresponds to the N
inputs, followed by the return value (if it exists). Each character must be one of the following: "i" for integer,
"f" for floating point, "s" for string, or "p" for SELF. Each character defines the type of the corresponding
input or output value of the Python function and affects the type conversion that is performed internally as
data is passed back and forth between LAMMPS and Python. Note that it is permissible to use a python-style
variable in a LAMMPS command that allows for an equal-style variable as an argument, but only if the output
of the Python function is flagged as a numeric value ("i" or "f") via the format keyword.
If the return keyword is used and the format keyword specifies the output as a string, then the default
maximum length of that string is 63 characters (64-1 for the string terminator). If you want to return a longer
string, the length keyword can be specified with its Nlen value set to a larger number (the code allocates space
for Nlen+1 to include the string terminator). If the Python function generates a string longer than the default
63 or the specified Nlen, it will be truncated.
Either the file, here, or exists keyword must be used, but only one of them. These keywords specify what
Python code to load into the Python interpreter. The file keyword gives the name of a file, which should end
with a ".py" suffix, which contains Python code. The code will be immediately loaded into and run in the
"main" module of the Python interpreter. Note that Python code which contains a function definition does not
"execute" the function when it is run; it simply defines the function so that it can be invoked later.
The here keyword does the same thing, except that the Python code follows as a single argument to the here
keyword. This can be done using triple quotes as delimiters, as in the examples above. This allows Python
code to be listed verbatim in your input script, with proper indentation, blank lines, and comments, as desired.
See Section 3.2, for an explanation of how triple quotes can be used as part of input script syntax.
The exists keyword takes no argument. It means that Python code containing the required Python function
defined by the func setting, is assumed to have been previously loaded by another python command.
Note that the Python code that is loaded and run must contain a function with the specified func name. To
operate properly when later invoked, the function code must match the input and return and format keywords
specified by the python command. Otherwise Python will generate an error.
This section describes how Python code can be written to work with LAMMPS.
Whether you load Python code from a file or directly from your input script, via the file and here keywords,
the code can be identical. It must be indented properly as Python requires. It can contain comments or blank
LAMMPS Users Manual
574
lines. If the code is in your input script, it cannot however contain triple-quoted Python strings, since that will
conflict with the triple-quote parsing that the LAMMPS input script performs.
All the Python code you specify via one or more python commands is loaded into the Python "main" module,
i.e. __main__. The code can define global variables or statements that are outside of function definitions. It
can contain multiple functions, only one of which matches the func setting in the python command. This
means you can use the file keyword once to load several functions, and the exists keyword thereafter in
subsequent python commands to access the other functions previously loaded.
A Python function you define (or more generally, the code you load) can import other Python modules or
classes, it can make calls to other system functions or functions you define, and it can access or modify global
variables (in the "main" module) which will persist between successive function calls. The latter can be
useful, for example, to prevent a function from being invoke multiple times per timestep by different
commands in a LAMMPS input script that access the returned python-style variable associated with the
function. For example, consider this function loaded with two global variables defined outside the function:
nsteplast = -1
nvaluelast = 0
def expensive(nstep):
global nsteplast,nvaluelast
if nstep == nsteplast: return nvaluelast
nsteplast = nstep
# perform complicated calculation
nvalue = ...
nvaluelast = nvalue
return nvalue
Nsteplast stores the previous timestep the function was invoked (passed as an argument to the function).
Nvaluelast stores the return value computed on the last function invocation. If the function is invoked again on
the same timestep, the previous value is simply returned, without re-computing it. The "global" statement
inside the Python function allows it to overwrite the global variables.
Note that if you load Python code multiple times (via multiple python commands), you can overwrite
previously loaded variables and functions if you are not careful. E.g. if the code above were loaded twice, the
global variables would be re-initialized, which might not be what you want. Likewise, if a function with the
same name exists in two chunks of Python code you load, the function loaded second will override the
function loaded first.
It's important to realize that if you are running LAMMPS in parallel, each MPI task will load the Python
interpreter and execute a local copy of the Python function(s) you define. There is no connection between the
Python interpreters running on different processors. This implies three important things.
First, if you put a print statement in your Python function, you will see P copies of the output, when running
on P processors. If the prints occur at (nearly) the same time, the P copies of the output may be mixed
together. Welcome to the world of parallel programming and debugging.
Second, if your Python code loads modules that are not pre-loaded by the Python library, then it will load the
module from disk. This may be a bottleneck if 1000s of processors try to load a module at the same time. On
some large supercomputers, loading of modules from disk by Python may be disabled. In this case you would
need to pre-build a Python library that has the required modules pre-loaded and link LAMMPS with that
library.
LAMMPS Users Manual
575

Third, if your Python code calls back to LAMMPS (discussed in the next section) and causes LAMMPS to
perform an MPI operation requires global communication (e.g. via MPI_Allreduce), such as computing the
global temperature of the system, then you must insure all your Python functions (running independently on
different processors) call back to LAMMPS. Otherwise the code may hang.
Your Python function can "call back" to LAMMPS through its library interface, if you use the SELF input to
pass Python a pointer to LAMMPS. The mechanism for doing this in your Python function is as follows:
def foo(lmpptr,...):
from lammps import lammps
lmp = lammps(ptr=lmpptr)
lmp.command('print "Hello from inside Python"')
...
The function definition must include a variable (lmpptr in this case) which corresponds to SELF in the python
command. The first line of the function imports the Python module lammps.py in the python dir of the
distribution. The second line creates a Python object "lmp" which wraps the instance of LAMMPS that called
the function. The "ptr=lmpptr" argument is what makes that happen. The thrid line invokes the command()
function in the LAMMPS library interface. It takes a single string argument which is a LAMMPS input script
command for LAMMPS to execute, the same as if it appeared in your input script. In this case, LAMMPS
should output
Hello from inside Python
to the screen and log file. Note that since the LAMMPS print command itself takes a string in quotes as its
argument, the Python string must be delimited with a different style of quotes.
Section 11.7 describes the syntax for how Python wraps the various functions included in the LAMMPS
library interface.
A more interesting example is in the examples/python/in.python script which loads and runs the following
function from examples/python/funcs.py:
def loop(N,cut0,thresh,lmpptr):
print "LOOP ARGS",N,cut0,thresh,lmpptr
from lammps import lammps
lmp = lammps(ptr=lmpptr)
natoms = lmp.get_natoms()
for i in range(N):
cut = cut0 + i*0.1
lmp.set_variable("cut",cut) # set a variable in LAMMPS
lmp.command("pair_style lj/cut ${cut}") # LAMMPS command
#lmp.command("pair_style lj/cut %d" % cut) # LAMMPS command option
lmp.command("pair_coeff * * 1.0 1.0") # ditto
lmp.command("run 10") # ditto
pe = lmp.extract_compute("thermo_pe",0,0) # extract total PE from LAMMPS
print "PE",pe/natoms,thresh
if pe/natoms <thresh: return
with these input script commands:
LAMMPS Users Manual
576

python loop input 4 10 1.0 -4.0 SELF format iffp file funcs.py
python loop invoke
This has the effect of looping over a series of 10 short runs (10 timesteps each) where the pair style cutoff is
increased from a value of 1.0 in distance units, in increments of 0.1. The looping stops when the per-atom
potential energy falls below a threshold of -4.0 in energy units. More generally, Python can be used to
implement a loop with complex logic, much more so than can be created using the LAMMPS jump and if
commands.
Several LAMMPS library functions are called from the loop function. Get_natoms() returns the number of
atoms in the simulation, so that it can be used to normalize the potential energy that is returned by
extract_compute() for the "thermo_pe" compute that is defined by default for LAMMPS thermodynamic
output. Set_variable() sets the value of a string variable defined in LAMMPS. This library function is a useful
way for a Python function to return multiple values to LAMMPS, more than the single value that can be
passed back via a return statement. This cutoff value in the "cut" variable is then substituted (by LAMMPS) in
the pair_style command that is executed next. Alternatively, the "LAMMPS command option" line could be
used in place of the 2 preceding lines, to have Python insert the value into the LAMMPS command string.
NOTE: When using the callback mechanism just described, recognize that there are some operations you
should not attempt because LAMMPS cannot execute them correctly. If the Python function is invoked
between runs in the LAMMPS input script, then it should be OK to invoke any LAMMPS input script
command via the library interface command() or file() functions, so long as the command would work if it
were executed in the LAMMPS input script directly at the same point.
However, a Python function can also be invoked during a run, whenever an associated LAMMPS variable it is
assigned to is evaluated. If the variable is an input argument to another LAMMPS command (e.g. fix
setforce), then the Python function will be invoked inside the class for that command, in one of its methods
that is invoked in the middle of a timestep. You cannot execute arbitrary input script commands from the
Python function (again, via the command() or file() functions) at that point in the run and expect it to work.
Other library functions such as those that invoke computes or other variables may have hidden side effects as
well. In these cases, LAMMPS has no simple way to check that something illogical is being attempted.
If you run Python code directly on your workstation, either interactively or by using Python to launch a
Python script stored in a file, and your code has an error, you will typically see informative error messages.
That is not the case when you run Python code from LAMMPS using an embedded Python interpreter. The
code will typically fail silently. LAMMPS will catch some errors but cannot tell you where in the Python code
the problem occurred. For example, if the Python code cannot be loaded and run because it has syntax or other
logic errors, you may get an error from Python pointing to the offending line, or you may get one of these
generic errors from LAMMPS:
Could not process Python file
Could not process Python string
When the Python function is invoked, if it does not return properly, you will typically get this generic error
from LAMMPS:
Python function evaluation failed
Here are three suggestions for debugging your Python code while running it under LAMMPS.
First, don't run it under LAMMPS, at least to start with! Debug it using plain Python. Load and invoke your
LAMMPS Users Manual
577

function, pass it arguments, check return values, etc.
Second, add Python print statements to the function to check how far it gets and intermediate values it
calculates. See the discussion above about printing from Python when running in parallel.
Third, use Python exception handling. For example, say this statement in your Python function is failing,
because you have not initialized the variable foo:
foo += 1
If you put one (or more) statements inside a "try" statement, like this:
import exceptions
print "Inside simple function"
try:
foo += 1 # one or more statements here
except Exception, e:
print "FOO error:",e
then you will get this message printed to the screen:
FOO error: local variable 'foo' referenced before assignment
If there is no error in the try statements, then nothing is printed. Either way the function continues on (unless
you put a return or sys.exit() in the except clause).
Restrictions:
This command is part of the PYTHON package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Building LAMMPS with the PYTHON package will link LAMMPS with the Python library on your system.
Settings to enable this are in the lib/python/Makefile.lammps file. See the lib/python/README file for
information on those settings.
If you use Python code which calls back to LAMMPS, via the SELF input argument explained above, there is
an extra step required when building LAMMPS. LAMMPS must also be built as a shared library and your
Python function must be able to to load the Python module in python/lammps.py that wraps the LAMMPS
library interface. These are the same steps required to use Python by itself to wrap LAMMPS. Details on these
steps are explained in Section python. Note that it is important that the stand-alone LAMMPS executable and
the LAMMPS shared library be consistent (built from the same source code files) in order for this to work. If
the two have been built at different times using different source files, problems may occur.
As described above, you can use the python command to invoke a Python function which calls back to
LAMMPS through its Python-wrapped library interface. However you cannot do the opposite. I.e. you cannot
call LAMMPS from Python and invoke the python command to "callback" to Python and execute a Python
function. LAMMPS will generate an error if you try to do that. Note that we think there actually should be a
way to do that, but haven't yet been able to figure out how to do it successfully.
Related commands:
shell, variable
LAMMPS Users Manual
578
Default: none
LAMMPS Users Manual
579

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
quit command
Syntax:
quit status
status = numerical exit status (optional)
Examples:
quit
if "$n > 10000" then "quit 1"
Description:
This command causes LAMMPS to exit, after shutting down all output cleanly.
It can be used as a debug statement in an input script, to terminate the script at some intermediate point.
It can also be used as an invoked command inside the "then" or "else" portion of an if command.
The optional status argument is an integer which signals the return status to a program calling LAMMPS. A
return status of 0 usually indicates success. A status != 0 is failure, where the specified value can be used to
distinguish the kind of error, e.g. where in the input script the quit was invoked. If not specified, a status of 0
is returned.
Restrictions: none
Related commands:
if
Default: none
LAMMPS Users Manual
580

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
read_data command
Syntax:
read_data file keyword args ...
file = name of data file to read in• zero or more keyword/arg pairs may be appended• keyword = add or offset or shift or extra/atom/types or extra/bond/types or extra/angle/types or
extra/dihedral/types or extra/improper/types or group or nocoeff or fix
add arg = append or Nstart or merge
append = add new atoms with IDs appended to current IDs
Nstart = add new atoms with IDs starting with Nstart
merge = add new atoms with their IDs unchanged
offset args = toff boff aoff doff ioff
toff = offset to add to atom types
boff = offset to add to bond types
aoff = offset to add to angle types
doff = offset to add to dihedral types
ioff = offset to add to improper types
shift args = Sx Sy Sz
Sx,Sy,Sz = distance to shift atoms when adding to system (distance units)
extra/atom/types arg = # of extra atom types
extra/bond/types arg = # of extra bond types
extra/angle/types arg = # of extra angle types
extra/dihedral/types arg = # of extra dihedral types
extra/improper/types arg = # of extra improper types
group args = groupID
groupID = add atoms in data file to this group
nocoeff = ignore force field parameters
fix args = fix-ID header-string section-string
fix-ID = ID of fix to process header lines and sections of data file
header-string = header lines containing this string will be passed to fix
section-string = section names with this string will be passed to fix
•
Examples:
read_data data.lj
read_data ../run7/data.polymer.gz
read_data data.protein fix mycmap crossterm CMAP
read_data data.water add append offset 3 1 1 1 1 shift 0.0 0.0 50.0
read_data data.water add merge 1 group solvent
Description:
Read in a data file containing information LAMMPS needs to run a simulation. The file can be ASCII text or
a gzipped text file (detected by a .gz suffix). This is one of 3 ways to specify initial atom coordinates; see the
read_restart and create_atoms commands for alternative methods. Also see the explanation of the -restart
command-line switch which can convert a restart file to a data file.
This command can be used multiple times to add new atoms and their properties to an existing system by
using the add, offset, and shift keywords. See more details below, which includes the use case for the extra
LAMMPS Users Manual
581

keywords.
The group keyword adds all the atoms in the data file to the specified group-ID. The group will be created if it
does not already exist. This is useful if you are reading multiple data files and wish to put sets of atoms into
different groups so they can be operated on later. E.g. a group of added atoms can be moved to new positions
via the displace_atoms command. Note that atoms read from the data file are also always added to the "all"
group. The group command discusses atom groups, as used in LAMMPS.
The nocoeff keyword tells read_data to ignore force field parameters. The various Coeff sections are still read
and have to have the correct number of lines, but they are not applied. This also allows to read a data file
without having any pair, bond, angle, dihedral or improper styles defined, or to read a data file for a different
force field.
The use of the fix keyword is discussed below.
Reading multiple data files
The read_data command can be used multiple times with the same or different data files to build up a complex
system from components contained in individual data files. For example one data file could contain fluid in a
confined domain; a second could contain wall atoms, and the second file could be read a third time to create a
wall on the other side of the fluid. The third set of atoms could be rotated to an opposing direction using the
displace_atoms command, after the third read_data command is used.
The add, offset, shift, extra, and group keywords are useful in this context.
If a simulation box does not yet exist, the add keyword cannot be used; the read_data command is being used
for the first time. If a simulation box does exist, due to using the create_box command, or a previous
read_data command, then the add keyword must be used.
NOTE: The simulation box size (xlo to xhi, ylo to yhi, zlo to zhi) in the new data file will be merged with the
existing simulation box to create a large enough box in each dimension to contain both the existing and new
atoms. Each box dimension never shrinks due to this merge operation, it only stays the same or grows. Care
must be used if you are growing the existing simulation box in a periodic dimension. If there are existing
atoms with bonds that straddle that periodic boundary, then the atoms may become far apart if the box size
grows. This will separate the atoms in the bond, which can lead to "lost" bond atoms or bad dynamics.
The three choices for the add argument affect how the IDs of atoms in the data file are treated. If append is
specified, atoms in the data file are added to the current system, with their atom IDs reset so that an atomID =
M in the data file becomes atomID = N+M, where N is the largest atom ID in the current system. This rule is
applied to all occurrences of atom IDs in the data file, e.g. in the Velocity or Bonds section. If Nstart is
specified, then Nstart is a numeric value is given, e.g. 1000, so that an atomID = M in the data file becomes
atomID = 1000+M. If merge is specified, the data file atoms are added to the current system without changing
their IDs. They are assumed to merge (without duplication) with the currently defined atoms. It is up to you to
insure there are no multiply defined atom IDs, as LAMMPS only performs an incomplete check that this is the
case by insuring the resulting max atomID >= the number of atoms.
The offset and shift keywords can only be used if the add keyword is also specified.
The offset keyword adds the specified offset values to the atom types, bond types, angle types, dihedral types,
and improper types as they are read from the data file. E.g. if toff = 2, and the file uses atom types 1,2,3, then
the added atoms will have atom types 3,4,5. These offsets apply to all occurrences of types in the data file, e.g.
LAMMPS Users Manual
582

for the Atoms or Masses or Pair Coeffs or Bond Coeffs sections. This makes it easy to use atoms and
molecules and their attributes from a data file in different simulations, where you want their types (atom,
bond, angle, etc) to be different depending on what other types already exist. All five offset values must be
specified, but individual values will be ignored if the data file does not use that attribute (e.g. no bonds).
The shift keyword can be used to specify an (Sx, Sy, Sz) displacement applied to the coordinates of each
atom. Sz must be 0.0 for a 2d simulation. This is a mechanism for adding structured collections of atoms at
different locations within the simulation box, to build up a complex geometry. It is up to you to insure atoms
do not end up overlapping unphysically which would lead to bad dynamics. Note that the displace_atoms
command can be used to move a subset of atoms after they have been read from a data file. Likewise, the
delete_atoms command can be used to remove overlapping atoms. Note that the shift values (Sx, Sy, Sz) are
also added to the simulation box information (xlo, xhi, ylo, yhi, zlo, zhi) in the data file to shift its boundaries.
E.g. xlo_new = xlo + Sx, xhi_new = xhi + Sx.
The extra keywords can only be used the first time the read_data command is used. They are useful if you
intend to add new atom, bond, angle, etc types later with additional read_data commands. This is because the
maximum number of allowed atom, bond, angle, etc types is set by LAMMPS when the system is first
initialized. If you do not use the extra keywords, then the number of these types will be limited to what
appears in the first data file you read. For example, if the first data file is a solid substrate of Si, it will likely
specify a single atom type. If you read a second data file with a different material (water molecules) that sit on
top of the substrate, you will want to use different atom types for those atoms. You can only do this if you set
the extra/atom/types keyword to a sufficiently large value when reading the substrate data file. Note that use
of the extra keywords also allows each data file to contain sections like Masses or Pair Coeffs or Bond Coeffs
which are sized appropriately for the number of types in that data file. If the offset keyword is used
appropriately when each data file is read, the values in those sections will be stored correctly in the larger data
structures allocated by the use of the extra keywords. E.g. the substrate file can list mass and pair coefficients
for type 1 silicon atoms. The water file can list mass and pair coefficients for type 1 and type 2 hydrogen and
oxygen atoms. Use of the extra and offset keywords will store those mass and pair coefficient values
appropriately in data structures that allow for 3 atom types (Si, H, O). Of course, you would still need to
specify coefficients for H/Si and O/Si interactions in your input script to have a complete pairwise interaction
model.
An alternative to using the extra keywords with the read_data command, is to use the create_box command to
initialize the simulation box and all the various type limits you need via its extra keywords. Then use the
read_data command one or more times to populate the system with atoms, bonds, angles, etc, using the offset
keyword if desired to alter types used in the various data files you read.
Format of a data file
The structure of the data file is important, though many settings and sections are optional or can come in any
order. See the examples directory for sample data files for different problems.
A data file has a header and a body. The header appears first. The first line of the header is always skipped; it
typically contains a description of the file. Then lines are read one at a time. Lines can have a trailing
comment starting with '#' that is ignored. If the line is blank (only whitespace after comment is deleted), it is
skipped. If the line contains a header keyword, the corresponding value(s) is read from the line. If it doesn't
contain a header keyword, the line begins the body of the file.
The body of the file contains zero or more sections. The first line of a section has only a keyword. This line
can have a trailing comment starting with '#' that is either ignored or can be used to check for a style match, as
described below. The next line is skipped. The remaining lines of the section contain values. The number of
LAMMPS Users Manual
583

lines depends on the section keyword as described below. Zero or more blank lines can be used between
sections. Sections can appear in any order, with a few exceptions as noted below.
The keyword fix can be used one or more times. Each usage specifies a fix that will be used to process a
specific portion of the data file. Any header line containing header-string and any section with a name
containing section-string will be passed to the specified fix. See the fix property/atom command for an
example of a fix that operates in this manner. The doc page for the fix defines the syntax of the header line(s)
and section(s) that it reads from the data file. Note that the header-string can be specified as NULL, in which
case no header lines are passed to the fix. This means that it can infer the length of its Section from standard
header settings, such as the number of atoms.
The formatting of individual lines in the data file (indentation, spacing between words and numbers) is not
important except that header and section keywords (e.g. atoms, xlo xhi, Masses, Bond Coeffs) must be
capitalized as shown and can't have extra white space between their words - e.g. two spaces or a tab between
the 2 words in "xlo xhi" or the 2 words in "Bond Coeffs", is not valid.
Format of the header of a data file
These are the recognized header keywords. Header lines can come in any order. The value(s) are read from
the beginning of the line. Thus the keyword atoms should be in a line like "1000 atoms"; the keyword ylo yhi
should be in a line like "-10.0 10.0 ylo yhi"; the keyword xy xz yz should be in a line like "0.0 5.0 6.0 xy xz
yz". All these settings have a default value of 0, except the lo/hi box size defaults are -0.5 and 0.5. A line need
only appear if the value is different than the default.
atoms = # of atoms in system• bonds = # of bonds in system• angles = # of angles in system• dihedrals = # of dihedrals in system• impropers = # of impropers in system• atom types = # of atom types in system• bond types = # of bond types in system• angle types = # of angle types in system• dihedral types = # of dihedral types in system• improper types = # of improper types in system• extra bond per atom = leave space for this many new bonds per atom• extra angle per atom = leave space for this many new angles per atom• extra dihedral per atom = leave space for this many new dihedrals per atom• extra improper per atom = leave space for this many new impropers per atom• extra special per atom = leave space for this many new special bonds per atom• ellipsoids = # of ellipsoids in system• lines = # of line segments in system• triangles = # of triangles in system• bodies = # of bodies in system• xlo xhi = simulation box boundaries in x dimension• ylo yhi = simulation box boundaries in y dimension• zlo zhi = simulation box boundaries in z dimension• xy xz yz = simulation box tilt factors for triclinic system•
The initial simulation box size is determined by the lo/hi settings. In any dimension, the system may be
periodic or non-periodic; see the boundary command. When the simulation box is created it is also partitioned
into a regular 3d grid of rectangular bricks, one per processor, based on the number of processors being used
LAMMPS Users Manual
584
and the settings of the processors command. The partitioning can later be changed by the balance or fix
balance commands.
If the xy xz yz line does not appear, LAMMPS will set up an axis-aligned (orthogonal) simulation box. If the
line does appear, LAMMPS creates a non-orthogonal simulation domain shaped as a parallelepiped with
triclinic symmetry. The parallelepiped has its "origin" at (xlo,ylo,zlo) and is defined by 3 edge vectors starting
from the origin given by A = (xhi-xlo,0,0); B = (xy,yhi-ylo,0); C = (xz,yz,zhi-zlo). Xy,xz,yz can be 0.0 or
positive or negative values and are called "tilt factors" because they are the amount of displacement applied to
faces of an originally orthogonal box to transform it into the parallelepiped.
By default, the tilt factors (xy,xz,yz) can not skew the box more than half the distance of the corresponding
parallel box length. For example, if xlo = 2 and xhi = 12, then the x box length is 10 and the xy tilt factor must
be between -5 and 5. Similarly, both xz and yz must be between -(xhi-xlo)/2 and +(yhi-ylo)/2. Note that this is
not a limitation, since if the maximum tilt factor is 5 (as in this example), then configurations with tilt = ...,
-15, -5, 5, 15, 25, ... are all geometrically equivalent. If you wish to define a box with tilt factors that exceed
these limits, you can use the box tilt command, with a setting of large; a setting of small is the default.
See Section 6.12 of the doc pages for a geometric description of triclinic boxes, as defined by LAMMPS, and
how to transform these parameters to and from other commonly used triclinic representations.
When a triclinic system is used, the simulation domain should normally be periodic in the dimension that the
tilt is applied to, which is given by the second dimension of the tilt factor (e.g. y for xy tilt). This is so that
pairs of atoms interacting across that boundary will have one of them shifted by the tilt factor. Periodicity is
set by the boundary command. For example, if the xy tilt factor is non-zero, then the y dimension should be
periodic. Similarly, the z dimension should be periodic if xz or yz is non-zero. LAMMPS does not require this
periodicity, but you may lose atoms if this is not the case.
Also note that if your simulation will tilt the box, e.g. via the fix deform command, the simulation box must
be setup to be triclinic, even if the tilt factors are initially 0.0. You can also change an orthogonal box to a
triclinic box or vice versa by using the change box command with its ortho and triclinic options.
For 2d simulations, the zlo zhi values should be set to bound the z coords for atoms that appear in the file; the
default of -0.5 0.5 is valid if all z coords are 0.0. For 2d triclinic simulations, the xz and yz tilt factors must be
0.0.
If the system is periodic (in a dimension), then atom coordinates can be outside the bounds (in that
dimension); they will be remapped (in a periodic sense) back inside the box. Note that if the add option is
being used to add atoms to a simulation box that already exists, this periodic remapping will be performed
using simulation box bounds that are the union of the existing box and the box boundaries in the new data file.
NOTE: If the system is non-periodic (in a dimension), then all atoms in the data file must have coordinates (in
that dimension) that are "greater than or equal to" the lo value and "less than or equal to" the hi value. If the
non-periodic dimension is of style "fixed" (see the boundary command), then the atom coords must be strictly
"less than" the hi value, due to the way LAMMPS assign atoms to processors. Note that you should not make
the lo/hi values radically smaller/larger than the extent of the atoms. For example, if your atoms extend from 0
to 50, you should not specify the box bounds as -10000 and 10000. This is because LAMMPS uses the
specified box size to layout the 3d grid of processors. A huge (mostly empty) box will be sub-optimal for
performance when using "fixed" boundary conditions (see the boundary command). When using
"shrink-wrap" boundary conditions (see the boundary command), a huge (mostly empty) box may cause a
parallel simulation to lose atoms when LAMMPS shrink-wraps the box around the atoms. The read_data
command will generate an error in this case.
LAMMPS Users Manual
585

The "extra bond per atom" setting (angle, dihedral, improper) is only needed if new bonds (angles, dihedrals,
impropers) will be added to the system when a simulation runs, e.g. by using the fix bond/create command.
This will pre-allocate space in LAMMPS data structures for storing the new bonds (angles, dihedrals,
impropers).
The "extra special per atom" setting is typically only needed if new bonds/angles/etc will be added to the
system, e.g. by using the fix bond/create command. Or if entire new molecules will be added to the system,
e.g. by using the fix deposit or fix pour commands, which will have more special 1-2,1-3,1-4 neighbors than
any other molecules defined in the data file. Using this setting will pre-allocate space in the LAMMPS data
structures for storing these neighbors. See the special_bonds and molecule doc pages for more discussion of
1-2,1-3,1-4 neighbors.
NOTE: All of the "extra" settings are only used if they appear in the first data file read; see the description of
the add keyword above for reading multiple data files. If they appear in later data files, they are ignored.
The "ellipsoids" and "lines" and "triangles" and "bodies" settings are only used with atom_style ellipsoid or
line or tri or body and specify how many of the atoms are finite-size ellipsoids or lines or triangles or bodies;
the remainder are point particles. See the discussion of ellipsoidflag and the Ellipsoids section below. See the
discussion of lineflag and the Lines section below. See the discussion of triangleflag and the Triangles section
below. See the discussion of bodyflag and the Bodies section below.
NOTE: For atom_style template, the molecular topology (bonds,angles,etc) is contained in the molecule
templates read-in by the molecule command. This means you cannot set the bonds, angles, etc header
keywords in the data file, nor can you define Bonds, Angles, etc sections as discussed below. You can set the
bond types, angle types, etc header keywords, though it is not necessary. If specified, they must match the
maximum values defined in any of the template molecules.
Format of the body of a data file
These are the section keywords for the body of the file.
Atoms, Velocities, Masses, Ellipsoids, Lines, Triangles, Bodies = atom-property sections• Bonds, Angles, Dihedrals, Impropers = molecular topology sections• Pair Coeffs, PairIJ Coeffs, Bond Coeffs, Angle Coeffs, Dihedral Coeffs, Improper Coeffs = force field
sections
•
BondBond Coeffs, BondAngle Coeffs, MiddleBondTorsion Coeffs, EndBondTorsion Coeffs,
AngleTorsion Coeffs, AngleAngleTorsion Coeffs, BondBond13 Coeffs, AngleAngle Coeffs = class 2
force field sections
•
These keywords will check an appended comment for a match with the currently defined style:
Atoms, Pair Coeffs, PairIJ Coeffs, Bond Coeffs, Angle Coeffs, Dihedral Coeffs, Improper Coeffs•
For example, these lines:
Atoms # sphere
Pair Coeffs # lj/cut
will check if the currently-defined atom_style is sphere, and the current pair_style is lj/cut. If not, LAMMPS
will issue a warning to indicate that the data file section likely does not contain the correct number or type of
parameters expected for the currently-defined style.
LAMMPS Users Manual
586

Each section is listed below in alphabetic order. The format of each section is described including the number
of lines it must contain and rules (if any) for where it can appear in the data file.
Any individual line in the various sections can have a trailing comment starting with "#" for annotation
purposes. E.g. in the Atoms section:
10 1 17 -1.0 10.0 5.0 6.0 # salt ion
Angle Coeffs section:
one line per angle type• line syntax: ID coeffs
ID = angle type (1-N)
coeffs = list of coeffs
•
example:
6 70 108.5 0 0
•
The number and meaning of the coefficients are specific to the defined angle style. See the angle_style and
angle_coeff commands for details. Coefficients can also be set via the angle_coeff command in the input
script.
AngleAngle Coeffs section:
one line per improper type• line syntax: ID coeffs
ID = improper type (1-N)
coeffs = list of coeffs (see improper_coeff)
•
AngleAngleTorsion Coeffs section:
one line per dihedral type• line syntax: ID coeffs
ID = dihedral type (1-N)
coeffs = list of coeffs (see dihedral_coeff)
•
Angles section:
one line per angle• line syntax: ID type atom1 atom2 atom3
ID = number of angle (1-Nangles)
type = angle type (1-Nangletype)
atom1,atom2,atom3 = IDs of 1st,2nd,3rd atoms in angle
example:
2 2 17 29 430
•
LAMMPS Users Manual
587
The 3 atoms are ordered linearly within the angle. Thus the central atom (around which the angle is
computed) is the atom2 in the list. E.g. H,O,H for a water molecule. The Angles section must appear after the
Atoms section. All values in this section must be integers (1, not 1.0).
AngleTorsion Coeffs section:
one line per dihedral type• line syntax: ID coeffs
ID = dihedral type (1-N)
coeffs = list of coeffs (see dihedral_coeff)
•
Atoms section:
one line per atom• line syntax: depends on atom style•
An Atoms section must appear in the data file if natoms > 0 in the header section. The atoms can be listed in
any order. These are the line formats for each atom style in LAMMPS. As discussed below, each line can
optionally have 3 flags (nx,ny,nz) appended to it, which indicate which image of a periodic simulation box the
atom is in. These may be important to include for some kinds of analysis.
angle atom-ID molecule-ID atom-type x y z
atomic atom-ID atom-type x y z
body atom-ID atom-type bodyflag mass x y z
bond atom-ID molecule-ID atom-type x y z
charge atom-ID atom-type q x y z
dipole atom-ID atom-type q x y z mux muy muz
dpd atom-ID atom-type theta x y z
electron atom-ID atom-type q spin eradius x y z
ellipsoid atom-ID atom-type ellipsoidflag density x y z
full atom-ID molecule-ID atom-type q x y z
line atom-ID molecule-ID atom-type lineflag density x y z
meso atom-ID atom-type rho e cv x y z
molecular atom-ID molecule-ID atom-type x y z
peri atom-ID atom-type volume density x y z
smd atom-ID atom-type molecule volume mass kernel-radius contact-radius x y z
sphere atom-ID atom-type diameter density x y z
template atom-ID molecule-ID template-index template-atom atom-type x y z
tri atom-ID molecule-ID atom-type triangleflag density x y z
wavepacket atom-ID atom-type charge spin eradius etag cs_re cs_im x y z
hybrid atom-ID atom-type x y z sub-style1 sub-style2 ...
The per-atom values have these meanings and units, listed alphabetically:
atom-ID = integer ID of atom• atom-type = type of atom (1-Ntype)•
LAMMPS Users Manual
588
bodyflag = 1 for body particles, 0 for point particles• contact-radius = ??? (distance units)• cs_re,cs_im = real/imaginary parts of wavepacket coefficients• cv = heat capacity (need units) for SPH particles• density = density of particle (mass/distance^3 or mass/distance^2 or mass/distance units, depending
on dimensionality of particle)
•
diameter = diameter of spherical atom (distance units)• e = energy (need units) for SPH particles• ellipsoidflag = 1 for ellipsoidal particles, 0 for point particles• eradius = electron radius (or fixed-core radius)• etag = integer ID of electron that each wavepacket belongs to• kernel-radius = ??? (distance units)• lineflag = 1 for line segment particles, 0 for point or spherical particles• mass = mass of particle (mass units)• molecule-ID = integer ID of molecule the atom belongs to• mux,muy,muz = components of dipole moment of atom (dipole units)• q = charge on atom (charge units)• rho = density (need units) for SPH particles• spin = electron spin (+1/-1), 0 = nuclei, 2 = fixed-core, 3 = pseudo-cores (i.e. ECP)• template-atom = which atom within a template molecule the atom is• template-index = which molecule within the molecule template the atom is part of• theta = internal temperature of a DPD particle• triangleflag = 1 for triangular particles, 0 for point or spherical particles• volume = volume of Peridynamic particle (distance^3 units)• x,y,z = coordinates of atom (distance units)•
The units for these quantities depend on the unit style; see the units command for details.
For 2d simulations specify z as 0.0, or a value within the zlo zhi setting in the data file header.
The atom-ID is used to identify the atom throughout the simulation and in dump files. Normally, it is a unique
value from 1 to Natoms for each atom. Unique values larger than Natoms can be used, but they will cause
extra memory to be allocated on each processor, if an atom map array is used, but not if an atom map hash is
used; see the atom_modify command for details. If an atom map is not used (e.g. an atomic system with no
bonds), and you don't care if unique atom IDs appear in dump files, then the atom-IDs can all be set to 0.
The molecule ID is a 2nd identifier attached to an atom. Normally, it is a number from 1 to N, identifying
which molecule the atom belongs to. It can be 0 if it is an unbonded atom or if you don't care to keep track of
molecule assignments.
The diameter specifies the size of a finite-size spherical particle. It can be set to 0.0, which means that atom is
a point particle.
The ellipsoidflag, lineflag, triangleflag, and bodyflag determine whether the particle is a finite-size ellipsoid
or line or triangle or body of finite size, or whether the particle is a point particle. Additional attributes must
be defined for each ellipsoid, line, triangle, or body in the corresponding Ellipsoids, Lines, Triangles, or
Bodies section.
The template-index and template-atom are only defined used by atom_style template. In this case the molecule
command is used to define a molecule template which contains one or more molecules. If an atom belongs to
one of those molecules, its template-index and template-atom are both set to positive integers; if not the values
LAMMPS Users Manual
589
are both 0. The template-index is which molecule (1 to Nmols) the atom belongs to. The template-atom is
which atom (1 to Natoms) within the molecule the atom is.
Some pair styles and fixes and computes that operate on finite-size particles allow for a mixture of finite-size
and point particles. See the doc pages of individual commands for details.
For finite-size particles, the density is used in conjunction with the particle volume to set the mass of each
particle as mass = density * volume. In this context, volume can be a 3d quantity (for spheres or ellipsoids), a
2d quantity (for triangles), or a 1d quantity (for line segments). If the volume is 0.0, meaning a point particle,
then the density value is used as the mass. One exception is for the body atom style, in which case the mass of
each particle (body or point particle) is specified explicitly. This is because the volume of the body is
unknown.
Note that for 2d simulations of spheres, this command will treat them as spheres when converting density to
mass. However, they can also be modeled as 2d discs (circles) if the set density/disc command is used to reset
their mass after the read_data command is used. A disc keyword can also be used with time integration fixes,
such as fix nve/sphere and fix nvt/sphere to time integrate their motion as 2d discs (not 3d spheres), by
changing their moment of inertia.
For atom_style hybrid, following the 5 initial values (ID,type,x,y,z), specific values for each sub-style must be
listed. The order of the sub-styles is the same as they were listed in the atom_style command. The sub-style
specific values are those that are not the 5 standard ones (ID,type,x,y,z). For example, for the "charge"
sub-style, a "q" value would appear. For the "full" sub-style, a "molecule-ID" and "q" would appear. These are
listed in the same order they appear as listed above. Thus if
atom_style hybrid charge sphere
were used in the input script, each atom line would have these fields:
atom-ID atom-type x y z q diameter density
Note that if a non-standard value is defined by multiple sub-styles, it must appear mutliple times in the atom
line. E.g. the atom line for atom_style hybrid dipole full would list "q" twice:
atom-ID atom-type x y z q mux muy myz molecule-ID q
Atom lines specify the (x,y,z) coordinates of atoms. These can be inside or outside the simulation box. When
the data file is read, LAMMPS wraps coordinates outside the box back into the box for dimensions that are
periodic. As discussed above, if an atom is outside the box in a non-periodic dimension, it will be lost.
LAMMPS always stores atom coordinates as values which are inside the simulation box. It also stores 3 flags
which indicate which image of the simulation box (in each dimension) the atom would be in if its coordinates
were unwrapped across periodic boundaries. An image flag of 0 means the atom is still inside the box when
unwrapped. A value of 2 means add 2 box lengths to get the unwrapped coordinate. A value of -1 means
subtract 1 box length to get the unwrapped coordinate. LAMMPS updates these flags as atoms cross periodic
boundaries during the simulation. The dump command can output atom atom coordinates in wrapped or
unwrapped form, as well as the 3 image flags.
In the data file, atom lines (all lines or none of them) can optionally list 3 trailing integer values (nx,ny,nz),
which are used to initialize the atom's image flags. If nx,ny,nz values are not listed in the data file, LAMMPS
initializes them to 0. Note that the image flags are immediately updated if an atom's coordinates need to
LAMMPS Users Manual
590

wrapped back into the simulation box.
It is only important to set image flags correctly in a data file if a simulation model relies on unwrapped
coordinates for some calculation; otherwise they can be left unspecified. Examples of LAMMPS commands
that use unwrapped coordinates internally are as follows:
Atoms in a rigid body (see fix rigid, fix rigid/small) must have consistent image flags, so that when
the atoms are unwrapped, they are near each other, i.e. as a single body.
•
If the replicate command is used to generate a larger system, image flags must be consistent for
bonded atoms when the bond crosses a periodic boundary. I.e. the values of the image flags should be
different by 1 (in the appropriate dimension) for the two atoms in such a bond.
•
If you plan to dump image flags and perform post-analysis that will unwrap atom coordinates, it may
be important that a continued run (restarted from a data file) begins with image flags that are
consistent with the previous run.
•
NOTE: If your system is an infinite periodic crystal with bonds then it is impossible to have fully consistent
image flags. This is because some bonds will cross periodic boundaries and connect two atoms with the same
image flag.
Atom velocities and other atom quantities not defined above are set to 0.0 when the Atoms section is read.
Velocities can be set later by a Velocities section in the data file or by a velocity or set command in the input
script.
Bodies section:
one or more lines per body• first line syntax: atom-ID Ninteger Ndouble
Ninteger = # of integer quantities for this particle
Ndouble = # of floating-point quantities for this particle
•
0 or more integer lines with total of Ninteger values• 0 or more double lines with total of Ndouble values• example:
12 3 6
2 3 2
1.0 2.0 3.0 1.0 2.0 4.0
•
example:
12 0 14
1.0 2.0 3.0 1.0 2.0 4.0 1.0
2.0 3.0 1.0 2.0 4.0 4.0 2.0
•
The Bodies section must appear if atom_style body is used and any atoms listed in the Atoms section have a
bodyflag = 1. The number of bodies should be specified in the header section via the "bodies" keyword.
Each body can have a variable number of integer and/or floating-point values. The number and meaning of the
values is defined by the body style, as described in the body doc page. The body style is given as an argument
to the atom_style body command.
The Ninteger and Ndouble values determine how many integer and floating-point values are specified for this
particle. Ninteger and Ndouble can be as large as needed and can be different for every body. Integer values
LAMMPS Users Manual
591

are then listed next on subsequent lines. Lines are read one at a time until Ninteger values are read.
Floating-point values follow on subsequent lines, Again lines are read one at a time until Ndouble values are
read. Note that if there are no values of a particular type, no lines appear for that type.
The Bodies section must appear after the Atoms section.
Bond Coeffs section:
one line per bond type• line syntax: ID coeffs
ID = bond type (1-N)
coeffs = list of coeffs
•
example:
4 250 1.49
•
The number and meaning of the coefficients are specific to the defined bond style. See the bond_style and
bond_coeff commands for details. Coefficients can also be set via the bond_coeff command in the input
script.
BondAngle Coeffs section:
one line per angle type• line syntax: ID coeffs
ID = angle type (1-N)
coeffs = list of coeffs (see class 2 section of angle_coeff)
•
BondBond Coeffs section:
one line per angle type• line syntax: ID coeffs
ID = angle type (1-N)
coeffs = list of coeffs (see class 2 section of angle_coeff)
•
BondBond13 Coeffs section:
one line per dihedral type• line syntax: ID coeffs
ID = dihedral type (1-N)
coeffs = list of coeffs (see class 2 section of dihedral_coeff)
•
Bonds section:
one line per bond• line syntax: ID type atom1 atom2
ID = bond number (1-Nbonds)
type = bond type (1-Nbondtype)
•
LAMMPS Users Manual
592

atom1,atom2 = IDs of 1st,2nd atoms in bond
example:
12 3 17 29
•
The Bonds section must appear after the Atoms section. All values in this section must be integers (1, not 1.0).
Dihedral Coeffs section:
one line per dihedral type• line syntax: ID coeffs
ID = dihedral type (1-N)
coeffs = list of coeffs
•
example:
3 0.6 1 0 1
•
The number and meaning of the coefficients are specific to the defined dihedral style. See the dihedral_style
and dihedral_coeff commands for details. Coefficients can also be set via the dihedral_coeff command in the
input script.
Dihedrals section:
one line per dihedral• line syntax: ID type atom1 atom2 atom3 atom4
ID = number of dihedral (1-Ndihedrals)
type = dihedral type (1-Ndihedraltype)
atom1,atom2,atom3,atom4 = IDs of 1st,2nd,3rd,4th atoms in dihedral
•
example:
12 4 17 29 30 21
•
The 4 atoms are ordered linearly within the dihedral. The Dihedrals section must appear after the Atoms
section. All values in this section must be integers (1, not 1.0).
Ellipsoids section:
one line per ellipsoid• line syntax: atom-ID shapex shapey shapez quatw quati quatj quatk
atom-ID = ID of atom which is an ellipsoid
shapex,shapey,shapez = 3 diameters of ellipsoid (distance units)
quatw,quati,quatj,quatk = quaternion components for orientation of atom
•
example:
12 1 2 1 1 0 0 0
•
The Ellipsoids section must appear if atom_style ellipsoid is used and any atoms are listed in the Atoms
section with an ellipsoidflag = 1. The number of ellipsoids should be specified in the header section via the
"ellipsoids" keyword.
LAMMPS Users Manual
593

The 3 shape values specify the 3 diameters or aspect ratios of a finite-size ellipsoidal particle, when it is
oriented along the 3 coordinate axes. They must all be non-zero values.
The values quatw, quati, quatj, and quatk set the orientation of the atom as a quaternion (4-vector). Note that
the shape attributes specify the aspect ratios of an ellipsoidal particle, which is oriented by default with its
x-axis along the simulation box's x-axis, and similarly for y and z. If this body is rotated (via the right-hand
rule) by an angle theta around a unit vector (a,b,c), then the quaternion that represents its new orientation is
given by (cos(theta/2), a*sin(theta/2), b*sin(theta/2), c*sin(theta/2)). These 4 components are quatw, quati,
quatj, and quatk as specified above. LAMMPS normalizes each atom's quaternion in case (a,b,c) is not
specified as a unit vector.
The Ellipsoids section must appear after the Atoms section.
EndBondTorsion Coeffs section:
one line per dihedral type• line syntax: ID coeffs
ID = dihedral type (1-N)
coeffs = list of coeffs (see class 2 section of dihedral_coeff)
•
Improper Coeffs section:
one line per improper type• line syntax: ID coeffs
ID = improper type (1-N)
coeffs = list of coeffs
•
example:
2 20 0.0548311
•
The number and meaning of the coefficients are specific to the defined improper style. See the improper_style
and improper_coeff commands for details. Coefficients can also be set via the improper_coeff command in
the input script.
Impropers section:
one line per improper• line syntax: ID type atom1 atom2 atom3 atom4
ID = number of improper (1-Nimpropers)
type = improper type (1-Nimpropertype)
atom1,atom2,atom3,atom4 = IDs of 1st,2nd,3rd,4th atoms in improper
•
example:
12 3 17 29 13 100
•
The ordering of the 4 atoms determines the definition of the improper angle used in the formula for each
improper style. See the doc pages for individual styles for details.
LAMMPS Users Manual
594

The Impropers section must appear after the Atoms section. All values in this section must be integers (1, not
1.0).
Lines section:
one line per line segment• line syntax: atom-ID x1 y1 x2 y2
atom-ID = ID of atom which is a line segment
x1,y1 = 1st end point
x2,y2 = 2nd end point
•
example:
12 1.0 0.0 2.0 0.0
•
The Lines section must appear if atom_style line is used and any atoms are listed in the Atoms section with a
lineflag = 1. The number of lines should be specified in the header section via the "lines" keyword.
The 2 end points are the end points of the line segment. The ordering of the 2 points should be such that using
a right-hand rule to cross the line segment with a unit vector in the +z direction, gives an "outward" normal
vector perpendicular to the line segment. I.e. normal = (c2-c1) x (0,0,1). This orientation may be important for
defining some interactions.
The Lines section must appear after the Atoms section.
Masses section:
one line per atom type• line syntax: ID mass
ID = atom type (1-N)
mass = mass value
•
example:
3 1.01
•
This defines the mass of each atom type. This can also be set via the mass command in the input script. This
section cannot be used for atom styles that define a mass for individual atoms - e.g. atom_style sphere.
MiddleBondTorsion Coeffs section:
one line per dihedral type• line syntax: ID coeffs
ID = dihedral type (1-N)
coeffs = list of coeffs (see class 2 section of dihedral_coeff)
•
Pair Coeffs section:
one line per atom type• line syntax: ID coeffs•
LAMMPS Users Manual
595

ID = atom type (1-N)
coeffs = list of coeffs
example:
3 0.022 2.35197 0.022 2.35197
•
The number and meaning of the coefficients are specific to the defined pair style. See the pair_style and
pair_coeff commands for details. Since pair coefficients for types I != J are not specified, these will be
generated automatically by the pair style's mixing rule. See the individual pair_style doc pages and the
pair_modify mix command for details. Pair coefficients can also be set via the pair_coeff command in the
input script.
PairIJ Coeffs section:
one line per pair of atom types for all I,J with I <= J• line syntax: ID1 ID2 coeffs
ID1 = atom type I = 1-N
ID2 = atom type J = I-N, with I <= J
coeffs = list of coeffs
•
examples:
3 3 0.022 2.35197 0.022 2.35197
3 5 0.022 2.35197 0.022 2.35197
•
This section must have N*(N+1)/2 lines where N = # of atom types. The number and meaning of the
coefficients are specific to the defined pair style. See the pair_style and pair_coeff commands for details.
Since pair coefficients for types I != J are all specified, these values will turn off the default mixing rule
defined by the pair style. See the individual pair_style doc pages and the pair_modify mix command for
details. Pair coefficients can also be set via the pair_coeff command in the input script.
Triangles section:
one line per triangle• line syntax: atom-ID x1 y1 z1 x2 y2 z2 x3 y3 z3
atom-ID = ID of atom which is a line segment
x1,y1,z1 = 1st corner point
x2,y2,z2 = 2nd corner point
x3,y3,z3 = 3rd corner point
•
example:
12 0.0 0.0 0.0 2.0 0.0 1.0 0.0 2.0 1.0
•
The Triangles section must appear if atom_style tri is used and any atoms are listed in the Atoms section with
a triangleflag = 1. The number of lines should be specified in the header section via the "triangles" keyword.
The 3 corner points are the corner points of the triangle. The ordering of the 3 points should be such that using
a right-hand rule to go from point1 to point2 to point3 gives an "outward" normal vector to the face of the
triangle. I.e. normal = (c2-c1) x (c3-c1). This orientation may be important for defining some interactions.
The Triangles section must appear after the Atoms section.
LAMMPS Users Manual
596

Velocities section:
one line per atom• line syntax: depends on atom style•
all styles except those listed atom-ID vx vy vz
electron atom-ID vx vy vz ervel
ellipsoid atom-ID vx vy vz lx ly lz
sphere atom-ID vx vy vz wx wy wz
hybrid atom-ID vx vy vz sub-style1 sub-style2 ...
where the keywords have these meanings:
vx,vy,vz = translational velocity of atom lx,ly,lz = angular momentum of aspherical atom wx,wy,wz = angular
velocity of spherical atom ervel = electron radial velocity (0 for fixed-core):ul
The velocity lines can appear in any order. This section can only be used after an Atoms section. This is
because the Atoms section must have assigned a unique atom ID to each atom so that velocities can be
assigned to them.
Vx, vy, vz, and ervel are in units of velocity. Lx, ly, lz are in units of angular momentum
(distance-velocity-mass). Wx, Wy, Wz are in units of angular velocity (radians/time).
For atom_style hybrid, following the 4 initial values (ID,vx,vy,vz), specific values for each sub-style must be
listed. The order of the sub-styles is the same as they were listed in the atom_style command. The sub-style
specific values are those that are not the 5 standard ones (ID,vx,vy,vz). For example, for the "sphere"
sub-style, "wx", "wy", "wz" values would appear. These are listed in the same order they appear as listed
above. Thus if
atom_style hybrid electron sphere
were used in the input script, each velocity line would have these fields:
atom-ID vx vy vz ervel wx wy wz
Translational velocities can also be set by the velocity command in the input script.
Restrictions:
To read gzipped data files, you must compile LAMMPS with the -DLAMMPS_GZIP option - see the Making
LAMMPS section of the documentation.
Related commands:
read_dump, read_restart, create_atoms, write_data
Default:
The default for all the extra keywords is 0.
LAMMPS Users Manual
597

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
read_dump command
Syntax:
read_dump file Nstep field1 field2 ... keyword values ...
file = name of dump file to read• Nstep = snapshot timestep to read from file• one or more fields may be appended
field = x or y or z or vx or vy or vz or q or ix or iy or iz or fx or fy or fz
x,y,z = atom coordinates
vx,vy,vz = velocity components
q = charge
ix,iy,iz = image flags in each dimension
fx,fy,fz = force components
•
zero or more keyword/value pairs may be appended• keyword = box or replace or purge or trim or add or label or scaled or wrapped or format
box value = yes or no = replace simulation box with dump box
replace value = yes or no = overwrite atoms with dump atoms
purge value = yes or no = delete all atoms before adding dump atoms
trim value = yes or no = trim atoms not in dump snapshot
add value = yes or no = add new dump atoms to system
label value = field column
field = one of the listed fields or id or type
column = label on corresponding column in dump file
scaled value = yes or no = coords in dump file are scaled/unscaled
wrapped value = yes or no = coords in dump file are wrapped/unwrapped
format values = format of dump file, must be last keyword if used
native = native LAMMPS dump file
xyz = XYZ file
molfile style path = VMD molfile plugin interface
style = dcd or xyz or others supported by molfile plugins
path = optional path for location of molfile plugins
•
Examples:
read_dump dump.file 5000 x y z
read_dump dump.xyz 5 x y z box no format xyz
read_dump dump.xyz 10 x y z box no format molfile xyz "../plugins"
read_dump dump.dcd 0 x y z box yes format molfile dcd
read_dump dump.file 1000 x y z vx vy vz box yes format molfile lammpstrj /usr/local/lib/vmd/plugins/LINUXAMD64/plugins/molfile
read_dump dump.file 5000 x y vx vy trim yes
read_dump ../run7/dump.file.gz 10000 x y z box yes
read_dump dump.xyz 10 x y z box no format molfile xyz ../plugins
read_dump dump.dcd 0 x y z format molfile dcd
read_dump dump.file 1000 x y z vx vy vz format molfile lammpstrj /usr/local/lib/vmd/plugins/LINUXAMD64/plugins/molfile
Description:
Read atom information from a dump file to overwrite the current atom coordinates, and optionally the atom
velocities and image flags and the simulation box dimensions. This is useful for restarting a run from a
particular snapshot in a dump file. See the read_restart and read_data commands for alternative methods to do
LAMMPS Users Manual
598

this. Also see the rerun command for a means of reading multiple snapshots from a dump file.
Note that a simulation box must already be defined before using the read_dump command. This can be done
by the create_box, read_data, or read_restart commands. The read_dump command can reset the simulation
box dimensions, as explained below.
Also note that reading per-atom information from a dump snapshot is limited to the atom coordinates,
velocities and image flags, as explained below. Other atom properties, which may be necessary to run a valid
simulation, such as atom charge, or bond topology information for a molecular system, are not read from (or
even contained in) dump files. Thus this auxiliary information should be defined in the usual way, e.g. in a
data file read in by a read_data command, before using the read_dump command, or by the set command,
after the dump snapshot is read.
If the dump filename specified as file ends with ".gz", the dump file is read in gzipped format. You cannot
(yet) read a dump file that was written in binary format with a ".bin" suffix, or to multiple files via the "%"
option in the dump file name. See the dump command for details.
The format of the dump file is selected through the format keyword. If specified, it must be the last keyword
used, since all remaining arguments are passed on to the dump reader. The native format is for native
LAMMPS dump files, written with a dump atom or dump custom command. The xyz format is for generic
XYZ formatted dump files. These formats take no additional values.
The molfile format supports reading data through using the VMD molfile plugin interface. This dump reader
format is only available, if the USER-MOLFILE package has been installed when compiling LAMMPS.
The molfile format takes one or two additional values. The style value determines the file format to be used
and can be any format that the molfile plugins support, such as DCD or XYZ. Note that DCD dump files can
be written by LAMMPS via the dump dcd command. The path value specifies a list of directories which
LAMMPS will search for the molfile plugins appropriate to the specified style. The syntax of the path value is
like other search paths: it can contain multiple directories separated by a colon (or semi-colon on windows).
The path keyword is optional and defaults to ".", i.e. the current directory.
Support for other dump format readers may be added in the future.
Global information is first read from the dump file, namely timestep and box information.
The dump file is scanned for a snapshot with a time stamp that matches the specified Nstep. This means the
LAMMPS timestep the dump file snapshot was written on for the native format. Note that the xyz and molfile
formats do not store the timestep. For these formats, timesteps are numbered logically, in a sequential manner,
starting from 0. Thus to access the 10th snapshot in an xyz or mofile formatted dump file, use Nstep = 9.
The dimensions of the simulation box for the selected snapshot are also read; see the box keyword discussion
below. For the native format, an error is generated if the snapshot is for a triclinic box and the current
simulation box is orthogonal or vice versa. A warning will be generated if the snapshot box boundary
conditions (periodic, shrink-wrapped, etc) do not match the current simulation boundary conditions, but the
boundary condition information in the snapshot is otherwise ignored. See the "boundary" command for more
details.
For the xyz format, no information about the box is available, so you must set the box flag to no. See details
below.
LAMMPS Users Manual
599

For the molfile format, reading simulation box information is typically supported, but the location of the
simulation box origin is lost and no explicit information about periodicity or orthogonal/triclinic box shape is
available. The USER-MOLFILE package makes a best effort to guess based on heuristics, but this may not
always work perfectly.
Per-atom information from the dump file snapshot is then read from the dump file snapshot. This corresponds
to the specified fields listed in the read_dump command. It is an error to specify a z-dimension field, namely z,
vz, or iz, for a 2d simulation.
For dump files in native format, each column of per-atom data has a text label listed in the file. A matching
label for each field must appear, e.g. the label "vy" for the field vy. For the x, y, z fields any of the following
labels are considered a match:
x, xs, xu, xsu for field x
y, ys, yu, ysu for field y
z, zs, zu, zsu for field z
The meaning of xs (scaled), xu (unwrapped), and xsu (scaled and unwrapped) is explained on the dump
command doc page. These labels are searched for in the list of column labels in the dump file, in order, until a
match is found.
The dump file must also contain atom IDs, with a column label of "id".
If the add keyword is specified with a value of yes, as discussed below, the dump file must contain atom
types, with a column label of "type".
If a column label you want to read from the dump file is not a match to a specified field, the label keyword
can be used to specify the specific column label from the dump file to associate with that field. An example is
if a time-averaged coordinate is written to the dump file via the fix ave/atom command. The column will then
have a label corresponding to the fix-ID rather than "x" or "xs". The label keyword can also be used to specify
new column labels for fields id and type.
For dump files in xyz format, only the x, y, and z fields are supported. The dump file does not store atom IDs,
so these are assigned consecutively to the atoms as they appear in the dump file, starting from 1. Thus you
should insure that order of atoms is consistent from snapshot to snapshot in the XYZ dump file. See the
dump_modify sort command if the XYZ dump file was written by LAMMPS.
For dump files in molfile format, the x, y, z, vx, vy, and vz fields can be specified. However, not all molfile
formats store velocities, or their respective plugins may not support reading of velocities. The molfile dump
files do not store atom IDs, so these are assigned consecutively to the atoms as they appear in the dump file,
starting from 1. Thus you should insure that order of atoms are consistent from snapshot to snapshot in the
molfile dump file. See the dump_modify sort command if the dump file was written by LAMMPS.
Information from the dump file snapshot is used to overwrite or replace properties of the current system.
There are various options for how this is done, determined by the specified fields and optional keywords.
The timestep of the snapshot becomes the current timestep for the simulation. See the reset_timestep
command if you wish to change this after the dump snapshot is read.
If the box keyword is specified with a yes value, then the current simulation box dimensions are replaced by
the dump snapshot box dimensions. If the box keyword is specified with a no value, the current simulation
LAMMPS Users Manual
600

box is unchanged.
If the purge keyword is specified with a yes value, then all current atoms in the system are deleted before any
of the operations invoked by the replace, trim, or add keywords take place.
If the replace keyword is specified with a yes value, then atoms with IDs that are in both the current system
and the dump snapshot have their properties overwritten by field values. If the replace keyword is specified
with a no value, atoms with IDs that are in both the current system and the dump snapshot are not modified.
If the trim keyword is specified with a yes value, then atoms with IDs that are in the current system but not in
the dump snapshot are deleted. These atoms are unaffected if the trim keyword is specified with a no value.
If the add keyword is specified with a yes value, then atoms with IDs that are in the dump snapshot, but not in
the current system are added to the system. These dump atoms are ignored if the add keyword is specified
with a no value.
Note that atoms added via the add keyword will have only the attributes read from the dump file due to the
field arguments. If x or y or z is not specified as a field, a value of 0.0 is used for added atoms. Added atoms
must have an atom type, so this value must appear in the dump file.
Any other attributes (e.g. charge or particle diameter for spherical particles) will be set to default values, the
same as if the create_atoms command were used.
Note that atom IDs are not preserved for new dump snapshot atoms added via the add keyword. The
procedure for assigning new atom IDS to added atoms is the same as is described for the create_atoms
command.
Atom coordinates read from the dump file are first converted into unscaled coordinates, relative to the box
dimensions of the snapshot. These coordinates are then be assigned to an existing or new atom in the current
simulation. The coordinates will then be remapped to the simulation box, whether it is the original box or the
dump snapshot box. If periodic boundary conditions apply, this means the atom will be remapped back into
the simulation box if necessary. If shrink-wrap boundary conditions apply, the new coordinates may change
the simulation box dimensions. If fixed boundary conditions apply, the atom will be lost if it is outside the
simulation box.
For native format dump files, the 3 xyz image flags for an atom in the dump file are set to the corresponding
values appearing in the dump file if the ix, iy, iz fields are specified. If not specified, the image flags for
replaced atoms are not changed and image flags for new atoms are set to default values. If coordinates read
from the dump file are in unwrapped format (e.g. xu) then the image flags for read-in atoms are also set to
default values. The remapping procedure described in the previous paragraph will then change images flags
for all atoms (old and new) if periodic boundary conditions are applied to remap an atom back into the
simulation box.
NOTE: If you get a warning about inconsistent image flags after reading in a dump snapshot, it means one or
more pairs of bonded atoms now have inconsistent image flags. As discussed in Section errors this may or
may not cause problems for subsequent simulations, One way this can happen is if you read image flag fields
from the dump file but do not also use the dump file box parameters.
LAMMPS knows how to compute unscaled and remapped coordinates for the snapshot column labels
discussed above, e.g. x, xs, xu, xsu. If another column label is assigned to the x or y or z field via the label
keyword, e.g. for coordinates output by the fix ave/atom command, then LAMMPS needs to know whether
LAMMPS Users Manual
601

the coordinate information in the dump file is scaled and/or wrapped. This can be set via the scaled and
wrapped keywords. Note that the value of the scaled and wrapped keywords is ignored for fields x or y or z if
the label keyword is not used to assign a column label to that field.
The scaled/unscaled and wrapped/unwrapped setting must be identical for any of the x, y, z fields that are
specified. Thus you cannot read xs and yu from the dump file. Also, if the dump file coordinates are scaled
and the simulation box is triclinic, then all 3 of the x, y, z fields must be specified, since they are all needed to
generate absolute, unscaled coordinates.
Restrictions:
To read gzipped dump files, you must compile LAMMPS with the -DLAMMPS_GZIP option - see the
Making LAMMPS section of the documentation.
The molfile dump file formats are part of the USER-MOLFILE package. They are only enabled if LAMMPS
was built with that packages. See the Making LAMMPS section for more info.
Related commands:
dump, dump molfile, read_data, read_restart, rerun
Default:
The option defaults are box = yes, replace = yes, purge = no, trim = no, add = no, scaled = no, wrapped = yes,
and format = native.
LAMMPS Users Manual
602

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
read_restart command
Syntax:
read_restart file flag
file = name of binary restart file to read in• flag = remap (optional)•
Examples:
read_restart save.10000
read_restart save.10000 remap
read_restart restart.*
read_restart restart.*.mpiio
read_restart poly.*.% remap
Description:
Read in a previously saved system configuration from a restart file. This allows continuation of a previous
run. Details about what information is stored (and not stored) in a restart file is given below. Basically this
operation will re-create the simulation box with all its atoms and their attributes as well as some related global
settings, at the point in time it was written to the restart file by a previous simulation. The simulation box will
be partitioned into a regular 3d grid of rectangular bricks, one per processor, based on the number of
processors in the current simulation and the settings of the processors command. The partitioning can later be
changed by the balance or fix balance commands.
NOTE: Normally, restart files are written by the restart or write_restart commands so that all atoms in the
restart file are inside the simulation box. If this is not the case, the read_restart command will print an error
that atoms were "lost" when the file is read. This error should be reported to the LAMMPS developers so the
invalid writing of the restart file can be fixed. If you still wish to use the restart file, the optional remap flag
can be appended to the read_restart command. This should avoid the error, by explicitly remapping each atom
back into the simulation box, updating image flags for the atom appropriately.
Restart files are saved in binary format to enable exact restarts, meaning that the trajectories of a restarted run
will precisely match those produced by the original run had it continued on.
Several things can prevent exact restarts due to round-off effects, in which case the trajectories in the 2 runs
will slowly diverge. These include running on a different number of processors or changing certain settings
such as those set by the newton or processors commands. LAMMPS will issue a warning in these cases.
Certain fixes will not restart exactly, though they should provide statistically similar results. These include fix
shake and fix langevin.
Certain pair styles will not restart exactly, though they should provide statistically similar results. This is
because the forces they compute depend on atom velocities, which are used at half-step values every timestep
when forces are computed. When a run restarts, forces are initially evaluated with a full-step velocity, which
is different than if the run had continued. These pair styles include granular pair styles, pair dpd, and pair
lubricate.
LAMMPS Users Manual
603

If a restarted run is immediately different than the run which produced the restart file, it could be a LAMMPS
bug, so consider reporting it if you think the behavior is wrong.
Because restart files are binary, they may not be portable to other machines. In this case, you can use the
-restart command-line switch to convert a restart file to a data file.
Similar to how restart files are written (see the write_restart and restart commands), the restart filename can
contain two wild-card characters. If a "*" appears in the filename, the directory is searched for all filenames
that match the pattern where "*" is replaced with a timestep value. The file with the largest timestep value is
read in. Thus, this effectively means, read the latest restart file. It's useful if you want your script to continue a
run from where it left off. See the run command and its "upto" option for how to specify the run command so
it doesn't need to be changed either.
If a "%" character appears in the restart filename, LAMMPS expects a set of multiple files to exist. The restart
and write_restart commands explain how such sets are created. Read_restart will first read a filename where
"%" is replaced by "base". This file tells LAMMPS how many processors created the set and how many files
are in it. Read_restart then reads the additional files. For example, if the restart file was specified as save.%
when it was written, then read_restart reads the files save.base, save.0, save.1, ... save.P-1, where P is the
number of processors that created the restart file.
Note that P could be the total number of processors in the previous simulation, or some subset of those
processors, if the fileper or nfile options were used when the restart file was written; see the restart and
write_restart commands for details. The processors in the current LAMMPS simulation share the work of
reading these files; each reads a roughly equal subset of the files. The number of processors which created the
set can be different the number of processors in the current LAMMPS simulation. This can be a fast mode of
input on parallel machines that support parallel I/O.
A restart file can also be read in parallel as one large binary file via the MPI-IO library, assuming it was also
written with MPI-IO. MPI-IO is part of the MPI standard for versions 2.0 and above. Using MPI-IO requires
two steps. First, build LAMMPS with its MPIIO package installed, e.g.
make yes-mpiio # installs the MPIIO package
make mpi # build LAMMPS for your platform
Second, use a restart filename which contains ".mpiio". Note that it does not have to end in ".mpiio", just
contain those characters. Unlike MPI-IO dump files, a particular restart file must be both written and read
using MPI-IO.
Here is the list of information included in a restart file, which means these quantities do not need to be
re-specified in the input script that reads the restart file, though you can redefine many of these settings after
the restart file is read.
units• newton bond (see discussion of newton command below)• atom style and atom_modify settings id, map, sort• comm style and comm_modify settings mode, cutoff, vel• timestep• simulation box size and shape and boundary settings• atom group definitions• per-type atom settings such as mass•
LAMMPS Users Manual
604
per-atom attributes including their group assignments and molecular topology attributes (bonds,
angles, etc)
•
force field styles (pair, bond, angle, etc)• force field coefficients (pair, bond, angle, etc) in some cases (see below)• pair_modify settings, except the compute option• special_bonds settings•
Here is a list of information not stored in a restart file, which means you must re-issue these commands in
your input script, after reading the restart file.
newton pair (see discussion of newton command below)• fix commands (see below)• compute commands (see below)• variable commands• region commands• neighbor list criteria including neigh_modify settings• kspace_style and kspace_modify settings• info for thermodynamic, dump, or restart output•
The newton command has two settings, one for pairwise interactions, the other for bonded. Both settings are
stored in the restart file. For the bond setting, the value in the file will overwrite the current value (at the time
the read_restart command is issued) and warn if the two values are not the same and the current value is not
the default. For the pair setting, the value in the file will not overwrite the current value (so that you can
override the previous run's value), but a warning is issued if the two values are not the same and the current
value is not the default.
Note that some force field styles (pair, bond, angle, etc) do not store their coefficient info in restart files.
Typically these are many-body or tabulated potentials which read their parameters from separate files. In these
cases you will need to re-specify the "pair pair_coeff, bond_coeff, etc commands in your restart input script.
The doc pages for individual force field styles mention if this is the case. This is also true of pair_style hybrid
(bond hybrid, angle hybrid, etc) commands; they do not store coefficient info.
As indicated in the above list, the fixes used for a simulation are not stored in the restart file. This means the
new input script should specify all fixes it will use. However, note that some fixes store an internal "state"
which is written to the restart file. This allows the fix to continue on with its calculations in a restarted
simulation. To re-enable such a fix, the fix command in the new input script must be of the same style and use
the same fix-ID as was used in the input script that wrote the restart file.
If a match is found, LAMMPS prints a message indicating that the fix is being re-enabled. If no match is
found before the first run or minimization is performed by the new script, the "state" information for the saved
fix is discarded. At the time the discard occurs, LAMMPS will also print a list of fixes for which the
information is being discarded. See the doc pages for individual fixes for info on which ones can be restarted
in this manner. Note that fixes which are created internally by other LAMMPS commands (computes, fixes,
etc) will have style names which are all-capitalized, and IDs which are generated internally.
Likewise, the computes used for a simulation are not stored in the restart file. This means the new input script
should specify all computes it will use. However, some computes create a fix internally to store "state"
information that persists from timestep to timestep. An example is the compute msd command which uses a
fix to store a reference coordinate for each atom, so that a displacement can be calculated at any later time. If
the compute command in the new input script uses the same compute-ID and group-ID as was used in the
input script that wrote the restart file, then it will create the same fix in the restarted run. This means the
LAMMPS Users Manual
605

re-created fix will be re-enabled with the stored state information as described in the previous paragraph, so
that the compute can continue its calculations in a consistent manner.
NOTE: There are a handful of commands which can be used before or between runs which may require a
system initialization. Examples include the "balance", "displace_atoms", "delete_atoms", "set" (some
options), and "velocity" (some options) commands. This is because they can migrate atoms to new processors.
Thus they will also discard unused "state" information from fixes. You will know the discard has occurred
because a list of discarded fixes will be printed to the screen and log file, as explained above. This means that
if you wish to retain that info in a restarted run, you must re-specify the relevant fixes and computes (which
create fixes) before those commands are used.
Some pair styles, like the granular pair styles, also use a fix to store "state" information that persists from
timestep to timestep. In the case of granular potentials, it is contact information between pairs of touching
particles. This info will also be re-enabled in the restart script, assuming you re-use the same granular pair
style.
LAMMPS allows bond interactions (angle, etc) to be turned off or deleted in various ways, which can affect
how their info is stored in a restart file.
If bonds (angles, etc) have been turned off by the fix shake or delete_bonds command, their info will be
written to a restart file as if they are turned on. This means they will need to be turned off again in a new run
after the restart file is read.
Bonds that are broken (e.g. by a bond-breaking potential) are written to the restart file as broken bonds with a
type of 0. Thus these bonds will still be broken when the restart file is read.
Bonds that have been broken by the fix bond/break command have disappeared from the system. No
information about these bonds is written to the restart file.
Restrictions:
To write and read restart files in parallel with MPI-IO, the MPIIO package must be installed.
Related commands:
read_data, read_dump, write_restart, restart
Default: none
LAMMPS Users Manual
606

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
region command
Syntax:
region ID style args keyword arg ...
ID = user-assigned name for the region• style = delete or block or cone or cylinder or plane or prism or sphere or union or intersect
delete = no args
block args = xlo xhi ylo yhi zlo zhi
xlo,xhi,ylo,yhi,zlo,zhi = bounds of block in all dimensions (distance units)
cone args = dim c1 c2 radlo radhi lo hi
dim = x or y or z = axis of cone
c1,c2 = coords of cone axis in other 2 dimensions (distance units)
radlo,radhi = cone radii at lo and hi end (distance units)
lo,hi = bounds of cone in dim (distance units)
cylinder args = dim c1 c2 radius lo hi
dim = x or y or z = axis of cylinder
c1,c2 = coords of cylinder axis in other 2 dimensions (distance units)
radius = cylinder radius (distance units)
radius can be a variable (see below)
lo,hi = bounds of cylinder in dim (distance units)
plane args = px py pz nx ny nz
px,py,pz = point on the plane (distance units)
nx,ny,nz = direction normal to plane (distance units)
prism args = xlo xhi ylo yhi zlo zhi xy xz yz
xlo,xhi,ylo,yhi,zlo,zhi = bounds of untilted prism (distance units)
xy = distance to tilt y in x direction (distance units)
xz = distance to tilt z in x direction (distance units)
yz = distance to tilt z in y direction (distance units)
sphere args = x y z radius
x,y,z = center of sphere (distance units)
radius = radius of sphere (distance units)
radius can be a variable (see below)
union args = N reg-ID1 reg-ID2 ...
N = # of regions to follow, must be 2 or greater
reg-ID1,reg-ID2, ... = IDs of regions to join together
intersect args = N reg-ID1 reg-ID2 ...
N = # of regions to follow, must be 2 or greater
reg-ID1,reg-ID2, ... = IDs of regions to intersect
•
zero or more keyword/arg pairs may be appended• keyword = side or units or move or rotate or open
side value = in or out
in = the region is inside the specified geometry
out = the region is outside the specified geometry
units value = lattice or box
lattice = the geometry is defined in lattice units
box = the geometry is defined in simulation box units
move args = v_x v_y v_z
v_x,v_y,v_z = equal-style variables for x,y,z displacement of region over time
rotate args = v_theta Px Py Pz Rx Ry Rz
v_theta = equal-style variable for rotaton of region over time (in radians)
Px,Py,Pz = origin for axis of rotation (distance units)
Rx,Ry,Rz = axis of rotation vector
•
LAMMPS Users Manual
607
open value = integer from 1-6 corresponding to face index (see below)
accelerated styles (with same args) = block/kk•
Examples:
region 1 block -3.0 5.0 INF 10.0 INF INF
region 2 sphere 0.0 0.0 0.0 5 side out
region void cylinder y 2 3 5 -5.0 EDGE units box
region 1 prism 0 10 0 10 0 10 2 0 0
region outside union 4 side1 side2 side3 side4
region 2 sphere 0.0 0.0 0.0 5 side out move v_left v_up NULL
region openbox block 0 10 0 10 0 10 open 5 open 6 units box
region funnel cone z 10 10 2 5 0 10 open 1 units box
Description:
This command defines a geometric region of space. Various other commands use regions. For example, the
region can be filled with atoms via the create_atoms command. Or a bounding box around the region, can be
used to define the simulation box via the create_box command. Or the atoms in the region can be identified as
a group via the group command, or deleted via the delete_atoms command. Or the surface of the region can be
used as a boundary wall via the fix wall/region command.
Commands which use regions typically test whether an atom's position is contained in the region or not. For
this purpose, coordinates exactly on the region boundary are considered to be interior to the region. This
means, for example, for a spherical region, an atom on the sphere surface would be part of the region if the
sphere were defined with the side in keyword, but would not be part of the region if it were defined using the
side out keyword. See more details on the side keyword below.
Normally, regions in LAMMPS are "static", meaning their geometric extent does not change with time. If the
move or rotate keyword is used, as described below, the region becomes "dynamic", meaning it's location or
orientation changes with time. This may be useful, for example, when thermostatting a region, via the
compute temp/region command, or when the fix wall/region command uses a region surface as a bounding
wall on particle motion, i.e. a rotating container.
The delete style removes the named region. Since there is little overhead to defining extra regions, there is
normally no need to do this, unless you are defining and discarding large numbers of regions in your input
script.
The lo/hi values for block or cone or cylinder or prism styles can be specified as EDGE or INF. EDGE means
they extend all the way to the global simulation box boundary. Note that this is the current box boundary; if
the box changes size during a simulation, the region does not. INF means a large negative or positive number
(1.0e20), so it should encompass the simulation box even if it changes size. If a region is defined before the
simulation box has been created (via create_box or read_data or read_restart commands), then an EDGE or
INF parameter cannot be used. For a prism region, a non-zero tilt factor in any pair of dimensions cannot be
used if both the lo/hi values in either of those dimensions are INF. E.g. if the xy tilt is non-zero, then xlo and
xhi cannot both be INF, nor can ylo and yhi.
NOTE: Regions in LAMMPS do not get wrapped across periodic boundaries, as specified by the boundary
command. For example, a spherical region that is defined so that it overlaps a periodic boundary is not treated
as 2 half-spheres, one on either side of the simulation box.
LAMMPS Users Manual
608

NOTE: Regions in LAMMPS are always 3d geometric objects, regardless of whether the dimension of a
simulation is 2d or 3d. Thus when using regions in a 2d simulation, you should be careful to define the region
so that its intersection with the 2d x-y plane of the simulation has the 2d geometric extent you want.
For style cone, an axis-aligned cone is defined which is like a cylinder except that two different radii (one at
each end) can be defined. Either of the radii (but not both) can be 0.0.
For style cone and cylinder, the c1,c2 params are coordinates in the 2 other dimensions besides the cylinder
axis dimension. For dim = x, c1/c2 = y/z; for dim = y, c1/c2 = x/z; for dim = z, c1/c2 = x/y. Thus the third
example above specifies a cylinder with its axis in the y-direction located at x = 2.0 and z = 3.0, with a radius
of 5.0, and extending in the y-direction from -5.0 to the upper box boundary.
For style plane, a plane is defined which contain the point (px,py,pz) and has a normal vector (nx,ny,nz). The
normal vector does not have to be of unit length. The "inside" of the plane is the half-space in the direction of
the normal vector; see the discussion of the side option below.
For style prism, a parallelepiped is defined (it's too hard to spell parallelepiped in an input script!). The
parallelepiped has its "origin" at (xlo,ylo,zlo) and is defined by 3 edge vectors starting from the origin given
by A = (xhi-xlo,0,0); B = (xy,yhi-ylo,0); C = (xz,yz,zhi-zlo). Xy,xz,yz can be 0.0 or positive or negative values
and are called "tilt factors" because they are the amount of displacement applied to faces of an originally
orthogonal box to transform it into the parallelepiped.
A prism region that will be used with the create_box command to define a triclinic simulation box must have
tilt factors (xy,xz,yz) that do not skew the box more than half the distance of corresponding the parallel box
length. For example, if xlo = 2 and xhi = 12, then the x box length is 10 and the xy tilt factor must be between
-5 and 5. Similarly, both xz and yz must be between -(xhi-xlo)/2 and +(yhi-ylo)/2. Note that this is not a
limitation, since if the maximum tilt factor is 5 (as in this example), then configurations with tilt = ..., -15, -5,
5, 15, 25, ... are all geometrically equivalent.
The radius value for style sphere and cylinder can be specified as an equal-style variable. If the value is a
variable, it should be specified as v_name, where name is the variable name. In this case, the variable will be
evaluated each timestep, and its value used to determine the radius of the region.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent radius.
See Section 6.12 of the doc pages for a geometric description of triclinic boxes, as defined by LAMMPS, and
how to transform these parameters to and from other commonly used triclinic representations.
The union style creates a region consisting of the volume of all the listed regions combined. The intersect
style creates a region consisting of the volume that is common to all the listed regions.
NOTE: The union and intersect regions operate by invoking methods from their list of sub-regions. Thus you
cannot delete the sub-regions after defining a union or intersection region.
The side keyword determines whether the region is considered to be inside or outside of the specified
geometry. Using this keyword in conjunction with union and intersect regions, complex geometries can be
built up. For example, if the interior of two spheres were each defined as regions, and a union style with side
= out was constructed listing the region-IDs of the 2 spheres, the resulting region would be all the volume in
the simulation box that was outside both of the spheres.
LAMMPS Users Manual
609

The units keyword determines the meaning of the distance units used to define the region for any argument
above listed as having distance units. It also affects the scaling of the velocity vector specified with the vel
keyword, the amplitude vector specified with the wiggle keyword, and the rotation point specified with the
rotate keyword, since they each involve a distance metric.
A box value selects standard distance units as defined by the units command, e.g. Angstroms for units = real
or metal. A lattice value means the distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacings which are used as follows:
For style block, the lattice spacing in dimension x is applied to xlo and xhi, similarly the spacings in
dimensions y,z are applied to ylo/yhi and zlo/zhi.
•
For style cone, the lattice spacing in argument dim is applied to lo and hi. The spacings in the two
radial dimensions are applied to c1 and c2. The two cone radii are scaled by the lattice spacing in the
dimension corresponding to c1.
•
For style cylinder, the lattice spacing in argument dim is applied to lo and hi. The spacings in the two
radial dimensions are applied to c1 and c2. The cylinder radius is scaled by the lattice spacing in the
dimension corresponding to c1.
•
For style plane, the lattice spacing in dimension x is applied to px and nx, similarly the spacings in
dimensions y,z are applied to py/ny and pz/nz.
•
For style prism, the lattice spacing in dimension x is applied to xlo and xhi, similarly for ylo/yhi and
zlo/zhi. The lattice spacing in dimension x is applied to xy and xz, and the spacing in dimension y to
yz.
•
For style sphere, the lattice spacing in dimensions x,y,z are applied to the sphere center x,y,z. The
spacing in dimension x is applied to the sphere radius.
•
If the move or rotate keywords are used, the region is "dynamic", meaning its location or orientation changes
with time. These keywords cannot be used with a union or intersect style region. Instead, the keywords should
be used to make the individual sub-regions of the union or intersect region dynamic. Normally, each
sub-region should be "dynamic" in the same manner (e.g. rotate around the same point), though this is not a
requirement.
The move keyword allows one or more equal-style variables to be used to specify the x,y,z displacement of
the region, typically as a function of time. A variable is specified as v_name, where name is the variable
name. Any of the three variables can be specified as NULL, in which case no displacement is calculated in
that dimension.
Note that equal-style variables can specify formulas with various mathematical functions, and include
thermo_style command keywords for the simulation box parameters and timestep and elapsed time. Thus it is
easy to specify a region displacement that change as a function of time or spans consecutive runs in a
continuous fashion. For the latter, see the start and stop keywords of the run command and the elaplong
keyword of thermo_style custom for details.
For example, these commands would displace a region from its initial position, in the positive x direction,
effectively at a constant velocity:
variable dx equal ramp(0,10)
region 2 sphere 10.0 10.0 0.0 5 move v_dx NULL NULL
Note that the initial displacement is 0.0, though that is not required.
Either of these variables would "wiggle" the region back and forth in the y direction:
LAMMPS Users Manual
610
variable dy equal swiggle(0,5,100)
variable dysame equal 5*sin(2*PI*elaplong*dt/100)
region 2 sphere 10.0 10.0 0.0 5 move NULL v_dy NULL
The rotate keyword rotates the region around a rotation axis R = (Rx,Ry,Rz) that goes thru a point P =
(Px,Py,Pz). The rotation angle is calculated, presumably as a function of time, by a variable specified as
v_theta, where theta is the variable name. The variable should generate its result in radians. The direction of
rotation for the region around the rotation axis is consistent with the right-hand rule: if your right-hand thumb
points along R, then your fingers wrap around the axis in the direction of rotation.
The move and rotate keywords can be used together. In this case, the displacement specified by the move
keyword is applied to the P point of the rotate keyword.
The open keyword can be used (multiple times) to indicate that one or more faces of the region are ignored for
purposes of particle/wall interactions. This keyword is only relevant for regions used by the fix wall/region
and fix wall/gran/region commands. It can be used to create "open" containers where only some of the region
faces are walls. For example, a funnel can be created with a cone style region that has an open face at the
smaller radius for particles to flow out, or at the larger radius for pouring particles into the cone, or both.
Note that using the open keyword partly overrides the side keyword, since both exterior and interior surfaces
of an open region are tested for particle contacts. The exception to this is a union or intersect region which
includes an open sub-region. In that case the side keyword is still used to define the union/intersect region
volume, and the open settings are only applied to the individual sub-regions that use them.
The indices specified as part of the open keyword have the following meanings:
For style block, indices 1-6 correspond to the xlo, xhi, ylo, yhi, zlo, zhi surfaces of the block. I.e. 1 is the yz
plane at x = xlo, 2 is the yz-plane at x = xhi, 3 is the xz plane at y = ylo, 4 is the xz plane at y = yhi, 5 is the xy
plane at z = zlo, 6 is the xy plane at z = zhi). In the second-to-last example above, the region is a box open at
both xy planes.
For style prism, values 1-6 have the same mapping as for style block. I.e. in an untilted prism, open indices
correspond to the xlo, xhi, ylo, yhi, zlo, zhi surfaces.
For style cylinder, index 1 corresponds to the flat end cap at the low coordinate along the cylinder axis, index
2 corresponds to the high-coordinate flat end cap along the cylinder axis, and index 3 is the curved cylinder
surface. For example, a cylinder region with open 1 open 2 keywords will be open at both ends (e.g. a section
of pipe), regardless of the cylinder orientation.
For style cone, the mapping is the same as for style cylinder. Index 1 is the low-coordinate flat end cap, index
2 is the high-coordinate flat end cap, and index 3 is the curved cone surface. In the last example above, a cone
region is defined along the z-axis that is open at the zlo value (e.g. for use as a funnel).
For all other styles, the open keyword is ignored. As indicated above, this includes the intersect and union
regions, though their sub-regions can be defined with the open keyword.
Styles with a kk suffix are functionally the same as the corresponding style without the suffix. They have been
optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The
accelerated styles take the same arguments and should produce the same results, except for round-off and
precision issues.
LAMMPS Users Manual
611

The code using the region (such as a fix or compute) must also be supported by Kokkos or no acceleration
will occur. Currently, only block style regions are supported by Kokkos.
These accelerated styles are part of the Kokkos package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
A prism cannot be of 0.0 thickness in any dimension; use a small z thickness for 2d simulations. For 2d
simulations, the xz and yz parameters must be 0.0.
Related commands:
lattice, create_atoms, delete_atoms, group
Default:
The option defaults are side = in, units = lattice, and no move or rotation.
LAMMPS Users Manual
612

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
replicate command
Syntax:
replicate nx ny nz
nx,ny,nz = replication factors in each dimension•
Examples:
replicate 2 3 2
Description:
Replicate the current simulation one or more times in each dimension. For example, replication factors of
2,2,2 will create a simulation with 8x as many atoms by doubling the simulation domain in each dimension. A
replication factor of 1 in a dimension leaves the simulation domain unchanged. When the new simulation box
is created it is also partitioned into a regular 3d grid of rectangular bricks, one per processor, based on the
number of processors being used and the settings of the processors command. The partitioning can later be
changed by the balance or fix balance commands.
All properties of the atoms are replicated, including their velocities, which may or may not be desirable. New
atom IDs are assigned to new atoms, as are molecule IDs. Bonds and other topology interactions are created
between pairs of new atoms as well as between old and new atoms. This is done by using the image flag for
each atom to "unwrap" it out of the periodic box before replicating it.
This means that any molecular bond you specify in the original data file that crosses a periodic boundary
should be between two atoms with image flags that differ by 1. This will allow the bond to be unwrapped
appropriately.
Restrictions:
A 2d simulation cannot be replicated in the z dimension.
If a simulation is non-periodic in a dimension, care should be used when replicating it in that dimension, as it
may put atoms nearly on top of each other.
NOTE: You cannot use the replicate command on a system which has a molecule that spans the box and is
bonded to itself across a periodic boundary, so that the molecule is effectively a loop. A simple example
would be a linear polymer chain that spans the simulation box and bonds back to itself across the periodic
boundary. More realistic examples would be a CNT (meant to be an infinitely long CNT) or a graphene sheet
or a bulk periodic crystal where there are explicit bonds specified between near neighbors. (Note that this only
applies to systems that have permanent bonds as specified in the data file. A CNT that is just atoms modeled
with the AIREBO potential has no such permanent bonds, so it can be replicated.) The reason replication does
not work with those systems is that the image flag settings described above cannot be made consistent. I.e. it
is not possible to define images flags so that when every pair of bonded atoms is unwrapped (using the image
flags), they will be close to each other. The only way the replicate command could work in this scenario is for
it to break a bond, insert more atoms, and re-connect the loop for the larger simulation box. But it is not clever
enough to do this. So you will have to construct a larger version of your molecule as a pre-processing step and
LAMMPS Users Manual
613
input a new data file to LAMMPS.
If the current simulation was read in from a restart file (before a run is performed), there must not be any fix
information stored in the file for individual atoms. Similarly, no fixes can be defined at the time the replicate
command is used that require vectors of atom information to be stored. This is because the replicate command
does not know how to replicate that information for new atoms it creates. To work around this restriction,
restart files may be converted into data files and fixes may be undefined via the unfix command before and
redefined after the replicate command.
Related commands: none
Default: none
LAMMPS Users Manual
614

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
rerun command
Syntax:
rerun file1 file2 ... keyword args ...
file1,file2,... = dump file(s) to read• one or more keywords may be appended, keyword dump must appear and be last
keyword = first or last or every or skip or start or stop or dump
first args = Nfirts
Nfirst = dump timestep to start on
last args = Nlast
Nlast = dumptimestep to stop on
every args = Nevery
Nevery = read snapshots matching every this many timesteps
skip args = Nskip
Nskip = read one out of every Nskip snapshots
start args = Nstart
Nstart = timestep on which pseudo run will start
stop args = Nstop
Nstop = timestep to which pseudo run will end
dump args = same as read_dump command starting with its field arguments
•
Examples:
rerun dump.file dump x y z vx vy vz
rerun dump1.txt dump2.txt first 10000 every 1000 dump x y z
rerun dump.vels dump x y z vx vy vz box yes format molfile lammpstrj
rerun dump.dcd dump x y z box no format molfile dcd
rerun ../run7/dump.file.gz skip 2 dump x y z box yes
Description:
Perform a pseudo simulation run where atom information is read one snapshot at a time from a dump file(s),
and energies and forces are computed on the shapshot to produce thermodynamic or other output.
This can be useful in the following kinds of scenarios, after an initial simulation produced the dump file:
Compute the energy and forces of snaphots using a different potential.• Calculate one or more diagnostic quantities on the snapshots that weren't computed in the initial run.
These can also be computed with settings not used in the initial run, e.g. computing an RDF via the
compute rdf command with a longer cutoff than was used initially.
•
Calculate the portion of per-atom forces resulting from a subset of the potential. E.g. compute only
Coulombic forces. This can be done by only defining only a Coulombic pair style in the rerun script.
Doing this in the original script would result in different (bad) dynamics.
•
Conceptually, using the rerun command is like running an input script that has a loop in it (see the next and
jump commands). Each iteration of the loop reads one snapshot from the dump file via the read_dump
command, sets the timestep to the appropriate value, and then invokes a run command for zero timesteps to
simply compute energy and forces, and any other thermodynamic output or diagnostic info you have defined.
This computation also invokes any fixes you have defined that apply constraints to the system, such as fix
LAMMPS Users Manual
615
shake or fix indent.
Note that a simulation box must already be defined before using the rerun command. This can be done by the
create_box, read_data, or read_restart commands.
Also note that reading per-atom information from dump snapshots is limited to the atom coordinates,
velocities and image flags as explained in the read_dump command. Other atom properties, which may be
necessary to compute energies and forces, such as atom charge, or bond topology information for a molecular
system, are not read from (or even contained in) dump files. Thus this auxiliary information should be defined
in the usual way, e.g. in a data file read in by a read_data command, before using the rerun command.
If more than one dump file is specified, the dump files are read one after the other. It is assumed that snapshot
timesteps will be in ascending order. If a snapshot is encountered that is not in ascending order, it will cause
the rerun command to complete.
The first, last, every, skip keywords determine which snapshots are read from the dump file(s). Snapshots are
skipped until they have a timestamp >= Nfirst. When a snapshot with a timestamp > Nlast is encountered, the
rerun command finishes. Note below that the defaults for first and last are to read all snapshots. If the every
keyword is set to a value > 0, then only snapshots with timestamps that are a multiple of Nevery are read (the
first snapshot is always read). If Nevery = 0, then this criterion is ignored, i.e. every snapshot is read that
meets the other criteria. If the skip keyword is used, then after the first snapshot is read, every Nth snapshot is
read, where N = Nskip. E.g. if Nskip = 3, then only 1 out of every 3 snapshots is read, assuming the snapshot
timestamp is also consistent with the other criteria.
The start and stop keywords do not affect which snapshots are read from the dump file(s). Rather, they have
the same meaning that they do for the run command. They only need to be defined if (a) you are using a fix
command that changes some value over time, and (b) you want the reference point for elapsed time (from start
to stop) to be different than the first and last settings. See the doc page for individual fixes to see which ones
can be used with the start/stop keywords. Note that if you define neither of the start/stop or first/last
keywords, then LAMMPS treats the pseudo run as going from 0 to a huge value (effectively infinity). This
means that any quantity that a fix scales as a fraction of elapsed time in the run, will essentially remain at its
initial value. Also note that an error will occur if you read a snapshot from the dump file with a timestep value
larger than the stop setting you have specified.
The dump keyword is required and must be the last keyword specified. Its arguments are passed internally to
the read_dump command. The first argument following the dump keyword should be the field1 argument of
the read_dump command. See the read_dump doc page for details on the various options it allows for
extracting information from the dump file snapshots, and for using that information to alter the LAMMPS
simulation.
In general, a LAMMPS input script that uses a rerun command can include and perform all the usual
operations of an input script that uses the run command. There are a few exceptions and points to consider, as
discussed here.
Fixes that perform time integration, such as fix nve or fix npt are not invoked, since no time integration is
performed. Fixes that perturb or constrain the forces on atoms will be invoked, just as they would during a
normal run. Examples are fix indent and fix langevin. So you should think carefully as to whether that makes
sense for the manner in which you are reprocessing the dump snapshots.
If you only want the rerun script to perform analyses that do not involve pair interactions, such as use compute
LAMMPS Users Manual
616

msd to calculated displacements over time, you do not need to define a pair style, which may also mean
neighbor lists will not need to be calculated which saves time. The comm_modify cutoff command can also be
used to insure ghost atoms are acquired from far enough away for operations like bond and angle evaluations,
if no pair style is being used.
Every time a snapshot is read, the timestep for the simulation is reset, as if the reset_timestep command were
used. This command has some restrictions as to what fixes can be defined. See its doc page for details. For
example, the fix deposit and fix dt/reset fixes are in this category. They also make no sense to use with a rerun
command.
If time-averaging fixes like fix ave/time are used, they are invoked on timesteps that are a function of their
Nevery, Nrepeat, and Nfreq settings. As an example, see the fix ave/time doc page for details. You must
insure those settings are consistent with the snapshot timestamps that are read from the dump file(s). If an
averaging fix is not invoked on a timestep it expects to be, LAMMPS will flag an error.
The various forms of LAMMPS output, as defined by the thermo_style, thermo, dump, and restart commands
occur on specific timesteps. If successive dump snapshots skip those timesteps, then no output will be
produced. E.g. if you request thermodynamic output every 100 steps, but the dump file snapshots are every
1000 steps, then you will only see thermodynamic output every 1000 steps.
Restrictions:
To read gzipped dump files, you must compile LAMMPS with the -DLAMMPS_GZIP option - see the
Making LAMMPS section of the documentation.
Related commands:
read_dump
Default:
The option defaults are first = 0, last = a huge value (effectively infinity), start = same as first, stop = same as
last, every = 0, skip = 1;
LAMMPS Users Manual
617

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
reset_timestep command
Syntax:
reset_timestep N
N = timestep number•
Examples:
reset_timestep 0
reset_timestep 4000000
Description:
Set the timestep counter to the specified value. This command normally comes after the timestep has been set
by reading a restart file via the read_restart command, or a previous simulation advanced the timestep.
The read_data and create_box commands set the timestep to 0; the read_restart command sets the timestep to
the value it had when the restart file was written.
Restrictions: none
This command cannot be used when any fixes are defined that keep track of elapsed time to perform certain
kinds of time-dependent operations. Examples are the fix deposit and fix dt/reset commands. The former adds
atoms on specific timesteps. The latter keeps track of accumulated time.
Various fixes use the current timestep to calculate related quantities. If the timestep is reset, this may produce
unexpected behavior, but LAMMPS allows the fixes to be defined even if the timestep is reset. For example,
commands which thermostat the system, e.g. fix nvt, allow you to specify a target temperature which ramps
from Tstart to Tstop which may persist over several runs. If you change the timestep, you may induce an
instantaneous change in the target temperature.
Resetting the timestep clears flags for computes that may have calculated some quantity from a previous run.
This means these quantity cannot be accessed by a variable in between runs until a new run is performed. See
the variable command for more details.
Related commands:
rerun
Default: none
LAMMPS Users Manual
618

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
restart command
Syntax:
restart 0
restart N root keyword value ...
restart N file1 file2 keyword value ...
N = write a restart file every this many timesteps• N can be a variable (see below)• root = filename to which timestep # is appended• file1,file2 = two full filenames, toggle between them when writing file• zero or more keyword/value pairs may be appended• keyword = fileper or nfile
fileper arg = Np
Np = write one file for every this many processors
nfile arg = Nf
Nf = write this many files, one from each of Nf processors
•
Examples:
restart 0
restart 1000 poly.restart
restart 1000 poly.restart.mpiio
restart 1000 restart.*.equil
restart 10000 poly.%.1 poly.%.2 nfile 10
restart v_mystep poly.restart
Description:
Write out a binary restart file with the current state of the simulation every so many timesteps, in either or
both of two modes, as a run proceeds. A value of 0 means do not write out any restart files. The two modes
are as follows. If one filename is specified, a series of filenames will be created which include the timestep in
the filename. If two filenames are specified, only 2 restart files will be created, with those names. LAMMPS
will toggle between the 2 names as it writes successive restart files.
Note that you can specify the restart command twice, once with a single filename and once with two
filenames. This would allow you, for example, to write out archival restart files every 100000 steps using a
single filenname, and more frequent temporary restart files every 1000 steps, using two filenames. Using
restart 0 will turn off both modes of output.
Similar to dump files, the restart filename(s) can contain two wild-card characters.
If a "*" appears in the single filename, it is replaced with the current timestep value. This is only recognized
when a single filename is used (not when toggling back and forth). Thus, the 3rd example above creates
restart files as follows: restart.1000.equil, restart.2000.equil, etc. If a single filename is used with no "*", then
the timestep value is appended. E.g. the 2nd example above creates restart files as follows: poly.restart.1000,
poly.restart.2000, etc.
LAMMPS Users Manual
619

If a "%" character appears in the restart filename(s), then one file is written for each processor and the "%"
character is replaced with the processor ID from 0 to P-1. An additional file with the "%" replaced by "base"
is also written, which contains global information. For example, the files written on step 1000 for filename
restart.% would be restart.base.1000, restart.0.1000, restart.1.1000, ..., restart.P-1.1000. This creates smaller
files and can be a fast mode of output and subsequent input on parallel machines that support parallel I/O. The
optional fileper and nfile keywords discussed below can alter the number of files written.
The restart file can also be written in parallel as one large binary file via the MPI-IO library, which is part of
the MPI standard for versions 2.0 and above. Using MPI-IO requires two steps. First, build LAMMPS with its
MPIIO package installed, e.g.
make yes-mpiio # installs the MPIIO package
make mpi # build LAMMPS for your platform
Second, use a restart filename which contains ".mpiio". Note that it does not have to end in ".mpiio", just
contain those characters. Unlike MPI-IO dump files, a particular restart file must be both written and read
using MPI-IO.
Restart files are written on timesteps that are a multiple of N but not on the first timestep of a run or
minimization. You can use the write_restart command to write a restart file before a run begins. A restart file
is not written on the last timestep of a run unless it is a multiple of N. A restart file is written on the last
timestep of a minimization if N > 0 and the minimization converges.
Instead of a numeric value, N can be specified as an equal-style variable, which should be specified as
v_name, where name is the variable name. In this case, the variable is evaluated at the beginning of a run to
determine the next timestep at which a restart file will be written out. On that timestep, the variable will be
evaluated again to determine the next timestep, etc. Thus the variable should return timestep values. See the
stagger() and logfreq() and stride() math functions for equal-style variables, as examples of useful functions to
use in this context. Other similar math functions could easily be added as options for equal-style variables.
For example, the following commands will write restart files every step from 1100 to 1200, and could be
useful for debugging a simulation where something goes wrong at step 1163:
variable s equal stride(1100,1200,1)
restart v_s tmp.restart
See the read_restart command for information about what is stored in a restart file.
Restart files can be read by a read_restart command to restart a simulation from a particular state. Because the
file is binary (to enable exact restarts), it may not be readable on another machine. In this case, you can use
the -r command-line switch to convert a restart file to a data file.
NOTE: Although the purpose of restart files is to enable restarting a simulation from where it left off, not all
information about a simulation is stored in the file. For example, the list of fixes that were specified during the
initial run is not stored, which means the new input script must specify any fixes you want to use. Even when
restart information is stored in the file, as it is for some fixes, commands may need to be re-specified in the
new input script, in order to re-use that information. See the read_restart command for information about what
is stored in a restart file.
The optional nfile or fileper keywords can be used in conjunction with the "%" wildcard character in the
specified restart file name(s). As explained above, the "%" character causes the restart file to be written in
LAMMPS Users Manual
620

pieces, one piece for each of P processors. By default P = the number of processors the simulation is running
on. The nfile or fileper keyword can be used to set P to a smaller value, which can be more efficient when
running on a large number of processors.
The nfile keyword sets P to the specified Nf value. For example, if Nf = 4, and the simulation is running on
100 processors, 4 files will be written, by processors 0,25,50,75. Each will collect information from itself and
the next 24 processors and write it to a restart file.
For the fileper keyword, the specified value of Np means write one file for every Np processors. For example,
if Np = 4, every 4th processor (0,4,8,12,etc) will collect information from itself and the next 3 processors and
write it to a restart file.
Restrictions:
To write and read restart files in parallel with MPI-IO, the MPIIO package must be installed.
Related commands:
write_restart, read_restart
Default:
restart 0
LAMMPS Users Manual
621

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
run command
Syntax:
run N keyword values ...
N = # of timesteps• zero or more keyword/value pairs may be appended• keyword = upto or start or stop or pre or post or every
upto value = none
start value = N1
N1 = timestep at which 1st run started
stop value = N2
N2 = timestep at which last run will end
pre value = no or yes
post value = no or yes
every values = M c1 c2 ...
M = break the run into M-timestep segments and invoke one or more commands between each segment
c1,c2,...,cN = one or more LAMMPS commands, each enclosed in quotes
c1 = NULL means no command will be invoked
•
Examples:
run 10000
run 1000000 upto
run 100 start 0 stop 1000
run 1000 pre no post yes
run 100000 start 0 stop 1000000 every 1000 "print 'Protein Rg = $r'"
run 100000 every 1000 NULL
Description:
Run or continue dynamics for a specified number of timesteps.
When the run style is respa, N refers to outer loop (largest) timesteps.
A value of N = 0 is acceptable; only the thermodynamics of the system are computed and printed without
taking a timestep.
The upto keyword means to perform a run starting at the current timestep up to the specified timestep. E.g. if
the current timestep is 10,000 and "run 100000 upto" is used, then an additional 90,000 timesteps will be run.
This can be useful for very long runs on a machine that allocates chunks of time and terminate your job when
time is exceeded. If you need to restart your script multiple times (reading in the last restart file), you can keep
restarting your script with the same run command until the simulation finally completes.
The start or stop keywords can be used if multiple runs are being performed and you want a fix command that
changes some value over time (e.g. temperature) to make the change across the entire set of runs and not just a
single run. See the doc page for individual fixes to see which ones can be used with the start/stop keywords.
For example, consider this fix followed by 10 run commands:
LAMMPS Users Manual
622
fix 1 all nvt 200.0 300.0 1.0
run 1000 start 0 stop 10000
run 1000 start 0 stop 10000
...
run 1000 start 0 stop 10000
The NVT fix ramps the target temperature from 200.0 to 300.0 during a run. If the run commands did not
have the start/stop keywords (just "run 1000"), then the temperature would ramp from 200.0 to 300.0 during
the 1000 steps of each run. With the start/stop keywords, the ramping takes place over the 10000 steps of all
runs together.
The pre and post keywords can be used to streamline the setup, clean-up, and associated output to the screen
that happens before and after a run. This can be useful if you wish to do many short runs in succession (e.g.
LAMMPS is being called as a library which is doing other computations between successive short LAMMPS
runs).
By default (pre and post = yes), LAMMPS creates neighbor lists, computes forces, and imposes fix constraints
before every run. And after every run it gathers and prints timings statistics. If a run is just a continuation of a
previous run (i.e. no settings are changed), the initial computation is not necessary; the old neighbor list is still
valid as are the forces. So if pre is specified as "no" then the initial setup is skipped, except for printing
thermodynamic info. Note that if pre is set to "no" for the very 1st run LAMMPS performs, then it is
overridden, since the initial setup computations must be done.
NOTE: If your input script changes the system between 2 runs, then the initial setup must be performed to
insure the change is recognized by all parts of the code that are affected. Examples are adding a fix or dump or
compute, changing a neighbor list parameter, or writing restart file which can migrate atoms between
processors. LAMMPS has no easy way to check if this has happened, but it is an error to use the pre no option
in this case.
If post is specified as "no", the full timing summary is skipped; only a one-line summary timing is printed.
The every keyword provides a means of breaking a LAMMPS run into a series of shorter runs. Optionally,
one or more LAMMPS commands (c1, c2, ..., cN) will be executed in between the short runs. If used, the
every keyword must be the last keyword, since it has a variable number of arguments. Each of the trailing
arguments is a single LAMMPS command, and each command should be enclosed in quotes, so that the entire
command will be treated as a single argument. This will also prevent any variables in the command from
being evaluated until it is executed multiple times during the run. Note that if a command itself needs one of
its arguments quoted (e.g. the print command), then you can use a combination of single and double quotes, as
in the example above or below.
The every keyword is a means to avoid listing a long series of runs and interleaving commands in your input
script. For example, a print command could be invoked or a fix could be redefined, e.g. to reset a thermostat
temperature. Or this could be useful for invoking a command you have added to LAMMPS that wraps some
other code (e.g. as a library) to perform a computation periodically during a long LAMMPS run. See this
section of the documentation for info about how to add new commands to LAMMPS. See this section of the
documentation for ideas about how to couple LAMMPS to other codes.
With the every option, N total steps are simulated, in shorter runs of M steps each. After each M-length run,
the specified commands are invoked. If only a single command is specified as NULL, then no command is
invoked. Thus these lines:
variable q equal x[100]
LAMMPS Users Manual
623
run 6000 every 2000 "print 'Coord = $q'"
are the equivalent of:
variable q equal x[100]
run 2000
print "Coord = $q"
run 2000
print "Coord = $q"
run 2000
print "Coord = $q"
which does 3 runs of 2000 steps and prints the x-coordinate of a particular atom between runs. Note that the
variable "$q" will be evaluated afresh each time the print command is executed.
Note that by using the line continuation character "&", the run every command can be spread across many
lines, though it is still a single command:
run 100000 every 1000 &
"print 'Minimum value = $a'" &
"print 'Maximum value = $b'" &
"print 'Temp = $c'" &
"print 'Press = $d'"
If the pre and post options are set to "no" when used with the every keyword, then the 1st run will do the full
setup and the last run will print the full timing summary, but these operations will be skipped for intermediate
runs.
NOTE: You might wish to specify a command that exits the run by jumping out of the loop, e.g.
variable t equal temp
run 10000 every 100 "if '$t <300.0' then 'jump SELF afterrun'"
However, this will not work. The run command simply executes each command one at a time each time it
pauses, then continues the run.
Instead, you should use the fix halt command, which has additional options for how to exit the run.
Restrictions:
When not using the upto keyword, the number of specified timesteps N must fit in a signed 32-bit integer, so
you are limited to slightly more than 2 billion steps (2^31) in a single run. When using upto, N can be larger
than a signed 32-bit integer, however the difference between N and the current timestep must still be no larger
than 2^31 steps.
However, with or without the upto keyword, you can perform successive runs to run a simulation for any
number of steps (ok, up to 2^63 total steps). I.e. the timestep counter within LAMMPS is a 64-bit signed
integer.
Related commands:
minimize, run_style, temper, fix halt
LAMMPS Users Manual
624
Default:
The option defaults are start = the current timestep, stop = current timestep + N, pre = yes, and post = yes.
LAMMPS Users Manual
625

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
run_style command
Syntax:
run_style style args
style = verlet or verlet/split or respa or respa/omp
verlet args = none
verlet/split args = none
respa args = N n1 n2 ... keyword values ...
N = # of levels of rRESPA
n1, n2, ... = loop factor between rRESPA levels (N-1 values)
zero or more keyword/value pairings may be appended to the loop factors
keyword = bond or angle or dihedral or improper or
pair or inner or middle or outer or hybrid or kspace
bond value = M
M = which level (1-N) to compute bond forces in
angle value = M
M = which level (1-N) to compute angle forces in
dihedral value = M
M = which level (1-N) to compute dihedral forces in
improper value = M
M = which level (1-N) to compute improper forces in
pair value = M
M = which level (1-N) to compute pair forces in
inner values = M cut1 cut2
M = which level (1-N) to compute pair inner forces in
cut1 = inner cutoff between pair inner and
pair middle or outer (distance units)
cut2 = outer cutoff between pair inner and
pair middle or outer (distance units)
middle values = M cut1 cut2
M = which level (1-N) to compute pair middle forces in
cut1 = inner cutoff between pair middle and pair outer (distance units)
cut2 = outer cutoff between pair middle and pair outer (distance units)
outer value = M
M = which level (1-N) to compute pair outer forces in
hybrid values = M1 M2 ... (as many values as there are hybrid sub-styles
M1 = which level (1-N) to compute the first pair_style hybrid sub-style in
M2 = which level (1-N) to compute the second pair_style hybrid sub-style in
M3,etc
kspace value = M
M = which level (1-N) to compute kspace forces in
•
Examples:
run_style verlet
run_style respa 4 2 2 2 bond 1 dihedral 2 pair 3 kspace 4
run_style respa 4 2 2 2 bond 1 dihedral 2 inner 3 5.0 6.0 outer 4 kspace 4
run_style respa 3 4 2 bond 1 hybrid 2 2 1 kspace 3
Description:
LAMMPS Users Manual
626

Choose the style of time integrator used for molecular dynamics simulations performed by LAMMPS.
The verlet style is a standard velocity-Verlet integrator.
The verlet/split style is also a velocity-Verlet integrator, but it splits the force calculation within each timestep
over 2 partitions of processors. See Section 2.7 for an explanation of the -partition command-line switch.
Specifically, this style performs all computation except the kspace_style portion of the force field on the 1st
partition. This include the pair style, bond style, neighbor list building, fixes including time integration, and
output. The kspace_style portion of the calculation is performed on the 2nd partition.
This is most useful for the PPPM kspace_style when its performance on a large number of processors
degrades due to the cost of communication in its 3d FFTs. In this scenario, splitting your P total processors
into 2 subsets of processors, P1 in the 1st partition and P2 in the 2nd partition, can enable your simulation to
run faster. This is because the long-range forces in PPPM can be calculated at the same time as pair-wise and
bonded forces are being calculated, and the FFTs can actually speed up when running on fewer processors.
To use this style, you must define 2 partitions where P1 is a multiple of P2. Typically having P1 be 3x larger
than P2 is a good choice. The 3d processor layouts in each partition must overlay in the following sense. If P1
is a Px1 by Py1 by Pz1 grid, and P2 = Px2 by Py2 by Pz2, then Px1 must be an integer multiple of Px2, and
similarly for Py1 a multiple of Py2, and Pz1 a multiple of Pz2.
Typically the best way to do this is to let the 1st partition choose its onn optimal layout, then require the 2nd
partition's layout to match the integer multiple constraint. See the processors command with its part keyword
for a way to control this, e.g.
procssors * * * part 1 2 multiple
You can also use the partition command to explicitly specify the processor layout on each partition. E.g. for 2
partitions of 60 and 15 processors each:
partition yes 1 processors 3 4 5
partition yes 2 processors 3 1 5
When you run in 2-partition mode with the verlet/split style, the thermodynamic data for the entire simulation
will be output to the log and screen file of the 1st partition, which are log.lammps.0 and screen.0 by default;
see the -plog and -pscreen command-line switches to change this. The log and screen file for the 2nd partition
will not contain thermodynamic output beyond the 1st timestep of the run.
See Section 5 of the manual for performance details of the speed-up offered by the verlet/split style. One
important performance consideration is the assignment of logical processors in the 2 partitions to the physical
cores of a parallel machine. The processors command has options to support this, and strategies are discussed
in Section 5 of the manual.
The respa style implements the rRESPA multi-timescale integrator (Tuckerman) with N hierarchical levels,
where level 1 is the innermost loop (shortest timestep) and level N is the outermost loop (largest timestep).
The loop factor arguments specify what the looping factor is between levels. N1 specifies the number of
iterations of level 1 for a single iteration of level 2, N2 is the iterations of level 2 per iteration of level 3, etc.
N-1 looping parameters must be specified.
LAMMPS Users Manual
627
The timestep command sets the timestep for the outermost rRESPA level. Thus if the example command
above for a 4-level rRESPA had an outer timestep of 4.0 fmsec, the inner timestep would be 8x smaller or 0.5
fmsec. All other LAMMPS commands that specify number of timesteps (e.g. neigh_modify parameters, dump
every N timesteps, etc) refer to the outermost timesteps.
The rRESPA keywords enable you to specify at what level of the hierarchy various forces will be computed.
If not specified, the defaults are that bond forces are computed at level 1 (innermost loop), angle forces are
computed where bond forces are, dihedral forces are computed where angle forces are, improper forces are
computed where dihedral forces are, pair forces are computed at the outermost level, and kspace forces are
computed where pair forces are. The inner, middle, outer forces have no defaults.
For fixes that support it, the rRESPA level at which a given fix is active, can be selected through the
fix_modify command.
The inner and middle keywords take additional arguments for cutoffs that are used by the pairwise force
computations. If the 2 cutoffs for inner are 5.0 and 6.0, this means that all pairs up to 6.0 apart are computed
by the inner force. Those between 5.0 and 6.0 have their force go ramped to 0.0 so the overlap with the next
regime (middle or outer) is smooth. The next regime (middle or outer) will compute forces for all pairs from
5.0 outward, with those from 5.0 to 6.0 having their value ramped in an inverse manner.
Only some pair potentials support the use of the inner and middle and outer keywords. If not, only the pair
keyword can be used with that pair style, meaning all pairwise forces are computed at the same rRESPA level.
See the doc pages for individual pair styles for details.i
Another option for using pair potentials with rRESPA is with the hybrid keyword, which requires the use of
the pair_style hybrid or hybrid/overlay command. In this scenario, different sub-styles of the hybrid pair style
are evaluated at different rRESPA levels. This can be useful, for example, to set different timesteps for hybrid
coarse-grained/all-atom models. The hybrid keyword requires as many level assignments as there are hybrid
substyles, which assigns each sub-style to a rRESPA level, following their order of definition in the pair_style
command. Since the hybrid keyword operates on pair style computations, it is mutually exclusive with either
the pair or the inner/middle/outer keywords.
When using rRESPA (or for any MD simulation) care must be taken to choose a timestep size(s) that insures
the Hamiltonian for the chosen ensemble is conserved. For the constant NVE ensemble, total energy must be
conserved. Unfortunately, it is difficult to know a priori how well energy will be conserved, and a fairly long
test simulation (~10 ps) is usually necessary in order to verify that no long-term drift in energy occurs with the
trial set of parameters.
With that caveat, a few rules-of-thumb may be useful in selecting respa settings. The following applies mostly
to biomolecular simulations using the CHARMM or a similar all-atom force field, but the concepts are
adaptable to other problems. Without SHAKE, bonds involving hydrogen atoms exhibit high-frequency
vibrations and require a timestep on the order of 0.5 fmsec in order to conserve energy. The relatively
inexpensive force computations for the bonds, angles, impropers, and dihedrals can be computed on this
innermost 0.5 fmsec step. The outermost timestep cannot be greater than 4.0 fmsec without risking energy
drift. Smooth switching of forces between the levels of the rRESPA hierarchy is also necessary to avoid drift,
and a 1-2 angstrom "healing distance" (the distance between the outer and inner cutoffs) works reasonably
well. We thus recommend the following settings for use of the respa style without SHAKE in biomolecular
simulations:
timestep 4.0
run_style respa 4 2 2 2 inner 2 4.5 6.0 middle 3 8.0 10.0 outer 4
LAMMPS Users Manual
628

With these settings, users can expect good energy conservation and roughly a 2.5 fold speedup over the verlet
style with a 0.5 fmsec timestep.
If SHAKE is used with the respa style, time reversibility is lost, but substantially longer time steps can be
achieved. For biomolecular simulations using the CHARMM or similar all-atom force field, bonds involving
hydrogen atoms exhibit high frequency vibrations and require a time step on the order of 0.5 fmsec in order to
conserve energy. These high frequency modes also limit the outer time step sizes since the modes are coupled.
It is therefore desirable to use SHAKE with respa in order to freeze out these high frequency motions and
increase the size of the time steps in the respa hierarchy. The following settings can be used for biomolecular
simulations with SHAKE and rRESPA:
fix 2 all shake 0.000001 500 0 m 1.0 a 1
timestep 4.0
run_style respa 2 2 inner 1 4.0 5.0 outer 2
With these settings, users can expect good energy conservation and roughly a 1.5 fold speedup over the verlet
style with SHAKE and a 2.0 fmsec timestep.
For non-biomolecular simulations, the respa style can be advantageous if there is a clear separation of time
scales - fast and slow modes in the simulation. Even a LJ system can benefit from rRESPA if the interactions
are divided by the inner, middle and outer keywords. A 2-fold or more speedup can be obtained while
maintaining good energy conservation. In real units, for a pure LJ fluid at liquid density, with a sigma of 3.0
angstroms, and epsilon of 0.1 Kcal/mol, the following settings seem to work well:
timestep 36.0
run_style respa 3 3 4 inner 1 3.0 4.0 middle 2 6.0 7.0 outer 3
The respa/omp styles is a variant of respa adapted for use with pair, bond, angle, dihedral, improper, or
kspace styles with an omp suffix. It is functionally equivalent to respa but performs additional operations
required for managing omp styles. For more on omp styles see the Section 5 of the manual. Accelerated styles
take the same arguments and should produce the same results, except for round-off and precision issues.
You can specify respa/omp explicitly in your input script, or you can use the -suffix command-line switch
when you invoke LAMMPS, or you can use the suffix command in your input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
The verlet/split style can only be used if LAMMPS was built with the REPLICA package. Correspondingly
the respa/omp style is available only if the USER-OMP package was included. See the Making LAMMPS
section for more info on packages.
Whenever using rRESPA, the user should experiment with trade-offs in speed and accuracy for their system,
and verify that they are conserving energy to adequate precision.
Related commands:
timestep, run
Default:
LAMMPS Users Manual
629

run_style verlet
(Tuckerman) Tuckerman, Berne and Martyna, J Chem Phys, 97, p 1990 (1992).
LAMMPS Users Manual
630

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
set command
Syntax:
set style ID keyword values ...
style = atom or type or mol or group or region• ID = atom ID range or type range or mol ID range or group ID or region ID• one or more keyword/value pairs may be appended• keyword = type or type/fraction or mol or x or y or z or charge or dipole or dipole/random or quat or
quat/random or diameter or shape or length or tri or theta or theta/random or angmom or omega or
mass or density or density/disc or volume or image or bond or angle or dihedral or improper or meso/e
or meso/cv or meso/rho or smd/contact/radius or smd/mass/density or dpd/theta or i_name or d_name
type value = atom type
value can be an atom-style variable (see below)
type/fraction values = type fraction seed
type = new atom type
fraction = fraction of selected atoms to set to new atom type
seed = random # seed (positive integer)
mol value = molecule ID
value can be an atom-style variable (see below)
x,y,z value = atom coordinate (distance units)
value can be an atom-style variable (see below)
charge value = atomic charge (charge units)
value can be an atom-style variable (see below)
dipole values = x y z
x,y,z = orientation of dipole moment vector
any of x,y,z can be an atom-style variable (see below)
dipole/random value = seed Dlen
seed = random # seed (positive integer) for dipole moment orientations
Dlen = magnitude of dipole moment (dipole units)
quat values = a b c theta
a,b,c = unit vector to rotate particle around via right-hand rule
theta = rotation angle (degrees)
any of a,b,c,theta can be an atom-style variable (see below)
quat/random value = seed
seed = random # seed (positive integer) for quaternion orientations
diameter value = diameter of spherical particle (distance units)
value can be an atom-style variable (see below)
shape value = Sx Sy Sz
Sx,Sy,Sz = 3 diameters of ellipsoid (distance units)
length value = len
len = length of line segment (distance units)
len can be an atom-style variable (see below)
tri value = side
side = side length of equilateral triangle (distance units)
side can be an atom-style variable (see below)
theta value = angle (degrees)
angle = orientation of line segment with respect to x-axis
angle can be an atom-style variable (see below)
theta/random value = seed
seed = random # seed (positive integer) for line segment orienations
angmom values = Lx Ly Lz
Lx,Ly,Lz = components of angular momentum vector (distance-mass-velocity units)
any of Lx,Ly,Lz can be an atom-style variable (see below)
•
LAMMPS Users Manual
631
omega values = Wx Wy Wz
Wx,Wy,Wz = components of angular velocity vector (radians/time units)
any of wx,wy,wz can be an atom-style variable (see below)
mass value = per-atom mass (mass units)
value can be an atom-style variable (see below)
density value = particle density for a sphere or ellipsoid (mass/distance^3 units), or for a triangle (mass/distance^2 units) or line (mass/distance units) particle
value can be an atom-style variable (see below)
density/disc value = particle density for a 2d disc or ellipse (mass/distance^2 units)
value can be an atom-style variable (see below)
volume value = particle volume for Peridynamic particle (distance^3 units)
value can be an atom-style variable (see below)
image nx ny nz
nx,ny,nz = which periodic image of the simulation box the atom is in
bond value = bond type for all bonds between selected atoms
angle value = angle type for all angles between selected atoms
dihedral value = dihedral type for all dihedrals between selected atoms
improper value = improper type for all impropers between selected atoms
meso/e value = energy of SPH particles (need units)
value can be an atom-style variable (see below)
meso/cv value = heat capacity of SPH particles (need units)
value can be an atom-style variable (see below)
meso/rho value = density of SPH particles (need units)
value can be an atom-style variable (see below)
smd/contact/radius = radius for short range interactions, i.e. contact and friction
value can be an atom-style variable (see below)
smd/mass/density = set particle mass based on volume by providing a mass density
value can be an atom-style variable (see below)
dpd/theta value = internal temperature of DPD particles (temperature units)
value can be an atom-style variable (see below)
value can be NULL which sets internal temp of each particle to KE temp
i_name value = value for custom integer vector with name
d_name value = value for custom floating-point vector with name
Examples:
set group solvent type 2
set group solvent type/fraction 2 0.5 12393
set group edge bond 4
set region half charge 0.5
set type 3 charge 0.5
set type 1*3 charge 0.5
set atom * charge v_atomfile
set atom 100*200 x 0.5 y 1.0
set atom 1492 type 3
Description:
Set one or more properties of one or more atoms. Since atom properties are initially assigned by the read_data,
read_restart or create_atoms commands, this command changes those assignments. This can be useful for
overriding the default values assigned by the create_atoms command (e.g. charge = 0.0). It can be useful for
altering pairwise and molecular force interactions, since force-field coefficients are defined in terms of types.
It can be used to change the labeling of atoms by atom type or molecule ID when they are output in dump
files. It can also be useful for debugging purposes; i.e. positioning an atom at a precise location to compute
subsequent forces or energy.
Note that the style and ID arguments determine which atoms have their properties reset. The remaining
keywords specify which properties to reset and what the new values are. Some strings like type or mol can be
used as a style and/or a keyword.
LAMMPS Users Manual
632

This section describes how to select which atoms to change the properties of, via the style and ID arguments.
The style atom selects all the atoms in a range of atom IDs. The style type selects all the atoms in a range of
types. The style mol selects all the atoms in a range of molecule IDs.
In each of the range cases, the range can be specified as a single numeric value, or a wildcard asterisk can be
used to specify a range of values. This takes the form "*" or "*n" or "n*" or "m*n". For example, for the style
type, if N = the number of atom types, then an asterisk with no numeric values means all types from 1 to N. A
leading asterisk means all types from 1 to n (inclusive). A trailing asterisk means all types from n to N
(inclusive). A middle asterisk means all types from m to n (inclusive). For all the styles except mol, the lowest
value for the wildcard is 1; for mol it is 0.
The style group selects all the atoms in the specified group. The style region selects all the atoms in the
specified geometric region. See the group and region commands for details of how to specify a group or
region.
This section describes the keyword options for which properties to change, for the selected atoms.
Note that except where explicitly prohibited below, all of the keywords allow an atom-style or atomfile-style
variable to be used as the specified value(s). If the value is a variable, it should be specified as v_name, where
name is the variable name. In this case, the variable will be evaluated, and its resulting per-atom value used to
determine the value assigned to each selected atom. Note that the per-atom value from the variable will be
ignored for atoms that are not selected via the style and ID settings explained above. A simple way to use
per-atom values from the variable to reset a property for all atoms is to use style atom with ID = "*"; this
selects all atom IDs.
Atom-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. They can also include
per-atom values, such as atom coordinates. Thus it is easy to specify a time-dependent or spatially-dependent
set of per-atom values. As explained on the variable doc page, atomfile-style variables can be used in place of
atom-style variables, and thus as arguments to the set command. Atomfile-style variables read their per-atoms
values from a file.
NOTE: Atom-style and atomfile-style variables return floating point per-atom values. If the values are
assigned to an integer variable, such as the molecule ID, then the floating point value is truncated to its integer
portion, e.g. a value of 2.6 would become 2.
Keyword type sets the atom type for all selected atoms. The specified value must be from 1 to ntypes, where
ntypes was set by the create_box command or the atom types field in the header of the data file read by the
read_data command.
Keyword type/fraction sets the atom type for a fraction of the selected atoms. The actual number of atoms
changed is not guaranteed to be exactly the requested fraction, but should be statistically close. Random
numbers are used in such a way that a particular atom is changed or not changed, regardless of how many
processors are being used. This keyword does not allow use of an atom-style variable.
Keyword mol sets the molecule ID for all selected atoms. The atom style being used must support the use of
molecule IDs.
LAMMPS Users Manual
633
Keywords x, y, z, and charge set the coordinates or charge of all selected atoms. For charge, the atom style
being used must support the use of atomic charge.
Keyword dipole uses the specified x,y,z values as components of a vector to set as the orientation of the
dipole moment vectors of the selected atoms. The magnitude of the dipole moment is set by the length of this
orientation vector.
Keyword dipole/random randomizes the orientation of the dipole moment vectors for the selected atoms and
sets the magnitude of each to the specified Dlen value. For 2d systems, the z component of the orientation is
set to 0.0. Random numbers are used in such a way that the orientation of a particular atom is the same,
regardless of how many processors are being used. This keyword does not allow use of an atom-style variable.
Keyword quat uses the specified values to create a quaternion (4-vector) that represents the orientation of the
selected atoms. The particles must define a quaternion for their orientation (e.g. ellipsoids, triangles, body
particles) as defined by the atom_style command. Note that particles defined by atom_style ellipsoid have 3
shape parameters. The 3 values must be non-zero for each particle set by this command. They are used to
specify the aspect ratios of an ellipsoidal particle, which is oriented by default with its x-axis along the
simulation box's x-axis, and similarly for y and z. If this body is rotated (via the right-hand rule) by an angle
theta around a unit rotation vector (a,b,c), then the quaternion that represents its new orientation is given by
(cos(theta/2), a*sin(theta/2), b*sin(theta/2), c*sin(theta/2)). The theta and a,b,c values are the arguments to the
quat keyword. LAMMPS normalizes the quaternion in case (a,b,c) was not specified as a unit vector. For 2d
systems, the a,b,c values are ignored, since a rotation vector of (0,0,1) is the only valid choice.
Keyword quat/random randomizes the orientation of the quaternion for the selected atoms. The particles must
define a quaternion for their orientation (e.g. ellipsoids, triangles, body particles) as defined by the atom_style
command. Random numbers are used in such a way that the orientation of a particular atom is the same,
regardless of how many processors are being used. For 2d systems, only orientations in the xy plane are
generated. As with keyword quat, for ellipsoidal particles, the 3 shape values must be non-zero for each
particle set by this command. This keyword does not allow use of an atom-style variable.
Keyword diameter sets the size of the selected atoms. The particles must be finite-size spheres as defined by
the atom_style sphere command. The diameter of a particle can be set to 0.0, which means they will be treated
as point particles. Note that this command does not adjust the particle mass, even if it was defined with a
density, e.g. via the read_data command.
Keyword shape sets the size and shape of the selected atoms. The particles must be ellipsoids as defined by
the atom_style ellipsoid command. The Sx, Sy, Sz settings are the 3 diameters of the ellipsoid in each
direction. All 3 can be set to the same value, which means the ellipsoid is effectively a sphere. They can also
all be set to 0.0 which means the particle will be treated as a point particle. Note that this command does not
adjust the particle mass, even if it was defined with a density, e.g. via the read_data command.
Keyword length sets the length of selected atoms. The particles must be line segments as defined by the
atom_style line command. If the specified value is non-zero the line segment is (re)set to a length = the
specified value, centered around the particle position, with an orientation along the x-axis. If the specified
value is 0.0, the particle will become a point particle. Note that this command does not adjust the particle
mass, even if it was defined with a density, e.g. via the read_data command.
Keyword tri sets the size of selected atoms. The particles must be triangles as defined by the atom_style tri
command. If the specified value is non-zero the triangle is (re)set to be an equilateral triangle in the xy plane
with side length = the specified value, with a centroid at the particle position, with its base parallel to the x
axis, and the y-axis running from the center of the base to the top point of the triangle. If the specified value is
LAMMPS Users Manual
634
0.0, the particle will become a point particle. Note that this command does not adjust the particle mass, even if
it was defined with a density, e.g. via the read_data command.
Keyword theta sets the orientation of selected atoms. The particles must be line segments as defined by the
atom_style line command. The specified value is used to set the orientation angle of the line segments with
respect to the x axis.
Keyword theta/random randomizes the orientation of theta for the selected atoms. The particles must be line
segments as defined by the atom_style line command. Random numbers are used in such a way that the
orientation of a particular atom is the same, regardless of how many processors are being used. This keyword
does not allow use of an atom-style variable.
Keyword angmom sets the angular momentum of selected atoms. The particles must be ellipsoids as defined
by the atom_style ellipsoid command or triangles as defined by the atom_style tri command. The angular
momentum vector of the particles is set to the 3 specified components.
Keyword omega sets the angular velocity of selected atoms. The particles must be spheres as defined by the
atom_style sphere atom_style.html command. The angular velocity vector of the particles is set to the 3
specified components.
Keyword mass sets the mass of all selected particles. The particles must have a per-atom mass attribute, as
defined by the atom_style command. See the "mass" command for how to set mass values on a per-type basis.
Keyword density or density/disc also sets the mass of all selected particles, but in a different way. The
particles must have a per-atom mass attribute, as defined by the atom_style command. If the atom has a radius
attribute (see atom_style sphere) and its radius is non-zero, its mass is set from the density and particle
volume for 3d systems (the input density is assumed to be in mass/distance^3 units). For 2d, the default is for
LAMMPS to model particles with a radius attribute as spheres. However, if the density/disc keyword is used,
then they can be modeled as 2d discs (circles). Their mass is set from the density and particle area (the input
density is assumed to be in mass/distance^2 units).
If the atom has a shape attribute (see atom_style ellipsoid) and its 3 shape parameters are non-zero, then its
mass is set from the density and particle volume (the input density is assumed to be in mass/distance^3 units).
The density/disc keyword has no effect; it does not (yet) treat 3d ellipsoids as 2d ellipses.
If the atom has a length attribute (see atom_style line) and its length is non-zero, then its mass is set from the
density and line segment length (the input density is assumed to be in mass/distance units). If the atom has an
area attribute (see atom_style tri) and its area is non-zero, then its mass is set from the density and triangle
area (the input density is assumed to be in mass/distance^2 units).
If none of these cases are valid, then the mass is set to the density value directly (the input density is assumed
to be in mass units).
Keyword volume sets the volume of all selected particles. Currently, only the atom_style peri command
defines particles with a volume attribute. Note that this command does not adjust the particle mass.
Keyword image sets which image of the simulation box the atom is considered to be in. An image of 0 means
it is inside the box as defined. A value of 2 means add 2 box lengths to get the true value. A value of -1 means
subtract 1 box length to get the true value. LAMMPS updates these flags as atoms cross periodic boundaries
during the simulation. The flags can be output with atom snapshots via the dump command. If a value of
NULL is specified for any of nx,ny,nz, then the current image value for that dimension is unchanged. For
LAMMPS Users Manual
635
non-periodic dimensions only a value of 0 can be specified. This keyword does not allow use of atom-style
variables. This command can be useful after a system has been equilibrated and atoms have diffused one or
more box lengths in various directions. This command can then reset the image values for atoms so that they
are effectively inside the simulation box, e.g if a diffusion coefficient is about to be measured via the compute
msd command. Care should be taken not to reset the image flags of two atoms in a bond to the same value if
the bond straddles a periodic boundary (rather they should be different by +/- 1). This will not affect the
dynamics of a simulation, but may mess up analysis of the trajectories if a LAMMPS diagnostic or your own
analysis relies on the image flags to unwrap a molecule which straddles the periodic box.
Keywords bond, angle, dihedral, and improper, set the bond type (angle type, etc) of all bonds (angles, etc) of
selected atoms to the specified value from 1 to nbondtypes (nangletypes, etc). All atoms in a particular bond
(angle, etc) must be selected atoms in order for the change to be made. The value of nbondtype (nangletypes,
etc) was set by the bond types (angle types, etc) field in the header of the data file read by the read_data
command. These keywords do not allow use of an atom-style variable.
Keywords meso/e, meso/cv, and meso/rho set the energy, heat capacity, and density of smoothed particle
hydrodynamics (SPH) particles. See this PDF guide to using SPH in LAMMPS.
Keyword smd/mass/density sets the mass of all selected particles, but it is only applicable to the Smooth Mach
Dynamics package USER-SMD. It assumes that the particle volume has already been correctly set and
calculates particle mass from the provided mass density value.
Keyword smd/contact/radius only applies to simulations with the Smooth Mach Dynamics package
USER-SMD. Itsets an interaction radius for computing short-range interactions, e.g. repulsive forces to
prevent different individual physical bodies from penetrating each other. Note that the SPH smoothing kernel
diameter used for computing long range, nonlocal interactions, is set using the diameter keyword.
Keyword dpd/theta sets the internal temperature of a DPD particle as defined by the USER-DPD package. If
the specified value is a number it must be >= 0.0. If the specified value is NULL, then the kinetic temperature
Tkin of each particle is computed as 3/2 k Tkin = KE = 1/2 m v^2 = 1/2 m (vx*vx+vy*vy+vz*vz). Each
particle's internal temperature is set to Tkin. If the specified value is an atom-style variable, then the variable
is evaluated for each particle. If a value >= 0.0, the internal temperature is set to that value. If it is < 0.0, the
computation of Tkin is performed and the internal temperature is set to that value.
Keywords i_name and d_name refer to custom integer and floating-point properties that have been added to
each atom via the fix property/atom command. When that command is used specific names are given to each
attribute which are what is specified as the "name" portion of i_name or d_name.
Restrictions:
You cannot set an atom attribute (e.g. mol or q or volume) if the atom_style does not have that attribute.
This command requires inter-processor communication to coordinate the setting of bond types (angle types,
etc). This means that your system must be ready to perform a simulation before using one of these keywords
(force fields set, atom mass set, etc). This is not necessary for other keywords.
Using the region style with the bond (angle, etc) keywords can give unpredictable results if there are bonds
(angles, etc) that straddle periodic boundaries. This is because the region may only extend up to the boundary
and partner atoms in the bond (angle, etc) may have coordinates outside the simulation box if they are ghost
atoms.
LAMMPS Users Manual
636

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
shell command
Syntax:
shell cmd args
cmd = cd or mkdir or mv or rm or rmdir or putenv or arbitrary command
cd arg = dir
dir = directory to change to
mkdir args = dir1 dir2 ...
dir1,dir2 = one or more directories to create
mv args = old new
old = old filename
new = new filename
rm args = file1 file2 ...
file1,file2 = one or more filenames to delete
rmdir args = dir1 dir2 ...
dir1,dir2 = one or more directories to delete
putenv args = var1=value1 var2=value2
var=value = one of more definitions of environment variables
anything else is passed as a command to the shell for direct execution
•
Examples:
shell cd sub1
shell cd ..
shell mkdir tmp1 tmp2 tmp3
shell rmdir tmp1
shell mv log.lammps hold/log.1
shell rm TMP/file1 TMP/file2
shell putenv LAMMPS_POTENTIALS=../../potentials
shell my_setup file1 10 file2
shell my_post_process 100 dump.out
Description:
Execute a shell command. A few simple file-based shell commands are supported directly, in Unix-style
syntax. Any command not listed above is passed as-is to the C-library system() call, which invokes the
command in a shell.
This is means to invoke other commands from your input script. For example, you can move files around in
preparation for the next section of the input script. Or you can run a program that pre-processes data for input
into LAMMPS. Or you can run a program that post-processes LAMMPS output data.
With the exception of cd, all commands, including ones invoked via a system() call, are executed by only a
single processor, so that files/directories are not being manipulated by multiple processors.
The cd cmd executes the Unix "cd" command to change the working directory. All subsequent LAMMPS
commands that read/write files will use the new directory. All processors execute this command.
The mkdir cmd executes the Unix "mkdir" command to create one or more directories.
LAMMPS Users Manual
638
The mv cmd executes the Unix "mv" command to rename a file and/or move it to a new directory.
The rm cmd executes the Unix "rm" command to remove one or more files.
The rmdir cmd executes the Unix "rmdir" command to remove one or more directories. A directory must be
empty to be successfully removed.
The putenv cmd defines or updates an environment variable directly. Since this command does not pass
through the shell, no shell variable expansion or globbing is performed, only the usual substitution for
LAMMPS variables defined with the variable command is performed. The resulting string is then used
literally.
Any other cmd is passed as-is to the shell along with its arguments as one string, invoked by the C-library
system() call. For example, these lines in your input script:
variable n equal 10
variable foo string file2
shell my_setup file1 $n ${foo}
would be the same as invoking
% my_setup file1 10 file2
from a command-line prompt. The executable program "my_setup" is run with 3 arguments: file1 10 file2.
Restrictions:
LAMMPS does not detect errors or print warnings when any of these commands execute. E.g. if the specified
directory does not exist, executing the cd command will silently do nothing.
Related commands: none
Default: none
LAMMPS Users Manual
639

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
special_bonds command
Syntax:
special_bonds keyword values ...
one or more keyword/value pairs may be appended• keyword = amber or charmm or dreiding or fene or lj/coul or lj or coul or angle or dihedral or extra
amber values = none
charmm values = none
dreiding values = none
fene values = none
lj/coul values = w1,w2,w3
w1,w2,w3 = weights (0.0 to 1.0) on pairwise Lennard-Jones and Coulombic interactions
lj values = w1,w2,w3
w1,w2,w3 = weights (0.0 to 1.0) on pairwise Lennard-Jones interactions
coul values = w1,w2,w3
w1,w2,w3 = weights (0.0 to 1.0) on pairwise Coulombic interactions
angle value = yes or no
dihedral value = yes or no
extra value = N
N = number of extra 1-2,1-3,1-4 interactions to save space for
•
Examples:
special_bonds amber
special_bonds charmm
special_bonds fene dihedral no
special_bonds lj/coul 0.0 0.0 0.5 angle yes dihedral yes
special_bonds lj 0.0 0.0 0.5 coul 0.0 0.0 0.0 dihedral yes
special_bonds lj/coul 0 1 1 extra 2
Description:
Set weighting coefficients for pairwise energy and force contributions between pairs of atoms that are also
permanently bonded to each other, either directly or via one or two intermediate bonds. These weighting
factors are used by nearly all pair styles in LAMMPS that compute simple pairwise interactions. Permanent
bonds between atoms are specified by defining the bond topology in the data file read by the read_data
command. Typically a bond_style command is also used to define a bond potential. The rationale for using
these weighting factors is that the interaction between a pair of bonded atoms is all (or mostly) specified by
the bond, angle, dihedral potentials, and thus the non-bonded Lennard-Jones or Coulombic interaction
between the pair of atoms should be excluded (or reduced by a weighting factor).
NOTE: These weighting factors are NOT used by pair styles that compute many-body interactions, since the
"bonds" that result from such interactions are not permanent, but are created and broken dynamically as atom
conformations change. Examples of pair styles in this category are EAM, MEAM, Stillinger-Weber, Tersoff,
COMB, AIREBO, and ReaxFF. In fact, it generally makes no sense to define permanent bonds between atoms
that interact via these potentials, though such bonds may exist elsewhere in your system, e.g. when using the
pair_style hybrid command. Thus LAMMPS ignores special_bonds settings when manybody potentials are
calculated.
LAMMPS Users Manual
640
NOTE: Unlike some commands in LAMMPS, you cannot use this command multiple times in an incremental
fashion: e.g. to first set the LJ settings and then the Coulombic ones. Each time you use this command it sets
all the coefficients to default values and only overrides the one you specify, so you should set all the options
you need each time you use it. See more details at the bottom of this page.
The Coulomb factors are applied to any Coulomb (charge interaction) term that the potential calculates. The
LJ factors are applied to the remaining terms that the potential calculates, whether they represent LJ
interactions or not. The weighting factors are a scaling pre-factor on the energy and force between the pair of
atoms. A value of 1.0 means include the full interaction; a value of 0.0 means exclude it completely.
The 1st of the 3 coefficients (LJ or Coulombic) is the weighting factor on 1-2 atom pairs, which are pairs of
atoms directly bonded to each other. The 2nd coefficient is the weighting factor on 1-3 atom pairs which are
those separated by 2 bonds (e.g. the two H atoms in a water molecule). The 3rd coefficient is the weighting
factor on 1-4 atom pairs which are those separated by 3 bonds (e.g. the 1st and 4th atoms in a dihedral
interaction). Thus if the 1-2 coefficient is set to 0.0, then the pairwise interaction is effectively turned off for
all pairs of atoms bonded to each other. If it is set to 1.0, then that interaction will be at full strength.
NOTE: For purposes of computing weighted pairwise interactions, 1-3 and 1-4 interactions are not defined
from the list of angles or dihedrals used by the simulation. Rather, they are inferred topologically from the set
of bonds specified when the simulation is defined from a data or restart file (see read_data or read_restart
commands). Thus the set of 1-2,1-3,1-4 interactions that the weights apply to is the same whether angle and
dihedral potentials are computed or not, and remains the same even if bonds are constrained, or turned off, or
removed during a simulation.
The two exceptions to this rule are (a) if the angle or dihedral keywords are set to yes (see below), or (b) if the
delete_bonds command is used with the special option that recomputes the 1-2,1-3,1-4 topologies after bonds
are deleted; see the delete_bonds command for more details.
The amber keyword sets the 3 coefficients to 0.0, 0.0, 0.5 for LJ interactions and to 0.0, 0.0, 0.8333 for
Coulombic interactions, which is the default for a commonly used version of the AMBER force field, where
the last value is really 5/6. See (Cornell) for a description of the AMBER force field.
The charmm keyword sets the 3 coefficients to 0.0, 0.0, 0.0 for both LJ and Coulombic interactions, which is
the default for a commonly used version of the CHARMM force field. Note that in pair styles
lj/charmm/coul/charmm and lj/charmm/coul/long the 1-4 coefficients are defined explicitly, and these
pairwise contributions are computed as part of the charmm dihedral style - see the pair_coeff and
dihedral_style commands for more information. See (MacKerell) for a description of the CHARMM force
field.
The dreiding keyword sets the 3 coefficients to 0.0, 0.0, 1.0 for both LJ and Coulombic interactions, which is
the default for the Dreiding force field, as discussed in (Mayo).
The fene keyword sets the 3 coefficients to 0.0, 1.0, 1.0 for both LJ and Coulombic interactions, which is
consistent with a coarse-grained polymer model with FENE bonds. See (Kremer) for a description of FENE
bonds.
The lj/coul, lj, and coul keywords allow the 3 coefficients to be set explicitly. The lj/coul keyword sets both
the LJ and Coulombic coefficients to the same 3 values. The lj and coul keywords only set either the LJ or
Coulombic coefficients. Use both of them if you wish to set the LJ coefficients to different values than the
Coulombic coefficients.
LAMMPS Users Manual
641

The angle keyword allows the 1-3 weighting factor to be ignored for individual atom pairs if they are not
listed as the first and last atoms in any angle defined in the simulation or as 1,3 or 2,4 atoms in any dihedral
defined in the simulation. For example, imagine the 1-3 weighting factor is set to 0.5 and you have a linear
molecule with 4 atoms and bonds as follows: 1-2-3-4. If your data file defines 1-2-3 as an angle, but does not
define 2-3-4 as an angle or 1-2-3-4 as a dihedral, then the pairwise interaction between atoms 1 and 3 will
always be weighted by 0.5, but different force fields use different rules for weighting the pairwise interaction
between atoms 2 and 4. If the angle keyword is specified as yes, then the pairwise interaction between atoms 2
and 4 will be unaffected (full weighting of 1.0). If the angle keyword is specified as no which is the default,
then the 2,4 interaction will also be weighted by 0.5.
The dihedral keyword allows the 1-4 weighting factor to be ignored for individual atom pairs if they are not
listed as the first and last atoms in any dihedral defined in the simulation. For example, imagine the 1-4
weighting factor is set to 0.5 and you have a linear molecule with 5 atoms and bonds as follows: 1-2-3-4-5. If
your data file defines 1-2-3-4 as a dihedral, but does not define 2-3-4-5 as a dihedral, then the pairwise
interaction between atoms 1 and 4 will always be weighted by 0.5, but different force fields use different rules
for weighting the pairwise interaction between atoms 2 and 5. If the dihedral keyword is specified as yes, then
the pairwise interaction between atoms 2 and 5 will be unaffected (full weighting of 1.0). If the dihedral
keyword is specified as no which is the default, then the 2,5 interaction will also be weighted by 0.5.
The extra keyword can be used when additional bonds will be created during a simulation run, e.g. by the fix
bond/create command. It can also be used if molecules will be added to the system, e.g. via the fix deposit, or
fix pour commands, which will have atoms with more special neighbors than any atom in the current system
has.
NOTE: LAMMPS stores and maintains a data structure with a list of the 1st, 2nd, and 3rd neighbors of each
atom (within the bond topology of the system). If new bonds are created (or molecules added containing
atoms with more special neighbors), the size of this list needs to grow. Note that adding a single bond always
adds a new 1st neighbor but may also induce *many* new 2nd and 3rd neighbors, depending on the molecular
topology of your system. Using the extra keyword leaves empty space in the list for this N additional 1st, 2nd,
or 3rd neighbors to be added. If you do not do this, you may get an error when bonds (or molecules) are
added.
NOTE: If you reuse this command in an input script, you should set all the options you need each time. This
command cannot be used a 2nd time incrementally, e.g. to add some extra storage locations via the extra
keyword. E.g. these two commands:
special_bonds lj 0.0 1.0 1.0 special_bonds coul 0.0 0.0 1.0
are not the same as
special_bonds lj 0.0 1.0 1.0 coul 0.0 0.0 1.0
In the first case you end up with (after the 2nd command):
LJ: 0.0 0.0 0.0 Coul: coul 0.0 0.0 1.0
because the LJ settings are reset to their default values each time the command is issued.
Likewise
special_bonds amber
LAMMPS Users Manual
642

special_bonds extra 2
is not the same as this single command:
special_bonds amber extra 2
since in the former case, the 2nd command will reset all the LJ and Coulombic weights to 0.0 (the default).
One exception to this rule is the extra option itself. It is not reset to its default value of 0 each time the
special_bonds command is invoked. This is because it can also be set by the read_data and create_box
commands, so this command will not override those settings unless you explicitly use extra as an option.
Restrictions: none
Related commands:
delete_bonds, fix bond/create
Default:
All 3 Lennard-Jones and 3 Coulombic weighting coefficients = 0.0, angle = no, dihedral = no, and extra = 0.
(Cornell) Cornell, Cieplak, Bayly, Gould, Merz, Ferguson, Spellmeyer, Fox, Caldwell, Kollman, JACS 117,
5179-5197 (1995).
(Kremer) Kremer, Grest, J Chem Phys, 92, 5057 (1990).
(MacKerell) MacKerell, Bashford, Bellott, Dunbrack, Evanseck, Field, Fischer, Gao, Guo, Ha, et al, J Phys
Chem, 102, 3586 (1998).
(Mayo) Mayo, Olfason, Goddard III, J Phys Chem, 94, 8897-8909 (1990).
LAMMPS Users Manual
643

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
suffix command
Syntax:
suffix style args
style = off or on or gpu or intel or kk or omp or opt or hybrid• args = for hybrid style, default suffix to be used and alternative suffix•
Examples:
suffix off
suffix on
suffix gpu
suffix intel
suffix hybrid intel omp
suffix kk
Description:
This command allows you to use variants of various styles if they exist. In that respect it operates the same as
the -suffix command-line switch. It also has options to turn off or back on any suffix setting made via the
command line.
The specified style can be gpu, intel, kk, omp, opt or hybrid. These refer to optional packages that LAMMPS
can be built with, as described in this section of the manual. The "gpu" style corresponds to the GPU package,
the "intel" style to the USER-INTEL package, the "kk" style to the KOKKOS package, the "omp" style to the
USER-OMP package, and the "opt" style to the OPT package.
These are the variants these packages provide:
GPU = a handful of pair styles and the PPPM kspace_style, optimized to run on one or more GPUs or
multicore CPU/GPU nodes
•
USER-INTEL = a collection of pair styles and neighbor routines optimized to run in single, mixed, or
double precision on CPUs and Intel(R) Xeon Phi(TM) coprocessors.
•
KOKKOS = a collection of atom, pair, and fix styles optimized to run using the Kokkos library on
various kinds of hardware, including GPUs via Cuda and many-core chips via OpenMP or threading.
•
USER-OMP = a collection of pair, bond, angle, dihedral, improper, kspace, compute, and fix styles
with support for OpenMP multi-threading
•
OPT = a handful of pair styles, cache-optimized for faster CPU performance• HYBRID = a combination of two packages can be specified (see below)•
As an example, all of the packages provide a pair_style lj/cut variant, with style names lj/cut/opt, lj/cut/omp,
lj/cut/gpu, lj/cut/intel, or lj/cut/kk. A variant styles can be specified explicitly in your input script, e.g.
pair_style lj/cut/gpu. If the suffix command is used with the appropriate style, you do not need to modify your
input script. The specified suffix (opt,omp,gpu,intel,kk) is automatically appended whenever your input script
command creates a new atom, pair, bond, angle, dihedral, improper, kspace, fix, compute, or run style. If the
variant version does not exist, the standard version is created.
LAMMPS Users Manual
644
For "hybrid", two packages are specified. The first is used whenever available. If a style with the first suffix is
not available, the style with the suffix for the second package will be used if available. For example, "hybrid
intel omp" will use styles from the USER-INTEL package as a first choice and styles from the USER-OMP
package as a second choice if no USER-INTEL variant is available.
If the specified style is off, then any previously specified suffix is temporarily disabled, whether it was
specified by a command-line switch or a previous suffix command. If the specified style is on, a disabled
suffix is turned back on. The use of these 2 commands lets your input script use a standard LAMMPS style
(i.e. a non-accelerated variant), which can be useful for testing or benchmarking purposes. Of course this is
also possible by not using any suffix commands, and explicitly appending or not appending the suffix to the
relevant commands in your input script.
NOTE: The default run_style verlet is invoked prior to reading the input script and is therefore not affected by
a suffix command in the input script. The KOKKOS package requires "run_style verlet/kk", so when using the
KOKKOS package it is necessary to either use the command line "-sf kk" command or add an explicit
"run_style verlet" command to the input script.
Restrictions: none
Related commands:
Command-line switch -suffix
Default: none
LAMMPS Users Manual
645

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
tad command
Syntax:
tad N t_event T_lo T_hi delta tmax compute-ID keyword value ...
N = # of timesteps to run (not including dephasing/quenching)• t_event = timestep interval between event checks• T_lo = temperature at which event times are desired• T_hi = temperature at which MD simulation is performed• delta = desired confidence level for stopping criterion• tmax = reciprocal of lowest expected preexponential factor (time units)• compute-ID = ID of the compute used for event detection• zero or more keyword/value pairs may be appended• keyword = min or neb or min_style or neb_style or neb_log
min values = etol ftol maxiter maxeval
etol = stopping tolerance for energy (energy units)
ftol = stopping tolerance for force (force units)
maxiter = max iterations of minimize
maxeval = max number of force/energy evaluations
neb values = ftol N1 N2 Nevery
etol = stopping tolerance for energy (energy units)
ftol = stopping tolerance for force (force units)
N1 = max # of iterations (timesteps) to run initial NEB
N2 = max # of iterations (timesteps) to run barrier-climbing NEB
Nevery = print NEB statistics every this many timesteps
neb_style value = quickmin or fire
neb_step value = dtneb
dtneb = timestep for NEB damped dynamics minimization
neb_log value = file where NEB statistics are printed
•
Examples:
tad 2000 50 1800 2300 0.01 0.01 event
tad 2000 50 1800 2300 0.01 0.01 event &
min 1e-05 1e-05 100 100 &
neb 0.0 0.01 200 200 20 &
min_style cg &
neb_style fire &
neb_log log.neb
Description:
Run a temperature accelerated dynamics (TAD) simulation. This method requires two or more partitions to
perform NEB transition state searches.
TAD is described in this paper by Art Voter. It is a method that uses accelerated dynamics at an elevated
temperature to generate results at a specified lower temperature. A good overview of accelerated dynamics
methods for such systems is given in this review paper from the same group. In general, these methods
assume that the long-time dynamics is dominated by infrequent events i.e. the system is is confined to low
energy basins for long periods, punctuated by brief, randomly-occurring transitions to adjacent basins. TAD is
LAMMPS Users Manual
646
suitable for infrequent-event systems, where in addition, the transition kinetics are well-approximated by
harmonic transition state theory (hTST). In hTST, the temperature dependence of transition rates follows the
Arrhenius relation. As a consequence a set of event times generated in a high-temperature simulation can be
mapped to a set of much longer estimated times in the low-temperature system. However, because this
mapping involves the energy barrier of the transition event, which is different for each event, the first event at
the high temperature may not be the earliest event at the low temperature. TAD handles this by first
generating a set of possible events from the current basin. After each event, the simulation is reflected
backwards into the current basin. This is repeated until the stopping criterion is satisfied, at which point the
event with the earliest low-temperature occurrence time is selected. The stopping criterion is that the
confidence measure be greater than 1-delta. The confidence measure is the probability that no earlier
low-temperature event will occur at some later time in the high-temperature simulation. hTST provides an
lower bound for this probability, based on the user-specified minimum pre-exponential factor (reciprocal of
tmax).
In order to estimate the energy barrier for each event, the TAD method invokes the NEB method. Each NEB
replica runs on a partition of processors. The current NEB implementation in LAMMPS restricts you to
having exactly one processor per replica. For more information, see the documentation for the neb command.
In the current LAMMPS implementation of TAD, all the non-NEB TAD operations are performed on the first
partition, while the other partitions remain idle. See Section 6.5 of the manual for further discussion of
multi-replica simulations.
A TAD run has several stages, which are repeated each time an event is performed. The logic for a TAD run
is as follows:
while (time remains):
while (time <tstop):
until (event occurs):
run dynamics for t_event steps
quench
run neb calculation using all replicas
compute tlo from energy barrier
update earliest event
update tstop
reflect back into current basin
execute earliest event
Before this outer loop begins, the initial potential energy basin is identified by quenching (an energy
minimization, see below) the initial state and storing the resulting coordinates for reference.
Inside the inner loop, dynamics is run continuously according to whatever integrator has been specified by the
user, stopping every t_event steps to check if a transition event has occurred. This check is performed by
quenching the system and comparing the resulting atom coordinates to the coordinates from the previous
basin.
A quench is an energy minimization and is performed by whichever algorithm has been defined by the
min_style command; its default is the CG minimizer. The tolerances and limits for each quench can be set by
the min keyword. Note that typically, you do not need to perform a highly-converged minimization to detect a
transition event.
The event check is performed by a compute with the specified compute-ID. Currently there is only one
compute that works with the TAD command, which is the compute event/displace command. Other
event-checking computes may be added. Compute event/displace checks whether any atom in the compute
group has moved further than a specified threshold distance. If so, an "event" has occurred.
LAMMPS Users Manual
647

The NEB calculation is similar to that invoked by the neb command, except that the final state is generated
internally, instead of being read in from a file. The style of minimization performed by NEB is determined by
the neb_style keyword and must be a damped dynamics minimizer. The tolerances and limits for each NEB
calculation can be set by the neb keyword. As discussed on the neb, it is often advantageous to use a larger
timestep for NEB than for normal dynamics. Since the size of the timestep set by the timestep command is
used by TAD for performing dynamics, there is a neb_step keyword which can be used to set a larger timestep
for each NEB calculation if desired.
A key aspect of the TAD method is setting the stopping criterion appropriately. If this criterion is too
conservative, then many events must be generated before one is finally executed. Conversely, if this criterion
is too aggressive, high-entropy high-barrier events will be over-sampled, while low-entropy low-barrier events
will be under-sampled. If the lowest pre-exponential factor is known fairly accurately, then it can be used to
estimate tmax, and the value of delta can be set to the desired confidence level e.g. delta = 0.05 corresponds to
95% confidence. However, for systems where the dynamics are not well characterized (the most common
case), it will be necessary to experiment with the values of delta and tmax to get a good trade-off between
accuracy and performance.
A second key aspect is the choice of t_hi. A larger value greatly increases the rate at which new events are
generated. However, too large a value introduces errors due to anharmonicity (not accounted for within
hTST). Once again, for any given system, experimentation is necessary to determine the best value of t_hi.
Five kinds of output can be generated during a TAD run: event statistics, NEB statistics, thermodynamic
output by each replica, dump files, and restart files.
Event statistics are printed to the screen and master log.lammps file each time an event is executed. The
quantities are the timestep, CPU time, global event number N, local event number M, event status, energy
barrier, time margin, t_lo and delt_lo. The timestep is the usual LAMMPS timestep, which corresponds to the
high-temperature time at which the event was detected, in units of timestep. The CPU time is the total
processor time since the start of the TAD run. The global event number N is a counter that increments with
each executed event. The local event number M is a counter that resets to zero upon entering each new basin.
The event status is E when an event is executed, and is D for an event that is detected, while DF is for a
detected event that is also the earliest (first) event at the low temperature.
The time margin is the ratio of the high temperature time in the current basin to the stopping time. This last
number can be used to judge whether the stopping time is too short or too long (see above).
t_lo is the low-temperature event time when the current basin was entered, in units of timestep. delt_lo is the
time of each detected event, measured relative to t_lo. delt_lo is equal to the high-temperature time since
entering the current basin, scaled by an exponential factor that depends on the hi/lo temperature ratio and the
energy barrier for that event.
On lines for executed events, with status E, the global event number is incremented by one, the local event
number and time margin are reset to zero, while the global event number, energy barrier, and delt_lo match
the last event with status DF in the immediately preceding block of detected events. The low-temperature
event time t_lo is incremented by delt_lo.
NEB statistics are written to the file specified by the neb_log keyword. If the keyword value is "none", then
no NEB statistics are printed out. The statistics are written every Nevery timesteps. See the neb command for
a full description of the NEB statistics. When invoked from TAD, NEB statistics are never printed to the
screen.
LAMMPS Users Manual
648

Because the NEB calculation must run on multiple partitions, LAMMPS produces additional screen and log
files for each partition, e.g. log.lammps.0, log.lammps.1, etc. For the TAD command, these contain the
thermodynamic output of each NEB replica. In addition, the log file for the first partition, log.lammps.0, will
contain thermodynamic output from short runs and minimizations corresponding to the dynamics and quench
operations, as well as a line for each new detected event, as described above.
After the TAD command completes, timing statistics for the TAD run are printed in each replica's log file,
giving a breakdown of how much CPU time was spent in each stage (NEB, dynamics, quenching, etc).
Any dump files defined in the input script will be written to during a TAD run at timesteps when an event is
executed. This means the requested dump frequency in the dump command is ignored. There will be one
dump file (per dump command) created for all partitions. The atom coordinates of the dump snapshot are
those of the minimum energy configuration resulting from quenching following the executed event. The
timesteps written into the dump files correspond to the timestep at which the event occurred and NOT the
clock. A dump snapshot corresponding to the initial minimum state used for event detection is written to the
dump file at the beginning of each TAD run.
If the restart command is used, a single restart file for all the partitions is generated, which allows a TAD run
to be continued by a new input script in the usual manner. The restart file is generated after an event is
executed. The restart file contains a snapshot of the system in the new quenched state, including the event
number and the low-temperature time. The restart frequency specified in the restart command is interpreted
differently when performing a TAD run. It does not mean the timestep interval between restart files. Instead it
means an event interval for executed events. Thus a frequency of 1 means write a restart file every time an
event is executed. A frequency of 10 means write a restart file every 10th executed event. When an input
script reads a restart file from a previous TAD run, the new script can be run on a different number of replicas
or processors.
Note that within a single state, the dynamics will typically temporarily continue beyond the event that is
ultimately chosen, until the stopping criterion is satisfied. When the event is eventually executed, the timestep
counter is reset to the value when the event was detected. Similarly, after each quench and NEB minimization,
the timestep counter is reset to the value at the start of the minimization. This means that the timesteps listed
in the replica log files do not always increase monotonically. However, the timestep values printed to the
master log file, dump files, and restart files are always monotonically increasing.
Restrictions:
This command can only be used if LAMMPS was built with the REPLICA package. See the Making
LAMMPS section for more info on packages.
N setting must be integer multiple of t_event.
Runs restarted from restart files written during a TAD run will only produce identical results if the
user-specified integrator supports exact restarts. So fix nvt will produce an exact restart, but fix langevin will
not.
This command cannot be used when any fixes are defined that keep track of elapsed time to perform
time-dependent operations. Examples include the "ave" fixes such as fix ave/chunk. Also fix dt/reset and fix
deposit.
Related commands:
LAMMPS Users Manual
649

compute event/displace, min_modify, min_style, run_style, minimize, temper, neb, prd
Default:
The option defaults are min = 0.1 0.1 40 50, neb = 0.01 100 100 10, neb_style = quickmin, neb_step = the
same timestep set by the timestep command, and neb_log = "none".
(Voter2000) Sorensen and Voter, J Chem Phys, 112, 9599 (2000)
(Voter2002) Voter, Montalenti, Germann, Annual Review of Materials Research 32, 321 (2002).
LAMMPS Users Manual
650

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
temper command
Syntax:
temper N M temp fix-ID seed1 seed2 index
N = total # of timesteps to run• M = attempt a tempering swap every this many steps• temp = initial temperature for this ensemble• fix-ID = ID of the fix that will control temperature during the run• seed1 = random # seed used to decide on adjacent temperature to partner with• seed2 = random # seed for Boltzmann factor in Metropolis swap• index = which temperature (0 to N-1) I am simulating (optional)•
Examples:
temper 100000 100 $t tempfix 0 58728
temper 40000 100 $t tempfix 0 32285 $w
Description:
Run a parallel tempering or replica exchange simulation using multiple replicas (ensembles) of a system. Two
or more replicas must be used.
Each replica runs on a partition of one or more processors. Processor partitions are defined at run-time using
the -partition command-line switch; see Section 2.7 of the manual. Note that if you have MPI installed, you
can run a multi-replica simulation with more replicas (partitions) than you have physical processors, e.g you
can run a 10-replica simulation on one or two processors. You will simply not get the performance speed-up
you would see with one or more physical processors per replica. See this section of the manual for further
discussion.
Each replica's temperature is controlled at a different value by a fix with fix-ID that controls temperature.
Most thermostat fix styles (with and without included time integration) are supported. The command will
print an error message and abort, if the chosen fix is unsupported. The desired temperature is specified by
temp, which is typically a variable previously set in the input script, so that each partition is assigned a
different temperature. See the variable command for more details. For example:
variable t world 300.0 310.0 320.0 330.0
fix myfix all nvt temp $t $t 100.0
temper 100000 100 $t myfix 3847 58382
would define 4 temperatures, and assign one of them to the thermostat used by each replica, and to the temper
command.
As the tempering simulation runs for N timesteps, a temperature swap between adjacent ensembles will be
attempted every M timesteps. If seed1 is 0, then the swap attempts will alternate between odd and even
pairings. If seed1 is non-zero then it is used as a seed in a random number generator to randomly choose an
odd or even pairing each time. Each attempted swap of temperatures is either accepted or rejected based on a
Boltzmann-weighted Metropolis criterion which uses seed2 in the random number generator.
LAMMPS Users Manual
651

As a tempering run proceeds, multiple log files and screen output files are created, one per replica. By default
these files are named log.lammps.M and screen.M where M is the replica number from 0 to N-1, with N = #
of replicas. See the section on command-line switches for info on how to change these names.
The main screen and log file (log.lammps) will list information about which temperature is assigned to each
replica at each thermodynamic output timestep. E.g. for a simulation with 16 replicas:
Running on 16 partitions of processors
Step T0 T1 T2 T3 T4 T5 T6 T7 T8 T9 T10 T11 T12 T13 T14 T15
0 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
500 1 0 3 2 5 4 6 7 8 9 10 11 12 13 14 15
1000 2 0 4 1 5 3 6 7 8 9 10 11 12 14 13 15
1500 2 1 4 0 5 3 6 7 9 8 10 11 12 14 13 15
2000 2 1 3 0 6 4 5 7 10 8 9 11 12 14 13 15
2500 2 1 3 0 6 4 5 7 11 8 9 10 12 14 13 15
...
The column headings T0 to TN-1 mean which temperature is currently assigned to the replica 0 to N-1. Thus
the columns represent replicas and the value in each column is its temperature (also numbered 0 to N-1). For
example, a 0 in the 4th column (column T3, step 2500) means that the 4th replica is assigned temperature 0,
i.e. the lowest temperature. You can verify this time sequence of temperature assignments for the Nth replica
by comparing the Nth column of screen output to the thermodynamic data in the corresponding log.lammps.N
or screen.N files as time proceeds.
You can have each replica create its own dump file in the following manner:
variable rep world 0 1 2 3 4 5 6 7
dump 1 all atom 1000 dump.temper.$rep
NOTE: Each replica's dump file will contain a continuous trajectory for its atoms where the temperature
varies over time as swaps take place involving that replica. If you want a series of dump files, each with
snapshots (from all replicas) that are all at a single temperature, then you will need to post-process the dump
files using the information from the log.lammps file. E.g. you could produce one dump file with snapshots at
300K (from all replicas), another with snapshots at 310K, etc. Note that these new dump files will not contain
"continuous trajectories" for individual atoms, because two successive snapshots (in time) may be from
different replicas.
The last argument index in the temper command is optional and is used when restarting a tempering run from
a set of restart files (one for each replica) which had previously swapped to new temperatures. The index
value (from 0 to N-1, where N is the # of replicas) identifies which temperature the replica was simulating on
the timestep the restart files were written. Obviously, this argument must be a variable so that each partition
has the correct value. Set the variable to the N values listed in the log file for the previous run for the replica
temperatures at that timestep. For example if the log file listed the following for a simulation with 5 replicas:
500000 2 4 0 1 3
then a setting of
variable w world 2 4 0 1 3
would be used to restart the run with a tempering command like the example above with $w as the last
argument.
LAMMPS Users Manual
652

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
temper/grem command
Syntax:
temper/grem N M lambda fix-ID thermostat-ID seed1 seed2 index
N = total # of timesteps to run• M = attempt a tempering swap every this many steps• lambda = initial lambda for this ensemble• fix-ID = ID of fix_grem• thermostat-ID = ID of the thermostat that controls kinetic temperature• seed1 = random # seed used to decide on adjacent temperature to partner with• seed2 = random # seed for Boltzmann factor in Metropolis swap• index = which temperature (0 to N-1) I am simulating (optional)•
Examples:
temper/grem 100000 1000 $lambda fxgREM fxnvt 0 58728
temper/grem 40000 100 $lambda fxgREM fxnpt 0 32285 $walkers
Description:
Run a parallel tempering or replica exchange simulation in LAMMPS partition mode using multiple
generalized replicas (ensembles) of a system defined by fix grem, which stands for the generalized replica
exchange method (gREM) originally developed by (Kim). It uses non-Boltzmann ensembles to sample over
first order phase transitions. The is done by defining replicas with an enthalpy dependent effective
temperature
Two or more replicas must be used. See the temper command for an explanation of how to run replicas on
multiple partitions of one or more processors.
This command is a modification of the temper command and has the same dependencies, restraints, and input
variables which are discussed there in greater detail.
Instead of temperature, this command performs replica exchanges in lambda as per the generalized ensemble
enforced by fix grem. The desired lambda is specified by lambda, which is typically a variable previously set
in the input script, so that each partition is assigned a different temperature. See the variable command for
more details. For example:
variable lambda world 400 420 440 460
fix fxnvt all nvt temp 300.0 300.0 100.0
fix fxgREM all grem $lambda -0.05 -50000 fxnvt
temper 100000 100 $lambda fxgREM fxnvt 3847 58382
would define 4 lambdas with constant kinetic temperature but unique generalized temperature, and assign one
of them to fix grem used by each replica, and to the grem command.
As the gREM simulation runs for N timesteps, a swap between adjacent ensembles will be attempted every M
timesteps. If seed1 is 0, then the swap attempts will alternate between odd and even pairings. If seed1 is
non-zero then it is used as a seed in a random number generator to randomly choose an odd or even pairing
LAMMPS Users Manual
654

each time. Each attempted swap of temperatures is either accepted or rejected based on a Metropolis criterion,
derived for gREM by (Kim), which uses seed2 in the random number generator.
File management works identical to the temper command. Dump files created by this fix contain continuous
trajectories and require post-processing to obtain per-replica information.
The last argument index in the grem command is optional and is used when restarting a run from a set of
restart files (one for each replica) which had previously swapped to new lambda. This is done using a variable.
For example if the log file listed the following for a simulation with 5 replicas:
500000 2 4 0 1 3
then a setting of
variable walkers world 2 4 0 1 3
would be used to restart the run with a grem command like the example above with $walkers as the last
argument. This functionality is identical to temper.
Restrictions:
This command can only be used if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info on packages.
This command must be used with fix grem.
Related commands:
fix grem, temper, variable
Default: none
(Kim) Kim, Keyes, Straub, J Chem Phys, 132, 224107 (2010).
LAMMPS Users Manual
655

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
thermo command
Syntax:
thermo N
N = output thermodynamics every N timesteps• N can be a variable (see below)•
Examples:
thermo 100
Description:
Compute and print thermodynamic info (e.g. temperature, energy, pressure) on timesteps that are a multiple of
N and at the beginning and end of a simulation. A value of 0 will only print thermodynamics at the beginning
and end.
The content and format of what is printed is controlled by the thermo_style and thermo_modify commands.
Instead of a numeric value, N can be specified as an equal-style variable, which should be specified as
v_name, where name is the variable name. In this case, the variable is evaluated at the beginning of a run to
determine the next timestep at which thermodynamic info will be written out. On that timestep, the variable
will be evaluated again to determine the next timestep, etc. Thus the variable should return timestep values.
See the stagger() and logfreq() and stride() math functions for equal-style variables, as examples of useful
functions to use in this context. Other similar math functions could easily be added as options for equal-style
variables.
For example, the following commands will output thermodynamic info at timesteps
0,10,20,30,100,200,300,1000,2000,etc:
variable s equal logfreq(10,3,10)
thermo v_s
Restrictions: none
Related commands:
thermo_style, thermo_modify
Default:
thermo 0
LAMMPS Users Manual
656

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
thermo_modify command
Syntax:
thermo_modify keyword value ...
one or more keyword/value pairs may be listed
keyword = lost or lost/bond or norm or flush or line or format or temp or press:l
lost value = error or warn or ignore
lost/bond value = error or warn or ignore
norm value = yes or no
flush value = yes or no
line value = one or multi
format values = line string, int string, float string, M string, or none
string = C-style format string
M = integer from 1 to N, where N = # of quantities being output
temp value = compute ID that calculates a temperature
press value = compute ID that calculates a pressure
•
Examples:
thermo_modify lost ignore flush yes
thermo_modify temp myTemp format 3 %15.8g
thermo_modify temp myTemp format line "%ld %g %g %15.8g"
thermo_modify line multi format float %g
Description:
Set options for how thermodynamic information is computed and printed by LAMMPS.
NOTE: These options apply to the currently defined thermo style. When you specify a thermo_style
command, all thermodynamic settings are restored to their default values, including those previously reset by
a thermo_modify command. Thus if your input script specifies a thermo_style command, you should use the
thermo_modify command after it.
The lost keyword determines whether LAMMPS checks for lost atoms each time it computes thermodynamics
and what it does if atoms are lost. An atom can be "lost" if it moves across a non-periodic simulation box
boundary or if it moves more than a box length outside the simulation domain (or more than a processor
sub-domain length) before reneighboring occurs. The latter case is typically due to bad dynamics, e.g. too
large a timestep or huge forces and velocities. If the value is ignore, LAMMPS does not check for lost atoms.
If the value is error or warn, LAMMPS checks and either issues an error or warning. The code will exit with
an error and continue with a warning. A warning will only be issued once, the first time an atom is lost. This
can be a useful debugging option.
The lost/bond keyword determines whether LAMMPS throws an error or not if an atom in a bonded
interaction (bond, angle, etc) cannot be found when it creates bonded neighbor lists. By default this is a fatal
error. However in some scenarios it may be desirable to only issue a warning or ignore it and skip the
computation of the missing bond, angle, etc. An example would be when gas molecules in a vapor are drifting
out of the box through a fixed boundary condition (see the boundary command). In this case one atom may be
deleted before the rest of the molecule is, on a later timestep.
LAMMPS Users Manual
657
The norm keyword determines whether various thermodynamic output values are normalized by the number
of atoms or not, depending on whether it is set to yes or no. Different unit styles have different defaults for
this setting (see below). Even if norm is set to yes, a value is only normalized if it is an "extensive" quantity,
meaning that it scales with the number of atoms in the system. For the thermo keywords described by the doc
page for the thermo_style command, all energy-related keywords are extensive, such as pe or ebond or
enthalpy. Other keywords such as temp or press are "intensive" meaning their value is independent (in a
statistical sense) of the number of atoms in the system and thus are never normalized. For thermodynamic
output values extracted from fixes and computes in a thermo_style custom command, the doc page for the
individual fix or compute lists whether the value is "extensive" or "intensive" and thus whether it is
normalized. Thermodynamic output values calculated by a variable formula are assumed to be "intensive" and
thus are never normalized. You can always include a divide by the number of atoms in the variable formula if
this is not the case.
The flush keyword invokes a flush operation after thermodynamic info is written to the log file. This insures
the output in that file is current (no buffering by the OS), even if LAMMPS halts before the simulation
completes.
The line keyword determines whether thermodynamics will be output as a series of numeric values on one
line or in a multi-line format with 3 quantities with text strings per line and a dashed-line header containing
the timestep and CPU time. This modify option overrides the one and multi thermo_style settings.
The format keyword can be used to change the default numeric format of any of quantities the thermo_style
command outputs. All the specified format strings are C-style formats, e.g. as used by the C/C++ printf()
command. The line keyword takes a single argument which is the format string for the entire line of thermo
output, with N fields, which you must enclose in quotes if it is more than one field. The int and float keywords
take a single format argument and are applied to all integer or floating-point quantities output. The setting for
M string also takes a single format argument which is used for the Mth value output in each line, e.g. the 5th
column is output in high precision for "format 5 %20.15g".
The format keyword can be used multiple times. The precedence is that for each value in a line of output, the
M format (if specified) is used, else the int or float setting (if specified) is used, else the line setting (if
specified) for that value is used, else the default setting is used. A setting of none clears all previous settings,
reverting all values to their default format.
NOTE: The thermo output values step and atoms are stored internally as 8-byte signed integers, rather than
the usual 4-byte signed integers. When specifying the format int option you can use a "%d"-style format
identifier in the format string and LAMMPS will convert this to the corresponding 8-byte form when it is
applied to those keywords. However, when specifying the line option or format M string option for step and
natoms, you should specify a format string appropriate for an 8-byte signed integer, e.g. one with "%ld".
The temp keyword is used to determine how thermodynamic temperature is calculated, which is used by all
thermo quantities that require a temperature ("temp", "press", "ke", "etotal", "enthalpy", "pxx", etc). The
specified compute ID must have been previously defined by the user via the compute command and it must be
a style of compute that calculates a temperature. As described in the thermo_style command, thermo output
uses a default compute for temperature with ID = thermo_temp. This option allows the user to override the
default.
The press keyword is used to determine how thermodynamic pressure is calculated, which is used by all
thermo quantities that require a pressure ("press", "enthalpy", "pxx", etc). The specified compute ID must
have been previously defined by the user via the compute command and it must be a style of compute that
calculates a pressure. As described in the thermo_style command, thermo output uses a default compute for
LAMMPS Users Manual
658
pressure with ID = thermo_press. This option allows the user to override the default.
NOTE: If both the temp and press keywords are used in a single thermo_modify command (or in two separate
commands), then the order in which the keywords are specified is important. Note that a pressure compute
defines its own temperature compute as an argument when it is specified. The temp keyword will override this
(for the pressure compute being used by thermodynamics), but only if the temp keyword comes after the press
keyword. If the temp keyword comes before the press keyword, then the new pressure compute specified by
the press keyword will be unaffected by the temp setting.
Restrictions: none
Related commands:
thermo, thermo_style
Default:
The option defaults are lost = error, norm = yes for unit style of lj, norm = no for unit style of real and metal,
flush = no, and temp/press = compute IDs defined by thermo_style.
The defaults for the line and format options depend on the thermo style. For styles "one" and "custom", the
line and format defaults are "one", "%8d", and "%12.8g". For style "multi", the line and format defaults are
"multi", "%8d", and "%14.4f".
LAMMPS Users Manual
659

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
thermo_style command
Syntax:
thermo_style style args
style = one or multi or custom• args = list of arguments for a particular style
one args = none
multi args = none
custom args = list of keywords
possible keywords = step, elapsed, elaplong, dt, time,
cpu, tpcpu, spcpu, cpuremain, part, timeremain,
atoms, temp, press, pe, ke, etotal, enthalpy,
evdwl, ecoul, epair, ebond, eangle, edihed, eimp,
emol, elong, etail,
vol, density, lx, ly, lz, xlo, xhi, ylo, yhi, zlo, zhi,
xy, xz, yz, xlat, ylat, zlat,
bonds, angles, dihedrals, impropers,
pxx, pyy, pzz, pxy, pxz, pyz,
fmax, fnorm, nbuild, ndanger,
cella, cellb, cellc, cellalpha, cellbeta, cellgamma,
c_ID, c_ID[I], c_ID[I][J],
f_ID, f_ID[I], f_ID[I][J],
v_name, v_name[I]
step = timestep
elapsed = timesteps since start of this run
elaplong = timesteps since start of initial run in a series of runs
dt = timestep size
time = simulation time
cpu = elapsed CPU time in seconds since start of this run
tpcpu = time per CPU second
spcpu = timesteps per CPU second
cpuremain = estimated CPU time remaining in run
part = which partition (0 to Npartition-1) this is
timeremain = remaining time in seconds on timer timeout.
atoms = # of atoms
temp = temperature
press = pressure
pe = total potential energy
ke = kinetic energy
etotal = total energy (pe + ke)
enthalpy = enthalpy (etotal + press*vol)
evdwl = VanderWaal pairwise energy (includes etail)
ecoul = Coulombic pairwise energy
epair = pairwise energy (evdwl + ecoul + elong)
ebond = bond energy
eangle = angle energy
edihed = dihedral energy
eimp = improper energy
emol = molecular energy (ebond + eangle + edihed + eimp)
elong = long-range kspace energy
etail = VanderWaal energy long-range tail correction
vol = volume
density = mass density of system
lx,ly,lz = box lengths in x,y,z
•
LAMMPS Users Manual
660
xlo,xhi,ylo,yhi,zlo,zhi = box boundaries
xy,xz,yz = box tilt for triclinic (non-orthogonal) simulation boxes
xlat,ylat,zlat = lattice spacings as calculated by lattice command
bonds,angles,dihedrals,impropers = # of these interactions defined
pxx,pyy,pzz,pxy,pxz,pyz = 6 components of pressure tensor
fmax = max component of force on any atom in any dimension
fnorm = length of force vector for all atoms
nbuild = # of neighbor list builds
ndanger = # of dangerous neighbor list builds
cella,cellb,cellc = periodic cell lattice constants a,b,c
cellalpha, cellbeta, cellgamma = periodic cell angles alpha,beta,gamma
c_ID = global scalar value calculated by a compute with ID
c_ID[I] = Ith component of global vector calculated by a compute with ID, I can include wildcard (see below)
c_ID[I][J] = I,J component of global array calculated by a compute with ID
f_ID = global scalar value calculated by a fix with ID
f_ID[I] = Ith component of global vector calculated by a fix with ID, I can include wildcard (see below)
f_ID[I][J] = I,J component of global array calculated by a fix with ID
v_name = value calculated by an equal-style variable with name
v_name[I] = value calculated by a vector-style variable with name
Examples:
thermo_style multi
thermo_style custom step temp pe etotal press vol
thermo_style custom step temp etotal c_myTemp v_abc
thermo_style custom step temp etotal c_myTemp[*] v_abc
Description:
Set the style and content for printing thermodynamic data to the screen and log file.
Style one prints a one-line summary of thermodynamic info that is the equivalent of "thermo_style custom
step temp epair emol etotal press". The line contains only numeric values.
Style multi prints a multiple-line listing of thermodynamic info that is the equivalent of "thermo_style custom
etotal ke temp pe ebond eangle edihed eimp evdwl ecoul elong press". The listing contains numeric values
and a string ID for each quantity.
Style custom is the most general setting and allows you to specify which of the keywords listed above you
want printed on each thermodynamic timestep. Note that the keywords c_ID, f_ID, v_name are references to
computes, fixes, and equal-style variables that have been defined elsewhere in the input script or can even be
new styles which users have added to LAMMPS (see the Section 10 section of the documentation). Thus the
custom style provides a flexible means of outputting essentially any desired quantity as a simulation proceeds.
All styles except custom have vol appended to their list of outputs if the simulation box volume changes
during the simulation.
The values printed by the various keywords are instantaneous values, calculated on the current timestep.
Time-averaged quantities, which include values from previous timesteps, can be output by using the f_ID
keyword and accessing a fix that does time-averaging such as the fix ave/time command.
Options invoked by the thermo_modify command can be used to set the one- or multi-line format of the
print-out, the normalization of thermodynamic output (total values versus per-atom values for extensive
quantities (ones which scale with the number of atoms in the system), and the numeric precision of each
printed value.
LAMMPS Users Manual
661

NOTE: When you use a "thermo_style" command, all thermodynamic settings are restored to their default
values, including those previously set by a thermo_modify command. Thus if your input script specifies a
thermo_style command, you should use the thermo_modify command after it.
Several of the thermodynamic quantities require a temperature to be computed: "temp", "press", "ke", "etotal",
"enthalpy", "pxx", etc. By default this is done by using a temperature compute which is created when
LAMMPS starts up, as if this command had been issued:
compute thermo_temp all temp
See the compute temp command for details. Note that the ID of this compute is thermo_temp and the group is
all. You can change the attributes of this temperature (e.g. its degrees-of-freedom) via the compute_modify
command. Alternatively, you can directly assign a new compute (that calculates temperature) which you have
defined, to be used for calculating any thermodynamic quantity that requires a temperature. This is done via
the thermo_modify command.
Several of the thermodynamic quantities require a pressure to be computed: "press", "enthalpy", "pxx", etc.
By default this is done by using a pressure compute which is created when LAMMPS starts up, as if this
command had been issued:
compute thermo_press all pressure thermo_temp
See the compute pressure command for details. Note that the ID of this compute is thermo_press and the
group is all. You can change the attributes of this pressure via the compute_modify command. Alternatively,
you can directly assign a new compute (that calculates pressure) which you have defined, to be used for
calculating any thermodynamic quantity that requires a pressure. This is done via the thermo_modify
command.
Several of the thermodynamic quantities require a potential energy to be computed: "pe", "etotal", "ebond",
etc. This is done by using a pe compute which is created when LAMMPS starts up, as if this command had
been issued:
compute thermo_pe all pe
See the compute pe command for details. Note that the ID of this compute is thermo_pe and the group is all.
You can change the attributes of this potential energy via the compute_modify command.
The kinetic energy of the system ke is inferred from the temperature of the system with 1/2 Kb T of energy for
each degree of freedom. Thus, using different compute commands for calculating temperature, via the
thermo_modify temp command, may yield different kinetic energies, since different computes that calculate
temperature can subtract out different non-thermal components of velocity and/or include different degrees of
freedom (translational, rotational, etc).
The potential energy of the system pe will include contributions from fixes if the fix_modify thermo option is
set for a fix that calculates such a contribution. For example, the fix wall/lj93 fix calculates the energy of
atoms interacting with the wall. See the doc pages for "individual fixes" to see which ones contribute.
A long-range tail correction etail for the VanderWaal pairwise energy will be non-zero only if the
pair_modify tail option is turned on. The etail contribution is included in evdwl, epair, pe, and etotal, and the
corresponding tail correction to the pressure is included in press and pxx, pyy, etc.
LAMMPS Users Manual
662
The step, elapsed, and elaplong keywords refer to timestep count. Step is the current timestep, or iteration
count when a minimization is being performed. Elapsed is the number of timesteps elapsed since the
beginning of this run. Elaplong is the number of timesteps elapsed since the beginning of an initial run in a
series of runs. See the start and stop keywords for the run for info on how to invoke a series of runs that keep
track of an initial starting time. If these keywords are not used, then elapsed and elaplong are the same value.
The dt keyword is the current timestep size in time units. The time keyword is the current elapsed simulation
time, also in time units, which is simply (step*dt) if the timestep size has not changed and the timestep has not
been reset. If the timestep has changed (e.g. via fix dt/reset) or the timestep has been reset (e.g. via the
"reset_timestep" command), then the simulation time is effectively a cumulative value up to the current point.
The cpu keyword is elapsed CPU seconds since the beginning of this run. The tpcpu and spcpu keywords are
measures of how fast your simulation is currently running. The tpcpu keyword is simulation time per CPU
second, where simulation time is in time units. E.g. for metal units, the tpcpu value would be picoseconds per
CPU second. The spcpu keyword is the number of timesteps per CPU second. Both quantities are on-the-fly
metrics, measured relative to the last time they were invoked. Thus if you are printing out thermodynamic
output every 100 timesteps, the two keywords will continually output the time and timestep rate for the last
100 steps. The tpcpu keyword does not attempt to track any changes in timestep size, e.g. due to using the fix
dt/reset command.
The cpuremain keyword estimates the CPU time remaining in the current run, based on the time elapsed thus
far. It will only be a good estimate if the CPU time/timestep for the rest of the run is similar to the preceding
timesteps. On the initial timestep the value will be 0.0 since there is no history to estimate from. For a
minimization run performed by the "minimize" command, the estimate is based on the maxiter parameter,
assuming the minimization will proceed for the maximum number of allowed iterations.
The part keyword is useful for multi-replica or multi-partition simulations to indicate which partition this
output and this file corresponds to, or for use in a variable to append to a filename for output specific to this
partition. See Section 2.7 of the manual for details on running in multi-partition mode.
The timeremain keyword returns the remaining seconds when a timeout has been configured via the timer
timeout command. If the timeout timer is inactive, the value of this keyword is 0.0 and if the timer is expired,
it is negative. This allows for example to exit loops cleanly, if the timeout is expired with:
if "$(timeremain) <0.0" then "quit 0"
The fmax and fnorm keywords are useful for monitoring the progress of an energy minimization. The fmax
keyword calculates the maximum force in any dimension on any atom in the system, or the infinity-norm of
the force vector for the system. The fnorm keyword calculates the 2-norm or length of the force vector.
The nbuild and ndanger keywords are useful for monitoring neighbor list builds during a run. Note that both
these values are also printed with the end-of-run statistics. The nbuild keyword is the number of re-builds
during the current run. The ndanger keyword is the number of re-builds that LAMMPS considered potentially
"dangerous". If atom movement triggered neighbor list rebuilding (see the neigh_modify command), then
dangerous reneighborings are those that were triggered on the first timestep atom movement was checked for.
If this count is non-zero you may wish to reduce the delay factor to insure no force interactions are missed by
atoms moving beyond the neighbor skin distance before a rebuild takes place.
The keywords cella, cellb, cellc, cellalpha, cellbeta, cellgamma, correspond to the usual crystallographic
quantities that define the periodic unit cell of a crystal. See this section of the doc pages for a geometric
description of triclinic periodic cells, including a precise definition of these quantities in terms of the internal
LAMMPS Users Manual
663

LAMMPS cell dimensions lx, ly, lz, yz, xz, xy.
For output values from a compute or fix, the bracketed index I used to index a vector, as in c_ID[I] or f_ID[I],
can be specified using a wildcard asterisk with the index to effectively specify multiple values. This takes the
form "*" or "*n" or "n*" or "m*n". If N = the size of the vector (for mode = scalar) or the number of columns
in the array (for mode = vector), then an asterisk with no numeric values means all indices from 1 to N. A
leading asterisk means all indices from 1 to n (inclusive). A trailing asterisk means all indices from n to N
(inclusive). A middle asterisk means all indices from m to n (inclusive).
Using a wildcard is the same as if the individual elements of the vector had been listed one by one. E.g. these
2 thermo_style commands are equivalent, since the compute temp command creates a global vector with 6
values.
compute myTemp all temp
thermo_style custom step temp etotal c_myTemp[*]
thermo_style custom step temp etotal &
c_myTemp[1] c_myTemp[2] c_myTemp[3] &
c_myTemp[4] c_myTemp[5] c_myTemp[6]
The c_ID and c_ID[I] and c_ID[I][J] keywords allow global values calculated by a compute to be output. As
discussed on the compute doc page, computes can calculate global, per-atom, or local values. Only global
values can be referenced by this command. However, per-atom compute values for an individual atom can be
referenced in a variable and the variable referenced by thermo_style custom, as discussed below. See the
discussion above for how the I in c_ID[I] can be specified with a wildcard asterisk to effectively specify
multiple values from a global compute vector.
The ID in the keyword should be replaced by the actual ID of a compute that has been defined elsewhere in
the input script. See the compute command for details. If the compute calculates a global scalar, vector, or
array, then the keyword formats with 0, 1, or 2 brackets will reference a scalar value from the compute.
Note that some computes calculate "intensive" global quantities like temperature; others calculate "extensive"
global quantities like kinetic energy that are summed over all atoms in the compute group. Intensive quantities
are printed directly without normalization by thermo_style custom. Extensive quantities may be normalized
by the total number of atoms in the simulation (NOT the number of atoms in the compute group) when output,
depending on the thermo_modify norm option being used.
The f_ID and f_ID[I] and f_ID[I][J] keywords allow global values calculated by a fix to be output. As
discussed on the fix doc page, fixes can calculate global, per-atom, or local values. Only global values can be
referenced by this command. However, per-atom fix values can be referenced for an individual atom in a
variable and the variable referenced by thermo_style custom, as discussed below. See the discussion above for
how the I in f_ID[I] can be specified with a wildcard asterisk to effectively specify multiple values from a
global fix vector.
The ID in the keyword should be replaced by the actual ID of a fix that has been defined elsewhere in the
input script. See the fix command for details. If the fix calculates a global scalar, vector, or array, then the
keyword formats with 0, 1, or 2 brackets will reference a scalar value from the fix.
Note that some fixes calculate "intensive" global quantities like timestep size; others calculate "extensive"
global quantities like energy that are summed over all atoms in the fix group. Intensive quantities are printed
directly without normalization by thermo_style custom. Extensive quantities may be normalized by the total
number of atoms in the simulation (NOT the number of atoms in the fix group) when output, depending on
LAMMPS Users Manual
664

the thermo_modify norm option being used.
The v_name keyword allow the current value of a variable to be output. The name in the keyword should be
replaced by the variable name that has been defined elsewhere in the input script. Only equal-style and
vector-style variables can be referenced; the latter requires a bracketed term to specify the Ith element of the
vector calculated by the variable. However, an atom-style variable can be referenced for an individual atom by
an equal-style variable and that variable referenced. See the variable command for details. Variables of style
equal and vector and atom define a formula which can reference per-atom properties or thermodynamic
keywords, or they can invoke other computes, fixes, or variables when evaluated, so this is a very general
means of creating thermodynamic output.
Note that equal-style and vector-style variables are assumed to produce "intensive" global quantities, which
are thus printed as-is, without normalization by thermo_style custom. You can include a division by "natoms"
in the variable formula if this is not the case.
Restrictions:
This command must come after the simulation box is defined by a read_data, read_restart, or create_box
command.
Related commands:
thermo, thermo_modify, fix_modify, compute temp, compute pressure
Default:
thermo_style one
LAMMPS Users Manual
665

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
timer command
Syntax:
timer args
args = one or more of off or loop or normal or full or sync or nosync or timeout or every
off = do not collect or print any timing information
loop = collect only the total time for the simulation loop
normal = collect timer information broken down by sections (default)
full = like normal but also include CPU and thread utilization
sync = explicitly synchronize MPI tasks between sections
nosync = do not synchronize MPI tasks between sections (default)
timeout elapse = set walltime limit to elapse
every Ncheck = perform timeout check every Ncheck steps
Examples:
timer full sync
timer timeout 2:00:00 every 100
timer loop
Description:
Select the level of detail at which LAMMPS performs its CPU timings. Multiple keywords can be specified
with the timer command. For keywords that are mutually exclusive, the last one specified takes precedence.
During a simulation run LAMMPS collects information about how much time is spent in different sections
of the code and thus can provide information for determining performance and load imbalance problems.
This can be done at different levels of detail and accuracy. For more information about the timing output,
see this discussion of screen output in Section 2.8.
The off setting will turn all time measurements off. The loop setting will only measure the total time for a
run and not collect any detailed per section information. With the normal setting, timing information for
portions of the timestep (pairwise calculations, neighbor list construction, output, etc) are collected as well
as information about load imbalances for those sections across processors. The full setting adds information
about CPU utilization and thread utilization, when multi-threading is enabled.
With the sync setting, all MPI tasks are synchronized at each timer call which measures load imbalance for
each section more accurately, though it can also slow down the simulation by prohibiting overlapping
independent computations on different MPI ranks Using the nosync setting (which is the default) turns this
synchronization off.
With the timeout keyword a walltime limit can be imposed, that affects the run and minimize commands.
This can be convenient when calculations have to comply with execution time limits, e.g. when running
under a batch system when you want to maximize the utilization of the batch time slot, especially for runs
where the time per timestep varies much and thus it becomes difficult to predict how many steps a
simulation can perform for a given walltime limit. This also applies for difficult to converge minimizations.
The timeout elapse value should be somewhat smaller than the maximum wall time requested from the
•
LAMMPS Users Manual
666
batch system, as there is usually some overhead to launch jobs, and it is advisable to write out a restart after
terminating a run due to a timeout.
The timeout timer starts when the command is issued. When the time limit is reached, the run or energy
minimization will exit on the next step or iteration that is a multiple of the Ncheck value which can be set
with the every keyword. Default is checking every 10 steps. After the timer timeout has expired all
subsequent run or minimize commands in the input script will be skipped. The remaining time or timer
status can be accessed with the thermo variable timeremain, which will be zero, if the timeout is inactive
(default setting), it will be negative, if the timeout time is expired and positive if there is time remaining
and in this case the value of the variable are the number of seconds remaining.
When the timeout key word is used a second time, the timer is restarted with a new time limit. The timeout
elapse value can be specified as off or unlimited to impose a no timeout condition (which is the default).
The elapse setting can be specified as a single number for seconds, two numbers separated by a colon
(MM:SS) for minutes and seconds, or as three numbers separated by colons for hours, minutes, and seconds
(H:MM:SS).
The every keyword sets how frequently during a run or energy minimization the wall clock will be checked.
This check count applies to the outer iterations or time steps during minimizations or r-RESPA runs,
respectively. Checking for timeout too often, can slow a calculation down. Checking too infrequently can
make the timeout measurement less accurate, with the run being stopped later than desired.
NOTE: Using the full and sync options provides the most detailed and accurate timing information, but can
also have a negative performance impact due to the overhead of the many required system calls. It is thus
recommended to use these settings only when testing tests to identify performance bottlenecks. For
calculations with few atoms or a very large number of processors, even the normal setting can have a
measurable negative performance impact. In those cases you can just use the loop or off setting.
Restrictions: none
Related commands:
run post no, kspace_modify fftbench
Default:
timer normal nosync
timer timeout off
timer every 10
LAMMPS Users Manual
667
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
timestep command
Syntax:
timestep dt
dt = timestep size (time units)•
Examples:
timestep 2.0
timestep 0.003
Description:
Set the timestep size for subsequent molecular dynamics simulations. See the units command for the time
units associated with each choice of units that LAMMPS supports.
The default value for the timestep size also depends on the choice of units for the simulation; see the default
values below.
When the run style is respa, dt is the timestep for the outer loop (largest) timestep.
Restrictions: none
Related commands:
fix dt/reset, run, run_style respa, units
Default:
choice of units time units default
timestep size
lj tau 0.005 tau
real fmsec 1.0 fmsec
metal psec 0.001 psec
si sec 1.0e-8 sec (10
nsec)
cgs sec 1.0e-8 sec (10
nsec)
electron fmsec 0.001 fmsec
micro usec 2.0 usec
nano nsec 0.00045 nsec
LAMMPS Users Manual
668

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
uncompute command
Syntax:
uncompute compute-ID
compute-ID = ID of a previously defined compute•
Examples:
uncompute 2
uncompute lower-boundary
Description:
Delete a compute that was previously defined with a compute command. This also wipes out any additional
changes made to the compute via the compute_modify command.
Restrictions: none
Related commands:
compute
Default: none
LAMMPS Users Manual
669

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
undump command
Syntax:
undump dump-ID
dump-ID = ID of previously defined dump•
Examples:
undump mine
undump 2
Description:
Turn off a previously defined dump so that it is no longer active. This closes the file associated with the
dump.
Restrictions: none
Related commands:
dump
Default: none
LAMMPS Users Manual
670

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
unfix command
Syntax:
unfix fix-ID
fix-ID = ID of a previously defined fix•
Examples:
unfix 2
unfix lower-boundary
Description:
Delete a fix that was previously defined with a fix command. This also wipes out any additional changes
made to the fix via the fix_modify command.
Restrictions: none
Related commands:
fix
Default: none
LAMMPS Users Manual
671
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
units command
Syntax:
units style
style = lj or real or metal or si or cgs or electron or micro or nano•
Examples:
units metal
units lj
Description:
This command sets the style of units used for a simulation. It determines the units of all quantities specified in
the input script and data file, as well as quantities output to the screen, log file, and dump files. Typically, this
command is used at the very beginning of an input script.
For all units except lj, LAMMPS uses physical constants from www.physics.nist.gov. For the definition of
Kcal in real units, LAMMPS uses the thermochemical calorie = 4.184 J.
The choice you make for units simply sets some internal conversion factors within LAMMPS. This means
that any simulation you perform for one choice of units can be duplicated with any other unit setting
LAMMPS supports. In this context "duplicate" means the particles will have identical trajectories and all
output generated by the simulation will be identical. This will be the case for some number of timesteps until
round-off effects accumulate, since the conversion factors for two different unit systems are not identical to
infinite precision.
To perform the same simulation in a different set of units you must change all the unit-based input parameters
in your input script and other input files (data file, potential files, etc) correctly to the new units. And you
must correctly convert all output from the new units to the old units when comparing to the original results.
That is often not simple to do.
For style lj, all quantities are unitless. Without loss of generality, LAMMPS sets the fundamental quantities
mass, sigma, epsilon, and the Boltzmann constant = 1. The masses, distances, energies you specify are
multiples of these fundamental values. The formulas relating the reduced or unitless quantity (with an
asterisk) to the same quantity with units is also given. Thus you can use the mass & sigma & epsilon values
for a specific material and convert the results from a unitless LJ simulation into physical quantities.
mass = mass or m• distance = sigma, where x* = x / sigma• time = tau, where t* = t (epsilon / m / sigma^2)^1/2• energy = epsilon, where E* = E / epsilon• velocity = sigma/tau, where v* = v tau / sigma• force = epsilon/sigma, where f* = f sigma / epsilon• torque = epsilon, where t* = t / epsilon• temperature = reduced LJ temperature, where T* = T Kb / epsilon• pressure = reduced LJ pressure, where P* = P sigma^3 / epsilon•
LAMMPS Users Manual
672
dynamic viscosity = reduced LJ viscosity, where eta* = eta sigma^3 / epsilon / tau• charge = reduced LJ charge, where q* = q / (4 pi perm0 sigma epsilon)^1/2• dipole = reduced LJ dipole, moment where *mu = mu / (4 pi perm0 sigma^3 epsilon)^1/2• electric field = force/charge, where E* = E (4 pi perm0 sigma epsilon)^1/2 sigma / epsilon• density = mass/volume, where rho* = rho sigma^dim•
Note that for LJ units, the default mode of thermodynamic output via the thermo_style command is to
normalize all extensive quantities by the number of atoms. E.g. potential energy is extensive because it is
summed over atoms, so it is output as energy/atom. Temperature is intensive since it is already normalized by
the number of atoms, so it is output as-is. This behavior can be changed via the thermo_modify norm
command.
For style real, these are the units:
mass = grams/mole• distance = Angstroms• time = femtoseconds• energy = Kcal/mole• velocity = Angstroms/femtosecond• force = Kcal/mole-Angstrom• torque = Kcal/mole• temperature = Kelvin• pressure = atmospheres• dynamic viscosity = Poise• charge = multiple of electron charge (1.0 is a proton)• dipole = charge*Angstroms• electric field = volts/Angstrom• density = gram/cm^dim•
For style metal, these are the units:
mass = grams/mole• distance = Angstroms• time = picoseconds• energy = eV• velocity = Angstroms/picosecond• force = eV/Angstrom• torque = eV• temperature = Kelvin• pressure = bars• dynamic viscosity = Poise• charge = multiple of electron charge (1.0 is a proton)• dipole = charge*Angstroms• electric field = volts/Angstrom• density = gram/cm^dim•
For style si, these are the units:
mass = kilograms• distance = meters• time = seconds•
LAMMPS Users Manual
673
energy = Joules• velocity = meters/second• force = Newtons• torque = Newton-meters• temperature = Kelvin• pressure = Pascals• dynamic viscosity = Pascal*second• charge = Coulombs (1.6021765e-19 is a proton)• dipole = Coulombs*meters• electric field = volts/meter• density = kilograms/meter^dim•
For style cgs, these are the units:
mass = grams• distance = centimeters• time = seconds• energy = ergs• velocity = centimeters/second• force = dynes• torque = dyne-centimeters• temperature = Kelvin• pressure = dyne/cm^2 or barye = 1.0e-6 bars• dynamic viscosity = Poise• charge = statcoulombs or esu (4.8032044e-10 is a proton)• dipole = statcoul-cm = 10^18 debye• electric field = statvolt/cm or dyne/esu• density = grams/cm^dim•
For style electron, these are the units:
mass = atomic mass units• distance = Bohr• time = femtoseconds• energy = Hartrees• velocity = Bohr/atomic time units [1.03275e-15 seconds]• force = Hartrees/Bohr• temperature = Kelvin• pressure = Pascals• charge = multiple of electron charge (1.0 is a proton)• dipole moment = Debye• electric field = volts/cm•
For style micro, these are the units:
mass = picograms• distance = micrometers• time = microseconds• energy = picogram-micrometer^2/microsecond^2• velocity = micrometers/microsecond• force = picogram-micrometer/microsecond^2•
LAMMPS Users Manual
674
torque = picogram-micrometer^2/microsecond^2• temperature = Kelvin• pressure = picogram/(micrometer-microsecond^2)• dynamic viscosity = picogram/(micrometer-microsecond)• charge = picocoulombs (1.6021765e-7 is a proton)• dipole = picocoulomb-micrometer• electric field = volt/micrometer• density = picograms/micrometer^dim•
For style nano, these are the units:
mass = attograms• distance = nanometers• time = nanoseconds• energy = attogram-nanometer^2/nanosecond^2• velocity = nanometers/nanosecond• force = attogram-nanometer/nanosecond^2• torque = attogram-nanometer^2/nanosecond^2• temperature = Kelvin• pressure = attogram/(nanometer-nanosecond^2)• dynamic viscosity = attogram/(nanometer-nanosecond)• charge = multiple of electron charge (1.0 is a proton)• dipole = charge-nanometer• electric field = volt/nanometer• density = attograms/nanometer^dim•
The units command also sets the timestep size and neighbor skin distance to default values for each style:
For style lj these are dt = 0.005 tau and skin = 0.3 sigma.• For style real these are dt = 1.0 fmsec and skin = 2.0 Angstroms.• For style metal these are dt = 0.001 psec and skin = 2.0 Angstroms.• For style si these are dt = 1.0e-8 sec and skin = 0.001 meters.• For style cgs these are dt = 1.0e-8 sec and skin = 0.1 cm.• For style electron these are dt = 0.001 fmsec and skin = 2.0 Bohr.• For style micro these are dt = 2.0 microsec and skin = 0.1 micrometers.• For style nano these are dt = 0.00045 nanosec and skin = 0.1 nanometers.•
Restrictions:
This command cannot be used after the simulation box is defined by a read_data or create_box command.
Related commands: none
Default:
units lj
LAMMPS Users Manual
675

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
variable command
Syntax:
variable name style args ...
name = name of variable to define• style = delete or index or loop or world or universe or uloop or string or format or getenv or file or
atomfile or python or internal or equal or vector or atom
delete = no args
index args = one or more strings
loop args = N
N = integer size of loop, loop from 1 to N inclusive
loop args = N pad
N = integer size of loop, loop from 1 to N inclusive
pad = all values will be same length, e.g. 001, 002, ..., 100
loop args = N1 N2
N1,N2 = loop from N1 to N2 inclusive
loop args = N1 N2 pad
N1,N2 = loop from N1 to N2 inclusive
pad = all values will be same length, e.g. 050, 051, ..., 100
world args = one string for each partition of processors
universe args = one or more strings
uloop args = N
N = integer size of loop
uloop args = N pad
N = integer size of loop
pad = all values will be same length, e.g. 001, 002, ..., 100
string arg = one string
format args = vname fstr
vname = name of equal-style variable to evaluate
fstr = C-style format string
getenv arg = one string
file arg = filename
atomfile arg = filename
python arg = function
internal arg = numeric value
equal or vector or atom args = one formula containing numbers, thermo keywords, math operations, group functions, atom values and vectors, compute/fix/variable references
numbers = 0.0, 100, -5.4, 2.8e-4, etc
constants = PI, version, on, off, true, false, yes, no
thermo keywords = vol, ke, press, etc from thermo_style
math operators = (), -x, x+y, x-y, x*y, x/y, x^y, x%y,
x == y, x != y, x <y, x <= y, x > y, x >= y, x && y, x || y, x |^ y, !x
math functions = sqrt(x), exp(x), ln(x), log(x), abs(x),
sin(x), cos(x), tan(x), asin(x), acos(x), atan(x), atan2(y,x),
random(x,y,z), normal(x,y,z), ceil(x), floor(x), round(x)
ramp(x,y), stagger(x,y), logfreq(x,y,z), logfreq2(x,y,z),
stride(x,y,z), stride2(x,y,z,a,b,c),
vdisplace(x,y), swiggle(x,y,z), cwiggle(x,y,z)
group functions = count(group), mass(group), charge(group),
xcm(group,dim), vcm(group,dim), fcm(group,dim),
bound(group,dir), gyration(group), ke(group),
angmom(group,dim), torque(group,dim),
inertia(group,dimdim), omega(group,dim)
region functions = count(group,region), mass(group,region), charge(group,region),
•
LAMMPS Users Manual
676
xcm(group,dim,region), vcm(group,dim,region), fcm(group,dim,region),
bound(group,dir,region), gyration(group,region), ke(group,reigon),
angmom(group,dim,region), torque(group,dim,region),
inertia(group,dimdim,region), omega(group,dim,region)
special functions = sum(x), min(x), max(x), ave(x), trap(x), slope(x), gmask(x), rmask(x), grmask(x,y), next(x)
feature functions = is_active(category,feature,exact), is_defined(category,id,exact)
atom value = id[i], mass[i], type[i], mol[i], x[i], y[i], z[i], vx[i], vy[i], vz[i], fx[i], fy[i], fz[i], q[i]
atom vector = id, mass, type, mol, x, y, z, vx, vy, vz, fx, fy, fz, q
compute references = c_ID, c_ID[i], c_ID[i][j], C_ID, C_ID[i]
fix references = f_ID, f_ID[i], f_ID[i][j], F_ID, F_ID[i]
variable references = v_name, v_name[i]
Examples:
variable x index run1 run2 run3 run4 run5 run6 run7 run8
variable LoopVar loop $n
variable beta equal temp/3.0
variable b1 equal x[234]+0.5*vol
variable b1 equal "x[234] + 0.5*vol"
variable b equal xcm(mol1,x)/2.0
variable b equal c_myTemp
variable b atom x*y/vol
variable foo string myfile
variable foo internal 3.5
variable myPy python increase
variable f file values.txt
variable temp world 300.0 310.0 320.0 ${Tfinal}
variable x universe 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
variable x uloop 15 pad
variable str format x %.6g
variable x delete
Description:
This command assigns one or more strings to a variable name for evaluation later in the input script or during
a simulation.
Variables can thus be useful in several contexts. A variable can be defined and then referenced elsewhere in
an input script to become part of a new input command. For variable styles that store multiple strings, the next
command can be used to increment which string is assigned to the variable. Variables of style equal store a
formula which when evaluated produces a single numeric value which can be output either directly (see the
print, fix print, and run every commands) or as part of thermodynamic output (see the thermo_style
command), or used as input to an averaging fix (see the fix ave/time command). Variables of style vector store
a formula which produces a vector of such values which can be used as input to various averaging fixes, or
elements of which can be part of thermodynamic output. Variables of style atom store a formula which when
evaluated produces one numeric value per atom which can be output to a dump file (see the dump custom
command) or used as input to an averaging fix (see the fix ave/chunk and fix ave/atom commands). Variables
of style atomfile can be used anywhere in an input script that atom-style variables are used; they get their
per-atom values from a file rather than from a formula. Variables of style python can be hooked to Python
functions using code you provide, so that the variable gets its value from the evaluation of the Python code.
Variables of style internal are used by a few commands which set their value directly.
NOTE: As discussed in Section 3.2 of the manual, an input script can use "immediate" variables, specified as
$(formula) with parenthesis, where the formula has the same syntax as equal-style variables described on this
page. This is a convenient way to evaluate a formula immediately without using the variable command to
LAMMPS Users Manual
677

define a named variable and then evaluate that variable. See below for a more detailed discussion of this
feature.
In the discussion that follows, the "name" of the variable is the arbitrary string that is the 1st argument in the
variable command. This name can only contain alphanumeric characters and underscores. The "string" is one
or more of the subsequent arguments. The "string" can be simple text as in the 1st example above, it can
contain other variables as in the 2nd example, or it can be a formula as in the 3rd example. The "value" is the
numeric quantity resulting from evaluation of the string. Note that the same string can generate different
values when it is evaluated at different times during a simulation.
NOTE: When an input script line is encountered that defines a variable of style equal or vector or atom or
python that contains a formula or Python code, the formula is NOT immediately evaluated. It will be
evaluated every time when the variable is used instead. If you simply want to evaluate a formula in place you
can use as so-called. See the section below about "Immediate Evaluation of Variables" for more details on the
topic. This is also true of a format style variable since it evaluates another variable when it is invoked.
Variables of style equal and vector and atom can be used as inputs to various other commands which evaluate
their formulas as needed, e.g. at different timesteps during a run.
Variables of style internal can be used in place of an equal-style variable, except by commands that set the
value stored by the internal-style variable. Thus any command that states it can use an equal-style variable as
an argument, can also use an internal-style variable. This means that when the command evaluates the
variable, it will use the value set (internally) by another command.
Variables of style python can be used in place of an equal-style variable so long as the associated Python
function, as defined by the python command, returns a numeric value. Thus any command that states it can
use an equal-style variable as an argument, can also use such a python-style variable. This means that when
the LAMMPS command evaluates the variable, the Python function will be executed.
NOTE: When a variable command is encountered in the input script and the variable name has already been
specified, the command is ignored. This means variables can NOT be re-defined in an input script (with two
exceptions, read further). This is to allow an input script to be processed multiple times without resetting the
variables; see the jump or include commands. It also means that using the command-line switch -var will
override a corresponding index variable setting in the input script.
There are two exceptions to this rule. First, variables of style string, getenv, internal, equal, vector, atom, and
python ARE redefined each time the command is encountered. This allows these style of variables to be
redefined multiple times in an input script. In a loop, this means the formula associated with an equal or atom
style variable can change if it contains a substitution for another variable, e.g. $x or v_x.
Second, as described below, if a variable is iterated on to the end of its list of strings via the next command, it
is removed from the list of active variables, and is thus available to be re-defined in a subsequent variable
command. The delete style does the same thing.
This section of the manual explains how occurrences of a variable name in an input script line are replaced by
the variable's string. The variable name can be referenced as $x if the name "x" is a single character, or as
${LoopVar} if the name "LoopVar" is one or more characters.
As described below, for variable styles index, loop, file, universe, and uloop, which string is assigned to a
variable can be incremented via the next command. When there are no more strings to assign, the variable is
exhausted and a flag is set that causes the next jump command encountered in the input script to be skipped.
LAMMPS Users Manual
678

This enables the construction of simple loops in the input script that are iterated over and then exited from.
As explained above, an exhausted variable can be re-used in an input script. The delete style also removes the
variable, the same as if it were exhausted, allowing it to be redefined later in the input script or when the input
script is looped over. This can be useful when breaking out of a loop via the if and jump commands before the
variable would become exhausted. For example,
label loop
variable a loop 5
print "A = $a"
if "$a > 2" then "jump in.script break"
next a
jump in.script loop
label break
variable a delete
This section describes how all the various variable styles are defined and what they store. Except for the equal
and vector and atom styles, which are explained in the next section.
Many of the styles store one or more strings. Note that a single string can contain spaces (multiple words), if it
is enclosed in quotes in the variable command. When the variable is substituted for in another input script
command, its returned string will then be interpreted as multiple arguments in the expanded command.
For the index style, one or more strings are specified. Initially, the 1st string is assigned to the variable. Each
time a next command is used with the variable name, the next string is assigned. All processors assign the
same string to the variable.
Index style variables with a single string value can also be set by using the command-line switch -var; see this
section for details.
The loop style is identical to the index style except that the strings are the integers from 1 to N inclusive, if
only one argument N is specified. This allows generation of a long list of runs (e.g. 1000) without having to
list N strings in the input script. Initially, the string "1" is assigned to the variable. Each time a next command
is used with the variable name, the next string ("2", "3", etc) is assigned. All processors assign the same string
to the variable. The loop style can also be specified with two arguments N1 and N2. In this case the loop runs
from N1 to N2 inclusive, and the string N1 is initially assigned to the variable. N1 <= N2 and N2 >= 0 is
required.
For the world style, one or more strings are specified. There must be one string for each processor partition or
"world". See this section of the manual for information on running LAMMPS with multiple partitions via the
"-partition" command-line switch. This variable command assigns one string to each world. All processors in
the world are assigned the same string. The next command cannot be used with equal style variables, since
there is only one value per world. This style of variable is useful when you wish to run different simulations
on different partitions, or when performing a parallel tempering simulation (see the temper command), to
assign different temperatures to different partitions.
For the universe style, one or more strings are specified. There must be at least as many strings as there are
processor partitions or "worlds". See this page for information on running LAMMPS with multiple partitions
via the "-partition" command-line switch. This variable command initially assigns one string to each world.
When a next command is encountered using this variable, the first processor partition to encounter it, is
assigned the next available string. This continues until all the variable strings are consumed. Thus, this
command can be used to run 50 simulations on 8 processor partitions. The simulations will be run one after
LAMMPS Users Manual
679
the other on whatever partition becomes available, until they are all finished. Universe style variables are
incremented using the files "tmp.lammps.variable" and "tmp.lammps.variable.lock" which you will see in
your directory during such a LAMMPS run.
The uloop style is identical to the universe style except that the strings are the integers from 1 to N. This
allows generation of long list of runs (e.g. 1000) without having to list N strings in the input script.
For the string style, a single string is assigned to the variable. The only difference between this and using the
index style with a single string is that a variable with string style can be redefined. E.g. by another command
later in the input script, or if the script is read again in a loop.
For the format style, an equal-style variable is specified along with a C-style format string, e.g. "%f" or
"%.10g", which must be appropriate for formatting a double-precision floating-point value. This allows an
equal-style variable to be formatted specifically for output as a string, e.g. by the print command, if the default
format "%.15g" has too much precision.
For the getenv style, a single string is assigned to the variable which should be the name of an environment
variable. When the variable is evaluated, it returns the value of the environment variable, or an empty string if
it not defined. This style of variable can be used to adapt the behavior of LAMMPS input scripts via
environment variable settings, or to retrieve information that has been previously stored with the shell putenv
command. Note that because environment variable settings are stored by the operating systems, they persist
beyond a clear command.
For the file style, a filename is provided which contains a list of strings to assign to the variable, one per line.
The strings can be numeric values if desired. See the discussion of the next() function below for equal-style
variables, which will convert the string of a file-style variable into a numeric value in a formula.
When a file-style variable is defined, the file is opened and the string on the first line is read and stored with
the variable. This means the variable can then be evaluated as many times as desired and will return that
string. There are two ways to cause the next string from the file to be read: use the next command or the next()
function in an equal- or atom-style variable, as discussed below.
The rules for formatting the file are as follows. A comment character "#" can be used anywhere on a line; text
starting with the comment character is stripped. Blank lines are skipped. The first "word" of a non-blank line,
delimited by white space, is the "string" assigned to the variable.
For the atomfile style, a filename is provided which contains one or more sets of values, to assign on a
per-atom basis to the variable. The format of the file is described below.
When an atomfile-style variable is defined, the file is opened and the first set of per-atom values are read and
stored with the variable. This means the variable can then be evaluated as many times as desired and will
return those values. There are two ways to cause the next set of per-atom values from the file to be read: use
the next command or the next() function in an atom-style variable, as discussed below.
The rules for formatting the file are as follows. Each time a set of per-atom values is read, a non-blank line is
searched for in the file. A comment character "#" can be used anywhere on a line; text starting with the
comment character is stripped. Blank lines are skipped. The first "word" of a non-blank line, delimited by
white space, is read as the count N of per-atom lines to immediately follow. N can be be the total number of
atoms in the system, or only a subset. The next N lines have the following format
ID value
LAMMPS Users Manual
680

where ID is an atom ID and value is the per-atom numeric value that will be assigned to that atom. IDs can be
listed in any order.
NOTE: Every time a set of per-atom lines is read, the value for all atoms is first set to 0.0. Thus values for
atoms whose ID does not appear in the set, will remain 0.0.
For the python style a Python function name is provided. This needs to match a function name specified in a
python command which returns a value to this variable as defined by its return keyword. For example these
two commands would be self-consistent:
variable foo python myMultiply
python myMultiply return v_foo format f file funcs.py
The two commands can appear in either order so long as both are specified before the Python function is
invoked for the first time.
Each time the variable is evaluated, the associated Python function is invoked, and the value it returns is also
returned by the variable. Since the Python function can use other LAMMPS variables as input, or query
interal LAMMPS quantities to perform its computation, this means the variable can return a different value
each time it is evaluated.
The type of value stored in the variable is determined by the format keyword of the python command. It can
be an integer (i), floating point (f), or string (s) value. As mentioned above, if it is a numeric value (integer or
floating point), then the python-style variable can be used in place of an equal-style variable anywhere in an
input script, e.g. as an argument to another command that allows for equal-style variables.
For the internal style a numeric value is provided. This value will be assigned to the variable until a
LAMMPS command sets it to a new value. There are currently only two LAMMPS commands that require
internal variables as inputs, because they reset them: create_atoms and fix controller. As mentioned above, an
internal-style variable can be used in place of an equal-style variable anywhere else in an input script, e.g. as
an argument to another command that allows for equal-style variables.
For the equal and vector and atom styles, a single string is specified which represents a formula that will be
evaluated afresh each time the variable is used. If you want spaces in the string, enclose it in double quotes so
the parser will treat it as a single argument. For equal-style variables the formula computes a scalar quantity,
which becomes the value of the variable whenever it is evaluated. For vector-style variables the formula must
compute a vector of quantities, which becomes the value of the variable whenever it is evaluated. The
calculated vector can be on length one, but it cannot be a simple scalar value like that produced by an
equal-style compute. I.e. the formula for a vector-style variable must have at least one quantity in it that refers
to a global vector produced by a compute, fix, or other vector-style variable. For atom-style variables the
formula computes one quantity for each atom whenever it is evaluated.
Note that equal, vector, and atom variables can produce different values at different stages of the input script
or at different times during a run. For example, if an equal variable is used in a fix print command, different
values could be printed each timestep it was invoked. If you want a variable to be evaluated immediately, so
that the result is stored by the variable instead of the string, see the section below on "Immediate Evaluation
of Variables".
The next command cannot be used with equal or vector or atom style variables, since there is only one string.
LAMMPS Users Manual
681

The formula for an equal, vector, or atom variable can contain a variety of quantities. The syntax for each
kind of quantity is simple, but multiple quantities can be nested and combined in various ways to build up
formulas of arbitrary complexity. For example, this is a valid (though strange) variable formula:
variable x equal "pe + c_MyTemp / vol^(1/3)"
Specifically, a formula can contain numbers, constants, thermo keywords, math operators, math functions,
group functions, region functions, atom values, atom vectors, compute references, fix references, and
references to other variables.
Number 0.2, 100, 1.0e20, -15.4, etc
Constant PI, version, on, off, true, false, yes, no
Thermo
keywords vol, pe, ebond, etc
Math
operators (), -x, x+y, x-y, x*y, x/y, x^y, x%y, x == y, x != y, x < y, x <= y, x > y, x >= y, x && y, x || y,
x |^ y, !x
Math
functions
sqrt(x), exp(x), ln(x), log(x), abs(x), sin(x), cos(x), tan(x), asin(x), acos(x), atan(x), atan2(y,x),
random(x,y,z), normal(x,y,z), ceil(x), floor(x), round(x), ramp(x,y), stagger(x,y),
logfreq(x,y,z), logfreq2(x,y,z), stride(x,y,z), stride2(x,y,z,a,b,c), vdisplace(x,y), swiggle(x,y,z),
cwiggle(x,y,z)
Group
functions count(ID), mass(ID), charge(ID), xcm(ID,dim), vcm(ID,dim), fcm(ID,dim), bound(ID,dir),
gyration(ID), ke(ID), angmom(ID,dim), torque(ID,dim), inertia(ID,dimdim), omega(ID,dim)
Region
functions
count(ID,IDR), mass(ID,IDR), charge(ID,IDR), xcm(ID,dim,IDR), vcm(ID,dim,IDR),
fcm(ID,dim,IDR), bound(ID,dir,IDR), gyration(ID,IDR), ke(ID,IDR), angmom(ID,dim,IDR),
torque(ID,dim,IDR), inertia(ID,dimdim,IDR), omega(ID,dim,IDR)
Special
functions sum(x), min(x), max(x), ave(x), trap(x), slope(x), gmask(x), rmask(x), grmask(x,y), next(x)
Atom values id[i], mass[i], type[i], mol[i], x[i], y[i], z[i], vx[i], vy[i], vz[i], fx[i], fy[i], fz[i], q[i]
Atom vectors id, mass, type, mol, x, y, z, vx, vy, vz, fx, fy, fz, q
Compute
references c_ID, c_ID[i], c_ID[i][j], C_ID, C_ID[i]
Fix
references f_ID, f_ID[i], f_ID[i][j], F_ID, F_ID[i]
Other
variables v_name, v_name[i]
Most of the formula elements produce a scalar value. Some produce a global or per-atom vector of values.
Global vectors can be produced by computes or fixes or by other vector-style variables. Per-atom vectors are
produced by atom vectors, compute references that represent a per-atom vector, fix references that represent a
per-atom vector, and variables that are atom-style variables. Math functions that operate on scalar values
produce a scalar value; math function that operate on global or per-atom vectors do so element-by-element
and produce a global or per-atom vector.
A formula for equal-style variables cannot use any formula element that produces a global or per-atom vector.
A formula for a vector-style variable can use formula elements that produce either a scalar value or a global
vector value, but cannot use a formula element that produces a per-atom vector. A formula for an atom-style
variable can use formula elements that produce either a scalar value or a per-atom vector, but not one that
produces a global vector. Atom-style variables are evaluated by other commands that define a group on which
they operate, e.g. a dump or compute or fix command. When they invoke the atom-style variable, only atoms
LAMMPS Users Manual
682
in the group are included in the formula evaluation. The variable evaluates to 0.0 for atoms not in the group.
Numbers, constants, and thermo keywords
Numbers can contain digits, scientific notation (3.0e20,3.0e-20,3.0E20,3.0E-20), and leading minus signs.
Constants are set at compile time and cannot be changed. PI will return the number
3.14159265358979323846; on, true or yes will return 1.0; off, false or no will return 0.0; version will return a
numeric version code of the current LAMMPS version (e.g. version 2 Sep 2015 will return the number
20150902). The corresponding value for newer versions of LAMMPS will be larger, for older versions of
LAMMPS will be smaller. This can be used to have input scripts adapt automatically to LAMMPS versions,
when non-backwards compatible syntax changes are introduced. Here is an illustrative example (which will
not work, since the version has been introduced more recently):
if $(version
The thermo keywords allowed in a formula are those defined by the thermo_style custom command. Thermo
keywords that require a compute to calculate their values such as "temp" or "press", use computes stored and
invoked by the thermo_style command. This means that you can only use those keywords in a variable if the
style you are using with the thermo_style command (and the thermo keywords associated with that style) also
define and use the needed compute. Note that some thermo keywords use a compute indirectly to calculate
their value (e.g. the enthalpy keyword uses temp, pe, and pressure). If a variable is evaluated directly in an
input script (not during a run), then the values accessed by the thermo keyword must be current. See the
discussion below about "Variable Accuracy".
Math Operators
Math operators are written in the usual way, where the "x" and "y" in the examples can themselves be
arbitrarily complex formulas, as in the examples above. In this syntax, "x" and "y" can be scalar values or
per-atom vectors. For example, "ke/natoms" is the division of two scalars, where "vy+vz" is the
element-by-element sum of two per-atom vectors of y and z velocities.
Operators are evaluated left to right and have the usual C-style precedence: unary minus and unary logical
NOT operator "!" have the highest precedence, exponentiation "^" is next; multiplication and division and the
modulo operator "%" are next; addition and subtraction are next; the 4 relational operators "", and ">=" are
next; the two remaining relational operators "==" and "!=" are next; then the logical AND operator "&&"; and
finally the logical OR operator "||" and logical XOR (exclusive or) operator "|^" have the lowest precedence.
Parenthesis can be used to group one or more portions of a formula and/or enforce a different order of
evaluation than what would occur with the default precedence.
NOTE: Because a unary minus is higher precedence than exponentiation, the formula "-2^2" will evaluate to
4, not -4. This convention is compatible with some programming languages, but not others. As mentioned,
this behavior can be easily overridden with parenthesis; the formula "-(2^2)" will evaluate to -4.
The 6 relational operators return either a 1.0 or 0.0 depending on whether the relationship between x and y is
TRUE or FALSE. For example the expression x
These relational and logical operators can be used as a masking or selection operation in a formula. For
example, the number of atoms whose properties satisfy one or more criteria could be calculated by taking the
returned per-atom vector of ones and zeroes and passing it to the compute reduce command.
LAMMPS Users Manual
683

Math Functions
Math functions are specified as keywords followed by one or more parenthesized arguments "x", "y", "z",
each of which can themselves be arbitrarily complex formulas. In this syntax, the arguments can represent
scalar values or global vectors or per-atom vectors. In the latter case, the math operation is performed on each
element of the vector. For example, "sqrt(natoms)" is the sqrt() of a scalar, where "sqrt(y*z)" yields a
per-atom vector with each element being the sqrt() of the product of one atom's y and z coordinates.
Most of the math functions perform obvious operations. The ln() is the natural log; log() is the base 10 log.
The random(x,y,z) function takes 3 arguments: x = lo, y = hi, and z = seed. It generates a uniform random
number between lo and hi. The normal(x,y,z) function also takes 3 arguments: x = mu, y = sigma, and z =
seed. It generates a Gaussian variate centered on mu with variance sigma^2. In both cases the seed is used the
first time the internal random number generator is invoked, to initialize it. For equal-style and vector-style
variables, every processor uses the same seed so that they each generate the same sequence of random
numbers. For atom-style variables, a unique seed is created for each processor, based on the specified seed.
This effectively generates a different random number for each atom being looped over in the atom-style
variable.
NOTE: Internally, there is just one random number generator for all equal-style and vector-style variables and
another one for all atom-style variables. If you define multiple variables (of each style) which use the
random() or normal() math functions, then the internal random number generators will only be initialized
once, which means only one of the specified seeds will determine the sequence of generated random numbers.
The ceil(), floor(), and round() functions are those in the C math library. Ceil() is the smallest integer not less
than its argument. Floor() if the largest integer not greater than its argument. Round() is the nearest integer to
its argument.
The ramp(x,y) function uses the current timestep to generate a value linearly interpolated between the
specified x,y values over the course of a run, according to this formula:
value = x + (y-x) * (timestep-startstep) / (stopstep-startstep)
The run begins on startstep and ends on stopstep. Startstep and stopstep can span multiple runs, using the start
and stop keywords of the run command. See the run command for details of how to do this.
The stagger(x,y) function uses the current timestep to generate a new timestep. X,y > 0 and x > y are required.
The generated timesteps increase in a staggered fashion, as the sequence x,x+y,2x,2x+y,3x,3x+y,etc. For any
current timestep, the next timestep in the sequence is returned. Thus if stagger(1000,100) is used in a variable
by the dump_modify every command, it will generate the sequence of output timesteps:
100,1000,1100,2000,2100,3000,etc
The logfreq(x,y,z) function uses the current timestep to generate a new timestep. X,y,z > 0 and y < z are
required. The generated timesteps are on a base-z logarithmic scale, starting with x, and the y value is how
many of the z-1 possible timesteps within one logarithmic interval are generated. I.e. the timesteps follow the
sequence x,2x,3x,...y*x,x*z,2x*z,3x*z,...y*x*z,x*z^2,2x*z^2,etc. For any current timestep, the next timestep
in the sequence is returned. Thus if logfreq(100,4,10) is used in a variable by the dump_modify every
command, it will generate this sequence of output timesteps:
LAMMPS Users Manual
684
100,200,300,400,1000,2000,3000,4000,10000,20000,etc
The logfreq2(x,y,z) function is similar to logfreq, except a single logarithmic interval is divided into y
equally-spaced timesteps and all of them are output. Y < z is not required. Thus, if logfreq2(100,18,10) is used
in a variable by the dump_modify every command, then the interval between 100 and 1000 is divided as
900/18 = 50 steps, and it will generate the sequence of output timesteps:
100,150,200,...950,1000,1500,2000,...9500,10000,15000,etc
The stride(x,y,z) function uses the current timestep to generate a new timestep. X,y >= 0 and z > 0 and x <= y
are required. The generated timesteps increase in increments of z, from x to y, i.e. it generates the sequence
x,x+z,x+2z,...,y. If y-x is not a multiple of z, then similar to the way a for loop operates, the last value will be
one that does not exceed y. For any current timestep, the next timestep in the sequence is returned. Thus if
stride(1000,2000,100) is used in a variable by the dump_modify every command, it will generate the sequence
of output timesteps:
1000,1100,1200, ... ,1900,2000
The stride2(x,y,z,a,b,c) function is similar to the stride() function except it generates two sets of strided
timesteps, one at a coarser level and one at a finer level. Thus it is useful for debugging, e.g. to produce output
every timestep at the point in simulation when a problem occurs. X,y >= 0 and z > 0 and x <= y are required,
as are a,b >= 0 and c > 0 and a < b. Also, a >= x and b <= y are required so that the second stride is inside the
first. The generated timesteps increase in increments of z, starting at x, until a is reached. At that point the
timestep increases in increments of c, from a to b, then after b, increments by z are resumed until y is reached.
For any current timestep, the next timestep in the sequence is returned. Thus if
stride(1000,2000,100,1350,1360,1) is used in a variable by the dump_modify every command, it will generate
the sequence of output timesteps:
1000,1100,1200,1300,1350,1351,1352, ... 1359,1360,1400,1500, ... ,2000
The vdisplace(x,y) function takes 2 arguments: x = value0 and y = velocity, and uses the elapsed time to
change the value by a linear displacement due to the applied velocity over the course of a run, according to
this formula:
value = value0 + velocity*(timestep-startstep)*dt
where dt = the timestep size.
The run begins on startstep. Startstep can span multiple runs, using the start keyword of the run command.
See the run command for details of how to do this. Note that the thermo_style keyword elaplong =
timestep-startstep.
The swiggle(x,y,z) and cwiggle(x,y,z) functions each take 3 arguments: x = value0, y = amplitude, z = period.
They use the elapsed time to oscillate the value by a sin() or cos() function over the course of a run, according
to one of these formulas, where omega = 2 PI / period:
value = value0 + Amplitude * sin(omega*(timestep-startstep)*dt)
value = value0 + Amplitude * (1 - cos(omega*(timestep-startstep)*dt))
where dt = the timestep size.
LAMMPS Users Manual
685

The run begins on startstep. Startstep can span multiple runs, using the start keyword of the run command.
See the run command for details of how to do this. Note that the thermo_style keyword elaplong =
timestep-startstep.
Group and Region Functions
Group functions are specified as keywords followed by one or two parenthesized arguments. The first
argument ID is the group-ID. The dim argument, if it exists, is x or y or z. The dir argument, if it exists, is
xmin, xmax, ymin, ymax, zmin, or zmax. The dimdim argument, if it exists, is xx or yy or zz or xy or yz or xz.
The group function count() is the number of atoms in the group. The group functions mass() and charge() are
the total mass and charge of the group. Xcm() and vcm() return components of the position and velocity of the
center of mass of the group. Fcm() returns a component of the total force on the group of atoms. Bound()
returns the min/max of a particular coordinate for all atoms in the group. Gyration() computes the
radius-of-gyration of the group of atoms. See the compute gyration command for a definition of the formula.
Angmom() returns components of the angular momentum of the group of atoms around its center of mass.
Torque() returns components of the torque on the group of atoms around its center of mass, based on current
forces on the atoms. Inertia() returns one of 6 components of the symmetric inertia tensor of the group of
atoms around its center of mass, ordered as Ixx,Iyy,Izz,Ixy,Iyz,Ixz. Omega() returns components of the
angular velocity of the group of atoms around its center of mass.
Region functions are specified exactly the same way as group functions except they take an extra final
argument IDR which is the region ID. The function is computed for all atoms that are in both the group and
the region. If the group is "all", then the only criteria for atom inclusion is that it be in the region.
Special Functions
Special functions take specific kinds of arguments, meaning their arguments cannot be formulas themselves.
The sum(x), min(x), max(x), ave(x), trap(x), and slope(x) functions each take 1 argument which is of the form
"c_ID" or "c_ID[N]" or "f_ID" or "f_ID[N]" or "v_name". The first two are computes and the second two are
fixes; the ID in the reference should be replaced by the ID of a compute or fix defined elsewhere in the input
script. The compute or fix must produce either a global vector or array. If it produces a global vector, then the
notation without "[N]" should be used. If it produces a global array, then the notation with "[N]" should be
used, when N is an integer, to specify which column of the global array is being referenced. The last form of
argument "v_name" is for a vector-style variable where "name" is replaced by the name of the variable.
These functions operate on a global vector of inputs and reduce it to a single scalar value. This is analogous to
the operation of the compute reduce command, which performs similar operations on per-atom and local
vectors.
The sum() function calculates the sum of all the vector elements. The min() and max() functions find the
minimum and maximum element respectively. The ave() function is the same as sum() except that it divides
the result by the length of the vector.
The trap() function is the same as sum() except the first and last elements are multiplied by a weighting factor
of 1/2 when performing the sum. This effectively implements an integration via the trapezoidal rule on the
global vector of data. I.e. consider a set of points, equally spaced by 1 in their x coordinate: (1,V1), (2,V2), ...,
(N,VN), where the Vi are the values in the global vector of length N. The integral from 1 to N of these points
is trap(). When appropriately normalized by the timestep size, this function is useful for calculating integrals
LAMMPS Users Manual
686

of time-series data, like that generated by the fix ave/correlate command.
The slope() function uses linear regression to fit a line to the set of points, equally spaced by 1 in their x
coordinate: (1,V1), (2,V2), ..., (N,VN), where the Vi are the values in the global vector of length N. The
returned value is the slope of the line. If the line has a single point or is vertical, it returns 1.0e20.
The gmask(x) function takes 1 argument which is a group ID. It can only be used in atom-style variables. It
returns a 1 for atoms that are in the group, and a 0 for atoms that are not.
The rmask(x) function takes 1 argument which is a region ID. It can only be used in atom-style variables. It
returns a 1 for atoms that are in the geometric region, and a 0 for atoms that are not.
The grmask(x,y) function takes 2 arguments. The first is a group ID, and the second is a region ID. It can only
be used in atom-style variables. It returns a 1 for atoms that are in both the group and region, and a 0 for
atoms that are not in both.
The next(x) function takes 1 argument which is a variable ID (not "v_foo", just "foo"). It must be for a
file-style or atomfile-style variable. Each time the next() function is invoked (i.e. each time the equal-style or
atom-style variable is evaluated), the following steps occur.
For file-style variables, the current string value stored by the file-style variable is converted to a numeric value
and returned by the function. And the next string value in the file is read and stored. Note that if the line
previously read from the file was not a numeric string, then it will typically evaluate to 0.0, which is likely not
what you want.
For atomfile-style variables, the current per-atom values stored by the atomfile-style variable are returned by
the function. And the next set of per-atom values in the file is read and stored.
Since file-style and atomfile-style variables read and store the first line of the file or first set of per-atoms
values when they are defined in the input script, these are the value(s) that will be returned the first time the
next() function is invoked. If next() is invoked more times than there are lines or sets of lines in the file, the
variable is deleted, similar to how the next command operates.
Feature Functions
Feature functions allow to probe the running LAMMPS executable for whether specific features are either
active, defined, or available. The functions take two arguments, a category and a corresponding argument.
The arguments are strings thus cannot be formulas themselves (only $-style immediate variable expansion is
possible). Return value is either 1.0 or 0.0 depending on whether the function evaluates to true or false,
respectively.
The is_active() function allows to query for active settings which are grouped by categories. Currently
supported categories and arguments are:
package (argument = cuda or gpu or intel or kokkos or omp)• newton (argument = pair or bond or any)• pair (argument = single or respa or manybody or tail or shift)• comm_style (argument = brick or tiled)• min_style (argument = any of the compiled in minimizer styles)• run_style (argument = any of the compiled in run styles)• atom_style (argument = any of the compiled in atom styles)•
LAMMPS Users Manual
687

pair_style (argument = any of the compiled in pair styles)• bond_style (argument = any of the compiled in bond styles)• angle_style (argument = any of the compiled in angle styles)• dihedral_style (argument = any of the compiled in dihedral styles)• improper_style (argument = any of the compiled in improper styles)• kspace_style (argument = any of the compiled in kspace styles)•
Most of the settings are self-explanatory, the single argument in the pair category allows to check whether a
pair style supports a Pair::single() function as needed by compute group/group and others features or
LAMMPS, respa allows to check whether the inner/middle/outer mode of r-RESPA is supported. In the
various style categories, the checking is also done using suffix flags, if available and enabled.
Example 1: disable use of suffix for pppm when using GPU package (i.e. run it on the CPU concurrently to
running the pair style on the GPU), but do use the suffix otherwise (e.g. with USER-OMP).
pair_style lj/cut/coul/long 14.0
if $(is_active(package,gpu)) then "suffix off"
kspace_style pppm
Example 2: use r-RESPA with inner/outer cutoff, if supported by pair style, otherwise fall back to using pair
and reducing the outer time step
timestep $(2.0*(1.0+*is_active(pair,respa))
if $(is_active(pair,respa)) then "run_style respa 4 3 2 2 improper 1 inner 2 5.5 7.0 outer 3 kspace 4" else "run_style respa 3 3 2 improper 1 pair 2 kspace 3"
The is_defined() function allows to query categories like compute, dump, fix, group, region, and variable
whether an entry with the provided name or id is defined.
The is_available(category,name) function allows to query whether a specific optional feature is available, i.e.
compiled in. This currently works for the following categories: command, compute, fix, pair_style and feature.
For all categories except command and feature also appending active suffixes is tried before reporting failure.
The feature category is used to check the availability of compiled in features such as GZIP support, PNG
support, JPEG support, FFMPEG support, and C++ exceptions for error handling. Corresponding values for
name are gzip, png, jpeg, ffmpeg and exceptions.
This enables writing input scripts which only dump using a given format if the compiled binary supports it.
if "$(is_available(feature,png))" then "print 'PNG supported'" else "print 'PNG not supported'"
if "$(is_available(feature,ffmpeg)" then "dump 3 all movie 25 movie.mp4 type type zoom 1.6 adiam 1.0"
Atom Values and Vectors
Atom values take an integer argument I from 1 to N, where I is the atom-ID, e.g. x[243], which means use the
x coordinate of the atom with ID = 243. Or they can take a variable name, specified as v_name, where name is
the name of the variable, like x[v_myIndex]. The variable can be of any style except vector or atom or
atomfile variables. The variable is evaluated and the result is expected to be numeric and is cast to an integer
(i.e. 3.4 becomes 3), to use an an index, which must be a value from 1 to N. Note that a "formula" cannot be
used as the argument between the brackets, e.g. x[243+10] or x[v_myIndex+1] are not allowed. To do this a
single variable can be defined that contains the needed formula.
LAMMPS Users Manual
688

Note that the 0 < atom-ID <= N, where N is the largest atom ID in the system. If an ID is specified for an
atom that does not currently exist, then the generated value is 0.0.
Atom vectors generate one value per atom, so that a reference like "vx" means the x-component of each
atom's velocity will be used when evaluating the variable.
The meaning of the different atom values and vectors is mostly self-explanatory. Mol refers to the molecule
ID of an atom, and is only defined if an atom_style is being used that defines molecule IDs.
Note that many other atom attributes can be used as inputs to a variable by using the compute property/atom
command and then specifying a quantity from that compute.
Compute References
Compute references access quantities calculated by a compute. The ID in the reference should be replaced by
the ID of a compute defined elsewhere in the input script. As discussed in the doc page for the compute
command, computes can produce global, per-atom, or local values. Only global and per-atom values can be
used in a variable. Computes can also produce a scalar, vector, or array.
An equal-style variable can only use scalar values, which means a global scalar, or an element of a global or
per-atom vector or array. A vector-style variable can use scalar values or a global vector of values, or a
column of a global array of values. Atom-style variables can use global scalar values. They can also use
per-atom vector values, or a column of a per-atom array. See the doc pages for individual computes to see
what kind of values they produce.
Examples of different kinds of compute references are as follows. There is typically no ambiguity (see
exception below) as to what a reference means, since computes only produce either global or per-atom
quantities, never both.
c_ID global scalar, or per-atom vector
c_ID[I] Ith element of global vector, or atom I's value in per-atom vector, or Ith column from per-atom
array
c_ID[I][J] I,J element of global array, or atom I's Jth value in per-atom array
For I and J indices, integers can be specified or a variable name, specified as v_name, where name is the name
of the variable. The rules for this syntax are the same as for the "Atom Values and Vectors" discussion above.
One source of ambiguity for compute references is when a vector-style variable refers to a compute that
produces both a global scalar and a global vector. Consider a compute with ID "foo" that does this, referenced
as follows by variable "a", where "myVec" is another vector-style variable:
variable a vector c_foo*v_myVec
The reference "c_foo" could refer to either the global scalar or global vector produced by compute "foo". In
this case, "c_foo" will always refer to the global scalar, and "C_foo" can be used to reference the global
vector. Similarly if the compute produces both a global vector and global array, then "c_foo[I]" will always
refer to an element of the global vector, and "C_foo[I]" can be used to reference the Ith column of the global
array.
Note that if a variable containing a compute is evaluated directly in an input script (not during a run), then the
values accessed by the compute must be current. See the discussion below about "Variable Accuracy".
LAMMPS Users Manual
689

Fix References
Fix references access quantities calculated by a fix. The ID in the reference should be replaced by the ID of a
fix defined elsewhere in the input script. As discussed in the doc page for the fix command, fixes can produce
global, per-atom, or local values. Only global and per-atom values can be used in a variable. Fixes can also
produce a scalar, vector, or array. An equal-style variable can only use scalar values, which means a global
scalar, or an element of a global or per-atom vector or array. Atom-style variables can use the same scalar
values. They can also use per-atom vector values. A vector value can be a per-atom vector itself, or a column
of an per-atom array. See the doc pages for individual fixes to see what kind of values they produce.
The different kinds of fix references are exactly the same as the compute references listed in the above table,
where "c_" is replaced by "f_". Again, there is typically no ambiguity (see exception below) as to what a
reference means, since fixes only produce either global or per-atom quantities, never both.
f_ID global scalar, or per-atom vector
f_ID[I] Ith element of global vector, or atom I's value in per-atom vector, or Ith column from per-atom
array
f_ID[I][J] I,J element of global array, or atom I's Jth value in per-atom array
For I and J indices, integers can be specified or a variable name, specified as v_name, where name is the name
of the variable. The rules for this syntax are the same as for the "Atom Values and Vectors" discussion above.
One source of ambiguity for fix references is the same ambiguity discussed for compute references above.
Namely when a vector-style variable refers to a fix that produces both a global scalar and a global vector. The
solution is the same as for compute references. For a fix with ID "foo", "f_foo" will always refer to the global
scalar, and "F_foo" can be used to reference the global vector. And similarly for distinguishing between a fix's
global vector versus global array with "f_foo[I]" versus "F_foo[I]".
Note that if a variable containing a fix is evaluated directly in an input script (not during a run), then the
values accessed by the fix should be current. See the discussion below about "Variable Accuracy".
Note that some fixes only generate quantities on certain timesteps. If a variable attempts to access the fix on
non-allowed timesteps, an error is generated. For example, the fix ave/time command may only generate
averaged quantities every 100 steps. See the doc pages for individual fix commands for details.
Variable References
Variable references access quantities stored or calculated by other variables, which will cause those variables
to be evaluated. The name in the reference should be replaced by the name of a variable defined elsewhere in
the input script.
As discussed on this doc page, equal-style variables generate a single global numeric value, vector-style
variables generate a vector of global numeric values, and atom-style and atomfile-style variables generate a
per-atom vector of numeric values. All other variables store one or more strings.
The formula for an equal-style variable can use any style of variable including a vector_style or atom-style or
atomfile-style. For these 3 styles, a subscript must be used to access a single value from the vector-, atom-, or
atomfile-style variable. If a string-storing variable is used, the string is converted to a numeric value. Note that
this will typically produce a 0.0 if the string is not a numeric string, which is likely not what you want.
LAMMPS Users Manual
690

The formula for a vector-style variable can use any style of variable, including atom-style or atomfile-style
variables. For these 2 styles, a subscript must be used to access a single value from the atom-, or
atomfile-style variable.
The formula for an atom-style variable can use any style of variable, including other atom-style or
atomfile-style variables. If it uses a vector-style variable, a subscript must be used to access a single value
from the vector-style variable.
Examples of different kinds of variable references are as follows. There is no ambiguity as to what a reference
means, since variables produce only a global scalar or global vector or per-atom vector.
v_name global scalar from equal-style variable
v_name global vector from vector-style variable
v_name per-atom vector from atom-style or atomfile-style variable
v_name[I] Ith element of a global vector from vector-style variable
v_name[I] value of atom with ID = I from atom-style or atomfile-style variable
For the I index, an integer can be specified or a variable name, specified as v_name, where name is the name
of the variable. The rules for this syntax are the same as for the "Atom Values and Vectors" discussion above.
Immediate Evaluation of Variables:
If you want an equal-style variable to be evaluated immediately, it may be the case that you do not need to
define a variable at all. See Section 3.2 of the manual, which describes the use of "immediate" variables in an
input script, specified as $(formula) with parenthesis, where the formula has the same syntax as equal-style
variables described on this page. This effectively evaluates a formula immediately without using the variable
command to define a named variable.
More generally, there is a difference between referencing a variable with a leading $ sign (e.g. $x or ${abc})
versus with a leading "v_" (e.g. v_x or v_abc). The former can be used in any input script command,
including a variable command. The input script parser evaluates the reference variable immediately and
substitutes its value into the command. As explained in Section 3.2 for "Parsing rules", you can also use
un-named "immediate" variables for this purpose. For example, a string like this $((xlo+xhi)/2+sqrt(v_area))
in an input script command evaluates the string between the parenthesis as an equal-style variable formula.
Referencing a variable with a leading "v_" is an optional or required kind of argument for some commands
(e.g. the fix ave/chunk or dump custom or thermo_style commands) if you wish it to evaluate a variable
periodically during a run. It can also be used in a variable formula if you wish to reference a second variable.
The second variable will be evaluated whenever the first variable is evaluated.
As an example, suppose you use this command in your input script to define the variable "v" as
variable v equal vol
before a run where the simulation box size changes. You might think this will assign the initial volume to the
variable "v". That is not the case. Rather it assigns a formula which evaluates the volume (using the
thermo_style keyword "vol") to the variable "v". If you use the variable "v" in some other command like fix
ave/time then the current volume of the box will be evaluated continuously during the run.
If you want to store the initial volume of the system, you can do it this way:
LAMMPS Users Manual
691

variable v equal vol
variable v0 equal $v
The second command will force "v" to be evaluated (yielding the initial volume) and assign that value to the
variable "v0". Thus the command
thermo_style custom step v_v v_v0
would print out both the current and initial volume periodically during the run.
Note that it is a mistake to enclose a variable formula in double quotes if it contains variables preceded by $
signs. For example,
variable vratio equal "${vfinal}/${v0}"
This is because the quotes prevent variable substitution (see this section on parsing input script commands),
and thus an error will occur when the formula for "vratio" is evaluated later.
Variable Accuracy:
Obviously, LAMMPS attempts to evaluate variables containing formulas (equal and atom style variables)
accurately whenever the evaluation is performed. Depending on what is included in the formula, this may
require invoking a compute, either directly or indirectly via a thermo keyword, or accessing a value previously
calculated by a compute, or accessing a value calculated and stored by a fix. If the compute is one that
calculates the pressure or energy of the system, then these quantities need to be tallied during the evaluation of
the interatomic potentials (pair, bond, etc) on timesteps that the variable will need the values.
LAMMPS keeps track of all of this during a run or energy minimization. An error will be generated if you
attempt to evaluate a variable on timesteps when it cannot produce accurate values. For example, if a
thermo_style custom command prints a variable which accesses values stored by a fix ave/time command and
the timesteps on which thermo output is generated are not multiples of the averaging frequency used in the fix
command, then an error will occur.
An input script can also request variables be evaluated before or after or in between runs, e.g. by including
them in a print command. In this case, if a compute is needed to evaluate a variable (either directly or
indirectly), LAMMPS will not invoke the compute, but it will use a value previously calculated by the
compute, and can do this only if it was invoked on the current timestep. Fixes will always provide a quantity
needed by a variable, but the quantity may or may not be current. This leads to one of three kinds of behavior:
(1) The variable may be evaluated accurately. If it contains references to a compute or fix, and these values
were calculated on the last timestep of a preceding run, then they will be accessed and used by the variable
and the result will be accurate.
(2) LAMMPS may not be able to evaluate the variable and will generate an error message stating so. For
example, if the variable requires a quantity from a compute that has not been invoked on the current timestep,
LAMMPS will generate an error. This means, for example, that such a variable cannot be evaluated before the
first run has occurred. Likewise, in between runs, a variable containing a compute cannot be evaluated unless
the compute was invoked on the last timestep of the preceding run, e.g. by thermodynamic output.
One way to get around this problem is to perform a 0-timestep run before using the variable. For example,
these commands
LAMMPS Users Manual
692
variable t equal temp
print "Initial temperature = $t"
run 1000
will generate an error if the run is the first run specified in the input script, because generating a value for the
"t" variable requires a compute for calculating the temperature to be invoked.
However, this sequence of commands would be fine:
run 0
variable t equal temp
print "Initial temperature = $t"
run 1000
The 0-timestep run initializes and invokes various computes, including the one for temperature, so that the
value it stores is current and can be accessed by the variable "t" after the run has completed. Note that a
0-timestep run does not alter the state of the system, so it does not change the input state for the 1000-timestep
run that follows. Also note that the 0-timestep run must actually use and invoke the compute in question (e.g.
via thermo or dump output) in order for it to enable the compute to be used in a variable after the run. Thus if
you are trying to print a variable that uses a compute you have defined, you can insure it is invoked on the last
timestep of the preceding run by including it in thermodynamic output.
Unlike computes, fixes will never generate an error if their values are accessed by a variable in between runs.
They always return some value to the variable. However, the value may not be what you expect if the fix has
not yet calculated the quantity of interest or it is not current. For example, the fix indent command stores the
force on the indenter. But this is not computed until a run is performed. Thus if a variable attempts to print
this value before the first run, zeroes will be output. Again, performing a 0-timestep run before printing the
variable has the desired effect.
(3) The variable may be evaluated incorrectly and LAMMPS may have no way to detect this has occurred.
Consider the following sequence of commands:
pair_coeff 1 1 1.0 1.0
run 1000
pair_coeff 1 1 1.5 1.0
variable e equal pe
print "Final potential energy = $e"
The first run is performed using one setting for the pairwise potential defined by the pair_style and pair_coeff
commands. The potential energy is evaluated on the final timestep and stored by the compute pe compute (this
is done by the thermo_style command). Then a pair coefficient is changed, altering the potential energy of the
system. When the potential energy is printed via the "e" variable, LAMMPS will use the potential energy
value stored by the compute pe compute, thinking it is current. There are many other commands which could
alter the state of the system between runs, causing a variable to evaluate incorrectly.
The solution to this issue is the same as for case (2) above, namely perform a 0-timestep run before the
variable is evaluated to insure the system is up-to-date. For example, this sequence of commands would print
a potential energy that reflected the changed pairwise coefficient:
pair_coeff 1 1 1.0 1.0
run 1000
pair_coeff 1 1 1.5 1.0
run 0
variable e equal pe
LAMMPS Users Manual
693

print "Final potential energy = $e"
Restrictions:
Indexing any formula element by global atom ID, such as an atom value, requires the atom style to use a
global mapping in order to look up the vector indices. By default, only atom styles with molecular information
create global maps. The atom_modify map command can override the default, e.g. for atomic-style atom
styles.
All universe- and uloop-style variables defined in an input script must have the same number of values.
Related commands:
next, jump, include, temper, fix print, print
Default: none
LAMMPS Users Manual
694

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
velocity command
Syntax:
velocity group-ID style args keyword value ...
group-ID = ID of group of atoms whose velocity will be changed• style = create or set or scale or ramp or zero
create args = temp seed
temp = temperature value (temperature units)
seed = random # seed (positive integer)
set args = vx vy vz
vx,vy,vz = velocity value or NULL (velocity units)
any of vx,vy,vz van be a variable (see below)
scale arg = temp
temp = temperature value (temperature units)
ramp args = vdim vlo vhi dim clo chi
vdim = vx or vy or vz
vlo,vhi = lower and upper velocity value (velocity units)
dim = x or y or z
clo,chi = lower and upper coordinate bound (distance units)
zero arg = linear or angular
linear = zero the linear momentum
angular = zero the angular momentum
•
zero or more keyword/value pairs may be appended• keyword = dist or sum or mom or rot or temp or bias or loop or units
dist value = uniform or gaussian
sum value = no or yes
mom value = no or yes
rot value = no or yes
temp value = temperature compute ID
bias value = no or yes
loop value = all or local or geom
rigid value = fix-ID
fix-ID = ID of rigid body fix
units value = box or lattice
•
Examples:
velocity all create 300.0 4928459 rot yes dist gaussian
velocity border set NULL 4.0 v_vz sum yes units box
velocity flow scale 300.0
velocity flow ramp vx 0.0 5.0 y 5 25 temp mytemp
velocity all zero linear
Description:
Set or change the velocities of a group of atoms in one of several styles. For each style, there are required
arguments and optional keyword/value parameters. Not all options are used by each style. Each option has a
default as listed below.
LAMMPS Users Manual
695

The create style generates an ensemble of velocities using a random number generator with the specified seed
as the specified temperature.
The set style sets the velocities of all atoms in the group to the specified values. If any component is specified
as NULL, then it is not set. Any of the vx,vy,vz velocity components can be specified as an equal-style or
atom-style variable. If the value is a variable, it should be specified as v_name, where name is the variable
name. In this case, the variable will be evaluated, and its value used to determine the velocity component.
Note that if a variable is used, the velocity it calculates must be in box units, not lattice units; see the
discussion of the units keyword below.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters or other parameters.
Atom-style variables can specify the same formulas as equal-style variables but can also include per-atom
values, such as atom coordinates. Thus it is easy to specify a spatially-dependent velocity field.
The scale style computes the current temperature of the group of atoms and then rescales the velocities to the
specified temperature.
The ramp style is similar to that used by the compute temp/ramp command. Velocities ramped uniformly
from vlo to vhi are applied to dimension vx, or vy, or vz. The value assigned to a particular atom depends on
its relative coordinate value (in dim) from clo to chi. For the example above, an atom with y-coordinate of 10
(1/4 of the way from 5 to 25), would be assigned a x-velocity of 1.25 (1/4 of the way from 0.0 to 5.0). Atoms
outside the coordinate bounds (less than 5 or greater than 25 in this case), are assigned velocities equal to vlo
or vhi (0.0 or 5.0 in this case).
The zero style adjusts the velocities of the group of atoms so that the aggregate linear or angular momentum is
zero. No other changes are made to the velocities of the atoms. If the rigid option is specified (see below),
then the zeroing is performed on individual rigid bodies, as defined by the fix rigid or fix rigid/small
commands. In other words, zero linear will set the linear momentum of each rigid body to zero, and zero
angular will set the angular momentum of each rigid body to zero. This is done by adjusting the velocities of
the atoms in each rigid body.
All temperatures specified in the velocity command are in temperature units; see the units command. The
units of velocities and coordinates depend on whether the units keyword is set to box or lattice, as discussed
below.
For all styles, no atoms are assigned z-component velocities if the simulation is 2d; see the dimension
command.
The keyword/value options are used in the following ways by the various styles.
The dist keyword is used by create. The ensemble of generated velocities can be a uniform distribution from
some minimum to maximum value, scaled to produce the requested temperature. Or it can be a gaussian
distribution with a mean of 0.0 and a sigma scaled to produce the requested temperature.
The sum keyword is used by all styles, except zero. The new velocities will be added to the existing ones if
sum = yes, or will replace them if sum = no.
The mom and rot keywords are used by create. If mom = yes, the linear momentum of the newly created
ensemble of velocities is zeroed; if rot = yes, the angular momentum is zeroed.
LAMMPS Users Manual
696

*line
If specified, the temp keyword is used by create and scale to specify a compute that calculates temperature in
a desired way, e.g. by first subtracting out a velocity bias, as discussed in Section 6.16 of the doc pages. If this
keyword is not specified, create and scale calculate temperature using a compute that is defined internally as
follows:
compute velocity_temp group-ID temp
where group-ID is the same ID used in the velocity command. i.e. the group of atoms whose velocity is being
altered. This compute is deleted when the velocity command is finished. See the compute temp command for
details. If the calculated temperature should have degrees-of-freedom removed due to fix constraints (e.g.
SHAKE or rigid-body constraints), then the appropriate fix command must be specified before the velocity
command is issued.
The bias keyword with a yes setting is used by create and scale, but only if the temp keyword is also used to
specify a compute that calculates temperature in a desired way. If the temperature compute also calculates a
velocity bias, the the bias is subtracted from atom velocities before the create and scale operations are
performed. After the operations, the bias is added back to the atom velocities. See Section 6.16 of the doc
pages for more discussion of temperature computes with biases. Note that the velocity bias is only applied to
atoms in the temperature compute specified with the temp keyword.
As an example, assume atoms are currently streaming in a flow direction (which could be separately
initialized with the ramp style), and you wish to initialize their thermal velocity to a desired temperature. In
this context thermal velocity means the per-particle velocity that remains when the streaming velocity is
subtracted. This can be done using the create style with the temp keyword specifying the ID of a compute
temp/ramp or compute temp/profile command, and the bias keyword set to a yes value.
The loop keyword is used by create in the following ways.
If loop = all, then each processor loops over all atoms in the simulation to create velocities, but only stores
velocities for atoms it owns. This can be a slow loop for a large simulation. If atoms were read from a data
file, the velocity assigned to a particular atom will be the same, independent of how many processors are
being used. This will not be the case if atoms were created using the create_atoms command, since atom IDs
will likely be assigned to atoms differently.
If loop = local, then each processor loops over only its atoms to produce velocities. The random number seed
is adjusted to give a different set of velocities on each processor. This is a fast loop, but the velocity assigned
to a particular atom will depend on which processor owns it. Thus the results will always be different when a
simulation is run on a different number of processors.
If loop = geom, then each processor loops over only its atoms. For each atom a unique random number seed is
created, based on the atom's xyz coordinates. A velocity is generated using that seed. This is a fast loop and
the velocity assigned to a particular atom will be the same, independent of how many processors are used.
However, the set of generated velocities may be more correlated than if the all or local keywords are used.
Note that the loop geom keyword will not necessarily assign identical velocities for two simulations run on
different machines. This is because the computations based on xyz coordinates are sensitive to tiny differences
in the double-precision value for a coordinate as stored on a particular machine.
LAMMPS Users Manual
697

The rigid keyword only has meaning when used with the zero style. It allows specification of a fix-ID for one
of the rigid-body fix variants which defines a set of rigid bodies. The zeroing of linear or angular momentum
is then performed for each rigid body defined by the fix, as described above.
The units keyword is used by set and ramp. If units = box, the velocities and coordinates specified in the
velocity command are in the standard units described by the units command (e.g. Angstroms/fmsec for real
units). If units = lattice, velocities are in units of lattice spacings per time (e.g. spacings/fmsec) and
coordinates are in lattice spacings. The lattice command must have been previously used to define the lattice
spacing.
Restrictions:
Assigning a temperature via the create style to a system with rigid bodies or SHAKE constraints may not
have the desired outcome for two reasons. First, the velocity command can be invoked before all of the
relevant fixes are created and initialized and the number of adjusted degrees of freedom (DOFs) is known.
Thus it is not possible to compute the target temperature correctly. Second, the assigned velocities may be
partially canceled when constraints are first enforced, leading to a different temperature than desired. A
workaround for this is to perform a run 0 command, which insures all DOFs are accounted for properly, and
then rescale the temperature to the desired value before performing a simulation. For example:
velocity all create 300.0 12345
run 0 # temperature may not be 300K
velocity all scale 300.0 # now it should be
Related commands:
fix rigid, fix shake, lattice
Default:
The keyword defaults are dist = uniform, sum = no, mom = yes, rot = no, bias = no, loop = all, and units =
lattice. The temp and rigid keywords are not defined by default.
LAMMPS Users Manual
698

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
write_coeff command
Syntax:
write_coeff file
file = name of data file to write out
Examples:
write_coeff polymer.coeff
Description:
Write a text format file with the currently defined force field coefficients in a way, that it can be read by
LAMMPS with the include command. In combination with the nocoeff option of write_data this can be used
to move the Coeffs sections from a data file into a separate file.
NOTE: The write_coeff command is not yet fully implemented in two respects. First, some pair styles do not
yet write their coefficient information into the coeff file. This means you will need to specify that information
in your input script that reads the data file, via the pair_coeff command.
Restrictions:
none
Related commands:
read_data, write_restart, write_data
LAMMPS Users Manual
699

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
write_data command
Syntax:
write_data file keyword value ...
file = name of data file to write out• zero or more keyword/value pairs may be appended• keyword = pair or nocoeff
nocoeff = do not write out force field info
pair value = ii or ij
ii = write one line of pair coefficient info per atom type
ij = write one line of pair coefficient info per IJ atom type pair
•
Examples:
write_data data.polymer
write_data data.*
Description:
Write a data file in text format of the current state of the simulation. Data files can be read by the read data
command to begin a simulation. The read_data command also describes their format.
Similar to dump files, the data filename can contain a "*" wild-card character. The "*" is replaced with the
current timestep value.
NOTE: The write-data command is not yet fully implemented in two respects. First, most pair styles do not
yet write their coefficient information into the data file. This means you will need to specify that information
in your input script that reads the data file, via the pair_coeff command. Second, a few of the atom styles
(body, ellipsoid, line, tri) that store auxiliary "bonus" information about aspherical particles, do not yet write
the bonus info into the data file. Both these functionalities will be added to the write_data command later.
Because a data file is in text format, if you use a data file written out by this command to restart a simulation,
the initial state of the new run will be slightly different than the final state of the old run (when the file was
written) which was represented internally by LAMMPS in binary format. A new simulation which reads the
data file will thus typically diverge from a simulation that continued in the original input script.
If you want to do more exact restarts, using binary files, see the restart, write_restart, and read_restart
commands. You can also convert binary restart files to text data files, after a simulation has run, using the -r
command-line switch.
NOTE: Only limited information about a simulation is stored in a data file. For example, no information about
atom groups and fixes are stored. Binary restart files store more information.
Bond interactions (angle, etc) that have been turned off by the fix shake or delete_bonds command will be
written to a data file as if they are turned on. This means they will need to be turned off again in a new run
after the data file is read.
LAMMPS Users Manual
700

Bonds that are broken (e.g. by a bond-breaking potential) are not written to the data file. Thus these bonds will
not exist when the data file is read.
The nocoeff keyword requests that no force field parameters should be written to the data file. This can be
very helpful, if one wants to make significant changes to the force field or if the parameters are read in
separately anyway, e.g. from an include file.
The pair keyword lets you specify in what format the pair coefficient information is written into the data file.
If the value is specified as ii, then one line per atom type is written, to specify the coefficients for each of the
I=J interactions. This means that no cross-interactions for I != J will be specified in the data file and the pair
style will apply its mixing rule, as documented on individual pair_style doc pages. Of course this behavior can
be overridden in the input script after reading the data file, by specifying additional pair_coeff commands for
any desired I,J pairs.
If the value is specified as ij, then one line of coefficients is written for all I,J pairs where I <= J. These
coefficients will include any specific settings made in the input script up to that point. The presence of these I
!= J coefficients in the data file will effectively turn off the default mixing rule for the pair style. Again, the
coefficient values in the data file can can be overridden in the input script after reading the data file, by
specifying additional pair_coeff commands for any desired I,J pairs.
Restrictions:
This command requires inter-processor communication to migrate atoms before the data file is written. This
means that your system must be ready to perform a simulation before using this command (force fields setup,
atom masses initialized, etc).
Related commands:
read_data, write_restart
Default:
The option defaults are pair = ii.
LAMMPS Users Manual
701

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
write_dump command
Syntax:
write_dump group-ID style file dump-args modify dump_modify-args
group-ID = ID of the group of atoms to be dumped• style = any of the supported dump styles• file = name of file to write dump info to• dump-args = any additional args needed for a particular dump style• modify = all args after this keyword are passed to dump_modify (optional)• dump-modify-args = args for dump_modify (optional)•
Examples:
write_dump all atom dump.atom
write_dump subgroup atom dump.run.bin
write_dump all custom dump.myforce.* id type x y vx fx
write_dump flow custom dump.%.myforce id type c_myF[3] v_ke modify sort id
write_dump all xyz system.xyz modify sort id element O H
write_dump all image snap*.jpg type type size 960 960 modify backcolor white
write_dump all image snap*.jpg element element &
bond atom 0.3 shiny 0.1 ssao yes 6345 0.2 size 1600 1600 &
modify backcolor white element C C O H N C C C O H H S O H
Description:
Dump a single snapshot of atom quantities to one or more files for the current state of the system. This is a
one-time immediate operation, in contrast to the dump command which will will set up a dump style to write
out snapshots periodically during a running simulation.
The syntax for this command is mostly identical to that of the dump and dump_modify commands as if they
were concatenated together, with the following exceptions: There is no need for a dump ID or dump
frequency and the keyword modify is added. The latter is so that the full range of dump_modify options can be
specified for the single snapshot, just as they can be for multiple snapshots. The modify keyword separates the
arguments that would normally be passed to the dump command from those that would be given the
dump_modify. Both support optional arguments and thus LAMMPS needs to be able to cleanly separate the
two sets of args.
Note that if the specified filename uses wildcard characters "*" or "%", as supported by the dump command,
they will operate in the same fashion to create the new filename(s). Normally, dump image files require a
filename with a "*" character for the timestep. That is not the case for the write_dump command; no wildcard
"*" character is necessary.
Restrictions:
All restrictions for the dump and dump_modify commands apply to this command as well, with the exception
of the dump image filename not requiring a wildcard "*" character, as noted above.
LAMMPS Users Manual
702
Since dumps are normally written during a run or energy minimization, the simulation has to be ready to run
before this command can be used. Similarly, if the dump requires information from a compute, fix, or
variable, the information needs to have been calculated for the current timestep (e.g. by a prior run), else
LAMMPS will generate an error message.
For example, it is not possible to dump per-atom energy with this command before a run has been performed,
since no energies and forces have yet been calculated. See the variable doc page section on Variable Accuracy
for more information on this topic.
Related commands:
dump, dump image, dump_modify
Default:
The defaults are listed on the doc pages for the dump and dump image and dump_modify commands.
LAMMPS Users Manual
703

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
write_restart command
Syntax:
write_restart file keyword value ...
file = name of file to write restart information to• zero or more keyword/value pairs may be appended• keyword = fileper or nfile
fileper arg = Np
Np = write one file for every this many processors
nfile arg = Nf
Nf = write this many files, one from each of Nf processors
•
Examples:
write_restart restart.equil
write_restart restart.equil.mpiio
write_restart poly.%.* nfile 10
Description:
Write a binary restart file of the current state of the simulation.
During a long simulation, the restart command is typically used to output restart files periodically. The
write_restart command is useful after a minimization or whenever you wish to write out a single current
restart file.
Similar to dump files, the restart filename can contain two wild-card characters. If a "*" appears in the
filename, it is replaced with the current timestep value. If a "%" character appears in the filename, then one
file is written by each processor and the "%" character is replaced with the processor ID from 0 to P-1. An
additional file with the "%" replaced by "base" is also written, which contains global information. For
example, the files written for filename restart.% would be restart.base, restart.0, restart.1, ... restart.P-1. This
creates smaller files and can be a fast mode of output and subsequent input on parallel machines that support
parallel I/O. The optional fileper and nfile keywords discussed below can alter the number of files written.
The restart file can also be written in parallel as one large binary file via the MPI-IO library, which is part of
the MPI standard for versions 2.0 and above. Using MPI-IO requires two steps. First, build LAMMPS with its
MPIIO package installed, e.g.
make yes-mpiio # installs the MPIIO package
make mpi # build LAMMPS for your platform
Second, use a restart filename which contains ".mpiio". Note that it does not have to end in ".mpiio", just
contain those characters. Unlike MPI-IO dump files, a particular restart file must be both written and read
using MPI-IO.
Restart files can be read by a read_restart command to restart a simulation from a particular state. Because the
file is binary (to enable exact restarts), it may not be readable on another machine. In this case, you can use
LAMMPS Users Manual
704

the -r command-line switch to convert a restart file to a data file.
NOTE: Although the purpose of restart files is to enable restarting a simulation from where it left off, not all
information about a simulation is stored in the file. For example, the list of fixes that were specified during the
initial run is not stored, which means the new input script must specify any fixes you want to use. Even when
restart information is stored in the file, as it is for some fixes, commands may need to be re-specified in the
new input script, in order to re-use that information. Details are usually given in the documentation of the
respective command. Also, see the read_restart command for general information about what is stored in a
restart file.
The optional nfile or fileper keywords can be used in conjunction with the "%" wildcard character in the
specified restart file name. As explained above, the "%" character causes the restart file to be written in
pieces, one piece for each of P processors. By default P = the number of processors the simulation is running
on. The nfile or fileper keyword can be used to set P to a smaller value, which can be more efficient when
running on a large number of processors.
The nfile keyword sets P to the specified Nf value. For example, if Nf = 4, and the simulation is running on
100 processors, 4 files will be written, by processors 0,25,50,75. Each will collect information from itself and
the next 24 processors and write it to a restart file.
For the fileper keyword, the specified value of Np means write one file for every Np processors. For example,
if Np = 4, every 4th processor (0,4,8,12,etc) will collect information from itself and the next 3 processors and
write it to a restart file.
Restrictions:
This command requires inter-processor communication to migrate atoms before the restart file is written. This
means that your system must be ready to perform a simulation before using this command (force fields setup,
atom masses initialized, etc).
To write and read restart files in parallel with MPI-IO, the MPIIO package must be installed.
Related commands:
restart, read_restart, write_data
Default: none
LAMMPS Users Manual
705

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix adapt command
Syntax:
fix ID group-ID adapt N attribute args ... keyword value ...
ID, group-ID are documented in fix command• adapt = style name of this fix command• N = adapt simulation settings every this many timesteps• one or more attribute/arg pairs may be appended• attribute = pair or kspace or atom
pair args = pstyle pparam I J v_name
pstyle = pair style name, e.g. lj/cut
pparam = parameter to adapt over time
I,J = type pair(s) to set parameter for
v_name = variable with name that calculates value of pparam
kspace arg = v_name
v_name = variable with name that calculates scale factor on K-space terms
atom args = aparam v_name
aparam = parameter to adapt over time
v_name = variable with name that calculates value of aparam
•
zero or more keyword/value pairs may be appended• keyword = scale or reset
scale value = no or yes
no = the variable value is the new setting
yes = the variable value multiplies the original setting
reset value = no or yes
no = values will remain altered at the end of a run
yes = reset altered values to their original values at the end of a run
•
Examples:
fix 1 all adapt 1 pair soft a 1 1 v_prefactor
fix 1 all adapt 1 pair soft a 2* 3 v_prefactor
fix 1 all adapt 1 pair lj/cut epsilon * * v_scale1 coul/cut scale 3 3 v_scale2 scale yes reset yes
fix 1 all adapt 10 atom diameter v_size
Description:
Change or adapt one or more specific simulation attributes or settings over time as a simulation runs. Pair
potential and K-space and atom attributes which can be varied by this fix are discussed below. Many other
fixes can also be used to time-vary simulation parameters, e.g. the "fix deform" command will change the
simulation box size/shape and the "fix move" command will change atom positions and velocities in a
prescribed manner. Also note that many commands allow variables as arguments for specific parameters, if
described in that manner on their doc pages. An equal-style variable can calculate a time-dependent quantity,
so this is another way to vary a simulation parameter over time.
If N is specified as 0, the specified attributes are only changed once, before the simulation begins. This is all
that is needed if the associated variables are not time-dependent. If N > 0, then changes are made every N
steps during the simulation, presumably with a variable that is time-dependent.
LAMMPS Users Manual
706
Depending on the value of the reset keyword, attributes changed by this fix will or will not be reset back to
their original values at the end of a simulation. Even if reset is specified as yes, a restart file written during a
simulation will contain the modified settings.
If the scale keyword is set to no, then the value the parameter is set to will be whatever the variable generates.
If the scale keyword is set to yes, then the value of the altered parameter will be the initial value of that
parameter multiplied by whatever the variable generates. I.e. the variable is now a "scale factor" applied in
(presumably) a time-varying fashion to the parameter.
Note that whether scale is no or yes, internally, the parameters themselves are actually altered by this fix.
Make sure you use the reset yes option if you want the parameters to be restored to their initial values after the
run.
The pair keyword enables various parameters of potentials defined by the pair_style command to be changed,
if the pair style supports it. Note that the pair_style and pair_coeff commands must be used in the usual
manner to specify these parameters initially; the fix adapt command simply overrides the parameters.
The pstyle argument is the name of the pair style. If pair_style hybrid or hybrid/overlay is used, pstyle should
be a sub-style name. If there are multiple sub-styles using the same pair style, then pstyle should be specified
as "style:N" where N is which instance of the pair style you wish to adapt, e.g. the first, second, etc. For
example, pstyle could be specified as "soft" or "lubricate" or "lj/cut:1" or "lj/cut:2". The pparam argument is
the name of the parameter to change. This is the current list of pair styles and parameters that can be varied by
this fix. See the doc pages for individual pair styles and their energy formulas for the meaning of these
parameters:
born a,b,c type pairs
buck a,c type pairs
coul/cut scale type pairs
coul/debye scale type pairs
coul/long scale type pairs
eam, eam/alloy, eam/fs scale type pairs
lj/cut epsilon,sigma type pairs
lj/expand epsilon,sigma,delta type pairs
lj/sf/dipole/sf epsilon,sigma,scale type pairs
lubricate mu global
gauss a type pairs
morse d0,r0,alpha type pairs
soft a type pairs
kim PARAM_FREE_*:i,j,... global
NOTE: It is easy to add new pairwise potentials and their parameters to this list. All it typically takes is
adding an extract() method to the pair_*.cpp file associated with the potential.
Some parameters are global settings for the pair style, e.g. the viscosity setting "mu" for pair_style lubricate.
For pair_kim, all free parameters supported by the KIM Model are available (e.g., PARAM_FREE_sigmas
provided by the LennardJones612_Universal__MO_826355984548_001 Model). If the free parameter
corresponds to an array, then the particular array element to be adapted must be specified (e.g.,
"PARAM_FREE_sigmas:10", to adapt the tenth entry of the sigmas array). Other parameters apply to atom
type pairs within the pair style, e.g. the prefactor "a" for pair_style soft.
LAMMPS Users Manual
707

Note that for many of the potentials, the parameter that can be varied is effectively a prefactor on the entire
energy expression for the potential, e.g. the lj/cut epsilon. The parameters listed as "scale" are exactly that,
since the energy expression for the coul/cut potential (for example) has no labeled prefactor in its formula. To
apply an effective prefactor to some potentials, multiple parameters need to be altered. For example, the
Buckingham potential needs both the A and C terms altered together. To scale the Buckingham potential, you
should thus list the pair style twice, once for A and once for C.
If a type pair parameter is specified, the I and J settings should be specified to indicate which type pairs to
apply it to. If a global parameter is specified, the I and J settings still need to be specified, but are ignored.
Similar to the pair_coeff command, I and J can be specified in one of two ways. Explicit numeric values can
be used for each, as in the 1st example above. I <= J is required. LAMMPS sets the coefficients for the
symmetric J,I interaction to the same values.
A wild-card asterisk can be used in place of or in conjunction with the I,J arguments to set the coefficients for
multiple pairs of atom types. This takes the form "*" or "*n" or "n*" or "m*n". If N = the number of atom
types, then an asterisk with no numeric values means all types from 1 to N. A leading asterisk means all types
from 1 to n (inclusive). A trailing asterisk means all types from n to N (inclusive). A middle asterisk means all
types from m to n (inclusive). Note that only type pairs with I <= J are considered; if asterisks imply type
pairs where J < I, they are ignored.
IMPROTANT NOTE: If pair_style hybrid or hybrid/overlay is being used, then the pstyle will be a sub-style
name. You must specify I,J arguments that correspond to type pair values defined (via the pair_coeff
command) for that sub-style.
The v_name argument for keyword pair is the name of an equal-style variable which will be evaluated each
time this fix is invoked to set the parameter to a new value. It should be specified as v_name, where name is
the variable name. Equal-style variables can specify formulas with various mathematical functions, and
include thermo_style command keywords for the simulation box parameters and timestep and elapsed time.
Thus it is easy to specify parameters that change as a function of time or span consecutive runs in a
continuous fashion. For the latter, see the start and stop keywords of the run command and the elaplong
keyword of thermo_style custom for details.
For example, these commands would change the prefactor coefficient of the pair_style soft potential from
10.0 to 30.0 in a linear fashion over the course of a simulation:
variable prefactor equal ramp(10,30)
fix 1 all adapt 1 pair soft a * * v_prefactor
The kspace keyword used the specified variable as a scale factor on the energy, forces, virial calculated by
whatever K-Space solver is defined by the kspace_style command. If the variable has a value of 1.0, then the
solver is unaltered.
The kspace keyword works this way whether the scale keyword is set to no or yes.
The atom keyword enables various atom properties to be changed. The aparam argument is the name of the
parameter to change. This is the current list of atom parameters that can be varied by this fix:
charge = charge on particle• diameter = diameter of particle•
LAMMPS Users Manual
708

The v_name argument of the atom keyword is the name of an equal-style variable which will be evaluated
each time this fix is invoked to set the parameter to a new value. It should be specified as v_name, where
name is the variable name. See the discussion above describing the formulas associated with equal-style
variables. The new value is assigned to the corresponding attribute for all atoms in the fix group.
NOTE: The atom keyword works this way whether the scale keyword is set to no or yes. I.e. the use of scale
yes is not yet supported by the atom keyword.
If the atom parameter is diameter and per-atom density and per-atom mass are defined for particles (e.g.
atom_style granular), then the mass of each particle is also changed when the diameter changes (density is
assumed to stay constant).
For example, these commands would shrink the diameter of all granular particles in the "center" group from
1.0 to 0.1 in a linear fashion over the course of a 1000-step simulation:
variable size equal ramp(1.0,0.1)
fix 1 center adapt 10 atom diameter v_size
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
For rRESPA time integration, this fix changes parameters on the outermost rRESPA level.
Restrictions: none
Related commands:
compute ti
Default:
The option defaults are scale = no, reset = no.
LAMMPS Users Manual
709

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix adapt/fep command
Syntax:
fix ID group-ID adapt/fep N attribute args ... keyword value ...
ID, group-ID are documented in fix command• adapt/fep = style name of this fix command• N = adapt simulation settings every this many timesteps• one or more attribute/arg pairs may be appended• attribute = pair or kspace or atom
pair args = pstyle pparam I J v_name
pstyle = pair style name, e.g. lj/cut
pparam = parameter to adapt over time
I,J = type pair(s) to set parameter for
v_name = variable with name that calculates value of pparam
kspace arg = v_name
v_name = variable with name that calculates scale factor on K-space terms
atom args = aparam v_name
aparam = parameter to adapt over time
I = type(s) to set parameter for
v_name = variable with name that calculates value of aparam
•
zero or more keyword/value pairs may be appended• keyword = scale or reset or after
scale value = no or yes
no = the variable value is the new setting
yes = the variable value multiplies the original setting
reset value = no or yes
no = values will remain altered at the end of a run
yes = reset altered values to their original values at the end
of a run
after value = no or yes
no = parameters are adapted at timestep N
yes = parameters are adapted one timestep after N
•
Examples:
fix 1 all adapt/fep 1 pair soft a 1 1 v_prefactor
fix 1 all adapt/fep 1 pair soft a 2* 3 v_prefactor
fix 1 all adapt/fep 1 pair lj/cut epsilon * * v_scale1 coul/cut scale 3 3 v_scale2 scale yes reset yes
fix 1 all adapt/fep 10 atom diameter 1 v_size
Description:
Change or adapt one or more specific simulation attributes or settings over time as a simulation runs.
This is an enhanced version of the fix adapt command with two differences,
It is possible to modify the charges of chosen atom types only, instead of scaling all the charges in the
system.
•
There is a new option after for better compatibility with "fix ave/time".•
LAMMPS Users Manual
710
This version is suited for free energy calculations using compute ti or compute fep.
If N is specified as 0, the specified attributes are only changed once, before the simulation begins. This is all
that is needed if the associated variables are not time-dependent. If N > 0, then changes are made every N
steps during the simulation, presumably with a variable that is time-dependent.
Depending on the value of the reset keyword, attributes changed by this fix will or will not be reset back to
their original values at the end of a simulation. Even if reset is specified as yes, a restart file written during a
simulation will contain the modified settings.
If the scale keyword is set to no, then the value the parameter is set to will be whatever the variable generates.
If the scale keyword is set to yes, then the value of the altered parameter will be the initial value of that
parameter multiplied by whatever the variable generates. I.e. the variable is now a "scale factor" applied in
(presumably) a time-varying fashion to the parameter. Internally, the parameters themselves are actually
altered; make sure you use the reset yes option if you want the parameters to be restored to their initial values
after the run.
If the after keyword is set to yes, then the parameters are changed one timestep after the multiple of N. In this
manner, if a fix such as "fix ave/time" is used to calculate averages at every N timesteps, all the contributions
to the average will be obtained with the same values of the parameters.
The pair keyword enables various parameters of potentials defined by the pair_style command to be changed,
if the pair style supports it. Note that the pair_style and pair_coeff commands must be used in the usual
manner to specify these parameters initially; the fix adapt command simply overrides the parameters.
The pstyle argument is the name of the pair style. If pair_style hybrid or hybrid/overlay is used, pstyle should
be a sub-style name. For example, pstyle could be specified as "soft" or "lubricate". The pparam argument is
the name of the parameter to change. This is the current list of pair styles and parameters that can be varied by
this fix. See the doc pages for individual pair styles and their energy formulas for the meaning of these
parameters:
born a,b,c type pairs
buck a,c type pairs
coul/cut scale type pairs
coul/debye scale type pairs
coul/long scale type pairs
lj/cut epsilon,sigma type pairs
lj/expand epsilon,sigma,delta type pairs
lubricate mu global
gauss a type pairs
soft a type pairs
NOTE: It is easy to add new potentials and their parameters to this list. All it typically takes is adding an
extract() method to the pair_*.cpp file associated with the potential.
Some parameters are global settings for the pair style, e.g. the viscosity setting "mu" for pair_style lubricate.
Other parameters apply to atom type pairs within the pair style, e.g. the prefactor "a" for pair_style soft.
Note that for many of the potentials, the parameter that can be varied is effectively a prefactor on the entire
LAMMPS Users Manual
711

energy expression for the potential, e.g. the lj/cut epsilon. The parameters listed as "scale" are exactly that,
since the energy expression for the coul/cut potential (for example) has no labeled prefactor in its formula. To
apply an effective prefactor to some potentials, multiple parameters need to be altered. For example, the
Buckingham potential needs both the A and C terms altered together. To scale the Buckingham potential, you
should thus list the pair style twice, once for A and once for C.
If a type pair parameter is specified, the I and J settings should be specified to indicate which type pairs to
apply it to. If a global parameter is specified, the I and J settings still need to be specified, but are ignored.
Similar to the pair_coeff command, I and J can be specified in one of two ways. Explicit numeric values can
be used for each, as in the 1st example above. I <= J is required. LAMMPS sets the coefficients for the
symmetric J,I interaction to the same values.
A wild-card asterisk can be used in place of or in conjunction with the I,J arguments to set the coefficients for
multiple pairs of atom types. This takes the form "*" or "*n" or "n*" or "m*n". If N = the number of atom
types, then an asterisk with no numeric values means all types from 1 to N. A leading asterisk means all types
from 1 to n (inclusive). A trailing asterisk means all types from n to N (inclusive). A middle asterisk means all
types from m to n (inclusive). Note that only type pairs with I <= J are considered; if asterisks imply type
pairs where J < I, they are ignored.
IMPROTANT NOTE: If pair_style hybrid or hybrid/overlay is being used, then the pstyle will be a sub-style
name. You must specify I,J arguments that correspond to type pair values defined (via the pair_coeff
command) for that sub-style.
The v_name argument for keyword pair is the name of an equal-style variable which will be evaluated each
time this fix is invoked to set the parameter to a new value. It should be specified as v_name, where name is
the variable name. Equal-style variables can specify formulas with various mathematical functions, and
include thermo_style command keywords for the simulation box parameters and timestep and elapsed time.
Thus it is easy to specify parameters that change as a function of time or span consecutive runs in a
continuous fashion. For the latter, see the start and stop keywords of the run command and the elaplong
keyword of thermo_style custom for details.
For example, these commands would change the prefactor coefficient of the pair_style soft potential from
10.0 to 30.0 in a linear fashion over the course of a simulation:
variable prefactor equal ramp(10,30)
fix 1 all adapt 1 pair soft a * * v_prefactor
The kspace keyword used the specified variable as a scale factor on the energy, forces, virial calculated by
whatever K-Space solver is defined by the kspace_style command. If the variable has a value of 1.0, then the
solver is unaltered.
The kspace keyword works this way whether the scale keyword is set to no or yes.
The atom keyword enables various atom properties to be changed. The aparam argument is the name of the
parameter to change. This is the current list of atom parameters that can be varied by this fix:
charge = charge on particle• diameter = diameter of particle•
LAMMPS Users Manual
712

The I argument indicates which atom types are affected. A wild-card asterisk can be used in place of or in
conjunction with the I argument to set the coefficients for multiple atom types.
The v_name argument of the atom keyword is the name of an equal-style variable which will be evaluated
each time this fix is invoked to set the parameter to a new value. It should be specified as v_name, where
name is the variable name. See the discussion above describing the formulas associated with equal-style
variables. The new value is assigned to the corresponding attribute for all atoms in the fix group.
If the atom parameter is diameter and per-atom density and per-atom mass are defined for particles (e.g.
atom_style granular), then the mass of each particle is also changed when the diameter changes (density is
assumed to stay constant).
For example, these commands would shrink the diameter of all granular particles in the "center" group from
1.0 to 0.1 in a linear fashion over the course of a 1000-step simulation:
variable size equal ramp(1.0,0.1)
fix 1 center adapt 10 atom diameter * v_size
For rRESPA time integration, this fix changes parameters on the outermost rRESPA level.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
compute fep, fix adapt, compute ti
Default:
The option defaults are scale = no, reset = no, after = no.
LAMMPS Users Manual
713

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix addforce command
Syntax:
fix ID group-ID addforce fx fy fz keyword value ...
ID, group-ID are documented in fix command• addforce = style name of this fix command• fx,fy,fz = force component values (force units)
any of fx,fy,fz can be a variable (see below)
•
zero or more keyword/value pairs may be appended to args• keyword = every or region or energy
every value = Nevery
Nevery = add force every this many timesteps
region value = region-ID
region-ID = ID of region atoms must be in to have added force
energy value = v_name
v_name = variable with name that calculates the potential energy of each atom in the added force field
•
Examples:
fix kick flow addforce 1.0 0.0 0.0
fix kick flow addforce 1.0 0.0 v_oscillate
fix ff boundary addforce 0.0 0.0 v_push energy v_espace
Description:
Add fx,fy,fz to the corresponding component of force for each atom in the group. This command can be used
to give an additional push to atoms in a simulation, such as for a simulation of Poiseuille flow in a channel.
Any of the 3 quantities defining the force components can be specified as an equal-style or atom-style
variable, namely fx, fy, fz. If the value is a variable, it should be specified as v_name, where name is the
variable name. In this case, the variable will be evaluated each timestep, and its value(s) used to determine the
force component.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent force field.
Atom-style variables can specify the same formulas as equal-style variables but can also include per-atom
values, such as atom coordinates. Thus it is easy to specify a spatially-dependent force field with optional
time-dependence as well.
If the every keyword is used, the Nevery setting determines how often the forces are applied. The default value
is 1, for every timestep.
If the region keyword is used, the atom must also be in the specified geometric region in order to have force
added to it.
LAMMPS Users Manual
714

Adding a force to atoms implies a change in their potential energy as they move due to the applied force field.
For dynamics via the "run" command, this energy can be optionally added to the system's potential energy for
thermodynamic output (see below). For energy minimization via the "minimize" command, this energy must
be added to the system's potential energy to formulate a self-consistent minimization problem (see below).
The energy keyword is not allowed if the added force is a constant vector F = (fx,fy,fz), with all components
defined as numeric constants and not as variables. This is because LAMMPS can compute the energy for each
atom directly as E = -x dot F = -(x*fx + y*fy + z*fz), so that -Grad(E) = F.
The energy keyword is optional if the added force is defined with one or more variables, and if you are
performing dynamics via the run command. If the keyword is not used, LAMMPS will set the energy to 0.0,
which is typically fine for dynamics.
The energy keyword is required if the added force is defined with one or more variables, and you are
performing energy minimization via the "minimize" command. The keyword specifies the name of an
atom-style variable which is used to compute the energy of each atom as function of its position. Like
variables used for fx, fy, fz, the energy variable is specified as v_name, where name is the variable name.
Note that when the energy keyword is used during an energy minimization, you must insure that the formula
defined for the atom-style variable is consistent with the force variable formulas, i.e. that -Grad(E) = F. For
example, if the force were a spring-like F = kx, then the energy formula should be E = -0.5kx^2. If you don't
do this correctly, the minimization will not converge properly.
Styles with a suffix are functionally the same as the corresponding style without the suffix. They have been
optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The
accelerated styles take the same arguments and should produce the same results, except for round-off and
precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the potential "energy" inferred by the added
force to the system's potential energy as part of thermodynamic output. This is a fictitious quantity but is
needed so that the minimize command can include the forces added by this fix in a consistent manner. I.e.
there is a decrease in potential energy when atoms move in the direction of the added force.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
LAMMPS Users Manual
715
This fix computes a global scalar and a global 3-vector of forces, which can be accessed by various output
commands. The scalar is the potential energy discussed above. The vector is the total force on the group of
atoms before the forces on individual atoms are changed by the fix. The scalar and vector values calculated by
this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
You should not specify force components with a variable that has time-dependence for use with a minimizer,
since the minimizer increments the timestep as the iteration count during the minimization.
NOTE: If you want the fictitious potential energy associated with the added forces to be included in the total
potential energy of the system (the quantity being minimized), you MUST enable the fix_modify energy
option for this fix.
Restrictions: none
Related commands:
fix setforce, fix aveforce
Default:
The option default for the every keyword is every = 1.
LAMMPS Users Manual
716
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix addtorque command
Syntax:
fix ID group-ID addtorque Tx Ty Tz
ID, group-ID are documented in fix command• addtorque = style name of this fix command• Tx,Ty,Tz = torque component values (torque units)• any of Tx,Ty,Tz can be a variable (see below)•
Examples:
fix kick bead addtorque 2.0 3.0 5.0
fix kick bead addtorque 0.0 0.0 v_oscillate
Description:
Add a set of forces to each atom in the group such that:
the components of the total torque applied on the group (around its center of mass) are Tx,Ty,Tz• the group would move as a rigid body in the absence of other forces.•
This command can be used to drive a group of atoms into rotation.
Any of the 3 quantities defining the torque components can be specified as an equal-style variable, namely Tx,
Ty, Tz. If the value is a variable, it should be specified as v_name, where name is the variable name. In this
case, the variable will be evaluated each timestep, and its value used to determine the torque component.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent torque.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the potential "energy" inferred by the added
forces to the system's potential energy as part of thermodynamic output. This is a fictitious quantity but is
needed so that the minimize command can include the forces added by this fix in a consistent manner. I.e.
there is a decrease in potential energy when atoms move in the direction of the added forces.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its torque. Default is the outermost level.
This fix computes a global scalar and a global 3-vector, which can be accessed by various output commands.
The scalar is the potential energy discussed above. The vector is the total torque on the group of atoms before
the forces on individual atoms are changed by the fix. The scalar and vector values calculated by this fix are
"extensive".
LAMMPS Users Manual
717
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
You should not specify force components with a variable that has time-dependence for use with a minimizer,
since the minimizer increments the timestep as the iteration count during the minimization.
Restrictions:
This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
fix addforce
Default: none
LAMMPS Users Manual
718

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix append/atoms command
Syntax:
fix ID group-ID append/atoms face ... keyword value ...
ID, group-ID are documented in fix command• append/atoms = style name of this fix command• face = zhi• zero or more keyword/value pairs may be appended• keyword = basis or size or freq or temp or random or units
basis values = M itype
M = which basis atom
itype = atom type (1-N) to assign to this basis atom
size args = Lz
Lz = z size of lattice region appended in a single event(distance units)
freq args = freq
freq = the number of timesteps between append events
temp args = target damp seed extent
target = target temperature for the region between zhi-extent and zhi (temperature units)
damp = damping parameter (time units)
seed = random number seed for langevin kicks
extent = extent of thermostated region (distance units)
random args = xmax ymax zmax seed
xmax, ymax, zmax = maximum displacement in particular direction (distance units)
seed = random number seed for random displacement
units value = lattice or box
lattice = the wall position is defined in lattice units
box = the wall position is defined in simulation box units
•
Examples:
fix 1 all append/atoms zhi size 5.0 freq 295 units lattice
fix 4 all append/atoms zhi size 15.0 freq 5 units box
fix A all append/atoms zhi size 1.0 freq 1000 units lattice
Description:
This fix creates atoms on a lattice, appended on the zhi edge of the system box. This can be useful when a
shock or wave is propagating from zlo. This allows the system to grow with time to accommodate an
expanding wave. A simulation box must already exist, which is typically created via the create_box command.
Before using this command, a lattice must also be defined using the lattice command.
This fix will automatically freeze atoms on the zhi edge of the system, so that overlaps are avoided when new
atoms are appended.
The basis keyword specifies an atom type that will be assigned to specific basis atoms as they are created. See
the lattice command for specifics on how basis atoms are defined for the unit cell of the lattice. By default, all
created atoms are assigned type = 1 unless this keyword specifies differently.
The size keyword defines the size in z of the chunk of material to be added.
LAMMPS Users Manual
719

The random keyword will give the atoms random displacements around their lattice points to simulate some
initial temperature.
The temp keyword will cause a region to be thermostated with a Langevin thermostat on the zhi boundary.
The size of the region is measured from zhi and is set with the extent argument.
The units keyword determines the meaning of the distance units used to define a wall position, but only when
a numeric constant is used. A box value selects standard distance units as defined by the units command, e.g.
Angstroms for units = real or metal. A lattice value means the distance units are in lattice spacings. The lattice
command must have been previously used to define the lattice spacings.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix style is part of the SHOCK package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
The boundary on which atoms are added with append/atoms must be shrink/minimum. The opposite boundary
may be any boundary type other than periodic.
Related commands:
fix wall/piston command
Default:
The keyword defaults are size = 0.0, freq = 0, units = lattice. All added atoms are of type 1 unless the basis
keyword is used.
LAMMPS Users Manual
720
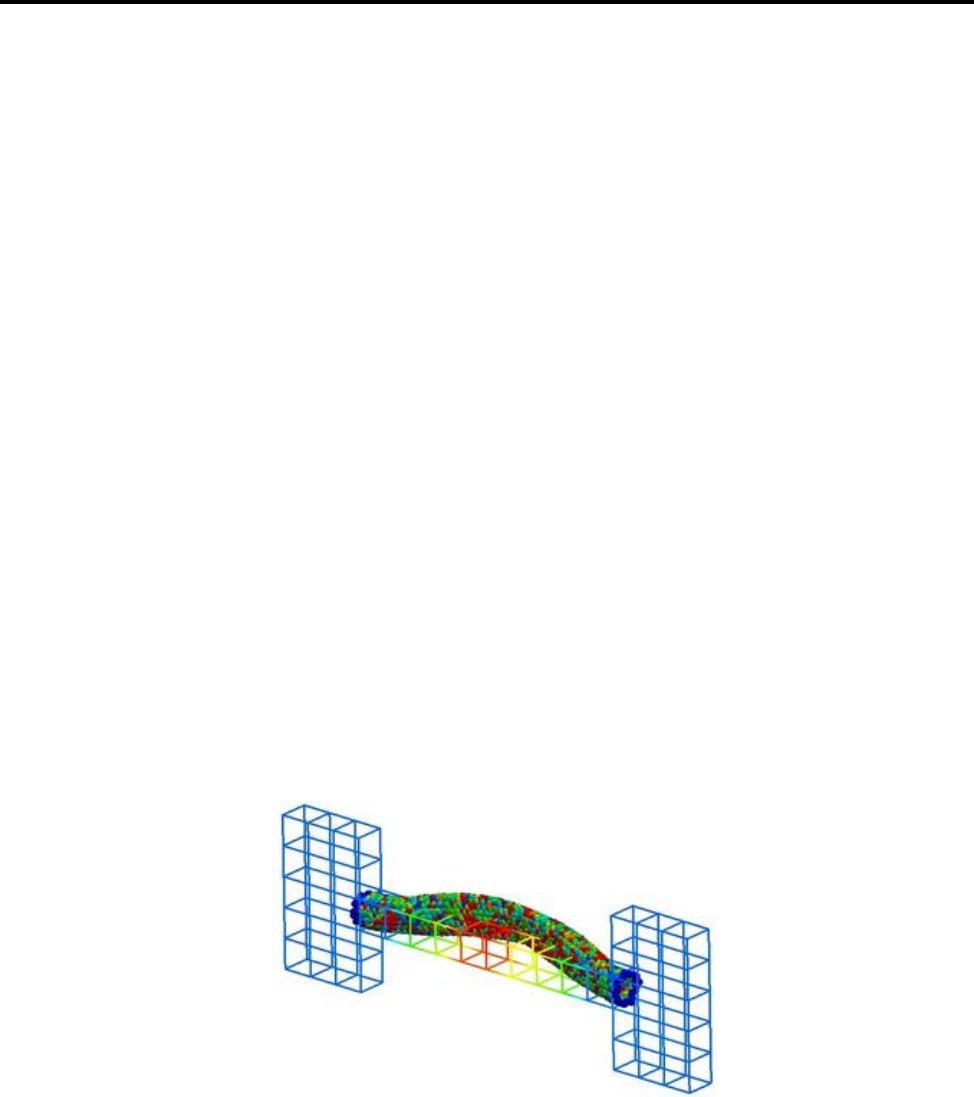
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix atc command
Syntax:
fix atc
fixID = name of fix• group = name of group fix is to be applied• type = thermal or two_temperature or hardy or field
thermal = thermal coupling with fields: temperature
two_temperature = electron-phonon coupling with field: temperature and electron_temperature
hardy = on-the-fly post-processing using kernel localization functions (see "related" section for possible fields)
field = on-the-fly post-processing using mesh-based localization functions (see "related" section for possible fields)
•
parameter_file = name of the file with material parameters. Note: Neither hardy nor field requires a
parameter file
Examples:
fix AtC internal atc thermal Ar_thermal.dat
fix AtC internal atc two_temperature Ar_ttm.mat
fix AtC internal atc hardy
fix AtC internal atc field
Description:
This fix is the beginning to creating a coupled FE/MD simulation and/or an on-the-fly estimation of
continuum fields. The coupled versions of this fix do Verlet integration and the post-processing does not.
After instantiating this fix, several other fix_modify commands will be needed to set up the problem, e.g.
define the finite element mesh and prescribe initial and boundary conditions.
The following coupling example is typical, but non-exhaustive:
# ... commands to create and initialize the MD system
# initial fix to designate coupling type and group to apply it to
# tag group physics material_file
fix AtC internal atc thermal Ar_thermal.mat
# create a uniform 12 x 2 x 2 mesh that covers region contain the group
•
LAMMPS Users Manual
721

# nx ny nz region periodicity
fix_modify AtC mesh create 12 2 2 mdRegion f p p
# specify the control method for the type of coupling
# physics control_type
fix_modify AtC thermal control flux
# specify the initial values for the empirical field "temperature"
# field node_group value
fix_modify AtC initial temperature all 30
# create an output stream for nodal fields
# filename output_frequency
fix_modify AtC output atc_fe_output 100
run 1000
likewise for this post-processing example:
# ... commands to create and initialize the MD system
# initial fix to designate post-processing and the group to apply it to
# no material file is allowed nor required
fix AtC internal atc hardy
# for hardy fix, specific kernel function (function type and range) to # be used as a localization function
fix AtC kernel quartic_sphere 10.0
# create a uniform 1 x 1 x 1 mesh that covers region contain the group
# with periodicity this effectively creats a system average
fix_modify AtC mesh create 1 1 1 box p p p
# change from default lagrangian map to eulerian
# refreshed every 100 steps
fix_modify AtC atom_element_map eulerian 100
# start with no field defined
# add mass density, potential energy density, stress and temperature
fix_modify AtC fields add density energy stress temperature
# create an output stream for nodal fields
# filename output_frequency
fix_modify AtC output nvtFE 100 text
run 1000
the mesh's linear interpolation functions can be used as the localization function by using the field option:
fix AtC internal atc field
fix_modify AtC mesh create 1 1 1 box p p p
...
Note coupling and post-processing can be combined in the same simulations using separate fixes.
LAMMPS Users Manual
722
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. The fix_modify options relevant to this fix
are listed below. No global scalar or vector or per-atom quantities are stored by this fix for access by
various output commands. No parameter of this fix can be used with the start/stop keywords of the run
command. This fix is not invoked during energy minimization.
Restrictions:
Thermal and two_temperature (coupling) types use a Verlet time-integration algorithm. The hardy type
does not contain its own time-integrator and must be used with a separate fix that does contain one, e.g.
nve, nvt, etc.
Currently,• - the coupling is restricted to thermal physics• - the FE computations are done in serial on each processor.•
Related commands:
After specifying this fix in your input script, several other fix_modify commands are used to setup the
problem, e.g. define the finite element mesh and prescribe initial and boundary conditions.
fix_modify commands for setup:
fix_modify AtC mesh create• fix_modify AtC mesh quadrature• fix_modify AtC mesh read• fix_modify AtC mesh write• fix_modify AtC mesh create_nodeset• fix_modify AtC mesh add_to_nodeset• fix_modify AtC mesh create_faceset box• fix_modify AtC mesh create_faceset plane• fix_modify AtC mesh create_elementset• fix_modify AtC mesh delete_elements• fix_modify AtC mesh nodeset_to_elementset• fix_modify AtC boundary• fix_modify AtC internal_quadrature• fix_modify AtC time_integration (thermal)• fix_modify AtC time_integration (momentum)• fix_modify AtC extrinsic electron_integration• fix_modify AtC internal_element_set• fix_modify AtC decomposition•
fix_modify commands for boundary and initial conditions:
fix_modify AtC initial• fix_modify AtC fix• fix_modify AtC unfix• fix_modify AtC fix_flux• fix_modify AtC unfix_flux• fix_modify AtC source•
LAMMPS Users Manual
723
fix_modify AtC remove_source•
fix_modify commands for control and filtering:
fix_modify AtC control• fix_modify AtC control thermal• fix_modify AtC control thermal correction_max_iterations• fix_modify AtC control momentum• fix_modify AtC control localized_lambda• fix_modify AtC control lumped_lambda_solve• fix_modify AtC control mask_direction control• fix_modify AtC filter• fix_modify AtC filter scale• fix_modify AtC filter type• fix_modify AtC equilibrium_start• fix_modify AtC extrinsic exchange• fix_modify AtC poisson_solver•
fix_modify commands for output:
fix_modify AtC output• fix_modify AtC output nodeset• fix_modify AtC output elementset• fix_modify AtC output boundary_integral• fix_modify AtC output contour_integral• fix_modify AtC mesh output• fix_modify AtC write_restart• fix_modify AtC read_restart•
fix_modify commands for post-processing:
fix_modify AtC kernel• fix_modify AtC fields• fix_modify AtC grdients• fix_modify AtC rates• fix_modify AtC computes• fix_modify AtC on_the_fly• fix_modify AtC pair_interactions/bond_interactions• fix_modify AtC sample_frequency• fix_modify AtC set•
miscellaneous fix_modify commands:
fix_modify AtC atom_element_map• fix_modify AtC atom_weight• fix_modify AtC write_atom_weights• fix_modify AtC reset_time• fix_modify AtC reset_atomic_reference_positions• fix_modify AtC fe_md_boundary• fix_modify AtC boundary_faceset• fix_modify AtC consistent_fe_initialization•
LAMMPS Users Manual
724

fix_modify AtC mass_matrix• fix_modify AtC material• fix_modify AtC atomic_charge• fix_modify AtC source_integration• fix_modify AtC temperature_definition• fix_modify AtC track_displacement• fix_modify AtC boundary_dynamics• fix_modify AtC add_species• fix_modify AtC add_molecule• fix_modify AtC remove_species• fix_modify AtC remove_molecule•
Note: a set of example input files with the attendant material files are included with this package
Default: None
For detailed exposition of the theory and algorithms please see:
(Wagner) Wagner, GJ; Jones, RE; Templeton, JA; Parks, MA, "An atomistic-to-continuum coupling
method for heat transfer in solids." Special Issue of Computer Methods and Applied Mechanics (2008)
197:3351.
(Zimmerman2004) Zimmerman, JA; Webb, EB; Hoyt, JJ;. Jones, RE; Klein, PA; Bammann, DJ,
"Calculation of stress in atomistic simulation." Special Issue of Modelling and Simulation in Materials
Science and Engineering (2004), 12:S319.
(Zimmerman2010) Zimmerman, JA; Jones, RE; Templeton, JA, "A material frame approach for
evaluating continuum variables in atomistic simulations." Journal of Computational Physics (2010),
229:2364.
(Templeton2010) Templeton, JA; Jones, RE; Wagner, GJ, "Application of a field-based method to
spatially varying thermal transport problems in molecular dynamics." Modelling and Simulation in
Materials Science and Engineering (2010), 18:085007.
(Jones) Jones, RE; Templeton, JA; Wagner, GJ; Olmsted, D; Modine, JA, "Electron transport enhanced
molecular dynamics for metals and semi-metals." International Journal for Numerical Methods in
Engineering (2010), 83:940.
(Templeton2011) Templeton, JA; Jones, RE; Lee, JW; Zimmerman, JA; Wong, BM, "A long-range
electric field solver for molecular dynamics based on atomistic-to-continuum modeling." Journal of
Chemical Theory and Computation (2011), 7:1736.
(Mandadapu) Mandadapu, KK; Templeton, JA; Lee, JW, "Polarization as a field variable from molecular
dynamics simulations." Journal of Chemical Physics (2013), 139:054115.
LAMMPS Users Manual
725
Please refer to the standard finite element (FE) texts, e.g. T.J.R Hughes " The finite element method ",
Dover 2003, for the basics of FE simulation.
LAMMPS Users Manual
726

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix atom/swap command
Syntax:
fix ID group-ID atom/swap N X seed T keyword values ...
ID, group-ID are documented in fix command• atom/swap = style name of this fix command• N = invoke this fix every N steps• X = number of swaps to attempt every N steps• seed = random # seed (positive integer)• T = scaling temperature of the MC swaps (temperature units)• one or more keyword/value pairs may be appended to args• keyword = types or mu or ke or semi-grand or region
types values = two or more atom types
mu values = chemical potential of swap types (energy units)
ke value = no or yes
no = no conservation of kinetic energy after atom swaps
yes = kinetic energy is conserved after atom swaps
semi-grand value = no or yes
no = particle type counts and fractions conserved
yes = semi-grand canonical ensemble, particle fractions not conserved
region value = region-ID
region-ID = ID of region to use as an exchange/move volume
•
Examples:
fix 2 all atom/swap 1 1 29494 300.0 ke no types 1 2
fix myFix all atom/swap 100 1 12345 298.0 region my_swap_region types 5 6
fix SGMC all atom/swap 1 100 345 1.0 semi-grand yes types 1 2 3 mu 0.0 4.3 -5.0
Description:
This fix performs Monte Carlo swaps of atoms of one given atom type with atoms of the other given atom
types. The specified T is used in the Metropolis criterion dictating swap probabilities.
Perform X swaps of atoms of one type with atoms of another type according to a Monte Carlo probability.
Swap candidates must be in the fix group, must be in the region (if specified), and must be of one of the listed
types. Swaps are attempted between candidates that are chosen randomly with equal probability among the
candidate atoms. Swaps are not attempted between atoms of the same type since nothing would happen.
All atoms in the simulation domain can be moved using regular time integration displacements, e.g. via fix
nvt, resulting in a hybrid MC+MD simulation. A smaller-than-usual timestep size may be needed when
running such a hybrid simulation, especially if the swapped atoms are not well equilibrated.
The types keyword is required. At least two atom types must be specified.
The ke keyword can be set to no to turn off kinetic energy conservation for swaps. The default is yes, which
means that swapped atoms have their velocities scaled by the ratio of the masses of the swapped atom types.
This ensures that the kinetic energy of each atom is the same after the swap as it was before the swap, even
LAMMPS Users Manual
727
though the atom masses have changed.
The semi-grand keyword can be set to yes to switch to the semi-grand canonical ensemble as discussed in
(Sadigh). This means that the total number of each particle type does not need to be conserved. The default is
no, which means that the only kind of swap allowed exchanges an atom of one type with an atom of a
different given type. In other words, the relative mole fractions of the swapped atoms remains constant.
Whereas in the semi-grand canonical ensemble, the composition of the system can change. Note that when
using semi-grand, atoms in the fix group whose type is not listed in the types keyword are ineligible for
attempted conversion. An attempt is made to switch the selected atom (if eligible) to one of the other listed
types with equal probability. Acceptance of each attempt depends upon the Metropolis criterion.
The mu keyword allows users to specify chemical potentials. This is required and allowed only when using
semi-grand. All chemical potentials are absolute, so there is one for each swap type listed following the types
keyword. In semi-grand canonical ensemble simulations the chemical composition of the system is controlled
by the difference in these values. So shifting all values by a constant amount will have no effect on the
simulation.
This command may optionally use the region keyword to define swap volume. The specified region must
have been previously defined with a region command. It must be defined with side = in. Swap attempts occur
only between atoms that are both within the specified region. Swaps are not otherwise attempted.
You should ensure you do not swap atoms belonging to a molecule, or LAMMPS will soon generate an error
when it tries to find those atoms. LAMMPS will warn you if any of the atoms eligible for swapping have a
non-zero molecule ID, but does not check for this at the time of swapping.
If not using semi-grand this fix checks to ensure all atoms of the given types have the same atomic charge.
LAMMPS doesn't enforce this in general, but it is needed for this fix to simplify the swapping procedure.
Successful swaps will swap the atom type and charge of the swapped atoms. Conversely, when using
semi-grand, it is assumed that all the atom types involved in switches have the same charge. Otherwise,
charge would not be conserved. As a consequence, no checks on atomic charges are performed, and
successful switches update the atom type but not the atom charge. While it is possible to use semi-grand with
groups of atoms that have different charges, these charges will not be changed when the atom types change.
Since this fix computes total potential energies before and after proposed swaps, so even complicated
potential energy calculations are OK, including the following:
long-range electrostatics (kspace)• many body pair styles• hybrid pair styles• eam pair styles• triclinic systems• need to include potential energy contributions from other fixes•
Some fixes have an associated potential energy. Examples of such fixes include: efield, gravity, addforce,
langevin, restrain, temp/berendsen, temp/rescale, and wall fixes. For that energy to be included in the total
potential energy of the system (the quantity used when performing GCMC moves), you MUST enable the
fix_modify energy option for that fix. The doc pages for individual fix commands specify if this should be
done.
Restart, fix_modify, output, run start/stop, minimize info:
LAMMPS Users Manual
728

This fix writes the state of the fix to binary restart files. This includes information about the random number
generator seed, the next timestep for MC exchanges, etc. See the read_restart command for info on how to
re-specify a fix in an input script that reads a restart file, so that the operation of the fix continues in an
uninterrupted fashion.
None of the fix_modify options are relevant to this fix.
This fix computes a global vector of length 2, which can be accessed by various output commands. The vector
values are the following global cumulative quantities:
1 = swap attempts• 2 = swap successes•
The vector values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the MC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
fix nvt, neighbor, fix deposit, fix evaporate, delete_atoms, fix gcmc
Default:
The option defaults are ke = yes, semi-grand = no, mu = 0.0 for all atom types.
(Sadigh) B Sadigh, P Erhart, A Stukowski, A Caro, E Martinez, and L Zepeda-Ruiz, Phys. Rev. B, 85,
184203 (2012).
LAMMPS Users Manual
729

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix ave/atom command
Syntax:
fix ID group-ID ave/atom Nevery Nrepeat Nfreq value1 value2 ...
ID, group-ID are documented in fix command• ave/atom = style name of this fix command• Nevery = use input values every this many timesteps• Nrepeat = # of times to use input values for calculating averages• Nfreq = calculate averages every this many timesteps one or more input values can be listed• value = x, y, z, vx, vy, vz, fx, fy, fz, c_ID, c_ID[i], f_ID, f_ID[i], v_name
x,y,z,vx,vy,vz,fx,fy,fz = atom attribute (position, velocity, force component)
c_ID = per-atom vector calculated by a compute with ID
c_ID[I] = Ith column of per-atom array calculated by a compute with ID, I can include wildcard (see below)
f_ID = per-atom vector calculated by a fix with ID
f_ID[I] = Ith column of per-atom array calculated by a fix with ID, I can include wildcard (see below)
v_name = per-atom vector calculated by an atom-style variable with name
•
Examples:
fix 1 all ave/atom 1 100 100 vx vy vz
fix 1 all ave/atom 10 20 1000 c_my_stress[1]
fix 1 all ave/atom 10 20 1000 c_my_stress[*]
Description:
Use one or more per-atom vectors as inputs every few timesteps, and average them atom by atom over longer
timescales. The resulting per-atom averages can be used by other output commands such as the fix ave/chunk
or dump custom commands.
The group specified with the command means only atoms within the group have their averages computed.
Results are set to 0.0 for atoms not in the group.
Each input value can be an atom attribute (position, velocity, force component) or can be the result of a
compute or fix or the evaluation of an atom-style variable. In the latter cases, the compute, fix, or variable
must produce a per-atom vector, not a global quantity or local quantity. If you wish to time-average global
quantities from a compute, fix, or variable, then see the fix ave/time command.
Each per-atom value of each input vector is averaged independently.
Computes that produce per-atom vectors or arrays are those which have the word atom in their style name.
See the doc pages for individual fixes to determine which ones produce per-atom vectors or arrays. Variables
of style atom are the only ones that can be used with this fix since they produce per-atom vectors.
Note that for values from a compute or fix, the bracketed index I can be specified using a wildcard asterisk
with the index to effectively specify multiple values. This takes the form "*" or "*n" or "n*" or "m*n". If N =
the size of the vector (for mode = scalar) or the number of columns in the array (for mode = vector), then an
asterisk with no numeric values means all indices from 1 to N. A leading asterisk means all indices from 1 to
LAMMPS Users Manual
730

n (inclusive). A trailing asterisk means all indices from n to N (inclusive). A middle asterisk means all indices
from m to n (inclusive).
Using a wildcard is the same as if the individual columns of the array had been listed one by one. E.g. these 2
fix ave/atom commands are equivalent, since the compute stress/atom command creates a per-atom array with
6 columns:
compute my_stress all stress/atom NULL
fix 1 all ave/atom 10 20 1000 c_my_stress[*]
fix 1 all ave/atom 10 20 1000 c_my_stress[1] c_my_stress[1] &
c_my_stress[3] c_my_stress[4] &
c_my_stress[5] c_my_stress[6]
The Nevery, Nrepeat, and Nfreq arguments specify on what timesteps the input values will be used in order to
contribute to the average. The final averaged quantities are generated on timesteps that are a multiple of
Nfreq. The average is over Nrepeat quantities, computed in the preceding portion of the simulation every
Nevery timesteps. Nfreq must be a multiple of Nevery and Nevery must be non-zero even if Nrepeat is 1. Also,
the timesteps contributing to the average value cannot overlap, i.e. Nrepeat*Nevery can not exceed Nfreq.
For example, if Nevery=2, Nrepeat=6, and Nfreq=100, then values on timesteps 90,92,94,96,98,100 will be
used to compute the final average on timestep 100. Similarly for timesteps 190,192,194,196,198,200 on
timestep 200, etc.
The atom attribute values (x,y,z,vx,vy,vz,fx,fy,fz) are self-explanatory. Note that other atom attributes can be
used as inputs to this fix by using the compute property/atom command and then specifying an input value
from that compute.
NOTE: The x,y,z attributes are values that are re-wrapped inside the periodic box whenever an atom crosses a
periodic boundary. Thus if you time average an atom that spends half its time on either side of the periodic
box, you will get a value in the middle of the box. If this is not what you want, consider averaging unwrapped
coordinates, which can be provided by the compute property/atom command via its xu,yu,zu attributes.
If a value begins with "c_", a compute ID must follow which has been previously defined in the input script. If
no bracketed term is appended, the per-atom vector calculated by the compute is used. If a bracketed term
containing an index I is appended, the Ith column of the per-atom array calculated by the compute is used.
Users can also write code for their own compute styles and add them to LAMMPS. See the discussion above
for how I can be specified with a wildcard asterisk to effectively specify multiple values.
If a value begins with "f_", a fix ID must follow which has been previously defined in the input script. If no
bracketed term is appended, the per-atom vector calculated by the fix is used. If a bracketed term containing
an index I is appended, the Ith column of the per-atom array calculated by the fix is used. Note that some fixes
only produce their values on certain timesteps, which must be compatible with Nevery, else an error will
result. Users can also write code for their own fix styles and add them to LAMMPS. See the discussion above
for how I can be specified with a wildcard asterisk to effectively specify multiple values.
If a value begins with "v_", a variable name must follow which has been previously defined in the input script
as an atom-style variable Variables of style atom can reference thermodynamic keywords, or invoke other
computes, fixes, or variables when they are evaluated, so this is a very general means of generating per-atom
quantities to time average.
Restart, fix_modify, output, run start/stop, minimize info:
LAMMPS Users Manual
731
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global scalar or vector quantities are stored by this fix for access by various output commands.
This fix produces a per-atom vector or array which can be accessed by various output commands. A vector is
produced if only a single quantity is averaged by this fix. If two or more quantities are averaged, then an array
of values is produced. The per-atom values can only be accessed on timesteps that are multiples of Nfreq since
that is when averaging is performed.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
compute, fix ave/histo, fix ave/chunk, fix ave/time, variable,
Default: none
LAMMPS Users Manual
732

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix ave/chunk command
Syntax:
fix ID group-ID ave/chunk Nevery Nrepeat Nfreq chunkID value1 value2 ... keyword args ...
ID, group-ID are documented in fix command• ave/chunk = style name of this fix command• Nevery = use input values every this many timesteps• Nrepeat = # of times to use input values for calculating averages• Nfreq = calculate averages every this many timesteps• chunkID = ID of compute chunk/atom command• one or more input values can be listed• value = vx, vy, vz, fx, fy, fz, density/mass, density/number, temp, c_ID, c_ID[I], f_ID, f_ID[I],
v_name
vx,vy,vz,fx,fy,fz = atom attribute (velocity, force component)
density/number, density/mass = number or mass density
temp = temperature
c_ID = per-atom vector calculated by a compute with ID
c_ID[I] = Ith column of per-atom array calculated by a compute with ID, I can include wildcard (see below)
f_ID = per-atom vector calculated by a fix with ID
f_ID[I] = Ith column of per-atom array calculated by a fix with ID, I can include wildcard (see below)
v_name = per-atom vector calculated by an atom-style variable with name
•
zero or more keyword/arg pairs may be appended• keyword = norm or ave or bias or adof or cdof or file or overwrite or title1 or title2 or title3
norm arg = all or sample or none = how output on Nfreq steps is normalized
all = output is sum of atoms across all Nrepeat samples, divided by atom count
sample = output is sum of Nrepeat sample averages, divided by Nrepeat
none = output is sum of Nrepeat sample sums, divided by Nrepeat
ave args = one or running or window M
one = output new average value every Nfreq steps
running = output cumulative average of all previous Nfreq steps
window M = output average of M most recent Nfreq steps
bias arg = bias-ID
bias-ID = ID of a temperature compute that removes a velocity bias for temperature calculation
adof value = dof_per_atom
dof_per_atom = define this many degrees-of-freedom per atom for temperature calculation
cdof value = dof_per_chunk
dof_per_chunk = define this many degrees-of-freedom per chunk for temperature calculation
file arg = filename
filename = file to write results to
overwrite arg = none = overwrite output file with only latest output
format arg = string
string = C-style format string
title1 arg = string
string = text to print as 1st line of output file
title2 arg = string
string = text to print as 2nd line of output file
title3 arg = string
string = text to print as 3rd line of output file
•
Examples:
LAMMPS Users Manual
733
fix 1 all ave/chunk 10000 1 10000 binchunk c_myCentro title1 "My output values"
fix 1 flow ave/chunk 100 10 1000 molchunk vx vz norm sample file vel.profile
fix 1 flow ave/chunk 100 5 1000 binchunk density/mass ave running
fix 1 flow ave/chunk 100 5 1000 binchunk density/mass ave running
NOTE:
If you are trying to replace a deprecated fix ave/spatial command with the newer, more flexible fix ave/chunk
and compute chunk/atom commands, you simply need to split the fix ave/spatial arguments across the two
new commands. For example, this command:
fix 1 flow ave/spatial 100 10 1000 y 0.0 1.0 vx vz norm sample file vel.profile
could be replaced by:
compute cc1 flow chunk/atom bin/1d y 0.0 1.0
fix 1 flow ave/chunk 100 10 1000 cc1 vx vz norm sample file vel.profile
Description:
Use one or more per-atom vectors as inputs every few timesteps, sum the values over the atoms in each chunk
at each timestep, then average the per-chunk values over longer timescales. The resulting chunk averages can
be used by other output commands such as thermo_style custom, and can also be written to a file.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
Note that only atoms in the specified group contribute to the summing and averaging calculations. The
compute chunk/atom command defines its own group as well as an optional region. Atoms will have a chunk
ID = 0, meaning they belong to no chunk, if they are not in that group or region. Thus you can specify the
"all" group for this command if you simply want to use the chunk definitions provided by chunkID.
Each specified per-atom value can be an atom attribute (position, velocity, force component), a mass or
number density, or the result of a compute or fix or the evaluation of an atom-style variable. In the latter
cases, the compute, fix, or variable must produce a per-atom quantity, not a global quantity. Note that the
compute property/atom command provides access to any attribute defined and stored by atoms. If you wish to
time-average global quantities from a compute, fix, or variable, then see the fix ave/time command.
The per-atom values of each input vector are summed and averaged independently of the per-atom values in
other input vectors.
Computes that produce per-atom quantities are those which have the word atom in their style name. See the
doc pages for individual fixes to determine which ones produce per-atom quantities. Variables of style atom
are the only ones that can be used with this fix since all other styles of variable produce global quantities.
Note that for values from a compute or fix, the bracketed index I can be specified using a wildcard asterisk
with the index to effectively specify multiple values. This takes the form "*" or "*n" or "n*" or "m*n". If N =
the size of the vector (for mode = scalar) or the number of columns in the array (for mode = vector), then an
asterisk with no numeric values means all indices from 1 to N. A leading asterisk means all indices from 1 to
LAMMPS Users Manual
734
n (inclusive). A trailing asterisk means all indices from n to N (inclusive). A middle asterisk means all indices
from m to n (inclusive).
Using a wildcard is the same as if the individual columns of the array had been listed one by one. E.g. these 2
fix ave/chunk commands are equivalent, since the compute property/atom command creates, in this case, a
per-atom array with 3 columns:
compute myAng all property/atom angmomx angmomy angmomz
fix 1 all ave/chunk 100 1 100 cc1 c_myAng[*] file tmp.angmom
fix 2 all ave/chunk 100 1 100 cc1 c_myAng[1] c_myAng[2] c_myAng[3] file tmp.angmom
NOTE: This fix works by creating an array of size Nchunk by Nvalues on each processor. Nchunk is the
number of chunks which is defined by the compute chunk/atom command. Nvalues is the number of input
values specified. Each processor loops over its atoms, tallying its values to the appropriate chunk. Then the
entire array is summed across all processors. This means that using a large number of chunks will incur an
overhead in memory and computational cost (summing across processors), so be careful to define a reasonable
number of chunks.
The Nevery, Nrepeat, and Nfreq arguments specify on what timesteps the input values will be accessed and
contribute to the average. The final averaged quantities are generated on timesteps that are a multiples of
Nfreq. The average is over Nrepeat quantities, computed in the preceding portion of the simulation every
Nevery timesteps. Nfreq must be a multiple of Nevery and Nevery must be non-zero even if Nrepeat is 1. Also,
the timesteps contributing to the average value cannot overlap, i.e. Nrepeat*Nevery can not exceed Nfreq.
For example, if Nevery=2, Nrepeat=6, and Nfreq=100, then values on timesteps 90,92,94,96,98,100 will be
used to compute the final average on timestep 100. Similarly for timesteps 190,192,194,196,198,200 on
timestep 200, etc. If Nrepeat=1 and Nfreq = 100, then no time averaging is done; values are simply generated
on timesteps 100,200,etc.
Each input value can also be averaged over the atoms in each chunk. The way the averaging is done across the
Nrepeat timesteps to produce output on the Nfreq timesteps, and across multiple Nfreq outputs, is determined
by the norm and ave keyword settings, as discussed below.
NOTE: To perform per-chunk averaging within a Nfreq time window, the number of chunks Nchunk defined
by the compute chunk/atom command must remain constant. If the ave keyword is set to running or window
then Nchunk must remain constant for the duration of the simulation. This fix forces the chunk/atom compute
specified by chunkID to hold Nchunk constant for the appropriate time windows, by not allowing it to
re-calculate Nchunk, which can also affect how it assigns chunk IDs to atoms. This is particularly important to
understand if the chunks defined by the compute chunk/atom command are spatial bins. If its units keyword is
set to box or lattice, then the number of bins Nchunk and size of each bin will be fixed over the Nfreq time
window, which can affect which atoms are discarded if the simulation box size changes. If its units keyword is
set to reduced, then the number of bins Nchunk will still be fixed, but the size of each bin can vary at each
timestep if the simulation box size changes, e.g. for an NPT simulation.
The atom attribute values (vx,vy,vz,fx,fy,fz) are self-explanatory. As noted above, any other atom attributes
can be used as input values to this fix by using the compute property/atom command and then specifying an
input value from that compute.
The density/number value means the number density is computed for each chunk, i.e. number/volume. The
density/mass value means the mass density is computed for each chunk, i.e. total-mass/volume. The output
values are in units of 1/volume or density (mass/volume). See the units command doc page for the definition
LAMMPS Users Manual
735

of density for each choice of units, e.g. gram/cm^3. If the chunks defined by the compute chunk/atom
command are spatial bins, the volume is the bin volume. Otherwise it is the volume of the entire simulation
box.
The temp value means the temperature is computed for each chunk, by the formula KE = DOF/2 k T, where
KE = total kinetic energy of the chunk of atoms (sum of 1/2 m v^2), DOF = the total number of degrees of
freedom for all atoms in the chunk, k = Boltzmann constant, and T = temperature.
The DOF is calculated as N*adof + cdof, where N = number of atoms in the chunk, adof = degrees of freedom
per atom, and cdof = degrees of freedom per chunk. By default adof = 2 or 3 = dimensionality of system, as
set via the dimension command, and cdof = 0.0. This gives the usual formula for temperature.
Note that currently this temperature only includes translational degrees of freedom for each atom. No
rotational degrees of freedom are included for finite-size particles. Also no degrees of freedom are subtracted
for any velocity bias or constraints that are applied, such as compute temp/partial, or fix shake or fix rigid.
This is because those degrees of freedom (e.g. a constrained bond) could apply to sets of atoms that are both
included and excluded from a specific chunk, and hence the concept is somewhat ill-defined. In some cases,
you can use the adof and cdof keywords to adjust the calculated degress of freedom appropriately, as
explained below.
Also note that a bias can be subtracted from atom velocities before they are used in the above formula for KE,
by using the bias keyword. This allows, for example, a thermal temperature to be computed after removal of a
flow velocity profile.
Note that the per-chunk temperature calculated by this fix and the compute temp/chunk command can be
different. The compute calculates the temperature for each chunk for a single snapshot. This fix can do that
but can also time average those values over many snapshots, or it can compute a temperature as if the atoms in
the chunk on different timesteps were collected together as one set of atoms to calculate their temperature.
The compute allows the center-of-mass velocity of each chunk to be subtracted before calculating the
temperature; this fix does not.
If a value begins with "c_", a compute ID must follow which has been previously defined in the input script. If
no bracketed integer is appended, the per-atom vector calculated by the compute is used. If a bracketed integer
is appended, the Ith column of the per-atom array calculated by the compute is used. Users can also write code
for their own compute styles and add them to LAMMPS. See the discussion above for how I can be specified
with a wildcard asterisk to effectively specify multiple values.
If a value begins with "f_", a fix ID must follow which has been previously defined in the input script. If no
bracketed integer is appended, the per-atom vector calculated by the fix is used. If a bracketed integer is
appended, the Ith column of the per-atom array calculated by the fix is used. Note that some fixes only
produce their values on certain timesteps, which must be compatible with Nevery, else an error results. Users
can also write code for their own fix styles and add them to LAMMPS. See the discussion above for how I can
be specified with a wildcard asterisk to effectively specify multiple values.
If a value begins with "v_", a variable name must follow which has been previously defined in the input
script. Variables of style atom can reference thermodynamic keywords and various per-atom attributes, or
invoke other computes, fixes, or variables when they are evaluated, so this is a very general means of
generating per-atom quantities to average within chunks.
Additional optional keywords also affect the operation of this fix and its outputs.
LAMMPS Users Manual
736
The norm keyword affects how averaging is done for the per-chunk values that are output every Nfreq
timesteps.
It the norm setting is all, which is the default, a chunk value is summed over all atoms in all Nrepeat samples,
as is the count of atoms in the chunk. The averaged output value for the chunk on the Nfreq timesteps is
Total-sum / Total-count. In other words it is an average over atoms across the entire Nfreq timescale. For the
density/number and density/mass values, the volume (bin volume or system volume) used in the final
normalization will be the volume at the final Nfreq timestep.
If the norm setting is sample, the chunk value is summed over atoms for each sample, as is the count, and an
"average sample value" is computed for each sample, i.e. Sample-sum / Sample-count. The output value for
the chunk on the Nfreq timesteps is the average of the Nrepeat "average sample values", i.e. the sum of
Nrepeat "average sample values" divided by Nrepeat. In other words it is an average of an average. For the
density/number and density/mass values, the volume (bin volume or system volume) used in the per-sample
normalization will be the current volume at each sampling step.
If the norm setting is none, a similar computation as for the sample setting is done, except the individual
"average sample values" are "summed sample values". A summed sample value is simply the chunk value
summed over atoms in the sample, without dividing by the number of atoms in the sample. The output value
for the chunk on the Nfreq timesteps is the average of the Nrepeat "summed sample values", i.e. the sum of
Nrepeat "summed sample values" divided by Nrepeat. For the density/number and density/mass values, the
volume (bin volume or system volume) used in the per-sample sum normalization will be the current volume
at each sampling step.
The ave keyword determines how the per-chunk values produced every Nfreq steps are averaged with values
produced on previous steps that were multiples of Nfreq, before they are accessed by another output command
or written to a file.
If the ave setting is one, which is the default, then the chunk values produced on timesteps that are multiples
of Nfreq are independent of each other; they are output as-is without further averaging.
If the ave setting is running, then the chunk values produced on timesteps that are multiples of Nfreq are
summed and averaged in a cumulative sense before being output. Each output chunk value is thus the average
of the chunk value produced on that timestep with all preceding values for the same chunk. This running
average begins when the fix is defined; it can only be restarted by deleting the fix via the unfix command, or
re-defining the fix by re-specifying it.
If the ave setting is window, then the chunk values produced on timesteps that are multiples of Nfreq are
summed and averaged within a moving "window" of time, so that the last M values for the same chunk are
used to produce the output. E.g. if M = 3 and Nfreq = 1000, then the output on step 10000 will be the average
of the individual chunk values on steps 8000,9000,10000. Outputs on early steps will average over less than
M values if they are not available.
The bias keyword specifies the ID of a temperature compute that removes a "bias" velocity from each atom,
specified as bias-ID. It is only used when the temp value is calculated, to compute the thermal temperature of
each chunk after the translational kinetic energy components have been altered in a prescribed way, e.g. to
remove a flow velocity profile. See the doc pages for individual computes that calculate a temperature to see
which ones implement a bias.
The adof and cdof keywords define the values used in the degree of freedom (DOF) formula described above
for for temperature calculation for each chunk. They are only used when the temp value is calculated. They
LAMMPS Users Manual
737
can be used to calculate a more appropriate temperature for some kinds of chunks. Here are 3 examples:
If spatially binned chunks contain some number of water molecules and fix shake is used to make each
molecule rigid, then you could calculate a temperature with 6 degrees of freedom (DOF) (3 translational, 3
rotational) per molecule by setting adof to 2.0.
If compute temp/partial is used with the bias keyword to only allow the x component of velocity to contribute
to the temperature, then adof = 1.0 would be appropriate.
If each chunk consists of a large molecule, with some number of its bonds constrained by fix shake or the
entire molecule by fix rigid/small, adof = 0.0 and cdof could be set to the remaining degrees of freedom for
the entire molecule (entire chunk in this case), e.g. 6 for 3d, or 3 for 2d, for a rigid molecule.
The file keyword allows a filename to be specified. Every Nfreq timesteps, a section of chunk info will be
written to a text file in the following format. A line with the timestep and number of chunks is written. Then
one line per chunk is written, containing the chunk ID (1-Nchunk), an optional original ID value, optional
coordinate values for chunks that represent spatial bins, the number of atoms in the chunk, and one or more
calculated values. More explanation of the optional values is given below. The number of values in each line
corresponds to the number of values specified in the fix ave/chunk command. The number of atoms and the
value(s) are summed or average quantities, as explained above.
The overwrite keyword will continuously overwrite the output file with the latest output, so that it only
contains one timestep worth of output. This option can only be used with the ave running setting.
The format keyword sets the numeric format of each value when it is printed to a file via the file keyword.
Note that all values are floating point quantities. The default format is %g. You can specify a higher precision
if desired, e.g. %20.16g.
The title1 and title2 and title3 keywords allow specification of the strings that will be printed as the first 3
lines of the output file, assuming the file keyword was used. LAMMPS uses default values for each of these,
so they do not need to be specified.
By default, these header lines are as follows:
# Chunk-averaged data for fix ID and group name
# Timestep Number-of-chunks
# Chunk (OrigID) (Coord1) (Coord2) (Coord3) Ncount value1 value2 ...
In the first line, ID and name are replaced with the fix-ID and group name. The second line describes the two
values that are printed at the first of each section of output. In the third line the values are replaced with the
appropriate value names, e.g. fx or c_myCompute2.
The words in parenthesis only appear with corresponding columns if the chunk style specified for the compute
chunk/atom command supports them. The OrigID column is only used if the compress keyword was set to yes
for the compute chunk/atom command. This means that the original chunk IDs (e.g. molecule IDs) will have
been compressed to remove chunk IDs with no atoms assigned to them. Thus a compressed chunk ID of 3
may correspond to an original chunk ID or molecule ID of 415. The OrigID column will list 415 for the 3rd
chunk.
The CoordN columns only appear if a binning style was used in the compute chunk/atom command. For
bin/1d, bin/2d, and bin/3d styles the column values are the center point of the bin in the corresponding
LAMMPS Users Manual
738

dimension. Just Coord1 is used for bin/1d, Coord2 is added for bin/2d, Coord3 is added for bin/3d. For
bin/sphere, just Coord1 is used, and it is the radial coordinate. For bin/cylinder, Coord1 and Coord2 are used.
Coord1 is the radial coordinate (away from the cylinder axis), and coord2 is the coordinate along the cylinder
axis.
Note that if the value of the units keyword used in the compute chunk/atom command is box or lattice, the
coordinate values will be in distance units. If the value of the units keyword is reduced, the coordinate values
will be in unitless reduced units (0-1). This is not true for the Coord1 value of style bin/sphere or bin/cylinder
which both represent radial dimensions. Those values are always in distance units.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global array of values which can be accessed by various output commands. The values
can only be accessed on timesteps that are multiples of Nfreq since that is when averaging is performed. The
global array has # of rows = the number of chunks Nchunk as calculated by the specified compute chunk/atom
command. The # of columns = M+1+Nvalues, where M = 1 to 4, depending on whether the optional columns
for OrigID and CoordN are used, as explained above. Following the optional columns, the next column
contains the count of atoms in the chunk, and the remaining columns are the Nvalue quantities. When the
array is accessed with a row I that exceeds the current number of chunks, than a 0.0 is returned by the fix
instead of an error, since the number of chunks can vary as a simulation runs depending on how that value is
computed by the compute chunk/atom command.
The array values calculated by this fix are treated as "intensive", since they are typically already normalized
by the count of atoms in each chunk.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
compute, fix ave/atom, fix ave/histo, fix ave/time, variable, fix ave/correlate
Default:
The option defaults are norm = all, ave = one, bias = none, no file output, and title 1,2,3 = strings as described
above.
LAMMPS Users Manual
739

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix ave/correlate command
Syntax:
fix ID group-ID ave/correlate Nevery Nrepeat Nfreq value1 value2 ... keyword args ...
ID, group-ID are documented in fix command• ave/correlate = style name of this fix command• Nevery = use input values every this many timesteps• Nrepeat = # of correlation time windows to accumulate• Nfreq = calculate time window averages every this many timesteps• one or more input values can be listed• value = c_ID, c_ID[N], f_ID, f_ID[N], v_name
c_ID = global scalar calculated by a compute with ID
c_ID[I] = Ith component of global vector calculated by a compute with ID, I can include wildcard (see below)
f_ID = global scalar calculated by a fix with ID
f_ID[I] = Ith component of global vector calculated by a fix with ID, I can include wildcard (see below)
v_name = global value calculated by an equal-style variable with name
v_name[I] = Ith component of a vector-style variable with name
•
zero or more keyword/arg pairs may be appended• keyword = type or ave or start or prefactor or file or overwrite or title1 or title2 or title3
type arg = auto or upper or lower or auto/upper or auto/lower or full
auto = correlate each value with itself
upper = correlate each value with each succeeding value
lower = correlate each value with each preceding value
auto/upper = auto + upper
auto/lower = auto + lower
full = correlate each value with every other value, including itself = auto + upper + lower
ave args = one or running
one = zero the correlation accumulation every Nfreq steps
running = accumulate correlations continuously
start args = Nstart
Nstart = start accumulating correlations on this timestep
prefactor args = value
value = prefactor to scale all the correlation data by
file arg = filename
filename = name of file to output correlation data to
overwrite arg = none = overwrite output file with only latest output
title1 arg = string
string = text to print as 1st line of output file
title2 arg = string
string = text to print as 2nd line of output file
title3 arg = string
string = text to print as 3rd line of output file
•
Examples:
fix 1 all ave/correlate 5 100 1000 c_myTemp file temp.correlate
fix 1 all ave/correlate 1 50 10000 &
c_thermo_press[1] c_thermo_press[2] c_thermo_press[3] &
type upper ave running title1 "My correlation data"
LAMMPS Users Manual
740

fix 1 all ave/correlate 1 50 10000 c_thermo_press[*]
Description:
Use one or more global scalar values as inputs every few timesteps, calculate time correlations between them
at varying time intervals, and average the correlation data over longer timescales. The resulting correlation
values can be time integrated by variables or used by other output commands such as thermo_style custom,
and can also be written to a file. See the fix ave/correlate/long command for an alternate method for
computing correlation functions efficiently over very long time windows.
The group specified with this command is ignored. However, note that specified values may represent
calculations performed by computes and fixes which store their own "group" definitions.
Each listed value can be the result of a compute or fix or the evaluation of an equal-style or vector-style
variable. In each case, the compute, fix, or variable must produce a global quantity, not a per-atom or local
quantity. If you wish to spatial- or time-average or histogram per-atom quantities from a compute, fix, or
variable, then see the fix ave/chunk, fix ave/atom, or fix ave/histo commands. If you wish to convert a
per-atom quantity into a single global value, see the compute reduce command.
The input values must either be all scalars. What kinds of correlations between input values are calculated is
determined by the type keyword as discussed below.
Computes that produce global quantities are those which do not have the word atom in their style name. Only
a few fixes produce global quantities. See the doc pages for individual fixes for info on which ones produce
such values. Variables of style equal and vector are the only ones that can be used with this fix. Variables of
style atom cannot be used, since they produce per-atom values.
Note that for values from a compute or fix, the bracketed index I can be specified using a wildcard asterisk
with the index to effectively specify multiple values. This takes the form "*" or "*n" or "n*" or "m*n". If N =
the size of the vector (for mode = scalar) or the number of columns in the array (for mode = vector), then an
asterisk with no numeric values means all indices from 1 to N. A leading asterisk means all indices from 1 to
n (inclusive). A trailing asterisk means all indices from n to N (inclusive). A middle asterisk means all indices
from m to n (inclusive).
Using a wildcard is the same as if the individual elements of the vector had been listed one by one. E.g. these
2 fix ave/correlate commands are equivalent, since the compute pressure command creates a global vector
with 6 values.
compute myPress all pressure NULL
fix 1 all ave/correlate 1 50 10000 c_myPress[*]
fix 1 all ave/correlate 1 50 10000 &
c_myPress[1] c_myPress[2] c_myPress[3] &
c_myPress[4] c_myPress[5] c_myPress[6]
The Nevery, Nrepeat, and Nfreq arguments specify on what timesteps the input values will be used to
calculate correlation data. The input values are sampled every Nevery timesteps. The correlation data for the
preceding samples is computed on timesteps that are a multiple of Nfreq. Consider a set of samples from some
initial time up to an output timestep. The initial time could be the beginning of the simulation or the last
output time; see the ave keyword for options. For the set of samples, the correlation value Cij is calculated as:
Cij(delta) = ave(Vi(t)*Vj(t+delta))
LAMMPS Users Manual
741
which is the correlation value between input values Vi and Vj, separated by time delta. Note that the second
value Vj in the pair is always the one sampled at the later time. The ave() represents an average over every
pair of samples in the set that are separated by time delta. The maximum delta used is of size
(Nrepeat-1)*Nevery. Thus the correlation between a pair of input values yields Nrepeat correlation datums:
Cij(0), Cij(Nevery), Cij(2*Nevery), ..., Cij((Nrepeat-1)*Nevery)
For example, if Nevery=5, Nrepeat=6, and Nfreq=100, then values on timesteps 0,5,10,15,...,100 will be used
to compute the final averages on timestep 100. Six averages will be computed: Cij(0), Cij(5), Cij(10), Cij(15),
Cij(20), and Cij(25). Cij(10) on timestep 100 will be the average of 19 samples, namely Vi(0)*Vj(10),
Vi(5)*Vj(15), Vi(10)*V j20), Vi(15)*Vj(25), ..., Vi(85)*Vj(95), Vi(90)*Vj(100).
Nfreq must be a multiple of Nevery; Nevery and Nrepeat must be non-zero. Also, if the ave keyword is set to
one which is the default, then Nfreq >= (Nrepeat-1)*Nevery is required.
If a value begins with "c_", a compute ID must follow which has been previously defined in the input script. If
no bracketed term is appended, the global scalar calculated by the compute is used. If a bracketed term is
appended, the Ith element of the global vector calculated by the compute is used. See the discussion above for
how I can be specified with a wildcard asterisk to effectively specify multiple values.
Note that there is a compute reduce command which can sum per-atom quantities into a global scalar or vector
which can thus be accessed by fix ave/correlate. Or it can be a compute defined not in your input script, but by
thermodynamic output or other fixes such as fix nvt or fix temp/rescale. See the doc pages for these
commands which give the IDs of these computes. Users can also write code for their own compute styles and
add them to LAMMPS.
If a value begins with "f_", a fix ID must follow which has been previously defined in the input script. If no
bracketed term is appended, the global scalar calculated by the fix is used. If a bracketed term is appended, the
Ith element of the global vector calculated by the fix is used. See the discussion above for how I can be
specified with a wildcard asterisk to effectively specify multiple values.
Note that some fixes only produce their values on certain timesteps, which must be compatible with Nevery,
else an error will result. Users can also write code for their own fix styles and add them to LAMMPS.
If a value begins with "v_", a variable name must follow which has been previously defined in the input
script. Only equal-style or vector-style variables can be referenced; the latter requires a bracketed term to
specify the Ith element of the vector calculated by the variable. See the variable command for details. Note
that variables of style equal or vector define a formula which can reference individual atom properties or
thermodynamic keywords, or they can invoke other computes, fixes, or variables when they are evaluated, so
this is a very general means of specifying quantities to time correlate.
Additional optional keywords also affect the operation of this fix.
The type keyword determines which pairs of input values are correlated with each other. For N input values
Vi, for i = 1 to N, let the number of pairs = Npair. Note that the second value in the pair Vi(t)*Vj(t+delta) is
always the one sampled at the later time.
If type is set to auto then each input value is correlated with itself. I.e. Cii = Vi*Vi, for i = 1 to N, so
Npair = N.
•
If type is set to upper then each input value is correlated with every succeeding value. I.e. Cij =
Vi*Vj, for i < j, so Npair = N*(N-1)/2.
•
LAMMPS Users Manual
742

If type is set to lower then each input value is correlated with every preceding value. I.e. Cij = Vi*Vj,
for i > j, so Npair = N*(N-1)/2.
•
If type is set to auto/upper then each input value is correlated with itself and every succeeding value.
I.e. Cij = Vi*Vj, for i >= j, so Npair = N*(N+1)/2.
•
If type is set to auto/lower then each input value is correlated with itself and every preceding value.
I.e. Cij = Vi*Vj, for i <= j, so Npair = N*(N+1)/2.
•
If type is set to full then each input value is correlated with itself and every other value. I.e. Cij =
Vi*Vj, for i,j = 1,N so Npair = N^2.
•
The ave keyword determines what happens to the accumulation of correlation samples every Nfreq timesteps.
If the ave setting is one, then the accumulation is restarted or zeroed every Nfreq timesteps. Thus the outputs
on successive Nfreq timesteps are essentially independent of each other. The exception is that the Cij(0) =
Vi(T)*Vj(T) value at a timestep T, where T is a multiple of Nfreq, contributes to the correlation output both at
time T and at time T+Nfreq.
If the ave setting is running, then the accumulation is never zeroed. Thus the output of correlation data at any
timestep is the average over samples accumulated every Nevery steps since the fix was defined. it can only be
restarted by deleting the fix via the unfix command, or by re-defining the fix by re-specifying it.
The start keyword specifies what timestep the accumulation of correlation samples will begin on. The default
is step 0. Setting it to a larger value can avoid adding non-equilibrated data to the correlation averages.
The prefactor keyword specifies a constant which will be used as a multiplier on the correlation data after it is
averaged. It is effectively a scale factor on Vi*Vj, which can be used to account for the size of the time
window or other unit conversions.
The file keyword allows a filename to be specified. Every Nfreq steps, an array of correlation data is written to
the file. The number of rows is Nrepeat, as described above. The number of columns is the Npair+2, also as
described above. Thus the file ends up to be a series of these array sections.
The overwrite keyword will continuously overwrite the output file with the latest output, so that it only
contains one timestep worth of output. This option can only be used with the ave running setting.
The title1 and title2 and title3 keywords allow specification of the strings that will be printed as the first 3
lines of the output file, assuming the file keyword was used. LAMMPS uses default values for each of these,
so they do not need to be specified.
By default, these header lines are as follows:
# Time-correlated data for fix ID
# TimeStep Number-of-time-windows
# Index TimeDelta Ncount valueI*valueJ valueI*valueJ ...
In the first line, ID is replaced with the fix-ID. The second line describes the two values that are printed at the
first of each section of output. In the third line the value pairs are replaced with the appropriate fields from the
fix ave/correlate command.
Let Sij = a set of time correlation data for input values I and J, namely the Nrepeat values:
Sij = Cij(0), Cij(Nevery), Cij(2*Nevery), ..., Cij(*Nrepeat-1)*Nevery)
LAMMPS Users Manual
743

As explained below, these datums are output as one column of a global array, which is effectively the
correlation matrix.
The trap function defined for equal-style variables can be used to perform a time integration of this vector of
datums, using a trapezoidal rule. This is useful for calculating various quantities which can be derived from
time correlation data. If a normalization factor is needed for the time integration, it can be included in the
variable formula or via the prefactor keyword.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global array of values which can be accessed by various output commands. The values
can only be accessed on timesteps that are multiples of Nfreq since that is when averaging is performed. The
global array has # of rows = Nrepeat and # of columns = Npair+2. The first column has the time delta (in
timesteps) between the pairs of input values used to calculate the correlation, as described above. The 2nd
column has the number of samples contributing to the correlation average, as described above. The remaining
Npair columns are for I,J pairs of the N input values, as determined by the type keyword, as described above.
For type = auto, the Npair = N columns are ordered: C11, C22, ..., CNN.• For type = upper, the Npair = N*(N-1)/2 columns are ordered: C12, C13, ..., C1N, C23, ..., C2N, C34,
..., CN-1N.
•
For type = lower, the Npair = N*(N-1)/2 columns are ordered: C21, C31, C32, C41, C42, C43, ...,
CN1, CN2, ..., CNN-1.
•
For type = auto/upper, the Npair = N*(N+1)/2 columns are ordered: C11, C12, C13, ..., C1N, C22,
C23, ..., C2N, C33, C34, ..., CN-1N, CNN.
•
For type = auto/lower, the Npair = N*(N+1)/2 columns are ordered: C11, C21, C22, C31, C32, C33,
C41, ..., C44, CN1, CN2, ..., CNN-1, CNN.
•
For type = full, the Npair = N^2 columns are ordered: C11, C12, ..., C1N, C21, C22, ..., C2N, C31, ...,
C3N, ..., CN1, ..., CNN-1, CNN.
•
The array values calculated by this fix are treated as intensive. If you need to divide them by the number of
atoms, you must do this in a later processing step, e.g. when using them in a variable.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
fix ave/correlate/long, compute, fix ave/time, fix ave/atom, fix ave/chunk, fix ave/histo, variable
Default: none
The option defaults are ave = one, type = auto, start = 0, no file output, title 1,2,3 = strings as described above,
and prefactor = 1.0.
LAMMPS Users Manual
744

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix ave/correlate/long command
Syntax:
fix ID group-ID ave/correlate/long Nevery Nfreq value1 value2 ... keyword args ...
ID, group-ID are documented in fix command• ave/correlate/long = style name of this fix command• Nevery = use input values every this many timesteps• Nfreq = save state of the time correlation functions every this many timesteps• one or more input values can be listed• value = c_ID, c_ID[N], f_ID, f_ID[N], v_name
c_ID = global scalar calculated by a compute with ID
c_ID[I] = Ith component of global vector calculated by a compute with ID
f_ID = global scalar calculated by a fix with ID
f_ID[I] = Ith component of global vector calculated by a fix with ID
v_name = global value calculated by an equal-style variable with name
•
zero or more keyword/arg pairs may be appended• keyword = type or start or file or overwrite or title1 or title2 or ncorr or p or m
type arg = auto or upper or lower or auto/upper or auto/lower or full
auto = correlate each value with itself
upper = correlate each value with each succeeding value
lower = correlate each value with each preceding value
auto/upper = auto + upper
auto/lower = auto + lower
full = correlate each value with every other value, including itself = auto + upper + lower
start args = Nstart
Nstart = start accumulating correlations on this timestep
file arg = filename
filename = name of file to output correlation data to
overwrite arg = none = overwrite output file with only latest output
title1 arg = string
string = text to print as 1st line of output file
title2 arg = string
string = text to print as 2nd line of output file
ncorr arg = Ncorrelators
Ncorrelators = number of correlators to store
nlen args = Nlen
Nlen = length of each correlator
ncount args = Ncount
Ncount = number of values over which succesive correlators are averaged
•
Examples:
fix 1 all ave/correlate/long 5 1000 c_myTemp file temp.correlate
fix 1 all ave/correlate/long 1 10000 &
c_thermo_press[1] c_thermo_press[2] c_thermo_press[3] &
type upper title1 "My correlation data" nlen 15 ncount 3
Description:
LAMMPS Users Manual
745
This fix is similar in spirit and syntax to the fix ave/correlate. However, this fix allows the efficient calculation
of time correlation functions on the fly over extremely long time windows without too much CPU overhead,
using a multiple-tau method (Ramirez) that decreases the resolution of the stored correlation function with
time.
The group specified with this command is ignored. However, note that specified values may represent
calculations performed by computes and fixes which store their own "group" definitions.
Each listed value can be the result of a compute or fix or the evaluation of an equal-style variable. See the fix
ave/correlate doc page for details.
The Nevery and Nfreq arguments specify on what timesteps the input values will be used to calculate
correlation data, and the frequency with which the time correlation functions will be output to a file. Note that
there is no Nrepeat argument, unlike the fix ave/correlate command.
The optional keywords ncorr, nlen, and ncount are unique to this command and determine the number of
correlation points calculated and the memory and CPU overhead used by this calculation. Nlen and ncount
determine the amount of averaging done at longer correlation times. The default values nlen=16, ncount=2
ensure that the systematic error of the multiple-tau correlator is always below the level of the statistical error
of a typical simulation (which depends on the ensemble size and the simulation length).
The maximum correlation time (in time steps) that can be reached is given by the formula (nlen-1) *
ncount^(ncorr-1). Longer correlation times are discarded and not calculated. With the default values of the
parameters (ncorr=20, nlen=16 and ncount=2), this corresponds to 7864320 time steps. If longer correlation
times are needed, the value of ncorr should be increased. Using nlen=16 and ncount=2, with ncorr=30, the
maximum number of steps that can be correlated is 80530636808. If ncorr=40, correlation times in excess of
8e12 time steps can be calculated.
The total memory needed for each correlation pair is roughly 4*ncorr*nlen*8 bytes. With the default values of
the parameters, this corresponds to about 10 KB.
For the meaning of the additional optional keywords, see the fix ave/correlate doc page.
Restart, fix_modify, output, run start/stop, minimize info:
Since this fix in intended for the calculation of time correlation functions over very long MD simulations, the
information about this fix is written automatically to binary restart files, so that the time correlation
calculation can continue in subsequent simulations. None of the fix_modify options are relevant to this fix.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This compute is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
fix ave/correlate
LAMMPS Users Manual
746

Default: none
The option defaults for keywords that are also keywords for the fix ave/correlate command are as follows:
type = auto, start = 0, no file output, title 1,2 = strings as described on the fix ave/correlate doc page.
The option defaults for keywords unique to this command are as follows: ncorr=20, nlen=16, ncount=2.
(Ramirez) J. Ramirez, S.K. Sukumaran, B. Vorselaars and A.E. Likhtman, J. Chem. Phys. 133, 154103
(2010).
LAMMPS Users Manual
747

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix ave/histo command
fix ave/histo/weight command
Syntax:
fix ID group-ID style Nevery Nrepeat Nfreq lo hi Nbin value1 value2 ... keyword args ...
ID, group-ID are documented in fix command• style = ave/histo or ave/histo/weight = style name of this fix command• Nevery = use input values every this many timesteps• Nrepeat = # of times to use input values for calculating histogram• Nfreq = calculate histogram every this many timesteps• lo,hi = lo/hi bounds within which to histogram• Nbin = # of histogram bins• one or more input values can be listed• value = x, y, z, vx, vy, vz, fx, fy, fz, c_ID, c_ID[N], f_ID, f_ID[N], v_name
x,y,z,vx,vy,vz,fx,fy,fz = atom attribute (position, velocity, force component)
c_ID = scalar or vector calculated by a compute with ID
c_ID[I] = Ith component of vector or Ith column of array calculated by a compute with ID, I can include wildcard (see below)
f_ID = scalar or vector calculated by a fix with ID
f_ID[I] = Ith component of vector or Ith column of array calculated by a fix with ID, I can include wildcard (see below)
v_name = value(s) calculated by an equal-style or vector-style or atom-style variable with name
v_name[I] = value calculated by a vector-style variable with name
•
zero or more keyword/arg pairs may be appended• keyword = mode or file or ave or start or beyond or overwrite or title1 or title2 or title3
mode arg = scalar or vector
scalar = all input values are scalars
vector = all input values are vectors
file arg = filename
filename = name of file to output histogram(s) to
ave args = one or running or window
one = output a new average value every Nfreq steps
running = output cumulative average of all previous Nfreq steps
window M = output average of M most recent Nfreq steps
start args = Nstart
Nstart = start averaging on this timestep
beyond arg = ignore or end or extra
ignore = ignore values outside histogram lo/hi bounds
end = count values outside histogram lo/hi bounds in end bins
extra = create 2 extra bins for value outside histogram lo/hi bounds
overwrite arg = none = overwrite output file with only latest output
title1 arg = string
string = text to print as 1st line of output file
title2 arg = string
string = text to print as 2nd line of output file
title3 arg = string
string = text to print as 3rd line of output file, only for vector mode
•
Examples:
LAMMPS Users Manual
748
fix 1 all ave/histo 100 5 1000 0.5 1.5 50 c_myTemp file temp.histo ave running
fix 1 all ave/histo 100 5 1000 -5 5 100 c_thermo_press[2] c_thermo_press[3] title1 "My output values"
fix 1 all ave/histo 100 5 1000 -5 5 100 c_thermo_press[*]
fix 1 all ave/histo 1 100 1000 -2.0 2.0 18 vx vy vz mode vector ave running beyond extra
fix 1 all ave/histo/weight 1 1 1 10 100 2000 c_XRD[1] c_XRD[2]
Description:
Use one or more values as inputs every few timesteps to create a single histogram. The histogram can then be
averaged over longer timescales. The resulting histogram can be used by other output commands, and can also
be written to a file. The fix ave/histo/weight command has identical syntax to fix ave/histo, except that exactly
two values must be specified. See details below.
The group specified with this command is ignored for global and local input values. For per-atom input
values, only atoms in the group contribute to the histogram. Note that regardless of the specified group,
specified values may represent calculations performed by computes and fixes which store their own "group"
definition.
A histogram is simply a count of the number of values that fall within a histogram bin. Nbins are defined, with
even spacing between lo and hi. Values that fall outside the lo/hi bounds can be treated in different ways; see
the discussion of the beyond keyword below.
Each input value can be an atom attribute (position, velocity, force component) or can be the result of a
compute or fix or the evaluation of an equal-style or vector-style or atom-style variable. The set of input
values can be either all global, all per-atom, or all local quantities. Inputs of different kinds (e.g. global and
per-atom) cannot be mixed. Atom attributes are per-atom vector values. See the doc page for individual
"compute" and "fix" commands to see what kinds of quantities they generate.
Note that the output of this command is a single histogram for all input values combined together, not one
histogram per input value. See below for details on the format of the output of this fix.
The input values must either be all scalars or all vectors (or arrays), depending on the setting of the mode
keyword.
If mode = scalar, then the input values must be scalars, or vectors with a bracketed term appended, indicating
the Ith value of the vector is used.
If mode = vector, then the input values must be vectors, or arrays with a bracketed term appended, indicating
the Ith column of the array is used.
Note that for values from a compute or fix, the bracketed index I can be specified using a wildcard asterisk
with the index to effectively specify multiple values. This takes the form "*" or "*n" or "n*" or "m*n". If N =
the size of the vector (for mode = scalar) or the number of columns in the array (for mode = vector), then an
asterisk with no numeric values means all indices from 1 to N. A leading asterisk means all indices from 1 to
n (inclusive). A trailing asterisk means all indices from n to N (inclusive). A middle asterisk means all indices
from m to n (inclusive).
Using a wildcard is the same as if the individual elements of the vector or columns of the array had been listed
one by one. E.g. these 2 fix ave/histo commands are equivalent, since the compute com/chunk command
creates a global array with 3 columns:
compute myCOM all com/chunk
LAMMPS Users Manual
749

fix 1 all ave/histo 100 1 100 c_myCOM[*] file tmp1.com mode vector
fix 2 all ave/histo 100 1 100 c_myCOM[1] c_myCOM[2] c_myCOM[3] file tmp2.com mode vector
If the fix ave/histo/weight command is used, exactly two values must be specified. If the values are vectors,
they must be the same length. The first value (a scalar or vector) is what is histogrammed into bins, in the
same manner the fix ave/histo command operates. The second value (a scalar or vector) is used as a "weight".
This means that instead of each value tallying a "1" to its bin, the corresponding weight is tallied. E.g. The
Nth entry (weight) in the second vector is tallied to the bin corresponding to the Nth entry in the first vector.
The Nevery, Nrepeat, and Nfreq arguments specify on what timesteps the input values will be used in order to
contribute to the histogram. The final histogram is generated on timesteps that are multiple of Nfreq. It is
averaged over Nrepeat histograms, computed in the preceding portion of the simulation every Nevery
timesteps. Nfreq must be a multiple of Nevery and Nevery must be non-zero even if Nrepeat is 1. Also, the
timesteps contributing to the histogram value cannot overlap, i.e. Nrepeat*Nevery can not exceed Nfreq.
For example, if Nevery=2, Nrepeat=6, and Nfreq=100, then input values on timesteps 90,92,94,96,98,100 will
be used to compute the final histogram on timestep 100. Similarly for timesteps 190,192,194,196,198,200 on
timestep 200, etc. If Nrepeat=1 and Nfreq = 100, then no time averaging of the histogram is done; a histogram
is simply generated on timesteps 100,200,etc.
The atom attribute values (x,y,z,vx,vy,vz,fx,fy,fz) are self-explanatory. Note that other atom attributes can be
used as inputs to this fix by using the compute property/atom command and then specifying an input value
from that compute.
If a value begins with "c_", a compute ID must follow which has been previously defined in the input script. If
mode = scalar, then if no bracketed term is appended, the global scalar calculated by the compute is used. If a
bracketed term is appended, the Ith element of the global vector calculated by the compute is used. If mode =
vector, then if no bracketed term is appended, the global or per-atom or local vector calculated by the compute
is used. If a bracketed term is appended, the Ith column of the global or per-atom or local array calculated by
the compute is used. See the discussion above for how I can be specified with a wildcard asterisk to
effectively specify multiple values.
Note that there is a compute reduce command which can sum per-atom quantities into a global scalar or vector
which can thus be accessed by fix ave/histo. Or it can be a compute defined not in your input script, but by
thermodynamic output or other fixes such as fix nvt or fix temp/rescale. See the doc pages for these
commands which give the IDs of these computes. Users can also write code for their own compute styles and
add them to LAMMPS.
If a value begins with "f_", a fix ID must follow which has been previously defined in the input script. If mode
= scalar, then if no bracketed term is appended, the global scalar calculated by the fix is used. If a bracketed
term is appended, the Ith element of the global vector calculated by the fix is used. If mode = vector, then if no
bracketed term is appended, the global or per-atom or local vector calculated by the fix is used. If a bracketed
term is appended, the Ith column of the global or per-atom or local array calculated by the fix is used. See the
discussion above for how I can be specified with a wildcard asterisk to effectively specify multiple values.
Note that some fixes only produce their values on certain timesteps, which must be compatible with Nevery,
else an error will result. Users can also write code for their own fix styles and add them to LAMMPS.
If a value begins with "v_", a variable name must follow which has been previously defined in the input
script. If mode = scalar, then only equal-style or vector-style variables can be used, which both produce global
values. In this mode, a vector-style variable requires a bracketed term to specify the Ith element of the vector
LAMMPS Users Manual
750

calculated by the variable. If mode = vector, then only vector-style or atom-style variables can be used, which
produce a global or per-atom vector respectively. The vector-style variable must be used without a bracketed
term. See the variable command for details.
Note that variables of style equal, vector, and atom define a formula which can reference individual atom
properties or thermodynamic keywords, or they can invoke other computes, fixes, or variables when they are
evaluated, so this is a very general means of specifying quantities to histogram.
Additional optional keywords also affect the operation of this fix.
If the mode keyword is set to scalar, then all input values must be global scalars, or elements of global
vectors. If the mode keyword is set to vector, then all input values must be global or per-atom or local vectors,
or columns of global or per-atom or local arrays.
The beyond keyword determines how input values that fall outside the lo to hi bounds are treated. Values such
that lo <= value <= hi are assigned to one bin. Values on a bin boundary are assigned to the lower of the 2
bins. If beyond is set to ignore then values < lo and values > hi are ignored, i.e. they are not binned. If beyond
is set to end then values < lo are counted in the first bin and values > hi are counted in the last bin. If beyond is
set to extend then two extra bins are created, so that there are Nbins+2 total bins. Values < lo are counted in
the first bin and values > hi are counted in the last bin (Nbins+1). Values between lo and hi (inclusive) are
counted in bins 2 thru Nbins+1. The "coordinate" stored and printed for these two extra bins is lo and hi.
The ave keyword determines how the histogram produced every Nfreq steps are averaged with histograms
produced on previous steps that were multiples of Nfreq, before they are accessed by another output command
or written to a file.
If the ave setting is one, then the histograms produced on timesteps that are multiples of Nfreq are
independent of each other; they are output as-is without further averaging.
If the ave setting is running, then the histograms produced on timesteps that are multiples of Nfreq are
summed and averaged in a cumulative sense before being output. Each bin value in the histogram is thus the
average of the bin value produced on that timestep with all preceding values for the same bin. This running
average begins when the fix is defined; it can only be restarted by deleting the fix via the unfix command, or
by re-defining the fix by re-specifying it.
If the ave setting is window, then the histograms produced on timesteps that are multiples of Nfreq are
summed within a moving "window" of time, so that the last M histograms are used to produce the output. E.g.
if M = 3 and Nfreq = 1000, then the output on step 10000 will be the combined histogram of the individual
histograms on steps 8000,9000,10000. Outputs on early steps will be sums over less than M histograms if they
are not available.
The start keyword specifies what timestep histogramming will begin on. The default is step 0. Often input
values can be 0.0 at time 0, so setting start to a larger value can avoid including a 0.0 in a running or
windowed histogram.
The file keyword allows a filename to be specified. Every Nfreq steps, one histogram is written to the file.
This includes a leading line that contains the timestep, number of bins, the total count of values contributing
to the histogram, the count of values that were not histogrammed (see the beyond keyword), the minimum
value encountered, and the maximum value encountered. The min/max values include values that were not
histogrammed. Following the leading line, one line per bin is written into the file. Each line contains the bin #,
the coordinate for the center of the bin (between lo and hi), the count of values in the bin, and the normalized
LAMMPS Users Manual
751

count. The normalized count is the bin count divided by the total count (not including values not
histogrammed), so that the normalized values sum to 1.0 across all bins.
The overwrite keyword will continuously overwrite the output file with the latest output, so that it only
contains one timestep worth of output. This option can only be used with the ave running setting.
The title1 and title2 and title3 keywords allow specification of the strings that will be printed as the first 3
lines of the output file, assuming the file keyword was used. LAMMPS uses default values for each of these,
so they do not need to be specified.
By default, these header lines are as follows:
# Histogram for fix ID
# TimeStep Number-of-bins Total-counts Missing-counts Min-value Max-value
# Bin Coord Count Count/Total
In the first line, ID is replaced with the fix-ID. The second line describes the six values that are printed at the
first of each section of output. The third describes the 4 values printed for each bin in the histogram.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix produces a global vector and global array which can be accessed by various output commands. The
values can only be accessed on timesteps that are multiples of Nfreq since that is when a histogram is
generated. The global vector has 4 values:
1 = total counts in the histogram• 2 = values that were not histogrammed (see beyond keyword)• 3 = min value of all input values, including ones not histogrammed• 4 = max value of all input values, including ones not histogrammed•
The global array has # of rows = Nbins and # of columns = 3. The first column has the bin coordinate, the 2nd
column has the count of values in that histogram bin, and the 3rd column has the bin count divided by the
total count (not including missing counts), so that the values in the 3rd column sum to 1.0.
The vector and array values calculated by this fix are all treated as intensive. If this is not the case, e.g. due to
histogramming per-atom input values, then you will need to account for that when interpreting the values
produced by this fix.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
compute, fix ave/atom, fix ave/chunk, fix ave/time, variable, fix ave/correlate,
Default: none
LAMMPS Users Manual
752
The option defaults are mode = scalar, ave = one, start = 0, no file output, beyond = ignore, and title 1,2,3 =
strings as described above.
LAMMPS Users Manual
753

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix ave/time command
Syntax:
fix ID group-ID ave/time Nevery Nrepeat Nfreq value1 value2 ... keyword args ...
ID, group-ID are documented in fix command• ave/time = style name of this fix command• Nevery = use input values every this many timesteps• Nrepeat = # of times to use input values for calculating averages• Nfreq = calculate averages every this many timesteps• one or more input values can be listed• value = c_ID, c_ID[N], f_ID, f_ID[N], v_name
c_ID = global scalar or vector calculated by a compute with ID
c_ID[I] = Ith component of global vector or Ith column of global array calculated by a compute with ID, I can include wildcard (see below)
f_ID = global scalar or vector calculated by a fix with ID
f_ID[I] = Ith component of global vector or Ith column of global array calculated by a fix with ID, I can include wildcard (see below)
v_name = value(s) calculated by an equal-style or vector-style variable with name
v_name[I] = value calculated by a vector-style variable with name
•
zero or more keyword/arg pairs may be appended• keyword = mode or file or ave or start or off or overwrite or title1 or title2 or title3
mode arg = scalar or vector
scalar = all input values are global scalars
vector = all input values are global vectors or global arrays
ave args = one or running or window M
one = output a new average value every Nfreq steps
running = output cumulative average of all previous Nfreq steps
window M = output average of M most recent Nfreq steps
start args = Nstart
Nstart = start averaging on this timestep
off arg = M = do not average this value
M = value # from 1 to Nvalues
file arg = filename
filename = name of file to output time averages to
overwrite arg = none = overwrite output file with only latest output
format arg = string
string = C-style format string
title1 arg = string
string = text to print as 1st line of output file
title2 arg = string
string = text to print as 2nd line of output file
title3 arg = string
string = text to print as 3rd line of output file, only for vector mode
•
Examples:
fix 1 all ave/time 100 5 1000 c_myTemp c_thermo_temp file temp.profile
fix 1 all ave/time 100 5 1000 c_thermo_press[2] ave window 20 &
title1 "My output values"
fix 1 all ave/time 100 5 1000 c_thermo_press[*]
fix 1 all ave/time 1 100 1000 f_indent f_indent[1] file temp.indent off 1
LAMMPS Users Manual
754

Description:
Use one or more global values as inputs every few timesteps, and average them over longer timescales. The
resulting averages can be used by other output commands such as thermo_style custom, and can also be
written to a file. Note that if no time averaging is done, this command can be used as a convenient way to
simply output one or more global values to a file.
The group specified with this command is ignored. However, note that specified values may represent
calculations performed by computes and fixes which store their own "group" definitions.
Each listed value can be the result of a compute or fix or the evaluation of an equal-style or vector-style
variable. In each case, the compute, fix, or variable must produce a global quantity, not a per-atom or local
quantity. If you wish to spatial- or time-average or histogram per-atom quantities from a compute, fix, or
variable, then see the fix ave/chunk, fix ave/atom, or fix ave/histo commands. If you wish to sum a per-atom
quantity into a single global quantity, see the compute reduce command.
Computes that produce global quantities are those which do not have the word atom in their style name. Only
a few fixes produce global quantities. See the doc pages for individual fixes for info on which ones produce
such values. Variables of style equal and vector are the only ones that can be used with this fix. Variables of
style atom cannot be used, since they produce per-atom values.
The input values must either be all scalars or all vectors depending on the setting of the mode keyword. In
both cases, the averaging is performed independently on each input value. I.e. each input scalar is averaged
independently or each element of each input vector is averaged independently.
If mode = scalar, then the input values must be scalars, or vectors with a bracketed term appended, indicating
the Ith value of the vector is used.
If mode = vector, then the input values must be vectors, or arrays with a bracketed term appended, indicating
the Ith column of the array is used. All vectors must be the same length, which is the length of the vector or
number of rows in the array.
Note that for values from a compute or fix, the bracketed index I can be specified using a wildcard asterisk
with the index to effectively specify multiple values. This takes the form "*" or "*n" or "n*" or "m*n". If N =
the size of the vector (for mode = scalar) or the number of columns in the array (for mode = vector), then an
asterisk with no numeric values means all indices from 1 to N. A leading asterisk means all indices from 1 to
n (inclusive). A trailing asterisk means all indices from n to N (inclusive). A middle asterisk means all indices
from m to n (inclusive).
Using a wildcard is the same as if the individual elements of the vector or columns of the array had been listed
one by one. E.g. these 2 fix ave/time commands are equivalent, since the compute rdf command creates, in
this case, a global array with 3 columns, each of length 50:
compute myRDF all rdf 50 1 2
fix 1 all ave/time 100 1 100 c_myRDF[*] file tmp1.rdf mode vector
fix 2 all ave/time 100 1 100 c_myRDF[1] c_myRDF[2] c_myRDF[3] file tmp2.rdf mode vector
The Nevery, Nrepeat, and Nfreq arguments specify on what timesteps the input values will be used in order to
contribute to the average. The final averaged quantities are generated on timesteps that are a mlutiple of
Nfreq. The average is over Nrepeat quantities, computed in the preceding portion of the simulation every
Nevery timesteps. Nfreq must be a multiple of Nevery and Nevery must be non-zero even if Nrepeat is 1. Also,
LAMMPS Users Manual
755
the timesteps contributing to the average value cannot overlap, i.e. Nrepeat*Nevery can not exceed Nfreq.
For example, if Nevery=2, Nrepeat=6, and Nfreq=100, then values on timesteps 90,92,94,96,98,100 will be
used to compute the final average on timestep 100. Similarly for timesteps 190,192,194,196,198,200 on
timestep 200, etc. If Nrepeat=1 and Nfreq = 100, then no time averaging is done; values are simply generated
on timesteps 100,200,etc.
If a value begins with "c_", a compute ID must follow which has been previously defined in the input script. If
mode = scalar, then if no bracketed term is appended, the global scalar calculated by the compute is used. If a
bracketed term is appended, the Ith element of the global vector calculated by the compute is used. If mode =
vector, then if no bracketed term is appended, the global vector calculated by the compute is used. If a
bracketed term is appended, the Ith column of the global array calculated by the compute is used. See the
discussion above for how I can be specified with a wildcard asterisk to effectively specify multiple values.
Note that there is a compute reduce command which can sum per-atom quantities into a global scalar or vector
which can thus be accessed by fix ave/time. Or it can be a compute defined not in your input script, but by
thermodynamic output or other fixes such as fix nvt or fix temp/rescale. See the doc pages for these
commands which give the IDs of these computes. Users can also write code for their own compute styles and
add them to LAMMPS.
If a value begins with "f_", a fix ID must follow which has been previously defined in the input script. If mode
= scalar, then if no bracketed term is appended, the global scalar calculated by the fix is used. If a bracketed
term is appended, the Ith element of the global vector calculated by the fix is used. If mode = vector, then if no
bracketed term is appended, the global vector calculated by the fix is used. If a bracketed term is appended,
the Ith column of the global array calculated by the fix is used. See the discussion above for how I can be
specified with a wildcard asterisk to effectively specify multiple values.
Note that some fixes only produce their values on certain timesteps, which must be compatible with Nevery,
else an error will result. Users can also write code for their own fix styles and add them to LAMMPS.
If a value begins with "v_", a variable name must follow which has been previously defined in the input
script. If mode = scalar, then only equal-style or vector-style variables can be used, which both produce global
values. In this mode, a vector-style variable requires a bracketed term to specify the Ith element of the vector
calculated by the variable. If mode = vector, then only a vector-style variable can be used, without a bracketed
term. See the variable command for details.
Note that variables of style equal and vector define a formula which can reference individual atom properties
or thermodynamic keywords, or they can invoke other computes, fixes, or variables when they are evaluated,
so this is a very general means of specifying quantities to time average.
Additional optional keywords also affect the operation of this fix.
If the mode keyword is set to scalar, then all input values must be global scalars, or elements of global
vectors. If the mode keyword is set to vector, then all input values must be global vectors, or columns of
global arrays. They can also be global arrays, which are converted into a series of global vectors (one per
column), as explained above.
The ave keyword determines how the values produced every Nfreq steps are averaged with values produced
on previous steps that were multiples of Nfreq, before they are accessed by another output command or
written to a file.
LAMMPS Users Manual
756
If the ave setting is one, then the values produced on timesteps that are multiples of Nfreq are independent of
each other; they are output as-is without further averaging.
If the ave setting is running, then the values produced on timesteps that are multiples of Nfreq are summed
and averaged in a cumulative sense before being output. Each output value is thus the average of the value
produced on that timestep with all preceding values. This running average begins when the fix is defined; it
can only be restarted by deleting the fix via the unfix command, or by re-defining the fix by re-specifying it.
If the ave setting is window, then the values produced on timesteps that are multiples of Nfreq are summed
and averaged within a moving "window" of time, so that the last M values are used to produce the output. E.g.
if M = 3 and Nfreq = 1000, then the output on step 10000 will be the average of the individual values on steps
8000,9000,10000. Outputs on early steps will average over less than M values if they are not available.
The start keyword specifies what timestep averaging will begin on. The default is step 0. Often input values
can be 0.0 at time 0, so setting start to a larger value can avoid including a 0.0 in a running or windowed
average.
The off keyword can be used to flag any of the input values. If a value is flagged, it will not be time averaged.
Instead the most recent input value will always be stored and output. This is useful if one of more of the
inputs produced by a compute or fix or variable are effectively constant or are simply current values. E.g. they
are being written to a file with other time-averaged values for purposes of creating well-formatted output.
The file keyword allows a filename to be specified. Every Nfreq steps, one quantity or vector of quantities is
written to the file for each input value specified in the fix ave/time command. For mode = scalar, this means a
single line is written each time output is performed. Thus the file ends up to be a series of lines, i.e. one
column of numbers for each input value. For mode = vector, an array of numbers is written each time output is
performed. The number of rows is the length of the input vectors, and the number of columns is the number of
values. Thus the file ends up to be a series of these array sections.
The overwrite keyword will continuously overwrite the output file with the latest output, so that it only
contains one timestep worth of output. This option can only be used with the ave running setting.
The format keyword sets the numeric format of each value when it is printed to a file via the file keyword.
Note that all values are floating point quantities. The default format is %g. You can specify a higher precision
if desired, e.g. %20.16g.
The title1 and title2 and title3 keywords allow specification of the strings that will be printed as the first 2 or 3
lines of the output file, assuming the file keyword was used. LAMMPS uses default values for each of these,
so they do not need to be specified.
By default, these header lines are as follows for mode = scalar:
# Time-averaged data for fix ID
# TimeStep value1 value2 ...
In the first line, ID is replaced with the fix-ID. In the second line the values are replaced with the appropriate
fields from the fix ave/time command. There is no third line in the header of the file, so the title3 setting is
ignored when mode = scalar.
By default, these header lines are as follows for mode = vector:
LAMMPS Users Manual
757

# Time-averaged data for fix ID
# TimeStep Number-of-rows
# Row value1 value2 ...
In the first line, ID is replaced with the fix-ID. The second line describes the two values that are printed at the
first of each section of output. In the third line the values are replaced with the appropriate fields from the fix
ave/time command.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix produces a global scalar or global vector or global array which can be accessed by various output
commands. The values can only be accessed on timesteps that are multiples of Nfreq since that is when
averaging is performed.
A scalar is produced if only a single input value is averaged and mode = scalar. A vector is produced if
multiple input values are averaged for mode = scalar, or a single input value for mode = vector. In the first
case, the length of the vector is the number of inputs. In the second case, the length of the vector is the same
as the length of the input vector. An array is produced if multiple input values are averaged and mode =
vector. The global array has # of rows = length of the input vectors and # of columns = number of inputs.
If the fix produces a scalar or vector, then the scalar and each element of the vector can be either "intensive"
or "extensive", depending on whether the values contributing to the scalar or vector element are "intensive" or
"extensive". If the fix produces an array, then all elements in the array must be the same, either "intensive" or
"extensive". If a compute or fix provides the value being time averaged, then the compute or fix determines
whether the value is intensive or extensive; see the doc page for that compute or fix for further info. Values
produced by a variable are treated as intensive.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
compute, fix ave/atom, fix ave/chunk, fix ave/histo, variable, fix ave/correlate,
Default:
The option defaults are mode = scalar, ave = one, start = 0, no file output, format = %g, title 1,2,3 = strings as
described above, and no off settings for any input values.
LAMMPS Users Manual
758

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix aveforce command
Syntax:
fix ID group-ID aveforce fx fy fz keyword value ...
ID, group-ID are documented in fix command• aveforce = style name of this fix command• fx,fy,fz = force component values (force units)
any of fx,fy,fz can be a variable (see below)
•
zero or more keyword/value pairs may be appended to args• keyword = region
region value = region-ID
region-ID = ID of region atoms must be in to have added force
•
Examples:
fix pressdown topwall aveforce 0.0 -1.0 0.0
fix 2 bottomwall aveforce NULL -1.0 0.0 region top
fix 2 bottomwall aveforce NULL -1.0 v_oscillate region top
Description:
Apply an additional external force to a group of atoms in such a way that every atom experiences the same
force. This is useful for pushing on wall or boundary atoms so that the structure of the wall does not change
over time.
The existing force is averaged for the group of atoms, component by component. The actual force on each
atom is then set to the average value plus the component specified in this command. This means each atom in
the group receives the same force.
Any of the fx,fy,fz values can be specified as NULL which means the force in that dimension is not changed.
Note that this is not the same as specifying a 0.0 value, since that sets all forces to the same average value
without adding in any additional force.
Any of the 3 quantities defining the force components can be specified as an equal-style variable, namely fx,
fy, fz. If the value is a variable, it should be specified as v_name, where name is the variable name. In this
case, the variable will be evaluated each timestep, and its value used to determine the average force.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent average force.
If the region keyword is used, the atom must also be in the specified geometric region in order to have force
added to it.
Styles with a suffix are functionally the same as the corresponding style without the suffix. They have been
LAMMPS Users Manual
759

optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The
accelerated styles take the same arguments and should produce the same results, except for round-off and
precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
This fix computes a global 3-vector of forces, which can be accessed by various output commands. This is the
total force on the group of atoms before the forces on individual atoms are changed by the fix. The vector
values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
You should not specify force components with a variable that has time-dependence for use with a minimizer,
since the minimizer increments the timestep as the iteration count during the minimization.
Restrictions: none
Related commands:
fix setforce, fix addforce
Default: none
LAMMPS Users Manual
760

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix balance command
Syntax:
fix ID group-ID balance Nfreq thresh style args keyword args ...
ID, group-ID are documented in fix command• balance = style name of this fix command• Nfreq = perform dynamic load balancing every this many steps• thresh = imbalance threshold that must be exceeded to perform a re-balance• style = shift or rcb
shift args = dimstr Niter stopthresh
dimstr = sequence of letters containing "x" or "y" or "z", each not more than once
Niter = # of times to iterate within each dimension of dimstr sequence
stopthresh = stop balancing when this imbalance threshold is reached
rcb args = none
•
zero or more keyword/arg pairs may be appended• keyword = weight or out
weight style args = use weighted particle counts for the balancing
style = group or neigh or time or var or store
group args = Ngroup group1 weight1 group2 weight2 ...
Ngroup = number of groups with assigned weights
group1, group2, ... = group IDs
weight1, weight2, ... = corresponding weight factors
neigh factor = compute weight based on number of neighbors
factor = scaling factor (> 0)
time factor = compute weight based on time spend computing
factor = scaling factor (> 0)
var name = take weight from atom-style variable
name = name of the atom-style variable
store name = store weight in custom atom property defined by fix property/atom command
name = atom property name (without d_ prefix)
out arg = filename
filename = write each processor's sub-domain to a file, at each re-balancing
•
Examples:
fix 2 all balance 1000 1.05 shift x 10 1.05
fix 2 all balance 100 0.9 shift xy 20 1.1 out tmp.balance
fix 2 all balance 100 0.9 shift xy 20 1.1 weight group 3 substrate 3.0 solvent 1.0 solute 0.8 out tmp.balance
fix 2 all balance 100 1.0 shift x 10 1.1 weight time 0.8
fix 2 all balance 100 1.0 shift xy 5 1.1 weight var myweight weight neigh 0.6 weight store allweight
fix 2 all balance 1000 1.1 rcb
Description:
This command adjusts the size and shape of processor sub-domains within the simulation box, to attempt to
balance the number of particles and thus the computational cost (load) evenly across processors. The load
balancing is "dynamic" in the sense that rebalancing is performed periodically during the simulation. To
perform "static" balancing, before or between runs, see the balance command.
LAMMPS Users Manual
761

Load-balancing is typically most useful if the particles in the simulation box have a spatially-varying density
distribution or where the computational cost varies significantly between different atoms. E.g. a model of a
vapor/liquid interface, or a solid with an irregular-shaped geometry containing void regions, or hybrid pair
style simulations which combine pair styles with different computational cost. In these cases, the LAMMPS
default of dividing the simulation box volume into a regular-spaced grid of 3d bricks, with one equal-volume
sub-domain per processor, may assign numbers of particles per processor in a way that the computational
effort varies significantly. This can lead to poor performance when the simulation is run in parallel.
The balancing can be performed with or without per-particle weighting. With no weighting, the balancing
attempts to assign an equal number of particles to each processor. With weighting, the balancing attempts to
assign an equal aggregate computational weight to each processor, which typically induces a different number
of atoms assigned to each processor.
NOTE: The weighting options listed above are documented with the balance command in this section of the
balance command doc page. That section describes the various weighting options and gives a few examples of
how they can be used. The weighting options are the same for both the fix balance and balance commands.
Note that the processors command allows some control over how the box volume is split across processors.
Specifically, for a Px by Py by Pz grid of processors, it allows choice of Px, Py, and Pz, subject to the
constraint that Px * Py * Pz = P, the total number of processors. This is sufficient to achieve good
load-balance for some problems on some processor counts. However, all the processor sub-domains will still
have the same shape and same volume.
On a particular timestep, a load-balancing operation is only performed if the current "imbalance factor" in
particles owned by each processor exceeds the specified thresh parameter. The imbalance factor is defined as
the maximum number of particles (or weight) owned by any processor, divided by the average number of
particles (or weight) per processor. Thus an imbalance factor of 1.0 is perfect balance.
As an example, for 10000 particles running on 10 processors, if the most heavily loaded processor has 1200
particles, then the factor is 1.2, meaning there is a 20% imbalance. Note that re-balances can be forced even if
the current balance is perfect (1.0) be specifying a thresh < 1.0.
NOTE: This command attempts to minimize the imbalance factor, as defined above. But depending on the
method a perfect balance (1.0) may not be achieved. For example, "grid" methods (defined below) that create
a logical 3d grid cannot achieve perfect balance for many irregular distributions of particles. Likewise, if a
portion of the system is a perfect lattice, e.g. the initial system is generated by the create_atoms command,
then "grid" methods may be unable to achieve exact balance. This is because entire lattice planes will be
owned or not owned by a single processor.
NOTE: The imbalance factor is also an estimate of the maximum speed-up you can hope to achieve by
running a perfectly balanced simulation versus an imbalanced one. In the example above, the 10000 particle
simulation could run up to 20% faster if it were perfectly balanced, versus when imbalanced. However,
computational cost is not strictly proportional to particle count, and changing the relative size and shape of
processor sub-domains may lead to additional computational and communication overheads, e.g. in the PPPM
solver used via the kspace_style command. Thus you should benchmark the run times of a simulation before
and after balancing.
The method used to perform a load balance is specified by one of the listed styles, which are described in
detail below. There are 2 kinds of styles.
LAMMPS Users Manual
762

The shift style is a "grid" method which produces a logical 3d grid of processors. It operates by changing the
cutting planes (or lines) between processors in 3d (or 2d), to adjust the volume (area in 2d) assigned to each
processor, as in the following 2d diagram where processor sub-domains are shown and atoms are colored by
the processor that owns them. The leftmost diagram is the default partitioning of the simulation box across
processors (one sub-box for each of 16 processors); the middle diagram is after a "grid" method has been
applied.
The rcb style is a "tiling" method which does not produce a logical 3d grid of processors. Rather it tiles the
simulation domain with rectangular sub-boxes of varying size and shape in an irregular fashion so as to have
equal numbers of particles (or weight) in each sub-box, as in the rightmost diagram above.
The "grid" methods can be used with either of the comm_style command options, brick or tiled. The "tiling"
methods can only be used with comm_style tiled.
When a "grid" method is specified, the current domain partitioning can be either a logical 3d grid or a tiled
partitioning. In the former case, the current logical 3d grid is used as a starting point and changes are made to
improve the imbalance factor. In the latter case, the tiled partitioning is discarded and a logical 3d grid is
created with uniform spacing in all dimensions. This is the starting point for the balancing operation.
When a "tiling" method is specified, the current domain partitioning ("grid" or "tiled") is ignored, and a new
partitioning is computed from scratch.
The group-ID is ignored. However the impact of balancing on different groups of atoms can be affected by
using the group weight style as described below.
The Nfreq setting determines how often a rebalance is performed. If Nfreq > 0, then rebalancing will occur
every Nfreq steps. Each time a rebalance occurs, a reneighboring is triggered, so Nfreq should not be too
small. If Nfreq = 0, then rebalancing will be done every time reneighboring normally occurs, as determined by
the the neighbor and neigh_modify command settings.
On rebalance steps, rebalancing will only be attempted if the current imbalance factor, as defined above,
exceeds the thresh setting.
The shift style invokes a "grid" method for balancing, as described above. It changes the positions of cutting
planes between processors in an iterative fashion, seeking to reduce the imbalance factor.
LAMMPS Users Manual
763

The dimstr argument is a string of characters, each of which must be an "x" or "y" or "z". Eacn character can
appear zero or one time, since there is no advantage to balancing on a dimension more than once. You should
normally only list dimensions where you expect there to be a density variation in the particles.
Balancing proceeds by adjusting the cutting planes in each of the dimensions listed in dimstr, one dimension
at a time. For a single dimension, the balancing operation (described below) is iterated on up to Niter times.
After each dimension finishes, the imbalance factor is re-computed, and the balancing operation halts if the
stopthresh criterion is met.
A rebalance operation in a single dimension is performed using a density-dependent recursive multisectioning
algorithm, where the position of each cutting plane (line in 2d) in the dimension is adjusted independently.
This is similar to a recursive bisectioning for a single value, except that the bounds used for each bisectioning
take advantage of information from neighboring cuts if possible, as well as counts of particles at the bounds
on either side of each cuts, which themselves were cuts in previous iterations. The latter is used to infer a
density of particles near each of the current cuts. At each iteration, the count of particles on either side of each
plane is tallied. If the counts do not match the target value for the plane, the position of the cut is adjusted
based on the local density. The low and high bounds are adjusted on each iteration, using new count
information, so that they become closer together over time. Thus as the recursion progresses, the count of
particles on either side of the plane gets closer to the target value.
The density-dependent part of this algorithm is often an advantage when you rebalance a system that is
already nearly balanced. It typically converges more quickly than the geometric bisectioning algorithm used
by the balance command. However, if can be a disadvantage if you attempt to rebalance a system that is far
from balanced, and converge more slowly. In this case you probably want to use the balance command before
starting a run, so that you begin the run with a balanced system.
Once the rebalancing is complete and final processor sub-domains assigned, particles migrate to their new
owning processor as part of the normal reneighboring procedure.
NOTE: At each rebalance operation, the bisectioning for each cutting plane (line in 2d) typically starts with
low and high bounds separated by the extent of a processor's sub-domain in one dimension. The size of this
bracketing region shrinks based on the local density, as described above, which should typically be 1/2 or
more every iteration. Thus if Niter is specified as 10, the cutting plane will typically be positioned to better
than 1 part in 1000 accuracy (relative to the perfect target position). For Niter = 20, it will be accurate to better
than 1 part in a million. Thus there is no need to set Niter to a large value. This is especially true if you are
rebalancing often enough that each time you expect only an incremental adjustment in the cutting planes is
necessary. LAMMPS will check if the threshold accuracy is reached (in a dimension) is less iterations than
Niter and exit early.
The rcb style invokes a "tiled" method for balancing, as described above. It performs a recursive coordinate
bisectioning (RCB) of the simulation domain. The basic idea is as follows.
The simulation domain is cut into 2 boxes by an axis-aligned cut in the longest dimension, leaving one new
box on either side of the cut. All the processors are also partitioned into 2 groups, half assigned to the box on
the lower side of the cut, and half to the box on the upper side. (If the processor count is odd, one side gets an
extra processor.) The cut is positioned so that the number of atoms in the lower box is exactly the number that
the processors assigned to that box should own for load balance to be perfect. This also makes load balance
for the upper box perfect. The positioning is done iteratively, by a bisectioning method. Note that counting
atoms on either side of the cut requires communication between all processors at each iteration.
LAMMPS Users Manual
764

That is the procedure for the first cut. Subsequent cuts are made recursively, in exactly the same manner. The
subset of processors assigned to each box make a new cut in the longest dimension of that box, splitting the
box, the subset of processors, and the atoms in the box in two. The recursion continues until every processor is
assigned a sub-box of the entire simulation domain, and owns the atoms in that sub-box.
The out keyword writes text to the specified filename with the results of each rebalancing operation. The file
contains the bounds of the sub-domain for each processor after the balancing operation completes. The format
of the file is compatible with the Pizza.py mdump tool which has support for manipulating and visualizing
mesh files. An example is shown here for a balancing by 4 processors for a 2d problem:
ITEM: TIMESTEP
0
ITEM: NUMBER OF NODES
16
ITEM: BOX BOUNDS
0 10
0 10
0 10
ITEM: NODES
1 1 0 0 0
2 1 5 0 0
3 1 5 5 0
4 1 0 5 0
5 1 5 0 0
6 1 10 0 0
7 1 10 5 0
8 1 5 5 0
9 1 0 5 0
10 1 5 5 0
11 1 5 10 0
12 1 10 5 0
13 1 5 5 0
14 1 10 5 0
15 1 10 10 0
16 1 5 10 0
ITEM: TIMESTEP
0
ITEM: NUMBER OF SQUARES
4
ITEM: SQUARES
1 1 1 2 3 4
2 1 5 6 7 8
3 1 9 10 11 12
4 1 13 14 15 16
The coordinates of all the vertices are listed in the NODES section, 5 per processor. Note that the 4
sub-domains share vertices, so there will be duplicate nodes in the list.
The "SQUARES" section lists the node IDs of the 4 vertices in a rectangle for each processor (1 to 4).
For a 3d problem, the syntax is similar with 8 vertices listed for each processor, instead of 4, and
"SQUARES" replaced by "CUBES".
Restart, fix_modify, output, run start/stop, minimize info:
LAMMPS Users Manual
765

No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global scalar which is the imbalance factor after the most recent rebalance and a global
vector of length 3 with additional information about the most recent rebalancing. The 3 values in the vector
are as follows:
1 = max # of particles per processor• 2 = total # iterations performed in last rebalance• 3 = imbalance factor right before the last rebalance was performed•
As explained above, the imbalance factor is the ratio of the maximum number of particles (or total weight) on
any processor to the average number of particles (or total weight) per processor.
These quantities can be accessed by various output commands. The scalar and vector values calculated by this
fix are "intensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
For 2d simulations, the z style cannot be used. Nor can a "z" appear in dimstr for the shift style.
Related commands:
group, processors, balance
Default: none
LAMMPS Users Manual
766

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix bond/break command
Syntax:
fix ID group-ID bond/break Nevery bondtype Rmax keyword values ...
ID, group-ID are documented in fix command• bond/break = style name of this fix command• Nevery = attempt bond breaking every this many steps• bondtype = type of bonds to break• Rmax = bond longer than Rmax can break (distance units)• zero or more keyword/value pairs may be appended to args• keyword = prob
prob values = fraction seed
fraction = break a bond with this probability if otherwise eligible
seed = random number seed (positive integer)
•
Examples:
fix 5 all bond/break 10 2 1.2
fix 5 polymer bond/break 1 1 2.0 prob 0.5 49829
Description:
Break bonds between pairs of atoms as a simulation runs according to specified criteria. This can be used to
model the dissolution of a polymer network due to stretching of the simulation box or other deformations. In
this context, a bond means an interaction between a pair of atoms computed by the bond_style command.
Once the bond is broken it will be permanently deleted, as will all angle, dihedral, and improper interactions
that bond is part of.
This is different than a pairwise bond-order potential such as Tersoff or AIREBO which infers bonds and
many-body interactions based on the current geometry of a small cluster of atoms and effectively creates and
destroys bonds and higher-order many-body interactions from timestep to timestep as atoms move.
A check for possible bond breakage is performed every Nevery timesteps. If two bonded atoms I,J are further
than a distance Rmax of each other, if the bond is of type bondtype, and if both I and J are in the specified fix
group, then I,J is labeled as a "possible" bond to break.
If several bonds involving an atom are stretched, it may have multiple possible bonds to break. Every atom
checks its list of possible bonds to break and labels the longest such bond as its "sole" bond to break. After
this is done, if atom I is bonded to atom J in its sole bond, and atom J is bonded to atom I in its sole bond, then
the I,J bond is "eligible" to be broken.
Note that these rules mean an atom will only be part of at most one broken bond on a given timestep. It also
means that if atom I chooses atom J as its sole partner, but atom J chooses atom K is its sole partner (due to
Rjk > Rij), then this means atom I will not be part of a broken bond on this timestep, even if it has other
possible bond partners.
LAMMPS Users Manual
767

The prob keyword can effect whether an eligible bond is actually broken. The fraction setting must be a value
between 0.0 and 1.0. A uniform random number between 0.0 and 1.0 is generated and the eligible bond is only
broken if the random number < fraction.
When a bond is broken, data structures within LAMMPS that store bond topology are updated to reflect the
breakage. Likewise, if the bond is part of a 3-body (angle) or 4-body (dihedral, improper) interaction, that
interaction is removed as well. These changes typically affect pairwise interactions between atoms that used to
be part of bonds, angles, etc.
NOTE: One data structure that is not updated when a bond breaks are the molecule IDs stored by each atom.
Even though one molecule becomes two molecules due to the broken bond, all atoms in both new molecules
retain their original molecule IDs.
Computationally, each timestep this fix operates, it loops over all the bonds in the system and computes
distances between pairs of bonded atoms. It also communicates between neighboring processors to coordinate
which bonds are broken. Moreover, if any bonds are broken, neighbor lists must be immediately updated on
the same timestep. This is to insure that any pairwise interactions that should be turned "on" due to a bond
breaking, because they are no longer excluded by the presence of the bond and the settings of the
special_bonds command, will be immediately recognized. All of these operations increase the cost of a
timestep. Thus you should be cautious about invoking this fix too frequently.
You can dump out snapshots of the current bond topology via the dump local command.
NOTE: Breaking a bond typically alters the energy of a system. You should be careful not to choose bond
breaking criteria that induce a dramatic change in energy. For example, if you define a very stiff harmonic
bond and break it when 2 atoms are separated by a distance far from the equilibribum bond length, then the 2
atoms will be dramatically released when the bond is broken. More generally, you may need to thermostat
your system to compensate for energy changes resulting from broken bonds (and angles, dihedrals,
impropers).
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes two statistics which it stores in a global vector of length 2, which can be accessed by
various output commands. The vector values calculated by this fix are "intensive".
These are the 2 quantities:
(1) # of bonds broken on the most recent breakage timestep• (2) cumulative # of bonds broken•
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the MC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
LAMMPS Users Manual
768

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix bond/create command
Syntax:
fix ID group-ID bond/create Nevery itype jtype Rmin bondtype keyword values ...
ID, group-ID are documented in fix command• bond/create = style name of this fix command• Nevery = attempt bond creation every this many steps• itype,jtype = atoms of itype can bond to atoms of jtype• Rmin = 2 atoms separated by less than Rmin can bond (distance units)• bondtype = type of created bonds• zero or more keyword/value pairs may be appended to args• keyword = iparam or jparam or prob or atype or dtype or itype
iparam values = maxbond, newtype
maxbond = max # of bonds of bondtype the itype atom can have
newtype = change the itype atom to this type when maxbonds exist
jparam values = maxbond, newtype
maxbond = max # of bonds of bondtype the jtype atom can have
newtype = change the jtype atom to this type when maxbonds exist
prob values = fraction seed
fraction = create a bond with this probability if otherwise eligible
seed = random number seed (positive integer)
atype value = angletype
angletype = type of created angles
dtype value = dihedraltype
dihedraltype = type of created dihedrals
itype value = impropertype
impropertype = type of created impropers
•
Examples:
fix 5 all bond/create 10 1 2 0.8 1
fix 5 all bond/create 1 3 3 0.8 1 prob 0.5 85784 iparam 2 3
fix 5 all bond/create 1 3 3 0.8 1 prob 0.5 85784 iparam 2 3 atype 1 dtype 2
Description:
Create bonds between pairs of atoms as a simulation runs according to specified criteria. This can be used to
model cross-linking of polymers, the formation of a percolation network, etc. In this context, a bond means an
interaction between a pair of atoms computed by the bond_style command. Once the bond is created it will be
permanently in place. Optionally, the creation of a bond can also create angle, dihedral, and improper
interactions that bond is part of. See the discussion of the atype, dtype, and itype keywords below.
This is different than a pairwise bond-order potential such as Tersoff or AIREBO which infers bonds and
many-body interactions based on the current geometry of a small cluster of atoms and effectively creates and
destroys bonds and higher-order many-body interactions from timestep to timestep as atoms move.
A check for possible new bonds is performed every Nevery timesteps. If two atoms I,J are within a distance
Rmin of each other, if I is of atom type itype, if J is of atom type jtype, if both I and J are in the specified fix
group, if a bond does not already exist between I and J, and if both I and J meet their respective maxbond
LAMMPS Users Manual
770
requirement (explained below), then I,J is labeled as a "possible" bond pair.
If several atoms are close to an atom, it may have multiple possible bond partners. Every atom checks its list
of possible bond partners and labels the closest such partner as its "sole" bond partner. After this is done, if
atom I has atom J as its sole partner, and atom J has atom I as its sole partner, then the I,J bond is "eligible" to
be formed.
Note that these rules mean an atom will only be part of at most one created bond on a given timestep. It also
means that if atom I chooses atom J as its sole partner, but atom J chooses atom K is its sole partner (due to
Rjk < Rij), then this means atom I will not form a bond on this timestep, even if it has other possible bond
partners.
It is permissible to have itype = jtype. Rmin must be <= the pairwise cutoff distance between itype and jtype
atoms, as defined by the pair_style command.
The iparam and jparam keywords can be used to limit the bonding functionality of the participating atoms.
Each atom keeps track of how many bonds of bondtype it already has. If atom I of itype already has maxbond
bonds (as set by the iparam keyword), then it will not form any more. Likewise for atom J. If maxbond is set
to 0, then there is no limit on the number of bonds that can be formed with that atom.
The newtype value for iparam and jparam can be used to change the atom type of atom I or J when it reaches
maxbond number of bonds of type bondtype. This means it can now interact in a pairwise fashion with other
atoms in a different way by specifying different pair_coeff coefficients. If you do not wish the atom type to
change, simply specify newtype as itype or jtype.
The prob keyword can also effect whether an eligible bond is actually created. The fraction setting must be a
value between 0.0 and 1.0. A uniform random number between 0.0 and 1.0 is generated and the eligible bond
is only created if the random number < fraction.
Any bond that is created is assigned a bond type of bondtype
When a bond is created, data structures within LAMMPS that store bond topology are updated to reflect the
creation. If the bond is part of new 3-body (angle) or 4-body (dihedral, improper) interactions, you can choose
to create new angles, dihedrals, impropers as well, using the atype, dtype, and itype keywords. All of these
changes typically affect pairwise interactions between atoms that are now part of new bonds, angles, etc.
NOTE: One data structure that is not updated when a bond breaks are the molecule IDs stored by each atom.
Even though two molecules become one molecule due to the created bond, all atoms in the new molecule
retain their original molecule IDs.
If the atype keyword is used and if an angle potential is defined via the angle_style command, then any new
3-body interactions inferred by the creation of a bond will create new angles of type angletype, with
parameters assigned by the corresponding angle_coeff command. Likewise, the dtype and itype keywords will
create new dihedrals and impropers of type dihedraltype and impropertype.
NOTE: To create a new bond, the internal LAMMPS data structures that store this information must have
space for it. When LAMMPS is initialized from a data file, the list of bonds is scanned and the maximum
number of bonds per atom is tallied. If some atom will acquire more bonds than this limit as this fix operates,
then the "extra bond per atom" parameter must be set to allow for it. Ditto for "extra angle per atom", "extra
dihedral per atom", and "extra improper per atom" if angles, dihedrals, or impropers are being added when
bonds are created. See the read_data or create_box command for more details. Note that a data file with no
LAMMPS Users Manual
771

atoms can be used if you wish to add unbonded atoms via the create atoms command, e.g. for a percolation
simulation.
NOTE: LAMMPS stores and maintains a data structure with a list of the 1st, 2nd, and 3rd neighbors of each
atom (within the bond topology of the system) for use in weighting pairwise interactions for bonded atoms.
Note that adding a single bond always adds a new 1st neighbor but may also induce *many* new 2nd and 3rd
neighbors, depending on the molecular topology of your system. The "extra special per atom" parameter must
typically be set to allow for the new maximum total size (1st + 2nd + 3rd neighbors) of this per-atom list.
There are 3 ways to do this. See the read_data or create_box or "special_bonds extra" commands for details.
NOTE: Even if you do not use the atype, dtype, or itype keywords, the list of topological neighbors is updated
for atoms affected by the new bond. This in turn affects which neighbors are considered for pairwise
interactions, using the weighting rules set by the special_bonds command. Consider a new bond created
between atoms I,J. If J has a bonded neighbor K, then K becomes a 2nd neighbor of I. Even if the atype
keyword is not used to create angle I-J-K, the pairwise interaction between I and K will be potentially turned
off or weighted by the 1-3 weighting specified by the special_bonds command. This is the case even if the
"angle yes" option was used with that command. The same is true for 3rd neighbors (1-4 interactions), the
dtype keyword, and the "dihedral yes" option used with the special_bonds command.
Note that even if your simulation starts with no bonds, you must define a bond_style and use the bond_coeff
command to specify coefficients for the bondtype. Similarly, if new atom types are specified by the iparam or
jparam keywords, they must be within the range of atom types allowed by the simulation and pairwise
coefficients must be specified for the new types.
Computationally, each timestep this fix operates, it loops over neighbor lists and computes distances between
pairs of atoms in the list. It also communicates between neighboring processors to coordinate which bonds are
created. Moreover, if any bonds are created, neighbor lists must be immediately updated on the same timestep.
This is to insure that any pairwise interactions that should be turned "off" due to a bond creation, because they
are now excluded by the presence of the bond and the settings of the special_bonds command, will be
immediately recognized. All of these operations increase the cost of a timestep. Thus you should be cautious
about invoking this fix too frequently.
You can dump out snapshots of the current bond topology via the dump local command.
NOTE: Creating a bond typically alters the energy of a system. You should be careful not to choose bond
creation criteria that induce a dramatic change in energy. For example, if you define a very stiff harmonic
bond and create it when 2 atoms are separated by a distance far from the equilibribum bond length, then the 2
atoms will oscillate dramatically when the bond is formed. More generally, you may need to thermostat your
system to compensate for energy changes resulting from created bonds (and angles, dihedrals, impropers).
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes two statistics which it stores in a global vector of length 2, which can be accessed by
various output commands. The vector values calculated by this fix are "intensive".
These are the 2 quantities:
(1) # of bonds created on the most recent creation timestep•
LAMMPS Users Manual
772
(2) cumulative # of bonds created•
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the MC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
fix bond/break, fix bond/swap, dump local, special_bonds
Default:
The option defaults are iparam = (0,itype), jparam = (0,jtype), and prob = 1.0.
LAMMPS Users Manual
773
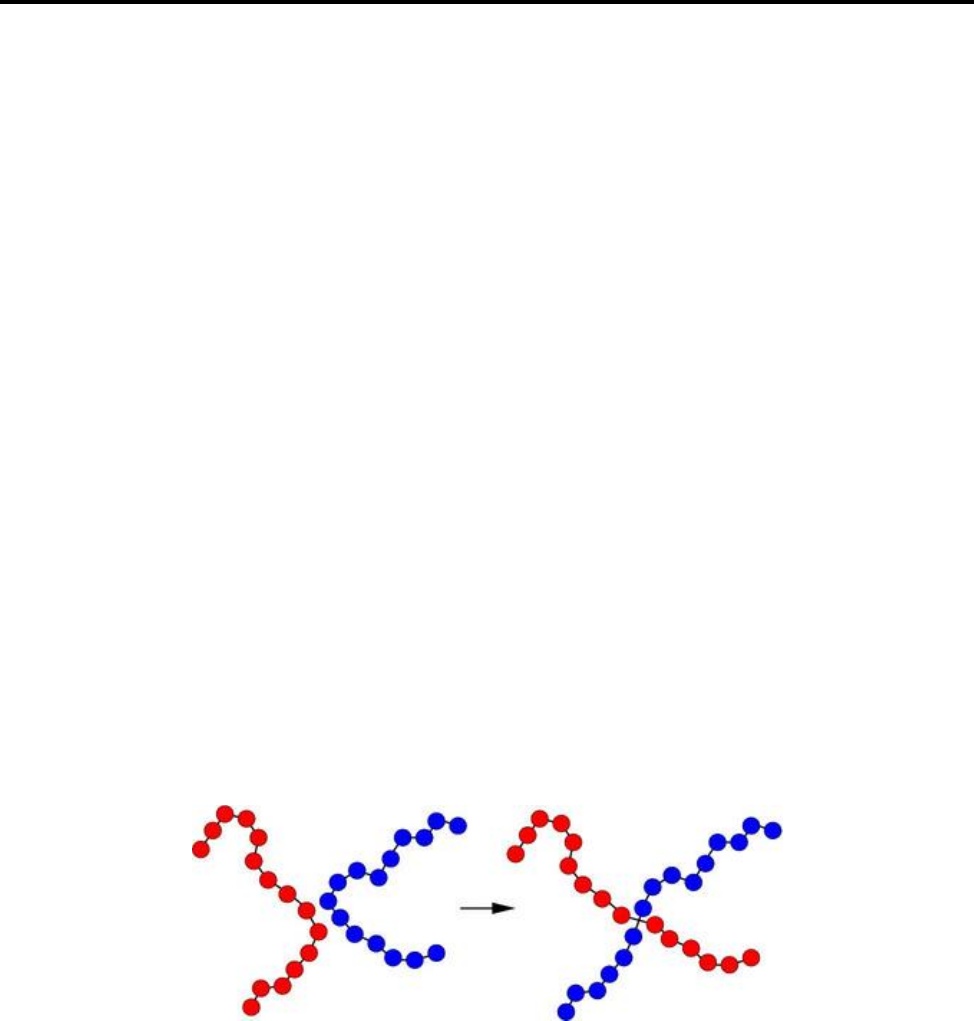
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix bond/swap command
Syntax:
fix ID group-ID bond/swap Nevery fraction cutoff seed
ID, group-ID are documented in fix command• bond/swap = style name of this fix command• Nevery = attempt bond swapping every this many steps• fraction = fraction of group atoms to consider for swapping• cutoff = distance at which swapping will be considered (distance units)• seed = random # seed (positive integer)•
Examples:
fix 1 all bond/swap 50 0.5 1.3 598934
Description:
In a simulation of polymer chains, this command attempts to swap bonds between two different chains,
effectively grafting the end of one chain onto another chain and vice versa. This is done via Monte Carlo rules
using the Boltzmann acceptance criterion. The purpose is to equilibrate the polymer chain conformations
more rapidly than dynamics alone would do it, by enabling instantaneous large conformational changes in a
dense polymer melt. The polymer chains should thus more rapidly converge to the proper end-to-end
distances and radii of gyration. It is designed for use with systems of FENE or harmonic bead-spring polymer
chains where each polymer is a linear chain of monomers, but LAMMPS does not enforce this requirement,
i.e. any bond_style can be used.
A schematic of the kinds of bond swaps that can occur is shown here:
On the left, the red and blue chains have two monomers A1 and B1 close to each other, which are currently
bonded to monomers A2 and B2 respectively within their own chains. The bond swap operation will attempt
to delete the A1-A2 and B1-B2 bonds and replace them with A1-B2 and B1-A2 bonds. If the swap is
energetically favorable, the two chains on the right are the result and each polymer chain has undergone a
dramatic conformational change. This reference, (Sides) provides more details on how the algorithm works
and its application:
The bond swapping operation is invoked every Nevery timesteps. If any bond is swapped, a re-build of the
neighbor lists is triggered, since a swap alters the list of which neighbors are considered for pairwise
interaction. At each invocation, each processor considers a random specified fraction of its atoms as potential
LAMMPS Users Manual
774

swapping monomers for this timestep. Choosing a small fraction value can reduce the likelihood of a reverse
swap occurring soon after an initial swap.
For each monomer A1, its neighbors are examined to find a possible B1 monomer. Both A1 and B1 must be
in the fix group, their separation must be less than the specified cutoff, and the molecule IDs of A1 and B1
must be the same (see below). If a suitable partner is found, the energy change due to swapping the 2 bonds is
computed. This includes changes in pairwise, bond, and angle energies due to the altered connectivity of the 2
chains. Dihedral and improper interactions are not allowed to be defined when this fix is used.
If the energy decreases due to the swap operation, the bond swap is accepted. If the energy increases it is
accepted with probability exp(-delta/kT) where delta is the increase in energy, k is the Boltzmann constant,
and T is the current temperature of the system. Whether the swap is accepted or rejected, no other swaps are
attempted by this processor on this timestep.
The criterion for matching molecule IDs is how bond swaps performed by this fix conserve chain length. To
use this features you must setup the molecule IDs for your polymer chains in a certain way, typically in the
data file, read by the read_data command. Consider a system of 6-mer chains. You have 2 choices. If the
molecule IDs for monomers on each chain are set to 1,2,3,4,5,6 then swaps will conserve chain length. For a
particular momoner there will be only one other monomer on another chain which is a potential swap partner.
If the molecule IDs for monomers on each chain are set to 1,2,3,3,2,1 then swaps will conserve chain length
but swaps will be able to occur at either end of a chain. Thus for a particular monomer there will be 2 possible
swap partners on another chain. In this scenario, swaps can also occur within a single chain, i.e. the two ends
of a chain swap with each other.
NOTE: If your simulation uses molecule IDs in the usual way, where all monomers on a single chain are
assigned the same ID (different for each chain), then swaps will only occur within the same chain. If you
assign the same molecule ID to all monomers in all chains then inter-chain swaps will occur, but they will not
conserve chain length. Neither of these scenarios is probably what you want for this fix.
NOTE: When a bond swap occurs the image flags of monomers in the new polymer chains can become
inconsistent. See the dump command for a discussion of image flags. This is not an issue for running
dynamics, but can affect calculation of some diagnostic quantities or the printing of unwrapped coordinates to
a dump file.
This fix computes a temperature each time it is invoked for use by the Boltzmann criterion. To do this, the fix
creates its own compute of style temp, as if this command had been issued:
compute fix-ID_temp all temp
See the compute temp command for details. Note that the ID of the new compute is the fix-ID with underscore
+ "temp" appended and the group for the new compute is "all", so that the temperature of the entire system is
used.
Note that this is NOT the compute used by thermodynamic output (see the thermo_style command) with ID =
thermo_temp. This means you can change the attributes of this fix's temperature (e.g. its degrees-of-freedom)
via the compute_modify command or print this temperature during thermodynamic output via the
thermo_style custom command using the appropriate compute-ID. It also means that changing attributes of
thermo_temp will have no effect on this fix.
Restart, fix_modify, thermo output, run start/stop, minimize info:
LAMMPS Users Manual
775

No information about this fix is written to binary restart files. Because the state of the random number
generator is not saved in restart files, this means you cannot do "exact" restarts with this fix, where the
simulation continues on the same as if no restart had taken place. However, in a statistical sense, a restarted
simulation should produce the same behavior. Also note that each processor generates possible swaps
independently of other processors. Thus if you repeat the same simulation on a different number of
processors, the specific swaps performed will be different.
The fix_modify temp option is supported by this fix. You can use it to assign a compute you have defined to
this fix which will be used to compute the temperature for the Boltzmann criterion.
This fix computes two statistical quantities as a global 2-vector of output, which can be accessed by various
output commands. The first component of the vector is the cumulative number of swaps performed by all
processors. The second component of the vector is the cumulative number of swaps attempted (whether
accepted or rejected). Note that a swap "attempt" only occurs when swap partners meeting the criteria
described above are found on a particular timestep. The vector values calculated by this fix are "intensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the MC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
The settings of the "special_bond" command must be 0,1,1 in order to use this fix, which is typical of
bead-spring chains with FENE or harmonic bonds. This means that pairwise interactions between bonded
atoms are turned off, but are turned on between atoms two or three hops away along the chain backbone.
Currently, energy changes in dihedral and improper interactions due to a bond swap are not considered. Thus
a simulation that uses this fix cannot use a dihedral or improper potential.
Related commands:
fix atom/swap
Default: none
(Sides) Sides, Grest, Stevens, Plimpton, J Polymer Science B, 42, 199-208 (2004).
LAMMPS Users Manual
776
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix box/relax command
Syntax:
fix ID group-ID box/relax keyword value ...
ID, group-ID are documented in fix command• box/relax = style name of this fix command
one or more keyword value pairs may be appended
keyword = iso or aniso or tri or x or y or z or xy or yz or xz or couple or nreset or vmax or dilate or scaleyz or scalexz or scalexy or fixedpoint
iso or aniso or tri value = Ptarget = desired pressure (pressure units)
x or y or z or xy or yz or xz value = Ptarget = desired pressure (pressure units)
couple = none or xyz or xy or yz or xz
nreset value = reset reference cell every this many minimizer iterations
vmax value = fraction = max allowed volume change in one iteration
dilate value = all or partial
scaleyz value = yes or no = scale yz with lz
scalexz value = yes or no = scale xz with lz
scalexy value = yes or no = scale xy with ly
fixedpoint values = x y z
x,y,z = perform relaxation dilation/contraction around this point (distance units)
•
Examples:
fix 1 all box/relax iso 0.0 vmax 0.001
fix 2 water box/relax aniso 0.0 dilate partial
fix 2 ice box/relax tri 0.0 couple xy nreset 100
Description:
Apply an external pressure or stress tensor to the simulation box during an energy minimization. This allows
the box size and shape to vary during the iterations of the minimizer so that the final configuration will be
both an energy minimum for the potential energy of the atoms, and the system pressure tensor will be close to
the specified external tensor. Conceptually, specifying a positive pressure is like squeezing on the simulation
box; a negative pressure typically allows the box to expand.
The external pressure tensor is specified using one or more of the iso, aniso, tri, x, y, z, xy, xz, yz, and couple
keywords. These keywords give you the ability to specify all 6 components of an external stress tensor, and to
couple various of these components together so that the dimensions they represent are varied together during
the minimization.
Orthogonal simulation boxes have 3 adjustable dimensions (x,y,z). Triclinic (non-orthogonal) simulation
boxes have 6 adjustable dimensions (x,y,z,xy,xz,yz). The create_box, read data, and read_restart commands
specify whether the simulation box is orthogonal or non-orthogonal (triclinic) and explain the meaning of the
xy,xz,yz tilt factors.
The target pressures Ptarget for each of the 6 components of the stress tensor can be specified independently
via the x, y, z, xy, xz, yz keywords, which correspond to the 6 simulation box dimensions. For example, if the y
keyword is used, the y-box length will change during the minimization. If the xy keyword is used, the xy tilt
factor will change. A box dimension will not change if that component is not specified.
LAMMPS Users Manual
777

Note that in order to use the xy, xz, or yz keywords, the simulation box must be triclinic, even if its initial tilt
factors are 0.0.
When the size of the simulation box changes, all atoms are re-scaled to new positions, unless the keyword
dilate is specified with a value of partial, in which case only the atoms in the fix group are re-scaled. This can
be useful for leaving the coordinates of atoms in a solid substrate unchanged and controlling the pressure of a
surrounding fluid.
The scaleyz, scalexz, and scalexy keywords control whether or not the corresponding tilt factors are scaled
with the associated box dimensions when relaxing triclinic periodic cells. The default values yes will turn on
scaling, which corresponds to adjusting the linear dimensions of the cell while preserving its shape. Choosing
no ensures that the tilt factors are not scaled with the box dimensions. See below for restrictions and default
values in different situations. In older versions of LAMMPS, scaling of tilt factors was not performed. The old
behavior can be recovered by setting all three scale keywords to no.
The fixedpoint keyword specifies the fixed point for cell relaxation. By default, it is the center of the box.
Whatever point is chosen will not move during the simulation. For example, if the lower periodic boundaries
pass through (0,0,0), and this point is provided to fixedpoint, then the lower periodic boundaries will remain at
(0,0,0), while the upper periodic boundaries will move twice as far. In all cases, the particle positions at each
iteration are unaffected by the chosen value, except that all particles are displaced by the same amount,
different on each iteration.
NOTE: Applying an external pressure to tilt dimensions xy, xz, yz can sometimes result in arbitrarily large
values of the tilt factors, i.e. a dramatically deformed simulation box. This typically indicates that there is
something badly wrong with how the simulation was constructed. The two most common sources of this error
are applying a shear stress to a liquid system or specifying an external shear stress tensor that exceeds the
yield stress of the solid. In either case the minimization may converge to a bogus conformation or not
converge at all. Also note that if the box shape tilts to an extreme shape, LAMMPS will run less efficiently,
due to the large volume of communication needed to acquire ghost atoms around a processor's
irregular-shaped sub-domain. For extreme values of tilt, LAMMPS may also lose atoms and generate an error.
NOTE: Performing a minimization with this fix is not a mathematically well-defined minimization problem.
This is because the objective function being minimized changes if the box size/shape changes. In practice this
means the minimizer can get "stuck" before you have reached the desired tolerance. The solution to this is to
restart the minimizer from the new adjusted box size/shape, since that creates a new objective function valid
for the new box size/shape. Repeat as necessary until the box size/shape has reached its new equilibrium.
The couple keyword allows two or three of the diagonal components of the pressure tensor to be "coupled"
together. The value specified with the keyword determines which are coupled. For example, xz means the Pxx
and Pzz components of the stress tensor are coupled. Xyz means all 3 diagonal components are coupled.
Coupling means two things: the instantaneous stress will be computed as an average of the corresponding
diagonal components, and the coupled box dimensions will be changed together in lockstep, meaning coupled
dimensions will be dilated or contracted by the same percentage every timestep. The Ptarget values for any
coupled dimensions must be identical. Couple xyz can be used for a 2d simulation; the z dimension is simply
ignored.
The iso, aniso, and tri keywords are simply shortcuts that are equivalent to specifying several other keywords
together.
LAMMPS Users Manual
778
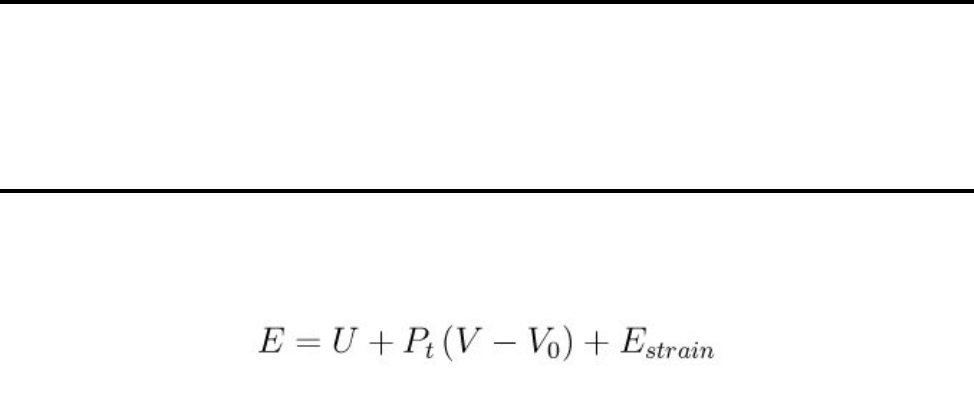
The keyword iso means couple all 3 diagonal components together when pressure is computed (hydrostatic
pressure), and dilate/contract the dimensions together. Using "iso Ptarget" is the same as specifying these 4
keywords:
x Ptarget
y Ptarget
z Ptarget
couple xyz
The keyword aniso means x, y, and z dimensions are controlled independently using the Pxx, Pyy, and Pzz
components of the stress tensor as the driving forces, and the specified scalar external pressure. Using "aniso
Ptarget" is the same as specifying these 4 keywords:
x Ptarget
y Ptarget
z Ptarget
couple none
The keyword tri means x, y, z, xy, xz, and yz dimensions are controlled independently using their individual
stress components as the driving forces, and the specified scalar pressure as the external normal stress. Using
"tri Ptarget" is the same as specifying these 7 keywords:
x Ptarget
y Ptarget
z Ptarget
xy 0.0
yz 0.0
xz 0.0
couple none
The vmax keyword can be used to limit the fractional change in the volume of the simulation box that can
occur in one iteration of the minimizer. If the pressure is not settling down during the minimization this can be
because the volume is fluctuating too much. The specified fraction must be greater than 0.0 and should be <<
1.0. A value of 0.001 means the volume cannot change by more than 1/10 of a percent in one iteration when
couple xyz has been specified. For any other case it means no linear dimension of the simulation box can
change by more than 1/10 of a percent.
With this fix, the potential energy used by the minimizer is augmented by an additional energy provided by
the fix. The overall objective function then is:
where U is the system potential energy, P_t is the desired hydrostatic pressure, V and V_0 are the system and
reference volumes, respectively. E_strain is the strain energy expression proposed by Parrinello and Rahman
(Parrinello1981). Taking derivatives of E w.r.t. the box dimensions, and setting these to zero, we find that at
the minimum of the objective function, the global system stress tensor P will satisfy the relation:
LAMMPS Users Manual
779

where I is the identity matrix, h_0 is the box dimension tensor of the reference cell, and h_0d is the diagonal
part of h_0. S_t is a symmetric stress tensor that is chosen by LAMMPS so that the upper-triangular
components of P equal the stress tensor specified by the user.
This equation only applies when the box dimensions are equal to those of the reference dimensions. If this is
not the case, then the converged stress tensor will not equal that specified by the user. We can resolve this
problem by periodically resetting the reference dimensions. The keyword nreset_ref controls how often this is
done. If this keyword is not used, or is given a value of zero, then the reference dimensions are set to those of
the initial simulation domain and are never changed. A value of nstep means that every nstep minimization
steps, the reference dimensions are set to those of the current simulation domain. Note that resetting the
reference dimensions changes the objective function and gradients, which sometimes causes the minimization
to fail. This can be resolved by changing the value of nreset, or simply continuing the minimization from a
restart file.
NOTE: As normally computed, pressure includes a kinetic- energy or temperature-dependent component; see
the compute pressure command. However, atom velocities are ignored during a minimization, and the applied
pressure(s) specified with this command are assumed to only be the virial component of the pressure (the
non-kinetic portion). Thus if atoms have a non-zero temperature and you print the usual thermodynamic
pressure, it may not appear the system is converging to your specified pressure. The solution for this is to
either (a) zero the velocities of all atoms before performing the minimization, or (b) make sure you are
monitoring the pressure without its kinetic component. The latter can be done by outputting the pressure from
the fix this command creates (see below) or a pressure fix you define yourself.
NOTE: Because pressure is often a very sensitive function of volume, it can be difficult for the minimizer to
equilibrate the system the desired pressure with high precision, particularly for solids. Some techniques that
seem to help are (a) use the "min_modify line quadratic" option when minimizing with box relaxations, (b)
minimize several times in succession if need be, to drive the pressure closer to the target pressure, (c) relax the
atom positions before relaxing the box, and (d) relax the box to the target hydrostatic pressure before relaxing
to a target shear stress state. Also note that some systems (e.g. liquids) will not sustain a non-hydrostatic
applied pressure, which means the minimizer will not converge.
This fix computes a temperature and pressure each timestep. The temperature is used to compute the kinetic
contribution to the pressure, even though this is subsequently ignored by default. To do this, the fix creates its
own computes of style "temp" and "pressure", as if these commands had been issued:
compute fix-ID_temp group-ID temp
compute fix-ID_press group-ID pressure fix-ID_temp virial
See the compute temp and compute pressure commands for details. Note that the IDs of the new computes are
the fix-ID + underscore + "temp" or fix_ID + underscore + "press", and the group for the new computes is the
same as the fix group. Also note that the pressure compute does not include a kinetic component.
Note that these are NOT the computes used by thermodynamic output (see the thermo_style command) with
ID = thermo_temp and thermo_press. This means you can change the attributes of this fix's temperature or
pressure via the compute_modify command or print this temperature or pressure during thermodynamic
output via the thermo_style custom command using the appropriate compute-ID. It also means that changing
LAMMPS Users Manual
780

attributes of thermo_temp or thermo_press will have no effect on this fix.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify temp and press options are supported by this fix. You can use them to assign a compute you
have defined to this fix which will be used in its temperature and pressure calculation, as described above.
Note that as described above, if you assign a pressure compute to this fix that includes a kinetic energy
component it will affect the minimization, most likely in an undesirable way.
NOTE: If both the temp and press keywords are used in a single thermo_modify command (or in two separate
commands), then the order in which the keywords are specified is important. Note that a pressure compute
defines its own temperature compute as an argument when it is specified. The temp keyword will override this
(for the pressure compute being used by fix npt), but only if the temp keyword comes after the press keyword.
If the temp keyword comes before the press keyword, then the new pressure compute specified by the press
keyword will be unaffected by the temp setting.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
pressure-volume energy, plus the strain energy, if it exists.
This fix computes a global scalar which can be accessed by various output commands. The scalar is given by
the energy expression shown above. The energy values reported at the end of a minimization run under
"Minimization stats" include this energy, and so differ from what LAMMPS normally reports as potential
energy. This fix does not support the fix_modify energy option, because that would result in double-counting
of the fix energy in the minimization energy. Instead, the fix energy can be explicitly added to the potential
energy using one of these two variants:
variable emin equal pe+f_1
variable emin equal pe+f_1/atoms
No parameter of this fix can be used with the start/stop keywords of the run command.
This fix is invoked during energy minimization, but not for the purpose of adding a contribution to the energy
or forces being minimized. Instead it alters the simulation box geometry as described above.
Restrictions:
Only dimensions that are available can be adjusted by this fix. Non-periodic dimensions are not available. z,
xz, and yz, are not available for 2D simulations. xy, xz, and yz are only available if the simulation domain is
non-orthogonal. The create_box, read data, and read_restart commands specify whether the simulation box is
orthogonal or non-orthogonal (triclinic) and explain the meaning of the xy,xz,yz tilt factors.
The scaleyz yes and scalexz yes keyword/value pairs can not be used for 2D simulations. scaleyz yes, scalexz
yes, and scalexy yes options can only be used if the 2nd dimension in the keyword is periodic, and if the tilt
factor is not coupled to the barostat via keywords tri, yz, xz, and xy.
Related commands:
fix npt, minimize
LAMMPS Users Manual
781

Default:
The keyword defaults are dilate = all, vmax = 0.0001, nreset = 0.
(Parrinello1981) Parrinello and Rahman, J Appl Phys, 52, 7182 (1981).
LAMMPS Users Manual
782

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix cmap command
Syntax:
fix ID group-ID cmap filename
ID, group-ID are documented in fix command• cmap = style name of this fix command• filename = force-field file with CMAP coefficients•
Examples:
fix myCMAP all cmap ../potentials/cmap36.data
read_data proteinX.data fix myCMAP crossterm CMAP
fix_modify myCMAP energy yes
Description:
This command enables CMAP crossterms to be added to simulations which use the CHARMM force field.
These are relevant for any CHARMM model of a peptide or protein sequences that is 3 or more amino-acid
residues long; see (Buck) and (Brooks) for details, including the analytic energy expressions for CMAP
interactions. The CMAP crossterms add additional potential energy contributions to pairs of overlapping
phi-psi dihedrals of amino-acids, which are important to properly represent their conformational behavior.
The examples/cmap directory has a sample input script and data file for a small peptide, that illustrates use of
the fix cmap command.
As in the example above, this fix should be used before reading a data file that contains a listing of CMAP
interactions. The filename specified should contain the CMAP parameters for a particular version of the
CHARMM force field. Two such files are including in the lammps/potentials directory: charmm22.cmap and
charmm36.cmap.
The data file read by the "read_data" must contain the topology of all the CMAP interactions, similar to the
topology data for bonds, angles, dihedrals, etc. Specially it should have a line like this in its header section:
N crossterms
where N is the number of CMAP crossterms. It should also have a section in the body of the data file like this
with N lines:
CMAP
1 1 8 10 12 18 20
2 5 18 20 22 25 27
[...]
N 3 314 315 317 318 330
The first column is an index from 1 to N to enumerate the CMAP terms; it is ignored by LAMMPS. The 2nd
column is the "type" of the interaction; it is an index into the CMAP force field file. The remaining 5 columns
are the atom IDs of the atoms in the two 4-atom dihedrals that overlap to create the CMAP 5-body interaction.
LAMMPS Users Manual
783
Note that the "crossterm" and "CMAP" keywords for the header and body sections match those specified in
the read_data command following the data file name; see the read_data doc page for more details.
A data file containing CMAP crossterms can be generated from a PDB file using the charmm2lammps.pl
script in the tools/ch2lmp directory of the LAMMPS distribution. The script must be invoked with the
optional "-cmap" flag to do this; see the tools/ch2lmp/README file for more information.
The potential energy associated with CMAP interactions can be output as described below. It can also be
included in the total potential energy of the system, as output by the thermo_style command, if the fix_modify
energy command is used, as in the example above. See the note below about how to include the CMAP
energy when performing an energy minimization.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the potential "energy" of the CMAP interactions
system's potential energy as part of thermodynamic output.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
potential energy discussed above. The scalar value calculated by this fix is "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
NOTE: If you want the potential energy associated with the CMAP terms forces to be included in the total
potential energy of the system (the quantity being minimized), you MUST enable the fix_modify energy
option for this fix.
Restrictions:
This fix can only be used if LAMMPS was built with the MOLECULE package. See the Making LAMMPS
section for more info on packages.
Related commands:
fix_modify, read_data
Default: none
(Buck) Buck, Bouguet-Bonnet, Pastor, MacKerell Jr., Biophys J, 90, L36 (2006).
(Brooks) Brooks, Brooks, MacKerell Jr., J Comput Chem, 30, 1545 (2009).
LAMMPS Users Manual
784

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix colvars command
Syntax:
fix ID group-ID colvars configfile keyword values ...
ID, group-ID are documented in fix command• colvars = style name of this fix command• configfile = the configuration file for the colvars module• keyword = input or output or seed or tstat
input arg = colvars.state file name or prefix or NULL (default: NULL)
output arg = output filename prefix (default: out)
seed arg = seed for random number generator (default: 1966)
unwrap arg = yes or no
use unwrapped coordinates in collective variables (default: yes)
tstat arg = fix id of a thermostat or NULL (default: NULL)
•
Examples:
fix mtd all colvars peptide.colvars.inp seed 2122 input peptide.colvars.state output peptide
fix abf all colvars colvars.inp tstat 1
Description:
This fix interfaces LAMMPS to the collective variables "Colvars" library, which allows to calculate potentials
of mean force (PMFs) for any set of colvars, using different sampling methods: currently implemented are the
Adaptive Biasing Force (ABF) method, metadynamics, Steered Molecular Dynamics (SMD) and Umbrella
Sampling (US) via a flexible harmonic restraint bias.
This documentation describes only the fix colvars command itself and LAMMPS specific parts of the code.
The full documentation of the colvars library is available as this supplementary PDF document
The Colvars library is developed at https://github.com/colvars/colvars A detailed discussion of its
implementation is in (Fiorin).
There are some example scripts for using this package with LAMMPS in the examples/USER/colvars
directory.
The only mandatory argument to the fix is the filename to the colvars input file that contains the input that is
independent from the MD program in which the colvars library has been integrated.
The group-ID entry is ignored. The collective variable module will always apply to the entire system and
there can only be one instance of the colvars fix at a time. The colvars fix will only communicate the
minimum information necessary and the colvars library supports multiple, completely independent collective
variables, so there is no restriction to functionality by limiting the number of colvars fixes.
The input keyword allows to specify a state file that would contain the restart information required in order to
continue a calculation from a prerecorded state. Fix colvars records it state in binary restart files, so when
using the read_restart command, this is usually not needed.
LAMMPS Users Manual
785

The output keyword allows to specify the output prefix. All output files generated will use this prefix
followed by the ".colvars." and a word like "state" or "traj".
The seed keyword contains the seed for the random number generator that will be used in the colvars module.
The unwrap keyword controls whether wrapped or unwrapped coordinates are passed to the colvars library for
calculation of the collective variables and the resulting forces. The default is yes, i.e. to use the image flags to
reconstruct the absolute atom positions. Setting this to no will use the current local coordinates that are
wrapped back into the simulation cell at each re-neighboring instead.
The tstat keyword can be either NULL or the label of a thermostating fix that thermostats all atoms in the fix
colvars group. This will be used to provide the colvars module with the current thermostat target temperature.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the current status of the colvars module into binary restart files. This is in addition to the text
mode status file that is written by the colvars module itself and the kind of information in both files is
identical.
The fix_modify energy option is supported by this fix to add the energy change from the biasing force added
by the fix to the system's potential energy as part of thermodynamic output.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
cumulative energy change due to this fix. The scalar value calculated by this fix is "extensive".
Restrictions:
This fix is part of the USER-COLVARS package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
There can only be one colvars fix active at a time. Since the interface communicates only the minimum
amount of information and colvars module itself can handle an arbitrary number of collective variables, this is
not a limitation of functionality.
Related commands:
fix smd
Default:
The default options are input = NULL, output = out, seed = 1966, unwrap yes, and tstat = NULL.
(Fiorin) Fiorin , Klein, Henin, Mol. Phys., DOI:10.1080/00268976.2013.813594
LAMMPS Users Manual
786

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix controller command
Syntax:
fix ID group-ID controller Nevery alpha Kp Ki Kd pvar setpoint cvar
ID, group-ID are documented in fix command• controller = style name of this fix command• Nevery = invoke controller every this many timesteps• alpha = coupling constant for PID equation (see units discussion below)• Kp = proportional gain in PID equation (unitless)• Ki = integral gain in PID equation (unitless)• Kd = derivative gain in PID equation (unitless)• pvar = process variable of form c_ID, c_ID[I], f_ID, f_ID[I], or v_name
c_ID = global scalar calculated by a compute with ID
c_ID[I] = Ith component of global vector calculated by a compute with ID
f_ID = global scalar calculated by a fix with ID
f_ID[I] = Ith component of global vector calculated by a fix with ID
v_name = value calculated by an equal-style variable with name
•
setpoint = desired value of process variable (same units as process variable)• cvar = name of control variable•
Examples:
fix 1 all controller 100 1.0 0.5 0.0 0.0 c_thermo_temp 1.5 tcontrol
fix 1 all controller 100 0.2 0.5 0 100.0 v_pxxwall 1.01325 xwall
fix 1 all controller 10000 0.2 0.5 0 2000 v_avpe -3.785 tcontrol
Description:
This fix enables control of a LAMMPS simulation using a control loop feedback mechanism known as a
proportional-integral-derivative (PID) controller. The basic idea is to define a "process variable" which is a
quantity that can be monitored during a running simulation. A desired target value is chosen for the process
variable. A "control variable" is also defined which is an adjustable attribute of the running simulation, which
the process variable will respond to. The PID controller continuously adjusts the control variable based on the
difference between the process variable and the target.
Here are examples of ways in which this fix can be used. The examples/pid directory contains a script that
implements the simple thermostat.
Goal process variable control variable
Simple thermostat instantaneous T thermostat target T
Find melting temperature average PE per atom thermostat target T
Control pressure in non-periodic system force on wall position of wall
NOTE: For this fix to work, the control variable must actually induce a change in a running LAMMPS
simulation. Typically this will only occur if there is some other command (e.g. a thermostat fix) which uses
the control variable as an input parameter. This could be done directly or indirectly, e.g. the other command
uses a variable as input whose formula uses the control variable. The other command should alter its behavior
LAMMPS Users Manual
787

dynamically as the variable changes.
NOTE: If there is a command you think could be used in this fashion, but does not currently allow a variable
as an input parameter, please notify the LAMMPS developers. It is often not difficult to enable a command to
use a variable as an input parameter.
The group specified with this command is ignored. However, note that the process variable may be defined by
calculations performed by computes and fixes which store their own "group" definitions.
The PID controller is invoked once each Nevery timesteps.
The PID controller is implemented as a discretized version of the following dynamic equation:
where c is the continuous time analog of the control variable, e=pvar-setpoint is the error in the process
variable, and alpha, Kp, Ki, and Kd are constants set by the corresponding keywords described above. The
discretized version of this equation is:
where tau = Nevery * timestep is the time interval between updates, and the subscripted variables indicate the
values of c and e at successive updates.
From the first equation, it is clear that if the three gain values Kp, Ki, Kd are dimensionless constants, then
alpha must have units of [unit cvar]/[unit pvar]/[unit time] e.g. [ eV/K/ps ]. The advantage of this unit scheme
is that the value of the constants should be invariant under a change of either the MD timestep size or the
value of Nevery. Similarly, if the LAMMPS unit style is changed, it should only be necessary to change the
value of alpha to reflect this, while leaving Kp, Ki, and Kd unaltered.
When choosing the values of the four constants, it is best to first pick a value and sign for alpha that is
consistent with the magnitudes and signs of pvar and cvar. The magnitude of Kp should then be tested over a
large positive range keeping Ki=Kd=0. A good value for Kp will produce a fast response in pvar, without
overshooting the setpoint. For many applications, proportional feedback is sufficient, and so Ki=Kd=0 can be
used. In cases where there is a substantial lag time in the response of pvar to a change in cvar, this can be
counteracted by increasing Kd. In situations where pvar plateaus without reaching setpoint, this can be
counteracted by increasing Ki. In the language of Charles Dickens, Kp represents the error of the present, Ki
the error of the past, and Kd the error yet to come.
Because this fix updates cvar, but does not initialize its value, the initial value is that assigned by the user in
the input script via the internal-style variable command. This value is used (by the other LAMMPS command
that used the variable) until this fix performs its first update of cvar after Nevery timesteps. On the first
update, the value of the derivative term is set to zero, because the value of e_n-1 is not yet defined.
LAMMPS Users Manual
788

The process variable pvar can be specified as the output of a compute or fix or the evaluation of a variable. In
each case, the compute, fix, or variable must produce a global quantity, not a per-atom or local quantity.
If pvar begins with "c_", a compute ID must follow which has been previously defined in the input script and
which generates a global scalar or vector. See the individual compute doc page for details. If no bracketed
integer is appended, the scalar calculated by the compute is used. If a bracketed integer is appended, the Ith
value of the vector calculated by the compute is used. Users can also write code for their own compute styles
and add them to LAMMPS.
If pvar begins with "f_", a fix ID must follow which has been previously defined in the input script and which
generates a global scalar or vector. See the individual fix doc page for details. Note that some fixes only
produce their values on certain timesteps, which must be compatible with when fix controller references the
values, or else an error results. If no bracketed integer is appended, the scalar calculated by the fix is used. If a
bracketed integer is appended, the Ith value of the vector calculated by the fix is used. Users can also write
code for their own fix style and add them to LAMMPS.
If pvar begins with "v_", a variable name must follow which has been previously defined in the input script.
Only equal-style variables can be referenced. See the variable command for details. Note that variables of
style equal define a formula which can reference individual atom properties or thermodynamic keywords, or
they can invoke other computes, fixes, or variables when they are evaluated, so this is a very general means of
specifying the process variable.
The target value setpoint for the process variable must be a numeric value, in whatever units pvar is defined
for.
The control variable cvar must be the name of an internal-style variable previously defined in the input script.
Note that it is not specified with a "v_" prefix, just the name of the variable. It must be an internal-style
variable, because this fix updates its value directly. Note that other commands can use an equal-style versus
internal-style variable interchangeably.
Restart, fix_modify, output, run start/stop, minimize info:
Currently, no information about this fix is written to binary restart files. None of the fix_modify options are
relevant to this fix.
This fix produces a global vector with 3 values which can be accessed by various output commands. The
values can be accessed on any timestep, though they are only updated on timesteps that are a multiple of
Nevery.
The three values are the most recent updates made to the control variable by each of the 3 terms in the PID
equation above. The first value is the proportional term, the second is the integral term, the third is the
derivative term.
The units of the vector values will be whatever units the control variable is in. The vector values calculated by
this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
LAMMPS Users Manual
789

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix deform command
fix deform/kk command
Syntax:
fix ID group-ID deform N parameter args ... keyword value ...
ID, group-ID are documented in fix command• deform = style name of this fix command• N = perform box deformation every this many timesteps• one or more parameter/arg pairs may be appended
parameter = x or y or z or xy or xz or yz
x, y, z args = style value(s)
style = final or delta or scale or vel or erate or trate or volume or wiggle or variable
final values = lo hi
lo hi = box boundaries at end of run (distance units)
delta values = dlo dhi
dlo dhi = change in box boundaries at end of run (distance units)
scale values = factor
factor = multiplicative factor for change in box length at end of run
vel value = V
V = change box length at this velocity (distance/time units),
effectively an engineering strain rate
erate value = R
R = engineering strain rate (1/time units)
trate value = R
R = true strain rate (1/time units)
volume value = none = adjust this dim to preserve volume of system
wiggle values = A Tp
A = amplitude of oscillation (distance units)
Tp = period of oscillation (time units)
variable values = v_name1 v_name2
v_name1 = variable with name1 for box length change as function of time
v_name2 = variable with name2 for change rate as function of time
xy, xz, yz args = style value
style = final or delta or vel or erate or trate or wiggle
final value = tilt
tilt = tilt factor at end of run (distance units)
delta value = dtilt
dtilt = change in tilt factor at end of run (distance units)
vel value = V
V = change tilt factor at this velocity (distance/time units),
effectively an engineering shear strain rate
erate value = R
R = engineering shear strain rate (1/time units)
trate value = R
R = true shear strain rate (1/time units)
wiggle values = A Tp
A = amplitude of oscillation (distance units)
Tp = period of oscillation (time units)
variable values = v_name1 v_name2
v_name1 = variable with name1 for tilt change as function of time
v_name2 = variable with name2 for change rate as function of time
•
LAMMPS Users Manual
791

zero or more keyword/value pairs may be appended• keyword = remap or flip or units
remap value = x or v or none
x = remap coords of atoms in group into deforming box
v = remap velocities of all atoms when they cross periodic boundaries
none = no remapping of x or v
flip value = yes or no
allow or disallow box flips when it becomes highly skewed
units value = lattice or box
lattice = distances are defined in lattice units
box = distances are defined in simulation box units
•
Examples:
fix 1 all deform 1 x final 0.0 9.0 z final 0.0 5.0 units box
fix 1 all deform 1 x trate 0.1 y volume z volume
fix 1 all deform 1 xy erate 0.001 remap v
fix 1 all deform 10 y delta -0.5 0.5 xz vel 1.0
Description:
Change the volume and/or shape of the simulation box during a dynamics run. Orthogonal simulation boxes
have 3 adjustable parameters (x,y,z). Triclinic (non-orthogonal) simulation boxes have 6 adjustable
parameters (x,y,z,xy,xz,yz). Any or all of them can be adjusted independently and simultaneously by this
command. This fix can be used to perform non-equilibrium MD (NEMD) simulations of a continuously
strained system. See the fix nvt/sllod and compute temp/deform commands for more details.
For the x, y, z parameters, the associated dimension cannot be shrink-wrapped. For the xy, yz, xz parameters,
the associated 2nd dimension cannot be shrink-wrapped. Dimensions not varied by this command can be
periodic or non-periodic. Dimensions corresponding to unspecified parameters can also be controlled by a fix
npt or fix nph command.
The size and shape of the simulation box at the beginning of the simulation run were either specified by the
create_box or read_data or read_restart command used to setup the simulation initially if it is the first run, or
they are the values from the end of the previous run. The create_box, read data, and read_restart commands
specify whether the simulation box is orthogonal or non-orthogonal (triclinic) and explain the meaning of the
xy,xz,yz tilt factors. If fix deform changes the xy,xz,yz tilt factors, then the simulation box must be triclinic,
even if its initial tilt factors are 0.0.
As described below, the desired simulation box size and shape at the end of the run are determined by the
parameters of the fix deform command. Every Nth timestep during the run, the simulation box is expanded,
contracted, or tilted to ramped values between the initial and final values.
For the x, y, and z parameters, this is the meaning of their styles and values.
The final, delta, scale, vel, and erate styles all change the specified dimension of the box via "constant
displacement" which is effectively a "constant engineering strain rate". This means the box dimension
changes linearly with time from its initial to final value.
For style final, the final lo and hi box boundaries of a dimension are specified. The values can be in lattice or
box distance units. See the discussion of the units keyword below.
LAMMPS Users Manual
792
For style delta, plus or minus changes in the lo/hi box boundaries of a dimension are specified. The values can
be in lattice or box distance units. See the discussion of the units keyword below.
For style scale, a multiplicative factor to apply to the box length of a dimension is specified. For example, if
the initial box length is 10, and the factor is 1.1, then the final box length will be 11. A factor less than 1.0
means compression.
For style vel, a velocity at which the box length changes is specified in units of distance/time. This is
effectively a "constant engineering strain rate", where rate = V/L0 and L0 is the initial box length. The
distance can be in lattice or box distance units. See the discussion of the units keyword below. For example, if
the initial box length is 100 Angstroms, and V is 10 Angstroms/psec, then after 10 psec, the box length will
have doubled. After 20 psec, it will have tripled.
The erate style changes a dimension of the box at a "constant engineering strain rate". The units of the
specified strain rate are 1/time. See the units command for the time units associated with different choices of
simulation units, e.g. picoseconds for "metal" units). Tensile strain is unitless and is defined as delta/L0,
where L0 is the original box length and delta is the change relative to the original length. The box length L as
a function of time will change as
L(t) = L0 (1 + erate*dt)
where dt is the elapsed time (in time units). Thus if erate R is specified as 0.1 and time units are picoseconds,
this means the box length will increase by 10% of its original length every picosecond. I.e. strain after 1 psec
= 0.1, strain after 2 psec = 0.2, etc. R = -0.01 means the box length will shrink by 1% of its original length
every picosecond. Note that for an "engineering" rate the change is based on the original box length, so
running with R = 1 for 10 picoseconds expands the box length by a factor of 11 (strain of 10), which is
different that what the trate style would induce.
The trate style changes a dimension of the box at a "constant true strain rate". Note that this is not an
"engineering strain rate", as the other styles are. Rather, for a "true" rate, the rate of change is constant, which
means the box dimension changes non-linearly with time from its initial to final value. The units of the
specified strain rate are 1/time. See the units command for the time units associated with different choices of
simulation units, e.g. picoseconds for "metal" units). Tensile strain is unitless and is defined as delta/L0,
where L0 is the original box length and delta is the change relative to the original length.
The box length L as a function of time will change as
L(t) = L0 exp(trate*dt)
where dt is the elapsed time (in time units). Thus if trate R is specified as ln(1.1) and time units are
picoseconds, this means the box length will increase by 10% of its current (not original) length every
picosecond. I.e. strain after 1 psec = 0.1, strain after 2 psec = 0.21, etc. R = ln(2) or ln(3) means the box length
will double or triple every picosecond. R = ln(0.99) means the box length will shrink by 1% of its current
length every picosecond. Note that for a "true" rate the change is continuous and based on the current length,
so running with R = ln(2) for 10 picoseconds does not expand the box length by a factor of 11 as it would with
erate, but by a factor of 1024 since the box length will double every picosecond.
Note that to change the volume (or cross-sectional area) of the simulation box at a constant rate, you can
change multiple dimensions via erate or trate. E.g. to double the box volume in a picosecond picosecond, you
could set "x erate M", "y erate M", "z erate M", with M = pow(2,1/3) - 1 = 0.26, since if each box dimension
grows by 26%, the box volume doubles. Or you could set "x trate M", "y trate M", "z trate M", with M =
LAMMPS Users Manual
793

ln(1.26) = 0.231, and the box volume would double every picosecond.
The volume style changes the specified dimension in such a way that the box volume remains constant while
other box dimensions are changed explicitly via the styles discussed above. For example, "x scale 1.1 y scale
1.1 z volume" will shrink the z box length as the x,y box lengths increase, to keep the volume constant
(product of x,y,z lengths). If "x scale 1.1 z volume" is specified and parameter y is unspecified, then the z box
length will shrink as x increases to keep the product of x,z lengths constant. If "x scale 1.1 y volume z
volume" is specified, then both the y,z box lengths will shrink as x increases to keep the volume constant
(product of x,y,z lengths). In this case, the y,z box lengths shrink so as to keep their relative aspect ratio
constant.
For solids or liquids, note that when one dimension of the box is expanded via fix deform (i.e. tensile strain),
it may be physically undesirable to hold the other 2 box lengths constant (unspecified by fix deform) since
that implies a density change. Using the volume style for those 2 dimensions to keep the box volume constant
may make more physical sense, but may also not be correct for materials and potentials whose Poisson ratio is
not 0.5. An alternative is to use fix npt aniso with zero applied pressure on those 2 dimensions, so that they
respond to the tensile strain dynamically.
The wiggle style oscillates the specified box length dimension sinusoidally with the specified amplitude and
period. I.e. the box length L as a function of time is given by
L(t) = L0 + A sin(2*pi t/Tp)
where L0 is its initial length. If the amplitude A is a positive number the box initially expands, then contracts,
etc. If A is negative then the box initially contracts, then expands, etc. The amplitude can be in lattice or box
distance units. See the discussion of the units keyword below.
The variable style changes the specified box length dimension by evaluating a variable, which presumably is
a function of time. The variable with name1 must be an equal-style variable and should calculate a change in
box length in units of distance. Note that this distance is in box units, not lattice units; see the discussion of
the units keyword below. The formula associated with variable name1 can reference the current timestep.
Note that it should return the "change" in box length, not the absolute box length. This means it should
evaluate to 0.0 when invoked on the initial timestep of the run following the definition of fix deform. It should
evaluate to a value > 0.0 to dilate the box at future times, or a value < 0.0 to compress the box.
The variable name2 must also be an equal-style variable and should calculate the rate of box length change, in
units of distance/time, i.e. the time-derivative of the name1 variable. This quantity is used internally by
LAMMPS to reset atom velocities when they cross periodic boundaries. It is computed internally for the other
styles, but you must provide it when using an arbitrary variable.
Here is an example of using the variable style to perform the same box deformation as the wiggle style
formula listed above, where we assume that the current timestep = 0.
variable A equal 5.0
variable Tp equal 10.0
variable displace equal "v_A * sin(2*PI * step*dt/v_Tp)"
variable rate equal "2*PI*v_A/v_Tp * cos(2*PI * step*dt/v_Tp)"
fix 2 all deform 1 x variable v_displace v_rate remap v
For the scale, vel, erate, trate, volume, wiggle, and variable styles, the box length is expanded or compressed
around its mid point.
LAMMPS Users Manual
794
For the xy, xz, and yz parameters, this is the meaning of their styles and values. Note that changing the tilt
factors of a triclinic box does not change its volume.
The final, delta, vel, and erate styles all change the shear strain at a "constant engineering shear strain rate".
This means the tilt factor changes linearly with time from its initial to final value.
For style final, the final tilt factor is specified. The value can be in lattice or box distance units. See the
discussion of the units keyword below.
For style delta, a plus or minus change in the tilt factor is specified. The value can be in lattice or box distance
units. See the discussion of the units keyword below.
For style vel, a velocity at which the tilt factor changes is specified in units of distance/time. This is
effectively an "engineering shear strain rate", where rate = V/L0 and L0 is the initial box length perpendicular
to the direction of shear. The distance can be in lattice or box distance units. See the discussion of the units
keyword below. For example, if the initial tilt factor is 5 Angstroms, and the V is 10 Angstroms/psec, then
after 1 psec, the tilt factor will be 15 Angstroms. After 2 psec, it will be 25 Angstroms.
The erate style changes a tilt factor at a "constant engineering shear strain rate". The units of the specified
shear strain rate are 1/time. See the units command for the time units associated with different choices of
simulation units, e.g. picoseconds for "metal" units). Shear strain is unitless and is defined as offset/length,
where length is the box length perpendicular to the shear direction (e.g. y box length for xy deformation) and
offset is the displacement distance in the shear direction (e.g. x direction for xy deformation) from the
unstrained orientation.
The tilt factor T as a function of time will change as
T(t) = T0 + L0*erate*dt
where T0 is the initial tilt factor, L0 is the original length of the box perpendicular to the shear direction (e.g.
y box length for xy deformation), and dt is the elapsed time (in time units). Thus if erate R is specified as 0.1
and time units are picoseconds, this means the shear strain will increase by 0.1 every picosecond. I.e. if the xy
shear strain was initially 0.0, then strain after 1 psec = 0.1, strain after 2 psec = 0.2, etc. Thus the tilt factor
would be 0.0 at time 0, 0.1*ybox at 1 psec, 0.2*ybox at 2 psec, etc, where ybox is the original y box length. R
= 1 or 2 means the tilt factor will increase by 1 or 2 every picosecond. R = -0.01 means a decrease in shear
strain by 0.01 every picosecond.
The trate style changes a tilt factor at a "constant true shear strain rate". Note that this is not an "engineering
shear strain rate", as the other styles are. Rather, for a "true" rate, the rate of change is constant, which means
the tilt factor changes non-linearly with time from its initial to final value. The units of the specified shear
strain rate are 1/time. See the units command for the time units associated with different choices of simulation
units, e.g. picoseconds for "metal" units). Shear strain is unitless and is defined as offset/length, where length
is the box length perpendicular to the shear direction (e.g. y box length for xy deformation) and offset is the
displacement distance in the shear direction (e.g. x direction for xy deformation) from the unstrained
orientation.
The tilt factor T as a function of time will change as
T(t) = T0 exp(trate*dt)
LAMMPS Users Manual
795

where T0 is the initial tilt factor and dt is the elapsed time (in time units). Thus if trate R is specified as ln(1.1)
and time units are picoseconds, this means the shear strain or tilt factor will increase by 10% every
picosecond. I.e. if the xy shear strain was initially 0.1, then strain after 1 psec = 0.11, strain after 2 psec =
0.121, etc. R = ln(2) or ln(3) means the tilt factor will double or triple every picosecond. R = ln(0.99) means
the tilt factor will shrink by 1% every picosecond. Note that the change is continuous, so running with R =
ln(2) for 10 picoseconds does not change the tilt factor by a factor of 10, but by a factor of 1024 since it
doubles every picosecond. Note that the initial tilt factor must be non-zero to use the trate option.
Note that shear strain is defined as the tilt factor divided by the perpendicular box length. The erate and trate
styles control the tilt factor, but assume the perpendicular box length remains constant. If this is not the case
(e.g. it changes due to another fix deform parameter), then this effect on the shear strain is ignored.
The wiggle style oscillates the specified tilt factor sinusoidally with the specified amplitude and period. I.e.
the tilt factor T as a function of time is given by
T(t) = T0 + A sin(2*pi t/Tp)
where T0 is its initial value. If the amplitude A is a positive number the tilt factor initially becomes more
positive, then more negative, etc. If A is negative then the tilt factor initially becomes more negative, then
more positive, etc. The amplitude can be in lattice or box distance units. See the discussion of the units
keyword below.
The variable style changes the specified tilt factor by evaluating a variable, which presumably is a function of
time. The variable with name1 must be an equal-style variable and should calculate a change in tilt in units of
distance. Note that this distance is in box units, not lattice units; see the discussion of the units keyword
below. The formula associated with variable name1 can reference the current timestep. Note that it should
return the "change" in tilt factor, not the absolute tilt factor. This means it should evaluate to 0.0 when
invoked on the initial timestep of the run following the definition of fix deform.
The variable name2 must also be an equal-style variable and should calculate the rate of tilt change, in units of
distance/time, i.e. the time-derivative of the name1 variable. This quantity is used internally by LAMMPS to
reset atom velocities when they cross periodic boundaries. It is computed internally for the other styles, but
you must provide it when using an arbitrary variable.
Here is an example of using the variable style to perform the same box deformation as the wiggle style
formula listed above, where we assume that the current timestep = 0.
variable A equal 5.0
variable Tp equal 10.0
variable displace equal "v_A * sin(2*PI * step*dt/v_Tp)"
variable rate equal "2*PI*v_A/v_Tp * cos(2*PI * step*dt/v_Tp)"
fix 2 all deform 1 xy variable v_displace v_rate remap v
All of the tilt styles change the xy, xz, yz tilt factors during a simulation. In LAMMPS, tilt factors (xy,xz,yz)
for triclinic boxes are normally bounded by half the distance of the parallel box length. See the discussion of
the flip keyword below, to allow this bound to be exceeded, if desired.
For example, if xlo = 2 and xhi = 12, then the x box length is 10 and the xy tilt factor must be between -5 and
5. Similarly, both xz and yz must be between -(xhi-xlo)/2 and +(yhi-ylo)/2. Note that this is not a limitation,
since if the maximum tilt factor is 5 (as in this example), then configurations with tilt = ..., -15, -5, 5, 15, 25,
... are all equivalent.
LAMMPS Users Manual
796

To obey this constraint and allow for large shear deformations to be applied via the xy, xz, or yz parameters,
the following algorithm is used. If prd is the associated parallel box length (10 in the example above), then if
the tilt factor exceeds the accepted range of -5 to 5 during the simulation, then the box is flipped to the other
limit (an equivalent box) and the simulation continues. Thus for this example, if the initial xy tilt factor was
0.0 and "xy final 100.0" was specified, then during the simulation the xy tilt factor would increase from 0.0 to
5.0, the box would be flipped so that the tilt factor becomes -5.0, the tilt factor would increase from -5.0 to
5.0, the box would be flipped again, etc. The flip occurs 10 times and the final tilt factor at the end of the
simulation would be 0.0. During each flip event, atoms are remapped into the new box in the appropriate
manner.
The one exception to this rule is if the 1st dimension in the tilt factor (x for xy) is non-periodic. In that case,
the limits on the tilt factor are not enforced, since flipping the box in that dimension does not change the atom
positions due to non-periodicity. In this mode, if you tilt the system to extreme angles, the simulation will
simply become inefficient due to the highly skewed simulation box.
Each time the box size or shape is changed, the remap keyword determines whether atom positions are
remapped to the new box. If remap is set to x (the default), atoms in the fix group are remapped; otherwise
they are not. Note that their velocities are not changed, just their positions are altered. If remap is set to v, then
any atom in the fix group that crosses a periodic boundary will have a delta added to its velocity equal to the
difference in velocities between the lo and hi boundaries. Note that this velocity difference can include tilt
components, e.g. a delta in the x velocity when an atom crosses the y periodic boundary. If remap is set to
none, then neither of these remappings take place.
Conceptually, setting remap to x forces the atoms to deform via an affine transformation that exactly matches
the box deformation. This setting is typically appropriate for solids. Note that though the atoms are effectively
"moving" with the box over time, it is not due to their having a velocity that tracks the box change, but only
due to the remapping. By contrast, setting remap to v is typically appropriate for fluids, where you want the
atoms to respond to the change in box size/shape on their own and acquire a velocity that matches the box
change, so that their motion will naturally track the box without explicit remapping of their coordinates.
NOTE: When non-equilibrium MD (NEMD) simulations are performed using this fix, the option "remap v"
should normally be used. This is because fix nvt/sllod adjusts the atom positions and velocities to induce a
velocity profile that matches the changing box size/shape. Thus atom coordinates should NOT be remapped
by fix deform, but velocities SHOULD be when atoms cross periodic boundaries, since that is consistent with
maintaining the velocity profile already created by fix nvt/sllod. LAMMPS will warn you if the remap setting
is not consistent with fix nvt/sllod.
NOTE: For non-equilibrium MD (NEMD) simulations using "remap v" it is usually desirable that the fluid (or
flowing material, e.g. granular particles) stream with a velocity profile consistent with the deforming box. As
mentioned above, using a thermostat such as fix nvt/sllod or fix lavgevin (with a bias provided by compute
temp/deform), will typically accomplish that. If you do not use a thermostat, then there is no driving force
pushing the atoms to flow in a manner consistent with the deforming box. E.g. for a shearing system the box
deformation velocity may vary from 0 at the bottom to 10 at the top of the box. But the stream velocity profile
of the atoms may vary from -5 at the bottom to +5 at the top. You can monitor these effects using the fix
ave/chunk, compute temp/deform, and compute temp/profile commands. One way to induce atoms to stream
consistent with the box deformation is to give them an initial velocity profile, via the velocity ramp command,
that matches the box deformation rate. This also typically helps the system come to equilibrium more quickly,
even if a thermostat is used.
NOTE: If a fix rigid is defined for rigid bodies, and remap is set to x, then the center-of-mass coordinates of
rigid bodies will be remapped to the changing simulation box. This will be done regardless of whether atoms
LAMMPS Users Manual
797

in the rigid bodies are in the fix deform group or not. The velocity of the centers of mass are not remapped
even if remap is set to v, since fix nvt/sllod does not currently do anything special for rigid particles. If you
wish to perform a NEMD simulation of rigid particles, you can either thermostat them independently or
include a background fluid and thermostat the fluid via fix nvt/sllod.
The flip keyword allows the tilt factors for a triclinic box to exceed half the distance of the parallel box length,
as discussed above. If the flip value is set to yes, the bound is enforced by flipping the box when it is
exceeded. If the flip value is set to no, the tilt will continue to change without flipping. Note that if you apply
large deformations, this means the box shape can tilt dramatically LAMMPS will run less efficiently, due to
the large volume of communication needed to acquire ghost atoms around a processor's irregular-shaped
sub-domain. For extreme values of tilt, LAMMPS may also lose atoms and generate an error.
The units keyword determines the meaning of the distance units used to define various arguments. A box
value selects standard distance units as defined by the units command, e.g. Angstroms for units = real or
metal. A lattice value means the distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacing. Note that the units choice also affects the vel style parameters
since it is defined in terms of distance/time. Also note that the units keyword does not affect the variable
style. You should use the xlat, ylat, zlat keywords of the thermo_style command if you want to include lattice
spacings in a variable formula.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands.
This fix can perform deformation over multiple runs, using the start and stop keywords of the run command.
See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
You cannot apply x, y, or z deformations to a dimension that is shrink-wrapped via the boundary command.
You cannot apply xy, yz, or xz deformations to a 2nd dimension (y in xy) that is shrink-wrapped via the
boundary command.
LAMMPS Users Manual
798

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix deposit command
Syntax:
fix ID group-ID deposit N type M seed keyword values ...
ID, group-ID are documented in fix command• deposit = style name of this fix command• N = # of atoms or molecules to insert• type = atom type to assign to inserted atoms (offset for molecule insertion)• M = insert a single atom or molecule every M steps• seed = random # seed (positive integer)• one or more keyword/value pairs may be appended to args• keyword = region or id or global or local or near or gaussian or attempt or rate or vx or vy or vz or
mol or rigid or shake or units
region value = region-ID
region-ID = ID of region to use as insertion volume
id value = max or next
max = atom ID for new atom(s) is max ID of all current atoms plus one
next = atom ID for new atom(s) increments by one for every deposition
global values = lo hi
lo,hi = put new atom/molecule a distance lo-hi above all other atoms (distance units)
local values = lo hi delta
lo,hi = put new atom/molecule a distance lo-hi above any nearby atom beneath it (distance units)
delta = lateral distance within which a neighbor is considered "nearby" (distance units)
near value = R
R = only insert atom/molecule if further than R from existing particles (distance units)
gaussian values = xmid ymid zmid sigma
xmid,ymid,zmid = center of the gaussian distribution (distance units)
sigma = width of gaussian distribution (distance units)
attempt value = Q
Q = attempt a single insertion up to Q times
rate value = V
V = z velocity (y in 2d) at which insertion volume moves (velocity units)
vx values = vxlo vxhi
vxlo,vxhi = range of x velocities for inserted atom/molecule (velocity units)
vy values = vylo vyhi
vylo,vyhi = range of y velocities for inserted atom/molecule (velocity units)
vz values = vzlo vzhi
vzlo,vzhi = range of z velocities for inserted atom/molecule (velocity units)
target values = tx ty tz
tx,ty,tz = location of target point (distance units)
mol value = template-ID
template-ID = ID of molecule template specified in a separate molecule command
molfrac values = f1 f2 ... fN
f1 to fN = relative probability of creating each of N molecules in template-ID
rigid value = fix-ID
fix-ID = ID of fix rigid/small command
shake value = fix-ID
fix-ID = ID of fix shake command
units value = lattice or box
lattice = the geometry is defined in lattice units
box = the geometry is defined in simulation box units
•
LAMMPS Users Manual
800
Examples:
fix 3 all deposit 1000 2 100 29494 region myblock local 1.0 1.0 1.0 units box
fix 2 newatoms deposit 10000 1 500 12345 region disk near 2.0 vz -1.0 -0.8
fix 4 sputter deposit 1000 2 500 12235 region sphere vz -1.0 -1.0 target 5.0 5.0 0.0 units lattice
fix 5 insert deposit 200 2 100 777 region disk gaussian 5.0 5.0 9.0 1.0 units box
Description:
Insert a single atom or molecule into the simulation domain every M timesteps until N atoms or molecules
have been inserted. This is useful for simulating deposition onto a surface. For the remainder of this doc page,
a single inserted atom or molecule is referred to as a "particle".
If inserted particles are individual atoms, they are assigned the specified atom type. If they are molecules, the
type of each atom in the inserted molecule is specified in the file read by the molecule command, and those
values are added to the specified atom type. E.g. if the file specifies atom types 1,2,3, and those are the atom
types you want for inserted molecules, then specify type = 0. If you specify type = 2, the in the inserted
molecule will have atom types 3,4,5.
All atoms in the inserted particle are assigned to two groups: the default group "all" and the group specified in
the fix deposit command (which can also be "all").
If you are computing temperature values which include inserted particles, you will want to use the
compute_modify dynamic option, which insures the current number of atoms is used as a normalizing factor
each time the temperature is computed.
Care must be taken that inserted particles are not too near existing atoms, using the options described below.
When inserting particles above a surface in a non-periodic box (see the boundary command), the possibility of
a particle escaping the surface and flying upward should be considered, since the particle may be lost or the
box size may grow infinitely large. A fix wall/reflect command can be used to prevent this behavior. Note that
if a shrink-wrap boundary is used, it is OK to insert the new particle outside the box, however the box will
immediately be expanded to include the new particle. When simulating a sputtering experiment it is probably
more realistic to ignore those atoms using the thermo_modify command with the lost ignore option and a
fixed boundary.
The fix deposit command must use the region keyword to define an insertion volume. The specified region
must have been previously defined with a region command. It must be defined with side = in.
NOTE: LAMMPS checks that the specified region is wholly inside the simulation box. It can do this correctly
for orthonormal simulation boxes. However for triclinic boxes, it only tests against the larger orthonormal box
that bounds the tilted simulation box. If the specified region includes volume outside the tilted box, then an
insertion will likely fail, leading to a "lost atoms" error. Thus for triclinic boxes you should insure the
specified region is wholly inside the simulation box.
The locations of inserted particles are taken from uniform distributed random numbers, unless the gaussian
keyword is used. Then the individual coordinates are taken from a gaussian distribution of width sigma
centered on xmid,ymid,zmid.
Individual atoms are inserted, unless the mol keyword is used. It specifies a template-ID previously defined
using the molecule command, which reads files that define one or more molecules. The coordinates, atom
types, charges, etc, as well as any bond/angle/etc and special neighbor information for the molecule can be
LAMMPS Users Manual
801
specified in the molecule file. See the molecule command for details. The only settings required to be in each
file are the coordinates and types of atoms in the molecule.
If the molecule template contains more than one molecule, the relative probability of depositing each
molecule can be specified by the molfrac keyword. N relative probabilities, each from 0.0 to 1.0, are
specified, where N is the number of molecules in the template. Each time a molecule is deposited, a random
number is used to sample from the list of relative probabilities. The N values must sum to 1.0.
If you wish to insert molecules via the mol keyword, that will be treated as rigid bodies, use the rigid
keyword, specifying as its value the ID of a separate fix rigid/small command which also appears in your
input script.
NOTE: If you wish the new rigid molecules (and other rigid molecules) to be thermostatted correctly via fix
rigid/small/nvt or fix rigid/small/npt, then you need to use the "fix_modify dynamic/dof yes" command for the
rigid fix. This is to inform that fix that the molecule count will vary dynamically.
If you wish to insert molecules via the mol keyword, that will have their bonds or angles constrained via
SHAKE, use the shake keyword, specifying as its value the ID of a separate fix shake command which also
appears in your input script.
Each timestep a particle is inserted, the coordinates for its atoms are chosen as follows. For insertion of
individual atoms, the "position" referred to in the following description is the coordinate of the atom. For
insertion of molecule, the "position" is the geometric center of the molecule; see the molecule doc page for
details. A random rotation of the molecule around its center point is performed, which determines the
coordinates all the individual atoms.
A random position within the region insertion volume is generated. If neither the global or local keyword is
used, the random position is the trial position. If the global keyword is used, the random x,y values are used,
but the z position of the new particle is set above the highest current atom in the simulation by a distance
randomly chosen between lo/hi. (For a 2d simulation, this is done for the y position.) If the local keyword is
used, the z position is set a distance between lo/hi above the highest current atom in the simulation that is
"nearby" the chosen x,y position. In this context, "nearby" means the lateral distance (in x,y) between the new
and old particles is less than the delta setting.
Once a trial x,y,z position has been selected, the insertion is only performed if no current atom in the
simulation is within a distance R of any atom in the new particle, including the effect of periodic boundary
conditions if applicable. R is defined by the near keyword. Note that the default value for R is 0.0, which will
allow atoms to strongly overlap if you are inserting where other atoms are present. This distance test is
performed independently for each atom in an inserted molecule, based on the randomly rotated configuration
of the molecule. If this test fails, a new random position within the insertion volume is chosen and another
trial is made. Up to Q attempts are made. If the particle is not successfully inserted, LAMMPS prints a
warning message.
NOTE: If you are inserting finite size particles or a molecule or rigid body consisting of finite-size particles,
then you should typically set R larger than the distance at which any inserted particle may overlap with either
a previously inserted particle or an existing particle. LAMMPS will issue a warning if R is smaller than this
value, based on the radii of existing and inserted particles.
The rate option moves the insertion volume in the z direction (3d) or y direction (2d). This enables particles to
be inserted from a successively higher height over time. Note that this parameter is ignored if the global or
local keywords are used, since those options choose a z-coordinate for insertion independently.
LAMMPS Users Manual
802
The vx, vy, and vz components of velocity for the inserted particle are set using the values specified for the
vx, vy, and vz keywords. Note that normally, new particles should be a assigned a negative vertical velocity so
that they move towards the surface. For molecules, the same velocity is given to every particle (no rotation or
bond vibration).
If the target option is used, the velocity vector of the inserted particle is changed so that it points from the
insertion position towards the specified target point. The magnitude of the velocity is unchanged. This can be
useful, for example, for simulating a sputtering process. E.g. the target point can be far away, so that all
incident particles strike the surface as if they are in an incident beam of particles at a prescribed angle.
The id keyword determines how atom IDs and molecule IDs are assigned to newly deposited particles.
Molecule IDs are only assigned if molecules are being inserted. For the max setting, the atom and molecule
IDs of all current atoms are checked. Atoms in the new particle are assigned IDs starting with the current
maximum plus one. If a molecule is inserted it is assigned an ID = current maximum plus one. This means
that if particles leave the system, the new IDs may replace the lost ones. For the next setting, the maximum ID
of any atom and molecule is stored at the time the fix is defined. Each time a new particle is added, this value
is incremented to assign IDs to the new atom(s) or molecule. Thus atom and molecule IDs for deposited
particles will be consecutive even if particles leave the system over time.
The units keyword determines the meaning of the distance units used for the other deposition parameters. A
box value selects standard distance units as defined by the units command, e.g. Angstroms for units = real or
metal. A lattice value means the distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacing. Note that the units choice affects all the keyword values that
have units of distance or velocity.
NOTE: If you are monitoring the temperature of a system where the atom count is changing due to adding
particles, you typically should use the compute_modify dynamic yes command for the temperature compute
you are using.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the deposition to binary restart files. This includes information about how many
particles have been depositied, the random number generator seed, the next timestep for deposition, etc. See
the read_restart command for info on how to re-specify a fix in an input script that reads a restart file, so that
the operation of the fix continues in an uninterrupted fashion.
None of the fix_modify options are relevant to this fix. No global or per-atom quantities are stored by this fix
for access by various output commands. No parameter of this fix can be used with the start/stop keywords of
the run command. This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the MISC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
The specified insertion region cannot be a "dynamic" region, as defined by the region command.
Related commands:
fix pour, region
LAMMPS Users Manual
803
Default:
Insertions are performed for individual atoms, i.e. no mol setting is defined. If the mol keyword is used, the
default for molfrac is an equal probabilities for all molecules in the template. Additional option defaults are id
= max, delta = 0.0, near = 0.0, attempt = 10, rate = 0.0, vx = 0.0 0.0, vy = 0.0 0.0, vz = 0.0 0.0, and units =
lattice.
LAMMPS Users Manual
804
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix dpd/energy command
Syntax:
fix ID group-ID dpd/energy
ID, group-ID are documented in fix command• dpd/energy = style name of this fix command•
Examples:
fix 1 all dpd/energy
Description:
Perform constant energy dissipative particle dynamics (DPD-E) integration. This fix updates the internal
energies for particles in the group at each timestep. It must be used in conjunction with a deterministic
integrator (e.g. fix nve) that updates the particle positions and velocities.
For fix dpd/energy, the particle internal temperature is related to the particle internal energy through a
mesoparticle equation of state. An additional fix must be specified that defines the equation of state for each
particle, e.g. fix eos/cv.
This fix must be used with the pair_style dpd/fdt/energy command.
Note that numerous variants of DPD can be specified by choosing an appropriate combination of the
integrator and pair_style dpd/fdt/energy command. DPD under isoenergetic conditions can be specified by
using fix dpd/energy, fix nve and pair_style dpd/fdt/energy. DPD under isoenthalpic conditions can be
specified by using fix dpd/energy, fix nph and pair_style dpd/fdt/energy. Examples of each DPD variant are
provided in the examples/USER/dpd directory.
Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This fix must be used with an additional fix that specifies time integration, e.g. fix nve.
The fix dpd/energy requires the dpd atom_style to be used in order to properly account for the particle internal
energies and temperature.
The fix dpd/energy must be used with an additional fix that specifies the mesoparticle equation of state for
each particle.
Related commands:
fix nve fix eos/cv
Default: none
LAMMPS Users Manual
805

(Lisal) M. Lisal, J.K. Brennan, J. Bonet Avalos, "Dissipative particle dynamics at isothermal, isobaric,
isoenergetic, and isoenthalpic conditions using Shardlow-like splitting algorithms.", J. Chem. Phys., 135,
204105 (2011).
(Larentzos) J.P. Larentzos, J.K. Brennan, J.D. Moore, and W.D. Mattson, "LAMMPS Implementation of
Constant Energy Dissipative Particle Dynamics (DPD-E)", ARL-TR-6863, U.S. Army Research Laboratory,
Aberdeen Proving Ground, MD (2014).
LAMMPS Users Manual
806

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix drag command
Syntax:
fix ID group-ID drag x y z fmag delta
ID, group-ID are documented in fix command• drag = style name of this fix command• x,y,z = coord to drag atoms towards• fmag = magnitude of force to apply to each atom (force units)• delta = cutoff distance inside of which force is not applied (distance units)•
Examples:
fix center small-molecule drag 0.0 10.0 0.0 5.0 2.0
Description:
Apply a force to each atom in a group to drag it towards the point (x,y,z). The magnitude of the force is
specified by fmag. If an atom is closer than a distance delta to the point, then the force is not applied.
Any of the x,y,z values can be specified as NULL which means do not include that dimension in the distance
calculation or force application.
This command can be used to steer one or more atoms to a new location in the simulation.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
This fix computes a global 3-vector of forces, which can be accessed by various output commands. This is the
total force on the group of atoms by the drag force. The vector values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
fix spring, fix spring/self, fix spring/rg, fix smd
Default: none
LAMMPS Users Manual
807

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix drude command
Syntax:
fix ID group-ID drude flag1 flag2 ... flagN
ID, group-ID are documented in fix command• drude = style name of this fix command• flag1 flag2 ... flagN = Drude flag for each atom type (1 to N) in the system•
Examples:
fix 1 all drude 1 1 0 1 0 2 2 2
fix 1 all drude C C N C N D D D
Description:
Assign each atom type in the system to be one of 3 kinds of atoms within the Drude polarization model. This
fix is designed to be used with the thermalized Drude oscillator model. Polarizable models in LAMMPS are
described in this Section.
The three possible types can be designated with an integer (0,1,2) or capital letter (N,C,D):
0 or N = non-polarizable atom (not part of Drude model)• 1 or C = Drude core• 2 or D = Drude electron•
Restrictions:
This fix should be invoked before any other commands that implement the Drude oscillator model, such as fix
langevin/drude, fix drude/transform, compute temp/drude, pair_style thole.
Related commands:
fix langevin/drude, fix drude/transform, compute temp/drude, pair_style thole
Default: None
LAMMPS Users Manual
808

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix drude/transform/direct command
fix drude/transform/inverse command
Syntax:
fix ID group-ID style keyword value ...
ID, group-ID are documented in fix command• style = drude/transform/direct or drude/transform/inverse•
Examples:
fix 3 all drude/transform/direct
fix 1 all drude/transform/inverse
Description:
Transform the coordinates of Drude oscillators from real to reduced and back for thermalizing the Drude
oscillators as described in (Lamoureux) using a Nose-Hoover thermostat. This fix is designed to be used with
the thermalized Drude oscillator model. Polarizable models in LAMMPS are described in this Section.
Drude oscillators are a pair of atoms representing a single polarizable atom. Ideally, the mass of Drude
particles would vanish and their positions would be determined self-consistently by iterative minimization of
the energy, the cores' positions being fixed. It is however more efficient and it yields comparable results, if the
Drude oscillators (the motion of the Drude particle relative to the core) are thermalized at a low temperature.
In that case, the Drude particles need a small mass.
The thermostats act on the reduced degrees of freedom, which are defined by the following equations. Note
that in these equations upper case denotes atomic or center of mass values and lower case denotes Drude
particle or dipole values. Primes denote the transformed (reduced) values, while bare letters denote the
original values.
Masses: \begin{equation} M' = M + m \end{equation} \begin{equation} m' = \frac {M\, m } {M'}
\end{equation} Positions: \begin{equation} X' = \frac {M\, X + m\, x} {M'} \end{equation} \begin{equation}
x' = x - X \end{equation} Velocities: \begin{equation} V' = \frac {M\, V + m\, v} {M'} \end{equation}
\begin{equation} v' = v - V \end{equation} Forces: \begin{equation} F' = F + f \end{equation}
\begin{equation} f' = \frac { M\, f - m\, F} {M'} \end{equation}
This transform conserves the total kinetic energy \begin{equation} \frac 1 2 \, (M\, V^2\ + m\, v^2) = \frac 1 2
\, (M'\, V'^2\ + m'\, v'^2) \end{equation} and the virial defined with absolute positions \begin{equation} X\, F
+ x\, f = X'\, F' + x'\, f' \end{equation}
This fix requires each atom know whether it is a Drude particle or not. You must therefore use the fix drude
command to specify the Drude status of each atom type.
NOTE: only the Drude core atoms need to be in the group specified for this fix. A Drude electron will be
transformed together with its core even if it is not itself in the group. It is safe to include Drude electrons or
non-polarizable atoms in the group. The non-polarizable atoms will simply not be transformed.
LAMMPS Users Manual
809

This fix does NOT perform time integration. It only transform masses, coordinates, velocities and forces.
Thus you must use separate time integration fixes, like fix nve or fix npt to actually update the velocities and
positions of atoms. In order to thermalize the reduced degrees of freedom at different temperatures, two
Nose-Hoover thermostats must be defined, acting on two distinct groups.
NOTE: The fix drude/transform/direct command must appear before any Nose-Hoover thermostating fixes.
The fix drude/transform/inverse command must appear after any Nose-Hoover thermostating fixes.
Example:
fix fDIRECT all drude/transform/direct
fix fNVT gCORES nvt temp 300.0 300.0 100
fix fNVT gDRUDES nvt temp 1.0 1.0 100
fix fINVERSE all drude/transform/inverse
compute TDRUDE all temp/drude
thermo_style custom step cpu etotal ke pe ebond ecoul elong press vol temp c_TDRUDE[1] c_TDRUDE[2]
In this example, gCORES is the group of the atom cores and gDRUDES is the group of the Drude particles
(electrons). The centers of mass of the Drude oscillators will be thermostated at 300.0 and the internal degrees
of freedom will be thermostated at 1.0. The temperatures of cores and Drude particles, in center-of-mass and
relative coordinates, are calculated using compute temp/drude
In addition, if you want to use a barostat to simulate a system at constant pressure, only one of the
Nose-Hoover fixes must be npt, the other one should be nvt. You must add a compute temp/com and a
fix_modify command so that the temperature of the npt fix be just that of its group (the Drude cores) but the
pressure be the overall pressure thermo_press.
Example:
compute cTEMP_CORE gCORES temp/com
fix fDIRECT all drude/transform/direct
fix fNPT gCORES npt temp 298.0 298.0 100 iso 1.0 1.0 500
fix_modify fNPT temp cTEMP_CORE press thermo_press
fix fNVT gDRUDES nvt temp 5.0 5.0 100
fix fINVERSE all drude/transform/inverse
In this example, gCORES is the group of the atom cores and gDRUDES is the group of the Drude particles.
The centers of mass of the Drude oscillators will be thermostated at 298.0 and the internal degrees of freedom
will be thermostated at 5.0. The whole system will be barostated at 1.0.
In order to avoid the flying ice cube problem (irreversible transfer of linear momentum to the center of mass
of the system), you may need to add a fix momentum command:
fix fMOMENTUM all momentum 100 linear 1 1 1
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
Restrictions: none
Related commands:
LAMMPS Users Manual
810


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix dt/reset command
Syntax:
fix ID group-ID dt/reset N Tmin Tmax Xmax keyword values ...
ID, group-ID are documented in fix command• dt/reset = style name of this fix command• N = recompute dt every N timesteps• Tmin = minimum dt allowed which can be NULL (time units)• Tmax = maximum dt allowed which can be NULL (time units)• Xmax = maximum distance for an atom to move in one timestep (distance units)• zero or more keyword/value pairs may be appended• keyword = units•
units value = lattice or box
lattice = Xmax is defined in lattice units
box = Xmax is defined in simulation box units
Examples:
fix 5 all dt/reset 10 1.0e-5 0.01 0.1
fix 5 all dt/reset 10 0.01 2.0 0.2 units box
Description:
Reset the timestep size every N steps during a run, so that no atom moves further than Xmax, based on current
atom velocities and forces. This can be useful when starting from a configuration with overlapping atoms,
where forces will be large. Or it can be useful when running an impact simulation where one or more
high-energy atoms collide with a solid, causing a damage cascade.
This fix overrides the timestep size setting made by the timestep command. The new timestep size dt is
computed in the following manner.
For each atom, the timestep is computed that would cause it to displace Xmax on the next integration step, as a
function of its current velocity and force. Since performing this calculation exactly would require the solution
to a quartic equation, a cheaper estimate is generated. The estimate is conservative in that the atom's
displacement is guaranteed not to exceed Xmax, though it may be smaller.
Given this putative timestep for each atom, the minimum timestep value across all atoms is computed. Then
the Tmin and Tmax bounds are applied, if specified. If one (or both) is specified as NULL, it is not applied.
When the run style is respa, this fix resets the outer loop (largest) timestep, which is the same timestep that
the timestep command sets.
Note that the cumulative simulation time (in time units), which accounts for changes in the timestep size as a
simulation proceeds, can be accessed by the thermo_style time keyword.
Restart, fix_modify, output, run start/stop, minimize info:
LAMMPS Users Manual
812
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global scalar which can be accessed by various output commands. The scalar stores the
last timestep on which the timestep was reset to a new value.
The scalar value calculated by this fix is "intensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
timestep
Default:
The option defaults is units = lattice.
LAMMPS Users Manual
813

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix efield command
Syntax:
fix ID group-ID efield ex ey ez keyword value ...
ID, group-ID are documented in fix command• efield = style name of this fix command• ex,ey,ez = E-field component values (electric field units)• any of ex,ey,ez can be a variable (see below)• zero or more keyword/value pairs may be appended to args• keyword = region or energy
region value = region-ID
region-ID = ID of region atoms must be in to have added force
energy value = v_name
v_name = variable with name that calculates the potential energy of each atom in the added E-field
•
Examples:
fix kick external-field efield 1.0 0.0 0.0
fix kick external-field efield 0.0 0.0 v_oscillate
Description:
Add a force F = qE to each charged atom in the group due to an external electric field being applied to the
system. If the system contains point-dipoles, also add a torque on the dipoles due to the external electric field.
For charges, any of the 3 quantities defining the E-field components can be specified as an equal-style or
atom-style variable, namely ex, ey, ez. If the value is a variable, it should be specified as v_name, where name
is the variable name. In this case, the variable will be evaluated each timestep, and its value used to determine
the E-field component.
For point-dipoles, equal-style variables can be used, but atom-style variables are not currently supported,
since they imply a spatial gradient in the electric field which means additional terms with gradients of the field
are required for the force and torque on dipoles.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent E-field.
Atom-style variables can specify the same formulas as equal-style variables but can also include per-atom
values, such as atom coordinates. Thus it is easy to specify a spatially-dependent E-field with optional
time-dependence as well.
If the region keyword is used, the atom must also be in the specified geometric region in order to have force
added to it.
Adding a force or torque to atoms implies a change in their potential energy as they move or rotate due to the
LAMMPS Users Manual
814

applied E-field.
For dynamics via the "run" command, this energy can be optionally added to the system's potential energy for
thermodynamic output (see below). For energy minimization via the "minimize" command, this energy must
be added to the system's potential energy to formulate a self-consistent minimization problem (see below).
The energy keyword is not allowed if the added field is a constant vector (ex,ey,ez), with all components
defined as numeric constants and not as variables. This is because LAMMPS can compute the energy for each
charged particle directly as E = -x dot qE = -q (x*ex + y*ey + z*ez), so that -Grad(E) = F. Similarly for
point-dipole particles the energy can be computed as E = -mu dot E = -(mux*ex + muy*ey + muz*ez).
The energy keyword is optional if the added force is defined with one or more variables, and if you are
performing dynamics via the run command. If the keyword is not used, LAMMPS will set the energy to 0.0,
which is typically fine for dynamics.
The energy keyword is required if the added force is defined with one or more variables, and you are
performing energy minimization via the "minimize" command for charged particles. It is not required for
point-dipoles, but a warning is issued since the minimizer in LAMMPS does not rotate dipoles, so you should
not expect to be able to minimize the orientation of dipoles in an applied electric field.
The energy keyword specifies the name of an atom-style variable which is used to compute the energy of each
atom as function of its position. Like variables used for ex, ey, ez, the energy variable is specified as v_name,
where name is the variable name.
Note that when the energy keyword is used during an energy minimization, you must insure that the formula
defined for the atom-style variable is consistent with the force variable formulas, i.e. that -Grad(E) = F. For
example, if the force due to the electric field were a spring-like F = kx, then the energy formula should be E =
-0.5kx^2. If you don't do this correctly, the minimization will not converge properly.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the potential "energy" inferred by the added
force due to the electric field to the system's potential energy as part of thermodynamic output. This is a
fictitious quantity but is needed so that the minimize command can include the forces added by this fix in a
consistent manner. I.e. there is a decrease in potential energy when atoms move in the direction of the added
force due to the electric field.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix adding its forces. Default is the outermost level.
This fix computes a global scalar and a global 3-vector of forces, which can be accessed by various output
commands. The scalar is the potential energy discussed above. The vector is the total force added to the group
of atoms. The scalar and vector values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
You should not specify force components with a variable that has time-dependence for use with a minimizer,
since the minimizer increments the timestep as the iteration count during the minimization.
LAMMPS Users Manual
815
NOTE: If you want the fictitious potential energy associated with the added forces to be included in the total
potential energy of the system (the quantity being minimized), you MUST enable the fix_modify energy
option for this fix.
Restrictions:
This fix is part of the MISC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
fix addforce
Default: none
LAMMPS Users Manual
816

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix ehex command
Syntax:
fix ID group-ID ehex nevery F keyword value
ID, group-ID are documented in fix command• ehex = style name of this fix command• nevery = add/subtract heat every this many timesteps• F = energy flux into the reservoir (energy/time units)• zero or more keyword/value pairs may be appended to args• keyword = region or constrain or com or hex
region value = region-ID
region-ID = ID of region (reservoir) atoms must be in for added thermostatting force
constrain value = none
apply the constraint algorithm (SHAKE or RATTLE) again at the end of the timestep
com value = none
rescale all sites of a constrained cluster of atom if its COM is in the reservoir
hex value = none
omit the coordinate correction to recover the HEX algorithm
•
Examples:
# Lennard-Jones, from examples/in.ehex.lj
fix fnve all nve
# specify regions rhot and rcold
...
fix fhot all ehex 1 0.15 region rhot
fix fcold all ehex 1 -0.15 region rcold
# SPC/E water, from examples/in.ehex.spce
fix fnve all nve
# specify regions rhot and rcold
...
fix fhot all ehex 1 0.075 region rhot constrain com
fix fcold all ehex 1 -0.075 region rcold constrain com
fix frattle all rattle 1e-10 400 0 b 1 a 1
Description:
This fix implements the asymmetric version of the enhanced heat exchange algorithm (Wirnsberger). The
eHEX algorithm is an extension of the heat exchange algorithm (Ikeshoji) and adds an additional coordinate
integration to account for higher-order truncation terms in the operator splitting. The original HEX algorithm
(implemented as fix heat) is known to exhibit a slight energy drift limiting the accessible simulation times to a
few nanoseconds. This issue is greatly improved by the new algorithm decreasing the energy drift by at least a
factor of a hundred (LJ and SPC/E water) with little computational overhead.
In both algorithms (non-translational) kinetic energy is constantly swapped between regions (reservoirs) to
impose a heat flux onto the system. The equations of motion are therefore modified if a particle \(i\) is located
inside a reservoir \(\Gamma_k\) where \(k>0\). We use \(\Gamma_0\) to label those parts of the simulation
LAMMPS Users Manual
817

box which are not thermostatted.) The input parameter region-ID of this fix corresponds to \(k\). The energy
swap is modelled by introducing an additional thermostatting force to the equations of motion, such that the
time evolution of coordinates and momenta of particle \(i\) becomes (Wirnsberger)
The thermostatting force is given by
where \(m_i\) is the mass and \(k(\mathbf r_i)\) maps the particle position to the respective reservoir. The
quantity \(F_{\Gamma_{k(\mathbf r_i)}}\) corresponds to the input parameter F, which is the energy flux
into the reservoir. Furthermore, \(K_{\Gamma_{k(\mathbf r_i)}}\) and \(v_{\Gamma_{k(\mathbf r_i)}}\)
denote the non-translational kinetic energy and the centre of mass velocity of that reservoir. The
thermostatting force does not affect the centre of mass velocities of the individual reservoirs and the entire
simulation box. A derivation of the equations and details on the numerical implementation with velocity
Verlet in LAMMPS can be found in reference "(Wirnsberger)"#_Wirnsberger.
NOTE: This fix only integrates the thermostatting force and must be combined with another integrator, such
as fix nve, to solve the full equations of motion.
This fix is different from a thermostat such as fix nvt or fix temp/rescale in that energy is added/subtracted
continually. Thus if there isn't another mechanism in place to counterbalance this effect, the entire system will
heat or cool continuously.
NOTE: If heat is subtracted from the system too aggressively so that the group's kinetic energy would go to
zero, then LAMMPS will halt with an error message. Increasing the value of nevery means that heat is
added/subtracted less frequently but in larger portions. The resulting temperature profile will therefore be the
same.
This fix will default to fix_heat (HEX algorithm) if the keyword hex is specified.
Compatibility with SHAKE and RATTLE (rigid molecules):
This fix is compatible with fix shake and fix rattle. If either of these constraining algorithms is specified in the
input script and the keyword constrain is set, the bond distances will be corrected a second time at the end of
the integration step. It is recommended to specify the keyword com in addition to the keyword constrain. With
this option all sites of a constrained cluster are rescaled, if its centre of mass is located inside the region.
Rescaling all sites of a cluster by the same factor does not introduce any velocity components along fixed
bonds. No rescaling takes place if the centre of mass lies outside the region.
LAMMPS Users Manual
818

NOTE: You can only use the keyword com along with constrain.
To achieve the highest accuracy it is recommended to use fix rattle with the keywords constrain and com as
shown in the second example. Only if RATTLE is employed, the velocity constraints will be satisfied.
NOTE: Even if RATTLE is used and the keywords com and constrain are both set, the coordinate constraints
will not necessarily be satisfied up to the target precision. The velocity constraints are satisfied as long as all
sites of a cluster are rescaled (keyword com) and the cluster does not span adjacent reservoirs. The current
implementation of the eHEX algorithm introduces a small error in the bond distances, which goes to zero with
order three in the timestep. For example, in a simulation of SPC/E water with a timestep of 2 fs the maximum
relative error in the bond distances was found to be on the order of \(10^{-7}\) for relatively large temperature
gradients. A higher precision can be achieved by decreasing the timestep.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the RIGID package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
fix heat, fix thermal/conductivity, compute temp, compute temp/region
Default: none
(Ikeshoji) Ikeshoji and Hafskjold, Molecular Physics, 81, 251-261 (1994).
(Wirnsberger) Wirnsberger, Frenkel, and Dellago, J Chem Phys, 143, 124104 (2015).
LAMMPS Users Manual
819

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix enforce2d command
Syntax:
fix ID group-ID enforce2d
ID, group-ID are documented in fix command• enforce2d = style name of this fix command•
Examples:
fix 5 all enforce2d
Description:
Zero out the z-dimension velocity and force on each atom in the group. This is useful when running a 2d
simulation to insure that atoms do not move from their initial z coordinate.
Styles with a suffix are functionally the same as the corresponding style without the suffix. They have been
optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The
accelerated styles take the same arguments and should produce the same results, except for round-off and
precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
Restrictions: none
Related commands: none
Default: none
LAMMPS Users Manual
820

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix eos/cv command
Syntax:
fix ID group-ID eos/cv cv
ID, group-ID are documented in fix command• eos/cv = style name of this fix command• cv = constant-volume heat capacity (energy/temperature units)•
Examples:
fix 1 all eos/cv 0.01
Description:
Fix eos/cv applies a mesoparticle equation of state to relate the particle internal energy (u_i) to the particle
internal temperature (dpdTheta_i). The eos/cv mesoparticle equation of state requires the constant-volume
heat capacity, and is defined as follows:
where Cv is the constant-volume heat capacity, u_cond is the internal conductive energy, and u_mech is the
internal mechanical energy. Note that alternative definitions of the mesoparticle equation of state are possible.
Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This command also requires use of the atom_style dpd command.
Related commands:
fix shardlow, pair dpd/fdt
Default: none
(Larentzos) J.P. Larentzos, J.K. Brennan, J.D. Moore, and W.D. Mattson, "LAMMPS Implementation of
Constant Energy Dissipative Particle Dynamics (DPD-E)", ARL-TR-6863, U.S. Army Research Laboratory,
Aberdeen Proving Ground, MD (2014).
LAMMPS Users Manual
821
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix eos/table command
Syntax:
fix ID group-ID eos/table style file N keyword
ID, group-ID are documented in fix command• eos/table = style name of this fix command• style = linear = method of interpolation• file = filename containing the tabulated equation of state• N = use N values in linear tables• keyword = name of table keyword corresponding to table file•
Examples:
fix 1 all eos/table linear eos.table 100000 KEYWORD
Description:
Fix eos/table applies a tabulated mesoparticle equation of state to relate the particle internal energy (u_i) to
the particle internal temperature (dpdTheta_i).
Fix eos/table creates interpolation tables of length N from internal energy values listed in a file as a function
of internal temperature.
The interpolation tables are created by fitting cubic splines to the file values and interpolating energy values at
each of N internal temperatures, and vice-versa. During a simulation, these tables are used to interpolate
internal energy or temperature values as needed. The interpolation is done with the linear style.
For the linear style, the internal temperature is used to find 2 surrounding table values from which an internal
energy is computed by linear interpolation, and vice-versa.
The filename specifies a file containing tabulated internal temperature and internal energy values. The
keyword specifies a section of the file. The format of this file is described below.
The format of a tabulated file is as follows (without the parenthesized comments):
# EOS TABLE (one or more comment or blank lines)
KEYWORD (keyword is first text on line)
N 500 (N parameter)
(blank)
1 1.00 0.000 (index, internal temperature, internal energy)
2 1.02 0.001
...
500 10.0 0.500
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections. The first line begins with a keyword which identifies the section.
The line can contain additional text, but the initial text must match the argument specified in the fix
LAMMPS Users Manual
822

command.
The next line lists the number of table entries. The parameter "N" is required and its value is the number of
table entries that follow. Note that this may be different than the N specified in the fix eos/table command. Let
Ntable = N in the fix command, and Nfile = "N" in the tabulated file. What LAMMPS does is a preliminary
interpolation by creating splines using the Nfile tabulated values as nodal points. It uses these to interpolate as
needed to generate energy and temperature values at Ntable different points. The resulting tables of length
Ntable are then used as described above, when computing energy and temperature relationships. This means
that if you want the interpolation tables of length Ntable to match exactly what is in the tabulated file (with
effectively no preliminary interpolation), you should set Ntable = Nfile.
Following a blank line, the next N lines list the tabulated values. On each line, the 1st value is the index from
1 to N, the 2nd value is the internal temperature (in temperature units), the 3rd value is the internal energy (in
energy units).
Note that the internal temperature and internal energy values must increase from one line to the next.
Note that one file can contain many sections, each with a tabulated potential. LAMMPS reads the file section
by section until it finds one that matches the specified keyword.
Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This command also requires use of the atom_style dpd command.
The equation of state must be a monotonically increasing function.
An error will occur if the internal temperature or internal energies are not within the table cutoffs.
Related commands:
fix shardlow, pair dpd/fdt
Default: none
LAMMPS Users Manual
823
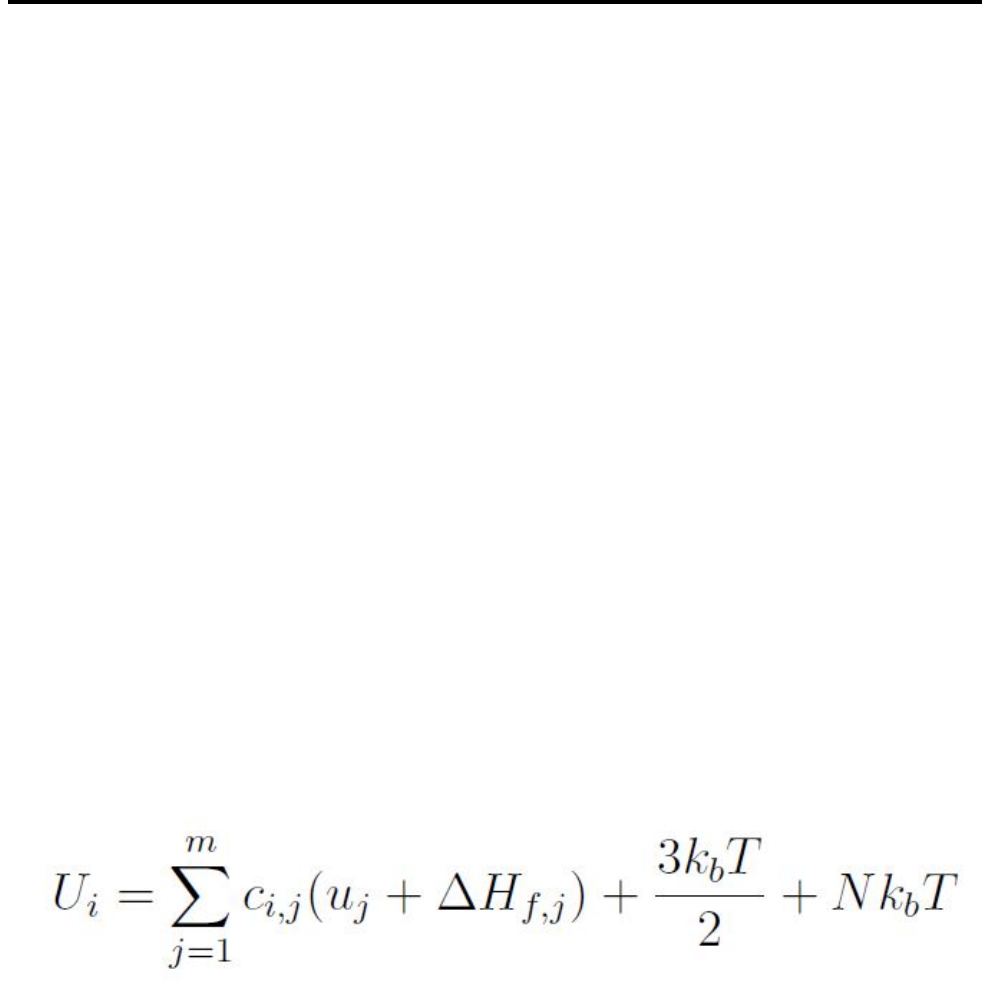
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix eos/table/rx command
Syntax:
fix ID group-ID eos/table/rx style file1 N keyword ...
ID, group-ID are documented in fix command• eos/table/rx = style name of this fix command• style = linear = method of interpolation• file1 = filename containing the tabulated equation of state• N = use N values in linear tables• keyword = name of table keyword corresponding to table file• file2 = filename containing the heats of formation of each species (optional)• deltaHf = heat of formation for a single species in energy units (optional)• energyCorr = energy correction in energy units (optional)• tempCorrCoeff = temperature correction coefficient (optional)•
Examples:
fix 1 all eos/table/rx linear eos.table 10000 KEYWORD thermo.table
fix 1 all eos/table/rx linear eos.table 10000 KEYWORD 1.5
fix 1 all eos/table/rx linear eos.table 10000 KEYWORD 1.5 0.025 0.0
Description:
Fix eos/table/rx applies a tabulated mesoparticle equation of state to relate the concentration-dependent
particle internal energy (u_i) to the particle internal temperature (dpdTheta_i).
The concentration-dependent particle internal energy (u_i) is computed according to the following relation:
where m is the number of species, c_i,j is the concentration of species j in particle i, u_j is the internal energy
of species j, DeltaH_f,j is the heat of formation of species j, N is the number of molecules represented by the
coarse-grained particle, kb is the Boltzmann constant, and T is the temperature of the system. Additionally, it
is possible to modify the concentration-dependent particle internal energy relation by adding an energy
correction, temperature-dependent correction, and/or a molecule-dependent correction. An energy correction
can be specified as a constant (in energy units). A temperature correction can be specified by multiplying a
temperature correction coefficient by the internal temperature. A molecular correction can be specified by by
multiplying a molecule correction coefficient by the average number of product gas particles in the
coarse-grain particle.
LAMMPS Users Manual
824
Fix eos/table/rx creates interpolation tables of length N from m internal energy values of each species u_j
listed in a file as a function of internal temperature. During a simulation, these tables are used to interpolate
internal energy or temperature values as needed. The interpolation is done with the linear style. For the linear
style, the internal temperature is used to find 2 surrounding table values from which an internal energy is
computed by linear interpolation. A secant solver is used to determine the internal temperature from the
internal energy.
The first filename specifies a file containing tabulated internal temperature and m internal energy values for
each species u_j. The keyword specifies a section of the file. The format of this file is described below.
The second filename specifies a file containing heat of formation DeltaH_f,j for each species.
In cases where the coarse-grain particle represents a single molecular species (i.e., no reactions occur and fix
rx is not present in the input file), fix eos/table/rx can be applied in a similar manner to fix eos/table within a
non-reactive DPD simulation. In this case, the heat of formation filename is replaced with the heat of
formation value for the single species. Additionally, the energy correction and temperature correction
coefficients may also be specified as fix arguments.
The format of a tabulated file is as follows (without the parenthesized comments):
# EOS TABLE (one or more comment or blank lines)
KEYWORD (keyword is first text on line)
N 500 h2 no2 n2 ... no (N parameter species1 species2 ... speciesN)
(blank)
1 1.00 0.000 ... 0.0000 (index, internal temperature, internal energy of species 1, ..., internal energy of species m)
2 1.02 0.001 ... 0.0002
...
500 10.0 0.500 ... 1.0000
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections. The first line begins with a keyword which identifies the section.
The line can contain additional text, but the initial text must match the argument specified in the fix
command.
The next line lists the number of table entries and the species names that correspond with all the species listed
in the reaction equations through the fix rx command. The parameter "N" is required and its value is the
number of table entries that follow. Let Nfile = "N" in the tabulated file. What LAMMPS does is a
preliminary interpolation by creating splines using the Nfile tabulated values as nodal points.
Following a blank line, the next N lines list the tabulated values. On each line, the 1st value is the index from
1 to N, the 2nd value is the internal temperature (in temperature units), the 3rd value until the m+3 value are
the internal energies of the m species (in energy units).
Note that all internal temperature and internal energy values must increase from one line to the next.
Note that one file can contain many sections, each with a tabulated potential. LAMMPS reads the file section
by section until it finds one that matches the specified keyword.
The format of a heat of formation file is as follows (without the parenthesized comments):
# HEAT OF FORMATION TABLE (one or more comment or blank lines)
LAMMPS Users Manual
825

(blank)
h2 0.00 (species name, heat of formation)
no2 0.34
n2 0.00
...
no 0.93
Note that the species can be listed in any order. The tag that is used as the species name must correspond with
the tags used to define the reactions with the fix rx command.
Alternatively, corrections to the EOS can be included by specifying three additional columns that correspond
to the energy correction, the temperature correction coefficient and molecule correction coefficient. In this
case, the format of the file is as follows:
# HEAT OF FORMATION TABLE (one or more comment or blank lines)
(blank)
h2 0.00 1.23 0.025 0.0 (species name, heat of formation, energy correction, temperature correction coefficient, molecule correction coefficient)
no2 0.34 0.00 0.000 -1.76
n2 0.00 0.00 0.000 -1.76
...
no 0.93 0.00 0.000 -1.76
Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This command also requires use of the atom_style dpd command.
The equation of state must be a monotonically increasing function.
An error will occur if the internal temperature or internal energies are not within the table cutoffs.
Related commands:
fix rx, pair dpd/fdt
Default: none
LAMMPS Users Manual
826

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix evaporate command
Syntax:
fix ID group-ID evaporate N M region-ID seed
ID, group-ID are documented in fix command• evaporate = style name of this fix command• N = delete atoms every this many timesteps• M = number of atoms to delete each time• region-ID = ID of region within which to perform deletions• seed = random number seed to use for choosing atoms to delete• zero or more keyword/value pairs may be appended
keyword = molecule
molecule value = no or yes
•
Examples:
fix 1 solvent evaporate 1000 10 surface 49892
fix 1 solvent evaporate 1000 10 surface 38277 molecule yes
Description:
Remove M atoms from the simulation every N steps. This can be used, for example, to model evaporation of
solvent particles or molecules (i.e. drying) of a system. Every N steps, the number of atoms in the fix group
and within the specified region are counted. M of these are chosen at random and deleted. If there are less than
M eligible particles, then all of them are deleted.
If the setting for the molecule keyword is no, then only single atoms are deleted. In this case, you should
insure you do not delete only a portion of a molecule (only some of its atoms), or LAMMPS will soon
generate an error when it tries to find those atoms. LAMMPS will warn you if any of the atoms eligible for
deletion have a non-zero molecule ID, but does not check for this at the time of deletion.
If the setting for the molecule keyword is yes, then when an atom is chosen for deletion, the entire molecule it
is part of is deleted. The count of deleted atoms is incremented by the number of atoms in the molecule, which
may make it exceed M. If the molecule ID of the chosen atom is 0, then it is assumed to not be part of a
molecule, and just the single atom is deleted.
As an example, if you wish to delete 10 water molecules every N steps, you should set M to 30. If only the
water's oxygen atoms were in the fix group, then two hydrogen atoms would be deleted when an oxygen atom
is selected for deletion, whether the hydrogens are inside the evaporation region or not.
Note that neighbor lists are re-built on timesteps that atoms are removed. Thus you should not remove atoms
too frequently or you will incur overhead due to the cost of building neighbor lists.
NOTE: If you are monitoring the temperature of a system where the atom count is changing due to
evaporation, you typically should use the compute_modify dynamic yes command for the temperature
compute you are using.
LAMMPS Users Manual
827
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global scalar, which can be accessed by various output commands. The scalar is the
cumulative number of deleted atoms. The scalar value calculated by this fix is "intensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the MISC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
fix deposit
Default:
The option defaults are molecule = no.
LAMMPS Users Manual
828
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix external command
Syntax:
fix ID group-ID external mode args
ID, group-ID are documented in fix command• external = style name of this fix command• mode = pf/callback or pf/array
pf/callback args = Ncall Napply
Ncall = make callback every Ncall steps
Napply = apply callback forces every Napply steps
pf/array args = Napply
Napply = apply array forces every Napply steps
•
Examples:
fix 1 all external pf/callback 1 1
fix 1 all external pf/callback 100 1
fix 1 all external pf/array 10
Description:
This fix allows external programs that are running LAMMPS through its library interface to modify certain
LAMMPS properties on specific timesteps, similar to the way other fixes do. The external driver can be a
C/C++ or Fortran program or a Python script.
If mode is pf/callback then the fix will make a callback every Ncall timesteps or minimization iterations to the
external program. The external program computes forces on atoms by setting values in an array owned by the
fix. The fix then adds these forces to each atom in the group, once every Napply steps, similar to the way the
fix addforce command works. Note that if Ncall > Napply, the force values produced by one callback will
persist, and be used multiple times to update atom forces.
The callback function "foo" is invoked by the fix as:
foo(void *ptr, bigint timestep, int nlocal, int *ids, double **x, double **fexternal);
The arguments are as follows:
ptr = pointer provided by and simply passed back to external driver• timestep = current LAMMPS timestep• nlocal = # of atoms on this processor• ids = list of atom IDs on this processor• x = coordinates of atoms on this processor• fexternal = forces to add to atoms on this processor•
Note that timestep is a "bigint" which is defined in src/lmptype.h, typically as a 64-bit integer.
Fexternal are the forces returned by the driver program.
LAMMPS Users Manual
829

The fix has a set_callback() method which the external driver can call to pass a pointer to its foo() function.
See the couple/lammps_quest/lmpqst.cpp file in the LAMMPS distribution for an example of how this is
done. This sample application performs classical MD using quantum forces computed by a density functional
code Quest.
If mode is pf/array then the fix simply stores force values in an array. The fix adds these forces to each atom
in the group, once every Napply steps, similar to the way the fix addforce command works.
The name of the public force array provided by the FixExternal class is
double **fexternal;
It is allocated by the FixExternal class as an (N,3) array where N is the number of atoms owned by a
processor. The 3 corresponds to the fx, fy, fz components of force.
It is up to the external program to set the values in this array to the desired quantities, as often as desired. For
example, the driver program might perform an MD run in stages of 1000 timesteps each. In between calls to
the LAMMPS run command, it could retrieve atom coordinates from LAMMPS, compute forces, set values in
fexternal, etc.
To use this fix during energy minimization, the energy corresponding to the added forces must also be set so
as to be consistent with the added forces. Otherwise the minimization will not converge correctly.
This can be done from the external driver by calling this public method of the FixExternal class:
void set_energy(double eng);
where eng is the potential energy. Eng is an extensive quantity, meaning it should be the sum over per-atom
energies of all affected atoms. It should also be provided in energy units consistent with the simulation. See
the details below for how to insure this energy setting is used appropriately in a minimization.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the potential "energy" set by the external driver
to the system's potential energy as part of thermodynamic output. This is a fictitious quantity but is needed so
that the minimize command can include the forces added by this fix in a consistent manner. I.e. there is a
decrease in potential energy when atoms move in the direction of the added force.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
potential energy discussed above. The scalar stored by this fix is "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
NOTE: If you want the fictitious potential energy associated with the added forces to be included in the total
potential energy of the system (the quantity being minimized), you MUST enable the fix_modify energy
option for this fix.
LAMMPS Users Manual
830
Restrictions: none
Related commands: none
Default: none
LAMMPS Users Manual
831

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix filter/corotate command
Syntax:
fix ID group-ID filter/corotate keyword value ...
ID, group-ID are documented in fix command• one or more constraint/value pairs are appended• constraint = b or a or t or m
b values = one or more bond types
a values = one or more angle types
t values = one or more atom types
m value = one or more mass values
•
Examples:
timestep 8
run_style respa 3 2 8 bond 1 pair 2 kspace 3
fix cor all filter/corotate m 1.0
fix cor all filter/corotate b 4 19 a 3 5 2
Description:
This fix implements a corotational filter for a mollified impulse method. In biomolecular simulations, it
allows the usage of larger timesteps for long-range electrostatic interactions. For details, see (Fath).
When using run_style respa for a biomolecular simulation with high-frequency covalent bonds, the outer
time-step is restricted to below ~ 4fs due to resonance problems. This fix filters the outer stage of the respa
and thus a larger (outer) time-step can be used. Since in large biomolecular simulations the computation of the
long-range electrostatic contributions poses a major bottleneck, this can significantly accelerate the
simulation.
The filter computes a cluster decomposition of the molecular structure following the criteria indicated by the
options a, b, t and m. This process is similar to the approach in fix shake, however, the clusters are not kept
contrained. Instead, the position is slightly modified only for the computation of long-range forces. A good
cluster decomposition constitutes in building clusters which contain the fastest covalent bonds inside clusters.
If the clusters are chosen suitably, the run_style respa is stable for outer time-steps of at least 8fs.
Restart, fix_modify, output, run start/stop, minimize info:
No information about these fixes is written to binary restart files. None of the fix_modify options are relevant
to these fixes. No global or per-atom quantities are stored by these fixes for access by various output
commands. No parameter of these fixes can be used with the start/stop keywords of the run command. These
fixes are not invoked during energy minimization.
Restrictions:
LAMMPS Users Manual
832

This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Currently, it does not support molecule templates.
Related commands:
Default: none
(Fath) Fath, Hochbruck, Singh, J Comp Phys, 333, 180-198 (2017).
LAMMPS Users Manual
833

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix flow/gauss command
Syntax:
fix ID group-ID flow/gauss xflag yflag zflag keyword
ID, group-ID are documented in fix command• flow/gauss = style name of this fix command• xflag,yflag,zflag = 0 or 1
0 = do not conserve current in this dimension
1 = conserve current in this dimension
•
zero or more keyword/value pairs may be appended• keyword = energy
energy value = no or yes
no = do not compute work done by this fix
yes = compute work done by this fix
•
Examples:
fix GD fluid flow/gauss 1 0 0
fix GD fluid flow/gauss 1 1 1 energy yes
Description:
This fix implements the Gaussian dynamics (GD) method to simulate a system at constant mass flux (Strong).
GD is a nonequilibrium molecular dynamics simulation method that can be used to study fluid flows through
pores, pipes, and channels. In its original implementation GD was used to compute the pressure required to
achieve a fixed mass flux through an opening. The flux can be conserved in any combination of the directions,
x, y, or z, using xflag,yflag,zflag. This fix does not initialize a net flux through a system, it only conserves the
center-of-mass momentum that is present when the fix is declared in the input script. Use the velocity
command to generate an initial center-of-mass momentum.
GD applies an external fluctuating gravitational field that acts as a driving force to keep the system away from
equilibrium. To maintain steady state, a profile-unbiased thermostat must be implemented to dissipate the heat
that is added by the driving force. Compute temp/profile can be used to implement a profile-unbiased
thermostat.
A common use of this fix is to compute a pressure drop across a pipe, pore, or membrane. The pressure profile
can be computed in LAMMPS with compute stress/atom and fix ave/chunk, or with the hardy method in fix
atc. Note that the simple compute stress/atom method is only accurate away from inhomogeneities in the
fluid, such as fixed wall atoms. Further, the computed pressure profile must be corrected for the acceleration
applied by GD before computing a pressure drop or comparing it to other methods, such as the pump method
(Zhu). The pressure correction is discussed and described in (Strong).
For a complete example including the considerations discussed above, see the examples/USER/flow_gauss
directory.
LAMMPS Users Manual
834

NOTE: Only the flux of the atoms in group-ID will be conserved. If the velocities of the group-ID atoms are
coupled to the velocities of other atoms in the simulation, the flux will not be conserved. For example, in a
simulation with fluid atoms and harmonically constrained wall atoms, if a single thermostat is applied to
group all, the fluid atom velocities will be coupled to the wall atom velocities, and the flux will not be
conserved. This issue can be avoided by thermostatting the fluid and wall groups separately.
Adding an acceleration to atoms does work on the system. This added energy can be optionally subtracted
from the potential energy for the thermodynamic output (see below) to check that the timestep is small enough
to conserve energy. Since the applied acceleration is fluctuating in time, the work cannot be computed from a
potential. As a result, computing the work is slightly more computationally expensive than usual, so it is not
performed by default. To invoke the work calculation, use the energy keyword. The fix_modify energy option
also invokes the work calculation, and overrides an energy no setting here. If neither energy yes or fix_modify
energy yes are set, the global scalar computed by the fix will return zero.
NOTE: In order to check energy conservation, any other fixes that do work on the system must have
fix_modify energy yes set as well. This includes thermostat fixes and any constraints that hold the positions of
wall atoms fixed, such as fix spring/self.
If this fix is used in a simulation with the rRESPA integrator, the applied acceleration must be computed and
applied at the same rRESPA level as the interactions between the flowing fluid and the obstacle. The rRESPA
level at which the acceleration is applied can be changed using the fix_modify respa option discussed below.
If the flowing fluid and the obstacle interact through multiple interactions that are computed at different
rRESPA levels, then there must be a separate flow/gauss fix for each level. For example, if the flowing fluid
and obstacle interact through pairwise and long-range Coulomb interactions, which are computed at rRESPA
levels 3 and 4, respectively, then there must be two separate flow/gauss fixes, one that specifies fix_modify
respa 3 and one with fix_modify respa 4.
Restart, fix_modify, output, run start/stop, minimize info:
This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to subtract the work done from the system's potential
energy as part of thermodynamic output.
The fix_modify respa option is supported by this fix. This allows the user to set at which level of the rRESPA
integrator the fix computes and adds the external acceleration. Default is the outermost level.
This fix computes a global scalar and a global 3-vector of forces, which can be accessed by various output
commands. The scalar is the negative of the work done on the system, see above discussion. The vector is the
total force that this fix applied to the group of atoms on the current timestep. The scalar and vector values
calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
Restrictions: none
Related commands:
LAMMPS Users Manual
835

fix addforce, compute temp/profile, velocity
Default:
The option default for the energy keyword is energy = no.
(Strong) Strong and Eaves, J. Phys. Chem. B 121, 189 (2017).
(Evans) Evans and Morriss, Phys. Rev. Lett. 56, 2172 (1986).
(Zhu) Zhu, Tajkhorshid, and Schulten, Biophys. J. 83, 154 (2002).
LAMMPS Users Manual
836
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix freeze command
Syntax:
fix ID group-ID freeze
ID, group-ID are documented in fix command• freeze = style name of this fix command•
Examples:
fix 2 bottom freeze
Description:
Zero out the force and torque on a granular particle. This is useful for preventing certain particles from
moving in a simulation. The granular pair styles also detect if this fix has been defined and compute
interactions between frozen and non-frozen particles appropriately, as if the frozen particle has infinite mass.
A similar functionality for normal (point) particles can be obtained using fix setforce.
Styles with a suffix are functionally the same as the corresponding style without the suffix. They have been
optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The
accelerated styles take the same arguments and should produce the same results, except for round-off and
precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global 3-vector of forces, which can be accessed by various output commands. This is the
total force on the group of atoms before the forces on individual atoms are changed by the fix. The vector
values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
LAMMPS Users Manual
837
This fix is part of the GRANULAR package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
There can only be a single freeze fix defined. This is because other the granular pair styles treat frozen
particles differently and need to be able to reference a single group to which this fix is applied.
Related commands:
atom_style sphere, fix setforce
Default: none
LAMMPS Users Manual
838

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix gcmc command
Syntax:
fix ID group-ID gcmc N X M type seed T mu displace keyword values ...
ID, group-ID are documented in fix command• gcmc = style name of this fix command• N = invoke this fix every N steps• X = average number of GCMC exchanges to attempt every N steps• M = average number of MC moves to attempt every N steps• type = atom type for inserted atoms (must be 0 if mol keyword used)• seed = random # seed (positive integer)• T = temperature of the ideal gas reservoir (temperature units)• mu = chemical potential of the ideal gas reservoir (energy units)• displace = maximum Monte Carlo translation distance (length units)• zero or more keyword/value pairs may be appended to args
keyword = mol, region, maxangle, pressure, fugacity_coeff, full_energy, charge, group, grouptype, intra_energy, tfac_insert, or overlap_cutoff
mol value = template-ID
template-ID = ID of molecule template specified in a separate molecule command
rigid value = fix-ID
fix-ID = ID of fix rigid/small command
shake value = fix-ID
fix-ID = ID of fix shake command
region value = region-ID
region-ID = ID of region where MC moves are allowed
maxangle value = maximum molecular rotation angle (degrees)
pressure value = pressure of the gas reservoir (pressure units)
fugacity_coeff value = fugacity coefficient of the gas reservoir (unitless)
full_energy = compute the entire system energy when performing MC moves
charge value = charge of inserted atoms (charge units)
group value = group-ID
group-ID = group-ID for inserted atoms (string)
grouptype values = type group-ID
type = atom type (int)
group-ID = group-ID for inserted atoms (string)
intra_energy value = intramolecular energy (energy units)
tfac_insert value = scale up/down temperature of inserted atoms (unitless)
overlap_cutoff value = maximum pair distance for overlap rejection (distance units)
•
Examples:
fix 2 gas gcmc 10 1000 1000 2 29494 298.0 -0.5 0.01
fix 3 water gcmc 10 100 100 0 3456543 3.0 -2.5 0.1 mol my_one_water maxangle 180 full_energy
fix 4 my_gas gcmc 1 10 10 1 123456543 300.0 -12.5 1.0 region disk
Description:
This fix performs grand canonical Monte Carlo (GCMC) exchanges of atoms or molecules of the given type
with an imaginary ideal gas reservoir at the specified T and chemical potential (mu) as discussed in (Frenkel).
If used with the fix nvt command, simulations in the grand canonical ensemble (muVT, constant chemical
potential, constant volume, and constant temperature) can be performed. Specific uses include computing
LAMMPS Users Manual
839
isotherms in microporous materials, or computing vapor-liquid coexistence curves.
Every N timesteps the fix attempts a number of GCMC exchanges (insertions or deletions) of gas atoms or
molecules of the given type between the simulation cell and the imaginary reservoir. It also attempts a number
of Monte Carlo moves (translations and molecule rotations) of gas of the given type within the simulation cell
or region. The average number of attempted GCMC exchanges is X. The average number of attempted MC
moves is M. M should typically be chosen to be approximately equal to the expected number of gas atoms or
molecules of the given type within the simulation cell or region, which will result in roughly one MC
translation per atom or molecule per MC cycle.
For MC moves of molecular gasses, rotations and translations are each attempted with 50% probability. For
MC moves of atomic gasses, translations are attempted 100% of the time. For MC exchanges of either
molecular or atomic gasses, deletions and insertions are each attempted with 50% probability.
All inserted particles are always assigned to two groups: the default group "all" and the group specified in the
fix gcmc command (which can also be "all"). In addition, particles are also added to any groups specified by
the group and grouptype keywords. If inserted particles are individual atoms, they are assigned the atom type
given by the type argument. If they are molecules, the type argument has no effect and must be set to zero.
Instead, the type of each atom in the inserted molecule is specified in the file read by the molecule command.
This fix cannot be used to perform MC insertions of gas atoms or molecules other than the exchanged type,
but MC deletions, translations, and rotations can be performed on any atom/molecule in the fix group. All
atoms in the simulation cell can be moved using regular time integration translations, e.g. via fix nvt, resulting
in a hybrid GCMC+MD simulation. A smaller-than-usual timestep size may be needed when running such a
hybrid simulation, especially if the inserted molecules are not well equilibrated.
This command may optionally use the region keyword to define an exchange and move volume. The specified
region must have been previously defined with a region command. It must be defined with side = in. Insertion
attempts occur only within the specified region. For non-rectangular regions, random trial points are generated
within the rectangular bounding box until a point is found that lies inside the region. If no valid point is
generated after 1000 trials, no insertion is performed, but it is counted as an attempted insertion. Move and
deletion attempt candidates are selected from gas atoms or molecules within the region. If there are no
candidates, no move or deletion is performed, but it is counted as an attempt move or deletion. If an attempted
move places the atom or molecule center-of-mass outside the specified region, a new attempted move is
generated. This process is repeated until the atom or molecule center-of-mass is inside the specified region.
If used with fix nvt, the temperature of the imaginary reservoir, T, should be set to be equivalent to the target
temperature used in fix nvt. Otherwise, the imaginary reservoir will not be in thermal equilibrium with the
simulation cell. Also, it is important that the temperature used by fix nvt be dynamic/dof, which can be
achieved as follows:
compute mdtemp mdatoms temp
compute_modify mdtemp dynamic/dof yes
fix mdnvt mdatoms nvt temp 300.0 300.0 10.0
fix_modify mdnvt temp mdtemp
Note that neighbor lists are re-built every timestep that this fix is invoked, so you should not set N to be too
small. However, periodic rebuilds are necessary in order to avoid dangerous rebuilds and missed interactions.
Specifically, avoid performing so many MC translations per timestep that atoms can move beyond the
neighbor list skin distance. See the neighbor command for details.
LAMMPS Users Manual
840
When an atom or molecule is to be inserted, its coordinates are chosen at a random position within the current
simulation cell or region, and new atom velocities are randomly chosen from the specified temperature
distribution given by T. The effective temperature for new atom velocities can be increased or decreased using
the optional keyword tfac_insert (see below). Relative coordinates for atoms in a molecule are taken from the
template molecule provided by the user. The center of mass of the molecule is placed at the insertion point.
The orientation of the molecule is chosen at random by rotating about this point.
Individual atoms are inserted, unless the mol keyword is used. It specifies a template-ID previously defined
using the molecule command, which reads a file that defines the molecule. The coordinates, atom types,
charges, etc, as well as any bond/angle/etc and special neighbor information for the molecule can be specified
in the molecule file. See the molecule command for details. The only settings required to be in this file are the
coordinates and types of atoms in the molecule.
When not using the mol keyword, you should ensure you do not delete atoms that are bonded to other atoms,
or LAMMPS will soon generate an error when it tries to find bonded neighbors. LAMMPS will warn you if
any of the atoms eligible for deletion have a non-zero molecule ID, but does not check for this at the time of
deletion.
If you wish to insert molecules via the mol keyword, that will be treated as rigid bodies, use the rigid
keyword, specifying as its value the ID of a separate fix rigid/small command which also appears in your
input script.
NOTE: If you wish the new rigid molecules (and other rigid molecules) to be thermostatted correctly via fix
rigid/small/nvt or fix rigid/small/npt, then you need to use the "fix_modify dynamic/dof yes" command for the
rigid fix. This is to inform that fix that the molecule count will vary dynamically.
If you wish to insert molecules via the mol keyword, that will have their bonds or angles constrained via
SHAKE, use the shake keyword, specifying as its value the ID of a separate fix shake command which also
appears in your input script.
Optionally, users may specify the maximum rotation angle for molecular rotations using the maxangle
keyword and specifying the angle in degrees. Rotations are performed by generating a random point on the
unit sphere and a random rotation angle on the range [0,maxangle). The molecule is then rotated by that angle
about an axis passing through the molecule center of mass. The axis is parallel to the unit vector defined by
the point on the unit sphere. The same procedure is used for randomly rotating molecules when they are
inserted, except that the maximum angle is 360 degrees.
Note that fix GCMC does not use configurational bias MC or any other kind of sampling of intramolecular
degrees of freedom. Inserted molecules can have different orientations, but they will all have the same
intramolecular configuration, which was specified in the molecule command input.
For atomic gasses, inserted atoms have the specified atom type, but deleted atoms are any atoms that have
been inserted or that belong to the user-specified fix group. For molecular gasses, exchanged molecules use
the same atom types as in the template molecule supplied by the user. In both cases, exchanged
atoms/molecules are assigned to two groups: the default group "all" and the group specified in the fix gcmc
command (which can also be "all").
The chemical potential is a user-specified input parameter defined as:
LAMMPS Users Manual
841
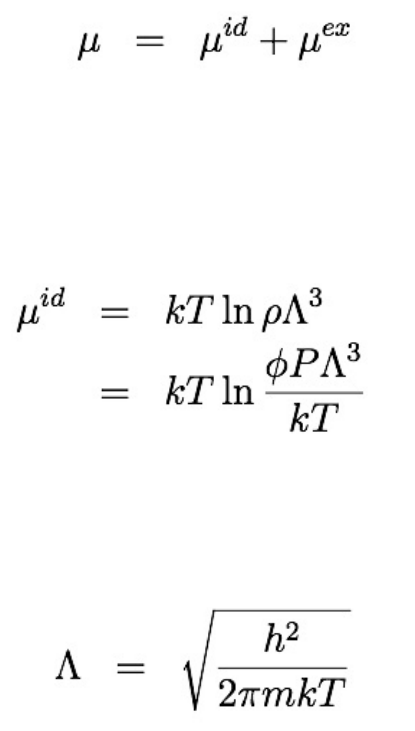
The second term mu_ex is the excess chemical potential due to energetic interactions and is formally zero for
the fictitious gas reservoir but is non-zero for interacting systems. So, while the chemical potential of the
reservoir and the simulation cell are equal, mu_ex is not, and as a result, the densities of the two are generally
quite different. The first term mu_id is the ideal gas contribution to the chemical potential. mu_id can be
related to the density or pressure of the fictitious gas reservoir by:
where k is Boltzman's constant, T is the user-specified temperature, rho is the number density, P is the
pressure, and phi is the fugacity coefficient. The constant Lambda is required for dimensional consistency. For
all unit styles except lj it is defined as the thermal de Broglie wavelength
where h is Planck's constant, and m is the mass of the exchanged atom or molecule. For unit style lj, Lambda
is simply set to the unity. Note that prior to March 2017, lambda for unit style lj was calculated using the
above formula with h set to the rather specific value of 0.18292026. Chemical potential under the old
definition can be converted to an equivalent value under the new definition by subtracting
3kTln(Lambda_old).
As an alternative to specifying mu directly, the ideal gas reservoir can be defined by its pressure P using the
pressure keyword, in which case the user-specified chemical potential is ignored. The user may also specify
the fugacity coefficient phi using the fugacity_coeff keyword, which defaults to unity.
The full_energy option means that fix GCMC will compute the total potential energy of the entire simulated
system. The total system energy before and after the proposed GCMC move is then used in the Metropolis
criterion to determine whether or not to accept the proposed GCMC move. By default, this option is off, in
which case only partial energies are computed to determine the difference in energy that would be caused by
the proposed GCMC move.
The full_energy option is needed for systems with complicated potential energy calculations, including the
following:
long-range electrostatics (kspace)• many-body pair styles• hybrid pair styles•
LAMMPS Users Manual
842
eam pair styles• tail corrections• need to include potential energy contributions from other fixes•
In these cases, LAMMPS will automatically apply the full_energy keyword and issue a warning message.
When the mol keyword is used, the full_energy option also includes the intramolecular energy of inserted and
deleted molecules. If this is not desired, the intra_energy keyword can be used to define an amount of energy
that is subtracted from the final energy when a molecule is inserted, and added to the initial energy when a
molecule is deleted. For molecules that have a non-zero intramolecular energy, this will ensure roughly the
same behavior whether or not the full_energy option is used.
Inserted atoms and molecules are assigned random velocities based on the specified temperature T. Because
the relative velocity of all atoms in the molecule is zero, this may result in inserted molecules that are
systematically too cold. In addition, the intramolecular potential energy of the inserted molecule may cause
the kinetic energy of the molecule to quickly increase or decrease after insertion. The tfac_insert keyword
allows the user to counteract these effects by changing the temperature used to assign velocities to inserted
atoms and molecules by a constant factor. For a particular application, some experimentation may be required
to find a value of tfac_insert that results in inserted molecules that equilibrate quickly to the correct
temperature.
Some fixes have an associated potential energy. Examples of such fixes include: efield, gravity, addforce,
langevin, restrain, temp/berendsen, temp/rescale, and wall fixes. For that energy to be included in the total
potential energy of the system (the quantity used when performing GCMC moves), you MUST enable the
fix_modify energy option for that fix. The doc pages for individual fix commands specify if this should be
done.
Use the charge option to insert atoms with a user-specified point charge. Note that doing so will cause the
system to become non-neutral. LAMMPS issues a warning when using long-range electrostatics (kspace) with
non-neutral systems. See the compute group/group documentation for more details about simulating
non-neutral systems with kspace on.
Use of this fix typically will cause the number of atoms to fluctuate, therefore, you will want to use the
compute_modify command to insure that the current number of atoms is used as a normalizing factor each
time temperature is computed. Here is the necessary command:
NOTE: If the density of the cell is initially very small or zero, and increases to a much larger density after a
period of equilibration, then certain quantities that are only calculated once at the start (kspace parameters, tail
corrections) may no longer be accurate. The solution is to start a new simulation after the equilibrium density
has been reached.
With some pair_styles, such as Buckingham, Born-Mayer-Huggins and ReaxFF, two atoms placed close to
each other may have an arbitrary large, negative potential energy due to the functional form of the potential.
While these unphysical configurations are inaccessible to typical dynamical trajectories, they can be generated
by Monte Carlo moves. The overlap_cutoff keyword suppresses these moves by effectively assigning an
infinite positive energy to all new configurations that place any pair of atoms closer than the specified overlap
cutoff distance.
compute_modify thermo_temp dynamic yes
LAMMPS Users Manual
843
If LJ units are used, note that a value of 0.18292026 is used by this fix as the reduced value for Planck's
constant. This value was derived from LJ parameters for argon, where h* = h/sqrt(sigma^2 * epsilon * mass),
sigma = 3.429 angstroms, epsilon/k = 121.85 K, and mass = 39.948 amu.
The group keyword assigns all inserted atoms to the group of the group-ID value. The grouptype keyword
assigns all inserted atoms of the specified type to the group of the group-ID value.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the fix to binary restart files. This includes information about the random number
generator seed, the next timestep for MC exchanges, etc. See the read_restart command for info on how to
re-specify a fix in an input script that reads a restart file, so that the operation of the fix continues in an
uninterrupted fashion.
None of the fix_modify options are relevant to this fix.
This fix computes a global vector of length 8, which can be accessed by various output commands. The vector
values are the following global cumulative quantities:
1 = translation attempts• 2 = translation successes• 3 = insertion attempts• 4 = insertion successes• 5 = deletion attempts• 6 = deletion successes• 7 = rotation attempts• 8 = rotation successes•
The vector values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the MC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Do not set "neigh_modify once yes" or else this fix will never be called. Reneighboring is required.
Can be run in parallel, but aspects of the GCMC part will not scale well in parallel. Only usable for 3D
simulations.
Note that very lengthy simulations involving insertions/deletions of billions of gas molecules may run out of
atom or molecule IDs and trigger an error, so it is better to run multiple shorter-duration simulations.
Likewise, very large molecules have not been tested and may turn out to be problematic.
Use of multiple fix gcmc commands in the same input script can be problematic if using a template molecule.
The issue is that the user-referenced template molecule in the second fix gcmc command may no longer exist
since it might have been deleted by the first fix gcmc command. An existing template molecule will need to
be referenced by the user for each subsequent fix gcmc command.
LAMMPS Users Manual
844

Related commands:
fix atom/swap, fix nvt, neighbor, fix deposit, fix evaporate, delete_atoms
Default:
The option defaults are mol = no, maxangle = 10, overlap_cutoff = 0.0, fugacity_coeff = 1, and full_energy =
no, except for the situations where full_energy is required, as listed above.
(Frenkel) Frenkel and Smit, Understanding Molecular Simulation, Academic Press, London, 2002.
LAMMPS Users Manual
845

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix gld command
Syntax:
fix ID group-ID gld Tstart Tstop N_k seed series c_1 tau_1 ... c_N_k tau_N_k keyword values ...
ID, group-ID are documented in fix command• gld = style name of this fix command• Tstart,Tstop = desired temperature at start/end of run (temperature units)• N_k = number of terms in the Prony series representation of the memory kernel• seed = random number seed to use for white noise (positive integer)• series = pprony is presently the only available option• c_k = the weight of the kth term in the Prony series (mass per time units)• tau_k = the time constant of the kth term in the Prony series (time units)• zero or more keyword/value pairs may be appended
keyword = frozen or zero
frozen value = no or yes
no = initialize extended variables using values drawn from equilibrium distribution at Tstart
yes = initialize extended variables to zero (i.e., from equilibrium distribution at zero temperature)
zero value = no or yes
no = do not set total random force to zero
yes = set total random force to zero
•
Examples:
fix 1 all gld 1.0 1.0 2 82885 pprony 0.5 1.0 1.0 2.0 frozen yes zero yes
fix 3 rouse gld 7.355 7.355 4 48823 pprony 107.1 0.02415 186.0 0.04294 428.6 0.09661 1714 0.38643
Description:
Applies Generalized Langevin Dynamics to a group of atoms, as described in (Baczewski). This is intended to
model the effect of an implicit solvent with a temporally non-local dissipative force and a colored Gaussian
random force, consistent with the Fluctuation-Dissipation Theorem. The functional form of the memory
kernel associated with the temporally non-local force is constrained to be a Prony series.
NOTE: While this fix bears many similarities to fix langevin, it has one significant difference. Namely, fix gld
performs time integration, whereas fix langevin does NOT. To this end, the specification of another fix to
perform time integration, such as fix nve, is NOT necessary.
With this fix active, the force on the jth atom is given as
LAMMPS Users Manual
846
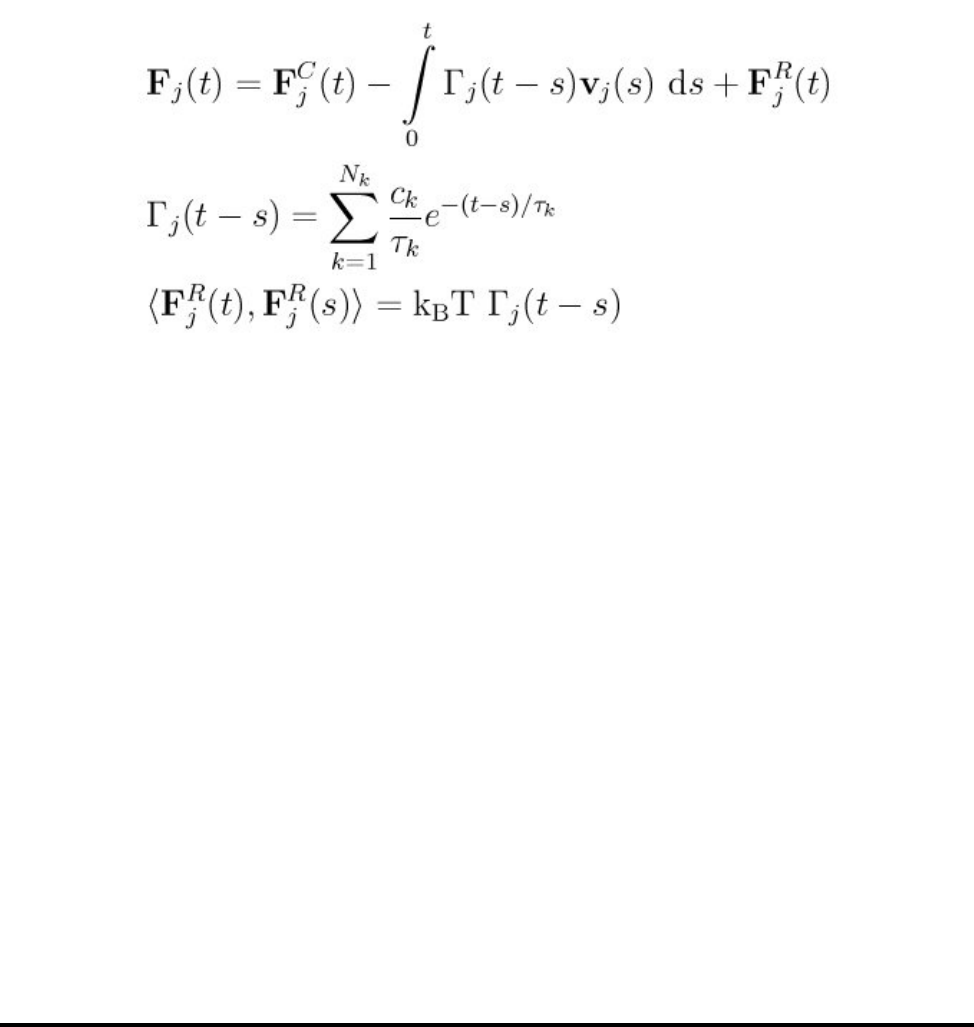
Here, the first term is representative of all conservative (pairwise, bonded, etc) forces external to this fix, the
second is the temporally non-local dissipative force given as a Prony series, and the third is the colored
Gaussian random force.
The Prony series form of the memory kernel is chosen to enable an extended variable formalism, with a
number of exemplary mathematical features discussed in (Baczewski). In particular, 3N_k extended variables
are added to each atom, which effect the action of the memory kernel without having to explicitly evaluate the
integral over time in the second term of the force. This also has the benefit of requiring the generation of
uncorrelated random forces, rather than correlated random forces as specified in the third term of the force.
Presently, the Prony series coefficients are limited to being greater than or equal to zero, and the time
constants are limited to being greater than zero. To this end, the value of series MUST be set to pprony, for
now. Future updates will allow for negative coefficients and other representations of the memory kernel. It is
with these updates in mind that the series option was included.
The units of the Prony series coefficients are chosen to be mass per time to ensure that the numerical
integration scheme stably approaches the Newtonian and Langevin limits. Details of these limits, and the
associated numerical concerns are discussed in (Baczewski).
The desired temperature at each timestep is ramped from Tstart to Tstop over the course of the next run.
The random # seed must be a positive integer. A Marsaglia random number generator is used. Each processor
uses the input seed to generate its own unique seed and its own stream of random numbers. Thus the dynamics
of the system will not be identical on two runs on different numbers of processors.
The keyword/value option pairs are used in the following ways.
The keyword frozen can be used to specify how the extended variables associated with the GLD memory
kernel are initialized. Specifying no (the default), the initial values are drawn at random from an equilibrium
distribution at Tstart, consistent with the Fluctuation-Dissipation Theorem. Specifying yes, initializes the
extended variables to zero.
The keyword zero can be used to eliminate drift due to the thermostat. Because the random forces on different
atoms are independent, they do not sum exactly to zero. As a result, this fix applies a small random force to
the entire system, and the center-of-mass of the system undergoes a slow random walk. If the keyword zero is
set to yes, the total random force is set exactly to zero by subtracting off an equal part of it from each atom in
LAMMPS Users Manual
847
the group. As a result, the center-of-mass of a system with zero initial momentum will not drift over time.
Restart, run start/stop, minimize info:
The instantaneous values of the extended variables are written to binary restart files. Because the state of the
random number generator is not saved in restart files, this means you cannot do "exact" restarts with this fix,
where the simulation continues on the same as if no restart had taken place. However, in a statistical sense, a
restarted simulation should produce the same behavior.
None of the fix_modify options are relevant to this fix. No global or per-atom quantities are stored by this fix
for access by various output commands.
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the MISC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
fix langevin, fix viscous, pair_style dpd/tstat
Default:
The option defaults are frozen = no, zero = no.
(Baczewski) A.D. Baczewski and S.D. Bond, J. Chem. Phys. 139, 044107 (2013).
LAMMPS Users Manual
848

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix gle command
Syntax:
fix ID id-group gle Ns Tstart Tstop seed Amatrix [noneq Cmatrix] [every stride]
ID, group-ID are documented in fix command• gle = style name of this fix command• Ns = number of additional fictitious momenta• Tstart, Tstop = temperature ramp during the run• Amatrix = file to read the drift matrix A from• seed = random number seed to use for generating noise (positive integer)• zero or more keyword/value pairs may be appended
keyword = noneq or every
noneq Cmatrix = file to read the non-equilibrium covariance matrix from
every stride = apply the GLE once every time steps. Reduces the accuracy
of the integration of the GLE, but has *no effect* on the accuracy of equilibrium
sampling. It might change sampling properties when used together with noneq.
•
Examples:
fix 3 boundary gle 6 300 300 31415 smart.A
fix 1 all gle 6 300 300 31415 qt-300k.A noneq qt-300k.C
Description:
Apply a Generalized Langevin Equation (GLE) thermostat as described in (Ceriotti). The formalism allows
one to obtain a number of different effects ranging from efficient sampling of all vibrational modes in the
system to inexpensive (approximate) modelling of nuclear quantum effects. Contrary to fix langevin, this fix
performs both thermostatting and evolution of the Hamiltonian equations of motion, so it should not be used
together with fix nve -- at least not on the same atom groups.
Each degree of freedom in the thermostatted group is supplemented with Ns additional degrees of freedom s,
and the equations of motion become
dq/dt=p/m
d(p,s)/dt=(F,0) - A(p,s) + B dW/dt
where F is the physical force, A is the drift matrix (that generalizes the friction in Langevin dynamics), B is
the diffusion term and dW/dt un-correlated Gaussian random forces. The A matrix couples the physical (q,p)
dynamics with that of the additional degrees of freedom, and makes it possible to obtain effectively a
history-dependent noise and friction kernel.
The drift matrix should be given as an external file Afile, as a (Ns+1 x Ns+1) matrix in inverse time units.
Matrices that are optimal for a given application and the system of choice can be obtained from (GLE4MD).
Equilibrium sampling a temperature T is obtained by specifying the target value as the Tstart and Tstop
arguments, so that the diffusion matrix that gives canonical sampling for a given A is computed automatically.
However, the GLE framework also allow for non-equilibrium sampling, that can be used for instance to model
LAMMPS Users Manual
849
inexpensively zero-point energy effects (Ceriotti2). This is achieved specifying the noneq keyword followed
by the name of the file that contains the static covariance matrix for the non-equilibrium dynamics.
Since integrating GLE dynamics can be costly when used together with simple potentials, one can use the
every optional keyword to apply the Langevin terms only once every several MD steps, in a multiple time-step
fashion. This should be used with care when doing non-equilibrium sampling, but should have no effect on
equilibrium averages when using canonical sampling.
The random number seed must be a positive integer. A Marsaglia random number generator is used. Each
processor uses the input seed to generate its own unique seed and its own stream of random numbers. Thus the
dynamics of the system will not be identical on two runs on different numbers of processors.
Note also that the Generalized Langevin Dynamics scheme that is implemented by the fix gld scheme is
closely related to the present one. In fact, it should be always possible to cast the Prony series form of the
memory kernel used by GLD into an appropriate input matrix for fix gle. While the GLE scheme is more
general, the form used by fix gld can be more directly related to the representation of an implicit solvent
environment.
Restart, fix_modify, output, run start/stop, minimize info:
The instantaneous values of the extended variables are written to binary restart files. Because the state of the
random number generator is not saved in restart files, this means you cannot do "exact" restarts with this fix,
where the simulation continues on the same as if no restart had taken place. However, in a statistical sense, a
restarted simulation should produce the same behavior. Note however that you should use a different seed
each time you restart, otherwise the same sequence of random numbers will be used each time, which might
lead to stochastic synchronization and subtle artefacts in the sampling.
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
The fix_modify energy option is supported by this fix to add the energy change induced by Langevin
thermostatting to the system's potential energy as part of thermodynamic output.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
cumulative energy change due to this fix. The scalar value calculated by this fix is "extensive".
Restrictions:
The GLE thermostat in its current implementation should not be used with rigid bodies, SHAKE or RATTLE.
It is expected that all the thermostatted degrees of freedom are fully flexible, and the sampled ensemble will
not be correct otherwise.
In order to perform constant-pressure simulations please use fix press/berendsen, rather than fix npt, to avoid
duplicate integration of the equations of motion.
This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
fix nvt, fix temp/rescale, fix viscous, fix nvt, pair_style dpd/tstat, fix gld
LAMMPS Users Manual
850


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix gravity command
fix gravity/omp command
Syntax:
fix ID group gravity magnitude style args
ID, group are documented in fix command• gravity = style name of this fix command• magnitude = size of acceleration (force/mass units)• magnitude can be a variable (see below)• style = chute or spherical or gradient or vector
chute args = angle
angle = angle in +x away from -z or -y axis in 3d/2d (in degrees)
angle can be a variable (see below)
spherical args = phi theta
phi = azimuthal angle from +x axis (in degrees)
theta = angle from +z or +y axis in 3d/2d (in degrees)
phi or theta can be a variable (see below)
vector args = x y z
x y z = vector direction to apply the acceleration
x or y or z can be a variable (see below)
•
Examples:
fix 1 all gravity 1.0 chute 24.0
fix 1 all gravity v_increase chute 24.0
fix 1 all gravity 1.0 spherical 0.0 -180.0
fix 1 all gravity 10.0 spherical v_phi v_theta
fix 1 all gravity 100.0 vector 1 1 0
Description:
Impose an additional acceleration on each particle in the group. This fix is typically used with granular
systems to include a "gravity" term acting on the macroscopic particles. More generally, it can represent any
kind of driving field, e.g. a pressure gradient inducing a Poiseuille flow in a fluid. Note that this fix operates
differently than the fix addforce command. The addforce fix adds the same force to each atom, independent of
its mass. This command imparts the same acceleration to each atom (force/mass).
The magnitude of the acceleration is specified in force/mass units. For granular systems (LJ units) this is
typically 1.0. See the units command for details.
Style chute is typically used for simulations of chute flow where the specified angle is the chute angle, with
flow occurring in the +x direction. For 3d systems, the tilt is away from the z axis; for 2d systems, the tilt is
away from the y axis.
Style spherical allows an arbitrary 3d direction to be specified for the acceleration vector. Phi and theta are
defined in the usual spherical coordinates. Thus for acceleration acting in the -z direction, theta would be
180.0 (or -180.0). Theta = 90.0 and phi = -90.0 would mean acceleration acts in the -y direction. For 2d
LAMMPS Users Manual
852

systems, phi is ignored and theta is an angle in the xy plane where theta = 0.0 is the y-axis.
Style vector imposes an acceleration in the vector direction given by (x,y,z). Only the direction of the vector is
important; it's length is ignored. For 2d systems, the z component is ignored.
Any of the quantities magnitude, angle, phi, theta, x, y, z which define the gravitational magnitude and
direction, can be specified as an equal-style variable. If the value is a variable, it should be specified as
v_name, where name is the variable name. In this case, the variable will be evaluated each timestep, and its
value used to determine the quantity. You should insure that the variable calculates a result in the appropriate
units, e.g. force/mass or degrees.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent gravitational field.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the gravitational potential energy of the system
to the system's potential energy as part of thermodynamic output.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
This fix computes a global scalar which can be accessed by various output commands. This scalar is the
gravitational potential energy of the particles in the defined field, namely mass * (g dot x) for each particles,
where x and mass are the particles position and mass, and g is the gravitational field. The scalar value
calculated by this fix is "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
LAMMPS Users Manual
853
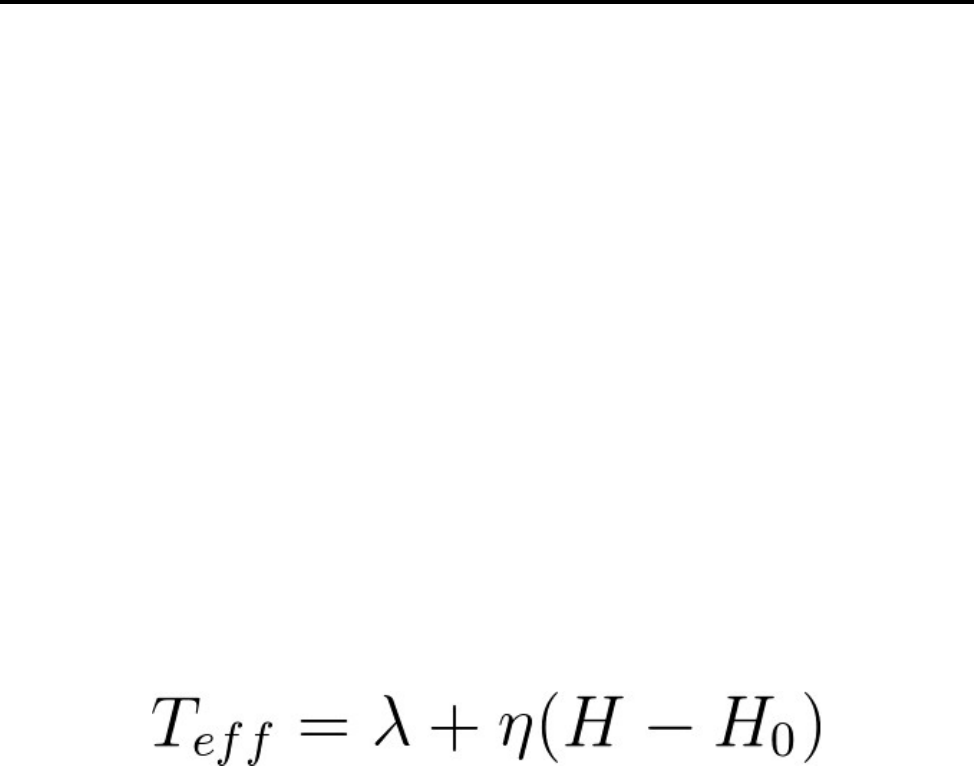
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix grem command
Syntax:
fix ID group-ID grem lambda eta H0 thermostat-ID
ID, group-ID are documented in fix command• grem = style name of this fix command• lambda = intercept parameter of linear effective temperature function• eta = slope parameter of linear effective temperature function• H0 = shift parameter of linear effective temperature function• thermostat-ID = ID of Nose-Hoover thermostat or barostat used in simulation•
Examples:
fix fxgREM all grem 400 -0.01 -30000 fxnpt
thermo_modify press fxgREM_press
fix fxgREM all grem 502 -0.15 -80000 fxnvt
Description:
This fix implements the molecular dynamics version of the generalized replica exchange method (gREM)
originally developed by (Kim), which uses non-Boltzmann ensembles to sample over first order phase
transitions. The is done by defining replicas with an enthalpy dependent effective temperature
with eta negative and steep enough to only intersect the characteristic microcanonical temperature (Ts) of the
system once, ensuring a unimodal enthalpy distribution in that replica. Lambda is the intercept and effects the
generalized ensemble similar to how temperature effects a Boltzmann ensemble. H0 is a reference enthalpy,
and is typically set as the lowest desired sampled enthalpy. Further explanation can be found in our recent
papers (Malolepsza).
This fix requires a Nose-Hoover thermostat fix reference passed to the grem as thermostat-ID. Two distinct
temperatures exist in this generalized ensemble, the effective temperature defined above, and a kinetic
temperature that controls the velocity distribution of particles as usual. Either constant volume or constant
pressure algorithms can be used.
The fix enforces a generalized ensemble in a single replica only. Typically, this ideaology is combined with
replica exchange with replicas differing by lambda only for simplicity, but this is not required. A multi-replica
simulation can be run within the LAMMPS environment using the temper/grem command. This utilizes
LAMMPS partition mode and requires the number of available processors be on the order of the number of
desired replicas. A 100-replica simulation would require at least 100 processors (1 per world at minimum). If
a many replicas are needed on a small number of processors, multi-replica runs can be run outside of
LAMMPS. An example of this can be found in examples/USER/misc/grem and has no limit on the number of
replicas per processor. However, this is very inefficient and error prone and should be avoided if possible.
LAMMPS Users Manual
855

In general, defining the generalized ensembles is unique for every system. When starting a many-replica
simulation without any knowledge of the underlying microcanonical temperature, there are several tricks we
have utilized to optimize the process. Choosing a less-steep eta yields broader distributions, requiring fewer
replicas to map the microcanonical temperature. While this likely struggles from the same sampling problems
gREM was built to avoid, it provides quick insight to Ts. Initially using an evenly-spaced lambda distribution
identifies regions where small changes in enthalpy lead to large temperature changes. Replicas are easily
added where needed.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The thermo_modify press option is supported by this fix to add the rescaled kinetic pressure as part of
thermodynamic output.
Restrictions:
This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
temper/grem, fix nvt, fix npt, thermo_modify
Default: none
(Kim) Kim, Keyes, Straub, J Chem. Phys, 132, 224107 (2010).
(Malolepsza) Malolepsza, Secor, Keyes, J Phys Chem B 119 (42), 13379-13384 (2015).
LAMMPS Users Manual
856

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix halt command
Syntax:
fix ID group-ID halt N attribute operator avalue keyword value ...
ID, group-ID are documented in fix command• halt = style name of this fix command• N = check halt condition every N steps• attribute = bondmax or tlimit or v_name
bondmax = length of longest bond in the system
tlimit = elapsed CPU time
v_name = name of equal-style variable
•
operator = "" or ">=" or "==" or "!=" or "|^"• avalue = numeric value to compare attribute to• zero or more keyword/value pairs may be appended• keyword = error or message
error value = hard or soft or continue
message value = yes or no
•
Examples:
fix 10 all halt 1 bondmax > 1.5
fix 10 all print 10 v_myCheck != 0 error soft
Description:
Check a condition every N steps during a simulation run. N must be >= 1. If the condition is met, exit the run
immediately. In this context a "run" can be dynamics or minimization iterations, as specified by the run or
minimize command.
The specified group-ID is ignored by this fix.
The specified attribute can be one of the options listed above, namely bondmax or tlimit, or an equal-style
variable referenced as v_name, where "name" is the name of a variable that has been defined previously in the
input script.
The bondmax attribute will loop over all bonds in the system, compute their current lengths, and set attribute
to the longest bond distance.
The tlimit attribute queries the elapsed CPU time (in seconds) since the current run began, and sets attribute to
that value. This is an alternative way to limit the length of a simulation run, similar to the timer timeout
command. There are two differences in using this method versus the timer command option. The first is that
the clock starts at the beginning of the current run (not when the timer or fix command is specified), so that
any setup time for the run is not included in the elapsed time. The second is that the timer invocation and
syncing across all processors (via MPI_Allreduce) is not performed once every N steps by this command.
Instead it is performed (typically) only a small number of times and the elapsed times are used to predict when
the end-of-the-run will be. Both of these attributes can be useful when performing benchmark calculations for
LAMMPS Users Manual
857

a desired length of time with minmimal overhead. For example, if a run is performing 1000s of timesteps/sec,
the overhead for syncing the timer frequently across a large number of processors may be non-negligble.
Equal-style variables evaluate to a numeric value. See the variable command for a description. They calculate
formulas which can involve mathematical operations, atom properties, group properties, thermodynamic
properties, global values calculated by a compute or fix, or references to other variables. Thus they are a very
general means of computing some attribute of the current system. For example, the following "bondmax"
variable will calculate the same quantity as the hstyle = bondmax option.
compute bdist all bond/local dist
compute bmax all reduce max c_bdist
variable bondmax equal c_bmax
Thus these two versions of a fix halt command will do the same thing:
fix 10 all halt 1 bondmax > 1.5
fix 10 all halt 1 v_bondmax > 1.5
The version with "bondmax" will just run somewhat faster, due to less overhead in computing bond lengths
and not storing them in a separate compute.
The choice of operators listed above are the usual comparison operators. The XOR operation (exclusive or) is
also included as "|^". In this context, XOR means that if either the attribute or avalue is 0.0 and the other is
non-zero, then the result is "true". Otherwise it is "false".
The specified avalue must be a numeric value.
The optional error keyword determines how the current run is halted. If its value is hard, then LAMMPS will
stop with an error message.
If its value is soft, LAMMPS will exit the current run, but continue to execute subsequent commands in the
input script. However, additional run or minimize commands will be skipped. For example, this allows a
script to output the current state of the system, e.g. via a write_dump or write_restart command.
If its value is continue, the behavior is the same as for soft, except subsequent subsequent run or minimize
commands are executed. This allows your script to remedy the condition that triggered the halt, if necessary.
Note that you may wish use the unfix command on the fix halt ID, so that the same condition is not
immediately triggered in a subsequent run.
The optional message keyword determines whether a message is printed to the screen and logfile when the
halt condition is triggered. If message is set to yes, a one line message with the values that triggered the halt is
printed. If message is set to no, no message is printed; the run simply exits. The latter may be desirable for
post-processing tools that extract thermodyanmic information from log files.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
LAMMPS Users Manual
858

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix heat command
Syntax:
fix ID group-ID heat N eflux
ID, group-ID are documented in fix command• heat = style name of this fix command• N = add/subtract heat every this many timesteps• eflux = rate of heat addition or subtraction (energy/time units)• eflux can be a variable (see below)• zero or more keyword/value pairs may be appended to args• keyword = region
region value = region-ID
region-ID = ID of region atoms must be in to have added force
•
Examples:
fix 3 qin heat 1 1.0
fix 3 qin heat 10 v_flux
fix 4 qout heat 1 -1.0 region top
Description:
Add non-translational kinetic energy (heat) to a group of atoms in a manner that conserves their aggregate
momentum. Two of these fixes can be used to establish a temperature gradient across a simulation domain by
adding heat (energy) to one group of atoms (hot reservoir) and subtracting heat from another (cold reservoir).
E.g. a simulation sampling from the McDLT ensemble.
If the region keyword is used, the atom must be in both the group and the specified geometric region in order
to have energy added or subtracted to it. If not specified, then the atoms in the group are affected wherever
they may move to.
Heat addition/subtraction is performed every N timesteps. The eflux parameter can be specified as a numeric
constant or as a variable (see below). If it is a numeric constant or equal-style variable which evaluates to a
scalar value, then the eflux determines the change in aggregate energy of the entire group of atoms per unit
time, e.g. in eV/psec for metal units. In this case it is an "extensive" quantity, meaning its magnitude should
be scaled with the number of atoms in the group. Note that since eflux has per-time units (i.e. it is a flux), this
means that a larger value of N will add/subtract a larger amount of energy each time the fix is invoked.
NOTE: The heat-exchange (HEX) algorithm implemented by this fix is known to exhibit a pronounced energy
drift. An improved algorithm (eHEX) is available as a fix ehex command and might be preferable if energy
conservation is important.
If eflux is specified as an atom-style variable (see below), then the variable computes one value per atom. In
this case, each value is the energy flux for a single atom, again in units of energy per unit time. In this case,
each value is an "intensive" quantity, which need not be scaled with the number of atoms in the group.
LAMMPS Users Manual
860
As mentioned above, the eflux parameter can be specified as an equal-style or atom_style variable. If the value
is a variable, it should be specified as v_name, where name is the variable name. In this case, the variable will
be evaluated each timestep, and its value(s) used to determine the flux.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent flux.
Atom-style variables can specify the same formulas as equal-style variables but can also include per-atom
values, such as atom coordinates. Thus it is easy to specify a spatially-dependent flux with optional
time-dependence as well.
NOTE: If heat is subtracted from the system too aggressively so that the group's kinetic energy would go to
zero, or any individual atom's kinetic energy would go to zero for the case where eflux is an atom-style
variable, then LAMMPS will halt with an error message.
Fix heat is different from a thermostat such as fix nvt or fix temp/rescale in that energy is added/subtracted
continually. Thus if there isn't another mechanism in place to counterbalance this effect, the entire system will
heat or cool continuously. You can use multiple heat fixes so that the net energy change is 0.0 or use fix
viscous to drain energy from the system.
This fix does not change the coordinates of its atoms; it only scales their velocities. Thus you must still use an
integration fix (e.g. fix nve) on the affected atoms. This fix should not normally be used on atoms that have
their temperature controlled by another fix - e.g. fix nvt or fix langevin fix.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global scalar which can be accessed by various output commands. This scalar is the most
recent value by which velocites were scaled. The scalar value calculated by this fix is "intensive". If eflux is
specified as an atom-style variable, this fix computes the average value by which the velocities were scaled
for all of the atoms that had their velocities scaled.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
fix ehex, compute temp, compute temp/region
Default: none
LAMMPS Users Manual
861

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix imd command
Syntax:
fix ID group-ID imd trate port keyword values ...
ID, group-ID are documented in fix command• imd = style name of this fix command• port = port number on which the fix listens for an IMD client• keyword = unwrap or fscale or trate
unwrap arg = on or off
off = coordinates are wrapped back into the principal unit cell (default)
on = "unwrapped" coordinates using the image flags used
fscale arg = factor
factor = floating point number to scale IMD forces (default: 1.0)
trate arg = transmission rate of coordinate data sets (default: 1)
nowait arg = on or off
off = LAMMPS waits to be connected to an IMD client before continuing (default)
on = LAMMPS listens for an IMD client, but continues with the run
•
Examples:
fix vmd all imd 5678
fix comm all imd 8888 trate 5 unwrap on fscale 10.0
Description:
This fix implements the "Interactive MD" (IMD) protocol which allows realtime visualization and
manipulation of MD simulations through the IMD protocol, as initially implemented in VMD and NAMD.
Specifically it allows LAMMPS to connect an IMD client, for example the VMD visualization program, so
that it can monitor the progress of the simulation and interactively apply forces to selected atoms.
If LAMMPS is compiled with the preprocessor flag -DLAMMPS_ASYNC_IMD then fix imd will use POSIX
threads to spawn a IMD communication thread on MPI rank 0 in order to offload data reading and writing
from the main execution thread and potentially lower the inferred latencies for slow communication links.
This feature has only been tested under linux.
There are example scripts for using this package with LAMMPS in examples/USER/imd. Additional
examples and a driver for use with the Novint Falcon game controller as haptic device can be found at:
http://sites.google.com/site/akohlmey/software/vrpn-icms.
The source code for this fix includes code developed by the Theoretical and Computational Biophysics Group
in the Beckman Institute for Advanced Science and Technology at the University of Illinois at
Urbana-Champaign. We thank them for providing a software interface that allows codes like LAMMPS to
hook to VMD.
Upon initialization of the fix, it will open a communication port on the node with MPI task 0 and wait for an
incoming connection. As soon as an IMD client is connected, the simulation will continue and the fix will
send the current coordinates of the fix's group to the IMD client at every trate MD step. When using r-RESPA,
LAMMPS Users Manual
862
trate applies to the steps of the outmost RESPA level. During a run with an active IMD connection also the
IMD client can request to apply forces to selected atoms of the fix group.
The port number selected must be an available network port number. On many machines, port numbers <
1024 are reserved for accounts with system manager privilege and specific applications. If multiple imd fixes
would be active at the same time, each needs to use a different port number.
The nowait keyword controls the behavior of the fix when no IMD client is connected. With the default
setting of off, LAMMPS will wait until a connection is made before continuing with the execution. Setting
nowait to on will have the LAMMPS code be ready to connect to a client, but continue with the simulation.
This can for example be used to monitor the progress of an ongoing calculation without the need to be
permanently connected or having to download a trajectory file.
The trate keyword allows to select how often the coordinate data is sent to the IMD client. It can also be
changed on request of the IMD client through an IMD protocol message. The unwrap keyword allows to send
"unwrapped" coordinates to the IMD client that undo the wrapping back of coordinates into the principle unit
cell, as done by default in LAMMPS. The fscale keyword allows to apply a scaling factor to forces
transmitted by the IMD client. The IMD protocols stipulates that forces are transferred in kcal/mol/angstrom
under the assumption that coordinates are given in angstrom. For LAMMPS runs with different units or as a
measure to tweak the forces generated by the manipulation of the IMD client, this option allows to make
adjustments.
To connect VMD to a listening LAMMPS simulation on the same machine with fix imd enabled, one needs to
start VMD and load a coordinate or topology file that matches the fix group. When the VMD command
prompts appears, one types the command line:
imd connect localhost 5678
This assumes that fix imd was started with 5678 as a port number for the IMD protocol.
The steps to do interactive manipulation of a running simulation in VMD are the following:
In the Mouse menu of the VMD Main window, select "Mouse -> Force -> Atom". You may alternately select
"Residue", or "Fragment" to apply forces to whole residues or fragments. Your mouse can now be used to
apply forces to your simulation. Click on an atom, residue, or fragment and drag to apply a force. Click
quickly without moving the mouse to turn the force off. You can also use a variety of 3D position trackers to
apply forces to your simulation. Game controllers or haptic devices with force-feedback such as the Novint
Falcon or Sensable PHANTOM allow you to feel the resistance due to inertia or interactions with neighbors
that the atoms experience you are trying to move, as if they were real objects. See the VMD IMD Homepage
and the VRPN-ICMS Homepage for more details.
If IMD control messages are received, a line of text describing the message and its effect will be printed to the
LAMMPS output screen, if screen output is active.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global scalar or vector or per-atom quantities are stored by this fix for access by various output
commands. No parameter of this fix can be used with the start/stop keywords of the run command. This fix is
not invoked during energy minimization.
LAMMPS Users Manual
863
Restrictions:
This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
When used in combination with VMD, a topology or coordinate file has to be loaded, which matches (in
number and ordering of atoms) the group the fix is applied to. The fix internally sorts atom IDs by ascending
integer value; in VMD (and thus the IMD protocol) those will be assigned 0-based consecutive index
numbers.
When using multiple active IMD connections at the same time, each needs to use a different port number.
Related commands: none
Default: none
LAMMPS Users Manual
864

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix indent command
Syntax:
fix ID group-ID indent K keyword values ...
ID, group-ID are documented in fix command• indent = style name of this fix command• K = force constant for indenter surface (force/distance^2 units)• one or more keyword/value pairs may be appended• keyword = sphere or cylinder or plane or side or units
sphere args = x y z R
x,y,z = initial position of center of indenter (distance units)
R = sphere radius of indenter (distance units)
any of x,y,z,R can be a variable (see below)
cylinder args = dim c1 c2 R
dim = x or y or z = axis of cylinder
c1,c2 = coords of cylinder axis in other 2 dimensions (distance units)
R = cylinder radius of indenter (distance units)
any of c1,c2,R can be a variable (see below)
plane args = dim pos side
dim = x or y or z = plane perpendicular to this dimension
pos = position of plane in dimension x, y, or z (distance units)
pos can be a variable (see below)
side = lo or hi
side value = in or out
in = the indenter acts on particles inside the sphere or cylinder
out = the indenter acts on particles outside the sphere or cylinder
units value = lattice or box
lattice = the geometry is defined in lattice units
box = the geometry is defined in simulation box units
•
Examples:
fix 1 all indent 10.0 sphere 0.0 0.0 15.0 3.0
fix 1 all indent 10.0 sphere v_x v_y 0.0 v_radius side in
fix 2 flow indent 10.0 cylinder z 0.0 0.0 10.0 units box
Description:
Insert an indenter within a simulation box. The indenter repels all atoms in the group that touch it, so it can be
used to push into a material or as an obstacle in a flow. Or it can be used as a constraining wall around a
simulation; see the discussion of the side keyword below.
The indenter can either be spherical or cylindrical or planar. You must set one of those 3 keywords.
A spherical indenter exerts a force of magnitude
F(r) = - K (r - R)^2
on each atom where K is the specified force constant, r is the distance from the atom to the center of the
indenter, and R is the radius of the indenter. The force is repulsive and F(r) = 0 for r > R.
LAMMPS Users Manual
865
A cylindrical indenter exerts the same force, except that r is the distance from the atom to the center axis of
the cylinder. The cylinder extends infinitely along its axis.
Spherical and cylindrical indenters account for periodic boundaries in two ways. First, the center point of a
spherical indenter (x,y,z) or axis of a cylindrical indenter (c1,c2) is remapped back into the simulation box, if
the box is periodic in a particular dimension. This occurs every timestep if the indenter geometry is specified
with a variable (see below), e.g. it is moving over time. Second, the calculation of distance to the indenter
center or axis accounts for periodic boundaries. Both of these mean that an indenter can effectively move
through and straddle one or more periodic boundaries.
A planar indenter is really an axis-aligned infinite-extent wall exerting the same force on atoms in the system,
where R is the position of the plane and r-R is the distance from the plane. If the side parameter of the plane is
specified as lo then it will indent from the lo end of the simulation box, meaning that atoms with a coordinate
less than the plane's current position will be pushed towards the hi end of the box and atoms with a coordinate
higher than the plane's current position will feel no force. Vice versa if side is specified as hi.
Any of the 4 quantities defining a spherical indenter's geometry can be specified as an equal-style variable,
namely x, y, z, or R. Similarly, for a cylindrical indenter, any of c1, c2, or R, can be a variable. For a planar
indenter, pos can be a variable. If the value is a variable, it should be specified as v_name, where name is the
variable name. In this case, the variable will be evaluated each timestep, and its value used to define the
indenter geometry.
Note that equal-style variables can specify formulas with various mathematical functions, and include
thermo_style command keywords for the simulation box parameters and timestep and elapsed time. Thus it is
easy to specify indenter properties that change as a function of time or span consecutive runs in a continuous
fashion. For the latter, see the start and stop keywords of the run command and the elaplong keyword of
thermo_style custom for details.
For example, if a spherical indenter's x-position is specified as v_x, then this variable definition will keep it's
center at a relative position in the simulation box, 1/4 of the way from the left edge to the right edge, even if
the box size changes:
variable x equal "xlo + 0.25*lx"
Similarly, either of these variable definitions will move the indenter from an initial position at 2.5 at a
constant velocity of 5:
variable x equal "2.5 + 5*elaplong*dt"
variable x equal vdisplace(2.5,5)
If a spherical indenter's radius is specified as v_r, then these variable definitions will grow the size of the
indenter at a specified rate.
variable r0 equal 0.0
variable rate equal 1.0
variable r equal "v_r0 + step*dt*v_rate"
If the side keyword is specified as out, which is the default, then particles outside the indenter are pushded
away from its outer surface, as described above. This only applies to spherical or cylindrical indenters. If the
side keyword is specified as in, the action of the indenter is reversed. Particles inside the indenter are pushed
away from its inner surface. In other words, the indenter is now a containing wall that traps the particles inside
it. If the radius shrinks over time, it will squeeze the particles.
LAMMPS Users Manual
866
The units keyword determines the meaning of the distance units used to define the indenter geometry. A box
value selects standard distance units as defined by the units command, e.g. Angstroms for units = real or
metal. A lattice value means the distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacing. The (x,y,z) coords of the indenter position are scaled by the x,y,z
lattice spacings respectively. The radius of a spherical or cylindrical indenter is scaled by the x lattice spacing.
Note that the units keyword only affects indenter geometry parameters specified directly with numbers, not
those specified as variables. In the latter case, you should use the xlat, ylat, zlat keywords of the thermo_style
command if you want to include lattice spacings in a variable formula.
The force constant K is not affected by the units keyword. It is always in force/distance^2 units where force
and distance are defined by the units command. If you wish K to be scaled by the lattice spacing, you can
define K with a variable whose formula contains xlat, ylat, zlat keywords of the thermo_style command, e.g.
variable k equal 100.0/xlat/xlat
fix 1 all indent $k sphere ...
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the energy of interaction between atoms and the
indenter to the system's potential energy as part of thermodynamic output. The energy of each particle
interacting with the indenter is K/3 (r - R)^3.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
This fix computes a global scalar energy and a global 3-vector of forces (on the indenter), which can be
accessed by various output commands. The scalar and vector values calculated by this fix are "extensive".
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
Note that if you define the indenter geometry with a variable using a time-dependent formula, LAMMPS uses
the iteration count in the minimizer as the timestep. But it is almost certainly a bad idea to have the indenter
change its position or size during a minimization. LAMMPS does not check if you have done this.
NOTE: If you want the atom/indenter interaction energy to be included in the total potential energy of the
system (the quantity being minimized), you must enable the fix_modify energy option for this fix.
Restrictions: none
Related commands: none
Default:
The option defaults are side = out and units = lattice.
LAMMPS Users Manual
867

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix ipi command
Syntax:
fix ID group-ID ipi address port [unix] [reset]
ID, group-ID are documented in fix command• ipi = style name of this fix command• address = internet address (FQDN or IP), or UNIX socket name• port = port number (ignored for UNIX sockets)• optional keyword = unix, if present uses a unix socket• optional keyword = reset, if present reset electrostatics at each call•
Examples:
fix 1 all ipi my.server.com 12345 fix 1 all ipi mysocket 666 unix reset
Description:
This fix enables LAMMPS to be run as a client for the i-PI Python wrapper (IPI) for performing a path
integral molecular dynamics (PIMD) simulation. The philosophy behind i-PI is described in the following
publication (IPI-CPC).
A version of the i-PI package, containing only files needed for use with LAMMPS, is provided in the
tools/i-pi directory. See the tools/i-pi/manual.pdf for an introduction to i-PI. The examples/USER/i-pi
directory contains example scripts for using i-PI with LAMMPS.
In brief, the path integral molecular dynamics is performed by the Python wrapper, while the client
(LAMMPS in this case) simply computes forces and energy for each configuration. The communication
between the two components takes place using sockets, and is reduced to the bare minimum. All the
parameters of the dynamics are specified in the input of i-PI, and all the parameters of the force field must be
specified as LAMMPS inputs, preceding the fix ipi command.
The server address must be specified by the address argument, and can be either the IP address, the
fully-qualified name of the server, or the name of a UNIX socket for local, faster communication. In the case
of internet sockets, the port argument specifies the port number on which i-PI is listening, while the unix
optional switch specifies that the socket is a UNIX socket.
Note that there is no check of data integrity, or that the atomic configurations make sense. It is assumed that
the species in the i-PI input are listed in the same order as in the data file of LAMMPS. The initial
configuration is ignored, as it will be substituted with the coordinates received from i-PI before forces are ever
evaluated.
A note of caution when using potentials that contain long-range electrostatics, or that contain parameters that
depend on box size: all of these options will be initialized based on the cell size in the LAMMPS-side initial
configuration and kept constant during the run. This is required to e.g. obtain reproducible and conserved
forces. If the cell varies too wildly, it may be advisable to reinitialize these interactions at each call. This
behavior can be requested by setting the reset switch.
LAMMPS Users Manual
868

Restart, fix_modify, output, run start/stop, minimize info:
There is no restart information associated with this fix, since all the dynamical parameters are dealt with by
i-PI.
Restrictions:
Using this fix on anything other than all atoms requires particular care, since i-PI will know nothing on atoms
that are not those whose coordinates are transferred. However, one could use this strategy to define an
external potential acting on the atoms that are moved by i-PI.
This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info. Because of the use of UNIX domain sockets, this fix will only
work in a UNIX environment.
Related commands:
fix nve
(IPI-CPC) Ceriotti, More and Manolopoulos, Comp Phys Comm, 185, 1019-1026 (2014).
(IPI) http://epfl-cosmo.github.io/gle4md/index.html?page=ipi
LAMMPS Users Manual
869

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix langevin command
fix langevin/kk command
Syntax:
fix ID group-ID langevin Tstart Tstop damp seed keyword values ...
ID, group-ID are documented in fix command• langevin = style name of this fix command• Tstart,Tstop = desired temperature at start/end of run (temperature units)• Tstart can be a variable (see below)• damp = damping parameter (time units)• seed = random number seed to use for white noise (positive integer)• zero or more keyword/value pairs may be appended• keyword = angmom or omega or scale or tally or zero
angmom value = no or factor
no = do not thermostat rotational degrees of freedom via the angular momentum
factor = do thermostat rotational degrees of freedom via the angular momentum and apply numeric scale factor as discussed below
gjf value = no or yes
no = use standard formulation
yes = use Gronbech-Jensen/Farago formulation
omega value = no or yes
no = do not thermostat rotational degrees of freedom via the angular velocity
yes = do thermostat rotational degrees of freedom via the angular velocity
scale values = type ratio
type = atom type (1-N)
ratio = factor by which to scale the damping coefficient
tally value = no or yes
no = do not tally the energy added/subtracted to atoms
yes = do tally the energy added/subtracted to atoms
zero value = no or yes
no = do not set total random force to zero
yes = set total random force to zero
•
Examples:
fix 3 boundary langevin 1.0 1.0 1000.0 699483
fix 1 all langevin 1.0 1.1 100.0 48279 scale 3 1.5
fix 1 all langevin 1.0 1.1 100.0 48279 angmom 3.333
Description:
Apply a Langevin thermostat as described in (Schneider) to a group of atoms which models an interaction
with a background implicit solvent. Used with fix nve, this command performs Brownian dynamics (BD),
since the total force on each atom will have the form:
F = Fc + Ff + Fr
Ff = - (m / damp) v
Fr is proportional to sqrt(Kb T m / (dt damp))
LAMMPS Users Manual
870
Fc is the conservative force computed via the usual inter-particle interactions (pair_style, bond_style, etc).
The Ff and Fr terms are added by this fix on a per-particle basis. See the pair_style dpd/tstat command for a
thermostatting option that adds similar terms on a pairwise basis to pairs of interacting particles.
Ff is a frictional drag or viscous damping term proportional to the particle's velocity. The proportionality
constant for each atom is computed as m/damp, where m is the mass of the particle and damp is the damping
factor specified by the user.
Fr is a force due to solvent atoms at a temperature T randomly bumping into the particle. As derived from the
fluctuation/dissipation theorem, its magnitude as shown above is proportional to sqrt(Kb T m / dt damp),
where Kb is the Boltzmann constant, T is the desired temperature, m is the mass of the particle, dt is the
timestep size, and damp is the damping factor. Random numbers are used to randomize the direction and
magnitude of this force as described in (Dunweg), where a uniform random number is used (instead of a
Gaussian random number) for speed.
Note that unless you use the omega or angmom keywords, the thermostat effect of this fix is applied to only
the translational degrees of freedom for the particles, which is an important consideration for finite-size
particles, which have rotational degrees of freedom, are being thermostatted. The translational degrees of
freedom can also have a bias velocity removed from them before thermostatting takes place; see the
description below.
NOTE: Unlike the fix nvt command which performs Nose/Hoover thermostatting AND time integration, this
fix does NOT perform time integration. It only modifies forces to effect thermostatting. Thus you must use a
separate time integration fix, like fix nve to actually update the velocities and positions of atoms using the
modified forces. Likewise, this fix should not normally be used on atoms that also have their temperature
controlled by another fix - e.g. by fix nvt or fix temp/rescale commands.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
The desired temperature at each timestep is a ramped value during the run from Tstart to Tstop.
Tstart can be specified as an equal-style or atom-style variable. In this case, the Tstop setting is ignored. If the
value is a variable, it should be specified as v_name, where name is the variable name. In this case, the
variable will be evaluated each timestep, and its value used to determine the target temperature.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent temperature.
Atom-style variables can specify the same formulas as equal-style variables but can also include per-atom
values, such as atom coordinates. Thus it is easy to specify a spatially-dependent temperature with optional
time-dependence as well.
Like other fixes that perform thermostatting, this fix can be used with compute commands that remove a
"bias" from the atom velocities. E.g. removing the center-of-mass velocity from a group of atoms or removing
the x-component of velocity from the calculation. This is not done by default, but only if the fix_modify
command is used to assign a temperature compute to this fix that includes such a bias term. See the doc pages
for individual compute commands to determine which ones include a bias. In this case, the thermostat works
in the following manner: bias is removed from each atom, thermostatting is performed on the remaining
LAMMPS Users Manual
871

thermal degrees of freedom, and the bias is added back in.
The damp parameter is specified in time units and determines how rapidly the temperature is relaxed. For
example, a value of 100.0 means to relax the temperature in a timespan of (roughly) 100 time units (tau or
fmsec or psec - see the units command). The damp factor can be thought of as inversely related to the
viscosity of the solvent. I.e. a small relaxation time implies a hi-viscosity solvent and vice versa. See the
discussion about gamma and viscosity in the documentation for the fix viscous command for more details.
The random # seed must be a positive integer. A Marsaglia random number generator is used. Each processor
uses the input seed to generate its own unique seed and its own stream of random numbers. Thus the dynamics
of the system will not be identical on two runs on different numbers of processors.
The keyword/value option pairs are used in the following ways.
The keyword angmom and omega keywords enable thermostatting of rotational degrees of freedom in
addition to the usual translational degrees of freedom. This can only be done for finite-size particles.
A simulation using atom_style sphere defines an omega for finite-size spheres. A simulation using atom_style
ellipsoid defines a finite size and shape for aspherical particles and an angular momentum. The Langevin
formulas for thermostatting the rotational degrees of freedom are the same as those above, where force is
replaced by torque, m is replaced by the moment of inertia I, and v is replaced by omega (which is derived
from the angular momentum in the case of aspherical particles).
The rotational temperature of the particles can be monitored by the compute temp/sphere and compute
temp/asphere commands with their rotate options.
For the omega keyword there is also a scale factor of 10.0/3.0 that is applied as a multiplier on the Ff
(damping) term in the equation above and of sqrt(10.0/3.0) as a multiplier on the Fr term. This does not affect
the thermostatting behaviour of the Langevin formalism but insures that the randomized rotational diffusivity
of spherical particles is correct.
For the angmom keyword a similar scale factor is needed which is 10.0/3.0 for spherical particles, but is
anisotropic for aspherical particles (e.g. ellipsoids). Currently LAMMPS only applies an isotropic scale factor,
and you can choose its magnitude as the specified value of the angmom keyword. If your aspherical particles
are (nearly) spherical than a value of 10.0/3.0 = 3.333 is a good choice. If they are highly aspherical, a value
of 1.0 is as good a choice as any, since the effects on rotational diffusivity of the particles will be incorrect
regardless. Note that for any reasonable scale factor, the thermostatting effect of the angmom keyword on the
rotational temperature of the aspherical particles should still be valid.
The keyword scale allows the damp factor to be scaled up or down by the specified factor for atoms of that
type. This can be useful when different atom types have different sizes or masses. It can be used multiple
times to adjust damp for several atom types. Note that specifying a ratio of 2 increases the relaxation time
which is equivalent to the solvent's viscosity acting on particles with 1/2 the diameter. This is the opposite
effect of scale factors used by the fix viscous command, since the damp factor in fix langevin is inversely
related to the gamma factor in fix viscous. Also note that the damping factor in fix langevin includes the
particle mass in Ff, unlike fix viscous. Thus the mass and size of different atom types should be accounted for
in the choice of ratio values.
The keyword tally enables the calculation of the cumulative energy added/subtracted to the atoms as they are
thermostatted. Effectively it is the energy exchanged between the infinite thermal reservoir and the particles.
As described below, this energy can then be printed out or added to the potential energy of the system to
LAMMPS Users Manual
872

monitor energy conservation.
NOTE: this accumulated energy does NOT include kinetic energy removed by the zero flag. LAMMPS will
print a warning when both options are active.
The keyword zero can be used to eliminate drift due to the thermostat. Because the random forces on different
atoms are independent, they do not sum exactly to zero. As a result, this fix applies a small random force to
the entire system, and the center-of-mass of the system undergoes a slow random walk. If the keyword zero is
set to yes, the total random force is set exactly to zero by subtracting off an equal part of it from each atom in
the group. As a result, the center-of-mass of a system with zero initial momentum will not drift over time.
The keyword gjf can be used to run the Gronbech-Jensen/Farago time-discretization of the Langevin model.
As described in the papers cited below, the purpose of this method is to enable longer timesteps to be used (up
to the numerical stability limit of the integrator), while still producing the correct Boltzmann distribution of
atom positions. It is implemented within LAMMPS, by changing how the random force is applied so that it is
composed of the average of two random forces representing half-contributions from the previous and current
time intervals.
In common with all methods based on Verlet integration, the discretized velocities generated by this method
in conjunction with velocity-Verlet time integration are not exactly conjugate to the positions. As a result the
temperature (computed from the discretized velocities) will be systematically lower than the target
temperature, by a small amount which grows with the timestep. Nonetheless, the distribution of atom
positions will still be consistent with the target temperature.
As an example of using the gjf keyword, for molecules containing C-H bonds, configurational properties
generated with dt = 2.5 fs and tdamp = 100 fs are indistinguishable from dt = 0.5 fs. Because the velocity
distribution systematically decreases with increasing timestep, the method should not be used to generate
properties that depend on the velocity distribution, such as the velocity autocorrelation function (VACF). In
this example, the velocity distribution at dt = 2.5fs generates an average temperature of 220 K, instead of 300
K.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. Because the state of the random number
generator is not saved in restart files, this means you cannot do "exact" restarts with this fix, where the
LAMMPS Users Manual
873

simulation continues on the same as if no restart had taken place. However, in a statistical sense, a restarted
simulation should produce the same behavior.
The fix_modify temp option is supported by this fix. You can use it to assign a temperature compute you have
defined to this fix which will be used in its thermostatting procedure, as described above. For consistency, the
group used by this fix and by the compute should be the same.
The fix_modify energy option is supported by this fix to add the energy change induced by Langevin
thermostatting to the system's potential energy as part of thermodynamic output. Note that use of this option
requires setting the tally keyword to yes.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
cumulative energy change due to this fix. The scalar value calculated by this fix is "extensive". Note that
calculation of this quantity requires setting the tally keyword to yes.
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions: none
Related commands:
fix nvt, fix temp/rescale, fix viscous, fix nvt, pair_style dpd/tstat
Default:
The option defaults are angmom = no, omega = no, scale = 1.0 for all types, tally = no, zero = no, gjf = no.
(Dunweg) Dunweg and Paul, Int J of Modern Physics C, 2, 817-27 (1991).
(Schneider) Schneider and Stoll, Phys Rev B, 17, 1302 (1978).
(Gronbech-Jensen) Gronbech-Jensen and Farago, Mol Phys, 111, 983 (2013); Gronbech-Jensen, Hayre, and
Farago, Comp Phys Comm, 185, 524 (2014)
LAMMPS Users Manual
874

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix langevin/drude command
Syntax:
fix ID group-ID langevin/drude Tcom damp_com seed_com Tdrude damp_drude seed_drude keyword values ...
ID, group-ID are documented in fix command• langevin/drude = style name of this fix command• Tcom = desired temperature of the centers of mass (temperature units)• damp_com = damping parameter for the thermostat on centers of mass (time units)• seed_com = random number seed to use for white noise of the thermostat on centers of mass (positive
integer)
•
Tdrude = desired temperature of the Drude oscillators (temperature units)• damp_drude = damping parameter for the thermostat on Drude oscillators (time units)• seed_drude = random number seed to use for white noise of the thermostat on Drude oscillators
(positive integer)
•
zero or more keyword/value pairs may be appended• keyword = zero
zero value = no or yes
no = do not set total random force on centers of mass to zero
yes = set total random force on centers of mass to zero
•
Examples:
fix 3 all langevin/drude 300.0 100.0 19377 1.0 20.0 83451
fix 1 all langevin/drude 298.15 100.0 19377 5.0 10.0 83451 zero yes
Description:
Apply two Langevin thermostats as described in (Jiang) for thermalizing the reduced degrees of freedom of
Drude oscillators. This link describes how to use the thermalized Drude oscillator model in LAMMPS and
polarizable models in LAMMPS are discussed in this Section.
Drude oscillators are a way to simulate polarizables atoms, by splitting them into a core and a Drude particle
bound by a harmonic bond. The thermalization works by transforming the particles degrees of freedom by
these equations. In these equations upper case denotes atomic or center of mass values and lower case denotes
Drude particle or dipole values. Primes denote the transformed (reduced) values, while bare letters denote the
original values.
Velocities: \begin{equation} V' = \frac {M\, V + m\, v} {M'} \end{equation} \begin{equation} v' = v - V
\end{equation} Masses: \begin{equation} M' = M + m \end{equation} \begin{equation} m' = \frac {M\, m }
{M'} \end{equation} The Langevin forces are computed as \begin{equation} F' = - \frac {M'}
{\mathtt{damp\_com}}\, V' + F_r' \end{equation} \begin{equation} f' = - \frac {m'}
{\mathtt{damp\_drude}}\, v' + f_r' \end{equation} \(F_r'\) is a random force proportional to \(\sqrt { \frac {2\,
k_B \mathtt{Tcom}\, m'} {\mathrm dt\, \mathtt{damp\_com} } } \).
\(f_r'\) is a random force proportional to \(\sqrt { \frac {2\, k_B \mathtt{Tdrude}\, m'} {\mathrm dt\,
\mathtt{damp\_drude} } } \).
LAMMPS Users Manual
875

Then the real forces acting on the particles are computed from the inverse transform: \begin{equation} F =
\frac M {M'}\, F' - f' \end{equation} \begin{equation} f = \frac m {M'}\, F' + f' \end{equation}
This fix also thermostates non-polarizable atoms in the group at temperature Tcom, as if they had a massless
Drude partner. The Drude particles themselves need not be in the group. The center of mass and the dipole are
thermostated iff the core atom is in the group.
Note that the thermostat effect of this fix is applied to only the translational degrees of freedom of the
particles, which is an important consideration if finite-size particles, which have rotational degrees of
freedom, are being thermostated. The translational degrees of freedom can also have a bias velocity removed
from them before thermostating takes place; see the description below.
NOTE: Like the fix langevin command, this fix does NOT perform time integration. It only modifies forces to
effect thermostating. Thus you must use a separate time integration fix, like fix nve or fix nph to actually
update the velocities and positions of atoms using the modified forces. Likewise, this fix should not normally
be used on atoms that also have their temperature controlled by another fix - e.g. by fix nvt or fix temp/rescale
commands.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostating.
This fix requires each atom know whether it is a Drude particle or not. You must therefore use the fix drude
command to specify the Drude status of each atom type.
NOTE: only the Drude core atoms need to be in the group specified for this fix. A Drude electron will be
transformed together with its cores even if it is not itself in the group. It is safe to include Drude electrons or
non-polarizable atoms in the group. The non-polarizable atoms will simply be thermostatted as if they had a
massless Drude partner (electron).
NOTE: Ghost atoms need to know their velocity for this fix to act correctly. You must use the comm_modify
command to enable this, e.g.
comm_modify vel yes
Tcom is the target temperature of the centers of mass, which would be used to thermostate the non-polarizable
atoms. Tdrude is the (normally low) target temperature of the core-Drude particle pairs (dipoles). Tcom and
Tdrude can be specified as an equal-style variable. If the value is a variable, it should be specified as v_name,
where name is the variable name. In this case, the variable will be evaluated each timestep, and its value used
to determine the target temperature.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent temperature.
Like other fixes that perform thermostating, this fix can be used with compute commands that remove a "bias"
from the atom velocities. E.g. removing the center-of-mass velocity from a group of atoms. This is not done
by default, but only if the fix_modify command is used to assign a temperature compute to this fix that
includes such a bias term. See the doc pages for individual compute commands to determine which ones
include a bias. In this case, the thermostat works in the following manner: bias is removed from each atom,
thermostating is performed on the remaining thermal degrees of freedom, and the bias is added back in.
NOTE: this feature has not been tested.
LAMMPS Users Manual
876
Note: The temperature thermostating the core-Drude particle pairs should be chosen low enough, so as to
mimic as closely as possible the self-consistent minimization. It must however be high enough, so that the
dipoles can follow the local electric field exerted by the neighbouring atoms. The optimal value probably
depends on the temperature of the centers of mass and on the mass of the Drude particles.
damp_com is the characteristic time for reaching thermal equilibrium of the centers of mass. For example, a
value of 100.0 means to relax the temperature of the centers of mass in a timespan of (roughly) 100 time units
(tau or fmsec or psec - see the units command). damp_drude is the characteristic time for reaching thermal
equilibrium of the dipoles. It is typically a few timesteps.
The number seed_com and seed_drude are positive integers. They set the seeds of the Marsaglia random
number generators used for generating the random forces on centers of mass and on the dipoles. Each
processor uses the input seed to generate its own unique seed and its own stream of random numbers. Thus the
dynamics of the system will not be identical on two runs on different numbers of processors.
The keyword zero can be used to eliminate drift due to the thermostat on centers of mass. Because the random
forces on different centers of mass are independent, they do not sum exactly to zero. As a result, this fix
applies a small random force to the entire system, and the momentum of the total center of mass of the system
undergoes a slow random walk. If the keyword zero is set to yes, the total random force on the centers of mass
is set exactly to zero by subtracting off an equal part of it from each center of mass in the group. As a result,
the total center of mass of a system with zero initial momentum will not drift over time.
The actual temperatures of cores and Drude particles, in center-of-mass and relative coordinates, respectively,
can be calculated using the compute temp/drude command.
Usage example for rigid bodies in the NPT ensemble:
comm_modify vel yes
fix TEMP all langevin/drude 300. 100. 1256 1. 20. 13977 zero yes
fix NPH ATOMS rigid/nph/small molecule iso 1. 1. 500.
fix NVE DRUDES nve
compute TDRUDE all temp/drude
thermo_style custom step cpu etotal ke pe ebond ecoul elong press vol temp c_TDRUDE[1] c_TDRUDE[2]
Comments:
Drude particles should not be in the rigid group, otherwise the Drude oscillators will be frozen and the
system will lose its polarizability.
•
zero yes avoids a drift of the center of mass of the system, but is a bit slower.• Use two different random seeds to avoid unphysical correlations.• Temperature is controlled by the fix langevin/drude, so the time-integration fixes do not thermostate.
Don't forget to time-integrate both cores and Drude particles.
•
Pressure is time-integrated only once by using nve for Drude particles and nph for atoms/cores (or
vice versa). Do not use nph for both.
•
The temperatures of cores and Drude particles are calculated by compute temp/drude• Contrary to the alternative thermostating using Nose-Hoover thermostat fix npt and fix
drude/transform, the fix_modify command is not required here, because the fix nph computes the
global pressure even if its group is ATOMS. This is what we want. If we thermostated ATOMS using
npt, the pressure should be the global one, but the temperature should be only that of the cores. That's
why the command fix_modify should be called in that case.
•
LAMMPS Users Manual
877

Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. Because the state of the random number
generator is not saved in restart files, this means you cannot do "exact" restarts with this fix, where the
simulation continues on the same as if no restart had taken place. However, in a statistical sense, a restarted
simulation should produce the same behavior.
The fix_modify temp option is supported by this fix. You can use it to assign a temperature compute you have
defined to this fix which will be used in its thermostating procedure, as described above. For consistency, the
group used by the compute should include the group of this fix and the Drude particles.
This fix is not invoked during energy minimization.
Restrictions: none
Related commands:
fix langevin, fix drude, fix drude/transform, compute temp/drude, pair_style thole
Default:
The option defaults are zero = no.
(Jiang) Jiang, Hardy, Phillips, MacKerell, Schulten, and Roux, J Phys Chem Lett, 2, 87-92 (2011).
LAMMPS Users Manual
878

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix langevin/eff command
Syntax:
fix ID group-ID langevin/eff Tstart Tstop damp seed keyword values ...
ID, group-ID are documented in fix command• langevin/eff = style name of this fix command• Tstart,Tstop = desired temperature at start/end of run (temperature units)• damp = damping parameter (time units)• seed = random number seed to use for white noise (positive integer)• zero or more keyword/value pairs may be appended
keyword = scale or tally or zero
scale values = type ratio
type = atom type (1-N)
ratio = factor by which to scale the damping coefficient
tally values = no or yes
no = do not tally the energy added/subtracted to atoms
yes = do tally the energy added/subtracted to atoms
zero value = no or yes
no = do not set total random force to zero
yes = set total random force to zero
•
Examples:
fix 3 boundary langevin/eff 1.0 1.0 10.0 699483
fix 1 all langevin/eff 1.0 1.1 10.0 48279 scale 3 1.5
Description:
Apply a Langevin thermostat as described in (Schneider) to a group of nuclei and electrons in the electron
force field model. Used with fix nve/eff, this command performs Brownian dynamics (BD), since the total
force on each atom will have the form:
F = Fc + Ff + Fr
Ff = - (m / damp) v
Fr is proportional to sqrt(Kb T m / (dt damp))
Fc is the conservative force computed via the usual inter-particle interactions (pair_style).
The Ff and Fr terms are added by this fix on a per-particle basis.
The operation of this fix is exactly like that described by the fix langevin command, except that the
thermostatting is also applied to the radial electron velocity for electron particles.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. Because the state of the random number
generator is not saved in restart files, this means you cannot do "exact" restarts with this fix, where the
LAMMPS Users Manual
879

simulation continues on the same as if no restart had taken place. However, in a statistical sense, a restarted
simulation should produce the same behavior.
The fix_modify temp option is supported by this fix. You can use it to assign a temperature compute you have
defined to this fix which will be used in its thermostatting procedure, as described above. For consistency, the
group used by this fix and by the compute should be the same.
The fix_modify energy option is supported by this fix to add the energy change induced by Langevin
thermostatting to the system's potential energy as part of thermodynamic output. Note that use of this option
requires setting the tally keyword to yes.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
cumulative energy change due to this fix. The scalar value calculated by this fix is "extensive". Note that
calculation of this quantity requires setting the tally keyword to yes.
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions: none
This fix is part of the USER-EFF package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
fix langevin
Default:
The option defaults are scale = 1.0 for all types and tally = no.
(Dunweg) Dunweg and Paul, Int J of Modern Physics C, 2, 817-27 (1991).
(Schneider) Schneider and Stoll, Phys Rev B, 17, 1302 (1978).
LAMMPS Users Manual
880

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix lb/fluid command
Syntax:
fix ID group-ID lb/fluid nevery LBtype viscosity density keyword values ...
ID, group-ID are documented in fix command• lb/fluid = style name of this fix command• nevery = update the lattice-Boltzmann fluid every this many timesteps• LBtype = 1 to use the standard finite difference LB integrator, 2 to use the LB integrator of Ollila et
al.
•
viscosity = the fluid viscosity (units of mass/(time*length)).• density = the fluid density.• zero or more keyword/value pairs may be appended• keyword = setArea or setGamma or scaleGamma or dx or dm or a0 or noise or calcforce or trilinear
or D3Q19 or read_restart or write_restart or zwall_velocity or bodyforce or printfluid
setArea values = type node_area
type = atom type (1-N)
node_area = portion of the surface area of the composite object associated with the particular atom type (used when the force coupling constant is set by default).
setGamma values = gamma
gamma = user set value for the force coupling constant.
scaleGamma values = type gammaFactor
type = atom type (1-N)
gammaFactor = factor to scale the setGamma gamma value by, for the specified atom type.
dx values = dx_LB = the lattice spacing.
dm values = dm_LB = the lattice-Boltzmann mass unit.
a0 values = a_0_real = the square of the speed of sound in the fluid.
noise values = Temperature seed
Temperature = fluid temperature.
seed = random number generator seed (positive integer)
calcforce values = N forcegroup-ID
N = output the force and torque every N timesteps
forcegroup-ID = ID of the particle group to calculate the force and torque of
trilinear values = none (used to switch from the default Peskin interpolation stencil to the trilinear stencil).
D3Q19 values = none (used to switch from the default D3Q15, 15 velocity lattice, to the D3Q19, 19 velocity lattice).
read_restart values = restart file = name of the restart file to use to restart a fluid run.
write_restart values = N = write a restart file every N MD timesteps.
zwall_velocity values = velocity_bottom velocity_top = velocities along the y-direction of the bottom and top walls (located at z=zmin and z=zmax).
bodyforce values = bodyforcex bodyforcey bodyforcez = the x,y and z components of a constant body force added to the fluid.
printfluid values = N = print the fluid density and velocity at each grid point every N timesteps.
•
Examples:
fix 1 all lb/fluid 1 2 1.0 1.0 setGamma 13.0 dx 4.0 dm 10.0 calcforce sphere1
fix 1 all lb/fluid 1 1 1.0 0.0009982071 setArea 1 1.144592082 dx 2.0 dm 0.3 trilinear noise 300.0 8979873
Description:
Implement a lattice-Boltzmann fluid on a uniform mesh covering the LAMMPS simulation domain. The MD
particles described by group-ID apply a velocity dependent force to the fluid.
The lattice-Boltzmann algorithm solves for the fluid motion governed by the Navier Stokes equations,
LAMMPS Users Manual
881

with,
where rho is the fluid density, u is the local fluid velocity, sigma is the stress tensor, F is a local external force,
and eta and Lambda are the shear and bulk viscosities respectively. Here, we have implemented
with a_0 set to 1/3 (dx/dt)^2 by default.
The algorithm involves tracking the time evolution of a set of partial distribution functions which evolve
according to a velocity discretized version of the Boltzmann equation,
where the first term on the right hand side represents a single time relaxation towards the equilibrium
distribution function, and tau is a parameter physically related to the viscosity. On a technical note, we have
implemented a 15 velocity model (D3Q15) as default; however, the user can switch to a 19 velocity model
(D3Q19) through the use of the D3Q19 keyword. This fix provides the user with the choice of two algorithms
to solve this equation, through the specification of the keyword LBtype. If LBtype is set equal to 1, the
standard finite difference LB integrator is used. If LBtype is set equal to 2, the algorithm of Ollila et al. is
used.
Physical variables are then defined in terms of moments of the distribution functions,
LAMMPS Users Manual
882
Full details of the lattice-Boltzmann algorithm used can be found in Mackay et al..
The fluid is coupled to the MD particles described by group-ID through a velocity dependent force. The
contribution to the fluid force on a given lattice mesh site j due to MD particle alpha is calculated as:
where v_n is the velocity of the MD particle, u_f is the fluid velocity interpolated to the particle location, and
gamma is the force coupling constant. Zeta is a weight assigned to the grid point, obtained by distributing the
particle to the nearest lattice sites. For this, the user has the choice between a trilinear stencil, which provides
a support of 8 lattice sites, or the immersed boundary method Peskin stencil, which provides a support of 64
lattice sites. While the Peskin stencil is seen to provide more stable results, the trilinear stencil may be better
suited for simulation of objects close to walls, due to its smaller support. Therefore, by default, the Peskin
stencil is used; however the user may switch to the trilinear stencil by specifying the keyword, trilinear.
By default, the force coupling constant, gamma, is calculated according to
Here, m_v is the mass of the MD particle, m_u is a representative fluid mass at the particle location, and
dt_collision is a collision time, chosen such that tau/dt_collision = 1 (see Mackay and Denniston for full
details). In order to calculate m_u, the fluid density is interpolated to the MD particle location, and multiplied
by a volume, node_area*dx_lb, where node_area represents the portion of the surface area of the composite
object associated with a given MD particle. By default, node_area is set equal to dx_lb*dx_lb; however
specific values for given atom types can be set using the setArea keyword.
The user also has the option of specifying their own value for the force coupling constant, for all the MD
particles associated with the fix, through the use of the setGamma keyword. This may be useful when
modelling porous particles. See Mackay et al. for a detailed description of the method by which the user can
choose an appropriate gamma value.
NOTE: while this fix applies the force of the particles on the fluid, it does not apply the force of the fluid to
the particles. When the force coupling constant is set using the default method, there is only one option to
include this hydrodynamic force on the particles, and that is through the use of the lb/viscous fix. This fix
adds the hydrodynamic force to the total force acting on the particles, after which any of the built-in
LAMMPS Users Manual
883

LAMMPS integrators can be used to integrate the particle motion. However, if the user specifies their own
value for the force coupling constant, as mentioned in Mackay et al., the built-in LAMMPS integrators may
prove to be unstable. Therefore, we have included our own integrators fix lb/rigid/pc/sphere, and fix lb/pc, to
solve for the particle motion in these cases. These integrators should not be used with the lb/viscous fix, as
they add hydrodynamic forces to the particles directly. In addition, they can not be used if the force coupling
constant has been set the default way.
NOTE: if the force coupling constant is set using the default method, and the lb/viscous fix is NOT used to
add the hydrodynamic force to the total force acting on the particles, this physically corresponds to a situation
in which an infinitely massive particle is moving through the fluid (since collisions between the particle and
the fluid do not act to change the particle's velocity). Therefore, the user should set the mass of the particle to
be significantly larger than the mass of the fluid at the particle location, in order to approximate an infinitely
massive particle (see the dragforce test run for an example).
Inside the fix, parameters are scaled by the lattice-Boltzmann timestep, dt, grid spacing, dx, and mass unit,
dm. dt is set equal to (nevery*dt_MD), where dt_MD is the MD timestep. By default, dm is set equal to 1.0,
and dx is chosen so that tau/(dt) = (3*eta*dt)/(rho*dx^2) is approximately equal to 1. However, the user has
the option of specifying their own values for dm, and dx, by using the optional keywords dm, and dx
respectively.
NOTE: Care must be taken when choosing both a value for dx, and a simulation domain size. This fix uses the
same subdivision of the simulation domain among processors as the main LAMMPS program. In order to
uniformly cover the simulation domain with lattice sites, the lengths of the individual LAMMPS subdomains
must all be evenly divisible by dx. If the simulation domain size is cubic, with equal lengths in all dimensions,
and the default value for dx is used, this will automatically be satisfied.
Physical parameters describing the fluid are specified through viscosity, density, and a0. If the force coupling
constant is set the default way, the surface area associated with the MD particles is specified using the setArea
keyword. If the user chooses to specify a value for the force coupling constant, this is set using the setGamma
keyword. These parameters should all be given in terms of the mass, distance, and time units chosen for the
main LAMMPS run, as they are scaled by the LB timestep, lattice spacing, and mass unit, inside the fix.
The setArea keyword allows the user to associate a surface area with a given atom type. For example if a
spherical composite object of radius R is represented as a spherical shell of N evenly distributed MD particles,
all of the same type, the surface area per particle associated with that atom type should be set equal to
4*pi*R^2/N. This keyword should only be used if the force coupling constant, gamma, is set the default way.
The setGamma keyword allows the user to specify their own value for the force coupling constant, gamma,
instead of using the default value.
The scaleGamma keyword should be used in conjunction with the setGamma keyword, when the user wishes
to specify different gamma values for different atom types. This keyword allows the user to scale the
setGamma gamma value by a factor, gammaFactor, for a given atom type.
The dx keyword allows the user to specify a value for the LB grid spacing.
The dm keyword allows the user to specify the LB mass unit.
If the a0 keyword is used, the value specified is used for the square of the speed of sound in the fluid. If this
keyword is not present, the speed of sound squared is set equal to (1/3)*(dx/dt)^2. Setting a0 > (dx/dt)^2 is not
LAMMPS Users Manual
884

allowed, as this may lead to instabilities.
If the noise keyword is used, followed by a positive temperature value, and a positive integer random number
seed, a thermal lattice-Boltzmann algorithm is used. If LBtype is set equal to 1 (i.e. the standard LB integrator
is chosen), the thermal LB algorithm of Adhikari et al. is used; however if LBtype is set equal to 2 both the LB
integrator, and thermal LB algorithm described in Ollila et al. are used.
If the calcforce keyword is used, both the fluid force and torque acting on the specified particle group are
printed to the screen every N timesteps.
If the keyword trilinear is used, the trilinear stencil is used to interpolate the particle nodes onto the fluid
mesh. By default, the immersed boundary method, Peskin stencil is used. Both of these interpolation methods
are described in Mackay et al..
If the keyword D3Q19 is used, the 19 velocity (D3Q19) lattice is used by the lattice-Boltzmann algorithm. By
default, the 15 velocity (D3Q15) lattice is used.
If the keyword write_restart is used, followed by a positive integer, N, a binary restart file is printed every N
LB timesteps. This restart file only contains information about the fluid. Therefore, a LAMMPS restart file
should also be written in order to print out full details of the simulation.
NOTE: When a large number of lattice grid points are used, the restart files may become quite large.
In order to restart the fluid portion of the simulation, the keyword read_restart is specified, followed by the
name of the binary lb_fluid restart file to be used.
If the zwall_velocity keyword is used y-velocities are assigned to the lower and upper walls. This keyword
requires the presence of walls in the z-direction. This is set by assigning fixed boundary conditions in the
z-direction. If fixed boundary conditions are present in the z-direction, and this keyword is not used, the walls
are assumed to be stationary.
If the bodyforce keyword is used, a constant body force is added to the fluid, defined by it's x, y and z
components.
If the printfluid keyword is used, followed by a positive integer, N, the fluid densities and velocities at each
lattice site are printed to the screen every N timesteps.
For further details, as well as descriptions and results of several test runs, see Mackay et al.. Please include a
citation to this paper if the lb_fluid fix is used in work contributing to published research.
Restart, fix_modify, output, run start/stop, minimize info:
Due to the large size of the fluid data, this fix writes it's own binary restart files, if requested, independent of
the main LAMMPS binary restart files; no information about lb_fluid is written to the main LAMMPS binary
restart files.
None of the fix_modify options are relevant to this fix. No global or per-atom quantities are stored by this fix
for access by various output commands. No parameter of this fix can be used with the start/stop keywords of
the run command. This fix is not invoked during energy minimization.
LAMMPS Users Manual
885

Restrictions:
This fix is part of the USER-LB package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix can only be used with an orthogonal simulation domain.
Walls have only been implemented in the z-direction. Therefore, the boundary conditions, as specified via the
main LAMMPS boundary command must be periodic for x and y, and either fixed or periodic for z.
Shrink-wrapped boundary conditions are not permitted with this fix.
This fix must be used before any of fix lb/viscous, fix lb/momentum, fix lb/rigid/pc/sphere, and/ or fix lb/pc ,
as the fluid needs to be initialized before any of these routines try to access its properties. In addition, in order
for the hydrodynamic forces to be added to the particles, this fix must be used in conjunction with the
lb/viscous fix if the force coupling constant is set by default, or either the lb/viscous fix or one of the
lb/rigid/pc/sphere or lb/pc integrators, if the user chooses to specify their own value for the force coupling
constant.
Related commands:
fix lb/viscous, fix lb/momentum, fix lb/rigid/pc/sphere, fix lb/pc
Default:
By default, the force coupling constant is set according to
and an area of dx_lb^2 per node, used to calculate the fluid mass at the particle node location, is assumed.
dx is chosen such that tau/(delta t_LB) = (3 eta dt_LB)/(rho dx_lb^2) is approximately equal to 1. dm is set
equal to 1.0. a0 is set equal to (1/3)*(dx_lb/dt_lb)^2. The Peskin stencil is used as the default interpolation
method. The D3Q15 lattice is used for the lattice-Boltzmann algorithm. If walls are present, they are assumed
to be stationary.
(Ollila et al.) Ollila, S.T.T., Denniston, C., Karttunen, M., and Ala-Nissila, T., Fluctuating lattice-Boltzmann
model for complex fluids, J. Chem. Phys. 134 (2011) 064902.
(Mackay et al.) Mackay, F. E., Ollila, S.T.T., and Denniston, C., Hydrodynamic Forces Implemented into
LAMMPS through a lattice-Boltzmann fluid, Computer Physics Communications 184 (2013) 2021-2031.
(Mackay and Denniston) Mackay, F. E., and Denniston, C., Coupling MD particles to a lattice-Boltzmann
fluid through the use of conservative forces, J. Comput. Phys. 237 (2013) 289-298.
LAMMPS Users Manual
886
(Adhikari et al.) Adhikari, R., Stratford, K., Cates, M. E., and Wagner, A. J., Fluctuating lattice Boltzmann,
Europhys. Lett. 71 (2005) 473-479.
LAMMPS Users Manual
887

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix lb/momentum command
Syntax:
fix ID group-ID lb/momentum nevery keyword values ...
ID, group-ID are documented in the fix command• lb/momentum = style name of this fix command• nevery = adjust the momentum every this many timesteps• zero or more keyword/value pairs may be appended• keyword = linear
linear values = xflag yflag zflag
xflag,yflag,zflag = 0/1 to exclude/include each dimension.
•
Examples:
fix 1 sphere lb/momentum
fix 1 all lb/momentum linear 1 1 0
Description:
This fix is based on the fix momentum command, and was created to be used in place of that command, when
a lattice-Boltzmann fluid is present.
Zero the total linear momentum of the system, including both the atoms specified by group-ID and the
lattice-Boltzmann fluid every nevery timesteps. This is accomplished by adjusting the particle velocities and
the fluid velocities at each lattice site.
NOTE: This fix only considers the linear momentum of the system.
By default, the subtraction is performed for each dimension. This can be changed by specifying the keyword
linear, along with a set of three flags set to 0/1 in order to exclude/ include the corresponding dimension.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
Can only be used if a lattice-Boltzmann fluid has been created via the fix lb/fluid command, and must come
after this command.
This fix is part of the USER-LB package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
LAMMPS Users Manual
888

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix lb/pc command
Syntax:
fix ID group-ID lb/pc
ID, group-ID are documented in the fix command• lb/pc = style name of this fix command•
Examples:
fix 1 all lb/pc
Description:
Update the positions and velocities of the individual particles described by group-ID, experiencing
velocity-dependent hydrodynamic forces, using the integration algorithm described in Mackay et al.. This
integration algorithm should only be used if a user-specified value for the force-coupling constant used in fix
lb/fluid has been set; do not use this integration algorithm if the force coupling constant has been set by
default.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the USER-LB package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Can only be used if a lattice-Boltzmann fluid has been created via the fix lb/fluid command, and must come
after this command.
Related commands:
fix lb/fluid fix lb/rigid/pc/sphere
Default: None.
(Mackay et al.) Mackay, F. E., Ollila, S.T.T., and Denniston, C., Hydrodynamic Forces Implemented into
LAMMPS through a lattice-Boltzmann fluid, Computer Physics Communications 184 (2013) 2021-2031.
LAMMPS Users Manual
890

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix lb/rigid/pc/sphere command
Syntax:
fix ID group-ID lb/rigid/pc/sphere bodystyle args keyword values ...
ID, group-ID are documented in fix command• lb/rigid/pc/sphere = style name of this fix command• bodystyle = single or molecule or group
single args = none
molecule args = none
group args = N groupID1 groupID2 ...
N = # of groups
•
zero or more keyword/value pairs may be appended• keyword = force or torque or innerNodes
force values = M xflag yflag zflag
M = which rigid body from 1-Nbody (see asterisk form below)
xflag,yflag,zflag = off/on if component of center-of-mass force is active
torque values = M xflag yflag zflag
M = which rigid body from 1-Nbody (see asterisk form below)
xflag,yflag,zflag = off/on if component of center-of-mass torque is active
innerNodes values = innergroup-ID
innergroup-ID = ID of the atom group which does not experience a hydrodynamic force from the lattice-Boltzmann fluid
•
Examples:
fix 1 spheres lb/rigid/pc/sphere
fix 1 all lb/rigid/pc/sphere force 1 0 0 innerNodes ForceAtoms
Description:
This fix is based on the fix rigid command, and was created to be used in place of that fix, to integrate the
equations of motion of spherical rigid bodies when a lattice-Boltzmann fluid is present with a user-specified
value of the force-coupling constant. The fix uses the integration algorithm described in Mackay et al. to
update the positions, velocities, and orientations of a set of spherical rigid bodies experiencing velocity
dependent hydrodynamic forces. The spherical bodies are assumed to rotate as solid, uniform density spheres,
with moments of inertia calculated using the combined sum of the masses of all the constituent particles
(which are assumed to be point particles).
By default, all of the atoms that this fix acts on experience a hydrodynamic force due to the presence of the
lattice-Boltzmann fluid. However, the innerNodes keyword allows the user to specify atoms belonging to a
rigid object which do not interact with the lattice-Boltzmann fluid (i.e. these atoms do not feel a
hydrodynamic force from the lattice-Boltzmann fluid). This can be used to distinguish between atoms on the
surface of a non-porous object, and those on the inside.
This feature can be used, for example, when implementing a hard sphere interaction between two spherical
objects. Instead of interactions occurring between the particles on the surfaces of the two spheres, it is
desirable simply to place an atom at the center of each sphere, which does not contribute to the hydrodynamic
force, and have these central atoms interact with one another.
LAMMPS Users Manual
891

Apart from the features described above, this fix is very similar to the rigid fix (although it includes fewer
optional arguments, and assumes the constituent atoms are point particles); see fix rigid for a complete
documentation.
Restart, fix_modify, output, run start/stop, minimize info:
No information about the rigid and rigid/nve fixes are written to binary restart files.
Similar to the fix rigid command: The rigid fix computes a global scalar which can be accessed by various
output commands. The scalar value calculated by these fixes is "intensive". The scalar is the current
temperature of the collection of rigid bodies. This is averaged over all rigid bodies and their translational and
rotational degrees of freedom. The translational energy of a rigid body is 1/2 m v^2, where m = total mass of
the body and v = the velocity of its center of mass. The rotational energy of a rigid body is 1/2 I w^2, where I
= the moment of inertia tensor of the body and w = its angular velocity. Degrees of freedom constrained by
the force and torque keywords are removed from this calculation.
All of these fixes compute a global array of values which can be accessed by various output commands. The
number of rows in the array is equal to the number of rigid bodies. The number of columns is 15. Thus for
each rigid body, 15 values are stored: the xyz coords of the center of mass (COM), the xyz components of the
COM velocity, the xyz components of the force acting on the COM, the xyz components of the torque acting
on the COM, and the xyz image flags of the COM, which have the same meaning as image flags for atom
positions (see the "dump" command). The force and torque values in the array are not affected by the force
and torque keywords in the fix rigid command; they reflect values before any changes are made by those
keywords.
The ordering of the rigid bodies (by row in the array) is as follows. For the single keyword there is just one
rigid body. For the molecule keyword, the bodies are ordered by ascending molecule ID. For the group
keyword, the list of group IDs determines the ordering of bodies.
The array values calculated by these fixes are "intensive", meaning they are independent of the number of
atoms in the simulation.
No parameter of these fixes can be used with the start/stop keywords of the run command. These fixes are not
invoked during energy minimization.
Restrictions:
This fix is part of the USER-LB package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Can only be used if a lattice-Boltzmann fluid has been created via the fix lb/fluid command, and must come
after this command. Should only be used if the force coupling constant used in fix lb/fluid has been set by the
user; this integration fix cannot be used if the force coupling constant is set by default.
Related commands:
fix lb/fluid, fix lb/pc
Default:
LAMMPS Users Manual
892

The defaults are force * on on on, and torque * on on on.
(Mackay et al.) Mackay, F. E., Ollila, S.T.T., and Denniston, C., Hydrodynamic Forces Implemented into
LAMMPS through a lattice-Boltzmann fluid, Computer Physics Communications 184 (2013) 2021-2031.
LAMMPS Users Manual
893

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix lb/viscous command
Syntax:
fix ID group-ID lb/viscous
ID, group-ID are documented in fix command• lb/viscous = style name of this fix command•
Examples:
fix 1 flow lb/viscous
Description:
This fix is similar to the fix viscous command, and is to be used in place of that command when a
lattice-Boltzmann fluid is present, and the user wishes to integrate the particle motion using one of the built in
LAMMPS integrators.
This fix adds a force, F = - Gamma*(velocity-fluid_velocity), to each atom, where Gamma is the force
coupling constant described in the fix lb/fluid command (which applies an equal and opposite force to the
fluid).
NOTE: This fix should only be used in conjunction with one of the built in LAMMPS integrators; it should
not be used with the fix lb/pc or fix lb/rigid/pc/sphere integrators, which already include the hydrodynamic
forces. These latter fixes should only be used if the force coupling constant has been set by the user (instead of
using the default value); if the default force coupling value is used, then this fix provides the only method for
adding the hydrodynamic forces to the particles.
For further details, as well as descriptions and results of several test runs, see Mackay et al.. Please include a
citation to this paper if this fix is used in work contributing to published research.
Restart, fix_modify, output, run start/stop, minimize info:
As described in the fix viscous documentation:
"No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
This fix should only be used with damped dynamics minimizers that allow for non-conservative forces. See
the min_style command for details."
Restrictions:
This fix is part of the USER-LB package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
LAMMPS Users Manual
894

Can only be used if a lattice-Boltzmann fluid has been created via the fix lb/fluid command, and must come
after this command.
This fix should not be used if either the fix lb/pc or fix lb/rigid/pc/sphere integrator is used.
Related commands:
fix lb/fluid, fix lb/pc, fix lb/rigid/pc/sphere
Default: none
(Mackay et al.) Mackay, F. E., Ollila, S.T.T., and Denniston, C., Hydrodynamic Forces Implemented into
LAMMPS through a lattice-Boltzmann fluid, Computer Physics Communications 184 (2013) 2021-2031.
LAMMPS Users Manual
895

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix lineforce command
Syntax:
fix ID group-ID lineforce x y z
ID, group-ID are documented in fix command• lineforce = style name of this fix command• x y z = direction of line as a 3-vector•
Examples:
fix hold boundary lineforce 0.0 1.0 1.0
Description:
Adjust the forces on each atom in the group so that only the component of force along the linear direction
specified by the vector (x,y,z) remains. This is done by subtracting out components of force in the plane
perpendicular to the line.
If the initial velocity of the atom is 0.0 (or along the line), then it should continue to move along the line
thereafter.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
Restrictions: none
Related commands:
fix planeforce
Default: none
LAMMPS Users Manual
896

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix manifoldforce command
Syntax:
fix ID group-ID manifoldforce manifold manifold-args ...
ID, group-ID are documented in fix command• manifold = name of the manifold• manifold-args = parameters for the manifold•
Examples:
fix constrain all manifoldforce sphere 5.0
Description:
This fix subtracts each time step from the force the component along the normal of the specified manifold.
This can be used in combination with minimize to remove overlap between particles while keeping them
(roughly) constrained to the given manifold, e.g. to set up a run with fix nve/manifold/rattle. I have found that
only hftn and quickmin with a very small time step perform adequately though.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is invoked during
energy minimization.
Restrictions:
This fix is part of the USER-MANIFOLD package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Only use this with min_style hftn or min_style quickmin. If not, the constraints will not be satisfied very well
at all. A warning is generated if the min_style is incompatible but no error.
Related commands:
fix nve/manifold/rattle, fix nvt/manifold/rattle
LAMMPS Users Manual
897

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix meso command
Syntax:
fix ID group-ID meso
ID, group-ID are documented in fix command• meso = style name of this fix command•
Examples:
fix 1 all meso
Description:
Perform time integration to update position, velocity, internal energy and local density for atoms in the group
each timestep. This fix is needed to time-integrate mesoscopic systems where particles carry internal variables
such as SPH or DPDE.
See this PDF guide to using SPH in LAMMPS.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the USER-SPH package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
"fix meso/stationary"
Default: none
LAMMPS Users Manual
898

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix meso/stationary command
Syntax:
fix ID group-ID meso/stationary
ID, group-ID are documented in fix command• meso = style name of this fix command•
Examples:
fix 1 boundary meso/stationary
Description:
Perform time integration to update internal energy and local density, but not position or velocity for atoms in
the group each timestep. This fix is needed for SPH simulations to correctly time-integrate fixed boundary
particles which constrain a fluid to a given region in space.
See this PDF guide to using SPH in LAMMPS.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the USER-SPH package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
"fix meso"
Default: none
LAMMPS Users Manual
899

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix momentum command
fix momentum/kk command
Syntax:
fix ID group-ID momentum N keyword values ...
ID, group-ID are documented in fix command• momentum = style name of this fix command• N = adjust the momentum every this many timesteps one or more keyword/value pairs may be
appended
•
keyword = linear or angular or rescale
linear values = xflag yflag zflag
xflag,yflag,zflag = 0/1 to exclude/include each dimension
angular values = none
rescale values = none
•
Examples:
fix 1 all momentum 1 linear 1 1 0
fix 1 all momentum 1 linear 1 1 1 rescale
fix 1 all momentum 100 linear 1 1 1 angular
Description:
Zero the linear and/or angular momentum of the group of atoms every N timesteps by adjusting the velocities
of the atoms. One (or both) of the linear or angular keywords must be specified.
If the linear keyword is used, the linear momentum is zeroed by subtracting the center-of-mass velocity of the
group from each atom. This does not change the relative velocity of any pair of atoms. One or more
dimensions can be excluded from this operation by setting the corresponding flag to 0.
If the angular keyword is used, the angular momentum is zeroed by subtracting a rotational component from
each atom.
This command can be used to insure the entire collection of atoms (or a subset of them) does not drift or rotate
during the simulation due to random perturbations (e.g. fix langevin thermostatting).
The rescale keyword enables conserving the kinetic energy of the group of atoms by rescaling the velocities
after the momentum was removed.
Note that the velocity command can be used to create initial velocities with zero aggregate linear and/or
angular momentum.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
LAMMPS Users Manual
900
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
fix recenter, velocity
Default: none
LAMMPS Users Manual
901

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix move command
Syntax:
fix ID group-ID move style args keyword values ...
ID, group-ID are documented in fix command• move = style name of this fix command• style = linear or wiggle or rotate or variable
linear args = Vx Vy Vz
Vx,Vy,Vz = components of velocity vector (velocity units), any component can be specified as NULL
wiggle args = Ax Ay Az period
Ax,Ay,Az = components of amplitude vector (distance units), any component can be specified as NULL
period = period of oscillation (time units)
rotate args = Px Py Pz Rx Ry Rz period
Px,Py,Pz = origin point of axis of rotation (distance units)
Rx,Ry,Rz = axis of rotation vector
period = period of rotation (time units)
variable args = v_dx v_dy v_dz v_vx v_vy v_vz
v_dx,v_dy,v_dz = 3 variable names that calculate x,y,z displacement as function of time, any component can be specified as NULL
v_vx,v_vy,v_vz = 3 variable names that calculate x,y,z velocity as function of time, any component can be specified as NULL
•
zero or more keyword/value pairs may be appended• keyword = units
units value = box or lattice
•
Examples:
fix 1 boundary move wiggle 3.0 0.0 0.0 1.0 units box
fix 2 boundary move rotate 0.0 0.0 0.0 0.0 0.0 1.0 5.0
fix 2 boundary move variable v_myx v_myy NULL v_VX v_VY NULL
Description:
Perform updates of position and velocity for atoms in the group each timestep using the specified settings or
formulas, without regard to forces on the atoms. This can be useful for boundary or other atoms, whose
movement can influence nearby atoms.
NOTE: The atoms affected by this fix should not normally be time integrated by other fixes (e.g. fix nve, fix
nvt), since that will change their positions and velocities twice.
NOTE: As atoms move due to this fix, they will pass thru periodic boundaries and be remapped to the other
side of the simulation box, just as they would during normal time integration (e.g. via the fix nve command).
It is up to you to decide whether periodic boundaries are appropriate with the kind of atom motion you are
prescribing with this fix.
NOTE: As discussed below, atoms are moved relative to their initial position at the time the fix is specified.
These initial coordinates are stored by the fix in "unwrapped" form, by using the image flags associated with
each atom. See the dump custom command for a discussion of "unwrapped" coordinates. See the Atoms
section of the read_data command for a discussion of image flags and how they are set for each atom. You
LAMMPS Users Manual
902

can reset the image flags (e.g. to 0) before invoking this fix by using the set image command.
The linear style moves atoms at a constant velocity, so that their position X = (x,y,z) as a function of time is
given in vector notation as
X(t) = X0 + V * delta
where X0 = (x0,y0,z0) is their position at the time the fix is specified, V is the specified velocity vector with
components (Vx,Vy,Vz), and delta is the time elapsed since the fix was specified. This style also sets the
velocity of each atom to V = (Vx,Vy,Vz). If any of the velocity components is specified as NULL, then the
position and velocity of that component is time integrated the same as the fix nve command would perform,
using the corresponding force component on the atom.
Note that the linear style is identical to using the variable style with an equal-style variable that uses the
vdisplace() function. E.g.
variable V equal 10.0
variable x equal vdisplace(0.0,$V)
fix 1 boundary move variable v_x NULL NULL v_V NULL NULL
The wiggle style moves atoms in an oscillatory fashion, so that their position X = (x,y,z) as a function of time
is given in vector notation as
X(t) = X0 + A sin(omega*delta)
where X0 = (x0,y0,z0) is their position at the time the fix is specified, A is the specified amplitude vector with
components (Ax,Ay,Az), omega is 2 PI / period, and delta is the time elapsed since the fix was specified. This
style also sets the velocity of each atom to the time derivative of this expression. If any of the amplitude
components is specified as NULL, then the position and velocity of that component is time integrated the
same as the fix nve command would perform, using the corresponding force component on the atom.
Note that the wiggle style is identical to using the variable style with equal-style variables that use the
swiggle() and cwiggle() functions. E.g.
variable A equal 10.0
variable T equal 5.0
variable omega equal 2.0*PI/$T
variable x equal swiggle(0.0,$A,$T)
variable v equal v_omega*($A-cwiggle(0.0,$A,$T))
fix 1 boundary move variable v_x NULL NULL v_v NULL NULL
The rotate style rotates atoms around a rotation axis R = (Rx,Ry,Rz) that goes thru a point P = (Px,Py,Pz).
The period of the rotation is also specified. The direction of rotation for the atoms around the rotation axis is
consistent with the right-hand rule: if your right-hand thumb points along R, then your fingers wrap around
the axis in the direction of rotation.
This style also sets the velocity of each atom to (omega cross Rperp) where omega is its angular velocity
around the rotation axis and Rperp is a perpendicular vector from the rotation axis to the atom. If the defined
atom_style assigns an angular velocity or angular momentum or orientation to each atom (atom styles sphere,
ellipsoid, line, tri, body), then those properties are also updated appropriately to correspond to the atom's
motion and rotation over time.
LAMMPS Users Manual
903

The variable style allows the position and velocity components of each atom to be set by formulas specified
via the variable command. Each of the 6 variables is specified as an argument to the fix as v_name, where
name is the variable name that is defined elsewhere in the input script.
Each variable must be of either the equal or atom style. Equal-style variables compute a single numeric
quantity, that can be a function of the timestep as well as of other simulation values. Atom-style variables
compute a numeric quantity for each atom, that can be a function per-atom quantities, such as the atom's
position, as well as of the timestep and other simulation values. Note that this fix stores the original
coordinates of each atom (see note below) so that per-atom quantity can be used in an atom-style variable
formula. See the variable command for details.
The first 3 variables (v_dx,v_dy,v_dz) specified for the variable style are used to calculate a displacement
from the atom's original position at the time the fix was specified. The second 3 variables (v_vx,v_vy,v_vz)
specified are used to compute a velocity for each atom.
Any of the 6 variables can be specified as NULL. If both the displacement and velocity variables for a
particular x,y,z component are specified as NULL, then the position and velocity of that component is time
integrated the same as the fix nve command would perform, using the corresponding force component on the
atom. If only the velocity variable for a component is specified as NULL, then the displacement variable will
be used to set the position of the atom, and its velocity component will not be changed. If only the
displacement variable for a component is specified as NULL, then the velocity variable will be used to set the
velocity of the atom, and the position of the atom will be time integrated using that velocity.
The units keyword determines the meaning of the distance units used to define the linear velocity and wiggle
amplitude and rotate origin. This setting is ignored for the variable style. A box value selects standard units as
defined by the units command, e.g. velocity in Angstroms/fmsec and amplitude and position in Angstroms for
units = real. A lattice value means the velocity units are in lattice spacings per time and the amplitude and
position are in lattice spacings. The lattice command must have been previously used to define the lattice
spacing. Each of these 3 quantities may be dependent on the x,y,z dimension, since the lattice spacings can be
different in x,y,z.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the original coordinates of moving atoms to binary restart files, as well as the initial timestep,
so that the motion can be continuous in a restarted simulation. See the read_restart command for info on how
to re-specify a fix in an input script that reads a restart file, so that the operation of the fix continues in an
uninterrupted fashion.
NOTE: Because the move positions are a function of the current timestep and the initial timestep, you cannot
reset the timestep to a different value after reading a restart file, if you expect a fix move command to work in
an uninterrupted fashion.
None of the fix_modify options are relevant to this fix.
This fix produces a per-atom array which can be accessed by various output commands. The number of
columns for each atom is 3, and the columns store the original unwrapped x,y,z coords of each atom. The
per-atom values can be accessed on any timestep.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
LAMMPS Users Manual
904

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix mscg command
Syntax:
fix ID group-ID mscg N keyword args ...
ID, group-ID are documented in fix command• mscg = style name of this fix command• N = envoke this fix every this many timesteps• zero or more keyword/value pairs may be appended• keyword = range or name or max
range arg = on or off
on = range finding functionality is performed
off = force matching functionality is performed
name args = name1 ... nameN
name1,...,nameN = string names for each atom type (1-Ntype)
max args = maxb maxa maxd
maxb,maxa,maxd = maximum bonds/angles/dihedrals per atom
•
Examples:
fix 1 all mscg 1
fix 1 all mscg 1 range name A B
fix 1 all mscg 1 max 4 8 20
Description:
This fix applies the Multi-Scale Coarse-Graining (MSCG) method to snapshots from a dump file to generate
potentials for coarse-grained simulations from all-atom simulations, using a force-matching technique
(Izvekov, Noid).
It makes use of the MS-CG library, written and maintained by Greg Voth's group at the University of
Chicago, which is freely available on their MS-CG GitHub site. See instructions on obtaining and installing
the MS-CG library in the src/MSCG/README file, which must be done before you build LAMMPS with this
fix command and use the command in a LAMMPS input script.
An example script using this fix is provided the examples/mscg directory.
The general workflow for using LAMMPS in conjunction with the MS-CG library to create a coarse-grained
model and run coarse-grained simulations is as follows:
Perform all-atom simulations on the system to be coarse grained.1. Generate a trajectory mapped to the coarse-grained model.2. Create input files for the MS-CG library.3. Run the range finder functionality of the MS-CG library.4. Run the force matching functionality of the MS-CG library.5. Check the results of the force matching.6. Run coarse-grained simulations using the new coarse-grained potentials.7.
LAMMPS Users Manual
906
This fix can perform the range finding and force matching steps 4 and 5 of the above workflow when used in
conjunction with the rerun command. It does not perform steps 1-3 and 6-7.
Step 2 can be performed using a Python script (what is the name?) provided with the MS-CG library which
defines the coarse-grained model and converts a standard LAMMPS dump file for an all-atom simulation
(step 1) into a LAMMPS dump file which has the positions of and forces on the coarse-grained beads.
In step 3, an input file named "control.in" is needed by the MS-CG library which sets parameters for the range
finding and force matching functionalities. See the examples/mscg/control.in file as an example. And see the
documentation provided with the MS-CG library for more info on this file.
When this fix is used to perform steps 4 and 5, the MS-CG library also produces additional output files. The
range finder functionality (step 4) outputs files defining pair and bonded interaction ranges. The force
matching functionality (step 5) outputs tabulated force files for every interaction in the system. Other
diagnostic files can also be output depending on the parameters in the MS-CG library input script. Again, see
the documentation provided with the MS-CG library for more info.
The range keyword specifies which MS-CG library functionality should be invoked. If on, the step 4 range
finder functionality is invoked. off, the step 5 force matching functionality is invoked.
If the name keyword is used, string names are defined to associate with the integer atom types in LAMMPS.
Ntype names must be provided, one for each atom type (1-Ntype).
The max keyword specifies the maximum number of bonds, angles, and dihedrals a bead can have in the
coarse-grained model.
Restrictions:
This fix is part of the MSCG package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
The MS-CG library uses C++11, which may not be supported by older compilers. The MS-CG library also
has some additional numeric library dependencies, which are described in its documentation.
Currently, the MS-CG library is not setup to run in parallel with MPI, so this fix can only be used in a serial
LAMMPS build and run on a single processor.
Related commands: none
Default:
The default keyword settings are range off, max 4 12 36.
(Izvekov) Izvekov, Voth, J Chem Phys 123, 134105 (2005).
(Noid) Noid, Chu, Ayton, Krishna, Izvekov, Voth, Das, Andersen, J Chem Phys 128, 134105 (2008).
LAMMPS Users Manual
907

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix msst command
Syntax:
fix ID group-ID msst dir shockvel keyword value ...
ID, group-ID are documented in fix command• msst = style name of this fix• dir = x or y or z• shockvel = shock velocity (strictly positive, distance/time units)• zero or more keyword value pairs may be appended• keyword = q or mu or p0 or v0 or e0 or tscale
q value = cell mass-like parameter (mass^2/distance^4 units)
mu value = artificial viscosity (mass/length/time units)
p0 value = initial pressure in the shock equations (pressure units)
v0 value = initial simulation cell volume in the shock equations (distance^3 units)
e0 value = initial total energy (energy units)
tscale value = reduction in initial temperature (unitless fraction between 0.0 and 1.0)
•
Examples:
fix 1 all msst y 100.0 q 1.0e5 mu 1.0e5
fix 2 all msst z 50.0 q 1.0e4 mu 1.0e4 v0 4.3419e+03 p0 3.7797e+03 e0 -9.72360e+02 tscale 0.01
Description:
This command performs the Multi-Scale Shock Technique (MSST) integration to update positions and
velocities each timestep to mimic a compressive shock wave passing over the system. See (Reed) for a
detailed description of this method. The MSST varies the cell volume and temperature in such a way as to
restrain the system to the shock Hugoniot and the Rayleigh line. These restraints correspond to the
macroscopic conservation laws dictated by a shock front. shockvel determines the steady shock velocity that
will be simulated.
To perform a simulation, choose a value of q that provides volume compression on the timescale of 100 fs to
1 ps. If the volume is not compressing, either the shock speed is chosen to be below the material sound speed
or p0 has been chosen inaccurately. Volume compression at the start can be sped up by using a non-zero value
of tscale. Use the smallest value of tscale that results in compression.
Under some special high-symmetry conditions, the pressure (volume) and/or temperature of the system may
oscillate for many cycles even with an appropriate choice of mass-like parameter q. Such oscillations have
physical significance in some cases. The optional mu keyword adds an artificial viscosity that helps break the
system symmetry to equilibrate to the shock Hugoniot and Rayleigh line more rapidly in such cases.
tscale is a factor between 0 and 1 that determines what fraction of thermal kinetic energy is converted to
compressive strain kinetic energy at the start of the simulation. Setting this parameter to a non-zero value may
assist in compression at the start of simulations where it is slow to occur.
If keywords e0, p0,or v0 are not supplied, these quantities will be calculated on the first step, after the energy
specified by tscale is removed. The value of e0 is not used in the dynamical equations, but is used in
LAMMPS Users Manual
908
calculating the deviation from the Hugoniot.
Values of shockvel less than a critical value determined by the material response will not have compressive
solutions. This will be reflected in lack of significant change of the volume in the MSST.
For all pressure styles, the simulation box stays orthogonal in shape. Parrinello-Rahman boundary conditions
(tilted box) are supported by LAMMPS, but are not implemented for MSST.
This fix computes a temperature and pressure each timestep. To do this, the fix creates its own computes of
style "temp" and "pressure", as if these commands had been issued:
compute fix-ID_temp group-ID temp
compute fix-ID_press group-ID pressure fix-ID_temp
See the compute temp and compute pressure commands for details. Note that the IDs of the new computes are
the fix-ID + underscore + "temp" or fix_ID + underscore + "press". The group for the new computes is "all".
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of all internal variables to binary restart files. See the read_restart command for info
on how to re-specify a fix in an input script that reads a restart file, so that the operation of the fix continues in
an uninterrupted fashion.
The progress of the MSST can be monitored by printing the global scalar and global vector quantities
computed by the fix.
The scalar is the cumulative energy change due to the fix. This is also the energy added to the potential energy
by the fix_modify energy command. With this command, the thermo keyword etotal prints the conserved
quantity of the MSST dynamic equations. This can be used to test if the MD timestep is sufficiently small for
accurate integration of the dynamic equations. See also thermo_style command.
The global vector contains four values in this order:
[dhugoniot, drayleigh, lagrangian_speed, lagrangian_position]
dhugoniot is the departure from the Hugoniot (temperature units).1. drayleigh is the departure from the Rayleigh line (pressure units).2. lagrangian_speed is the laboratory-frame Lagrangian speed (particle velocity) of the computational
cell (velocity units).
3.
lagrangian_position is the computational cell position in the reference frame moving at the shock
speed. This is usually a good estimate of distance of the computational cell behind the shock front.
4.
To print these quantities to the log file with descriptive column headers, the following LAMMPS commands
are suggested:
fix msst all msst z
fix_modify msst energy yes
variable dhug equal f_msst[1]
variable dray equal f_msst[2]
variable lgr_vel equal f_msst[3]
variable lgr_pos equal f_msst[4]
thermo_style custom step temp ke pe lz pzz etotal v_dhug v_dray v_lgr_vel v_lgr_pos f_msst
LAMMPS Users Manual
909

These fixes compute a global scalar and a global vector of 4 quantities, which can be accessed by various
output commands. The scalar values calculated by this fix are "extensive"; the vector values are "intensive".
Restrictions:
This fix style is part of the SHOCK package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
All cell dimensions must be periodic. This fix can not be used with a triclinic cell. The MSST fix has been
tested only for the group-ID all.
Related commands:
fix nphug, fix deform
Default:
The keyword defaults are q = 10, mu = 0, tscale = 0.01. p0, v0, and e0 are calculated on the first step.
(Reed) Reed, Fried, and Joannopoulos, Phys. Rev. Lett., 90, 235503 (2003).
LAMMPS Users Manual
910

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix neb command
Syntax:
fix ID group-ID neb Kspring
ID, group-ID are documented in fix command• neb = style name of this fix command• Kspring = inter-replica spring constant (force/distance units)•
Examples:
fix 1 active neb 10.0
Description:
Add inter-replica forces to atoms in the group for a multi-replica simulation run via the neb command to
perform a nudged elastic band (NEB) calculation for transition state finding. Hi-level explanations of NEB are
given with the neb command and in Section 6.5 of the manual. The fix neb command must be used with the
"neb" command to define how inter-replica forces are computed.
Only the N atoms in the fix group experience inter-replica forces. Atoms in the two end-point replicas do not
experience these forces, but those in intermediate replicas do. During the initial stage of NEB, the 3N-length
vector of interatomic forces Fi = -Grad(V) acting on the atoms of each intermediate replica I is altered, as
described in the (Henkelman1) paper, to become:
Fi = -Grad(V) + (Grad(V) dot That) That + Kspring (| Ri+i - Ri | - | Ri - Ri-1 |) That
Ri are the atomic coordinates of replica I; Ri-1 and Ri+1 are the coordinates of its neighbor replicas. That (t
with a hat over it) is the unit "tangent" vector for replica I which is a function of Ri, Ri-1, Ri+1, and the
potential energy of the 3 replicas; it points roughly in the direction of (Ri+i - Ri-1); see the (Henkelman1)
paper for details.
The first two terms in the above equation are the component of the interatomic forces perpendicular to the
tangent vector. The last term is a spring force between replica I and its neighbors, parallel to the tangent
vector direction with the specified spring constant Kspring.
The effect of the first two terms is to push the atoms of each replica toward the minimum energy path (MEP)
of conformational states that transition over the energy barrier. The MEP for an energy barrier is defined as a
sequence of 3N-dimensional states which cross the barrier at its saddle point, each of which has a potential
energy gradient parallel to the MEP itself.
The effect of the last term is to push each replica away from its two neighbors in a direction along the MEP,
so that the final set of states are equidistant from each other.
During the second stage of NEB, the forces on the N atoms in the replica nearest the top of the energy barrier
are altered so that it climbs to the top of the barrier and finds the saddle point. The forces on atoms in this
replica are described in the (Henkelman2) paper, and become:
LAMMPS Users Manual
911
Fi = -Grad(V) + 2 (Grad(V) dot That) That
The inter-replica forces for the other replicas are unchanged from the first equation.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, as invoked by the minimize command
via the neb command.
Restrictions:
This command can only be used if LAMMPS was built with the REPLICA package. See the Making
LAMMPS section for more info on packages.
Related commands:
neb
Default: none
(Henkelman1) Henkelman and Jonsson, J Chem Phys, 113, 9978-9985 (2000).
(Henkelman2) Henkelman, Uberuaga, Jonsson, J Chem Phys, 113, 9901-9904 (2000).
LAMMPS Users Manual
912

<LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nvt command
fix nvt/intel command
fix nvt/kk command
fix nvt/omp command
fix npt command
fix npt/intel command
fix npt/kk command
fix npt/omp command
fix nph command
fix nph/kk command
fix nph/omp command
Syntax:
fix ID group-ID style_name keyword value ...
ID, group-ID are documented in fix command• style_name = nvt or npt or nph• one or more keyword/value pairs may be appended
keyword = temp or iso or aniso or tri or x or y or z or xy or yz or xz or couple or tchain or pchain or mtk or tloop or ploop or nreset or drag or dilate or scalexy or scaleyz or scalexz or flip or fixedpoint or update
temp values = Tstart Tstop Tdamp
Tstart,Tstop = external temperature at start/end of run
Tdamp = temperature damping parameter (time units)
iso or aniso or tri values = Pstart Pstop Pdamp
Pstart,Pstop = scalar external pressure at start/end of run (pressure units)
Pdamp = pressure damping parameter (time units)
x or y or z or xy or yz or xz values = Pstart Pstop Pdamp
Pstart,Pstop = external stress tensor component at start/end of run (pressure units)
Pdamp = stress damping parameter (time units)
couple = none or xyz or xy or yz or xz
tchain value = N
N = length of thermostat chain (1 = single thermostat)
pchain values = N
N length of thermostat chain on barostat (0 = no thermostat)
mtk value = yes or no = add in MTK adjustment term or not
tloop value = M
M = number of sub-cycles to perform on thermostat
ploop value = M
M = number of sub-cycles to perform on barostat thermostat
•
LAMMPS Users Manual
913

nreset value = reset reference cell every this many timesteps
drag value = Df
Df = drag factor added to barostat/thermostat (0.0 = no drag)
dilate value = dilate-group-ID
dilate-group-ID = only dilate atoms in this group due to barostat volume changes
scalexy value = yes or no = scale xy with ly
scaleyz value = yes or no = scale yz with lz
scalexz value = yes or no = scale xz with lz
flip value = yes or no = allow or disallow box flips when it becomes highly skewed
fixedpoint values = x y z
x,y,z = perform barostat dilation/contraction around this point (distance units)
update value = dipole or dipole/dlm
dipole = update dipole orientation (only for sphere variants)
dipole/dlm = use DLM integrator to update dipole orientation (only for sphere variants)
Examples:
fix 1 all nvt temp 300.0 300.0 100.0
fix 1 water npt temp 300.0 300.0 100.0 iso 0.0 0.0 1000.0
fix 2 jello npt temp 300.0 300.0 100.0 tri 5.0 5.0 1000.0
fix 2 ice nph x 1.0 1.0 0.5 y 2.0 2.0 0.5 z 3.0 3.0 0.5 yz 0.1 0.1 0.5 xz 0.2 0.2 0.5 xy 0.3 0.3 0.5 nreset 1000
Description:
These commands perform time integration on Nose-Hoover style non-Hamiltonian equations of motion which
are designed to generate positions and velocities sampled from the canonical (nvt), isothermal-isobaric (npt),
and isenthalpic (nph) ensembles. This updates the position and velocity for atoms in the group each timestep.
The thermostatting and barostatting is achieved by adding some dynamic variables which are coupled to the
particle velocities (thermostatting) and simulation domain dimensions (barostatting). In addition to basic
thermostatting and barostatting, these fixes can also create a chain of thermostats coupled to the particle
thermostat, and another chain of thermostats coupled to the barostat variables. The barostat can be coupled to
the overall box volume, or to individual dimensions, including the xy, xz and yz tilt dimensions. The external
pressure of the barostat can be specified as either a scalar pressure (isobaric ensemble) or as components of a
symmetric stress tensor (constant stress ensemble). When used correctly, the time-averaged temperature and
stress tensor of the particles will match the target values specified by Tstart/Tstop and Pstart/Pstop.
The equations of motion used are those of Shinoda et al in (Shinoda), which combine the hydrostatic
equations of Martyna, Tobias and Klein in (Martyna) with the strain energy proposed by Parrinello and
Rahman in (Parrinello). The time integration schemes closely follow the time-reversible measure-preserving
Verlet and rRESPA integrators derived by Tuckerman et al in (Tuckerman).
The thermostat parameters for fix styles nvt and npt is specified using the temp keyword. Other
thermostat-related keywords are tchain, tloop and drag, which are discussed below.
The thermostat is applied to only the translational degrees of freedom for the particles. The translational
degrees of freedom can also have a bias velocity removed before thermostatting takes place; see the
description below. The desired temperature at each timestep is a ramped value during the run from Tstart to
Tstop. The Tdamp parameter is specified in time units and determines how rapidly the temperature is relaxed.
For example, a value of 10.0 means to relax the temperature in a timespan of (roughly) 10 time units (e.g. tau
or fmsec or psec - see the units command). The atoms in the fix group are the only ones whose velocities and
positions are updated by the velocity/position update portion of the integration.
LAMMPS Users Manual
914

NOTE: A Nose-Hoover thermostat will not work well for arbitrary values of Tdamp. If Tdamp is too small,
the temperature can fluctuate wildly; if it is too large, the temperature will take a very long time to equilibrate.
A good choice for many models is a Tdamp of around 100 timesteps. Note that this is NOT the same as 100
time units for most units settings.
The barostat parameters for fix styles npt and nph is specified using one or more of the iso, aniso, tri, x, y, z,
xy, xz, yz, and couple keywords. These keywords give you the ability to specify all 6 components of an
external stress tensor, and to couple various of these components together so that the dimensions they
represent are varied together during a constant-pressure simulation.
Other barostat-related keywords are pchain, mtk, ploop, nreset, drag, and dilate, which are discussed below.
Orthogonal simulation boxes have 3 adjustable dimensions (x,y,z). Triclinic (non-orthogonal) simulation
boxes have 6 adjustable dimensions (x,y,z,xy,xz,yz). The create_box, read data, and read_restart commands
specify whether the simulation box is orthogonal or non-orthogonal (triclinic) and explain the meaning of the
xy,xz,yz tilt factors.
The target pressures for each of the 6 components of the stress tensor can be specified independently via the x,
y, z, xy, xz, yz keywords, which correspond to the 6 simulation box dimensions. For each component, the
external pressure or tensor component at each timestep is a ramped value during the run from Pstart to Pstop.
If a target pressure is specified for a component, then the corresponding box dimension will change during a
simulation. For example, if the y keyword is used, the y-box length will change. If the xy keyword is used, the
xy tilt factor will change. A box dimension will not change if that component is not specified, although you
have the option to change that dimension via the fix deform command.
Note that in order to use the xy, xz, or yz keywords, the simulation box must be triclinic, even if its initial tilt
factors are 0.0.
For all barostat keywords, the Pdamp parameter operates like the Tdamp parameter, determining the time
scale on which pressure is relaxed. For example, a value of 10.0 means to relax the pressure in a timespan of
(roughly) 10 time units (e.g. tau or fmsec or psec - see the units command).
NOTE: A Nose-Hoover barostat will not work well for arbitrary values of Pdamp. If Pdamp is too small, the
pressure and volume can fluctuate wildly; if it is too large, the pressure will take a very long time to
equilibrate. A good choice for many models is a Pdamp of around 1000 timesteps. However, note that Pdamp
is specified in time units, and that timesteps are NOT the same as time units for most units settings.
Regardless of what atoms are in the fix group (the only atoms which are time integrated), a global pressure or
stress tensor is computed for all atoms. Similarly, when the size of the simulation box is changed, all atoms
are re-scaled to new positions, unless the keyword dilate is specified with a dilate-group-ID for a group that
represents a subset of the atoms. This can be useful, for example, to leave the coordinates of atoms in a solid
substrate unchanged and controlling the pressure of a surrounding fluid. This option should be used with care,
since it can be unphysical to dilate some atoms and not others, because it can introduce large, instantaneous
displacements between a pair of atoms (one dilated, one not) that are far from the dilation origin. Also note
that for atoms not in the fix group, a separate time integration fix like fix nve or fix nvt can be used on them,
independent of whether they are dilated or not.
The couple keyword allows two or three of the diagonal components of the pressure tensor to be "coupled"
together. The value specified with the keyword determines which are coupled. For example, xz means the Pxx
and Pzz components of the stress tensor are coupled. Xyz means all 3 diagonal components are coupled.
LAMMPS Users Manual
915
Coupling means two things: the instantaneous stress will be computed as an average of the corresponding
diagonal components, and the coupled box dimensions will be changed together in lockstep, meaning coupled
dimensions will be dilated or contracted by the same percentage every timestep. The Pstart, Pstop, Pdamp
parameters for any coupled dimensions must be identical. Couple xyz can be used for a 2d simulation; the z
dimension is simply ignored.
The iso, aniso, and tri keywords are simply shortcuts that are equivalent to specifying several other keywords
together.
The keyword iso means couple all 3 diagonal components together when pressure is computed (hydrostatic
pressure), and dilate/contract the dimensions together. Using "iso Pstart Pstop Pdamp" is the same as
specifying these 4 keywords:
x Pstart Pstop Pdamp
y Pstart Pstop Pdamp
z Pstart Pstop Pdamp
couple xyz
The keyword aniso means x, y, and z dimensions are controlled independently using the Pxx, Pyy, and Pzz
components of the stress tensor as the driving forces, and the specified scalar external pressure. Using "aniso
Pstart Pstop Pdamp" is the same as specifying these 4 keywords:
x Pstart Pstop Pdamp
y Pstart Pstop Pdamp
z Pstart Pstop Pdamp
couple none
The keyword tri means x, y, z, xy, xz, and yz dimensions are controlled independently using their individual
stress components as the driving forces, and the specified scalar pressure as the external normal stress. Using
"tri Pstart Pstop Pdamp" is the same as specifying these 7 keywords:
x Pstart Pstop Pdamp
y Pstart Pstop Pdamp
z Pstart Pstop Pdamp
xy 0.0 0.0 Pdamp
yz 0.0 0.0 Pdamp
xz 0.0 0.0 Pdamp
couple none
In some cases (e.g. for solids) the pressure (volume) and/or temperature of the system can oscillate
undesirably when a Nose/Hoover barostat and thermostat is applied. The optional drag keyword will damp
these oscillations, although it alters the Nose/Hoover equations. A value of 0.0 (no drag) leaves the
Nose/Hoover formalism unchanged. A non-zero value adds a drag term; the larger the value specified, the
greater the damping effect. Performing a short run and monitoring the pressure and temperature is the best
way to determine if the drag term is working. Typically a value between 0.2 to 2.0 is sufficient to damp
oscillations after a few periods. Note that use of the drag keyword will interfere with energy conservation and
will also change the distribution of positions and velocities so that they do not correspond to the nominal
NVT, NPT, or NPH ensembles.
An alternative way to control initial oscillations is to use chain thermostats. The keyword tchain determines
the number of thermostats in the particle thermostat. A value of 1 corresponds to the original Nose-Hoover
thermostat. The keyword pchain specifies the number of thermostats in the chain thermostatting the barostat
degrees of freedom. A value of 0 corresponds to no thermostatting of the barostat variables.
LAMMPS Users Manual
916

The mtk keyword controls whether or not the correction terms due to Martyna, Tuckerman, and Klein are
included in the equations of motion (Martyna). Specifying no reproduces the original Hoover barostat, whose
volume probability distribution function differs from the true NPT and NPH ensembles by a factor of 1/V.
Hence using yes is more correct, but in many cases the difference is negligible.
The keyword tloop can be used to improve the accuracy of integration scheme at little extra cost. The initial
and final updates of the thermostat variables are broken up into tloop substeps, each of length dt/tloop. This
corresponds to using a first-order Suzuki-Yoshida scheme (Tuckerman). The keyword ploop does the same
thing for the barostat thermostat.
The keyword nreset controls how often the reference dimensions used to define the strain energy are reset. If
this keyword is not used, or is given a value of zero, then the reference dimensions are set to those of the
initial simulation domain and are never changed. If the simulation domain changes significantly during the
simulation, then the final average pressure tensor will differ significantly from the specified values of the
external stress tensor. A value of nstep means that every nstep timesteps, the reference dimensions are set to
those of the current simulation domain.
The scaleyz, scalexz, and scalexy keywords control whether or not the corresponding tilt factors are scaled
with the associated box dimensions when barostatting triclinic periodic cells. The default values yes will turn
on scaling, which corresponds to adjusting the linear dimensions of the cell while preserving its shape.
Choosing no ensures that the tilt factors are not scaled with the box dimensions. See below for restrictions and
default values in different situations. In older versions of LAMMPS, scaling of tilt factors was not performed.
The old behavior can be recovered by setting all three scale keywords to no.
The flip keyword allows the tilt factors for a triclinic box to exceed half the distance of the parallel box length,
as discussed below. If the flip value is set to yes, the bound is enforced by flipping the box when it is
exceeded. If the flip value is set to no, the tilt will continue to change without flipping. Note that if applied
stress induces large deformations (e.g. in a liquid), this means the box shape can tilt dramatically and
LAMMPS will run less efficiently, due to the large volume of communication needed to acquire ghost atoms
around a processor's irregular-shaped sub-domain. For extreme values of tilt, LAMMPS may also lose atoms
and generate an error.
The fixedpoint keyword specifies the fixed point for barostat volume changes. By default, it is the center of
the box. Whatever point is chosen will not move during the simulation. For example, if the lower periodic
boundaries pass through (0,0,0), and this point is provided to fixedpoint, then the lower periodic boundaries
will remain at (0,0,0), while the upper periodic boundaries will move twice as far. In all cases, the particle
trajectories are unaffected by the chosen value, except for a time-dependent constant translation of positions.
If the update keyword is used with the dipole value, then the orientation of the dipole moment of each particle
is also updated during the time integration. This option should be used for models where a dipole moment is
assigned to finite-size particles, e.g. spheroids via use of the atom_style hybrid sphere dipole command.
The default dipole orientation integrator can be changed to the Dullweber-Leimkuhler-McLachlan integration
scheme (Dullweber) when using update with the value dipole/dlm. This integrator is symplectic and
time-reversible, giving better energy conservation and allows slightly longer timesteps at only a small
additional computational cost.
NOTE: Using a barostat coupled to tilt dimensions xy, xz, yz can sometimes result in arbitrarily large values of
the tilt dimensions, i.e. a dramatically deformed simulation box. LAMMPS allows the tilt factors to grow a
small amount beyond the normal limit of half the box length (0.6 times the box length), and then performs a
box "flip" to an equivalent periodic cell. See the discussion of the flip keyword above, to allow this bound to
LAMMPS Users Manual
917

be exceeded, if desired.
The flip operation is described in more detail in the doc page for fix deform. Both the barostat dynamics and
the atom trajectories are unaffected by this operation. However, if a tilt factor is incremented by a large
amount (1.5 times the box length) on a single timestep, LAMMPS can not accomodate this event and will
terminate the simulation with an error. This error typically indicates that there is something badly wrong with
how the simulation was constructed, such as specifying values of Pstart that are too far from the current stress
value, or specifying a timestep that is too large. Triclinic barostatting should be used with care. This also is
true for other barostat styles, although they tend to be more forgiving of insults. In particular, it is important to
recognize that equilibrium liquids can not support a shear stress and that equilibrium solids can not support
shear stresses that exceed the yield stress.
One exception to this rule is if the 1st dimension in the tilt factor (x for xy) is non-periodic. In that case, the
limits on the tilt factor are not enforced, since flipping the box in that dimension does not change the atom
positions due to non-periodicity. In this mode, if you tilt the system to extreme angles, the simulation will
simply become inefficient due to the highly skewed simulation box.
NOTE: Unlike the fix temp/berendsen command which performs thermostatting but NO time integration,
these fixes perform thermostatting/barostatting AND time integration. Thus you should not use any other time
integration fix, such as fix nve on atoms to which this fix is applied. Likewise, fix nvt and fix npt should not
normally be used on atoms that also have their temperature controlled by another fix - e.g. by fix langevin or
fix temp/rescale commands.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting and barostatting.
These fixes compute a temperature and pressure each timestep. To do this, the fix creates its own computes of
style "temp" and "pressure", as if one of these two sets of commands had been issued:
compute fix-ID_temp group-ID temp
compute fix-ID_press group-ID pressure fix-ID_temp
compute fix-ID_temp all temp
compute fix-ID_press all pressure fix-ID_temp
See the compute temp and compute pressure commands for details. Note that the IDs of the new computes are
the fix-ID + underscore + "temp" or fix_ID + underscore + "press". For fix nvt, the group for the new
computes is the same as the fix group. For fix nph and fix npt, the group for the new computes is "all" since
pressure is computed for the entire system.
Note that these are NOT the computes used by thermodynamic output (see the thermo_style command) with
ID = thermo_temp and thermo_press. This means you can change the attributes of this fix's temperature or
pressure via the compute_modify command or print this temperature or pressure during thermodynamic
output via the thermo_style custom command using the appropriate compute-ID. It also means that changing
attributes of thermo_temp or thermo_press will have no effect on this fix.
Like other fixes that perform thermostatting, fix nvt and fix npt can be used with compute commands that
calculate a temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass
velocity from a group of atoms or only calculating temperature on the x-component of velocity or only
calculating temperature for atoms in a geometric region. This is not done by default, but only if the
fix_modify command is used to assign a temperature compute to this fix that includes such a bias term. See
LAMMPS Users Manual
918

the doc pages for individual compute commands to determine which ones include a bias. In this case, the
thermostat works in the following manner: the current temperature is calculated taking the bias into account,
bias is removed from each atom, thermostatting is performed on the remaining thermal degrees of freedom,
and the bias is added back in.
These fixes can be used with either the verlet or respa integrators. When using one of the barostat fixes with
respa, LAMMPS uses an integrator constructed according to the following factorization of the Liouville
propagator (for two rRESPA levels):
This factorization differs somewhat from that of Tuckerman et al, in that the barostat is only updated at the
outermost rRESPA level, whereas Tuckerman's factorization requires splitting the pressure into pieces
corresponding to the forces computed at each rRESPA level. In theory, the latter method will exhibit better
numerical stability. In practice, because Pdamp is normally chosen to be a large multiple of the outermost
rRESPA timestep, the barostat dynamics are not the limiting factor for numerical stability. Both factorizations
are time-reversible and can be shown to preserve the phase space measure of the underlying non-Hamiltonian
equations of motion.
NOTE: This implementation has been shown to conserve linear momentum up to machine precision under
NVT dynamics. Under NPT dynamics, for a system with zero initial total linear momentum, the total
momentum fluctuates close to zero. It may occasionally undergo brief excursions to non-negligible values,
before returning close to zero. Over long simulations, this has the effect of causing the center-of-mass to
undergo a slow random walk. This can be mitigated by resetting the momentum at infrequent intervals using
the fix momentum command.
The fix npt and fix nph commands can be used with rigid bodies or mixtures of rigid bodies and non-rigid
particles (e.g. solvent). But there are also fix rigid/npt and fix rigid/nph commands, which are typically a more
natural choice. See the doc page for those commands for more discussion of the various ways to do this.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
LAMMPS Users Manual
919

section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
These fixes writes the state of all the thermostat and barostat variables to binary restart files. See the
read_restart command for info on how to re-specify a fix in an input script that reads a restart file, so that the
operation of the fix continues in an uninterrupted fashion.
The fix_modify temp and press options are supported by these fixes. You can use them to assign a compute
you have defined to this fix which will be used in its thermostatting or barostatting procedure, as described
above. If you do this, note that the kinetic energy derived from the compute temperature should be consistent
with the virial term computed using all atoms for the pressure. LAMMPS will warn you if you choose to
compute temperature on a subset of atoms.
NOTE: If both the temp and press keywords are used in a single thermo_modify command (or in two separate
commands), then the order in which the keywords are specified is important. Note that a pressure compute
defines its own temperature compute as an argument when it is specified. The temp keyword will override this
(for the pressure compute being used by fix npt), but only if the temp keyword comes after the press keyword.
If the temp keyword comes before the press keyword, then the new pressure compute specified by the press
keyword will be unaffected by the temp setting.
The fix_modify energy option is supported by these fixes to add the energy change induced by Nose/Hoover
thermostatting and barostatting to the system's potential energy as part of thermodynamic output.
These fixes compute a global scalar and a global vector of quantities, which can be accessed by various output
commands. The scalar value calculated by these fixes is "extensive"; the vector values are "intensive".
The scalar is the cumulative energy change due to the fix.
The vector stores internal Nose/Hoover thermostat and barostat variables. The number and meaning of the
vector values depends on which fix is used and the settings for keywords tchain and pchain, which specify the
number of Nose/Hoover chains for the thermostat and barostat. If no thermostatting is done, then tchain is 0.
If no barostatting is done, then pchain is 0. In the following list, "ndof" is 0, 1, 3, or 6, and is the number of
degrees of freedom in the barostat. Its value is 0 if no barostat is used, else its value is 6 if any off-diagonal
stress tensor component is barostatted, else its value is 1 if couple xyz is used or couple xy for a 2d simulation,
otherwise its value is 3.
The order of values in the global vector and their meaning is as follows. The notation means there are tchain
values for eta, followed by tchain for eta_dot, followed by ndof for omega, etc:
eta[tchain] = particle thermostat displacements (unitless)• eta_dot[tchain] = particle thermostat velocities (1/time units)• omega[ndof] = barostat displacements (unitless)• omega_dot[ndof] = barostat velocities (1/time units)• etap[pchain] = barostat thermostat displacements (unitless)•
LAMMPS Users Manual
920
etap_dot[pchain] = barostat thermostat velocities (1/time units)• PE_eta[tchain] = potential energy of each particle thermostat displacement (energy units)• KE_eta_dot[tchain] = kinetic energy of each particle thermostat velocity (energy units)• PE_omega[ndof] = potential energy of each barostat displacement (energy units)• KE_omega_dot[ndof] = kinetic energy of each barostat velocity (energy units)• PE_etap[pchain] = potential energy of each barostat thermostat displacement (energy units)• KE_etap_dot[pchain] = kinetic energy of each barostat thermostat velocity (energy units)• PE_strain[1] = scalar strain energy (energy units)•
These fixes can ramp their external temperature and pressure over multiple runs, using the start and stop
keywords of the run command. See the run command for details of how to do this.
These fixes are not invoked during energy minimization.
Restrictions:
X, y, z cannot be barostatted if the associated dimension is not periodic. Xy, xz, and yz can only be barostatted
if the simulation domain is triclinic and the 2nd dimension in the keyword (y dimension in xy) is periodic. Z,
xz, and yz, cannot be barostatted for 2D simulations. The create_box, read data, and read_restart commands
specify whether the simulation box is orthogonal or non-orthogonal (triclinic) and explain the meaning of the
xy,xz,yz tilt factors.
For the temp keyword, the final Tstop cannot be 0.0 since it would make the external T = 0.0 at some timestep
during the simulation which is not allowed in the Nose/Hoover formulation.
The scaleyz yes and scalexz yes keyword/value pairs can not be used for 2D simulations. scaleyz yes, scalexz
yes, and scalexy yes options can only be used if the 2nd dimension in the keyword is periodic, and if the tilt
factor is not coupled to the barostat via keywords tri, yz, xz, and xy.
These fixes can be used with dynamic groups as defined by the group command. Likewise they can be used
with groups to which atoms are added or deleted over time, e.g. a deposition simulation. However, the
conservation properties of the thermostat and barostat are defined for systems with a static set of atoms. You
may observe odd behavior if the atoms in a group vary dramatically over time or the atom count becomes very
small.
Related commands:
fix nve, fix_modify, run_style
Default:
The keyword defaults are tchain = 3, pchain = 3, mtk = yes, tloop = ploop = 1, nreset = 0, drag = 0.0, dilate =
all, couple = none, scaleyz = scalexz = scalexy = yes if periodic in 2nd dimension and not coupled to barostat,
otherwise no.
(Martyna) Martyna, Tobias and Klein, J Chem Phys, 101, 4177 (1994).
(Parrinello) Parrinello and Rahman, J Appl Phys, 52, 7182 (1981).
LAMMPS Users Manual
921
(Tuckerman) Tuckerman, Alejandre, Lopez-Rendon, Jochim, and Martyna, J Phys A: Math Gen, 39, 5629
(2006).
(Shinoda) Shinoda, Shiga, and Mikami, Phys Rev B, 69, 134103 (2004).
(Dullweber) Dullweber, Leimkuhler and McLachlan, J Chem Phys, 107, 5840 (1997).
LAMMPS Users Manual
922

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nvt/eff command
fix npt/eff command
fix nph/eff command
Syntax:
fix ID group-ID style_name keyword value ...
ID, group-ID are documented in fix command• style_name = nvt/eff or npt/eff or nph/eff
one or more keyword value pairs may be appended
keyword = temp or iso or aniso or tri or x or y or z or xy or yz or xz or couple or tchain or pchain or mtk or tloop or ploop or nreset or drag or dilate
temp values = Tstart Tstop Tdamp
Tstart,Tstop = external temperature at start/end of run
Tdamp = temperature damping parameter (time units)
iso or aniso or tri values = Pstart Pstop Pdamp
Pstart,Pstop = scalar external pressure at start/end of run (pressure units)
Pdamp = pressure damping parameter (time units)
x or y or z or xy or yz or xz values = Pstart Pstop Pdamp
Pstart,Pstop = external stress tensor component at start/end of run (pressure units)
Pdamp = stress damping parameter (time units)
couple = none or xyz or xy or yz or xz
tchain value = length of thermostat chain (1 = single thermostat)
pchain values = length of thermostat chain on barostat (0 = no thermostat)
mtk value = yes or no = add in MTK adjustment term or not
tloop value = number of sub-cycles to perform on thermostat
ploop value = number of sub-cycles to perform on barostat thermostat
nreset value = reset reference cell every this many timesteps
drag value = drag factor added to barostat/thermostat (0.0 = no drag)
dilate value = all or partial
•
Examples:
fix 1 all nvt/eff temp 300.0 300.0 0.1
fix 1 part npt/eff temp 300.0 300.0 0.1 iso 0.0 0.0 1.0
fix 2 part npt/eff temp 300.0 300.0 0.1 tri 5.0 5.0 1.0
fix 2 ice nph/eff x 1.0 1.0 0.5 y 2.0 2.0 0.5 z 3.0 3.0 0.5 yz 0.1 0.1 0.5 xz 0.2 0.2 0.5 xy 0.3 0.3 0.5 nreset 1000
Description:
These commands perform time integration on Nose-Hoover style non-Hamiltonian equations of motion for
nuclei and electrons in the group for the electron force field model. The fixes are designed to generate
positions and velocities sampled from the canonical (nvt), isothermal-isobaric (npt), and isenthalpic (nph)
ensembles. This is achieved by adding some dynamic variables which are coupled to the particle velocities
(thermostatting) and simulation domain dimensions (barostatting). In addition to basic thermostatting and
barostatting, these fixes can also create a chain of thermostats coupled to the particle thermostat, and another
chain of thermostats coupled to the barostat variables. The barostat can be coupled to the overall box volume,
or to individual dimensions, including the xy, xz and yz tilt dimensions. The external pressure of the barostat
can be specified as either a scalar pressure (isobaric ensemble) or as components of a symmetric stress tensor
LAMMPS Users Manual
923

(constant stress ensemble). When used correctly, the time-averaged temperature and stress tensor of the
particles will match the target values specified by Tstart/Tstop and Pstart/Pstop.
The operation of these fixes is exactly like that described by the fix nvt, npt, and nph commands, except that
the radius and radial velocity of electrons are also updated. Likewise the temperature and pressure calculated
by the fix, using the computes it creates (as discussed in the fix nvt, npt, and nph doc page), are performed
with computes that include the eFF contribution to the temperature or kinetic energy from the electron radial
velocity.
NOTE: there are two different pressures that can be reported for eFF when defining the pair_style (see pair
eff/cut to understand these settings), one (default) that considers electrons do not contribute radial virial
components (i.e. electrons treated as incompressible 'rigid' spheres) and one that does. The radial electronic
contributions to the virials are only tallied if the flexible pressure option is set, and this will affect both global
and per-atom quantities. In principle, the true pressure of a system is somewhere in between the rigid and the
flexible eFF pressures, but, for most cases, the difference between these two pressures will not be significant
over long-term averaged runs (i.e. even though the energy partitioning changes, the total energy remains
similar).
NOTE: currently, there is no available option for the user to set or create temperature distributions that include
the radial electronic degrees of freedom with the velocity command, so the the user must allow for these
degrees of freedom to equilibrate (i.e. equi-partitioning of energy) through time integration.
Restart, fix_modify, output, run start/stop, minimize info:
See the doc page for the fix nvt, npt, and nph commands for details.
Restrictions:
This fix is part of the USER-EFF package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Other restriction discussed on the doc page for the fix nvt, npt, and nph commands also apply.
NOTE: The temperature for systems (regions or groups) with only electrons and no nuclei is 0.0 (i.e. not
defined) in the current temperature calculations, a practical example would be a uniform electron gas or a very
hot plasma, where electrons remain delocalized from the nuclei. This is because, even though electron virials
are included in the temperature calculation, these are averaged over the nuclear degrees of freedom only. In
such cases a corrective term must be added to the pressure to get the correct kinetic contribution.
Related commands:
fix nvt, fix nph, fix npt, fix_modify, run_style
Default:
The keyword defaults are tchain = 3, pchain = 3, mtk = yes, tloop = ploop = 1, nreset = 0, drag = 0.0, dilate =
all, and couple = none.
(Martyna) Martyna, Tobias and Klein, J Chem Phys, 101, 4177 (1994).
LAMMPS Users Manual
924
(Parrinello) Parrinello and Rahman, J Appl Phys, 52, 7182 (1981).
(Tuckerman) Tuckerman, Alejandre, Lopez-Rendon, Jochim, and Martyna, J Phys A: Math Gen, 39, 5629
(2006).
(Shinoda) Shinoda, Shiga, and Mikami, Phys Rev B, 69, 134103 (2004).
LAMMPS Users Manual
925

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nph/asphere command
fix nph/asphere/omp command
Syntax:
fix ID group-ID nph/asphere args keyword value ...
ID, group-ID are documented in fix command• nph/asphere = style name of this fix command• additional barostat related keyword/value pairs from the fix nph command can be appended•
Examples:
fix 1 all nph/asphere iso 0.0 0.0 1000.0
fix 2 all nph/asphere x 5.0 5.0 1000.0
fix 2 all nph/asphere x 5.0 5.0 1000.0 drag 0.2
fix 2 water nph/asphere aniso 0.0 0.0 1000.0 dilate partial
Description:
Perform constant NPH integration to update position, velocity, orientation, and angular velocity each timestep
for aspherical or ellipsoidal particles in the group using a Nose/Hoover pressure barostat. P is pressure; H is
enthalpy. This creates a system trajectory consistent with the isenthalpic ensemble.
This fix differs from the fix nph command, which assumes point particles and only updates their position and
velocity.
Additional parameters affecting the barostat are specified by keywords and values documented with the fix
nph command. See, for example, discussion of the aniso, and dilate keywords.
The particles in the fix group are the only ones whose velocities and positions are updated by the
velocity/position update portion of the NPH integration.
Regardless of what particles are in the fix group, a global pressure is computed for all particles. Similarly,
when the size of the simulation box is changed, all particles are re-scaled to new positions, unless the keyword
dilate is specified with a value of partial, in which case only the particles in the fix group are re-scaled. The
latter can be useful for leaving the coordinates of particles in a solid substrate unchanged and controlling the
pressure of a surrounding fluid.
This fix computes a temperature and pressure each timestep. To do this, the fix creates its own computes of
style "temp/asphere" and "pressure", as if these commands had been issued:
compute fix-ID_temp all temp/asphere
compute fix-ID_press all pressure fix-ID_temp
See the compute temp/asphere and compute pressure commands for details. Note that the IDs of the new
computes are the fix-ID + underscore + "temp" or fix_ID + underscore + "press", and the group for the new
computes is "all" since pressure is computed for the entire system.
LAMMPS Users Manual
926

Note that these are NOT the computes used by thermodynamic output (see the thermo_style command) with
ID = thermo_temp and thermo_press. This means you can change the attributes of this fix's temperature or
pressure via the compute_modify command or print this temperature or pressure during thermodynamic
output via the thermo_style custom command using the appropriate compute-ID. It also means that changing
attributes of thermo_temp or thermo_press will have no effect on this fix.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover barostat to binary restart files. See the read_restart command for
info on how to re-specify a fix in an input script that reads a restart file, so that the operation of the fix
continues in an uninterrupted fashion.
The fix_modify temp and press options are supported by this fix. You can use them to assign a compute you
have defined to this fix which will be used in its thermostatting or barostatting procedure. If you do this, note
that the kinetic energy derived from the compute temperature should be consistent with the virial term
computed using all atoms for the pressure. LAMMPS will warn you if you choose to compute temperature on
a subset of atoms.
The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
barostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix nph command.
This fix can ramp its target pressure over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that atoms store torque and angular momentum and a quaternion as defined by the
atom_style ellipsoid command.
LAMMPS Users Manual
927

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nph/body command
Syntax:
fix ID group-ID nph/body args keyword value ...
ID, group-ID are documented in fix command• nph/body = style name of this fix command• additional barostat related keyword/value pairs from the fix nph command can be appended•
Examples:
fix 1 all nph/body iso 0.0 0.0 1000.0
fix 2 all nph/body x 5.0 5.0 1000.0
fix 2 all nph/body x 5.0 5.0 1000.0 drag 0.2
fix 2 water nph/body aniso 0.0 0.0 1000.0 dilate partial
Description:
Perform constant NPH integration to update position, velocity, orientation, and angular velocity each timestep
for body particles in the group using a Nose/Hoover pressure barostat. P is pressure; H is enthalpy. This
creates a system trajectory consistent with the isenthalpic ensemble.
This fix differs from the fix nph command, which assumes point particles and only updates their position and
velocity.
Additional parameters affecting the barostat are specified by keywords and values documented with the fix
nph command. See, for example, discussion of the aniso, and dilate keywords.
The particles in the fix group are the only ones whose velocities and positions are updated by the
velocity/position update portion of the NPH integration.
Regardless of what particles are in the fix group, a global pressure is computed for all particles. Similarly,
when the size of the simulation box is changed, all particles are re-scaled to new positions, unless the keyword
dilate is specified with a value of partial, in which case only the particles in the fix group are re-scaled. The
latter can be useful for leaving the coordinates of particles in a solid substrate unchanged and controlling the
pressure of a surrounding fluid.
This fix computes a temperature and pressure each timestep. To do this, the fix creates its own computes of
style "temp/body" and "pressure", as if these commands had been issued:
compute fix-ID_temp all temp/body
compute fix-ID_press all pressure fix-ID_temp
See the compute temp/body and compute pressure commands for details. Note that the IDs of the new
computes are the fix-ID + underscore + "temp" or fix_ID + underscore + "press", and the group for the new
computes is "all" since pressure is computed for the entire system.
Note that these are NOT the computes used by thermodynamic output (see the thermo_style command) with
LAMMPS Users Manual
929

ID = thermo_temp and thermo_press. This means you can change the attributes of this fix's temperature or
pressure via the compute_modify command or print this temperature or pressure during thermodynamic
output via the thermo_style custom command using the appropriate compute-ID. It also means that changing
attributes of thermo_temp or thermo_press will have no effect on this fix.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover barostat to binary restart files. See the read_restart command for
info on how to re-specify a fix in an input script that reads a restart file, so that the operation of the fix
continues in an uninterrupted fashion.
The fix_modify temp and press options are supported by this fix. You can use them to assign a compute you
have defined to this fix which will be used in its thermostatting or barostatting procedure. If you do this, note
that the kinetic energy derived from the compute temperature should be consistent with the virial term
computed using all atoms for the pressure. LAMMPS will warn you if you choose to compute temperature on
a subset of atoms.
The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
barostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix nph command.
This fix can ramp its target pressure over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the BODY package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that atoms store torque and angular momentum and a quaternion as defined by the
atom_style body command.
Related commands:
LAMMPS Users Manual
930

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nph/sphere command
fix nph/sphere/omp command
Syntax:
fix ID group-ID nph/sphere args keyword value ...
ID, group-ID are documented in fix command• nph/sphere = style name of this fix command• keyword = disc
disc value = none = treat particles as 2d discs, not spheres
•
additional barostat related keyword/value pairs from the fix nph command can be appended•
Examples:
fix 1 all nph/sphere iso 0.0 0.0 1000.0
fix 2 all nph/sphere x 5.0 5.0 1000.0
fix 2 all nph/sphere x 5.0 5.0 1000.0 disc
fix 2 all nph/sphere x 5.0 5.0 1000.0 drag 0.2
fix 2 water nph/sphere aniso 0.0 0.0 1000.0 dilate partial
Description:
Perform constant NPH integration to update position, velocity, and angular velocity each timestep for
finite-size spherical particles in the group using a Nose/Hoover pressure barostat. P is pressure; H is enthalpy.
This creates a system trajectory consistent with the isenthalpic ensemble.
This fix differs from the fix nph command, which assumes point particles and only updates their position and
velocity.
If the disc keyword is used, then each particle is treated as a 2d disc (circle) instead of as a sphere. This is only
possible for 2d simulations, as defined by the dimension keyword. The only difference between discs and
spheres in this context is their moment of inertia, as used in the time integration.
Additional parameters affecting the barostat are specified by keywords and values documented with the fix
nph command. See, for example, discussion of the aniso, and dilate keywords.
The particles in the fix group are the only ones whose velocities and positions are updated by the
velocity/position update portion of the NPH integration.
Regardless of what particles are in the fix group, a global pressure is computed for all particles. Similarly,
when the size of the simulation box is changed, all particles are re-scaled to new positions, unless the keyword
dilate is specified with a value of partial, in which case only the particles in the fix group are re-scaled. The
latter can be useful for leaving the coordinates of particles in a solid substrate unchanged and controlling the
pressure of a surrounding fluid.
LAMMPS Users Manual
932

This fix computes a temperature and pressure each timestep. To do this, the fix creates its own computes of
style "temp/sphere" and "pressure", as if these commands had been issued:
compute fix-ID_temp all temp/sphere
compute fix-ID_press all pressure fix-ID_temp
See the compute temp/sphere and compute pressure commands for details. Note that the IDs of the new
computes are the fix-ID + underscore + "temp" or fix_ID + underscore + "press", and the group for the new
computes is "all" since pressure is computed for the entire system.
Note that these are NOT the computes used by thermodynamic output (see the thermo_style command) with
ID = thermo_temp and thermo_press. This means you can change the attributes of this fix's temperature or
pressure via the compute_modify command or print this temperature or pressure during thermodynamic
output via the thermo_style custom command using the appropriate compute-ID. It also means that changing
attributes of thermo_temp or thermo_press will have no effect on this fix.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover barostat to binary restart files. See the read_restart command for
info on how to re-specify a fix in an input script that reads a restart file, so that the operation of the fix
continues in an uninterrupted fashion.
The fix_modify temp and press options are supported by this fix. You can use them to assign a compute you
have defined to this fix which will be used in its thermostatting or barostatting procedure. If you do this, note
that the kinetic energy derived from the compute temperature should be consistent with the virial term
computed using all atoms for the pressure. LAMMPS will warn you if you choose to compute temperature on
a subset of atoms.
The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
barostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix nph command.
This fix can ramp its target pressure over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
LAMMPS Users Manual
933
This fix is not invoked during energy minimization.
Restrictions:
This fix requires that atoms store torque and angular velocity (omega) and a radius as defined by the
atom_style sphere command.
All particles in the group must be finite-size spheres. They cannot be point particles.
Use of the disc keyword is only allowed for 2d simulations, as defined by the dimension keyword.
Related commands:
fix nph, fix nve_sphere, fix nvt_sphere, fix npt_sphere, fix_modify
Default: none
LAMMPS Users Manual
934

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nphug command
fix nphug/omp command
Syntax:
fix ID group-ID nphug keyword value ...
ID, group-ID are documented in fix command
one or more keyword value pairs may be appended
keyword = temp or iso or aniso or tri or x or y or z or couple or tchain or pchain or mtk or tloop or ploop or nreset or drag or dilate or scaleyz or scalexz or scalexy
temp values = Value1 Value2 Tdamp
Value1, Value2 = Nose-Hoover target temperatures, ignored by Hugoniostat
Tdamp = temperature damping parameter (time units)
iso or aniso or tri values = Pstart Pstop Pdamp
Pstart,Pstop = scalar external pressures, must be equal (pressure units)
Pdamp = pressure damping parameter (time units)
x or y or z or xy or yz or xz values = Pstart Pstop Pdamp
Pstart,Pstop = external stress tensor components, must be equal (pressure units)
Pdamp = stress damping parameter (time units)
couple = none or xyz or xy or yz or xz
tchain value = length of thermostat chain (1 = single thermostat)
pchain values = length of thermostat chain on barostat (0 = no thermostat)
mtk value = yes or no = add in MTK adjustment term or not
tloop value = number of sub-cycles to perform on thermostat
ploop value = number of sub-cycles to perform on barostat thermostat
nreset value = reset reference cell every this many timesteps
drag value = drag factor added to barostat/thermostat (0.0 = no drag)
dilate value = all or partial
scaleyz value = yes or no = scale yz with lz
scalexz value = yes or no = scale xz with lz
scalexy value = yes or no = scale xy with ly
•
Examples:
fix myhug all nphug temp 1.0 1.0 10.0 z 40.0 40.0 70.0
fix myhug all nphug temp 1.0 1.0 10.0 iso 40.0 40.0 70.0 drag 200.0 tchain 1 pchain 0
Description:
This command is a variant of the Nose-Hoover fix npt fix style. It performs time integration of the
Hugoniostat equations of motion developed by Ravelo et al. (Ravelo). These equations compress the system
to a state with average axial stress or pressure equal to the specified target value and that satisfies the
Rankine-Hugoniot (RH) jump conditions for steady shocks.
The compression can be performed either hydrostatically (using keyword iso, aniso, or tri) or uniaxially
(using keywords x, y, or z). In the hydrostatic case, the cell dimensions change dynamically so that the average
axial stress in all three directions converges towards the specified target value. In the uniaxial case, the chosen
cell dimension changes dynamically so that the average axial stress in that direction converges towards the
target value. The other two cell dimensions are kept fixed (zero lateral strain).
LAMMPS Users Manual
935

This leads to the following additional restrictions on the keywords:
One and only one of the following keywords should be used: iso, aniso, tri, x, y, z• The specified initial and final target pressures must be the same.• The keywords xy, xz, yz may not be used.• The only admissible value for the couple keyword is xyz, which has the same effect as keyword iso• The temp keyword must be used to specify the time constant for kinetic energy relaxation, but initial
and final target temperature values are ignored.
•
Essentially, a Hugoniostat simulation is an NPT simulation in which the user-specified target temperature is
replaced with a time-dependent target temperature Tt obtained from the following equation:
where T and Tt are the instantaneous and target temperatures, P and P0 are the instantaneous and reference
pressures or axial stresses, depending on whether hydrostatic or uniaxial compression is being performed, V
and V0 are the instantaneous and reference volumes, E and E0 are the instantaneous and reference internal
energy (potential plus kinetic), Ndof is the number of degrees of freedom used in the definition of
temperature, and kB is the Boltzmann constant. Delta is the negative deviation of the instantaneous
temperature from the target temperature. When the system reaches a stable equilibrium, the value of Delta
should fluctuate about zero.
The values of E0, V0, and P0 are the instantaneous values at the start of the simulation. These can be
overridden using the fix_modify keywords e0, v0, and p0 described below.
NOTE: Unlike the fix temp/berendsen command which performs thermostatting but NO time integration, this
fix performs thermostatting/barostatting AND time integration. Thus you should not use any other time
integration fix, such as fix nve on atoms to which this fix is applied. Likewise, this fix should not be used on
atoms that have their temperature controlled by another fix - e.g. by fix langevin or fix temp/rescale
commands.
This fix computes a temperature and pressure at each timestep. To do this, the fix creates its own computes of
style "temp" and "pressure", as if one of these two sets of commands had been issued:
compute fix-ID_temp group-ID temp
compute fix-ID_press group-ID pressure fix-ID_temp
compute fix-ID_temp all temp
compute fix-ID_press all pressure fix-ID_temp
See the compute temp and compute pressure commands for details. Note that the IDs of the new computes are
the fix-ID + underscore + "temp" or fix_ID + underscore + "press". The group for the new computes is "all"
since pressure is computed for the entire system.
Note that these are NOT the computes used by thermodynamic output (see the thermo_style command) with
ID = thermo_temp and thermo_press. This means you can change the attributes of this fix's temperature or
LAMMPS Users Manual
936

pressure via the compute_modify command or print this temperature or pressure during thermodynamic
output via the thermo_style custom command using the appropriate compute-ID. It also means that changing
attributes of thermo_temp or thermo_press will have no effect on this fix.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the values of E0, V0, and P0, as well as the state of all the thermostat and barostat variables to
binary restart files. See the read_restart command for info on how to re-specify a fix in an input script that
reads a restart file, so that the operation of the fix continues in an uninterrupted fashion.
The fix_modify e0, v0 and p0 keywords can be used to define the values of E0, V0, and P0. Note the the
values for e0 and v0 are extensive, and so must correspond to the total energy and volume of the entire
system, not energy and volume per atom. If any of these quantities are not specified, then the instantaneous
value in the system at the start of the simulation is used.
The fix_modify temp and press options are supported by these fixes. You can use them to assign a compute
you have defined to this fix which will be used in its thermostatting or barostatting procedure, as described
above. If you do this, note that the kinetic energy derived from the compute temperature should be consistent
with the virial term computed using all atoms for the pressure. LAMMPS will warn you if you choose to
compute temperature on a subset of atoms.
The fix_modify energy option is supported by these fixes to add the energy change induced by Nose/Hoover
thermostatting and barostatting to the system's potential energy as part of thermodynamic output. Either way,
this energy is *not* included in the definition of internal energy E when calculating the value of Delta in the
above equation.
These fixes compute a global scalar and a global vector of quantities, which can be accessed by various output
commands. The scalar value calculated by these fixes is "extensive"; the vector values are "intensive".
The scalar is the cumulative energy change due to the fix.
The vector stores three quantities unique to this fix (Delta, Us, and up), followed by all the internal
Nose/Hoover thermostat and barostat variables defined for fix npt. Delta is the deviation of the temperature
from the target temperature, given by the above equation. Us and up are the shock and particle velocity
corresponding to a steady shock calculated from the RH conditions. They have units of distance/time.
LAMMPS Users Manual
937

Restrictions:
This fix style is part of the SHOCK package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
All the usual restrictions for fix npt apply, plus the additional ones mentioned above.
Related commands:
fix msst, fix npt, fix_modify
Default:
The keyword defaults are the same as those for fix npt
(Ravelo) Ravelo, Holian, Germann and Lomdahl, Phys Rev B, 70, 014103 (2004).
LAMMPS Users Manual
938

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix npt/asphere command
fix npt/asphere/omp command
Syntax:
fix ID group-ID npt/asphere keyword value ...
ID, group-ID are documented in fix command• npt/asphere = style name of this fix command• additional thermostat and barostat related keyword/value pairs from the fix npt command can be
appended
•
Examples:
fix 1 all npt/asphere temp 300.0 300.0 100.0 iso 0.0 0.0 1000.0
fix 2 all npt/asphere temp 300.0 300.0 100.0 x 5.0 5.0 1000.0
fix 2 all npt/asphere temp 300.0 300.0 100.0 x 5.0 5.0 1000.0 drag 0.2
fix 2 water npt/asphere temp 300.0 300.0 100.0 aniso 0.0 0.0 1000.0 dilate partial
Description:
Perform constant NPT integration to update position, velocity, orientation, and angular velocity each timestep
for aspherical or ellipsoidal particles in the group using a Nose/Hoover temperature thermostat and
Nose/Hoover pressure barostat. P is pressure; T is temperature. This creates a system trajectory consistent
with the isothermal-isobaric ensemble.
This fix differs from the fix npt command, which assumes point particles and only updates their position and
velocity.
The thermostat is applied to both the translational and rotational degrees of freedom for the aspherical
particles, assuming a compute is used which calculates a temperature that includes the rotational degrees of
freedom (see below). The translational degrees of freedom can also have a bias velocity removed from them
before thermostatting takes place; see the description below.
Additional parameters affecting the thermostat and barostat are specified by keywords and values documented
with the fix npt command. See, for example, discussion of the temp, iso, aniso, and dilate keywords.
The particles in the fix group are the only ones whose velocities and positions are updated by the
velocity/position update portion of the NPT integration.
Regardless of what particles are in the fix group, a global pressure is computed for all particles. Similarly,
when the size of the simulation box is changed, all particles are re-scaled to new positions, unless the keyword
dilate is specified with a value of partial, in which case only the particles in the fix group are re-scaled. The
latter can be useful for leaving the coordinates of particles in a solid substrate unchanged and controlling the
pressure of a surrounding fluid.
This fix computes a temperature and pressure each timestep. To do this, the fix creates its own computes of
style "temp/asphere" and "pressure", as if these commands had been issued:
LAMMPS Users Manual
939

compute fix-ID_temp all temp/asphere
compute fix-ID_press all pressure fix-ID_temp
See the compute temp/asphere and compute pressure commands for details. Note that the IDs of the new
computes are the fix-ID + underscore + "temp" or fix_ID + underscore + "press", and the group for the new
computes is "all" since pressure is computed for the entire system.
Note that these are NOT the computes used by thermodynamic output (see the thermo_style command) with
ID = thermo_temp and thermo_press. This means you can change the attributes of this fix's temperature or
pressure via the compute_modify command or print this temperature or pressure during thermodynamic
output via the thermo_style custom command using the appropriate compute-ID. It also means that changing
attributes of thermo_temp or thermo_press will have no effect on this fix.
Like other fixes that perform thermostatting, this fix can be used with compute commands that calculate a
temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass velocity from
a group of atoms or only calculating temperature on the x-component of velocity or only calculating
temperature for atoms in a geometric region. This is not done by default, but only if the fix_modify command
is used to assign a temperature compute to this fix that includes such a bias term. See the doc pages for
individual compute commands to determine which ones include a bias. In this case, the thermostat works in
the following manner: the current temperature is calculated taking the bias into account, bias is removed from
each atom, thermostatting is performed on the remaining thermal degrees of freedom, and the bias is added
back in.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover thermostat and barostat to binary restart files. See the read_restart
command for info on how to re-specify a fix in an input script that reads a restart file, so that the operation of
the fix continues in an uninterrupted fashion.
The fix_modify temp and press options are supported by this fix. You can use them to assign a compute you
have defined to this fix which will be used in its thermostatting or barostatting procedure. If you do this, note
that the kinetic energy derived from the compute temperature should be consistent with the virial term
computed using all atoms for the pressure. LAMMPS will warn you if you choose to compute temperature on
a subset of atoms.
LAMMPS Users Manual
940
The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
thermostatting and barostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix npt command.
This fix can ramp its target temperature and pressure over multiple runs, using the start and stop keywords of
the run command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that atoms store torque and angular momentum and a quaternion as defined by the
atom_style ellipsoid command.
All particles in the group must be finite-size. They cannot be point particles, but they can be aspherical or
spherical as defined by their shape attribute.
Related commands:
fix npt, fix nve_asphere, fix nvt_asphere, fix_modify
Default: none
LAMMPS Users Manual
941

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix npt/body command
Syntax:
fix ID group-ID npt/body keyword value ...
ID, group-ID are documented in fix command• npt/body = style name of this fix command• additional thermostat and barostat related keyword/value pairs from the fix npt command can be
appended
•
Examples:
fix 1 all npt/body temp 300.0 300.0 100.0 iso 0.0 0.0 1000.0
fix 2 all npt/body temp 300.0 300.0 100.0 x 5.0 5.0 1000.0
fix 2 all npt/body temp 300.0 300.0 100.0 x 5.0 5.0 1000.0 drag 0.2
fix 2 water npt/body temp 300.0 300.0 100.0 aniso 0.0 0.0 1000.0 dilate partial
Description:
Perform constant NPT integration to update position, velocity, orientation, and angular velocity each timestep
for body particles in the group using a Nose/Hoover temperature thermostat and Nose/Hoover pressure
barostat. P is pressure; T is temperature. This creates a system trajectory consistent with the
isothermal-isobaric ensemble.
This fix differs from the fix npt command, which assumes point particles and only updates their position and
velocity.
The thermostat is applied to both the translational and rotational degrees of freedom for the body particles,
assuming a compute is used which calculates a temperature that includes the rotational degrees of freedom
(see below). The translational degrees of freedom can also have a bias velocity removed from them before
thermostatting takes place; see the description below.
Additional parameters affecting the thermostat and barostat are specified by keywords and values documented
with the fix npt command. See, for example, discussion of the temp, iso, aniso, and dilate keywords.
The particles in the fix group are the only ones whose velocities and positions are updated by the
velocity/position update portion of the NPT integration.
Regardless of what particles are in the fix group, a global pressure is computed for all particles. Similarly,
when the size of the simulation box is changed, all particles are re-scaled to new positions, unless the keyword
dilate is specified with a value of partial, in which case only the particles in the fix group are re-scaled. The
latter can be useful for leaving the coordinates of particles in a solid substrate unchanged and controlling the
pressure of a surrounding fluid.
This fix computes a temperature and pressure each timestep. To do this, the fix creates its own computes of
style "temp/body" and "pressure", as if these commands had been issued:
compute fix-ID_temp all temp/body
compute fix-ID_press all pressure fix-ID_temp
LAMMPS Users Manual
942

See the compute temp/body and compute pressure commands for details. Note that the IDs of the new
computes are the fix-ID + underscore + "temp" or fix_ID + underscore + "press", and the group for the new
computes is "all" since pressure is computed for the entire system.
Note that these are NOT the computes used by thermodynamic output (see the thermo_style command) with
ID = thermo_temp and thermo_press. This means you can change the attributes of this fix's temperature or
pressure via the compute_modify command or print this temperature or pressure during thermodynamic
output via the thermo_style custom command using the appropriate compute-ID. It also means that changing
attributes of thermo_temp or thermo_press will have no effect on this fix.
Like other fixes that perform thermostatting, this fix can be used with compute commands that calculate a
temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass velocity from
a group of atoms or only calculating temperature on the x-component of velocity or only calculating
temperature for atoms in a geometric region. This is not done by default, but only if the fix_modify command
is used to assign a temperature compute to this fix that includes such a bias term. See the doc pages for
individual compute commands to determine which ones include a bias. In this case, the thermostat works in
the following manner: the current temperature is calculated taking the bias into account, bias is removed from
each atom, thermostatting is performed on the remaining thermal degrees of freedom, and the bias is added
back in.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover thermostat and barostat to binary restart files. See the read_restart
command for info on how to re-specify a fix in an input script that reads a restart file, so that the operation of
the fix continues in an uninterrupted fashion.
The fix_modify temp and press options are supported by this fix. You can use them to assign a compute you
have defined to this fix which will be used in its thermostatting or barostatting procedure. If you do this, note
that the kinetic energy derived from the compute temperature should be consistent with the virial term
computed using all atoms for the pressure. LAMMPS will warn you if you choose to compute temperature on
a subset of atoms.
The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
thermostatting and barostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix npt command.
LAMMPS Users Manual
943
This fix can ramp its target temperature and pressure over multiple runs, using the start and stop keywords of
the run command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the BODY package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that atoms store torque and angular momentum and a quaternion as defined by the
atom_style body command.
Related commands:
fix npt, fix nve_body, fix nvt_body, fix_modify
Default: none
LAMMPS Users Manual
944

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix npt/sphere command
fix npt/sphere/omp command
Syntax:
fix ID group-ID npt/sphere keyword value ...
ID, group-ID are documented in fix command npt/sphere = style name of this fix command zero or more
keyword/value pairs may be appended
•
keyword = disc
disc value = none = treat particles as 2d discs, not spheres
•
additional thermostat and barostat related keyword/value pairs from the fix npt command can be appended
Examples:
fix 1 all npt/sphere temp 300.0 300.0 100.0 iso 0.0 0.0 1000.0
fix 2 all npt/sphere temp 300.0 300.0 100.0 x 5.0 5.0 1000.0
fix 2 all npt/sphere temp 300.0 300.0 100.0 x 5.0 5.0 1000.0 disc
fix 2 all npt/sphere temp 300.0 300.0 100.0 x 5.0 5.0 1000.0 drag 0.2
fix 2 water npt/sphere temp 300.0 300.0 100.0 aniso 0.0 0.0 1000.0 dilate partial
Description:
Perform constant NPT integration to update position, velocity, and angular velocity each timestep for
finite-sizex spherical particles in the group using a Nose/Hoover temperature thermostat and Nose/Hoover
pressure barostat. P is pressure; T is temperature. This creates a system trajectory consistent with the
isothermal-isobaric ensemble.
This fix differs from the fix npt command, which assumes point particles and only updates their position
and velocity.
The thermostat is applied to both the translational and rotational degrees of freedom for the spherical
particles, assuming a compute is used which calculates a temperature that includes the rotational degrees of
freedom (see below). The translational degrees of freedom can also have a bias velocity removed from
them before thermostatting takes place; see the description below.
If the disc keyword is used, then each particle is treated as a 2d disc (circle) instead of as a sphere. This is
only possible for 2d simulations, as defined by the dimension keyword. The only difference between discs
and spheres in this context is their moment of inertia, as used in the time integration.
Additional parameters affecting the thermostat and barostat are specified by keywords and values
documented with the fix npt command. See, for example, discussion of the temp, iso, aniso, and dilate
keywords.
The particles in the fix group are the only ones whose velocities and positions are updated by the
velocity/position update portion of the NPT integration.
•
LAMMPS Users Manual
945
Regardless of what particles are in the fix group, a global pressure is computed for all particles. Similarly,
when the size of the simulation box is changed, all particles are re-scaled to new positions, unless the
keyword dilate is specified with a value of partial, in which case only the particles in the fix group are
re-scaled. The latter can be useful for leaving the coordinates of particles in a solid substrate unchanged and
controlling the pressure of a surrounding fluid.
This fix computes a temperature and pressure each timestep. To do this, the fix creates its own computes of
style "temp/sphere" and "pressure", as if these commands had been issued:
compute fix-ID_temp all temp/sphere
compute fix-ID_press all pressure fix-ID_temp
See the compute temp/sphere and compute pressure commands for details. Note that the IDs of the new
computes are the fix-ID + underscore + "temp" or fix_ID + underscore + "press", and the group for the new
computes is "all" since pressure is computed for the entire system.
Note that these are NOT the computes used by thermodynamic output (see the thermo_style command)
with ID = thermo_temp and thermo_press. This means you can change the attributes of this fix's
temperature or pressure via the compute_modify command or print this temperature or pressure during
thermodynamic output via the thermo_style custom command using the appropriate compute-ID. It also
means that changing attributes of thermo_temp or thermo_press will have no effect on this fix.
Like other fixes that perform thermostatting, this fix can be used with compute commands that calculate a
temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass velocity
from a group of atoms or only calculating temperature on the x-component of velocity or only calculating
temperature for atoms in a geometric region. This is not done by default, but only if the fix_modify
command is used to assign a temperature compute to this fix that includes such a bias term. See the doc
pages for individual compute commands to determine which ones include a bias. In this case, the thermostat
works in the following manner: the current temperature is calculated taking the bias into account, bias is
removed from each atom, thermostatting is performed on the remaining thermal degrees of freedom, and
the bias is added back in.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without
the suffix. They have been optimized to run faster, depending on your available hardware, as discussed in
Section 5 of the manual. The accelerated styles take the same arguments and should produce the same
results, except for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can
use the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in
your input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover thermostat and barostat to binary restart files. See the
read_restart command for info on how to re-specify a fix in an input script that reads a restart file, so that
LAMMPS Users Manual
946
the operation of the fix continues in an uninterrupted fashion.
The fix_modify temp and press options are supported by this fix. You can use them to assign a compute
you have defined to this fix which will be used in its thermostatting or barostatting procedure. If you do
this, note that the kinetic energy derived from the compute temperature should be consistent with the virial
term computed using all atoms for the pressure. LAMMPS will warn you if you choose to compute
temperature on a subset of atoms.
The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
thermostatting and barostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix npt command.
This fix can ramp its target temperature and pressure over multiple runs, using the start and stop keywords
of the run command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix requires that atoms store torque and angular velocity (omega) and a radius as defined by the
atom_style sphere command.
All particles in the group must be finite-size spheres. They cannot be point particles.
Use of the disc keyword is only allowed for 2d simulations, as defined by the dimension keyword.
Related commands:
fix npt, fix nve_sphere, fix nvt_sphere, fix npt_asphere, fix_modify
Default: none
LAMMPS Users Manual
947

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve command
fix nve/intel command
fix nve/kk command
fix nve/omp command
Syntax:
fix ID group-ID nve
ID, group-ID are documented in fix command• nve = style name of this fix command•
Examples:
fix 1 all nve
Description:
Perform constant NVE integration to update position and velocity for atoms in the group each timestep. V is
volume; E is energy. This creates a system trajectory consistent with the microcanonical ensemble.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
LAMMPS Users Manual
948

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/asphere command
fix nve/asphere/intel command
Syntax:
fix ID group-ID nve/asphere
ID, group-ID are documented in fix command• nve/asphere = style name of this fix command•
Examples:
fix 1 all nve/asphere
Description:
Perform constant NVE integration to update position, velocity, orientation, and angular velocity for aspherical
particles in the group each timestep. V is volume; E is energy. This creates a system trajectory consistent with
the microcanonical ensemble.
This fix differs from the fix nve command, which assumes point particles and only updates their position and
velocity.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
950
This fix is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that atoms store torque and angular momentum and a quaternion as defined by the
atom_style ellipsoid command.
All particles in the group must be finite-size. They cannot be point particles, but they can be aspherical or
spherical as defined by their shape attribute.
Related commands:
fix nve, fix nve/sphere
Default: none
LAMMPS Users Manual
951
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/asphere/noforce command
Syntax:
fix ID group-ID nve/asphere/noforce
ID, group-ID are documented in fix command• nve/asphere/noforce = style name of this fix command•
Examples:
fix 1 all nve/asphere/noforce
Description:
Perform updates of position and orientation, but not velocity or angular momentum for atoms in the group
each timestep. In other words, the force and torque on the atoms is ignored and their velocity and angular
momentum are not updated. The atom velocities and angular momenta are used to update their positions and
orientation.
This is useful as an implicit time integrator for Fast Lubrication Dynamics, since the velocity and angular
momentum are updated by the pair_style lubricuteU command.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that atoms store torque and angular momentum and a quaternion as defined by the
atom_style ellipsoid command.
All particles in the group must be finite-size. They cannot be point particles, but they can be aspherical or
spherical as defined by their shape attribute.
Related commands:
fix nve/noforce, fix nve/asphere
Default: none
LAMMPS Users Manual
952

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/body command
Syntax:
fix ID group-ID nve/body
ID, group-ID are documented in fix command• nve/body = style name of this fix command•
Examples:
fix 1 all nve/body
Description:
Perform constant NVE integration to update position, velocity, orientation, and angular velocity for body
particles in the group each timestep. V is volume; E is energy. This creates a system trajectory consistent with
the microcanonical ensemble. See Section 6.14 of the manual and the body doc page for more details on using
body particles.
This fix differs from the fix nve command, which assumes point particles and only updates their position and
velocity.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the BODY package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that atoms store torque and angular momentum and a quaternion as defined by the
atom_style body command.
All particles in the group must be body particles. They cannot be point particles.
Related commands:
fix nve, fix nve/sphere, fix nve/asphere
Default: none
LAMMPS Users Manual
953

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/dot command
Syntax:
fix ID group-ID nve/dot
ID, group-ID are documented in fix command• nve/dot = style name of this fix command•
Examples:
fix 1 all nve/dot
Description:
Apply a rigid-body integrator as described in (Davidchack) to a group of atoms, but without Langevin
dynamics. This command performs Molecular dynamics (MD) via a velocity-Verlet algorithm and an
evolution operator that rotates the quaternion degrees of freedom, similar to the scheme outlined in (Miller).
This command is the equivalent of the fix nve/dotc/langevin without damping and noise and can be used to
determine the stability range in a NVE ensemble prior to using the Langevin-type DOTC-integrator (see also
fix nve/dotc/langevin). The command is equivalent to the fix nve. The particles are always considered to have
a finite size.
An example input file can be found in /examples/USER/cgdna/examples/duplex1/. A technical report with
more information on this integrator can be found here.
Restrictions:
These pair styles can only be used if LAMMPS was built with the USER-CGDNA package and the
MOLECULE and ASPHERE package. See the Making LAMMPS section for more info on packages.
Related commands:
fix nve/dotc/langevin, fix nve
Default: none
(Davidchack) R.L Davidchack, T.E. Ouldridge, and M.V. Tretyakov. J. Chem. Phys. 142, 144114 (2015).
(Miller) T. F. Miller III, M. Eleftheriou, P. Pattnaik, A. Ndirango, G. J. Martyna, J. Chem. Phys., 116,
8649-8659 (2002).
LAMMPS Users Manual
954

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/dotc/langevin command
Syntax:
fix ID group-ID nve/dotc/langevin Tstart Tstop damp seed keyword value
ID, group-ID are documented in fix command• nve/dotc/langevin = style name of this fix command• Tstart,Tstop = desired temperature at start/end of run (temperature units)• damp = damping parameter (time units)• seed = random number seed to use for white noise (positive integer)• keyword = angmom
angmom value = factor
factor = do thermostat rotational degrees of freedom via the angular momentum and apply numeric scale factor as discussed below
•
Examples:
fix 1 all nve/dotc/langevin 1.0 1.0 0.03 457145 angmom 10
Description:
Apply a rigid-body Langevin-type integrator of the kind "Langevin C" as described in (Davidchack) to a
group of atoms, which models an interaction with an implicit background solvent. This command performs
Brownian dynamics (BD) via a technique that splits the integration into a deterministic Hamiltonian part and
the Ornstein-Uhlenbeck process for noise and damping. The quaternion degrees of freedom are updated
though an evolution operator which performs a rotation in quaternion space, preserves the quaternion norm
and is akin to (Miller).
In terms of syntax this command has been closely modelled on the fix langevin and its angmom option. But it
combines the fix nve and the fix langevin in one single command. The main feature is improved stability over
the standard integrator, permitting slightly larger timestep sizes.
NOTE: Unlike the fix langevin this command performs also time integration of the translational and
quaternion degrees of freedom.
The total force on each atom will have the form:
F = Fc + Ff + Fr
Ff = - (m / damp) v
Fr is proportional to sqrt(Kb T m / (dt damp))
Fc is the conservative force computed via the usual inter-particle interactions (pair_style, bond_style, etc).
The Ff and Fr terms are implicitly taken into account by this fix on a per-particle basis.
Ff is a frictional drag or viscous damping term proportional to the particle's velocity. The proportionality
constant for each atom is computed as m/damp, where m is the mass of the particle and damp is the damping
factor specified by the user.
LAMMPS Users Manual
955

Fr is a force due to solvent atoms at a temperature T randomly bumping into the particle. As derived from the
fluctuation/dissipation theorem, its magnitude as shown above is proportional to sqrt(Kb T m / dt damp),
where Kb is the Boltzmann constant, T is the desired temperature, m is the mass of the particle, dt is the
timestep size, and damp is the damping factor. Random numbers are used to randomize the direction and
magnitude of this force as described in (Dunweg), where a uniform random number is used (instead of a
Gaussian random number) for speed.
Tstart and Tstop have to be constant values, i.e. they cannot be variables.
The damp parameter is specified in time units and determines how rapidly the temperature is relaxed. For
example, a value of 0.03 means to relax the temperature in a timespan of (roughly) 0.03 time units tau (see the
units command). The damp factor can be thought of as inversely related to the viscosity of the solvent, i.e. a
small relaxation time implies a hi-viscosity solvent and vice versa. See the discussion about gamma and
viscosity in the documentation for the fix viscous command for more details.
The random # seed must be a positive integer. A Marsaglia random number generator is used. Each processor
uses the input seed to generate its own unique seed and its own stream of random numbers. Thus the dynamics
of the system will not be identical on two runs on different numbers of processors.
The keyword/value option has to be used in the following way:
This fix has to be used together with the angmom keyword. The particles are always considered to have a
finite size. The keyword angmom enables thermostatting of the rotational degrees of freedom in addition to
the usual translational degrees of freedom.
The scale factor after the angmom keyword gives the ratio of the rotational to the translational friction
coefficient.
An example input file can be found in /examples/USER/cgdna/examples/duplex2/. A technical report with
more information on this integrator can be found here.
Restrictions:
These pair styles can only be used if LAMMPS was built with the USER-CGDNA package and the
MOLECULE and ASPHERE package. See the Making LAMMPS section for more info on packages.
Related commands:
fix nve, fix langevin, fix nve/dot,
Default: none
(Davidchack) R.L Davidchack, T.E. Ouldridge, M.V. Tretyakov. J. Chem. Phys. 142, 144114 (2015).
(Miller) T. F. Miller III, M. Eleftheriou, P. Pattnaik, A. Ndirango, G. J. Martyna, J. Chem. Phys., 116,
8649-8659 (2002).
(Dunweg) B. Dunweg, W. Paul, Int. J. Mod. Phys. C, 2, 817-27 (1991).
LAMMPS Users Manual
956

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/eff command
Syntax:
fix ID group-ID nve/eff
ID, group-ID are documented in fix command• nve/eff = style name of this fix command•
Examples:
fix 1 all nve/eff
Description:
Perform constant NVE integration to update position and velocity for nuclei and electrons in the group for the
electron force field model. V is volume; E is energy. This creates a system trajectory consistent with the
microcanonical ensemble.
The operation of this fix is exactly like that described by the fix nve command, except that the radius and
radial velocity of electrons are also updated.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the USER-EFF package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
fix nve, fix nvt/eff, fix npt/eff
Default: none
LAMMPS Users Manual
957

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/limit command
Syntax:
fix ID group-ID nve/limit xmax
ID, group-ID are documented in fix command• nve = style name of this fix command• xmax = maximum distance an atom can move in one timestep (distance units)•
Examples:
fix 1 all nve/limit 0.1
Description:
Perform constant NVE updates of position and velocity for atoms in the group each timestep. A limit is
imposed on the maximum distance an atom can move in one timestep. This is useful when starting a
simulation with a configuration containing highly overlapped atoms. Normally this would generate huge
forces which would blow atoms out of the simulation box, causing LAMMPS to stop with an error.
Using this fix can overcome that problem. Forces on atoms must still be computable (which typically means 2
atoms must have a separation distance > 0.0). But large velocities generated by large forces are reset to a
value that corresponds to a displacement of length xmax in a single timestep. Xmax is specified in distance
units; see the units command for details. The value of xmax should be consistent with the neighbor skin
distance and the frequency of neighbor list re-building, so that pairwise interactions are not missed on
successive timesteps as atoms move. See the neighbor and neigh_modify commands for details.
Note that if a velocity reset occurs the integrator will not conserve energy. On steps where no velocity resets
occur, this integrator is exactly like the fix nve command. Since forces are unaltered, pressures computed by
thermodynamic output will still be very large for overlapped configurations.
NOTE: You should not use fix shake in conjunction with this fix. That is because fix shake applies constraint
forces based on the predicted positions of atoms after the next timestep. It has no way of knowing the timestep
may change due to this fix, which will cause the constraint forces to be invalid. A better strategy is to turn off
fix shake when performing initial dynamics that need this fix, then turn fix shake on when doing normal
dynamics with a fixed-size timestep.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the count
of how many updates of atom's velocity/position were limited by the maximum distance criterion. This should
be roughly the number of atoms so affected, except that updates occur at both the beginning and end of a
timestep in a velocity Verlet timestepping algorithm. This is a cumulative quantity for the current run, but is
re-initialized to zero each time a run is performed. The scalar value calculated by this fix is "extensive".
LAMMPS Users Manual
958

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/line command
Syntax:
fix ID group-ID nve/line
ID, group-ID are documented in fix command• nve/line = style name of this fix command•
Examples:
fix 1 all nve/line
Description:
Perform constant NVE integration to update position, velocity, orientation, and angular velocity for line
segment particles in the group each timestep. V is volume; E is energy. This creates a system trajectory
consistent with the microcanonical ensemble. See Section 6.14 of the manual for an overview of using line
segment particles.
This fix differs from the fix nve command, which assumes point particles and only updates their position and
velocity.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that particles be line segments as defined by the atom_style line command.
Related commands:
fix nve, fix nve/asphere
Default: none
LAMMPS Users Manual
960

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/manifold/rattle command
Syntax:
fix ID group-ID nve/manifold/rattle tol maxit manifold manifold-args keyword value ...
ID, group-ID are documented in fix command• nve/manifold/rattle = style name of this fix command• tol = tolerance to which Newton iteration must converge• maxit = maximum number of iterations to perform• manifold = name of the manifold• manifold-args = parameters for the manifold• one or more keyword/value pairs may be appended
keyword = every
every values = N
N = print info about iteration every N steps. N = 0 means no output
•
Examples:
fix 1 all nve/manifold/rattle 1e-4 10 sphere 5.0
fix step all nve/manifold/rattle 1e-8 100 ellipsoid 2.5 2.5 5.0 every 25
Description:
Perform constant NVE integration to update position and velocity for atoms constrained to a curved surface
(manifold) in the group each timestep. The constraint is handled by RATTLE (Andersen) written out for the
special case of single-particle constraints as explained in (Paquay). V is volume; E is energy. This way, the
dynamics of particles constrained to curved surfaces can be studied. If combined with fix langevin, this
generates Brownian motion of particles constrained to a curved surface. For a list of currently supported
manifolds and their parameters, see manifolds.
Note that the particles must initially be close to the manifold in question. If not, RATTLE will not be able to
iterate until the constraint is satisfied, and an error is generated. For simple manifolds this can be achieved
with region and create_atoms commands, but for more complex surfaces it might be more useful to write a
script.
The manifold args may be equal-style variables, like so:
variable R equal "ramp(5.0,3.0)"
fix shrink_sphere all nve/manifold/rattle 1e-4 10 sphere v_R
In this case, the manifold parameter will change in time according to the variable. This is not a problem for
the time integrator as long as the change of the manifold is slow with respect to the dynamics of the particles.
Note that if the manifold has to exert work on the particles because of these changes, the total energy might
not be conserved.
Restart, fix_modify, output, run start/stop, minimize info:
LAMMPS Users Manual
961

No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the USER-MANIFOLD package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Related commands:
fix nvt/manifold/rattle, fix manifoldforce
Default: every = 0, tchain = 3
(Andersen) Andersen, J. Comp. Phys. 52, 24, (1983).
(Paquay) Paquay and Kusters, Biophys. J., 110, 6, (2016). preprint available at arXiv:1411.3019.
LAMMPS Users Manual
962

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/noforce command
Syntax:
fix ID group-ID nve
ID, group-ID are documented in fix command• nve/noforce = style name of this fix command•
Examples:
fix 3 wall nve/noforce
Description:
Perform updates of position, but not velocity for atoms in the group each timestep. In other words, the force
on the atoms is ignored and their velocity is not updated. The atom velocities are used to update their
positions.
This can be useful for wall atoms, when you set their velocities, and want the wall to move (or stay stationary)
in a prescribed fashion.
This can also be accomplished via the fix setforce command, but with fix nve/noforce, the forces on the wall
atoms are unchanged, and can thus be printed by the dump command or queried with an equal-style variable
that uses the fcm() group function to compute the total force on the group of atoms.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
fix nve
Default: none
LAMMPS Users Manual
963

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/sphere command
fix nve/sphere/omp command
Syntax:
fix ID group-ID nve/sphere
ID, group-ID are documented in fix command• nve/sphere = style name of this fix command• zero or more keyword/value pairs may be appended• keyword = update or disc
update value = dipole or dipole/dlm
dipole = update orientation of dipole moment during integration
dipole/dlm = use DLM integrator to update dipole orientation
disc value = none = treat particles as 2d discs, not spheres
•
Examples:
fix 1 all nve/sphere
fix 1 all nve/sphere update dipole
fix 1 all nve/sphere disc
fix 1 all nve/sphere update dipole/dlm
Description:
Perform constant NVE integration to update position, velocity, and angular velocity for finite-size spherical
particles in the group each timestep. V is volume; E is energy. This creates a system trajectory consistent with
the microcanonical ensemble.
This fix differs from the fix nve command, which assumes point particles and only updates their position and
velocity.
If the update keyword is used with the dipole value, then the orientation of the dipole moment of each particle
is also updated during the time integration. This option should be used for models where a dipole moment is
assigned to finite-size particles, e.g. spheroids via use of the atom_style hybrid sphere dipole command.
The default dipole orientation integrator can be changed to the Dullweber-Leimkuhler-McLachlan integration
scheme (Dullweber) when using update with the value dipole/dlm. This integrator is symplectic and
time-reversible, giving better energy conservation and allows slightly longer timesteps at only a small
additional computational cost.
If the disc keyword is used, then each particle is treated as a 2d disc (circle) instead of as a sphere. This is only
possible for 2d simulations, as defined by the dimension keyword. The only difference between discs and
spheres in this context is their moment of inertia, as used in the time integration.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
LAMMPS Users Manual
964
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix requires that atoms store torque and angular velocity (omega) and a radius as defined by the
atom_style sphere command. If the dipole keyword is used, then they must also store a dipole moment as
defined by the atom_style dipole command.
All particles in the group must be finite-size spheres. They cannot be point particles.
Use of the disc keyword is only allowed for 2d simulations, as defined by the dimension keyword.
Related commands:
fix nve, fix nve/asphere
Default: none
(Dullweber) Dullweber, Leimkuhler and McLachlan, J Chem Phys, 107, 5840 (1997).
LAMMPS Users Manual
965

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nve/tri command
Syntax:
fix ID group-ID nve/tri
ID, group-ID are documented in fix command• nve/tri = style name of this fix command•
Examples:
fix 1 all nve/tri
Description:
Perform constant NVE integration to update position, velocity, orientation, and angular momentum for
triangular particles in the group each timestep. V is volume; E is energy. This creates a system trajectory
consistent with the microcanonical ensemble. See Section 6.14 of the manual for an overview of using
triangular particles.
This fix differs from the fix nve command, which assumes point particles and only updates their position and
velocity.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that particles be triangles as defined by the atom_style tri command.
Related commands:
fix nve, fix nve/asphere
Default: none
LAMMPS Users Manual
966

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nvk command
Syntax:
fix ID group-ID nvk
ID, group-ID are documented in fix command• nvk = style name of this fix command•
Examples:
fix 1 all nvk
Description:
Perform constant kinetic energy integration using the Gaussian thermostat to update position and velocity for
atoms in the group each timestep. V is volume; K is kinetic energy. This creates a system trajectory consistent
with the isokinetic ensemble.
The equations of motion used are those of Minary et al in (Minary), a variant of those initially given by Zhang
in (Zhang).
The kinetic energy will be held constant at its value given when fix nvk is initiated. If a different kinetic
energy is desired, the velocity command should be used to change the kinetic energy prior to this fix.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
The Gaussian thermostat only works when it is applied to all atoms in the simulation box. Therefore, the
group must be set to all.
This fix has not yet been implemented to work with the RESPA integrator.
This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands: none
Default: none
(Minary) Minary, Martyna, and Tuckerman, J Chem Phys, 18, 2510 (2003).
LAMMPS Users Manual
967
(Zhang) Zhang, J Chem Phys, 106, 6102 (1997).
LAMMPS Users Manual
968

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nvt/asphere command
fix nvt/asphere/omp command
Syntax:
fix ID group-ID nvt/asphere keyword value ...
ID, group-ID are documented in fix command• nvt/asphere = style name of this fix command• additional thermostat related keyword/value pairs from the fix nvt command can be appended•
Examples:
fix 1 all nvt/asphere temp 300.0 300.0 100.0
fix 1 all nvt/asphere temp 300.0 300.0 100.0 drag 0.2
Description:
Perform constant NVT integration to update position, velocity, orientation, and angular velocity each timestep
for aspherical or ellipsoidal particles in the group using a Nose/Hoover temperature thermostat. V is volume;
T is temperature. This creates a system trajectory consistent with the canonical ensemble.
This fix differs from the fix nvt command, which assumes point particles and only updates their position and
velocity.
The thermostat is applied to both the translational and rotational degrees of freedom for the aspherical
particles, assuming a compute is used which calculates a temperature that includes the rotational degrees of
freedom (see below). The translational degrees of freedom can also have a bias velocity removed from them
before thermostatting takes place; see the description below.
Additional parameters affecting the thermostat are specified by keywords and values documented with the fix
nvt command. See, for example, discussion of the temp and drag keywords.
This fix computes a temperature each timestep. To do this, the fix creates its own compute of style
"temp/asphere", as if this command had been issued:
compute fix-ID_temp group-ID temp/asphere
See the compute temp/asphere command for details. Note that the ID of the new compute is the fix-ID +
underscore + "temp", and the group for the new compute is the same as the fix group.
Note that this is NOT the compute used by thermodynamic output (see the thermo_style command) with ID =
thermo_temp. This means you can change the attributes of this fix's temperature (e.g. its degrees-of-freedom)
via the compute_modify command or print this temperature during thermodynamic output via the
thermo_style custom command using the appropriate compute-ID. It also means that changing attributes of
thermo_temp will have no effect on this fix.
LAMMPS Users Manual
969

Like other fixes that perform thermostatting, this fix can be used with compute commands that calculate a
temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass velocity from
a group of atoms or only calculating temperature on the x-component of velocity or only calculating
temperature for atoms in a geometric region. This is not done by default, but only if the fix_modify command
is used to assign a temperature compute to this fix that includes such a bias term. See the doc pages for
individual compute commands to determine which ones include a bias. In this case, the thermostat works in
the following manner: the current temperature is calculated taking the bias into account, bias is removed from
each atom, thermostatting is performed on the remaining thermal degrees of freedom, and the bias is added
back in.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover thermostat to binary restart files. See the read_restart command
for info on how to re-specify a fix in an input script that reads a restart file, so that the operation of the fix
continues in an uninterrupted fashion.
The fix_modify temp option is supported by this fix. You can use it to assign a compute you have defined to
this fix which will be used in its thermostatting procedure.
The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
thermostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix nvt command.
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that atoms store torque and angular momentum and a quaternion as defined by the
atom_style ellipsoid command.
LAMMPS Users Manual
970

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nvt/body command
Syntax:
fix ID group-ID nvt/body keyword value ...
ID, group-ID are documented in fix command• nvt/body = style name of this fix command• additional thermostat related keyword/value pairs from the fix nvt command can be appended•
Examples:
fix 1 all nvt/body temp 300.0 300.0 100.0
fix 1 all nvt/body temp 300.0 300.0 100.0 drag 0.2
Description:
Perform constant NVT integration to update position, velocity, orientation, and angular velocity each timestep
for body particles in the group using a Nose/Hoover temperature thermostat. V is volume; T is temperature.
This creates a system trajectory consistent with the canonical ensemble.
This fix differs from the fix nvt command, which assumes point particles and only updates their position and
velocity.
The thermostat is applied to both the translational and rotational degrees of freedom for the body particles,
assuming a compute is used which calculates a temperature that includes the rotational degrees of freedom
(see below). The translational degrees of freedom can also have a bias velocity removed from them before
thermostatting takes place; see the description below.
Additional parameters affecting the thermostat are specified by keywords and values documented with the fix
nvt command. See, for example, discussion of the temp and drag keywords.
This fix computes a temperature each timestep. To do this, the fix creates its own compute of style
"temp/body", as if this command had been issued:
compute fix-ID_temp group-ID temp/body
See the compute temp/body command for details. Note that the ID of the new compute is the fix-ID +
underscore + "temp", and the group for the new compute is the same as the fix group.
Note that this is NOT the compute used by thermodynamic output (see the thermo_style command) with ID =
thermo_temp. This means you can change the attributes of this fix's temperature (e.g. its degrees-of-freedom)
via the compute_modify command or print this temperature during thermodynamic output via the
thermo_style custom command using the appropriate compute-ID. It also means that changing attributes of
thermo_temp will have no effect on this fix.
Like other fixes that perform thermostatting, this fix can be used with compute commands that calculate a
temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass velocity from
a group of atoms or only calculating temperature on the x-component of velocity or only calculating
LAMMPS Users Manual
972

temperature for atoms in a geometric region. This is not done by default, but only if the fix_modify command
is used to assign a temperature compute to this fix that includes such a bias term. See the doc pages for
individual compute commands to determine which ones include a bias. In this case, the thermostat works in
the following manner: the current temperature is calculated taking the bias into account, bias is removed from
each atom, thermostatting is performed on the remaining thermal degrees of freedom, and the bias is added
back in.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover thermostat to binary restart files. See the read_restart command
for info on how to re-specify a fix in an input script that reads a restart file, so that the operation of the fix
continues in an uninterrupted fashion.
The fix_modify temp option is supported by this fix. You can use it to assign a compute you have defined to
this fix which will be used in its thermostatting procedure.
The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
thermostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix nvt command.
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the BODY package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix requires that atoms store torque and angular momentum and a quaternion as defined by the
atom_style body command.
Related commands:
LAMMPS Users Manual
973

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nvt/manifold/rattle command
Syntax:
fix ID group-ID nvt/manifold/rattle tol maxit manifold manifold-args keyword value ...
ID, group-ID are documented in fix command• nvt/manifold/rattle = style name of this fix command• tol = tolerance to which Newton iteration must converge• maxit = maximum number of iterations to perform• manifold = name of the manifold• manifold-args = parameters for the manifold• one or more keyword/value pairs may be appended
keyword = temp or tchain or every
temp values = Tstart Tstop Tdamp
Tstart, Tstop = external temperature at start/end of run
Tdamp = temperature damping parameter (time units)
tchain value = N
N = length of thermostat chain (1 = single thermostat)
every value = N
N = print info about iteration every N steps. N = 0 means no output
•
Examples:
fix 1 all nvt/manifold/rattle 1e-4 10 cylinder 3.0 temp 1.0 1.0 10.0
Description:
This fix combines the RATTLE-based (Andersen) time integrator of fix nve/manifold/rattle (Paquay) with a
Nose-Hoover-chain thermostat to sample the canonical ensemble of particles constrained to a curved surface
(manifold). This sampling does suffer from discretization bias of O(dt). For a list of currently supported
manifolds and their parameters, see manifolds
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the USER-MANIFOLD package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Related commands:
fix nve/manifold/rattle, fix manifoldforce Default: every = 0
LAMMPS Users Manual
975


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nvt/sllod command
fix nvt/sllod/intel command
fix nvt/sllod/omp command
Syntax:
fix ID group-ID nvt/sllod keyword value ...
ID, group-ID are documented in fix command• nvt/sllod = style name of this fix command• additional thermostat related keyword/value pairs from the fix nvt command can be appended•
Examples:
fix 1 all nvt/sllod temp 300.0 300.0 100.0
fix 1 all nvt/sllod temp 300.0 300.0 100.0 drag 0.2
Description:
Perform constant NVT integration to update positions and velocities each timestep for atoms in the group
using a Nose/Hoover temperature thermostat. V is volume; T is temperature. This creates a system trajectory
consistent with the canonical ensemble.
This thermostat is used for a simulation box that is changing size and/or shape, for example in a
non-equilibrium MD (NEMD) simulation. The size/shape change is induced by use of the fix deform
command, so each point in the simulation box can be thought of as having a "streaming" velocity. This
position-dependent streaming velocity is subtracted from each atom's actual velocity to yield a thermal
velocity which is used for temperature computation and thermostatting. For example, if the box is being
sheared in x, relative to y, then points at the bottom of the box (low y) have a small x velocity, while points at
the top of the box (hi y) have a large x velocity. These velocities do not contribute to the thermal
"temperature" of the atom.
NOTE: Fix deform has an option for remapping either atom coordinates or velocities to the changing
simulation box. To use fix nvt/sllod, fix deform should NOT remap atom positions, because fix nvt/sllod
adjusts the atom positions and velocities to create a velocity profile that matches the changing box size/shape.
Fix deform SHOULD remap atom velocities when atoms cross periodic boundaries since that is consistent
with maintaining the velocity profile created by fix nvt/sllod. LAMMPS will give an error if this setting is not
consistent.
The SLLOD equations of motion, originally proposed by Hoover and Ladd (see (Evans and Morriss)), were
proven to be equivalent to Newton's equations of motion for shear flow by (Evans and Morriss). They were
later shown to generate the desired velocity gradient and the correct production of work by stresses for all
forms of homogeneous flow by (Daivis and Todd). As implemented in LAMMPS, they are coupled to a
Nose/Hoover chain thermostat in a velocity Verlet formulation, closely following the implementation used for
the fix nvt command.
LAMMPS Users Manual
977

Additional parameters affecting the thermostat are specified by keywords and values documented with the fix
nvt command. See, for example, discussion of the temp and drag keywords.
This fix computes a temperature each timestep. To do this, the fix creates its own compute of style
"temp/deform", as if this command had been issued:
compute fix-ID_temp group-ID temp/deform
See the compute temp/deform command for details. Note that the ID of the new compute is the fix-ID +
underscore + "temp", and the group for the new compute is the same as the fix group.
Note that this is NOT the compute used by thermodynamic output (see the thermo_style command) with ID =
thermo_temp. This means you can change the attributes of this fix's temperature (e.g. its degrees-of-freedom)
via the compute_modify command or print this temperature during thermodynamic output via the
thermo_style custom command using the appropriate compute-ID. It also means that changing attributes of
thermo_temp will have no effect on this fix.
Like other fixes that perform thermostatting, this fix can be used with compute commands that calculate a
temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass velocity from
a group of atoms or only calculating temperature on the x-component of velocity or only calculating
temperature for atoms in a geometric region. This is not done by default, but only if the fix_modify command
is used to assign a temperature compute to this fix that includes such a bias term. See the doc pages for
individual compute commands to determine which ones include a bias. In this case, the thermostat works in
the following manner: the current temperature is calculated taking the bias into account, bias is removed from
each atom, thermostatting is performed on the remaining thermal degrees of freedom, and the bias is added
back in.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover thermostat to binary restart files. See the read_restart command
for info on how to re-specify a fix in an input script that reads a restart file, so that the operation of the fix
continues in an uninterrupted fashion.
The fix_modify temp option is supported by this fix. You can use it to assign a compute you have defined to
this fix which will be used in its thermostatting procedure.
LAMMPS Users Manual
978

The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
thermostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix nvt command.
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix works best without Nose-Hoover chain thermostats, i.e. using tchain = 1. Setting tchain to larger
values can result in poor equilibration.
Related commands:
fix nve, fix nvt, fix temp/rescale, fix langevin, fix_modify, compute temp/deform
Default:
Same as fix nvt, except tchain = 1.
(Evans and Morriss) Evans and Morriss, Phys Rev A, 30, 1528 (1984).
(Daivis and Todd) Daivis and Todd, J Chem Phys, 124, 194103 (2006).
LAMMPS Users Manual
979

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nvt/sllod/eff command
Syntax:
fix ID group-ID nvt/sllod/eff keyword value ...
ID, group-ID are documented in fix command• nvt/sllod/eff = style name of this fix command• additional thermostat related keyword/value pairs from the fix nvt/eff command can be appended•
Examples:
fix 1 all nvt/sllod/eff temp 300.0 300.0 0.1
fix 1 all nvt/sllod/eff temp 300.0 300.0 0.1 drag 0.2
Description:
Perform constant NVT integration to update positions and velocities each timestep for nuclei and electrons in
the group for the electron force field model, using a Nose/Hoover temperature thermostat. V is volume; T is
temperature. This creates a system trajectory consistent with the canonical ensemble.
The operation of this fix is exactly like that described by the fix nvt/sllod command, except that the radius and
radial velocity of electrons are also updated and thermostatted. Likewise the temperature calculated by the fix,
using the compute it creates (as discussed in the fix nvt, npt, and nph doc page), is performed with a compute
temp/deform/eff command that includes the eFF contribution to the temperature from the electron radial
velocity.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover thermostat to binary restart files. See the read_restart command
for info on how to re-specify a fix in an input script that reads a restart file, so that the operation of the fix
continues in an uninterrupted fashion.
The fix_modify temp option is supported by this fix. You can use it to assign a compute you have defined to
this fix which will be used in its thermostatting procedure.
The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
thermostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix nvt/eff command.
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
LAMMPS Users Manual
980

This fix is part of the USER-EFF package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix works best without Nose-Hoover chain thermostats, i.e. using tchain = 1. Setting tchain to larger
values can result in poor equilibration.
Related commands:
fix nve/eff, fix nvt/eff, fix langevin/eff, fix nvt/sllod, fix_modify, compute temp/deform/eff
Default:
Same as fix nvt/eff, except tchain = 1.
(Tuckerman) Tuckerman, Mundy, Balasubramanian, Klein, J Chem Phys, 106, 5615 (1997).
LAMMPS Users Manual
981

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix nvt/sphere command
fix nvt/sphere/omp command
Syntax:
fix ID group-ID nvt/sphere keyword value ...
ID, group-ID are documented in fix command• nvt/sphere = style name of this fix command• zero or more keyword/value pairs may be appended• keyword = disc
disc value = none = treat particles as 2d discs, not spheres
•
additional thermostat related keyword/value pairs from the fix nvt command can be appended•
Examples:
fix 1 all nvt/sphere temp 300.0 300.0 100.0
fix 1 all nvt/sphere temp 300.0 300.0 100.0 disc
fix 1 all nvt/sphere temp 300.0 300.0 100.0 drag 0.2
Description:
Perform constant NVT integration to update position, velocity, and angular velocity each timestep for
finite-size spherical particles in the group using a Nose/Hoover temperature thermostat. V is volume; T is
temperature. This creates a system trajectory consistent with the canonical ensemble.
This fix differs from the fix nvt command, which assumes point particles and only updates their position and
velocity.
The thermostat is applied to both the translational and rotational degrees of freedom for the spherical particles,
assuming a compute is used which calculates a temperature that includes the rotational degrees of freedom
(see below). The translational degrees of freedom can also have a bias velocity removed from them before
thermostatting takes place; see the description below.
If the disc keyword is used, then each particle is treated as a 2d disc (circle) instead of as a sphere. This is only
possible for 2d simulations, as defined by the dimension keyword. The only difference between discs and
spheres in this context is their moment of inertia, as used in the time integration.
Additional parameters affecting the thermostat are specified by keywords and values documented with the fix
nvt command. See, for example, discussion of the temp and drag keywords.
This fix computes a temperature each timestep. To do this, the fix creates its own compute of style
"temp/sphere", as if this command had been issued:
compute fix-ID_temp group-ID temp/sphere
LAMMPS Users Manual
982

See the compute temp/sphere command for details. Note that the ID of the new compute is the fix-ID +
underscore + "temp", and the group for the new compute is the same as the fix group.
Note that this is NOT the compute used by thermodynamic output (see the thermo_style command) with ID =
thermo_temp. This means you can change the attributes of this fix's temperature (e.g. its degrees-of-freedom)
via the compute_modify command or print this temperature during thermodynamic output via the
thermo_style custom command using the appropriate compute-ID. It also means that changing attributes of
thermo_temp will have no effect on this fix.
Like other fixes that perform thermostatting, this fix can be used with compute commands that calculate a
temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass velocity from
a group of atoms or only calculating temperature on the x-component of velocity or only calculating
temperature for atoms in a geometric region. This is not done by default, but only if the fix_modify command
is used to assign a temperature compute to this fix that includes such a bias term. See the doc pages for
individual compute commands to determine which ones include a bias. In this case, the thermostat works in
the following manner: the current temperature is calculated taking the bias into account, bias is removed from
each atom, thermostatting is performed on the remaining thermal degrees of freedom, and the bias is added
back in.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the state of the Nose/Hoover thermostat to binary restart files. See the read_restart command
for info on how to re-specify a fix in an input script that reads a restart file, so that the operation of the fix
continues in an uninterrupted fashion.
The fix_modify temp option is supported by this fix. You can use it to assign a compute you have defined to
this fix which will be used in its thermostatting procedure.
The fix_modify energy option is supported by this fix to add the energy change induced by Nose/Hoover
thermostatting to the system's potential energy as part of thermodynamic output.
This fix computes the same global scalar and global vector of quantities as does the fix nvt command.
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
LAMMPS Users Manual
983
This fix is not invoked during energy minimization.
Restrictions:
This fix requires that atoms store torque and angular velocity (omega) and a radius as defined by the
atom_style sphere command.
All particles in the group must be finite-size spheres. They cannot be point particles.
Use of the disc keyword is only allowed for 2d simulations, as defined by the dimension keyword.
Related commands:
fix nvt, fix nve_sphere, fix nvt_asphere, fix npt_sphere, fix_modify
Default: none
LAMMPS Users Manual
984

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix oneway command
Syntax:
fix ID group-ID oneway N region-ID direction
ID, group-ID are documented in fix command• oneway = style name of this fix command• N = apply this fix every this many timesteps• region-ID = ID of region where fix is active• direction = x or -x or y or -y or z or -z = coordinate and direction of the oneway constraint•
Examples:
fix ions oneway 10 semi -x
fix all oneway 1 left -z
fix all oneway 1 right z
Description:
Enforce that particles in the group and in a given region can only move in one direction. This is done by
reversing a particle's velocity component, if it has the wrong sign in the specified dimension. The effect is that
the particle moves in one direction only.
This can be used, for example, as a simple model of a semi-permeable membrane, or as an implementation of
Maxwell's demon.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
fix wall/reflect command
Default: none
LAMMPS Users Manual
985

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix orient/fcc command
fix orient/bcc command
fix ID group-ID orient/fcc nstats dir alat dE cutlo cuthi file0 file1
fix ID group-ID orient/bcc nstats dir alat dE cutlo cuthi file0 file1
ID, group-ID are documented in fix command• nstats = print stats every this many steps, 0 = never• dir = 0/1 for which crystal is used as reference• alat = fcc/bcc cubic lattice constant (distance units)• dE = energy added to each atom (energy units)• cutlo,cuthi = values between 0.0 and 1.0, cutlo < cuthi• file0,file1 = files that specify orientation of each grain•
Examples:
fix gb all orient/fcc 0 1 4.032008 0.001 0.25 0.75 xi.vec chi.vec
fix gb all orient/bcc 0 1 2.882 0.001 0.25 0.75 ngb.left ngb.right
Description:
The fix applies an orientation-dependent force to atoms near a planar grain boundary which can be used to
induce grain boundary migration (in the direction perpendicular to the grain boundary plane). The motivation
and explanation of this force and its application are described in (Janssens). The adaptation to bcc crystals is
described in (Wicaksono1). The computed force is only applied to atoms in the fix group.
The basic idea is that atoms in one grain (on one side of the boundary) have a potential energy dE added to
them. Atoms in the other grain have 0.0 potential energy added. Atoms near the boundary (whose neighbor
environment is intermediate between the two grain orientations) have an energy between 0.0 and dE added.
This creates an effective driving force to reduce the potential energy of atoms near the boundary by pushing
them towards one of the grain orientations. For dir = 1 and dE > 0, the boundary will thus move so that the
grain described by file0 grows and the grain described by file1 shrinks. Thus this fix is designed for
simulations of two-grain systems, either with one grain boundary and free surfaces parallel to the boundary, or
a system with periodic boundary conditions and two equal and opposite grain boundaries. In either case, the
entire system can displace during the simulation, and such motion should be accounted for in measuring the
grain boundary velocity.
The potential energy added to atom I is given by these formulas
LAMMPS Users Manual
986
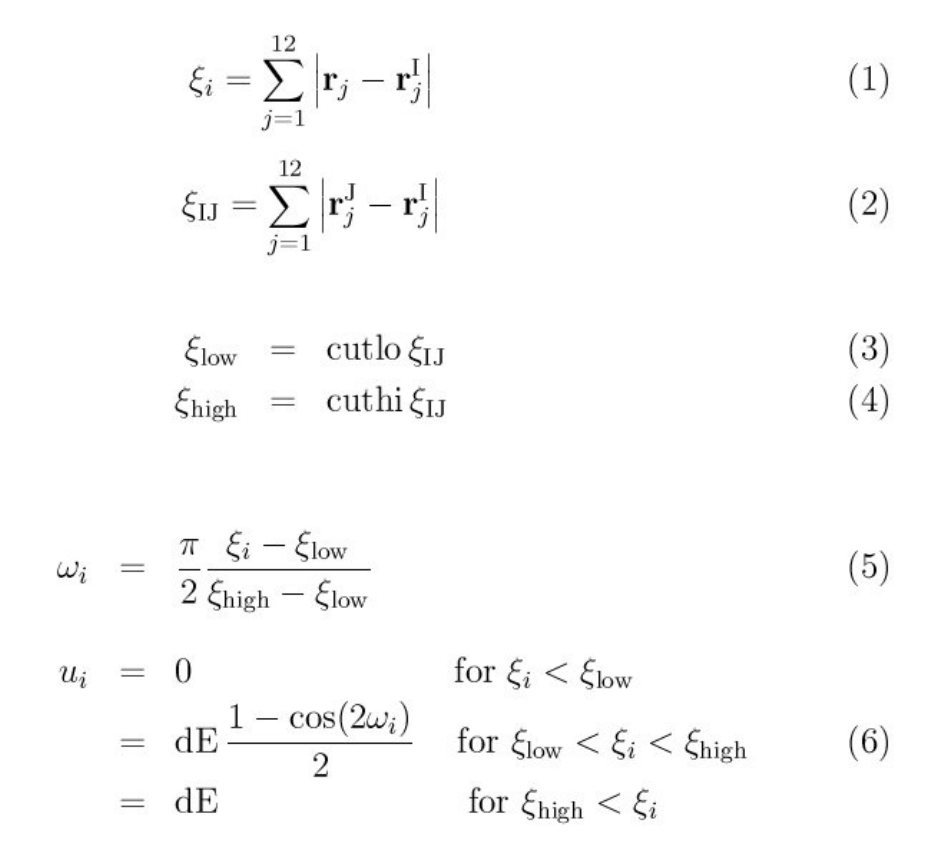
which are fully explained in (Janssens). For fcc crystals this order parameter Xi for atom I in equation (1) is a
sum over the 12 nearest neighbors of atom I. For bcc crystals it is the corresponding sum of the 8 nearest
neighbors. Rj is the vector from atom I to its neighbor J, and RIj is a vector in the reference (perfect) crystal.
That is, if dir = 0/1, then RIj is a vector to an atom coord from file 0/1. Equation (2) gives the expected value
of the order parameter XiIJ in the other grain. Hi and lo cutoffs are defined in equations (3) and (4), using the
input parameters cutlo and cuthi as thresholds to avoid adding grain boundary energy when the deviation in
the order parameter from 0 or 1 is small (e.g. due to thermal fluctuations in a perfect crystal). The added
potential energy Ui for atom I is given in equation (6) where it is interpolated between 0 and dE using the two
threshold Xi values and the Wi value of equation (5).
The derivative of this energy expression gives the force on each atom which thus depends on the orientation
of its neighbors relative to the 2 grain orientations. Only atoms near the grain boundary feel a net force which
tends to drive them to one of the two grain orientations.
In equation (1), the reference vector used for each neighbor is the reference vector closest to the actual
neighbor position. This means it is possible two different neighbors will use the same reference vector. In
such cases, the atom in question is far from a perfect orientation and will likely receive the full dE addition, so
the effect of duplicate reference vector usage is small.
LAMMPS Users Manual
987
The dir parameter determines which grain wants to grow at the expense of the other. A value of 0 means the
first grain will shrink; a value of 1 means it will grow. This assumes that dE is positive. The reverse will be
true if dE is negative.
The alat parameter is the cubic lattice constant for the fcc or bcc material and is only used to compute a cutoff
distance of 1.57 * alat / sqrt(2) for finding the 12 or 8 nearest neighbors of each atom (which should be valid
for an fcc or bcc crystal). A longer/shorter cutoff can be imposed by adjusting alat. If a particular atom has
less than 12 or 8 neighbors within the cutoff, the order parameter of equation (1) is effectively multiplied by
12 or 8 divided by the actual number of neighbors within the cutoff.
The dE parameter is the maximum amount of additional energy added to each atom in the grain which wants
to shrink.
The cutlo and cuthi parameters are used to reduce the force added to bulk atoms in each grain far away from
the boundary. An atom in the bulk surrounded by neighbors at the ideal grain orientation would compute an
order parameter of 0 or 1 and have no force added. However, thermal vibrations in the solid will cause the
order parameters to be greater than 0 or less than 1. The cutoff parameters mask this effect, allowing forces to
only be added to atoms with order-parameters between the cutoff values.
File0 and file1 are filenames for the two grains which each contain 6 vectors (6 lines with 3 values per line)
which specify the grain orientations. Each vector is a displacement from a central atom (0,0,0) to a nearest
neighbor atom in an fcc lattice at the proper orientation. The vector lengths should all be identical since an fcc
lattice has a coordination number of 12. Only 6 are listed due to symmetry, so the list must include one from
each pair of equal-and-opposite neighbors. A pair of orientation files for a Sigma=5 tilt boundary are shown
below. A tutorial that can help for writing the orientation files is given in (Wicaksono2)
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the potential energy of atom interactions with the
grain boundary driving force to the system's potential energy as part of thermodynamic output.
The fix_modify respa option is supported by these fixes. This allows to set at which level of the r-RESPA
integrator a fix is adding its forces. Default is the outermost level.
This fix calculates a global scalar which can be accessed by various output commands. The scalar is the
potential energy change due to this fix. The scalar value calculated by this fix is "extensive".
This fix also calculates a per-atom array which can be accessed by various output commands. The array stores
the order parameter Xi and normalized order parameter (0 to 1) for each atom. The per-atom values can be
accessed on any timestep.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the MISC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
LAMMPS Users Manual
988

This fix should only be used with fcc or bcc lattices.
Related commands:
fix_modify
Default: none
(Janssens) Janssens, Olmsted, Holm, Foiles, Plimpton, Derlet, Nature Materials, 5, 124-127 (2006).
(Wicaksono1) Wicaksono, Sinclair, Militzer, Computational Materials Science, 117, 397-405 (2016).
(Wicaksono2) Wicaksono, figshare, https://dx.doi.org/10.6084/m9.figshare.1488628.v1 (2015).
For illustration purposes, here are example files that specify a Sigma=5 tilt boundary. This is for a lattice
constant of 3.5706 Angs.
file0:
0.798410432046075 1.785300000000000 1.596820864092150
-0.798410432046075 1.785300000000000 -1.596820864092150
2.395231296138225 0.000000000000000 0.798410432046075
0.798410432046075 0.000000000000000 -2.395231296138225
1.596820864092150 1.785300000000000 -0.798410432046075
1.596820864092150 -1.785300000000000 -0.798410432046075
file1:
-0.798410432046075 1.785300000000000 1.596820864092150
0.798410432046075 1.785300000000000 -1.596820864092150
0.798410432046075 0.000000000000000 2.395231296138225
2.395231296138225 0.000000000000000 -0.798410432046075
1.596820864092150 1.785300000000000 0.798410432046075
1.596820864092150 -1.785300000000000 0.798410432046075
LAMMPS Users Manual
989

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix phonon command
Syntax:
fix ID group-ID phonon N Noutput Nwait map_file prefix keyword values ...
ID, group-ID are documented in fix command• phonon = style name of this fix command• N = measure the Green's function every this many timesteps• Noutput = output the dynamical matrix every this many measurements• Nwait = wait this many timesteps before measuring• map_file = file or GAMMA
file is the file that contains the mapping info between atom ID and the lattice indices.
GAMMA flags to treate the whole simulation box as a unit cell, so that the mapping
info can be generated internally. In this case, dynamical matrix at only the gamma-point
will/can be evaluated.
•
prefix = prefix for output files• one or none keyword/value pairs may be appended• keyword = sysdim or nasr
sysdim value = d
d = dimension of the system, usually the same as the MD model dimension
nasr value = n
n = number of iterations to enforce the acoustic sum rule
•
Examples:
fix 1 all phonon 20 5000 200000 map.in LJ1D sysdim 1
fix 1 all phonon 20 5000 200000 map.in EAM3D
fix 1 all phonon 10 5000 500000 GAMMA EAM0D nasr 100
Description:
Calculate the dynamical matrix from molecular dynamics simulations based on fluctuation-dissipation theory
for a group of atoms.
Consider a crystal with \(N\) unit cells in three dimensions labeled \(l = (l_1, l_2, l_3)\) where \(l_i\) are
integers. Each unit cell is defined by three linearly independent vectors \(\mathbf{a}_1\), \(\mathbf{a}_2\),
\(\mathbf{a}_3\) forming a parallelipiped, containing \(K\) basis atoms labeled \(k\).
Based on fluctuation-dissipation theory, the force constant coefficients of the system in reciprocal space are
given by (Campana , Kong)
\begin{equation} \mathbf{\Phi}_{k\alpha,k^\prime \beta}(\mathbf{q}) = k_B T
\mathbf{G}^{-1}_{k\alpha,k^\prime \beta}(\mathbf{q}) \end{equation}
where \(\mathbf{G}\) is the Green's functions coefficients given by
LAMMPS Users Manual
990
\begin{equation} \mathbf{G}_{k\alpha,k^\prime \beta}(\mathbf{q}) = \left<
\mathbf{u}_{k\alpha}(\mathbf{q}) \bullet \mathbf{u}_{k^\prime \beta}^*(\mathbf{q}) \right>
\end{equation}
where \(\left< \ldots \right>\) denotes the ensemble average, and
\begin{equation} \mathbf{u}_{k\alpha}(\mathbf{q}) = \sum_l \mathbf{u}_{l k \alpha}
\exp{(i\mathbf{qr}_l)} \end{equation}
is the \(\alpha\) component of the atomic displacement for the \(k\) th atom in the unit cell in reciprocal space
at \(\mathbf{q}\). In practice, the Green's functions coefficients can also be measured according to the
following formula,
\begin{equation} \mathbf{G}_{k\alpha,k^\prime \beta}(\mathbf{q}) = \left< \mathbf{R}_{k
\alpha}(\mathbf{q}) \bullet \mathbf{R}^*_{k^\prime \beta}(\mathbf{q}) \right> - \left_{k
\alpha}(\mathbf{q}) \bullet \left^*_{k^\prime \beta}(\mathbf{q}) \end{equation}
where \(\mathbf{R}\) is the instantaneous positions of atoms, and \(\left\) is the averaged atomic positions. It
gives essentially the same results as the displacement method and is easier to implement in an MD code.
Once the force constant matrix is known, the dynamical matrix \(\mathbf{D}\) can then be obtained by
\begin{equation} \mathbf{D}_{k\alpha, k^\prime\beta}(\mathbf{q}) = (m_k m_{k^\prime})^{-\frac{1}{2}}
\mathbf{\Phi}_{k \alpha, k^\prime \beta}(\mathbf{q}) \end{equation}
whose eigenvalues are exactly the phonon frequencies at \(\mathbf{q}\).
This fix uses positions of atoms in the specified group and calculates two-point correlations. To achieve this.
the positions of the atoms are examined every Nevery steps and are Fourier-transformed into reciprocal space,
where the averaging process and correlation computation is then done. After every Noutput measurements, the
matrix \(\mathbf{G}(\mathbf{q})\) is calculated and inverted to obtain the elastic stiffness coefficients. The
dynamical matrices are then constructed and written to prefix.bin.timestep files in binary format and to the file
prefix.log for each wavevector \(\mathbf{q}\).
A detailed description of this method can be found in (Kong2011).
The sysdim keyword is optional. If specified with a value smaller than the dimensionality of the LAMMPS
simulation, its value is used for the dynamical matrix calculation. For example, using LAMMPS ot model a
2D or 3D system, the phonon dispersion of a 1D atomic chain can be computed using sysdim = 1.
The nasr keyword is optional. An iterative procedure is employed to enforce the acoustic sum rule on \(\Phi\)
at \(\Gamma\), and the number provided by keyword nasr gives the total number of iterations. For a system
whose unit cell has only one atom, nasr = 1 is sufficient; for other systems, nasr = 10 is typically sufficient.
The map_file contains the mapping information between the lattice indices and the atom IDs, which tells the
code which atom sits at which lattice point; the lattice indices start from 0. An auxiliary code, latgen, can be
employed to generate the compatible map file for various crystals.
In case one simulates an aperiodic system, where the whole simulation box is treated as a unit cell, one can set
map_file as GAMMA, so that the mapping info will be generated internally and a file is not needed. In this
case, the dynamical matrix at only the gamma-point will/can be evaluated. Please keep in mind that
LAMMPS Users Manual
991

fix-phonon is designed for cyrstals, it will be inefficient and even degrade the performance of lammps in case
the unit cell is too large.
The calculated dynamical matrix elements are written out in energy/distance^2/mass units. The coordinates
for q points in the log file is in the units of the basis vectors of the corresponding reciprocal lattice.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify temp option is supported by this fix. You can use it to change the temperature compute from
thermo_temp to the one that reflects the true temperature of atoms in the group.
No global scalar or vector or per-atom quantities are stored by this fix for access by various output commands.
Instead, this fix outputs its initialization information (including mapping information) and the calculated
dynamical matrices to the file prefix.log, with the specified prefix. The dynamical matrices are also written to
files prefix.bin.timestep in binary format. These can be read by the post-processing tool in tools/phonon to
compute the phonon density of states and/or phonon dispersion curves.
No parameter of this fix can be used with the start/stop keywords of the run command.
This fix is not invoked during energy minimization.
Restrictions:
This fix assumes a crystalline system with periodical lattice. The temperature of the system should not exceed
the melting temperature to keep the system in its solid state.
This fix is part of the USER-PHONON package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This fix requires LAMMPS be built with an FFT library. See the Making LAMMPS section for more info.
Related commands:
compute msd
Default:
The option defaults are sysdim = the same dimension as specified by the dimension command, and nasr = 20.
(Campana) C. Campana and M. H. Muser, Practical Green's function approach to the simulation of elastic
semi-infinite solids, Phys. Rev. B [74], 075420 (2006)
(Kong) L.T. Kong, G. Bartels, C. Campana, C. Denniston, and Martin H. Muser, Implementation of Green's
function molecular dynamics: An extension to LAMMPS, Computer Physics Communications
[180](6):1004-1010 (2009).
LAMMPS Users Manual
992
L.T. Kong, C. Denniston, and Martin H. Muser, An improved version of the Green's function molecular
dynamics method, Computer Physics Communications [182](2):540-541 (2011).
(Kong2011) L.T. Kong, Phonon dispersion measured directly from molecular dynamics simulations,
Computer Physics Communications [182](10):2201-2207, (2011).
LAMMPS Users Manual
993
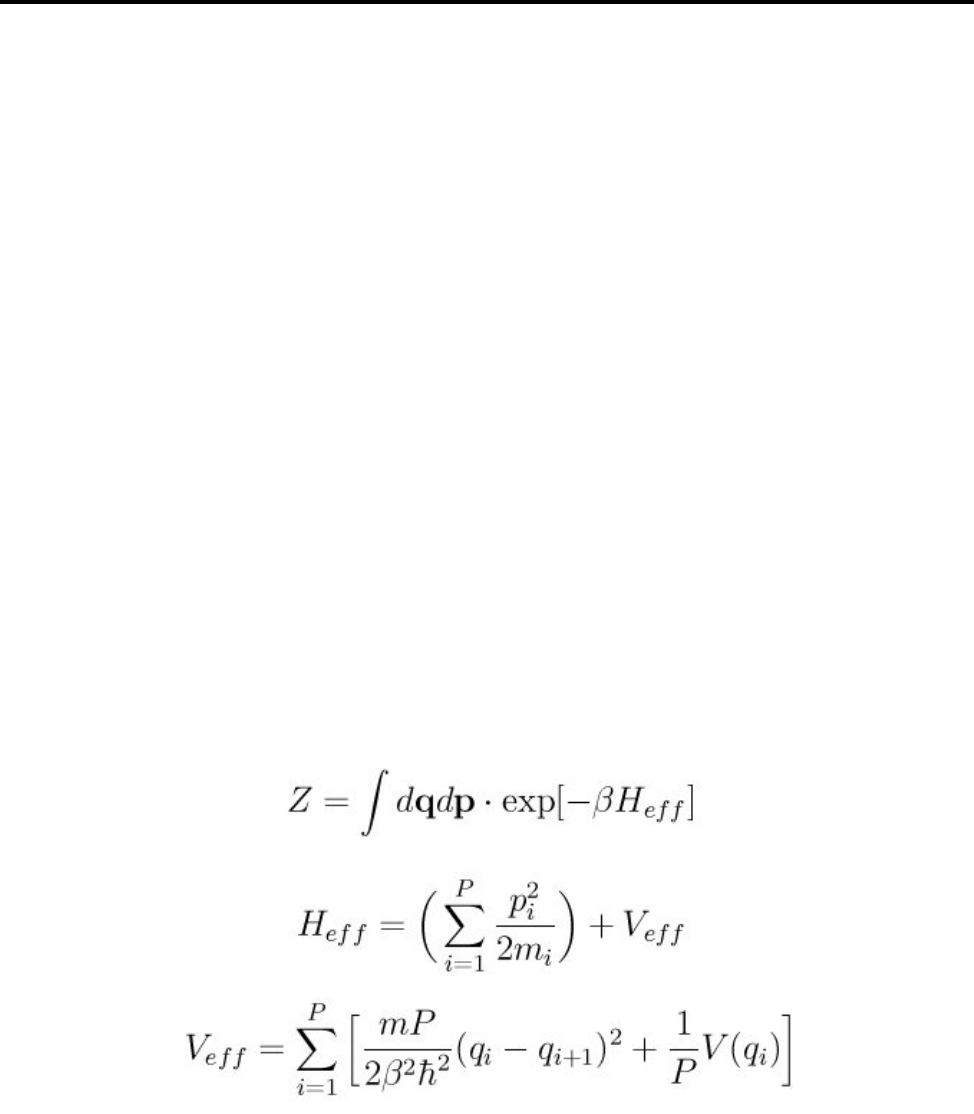
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix pimd command
Syntax:
fix ID group-ID pimd keyword value ...
ID, group-ID are documented in fix command• pimd = style name of this fix command• zero or more keyword/value pairs may be appended• keyword = method or fmass or sp or temp or nhc
method value = pimd or nmpimd or cmd
fmass value = scaling factor on mass
sp value = scaling factor on Planck constant
temp value = temperature (temperarate units)
nhc value = Nc = number of chains in Nose-Hoover thermostat
•
Examples:
fix 1 all pimd method nmpimd fmass 1.0 sp 2.0 temp 300.0 nhc 4
Description:
This command performs quantum molecular dynamics simulations based on the Feynman path integral to
include effects of tunneling and zero-point motion. In this formalism, the isomorphism of a quantum partition
function for the original system to a classical partition function for a ring-polymer system is exploited, to
efficiently sample configurations from the canonical ensemble (Feynman). The classical partition function and
its components are given by the following equations:
The interested user is referred to any of the numerous references on this methodology, but briefly, each
quantum particle in a path integral simulation is represented by a ring-polymer of P quasi-beads, labeled from
1 to P. During the simulation, each quasi-bead interacts with beads on the other ring-polymers with the same
imaginary time index (the second term in the effective potential above). The quasi-beads also interact with the
two neighboring quasi-beads through the spring potential in imaginary-time space (first term in effective
potential). To sample the canonical ensemble, a Nose-Hoover massive chain thermostat is applied
LAMMPS Users Manual
994
(Tuckerman). With the massive chain algorithm, a chain of NH thermostats is coupled to each degree of
freedom for each quasi-bead. The keyword temp sets the target temperature for the system and the keyword
nhc sets the number Nc of thermostats in each chain. For example, for a simulation of N particles with P beads
in each ring-polymer, the total number of NH thermostats would be 3 x N x P x Nc.
NOTE: This fix implements a complete velocity-verlet integrator combined with NH massive chain
thermostat, so no other time integration fix should be used.
The method keyword determines what style of PIMD is performed. A value of pimd is standard PIMD. A
value of nmpimd is for normal-mode PIMD. A value of cmd is for centroid molecular dynamics (CMD). The
difference between the styles is as follows.
In standard PIMD, the value used for a bead's fictitious mass is arbitrary. A common choice is to use Mi =
m/P, which results in the mass of the entire ring-polymer being equal to the real quantum particle. But it can
be difficult to efficiently integrate the equations of motion for the stiff harmonic interactions in the ring
polymers.
A useful way to resolve this issue is to integrate the equations of motion in a normal mode representation,
using Normal Mode Path-Integral Molecular Dynamics (NMPIMD) (Cao1). In NMPIMD, the NH chains are
attached to each normal mode of the ring-polymer and the fictitious mass of each mode is chosen as Mk = the
eigenvalue of the Kth normal mode for k > 0. The k = 0 mode, referred to as the zero-frequency mode or
centroid, corresponds to overall translation of the ring-polymer and is assigned the mass of the real particle.
Motion of the centroid can be effectively uncoupled from the other normal modes by scaling the fictitious
masses to achieve a partial adiabatic separation. This is called a Centroid Molecular Dynamics (CMD)
approximation (Cao2). The time-evolution (and resulting dynamics) of the quantum particles can be used to
obtain centroid time correlation functions, which can be further used to obtain the true quantum correlation
function for the original system. The CMD method also uses normal modes to evolve the system, except only
the k > 0 modes are thermostatted, not the centroid degrees of freedom.
The keyword fmass sets a further scaling factor for the fictitious masses of beads, which can be used for the
Partial Adiabatic CMD (Hone), or to be set as P, which results in the fictitious masses to be equal to the real
particle masses.
The keyword sp is a scaling factor on Planck's constant, which can be useful for debugging or other purposes.
The default value of 1.0 is appropriate for most situations.
The PIMD algorithm in LAMMPS is implemented as a hyper-parallel scheme as described in (Calhoun). In
LAMMPS this is done by using multi-replica feature in LAMMPS, where each quasi-particle system is stored
and simulated on a separate partition of processors. The following diagram illustrates this approach. The
original system with 2 ring polymers is shown in red. Since each ring has 4 quasi-beads (imaginary time
slices), there are 4 replicas of the system, each running on one of the 4 partitions of processors. Each replica
(shown in green) owns one quasi-bead in each ring.
LAMMPS Users Manual
995
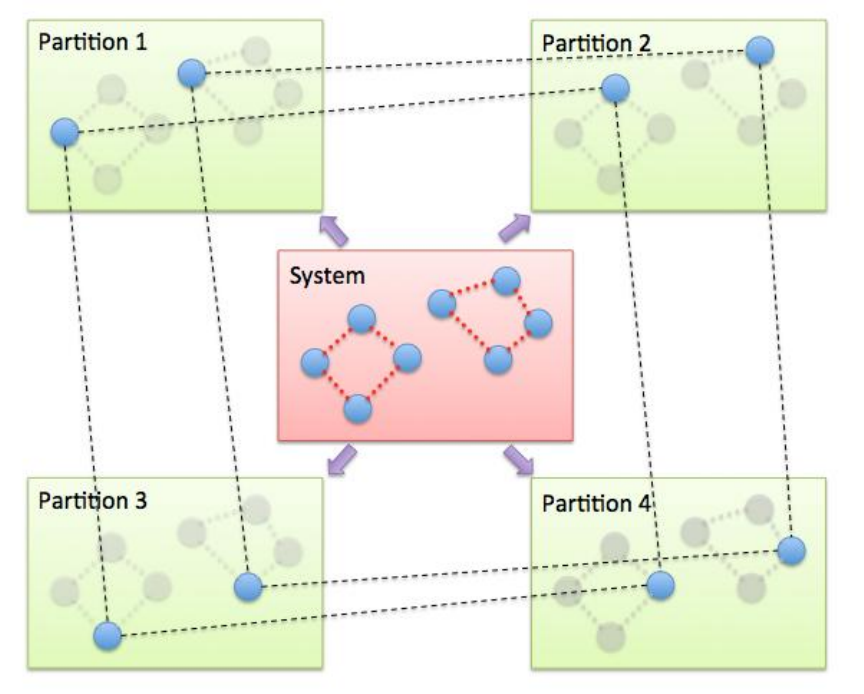
To run a PIMD simulation with M quasi-beads in each ring polymer using N MPI tasks for each partition's
domain-decomposition, you would use P = MxN processors (cores) and run the simulation as follows:
mpirun -np P lmp_mpi -partition MxN -in script
Note that in the LAMMPS input script for a multi-partition simulation, it is often very useful to define a
uloop-style variable such as
variable ibead uloop M pad
where M is the number of quasi-beads (partitions) used in the calculation. The uloop variable can then be used
to manage I/O related tasks for each of the partitions, e.g.
dump dcd all dcd 10 system_${ibead}.dcd
restart 1000 system_${ibead}.restart1 system_${ibead}.restart2
read_restart system_${ibead}.restart2
Restrictions:
This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
A PIMD simulation can be initialized with a single data file read via the read_data command. However, this
means all quasi-beads in a ring polymer will have identical positions and velocities, resulting in identical
LAMMPS Users Manual
996

trajectories for all quasi-beads. To avoid this, users can simply initialize velocities with different random
number seeds assigned to each partition, as defined by the uloop variable, e.g.
velocity all create 300.0 1234${ibead} rot yes dist gaussian
Default:
The keyword defaults are method = pimd, fmass = 1.0, sp = 1.0, temp = 300.0, and nhc = 2.
(Feynman) R. Feynman and A. Hibbs, Chapter 7, Quantum Mechanics and Path Integrals, McGraw-Hill,
New York (1965).
(Tuckerman) M. Tuckerman and B. Berne, J Chem Phys, 99, 2796 (1993).
(Cao1) J. Cao and B. Berne, J Chem Phys, 99, 2902 (1993).
(Cao2) J. Cao and G. Voth, J Chem Phys, 100, 5093 (1994).
(Hone) T. Hone, P. Rossky, G. Voth, J Chem Phys, 124, 154103 (2006).
(Calhoun) A. Calhoun, M. Pavese, G. Voth, Chem Phys Letters, 262, 415 (1996).
LAMMPS Users Manual
997

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix planeforce command
Syntax:
fix ID group-ID planeforce x y z
ID, group-ID are documented in fix command• planeforce = style name of this fix command• x y z = 3-vector that is normal to the plane•
Examples:
fix hold boundary planeforce 1.0 0.0 0.0
Description:
Adjust the forces on each atom in the group so that only the components of force in the plane specified by the
normal vector (x,y,z) remain. This is done by subtracting out the component of force perpendicular to the
plane.
If the initial velocity of the atom is 0.0 (or in the plane), then it should continue to move in the plane
thereafter.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
Restrictions: none
Related commands:
fix lineforce
Default: none
LAMMPS Users Manual
998

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix poems
Syntax:
fix ID group-ID poems keyword values
ID, group-ID are documented in fix command• poems = style name of this fix command• keyword = group or file or molecule
group values = list of group IDs
molecule values = none
file values = filename
•
Examples:
fix 3 fluid poems group clump1 clump2 clump3
fix 3 fluid poems file cluster.list
Description:
Treats one or more sets of atoms as coupled rigid bodies. This means that each timestep the total force and
torque on each rigid body is computed and the coordinates and velocities of the atoms are updated so that the
collection of bodies move as a coupled set. This can be useful for treating a large biomolecule as a collection
of connected, coarse-grained particles.
The coupling, associated motion constraints, and time integration is performed by the software package
Parallelizable Open source Efficient Multibody Software (POEMS) which computes the constrained
rigid-body motion of articulated (jointed) multibody systems (Anderson). POEMS was written and is
distributed by Prof Kurt Anderson, his graduate student Rudranarayan Mukherjee, and other members of his
group at Rensselaer Polytechnic Institute (RPI). Rudranarayan developed the LAMMPS/POEMS interface.
For copyright information on POEMS and other details, please refer to the documents in the poems directory
distributed with LAMMPS.
This fix updates the positions and velocities of the rigid atoms with a constant-energy time integration, so you
should not update the same atoms via other fixes (e.g. nve, nvt, npt, temp/rescale, langevin).
Each body must have a non-degenerate inertia tensor, which means if must contain at least 3 non-collinear
atoms. Which atoms are in which bodies can be defined via several options.
For option group, each of the listed groups is treated as a rigid body. Note that only atoms that are also in the
fix group are included in each rigid body.
For option molecule, each set of atoms in the group with a different molecule ID is treated as a rigid body.
For option file, sets of atoms are read from the specified file and each set is treated as a rigid body. Each line
of the file specifies a rigid body in the following format:
ID type atom1-ID atom2-ID atom3-ID ...
LAMMPS Users Manual
999

ID as an integer from 1 to M (the number of rigid bodies). Type is any integer; it is not used by the fix poems
command. The remaining arguments are IDs of atoms in the rigid body, each typically from 1 to N (the
number of atoms in the system). Only atoms that are also in the fix group are included in each rigid body.
Blank lines and lines that begin with '#' are skipped.
A connection between a pair of rigid bodies is inferred if one atom is common to both bodies. The POEMS
solver treats that atom as a spherical joint with 3 degrees of freedom. Currently, a collection of bodies can
only be connected by joints as a linear chain. The entire collection of rigid bodies can represent one or more
chains. Other connection topologies (tree, ring) are not allowed, but will be added later. Note that if no joints
exist, it is more efficient to use the fix rigid command to simulate the system.
When the poems fix is defined, it will print out statistics on the total # of clusters, bodies, joints, atoms
involved. A cluster in this context means a set of rigid bodies connected by joints.
For computational efficiency, you should turn off pairwise and bond interactions within each rigid body, as
they no longer contribute to the motion. The "neigh_modify exclude" and "delete_bonds" commands can be
used to do this if each rigid body is a group.
For computational efficiency, you should only define one fix poems which includes all the desired rigid
bodies. LAMMPS will allow multiple poems fixes to be defined, but it is more expensive.
The degrees-of-freedom removed by coupled rigid bodies are accounted for in temperature and pressure
computations. Similarly, the rigid body contribution to the pressure virial is also accounted for. The latter is
only correct if forces within the bodies have been turned off, and there is only a single fix poems defined.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the POEMS package. It is only enabled if LAMMPS was built with that package, which
also requires the POEMS library be built and linked with LAMMPS. See the Making LAMMPS section for
more info.
Related commands:
fix rigid, delete_bonds, neigh_modify exclude
Default: none
(Anderson) Anderson, Mukherjee, Critchley, Ziegler, and Lipton "POEMS: Parallelizable Open-source
Efficient Multibody Software ", Engineering With Computers (2006). (link to paper)
LAMMPS Users Manual
1000

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix pour command
Syntax:
fix ID group-ID pour N type seed keyword values ...
ID, group-ID are documented in fix command• pour = style name of this fix command• N = # of particles to insert• type = atom type to assign to inserted particles (offset for molecule insertion)• seed = random # seed (positive integer)• one or more keyword/value pairs may be appended to args• keyword = region or diam or vol or rate or dens or vel or mol or rigid or shake or ignore
region value = region-ID
region-ID = ID of region to use as insertion volume
diam values = dstyle args
dstyle = one or range or poly
one args = D
D = single diameter for inserted particles (distance units)
range args = Dlo Dhi
Dlo,Dhi = range of diameters for inserted particles (distance units)
poly args = Npoly D1 P1 D2 P2 ...
Npoly = # of (D,P) pairs
D1,D2,... = diameter for subset of inserted particles (distance units)
P1,P2,... = percentage of inserted particles with this diameter (0-1)
id values = idflag
idflag = max or next = how to choose IDs for inserted particles and molecules
vol values = fraction Nattempt
fraction = desired volume fraction for filling insertion volume
Nattempt = max # of insertion attempts per particle
rate value = V
V = z velocity (3d) or y velocity (2d) at which
insertion volume moves (velocity units)
dens values = Rholo Rhohi
Rholo,Rhohi = range of densities for inserted particles (mass/volume units)
vel values (3d) = vxlo vxhi vylo vyhi vz
vel values (2d) = vxlo vxhi vy
vxlo,vxhi = range of x velocities for inserted particles (velocity units)
vylo,vyhi = range of y velocities for inserted particles (velocity units)
vz = z velocity (3d) assigned to inserted particles (velocity units)
vy = y velocity (2d) assigned to inserted particles (velocity units)
mol value = template-ID
template-ID = ID of molecule template specified in a separate molecule command
molfrac values = f1 f2 ... fN
f1 to fN = relative probability of creating each of N molecules in template-ID
rigid value = fix-ID
fix-ID = ID of fix rigid/small command
shake value = fix-ID
fix-ID = ID of fix shake command
ignore value = none
skip any line or triangle particles when detecting possible
overlaps with inserted particles
•
Examples:
LAMMPS Users Manual
1001
fix 3 all pour 1000 2 29494 region myblock
fix 2 all pour 10000 1 19985583 region disk vol 0.33 100 rate 1.0 diam range 0.9 1.1
fix 2 all pour 10000 1 19985583 region disk diam poly 2 0.7 0.4 1.5 0.6
fix ins all pour 500 1 4767548 vol 0.8 10 region slab mol object rigid myRigid
Description:
Insert finite-size particles or molecules into the simulation box every few timesteps within a specified region
until N particles or molecules have been inserted. This is typically used to model the pouring of granular
particles into a container under the influence of gravity. For the remainder of this doc page, a single inserted
atom or molecule is referred to as a "particle".
If inserted particles are individual atoms, they are assigned the specified atom type. If they are molecules, the
type of each atom in the inserted molecule is specified in the file read by the molecule command, and those
values are added to the specified atom type. E.g. if the file specifies atom types 1,2,3, and those are the atom
types you want for inserted molecules, then specify type = 0. If you specify type = 2, the in the inserted
molecule will have atom types 3,4,5.
All atoms in the inserted particle are assigned to two groups: the default group "all" and the group specified in
the fix pour command (which can also be "all").
This command must use the region keyword to define an insertion volume. The specified region must have
been previously defined with a region command. It must be of type block or a z-axis cylinder and must be
defined with side = in. The cylinder style of region can only be used with 3d simulations.
Individual atoms are inserted, unless the mol keyword is used. It specifies a template-ID previously defined
using the molecule command, which reads a file that defines the molecule. The coordinates, atom types,
center-of-mass, moments of inertia, etc, as well as any bond/angle/etc and special neighbor information for the
molecule can be specified in the molecule file. See the molecule command for details. The only settings
required to be in this file are the coordinates and types of atoms in the molecule.
If the molecule template contains more than one molecule, the relative probability of depositing each
molecule can be specified by the molfrac keyword. N relative probabilities, each from 0.0 to 1.0, are
specified, where N is the number of molecules in the template. Each time a molecule is inserted, a random
number is used to sample from the list of relative probabilities. The N values must sum to 1.0.
If you wish to insert molecules via the mol keyword, that will be treated as rigid bodies, use the rigid
keyword, specifying as its value the ID of a separate fix rigid/small command which also appears in your
input script.
NOTE: If you wish the new rigid molecules (and other rigid molecules) to be thermostatted correctly via fix
rigid/small/nvt or fix rigid/small/npt, then you need to use the "fix_modify dynamic/dof yes" command for the
rigid fix. This is to inform that fix that the molecule count will vary dynamically.
If you wish to insert molecules via the mol keyword, that will have their bonds or angles constrained via
SHAKE, use the shake keyword, specifying as its value the ID of a separate fix shake command which also
appears in your input script.
Each timestep particles are inserted, they are placed randomly inside the insertion volume so as to mimic a
stream of poured particles. If they are molecules they are also oriented randomly. Each atom in the particle is
tested for overlaps with existing particles, including effects due to periodic boundary conditions if applicable.
LAMMPS Users Manual
1002

If an overlap is detected, another random insertion attempt is made; see the vol keyword discussion below.
The larger the volume of the insertion region, the more particles that can be inserted at any one timestep.
Particles are inserted again after enough time has elapsed that the previously inserted particles fall out of the
insertion volume under the influence of gravity. Insertions continue every so many timesteps until the desired
# of particles has been inserted.
NOTE: If you are monitoring the temperature of a system where the particle count is changing due to adding
particles, you typically should use the compute_modify dynamic yes command for the temperature compute
you are using.
All other keywords are optional with defaults as shown below.
The diam option is only used when inserting atoms and specifies the diameters of inserted particles. There are
3 styles: one, range, or poly. For one, all particles will have diameter D. For range, the diameter of each
particle will be chosen randomly and uniformly between the specified Dlo and Dhi bounds. For poly, a series
of Npoly diameters is specified. For each diameter a percentage value from 0.0 to 1.0 is also specified. The
Npoly percentages must sum to 1.0. For the example shown above with "diam 2 0.7 0.4 1.5 0.6", all inserted
particles will have a diameter of 0.7 or 1.5. 40% of the particles will be small; 60% will be large.
Note that for molecule insertion, the diameters of individual atoms in the molecule can be specified in the file
read by the molecule command. If not specified, the diameter of each atom in the molecule has a default
diameter of 1.0.
The id option has two settings which are used to determine the atom or molecule IDs to assign to inserted
particles/molecules. In both cases a check is done of the current system to find the maximum current atom and
molecule ID of any existing particle. Newly inserted particles and molecules are assigned IDs that increment
those max values. For the max setting, which is the default, this check is done at every insertion step, which
allows for particles to leave the system, and their IDs to potentially be re-used. For the next setting this check
is done only once when the fix is specified, which can be more efficient if you are sure particles will not be
added in some other way.
The vol option specifies what volume fraction of the insertion volume will be filled with particles. For
particles with a size specified by the diam range keyword, they are assumed to all be of maximum diameter
Dhi for purposes of computing their contribution to the volume fraction.
The higher the volume fraction value, the more particles are inserted each timestep. Since inserted particles
cannot overlap, the maximum volume fraction should be no higher than about 0.6. Each timestep particles are
inserted, LAMMPS will make up to a total of M tries to insert the new particles without overlaps, where M =
# of inserted particles * Nattempt. If LAMMPS is unsuccessful at completing all insertions, it prints a
warning.
The dens and vel options enable inserted particles to have a range of densities or xy velocities. The specific
values for a particular inserted particle will be chosen randomly and uniformly between the specified bounds.
Internally, the density value for a particle is converted to a mass, based on the radius (volume) of the particle.
The vz or vy value for option vel assigns a z-velocity (3d) or y-velocity (2d) to each inserted particle.
The rate option moves the insertion volume in the z direction (3d) or y direction (2d). This enables pouring
particles from a successively higher height over time.
The ignore option is useful when running a simulation that used line segment (2d) or triangle (3d) particles,
typically to define boundaries for spherical granular particles to interact with. See the atom_style line or tri
LAMMPS Users Manual
1003

command for details. Lines and triangles store their size, and if the size is large it may overlap (in a spherical
sense) with the insertion region, even if the line/triangle is oriented such that there is no actual overlap. This
can prevent particles from being inserted. The ignore keyword causes the overlap check to skip any line or
triangle particles. Obviously you should only use it if there is in fact no overlap of the line or triangle particles
with the insertion region.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. This means you must be careful when restarting
a pouring simulation, when the restart file was written in the middle of the pouring operation. Specifically,
you should use a new fix pour command in the input script for the restarted simulation that continues the
operation. You will need to adjust the arguments of the original fix pour command to do this.
Also note that because the state of the random number generator is not saved in restart files, you cannot do
"exact" restarts with this fix, where the simulation continues on the same as if no restart had taken place.
However, in a statistical sense, a restarted simulation should produce the same behavior if you adjust the fix
pour parameters appropriately.
None of the fix_modify options are relevant to this fix. No global or per-atom quantities are stored by this fix
for access by various output commands. No parameter of this fix can be used with the start/stop keywords of
the run command. This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the GRANULAR package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
For 3d simulations, a gravity fix in the -z direction must be defined for use in conjunction with this fix. For 2d
simulations, gravity must be defined in the -y direction.
The specified insertion region cannot be a "dynamic" region, as defined by the region command.
Related commands:
fix deposit, fix gravity, region
Default:
Insertions are performed for individual particles, i.e. no mol setting is defined. If the mol keyword is used, the
default for molfrac is an equal probabilities for all molecules in the template. Additional option defaults are
diam = one 1.0, dens = 1.0 1.0, vol = 0.25 50, rate = 0.0, vel = 0.0 0.0 0.0 0.0 0.0 (for 3d), vel = 0.0 0.0 0.0
(for 2d), and id = max.
LAMMPS Users Manual
1004

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix press/berendsen command
Syntax:
fix ID group-ID press/berendsen keyword value ...
ID, group-ID are documented in fix command• press/berendsen = style name of this fix command
one or more keyword value pairs may be appended
keyword = iso or aniso or x or y or z or couple or dilate or modulus
iso or aniso values = Pstart Pstop Pdamp
Pstart,Pstop = scalar external pressure at start/end of run (pressure units)
Pdamp = pressure damping parameter (time units)
x or y or z values = Pstart Pstop Pdamp
Pstart,Pstop = external stress tensor component at start/end of run (pressure units)
Pdamp = stress damping parameter (time units)
couple = none or xyz or xy or yz or xz
modulus value = bulk modulus of system (pressure units)
dilate value = all or partial
•
Examples:
fix 1 all press/berendsen iso 0.0 0.0 1000.0
fix 2 all press/berendsen aniso 0.0 0.0 1000.0 dilate partial
Description:
Reset the pressure of the system by using a Berendsen barostat (Berendsen), which rescales the system
volume and (optionally) the atoms coordinates within the simulation box every timestep.
Regardless of what atoms are in the fix group, a global pressure is computed for all atoms. Similarly, when the
size of the simulation box is changed, all atoms are re-scaled to new positions, unless the keyword dilate is
specified with a value of partial, in which case only the atoms in the fix group are re-scaled. The latter can be
useful for leaving the coordinates of atoms in a solid substrate unchanged and controlling the pressure of a
surrounding fluid.
NOTE: Unlike the fix npt or fix nph commands which perform Nose/Hoover barostatting AND time
integration, this fix does NOT perform time integration. It only modifies the box size and atom coordinates to
effect barostatting. Thus you must use a separate time integration fix, like fix nve or fix nvt to actually update
the positions and velocities of atoms. This fix can be used in conjunction with thermostatting fixes to control
the temperature, such as fix nvt or fix langevin or fix temp/berendsen.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting and barostatting.
The barostat is specified using one or more of the iso, aniso, x, y, z, and couple keywords. These keywords
give you the ability to specify the 3 diagonal components of an external stress tensor, and to couple various of
these components together so that the dimensions they represent are varied together during a constant-pressure
simulation. Unlike the fix npt and fix nph commands, this fix cannot be used with triclinic (non-orthogonal)
simulation boxes to control all 6 components of the general pressure tensor.
LAMMPS Users Manual
1005

The target pressures for each of the 3 diagonal components of the stress tensor can be specified independently
via the x, y, z, keywords, which correspond to the 3 simulation box dimensions. For each component, the
external pressure or tensor component at each timestep is a ramped value during the run from Pstart to Pstop.
If a target pressure is specified for a component, then the corresponding box dimension will change during a
simulation. For example, if the y keyword is used, the y-box length will change. A box dimension will not
change if that component is not specified, although you have the option to change that dimension via the fix
deform command.
For all barostat keywords, the Pdamp parameter determines the time scale on which pressure is relaxed. For
example, a value of 10.0 means to relax the pressure in a timespan of (roughly) 10 time units (tau or fmsec or
psec - see the units command).
NOTE: A Berendsen barostat will not work well for arbitrary values of Pdamp. If Pdamp is too small, the
pressure and volume can fluctuate wildly; if it is too large, the pressure will take a very long time to
equilibrate. A good choice for many models is a Pdamp of around 1000 timesteps. However, note that Pdamp
is specified in time units, and that timesteps are NOT the same as time units for most units settings.
NOTE: The relaxation time is actually also a function of the bulk modulus of the system (inverse of
isothermal compressibility). The bulk modulus has units of pressure and is the amount of pressure that would
need to be applied (isotropically) to reduce the volume of the system by a factor of 2 (assuming the bulk
modulus was a constant, independent of density, which it's not). The bulk modulus can be set via the keyword
modulus. The Pdamp parameter is effectively multiplied by the bulk modulus, so if the pressure is relaxing
faster than expected or desired, increasing the bulk modulus has the same effect as increasing Pdamp. The
converse is also true. LAMMPS does not attempt to guess a correct value of the bulk modulus; it just uses
10.0 as a default value which gives reasonable relaxation for a Lennard-Jones liquid, but will be way off for
other materials and way too small for solids. Thus you should experiment to find appropriate values of Pdamp
and/or the modulus when using this fix.
The couple keyword allows two or three of the diagonal components of the pressure tensor to be "coupled"
together. The value specified with the keyword determines which are coupled. For example, xz means the Pxx
and Pzz components of the stress tensor are coupled. Xyz means all 3 diagonal components are coupled.
Coupling means two things: the instantaneous stress will be computed as an average of the corresponding
diagonal components, and the coupled box dimensions will be changed together in lockstep, meaning coupled
dimensions will be dilated or contracted by the same percentage every timestep. The Pstart, Pstop, Pdamp
parameters for any coupled dimensions must be identical. Couple xyz can be used for a 2d simulation; the z
dimension is simply ignored.
The iso and aniso keywords are simply shortcuts that are equivalent to specifying several other keywords
together.
The keyword iso means couple all 3 diagonal components together when pressure is computed (hydrostatic
pressure), and dilate/contract the dimensions together. Using "iso Pstart Pstop Pdamp" is the same as
specifying these 4 keywords:
x Pstart Pstop Pdamp
y Pstart Pstop Pdamp
z Pstart Pstop Pdamp
couple xyz
The keyword aniso means x, y, and z dimensions are controlled independently using the Pxx, Pyy, and Pzz
components of the stress tensor as the driving forces, and the specified scalar external pressure. Using "aniso
LAMMPS Users Manual
1006

Pstart Pstop Pdamp" is the same as specifying these 4 keywords:
x Pstart Pstop Pdamp
y Pstart Pstop Pdamp
z Pstart Pstop Pdamp
couple none
This fix computes a temperature and pressure each timestep. To do this, the fix creates its own computes of
style "temp" and "pressure", as if these commands had been issued:
compute fix-ID_temp group-ID temp
compute fix-ID_press group-ID pressure fix-ID_temp
See the compute temp and compute pressure commands for details. Note that the IDs of the new computes are
the fix-ID + underscore + "temp" or fix_ID + underscore + "press", and the group for the new computes is the
same as the fix group.
Note that these are NOT the computes used by thermodynamic output (see the thermo_style command) with
ID = thermo_temp and thermo_press. This means you can change the attributes of this fix's temperature or
pressure via the compute_modify command or print this temperature or pressure during thermodynamic
output via the thermo_style custom command using the appropriate compute-ID. It also means that changing
attributes of thermo_temp or thermo_press will have no effect on this fix.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify temp and press options are supported by this fix. You can use them to assign a compute you
have defined to this fix which will be used in its temperature and pressure calculations. If you do this, note
that the kinetic energy derived from the compute temperature should be consistent with the virial term
computed using all atoms for the pressure. LAMMPS will warn you if you choose to compute temperature on
a subset of atoms.
No global or per-atom quantities are stored by this fix for access by various output commands.
This fix can ramp its target pressure over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
Any dimension being adjusted by this fix must be periodic.
Related commands:
fix nve, fix nph, fix npt, fix temp/berendsen, fix_modify
Default:
The keyword defaults are dilate = all, modulus = 10.0 in units of pressure for whatever units are defined.
LAMMPS Users Manual
1007

(Berendsen) Berendsen, Postma, van Gunsteren, DiNola, Haak, J Chem Phys, 81, 3684 (1984).
LAMMPS Users Manual
1008

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix print command
Syntax:
fix ID group-ID print N string keyword value ...
ID, group-ID are documented in fix command• print = style name of this fix command• N = print every N steps• string = text string to print with optional variable names• zero or more keyword/value pairs may be appended• keyword = file or append or screen or title
file value = filename
append value = filename
screen value = yes or no
title value = string
string = text to print as 1st line of output file
•
Examples:
fix extra all print 100 "Coords of marker atom = $x $y $z"
fix extra all print 100 "Coords of marker atom = $x $y $z" file coord.txt
Description:
Print a text string every N steps during a simulation run. This can be used for diagnostic purposes or as a
debugging tool to monitor some quantity during a run. The text string must be a single argument, so it should
be enclosed in double quotes if it is more than one word. If it contains variables it must be enclosed in double
quotes to insure they are not evaluated when the input script line is read, but will instead be evaluated each
time the string is printed.
The specified group-ID is ignored by this fix.
See the variable command for a description of equal style variables which are the most useful ones to use with
the fix print command, since they are evaluated afresh each timestep that the fix print line is output.
Equal-style variables calculate formulas involving mathematical operations, atom properties, group properties,
thermodynamic properties, global values calculated by a compute or fix, or references to other variables.
If the file or append keyword is used, a filename is specified to which the output generated by this fix will be
written. If file is used, then the filename is overwritten if it already exists. If append is used, then the filename
is appended to if it already exists, or created if it does not exist.
If the screen keyword is used, output by this fix to the screen and logfile can be turned on or off as desired.
The title keyword allow specification of the string that will be printed as the first line of the output file,
assuming the file keyword was used. By default, the title line is as follows:
# Fix print output for fix ID
LAMMPS Users Manual
1009
where ID is replaced with the fix-ID.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
variable, print
Default:
The option defaults are no file output, screen = yes, and title string as described above.
LAMMPS Users Manual
1010

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix property/atom command
Syntax:
fix ID group-ID property/atom vec1 vec2 ... keyword value ...
ID, group-ID are documented in fix command• property/atom = style name of this fix command• vec1,vec2,... = mol or q or rmass or i_name or d_name
mol = molecule IDs
q = charge
rmass = per-atom mass
i_name = new integer vector referenced by name
d_name = new floating-point vector referenced by name
•
zero of more keyword/value pairs may be appended• keyword = ghost
ghost value = no or yes for whether ghost atom info in communicated
•
Examples:
fix 1 all property/atom mol
fix 1 all property/atom i_myflag1 i_myflag2
fix 1 all property/atom d_sx d_sy d_sz
Description:
Create one or more additional per-atom vectors to store information about atoms and to use during a
simulation. The specified group-ID is ignored by this fix.
The atom style used for a simulation defines a set of per-atom properties, as explained on the atom_style and
read_data doc pages. The latter command allows these properties to be defined for each atom in the system
when a data file is read. This fix will augment the set of properties with new custom ones. This can be useful
in several scenarios.
If the atom style does not define molecule IDs, per-atom charge, or per-atom mass, they can be added using
the mol, q or rmass keywords. This can be useful, e.g, to define "molecules" to use as rigid bodies with the fix
rigid command, or just to carry around an extra flag with the atoms (stored as a molecule ID) that can be used
to group atoms without having to use the group command (which is limited to a total of 32 groups including
all).
Another application would be to use the rmass flag in order to have per-atom masses instead of per-type
masses, for example this can be useful to study isotope effects with partial isotope substitution. Please see
below for an example of simulating a mixture of light and heavy water with the TIP4P water potential.
An alternative to using fix property/atom in these ways is to use an atom style that does define molecule IDs
or charge or per-atom mass (indirectly via diameter and density) or to use a hybrid atom style that combines
two or more atom styles to provide the union of all atom properties. However, this has two practical
drawbacks: first, it typically necessitates changing the format of the data file, which can be tedious for large
LAMMPS Users Manual
1011

systems; and second, it may define additional properties that are not needed such as bond lists, which has
some overhead when there are no bonds.
In the future, we may add additional per-atom properties similar to mol, q or rmass, which "turn-on" specific
properties defined by some atom styles, so they can be used by atom styles that do not define them.
More generally, the i_name and d_name vectors allow one or more new custom per-atom properties to be
defined. Each name must be unique and can use alphanumeric or underscore characters. These vectors can
store whatever values you decide are useful in your simulation. As explained below there are several ways to
initialize and access and output these values, both via input script commands and in new code that you add to
LAMMPS.
This is effectively a simple way to add per-atom properties to a model without needing to write code for a new
atom style that defines the properties. Note however that implementing a new atom style allows new atom
properties to be more tightly and seamlessly integrated with the rest of the code.
The new atom properties encode values that migrate with atoms to new processors and are written to restart
files. If you want the new properties to also be defined for ghost atoms, then use the ghost keyword with a
value of yes. This will invoke extra communication when ghost atoms are created (at every re-neighboring) to
insure the new properties are also defined for the ghost atoms.
NOTE: If you use this command with the mol, q or rmass vectors, then you most likely want to set ghost yes,
since these properties are stored with ghost atoms if you use an atom_style that defines them, and many
LAMMPS operations that use molecule IDs or charge, such as neighbor lists and pair styles, will expect ghost
atoms to have these valuse. LAMMPS will issue a warning it you define those vectors but do not set ghost
yes.
NOTE: The properties for ghost atoms are not updated every timestep, but only once every few steps when
neighbor lists are re-built. Thus the ghost keyword is suitable for static properties, like molecule IDs, but not
for dynamic properties that change every step. For the latter, the code you add to LAMMPS to change the
properties will also need to communicate their new values to/from ghost atoms, an operation that can be
invoked from within a pair style or fix or compute that you write.
This fix is one of a small number that can be defined in an input script before the simulation box is created or
atoms are defined. This is so it can be used with the read_data command as described below.
Per-atom properties that are defined by the atom style are initialized when atoms are created, e.g. by the
read_data or create_atoms commands. The per-atom properties defined by this fix are not. So you need to
initialize them explicitly. This can be done by the read_data command, using its fix keyword and passing it the
fix-ID of this fix.
Thus these commands:
fix prop all property/atom mol d_flag
read_data data.txt fix prop NULL Molecules
would allow a data file to have a section like this:
Molecules
1 4 1.5
2 4 3.0
LAMMPS Users Manual
1012

3 10 1.0
4 10 1.0
5 10 1.0
...
N 763 4.5
where N is the number of atoms, and the first field on each line is the atom-ID, followed by a molecule-ID
and a floating point value that will be stored in a new property called "flag". Note that the list of per-atom
properties can be in any order.
Another way of initializing the new properties is via the set command. For example, if you wanted molecules
defined for every set of 10 atoms, based on their atom-IDs, these commands could be used:
fix prop all property/atom mol
variable cluster atom ((id-1)/10)+1
set id * mol v_cluster
The atom-style variable will create values for atoms with IDs 31,32,33,...40 that are 4.0,4.1,4.2,...,4.9. When
the set commands assigns them to the molecule ID for each atom, they will be truncated to an integer value, so
atoms 31-40 will all be assigned a molecule ID of 4.
Note that atomfile-style variables can also be used in place of atom-style variables, which means in this case
that the molecule IDs could be read-in from a separate file and assinged by the set command. This allows you
to initialize new per-atom properties in a completely general fashion.
For new atom properties specified as i_name or d_name, the compute property/atom command can access
their values. This means that the values can be output via the dump custom command, accessed by fixes like
fix ave/atom, accessed by other computes like compute reduce, or used in atom-style variables.
For example, these commands will output two new properties to a custom dump file:
fix prop all property/atom i_flag1 d_flag2
compute 1 all property/atom i_flag1 d_flag2
dump 1 all custom 100 tmp.dump id x y z c_1[1] c_1[2]
If you wish to add new pair styles, fixes, or computes that use the per-atom properties defined by this fix, see
Section modify of the manual which has some details on how the properties can be accessed from added
classes.
Example for using per-atom masses with TIP4P water to study isotope effects. When setting up simulations
with the TIP4P pair styles for water, you have to provide exactly one atom type each to identify the water
oxygen and hydrogen atoms. Since the atom mass is normally tied to the atom type, this makes it impossible
to study multiple isotopes in the same simulation. With fix property/atom rmass however, the per-type masses
are replaced by per-atom masses. Asumming you have a working input deck for regular TIP4P water, where
water oxygen is atom type 1 and water hydrogen is atom type 2, the following lines of input script convert this
to using per-atom masses:
fix Isotopes all property/atom rmass ghost yes
set type 1 mass 15.9994
set type 2 mass 1.008
LAMMPS Users Manual
1013

When writing out the system data with the write_data command, there will be a new section named with the
fix-ID (i.e. Isotopes in this case). Alternatively, you can take an existing data file and just add this Isotopes
section with one line per atom containing atom-ID and mass. Either way, the extended data file can be read
back with:
fix Isotopes all property/atom rmass ghost yes
read_data tip4p-isotopes.data fix Isotopes NULL Isotopes
Please note that the first Isotopes refers to the fix-ID and the second to the name of the section. The following
input script code will now change the first 100 water molecules in this example to heavy water:
group hwat id 2:300:3
group hwat id 3:300:3
set group hwat mass 2.0141018
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the per-atom values it stores to binary restart files, so that the values can be restored when a
simulation is restarted. See the read_restart command for info on how to re-specify a fix in an input script that
reads a restart file, so that the operation of the fix continues in an uninterrupted fashion.
None of the fix_modify options are relevant to this fix. No global or per-atom quantities are stored by this fix
for access by various output commands. No parameter of this fix can be used with the start/stop keywords of
the run command. This fix is not invoked during energy minimization.
Restrictions: none
Related commands:
read_data, set, compute property/atom
Default:
The default keyword values are ghost = no.
LAMMPS Users Manual
1014
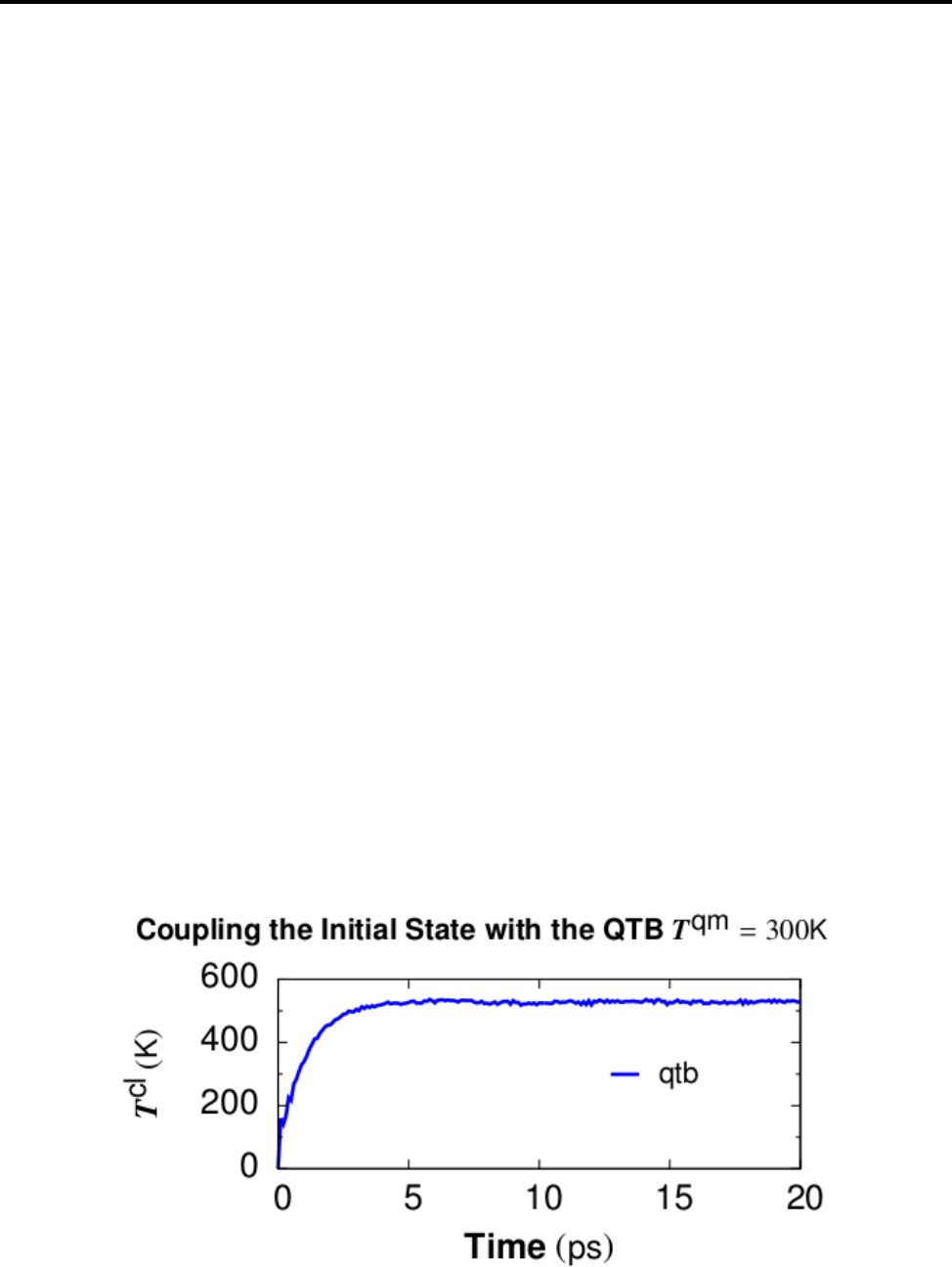
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix qbmsst command
Syntax:
fix ID group-ID qbmsst dir shockvel keyword value ...
ID, group-ID are documented in fix command• qbmsst = style name of this fix• dir = x or y or z• shockvel = shock velocity (strictly positive, velocity units)• zero or more keyword/value pairs may be appended• keyword = q or mu or p0 or v0 or e0 or tscale or damp or seedor f_max or N_f or eta or beta or T_init
q value = cell mass-like parameter (mass^2/distance^4 units)
mu value = artificial viscosity (mass/distance/time units)
p0 value = initial pressure in the shock equations (pressure units)
v0 value = initial simulation cell volume in the shock equations (distance^3 units)
e0 value = initial total energy (energy units)
tscale value = reduction in initial temperature (unitless fraction between 0.0 and 1.0)
damp value = damping parameter (time units) inverse of friction γ
seed value = random number seed (positive integer)
f_max value = upper cutoff frequency of the vibration spectrum (1/time units)
N_f value = number of frequency bins (positive integer)
eta value = coupling constant between the shock system and the quantum thermal bath (positive unitless)
beta value = the quantum temperature is updated every beta time steps (positive integer)
T_init value = quantum temperature for the initial state (temperature units)
•
Examples:
fix 1 all qbmsst z 0.122 q 25 mu 0.9 tscale 0.01 damp 200 seed 35082 f_max 0.3 N_f 100 eta 1 beta 400 T_init 110 (liquid methane modeled with the REAX force field, real units)
fix 2 all qbmsst z 72 q 40 tscale 0.05 damp 1 seed 47508 f_max 120.0 N_f 100 eta 1.0 beta 500 T_init 300 (quartz modeled with the BKS force field, metal units)
Two example input scripts are given, including shocked alpha quartz and shocked liquid methane. The input
script first equilibrate an initial state with the quantum thermal bath at the target temperature and then apply
the qbmsst to simulate shock compression with quantum nuclear correction. The following two figures plot
related quantities for shocked alpha quartz.
LAMMPS Users Manual
1015
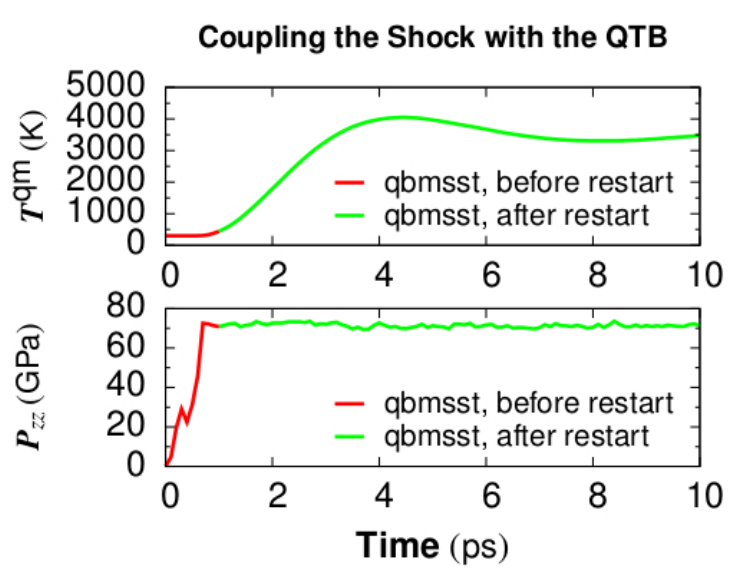
Figure 1. Classical temperature Tcl = ∑ mivi2/3NkB vs. time for coupling the alpha quartz initial state with the
quantum thermal bath at target quantum temperature Tqm = 300 K. The NpH ensemble is used for time
integration while QTB provides the colored random force. Tcl converges at the timescale of damp which is set
to be 1 ps.
Figure 2. Quantum temperature and pressure vs. time for simulating shocked alpha quartz with the QBMSST.
The shock propagates along the z direction. Restart of the QBMSST command is demonstrated in the example
input script. Thermodynamic quantities stay continuous before and after the restart.
Description:
This command performs the Quantum-Bath coupled Multi-Scale Shock Technique (QBMSST) integration.
See (Qi) for a detailed description of this method. The QBMSST provides description of the thermodynamics
and kinetics of shock processes while incorporating quantum nuclear effects. The shockvel setting determines
the steady shock velocity that will be simulated along direction dir.
Quantum nuclear effects (fix qtb) can be crucial especially when the temperature of the initial state is below
the classical limit or there is a great change in the zero point energies between the initial and final states.
Theoretical post processing quantum corrections of shock compressed water and methane have been reported
as much as 30% of the temperatures (Goldman). A self-consistent method that couples the shock to a quantum
thermal bath described by a colored noise Langevin thermostat has been developed by Qi et al (Qi) and
applied to shocked methane. The onset of chemistry is reported to be at a pressure on the shock Hugoniot that
is 40% lower than observed with classical molecular dynamics.
It is highly recommended that the system be already in an equilibrium state with a quantum thermal bath at
temperature of T_init. The fix command fix qtb at constant temperature T_init could be used before applying
this command to introduce self-consistent quantum nuclear effects into the initial state.
LAMMPS Users Manual
1016

The parameters q, mu, e0, p0, v0 and tscale are described in the command fix msst. The values of e0, p0, or v0
will be calculated on the first step if not specified. The parameter of damp, f_max, and N_f are described in the
command fix qtb.
The fix qbmsst command couples the shock system to a quantum thermal bath with a rate that is proportional
to the change of the total energy of the shock system, etot - etot0. Here etot consists of both the system energy
and a thermal term, see (Qi), and etot0 = e0 is the initial total energy.
The eta (η) parameter is a unitless coupling constant between the shock system and the quantum thermal bath.
A small eta value cannot adjust the quantum temperature fast enough during the temperature ramping period
of shock compression while large eta leads to big temperature oscillation. A value of eta between 0.3 and 1 is
usually appropriate for simulating most systems under shock compression. We observe that different values of
eta lead to almost the same final thermodynamic state behind the shock, as expected.
The quantum temperature is updated every beta (β) steps with an integration time interval beta times longer
than the simulation time step. In that case, etot is taken as its average over the past beta steps. The temperature
of the quantum thermal bath Tqm changes dynamically according to the following equation where ∆t is the
MD time step and γ is the friction constant which is equal to the inverse of the damp parameter.
dTqm/dt = γη∑βl = 1[etot(t-l∆t)-etot0]/3βNkB
The parameter T_init is the initial temperature of the quantum thermal bath and the system before shock
loading.
For all pressure styles, the simulation box stays orthorhombic in shape. Parrinello-Rahman boundary
conditions (tilted box) are supported by LAMMPS, but are not implemented for QBMSST.
Restart, fix_modify, output, run start/stop, minimize info:
Because the state of the random number generator is not written to binary restart files, this fix cannot be
restarted "exactly" in an uninterrupted fashion. However, in a statistical sense, a restarted simulation should
produce similar behaviors of the system as if it is not interrupted. To achieve such a restart, one should write
explicitly the same value for q, mu, damp, f_max, N_f, eta, and beta and set tscale = 0 if the system is
compressed during the first run.
The progress of the QBMSST can be monitored by printing the global scalar and global vector quantities
computed by the fix. The global vector contains five values in this order:
[dhugoniot, drayleigh, lagrangian_speed, lagrangian_position, quantum_temperature]
dhugoniot is the departure from the Hugoniot (temperature units).1. drayleigh is the departure from the Rayleigh line (pressure units).2. lagrangian_speed is the laboratory-frame Lagrangian speed (particle velocity) of the computational
cell (velocity units).
3.
lagrangian_position is the computational cell position in the reference frame moving at the shock
speed. This is the distance of the computational cell behind the shock front.
4.
quantum_temperature is the temperature of the quantum thermal bath Tqm.5.
To print these quantities to the log file with descriptive column headers, the following LAMMPS commands
are suggested. Here the fix_modify energy command is also enabled to allow the thermo keyword etotal to
LAMMPS Users Manual
1017
print the quantity etot. See also the thermo_style command.
fix fix_id all msst z
fix_modify fix_id energy yes
variable dhug equal f_fix_id[1]
variable dray equal f_fix_id[2]
variable lgr_vel equal f_fix_id[3]
variable lgr_pos equal f_fix_id[4]
variable T_qm equal f_fix_id[5]
thermo_style custom step temp ke pe lz pzz etotal v_dhug v_dray v_lgr_vel v_lgr_pos v_T_qm f_fix_id
The global scalar under the entry f_fix_id is the quantity of thermo energy as an extra part of etot. This global
scalar and the vector of 5 quantities can be accessed by various output commands. It is worth noting that the
temp keyword under the thermo_style command print the instantaneous classical temperature Tcl as described
in the command fix qtb.
Restrictions:
This fix style is part of the USER-QTB package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
All cell dimensions must be periodic. This fix can not be used with a triclinic cell. The QBMSST fix has been
tested only for the group-ID all.
Related commands:
fix qtb, fix msst
Default:
The keyword defaults are q = 10, mu = 0, tscale = 0.01, damp = 1, seed = 880302, f_max = 200.0, N_f = 100,
eta = 1.0, beta = 100, and T_init=300.0. e0, p0, and v0 are calculated on the first step.
(Goldman) Goldman, Reed and Fried, J. Chem. Phys. 131, 204103 (2009)
(Qi) Qi and Reed, J. Phys. Chem. A 116, 10451 (2012).
LAMMPS Users Manual
1018

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix qeq/point command
fix qeq/shielded command
fix qeq/slater command
fix qeq/dynamic command
fix qeq/fire command
Syntax:
fix ID group-ID style Nevery cutoff tolerance maxiter qfile keyword ...
ID, group-ID are documented in fix command• style = qeq/point or qeq/shielded or qeq/slater or qeq/dynamic or qeq/fire• Nevery = perform charge equilibration every this many steps• cutoff = global cutoff for charge-charge interactions (distance unit)• tolerance = precision to which charges will be equilibrated• maxiter = maximum iterations to perform charge equilibration• qfile = a filename with QEq parameters• zero or more keyword/value pairs may be appended• keyword = alpha or qdamp or qstep
alpha value = Slater type orbital exponent (qeq/slater only)
qdamp value = damping factor for damped dynamics charge solver (qeq/dynamic and qeq/fire only)
qstep value = time step size for damped dynamics charge solver (qeq/dynamic and qeq/fire only)
•
Examples:
fix 1 all qeq/point 1 10 1.0e-6 200 param.qeq1
fix 1 qeq qeq/shielded 1 8 1.0e-6 100 param.qeq2
fix 1 all qeq/slater 5 10 1.0e-6 100 params alpha 0.2
fix 1 qeq qeq/dynamic 1 12 1.0e-3 100 my_qeq
fix 1 all qeq/fire 1 10 1.0e-3 100 my_qeq qdamp 0.2 qstep 0.1
Description:
Perform the charge equilibration (QEq) method as described in (Rappe and Goddard) and formulated in
(Nakano) (also known as the matrix inversion method) and in (Rick and Stuart) (also known as the extended
Lagrangian method) based on the electronegativity equilization principle.
These fixes can be used with any pair style in LAMMPS, so long as per-atom charges are defined. The most
typical use-case is in conjunction with a pair style that performs charge equilibration periodically (e.g. every
timestep), such as the ReaxFF or Streitz-Mintmire potential. But these fixes can also be used with potentials
that normally assume per-atom charges are fixed, e.g. a Buckingham or LJ/Coulombic potential.
Because the charge equilibration calculation is effectively independent of the pair style, these fixes can also be
used to perform a one-time assignment of charges to atoms. For example, you could define the QEq fix,
LAMMPS Users Manual
1019
perform a zero-timestep run via the run command without any pair style defined which would set per-atom
charges (based on the current atom configuration), then remove the fix via the unfix command before
performing further dynamics.
NOTE: Computing and using charge values different from published values defined for a fixed-charge
potential like Buckingham or CHARMM or AMBER, can have a strong effect on energies and forces, and
produces a different model than the published versions.
NOTE: The fix qeq/comb command must still be used to perform charge equilibration with the COMB
potential. The fix qeq/reax command can be used to perform charge equilibration with the ReaxFF force field,
although fix qeq/shielded yields the same results as fix qeq/reax if Nevery, cutoff, and tolerance are the same.
Eventually the fix qeq/reax command will be deprecated.
The QEq method minimizes the electrostatic energy of the system (or equalizes the derivative of energy with
respect to charge of all the atoms) by adjusting the partial charge on individual atoms based on interactions
with their neighbors within cutoff. It requires a few parameters, in metal units, for each atom type which
provided in a file specified by qfile. The file has the following format
1 chi eta gamma zeta qcore
2 chi eta gamma zeta qcore
...
Ntype chi eta gamma zeta qcore
There is one line per atom type with the following parameters. Only a subset of the parameters is used by each
QEq style as described below, thus the others can be set to 0.0 if desired.
chi = electronegativity in energy units• eta = self-Coulomb potential in energy units• gamma = shielded Coulomb constant defined by ReaxFF force field in distance units• zeta = Slater type orbital exponent defined by the Streitz-Mintmire potential in reverse distance units• qcore = charge of the nucleus defined by the Streitz-Mintmire potential potential in charge units•
The qeq/point style describes partial charges on atoms as point charges. Interaction between a pair of charged
particles is 1/r, which is the simplest description of the interaction between charges. Only the chi and eta
parameters from the qfile file are used. Note that Coulomb catastrophe can occur if repulsion between the pair
of charged particles is too weak. This style solves partial charges on atoms via the matrix inversion method. A
tolerance of 1.0e-6 is usually a good number.
The qeq/shielded style describes partial charges on atoms also as point charges, but uses a shielded Coulomb
potential to describe the interaction between a pair of charged particles. Interaction through the shielded
Coulomb is given by equation (13) of the ReaxFF force field paper. The shielding accounts for charge overlap
between charged particles at small separation. This style is the same as fix qeq/reax, and can be used with
pair_style reax/c. Only the chi, eta, and gamma parameters from the qfile file are used. This style solves
partial charges on atoms via the matrix inversion method. A tolerance of 1.0e-6 is usually a good number.
The qeq/slater style describes partial charges on atoms as spherical charge densities centered around atoms
via the Slater 1s orbital, so that the interaction between a pair of charged particles is the product of two Slater
1s orbitals. The expression for the Slater 1s orbital is given under equation (6) of the Streitz-Mintmire paper.
Only the chi, eta, zeta, and qcore parameters from the qfile file are used. This style solves partial charges on
atoms via the matrix inversion method. A tolerance of 1.0e-6 is usually a good number. Keyword alpha can
be used to change the Slater type orbital exponent.
LAMMPS Users Manual
1020

The qeq/dynamic style describes partial charges on atoms as point charges that interact through 1/r, but the
extended Lagrangian method is used to solve partial charges on atoms. Only the chi and eta parameters from
the qfile file are used. Note that Coulomb catastrophe can occur if repulsion between the pair of charged
particles is too weak. A tolerance of 1.0e-3 is usually a good number. Keyword qdamp can be used to change
the damping factor, while keyword qstep can be used to change the time step size.
The qeq/fire style describes the same charge model and charge solver as the qeq/dynamic style, but employs a
FIRE minimization algorithm to solve for equilibrium charges. Keyword qdamp can be used to change the
damping factor, while keyword qstep can be used to change the time step size.
Note that qeq/point, qeq/shielded, and qeq/slater describe different charge models, whereas the matrix
inversion method and the extended Lagrangian method (qeq/dynamic and qeq/fire) are different solvers.
Note that qeq/point, qeq/dynamic and qeq/fire styles all describe charges as point charges that interact through
1/r relationship, but solve partial charges on atoms using different solvers. These three styles should yield
comparable results if the QEq parameters and Nevery, cutoff, and tolerance are the same. Style qeq/point is
typically faster, qeq/dynamic scales better on larger sizes, and qeq/fire is faster than qeq/dynamic.
NOTE: To avoid the evaluation of the derivative of charge with respect to position, which is typically
ill-defined, the system should have a zero net charge.
NOTE: Developing QEq parameters (chi, eta, gamma, zeta, and qcore) is non-trivial. Charges on atoms are
not guaranteed to equilibrate with arbitrary choices of these parameters. We do not develop these QEq
parameters. See the examples/qeq directory for some examples.
Restart, fix_modify, output, run start/stop, minimize info:
No information about these fixes is written to binary restart files. No global scalar or vector or per-atom
quantities are stored by these fixes for access by various output commands. No parameter of these fixes can be
used with the start/stop keywords of the run command.
Thexe fixes are invoked during energy minimization.
Restrictions:
These fixes are part of the QEQ package. They are only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
fix qeq/reax, fix qeq/comb
Default: none
(Rappe and Goddard) A. K. Rappe and W. A. Goddard III, J Physical Chemistry, 95, 3358-3363 (1991).
(Nakano) A. Nakano, Computer Physics Communications, 104, 59-69 (1997).
LAMMPS Users Manual
1021
(Rick and Stuart) S. W. Rick, S. J. Stuart, B. J. Berne, J Chemical Physics 101, 16141 (1994).
(Streitz-Mintmire) F. H. Streitz, J. W. Mintmire, Physical Review B, 50, 16, 11996 (1994)
(ReaxFF) A. C. T. van Duin, S. Dasgupta, F. Lorant, W. A. Goddard III, J Physical Chemistry, 105,
9396-9049 (2001)
(QEq/Fire) T.-R. Shan, A. P. Thompson, S. J. Plimpton, in preparation
LAMMPS Users Manual
1022

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix qeq/comb command
fix qeq/comb/omp command
Syntax:
fix ID group-ID qeq/comb Nevery precision keyword value ...
ID, group-ID are documented in fix command• qeq/comb = style name of this fix command• Nevery = perform charge equilibration every this many steps• precision = convergence criterion for charge equilibration• zero or more keyword/value pairs may be appended• keyword = file
file value = filename
filename = name of file to write QEQ equilibration info to
•
Examples:
fix 1 surface qeq/comb 10 0.0001
Description:
Perform charge equilibration (QeQ) in conjunction with the COMB (Charge-Optimized Many-Body) potential
as described in (COMB_1) and (COMB_2). It performs the charge equilibration portion of the calculation
using the so-called QEq method, whereby the charge on each atom is adjusted to minimize the energy of the
system. This fix can only be used with the COMB potential; see the fix qeq/reax command for a QeQ
calculation that can be used with any potential.
Only charges on the atoms in the specified group are equilibrated. The fix relies on the pair style (COMB in
this case) to calculate the per-atom electronegativity (effective force on the charges). An electronegativity
equalization calculation (or QEq) is performed in an iterative fashion, which in parallel requires
communication at each iteration for processors to exchange charge information about nearby atoms with each
other. See Rappe_and_Goddard and Rick_and_Stuart for details.
During a run, charge equilibration is performed every Nevery time steps. Charge equilibration is also always
enforced on the first step of each run. The precision argument controls the tolerance for the difference in
electronegativity for all atoms during charge equilibration. Precision is a trade-off between the cost of
performing charge equilibration (more iterations) and accuracy.
If the file keyword is used, then information about each equilibration calculation is written to the specified
file.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
LAMMPS Users Manual
1023
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is performing charge equilibration. Default is the outermost level.
This fix produces a per-atom vector which can be accessed by various output commands. The vector stores the
gradient of the charge on each atom. The per-atom values be accessed on any timestep.
No parameter of this fix can be used with the start/stop keywords of the run command.
This fix can be invoked during energy minimization.
Restrictions:
This fix command currently only supports pair style comb.
Related commands:
pair_style comb
Default:
No file output is performed.
(COMB_1) J. Yu, S. B. Sinnott, S. R. Phillpot, Phys Rev B, 75, 085311 (2007),
(COMB_2) T.-R. Shan, B. D. Devine, T. W. Kemper, S. B. Sinnott, S. R. Phillpot, Phys Rev B, 81, 125328
(2010).
(Rappe_and_Goddard) A. K. Rappe, W. A. Goddard, J Phys Chem 95, 3358 (1991).
(Rick_and_Stuart) S. W. Rick, S. J. Stuart, B. J. Berne, J Chem Phys 101, 16141 (1994).
LAMMPS Users Manual
1024

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix qeq/reax command
fix qeq/reax/kk command
Syntax:
fix ID group-ID qeq/reax Nevery cutlo cuthi tolerance params
ID, group-ID are documented in fix command• qeq/reax = style name of this fix command• Nevery = perform QEq every this many steps• cutlo,cuthi = lo and hi cutoff for Taper radius• tolerance = precision to which charges will be equilibrated• params = reax/c or a filename•
Examples:
fix 1 all qeq/reax 1 0.0 10.0 1.0e-6 reax/c
fix 1 all qeq/reax 1 0.0 10.0 1.0e-6 param.qeq
Description:
Perform the charge equilibration (QEq) method as described in (Rappe and Goddard) and formulated in
(Nakano). It is typically used in conjunction with the ReaxFF force field model as implemented in the
pair_style reax/c command, but it can be used with any potential in LAMMPS, so long as it defines and uses
charges on each atom. The fix qeq/comb command should be used to perform charge equilibration with the
COMB potential. For more technical details about the charge equilibration performed by fix qeq/reax, see the
(Aktulga) paper.
The QEq method minimizes the electrostatic energy of the system by adjusting the partial charge on
individual atoms based on interactions with their neighbors. It requires some parameters for each atom type. If
the params setting above is the word "reax/c", then these are extracted from the pair_style reax/c command
and the ReaxFF force field file it reads in. If a file name is specified for params, then the parameters are taken
from the specified file and the file must contain one line for each atom type. The latter form must be used
when performing QeQ with a non-ReaxFF potential. Each line should be formatted as follows:
itype chi eta gamma
where itype is the atom type from 1 to Ntypes, chi denotes the electronegativity in eV, eta denotes the
self-Coulomb potential in eV, and gamma denotes the valence orbital exponent. Note that these 3 quantities
are also in the ReaxFF potential file, except that eta is defined here as twice the eta value in the ReaxFF file.
Note that unlike the rest of LAMMPS, the units of this fix are hard-coded to be A, eV, and electronic charge.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. No global scalar or vector or per-atom quantities
are stored by this fix for access by various output commands. No parameter of this fix can be used with the
start/stop keywords of the run command.
LAMMPS Users Manual
1025

This fix is invoked during energy minimization.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This fix is part of the USER-REAXC package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This fix does not correctly handle interactions involving multiple periodic images of the same atom. Hence, it
should not be used for periodic cell dimensions less than 10 angstroms.
Related commands:
pair_style reax/c
Default: none
(Rappe) Rappe and Goddard III, Journal of Physical Chemistry, 95, 3358-3363 (1991).
(Nakano) Nakano, Computer Physics Communications, 104, 59-69 (1997).
(Aktulga) Aktulga, Fogarty, Pandit, Grama, Parallel Computing, 38, 245-259 (2012).
LAMMPS Users Manual
1026

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix qmmm command
Syntax:
fix ID group-ID qmmm
ID, group-ID are documented in fix command• qmmm = style name of this fix command•
Examples:
fix 1 qmol qmmm
Description:
This fix provides functionality to enable a quantum mechanics/molecular mechanice (QM/MM) coupling of
LAMMPS to a quantum mechanical code. The current implementation only supports an ONIOM style
mechanical coupling to the Quantum ESPRESSO plane wave DFT package. Electrostatic coupling is in
preparation and the interface has been written in a manner that coupling to other QM codes should be possible
without changes to LAMMPS itself.
The interface code for this is in the lib/qmmm directory of the LAMMPS distribution and is being made
available at this early stage of development in order to encourage contributions for interfaces to other QM
codes. This will allow the LAMMPS side of the implementation to be adapted if necessary before being
finalized.
Details about how to use this fix are currently documented in the description of the QM/MM interface code
itself in lib/qmmm/README.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global scalar or vector or per-atom quantities are stored by this fix for access by various output
commands. No parameter of this fix can be used with the start/stop keywords of the run command. This fix is
not invoked during energy minimization.
Restrictions:
This fix is part of the USER-QMMM package. It is only enabled if LAMMPS was built with that package. It
also requires building a library provided with LAMMPS. See the Making LAMMPS section for more info.
The fix is only functional when LAMMPS is built as a library and linked with a compatible QM program and
a QM/MM frontend into a QM/MM executable. See the lib/qmmm/README file for details.
Related commands: none
Default: none
LAMMPS Users Manual
1027

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix qtb command
Syntax:
fix ID group-ID qtb keyword value ...
ID, group-ID are documented in fix command• qtb = style name of this fix• zero or more keyword/value pairs may be appended• keyword = temp or damp or seed or f_max or N_f
temp value = target quantum temperature (temperature units)
damp value = damping parameter (time units) inverse of friction γ
seed value = random number seed (positive integer)
f_max value = upper cutoff frequency of the vibration spectrum (1/time units)
N_f value = number of frequency bins (positive integer)
•
Examples:
fix 1 all nve
fix 1 all qtb temp 110 damp 200 seed 35082 f_max 0.3 N_f 100 (liquid methane modeled with the REAX force field, real units)
fix 2 all nph iso 1.01325 1.01325 1
fix 2 all qtb temp 300 damp 1 seed 47508 f_max 120.0 N_f 100 (quartz modeled with the BKS force field, metal units)
Description:
This command performs the quantum thermal bath scheme proposed by (Dammak) to include self-consistent
quantum nuclear effects, when used in conjunction with the fix nve or fix nph commands.
Classical molecular dynamics simulation does not include any quantum nuclear effect. Quantum treatment of
the vibrational modes will introduce zero point energy into the system, alter the energy power spectrum and
bias the heat capacity from the classical limit. Missing all the quantum nuclear effects, classical MD cannot
model systems at temperatures lower than their classical limits. This effect is especially important for
materials with a large population of hydrogen atoms and thus higher classical limits.
The equation of motion implemented by this command follows a Langevin form:
miai = fi + Ri - miγvi.
Here mi, ai, fi , Ri, γ and vi represent mass, acceleration, force exerted by all other atoms, random force,
frictional coefficient (the inverse of damping parameter damp), and velocity. The random force Ri is "colored"
so that any vibrational mode with frequency ω will have a temperature-sensitive energy θ(ω,T) which
resembles the energy expectation for a quantum harmonic oscillator with the same natural frequency:
θ(ω,T) = ℏω/2 + ℏω[exp(ℏω/kBT)-1]-1
To efficiently generate the random forces, we employ the method of (Barrat), that circumvents the need to
generate all random forces for all times before the simulation. The memory requirement of this approach is
less demanding and independent of the simulation duration. Since the total random force Rtot does not
necessarily vanish for a finite number of atoms, Ri is replaced by Ri - Rtot/Ntot to avoid collective motion of the
LAMMPS Users Manual
1028

system.
The temp parameter sets the target quantum temperature. LAMMPS will still have an output temperature in its
thermo style. That is the instantaneous classical temperature Tcl derived from the atom velocities at thermal
equilibrium. A non-zero Tcl will be present even when the quantum temperature approaches zero. This is
associated with zero-point energy at low temperatures.
Tcl = ∑ mivi2/3NkB
The damp parameter is specified in time units, and it equals the inverse of the frictional coefficient γ. γ
should be as small as possible but slightly larger than the timescale of anharmonic coupling in the system
which is about 10 ps to 100 ps. When γ is too large, it gives an energy spectrum that differs from the desired
Bose-Einstein spectrum. When γ is too small, the quantum thermal bath coupling to the system will be less
significant than anharmonic effects, reducing to a classical limit. We find that setting γ between 5 THz and 1
THz could be appropriate depending on the system.
The random number seed is a positive integer used to initiate a Marsaglia random number generator. Each
processor uses the input seed to generate its own unique seed and its own stream of random numbers. Thus the
dynamics of the system will not be identical on two runs on different numbers of processors.
The f_max parameter truncate the noise frequency domain so that vibrational modes with frequencies higher
than f_max will not be modulated. If we denote ∆t as the time interval for the MD integration, f_max is always
reset by the code to make α = (int)(2f_max∆t)-1 a positive integer and print out relative information. An
appropriate value for the cutoff frequency f_max would be around 2~3 fD, where fD is the Debye frequency.
The N_f parameter is the frequency grid size, the number of points from 0 to f_max in the frequency domain
that will be sampled. 3×2 N_f per-atom random numbers are required in the random force generation and
there could be as many atoms as in the whole simulation that can migrate into every individual processor. A
larger N_f provides a more accurate sampling of the spectrum while consumes more memory. With fixed
f_max and γ, N_f should be big enough to converge the classical temperature Tcl as a function of target
quantum bath temperature. Memory usage per processor could be from 10 to 100 Mbytes.
NOTE: Unlike the fix nvt command which performs Nose/Hoover thermostatting AND time integration, this
fix does NOT perform time integration. It only modifies forces to a colored thermostat. Thus you must use a
separate time integration fix, like fix nve or fix nph to actually update the velocities and positions of atoms (as
shown in the examples). Likewise, this fix should not normally be used with other fixes or commands that
also specify system temperatures , e.g. fix nvt and fix temp/rescale.
Restart, fix_modify, output, run start/stop, minimizie info:
No information about this fix is written to binary restart files. Because the state of the random number
generator is not saved in restart files, this means you cannot do "exact" restarts with this fix. However, in a
statistical sense, a restarted simulation should produce similar behaviors of the system.
This fix is not invoked during energy minimization.
Restrictions:
This fix style is part of the USER-QTB package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
LAMMPS Users Manual
1029

Related commands:
fix nve, fix nph, fix langevin, fix qbmsst
Default:
The keyword defaults are temp = 300, damp = 1, seed = 880302, f_max=200.0 and N_f = 100.
(Dammak) Dammak, Chalopin, Laroche, Hayoun, and Greffet, Phys Rev Lett, 103, 190601 (2009).
(Barrat) Barrat and Rodney, J. Stat. Phys, 144, 679 (2011).
LAMMPS Users Manual
1030

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix reax/bonds command
fix reax/c/bonds command
fix reax/c/bonds/kk command
Syntax:
fix ID group-ID reax/bonds Nevery filename
ID, group-ID are documented in fix command• reax/bonds = style name of this fix command• Nevery = output interval in timesteps• filename = name of output file•
Examples:
fix 1 all reax/bonds 100 bonds.tatb
fix 1 all reax/c/bonds 100 bonds.reaxc
Description:
Write out the bond information computed by the ReaxFF potential specified by pair_style reax or pair_style
reax/c in the exact same format as the original stand-alone ReaxFF code of Adri van Duin. The bond
information is written to filename on timesteps that are multiples of Nevery, including timestep 0. For
time-averaged chemical species analysis, please see the fix reaxc/c/species command.
The format of the output file should be self-explanatory.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in
Section_accelerate of the manual. The accelerated styles take the same arguments and should produce the
same results, except for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
LAMMPS Users Manual
1031

See Section_accelerate of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
The fix reax/bonds command requires that the pair_style reax be invoked. This fix is part of the REAX
package. It is only enabled if LAMMPS was built with that package, which also requires the REAX library be
built and linked with LAMMPS. The fix reax/c/bonds command requires that the pair_style reax/c be invoked.
This fix is part of the USER-REAXC package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
pair_style reax, pair_style reax/c, fix reax/c/species
Default: none
LAMMPS Users Manual
1032

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix reax/c/species command
fix reax/c/species/kk command
Syntax:
fix ID group-ID reax/c/species Nevery Nrepeat Nfreq filename keyword value ...
ID, group-ID are documented in fix command• reax/c/species = style name of this command• Nevery = sample bond-order every this many timesteps• Nrepeat = # of bond-order samples used for calculating averages• Nfreq = calculate average bond-order every this many timesteps• filename = name of output file• zero or more keyword/value pairs may be appended• keyword = cutoff or element or position
cutoff value = I J Cutoff
I, J = atom types
Cutoff = Bond-order cutoff value for this pair of atom types
element value = Element1, Element2, ...
position value = posfreq filepos
posfreq = write position files every this many timestep
filepos = name of position output file
•
Examples:
fix 1 all reax/c/species 10 10 100 species.out
fix 1 all reax/c/species 1 2 20 species.out cutoff 1 1 0.40 cutoff 1 2 0.55
fix 1 all reax/c/species 1 100 100 species.out element Au O H position 1000 AuOH.pos
Description:
Write out the chemical species information computed by the ReaxFF potential specified by pair_style reax/c.
Bond-order values (either averaged or instantaneous, depending on value of Nrepeat) are used to determine
chemical bonds. Every Nfreq timesteps, chemical species information is written to filename as a two line
output. The first line is a header containing labels. The second line consists of the following: timestep, total
number of molecules, total number of distinct species, number of molecules of each species. In this context,
"species" means a unique molecule. The chemical formula of each species is given in the first line.
Optional keyword cutoff can be assigned to change the minimum bond-order values used in identifying
chemical bonds between pairs of atoms. Bond-order cutoffs should be carefully chosen, as bond-order cutoffs
that are too small may include too many bonds (which will result in an error), while cutoffs that are too large
will result in fragmented molecules. The default cutoff of 0.3 usually gives good results.
The optional keyword element can be used to specify the chemical symbol printed for each LAMMPS atom
type. The number of symbols must match the number of LAMMPS atom types and each symbol must consist
of 1 or 2 alphanumeric characters. Normally, these symbols should be chosen to match the chemical identity
of each LAMMPS atom type, as specified using the reax/c pair_coeff command and the ReaxFF force field
file.
LAMMPS Users Manual
1033

The optional keyword position writes center-of-mass positions of each identified molecules to file filepos
every posfreq timesteps. The first line contains information on timestep, total number of molecules, total
number of distinct species, and box dimensions. The second line is a header containing labels. From the third
line downward, each molecule writes a line of output containing the following information: molecule ID,
number of atoms in this molecule, chemical formula, total charge, and center-of-mass xyz positions of this
molecule. The xyz positions are in fractional coordinates relative to the box dimensions.
For the keyword position, the filepos is the name of the output file. It can contain the wildcard character "*".
If the "*" character appears in filepos, then one file per snapshot is written at posfreq and the "*" character is
replaced with the timestep value. For example, AuO.pos.* becomes AuO.pos.0, AuO.pos.1000, etc.
The Nevery, Nrepeat, and Nfreq arguments specify on what timesteps the bond-order values are sampled to
get the average bond order. The species analysis is performed using the average bond-order on timesteps that
are a multiple of Nfreq. The average is over Nrepeat bond-order samples, computed in the preceding portion
of the simulation every Nevery timesteps. Nfreq must be a multiple of Nevery and Nevery must be non-zero
even if Nrepeat is 1. Also, the timesteps contributing to the average bond-order cannot overlap, i.e.
Nrepeat*Nevery can not exceed Nfreq.
For example, if Nevery=2, Nrepeat=6, and Nfreq=100, then bond-order values on timesteps
90,92,94,96,98,100 will be used to compute the average bond-order for the species analysis output on timestep
100.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes both a global vector of length 2 and a per-atom vector, either of which can be accessed by
various output commands. The values in the global vector are "intensive".
The 2 values in the global vector are as follows:
1 = total number of molecules• 2 = total number of distinct species•
The per-atom vector stores the molecule ID for each atom as identified by the fix. If an atom is not in a
molecule, its ID will be 0. For atoms in the same molecule, the molecule ID for all of them will be the same
and will be equal to the smallest atom ID of any atom in the molecule.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in
Section_accelerate of the manual. The accelerated styles take the same arguments and should produce the
same results, except for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
LAMMPS Users Manual
1034

You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section_accelerate of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
The fix species currently only works with pair_style reax/c and it requires that the pair_style reax/c be
invoked. This fix is part of the USER-REAXC package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
It should be possible to extend it to other reactive pair_styles (such as rebo, airebo, comb, and bop), but this
has not yet been done.
Related commands:
pair_style reax/c, fix reax/bonds
Default:
The default values for bond-order cutoffs are 0.3 for all I-J pairs. The default element symbols are C, H, O, N.
Position files are not written by default.
LAMMPS Users Manual
1035

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix recenter command
Syntax:
fix ID group-ID recenter x y z keyword value ...
ID, group-ID are documented in fix command• recenter = style name of this fix command• x,y,z = constrain center-of-mass to these coords (distance units), any coord can also be NULL or INIT
(see below)
•
zero or more keyword/value pairs may be appended• keyword = shift or units
shift value = group-ID
group-ID = group of atoms whose coords are shifted
units value = box or lattice or fraction
•
Examples:
fix 1 all recenter 0.0 0.5 0.0
fix 1 all recenter INIT INIT NULL
fix 1 all recenter INIT 0.0 0.0 units box
Description:
Constrain the center-of-mass position of a group of atoms by adjusting the coordinates of the atoms every
timestep. This is simply a small shift that does not alter the dynamics of the system or change the relative
coordinates of any pair of atoms in the group. This can be used to insure the entire collection of atoms (or a
portion of them) do not drift during the simulation due to random perturbations (e.g. fix langevin
thermostatting).
Distance units for the x,y,z values are determined by the setting of the units keyword, as discussed below. One
or more x,y,z values can also be specified as NULL, which means exclude that dimension from this operation.
Or it can be specified as INIT which means to constrain the center-of-mass to its initial value at the beginning
of the run.
The center-of-mass (COM) is computed for the group specified by the fix. If the current COM is different
than the specified x,y,z, then a group of atoms has their coordinates shifted by the difference. By default the
shifted group is also the group specified by the fix. A different group can be shifted by using the shift
keyword. For example, the COM could be computed on a protein to keep it in the center of the simulation
box. But the entire system (protein + water) could be shifted.
If the units keyword is set to box, then the distance units of x,y,z are defined by the units command - e.g.
Angstroms for real units. A lattice value means the distance units are in lattice spacings. The lattice command
must have been previously used to define the lattice spacing. A fraction value means a fractional distance
between the lo/hi box boundaries, e.g. 0.5 = middle of the box. The default is to use lattice units.
Note that the velocity command can be used to create velocities with zero aggregate linear and/or angular
momentum.
LAMMPS Users Manual
1036
NOTE: This fix performs its operations at the same point in the timestep as other time integration fixes, such
as fix nve, fix nvt, or fix npt. Thus fix recenter should normally be the last such fix specified in the input
script, since the adjustments it makes to atom coordinates should come after the changes made by time
integration. LAMMPS will warn you if your fixes are not ordered this way.
NOTE: If you use this fix on a small group of atoms (e.g. a molecule in solvent) without using the shift
keyword to adjust the positions of all atoms in the system, then the results can be unpredictable. For example,
if the molecule is pushed consistently in one direction by a flowing solvent, its velocity will increase. But its
coordinates will be recentered, meaning it is moved back towards the force. Thus over time, the velocity and
effective temperature of the molecule could become very large, though it won't actually be moving due to the
recentering. If you are thermostatting the entire system, then the solvent would be cooled to compensate. A
better solution for this simulation scenario is to use the fix spring command to tether the molecule in place.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
distance the group is moved by fix recenter.
This fix also computes global 3-vector which can be accessed by various output commands. The 3 quantities
in the vector are xyz components of displacement applied to the group of atoms by the fix.
The scalar and vector values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix should not be used with an x,y,z setting that causes a large shift in the system on the 1st timestep, due
to the requested COM being very different from the initial COM. This could cause atoms to be lost, especially
in parallel. Instead, use the displace_atoms command, which can be used to move atoms a large distance.
Related commands:
fix momentum, velocity
Default:
The option defaults are shift = fix group-ID, and units = lattice.
LAMMPS Users Manual
1037

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix restrain command
Syntax:
fix ID group-ID restrain keyword args ...
ID, group-ID are documented in fix command• restrain = style name of this fix command• one or more keyword/arg pairs may be appended• keyword = bond or angle or dihedral
bond args = atom1 atom2 Kstart Kstop r0
atom1,atom2 = IDs of 2 atoms in bond
Kstart,Kstop = restraint coefficients at start/end of run (energy units)
r0 = equilibrium bond distance (distance units)
angle args = atom1 atom2 atom3 Kstart Kstop theta0
atom1,atom2,atom3 = IDs of 3 atoms in angle, atom2 = middle atom
Kstart,Kstop = restraint coefficients at start/end of run (energy units)
theta0 = equilibrium angle theta (degrees)
dihedral args = atom1 atom2 atom3 atom4 Kstart Kstop phi0
atom1,atom2,atom3,atom4 = IDs of 4 atoms in dihedral in linear order
Kstart,Kstop = restraint coefficients at start/end of run (energy units)
phi0 = equilibrium dihedral angle phi (degrees)
•
Examples:
fix holdem all restrain bond 45 48 2000.0 2000.0 2.75
fix holdem all restrain dihedral 1 2 3 4 2000.0 2000.0 120.0
fix holdem all restrain bond 45 48 2000.0 2000.0 2.75 dihedral 1 2 3 4 2000.0 2000.0 120.0
fix texas_holdem all restrain dihedral 1 2 3 4 0.0 2000.0 120.0 dihedral 1 2 3 5 0.0 2000.0 -120.0 dihedral 1 2 3 6 0.0 2000.0 0.0
Description:
Restrain the motion of the specified sets of atoms by making them part of a bond or angle or dihedral
interaction whose strength can vary over time during a simulation. This is functionally similar to creating a
bond or angle or dihedral for the same atoms in a data file, as specified by the read_data command, albeit with
a time-varying pre-factor coefficient, and except for exclusion rules, as explained below.
For the purpose of forcefield parameter-fitting or mapping a molecular potential energy surface, this fix
reduces the hassle and risk associated with modifying data files. In other words, use this fix to temporarily
force a molecule to adopt a particular conformation. To create a permanent bond or angle or dihedral, you
should modify the data file.
NOTE: Adding a bond/angle/dihedral with this command does not apply the exclusion rules and weighting
factors specified by the special_bonds command to atoms in the restraint that are now bonded (1-2,1-3,1-4
neighbors) as a result. If they are close enough to interact in a pair_style sense (non-bonded interaction), then
the bond/angle/dihedral restraint interaction will simply be superposed on top of that interaction.
The group-ID specified by this fix is ignored.
LAMMPS Users Manual
1038
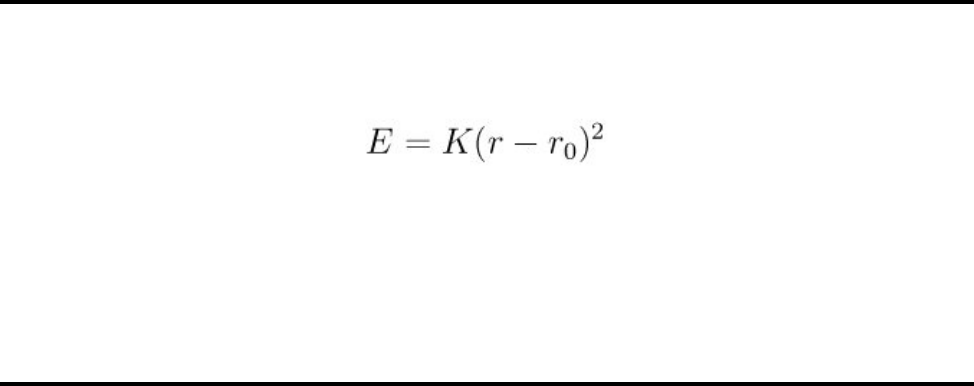
The second example above applies a restraint to hold the dihedral angle formed by atoms 1, 2, 3, and 4 near
120 degrees using a constant restraint coefficient. The fourth example applies similar restraints to multiple
dihedral angles using a restraint coefficient that increases from 0.0 to 2000.0 over the course of the run.
NOTE: Adding a force to atoms implies a change in their potential energy as they move due to the applied
force field. For dynamics via the run command, this energy can be added to the system's potential energy for
thermodynamic output (see below). For energy minimization via the minimize command, this energy must be
added to the system's potential energy to formulate a self-consistent minimization problem (see below).
In order for a restraint to be effective, the restraint force must typically be significantly larger than the forces
associated with conventional forcefield terms. If the restraint is applied during a dynamics run (as opposed to
during an energy minimization), a large restraint coefficient can significantly reduce the stable timestep size,
especially if the atoms are initially far from the preferred conformation. You may need to experiment to
determine what value of K works best for a given application.
For the case of finding a minimum energy structure for a single molecule with particular restraints (e.g. for
fitting forcefield parameters or constructing a potential energy surface), commands such as the following may
be useful:
# minimize molecule energy with restraints
velocity all create 600.0 8675309 mom yes rot yes dist gaussian
fix NVE all nve
fix TFIX all langevin 600.0 0.0 100 24601
fix REST all restrain dihedral 2 1 3 8 0.0 5000.0 ${angle1} dihedral 3 1 2 9 0.0 5000.0 ${angle2}
fix_modify REST energy yes
run 10000
fix TFIX all langevin 0.0 0.0 100 24601
fix REST all restrain dihedral 2 1 3 8 5000.0 5000.0 ${angle1} dihedral 3 1 2 9 5000.0 5000.0 ${angle2}
fix_modify REST energy yes
run 10000
# sanity check for convergence
minimize 1e-6 1e-9 1000 100000
# report unrestrained energies
unfix REST
run 0
The bond keyword applies a bond restraint to the specified atoms using the same functional form used by the
bond_style harmonic command. The potential associated with the restraint is
with the following coefficients:
K (energy/distance^2)• r0 (distance)•
K and r0 are specified with the fix. Note that the usual 1/2 factor is included in K.
The angle keyword applies an angle restraint to the specified atoms using the same functional form used by
the angle_style harmonic command. The potential associated with the restraint is
LAMMPS Users Manual
1039

with the following coefficients:
K (energy/radian^2)• theta0 (degrees)•
K and theta0 are specified with the fix. Note that the usual 1/2 factor is included in K.
The dihedral keyword applies a dihedral restraint to the specified atoms using a simplified form of the
function used by the dihedral_style charmm command. The potential associated with the restraint is
with the following coefficients:
K (energy)• n = 1• d (degrees) = phi0 + 180•
K and phi0 are specified with the fix. Note that the value of n is hard-wired to 1. Also note that the energy will
be a minimum when the current dihedral angle phi is equal to phi0.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the potential energy associated with this fix to
the system's potential energy as part of thermodynamic output.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
NOTE: If you want the fictitious potential energy associated with the added forces to be included in the total
potential energy of the system (the quantity being minimized), you MUST enable the fix_modify energy
option for this fix.
This fix computes a global scalar, which can be accessed by various output commands. The scalar is the
potential energy for all the restraints as discussed above. The scalar value calculated by this fix is "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
Restrictions: none
Related commands: none
LAMMPS Users Manual
1040
Default: none
LAMMPS Users Manual
1041

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix rigid command
fix rigid/nve command
fix rigid/nvt command
fix rigid/npt command
fix rigid/nph command
fix rigid/small command
fix rigid/nve/small command
fix rigid/nvt/small command
fix rigid/npt/small command
fix rigid/nph/small command
Syntax:
fix ID group-ID style bodystyle args keyword values ...
ID, group-ID are documented in fix command• style = rigid or rigid/nve or rigid/nvt or rigid/npt or rigid/nph or rigid/small or rigid/nve/small or
rigid/nvt/small or rigid/npt/small or rigid/nph/small
•
bodystyle = single or molecule or group
single args = none
molecule args = none
group args = N groupID1 groupID2 ...
N = # of groups
groupID1, groupID2, ... = list of N group IDs
•
zero or more keyword/value pairs may be appended• keyword = langevin or temp or iso or aniso or x or y or z or couple or tparam or pchain or dilate or
force or torque or infile
langevin values = Tstart Tstop Tperiod seed
Tstart,Tstop = desired temperature at start/stop of run (temperature units)
Tdamp = temperature damping parameter (time units)
seed = random number seed to use for white noise (positive integer)
temp values = Tstart Tstop Tdamp
Tstart,Tstop = desired temperature at start/stop of run (temperature units)
Tdamp = temperature damping parameter (time units)
iso or aniso values = Pstart Pstop Pdamp
Pstart,Pstop = scalar external pressure at start/end of run (pressure units)
Pdamp = pressure damping parameter (time units)
x or y or z values = Pstart Pstop Pdamp
Pstart,Pstop = external stress tensor component at start/end of run (pressure units)
•
LAMMPS Users Manual
1042
Pdamp = stress damping parameter (time units)
couple = none or xyz or xy or yz or xz
tparam values = Tchain Titer Torder
Tchain = length of Nose/Hoover thermostat chain
Titer = number of thermostat iterations performed
Torder = 3 or 5 = Yoshida-Suzuki integration parameters
pchain values = Pchain
Pchain = length of the Nose/Hoover thermostat chain coupled with the barostat
dilate value = dilate-group-ID
dilate-group-ID = only dilate atoms in this group due to barostat volume changes
force values = M xflag yflag zflag
M = which rigid body from 1-Nbody (see asterisk form below)
xflag,yflag,zflag = off/on if component of center-of-mass force is active
torque values = M xflag yflag zflag
M = which rigid body from 1-Nbody (see asterisk form below)
xflag,yflag,zflag = off/on if component of center-of-mass torque is active
infile filename
filename = file with per-body values of mass, center-of-mass, moments of inertia
mol value = template-ID
template-ID = ID of molecule template specified in a separate molecule command
Examples:
fix 1 clump rigid single
fix 1 clump rigid/small molecule
fix 1 clump rigid single force 1 off off on langevin 1.0 1.0 1.0 428984
fix 1 polychains rigid/nvt molecule temp 1.0 1.0 5.0
fix 1 polychains rigid molecule force 1*5 off off off force 6*10 off off on
fix 1 polychains rigid/small molecule langevin 1.0 1.0 1.0 428984
fix 2 fluid rigid group 3 clump1 clump2 clump3 torque * off off off
fix 1 rods rigid/npt molecule temp 300.0 300.0 100.0 iso 0.5 0.5 10.0
fix 1 particles rigid/npt molecule temp 1.0 1.0 5.0 x 0.5 0.5 1.0 z 0.5 0.5 1.0 couple xz
fix 1 water rigid/nph molecule iso 0.5 0.5 1.0
fix 1 particles rigid/npt/small molecule temp 1.0 1.0 1.0 iso 0.5 0.5 1.0
Description:
Treat one or more sets of atoms as independent rigid bodies. This means that each timestep the total force and
torque on each rigid body is computed as the sum of the forces and torques on its constituent particles. The
coordinates, velocities, and orientations of the atoms in each body are then updated so that the body moves
and rotates as a single entity.
Examples of large rigid bodies are a colloidal particle, or portions of a biomolecule such as a protein.
Example of small rigid bodies are patchy nanoparticles, such as those modeled in this paper by Sharon
Glotzer's group, clumps of granular particles, lipid molecules consiting of one or more point dipoles
connected to other spheroids or ellipsoids, irregular particles built from line segments (2d) or triangles (3d),
and coarse-grain models of nano or colloidal particles consisting of a small number of constituent particles.
Note that the fix shake command can also be used to rigidify small molecules of 2, 3, or 4 atoms, e.g. water
molecules. That fix treats the constituent atoms as point masses.
These fixes also update the positions and velocities of the atoms in each rigid body via time integration, in the
NVE, NVT, NPT, or NPH ensemble, as described below.
There are two main variants of this fix, fix rigid and fix rigid/small. The NVE/NVT/NPT/NHT versions
belong to one of the two variants, as their style names indicate.
LAMMPS Users Manual
1043

NOTE: Not all of the bodystyle options and keyword/value options are available for both the rigid and
rigid/small variants. See details below.
The rigid styles are typically the best choice for a system with a small number of large rigid bodies, each of
which can extend across the domain of many processors. It operates by creating a single global list of rigid
bodies, which all processors contribute to. MPI_Allreduce operations are performed each timestep to sum the
contributions from each processor to the force and torque on all the bodies. This operation will not scale well
in parallel if large numbers of rigid bodies are simulated.
The rigid/small styles are typically best for a system with a large number of small rigid bodies. Each body is
assigned to the atom closest to the geometrical center of the body. The fix operates using local lists of rigid
bodies owned by each processor and information is exchanged and summed via local communication between
neighboring processors when ghost atom info is accumulated.
NOTE: To use the rigid/small styles the ghost atom cutoff must be large enough to span the distance between
the atom that owns the body and every other atom in the body. This distance value is printed out when the
rigid bodies are defined. If the pair_style cutoff plus neighbor skin does not span this distance, then you
should use the comm_modify cutoff command with a setting epsilon larger than the distance.
Which of the two variants is faster for a particular problem is hard to predict. The best way to decide is to
perform a short test run. Both variants should give identical numerical answers for short runs. Long runs
should give statistically similar results, but round-off differences may accumulate to produce divergent
trajectories.
NOTE: You should not update the atoms in rigid bodies via other time-integration fixes (e.g. fix nve, fix nvt,
fix npt), or you will be integrating their motion more than once each timestep. When performing a hybrid
simulation with some atoms in rigid bodies, and some not, a separate time integration fix like fix nve or fix
nvt should be used for the non-rigid particles.
NOTE: These fixes are overkill if you simply want to hold a collection of atoms stationary or have them move
with a constant velocity. A simpler way to hold atoms stationary is to not include those atoms in your time
integration fix. E.g. use "fix 1 mobile nve" instead of "fix 1 all nve", where "mobile" is the group of atoms
that you want to move. You can move atoms with a constant velocity by assigning them an initial velocity
(via the velocity command), setting the force on them to 0.0 (via the fix setforce command), and integrating
them as usual (e.g. via the fix nve command).
NOTE: The aggregate properties of each rigid body are calculated one time at the start of the first simulation
run after these fixes are specified. The properties include the position and velocity of the center-of-mass of the
body, its moments of inertia, and its angular momentum. This is done using the properties of the constituent
atoms of the body at that point in time (or see the infile keyword option). Thereafter, changing properties of
individual atoms in the body will have no effect on a rigid body's dynamics, unless they affect the pair_style
interactions that individual particles are part of. For example, you might think you could displace the atoms in
a body or add a large velocity to each atom in a body to make it move in a desired direction before a 2nd run
is performed, using the set or displace_atoms or velocity command. But these commands will not affect the
internal attributes of the body, and the position and velocity of individual atoms in the body will be reset when
time integration starts.
Each rigid body must have two or more atoms. An atom can belong to at most one rigid body. Which atoms
are in which bodies can be defined via several options.
LAMMPS Users Manual
1044
NOTE: With the rigid/small styles, which require that bodystyle be specified as molecule, you can define a
system that has no rigid bodies initially. This is useful when you are using the mol keyword in conjunction
with another fix that is adding rigid bodies on-the-fly as molecules, such as fix deposit or fix pour.
For bodystyle single the entire fix group of atoms is treated as one rigid body. This option is only allowed for
the rigid styles.
For bodystyle molecule, each set of atoms in the fix group with a different molecule ID is treated as a rigid
body. This option is allowed for both the rigid and rigid/small styles. Note that atoms with a molecule ID = 0
will be treated as a single rigid body. For a system with atomic solvent (typically this is atoms with molecule
ID = 0) surrounding rigid bodies, this may not be what you want. Thus you should be careful to use a fix
group that only includes atoms you want to be part of rigid bodies.
For bodystyle group, each of the listed groups is treated as a separate rigid body. Only atoms that are also in
the fix group are included in each rigid body. This option is only allowed for the rigid styles.
NOTE: To compute the initial center-of-mass position and other properties of each rigid body, the image flags
for each atom in the body are used to "unwrap" the atom coordinates. Thus you must insure that these image
flags are consistent so that the unwrapping creates a valid rigid body (one where the atoms are close together),
particularly if the atoms in a single rigid body straddle a periodic boundary. This means the input data file or
restart file must define the image flags for each atom consistently or that you have used the set command to
specify them correctly. If a dimension is non-periodic then the image flag of each atom must be 0 in that
dimension, else an error is generated.
The force and torque keywords discussed next are only allowed for the rigid styles.
By default, each rigid body is acted on by other atoms which induce an external force and torque on its center
of mass, causing it to translate and rotate. Components of the external center-of-mass force and torque can be
turned off by the force and torque keywords. This may be useful if you wish a body to rotate but not translate,
or vice versa, or if you wish it to rotate or translate continuously unaffected by interactions with other
particles. Note that if you expect a rigid body not to move or rotate by using these keywords, you must insure
its initial center-of-mass translational or angular velocity is 0.0. Otherwise the initial translational or angular
momentum the body has will persist.
An xflag, yflag, or zflag set to off means turn off the component of force of torque in that dimension. A setting
of on means turn on the component, which is the default. Which rigid body(s) the settings apply to is
determined by the first argument of the force and torque keywords. It can be an integer M from 1 to Nbody,
where Nbody is the number of rigid bodies defined. A wild-card asterisk can be used in place of, or in
conjunction with, the M argument to set the flags for multiple rigid bodies. This takes the form "*" or "*n" or
"n*" or "m*n". If N = the number of rigid bodies, then an asterisk with no numeric values means all bodies
from 1 to N. A leading asterisk means all bodies from 1 to n (inclusive). A trailing asterisk means all bodies
from n to N (inclusive). A middle asterisk means all types from m to n (inclusive). Note that you can use the
force or torque keywords as many times as you like. If a particular rigid body has its component flags set
multiple times, the settings from the final keyword are used.
NOTE: For computational efficiency, you may wish to turn off pairwise and bond interactions within each
rigid body, as they no longer contribute to the motion. The neigh_modify exclude and delete_bonds
commands are used to do this. If the rigid bodies have strongly overlapping atoms, you may need to turn off
these interactions to avoid numerical problems due to large equal/opposite intra-body forces swamping the
contribution of small inter-body forces.
LAMMPS Users Manual
1045

For computational efficiency, you should typically define one fix rigid or fix rigid/small command which
includes all the desired rigid bodies. LAMMPS will allow multiple rigid fixes to be defined, but it is more
expensive.
The constituent particles within a rigid body can be point particles (the default in LAMMPS) or finite-size
particles, such as spheres or ellipsoids or line segments or triangles. See the atom_style sphere and ellipsoid
and line and tri commands for more details on these kinds of particles. Finite-size particles contribute
differently to the moment of inertia of a rigid body than do point particles. Finite-size particles can also
experience torque (e.g. due to frictional granular interactions) and have an orientation. These contributions are
accounted for by these fixes.
Forces between particles within a body do not contribute to the external force or torque on the body. Thus for
computational efficiency, you may wish to turn off pairwise and bond interactions between particles within
each rigid body. The neigh_modify exclude and delete_bonds commands are used to do this. For finite-size
particles this also means the particles can be highly overlapped when creating the rigid body.
The rigid, rigid/nve, rigid/small, and rigid/small/nve styles perform constant NVE time integration. They are
referred to below as the 4 NVE rigid styles. The only difference is that the rigid and rigid/small styles use an
integration technique based on Richardson iterations. The rigid/nve and rigid/small/nve styles uses the
methods described in the paper by Miller, which are thought to provide better energy conservation than an
iterative approach.
The rigid/nvt and rigid/nvt/small styles performs constant NVT integration using a Nose/Hoover thermostat
with chains as described originally in (Hoover) and (Martyna), which thermostats both the translational and
rotational degrees of freedom of the rigid bodies. They are referred to below as the 2 NVT rigid styles. The
rigid-body algorithm used by rigid/nvt is described in the paper by Kamberaj.
The rigid/npt, rigid/nph, rigid/npt/small, and rigid/nph/small styles perform constant NPT or NPH integration
using a Nose/Hoover barostat with chains. They are referred to below as the 4 NPT and NPH rigid styles. For
the NPT case, the same Nose/Hoover thermostat is also used as with rigid/nvt and rigid/nvt/small.
The barostat parameters are specified using one or more of the iso, aniso, x, y, z and couple keywords. These
keywords give you the ability to specify 3 diagonal components of the external stress tensor, and to couple
these components together so that the dimensions they represent are varied together during a constant-pressure
simulation. The effects of these keywords are similar to those defined in fix npt/nph
NOTE: Currently the rigid/npt, rigid/nph, rigid/npt/small, and rigid/nph/small styles do not support triclinic
(non-orthogonal) boxes.
The target pressures for each of the 6 components of the stress tensor can be specified independently via the x,
y, z keywords, which correspond to the 3 simulation box dimensions. For each component, the external
pressure or tensor component at each timestep is a ramped value during the run from Pstart to Pstop. If a
target pressure is specified for a component, then the corresponding box dimension will change during a
simulation. For example, if the y keyword is used, the y-box length will change. A box dimension will not
change if that component is not specified, although you have the option to change that dimension via the fix
deform command.
For all barostat keywords, the Pdamp parameter operates like the Tdamp parameter, determining the time
scale on which pressure is relaxed. For example, a value of 10.0 means to relax the pressure in a timespan of
(roughly) 10 time units (e.g. tau or fmsec or psec - see the units command).
LAMMPS Users Manual
1046

Regardless of what atoms are in the fix group (the only atoms which are time integrated), a global pressure or
stress tensor is computed for all atoms. Similarly, when the size of the simulation box is changed, all atoms
are re-scaled to new positions, unless the keyword dilate is specified with a dilate-group-ID for a group that
represents a subset of the atoms. This can be useful, for example, to leave the coordinates of atoms in a solid
substrate unchanged and controlling the pressure of a surrounding fluid. Another example is a system
consisting of rigid bodies and point particles where the barostat is only coupled with the rigid bodies. This
option should be used with care, since it can be unphysical to dilate some atoms and not others, because it can
introduce large, instantaneous displacements between a pair of atoms (one dilated, one not) that are far from
the dilation origin.
The couple keyword allows two or three of the diagonal components of the pressure tensor to be "coupled"
together. The value specified with the keyword determines which are coupled. For example, xz means the Pxx
and Pzz components of the stress tensor are coupled. Xyz means all 3 diagonal components are coupled.
Coupling means two things: the instantaneous stress will be computed as an average of the corresponding
diagonal components, and the coupled box dimensions will be changed together in lockstep, meaning coupled
dimensions will be dilated or contracted by the same percentage every timestep. The Pstart, Pstop, Pdamp
parameters for any coupled dimensions must be identical. Couple xyz can be used for a 2d simulation; the z
dimension is simply ignored.
The iso and aniso keywords are simply shortcuts that are equivalent to specifying several other keywords
together.
The keyword iso means couple all 3 diagonal components together when pressure is computed (hydrostatic
pressure), and dilate/contract the dimensions together. Using "iso Pstart Pstop Pdamp" is the same as
specifying these 4 keywords:
x Pstart Pstop Pdamp
y Pstart Pstop Pdamp
z Pstart Pstop Pdamp
couple xyz
The keyword aniso means x, y, and z dimensions are controlled independently using the Pxx, Pyy, and Pzz
components of the stress tensor as the driving forces, and the specified scalar external pressure. Using "aniso
Pstart Pstop Pdamp" is the same as specifying these 4 keywords:
x Pstart Pstop Pdamp
y Pstart Pstop Pdamp
z Pstart Pstop Pdamp
couple none
The keyword/value option pairs are used in the following ways.
The langevin and temp and tparam keywords perform thermostatting of the rigid bodies, altering both their
translational and rotational degrees of freedom. What is meant by "temperature" of a collection of rigid bodies
and how it can be monitored via the fix output is discussed below.
The langevin keyword applies a Langevin thermostat to the constant NVE time integration performed by any
of the 4 NVE rigid styles: rigid, rigid/nve, rigid/small, rigid/small/nve. It cannot be used with the 2 NVT rigid
styles: rigid/nvt, rigid/small/nvt. The desired temperature at each timestep is a ramped value during the run
from Tstart to Tstop. The Tdamp parameter is specified in time units and determines how rapidly the
temperature is relaxed. For example, a value of 100.0 means to relax the temperature in a timespan of
(roughly) 100 time units (tau or fmsec or psec - see the units command). The random # seed must be a
LAMMPS Users Manual
1047

positive integer.
The way that Langevin thermostatting operates is explained on the fix langevin doc page. If you wish to
simply viscously damp the rotational motion without thermostatting, you can set Tstart and Tstop to 0.0,
which means only the viscous drag term in the Langevin thermostat will be applied. See the discussion on the
fix viscous doc page for details.
NOTE: When the langevin keyword is used with fix rigid versus fix rigid/small, different dynamics will result
for parallel runs. This is because of the way random numbers are used in the two cases. The dynamics for the
two cases should be statistically similar, but will not be identical, even for a single timestep.
The temp and tparam keywords apply a Nose/Hoover thermostat to the NVT time integration performed by
the 2 NVT rigid styles. They cannot be used with the 4 NVE rigid styles. The desired temperature at each
timestep is a ramped value during the run from Tstart to Tstop. The Tdamp parameter is specified in time
units and determines how rapidly the temperature is relaxed. For example, a value of 100.0 means to relax the
temperature in a timespan of (roughly) 100 time units (tau or fmsec or psec - see the units command).
Nose/Hoover chains are used in conjunction with this thermostat. The tparam keyword can optionally be used
to change the chain settings used. Tchain is the number of thermostats in the Nose Hoover chain. This value,
along with Tdamp can be varied to dampen undesirable oscillations in temperature that can occur in a
simulation. As a rule of thumb, increasing the chain length should lead to smaller oscillations. The keyword
pchain specifies the number of thermostats in the chain thermostatting the barostat degrees of freedom.
NOTE: There are alternate ways to thermostat a system of rigid bodies. You can use fix langevin to treat the
individual particles in the rigid bodies as effectively immersed in an implicit solvent, e.g. a Brownian
dynamics model. For hybrid systems with both rigid bodies and solvent particles, you can thermostat only the
solvent particles that surround one or more rigid bodies by appropriate choice of groups in the compute and
fix commands for temperature and thermostatting. The solvent interactions with the rigid bodies should then
effectively thermostat the rigid body temperature as well without use of the Langevin or Nose/Hoover options
associated with the fix rigid commands.
The mol keyword can only be used with the rigid/small styles. It must be used when other commands, such as
fix deposit or fix pour, add rigid bodies on-the-fly during a simulation. You specify a template-ID previously
defined using the molecule command, which reads a file that defines the molecule. You must use the same
template-ID that the other fix which is adding rigid bodies uses. The coordinates, atom types, atom diameters,
center-of-mass, and moments of inertia can be specified in the molecule file. See the molecule command for
details. The only settings required to be in this file are the coordinates and types of atoms in the molecule, in
which case the molecule command calculates the other quantities itself.
Note that these other fixes create new rigid bodies, in addition to those defined initially by this fix via the
bodystyle setting.
Also note that when using the mol keyword, extra restart information about all rigid bodies is written out
whenever a restart file is written out. See the NOTE in the next section for details.
The infile keyword allows a file of rigid body attributes to be read in from a file, rather then having LAMMPS
compute them. There are 5 such attributes: the total mass of the rigid body, its center-of-mass position, its 6
moments of inertia, its center-of-mass velocity, and the 3 image flags of the center-of-mass position. For rigid
bodies consisting of point particles or non-overlapping finite-size particles, LAMMPS can compute these
values accurately. However, for rigid bodies consisting of finite-size particles which overlap each other,
LAMMPS Users Manual
1048

LAMMPS will ignore the overlaps when computing these 4 attributes. The amount of error this induces
depends on the amount of overlap. To avoid this issue, the values can be pre-computed (e.g. using Monte
Carlo integration).
The format of the file is as follows. Note that the file does not have to list attributes for every rigid body
integrated by fix rigid. Only bodies which the file specifies will have their computed attributes overridden.
The file can contain initial blank lines or comment lines starting with "#" which are ignored. The first
non-blank, non-comment line should list N = the number of lines to follow. The N successive lines contain the
following information:
ID1 masstotal xcm ycm zcm ixx iyy izz ixy ixz iyz vxcm vycm vzcm lx ly lz ixcm iycm izcm
ID2 masstotal xcm ycm zcm ixx iyy izz ixy ixz iyz vxcm vycm vzcm lx ly lz ixcm iycm izcm
...
IDN masstotal xcm ycm zcm ixx iyy izz ixy ixz iyz vxcm vycm vzcm lx ly lz ixcm iycm izcm
The rigid body IDs are all positive integers. For the single bodystyle, only an ID of 1 can be used. For the
group bodystyle, IDs from 1 to Ng can be used where Ng is the number of specified groups. For the molecule
bodystyle, use the molecule ID for the atoms in a specific rigid body as the rigid body ID.
The masstotal and center-of-mass coordinates (xcm,ycm,zcm) are self-explanatory. The center-of-mass should
be consistent with what is calculated for the position of the rigid body with all its atoms unwrapped by their
respective image flags. If this produces a center-of-mass that is outside the simulation box, LAMMPS wraps it
back into the box.
The 6 moments of inertia (ixx,iyy,izz,ixy,ixz,iyz) should be the values consistent with the current orientation
of the rigid body around its center of mass. The values are with respect to the simulation box XYZ axes, not
with respect to the principal axes of the rigid body itself. LAMMPS performs the latter calculation internally.
The (vxcm,vycm,vzcm) values are the velocity of the center of mass. The (lx,ly,lz) values are the angular
momentum of the body. The (vxcm,vycm,vzcm) and (lx,ly,lz) values can simply be set to 0 if you wish the
body to have no initial motion.
The (ixcm,iycm,izcm) values are the image flags of the center of mass of the body. For periodic dimensions,
they specify which image of the simulation box the body is considered to be in. An image of 0 means it is
inside the box as defined. A value of 2 means add 2 box lengths to get the true value. A value of -1 means
subtract 1 box length to get the true value. LAMMPS updates these flags as the rigid bodies cross periodic
boundaries during the simulation.
NOTE: If you use the infile or mol keywords and write restart files during a simulation, then each time a
restart file is written, the fix also write an auxiliary restart file with the name rfile.rigid, where "rfile" is the
name of the restart file, e.g. tmp.restart.10000 and tmp.restart.10000.rigid. This auxiliary file is in the same
format described above. Thus it can be used in a new input script that restarts the run and re-specifies a rigid
fix using an infile keyword and the appropriate filename. Note that the auxiliary file will contain one line for
every rigid body, even if the original file only listed a subset of the rigid bodies.
If you use a temperature compute with a group that includes particles in rigid bodies, the degrees-of-freedom
removed by each rigid body are accounted for in the temperature (and pressure) computation, but only if the
temperature group includes all the particles in a particular rigid body.
A 3d rigid body has 6 degrees of freedom (3 translational, 3 rotational), except for a collection of point
particles lying on a straight line, which has only 5, e.g a dimer. A 2d rigid body has 3 degrees of freedom (2
LAMMPS Users Manual
1049
translational, 1 rotational).
NOTE: You may wish to explicitly subtract additional degrees-of-freedom if you use the force and torque
keywords to eliminate certain motions of one or more rigid bodies. LAMMPS does not do this automatically.
The rigid body contribution to the pressure of the system (virial) is also accounted for by this fix.
If your simulation is a hybrid model with a mixture of rigid bodies and non-rigid particles (e.g. solvent) there
are several ways these rigid fixes can be used in tandem with fix nve, fix nvt, fix npt, and fix nph.
If you wish to perform NVE dynamics (no thermostatting or barostatting), use one of 4 NVE rigid styles to
integrate the rigid bodies, and fix nve to integrate the non-rigid particles.
If you wish to perform NVT dynamics (thermostatting, but no barostatting), you can use one of the 2 NVT
rigid styles for the rigid bodies, and any thermostatting fix for the non-rigid particles (fix nvt, fix langevin, fix
temp/berendsen). You can also use one of the 4 NVE rigid styles for the rigid bodies and thermostat them
using fix langevin on the group that contains all the particles in the rigid bodies. The net force added by fix
langevin to each rigid body effectively thermostats its translational center-of-mass motion. Not sure how well
it does at thermostatting its rotational motion.
If you with to perform NPT or NPH dynamics (barostatting), you cannot use both fix npt and the NPT or NPH
rigid styles. This is because there can only be one fix which monitors the global pressure and changes the
simulation box dimensions. So you have 3 choices:
Use one of the 4 NPT or NPH styles for the rigid bodies. Use the dilate all option so that it will dilate
the positions of the non-rigid particles as well. Use fix nvt (or any other thermostat) for the non-rigid
particles.
•
Use fix npt for the group of non-rigid particles. Use the dilate all option so that it will dilate the
center-of-mass positions of the rigid bodies as well. Use one of the 4 NVE or 2 NVT rigid styles for
the rigid bodies.
•
Use fix press/berendsen to compute the pressure and change the box dimensions. Use one of the 4
NVE or 2 NVT rigid styles for the rigid bodies. Use fix nvt (or any other thermostat) for the non-rigid
particles.
•
In all case, the rigid bodies and non-rigid particles both contribute to the global pressure and the box is scaled
the same by any of the barostatting fixes.
You could even use the 2nd and 3rd options for a non-hybrid simulation consisting of only rigid bodies,
assuming you give fix npt an empty group, though it's an odd thing to do. The barostatting fixes (fix npt and
fix press/berensen) will monitor the pressure and change the box dimensions, but not time integrate any
particles. The integration of the rigid bodies will be performed by fix rigid/nvt.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
LAMMPS Users Manual
1050

You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about the 4 NVE rigid styles is written to binary restart files. The exception is if the infile or
mol keyword is used, in which case an auxiliary file is written out with rigid body information each time a
restart file is written, as explained above for the infile keyword. For the 2 NVT rigid styles, the state of the
Nose/Hoover thermostat is written to binary restart files. Ditto for the 4 NPT and NPH rigid styles, and the
state of the Nose/Hoover barostat. See the read_restart command for info on how to re-specify a fix in an
input script that reads a restart file, so that the operation of the fix continues in an uninterrupted fashion.
The fix_modify energy option is supported by the 6 NVT, NPT, NPH rigid styles to add the energy change
induced by the thermostatting to the system's potential energy as part of thermodynamic output.
The fix_modify temp and press options are supported by the 4 NPT and NPH rigid styles to change the
computes used to calculate the instantaneous pressure tensor. Note that the 2 NVT rigid fixes do not use any
external compute to compute instantaneous temperature.
The 2 NVE rigid fixes compute a global scalar which can be accessed by various output commands. The
scalar value calculated by these fixes is "intensive". The scalar is the current temperature of the collection of
rigid bodies. This is averaged over all rigid bodies and their translational and rotational degrees of freedom.
The translational energy of a rigid body is 1/2 m v^2, where m = total mass of the body and v = the velocity of
its center of mass. The rotational energy of a rigid body is 1/2 I w^2, where I = the moment of inertia tensor of
the body and w = its angular velocity. Degrees of freedom constrained by the force and torque keywords are
removed from this calculation, but only for the rigid and rigid/nve fixes.
The 6 NVT, NPT, NPH rigid fixes compute a global scalar which can be accessed by various output
commands. The scalar value calculated by these fixes is "extensive". The scalar is the cumulative energy
change due to the thermostatting and barostatting the fix performs.
All of the rigid styles (not the rigid/small styles) compute a global array of values which can be accessed by
various output commands. Similar information about the bodies defined by the rigid/small styles can be
accessed via the compute rigid/local command.
The number of rows in the array is equal to the number of rigid bodies. The number of columns is 15. Thus
for each rigid body, 15 values are stored: the xyz coords of the center of mass (COM), the xyz components of
the COM velocity, the xyz components of the force acting on the COM, the xyz components of the torque
acting on the COM, and the xyz image flags of the COM.
The center of mass (COM) for each body is similar to unwrapped coordinates written to a dump file. It will
always be inside (or slightly outside) the simulation box. The image flags have the same meaning as image
flags for atom positions (see the "dump" command). This means you can calculate the unwrapped COM by
applying the image flags to the COM, the same as when unwrapped coordinates are written to a dump file.
The force and torque values in the array are not affected by the force and torque keywords in the fix rigid
command; they reflect values before any changes are made by those keywords.
LAMMPS Users Manual
1051
The ordering of the rigid bodies (by row in the array) is as follows. For the single keyword there is just one
rigid body. For the molecule keyword, the bodies are ordered by ascending molecule ID. For the group
keyword, the list of group IDs determines the ordering of bodies.
The array values calculated by these fixes are "intensive", meaning they are independent of the number of
atoms in the simulation.
No parameter of these fixes can be used with the start/stop keywords of the run command. These fixes are not
invoked during energy minimization.
Restrictions:
These fixes are all part of the RIGID package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Assigning a temperature via the velocity create command to a system with rigid bodies may not have the
desired outcome for two reasons. First, the velocity command can be invoked before the rigid-body fix is
invoked or initialized and the number of adjusted degrees of freedom (DOFs) is known. Thus it is not possible
to compute the target temperature correctly. Second, the assigned velocities may be partially canceled when
constraints are first enforced, leading to a different temperature than desired. A workaround for this is to
perform a run 0 command, which insures all DOFs are accounted for properly, and then rescale the
temperature to the desired value before performing a simulation. For example:
velocity all create 300.0 12345
run 0 # temperature may not be 300K
velocity all scale 300.0 # now it should be
Related commands:
delete_bonds, neigh_modify exclude, fix shake
Default:
The option defaults are force * on on on and torque * on on on, meaning all rigid bodies are acted on by
center-of-mass force and torque. Also Tchain = Pchain = 10, Titer = 1, Torder = 3.
(Hoover) Hoover, Phys Rev A, 31, 1695 (1985).
(Kamberaj) Kamberaj, Low, Neal, J Chem Phys, 122, 224114 (2005).
(Martyna) Martyna, Klein, Tuckerman, J Chem Phys, 97, 2635 (1992); Martyna, Tuckerman, Tobias, Klein,
Mol Phys, 87, 1117.
(Miller) Miller, Eleftheriou, Pattnaik, Ndirango, and Newns, J Chem Phys, 116, 8649 (2002).
(Zhang) Zhang, Glotzer, Nanoletters, 4, 1407-1413 (2004).
LAMMPS Users Manual
1052
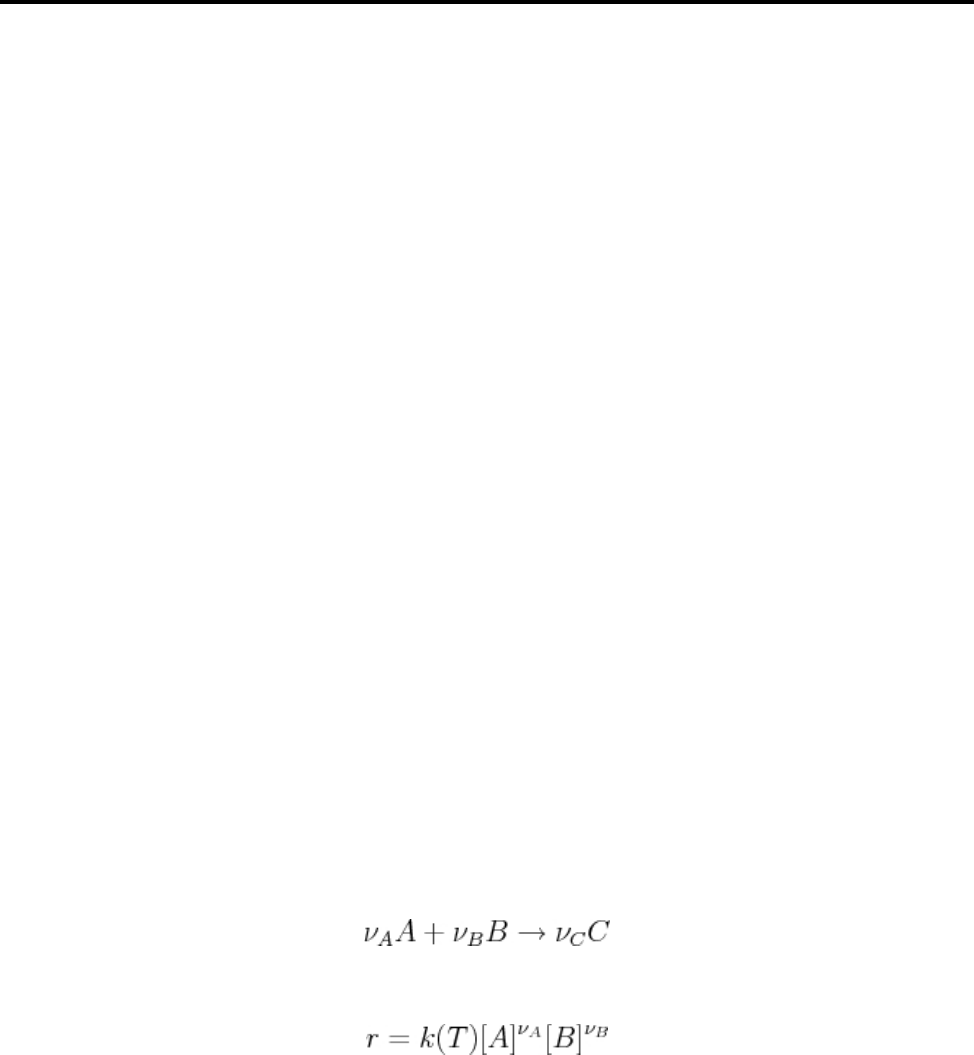
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix rx command
Syntax:
fix ID group-ID rx file localTemp matrix solver minSteps ...
ID, group-ID are documented in fix command• rx = style name of this fix command• file = filename containing the reaction kinetic equations and Arrhenius parameters• localTemp = none,lucy = no local temperature averaging or local temperature defined through Lucy
weighting function
•
matrix = sparse, dense format for the stoichiometric matrix• solver = lammps_rk4,rkf45 = rk4 is an explicit 4th order Runge-Kutta method; rkf45 is an adaptive
4th-order Runge-Kutta-Fehlberg method
•
minSteps = # of steps for rk4 solver or minimum # of steps for rkf45 (rk4 or rkf45)• maxSteps = maximum number of steps for the rkf45 solver (rkf45 only)• relTol = relative tolerance for the rkf45 solver (rkf45 only)• absTol = absolute tolerance for the rkf45 solver (rkf45 only)• diag = Diagnostics frequency for the rkf45 solver (optional, rkf45 only)•
Examples:
fix 1 all rx kinetics.rx none dense lammps_rk4
fix 1 all rx kinetics.rx none sparse lammps_rk4 1
fix 1 all rx kinetics.rx lucy sparse lammps_rk4 10
fix 1 all rx kinetics.rx none dense rkf45 1 100 1e-6 1e-8
fix 1 all rx kinetics.rx none dense rkf45 1 100 1e-6 1e-8 -1
Description:
Fix rx solves the reaction kinetic ODEs for a given reaction set that is defined within the file associated with
this command.
For a general reaction such that
the reaction rate equation is defined to be of the form
In the current implementation, the exponents are defined to be equal to the stoichiometric coefficients. A
given reaction set consisting of n reaction equations will contain a total of m species. A set of m ordinary
differential equations (ODEs) that describe the change in concentration of a given species as a function of
time are then constructed based on the n reaction rate equations.
The ODE systems are solved over the full DPD timestep dt using either a 4th order Runge-Kutta rk4 method
with a fixed step-size h, specified by the lammps_rk4 keyword, or a 4th order Runge-Kutta-Fehlberg (rkf45)
method with an adaptive step-size for h. The number of ODE steps per DPD timestep for the rk4 method is
LAMMPS Users Manual
1053
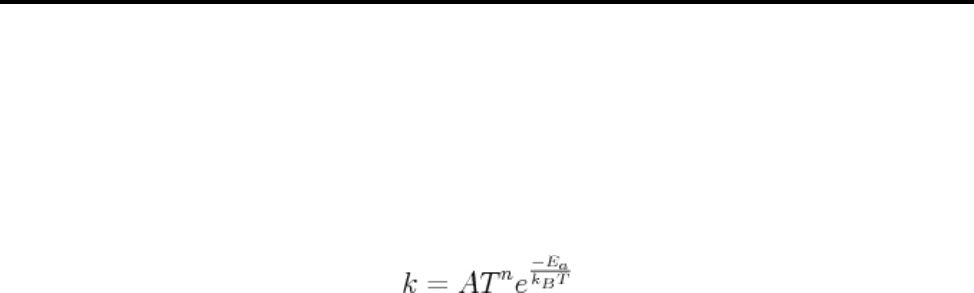
optionally specified immediately after the rk4 keyword. The ODE step-size is set as dt/num_steps. Smaller
step-sizes tend to yield more accurate results but there is not control on the error. For error control, use the
rkf45 ODE solver.
The rkf45 method adjusts the step-size so that the local truncation error is held within the specified absolute
and relative tolerances. The initial step-size h0 can be specified by the user or estimated internally. It is
recommended that the user specify h0 since this will generally reduced the number of ODE integration steps
required. h0 is defined as dt / min_steps if min_steps >= 1. If min_steps == 0, h0 is estimated such that an
explicit Euler method would likely produce an acceptable solution. This is generally overly conservative for
the 4th-order method and users are advised to specify h0 as some fraction of the DPD timestep. For small
DPD timesteps, only one step may be necessary depending upon the tolerances. Note that more than
min_steps ODE steps may be taken depending upon the ODE stiffness but no more than max_steps will be
taken. If max_steps is reached, an error warning is printed and the simulation is stopped.
After each ODE step, the solution error e is tested and weighted using the absTol and relTol values. The error
vector is weighted as e / (relTol * |u| + absTol) where u is the solution vector. If the norm of the error is <= 1,
the solution is accepted, h is increased by a proportional amount, and the next ODE step is begun. Otherwise,
h is shrunk and the ODE step is repeated.
Run-time diagnostics are available for the rkf45 ODE solver. The frequency (in time-steps) that diagnostics
are reported is controlled by the last (optional) 12th argument. A negative frequency means that diagnostics
are reported once at the end of each run. A positive value N means that the diagnostics are reported once per
N time-steps.
The diagnostics report the average # of integrator steps and RHS function evaluations and run-time per ODE
as well as the average/RMS/min/max per process. If the reporting frequency is 1, the RMS/min/max per ODE
are also reported. The per ODE statistics can be used to adjust the tolerance and min/max step parameters. The
statistics per MPI process can be useful to examine any load imbalance caused by the adaptive ODE solver.
(Some DPD particles can take longer to solve than others. This can lead to an imbalance across the MPI
processes.)
The filename specifies a file that contains the entire set of reaction kinetic equations and corresponding
Arrhenius parameters. The format of this file is described below.
There is no restriction on the total number or reaction equations that are specified. The species names are
arbitrary string names that are associated with the species concentrations. Each species in a given reaction
must be preceded by it's stoichiometric coefficient. The only delimiters that are recognized between the
species are either a + or = character. The = character corresponds to an irreversible reaction. After specifying
the reaction, the reaction rate constant is determined through the temperature dependent Arrhenius equation:
where A is the Arrhenius factor in time units or concentration/time units, n is the unitless exponent of the
temperature dependence, and E_a is the activation energy in energy units. The temperature dependence can be
removed by specifying the exponent as zero.
The internal temperature of the coarse-grained particles can be used in constructing the reaction rate constants
at every DPD timestep by specifying the keyword none. Alternatively, the keyword lucy can be specified to
compute a local-average particle internal temperature for use in the reaction rate constant expressions. The
local-average particle internal temperature is defined as:
LAMMPS Users Manual
1054
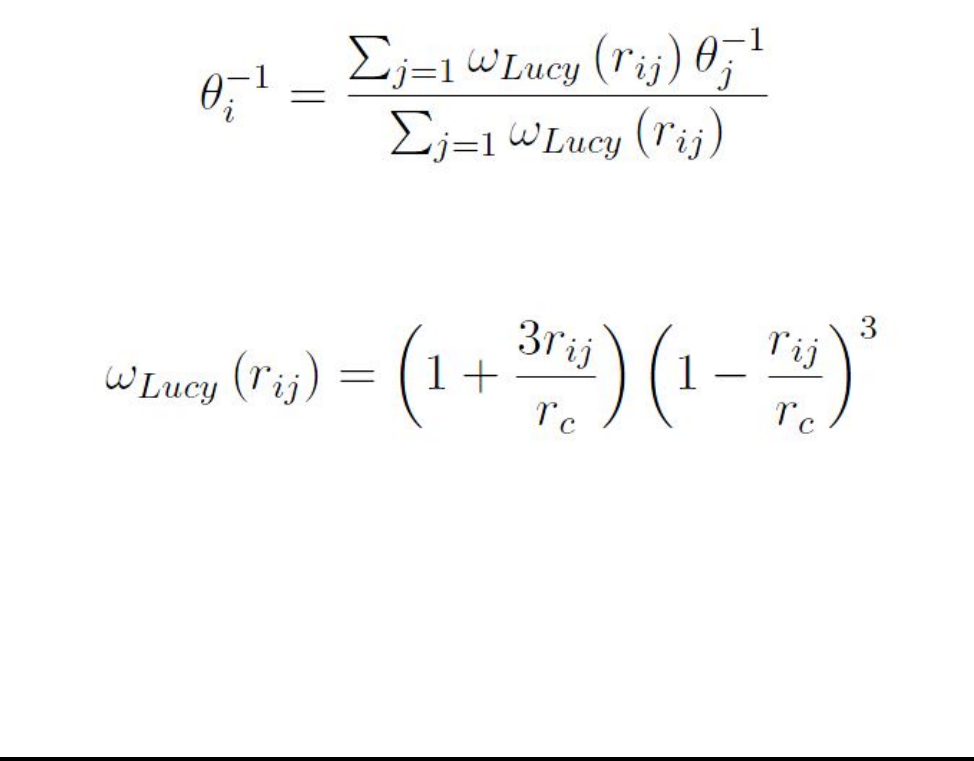
where the Lucy function is expressed as:
The self-particle interaction is included in the above equation.
The stoichiometric coefficients for the reaction mechanism are stored in either a sparse or dense matrix
format. The dense matrix should only be used for small reaction mechanisms. The sparse matrix should be
used when there are many reactions (e.g., more than 5). This allows the number of reactions and species to
grow while keeping the computational cost tractable. The matrix format can be specified as using either the
sparse or dense keywords. If all stoichiometric coefficients for a reaction are small integers (whole numbers
<= 3), a fast exponential function is used. This can save significant computational time so users are
encouraged to use integer coefficients where possible.
The format of a tabulated file is as follows (without the parenthesized comments):
# Rxn equations and parameters (one or more comment or blank lines)
1.0 hcn + 1.0 no2 = 1.0 no + 0.5 n2 + 0.5 h2 + 1.0 co 2.49E+01 0.0 1.34 (rxn equation, A, n, Ea)
1.0 hcn + 1.0 no = 1.0 co + 1.0 n2 + 0.5 h2 2.16E+00 0.0 1.52
...
1.0 no + 1.0 co = 0.5 n2 + 1.0 co2 1.66E+06 0.0 0.69
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections.
Following a blank line, the next N lines list the N reaction equations. Each species within the reaction
equation is specified through its stoichiometric coefficient and a species tag. Reactant species are specified on
the left-hand side of the equation and product species are specified on the right-hand side of the equation.
After specifying the reactant and product species, the final three arguments of each line represent the
Arrhenius parameter A, the temperature exponent n, and the activation energy Ea.
Note that the species tags that are defined in the reaction equations are used by the fix eos/table/rx command
to define the thermodynamic properties of each species. Furthermore, the number of species molecules (i.e.,
concentration) can be specified either with the set command using the "d_" prefix or by reading directly the
concentrations from a data file. For the latter case, the read_data command with the fix keyword should be
LAMMPS Users Manual
1055

specified, where the fix-ID will be the "fix rx ID with a suffix, e.g.
fix foo all rx reaction.file ... read_data data.dpd fix foo_SPECIES NULL Species
Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This command also requires use of the atom_style dpd command.
This command can only be used with a constant energy or constant enthalpy DPD simulation.
Related commands:
fix eos/table/rx, fix shardlow, pair dpd/fdt/energy
Default: none
LAMMPS Users Manual
1056

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix saed/vtk command
Syntax:
fix ID group-ID saed/vtk Nevery Nrepeat Nfreak c_ID attribute args ... keyword args ...
ID, group-ID are documented in fix command• saed/vtk = style name of this fix command• Nevery = use input values every this many timesteps• Nrepeat = # of times to use input values for calculating averages• Nfreq = calculate averages every this many timesteps• c_ID = saed compute ID
keyword = file or ave or start or file or overwrite:l
ave args = one or running or window M
one = output a new average value every Nfreq steps
running = output cumulative average of all previous Nfreq steps
window M = output average of M most recent Nfreq steps
start args = Nstart
Nstart = start averaging on this timestep
file arg = filename
filename = name of file to output time averages to
overwrite arg = none = overwrite output file with only latest output
•
Examples:
compute 1 all saed 0.0251 Al O Kmax 1.70 Zone 0 0 1 dR_Ewald 0.01 c 0.5 0.5 0.5
compute 2 all saed 0.0251 Ni Kmax 1.70 Zone 0 0 0 c 0.05 0.05 0.05 manual echo
fix saed/vtk 1 1 1 c_1 file Al2O3_001.saed
fix saed/vtk 1 1 1 c_2 file Ni_000.saed
Description:
Time average computed intensities from compute saed and write output to a file in the 3rd generation vtk
image data format for visualization directly in parallelized visualization software packages like ParaView and
VisIt. Note that if no time averaging is done, this command can be used as a convenient way to simply output
diffraction intensities at a single snapshot.
To produce output in the image data vtk format ghost data is added outside the Kmax range assigned in the
compute saed. The ghost data is assigned a value of -1 and can be removed setting a minimum isovolume of 0
within the visualization software. SAED images can be created by visualizing a spherical slice of the data that
is centered at R_Ewald*[h k l]/norm([h k l]), where R_Ewald=1/lambda.
The group specified within this command is ignored. However, note that specified values may represent
calculations performed by saed computes which store their own "group" definitions.
Fix saed/vtk is designed to work only with compute saed values, e.g.
compute 3 top saed 0.0251 Al O
fix saed/vtk 1 1 1 c_3 file Al2O3_001.saed
LAMMPS Users Manual
1057

The Nevery, Nrepeat, and Nfreq arguments specify on what timesteps the input values will be used in order to
contribute to the average. The final averaged quantities are generated on timesteps that are a multiple of
Nfreq. The average is over Nrepeat quantities, computed in the preceding portion of the simulation every
Nevery timesteps. Nfreq must be a multiple of Nevery and Nevery must be non-zero even if Nrepeat is 1. Also,
the timesteps contributing to the average value cannot overlap, i.e. Nrepeat*Nevery can not exceed Nfreq.
For example, if Nevery=2, Nrepeat=6, and Nfreq=100, then values on timesteps 90,92,94,96,98,100 will be
used to compute the final average on timestep 100. Similarly for timesteps 190,192,194,196,198,200 on
timestep 200, etc. If Nrepeat=1 and Nfreq = 100, then no time averaging is done; values are simply generated
on timesteps 100,200,etc.
The output for fix ave/time/saed is a file written with the 3rd generation vtk image data formatting. The
filename assigned by the file keyword is appended with _N.vtk where N is an index (0,1,2...) to account for
multiple diffraction intensity outputs.
By default the header contains the following information (with example data):
# vtk DataFile Version 3.0 c_SAED
Image data set
ASCII
DATASET STRUCTURED_POINTS
DIMENSIONS 337 219 209
ASPECT_RATIO 0.00507953 0.00785161 0.00821458
ORIGIN -0.853361 -0.855826 -0.854316
POINT_DATA 15424827
SCALARS intensity float
LOOKUP_TABLE default
...data
In this example, kspace is sampled across a 337 x 219 x 209 point mesh where the mesh spacing is
approximately 0.005, 0.007, and 0.008 inv(length) units in the k1, k2, and k3 directions, respectively. The
data is shifted by -0.85, -0.85, -0.85 inv(length) units so that the origin will lie at 0, 0, 0. Here, 15,424,827
kspace points are sampled in total.
Additional optional keywords also affect the operation of this fix.
The ave keyword determines how the values produced every Nfreq steps are averaged with values produced
on previous steps that were multiples of Nfreq, before they are accessed by another output command or
written to a file.
If the ave setting is one, then the values produced on timesteps that are multiples of Nfreq are independent of
each other; they are output as-is without further averaging.
If the ave setting is running, then the values produced on timesteps that are multiples of Nfreq are summed
and averaged in a cumulative sense before being output. Each output value is thus the average of the value
produced on that timestep with all preceding values. This running average begins when the fix is defined; it
can only be restarted by deleting the fix via the unfix command, or by re-defining the fix by re-specifying it.
If the ave setting is window, then the values produced on timesteps that are multiples of Nfreq are summed
and averaged within a moving "window" of time, so that the last M values are used to produce the output. E.g.
if M = 3 and Nfreq = 1000, then the output on step 10000 will be the average of the individual values on steps
LAMMPS Users Manual
1058

8000,9000,10000. Outputs on early steps will average over less than M values if they are not available.
The start keyword specifies what timestep averaging will begin on. The default is step 0. Often input values
can be 0.0 at time 0, so setting start to a larger value can avoid including a 0.0 in a running or windowed
average.
The file keyword allows a filename to be specified. Every Nfreq steps, the vector of saed intensity data is
written to a new file using the 3rd generation vtk format. The base of each file is assigned by the file keyword
and this string is appended with _N.vtk where N is an index (0,1,2...) to account for situations with multiple
diffraction intensity outputs.
The overwrite keyword will continuously overwrite the output file with the latest output, so that it only
contains one timestep worth of output. This option can only be used with the ave running setting.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
The attributes for fix_saed_vtk must match the values assigned in the associated compute_saed command.
Related commands:
compute_saed
Default:
The option defaults are ave = one, start = 0, no file output.
(Coleman) Coleman, Spearot, Capolungo, MSMSE, 21, 055020 (2013).
LAMMPS Users Manual
1059

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix setforce command
fix setforce/kk command
Syntax:
fix ID group-ID setforce fx fy fz keyword value ...
ID, group-ID are documented in fix command• setforce = style name of this fix command• fx,fy,fz = force component values• any of fx,fy,fz can be a variable (see below)• zero or more keyword/value pairs may be appended to args• keyword = region
region value = region-ID
region-ID = ID of region atoms must be in to have added force
•
Examples:
fix freeze indenter setforce 0.0 0.0 0.0
fix 2 edge setforce NULL 0.0 0.0
fix 2 edge setforce NULL 0.0 v_oscillate
Description:
Set each component of force on each atom in the group to the specified values fx,fy,fz. This erases all
previously computed forces on the atom, though additional fixes could add new forces. This command can be
used to freeze certain atoms in the simulation by zeroing their force, either for running dynamics or
performing an energy minimization. For dynamics, this assumes their initial velocity is also zero.
Any of the fx,fy,fz values can be specified as NULL which means do not alter the force component in that
dimension.
Any of the 3 quantities defining the force components can be specified as an equal-style or atom-style
variable, namely fx, fy, fz. If the value is a variable, it should be specified as v_name, where name is the
variable name. In this case, the variable will be evaluated each timestep, and its value used to determine the
force component.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent force field.
Atom-style variables can specify the same formulas as equal-style variables but can also include per-atom
values, such as atom coordinates. Thus it is easy to specify a spatially-dependent force field with optional
time-dependence as well.
If the region keyword is used, the atom must also be in the specified geometric region in order to have force
added to it.
LAMMPS Users Manual
1060

Styles with a r kk suffix are functionally the same as the corresponding style without the suffix. They have
been optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual.
The accelerated styles take the same arguments and should produce the same results, except for round-off and
precision issues.
The region keyword is also supported by Kokkos, but a Kokkos-enabled region must be used. See the region
region command for more information.
These accelerated styles are part of the r Kokkos package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is setting the forces to the desired values; on all other levels, the force is set to 0.0 for the
atoms in the fix group, so that setforce values are not counted multiple times. Default is to to override forces
at the outermost level.
This fix computes a global 3-vector of forces, which can be accessed by various output commands. This is the
total force on the group of atoms before the forces on individual atoms are changed by the fix. The vector
values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command, but
you cannot set forces to any value besides zero when performing a minimization. Use the fix addforce
command if you want to apply a non-zero force to atoms during a minimization.
Restrictions: none
Related commands:
fix addforce, fix aveforce
Default: none
LAMMPS Users Manual
1061

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix shake command
fix rattle command
Syntax:
fix ID group-ID style tol iter N constraint values ... keyword value ...
ID, group-ID are documented in fix command• style = shake or rattle = style name of this fix command• tol = accuracy tolerance of SHAKE solution• iter = max # of iterations in each SHAKE solution• N = print SHAKE statistics every this many timesteps (0 = never)• one or more constraint/value pairs are appended• constraint = b or a or t or m
b values = one or more bond types
a values = one or more angle types
t values = one or more atom types
m value = one or more mass values
•
zero or more keyword/value pairs may be appended• keyword = mol
mol value = template-ID
template-ID = ID of molecule template specified in a separate molecule command
•
Examples:
fix 1 sub shake 0.0001 20 10 b 4 19 a 3 5 2
fix 1 sub shake 0.0001 20 10 t 5 6 m 1.0 a 31
fix 1 sub shake 0.0001 20 10 t 5 6 m 1.0 a 31 mol myMol
fix 1 sub rattle 0.0001 20 10 t 5 6 m 1.0 a 31
fix 1 sub rattle 0.0001 20 10 t 5 6 m 1.0 a 31 mol myMol
Description:
Apply bond and angle constraints to specified bonds and angles in the simulation by either the SHAKE or
RATTLE algorithms. This typically enables a longer timestep.
SHAKE vs RATTLE:
The SHAKE algorithm was invented for schemes such as standard Verlet timestepping, where only the
coordinates are integrated and the velocities are approximated as finite differences to the trajectories
(Ryckaert et al. (1977)). If the velocities are integrated explicitly, as with velocity Verlet which is what
LAMMPS uses as an integration method, a second set of constraining forces is required in order to eliminate
velocity components along the bonds (Andersen (1983)).
In order to formulate individual constraints for SHAKE and RATTLE, focus on a single molecule whose
bonds are constrained. Let Ri and Vi be the position and velocity of atom i at time n, for i=1,...,N, where N is
the number of sites of our reference molecule. The distance vector between sites i and j is given by
LAMMPS Users Manual
1062
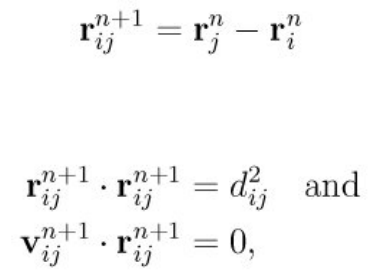
The constraints can then be formulated as
The SHAKE algorithm satisfies the first condition, i.e. the sites at time n+1 will have the desired separations
Dij immediately after the coordinates are integrated. If we also enforce the second condition, the velocity
components along the bonds will vanish. RATTLE satisfies both conditions. As implemented in LAMMPS,
fix rattle uses fix shake for satisfying the coordinate constraints. Therefore the settings and optional keywords
are the same for both fixes, and all the information below about SHAKE is also relevant for RATTLE.
SHAKE:
Each timestep the specified bonds and angles are reset to their equilibrium lengths and angular values via the
SHAKE algorithm (Ryckaert et al. (1977)). This is done by applying an additional constraint force so that the
new positions preserve the desired atom separations. The equations for the additional force are solved via an
iterative method that typically converges to an accurate solution in a few iterations. The desired tolerance (e.g.
1.0e-4 = 1 part in 10000) and maximum # of iterations are specified as arguments. Setting the N argument will
print statistics to the screen and log file about regarding the lengths of bonds and angles that are being
constrained. Small delta values mean SHAKE is doing a good job.
In LAMMPS, only small clusters of atoms can be constrained. This is so the constraint calculation for a
cluster can be performed by a single processor, to enable good parallel performance. A cluster is defined as a
central atom connected to others in the cluster by constrained bonds. LAMMPS allows for the following kinds
of clusters to be constrained: one central atom bonded to 1 or 2 or 3 atoms, or one central atom bonded to 2
others and the angle between the 3 atoms also constrained. This means water molecules or CH2 or CH3
groups may be constrained, but not all the C-C backbone bonds of a long polymer chain.
The b constraint lists bond types that will be constrained. The t constraint lists atom types. All bonds
connected to an atom of the specified type will be constrained. The m constraint lists atom masses. All bonds
connected to atoms of the specified masses will be constrained (within a fudge factor of MASSDELTA
specified in fix_shake.cpp). The a constraint lists angle types. If both bonds in the angle are constrained then
the angle will also be constrained if its type is in the list.
For all constraints, a particular bond is only constrained if both atoms in the bond are in the group specified
with the SHAKE fix.
The degrees-of-freedom removed by SHAKE bonds and angles are accounted for in temperature and pressure
computations. Similarly, the SHAKE contribution to the pressure of the system (virial) is also accounted for.
NOTE: This command works by using the current forces on atoms to calculate an additional constraint force
which when added will leave the atoms in positions that satisfy the SHAKE constraints (e.g. bond length)
after the next time integration step. If you define fixes (e.g. fix efield) that add additional force to the atoms
after fix shake operates, then this fix will not take them into account and the time integration will typically not
LAMMPS Users Manual
1063

satisfy the SHAKE constraints. The solution for this is to make sure that fix shake is defined in your input
script after any other fixes which add or change forces (to atoms that fix shake operates on).
The mol keyword should be used when other commands, such as fix deposit or fix pour, add molecules
on-the-fly during a simulation, and you wish to constrain the new molecules via SHAKE. You specify a
template-ID previously defined using the molecule command, which reads a file that defines the molecule.
You must use the same template-ID that the command adding molecules uses. The coordinates, atom types,
special bond restrictions, and SHAKE info can be specified in the molecule file. See the molecule command
for details. The only settings required to be in this file (by this command) are the SHAKE info of atoms in the
molecule.
Styles with a suffix are functionally the same as the corresponding style without the suffix. They have been
optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The
accelerated styles take the same arguments and should produce the same results, except for round-off and
precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
RATTLE:
The velocity constraints lead to a linear system of equations which can be solved analytically. The
implementation of the algorithm in LAMMPS closely follows (Andersen (1983)).
NOTE: The fix rattle command modifies forces and velocities and thus should be defined after all other
integration fixes in your input script. If you define other fixes that modify velocities or forces after fix rattle
operates, then fix rattle will not take them into account and the overall time integration will typically not
satisfy the RATTLE constraints. You can check whether the constraints work correctly by setting the value of
RATTLE_DEBUG in src/fix_rattle.cpp to 1 and recompiling LAMMPS.
Restart, fix_modify, output, run start/stop, minimize info:
No information about these fixes is written to binary restart files. None of the fix_modify options are relevant
to these fixes. No global or per-atom quantities are stored by these fixes for access by various output
commands. No parameter of these fixes can be used with the start/stop keywords of the run command. These
fixes are not invoked during energy minimization.
Restrictions:
These fixes are part of the RIGID package. They are only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
For computational efficiency, there can only be one shake or rattle fix defined in a simulation.
LAMMPS Users Manual
1064

If you use a tolerance that is too large or a max-iteration count that is too small, the constraints will not be
enforced very strongly, which can lead to poor energy conservation. You can test for this in your system by
running a constant NVE simulation with a particular set of SHAKE parameters and monitoring the energy
versus time.
SHAKE or RATTLE should not be used to constrain an angle at 180 degrees (e.g. linear CO2 molecule). This
causes numeric difficulties.
Related commands: none
Default: none
(Ryckaert) J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J of Comp Phys, 23, 327-341 (1977).
(Andersen) H. Andersen, J of Comp Phys, 52, 24-34 (1983).
LAMMPS Users Manual
1065
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix shardlow command
Syntax:
fix ID group-ID shardlow
ID, group-ID are documented in fix command• shardlow = style name of this fix command•
Examples:
fix 1 all shardlow
Description:
Specifies that the Shardlow splitting algorithm (SSA) is to be used to integrate the DPD equations of motion.
The SSA splits the integration into a stochastic and deterministic integration step. The fix shardlow performs
the stochastic integration step and must be used in conjunction with a deterministic integrator (e.g. fix nve or
fix nph). The stochastic integration of the dissipative and random forces is performed prior to the
deterministic integration of the conservative force. Further details regarding the method are provided in
(Lisal) and (Larentzos1).
The fix shardlow must be used with the pair_style dpd/fdt or pair_style dpd/fdt/energy command to properly
initialize the fluctuation-dissipation theorem parameter(s) sigma (and kappa, if necessary).
Note that numerous variants of DPD can be specified by choosing an appropriate combination of the
integrator and pair_style dpd/fdt command. DPD under isothermal conditions can be specified by using fix
shardlow, fix nve and pair_style dpd/fdt. DPD under isoenergetic conditions can be specified by using fix
shardlow, fix nve and pair_style dpd/fdt/energy. DPD under isobaric conditions can be specified by using fix
shardlow, fix nph and pair_style dpd/fdt. DPD under isoenthalpic conditions can be specified by using fix
shardlow, fix nph and pair_style dpd/fdt/energy. Examples of each DPD variant are provided in the
examples/USER/dpd directory.
Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This fix is currently limited to orthogonal simulation cell geometries.
This fix must be used with an additional fix that specifies time integration, e.g. fix nve or fix nph.
The Shardlow splitting algorithm requires the sizes of the sub-domain lengths to be larger than twice the
cutoff+skin. Generally, the domain decomposition is dependent on the number of processors requested.
Related commands:
pair_style dpd/fdt, fix eos/cv
LAMMPS Users Manual
1066

Default: none
(Lisal) M. Lisal, J.K. Brennan, J. Bonet Avalos, "Dissipative particle dynamics as isothermal, isobaric,
isoenergetic, and isoenthalpic conditions using Shardlow-like splitting algorithms.", J. Chem. Phys., 135,
204105 (2011).
(Larentzos1) J.P. Larentzos, J.K. Brennan, J.D. Moore, M. Lisal and W.D. Mattson, "Parallel Implementation
of Isothermal and Isoenergetic Dissipative Particle Dynamics Using Shardlow-Like Splitting Algorithms",
Comput. Phys. Commun., 185, 1987-1998 (2014).
(Larentzos2) J.P. Larentzos, J.K. Brennan, J.D. Moore, and W.D. Mattson, "LAMMPS Implementation of
Constant Energy Dissipative Particle Dynamics (DPD-E)", ARL-TR-6863, U.S. Army Research Laboratory,
Aberdeen Proving Ground, MD (2014).
LAMMPS Users Manual
1067

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix smd command
Syntax:
fix ID group-ID smd type values keyword values
ID, group-ID are documented in fix command• smd = style name of this fix command• mode = cvel or cfor to select constant velocity or constant force SMD
cvel values = K vel
K = spring constant (force/distance units)
vel = velocity of pulling (distance/time units)
cfor values = force
force = pulling force (force units)
•
keyword = tether or couple
tether values = x y z R0
x,y,z = point to which spring is tethered
R0 = distance of end of spring from tether point (distance units)
couple values = group-ID2 x y z R0
group-ID2 = 2nd group to couple to fix group with a spring
x,y,z = direction of spring, automatically computed with 'auto'
R0 = distance of end of spring (distance units)
•
Examples:
fix pull cterm smd cvel 20.0 -0.00005 tether NULL NULL 100.0 0.0
fix pull cterm smd cvel 20.0 -0.0001 tether 25.0 25 25.0 0.0
fix stretch cterm smd cvel 20.0 0.0001 couple nterm auto auto auto 0.0
fix pull cterm smd cfor 5.0 tether 25.0 25.0 25.0 0.0
Description:
This fix implements several options of steered MD (SMD) as reviewed in (Izrailev), which allows to induce
conformational changes in systems and to compute the potential of mean force (PMF) along the assumed
reaction coordinate (Park) based on Jarzynski's equality (Jarzynski). This fix borrows a lot from fix spring and
fix setforce.
You can apply a moving spring force to a group of atoms (tether style) or between two groups of atoms
(couple style). The spring can then be used in either constant velocity (cvel) mode or in constant force (cfor)
mode to induce transitions in your systems. When running in tether style, you may need some way to fix some
other part of the system (e.g. via fix spring/self)
The tether style attaches a spring between a point at a distance of R0 away from a fixed point x,y,z and the
center of mass of the fix group of atoms. A restoring force of magnitude K (R - R0) Mi / M is applied to each
atom in the group where K is the spring constant, Mi is the mass of the atom, and M is the total mass of all
atoms in the group. Note that K thus represents the total force on the group of atoms, not a per-atom force.
In cvel mode the distance R is incremented or decremented monotonously according to the pulling (or
pushing) velocity. In cfor mode a constant force is added and the actual distance in direction of the spring is
LAMMPS Users Manual
1068

recorded.
The couple style links two groups of atoms together. The first group is the fix group; the second is specified
by group-ID2. The groups are coupled together by a spring that is at equilibrium when the two groups are
displaced by a vector in direction x,y,z with respect to each other and at a distance R0 from that displacement.
Note that x,y,z only provides a direction and will be internally normalized. But since it represents the absolute
displacement of group-ID2 relative to the fix group, (1,1,0) is a different spring than (-1,-1,0). For each vector
component, the displacement can be described with the auto parameter. In this case the direction is
recomputed in every step, which can be useful for steering a local process where the whole object undergoes
some other change. When the relative positions and distance between the two groups are not in equilibrium,
the same spring force described above is applied to atoms in each of the two groups.
For both the tether and couple styles, any of the x,y,z values can be specified as NULL which means do not
include that dimension in the distance calculation or force application.
For constant velocity pulling (cvel mode), the running integral over the pulling force in direction of the spring
is recorded and can then later be used to compute the potential of mean force (PMF) by averaging over
multiple independent trajectories along the same pulling path.
Restart, fix_modify, output, run start/stop, minimize info:
The fix stores the direction of the spring, current pulling target distance and the running PMF to binary restart
files. See the read_restart command for info on how to re-specify a fix in an input script that reads a restart
file, so that the operation of the fix continues in an uninterrupted fashion.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
This fix computes a vector list of 7 quantities, which can be accessed by various output commands. The
quantities in the vector are in this order: the x-, y-, and z-component of the pulling force, the total force in
direction of the pull, the equilibrium distance of the spring, the distance between the two reference points, and
finally the accumulated PMF (the sum of pulling forces times displacement).
The force is the total force on the group of atoms by the spring. In the case of the couple style, it is the force
on the fix group (group-ID) or the negative of the force on the 2nd group (group-ID2). The vector values
calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
fix drag, fix spring, fix spring/self, fix spring/rg
Default: none
LAMMPS Users Manual
1069
(Izrailev) Izrailev, Stepaniants, Isralewitz, Kosztin, Lu, Molnar, Wriggers, Schulten. Computational
Molecular Dynamics: Challenges, Methods, Ideas, volume 4 of Lecture Notes in Computational Science and
Engineering, pp. 39-65. Springer-Verlag, Berlin, 1998.
(Park) Park, Schulten, J. Chem. Phys. 120 (13), 5946 (2004)
(Jarzynski) Jarzynski, Phys. Rev. Lett. 78, 2690 (1997)
LAMMPS Users Manual
1070

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix smd/adjust_dt command
Syntax:
fix ID group-ID smd/adjust_dt arg
ID, group-ID are documented in fix command• smd/adjust_dt = style name of this fix command• arg = s_fact
s_fact = safety factor
•
Examples:
fix 1 all smd/adjust_dt 0.1
Description:
The fix calculates a new stable time increment for use with the SMD time integrators.
The stable time increment is based on multiple conditions. For the SPH pair styles, a CFL criterion (Courant,
Friedrichs & Lewy, 1928) is evaluated, which determines the speed of sound cannot propagate further than a
typical spacing between particles within a single time step to ensure no information is lost. For the contact
pair styles, a linear analysis of the pair potential determines a stable maximum time step.
This fix inquires the minimum stable time increment across all particles contained in the group for which this
fix is defined. An additional safety factor s_fact is applied to the time increment.
See this PDF guide to use Smooth Mach Dynamics in LAMMPS.
Restart, fix_modify, output, run start/stop, minimize info:
Currently, no part of USER-SMD supports restarting nor minimization.
Restrictions:
This fix is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
smd/tlsph_dt
Default: none
LAMMPS Users Manual
1071

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix smd/integrate_tlsph command
Syntax:
fix ID group-ID smd/integrate_tlsph keyword values
ID, group-ID are documented in fix command• smd/integrate_tlsph = style name of this fix command• zero or more keyword/value pairs may be appended•
keyword = limit_velocity
limit_velocity value = max_vel
max_vel = maximum allowed velocity
Examples:
fix 1 all smd/integrate_tlsph
fix 1 all smd/integrate_tlsph limit_velocity 1000
Description:
The fix performs explicit time integration for particles which interact according with the Total-Lagrangian
SPH pair style.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
The limit_velocity keyword will control the velocity, scaling the norm of the velocity vector to max_vel in
case it exceeds this velocity limit.
Restart, fix_modify, output, run start/stop, minimize info:
Currently, no part of USER-SMD supports restarting nor minimization. This fix has no outputs.
Restrictions:
This fix is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
smd/integrate_ulsph
Default: none
•
LAMMPS Users Manual
1072

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix smd/integrate_ulsph command
Syntax:
fix ID group-ID smd/integrate_ulsph keyword
ID, group-ID are documented in fix command• smd/integrate_ulsph = style name of this fix command• zero or more keyword/value pairs may be appended•
keyword = adjust_radius or limit_velocity
adjust_radius values = adjust_radius_factor min_nn max_nn adjust_radius_factor = factor which scale the
smooth/kernel radius min_nn = minimum number of neighbors max_nn = maximum number of neighbors
limit_velocity values = max_velocity max_velocity = maximum allowed velocity.
Examples:
fix 1 all smd/integrate_ulsph adjust_radius 1.02 25 50
fix 1 all smd/integrate_ulsph limit_velocity 1000
Description:
The fix performs explicit time integration for particles which interact with the updated Lagrangian SPH pair
style. See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
The adjust_radius keyword activates dynamic adjustment of the per-particle SPH smoothing kernel radius
such that the number of neighbors per particles remains within the interval min_nn to max_nn. The parameter
adjust_radius_factor determines the amount of adjustment per timestep. Typical values are
adjust_radius_factor=1.02, min_nn=15, and max_nn=20.
The limit_velocity keyword will control the velocity, scaling the norm of the velocity vector to max_vel in
case it exceeds this velocity limit.
Restart, fix_modify, output, run start/stop, minimize info:
Currently, no part of USER-SMD supports restarting nor minimization. This fix has no outputs.
Restrictions:
This fix is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
Default: none
LAMMPS Users Manual
1073

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix smd/move_tri_surf command
Syntax:
fix ID group-ID smd/move_tri_surf keyword
ID, group-ID are documented in fix command• smd/move_tri_surf keyword = style name of this fix command• keyword = *LINEAR or *WIGGLE or *ROTATE
*LINEAR args = Vx Vy Vz
Vx,Vy,Vz = components of velocity vector (velocity units), any component can be specified as NULL
*WIGGLE args = Vx Vy Vz max_travel
vx,vy,vz = components of velocity vector (velocity units), any component can be specified as NULL
max_travel = wiggle amplitude
*ROTATE args = Px Py Pz Rx Ry Rz period
Px,Py,Pz = origin point of axis of rotation (distance units)
Rx,Ry,Rz = axis of rotation vector
period = period of rotation (time units)
•
Examples:
fix 1 tool smd/move_tri_surf *LINEAR 20 20 10
fix 2 tool smd/move_tri_surf *WIGGLE 20 20 10
fix 2 tool smd/move_tri_surf *ROTATE 0 0 0 5 2 1
Description:
This fix applies only to rigid surfaces read from .STL files via fix smd/wall_surface . It updates position and
velocity for the particles in the group each timestep without regard to forces on the particles. The rigid
surfaces can thus be moved along simple trajectories during the simulation.
The *LINEAR style moves particles with the specified constant velocity vector V = (Vx,Vy,Vz). This style
also sets the velocity of each particle to V = (Vx,Vy,Vz).
The *WIGGLE style moves particles in an oscillatory fashion. Particles are moved along (vx, vy, vz) with
constant velocity until a displacement of max_travel is reached. Then, the velocity vector is reversed. This
process is repeated.
The *ROTATE style rotates particles around a rotation axis R = (Rx,Ry,Rz) that goes through a point P =
(Px,Py,Pz). The period of the rotation is also specified. This style also sets the velocity of each particle to
(omega cross Rperp) where omega is its angular velocity around the rotation axis and Rperp is a perpendicular
vector from the rotation axis to the particle.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Restart, fix_modify, output, run start/stop, minimize info:
Currently, no part of USER-SMD supports restarting nor minimization. This fix has no outputs.
Restrictions:
LAMMPS Users Manual
1074

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix smd/setvel command
Syntax:
fix ID group-ID smd/setvel vx vy vz keyword value ...
ID, group-ID are documented in fix command• smd/setvel = style name of this fix command• vx,vy,vz = velocity component values• any of vx,vy,vz can be a variable (see below)• zero or more keyword/value pairs may be appended to args• keyword = region
region value = region-ID
region-ID = ID of region particles must be in to have their velocities set
•
Examples:
fix top_velocity top_group setvel 1.0 0.0 0.0
Description:
Set each component of velocity on each particle in the group to the specified values vx,vy,vz, regardless of the
forces acting on the particle. This command can be used to impose velocity boundary conditions.
Any of the vx,vy,vz values can be specified as NULL which means do not alter the velocity component in that
dimension.
This fix is indented to be used together with a time integration fix.
Any of the 3 quantities defining the velocity components can be specified as an equal-style or atom-style
variable, namely vx, vy, vz. If the value is a variable, it should be specified as v_name, where name is the
variable name. In this case, the variable will be evaluated each timestep, and its value used to determine the
force component.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent velocity field.
Atom-style variables can specify the same formulas as equal-style variables but can also include per-atom
values, such as atom coordinates. Thus it is easy to specify a spatially-dependent velocity field with optional
time-dependence as well.
If the region keyword is used, the particle must also be in the specified geometric region in order to have its
velocity set by this command.
Restart, fix_modify, output, run start/stop, minimize info:
LAMMPS Users Manual
1076
Currently, no part of USER-SMD supports restarting nor minimization None of the fix_modify options are
relevant to this fix.
This fix computes a global 3-vector of forces, which can be accessed by various output commands. This is the
total force on the group of atoms. The vector values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
Restrictions:
This fix is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands: none
Default: none
LAMMPS Users Manual
1077

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix smd/wall_surface command
Syntax:
fix ID group-ID smd/wall_surface arg type mol-ID
ID, group-ID are documented in fix command• smd/wall_surface = style name of this fix command• arg = file
file = file name of a triangular mesh in stl format
•
type = particle type to be given to the new particles created by this fix• mol-ID = molecule-ID to be given to the new particles created by this fix (must be >= 65535)•
Examples:
fix stl_surf all smd/wall_surface tool.stl 2 65535
Description:
This fix creates reads a triangulated surface from a file in .STL format. For each triangle, a new particle is
created which stores the barycenter of the triangle and the vertex positions. The radius of the new particle is
that of the minimum circle which encompasses the triangle vertices.
The triangulated surface can be used as a complex rigid wall via the smd/tri_surface pair style. It is possible to
move the triangulated surface via the smd/move_tri_surf fix style.
Immediately after a .STL file has been read, the simulation needs to be run for 0 timesteps in order to properly
register the new particles in the system. See the "funnel_flow" example in the USER-SMD examples
directory.
See this PDF guide to use Smooth Mach Dynamics in LAMMPS.
Restart, fix_modify, output, run start/stop, minimize info:
Currently, no part of USER-SMD supports restarting nor minimization. This fix has no outputs.
Restrictions:
This fix is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info. The molecule ID given to the particles created by this fix have to
be equal to or larger than 65535.
Within each .STL file, only a single triangulated object must be present, even though the STL format allows
for the possibility of multiple objects in one file.
Related commands:
smd/triangle_mesh_vertices, smd/move_tri_surf, smd/tri_surface
LAMMPS Users Manual
1078
Default: none
LAMMPS Users Manual
1079

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix spring command
Syntax:
fix ID group-ID spring keyword values
ID, group-ID are documented in fix command• spring = style name of this fix command• keyword = tether or couple
tether values = K x y z R0
K = spring constant (force/distance units)
x,y,z = point to which spring is tethered
R0 = equilibrium distance from tether point (distance units)
couple values = group-ID2 K x y z R0
group-ID2 = 2nd group to couple to fix group with a spring
K = spring constant (force/distance units)
x,y,z = direction of spring
R0 = equilibrium distance of spring (distance units)
•
Examples:
fix pull ligand spring tether 50.0 0.0 0.0 0.0 0.0
fix pull ligand spring tether 50.0 0.0 0.0 0.0 5.0
fix pull ligand spring tether 50.0 NULL NULL 2.0 3.0
fix 5 bilayer1 spring couple bilayer2 100.0 NULL NULL 10.0 0.0
fix longitudinal pore spring couple ion 100.0 NULL NULL -20.0 0.0
fix radial pore spring couple ion 100.0 0.0 0.0 NULL 5.0
Description:
Apply a spring force to a group of atoms or between two groups of atoms. This is useful for applying an
umbrella force to a small molecule or lightly tethering a large group of atoms (e.g. all the solvent or a large
molecule) to the center of the simulation box so that it doesn't wander away over the course of a long
simulation. It can also be used to hold the centers of mass of two groups of atoms at a given distance or
orientation with respect to each other.
The tether style attaches a spring between a fixed point x,y,z and the center of mass of the fix group of atoms.
The equilibrium position of the spring is R0. At each timestep the distance R from the center of mass of the
group of atoms to the tethering point is computed, taking account of wrap-around in a periodic simulation
box. A restoring force of magnitude K (R - R0) Mi / M is applied to each atom in the group where K is the
spring constant, Mi is the mass of the atom, and M is the total mass of all atoms in the group. Note that K thus
represents the spring constant for the total force on the group of atoms, not for a spring applied to each atom.
The couple style links two groups of atoms together. The first group is the fix group; the second is specified
by group-ID2. The groups are coupled together by a spring that is at equilibrium when the two groups are
displaced by a vector x,y,z with respect to each other and at a distance R0 from that displacement. Note that
x,y,z is the equilibrium displacement of group-ID2 relative to the fix group. Thus (1,1,0) is a different spring
than (-1,-1,0). When the relative positions and distance between the two groups are not in equilibrium, the
same spring force described above is applied to atoms in each of the two groups.
LAMMPS Users Manual
1080
For both the tether and couple styles, any of the x,y,z values can be specified as NULL which means do not
include that dimension in the distance calculation or force application.
The first example above pulls the ligand towards the point (0,0,0). The second example holds the ligand near
the surface of a sphere of radius 5 around the point (0,0,0). The third example holds the ligand a distance 3
away from the z=2 plane (on either side).
The fourth example holds 2 bilayers a distance 10 apart in z. For the last two examples, imagine a pore (a slab
of atoms with a cylindrical hole cut out) oriented with the pore axis along z, and an ion moving within the
pore. The fifth example holds the ion a distance of -20 below the z = 0 center plane of the pore (umbrella
sampling). The last example holds the ion a distance 5 away from the pore axis (assuming the center-of-mass
of the pore in x,y is the pore axis).
NOTE: The center of mass of a group of atoms is calculated in "unwrapped" coordinates using atom image
flags, which means that the group can straddle a periodic boundary. See the dump doc page for a discussion of
unwrapped coordinates. It also means that a spring connecting two groups or a group and the tether point can
cross a periodic boundary and its length be calculated correctly.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the energy stored in the spring to the system's
potential energy as part of thermodynamic output.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the spring
energy = 0.5 * K * r^2.
This fix also computes global 4-vector which can be accessed by various output commands. The first 3
quantities in the vector are xyz components of the total force added to the group of atoms by the spring. In the
case of the couple style, it is the force on the fix group (group-ID) or the negative of the force on the 2nd
group (group-ID2). The 4th quantity in the vector is the magnitude of the force added by the spring, as a
positive value if (r-R0) > 0 and a negative value if (r-R0) < 0. This sign convention can be useful when using
the spring force to compute a potential of mean force (PMF).
The scalar and vector values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
NOTE: If you want the spring energy to be included in the total potential energy of the system (the quantity
being minimized), you MUST enable the fix_modify energy option for this fix.
Restrictions: none
Related commands:
LAMMPS Users Manual
1081

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix spring/chunk command
Syntax:
fix ID group-ID spring/chunk K chunkID comID
ID, group-ID are documented in fix command• spring/chunk = style name of this fix command• K = spring constant for each chunk (force/distance units)• chunkID = ID of compute chunk/atom command• comID = ID of compute com/chunk command•
Examples:
fix restrain all spring/chunk 100 chunkID comID
Description:
Apply a spring force to the center-of-mass (COM) of chunks of atoms as defined by the compute chunk/atom
command. Chunks can be molecules or spatial bins or other groupings of atoms. This is a way of tethering
each chunk to its initial COM coordinates.
The chunkID is the ID of a compute chunk/atom command defined in the input script. It is used to define the
chunks. The comID is the ID of a compute com/chunk command defined in the input script. It is used to
compute the COMs of each chunk.
At the beginning of the first run or minimize command after this fix is defined, the initial COM of each chunk
is calculated and stored as R0m, where M is the chunk number. Thereafter, at every timestep (or minimization
iteration), the current COM of each chunk is calculated as Rm. A restoring force of magnitude K (Rm - R0m)
Mi / Mm is applied to each atom in each chunk where K is the specified spring constant, Mi is the mass of the
atom, and Mm is the total mass of all atoms in the chunk. Note that K thus represents the spring constant for
the total force on each chunk of atoms, not for a spring applied to each atom.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the energy stored in all the springs to the
system's potential energy as part of thermodynamic output.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
energy of all the springs, i.e. 0.5 * K * r^2 per-spring.
The scalar value calculated by this fix is "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
LAMMPS Users Manual
1083
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
NOTE: If you want the spring energies to be included in the total potential energy of the system (the quantity
being minimized), you MUST enable the fix_modify energy option for this fix.
Restrictions: none
Related commands:
fix spring, fix spring/self, fix spring/rg
Default: none
LAMMPS Users Manual
1084

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix spring/rg command
Syntax:
fix ID group-ID spring/rg K RG0
ID, group-ID are documented in fix command• spring/rg = style name of this fix command• K = harmonic force constant (force/distance units)• RG0 = target radius of gyration to constrain to (distance units)•
if RG0 = NULL, use the current RG as the target value
Examples:
fix 1 protein spring/rg 5.0 10.0
fix 2 micelle spring/rg 5.0 NULL
Description:
Apply a harmonic restraining force to atoms in the group to affect their central moment about the center of
mass (radius of gyration). This fix is useful to encourage a protein or polymer to fold/unfold and also when
sampling along the radius of gyration as a reaction coordinate (i.e. for protein folding).
The radius of gyration is defined as RG in the first formula. The energy of the constraint and associated force
on each atom is given by the second and third formulas, when the group is at a different RG than the target
value RG0.
The (xi - center-of-mass) term is computed taking into account periodic boundary conditions, m_i is the mass
of the atom, and M is the mass of the entire group. Note that K is thus a force constant for the aggregate force
on the group of atoms, not a per-atom force.
LAMMPS Users Manual
1085
If RG0 is specified as NULL, then the RG of the group is computed at the time the fix is specified, and that
value is used as the target.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
Restrictions: none
Related commands:
fix spring, fix spring/self fix drag, fix smd
Default: none
LAMMPS Users Manual
1086

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix spring/self command
Syntax:
fix ID group-ID spring/self K dir
ID, group-ID are documented in fix command• spring/self = style name of this fix command• K = spring constant (force/distance units)• dir = xyz, xy, xz, yz, x, y, or z (optional, default: xyz)•
Examples:
fix tether boundary-atoms spring/self 10.0
fix zrest move spring/self 10.0 z
Description:
Apply a spring force independently to each atom in the group to tether it to its initial position. The initial
position for each atom is its location at the time the fix command was issued. At each timestep, the magnitude
of the force on each atom is -Kr, where r is the displacement of the atom from its current position to its initial
position. The distance r correctly takes into account any crossings of periodic boundary by the atom since it
was in its initial position.
With the (optional) dir flag, one can select in which direction the spring force is applied. By default, the
restraint is applied in all directions, but it can be limited to the xy-, xz-, yz-plane and the x-, y-, or z-direction,
thus restraining the atoms to a line or a plane, respectively.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the original coordinates of tethered atoms to binary restart files, so that the spring effect will be
the same in a restarted simulation. See the read_restart command for info on how to re-specify a fix in an
input script that reads a restart file, so that the operation of the fix continues in an uninterrupted fashion.
The fix_modify energy option is supported by this fix to add the energy stored in the per-atom springs to the
system's potential energy as part of thermodynamic output.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
This fix computes a global scalar which can be accessed by various output commands. The scalar is an energy
which is the sum of the spring energy for each atom, where the per-atom energy is 0.5 * K * r^2. The scalar
value calculated by this fix is "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
LAMMPS Users Manual
1087
NOTE: If you want the per-atom spring energy to be included in the total potential energy of the system (the
quantity being minimized), you MUST enable the fix_modify energy option for this fix.
Restrictions: none
Related commands:
fix drag, fix spring, fix smd, fix spring/rg
Default: none
LAMMPS Users Manual
1088

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix srd command
Syntax:
fix ID group-ID srd N groupbig-ID Tsrd hgrid seed keyword value ...
ID, group-ID are documented in fix command• srd = style name of this fix command• N = reset SRD particle velocities every this many timesteps• groupbig-ID = ID of group of large particles that SRDs interact with• Tsrd = temperature of SRD particles (temperature units)• hgrid = grid spacing for SRD grouping (distance units)• seed = random # seed (positive integer)•
zero or more keyword/value pairs may be appended• keyword = lamda or collision or overlap or inside or exact or radius or bounce or search or cubic or
shift or tstat or rescale
lamda value = mean free path of SRD particles (distance units)
collision value = noslip or slip = collision model
overlap value = yes or no = whether big particles may overlap
inside value = error or warn or ignore = how SRD particles which end up inside a big particle are treated
exact value = yes or no
radius value = rfactor = scale collision radius by this factor
bounce value = Nbounce = max # of collisions an SRD particle can undergo in one timestep
search value = sgrid = grid spacing for collision partner searching (distance units)
cubic values = style tolerance
style = error or warn
tolerance = fractional difference allowed (0 <= tol <= 1)
shift values = flag shiftseed
flag = yes or no or possible = SRD bin shifting for better statistics
yes = perform bin shifting each time SRD velocities are rescaled
no = no shifting
possible = shift depending on mean free path and bin size
shiftseed = random # seed (positive integer)
tstat value = yes or no = thermostat SRD particles or not
rescale value = yes or no or rotate or collide = rescaling of SRD velocities
yes = rescale during velocity rotation and collisions
no = no rescaling
rotate = rescale during velocity rotation, but not collisions
collide = rescale during collisions, but not velocity rotation
•
Examples:
fix 1 srd srd 10 big 1.0 0.25 482984
fix 1 srd srd 10 big 0.5 0.25 482984 collision slip search 0.5
Description:
Treat a group of particles as stochastic rotation dynamics (SRD) particles that serve as a background solvent
when interacting with big (colloidal) particles in groupbig-ID. The SRD formalism is described in (Hecht).
The key idea behind using SRD particles as a cheap coarse-grained solvent is that SRD particles do not
interact with each other, but only with the solute particles, which in LAMMPS can be spheroids, ellipsoids, or
LAMMPS Users Manual
1089
line segments, or triangles, or rigid bodies containing multiple spheriods or ellipsoids or line segments or
triangles. The collision and rotation properties of the model imbue the SRD particles with fluid-like
properties, including an effective viscosity. Thus simulations with large solute particles can be run more
quickly, to measure solute properties like diffusivity and viscosity in a background fluid. The usual LAMMPS
fixes for such simulations, such as fix deform, fix viscosity, and fix nvt/sllod, can be used in conjunction with
the SRD model.
For more details on how the SRD model is implemented in LAMMPS, this paper describes the
implementation and usage of pure SRD fluids. This paper, which is nearly complete, describes the
implementation and usage of mixture systems (solute particles in an SRD fluid). See the examples/srd
directory for sample input scripts using SRD particles in both settings.
This fix does 2 things:
(1) It advects the SRD particles, performing collisions between SRD and big particles or walls every timestep,
imparting force and torque to the big particles. Collisions also change the position and velocity of SRD
particles.
(2) It resets the velocity distribution of SRD particles via random rotations every N timesteps.
SRD particles have a mass, temperature, characteristic timestep dt_SRD, and mean free path between
collisions (lamda). The fundamental equation relating these 4 quantities is
lamda = dt_SRD * sqrt(Kboltz * Tsrd / mass)
The mass of SRD particles is set by the mass command elsewhere in the input script. The SRD timestep
dt_SRD is N times the step dt defined by the timestep command. Big particles move in the normal way via a
time integration fix with a short timestep dt. SRD particles advect with a large timestep dt_SRD >= dt.
If the lamda keyword is not specified, the SRD temperature Tsrd is used in the above formula to compute
lamda. If the lamda keyword is specified, then the Tsrd setting is ignored and the above equation is used to
compute the SRD temperature.
The characteristic length scale for the SRD fluid is set by hgrid which is used to bin SRD particles for
purposes of resetting their velocities. Normally hgrid is set to be 1/4 of the big particle diameter or smaller, to
adequately resolve fluid properties around the big particles.
Lamda cannot be smaller than 0.6 * hgrid, else an error is generated (unless the shift keyword is used, see
below). The velocities of SRD particles are bounded by Vmax, which is set so that an SRD particle will not
advect further than Dmax = 4*lamda in dt_SRD. This means that roughly speaking, Dmax should not be
larger than a big particle diameter, else SRDs may pass thru big particles without colliding. A warning is
generated if this is the case.
Collisions between SRD particles and big particles or walls are modeled as a lightweight SRD point particle
hitting a heavy big particle of given diameter or a wall at a point on its surface and bouncing off with a new
velocity. The collision changes the momentum of the SRD particle. It imparts a force and torque to the big
particle. It imparts a force to a wall. Static or moving SRD walls are setup via the fix wall/srd command. For
the remainder of this doc page, a collision of an SRD particle with a wall can be viewed as a collision with a
big particle of infinite radius and mass.
LAMMPS Users Manual
1090
The collision keyword sets the style of collisions. The slip style means that the tangential component of the
SRD particle momentum is preserved. Thus a force is imparted to a big particle, but no torque. The normal
component of the new SRD velocity is sampled from a Gaussian distribution at temperature Tsrd.
For the noslip style, both the normal and tangential components of the new SRD velocity are sampled from a
Gaussian distribution at temperature Tsrd. Additionally, a new tangential direction for the SRD velocity is
chosen randomly. This collision style imparts torque to a big particle. Thus a time integrator fix that rotates
the big particles appropriately should be used.
The overlap keyword should be set to yes if two (or more) big particles can ever overlap. This depends on the
pair potential interaction used for big-big interactions, or could be the case if multiple big particles are held
together as rigid bodies via the fix rigid command. If the overlap keyword is no and big particles do in fact
overlap, then SRD/big collisions can generate an error if an SRD ends up inside two (or more) big particles at
once. How this error is treated is determined by the inside keyword. Running with overlap set to no allows for
faster collision checking, so it should only be set to yes if needed.
The inside keyword determines how a collision is treated if the computation determines that the timestep
started with the SRD particle already inside a big particle. If the setting is error then this generates an error
message and LAMMPS stops. If the setting is warn then this generates a warning message and the code
continues. If the setting is ignore then no message is generated. One of the output quantities logged by the fix
(see below) tallies the number of such events, so it can be monitored. Note that once an SRD particle is inside
a big particle, it may remain there for several steps until it drifts outside the big particle.
The exact keyword determines how accurately collisions are computed. A setting of yes computes the time
and position of each collision as SRD and big particles move together. A setting of no estimates the position
of each collision based on the end-of-timestep positions of the SRD and big particle. If overlap is set to yes,
the setting of the exact keyword is ignored since time-accurate collisions are needed.
The radius keyword scales the effective size of big particles. If big particles will overlap as they undergo
dynamics, then this keyword can be used to scale down their effective collision radius by an amount rfactor,
so that SRD particle will only collide with one big particle at a time. For example, in a Lennard-Jones system
at a temperature of 1.0 (in reduced LJ units), the minimum separation between two big particles is as small as
about 0.88 sigma. Thus an rfactor value of 0.85 should prevent dual collisions.
The bounce keyword can be used to limit the maximum number of collisions an SRD particle undergoes in a
single timestep as it bounces between nearby big particles. Note that if the limit is reached, the SRD can be
left inside a big particle. A setting of 0 is the same as no limit.
There are 2 kinds of bins created and maintained when running an SRD simulation. The first are "SRD bins"
which are used to bin SRD particles and reset their velocities, as discussed above. The second are "search
bins" which are used to identify SRD/big particle collisions.
The search keyword can be used to choose a search bin size for identifying SRD/big particle collisions. The
default is to use the hgrid parameter for SRD bins as the search bin size. Choosing a smaller or large value
may be more efficient, depending on the problem. But, in a statistical sense, it should not change the
simulation results.
The cubic keyword can be used to generate an error or warning when the bin size chosen by LAMMPS
creates SRD bins that are non-cubic or different than the requested value of hgrid by a specified tolerance.
Note that using non-cubic SRD bins can lead to undetermined behavior when rotating the velocities of SRD
LAMMPS Users Manual
1091
particles, hence LAMMPS tries to protect you from this problem.
LAMMPS attempts to set the SRD bin size to exactly hgrid. However, there must be an integer number of
bins in each dimension of the simulation box. Thus the actual bin size will depend on the size and shape of the
overall simulation box. The actual bin size is printed as part of the SRD output when a simulation begins.
If the actual bin size in non-cubic by an amount exceeding the tolerance, an error or warning is printed,
depending on the style of the cubic keyword. Likewise, if the actual bin size differs from the requested hgrid
value by an amount exceeding the tolerance, then an error or warning is printed. The tolerance is a fractional
difference. E.g. a tolerance setting of 0.01 on the shape means that if the ratio of any 2 bin dimensions exceeds
(1 +/- tolerance) then an error or warning is generated. Similarly, if the ratio of any bin dimension with hgrid
exceeds (1 +/- tolerance), then an error or warning is generated.
NOTE: The fix srd command can be used with simulations the size and/or shape of the simulation box
changes. This can be due to non-periodic boundary conditions or the use of fixes such as the fix deform or fix
wall/srd commands to impose a shear on an SRD fluid or an interaction with an external wall. If the box size
changes then the size of SRD bins must be recalculated every reneighboring. This is not necessary if only the
box shape changes. This re-binning is always done so as to fit an integer number of bins in the current box
dimension, whether it be a fixed, shrink-wrapped, or periodic boundary, as set by the boundary command. If
the box size or shape changes, then the size of the search bins must be recalculated every reneighboring. Note
that changing the SRD bin size may alter the properties of the SRD fluid, such as its viscosity.
The shift keyword determines whether the coordinates of SRD particles are randomly shifted when binned for
purposes of rotating their velocities. When no shifting is performed, SRD particles are binned and the velocity
distribution of the set of SRD particles in each bin is adjusted via a rotation operator. This is a statistically
valid operation if SRD particles move sufficiently far between successive rotations. This is determined by
their mean-free path lamda. If lamda is less than 0.6 of the SRD bin size, then shifting is required. A shift
means that all of the SRD particles are shifted by a vector whose coordinates are chosen randomly in the
range [-1/2 bin size, 1/2 bin size]. Note that all particles are shifted by the same vector. The specified random
number shiftseed is used to generate these vectors. This operation sufficiently randomizes which SRD
particles are in the same bin, even if lamda is small.
If the shift flag is set to no, then no shifting is performed, but bin data will be communicated if bins overlap
processor boundaries. An error will be generated if lamda < 0.6 of the SRD bin size. If the shift flag is set to
possible, then shifting is performed only if lamda < 0.6 of the SRD bin size. A warning is generated to let you
know this is occurring. If the shift flag is set to yes then shifting is performed regardless of the magnitude of
lamda. Note that the shiftseed is not used if the shift flag is set to no, but must still be specified.
Note that shifting of SRD coordinates requires extra communication, hence it should not normally be enabled
unless required.
The tstat keyword will thermostat the SRD particles to the specified Tsrd. This is done every N timesteps,
during the velocity rotation operation, by rescaling the thermal velocity of particles in each SRD bin to the
desired temperature. If there is a streaming velocity associated with the system, e.g. due to use of the fix
deform command to perform a simulation undergoing shear, then that is also accounted for. The mean
velocity of each bin of SRD particles is set to the position-dependent streaming velocity, based on the
coordinates of the center of the SRD bin. Note that collisions of SRD particles with big particles or walls has a
thermostatting effect on the colliding particles, so it may not be necessary to thermostat the SRD particles on a
bin by bin basis in that case. Also note that for streaming simulations, if no thermostatting is performed (the
default), then it may take a long time for the SRD fluid to come to equilibrium with a velocity profile that
matches the simulation box deformation.
LAMMPS Users Manual
1092

The rescale keyword enables rescaling of an SRD particle's velocity if it would travel more than 4 mean-free
paths in an SRD timestep. If an SRD particle exceeds this velocity it is possible it will be lost when migrating
to other processors or that collisions with big particles will be missed, either of which will generate errors.
Thus the safest mode is to run with rescaling enabled. However rescaling removes kinetic energy from the
system (the particle's velocity is reduced). The latter will not typically be a problem if thermostatting is
enabled via the tstat keyword or if SRD collisions with big particles or walls effectively thermostat the
system. If you wish to turn off rescaling (on is the default), e.g. for a pure SRD system with no thermostatting
so that the temperature does not decline over time, the rescale keyword can be used. The no value turns
rescaling off during collisions and the per-bin velocity rotation operation. The collide and rotate values turn it
on for one of the operations and off for the other.
NOTE: This fix is normally used for simulations with a huge number of SRD particles relative to the number
of big particles, e.g. 100 to 1. In this scenario, computations that involve only big particles (neighbor list
creation, communication, time integration) can slow down dramatically due to the large number of
background SRD particles.
Three other input script commands will largely overcome this effect, speeding up an SRD simulation by a
significant amount. These are the atom_modify first, neigh_modify include, and comm_modify group
commands. Each takes a group-ID as an argument, which in this case should be the group-ID of the big solute
particles.
Additionally, when a pair_style for big/big particle interactions is specified, the pair_coeff command should
be used to turn off big/SRD interactions, e.g. by setting their epsilon or cutoff length to 0.0.
The "delete_atoms overlap" command may be useful in setting up an SRD simulation to insure there are no
initial overlaps between big and SRD particles.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix tabulates several SRD statistics which are stored in a vector of length 12, which can be accessed by
various output commands. The vector values calculated by this fix are "intensive", meaning they do not scale
with the size of the simulation. Technically, the first 8 do scale with the size of the simulation, but treating
them as intensive means they are not scaled when printed as part of thermodynamic output.
These are the 12 quantities. All are values for the current timestep, except for quantity 5 and the last three,
each of which are cumulative quantities since the beginning of the run.
(1) # of SRD/big collision checks performed• (2) # of SRDs which had a collision• (3) # of SRD/big collisions (including multiple bounces)• (4) # of SRD particles inside a big particle• (5) # of SRD particles whose velocity was rescaled to be < Vmax• (6) # of bins for collision searching• (7) # of bins for SRD velocity rotation• (8) # of bins in which SRD temperature was computed• (9) SRD temperature• (10) # of SRD particles which have undergone max # of bounces•
LAMMPS Users Manual
1093

(11) max # of bounces any SRD particle has had in a single step• (12) # of reneighborings due to SRD particles moving too far•
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This command can only be used if LAMMPS was built with the SRD package. See the Making LAMMPS
section for more info on packages.
Related commands:
fix wall/srd
Default:
The option defaults are lamda inferred from Tsrd, collision = noslip, overlap = no, inside = error, exact = yes,
radius = 1.0, bounce = 0, search = hgrid, cubic = error 0.01, shift = no, tstat = no, and rescale = yes.
(Hecht) Hecht, Harting, Ihle, Herrmann, Phys Rev E, 72, 011408 (2005).
(Petersen) Petersen, Lechman, Plimpton, Grest, in' t Veld, Schunk, J Chem Phys, 132, 174106 (2010).
(Lechman) Lechman, et al, in preparation (2010).
LAMMPS Users Manual
1094

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix store/force command
Syntax:
fix ID group-ID store/force
ID, group-ID are documented in fix command• store/force = style name of this fix command•
Examples:
fix 1 all store/force
Description:
Store the forces on atoms in the group at the point during each timestep when the fix is invoked, as described
below. This is useful for storing forces before constraints or other boundary conditions are computed which
modify the forces, so that unmodified forces can be written to a dump file or accessed by other output
commands that use per-atom quantities.
This fix is invoked at the point in the velocity-Verlet timestepping immediately after pair, bond, angle,
dihedral, improper, and long-range forces have been calculated. It is the point in the timestep when various
fixes that compute constraint forces are calculated and potentially modify the force on each atom. Examples
of such fixes are fix shake, fix wall, and fix indent.
NOTE: The order in which various fixes are applied which operate at the same point during the timestep, is
the same as the order they are specified in the input script. Thus normally, if you want to store per-atom forces
due to force field interactions, before constraints are applied, you should list this fix first within that set of
fixes, i.e. before other fixes that apply constraints. However, if you wish to include certain constraints (e.g. fix
shake) in the stored force, then it could be specified after some fixes and before others.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix produces a per-atom array which can be accessed by various output commands. The number of
columns for each atom is 3, and the columns store the x,y,z forces on each atom. The per-atom values be
accessed on any timestep.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
fix store_state
LAMMPS Users Manual
1095
Default: none
LAMMPS Users Manual
1096

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix store/state command
Syntax:
fix ID group-ID store/state N input1 input2 ... keyword value ...
ID, group-ID are documented in fix command• store/state = style name of this fix command• N = store atom attributes every N steps, N = 0 for initial store only• input = one or more atom attributes
possible attributes = id, mol, type, mass,
x, y, z, xs, ys, zs, xu, yu, zu, xsu, ysu, zsu, ix, iy, iz,
vx, vy, vz, fx, fy, fz,
q, mux, muy, muz, mu,
radius, diameter, omegax, omegay, omegaz,
angmomx, angmomy, angmomz, tqx, tqy, tqz,
c_ID, c_ID[N], f_ID, f_ID[N], v_name,
d_name, i_name
id = atom ID
mol = molecule ID
type = atom type
mass = atom mass
x,y,z = unscaled atom coordinates
xs,ys,zs = scaled atom coordinates
xu,yu,zu = unwrapped atom coordinates
xsu,ysu,zsu = scaled unwrapped atom coordinates
ix,iy,iz = box image that the atom is in
vx,vy,vz = atom velocities
fx,fy,fz = forces on atoms
q = atom charge
mux,muy,muz = orientation of dipolar atom
mu = magnitued of dipole moment of atom
radius,diameter = radius.diameter of spherical particle
omegax,omegay,omegaz = angular velocity of spherical particle
angmomx,angmomy,angmomz = angular momentum of aspherical particle
tqx,tqy,tqz = torque on finite-size particles
c_ID = per-atom vector calculated by a compute with ID
c_ID[I] = Ith column of per-atom array calculated by a compute with ID
f_ID = per-atom vector calculated by a fix with ID
f_ID[I] = Ith column of per-atom array calculated by a fix with ID
v_name = per-atom vector calculated by an atom-style variable with name
d_name = per-atom floating point vector name, managed by fix property/atom
i_name = per-atom integer vector name, managed by fix property/atom
•
zero or more keyword/value pairs may be appended• keyword = com
com value = yes or no
•
Examples:
fix 1 all store/state 0 x y z
fix 1 all store/state 0 xu yu zu com yes
fix 2 all store/state 1000 vx vy vz
LAMMPS Users Manual
1097
Description:
Define a fix that stores attributes for each atom in the group at the time the fix is defined. If N is 0, then the
values are never updated, so this is a way of archiving an atom attribute at a given time for future use in a
calculation or output. See the discussion of output commands that take fixes as inputs.
If N is not zero, then the attributes will be updated every N steps.
NOTE: Actually, only atom attributes specified by keywords like xu or vy or radius are initially stored
immediately at the point in your input script when the fix is defined. Attributes specified by a compute, fix, or
variable are not initially stored until the first run following the fix definition begins. This is because
calculating those attributes may require quantities that are not defined in between runs.
The list of possible attributes is the same as that used by the dump custom command, which describes their
meaning.
If the com keyword is set to yes then the xu, yu, and zu inputs store the position of each atom relative to the
center-of-mass of the group of atoms, instead of storing the absolute position.
The requested values are stored in a per-atom vector or array as discussed below. Zeroes are stored for atoms
not in the specified group.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the per-atom values it stores to binary restart files, so that the values can be restored when a
simulation is restarted. See the read_restart command for info on how to re-specify a fix in an input script that
reads a restart file, so that the operation of the fix continues in an uninterrupted fashion.
None of the fix_modify options are relevant to this fix.
If a single input is specified, this fix produces a per-atom vector. If multiple inputs are specified, a per-atom
array is produced where the number of columns for each atom is the number of inputs. These can be accessed
by various output commands. The per-atom values be accessed on any timestep.
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions: none
Related commands:
dump custom, compute property/atom, fix property/atom, variable
Default:
The option default is com = no.
LAMMPS Users Manual
1098

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix temp/berendsen command
Syntax:
fix ID group-ID temp/berendsen Tstart Tstop Tdamp
ID, group-ID are documented in fix command• temp/berendsen = style name of this fix command• Tstart,Tstop = desired temperature at start/end of run
Tstart can be a variable (see below)
•
Tdamp = temperature damping parameter (time units)•
Examples:
fix 1 all temp/berendsen 300.0 300.0 100.0
Description:
Reset the temperature of a group of atoms by using a Berendsen thermostat (Berendsen), which rescales their
velocities every timestep.
The thermostat is applied to only the translational degrees of freedom for the particles, which is an important
consideration for finite-size particles which have rotational degrees of freedom are being thermostatted with
this fix. The translational degrees of freedom can also have a bias velocity removed from them before
thermostatting takes place; see the description below.
The desired temperature at each timestep is a ramped value during the run from Tstart to Tstop. The Tdamp
parameter is specified in time units and determines how rapidly the temperature is relaxed. For example, a
value of 100.0 means to relax the temperature in a timespan of (roughly) 100 time units (tau or fmsec or psec -
see the units command).
Tstart can be specified as an equal-style variable. In this case, the Tstop setting is ignored. If the value is a
variable, it should be specified as v_name, where name is the variable name. In this case, the variable will be
evaluated each timestep, and its value used to determine the target temperature.
NOTE: This thermostat will generate an error if the current temperature is zero at the end of a timestep. It
cannot rescale a zero temperature.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent temperature.
NOTE: Unlike the fix nvt command which performs Nose/Hoover thermostatting AND time integration, this
fix does NOT perform time integration. It only modifies velocities to effect thermostatting. Thus you must use
a separate time integration fix, like fix nve to actually update the positions of atoms using the modified
velocities. Likewise, this fix should not normally be used on atoms that also have their temperature controlled
by another fix - e.g. by fix nvt or fix langevin commands.
LAMMPS Users Manual
1099

See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
This fix computes a temperature each timestep. To do this, the fix creates its own compute of style "temp", as
if this command had been issued:
compute fix-ID_temp group-ID temp
See the compute temp command for details. Note that the ID of the new compute is the fix-ID + underscore +
"temp", and the group for the new compute is the same as the fix group.
Note that this is NOT the compute used by thermodynamic output (see the thermo_style command) with ID =
thermo_temp. This means you can change the attributes of this fix's temperature (e.g. its degrees-of-freedom)
via the compute_modify command or print this temperature during thermodynamic output via the
thermo_style custom command using the appropriate compute-ID. It also means that changing attributes of
thermo_temp will have no effect on this fix.
Like other fixes that perform thermostatting, this fix can be used with compute commands that calculate a
temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass velocity from
a group of atoms or only calculating temperature on the x-component of velocity or only calculating
temperature for atoms in a geometric region. This is not done by default, but only if the fix_modify command
is used to assign a temperature compute to this fix that includes such a bias term. See the doc pages for
individual compute commands to determine which ones include a bias. In this case, the thermostat works in
the following manner: the current temperature is calculated taking the bias into account, bias is removed from
each atom, thermostatting is performed on the remaining thermal degrees of freedom, and the bias is added
back in.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify temp option is supported by this fix. You can use it to assign a temperature compute you have
defined to this fix which will be used in its thermostatting procedure, as described above. For consistency, the
group used by this fix and by the compute should be the same.
The fix_modify energy option is supported by this fix to add the energy change implied by a velocity
rescaling to the system's potential energy as part of thermodynamic output.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
cumulative energy change due to this fix. The scalar value calculated by this fix is "extensive".
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix can be used with dynamic groups as defined by the group command. Likewise it can be used with
groups to which atoms are added or deleted over time, e.g. a deposition simulation. However, the conservation
properties of the thermostat and barostat are defined for systems with a static set of atoms. You may observe
LAMMPS Users Manual
1100

odd behavior if the atoms in a group vary dramatically over time or the atom count becomes very small.
Related commands:
fix nve, fix nvt, fix temp/rescale, fix langevin, fix_modify, compute temp, fix press/berendsen
Default: none
(Berendsen) Berendsen, Postma, van Gunsteren, DiNola, Haak, J Chem Phys, 81, 3684 (1984).
LAMMPS Users Manual
1101

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix temp/csvr command
fix temp/csld command
Syntax:
fix ID group-ID temp/csvr Tstart Tstop Tdamp seed
fix ID group-ID temp/csld Tstart Tstop Tdamp seed
ID, group-ID are documented in fix command• temp/csvr or temp/csld = style name of this fix command• Tstart,Tstop = desired temperature at start/end of run
Tstart can be a variable (see below)
•
Tdamp = temperature damping parameter (time units)• seed = random number seed to use for white noise (positive integer)•
Examples:
fix 1 all temp/csvr 300.0 300.0 100.0 54324
fix 1 all temp/csld 100.0 300.0 10.0 123321
Description:
Adjust the temperature with a canonical sampling thermostat that uses global velocity rescaling with
Hamiltonian dynamics (temp/csvr) (Bussi1), or Langevin dynamics (temp/csld) (Bussi2). In the case of
temp/csvr the thermostat is similar to the empirical Berendsen thermostat in temp/berendsen, but chooses the
actual scaling factor from a suitably chosen (gaussian) distribution rather than having it determined from the
time constant directly. In the case of temp/csld the velocities are updated to a linear combination of the current
velocities with a gaussian distribution of velocities at the desired temperature. Both thermostats are applied
every timestep.
The thermostat is applied to only the translational degrees of freedom for the particles, which is an important
consideration for finite-size particles which have rotational degrees of freedom are being thermostatted with
these fixes. The translational degrees of freedom can also have a bias velocity removed from them before
thermostatting takes place; see the description below.
The desired temperature at each timestep is a ramped value during the run from Tstart to Tstop. The Tdamp
parameter is specified in time units and determines how rapidly the temperature is relaxed. For example, a
value of 100.0 means to relax the temperature in a timespan of (roughly) 100 time units (tau or fmsec or psec -
see the units command).
Tstart can be specified as an equal-style variable. In this case, the Tstop setting is ignored. If the value is a
variable, it should be specified as v_name, where name is the variable name. In this case, the variable will be
evaluated each timestep, and its value used to determine the target temperature.
LAMMPS Users Manual
1102

Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent temperature.
NOTE: Unlike the fix nvt command which performs Nose/Hoover thermostatting AND time integration, these
fixes do NOT perform time integration. They only modify velocities to effect thermostatting. Thus you must
use a separate time integration fix, like fix nve to actually update the positions of atoms using the modified
velocities. Likewise, these fixes should not normally be used on atoms that also have their temperature
controlled by another fix - e.g. by fix nvt or fix langevin commands.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
These fixes compute a temperature each timestep. To do this, the fix creates its own compute of style "temp",
as if this command had been issued:
compute fix-ID_temp group-ID temp
See the compute temp command for details. Note that the ID of the new compute is the fix-ID + underscore +
"temp", and the group for the new compute is the same as the fix group.
Note that this is NOT the compute used by thermodynamic output (see the thermo_style command) with ID =
thermo_temp. This means you can change the attributes of this fix's temperature (e.g. its degrees-of-freedom)
via the compute_modify command or print this temperature during thermodynamic output via the
thermo_style custom command using the appropriate compute-ID. It also means that changing attributes of
thermo_temp will have no effect on this fix.
Like other fixes that perform thermostatting, these fixes can be used with compute commands that calculate a
temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass velocity from
a group of atoms or only calculating temperature on the x-component of velocity or only calculating
temperature for atoms in a geometric region. This is not done by default, but only if the fix_modify command
is used to assign a temperature compute to this fix that includes such a bias term. See the doc pages for
individual compute commands to determine which ones include a bias. In this case, the thermostat works in
the following manner: the current temperature is calculated taking the bias into account, bias is removed from
each atom, thermostatting is performed on the remaining thermal degrees of freedom, and the bias is added
back in.
Restart, fix_modify, output, run start/stop, minimize info:
No information about these fixes are written to binary restart files.
The fix_modify temp option is supported by these fixes. You can use it to assign a temperature compute you
have defined to these fixes which will be used in its thermostatting procedure, as described above. For
consistency, the group used by these fixes and by the compute should be the same.
These fixes can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
These fixes are not invoked during energy minimization.
LAMMPS Users Manual
1103

These fixes compute a global scalar which can be accessed by various output commands. The scalar is the
cumulative energy change due to the fix. The scalar value calculated by this fix is "extensive".
Restrictions:
These fixes are not compatible with fix shake.
The fix can be used with dynamic groups as defined by the group command. Likewise it can be used with
groups to which atoms are added or deleted over time, e.g. a deposition simulation. However, the conservation
properties of the thermostat and barostat are defined for systems with a static set of atoms. You may observe
odd behavior if the atoms in a group vary dramatically over time or the atom count becomes very small.
Related commands:
fix nve, fix nvt, fix temp/rescale, fix langevin, fix_modify, compute temp, fix temp/berendsen
Default: none
(Bussi1) Bussi, Donadio and Parrinello, J. Chem. Phys. 126, 014101(2007)
(Bussi2) Bussi and Parrinello, Phys. Rev. E 75, 056707 (2007)
LAMMPS Users Manual
1104

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix temp/rescale command
Syntax:
fix ID group-ID temp/rescale N Tstart Tstop window fraction
ID, group-ID are documented in fix command• temp/rescale = style name of this fix command• N = perform rescaling every N steps• Tstart,Tstop = desired temperature at start/end of run (temperature units)
Tstart can be a variable (see below)
•
window = only rescale if temperature is outside this window (temperature units)• fraction = rescale to target temperature by this fraction•
Examples:
fix 3 flow temp/rescale 100 1.0 1.1 0.02 0.5
fix 3 boundary temp/rescale 1 1.0 1.5 0.05 1.0
fix 3 boundary temp/rescale 1 1.0 1.5 0.05 1.0
Description:
Reset the temperature of a group of atoms by explicitly rescaling their velocities.
The rescaling is applied to only the translational degrees of freedom for the particles, which is an important
consideration if finite-size particles which have rotational degrees of freedom are being thermostatted with
this fix. The translational degrees of freedom can also have a bias velocity removed from them before
thermostatting takes place; see the description below.
Rescaling is performed every N timesteps. The target temperature is a ramped value between the Tstart and
Tstop temperatures at the beginning and end of the run.
NOTE: This thermostat will generate an error if the current temperature is zero at the end of a timestep it is
invoked on. It cannot rescale a zero temperature.
Tstart can be specified as an equal-style variable. In this case, the Tstop setting is ignored. If the value is a
variable, it should be specified as v_name, where name is the variable name. In this case, the variable will be
evaluated each timestep, and its value used to determine the target temperature.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent temperature.
Rescaling is only performed if the difference between the current and desired temperatures is greater than the
window value. The amount of rescaling that is applied is a fraction (from 0.0 to 1.0) of the difference between
the actual and desired temperature. E.g. if fraction = 1.0, the temperature is reset to exactly the desired value.
LAMMPS Users Manual
1105

NOTE: Unlike the fix nvt command which performs Nose/Hoover thermostatting AND time integration, this
fix does NOT perform time integration. It only modifies velocities to effect thermostatting. Thus you must use
a separate time integration fix, like fix nve to actually update the positions of atoms using the modified
velocities. Likewise, this fix should not normally be used on atoms that also have their temperature controlled
by another fix - e.g. by fix nvt or fix langevin commands.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
This fix computes a temperature each timestep. To do this, the fix creates its own compute of style "temp", as
if one of this command had been issued:
compute fix-ID_temp group-ID temp
See the compute temp for details. Note that the ID of the new compute is the fix-ID + underscore + "temp",
and the group for the new compute is the same as the fix group.
Note that this is NOT the compute used by thermodynamic output (see the thermo_style command) with ID =
thermo_temp. This means you can change the attributes of this fix's temperature (e.g. its degrees-of-freedom)
via the compute_modify command or print this temperature during thermodynamic output via the
thermo_style custom command using the appropriate compute-ID. It also means that changing attributes of
thermo_temp will have no effect on this fix.
Like other fixes that perform thermostatting, this fix can be used with compute commands that calculate a
temperature after removing a "bias" from the atom velocities. E.g. removing the center-of-mass velocity from
a group of atoms or only calculating temperature on the x-component of velocity or only calculating
temperature for atoms in a geometric region. This is not done by default, but only if the fix_modify command
is used to assign a temperature compute to this fix that includes such a bias term. See the doc pages for
individual compute commands to determine which ones include a bias. In this case, the thermostat works in
the following manner: the current temperature is calculated taking the bias into account, bias is removed from
each atom, thermostatting is performed on the remaining thermal degrees of freedom, and the bias is added
back in.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify temp option is supported by this fix. You can use it to assign a temperature compute you have
defined to this fix which will be used in its thermostatting procedure, as described above. For consistency, the
group used by this fix and by the compute should be the same.
The fix_modify energy option is supported by this fix to add the energy change implied by a velocity
rescaling to the system's potential energy as part of thermodynamic output.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
cumulative energy change due to this fix. The scalar value calculated by this fix is "extensive".
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
LAMMPS Users Manual
1106

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix temp/rescale/eff command
Syntax:
fix ID group-ID temp/rescale/eff N Tstart Tstop window fraction
ID, group-ID are documented in fix command• temp/rescale/eff = style name of this fix command• N = perform rescaling every N steps• Tstart,Tstop = desired temperature at start/end of run (temperature units)• window = only rescale if temperature is outside this window (temperature units)• fraction = rescale to target temperature by this fraction•
Examples:
fix 3 flow temp/rescale/eff 10 1.0 100.0 0.02 1.0
Description:
Reset the temperature of a group of nuclei and electrons in the electron force field model by explicitly
rescaling their velocities.
The operation of this fix is exactly like that described by the fix temp/rescale command, except that the
rescaling is also applied to the radial electron velocity for electron particles.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify temp option is supported by this fix. You can use it to assign a temperature compute you have
defined to this fix which will be used in its thermostatting procedure, as described above. For consistency, the
group used by this fix and by the compute should be the same.
The fix_modify energy option is supported by this fix to add the energy change implied by a velocity
rescaling to the system's potential energy as part of thermodynamic output.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
cumulative energy change due to this fix. The scalar value calculated by this fix is "extensive".
This fix can ramp its target temperature over multiple runs, using the start and stop keywords of the run
command. See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the USER-EFF package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
LAMMPS Users Manual
1108

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix tfmc command
Syntax:
fix ID group-ID tfmc Delta Temp seed keyword value
ID, group-ID are documented in fix command• tfmc = style name of this fix command• Delta = maximal displacement length (distance units)• Temp = imposed temperature of the system• seed = random number seed (positive integer)• zero or more keyword/arg pairs may be appended• keyword = com or rot
com args = xflag yflag zflag
xflag,yflag,zflag = 0/1 to exclude/include each dimension
rot args = none
•
Examples:
fix 1 all tfmc 0.1 1000.0 159345
fix 1 all tfmc 0.05 600.0 658943 com 1 1 0
fix 1 all tfmc 0.1 750.0 387068 com 1 1 1 rot
Description:
Perform uniform-acceptance force-bias Monte Carlo (fbMC) simulations, using the time-stamped force-bias
Monte Carlo (tfMC) algorithm described in (Mees) and (Bal).
One successful use case of force-bias Monte Carlo methods is that they can be used to extend the time scale of
atomistic simulations, in particular when long time scale relaxation effects must be considered; some
interesting examples are given in the review by (Neyts). An example of a typical use case would be the
modelling of chemical vapor deposition (CVD) processes on a surface, in which impacts by gas-phase species
can be performed using MD, but subsequent relaxation of the surface is too slow to be done using MD only.
Using tfMC can allow for a much faster relaxation of the surface, so that higher fluxes can be used, effectively
extending the time scale of the simulation. (Such an alternating simulation approach could be set up using a
loop.)
The initial version of tfMC algorithm in (Mees) contained an estimation of the effective time scale of such a
simulation, but it was later shown that the speed-up one can gain from a tfMC simulation is system- and
process-dependent, ranging from none to several orders of magnitude. In general, solid-state processes such as
(re)crystallisation or growth can be accelerated by up to two or three orders of magnitude, whereas diffusion
in the liquid phase is not accelerated at all. The observed pseudodynamics when using the tfMC method is not
the actual dynamics one would obtain using MD, but the relative importance of processes can match the actual
relative dynamics of the system quite well, provided Delta is chosen with care. Thus, the system's equilibrium
is reached faster than in MD, along a path that is generally roughly similar to a typical MD simulation (but not
necessarily so). See (Bal) for details.
Each step, all atoms in the selected group are displaced using the stochastic tfMC algorithm, which is
designed to sample the canonical (NVT) ensemble at the temperature Temp. Although tfMC is a Monte Carlo
LAMMPS Users Manual
1110

algorithm and thus strictly speaking does not perform time integration, it is similar in the sense that it uses the
forces on all atoms in order to update their positions. Therefore, it is implemented as a time integration fix,
and no other fixes of this type (such as fix nve) should be used at the same time. Because velocities do not
play a role in this kind of Monte Carlo simulations, instantaneous temperatures as calculated by temperature
computes or thermodynamic output have no meaning: the only relevant temperature is the sampling
temperature Temp. Similarly, performing tfMC simulations does not require setting a timestep and the
simulated time as calculated by LAMMPS is meaningless.
The critical parameter determining the success of a tfMC simulation is Delta, the maximal displacement
length of the lightest element in the system: the larger it is, the longer the effective time scale of the
simulation will be (there is an approximately quadratic dependence). However, Delta must also be chosen
sufficiently small in order to comply with detailed balance; in general values between 5 and 10 % of the
nearest neighbor distance are found to be a good choice. For a more extensive discussion with specific
examples, please refer to (Bal), which also describes how the code calculates element-specific maximal
displacements from Delta, based on the fourth root of their mass.
Because of the uncorrelated movements of the atoms, the center-of-mass of the fix group will not necessarily
be stationary, just like its orientation. When the com keyword is used, all atom positions will be shifted (after
every tfMC iteration) in order to fix the position of the center-of-mass along the included directions, by setting
the corresponding flag to 1. The rot keyword does the same for the rotational component of the tfMC
displacements after every iteration.
NOTE: the com and rot keywords should not be used if an external force is acting on the specified fix group,
along the included directions. This can be either a true external force (e.g. through fix wall) or forces due to
the interaction with atoms not included in the fix group. This is because in such cases, translations or rotations
of the fix group could be induced by these external forces, and removing them will lead to a violation of
detailed balance.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
None of the fix_modify options are relevant to this fix.
This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the MC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This fix is not compatible with fix shake.
Related commands:
fix gcmc, fix nvt
Default:
The option default is com = 0 0 0
LAMMPS Users Manual
1111

(Bal) K. M Bal and E. C. Neyts, J. Chem. Phys. 141, 204104 (2014).
(Mees) M. J. Mees, G. Pourtois, E. C. Neyts, B. J. Thijsse, and A. Stesmans, Phys. Rev. B 85, 134301 (2012).
(Neyts) E. C. Neyts and A. Bogaerts, Theor. Chem. Acc. 132, 1320 (2013).
LAMMPS Users Manual
1112

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix thermal/conductivity command
Syntax:
fix ID group-ID thermal/conductivity N edim Nbin keyword value ...
ID, group-ID are documented in fix command• thermal/conductivity = style name of this fix command• N = perform kinetic energy exchange every N steps• edim = x or y or z = direction of kinetic energy transfer• Nbin = # of layers in edim direction (must be even number)• zero or more keyword/value pairs may be appended• keyword = swap
swap value = Nswap = number of swaps to perform every N steps
•
Examples:
fix 1 all thermal/conductivity 100 z 20
fix 1 all thermal/conductivity 50 z 20 swap 2
Description:
Use the Muller-Plathe algorithm described in this paper to exchange kinetic energy between two particles in
different regions of the simulation box every N steps. This induces a temperature gradient in the system. As
described below this enables the thermal conductivity of a material to be calculated. This algorithm is
sometimes called a reverse non-equilibrium MD (reverse NEMD) approach to computing thermal
conductivity. This is because the usual NEMD approach is to impose a temperature gradient on the system
and measure the response as the resulting heat flux. In the Muller-Plathe method, the heat flux is imposed, and
the temperature gradient is the system's response.
See the compute heat/flux command for details on how to compute thermal conductivity in an alternate way,
via the Green-Kubo formalism.
The simulation box is divided into Nbin layers in the edim direction, where the layer 1 is at the low end of that
dimension and the layer Nbin is at the high end. Every N steps, Nswap pairs of atoms are chosen in the
following manner. Only atoms in the fix group are considered. The hottest Nswap atoms in layer 1 are
selected. Similarly, the coldest Nswap atoms in the "middle" layer (see below) are selected. The two sets of
Nswap atoms are paired up and their velocities are exchanged. This effectively swaps their kinetic energies,
assuming their masses are the same. If the masses are different, an exchange of velocities relative to center of
mass motion of the 2 atoms is performed, to conserve kinetic energy. Over time, this induces a temperature
gradient in the system which can be measured using commands such as the following, which writes the
temperature profile (assuming z = edim) to the file tmp.profile:
compute ke all ke/atom
variable temp atom c_ke/1.5
compute layers all chunk/atom bin/1d z lower 0.05 units reduced
fix 3 all ave/chunk 10 100 1000 layers v_temp file tmp.profile
LAMMPS Users Manual
1113
Note that by default, Nswap = 1, though this can be changed by the optional swap keyword. Setting this
parameter appropriately, in conjunction with the swap rate N, allows the heat flux to be adjusted across a wide
range of values, and the kinetic energy to be exchanged in large chunks or more smoothly.
The "middle" layer for velocity swapping is defined as the Nbin/2 + 1 layer. Thus if Nbin = 20, the two
swapping layers are 1 and 11. This should lead to a symmetric temperature profile since the two layers are
separated by the same distance in both directions in a periodic sense. This is why Nbin is restricted to being an
even number.
As described below, the total kinetic energy transferred by these swaps is computed by the fix and can be
output. Dividing this quantity by time and the cross-sectional area of the simulation box yields a heat flux.
The ratio of heat flux to the slope of the temperature profile is proportional to the thermal conductivity of the
fluid, in appropriate units. See the Muller-Plathe paper for details.
NOTE: If your system is periodic in the direction of the heat flux, then the flux is going in 2 directions. This
means the effective heat flux in one direction is reduced by a factor of 2. You will see this in the equations for
thermal conductivity (kappa) in the Muller-Plathe paper. LAMMPS is simply tallying kinetic energy which
does not account for whether or not your system is periodic; you must use the value appropriately to yield a
kappa for your system.
NOTE: After equilibration, if the temperature gradient you observe is not linear, then you are likely swapping
energy too frequently and are not in a regime of linear response. In this case you cannot accurately infer a
thermal conductivity and should try increasing the Nevery parameter.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
cumulative kinetic energy transferred between the bottom and middle of the simulation box (in the edim
direction) is stored as a scalar quantity by this fix. This quantity is zeroed when the fix is defined and
accumulates thereafter, once every N steps. The units of the quantity are energy; see the units command for
details. The scalar value calculated by this fix is "intensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the MISC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Swaps conserve both momentum and kinetic energy, even if the masses of the swapped atoms are not equal.
Thus you should not need to thermostat the system. If you do use a thermostat, you may want to apply it only
to the non-swapped dimensions (other than vdim).
LAMMPS does not check, but you should not use this fix to swap the kinetic energy of atoms that are in
constrained molecules, e.g. via fix shake or fix rigid. This is because application of the constraints will alter
the amount of transferred momentum. You should, however, be able to use flexible molecules. See the Zhang
paper for a discussion and results of this idea.
LAMMPS Users Manual
1114

When running a simulation with large, massive particles or molecules in a background solvent, you may want
to only exchange kinetic energy between solvent particles.
Related commands:
fix ehex, fix heat, fix ave/chunk, fix viscosity, compute heat/flux
Default:
The option defaults are swap = 1.
(Muller-Plathe) Muller-Plathe, J Chem Phys, 106, 6082 (1997).
(Zhang) Zhang, Lussetti, de Souza, Muller-Plathe, J Phys Chem B, 109, 15060-15067 (2005).
LAMMPS Users Manual
1115

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix ti/spring command
Syntax:
fix ID group-ID ti/spring k t_s t_eq keyword value ...
ID, group-ID are documented in fix command• ti/spring = style name of this fix command• k = spring constant (force/distance units)• t_eq = number of steps for the equilibration procedure• t_s = number of steps for the switching procedure• zero or more keyword/value pairs may be appended to args• keyword = function
function value = function-ID
function-ID = ID of the switching function (1 or 2)
•
Example:
fix 1 all ti/spring 50.0 2000 1000 function 2
Description:
This fix allows you to compute the free energy of crystalline solids by performing a nonequilibrium
thermodynamic integration between the solid of interest and an Einstein crystal. A detailed explanation of
how to use this command and choose its parameters for optimal performance and accuracy is given in the
paper by Freitas. The paper also presents a short summary of the theory of nonequilibrium thermodynamic
integrations.
The thermodynamic integration procedure is performed by rescaling the force on each atom. Given an atomic
configuration the force (F) on each atom is given by
where F_solid is the force that acts on an atom due to an interatomic potential (e.g. EAM potential), F_harm is
the force due to the Einstein crystal harmonic spring, and lambda is the coupling parameter of the
thermodynamic integration. An Einstein crystal is a solid where each atom is attached to its equilibrium
position by a harmonic spring with spring constant k. With this fix a spring force is applied independently to
each atom in the group defined by the fix to tether it to its initial position. The initial position of each atom is
its position at the time the fix command was issued.
The fix acts as follows: during the first t_eq steps after the fix is defined the value of lambda is zero. This is
the period to equilibrate the system in the lambda = 0 state. After this the value of lambda changes
dynamically during the simulation from 0 to 1 according to the function defined using the keyword function
(described below), this switching from lambda from 0 to 1 is done in t_s steps. Then comes the second
equilibration period of t_eq to equilibrate the system in the lambda = 1 state. After that, the switching back to
the lambda = 0 state is made using t_s timesteps and following the same switching function. After this period
LAMMPS Users Manual
1116
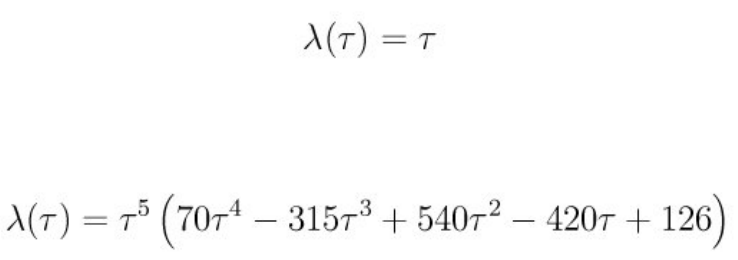
the value of lambda is kept equal to zero and the fix has no other effect on the dynamics of the system.
The processes described above is known as nonequilibrium thermodynamic integration and is has been shown
(Freitas) to present a much superior efficiency when compared to standard equilibrium methods. The reason
why the switching it is made in both directions (potential to Einstein crystal and back) is to eliminate the
dissipated heat due to the nonequilibrium process. Further details about nonequilibrium thermodynamic
integration and its implementation in LAMMPS is available in Freitas.
The function keyword allows the use of two different lambda paths. Option 1 results in a constant rate of
change of lambda with time:
where tau is the scaled time variable t/t_s. The option 2 performs the lambda switching at a rate defined by the
following switching function
This function has zero slope as lambda approaches its extreme values (0 and 1), according to de Koning this
results in smaller fluctuations on the integral to be computed on the thermodynamic integration. The use of
option 2 is recommended since it results in better accuracy and less dissipation without any increase in
computational resources cost.
NOTE: As described in Freitas, it is important to keep the center-of-mass fixed during the thermodynamic
integration. A nonzero total velocity will result in divergences during the integration due to the fact that the
atoms are 'attached' to their equilibrium positions by the Einstein crystal. Check the option zero of fix
langevin and velocity. The use of the Nose-Hoover thermostat (fix nvt) is NOT recommended due to its well
documented issues with the canonical sampling of harmonic degrees of freedom (notice that the chain option
will NOT solve this problem). The Langevin thermostat (fix langevin) correctly thermostats the system and
we advise its usage with ti/spring command.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the original coordinates of tethered atoms to binary restart files, so that the spring effect will be
the same in a restarted simulation. See the read restart command for info on how to re-specify a fix in an input
script that reads a restart file, so that the operation of the fix continues in an uninterrupted fashion.
The fix modify energy option is supported by this fix to add the energy stored in the per-atom springs to the
system's potential energy as part of thermodynamic output.
This fix computes a global scalar and a global vector quantities which can be accessed by various output
commands. The scalar is an energy which is the sum of the spring energy for each atom, where the per-atom
energy is 0.5 * k * r^2. The vector has 2 positions, the first one is the coupling parameter lambda and the
second one is the time derivative of lambda. The scalar and vector values calculated by this fix are
"extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
LAMMPS Users Manual
1117

The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
NOTE: If you want the per-atom spring energy to be included in the total potential energy of the system (the
quantity being minimized), you MUST enable the fix modify energy option for this fix.
Related commands:
fix spring, fix adapt
Restrictions: none
Default:
The keyword default is function = 1.
(Freitas) Freitas, Asta, and de Koning, Computational Materials Science, 112, 333 (2016).
(de Koning) de Koning and Antonelli, Phys Rev E, 53, 465 (1996).
LAMMPS Users Manual
1118

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix tmd command
Syntax:
fix ID group-ID tmd rho_final file1 N file2
ID, group-ID are documented in fix command• tmd = style name of this fix command• rho_final = desired value of rho at the end of the run (distance units)• file1 = filename to read target structure from• N = dump TMD statistics every this many timesteps, 0 = no dump• file2 = filename to write TMD statistics to (only needed if N > 0)•
Examples:
fix 1 all nve
fix 2 tmdatoms tmd 1.0 target_file 100 tmd_dump_file
Description:
Perform targeted molecular dynamics (TMD) on a group of atoms. A holonomic constraint is used to force the
atoms to move towards (or away from) the target configuration. The parameter "rho" is monotonically
decreased (or increased) from its initial value to rho_final at the end of the run.
Rho has distance units and is a measure of the root-mean-squared distance (RMSD) between the current
configuration of the atoms in the group and the target coordinates listed in file1. Thus a value of rho_final =
0.0 means move the atoms all the way to the final structure during the course of the run.
The target file1 can be ASCII text or a gzipped text file (detected by a .gz suffix). The format of the target
file1 is as follows:
0.0 25.0 xlo xhi
0.0 25.0 ylo yhi
0.0 25.0 zlo zhi
125 24.97311 1.69005 23.46956 0 0 -1
126 1.94691 2.79640 1.92799 1 0 0
127 0.15906 3.46099 0.79121 1 0 0
...
The first 3 lines may or may not be needed, depending on the format of the atoms to follow. If image flags are
included with the atoms, the 1st 3 lo/hi lines must appear in the file. If image flags are not included, the 1st 3
lines should not appear. The 3 lines contain the simulation box dimensions for the atom coordinates, in the
same format as in a LAMMPS data file (see the read_data command).
The remaining lines each contain an atom ID and its target x,y,z coordinates. The atom lines (all or none of
them) can optionally be followed by 3 integer values: nx,ny,nz. For periodic dimensions, they specify which
image of the box the atom is considered to be in, i.e. a value of N (positive or negative) means add N times the
box length to the coordinate to get the true value.
LAMMPS Users Manual
1119

The atom lines can be listed in any order, but every atom in the group must be listed in the file. Atoms not in
the fix group may also be listed; they will be ignored.
TMD statistics are written to file2 every N timesteps, unless N is specified as 0, which means no statistics.
The atoms in the fix tmd group should be integrated (via a fix nve, nvt, npt) along with other atoms in the
system.
Restarts can be used with a fix tmd command. For example, imagine a 10000 timestep run with a rho_initial =
11 and a rho_final = 1. If a restart file was written after 2000 time steps, then the configuration in the file
would have a rho value of 9. A new 8000 time step run could be performed with the same rho_final = 1 to
complete the conformational change at the same transition rate. Note that for restarted runs, the name of the
TMD statistics file should be changed to prevent it being overwritten.
For more information about TMD, see (Schlitter1) and (Schlitter2).
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands.
This fix can ramp its rho parameter over multiple runs, using the start and stop keywords of the run command.
See the run command for details of how to do this.
This fix is not invoked during energy minimization.
Restrictions:
All TMD fixes must be listed in the input script after all integrator fixes (nve, nvt, npt) are applied. This
ensures that atoms are moved before their positions are corrected to comply with the constraint.
Atoms that have a TMD fix applied should not be part of a group to which a SHAKE fix is applied. This is
because LAMMPS assumes there are not multiple competing holonomic constraints applied to the same
atoms.
To read gzipped target files, you must compile LAMMPS with the -DLAMMPS_GZIP option - see the
Making LAMMPS section of the documentation.
Related commands: none
Default: none
(Schlitter1) Schlitter, Swegat, Mulders, "Distance-type reaction coordinates for modelling activated
processes", J Molecular Modeling, 7, 171-177 (2001).
(Schlitter2) Schlitter and Klahn, "The free energy of a reaction coordinate at multiple constraints: a concise
formulation", Molecular Physics, 101, 3439-3443 (2003).
LAMMPS Users Manual
1120

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix ttm command
fix ttm/mod command
Syntax:
fix ID group-ID ttm seed C_e rho_e kappa_e gamma_p gamma_s v_0 Nx Ny Nz T_infile N T_outfile
fix ID group-ID ttm/mod seed init_file Nx Ny Nz T_infile N T_outfile
ID, group-ID are documented in fix command• style = ttm or ttm_mod• seed = random number seed to use for white noise (positive integer)• remaining arguments for fix ttm:
C_e = electronic specific heat (energy/(electron*temperature) units)
rho_e = electronic density (electrons/volume units)
kappa_e = electronic thermal conductivity (energy/(time*distance*temperature) units)
gamma_p = friction coefficient due to electron-ion interactions (mass/time units)
gamma_s = friction coefficient due to electronic stopping (mass/time units)
v_0 = electronic stopping critical velocity (velocity units)
Nx = number of thermal solve grid points in the x-direction (positive integer)
Ny = number of thermal solve grid points in the y-direction (positive integer)
Nz = number of thermal solve grid points in the z-direction (positive integer)
T_infile = filename to read initial electronic temperature from
N = dump TTM temperatures every this many timesteps, 0 = no dump
T_outfile = filename to write TTM temperatures to (only needed if N > 0)
•
remaining arguments for fix ttm/mod:
init_file = file with the parameters to TTM
Nx = number of thermal solve grid points in the x-direction (positive integer)
Ny = number of thermal solve grid points in the y-direction (positive integer)
Nz = number of thermal solve grid points in the z-direction (positive integer)
T_infile = filename to read initial electronic temperature from
N = dump TTM temperatures every this many timesteps, 0 = no dump
T_outfile = filename to write TTM temperatures to (only needed if N > 0)
•
Examples:
fix 2 all ttm 699489 1.0 1.0 10 0.1 0.0 2.0 1 12 1 initialTs 1000 T.out
fix 2 all ttm 123456 1.0 1.0 1.0 1.0 1.0 5.0 5 5 5 Te.in 1 Te.out
fix 2 all ttm/mod 34277 parameters.txt 5 5 5 T_init 10 T_out
Description:
Use a two-temperature model (TTM) to represent heat transfer through and between electronic and atomic
subsystems. LAMMPS models the atomic subsystem as usual with a molecular dynamics model and the
classical force field specified by the user, but the electronic subsystem is modeled as a continuum, or a
background "gas", on a regular grid. Energy can be transferred spatially within the grid representing the
electrons. Energy can also be transferred between the electronic and the atomic subsystems. The algorithm
underlying this fix was derived by D. M. Duffy and A. M. Rutherford and is discussed in two J Physics:
Condensed Matter papers: (Duffy) and (Rutherford). They used this algorithm in cascade simulations where a
primary knock-on atom (PKA) was initialized with a high velocity to simulate a radiation event.
LAMMPS Users Manual
1121
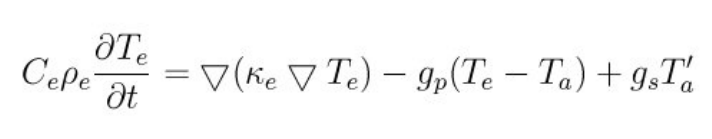
The description in this sub-section applies to both fix ttm and fix ttm/mod. Fix ttm/mod adds options to
account for external heat sources (e.g. at a surface) and for specifying parameters that allow the electronic
heat capacity to depend strongly on electronic temperature. It is more expensive computationally than fix ttm
because it treats the thermal diffusion equation as non-linear. More details on fix ttm/mod are given below.
Heat transfer between the electronic and atomic subsystems is carried out via an inhomogeneous Langevin
thermostat. This thermostat differs from the regular Langevin thermostat (fix langevin) in three important
ways. First, the Langevin thermostat is applied uniformly to all atoms in the user-specified group for a single
target temperature, whereas the TTM fix applies Langevin thermostatting locally to atoms within the volumes
represented by the user-specified grid points with a target temperature specific to that grid point. Second, the
Langevin thermostat couples the temperature of the atoms to an infinite heat reservoir, whereas the heat
reservoir for fix TTM is finite and represents the local electrons. Third, the TTM fix allows users to specify
not just one friction coefficient, but rather two independent friction coefficients: one for the electron-ion
interactions (gamma_p), and one for electron stopping (gamma_s).
When the friction coefficient due to electron stopping, gamma_s, is non-zero, electron stopping effects are
included for atoms moving faster than the electron stopping critical velocity, v_0. For further details about this
algorithm, see (Duffy) and (Rutherford).
Energy transport within the electronic subsystem is solved according to the heat diffusion equation with added
source terms for heat transfer between the subsystems:
where C_e is the specific heat, rho_e is the density, kappa_e is the thermal conductivity, T is temperature, the
"e" and "a" subscripts represent electronic and atomic subsystems respectively, g_p is the coupling constant
for the electron-ion interaction, and g_s is the electron stopping coupling parameter. C_e, rho_e, and kappa_e
are specified as parameters to the fix. The other quantities are derived. The form of the heat diffusion equation
used here is almost the same as that in equation 6 of (Duffy), with the exception that the electronic density is
explicitly represented, rather than being part of the specific heat parameter.
Currently, fix ttm assumes that none of the user-supplied parameters will vary with temperature. Note that
(Duffy) used a tanh() functional form for the temperature dependence of the electronic specific heat, but
ignored temperature dependencies of any of the other parameters. See more discussion below for fix ttm/mod.
These fixes require use of periodic boundary conditions and a 3D simulation. Periodic boundary conditions
are also used in the heat equation solve for the electronic subsystem. This varies from the approach of
(Rutherford) where the atomic subsystem was embedded within a larger continuum representation of the
electronic subsystem.
The initial electronic temperature input file, T_infile, is a text file LAMMPS reads in with no header and with
four numeric columns (ix,iy,iz,Temp) and with a number of rows equal to the number of user-specified grid
points (Nx by Ny by Nz). The ix,iy,iz are node indices from 0 to nxnodes-1, etc. For example, the initial
electronic temperatures on a 1 by 2 by 3 grid could be specified in a T_infile as follows:
0 0 0 1.0
0 0 1 1.0
LAMMPS Users Manual
1122
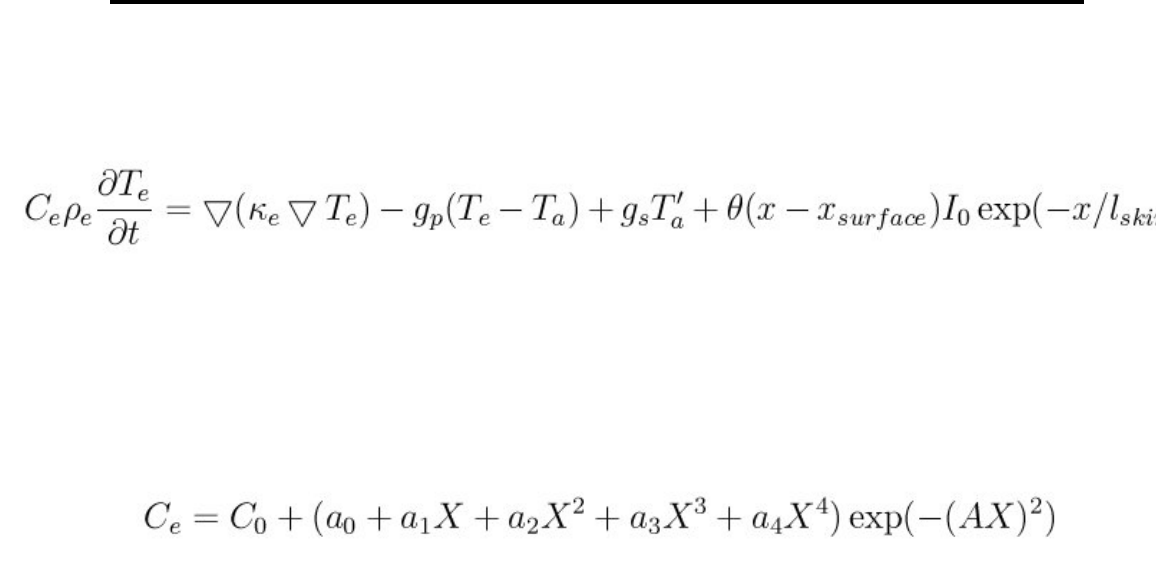
0 0 2 1.0
0 1 0 2.0
0 1 1 2.0
0 1 2 2.0
where the electronic temperatures along the y=0 plane have been set to 1.0, and the electronic temperatures
along the y=1 plane have been set to 2.0. The order of lines in this file is no important. If all the nodal values
are not specified, LAMMPS will generate an error.
The temperature output file, T_oufile, is created and written by this fix. Temperatures for both the electronic
and atomic subsystems at every node and every N timesteps are output. If N is specified as zero, no output is
generated, and no output filename is needed. The format of the output is as follows. One long line is written
every output timestep. The timestep itself is given in the first column. The next Nx*Ny*Nz columns contain
the temperatures for the atomic subsystem, and the final Nx*Ny*Nz columns contain the temperatures for the
electronic subsystem. The ordering of the Nx*Ny*Nz columns is with the z index varying fastest, y the next
fastest, and x the slowest.
These fixes do not change the coordinates of their atoms; they only scales their velocities. Thus a time
integration fix (e.g. fix nve) should still be used to time integrate the affected atoms. The fixes should not
normally be used on atoms that have their temperature controlled by another fix - e.g. fix nvt or fix langevin.
NOTE: The current implementations of these fixes create a copy of the electron grid that overlays the entire
simulation domain, for each processor. Values on the grid are summed across all processors. Thus you should
insure that this grid is not too large, else your simulation could incur high memory and communication costs.
Additional details for fix ttm/mod
Fix ttm/mod uses the heat diffusion equation with possible external heat sources (e.g. laser heating in ablation
simulations):
where theta is the Heaviside step function, I_0 is the (absorbed) laser pulse intensity for ablation simulations,
l_skin is the depth of skin-layer, and all other designations have the same meaning as in the former equation.
The duration of the pulse is set by the parameter tau in the init_file.
Fix ttm/mod also allows users to specify the dependencies of C_e and kappa_e on the electronic temperature.
The specific heat is expressed as
where X = T_e/1000, and the thermal conductivity is defined as kappa_e = D_e*rho_e*C_e, where D_e is the
thermal diffusion coefficient.
Electronic pressure effects are included in the TTM model to account for the blast force acting on ions
because of electronic pressure gradient (see (Chen), (Norman)). The total force acting on an ion is:
LAMMPS Users Manual
1123

where F_langevin is a force from Langevin thermostat simulating electron-phonon coupling, and nabla
P_e/n_ion is the electron blast force.
The electronic pressure is taken to be P_e = B*rho_e*C_e*T_e
The current fix ttm/mod implementation allows TTM simulations with a vacuum. The vacuum region is
defined as the grid cells with zero electronic temperature. The numerical scheme does not allow energy
exchange with such cells. Since the material can expand to previously unoccupied region in some simulations,
the vacuum border can be allowed to move. It is controlled by the surface_movement parameter in the
init_file. If it is set to 1, then "vacuum" cells can be changed to "electron-filled" cells with the temperature
T_e_min if atoms move into them (currently only implemented for the case of 1-dimensional motion of flat
surface normal to the X axis). The initial borders of vacuum can be set in the init_file via lsurface and
rsurface parameters. In this case, electronic pressure gradient is calculated as
where lambda is the electron mean free path (see (Norman), (Pisarev))
The fix ttm/mod parameter file init_file has the following syntax/ Every line with the odd number is
considered as a comment and ignored. The lines with the even numbers are treated as follows:
a_0, energy/(temperature*electron) units
a_1, energy/(temperature^2*electron) units
a_2, energy/(temperature^3*electron) units
a_3, energy/(temperature^4*electron) units
a_4, energy/(temperature^5*electron) units
C_0, energy/(temperature*electron) units
A, 1/temperature units
rho_e, electrons/volume units
D_e, length^2/time units
gamma_p, mass/time units
gamma_s, mass/time units
v_0, length/time units
I_0, energy/(time*length^2) units
lsurface, electron grid units (positive integer)
rsurface, electron grid units (positive integer)
l_skin, length units
tau, time units
B, dimensionless
lambda, length units
n_ion, ions/volume units
surface_movement: 0 to disable tracking of surface motion, 1 to enable
T_e_min, temperature units
Restart, fix_modify, output, run start/stop, minimize info:
LAMMPS Users Manual
1124

These fixes write the state of the electronic subsystem and the energy exchange between the subsystems to
binary restart files. See the read_restart command for info on how to re-specify a fix in an input script that
reads a restart file, so that the operation of the fix continues in an uninterrupted fashion.
Because the state of the random number generator is not saved in the restart files, this means you cannot do
"exact" restarts with this fix, where the simulation continues on the same as if no restart had taken place.
However, in a statistical sense, a restarted simulation should produce the same behavior.
None of the fix_modify options are relevant to these fixes.
Both fixes compute 2 output quantities stored in a vector of length 2, which can be accessed by various output
commands. The first quantity is the total energy of the electronic subsystem. The second quantity is the
energy transferred from the electronic to the atomic subsystem on that timestep. Note that the velocity verlet
integrator applies the fix ttm forces to the atomic subsystem as two half-step velocity updates: one on the
current timestep and one on the subsequent timestep. Consequently, the change in the atomic subsystem
energy is lagged by half a timestep relative to the change in the electronic subsystem energy. As a result of
this, users may notice slight fluctuations in the sum of the atomic and electronic subsystem energies reported
at the end of the timestep.
The vector values calculated are "extensive".
No parameter of the fixes can be used with the start/stop keywords of the run command. The fixes are not
invoked during energy minimization.
Restrictions:
Fix ttm is part of the MISC package. It is only enabled if LAMMPS was built with that package. Fix ttm/mod
is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
These fixes can only be used for 3d simulations and orthogonal simulation boxes. You must also use periodic
boundary conditions.
Related commands:
fix langevin, fix dt/reset
Default: none
(Duffy) D M Duffy and A M Rutherford, J. Phys.: Condens. Matter, 19, 016207-016218 (2007).
(Rutherford) A M Rutherford and D M Duffy, J. Phys.: Condens. Matter, 19, 496201-496210 (2007).
(Chen) J Chen, D Tzou and J Beraun, Int. J. Heat Mass Transfer, 49, 307-316 (2006).
(Norman) G E Norman, S V Starikov, V V Stegailov et al., Contrib. Plasma Phys., 53, 129-139 (2013).
LAMMPS Users Manual
1125
(Pisarev) V V Pisarev and S V Starikov, J. Phys.: Condens. Matter, 26, 475401 (2014).
LAMMPS Users Manual
1126

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix tune/kspace command
Syntax:
fix ID group-ID tune/kspace N
ID, group-ID are documented in fix command• tune/kspace = style name of this fix command• N = invoke this fix every N steps•
Examples:
fix 2 all tune/kspace 100
Description:
This fix tests each kspace style (Ewald, PPPM, and MSM), and automatically selects the fastest style to use
for the remainder of the run. If the fastest style is Ewald or PPPM, the fix also adjusts the coulomb cutoff
towards optimal speed. Future versions of this fix will automatically select other kspace parameters to use for
maximum simulation speed. The kspace parameters may include the style, cutoff, grid points in each
direction, order, Ewald parameter, MSM parallelization cut-point, MPI tasks to use, etc.
The rationale for this fix is to provide the user with as-fast-as-possible simulations that include long-range
electrostatics (kspace) while meeting the user-prescribed accuracy requirement. A simple heuristic could
never capture the optimal combination of parameters for every possible run-time scenario. But by performing
short tests of various kspace parameter sets, this fix allows parameters to be tailored specifically to the user's
machine, MPI ranks, use of threading or accelerators, the simulated system, and the simulation details. In
addition, it is possible that parameters could be evolved with the simulation on-the-fly, which is useful for
systems that are dynamically evolving (e.g. changes in box size/shape or number of particles).
When this fix is invoked, LAMMPS will perform short timed tests of various parameter sets to determine the
optimal parameters. Tests are performed on-the-fly, with a new test initialized every N steps. N should be
chosen large enough so that adequate CPU time lapses between tests, thereby providing statistically
significant timings. But N should not be chosen to be so large that an unfortunate parameter set test takes an
inordinate amount of wall time to complete. An N of 100 for most problems seems reasonable. Once an
optimal parameter set is found, that set is used for the remainder of the run.
This fix uses heristics to guide it's selection of parameter sets to test, but the actual timed results will be used
to decide which set to use in the simulation.
It is not necessary to discard trajectories produced using sub-optimal parameter sets, or a mix of various
parameter sets, since the user-prescribed accuracy will have been maintained throughout. However, some
users may prefer to use this fix only to discover the optimal parameter set for a given setup that can then be
used on subsequent production runs.
This fix starts with kspace parameters that are set by the user with the kspace_style and kspace_modify
commands. The prescribed accuracy will be maintained by this fix throughout the simulation.
None of the fix_modify options are relevant to this fix.
LAMMPS Users Manual
1127
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the KSPACE package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Do not set "neigh_modify once yes" or else this fix will never be called. Reneighboring is required.
Related commands:
kspace_style, boundary kspace_modify, pair_style lj/cut/coul/long, pair_style lj/charmm/coul/long, pair_style
lj/long, pair_style lj/long/coul/long, pair_style buck/coul/long
Default:
LAMMPS Users Manual
1128

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix vector command
Syntax:
fix ID group-ID vector Nevery value1 value2 ...
ID, group-ID are documented in fix command• vector = style name of this fix command• Nevery = use input values every this many timesteps• one or more input values can be listed• value = c_ID, c_ID[N], f_ID, f_ID[N], v_name
c_ID = global scalar calculated by a compute with ID
c_ID[I] = Ith component of global vector calculated by a compute with ID
f_ID = global scalar calculated by a fix with ID
f_ID[I] = Ith component of global vector calculated by a fix with ID
v_name = value calculated by an equal-style variable with name
v_name[I] = Ith component of vector-style variable with name
•
Examples:
fix 1 all vector 100 c_myTemp
fix 1 all vector 5 c_myTemp v_integral
Description:
Use one or more global values as inputs every few timesteps, and simply store them. For a single specified
value, the values are stored as a global vector of growing length. For multiple specified values, they are stored
as rows in a global array, whose number of rows is growing. The resulting vector or array can be used by
other output commands.
One way to to use this command is to accumulate a vector that is time-integrated using the variable trap()
function. For example the velocity auto-correlation function (VACF) can be time-integrated, to yield a
diffusion coefficient, as follows:
compute 2 all vacf
fix 5 all vector 1 c_2[4]
variable diff equal dt*trap(f_5)
thermo_style custom step v_diff
The group specified with this command is ignored. However, note that specified values may represent
calculations performed by computes and fixes which store their own "group" definitions.
Each listed value can be the result of a compute or fix or the evaluation of an equal-style or vector-style
variable. In each case, the compute, fix, or variable must produce a global quantity, not a per-atom or local
quantity. And the global quantity must be a scalar, not a vector or array.
Computes that produce global quantities are those which do not have the word atom in their style name. Only
a few fixes produce global quantities. See the doc pages for individual fixes for info on which ones produce
such values. Variables of style equal or vector are the only ones that can be used with this fix. Variables of
style atom cannot be used, since they produce per-atom values.
LAMMPS Users Manual
1129
The Nevery argument specifies on what timesteps the input values will be used in order to be stored. Only
timesteps that are a multiple of Nevery, including timestep 0, will contribute values.
Note that if you perform multiple runs, using the "pre no" option of the run command to avoid initialization on
subsequent runs, then you need to use the stop keyword with the first run command with a timestep value that
encompasses all the runs. This is so that the vector or array stored by this fix can be allocated to a sufficient
size.
If a value begins with "c_", a compute ID must follow which has been previously defined in the input script. If
no bracketed term is appended, the global scalar calculated by the compute is used. If a bracketed term is
appended, the Ith element of the global vector calculated by the compute is used.
Note that there is a compute reduce command which can sum per-atom quantities into a global scalar or vector
which can thus be accessed by fix vector. Or it can be a compute defined not in your input script, but by
thermodynamic output or other fixes such as fix nvt or fix temp/rescale. See the doc pages for these
commands which give the IDs of these computes. Users can also write code for their own compute styles and
add them to LAMMPS.
If a value begins with "f_", a fix ID must follow which has been previously defined in the input script. If no
bracketed term is appended, the global scalar calculated by the fix is used. If a bracketed term is appended, the
Ith element of the global vector calculated by the fix is used.
Note that some fixes only produce their values on certain timesteps, which must be compatible with Nevery,
else an error will result. Users can also write code for their own fix styles and add them to LAMMPS.
If a value begins with "v_", a variable name must follow which has been previously defined in the input
script. An equal-style or vector-style variable can be referenced; the latter requires a bracketed term to specify
the Ith element of the vector calculated by the variable. See the variable command for details. Note that
variables of style equal and vector define a formula which can reference individual atom properties or
thermodynamic keywords, or they can invoke other computes, fixes, or variables when they are evaluated, so
this is a very general means of specifying quantities to be stored by fix vector.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix produces a global vector or global array which can be accessed by various output commands. The
values can only be accessed on timesteps that are multiples of Nevery.
A vector is produced if only a single input value is specified. An array is produced if multiple input values are
specified. The length of the vector or the number of rows in the array grows by 1 every Nevery timesteps.
If the fix produces a vector, then the entire vector will be either "intensive" or "extensive", depending on
whether the values stored in the vector are "intensive" or "extensive". If the fix produces an array, then all
elements in the array must be the same, either "intensive" or "extensive". If a compute or fix provides the
value stored, then the compute or fix determines whether the value is intensive or extensive; see the doc page
for that compute or fix for further info. Values produced by a variable are treated as intensive.
This fix can allocate storage for stored values accumulated over multiple runs, using the start and stop
LAMMPS Users Manual
1130
keywords of the run command. See the run command for details of how to do this. If using the run pre no
command option, this is required to allow the fix to allocate sufficient storage for stored values.
This fix is not invoked during energy minimization.
Restrictions: none
Related commands:
compute, variable
Default: none
LAMMPS Users Manual
1131

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix viscosity command
Syntax:
fix ID group-ID viscosity N vdim pdim Nbin keyword value ...
ID, group-ID are documented in fix command• viscosity = style name of this fix command• N = perform momentum exchange every N steps• vdim = x or y or z = which momentum component to exchange• pdim = x or y or z = direction of momentum transfer• Nbin = # of layers in pdim direction (must be even number)• zero or more keyword/value pairs may be appended• keyword = swap or target
swap value = Nswap = number of swaps to perform every N steps
vtarget value = V or INF = target velocity of swap partners (velocity units)
•
Examples:
fix 1 all viscosity 100 x z 20
fix 1 all viscosity 50 x z 20 swap 2 vtarget 1.5
Description:
Use the Muller-Plathe algorithm described in this paper to exchange momenta between two particles in
different regions of the simulation box every N steps. This induces a shear velocity profile in the system. As
described below this enables a viscosity of the fluid to be calculated. This algorithm is sometimes called a
reverse non-equilibrium MD (reverse NEMD) approach to computing viscosity. This is because the usual
NEMD approach is to impose a shear velocity profile on the system and measure the response via an
off-diagonal component of the stress tensor, which is proportional to the momentum flux. In the Muller-Plathe
method, the momentum flux is imposed, and the shear velocity profile is the system's response.
The simulation box is divided into Nbin layers in the pdim direction, where the layer 1 is at the low end of that
dimension and the layer Nbin is at the high end. Every N steps, Nswap pairs of atoms are chosen in the
following manner. Only atoms in the fix group are considered. Nswap atoms in layer 1 with positive velocity
components in the vdim direction closest to the target value V are selected. Similarly, Nswap atoms in the
"middle" layer (see below) with negative velocity components in the vdim direction closest to the negative of
the target value V are selected. The two sets of Nswap atoms are paired up and their vdim momenta
components are swapped within each pair. This resets their velocities, typically in opposite directions. Over
time, this induces a shear velocity profile in the system which can be measured using commands such as the
following, which writes the profile to the file tmp.profile:
compute layers all chunk/atom bin/1d z lower 0.05 units reduced
fix f1 all ave/chunk 100 10 1000 layers vx file tmp.profile
Note that by default, Nswap = 1 and vtarget = INF, though this can be changed by the optional swap and
vtarget keywords. When vtarget = INF, one or more atoms with the most positive and negative velocity
components are selected. Setting these parameters appropriately, in conjunction with the swap rate N, allows
the momentum flux rate to be adjusted across a wide range of values, and the momenta to be exchanged in
LAMMPS Users Manual
1132
large chunks or more smoothly.
The "middle" layer for momenta swapping is defined as the Nbin/2 + 1 layer. Thus if Nbin = 20, the two
swapping layers are 1 and 11. This should lead to a symmetric velocity profile since the two layers are
separated by the same distance in both directions in a periodic sense. This is why Nbin is restricted to being an
even number.
As described below, the total momentum transferred by these velocity swaps is computed by the fix and can
be output. Dividing this quantity by time and the cross-sectional area of the simulation box yields a
momentum flux. The ratio of momentum flux to the slope of the shear velocity profile is proportional to the
viscosity of the fluid, in appropriate units. See the Muller-Plathe paper for details.
NOTE: If your system is periodic in the direction of the momentum flux, then the flux is going in 2 directions.
This means the effective momentum flux in one direction is reduced by a factor of 2. You will see this in the
equations for viscosity in the Muller-Plathe paper. LAMMPS is simply tallying momentum which does not
account for whether or not your system is periodic; you must use the value appropriately to yield a viscosity
for your system.
NOTE: After equilibration, if the velocity profile you observe is not linear, then you are likely swapping
momentum too frequently and are not in a regime of linear response. In this case you cannot accurately infer a
viscosity and should try increasing the Nevery parameter.
An alternative method for calculating a viscosity is to run a NEMD simulation, as described in Section 6.13 of
the manual. NEMD simulations deform the simulation box via the fix deform command. Thus they cannot be
run on a charged system using a PPPM solver since PPPM does not currently support non-orthogonal boxes.
Using fix viscosity keeps the box orthogonal; thus it does not suffer from this limitation.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global scalar which can be accessed by various output commands. The scalar is the
cumulative momentum transferred between the bottom and middle of the simulation box (in the pdim
direction) is stored as a scalar quantity by this fix. This quantity is zeroed when the fix is defined and
accumulates thereafter, once every N steps. The units of the quantity are momentum = mass*velocity. The
scalar value calculated by this fix is "intensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix is part of the MISC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Swaps conserve both momentum and kinetic energy, even if the masses of the swapped atoms are not equal.
Thus you should not need to thermostat the system. If you do use a thermostat, you may want to apply it only
to the non-swapped dimensions (other than vdim).
LAMMPS Users Manual
1133

LAMMPS does not check, but you should not use this fix to swap velocities of atoms that are in constrained
molecules, e.g. via fix shake or fix rigid. This is because application of the constraints will alter the amount of
transferred momentum. You should, however, be able to use flexible molecules. See the Maginn paper for an
example of using this algorithm in a computation of alcohol molecule properties.
When running a simulation with large, massive particles or molecules in a background solvent, you may want
to only exchange momenta between solvent particles.
Related commands:
fix ave/chunk, fix thermal/conductivity
Default:
The option defaults are swap = 1 and vtarget = INF.
(Muller-Plathe) Muller-Plathe, Phys Rev E, 59, 4894-4898 (1999).
(Maginn) Kelkar, Rafferty, Maginn, Siepmann, Fluid Phase Equilibria, 260, 218-231 (2007).
LAMMPS Users Manual
1134

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix viscous command
Syntax:
fix ID group-ID viscous gamma keyword values ...
ID, group-ID are documented in fix command• viscous = style name of this fix command• gamma = damping coefficient (force/velocity units)• zero or more keyword/value pairs may be appended
keyword = scale
scale values = type ratio
type = atom type (1-N)
ratio = factor to scale the damping coefficient by
•
Examples:
fix 1 flow viscous 0.1
fix 1 damp viscous 0.5 scale 3 2.5
Description:
Add a viscous damping force to atoms in the group that is proportional to the velocity of the atom. The added
force can be thought of as a frictional interaction with implicit solvent, i.e. the no-slip Stokes drag on a
spherical particle. In granular simulations this can be useful for draining the kinetic energy from the system in
a controlled fashion. If used without additional thermostatting (to add kinetic energy to the system), it has the
effect of slowly (or rapidly) freezing the system; hence it can also be used as a simple energy minimization
technique.
The damping force F is given by F = - gamma * velocity. The larger the coefficient, the faster the kinetic
energy is reduced. If the optional keyword scale is used, gamma can scaled up or down by the specified factor
for atoms of that type. It can be used multiple times to adjust gamma for several atom types.
NOTE: You should specify gamma in force/velocity units. This is not the same as mass/time units, at least for
some of the LAMMPS units options like "real" or "metal" that are not self-consistent.
In a Brownian dynamics context, gamma = Kb T / D, where Kb = Boltzmann's constant, T = temperature, and
D = particle diffusion coefficient. D can be written as Kb T / (3 pi eta d), where eta = dynamic viscosity of the
frictional fluid and d = diameter of particle. This means gamma = 3 pi eta d, and thus is proportional to the
viscosity of the fluid and the particle diameter.
In the current implementation, rather than have the user specify a viscosity, gamma is specified directly in
force/velocity units. If needed, gamma can be adjusted for atoms of different sizes (i.e. sigma) by using the
scale keyword.
Note that Brownian dynamics models also typically include a randomized force term to thermostat the system
at a chosen temperature. The fix langevin command does this. It has the same viscous damping term as fix
viscous and adds a random force to each atom. The random force term is proportional to the sqrt of the chosen
thermostatting temperature. Thus if you use fix langevin with a target T = 0, its random force term is zero, and
LAMMPS Users Manual
1135

you are essentially performing the same operation as fix viscous. Also note that the gamma of fix viscous is
related to the damping parameter of fix langevin, however the former is specified in units of force/velocity
and the latter in units of time, so that it can more easily be used as a thermostat.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is modifying forces. Default is the outermost level.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
This fix should only be used with damped dynamics minimizers that allow for non-conservative forces. See
the min_style command for details.
Restrictions: none
Related commands:
fix langevin
Default: none
LAMMPS Users Manual
1136

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix wall/lj93 command
fix wall/lj126 command
fix wall/lj1043 command
fix wall/colloid command
fix wall/harmonic command
Syntax:
fix ID group-ID style face args ... keyword value ...
ID, group-ID are documented in fix command• style = wall/lj93 or wall/lj126 or wall/lj1043 or wall/colloid or wall/harmonic• one or more face/arg pairs may be appended• face = xlo or xhi or ylo or yhi or zlo or zhi
args = coord epsilon sigma cutoff
coord = position of wall = EDGE or constant or variable
EDGE = current lo or hi edge of simulation box
constant = number like 0.0 or -30.0 (distance units)
variable = equal-style variable like v_x or v_wiggle
epsilon = strength factor for wall-particle interaction (energy or energy/distance^2 units)
epsilon can be a variable (see below)
sigma = size factor for wall-particle interaction (distance units)
sigma can be a variable (see below)
cutoff = distance from wall at which wall-particle interaction is cut off (distance units)
•
zero or more keyword/value pairs may be appended• keyword = units or fld
units value = lattice or box
lattice = the wall position is defined in lattice units
box = the wall position is defined in simulation box units
fld value = yes or no
yes = invoke the wall constraint to be compatible with implicit FLD
no = invoke the wall constraint in the normal way
pbc value = yes or no
yes = allow periodic boundary in a wall dimension
no = require non-perioidic boundaries in any wall dimension
•
Examples:
fix wallhi all wall/lj93 xlo -1.0 1.0 1.0 2.5 units box
fix wallhi all wall/lj93 xhi EDGE 1.0 1.0 2.5
fix wallhi all wall/lj126 v_wiggle 23.2 1.0 1.0 2.5
fix zwalls all wall/colloid zlo 0.0 1.0 1.0 0.858 zhi 40.0 1.0 1.0 0.858
Description:
LAMMPS Users Manual
1137
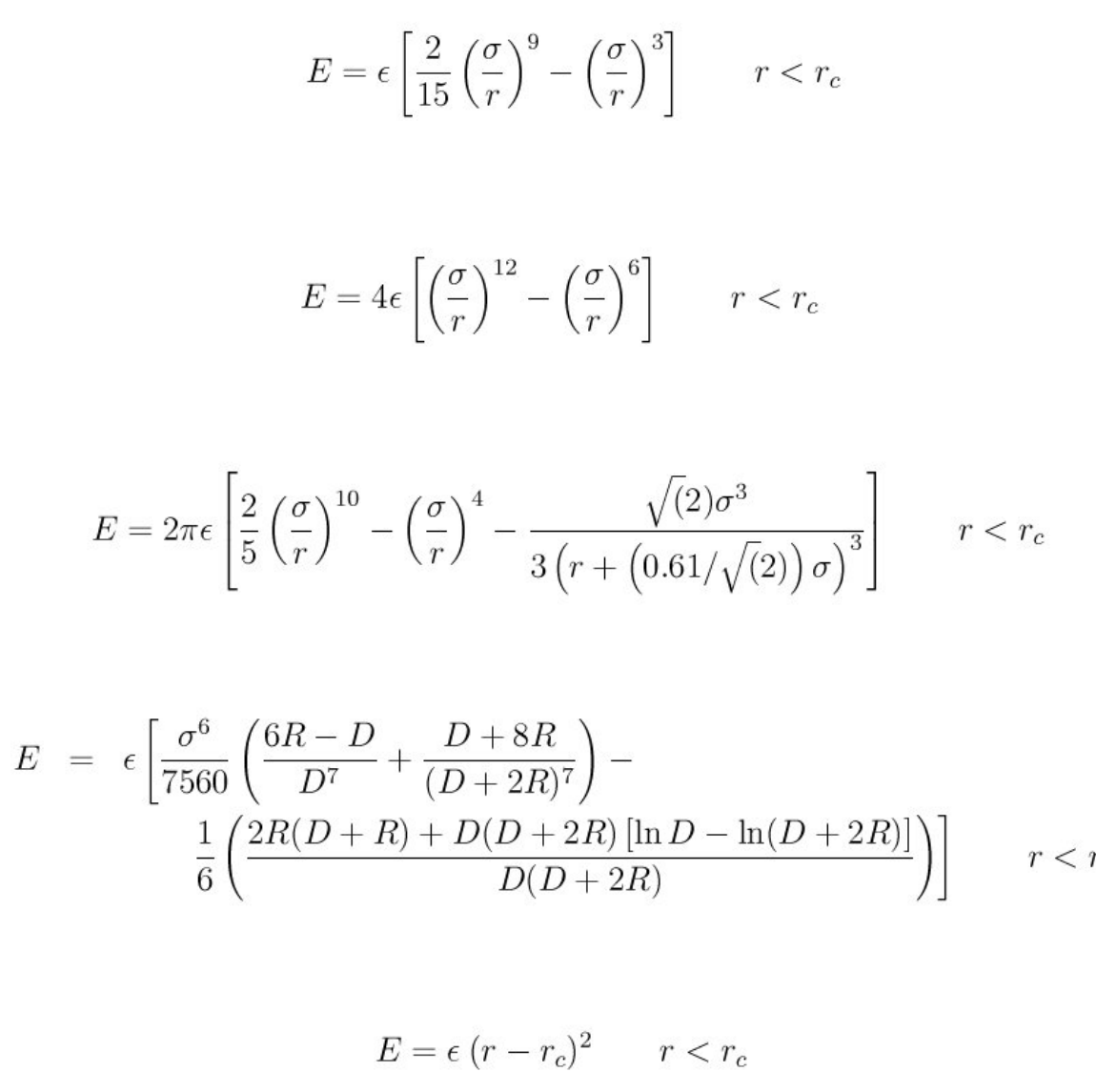
Bound the simulation domain on one or more of its faces with a flat wall that interacts with the atoms in the
group by generating a force on the atom in a direction perpendicular to the wall. The energy of wall-particle
interactions depends on the style.
For style wall/lj93, the energy E is given by the 9/3 potential:
For style wall/lj126, the energy E is given by the 12/6 potential:
For style wall/lj1043, the energy E is given by the 10/4/3 potential:
For style wall/colloid, the energy E is given by an integrated form of the pair_style colloid potential:
For style wall/harmonic, the energy E is given by a harmonic spring potential:
LAMMPS Users Manual
1138
In all cases, r is the distance from the particle to the wall at position coord, and Rc is the cutoff distance at
which the particle and wall no longer interact. The energy of the wall potential is shifted so that the
wall-particle interaction energy is 0.0 at the cutoff distance.
Up to 6 walls or faces can be specified in a single command: xlo, xhi, ylo, yhi, zlo, zhi. A lo face interacts with
particles near the lower side of the simulation box in that dimension. A hi face interacts with particles near the
upper side of the simulation box in that dimension.
The position of each wall can be specified in one of 3 ways: as the EDGE of the simulation box, as a constant
value, or as a variable. If EDGE is used, then the corresponding boundary of the current simulation box is
used. If a numeric constant is specified then the wall is placed at that position in the appropriate dimension (x,
y, or z). In both the EDGE and constant cases, the wall will never move. If the wall position is a variable, it
should be specified as v_name, where name is an equal-style variable name. In this case the variable is
evaluated each timestep and the result becomes the current position of the reflecting wall. Equal-style
variables can specify formulas with various mathematical functions, and include thermo_style command
keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to specify a
time-dependent wall position. See examples below.
For the wall/lj93 and wall/lj126 and wall/lj1043 styles, epsilon and sigma are the usual Lennard-Jones
parameters, which determine the strength and size of the particle as it interacts with the wall. Epsilon has
energy units. Note that this epsilon and sigma may be different than any epsilon or sigma values defined for a
pair style that computes particle-particle interactions.
The wall/lj93 interaction is derived by integrating over a 3d half-lattice of Lennard-Jones 12/6 particles. The
wall/lj126 interaction is effectively a harder, more repulsive wall interaction. The wall/lj1043 interaction is
yet a different form of wall interaction, described in Magda et al in (Magda).
For the wall/colloid style, R is the radius of the colloid particle, D is the distance from the surface of the
colloid particle to the wall (r-R), and sigma is the size of a constituent LJ particle inside the colloid particle
and wall. Note that the cutoff distance Rc in this case is the distance from the colloid particle center to the
wall. The prefactor epsilon can be thought of as an effective Hamaker constant with energy units for the
strength of the colloid-wall interaction. More specifically, the epsilon pre-factor = 4 * pi^2 * rho_wall *
rho_colloid * epsilon * sigma^6, where epsilon and sigma are the LJ parameters for the constituent LJ
particles. Rho_wall and rho_colloid are the number density of the constituent particles, in the wall and colloid
respectively, in units of 1/volume.
The wall/colloid interaction is derived by integrating over constituent LJ particles of size sigma within the
colloid particle and a 3d half-lattice of Lennard-Jones 12/6 particles of size sigma in the wall. As mentioned in
the preceding paragraph, the density of particles in the wall and colloid can be different, as specified by the
epsilon pre-factor.
For the wall/harmonic style, epsilon is effectively the spring constant K, and has units (energy/distance^2).
The input parameter sigma is ignored. The minimum energy position of the harmonic spring is at the cutoff.
This is a repulsive-only spring since the interaction is truncated at the cutoff
For any wall, the epsilon and/or sigma parameter can be specified as an equal-style variable, in which case it
should be specified as v_name, where name is the variable name. As with a variable wall position, the
variable is evaluated each timestep and the result becomes the current epsilon or sigma of the wall.
Equal-style variables can specify formulas with various mathematical functions, and include thermo_style
command keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to
specify a time-dependent wall interaction.
LAMMPS Users Manual
1139

NOTE: For all of the styles, you must insure that r is always > 0 for all particles in the group, or LAMMPS
will generate an error. This means you cannot start your simulation with particles at the wall position coord (r
= 0) or with particles on the wrong side of the wall (r < 0). For the wall/lj93 and wall/lj126 styles, the energy
of the wall/particle interaction (and hence the force on the particle) blows up as r -> 0. The wall/colloid style
is even more restrictive, since the energy blows up as D = r-R -> 0. This means the finite-size particles of
radius R must be a distance larger than R from the wall position coord. The harmonic style is a softer
potential and does not blow up as r -> 0, but you must use a large enough epsilon that particles always reamin
on the correct side of the wall (r > 0).
The units keyword determines the meaning of the distance units used to define a wall position, but only when
a numeric constant or variable is used. It is not relevant when EDGE is used to specify a face position. In the
variable case, the variable is assumed to produce a value compatible with the units setting you specify.
A box value selects standard distance units as defined by the units command, e.g. Angstroms for units = real
or metal. A lattice value means the distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacings.
The fld keyword can be used with a yes setting to invoke the wall constraint before pairwise interactions are
computed. This allows an implicit FLD model using pair_style lubricateU to include the wall force in its
calculations. If the setting is no, wall forces are imposed after pairwise interactions, in the usual manner.
The pbc keyword can be used with a yes setting to allow walls to be specified in a periodic dimension. See the
boundary command for options on simulation box boundaries. The default for pbc is no, which means the
system must be non-periodic when using a wall. But you may wish to use a periodic box. E.g. to allow some
particles to interact with the wall via the fix group-ID, and others to pass through it and wrap around a
periodic box. In this case you should insure that the wall if sufficiently far enough away from the box
boundary. If you do not, then particles may interact with both the wall and with periodic images on the other
side of the box, which is probably not what you want.
Here are examples of variable definitions that move the wall position in a time-dependent fashion using
equal-style variables. The wall interaction parameters (epsilon, sigma) could be varied with additional
variable definitions.
variable ramp equal ramp(0,10)
fix 1 all wall xlo v_ramp 1.0 1.0 2.5
variable linear equal vdisplace(0,20)
fix 1 all wall xlo v_linear 1.0 1.0 2.5
variable wiggle equal swiggle(0.0,5.0,3.0)
fix 1 all wall xlo v_wiggle 1.0 1.0 2.5
variable wiggle equal cwiggle(0.0,5.0,3.0)
fix 1 all wall xlo v_wiggle 1.0 1.0 2.5
The ramp(lo,hi) function adjusts the wall position linearly from lo to hi over the course of a run. The
vdisplace(c0,velocity) function does something similar using the equation position = c0 + velocity*delta,
where delta is the elapsed time.
The swiggle(c0,A,period) function causes the wall position to oscillate sinusoidally according to this equation,
where omega = 2 PI / period:
LAMMPS Users Manual
1140
position = c0 + A sin(omega*delta)
The cwiggle(c0,A,period) function causes the wall position to oscillate sinusoidally according to this
equation, which will have an initial wall velocity of 0.0, and thus may impose a gentler perturbation on the
particles:
position = c0 + A (1 - cos(omega*delta))
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the energy of interaction between atoms and each
wall to the system's potential energy as part of thermodynamic output.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
This fix computes a global scalar energy and a global vector of forces, which can be accessed by various
output commands. Note that the scalar energy is the sum of interactions with all defined walls. If you want the
energy on a per-wall basis, you need to use multiple fix wall commands. The length of the vector is equal to
the number of walls defined by the fix. Each vector value is the normal force on a specific wall. Note that an
outward force on a wall will be a negative value for lo walls and a positive value for hi walls. The scalar and
vector values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
NOTE: If you want the atom/wall interaction energy to be included in the total potential energy of the system
(the quantity being minimized), you MUST enable the fix_modify energy option for this fix.
Restrictions: none
Related commands:
fix wall/reflect, fix wall/gran, fix wall/region
Default:
The option defaults units = lattice, fld = no, and pbc = no.
(Magda) Magda, Tirrell, Davis, J Chem Phys, 83, 1888-1901 (1985); erratum in JCP 84, 2901 (1986).
LAMMPS Users Manual
1141

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix wall/gran command
Syntax:
fix ID group-ID wall/gran fstyle Kn Kt gamma_n gamma_t xmu dampflag wallstyle args keyword values ...
ID, group-ID are documented in fix command• wall/gran = style name of this fix command• fstyle = style of force interactions between particles and wall
possible choices: hooke, hooke/history, hertz/history
•
Kn = elastic constant for normal particle repulsion (force/distance units or pressure units - see
discussion below)
•
Kt = elastic constant for tangential contact (force/distance units or pressure units - see discussion
below)
•
gamma_n = damping coefficient for collisions in normal direction (1/time units or 1/time-distance
units - see discussion below)
•
gamma_t = damping coefficient for collisions in tangential direction (1/time units or 1/time-distance
units - see discussion below)
•
xmu = static yield criterion (unitless value between 0.0 and 1.0e4)• dampflag = 0 or 1 if tangential damping force is excluded or included• wallstyle = xplane or yplane or zplane or zcylinder• args = list of arguments for a particular style
xplane or yplane or zplane args = lo hi
lo,hi = position of lower and upper plane (distance units), either can be NULL)
zcylinder args = radius
radius = cylinder radius (distance units)
•
zero or more keyword/value pairs may be appended to args• keyword = wiggle or shear
wiggle values = dim amplitude period
dim = x or y or z
amplitude = size of oscillation (distance units)
period = time of oscillation (time units)
shear values = dim vshear
dim = x or y or z
vshear = magnitude of shear velocity (velocity units)
•
Examples:
fix 1 all wall/gran hooke 200000.0 NULL 50.0 NULL 0.5 0 xplane -10.0 10.0
fix 1 all wall/gran hooke/history 200000.0 NULL 50.0 NULL 0.5 0 zplane 0.0 NULL
fix 2 all wall/gran hooke 100000.0 20000.0 50.0 30.0 0.5 1 zcylinder 15.0 wiggle z 3.0 2.0
Description:
Bound the simulation domain of a granular system with a frictional wall. All particles in the group interact
with the wall when they are close enough to touch it.
LAMMPS Users Manual
1142
The nature of the wall/particle interactions are determined by the fstyle setting. It can be any of the styles
defined by the pair_style granular commands. Currently this is hooke, hooke/history, or hertz/history. The
equation for the force between the wall and particles touching it is the same as the corresponding equation on
the pair_style granular doc page, in the limit of one of the two particles going to infinite radius and mass (flat
wall). Specifically, delta = radius - r = overlap of particle with wall, m_eff = mass of particle, and the
effective radius of contact = RiRj/Ri+Rj is just the radius of the particle.
The parameters Kn, Kt, gamma_n, gamma_t, xmu and dampflag have the same meaning and units as those
specified with the pair_style granular commands. This means a NULL can be used for either Kt or gamma_t
as described on that page. If a NULL is used for Kt, then a default value is used where Kt = 2/7 Kn. If a NULL
is used for gamma_t, then a default value is used where gamma_t = 1/2 gamma_n.
Note that you can choose a different force styles and/or different values for the 6 wall/particle coefficients
than for particle/particle interactions. E.g. if you wish to model the wall as a different material.
NOTE: As discussed on the doc page for pair_style granular, versions of LAMMPS before 9Jan09 used a
different equation for Hertzian interactions. This means Hertizian wall/particle interactions have also changed.
They now include a sqrt(radius) term which was not present before. Also the previous versions used Kn and
Kt from the pairwise interaction and hardwired dampflag to 1, rather than letting them be specified directly.
This means you can set the values of the wall/particle coefficients appropriately in the current code to
reproduce the results of a previous Hertzian monodisperse calculation. For example, for the common case of a
monodisperse system with particles of diameter 1, Kn, Kt, gamma_n, and gamma_s should be set sqrt(2.0)
larger than they were previously.
The effective mass m_eff in the formulas listed on the pair_style granular doc page is the mass of the particle
for particle/wall interactions (mass of wall is infinite). If the particle is part of a rigid body, its mass is
replaced by the mass of the rigid body in those formulas. This is determined by searching for a fix rigid
command (or its variants).
The wallstyle can be planar or cylindrical. The 3 planar options specify a pair of walls in a dimension. Wall
positions are given by lo and hi. Either of the values can be specified as NULL if a single wall is desired. For
a zcylinder wallstyle, the cylinder's axis is at x = y = 0.0, and the radius of the cylinder is specified.
Optionally, the wall can be moving, if the wiggle or shear keywords are appended. Both keywords cannot be
used together.
For the wiggle keyword, the wall oscillates sinusoidally, similar to the oscillations of particles which can be
specified by the fix move command. This is useful in packing simulations of granular particles. The
arguments to the wiggle keyword specify a dimension for the motion, as well as it's amplitude and period.
Note that if the dimension is in the plane of the wall, this is effectively a shearing motion. If the dimension is
perpendicular to the wall, it is more of a shaking motion. A zcylinder wall can only be wiggled in the z
dimension.
Each timestep, the position of a wiggled wall in the appropriate dim is set according to this equation:
position = coord + A - A cos (omega * delta)
where coord is the specified initial position of the wall, A is the amplitude, omega is 2 PI / period, and delta is
the time elapsed since the fix was specified. The velocity of the wall is set to the derivative of this expression.
LAMMPS Users Manual
1143
For the shear keyword, the wall moves continuously in the specified dimension with velocity vshear. The
dimension must be tangential to walls with a planar wallstyle, e.g. in the y or z directions for an xplane wall.
For zcylinder walls, a dimension of z means the cylinder is moving in the z-direction along it's axis. A
dimension of x or y means the cylinder is spinning around the z-axis, either in the clockwise direction for
vshear > 0 or counter-clockwise for vshear < 0. In this case, vshear is the tangential velocity of the wall at
whatever radius has been defined.
Restart, fix_modify, output, run start/stop, minimize info:
This fix writes the shear friction state of atoms interacting with the wall to binary restart files, so that a
simulation can continue correctly if granular potentials with shear "history" effects are being used. See the
read_restart command for info on how to re-specify a fix in an input script that reads a restart file, so that the
operation of the fix continues in an uninterrupted fashion.
None of the fix_modify options are relevant to this fix. No global or per-atom quantities are stored by this fix
for access by various output commands. No parameter of this fix can be used with the start/stop keywords of
the run command. This fix is not invoked during energy minimization.
Restrictions:
This fix is part of the GRANULAR package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Any dimension (xyz) that has a granular wall must be non-periodic.
Related commands:
fix move, fix wall/gran/region, pair_style granular
Default: none
LAMMPS Users Manual
1144

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix wall/gran/region command
Syntax:
fix ID group-ID wall/gran/region fstyle Kn Kt gamma_n gamma_t xmu dampflag wallstyle regionID
ID, group-ID are documented in fix command• wall/region = style name of this fix command• fstyle = style of force interactions between particles and wall
possible choices: hooke, hooke/history, hertz/history
•
Kn = elastic constant for normal particle repulsion (force/distance units or pressure units - see
discussion below)
•
Kt = elastic constant for tangential contact (force/distance units or pressure units - see discussion
below)
•
gamma_n = damping coefficient for collisions in normal direction (1/time units or 1/time-distance
units - see discussion below)
•
gamma_t = damping coefficient for collisions in tangential direction (1/time units or 1/time-distance
units - see discussion below)
•
xmu = static yield criterion (unitless value between 0.0 and 1.0e4)• dampflag = 0 or 1 if tangential damping force is excluded or included• wallstyle = region (see fix wall/gran for options for other kinds of walls)• region-ID = region whose boundary will act as wall•
Examples:
fix wall all wall/gran/region hooke/history 1000.0 200.0 200.0 100.0 0.5 1 region myCone
Description:
Treat the surface of the geometric region defined by the region-ID as a bounding frictional wall which
interacts with nearby finite-size granular particles when they are close enough to touch the wall. See the fix
wall/region and fix wall/gran commands for related kinds of walls for non-granular particles and simpler wall
geometries, respectively.
Here are snapshots of example models using this command. Corresponding input scripts can be found in
examples/granregion. Click on the images to see a bigger picture. Movies of these simulations are here on the
Movies page of the LAMMPS web site.
LAMMPS Users Manual
1145

The distance between a particle and the region boundary is the distance to the nearest point on the region
surface. The force the wall exerts on the particle is along the direction between that point and the particle
center, which is the direction normal to the surface at that point. Note that if the region surface is comprised of
multiple "faces", then each face can exert a force on the particle if it is close enough. E.g. for region_style
block, a particle in the interior, near a corner of the block, could feel wall forces from 1, 2, or 3 faces of the
block.
Regions are defined using the region command. Note that the region volume can be interior or exterior to the
bounding surface, which will determine in which direction the surface interacts with particles, i.e. the
direction of the surface normal. The exception to this is if one or more open options are specified for the
region command, in which case particles interact with both the interior and exterior surfaces of regions.
Regions can either be primitive shapes (block, sphere, cylinder, etc) or combinations of primitive shapes
specified via the union or intersect region styles. These latter styles can be used to construct particle
containers with complex shapes. Regions can also move dynamically via the region command keywords
(move) and rotate, or change their shape by use of variables as inputs to the region command. If such a region
is used with this fix, then the region surface will move in time in the corresponding manner.
NOTE: As discussed on the region command doc page, regions in LAMMPS do not get wrapped across
periodic boundaries. It is up to you to ensure that the region location with respect to periodic or non-periodic
boundaries is specified appropriately via the region and boundary commands when using a region as a wall
that bounds particle motion.
NOTE: For primitive regions with sharp corners and/or edges (e.g. a block or cylinder), wall/particle forces
are computed accurately for both interior and exterior regions. For union and intersect regions, additional
sharp corners and edges may be present due to the intersection of the surfaces of 2 or more primitive volumes.
These corners and edges can be of two types: concave or convex. Concave points/edges are like the corners of
a cube as seen by particles in the interior of a cube. Wall/particle forces around these features are computed
correctly. Convex points/edges are like the corners of a cube as seen by particles exterior to the cube, i.e. the
points jut into the volume where particles are present. LAMMPS does NOT compute the location of these
convex points directly, and hence wall/particle forces in the cutoff volume around these points suffer from
inaccuracies. The basic problem is that the outward normal of the surface is not continuous at these points.
This can cause particles to feel no force (they don't "see" the wall) when in one location, then move a distance
epsilon, and suddenly feel a large force because they now "see" the wall. In a worst-case scenario, this can
blow particles out of the simulation box. Thus, as a general rule you should not use the fix wall/gran/region
command with union or interesect regions that have convex points or edges resulting from the
union/intersection (convex points/edges in the union/intersection due to a single sub-region are still OK).
LAMMPS Users Manual
1146
NOTE: Similarly, you should not define union or intersert regions for use with this command that share an
overlapping common face that is part of the overall outer boundary (interior boundary is OK), even if the face
is smooth. E.g. two regions of style block in a union region, where the two blocks overlap on one or more of
their faces. This is because LAMMPS discards points that are part of multiple sub-regions when calculating
wall/particle interactions, to avoid double-counting the interaction. Having two coincident faces could cause
the face to become invisible to the particles. The solution is to make the two faces differ by epsilon in their
position.
The nature of the wall/particle interactions are determined by the fstyle setting. It can be any of the styles
defined by the pair_style granular commands. Currently this is hooke, hooke/history, or hertz/history. The
equation for the force between the wall and particles touching it is the same as the corresponding equation on
the pair_style granular doc page, but the effective radius is calculated using the radius of the particle and the
radius of curvature of the wall at the contact point.
Specifically, delta = radius - r = overlap of particle with wall, m_eff = mass of particle, and RiRj/Ri+Rj is the
effective radius, with Rj replaced by the radius of curvature of the wall at the contact point. The radius of
curvature can be negative for a concave wall section, e.g. the interior of cylinder. For a flat wall, delta = radius
- r = overlap of particle with wall, m_eff = mass of particle, and the effective radius of contact is just the
radius of the particle.
The parameters Kn, Kt, gamma_n, gamma_t, xmu and dampflag have the same meaning and units as those
specified with the pair_style granular commands. This means a NULL can be used for either Kt or gamma_t
as described on that page. If a NULL is used for Kt, then a default value is used where Kt = 2/7 Kn. If a NULL
is used for gamma_t, then a default value is used where gamma_t = 1/2 gamma_n.
Note that you can choose a different force styles and/or different values for the 6 wall/particle coefficients
than for particle/particle interactions. E.g. if you wish to model the wall as a different material.
Restart, fix_modify, output, run start/stop, minimize info:
Similar to fix wall/gran command, this fix writes the shear friction state of atoms interacting with the wall to
binary restart files, so that a simulation can continue correctly if granular potentials with shear "history"
effects are being used. This fix also includes info about a moving region in the restart file. See the read_restart
command for info on how to re-specify a fix in an input script that reads a restart file, so that the operation of
the fix continues in an uninterrupted fashion.
NOTE: Information about region definitions is NOT included in restart files, as discussed on the read_restart
doc page. So you must re-define your region and if it is a moving region, define its motion attributes in a way
that is consistent with the simulation that wrote the restart file. In particular, if you want to change the region
motion attributes (e.g. its velocity), then you should ensure the position/orientation of the region at the initial
restart timestep is the same as it was on the timestep the restart file was written. If this is not possible, you
may need to ignore info in the restart file by defining a new fix wall/gran/region command in your restart
script, e.g. with a different fix ID. Or if you want to keep the shear history info but discard the region motion
information, you can use the same fix ID for fix wall/gran/region, but assign it a region with a different region
ID.
None of the fix_modify options are relevant to this fix. No global or per-atom quantities are stored by this fix
for access by various output commands. No parameter of this fix can be used with the start/stop keywords of
the run command. This fix is not invoked during energy minimization.
Restrictions:
LAMMPS Users Manual
1147

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix wall/piston command
Syntax:
fix ID group-ID wall/piston face ... keyword value ...
ID, group-ID are documented in fix command• wall/piston = style name of this fix command• face = zlo• zero or more keyword/value pairs may be appended• keyword = pos or vel or ramp or units
pos args = z
z = z coordinate at which the piston begins (distance units)
vel args = vz
vz = final velocity of the piston (velocity units)
ramp = use a linear velocity ramp from 0 to vz
temp args = target damp seed extent
target = target velocity for region immediately ahead of the piston
damp = damping parameter (time units)
seed = random number seed for langevin kicks
extent = extent of thermostated region (distance units)
units value = lattice or box
lattice = the wall position is defined in lattice units
box = the wall position is defined in simulation box units
•
Examples:
fix xwalls all wall/piston zlo
fix walls all wall/piston zlo pos 1.0 vel 10.0 units box
fix top all wall/piston zlo vel 10.0 ramp
Description:
Bound the simulation with a moving wall which reflect particles in the specified group and drive the system
with an effective infinite-mass piston capable of driving shock waves.
A momentum mirror technique is used, which means that if an atom (or the wall) moves such that an atom is
outside the wall on a timestep by a distance delta (e.g. due to fix nve), then it is put back inside the face by the
same delta, and the velocity relative to the moving wall is flipped in z. For instance, a stationary particle hit
with a piston wall with velocity vz, will end the timestep with a velocity of 2*vz.
Currently the face keyword can only be zlo. This creates a piston moving in the positive z direction. Particles
with z coordinate less than the wall position are reflected to a z coordinate greater than the wall position. If the
piston velocity is vpz and the particle velocity before reflection is vzi, the particle velocity after reflection is
-vzi + 2*vpz.
The initial position of the wall can be specified by the pos keyword.
The final velocity of the wall can be specified by the vel keyword
LAMMPS Users Manual
1149

The ramp keyword will cause the wall/piston to adjust the velocity linearly from zero velocity to vel over the
course of the run. If the ramp keyword is omitted then the wall/piston moves at a constant velocity defined by
vel.
The temp keyword will cause the region immediately in front of the wall/piston to be thermostated with a
Langevin thermostat. This region moves with the piston. The damping and kicking are measured in the
reference frame of the piston. So, a temperature of zero would mean all particles were moving at exactly the
speed of the wall/piston.
The units keyword determines the meaning of the distance units used to define a wall position, but only when
a numeric constant is used.
A box value selects standard distance units as defined by the units command, e.g. Angstroms for units = real
or metal. A lattice value means the distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacings.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
This fix style is part of the SHOCK package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
The face that has the wall/piston must be boundary type 's' (shrink-wrapped). The opposing face can be any
boundary type other than periodic.
A wall/piston should not be used with rigid bodies such as those defined by a "fix rigid" command. This is
because the wall/piston displaces atoms directly rather than exerting a force on them.
Related commands:
fix wall/reflect command, fix append/atoms command
Default:
The keyword defaults are pos = 0, vel = 0, units = lattice.
LAMMPS Users Manual
1150

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix wall/reflect command
fix wall/reflect/kk command
Syntax:
fix ID group-ID wall/reflect face arg ... keyword value ...
ID, group-ID are documented in fix command• wall/reflect = style name of this fix command• one or more face/arg pairs may be appended• face = xlo or xhi or ylo or yhi or zlo or zhi
xlo,ylo,zlo arg = EDGE or constant or variable
EDGE = current lo edge of simulation box
constant = number like 0.0 or -30.0 (distance units)
variable = equal-style variable like v_x or v_wiggle
xhi,yhi,zhi arg = EDGE or constant or variable
EDGE = current hi edge of simulation box
constant = number like 50.0 or 100.3 (distance units)
variable = equal-style variable like v_x or v_wiggle
•
zero or more keyword/value pairs may be appended• keyword = units
units value = lattice or box
lattice = the wall position is defined in lattice units
box = the wall position is defined in simulation box units
•
Examples:
fix xwalls all wall/reflect xlo EDGE xhi EDGE
fix walls all wall/reflect xlo 0.0 ylo 10.0 units box
fix top all wall/reflect zhi v_pressdown
Description:
Bound the simulation with one or more walls which reflect particles in the specified group when they attempt
to move thru them.
Reflection means that if an atom moves outside the wall on a timestep by a distance delta (e.g. due to fix nve),
then it is put back inside the face by the same delta, and the sign of the corresponding component of its
velocity is flipped.
When used in conjunction with fix nve and run_style verlet, the resultant time-integration algorithm is
equivalent to the primitive splitting algorithm (PSA) described by Bond. Because each reflection event
divides the corresponding timestep asymmetrically, energy conservation is only satisfied to O(dt), rather than
to O(dt^2) as it would be for velocity-Verlet integration without reflective walls.
Up to 6 walls or faces can be specified in a single command: xlo, xhi, ylo, yhi, zlo, zhi. A lo face reflects
particles that move to a coordinate less than the wall position, back in the hi direction. A hi face reflects
particles that move to a coordinate higher than the wall position, back in the lo direction.
LAMMPS Users Manual
1151
The position of each wall can be specified in one of 3 ways: as the EDGE of the simulation box, as a constant
value, or as a variable. If EDGE is used, then the corresponding boundary of the current simulation box is
used. If a numeric constant is specified then the wall is placed at that position in the appropriate dimension (x,
y, or z). In both the EDGE and constant cases, the wall will never move. If the wall position is a variable, it
should be specified as v_name, where name is an equal-style variable name. In this case the variable is
evaluated each timestep and the result becomes the current position of the reflecting wall. Equal-style
variables can specify formulas with various mathematical functions, and include thermo_style command
keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to specify a
time-dependent wall position.
The units keyword determines the meaning of the distance units used to define a wall position, but only when
a numeric constant or variable is used. It is not relevant when EDGE is used to specify a face position. In the
variable case, the variable is assumed to produce a value compatible with the units setting you specify.
A box value selects standard distance units as defined by the units command, e.g. Angstroms for units = real
or metal. A lattice value means the distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacings.
Here are examples of variable definitions that move the wall position in a time-dependent fashion using
equal-style variables.
variable ramp equal ramp(0,10)
fix 1 all wall/reflect xlo v_ramp
variable linear equal vdisplace(0,20)
fix 1 all wall/reflect xlo v_linear
variable wiggle equal swiggle(0.0,5.0,3.0)
fix 1 all wall/reflect xlo v_wiggle
variable wiggle equal cwiggle(0.0,5.0,3.0)
fix 1 all wall/reflect xlo v_wiggle
The ramp(lo,hi) function adjusts the wall position linearly from lo to hi over the course of a run. The
vdisplace(c0,velocity) function does something similar using the equation position = c0 + velocity*delta,
where delta is the elapsed time.
The swiggle(c0,A,period) function causes the wall position to oscillate sinusoidally according to this equation,
where omega = 2 PI / period:
position = c0 + A sin(omega*delta)
The cwiggle(c0,A,period) function causes the wall position to oscillate sinusoidally according to this
equation, which will have an initial wall velocity of 0.0, and thus may impose a gentler perturbation on the
particles:
position = c0 + A (1 - cos(omega*delta))
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
LAMMPS Users Manual
1152

These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix. No global or per-atom quantities are stored by this fix for access by various output commands. No
parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
Any dimension (xyz) that has a reflecting wall must be non-periodic.
A reflecting wall should not be used with rigid bodies such as those defined by a "fix rigid" command. This is
because the wall/reflect displaces atoms directly rather than exerts a force on them. For rigid bodies, use a soft
wall instead, such as fix wall/lj93. LAMMPS will flag the use of a rigid fix with fix wall/reflect with a
warning, but will not generate an error.
Related commands:
fix wall/lj93, fix oneway
Default: none
(Bond) Bond and Leimkuhler, SIAM J Sci Comput, 30, p 134 (2007).
LAMMPS Users Manual
1153

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix wall/region command
Syntax:
fix ID group-ID wall/region region-ID style epsilon sigma cutoff
ID, group-ID are documented in fix command• wall/region = style name of this fix command• region-ID = region whose boundary will act as wall• style = lj93 or lj126 or colloid or harmonic• epsilon = strength factor for wall-particle interaction (energy or energy/distance^2 units)• sigma = size factor for wall-particle interaction (distance units)• cutoff = distance from wall at which wall-particle interaction is cut off (distance units)•
Examples:
fix wall all wall/region mySphere lj93 1.0 1.0 2.5
Description:
Treat the surface of the geometric region defined by the region-ID as a bounding wall which interacts with
nearby particles according to the specified style.
The distance between a particle and the surface is the distance to the nearest point on the surface and the force
the wall exerts on the particle is along the direction between that point and the particle, which is the direction
normal to the surface at that point. Note that if the region surface is comprised of multiple "faces", then each
face can exert a force on the particle if it is close enough. E.g. for region_style block, a particle in the interior,
near a corner of the block, could feel wall forces from 1, 2, or 3 faces of the block.
Regions are defined using the region command. Note that the region volume can be interior or exterior to the
bounding surface, which will determine in which direction the surface interacts with particles, i.e. the
direction of the surface normal. The surface of the region only exerts forces on particles "inside" the region; if
a particle is "outside" the region it will generate an error, because it has moved through the wall.
Regions can either be primitive shapes (block, sphere, cylinder, etc) or combinations of primitive shapes
specified via the union or intersect region styles. These latter styles can be used to construct particle
containers with complex shapes. Regions can also change over time via the region command keywords
(move) and rotate. If such a region is used with this fix, then the of region surface will move over time in the
corresponding manner.
NOTE: As discussed on the region command doc page, regions in LAMMPS do not get wrapped across
periodic boundaries. It is up to you to insure that periodic or non-periodic boundaries are specified
appropriately via the boundary command when using a region as a wall that bounds particle motion. This also
means that if you embed a region in your simulation box and want it to repulse particles from its surface
(using the "side out" option in the region command), that its repulsive force will not be felt across a periodic
boundary.
NOTE: For primitive regions with sharp corners and/or edges (e.g. a block or cylinder), wall/particle forces
are computed accurately for both interior and exterior regions. For union and intersect regions, additional
LAMMPS Users Manual
1154
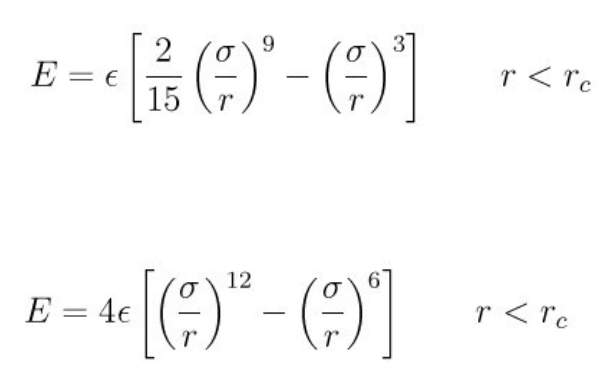
sharp corners and edges may be present due to the intersection of the surfaces of 2 or more primitive volumes.
These corners and edges can be of two types: concave or convex. Concave points/edges are like the corners of
a cube as seen by particles in the interior of a cube. Wall/particle forces around these features are computed
correctly. Convex points/edges are like the corners of a cube as seen by particles exterior to the cube, i.e. the
points jut into the volume where particles are present. LAMMPS does NOT compute the location of these
convex points directly, and hence wall/particle forces in the cutoff volume around these points suffer from
inaccuracies. The basic problem is that the outward normal of the surface is not continuous at these points.
This can cause particles to feel no force (they don't "see" the wall) when in one location, then move a distance
epsilon, and suddenly feel a large force because they now "see" the wall. In a worst-case scenario, this can
blow particles out of the simulation box. Thus, as a general rule you should not use the fix wall/gran/region
command with union or interesect regions that have convex points or edges resulting from the
union/intersection (convex points/edges in the union/intersection due to a single sub-region are still OK).
NOTE: Similarly, you should not define union or intersert regions for use with this command that share an
overlapping common face that is part of the overall outer boundary (interior boundary is OK), even if the face
is smooth. E.g. two regions of style block in a union region, where the two blocks overlap on one or more of
their faces. This is because LAMMPS discards points that are part of multiple sub-regions when calculating
wall/particle interactions, to avoid double-counting the interaction. Having two coincident faces could cause
the face to become invisible to the particles. The solution is to make the two faces differ by epsilon in their
position.
The energy of wall-particle interactions depends on the specified style.
For style lj93, the energy E is given by the 9/3 potential:
For style lj126, the energy E is given by the 12/6 potential:
For style colloid, the energy E is given by an integrated form of the pair_style colloid potential:
LAMMPS Users Manual
1155
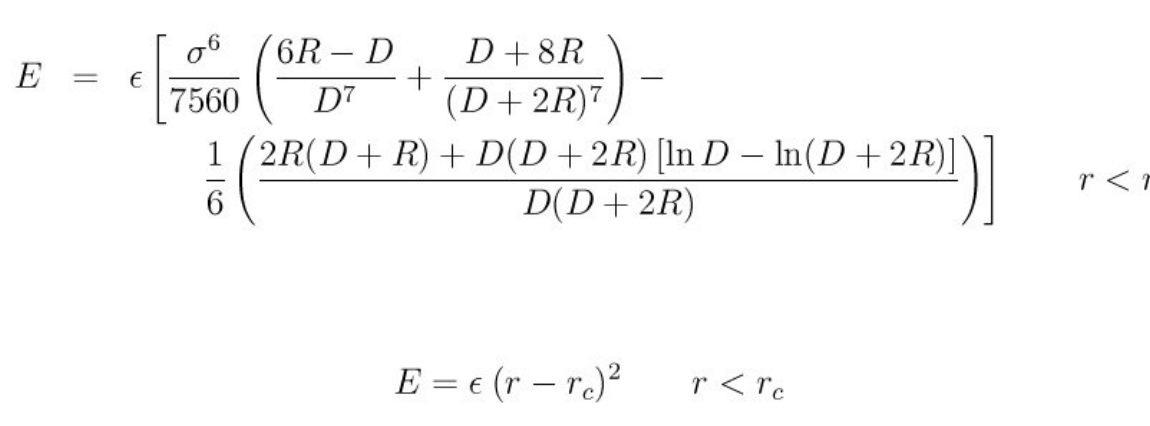
For style wall/harmonic, the energy E is given by a harmonic spring potential:
In all cases, r is the distance from the particle to the region surface, and Rc is the cutoff distance at which the
particle and surface no longer interact. The energy of the wall potential is shifted so that the wall-particle
interaction energy is 0.0 at the cutoff distance.
For the lj93 and lj126 styles, epsilon and sigma are the usual Lennard-Jones parameters, which determine the
strength and size of the particle as it interacts with the wall. Epsilon has energy units. Note that this epsilon
and sigma may be different than any epsilon or sigma values defined for a pair style that computes
particle-particle interactions.
The lj93 interaction is derived by integrating over a 3d half-lattice of Lennard-Jones 12/6 particles. The lj126
interaction is effectively a harder, more repulsive wall interaction.
For the colloid style, epsilon is effectively a Hamaker constant with energy units for the colloid-wall
interaction, R is the radius of the colloid particle, D is the distance from the surface of the colloid particle to
the wall (r-R), and sigma is the size of a constituent LJ particle inside the colloid particle. Note that the cutoff
distance Rc in this case is the distance from the colloid particle center to the wall.
The colloid interaction is derived by integrating over constituent LJ particles of size sigma within the colloid
particle and a 3d half-lattice of Lennard-Jones 12/6 particles of size sigma in the wall.
For the wall/harmonic style, epsilon is effectively the spring constant K, and has units (energy/distance^2).
The input parameter sigma is ignored. The minimum energy position of the harmonic spring is at the cutoff.
This is a repulsive-only spring since the interaction is truncated at the cutoff
NOTE: For all of the styles, you must insure that r is always > 0 for all particles in the group, or LAMMPS
will generate an error. This means you cannot start your simulation with particles on the region surface (r = 0)
or with particles on the wrong side of the region surface (r < 0). For the wall/lj93 and wall/lj126 styles, the
energy of the wall/particle interaction (and hence the force on the particle) blows up as r -> 0. The
wall/colloid style is even more restrictive, since the energy blows up as D = r-R -> 0. This means the
finite-size particles of radius R must be a distance larger than R from the region surface. The harmonic style is
a softer potential and does not blow up as r -> 0, but you must use a large enough epsilon that particles always
reamin on the correct side of the region surface (r > 0).
Restart, fix_modify, output, run start/stop, minimize info:
LAMMPS Users Manual
1156
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the energy of interaction between atoms and the
wall to the system's potential energy as part of thermodynamic output.
The fix_modify respa option is supported by this fix. This allows to set at which level of the r-RESPA
integrator the fix is adding its forces. Default is the outermost level.
This fix computes a global scalar energy and a global 3-length vector of forces, which can be accessed by
various output commands. The scalar energy is the sum of energy interactions for all particles interacting with
the wall represented by the region surface. The 3 vector quantities are the x,y,z components of the total force
acting on the wall due to the particles. The scalar and vector values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
NOTE: If you want the atom/wall interaction energy to be included in the total potential energy of the system
(the quantity being minimized), you MUST enable the fix_modify energy option for this fix.
Restrictions: none
Related commands:
fix wall/lj93, fix wall/lj126, fix wall/colloid, fix wall/gran
Default: none
LAMMPS Users Manual
1157

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
fix wall/srd command
Syntax:
fix ID group-ID wall/srd face arg ... keyword value ...
ID, group-ID are documented in fix command• wall/srd = style name of this fix command• one or more face/arg pairs may be appended• face = xlo or xhi or ylo or yhi or zlo or zhi
xlo,ylo,zlo arg = EDGE or constant or variable
EDGE = current lo edge of simulation box
constant = number like 0.0 or -30.0 (distance units)
variable = equal-style variable like v_x or v_wiggle
xhi,yhi,zhi arg = EDGE or constant or variable
EDGE = current hi edge of simulation box
constant = number like 50.0 or 100.3 (distance units)
variable = equal-style variable like v_x or v_wiggle
•
zero or more keyword/value pairs may be appended• keyword = units
units value = lattice or box
lattice = the wall position is defined in lattice units
box = the wall position is defined in simulation box units
•
Examples:
fix xwalls all wall/srd xlo EDGE xhi EDGE
fix walls all wall/srd xlo 0.0 ylo 10.0 units box
fix top all wall/srd zhi v_pressdown
Description:
Bound the simulation with one or more walls which interact with stochastic reaction dynamics (SRD)
particles as slip (smooth) or no-slip (rough) flat surfaces. The wall interaction is actually invoked via the fix
srd command, only on the group of SRD particles it defines, so the group setting for the fix wall/srd command
is ignored.
A particle/wall collision occurs if an SRD particle moves outside the wall on a timestep. This alters the
position and velocity of the SRD particle and imparts a force to the wall.
The collision and Tsrd settings specified via the fix srd command affect the SRD/wall collisions. A slip
setting for the collision keyword means that the tangential component of the SRD particle momentum is
preserved. Thus only a normal force is imparted to the wall. The normal component of the new SRD velocity
is sampled from a Gaussian distribution at temperature Tsrd.
For a noslip setting of the collision keyword, both the normal and tangential components of the new SRD
velocity are sampled from a Gaussian distribution at temperature Tsrd. Additionally, a new tangential
direction for the SRD velocity is chosen randomly. This collision style imparts both a normal and tangential
force to the wall.
LAMMPS Users Manual
1158

Up to 6 walls or faces can be specified in a single command: xlo, xhi, ylo, yhi, zlo, zhi. A lo face reflects
particles that move to a coordinate less than the wall position, back in the hi direction. A hi face reflects
particles that move to a coordinate higher than the wall position, back in the lo direction.
The position of each wall can be specified in one of 3 ways: as the EDGE of the simulation box, as a constant
value, or as a variable. If EDGE is used, then the corresponding boundary of the current simulation box is
used. If a numeric constant is specified then the wall is placed at that position in the appropriate dimension (x,
y, or z). In both the EDGE and constant cases, the wall will never move. If the wall position is a variable, it
should be specified as v_name, where name is an equal-style variable name. In this case the variable is
evaluated each timestep and the result becomes the current position of the reflecting wall. Equal-style
variables can specify formulas with various mathematical functions, and include thermo_style command
keywords for the simulation box parameters and timestep and elapsed time. Thus it is easy to specify a
time-dependent wall position.
NOTE: Because the trajectory of the SRD particle is tracked as it collides with the wall, you must insure that r
= distance of the particle from the wall, is always > 0 for SRD particles, or LAMMPS will generate an error.
This means you cannot start your simulation with SRD particles at the wall position coord (r = 0) or with
particles on the wrong side of the wall (r < 0).
NOTE: If you have 2 or more walls that come together at an edge or corner (e.g. walls in the x and y
dimensions), then be sure to set the overlap keyword to yes in the fix srd command, since the walls effectively
overlap when SRD particles collide with them. LAMMPS will issue a warning if you do not do this.
NOTE: The walls of this fix only interact with SRD particles, as defined by the fix srd command. If you are
simulating a mixture containing other kinds of particles, then you should typically use another wall command
to act on the other particles. Since SRD particles will be colliding both with the walls and the other particles,
it is important to insure that the other particle's finite extent does not overlap an SRD wall. If you do not do
this, you may generate errors when SRD particles end up "inside" another particle or a wall at the beginning
of a collision step.
The units keyword determines the meaning of the distance units used to define a wall position, but only when
a numeric constant is used. It is not relevant when EDGE or a variable is used to specify a face position.
A box value selects standard distance units as defined by the units command, e.g. Angstroms for units = real
or metal. A lattice value means the distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacings.
Here are examples of variable definitions that move the wall position in a time-dependent fashion using
equal-style variables.
variable ramp equal ramp(0,10)
fix 1 all wall/srd xlo v_ramp
variable linear equal vdisplace(0,20)
fix 1 all wall/srd xlo v_linear
variable wiggle equal swiggle(0.0,5.0,3.0)
fix 1 all wall/srd xlo v_wiggle
variable wiggle equal cwiggle(0.0,5.0,3.0)
fix 1 all wall/srd xlo v_wiggle
LAMMPS Users Manual
1159

The ramp(lo,hi) function adjusts the wall position linearly from lo to hi over the course of a run. The
displace(c0,velocity) function does something similar using the equation position = c0 + velocity*delta, where
delta is the elapsed time.
The swiggle(c0,A,period) function causes the wall position to oscillate sinusoidally according to this equation,
where omega = 2 PI / period:
position = c0 + A sin(omega*delta)
The cwiggle(c0,A,period) function causes the wall position to oscillate sinusoidally according to this
equation, which will have an initial wall velocity of 0.0, and thus may impose a gentler perturbation on the
particles:
position = c0 + A (1 - cos(omega*delta))
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files. None of the fix_modify options are relevant to
this fix.
This fix computes a global array of values which can be accessed by various output commands. The number
of rows in the array is equal to the number of walls defined by the fix. The number of columns is 3, for the
x,y,z components of force on each wall.
Note that an outward normal force on a wall will be a negative value for lo walls and a positive value for hi
walls. The array values calculated by this fix are "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command. This fix is not invoked
during energy minimization.
Restrictions:
Any dimension (xyz) that has an SRD wall must be non-periodic.
Related commands:
fix srd
Default: none
LAMMPS Users Manual
1160

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute ackland/atom command
Syntax:
compute ID group-ID ackland/atom
ID, group-ID are documented in compute command• ackland/atom = style name of this compute command•
Examples:
compute 1 all ackland/atom
Description:
Defines a computation that calculates the local lattice structure according to the formulation given in
(Ackland).
In contrast to the centro-symmetry parameter this method is stable against temperature boost, because it is
based not on the distance between particles but the angles. Therefore statistical fluctuations are averaged out a
little more. A comparison with the Common Neighbor Analysis metric is made in the paper.
The result is a number which is mapped to the following different lattice structures:
0 = UNKNOWN• 1 = BCC• 2 = FCC• 3 = HCP• 4 = ICO•
The neighbor list needed to compute this quantity is constructed each time the calculation is performed (i.e.
each time a snapshot of atoms is dumped). Thus it can be inefficient to compute/dump this quantity too
frequently or to have multiple compute/dump commands, each of which computes this quantity.-
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
Restrictions:
This compute is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
The per-atom vector values will be unitless since they are the integers defined above.
Related commands:
compute centro/atom
LAMMPS Users Manual
1161

Default: none
(Ackland) Ackland, Jones, Phys Rev B, 73, 054104 (2006).
LAMMPS Users Manual
1162

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute angle command
Syntax:
compute ID group-ID angle
ID, group-ID are documented in compute command• angle = style name of this compute command•
Examples:
compute 1 all angle
Description:
Define a computation that extracts the angle energy calculated by each of the angle sub-styles used in the
"angle_style hybrid" angle_hybrid.html command. These values are made accessible for output or further
processing by other commands. The group specified for this command is ignored.
This compute is useful when using angle_style hybrid if you want to know the portion of the total energy
contributed by one or more of the hybrid sub-styles.
Output info:
This compute calculates a global vector of length N where N is the number of sub_styles defined by the
angle_style hybrid command, which can be accessed by indices 1-N. These values can be used by any
command that uses global scalar or vector values from a compute as input. See this section for an overview of
LAMMPS output options.
The vector values are "extensive" and will be in energy units.
Restrictions: none
Related commands:
compute pe, compute pair
Default: none
LAMMPS Users Manual
1163

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute angle/local command
Syntax:
compute ID group-ID angle/local value1 value2 ...
ID, group-ID are documented in compute command• angle/local = style name of this compute command• one or more values may be appended• value = theta or eng
theta = tabulate angles
eng = tabulate angle energies
•
Examples:
compute 1 all angle/local theta
compute 1 all angle/local eng theta
Description:
Define a computation that calculates properties of individual angle interactions. The number of datums
generated, aggregated across all processors, equals the number of angles in the system, modified by the group
parameter as explained below.
The value theta is the angle for the 3 atoms in the interaction.
The value eng is the interaction energy for the angle.
The local data stored by this command is generated by looping over all the atoms owned on a processor and
their angles. An angle will only be included if all 3 atoms in the angle are in the specified compute group. Any
angles that have been broken (see the angle_style command) by setting their angle type to 0 are not included.
Angles that have been turned off (see the fix shake or delete_bonds commands) by setting their angle type
negative are written into the file, but their energy will be 0.0.
Note that as atoms migrate from processor to processor, there will be no consistent ordering of the entries
within the local vector or array from one timestep to the next. The only consistency that is guaranteed is that
the ordering on a particular timestep will be the same for local vectors or arrays generated by other compute
commands. For example, angle output from the compute property/local command can be combined with data
from this command and output by the dump local command in a consistent way.
Here is an example of how to do this:
compute 1 all property/local atype aatom1 aatom2 aatom3
compute 2 all angle/local theta eng
dump 1 all local 1000 tmp.dump index c_1[1] c_1[2] c_1[3] c_1[4] c_2[1] c_2[2]
Output info:
LAMMPS Users Manual
1164
This compute calculates a local vector or local array depending on the number of keywords. The length of the
vector or number of rows in the array is the number of angles. If a single keyword is specified, a local vector
is produced. If two or more keywords are specified, a local array is produced where the number of columns =
the number of keywords. The vector or array can be accessed by any command that uses local values from a
compute as input. See this section for an overview of LAMMPS output options.
The output for theta will be in degrees. The output for eng will be in energy units.
Restrictions: none
Related commands:
dump local, compute property/local
Default: none
LAMMPS Users Manual
1165

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute angmom/chunk command
Syntax:
compute ID group-ID angmom/chunk chunkID
ID, group-ID are documented in compute command• angmom/chunk = style name of this compute command• chunkID = ID of compute chunk/atom command•
Examples:
compute 1 fluid angmom/chunk molchunk
Description:
Define a computation that calculates the angular momentum of multiple chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
This compute calculates the 3 components of the angular momentum vector for each chunk, due to the
velocity/momentum of the individual atoms in the chunk around the center-of-mass of the chunk. The
calculation includes all effects due to atoms passing thru periodic boundaries.
Note that only atoms in the specified group contribute to the calculation. The compute chunk/atom command
defines its own group; atoms will have a chunk ID = 0 if they are not in that group, signifying they are not
assigned to a chunk, and will thus also not contribute to this calculation. You can specify the "all" group for
this command if you simply want to include atoms with non-zero chunk IDs.
NOTE: The coordinates of an atom contribute to the chunk's angular momentum in "unwrapped" form, by
using the image flags associated with each atom. See the dump custom command for a discussion of
"unwrapped" coordinates. See the Atoms section of the read_data command for a discussion of image flags
and how they are set for each atom. You can reset the image flags (e.g. to 0) before invoking this compute by
using the set image command.
The simplest way to output the results of the compute angmom/chunk calculation to a file is to use the fix
ave/time command, for example:
compute cc1 all chunk/atom molecule
compute myChunk all angmom/chunk cc1
fix 1 all ave/time 100 1 100 c_myChunk[*] file tmp.out mode vector
Output info:
LAMMPS Users Manual
1166
This compute calculates a global array where the number of rows = the number of chunks Nchunk as
calculated by the specified compute chunk/atom command. The number of columns = 3 for the 3 xyz
components of the angular momentum for each chunk. These values can be accessed by any command that
uses global array values from a compute as input. See Section 6.15 for an overview of LAMMPS output
options.
The array values are "intensive". The array values will be in mass-velocity-distance units.
Restrictions: none
Related commands:
variable angmom() function
Default: none
LAMMPS Users Manual
1167

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute basal/atom command
Syntax:
compute ID group-ID basal/atom
ID, group-ID are documented in compute command• basal/atom = style name of this compute command•
Examples:
compute 1 all basal/atom
Description:
Defines a computation that calculates the hexagonal close-packed "c" lattice vector for each atom in the
group. It does this by calculating the normal unit vector to the basal plane for each atom. The results enable
efficient identification and characterization of twins and grains in hexagonal close-packed structures.
The output of the compute is thus the 3 components of a unit vector associdate with each atom. The
components are set to 0.0 for atoms not in the group.
Details of the calculation are given in (Barrett).
The neighbor list needed to compute this quantity is constructed each time the calculation is performed (i.e.
each time a snapshot of atoms is dumped). Thus it can be inefficient to compute/dump this quantity too
frequently or to have multiple compute/dump commands, each of which computes this quantity.
An example input script that uses this compute is provided in examples/USER/misc/basal.
Output info:
This compute calculates a per-atom array with 3 columns, which can be accessed by indices 1-3 by any
command that uses per-atom values from a compute as input. See Section 6.15 for an overview of LAMMPS
output options.
The per-atom vector values are unitless since the 3 columns represent components of a unit vector.
Restrictions:
This compute is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
The output of this compute will be meaningless unless the atoms are on (or near) hcp lattice sites, since the
calculation assumes a well-defined basal plane.
Related commands:
compute centro/atom, compute ackland/atom
LAMMPS Users Manual
1168

Default: none
(Barrett) Barrett, Tschopp, El Kadiri, Scripta Mat. 66, p.666 (2012).
LAMMPS Users Manual
1169

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute body/local command
Syntax:
compute ID group-ID body/local input1 input2 ...
ID, group-ID are documented in compute command• body/local = style name of this compute command• one or more keywords may be appended• keyword = id or type or integer
id = atom ID of the body particle
type = atom type of the body particle
integer = 1,2,3,etc = index of fields defined by body style
•
Examples:
compute 1 all body/local type 1 2 3
compute 1 all body/local 3 6
Description:
Define a computation that calculates properties of individual body sub-particles. The number of datums
generated, aggregated across all processors, equals the number of body sub-particles plus the number of
non-body particles in the system, modified by the group parameter as explained below. See Section 6.14 of
the manual and the body doc page for more details on using body particles.
The local data stored by this command is generated by looping over all the atoms. An atom will only be
included if it is in the group. If the atom is a body particle, then its N sub-particles will be looped over, and it
will contribute N datums to the count of datums. If it is not a body particle, it will contribute 1 datum.
For both body particles and non-body particles, the id keyword will store the ID of the particle.
For both body particles and non-body particles, the type keyword will store the type of the particle.
The integer keywords mean different things for body and non-body particles. If the atom is not a body
particle, only its x, y, z coordinates can be referenced, using the integer keywords 1,2,3. Note that this means
that if you want to access more fields than this for body particles, then you cannot include non-body particles
in the group.
For a body particle, the integer keywords refer to fields calculated by the body style for each sub-particle. The
body style, as specified by the atom_style body, determines how many fields exist and what they are. See the
body doc page for details of the different styles.
Here is an example of how to output body information using the dump local command with this compute. If
fields 1,2,3 for the body sub-particles are x,y,z coordinates, then the dump file will be formatted similar to the
output of a dump atom or custom command.
compute 1 all body/local type 1 2 3
dump 1 all local 1000 tmp.dump index c_1[1] c_1[2] c_1[3] c_1[4]
LAMMPS Users Manual
1170
Output info:
This compute calculates a local vector or local array depending on the number of keywords. The length of the
vector or number of rows in the array is the number of datums as described above. If a single keyword is
specified, a local vector is produced. If two or more keywords are specified, a local array is produced where
the number of columns = the number of keywords. The vector or array can be accessed by any command that
uses local values from a compute as input. See this section for an overview of LAMMPS output options.
The units for output values depend on the body style.
Restrictions: none
Related commands:
dump local
Default: none
LAMMPS Users Manual
1171

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute bond command
Syntax:
compute ID group-ID bond
ID, group-ID are documented in compute command• bond = style name of this compute command•
Examples:
compute 1 all bond
Description:
Define a computation that extracts the bond energy calculated by each of the bond sub-styles used in the
bond_style hybrid command. These values are made accessible for output or further processing by other
commands. The group specified for this command is ignored.
This compute is useful when using bond_style hybrid if you want to know the portion of the total energy
contributed by one or more of the hybrid sub-styles.
Output info:
This compute calculates a global vector of length N where N is the number of sub_styles defined by the
bond_style hybrid command, which can be accessed by indices 1-N. These values can be used by any
command that uses global scalar or vector values from a compute as input. See this section for an overview of
LAMMPS output options.
The vector values are "extensive" and will be in energy units.
Restrictions: none
Related commands:
compute pe, compute pair
Default: none
LAMMPS Users Manual
1172

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute bond/local command
Syntax:
compute ID group-ID bond/local value1 value2 ...
ID, group-ID are documented in compute command• bond/local = style name of this compute command• one or more values may be appended• value = dist or engpot or force or engvib or engrot or engtrans or omega or velvib
dist = bond distance
engpot = bond potential energy
force = bond force
engvib = bond kinetic energy of vibration
engrot = bond kinetic energy of rotation
engtrans = bond kinetic energy of translation
omega = magnitude of bond angular velocity
velvib = vibrational velocity along the bond length
•
Examples:
compute 1 all bond/local engpot
compute 1 all bond/local dist engpot force
Description:
Define a computation that calculates properties of individual bond interactions. The number of datums
generated, aggregated across all processors, equals the number of bonds in the system, modified by the group
parameter as explained below.
The value dist is the current length of the bond.
The value engpot is the potential energy for the bond, based on the current separation of the pair of atoms in
the bond.
The value force is the magnitude of the force acting between the pair of atoms in the bond.
The remaining properties are all computed for motion of the two atoms relative to the center of mass (COM)
velocity of the 2 atoms in the bond.
The value engvib is the vibrational kinetic energy of the two atoms in the bond, which is simply 1/2 m1 v1^2
+ 1/2 m2 v2^2, where v1 and v2 are the magnitude of the velocity of the 2 atoms along the bond direction,
after the COM velocity has been subtracted from each.
The value engrot is the rotationsl kinetic energy of the two atoms in the bond, which is simply 1/2 m1 v1^2 +
1/2 m2 v2^2, where v1 and v2 are the magnitude of the velocity of the 2 atoms perpendicular to the bond
direction, after the COM velocity has been subtracted from each.
LAMMPS Users Manual
1173
The value engtrans is the translational kinetic energy associated with the motion of the COM of the system
itself, namely 1/2 (m1+m2) Vcm^2 where Vcm = magnitude of the velocity of the COM.
Note that these 3 kinetic energy terms are simply a partitioning of the summed kinetic energy of the 2 atoms
themselves. I.e. total KE = 1/2 m1 v1^2 + 1/2 m2 v2^2 = engvib + engrot + engtrans, where v1,v2 are the
magnitude of the velocities of the 2 atoms, without any adjustment for the COM velocity.
The value omega is the magnitude of the angular velocity of the two atoms around their COM position.
The value velvib is the magnitude of the relative velocity of the two atoms in the bond towards each other. A
negative value means the 2 atoms are moving toward each other; a positive value means they are moving
apart.
Note that all these properties are computed for the pair of atoms in a bond, whether the 2 atoms represent a
simple diatomic molecule, or are part of some larger molecule.
The local data stored by this command is generated by looping over all the atoms owned on a processor and
their bonds. A bond will only be included if both atoms in the bond are in the specified compute group. Any
bonds that have been broken (see the bond_style command) by setting their bond type to 0 are not included.
Bonds that have been turned off (see the fix shake or delete_bonds commands) by setting their bond type
negative are written into the file, but their energy will be 0.0.
Note that as atoms migrate from processor to processor, there will be no consistent ordering of the entries
within the local vector or array from one timestep to the next. The only consistency that is guaranteed is that
the ordering on a particular timestep will be the same for local vectors or arrays generated by other compute
commands. For example, bond output from the compute property/local command can be combined with data
from this command and output by the dump local command in a consistent way.
Here is an example of how to do this:
compute 1 all property/local btype batom1 batom2
compute 2 all bond/local dist engpot
dump 1 all local 1000 tmp.dump index c_1[*] c_2[*]
Output info:
This compute calculates a local vector or local array depending on the number of keywords. The length of the
vector or number of rows in the array is the number of bonds. If a single keyword is specified, a local vector is
produced. If two or more keywords are specified, a local array is produced where the number of columns =
the number of keywords. The vector or array can be accessed by any command that uses local values from a
compute as input. See this section for an overview of LAMMPS output options.
The output for dist will be in distance units. The output for velvib will be in velocity units. The output for
omega will be in velocity/distance units. The output for engtrans, engvib, engrot, and engpot will be in energy
units. The output for force will be in force units.
Restrictions: none
Related commands:
dump local, compute property/local
LAMMPS Users Manual
1174
Default: none
LAMMPS Users Manual
1175

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute centro/atom command
Syntax:
compute ID group-ID centro/atom lattice keyword value ...
ID, group-ID are documented in compute command centro/atom = style name of this compute command
lattice = fcc or bcc or N = # of neighbors per atom to include
•
zero or more keyword/value pairs may be appended• keyword = axes
axes value = no or yes
no = do not calculate 3 symmetry axes
yes = calculate 3 symmetry axes
Examples:
compute 1 all centro/atom fcc
compute 1 all centro/atom 8
Description:
Define a computation that calculates the centro-symmetry parameter for each atom in the group, for either
FCC or BCC lattices, depending on the choice of the lattice argument. In solid-state systems the
centro-symmetry parameter is a useful measure of the local lattice disorder around an atom and can be used
to characterize whether the atom is part of a perfect lattice, a local defect (e.g. a dislocation or stacking
fault), or at a surface.
The value of the centro-symmetry parameter will be 0.0 for atoms not in the specified compute group.
This parameter is computed using the following formula from (Kelchner)
where the N nearest neighbors of each atom are identified and Ri and Ri+N/2 are vectors from the central
atom to a particular pair of nearest neighbors. There are N*(N-1)/2 possible neighbor pairs that can
contribute to this formula. The quantity in the sum is computed for each, and the N/2 smallest are used.
This will typically be for pairs of atoms in symmetrically opposite positions with respect to the central
atom; hence the i+N/2 notation.
N is an input parameter, which should be set to correspond to the number of nearest neighbors in the
underlying lattice of atoms. If the keyword fcc or bcc is used, N is set to 12 and 8 respectively. More
generally, N can be set to a positive, even integer.
•
LAMMPS Users Manual
1176
For an atom on a lattice site, surrounded by atoms on a perfect lattice, the centro-symmetry parameter will
be 0. It will be near 0 for small thermal perturbations of a perfect lattice. If a point defect exists, the
symmetry is broken, and the parameter will be a larger positive value. An atom at a surface will have a
large positive parameter. If the atom does not have N neighbors (within the potential cutoff), then its
centro-symmetry parameter is set to 0.0.
If the keyword axes has the setting yes, then this compute also estimates three symmetry axes for each
atom's local neighborhood. The first two of these are the vectors joining the two pairs of neighbor atoms
with smallest contributions to the centrosymmetry parameter, i.e. the two most symmetric pairs of atoms.
The third vector is normal to the first two by the right-hand rule. All three vectors are normalized to unit
length. For FCC crystals, the first two vectors will lie along a direction, while the third vector will lie along
either a or direction. For HCP crystals, the first two vectors will lie along directions, while the third vector
will lie along . This provides a simple way to measure local orientation in HCP structures. In general, the
axes keyword can be used to estimate the orientation of symmetry axes in the neighborhood of any atom.
Only atoms within the cutoff of the pairwise neighbor list are considered as possible neighbors. Atoms not
in the compute group are included in the N neighbors used in this calculation.
The neighbor list needed to compute this quantity is constructed each time the calculation is performed (e.g.
each time a snapshot of atoms is dumped). Thus it can be inefficient to compute/dump this quantity too
frequently or to have multiple compute/dump commands, each with a centro/atom style.
Output info:
By default, this compute calculates the centrosymmetry value for each atom as a per-atom vector, which
can be accessed by any command that uses per-atom values from a compute as input. See Section 6.15 for
an overview of LAMMPS output options.
If the axes keyword setting is yes, then a per-atom array is calculated. The first column is the
centrosymmetry parameter. The next three columns are the x, y, and z components of the first symmetry
axis, followed by the second, and third symmetry axes in columns 5-7 and 8-10.
The centrosymmetry values are unitless values >= 0.0. Their magnitude depends on the lattice style due to
the number of contributing neighbor pairs in the summation in the formula above. And it depends on the
local defects surrounding the central atom, as described above. For the axes yes case, the vector
components are also unitless, since they represent spatial directions.
Here are typical centro-symmetry values, from a nanoindentation simulation into gold (FCC). These were
provided by Jon Zimmerman (Sandia):
Bulk lattice = 0
Dislocation core ~ 1.0 (0.5 to 1.25)
Stacking faults ~ 5.0 (4.0 to 6.0)
Free surface ~ 23.0
These values are *not* normalized by the square of the lattice parameter. If they were, normalized values
would be:
Bulk lattice = 0
Dislocation core ~ 0.06 (0.03 to 0.075)
Stacking faults ~ 0.3 (0.24 to 0.36)
Free surface ~ 1.38
LAMMPS Users Manual
1177

For BCC materials, the values for dislocation cores and free surfaces would be somewhat different, due to
their being only 8 neighbors instead of 12.
Restrictions: none
Related commands:
compute cna/atom
Default:
The default value for the optional keyword is axes = no.
(Kelchner) Kelchner, Plimpton, Hamilton, Phys Rev B, 58, 11085 (1998).
LAMMPS Users Manual
1178

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute chunk/atom command
Syntax:
compute ID group-ID chunk/atom style args keyword values ...
ID, group-ID are documented in compute command• chunk/atom = style name of this compute command
style = bin/1d or bin/2d or bin/3d or bin/sphere or type or molecule or compute/fix/variable
bin/1d args = dim origin delta
dim = x or y or z
origin = lower or center or upper or coordinate value (distance units)
delta = thickness of spatial bins in dim (distance units)
bin/2d args = dim origin delta dim origin delta
dim = x or y or z
origin = lower or center or upper or coordinate value (distance units)
delta = thickness of spatial bins in dim (distance units)
bin/3d args = dim origin delta dim origin delta dim origin delta
dim = x or y or z
origin = lower or center or upper or coordinate value (distance units)
delta = thickness of spatial bins in dim (distance units)
bin/sphere args = xorig yorig zorig rmin rmax nsbin
xorig,yorig,zorig = center point of sphere
srmin,srmax = bin from sphere radius rmin to rmax
nsbin = # of spherical shell bins between rmin and rmax
bin/cylinder args = dim origin delta c1 c2 rmin rmax ncbin
dim = x or y or z = axis of cylinder axis
origin = lower or center or upper or coordinate value (distance units)
delta = thickness of spatial bins in dim (distance units)
c1,c2 = coords of cylinder axis in other 2 dimensions (distance units)
crmin,crmax = bin from cylinder radius rmin to rmax (distance units)
ncbin = # of concentric circle bins between rmin and rmax
type args = none
molecule args = none
compute/fix/variable = c_ID, c_ID[I], f_ID, f_ID[I], v_name with no args
c_ID = per-atom vector calculated by a compute with ID
c_ID[I] = Ith column of per-atom array calculated by a compute with ID
f_ID = per-atom vector calculated by a fix with ID
f_ID[I] = Ith column of per-atom array calculated by a fix with ID
v_name = per-atom vector calculated by an atom-style variable with name
•
zero or more keyword/values pairs may be appended• keyword = region or nchunk or static or compress or bound or discard or pbc or units
region value = region-ID
region-ID = ID of region atoms must be in to be part of a chunk
nchunk value = once or every
once = only compute the number of chunks once
every = re-compute the number of chunks whenever invoked
limit values = 0 or Nc max or Nc exact
0 = no limit on the number of chunks
Nc max = limit number of chunks to be <= Nc
Nc exact = set number of chunks to exactly Nc
ids value = once or nfreq or every
once = assign chunk IDs to atoms only once, they persist thereafter
nfreq = assign chunk IDs to atoms only once every Nfreq steps (if invoked by fix ave/chunk which sets Nfreq)
•
LAMMPS Users Manual
1179
every = assign chunk IDs to atoms whenever invoked
compress value = yes or no
yes = compress chunk IDs to eliminate IDs with no atoms
no = do not compress chunk IDs even if some IDs have no atoms
discard value = yes or no or mixed
yes = discard atoms with out-of-range chunk IDs by assigning a chunk ID = 0
no = keep atoms with out-of-range chunk IDs by assigning a valid chunk ID
mixed = keep or discard such atoms according to spatial binning rule
bound values = x/y/z lo hi
x/y/z = x or y or z to bound sptial bins in this dimension
lo = lower or coordinate value (distance units)
hi = upper or coordinate value (distance units)
pbc value = no or yes
yes = use periodic distance for bin/sphere and bin/cylinder styles
units value = box or lattice or reduced
Examples:
compute 1 all chunk/atom type
compute 1 all chunk/atom bin/1d z lower 0.02 units reduced
compute 1 all chunk/atom bin/2d z lower 1.0 y 0.0 2.5
compute 1 all chunk/atom molecule region sphere nchunk once ids once compress yes
compute 1 all chunk/atom bin/sphere 5 5 5 2.0 5.0 5 discard yes
compute 1 all chunk/atom bin/cylinder z lower 2 10 10 2.0 5.0 3 discard yes
Description:
Define a computation that calculates an integer chunk ID from 1 to Nchunk for each atom in the group.
Values of chunk IDs are determined by the style of chunk, which can be based on atom type or molecule ID or
spatial binning or a per-atom property or value calculated by another compute, fix, or atom-style variable.
Per-atom chunk IDs can be used by other computes with "chunk" in their style name, such as compute
com/chunk or compute msd/chunk. Or they can be used by the fix ave/chunk command to sum and time
average a variety of per-atom properties over the atoms in each chunk. Or they can simply be accessed by any
command that uses per-atom values from a compute as input, as discussed in Section 6.15.
See Section 6.23 for an overview of how this compute can be used with a variety of other commands to
tabulate properties of a simulation. The howto section gives several examples of input script commands that
can be used to calculate interesting properties.
Conceptually it is important to realize that this compute does two simple things. First, it sets the value of
Nchunk = the number of chunks, which can be a constant value or change over time. Second, it assigns each
atom to a chunk via a chunk ID. Chunk IDs range from 1 to Nchunk inclusive; some chunks may have no
atoms assigned to them. Atoms that do not belong to any chunk are assigned a value of 0. Note that the two
operations are not always performed together. For example, spatial bins can be setup once (which sets
Nchunk), and atoms assigned to those bins many times thereafter (setting their chunk IDs).
All other commands in LAMMPS that use chunk IDs assume there are Nchunk number of chunks, and that
every atom is assigned to one of those chunks, or not assigned to any chunk.
There are many options for specifying for how and when Nchunk is calculated, and how and when chunk IDs
are assigned to atoms. The details depend on the chunk style and its args, as well as optional keyword
settings. They can also depend on whether a fix ave/chunk command is using this compute, since that
command requires Nchunk to remain static across windows of timesteps it specifies, while it accumulates
per-chunk averages.
LAMMPS Users Manual
1180

The details are described below.
The different chunk styles operate as follows. For each style, how it calculates Nchunk and assigns chunk IDs
to atoms is explained. Note that using the optional keywords can change both of those actions, as described
further below where the keywords are discussed.
The binning styles perform a spatial binning of atoms, and assign an atom the chunk ID corresponding to the
bin number it is in. Nchunk is set to the number of bins, which can change if the simulation box size changes.
This also depends on the setting of the units keyword; e.g. for reduced units the number of chunks may not
change even if the box size does.
The bin/1d, bin/2d, and bin/3d styles define bins as 1d layers (slabs), 2d pencils, or 3d boxes. The dim, origin,
and delta settings are specified 1, 2, or 3 times. For 2d or 3d bins, there is no restriction on specifying dim = x
before dim = y or z, or dim = y before dim = z. Bins in a particular dim have a bin size in that dimension given
by delta. In each dimension, bins are defined relative to a specified origin, which may be the lower/upper
edge of the simulation box (in that dimension), or its center point, or a specified coordinate value. Starting at
the origin, sufficient bins are created in both directions to completely span the simulation box or the bounds
specified by the optional bounds keyword.
For orthogonal simulation boxes, the bins are layers, pencils, or boxes aligned with the xyz coordinate axes.
For triclinic (non-orthogonal) simulation boxes, the bin faces are parallel to the tilted faces of the simulation
box. See this section of the manual for a discussion of the geometry of triclinic boxes in LAMMPS. As
described there, a tilted simulation box has edge vectors a,b,c. In that nomenclature, bins in the x dimension
have faces with normals in the "b" cross "c" direction. Bins in y have faces normal to the "a" cross "c"
direction. And bins in z have faces normal to the "a" cross "b" direction. Note that in order to define the size
and position of these bins in an unambiguous fashion, the units option must be set to reduced when using a
triclinic simulation box, as noted below.
The meaning of origin and delta for triclinic boxes is as follows. Consider a triclinic box with bins that are 1d
layers or slabs in the x dimension. No matter how the box is tilted, an origin of 0.0 means start layers at the
lower "b" cross "c" plane of the simulation box and an origin of 1.0 means to start layers at the upper "b"
cross "c" face of the box. A delta value of 0.1 in reduced units means there will be 10 layers from 0.0 to 1.0,
regardless of the current size or shape of the simulation box.
The bin/sphere style defines a set of spherical shell bins around the origin (xorig,yorig,zorig), using nsbin bins
with radii equally spaced between srmin and srmax. This is effectively a 1d vector of bins. For example, if
srmin = 1.0 and srmax = 10.0 and nsbin = 9, then the first bin spans 1.0 < r < 2.0, and the last bin spans 9.0 < r
10.0. The geometry of the bins is the same whether the simulation box is orthogonal or triclinic; i.e. the
spherical shells are not tilted or scaled differently in different dimensions to transform them into ellipsoidal
shells.
The bin/cylinder style defines bins for a cylinder oriented along the axis dim with the axis coordinates in the
other two radial dimensions at (c1,c2). For dim = x, c1/c2 = y/z; for dim = y, c1/c2 = x/z; for dim = z, c1/c2 =
x/y. This is effectively a 2d array of bins. The first dimension is along the cylinder axis, the second dimension
is radially outward from the cylinder axis. The bin size and positions along the cylinder axis are specified by
the origin and delta values, the same as for the bin/1d, bin/2d, and bin/3d styles. There are ncbin concentric
circle bins in the radial direction from the cylinder axis with radii equally spaced between crmin and crmax.
For example, if crmin = 1.0 and crmax = 10.0 and ncbin = 9, then the first bin spans 1.0 < r < 2.0, and the last
bin spans 9.0 < r 10.0. The geometry of the bins in the radial dimensions is the same whether the simulation
LAMMPS Users Manual
1181

box is orthogonal or triclinic; i.e. the concetric circles are not tilted or scaled differently in the two different
dimensions to transform them into ellipses.
The created bins (and hence the chunk IDs) are numbered consecutively from 1 to the number of bins =
Nchunk. For bin2d and bin3d, the numbering varies most rapidly in the first dimension (which could be x, y,
or z), next rapidly in the 2nd dimension, and most slowly in the 3rd dimension. For bin/sphere, the bin with
smallest radii is chunk 1 and the bni with largest radii is chunk Nchunk = ncbin. For bin/cylinder, the
numbering varies most rapidly in the dimension along the cylinder axis and most slowly in the radial
direction.
Each time this compute is invoked, each atom is mapped to a bin based on its current position. Note that
between reneighboring timesteps, atoms can move outside the current simulation box. If the box is periodic
(in that dimension) the atom is remapping into the periodic box for purposes of binning. If the box in not
periodic, the atom may have moved outside the bounds of all bins. If an atom is not inside any bin, the discard
keyword is used to determine how a chunk ID is assigned to the atom.
The type style uses the atom type as the chunk ID. Nchunk is set to the number of atom types defined for the
simulation, e.g. via the create_box or read_data commands.
The molecule style uses the molecule ID of each atom as its chunk ID. Nchunk is set to the largest chunk ID.
Note that this excludes molecule IDs for atoms which are not in the specified group or optional region.
There is no requirement that all atoms in a particular molecule are assigned the same chunk ID (zero or
non-zero), though you probably want that to be the case, if you wish to compute a per-molecule property.
LAMMPS will issue a warning if that is not the case, but only the first time that Nchunk is calculated.
Note that atoms with a molecule ID = 0, which may be non-molecular solvent atoms, have an out-of-range
chunk ID. These atoms are discarded (not assigned to any chunk) or assigned to Nchunk, depending on the
value of the discard keyword.
The compute/fix/variable styles set the chunk ID of each atom based on a quantity calculated and stored by a
compute, fix, or variable. In each case, it must be a per-atom quantity. In each case the referenced floating
point values are converted to an integer chunk ID as follows. The floating point value is truncated (rounded
down) to an integer value. If the integer value is <= 0, then a chunk ID of 0 is assigned to the atom. If the
integer value is > 0, it becomes the chunk ID to the atom. Nchunk is set to the largest chunk ID. Note that this
excludes atoms which are not in the specified group or optional region.
If the style begins with "c_", a compute ID must follow which has been previously defined in the input script.
If no bracketed integer is appended, the per-atom vector calculated by the compute is used. If a bracketed
integer is appended, the Ith column of the per-atom array calculated by the compute is used. Users can also
write code for their own compute styles and add them to LAMMPS.
If the style begins with "f_", a fix ID must follow which has been previously defined in the input script. If no
bracketed integer is appended, the per-atom vector calculated by the fix is used. If a bracketed integer is
appended, the Ith column of the per-atom array calculated by the fix is used. Note that some fixes only
produce their values on certain timesteps, which must be compatible with the timestep on which this compute
accesses the fix, else an error results. Users can also write code for their own fix styles and add them to
LAMMPS.
LAMMPS Users Manual
1182

If a value begins with "v_", a variable name for an atom or atomfile style variable must follow which has been
previously defined in the input script. Variables of style atom can reference thermodynamic keywords and
various per-atom attributes, or invoke other computes, fixes, or variables when they are evaluated, so this is a
very general means of generating per-atom quantities to treat as a chunk ID.
Normally, Nchunk = the number of chunks, is re-calculated every time this fix is invoked, though the value
may or may not change. As explained below, the nchunk keyword can be set to once which means Nchunk
will never change.
If a fix ave/chunk command uses this compute, it can also turn off the re-calculation of Nchunk for one or
more windows of timesteps. The extent of the windows, during which Nchunk is held constant, are
determined by the Nevery, Nrepeat, Nfreq values and the ave keyword setting that are used by the fix
ave/chunk command.
Specifically, if ave = one, then for each span of Nfreq timesteps, Nchunk is held constant between the first
timestep when averaging is done (within the Nfreq-length window), and the last timestep when averaging is
done (multiple of Nfreq). If ave = running or window, then Nchunk is held constant forever, starting on the
first timestep when the fix ave/chunk command invokes this compute.
Note that multiple fix ave/chunk commands can use the same compute chunk/atom compute. However, the
time windows they induce for holding Nchunk constant must be identical, else an error will be generated.
The various optional keywords operate as follows. Note that some of them function differently or are ignored
by different chunk styles. Some of them also have different default values, depending on the chunk style, as
listed below.
The region keyword applies to all chunk styles. If used, an atom must be in both the specified group and the
specified geometric region to be assigned to a chunk.
The nchunk keyword applies to all chunk styles. It specifies how often Nchunk is recalculated, which in turn
can affect the chunk IDs assigned to individual atoms.
If nchunk is set to once, then Nchunk is only calculated once, the first time this compute is invoked. If nchunk
is set to every, then Nchunk is re-calculated every time the compute is invoked. Note that, as described above,
the use of this compute by the fix ave/chunk command can override the every setting.
The default values for nchunk are listed below and depend on the chunk style and other system and keyword
settings. They attempt to represent typical use cases for the various chunk styles. The nchunk value can
always be set explicitly if desired.
The limit keyword can be used to limit the calculated value of Nchunk = the number of chunks. The limit is
applied each time Nchunk is calculated, which also limits the chunk IDs assigned to any atom. The limit
keyword is used by all chunk styles except the binning styles, which ignore it. This is because the number of
bins can be tailored using the bound keyword (described below) which effectively limits the size of Nchunk.
If limit is set to Nc = 0, then no limit is imposed on Nchunk, though the compress keyword can still be used to
reduce Nchunk, as described below.
LAMMPS Users Manual
1183

If Nc > 0, then the effect of the limit keyword depends on whether the compress keyword is also used with a
setting of yes, and whether the compress keyword is specified before the limit keyword or after.
In all cases, Nchunk is first calculated in the usual way for each chunk style, as described above.
First, here is what occurs if compress yes is not set. If limit is set to Nc max, then Nchunk is reset to the
smaller of Nchunk and Nc. If limit is set to Nc exact, then Nchunk is reset to Nc, whether the original Nchunk
was larger or smaller than Nc. If Nchunk shrank due to the limit setting, then atom chunk IDs > Nchunk will
be reset to 0 or Nchunk, depending on the setting of the discard keyword. If Nchunk grew, there will simply
be some chunks with no atoms assigned to them.
If compress yes is set, and the compress keyword comes before the limit keyword, the compression operation
is performed first, as described below, which resets Nchunk. The limit keyword is then applied to the new
Nchunk value, exactly as described in the preceding paragraph. Note that in this case, all atoms will end up
with chunk IDs <= Nc, but their original values (e.g. molecule ID or compute/fix/variable value) may have
been > Nc, because of the compression operation.
If compress yes is set, and the compress keyword comes after the limit keyword, then the limit value of Nc is
applied first to the uncompressed value of Nchunk, but only if Nc < Nchunk (whether Nc max or Nc exact is
used). This effectively means all atoms with chunk IDs > Nc have their chunk IDs reset to 0 or Nc, depending
on the setting of the discard keyword. The compression operation is then performed, which may shrink
Nchunk further. If the new Nchunk < Nc and limit = Nc exact is specified, then Nchunk is reset to Nc, which
results in extra chunks with no atoms assigned to them. Note that in this case, all atoms will end up with
chunk IDs <= Nc, and their original values (e.g. molecule ID or compute/fix/variable value) will also have
been <= Nc.
The ids keyword applies to all chunk styles. If the setting is once then the chunk IDs assigned to atoms the
first time this compute is invoked will be permanent, and never be re-computed.
If the setting is nfreq and if a fix ave/chunk command is using this compute, then in each of the Nchunk =
constant time windows (discussed above), the chunk ID's assigned to atoms on the first step of the time
window will persist until the end of the time window.
If the setting is every, which is the default, then chunk IDs are re-calculated on any timestep this compute is
invoked.
NOTE: If you want the persistent chunk-IDs calculated by this compute to be continuous when running from
a restart file, then you should use the same ID for this compute, as in the original run. This is so that the fix
this compute creates to store per-atom quantities will also have the same ID, and thus be initialized correctly
with chunk IDs from the restart file.
The compress keyword applies to all chunk styles and affects how Nchunk is calculated, which in turn affects
the chunk IDs assigned to each atom. It is useful for converting a "sparse" set of chunk IDs (with many IDs
that have no atoms assigned to them), into a "dense" set of IDs, where every chunk has one or more atoms
assigned to it.
Two possible use cases are as follows. If a large simulation box is mostly empty space, then the binning style
may produce many bins with no atoms. If compress is set to yes, only bins with atoms will be contribute to
Nchunk. Likewise, the molecule or compute/fix/variable styles may produce large Nchunk values. For
example, the compute cluster/atom command assigns every atom an atom ID for one of the atoms it is
LAMMPS Users Manual
1184

clustered with. For a million-atom system with 5 clusters, there would only be 5 unique chunk IDs, but the
largest chunk ID might be 1 million, resulting in Nchunk = 1 million. If compress is set to yes, Nchunk will be
reset to 5.
If compress is set to no, which is the default, no compression is done. If it is set to yes, all chunk IDs with no
atoms are removed from the list of chunk IDs, and the list is sorted. The remaining chunk IDs are renumbered
from 1 to Nchunk where Nchunk is the new length of the list. The chunk IDs assigned to each atom reflect the
new renumbering from 1 to Nchunk.
The original chunk IDs (before renumbering) can be accessed by the compute property/chunk command and
its id keyword, or by the fix ave/chunk command which outputs the original IDs as one of the columns in its
global output array. For example, using the "compute cluster/atom" command discussed above, the original 5
unique chunk IDs might be atom IDs (27,4982,58374,857838,1000000). After compression, these will be
renumbered to (1,2,3,4,5). The original values (27,...,1000000) can be output to a file by the fix ave/chunk
command, or by using the fix ave/time command in conjunction with the compute property/chunk command.
NOTE: The compression operation requires global communication across all processors to share their chunk
ID values. It can require large memory on every processor to store them, even after they are compressed, if
there are are a large number of unique chunk IDs with atoms assigned to them. It uses a STL map to find
unique chunk IDs and store them in sorted order. Each time an atom is assigned a compressed chunk ID, it
must access the STL map. All of this means that compression can be expensive, both in memory and CPU
time. The use of the limit keyword in conjunction with the compress keyword can affect these costs,
depending on which keyword is used first. So use this option with care.
The discard keyword applies to all chunk styles. It affects what chunk IDs are assigned to atoms that do not
match one of the valid chunk IDs from 1 to Nchunk. Note that it does not apply to atoms that are not in the
specified group or optionally specified region. Those atoms are always assigned a chunk ID = 0.
If the calculated chunk ID for an atom is not within the range 1 to Nchunk then it is a "discard" atom. Note
that Nchunk may have been shrunk by the limit keyword. Or the compress keyword may have eliminated
chunk IDs that were valid before the compression took place, and are now not in the compressed list. Also
note that for the molecule chunk style, if new molecules are added to the system, their chunk IDs may exceed
a previously calculated Nchunk. Likewise, evaluation of a compute/fix/variable on a later timestep may return
chunk IDs that are invalid for the previously calculated Nchunk.
All the chunk styles except the binning styles, must use discard set to either yes or no. If discard is set to yes,
which is the default, then every "discard" atom has its chunk ID set to 0. If discard is set to no, every
"discard" atom has its chunk ID set to Nchunk. I.e. it becomes part of the last chunk.
The binning styles use the discard keyword to decide whether to discard atoms outside the spatial domain
covered by bins, or to assign them to the bin they are nearest to.
For the bin/1d, bin/2d, bin/3d styles the details are as follows. If discard is set to yes, an out-of-domain atom
will have its chunk ID set to 0. If discard is set to no, the atom will have its chunk ID set to the first or last bin
in that dimension. If discard is set to mixed, which is the default, it will only have its chunk ID set to the first
or last bin if bins extend to the simulation box boundary in that dimension. This is the case if the bound
keyword settings are lower and upper, which is the default. If the bound keyword settings are numeric values,
then the atom will have its chunk ID set to 0 if it is outside the bounds of any bin. Note that in this case, it is
possible that the first or last bin extends beyond the numeric bounds settings, depending on the specified
origin. If this is the case, the chunk ID of the atom is only set to 0 if it is outside the first or last bin, not if it is
simply outside the numeric bounds setting.
LAMMPS Users Manual
1185

For the bin/sphere style the details are as follows. If discard is set to yes, an out-of-domain atom will have its
chunk ID set to 0. If discard is set to no or mixed, the atom will have its chunk ID set to the first or last bin,
i.e. the innermost or outermost spherical shell. If the distance of the atom from the origin is less than rmin, it
will be assigned to the first bin. If the distance of the atom from the origin is greater than rmax, it will be
assigned to the last bin.
For the bin/cylinder style the details are as follows. If discard is set to yes, an out-of-domain atom will have
its chunk ID set to 0. If discard is set to no, the atom will have its chunk ID set to the first or last bin in both
the radial and axis dimensions. If discard is set to mixed, which is the default, the radial dimension is treated
the same as for discard = no. But for the axis dimension, it will only have its chunk ID set to the first or last
bin if bins extend to the simulation box boundary in the axis dimension. This is the case if the bound keyword
settings are lower and upper, which is the default. If the bound keyword settings are numeric values, then the
atom will have its chunk ID set to 0 if it is outside the bounds of any bin. Note that in this case, it is possible
that the first or last bin extends beyond the numeric bounds settings, depending on the specified origin. If this
is the case, the chunk ID of the atom is only set to 0 if it is outside the first or last bin, not if it is simply
outside the numeric bounds setting.
If discard is set to no or mixed, the atom will have its chunk ID set to the first or last bin, i.e. the innermost or
outermost spherical shell. If the distance of the atom from the origin is less than rmin, it will be assigned to
the first bin. If the distance of the atom from the origin is greater than rmax, it will be assigned to the last bin.
The bound keyword only applies to the bin/1d, bin/2d, bin/3d styles and to the axis dimension of the
bin/cylinder style; otherwise it is ignored. It can be used one or more times to limit the extent of bin coverage
in a specified dimension, i.e. to only bin a portion of the box. If the lo setting is lower or the hi setting is
upper, the bin extent in that direction extends to the box boundary. If a numeric value is used for lo and/or hi,
then the bin extent in the lo or hi direction extends only to that value, which is assumed to be inside (or at
least near) the simulation box boundaries, though LAMMPS does not check for this. Note that using the
bound keyword typically reduces the total number of bins and thus the number of chunks Nchunk.
The pbc keyword only applies to the bin/sphere and bin/cylinder styles. If set to yes, the distance an atom is
from the sphere origin or cylinder axis is calculated in a minimum image sense with respect to periodic
dimensions, when determining which bin the atom is in. I.e. if x is a periodic dimension and the distance
between the atom and the sphere center in the x dimension is greater than 0.5 * simulation box length in x,
then a box length is subtracted to give a distance < 0.5 * simulation box length. This allosws the sphere or
cylinder center to be near a box edge, and atoms on the other side of the periodic box will still be close to the
center point/axis. Note that with a setting of yes, the outer sphere or cylinder radius must also be <= 0.5 *
simulation box length in any periodic dimension except for the cylinder axis dimension, or an error is
generated.
The units keyword only applies to the binning styles; otherwise it is ignored. For the bin/1d, bin/2d, bin/3d
styles, it determines the meaning of the distance units used for the bin sizes delta and for origin and bounds
values if they are coordinate values. For the bin/sphere style it determines the meaning of the distance units
used for xorig,yorig,zorig and the radii srmin and srmax. For the bin/cylinder style it determines the meaning
of the distance units used for delta,c1,c2 and the radii crmin and crmax.
For orthogonal simulation boxes, any of the 3 options may be used. For non-orthogonal (triclinic) simulation
boxes, only the reduced option may be used.
A box value selects standard distance units as defined by the units command, e.g. Angstroms for units = real
or metal. A lattice value means the distance units are in lattice spacings. The lattice command must have been
previously used to define the lattice spacing. A reduced value means normalized unitless values between 0
LAMMPS Users Manual
1186

and 1, which represent the lower and upper faces of the simulation box respectively. Thus an origin value of
0.5 means the center of the box in any dimension. A delta value of 0.1 means 10 bins span the box in that
dimension.
Note that for the bin/sphere style, the radii srmin and srmax are scaled by the lattice spacing or reduced value
of the x dimension.
Note that for the bin/cylinder style, the radii crmin and crmax are scaled by the lattice spacing or reduced
value of the 1st dimension perpendicular to the cylinder axis. E.g. y for an x-axis cylinder, x for a y-axis
cylinder, and x for a z-axis cylinder.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values are unitless chunk IDs, ranging from 1 to Nchunk (inclusive) for atoms assigned to
chunks, and 0 for atoms not belonging to a chunk.
Restrictions:
Even if the nchunk keyword is set to once, the chunk IDs assigned to each atom are not stored in a restart files.
This means you cannot expect those assignments to persist in a restarted simulation. Instead you must
re-specify this command and assign atoms to chunks when the restarted simulation begins.
Related commands:
fix ave/chunk, compute global/atom
Default:
The option defaults are as follows:
region = none• nchunk = every, if compress is yes, overriding other defaults listed here• nchunk = once, for type style• nchunk = once, for mol style if region is none• nchunk = every, for mol style if region is set• nchunk = once, for binning style if the simulation box size is static or units = reduced• nchunk = every, for binning style if the simulation box size is dynamic and units is lattice or box• nchunk = every, for compute/fix/variable style• limit = 0• ids = every• compress = no• discard = yes, for all styles except binning• discard = mixed, for binning styles• bound = lower and upper in all dimensions• pbc = no• units = lattice•
LAMMPS Users Manual
1187

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute cluster/atom command
Syntax:
compute ID group-ID cluster/atom cutoff
ID, group-ID are documented in compute command• cluster/atom = style name of this compute command• cutoff = distance within which to label atoms as part of same cluster (distance units)•
Examples:
compute 1 all cluster/atom 1.0
Description:
Define a computation that assigns each atom a cluster ID.
A cluster is defined as a set of atoms, each of which is within the cutoff distance from one or more other
atoms in the cluster. If an atom has no neighbors within the cutoff distance, then it is a 1-atom cluster. The ID
of every atom in the cluster will be the smallest atom ID of any atom in the cluster.
Only atoms in the compute group are clustered and assigned cluster IDs. Atoms not in the compute group are
assigned a cluster ID = 0.
The neighbor list needed to compute this quantity is constructed each time the calculation is performed (i.e.
each time a snapshot of atoms is dumped). Thus it can be inefficient to compute/dump this quantity too
frequently or to have multiple compute/dump commands, each of a cluster/atom style.
NOTE: If you have a bonded system, then the settings of special_bonds command can remove pairwise
interactions between atoms in the same bond, angle, or dihedral. This is the default setting for the
special_bonds command, and means those pairwise interactions do not appear in the neighbor list. Because
this fix uses the neighbor list, it also means those pairs will not be included when computing the clusters. This
does not apply when using long-range coulomb (coul/long, coul/msm, coul/wolf or similar. One way to get
around this would be to set special_bond scaling factors to very tiny numbers that are not exactly zero (e.g.
1.0e-50). Another workaround is to write a dump file, and use the rerun command to compute the clusters for
snapshots in the dump file. The rerun script can use a special_bonds command that includes all pairs in the
neighbor list.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values will be an ID > 0, as explained above.
Restrictions: none
Related commands:
LAMMPS Users Manual
1188

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute cna/atom command
Syntax:
compute ID group-ID cna/atom cutoff
ID, group-ID are documented in compute command• cna/atom = style name of this compute command• cutoff = cutoff distance for nearest neighbors (distance units)•
Examples:
compute 1 all cna/atom 3.08
Description:
Define a computation that calculates the CNA (Common Neighbor Analysis) pattern for each atom in the
group. In solid-state systems the CNA pattern is a useful measure of the local crystal structure around an
atom. The CNA methodology is described in (Faken) and (Tsuzuki).
Currently, there are five kinds of CNA patterns LAMMPS recognizes:
fcc = 1• hcp = 2• bcc = 3• icosohedral = 4• unknown = 5•
The value of the CNA pattern will be 0 for atoms not in the specified compute group. Note that normally a
CNA calculation should only be performed on mono-component systems.
The CNA calculation can be sensitive to the specified cutoff value. You should insure the appropriate nearest
neighbors of an atom are found within the cutoff distance for the presumed crystal structure. E.g. 12 nearest
neighbor for perfect FCC and HCP crystals, 14 nearest neighbors for perfect BCC crystals. These formulas
can be used to obtain a good cutoff distance:
LAMMPS Users Manual
1190

where a is the lattice constant for the crystal structure concerned and in the HCP case, x = (c/a) / 1.633, where
1.633 is the ideal c/a for HCP crystals.
Also note that since the CNA calculation in LAMMPS uses the neighbors of an owned atom to find the
nearest neighbors of a ghost atom, the following relation should also be satisfied:
where Rc is the cutoff distance of the potential, Rs is the skin distance as specified by the neighbor command,
and cutoff is the argument used with the compute cna/atom command. LAMMPS will issue a warning if this
is not the case.
The neighbor list needed to compute this quantity is constructed each time the calculation is performed (e.g.
each time a snapshot of atoms is dumped). Thus it can be inefficient to compute/dump this quantity too
frequently or to have multiple compute/dump commands, each with a cna/atom style.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values will be a number from 0 to 5, as explained above.
Restrictions: none
Related commands:
compute centro/atom
Default: none
(Faken) Faken, Jonsson, Comput Mater Sci, 2, 279 (1994).
(Tsuzuki) Tsuzuki, Branicio, Rino, Comput Phys Comm, 177, 518 (2007).
LAMMPS Users Manual
1191

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute com command
Syntax:
compute ID group-ID com
ID, group-ID are documented in compute command• com = style name of this compute command•
Examples:
compute 1 all com
Description:
Define a computation that calculates the center-of-mass of the group of atoms, including all effects due to
atoms passing thru periodic boundaries.
A vector of three quantities is calculated by this compute, which are the x,y,z coordinates of the center of
mass.
NOTE: The coordinates of an atom contribute to the center-of-mass in "unwrapped" form, by using the image
flags associated with each atom. See the dump custom command for a discussion of "unwrapped" coordinates.
See the Atoms section of the read_data command for a discussion of image flags and how they are set for each
atom. You can reset the image flags (e.g. to 0) before invoking this compute by using the set image command.
Output info:
This compute calculates a global vector of length 3, which can be accessed by indices 1-3 by any command
that uses global vector values from a compute as input. See this section for an overview of LAMMPS output
options.
The vector values are "intensive". The vector values will be in distance units.
Restrictions: none
Related commands:
compute com/chunk
Default: none
LAMMPS Users Manual
1192

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute com/chunk command
Syntax:
compute ID group-ID com/chunk chunkID
ID, group-ID are documented in compute command• com/chunk = style name of this compute command• chunkID = ID of compute chunk/atom command•
Examples:
compute 1 fluid com/chunk molchunk
Description:
Define a computation that calculates the center-of-mass for multiple chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
This compute calculates the x,y,z coordinates of the center-of-mass for each chunk, which includes all effects
due to atoms passing thru periodic boundaries.
Note that only atoms in the specified group contribute to the calculation. The compute chunk/atom command
defines its own group; atoms will have a chunk ID = 0 if they are not in that group, signifying they are not
assigned to a chunk, and will thus also not contribute to this calculation. You can specify the "all" group for
this command if you simply want to include atoms with non-zero chunk IDs.
NOTE: The coordinates of an atom contribute to the chunk's center-of-mass in "unwrapped" form, by using
the image flags associated with each atom. See the dump custom command for a discussion of "unwrapped"
coordinates. See the Atoms section of the read_data command for a discussion of image flags and how they
are set for each atom. You can reset the image flags (e.g. to 0) before invoking this compute by using the set
image command.
The simplest way to output the results of the compute com/chunk calculation to a file is to use the fix ave/time
command, for example:
compute cc1 all chunk/atom molecule
compute myChunk all com/chunk cc1
fix 1 all ave/time 100 1 100 c_myChunk[*] file tmp.out mode vector
Output info:
This compute calculates a global array where the number of rows = the number of chunks Nchunk as
calculated by the specified compute chunk/atom command. The number of columns = 3 for the x,y,z
LAMMPS Users Manual
1193
center-of-mass coordinates of each chunk. These values can be accessed by any command that uses global
array values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The array values are "intensive". The array values will be in distance units.
Restrictions: none
Related commands:
compute com
Default: none
LAMMPS Users Manual
1194

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute contact/atom command
Syntax:
compute ID group-ID contact/atom
ID, group-ID are documented in compute command• contact/atom = style name of this compute command•
Examples:
compute 1 all contact/atom
Description:
Define a computation that calculates the number of contacts for each atom in a group.
The contact number is defined for finite-size spherical particles as the number of neighbor atoms which
overlap the central particle, meaning that their distance of separation is less than or equal to the sum of the
radii of the two particles.
The value of the contact number will be 0.0 for atoms not in the specified compute group.
Output info:
This compute calculates a per-atom vector, whose values can be accessed by any command that uses per-atom
values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values will be a number >= 0.0, as explained above.
Restrictions:
This compute requires that atoms store a radius as defined by the atom_style sphere command.
Related commands:
compute coord/atom
Default: none
LAMMPS Users Manual
1195

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute coord/atom command
Syntax:
compute ID group-ID coord/atom cstyle args ...
ID, group-ID are documented in compute command• coord/atom = style name of this compute command• cstyle = cutoff or orientorder
cutoff args = cutoff typeN
cutoff = distance within which to count coordination neighbors (distance units)
typeN = atom type for Nth coordination count (see asterisk form below)
orientorder args = orientorderID threshold
orientorderID = ID of an orientorder/atom compute
threshold = minimum value of the product of two "connected" atoms
•
Examples:
compute 1 all coord/atom cutoff 2.0
compute 1 all coord/atom cutoff 6.0 1 2
compute 1 all coord/atom cutoff 6.0 2*4 5*8 *
compute 1 all coord/atom orientorder 2 0.5
Description:
This compute performs calculations between neighboring atoms to determine a coordination value. The
specific calculation and the meaning of the resulting value depend on the cstyle keyword used.
The cutoff cstyle calculates one or more traditional coordination numbers for each atom. A coordination
number is defined as the number of neighbor atoms with specified atom type(s) that are within the specified
cutoff distance from the central atom. Atoms not in the specified group are included in the coordination
number tally.
The typeN keywords allow specification of which atom types contribute to each coordination number. One
coordination number is computed for each of the typeN keywords listed. If no typeN keywords are listed, a
single coordination number is calculated, which includes atoms of all types (same as the "*" format, see
below).
The typeN keywords can be specified in one of two ways. An explicit numeric value can be used, as in the 2nd
example above. Or a wild-card asterisk can be used to specify a range of atom types. This takes the form "*"
or "*n" or "n*" or "m*n". If N = the number of atom types, then an asterisk with no numeric values means all
types from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A trailing asterisk means all
types from n to N (inclusive). A middle asterisk means all types from m to n (inclusive).
The orientorder cstyle calculates the number of "connected" neighbor atoms J around each central atom I. For
this cstyle, connected is defined by the orientational order parameter calculated by the compute
orientorder/atom command. This cstyle thus allows one to apply the ten Wolde's criterion to identify
crystal-like atoms in a system, as discussed in ten Wolde.
LAMMPS Users Manual
1196

The ID of the previously specified compute orientorder/atom command is specified as orientorderID. The
compute must invoke its components option to calculate components of the Ybar_lm vector for each atoms, as
described in its documentation. Note that orientorder/atom compute defines its own criteria for identifying
neighboring atoms. If the scalar product (Ybar_lm(i),Ybar_lm(j)), calculated by the orientorder/atom compute
is larger than the specified threshold, then I and J are connected, and the coordination value of I is
incremented by one.
For all cstyle settings, all coordination values will be 0.0 for atoms not in the specified compute group.
The neighbor list needed to compute this quantity is constructed each time the calculation is performed (i.e.
each time a snapshot of atoms is dumped). Thus it can be inefficient to compute/dump this quantity too
frequently.
NOTE: If you have a bonded system, then the settings of special_bonds command can remove pairwise
interactions between atoms in the same bond, angle, or dihedral. This is the default setting for the
special_bonds command, and means those pairwise interactions do not appear in the neighbor list. Because
this fix uses the neighbor list, it also means those pairs will not be included in the coordination count. One
way to get around this, is to write a dump file, and use the rerun command to compute the coordination for
snapshots in the dump file. The rerun script can use a special_bonds command that includes all pairs in the
neighbor list.
Output info:
For cstyle cutoff, this compute can calculate a per-atom vector or array. If single type1 keyword is specified
(or if none are specified), this compute calculates a per-atom vector. If multiple typeN keywords are specified,
this compute calculates a per-atom array, with N columns.
For cstyle orientorder, this compute calculates a per-atom vector.
These values can be accessed by any command that uses per-atom values from a compute as input. See
Section 6.15 for an overview of LAMMPS output options.
The per-atom vector or array values will be a number >= 0.0, as explained above.
Restrictions: none
Related commands:
compute cluster/atom compute orientorder/atom
Default: none
(tenWolde) P. R. ten Wolde, M. J. Ruiz-Montero, D. Frenkel, J. Chem. Phys. 104, 9932 (1996).
LAMMPS Users Manual
1197

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute damage/atom command
Syntax:
compute ID group-ID damage/atom
ID, group-ID are documented in compute command• damage/atom = style name of this compute command•
Examples:
compute 1 all damage/atom
Description:
Define a computation that calculates the per-atom damage for each atom in a group. This is a quantity relevant
for Peridynamics models. See this document for an overview of LAMMPS commands for Peridynamics
modeling.
The "damage" of a Peridymaics particles is based on the bond breakage between the particle and its neighbors.
If all the bonds are broken the particle is considered to be fully damaged.
See the PDLAMMPS user guide for a formal definition of "damage" and more details about Peridynamics as
it is implemented in LAMMPS.
This command can be used with all the Peridynamic pair styles.
The damage value will be 0.0 for atoms not in the specified compute group.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values are unitless numbers (damage) >= 0.0.
Restrictions:
This compute is part of the PERI package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
compute dilatation/atom, compute plasticity/atom
Default: none
LAMMPS Users Manual
1198

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute dihedral command
Syntax:
compute ID group-ID dihedral
ID, group-ID are documented in compute command• dihedral = style name of this compute command•
Examples:
compute 1 all dihedral
Description:
Define a computation that extracts the dihedral energy calculated by each of the dihedral sub-styles used in
the dihedral_style hybrid command. These values are made accessible for output or further processing by
other commands. The group specified for this command is ignored.
This compute is useful when using dihedral_style hybrid if you want to know the portion of the total energy
contributed by one or more of the hybrid sub-styles.
Output info:
This compute calculates a global vector of length N where N is the number of sub_styles defined by the
dihedral_style hybrid command. which can be accessed by indices 1-N. These values can be used by any
command that uses global scalar or vector values from a compute as input. See this section for an overview of
LAMMPS output options.
The vector values are "extensive" and will be in energy units.
Restrictions: none
Related commands:
compute pe, compute pair
Default: none
LAMMPS Users Manual
1199

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute dihedral/local command
Syntax:
compute ID group-ID dihedral/local value1 value2 ...
ID, group-ID are documented in compute command• dihedral/local = style name of this compute command• one or more values may be appended• value = phi
phi = tabulate dihedral angles
•
Examples:
compute 1 all dihedral/local phi
Description:
Define a computation that calculates properties of individual dihedral interactions. The number of datums
generated, aggregated across all processors, equals the number of angles in the system, modified by the group
parameter as explained below.
The value phi is the dihedral angle, as defined in the diagram on the dihedral_style doc page.
The local data stored by this command is generated by looping over all the atoms owned on a processor and
their dihedrals. A dihedral will only be included if all 4 atoms in the dihedral are in the specified compute
group.
Note that as atoms migrate from processor to processor, there will be no consistent ordering of the entries
within the local vector or array from one timestep to the next. The only consistency that is guaranteed is that
the ordering on a particular timestep will be the same for local vectors or arrays generated by other compute
commands. For example, dihedral output from the compute property/local command can be combined with
data from this command and output by the dump local command in a consistent way.
Here is an example of how to do this:
compute 1 all property/local dtype datom1 datom2 datom3 datom4
compute 2 all dihedral/local phi
dump 1 all local 1000 tmp.dump index c_1[1] c_1[2] c_1[3] c_1[4] c_1[5] c_2[1]
Output info:
This compute calculates a local vector or local array depending on the number of keywords. The length of the
vector or number of rows in the array is the number of dihedrals. If a single keyword is specified, a local
vector is produced. If two or more keywords are specified, a local array is produced where the number of
columns = the number of keywords. The vector or array can be accessed by any command that uses local
values from a compute as input. See this section for an overview of LAMMPS output options.
LAMMPS Users Manual
1200

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute dilatation/atom command
Syntax:
compute ID group-ID dilatation/atom
ID, group-ID are documented in compute command• dilation/atom = style name of this compute command•
Examples:
compute 1 all dilatation/atom
Description:
Define a computation that calculates the per-atom dilatation for each atom in a group. This is a quantity
relevant for Peridynamics models. See this document for an overview of LAMMPS commands for
Peridynamics modeling.
For small deformation, dilatation of is the measure of the volumetric strain.
The dilatation "theta" for each peridynamic particle I is calculated as a sum over its neighbors with unbroken
bonds, where the contribution of the IJ pair is a function of the change in bond length (versus the initial length
in the reference state), the volume fraction of the particles and an influence function. See the PDLAMMPS
user guide for a formal definition of dilatation.
This command can only be used with a subset of the Peridynamic pair styles: peri/lps, peri/ves and peri/eps.
The dilatation value will be 0.0 for atoms not in the specified compute group.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section_howto 15 for an overview of LAMMPS output options.
The per-atom vector values are unitless numbers (theta) >= 0.0.
Restrictions:
This compute is part of the PERI package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
compute damage/atom, compute plasticity/atom
Default: none
LAMMPS Users Manual
1202

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute dipole/chunk command
Syntax:
compute ID group-ID dipole/chunk chunkID charge-correction
ID, group-ID are documented in compute command• dipole/chunk = style name of this compute command• chunkID = ID of compute chunk/atom command• charge-correction = mass or geometry, use COM or geometric center for charged chunk correction
(optional)
•
Examples:
compute 1 fluid dipole/chunk molchunk
compute dw water dipole/chunk 1 geometry
Description:
Define a computation that calculates the dipole vector and total dipole for multiple chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
This compute calculates the x,y,z coordinates of the dipole vector and the total dipole moment for each chunk,
which includes all effects due to atoms passing thru periodic boundaries. For chunks with a net charge the
resulting dipole is made position independent by subtracting the position vector of the center of mass or
geometric center times the net charge from the computed dipole vector.
Note that only atoms in the specified group contribute to the calculation. The compute chunk/atom command
defines its own group; atoms will have a chunk ID = 0 if they are not in that group, signifying they are not
assigned to a chunk, and will thus also not contribute to this calculation. You can specify the "all" group for
this command if you simply want to include atoms with non-zero chunk IDs.
NOTE: The coordinates of an atom contribute to the chunk's dipole in "unwrapped" form, by using the image
flags associated with each atom. See the dump custom command for a discussion of "unwrapped" coordinates.
See the Atoms section of the read_data command for a discussion of image flags and how they are set for each
atom. You can reset the image flags (e.g. to 0) before invoking this compute by using the set image command.
The simplest way to output the results of the compute com/chunk calculation to a file is to use the fix ave/time
command, for example:
compute cc1 all chunk/atom molecule
compute myChunk all dipole/chunk cc1
fix 1 all ave/time 100 1 100 c_myChunk[*] file tmp.out mode vector
LAMMPS Users Manual
1203
Output info:
This compute calculates a global array where the number of rows = the number of chunks Nchunk as
calculated by the specified compute chunk/atom command. The number of columns = 4 for the x,y,z dipole
vector components and the total dipole of each chunk. These values can be accessed by any command that
uses global array values from a compute as input. See Section 6.15 for an overview of LAMMPS output
options.
The array values are "intensive". The array values will be in dipole units, i.e. charge units times distance units.
Restrictions: none
Related commands:
compute com/chunk
Default: none
LAMMPS Users Manual
1204

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute displace/atom command
Syntax:
compute ID group-ID displace/atom
ID, group-ID are documented in compute command• displace/atom = style name of this compute command•
Examples:
compute 1 all displace/atom
Description:
Define a computation that calculates the current displacement of each atom in the group from its original
coordinates, including all effects due to atoms passing thru periodic boundaries.
A vector of four quantities per atom is calculated by this compute. The first 3 elements of the vector are the
dx,dy,dz displacements. The 4th component is the total displacement, i.e. sqrt(dx*dx + dy*dy + dz*dz).
The displacement of an atom is from its original position at the time the compute command was issued. The
value of the displacement will be 0.0 for atoms not in the specified compute group.
NOTE: Initial coordinates are stored in "unwrapped" form, by using the image flags associated with each
atom. See the dump custom command for a discussion of "unwrapped" coordinates. See the Atoms section of
the read_data command for a discussion of image flags and how they are set for each atom. You can reset the
image flags (e.g. to 0) before invoking this compute by using the set image command.
NOTE: If you want the quantities calculated by this compute to be continuous when running from a restart
file, then you should use the same ID for this compute, as in the original run. This is so that the fix this
compute creates to store per-atom quantities will also have the same ID, and thus be initialized correctly with
time=0 atom coordinates from the restart file.
Output info:
This compute calculates a per-atom array with 4 columns, which can be accessed by indices 1-4 by any
command that uses per-atom values from a compute as input. See Section 6.15 for an overview of LAMMPS
output options.
The per-atom array values will be in distance units.
Restrictions: none
Related commands:
compute msd, dump custom, fix store/state
Default: none
LAMMPS Users Manual
1205
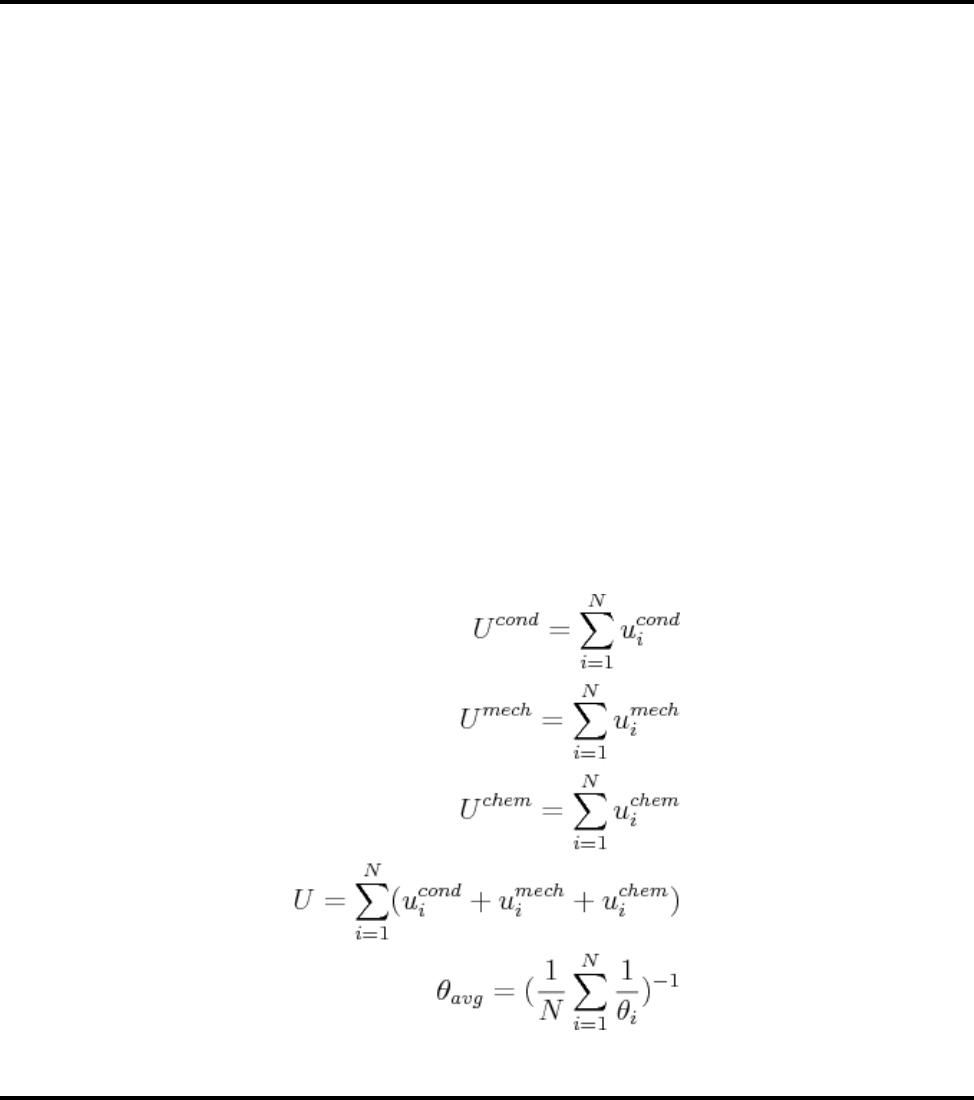
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute dpd command
Syntax:
compute ID group-ID dpd
ID, group-ID are documented in compute command• dpd = style name of this compute command•
Examples:
compute 1 all dpd
Description:
Define a computation that accumulates the total internal conductive energy (U_cond), the total internal
mechanical energy (U_mech), the total chemical energy (U_chem) and the harmonic average of the internal
temperature (dpdTheta) for the entire system of particles. See the compute dpd/atom command if you want
per-particle internal energies and internal temperatures.
The system internal properties are computed according to the following relations:
where N is the number of particles in the system
Output info:
This compute calculates a global vector of length 5 (U_cond, U_mech, U_chem, dpdTheta, N_particles),
which can be accessed by indices 1-5. See this section for an overview of LAMMPS output options.
The vector values will be in energy and temperature units.
LAMMPS Users Manual
1206

Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This command also requires use of the atom_style dpd command.
Related commands:
compute dpd/atom, thermo_style
Default: none
(Larentzos) J.P. Larentzos, J.K. Brennan, J.D. Moore, and W.D. Mattson, "LAMMPS Implementation of
Constant Energy Dissipative Particle Dynamics (DPD-E)", ARL-TR-6863, U.S. Army Research Laboratory,
Aberdeen Proving Ground, MD (2014).
LAMMPS Users Manual
1207

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute dpd/atom command
Syntax:
compute ID group-ID dpd/atom
ID, group-ID are documented in compute command• dpd/atom = style name of this compute command•
Examples:
compute 1 all dpd/atom
Description:
Define a computation that accesses the per-particle internal conductive energy (u_cond), internal mechanical
energy (u_mech), internal chemical energy (u_chem) and internal temperatures (dpdTheta) for each particle in
a group. See the compute dpd command if you want the total internal conductive energy, the total internal
mechanical energy, the total chemical energy and average internal temperature of the entire system or group
of dpd particles.
Output info:
This compute calculates a per-particle array with 4 columns (u_cond, u_mech, u_chem, dpdTheta), which can
be accessed by indices 1-4 by any command that uses per-particle values from a compute as input. See
Section 6.15 for an overview of LAMMPS output options.
The per-particle array values will be in energy (u_cond, u_mech, u_chem) and temperature (dpdTheta) units.
Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This command also requires use of the atom_style dpd command.
Related commands:
dump custom, compute dpd
Default: none
(Larentzos) J.P. Larentzos, J.K. Brennan, J.D. Moore, and W.D. Mattson, "LAMMPS Implementation of
Constant Energy Dissipative Particle Dynamics (DPD-E)", ARL-TR-6863, U.S. Army Research Laboratory,
Aberdeen Proving Ground, MD (2014).
LAMMPS Users Manual
1208

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute erotate/asphere command
Syntax:
compute ID group-ID erotate/asphere
ID, group-ID are documented in compute command• erotate/asphere = style name of this compute command•
Examples:
compute 1 all erotate/asphere
Description:
Define a computation that calculates the rotational kinetic energy of a group of aspherical particles. The
aspherical particles can be ellipsoids, or line segments, or triangles. See the atom_style and read_data
commands for descriptions of these options.
For all 3 types of particles, the rotational kinetic energy is computed as 1/2 I w^2, where I is the inertia tensor
for the aspherical particle and w is its angular velocity, which is computed from its angular momentum if
needed.
NOTE: For 2d models, ellipsoidal particles are treated as ellipsoids, not ellipses, meaning their moments of
inertia will be the same as in 3d.
Output info:
This compute calculates a global scalar (the KE). This value can be used by any command that uses a global
scalar value from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The scalar value calculated by this compute is "extensive". The scalar value will be in energy units.
Restrictions:
This compute requires that ellipsoidal particles atoms store a shape and quaternion orientation and angular
momentum as defined by the atom_style ellipsoid command.
This compute requires that line segment particles atoms store a length and orientation and angular velocity as
defined by the atom_style line command.
This compute requires that triangular particles atoms store a size and shape and quaternion orientation and
angular momentum as defined by the atom_style tri command.
All particles in the group must be finite-size. They cannot be point particles.
Related commands: none
compute erotate/sphere
LAMMPS Users Manual
1209
Default: none
LAMMPS Users Manual
1210

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute erotate/rigid command
Syntax:
compute ID group-ID erotate/rigid fix-ID
ID, group-ID are documented in compute command• erotate/rigid = style name of this compute command• fix-ID = ID of rigid body fix•
Examples:
compute 1 all erotate/rigid myRigid
Description:
Define a computation that calculates the rotational kinetic energy of a collection of rigid bodies, as defined by
one of the fix rigid command variants.
The rotational energy of each rigid body is computed as 1/2 I Wbody^2, where I is the inertia tensor for the
rigid body, and Wbody is its angular velocity vector. Both I and Wbody are in the frame of reference of the
rigid body, i.e. I is diagonalized.
The fix-ID should be the ID of one of the fix rigid commands which defines the rigid bodies. The group
specified in the compute command is ignored. The rotational energy of all the rigid bodies defined by the fix
rigid command in included in the calculation.
Output info:
This compute calculates a global scalar (the summed rotational energy of all the rigid bodies). This value can
be used by any command that uses a global scalar value from a compute as input. See Section 6.15 for an
overview of LAMMPS output options.
The scalar value calculated by this compute is "extensive". The scalar value will be in energy units.
Restrictions:
This compute is part of the RIGID package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
compute ke/rigid
Default: none
LAMMPS Users Manual
1211

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute erotate/sphere command
Syntax:
compute ID group-ID erotate/sphere
ID, group-ID are documented in compute command• erotate/sphere = style name of this compute command•
Examples:
compute 1 all erotate/sphere
Description:
Define a computation that calculates the rotational kinetic energy of a group of spherical particles.
The rotational energy is computed as 1/2 I w^2, where I is the moment of inertia for a sphere and w is the
particle's angular velocity.
NOTE: For 2d models, particles are treated as spheres, not disks, meaning their moment of inertia will be the
same as in 3d.
Output info:
This compute calculates a global scalar (the KE). This value can be used by any command that uses a global
scalar value from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The scalar value calculated by this compute is "extensive". The scalar value will be in energy units.
Restrictions:
This compute requires that atoms store a radius and angular velocity (omega) as defined by the atom_style
sphere command.
All particles in the group must be finite-size spheres or point particles. They cannot be aspherical. Point
particles will not contribute to the rotational energy.
Related commands:
compute erotate/asphere
Default: none
LAMMPS Users Manual
1212

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute erotate/sphere/atom command
Syntax:
compute ID group-ID erotate/sphere/atom
ID, group-ID are documented in compute command• erotate/sphere/atom = style name of this compute command•
Examples:
compute 1 all erotate/sphere/atom
Description:
Define a computation that calculates the rotational kinetic energy for each particle in a group.
The rotational energy is computed as 1/2 I w^2, where I is the moment of inertia for a sphere and w is the
particle's angular velocity.
NOTE: For 2d models, particles are treated as spheres, not disks, meaning their moment of inertia will be the
same as in 3d.
The value of the rotational kinetic energy will be 0.0 for atoms not in the specified compute group or for point
particles with a radius = 0.0.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values will be in energy units.
Restrictions: none
Related commands:
dump custom
Default: none
LAMMPS Users Manual
1213

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute event/displace command
Syntax:
compute ID group-ID event/displace threshold
ID, group-ID are documented in compute command• event/displace = style name of this compute command• threshold = minimum distance any particle must move to trigger an event (distance units)•
Examples:
compute 1 all event/displace 0.5
Description:
Define a computation that flags an "event" if any particle in the group has moved a distance greater than the
specified threshold distance when compared to a previously stored reference state (i.e. the previous event).
This compute is typically used in conjunction with the prd and tad commands, to detect if a transition to a new
minimum energy basin has occurred.
This value calculated by the compute is equal to 0 if no particle has moved far enough, and equal to 1 if one or
more particles have moved further than the threshold distance.
NOTE: If the system is undergoing significant center-of-mass motion, due to thermal motion, an external
force, or an initial net momentum, then this compute will not be able to distinguish that motion from local
atom displacements and may generate "false positives."
Output info:
This compute calculates a global scalar (the flag). This value can be used by any command that uses a global
scalar value from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The scalar value will be a 0 or 1 as explained
above.
Restrictions:
This command can only be used if LAMMPS was built with the REPLICA package. See the Making
LAMMPS section for more info on packages.
Related commands:
prd, tad
Default: none
LAMMPS Users Manual
1214

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute fep command
Syntax:
compute ID group-ID fep temp attribute args ... keyword value ...
ID, group-ID are documented in the compute command• fep = name of this compute command• temp = external temperature (as specified for constant-temperature run)• one or more attributes with args may be appended• attribute = pair or atom
pair args = pstyle pparam I J v_delta
pstyle = pair style name, e.g. lj/cut
pparam = parameter to perturb
I,J = type pair(s) to set parameter for
v_delta = variable with perturbation to apply (in the units of the parameter)
atom args = aparam I v_delta
aparam = parameter to perturb
I = type to set parameter for
v_delta = variable with perturbation to apply (in the units of the parameter)
•
zero or more keyword/value pairs may be appended• keyword = tail or volume
tail value = no or yes
no = ignore tail correction to pair energies (usually small in fep)
yes = include tail correction to pair energies
volume value = no or yes
no = ignore volume changes (e.g. in NVE or NVT trajectories)
yes = include volume changes (e.g. in NpT trajectories)
•
Examples:
compute 1 all fep 298 pair lj/cut epsilon 1 * v_delta pair lj/cut sigma 1 * v_delta volume yes
compute 1 all fep 300 atom charge 2 v_delta
Description:
Apply a perturbation to parameters of the interaction potential and recalculate the pair potential energy
without changing the atomic coordinates from those of the reference, unperturbed system. This compute can
be used to calculate free energy differences using several methods, such as free-energy perturbation (FEP),
finite-difference thermodynamic integration (FDTI) or Bennet's acceptance ratio method (BAR).
The potential energy of the system is decomposed in three terms: a background term corresponding to
interaction sites whose parameters remain constant, a reference term \(U_0\) corresponding to the initial
interactions of the atoms that will undergo perturbation, and a term \(U_1\) corresponding to the final
interactions of these atoms:
A coupling parameter \(\lambda\) varying from 0 to 1 connects the reference and perturbed systems:
LAMMPS Users Manual
1215

It is possible but not necessary that the coupling parameter (or a function thereof) appears as a multiplication
factor of the potential energy. Therefore, this compute can apply perturbations to interaction parameters that
are not directly proportional to the potential energy (e.g. \(\sigma\) in Lennard-Jones potentials).
This command can be combined with fix adapt to perform multistage free-energy perturbation calculations
along stepwise alchemical transformations during a simulation run:
This compute is suitable for the finite-difference thermodynamic integration (FDTI) method (Mezei), which is
based on an evaluation of the numerical derivative of the free energy by a perturbation method using a very
small \(\delta\):
where \(w_i\) are weights of a numerical quadrature. The fix adapt command can be used to define the stages
of \(\lambda\) at which the derivative is calculated and averaged.
The compute fep calculates the exponential Boltzmann term and also the potential energy difference \(U_1
-U_0\). By choosing a very small perturbation \(\delta\) the thermodynamic integration method can be
implemented using a numerical evaluation of the derivative of the potential energy with respect to \(\lambda\):
Another technique to calculate free energy differences is the acceptance ratio method (Bennet), which can be
implemented by calculating the potential energy differences with \(\delta\) = 1.0 on both the forward and
reverse routes:
The value of the free energy difference is determined by numerical root finding to establish the equality.
Concerning the choice of how the atomic parameters are perturbed in order to setup an alchemical
transformation route, several strategies are available, such as single-topology or double-topology strategies
(Pearlman). The latter does not require modification of bond lengths, angles or other internal coordinates.
NOTES: This compute command does not take kinetic energy into account, therefore the masses of the
particles should not be modified between the reference and perturbed states, or along the alchemical
transformation route. This compute command does not change bond lengths or other internal coordinates
(Boresch, Karplus).
LAMMPS Users Manual
1216

The pair attribute enables various parameters of potentials defined by the pair_style and pair_coeff commands
to be changed, if the pair style supports it.
The pstyle argument is the name of the pair style. For example, pstyle could be specified as "lj/cut". The
pparam argument is the name of the parameter to change. This is a (non-exclusive) list of pair styles and
parameters that can be used with this compute. See the doc pages for individual pair styles and their energy
formulas for the meaning of these parameters:
lj/cut epsilon,sigma type pairs
lj/cut/coul/cut epsilon,sigma type pairs
lj/cut/coul/long epsilon,sigma type pairs
lj/cut/soft epsilon,sigma,lambda type pairs
coul/cut/soft lambda type pairs
coul/long/soft lambda type pairs
lj/cut/coul/cut/soft epsilon,sigma,lambda type pairs
lj/cut/coul/long/soft epsilon,sigma,lambda type pairs
lj/cut/tip4p/long/soft epsilon,sigma,lambda type pairs
tip4p/long/soft lambda type pairs
lj/charmm/coul/long/soft epsilon,sigma,lambda type pairs
born a,b,c type pairs
buck a,c type pairs
Note that it is easy to add new potentials and their parameters to this list. All it typically takes is adding an
extract() method to the pair_*.cpp file associated with the potential.
Similar to the pair_coeff command, I and J can be specified in one of two ways. Explicit numeric values can
be used for each, as in the 1st example above. I <= J is required. LAMMPS sets the coefficients for the
symmetric J,I interaction to the same values. A wild-card asterisk can be used in place of or in conjunction
with the I,J arguments to set the coefficients for multiple pairs of atom types. This takes the form "*" or "*n"
or "n*" or "m*n". If N = the number of atom types, then an asterisk with no numeric values means all types
from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A trailing asterisk means all types
from n to N (inclusive). A middle asterisk means all types from m to n (inclusive). Note that only type pairs
with I <= J are considered; if asterisks imply type pairs where J < I, they are ignored.
If pair_style hybrid or hybrid/overlay is being used, then the pstyle will be a sub-style name. You must specify
I,J arguments that correspond to type pair values defined (via the pair_coeff command) for that sub-style.
The v_name argument for keyword pair is the name of an equal-style variable which will be evaluated each
time this compute is invoked. It should be specified as v_name, where name is the variable name.
The atom attribute enables atom properties to be changed. The aparam argument is the name of the parameter
to change. This is the current list of atom parameters that can be used with this compute:
charge = charge on particle•
The v_name argument for keyword pair is the name of an equal-style variable which will be evaluated each
time this compute is invoked. It should be specified as v_name, where name is the variable name.
LAMMPS Users Manual
1217

The tail keyword controls the calculation of the tail correction to "van der Waals" pair energies beyond the
cutoff, if this has been activated via the pair_modify command. If the perturbation is small, the tail
contribution to the energy difference between the reference and perturbed systems should be negligible.
If the keyword volume = yes, then the Boltzmann term is multiplied by the volume so that correct ensemble
averaging can be performed over trajectories during which the volume fluctuates or changes (Allen and
Tildesley):
Output info:
This compute calculates a global vector of length 3 which contains the energy difference ( \(U_1-U_0\) ) as
c_ID[1], the Boltzmann factor \(\exp(-(U_1-U_0)/kT)\), or \(V \exp(-(U_1-U_0)/kT)\), as c_ID[2] and the
volume of the simulation box \(V\) as c_ID[3]. \(U_1\) is the pair potential energy obtained with the perturbed
parameters and \(U_0\) is the pair potential energy obtained with the unperturbed parameters. The energies
include kspace terms if these are used in the simulation.
These output results can be used by any command that uses a global scalar or vector from a compute as input.
See Section 6.15 for an overview of LAMMPS output options. For example, the computed values can be
averaged using fix ave/time.
The values calculated by this compute are "extensive".
Restrictions:
This compute is distributed as the USER-FEP package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Related commands:
fix adapt/fep, fix ave/time, pair_style lj/soft/coul/soft
Default:
The option defaults are tail = no, volume = no.
(Pearlman) Pearlman, J Chem Phys, 98, 1487 (1994)
(Mezei) Mezei, J Chem Phys, 86, 7084 (1987)
(Bennet) Bennet, J Comput Phys, 22, 245 (1976)
LAMMPS Users Manual
1218
(BoreschKarplus) Boresch and Karplus, J Phys Chem A, 103, 103 (1999)
(AllenTildesley) Allen and Tildesley, Computer Simulation of Liquids, Oxford University Press (1987)
LAMMPS Users Manual
1219

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute global/atom command
Syntax:
compute ID group-ID style index input1 input2 ...
ID, group-ID are documented in compute command• global/atom = style name of this compute command• index = c_ID, c_ID[N], f_ID, f_ID[N], v_name
c_ID = per-atom vector calculated by a compute with ID
c_ID[I] = Ith column of per-atom array calculated by a compute with ID
f_ID = per-atom vector calculated by a fix with ID
f_ID[I] = Ith column of per-atom array calculated by a fix with ID
v_name = per-atom vector calculated by an atom-style variable with name
•
one or more inputs can be listed• input = c_ID, c_ID[N], f_ID, f_ID[N], v_name
c_ID = global vector calculated by a compute with ID
c_ID[I] = Ith column of global array calculated by a compute with ID, I can include wildcard (see below)
f_ID = global vector calculated by a fix with ID
f_ID[I] = Ith column of global array calculated by a fix with ID, I can include wildcard (see below)
v_name = global vector calculated by a vector-style variable with name
•
Examples:
compute 1 all global/atom c_chunk c_com[1\] c_com[2\] c_com[3\]
compute 1 all global/atom c_chunk c_com[*\]
Description:
Define a calculation that assigns global values to each atom from vectors or arrays of global values. The
specified index parameter is used to determine which global value is assigned to each atom.
The index parameter must reference a per-atom vector or array from a compute or fix or the evaluation of an
atom-style variable. Each input value must reference a global vector or array from a compute or fix or the
evaluation of an vector-style variable. Details are given below.
The index value for an atom is used as a index I (from 1 to N) into the vector associated with each of the input
values. The Ith value from the input vector becomes one output value for that atom. If the atom is not in the
specified group, or the index I < 1 or I > M, where M is the actual length of the input vector, then an output
value of 0.0 is assigned to the atom.
An example of how this command is useful, is in the context of "chunks" which are static or dynamic subsets
of atoms. The compute chunk/atom command assigns unique chunk IDs to each atom. It's output can be used
as the index parameter for this command. Various other computes with "chunk" in their style name, such as
compute com/chunk or compute msd/chunk, calculate properties for each chunk. The output of these
commands are global vectors or arrays, with one or more values per chunk, and can be used as input values
for this command. This command will then assign the global chunk value to each atom in the chunk,
producing a per-atom vector or per-atom array as output. The per-atom values can then be output to a dump
file or used by any command that uses per-atom values from a compute as input, as discussed in Section 6.15.
LAMMPS Users Manual
1220

As a concrete example, these commands will calculate the displacement of each atom from the center-of-mass
of the molecule it is in, and dump those values to a dump file. In this case, each molecule is a chunk.
compute cc1 all chunk/atom molecule
compute myChunk all com/chunk cc1
compute prop all property/atom xu yu zu
compute glob all global/atom c_cc1 c_myChunk[*]
variable dx atom c_prop[1]-c_glob[1]
variable dy atom c_prop[2]-c_glob[2]
variable dz atom c_prop[3]-c_glob[3]
variable dist atom sqrt(v_dx*v_dx+v_dy*v_dy+v_dz*v_dz)
dump 1 all custom 100 tmp.dump id xu yu zu c_glob[1] c_glob[2] c_glob[3] &
v_dx v_dy v_dz v_dist
dump_modify 1 sort id
You can add these commands to the bench/in.chain script to see how they work.
Note that for input values from a compute or fix, the bracketed index I can be specified using a wildcard
asterisk with the index to effectively specify multiple values. This takes the form "*" or "*n" or "n*" or
"m*n". If N = the size of the vector (for mode = scalar) or the number of columns in the array (for mode =
vector), then an asterisk with no numeric values means all indices from 1 to N. A leading asterisk means all
indices from 1 to n (inclusive). A trailing asterisk means all indices from n to N (inclusive). A middle asterisk
means all indices from m to n (inclusive).
Using a wildcard is the same as if the individual columns of the array had been listed one by one. E.g. these 2
compute global/atom commands are equivalent, since the compute com/chunk command creates a global
array with 3 columns:
compute cc1 all chunk/atom molecule
compute com all com/chunk cc1
compute 1 all global/atom c_cc1 c_com[1] c_com[2] c_com[3]
compute 1 all global/atom c_cc1 c_com[*]
This section explains the index parameter. Note that it must reference per-atom values, as contrasted with the
input values which must reference global values.
Note that all of these options generate floating point values. When they are used as an index into the specified
input vectors, they simple rounded down to convert the value to integer indices. The final values should range
from 1 to N (inclusive), since they are used to access values from N-length vectors.
If index begins with "c_", a compute ID must follow which has been previously defined in the input script.
The compute must generate per-atom quantities. See the individual compute doc page for details. If no
bracketed integer is appended, the per-atom vector calculated by the compute is used. If a bracketed integer is
appended, the Ith column of the per-atom array calculated by the compute is used. Users can also write code
for their own compute styles and add them to LAMMPS. See the discussion above for how I can be specified
with a wildcard asterisk to effectively specify multiple values.
If index begins with "f_", a fix ID must follow which has been previously defined in the input script. The Fix
must generate per-atom quantities. See the individual fix doc page for details. Note that some fixes only
produce their values on certain timesteps, which must be compatible with when compute global/atom
references the values, else an error results. If no bracketed integer is appended, the per-atom vector calculated
by the fix is used. If a bracketed integer is appended, the Ith column of the per-atom array calculated by the
fix is used. Users can also write code for their own fix style and add them to LAMMPS. See the discussion
LAMMPS Users Manual
1221
above for how I can be specified with a wildcard asterisk to effectively specify multiple values.
If index begins with "v_", a variable name must follow which has been previously defined in the input script.
It must be an atom-style variable. Atom-style variables can reference thermodynamic keywords and various
per-atom attributes, or invoke other computes, fixes, or variables when they are evaluated, so this is a very
general means of generating per-atom quantities to use as index.
This section explains the kinds of input values that can be used. Note that inputs reference global values, as
contrasted with the index parameter which must reference per-atom values.
If a value begins with "c_", a compute ID must follow which has been previously defined in the input script.
The compute must generate a global vector or array. See the individual compute doc page for details. If no
bracketed integer is appended, the vector calculated by the compute is used. If a bracketed integer is
appended, the Ith column of the array calculated by the compute is used. Users can also write code for their
own compute styles and add them to LAMMPS. See the discussion above for how I can be specified with a
wildcard asterisk to effectively specify multiple values.
If a value begins with "f_", a fix ID must follow which has been previously defined in the input script. The fix
must generate a global vector or array. See the individual fix doc page for details. Note that some fixes only
produce their values on certain timesteps, which must be compatible with when compute global/atom
references the values, else an error results. If no bracketed integer is appended, the vector calculated by the fix
is used. If a bracketed integer is appended, the Ith column of the array calculated by the fix is used. Users can
also write code for their own fix style and add them to LAMMPS. See the discussion above for how I can be
specified with a wildcard asterisk to effectively specify multiple values.
If a value begins with "v_", a variable name must follow which has been previously defined in the input
script. It must be a vector-style variable. Vector-style variables can reference thermodynamic keywords and
various other attributes of atoms, or invoke other computes, fixes, or variables when they are evaluated, so
this is a very general means of generating a vector of global quantities which the index parameter will
reference for assignment of global values to atoms.
Output info:
If a single input is specified this compute produces a per-atom vector. If multiple inputs are specified, this
compute produces a per-atom array values, where the number of columns is equal to the number of inputs
specified. These values can be used by any command that uses per-atom vector or array values from a
compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector or array values will be in whatever units the corresponding input values are in.
Restrictions: none
Related commands:
compute, fix, variable, compute chunk/atom, compute reduce
Default: none
LAMMPS Users Manual
1222

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute group/group command
Syntax:
compute ID group-ID group/group group2-ID keyword value ...
ID, group-ID are documented in compute command• group/group = style name of this compute command• group2-ID = group ID of second (or same) group• zero or more keyword/value pairs may be appended• keyword = pair or kspace or boundary or molecule
pair value = yes or no
kspace value = yes or no
boundary value = yes or no
molecule value = off or inter or intra
•
Examples:
compute 1 lower group/group upper
compute 1 lower group/group upper kspace yes
compute mine fluid group/group wall
Description:
Define a computation that calculates the total energy and force interaction between two groups of atoms: the
compute group and the specified group2. The two groups can be the same.
If the pair keyword is set to yes, which is the default, then the the interaction energy will include a pair
component which is defined as the pairwise energy between all pairs of atoms where one atom in the pair is in
the first group and the other is in the second group. Likewise, the interaction force calculated by this compute
will include the force on the compute group atoms due to pairwise interactions with atoms in the specified
group2.
NOTE: The energies computed by the pair keyword do not include tail corrections, even if they are enabled
via the pair_modify command.
If the molecule keyword is set to inter or intra than an additional check is made based on the molecule IDs of
the two atoms in each pair before including their pairwise interaction energy and force. For the inter setting,
the two atoms must be in different molecules. For the intra setting, the two atoms must be in the same
molecule.
If the kspace keyword is set to yes, which is not the default, and if a kspace_style is defined, then the
interaction energy will include a Kspace component which is the long-range Coulombic energy between all
the atoms in the first group and all the atoms in the 2nd group. Likewise, the interaction force calculated by
this compute will include the force on the compute group atoms due to long-range Coulombic interactions
with atoms in the specified group2.
Normally the long-range Coulombic energy converges only when the net charge of the unit cell is zero.
However, one can assume the net charge of the system is neutralized by a uniform background plasma, and a
LAMMPS Users Manual
1223

correction to the system energy can be applied to reduce artifacts. For more information see (Bogusz). If the
boundary keyword is set to yes, which is the default, and kspace contributions are included, then this energy
correction term will be added to the total group-group energy. This correction term does not affect the force
calculation and will be zero if one or both of the groups are charge neutral. This energy correction term is the
same as that included in the regular Ewald and PPPM routines.
NOTE: The molecule setting only affects the group/group contributions calculated by the pair keyword. It
does not affect the group/group contributions calculated by the kspace keyword.
This compute does not calculate any bond or angle or dihedral or improper interactions between atoms in the
two groups.
The pairwise contributions to the group-group interactions are calculated by looping over a neighbor list. The
Kspace contribution to the group-group interactions require essentially the same amount of work (FFTs,
Ewald summation) as computing long-range forces for the entire system. Thus it can be costly to invoke this
compute too frequently.
NOTE: If you have a bonded system, then the settings of special_bonds command can remove pairwise
interactions between atoms in the same bond, angle, or dihedral. This is the default setting for the
special_bonds command, and means those pairwise interactions do not appear in the neighbor list. Because
this compute uses a neighbor list, it also means those pairs will not be included in the group/group interaction.
This does not apply when using long-range coulomb interactions (coul/long, coul/msm, coul/wolf or similar.
One way to get around this would be to set special_bond scaling factors to very tiny numbers that are not
exactly zero (e.g. 1.0e-50). Another workaround is to write a dump file, and use the rerun command to
compute the group/group interactions for snapshots in the dump file. The rerun script can use a special_bonds
command that includes all pairs in the neighbor list.
If you desire a breakdown of the interactions into a pairwise and Kspace component, simply invoke the
compute twice with the appropriate yes/no settings for the pair and kspace keywords. This is no more costly
than using a single compute with both keywords set to yes. The individual contributions can be summed in a
variable if desired.
This document describes how the long-range group-group calculations are performed.
Output info:
This compute calculates a global scalar (the energy) and a global vector of length 3 (force), which can be
accessed by indices 1-3. These values can be used by any command that uses global scalar or vector values
from a compute as input. See this section for an overview of LAMMPS output options.
Both the scalar and vector values calculated by this compute are "extensive". The scalar value will be in
energy units. The vector values will be in force units.
Restrictions:
Not all pair styles can be evaluated in a pairwise mode as required by this compute. For example, 3-body and
other many-body potentials, such as Tersoff and Stillinger-Weber cannot be used. EAM potentials only
include the pair potential portion of the EAM interaction when used by this compute, not the embedding term.
Not all Kspace styles support calculation of group/group interactions. The ewald and pppm styles do.
LAMMPS Users Manual
1224

Related commands: none
Default:
The option defaults are pair = yes, kspace = no, boundary = yes, molecule = off.
Bogusz et al, J Chem Phys, 108, 7070 (1998)
LAMMPS Users Manual
1225

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute gyration command
Syntax:
compute ID group-ID gyration
ID, group-ID are documented in compute command• gyration = style name of this compute command•
Examples:
compute 1 molecule gyration
Description:
Define a computation that calculates the radius of gyration Rg of the group of atoms, including all effects due
to atoms passing thru periodic boundaries.
Rg is a measure of the size of the group of atoms, and is computed as the square root of the Rg^2 value in this
formula
where M is the total mass of the group, Rcm is the center-of-mass position of the group, and the sum is over
all atoms in the group.
A Rg^2 tensor, stored as a 6-element vector, is also calculated by this compute. The formula for the
components of the tensor is the same as the above formula, except that (Ri - Rcm)^2 is replaced by (Rix -
Rcmx) * (Riy - Rcmy) for the xy component, etc. The 6 components of the vector are ordered xx, yy, zz, xy,
xz, yz. Note that unlike the scalar Rg, each of the 6 values of the tensor is effectively a "squared" value, since
the cross-terms may be negative and taking a sqrt() would be invalid.
NOTE: The coordinates of an atom contribute to Rg in "unwrapped" form, by using the image flags associated
with each atom. See the dump custom command for a discussion of "unwrapped" coordinates. See the Atoms
section of the read_data command for a discussion of image flags and how they are set for each atom. You
can reset the image flags (e.g. to 0) before invoking this compute by using the set image command.
Output info:
This compute calculates a global scalar (Rg) and a global vector of length 6 (Rg^2 tensor), which can be
accessed by indices 1-6. These values can be used by any command that uses a global scalar value or vector
values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The scalar and vector values calculated by this compute are "intensive". The scalar and vector values will be
in distance and distance^2 units respectively.
LAMMPS Users Manual
1226

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute gyration/chunk command
Syntax:
compute ID group-ID gyration/chunk chunkID keyword value ...
ID, group-ID are documented in compute command• gyration/chunk = style name of this compute command• chunkID = ID of compute chunk/atom command• zero or more keyword/value pairs may be appended• keyword = tensor
tensor value = none
•
Examples:
compute 1 molecule gyration/chunk molchunk
compute 2 molecule gyration/chunk molchunk tensor
Description:
Define a computation that calculates the radius of gyration Rg for multiple chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
This compute calculates the radius of gyration Rg for each chunk, which includes all effects due to atoms
passing thru periodic boundaries.
Rg is a measure of the size of a chunk, and is computed by this formula
where M is the total mass of the chunk, Rcm is the center-of-mass position of the chunk, and the sum is over
all atoms in the chunk.
Note that only atoms in the specified group contribute to the calculation. The compute chunk/atom command
defines its own group; atoms will have a chunk ID = 0 if they are not in that group, signifying they are not
assigned to a chunk, and will thus also not contribute to this calculation. You can specify the "all" group for
this command if you simply want to include atoms with non-zero chunk IDs.
LAMMPS Users Manual
1228
If the tensor keyword is specified, then the scalar Rg value is not calculated, but an Rg tensor is instead
calculated for each chunk. The formula for the components of the tensor is the same as the above formula,
except that (Ri - Rcm)^2 is replaced by (Rix - Rcmx) * (Riy - Rcmy) for the xy component, etc. The 6
components of the tensor are ordered xx, yy, zz, xy, xz, yz.
NOTE: The coordinates of an atom contribute to Rg in "unwrapped" form, by using the image flags associated
with each atom. See the dump custom command for a discussion of "unwrapped" coordinates. See the Atoms
section of the read_data command for a discussion of image flags and how they are set for each atom. You
can reset the image flags (e.g. to 0) before invoking this compute by using the set image command.
The simplest way to output the results of the compute gyration/chunk calculation to a file is to use the fix
ave/time command, for example:
compute cc1 all chunk/atom molecule
compute myChunk all gyration/chunk cc1
fix 1 all ave/time 100 1 100 c_myChunk file tmp.out mode vector
Output info:
This compute calculates a global vector if the tensor keyword is not specified and a global array if it is. The
length of the vector or number of rows in the array = the number of chunks Nchunk as calculated by the
specified compute chunk/atom command. If the tensor keyword is specified, the global array has 6 columns.
The vector or array can be accessed by any command that uses global values from a compute as input. See
this section for an overview of LAMMPS output options.
All the vector or array values calculated by this compute are "intensive". The vector or array values will be in
distance units, since they are the square root of values represented by the formula above.
Restrictions: none
Related commands: none
compute gyration
Default: none
LAMMPS Users Manual
1229

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute heat/flux command
Syntax:
compute ID group-ID heat/flux ke-ID pe-ID stress-ID
ID, group-ID are documented in compute command• heat/flux = style name of this compute command• ke-ID = ID of a compute that calculates per-atom kinetic energy• pe-ID = ID of a compute that calculates per-atom potential energy• stress-ID = ID of a compute that calculates per-atom stress•
Examples:
compute myFlux all heat/flux myKE myPE myStress
Description:
Define a computation that calculates the heat flux vector based on contributions from atoms in the specified
group. This can be used by itself to measure the heat flux into or out of a reservoir of atoms, or to calculate a
thermal conductivity using the Green-Kubo formalism.
See the fix thermal/conductivity command for details on how to compute thermal conductivity in an alternate
way, via the Muller-Plathe method. See the fix heat command for a way to control the heat added or
subtracted to a group of atoms.
The compute takes three arguments which are IDs of other computes. One calculates per-atom kinetic energy
(ke-ID), one calculates per-atom potential energy (pe-ID), and the third calculates per-atom stress (stress-ID).
NOTE: These other computes should provide values for all the atoms in the group this compute specifies.
That means the other computes could use the same group as this compute, or they can just use group "all" (or
any group whose atoms are superset of the atoms in this compute's group). LAMMPS does not check for this.
The Green-Kubo formulas relate the ensemble average of the auto-correlation of the heat flux J to the thermal
conductivity kappa:
LAMMPS Users Manual
1230

Ei in the first term of the equation for J is the per-atom energy (potential and kinetic). This is calculated by the
computes ke-ID and pe-ID. Si in the second term of the equation for J is the per-atom stress tensor calculated
by the compute stress-ID. The tensor multiplies Vi as a 3x3 matrix-vector multiply to yield a vector. Note that
as discussed below, the 1/V scaling factor in the equation for J is NOT included in the calculation performed
by this compute; you need to add it for a volume appropriate to the atoms included in the calculation.
NOTE: The compute pe/atom and compute stress/atom commands have options for which terms to include in
their calculation (pair, bond, etc). The heat flux calculation will thus include exactly the same terms. Normally
you should use compute stress/atom virial so as not to include a kinetic energy term in the heat flux.
This compute calculates 6 quantities and stores them in a 6-component vector. The first 3 components are the
x, y, z components of the full heat flux vector, i.e. (Jx, Jy, Jz). The next 3 components are the x, y, z
components of just the convective portion of the flux, i.e. the first term in the equation for J above.
The heat flux can be output every so many timesteps (e.g. via the thermo_style custom command). Then as a
post-processing operation, an autocorrelation can be performed, its integral estimated, and the Green-Kubo
formula above evaluated.
The fix ave/correlate command can calculate the autocorrelation. The trap() function in the variable command
can calculate the integral.
An example LAMMPS input script for solid Ar is appended below. The result should be: average conductivity
~0.29 in W/mK.
Output info:
This compute calculates a global vector of length 6 (total heat flux vector, followed by convective heat flux
vector), which can be accessed by indices 1-6. These values can be used by any command that uses global
vector values from a compute as input. See this section for an overview of LAMMPS output options.
LAMMPS Users Manual
1231

The vector values calculated by this compute are "extensive", meaning they scale with the number of atoms in
the simulation. They can be divided by the appropriate volume to get a flux, which would then be an
"intensive" value, meaning independent of the number of atoms in the simulation. Note that if the compute is
"all", then the appropriate volume to divide by is the simulation box volume. However, if a sub-group is used,
it should be the volume containing those atoms.
The vector values will be in energy*velocity units. Once divided by a volume the units will be that of flux,
namely energy/area/time units
Restrictions: none
Related commands:
fix thermal/conductivity, fix ave/correlate, variable
Default: none
# Sample LAMMPS input script for thermal conductivity of solid Ar
units real
variable T equal 70
variable V equal vol
variable dt equal 4.0
variable p equal 200 # correlation length
variable s equal 10 # sample interval
variable d equal $p*$s # dump interval
# convert from LAMMPS real units to SI
variable kB equal 1.3806504e-23 # [J/K] Boltzmann
variable kCal2J equal 4186.0/6.02214e23
variable A2m equal 1.0e-10
variable fs2s equal 1.0e-15
variable convert equal ${kCal2J}*${kCal2J}/${fs2s}/${A2m}
# setup problem
dimension 3
boundary p p p
lattice fcc 5.376 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
region box block 0 4 0 4 0 4
create_box 1 box
create_atoms 1 box
mass 1 39.948
pair_style lj/cut 13.0
pair_coeff * * 0.2381 3.405
timestep ${dt}
thermo $d
# equilibration and thermalization
velocity all create $T 102486 mom yes rot yes dist gaussian
fix NVT all nvt temp $T $T 10 drag 0.2
run 8000
# thermal conductivity calculation, switch to NVE if desired
LAMMPS Users Manual
1232
#unfix NVT
#fix NVE all nve
reset_timestep 0
compute myKE all ke/atom
compute myPE all pe/atom
compute myStress all stress/atom NULL virial
compute flux all heat/flux myKE myPE myStress
variable Jx equal c_flux[1]/vol
variable Jy equal c_flux[2]/vol
variable Jz equal c_flux[3]/vol
fix JJ all ave/correlate $s $p $d &
c_flux[1] c_flux[2] c_flux[3] type auto file J0Jt.dat ave running
variable scale equal ${convert}/${kB}/$T/$T/$V*$s*${dt}
variable k11 equal trap(f_JJ[3])*${scale}
variable k22 equal trap(f_JJ[4])*${scale}
variable k33 equal trap(f_JJ[5])*${scale}
thermo_style custom step temp v_Jx v_Jy v_Jz v_k11 v_k22 v_k33
run 100000
variable k equal (v_k11+v_k22+v_k33)/3.0
variable ndens equal count(all)/vol
print "average conductivity: $k[W/mK] @ $T K, ${ndens} /A^3"
LAMMPS Users Manual
1233
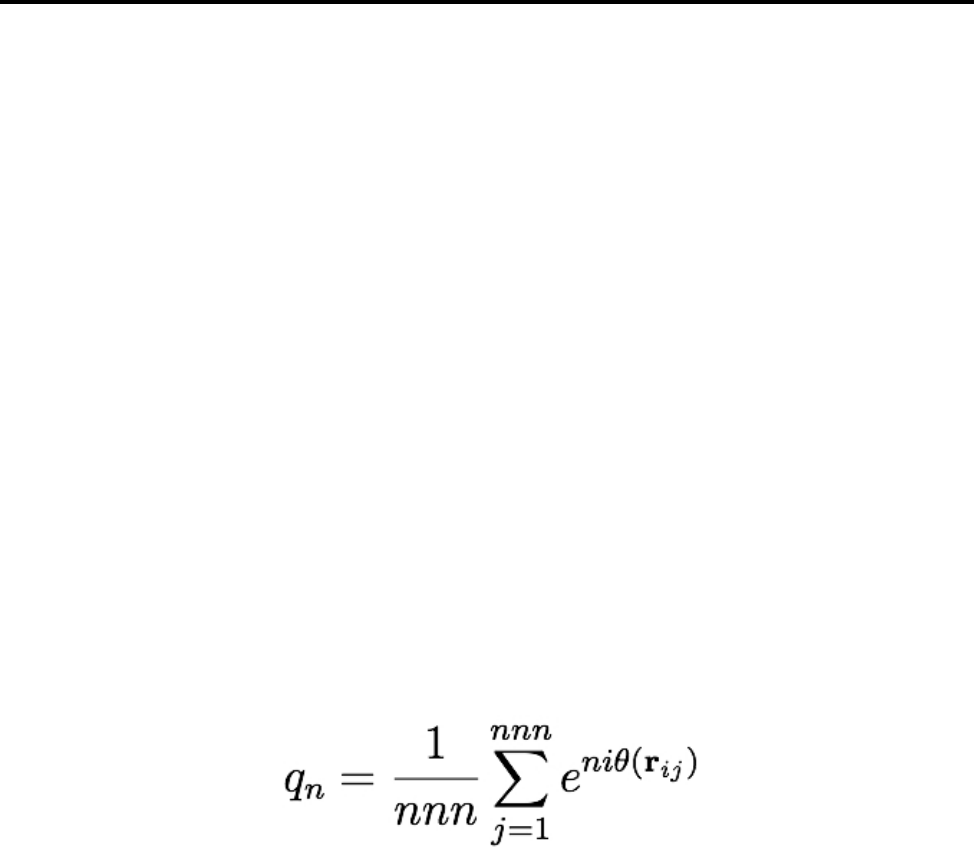
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute hexorder/atom command
Syntax:
compute ID group-ID hexorder/atom keyword values ...
ID, group-ID are documented in compute command• hexorder/atom = style name of this compute command• one or more keyword/value pairs may be appended
keyword = degree or nnn or cutoff
cutoff value = distance cutoff
nnn value = number of nearest neighbors
degree value = degree n of order parameter
•
Examples:
compute 1 all hexorder/atom
compute 1 all hexorder/atom degree 4 nnn 4 cutoff 1.2
Description:
Define a computation that calculates qn the bond-orientational order parameter for each atom in a group. The
hexatic (n = 6) order parameter was introduced by Nelson and Halperin as a way to detect hexagonal
symmetry in two-dimensional systems. For each atom, qn is a complex number (stored as two real numbers)
defined as follows:
where the sum is over the nnn nearest neighbors of the central atom. The angle theta is formed by the bond
vector rij and the x axis. theta is calculated only using the x and y components, whereas the distance from the
central atom is calculated using all three x, y, and z components of the bond vector. Neighbor atoms not in the
group are included in the order parameter of atoms in the group.
The optional keyword cutoff defines the distance cutoff used when searching for neighbors. The default value,
also the maximum allowable value, is the cutoff specified by the pair style.
The optional keyword nnn defines the number of nearest neighbors used to calculate qn. The default value is
6. If the value is NULL, then all neighbors up to the distance cutoff are used.
The optional keyword degree sets the degree n of the order parameter. The default value is 6. For a perfect
hexagonal lattice with nnn = 6, q6 = exp(6 i phi) for all atoms, where the constant 0 < phi < pi/3 depends only
on the orientation of the lattice relative to the x axis. In an isotropic liquid, local neighborhoods may still
exhibit weak hexagonal symmetry, but because the orientational correlation decays quickly with distance, the
value of phi will be different for different atoms, and so when q6 is averaged over all the atoms in the system,
LAMMPS Users Manual
1234

\|<q6>\| << 1.
The value of qn is set to zero for atoms not in the specified compute group, as well as for atoms that have less
than nnn neighbors within the distance cutoff.
The neighbor list needed to compute this quantity is constructed each time the calculation is performed (i.e.
each time a snapshot of atoms is dumped). Thus it can be inefficient to compute/dump this quantity too
frequently.
NOTE: If you have a bonded system, then the settings of special_bonds command can remove pairwise
interactions between atoms in the same bond, angle, or dihedral. This is the default setting for the
special_bonds command, and means those pairwise interactions do not appear in the neighbor list. Because
this fix uses the neighbor list, it also means those pairs will not be included in the order parameter. This
difficulty can be circumvented by writing a dump file, and using the rerun command to compute the order
parameter for snapshots in the dump file. The rerun script can use a special_bonds command that includes all
pairs in the neighbor list.
Output info:
This compute calculates a per-atom array with 2 columns, giving the real and imaginary parts qn, a complex
number restricted to the unit disk of the complex plane i.e. Re(qn)^2 + Im(qn)^2 <= 1 .
These values can be accessed by any command that uses per-atom values from a compute as input. See
Section 6.15 for an overview of LAMMPS output options.
Restrictions: none
Related commands:
compute orientorder/atom, compute coord/atom, compute centro/atom
Default:
The option defaults are cutoff = pair style cutoff, nnn = 6, degree = 6
(Nelson) Nelson, Halperin, Phys Rev B, 19, 2457 (1979).
LAMMPS Users Manual
1235

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute improper command
Syntax:
compute ID group-ID improper
ID, group-ID are documented in compute command• improper = style name of this compute command•
Examples:
compute 1 all improper
Description:
Define a computation that extracts the improper energy calculated by each of the improper sub-styles used in
the improper_style hybrid command. These values are made accessible for output or further processing by
other commands. The group specified for this command is ignored.
This compute is useful when using improper_style hybrid if you want to know the portion of the total energy
contributed by one or more of the hybrid sub-styles.
Output info:
This compute calculates a global vector of length N where N is the number of sub_styles defined by the
improper_style hybrid command. which can be accessed by indices 1-N. These values can be used by any
command that uses global scalar or vector values from a compute as input. See this section for an overview of
LAMMPS output options.
The vector values are "extensive" and will be in energy units.
Restrictions: none
Related commands:
compute pe, compute pair
Default: none
LAMMPS Users Manual
1236

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute improper/local command
Syntax:
compute ID group-ID improper/local value1 value2 ...
ID, group-ID are documented in compute command• improper/local = style name of this compute command• one or more values may be appended• value = chi
chi = tabulate improper angles
•
Examples:
compute 1 all improper/local chi
Description:
Define a computation that calculates properties of individual improper interactions. The number of datums
generated, aggregated across all processors, equals the number of impropers in the system, modified by the
group parameter as explained below.
The value chi is the improper angle, as defined in the doc pages for the individual improper styles listed on
improper_style doc page.
The local data stored by this command is generated by looping over all the atoms owned on a processor and
their impropers. An improper will only be included if all 4 atoms in the improper are in the specified compute
group.
Note that as atoms migrate from processor to processor, there will be no consistent ordering of the entries
within the local vector or array from one timestep to the next. The only consistency that is guaranteed is that
the ordering on a particular timestep will be the same for local vectors or arrays generated by other compute
commands. For example, improper output from the compute property/local command can be combined with
data from this command and output by the dump local command in a consistent way.
Here is an example of how to do this:
compute 1 all property/local itype iatom1 iatom2 iatom3 iatom4
compute 2 all improper/local chi
dump 1 all local 1000 tmp.dump index c_1[1] c_1[2] c_1[3] c_1[4] c_1[5] c_2[1]
Output info:
This compute calculates a local vector or local array depending on the number of keywords. The length of the
vector or number of rows in the array is the number of impropers. If a single keyword is specified, a local
vector is produced. If two or more keywords are specified, a local array is produced where the number of
columns = the number of keywords. The vector or array can be accessed by any command that uses local
values from a compute as input. See this section for an overview of LAMMPS output options.
LAMMPS Users Manual
1237

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute inertia/chunk command
Syntax:
compute ID group-ID inertia/chunk chunkID
ID, group-ID are documented in compute command• inertia/chunk = style name of this compute command• chunkID = ID of compute chunk/atom command•
Examples:
compute 1 fluid inertia/chunk molchunk
Description:
Define a computation that calculates the inertia tensor for multiple chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
This compute calculates the 6 components of the symmetric inertia tensor for each chunk, ordered
Ixx,Iyy,Izz,Ixy,Iyz,Ixz. The calculation includes all effects due to atoms passing thru periodic boundaries.
Note that only atoms in the specified group contribute to the calculation. The compute chunk/atom command
defines its own group; atoms will have a chunk ID = 0 if they are not in that group, signifying they are not
assigned to a chunk, and will thus also not contribute to this calculation. You can specify the "all" group for
this command if you simply want to include atoms with non-zero chunk IDs.
NOTE: The coordinates of an atom contribute to the chunk's inertia tensor in "unwrapped" form, by using the
image flags associated with each atom. See the dump custom command for a discussion of "unwrapped"
coordinates. See the Atoms section of the read_data command for a discussion of image flags and how they
are set for each atom. You can reset the image flags (e.g. to 0) before invoking this compute by using the set
image command.
The simplest way to output the results of the compute inertia/chunk calculation to a file is to use the fix
ave/time command, for example:
compute cc1 all chunk/atom molecule
compute myChunk all inertia/chunk cc1
fix 1 all ave/time 100 1 100 c_myChunk[*] file tmp.out mode vector
Output info:
This compute calculates a global array where the number of rows = the number of chunks Nchunk as
calculated by the specified compute chunk/atom command. The number of columns = 6 for the 6 components
LAMMPS Users Manual
1239
of the inertia tensor for each chunk, ordered as listed above. These values can be accessed by any command
that uses global array values from a compute as input. See Section 6.15 for an overview of LAMMPS output
options.
The array values are "intensive". The array values will be in mass*distance^2 units.
Restrictions: none
Related commands:
variable inertia() function
Default: none
LAMMPS Users Manual
1240

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute ke command
Syntax:
compute ID group-ID ke
ID, group-ID are documented in compute command• ke = style name of this compute command•
Examples:
compute 1 all ke
Description:
Define a computation that calculates the translational kinetic energy of a group of particles.
The kinetic energy of each particle is computed as 1/2 m v^2, where m and v are the mass and velocity of the
particle.
There is a subtle difference between the quantity calculated by this compute and the kinetic energy calculated
by the ke or etotal keyword used in thermodynamic output, as specified by the thermo_style command. For
this compute, kinetic energy is "translational" kinetic energy, calculated by the simple formula above. For
thermodynamic output, the ke keyword infers kinetic energy from the temperature of the system with 1/2 Kb
T of energy for each degree of freedom. For the default temperature computation via the compute temp
command, these are the same. But different computes that calculate temperature can subtract out different
non-thermal components of velocity and/or include different degrees of freedom (translational, rotational,
etc).
Output info:
This compute calculates a global scalar (the summed KE). This value can be used by any command that uses a
global scalar value from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The scalar value calculated by this compute is "extensive". The scalar value will be in energy units.
Restrictions: none
Related commands:
compute erotate/sphere
Default: none
LAMMPS Users Manual
1241

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute ke/atom command
Syntax:
compute ID group-ID ke/atom
ID, group-ID are documented in compute command• ke/atom = style name of this compute command•
Examples:
compute 1 all ke/atom
Description:
Define a computation that calculates the per-atom translational kinetic energy for each atom in a group.
The kinetic energy is simply 1/2 m v^2, where m is the mass and v is the velocity of each atom.
The value of the kinetic energy will be 0.0 for atoms not in the specified compute group.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values will be in energy units.
Restrictions: none
Related commands:
dump custom
Default: none
LAMMPS Users Manual
1242

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute ke/atom/eff command
Syntax:
compute ID group-ID ke/atom/eff
ID, group-ID are documented in compute command• ke/atom/eff = style name of this compute command•
Examples:
compute 1 all ke/atom/eff
Description:
Define a computation that calculates the per-atom translational (nuclei and electrons) and radial kinetic energy
(electron only) in a group. The particles are assumed to be nuclei and electrons modeled with the electronic
force field.
The kinetic energy for each nucleus is computed as 1/2 m v^2, where m corresponds to the corresponding
nuclear mass, and the kinetic energy for each electron is computed as 1/2 (me v^2 + 3/4 me s^2), where me
and v correspond to the mass and translational velocity of each electron, and s to its radial velocity,
respectively.
There is a subtle difference between the quantity calculated by this compute and the kinetic energy calculated
by the ke or etotal keyword used in thermodynamic output, as specified by the thermo_style command. For
this compute, kinetic energy is "translational" plus electronic "radial" kinetic energy, calculated by the simple
formula above. For thermodynamic output, the ke keyword infers kinetic energy from the temperature of the
system with 1/2 Kb T of energy for each (nuclear-only) degree of freedom in eFF.
NOTE: The temperature in eFF should be monitored via the compute temp/eff command, which can be
printed with thermodynamic output by using the thermo_modify command, as shown in the following
example:
compute effTemp all temp/eff
thermo_style custom step etotal pe ke temp press
thermo_modify temp effTemp
The value of the kinetic energy will be 0.0 for atoms (nuclei or electrons) not in the specified compute group.
Output info:
This compute calculates a scalar quantity for each atom, which can be accessed by any command that uses
per-atom computes as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values will be in energy units.
Restrictions:
LAMMPS Users Manual
1243

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute ke/eff command
Syntax:
compute ID group-ID ke/eff
ID, group-ID are documented in compute command• ke/eff = style name of this compute command•
Examples:
compute 1 all ke/eff
Description:
Define a computation that calculates the kinetic energy of motion of a group of eFF particles (nuclei and
electrons), as modeled with the electronic force field.
The kinetic energy for each nucleus is computed as 1/2 m v^2 and the kinetic energy for each electron is
computed as 1/2(me v^2 + 3/4 me s^2), where m corresponds to the nuclear mass, me to the electron mass, v
to the translational velocity of each particle, and s to the radial velocity of the electron, respectively.
There is a subtle difference between the quantity calculated by this compute and the kinetic energy calculated
by the ke or etotal keyword used in thermodynamic output, as specified by the thermo_style command. For
this compute, kinetic energy is "translational" and "radial" (only for electrons) kinetic energy, calculated by
the simple formula above. For thermodynamic output, the ke keyword infers kinetic energy from the
temperature of the system with 1/2 Kb T of energy for each degree of freedom. For the eFF temperature
computation via the compute temp_eff command, these are the same. But different computes that calculate
temperature can subtract out different non-thermal components of velocity and/or include other degrees of
freedom.
IMPRORTANT NOTE: The temperature in eFF models should be monitored via the compute temp/eff
command, which can be printed with thermodynamic output by using the thermo_modify command, as shown
in the following example:
compute effTemp all temp/eff
thermo_style custom step etotal pe ke temp press
thermo_modify temp effTemp
See compute temp/eff.
Output info:
This compute calculates a global scalar (the KE). This value can be used by any command that uses a global
scalar value from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The scalar value calculated by this compute is "extensive". The scalar value will be in energy units.
Restrictions:
LAMMPS Users Manual
1245

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute ke/rigid command
Syntax:
compute ID group-ID ke/rigid fix-ID
ID, group-ID are documented in compute command• ke = style name of this compute command• fix-ID = ID of rigid body fix•
Examples:
compute 1 all ke/rigid myRigid
Description:
Define a computation that calculates the translational kinetic energy of a collection of rigid bodies, as defined
by one of the fix rigid command variants.
The kinetic energy of each rigid body is computed as 1/2 M Vcm^2, where M is the total mass of the rigid
body, and Vcm is its center-of-mass velocity.
The fix-ID should be the ID of one of the fix rigid commands which defines the rigid bodies. The group
specified in the compute command is ignored. The kinetic energy of all the rigid bodies defined by the fix
rigid command in included in the calculation.
Output info:
This compute calculates a global scalar (the summed KE of all the rigid bodies). This value can be used by
any command that uses a global scalar value from a compute as input. See Section 6.15 for an overview of
LAMMPS output options.
The scalar value calculated by this compute is "extensive". The scalar value will be in energy units.
Restrictions:
This compute is part of the RIGID package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
compute erotate/rigid
Default: none
LAMMPS Users Manual
1247

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute meso/e/atom command
Syntax:
compute ID group-ID meso/e/atom
ID, group-ID are documented in compute command• meso/e/atom = style name of this compute command•
Examples:
compute 1 all meso/e/atom
Description:
Define a computation that calculates the per-atom internal energy for each atom in a group.
The internal energy is the energy associated with the internal degrees of freedom of a mesoscopic particles,
e.g. a Smooth-Particle Hydrodynamics particle.
See this PDF guide to using SPH in LAMMPS.
The value of the internal energy will be 0.0 for atoms not in the specified compute group.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values will be in energy units.
Restrictions:
This compute is part of the USER-SPH package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
dump custom
Default: none
LAMMPS Users Manual
1248

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute meso/rho/atom command
Syntax:
compute ID group-ID meso/rho/atom
ID, group-ID are documented in compute command• meso/rho/atom = style name of this compute command•
Examples:
compute 1 all meso/rho/atom
Description:
Define a computation that calculates the per-atom mesoscopic density for each atom in a group.
The mesoscopic density is the mass density of a mesoscopic particle, calculated by kernel function
interpolation using "pair style sph/rhosum".
See this PDF guide to using SPH in LAMMPS.
The value of the mesoscopic density will be 0.0 for atoms not in the specified compute group.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values will be in mass/volume units.
Restrictions:
This compute is part of the USER-SPH package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
dump custom
Default: none
LAMMPS Users Manual
1249

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute meso/t/atom command
Syntax:
compute ID group-ID meso/t/atom
ID, group-ID are documented in compute command• meso/t/atom = style name of this compute command•
Examples:
compute 1 all meso/t/atom
Description:
Define a computation that calculates the per-atom internal temperature for each atom in a group.
The internal temperature is the ratio of internal energy over the heat capacity associated with the internal
degrees of freedom of a mesoscopic particles, e.g. a Smooth-Particle Hydrodynamics particle.
T_int = E_int / C_V, int
See this PDF guide to using SPH in LAMMPS.
The value of the internal energy will be 0.0 for atoms not in the specified compute group.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values will be in temperature units.
Restrictions:
This compute is part of the USER-SPH package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
dump custom
Default: none
LAMMPS Users Manual
1250

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute msd command
Syntax:
compute ID group-ID msd keyword values ...
ID, group-ID are documented in compute command• msd = style name of this compute command• zero or more keyword/value pairs may be appended• keyword = com or average
com value = yes or no
average value = yes or no
•
Examples:
compute 1 all msd
compute 1 upper msd com yes average yes
Description:
Define a computation that calculates the mean-squared displacement (MSD) of the group of atoms, including
all effects due to atoms passing thru periodic boundaries. For computation of the non-Gaussian parameter of
mean-squared displacement, see the compute msd/nongauss command.
A vector of four quantities is calculated by this compute. The first 3 elements of the vector are the squared
dx,dy,dz displacements, summed and averaged over atoms in the group. The 4th element is the total squared
displacement, i.e. (dx*dx + dy*dy + dz*dz), summed and averaged over atoms in the group.
The slope of the mean-squared displacement (MSD) versus time is proportional to the diffusion coefficient of
the diffusing atoms.
The displacement of an atom is from its reference position. This is normally the original position at the time
the compute command was issued, unless the average keyword is set to yes. The value of the displacement
will be 0.0 for atoms not in the specified compute group.
If the com option is set to yes then the effect of any drift in the center-of-mass of the group of atoms is
subtracted out before the displacment of each atom is calculated.
If the average option is set to yes then the reference position of an atom is based on the average position of
that atom, corrected for center-of-mass motion if requested. The average position is a running average over all
previous calls to the compute, including the current call. So on the first call it is current position, on the
second call it is the arithmetic average of the current position and the position on the first call, and so on. Note
that when using this option, the precise value of the mean square displacement will depend on the number of
times the compute is called. So, for example, changing the frequency of thermo output may change the
computed displacement. Also, the precise values will be changed if a single simulation is broken up into two
parts, using either multiple run commands or a restart file. It only makes sense to use this option if the atoms
are not diffusing, so that their average positions relative to the center of mass of the system are stationary. The
most common case is crystalline solids undergoing thermal motion.
LAMMPS Users Manual
1251
NOTE: Initial coordinates are stored in "unwrapped" form, by using the image flags associated with each
atom. See the dump custom command for a discussion of "unwrapped" coordinates. See the Atoms section of
the read_data command for a discussion of image flags and how they are set for each atom. You can reset the
image flags (e.g. to 0) before invoking this compute by using the set image command.
NOTE: If you want the quantities calculated by this compute to be continuous when running from a restart
file, then you should use the same ID for this compute, as in the original run. This is so that the fix this
compute creates to store per-atom quantities will also have the same ID, and thus be initialized correctly with
atom reference positions from the restart file. When average is set to yes, then the atom reference positions
are restored correctly, but not the number of samples used obtain them. As a result, the reference positions
from the restart file are combined with subsequent positions as if they were from a single sample, instead of
many, which will change the values of msd somewhat.
Output info:
This compute calculates a global vector of length 4, which can be accessed by indices 1-4 by any command
that uses global vector values from a compute as input. See this section for an overview of LAMMPS output
options.
The vector values are "intensive". The vector values will be in distance^2 units.
Restrictions: none
Related commands:
compute msd/nongauss, compute displace_atom, fix store/state, compute msd/chunk
Default:
The option default are com = no, average = no.
LAMMPS Users Manual
1252

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute msd/chunk command
Syntax:
compute ID group-ID msd/chunk chunkID
ID, group-ID are documented in compute command• msd/chunk = style name of this compute command• chunkID = ID of compute chunk/atom command•
Examples:
compute 1 all msd/chunk molchunk
Description:
Define a computation that calculates the mean-squared displacement (MSD) for multiple chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
Four quantities are calculated by this compute for each chunk. The first 3 quantities are the squared dx,dy,dz
displacements of the center-of-mass. The 4th component is the total squared displacement, i.e. (dx*dx +
dy*dy + dz*dz) of the center-of-mass. These calculations include all effects due to atoms passing thru
periodic boundaries.
Note that only atoms in the specified group contribute to the calculation. The compute chunk/atom command
defines its own group; atoms will have a chunk ID = 0 if they are not in that group, signifying they are not
assigned to a chunk, and will thus also not contribute to this calculation. You can specify the "all" group for
this command if you simply want to include atoms with non-zero chunk IDs.
The slope of the mean-squared displacement (MSD) versus time is proportional to the diffusion coefficient of
the diffusing chunks.
The displacement of the center-of-mass of the chunk is from its original center-of-mass position, calculated on
the timestep this compute command was first invoked.
NOTE: The number of chunks Nchunk calculated by the compute chunk/atom command must remain constant
each time this compute is invoked, so that the displacement for each chunk from its original position can be
computed consistently. If Nchunk does not remain constant, an error will be generated. If needed, you can
enforce a constant Nchunk by using the nchunk once or ids once options when specifying the compute
chunk/atom command.
NOTE: This compute stores the original position (of the center-of-mass) of each chunk. When a displacement
is calculated on a later timestep, it is assumed that the same atoms are assigned to the same chunk ID.
However LAMMPS has no simple way to insure this is the case, though you can use the ids once option when
LAMMPS Users Manual
1253
specifying the compute chunk/atom command. Note that if this is not the case, the MSD calculation does not
have a sensible meaning.
NOTE: The initial coordinates of the atoms in each chunk are stored in "unwrapped" form, by using the image
flags associated with each atom. See the dump custom command for a discussion of "unwrapped" coordinates.
See the Atoms section of the read_data command for a discussion of image flags and how they are set for each
atom. You can reset the image flags (e.g. to 0) before invoking this compute by using the set image command.
NOTE: If you want the quantities calculated by this compute to be continuous when running from a restart
file, then you should use the same ID for this compute, as in the original run. This is so that the fix this
compute creates to store per-chunk quantities will also have the same ID, and thus be initialized correctly with
chunk reference positions from the restart file.
The simplest way to output the results of the compute com/msd calculation to a file is to use the fix ave/time
command, for example:
compute cc1 all chunk/atom molecule
compute myChunk all com/msd cc1
fix 1 all ave/time 100 1 100 c_myChunk[*] file tmp.out mode vector
Output info:
This compute calculates a global array where the number of rows = the number of chunks Nchunk as
calculated by the specified compute chunk/atom command. The number of columns = 4 for dx,dy,dz and the
total displacement. These values can be accessed by any command that uses global array values from a
compute as input. See this section for an overview of LAMMPS output options.
The array values are "intensive". The array values will be in distance^2 units.
Restrictions: none
Related commands:
compute msd
Default: none
LAMMPS Users Manual
1254
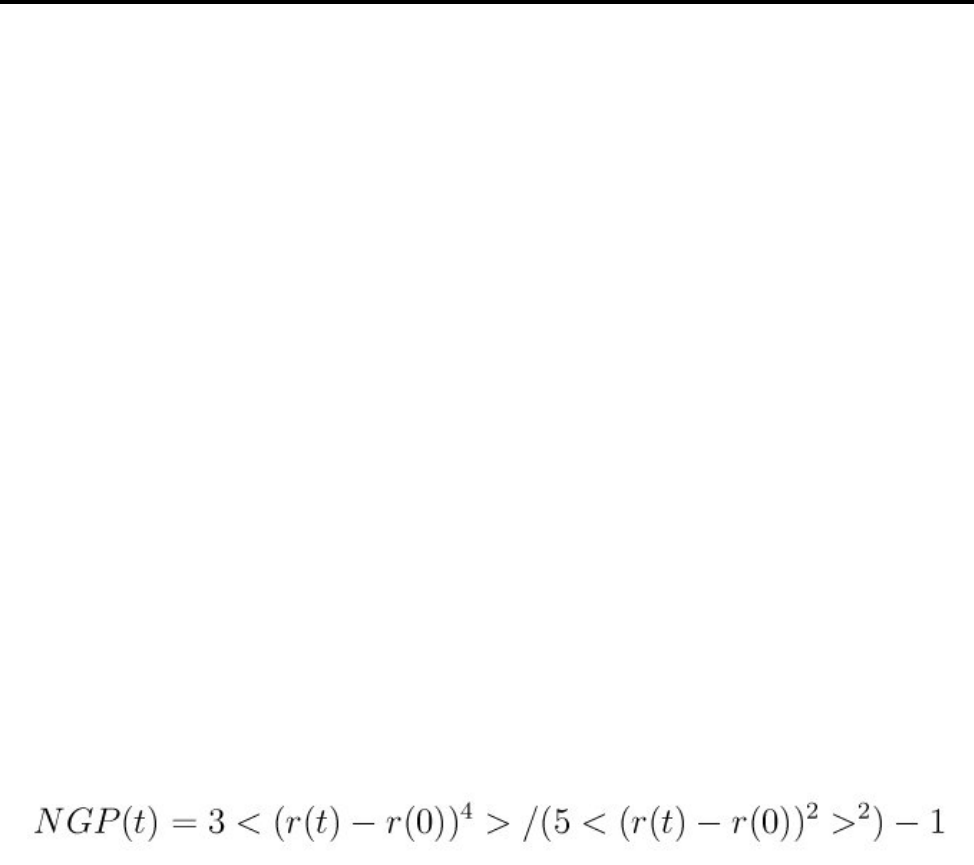
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute msd/nongauss command
Syntax:
compute ID group-ID msd/nongauss keyword values ...
ID, group-ID are documented in compute command• msd/nongauss = style name of this compute command• zero or more keyword/value pairs may be appended• keyword = com
com value = yes or no
•
Examples:
compute 1 all msd/nongauss
compute 1 upper msd/nongauss com yes
Description:
Define a computation that calculates the mean-squared displacement (MSD) and non-Gaussian parameter
(NGP) of the group of atoms, including all effects due to atoms passing thru periodic boundaries.
A vector of three quantities is calculated by this compute. The first element of the vector is the total squared
dx,dy,dz displacements drsquared = (dx*dx + dy*dy + dz*dz) of atoms, and the second is the fourth power of
these displacements drfourth = (dx*dx + dy*dy + dz*dz)*(dx*dx + dy*dy + dz*dz), summed and averaged
over atoms in the group. The 3rd component is the nonGaussian diffusion parameter NGP =
3*drfourth/(5*drsquared*drsquared), i.e.
The NGP is a commonly used quantity in studies of dynamical heterogeneity. Its minimum theoretical value
(-0.4) occurs when all atoms have the same displacement magnitude. NGP=0 for Brownian diffusion, while
NGP > 0 when some mobile atoms move faster than others.
If the com option is set to yes then the effect of any drift in the center-of-mass of the group of atoms is
subtracted out before the displacment of each atom is calculated.
See the compute msd doc page for further important NOTEs, which also apply to this compute.
Output info:
This compute calculates a global vector of length 3, which can be accessed by indices 1-3 by any command
that uses global vector values from a compute as input. See this section for an overview of LAMMPS output
options.
LAMMPS Users Manual
1255
The vector values are "intensive". The first vector value will be in distance^2 units, the second is in distance^4
units, and the 3rd is dimensionless.
Restrictions:
This compute is part of the MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
compute msd
Default:
The option default is com = no.
LAMMPS Users Manual
1256

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute omega/chunk command
Syntax:
compute ID group-ID omega/chunk chunkID
ID, group-ID are documented in compute command• omega/chunk = style name of this compute command• chunkID = ID of compute chunk/atom command•
Examples:
compute 1 fluid omega/chunk molchunk
Description:
Define a computation that calculates the angular velocity (omega) of multiple chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
This compute calculates the 3 components of the angular velocity vector for each chunk, via the formula L =
Iw where L is the angular momentum vector of the chunk, I is its moment of inertia tensor, and w is omega =
angular velocity of the chunk. The calculation includes all effects due to atoms passing thru periodic
boundaries.
Note that only atoms in the specified group contribute to the calculation. The compute chunk/atom command
defines its own group; atoms will have a chunk ID = 0 if they are not in that group, signifying they are not
assigned to a chunk, and will thus also not contribute to this calculation. You can specify the "all" group for
this command if you simply want to include atoms with non-zero chunk IDs.
NOTE: The coordinates of an atom contribute to the chunk's angular velocity in "unwrapped" form, by using
the image flags associated with each atom. See the dump custom command for a discussion of "unwrapped"
coordinates. See the Atoms section of the read_data command for a discussion of image flags and how they
are set for each atom. You can reset the image flags (e.g. to 0) before invoking this compute by using the set
image command.
The simplest way to output the results of the compute omega/chunk calculation to a file is to use the fix
ave/time command, for example:
compute cc1 all chunk/atom molecule
compute myChunk all omega/chunk cc1
fix 1 all ave/time 100 1 100 c_myChunk[*] file tmp.out mode vector
Output info:
LAMMPS Users Manual
1257
This compute calculates a global array where the number of rows = the number of chunks Nchunk as
calculated by the specified compute chunk/atom command. The number of columns = 3 for the 3 xyz
components of the angular velocity for each chunk. These values can be accessed by any command that uses
global array values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The array values are "intensive". The array values will be in velocity/distance units.
Restrictions: none
Related commands:
variable omega() function
Default: none
LAMMPS Users Manual
1258
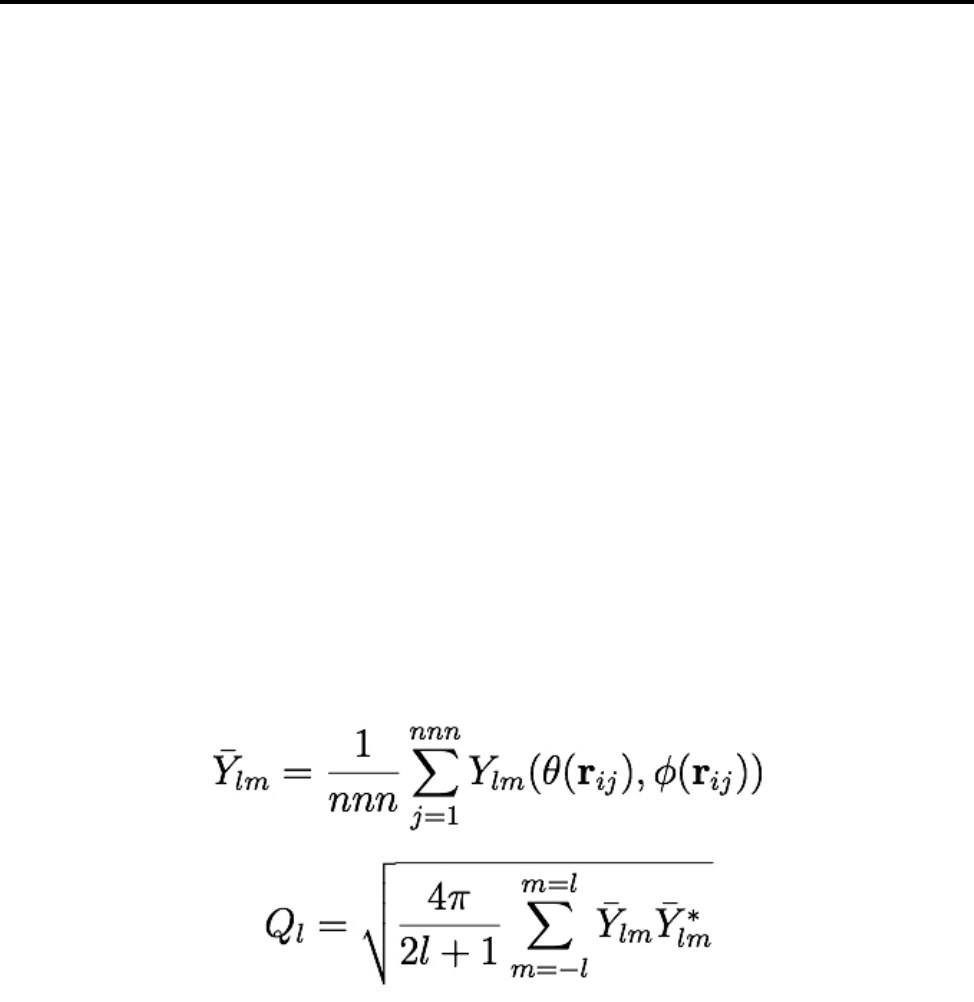
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute orientorder/atom command
Syntax:
compute ID group-ID orientorder/atom keyword values ...
ID, group-ID are documented in compute command• orientorder/atom = style name of this compute command• one or more keyword/value pairs may be appended
keyword = cutoff or nnn or degrees or components
cutoff value = distance cutoff
nnn value = number of nearest neighbors
degrees values = nlvalues, l1, l2,...
components value = ldegree
•
Examples:
compute 1 all orientorder/atom
compute 1 all orientorder/atom degrees 5 4 6 8 10 12 nnn NULL cutoff 1.5
compute 1 all orientorder/atom degrees 4 6 components 6 nnn NULL cutoff 3.0
Description:
Define a computation that calculates a set of bond-orientational order parameters Ql for each atom in a group.
These order parameters were introduced by Steinhardt et al. as a way to characterize the local orientational
order in atomic structures. For each atom, Ql is a real number defined as follows:
The first equation defines the spherical harmonic order parameters. These are complex number components of
the 3D analog of the 2D order parameter qn, which is implemented as LAMMPS compute hexorder/atom. The
summation is over the nnn nearest neighbors of the central atom. The angles theta and phi are the standard
spherical polar angles defining the direction of the bond vector rij. The second equation defines Ql, which is a
rotationally invariant scalar quantity obtained by summing over all the components of degree l.
The optional keyword cutoff defines the distance cutoff used when searching for neighbors. The default value,
also the maximum allowable value, is the cutoff specified by the pair style.
LAMMPS Users Manual
1259
The optional keyword nnn defines the number of nearest neighbors used to calculate Ql. The default value is
12. If the value is NULL, then all neighbors up to the specified distance cutoff are used.
The optional keyword degrees defines the list of order parameters to be computed. The first argument
nlvalues is the number of order parameters. This is followed by that number of integers giving the degree of
each order parameter. Because Q2 and all odd-degree order parameters are zero for atoms in cubic crystals
(see Steinhardt), the default order parameters are Q4, Q6, Q8, Q10, and Q12. For the FCC crystal with
nnn=12, Q4 = sqrt(7/3)/8 = 0.19094.... The numerical values of all order parameters up to Q12 for a range of
commonly encountered high-symmetry structures are given in Table I of Mickel et al..
The optional keyword components will output the components of the normalized complex vector Ybar_lm of
degree ldegree, which must be explicitly included in the keyword degrees. This option can be used in
conjunction with compute coord_atom to calculate the ten Wolde's criterion to identify crystal-like particles,
as discussed in ten Wolde.
The value of Ql is set to zero for atoms not in the specified compute group, as well as for atoms that have less
than nnn neighbors within the distance cutoff.
The neighbor list needed to compute this quantity is constructed each time the calculation is performed (i.e.
each time a snapshot of atoms is dumped). Thus it can be inefficient to compute/dump this quantity too
frequently.
NOTE: If you have a bonded system, then the settings of special_bonds command can remove pairwise
interactions between atoms in the same bond, angle, or dihedral. This is the default setting for the
special_bonds command, and means those pairwise interactions do not appear in the neighbor list. Because
this fix uses the neighbor list, it also means those pairs will not be included in the order parameter. This
difficulty can be circumvented by writing a dump file, and using the rerun command to compute the order
parameter for snapshots in the dump file. The rerun script can use a special_bonds command that includes all
pairs in the neighbor list.
Output info:
This compute calculates a per-atom array with nlvalues columns, giving the Ql values for each atom, which
are real numbers on the range 0 <= Ql <= 1.
If the keyword components is set, then the real and imaginary parts of each component of (normalized)
Ybar_lm will be added to the output array in the following order: Re(Ybar_-m) Im(Ybar_-m) Re(Ybar_-m+1)
Im(Ybar_-m+1) ... Re(Ybar_m) Im(Ybar_m). This way, the per-atom array will have a total of
nlvalues+2*(2l+1) columns.
These values can be accessed by any command that uses per-atom values from a compute as input. See
Section 6.15 for an overview of LAMMPS output options.
Restrictions: none
Related commands:
compute coord/atom, compute centro/atom, compute hexorder/atom
Default:
LAMMPS Users Manual
1260

The option defaults are cutoff = pair style cutoff, nnn = 12, degrees = 5 4 6 8 10 12 i.e. Q4, Q6, Q8, Q10, and
Q12.
(Steinhardt) P. Steinhardt, D. Nelson, and M. Ronchetti, Phys. Rev. B 28, 784 (1983).
(Mickel) W. Mickel, S. C. Kapfer, G. E. Schroeder-Turkand, K. Mecke, J. Chem. Phys. 138, 044501 (2013).
(tenWolde) P. R. ten Wolde, M. J. Ruiz-Montero, D. Frenkel, J. Chem. Phys. 104, 9932 (1996).
LAMMPS Users Manual
1261

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute pair command
Syntax:
compute ID group-ID pair pstyle evalue
ID, group-ID are documented in compute command• pair = style name of this compute command• pstyle = style name of a pair style that calculates additional values• evalue = epair or evdwl or ecoul or blank (optional setting)•
Examples:
compute 1 all pair gauss
compute 1 all pair lj/cut/coul/cut ecoul
compute 1 all pair reax
Description:
Define a computation that extracts additional values calculated by a pair style, and makes them accessible for
output or further processing by other commands. The group specified for this command is ignored.
The specified pstyle must be a pair style used in your simulation either by itself or as a sub-style in a
pair_style hybrid or hybrid/overlay command.
The evalue setting is optional; it may be left off the command. All pair styles tally a potential energy epair
which may be broken into two parts: evdwl and ecoul such that epair = evdwl + ecoul. If the pair style
calculates Coulombic interactions, their energy will be tallied in ecoul. Everything else (whether it is a
Lennard-Jones style van der Waals interaction or not) is tallied in evdwl. If evalue is specified as epair or left
out, then epair is stored as a global scalar by this compute. This is useful when using pair_style hybrid if you
want to know the portion of the total energy contributed by one sub-style. If evalue is specified as evdwl or
ecoul, then just that portion of the energy is stored as a global scalar.
NOTE: The energy returned by the evdwl keyword does not include tail corrections, even if they are enabled
via the pair_modify command.
Some pair styles tally additional quantities, e.g. a breakdown of potential energy into a dozen or so
components is tallied by the pair_style reax command. These values (1 or more) are stored as a global vector
by this compute. See the doc page for individual pair styles for info on these values.
Output info:
This compute calculates a global scalar which is epair or evdwl or ecoul. If the pair style supports it, it also
calculates a global vector of length >= 1, as determined by the pair style. These values can be used by any
command that uses global scalar or vector values from a compute as input. See this section for an overview of
LAMMPS output options.
The scalar and vector values calculated by this compute are "extensive".
LAMMPS Users Manual
1262

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute pair/local command
Syntax:
compute ID group-ID pair/local value1 value2 ... keyword args ...
ID, group-ID are documented in compute command• pair/local = style name of this compute command• one or more values may be appended• value = dist or eng or force or fx or fy or fz or pN
dist = pairwise distance
eng = pairwise energy
force = pairwise force
fx,fy,fz = components of pairwise force
pN = pair style specific quantities for allowed N values
•
zero or more keyword/arg pairs may be appended• keyword = cutoff
cutoff arg = type or radius
•
Examples:
compute 1 all pair/local eng
compute 1 all pair/local dist eng force
compute 1 all pair/local dist eng fx fy fz
compute 1 all pair/local dist fx fy fz p1 p2 p3
Description:
Define a computation that calculates properties of individual pairwise interactions. The number of datums
generated, aggregated across all processors, equals the number of pairwise interactions in the system.
The local data stored by this command is generated by looping over the pairwise neighbor list. Info about an
individual pairwise interaction will only be included if both atoms in the pair are in the specified compute
group, and if the current pairwise distance is less than the force cutoff distance for that interaction, as defined
by the pair_style and pair_coeff commands.
The value dist is the distance between the pair of atoms.
The value eng is the interaction energy for the pair of atoms.
The value force is the force acting between the pair of atoms, which is positive for a repulsive force and
negative for an attractive force. The values fx, fy, and fz are the xyz components of force on atom I.
A pair style may define additional pairwise quantities which can be accessed as p1 to pN, where N is defined
by the pair style. Most pair styles do not define any additional quantities, so N = 0. An example of ones that
do are the granular pair styles which calculate the tangential force between two particles and return its
components and magnitude acting on atom I for N = 1,2,3,4. See individual pair styles for detils.
LAMMPS Users Manual
1264
The value dist will be in distance units. The value eng will be in energy units. The values force, fx, fy, and fz
will be in force units. The values pN will be in whatever units the pair style defines.
The optional cutoff keyword determines how the force cutoff distance for an interaction is determined. For the
default setting of type, the pairwise cutoff defined by the pair_style command for the types of the two atoms is
used. For the radius setting, the sum of the radii of the two particles is used as a cutoff. For example, this is
appropriate for granular particles which only interact when they are overlapping, as computed by granular pair
styles.
Note that as atoms migrate from processor to processor, there will be no consistent ordering of the entries
within the local vector or array from one timestep to the next. The only consistency that is guaranteed is that
the ordering on a particular timestep will be the same for local vectors or arrays generated by other compute
commands. For example, pair output from the compute property/local command can be combined with data
from this command and output by the dump local command in a consistent way.
Here is an example of how to do this:
compute 1 all property/local patom1 patom2
compute 2 all pair/local dist eng force
dump 1 all local 1000 tmp.dump index c_1[1] c_1[2] c_2[1] c_2[2] c_2[3]
NOTE: For pairs, if two atoms I,J are involved in 1-2, 1-3, 1-4 interactions within the molecular topology,
their pairwise interaction may be turned off, and thus they may not appear in the neighbor list, and will not be
part of the local data created by this command. More specifically, this will be true of I,J pairs with a weighting
factor of 0.0; pairs with a non-zero weighting factor are included. The weighting factors for 1-2, 1-3, and 1-4
pairwise interactions are set by the special_bonds command. An exception is if long-range Coulombics are
being computed via the kspace_style command, then atom pairs with weighting factors of zero are still
included in the neighbor list, so that a portion of the long-range interaction contribution can be computed in
the pair style. Hence in that case, those atom pairs will be part of the local data created by this command.
Output info:
This compute calculates a local vector or local array depending on the number of keywords. The length of the
vector or number of rows in the array is the number of pairs. If a single keyword is specified, a local vector is
produced. If two or more keywords are specified, a local array is produced where the number of columns =
the number of keywords. The vector or array can be accessed by any command that uses local values from a
compute as input. See this section for an overview of LAMMPS output options.
The output for dist will be in distance units. The output for eng will be in energy units. The output for force,
fx, fy, and fz will be in force units. The outpur for pN will be in whatever units the pair style defines.
Restrictions: none
Related commands:
dump local, compute property/local
Default:
The keyword default is cutoff = type.
LAMMPS Users Manual
1265

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute pe command
Syntax:
compute ID group-ID pe keyword ...
ID, group-ID are documented in compute command• pe = style name of this compute command• zero or more keywords may be appended• keyword = pair or bond or angle or dihedral or improper or kspace or fix•
Examples:
compute 1 all pe
compute molPE all pe bond angle dihedral improper
Description:
Define a computation that calculates the potential energy of the entire system of atoms. The specified group
must be "all". See the compute pe/atom command if you want per-atom energies. These per-atom values could
be summed for a group of atoms via the compute reduce command.
The energy is calculated by the various pair, bond, etc potentials defined for the simulation. If no extra
keywords are listed, then the potential energy is the sum of pair, bond, angle, dihedral, improper, kspace
(long-range), and fix energy. I.e. it is as if all the keywords were listed. If any extra keywords are listed, then
only those components are summed to compute the potential energy.
The Kspace contribution requires 1 extra FFT each timestep the energy is calculated, if using the PPPM solver
via the kspace_style pppm command. Thus it can increase the cost of the PPPM calculation if it is needed on a
large fraction of the simulation timesteps.
Various fixes can contribute to the total potential energy of the system if the fix contribution is included. See
the doc pages for individual fixes for details of which ones compute a potential energy.
NOTE: The fix_modify energy yes command must also be specified if a fix is to contribute potential energy to
this command.
A compute of this style with the ID of "thermo_pe" is created when LAMMPS starts up, as if this command
were in the input script:
compute thermo_pe all pe
See the "thermo_style" command for more details.
Output info:
This compute calculates a global scalar (the potential energy). This value can be used by any command that
uses a global scalar value from a compute as input. See Section 6.15 for an overview of LAMMPS output
options.
LAMMPS Users Manual
1266

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute pe/atom command
Syntax:
compute ID group-ID pe/atom keyword ...
ID, group-ID are documented in compute command• pe/atom = style name of this compute command• zero or more keywords may be appended• keyword = pair or bond or angle or dihedral or improper or kspace or fix•
Examples:
compute 1 all pe/atom
compute 1 all pe/atom pair
compute 1 all pe/atom pair bond
Description:
Define a computation that computes the per-atom potential energy for each atom in a group. See the compute
pe command if you want the potential energy of the entire system.
The per-atom energy is calculated by the various pair, bond, etc potentials defined for the simulation. If no
extra keywords are listed, then the potential energy is the sum of pair, bond, angle, dihedral,improper, kspace
(long-range), and fix energy. I.e. it is as if all the keywords were listed. If any extra keywords are listed, then
only those components are summed to compute the potential energy.
Note that the energy of each atom is due to its interaction with all other atoms in the simulation, not just with
other atoms in the group.
For an energy contribution produced by a small set of atoms (e.g. 4 atoms in a dihedral or 3 atoms in a Tersoff
3-body interaction), that energy is assigned in equal portions to each atom in the set. E.g. 1/4 of the dihedral
energy to each of the 4 atoms.
The dihedral_style charmm style calculates pairwise interactions between 1-4 atoms. The energy contribution
of these terms is included in the pair energy, not the dihedral energy.
The KSpace contribution is calculated using the method in (Heyes) for the Ewald method and a related
method for PPPM, as specified by the kspace_style pppm command. For PPPM, the calculation requires 1
extra FFT each timestep that per-atom energy is calculated. This document describes how the long-range
per-atom energy calculation is performed.
Various fixes can contribute to the per-atom potential energy of the system if the fix contribution is included.
See the doc pages for individual fixes for details of which ones compute a per-atom potential energy.
NOTE: The fix_modify energy yes command must also be specified if a fix is to contribute per-atom potential
energy to this command.
LAMMPS Users Manual
1268

As an example of per-atom potential energy compared to total potential energy, these lines in an input script
should yield the same result in the last 2 columns of thermo output:
compute peratom all pe/atom
compute pe all reduce sum c_peratom
thermo_style custom step temp etotal press pe c_pe
NOTE: The per-atom energy does not include any Lennard-Jones tail corrections to the energy added by the
pair_modify tail yes command, since those are contributions to the global system energy.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-atom vector values will be in energy units.
Restrictions:
Related commands:
compute pe, compute stress/atom
Default: none
(Heyes) Heyes, Phys Rev B 49, 755 (1994),
LAMMPS Users Manual
1269

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute plasticity/atom command
Syntax:
compute ID group-ID plasticity/atom
ID, group-ID are documented in compute command• plasticity/atom = style name of this compute command•
Examples:
compute 1 all plasticity/atom
Description:
Define a computation that calculates the per-atom plasticity for each atom in a group. This is a quantity
relevant for Peridynamics models. See this document for an overview of LAMMPS commands for
Peridynamics modeling.
The plasticity for a Peridynamic particle is the so-called consistency parameter (lambda). For elastic
deformation lambda = 0, otherwise lambda > 0 for plastic deformation. For details, see (Mitchell) and the
PDF doc included in the LAMMPS distro in doc/PDF/PDLammps_EPS.pdf.
This command can be invoked for one of the Peridynamic pair styles: peri/eps.
The plasticity value will be 0.0 for atoms not in the specified compute group.
Output info:
This compute calculates a per-atom vector, which can be accessed by any command that uses per-atom values
from a compute as input. See Section_howto 15 for an overview of LAMMPS output options.
The per-atom vector values are unitless numbers (lambda) >= 0.0.
Restrictions:
This compute is part of the PERI package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
compute damage/atom, compute dilatation/atom
Default: none
(Mitchell) Mitchell, "A non-local, ordinary-state-based viscoelasticity model for peridynamics", Sandia
National Lab Report, 8064:1-28 (2011).
LAMMPS Users Manual
1270

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute pressure command
Syntax:
compute ID group-ID pressure temp-ID keyword ...
ID, group-ID are documented in compute command• pressure = style name of this compute command• temp-ID = ID of compute that calculates temperature, can be NULL if not needed• zero or more keywords may be appended• keyword = ke or pair or bond or angle or dihedral or improper or kspace or fix or virial•
Examples:
compute 1 all pressure thermo_temp
compute 1 all pressure NULL pair bond
Description:
Define a computation that calculates the pressure of the entire system of atoms. The specified group must be
"all". See the compute stress/atom command if you want per-atom pressure (stress). These per-atom values
could be summed for a group of atoms via the compute reduce command.
The pressure is computed by the formula
where N is the number of atoms in the system (see discussion of DOF below), Kb is the Boltzmann constant,
T is the temperature, d is the dimensionality of the system (2 or 3 for 2d/3d), and V is the system volume (or
area in 2d). The second term is the virial, equal to -dU/dV, computed for all pairwise as well as 2-body,
3-body, 4-body, manybody, and long-range interactions, where r_i and f_i are the position and force vector of
atom i, and the black dot indicates a dot product. When periodic boundary conditions are used, N' necessarily
includes periodic image (ghost) atoms outside the central box, and the position and force vectors of ghost
atoms are thus included in the summation. When periodic boundary conditions are not used, N' = N = the
number of atoms in the system. Fixes that impose constraints (e.g. the fix shake command) also contribute to
the virial term.
A symmetric pressure tensor, stored as a 6-element vector, is also calculated by this compute. The 6
components of the vector are ordered xx, yy, zz, xy, xz, yz. The equation for the I,J components (where I and
J = x,y,z) is similar to the above formula, except that the first term uses components of the kinetic energy
tensor and the second term uses components of the virial tensor:
LAMMPS Users Manual
1271

If no extra keywords are listed, the entire equations above are calculated. This includes a kinetic energy
(temperature) term and the virial as the sum of pair, bond, angle, dihedral, improper, kspace (long-range), and
fix contributions to the force on each atom. If any extra keywords are listed, then only those components are
summed to compute temperature or ke and/or the virial. The virial keyword means include all terms except
the kinetic energy ke.
Details of how LAMMPS computes the virial efficiently for the entire system, including for manybody
potentials and accounting for the effects of periodic boundary conditions are discussed in (Thompson).
The temperature and kinetic energy tensor is not calculated by this compute, but rather by the temperature
compute specified with the command. If the kinetic energy is not included in the pressure, than the
temperature compute is not used and can be specified as NULL. Normally the temperature compute used by
compute pressure should calculate the temperature of all atoms for consistency with the virial term, but any
compute style that calculates temperature can be used, e.g. one that excludes frozen atoms or other degrees of
freedom.
Note that if desired the specified temperature compute can be one that subtracts off a bias to calculate a
temperature using only the thermal velocity of the atoms, e.g. by subtracting a background streaming velocity.
See the doc pages for individual compute commands to determine which ones include a bias.
Also note that the N in the first formula above is really degrees-of-freedom divided by d = dimensionality,
where the DOF value is calculated by the temperature compute. See the various compute temperature styles
for details.
A compute of this style with the ID of "thermo_press" is created when LAMMPS starts up, as if this
command were in the input script:
compute thermo_press all pressure thermo_temp
where "thermo_temp" is the ID of a similarly defined compute of style "temp". See the "thermo_style"
command for more details.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
LAMMPS Users Manual
1272

See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Output info:
This compute calculates a global scalar (the pressure) and a global vector of length 6 (pressure tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar and vector values calculated by this compute are "intensive". The scalar and vector values will be
in pressure units.
Restrictions: none
Related commands:
compute temp, compute stress/atom, thermo_style,
Default: none
(Thompson) Thompson, Plimpton, Mattson, J Chem Phys, 131, 154107 (2009).
LAMMPS Users Manual
1273

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute property/atom command
Syntax:
compute ID group-ID property/atom input1 input2 ...
ID, group-ID are documented in compute command• property/atom = style name of this compute command• input = one or more atom attributes
possible attributes = id, mol, proc, type, mass,
x, y, z, xs, ys, zs, xu, yu, zu, ix, iy, iz,
vx, vy, vz, fx, fy, fz,
q, mux, muy, muz, mu,
radius, diameter, omegax, omegay, omegaz,
angmomx, angmomy, angmomz,
shapex,shapey, shapez,
quatw, quati, quatj, quatk, tqx, tqy, tqz,
end1x, end1y, end1z, end2x, end2y, end2z,
corner1x, corner1y, corner1z,
corner2x, corner2y, corner2z,
corner3x, corner3y, corner3z,
nbonds,
vfrac, s0,
spin, eradius, ervel, erforce,
rho, drho, e, de, cv,
i_name, d_name
id = atom ID
mol = molecule ID
proc = ID of processor that owns atom
type = atom type
mass = atom mass
x,y,z = unscaled atom coordinates
xs,ys,zs = scaled atom coordinates
xu,yu,zu = unwrapped atom coordinates
ix,iy,iz = box image that the atom is in
vx,vy,vz = atom velocities
fx,fy,fz = forces on atoms
q = atom charge
mux,muy,muz = orientation of dipole moment of atom
mu = magnitude of dipole moment of atom
radius,diameter = radius,diameter of spherical particle
omegax,omegay,omegaz = angular velocity of spherical particle
angmomx,angmomy,angmomz = angular momentum of aspherical particle
shapex,shapey,shapez = 3 diameters of aspherical particle
quatw,quati,quatj,quatk = quaternion components for aspherical or body particles
tqx,tqy,tqz = torque on finite-size particles
end12x, end12y, end12z = end points of line segment
corner123x, corner123y, corner123z = corner points of triangle
nbonds = number of bonds assigned to an atom
PERI package per-atom properties:
vfrac = ???
s0 = ???
•
LAMMPS Users Manual
1274
USER-EFF and USER-AWPMD package per-atom properties:
spin = electron spin
eradius = electron radius
ervel = electron radial velocity
erforce = electron radial force
USER-SPH package per-atom properties:
rho = ???
drho = ???
e = ???
de = ???
cv = ???
fix property/atom per-atom properties:
i_name = custom integer vector with name
d_name = custom integer vector with name
Examples:
compute 1 all property/atom xs vx fx mux
compute 2 all property/atom type
compute 1 all property/atom ix iy iz
Description:
Define a computation that simply stores atom attributes for each atom in the group. This is useful so that the
values can be used by other output commands that take computes as inputs. See for example, the compute
reduce, fix ave/atom, fix ave/histo, fix ave/chunk, and atom-style variable commands.
The list of possible attributes is the same as that used by the dump custom command, which describes their
meaning, with some additional quantities that are only defined for certain atom styles. Basically, this
augmented list gives an input script access to any per-atom quantity stored by LAMMPS.
The values are stored in a per-atom vector or array as discussed below. Zeroes are stored for atoms not in the
specified group or for quantities that are not defined for a particular particle in the group (e.g. shapex if the
particle is not an ellipsoid).
The additional quantities only accessible via this command, and not directly via the dump custom command,
are as follows.
Shapex, shapey, and shapez are defined for ellipsoidal particles and define the 3d shape of each particle.
Quatw, quati, quatj, and quatk are defined for ellipsoidal particles and body particles and store the 4-vector
quaternion representing the orientation of each particle. See the set command for an explanation of the
quaternion vector.
End1x, end1y, end1z, end2x, end2y, end2z, are defined for line segment particles and define the end points of
each line segment.
Corner1x, corner1y, corner1z, corner2x, corner2y, corner2z, corner3x, corner3y, corner3z, are defined for
triangular particles and define the corner points of each triangle.
Nbonds is available for all molecular atom styles and refers to the number of explicit bonds assigned to an
atom. Note that if the newton bond command is set to on, which is the default, then every bond in the system
LAMMPS Users Manual
1275
is assigned to only one of the two atoms in the bond. Thus a bond between atoms I,J may be tallied for either
atom I or atom J. If newton bond off is set, it will be tallied with both atom I and atom J.
The i_name and d_name attributes refer to custom integer and floating-point properties that have been added
to each atom via the fix property/atom command. When that command is used specific names are given to
each attribute which are what is specified as the "name" portion of i_name or d_name.
Output info:
This compute calculates a per-atom vector or per-atom array depending on the number of input values. If a
single input is specified, a per-atom vector is produced. If two or more inputs are specified, a per-atom array is
produced where the number of columns = the number of inputs. The vector or array can be accessed by any
command that uses per-atom values from a compute as input. See this section for an overview of LAMMPS
output options.
The vector or array values will be in whatever units the corresponding attribute is in, e.g. velocity units for vx,
charge units for q, etc.
Restrictions: none
Related commands:
dump custom, compute reduce, fix ave/atom, fix ave/chunk, fix property/atom
Default: none
LAMMPS Users Manual
1276

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute property/chunk command
Syntax:
compute ID group-ID property/chunk chunkID input1 input2 ...
ID, group-ID are documented in compute command• property/chunk = style name of this compute command• input = one or more attributes
attributes = count, id, coord1, coord2, coord3
count = # of atoms in chunk
id = original chunk IDs before compression by compute chunk/atom
coord123 = coordinates for spatial bins calculated by compute chunk/atom
•
Examples:
compute 1 all property/chunk count
compute 1 all property/chunk ID coord1
Description:
Define a computation that stores the specified attributes of chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
This compute calculates and stores the specified attributes of chunks as global data so they can be accessed by
other output commands and used in conjunction with other commands that generate per-chunk data, such as
compute com/chunk or compute msd/chunk.
Note that only atoms in the specified group contribute to the calculation of the count attribute. The compute
chunk/atom command defines its own group; atoms will have a chunk ID = 0 if they are not in that group,
signifying they are not assigned to a chunk, and will thus also not contribute to this calculation. You can
specify the "all" group for this command if you simply want to include atoms with non-zero chunk IDs.
The count attribute is the number of atoms in the chunk.
The id attribute stores the original chunk ID for each chunk. It can only be used if the compress keyword was
set to yes for the compute chunk/atom command referenced by chunkID. This means that the original chunk
IDs (e.g. molecule IDs) will have been compressed to remove chunk IDs with no atoms assigned to them.
Thus a compressed chunk ID of 3 may correspond to an original chunk ID (molecule ID in this case) of 415.
The id attribute will then be 415 for the 3rd chunk.
The coordN attributes can only be used if a binning style was used in the compute chunk/atom command
referenced by chunkID. For bin/1d, bin/2d, and bin/3d styles the attribute is the center point of the bin in the
corresponding dimension. Style bin/1d only defines a coord1 attribute. Style bin/2d adds a coord2 attribute.
LAMMPS Users Manual
1277
Style bin/3d adds a coord3 attribute.
Note that if the value of the units keyword used in the compute chunk/atom command is box or lattice, the
coordN attributes will be in distance units. If the value of the units keyword is reduced, the coordN attributes
will be in unitless reduced units (0-1).
The simplest way to output the results of the compute property/chunk calculation to a file is to use the fix
ave/time command, for example:
compute cc1 all chunk/atom molecule
compute myChunk1 all property/chunk cc1 count
compute myChunk2 all com/chunk cc1
fix 1 all ave/time 100 1 100 c_myChunk1 c_myChunk2[*] file tmp.out mode vector
Output info:
This compute calculates a global vector or global array depending on the number of input values. The length
of the vector or number of rows in the array is the number of chunks.
This compute calculates a global vector or global array where the number of rows = the number of chunks
Nchunk as calculated by the specified compute chunk/atom command. If a single input is specified, a global
vector is produced. If two or more inputs are specified, a global array is produced where the number of
columns = the number of inputs. The vector or array can be accessed by any command that uses global values
from a compute as input. See this section for an overview of LAMMPS output options.
The vector or array values are "intensive". The values will be unitless or in the units discussed above.
Restrictions: none
Related commands:
fix ave/chunk
Default: none
LAMMPS Users Manual
1278

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute property/local command
Syntax:
compute ID group-ID property/local attribute1 attribute2 ... keyword args ...
ID, group-ID are documented in compute command• property/local = style name of this compute command• one or more attributes may be appended
possible attributes = natom1 natom2 ntype1 ntype2
patom1 patom2 ptype1 ptype2
batom1 batom2 btype
aatom1 aatom2 aatom3 atype
datom1 datom2 datom3 dtype
iatom1 iatom2 iatom3 itype
natom1, natom2 = IDs of 2 atoms in each pair (within neighbor cutoff)
ntype1, ntype2 = type of 2 atoms in each pair (within neighbor cutoff)
patom1, patom2 = IDs of 2 atoms in each pair (within force cutoff)
ptype1, ptype2 = type of 2 atoms in each pair (within force cutoff)
batom1, batom2 = IDs of 2 atoms in each bond
btype = bond type of each bond
aatom1, aatom2, aatom3 = IDs of 3 atoms in each angle
atype = angle type of each angle
datom1, datom2, datom3, datom4 = IDs of 4 atoms in each dihedral
dtype = dihedral type of each dihedral
iatom1, iatom2, iatom3, iatom4 = IDs of 4 atoms in each improper
itype = improper type of each improper
•
zero or more keyword/arg pairs may be appended• keyword = cutoff
cutoff arg = type or radius
•
Examples:
compute 1 all property/local btype batom1 batom2
compute 1 all property/local atype aatom2
Description:
Define a computation that stores the specified attributes as local data so it can be accessed by other output
commands. If the input attributes refer to bond information, then the number of datums generated, aggregated
across all processors, equals the number of bonds in the system. Ditto for pairs, angles, etc.
If multiple attributes are specified then they must all generate the same amount of information, so that the
resulting local array has the same number of rows for each column. This means that only bond attributes can
be specified together, or angle attributes, etc. Bond and angle attributes can not be mixed in the same compute
property/local command.
If the inputs are pair attributes, the local data is generated by looping over the pairwise neighbor list. Info
about an individual pairwise interaction will only be included if both atoms in the pair are in the specified
LAMMPS Users Manual
1279
compute group. For natom1 and natom2, all atom pairs in the neighbor list are considered (out to the neighbor
cutoff = force cutoff + neighbor skin). For patom1 and patom2, the distance between the atoms must be less
than the force cutoff distance for that pair to be included, as defined by the pair_style and pair_coeff
commands.
The optional cutoff keyword determines how the force cutoff distance for an interaction is determined for the
patom1 and patom2 attributes. For the default setting of type, the pairwise cutoff defined by the pair_style
command for the types of the two atoms is used. For the radius setting, the sum of the radii of the two
particles is used as a cutoff. For example, this is appropriate for granular particles which only interact when
they are overlapping, as computed by granular pair styles.
If the inputs are bond, angle, etc attributes, the local data is generated by looping over all the atoms owned on
a processor and extracting bond, angle, etc info. For bonds, info about an individual bond will only be
included if both atoms in the bond are in the specified compute group. Likewise for angles, dihedrals, etc.
For bonds and angles, a bonds/angles that have been broken by setting their bond/angle type to 0 will not be
included. Bonds/angles that have been turned off (see the fix shake or delete_bonds commands) by setting
their bond/angle type negative are written into the file. This is consistent with the compute bond/local and
compute angle/local commands
Note that as atoms migrate from processor to processor, there will be no consistent ordering of the entries
within the local vector or array from one timestep to the next. The only consistency that is guaranteed is that
the ordering on a particular timestep will be the same for local vectors or arrays generated by other compute
commands. For example, output from the compute bond/local command can be combined with bond atom
indices from this command and output by the dump local command in a consistent way.
The natom1 and natom2, or patom1 and patom2 attributes refer to the atom IDs of the 2 atoms in each
pairwise interaction computed by the pair_style command. The ntype1 and ntype2, or ptype1 and ptype2
attributes refer to the atom types of the 2 atoms in each pairwise interaction.
NOTE: For pairs, if two atoms I,J are involved in 1-2, 1-3, 1-4 interactions within the molecular topology,
their pairwise interaction may be turned off, and thus they may not appear in the neighbor list, and will not be
part of the local data created by this command. More specifically, this may be true of I,J pairs with a
weighting factor of 0.0; pairs with a non-zero weighting factor are included. The weighting factors for 1-2,
1-3, and 1-4 pairwise interactions are set by the special_bonds command.
The batom1 and batom2 attributes refer to the atom IDs of the 2 atoms in each bond. The btype attribute refers
to the type of the bond, from 1 to Nbtypes = # of bond types. The number of bond types is defined in the data
file read by the read_data command.
The attributes that start with "a", "d", "i", refer to similar values for angles, dihedrals, and impropers.
Output info:
This compute calculates a local vector or local array depending on the number of input values. The length of
the vector or number of rows in the array is the number of bonds, angles, etc. If a single input is specified, a
local vector is produced. If two or more inputs are specified, a local array is produced where the number of
columns = the number of inputs. The vector or array can be accessed by any command that uses local values
from a compute as input. See this section for an overview of LAMMPS output options.
The vector or array values will be integers that correspond to the specified attribute.
LAMMPS Users Manual
1280

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute rdf command
Syntax:
compute ID group-ID rdf Nbin itype1 jtype1 itype2 jtype2 ... keyword/value ...
ID, group-ID are documented in compute command• rdf = style name of this compute command• Nbin = number of RDF bins• itypeN = central atom type for Nth RDF histogram (see asterisk form below)• jtypeN = distribution atom type for Nth RDF histogram (see asterisk form below)• zero or more keyword/value pairs may be appended• keyword = cutoff
cutoff value = Rcut
Rcut = cutoff distance for RDF computation (distance units)
•
Examples:
compute 1 all rdf 100
compute 1 all rdf 100 1 1
compute 1 all rdf 100 * 3 cutoff 5.0
compute 1 fluid rdf 500 1 1 1 2 2 1 2 2
compute 1 fluid rdf 500 1*3 2 5 *10 cutoff 3.5
Description:
Define a computation that calculates the radial distribution function (RDF), also called g(r), and the
coordination number for a group of particles. Both are calculated in histogram form by binning pairwise
distances into Nbin bins from 0.0 to the maximum force cutoff defined by the pair_style command or the
cutoff distance Rcut specified via the cutoff keyword. The bins are of uniform size in radial distance. Thus a
single bin encompasses a thin shell of distances in 3d and a thin ring of distances in 2d.
NOTE: If you have a bonded system, then the settings of special_bonds command can remove pairwise
interactions between atoms in the same bond, angle, or dihedral. This is the default setting for the
special_bonds command, and means those pairwise interactions do not appear in the neighbor list. Because
this fix uses a neighbor list, it also means those pairs will not be included in the RDF. This does not apply
when using long-range coulomb interactions (coul/long, coul/msm, coul/wolf or similar. One way to get
around this would be to set special_bond scaling factors to very tiny numbers that are not exactly zero (e.g.
1.0e-50). Another workaround is to write a dump file, and use the rerun command to compute the RDF for
snapshots in the dump file. The rerun script can use a special_bonds command that includes all pairs in the
neighbor list.
By default the RDF is computed out to the maximum force cutoff defined by the pair_style command. If the
cutoff keyword is used, then the RDF is computed accurately out to the Rcut > 0.0 distance specified.
NOTE: Normally, you should only use the cutoff keyword if no pair style is defined, e.g. the rerun command
is being used to post-process a dump file of snapshots. Or if you really want the RDF for distances beyond the
pair_style force cutoff and cannot easily post-process a dump file to calculate it. This is because using the
cutoff keyword incurs extra computation and possibly communication, which may slow down your
LAMMPS Users Manual
1282
simulation. If you specify a Rcut <= force cutoff, you will force an additional neighbor list to be built at every
timestep this command is invoked (or every reneighboring timestep, whichever is less frequent), which is
inefficient. LAMMPS will warn you if this is the case. If you specify a Rcut > force cutoff, you must insure
ghost atom information out to Rcut + skin is communicated, via the comm_modify cutoff command, else the
RDF computation cannot be performed, and LAMMPS will give an error message. The skin value is what is
specified with the neighbor command. In this case, you are forcing a large neighbor list to be built just for the
RDF computation, and extra communication to be performed every timestep.
The itypeN and jtypeN arguments are optional. These arguments must come in pairs. If no pairs are listed, then
a single histogram is computed for g(r) between all atom types. If one or more pairs are listed, then a separate
histogram is generated for each itype,jtype pair.
The itypeN and jtypeN settings can be specified in one of two ways. An explicit numeric value can be used, as
in the 4th example above. Or a wild-card asterisk can be used to specify a range of atom types. This takes the
form "*" or "*n" or "n*" or "m*n". If N = the number of atom types, then an asterisk with no numeric values
means all types from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A trailing asterisk
means all types from n to N (inclusive). A middle asterisk means all types from m to n (inclusive).
If both itypeN and jtypeN are single values, as in the 4th example above, this means that a g(r) is computed
where atoms of type itypeN are the central atom, and atoms of type jtypeN are the distribution atom. If either
itypeN and jtypeN represent a range of values via the wild-card asterisk, as in the 5th example above, this
means that a g(r) is computed where atoms of any of the range of types represented by itypeN are the central
atom, and atoms of any of the range of types represented by jtypeN are the distribution atom.
Pairwise distances are generated by looping over a pairwise neighbor list, just as they would be in a pair_style
computation. The distance between two atoms I and J is included in a specific histogram if the following
criteria are met:
atoms I,J are both in the specified compute group• the distance between atoms I,J is less than the maximum force cutoff• the type of the I atom matches itypeN (one or a range of types)• the type of the J atom matches jtypeN (one or a range of types)•
It is OK if a particular pairwise distance is included in more than one individual histogram, due to the way the
itypeN and jtypeN arguments are specified.
The g(r) value for a bin is calculated from the histogram count by scaling it by the idealized number of how
many counts there would be if atoms of type jtypeN were uniformly distributed. Thus it involves the count of
itypeN atoms, the count of jtypeN atoms, the volume of the entire simulation box, and the volume of the bin's
thin shell in 3d (or the area of the bin's thin ring in 2d).
A coordination number coord(r) is also calculated, which is the number of atoms of type jtypeN within the
current bin or closer, averaged over atoms of type itypeN. This is calculated as the area- or volume-weighted
sum of g(r) values over all bins up to and including the current bin, multiplied by the global average volume
density of atoms of type jtypeN.
The simplest way to output the results of the compute rdf calculation to a file is to use the fix ave/time
command, for example:
compute myRDF all rdf 50
fix 1 all ave/time 100 1 100 c_myRDF[*] file tmp.rdf mode vector
LAMMPS Users Manual
1283
Output info:
This compute calculates a global array with the number of rows = Nbins, and the number of columns = 1 +
2*Npairs, where Npairs is the number of I,J pairings specified. The first column has the bin coordinate (center
of the bin), Each successive set of 2 columns has the g(r) and coord(r) values for a specific set of itypeN
versus jtypeN interactions, as described above. These values can be used by any command that uses a global
values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The array values calculated by this compute are all "intensive".
The first column of array values will be in distance units. The g(r) columns of array values are normalized
numbers >= 0.0. The coordination number columns of array values are also numbers >= 0.0.
Restrictions:
The RDF is not computed for distances longer than the force cutoff, since processors (in parallel) don't know
about atom coordinates for atoms further away than that distance. If you want an RDF for larger distances,
you can use the rerun command to post-process a dump file and set the cutoff for the potential to be longer in
the rerun script. Note that in the rerun context, the force cutoff is arbitrary, since you aren't running dynamics
and thus are not changing your model. The definition of g(r) used by LAMMPS is only appropriate for
characterizing atoms that are uniformly distributed throughout the simulation cell. In such cases, the
coordination number is still correct and meaningful. As an example, if a large simulation cell contains only
one atom of type itypeN and one of jtypeN, then g(r) will register an arbitrarily large spike at whatever
distance they happen to be at, and zero everywhere else. Coord(r) will show a step change from zero to one at
the location of the spike in g(r).
Related commands:
fix ave/time
Default:
The keyword defaults are cutoff = 0.0 (use the pairwise force cutoff).
LAMMPS Users Manual
1284

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute reduce command
compute reduce/region command
Syntax:
compute ID group-ID style arg mode input1 input2 ... keyword args ...
ID, group-ID are documented in compute command• style = reduce or reduce/region
reduce arg = none
reduce/region arg = region-ID
region-ID = ID of region to use for choosing atoms
•
mode = sum or min or max or ave or sumsq or avesq• one or more inputs can be listed• input = x, y, z, vx, vy, vz, fx, fy, fz, c_ID, c_ID[N], f_ID, f_ID[N], v_name
x,y,z,vx,vy,vz,fx,fy,fz = atom attribute (position, velocity, force component)
c_ID = per-atom or local vector calculated by a compute with ID
c_ID[I] = Ith column of per-atom or local array calculated by a compute with ID, I can include wildcard (see below)
f_ID = per-atom or local vector calculated by a fix with ID
f_ID[I] = Ith column of per-atom or local array calculated by a fix with ID, I can include wildcard (see below)
v_name = per-atom vector calculated by an atom-style variable with name
•
zero or more keyword/args pairs may be appended• keyword = replace
replace args = vec1 vec2
vec1 = reduced value from this input vector will be replaced
vec2 = replace it with vec1[N] where N is index of max/min value from vec2
•
Examples:
compute 1 all reduce sum c_force
compute 1 all reduce/region subbox sum c_force
compute 2 all reduce min c_press[2] f_ave v_myKE
compute 2 all reduce min c_press[*] f_ave v_myKE
compute 3 fluid reduce max c_index[1] c_index[2] c_dist replace 1 3 replace 2 3
Description:
Define a calculation that "reduces" one or more vector inputs into scalar values, one per listed input. The
inputs can be per-atom or local quantities; they cannot be global quantities. Atom attributes are per-atom
quantities, computes and fixes may generate any of the three kinds of quantities, and atom-style variables
generate per-atom quantities. See the variable command and its special functions which can perform the same
operations as the compute reduce command on global vectors.
The reduction operation is specified by the mode setting. The sum option adds the values in the vector into a
global total. The min or max options find the minimum or maximum value across all vector values. The ave
setting adds the vector values into a global total, then divides by the number of values in the vector. The
sumsq option sums the square of the values in the vector into a global total. The avesq setting does the same
LAMMPS Users Manual
1285

as sumsq, then divdes the sum of squares by the number of values. The last two options can be useful for
calculating the variance of some quantity, e.g. variance = sumsq - ave^2.
Each listed input is operated on independently. For per-atom inputs, the group specified with this command
means only atoms within the group contribute to the result. For per-atom inputs, if the compute reduce/region
command is used, the atoms must also currently be within the region. Note that an input that produces
per-atom quantities may define its own group which affects the quantities it returns. For example, if a
compute is used as an input which generates a per-atom vector, it will generate values of 0.0 for atoms that are
not in the group specified for that compute.
Each listed input can be an atom attribute (position, velocity, force component) or can be the result of a
compute or fix or the evaluation of an atom-style variable.
Note that for values from a compute or fix, the bracketed index I can be specified using a wildcard asterisk
with the index to effectively specify multiple values. This takes the form "*" or "*n" or "n*" or "m*n". If N =
the size of the vector (for mode = scalar) or the number of columns in the array (for mode = vector), then an
asterisk with no numeric values means all indices from 1 to N. A leading asterisk means all indices from 1 to
n (inclusive). A trailing asterisk means all indices from n to N (inclusive). A middle asterisk means all indices
from m to n (inclusive).
Using a wildcard is the same as if the individual columns of the array had been listed one by one. E.g. these 2
compute reduce commands are equivalent, since the compute stress/atom command creates a per-atom array
with 6 columns:
compute myPress all stress/atom NULL
compute 2 all reduce min myPress[*]
compute 2 all reduce min myPress[1] myPress[2] myPress[3] &
myPress[4] myPress[5] myPress[6]
The atom attribute values (x,y,z,vx,vy,vz,fx,fy,fz) are self-explanatory. Note that other atom attributes can be
used as inputs to this fix by using the compute property/atom command and then specifying an input value
from that compute.
If a value begins with "c_", a compute ID must follow which has been previously defined in the input script.
Computes can generate per-atom or local quantities. See the individual compute doc page for details. If no
bracketed integer is appended, the vector calculated by the compute is used. If a bracketed integer is
appended, the Ith column of the array calculated by the compute is used. Users can also write code for their
own compute styles and add them to LAMMPS. See the discussion above for how I can be specified with a
wildcard asterisk to effectively specify multiple values.
If a value begins with "f_", a fix ID must follow which has been previously defined in the input script. Fixes
can generate per-atom or local quantities. See the individual fix doc page for details. Note that some fixes only
produce their values on certain timesteps, which must be compatible with when compute reduce references the
values, else an error results. If no bracketed integer is appended, the vector calculated by the fix is used. If a
bracketed integer is appended, the Ith column of the array calculated by the fix is used. Users can also write
code for their own fix style and add them to LAMMPS. See the discussion above for how I can be specified
with a wildcard asterisk to effectively specify multiple values.
If a value begins with "v_", a variable name must follow which has been previously defined in the input
script. It must be an atom-style variable. Atom-style variables can reference thermodynamic keywords and
various per-atom attributes, or invoke other computes, fixes, or variables when they are evaluated, so this is a
LAMMPS Users Manual
1286

very general means of generating per-atom quantities to reduce.
If the replace keyword is used, two indices vec1 and vec2 are specified, where each index ranges from 1 to the
# of input values. The replace keyword can only be used if the mode is min or max. It works as follows. A
min/max is computed as usual on the vec2 input vector. The index N of that value within vec2 is also stored.
Then, instead of performing a min/max on the vec1 input vector, the stored index is used to select the Nth
element of the vec1 vector.
Thus, for example, if you wish to use this compute to find the bond with maximum stretch, you can do it as
follows:
compute 1 all property/local batom1 batom2
compute 2 all bond/local dist
compute 3 all reduce max c_1[1] c_1[2] c_2 replace 1 3 replace 2 3
thermo_style custom step temp c_3[1] c_3[2] c_3[3]
The first two input values in the compute reduce command are vectors with the IDs of the 2 atoms in each
bond, using the compute property/local command. The last input value is bond distance, using the compute
bond/local command. Instead of taking the max of the two atom ID vectors, which does not yield useful
information in this context, the replace keywords will extract the atom IDs for the two atoms in the bond of
maximum stretch. These atom IDs and the bond stretch will be printed with thermodynamic output.
If a single input is specified this compute produces a global scalar value. If multiple inputs are specified, this
compute produces a global vector of values, the length of which is equal to the number of inputs specified.
As discussed below, for the sum and sumsq modes, the value(s) produced by this compute are all "extensive",
meaning their value scales linearly with the number of atoms involved. If normalized values are desired, this
compute can be accessed by the thermo_style custom command with thermo_modify norm yes set as an
option. Or it can be accessed by a variable that divides by the appropriate atom count.
Output info:
This compute calculates a global scalar if a single input value is specified or a global vector of length N where
N is the number of inputs, and which can be accessed by indices 1 to N. These values can be used by any
command that uses global scalar or vector values from a compute as input. See Section 6.15 for an overview
of LAMMPS output options.
All the scalar or vector values calculated by this compute are "intensive", except when the sum or sumsq
modes are used on per-atom or local vectors, in which case the calculated values are "extensive".
The scalar or vector values will be in whatever units the quantities being reduced are in.
Restrictions: none
Related commands:
compute, fix, variable
Default: none
LAMMPS Users Manual
1287

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute rigid/local command
Syntax:
compute ID group-ID rigid/local rigidID input1 input2 ...
ID, group-ID are documented in compute command• rigid/local = style name of this compute command• rigidID = ID of fix rigid/small command or one of its variants• input = one or more rigid body attributes
possible attributes = id, mol, mass,
x, y, z, xu, yu, zu, ix, iy, iz
vx, vy, vz, fx, fy, fz,
omegax, omegay, omegaz,
angmomx, angmomy, angmomz,
quatw, quati, quatj, quatk,
tqx, tqy, tqz,
inertiax, inertiay, inertiaz
id = atom ID of atom within body which owns body properties
mol = molecule ID used to define body in fix rigid/small command
mass = total mass of body
x,y,z = center of mass coords of body
xu,yu,zu = unwrapped center of mass coords of body
ix,iy,iz = box image that the center of mass is in
vx,vy,vz = center of mass velocities
fx,fy,fz = force of center of mass
omegax,omegay,omegaz = angular velocity of body
angmomx,angmomy,angmomz = angular momentum of body
quatw,quati,quatj,quatk = quaternion components for body
tqx,tqy,tqz = torque on body
inertiax,inertiay,inertiaz = diagonalized moments of inertia of body
•
Examples:
compute 1 all rigid/local myRigid mol x y z
Description:
Define a computation that simply stores rigid body attributes for rigid bodies defined by the fix rigid/small
command or one of its NVE, NVT, NPT, NPH variants. The data is stored as local data so it can be accessed
by other output commands that process local data, such as the compute reduce or dump local commands.
Note that this command only works with the fix rigid/small command or its variants, not the fix rigid
command and its variants. The ID of the fix rigid/small command used to define rigid bodies must be
specified as rigidID. The fix rigid command is typically used to define a handful of (potentially very large)
rigid bodies. It outputs similar per-body information as this command directly from the fix as global data; see
the fix rigid doc page for details
The local data stored by this command is generated by looping over all the atoms owned on a processor. If the
atom is not in the specified group-ID or is not part of a rigid body it is skipped. If it is not the atom within a
body that is assigned to store the body information it is skipped (only one atom per body is so assigned). If it
LAMMPS Users Manual
1288

is the assigned atom, then the info for that body is output. This means that information for N bodies is
generated. N may be less than the # of bodies defined by the fix rigid command, if the atoms in some bodies
are not in the group-ID.
NOTE: Which atom in a body owns the body info is determined internal to LAMMPS; it's the one nearest the
geometric center of the body. Typically you should avoid this complication, by defining the group associated
with this fix to include/exclude entire bodies.
Note that as atoms and bodies migrate from processor to processor, there will be no consistent ordering of the
entries within the local vector or array from one timestep to the next.
Here is an example of how to use this compute to dump rigid body info to a file:
compute 1 all rigid/local myRigid mol x y z fx fy fz
dump 1 all local 1000 tmp.dump index c_1[1] c_1[2] c_1[3] c_1[4] c_1[5] c_1[6] c_1[7]
This section explains the rigid body attributes that can be specified.
The id attribute is the atomID of the atom which owns the rigid body, which is assigned by the fix rigid/small
command.
The mol attribute is the molecule ID of the rigid body. It should be the molecule ID which all of the atoms in
the body belong to, since that is how the fix rigid/small command defines its rigid bodies.
The mass attribute is the total mass of the rigid body.
There are two options for outputting the coordinates of the center of mass (COM) of the body. The x, y, z
attributes write the COM "unscaled", in the appropriate distance units (Angstroms, sigma, etc). Use xu, yu, zu
if you want the COM "unwrapped" by the image flags for each atobody. Unwrapped means that if the body
COM has passed thru a periodic boundary one or more times, the value is generated what the COM coordinate
would be if it had not been wrapped back into the periodic box.
The image flags for the body can be generated directly using the ix, iy, iz attributes. For periodic dimensions,
they specify which image of the simulation box the COM is considered to be in. An image of 0 means it is
inside the box as defined. A value of 2 means add 2 box lengths to get the true value. A value of -1 means
subtract 1 box length to get the true value. LAMMPS updates these flags as the rigid body COMs cross
periodic boundaries during the simulation.
The vx, vy, vz, fx, fy, fz attributes are components of the COM velocity and force on the COM of the body.
The omegax, omegay, and omegaz attributes are the angular velocity components of the body around its
COM.
The angmomx, angmomy, and angmomz attributes are the angular momentum components of the body around
its COM.
The quatw, quati, quatj, and quatk attributes are the components of the 4-vector quaternion representing the
orientation of the rigid body. See the set command for an explanation of the quaternion vector.
The angmomx, angmomy, and angmomz attributes are the angular momentum components of the body around
its COM.
LAMMPS Users Manual
1289

The tqx, tqy, tqz attributes are components of the torque acting on the body around its COM.
The inertiax, inertiay, inertiaz attributes are components of diagonalized inertia tensor for the body, i.e the 3
moments of inertia for the body around its principal axes, as computed internally by LAMMPS.
Output info:
This compute calculates a local vector or local array depending on the number of keywords. The length of the
vector or number of rows in the array is the number of rigid bodies. If a single keyword is specified, a local
vector is produced. If two or more keywords are specified, a local array is produced where the number of
columns = the number of keywords. The vector or array can be accessed by any command that uses local
values from a compute as input. See this section for an overview of LAMMPS output options.
The vector or array values will be in whatever units the corresponding attribute is in:
id,mol = unitless• mass = mass units• x,y,z and xy,yu,zu = distance units• vx,vy,vz = velocity units• fx,fy,fz = force units• omegax,omegay,omegaz = radians/time units• angmomx,angmomy,angmomz = mass*distance^2/time units• quatw,quati,quatj,quatk = unitless• tqx,tqy,tqz = torque units• inertiax,inertiay,inertiaz = mass*distance^2 units•
Restrictions:
This compute is part of the RIGID package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
dump local, compute reduce
Default: none
LAMMPS Users Manual
1290

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute saed command
Syntax:
compute ID group-ID saed lambda type1 type2 ... typeN keyword value ...
ID, group-ID are documented in compute command• saed = style name of this compute command• lambda = wavelength of incident radiation (length units)• type1 type2 ... typeN = chemical symbol of each atom type (see valid options below)• zero or more keyword/value pairs may be appended• keyword = Kmax or Zone or dR_Ewald or c or manual or echo
Kmax value = Maximum distance explored from reciprocal space origin
(inverse length units)
Zone values = z1 z2 z3
z1,z2,z3 = Zone axis of incident radiation. If z1=z2=z3=0 all
reciprocal space will be meshed up to Kmax
dR_Ewald value = Thickness of Ewald sphere slice intercepting
reciprocal space (inverse length units)
c values = c1 c2 c3
c1,c2,c3 = parameters to adjust the spacing of the reciprocal
lattice nodes in the h, k, and l directions respectively
manual = flag to use manual spacing of reciprocal lattice points
based on the values of the c parameters
echo = flag to provide extra output for debugging purposes
•
Examples:
compute 1 all saed 0.0251 Al O Kmax 1.70 Zone 0 0 1 dR_Ewald 0.01 c 0.5 0.5 0.5
compute 2 all saed 0.0251 Ni Kmax 1.70 Zone 0 0 0 c 0.05 0.05 0.05 manual echo
fix saed/vtk 1 1 1 c_1 file Al2O3_001.saed
fix saed/vtk 1 1 1 c_2 file Ni_000.saed
Description:
Define a computation that calculates electron diffraction intensity as described in (Coleman) on a mesh of
reciprocal lattice nodes defined by the entire simulation domain (or manually) using simulated radiation of
wavelength lambda.
The electron diffraction intensity I at each reciprocal lattice point is computed from the structure factor F
using the equations:
LAMMPS Users Manual
1291
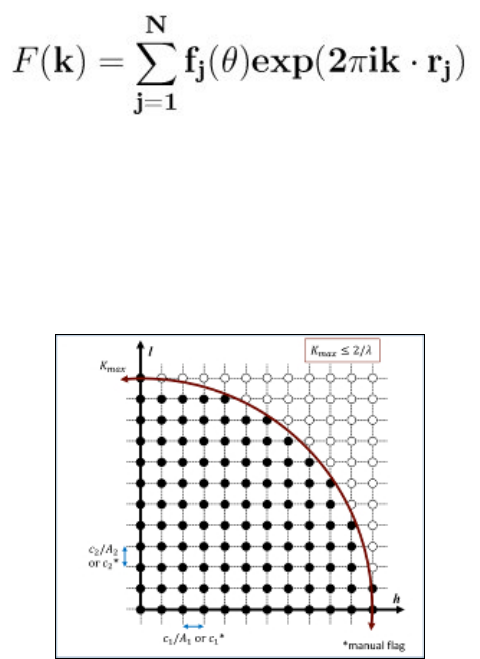
Here, K is the location of the reciprocal lattice node, rj is the position of each atom, fj are atomic scattering
factors.
Diffraction intensities are calculated on a three-dimensional mesh of reciprocal lattice nodes. The mesh
spacing is defined either (a) by the entire simulation domain or (b) manually using selected values as shown in
the 2D diagram below.
For a mesh defined by the simulation domain, a rectilinear grid is constructed with spacing c*inv(A) along
each reciprocal lattice axis. Where A are the vectors corresponding to the edges of the simulation cell. If one
or two directions has non-periodic boundary conditions, then the spacing in these directions is defined from
the average of the (inversed) box lengths with periodic boundary conditions. Meshes defined by the
simulation domain must contain at least one periodic boundary.
If the manual flag is included, the mesh of reciprocal lattice nodes will defined using the c values for the
spacing along each reciprocal lattice axis. Note that manual mapping of the reciprocal space mesh is good for
comparing diffraction results from multiple simulations; however it can reduce the likelihood that Bragg
reflections will be satisfied unless small spacing parameters
The limits of the reciprocal lattice mesh are determined by the use of the Kmax, Zone, and dR_Ewald
parameters. The rectilinear mesh created about the origin of reciprocal space is terminated at the boundary of
a sphere of radius Kmax centered at the origin. If Zone parameters z1=z2=z3=0 are used, diffraction
intensities are computed throughout the entire spherical volume - note this can greatly increase the cost of
computation. Otherwise, Zone parameters will denote the z1=h, z2=k, and z3=l (in a global since) zone axis of
an intersecting Ewald sphere. Diffraction intensities will only be computed at the intersection of the reciprocal
lattice mesh and a dR_Ewald thick surface of the Ewald sphere. See the example 3D intensity data and the
intersection of a [010] zone axis in the below image.
LAMMPS Users Manual
1292
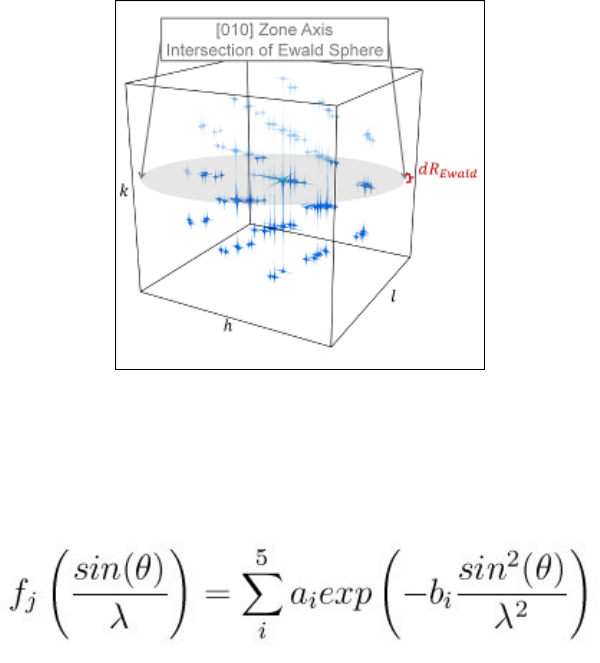
The atomic scattering factors, fj, accounts for the reduction in diffraction intensity due to Compton scattering.
Compute saed uses analytical approximations of the atomic scattering factors that vary for each atom type
(type1 type2 ... typeN) and angle of diffraction. The analytic approximation is computed using the formula
(Brown):
Coefficients parameterized by (Fox) are assigned for each atom type designating the chemical symbol and
charge of each atom type. Valid chemical symbols for compute saed are:
H: He: Li: Be: B: C: N: O: F: Ne: Na: Mg: Al: Si: P: S: Cl: Ar: K: Ca: Sc: Ti: V: Cr: Mn: Fe: Co: Ni: Cu: Zn:
Ga: Ge: As: Se: Br: Kr: Rb: Sr: Y: Zr: Nb: Mo: Tc: Ru: Rh: Pd: Ag: Cd: In: Sn: Sb: Te: I: Xe: Cs: Ba: La: Ce:
Pr: Nd: Pm: Sm: Eu: Gd: Tb: Dy: Ho: Er: Tm: Yb: Lu: Hf: Ta: W: Re: Os: Ir: Pt: Au: Hg: Tl: Pb: Bi: Po: At:
Rn: Fr: Ra: Ac: Th: Pa: U: Np: Pu: Am: Cm: Bk: Cf:tb(c=5,s=:)
If the echo keyword is specified, compute saed will provide extra reporting information to the screen.
Output info:
This compute calculates a global vector. The length of the vector is the number of reciprocal lattice nodes that
are explored by the mesh. The entries of the global vector are the computed diffraction intensities as described
above.
The vector can be accessed by any command that uses global values from a compute as input. See this section
for an overview of LAMMPS output options.
All array values calculated by this compute are "intensive".
Restrictions:
This compute is part of the USER-DIFFRACTION package. It is only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info.
LAMMPS Users Manual
1293

The compute_saed command does not work for triclinic cells.
Related commands:
fix saed_vtk, compute xrd
Default:
The option defaults are Kmax = 1.70, Zone 1 0 0, c 1 1 1, dR_Ewald = 0.01.
(Coleman) Coleman, Spearot, Capolungo, MSMSE, 21, 055020 (2013).
(Brown) Brown et al. International Tables for Crystallography Volume C: Mathematical and Chemical
Tables, 554-95 (2004).
(Fox) Fox, O'Keefe, Tabbernor, Acta Crystallogr. A, 45, 786-93 (1989).
LAMMPS Users Manual
1294

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute slice command
Syntax:
compute ID group-ID slice Nstart Nstop Nskip input1 input2 ...
ID, group-ID are documented in compute command• slice = style name of this compute command• Nstart = starting index within input vector(s)• Nstop = stopping index within input vector(s)• Nskip = extract every Nskip elements from input vector(s)• input = c_ID, c_ID[N], f_ID, f_ID[N]
c_ID = global vector calculated by a compute with ID
c_ID[I] = Ith column of global array calculated by a compute with ID
f_ID = global vector calculated by a fix with ID
f_ID[I] = Ith column of global array calculated by a fix with ID
v_name = vector calculated by an vector-style variable with name
•
Examples:
compute 1 all slice 1 100 10 c_msdmol[4]
compute 1 all slice 301 400 1 c_msdmol[4] v_myVec
Description:
Define a calculation that "slices" one or more vector inputs into smaller vectors, one per listed input. The
inputs can be global quantities; they cannot be per-atom or local quantities. Computes and fixes and
vector-style variables can generate such global quantities. The group specified with this command is ignored.
The values extracted from the input vector(s) are determined by the Nstart, Nstop, and Nskip parameters. The
elements of an input vector of length N are indexed from 1 to N. Starting at element Nstart, every Mth
element is extracted, where M = Nskip, until element Nstop is reached. The extracted quantities are stored as a
vector, which is typically shorter than the input vector.
Each listed input is operated on independently to produce one output vector. Each listed input must be a
global vector or column of a global array calculated by another compute or fix.
If an input value begins with "c_", a compute ID must follow which has been previously defined in the input
script and which generates a global vector or array. See the individual compute doc page for details. If no
bracketed integer is appended, the vector calculated by the compute is used. If a bracketed integer is
appended, the Ith column of the array calculated by the compute is used. Users can also write code for their
own compute styles and add them to LAMMPS.
If a value begins with "f_", a fix ID must follow which has been previously defined in the input script and
which generates a global vector or array. See the individual fix doc page for details. Note that some fixes only
produce their values on certain timesteps, which must be compatible with when compute slice references the
values, else an error results. If no bracketed integer is appended, the vector calculated by the fix is used. If a
bracketed integer is appended, the Ith column of the array calculated by the fix is used. Users can also write
code for their own fix style and add them to LAMMPS.
LAMMPS Users Manual
1295

If an input value begins with "v_", a variable name must follow which has been previously defined in the
input script. Only vector-style variables can be referenced. See the variable command for details. Note that
variables of style vector define a formula which can reference individual atom properties or thermodynamic
keywords, or they can invoke other computes, fixes, or variables when they are evaluated, so this is a very
general means of specifying quantities to slice.
If a single input is specified this compute produces a global vector, even if the length of the vector is 1. If
multiple inputs are specified, then a global array of values is produced, with the number of columns equal to
the number of inputs specified.
Output info:
This compute calculates a global vector if a single input value is specified or a global array with N columns
where N is the number of inputs. The length of the vector or the number of rows in the array is equal to the
number of values extracted from each input vector. These values can be used by any command that uses
global vector or array values from a compute as input. See this section for an overview of LAMMPS output
options.
The vector or array values calculated by this compute are simply copies of values generated by computes or
fixes or variables that are input vectors to this compute. If there is a single input vector of intensive and/or
extensive values, then each value in the vector of values calculated by this compute will be "intensive" or
"extensive", depending on the corresponding input value. If there are multiple input vectors, and all the values
in them are intensive, then the array values calculated by this compute are "intensive". If there are multiple
input vectors, and any value in them is extensive, then the array values calculated by this compute are
"extensive". Values produced by a variable are treated as intensive.
The vector or array values will be in whatever units the input quantities are in.
Restrictions: none
Related commands:
compute, fix, compute reduce
Default: none
LAMMPS Users Manual
1296

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/contact/radius command
Syntax:
compute ID group-ID smd/contact/radius
ID, group-ID are documented in compute command• smd/contact/radius = style name of this compute command•
Examples:
compute 1 all smd/contact/radius
Description:
Define a computation which outputs the contact radius, i.e., the radius used to prevent particles from
penetrating each other. The contact radius is used only to prevent particles belonging to different physical
bodies from penetrating each other. It is used by the contact pair styles, e.g., smd/hertz and smd/tri_surface.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
The value of the contact radius will be 0.0 for particles not in the specified compute group.
Output info:
This compute calculates a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-particle vector values will be in distance units.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
dump custom smd/hertz smd/tri_surface
Default: none
LAMMPS Users Manual
1297

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/damage command
Syntax:
compute ID group-ID smd/damage
ID, group-ID are documented in compute command• smd/damage = style name of this compute command•
Examples:
compute 1 all smd/damage
Description:
Define a computation that calculates the damage status of SPH particles according to the damage model which
is defined via the SMD SPH pair styles, e.g., the maximum plastic strain failure criterion.
See this PDF guide to use Smooth Mach Dynamics in LAMMPS.
Output Info:
This compute calculates a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See How-to discussions, section 6.15 for an overview of LAMMPS output
options.
The per-particle values are dimensionless an in the range of zero to one.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
smd/plastic_strain, smd/tlsph_stress
Default: none
LAMMPS Users Manual
1298

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/hourglass/error command
Syntax:
compute ID group-ID smd/hourglass/error
ID, group-ID are documented in compute command• smd/hourglass/error = style name of this compute command•
Examples:
compute 1 all smd/hourglass/error
Description:
Define a computation which outputs the error of the approximated relative separation with respect to the
actual relative separation of the particles i and j. Ideally, if the deformation gradient is exact, and there exists a
unique mapping between all particles' positions within the neighborhood of the central node and the
deformation gradient, the approximated relative separation will coincide with the actual relative separation of
the particles i and j in the deformed configuration. This compute is only really useful for debugging the
hourglass control mechanim which is part of the Total-Lagrangian SPH pair style.
See this PDF guide to use Smooth Mach Dynamics in LAMMPS.
Output Info:
This compute calculates a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See How-to discussions, section 6.15 for an overview of LAMMPS output
options.
The per-particle vector values will are dimensionless. See units.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This quantity will be computed only for particles which interact with tlsph pair style.
Related Commands:
smd/tlsph_defgrad
Default:
LAMMPS Users Manual
1299

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/internal/energy command
Syntax:
compute ID group-ID smd/internal/energy
ID, group-ID are documented in compute command• smd/smd/internal/energy = style name of this compute command•
Examples:
compute 1 all smd/internal/energy
Description:
Define a computation which outputs the per-particle enthalpy, i.e., the sum of potential energy and heat.
See this PDF guide to use Smooth Mach Dynamics in LAMMPS.
Output Info:
This compute calculates a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See How-to discussions, section 6.15 for an overview of LAMMPS output
options.
The per-particle vector values will be given in units of energy.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info. This compute can only be used for particles which interact
via the updated Lagrangian or total Lagrangian SPH pair styles.
Related Commands:
Default:
LAMMPS Users Manual
1300

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/plastic/strain command
Syntax:
compute ID group-ID smd/plastic/strain
ID, group-ID are documented in compute command• smd/plastic/strain = style name of this compute command•
Examples:
compute 1 all smd/plastic/strain
Description:
Define a computation that outputs the equivalent plastic strain per particle. This command is only meaningful
if a material model with plasticity is defined.
See this PDF guide to use Smooth Mach Dynamics in LAMMPS.
Output Info:
This compute calculates a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See How-to discussions, section 6.15 for an overview of LAMMPS output
options.
The per-particle values will be given dimensionless. See units.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info. This compute can only be used for particles which interact
via the updated Lagrangian or total Lagrangian SPH pair styles.
Related commands:
smd/plastic/strain/rate, smd/tlsph/strain/rate, smd/tlsph/strain
Default: none
LAMMPS Users Manual
1301

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/plastic/strain/rate command
Syntax:
compute ID group-ID smd/plastic/strain/rate
ID, group-ID are documented in compute command• smd/plastic/strain/rate = style name of this compute command•
Examples:
compute 1 all smd/plastic/strain/rate
Description:
Define a computation that outputs the time rate of the equivalent plastic strain. This command is only
meaningful if a material model with plasticity is defined.
See this PDF guide to use Smooth Mach Dynamics in LAMMPS.
Output Info:
This compute calculates a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See How-to discussions, section 6.15 for an overview of LAMMPS output
options.
The per-particle values will be given in units of one over time.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info. This compute can only be used for particles which interact
via the updated Lagrangian or total Lagrangian SPH pair styles.
Related commands:
smd/plastic/strain, smd/tlsph/strain/rate, smd/tlsph/strain
Default: none
LAMMPS Users Manual
1302

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/rho command
Syntax:
compute ID group-ID smd/rho
ID, group-ID are documented in compute command• smd/rho = style name of this compute command•
Examples:
compute 1 all smd/rho
Description:
Define a computation that calculates the per-particle mass density. The mass density is the mass of a particle
which is constant during the course of a simulation, divided by its volume, which can change due to
mechanical deformation.
See this PDF guide to use Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See How-to discussions, section 6.15 for an overview of LAMMPS output
options.
The per-particle values will be in units of mass over volume.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
compute smd/vol
Default: none
LAMMPS Users Manual
1303

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/tlsph/defgrad command
Syntax:
compute ID group-ID smd/tlsph/defgrad
ID, group-ID are documented in compute command• smd/tlsph/defgrad = style name of this compute command•
Examples:
compute 1 all smd/tlsph/defgrad
Description:
Define a computation that calculates the deformation gradient. It is only meaningful for particles which
interact according to the Total-Lagrangian SPH pair style.
See this PDF guide to use Smooth Mach Dynamics in LAMMPS.
Output info:
This compute outputss a per-particle vector of vectors (tensors), which can be accessed by any command that
uses per-particle values from a compute as input. See How-to discussions, section 6.15 for an overview of
LAMMPS output options.
The per-particle vector values will be given dimensionless. See units. The per-particle vector has 10 entries.
The first nine entries correspond to the xx, xy, xz, yx, yy, yz, zx, zy, zz components of the asymmetric
deformation gradient tensor. The tenth entry is the determinant of the deformation gradient.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info. TThis compute can only be used for particles which interact
via the total Lagrangian SPH pair style.
Related commands:
smd/hourglass/error
Default: none
LAMMPS Users Manual
1304

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/tlsph/dt command
Syntax:
compute ID group-ID smd/tlsph/dt
ID, group-ID are documented in compute command• smd/tlsph/dt = style name of this compute command•
Examples:
compute 1 all smd/tlsph/dt
Description:
Define a computation that outputs the CFL-stable time increment per particle. This time increment is
essentially given by the speed of sound, divided by the SPH smoothing length. Because both the speed of
sound and the smoothing length typically change during the course of a simulation, the stable time increment
needs to be recomputed every time step. This calculation is performed automatically in the relevant SPH pair
styles and this compute only serves to make the stable time increment accessible for output purposes.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See How-to discussions, section 6.15 for an overview of LAMMPS output
options.
The per-particle values will be given in units of time.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This compute can only be used for particles interacting with the Total-Lagrangian SPH pair style.
Related commands:
smd/adjust/dt
Default: none
LAMMPS Users Manual
1305

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/tlsph/num/neighs command
Syntax:
compute ID group-ID smd/tlsph/num/neighs
ID, group-ID are documented in compute command• smd/tlsph/num/neighs = style name of this compute command•
Examples:
compute 1 all smd/tlsph/num/neighs
Description:
Define a computation that calculates the number of particles inside of the smoothing kernel radius for particles
interacting via the Total-Lagrangian SPH pair style.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See How-to discussions, section 6.15 for an overview of LAMMPS output
options.
The per-particle values are dimensionless. See units.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This quantity will be computed only for particles which interact with the Total-Lagrangian pair style.
Related commands:
smd/ulsph/num/neighs
Default: none
LAMMPS Users Manual
1306

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/tlsph/shape command
Syntax:
compute ID group-ID smd/tlsph/shape
ID, group-ID are documented in compute command• smd/tlsph/shape = style name of this compute command•
Examples:
compute 1 all smd/tlsph/shape
Description:
Define a computation that outputs the current shape of the volume associated with a particle as a rotated
ellipsoid. It is only meaningful for particles which interact according to the Total-Lagrangian SPH pair style.
See this PDF guide to use Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle vector of vectors, which can be accessed by any command that uses
per-particle values from a compute as input. See How-to discussions, section 6.15 for an overview of
LAMMPS output options.
The per-particle vector has 7 entries. The first three entries correspond to the lengths of the ellipsoid's axes
and have units of length. These axis valus are computed as the contact radius times the xx, yy, or zz
components of the Green-Lagrange strain tensor associated with the particle. The next 4 values are
quaternions (order: q, x, y, z) which describe the spatial rotation of the particle relative to its initial state.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This quantity will be computed only for particles which interact with the Total-Lagrangian SPH pair style.
Related commands:
smd/contact/radius
Default: none
LAMMPS Users Manual
1307

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/tlsph/strain command
Syntax:
compute ID group-ID smd/tlsph/strain
ID, group-ID are documented in compute command• smd/tlsph/strain = style name of this compute command•
Examples:
compute 1 all smd/tlsph/strain
Description:
Define a computation that calculates the Green-Lagrange strain tensor for particles interacting via the
Total-Lagrangian SPH pair style.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle vector of vectors (tensors), which can be accessed by any command
that uses per-particle values from a compute as input. See How-to discussions, section 6.15 for an overview of
LAMMPS output options.
The per-particle tensor values will be given dimensionless. See units.
The per-particle vector has 6 entries, corresponding to the xx, yy, zz, xy, xz, yz components of the symmetric
strain tensor.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This quantity will be computed only for particles which interact with the Total-Lagrangian SPH pair style.
Related commands:
smd/tlsph/strain/rate, smd/tlsph/stress
Default: none
LAMMPS Users Manual
1308

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/tlsph/strain/rate command
Syntax:
compute ID group-ID smd/tlsph/strain/rate
ID, group-ID are documented in compute command• smd/tlsph/strain/rate = style name of this compute command•
Examples:
compute 1 all smd/tlsph/strain/rate
Description:
Define a computation that calculates the rate of the strain tensor for particles interacting via the
Total-Lagrangian SPH pair style.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle vector of vectors (tensors), which can be accessed by any command
that uses per-particle values from a compute as input. See How-to discussions, section 6.15 for an overview of
LAMMPS output options.
The values will be given in units of one over time.
The per-particle vector has 6 entries, corresponding to the xx, yy, zz, xy, xz, yz components of the symmetric
strain rate tensor.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This quantity will be computed only for particles which interact with Total-Lagrangian SPH pair style.
Related commands:
compute smd/tlsph/strain, compute smd/tlsph/stress
Default: none
LAMMPS Users Manual
1309

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/tlsph/stress command
Syntax:
compute ID group-ID smd/tlsph/stress
ID, group-ID are documented in compute command• smd/tlsph/stress = style name of this compute command•
Examples:
compute 1 all smd/tlsph/stress
Description:
Define a computation that outputs the Cauchy stress tensor for particles interacting via the Total-Lagrangian
SPH pair style.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle vector of vectors (tensors), which can be accessed by any command
that uses per-particle values from a compute as input. See How-to discussions, section 6.15 for an overview of
LAMMPS output options.
The values will be given in units of pressure.
The per-particle vector has 7 entries. The first six entries correspond to the xx, yy, zz, xy, xz and yz
components of the symmetric Cauchy stress tensor. The seventh entry is the second invariant of the stress
tensor, i.e., the von Mises equivalent stress.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This quantity will be computed only for particles which interact with the Total-Lagrangian SPH pair style.
Related commands:
compute smd/tlsph/strain, cmopute smd/tlsph/strain/rate
Default: none
LAMMPS Users Manual
1310

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/triangle/mesh/vertices
Syntax:
compute ID group-ID smd/triangle/mesh/vertices
ID, group-ID are documented in compute command• smd/triangle/mesh/vertices = style name of this compute command•
Examples:
compute 1 all smd/triangle/mesh/vertices
Description:
Define a computation that returns the coordinates of the vertices corresponding to the triangle-elements of a
mesh created by the fix smd/wall_surface.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute returns a per-particle vector of vectors, which can be accessed by any command that uses
per-particle values from a compute as input. See How-to discussions, section 6.15 for an overview of
LAMMPS output options.
The per-particle vector has nine entries, (x1/y1/z1), (x2/y2/z2), and (x3/y3/z3) corresponding to the first,
second, and third vertex of each triangle.
It is only meaningful to use this compute for a group of particles which is created via the fix smd/wall_surface
command.
The output of this compute can be used with the dump2vtk_tris tool to generate a VTK representation of the
smd/wall_surface mesh for visualization purposes.
The values will be given in units of distance.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
fix smd/move/tri/surf, fix smd/wall_surface
Default: none
LAMMPS Users Manual
1311

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/ulsph/num/neighs command
Syntax:
compute ID group-ID smd/ulsph/num/neighs
ID, group-ID are documented in compute command• smd/ulsph/num/neighs = style name of this compute command•
Examples:
compute 1 all smd/ulsph/num/neighs
Description:
Define a computation that returns the number of neighbor particles inside of the smoothing kernel radius for
particles interacting via the updated Lagrangian SPH pair style.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute returns a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-particle values will be given dimensionless, see units.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info. This compute can only be used for particles which interact
with the updated Lagrangian SPH pair style.
Related commands:
compute smd/tlsph/num/neighs
Default: none
LAMMPS Users Manual
1312

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/ulsph/strain command
Syntax:
compute ID group-ID smd/ulsph/strain
ID, group-ID are documented in compute command• smd/ulsph/strain = style name of this compute command•
Examples:
compute 1 all smd/ulsph/strain
Description:
Define a computation that outputs the logarithmic strain tensor. for particles interacting via the updated
Lagrangian SPH pair style.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle tensor, which can be accessed by any command that uses per-particle
values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The per-particle vector has 6 entries, corresponding to the xx, yy, zz, xy, xz, yz components of the symmetric
strain rate tensor.
The per-particle tensor values will be given dimensionless, see units.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info. This compute can only be used for particles which interact
with the updated Lagrangian SPH pair style.
Related commands:
compute smd/tlsph/strain
Default: none
LAMMPS Users Manual
1313

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/ulsph/strain/rate command
Syntax:
compute ID group-ID smd/ulsph/strain/rate
ID, group-ID are documented in compute command• smd/ulsph/strain/rate = style name of this compute command•
Examples:
compute 1 all smd/ulsph/strain/rate
Description:
Define a computation that outputs the rate of the logarithmic strain tensor for particles interacting via the
updated Lagrangian SPH pair style.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle vector of vectors (tensors), which can be accessed by any command
that uses per-particle values from a compute as input. See Section 6.15 for an overview of LAMMPS output
options.
The values will be given in units of one over time.
The per-particle vector has 6 entries, corresponding to the xx, yy, zz, xy, xz, yz components of the symmetric
strain rate tensor.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info. This compute can only be used for particles which interact
with the updated Lagrangian SPH pair style.
Related commands:
compute smd/tlsph/strain/rate
Default: none
LAMMPS Users Manual
1314

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/ulsph/stress command
Syntax:
compute ID group-ID smd/ulsph/stress
ID, group-ID are documented in compute command• smd/ulsph/stress = style name of this compute command•
Examples:
compute 1 all smd/ulsph/stress
Description:
Define a computation that outputs the Cauchy stress tensor.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle vector of vectors (tensors), which can be accessed by any command
that uses per-particle values from a compute as input. See Section 6.15 for an overview of LAMMPS output
options.
The values will be given in units of pressure.
The per-particle vector has 7 entries. The first six entries correspond to the xx, yy, zz, xy, xz, yz components
of the symmetric Cauchy stress tensor. The seventh entry is the second invariant of the stress tensor, i.e., the
von Mises equivalent stress.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info. This compute can only be used for particles which interact
with the updated Lagrangian SPH pair style.
Related commands:
compute smd/ulsph/strain, compute smd/ulsph/strain/rate compute smd/tlsph/stress
Default: none
LAMMPS Users Manual
1315

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute smd/vol command
Syntax:
compute ID group-ID smd/vol
ID, group-ID are documented in compute command• smd/vol = style name of this compute command•
Examples:
compute 1 all smd/vol
Description:
Define a computation that provides the per-particle volume and the sum of the per-particle volumes of the
group for which the fix is defined.
See this PDF guide to using Smooth Mach Dynamics in LAMMPS.
Output info:
This compute calculates a per-particle vector, which can be accessed by any command that uses per-particle
values from a compute as input. See How-to discussions, section 6.15 for an overview of LAMMPS output
options.
The per-particle vector values will be given in units of volume.
Additionally, the compute returns a scalar, which is the sum of the per-particle volumes of the group for
which the fix is defined.
Restrictions:
This compute is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
compute smd/rho
Default: none
LAMMPS Users Manual
1316

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute sna/atom command
compute snad/atom command
compute snav/atom command
Syntax:
compute ID group-ID sna/atom rcutfac rfac0 twojmax R_1 R_2 ... w_1 w_2 ... keyword values ...
compute ID group-ID snad/atom rcutfac rfac0 twojmax R_1 R_2 ... w_1 w_2 ... keyword values ...
compute ID group-ID snav/atom rcutfac rfac0 twojmax R_1 R_2 ... w_1 w_2 ... keyword values ...
ID, group-ID are documented in compute command• sna/atom = style name of this compute command• rcutfac = scale factor applied to all cutoff radii (positive real)• rfac0 = parameter in distance to angle conversion (0 < rcutfac < 1)• twojmax = band limit for bispectrum components (non-negative integer)• R_1, R_2,... = list of cutoff radii, one for each type (distance units)• w_1, w_2,... = list of neighbor weights, one for each type• zero or more keyword/value pairs may be appended• keyword = diagonal or rmin0 or switchflag or bzeroflag
diagonal value = 0 or 1 or 2 or 3
0 = all j1, j2, j <= twojmax, j2 <= j1
1 = subset satisfying j1 == j2
2 = subset satisfying j1 == j2 == j3
3 = subset satisfying j2 <= j1 <= j
rmin0 value = parameter in distance to angle conversion (distance units)
switchflag value = 0 or 1
0 = do not use switching function
1 = use switching function
bzeroflag value = 0 or 1
0 = do not subtract B0
1 = subtract B0
•
Examples:
compute b all sna/atom 1.4 0.99363 6 2.0 2.4 0.75 1.0 diagonal 3 rmin0 0.0
compute db all sna/atom 1.4 0.95 6 2.0 1.0
compute vb all sna/atom 1.4 0.95 6 2.0 1.0
Description:
Define a computation that calculates a set of bispectrum components for each atom in a group.
Bispectrum components of an atom are order parameters characterizing the radial and angular distribution of
neighbor atoms. The detailed mathematical definition is given in the paper by Thompson et al. (Thompson)
The position of a neighbor atom i' relative to a central atom i is a point within the 3D ball of radius R_ii' =
rcutfac*(R_i + R_i')
LAMMPS Users Manual
1317

Bartok et al. (Bartok), proposed mapping this 3D ball onto the 3-sphere, the surface of the unit ball in a
four-dimensional space. The radial distance r within R_ii' is mapped on to a third polar angle theta0 defined
by,
In this way, all possible neighbor positions are mapped on to a subset of the 3-sphere. Points south of the
latitude theta0max=rfac0*Pi are excluded.
The natural basis for functions on the 3-sphere is formed by the 4D hyperspherical harmonics U^j_m,m'(theta,
phi, theta0). These functions are better known as D^j_m,m', the elements of the Wigner D-matrices
(Meremianin, Varshalovich).
The density of neighbors on the 3-sphere can be written as a sum of Dirac-delta functions, one for each
neighbor, weighted by species and radial distance. Expanding this density function as a generalized Fourier
series in the basis functions, we can write each Fourier coefficient as
The w_i' neighbor weights are dimensionless numbers that are chosen to distinguish atoms of different types,
while the central atom is arbitrarily assigned a unit weight. The function fc(r) ensures that the contribution of
each neighbor atom goes smoothly to zero at R_ii':
The expansion coefficients u^j_m,m' are complex-valued and they are not directly useful as descriptors,
because they are not invariant under rotation of the polar coordinate frame. However, the following scalar
triple products of expansion coefficients can be shown to be real-valued and invariant under rotation (Bartok).
The constants H^jmm'_j1m1m1'_j2m2m2' are coupling coefficients, analogous to Clebsch-Gordan coefficients
for rotations on the 2-sphere. These invariants are the components of the bispectrum and these are the
LAMMPS Users Manual
1318
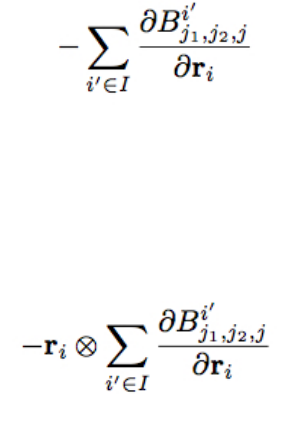
quantities calculated by the compute sna/atom. They characterize the strength of density correlations at three
points on the 3-sphere. The j2=0 subset form the power spectrum, which characterizes the correlations of two
points. The lowest-order components describe the coarsest features of the density function, while higher-order
components reflect finer detail. Note that the central atom is included in the expansion, so three
point-correlations can be either due to three neighbors, or two neighbors and the central atom.
Compute snad/atom calculates the derivative of the bispectrum components summed separately for each atom
type:
The sum is over all atoms i' of atom type I. For each atom i, this compute evaluates the above expression for
each direction, each atom type, and each bispectrum component. See section below on output for a detailed
explanation.
Compute snav/atom calculates the virial contribution due to the derivatives:
Again, the sum is over all atoms i' of atom type I. For each atom i, this compute evaluates the above
expression for each of the six virial components, each atom type, and each bispectrum component. See section
below on output for a detailed explanation.
The value of all bispectrum components will be zero for atoms not in the group. Neighbor atoms not in the
group do not contribute to the bispectrum of atoms in the group.
The neighbor list needed to compute this quantity is constructed each time the calculation is performed (i.e.
each time a snapshot of atoms is dumped). Thus it can be inefficient to compute/dump this quantity too
frequently.
The argument rcutfac is a scale factor that controls the ratio of atomic radius to radial cutoff distance.
The argument rfac0 and the optional keyword rmin0 define the linear mapping from radial distance to polar
angle theta0 on the 3-sphere.
The argument twojmax and the keyword diagonal define which bispectrum components are generated. See
section below on output for a detailed explanation of the number of bispectrum components and the ordered in
which they are listed
The keyword switchflag can be used to turn off the switching function.
The keyword bzeroflag determines whether or not B0, the bispectrum components of an atom with no
neighbors, are subtracted from the calculated bispectrum components. This optional keyword is only available
for compute sna/atom, as snad/atom and snav/atom are unaffected by the removal of constant terms.
LAMMPS Users Manual
1319
NOTE: If you have a bonded system, then the settings of special_bonds command can remove pairwise
interactions between atoms in the same bond, angle, or dihedral. This is the default setting for the
special_bonds command, and means those pairwise interactions do not appear in the neighbor list. Because
this fix uses the neighbor list, it also means those pairs will not be included in the calculation. One way to get
around this, is to write a dump file, and use the rerun command to compute the bispectrum components for
snapshots in the dump file. The rerun script can use a special_bonds command that includes all pairs in the
neighbor list.
;line
Output info:
Compute sna/atom calculates a per-atom array, each column corresponding to a particular bispectrum
component. The total number of columns and the identities of the bispectrum component contained in each
column depend on the values of twojmax and diagonal, as described by the following piece of python code:
for j1 in range(0,twojmax+1):
if(diagonal==2):
print j1/2.,j1/2.,j1/2.
elif(diagonal==1):
for j in range(0,min(twojmax,2*j1)+1,2):
print j1/2.,j1/2.,j/2.
elif(diagonal==0):
for j2 in range(0,j1+1):
for j in range(j1-j2,min(twojmax,j1+j2)+1,2):
print j1/2.,j2/2.,j/2.
elif(diagonal==3):
for j2 in range(0,j1+1):
for j in range(j1-j2,min(twojmax,j1+j2)+1,2):
if (j>=j1): print j1/2.,j2/2.,j/2.
Compute snad/atom evaluates a per-atom array. The columns are arranged into ntypes blocks, listed in order
of atom type I. Each block contains three sub-blocks corresponding to the x, y, and z components of the atom
position. Each of these sub-blocks contains one column for each bispectrum component, the same as for
compute sna/atom
Compute snav/atom evaluates a per-atom array. The columns are arranged into ntypes blocks, listed in order
of atom type I. Each block contains six sub-blocks corresponding to the xx, yy, zz, yz, xz, and xy components
of the virial tensor in Voigt notation. Each of these sub-blocks contains one column for each bispectrum
component, the same as for compute sna/atom
These values can be accessed by any command that uses per-atom values from a compute as input. See
Section 6.15 for an overview of LAMMPS output options.
Restrictions:
These computes are part of the SNAP package. They are only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Related commands:
pair_style snap
LAMMPS Users Manual
1320

Default:
The optional keyword defaults are diagonal = 0, rmin0 = 0, switchflag = 1, bzeroflag = 0.
(Thompson) Thompson, Swiler, Trott, Foiles, Tucker, under review, preprint available at arXiv:1409.3880
(Bartok) Bartok, Payne, Risi, Csanyi, Phys Rev Lett, 104, 136403 (2010).
(Meremianin) Meremianin, J. Phys. A, 39, 3099 (2006).
(Varshalovich) Varshalovich, Moskalev, Khersonskii, Quantum Theory of Angular Momentum, World
Scientific, Singapore (1987).
LAMMPS Users Manual
1321

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute stress/atom command
Syntax:
compute ID group-ID stress/atom temp-ID keyword ...
ID, group-ID are documented in compute command• stress/atom = style name of this compute command• temp-ID = ID of compute that calculates temperature, can be NULL if not needed• zero or more keywords may be appended• keyword = ke or pair or bond or angle or dihedral or improper or kspace or fix or virial•
Examples:
compute 1 mobile stress/atom NULL
compute 1 mobile stress/atom myRamp
compute 1 all stress/atom NULL pair bond
Description:
Define a computation that computes the symmetric per-atom stress tensor for each atom in a group. The tensor
for each atom has 6 components and is stored as a 6-element vector in the following order: xx, yy, zz, xy, xz,
yz. See the compute pressure command if you want the stress tensor (pressure) of the entire system.
The stress tensor for atom I is given by the following formula, where a and b take on values x,y,z to generate
the 6 components of the symmetric tensor:
The first term is a kinetic energy contribution for atom I. See details below on how the specified temp-ID can
affect the velocities used in this calculation. The second term is a pairwise energy contribution where n loops
over the Np neighbors of atom I, r1 and r2 are the positions of the 2 atoms in the pairwise interaction, and F1
and F2 are the forces on the 2 atoms resulting from the pairwise interaction. The third term is a bond
contribution of similar form for the Nb bonds which atom I is part of. There are similar terms for the Na angle,
Nd dihedral, and Ni improper interactions atom I is part of. There is also a term for the KSpace contribution
from long-range Coulombic interactions, if defined. Finally, there is a term for the Nf fixes that apply internal
constraint forces to atom I. Currently, only the fix shake and fix rigid commands contribute to this term.
LAMMPS Users Manual
1322

As the coefficients in the formula imply, a virial contribution produced by a small set of atoms (e.g. 4 atoms
in a dihedral or 3 atoms in a Tersoff 3-body interaction) is assigned in equal portions to each atom in the set.
E.g. 1/4 of the dihedral virial to each of the 4 atoms, or 1/3 of the fix virial due to SHAKE constraints applied
to atoms in a water molecule via the fix shake command.
If no extra keywords are listed, all of the terms in this formula are included in the per-atom stress tensor. If
any extra keywords are listed, only those terms are summed to compute the tensor. The virial keyword means
include all terms except the kinetic energy ke.
Note that the stress for each atom is due to its interaction with all other atoms in the simulation, not just with
other atoms in the group.
Details of how LAMMPS computes the virial for individual atoms for either pairwise or manybody potentials,
and including the effects of periodic boundary conditions is discussed in (Thompson). The basic idea for
manybody potentials is to treat each component of the force computation between a small cluster of atoms in
the same manner as in the formula above for bond, angle, dihedral, etc interactions. Namely the quantity R dot
F is summed over the atoms in the interaction, with the R vectors unwrapped by periodic boundaries so that
the cluster of atoms is close together. The total contribution for the cluster interaction is divided evenly among
those atoms.
The dihedral_style charmm style calculates pairwise interactions between 1-4 atoms. The virial contribution
of these terms is included in the pair virial, not the dihedral virial.
The KSpace contribution is calculated using the method in (Heyes) for the Ewald method and by the
methodology described in (Sirk) for PPPM. The choice of KSpace solver is specified by the kspace_style
pppm command. Note that for PPPM, the calculation requires 6 extra FFTs each timestep that per-atom stress
is calculated. Thus it can significantly increase the cost of the PPPM calculation if it is needed on a large
fraction of the simulation timesteps.
The temp-ID argument can be used to affect the per-atom velocities used in the kinetic energy contribution to
the total stress. If the kinetic energy is not included in the stress, than the temperature compute is not used and
can be specified as NULL. If the kinetic energy is included and you wish to use atom velocities as-is, then
temp-ID can also be specified as NULL. If desired, the specified temperature compute can be one that
subtracts off a bias to leave each atom with only a thermal velocity to use in the formula above, e.g. by
subtracting a background streaming velocity. See the doc pages for individual compute commands to
determine which ones include a bias.
Note that as defined in the formula, per-atom stress is the negative of the per-atom pressure tensor. It is also
really a stress*volume formulation, meaning the computed quantity is in units of pressure*volume. It would
need to be divided by a per-atom volume to have units of stress (pressure), but an individual atom's volume is
not well defined or easy to compute in a deformed solid or a liquid. See the compute voronoi/atom command
for one possible way to estimate a per-atom volume.
Thus, if the diagonal components of the per-atom stress tensor are summed for all atoms in the system and the
sum is divided by dV, where d = dimension and V is the volume of the system, the result should be -P, where
P is the total pressure of the system.
These lines in an input script for a 3d system should yield that result. I.e. the last 2 columns of thermo output
will be the same:
compute peratom all stress/atom NULL
LAMMPS Users Manual
1323

compute p all reduce sum c_peratom[1] c_peratom[2] c_peratom[3]
variable press equal -(c_p[1]+c_p[2]+c_p[3])/(3*vol)
thermo_style custom step temp etotal press v_press
NOTE: The per-atom stress does not include any Lennard-Jones tail corrections to the pressure added by the
pair_modify tail yes command, since those are contributions to the global system pressure.
Output info:
This compute calculates a per-atom array with 6 columns, which can be accessed by indices 1-6 by any
command that uses per-atom values from a compute as input. See Section 6.15 for an overview of LAMMPS
output options.
The per-atom array values will be in pressure*volume units as discussed above.
Restrictions: none
Related commands:
compute pe, compute pressure
Default: none
(Heyes) Heyes, Phys Rev B 49, 755 (1994),
(Sirk) Sirk, Moore, Brown, J Chem Phys, 138, 064505 (2013).
(Thompson) Thompson, Plimpton, Mattson, J Chem Phys, 131, 154107 (2009).
LAMMPS Users Manual
1324

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute force/tally command
compute heat/flux/tally command
compute pe/tally command
compute pe/mol/tally command
compute stress/tally command
Syntax:
compute ID group-ID style group2-ID
ID, group-ID are documented in compute command• style = force/tally or pe/tally or pe/mol/tally or stress/tally• group2-ID = group ID of second (or same) group•
Examples:
compute 1 lower force/tally upper
compute 1 left pe/tally right
compute 1 lower stress/tally lower
Description:
Define a computation that calculates properties between two groups of atoms by accumulating them from
pairwise non-bonded computations. The two groups can be the same. This is similar to compute group/group
only that the data is accumulated directly during the non-bonded force computation. The computes force/tally,
pe/tally, stress/tally, and heat/flux/tally are primarily provided as example how to program additional, more
sophisticated computes using the tally callback mechanism. Compute pe/mol/tally is one such style, that can -
through using this mechanism - separately tally intermolecular and intramolecular energies. Something that
would otherwise be impossible without integrating this as a core functionality into the based classes of
LAMMPS.
The pairwise contributions are computing via a callback that the compute registers with the non-bonded
pairwise force computation. This limits the use to systems that have no bonds, no Kspace, and no manybody
interactions. On the other hand, the computation does not have to compute forces or energies a second time
and thus can be much more efficient. The callback mechanism allows to write more complex pairwise
property computations.
Output info:
Compute pe/tally calculates a global scalar (the energy) and a per atom scalar (the contributions of the single
atom to the global scalar). Compute pe/mol/tally calculates a global 4-element vector containing (in this
order): evdwl and ecoul for intramolecular pairs and evdwl and ecoul for intermolecular pairs. Since molecules
are identified by their molecule IDs, the partitioning does not have to be related to molecules, but the energies
are tallied into the respective slots depending on whether the molecule IDs of a pair are the same or different.
LAMMPS Users Manual
1325
Compute force/tally calculates a global scalar (the force magnitude) and a per atom 3-element vector (force
contribution from each atom). Compute stress/tally calculates a global scalar (average of the diagonal
elements of the stress tensor) and a per atom vector (the 6 elements of stress tensor contributions from the
individual atom).
Both the scalar and vector values calculated by this compute are "extensive".
Restrictions:
This compute is part of the USER-TALLY package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Not all pair styles can be evaluated in a pairwise mode as required by this compute. For example, 3-body and
other many-body potentials, such as Tersoff and Stillinger-Weber cannot be used. EAM potentials only
include the pair potential portion of the EAM interaction when used by this compute, not the embedding term.
Also bonded or Kspace interactions do not contribute to this compute.
Related commands:
compute group/group_compute_group_group.html, compute heat/flux_compute_heat_flux.html
Default: none
LAMMPS Users Manual
1326

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp command
compute temp/kk command
Syntax:
compute ID group-ID temp
ID, group-ID are documented in compute command• temp = style name of this compute command•
Examples:
compute 1 all temp
compute myTemp mobile temp
Description:
Define a computation that calculates the temperature of a group of atoms. A compute of this style can be used
by any command that computes a temperature, e.g. thermo_modify, fix temp/rescale, fix npt, etc.
The temperature is calculated by the formula KE = dim/2 N k T, where KE = total kinetic energy of the group
of atoms (sum of 1/2 m v^2), dim = 2 or 3 = dimensionality of the simulation, N = number of atoms in the
group, k = Boltzmann constant, and T = temperature.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute for use in the
computation of a pressure tensor. The formula for the components of the tensor is the same as the above
formula, except that v^2 is replaced by vx*vy for the xy component, etc. The 6 components of the vector are
ordered xx, yy, zz, xy, xz, yz.
The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
This compute subtracts out degrees-of-freedom due to fixes that constrain molecular motion, such as fix shake
and fix rigid. This means the temperature of groups of atoms that include these constraints will be computed
correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option of the
compute_modify command.
A compute of this style with the ID of "thermo_temp" is created when LAMMPS starts up, as if this command
were in the input script:
compute thermo_temp all temp
See the "thermo_style" command for more details.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
LAMMPS Users Manual
1327

Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions: none
Related commands:
compute temp/partial, compute temp/region, compute pressure
Default: none
LAMMPS Users Manual
1328

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/asphere command
Syntax:
compute ID group-ID temp/asphere keyword value ...
ID, group-ID are documented in compute command• temp/asphere = style name of this compute command• zero or more keyword/value pairs may be appended• keyword = bias or dof
bias value = bias-ID
bias-ID = ID of a temperature compute that removes a velocity bias
dof value = all or rotate
all = compute temperature of translational and rotational degrees of freedom
rotate = compute temperature of just rotational degrees of freedom
•
Examples:
compute 1 all temp/asphere
compute myTemp mobile temp/asphere bias tempCOM
compute myTemp mobile temp/asphere dof rotate
Description:
Define a computation that calculates the temperature of a group of aspherical particles, including a
contribution from both their translational and rotational kinetic energy. This differs from the usual compute
temp command, which assumes point particles with only translational kinetic energy.
Only finite-size particles (aspherical or spherical) can be included in the group. For 3d finite-size particles,
each has 6 degrees of freedom (3 translational, 3 rotational). For 2d finite-size particles, each has 3 degrees of
freedom (2 translational, 1 rotational).
NOTE: This choice for degrees of freedom (dof) assumes that all finite-size aspherical or spherical particles in
your model will freely rotate, sampling all their rotational dof. It is possible to use a combination of
interaction potentials and fixes that induce no torque or otherwise constrain some of all of your particles so
that this is not the case. Then there are less dof and you should use the compute_modify extra command to
adjust the dof accordingly.
For example, an aspherical particle with all three of its shape parameters the same is a sphere. If it does not
rotate, then it should have 3 dof instead of 6 in 3d (or 2 instead of 3 in 2d). A uniaxial aspherical particle has
two of its three shape parameters the same. If it does not rotate around the axis perpendicular to its circular
cross section, then it should have 5 dof instead of 6 in 3d. The latter is the case for uniaxial ellipsoids in a
GayBerne model since there is no induced torque around the optical axis. It will also be the case for biaxial
ellipsoids when exactly two of the semiaxes have the same length and the corresponding relative well depths
are equal.
The translational kinetic energy is computed the same as is described by the compute temp command. The
rotational kinetic energy is computed as 1/2 I w^2, where I is the inertia tensor for the aspherical particle and
w is its angular velocity, which is computed from its angular momentum.
LAMMPS Users Manual
1329

NOTE: For 2d models, particles are treated as ellipsoids, not ellipses, meaning their moments of inertia will
be the same as in 3d.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute. The formula for the
components of the tensor is the same as the above formula, except that v^2 and w^2 are replaced by vx*vy
and wx*wy for the xy component, and the appropriate elements of the inertia tensor are used. The 6
components of the vector are ordered xx, yy, zz, xy, xz, yz.
The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
This compute subtracts out translational degrees-of-freedom due to fixes that constrain molecular motion,
such as fix shake and fix rigid. This means the temperature of groups of atoms that include these constraints
will be computed correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option
of the compute_modify command.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
The keyword/value option pairs are used in the following ways.
For the bias keyword, bias-ID refers to the ID of a temperature compute that removes a "bias" velocity from
each atom. This allows compute temp/sphere to compute its thermal temperature after the translational kinetic
energy components have been altered in a prescribed way, e.g. to remove a flow velocity profile. Thermostats
that use this compute will work with this bias term. See the doc pages for individual computes that calculate a
temperature and the doc pages for fixes that perform thermostatting for more details.
For the dof keyword, a setting of all calculates a temperature that includes both translational and rotational
degrees of freedom. A setting of rotate calculates a temperature that includes only rotational degrees of
freedom.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions:
This compute is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
This compute requires that atoms store angular momentum and a quaternion as defined by the atom_style
ellipsoid command.
All particles in the group must be finite-size. They cannot be point particles, but they can be aspherical or
LAMMPS Users Manual
1330

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/body command
Syntax:
compute ID group-ID temp/body keyword value ...
ID, group-ID are documented in compute command• temp/body = style name of this compute command• zero or more keyword/value pairs may be appended• keyword = bias or dof
bias value = bias-ID
bias-ID = ID of a temperature compute that removes a velocity bias
dof value = all or rotate
all = compute temperature of translational and rotational degrees of freedom
rotate = compute temperature of just rotational degrees of freedom
•
Examples:
compute 1 all temp/body
compute myTemp mobile temp/body bias tempCOM
compute myTemp mobile temp/body dof rotate
Description:
Define a computation that calculates the temperature of a group of body particles, including a contribution
from both their translational and rotational kinetic energy. This differs from the usual compute temp
command, which assumes point particles with only translational kinetic energy.
Only body particles can be included in the group. For 3d particles, each has 6 degrees of freedom (3
translational, 3 rotational). For 2d body particles, each has 3 degrees of freedom (2 translational, 1 rotational).
NOTE: This choice for degrees of freedom (dof) assumes that all body particles in your model will freely
rotate, sampling all their rotational dof. It is possible to use a combination of interaction potentials and fixes
that induce no torque or otherwise constrain some of all of your particles so that this is not the case. Then
there are less dof and you should use the compute_modify extra command to adjust the dof accordingly.
The translational kinetic energy is computed the same as is described by the compute temp command. The
rotational kinetic energy is computed as 1/2 I w^2, where I is the inertia tensor for the aspherical particle and
w is its angular velocity, which is computed from its angular momentum.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute. The formula for the
components of the tensor is the same as the above formula, except that v^2 and w^2 are replaced by vx*vy
and wx*wy for the xy component, and the appropriate elements of the inertia tensor are used. The 6
components of the vector are ordered xx, yy, zz, xy, xz, yz.
The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
LAMMPS Users Manual
1332

This compute subtracts out translational degrees-of-freedom due to fixes that constrain molecular motion,
such as fix shake and fix rigid. This means the temperature of groups of atoms that include these constraints
will be computed correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option
of the compute_modify command.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
The keyword/value option pairs are used in the following ways.
For the bias keyword, bias-ID refers to the ID of a temperature compute that removes a "bias" velocity from
each atom. This allows compute temp/sphere to compute its thermal temperature after the translational kinetic
energy components have been altered in a prescribed way, e.g. to remove a flow velocity profile. Thermostats
that use this compute will work with this bias term. See the doc pages for individual computes that calculate a
temperature and the doc pages for fixes that perform thermostatting for more details.
For the dof keyword, a setting of all calculates a temperature that includes both translational and rotational
degrees of freedom. A setting of rotate calculates a temperature that includes only rotational degrees of
freedom.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions:
This compute is part of the BODY package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
This compute requires that atoms store angular momentum and a quaternion as defined by the atom_style
body command.
Related commands:
compute temp
Default: none
LAMMPS Users Manual
1333

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/chunk command
Syntax:
compute ID group-ID temp/chunk chunkID value1 value2 ... keyword value ...
ID, group-ID are documented in compute command• temp/chunk = style name of this compute command• chunkID = ID of compute chunk/atom command• zero or more values can be listed as value1,value2,etc• value = temp or kecom or internal
temp = temperature of each chunk
kecom = kinetic energy of each chunk based on velocity of center of mass
internal = internal kinetic energy of each chunk
•
zero or more keyword/value pairs may be appended• keyword = com or bias or adof or cdof
com value = yes or no
yes = subtract center-of-mass velocity from each chunk before calculating temperature
no = do not subtract center-of-mass velocity
bias value = bias-ID
bias-ID = ID of a temperature compute that removes a velocity bias
adof value = dof_per_atom
dof_per_atom = define this many degrees-of-freedom per atom
cdof value = dof_per_chunk
dof_per_chunk = define this many degrees-of-freedom per chunk
•
Examples:
compute 1 fluid temp/chunk molchunk
compute 1 fluid temp/chunk molchunk temp internal
compute 1 fluid temp/chunk molchunk bias tpartial adof 2.0
Description:
Define a computation that calculates the temperature of a group of atoms that are also in chunks, after
optionally subtracting out the center-of-mass velocity of each chunk. By specifying optional values, it can also
calculate the per-chunk temperature or energies of the multiple chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
The temperature is calculated by the formula KE = DOF/2 k T, where KE = total kinetic energy of all atoms
assigned to chunks (sum of 1/2 m v^2), DOF = the total number of degrees of freedom for those atoms, k =
Boltzmann constant, and T = temperature.
LAMMPS Users Manual
1334

The DOF is calculated as N*adof + Nchunk*cdof, where N = number of atoms contributing to the KE, adof =
degrees of freedom per atom, and cdof = degrees of freedom per chunk. By default adof = 2 or 3 =
dimensionality of system, as set via the dimension command, and cdof = 0.0. This gives the usual formula for
temperature.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute for use in the
computation of a pressure tensor. The formula for the components of the tensor is the same as the above
formula, except that v^2 is replaced by vx*vy for the xy component, etc. The 6 components of the vector are
ordered xx, yy, zz, xy, xz, yz.
Note that the number of atoms contributing to the temperature is calculated each time the temperature is
evaluated since it is assumed the atoms may be dynamically assigned to chunks. Thus there is no need to use
the dynamic option of the compute_modify command for this compute style.
If any optional values are specified, then per-chunk quantities are also calculated and stored in a global array,
as described below.
The temp value calculates the temperature for each chunk by the formula KE = DOF/2 k T, where KE = total
kinetic energy of the chunk of atoms (sum of 1/2 m v^2), DOF = the total number of degrees of freedom for
all atoms in the chunk, k = Boltzmann constant, and T = temperature.
The DOF in this case is calculated as N*adof + cdof, where N = number of atoms in the chunk, adof = degrees
of freedom per atom, and cdof = degrees of freedom per chunk. By default adof = 2 or 3 = dimensionality of
system, as set via the dimension command, and cdof = 0.0. This gives the usual formula for temperature.
The kecom value calculates the kinetic energy of each chunk as if all its atoms were moving with the velocity
of the center-of-mass of the chunk.
The internal value calculates the internal kinetic energy of each chunk. The interal KE is summed over the
atoms in the chunk using an internal "thermal" velocity for each atom, which is its velocity minus the
center-of-mass velocity of the chunk.
Note that currently the global and per-chunk temperatures calculated by this compute only include
translational degrees of freedom for each atom. No rotational degrees of freedom are included for finite-size
particles. Also no degrees of freedom are subtracted for any velocity bias or constraints that are applied, such
as compute temp/partial, or fix shake or fix rigid. This is because those degrees of freedom (e.g. a constrained
bond) could apply to sets of atoms that are both included and excluded from a specific chunk, and hence the
concept is somewhat ill-defined. In some cases, you can use the adof and cdof keywords to adjust the
calculated degress of freedom appropriately, as explained below.
Note that the per-chunk temperature calculated by this compute and the fix ave/chunk temp command can be
different. This compute calculates the temperature for each chunk for a single snapshot. Fix ave/chunk can do
that but can also time average those values over many snapshots, or it can compute a temperature as if the
atoms in the chunk on different timesteps were collected together as one set of atoms to calculate their
temperature. This compute allows the center-of-mass velocity of each chunk to be subtracted before
calculating the temperature; fix ave/chunk does not.
NOTE: Only atoms in the specified group contribute to the calculations performed by this compute. The
compute chunk/atom command defines its own group; atoms will have a chunk ID = 0 if they are not in that
group, signifying they are not assigned to a chunk, and will thus also not contribute to this calculation. You
can specify the "all" group for this command if you simply want to include atoms with non-zero chunk IDs.
LAMMPS Users Manual
1335
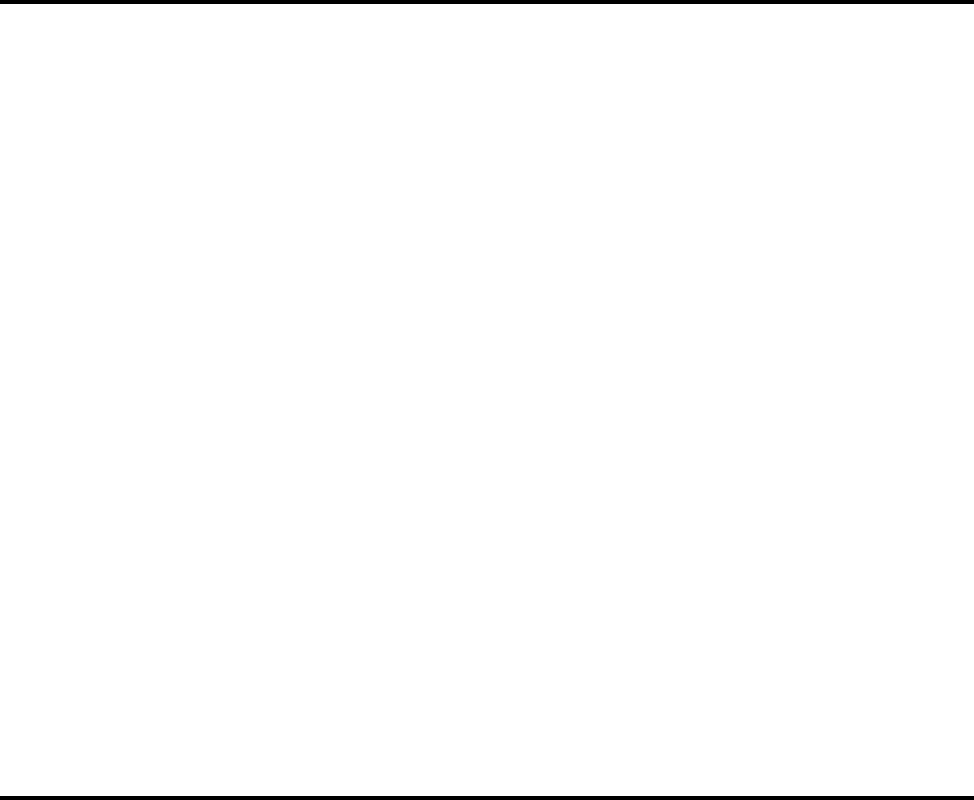
The simplest way to output the per-chunk results of the compute temp/chunk calculation to a file is to use the
fix ave/time command, for example:
compute cc1 all chunk/atom molecule
compute myChunk all temp/chunk cc1 temp
fix 1 all ave/time 100 1 100 c_myChunk file tmp.out mode vector
The keyword/value option pairs are used in the following ways.
The com keyword can be used with a value of yes to subtract the velocity of the center-of-mass for each chunk
from the velocity of the atoms in that chunk, before calculating either the global or per-chunk temperature.
This can be useful if the atoms are streaming or otherwise moving collectively, and you wish to calculate only
the thermal temperature.
For the bias keyword, bias-ID refers to the ID of a temperature compute that removes a "bias" velocity from
each atom. This also allows calculation of the global or per-chunk temperature using only the thermal
temperature of atoms in each chunk after the translational kinetic energy components have been altered in a
prescribed way, e.g. to remove a velocity profile. It also applies to the calculation of the other per-chunk
values, such as kecom or internal, which involve the center-of-mass velocity of each chunk, which is
calculated after the velocity bias is removed from each atom. Note that the temperature compute will apply its
bias globally to the entire system, not on a per-chunk basis.
The adof and cdof keywords define the values used in the degree of freedom (DOF) formulas used for the
global or per-chunk temperature, as described above. They can be used to calculate a more appropriate
temperature for some kinds of chunks. Here are 3 examples:
If spatially binned chunks contain some number of water molecules and fix shake is used to make each
molecule rigid, then you could calculate a temperature with 6 degrees of freedom (DOF) (3 translational, 3
rotational) per molecule by setting adof to 2.0.
If compute temp/partial is used with the bias keyword to only allow the x component of velocity to contribute
to the temperature, then adof = 1.0 would be appropriate.
If each chunk consists of a large molecule, with some number of its bonds constrained by fix shake or the
entire molecule by fix rigid/small, adof = 0.0 and cdof could be set to the remaining degrees of freedom for
the entire molecule (entire chunk in this case), e.g. 6 for 3d, or 3 for 2d, for a rigid molecule.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
This compute also optionally calculates a global array, if one or more of the optional values are specified. The
number of rows in the array = the number of chunks Nchunk as calculated by the specified compute
chunk/atom command. The number of columns is the number of specified values (1 or more). These values
can be accessed by any command that uses global array values from a compute as input. Again, see Section
6.15 for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive". The array values
are "intensive".
LAMMPS Users Manual
1336
The scalar value will be in temperature units. The vector values will be in energy units. The array values will
be in temperature units for the temp value, and in energy units for the kecom and internal values.
Restrictions:
The com and bias keywords cannot be used together.
Related commands:
compute temp, fix ave/chunk temp
Default:
The option defaults are com no, no bias, adof = dimensionality of the system (2 or 3), and cdof = 0.0.
LAMMPS Users Manual
1337

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/com command
Syntax:
compute ID group-ID temp/com
ID, group-ID are documented in compute command• temp/com = style name of this compute command•
Examples:
compute 1 all temp/com
compute myTemp mobile temp/com
Description:
Define a computation that calculates the temperature of a group of atoms, after subtracting out the
center-of-mass velocity of the group. This is useful if the group is expected to have a non-zero net velocity for
some reason. A compute of this style can be used by any command that computes a temperature, e.g.
thermo_modify, fix temp/rescale, fix npt, etc.
After the center-of-mass velocity has been subtracted from each atom, the temperature is calculated by the
formula KE = dim/2 N k T, where KE = total kinetic energy of the group of atoms (sum of 1/2 m v^2), dim =
2 or 3 = dimensionality of the simulation, N = number of atoms in the group, k = Boltzmann constant, and T =
temperature.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute for use in the
computation of a pressure tensor. The formula for the components of the tensor is the same as the above
formula, except that v^2 is replaced by vx*vy for the xy component, etc. The 6 components of the vector are
ordered xx, yy, zz, xy, xz, yz.
The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
The removal of the center-of-mass velocity by this fix is essentially computing the temperature after a "bias"
has been removed from the velocity of the atoms. If this compute is used with a fix command that performs
thermostatting then this bias will be subtracted from each atom, thermostatting of the remaining thermal
velocity will be performed, and the bias will be added back in. Thermostatting fixes that work in this way
include fix nvt, fix temp/rescale, fix temp/berendsen, and fix langevin.
This compute subtracts out degrees-of-freedom due to fixes that constrain molecular motion, such as fix shake
and fix rigid. This means the temperature of groups of atoms that include these constraints will be computed
correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option of the
compute_modify command.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
Output info:
LAMMPS Users Manual
1338
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions: none
Related commands:
compute temp
Default: none
LAMMPS Users Manual
1339

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/cs command
Syntax:
compute ID group-ID temp/cs group1 group2
ID, group-ID are documented in compute command• temp/cs = style name of this compute command• group1 = group-ID of either cores or shells• group2 = group-ID of either shells or cores•
Examples:
compute oxygen_c-s all temp/cs O_core O_shell
compute core_shells all temp/cs cores shells
Description:
Define a computation that calculates the temperature of a system based on the center-of-mass velocity of atom
pairs that are bonded to each other. This compute is designed to be used with the adiabatic core/shell model of
(Mitchell and Finchham). See Section 6.25 of the manual for an overview of the model as implemented in
LAMMPS. Specifically, this compute enables correct temperature calculation and thermostatting of core/shell
pairs where it is desirable for the internal degrees of freedom of the core/shell pairs to not be influenced by a
thermostat. A compute of this style can be used by any command that computes a temperature via fix_modify
e.g. fix temp/rescale, fix npt, etc.
Note that this compute does not require all ions to be polarized, hence defined as core/shell pairs. One can mix
core/shell pairs and ions without a satellite particle if desired. The compute will consider the non-polarized
ions according to the physical system.
For this compute, core and shell particles are specified by two respective group IDs, which can be defined
using the group command. The number of atoms in the two groups must be the same and there should be one
bond defined between a pair of atoms in the two groups. Non-polarized ions which might also be included in
the treated system should not be included into either of these groups, they are taken into account by the
group-ID (2nd argument) of the compute.
The temperature is calculated by the formula KE = dim/2 N k T, where KE = total kinetic energy of the group
of atoms (sum of 1/2 m v^2), dim = 2 or 3 = dimensionality of the simulation, N = number of atoms in the
group, k = Boltzmann constant, and T = temperature. Note that the velocity of each core or shell atom used in
the KE calculation is the velocity of the center-of-mass (COM) of the core/shell pair the atom is part of.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute for use in the
computation of a pressure tensor. The formula for the components of the tensor is the same as the above
formula, except that v^2 is replaced by vx*vy for the xy component, etc. The 6 components of the vector are
ordered xx, yy, zz, xy, xz, yz. In contrast to the temperature, the velocity of each core or shell atom is taken
individually.
The change this fix makes to core/shell atom velocities is essentially computing the temperature after a "bias"
has been removed from the velocity of the atoms. This "bias" is the velocity of the atom relative to the COM
LAMMPS Users Manual
1340

velocity of the core/shell pair. If this compute is used with a fix command that performs thermostatting then
this bias will be subtracted from each atom, thermostatting of the remaining COM velocity will be performed,
and the bias will be added back in. This means the thermostating will effectively be performed on the
core/shell pairs, instead of on the individual core and shell atoms. Thermostatting fixes that work in this way
include fix nvt, fix temp/rescale, fix temp/berendsen, and fix langevin.
The internal energy of core/shell pairs can be calculated by the compute temp/chunk command, if chunks are
defined as core/shell pairs. See Section 6.25 for more discussion on how to do this.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions:
The number of core/shell pairs contributing to the temperature is assumed to be constant for the duration of
the run. No fixes should be used which generate new molecules or atoms during a simulation.
Related commands:
compute temp, compute temp/chunk
Default: none
(Mitchell and Finchham) Mitchell, Finchham, J Phys Condensed Matter, 5, 1031-1038 (1993).
LAMMPS Users Manual
1341

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/deform command
Syntax:
compute ID group-ID temp/deform
ID, group-ID are documented in compute command• temp/deform = style name of this compute command•
Examples:
compute myTemp all temp/deform
Description:
Define a computation that calculates the temperature of a group of atoms, after subtracting out a streaming
velocity induced by the simulation box changing size and/or shape, for example in a non-equilibrium MD
(NEMD) simulation. The size/shape change is induced by use of the fix deform command. A compute of this
style is created by the fix nvt/sllod command to compute the thermal temperature of atoms for thermostatting
purposes. A compute of this style can also be used by any command that computes a temperature, e.g.
thermo_modify, fix temp/rescale, fix npt, etc.
The deformation fix changes the box size and/or shape over time, so each atom in the simulation box can be
thought of as having a "streaming" velocity. For example, if the box is being sheared in x, relative to y, then
atoms at the bottom of the box (low y) have a small x velocity, while atoms at the top of the box (hi y) have a
large x velocity. This position-dependent streaming velocity is subtracted from each atom's actual velocity to
yield a thermal velocity which is used to compute the temperature.
NOTE: Fix deform has an option for remapping either atom coordinates or velocities to the changing
simulation box. When using this compute in conjunction with a deforming box, fix deform should NOT
remap atom positions, but rather should let atoms respond to the changing box by adjusting their own
velocities (or let fix deform remap the atom velocities, see it's remap option). If fix deform does remap atom
positions, then they appear to move with the box but their velocity is not changed, and thus they do NOT have
the streaming velocity assumed by this compute. LAMMPS will warn you if fix deform is defined and its
remap setting is not consistent with this compute.
After the streaming velocity has been subtracted from each atom, the temperature is calculated by the formula
KE = dim/2 N k T, where KE = total kinetic energy of the group of atoms (sum of 1/2 m v^2), dim = 2 or 3 =
dimensionality of the simulation, N = number of atoms in the group, k = Boltzmann constant, and T =
temperature. Note that v in the kinetic energy formula is the atom's thermal velocity.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute for use in the
computation of a pressure tensor. The formula for the components of the tensor is the same as the above
formula, except that v^2 is replaced by vx*vy for the xy component, etc. The 6 components of the vector are
ordered xx, yy, zz, xy, xz, yz.
The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
LAMMPS Users Manual
1342
The removal of the box deformation velocity component by this fix is essentially computing the temperature
after a "bias" has been removed from the velocity of the atoms. If this compute is used with a fix command
that performs thermostatting then this bias will be subtracted from each atom, thermostatting of the remaining
thermal velocity will be performed, and the bias will be added back in. Thermostatting fixes that work in this
way include fix nvt, fix temp/rescale, fix temp/berendsen, and fix langevin.
NOTE: The temperature calculated by this compute is only accurate if the atoms are indeed moving with a
stream velocity profile that matches the box deformation. If not, then the compute will subtract off an
incorrect stream velocity, yielding a bogus thermal temperature. You should NOT assume that your atoms are
streaming at the same rate the box is deforming. Rather, you should monitor their velocity profile, e.g. via the
fix ave/chunk command. And you can compare the results of this compute to compute temp/profile, which
actually calculates the stream profile before subtracting it. If the two computes do not give roughly the same
temperature, then your atoms are not streaming consistent with the box deformation. See the fix deform
command for more details on ways to get atoms to stream consistently with the box deformation.
This compute subtracts out degrees-of-freedom due to fixes that constrain molecular motion, such as fix shake
and fix rigid. This means the temperature of groups of atoms that include these constraints will be computed
correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option of the
compute_modify command.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions: none
Related commands:
compute temp/ramp, compute temp/profile, fix deform, fix nvt/sllod
Default: none
LAMMPS Users Manual
1343

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/deform/eff command
Syntax:
compute ID group-ID temp/deform/eff
ID, group-ID are documented in compute command• temp/deform/eff = style name of this compute command•
Examples:
compute myTemp all temp/deform/eff
Description:
Define a computation that calculates the temperature of a group of nuclei and electrons in the electron force
field model, after subtracting out a streaming velocity induced by the simulation box changing size and/or
shape, for example in a non-equilibrium MD (NEMD) simulation. The size/shape change is induced by use of
the fix deform command. A compute of this style is created by the fix nvt/sllod/eff command to compute the
thermal temperature of atoms for thermostatting purposes. A compute of this style can also be used by any
command that computes a temperature, e.g. thermo_modify, fix npt/eff, etc.
The calculation performed by this compute is exactly like that described by the compute temp/deform
command, except that the formula for the temperature includes the radial electron velocity contributions, as
discussed by the compute temp/eff command. Note that only the translational degrees of freedom for each
nuclei or electron are affected by the streaming velocity adjustment. The radial velocity component of the
electrons is not affected.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions:
This compute is part of the USER-EFF package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
compute temp/ramp, fix deform, fix nvt/sllod/eff
Default: none
LAMMPS Users Manual
1344

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/drude command
Syntax:
compute ID group-ID temp/drude
ID, group-ID are documented in compute command• temp/drude = style name of this compute command•
Examples:
compute TDRUDE all temp/drude
Description:
Define a computation that calculates the temperatures of core-Drude pairs. This compute is designed to be
used with the thermalized Drude oscillator model. Polarizable models in LAMMPS are described in this
Section.
Drude oscillators consist of a core particle and a Drude particle connected by a harmonic bond, and the
relative motion of these Drude oscillators is usually maintained cold by a specific thermostat that acts on the
relative motion of the core-Drude particle pairs. Therefore, because LAMMPS considers Drude particles as
normal atoms in its default temperature compute (compute temp command), the reduced temperature of the
core-Drude particle pairs is not calculated correctly.
By contrast, this compute calculates the temperature of the cores using center-of-mass velocities of the
core-Drude pairs, and the reduced temperature of the Drude particles using the relative velocities of the Drude
particles with respect to their cores. Non-polarizable atoms are considered as cores. Their velocities contribute
to the temperature of the cores.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6, which can be
accessed by indices 1-6, whose components are
temperature of the centers of mass (temperature units)1. temperature of the dipoles (temperature units)2. number of degrees of freedom of the centers of mass3. number of degrees of freedom of the dipoles4. kinetic energy of the centers of mass (energy units)5. kinetic energy of the dipoles (energy units)6.
These values can be used by any command that uses global scalar or vector values from a compute as input.
See this section for an overview of LAMMPS output options.
Both the scalar value and the first two values of the vector calculated by this compute are "intensive". The
other 4 vector values are "extensive".
Restrictions:
LAMMPS Users Manual
1345
The number of degrees of freedom contributing to the temperature is assumed to be constant for the duration
of the run unless the fix_modify command sets the option dynamic yes.
Related commands:
fix drude, fix langevin/drude, fix drude/transform, pair_style thole, compute temp
Default: none
LAMMPS Users Manual
1346

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/eff command
Syntax:
compute ID group-ID temp/eff
ID, group-ID are documented in compute command• temp/eff = style name of this compute command•
Examples:
compute 1 all temp/eff
compute myTemp mobile temp/eff
Description:
Define a computation that calculates the temperature of a group of nuclei and electrons in the electron force
field model. A compute of this style can be used by commands that compute a temperature, e.g.
thermo_modify, fix npt/eff, etc.
The temperature is calculated by the formula KE = dim/2 N k T, where KE = total kinetic energy of the group
of atoms (sum of 1/2 m v^2 for nuclei and sum of 1/2 (m v^2 + 3/4 m s^2) for electrons, where s includes the
radial electron velocity contributions), dim = 2 or 3 = dimensionality of the simulation, N = number of atoms
(only total number of nuclei in the eFF (see the pair_eff command) in the group, k = Boltzmann constant, and
T = temperature. This expression is summed over all nuclear and electronic degrees of freedom, essentially by
setting the kinetic contribution to the heat capacity to 3/2k (where only nuclei contribute). This subtlety is
valid for temperatures well below the Fermi temperature, which for densities two to five times the density of
liquid H2 ranges from 86,000 to 170,000 K.
NOTE: For eFF models, in order to override the default temperature reported by LAMMPS in the
thermodynamic quantities reported via the thermo command, the user should apply a thermo_modify
command, as shown in the following example:
compute effTemp all temp/eff
thermo_style custom step etotal pe ke temp press
thermo_modify temp effTemp
A 6-component kinetic energy tensor is also calculated by this compute for use in the computation of a
pressure tensor. The formula for the components of the tensor is the same as the above formula, except that
v^2 is replaced by vx * vy for the xy component, etc. For the eFF, again, the radial electronic velocities are
also considered.
The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
This compute subtracts out degrees-of-freedom due to fixes that constrain molecular motion, such as fix shake
and fix rigid. This means the temperature of groups of atoms that include these constraints will be computed
correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option of the
compute_modify command.
LAMMPS Users Manual
1347
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
Output info:
The scalar value calculated by this compute is "intensive", meaning it is independent of the number of atoms
in the simulation. The vector values are "extensive", meaning they scale with the number of atoms in the
simulation.
Restrictions:
This compute is part of the USER-EFF package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
compute temp/partial, compute temp/region, compute pressure
Default: none
LAMMPS Users Manual
1348

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/partial command
Syntax:
compute ID group-ID temp/partial xflag yflag zflag
ID, group-ID are documented in compute command• temp/partial = style name of this compute command• xflag,yflag,zflag = 0/1 for whether to exclude/include this dimension•
Examples:
compute newT flow temp/partial 1 1 0
Description:
Define a computation that calculates the temperature of a group of atoms, after excluding one or more velocity
components. A compute of this style can be used by any command that computes a temperature, e.g.
thermo_modify, fix temp/rescale, fix npt, etc.
The temperature is calculated by the formula KE = dim/2 N k T, where KE = total kinetic energy of the group
of atoms (sum of 1/2 m v^2), dim = dimensionality of the simulation, N = number of atoms in the group, k =
Boltzmann constant, and T = temperature. The calculation of KE excludes the x, y, or z dimensions if xflag,
yflag, or zflag = 0. The dim parameter is adjusted to give the correct number of degrees of freedom.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute for use in the
calculation of a pressure tensor. The formula for the components of the tensor is the same as the above
formula, except that v^2 is replaced by vx*vy for the xy component, etc. The 6 components of the vector are
ordered xx, yy, zz, xy, xz, yz.
The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
The removal of velocity components by this fix is essentially computing the temperature after a "bias" has
been removed from the velocity of the atoms. If this compute is used with a fix command that performs
thermostatting then this bias will be subtracted from each atom, thermostatting of the remaining thermal
velocity will be performed, and the bias will be added back in. Thermostatting fixes that work in this way
include fix nvt, fix temp/rescale, fix temp/berendsen, and fix langevin.
This compute subtracts out degrees-of-freedom due to fixes that constrain molecular motion, such as fix shake
and fix rigid. This means the temperature of groups of atoms that include these constraints will be computed
correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option of the
compute_modify command.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
LAMMPS Users Manual
1349

5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions: none
Related commands:
compute temp, compute temp/region, compute pressure
Default: none
LAMMPS Users Manual
1350

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/profile command
Syntax:
compute ID group-ID temp/profile xflag yflag zflag binstyle args
ID, group-ID are documented in compute command• temp/profile = style name of this compute command• xflag,yflag,zflag = 0/1 for whether to exclude/include this dimension• binstyle = x or y or z or xy or yz or xz or xyz
x arg = Nx
y arg = Ny
z arg = Nz
xy args = Nx Ny
yz args = Ny Nz
xz args = Nx Nz
xyz args = Nx Ny Nz
Nx,Ny,Nz = number of velocity bins in x,y,z dimensions
•
zero or more keyword/value pairs may be appended• keyword = out
out value = tensor or bin
•
Examples:
compute myTemp flow temp/profile 1 1 1 x 10
compute myTemp flow temp/profile 1 1 1 x 10 out bin
compute myTemp flow temp/profile 0 1 1 xyz 20 20 20
Description:
Define a computation that calculates the temperature of a group of atoms, after subtracting out a
spatially-averaged center-of-mass velocity field, before computing the kinetic energy. This can be useful for
thermostatting a collection of atoms undergoing a complex flow, e.g. via a profile-unbiased thermostat (PUT)
as described in (Evans). A compute of this style can be used by any command that computes a temperature,
e.g. thermo_modify, fix temp/rescale, fix npt, etc.
The xflag, yflag, zflag settings determine which components of average velocity are subtracted out.
The binstyle setting and its Nx, Ny, Nz arguments determine how bins are setup to perform spatial averaging.
"Bins" can be 1d slabs, 2d pencils, or 3d bricks depending on which binstyle is used. The simulation box is
partitioned conceptually into Nx by Ny by Nz bins. Depending on the binstyle, you may only specify one or
two of these values; the others are effectively set to 1 (no binning in that dimension). For non-orthogonal
(triclinic) simulation boxes, the bins are "tilted" slabs or pencils or bricks that are parallel to the tilted faces of
the box. See the region prism command for a discussion of the geometry of tilted boxes in LAMMPS.
When a temperature is computed, the center-of-mass velocity for the set of atoms that are both in the compute
group and in the same spatial bin is calculated. This bias velocity is then subtracted from the velocities of
individual atoms in the bin to yield a thermal velocity for each atom. Note that if there is only one atom in the
bin, its thermal velocity will thus be 0.0.
LAMMPS Users Manual
1351
After the spatially-averaged velocity field has been subtracted from each atom, the temperature is calculated
by the formula KE = (dim*N - dim*Nx*Ny*Nz) k T/2, where KE = total kinetic energy of the group of atoms
(sum of 1/2 m v^2), dim = 2 or 3 = dimensionality of the simulation, N = number of atoms in the group, k =
Boltzmann constant, and T = temperature. The dim*Nx*Ny*Nz term are degrees of freedom subtracted to
adjust for the removal of the center-of-mass velocity in each of Nx*Ny*Nz bins, as discussed in the (Evans)
paper.
If the out keyword is used with a tensor value, which is the default, a kinetic energy tensor, stored as a
6-element vector, is also calculated by this compute for use in the computation of a pressure tensor. The
formula for the components of the tensor is the same as the above formula, except that v^2 is replaced by
vx*vy for the xy component, etc. The 6 components of the vector are ordered xx, yy, zz, xy, xz, yz.
If the out keyword is used with a bin value, the count of atoms and computed temperature for each bin are
stored for output, as an array of values, as described below. The temperature of each bin is calculated as
described above, where the bias velocity is subtracted and only the remaining thermal velocity of atoms in the
bin contributes to the temperature. See the note below for how the temperature is normalized by the
degrees-of-freedom of atoms in the bin.
The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
The removal of the spatially-averaged velocity field by this fix is essentially computing the temperature after a
"bias" has been removed from the velocity of the atoms. If this compute is used with a fix command that
performs thermostatting then this bias will be subtracted from each atom, thermostatting of the remaining
thermal velocity will be performed, and the bias will be added back in. Thermostatting fixes that work in this
way include fix nvt, fix temp/rescale, fix temp/berendsen, and fix langevin.
This compute subtracts out degrees-of-freedom due to fixes that constrain molecular motion, such as fix shake
and fix rigid. This means the temperature of groups of atoms that include these constraints will be computed
correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option of the
compute_modify command.
NOTE: When using the out keyword with a value of bin, the calculated temperature for each bin does not
include the degrees-of-freedom adjustment described in the preceding paragraph, for fixes that constrain
molecular motion. It does include the adjustment due to the extra option, which is applied to each bin.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting. Using this compute in conjunction with a thermostatting fix, as explained there, will
effectively implement a profile-unbiased thermostat (PUT), as described in (Evans).
Output info:
This compute calculates a global scalar (the temperature). Depending on the setting of the out keyword, it also
calculates a global vector or array. For out = tensor, it calculates a vector of length 6 (KE tensor), which can
be accessed by indices 1-6. For out = bin it calculates a global array which has 2 columns and N rows, where
N is the number of bins. The first column contains the number of atoms in that bin. The second contains the
temperature of that bin, calculated as described above. The ordering of rows in the array is as follows. Bins in
x vary fastest, then y, then z. Thus for a 10x10x10 3d array of bins, there will be 1000 rows. The bin with
indices ix,iy,iz = 2,3,4 would map to row M = (iz-1)*10*10 + (iy-1)*10 + ix = 322, where the rows are
numbered from 1 to 1000 and the bin indices are numbered from 1 to 10 in each dimension.
LAMMPS Users Manual
1352

These values can be used by any command that uses global scalar or vector or array values from a compute as
input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive". The array values
are "intensive".
The scalar value will be in temperature units. The vector values will be in energy units. The first column of
array values are counts; the values in the second column will be in temperature units.
Restrictions:
You should not use too large a velocity-binning grid, especially in 3d. In the current implementation, the
binned velocity averages are summed across all processors, so this will be inefficient if the grid is too large,
and the operation is performed every timestep, as it will be for most thermostats.
Related commands:
compute temp, compute temp/ramp, compute temp/deform, compute pressure
Default:
The option default is out = tensor.
(Evans) Evans and Morriss, Phys Rev Lett, 56, 2172-2175 (1986).
LAMMPS Users Manual
1353

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/ramp command
Syntax:
compute ID group-ID temp/ramp vdim vlo vhi dim clo chi keyword value ...
ID, group-ID are documented in compute command• temp/ramp = style name of this compute command• vdim = vx or vy or vz• vlo,vhi = subtract velocities between vlo and vhi (velocity units)• dim = x or y or z• clo,chi = lower and upper bound of domain to subtract from (distance units)• zero or more keyword/value pairs may be appended• keyword = units•
units value = lattice or box
Examples:
compute 2nd middle temp/ramp vx 0 8 y 2 12 units lattice
Description:
Define a computation that calculates the temperature of a group of atoms, after subtracting out an ramped
velocity profile before computing the kinetic energy. A compute of this style can be used by any command
that computes a temperature, e.g. thermo_modify, fix temp/rescale, fix npt, etc.
The meaning of the arguments for this command which define the velocity ramp are the same as for the
velocity ramp command which was presumably used to impose the velocity.
After the ramp velocity has been subtracted from the specified dimension for each atom, the temperature is
calculated by the formula KE = dim/2 N k T, where KE = total kinetic energy of the group of atoms (sum of
1/2 m v^2), dim = 2 or 3 = dimensionality of the simulation, N = number of atoms in the group, k =
Boltzmann constant, and T = temperature.
The units keyword determines the meaning of the distance units used for coordinates (c1,c2) and velocities
(vlo,vhi). A box value selects standard distance units as defined by the units command, e.g. Angstroms for
units = real or metal. A lattice value means the distance units are in lattice spacings; e.g. velocity = lattice
spacings / tau. The lattice command must have been previously used to define the lattice spacing.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute for use in the
computation of a pressure tensor. The formula for the components of the tensor is the same as the above
formula, except that v^2 is replaced by vx*vy for the xy component, etc. The 6 components of the vector are
ordered xx, yy, zz, xy, xz, yz.
The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
LAMMPS Users Manual
1354
The removal of the ramped velocity component by this fix is essentially computing the temperature after a
"bias" has been removed from the velocity of the atoms. If this compute is used with a fix command that
performs thermostatting then this bias will be subtracted from each atom, thermostatting of the remaining
thermal velocity will be performed, and the bias will be added back in. Thermostatting fixes that work in this
way include fix nvt, fix temp/rescale, fix temp/berendsen, and fix langevin.
This compute subtracts out degrees-of-freedom due to fixes that constrain molecular motion, such as fix shake
and fix rigid. This means the temperature of groups of atoms that include these constraints will be computed
correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option of the
compute_modify command.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions: none
Related commands:
compute temp, compute temp/profie, compute temp/deform, compute pressure
Default:
The option default is units = lattice.
LAMMPS Users Manual
1355

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/region command
Syntax:
compute ID group-ID temp/region region-ID
ID, group-ID are documented in compute command• temp/region = style name of this compute command• region-ID = ID of region to use for choosing atoms•
Examples:
compute mine flow temp/region boundary
Description:
Define a computation that calculates the temperature of a group of atoms in a geometric region. This can be
useful for thermostatting one portion of the simulation box. E.g. a McDLT simulation where one side is
cooled, and the other side is heated. A compute of this style can be used by any command that computes a
temperature, e.g. thermo_modify, fix temp/rescale, etc.
Note that a region-style temperature can be used to thermostat with fix temp/rescale or fix langevin, but
should probably not be used with Nose/Hoover style fixes (fix nvt, fix npt, or fix nph), if the
degrees-of-freedom included in the computed T varies with time.
The temperature is calculated by the formula KE = dim/2 N k T, where KE = total kinetic energy of the group
of atoms (sum of 1/2 m v^2), dim = 2 or 3 = dimensionality of the simulation, N = number of atoms in both
the group and region, k = Boltzmann constant, and T = temperature.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute for use in the
computation of a pressure tensor. The formula for the components of the tensor is the same as the above
formula, except that v^2 is replaced by vx*vy for the xy component, etc. The 6 components of the vector are
ordered xx, yy, zz, xy, xz, yz.
The number of atoms contributing to the temperature is calculated each time the temperature is evaluated
since it is assumed atoms can enter/leave the region. Thus there is no need to use the dynamic option of the
compute_modify command for this compute style.
The removal of atoms outside the region by this fix is essentially computing the temperature after a "bias" has
been removed, which in this case is the velocity of any atoms outside the region. If this compute is used with a
fix command that performs thermostatting then this bias will be subtracted from each atom, thermostatting of
the remaining thermal velocity will be performed, and the bias will be added back in. Thermostatting fixes
that work in this way include fix nvt, fix temp/rescale, fix temp/berendsen, and fix langevin. This means that
when this compute is used to calculate the temperature for any of the thermostatting fixes via the fix modify
temp command, the thermostat will operate only on atoms that are currently in the geometric region.
Unlike other compute styles that calculate temperature, this compute does not subtract out degrees-of-freedom
due to fixes that constrain motion, such as fix shake and fix rigid. This is because those degrees of freedom
(e.g. a constrained bond) could apply to sets of atoms that straddle the region boundary, and hence the concept
LAMMPS Users Manual
1356
is somewhat ill-defined. If needed the number of subtracted degrees-of-freedom can be set explicitly using the
extra option of the compute_modify command.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions: none
Related commands:
compute temp, compute pressure
Default: none
LAMMPS Users Manual
1357

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/region/eff command
Syntax:
compute ID group-ID temp/region/eff region-ID
ID, group-ID are documented in compute command• temp/region/eff = style name of this compute command• region-ID = ID of region to use for choosing atoms•
Examples:
compute mine flow temp/region/eff boundary
Description:
Define a computation that calculates the temperature of a group of nuclei and electrons in the electron force
field model, within a geometric region using the electron force field. A compute of this style can be used by
commands that compute a temperature, e.g. thermo_modify.
The operation of this compute is exactly like that described by the compute temp/region command, except that
the formula for the temperature itself includes the radial electron velocity contributions, as discussed by the
compute temp/eff command.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions:
This compute is part of the USER-EFF package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
compute temp/region, compute temp/eff, compute pressure
Default: none
LAMMPS Users Manual
1358

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/rotate command
Syntax:
compute ID group-ID temp/rotate
ID, group-ID are documented in compute command• temp/rotate = style name of this compute command•
Examples:
compute Tbead bead temp/rotate
Description:
Define a computation that calculates the temperature of a group of atoms, after subtracting out the
center-of-mass velocity and angular velocity of the group. This is useful if the group is expected to have a
non-zero net velocity and/or global rotation motion for some reason. A compute of this style can be used by
any command that computes a temperature, e.g. thermo_modify, fix temp/rescale, fix npt, etc.
After the center-of-mass velocity and angular velocity has been subtracted from each atom, the temperature is
calculated by the formula KE = dim/2 N k T, where KE = total kinetic energy of the group of atoms (sum of
1/2 m v^2), dim = 2 or 3 = dimensionality of the simulation, N = number of atoms in the group, k =
Boltzmann constant, and T = temperature.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute for use in the
computation of a pressure tensor. The formula for the components of the tensor is the same as the above
formula, except that v^2 is replaced by vx*vy for the xy component, etc. The 6 components of the vector are
ordered xx, yy, zz, xy, xz, yz.
The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
The removal of the center-of-mass velocity and angular velocity by this fix is essentially computing the
temperature after a "bias" has been removed from the velocity of the atoms. If this compute is used with a fix
command that performs thermostatting then this bias will be subtracted from each atom, thermostatting of the
remaining thermal velocity will be performed, and the bias will be added back in. Thermostatting fixes that
work in this way include fix nvt, fix temp/rescale, fix temp/berendsen, and fix langevin.
This compute subtracts out degrees-of-freedom due to fixes that constrain molecular motion, such as fix shake
and fix rigid. This means the temperature of groups of atoms that include these constraints will be computed
correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option of the
compute_modify command.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
Output info:
LAMMPS Users Manual
1359
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions:
This compute is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
compute temp
Default: none
LAMMPS Users Manual
1360

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute temp/sphere command
Syntax:
compute ID group-ID temp/sphere keyword value ...
ID, group-ID are documented in compute command• temp/sphere = style name of this compute command• zero or more keyword/value pairs may be appended• keyword = bias or dof
bias value = bias-ID
bias-ID = ID of a temperature compute that removes a velocity bias
dof value = all or rotate
all = compute temperature of translational and rotational degrees of freedom
rotate = compute temperature of just rotational degrees of freedom
•
Examples:
compute 1 all temp/sphere
compute myTemp mobile temp/sphere bias tempCOM
compute myTemp mobile temp/sphere dof rotate
Description:
Define a computation that calculates the temperature of a group of spherical particles, including a contribution
from both their translational and rotational kinetic energy. This differs from the usual compute temp
command, which assumes point particles with only translational kinetic energy.
Both point and finite-size particles can be included in the group. Point particles do not rotate, so they have
only 3 translational degrees of freedom. For 3d spherical particles, each has 6 degrees of freedom (3
translational, 3 rotational). For 2d spherical particles, each has 3 degrees of freedom (2 translational, 1
rotational).
NOTE: This choice for degrees of freedom (dof) assumes that all finite-size spherical particles in your model
will freely rotate, sampling all their rotational dof. It is possible to use a combination of interaction potentials
and fixes that induce no torque or otherwise constrain some of all of your particles so that this is not the case.
Then there are less dof and you should use the compute_modify extra command to adjust the dof accordingly.
The translational kinetic energy is computed the same as is described by the compute temp command. The
rotational kinetic energy is computed as 1/2 I w^2, where I is the moment of inertia for a sphere and w is the
particle's angular velocity.
NOTE: For 2d models, particles are treated as spheres, not disks, meaning their moment of inertia will be the
same as in 3d.
A kinetic energy tensor, stored as a 6-element vector, is also calculated by this compute. The formula for the
components of the tensor is the same as the above formulas, except that v^2 and w^2 are replaced by vx*vy
and wx*wy for the xy component. The 6 components of the vector are ordered xx, yy, zz, xy, xz, yz.
LAMMPS Users Manual
1361

The number of atoms contributing to the temperature is assumed to be constant for the duration of the run; use
the dynamic option of the compute_modify command if this is not the case.
This compute subtracts out translational degrees-of-freedom due to fixes that constrain molecular motion,
such as fix shake and fix rigid. This means the temperature of groups of atoms that include these constraints
will be computed correctly. If needed, the subtracted degrees-of-freedom can be altered using the extra option
of the compute_modify command.
See this howto section of the manual for a discussion of different ways to compute temperature and perform
thermostatting.
The keyword/value option pairs are used in the following ways.
For the bias keyword, bias-ID refers to the ID of a temperature compute that removes a "bias" velocity from
each atom. This allows compute temp/sphere to compute its thermal temperature after the translational kinetic
energy components have been altered in a prescribed way, e.g. to remove a flow velocity profile. Thermostats
that use this compute will work with this bias term. See the doc pages for individual computes that calculate a
temperature and the doc pages for fixes that perform thermostatting for more details.
For the dof keyword, a setting of all calculates a temperature that includes both translational and rotational
degrees of freedom. A setting of rotate calculates a temperature that includes only rotational degrees of
freedom.
Output info:
This compute calculates a global scalar (the temperature) and a global vector of length 6 (KE tensor), which
can be accessed by indices 1-6. These values can be used by any command that uses global scalar or vector
values from a compute as input. See this section for an overview of LAMMPS output options.
The scalar value calculated by this compute is "intensive". The vector values are "extensive".
The scalar value will be in temperature units. The vector values will be in energy units.
Restrictions:
This fix requires that atoms store torque and angular velocity (omega) and a radius as defined by the
atom_style sphere command.
All particles in the group must be finite-size spheres, or point particles with radius = 0.0.
Related commands:
compute temp, compute temp/asphere
Default:
The option defaults are no bias and dof = all.
LAMMPS Users Manual
1362

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute ti command
Syntax:
compute ID group ti keyword args ...
ID, group-ID are documented in compute command• ti = style name of this compute command• one or more attribute/arg pairs may be appended• keyword = pair style (lj/cut, gauss, born, etc) or tail or kspace
pair style args = atype v_name1 v_name2
atype = atom type (see asterisk form below)
v_name1 = variable with name1 that is energy scale factor and function of lambda
v_name2 = variable with name2 that is derivative of v_name1 with respect to lambda
tail args = atype v_name1 v_name2
atype = atom type (see asterisk form below)
v_name1 = variable with name1 that is energy tail correction scale factor and function of lambda
v_name2 = variable with name2 that is derivative of v_name1 with respect to lambda
kspace args = atype v_name1 v_name2
atype = atom type (see asterisk form below)
v_name1 = variable with name1 that is K-Space scale factor and function of lambda
v_name2 = variable with name2 that is derivative of v_name1 with respect to lambda
•
Examples:
compute 1 all ti lj/cut 1 v_lj v_dlj coul/long 2 v_c v_dc kspace 1 v_ks v_dks
compute 1 all ti lj/cut 1*3 v_lj v_dlj coul/long * v_c v_dc kspace * v_ks v_dks
Description:
Define a computation that calculates the derivative of the interaction potential with respect to lambda, the
coupling parameter used in a thermodynamic integration. This derivative can be used to infer a free energy
difference resulting from an alchemical simulation, as described in Eike.
Typically this compute will be used in conjunction with the fix adapt command which can perform alchemical
transformations by adusting the strength of an interaction potential as a simulation runs, as defined by one or
more pair_style or kspace_style commands. This scaling is done via a prefactor on the energy, forces, virial
calculated by the pair or K-Space style. The prefactor is often a function of a lambda parameter which may be
adjusted from 0 to 1 (or vice versa) over the course of a run. The time-dependent adjustment is what the fix
adapt command does.
Assume that the unscaled energy of a pair_style or kspace_style is given by U. Then the scaled energy is
Us = f(lambda) U
where f() is some function of lambda. What this compute calculates is
dUs / d(lambda) = U df(lambda)/dlambda = Us / f(lambda) df(lambda)/dlambda
which is the derivative of the system's scaled potential energy Us with respect to lambda.
LAMMPS Users Manual
1363

To perform this calculation, you provide one or more atom types as atype. Atype can be specified in one of
two ways. An explicit numeric values can be used, as in the 1st example above. Or a wildcard asterisk can be
used in place of or in conjunction with the atype argument to select multiple atom types. This takes the form
"*" or "*n" or "n*" or "m*n". If N = the number of atom types, then an asterisk with no numeric values means
all types from 1 to N. A leading asterisk means all types from 1 to n (inclusive). A trailing asterisk means all
types from n to N (inclusive). A middle asterisk means all types from m to n (inclusive).
You also specify two functions, as equal-style variables. The first is specified as v_name1, where name1 is the
name of the variable, and is f(lambda) in the notation above. The second is specified as v_name2, where
name2 is the name of the variable, and is df(lambda) / dlambda in the notation above. I.e. it is the analytic
derivative of f() with respect to lambda. Note that the name1 variable is also typically given as an argument to
the fix adapt command.
An alchemical simulation may use several pair potentials together, invoked via the pair_style hybrid or
hybrid/overlay command. The total dUs/dlambda for the overall system is calculated as the sum of each
contributing term as listed by the keywords in the compute ti command. Individual pair potentials can be
listed, which will be sub-styles in the hybrid case. You can also include a K-space term via the kspace
keyword. You can also include a pairwise long-range tail correction to the energy via the tail keyword.
For each term you can specify a different (or the same) scale factor by the two variables that you list. Again,
these will typically correspond toe the scale factors applied to these various potentials and the K-Space
contribution via the fix adapt command.
More details about the exact functional forms for the computation of du/dl can be found in the paper by Eike.
Output info:
This compute calculates a global scalar, namely dUs/dlambda. This value can be used by any command that
uses a global scalar value from a compute as input. See Section 6.15 for an overview of LAMMPS output
options.
The scalar value calculated by this compute is "extensive".
The scalar value will be in energy units.
Restrictions:
This compute is part of the MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
fix adapt
Default: none
(Eike) Eike and Maginn, Journal of Chemical Physics, 124, 164503 (2006).
LAMMPS Users Manual
1364

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute torque/chunk command
Syntax:
compute ID group-ID torque/chunk chunkID
ID, group-ID are documented in compute command• torque/chunk = style name of this compute command• chunkID = ID of compute chunk/atom command•
Examples:
compute 1 fluid torque/chunk molchunk
Description:
Define a computation that calculates the torque on multiple chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
This compute calculates the 3 components of the torque vector for eqch chunk, due to the forces on the
individual atoms in the chunk around the center-of-mass of the chunk. The calculation includes all effects due
to atoms passing thru periodic boundaries.
Note that only atoms in the specified group contribute to the calculation. The compute chunk/atom command
defines its own group; atoms will have a chunk ID = 0 if they are not in that group, signifying they are not
assigned to a chunk, and will thus also not contribute to this calculation. You can specify the "all" group for
this command if you simply want to include atoms with non-zero chunk IDs.
NOTE: The coordinates of an atom contribute to the chunk's torque in "unwrapped" form, by using the image
flags associated with each atom. See the dump custom command for a discussion of "unwrapped" coordinates.
See the Atoms section of the read_data command for a discussion of image flags and how they are set for each
atom. You can reset the image flags (e.g. to 0) before invoking this compute by using the set image command.
The simplest way to output the results of the compute torque/chunk calculation to a file is to use the fix
ave/time command, for example:
compute cc1 all chunk/atom molecule
compute myChunk all torque/chunk cc1
fix 1 all ave/time 100 1 100 c_myChunk[*] file tmp.out mode vector
Output info:
This compute calculates a global array where the number of rows = the number of chunks Nchunk as
calculated by the specified compute chunk/atom command. The number of columns = 3 for the 3 xyz
LAMMPS Users Manual
1365
components of the torque for each chunk. These values can be accessed by any command that uses global
array values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The array values are "intensive". The array values will be in force-distance units.
Restrictions: none
Related commands:
variable torque() function
Default: none
LAMMPS Users Manual
1366

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute vacf command
Syntax:
compute ID group-ID vacf
ID, group-ID are documented in compute command• vacf = style name of this compute command•
Examples:
compute 1 all vacf
compute 1 upper vacf
Description:
Define a computation that calculates the velocity auto-correlation function (VACF), averaged over a group of
atoms. Each atom's contribution to the VACF is its current velocity vector dotted into its initial velocity vector
at the time the compute was specified.
A vector of four quantities is calculated by this compute. The first 3 elements of the vector are vx * vx0 (and
similarly for the y and z components), summed and averaged over atoms in the group. Vx is the current
x-component of velocity for the atom, vx0 is the initial x-component of velocity for the atom. The 4th element
of the vector is the total VACF, i.e. (vx*vx0 + vy*vy0 + vz*vz0), summed and averaged over atoms in the
group.
The integral of the VACF versus time is proportional to the diffusion coefficient of the diffusing atoms. This
can be computed in the following manner, using the variable trap() function:
compute 2 all vacf
fix 5 all vector 1 c_2[4]
variable diff equal dt*trap(f_5)
thermo_style custom step v_diff
NOTE: If you want the quantities calculated by this compute to be continuous when running from a restart
file, then you should use the same ID for this compute, as in the original run. This is so that the fix this
compute creates to store per-atom quantities will also have the same ID, and thus be initialized correctly with
time=0 atom velocities from the restart file.
Output info:
This compute calculates a global vector of length 4, which can be accessed by indices 1-4 by any command
that uses global vector values from a compute as input. See this section for an overview of LAMMPS output
options.
The vector values are "intensive". The vector values will be in velocity^2 units.
Restrictions: none
Related commands:
LAMMPS Users Manual
1367

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute vcm/chunk command
Syntax:
compute ID group-ID vcm/chunk chunkID
ID, group-ID are documented in compute command• vcm/chunk = style name of this compute command• chunkID = ID of compute chunk/atom command•
Examples:
compute 1 fluid vcm/chunk molchunk
Description:
Define a computation that calculates the center-of-mass velocity for multiple chunks of atoms.
In LAMMPS, chunks are collections of atoms defined by a compute chunk/atom command, which assigns
each atom to a single chunk (or no chunk). The ID for this command is specified as chunkID. For example, a
single chunk could be the atoms in a molecule or atoms in a spatial bin. See the compute chunk/atom doc page
and Section 6.23 for details of how chunks can be defined and examples of how they can be used to measure
properties of a system.
This compute calculates the x,y,z components of the center-of-mass velocity for each chunk. This is done by
summing mass*velocity for each atom in the chunk and dividing the sum by the total mass of the chunk.
Note that only atoms in the specified group contribute to the calculation. The compute chunk/atom command
defines its own group; atoms will have a chunk ID = 0 if they are not in that group, signifying they are not
assigned to a chunk, and will thus also not contribute to this calculation. You can specify the "all" group for
this command if you simply want to include atoms with non-zero chunk IDs.
The simplest way to output the results of the compute vcm/chunk calculation to a file is to use the fix ave/time
command, for example:
compute cc1 all chunk/atom molecule
compute myChunk all vcm/chunk cc1
fix 1 all ave/time 100 1 100 c_myChunk[*] file tmp.out mode vector
Output info:
This compute calculates a global array where the number of rows = the number of chunks Nchunk as
calculated by the specified compute chunk/atom command. The number of columns = 3 for the x,y,z
center-of-mass velocity coordinates of each chunk. These values can be accessed by any command that uses
global array values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
The array values are "intensive". The array values will be in velocity units.
Restrictions: none
LAMMPS Users Manual
1369
Related commands: none
Default: none
LAMMPS Users Manual
1370

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute voronoi/atom command
Syntax:
compute ID group-ID voronoi/atom keyword arg ...
ID, group-ID are documented in compute command• voronoi/atom = style name of this compute command• zero or more keyword/value pairs may be appended• keyword = only_group or surface or radius or edge_histo or edge_threshold or face_threshold or
neighbors or peratom
only_group = no arg
occupation = no arg
surface arg = sgroup-ID
sgroup-ID = compute the dividing surface between group-ID and sgroup-ID
this keyword adds a third column to the compute output
radius arg = v_r
v_r = radius atom style variable for a poly-disperse Voronoi tessellation
edge_histo arg = maxedge
maxedge = maximum number of Voronoi cell edges to be accounted in the histogram
edge_threshold arg = minlength
minlength = minimum length for an edge to be counted
face_threshold arg = minarea
minarea = minimum area for a face to be counted
neighbors value = yes or no = store list of all neighbors or no
peratom value = yes or no = per-atom quantities accessible or no
•
Examples:
compute 1 all voronoi/atom
compute 2 precipitate voronoi/atom surface matrix
compute 3b precipitate voronoi/atom radius v_r
compute 4 solute voronoi/atom only_group
compute 5 defects voronoi/atom occupation
compute 6 all voronoi/atom neighbors yes
Description:
Define a computation that calculates the Voronoi tessellation of the atoms in the simulation box. The
tessellation is calculated using all atoms in the simulation, but non-zero values are only stored for atoms in the
group.
By default two per-atom quantities are calculated by this compute. The first is the volume of the Voronoi cell
around each atom. Any point in an atom's Voronoi cell is closer to that atom than any other. The second is the
number of faces of the Voronoi cell. This is equal to the number of nearest neighbors of the central atom, plus
any exterior faces (see note below). If the peratom keyword is set to "no", the per-atom quantities are still
calculated, but they are not accessible.
If the only_group keyword is specified the tessellation is performed only with respect to the atoms contained
in the compute group. This is equivalent to deleting all atoms not contained in the group prior to evaluating
the tessellation.
LAMMPS Users Manual
1371
If the surface keyword is specified a third quantity per atom is computed: the Voronoi cell surface of the
given atom. surface takes a group ID as an argument. If a group other than all is specified, only the Voronoi
cell facets facing a neighbor atom from the specified group are counted towards the surface area.
In the example above, a precipitate embedded in a matrix, only atoms at the surface of the precipitate will
have non-zero surface area, and only the outward facing facets of the Voronoi cells are counted (the hull of
the precipitate). The total surface area of the precipitate can be obtained by running a "reduce sum" compute
on c_2[3]
If the radius keyword is specified with an atom style variable as the argument, a poly-disperse Voronoi
tessellation is performed. Examples for radius variables are
variable r1 atom (type==1)*0.1+(type==2)*0.4
compute radius all property/atom radius
variable r2 atom c_radius
Here v_r1 specifies a per-type radius of 0.1 units for type 1 atoms and 0.4 units for type 2 atoms, and v_r2
accesses the radius property present in atom_style sphere for granular models.
The edge_histo keyword activates the compilation of a histogram of number of edges on the faces of the
Voronoi cells in the compute group. The argument maxedge of the this keyword is the largest number of
edges on a single Voronoi cell face expected to occur in the sample. This keyword adds the generation of a
global vector with maxedge+1 entries. The last entry in the vector contains the number of faces with with
more than maxedge edges. Since the polygon with the smallest amount of edges is a triangle, entries 1 and 2
of the vector will always be zero.
The edge_threshold and face_threshold keywords allow the suppression of edges below a given minimum
length and faces below a given minimum area. Ultra short edges and ultra small faces can occur as artifacts of
the Voronoi tessellation. These keywords will affect the neighbor count and edge histogram outputs.
If the occupation keyword is specified the tessellation is only performed for the first invocation of the
compute and then stored. For all following invocations of the compute the number of atoms in each Voronoi
cell in the stored tessellation is counted. In this mode the compute returns a per-atom array with 2 columns.
The first column is the number of atoms currently in the Voronoi volume defined by this atom at the time of
the first invocation of the compute (note that the atom may have moved significantly). The second column
contains the total number of atoms sharing the Voronoi cell of the stored tessellation at the location of the
current atom. Numbers in column one can be any positive integer including zero, while column two values
will always be greater than zero. Column one data can be used to locate vacancies (the coordinates are given
by the atom coordinates at the time step when the compute was first invoked), while column two data can be
used to identify interstitial atoms.
If the neighbors value is set to yes, then this compute creates a local array with 3 columns. There is one row
for each face of each Voronoi cell. The 3 columns are the atom ID of the atom that owns the cell, the atom ID
of the atom in the neighboring cell (or zero if the face is external), and the area of the face. The array can be
accessed by any command that uses local values from a compute as input. See this section for an overview of
LAMMPS output options. More specifically, the array can be accessed by a dump local command to write a
file containing all the Voronoi neighbors in a system:
compute 6 all voronoi/atom neighbors yes
dump d2 all local 1 dump.neighbors index c_6[1] c_6[2] c_6[3]
If the face_threshold keyword is used, then only faces with areas greater than the threshold are stored.
LAMMPS Users Manual
1372

The Voronoi calculation is performed by the freely available Voro++ package, written by Chris Rycroft at UC
Berkeley and LBL, which must be installed on your system when building LAMMPS for use with this
compute. See instructions on obtaining and installing the Voro++ software in the src/VORONOI/README
file.
NOTE: The calculation of Voronoi volumes is performed by each processor for the atoms it owns, and
includes the effect of ghost atoms stored by the processor. This assumes that the Voronoi cells of owned
atoms are not affected by atoms beyond the ghost atom cut-off distance. This is usually a good assumption for
liquid and solid systems, but may lead to underestimation of Voronoi volumes in low density systems. By
default, the set of ghost atoms stored by each processor is determined by the cutoff used for pair_style
interactions. The cutoff can be set explicitly via the comm_modify cutoff command. The Voronoi cells for
atoms adjacent to empty regions will extend into those regions up to the communication cutoff in x, y, or z. In
that situation, an exterior face is created at the cutoff distance normal to the x, y, or z direction. For triclinic
systems, the exterior face is parallel to the corresponding reciprocal lattice vector.
NOTE: The Voro++ package performs its calculation in 3d. This will still work for a 2d LAMMPS
simulation, provided all the atoms have the same z coordinate. The Voronoi cell of each atom will be a
columnar polyhedron with constant cross-sectional area along the z direction and two exterior faces at the top
and bottom of the simulation box. If the atoms do not all have the same z coordinate, then the columnar cells
will be accordingly distorted. The cross-sectional area of each Voronoi cell can be obtained by dividing its
volume by the z extent of the simulation box. Note that you define the z extent of the simulation box for 2d
simulations when using the create_box or read_data commands.
Output info:
By default, this compute calculates a per-atom array with 2 columns. In regular dynamic tessellation mode the
first column is the Voronoi volume, the second is the neighbor count, as described above (read above for the
output data in case the occupation keyword is specified). These values can be accessed by any command that
uses per-atom values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
If the peratom keyword is set to "no", the per-atom array is still created, but it is not accessible.
If the edge_histo keyword is used, then this compute generates a global vector of length maxedge+1,
containing a histogram of the number of edges per face.
If the neighbors value is set to yes, then this compute calculates a local array with 3 columns. There is one
row for each face of each Voronoi cell.
NOTE: Some LAMMPS commands such as the compute reduce command can accept either a per-atom or
local quantity. If this compute produces both quantities, the command may access the per-atom quantity, even
if you want to access the local quantity. This effect can be eliminated by using the peratom keyword to turn
off the production of the per-atom quantities. For the default value yes both quantities are produced. For the
value no, only the local array is produced.
The Voronoi cell volume will be in distance units cubed. The Voronoi face area will be in distance units
squared.
Restrictions:
This compute is part of the VORONOI package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
LAMMPS Users Manual
1373

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
compute xrd command
Syntax:
compute ID group-ID xrd lambda type1 type2 ... typeN keyword value ...
ID, group-ID are documented in compute command• xrd = style name of this compute command• lambda = wavelength of incident radiation (length units)• type1 type2 ... typeN = chemical symbol of each atom type (see valid options below)• zero or more keyword/value pairs may be appended• keyword = 2Theta or c or LP or manual or echo
2Theta values = Min2Theta Max2Theta
Min2Theta,Max2Theta = minimum and maximum 2 theta range to explore
(radians or degrees)
c values = c1 c2 c3
c1,c2,c3 = parameters to adjust the spacing of the reciprocal
lattice nodes in the h, k, and l directions respectively
LP value = switch to apply Lorentz-polarization factor
0/1 = off/on
manual = flag to use manual spacing of reciprocal lattice points
based on the values of the c parameters
echo = flag to provide extra output for debugging purposes
•
Examples:
compute 1 all xrd 1.541838 Al O 2Theta 0.087 0.87 c 1 1 1 LP 1 echo
compute 2 all xrd 1.541838 Al O 2Theta 10 100 c 0.05 0.05 0.05 LP 1 manual
fix 1 all ave/histo/weight 1 1 1 0.087 0.87 250 c_1[1] c_1[2] mode vector file Rad2Theta.xrd
fix 2 all ave/histo/weight 1 1 1 10 100 250 c_2[1] c_2[2] mode vector file Deg2Theta.xrd
Description:
Define a computation that calculates x-ray diffraction intensity as described in (Coleman) on a mesh of
reciprocal lattice nodes defined by the entire simulation domain (or manually) using a simulated radiation of
wavelength lambda.
The x-ray diffraction intensity, I, at each reciprocal lattice point, k, is computed from the structure factor, F,
using the equations:
LAMMPS Users Manual
1375
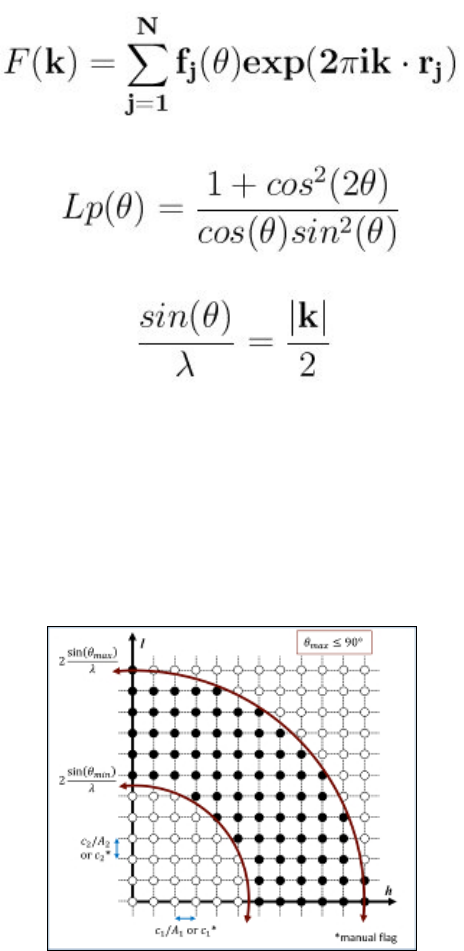
Here, K is the location of the reciprocal lattice node, rj is the position of each atom, fj are atomic scattering
factors, LP is the Lorentz-polarization factor, and theta is the scattering angle of diffraction. The
Lorentz-polarization factor can be turned off using the optional LP keyword.
Diffraction intensities are calculated on a three-dimensional mesh of reciprocal lattice nodes. The mesh
spacing is defined either (a) by the entire simulation domain or (b) manually using selected values as shown in
the 2D diagram below.
For a mesh defined by the simulation domain, a rectilinear grid is constructed with spacing c*inv(A) along
each reciprocal lattice axis. Where A are the vectors corresponding to the edges of the simulation cell. If one
or two directions has non-periodic boundary conditions, then the spacing in these directions is defined from
the average of the (inversed) box lengths with periodic boundary conditions. Meshes defined by the
simulation domain must contain at least one periodic boundary.
If the manual flag is included, the mesh of reciprocal lattice nodes will defined using the c values for the
spacing along each reciprocal lattice axis. Note that manual mapping of the reciprocal space mesh is good for
comparing diffraction results from multiple simulations; however it can reduce the likelihood that Bragg
reflections will be satisfied unless small spacing parameters (< 0.05 Angstrom^(-1)) are implemented. Meshes
with manual spacing do not require a periodic boundary.
The limits of the reciprocal lattice mesh are determined by range of scattering angles explored. The 2Theta
parameters allows the user to reduce the scattering angle range to only the region of interest which reduces the
LAMMPS Users Manual
1376

cost of the computation.
The atomic scattering factors, fj, accounts for the reduction in diffraction intensity due to Compton scattering.
Compute xrd uses analytical approximations of the atomic scattering factors that vary for each atom type
(type1 type2 ... typeN) and angle of diffraction. The analytic approximation is computed using the formula
(Colliex):
Coefficients parameterized by (Peng) are assigned for each atom type designating the chemical symbol and
charge of each atom type. Valid chemical symbols for compute xrd are:
H He1- He Li Li1+
Be Be2+ B C Cval
N O O1- F F1-
Ne Na Na1+ Mg Mg2+
Al Al3+ Si Sival Si4+
P S Cl Cl1- Ar
K Ca Ca2+ Sc Sc3+
Ti Ti2+ Ti3+ Ti4+ V
V2+ V3+ V5+ Cr Cr2+
Cr3+ Mn Mn2+ Mn3+ Mn4+
Fe Fe2+ Fe3+ Co Co2+
Co Ni Ni2+ Ni3+ Cu
Cu1+ Cu2+ Zn Zn2+ Ga
Ga3+ Ge Ge4+ As Se
Br Br1- Kr Rb Rb1+
Sr Sr2+ Y Y3+ Zr
Zr4+ Nb Nb3+ Nb5+ Mo
Mo3+ Mo5+ Mo6+ Tc Ru
Ru3+ Ru4+ Rh Rh3+ Rh4+
Pd Pd2+ Pd4+ Ag Ag1+
Ag2+ Cd Cd2+ In In3+
Sn Sn2+ Sn4+ Sb Sb3+
Sb5+ Te I I1- Xe
Cs Cs1+ Ba Ba2+ La
La3+ Ce Ce3+ Ce4+ Pr
Pr3+ Pr4+ Nd Nd3+ Pm
Pm3+ Sm Sm3+ Eu Eu2+
Eu3+ Gd Gd3+ Tb Tb3+
Dy Dy3+ Ho Ho3+ Er
LAMMPS Users Manual
1377
Er3+ Tm Tm3+ Yb Yb2+
Yb3+ Lu Lu3+ Hf Hf4+
Ta Ta5+ W W6+ Re
Os Os4+ Ir Ir3+ Ir4+
Pt Pt2+ Pt4+ Au Au1+
Au3+ Hg Hg1+ Hg2+ Tl
Tl1+ Tl3+ Pb Pb2+ Pb4+
Bi Bi3+ Bi5+ Po At
Rn Fr Ra Ra2+ Ac
Ac3+ Th Th4+ Pa U
U3+ U4+ U6+ Np Np3+
Np4+ Np6+ Pu Pu3+ Pu4+
Pu6+ Am Cm Bk Cf
If the echo keyword is specified, compute xrd will provide extra reporting information to the screen.
Output info:
This compute calculates a global array. The number of rows in the array is the number of reciprocal lattice
nodes that are explored which by the mesh. The global array has 2 columns.
The first column contains the diffraction angle in the units (radians or degrees) provided with the 2Theta
values. The second column contains the computed diffraction intensities as described above.
The array can be accessed by any command that uses global values from a compute as input. See this section
for an overview of LAMMPS output options.
All array values calculated by this compute are "intensive".
Restrictions:
This compute is part of the USER-DIFFRACTION package. It is only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info.
The compute_xrd command does not work for triclinic cells.
Related commands:
fix ave/histo, compute saed
Default:
The option defaults are 2Theta = 1 179 (degrees), c = 1 1 1, LP = 1, no manual flag, no echo flag.
(Coleman) Coleman, Spearot, Capolungo, MSMSE, 21, 055020 (2013).
LAMMPS Users Manual
1378
(Colliex) Colliex et al. International Tables for Crystallography Volume C: Mathematical and Chemical
Tables, 249-429 (2004).
(Peng) Peng, Ren, Dudarev, Whelan, Acta Crystallogr. A, 52, 257-76 (1996).
LAMMPS Users Manual
1379
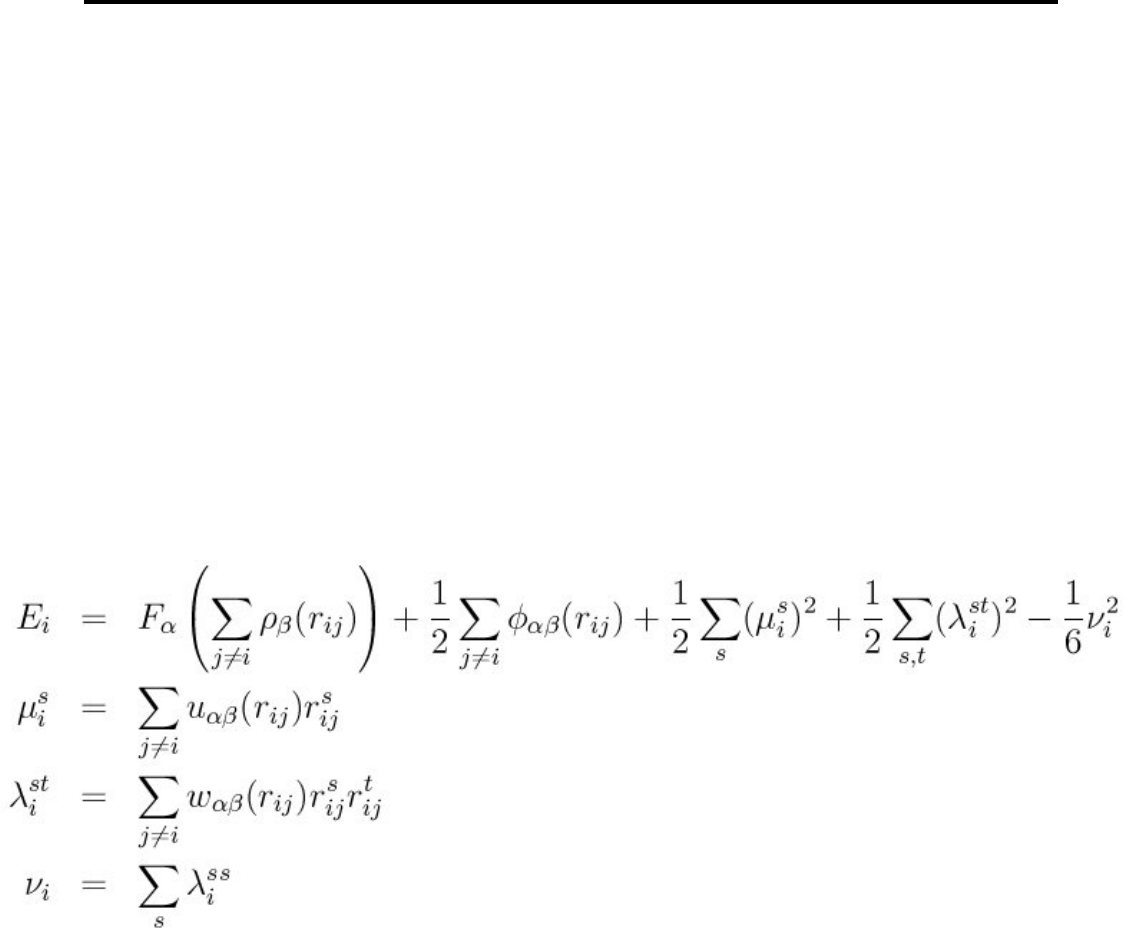
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style adp command
pair_style adp/omp command
Syntax:
pair_style adp
Examples:
pair_style adp
pair_coeff * * Ta.adp Ta
pair_coeff * * ../potentials/AlCu.adp Al Al Cu
Description:
Style adp computes pairwise interactions for metals and metal alloys using the angular dependent potential
(ADP) of (Mishin), which is a generalization of the embedded atom method (EAM) potential. The LAMMPS
implementation is discussed in (Singh). The total energy Ei of an atom I is given by
where F is the embedding energy which is a function of the atomic electron density rho, phi is a pair potential
interaction, alpha and beta are the element types of atoms I and J, and s and t = 1,2,3 and refer to the cartesian
coordinates. The mu and lambda terms represent the dipole and quadruple distortions of the local atomic
environment which extend the original EAM framework by introducing angular forces.
Note that unlike for other potentials, cutoffs for ADP potentials are not set in the pair_style or pair_coeff
command; they are specified in the ADP potential files themselves. Likewise, the ADP potential files list
atomic masses; thus you do not need to use the mass command to specify them.
The NIST WWW site distributes and documents ADP potentials:
http://www.ctcms.nist.gov/potentials
LAMMPS Users Manual
1380

Note that these must be converted into the extended DYNAMO setfl format discussed below.
The NIST site is maintained by Chandler Becker (cbecker at nist.gov) who is good resource for info on
interatomic potentials and file formats.
Only a single pair_coeff command is used with the adp style which specifies an extended DYNAMO setfl file,
which contains information for M elements. These are mapped to LAMMPS atom types by specifying N
additional arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom
types:
filename• N element names = mapping of extended setfl elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, the potentials/AlCu.adp file, included in the potentials directory of the LAMMPS distribution,
is an extended setfl file which has tabulated ADP values for w elements and their alloy interactions: Cu and
Al. If your LAMMPS simulation has 4 atoms types and you want the 1st 3 to be Al, and the 4th to be Cu, you
would use the following pair_coeff command:
pair_coeff * * AlCu.adp Al Al Al Cu
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Al arguments map
LAMMPS atom types 1,2,3 to the Al element in the extended setfl file. The final Cu argument maps
LAMMPS atom type 4 to the Al element in the extended setfl file. Note that there is no requirement that your
simulation use all the elements specified by the extended setfl file.
If a mapping value is specified as NULL, the mapping is not performed. This can be used when an adp
potential is used as part of the hybrid pair style. The NULL values are placeholders for atom types that will be
used with other potentials.
Adp files in the potentials directory of the LAMMPS distribution have an ".adp" suffix. A DYNAMO setfl file
extended for ADP is formatted as follows. Basically it is the standard setfl format with additional tabulated
functions u and w added to the file after the tabulated pair potentials. See the pair_eam command for further
details on the setfl format.
lines 1,2,3 = comments (ignored)• line 4: Nelements Element1 Element2 ... ElementN• line 5: Nrho, drho, Nr, dr, cutoff•
Following the 5 header lines are Nelements sections, one for each element, each with the following format:
line 1 = atomic number, mass, lattice constant, lattice type (e.g. FCC)• embedding function F(rho) (Nrho values)• density function rho(r) (Nr values)•
Following the Nelements sections, Nr values for each pair potential phi(r) array are listed for all i,j element
pairs in the same format as other arrays. Since these interactions are symmetric (i,j = j,i) only phi arrays with i
>= j are listed, in the following order: i,j = (1,1), (2,1), (2,2), (3,1), (3,2), (3,3), (4,1), ..., (Nelements,
Nelements). The tabulated values for each phi function are listed as r*phi (in units of eV-Angstroms), since
they are for atom pairs, the same as for other EAM files.
LAMMPS Users Manual
1381
After the phi(r) arrays, each of the u(r) arrays are listed in the same order with the same assumptions of
symmetry. Directly following the u(r), the w(r) arrays are listed. Note that phi(r) is the only array tabulated
with a scaling by r.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, where types I and J correspond to two different element types, no special
mixing rules are needed, since the ADP potential files specify alloy interactions explicitly.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in tabulated potential files.
Thus, you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the MANYBODY package. It is only enabled if LAMMPS was built with that
package.
Related commands:
pair_coeff, pair_eam
Default: none
(Mishin) Mishin, Mehl, and Papaconstantopoulos, Acta Mater, 53, 4029 (2005).
(Singh) Singh and Warner, Acta Mater, 58, 5797-5805 (2010),
LAMMPS Users Manual
1382

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style agni command
pair_style agni/omp command
Syntax:
pair_style agni
Examples: pair_style agni pair_coeff * * Al.agni Al
Description:
Style agni style computes the manybody vectorial force components for an atom as
u labels the individual components, i.e. x, y or z, and V is the corresponding atomic fingerprint. d is the
Euclidean distance between any two atomic fingerprints. A total of N_t reference atomic environments are
considered to construct the force field file. alpha_t and l are the weight coefficients and length scale parameter
of the non-linear regression model.
The method implements the recently proposed machine learning access to atomic forces as discussed
extensively in the following publications - (Botu1) and (Botu2). The premise of the method is to map the
atomic environment numerically into a fingerprint, and use machine learning methods to create a mapping to
the vectorial atomic forces.
Only a single pair_coeff command is used with the agni style which specifies an AGNI potential file
containing the parameters of the force field for the needed elements. These are mapped to LAMMPS atom
types by specifying N additional arguments after the filename in the pair_coeff command, where N is the
number of LAMMPS atom types:
filename• N element names = mapping of AGNI elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the force field file.
LAMMPS Users Manual
1383
An AGNI force field is fully specified by the filename which contains the parameters of the force field, i.e.,
the reference training environments used to construct the machine learning force field. Example force field
and input files are provided in the examples/USER/misc/agni directory.
Styles with omp suffix is functionally the same as the corresponding style without the suffix. They have been
optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The
accelerated style takes the same arguments and should produce the same results, except for round-off and
precision issues.
The accelerated style is part of the USER-OMP. They are only enabled if LAMMPS was built with those
packages. See the Making LAMMPS section for more info.
You can specify the accelerated style explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
Currently, only elemental systems are implemented. Also, the method only provides access to the forces and
not energies or stresses. However, one can access the energy via thermodynamic integration of the forces as
discussed in (Botu3). This pair style is part of the USER-MISC package. It is only enabled if LAMMPS was
built with that package. See the Making LAMMPS section for more info.
The AGNI force field files provided with LAMMPS (see the potentials directory) are parameterized for metal
units. You can use the AGNI potential with any LAMMPS units, but you would need to create your own
AGNI potential file with coefficients listed in the appropriate units if your simulation doesn't use "metal"
units.
Related commands:
pair_coeff
Default: none
(Botu1) V. Botu and R. Ramprasad, Int. J. Quant. Chem., 115(16), 1074 (2015).
LAMMPS Users Manual
1384
(Botu2) V. Botu and R. Ramprasad, Phys. Rev. B, 92(9), 094306 (2015).
(Botu3) V. Botu, R. Batra, J. Chapman and R. Ramprasad, https://arxiv.org/abs/1610.02098 (2016).
LAMMPS Users Manual
1385

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style airebo command
pair_style airebo/omp command
pair_style airebo/morse command
pair_style airebo/morse/omp command
pair_style rebo command
pair_style rebo/omp command
Syntax:
pair_style style cutoff LJ_flag TORSION_flag
style = airebo or airebo/morse or rebo• cutoff = LJ or Morse cutoff (sigma scale factor) (AIREBO and AIREBO-M only)• LJ_flag = 0/1 to turn off/on the LJ or Morse term (AIREBO and AIREBO-M only, optional)• TORSION_flag = 0/1 to turn off/on the torsion term (AIREBO and AIREBO-M only, optional)•
Examples:
pair_style airebo 3.0
pair_style airebo 2.5 1 0
pair_coeff * * ../potentials/CH.airebo H C
pair_style airebo/morse 3.0
pair_coeff * * ../potentials/CH.airebo-m H C
pair_style rebo
pair_coeff * * ../potentials/CH.airebo H C
Description:
The airebo pair style computes the Adaptive Intermolecular Reactive Empirical Bond Order (AIREBO)
Potential of (Stuart) for a system of carbon and/or hydrogen atoms. Note that this is the initial formulation of
AIREBO from 2000, not the later formulation.
The airebo/morse pair style computes the AIREBO-M potential, which is equivalent to AIREBO, but replaces
the LJ term with a Morse potential. The Morse potentials are parameterized by high-quality quantum
chemistry (MP2) calculations and do not diverge as quickly as particle density increases. This allows
AIREBO-M to retain accuracy to much higher pressures than AIREBO (up to 40 GPa for Polyethylene).
Details for this potential and its parameterization are given in (O'Conner).
The rebo pair style computes the Reactive Empirical Bond Order (REBO) Potential of (Brenner). Note that
this is the so-called 2nd generation REBO from 2002, not the original REBO from 1990. As discussed below,
2nd generation REBO is closely related to the initial AIREBO; it is just a subset of the potential energy terms.
LAMMPS Users Manual
1386

The AIREBO potential consists of three terms:
By default, all three terms are included. For the airebo style, if the two optional flag arguments to the
pair_style command are included, the LJ and torsional terms can be turned off. Note that both or neither of the
flags must be included. If both of the LJ an torsional terms are turned off, it becomes the 2nd-generation
REBO potential, with a small caveat on the spline fitting procedure mentioned below. This can be specified
directly as pair_style rebo with no additional arguments.
The detailed formulas for this potential are given in (Stuart); here we provide only a brief description.
The E_REBO term has the same functional form as the hydrocarbon REBO potential developed in (Brenner).
The coefficients for E_REBO in AIREBO are essentially the same as Brenner's potential, but a few fitted
spline values are slightly different. For most cases the E_REBO term in AIREBO will produce the same
energies, forces and statistical averages as the original REBO potential from which it was derived. The
E_REBO term in the AIREBO potential gives the model its reactive capabilities and only describes
short-ranged C-C, C-H and H-H interactions (r < 2 Angstroms). These interactions have strong
coordination-dependence through a bond order parameter, which adjusts the attraction between the I,J atoms
based on the position of other nearby atoms and thus has 3- and 4-body dependence.
The E_LJ term adds longer-ranged interactions (2 < r < cutoff) using a form similar to the standard Lennard
Jones potential. The E_LJ term in AIREBO contains a series of switching functions so that the short-ranged
LJ repulsion (1/r^12) does not interfere with the energetics captured by the E_REBO term. The extent of the
E_LJ interactions is determined by the cutoff argument to the pair_style command which is a scale factor. For
each type pair (C-C, C-H, H-H) the cutoff is obtained by multiplying the scale factor by the sigma value
defined in the potential file for that type pair. In the standard AIREBO potential, sigma_CC = 3.4 Angstroms,
so with a scale factor of 3.0 (the argument in pair_style), the resulting E_LJ cutoff would be 10.2 Angstroms.
The E_TORSION term is an explicit 4-body potential that describes various dihedral angle preferences in
hydrocarbon configurations.
Only a single pair_coeff command is used with the airebo, airebo or rebo style which specifies an AIREBO
or AIREBO-M potential file with parameters for C and H. Note that the rebo style in LAMMPS uses the same
AIREBO-formatted potential file. These are mapped to LAMMPS atom types by specifying N additional
arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N element names = mapping of AIREBO elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, if your LAMMPS simulation has 4 atom types and you want the 1st 3 to be C, and the 4th to
be H, you would use the following pair_coeff command:
pair_coeff * * CH.airebo C C C H
LAMMPS Users Manual
1387

The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three C arguments map
LAMMPS atom types 1,2,3 to the C element in the AIREBO file. The final H argument maps LAMMPS atom
type 4 to the H element in the SW file. If a mapping value is specified as NULL, the mapping is not
performed. This can be used when a airebo potential is used as part of the hybrid pair style. The NULL values
are placeholders for atom types that will be used with other potentials.
The parameters/coefficients for the AIREBO potentials are listed in the CH.airebo file to agree with the
original (Stuart) paper. Thus the parameters are specific to this potential and the way it was fit, so modifying
the file should be done cautiously.
Similarly the parameters/coefficients for the AIREBO-M potentials are listed in the CH.airebo-m file to agree
with the (O'Connor) paper. Thus the parameters are specific to this potential and the way it was fit, so
modifying the file should be done cautiously. The AIREBO-M Morse potentials were parameterized using a
cutoff of 3.0 (sigma). Modifying this cutoff may impact simulation accuracy.
This pair style tallies a breakdown of the total AIREBO potential energy into sub-categories, which can be
accessed via the compute pair command as a vector of values of length 3. The 3 values correspond to the
following sub-categories:
E_REBO = REBO energy1. E_LJ = Lennard-Jones energy2. E_TORSION = Torsion energy3.
To print these quantities to the log file (with descriptive column headings) the following commands could be
included in an input script:
compute 0 all pair airebo
variable REBO equal c_0[1]
variable LJ equal c_0[2]
variable TORSION equal c_0[3]
thermo_style custom step temp epair v_REBO v_LJ v_TORSION
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
These pair styles do not support the pair_modify mix, shift, table, and tail options.
LAMMPS Users Manual
1388

These pair styles do not write their information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
These pair styles can only be used via the pair keyword of the run_style respa command. They do not support
the inner, middle, outer keywords.
Restrictions:
These pair styles are part of the MANYBODY package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info.
These pair potentials require the newton setting to be "on" for pair interactions.
The CH.airebo and CH.airebo-m potential files provided with LAMMPS (see the potentials directory) are
parameterized for metal units. You can use the AIREBO, AIREBO-M or REBO potential with any LAMMPS
units, but you would need to create your own AIREBO or AIREBO-M potential file with coefficients listed in
the appropriate units, if your simulation doesn't use "metal" units.
Related commands:
pair_coeff
Default: none
(Stuart) Stuart, Tutein, Harrison, J Chem Phys, 112, 6472-6486 (2000).
(Brenner) Brenner, Shenderova, Harrison, Stuart, Ni, Sinnott, J Physics: Condensed Matter, 14, 783-802
(2002).
(O'Connor) O'Connor et al., J. Chem. Phys. 142, 024903 (2015).
LAMMPS Users Manual
1389

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style awpmd/cut command
Syntax:
pair_style awpmd/cut Rc keyword value ...
Rc = global cutoff, -1 means cutoff of half the shortest box length• zero or more keyword/value pairs may be appended• keyword = hartree or dproduct or uhf or free or pbc or fix or harm or ermscale or flex_press
hartree value = none
dproduct value = none
uhf value = none
free value = none
pbc value = Plen
Plen = periodic width of electron = -1 or positive value (distance units)
fix value = Flen
Flen = fixed width of electron = -1 or positive value (distance units)
harm value = width
width = harmonic width constraint
ermscale value = factor
factor = scaling between electron mass and width variable mass
flex_press value = none
•
Examples:
pair_style awpmd/cut -1
pair_style awpmd/cut 40.0 uhf free
pair_coeff * *
pair_coeff 2 2 20.0
Description:
This pair style contains an implementation of the Antisymmetrized Wave Packet Molecular Dynamics
(AWPMD) method. Need citation here. Need basic formulas here. Could be links to other documents.
Rc is the cutoff.
The pair_style command allows for several optional keywords to be specified.
The hartree, dproduct, and uhf keywords specify the form of the initial trial wave function for the system. If
the hartree keyword is used, then a Hartree multielectron trial wave function is used. If the dproduct keyword
is used, then a trial function which is a product of two determinants for each spin type is used. If the uhf
keyword is used, then an unrestricted Hartree-Fock trial wave function is used.
The free, pbc, and fix keywords specify a width constraint on the electron wavepackets. If the free keyword is
specified, then there is no constraint. If the pbc keyword is used and Plen is specified as -1, then the maximum
width is half the shortest box length. If Plen is a positive value, then the value is the maximum width. If the fix
keyword is used and Flen is specified as -1, then electrons have a constant width that is read from the data
file. If Flen is a positive value, then the constant width for all electrons is set to Flen.
LAMMPS Users Manual
1390

The harm keyword allow oscillations in the width of the electron wavepackets. More details are needed.
The ermscale keyword specifies a unitless scaling factor between the electron masses and the width variable
mass. More details needed.
If the flex_press keyword is used, then a contribution from the electrons is added to the total virial and
pressure of the system.
This potential is designed to be used with atom_style wavepacket definitions, in order to handle the
description of systems with interacting nuclei and explicit electrons.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
cutoff (distance units)•
For awpmd/cut, the cutoff coefficient is optional. If it is not used (as in some of the examples above), the
default global value specified in the pair_style command is used.
Mixing, shift, table, tail correction, restart, rRESPA info:
The pair_modify mix, shift, table, and tail options are not relevant for this pair style.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_coeff
Default:
These are the defaults for the pair_style keywords: hartree for the initial wavefunction, free for the
wavepacket width.
LAMMPS Users Manual
1391

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style beck command
pair_style beck/gpu command
pair_style beck/omp command
Syntax:
pair_style beck Rc
Rc = cutoff for interactions (distance units)•
Examples:
pair_style beck 8.0
pair_coeff * * 399.671876712 0.0000867636112694 0.675 4.390 0.0003746
pair_coeff 1 1 399.671876712 0.0000867636112694 0.675 4.390 0.0003746 6.0
Description:
Style beck computes interactions based on the potential by (Beck), originally designed for simulation of
Helium. It includes truncation at a cutoff distance Rc.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands.
A (energy units)• B (energy-distance^6 units)• a (distance units)• alpha (1/distance units)• beta (1/distance^6 units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global cutoff Rc is used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
LAMMPS Users Manual
1392

section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, coefficients must be specified. No default mixing rules are used.
This pair style does not support the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_coeff
Default: none
(Beck) Beck, Molecular Physics, 14, 311 (1968).
LAMMPS Users Manual
1393

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style body command
Syntax:
pair_style body cutoff
cutoff = global cutoff for interactions (distance units)
Examples:
pair_style body 3.0
pair_coeff * * 1.0 1.0
pair_coeff 1 1 1.0 1.5 2.5
Description:
Style body is for use with body particles and calculates pairwise body/body interactions as well as interactions
between body and point-particles. See Section 6.14 of the manual and the body doc page for more details on
using body particles.
This pair style is designed for use with the "nparticle" body style, which is specified as an argument to the
"atom-style body" command. See the body doc page for more details about the body styles LAMMPS
supports. The "nparticle" style treats a body particle as a rigid body composed of N sub-particles.
The coordinates of a body particle are its center-of-mass (COM). If the COMs of a pair of body particles are
within the cutoff (global or type-specific, as specified above), then all interactions between pairs of
sub-particles in the two body particles are computed. E.g. if the first body particle has 3 sub-particles, and the
second has 10, then 30 interactions are computed and summed to yield the total force and torque on each body
particle.
NOTE: In the example just described, all 30 interactions are computed even if the distance between a
particular pair of sub-particles is greater than the cutoff. Likewise, no interaction between two body particles
is computed if the two COMs are further apart than the cutoff, even if the distance between some pairs of their
sub-particles is within the cutoff. Thus care should be used in defining the cutoff distances for body particles,
depending on their shape and size.
Similar rules apply for a body particle interacting with a point particle. The distance between the two particles
is calculated using the COM of the body particle and the position of the point particle. If the distance is within
the cutoff and the body particle has N sub-particles, then N interactions with the point particle are computed
and summed. If the distance is not within the cutoff, no interactions between the body and point particle are
computed.
The interaction between two sub-particles, or a sub-particle and point particle, or between two point particles
is computed as a Lennard-Jones interaction, using the standard formula
LAMMPS Users Manual
1394

where Rc is the cutoff. As explained above, an interaction involving one or two body sub-particles may be
computed even for r > Rc.
For style body, the following coefficients must be defined for each pair of atoms types via the pair_coeff
command as in the examples above, or in the data file or restart files read by the read_data or read_restart
commands:
epsilon (energy units)• sigma (distance units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global cutoff is used.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of this pair
style can be mixed. The default mix value is geometric. See the "pair_modify" command for details.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This style is part of the BODY package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Defining particles to be bodies so they participate in body/body or body/particle interactions requires the use
of the atom_style body command.
Related commands:
pair_coeff, fix rigid
Default: none
LAMMPS Users Manual
1395

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style bop command
Syntax:
pair_style bop keyword ...
zero or more keywords may be appended• keyword = save
save = pre-compute and save some values
Examples:
pair_style bop
pair_coeff * * ../potentials/CdTe_bop Cd Te
pair_style bop save
pair_coeff * * ../potentials/CdTe.bop.table Cd Te Te
comm_modify cutoff 14.70
Description:
The bop pair style computes Bond-Order Potentials (BOP) based on quantum mechanical theory
incorporating both sigma and pi bondings. By analytically deriving the BOP from quantum mechanical
theory its transferability to different phases can approach that of quantum mechanical methods. This
potential is similar to the original BOP developed by Pettifor (Pettifor_1, Pettifor_2, Pettifor_3) and later
updated by Murdick, Zhou, and Ward (Murdick, Ward). Currently, BOP potential files for these systems
are provided with LAMMPS: AlCu, CCu, CdTe, CdTeSe, CdZnTe, CuH, GaAs. A system with only a
subset of these elements, including a single element (e.g. C or Cu or Al or Ga or Zn or CdZn), can also be
modeled by using the appropriate alloy file and assigning all atom types to the single element or subset of
elements via the pair_coeff command, as discussed below.
The BOP potential consists of three terms:
where phi_ij(r_ij) is a short-range two-body function representing the repulsion between a pair of ion cores,
beta_(sigma,ij)(r_ij) and beta_(sigma,ij)(r_ij) are respectively sigma and pi bond integrals,
THETA_(sigma,ij) and THETA_(pi,ij) are sigma and pi bond-orders, and U_prom is the promotion energy
for sp-valent systems.
The detailed formulas for this potential are given in Ward (Ward); here we provide only a brief description.
The repulsive energy phi_ij(r_ij) and the bond integrals beta_(sigma,ij)(r_ij) and beta_(phi,ij)(r_ij) are
functions of the interatomic distance r_ij between atom i and j. Each of these potentials has a smooth cutoff
at a radius of r_(cut,ij). These smooth cutoffs ensure stable behavior at situations with high sampling near
the cutoff such as melts and surfaces.
•
LAMMPS Users Manual
1396

The bond-orders can be viewed as environment-dependent local variables that are ij bond specific. The
maximum value of the sigma bond-order (THETA_sigma) is 1, while that of the pi bond-order
(THETA_pi) is 2, attributing to a maximum value of the total bond-order (THETA_sigma+THETA_pi) of
3. The sigma and pi bond-orders reflect the ubiquitous single-, double-, and triple- bond behavior of
chemistry. Their analytical expressions can be derived from tight- binding theory by recursively expanding
an inter-site Green's function as a continued fraction. To accurately represent the bonding with a
computationally efficient potential formulation suitable for MD simulations, the derived BOP only takes
(and retains) the first two levels of the recursive representations for both the sigma and the pi bond-orders.
Bond-order terms can be understood in terms of molecular orbital hopping paths based upon the
Cyrot-Lackmann theorem (Pettifor_1). The sigma bond-order with a half-full valence shell is used to
interpolate the bond-order expression that incorporated explicit valance band filling. This pi bond-order
expression also contains also contains a three-member ring term that allows implementation of an
asymmetric density of states, which helps to either stabilize or destabilize close-packed structures. The pi
bond-order includes hopping paths of length 4. This enables the incorporation of dihedral angles effects.
NOTE: Note that unlike for other potentials, cutoffs for BOP potentials are not set in the pair_style or
pair_coeff command; they are specified in the BOP potential files themselves. Likewise, the BOP potential
files list atomic masses; thus you do not need to use the mass command to specify them. Note that for BOP
potentials with hydrogen, you will likely want to set the mass of H atoms to be 10x or 20x larger to avoid
having to use a tiny timestep. You can do this by using the mass command after using the pair_coeff
command to read the BOP potential file.
One option can be specified as a keyword with the pair_style command.
The save keyword gives you the option to calculate in advance and store a set of distances, angles, and
derivatives of angles. The default is to not do this, but to calculate them on-the-fly each time they are
needed. The former may be faster, but takes more memory. The latter requires less memory, but may be
slower. It is best to test this option to optimize the speed of BOP for your particular system configuration.
Only a single pair_coeff command is used with the bop style which specifies a BOP potential file, with
parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional
arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N element names = mapping of BOP elements to atom types•
As an example, imagine the CdTe.bop file has BOP values for Cd and Te. If your LAMMPS simulation has
4 atoms types and you want the 1st 3 to be Cd, and the 4th to be Te, you would use the following pair_coeff
command:
pair_coeff * * CdTe Cd Cd Cd Te
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Cd arguments map
LAMMPS atom types 1,2,3 to the Cd element in the BOP file. The final Te argument maps LAMMPS atom
type 4 to the Te element in the BOP file.
BOP files in the potentials directory of the LAMMPS distribution have a ".bop" suffix. The potentials are
in tabulated form containing pre-tabulated pair functions for phi_ij(r_ij), beta_(sigma,ij)(r_ij), and
beta_pi,ij)(r_ij).
LAMMPS Users Manual
1397

The parameters/coefficients format for the different kinds of BOP files are given below with variables
matching the formulation of Ward (Ward) and Zhou (Zhou). Each header line containing a ":" is preceded
by a blank line.
No angular table file format:
The parameters/coefficients format for the BOP potentials input file containing pre-tabulated functions of g
is given below with variables matching the formulation of Ward (Ward). This format also assumes the
angular functions have the formulation of (Ward).
Line 1: # elements N•
The first line is followed by N lines containing the atomic number, mass, and element symbol of each
element.
Following the definition of the elements several global variables for the tabulated functions are given.
Line 1: nr, nBOt (nr is the number of divisions the radius is broken into for function tables and
MUST be a factor of 5; nBOt is the number of divisions for the tabulated values of THETA_(S,ij)
•
Line 2: delta_1-delta_7 (if all are not used in the particular• formulation, set unused values to 0.0)•
Following this N lines for e_1-e_N containing p_pi.
Line 3: p_pi (for e_1)• Line 4: p_pi (for e_2 and continues to e_N)•
The next section contains several pair constants for the number of interaction types e_i-e_j, with i=1->N,
j=i->N
Line 1: r_cut (for e_1-e_1 interactions)• Line 2: c_sigma, a_sigma, c_pi, a_pi• Line 3: delta_sigma, delta_pi• Line 4: f_sigma, k_sigma, delta_3 (This delta_3 is similar to that of the previous section but is
interaction type dependent)
•
The next section contains a line for each three body interaction type e_j-e_i-e_k with i=0->N, j=0->N,
k=j->N
Line 1: g_(sigma0), g_(sigma1), g_(sigma2) (These are coefficients for g_(sigma,jik)(THETA_ijk)
for e_1-e_1-e_1 interaction. Ward contains the full expressions for the constants as functions of
b_(sigma,ijk), p_(sigma,ijk), u_(sigma,ijk))
•
Line 2: g_(sigma0), g_(sigma1), g_(sigma2) (for e_1-e_1-e_2)•
The next section contains a block for each interaction type for the phi_ij(r_ij). Each block has nr entries
with 5 entries per line.
Line 1: phi(r1), phi(r2), phi(r3), phi(r4), phi(r5) (for the e_1-e_1 interaction type)• Line 2: phi(r6), phi(r7), phi(r8), phi(r9), phi(r10) (this continues until nr)• ...• Line nr/5_1: phi(r1), phi(r2), phi(r3), phi(r4), phi(r5), (for the e_1-e_1 interaction type)•
LAMMPS Users Manual
1398

The next section contains a block for each interaction type for the beta_(sigma,ij)(r_ij). Each block has nr
entries with 5 entries per line.
Line 1: beta_sigma(r1), beta_sigma(r2), beta_sigma(r3), beta_sigma(r4), beta_sigma(r5) (for the
e_1-e_1 interaction type)
•
Line 2: beta_sigma(r6), beta_sigma(r7), beta_sigma(r8), beta_sigma(r9), beta_sigma(r10) (this
continues until nr)
•
...• Line nr/5+1: beta_sigma(r1), beta_sigma(r2), beta_sigma(r3), beta_sigma(r4), beta_sigma(r5) (for
the e_1-e_2 interaction type)
•
The next section contains a block for each interaction type for beta_(pi,ij)(r_ij). Each block has nr entries
with 5 entries per line.
Line 1: beta_pi(r1), beta_pi(r2), beta_pi(r3), beta_pi(r4), beta_pi(r5) (for the e_1-e_1 interaction
type)
•
Line 2: beta_pi(r6), beta_pi(r7), beta_pi(r8), beta_pi(r9), beta_pi(r10) (this continues until nr)• ...• Line nr/5+1: beta_pi(r1), beta_pi(r2), beta_pi(r3), beta_pi(r4), beta_pi(r5) (for the e_1-e_2
interaction type)
•
The next section contains a block for each interaction type for the
THETA_(S,ij)((THETA_(sigma,ij))^(1/2), f_(sigma,ij)). Each block has nBOt entries with 5 entries per
line.
Line 1: THETA_(S,ij)(r1), THETA_(S,ij)(r2), THETA_(S,ij)(r3), THETA_(S,ij)(r4),
THETA_(S,ij)(r5) (for the e_1-e_2 interaction type)
•
Line 2: THETA_(S,ij)(r6), THETA_(S,ij)(r7), THETA_(S,ij)(r8), THETA_(S,ij)(r9),
THETA_(S,ij)(r10) (this continues until nBOt)
•
...• Line nBOt/5+1: THETA_(S,ij)(r1), THETA_(S,ij)(r2), THETA_(S,ij)(r3), THETA_(S,ij)(r4),
THETA_(S,ij)(r5) (for the e_1-e_2 interaction type)
•
The next section contains a block of N lines for e_1-e_N
Line 1: delta^mu (for e_1)• Line 2: delta^mu (for e_2 and repeats to e_N)•
The last section contains more constants for e_i-e_j interactions with i=0->N, j=i->N
Line 1: (A_ij)^(mu*nu) (for e1-e1)• Line 2: (A_ij)^(mu*nu) (for e1-e2 and repeats as above)•
Angular spline table file format:
The parameters/coefficients format for the BOP potentials input file containing pre-tabulated functions of g
is given below with variables matching the formulation of Ward (Ward). This format also assumes the
angular functions have the formulation of (Zhou).
Line 1: # elements N•
LAMMPS Users Manual
1399

The first line is followed by N lines containing the atomic number, mass, and element symbol of each
element.
Following the definition of the elements several global variables for the tabulated functions are given.
Line 1: nr, ntheta, nBOt (nr is the number of divisions the radius is broken into for function tables
and MUST be a factor of 5; ntheta is the power of the power of the spline used to fit the angular
function; nBOt is the number of divisions for the tabulated values of THETA_(S,ij)
•
Line 2: delta_1-delta_7 (if all are not used in the particular• formulation, set unused values to 0.0)•
Following this N lines for e_1-e_N containing p_pi.
Line 3: p_pi (for e_1)• Line 4: p_pi (for e_2 and continues to e_N)•
The next section contains several pair constants for the number of interaction types e_i-e_j, with i=1->N,
j=i->N
Line 1: r_cut (for e_1-e_1 interactions)• Line 2: c_sigma, a_sigma, c_pi, a_pi• Line 3: delta_sigma, delta_pi• Line 4: f_sigma, k_sigma, delta_3 (This delta_3 is similar to that of the previous section but is
interaction type dependent)
•
The next section contains a line for each three body interaction type e_j-e_i-e_k with i=0->N, j=0->N,
k=j->N
Line 1: g0, g1, g2... (These are coefficients for the angular spline of the g_(sigma,jik)(THETA_ijk) for
e_1-e_1-e_1 interaction. The function can contain up to 10 term thus 10 constants. The first line can contain
up to five constants. If the spline has more than five terms the second line will contain the remaining
constants The following lines will then contain the constants for the remaining g0, g1, g2... (for
e_1-e_1-e_2) and the other three body interactions
The rest of the table has the same structure as the previous section (see above).
Angular no-spline table file format:
The parameters/coefficients format for the BOP potentials input file containing pre-tabulated functions of g
is given below with variables matching the formulation of Ward (Ward). This format also assumes the
angular functions have the formulation of (Zhou).
Line 1: # elements N•
The first two lines are followed by N lines containing the atomic number, mass, and element symbol of
each element.
Following the definition of the elements several global variables for the tabulated functions are given.
Line 1: nr, ntheta, nBOt (nr is the number of divisions the radius is broken into for function tables
and MUST be a factor of 5; ntheta is the number of divisions for the tabulated values of the g
angular function; nBOt is the number of divisions for the tabulated values of THETA_(S,ij)
•
•
LAMMPS Users Manual
1400

Line 2: delta_1-delta_7 (if all are not used in the particular• formulation, set unused values to 0.0)•
Following this N lines for e_1-e_N containing p_pi.
Line 3: p_pi (for e_1)• Line 4: p_pi (for e_2 and continues to e_N)•
The next section contains several pair constants for the number of interaction types e_i-e_j, with i=1->N,
j=i->N
Line 1: r_cut (for e_1-e_1 interactions)• Line 2: c_sigma, a_sigma, c_pi, a_pi• Line 3: delta_sigma, delta_pi• Line 4: f_sigma, k_sigma, delta_3 (This delta_3 is similar to that of the previous section but is
interaction type dependent)
•
The next section contains a line for each three body interaction type e_j-e_i-e_k with i=0->N, j=0->N,
k=j->N
Line 1: g(theta1), g(theta2), g(theta3), g(theta4), g(theta5) (for the e_1-e_1-e_1 interaction type)• Line 2: g(theta6), g(theta7), g(theta8), g(theta9), g(theta10) (this continues until ntheta)• ...• Line ntheta/5+1: g(theta1), g(theta2), g(theta3), g(theta4), g(theta5), (for the e_1-e_1-e_2
interaction type)
•
The rest of the table has the same structure as the previous section (see above).
Mixing, shift, table tail correction, restart:
This pair style does not support the pair_modify mix, shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support
the inner, middle, outer keywords.
Restrictions:
These pair styles are part of the MANYBODY package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info.
These pair potentials require the newtion setting to be "on" for pair interactions.
The CdTe.bop and GaAs.bop potential files provided with LAMMPS (see the potentials directory) are
parameterized for metal units. You can use the BOP potential with any LAMMPS units, but you would
need to create your own BOP potential file with coefficients listed in the appropriate units if your
simulation does not use "metal" units.
LAMMPS Users Manual
1401

Related commands:
pair_coeff
Default:
non-tabulated potential file, a_0 is non-zero.
(Pettifor_1) D.G. Pettifor and I.I. Oleinik, Phys. Rev. B, 59, 8487 (1999).
(Pettifor_2) D.G. Pettifor and I.I. Oleinik, Phys. Rev. Lett., 84, 4124 (2000).
(Pettifor_3) D.G. Pettifor and I.I. Oleinik, Phys. Rev. B, 65, 172103 (2002).
(Murdick) D.A. Murdick, X.W. Zhou, H.N.G. Wadley, D. Nguyen-Manh, R. Drautz, and D.G. Pettifor,
Phys. Rev. B, 73, 45206 (2006).
(Ward) D.K. Ward, X.W. Zhou, B.M. Wong, F.P. Doty, and J.A. Zimmerman, Phys. Rev. B, 85,115206
(2012).
(Zhou) X.W. Zhou, D.K. Ward, M. Foster (TBP).
LAMMPS Users Manual
1402

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style born command
pair_style born/omp command
pair_style born/gpu command
pair_style born/coul/long command
pair_style born/coul/long/cs command
pair_style born/coul/long/gpu command
pair_style born/coul/long/omp command
pair_style born/coul/msm command
pair_style born/coul/msm/omp command
pair_style born/coul/wolf command
pair_style born/coul/wolf/gpu command
pair_style born/coul/wolf/omp command
pair_style born/coul/dsf command
pair_style born/coul/dsf/cs command
Syntax:
pair_style style args
style = born or born/coul/long or born/coul/long/cs or born/coul/msm or born/coul/wolf• args = list of arguments for a particular style•
born args = cutoff
cutoff = global cutoff for non-Coulombic interactions (distance units)
born/coul/long or born/coul/long/cs args = cutoff (cutoff2)
cutoff = global cutoff for non-Coulombic (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
born/coul/msm args = cutoff (cutoff2)
cutoff = global cutoff for non-Coulombic (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
born/coul/wolf args = alpha cutoff (cutoff2)
alpha = damping parameter (inverse distance units)
cutoff = global cutoff for non-Coulombic (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
born/coul/dsf or born/coul/dsf/cs args = alpha cutoff (cutoff2)
LAMMPS Users Manual
1403
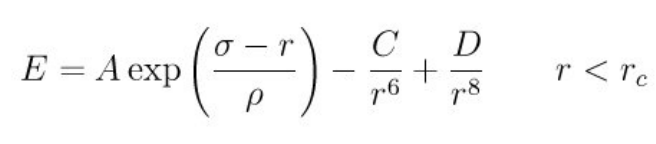
alpha = damping parameter (inverse distance units)
cutoff = global cutoff for non-Coulombic (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (distance units)
Examples:
pair_style born 10.0
pair_coeff * * 6.08 0.317 2.340 24.18 11.51
pair_coeff 1 1 6.08 0.317 2.340 24.18 11.51
pair_style born/coul/long 10.0
pair_style born/coul/long/cs 10.0
pair_style born/coul/long 10.0 8.0
pair_style born/coul/long/cs 10.0 8.0
pair_coeff * * 6.08 0.317 2.340 24.18 11.51
pair_coeff 1 1 6.08 0.317 2.340 24.18 11.51
pair_style born/coul/msm 10.0
pair_style born/coul/msm 10.0 8.0
pair_coeff * * 6.08 0.317 2.340 24.18 11.51
pair_coeff 1 1 6.08 0.317 2.340 24.18 11.51
pair_style born/coul/wolf 0.25 10.0
pair_style born/coul/wolf 0.25 10.0 9.0
pair_coeff * * 6.08 0.317 2.340 24.18 11.51
pair_coeff 1 1 6.08 0.317 2.340 24.18 11.51
pair_style born/coul/dsf 0.1 10.0 12.0
pair_coeff * * 0.0 1.00 0.00 0.00 0.00
pair_coeff 1 1 480.0 0.25 0.00 1.05 0.50
Description:
The born style computes the Born-Mayer-Huggins or Tosi/Fumi potential described in (Fumi and Tosi), given
by
where sigma is an interaction-dependent length parameter, rho is an ionic-pair dependent length parameter,
and Rc is the cutoff.
The styles with coul/long or coul/msm add a Coulombic term as described for the lj/cut pair styles. An
additional damping factor is applied to the Coulombic term so it can be used in conjunction with the
kspace_style command and its ewald or pppm of msm option. The Coulombic cutoff specified for this style
means that pairwise interactions within this distance are computed directly; interactions outside that distance
are computed in reciprocal space.
If one cutoff is specified for the born/coul/long and born/coul/msm style, it is used for both the A,C,D and
Coulombic terms. If two cutoffs are specified, the first is used as the cutoff for the A,C,D terms, and the
second is the cutoff for the Coulombic term.
LAMMPS Users Manual
1404

The born/coul/wolf style adds a Coulombic term as described for the Wolf potential in the coul/wolf pair
style.
The born/coul/dsf style computes the Coulomb contribution with the damped shifted force model as in the
coul/dsf style.
Style born/coul/long/cs is identical to born/coul/long except that a term is added for the core/shell model to
allow charges on core and shell particles to be separated by r = 0.0. The same correction is introduced for
born/coul/dsf/cs style which is identical to born/coul/dsf.
Note that these potentials are related to the Buckingham potential.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
A (energy units)• rho (distance units)• sigma (distance units)• C (energy units * distance units^6)• D (energy units * distance units^8)• cutoff (distance units)•
The second coefficient, rho, must be greater than zero.
The last coefficient is optional. If not specified, the global A,C,D cutoff specified in the pair_style command
is used.
For born/coul/long, born/coul/wolf and born/coul/dsf no Coulombic cutoff can be specified for an individual
I,J type pair. All type pairs use the same global Coulombic cutoff specified in the pair_style command.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
These pair styles do not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
LAMMPS Users Manual
1405

These styles support the pair_modify shift option for the energy of the exp(), 1/r^6, and 1/r^8 portion of the
pair interaction.
The born/coul/long pair style supports the pair_modify table option ti tabulate the short-range portion of the
long-range Coulombic interaction.
These styles support the pair_modify tail option for adding long-range tail corrections to energy and pressure.
Thess styles writes thei information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
These styles can only be used via the pair keyword of the run_style respa command. They do not support the
inner, middle, outer keywords.
Restrictions:
The born/coul/long style is part of the KSPACE package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Related commands:
pair_coeff, pair_style buck
Default: none
Fumi and Tosi, J Phys Chem Solids, 25, 31 (1964), Fumi and Tosi, J Phys Chem Solids, 25, 45 (1964).
LAMMPS Users Manual
1406

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style brownian command
pair_style brownian/omp command
pair_style brownian/poly command
pair_style brownian/poly/omp command
Syntax:
pair_style style mu flaglog flagfld cutinner cutoff t_target seed flagHI flagVF
style = brownian or brownian/poly• mu = dynamic viscosity (dynamic viscosity units)• flaglog = 0/1 log terms in the lubrication approximation on/off• flagfld = 0/1 to include/exclude Fast Lubrication Dynamics effects• cutinner = inner cutoff distance (distance units)• cutoff = outer cutoff for interactions (distance units)• t_target = target temp of the system (temperature units)• seed = seed for the random number generator (positive integer)• flagHI (optional) = 0/1 to include/exclude 1/r hydrodynamic interactions• flagVF (optional) = 0/1 to include/exclude volume fraction corrections in the long-range isotropic
terms
•
Examples:
pair_style brownian 1.5 1 1 2.01 2.5 2.0 5878567 (assuming radius = 1)
pair_coeff 1 1 2.05 2.8
pair_coeff * *
Description:
Styles brownian and brownian/poly compute Brownian forces and torques on finite-size spherical particles.
The former requires monodisperse spherical particles; the latter allows for polydisperse spherical particles.
These pair styles are designed to be used with either the pair_style lubricate or pair_style lubricateU
commands to provide thermostatting when dissipative lubrication forces are acting. Thus the parameters mu,
flaglog, flagfld, cutinner, and cutoff should be specified consistent with the settings in the lubrication pair
styles. For details, refer to either of the lubrication pair styles.
The t_target setting is used to specify the target temperature of the system. The random number seed is used
to generate random numbers for the thermostatting procedure.
The flagHI and flagVF settings are optional. Neither should be used, or both must be defined.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
LAMMPS Users Manual
1407

cutinner (distance units)• cutoff (distance units)•
The two coefficients are optional. If neither is specified, the two cutoffs specified in the pair_style command
are used. Otherwise both must be specified.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in this
section of the manual. The accelerated styles take the same arguments and should produce the same results,
except for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See this section of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the two cutoff distances for this pair style can be mixed. The default mix
value is geometric. See the "pair_modify" command for details.
This pair style does not support the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
These styles are part of the COLLOID package. They are only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Only spherical monodisperse particles are allowed for pair_style brownian.
Only spherical particles are allowed for pair_style brownian/poly.
Related commands:
LAMMPS Users Manual
1408

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style buck command
pair_style buck/gpu command
pair_style buck/intel command
pair_style buck/kk command
pair_style buck/omp command
pair_style buck/coul/cut command
pair_style buck/coul/cut/gpu command
pair_style buck/coul/cut/intel command
pair_style buck/coul/cut/kk command
pair_style buck/coul/cut/omp command
pair_style buck/coul/long command
pair_style buck/coul/long/cs command
pair_style buck/coul/long/gpu command
pair_style buck/coul/long/intel command
pair_style buck/coul/long/kk command
pair_style buck/coul/long/omp command
pair_style buck/coul/msm command
pair_style buck/coul/msm/omp command
Syntax:
pair_style style args
style = buck or buck/coul/cut or buck/coul/long or buck/coul/long/cs or buck/coul/msm• args = list of arguments for a particular style•
buck args = cutoff
cutoff = global cutoff for Buckingham interactions (distance units)
buck/coul/cut args = cutoff (cutoff2)
LAMMPS Users Manual
1410
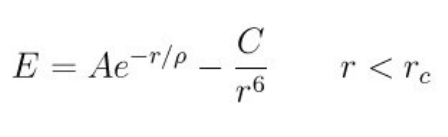
cutoff = global cutoff for Buckingham (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
buck/coul/long or buck/coul/long/cs args = cutoff (cutoff2)
cutoff = global cutoff for Buckingham (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
buck/coul/msm args = cutoff (cutoff2)
cutoff = global cutoff for Buckingham (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
Examples:
pair_style buck 2.5
pair_coeff * * 100.0 1.5 200.0
pair_coeff * * 100.0 1.5 200.0 3.0
pair_style buck/coul/cut 10.0
pair_style buck/coul/cut 10.0 8.0
pair_coeff * * 100.0 1.5 200.0
pair_coeff 1 1 100.0 1.5 200.0 9.0
pair_coeff 1 1 100.0 1.5 200.0 9.0 8.0
pair_style buck/coul/long 10.0
pair_style buck/coul/long/cs 10.0
pair_style buck/coul/long 10.0 8.0
pair_style buck/coul/long/cs 10.0 8.0
pair_coeff * * 100.0 1.5 200.0
pair_coeff 1 1 100.0 1.5 200.0 9.0
pair_style buck/coul/msm 10.0
pair_style buck/coul/msm 10.0 8.0
pair_coeff * * 100.0 1.5 200.0
pair_coeff 1 1 100.0 1.5 200.0 9.0
Description:
The buck style computes a Buckingham potential (exp/6 instead of Lennard-Jones 12/6) given by
where rho is an ionic-pair dependent length parameter, and Rc is the cutoff on both terms.
The styles with coul/cut or coul/long or coul/msm add a Coulombic term as described for the lj/cut pair styles.
For buck/coul/long and buc/coul/msm, an additional damping factor is applied to the Coulombic term so it can
be used in conjunction with the kspace_style command and its ewald or pppm or msm option. The Coulombic
cutoff specified for this style means that pairwise interactions within this distance are computed directly;
interactions outside that distance are computed in reciprocal space.
If one cutoff is specified for the born/coul/cut and born/coul/long and born/coul/msm styles, it is used for both
the A,C and Coulombic terms. If two cutoffs are specified, the first is used as the cutoff for the A,C terms, and
the second is the cutoff for the Coulombic term.
LAMMPS Users Manual
1411

Style buck/coul/long/cs is identical to buck/coul/long except that a term is added for the core/shell model to
allow charges on core and shell particles to be separated by r = 0.0.
Note that these potentials are related to the Born-Mayer-Huggins potential.
NOTE: For all these pair styles, the terms with A and C are always cutoff. The additional Coulombic term can
be cutoff or long-range (no cutoff) depending on whether the style name includes coul/cut or coul/long or
coul/msm. If you wish the C/r^6 term to be long-range (no cutoff), then see the pair_style buck/long/coul/long
command.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands:
A (energy units)• rho (distance units)• C (energy-distance^6 units)• cutoff (distance units)• cutoff2 (distance units)•
The second coefficient, rho, must be greater than zero.
The latter 2 coefficients are optional. If not specified, the global A,C and Coulombic cutoffs are used. If only
one cutoff is specified, it is used as the cutoff for both A,C and Coulombic interactions for this type pair. If
both coefficients are specified, they are used as the A,C and Coulombic cutoffs for this type pair. You cannot
specify 2 cutoffs for style buck, since it has no Coulombic terms.
For buck/coul/long only the LJ cutoff can be specified since a Coulombic cutoff cannot be specified for an
individual I,J type pair. All type pairs use the same global Coulombic cutoff specified in the pair_style
command.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
These pair styles do not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
These styles support the pair_modify shift option for the energy of the exp() and 1/r^6 portion of the pair
LAMMPS Users Manual
1412
interaction.
The buck/coul/long pair style supports the pair_modify table option to tabulate the short-range portion of the
long-range Coulombic interaction.
These styles support the pair_modify tail option for adding long-range tail corrections to energy and pressure
for the A,C terms in the pair interaction.
These styles write their information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
These styles can only be used via the pair keyword of the run_style respa command. They do not support the
inner, middle, outer keywords.
Restrictions:
The buck/coul/long style is part of the KSPACE package. The buck/coul/long/cs style is part of the
CORESHELL package. They are only enabled if LAMMPS was built with that package. See the Making
LAMMPS section for more info.
Related commands:
pair_coeff, pair_style born
Default: none
LAMMPS Users Manual
1413

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style buck/long/coul/long command
pair_style buck/long/coul/long/omp command
Syntax:
pair_style buck/long/coul/long flag_buck flag_coul cutoff (cutoff2)
flag_buck = long or cut
long = use Kspace long-range summation for the dispersion term 1/r^6
cut = use a cutoff
•
flag_coul = long or off
long = use Kspace long-range summation for the Coulombic term 1/r
off = omit the Coulombic term
•
cutoff = global cutoff for Buckingham (and Coulombic if only 1 cutoff) (distance units)• cutoff2 = global cutoff for Coulombic (optional) (distance units)•
Examples:
pair_style buck/long/coul/long cut off 2.5
pair_style buck/long/coul/long cut long 2.5 4.0
pair_style buck/long/coul/long long long 4.0
pair_coeff * * 1 1
pair_coeff 1 1 1 3 4
Description:
The buck/long/coul/long style computes a Buckingham potential (exp/6 instead of Lennard-Jones 12/6) and
Coulombic potential, given by
Rc is the cutoff. If one cutoff is specified in the pair_style command, it is used for both the Buckingham and
Coulombic terms. If two cutoffs are specified, they are used as cutoffs for the Buckingham and Coulombic
terms respectively.
The purpose of this pair style is to capture long-range interactions resulting from both attractive 1/r^6
Buckingham and Coulombic 1/r interactions. This is done by use of the flag_buck and flag_coul settings. The
LAMMPS Users Manual
1414

Ismail paper has more details on when it is appropriate to include long-range 1/r^6 interactions, using this
potential.
If flag_buck is set to long, no cutoff is used on the Buckingham 1/r^6 dispersion term. The long-range portion
can be calculated by using the kspace_style ewald/disp or pppm/disp commands. The specified Buckingham
cutoff then determines which portion of the Buckingham interactions are computed directly by the pair
potential versus which part is computed in reciprocal space via the Kspace style. If flag_buck is set to cut, the
Buckingham interactions are simply cutoff, as with pair_style buck.
If flag_coul is set to long, no cutoff is used on the Coulombic interactions. The long-range portion can
calculated by using any of several kspace_style command options such as pppm or ewald. Note that if
flag_buck is also set to long, then the ewald/disp or pppm/disp Kspace style needs to be used to perform the
long-range calculations for both the Buckingham and Coulombic interactions. If flag_coul is set to off,
Coulombic interactions are not computed.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands:
A (energy units)• rho (distance units)• C (energy-distance^6 units)• cutoff (distance units)• cutoff2 (distance units)•
The second coefficient, rho, must be greater than zero.
The latter 2 coefficients are optional. If not specified, the global Buckingham and Coulombic cutoffs specified
in the pair_style command are used. If only one cutoff is specified, it is used as the cutoff for both
Buckingham and Coulombic interactions for this type pair. If both coefficients are specified, they are used as
the Buckingham and Coulombic cutoffs for this type pair. Note that if you are using flag_buck set to long, you
cannot specify a Buckingham cutoff for an atom type pair, since only one global Buckingham cutoff is
allowed. Similarly, if you are using flag_coul set to long, you cannot specify a Coulombic cutoff for an atom
type pair, since only one global Coulombic cutoff is allowed.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
LAMMPS Users Manual
1415

This pair styles does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
This pair style supports the pair_modify shift option for the energy of the exp() and 1/r^6 portion of the pair
interaction, assuming flag_buck is cut.
This pair style does not support the pair_modify shift option for the energy of the Buckingham portion of the
pair interaction.
This pair style supports the pair_modify table and table/disp options since they can tabulate the short-range
portion of the long-range Coulombic and dispersion interactions.
This pair style write its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style supports the use of the inner, middle, and outer keywords of the run_style respa command,
meaning the pairwise forces can be partitioned by distance at different levels of the rRESPA hierarchy. See
the run_style command for details.
Restrictions:
This style is part of the KSPACE package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info. Note that the KSPACE package is installed by default.
Related commands:
pair_coeff
Default: none
(Ismail) Ismail, Tsige, In 't Veld, Grest, Molecular Physics (accepted) (2007).
LAMMPS Users Manual
1416

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/charmm/coul/charmm command
pair_style lj/charmm/coul/charmm/omp command
pair_style lj/charmm/coul/charmm/implicit command
pair_style lj/charmm/coul/charmm/implicit/omp command
pair_style lj/charmm/coul/long command
pair_style lj/charmm/coul/long/gpu command
pair_style lj/charmm/coul/long/intel command
pair_style lj/charmm/coul/long/opt command
pair_style lj/charmm/coul/long/omp command
pair_style lj/charmm/coul/msm command
pair_style lj/charmm/coul/msm/omp command
pair_style lj/charmmfsw/coul/charmmfsh command
pair_style lj/charmmfsw/coul/long command
Syntax:
pair_style style args
style = lj/charmm/coul/charmm or lj/charmm/coul/charmm/implicit or lj/charmm/coul/long or
lj/charmm/coul/msm or lj/charmmfsw/coul/charmmfsh or lj/charmmfsw/coul/long
•
args = list of arguments for a particular style•
lj/charmm/coul/charmm args = inner outer (inner2) (outer2)
inner, outer = global switching cutoffs for Lennard Jones (and Coulombic if only 2 args)
inner2, outer2 = global switching cutoffs for Coulombic (optional)
lj/charmm/coul/charmm/implicit args = inner outer (inner2) (outer2)
inner, outer = global switching cutoffs for LJ (and Coulombic if only 2 args)
inner2, outer2 = global switching cutoffs for Coulombic (optional)
lj/charmm/coul/long args = inner outer (cutoff)
inner, outer = global switching cutoffs for LJ (and Coulombic if only 2 args)
cutoff = global cutoff for Coulombic (optional, outer is Coulombic cutoff if only 2 args)
lj/charmm/coul/msm args = inner outer (cutoff)
inner, outer = global switching cutoffs for LJ (and Coulombic if only 2 args)
cutoff = global cutoff for Coulombic (optional, outer is Coulombic cutoff if only 2 args)
lj/charmmfsw/coul/charmmfsh args = inner outer (cutoff)
inner, outer = global cutoffs for LJ (and Coulombic if only 2 args)
cutoff = global cutoff for Coulombic (optional, outer is Coulombic cutoff if only 2 args)
LAMMPS Users Manual
1417
lj/charmmfsw/coul/long args = inner outer (cutoff)
inner, outer = global cutoffs for LJ (and Coulombic if only 2 args)
cutoff = global cutoff for Coulombic (optional, outer is Coulombic cutoff if only 2 args)
Examples:
pair_style lj/charmm/coul/charmm 8.0 10.0
pair_style lj/charmm/coul/charmm 8.0 10.0 7.0 9.0
pair_style lj/charmmfsw/coul/charmmfsh 10.0 12.0
pair_style lj/charmmfsw/coul/charmmfsh 10.0 12.0 9.0
pair_coeff * * 100.0 2.0
pair_coeff 1 1 100.0 2.0 150.0 3.5
pair_style lj/charmm/coul/charmm/implicit 8.0 10.0
pair_style lj/charmm/coul/charmm/implicit 8.0 10.0 7.0 9.0
pair_coeff * * 100.0 2.0
pair_coeff 1 1 100.0 2.0 150.0 3.5
pair_style lj/charmm/coul/long 8.0 10.0
pair_style lj/charmm/coul/long 8.0 10.0 9.0
pair_style lj/charmmfsw/coul/long 8.0 10.0
pair_style lj/charmmfsw/coul/long 8.0 10.0 9.0
pair_coeff * * 100.0 2.0
pair_coeff 1 1 100.0 2.0 150.0 3.5
pair_style lj/charmm/coul/msm 8.0 10.0
pair_style lj/charmm/coul/msm 8.0 10.0 9.0
pair_coeff * * 100.0 2.0
pair_coeff 1 1 100.0 2.0 150.0 3.5
Description:
These pair styles compute Lennard Jones (LJ) and Coulombic interactions with additional switching or
shifting functions that ramp the energy and/or force smoothly to zero between an inner and outer cutoff. They
are implementations of the widely used CHARMM force field used in the CHARMM MD code (and others).
See (MacKerell) for a description of the CHARMM force field.
The styles with charmm (not charmmfsw or charmmfsh) in their name are the older, original LAMMPS
implementations. They compute the LJ and Coulombic interactions with an energy switching function (esw,
shown in the formula below as S(r)), which ramps the energy smoothly to zero between the inner and outer
cutoff. This can cause irregularities in pair-wise forces (due to the discontinuous 2nd derivative of energy at
the boundaries of the switching region), which in some cases can result in detectable artifacts in an MD
simulation.
The newer styles with charmmfsw or charmmfsh in their name replace the energy switching with force
switching (fsw) and force shifting (fsh) functions, for LJ and Coulombic interactions respectively. These
follow the formulas and description given in (Steinbach) and (Brooks) to minimize these artifacts.
NOTE: The newer charmmfsw or charmmfsh styles were released in March 2017. We recommend they be
used instead of the older charmm styles. Eventually code from the new styles will propagate into the related
pair styles (e.g. implicit, accelerator, free energy variants).
The general CHARMM formulas are as follows
LAMMPS Users Manual
1418
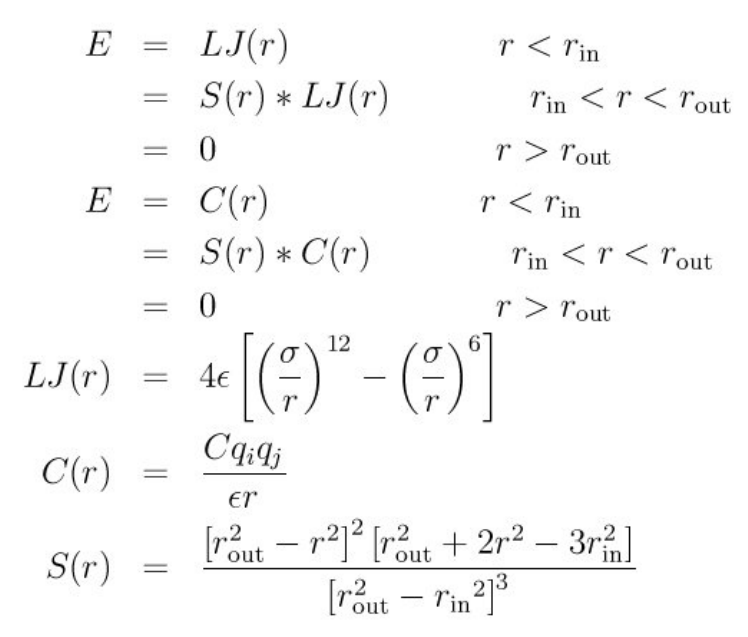
where S(r) is the energy switching function mentioned above for the charmm styles. See the (Steinbach) paper
for the functional forms of the force switching and force shifting functions used in the charmmfsw and
charmmfsh styles.
When using the lj/charmm/coul/charmm styles, both the LJ and Coulombic terms require an inner and outer
cutoff. They can be the same for both formulas or different depending on whether 2 or 4 arguments are used
in the pair_style command. For the lj/charmmfsw/coul/charmmfsh style, the LJ term requires both an inner
and outer cutoff, while the Coulombic term requires only one cutoff. If the Coulomb cutoff is not specified (2
instead of 3 arguments), the LJ outer cutoff is used for the Coulombic cutoff. In all cases where an inner and
outer cutoff are specified, the inner cutoff distance must be less than the outer cutoff. It is typical to make the
difference between the inner and outer cutoffs about 2.0 Angstroms.
Style lj/charmm/coul/charmm/implicit computes the same formulas as style lj/charmm/coul/charmm except
that an additional 1/r term is included in the Coulombic formula. The Coulombic energy thus varies as 1/r^2.
This is effectively a distance-dependent dielectric term which is a simple model for an implicit solvent with
additional screening. It is designed for use in a simulation of an unsolvated biomolecule (no explicit water
molecules).
Styles lj/charmm/coul/long and lj/charmm/coul/msm compute the same formulas as style
lj/charmm/coul/charmm and style lj/charmmfsw/coul/long computes the same formulas as style
lj/charmmfsw/coul/charmmfsh, except that an additional damping factor is applied to the Coulombic term, so
it can be used in conjunction with the kspace_style command and its ewald or pppm or msm option. Only one
Coulombic cutoff is specified for these styles; if only 2 arguments are used in the pair_style command, then
the outer LJ cutoff is used as the single Coulombic cutoff. The Coulombic cutoff specified for these styles
means that pairwise interactions within this distance are computed directly; interactions outside that distance
are computed in reciprocal space.
LAMMPS Users Manual
1419

The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• epsilon_14 (energy units)• sigma_14 (distance units)•
Note that sigma is defined in the LJ formula as the zero-crossing distance for the potential, not as the energy
minimum at 2^(1/6) sigma.
The latter 2 coefficients are optional. If they are specified, they are used in the LJ formula between 2 atoms of
these types which are also first and fourth atoms in any dihedral. No cutoffs are specified because the
CHARMM force field does not allow varying cutoffs for individual atom pairs; all pairs use the global
cutoff(s) specified in the pair_style command.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon, sigma, epsilon_14, and sigma_14 coefficients for all of the
lj/charmm pair styles can be mixed. The default mix value is arithmetic to coincide with the usual settings for
the CHARMM force field. See the "pair_modify" command for details.
None of the lj/charmm or lj/charmmfsw pair styles support the pair_modify shift option, since the
Lennard-Jones portion of the pair interaction is smoothed to 0.0 at the cutoff.
The lj/charmm/coul/long and lj/charmmfsw/coul/long styles support the pair_modify table option since they
can tabulate the short-range portion of the long-range Coulombic interaction.
None of the lj/charmm or lj/charmmfsw pair styles support the pair_modify tail option for adding long-range
tail corrections to energy and pressure, since the Lennard-Jones portion of the pair interaction is smoothed to
0.0 at the cutoff.
All of the lj/charmm and lj/charmmfsw pair styles write their information to binary restart files, so pair_style
and pair_coeff commands do not need to be specified in an input script that reads a restart file.
LAMMPS Users Manual
1420

The lj/charmm/coul/long and lj/charmmfsw/coul/long pair styles support the use of the inner, middle, and
outer keywords of the run_style respa command, meaning the pairwise forces can be partitioned by distance at
different levels of the rRESPA hierarchy. The other styles only support the pair keyword of run_style respa.
See the run_style command for details.
Restrictions:
All the styles with coul/charmm or coul/charmmfsh styles are part of the MOLECULE package. All the styles
with coul/long style are part of the KSPACE package. They are only enabled if LAMMPS was built with
those packages. See the Making LAMMPS section for more info. Note that the MOLECULE and KSPACE
packages are installed by default.
Related commands:
pair_coeff
Default: none
(Brooks) Brooks, et al, J Comput Chem, 30, 1545 (2009).
(MacKerell) MacKerell, Bashford, Bellott, Dunbrack, Evanseck, Field, Fischer, Gao, Guo, Ha, et al, J Phys
Chem, 102, 3586 (1998).
(Steinbach) Steinbach, Brooks, J Comput Chem, 15, 667 (1994).
LAMMPS Users Manual
1421

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/class2 command
pair_style lj/class2/gpu command
pair_style lj/class2/kk command
pair_style lj/class2/omp command
pair_style lj/class2/coul/cut command
pair_style lj/class2/coul/cut/kk command
pair_style lj/class2/coul/cut/omp command
pair_style lj/class2/coul/long command
pair_style lj/class2/coul/long/gpu command
pair_style lj/class2/coul/long/kk command
pair_style lj/class2/coul/long/omp command
Syntax:
pair_style style args
style = lj/class2 or lj/class2/coul/cut or lj/class2/coul/long• args = list of arguments for a particular style•
lj/class2 args = cutoff
cutoff = global cutoff for class 2 interactions (distance units)
lj/class2/coul/cut args = cutoff (cutoff2)
cutoff = global cutoff for class 2 (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
lj/class2/coul/long args = cutoff (cutoff2)
cutoff = global cutoff for class 2 (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
Examples:
pair_style lj/class2 10.0
pair_coeff * * 100.0 2.5
pair_coeff 1 2* 100.0 2.5 9.0
pair_style lj/class2/coul/cut 10.0
pair_style lj/class2/coul/cut 10.0 8.0
pair_coeff * * 100.0 3.0
pair_coeff 1 1 100.0 3.5 9.0
pair_coeff 1 1 100.0 3.5 9.0 9.0
LAMMPS Users Manual
1422

pair_style lj/class2/coul/long 10.0
pair_style lj/class2/coul/long 10.0 8.0
pair_coeff * * 100.0 3.0
pair_coeff 1 1 100.0 3.5 9.0
Description:
The lj/class2 styles compute a 6/9 Lennard-Jones potential given by
Rc is the cutoff.
The lj/class2/coul/cut and lj/class2/coul/long styles add a Coulombic term as described for the lj/cut pair
styles.
See (Sun) for a description of the COMPASS class2 force field.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• cutoff1 (distance units)• cutoff2 (distance units)•
The latter 2 coefficients are optional. If not specified, the global class 2 and Coulombic cutoffs are used. If
only one cutoff is specified, it is used as the cutoff for both class 2 and Coulombic interactions for this type
pair. If both coefficients are specified, they are used as the class 2 and Coulombic cutoffs for this type pair.
You cannot specify 2 cutoffs for style lj/class2, since it has no Coulombic terms.
For lj/class2/coul/long only the class 2 cutoff can be specified since a Coulombic cutoff cannot be specified
for an individual I,J type pair. All type pairs use the same global Coulombic cutoff specified in the pair_style
command.
If the pair_coeff command is not used to define coefficients for a particular I != J type pair, the mixing rule
for epsilon and sigma for all class2 potentials is to use the sixthpower formulas documented by the
pair_modify command. The pair_modify mix setting is thus ignored for class2 potentials for epsilon and
sigma. However it is still followed for mixing the cutoff distance.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
LAMMPS Users Manual
1423
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of the lj/class2
pair styles can be mixed. Epsilon and sigma are always mixed with the value sixthpower. The cutoff distance
is mixed by whatever option is set by the pair_modify command (default = geometric). See the "pair_modify"
command for details.
All of the lj/class2 pair styles support the pair_modify shift option for the energy of the Lennard-Jones portion
of the pair interaction.
The lj/class2/coul/long pair style does not support the pair_modify table option since a tabulation capability
has not yet been added to this potential.
All of the lj/class2 pair styles support the pair_modify tail option for adding a long-range tail correction to the
energy and pressure of the Lennard-Jones portion of the pair interaction.
All of the lj/class2 pair styles write their information to binary restart files, so pair_style and pair_coeff
commands do not need to be specified in an input script that reads a restart file.
All of the lj/class2 pair styles can only be used via the pair keyword of the run_style respa command. They do
not support the inner, middle, outer keywords.
Restrictions:
These styles are part of the CLASS2 package. They are only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Related commands:
pair_coeff
Default: none
(Sun) Sun, J Phys Chem B 102, 7338-7364 (1998).
LAMMPS Users Manual
1424

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style colloid command
pair_style colloid/gpu command
pair_style colloid/omp command
Syntax:
pair_style colloid cutoff
cutoff = global cutoff for colloidal interactions (distance units)•
Examples:
pair_style colloid 10.0
pair_coeff * * 25 1.0 10.0 10.0
pair_coeff 1 1 144 1.0 0.0 0.0 3.0
pair_coeff 1 2 75.398 1.0 0.0 10.0 9.0
pair_coeff 2 2 39.478 1.0 10.0 10.0 25.0
Description:
Style colloid computes pairwise interactions between large colloidal particles and small solvent particles using
3 formulas. A colloidal particle has a size > sigma; a solvent particle is the usual Lennard-Jones particle of
size sigma.
The colloid-colloid interaction energy is given by
LAMMPS Users Manual
1425

where A_cc is the Hamaker constant, a1 and a2 are the radii of the two colloidal particles, and Rc is the
cutoff. This equation results from describing each colloidal particle as an integrated collection of
Lennard-Jones particles of size sigma and is derived in (Everaers).
The colloid-solvent interaction energy is given by
where A_cs is the Hamaker constant, a is the radius of the colloidal particle, and Rc is the cutoff. This formula
is derived from the colloid-colloid interaction, letting one of the particle sizes go to zero.
The solvent-solvent interaction energy is given by the usual Lennard-Jones formula
LAMMPS Users Manual
1426
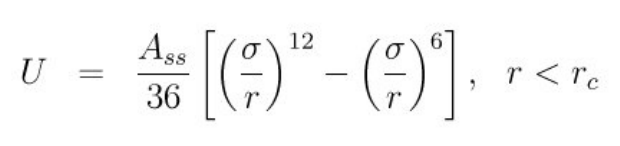
with A_ss set appropriately, which results from letting both particle sizes go to zero.
When used in combination with pair_style yukawa/colloid, the two terms become the so-called DLVO
potential, which combines electrostatic repulsion and van der Waals attraction.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
A (energy units)• sigma (distance units)• d1 (distance units)• d2 (distance units)• cutoff (distance units)•
A is the Hamaker energy prefactor and should typically be set as follows:
A_cc = colloid/colloid = 4 pi^2 = 39.5• A_cs = colloid/solvent = sqrt(A_cc*A_ss)• A_ss = solvent/solvent = 144 (assuming epsilon = 1, so that 144/36 = 4)•
Sigma is the size of the solvent particle or the constituent particles integrated over in the colloidal particle and
should typically be set as follows:
Sigma_cc = colloid/colloid = 1.0• Sigma_cs = colloid/solvent = arithmetic mixing between colloid sigma and solvent sigma• Sigma_ss = solvent/solvent = 1.0 or whatever size the solvent particle is•
Thus typically Sigma_cs = 1.0, unless the solvent particle's size != 1.0.
D1 and d2 are particle diameters, so that d1 = 2*a1 and d2 = 2*a2 in the formulas above. Both d1 and d2 must
be values >= 0. If d1 > 0 and d2 > 0, then the pair interacts via the colloid-colloid formula above. If d1 = 0
and d2 = 0, then the pair interacts via the solvent-solvent formula. I.e. a d value of 0 is a Lennard-Jones
particle of size sigma. If either d1 = 0 or d2 = 0 and the other is larger, then the pair interacts via the
colloid-solvent formula.
Note that the diameter of a particular particle type may appear in multiple pair_coeff commands, as it interacts
with other particle types. You should insure the particle diameter is specified consistently each time it appears.
The last coefficient is optional. If not specified, the global cutoff specified in the pair_style command is used.
However, you typically want different cutoffs for interactions between different particle sizes. E.g. if colloidal
particles of diameter 10 are used with solvent particles of diameter 1, then a solvent-solvent cutoff of 2.5
would correspond to a colloid-colloid cutoff of 25. A good rule-of-thumb is to use a colloid-solvent cutoff that
is half the big diameter + 4 times the small diameter. I.e. 9 = 5 + 4 for the colloid-solvent cutoff in this case.
LAMMPS Users Manual
1427

NOTE: When using pair_style colloid for a mixture with 2 (or more) widely different particles sizes (e.g.
sigma=10 colloids in a background sigma=1 LJ fluid), you will likely want to use these commands for
efficiency: neighbor multi and comm_modify multi.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the A, sigma, d1, and d2 coefficients and cutoff distance for this pair style
can be mixed. A is an energy value mixed like a LJ epsilon. D1 and d2 are distance values and are mixed like
sigma. The default mix value is geometric. See the "pair_modify" command for details.
This pair style supports the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This style is part of the COLLOID package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Normally, this pair style should be used with finite-size particles which have a diameter, e.g. see the
atom_style sphere command. However, this is not a requirement, since the only definition of particle size is
via the pair_coeff parameters for each type. In other words, the physical radius of the particle is ignored. Thus
you should insure that the d1,d2 parameters you specify are consistent with the physical size of the particles of
that type.
LAMMPS Users Manual
1428


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style comb command
pair_style comb/omp command
pair_style comb3 command
Syntax:
pair_style comb
pair_style comb3 keyword
keyword = polar
polar value = polar_on or polar_off = whether or not to include atomic polarization
Examples:
pair_style comb
pair_coeff * * ../potentials/ffield.comb Si
pair_coeff * * ../potentials/ffield.comb Hf Si O
pair_style comb3 polar_off
pair_coeff * * ../potentials/ffield.comb3 O Cu N C O
Description:
Style comb computes the second-generation variable charge COMB (Charge-Optimized Many-Body)
potential. Style comb3 computes the third-generation COMB potential. These COMB potentials are described
in (COMB) and (COMB3). Briefly, the total energy ET of a system of atoms is given by
where Eiself is the self-energy of atom i (including atomic ionization energies and electron affinities), Eijshort is
the bond-order potential between atoms i and j, EijCoul is the Coulomb interactions, Epolar is the polarization
term for organic systems (style comb3 only), EvdW is the van der Waals energy (style comb3 only), Ebarr is a
charge barrier function, and Ecorr are angular correction terms.
The COMB potentials (styles comb and comb3) are variable charge potentials. The equilibrium charge on
each atom is calculated by the electronegativity equalization (QEq) method. See Rick for further details. This
is implemented by the fix qeq/comb command, which should normally be specified in the input script when
running a model with the COMB potential. The fix qeq/comb command has options that determine how often
charge equilibration is performed, its convergence criterion, and which atoms are included in the calculation.
LAMMPS Users Manual
1430

Only a single pair_coeff command is used with the comb and comb3 styles which specifies the COMB
potential file with parameters for all needed elements. These are mapped to LAMMPS atom types by
specifying N additional arguments after the potential file in the pair_coeff command, where N is the number
of LAMMPS atom types.
For example, if your LAMMPS simulation of a Si/SiO2/ HfO2 interface has 4 atom types, and you want the
1st and last to be Si, the 2nd to be Hf, and the 3rd to be O, and you would use the following pair_coeff
command:
pair_coeff * * ../potentials/ffield.comb Si Hf O Si
The first two arguments must be * * so as to span all LAMMPS atom types. The first and last Si arguments
map LAMMPS atom types 1 and 4 to the Si element in the ffield.comb file. The second Hf argument maps
LAMMPS atom type 2 to the Hf element, and the third O argument maps LAMMPS atom type 3 to the O
element in the potential file. If a mapping value is specified as NULL, the mapping is not performed. This can
be used when a comb potential is used as part of the hybrid pair style. The NULL values are placeholders for
atom types that will be used with other potentials.
For style comb, the provided potential file ffield.comb contains all currently-available 2nd generation COMB
parameterizations: for Si, Cu, Hf, Ti, O, their oxides and Zr, Zn and U metals. For style comb3, the potential
file ffield.comb3 contains all currently-available 3rd generation COMB parameterizations: O, Cu, N, C, H, Ti,
Zn and Zr. The status of the optimization of the compounds, for example Cu2O, TiN and hydrocarbons, are
given in the following table:
For style comb3, in addition to ffield.comb3, a special parameter file, lib.comb3, that is exclusively used for
C/O/H systems, will be automatically loaded if carbon atom is detected in LAMMPS input structure. This file
must be in your working directory or in the directory pointed to by the environment variable
LAMMPS_POTENTIALS, as described on the pair_coeff command doc page.
Keyword polar indicates whether the force field includes the atomic polarization. Since the equilibration of
the polarization has not yet been implemented, it can only set polar_off at present.
LAMMPS Users Manual
1431
NOTE: You can not use potential file ffield.comb with style comb3, nor file ffield.comb3 with style comb.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, where types I and J correspond to two different element types, mixing is
performed by LAMMPS as described above from values in the potential file.
These pair styles does not support the pair_modify shift, table, and tail options.
These pair styles do not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style, pair_coeff, and fix qeq/comb commands in an input script that reads a
restart file.
These pair styles can only be used via the pair keyword of the run_style respa command. It does not support
the inner, middle, outer keywords.
Restrictions:
These pair styles are part of the MANYBODY package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
These pair styles requires the newton setting to be "on" for pair interactions.
The COMB potentials in the ffield.comb and ffield.comb3 files provided with LAMMPS (see the potentials
directory) are parameterized for metal units. You can use the COMB potential with any LAMMPS units, but
you would need to create your own COMB potential file with coefficients listed in the appropriate units if
your simulation doesn't use "metal" units.
Related commands:
pair_style, pair_coeff, fix qeq/comb
Default: none
LAMMPS Users Manual
1432
(COMB) T.-R. Shan, B. D. Devine, T. W. Kemper, S. B. Sinnott, and S. R. Phillpot, Phys. Rev. B 81, 125328
(2010)
(COMB3) T. Liang, T.-R. Shan, Y.-T. Cheng, B. D. Devine, M. Noordhoek, Y. Li, Z. Lu, S. R. Phillpot, and
S. B. Sinnott, Mat. Sci. & Eng: R 74, 255-279 (2013).
(Rick) S. W. Rick, S. J. Stuart, B. J. Berne, J Chem Phys 101, 6141 (1994).
LAMMPS Users Manual
1433

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style coul/cut command
pair_style coul/cut/gpu command
pair_style coul/cut/kk command
pair_style coul/cut/omp command
pair_style coul/debye command
pair_style coul/debye/gpu command
pair_style coul/debye/kk command
pair_style coul/debye/omp command
pair_style coul/dsf command
pair_style coul/dsf/gpu command
pair_style coul/dsf/kk command
pair_style coul/dsf/omp command
pair_style coul/long command
pair_style coul/long/cs command
pair_style coul/long/omp command
pair_style coul/long/gpu command
pair_style coul/long/kk command
pair_style coul/msm command
pair_style coul/msm/omp command
pair_style coul/streitz command
pair_style coul/wolf command
LAMMPS Users Manual
1434
pair_style coul/wolf/kk command
pair_style coul/wolf/omp command
pair_style tip4p/cut command
pair_style tip4p/long command
pair_style tip4p/cut/omp command
pair_style tip4p/long/omp command
Syntax:
pair_style coul/cut cutoff
pair_style coul/debye kappa cutoff
pair_style coul/dsf alpha cutoff
pair_style coul/long cutoff
pair_style coul/long/cs cutoff
pair_style coul/long/gpu cutoff
pair_style coul/wolf alpha cutoff
pair_style coul/streitz cutoff keyword alpha
pair_style tip4p/cut otype htype btype atype qdist cutoff
pair_style tip4p/long otype htype btype atype qdist cutoff
cutoff = global cutoff for Coulombic interactions• kappa = Debye length (inverse distance units)• alpha = damping parameter (inverse distance units)•
Examples:
pair_style coul/cut 2.5
pair_coeff * *
pair_coeff 2 2 3.5
pair_style coul/debye 1.4 3.0
pair_coeff * *
pair_coeff 2 2 3.5
pair_style coul/dsf 0.05 10.0
pair_coeff * *
pair_style coul/long 10.0
pair_style coul/long/cs 10.0
pair_coeff * *
pair_style coul/msm 10.0
pair_coeff * *
pair_style coul/wolf 0.2 9.0
pair_coeff * *
pair_style coul/streitz 12.0 ewald
pair_style coul/streitz 12.0 wolf 0.30
LAMMPS Users Manual
1435

pair_coeff * * AlO.streitz Al O
pair_style tip4p/cut 1 2 7 8 0.15 12.0
pair_coeff * *
pair_style tip4p/long 1 2 7 8 0.15 10.0
pair_coeff * *
Description:
The coul/cut style computes the standard Coulombic interaction potential given by
where C is an energy-conversion constant, Qi and Qj are the charges on the 2 atoms, and epsilon is the
dielectric constant which can be set by the dielectric command. The cutoff Rc truncates the interaction
distance.
Style coul/debye adds an additional exp() damping factor to the Coulombic term, given by
where kappa is the Debye length. This potential is another way to mimic the screening effect of a polar
solvent.
Style coul/dsf computes Coulombic interactions via the damped shifted force model described in Fennell,
given by:
where alpha is the damping parameter and erfc() is the complementary error-function. The potential corrects
issues in the Wolf model (described below) to provide consistent forces and energies (the Wolf potential is not
differentiable at the cutoff) and smooth decay to zero.
Style coul/wolf computes Coulombic interactions via the Wolf summation method, described in Wolf, given
by:
LAMMPS Users Manual
1436

where alpha is the damping parameter, and erc() and erfc() are error-function and complementary
error-function terms. This potential is essentially a short-range, spherically-truncated, charge-neutralized,
shifted, pairwise 1/r summation. With a manipulation of adding and subtracting a self term (for i = j) to the
first and second term on the right-hand-side, respectively, and a small enough alpha damping parameter, the
second term shrinks and the potential becomes a rapidly-converging real-space summation. With a long
enough cutoff and small enough alpha parameter, the energy and forces calculated by the Wolf summation
method approach those of the Ewald sum. So it is a means of getting effective long-range interactions with a
short-range potential.
Style coul/streitz is the Coulomb pair interaction defined as part of the Streitz-Mintmire potential, as
described in this paper, in which charge distribution about an atom is modeled as a Slater 1s orbital. More
details can be found in the referenced paper. To fully reproduce the published Streitz-Mintmire potential,
which is a variable charge potential, style coul/streitz must be used with pair_style eam/alloy (or some other
short-range potential that has been parameterized appropriately) via the pair_style hybrid/overlay command.
Likewise, charge equilibration must be performed via the fix qeq/slater command. For example:
pair_style hybrid/overlay coul/streitz 12.0 wolf 0.31 eam/alloy
pair_coeff * * coul/streitz AlO.streitz Al O
pair_coeff * * eam/alloy AlO.eam.alloy Al O
fix 1 all qeq/slater 1 12.0 1.0e-6 100 coul/streitz
The keyword wolf in the coul/streitz command denotes computing Coulombic interactions via Wolf
summation. An additional damping parameter is required for the Wolf summation, as described for the
coul/wolf potential above. Alternatively, Coulombic interactions can be computed via an Ewald summation.
For example:
pair_style hybrid/overlay coul/streitz 12.0 ewald eam/alloy
kspace_style ewald 1e-6
Keyword ewald does not need a damping parameter, but a kspace_style must be defined, which can be style
ewald or pppm. The Ewald method was used in Streitz and Mintmire's original paper, but a Wolf summation
offers a speed-up in some cases.
For the fix qeq/slater command, the qfile can be a filename that contains QEq parameters as discussed on the
fix qeq command doc page. Alternatively qfile can be replaced by "coul/streitz", in which case the fix will
extract QEq parameters from the coul/streitz pair style itself.
See the examples/strietz directory for an example input script that uses the Streitz-Mintmire potential. The
potentials directory has the AlO.eam.alloy and AlO.streitz potential files used by the example.
Note that the Streiz-Mintmire potential is generally used for oxides, but there is no conceptual problem with
extending it to nitrides and carbides (such as SiC, TiN). Pair coul/strietz used by itself or with any other pair
style such as EAM, MEAM, Tersoff, or LJ in hybrid/overlay mode. To do this, you would need to provide a
Streitz-Mintmire parameterization for the material being modeled.
LAMMPS Users Manual
1437

Styles coul/long and coul/msm compute the same Coulombic interactions as style coul/cut except that an
additional damping factor is applied so it can be used in conjunction with the kspace_style command and its
ewald or pppm option. The Coulombic cutoff specified for this style means that pairwise interactions within
this distance are computed directly; interactions outside that distance are computed in reciprocal space.
Style coul/long/cs is identical to coul/long except that a term is added for the core/shell model to allow
charges on core and shell particles to be separated by r = 0.0.
Styles tip4p/cut and tip4p/long implement the coulomb part of the TIP4P water model of (Jorgensen), which
introduces a massless site located a short distance away from the oxygen atom along the bisector of the HOH
angle. The atomic types of the oxygen and hydrogen atoms, the bond and angle types for OH and HOH
interactions, and the distance to the massless charge site are specified as pair_style arguments. Style tip4p/cut
uses a global cutoff for Coulomb interactions; style tip4p/long is for use with a long-range Coulombic solver
(Ewald or PPPM).
NOTE: For each TIP4P water molecule in your system, the atom IDs for the O and 2 H atoms must be
consecutive, with the O atom first. This is to enable LAMMPS to "find" the 2 H atoms associated with each O
atom. For example, if the atom ID of an O atom in a TIP4P water molecule is 500, then its 2 H atoms must
have IDs 501 and 502.
See the howto section for more information on how to use the TIP4P pair styles and lists of parameters to set.
Note that the neighbor list cutoff for Coulomb interactions is effectively extended by a distance 2*qdist when
using the TIP4P pair style, to account for the offset distance of the fictitious charges on O atoms in water
molecules. Thus it is typically best in an efficiency sense to use a LJ cutoff >= Coulomb cutoff + 2*qdist, to
shrink the size of the neighbor list. This leads to slightly larger cost for the long-range calculation, so you can
test the trade-off for your model.
Note that these potentials are designed to be combined with other pair potentials via the pair_style
hybrid/overlay command. This is because they have no repulsive core. Hence if they are used by themselves,
there will be no repulsion to keep two oppositely charged particles from moving arbitrarily close to each
other.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
cutoff (distance units)•
For coul/cut and coul/debye, the cutoff coefficient is optional. If it is not used (as in some of the examples
above), the default global value specified in the pair_style command is used.
For coul/long and coul/msm no cutoff can be specified for an individual I,J type pair via the pair_coeff
command. All type pairs use the same global Coulombic cutoff specified in the pair_style command.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
LAMMPS Users Manual
1438

respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the cutoff distance for the coul/cut style can be mixed. The default mix
value is geometric. See the "pair_modify" command for details.
The pair_modify shift option is not relevant for these pair styles.
The coul/long style supports the pair_modify table option for tabulation of the short-range portion of the
long-range Coulombic interaction.
These pair styles do not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
These pair styles write their information to binary restart files, so pair_style and pair_coeff commands do not
need to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
The coul/long, coul/msm and tip4p/long styles are part of the KSPACE package. The coul/long/cs style is part
of the CORESHELL package. They are only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
pair_coeff, pair_style, hybrid/overlay, kspace_style
Default: none
(Wolf) D. Wolf, P. Keblinski, S. R. Phillpot, J. Eggebrecht, J Chem Phys, 110, 8254 (1999).
(Fennell) C. J. Fennell, J. D. Gezelter, J Chem Phys, 124, 234104 (2006).
(Streitz) F. H. Streitz, J. W. Mintmire, Phys Rev B, 50, 11996-12003 (1994).
LAMMPS Users Manual
1439
(Jorgensen) Jorgensen, Chandrasekhar, Madura, Impey, Klein, J Chem Phys, 79, 926 (1983).
LAMMPS Users Manual
1440

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style coul/diel command
pair_style coul/diel/omp command
Syntax:
pair_style coul/diel cutoff
cutoff = global cutoff (distance units)
Examples:
pair_style coul/diel 3.5
pair_coeff 1 4 78. 1.375 0.112
Description:
Style coul/diel computes a Coulomb correction for implicit solvent ion interactions in which the dielectric
permittivity is distance dependent. The dielectric permittivity epsilon_D(r) connects to limiting regimes: One
limit is defined by a small dielectric permittivity (close to vacuum) at or close to contact separation between
the ions. At larger separations the dielectric permittivity reaches a bulk value used in the regular Coulomb
interaction coul/long or coul/cut. The transition is modeled by a hyperbolic function which is incorporated in
the Coulomb correction term for small ion separations as follows
where r_me is the inflection point of epsilon_D(r) and sigma_e is a slope defining length scale. C is the same
Coulomb conversion factor as in the pair_styles coul/cut, coul/long, and coul/debye. In this way the Coulomb
interaction between ions is corrected at small distances r. The lower limit of epsilon_D(r->0)=5.2 due to
dielectric saturation (Stiles) while the Coulomb interaction reaches its bulk limit by setting
epsilon_D(r->\infty)=epsilon, the bulk value of the solvent which is 78 for water at 298K.
Examples of the use of this type of Coulomb interaction include implicit solvent simulations of salt ions
(Lenart) and of ionic surfactants (Jusufi). Note that this potential is only reasonable for implicit solvent
simulations and in combination with coul/cut or coul/long. It is also usually combined with gauss/cut, see
(Lenart) or (Jusufi).
The following coefficients must be defined for each pair of atom types via the pair_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
epsilon (no units)•
LAMMPS Users Manual
1441
r_me (distance units)• sigma_e (distance units)•
The global cutoff (r_c) specified in the pair_style command is used.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support parameter mixing. Coefficients must be given explicitly for each type of
particle pairs.
This pair style supports the pair_modify shift option for the energy of the Gauss-potential portion of the pair
interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This style is part of the "user-misc" package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
pair_coeff pair_style gauss/cut
Default: none
(Stiles) Stiles , Hubbard, and Kayser, J Chem Phys, 77, 6189 (1982).
(Lenart) Lenart , Jusufi, and Panagiotopoulos, J Chem Phys, 126, 044509 (2007).
(Jusufi) Jusufi, Hynninen, and Panagiotopoulos, J Phys Chem B, 112, 13783 (2008).
LAMMPS Users Manual
1442

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style born/coul/long/cs command
pair_style buck/coul/long/cs command
pair_style born/coul/dsf/cs command
Syntax:
pair_style style args
style = born/coul/long/cs or buck/coul/long/cs or born/coul/dsf/cs• args = list of arguments for a particular style•
born/coul/long/cs args = cutoff (cutoff2)
cutoff = global cutoff for non-Coulombic (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
buck/coul/long/cs args = cutoff (cutoff2)
cutoff = global cutoff for Buckingham (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
born/coul/dsf/cs args = alpha cutoff (cutoff2)
alpha = damping parameter (inverse distance units)
cutoff = global cutoff for non-Coulombic (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (distance units)
Examples:
pair_style born/coul/long/cs 10.0 8.0
pair_coeff 1 1 6.08 0.317 2.340 24.18 11.51
pair_style buck/coul/long/cs 10.0
pair_style buck/coul/long/cs 10.0 8.0
pair_coeff * * 100.0 1.5 200.0
pair_coeff 1 1 100.0 1.5 200.0 9.0
pair_style born/coul/dsf/cs 0.1 10.0 12.0
pair_coeff * * 0.0 1.00 0.00 0.00 0.00
pair_coeff 1 1 480.0 0.25 0.00 1.05 0.50
Description:
These pair styles are designed to be used with the adiabatic core/shell model of (Mitchell and Finchham). See
Section 6.25 of the manual for an overview of the model as implemented in LAMMPS.
The styles with a coul/long term are identical to the pair_style born/coul/long and pair_style buck/coul/long
styles, except they correctly treat the special case where the distance between two charged core and shell
atoms in the same core/shell pair approach r = 0.0. This needs special treatment when a long-range solver for
Coulombic interactions is also used, i.e. via the kspace_style command.
More specifically, the short-range Coulomb interaction between a core and its shell should be turned off using
the special_bonds command by setting the 1-2 weight to 0.0, which works because the core and shell atoms
are bonded to each other. This induces a long-range correction approximation which fails at small distances
LAMMPS Users Manual
1443

(~< 10e-8). Therefore, the Coulomb term which is used to calculate the correction factor is extended by a
minimal distance (r_min = 1.0-6) when the interaction between a core/shell pair is treated, as follows
where C is an energy-conversion constant, Qi and Qj are the charges on the core and shell, epsilon is the
dielectric constant and r_min is the minimal distance.
The pair style born/coul/dsf/cs is identical to the pair_style born/coul/dsf style, which uses the the damped
shifted force model as in coul/dsf to compute the Coulomb contribution. This approach does not require a
long-range solver, thus the only correction is the addition of a minimal distance to avoid the possible r = 0.0
case for a core/shell pair.
Restrictions:
These pair styles are part of the CORESHELL package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info.
Related commands:
pair_coeff, pair_style born, pair_style buck
Default: none
(Mitchell and Finchham) Mitchell, Finchham, J Phys Condensed Matter, 5, 1031-1038 (1993).
LAMMPS Users Manual
1444

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/cut/dipole/cut command
pair_style lj/cut/dipole/cut/gpu command
pair_style lj/cut/dipole/cut/omp command
pair_style lj/sf/dipole/sf command
pair_style lj/sf/dipole/sf/gpu command
pair_style lj/sf/dipole/sf/omp command
pair_style lj/cut/dipole/long command
pair_style lj/long/dipole/long command
Syntax:
pair_style lj/cut/dipole/cut cutoff (cutoff2)
pair_style lj/sf/dipole/sf cutoff (cutoff2)
pair_style lj/cut/dipole/long cutoff (cutoff2)
pair_style lj/long/dipole/long flag_lj flag_coul cutoff (cutoff2)
cutoff = global cutoff LJ (and Coulombic if only 1 arg) (distance units)• cutoff2 = global cutoff for Coulombic and dipole (optional) (distance units)• flag_lj = long or cut or off
long = use long-range damping on dispersion 1/r^6 term
cut = use a cutoff on dispersion 1/r^6 term
off = omit disperion 1/r^6 term entirely
•
flag_coul = long or off
long = use long-range damping on Coulombic 1/r and point-dipole terms
off = omit Coulombic and point-dipole terms entirely
•
Examples:
pair_style lj/cut/dipole/cut 10.0
pair_coeff * * 1.0 1.0
pair_coeff 2 3 1.0 1.0 2.5 4.0
pair_style lj/sf/dipole/sf 9.0
pair_coeff * * 1.0 1.0
pair_coeff 2 3 1.0 1.0 2.5 4.0 scale 0.5
pair_coeff 2 3 1.0 1.0 2.5 4.0
pair_style lj/cut/dipole/long 10.0
pair_coeff * * 1.0 1.0
pair_coeff 2 3 1.0 1.0 2.5 4.0
LAMMPS Users Manual
1445
pair_style lj/long/dipole/long long long 3.5 10.0
pair_coeff * * 1.0 1.0
pair_coeff 2 3 1.0 1.0 2.5 4.0
Description:
Style lj/cut/dipole/cut computes interactions between pairs of particles that each have a charge and/or a point
dipole moment. In addition to the usual Lennard-Jones interaction between the particles (Elj) the
charge-charge (Eqq), charge-dipole (Eqp), and dipole-dipole (Epp) interactions are computed by these
formulas for the energy (E), force (F), and torque (T) between particles I and J.
LAMMPS Users Manual
1446
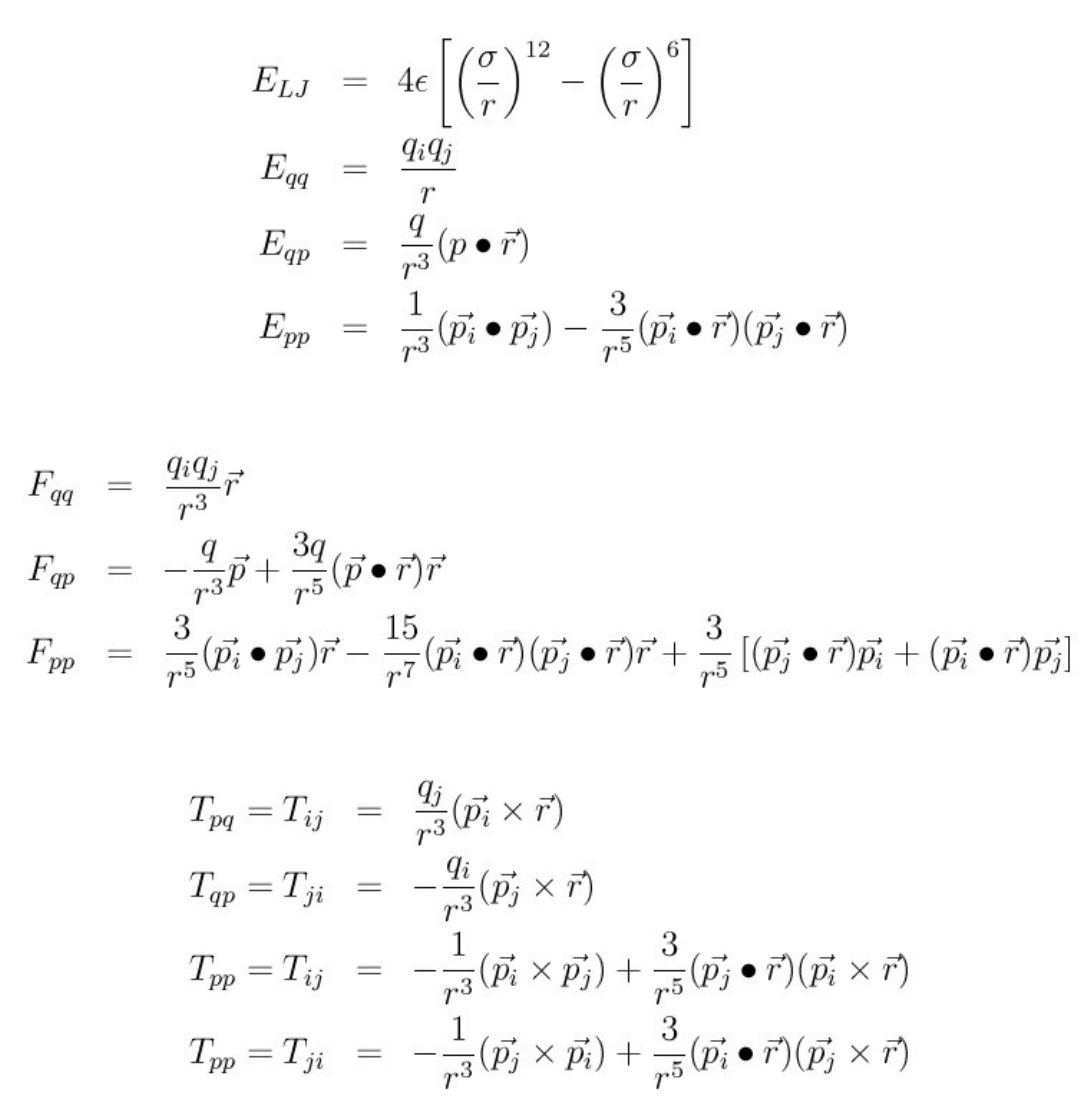
where qi and qj are the charges on the two particles, pi and pj are the dipole moment vectors of the two
particles, r is their separation distance, and the vector r = Ri - Rj is the separation vector between the two
particles. Note that Eqq and Fqq are simply Coulombic energy and force, Fij = -Fji as symmetric forces, and
Tij != -Tji since the torques do not act symmetrically. These formulas are discussed in (Allen) and in
(Toukmaji).
Style lj/sf/dipole/sf computes "shifted-force" interactions between pairs of particles that each have a charge
and/or a point dipole moment. In general, a shifted-force potential is a (sligthly) modified potential containing
LAMMPS Users Manual
1447
extra terms that make both the energy and its derivative go to zero at the cutoff distance; this removes
(cutoff-related) problems in energy conservation and any numerical instability in the equations of motion
(Allen). Shifted-force interactions for the Lennard-Jones (E_LJ), charge-charge (Eqq), charge-dipole (Eqp),
dipole-charge (Epq) and dipole-dipole (Epp) potentials are computed by these formulas for the energy (E),
force (F), and torque (T) between particles I and J:
LAMMPS Users Manual
1448
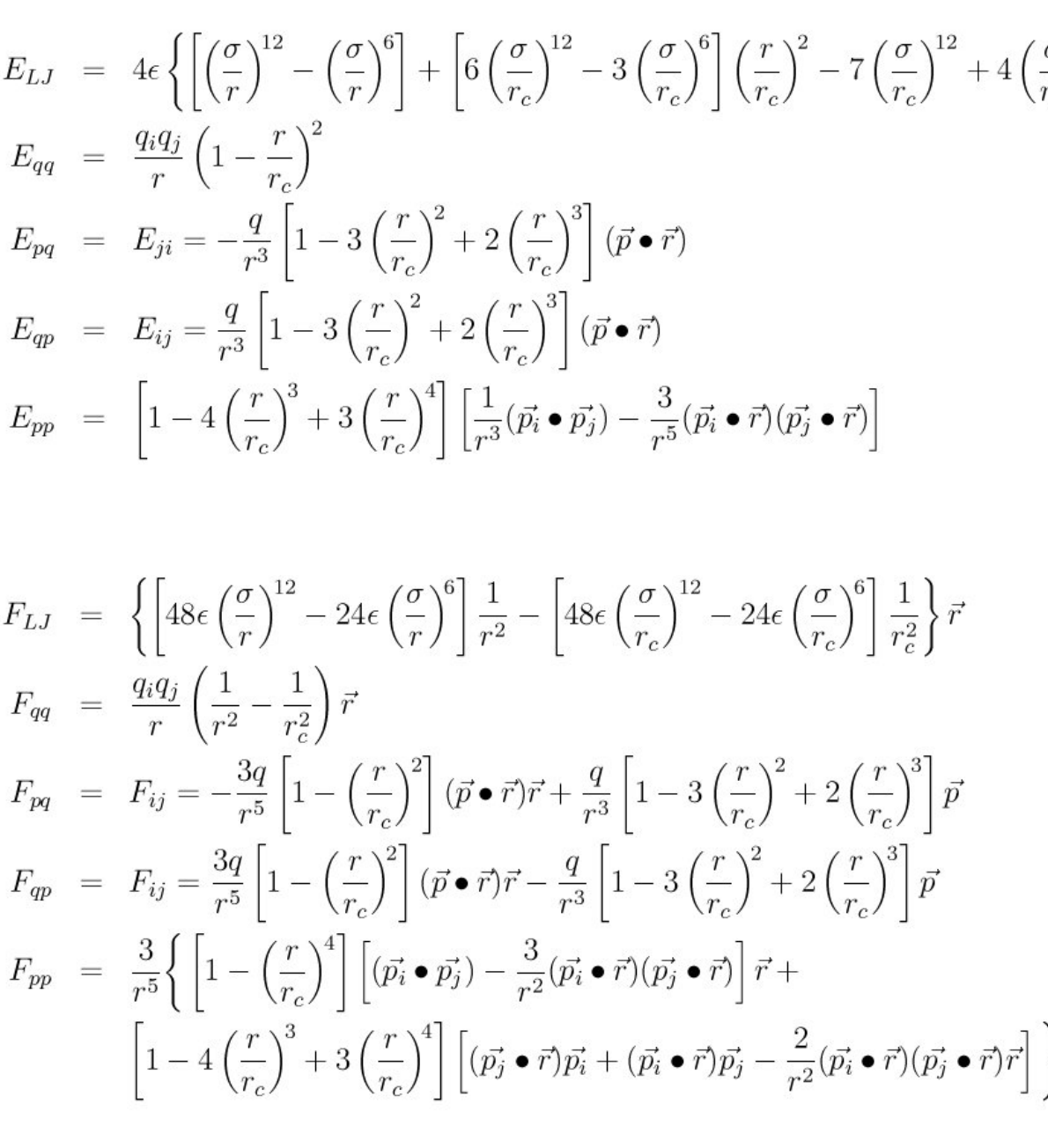
LAMMPS Users Manual
1449
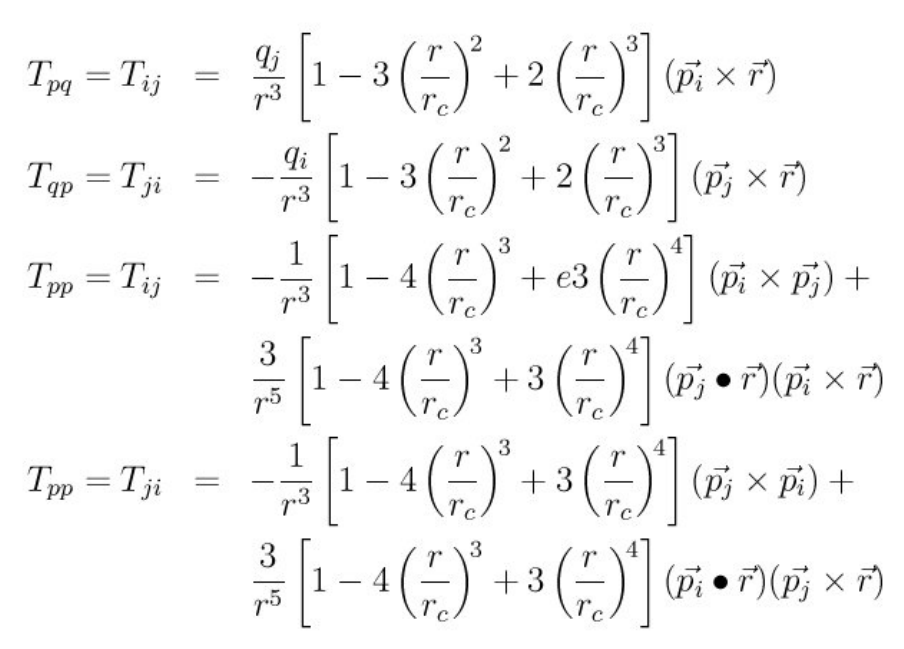
where epsilon and sigma are the standard LJ parameters, r_c is the cutoff, qi and qj are the charges on the two
particles, pi and pj are the dipole moment vectors of the two particles, r is their separation distance, and the
vector r = Ri - Rj is the separation vector between the two particles. Note that Eqq and Fqq are simply
Coulombic energy and force, Fij = -Fji as symmetric forces, and Tij != -Tji since the torques do not act
symmetrically. The shifted-force formula for the Lennard-Jones potential is reported in (Stoddard). The
original (unshifted) formulas for the electrostatic potentials, forces and torques can be found in (Price). The
shifted-force electrostatic potentials have been obtained by applying equation 5.13 of (Allen). The formulas
for the corresponding forces and torques have been obtained by applying the 'chain rule' as in appendix C.3 of
(Allen).
If one cutoff is specified in the pair_style command, it is used for both the LJ and Coulombic (q,p) terms. If
two cutoffs are specified, they are used as cutoffs for the LJ and Coulombic (q,p) terms respectively. This pair
style also supports an optional scale keyword as part of a pair_coeff statement, where the interactions can be
scaled according to this factor. This scale factor is also made available for use with fix adapt.
Style lj/cut/dipole/long computes long-range point-dipole interactions as discussed in (Toukmaji).
Dipole-dipole, dipole-charge, and charge-charge interactions are all supported, along with the standard 12/6
Lennard-Jones interactions, which are computed with a cutoff. A kspace_style must be defined to use this pair
style. Currently, only kspace_style ewald/disp support long-range point-dipole interactions.
Style lj/long/dipole/long also computes point-dipole interactions as discussed in (Toukmaji). Long-range
dipole-dipole, dipole-charge, and charge-charge interactions are all supported, along with the standard 12/6
Lennard-Jones interactions. LJ interactions can be cutoff or long-ranged.
LAMMPS Users Manual
1450

For style lj/long/dipole/long, if flag_lj is set to long, no cutoff is used on the LJ 1/r^6 dispersion term. The
long-range portion is calculated by using the kspace_style ewald_disp command. The specified LJ cutoff then
determines which portion of the LJ interactions are computed directly by the pair potential versus which part
is computed in reciprocal space via the Kspace style. If flag_lj is set to cut, the LJ interactions are simply
cutoff, as with pair_style lj/cut. If flag_lj is set to off, LJ interactions are not computed at all.
If flag_coul is set to long, no cutoff is used on the Coulombic or dipole interactions. The long-range portion is
calculated by using ewald_disp of the kspace_style command. If flag_coul is set to off, Coulombic and dipole
interactions are not computed at all.
Atoms with dipole moments should be integrated using the fix nve/sphere update dipole or the fix nvt/sphere
update dipole command to rotate the dipole moments. The omega option on the fix langevin command can be
used to thermostat the rotational motion. The compute temp/sphere command can be used to monitor the
temperature, since it includes rotational degrees of freedom. The atom_style hybrid dipole sphere command
should be used since it defines the point dipoles and their rotational state. The magnitude and orientation of
the dipole moment for each particle can be defined by the set command or in the "Atoms" section of the data
file read in by the read_data command.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• cutoff1 (distance units)• cutoff2 (distance units)•
The latter 2 coefficients are optional. If not specified, the global LJ and Coulombic cutoffs specified in the
pair_style command are used. If only one cutoff is specified, it is used as the cutoff for both LJ and
Coulombic interactions for this type pair. If both coefficients are specified, they are used as the LJ and
Coulombic cutoffs for this type pair.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distances for this pair style can
LAMMPS Users Manual
1451

be mixed. The default mix value is geometric. See the "pair_modify" command for details.
For atom type pairs I,J and I != J, the A, sigma, d1, and d2 coefficients and cutoff distance for this pair style
can be mixed. A is an energy value mixed like a LJ epsilon. D1 and d2 are distance values and are mixed like
sigma. The default mix value is geometric. See the "pair_modify" command for details.
This pair style does not support the pair_modify shift option for the energy of the Lennard-Jones portion of
the pair interaction; such energy goes to zero at the cutoff by construction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
The lj/cut/dipole/cut, lj/cut/dipole/long, and lj/long/dipole/long styles are part of the DIPOLE package. They
are only enabled if LAMMPS was built with that package. See the Making LAMMPS section for more info.
The lj/sf/dipole/sf style is part of the USER-MISC package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Using dipole pair styles with electron units is not currently supported.
Related commands:
pair_coeff, set, read_data, fix nve/sphere, fix nvt/sphere
Default: none
(Allen) Allen and Tildesley, Computer Simulation of Liquids, Clarendon Press, Oxford, 1987.
(Toukmaji) Toukmaji, Sagui, Board, and Darden, J Chem Phys, 113, 10913 (2000).
(Stoddard) Stoddard and Ford, Phys Rev A, 8, 1504 (1973).
(Price) Price, Stone and Alderton, Mol Phys, 52, 987 (1984).
LAMMPS Users Manual
1452

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style dpd command
pair_style dpd/gpu command
pair_style dpd/omp command
pair_style dpd/tstat command
pair_style dpd/tstat/gpu command
pair_style dpd/tstat/omp command
Syntax:
pair_style dpd T cutoff seed
pair_style dpd/tstat Tstart Tstop cutoff seed
T = temperature (temperature units)• Tstart,Tstop = desired temperature at start/end of run (temperature units)• cutoff = global cutoff for DPD interactions (distance units)• seed = random # seed (positive integer)•
Examples:
pair_style dpd 1.0 2.5 34387
pair_coeff * * 3.0 1.0
pair_coeff 1 1 3.0 1.0 1.0
pair_style dpd/tstat 1.0 1.0 2.5 34387
pair_coeff * * 1.0
pair_coeff 1 1 1.0 1.0
Description:
Style dpd computes a force field for dissipative particle dynamics (DPD) following the exposition in (Groot).
Style dpd/tstat invokes a DPD thermostat on pairwise interactions, which is equivalent to the
non-conservative portion of the DPD force field. This pair-wise thermostat can be used in conjunction with
any pair style, and in leiu of per-particle thermostats like fix langevin or ensemble thermostats like Nose
Hoover as implemented by fix nvt. To use dpd/tstat as a thermostat for another pair style, use the pair_style
hybrid/overlay command to compute both the desired pair interaction and the thermostat for each pair of
particles.
For style dpd, the force on atom I due to atom J is given as a sum of 3 terms
LAMMPS Users Manual
1453
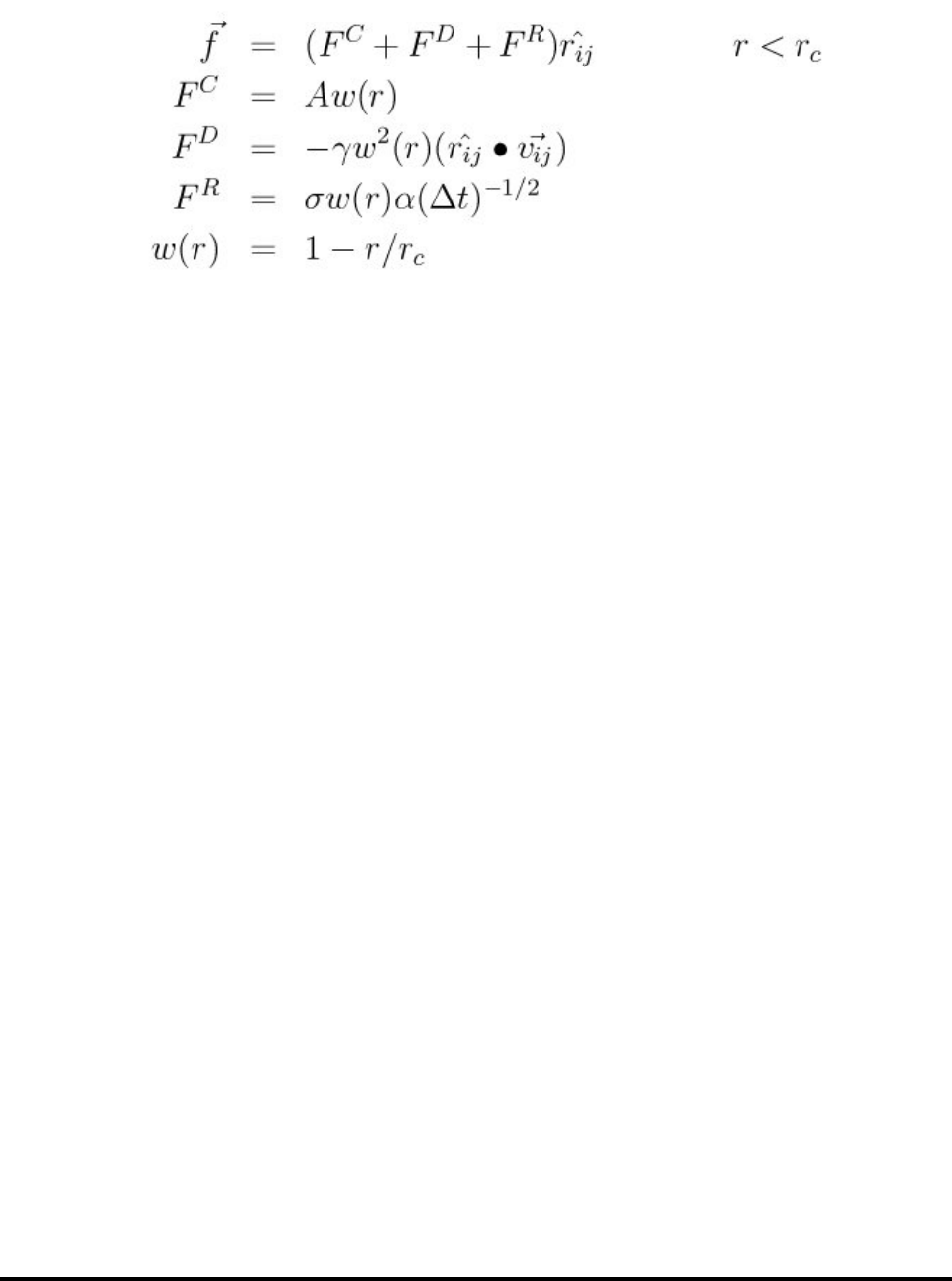
where Fc is a conservative force, Fd is a dissipative force, and Fr is a random force. Rij is a unit vector in the
direction Ri - Rj, Vij is the vector difference in velocities of the two atoms = Vi - Vj, alpha is a Gaussian
random number with zero mean and unit variance, dt is the timestep size, and w(r) is a weighting factor that
varies between 0 and 1. Rc is the cutoff. Sigma is set equal to sqrt(2 Kb T gamma), where Kb is the
Boltzmann constant and T is the temperature parameter in the pair_style command.
For style dpd/tstat, the force on atom I due to atom J is the same as the above equation, except that the
conservative Fc term is dropped. Also, during the run, T is set each timestep to a ramped value from Tstart to
Tstop.
For style dpd, the pairwise energy associated with style dpd is only due to the conservative force term Fc, and
is shifted to be zero at the cutoff distance Rc. The pairwise virial is calculated using all 3 terms. For style
dpd/tstat there is no pairwise energy, but the last two terms of the formula make a contribution to the virial.
For style dpd, the following coefficients must be defined for each pair of atoms types via the pair_coeff
command as in the examples above, or in the data file or restart files read by the read_data or read_restart
commands:
A (force units)• gamma (force/velocity units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global DPD cutoff is used. Note that sigma is set equal to
sqrt(2 T gamma), where T is the temperature set by the pair_style command so it does not need to be
specified.
For style dpd/tstat, the coefficients defined for each pair of atoms types via the pair_coeff command is the
same, except that A is not included.
The GPU-accelerated versions of these styles are implemented based on the work of (Afshar) and (Phillips).
NOTE: If you are modeling DPD polymer chains, you may want to use the pair_style srp command in
conjuction with these pair styles. It is a soft segmental repulsive potential (SRP) that can prevent DPD
polymer chains from crossing each other.
NOTE: The virial calculation for pressure when using this pair style includes all the components of force
listed above, including the random force.
LAMMPS Users Manual
1454
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
These pair styles do not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
These pair styles do not support the pair_modify shift option for the energy of the pair interaction. Note that as
discussed above, the energy due to the conservative Fc term is already shifted to be 0.0 at the cutoff distance
Rc.
The pair_modify table option is not relevant for these pair styles.
These pair style do not support the pair_modify tail option for adding long-range tail corrections to energy and
pressure.
These pair styles writes their information to binary restart files, so pair_style and pair_coeff commands do not
need to be specified in an input script that reads a restart file. Note that the user-specified random number seed
is stored in the restart file, so when a simulation is restarted, each processor will re-initialize its random
number generator the same way it did initially. This means the random forces will be random, but will not be
the same as they would have been if the original simulation had continued past the restart time.
These pair styles can only be used via the pair keyword of the run_style respa command. They do not support
the inner, middle, outer keywords.
The dpd/tstat style can ramp its target temperature over multiple runs, using the start and stop keywords of the
run command. See the run command for details of how to do this.
Restrictions:
The default frequency for rebuilding neighbor lists is every 10 steps (see the neigh_modify command). This
may be too infrequent for style dpd simulations since particles move rapidly and can overlap by large
amounts. If this setting yields a non-zero number of "dangerous" reneighborings (printed at the end of a
simulation), you should experiment with forcing reneighboring more often and see if system
energies/trajectories change.
These pair styles requires you to use the comm_modify vel yes command so that velocites are stored by ghost
atoms.
LAMMPS Users Manual
1455

These pair styles will not restart exactly when using the read_restart command, though they should provide
statistically similar results. This is because the forces they compute depend on atom velocities. See the
read_restart command for more details.
Related commands:
pair_coeff, fix nvt, fix langevin, pair_style srp
Default: none
(Groot) Groot and Warren, J Chem Phys, 107, 4423-35 (1997).
(Afshar) Afshar, F. Schmid, A. Pishevar, S. Worley, Comput Phys Comm, 184, 1119-1128 (2013).
(Phillips) C. L. Phillips, J. A. Anderson, S. C. Glotzer, Comput Phys Comm, 230, 7191-7201 (2011).
LAMMPS Users Manual
1456

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style dpd/fdt command
pair_style dpd/fdt/energy command
Syntax:
pair_style style args
style = dpd/fdt or dpd/fdt/energy• args = list of arguments for a particular style•
dpd/fdt args = T cutoff seed
T = temperature (temperature units)
cutoff = global cutoff for DPD interactions (distance units)
seed = random # seed (positive integer)
dpd/fdt/energy args = cutoff seed
cutoff = global cutoff for DPD interactions (distance units)
seed = random # seed (positive integer)
Examples:
pair_style dpd/fdt 300.0 2.5 34387
pair_coeff * * 3.0 1.0 2.5
pair_style dpd/fdt/energy 2.5 34387
pair_coeff * * 3.0 1.0 0.1 2.5
Description:
Styles dpd/fdt and dpd/fdt/energy compute the force for dissipative particle dynamics (DPD) simulations. The
dpd/fdt style is used to perform DPD simulations under isothermal and isobaric conditions, while the
dpd/fdt/energy style is used to perform DPD simulations under isoenergetic and isoenthalpic conditions (see
(Lisal)). For DPD simulations in general, the force on atom I due to atom J is given as a sum of 3 terms
where Fc is a conservative force, Fd is a dissipative force, and Fr is a random force. Rij is a unit vector in the
direction Ri - Rj, Vij is the vector difference in velocities of the two atoms = Vi - Vj, alpha is a Gaussian
random number with zero mean and unit variance, dt is the timestep size, and w(r) is a weighting factor that
varies between 0 and 1. Rc is the cutoff. The weighting factor, omega_ij, varies between 0 and 1, and is
LAMMPS Users Manual
1457

chosen to have the following functional form:
Note that alternative definitions of the weighting function exist, but would have to be implemented as a
separate pair style command.
For style dpd/fdt, the fluctuation-dissipation theorem defines gamma to be set equal to sigma*sigma/(2 T),
where T is the set point temperature specified as a pair style parameter in the above examples. The following
coefficients must be defined for each pair of atoms types via the pair_coeff command as in the examples
above, or in the data file or restart files read by the read_data or read_restart commands:
A (force units)• sigma (force*time^(1/2) units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global DPD cutoff is used.
Style dpd/fdt/energy is used to perform DPD simulations under isoenergetic and isoenthalpic conditions. The
fluctuation-dissipation theorem defines gamma to be set equal to sigma*sigma/(2 dpdTheta), where dpdTheta
is the average internal temperature for the pair. The particle internal temperature is related to the particle
internal energy through a mesoparticle equation of state (see fix eos). The differential internal conductive and
mechanical energies are computed within style dpd/fdt/energy as:
where
Zeta_ij^q is a second Gaussian random number with zero mean and unit variance that is used to compute the
internal conductive energy. The fluctuation-dissipation theorem defines alpha*alpha to be set equal to
2*kB*kappa, where kappa is the mesoparticle thermal conductivity parameter. The following coefficients
must be defined for each pair of atoms types via the pair_coeff command as in the examples above, or in the
data file or restart files read by the read_data or read_restart commands:
A (force units)• sigma (force*time^(1/2) units)•
LAMMPS Users Manual
1458

kappa (energy*temperature/time units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global DPD cutoff is used.
The pairwise energy associated with styles dpd/fdt and dpd/fdt/energy is only due to the conservative force
term Fc, and is shifted to be zero at the cutoff distance Rc. The pairwise virial is calculated using only the
conservative term.
The forces computed through the dpd/fdt and dpd/fdt/energy styles can be integrated with the velocity-Verlet
integration scheme or the Shardlow splitting integration scheme described by (Lisal). In the cases when these
pair styles are combined with the fix shardlow, these pair styles differ from the other dpd styles in that the
dissipative and random forces are split from the force calculation and are not computed within the pair style.
Thus, only the conservative force is computed by the pair style, while the stochastic integration of the
dissipative and random forces are handled through the Shardlow splitting algorithm approach. The Shardlow
splitting algorithm is advantageous, especially when performing DPD under isoenergetic conditions, as it
allows significantly larger timesteps to be taken.
Restrictions:
These commands are part of the USER-DPD package. They are only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Pair styles dpd/fdt and dpd/fdt/energy require use of the comm_modify vel yes option so that velocites are
stored by ghost atoms.
Pair style dpd/fdt/energy requires atom_style dpd to be used in order to properly account for the particle
internal energies and temperatures.
Related commands:
pair_coeff, fix shardlow
Default: none
(Lisal) M. Lisal, J.K. Brennan, J. Bonet Avalos, "Dissipative particle dynamics at isothermal, isobaric,
isoenergetic, and isoenthalpic conditions using Shardlow-like splitting algorithms.", J. Chem. Phys., 135,
204105 (2011).
LAMMPS Users Manual
1459

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style dsmc command
Syntax:
pair_style dsmc max_cell_size seed weighting Tref Nrecompute Nsample
max_cell_size = global maximum cell size for DSMC interactions (distance units)• seed = random # seed (positive integer)• weighting = macroparticle weighting• Tref = reference temperature (temperature units)• Nrecompute = recompute v*sigma_max every this many timesteps (timesteps)• Nsample = sample this many times in recomputing v*sigma_max•
Examples:
pair_style dsmc 2.5 34387 10 1.0 100 20
pair_coeff * * 1.0
pair_coeff 1 1 1.0
Description:
Style dsmc computes collisions between pairs of particles for a direct simulation Monte Carlo (DSMC) model
following the exposition in (Bird). Each collision resets the velocities of the two particles involved. The
number of pairwise collisions for each pair or particle types and the length scale within which they occur are
determined by the parameters of the pair_style and pair_coeff commands.
Stochastic collisions are performed using the variable hard sphere (VHS) approach, with the user-defined
max_cell_size value used as the maximum DSMC cell size, and reference cross-sections for collisions given
using the pair_coeff command.
There is no pairwise energy or virial contributions associated with this pair style.
The following coefficient must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands:
sigma (area units, i.e. distance-squared)•
The global DSMC max_cell_size determines the maximum cell length used in the DSMC calculation. A
structured mesh is overlayed on the simulation box such that an integer number of cells are created in each
direction for each processor's sub-domain. Cell lengths are adjusted up to the user-specified maximum cell
size.
To perform a DSMC simulation with LAMMPS, several additional options should be set in your input script,
though LAMMPS does not check for these settings.
Since this pair style does not compute particle forces, you should use the "fix nve/noforce" time integration fix
for the DSMC particles, e.g.
fix 1 all nve/noforce
LAMMPS Users Manual
1460

This pair style assumes that all particles will communicated to neighboring processors every timestep as they
move. This makes it possible to perform all collisions between pairs of particles that are on the same
processor. To ensure this occurs, you should use these commands:
neighbor 0.0 bin
neigh_modify every 1 delay 0 check no
atom_modify sort 0 0.0
communicate single cutoff 0.0
These commands ensure that LAMMPS communicates particles to neighboring processors every timestep and
that no ghost atoms are created. The output statistics for a simulation run should indicate there are no ghost
particles or neighbors.
In order to get correct DSMC collision statistics, users should specify a Gaussian velocity distribution when
populating the simulation domain. Note that the default velocity distribution is uniform, which will not give
good DSMC collision rates. Specify "dist gaussian" when using the velocity command as in the following:
velocity all create 594.6 87287 loop geom dist gaussian
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
This pair style does not support the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file. Note that the user-specified random number seed is
stored in the restart file, so when a simulation is restarted, each processor will re-initialize its random number
generator the same way it did initially. This means the random forces will be random, but will not be the same
as they would have been if the original simulation had continued past the restart time.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This style is part of the MC package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
pair_coeff, fix nve/noforce, neigh_modify, neighbor, comm_modify
Default: none
LAMMPS Users Manual
1461
(Bird) G. A. Bird, "Molecular Gas Dynamics and the Direct Simulation of Gas Flows" (1994).
LAMMPS Users Manual
1462

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style eam command
pair_style eam/gpu command
pair_style eam/intel command
pair_style eam/kk command
pair_style eam/omp command
pair_style eam/opt command
pair_style eam/alloy command
pair_style eam/alloy/gpu command
pair_style eam/alloy/kk command
pair_style eam/alloy/omp command
pair_style eam/alloy/opt command
pair_style eam/cd command
pair_style eam/cd/omp command
pair_style eam/fs command
pair_style eam/fs/gpu command
pair_style eam/fs/kk command
pair_style eam/fs/omp command
pair_style eam/fs/opt command
Syntax:
pair_style style
style = eam or eam/alloy or eam/cd or eam/fs•
Examples:
pair_style eam
pair_coeff * * cuu3
LAMMPS Users Manual
1463
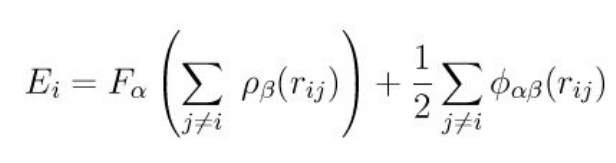
pair_coeff 1*3 1*3 niu3.eam
pair_style eam/alloy
pair_coeff * * ../potentials/NiAlH_jea.eam.alloy Ni Al Ni Ni
pair_style eam/cd
pair_coeff * * ../potentials/FeCr.cdeam Fe Cr
pair_style eam/fs
pair_coeff * * NiAlH_jea.eam.fs Ni Al Ni Ni
Description:
Style eam computes pairwise interactions for metals and metal alloys using embedded-atom method (EAM)
potentials (Daw). The total energy Ei of an atom I is given by
where F is the embedding energy which is a function of the atomic electron density rho, phi is a pair potential
interaction, and alpha and beta are the element types of atoms I and J. The multi-body nature of the EAM
potential is a result of the embedding energy term. Both summations in the formula are over all neighbors J of
atom I within the cutoff distance.
The cutoff distance and the tabulated values of the functionals F, rho, and phi are listed in one or more files
which are specified by the pair_coeff command. These are ASCII text files in a DYNAMO-style format
which is described below. DYNAMO was the original serial EAM MD code, written by the EAM originators.
Several DYNAMO potential files for different metals are included in the "potentials" directory of the
LAMMPS distribution. All of these files are parameterized in terms of LAMMPS metal units.
NOTE: The eam style reads single-element EAM potentials in the DYNAMO funcfl format. Either single
element or alloy systems can be modeled using multiple funcfl files and style eam. For the alloy case
LAMMPS mixes the single-element potentials to produce alloy potentials, the same way that DYNAMO
does. Alternatively, a single DYNAMO setfl file or Finnis/Sinclair EAM file can be used by LAMMPS to
model alloy systems by invoking the eam/alloy or eam/cd or eam/fs styles as described below. These files
require no mixing since they specify alloy interactions explicitly.
NOTE: Note that unlike for other potentials, cutoffs for EAM potentials are not set in the pair_style or
pair_coeff command; they are specified in the EAM potential files themselves. Likewise, the EAM potential
files list atomic masses; thus you do not need to use the mass command to specify them.
There are several WWW sites that distribute and document EAM potentials stored in DYNAMO or other
formats:
http://www.ctcms.nist.gov/potentials
http://cst-www.nrl.navy.mil/ccm6/ap
http://enpub.fulton.asu.edu/cms/potentials/main/main.htm
LAMMPS Users Manual
1464

These potentials should be usable with LAMMPS, though the alternate formats would need to be converted to
the DYNAMO format used by LAMMPS and described on this page. The NIST site is maintained by
Chandler Becker (cbecker at nist.gov) who is good resource for info on interatomic potentials and file formats.
For style eam, potential values are read from a file that is in the DYNAMO single-element funcfl format. If
the DYNAMO file was created by a Fortran program, it cannot have "D" values in it for exponents. C only
recognizes "e" or "E" for scientific notation.
Note that unlike for other potentials, cutoffs for EAM potentials are not set in the pair_style or pair_coeff
command; they are specified in the EAM potential files themselves.
For style eam a potential file must be assigned to each I,I pair of atom types by using one or more pair_coeff
commands, each with a single argument:
filename•
Thus the following command
pair_coeff *2 1*2 cuu3.eam
will read the cuu3 potential file and use the tabulated Cu values for F, phi, rho that it contains for type pairs
1,1 and 2,2 (type pairs 1,2 and 2,1 are ignored). See the pair_coeff doc page for alternate ways to specify the
path for the potential file. In effect, this makes atom types 1 and 2 in LAMMPS be Cu atoms. Different
single-element files can be assigned to different atom types to model an alloy system. The mixing to create
alloy potentials for type pairs with I != J is done automatically the same way that the serial DYNAMO code
originally did it; you do not need to specify coefficients for these type pairs.
Funcfl files in the potentials directory of the LAMMPS distribution have an ".eam" suffix. A DYNAMO
single-element funcfl file is formatted as follows:
line 1: comment (ignored)• line 2: atomic number, mass, lattice constant, lattice type (e.g. FCC)• line 3: Nrho, drho, Nr, dr, cutoff•
On line 2, all values but the mass are ignored by LAMMPS. The mass is in mass units, e.g. mass number or
grams/mole for metal units. The cubic lattice constant is in Angstroms. On line 3, Nrho and Nr are the number
of tabulated values in the subsequent arrays, drho and dr are the spacing in density and distance space for the
values in those arrays, and the specified cutoff becomes the pairwise cutoff used by LAMMPS for the
potential. The units of dr are Angstroms; I'm not sure of the units for drho - some measure of electron density.
Following the three header lines are three arrays of tabulated values:
embedding function F(rho) (Nrho values)• effective charge function Z(r) (Nr values)• density function rho(r) (Nr values)•
The values for each array can be listed as multiple values per line, so long as each array starts on a new line.
For example, the individual Z(r) values are for r = 0,dr,2*dr, ... (Nr-1)*dr.
The units for the embedding function F are eV. The units for the density function rho are the same as for drho
(see above, electron density). The units for the effective charge Z are "atomic charge" or sqrt(Hartree *
LAMMPS Users Manual
1465

Bohr-radii). For two interacting atoms i,j this is used by LAMMPS to compute the pair potential term in the
EAM energy expression as r*phi, in units of eV-Angstroms, via the formula
r*phi = 27.2 * 0.529 * Zi * Zj
where 1 Hartree = 27.2 eV and 1 Bohr = 0.529 Angstroms.
Style eam/alloy computes pairwise interactions using the same formula as style eam. However the associated
pair_coeff command reads a DYNAMO setfl file instead of a funcfl file. Setfl files can be used to model a
single-element or alloy system. In the alloy case, as explained above, setfl files contain explicit tabulated
values for alloy interactions. Thus they allow more generality than funcfl files for modeling alloys.
For style eam/alloy, potential values are read from a file that is in the DYNAMO multi-element setfl format,
except that element names (Ni, Cu, etc) are added to one of the lines in the file. If the DYNAMO file was
created by a Fortran program, it cannot have "D" values in it for exponents. C only recognizes "e" or "E" for
scientific notation.
Only a single pair_coeff command is used with the eam/alloy style which specifies a DYNAMO setfl file,
which contains information for M elements. These are mapped to LAMMPS atom types by specifying N
additional arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom
types:
filename• N element names = mapping of setfl elements to atom types•
As an example, the potentials/NiAlH_jea.eam.alloy file is a setfl file which has tabulated EAM values for 3
elements and their alloy interactions: Ni, Al, and H. See the pair_coeff doc page for alternate ways to specify
the path for the potential file. If your LAMMPS simulation has 4 atoms types and you want the 1st 3 to be Ni,
and the 4th to be Al, you would use the following pair_coeff command:
pair_coeff * * NiAlH_jea.eam.alloy Ni Ni Ni Al
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Ni arguments map
LAMMPS atom types 1,2,3 to the Ni element in the setfl file. The final Al argument maps LAMMPS atom
type 4 to the Al element in the setfl file. Note that there is no requirement that your simulation use all the
elements specified by the setfl file.
If a mapping value is specified as NULL, the mapping is not performed. This can be used when an eam/alloy
potential is used as part of the hybrid pair style. The NULL values are placeholders for atom types that will be
used with other potentials.
Setfl files in the potentials directory of the LAMMPS distribution have an ".eam.alloy" suffix. A DYNAMO
multi-element setfl file is formatted as follows:
lines 1,2,3 = comments (ignored)• line 4: Nelements Element1 Element2 ... ElementN• line 5: Nrho, drho, Nr, dr, cutoff•
In a DYNAMO setfl file, line 4 only lists Nelements = the # of elements in the setfl file. For LAMMPS, the
element name (Ni, Cu, etc) of each element must be added to the line, in the order the elements appear in the
file.
LAMMPS Users Manual
1466

The meaning and units of the values in line 5 is the same as for the funcfl file described above. Note that the
cutoff (in Angstroms) is a global value, valid for all pairwise interactions for all element pairings.
Following the 5 header lines are Nelements sections, one for each element, each with the following format:
line 1 = atomic number, mass, lattice constant, lattice type (e.g. FCC)• embedding function F(rho) (Nrho values)• density function rho(r) (Nr values)•
As with the funcfl files, only the mass (in mass units, e.g. mass number or grams/mole for metal units) is used
by LAMMPS from the 1st line. The cubic lattice constant is in Angstroms. The F and rho arrays are unique to
a single element and have the same format and units as in a funcfl file.
Following the Nelements sections, Nr values for each pair potential phi(r) array are listed for all i,j element
pairs in the same format as other arrays. Since these interactions are symmetric (i,j = j,i) only phi arrays with i
>= j are listed, in the following order: i,j = (1,1), (2,1), (2,2), (3,1), (3,2), (3,3), (4,1), ..., (Nelements,
Nelements). Unlike the effective charge array Z(r) in funcfl files, the tabulated values for each phi function are
listed in setfl files directly as r*phi (in units of eV-Angstroms), since they are for atom pairs.
Style eam/cd is similar to the eam/alloy style, except that it computes alloy pairwise interactions using the
concentration-dependent embedded-atom method (CD-EAM). This model can reproduce the enthalpy of
mixing of alloys over the full composition range, as described in (Stukowski).
The pair_coeff command is specified the same as for the eam/alloy style. However the DYNAMO setfl file
must has two lines added to it, at the end of the file:
line 1: Comment line (ignored)• line 2: N Coefficient0 Coefficient1 ... CoefficientN•
The last line begins with the degree N of the polynomial function h(x) that modifies the cross interaction
between A and B elements. Then N+1 coefficients for the terms of the polynomial are then listed.
Modified EAM setfl files used with the eam/cd style must contain exactly two elements, i.e. in the current
implementation the eam/cd style only supports binary alloys. The first and second elements in the input EAM
file are always taken as the A and B species.
CD-EAM files in the potentials directory of the LAMMPS distribution have a ".cdeam" suffix.
Style eam/fs computes pairwise interactions for metals and metal alloys using a generalized form of EAM
potentials due to Finnis and Sinclair (Finnis). The total energy Ei of an atom I is given by
This has the same form as the EAM formula above, except that rho is now a functional specific to the atomic
types of both atoms I and J, so that different elements can contribute differently to the total electron density at
LAMMPS Users Manual
1467

an atomic site depending on the identity of the element at that atomic site.
The associated pair_coeff command for style eam/fs reads a DYNAMO setfl file that has been extended to
include additional rho_alpha_beta arrays of tabulated values. A discussion of how FS EAM differs from
conventional EAM alloy potentials is given in (Ackland1). An example of such a potential is the same
author's Fe-P FS potential (Ackland2). Note that while FS potentials always specify the embedding energy
with a square root dependence on the total density, the implementation in LAMMPS does not require that; the
user can tabulate any functional form desired in the FS potential files.
For style eam/fs, the form of the pair_coeff command is exactly the same as for style eam/alloy, e.g.
pair_coeff * * NiAlH_jea.eam.fs Ni Ni Ni Al
where there are N additional arguments after the filename, where N is the number of LAMMPS atom types.
See the pair_coeff doc page for alternate ways to specify the path for the potential file. The N values
determine the mapping of LAMMPS atom types to EAM elements in the file, as described above for style
eam/alloy. As with eam/alloy, if a mapping value is NULL, the mapping is not performed. This can be used
when an eam/fs potential is used as part of the hybrid pair style. The NULL values are used as placeholders
for atom types that will be used with other potentials.
FS EAM files include more information than the DYNAMO setfl format files read by eam/alloy, in that i,j
density functionals for all pairs of elements are included as needed by the Finnis/Sinclair formulation of the
EAM.
FS EAM files in the potentials directory of the LAMMPS distribution have an ".eam.fs" suffix. They are
formatted as follows:
lines 1,2,3 = comments (ignored)• line 4: Nelements Element1 Element2 ... ElementN• line 5: Nrho, drho, Nr, dr, cutoff•
The 5-line header section is identical to an EAM setfl file.
Following the header are Nelements sections, one for each element I, each with the following format:
line 1 = atomic number, mass, lattice constant, lattice type (e.g. FCC)• embedding function F(rho) (Nrho values)• density function rho(r) for element I at element 1 (Nr values)• density function rho(r) for element I at element 2• ...• density function rho(r) for element I at element Nelement•
The units of these quantities in line 1 are the same as for setfl files. Note that the rho(r) arrays in
Finnis/Sinclair can be asymmetric (i,j != j,i) so there are Nelements^2 of them listed in the file.
Following the Nelements sections, Nr values for each pair potential phi(r) array are listed in the same manner
(r*phi, units of eV-Angstroms) as in EAM setfl files. Note that in Finnis/Sinclair, the phi(r) arrays are still
symmetric, so only phi arrays for i >= j are listed.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
LAMMPS Users Manual
1468

5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, where types I and J correspond to two different element types, mixing is
performed by LAMMPS as described above with the individual styles. You never need to specify a pair_coeff
command with I != J arguments for the eam styles.
This pair style does not support the pair_modify shift, table, and tail options.
The eam pair styles do not write their information to binary restart files, since it is stored in tabulated potential
files. Thus, you need to re-specify the pair_style and pair_coeff commands in an input script that reads a
restart file.
The eam pair styles can only be used via the pair keyword of the run_style respa command. They do not
support the inner, middle, outer keywords.
Restrictions:
All of these styles except the eam/cd style are part of the MANYBODY package. They are only enabled if
LAMMPS was built with that package. See the Making LAMMPS section for more info.
The eam/cd style is part of the USER-MISC package and also requires the MANYBODY package. It is only
enabled if LAMMPS was built with those packages. See the Making LAMMPS section for more info.
Related commands:
pair_coeff
Default: none
(Ackland1) Ackland, Condensed Matter (2005).
(Ackland2) Ackland, Mendelev, Srolovitz, Han and Barashev, Journal of Physics: Condensed Matter, 16,
S2629 (2004).
LAMMPS Users Manual
1469
(Daw) Daw, Baskes, Phys Rev Lett, 50, 1285 (1983). Daw, Baskes, Phys Rev B, 29, 6443 (1984).
(Finnis) Finnis, Sinclair, Philosophical Magazine A, 50, 45 (1984).
(Stukowski) Stukowski, Sadigh, Erhart, Caro; Modeling Simulation Materials Science & Engineering, 7,
075005 (2009).
LAMMPS Users Manual
1470

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style edip command
Syntax:
pair_style edip
pair_style edip/omp
Examples:
pair_style edip pair_coeff * * Si.edip Si
Description:
The edip style computes a 3-body EDIP potential which is popular for modeling silicon materials where it can
have advantages over other models such as the Stillinger-Weber or Tersoff potentials. In EDIP, the energy E
of a system of atoms is
where phi2 is a two-body term and phi3 is a three-body term. The summations in the formula are over all
neighbors J and K of atom I within a cutoff distance = a. Both terms depend on the local environment of atom
I through its effective coordination number defined by Z, which is unity for a cutoff distance < c and gently
goes to 0 at distance = a.
LAMMPS Users Manual
1471
Only a single pair_coeff command is used with the edip style which specifies a EDIP potential file with
parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional
arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N element names = mapping of EDIP elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, imagine a file Si.edip has EDIP values for Si.
EDIP files in the potentials directory of the LAMMPS distribution have a ".edip" suffix. Lines that are not
blank or comments (starting with #) define parameters for a triplet of elements. The parameters in a single
entry correspond to the two-body and three-body coefficients in the formula above:
element 1 (the center atom in a 3-body interaction)• element 2• element 3• A (energy units)• B (distance units)• cutoffA (distance units)• cutoffC (distance units)• alpha• beta• eta• gamma (distance units)• lambda (energy units)• mu• tho• sigma (distance units)• Q0• u1• u2• u3• u4•
The A, B, beta, sigma parameters are used only for two-body interactions. The eta, gamma, lambda, mu, Q0
and all u1 to u4 parameters are used only for three-body interactions. The alpha and cutoffC parameters are
used for the coordination environment function only.
The EDIP potential file must contain entries for all the elements listed in the pair_coeff command. It can also
contain entries for additional elements not being used in a particular simulation; LAMMPS ignores those
entries.
For a single-element simulation, only a single entry is required (e.g. SiSiSi). For a two-element simulation, the
file must contain 8 entries (for SiSiSi, SiSiC, SiCSi, SiCC, CSiSi, CSiC, CCSi, CCC), that specify EDIP
parameters for all permutations of the two elements interacting in three-body configurations. Thus for 3
elements, 27 entries would be required, etc.
At the moment, only a single element parametrization is implemented. However, the author is not aware of
other multi-element EDIP parameterization. If you know any and you are interest in that, please contact the
LAMMPS Users Manual
1472
author of the EDIP package.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This angle style can only be used if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info on packages.
This pair style requires the newton setting to be "on" for pair interactions.
The EDIP potential files provided with LAMMPS (see the potentials directory) are parameterized for metal
units. You can use the SW potential with any LAMMPS units, but you would need to create your own EDIP
potential file with coefficients listed in the appropriate units if your simulation doesn't use "metal" units.
Related commands:
pair_coeff
Default: none
(EDIP) J. F. Justo et al., Phys. Rev. B 58, 2539 (1998).
LAMMPS Users Manual
1473

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style eff/cut command
Syntax:
pair_style eff/cut cutoff keyword args ...
cutoff = global cutoff for Coulombic interactions• zero or more keyword/value pairs may be appended
keyword = limit/eradius or pressure/evirials or ecp
limit/eradius args = none
pressure/evirials args = none
ecp args = type element type element ...
type = LAMMPS atom type (1 to Ntypes)
element = element symbol (e.g. H, Si)
•
Examples:
pair_style eff/cut 39.7
pair_style eff/cut 40.0 limit/eradius
pair_style eff/cut 40.0 limit/eradius pressure/evirials
pair_style eff/cut 40.0 ecp 1 Si 3 C
pair_coeff * *
pair_coeff 2 2 20.0
pair_coeff 1 s 0.320852 2.283269 0.814857
pair_coeff 3 p 22.721015 0.728733 1.103199 17.695345 6.693621
Description:
This pair style contains a LAMMPS implementation of the electron Force Field (eFF) potential currently
under development at Caltech, as described in (Jaramillo-Botero). The eFF for Z(Su) in 2007. It has been
extended to higher Zs by using effective core potentials (ECPs) that now cover up to 2nd and 3rd row p-block
elements of the periodic table.
eFF can be viewed as an approximation to QM wave packet dynamics and Fermionic molecular dynamics,
combining the ability of electronic structure methods to describe atomic structure, bonding, and chemistry in
materials, and of plasma methods to describe nonequilibrium dynamics of large systems with a large number
of highly excited electrons. Yet, eFF relies on a simplification of the electronic wavefunction in which
electrons are described as floating Gaussian wave packets whose position and size respond to the various
dynamic forces between interacting classical nuclear particles and spherical Gaussian electron wavepackets.
The wavefunction is taken to be a Hartree product of the wave packets. To compensate for the lack of explicit
antisymmetry in the resulting wavefunction, a spin-dependent Pauli potential is included in the Hamiltonian.
Substituting this wavefunction into the time-dependent Schrodinger equation produces equations of motion
that correspond - to second order - to classical Hamiltonian relations between electron position and size, and
their conjugate momenta. The N-electron wavefunction is described as a product of one-electron Gaussian
functions, whose size is a dynamical variable and whose position is not constrained to a nuclear center. This
form allows for straightforward propagation of the wavefunction, with time, using a simple formulation from
which the equations of motion are then integrated with conventional MD algorithms. In addition to this
spin-dependent Pauli repulsion potential term between Gaussians, eFF includes the electron kinetic energy
from the Gaussians. These two terms are based on first-principles quantum mechanics. On the other hand,
nuclei are described as point charges, which interact with other nuclei and electrons through standard
LAMMPS Users Manual
1474
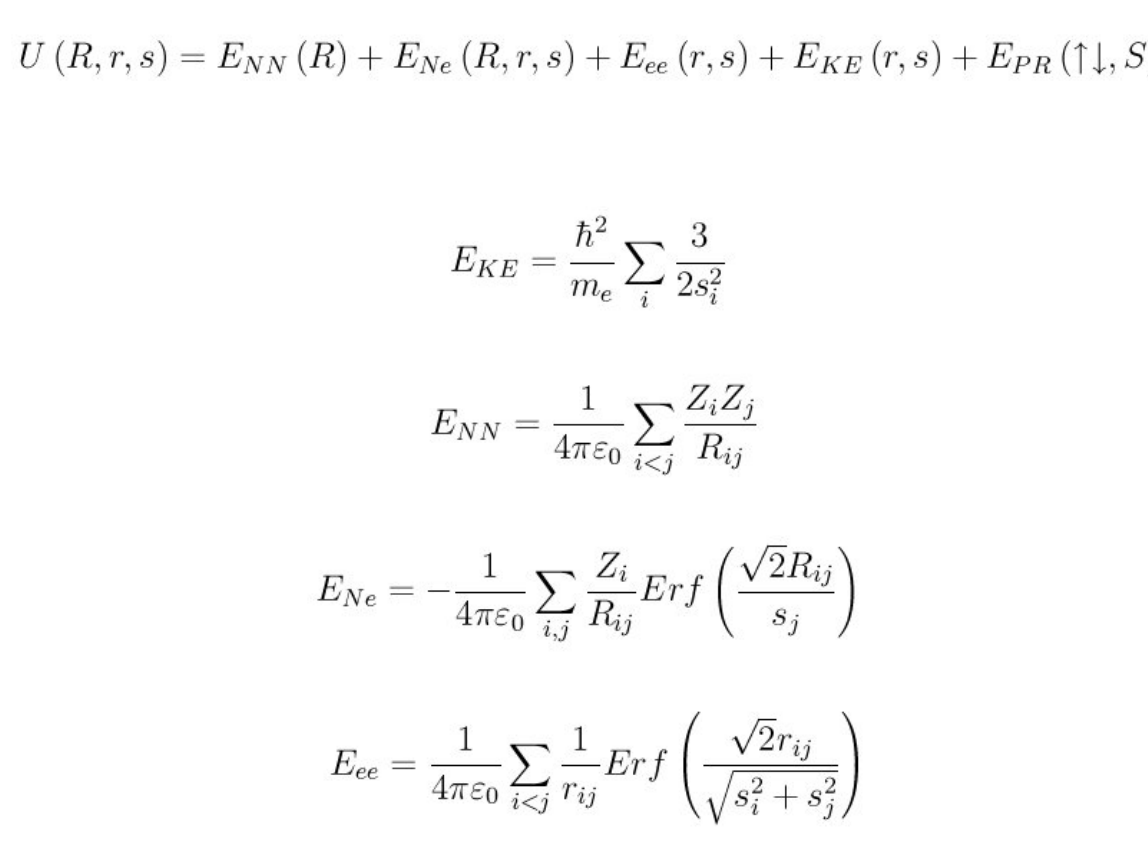
electrostatic potential forms.
The full Hamiltonian (shown below), contains then a standard description for electrostatic interactions
between a set of delocalized point and Gaussian charges which include, nuclei-nuclei (NN), electron-electron
(ee), and nuclei-electron (Ne). Thus, eFF is a mixed QM-classical mechanics method rather than a
conventional force field method (in which electron motions are averaged out into ground state nuclear
motions, i.e a single electronic state, and particle interactions are described via empirically parameterized
interatomic potential functions). This makes eFF uniquely suited to simulate materials over a wide range of
temperatures and pressures where electronically excited and ionized states of matter can occur and coexist.
Furthermore, the interactions between particles -nuclei and electrons- reduce to the sum of a set of effective
pairwise potentials in the eFF formulation. The eff/cut style computes the pairwise Coulomb interactions
between nuclei and electrons (E_NN,E_Ne,E_ee), and the quantum-derived Pauli (E_PR) and Kinetic energy
interactions potentials between electrons (E_KE) for a total energy expression given as,
The individual terms are defined as follows:
LAMMPS Users Manual
1475

where, s_i correspond to the electron sizes, the sigmas i's to the fixed spins of the electrons, Z_i to the charges
on the nuclei, R_ij to the distances between the nuclei or the nuclei and electrons, and r_ij to the distances
between electrons. For additional details see (Jaramillo-Botero).
The overall electrostatics energy is given in Hartree units of energy by default and can be modified by an
energy-conversion constant, according to the units chosen (see electron_units). The cutoff Rc, given in Bohrs
(by default), truncates the interaction distance. The recommended cutoff for this pair style should follow the
minimum image criterion, i.e. half of the minimum unit cell length.
Style eff/long (not yet available) computes the same interactions as style eff/cut except that an additional
damping factor is applied so it can be used in conjunction with the kspace_style command and its ewald or
pppm option. The Coulombic cutoff specified for this style means that pairwise interactions within this
distance are computed directly; interactions outside that distance are computed in reciprocal space.
This potential is designed to be used with atom_style electron definitions, in order to handle the description of
systems with interacting nuclei and explicit electrons.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
cutoff (distance units)•
For eff/cut, the cutoff coefficient is optional. If it is not used (as in some of the examples above), the default
global value specified in the pair_style command is used.
For eff/long (not yet available) no cutoff will be specified for an individual I,J type pair via the pair_coeff
command. All type pairs use the same global cutoff specified in the pair_style command.
The limit/eradius and pressure/evirials keywords are optional. Neither or both must be specified. If not
specified they are unset.
The limit/eradius keyword is used to restrain electron size from becoming excessively diffuse at very high
temperatures were the Gaussian wave packet representation breaks down, and from expanding as free
particles to infinite size. If unset, electron radius is free to increase without bounds. If set, a restraining
harmonic potential of the form E = 1/2k_ss^2 for s > L_box/2, where k_s = 1 Hartrees/Bohr^2, is applied on
the electron radius.
The pressure/evirials keyword is used to control between two types of pressure computation: if unset, the
computed pressure does not include the electronic radial virials contributions to the total pressure (scalar or
tensor). If set, the computed pressure will include the electronic radial virial contributions to the total pressure
(scalar and tensor).
The ecp keyword is used to associate an ECP representation for a particular atom type. The ECP captures the
orbital overlap between a core pseudo particle and valence electrons within the Pauli repulsion. A list of
LAMMPS Users Manual
1476
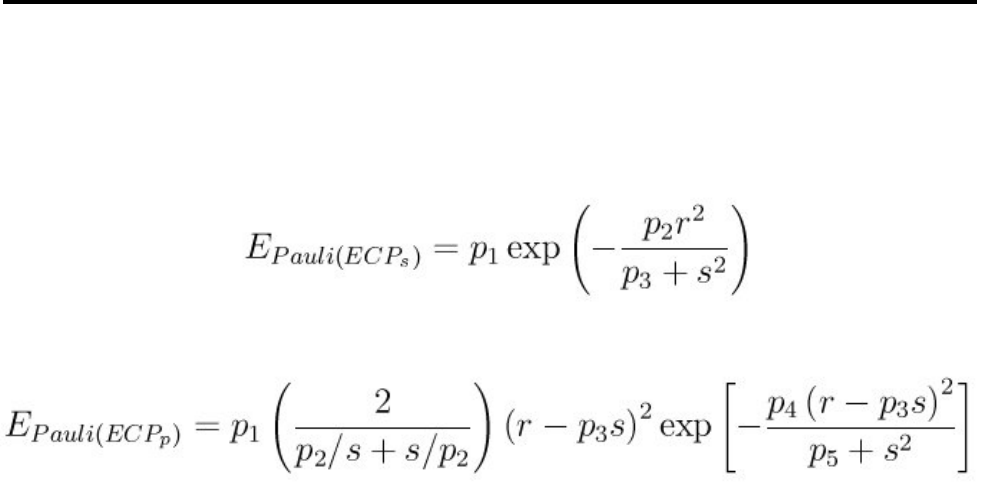
type:element-symbol pairs may be provided for all ECP representations, after the "ecp" keyword.
NOTE: Default ECP parameters are provided for C, N, O, Al, and Si. Users can modify these using the
pair_coeff command as exemplified above. For this, the User must distinguish between two different
functional forms supported, one that captures the orbital overlap assuming the s-type core interacts with an
s-like valence electron (s-s) and another that assumes the interaction is s-p. For systems that exhibit significant
p-character (e.g. C, N, O) the s-p form is recommended. The "s" ECP form requires 3 parameters and the "p"
5 parameters.
NOTE: there are two different pressures that can be reported for eFF when defining this pair_style, one
(default) that considers electrons do not contribute radial virial components (i.e. electrons treated as
incompressible 'rigid' spheres) and one that does. The radial electronic contributions to the virials are only
tallied if the flexible pressure option is set, and this will affect both global and per-atom quantities. In
principle, the true pressure of a system is somewhere in between the rigid and the flexible eFF pressures, but,
for most cases, the difference between these two pressures will not be significant over long-term averaged
runs (i.e. even though the energy partitioning changes, the total energy remains similar).
NOTE: This implementation of eFF gives a reasonably accurate description for systems containing nuclei
from Z = 1-6 in "all electron" representations. For systems with increasingly non-spherical electrons, Users
should use the ECP representations. ECPs are now supported and validated for most of the 2nd and 3rd row
elements of the p-block. Predefined parameters are provided for C, N, O, Al, and Si. The ECP captures the
orbital overlap between the core and valence electrons (i.e. Pauli repulsion) with one of the functional forms:
Where the 1st form correspond to core interactions with s-type valence electrons and the 2nd to core
interactions with p-type valence electrons.
The current version adds full support for models with fixed-core and ECP definitions. to enable larger
timesteps (i.e. by avoiding the high frequency vibrational modes -translational and radial- of the 2 s electrons),
and in the ECP case to reduce the increased orbital complexity in higher Z elements (up to Z
In general, eFF excels at computing the properties of materials in extreme conditions and tracing the system
dynamics over multi-picosend timescales; this is particularly relevant where electron excitations can change
significantly the nature of bonding in the system. It can capture with surprising accuracy the behavior of such
systems because it describes consistently and in an unbiased manner many different kinds of bonds, including
covalent, ionic, multicenter, ionic, and plasma, and how they interconvert and/or change when they become
excited. eFF also excels in computing the relative thermochemistry of isodemic reactions and conformational
changes, where the bonds of the reactants are of the same type as the bonds of the products. eFF assumes that
LAMMPS Users Manual
1477

kinetic energy differences dominate the overall exchange energy, which is true when the electrons present are
nearly spherical and nodeless and valid for covalent compounds such as dense hydrogen, hydrocarbons, and
diamond; alkali metals (e.g. lithium), alkali earth metals (e.g. beryllium) and semimetals such as boron; and
various compounds containing ionic and/or multicenter bonds, such as boron dihydride.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the cutoff distance for the eff/cut style can be mixed. The default mix value
is geometric. See the "pair_modify" command for details.
The pair_modify shift option is not relevant for these pair styles.
The eff/long (not yet available) style supports the pair_modify table option for tabulation of the short-range
portion of the long-range Coulombic interaction.
These pair styles do not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
These pair styles write their information to binary restart files, so pair_style and pair_coeff commands do not
need to be specified in an input script that reads a restart file.
These pair styles can only be used via the pair keyword of the run_style respa command. They do not support
the inner, middle, outer keywords.
Restrictions:
These pair styles will only be enabled if LAMMPS is built with the USER-EFF package. It will only be
enabled if LAMMPS was built with that package. See the Making LAMMPS section for more info.
These pair styles require that particles store electron attributes such as radius, radial velocity, and radial force,
as defined by the atom_style. The electron atom style does all of this.
Thes pair styles require you to use the comm_modify vel yes command so that velocites are stored by ghost
atoms.
Related commands:
pair_coeff
Default:
If not specified, limit_eradius = 0 and pressure_with_evirials = 0.
(Su) Su and Goddard, Excited Electron Dynamics Modeling of Warm Dense Matter, Phys Rev Lett,
99:185003 (2007).
(Jaramillo-Botero) Jaramillo-Botero, Su, Qi, Goddard, Large-scale, Long-term Non-adiabatic Electron
Molecular Dynamics for Describing Material Properties and Phenomena in Extreme Environments, J Comp
LAMMPS Users Manual
1478
Chem, 32, 497-512 (2011).
LAMMPS Users Manual
1479

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style eim command
pair_style eim/omp command
Syntax:
pair_style style
style = eim•
Examples:
pair_style eim
pair_coeff * * Na Cl ../potentials/ffield.eim Na Cl
pair_coeff * * Na Cl ffield.eim Na Na Na Cl
pair_coeff * * Na Cl ../potentials/ffield.eim Cl NULL Na
Description:
Style eim computes pairwise interactions for ionic compounds using embedded-ion method (EIM) potentials
(Zhou). The energy of the system E is given by
The first term is a double pairwise sum over the J neighbors of all I atoms, where phi_ij is a pair potential. The
second term sums over the embedding energy E_i of atom I, which is a function of its charge q_i and the
electrical potential sigma_i at its location. E_i, q_i, and sigma_i are calculated as
where eta_ji is a pairwise function describing electron flow from atom I to atom J, and psi_ij is another
pairwise function. The multi-body nature of the EIM potential is a result of the embedding energy term. A
LAMMPS Users Manual
1480

complete list of all the pair functions used in EIM is summarized below
Here E_b, r_e, r_(c,phi), alpha, beta, A_(psi), zeta, r_(s,psi), r_(c,psi), A_(eta), r_(s,eta), r_(c,eta), chi, and
pair function type p are parameters, with subscripts ij indicating the two species of atoms in the atomic pair.
NOTE: Even though the EIM potential is treating atoms as charged ions, you should not use a LAMMPS
atom_style that stores a charge on each atom and thus requires you to assign a charge to each atom, e.g. the
charge or full atom styles. This is because the EIM potential infers the charge on an atom from the equation
above for q_i; you do not assign charges explicitly.
All the EIM parameters are listed in a potential file which is specified by the pair_coeff command. This is an
ASCII text file in a format described below. The "ffield.eim" file included in the "potentials" directory of the
LAMMPS distribution currently includes nine elements Li, Na, K, Rb, Cs, F, Cl, Br, and I. A system with any
combination of these elements can be modeled. This file is parameterized in terms of LAMMPS metal units.
Note that unlike other potentials, cutoffs for EIM potentials are not set in the pair_style or pair_coeff
command; they are specified in the EIM potential file itself. Likewise, the EIM potential file lists atomic
masses; thus you do not need to use the mass command to specify them.
Only a single pair_coeff command is used with the eim style which specifies an EIM potential file and the
element(s) to extract information for. The EIM elements are mapped to LAMMPS atom types by specifying N
additional arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom
types:
Elem1, Elem2, ...• EIM potential file• N element names = mapping of EIM elements to atom types•
LAMMPS Users Manual
1481

See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example like one of those above, suppose you want to model a system with Na and Cl atoms. If your
LAMMPS simulation has 4 atoms types and you want the 1st 3 to be Na, and the 4th to be Cl, you would use
the following pair_coeff command:
pair_coeff * * Na Cl ffield.eim Na Na Na Cl
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The filename is the EIM potential
file. The Na and Cl arguments (before the file name) are the two elements for which info will be extracted
from the potential file. The first three trailing Na arguments map LAMMPS atom types 1,2,3 to the EIM Na
element. The final Cl argument maps LAMMPS atom type 4 to the EIM Cl element.
If a mapping value is specified as NULL, the mapping is not performed. This can be used when an eim
potential is used as part of the hybrid pair style. The NULL values are placeholders for atom types that will be
used with other potentials.
The ffield.eim file in the potentials directory of the LAMMPS distribution is formatted as follows:
Lines starting with # are comments and are ignored by LAMMPS. Lines starting with "global:" include three
global values. The first value divides the cations from anions, i.e., any elements with electronegativity above
this value are viewed as anions, and any elements with electronegativity below this value are viewed as
cations. The second and third values are related to the cutoff function - i.e. the 0.510204, 1.64498, and
0.010204 shown in the above equation can be derived from these values.
Lines starting with "element:" are formatted as follows: name of element, atomic number, atomic mass,
electronic negativity, atomic radius (LAMMPS ignores it), ionic radius (LAMMPS ignores it), cohesive
energy (LAMMPS ignores it), and q0 (must be 0).
Lines starting with "pair:" are entered as: element 1, element 2, r_(c,phi), r_(c,phi) (redundant for historical
reasons), E_b, r_e, alpha, beta, r_(c,eta), A_(eta), r_(s,eta), r_(c,psi), A_(psi), zeta, r_(s,psi), and p.
The lines in the file can be in any order; LAMMPS extracts the info it needs.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
1482


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style exp6/rx command
Syntax:
pair_style exp6/rx cutoff ...
cutoff = global cutoff for DPD interactions (distance units)• weighting = fractional or molecular (optional)•
Examples:
pair_style exp6/rx 10.0
pair_style exp6/rx 10.0 fractional
pair_style exp6/rx 10.0 molecular
pair_coeff * * exp6.params h2o h2o exponent 1.0 1.0 10.0
pair_coeff * * exp6.params h2o 1fluid exponent 1.0 1.0 10.0
pair_coeff * * exp6.params 1fluid 1fluid exponent 1.0 1.0 10.0
pair_coeff * * exp6.params 1fluid 1fluid none 10.0
pair_coeff * * exp6.params 1fluid 1fluid polynomial filename 10.0
Description:
Style exp6/rx is used in reaction DPD simulations, where the coarse-grained (CG) particles are composed of m
species whose reaction rate kinetics are determined from a set of n reaction rate equations through the fix rx
command. The species of one CG particle can interact with a species in a neighboring CG particle through a
site-site interaction potential model. The exp6/rx style computes an exponential-6 potential given by
where the epsilon parameter determines the depth of the potential minimum located at Rm, and alpha
determines the softness of the repulsion.
The coefficients must be defined for each species in a given particle type via the pair_coeff command as in the
examples above, where the first argument is the filename that includes the exponential-6 parameters for each
species. The file includes the species tag followed by the alpha, epsilon and Rm parameters. The format of the
file is described below.
The second and third arguments specify the site-site interaction potential between two species contained
within two different particles. The species tags must either correspond to the species defined in the reaction
kinetics files specified with the fix rx command or they must correspond to the tag "1fluid", signifying
interaction with a product species mixture determined through a one-fluid approximation. The interaction
potential is weighted by the geometric average of either the mole fraction concentrations or the number of
molecules associated with the interacting coarse-grained particles (see the fractional or molecular weighting
pair style options). The coarse-grained potential is stored before and after the reaction kinetics solver is
applied, where the difference is defined to be the internal chemical energy (uChem).
The fourth argument specifies the type of scaling that will be used to scale the EXP-6 parameters as reactions
occur. Currently, there are three scaling options: exponent, polynomial and none.
LAMMPS Users Manual
1484

Exponent scaling requires two additional arguments for scaling the Rm and epsilon parameters, respectively.
The scaling factor is computed by phi^exponent, where phi is the number of molecules represented by the
coarse-grain particle and exponent is specified as a pair coefficient argument for Rm and epsilon, respectively.
The Rm and epsilon parameters are multiplied by the scaling factor to give the scaled interaction parameters
for the CG particle.
Polynomial scaling requires a filename to be specified as a pair coeff argument. The file contains the
coefficients to a fifth order polynomial for the alpha, epsilon and Rm parameters that depend upon phi (the
number of molecules represented by the CG particle). The format of a polynomial file is provided below.
The none option to the scaling does not have any additional pair coeff arguments. This is equivalent to
specifying the exponent option with Rm and epsilon exponents of 0.0 and 0.0, respectively.
The final argument specifies the interaction cutoff (optional).
The format of a tabulated file is as follows (without the parenthesized comments):
# exponential-6 parameters for various species (one or more comment or blank lines)
h2o exp6 11.00 0.02 3.50 (species, exp6, alpha, Rm, epsilon)
no2 exp6 13.60 0.01 3.70
...
co2 exp6 13.00 0.03 3.20
The format of the polynomial scaling file as follows (without the parenthesized comments):
# POLYNOMIAL FILE (one or more comment or blank lines)
# General Functional Form:
# A*phi^5 + B*phi^4 + C*phi^3 + D*phi^2 + E*phi + F
#
# Parameter A B C D E F
(blank)
alpha 0.0000 0.00000 0.00008 0.04955 -0.73804 13.63201
epsilon 0.0000 0.00478 -0.06283 0.24486 -0.33737 2.60097
rm 0.0001 -0.00118 -0.00253 0.05812 -0.00509 1.50106
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections.
Following a blank line, the next N lines list the species and their corresponding parameters. The first argument
is the species tag, the second argument is the exp6 tag, the 3rd argument is the alpha parameter (energy units),
the 4th argument is the epsilon parameter (energy-distance^6 units), and the 5th argument is the Rm parameter
(distance units). If a species tag of "1fluid" is listed as a pair coefficient, a one-fluid approximation is
specified where a concentration-dependent combination of the parameters is computed through the following
equations:
LAMMPS Users Manual
1485

where
and xa and xb are the mole fractions of a and b, respectively, which comprise the gas mixture.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
This style does not support the pair_modify shift option for the energy of the exp() and 1/r^6 portion of the
pair interaction.
This style does not support the pair_modify tail option for adding long-range tail corrections to energy and
pressure for the A,C terms in the pair interaction.
Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
pair_coeff
Default: fractional weighting
LAMMPS Users Manual
1486

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style gauss command
pair_style gauss/gpu command
pair_style gauss/omp command
pair_style gauss/cut command
pair_style gauss/cut/omp command
Syntax:
pair_style gauss cutoff
pair_style gauss/cut cutoff
cutoff = global cutoff for Gauss interactions (distance units)•
Examples:
pair_style gauss 12.0
pair_coeff * * 1.0 0.9
pair_coeff 1 4 1.0 0.9 10.0
pair_style gauss/cut 3.5
pair_coeff 1 4 0.2805 1.45 0.112
Description:
Style gauss computes a tethering potential of the form
between an atom and its corresponding tether site which will typically be a frozen atom in the simulation. Rc
is the cutoff.
The following coefficients must be defined for each pair of atom types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands:
A (energy units)• B (1/distance^2 units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global cutoff is used.
Style gauss/cut computes a generalized Gaussian interaction potential between pairs of particles:
LAMMPS Users Manual
1487

where H determines together with the standard deviation sigma_h the peak height of the Gaussian function,
and r_mh the peak position. Examples of the use of the Gaussian potentials include implicit solvent
simulations of salt ions (Lenart) and of surfactants (Jusufi). In these instances the Gaussian potential mimics
the hydration barrier between a pair of particles. The hydration barrier is located at r_mh and has a width of
sigma_h. The prefactor determines the height of the potential barrier.
The following coefficients must be defined for each pair of atom types via the pair_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
H (energy * distance units)• r_mh (distance units)• sigma_h (distance units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global cutoff is used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the "-suffix command-line switch7_Section_start.html#start_6 when you invoke LAMMPS, or you can use
the suffix command in your input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the A, B, H, sigma_h, r_mh parameters, and the cutoff distance for these
pair styles can be mixed: A (energy units) sqrt(1/B) (distance units, see below) H (energy units) sigma_h
(distance units) r_mh (distance units) cutoff (distance units):ul
The default mix value is geometric. Only arithmetic and geometric mix values are supported. See the
"pair_modify" command for details.
The A and H parameters are mixed using the same rules normally used to mix the "epsilon" parameter in a
Lennard Jones interaction. The sigma_h, r_mh, and the cutoff distance are mixed using the same rules used to
mix the "sigma" parameter in a Lennard Jones interaction. The B parameter is converted to a distance (sigma),
before mixing (using sigma=B^-0.5), and converted back to a coefficient afterwards (using B=sigma^2).
LAMMPS Users Manual
1488

Negative A values are converted to positive A values (using abs(A)) before mixing, and converted back after
mixing (by multiplying by sign(Ai)*sign(Aj)). This way, if either particle is repulsive (if Ai
The gauss style does not support the pair_modify shift option. There is no effect due to the Gaussian well
beyond the cutoff; hence reasonable cutoffs need to be specified.
The gauss/cut style supports the pair_modify shift option for the energy of the Gauss-potential portion of the
pair interaction.
The pair_modify table and tail options are not relevant for these pair styles.
These pair styles write their information to binary restart files, so pair_style and pair_coeff commands do not
need to be specified in an input script that reads a restart file.
These pair styles can only be used via the pair keyword of the run_style respa command. They do not support
the inner, middle, outer keywords.
The gauss pair style tallies an "occupancy" count of how many Gaussian-well sites have an atom within the
distance at which the force is a maximum = sqrt(0.5/b). This quantity can be accessed via the compute pair
command as a vector of values of length 1.
To print this quantity to the log file (with a descriptive column heading) the following commands could be
included in an input script:
compute gauss all pair gauss
variable occ equal c_gauss[1]
thermo_style custom step temp epair v_occ
Restrictions:
The gauss/cut style is part of the "user-misc" package. It is only enabled if LAMMPS is build with that
package. See the Making of LAMMPS section for more info.
Related commands:
pair_coeff, pair_style coul/diel
Default: none
(Lenart) Lenart , Jusufi, and Panagiotopoulos, J Chem Phys, 126, 044509 (2007).
(Jusufi) Jusufi, Hynninen, and Panagiotopoulos, J Phys Chem B, 112, 13783 (2008).
LAMMPS Users Manual
1489

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style gayberne command
pair_style gayberne/gpu command
pair_style gayberne/intel command
pair_style gayberne/omp command
Syntax:
pair_style gayberne gamma upsilon mu cutoff
gamma = shift for potential minimum (typically 1)• upsilon = exponent for eta orientation-dependent energy function• mu = exponent for chi orientation-dependent energy function• cutoff = global cutoff for interactions (distance units)•
Examples:
pair_style gayberne 1.0 1.0 1.0 10.0
pair_coeff * * 1.0 1.7 1.7 3.4 3.4 1.0 1.0 1.0
Description:
The gayberne styles compute a Gay-Berne anisotropic LJ interaction (Berardi) between pairs of ellipsoidal
particles or an ellipsoidal and spherical particle via the formulas
where A1 and A2 are the transformation matrices from the simulation box frame to the body frame and r12 is
the center to center vector between the particles. Ur controls the shifted distance dependent interaction based
on the distance of closest approach of the two particles (h12) and the user-specified shift parameter gamma.
When both particles are spherical, the formula reduces to the usual Lennard-Jones interaction (see details
below for when Gay-Berne treats a particle as "spherical").
LAMMPS Users Manual
1490
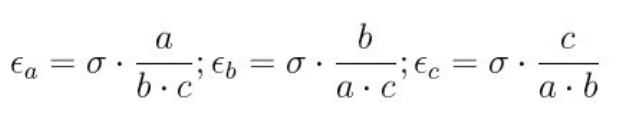
For large uniform molecules it has been shown that the energy parameters are approximately representable in
terms of local contact curvatures (Everaers):
The variable names utilized as potential parameters are for the most part taken from (Everaers) in order to be
consistent with the RE-squared pair potential. Details on the upsilon and mu parameters are given here.
More details of the Gay-Berne formulation are given in the references listed below and in this supplementary
document.
Use of this pair style requires the NVE, NVT, or NPT fixes with the asphere extension (e.g. fix nve/asphere)
in order to integrate particle rotation. Additionally, atom_style ellipsoid should be used since it defines the
rotational state and the size and shape of each ellipsoidal particle.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon = well depth (energy units)• sigma = minimum effective particle radii (distance units)• epsilon_i_a = relative well depth of type I for side-to-side interactions• epsilon_i_b = relative well depth of type I for face-to-face interactions• epsilon_i_c = relative well depth of type I for end-to-end interactions• epsilon_j_a = relative well depth of type J for side-to-side interactions• epsilon_j_b = relative well depth of type J for face-to-face interactions• epsilon_j_c = relative well depth of type J for end-to-end interactions• cutoff (distance units)•
The last coefficient is optional. If not specified, the global cutoff specified in the pair_style command is used.
It is typical with the Gay-Berne potential to define sigma as the minimum of the 3 shape diameters of the
particles involved in an I,I interaction, though this is not required. Note that this is a different meaning for
sigma than the pair_style resquared potential uses.
The epsilon_i and epsilon_j coefficients are actually defined for atom types, not for pairs of atom types. Thus,
in a series of pair_coeff commands, they only need to be specified once for each atom type.
Specifically, if any of epsilon_i_a, epsilon_i_b, epsilon_i_c are non-zero, the three values are assigned to
atom type I. If all the epsilon_i values are zero, they are ignored. If any of epsilon_j_a, epsilon_j_b,
epsilon_j_c are non-zero, the three values are assigned to atom type J. If all three epsilon_j values are zero,
they are ignored. Thus the typical way to define the epsilon_i and epsilon_j coefficients is to list their values
in "pair_coeff I J" commands when I = J, but set them to 0.0 when I != J. If you do list them when I != J, you
should insure they are consistent with their values in other pair_coeff commands, since only the last setting
will be in effect.
LAMMPS Users Manual
1491

Note that if this potential is being used as a sub-style of pair_style hybrid, and there is no "pair_coeff I I"
setting made for Gay-Berne for a particular type I (because I-I interactions are computed by another hybrid
pair potential), then you still need to insure the epsilon a,b,c coefficients are assigned to that type. e.g. in a
"pair_coeff I J" command.
NOTE: If the epsilon a = b = c for an atom type, and if the shape of the particle itself is spherical, meaning its
3 shape parameters are all the same, then the particle is treated as an LJ sphere by the Gay-Berne potential.
This is significant because if two LJ spheres interact, then the simple Lennard-Jones formula is used to
compute their interaction energy/force using the specified epsilon and sigma as the standard LJ parameters.
This is much cheaper to compute than the full Gay-Berne formula. To treat the particle as a LJ sphere with
sigma = D, you should normally set epsilon a = b = c = 1.0, set the pair_coeff sigma = D, and also set the 3
shape parameters for the particle to D. The one exception is that if the 3 shape parameters are set to 0.0, which
is a valid way in LAMMPS to specify a point particle, then the Gay-Berne potential will treat that as shape
parameters of 1.0 (i.e. a LJ particle with sigma = 1), since it requires finite-size particles. In this case you
should still set the pair_coeff sigma to 1.0 as well.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for this pair style can
be mixed. The default mix value is geometric. See the "pair_modify" command for details.
This pair styles supports the pair_modify shift option for the energy of the Lennard-Jones portion of the pair
interaction, but only for sphere-sphere interactions. There is no shifting performed for ellipsoidal interactions
due to the anisotropic dependence of the interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
LAMMPS Users Manual
1492
Restrictions:
The gayberne style is part of the ASPHERE package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
These pair style require that atoms store torque and a quaternion to represent their orientation, as defined by
the atom_style. It also require they store a per-type shape. The particles cannot store a per-particle diameter.
This pair style requires that atoms be ellipsoids as defined by the atom_style ellipsoid command.
Particles acted on by the potential can be finite-size aspherical or spherical particles, or point particles.
Spherical particles have all 3 of their shape parameters equal to each other. Point particles have all 3 of their
shape parameters equal to 0.0.
The Gay-Berne potential does not become isotropic as r increases (Everaers). The
distance-of-closest-approach approximation used by LAMMPS becomes less accurate when high-aspect ratio
ellipsoids are used.
Related commands:
pair_coeff, fix nve/asphere, compute temp/asphere, pair_style resquared
Default: none
(Everaers) Everaers and Ejtehadi, Phys Rev E, 67, 041710 (2003).
(Berardi) Berardi, Fava, Zannoni, Chem Phys Lett, 297, 8-14 (1998). Berardi, Muccioli, Zannoni, J Chem
Phys, 128, 024905 (2008).
(Perram) Perram and Rasmussen, Phys Rev E, 54, 6565-6572 (1996).
(Allen) Allen and Germano, Mol Phys 104, 3225-3235 (2006).
LAMMPS Users Manual
1493

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style gran/hooke command
pair_style gran/omp command
pair_style gran/hooke/history command
pair_style gran/hooke/history/omp command
pair_style gran/hertz/history command
pair_style gran/hertz/history/omp command
Syntax:
pair_style style Kn Kt gamma_n gamma_t xmu dampflag
style = gran/hooke or gran/hooke/history or gran/hertz/history• Kn = elastic constant for normal particle repulsion (force/distance units or pressure units - see
discussion below)
•
Kt = elastic constant for tangential contact (force/distance units or pressure units - see discussion
below)
•
gamma_n = damping coefficient for collisions in normal direction (1/time units or 1/time-distance
units - see discussion below)
•
gamma_t = damping coefficient for collisions in tangential direction (1/time units or 1/time-distance
units - see discussion below)
•
xmu = static yield criterion (unitless value between 0.0 and 1.0e4)• dampflag = 0 or 1 if tangential damping force is excluded or included•
NOTE: Versions of LAMMPS before 9Jan09 had different style names for granular force fields. This is to
emphasize the fact that the Hertzian equation has changed to model polydispersity more accurately. A side
effect of the change is that the Kn, Kt, gamma_n, and gamma_t coefficients in the pair_style command must
be specified with different values in order to reproduce calculations made with earlier versions of LAMMPS,
even for monodisperse systems. See the NOTE below for details.
Examples:
pair_style gran/hooke/history 200000.0 NULL 50.0 NULL 0.5 1
pair_style gran/hooke 200000.0 70000.0 50.0 30.0 0.5 0
Description:
The gran styles use the following formulas for the frictional force between two granular particles, as
described in (Brilliantov), (Silbert), and (Zhang), when the distance r between two particles of radii Ri and Rj
is less than their contact distance d = Ri + Rj. There is no force between the particles when r > d.
The two Hookean styles use this formula:
LAMMPS Users Manual
1494
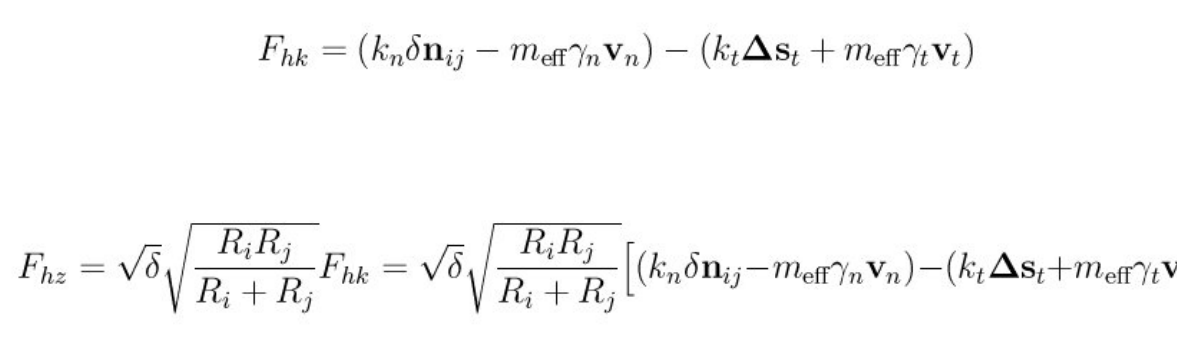
The Hertzian style uses this formula:
In both equations the first parenthesized term is the normal force between the two particles and the second
parenthesized term is the tangential force. The normal force has 2 terms, a contact force and a damping force.
The tangential force also has 2 terms: a shear force and a damping force. The shear force is a "history" effect
that accounts for the tangential displacement between the particles for the duration of the time they are in
contact. This term is included in pair styles hooke/history and hertz/history, but is not included in pair style
hooke. The tangential damping force term is included in all three pair styles if dampflag is set to 1; it is not
included if dampflag is set to 0.
The other quantities in the equations are as follows:
delta = d - r = overlap distance of 2 particles• Kn = elastic constant for normal contact• Kt = elastic constant for tangential contact• gamma_n = viscoelastic damping constant for normal contact• gamma_t = viscoelastic damping constant for tangential contact• m_eff = Mi Mj / (Mi + Mj) = effective mass of 2 particles of mass Mi and Mj• Delta St = tangential displacement vector between 2 particles which is truncated to satisfy a frictional
yield criterion
•
n_ij = unit vector along the line connecting the centers of the 2 particles• Vn = normal component of the relative velocity of the 2 particles• Vt = tangential component of the relative velocity of the 2 particles•
The Kn, Kt, gamma_n, and gamma_t coefficients are specified as parameters to the pair_style command. If a
NULL is used for Kt, then a default value is used where Kt = 2/7 Kn. If a NULL is used for gamma_t, then a
default value is used where gamma_t = 1/2 gamma_n.
The interpretation and units for these 4 coefficients are different in the Hookean versus Hertzian equations.
The Hookean model is one where the normal push-back force for two overlapping particles is a linear function
of the overlap distance. Thus the specified Kn is in units of (force/distance). Note that this push-back force is
independent of absolute particle size (in the monodisperse case) and of the relative sizes of the two particles
(in the polydisperse case). This model also applies to the other terms in the force equation so that the specified
gamma_n is in units of (1/time), Kt is in units of (force/distance), and gamma_t is in units of (1/time).
The Hertzian model is one where the normal push-back force for two overlapping particles is proportional to
the area of overlap of the two particles, and is thus a non-linear function of overlap distance. Thus Kn has
units of force per area and is thus specified in units of (pressure). The effects of absolute particle size
LAMMPS Users Manual
1495

(monodispersity) and relative size (polydispersity) are captured in the radii-dependent pre-factors. When these
pre-factors are carried through to the other terms in the force equation it means that the specified gamma_n is
in units of (1/(time*distance)), Kt is in units of (pressure), and gamma_t is in units of (1/(time*distance)).
Note that in the Hookean case, Kn can be thought of as a linear spring constant with units of force/distance. In
the Hertzian case, Kn is like a non-linear spring constant with units of force/area or pressure, and as shown in
the (Zhang) paper, Kn = 4G / (3(1-nu)) where nu = the Poisson ratio, G = shear modulus = E / (2(1+nu)), and
E = Young's modulus. Similarly, Kt = 4G / (2-nu). (NOTE: in an earlier version of the manual, we incorrectly
stated that Kt = 8G / (2-nu).)
Thus in the Hertzian case Kn and Kt can be set to values that corresponds to properties of the material being
modeled. This is also true in the Hookean case, except that a spring constant must be chosen that is
appropriate for the absolute size of particles in the model. Since relative particle sizes are not accounted for,
the Hookean styles may not be a suitable model for polydisperse systems.
NOTE: In versions of LAMMPS before 9Jan09, the equation for Hertzian interactions did not include the
sqrt(RiRj/Ri+Rj) term and thus was not as accurate for polydisperse systems. For monodisperse systems,
sqrt(RiRj/Ri+Rj) is a constant factor that effectively scales all 4 coefficients: Kn, Kt, gamma_n, gamma_t.
Thus you can set the values of these 4 coefficients appropriately in the current code to reproduce the results of
a previous Hertzian monodisperse calculation. For example, for the common case of a monodisperse system
with particles of diameter 1, all 4 of these coefficients should now be set 2x larger than they were previously.
Xmu is also specified in the pair_style command and is the upper limit of the tangential force through the
Coulomb criterion Ft = xmu*Fn, where Ft and Fn are the total tangential and normal force components in the
formulas above. Thus in the Hookean case, the tangential force between 2 particles grows according to a
tangential spring and dash-pot model until Ft/Fn = xmu and is then held at Ft = Fn*xmu until the particles lose
contact. In the Hertzian case, a similar analogy holds, though the spring is no longer linear.
NOTE: Normally, xmu should be specified as a fractional value between 0.0 and 1.0, however LAMMPS
allows large values (up to 1.0e4) to allow for modeling of systems which can sustain very large tangential
forces.
The effective mass m_eff is given by the formula above for two isolated particles. If either particle is part of a
rigid body, its mass is replaced by the mass of the rigid body in the formula above. This is determined by
searching for a fix rigid command (or its variants).
For granular styles there are no additional coefficients to set for each pair of atom types via the pair_coeff
command. All settings are global and are made via the pair_style command. However you must still use the
pair_coeff for all pairs of granular atom types. For example the command
pair_coeff * *
should be used if all atoms in the simulation interact via a granular potential (i.e. one of the pair styles above
is used). If a granular potential is used as a sub-style of pair_style hybrid, then specific atom types can be used
in the pair_coeff command to determine which atoms interact via a granular potential.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
LAMMPS Users Manual
1496

These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
The pair_modify mix, shift, table, and tail options are not relevant for granular pair styles.
These pair styles write their information to binary restart files, so a pair_style command does not need to be
specified in an input script that reads a restart file.
These pair styles can only be used via the pair keyword of the run_style respa command. They do not support
the inner, middle, outer keywords.
The single() function of these pair styles returns 0.0 for the energy of a pairwise interaction, since energy is
not conserved in these dissipative potentials. It also returns only the normal component of the pairwise
interaction force. However, the single() function also calculates 10 extra pairwise quantities. The first 3 are
the components of the tangential force between particles I and J, acting on particle I. The 4th is the magnitude
of this tangential force. The next 3 (5-7) are the components of the relative velocity in the normal direction
(along the line joining the 2 sphere centers). The last 3 (8-10) the components of the relative velocity in the
tangential direction.
These extra quantities can be accessed by the compute pair/local command, as p1, p2, ..., p10.
Restrictions:
All the granular pair styles are part of the GRANULAR package. It is only enabled if LAMMPS was built
with that package. See the Making LAMMPS section for more info.
These pair styles require that atoms store torque and angular velocity (omega) as defined by the atom_style.
They also require a per-particle radius is stored. The sphere atom style does all of this.
This pair style requires you to use the comm_modify vel yes command so that velocites are stored by ghost
atoms.
These pair styles will not restart exactly when using the read_restart command, though they should provide
statistically similar results. This is because the forces they compute depend on atom velocities. See the
read_restart command for more details.
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1497

(Brilliantov) Brilliantov, Spahn, Hertzsch, Poschel, Phys Rev E, 53, p 5382-5392 (1996).
(Silbert) Silbert, Ertas, Grest, Halsey, Levine, Plimpton, Phys Rev E, 64, p 051302 (2001).
(Zhang) Zhang and Makse, Phys Rev E, 72, p 011301 (2005).
LAMMPS Users Manual
1498

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/gromacs command
pair_style lj/gromacs/gpu command
pair_style lj/gromacs/omp command
pair_style lj/gromacs/coul/gromacs command
pair_style lj/gromacs/coul/gromacs/omp command
Syntax:
pair_style style args
style = lj/gromacs or lj/gromacs/coul/gromacs• args = list of arguments for a particular style•
lj/gromacs args = inner outer
inner, outer = global switching cutoffs for Lennard Jones
lj/gromacs/coul/gromacs args = inner outer (inner2) (outer2)
inner, outer = global switching cutoffs for Lennard Jones (and Coulombic if only 2 args)
inner2, outer2 = global switching cutoffs for Coulombic (optional)
Examples:
pair_style lj/gromacs 9.0 12.0
pair_coeff * * 100.0 2.0
pair_coeff 2 2 100.0 2.0 8.0 10.0
pair_style lj/gromacs/coul/gromacs 9.0 12.0
pair_style lj/gromacs/coul/gromacs 8.0 10.0 7.0 9.0
pair_coeff * * 100.0 2.0
Description:
The lj/gromacs styles compute shifted LJ and Coulombic interactions with an additional switching function
S(r) that ramps the energy and force smoothly to zero between an inner and outer cutoff. It is a commonly
used potential in the GROMACS MD code and for the coarse-grained models of (Marrink).
LAMMPS Users Manual
1499
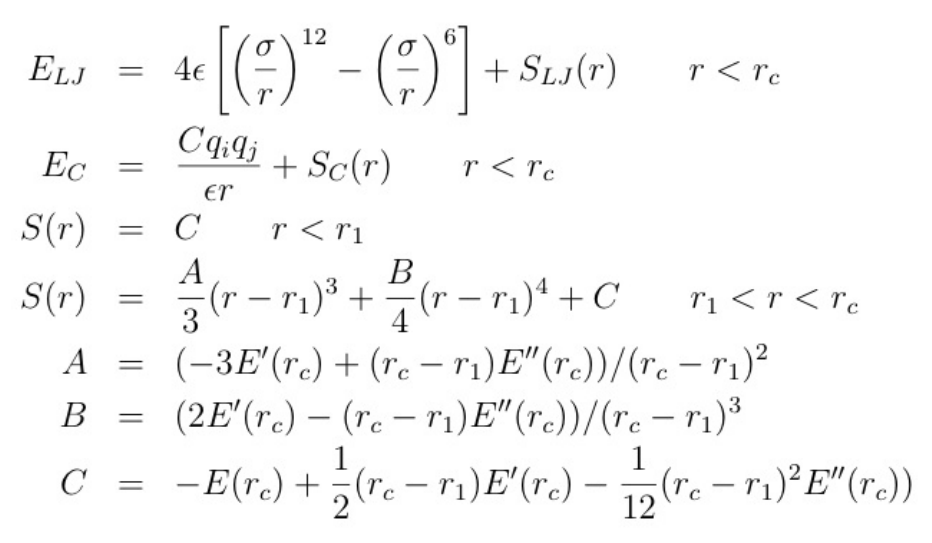
r1 is the inner cutoff; rc is the outer cutoff. The coefficients A, B, and C are computed by LAMMPS to
perform the shifting and smoothing. The function S(r) is actually applied once to each term of the LJ formula
and once to the Coulombic formula, so there are 2 or 3 sets of A,B,C coefficients depending on which
pair_style is used. The boundary conditions applied to the smoothing function are as follows: S'(r1) = S''(r1) =
0, S(rc) = -E(rc), S'(rc) = -E'(rc), and S''(rc) = -E''(rc), where E(r) is the corresponding term in the LJ or
Coulombic potential energy function. Single and double primes denote first and second derivatives with
respect to r, respectively.
The inner and outer cutoff for the LJ and Coulombic terms can be the same or different depending on whether
2 or 4 arguments are used in the pair_style command. The inner LJ cutoff must be > 0, but the inner
Coulombic cutoff can be >= 0.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• inner (distance units)• outer (distance units)•
Note that sigma is defined in the LJ formula as the zero-crossing distance for the potential, not as the energy
minimum at 2^(1/6) sigma.
The last 2 coefficients are optional inner and outer cutoffs for style lj/gromacs. If not specified, the global
inner and outer values are used.
The last 2 coefficients cannot be used with style lj/gromacs/coul/gromacs because this force field does not
allow varying cutoffs for individual atom pairs; all pairs use the global cutoff(s) specified in the pair_style
LAMMPS Users Manual
1500
command.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of the lj/cut
pair styles can be mixed. The default mix value is geometric. See the "pair_modify" command for details.
None of the GROMACS pair styles support the pair_modify shift option, since the Lennard-Jones portion of
the pair interaction is already smoothed to 0.0 at the cutoff.
The pair_modify table option is not relevant for this pair style.
None of the GROMACS pair styles support the pair_modify tail option for adding long-range tail corrections
to energy and pressure, since there are no corrections for a potential that goes to 0.0 at the cutoff.
All of the GROMACS pair styles write their information to binary restart files, so pair_style and pair_coeff
commands do not need to be specified in an input script that reads a restart file.
All of the GROMACS pair styles can only be used via the pair keyword of the run_style respa command.
They do not support the inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_coeff
Default: none
(Marrink) Marrink, de Vries, Mark, J Phys Chem B, 108, 750-760 (2004).
LAMMPS Users Manual
1501

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style hbond/dreiding/lj command
pair_style hbond/dreiding/lj/omp command
pair_style hbond/dreiding/morse command
pair_style hbond/dreiding/morse/omp command
Syntax:
pair_style style N inner_distance_cutoff outer_distance_cutoff angle_cutof
style = hbond/dreiding/lj or hbond/dreiding/morse• n = cosine angle periodicity• inner_distance_cutoff = global inner cutoff for Donor-Acceptor interactions (distance units)• outer_distance_cutoff = global cutoff for Donor-Acceptor interactions (distance units)• angle_cutoff = global angle cutoff for Acceptor-Hydrogen-Donor• interactions (degrees)•
Examples:
pair_style hybrid/overlay lj/cut 10.0 hbond/dreiding/lj 4 9.0 11.0 90
pair_coeff 1 2 hbond/dreiding/lj 3 i 9.5 2.75 4 9.0 11.0 90.0
pair_style hybrid/overlay lj/cut 10.0 hbond/dreiding/morse 2 9.0 11.0 90
pair_coeff 1 2 hbond/dreiding/morse 3 i 3.88 1.7241379 2.9 2 9 11 90
Description:
The hbond/dreiding styles compute the Acceptor-Hydrogen-Donor (AHD) 3-body hydrogen bond interaction
for the DREIDING force field, given by:
LAMMPS Users Manual
1502
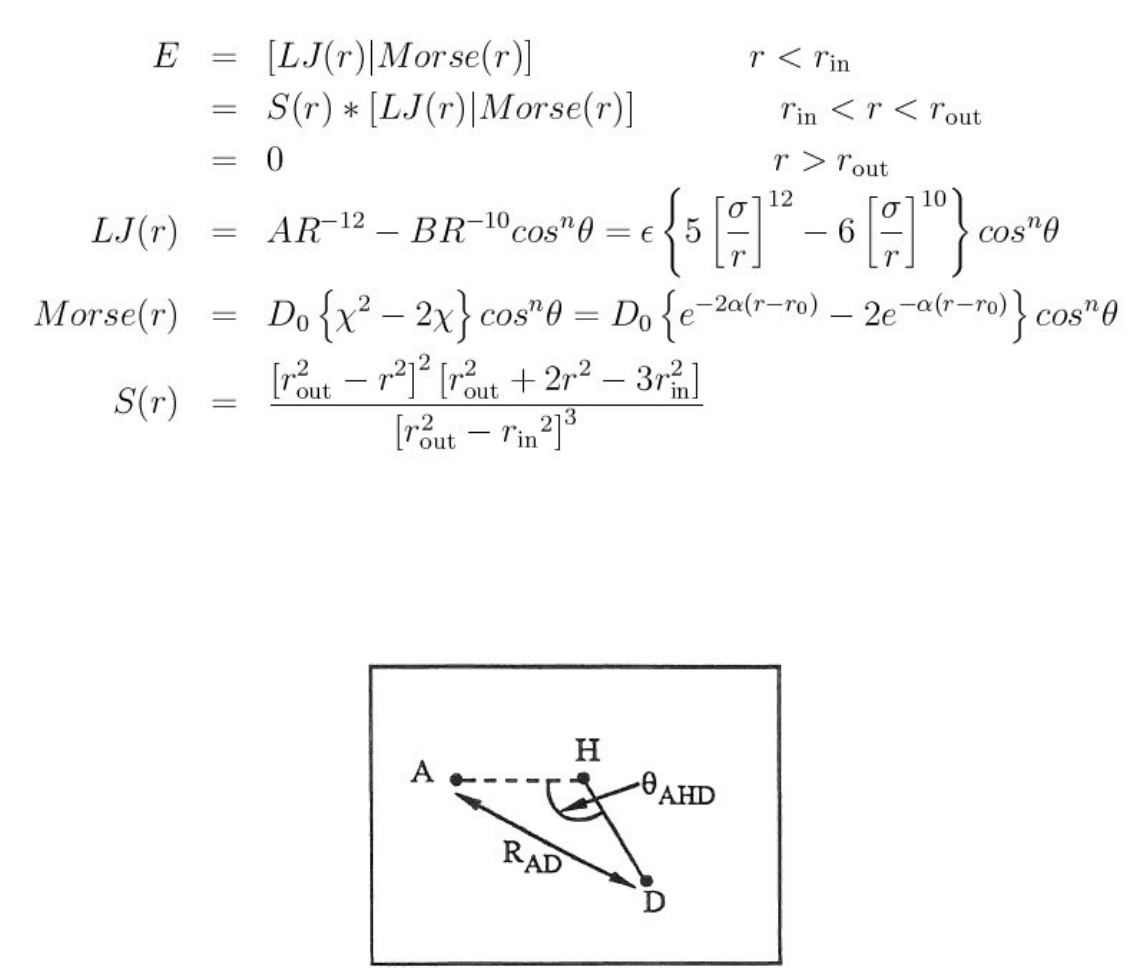
where Rin is the inner spline distance cutoff, Rout is the outer distance cutoff, theta_c is the angle cutoff, and
n is the cosine periodicity.
Here, r is the radial distance between the donor (D) and acceptor (A) atoms and theta is the bond angle
between the acceptor, the hydrogen (H) and the donor atoms:
These 3-body interactions can be defined for pairs of acceptor and donor atoms, based on atom types. For
each donor/acceptor atom pair, the 3rd atom in the interaction is a hydrogen permanently bonded to the donor
atom, e.g. in a bond list read in from a data file via the read_data command. The atom types of possible
hydrogen atoms for each donor/acceptor type pair are specified by the pair_coeff command (see below).
Style hbond/dreiding/lj is the original DREIDING potential of (Mayo). It uses a LJ 12/10 functional for the
Donor-Acceptor interactions. To match the results in the original paper, use n = 4.
Style hbond/dreiding/morse is an improved version using a Morse potential for the Donor-Acceptor
interactions. (Liu) showed that the Morse form gives improved results for Dendrimer simulations, when n = 2.
See this howto section of the manual for more information on the DREIDING forcefield.
LAMMPS Users Manual
1503

NOTE: Because the Dreiding hydrogen bond potential is only one portion of an overall force field which
typically includes other pairwise interactions, it is common to use it as a sub-style in a pair_style
hybrid/overlay command, where another pair style provides the repulsive core interaction between pairs of
atoms, e.g. a 1/r^12 Lennard-Jones repulsion.
NOTE: When using the hbond/dreiding pair styles with pair_style hybrid/overlay, you should explicitly define
pair interactions between the donor atom and acceptor atoms, (as well as between these atoms and ALL other
atoms in your system). Whenever pair_style hybrid/overlay is used, ordinary mixing rules are not applied to
atoms like the donor and acceptor atoms because they are typically referenced in multiple pair styles.
Neglecting to do this can cause difficult-to-detect physics problems.
NOTE: In the original Dreiding force field paper 1-4 non-bonded interactions ARE allowed. If this is desired
for your model, use the special_bonds command (e.g. "special_bonds lj 0.0 0.0 1.0") to turn these interactions
on.
The following coefficients must be defined for pairs of eligible donor/acceptor types via the pair_coeff
command as in the examples above.
NOTE: Unlike other pair styles and their associated pair_coeff commands, you do not need to specify
pair_coeff settings for all possible I,J type pairs. Only I,J type pairs for atoms which act as joint
donors/acceptors need to be specified; all other type pairs are assumed to be inactive.
NOTE: A pair_coeff command can be specified multiple times for the same donor/acceptor type pair. This
enables multiple hydrogen types to be assigned to the same donor/acceptor type pair. For other pair_styles, if
the pair_coeff command is re-used for the same I.J type pair, the settings for that type pair are overwritten. For
the hydrogen bond potentials this is not the case; the settings are cumulative. This means the only way to turn
off a previous setting, is to re-use the pair_style command and start over.
For the hbond/dreiding/lj style the list of coefficients is as follows:
K = hydrogen atom type = 1 to Ntypes• donor flag = i or j• epsilon (energy units)• sigma (distance units)• n = exponent in formula above• distance cutoff Rin (distance units)• distance cutoff Rout (distance units)• angle cutoff (degrees)•
For the hbond/dreiding/morse style the list of coefficients is as follows:
K = hydrogen atom type = 1 to Ntypes• donor flag = i or j• D0 (energy units)• alpha (1/distance units)• r0 (distance units)• n = exponent in formula above• distance cutoff Rin (distance units)• distance cutoff Rout (distance units)• angle cutoff (degrees)•
LAMMPS Users Manual
1504

A single hydrogen atom type K can be specified, or a wild-card asterisk can be used in place of or in
conjunction with the K arguments to select multiple types as hydrogens. This takes the form "*" or "*n" or
"n*" or "m*n". See the pair_coeff command doc page for details.
If the donor flag is i, then the atom of type I in the pair_coeff command is treated as the donor, and J is the
acceptor. If the donor flag is j, then the atom of type J in the pair_coeff command is treated as the donor and I
is the donor. This option is required because the pair_coeff command requires that I <= J.
Epsilon and sigma are settings for the hydrogen bond potential based on a Lennard-Jones functional form.
Note that sigma is defined as the zero-crossing distance for the potential, not as the energy minimum at
2^(1/6) sigma.
D0 and alpha and r0 are settings for the hydrogen bond potential based on a Morse functional form.
The last 3 coefficients for both styles are optional. If not specified, the global n, distance cutoff, and angle
cutoff specified in the pair_style command are used. If you wish to only override the 2nd or 3rd optional
parameter, you must also specify the preceding optional parameters.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
These pair styles do not support mixing. You must explicitly identify each donor/acceptor type pair.
These styles do not support the pair_modify shift option for the energy of the interactions.
The pair_modify table option is not relevant for these pair styles.
These pair styles do not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
These pair styles do not write their information to binary restart files, so pair_style and pair_coeff commands
need to be re-specified in an input script that reads a restart file.
These pair styles can only be used via the pair keyword of the run_style respa command. They do not support
the inner, middle, outer keywords.
LAMMPS Users Manual
1505

These pair styles tally a count of how many hydrogen bonding interactions they calculate each timestep and
the hbond energy. These quantities can be accessed via the compute pair command as a vector of values of
length 2.
To print these quantities to the log file (with a descriptive column heading) the following commands could be
included in an input script:
compute hb all pair hbond/dreiding/lj
variable n_hbond equal c_hb[1] #number hbonds
variable E_hbond equal c_hb[2] #hbond energy
thermo_style custom step temp epair v_E_hbond
Restrictions: none
Related commands:
pair_coeff
Default: none
(Mayo) Mayo, Olfason, Goddard III, J Phys Chem, 94, 8897-8909 (1990).
(Liu) Liu, Bryantsev, Diallo, Goddard III, J. Am. Chem. Soc 131 (8) 2798 (2009)
LAMMPS Users Manual
1506

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style hybrid command
pair_style hybrid/omp command
pair_style hybrid/overlay command
pair_style hybrid/overlay/omp command
Syntax:
pair_style hybrid style1 args style2 args ...
pair_style hybrid/overlay style1 args style2 args ...
style1,style2 = list of one or more pair styles and their arguments•
Examples:
pair_style hybrid lj/cut/coul/cut 10.0 eam lj/cut 5.0
pair_coeff 1*2 1*2 eam niu3
pair_coeff 3 3 lj/cut/coul/cut 1.0 1.0
pair_coeff 1*2 3 lj/cut 0.5 1.2
pair_style hybrid/overlay lj/cut 2.5 coul/long 2.0
pair_coeff * * lj/cut 1.0 1.0
pair_coeff * * coul/long
Description:
The hybrid and hybrid/overlay styles enable the use of multiple pair styles in one simulation. With the hybrid
style, exactly one pair style is assigned to each pair of atom types. With the hybrid/overlay style, one or more
pair styles can be assigned to each pair of atom types. The assignment of pair styles to type pairs is made via
the pair_coeff command.
Here are two examples of hybrid simulations. The hybrid style could be used for a simulation of a metal
droplet on a LJ surface. The metal atoms interact with each other via an eam potential, the surface atoms
interact with each other via a lj/cut potential, and the metal/surface interaction is also computed via a lj/cut
potential. The hybrid/overlay style could be used as in the 2nd example above, where multiple potentials are
superposed in an additive fashion to compute the interaction between atoms. In this example, using lj/cut and
coul/long together gives the same result as if the lj/cut/coul/long potential were used by itself. In this case, it
would be more efficient to use the single combined potential, but in general any combination of pair potentials
can be used together in to produce an interaction that is not encoded in any single pair_style file, e.g. adding
Coulombic forces between granular particles.
All pair styles that will be used are listed as "sub-styles" following the hybrid or hybrid/overlay keyword, in
any order. Each sub-style's name is followed by its usual arguments, as illustrated in the example above. See
the doc pages of individual pair styles for a listing and explanation of the appropriate arguments.
Note that an individual pair style can be used multiple times as a sub-style. For efficiency this should only be
done if your model requires it. E.g. if you have different regions of Si and C atoms and wish to use a Tersoff
LAMMPS Users Manual
1507
potential for pure Si for one set of atoms, and a Tersoff potential for pure C for the other set (presumably with
some 3rd potential for Si-C interactions), then the sub-style tersoff could be listed twice. But if you just want
to use a Lennard-Jones or other pairwise potential for several different atom type pairs in your model, then
you should just list the sub-style once and use the pair_coeff command to assign parameters for the different
type pairs.
NOTE: There are two exceptions to this option to list an individual pair style multiple times. The first is for
pair styles implemented as Fortran libraries: pair_style meam and pair_style reax (pair_style reax/c is OK).
This is because unlike a C++ class, they can not be instantiated multiple times, due to the manner in which
they were coded in Fortran. The second is for GPU-enabled pair styles in the GPU package. This is b/c the
GPU package also currently assumes that only one instance of a pair style is being used.
In the pair_coeff commands, the name of a pair style must be added after the I,J type specification, with the
remaining coefficients being those appropriate to that style. If the pair style is used multiple times in the
pair_style command, then an additional numeric argument must also be specified which is a number from 1 to
M where M is the number of times the sub-style was listed in the pair style command. The extra number
indicates which instance of the sub-style these coefficients apply to.
For example, consider a simulation with 3 atom types: types 1 and 2 are Ni atoms, type 3 are LJ atoms with
charges. The following commands would set up a hybrid simulation:
pair_style hybrid eam/alloy lj/cut/coul/cut 10.0 lj/cut 8.0
pair_coeff * * eam/alloy nialhjea Ni Ni NULL
pair_coeff 3 3 lj/cut/coul/cut 1.0 1.0
pair_coeff 1*2 3 lj/cut 0.8 1.3
As an example of using the same pair style multiple times, consider a simulation with 2 atom types. Type 1 is
Si, type 2 is C. The following commands would model the Si atoms with Tersoff, the C atoms with Tersoff,
and the cross-interactions with Lennard-Jones:
pair_style hybrid lj/cut 2.5 tersoff tersoff
pair_coeff * * tersoff 1 Si.tersoff Si NULL
pair_coeff * * tersoff 2 C.tersoff NULL C
pair_coeff 1 2 lj/cut 1.0 1.5
If pair coefficients are specified in the data file read via the read_data command, then the same rule applies.
E.g. "eam/alloy" or "lj/cut" must be added after the atom type, for each line in the "Pair Coeffs" section, e.g.
Pair Coeffs
1 lj/cut/coul/cut 1.0 1.0
...
Note that the pair_coeff command for some potentials such as pair_style eam/alloy includes a mapping
specification of elements to all atom types, which in the hybrid case, can include atom types not assigned to
the eam/alloy potential. The NULL keyword is used by many such potentials (eam/alloy, Tersoff, AIREBO,
etc), to denote an atom type that will be assigned to a different sub-style.
For the hybrid style, each atom type pair I,J is assigned to exactly one sub-style. Just as with a simulation
using a single pair style, if you specify the same atom type pair in a second pair_coeff command, the previous
assignment will be overwritten.
LAMMPS Users Manual
1508

For the hybrid/overlay style, each atom type pair I,J can be assigned to one or more sub-styles. If you specify
the same atom type pair in a second pair_coeff command with a new sub-style, then the second sub-style is
added to the list of potentials that will be calculated for two interacting atoms of those types. If you specify
the same atom type pair in a second pair_coeff command with a sub-style that has already been defined for
that pair of atoms, then the new pair coefficients simply override the previous ones, as in the normal usage of
the pair_coeff command. E.g. these two sets of commands are the same:
pair_style lj/cut 2.5
pair_coeff * * 1.0 1.0
pair_coeff 2 2 1.5 0.8
pair_style hybrid/overlay lj/cut 2.5
pair_coeff * * lj/cut 1.0 1.0
pair_coeff 2 2 lj/cut 1.5 0.8
Coefficients must be defined for each pair of atoms types via the pair_coeff command as described above, or
in the data file or restart files read by the read_data or read_restart commands, or by mixing as described
below.
For both the hybrid and hybrid/overlay styles, every atom type pair I,J (where I <= J) must be assigned to at
least one sub-style via the pair_coeff command as in the examples above, or in the data file read by the
read_data, or by mixing as described below.
If you want there to be no interactions between a particular pair of atom types, you have 3 choices. You can
assign the type pair to some sub-style and use the neigh_modify exclude type command. You can assign it to
some sub-style and set the coefficients so that there is effectively no interaction (e.g. epsilon = 0.0 in a LJ
potential). Or, for hybrid and hybrid/overlay simulations, you can use this form of the pair_coeff command in
your input script:
pair_coeff 2 3 none
or this form in the "Pair Coeffs" section of the data file:
3 none
If an assignment to none is made in a simulation with the hybrid/overlay pair style, it wipes out all previous
assignments of that atom type pair to sub-styles.
Note that you may need to use an atom_style hybrid command in your input script, if atoms in the simulation
will need attributes from several atom styles, due to using multiple pair potentials.
Different force fields (e.g. CHARMM vs AMBER) may have different rules for applying weightings that
change the strength of pairwise interactions between pairs of atoms that are also 1-2, 1-3, and 1-4 neighbors in
the molecular bond topology, as normally set by the special_bonds command. Different weights can be
assigned to different pair hybrid sub-styles via the pair_modify special command. This allows multiple force
fields to be used in a model of a hybrid system, however, there is no consistent approach to determine
parameters automatically for the interactions between the two force fields, this is only recommended when
particles described by the different force fields do not mix.
Here is an example for mixing CHARMM and AMBER: The global amber setting sets the 1-4 interactions to
non-zero scaling factors and then overrides them with 0.0 only for CHARMM:
LAMMPS Users Manual
1509

special_bonds amber
pair_hybrid lj/charmm/coul/long 8.0 10.0 lj/cut/coul/long 10.0
pair_modify pair lj/charmm/coul/long special lj/coul 0.0 0.0 0.0
The this input achieves the same effect:
special_bonds 0.0 0.0 0.1
pair_hybrid lj/charmm/coul/long 8.0 10.0 lj/cut/coul/long 10.0
pair_modify pair lj/cut/coul/long special lj 0.0 0.0 0.5
pair_modify pair lj/cut/coul/long special coul 0.0 0.0 0.83333333
pair_modify pair lj/charmm/coul/long special lj/coul 0.0 0.0 0.0
Here is an example for mixing Tersoff with OPLS/AA based on a data file that defines bonds for all atoms
where for the Tersoff part of the system the force constants for the bonded interactions have been set to 0.
Note the global settings are effectively lj/coul 0.0 0.0 0.5 as required for OPLS/AA:
special_bonds lj/coul 1e-20 1e-20 0.5
pair_hybrid tersoff lj/cut/coul/long 12.0
pair_modify pair tersoff special lj/coul 1.0 1.0 1.0
See the pair_modify doc page for details on the specific syntax, requirements and restrictions.
The potential energy contribution to the overall system due to an individual sub-style can be accessed and
output via the compute pair command.
NOTE: Several of the potentials defined via the pair_style command in LAMMPS are really many-body
potentials, such as Tersoff, AIREBO, MEAM, ReaxFF, etc. The way to think about using these potentials in a
hybrid setting is as follows.
A subset of atom types is assigned to the many-body potential with a single pair_coeff command, using "* *"
to include all types and the NULL keywords described above to exclude specific types not assigned to that
potential. If types 1,3,4 were assigned in that way (but not type 2), this means that all many-body interactions
between all atoms of types 1,3,4 will be computed by that potential. Pair_style hybrid allows interactions
between type pairs 2-2, 1-2, 2-3, 2-4 to be specified for computation by other pair styles. You could even add
a second interaction for 1-1 to be computed by another pair style, assuming pair_style hybrid/overlay is used.
But you should not, as a general rule, attempt to exclude the many-body interactions for some subset of the
type pairs within the set of 1,3,4 interactions, e.g. exclude 1-1 or 1-3 interactions. That is not conceptually
well-defined for many-body interactions, since the potential will typically calculate energies and foces for
small groups of atoms, e.g. 3 or 4 atoms, using the neighbor lists of the atoms to find the additional atoms in
the group. It is typically non-physical to think of excluding an interaction between a particular pair of atoms
when the potential computes 3-body or 4-body interactions.
However, you can still use the pair_coeff none setting or the neigh_modify exclude command to exclude
certain type pairs from the neighbor list that will be passed to a manybody sub-style. This will alter the
calculations made by a many-body potential, since it builds its list of 3-body, 4-body, etc interactions from the
pair list. You will need to think carefully as to whether it produces a physically meaningful result for your
model.
For example, imagine you have two atom types in your model, type 1 for atoms in one surface, and type 2 for
atoms in the other, and you wish to use a Tersoff potential to compute interactions within each surface, but not
between surfaces. Then either of these two command sequences would implement that model:
LAMMPS Users Manual
1510

pair_style hybrid tersoff
pair_coeff * * tersoff SiC.tersoff C C
pair_coeff 1 2 none
pair_style tersoff
pair_coeff * * SiC.tersoff C C
neigh_modify exclude type 1 2
Either way, only neighbor lists with 1-1 or 2-2 interactions would be passed to the Tersoff potential, which
means it would compute no 3-body interactions containing both type 1 and 2 atoms.
Here is another example, using hybrid/overlay, to use 2 many-body potentials together, in an overlapping
manner. Imagine you have CNT (C atoms) on a Si surface. You want to use Tersoff for Si/Si and Si/C
interactions, and AIREBO for C/C interactions. Si atoms are type 1; C atoms are type 2. Something like this
will work:
pair_style hybrid/overlay tersoff airebo 3.0
pair_coeff * * tersoff SiC.tersoff.custom Si C
pair_coeff * * airebo CH.airebo NULL C
Note that to prevent the Tersoff potential from computing C/C interactions, you would need to modify the
SiC.tersoff file to turn off C/C interaction, i.e. by setting the appropriate coefficients to 0.0.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual.
Since the hybrid and hybrid/overlay styles delegate computation to the individual sub-styles, the suffix
versions of the hybrid and hybrid/overlay styles are used to propagate the corresponding suffix to all
sub-styles, if those versions exist. Otherwise the non-accelerated version will be used.
The individual accelerated sub-styles are part of the GPU, USER-OMP and OPT packages, respectively. They
are only enabled if LAMMPS was built with those packages. See the Making LAMMPS section for more
info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
Any pair potential settings made via the pair_modify command are passed along to all sub-styles of the hybrid
potential.
For atom type pairs I,J and I != J, if the sub-style assigned to I,I and J,J is the same, and if the sub-style allows
for mixing, then the coefficients for I,J can be mixed. This means you do not have to specify a pair_coeff
command for I,J since the I,J type pair will be assigned automatically to the sub-style defined for both I,I and
J,J and its coefficients generated by the mixing rule used by that sub-style. For the hybrid/overlay style, there
is an additional requirement that both the I,I and J,J pairs are assigned to a single sub-style. See the
"pair_modify" command for details of mixing rules. See the See the doc page for the sub-style to see if allows
LAMMPS Users Manual
1511
for mixing.
The hybrid pair styles supports the pair_modify shift, table, and tail options for an I,J pair interaction, if the
associated sub-style supports it.
For the hybrid pair styles, the list of sub-styles and their respective settings are written to binary restart files,
so a pair_style command does not need to specified in an input script that reads a restart file. However, the
coefficient information is not stored in the restart file. Thus, pair_coeff commands need to be re-specified in
the restart input script.
These pair styles support the use of the inner, middle, and outer keywords of the run_style respa command, if
their sub-styles do.
Restrictions:
When using a long-range Coulombic solver (via the kspace_style command) with a hybrid pair_style, one or
more sub-styles will be of the "long" variety, e.g. lj/cut/coul/long or buck/coul/long. You must insure that the
short-range Coulombic cutoff used by each of these long pair styles is the same or else LAMMPS will
generate an error.
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1512

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style kim command
Syntax:
pair_style kim virialmode model printflag
virialmode = KIMvirial or LAMMPSvirial• model = name of KIM model (potential)• printflag = 1/0 do or do not print KIM descriptor file, optional•
Examples:
pair_style kim KIMvirial model_Ar_P_Morse
pair_coeff * * Ar Ar
pair_style kim KIMvirial model_Ar_P_Morse 1
pair_coeff * * Ar Ar
Description:
This pair style is a wrapper on the Knowledge Base for Interatomic Models (KIM) repository of interatomic
potentials, so that they can be used by LAMMPS scripts.
In KIM lingo, a potential is a "model" and a model contains both the analytic formulas that define the
potential as well as the parameters needed to run it for one or more materials, including coefficients and
cutoffs.
The argument virialmode determines how the global virial is calculated. If KIMvirial is specified, the KIM
model performs the global virial calculation (if it knows how). If LAMMPSvirial is specified, LAMMPS
computes the global virial using its fdotr mechanism.
The argument model is the name of the KIM model for a specific potential as KIM defines it. In principle,
LAMMPS can invoke any KIM model. You should get an error or warning message from either LAMMPS or
KIM if there is an incompatibility.
The argument printflag is optional. If it is set to a non-zero value then a KIM descriptor file is printed when
KIM is invoked. This can be useful for debugging. The default is to not print this file.
Only a single pair_coeff command is used with the kim style which specifies the mapping of LAMMPS atom
types to KIM elements. This is done by specifying N additional arguments after the * * in the pair_coeff
command, where N is the number of LAMMPS atom types:
N element names = mapping of KIM elements to atom types•
As an example, imagine the KIM model supports Si and C atoms. If your LAMMPS simulation has 4 atom
types and you want the 1st 3 to be Si, and the 4th to be C, you would use the following pair_coeff command:
pair_coeff * * Si Si Si C
LAMMPS Users Manual
1513

The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Si arguments map
LAMMPS atom types 1,2,3 to Si as defined within KIM. The final C argument maps LAMMPS atom type 4
to C as defined within KIM. If a mapping value is specified as NULL, the mapping is not performed. This can
only be used when a kim potential is used as part of the hybrid pair style. The NULL values are placeholders
for atom types that will be used with other potentials.
In addition to the usual LAMMPS error messages, the KIM library itself may generate errors, which should be
printed to the screen. In this case it is also useful to check the kim.log file for additional error information.
This file kim.log should be generated in the same directory where LAMMPS is running.
To download, build, and install the KIM library on your system, see the lib/kim/README file. Once you
have done this and built LAMMPS with the KIM package installed you can run the example input scripts in
examples/kim.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify mix, shift, table, and tail options.
This pair style does not write its information to binary restart files, since KIM stores the potential parameters.
Thus, you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the KIM package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
This current version of pair_style kim is compatible with the kim-api package version 1.6.0 and higher.
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1514

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style kolmogorov/crespi/z command
Syntax:
pair_style hybrid/overlay kolmogorov/crespi/z cutoff
Examples:
pair_style hybrid/overlay kolmogorov/crespi/z 20.0
pair_coeff * * none
pair_coeff 1 2 kolmogorov/crespi/z CC.KC C C
pair_style hybrid/overlay rebo kolmogorov/crespi/z 14.0
pair_coeff * * rebo CH.airebo C C
pair_coeff 1 2 kolmogorov/crespi/z CC.KC C C
Description:
The kolmogorov/crespi/z style computes the Kolmogorov-Crespi interaction potential as described in (KC05).
An important simplification is made, which is to take all normals along the z-axis.
It is important to have a suffiently large cutoff to ensure smooth forces. Energies are shifted so that they go
continously to zero at the cutoff assuming that the exponential part of Vij (first term) decays sufficiently fast.
This shift is achieved by the last term in the equation for Vij above.
This potential is intended for interactions between two layers of graphene. Therefore, to avoid interaction
between layers in multi-layered materials, each layer should have a separate atom type and interactions should
only be computed between atom types of neighbouring layers.
The parameter file (e.g. CC.KC), is intended for use with metal units, with energies in meV. An additional
parameter, S, is available to facilitate scaling of energies in accordance with (vanWijk).
This potential must be used in combination with hybrid/overlay. Other interactions can be set to zero using
pair_style none.
LAMMPS Users Manual
1515

Restrictions:
This fix is part of the USER-MISC package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
pair_coeff pair_none pair_style hybrid/overlay
Default: none
(KC05) A. N. Kolmogorov, V. H. Crespi, Phys. Rev. B 71, 235415 (2005)
(vanWijk) M. M. van Wijk, A. Schuring, M. I. Katsnelson, and A. Fasolino, Physical Review Letters, 113,
135504 (2014)
LAMMPS Users Manual
1516

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lcbop command
Syntax:
pair_style lcbop
Examples:
pair_style lcbop
pair_coeff * * ../potentials/C.lcbop C
Description:
The lcbop pair style computes the long-range bond-order potential for carbon (LCBOP) of (Los and Fasolino).
See section II in that paper for the analytic equations associated with the potential.
Only a single pair_coeff command is used with the lcbop style which specifies an LCBOP potential file with
parameters for specific elements. These are mapped to LAMMPS atom types by specifying N additional
arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N element names = mapping of LCBOP elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, if your LAMMPS simulation has 4 atom types and you want the 1st 3 to be C you would use
the following pair_coeff command:
pair_coeff * * C.lcbop C C C NULL
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first C argument maps
LAMMPS atom type 1 to the C element in the LCBOP file. If a mapping value is specified as NULL, the
mapping is not performed. This can be used when a lcbop potential is used as part of the hybrid pair style. The
NULL values are placeholders for atom types that will be used with other potentials.
The parameters/coefficients for the LCBOP potential as applied to C are listed in the C.lcbop file to agree
with the original (Los and Fasolino) paper. Thus the parameters are specific to this potential and the way it
was fit, so modifying the file should be done carefully.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify mix, shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
LAMMPS Users Manual
1517

Restrictions:
This pair styles is part of the MANYBODY package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
This pair potential requires the newton setting to be "on" for pair interactions.
The C.lcbop potential file provided with LAMMPS (see the potentials directory) is parameterized for metal
units. You can use the LCBOP potential with any LAMMPS units, but you would need to create your own
LCBOP potential file with coefficients listed in the appropriate units if your simulation doesn't use "metal"
units.
Related commands:
pair_airebo, pair_coeff
Default: none
(Los and Fasolino) J. H. Los and A. Fasolino, Phys. Rev. B 68, 024107 (2003).
LAMMPS Users Manual
1518

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style line/lj command
Syntax:
pair_style line/lj cutoff
cutoff = global cutoff for interactions (distance units)
Examples:
pair_style line/lj 3.0
pair_coeff * * 1.0 1.0 1.0 0.8 1.12
pair_coeff 1 2 1.0 2.0 1.0 1.5 1.12 5.0
pair_coeff 1 2 1.0 0.0 1.0 1.0 2.5
Description:
Style line/lj treats particles which are line segments as a set of small spherical particles that tile the line
segment length as explained below. Interactions between two line segments, each with N1 and N2 spherical
particles, are calculated as the pairwise sum of N1*N2 Lennard-Jones interactions. Interactions between a line
segment with N spherical particles and a point particle are treated as the pairwise sum of N Lennard-Jones
interactions. See the pair_style lj/cut doc page for the definition of Lennard-Jones interactions.
The set of non-overlapping spherical sub-particles that represent a line segment are generated in the following
manner. Their size is a function of the line segment length and the specified sub-particle size for that particle
type. If a line segment has a length L and is of type I, then the number of spheres N that represent the segment
is calculated as N = L/sizeI, rounded up to an integer value. Thus if L is not evenly divisible by sizeI, N is
incremented to include one extra sphere. The centers of the spheres are spaced equally along the line segment.
Imagine N+1 equally-space points, which include the 2 end points of the segment. The sphere centers are
halfway between each pair of points.
The LJ interaction between 2 spheres on different line segments (or a sphere on a line segment and a point
particles) is computed with sub-particle epsilon, sigma, and cutoff values that are set by the pair_coeff
command, as described below. If the distance between the 2 spheres is greater than the sub-particle cutoff,
there is no interaction. This means that some pairs of sub-particles on 2 line segments may interact, but others
may not.
For purposes of creating the neighbor list for pairs of interacting line segments or lines/point particles, a
regular particle-particle cutoff is used, as defined by the cutoff setting above in the pair_style command or
overridden with an optional argument in the pair_coeff command for a type pair as discussed below. The
distance between the centers of 2 line segments, or the center of a line segment and a point particle, must be
less than this distance (plus the neighbor skin; see the neighbor command), for the pair of particles to be
included in the neighbor list.
NOTE: This means that a too-short value for the cutoff setting can exclude a pair of particles from the
neighbor list even if pairs of their sub-particle spheres would interact, based on the sub-particle cutoff
specified in the pair_coeff command. E.g. sub-particles at the ends of the line segments that are close to each
other. Which may not be what you want, since it means the ends of 2 line segments could pass through each
other. It is up to you to specify a cutoff setting that is consistent with the length of the line segments you are
LAMMPS Users Manual
1519

using and the sub-particle cutoff settings.
For style line/lj, the following coefficients must be defined for each pair of atom types via the pair_coeff
command as in the examples above, or in the data file or restart files read by the read_data or read_restart
commands:
sizeI (distance units)• sizeJ (distance units)• epsilon (energy units)• sigma (distance units)• subcutoff (distance units)• cutoff (distance units)•
The sizeI and sizeJ coefficients are the sub-particle sizes for line particles of type I and type J. They are used
to define the N sub-particles per segment as described above. These coefficients are actually stored on a
per-type basis. Thus if there are multiple pair_coeff commands that involve type I, as either the first or second
atom type, you should use consistent values for sizeI or sizeJ in all of them. If you do not do this, the last
value specified for sizeI will apply to all segments of type I. If typeI or typeJ refers to point particles, the
corresponding sizeI or sizeJ is ignored; it can be set to 0.0.
The epsilon, sigma, and subcutoff coefficients are used to compute an LJ interactions between a pair of
sub-particles on 2 line segments (of type I and J), or between a sub particle/point particle pair. As discussed
above, the subcutoff and cutoff params are different. The latter is only used for building the neighbor list when
the distance between centers of two line segments or one segment and a point particle is calculated.
The cutoff coefficient is optional. If not specified, the global cutoff is used.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, coefficients must be specified. No default mixing rules are used.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This style is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Defining particles to be line segments so they participate in line/line or line/particle interactions requires the
use the atom_style line command.
Related commands:
pair_coeff, pair_style tri/lj
LAMMPS Users Manual
1520
Default: none
LAMMPS Users Manual
1521

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style list command
Syntax:
pair_style list listfile cutoff keyword
listfile = name of file with list of pairwise interactions• cutoff = global cutoff (distance units)• keyword = optional flag nocheck or check (default is check)•
Examples:
pair_style list restraints.txt 200.0
pair_coeff * *
pair_style hybrid/overlay lj/cut 1.1225 list pair_list.txt 300.0
pair_coeff * * lj/cut 1.0 1.0
pair_coeff 3* 3* list
Description:
Style list computes interactions between explicitly listed pairs of atoms with the option to select functional
form and parameters for each individual pair. Because the parameters are set in the list file, the pair_coeff
command has no parameters (but still needs to be provided). The check and nocheck keywords enable/disable
a test that checks whether all listed bonds were present and computed.
This pair style can be thought of as a hybrid between bonded, non-bonded, and restraint interactions. It will
typically be used as an additional interaction within the hybrid/overlay pair style. It currently supports three
interaction styles: a 12-6 Lennard-Jones, a Morse and a harmonic potential.
The format of the list file is as follows:
one line per pair of atoms• empty lines will be ignored• comment text starts with a '#' character• line syntax: ID1 ID2 style coeffs cutoff
ID1 = atom ID of first atom
ID2 = atom ID of second atom
style = style of interaction
coeffs = list of coeffs
cutoff = cutoff for interaction (optional)
•
The cutoff parameter is optional. If not specified, the global cutoff is used.
Here is an example file:
# this is a comment
15 259 lj126 1.0 1.0 50.0
15 603 morse 10.0 1.2 2.0 10.0 # and another comment
LAMMPS Users Manual
1522
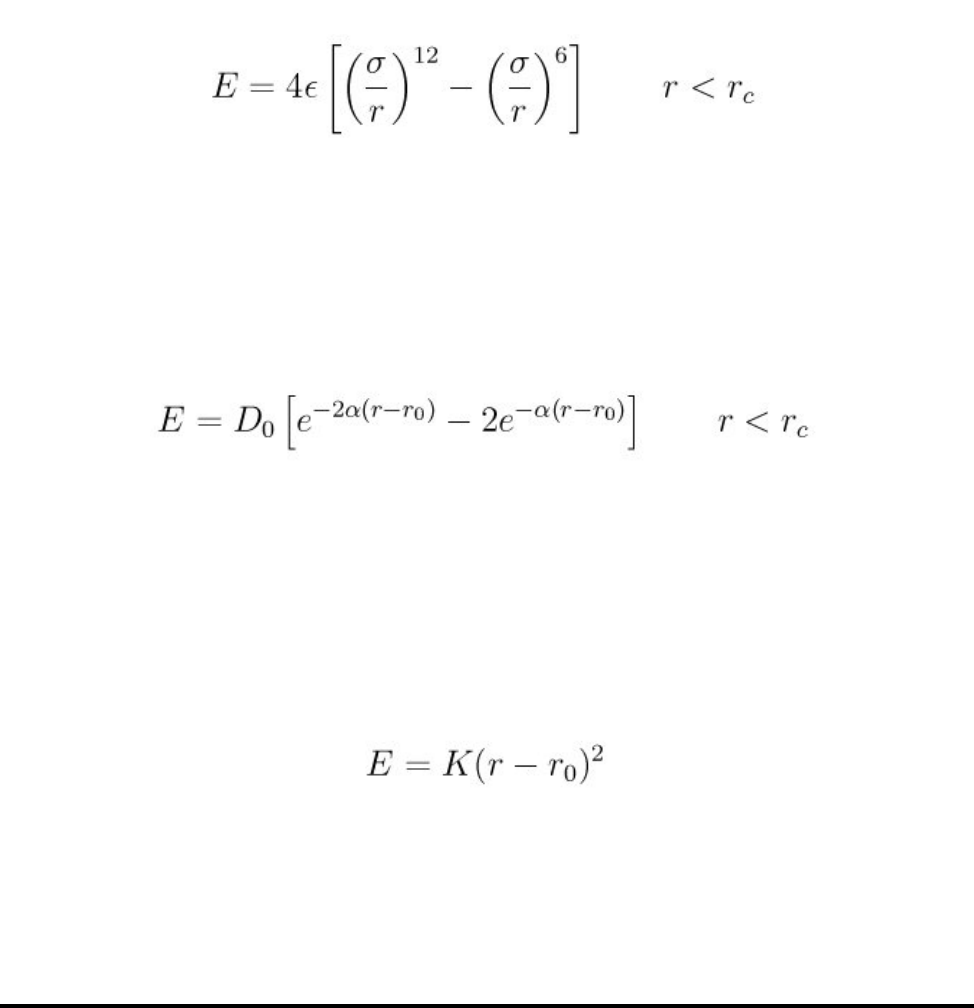
18 470 harmonic 50.0 1.2 5.0
The style lj126 computes pairwise interactions with the formula
and the coefficients:
epsilon (energy units)• sigma (distance units)•
The style morse computes pairwise interactions with the formula
and the coefficients:
D0 (energy units)• alpha (1/distance units)• r0 (distance units)•
The style harmonic computes pairwise interactions with the formula
and the coefficients:
K (energy units)• r0 (distance units)•
Note that the usual 1/2 factor is included in K.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support mixing since all parameters are explicit for each pair.
The pair_modify shift option is supported by this pair style.
The pair_modify table and tail options are not relevant for this pair style.
LAMMPS Users Manual
1523

This pair style does not write its information to binary restart files, so pair_style and pair_coeff commands
need to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style does not use a neighbor list and instead identifies atoms by their IDs. This has two
consequences: 1) The cutoff has to be chosen sufficiently large, so that the second atom of a pair has to be a
ghost atom on the same node on which the first atom is local; otherwise the interaction will be skipped. You
can use the check option to detect, if interactions are missing. 2) Unlike other pair styles in LAMMPS, an
atom I will not interact with multiple images of atom J (assuming the images are within the cutoff distance),
but only with the nearest image.
This style is part of the USER-MISC package. It is only enabled if LAMMPS is build with that package. See
the Making of LAMMPS section for more info.
Related commands:
pair_coeff, pair_style hybrid/overlay, pair_style lj/cut, pair_style morse, bond_style harmonic
Default: none
LAMMPS Users Manual
1524

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/cut command
pair_style lj/cut/gpu command
pair_style lj/cut/intel command
pair_style lj/cut/kk command
pair_style lj/cut/opt command
pair_style lj/cut/omp command
pair_style lj/cut/coul/cut command
pair_style lj/cut/coul/cut/gpu command
pair_style lj/cut/coul/cut/omp command
pair_style lj/cut/coul/debye command
pair_style lj/cut/coul/debye/gpu command
pair_style lj/cut/coul/debye/kk command
pair_style lj/cut/coul/debye/omp command
pair_style lj/cut/coul/dsf command
pair_style lj/cut/coul/dsf/gpu command
pair_style lj/cut/coul/dsf/kk command
pair_style lj/cut/coul/dsf/omp command
pair_style lj/cut/coul/long command
pair_style lj/cut/coul/long/cs command
pair_style lj/cut/coul/long/gpu command
pair_style lj/cut/coul/long/intel command
LAMMPS Users Manual
1525
pair_style lj/cut/coul/long/opt command
pair_style lj/cut/coul/long/omp command
pair_style lj/cut/coul/msm command
pair_style lj/cut/coul/msm/gpu command
pair_style lj/cut/coul/msm/omp command
pair_style lj/cut/tip4p/cut command
pair_style lj/cut/tip4p/cut/omp command
pair_style lj/cut/tip4p/long command
pair_style lj/cut/tip4p/long/omp command
pair_style lj/cut/tip4p/long/opt command
Syntax:
pair_style style args
style = lj/cut or lj/cut/coul/cut or lj/cut/coul/debye or lj/cut/coul/dsf or lj/cut/coul/long or
lj/cut/coul/long/cs or lj/cut/coul/msm or lj/cut/tip4p/long
•
args = list of arguments for a particular style•
lj/cut args = cutoff
cutoff = global cutoff for Lennard Jones interactions (distance units)
lj/cut/coul/cut args = cutoff (cutoff2)
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
lj/cut/coul/debye args = kappa cutoff (cutoff2)
kappa = inverse of the Debye length (inverse distance units)
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
lj/cut/coul/dsf args = alpha cutoff (cutoff2)
alpha = damping parameter (inverse distance units)
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (distance units)
lj/cut/coul/long args = cutoff (cutoff2)
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
lj/cut/coul/msm args = cutoff (cutoff2)
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
lj/cut/tip4p/cut args = otype htype btype atype qdist cutoff (cutoff2)
otype,htype = atom types for TIP4P O and H
btype,atype = bond and angle types for TIP4P waters
qdist = distance from O atom to massless charge (distance units)
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
LAMMPS Users Manual
1526
lj/cut/tip4p/long args = otype htype btype atype qdist cutoff (cutoff2)
otype,htype = atom types for TIP4P O and H
btype,atype = bond and angle types for TIP4P waters
qdist = distance from O atom to massless charge (distance units)
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
Examples:
pair_style lj/cut 2.5
pair_coeff * * 1 1
pair_coeff 1 1 1 1.1 2.8
pair_style lj/cut/coul/cut 10.0
pair_style lj/cut/coul/cut 10.0 8.0
pair_coeff * * 100.0 3.0
pair_coeff 1 1 100.0 3.5 9.0
pair_coeff 1 1 100.0 3.5 9.0 9.0
pair_style lj/cut/coul/debye 1.5 3.0
pair_style lj/cut/coul/debye 1.5 2.5 5.0
pair_coeff * * 1.0 1.0
pair_coeff 1 1 1.0 1.5 2.5
pair_coeff 1 1 1.0 1.5 2.5 5.0
pair_style lj/cut/coul/dsf 0.05 2.5 10.0
pair_coeff * * 1.0 1.0
pair_coeff 1 1 1.0 1.0 2.5
pair_style lj/cut/coul/long 10.0
pair_style lj/cut/coul/long/cs 10.0
pair_style lj/cut/coul/long 10.0 8.0
pair_style lj/cut/coul/long/cs 10.0 8.0
pair_coeff * * 100.0 3.0
pair_coeff 1 1 100.0 3.5 9.0
pair_style lj/cut/coul/msm 10.0
pair_style lj/cut/coul/msm 10.0 8.0
pair_coeff * * 100.0 3.0
pair_coeff 1 1 100.0 3.5 9.0
pair_style lj/cut/tip4p/cut 1 2 7 8 0.15 12.0
pair_style lj/cut/tip4p/cut 1 2 7 8 0.15 12.0 10.0
pair_coeff * * 100.0 3.0
pair_coeff 1 1 100.0 3.5 9.0
pair_style lj/cut/tip4p/long 1 2 7 8 0.15 12.0
pair_style lj/cut/tip4p/long 1 2 7 8 0.15 12.0 10.0
pair_coeff * * 100.0 3.0
pair_coeff 1 1 100.0 3.5 9.0
Description:
The lj/cut styles compute the standard 12/6 Lennard-Jones potential, given by
LAMMPS Users Manual
1527

Rc is the cutoff.
Style lj/cut/coul/cut adds a Coulombic pairwise interaction given by
where C is an energy-conversion constant, Qi and Qj are the charges on the 2 atoms, and epsilon is the
dielectric constant which can be set by the dielectric command. If one cutoff is specified in the pair_style
command, it is used for both the LJ and Coulombic terms. If two cutoffs are specified, they are used as cutoffs
for the LJ and Coulombic terms respectively.
Style lj/cut/coul/debye adds an additional exp() damping factor to the Coulombic term, given by
where kappa is the inverse of the Debye length. This potential is another way to mimic the screening effect of
a polar solvent.
Style lj/cut/coul/dsf computes the Coulombic term via the damped shifted force model described in Fennell,
given by:
where alpha is the damping parameter and erfc() is the complementary error-function. This potential is
essentially a short-range, spherically-truncated, charge-neutralized, shifted, pairwise 1/r summation. The
potential is based on Wolf summation, proposed as an alternative to Ewald summation for condensed phase
systems where charge screening causes electrostatic interactions to become effectively short-ranged. In order
for the electrostatic sum to be absolutely convergent, charge neutralization within the cutoff radius is enforced
by shifting the potential through placement of image charges on the cutoff sphere. Convergence can often be
improved by setting alpha to a small non-zero value.
LAMMPS Users Manual
1528

Styles lj/cut/coul/long and lj/cut/coul/msm compute the same Coulombic interactions as style lj/cut/coul/cut
except that an additional damping factor is applied to the Coulombic term so it can be used in conjunction
with the kspace_style command and its ewald or pppm option. The Coulombic cutoff specified for this style
means that pairwise interactions within this distance are computed directly; interactions outside that distance
are computed in reciprocal space.
Style lj/cut/coul/long/cs is identical to lj/cut/coul/long except that a term is added for the core/shell model to
allow charges on core and shell particles to be separated by r = 0.0.
Styles lj/cut/tip4p/cut and lj/cut/tip4p/long implement the TIP4P water model of (Jorgensen), which
introduces a massless site located a short distance away from the oxygen atom along the bisector of the HOH
angle. The atomic types of the oxygen and hydrogen atoms, the bond and angle types for OH and HOH
interactions, and the distance to the massless charge site are specified as pair_style arguments. Style
lj/cut/tip4p/cut uses a cutoff for Coulomb interactions; style lj/cut/tip4p/long is for use with a long-range
Coulombic solver (Ewald or PPPM).
NOTE: For each TIP4P water molecule in your system, the atom IDs for the O and 2 H atoms must be
consecutive, with the O atom first. This is to enable LAMMPS to "find" the 2 H atoms associated with each O
atom. For example, if the atom ID of an O atom in a TIP4P water molecule is 500, then its 2 H atoms must
have IDs 501 and 502.
See the howto section for more information on how to use the TIP4P pair styles and lists of parameters to set.
Note that the neighbor list cutoff for Coulomb interactions is effectively extended by a distance 2*qdist when
using the TIP4P pair style, to account for the offset distance of the fictitious charges on O atoms in water
molecules. Thus it is typically best in an efficiency sense to use a LJ cutoff >= Coulomb cutoff + 2*qdist, to
shrink the size of the neighbor list. This leads to slightly larger cost for the long-range calculation, so you can
test the trade-off for your model.
For all of the lj/cut pair styles, the following coefficients must be defined for each pair of atoms types via the
pair_coeff command as in the examples above, or in the data file or restart files read by the read_data or
read_restart commands, or by mixing as described below:
epsilon (energy units)• sigma (distance units)• cutoff1 (distance units)• cutoff2 (distance units)•
Note that sigma is defined in the LJ formula as the zero-crossing distance for the potential, not as the energy
minimum at 2^(1/6) sigma.
The latter 2 coefficients are optional. If not specified, the global LJ and Coulombic cutoffs specified in the
pair_style command are used. If only one cutoff is specified, it is used as the cutoff for both LJ and
Coulombic interactions for this type pair. If both coefficients are specified, they are used as the LJ and
Coulombic cutoffs for this type pair. You cannot specify 2 cutoffs for style lj/cut, since it has no Coulombic
terms.
For lj/cut/coul/long and lj/cut/coul/msm and lj/cut/tip4p/cut and lj/cut/tip4p/long only the LJ cutoff can be
specified since a Coulombic cutoff cannot be specified for an individual I,J type pair. All type pairs use the
same global Coulombic cutoff specified in the pair_style command.
LAMMPS Users Manual
1529

Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of the lj/cut
pair styles can be mixed. The default mix value is geometric. See the "pair_modify" command for details.
All of the lj/cut pair styles support the pair_modify shift option for the energy of the Lennard-Jones portion of
the pair interaction.
The lj/cut/coul/long and lj/cut/tip4p/long pair styles support the pair_modify table option since they can
tabulate the short-range portion of the long-range Coulombic interaction.
All of the lj/cut pair styles support the pair_modify tail option for adding a long-range tail correction to the
energy and pressure for the Lennard-Jones portion of the pair interaction.
All of the lj/cut pair styles write their information to binary restart files, so pair_style and pair_coeff
commands do not need to be specified in an input script that reads a restart file.
The lj/cut and lj/cut/coul/long pair styles support the use of the inner, middle, and outer keywords of the
run_style respa command, meaning the pairwise forces can be partitioned by distance at different levels of the
rRESPA hierarchy. The other styles only support the pair keyword of run_style respa. See the run_style
command for details.
Restrictions:
The lj/cut/coul/long and lj/cut/tip4p/long styles are part of the KSPACE package. The lj/cut/tip4p/cut style is
part of the MOLECULE package. These styles are only enabled if LAMMPS was built with those packages.
See the Making LAMMPS section for more info. Note that the KSPACE and MOLECULE packages are
installed by default.
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1530
(Jorgensen) Jorgensen, Chandrasekhar, Madura, Impey, Klein, J Chem Phys, 79, 926 (1983).
(Fennell) C. J. Fennell, J. D. Gezelter, J Chem Phys, 124, 234104 (2006).
LAMMPS Users Manual
1531
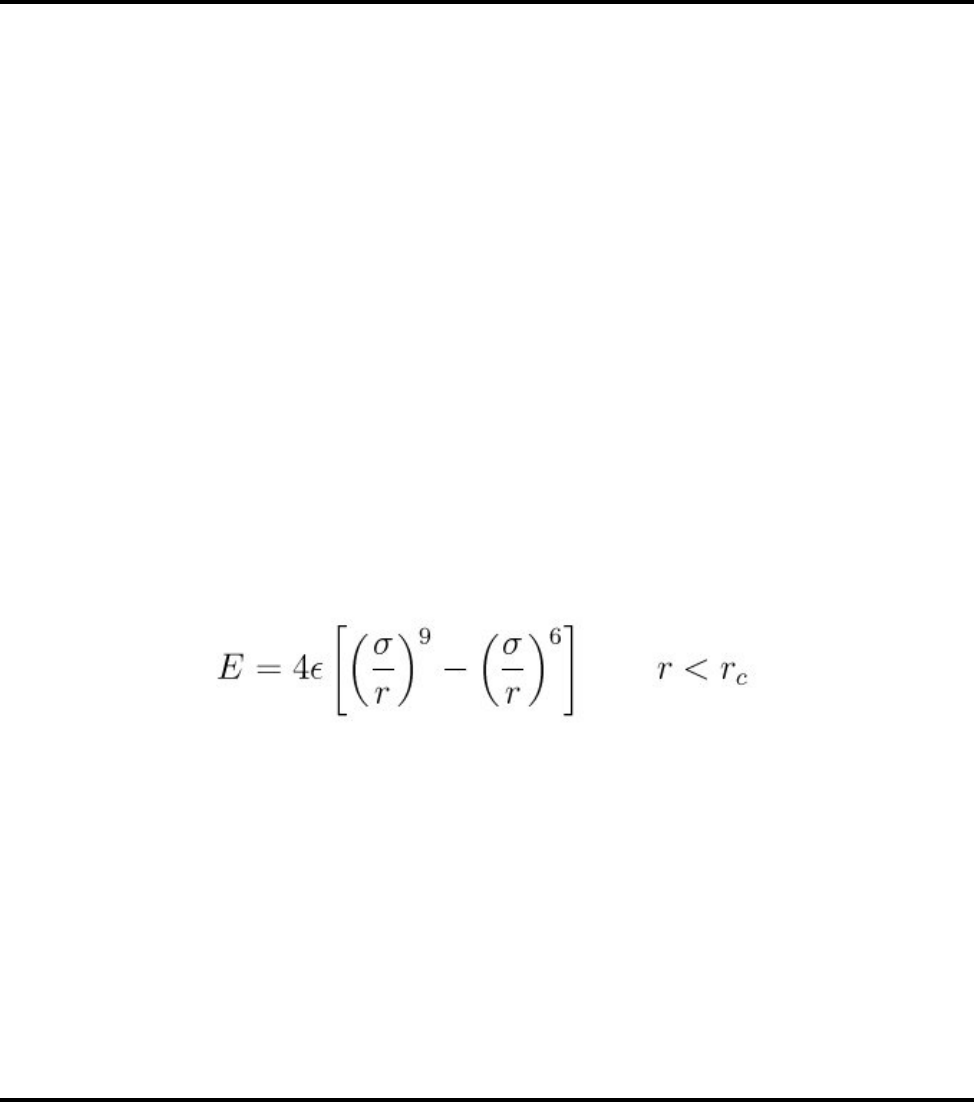
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj96/cut command
pair_style lj96/cut/gpu command
pair_style lj96/cut/omp command
Syntax:
pair_style lj96/cut cutoff
cutoff = global cutoff for lj96/cut interactions (distance units)•
Examples:
pair_style lj96/cut 2.5
pair_coeff * * 1.0 1.0 4.0
pair_coeff 1 1 1.0 1.0
Description:
The lj96/cut style compute a 9/6 Lennard-Jones potential, instead of the standard 12/6 potential, given by
Rc is the cutoff.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global LJ cutoff specified in the pair_style command is
used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
LAMMPS Users Manual
1532

section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of the lj/cut
pair styles can be mixed. The default mix value is geometric. See the "pair_modify" command for details.
This pair style supports the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style supports the pair_modify tail option for adding a long-range tail correction to the energy and
pressure of the pair interaction.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style supports the use of the inner, middle, and outer keywords of the run_style respa command,
meaning the pairwise forces can be partitioned by distance at different levels of the rRESPA hierarchy. See
the run_style command for details.
Restrictions: none
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1533
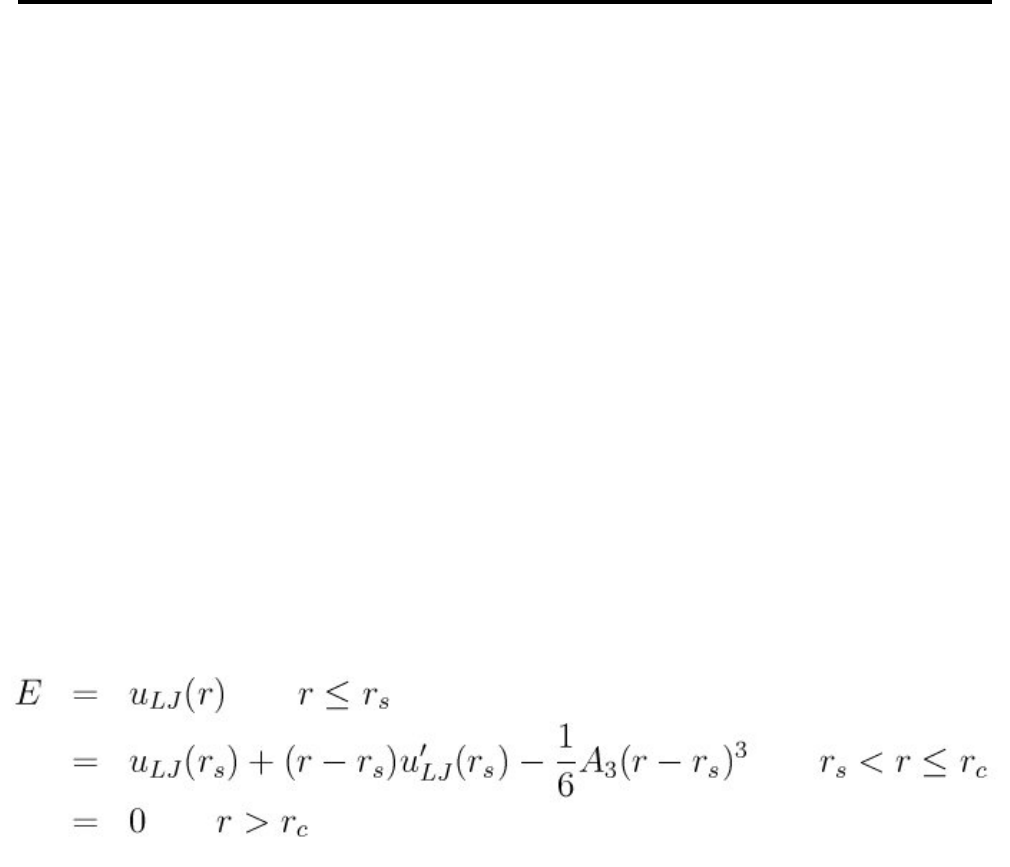
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/cubic command
pair_style lj/cubic/gpu command
pair_style lj/cubic/omp command
Syntax:
pair_style lj/cubic
Examples:
pair_style lj/cubic
pair_coeff * * 1.0 0.8908987
Description:
The lj/cubic style computes a truncated LJ interaction potential whose energy and force are continuous
everywhere. Inside the inflection point the interaction is identical to the standard 12/6 Lennard-Jones
potential. The LJ function outside the inflection point is replaced with a cubic function of distance. The
energy, force, and second derivative are continuous at the inflection point. The cubic coefficient A3 is chosen
so that both energy and force go to zero at the cutoff distance. Outside the cutoff distance the energy and force
are zero.
The location of the inflection point rs is defined by the LJ diameter, rs/sigma = (26/7)^1/6. The cutoff distance
is defined by rc/rs = 67/48 or rc/sigma = 1.737.... The analytic expression for the the cubic coefficient
A3*rmin^3/epsilon = 27.93... is given in the paper by Holian and Ravelo (Holian).
This potential is commonly used to study the shock mechanics of FCC solids, as in Ravelo et al. (Ravelo).
The following coefficients must be defined for each pair of atom types via the pair_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)•
Note that sigma is defined in the LJ formula as the zero-crossing distance for the potential, not as the energy
minimum, which is located at rmin = 2^(1/6)*sigma. In the above example, sigma = 0.8908987, so rmin = 1.
LAMMPS Users Manual
1534
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of the lj/cut
pair styles can be mixed. The default mix value is geometric. See the "pair_modify" command for details.
The lj/cubic pair style does not support the pair_modify shift option, since pair interaction is already smoothed
to 0.0 at the cutoff.
The pair_modify table option is not relevant for this pair style.
The lj/cubic pair style does not support the pair_modify tail option for adding long-range tail corrections to
energy and pressure, since there are no corrections for a potential that goes to 0.0 at the cutoff.
The lj/cubic pair style writes its information to binary restart files, so pair_style and pair_coeff commands do
not need to be specified in an input script that reads a restart file.
The lj/cubic pair style can only be used via the pair keyword of the run_style respa command. It does not
support the inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_coeff
Default: none
(Holian) Holian and Ravelo, Phys Rev B, 51, 11275 (1995).
(Ravelo) Ravelo, Holian, Germann and Lomdahl, Phys Rev B, 70, 014103 (2004).
LAMMPS Users Manual
1535
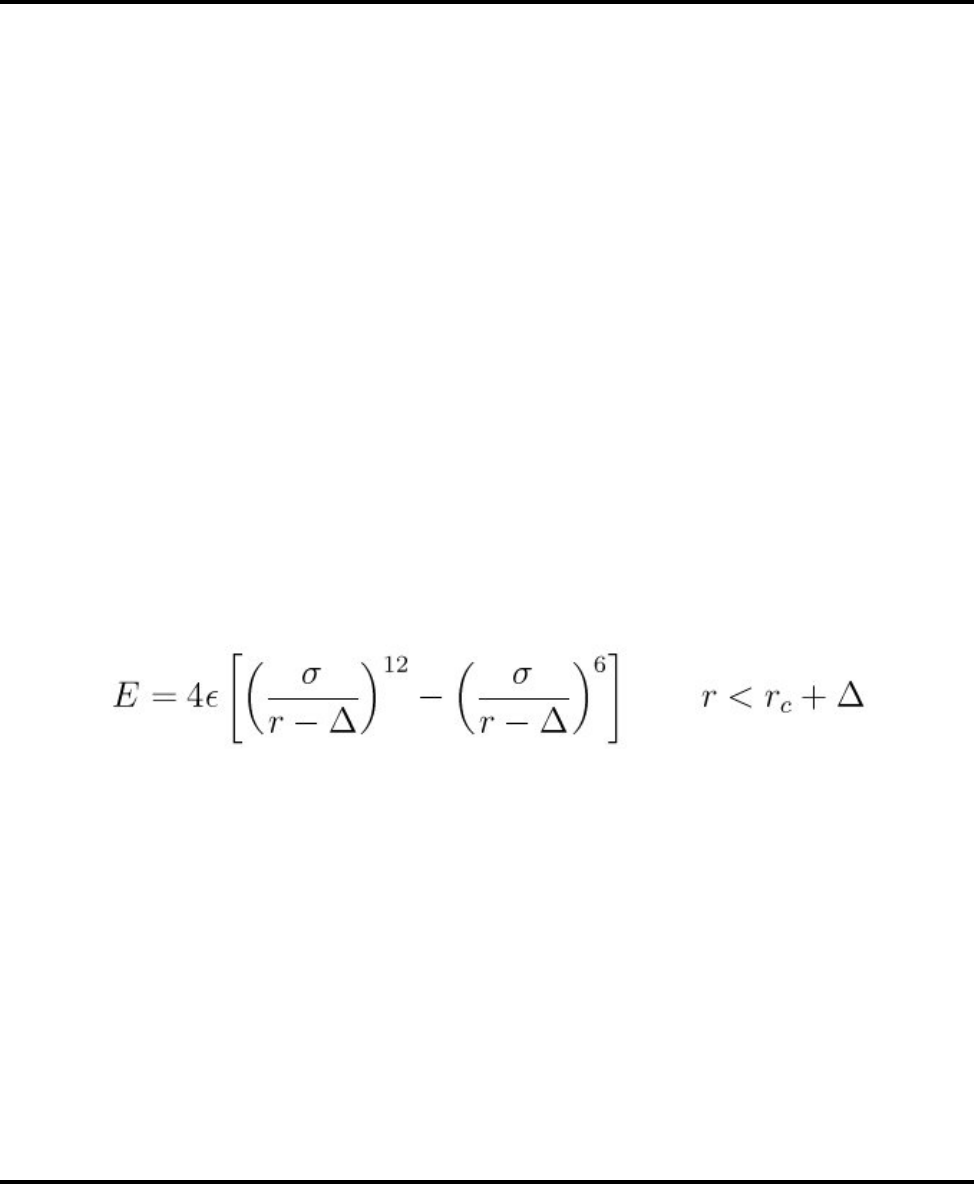
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/expand command
pair_style lj/expand/gpu command
pair_style lj/expand/omp command
Syntax:
pair_style lj/expand cutoff
cutoff = global cutoff for lj/expand interactions (distance units)•
Examples:
pair_style lj/expand 2.5
pair_coeff * * 1.0 1.0 0.5
pair_coeff 1 1 1.0 1.0 -0.2 2.0
Description:
Style lj/expand computes a LJ interaction with a distance shifted by delta which can be useful when particles
are of different sizes, since it is different that using different sigma values in a standard LJ formula:
Rc is the cutoff which does not include the delta distance. I.e. the actual force cutoff is the sum of cutoff +
delta.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• delta (distance units)• cutoff (distance units)•
The delta values can be positive or negative. The last coefficient is optional. If not specified, the global LJ
cutoff is used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
LAMMPS Users Manual
1536

These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon, sigma, and shift coefficients and cutoff distance for this pair
style can be mixed. Shift is always mixed via an arithmetic rule. The other coefficients are mixed according to
the pair_modify mix value. The default mix value is geometric. See the "pair_modify" command for details.
This pair style supports the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style supports the pair_modify tail option for adding a long-range tail correction to the energy and
pressure of the pair interaction.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1537

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/long/coul/long command
pair_style lj/long/coul/long/omp command
pair_style lj/long/coul/long/opt command
pair_style lj/long/tip4p/long command
Syntax:
pair_style style args
style = lj/long/coul/long or lj/long/tip4p/long• args = list of arguments for a particular style•
lj/long/coul/long args = flag_lj flag_coul cutoff (cutoff2)
flag_lj = long or cut or off
long = use Kspace long-range summation for dispersion 1/r^6 term
cut = use a cutoff on dispersion 1/r^6 term
off = omit disperion 1/r^6 term entirely
flag_coul = long or off
long = use Kspace long-range summation for Coulombic 1/r term
off = omit Coulombic term
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
lj/long/tip4p/long args = flag_lj flag_coul otype htype btype atype qdist cutoff (cutoff2)
flag_lj = long or cut
long = use Kspace long-range summation for dispersion 1/r^6 term
cut = use a cutoff
flag_coul = long or off
long = use Kspace long-range summation for Coulombic 1/r term
off = omit Coulombic term
otype,htype = atom types for TIP4P O and H
btype,atype = bond and angle types for TIP4P waters
qdist = distance from O atom to massless charge (distance units)
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
Examples:
pair_style lj/long/coul/long cut off 2.5
pair_style lj/long/coul/long cut long 2.5 4.0
pair_style lj/long/coul/long long long 2.5 4.0
pair_coeff * * 1 1
pair_coeff 1 1 1 3 4
pair_style lj/long/tip4p/long long long 1 2 7 8 0.15 12.0
pair_style lj/long/tip4p/long long long 1 2 7 8 0.15 12.0 10.0
pair_coeff * * 100.0 3.0
pair_coeff 1 1 100.0 3.5 9.0
Description:
LAMMPS Users Manual
1538
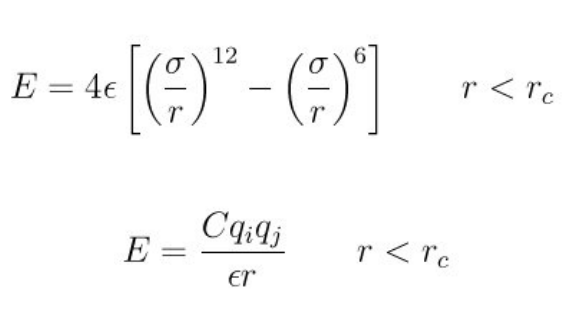
Style lj/long/coul/long computes the standard 12/6 Lennard-Jones and Coulombic potentials, given by
where C is an energy-conversion constant, Qi and Qj are the charges on the 2 atoms, epsilon is the dielectric
constant which can be set by the dielectric command, and Rc is the cutoff. If one cutoff is specified in the
pair_style command, it is used for both the LJ and Coulombic terms. If two cutoffs are specified, they are used
as cutoffs for the LJ and Coulombic terms respectively.
The purpose of this pair style is to capture long-range interactions resulting from both attractive 1/r^6
Lennard-Jones and Coulombic 1/r interactions. This is done by use of the flag_lj and flag_coul settings. The
In 't Veld paper has more details on when it is appropriate to include long-range 1/r^6 interactions, using this
potential.
Style lj/long/tip4p/long implements the TIP4P water model of (Jorgensen), which introduces a massless site
located a short distance away from the oxygen atom along the bisector of the HOH angle. The atomic types of
the oxygen and hydrogen atoms, the bond and angle types for OH and HOH interactions, and the distance to
the massless charge site are specified as pair_style arguments.
NOTE: For each TIP4P water molecule in your system, the atom IDs for the O and 2 H atoms must be
consecutive, with the O atom first. This is to enable LAMMPS to "find" the 2 H atoms associated with each O
atom. For example, if the atom ID of an O atom in a TIP4P water molecule is 500, then its 2 H atoms must
have IDs 501 and 502.
See the howto section for more information on how to use the TIP4P pair style. Note that the neighbor list
cutoff for Coulomb interactions is effectively extended by a distance 2*qdist when using the TIP4P pair style,
to account for the offset distance of the fictitious charges on O atoms in water molecules. Thus it is typically
best in an efficiency sense to use a LJ cutoff >= Coulomb cutoff + 2*qdist, to shrink the size of the neighbor
list. This leads to slightly larger cost for the long-range calculation, so you can test the trade-off for your
model.
If flag_lj is set to long, no cutoff is used on the LJ 1/r^6 dispersion term. The long-range portion can be
calculated by using the kspace_style ewald/disp or pppm/disp commands. The specified LJ cutoff then
determines which portion of the LJ interactions are computed directly by the pair potential versus which part
is computed in reciprocal space via the Kspace style. If flag_lj is set to cut, the LJ interactions are simply
cutoff, as with pair_style lj/cut.
If flag_coul is set to long, no cutoff is used on the Coulombic interactions. The long-range portion can
calculated by using any of several kspace_style command options such as pppm or ewald. Note that if flag_lj
is also set to long, then the ewald/disp or pppm/disp Kspace style needs to be used to perform the long-range
LAMMPS Users Manual
1539

calculations for both the LJ and Coulombic interactions. If flag_coul is set to off, Coulombic interactions are
not computed.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• cutoff1 (distance units)• cutoff2 (distance units)•
Note that sigma is defined in the LJ formula as the zero-crossing distance for the potential, not as the energy
minimum at 2^(1/6) sigma.
The latter 2 coefficients are optional. If not specified, the global LJ and Coulombic cutoffs specified in the
pair_style command are used. If only one cutoff is specified, it is used as the cutoff for both LJ and
Coulombic interactions for this type pair. If both coefficients are specified, they are used as the LJ and
Coulombic cutoffs for this type pair.
Note that if you are using flag_lj set to long, you cannot specify a LJ cutoff for an atom type pair, since only
one global LJ cutoff is allowed. Similarly, if you are using flag_coul set to long, you cannot specify a
Coulombic cutoff for an atom type pair, since only one global Coulombic cutoff is allowed.
For lj/long/tip4p/long only the LJ cutoff can be specified since a Coulombic cutoff cannot be specified for an
individual I,J type pair. All type pairs use the same global Coulombic cutoff specified in the pair_style
command.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of the lj/long
pair styles can be mixed. The default mix value is geometric. See the "pair_modify" command for details.
These pair styles support the pair_modify shift option for the energy of the Lennard-Jones portion of the pair
interaction, assuming flag_lj is cut.
LAMMPS Users Manual
1540

These pair styles support the pair_modify table and table/disp options since they can tabulate the short-range
portion of the long-range Coulombic and dispersion interactions.
Thes pair styles do not support the pair_modify tail option for adding a long-range tail correction to the
Lennard-Jones portion of the energy and pressure.
These pair styles write their information to binary restart files, so pair_style and pair_coeff commands do not
need to be specified in an input script that reads a restart file.
The pair lj/long/coul/long styles support the use of the inner, middle, and outer keywords of the run_style
respa command, meaning the pairwise forces can be partitioned by distance at different levels of the rRESPA
hierarchy. See the run_style command for details.
Restrictions:
These styles are part of the KSPACE package. They are only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info. Note that the KSPACE package is installed by
default.
Related commands:
pair_coeff
Default: none
(In 't Veld) In 't Veld, Ismail, Grest, J Chem Phys (accepted) (2007).
(Jorgensen) Jorgensen, Chandrasekhar, Madura, Impey, Klein, J Chem Phys, 79, 926 (1983).
LAMMPS Users Manual
1541

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/sf command
pair_style lj/sf/omp command
Syntax:
pair_style lj/sf cutoff
cutoff = global cutoff for Lennard-Jones interactions (distance units)•
Examples:
pair_style lj/sf 2.5
pair_coeff * * 1.0 1.0
pair_coeff 1 1 1.0 1.0 3.0
Description:
Style lj/sf computes a truncated and force-shifted LJ interaction (Shifted Force Lennard-Jones), so that both
the potential and the force go continuously to zero at the cutoff (Toxvaerd):
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global LJ cutoff specified in the pair_style command is
used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
LAMMPS Users Manual
1542

section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for this pair style can
be mixed. Rin is a cutoff value and is mixed like the cutoff. The default mix value is geometric. See the
"pair_modify" command for details.
The pair_modify shift option is not relevant for this pair style, since the pair interaction goes to 0.0 at the
cutoff.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure, since the energy of the pair interaction is smoothed to 0.0 at the cutoff.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the USER-MISC package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Related commands:
pair_coeff
Default: none
(Toxvaerd) Toxvaerd, Dyre, J Chem Phys, 134, 081102 (2011).
LAMMPS Users Manual
1543
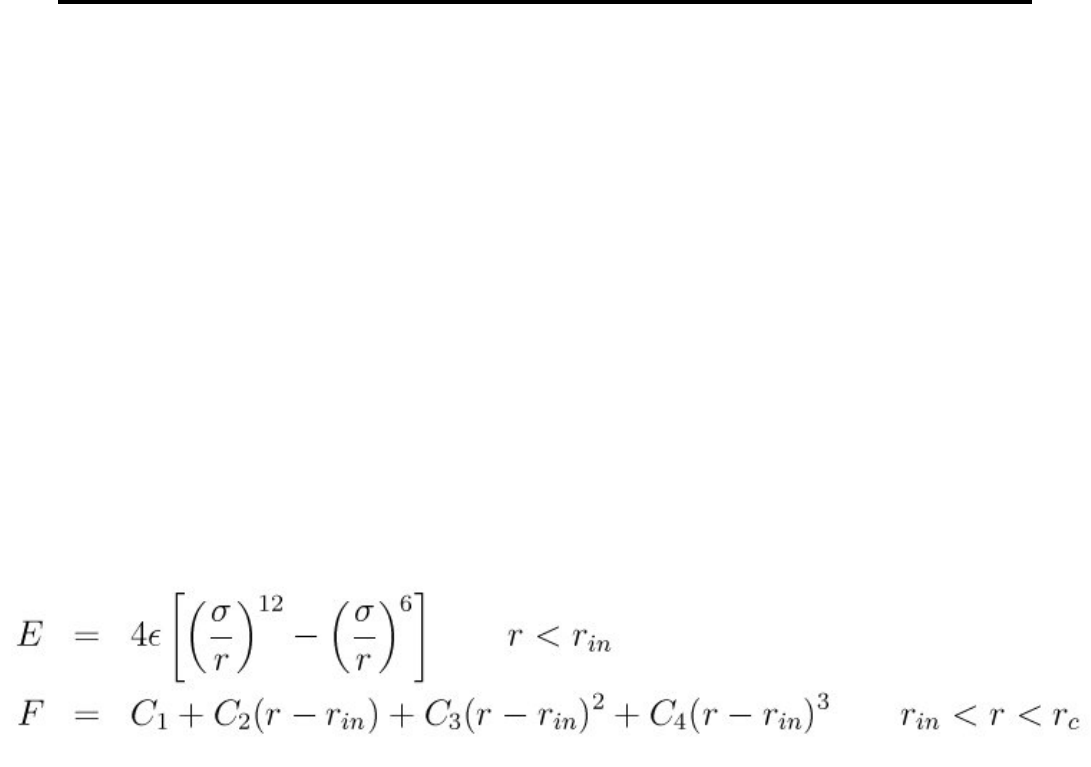
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/smooth command
pair_style lj/smooth/omp command
Syntax:
pair_style lj/smooth Rin Rc
Rin = inner cutoff beyond which force smoothing will be applied (distance units)• Rc = outer cutoff for lj/smooth interactions (distance units)•
Examples:
pair_style lj/smooth 8.0 10.0
pair_coeff * * 10.0 1.5
pair_coeff 1 1 20.0 1.3 7.0 9.0
Description:
Style lj/smooth computes a LJ interaction with a force smoothing applied between the inner and outer cutoff.
The polynomial coefficients C1, C2, C3, C4 are computed by LAMMPS to cause the force to vary smoothly
from the inner cutoff Rin to the outer cutoff Rc.
At the inner cutoff the force and its 1st derivative will match the unsmoothed LJ formula. At the outer cutoff
the force and its 1st derivative will be 0.0. The inner cutoff cannot be 0.0.
NOTE: this force smoothing causes the energy to be discontinuous both in its values and 1st derivative. This
can lead to poor energy conservation and may require the use of a thermostat. Plot the energy and force
resulting from this formula via the pair_write command to see the effect.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• inner (distance units)• outer (distance units)•
LAMMPS Users Manual
1544

The last 2 coefficients are optional inner and outer cutoffs. If not specified, the global values for Rin and Rc
are used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon, sigma, Rin coefficients and the cutoff distance for this pair style
can be mixed. Rin is a cutoff value and is mixed like the cutoff. The other coefficients are mixed according to
the pair_modify mix option. The default mix value is geometric. See the "pair_modify" command for details.
This pair style supports the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure, since the energy of the pair interaction is smoothed to 0.0 at the cutoff.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_coeff, pair lj/smooth/linear
Default: none
LAMMPS Users Manual
1545
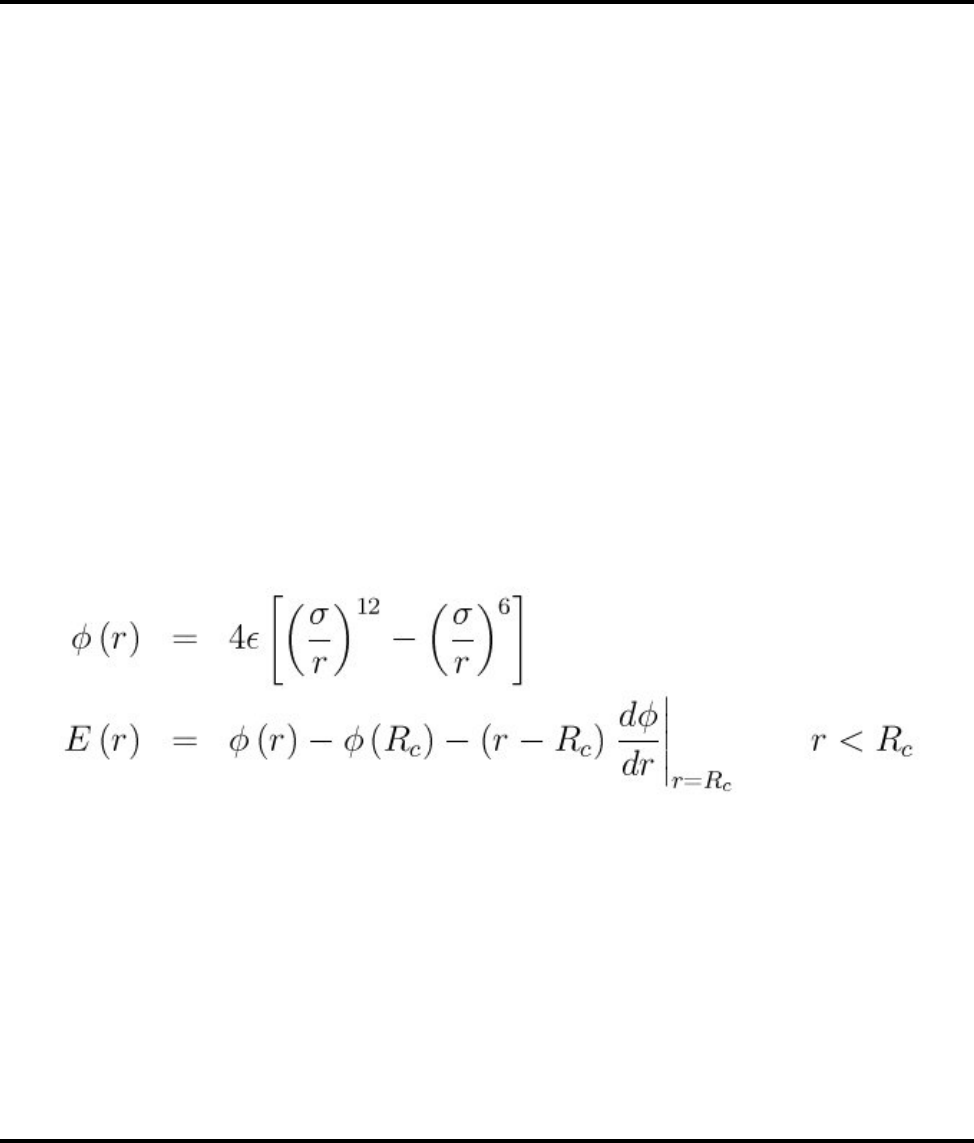
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/smooth/linear command
pair_style lj/smooth/linear/omp command
Syntax:
pair_style lj/smooth/linear Rc
Rc = cutoff for lj/smooth/linear interactions (distance units)•
Examples:
pair_style lj/smooth/linear 5.456108274435118
pair_coeff * * 0.7242785984051078 2.598146797350056
pair_coeff 1 1 20.0 1.3 9.0
Description:
Style lj/smooth/linear computes a LJ interaction that combines the standard 12/6 Lennard-Jones function and
subtracts a linear term that includes the cutoff distance Rc, as in this formula:
At the cutoff Rc, the energy and force (its 1st derivative) will be 0.0.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global value for Rc is used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
LAMMPS Users Manual
1546

These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance can be mixed. The
default mix value is geometric. See the "pair_modify" command for details.
This pair style does not support the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure, since the energy of the pair interaction is smoothed to 0.0 at the cutoff.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_coeff, pair lj/smooth
Default: none
LAMMPS Users Manual
1547

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/cut/soft command
pair_style lj/cut/soft/omp command
pair_style lj/cut/coul/cut/soft command
pair_style lj/cut/coul/cut/soft/omp command
pair_style lj/cut/coul/long/soft command
pair_style lj/cut/coul/long/soft/omp command
pair_style lj/cut/tip4p/long/soft command
pair_style lj/cut/tip4p/long/soft/omp command
pair_style lj/charmm/coul/long/soft command
pair_style lj/charmm/coul/long/soft/omp command
pair_style coul/cut/soft command
pair_style coul/cut/soft/omp command
pair_style coul/long/soft command
pair_style coul/long/soft/omp command
pair_style tip4p/long/soft command
pair_style tip4p/long/soft/omp command
Syntax:
pair_style style args
style = lj/cut/soft or lj/cut/coul/cut/soft or lj/cut/coul/long/soft or lj/cut/tip4p/long/soft or
lj/charmm/coul/long/soft or coul/cut/soft or coul/long/soft or tip4p/long/soft
•
args = list of arguments for a particular style•
lj/cut/soft args = n alpha_lj cutoff
n, alpha_LJ = parameters of soft-core potential
cutoff = global cutoff for Lennard-Jones interactions (distance units)
lj/cut/coul/cut/soft args = n alpha_LJ alpha_C cutoff (cutoff2)
n, alpha_LJ, alpha_C = parameters of soft-core potential
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
LAMMPS Users Manual
1548
lj/cut/coul/long/soft args = n alpha_LJ alpha_C cutoff
n, alpha_LJ, alpha_C = parameters of the soft-core potential
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
lj/cut/tip4p/long/soft args = otype htype btype atype qdist n alpha_LJ alpha_C cutoff (cutoff2)
otype,htype = atom types for TIP4P O and H
btype,atype = bond and angle types for TIP4P waters
qdist = distance from O atom to massless charge (distance units)
n, alpha_LJ, alpha_C = parameters of the soft-core potential
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
lj/charmm/coul/long/soft args = n alpha_LJ alpha_C inner outer (cutoff)
n, alpha_LJ, alpha_C = parameters of the soft-core potential
inner, outer = global switching cutoffs for LJ (and Coulombic if only 5 args)
cutoff = global cutoff for Coulombic (optional, outer is Coulombic cutoff if only 5 args)
coul/cut/soft args = n alpha_C cutoff
n, alpha_C = parameters of the soft-core potential
cutoff = global cutoff for Coulomb interactions (distance units)
coul/long/soft args = n alpha_C cutoff
n, alpha_C = parameters of the soft-core potential
cutoff = global cutoff for Coulomb interactions (distance units)
tip4p/long/soft args = otype htype btype atype qdist n alpha_C cutoff
otype,htype = atom types for TIP4P O and H
btype,atype = bond and angle types for TIP4P waters
qdist = distance from O atom to massless charge (distance units)
n, alpha_C = parameters of the soft-core potential
cutoff = global cutoff for Coulomb interactions (distance units)
Examples:
pair_style lj/cut/soft 2.0 0.5 9.5
pair_coeff * * 0.28 3.1 1.0
pair_coeff 1 1 0.28 3.1 1.0 9.5
pair_style lj/cut/coul/cut/soft 2.0 0.5 10.0 9.5
pair_style lj/cut/coul/cut/soft 2.0 0.5 10.0 9.5 9.5
pair_coeff * * 0.28 3.1 1.0
pair_coeff 1 1 0.28 3.1 0.5 10.0
pair_coeff 1 1 0.28 3.1 0.5 10.0 9.5
pair_style lj/cut/coul/long/soft 2.0 0.5 10.0 9.5
pair_style lj/cut/coul/long/soft 2.0 0.5 10.0 9.5 9.5
pair_coeff * * 0.28 3.1 1.0
pair_coeff 1 1 0.28 3.1 0.0 10.0
pair_coeff 1 1 0.28 3.1 0.0 10.0 9.5
pair_style lj/cut/tip4p/long/soft 1 2 7 8 0.15 2.0 0.5 10.0 9.8
pair_style lj/cut/tip4p/long/soft 1 2 7 8 0.15 2.0 0.5 10.0 9.8 9.5
pair_coeff * * 0.155 3.1536 1.0
pair_coeff 1 1 0.155 3.1536 1.0 9.5
pair_style lj/charmm/coul/long 2.0 0.5 10.0 8.0 10.0
pair_style lj/charmm/coul/long 2.0 0.5 10.0 8.0 10.0 9.0
pair_coeff * * 0.28 3.1 1.0
pair_coeff 1 1 0.28 3.1 1.0 0.14 3.1
pair_style coul/long/soft 1.0 10.0 9.5
pair_coeff * * 1.0
pair_coeff 1 1 1.0 9.5
LAMMPS Users Manual
1549
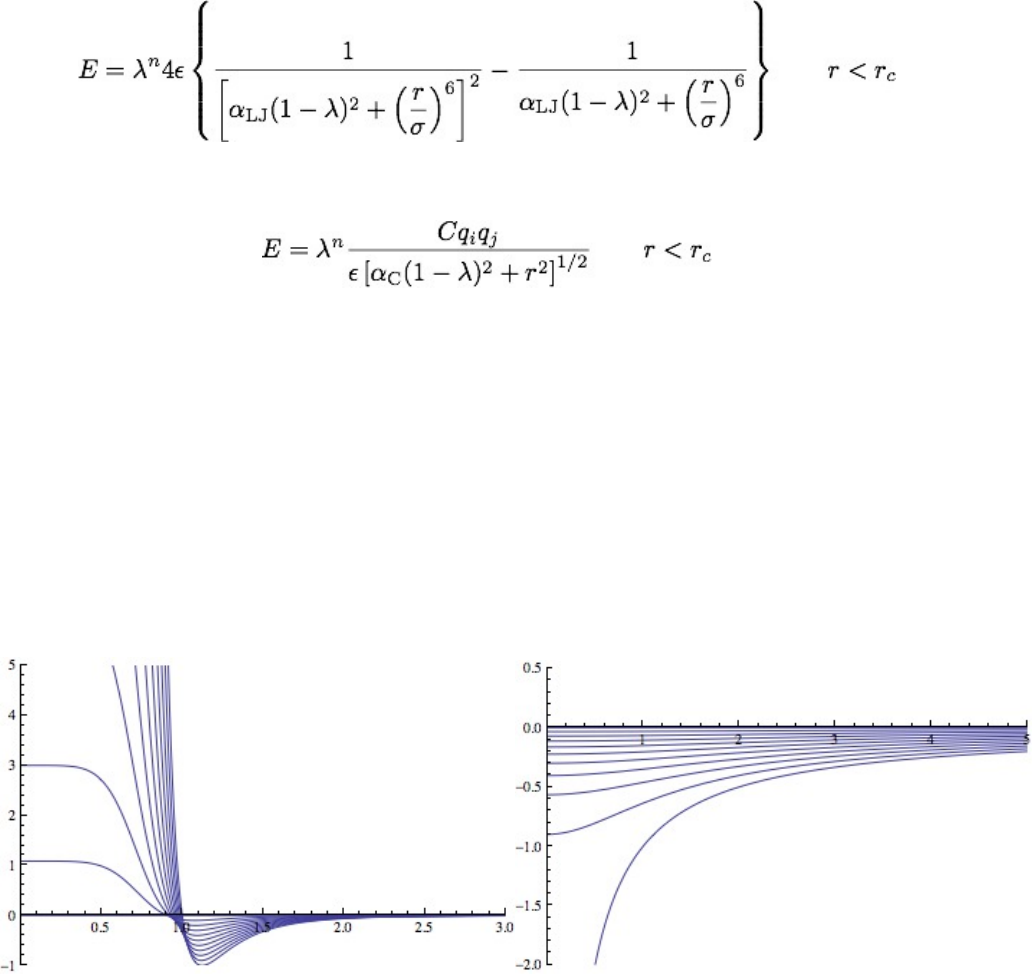
pair_style tip4p/long/soft 1 2 7 8 0.15 2.0 0.5 10.0 9.8
pair_coeff * * 1.0
pair_coeff 1 1 1.0 9.5
Description:
The lj/cut/soft style and substyles compute the 12/6 Lennard-Jones and Coulomb potential modified by a soft
core, in order to avoid singularities during free energy calculations when sites are created or annihilated
(Beutler),
Coulomb interactions are also damped with a soft core at short distance,
In the Coulomb part C is an energy-conversion constant, q_i and q_j are the charges on the 2 atoms, and
epsilon is the dielectric constant which can be set by the dielectric command.
The coefficient lambda is an activation parameter. When lambda = 1 the pair potential is identical to a
Lennard-Jones term or a Coulomb term or a combination of both. When lambda = 0 the interactions are
deactivated. The transition between these two extrema is smoothed by a soft repulsive core in order to avoid
singularities in potential energy and forces when sites are created or annihilated and can overlap (Beutler).
The parameters n, alpha_LJ and alpha_C are set in the pair_style command, before the cutoffs. Usual choices
for the exponent are n = 2 or n = 1. For the remaining coefficients alpha_LJ = 0.5 and alpha_C = 10
Angstrom^2 are appropriate choices. Plots of the LJ and Coulomb terms are shown below, for lambda ranging
from 1 to 0 every 0.1.
For the lj/cut/coul/cut/soft or lj/cut/coul/long/soft pair styles, the following coefficients must be defined for
each pair of atoms types via the pair_coeff command as in the examples above, or in the data file or restart
files read by the read_data or read_restart commands, or by mixing as described below:
LAMMPS Users Manual
1550

epsilon (energy units)• sigma (distance units)• lambda (activation parameter between 0 and 1)• cutoff1 (distance units)• cutoff2 (distance units)•
The latter two coefficients are optional. If not specified, the global LJ and Coulombic cutoffs specified in the
pair_style command are used. If only one cutoff is specified, it is used as the cutoff for both LJ and
Coulombic interactions for this type pair. If both coefficients are specified, they are used as the LJ and
Coulombic cutoffs for this type pair. You cannot specify 2 cutoffs for style lj/cut/soft, since it has no
Coulombic terms. For the coul/cut/soft and coul/long/soft only lambda and the optional cutoff2 are to be
specified.
Style lj/cut/tip4p/long/soft implements a soft-core version of the TIP4P water model. The usage of this pair
style is documented in the pair_lj styles. The soft-core version introduces the lambda parameter to the list of
arguments, after epsilon and sigma in the pair_coeff command. The parameters n, alpha_LJ and alpha_C are
set in the pair_style command, before the cutoffs.
Style lj/charmm/coul/long/soft implements a soft-core version of the CHARMM version of LJ interactions
with an additional switching function S(r) that ramps the energy and force smoothly to zero between an inner
and outer cutoff. The usage of this pair style is documented in the pair_charmm styles. The soft-core version
introduces the lambda parameter to the list of arguments, after epsilon and sigma in the pair_coeff command
(and before the optional eps14 and sigma14). The parameters n, alpha_LJ and alpha_C are set in the pair_style
command, before the cutoffs.
The coul/cut/soft, coul/long/soft and tip4p/long/soft substyles are designed to be combined with other pair
potentials via the pair_style hybrid/overlay command. This is because they have no repulsive core. Hence, if
used by themselves, there will be no repulsion to keep two oppositely charged particles from overlapping each
other. In this case, if lambda = 1, a singularity may occur. These substyles are suitable to represent charges
embedded in the Lennard-Jones radius of another site (for example hydrogen atoms in several water models).
NOTES: When using the core-softed Coulomb potentials with long-range solvers (coul/long/soft,
lj/cut/coul/long/soft, etc.) in a free energy calculation in which sites holding electrostatic charges are being
created or annihilated (using fix adapt/fep and compute fep) it is important to adapt both the lambda activation
parameter (from 0 to 1, or the reverse) and the value of the charge (from 0 to its final value, or the reverse).
This ensures that long-range electrostatic terms (kspace) are correct. It is not necessary to use core-softed
Coulomb potentials if the van der Waals site is present during the free-energy route, thus avoiding overlap of
the charges. Examples are provided in the LAMMPS source directory tree, under examples/USER/fep.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
LAMMPS Users Manual
1551
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, tail correction, restart info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for this pair style can
be mixed. The default mix value is geometric. See the "pair_modify" command for details.
These pair styles support the pair_modify shift option for the energy of the Lennard-Jones portion of the pair
interaction.
These pair styles support the pair_modify tail option for adding a long-range tail correction to the energy and
pressure for the Lennard-Jones portion of the pair interaction.
These pair styles write information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
Restrictions:
To avoid division by zero do not set sigma = 0; use the lambda parameter instead to activate/deactivate
interactions, or use epsilon = 0 and sigma = 1. Alternatively, when sites do not interact though the
Lennard-Jones term the coul/long/soft or similar substyle can be used via the pair_style hybrid/overlay
command.
All of the plain soft pair styles are part of the USER-FEP package. The long styles also requires the KSPACE
package to be installed. They are only enabled if LAMMPS was built with those packages. See the Making
LAMMPS section for more info.
Related commands:
pair_coeff, fix adapt, fix adapt/fep, compute fep
Default: none
(Beutler) Beutler, Mark, van Schaik, Gerber, van Gunsteren, Chem Phys Lett, 222, 529 (1994).
LAMMPS Users Manual
1552
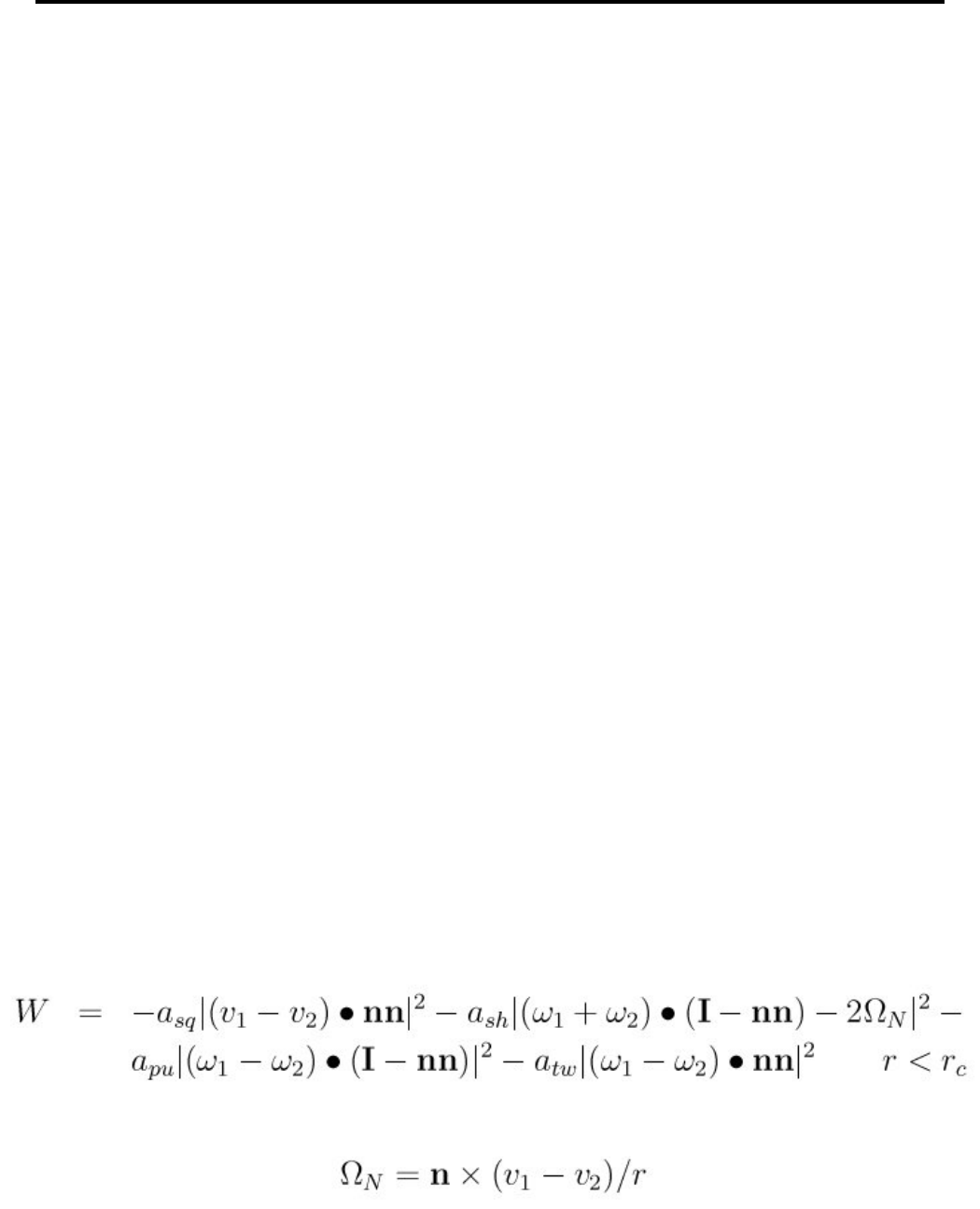
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lubricate command
pair_style lubricate/omp command
pair_style lubricate/poly command
pair_style lubricate/poly/omp command
Syntax:
pair_style style mu flaglog flagfld cutinner cutoff flagHI flagVF
style = lubricate or lubricate/poly• mu = dynamic viscosity (dynamic viscosity units)• flaglog = 0/1 to exclude/include log terms in the lubrication approximation• flagfld = 0/1 to exclude/include Fast Lubrication Dynamics (FLD) effects• cutinner = inner cutoff distance (distance units)• cutoff = outer cutoff for interactions (distance units)• flagHI (optional) = 0/1 to exclude/include 1/r hydrodynamic interactions• flagVF (optional) = 0/1 to exclude/include volume fraction corrections in the long-range isotropic
terms
•
Examples: (all assume radius = 1)
pair_style lubricate 1.5 1 1 2.01 2.5
pair_coeff 1 1 2.05 2.8
pair_coeff * *
pair_style lubricate 1.5 1 1 2.01 2.5
pair_coeff * *
variable mu equal ramp(1,2)
fix 1 all adapt 1 pair lubricate mu * * v_mu
Description:
Styles lubricate and lubricate/poly compute hydrodynamic interactions between mono-disperse finite-size
spherical particles in a pairwise fashion. The interactions have 2 components. The first is Ball-Melrose
lubrication terms via the formulas in (Ball and Melrose)
LAMMPS Users Manual
1553
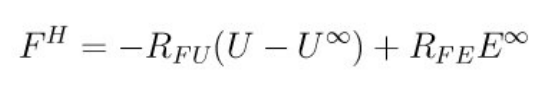
which represents the dissipation W between two nearby particles due to their relative velocities in the
presence of a background solvent with viscosity mu. Note that this is dynamic viscosity which has units of
mass/distance/time, not kinematic viscosity.
The Asq (squeeze) term is the strongest and is included if flagHI is set to 1 (default). It scales as 1/gap where
gap is the separation between the surfaces of the 2 particles. The Ash (shear) and Apu (pump) terms are only
included if flaglog is set to 1. They are the next strongest interactions, and the only other singular interaction,
and scale as log(gap). Note that flaglog = 1 and flagHI = 0 is invalid, and will result in a warning message,
after which flagHI will be set to 1. The Atw (twist) term is currently not included. It is typically a very small
contribution to the lubrication forces.
The flagHI and flagVF settings are optional. Neither should be used, or both must be defined.
Cutinner sets the minimum center-to-center separation that will be used in calculations irrespective of the
actual separation. Cutoff is the maximum center-to-center separation at which an interaction is computed.
Using a cutoff less than 3 radii is recommended if flaglog is set to 1.
The other component is due to the Fast Lubrication Dynamics (FLD) approximation, described in (Kumar),
which can be represented by the following equation
where U represents the velocities and angular velocities of the particles, U^infty represents the velocity and
the angular velocity of the undisturbed fluid, and E^infty represents the rate of strain tensor of the undisturbed
fluid with viscosity mu. Again, note that this is dynamic viscosity which has units of mass/distance/time, not
kinematic viscosity. Volume fraction corrections to R_FU are included as long as flagVF is set to 1 (default).
NOTE: When using the FLD terms, these pair styles are designed to be used with explicit time integration and
a correspondingly small timestep. Thus either fix nve/sphere or fix nve/asphere should be used for time
integration. To perform implicit FLD, see the pair_style lubricateU command.
Style lubricate requires monodisperse spherical particles; style lubricate/poly allows for polydisperse
spherical particles.
The viscosity mu can be varied in a time-dependent manner over the course of a simulation, in which case in
which case the pair_style setting for mu will be overridden. See the fix adapt command for details.
If the suspension is sheared via the fix deform command then the pair style uses the shear rate to adjust the
hydrodynamic interactions accordingly. Volume changes due to fix deform are accounted for when computing
the volume fraction corrections to R_FU.
When computing the volume fraction corrections to R_FU, the presence of walls (whether moving or
stationary) will affect the volume fraction available to colloidal particles. This is currently accounted for with
the following types of walls: wall/lj93, wall/lj126, wall/colloid, and wall/harmonic. For these wall styles, the
correct volume fraction will be used when walls do not coincide with the box boundary, as well as when walls
move and thereby cause a change in the volume fraction. Other wall styles will still work, but they will result
in the volume fraction being computed based on the box boundaries.
LAMMPS Users Manual
1554

Since lubrication forces are dissipative, it is usually desirable to thermostat the system at a constant
temperature. If Brownian motion (at a constant temperature) is desired, it can be set using the pair_style
brownian command. These pair styles and the brownian style should use consistent parameters for mu,
flaglog, flagfld, cutinner, cutoff, flagHI and flagVF.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
cutinner (distance units)• cutoff (distance units)•
The two coefficients are optional. If neither is specified, the two cutoffs specified in the pair_style command
are used. Otherwise both must be specified.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in this
section of the manual. The accelerated styles take the same arguments and should produce the same results,
except for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See this section of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the two cutoff distances for this pair style can be mixed. The default mix
value is geometric. See the "pair_modify" command for details.
This pair style does not support the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
LAMMPS Users Manual
1555

These styles are part of the COLLOID package. They are only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Only spherical monodisperse particles are allowed for pair_style lubricate.
Only spherical particles are allowed for pair_style lubricate/poly.
These pair styles will not restart exactly when using the read_restart command, though they should provide
statistically similar results. This is because the forces they compute depend on atom velocities. See the
read_restart command for more details.
Related commands:
pair_coeff, pair_style lubricateU
Default:
The default settings for the optional args are flagHI = 1 and flagVF = 1.
(Ball) Ball and Melrose, Physica A, 247, 444-472 (1997).
(Kumar) Kumar and Higdon, Phys Rev E, 82, 051401 (2010). See also his thesis for more details: A. Kumar,
"Microscale Dynamics in Suspensions of Non-spherical Particles", Thesis, University of Illinois
Urbana-Champaign, (2010). (https://www.ideals.illinois.edu/handle/2142/16032)
LAMMPS Users Manual
1556
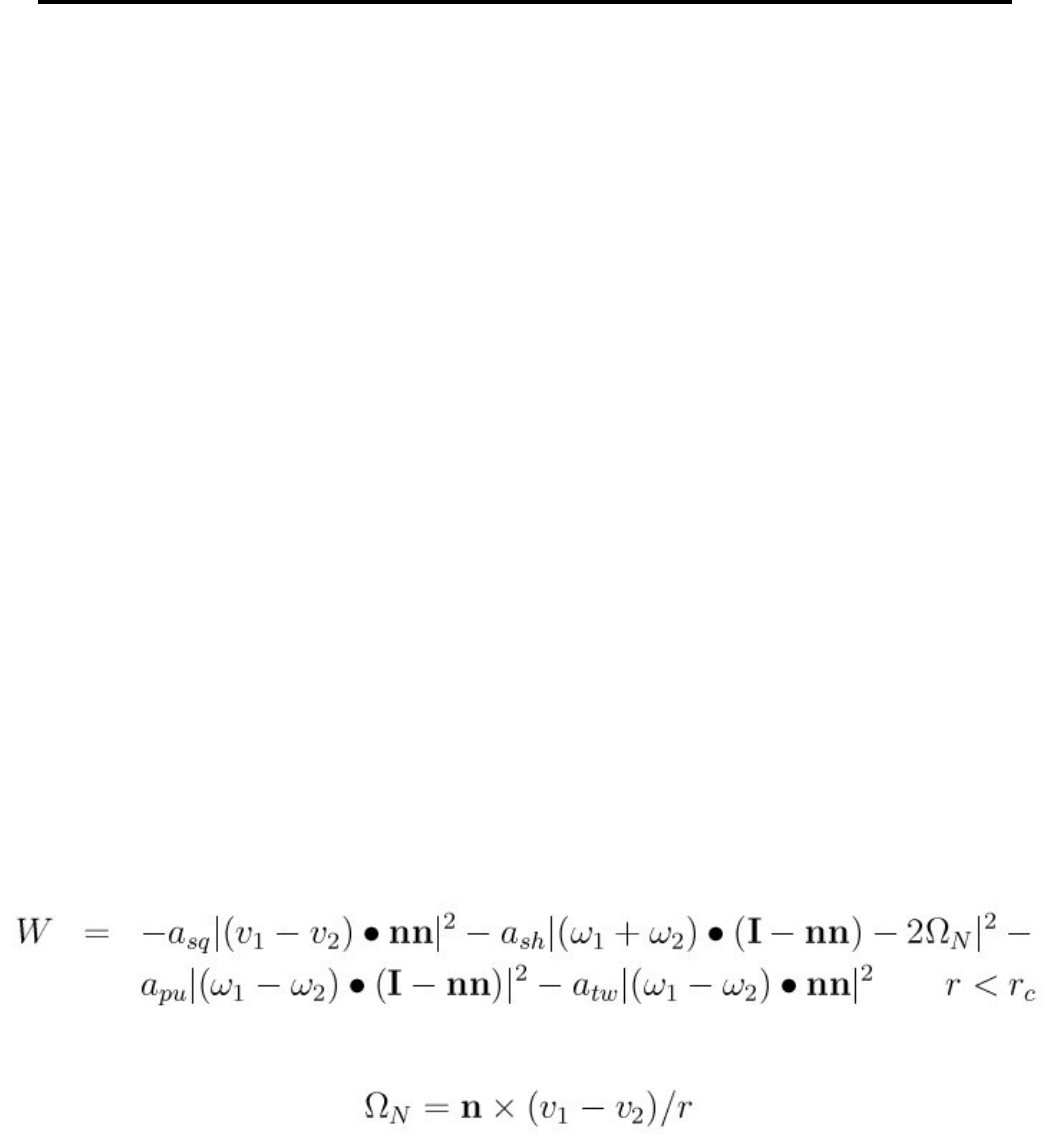
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lubricateU command
pair_style lubricateU/poly command
Syntax:
pair_style style mu flaglog cutinner cutoff gdot flagHI flagVF
style = lubricateU or lubricateU/poly• mu = dynamic viscosity (dynamic viscosity units)• flaglog = 0/1 to exclude/include log terms in the lubrication approximation• cutinner = inner cut off distance (distance units)• cutoff = outer cutoff for interactions (distance units)• gdot = shear rate (1/time units)• flagHI (optional) = 0/1 to exclude/include 1/r hydrodynamic interactions• flagVF (optional) = 0/1 to exclude/include volume fraction corrections in the long-range isotropic
terms
•
Examples: (all assume radius = 1)
pair_style lubricateU 1.5 1 2.01 2.5 0.01 1 1
pair_coeff 1 1 2.05 2.8
pair_coeff * *
Description:
Styles lubricateU and lubricateU/poly compute velocities and angular velocities for finite-size spherical
particles such that the hydrodynamic interaction balances the force and torque due to all other types of
interactions.
The interactions have 2 components. The first is Ball-Melrose lubrication terms via the formulas in (Ball and
Melrose)
which represents the dissipation W between two nearby particles due to their relative velocities in the
presence of a background solvent with viscosity mu. Note that this is dynamic viscosity which has units of
mass/distance/time, not kinematic viscosity.
LAMMPS Users Manual
1557
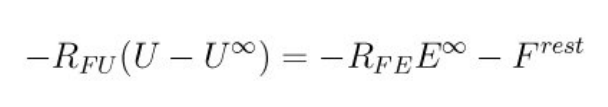
The Asq (squeeze) term is the strongest and is included as long as flagHI is set to 1 (default). It scales as 1/gap
where gap is the separation between the surfaces of the 2 particles. The Ash (shear) and Apu (pump) terms are
only included if flaglog is set to 1. They are the next strongest interactions, and the only other singular
interaction, and scale as log(gap). Note that flaglog = 1 and flagHI = 0 is invalid, and will result in a warning
message, after which flagHI will be set to 1. The Atw (twist) term is currently not included. It is typically a
very small contribution to the lubrication forces.
The flagHI and flagVF settings are optional. Neither should be used, or both must be defined.
Cutinner sets the minimum center-to-center separation that will be used in calculations irrespective of the
actual separation. Cutoff is the maximum center-to-center separation at which an interaction is computed.
Using a cutoff less than 3 radii is recommended if flaglog is set to 1.
The other component is due to the Fast Lubrication Dynamics (FLD) approximation, described in (Kumar).
The equation being solved to balance the forces and torques is
where U represents the velocities and angular velocities of the particles, U^infty represents the velocities and
the angular velocities of the undisturbed fluid, and E^infty represents the rate of strain tensor of the
undisturbed fluid flow with viscosity mu. Again, note that this is dynamic viscosity which has units of
mass/distance/time, not kinematic viscosity. Volume fraction corrections to R_FU are included if flagVF is set
to 1 (default).
Frest represents the forces and torques due to all other types of interactions, e.g. Brownian, electrostatic etc.
Note that this algorithm neglects the inertial terms, thereby removing the restriction of resolving the small
interial time scale, which may not be of interest for colloidal particles. This pair style solves for the velocity
such that the hydrodynamic force balances all other types of forces, thereby resulting in a net zero force (zero
inertia limit). When defining this pair style, it must be defined last so that when this style is invoked all other
types of forces have already been computed. For the same reason, it won't work if additional non-pair styles
are defined (such as bond or Kspace forces) as they are calculated in LAMMPS after the pairwise interactions
have been computed.
NOTE: When using these styles, the these pair styles are designed to be used with implicit time integration
and a correspondingly larger timestep. Thus either fix nve/noforce should be used for spherical particles
defined via atom_style sphere or fix nve/asphere/noforce should be used for spherical particles defined via
atom_style ellipsoid. This is because the velocity and angular momentum of each particle is set by the pair
style, and should not be reset by the time integration fix.
Style lubricateU requires monodisperse spherical particles; style lubricateU/poly allows for polydisperse
spherical particles.
If the suspension is sheared via the fix deform command then the pair style uses the shear rate to adjust the
hydrodynamic interactions accordingly. Volume changes due to fix deform are accounted for when computing
the volume fraction corrections to R_FU.
When computing the volume fraction corrections to R_FU, the presence of walls (whether moving or
stationary) will affect the volume fraction available to colloidal particles. This is currently accounted for with
LAMMPS Users Manual
1558

the following types of walls: wall/lj93, wall/lj126, wall/colloid, and wall/harmonic. For these wall styles, the
correct volume fraction will be used when walls do not coincide with the box boundary, as well as when walls
move and thereby cause a change in the volume fraction. To use these wall styles with pair_style lubricateU
or lubricateU/poly, the fld yes option must be specified in the fix wall command.
Since lubrication forces are dissipative, it is usually desirable to thermostat the system at a constant
temperature. If Brownian motion (at a constant temperature) is desired, it can be set using the pair_style
brownian command. These pair styles and the brownian style should use consistent parameters for mu,
flaglog, flagfld, cutinner, cutoff, flagHI and flagVF.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
cutinner (distance units)• cutoff (distance units)•
The two coefficients are optional. If neither is specified, the two cutoffs specified in the pair_style command
are used. Otherwise both must be specified.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the two cutoff distances for this pair style can be mixed. The default mix
value is geometric. See the "pair_modify" command for details.
This pair style does not support the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
These styles are part of the COLLOID package. They are only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Currently, these pair styles assume that all other types of forces/torques on the particles have been already
been computed when it is invoked. This requires this style to be defined as the last of the pair styles, and that
no fixes apply additional constraint forces. One exception is the fix wall/colloid commands, which has an
"fld" option to apply their wall forces correctly.
Only spherical monodisperse particles are allowed for pair_style lubricateU.
LAMMPS Users Manual
1559

Only spherical particles are allowed for pair_style lubricateU/poly.
For sheared suspensions, it is assumed that the shearing is done in the xy plane, with x being the velocity
direction and y being the velocity-gradient direction. In this case, one must use fix deform with the same rate
of shear (erate).
Related commands:
pair_coeff, pair_style lubricate
Default:
The default settings for the optional args are flagHI = 1 and flagVF = 1.
(Ball) Ball and Melrose, Physica A, 247, 444-472 (1997).
(Kumar) Kumar and Higdon, Phys Rev E, 82, 051401 (2010).
LAMMPS Users Manual
1560

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/mdf command
pair_style buck/mdf command
pair_style lennard/mdf command
Syntax:
pair_style style args
style = lj/mdf or buck/mdf or lennard/mdf• args = list of arguments for a particular style
lj/mdf args = cutoff1 cutoff2
cutoff1 = inner cutoff for the start of the tapering function
cutoff1 = out cutoff for the end of the tapering function
buck/mdf args = cutoff1 cutoff2
cutoff1 = inner cutoff for the start of the tapering function
cutoff1 = out cutoff for the end of the tapering function
lennard/mdf args = cutoff1 cutoff2
cutoff1 = inner cutoff for the start of the tapering function
cutoff1 = out cutoff for the end of the tapering function
•
Examples:
pair_style lj/mdf 2.5 3.0
pair_coeff * * 1 1
pair_coeff 1 1 1 1.1 2.8 3.0 3.2
pair_style buck 2.5 3.0
pair_coeff * * 100.0 1.5 200.0
pair_coeff * * 100.0 1.5 200.0 3.0 3.5
pair_style lennard/mdf 2.5 3.0
pair_coeff * * 1 1
pair_coeff 1 1 1 1.1 2.8 3.0 3.2
Description:
The lj/mdf, buck/mdf and lennard/mdf compute the standard 12-6 Lennard-Jones and Buckingham potential
with the addition of a taper function that ramps the energy and force smoothly to zero between an inner and
outer cutoff.
The tapering, f(r), is done by using the Mei, Davenport, Fernando function (Mei).
LAMMPS Users Manual
1561

where
Here r_m is the inner cutoff radius and r_cut is the outer cutoff radius.
For the lj/mdf pair_style, the potential energy, E(r), is the standard 12-6 Lennard-Jones written in the
epsilon/sigma form:
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• r_m (distance units)• r_cut (distance units)•
For the buck/mdf pair_style, the potential energy, E(r), is the standard Buckingham potential:
A (energy units)• \rho (distance units)• C (energy-distance^6 units)• r_m (distance units)• r_cut$ (distance units)•
For the lennard/mdf pair_style, the potential energy, E(r), is the standard 12-6 Lennard-Jones written in the
$A/B$ form:
LAMMPS Users Manual
1562

The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
A (energy-distance^12 units)• B (energy-distance^6 units)• r_m (distance units)• r_cut (distance units)•
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of the lj/cut
pair styles can be mixed. The default mix value is geometric. See the "pair_modify" command for details.
All of the lj/cut pair styles support the pair_modify shift option for the energy of the Lennard-Jones portion of
the pair interaction.
The lj/cut/coul/long and lj/cut/tip4p/long pair styles support the pair_modify table option since they can
tabulate the short-range portion of the long-range Coulombic interaction.
All of the lj/cut pair styles support the pair_modify tail option for adding a long-range tail correction to the
energy and pressure for the Lennard-Jones portion of the pair interaction.
All of the lj/cut pair styles write their information to binary restart files, so pair_style and pair_coeff
commands do not need to be specified in an input script that reads a restart file.
The lj/cut and lj/cut/coul/long pair styles support the use of the inner, middle, and outer keywords of the
run_style respa command, meaning the pairwise forces can be partitioned by distance at different levels of the
rRESPA hierarchy. The other styles only support the pair keyword of run_style respa. See the run_style
command for details.
Restrictions:
These pair styles can only be used if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info on packages.
Related commands:
pair_coeff
Default: none
(Mei) Mei, Davenport, Fernando, Phys Rev B, 43 4653 (1991)
LAMMPS Users Manual
1563

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style meam command
Syntax:
pair_style meam
Examples:
pair_style meam
pair_coeff * * ../potentials/library.meam Si ../potentials/si.meam Si
pair_coeff * * ../potentials/library.meam Ni Al NULL Ni Al Ni Ni
Description:
NOTE: The behavior of the MEAM potential for alloy systems has changed as of November 2010; see
description below of the mixture_ref_t parameter
Style meam computes pairwise interactions for a variety of materials using modified embedded-atom method
(MEAM) potentials (Baskes). Conceptually, it is an extension to the original EAM potentials which adds
angular forces. It is thus suitable for modeling metals and alloys with fcc, bcc, hcp and diamond cubic
structures, as well as covalently bonded materials like silicon and carbon.
In the MEAM formulation, the total energy E of a system of atoms is given by:
where F is the embedding energy which is a function of the atomic electron density rho, and phi is a pair
potential interaction. The pair interaction is summed over all neighbors J of atom I within the cutoff distance.
As with EAM, the multi-body nature of the MEAM potential is a result of the embedding energy term. Details
of the computation of the embedding and pair energies, as implemented in LAMMPS, are given in (Gullet)
and references therein.
The various parameters in the MEAM formulas are listed in two files which are specified by the pair_coeff
command. These are ASCII text files in a format consistent with other MD codes that implement MEAM
potentials, such as the serial DYNAMO code and Warp. Several MEAM potential files with parameters for
different materials are included in the "potentials" directory of the LAMMPS distribution with a ".meam"
suffix. All of these are parameterized in terms of LAMMPS metal units.
Note that unlike for other potentials, cutoffs for MEAM potentials are not set in the pair_style or pair_coeff
command; they are specified in the MEAM potential files themselves.
Only a single pair_coeff command is used with the meam style which specifies two MEAM files and the
element(s) to extract information for. The MEAM elements are mapped to LAMMPS atom types by
LAMMPS Users Manual
1564
specifying N additional arguments after the 2nd filename in the pair_coeff command, where N is the number
of LAMMPS atom types:
MEAM library file• Elem1, Elem2, ...• MEAM parameter file• N element names = mapping of MEAM elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential files.
As an example, the potentials/library.meam file has generic MEAM settings for a variety of elements. The
potentials/sic.meam file has specific parameter settings for a Si and C alloy system. If your LAMMPS
simulation has 4 atoms types and you want the 1st 3 to be Si, and the 4th to be C, you would use the following
pair_coeff command:
pair_coeff * * library.meam Si C sic.meam Si Si Si C
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The two filenames are for the library
and parameter file respectively. The Si and C arguments (between the file names) are the two elements for
which info will be extracted from the library file. The first three trailing Si arguments map LAMMPS atom
types 1,2,3 to the MEAM Si element. The final C argument maps LAMMPS atom type 4 to the MEAM C
element.
If the 2nd filename is specified as NULL, no parameter file is read, which simply means the generic
parameters in the library file are used. Use of the NULL specification for the parameter file is discouraged for
systems with more than a single element type (e.g. alloys), since the parameter file is expected to set element
interaction terms that are not captured by the information in the library file.
If a mapping value is specified as NULL, the mapping is not performed. This can be used when a meam
potential is used as part of the hybrid pair style. The NULL values are placeholders for atom types that will be
used with other potentials.
The MEAM library file provided with LAMMPS has the name potentials/library.meam. It is the "meamf" file
used by other MD codes. Aside from blank and comment lines (start with #) which can appear anywhere, it is
formatted as a series of entries, each of which has 19 parameters and can span multiple lines:
elt, lat, z, ielement, atwt, alpha, b0, b1, b2, b3, alat, esub, asub, t0, t1, t2, t3, rozero, ibar
The "elt" and "lat" parameters are text strings, such as elt = Si or Cu and lat = dia or fcc. Because the library
file is used by Fortran MD codes, these strings may be enclosed in single quotes, but this is not required. The
other numeric parameters match values in the formulas above. The value of the "elt" string is what is used in
the pair_coeff command to identify which settings from the library file you wish to read in. There can be
multiple entries in the library file with the same "elt" value; LAMMPS reads the 1st matching entry it finds
and ignores the rest.
Other parameters in the MEAM library file correspond to single-element potential parameters:
lat = lattice structure of reference configuration
z = number of nearest neighbors in the reference structure
ielement = atomic number
atwt = atomic weight
alat = lattice constant of reference structure
LAMMPS Users Manual
1565
esub = energy per atom (eV) in the reference structure at equilibrium
asub = "A" parameter for MEAM (see e.g. (Baskes))
The alpha, b0, b1, b2, b3, t0, t1, t2, t3 parameters correspond to the standard MEAM parameters in the
literature (Baskes) (the b parameters are the standard beta parameters). The rozero parameter is an
element-dependent density scaling that weights the reference background density (see e.g. equation 4.5 in
(Gullet)) and is typically 1.0 for single-element systems. The ibar parameter selects the form of the function
G(Gamma) used to compute the electron density; options are
0 => G = sqrt(1+Gamma)
1 => G = exp(Gamma/2)
2 => not implemented
3 => G = 2/(1+exp(-Gamma))
4 => G = sqrt(1+Gamma)
-5 => G = +-sqrt(abs(1+Gamma))
If used, the MEAM parameter file contains settings that override or complement the library file settings.
Examples of such parameter files are in the potentials directory with a ".meam" suffix. Their format is the
same as is read by other Fortran MD codes. Aside from blank and comment lines (start with #) which can
appear anywhere, each line has one of the following forms. Each line can also have a trailing comment
(starting with #) which is ignored.
keyword = value
keyword(I) = value
keyword(I,J) = value
keyword(I,J,K) = value
The recognized keywords are as follows:
Ec, alpha, rho0, delta, lattce, attrac, repuls, nn2, Cmin, Cmax, rc, delr, augt1, gsmooth_factor, re
where
rc = cutoff radius for cutoff function; default = 4.0
delr = length of smoothing distance for cutoff function; default = 0.1
rho0(I) = relative density for element I (overwrites value
read from meamf file)
Ec(I,J) = cohesive energy of reference structure for I-J mixture
delta(I,J) = heat of formation for I-J alloy; if Ec_IJ is input as
zero, then LAMMPS sets Ec_IJ = (Ec_II + Ec_JJ)/2 - delta_IJ
alpha(I,J) = alpha parameter for pair potential between I and J (can
be computed from bulk modulus of reference structure
re(I,J) = equilibrium distance between I and J in the reference
structure
Cmax(I,J,K) = Cmax screening parameter when I-J pair is screened
by K (I<=J); default = 2.8
Cmin(I,J,K) = Cmin screening parameter when I-J pair is screened
by K (I<=J); default = 2.0
lattce(I,J) = lattice structure of I-J reference structure:
dia = diamond (interlaced fcc for alloy)
fcc = face centered cubic
bcc = body centered cubic
dim = dimer
b1 = rock salt (NaCl structure)
hcp = hexagonal close-packed
c11 = MoSi2 structure
l12 = Cu3Au structure (lower case L, followed by 12)
LAMMPS Users Manual
1566
b2 = CsCl structure (interpenetrating simple cubic)
nn2(I,J) = turn on second-nearest neighbor MEAM formulation for
I-J pair (see for example (Lee)).
0 = second-nearest neighbor formulation off
1 = second-nearest neighbor formulation on
default = 0
attrac(I,J) = additional cubic attraction term in Rose energy I-J pair potential
default = 0
repuls(I,J) = additional cubic repulsive term in Rose energy I-J pair potential
default = 0
zbl(I,J) = blend the MEAM I-J pair potential with the ZBL potential for small
atom separations (ZBL)
default = 1
gsmooth_factor = factor determining the length of the G-function smoothing
region; only significant for ibar=0 or ibar=4.
99.0 = short smoothing region, sharp step
0.5 = long smoothing region, smooth step
default = 99.0
augt1 = integer flag for whether to augment t1 parameter by
3/5*t3 to account for old vs. new meam formulations;
0 = don't augment t1
1 = augment t1
default = 1
ialloy = integer flag to use alternative averaging rule for t parameters,
for comparison with the DYNAMO MEAM code
0 = standard averaging (matches ialloy=0 in DYNAMO)
1 = alternative averaging (matches ialloy=1 in DYNAMO)
2 = no averaging of t (use single-element values)
default = 0
mixture_ref_t = integer flag to use mixture average of t to compute the background
reference density for alloys, instead of the single-element values
(see description and warning elsewhere in this doc page)
0 = do not use mixture averaging for t in the reference density
1 = use mixture averaging for t in the reference density
default = 0
erose_form = integer value to select the form of the Rose energy function
(see description below).
default = 0
emb_lin_neg = integer value to select embedding function for negative densities
0 = F(rho)=0
1 = F(rho) = -asub*esub*rho (linear in rho, matches DYNAMO)
default = 0
bkgd_dyn = integer value to select background density formula
0 = rho_bkgd = rho_ref_meam(a) (as in the reference structure)
1 = rho_bkgd = rho0_meam(a)*Z_meam(a) (matches DYNAMO)
default = 0
Rc, delr, re are in distance units (Angstroms in the case of metal units). Ec and delta are in energy units (eV in
the case of metal units).
Each keyword represents a quantity which is either a scalar, vector, 2d array, or 3d array and must be
specified with the correct corresponding array syntax. The indices I,J,K each run from 1 to N where N is the
number of MEAM elements being used.
Thus these lines
rho0(2) = 2.25
alpha(1,2) = 4.37
LAMMPS Users Manual
1567

set rho0 for the 2nd element to the value 2.25 and set alpha for the alloy interaction between elements 1 and 2
to 4.37.
The augt1 parameter is related to modifications in the MEAM formulation of the partial electron density
function. In recent literature, an extra term is included in the expression for the third-order density in order to
make the densities orthogonal (see for example (Wang), equation 3d); this term is included in the MEAM
implementation in lammps. However, in earlier published work this term was not included when deriving
parameters, including most of those provided in the library.meam file included with lammps, and to account
for this difference the parameter t1 must be augmented by 3/5*t3. If augt1=1, the default, this augmentation is
done automatically. When parameter values are fit using the modified density function, as in more recent
literature, augt1 should be set to 0.
The mixture_ref_t parameter is available to match results with those of previous versions of lammps (before
January 2011). Newer versions of lammps, by default, use the single-element values of the t parameters to
compute the background reference density. This is the proper way to compute these parameters. Earlier
versions of lammps used an alloy mixture averaged value of t to compute the background reference density.
Setting mixture_ref_t=1 gives the old behavior. WARNING: using mixture_ref_t=1 will give results that are
demonstrably incorrect for second-neighbor MEAM, and non-standard for first-neighbor MEAM; this option
is included only for matching with previous versions of lammps and should be avoided if possible.
The parameters attrac and repuls, along with the integer selection parameter erose_form, can be used to
modify the Rose energy function used to compute the pair potential. This function gives the energy of the
reference state as a function of interatomic spacing. The form of this function is:
astar = alpha * (r/re - 1.d0)
if erose_form = 0: erose = -Ec*(1+astar+a3*(astar**3)/(r/re))*exp(-astar)
if erose_form = 1: erose = -Ec*(1+astar+(-attrac+repuls/r)*(astar**3))*exp(-astar)
if erose_form = 2: erose = -Ec*(1 +astar + a3*(astar**3))*exp(-astar)
a3 = repuls, astar <0
a3 = attrac, astar >= 0
Most published MEAM parameter sets use the default values attrac=repulse=0. Setting repuls=attrac=delta
corresponds to the form used in several recent published MEAM parameter sets, such as (Valone)
NOTE: The default form of the erose expression in LAMMPS was corrected in March 2009. The current
version is correct, but may show different behavior compared with earlier versions of lammps with the attrac
and/or repuls parameters are non-zero. To obtain the previous default form, use erose_form = 1 (this form
does not seem to appear in the literature). An alternative form (see e.g. (Lee2)) is available using erose_form
= 2.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, where types I and J correspond to two different element types, mixing is
performed by LAMMPS with user-specifiable parameters as described above. You never need to specify a
pair_coeff command with I != J arguments for this style.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
LAMMPS Users Manual
1568

This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This style is part of the MEAM package. It is only enabled if LAMMPS was built with that package, which
also requires the MEAM library be built and linked with LAMMPS. See the Making LAMMPS section for
more info.
Related commands:
pair_coeff, pair_style eam, pair_style meam/spline
Default: none
(Baskes) Baskes, Phys Rev B, 46, 2727-2742 (1992).
(Gullet) Gullet, Wagner, Slepoy, SANDIA Report 2003-8782 (2003). This report may be accessed on-line via
this link.
(Lee) Lee, Baskes, Phys. Rev. B, 62, 8564-8567 (2000).
(Lee2) Lee, Baskes, Kim, Cho. Phys. Rev. B, 64, 184102 (2001).
(Valone) Valone, Baskes, Martin, Phys. Rev. B, 73, 214209 (2006).
(Wang) Wang, Van Hove, Ross, Baskes, J. Chem. Phys., 121, 5410 (2004).
(ZBL) J.F. Ziegler, J.P. Biersack, U. Littmark, "Stopping and Ranges of Ions in Matter", Vol 1, 1985,
Pergamon Press.
LAMMPS Users Manual
1569
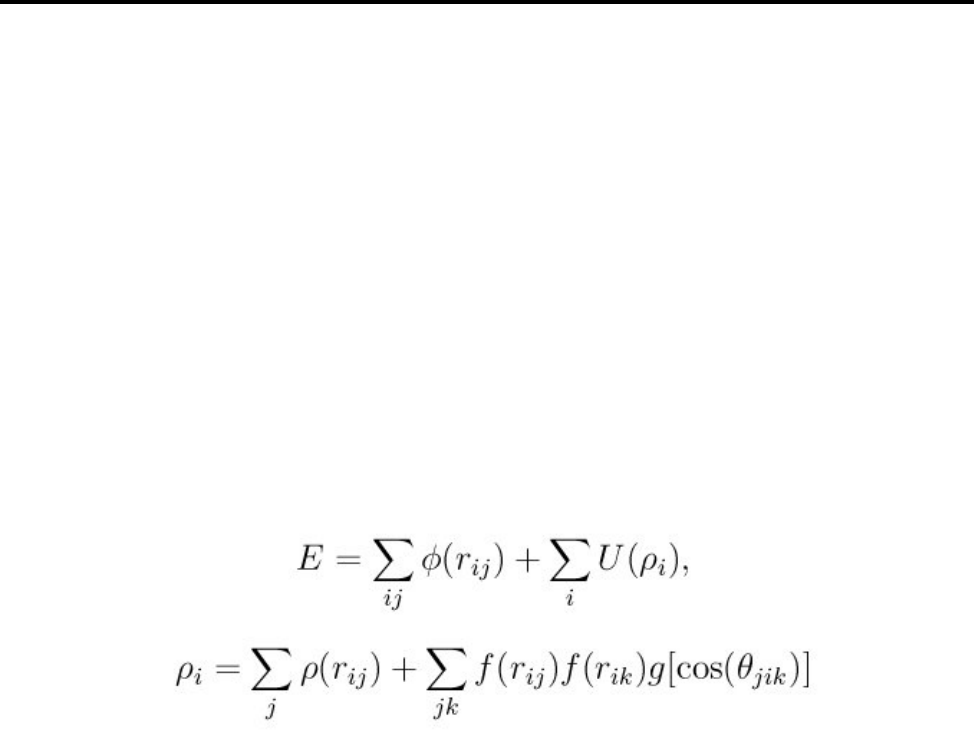
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style meam/spline
pair_style meam/spline/omp
Syntax:
pair_style meam/spline
Examples:
pair_style meam/spline
pair_coeff * * Ti.meam.spline Ti
pair_coeff * * Ti.meam.spline Ti Ti Ti
Description:
The meam/spline style computes pairwise interactions for metals using a variant of modified embedded-atom
method (MEAM) potentials (Lenosky). The total energy E is given by
where rho_i is the density at atom I, theta_jik is the angle between atoms J, I, and K centered on atom I. The
five functions Phi, U, rho, f, and g are represented by cubic splines.
The cutoffs and the coefficients for these spline functions are listed in a parameter file which is specified by
the pair_coeff command. Parameter files for different elements are included in the "potentials" directory of the
LAMMPS distribution and have a ".meam.spline" file suffix. All of these files are parameterized in terms of
LAMMPS metal units.
Note that unlike for other potentials, cutoffs for spline-based MEAM potentials are not set in the pair_style or
pair_coeff command; they are specified in the potential files themselves.
Unlike the EAM pair style, which retrieves the atomic mass from the potential file, the spline-based MEAM
potentials do not include mass information; thus you need to use the mass command to specify it.
Only a single pair_coeff command is used with the meam/spline style which specifies a potential file with
parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional
arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N element names = mapping of spline-based MEAM elements to atom types•
LAMMPS Users Manual
1570

See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, imagine the Ti.meam.spline file has values for Ti. If your LAMMPS simulation has 3 atoms
types and they are all to be treated with this potentials, you would use the following pair_coeff command:
pair_coeff * * Ti.meam.spline Ti Ti Ti
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The three Ti arguments map
LAMMPS atom types 1,2,3 to the Ti element in the potential file. If a mapping value is specified as NULL,
the mapping is not performed. This can be used when a meam/spline potential is used as part of the hybrid
pair style. The NULL values are placeholders for atom types that will be used with other potentials.
NOTE: The meam/spline style currently supports only single-element MEAM potentials. It may be extended
for alloy systems in the future.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
The current version of this pair style does not support multiple element types or mixing. It has been designed
for pure elements only.
This pair style does not support the pair_modify shift, table, and tail options.
The meam/spline pair style does not write its information to binary restart files, since it is stored in an external
potential parameter file. Thus, you need to re-specify the pair_style and pair_coeff commands in an input
script that reads a restart file.
The meam/spline pair style can only be used via the pair keyword of the run_style respa command. They do
not support the inner, middle, outer keywords.
Restrictions:
This pair style requires the newton setting to be "on" for pair interactions.
This pair style is only enabled if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info.
LAMMPS Users Manual
1571


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style meam/sw/spline
pair_style meam/sw/spline/omp
Syntax:
pair_style meam/sw/spline
Examples:
pair_style meam/sw/spline
pair_coeff * * Ti.meam.sw.spline Ti
pair_coeff * * Ti.meam.sw.spline Ti Ti Ti
Description:
The meam/sw/spline style computes pairwise interactions for metals using a variant of modified
embedded-atom method (MEAM) potentials (Lenosky) with an additional Stillinger-Weber (SW) term
(Stillinger) in the energy. This form of the potential was first proposed by Nicklas, Fellinger, and Park
(Nicklas). We refer to it as MEAM+SW. The total energy E is given by
where rho_I is the density at atom I, theta_JIK is the angle between atoms J, I, and K centered on atom I. The
seven functions Phi, F, G, U, rho, f, and g are represented by cubic splines.
The cutoffs and the coefficients for these spline functions are listed in a parameter file which is specified by
the pair_coeff command. Parameter files for different elements are included in the "potentials" directory of the
LAMMPS distribution and have a ".meam.sw.spline" file suffix. All of these files are parameterized in terms
of LAMMPS metal units.
Note that unlike for other potentials, cutoffs for spline-based MEAM+SW potentials are not set in the
pair_style or pair_coeff command; they are specified in the potential files themselves.
Unlike the EAM pair style, which retrieves the atomic mass from the potential file, the spline-based
MEAM+SW potentials do not include mass information; thus you need to use the mass command to specify
it.
LAMMPS Users Manual
1573

Only a single pair_coeff command is used with the meam/sw/spline style which specifies a potential file with
parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional
arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N element names = mapping of spline-based MEAM+SW elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, imagine the Ti.meam.sw.spline file has values for Ti. If your LAMMPS simulation has 3
atoms types and they are all to be treated with this potential, you would use the following pair_coeff
command:
pair_coeff * * Ti.meam.sw.spline Ti Ti Ti
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The three Ti arguments map
LAMMPS atom types 1,2,3 to the Ti element in the potential file. If a mapping value is specified as NULL,
the mapping is not performed. This can be used when a meam/sw/spline potential is used as part of the hybrid
pair style. The NULL values are placeholders for atom types that will be used with other potentials.
NOTE: The meam/sw/spline style currently supports only single-element MEAM+SW potentials. It may be
extended for alloy systems in the future.
Example input scripts that use this pair style are provided in the examples/USER/misc/meam_sw_spline
directory.
Mixing, shift, table, tail correction, restart, rRESPA info:
The pair style does not support multiple element types or mixing. It has been designed for pure elements only.
This pair style does not support the pair_modify shift, table, and tail options.
The meam/sw/spline pair style does not write its information to binary restart files, since it is stored in an
external potential parameter file. Thus, you need to re-specify the pair_style and pair_coeff commands in an
input script that reads a restart file.
The meam/sw/spline pair style can only be used via the pair keyword of the run_style respa command. They
do not support the inner, middle, outer keywords.
Restrictions:
This pair style requires the newton setting to be "on" for pair interactions.
This pair style is only enabled if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info.
Related commands:
pair_coeff, pair_style meam, pair_style meam/spline
LAMMPS Users Manual
1574

Default: none
(Lenosky) Lenosky, Sadigh, Alonso, Bulatov, de la Rubia, Kim, Voter, Kress, Modell. Simul. Mater. Sci.
Eng. 8, 825 (2000).
(Stillinger) Stillinger, Weber, Phys. Rev. B 31, 5262 (1985).
(Nicklas) The spline-based MEAM+SW format was first devised and used to develop potentials for bcc
transition metals by Jeremy Nicklas, Michael Fellinger, and Hyoungki Park at The Ohio State University.
LAMMPS Users Manual
1575
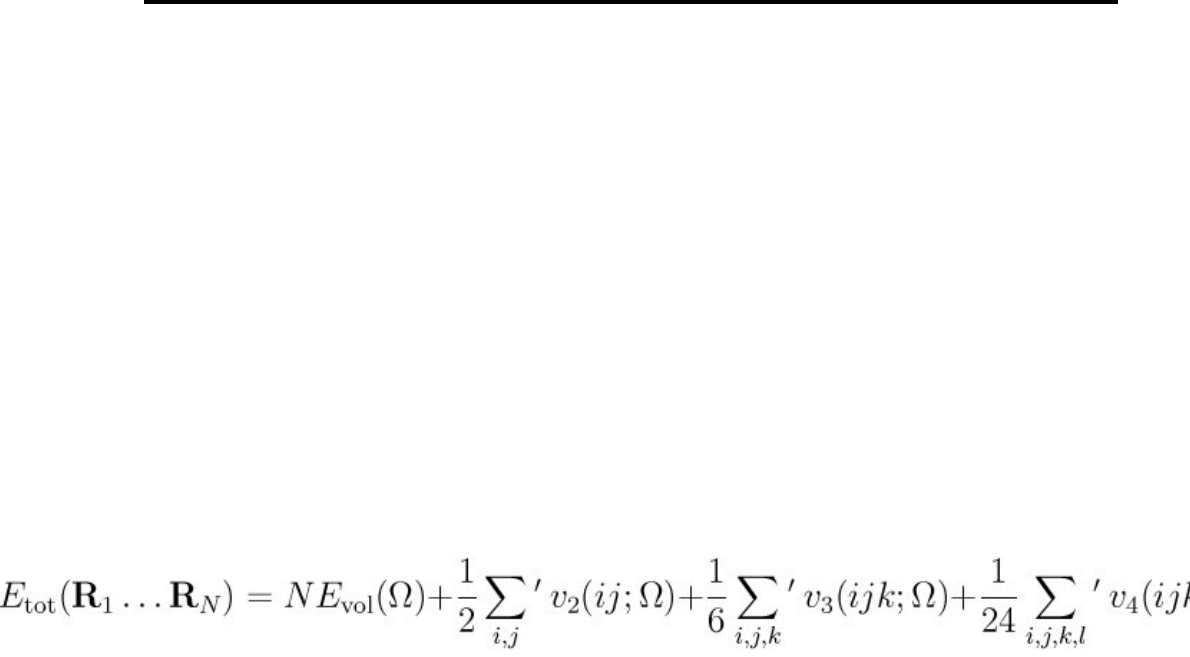
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style mgpt command
Syntax:
pair_style mgpt
Examples:
pair_style mgpt
pair_coeff * * Ta6.8x.mgpt.parmin Ta6.8x.mgpt.potin Omega
cp ~/lammps/potentials/Ta6.8x.mgpt.parmin parmin
cp ~/lammps/potentials/Ta6.8x.mgpt.potin potin
pair_coeff * * parmin potin Omega volpress yes nbody 1234 precision double
pair_coeff * * parmin potin Omega volpress yes nbody 12
Description:
Within DFT quantum mechanics, generalized pseudopotential theory (GPT) (Moriarty1) provides a
first-principles approach to multi-ion interatomic potentials in d-band transition metals, with a
volume-dependent, real-space total-energy functional for the N-ion elemental bulk material in the form
where the prime on each summation sign indicates the exclusion of all self-interaction terms from the
summation. The leading volume term E_vol as well as the two-ion central-force pair potential v_2 and the
three- and four-ion angular-force potentials, v_3 and v_4, depend explicitly on the atomic volume Omega, but
are structure independent and transferable to all bulk ion configurations, either ordered or disordered, and with
of without the presence of point and line defects. The simplified model GPT or MGPT (Moriarty2,
Moriarty3), which retains the form of E_tot and permits more efficient large-scale atomistic simulations,
derives from the GPT through a series of systematic approximations applied to E_vol and the potentials v_n
that are valid for mid-period transition metals with nearly half-filled d bands.
Both analytic (Moriarty2) and matrix (Moriarty3) representations of MGPT have been developed. In the more
general matrix representation, which can also be applied to f-band actinide metals and permits both canonical
and non-canonical d/f bands, the multi-ion potentials are evaluated on the fly during a simulation through d-
or f-state matrix multiplication, and the forces that move the ions are determined analytically. Fast
matrix-MGPT algorithms have been developed independently by Glosli (Glosli, Moriarty3) and by
Oppelstrup (Oppelstrup)
The mgpt pair style calculates forces, energies, and the total energy per atom, E_tot/N, using the Oppelstrup
matrix-MGPT algorithm. Input potential and control data are entered through the pair_coeff command. Each
material treated requires input parmin and potin potential files, as shown in the above examples, as well as
specification by the user of the initial atomic volume Omega through pair_coeff. At the beginning of a time
step in any simulation, the total volume of the simulation cell V should always be equal to Omega*N, where
N is the number of metal ions present, taking into account the presence of any vacancies and/or interstitials in
the case of a solid. In a constant-volume simulation, which is the normal mode of operation for the mgpt pair
LAMMPS Users Manual
1576

style, Omega, V and N all remain constant throughout the simulation and thus are equal to their initial values.
In a constant-stress simulation, the cell volume V will change (slowly) as the simulation proceeds. After each
time step, the atomic volume should be updated by the code as Omega = V/N. In addition, the volume term
E_vol and the potentials v_2, v_3 and v_4 have to be removed at the end of the time step, and then respecified
at the new value of Omega. In all simulations, Omega must remain within the defined volume range for E_vol
and the potentials for the given material.
The default option volpress yes in the pair_coeff command includes all volume derivatives of E_tot required
to calculate the stress tensor and pressure correctly. The option volpress no disregards the pressure
contribution resulting from the volume term E_vol, and can be used for testing and analysis purposes. The
additional optional variable nbody controls the specific terms in E_tot that are calculated. The default option
and the normal option for mid-period transition and actinide metals is nbody 1234 for which all four terms in
E_tot are retained. The option nbody 12, for example, retains only the volume term and the two-ion pair
potential term and can be used for GPT series-end transition metals that can be well described without v_3
and v_4. The nbody option can also be used to test or analyze the contribution of any of the four terms in
E_tot to a given calculated property.
The mgpt pair style makes extensive use of matrix algebra and includes optimized kernels for the BlueGene/Q
architecture and the Intel/AMD (x86) architectures. When compiled with the appropriate compiler and
compiler switches (-msse3 on x86, and using the IBM XL compiler on BG/Q), these optimized routines are
used automatically. For BG/Q machines, building with the default Makefile for that architecture (e.g., "make
bgq") should enable the optimized algebra routines. For x-86 machines, there is a provided Makefile.mgptfast
which enables the fast algebra routines, i.e. build LAMMPS with "make mgptfast". The user will be informed
in the output files of the matrix kernels in use. To further improve speed, on x86 the option precision single
can be added to the pair_coeff command line, which improves speed (up to a factor of two) at the cost of
doing matrix calculations with 7 digit precision instead of the default 16. For consistency the default option
can be specified explicitly by the option precision double.
All remaining potential and control data are contained with the parmin and potin files, including cutoffs,
atomic mass, and other basic MGPT variables. Specific MGPT potential data for the transition metals
tantalum (Ta4 and Ta6.8x potentials), molybdenum (Mo5.2 potentials), and vanadium (V6.1 potentials) are
contained in the LAMMPS potentials directory. The stored files are, respectively, Ta4.mgpt.parmin,
Ta4.mgpt.potin, Ta6.8x.mgpt.parmin, Ta6.8x.mgpt.potin, Mo5.2.mgpt.parmin, Mo5.2.mgpt.potin,
V6.1.mgpt.parmin, and V6.1.mgpt.potin . Useful corresponding informational "README" files on the Ta4,
Ta6.8x, Mo5.2 and V6.1 potentials are also included in the potentials directory. These latter files indicate the
volume mesh and range for each potential and give appropriate references for the potentials. It is expected that
MGPT potentials for additional materials will be added over time.
Useful example MGPT scripts are given in the examples/USER/mgpt directory. These scripts show the
necessary steps to perform constant-volume calculations and simulations. It is strongly recommended that the
user work through and understand these examples before proceeding to more complex simulations.
NOTE: For good performance, LAMMPS should be built with the compiler flags "-O3 -msse3 -funroll-loops"
when including this pair style. The src/MAKE/OPTIONS/Makefile.mgptfast is an example machine Makefile
with these options included as part of a standard MPI build. Note that it as provided, it will build with
whatever low-level compiler (g++, icc, etc) is the default for your MPI installation.
Mixing, shift, table tail correction, restart:
This pair style does not support the pair_modify mix, shift, table, and tail options.
LAMMPS Users Manual
1577
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you needs to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the USER-MGPT package and is only enabled if LAMMPS is built with that
package. See the Making LAMMPS section for more info.
The MGPT potentials require the newtion setting to be "on" for pair style interactions.
The stored parmin and potin potential files provided with LAMMPS in the "potentials" directory are written in
Rydberg atomic units, with energies in Rydbergs and distances in Bohr radii. The mgpt pair style converts
Rydbergs to Hartrees to make the potential files compatible with LAMMPS electron units.
The form of E_tot used in the mgpt pair style is only appropriate for elemental bulk solids and liquids. This
includes solids with point and extended defects such as vacancies, interstitials, grain boundaries and
dislocations. Alloys and free surfaces, however, require significant modifications, which are not included in
the mgpt pair style. Likewise, the hybrid pair style is not allowed, where MGPT would be used for some
atoms but not for others.
Electron-thermal effects are not included in the standard MGPT potentials provided in the "potentials"
directory, where the potentials have been constructed at zero electron temperature. Physically,
electron-thermal effects may be important in 3d (e.g., V) and 4d (e.g., Mo) transition metals at high
temperatures near melt and above. It is expected that temperature-dependent MGPT potentials for such cases
will be added over time.
Related commands:
pair_coeff
Default:
The options defaults for the pair_coeff command are volpress yes, nbody 1234, and precision double.
(Moriarty1) Moriarty, Physical Review B, 38, 3199 (1988).
(Moriarty2) Moriarty, Physical Review B, 42, 1609 (1990). Moriarty, Physical Review B 49, 12431 (1994).
(Moriarty3) Moriarty, Benedict, Glosli, Hood, Orlikowski, Patel, Soderlind, Streitz, Tang, and Yang, Journal
of Materials Research, 21, 563 (2006).
(Glosli) Glosli, unpublished, 2005. Streitz, Glosli, Patel, Chan, Yates, de Supinski, Sexton and Gunnels,
Journal of Physics: Conference Series, 46, 254 (2006).
LAMMPS Users Manual
1578
(Oppelstrup) Oppelstrup, unpublished, 2015. Oppelstrup and Moriarty, to be published.
LAMMPS Users Manual
1579

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style mie/cut command
pair_style mie/cut/gpu command
Syntax:
pair_style mie/cut cutoff
cutoff = global cutoff for mie/cut interactions (distance units)•
Examples:
pair_style mie/cut 10.0
pair_coeff 1 1 0.72 3.40 23.00 6.66
pair_coeff 2 2 0.30 3.55 12.65 6.00
pair_coeff 1 2 0.46 3.32 16.90 6.31
Description:
The mie/cut style computes the Mie potential, given by
Rc is the cutoff and C is a function that depends on the repulsive and attractive exponents, given by:
Note that for 12/6 exponents, C is equal to 4 and the formula is the same as the standard Lennard-Jones
potential.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
epsilon (energy units)• sigma (distance units)• gammaR• gammaA• cutoff (distance units)•
The last coefficient is optional. If not specified, the global cutoff specified in the pair_style command is used.
LAMMPS Users Manual
1580

Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of the mie/cut
pair styles can be mixed. If not explicitly defined, both the repulsive and attractive gamma exponents for
different atoms will be calculated following the same mixing rule defined for distances. The default mix value
is geometric. See the "pair_modify" command for details.
This pair style supports the pair_modify shift option for the energy of the pair interaction.
This pair style supports the pair_modify tail option for adding a long-range tail correction to the energy and
pressure of the pair interaction.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style supports the use of the inner, middle, and outer keywords of the run_style respa command,
meaning the pairwise forces can be partitioned by distance at different levels of the rRESPA hierarchy. See
the run_style command for details.
Restrictions: none
Related commands:
pair_coeff
Default: none
(Mie) G. Mie, Ann Phys, 316, 657 (1903).
(Avendano) C. Avendano, T. Lafitte, A. Galindo, C. S. Adjiman, G. Jackson, E. Muller, J Phys Chem B, 115,
11154 (2011).
LAMMPS Users Manual
1581
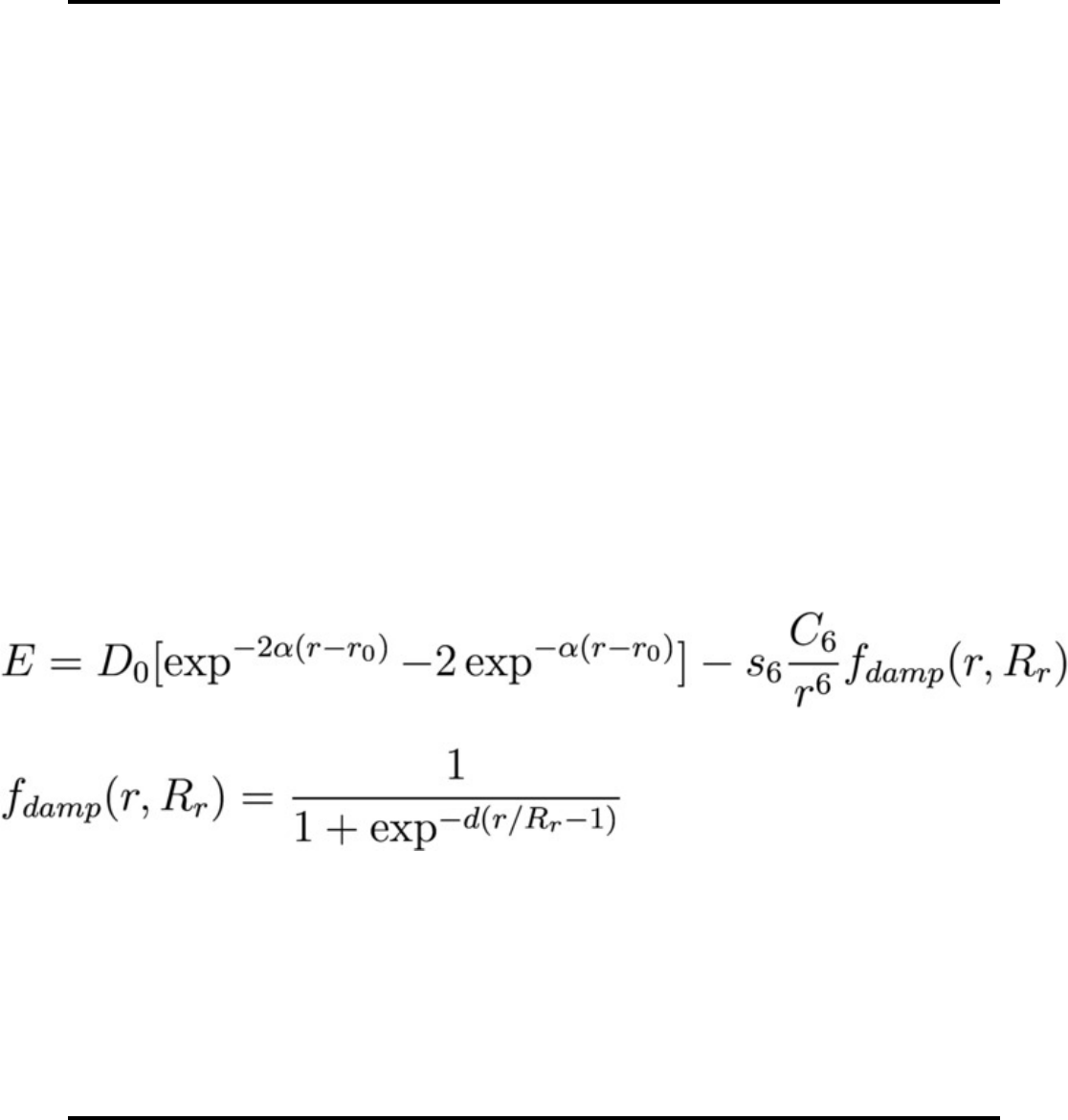
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style momb command
Syntax:
pair_style momb cutoff s6 d
cutoff = global cutoff (distance units)• s6 = global scaling factor of the exchange-correlation functional used (unitless)• d = damping scaling factor of Grimme's method (unitless)•
Examples:
pair_style momb 12.0 0.75 20.0
pair_style hybrid/overlay eam/fs lj/charmm/coul/long 10.0 12.0 momb 12.0 0.75 20.0 morse 5.5
pair_coeff 1 2 momb 0.0 1.0 1.0 10.2847 2.361
Description:
Style momb computes pairwise van der Waals (vdW) and short-range interactions using the Morse potential
and (Grimme) method implemented in the Many-Body Metal-Organic (MOMB) force field described
comprehensively in (Fichthorn) and (Zhou). Grimme's method is widely used to correct for dispersion in
density functional theory calculations.
For the momb pair style, the following coefficients must be defined for each pair of atoms types via the
pair_coeff command as in the examples above, or in the data file or restart files read by the read_data as
described below:
D0 (energy units)• alpha (1/distance units)• r0 (distance units)• C6 (energy*distance^6 units)• Rr (distance units, typically sum of atomic vdW radii)•
Restrictions:
This style is part of the USER-MISC package. It is only enabled if LAMMPS is built with that package. See
the Making of LAMMPS section for more info.
LAMMPS Users Manual
1582


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style morse command
pair_style morse/gpu command
pair_style morse/omp command
pair_style morse/opt command
pair_style morse/smooth/linear command
pair_style morse/smooth/linear/omp command
pair_style morse/soft command
pair_style morse/kk command
Syntax:
pair_style style args
style = morse or morse/smooth/linear or morse/soft• args = list of arguments for a particular style•
morse args = cutoff
cutoff = global cutoff for Morse interactions (distance units)
morse/smooth/linear args = cutoff
cutoff = global cutoff for Morse interactions (distance units)
morse/soft args = n lf cutoff
n = soft-core parameter
lf = transformation range is lf <lambda <1
cutoff = global cutoff for Morse interactions (distance units)
Examples:
pair_style morse 2.5 pair_style morse/smooth/linear 2.5 pair_coeff * * 100.0 2.0 1.5 pair_coeff 1 1 100.0 2.0
1.5 3.0
pair_style morse/soft 4 0.9 10.0
pair_coeff * * 100.0 2.0 1.5 1.0
pair_coeff 1 1 100.0 2.0 1.5 1.0 3.0
Description:
Style morse computes pairwise interactions with the formula
LAMMPS Users Manual
1584

Rc is the cutoff.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands:
D0 (energy units)• alpha (1/distance units)• r0 (distance units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global morse cutoff is used.
The morse/smooth/linear variant is similar to the lj/smooth/linear variant in that it adds to the potential a shift
and a linear term so that both, potential energy and force, go to zero at the cut-off:
The syntax of the pair_style and pair_coeff commands are the same for the morse and morse/smooth/linear
styles.
The morse/soft variant is similar to the lj/cut/soft pair style in that it modifies the potential at short range to
have a soft core. This helps to avoid singularities during free energy calculation in which sites are created or
annihilated. The formula differs from that of lj/cut/soft, and is instead given by:
The morse/soft style requires the following pair coefficients:
D0 (energy units)• alpha (1/distance units)• r0 (distance units)• lamda (unitless, between 0.0 and 1.0)•
LAMMPS Users Manual
1585

cutoff (distance units)•
The last coefficient is optional. If not specified, the global morse cutoff is used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
None of these pair styles support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
All of these pair styles support the pair_modify shift option for the energy of the pair interaction.
The pair_modify table options is not relevant for the Morse pair styles.
None of these pair styles support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
All of these pair styles write their information to binary restart files, so pair_style and pair_coeff commands
do not need to be specified in an input script that reads a restart file.
These pair styles can only be used via the pair keyword of the run_style respa command. They do not support
the inner, middle, outer keywords.
Restrictions:
The morse/smooth/linear pair style is only enabled if LAMMPS was built with the USER-MISC package. See
the Making LAMMPS section for more info.
The morse/soft pair style is only enabled if LAMMPS was built with the USER-FEP package. See the Making
LAMMPS section for more info.
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1586
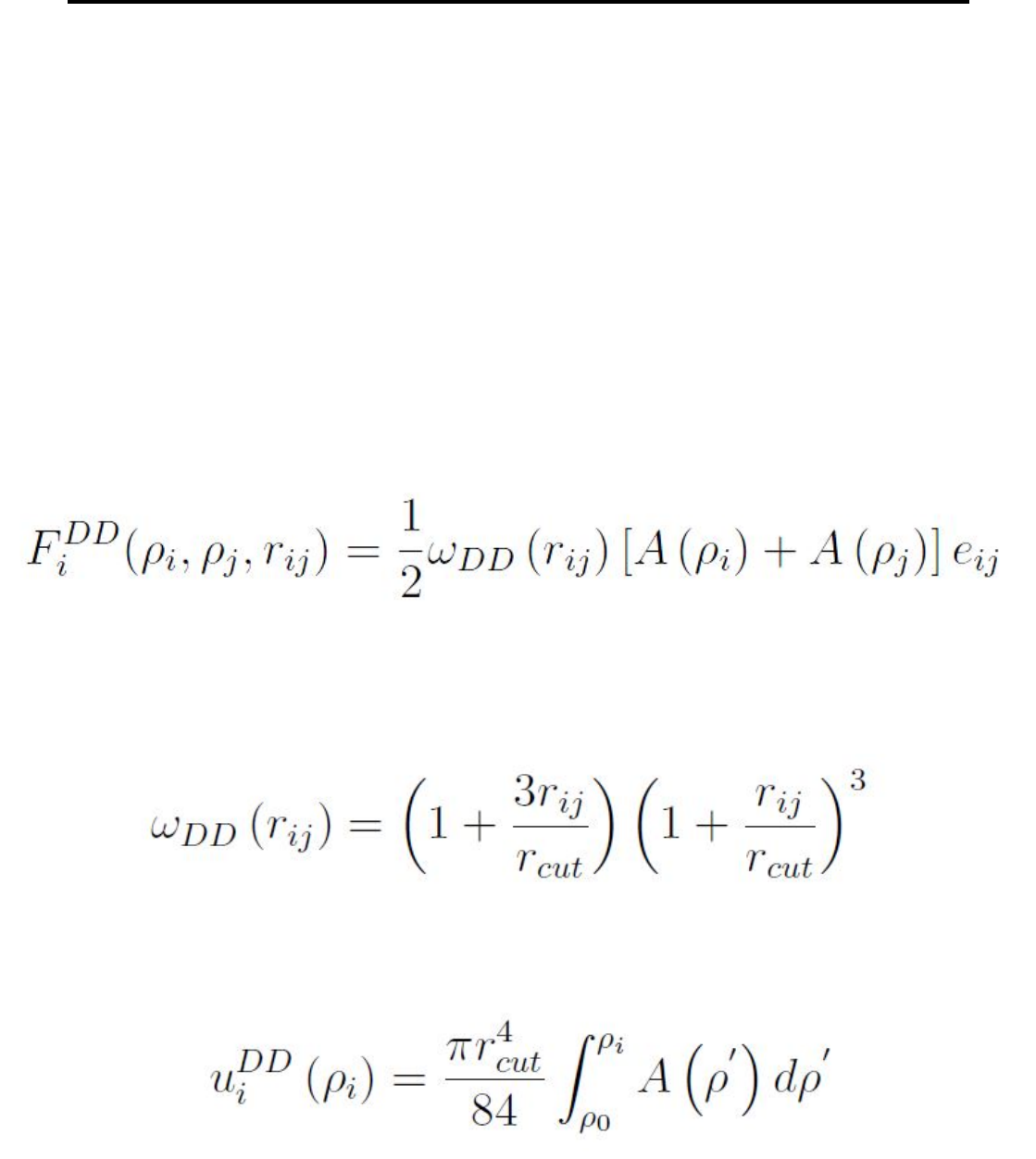
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style multi/lucy command
Syntax:
pair_style multi/lucy style N keyword ...
style = lookup or linear = method of interpolation• N = use N values in lookup, linear tables•
Examples:
pair_style multi/lucy linear 1000
pair_coeff * * multibody.table ENTRY1 7.0
Description:
Style multi/lucy computes a density-dependent force following from the many-body form described in
(Moore) and (Warren) as
which consists of a density-dependent function, A(rho), and a radial-dependent weight function,
omegaDD(rij). The radial-dependent weight function, omegaDD(rij), is taken as the Lucy function:
The density-dependent energy for a given particle is given by:
LAMMPS Users Manual
1587

See the supporting information of (Brennan) or the publication by (Moore) for more details on the functional
form.
An interpolation table is used to evaluate the density-dependent energy (Integral(A(rho)drho) and force
(A(rho)). Note that the pre-factor to the energy is computed after the interpolation, thus the
Integral(A(rho)drho will have units of energy / length^4.
The interpolation table is created as a pre-computation by fitting cubic splines to the file values and
interpolating the density-dependent energy and force at each of N densities. During a simulation, the tables are
used to interpolate the density-dependent energy and force as needed for each pair of particles separated by a
distance R. The interpolation is done in one of 2 styles: lookup and linear.
For the lookup style, the density is used to find the nearest table entry, which is the density-dependent energy
and force.
For the linear style, the density is used to find the 2 surrounding table values from which the
density-dependent energy and force are computed by linear interpolation.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above.
filename• keyword• cutoff (distance units)•
The filename specifies a file containing the tabulated density-dependent energy and force. The keyword
specifies a section of the file. The cutoff is an optional coefficient. If not specified, the outer cutoff in the table
itself (see below) will be used to build an interpolation table that extend to the largest tabulated distance. If
specified, only file values up to the cutoff are used to create the interpolation table. The format of this file is
described below.
The format of a tabulated file is a series of one or more sections, defined as follows (without the parenthesized
comments):
# Density-dependent function (one or more comment or blank lines)
DD-FUNCTION (keyword is first text on line)
N 500 R 1.0 10.0 (N, R, RSQ parameters)
(blank)
1 1.0 25.5 102.34 (index, density, energy/r^4, force)
2 1.02 23.4 98.5
...
500 10.0 0.001 0.003
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections. The first line begins with a keyword which identifies the section.
The line can contain additional text, but the initial text must match the argument specified in the pair_coeff
command. The next line lists (in any order) one or more parameters for the table. Each parameter is a keyword
followed by one or more numeric values.
The parameter "N" is required and its value is the number of table entries that follow. Note that this may be
different than the N specified in the pair_style multi/lucy command. Let Ntable = N in the pair_style
LAMMPS Users Manual
1588

command, and Nfile = "N" in the tabulated file. What LAMMPS does is a preliminary interpolation by
creating splines using the Nfile tabulated values as nodal points. It uses these to interpolate the
density-dependent energy and force at Ntable different points. The resulting tables of length Ntable are then
used as described above, when computing the density-dependent energy and force. This means that if you
want the interpolation tables of length Ntable to match exactly what is in the tabulated file (with effectively no
preliminary interpolation), you should set Ntable = Nfile, and use the "RSQ" parameter. This is because the
internal table abscissa is always RSQ (separation distance squared), for efficient lookup.
All other parameters are optional. If "R" or "RSQ" does not appear, then the distances in each line of the table
are used as-is to perform spline interpolation. In this case, the table values can be spaced in density uniformly
or however you wish to position table values in regions of large gradients.
If used, the parameters "R" or "RSQ" are followed by 2 values rlo and rhi. If specified, the density associated
with each density-dependent energy and force value is computed from these 2 values (at high accuracy),
rather than using the (low-accuracy) value listed in each line of the table. The density values in the table file
are ignored in this case. For "R", distances uniformly spaced between rlo and rhi are computed; for "RSQ",
squared distances uniformly spaced between rlo*rlo and rhi*rhi are computed.
NOTE: If you use "R" or "RSQ", the tabulated distance values in the file are effectively ignored, and replaced
by new values as described in the previous paragraph. If the density value in the table is not very close to the
new value (i.e. round-off difference), then you will be assigning density-dependent energy and force values to
a different density, which is probably not what you want. LAMMPS will warn if this is occurring.
Following a blank line, the next N lines list the tabulated values. On each line, the 1st value is the index from
1 to N, the 2nd value is r (in density units), the 3rd value is the density-dependent function value (in energy
units / length^4), and the 4th is the force (in force units). The density values must increase from one line to the
next.
Note that one file can contain many sections, each with a tabulated potential. LAMMPS reads the file section
by section until it finds one that matches the specified keyword.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
The pair_modify shift, table, and tail options are not relevant for this pair style.
This pair style writes the settings for the "pair_style multi/lucy" command to binary restart files, so a
pair_style command does not need to specified in an input script that reads a restart file. However, the
coefficient information is not stored in the restart file, since it is tabulated in the potential files. Thus,
pair_coeff commands do need to be specified in the restart input script.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
LAMMPS Users Manual
1589

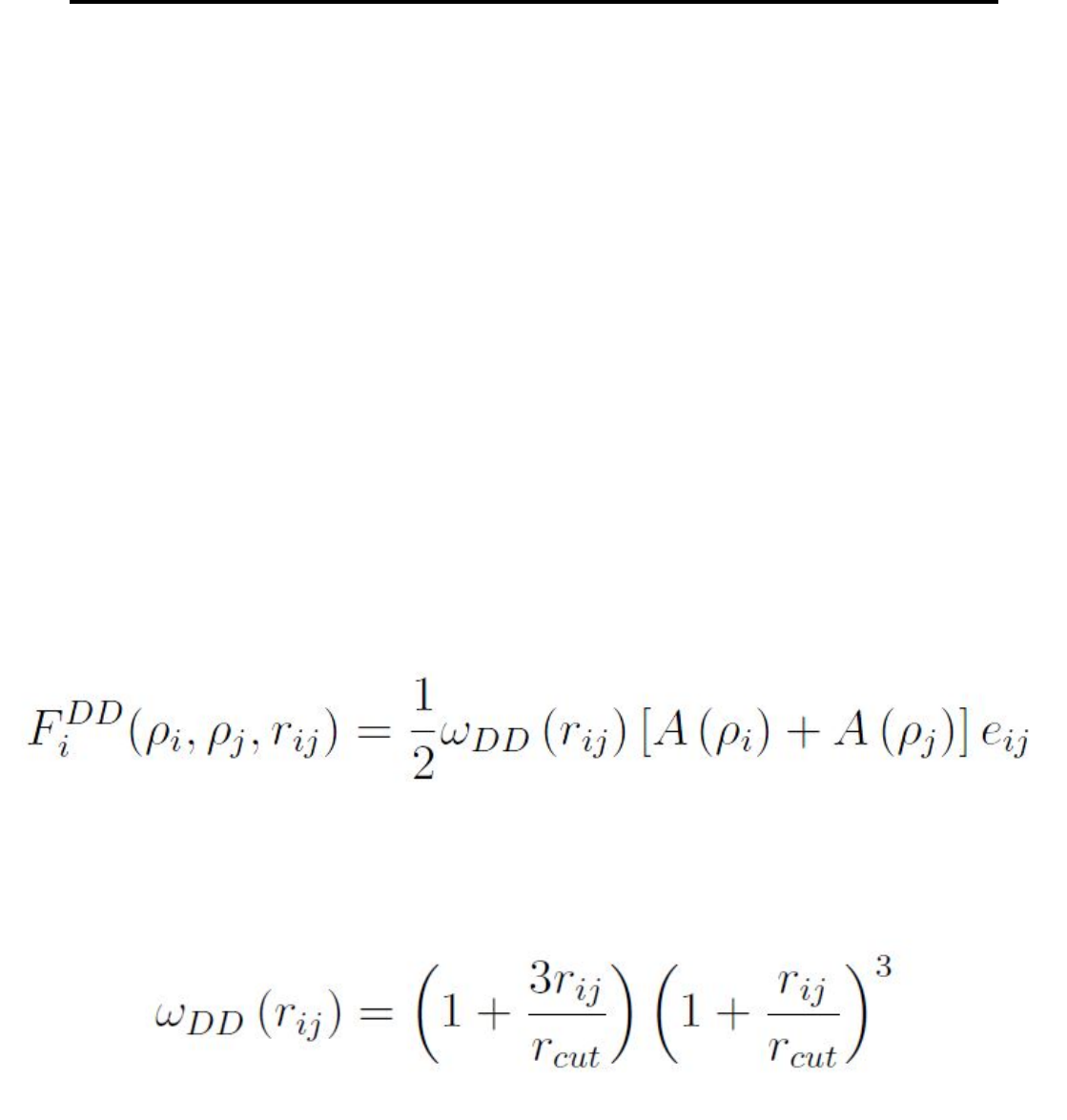
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style multi/lucy/rx command
Syntax:
pair_style multi/lucy/rx style N keyword ...
style = lookup or linear = method of interpolation• N = use N values in lookup, linear tables• weighting = fractional or molecular (optional)•
Examples:
pair_style multi/lucy/rx linear 1000
pair_style multi/lucy/rx linear 1000 fractional
pair_style multi/lucy/rx linear 1000 molecular
pair_coeff * * multibody.table ENTRY1 h2o h2o 7.0
pair_coeff * * multibody.table ENTRY1 h2o 1fluid 7.0
Description:
Style multi/lucy/rx is used in reaction DPD simulations, where the coarse-grained (CG) particles are
composed of m species whose reaction rate kinetics are determined from a set of n reaction rate equations
through the fix rx command. The species of one CG particle can interact with a species in a neighboring CG
particle through a site-site interaction potential model. Style multi/lucy/rx computes the site-site
density-dependent force following from the many-body form described in (Moore) and (Warren) as
which consists of a density-dependent function, A(rho), and a radial-dependent weight function,
omegaDD(rij). The radial-dependent weight function, omegaDD(rij), is taken as the Lucy function:
The density-dependent energy for a given particle is given by:
LAMMPS Users Manual
1591

See the supporting information of (Brennan) or the publication by (Moore) for more details on the functional
form.
An interpolation table is used to evaluate the density-dependent energy (Integral(A(rho)drho) and force
(A(rho)). Note that the pre-factor to the energy is computed after the interpolation, thus the
Integral(A(rho)drho will have units of energy / length^4.
The interpolation table is created as a pre-computation by fitting cubic splines to the file values and
interpolating the density-dependent energy and force at each of N densities. During a simulation, the tables are
used to interpolate the density-dependent energy and force as needed for each pair of particles separated by a
distance R. The interpolation is done in one of 2 styles: lookup and linear.
For the lookup style, the density is used to find the nearest table entry, which is the density-dependent energy
and force.
For the linear style, the density is used to find the 2 surrounding table values from which the
density-dependent energy and force are computed by linear interpolation.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above.
filename• keyword• species1• species2• cutoff (distance units)•
The filename specifies a file containing the tabulated density-dependent energy and force. The keyword
specifies a section of the file. The cutoff is an optional coefficient. If not specified, the outer cutoff in the table
itself (see below) will be used to build an interpolation table that extend to the largest tabulated distance. If
specified, only file values up to the cutoff are used to create the interpolation table. The format of this file is
described below.
The species tags define the site-site interaction potential between two species contained within two different
particles. The species tags must either correspond to the species defined in the reaction kinetics files specified
with the fix rx command or they must correspond to the tag "1fluid", signifying interaction with a product
species mixture determined through a one-fluid approximation. The interaction potential is weighted by the
geometric average of either the mole fraction concentrations or the number of molecules associated with the
interacting coarse-grained particles (see the fractional or molecular weighting pair style options). The
coarse-grained potential is stored before and after the reaction kinetics solver is applied, where the difference
is defined to be the internal chemical energy (uChem).
LAMMPS Users Manual
1592

The format of a tabulated file is a series of one or more sections, defined as follows (without the parenthesized
comments):
# Density-dependent function (one or more comment or blank lines)
DD-FUNCTION (keyword is first text on line)
N 500 R 1.0 10.0 (N, R, RSQ parameters)
(blank)
1 1.0 25.5 102.34 (index, density, energy/r^4, force)
2 1.02 23.4 98.5
...
500 10.0 0.001 0.003
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections. The first line begins with a keyword which identifies the section.
The line can contain additional text, but the initial text must match the argument specified in the pair_coeff
command. The next line lists (in any order) one or more parameters for the table. Each parameter is a keyword
followed by one or more numeric values.
The parameter "N" is required and its value is the number of table entries that follow. Note that this may be
different than the N specified in the pair_style multi/lucy/rx command. Let Ntable = N in the pair_style
command, and Nfile = "N" in the tabulated file. What LAMMPS does is a preliminary interpolation by
creating splines using the Nfile tabulated values as nodal points. It uses these to interpolate the
density-dependent energy and force at Ntable different points. The resulting tables of length Ntable are then
used as described above, when computing the density-dependent energy and force. This means that if you
want the interpolation tables of length Ntable to match exactly what is in the tabulated file (with effectively no
preliminary interpolation), you should set Ntable = Nfile, and use the "RSQ" parameter. This is because the
internal table abscissa is always RSQ (separation distance squared), for efficient lookup.
All other parameters are optional. If "R" or "RSQ" does not appear, then the distances in each line of the table
are used as-is to perform spline interpolation. In this case, the table values can be spaced in density uniformly
or however you wish to position table values in regions of large gradients.
If used, the parameters "R" or "RSQ" are followed by 2 values rlo and rhi. If specified, the density associated
with each density-dependent energy and force value is computed from these 2 values (at high accuracy),
rather than using the (low-accuracy) value listed in each line of the table. The density values in the table file
are ignored in this case. For "R", distances uniformly spaced between rlo and rhi are computed; for "RSQ",
squared distances uniformly spaced between rlo*rlo and rhi*rhi are computed.
NOTE: If you use "R" or "RSQ", the tabulated distance values in the file are effectively ignored, and replaced
by new values as described in the previous paragraph. If the density value in the table is not very close to the
new value (i.e. round-off difference), then you will be assigning density-dependent energy and force values to
a different density, which is probably not what you want. LAMMPS will warn if this is occurring.
Following a blank line, the next N lines list the tabulated values. On each line, the 1st value is the index from
1 to N, the 2nd value is r (in density units), the 3rd value is the density-dependent function value (in energy
units / length^4), and the 4th is the force (in force units). The density values must increase from one line to the
next.
Note that one file can contain many sections, each with a tabulated potential. LAMMPS reads the file section
by section until it finds one that matches the specified keyword.
LAMMPS Users Manual
1593

Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
The pair_modify shift, table, and tail options are not relevant for this pair style.
This pair style writes the settings for the "pair_style multi/lucy/rx" command to binary restart files, so a
pair_style command does not need to specified in an input script that reads a restart file. However, the
coefficient information is not stored in the restart file, since it is tabulated in the potential files. Thus,
pair_coeff commands do need to be specified in the restart input script.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This command is part of the USER-DPD package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
pair_coeff
Default: fractional weighting
(Warren) Warren, Phys Rev E, 68, 066702 (2003).
(Brennan) Brennan, J Chem Phys Lett, 5, 2144-2149 (2014).
(Moore) Moore, J Chem Phys, 144, 104501 (2016).
LAMMPS Users Manual
1594
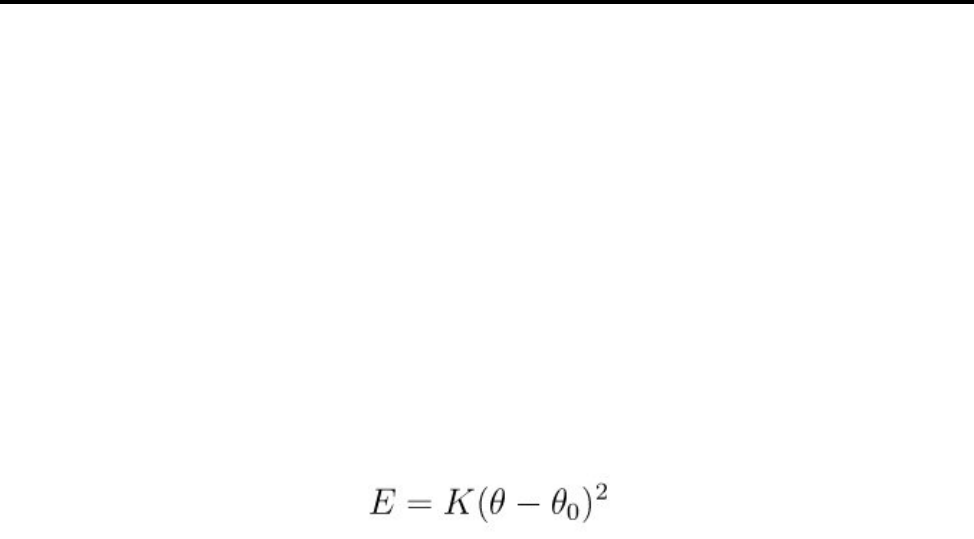
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style nb3b/harmonic command
pair_style nb3b/harmonic/omp command
Syntax:
pair_style nb3b/harmonic
Examples:
pair_style nb3b/harmonic
pair_coeff * * MgOH.nb3bharmonic Mg O H
Description:
This pair style computes a nonbonded 3-body harmonic potential for the energy E of a system of atoms as
where theta_0 is the equilibrium value of the angle and K is a prefactor. Note that the usual 1/2 factor is
included in K. The form of the potential is identical to that used in angle_style harmonic, but in this case, the
atoms do not need to be explicitly bonded.
Only a single pair_coeff command is used with this style which specifies a potential file with parameters for
specified elements. These are mapped to LAMMPS atom types by specifying N additional arguments after the
filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N element names = mapping of elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, imagine a file SiC.nb3b.harmonic has potential values for Si and C. If your LAMMPS
simulation has 4 atoms types and you want the 1st 3 to be Si, and the 4th to be C, you would use the following
pair_coeff command:
pair_coeff * * SiC.nb3b.harmonic Si Si Si C
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Si arguments map
LAMMPS atom types 1,2,3 to the Si element in the potential file. The final C argument maps LAMMPS atom
type 4 to the C element in the potential file. If a mapping value is specified as NULL, the mapping is not
performed. This can be used when the potential is used as part of the hybrid pair style. The NULL values are
placeholders for atom types that will be used with other potentials. An example of a pair_coeff command for
use with the hybrid pair style is:
pair_coeff * * nb3b/harmonic MgOH.nb3b.harmonic Mg O H
LAMMPS Users Manual
1595

Three-body nonbonded harmonic files in the potentials directory of the LAMMPS distribution have a
".nb3b.harmonic" suffix. Lines that are not blank or comments (starting with #) define parameters for a triplet
of elements.
Each entry has six arguments. The first three are atom types as referenced in the LAMMPS input file. The
first argument specifies the central atom. The fourth argument indicates the K parameter. The fifth argument
indicates theta_0. The sixth argument indicates a separation cutoff in Angstroms.
For a given entry, if the second and third arguments are identical, then the entry is for a cutoff for the distance
between types 1 and 2 (values for K and theta_0 are irrelevant in this case).
For a given entry, if the first three arguments are all different, then the entry is for the K and theta_0
parameters (the cutoff in this case is irrelevant).
It is not required that the potential file contain entries for all of the elements listed in the pair_coeff command.
It can also contain entries for additional elements not being used in a particular simulation; LAMMPS ignores
those entries.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This pair style can only be used if LAMMPS was built with the MANYBODY package. See the Making
LAMMPS section for more info on packages.
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1596

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style nm/cut command
pair_style nm/cut/coul/cut command
pair_style nm/cut/coul/long command
pair_style nm/cut/omp command
pair_style nm/cut/coul/cut/omp command
pair_style nm/cut/coul/long/omp command
Syntax:
pair_style style args
style = nm/cut or nm/cut/coul/cut or nm/cut/coul/long• args = list of arguments for a particular style
nm/cut args = cutoff
cutoff = global cutoff for Pair interactions (distance units)
nm/cut/coul/cut args = cutoff (cutoff2)
cutoff = global cutoff for Pair (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
nm/cut/coul/long args = cutoff (cutoff2)
cutoff = global cutoff for Pair (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
•
Examples:
pair_style nm/cut 12.0
pair_coeff * * 0.01 5.4 8.0 7.0
pair_coeff 1 1 0.01 4.4 7.0 6.0
pair_style nm/cut/coul/cut 12.0 15.0
pair_coeff * * 0.01 5.4 8.0 7.0
pair_coeff 1 1 0.01 4.4 7.0 6.0
pair_style nm/cut/coul/long 12.0 15.0
pair_coeff * * 0.01 5.4 8.0 7.0
pair_coeff 1 1 0.01 4.4 7.0 6.0
Description:
Style nm computes site-site interactions based on the N-M potential by Clarke, mainly used for ionic liquids.
A site can represent a single atom or a united-atom site. The energy of an interaction has the following form:
LAMMPS Users Manual
1597

Rc is the cutoff.
Style nm/cut/coul/cut adds a Coulombic pairwise interaction given by
where C is an energy-conversion constant, Qi and Qj are the charges on the 2 atoms, and epsilon is the
dielectric constant which can be set by the dielectric command. If one cutoff is specified in the pair_style
command, it is used for both the NM and Coulombic terms. If two cutoffs are specified, they are used as
cutoffs for the NM and Coulombic terms respectively.
Styles nm/cut/coul/long compute the same Coulombic interactions as style nm/cut/coul/cut except that an
additional damping factor is applied to the Coulombic term so it can be used in conjunction with the
kspace_style command and its ewald or pppm option. The Coulombic cutoff specified for this style means that
pairwise interactions within this distance are computed directly; interactions outside that distance are
computed in reciprocal space.
For all of the nm pair styles, the following coefficients must be defined for each pair of atoms types via the
pair_coeff command as in the examples above, or in the data file or restart files read by the read_data or
read_restart commands.
E0 (energy units)• r0 (distance units)• n (unitless)• m (unitless)• cutoff1 (distance units)• cutoff2 (distance units)•
The latter 2 coefficients are optional. If not specified, the global NM and Coulombic cutoffs specified in the
pair_style command are used. If only one cutoff is specified, it is used as the cutoff for both NM and
Coulombic interactions for this type pair. If both coefficients are specified, they are used as the NM and
Coulombic cutoffs for this type pair. You cannot specify 2 cutoffs for style nm, since it has no Coulombic
terms.
For nm/cut/coul/long only the NM cutoff can be specified since a Coulombic cutoff cannot be specified for an
individual I,J type pair. All type pairs use the same global Coulombic cutoff specified in the pair_style
command.
Mixing, shift, table, tail correction, restart, rRESPA info:
These pair styles do not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
LAMMPS Users Manual
1598
All of the nm pair styles supports the pair_modify shift option for the energy of the pair interaction.
The nm/cut/coul/long pair styles support the pair_modify table option since they can tabulate the short-range
portion of the long-range Coulombic interaction.
All of the nm pair styles support the pair_modify tail option for adding a long-range tail correction to the
energy and pressure for the NM portion of the pair interaction.
All of the nm pair styles write their information to binary restart files, so pair_style and pair_coeff commands
do not need to be specified in an input script that reads a restart file.
All of the nm pair styles can only be used via the pair keyword of the run_style respa command. They do not
support the inner, middle, outer keywords.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
These pair styles are part of the MISC package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
pair_coeff
Default: none
(Clarke) Clarke and Smith, J Chem Phys, 84, 2290 (1986).
LAMMPS Users Manual
1599

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style none command
Syntax:
pair_style none
Examples:
pair_style none
Description:
Using a pair style of none means pair forces and energies are not computed.
With this choice, the force cutoff is 0.0, which means that only atoms within the neighbor skin distance (see
the neighbor command) are communicated between processors. You must insure the skin distance is large
enough to acquire atoms needed for computing bonds, angles, etc.
A pair style of none will also prevent pairwise neighbor lists from being built. However if the neighbor style
is bin, data structures for binning are still allocated. If the neighbor skin distance is small, then these data
structures can consume a large amount of memory. So you should either set the neighbor style to nsq or set
the skin distance to a larger value.
See the pair_style zero for a way to trigger the building of a neighbor lists, but compute no pairwise
interactions.
Restrictions: none
Related commands:
pair_style zero
Default: none
LAMMPS Users Manual
1600

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style oxdna/excv command
pair_style oxdna/stk command
pair_style oxdna/hbond command
pair_style oxdna/xstk command
pair_style oxdna/coaxstk command
Syntax:
pair_style style1
pair_coeff * * style2 args
style1 = hybrid/overlay oxdna/excv oxdna/stk oxdna/hbond oxdna/xstk oxdna/coaxstk•
style2 = oxdna/stk• args = list of arguments for these two particular styles•
oxdna2/stk args = T 6.0 0.4 0.9 0.32 0.6 1.3 0 0.8 0.9 0 0.95 0.9 0 0.95 2.0 0.65 2.0 0.65
T = temperature (oxDNA units, 0.1 = 300 K)
Examples:
pair_style hybrid/overlay oxdna/excv oxdna/stk oxdna/hbond oxdna/xstk oxdna/coaxstk
pair_coeff * * oxdna/excv 2.0 0.7 0.675 2.0 0.515 0.5 2.0 0.33 0.32
pair_coeff * * oxdna/stk 0.1 6.0 0.4 0.9 0.32 0.6 1.3 0 0.8 0.9 0 0.95 0.9 0 0.95 2.0 0.65 2.0 0.65
pair_coeff * * oxdna/hbond 0.0 8.0 0.4 0.75 0.34 0.7 1.5 0 0.7 1.5 0 0.7 1.5 0 0.7 0.46 3.141592653589793 0.7 4.0 1.5707963267948966 0.45 4.0 1.5707963267948966 0.45
pair_coeff 1 4 oxdna/hbond 1.077 8.0 0.4 0.75 0.34 0.7 1.5 0 0.7 1.5 0 0.7 1.5 0 0.7 0.46 3.141592653589793 0.7 4.0 1.5707963267948966 0.45 4.0 1.5707963267948966 0.45
pair_coeff 2 3 oxdna/hbond 1.077 8.0 0.4 0.75 0.34 0.7 1.5 0 0.7 1.5 0 0.7 1.5 0 0.7 0.46 3.141592653589793 0.7 4.0 1.5707963267948966 0.45 4.0 1.5707963267948966 0.45
pair_coeff * * oxdna/xstk 47.5 0.575 0.675 0.495 0.655 2.25 0.791592653589793 0.58 1.7 1.0 0.68 1.7 1.0 0.68 1.5 0 0.65 1.7 0.875 0.68 1.7 0.875 0.68
pair_coeff * * oxdna/coaxstk 46.0 0.4 0.6 0.22 0.58 2.0 2.541592653589793 0.65 1.3 0 0.8 0.9 0 0.95 0.9 0 0.95 2.0 -0.65 2.0 -0.65
Description:
The oxdna pair styles compute the pairwise-additive parts of the oxDNA force field for coarse-grained
modelling of DNA. The effective interaction between the nucleotides consists of potentials for the excluded
volume interaction oxdna/excv, the stacking oxdna/stk, cross-stacking oxdna/xstk and coaxial stacking
interaction oxdna/coaxstk as well as the hydrogen-bonding interaction oxdna/hbond between complementary
pairs of nucleotides on opposite strands.
The exact functional form of the pair styles is rather complex, which manifests itself in the 144 coefficients in
the above example. The individual potentials consist of products of modulation factors, which themselves are
constructed from a number of more basic potentials (Morse, Lennard-Jones, harmonic angle and distance) as
well as quadratic smoothing and modulation terms. We refer to (Ouldridge-DPhil) and (Ouldridge) for a
detailed description of the oxDNA force field.
LAMMPS Users Manual
1601

NOTE: These pair styles have to be used together with the related oxDNA bond style oxdna/fene for the
connectivity of the phosphate backbone (see also documentation of bond_style oxdna/fene). With one
exception the coefficients in the above example have to be kept fixed and cannot be changed without
reparametrizing the entire model. The exception is the first coefficient after oxdna/stk (T=0.1 in the above
example). When using a Langevin thermostat, e.g. through fix langevin or fix nve/dotc/langevin the
temperature coefficients have to be matched to the one used in the fix.
Example input and data files for DNA duplexes can be found in examples/USER/cgdna/examples/oxDNA/
and /oxDNA2/. A simple python setup tool which creates single straight or helical DNA strands, DNA
duplexes or arrays of DNA duplexes can be found in examples/USER/cgdna/util/. A technical report with
more information on the model, the structure of the input file, the setup tool and the performance of the
LAMMPS-implementation of oxDNA can be found here.
Restrictions:
These pair styles can only be used if LAMMPS was built with the USER-CGDNA package and the
MOLECULE and ASPHERE package. See the Making LAMMPS section for more info on packages.
Related commands:
bond_style oxdna/fene, fix nve/dotc/langevin, pair_coeff, bond_style oxdna2/fene, pair_style oxdna2/excv
Default: none
(Ouldrigde-DPhil) T.E. Ouldridge, Coarse-grained modelling of DNA and DNA self-assembly, DPhil.
University of Oxford (2011).
(Ouldridge) T.E. Ouldridge, A.A. Louis, J.P.K. Doye, J. Chem. Phys. 134, 085101 (2011).
LAMMPS Users Manual
1602

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style oxdna2/excv command
pair_style oxdna2/stk command
pair_style oxdna2/hbond command
pair_style oxdna2/xstk command
pair_style oxdna2/coaxstk command
pair_style oxdna2/dh command
Syntax:
pair_style style1
pair_coeff * * style2 args
style1 = hybrid/overlay oxdna2/excv oxdna2/stk oxdna2/hbond oxdna2/xstk oxdna2/coaxstk
oxdna2/dh
•
style2 = oxdna2/stk or oxdna2/dh• args = list of arguments for these two particular styles•
oxdna2/stk args = T 6.0 0.4 0.9 0.32 0.6 1.3 0 0.8 0.9 0 0.95 0.9 0 0.95 2.0 0.65 2.0 0.65
T = temperature (oxDNA units, 0.1 = 300 K)
oxdna2/dh args = T rhos qeff
T = temperature (oxDNA units, 0.1 = 300 K)
rhos = salt concentration (mole per litre)
qeff = effective charge (elementary charges)
Examples:
pair_style hybrid/overlay oxdna2/excv oxdna2/stk oxdna2/hbond oxdna2/xstk oxdna2/coaxstk oxdna2/dh
pair_coeff * * oxdna2/excv 2.0 0.7 0.675 2.0 0.515 0.5 2.0 0.33 0.32
pair_coeff * * oxdna2/stk 0.1 6.0 0.4 0.9 0.32 0.6 1.3 0 0.8 0.9 0 0.95 0.9 0 0.95 2.0 0.65 2.0 0.65
pair_coeff * * oxdna2/hbond 0.0 8.0 0.4 0.75 0.34 0.7 1.5 0 0.7 1.5 0 0.7 1.5 0 0.7 0.46 3.141592653589793 0.7 4.0 1.5707963267948966 0.45 4.0 1.5707963267948966 0.45
pair_coeff 1 4 oxdna2/hbond 1.0678 8.0 0.4 0.75 0.34 0.7 1.5 0 0.7 1.5 0 0.7 1.5 0 0.7 0.46 3.141592653589793 0.7 4.0 1.5707963267948966 0.45 4.0 1.5707963267948966 0.45
pair_coeff 2 3 oxdna2/hbond 1.0678 8.0 0.4 0.75 0.34 0.7 1.5 0 0.7 1.5 0 0.7 1.5 0 0.7 0.46 3.141592653589793 0.7 4.0 1.5707963267948966 0.45 4.0 1.5707963267948966 0.45
pair_coeff * * oxdna2/xstk 47.5 0.575 0.675 0.495 0.655 2.25 0.791592653589793 0.58 1.7 1.0 0.68 1.7 1.0 0.68 1.5 0 0.65 1.7 0.875 0.68 1.7 0.875 0.68
pair_coeff * * oxdna2/coaxstk 58.5 0.4 0.6 0.22 0.58 2.0 2.891592653589793 0.65 1.3 0 0.8 0.9 0 0.95 0.9 0 0.95 40.0 3.116592653589793
pair_coeff * * oxdna2/dh 0.1 1.0 0.815
Description:
The oxdna2 pair styles compute the pairwise-additive parts of the oxDNA force field for coarse-grained
modelling of DNA. The effective interaction between the nucleotides consists of potentials for the excluded
volume interaction oxdna2/excv, the stacking oxdna2/stk, cross-stacking oxdna2/xstk and coaxial stacking
interaction oxdna2/coaxstk, electrostatic Debye-Hueckel interaction oxdna2/dh as well as the
hydrogen-bonding interaction oxdna2/hbond between complementary pairs of nucleotides on opposite strands.
LAMMPS Users Manual
1603

The exact functional form of the pair styles is rather complex. The individual potentials consist of products of
modulation factors, which themselves are constructed from a number of more basic potentials (Morse,
Lennard-Jones, harmonic angle and distance) as well as quadratic smoothing and modulation terms. We refer
to (Snodin) and the original oxDNA publications (Ouldridge-DPhil) and (Ouldridge) for a detailed description
of the oxDNA2 force field.
NOTE: These pair styles have to be used together with the related oxDNA2 bond style oxdna2/fene for the
connectivity of the phosphate backbone (see also documentation of bond_style oxdna2/fene). Almost all
coefficients in the above example have to be kept fixed and cannot be changed without reparametrizing the
entire model. Exceptions are the first coefficient after oxdna2/stk (T=0.1 in the above example) and the
coefficients after oxdna2/dh (T=0.1, rhos=1.0, qeff=0.815 in the above example). When using a Langevin
thermostat e.g. through fix langevin or fix nve/dotc/langevin the temperature coefficients have to be matched
to the one used in the fix.
Example input and data files for DNA duplexes can be found in examples/USER/cgdna/examples/oxDNA/
and /oxDNA2/. A simple python setup tool which creates single straight or helical DNA strands, DNA
duplexes or arrays of DNA duplexes can be found in examples/USER/cgdna/util/. A technical report with
more information on the model, the structure of the input file, the setup tool and the performance of the
LAMMPS-implementation of oxDNA can be found here.
Restrictions:
These pair styles can only be used if LAMMPS was built with the USER-CGDNA package and the
MOLECULE and ASPHERE package. See the Making LAMMPS section for more info on packages.
Related commands:
bond_style oxdna2/fene, fix nve/dotc/langevin, pair_coeff, bond_style oxdna/fene, pair_style oxdna/excv
Default: none
(Snodin) B.E. Snodin, F. Randisi, M. Mosayebi, et al., J. Chem. Phys. 142, 234901 (2015).
(Ouldrigde-DPhil) T.E. Ouldridge, Coarse-grained modelling of DNA and DNA self-assembly, DPhil.
University of Oxford (2011).
(Ouldridge) T.E. Ouldridge, A.A. Louis, J.P.K. Doye, J. Chem. Phys. 134, 085101 (2011).
LAMMPS Users Manual
1604

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style peri/pmb command
pair_style peri/pmb/omp command
pair_style peri/lps command
pair_style peri/lps/omp command
pair_style peri/ves command
pair_style peri/eps command
Syntax:
pair_style style
style = peri/pmb or peri/lps or peri/ves or peri/eps•
Examples:
pair_style peri/pmb
pair_coeff * * 1.6863e22 0.0015001 0.0005 0.25
pair_style peri/lps
pair_coeff * * 14.9e9 14.9e9 0.0015001 0.0005 0.25
pair_style peri/ves
pair_coeff * * 14.9e9 14.9e9 0.0015001 0.0005 0.25 0.5 0.001
pair_style peri/eps
pair_coeff * * 14.9e9 14.9e9 0.0015001 0.0005 0.25 118.43
Description:
The peridynamic pair styles implement material models that can be used at the mescscopic and macroscopic
scales. See this document for an overview of LAMMPS commands for Peridynamics modeling.
Style peri/pmb implements the Peridynamic bond-based prototype microelastic brittle (PMB) model.
Style peri/lps implements the Peridynamic state-based linear peridynamic solid (LPS) model.
Style peri/ves implements the Peridynamic state-based linear peridynamic viscoelastic solid (VES) model.
Style peri/eps implements the Peridynamic state-based elastic-plastic solid (EPS) model.
The canonical papers on Peridynamics are (Silling 2000) and (Silling 2007). The implementation of
Peridynamics in LAMMPS is described in (Parks). Also see the PDLAMMPS user guide for more details
about its implementation.
LAMMPS Users Manual
1605
The peridynamic VES and EPS models in PDLAMMPS were implemented by R. Rahman and J. T. Foster at
University of Texas at San Antonio. The original VES formulation is described in "(Mitchell2011)" and the
original EPS formulation is in "(Mitchell2011a)". Additional PDF docs that describe the VES and EPS
implementations are include in the LAMMPS distro in doc/PDF/PDLammps_VES.pdf and
doc/PDF/PDLammps_EPS.pdf. For questions regarding the VES and EPS models in LAMMPS you can
contact R. Rahman (rezwanur.rahman at utsa.edu).
The following coefficients must be defined for each pair of atom types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below.
For the peri/pmb style:
c (energy/distance/volume^2 units)• horizon (distance units)• s00 (unitless)• alpha (unitless)•
C is the effectively a spring constant for Peridynamic bonds, the horizon is a cutoff distance for truncating
interactions, and s00 and alpha are used as a bond breaking criteria. The units of c are such that c/distance =
stiffness/volume^2, where stiffness is energy/distance^2 and volume is distance^3. See the users guide for
more details.
For the peri/lps style:
K (force/area units)• G (force/area units)• horizon (distance units)• s00 (unitless)• alpha (unitless)•
K is the bulk modulus and G is the shear modulus. The horizon is a cutoff distance for truncating interactions,
and s00 and alpha are used as a bond breaking criteria. See the users guide for more details.
For the peri/ves style:
K (force/area units)• G (force/area units)• horizon (distance units)• s00 (unitless)• alpha (unitless)• m_lambdai (unitless)• m_taubi (unitless)•
K is the bulk modulus and G is the shear modulus. The horizon is a cutoff distance for truncating interactions,
and s00 and alpha are used as a bond breaking criteria. m_lambdai and m_taubi are the viscoelastic relaxation
parameter and time constant, respectively. m_lambdai varies within zero to one. For very small values of
m_lambdai the viscoelastic model responds very similar to a linear elastic model. For details please see the
description in "(Mtchell2011)".
For the peri/eps style:
LAMMPS Users Manual
1606
K (force/area units) G (force/area units) horizon (distance units) s00 (unitless) alpha (unitless) m_yield_stress
(force/area units)
K is the bulk modulus and G is the shear modulus. The horizon is a cutoff distance and s00 and alpha are used
as a bond breaking criteria. m_yield_stress is the yield stress of the material. For details please see the
description in "(Mtchell2011a)".
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
These pair styles do not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
These pair styles do not support the pair_modify shift option.
The pair_modify table and tail options are not relevant for these pair styles.
These pair styles write their information to binary restart files, so pair_style and pair_coeff commands do not
need to be specified in an input script that reads a restart file.
These pair styles can only be used via the pair keyword of the run_style respa command. They do not support
the inner, middle, outer keywords.
Restrictions:
All of these styles are part of the PERI package. They are only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
Related commands:
pair_coeff
Default: none
(Parks) Parks, Lehoucq, Plimpton, Silling, Comp Phys Comm, 179(11), 777-783 (2008).
LAMMPS Users Manual
1607
(Silling 2000) Silling, J Mech Phys Solids, 48, 175-209 (2000).
(Silling 2007) Silling, Epton, Weckner, Xu, Askari, J Elasticity, 88, 151-184 (2007).
(Mitchell2011) Mitchell. A non-local, ordinary-state-based viscoelasticity model for peridynamics. Sandia
National Lab Report, 8064:1-28 (2011).
(Mitchell2011a) Mitchell. A Nonlocal, Ordinary, State-Based Plasticity Model for Peridynamics. Sandia
National Lab Report, 3166:1-34 (2011).
LAMMPS Users Manual
1608

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style polymorphic command
Syntax:
pair_style polymorphic
style = polymorphic
Examples:
pair_style polymorphic
pair_coeff * * TlBr_msw.polymorphic Tl Br
pair_coeff * * AlCu_eam.polymorphic Al Cu
pair_coeff * * GaN_tersoff.polymorphic Ga N
pair_coeff * * GaN_sw.polymorphic GaN
Description:
The polymorphic pair style computes a 3-body free-form potential (Zhou) for the energy E of a system of
atoms as
where I, J, K represent species of atoms i, j, and k, i_1, ..., i_N represents a list of i's neighbors, delta_ij is a
Direc constant (i.e., delta_ij = 1 when i = j, and delta_ij = 0 otherwise), eta_ij is similar constant that can be
set either to eta_ij = delta_ij or eta_ij = 1 - delta_ij depending on the potential type, U_IJ(r_ij), V_IJ(r_ij),
W_IK(r_ik) are pair functions, G_JIK(cos(theta)) is an angular function, P_IK(delta r_jik) is a function of
atomic spacing differential delta r_jik = r_ij - xi_IJ*r_ik with xi_IJ being a pair-dependent parameter, and
F_IJ(X_ij) is a function of the local environment variable X_ij. This generic potential is fully defined once the
constants eta_ij and xi_IJ, and the six functions U_IJ(r_ij), V_IJ(r_ij), W_IK(r_ik), G_JIK(cos(theta)),
P_IK(delta r_jik), and F_IJ(X_ij) are given. Note that these six functions are all one dimensional, and hence
can be provided in an analytic or tabular form. This allows users to design different potentials solely based on
a manipulation of these functions. For instance, the potential reduces to Stillinger-Weber potential (SW) if we
set
LAMMPS Users Manual
1609

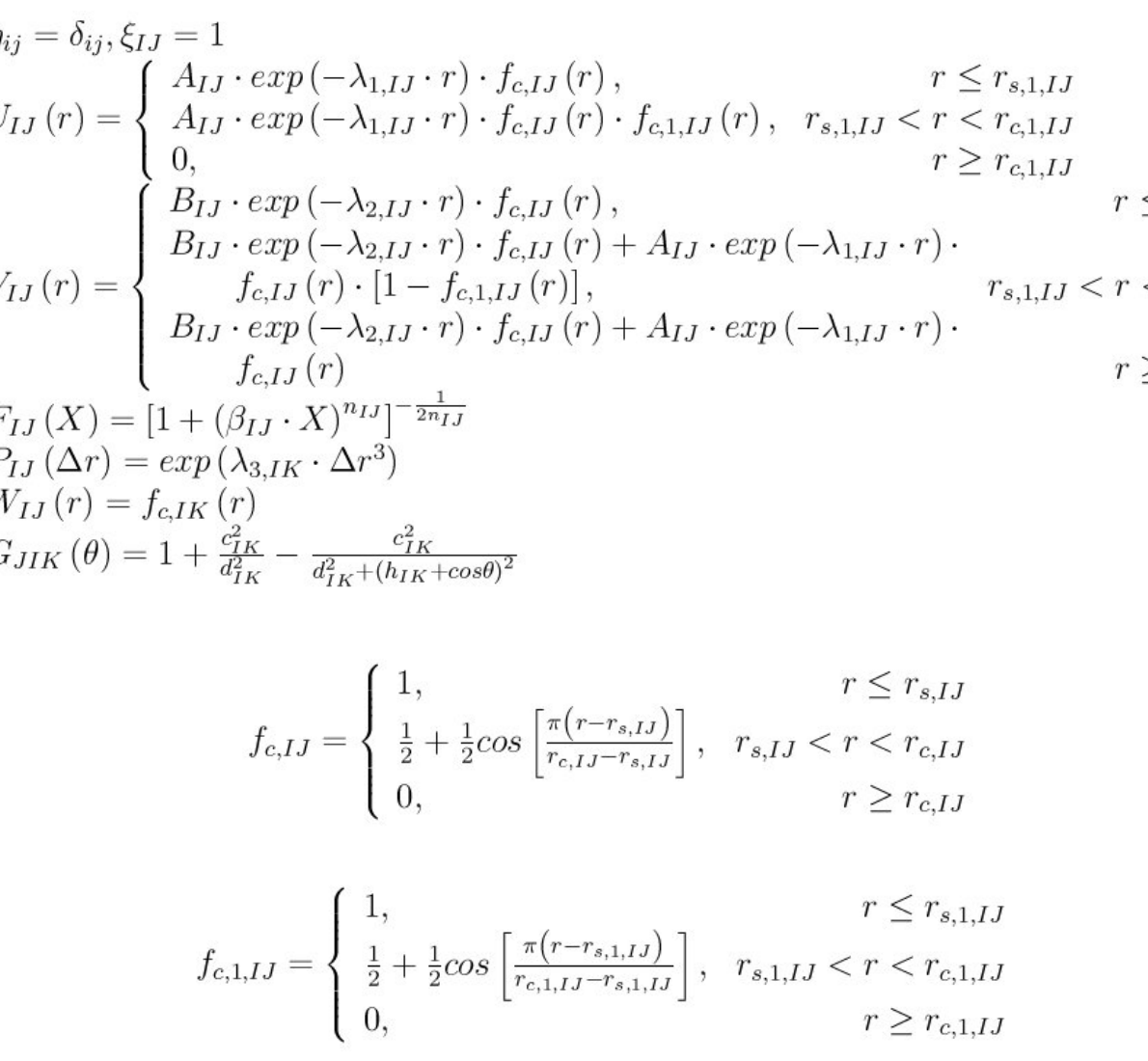
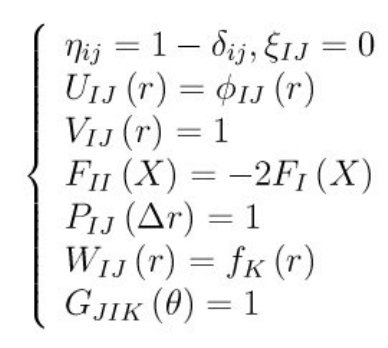
In the embedded atom method case, phi_IJ(r_ij) is the pair energy, F_I(X) is the embedding energy, X is the
local electron density, and f_K(r) is the atomic electron density function.
If the tabulated functions are created using the parameters of sw, tersoff, and eam potentials, the polymorphic
pair style will produce the same global properties (energies and stresses) and the same forces as the sw,
tersoff, and eam pair styles. The polymorphic pair style also produces the same atom properties (energies and
stresses) as the corresponding tersoff and eam pair styles. However, due to a different partition of global
properties to atom properties, the polymorphic pair style will produce different atom properties (energies and
stresses) as the sw pair style. This does not mean that polymorphic pair style is different from the sw pair style
in this case. It just means that the definitions of the atom energies and atom stresses are different.
Only a single pair_coeff command is used with the polymorphic style which specifies an potential file for all
needed elements. These are mapped to LAMMPS atom types by specifying N additional arguments after the
filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N element names = mapping of Tersoff elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file. Several files for
polymorphic potentials are included in the potentials dir of the LAMMPS distro. They have a "poly" suffix.
As an example, imagine the SiC_tersoff.polymorphic file has tabulated functions for Si-C tersoff potential. If
your LAMMPS simulation has 4 atoms types and you want the 1st 3 to be Si, and the 4th to be C, you would
use the following pair_coeff command:
pair_coeff * * SiC_tersoff.polymorphic Si Si Si C
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Si arguments map
LAMMPS atom types 1,2,3 to the Si element in the polymorphic file. The final C argument maps LAMMPS
atom type 4 to the C element in the polymorphic file. If a mapping value is specified as NULL, the mapping is
not performed. This can be used when an polymorphic potential is used as part of the hybrid pair style. The
NULL values are placeholders for atom types that will be used with other potentials.
Potential files in the potentials directory of the LAMMPS distribution have a ".poly" suffix. At the beginning
of the files, an unlimited number of lines starting with '#' are used to describe the potential and are ignored by
LAMMPS. The next line lists two numbers:
LAMMPS Users Manual
1612

ntypes eta
Here ntypes represent total number of species defined in the potential file, and eta = 0 or 1. The number
ntypes must equal the total number of different species defined in the pair_coeff command. When eta = 1,
eta_ij defined in the potential functions above is set to 1 - delta_ij, otherwise eta_ij is set to delta_ij. The next
ntypes lines each lists two numbers and a character string representing atomic number, atomic mass, and name
of the species of the ntypes elements:
atomic_number atomic-mass element (1)
atomic_number atomic-mass element (2)
...
atomic_number atomic-mass element (ntypes)
The next ntypes*(ntypes+1)/2 lines contain two numbers:
cut xi (1)
cut xi (2)
...
cut xi (ntypes*(ntypes+1)/2)
Here cut means the cutoff distance of the pair functions, xi is the same as defined in the potential functions
above. The ntypes*(ntypes+1)/2 lines are related to the pairs according to the sequence of first ii (self) pairs, i
= 1, 2, ..., ntypes, and then then ij (cross) pairs, i = 1, 2, ..., ntypes-1, and j = i+1, i+2, ..., ntypes (i.e., the
sequence of the ij pairs follows 11, 22, ..., 12, 13, 14, ..., 23, 24, ...).
The final blocks of the potential file are the U, V, W, P, G, and F functions are listed sequentially. First, U
functions are given for each of the ntypes*(ntypes+1)/2 pairs according to the sequence described above. For
each of the pairs, nr values are listed. Next, similar arrays are given for V, W, and P functions. Then G
functions are given for all the ntypes*ntypes*ntypes ijk triplets in a natural sequence i from 1 to ntypes, j from
1 to ntypes, and k from 1 to ntypes (i.e., ijk = 111, 112, 113, ..., 121, 122, 123 ..., 211, 212, ...). Each of the ijk
functions contains ng values. Finally, the F functions are listed for all ntypes*(ntypes+1)/2 pairs, each
containing nx values. Either analytic or tabulated functions can be specified. Currently, constant, exponential,
sine and cosine analytic functions are available which are specified with: constant c1 , where f(x) = c1
exponential c1 c2 , where f(x) = c1 exp(c2*x) sine c1 c2 , where f(x) = c1 sin(c2*x) cos c1 c2 , where f(x) =
c1 cos(c2*x) Tabulated functions are specified by spline n x1 x2, where n=number of point, (x1,x2)=range
and then followed by n values evaluated uniformly over these argument ranges. The valid argument ranges of
the functions are between 0 <= r <= cut for the U(r), V(r), W(r) functions, -cutmax <= delta_r <= cutmax for
the P(delta_r) functions, -1 <= costheta <= 1 for the G(costheta) functions, and 0 <= X <= maxX for the F(X)
functions.
Mixing, shift, table tail correction, restart:
This pair styles does not support the pair_modify shift, table, and tail options.
This pair style does not write their information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
Restrictions:
If using create_atoms command, atomic masses must be defined in the input script. If using read_data, atomic
masses must be defined in the atomic structure data file.
LAMMPS Users Manual
1613

This pair style is part of the MANYBODY package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
This pair potential requires the newtion setting to be "on" for pair interactions.
The potential files provided with LAMMPS (see the potentials directory) are parameterized for metal units.
You can use any LAMMPS units, but you would need to create your own potential files.
Related commands:
pair_coeff
(Zhou) X. W. Zhou, M. E. Foster, R. E. Jones, P. Yang, H. Fan, and F. P. Doty, J. Mater. Sci. Res., 4, 15
(2015).
(SW) F. H. Stillinger-Weber, and T. A. Weber, Phys. Rev. B, 31, 5262 (1985).
(Tersoff) J. Tersoff, Phys. Rev. B, 39, 5566 (1989).
(Albe) K. Albe, K. Nordlund, J. Nord, and A. Kuronen, Phys. Rev. B, 66, 035205 (2002).
(Wang) J. Wang, and A. Rockett, Phys. Rev. B, 43, 12571 (1991).
(Daw) M. S. Daw, and M. I. Baskes, Phys. Rev. B, 29, 6443 (1984).
LAMMPS Users Manual
1614

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style quip command
Syntax:
pair_style quip
Examples:
pair_style quip
pair_coeff * * gap_example.xml "Potential xml_label=GAP_2014_5_8_60_17_10_38_466" 14
pair_coeff * * sw_example.xml "IP SW" 14
Description:
Style quip provides an interface for calling potential routines from the QUIP package. QUIP is built
separately, and then linked to LAMMPS. The most recent version of the QUIP package can be downloaded
from GitHub: https://github.com/libAtoms/QUIP. The interface is chiefly intended to be used to run Gaussian
Approximation Potentials (GAP), which are described in the following publications: (Bartok et al) and (PhD
thesis of Bartok).
Only a single pair_coeff command is used with the quip style that specifies a QUIP potential file containing
the parameters of the potential for all needed elements in XML format. This is followed by a QUIP
initialization string. Finally, the QUIP elements are mapped to LAMMPS atom types by specifying N atomic
numbers, where N is the number of LAMMPS atom types:
QUIP filename• QUIP initialization string• N atomic numbers = mapping of QUIP elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
A QUIP potential is fully specified by the filename which contains the parameters of the potential in XML
format, the initialization string, and the map of atomic numbers.
GAP potentials can be obtained from the Data repository section of http://www.libatoms.org, where the
appropriate initialization strings are also advised. The list of atomic numbers must be matched to the
LAMMPS atom types specified in the LAMMPS data file or elsewhere.
Two examples input scripts are provided in the examples/USER/quip directory.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify mix, shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
LAMMPS Users Manual
1615

Restrictions:
This pair style is part of the USER-QUIP package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
QUIP potentials are parametrized in electron-volts and Angstroms and therefore should be used with
LAMMPS metal units.
Related commands:
pair_coeff
(Bartok_2010) AP Bartok, MC Payne, R Kondor, and G Csanyi, Physical Review Letters 104, 136403
(2010).
(Bartok_PhD) A Bartok-Partay, PhD Thesis, University of Cambridge, (2010).
LAMMPS Users Manual
1616

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style reax command
Syntax:
pair_style reax hbcut hbnewflag tripflag precision
hbcut = hydrogen-bond cutoff (optional) (distance units)• hbnewflag = use old or new hbond function style (0 or 1) (optional)• tripflag = apply stabilization to all triple bonds (0 or 1) (optional)• precision = precision for charge equilibration (optional)•
Examples:
pair_style reax
pair_style reax 10.0 0 1 1.0e-5
pair_coeff * * ffield.reax 3 1 2 2
pair_coeff * * ffield.reax 3 NULL NULL 3
Description:
Style reax computes the ReaxFF potential of van Duin, Goddard and co-workers. ReaxFF uses
distance-dependent bond-order functions to represent the contributions of chemical bonding to the potential
energy. There is more than one version of ReaxFF. The version implemented in LAMMPS uses the functional
forms documented in the supplemental information of the following paper: (Chenoweth). The version
integrated into LAMMPS matches the most up-to-date version of ReaxFF as of summer 2010.
WARNING: pair style reax is now deprecated and will soon be retired. Users should switch to pair_style
reax/c. The reax style differs from the reax/c style in the lo-level implementation details. The reax style is a
Fortran library, linked to LAMMPS. The reax/c style was initially implemented as stand-alone C code and is
now integrated into LAMMPS as a package.
LAMMPS requires that a file called ffield.reax be provided, containing the ReaxFF parameters for each atom
type, bond type, etc. The format is identical to the ffield file used by van Duin and co-workers. The filename
is required as an argument in the pair_coeff command. Any value other than "ffield.reax" will be rejected (see
below).
LAMMPS provides several different versions of ffield.reax in its potentials dir, each called
potentials/ffield.reax.label. These are documented in potentials/README.reax. The default ffield.reax
contains parameterizations for the following elements: C, H, O, N.
NOTE: We do not distribute a wide variety of ReaxFF force field files with LAMMPS. Adri van Duin's group
at PSU is the central repository for this kind of data as they are continuously deriving and updating
parameterizations for different classes of materials. You can submit a contact request at the Materials
Computation Center (MCC) website https://www.mri.psu.edu/materials-computation-center/connect-mcc,
describing the material(s) you are interested in modeling with ReaxFF. They can tell you what is currently
available or what it would take to create a suitable ReaxFF parameterization.
The format of these files is identical to that used originally by van Duin. We have tested the accuracy of
pair_style reax potential against the original ReaxFF code for the systems mentioned above. You can use
LAMMPS Users Manual
1617
other ffield files for specific chemical systems that may be available elsewhere (but note that their accuracy
may not have been tested).
The hbcut, hbnewflag, tripflag, and precision settings are optional arguments. If none are provided, default
settings are used: hbcut = 6 (which is Angstroms in real units), hbnewflag = 1 (use new hbond function style),
tripflag = 1 (apply stabilization to all triple bonds), and precision = 1.0e-6 (one part in 10^6). If you wish to
override any of these defaults, then all of the settings must be specified.
Two examples using pair_style reax are provided in the examples/reax sub-directory, along with
corresponding examples for pair_style reax/c. Note that while the energy and force calculated by both of these
pair styles match very closely, the contributions due to the valence angles differ slightly due to the fact that
with pair_style reax/c the default value of thb_cutoff_sq is 0.00001, while for pair_style reax it is hard-coded
to be 0.001.
Use of this pair style requires that a charge be defined for every atom since the reax pair style performs a
charge equilibration (QEq) calculation. See the atom_style and read_data commands for details on how to
specify charges.
The thermo variable evdwl stores the sum of all the ReaxFF potential energy contributions, with the exception
of the Coulombic and charge equilibration contributions which are stored in the thermo variable ecoul. The
output of these quantities is controlled by the thermo command.
This pair style tallies a breakdown of the total ReaxFF potential energy into sub-categories, which can be
accessed via the compute pair command as a vector of values of length 14. The 14 values correspond to the
following sub-categories (the variable names in italics match those used in the ReaxFF FORTRAN library):
eb = bond energy1. ea = atom energy2. elp = lone-pair energy3. emol = molecule energy (always 0.0)4. ev = valence angle energy5. epen = double-bond valence angle penalty6. ecoa = valence angle conjugation energy7. ehb = hydrogen bond energy8. et = torsion energy9. eco = conjugation energy10. ew = van der Waals energy11. ep = Coulomb energy12. efi = electric field energy (always 0.0)13. eqeq = charge equilibration energy14.
To print these quantities to the log file (with descriptive column headings) the following commands could be
included in an input script:
compute reax all pair reax
variable eb equal c_reax[1]
variable ea equal c_reax[2]
...
variable eqeq equal c_reax[14]
thermo_style custom step temp epair v_eb v_ea ... v_eqeq
LAMMPS Users Manual
1618

Only a single pair_coeff command is used with the reax style which specifies a ReaxFF potential file with
parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional
arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N indices = mapping of ReaxFF elements to atom types•
The specification of the filename and the mapping of LAMMPS atom types recognized by the ReaxFF is done
differently than for other LAMMPS potentials, due to the non-portable difficulty of passing character strings
(e.g. filename, element names) between C++ and Fortran.
The filename has to be "ffield.reax" and it has to exist in the directory you are running LAMMPS in. This
means you cannot prepend a path to the file in the potentials dir. Rather, you should copy that file into the
directory you are running from. If you wish to use another ReaxFF potential file, then name it "ffield.reax"
and put it in the directory you run from.
In the ReaxFF potential file, near the top, after the general parameters, is the atomic parameters section that
contains element names, each with a couple dozen numeric parameters. If there are M elements specified in
the ffield file, think of these as numbered 1 to M. Each of the N indices you specify for the N atom types of
LAMMPS atoms must be an integer from 1 to M. Atoms with LAMMPS type 1 will be mapped to whatever
element you specify as the first index value, etc. If a mapping value is specified as NULL, the mapping is not
performed. This can be used when a ReaxFF potential is used as part of the hybrid pair style. The NULL
values are placeholders for atom types that will be used with other potentials.
NOTE: Currently the reax pair style cannot be used as part of the hybrid pair style. Some additional changes
still need to be made to enable this.
As an example, say your LAMMPS simulation has 4 atom types and the elements are ordered as C, H, O, N in
the ffield file. If you want the LAMMPS atom type 1 and 2 to be C, type 3 to be N, and type 4 to be H, you
would use the following pair_coeff command:
pair_coeff * * ffield.reax 1 1 4 2
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify mix, shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
The ReaxFF potential files provided with LAMMPS in the potentials directory are parameterized for real
units. You can use the ReaxFF potential with any LAMMPS units, but you would need to create your own
potential file with coefficients listed in the appropriate units if your simulation doesn't use "real" units.
Related commands:
LAMMPS Users Manual
1619


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style reax/c command
pair_style reax/c/kk command
Syntax:
pair_style reax/c cfile keyword value
cfile = NULL or name of a control file• zero or more keyword/value pairs may be appended
keyword = checkqeq or lgvdw or safezone or mincap
checkqeq value = yes or no = whether or not to require qeq/reax fix
lgvdw value = yes or no = whether or not to use a low gradient vdW correction
safezone = factor used for array allocation
mincap = minimum size for array allocation
•
Examples:
pair_style reax/c NULL
pair_style reax/c controlfile checkqeq no
pair_style reax/c NULL lgvdw yes
pair_style reax/c NULL safezone 1.6 mincap 100
pair_coeff * * ffield.reax C H O N
Description:
Style reax/c computes the ReaxFF potential of van Duin, Goddard and co-workers. ReaxFF uses
distance-dependent bond-order functions to represent the contributions of chemical bonding to the potential
energy. There is more than one version of ReaxFF. The version implemented in LAMMPS uses the functional
forms documented in the supplemental information of the following paper: (Chenoweth et al., 2008). The
version integrated into LAMMPS matches the most up-to-date version of ReaxFF as of summer 2010. For
more technical details about the pair reax/c implementation of ReaxFF, see the (Aktulga) paper. The reax/c
style was initially implemented as a stand-alone C code and is now integrated into LAMMPS as a package.
The reax/c/kk style is a Kokkos version of the ReaxFF potential that is derived from the reax/c style. The
Kokkos version can run on GPUs and can also use OpenMP multithreading. For more information about the
Kokkos package, see Section 4 and Section 5.3.3. One important consideration when using the reax/c/kk style
is the choice of either half or full neighbor lists. This setting can be changed using the Kokkos package
command.
The reax/c style differs from the pair_style reax command in the lo-level implementation details. The reax
style is a Fortran library, linked to LAMMPS. The reax/c style was initially implemented as stand-alone C
code and is now integrated into LAMMPS as a package.
LAMMPS provides several different versions of ffield.reax in its potentials dir, each called
potentials/ffield.reax.label. These are documented in potentials/README.reax. The default ffield.reax
contains parameterizations for the following elements: C, H, O, N.
LAMMPS Users Manual
1621
The format of these files is identical to that used originally by van Duin. We have tested the accuracy of
pair_style reax/c potential against the original ReaxFF code for the systems mentioned above. You can use
other ffield files for specific chemical systems that may be available elsewhere (but note that their accuracy
may not have been tested).
NOTE: We do not distribute a wide variety of ReaxFF force field files with LAMMPS. Adri van Duin's group
at PSU is the central repository for this kind of data as they are continuously deriving and updating
parameterizations for different classes of materials. You can submit a contact request at the Materials
Computation Center (MCC) website https://www.mri.psu.edu/materials-computation-center/connect-mcc,
describing the material(s) you are interested in modeling with ReaxFF. They can tell you what is currently
available or what it would take to create a suitable ReaxFF parameterization.
The cfile setting can be specified as NULL, in which case default settings are used. A control file can be
specified which defines values of control variables. Some control variables are global parameters for the
ReaxFF potential. Others define certain performance and output settings. Each line in the control file specifies
the value for a control variable. The format of the control file is described below.
NOTE: The LAMMPS default values for the ReaxFF global parameters correspond to those used by Adri van
Duin's stand-alone serial code. If these are changed by setting control variables in the control file, the results
from LAMMPS and the serial code will not agree.
Two examples using pair_style reax/c are provided in the examples/reax sub-directory, along with
corresponding examples for pair_style reax.
Use of this pair style requires that a charge be defined for every atom. See the atom_style and read_data
commands for details on how to specify charges.
The ReaxFF parameter files provided were created using a charge equilibration (QEq) model for handling the
electrostatic interactions. Therefore, by default, LAMMPS requires that the fix qeq/reax command be used
with pair_style reax/c when simulating a ReaxFF model, to equilibrate charge each timestep. Using the
keyword checkqeq with the value no turns off the check for fix qeq/reax, allowing a simulation to be run
without charge equilibration. In this case, the static charges you assign to each atom will be used for
computing the electrostatic interactions in the system. See the fix qeq/reax command for details.
Using the optional keyword lgvdw with the value yes turns on the low-gradient correction of the ReaxFF/C for
long-range London Dispersion, as described in the (Liu) paper. Force field file ffield.reax.lg is designed for
this correction, and is trained for several energetic materials (see "Liu"). When using lg-correction,
recommended value for parameter thb is 0.01, which can be set in the control file. Note: Force field files are
different for the original or lg corrected pair styles, using wrong ffield file generates an error message.
Optional keywords safezone and mincap are used for allocating reax/c arrays. Increasing these values can
avoid memory problems, such as segmentation faults and bondchk failed errors, that could occur under certain
conditions. These keywords aren't used by the Kokkos version, which instead uses a more robust memory
allocation scheme that checks if the sizes of the arrays have been exceeded and automatically allocates more
memory.
The thermo variable evdwl stores the sum of all the ReaxFF potential energy contributions, with the exception
of the Coulombic and charge equilibration contributions which are stored in the thermo variable ecoul. The
output of these quantities is controlled by the thermo command.
LAMMPS Users Manual
1622
This pair style tallies a breakdown of the total ReaxFF potential energy into sub-categories, which can be
accessed via the compute pair command as a vector of values of length 14. The 14 values correspond to the
following sub-categories (the variable names in italics match those used in the original FORTRAN ReaxFF
code):
eb = bond energy1. ea = atom energy2. elp = lone-pair energy3. emol = molecule energy (always 0.0)4. ev = valence angle energy5. epen = double-bond valence angle penalty6. ecoa = valence angle conjugation energy7. ehb = hydrogen bond energy8. et = torsion energy9. eco = conjugation energy10. ew = van der Waals energy11. ep = Coulomb energy12. efi = electric field energy (always 0.0)13. eqeq = charge equilibration energy14.
To print these quantities to the log file (with descriptive column headings) the following commands could be
included in an input script:
compute reax all pair reax/c
variable eb equal c_reax[1]
variable ea equal c_reax[2]
[...]
variable eqeq equal c_reax[14]
thermo_style custom step temp epair v_eb v_ea [...] v_eqeq
Only a single pair_coeff command is used with the reax/c style which specifies a ReaxFF potential file with
parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional
arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N indices = ReaxFF elements•
The filename is the ReaxFF potential file. Unlike for the reax pair style, any filename can be used.
In the ReaxFF potential file, near the top, after the general parameters, is the atomic parameters section that
contains element names, each with a couple dozen numeric parameters. If there are M elements specified in
the ffield file, think of these as numbered 1 to M. Each of the N indices you specify for the N atom types of
LAMMPS atoms must be an integer from 1 to M. Atoms with LAMMPS type 1 will be mapped to whatever
element you specify as the first index value, etc. If a mapping value is specified as NULL, the mapping is not
performed. This can be used when the reax/c style is used as part of the hybrid pair style. The NULL values
are placeholders for atom types that will be used with other potentials.
As an example, say your LAMMPS simulation has 4 atom types and the elements are ordered as C, H, O, N in
the ffield file. If you want the LAMMPS atom type 1 and 2 to be C, type 3 to be N, and type 4 to be H, you
would use the following pair_coeff command:
pair_coeff * * ffield.reax C C N H
LAMMPS Users Manual
1623

The format of a line in the control file is as follows:
variable_name value
and it may be followed by an "!" character and a trailing comment.
If the value of a control variable is not specified, then default values are used. What follows is the list of
variables along with a brief description of their use and default values.
simulation_name: Output files produced by pair_style reax/c carry this name + extensions specific to their
contents. Partial energies are reported with a ".pot" extension, while the trajectory file has ".trj" extension.
tabulate_long_range: To improve performance, long range interactions can optionally be tabulated (0 means
no tabulation). Value of this variable denotes the size of the long range interaction table. The range from 0 to
long range cutoff (defined in the ffield file) is divided into tabulate_long_range points. Then at the start of
simulation, we fill in the entries of the long range interaction table by computing the energies and forces
resulting from van der Waals and Coulomb interactions between every possible atom type pairs present in the
input system. During the simulation we consult to the long range interaction table to estimate the energy and
forces between a pair of atoms. Linear interpolation is used for estimation. (default value = 0)
energy_update_freq: Denotes the frequency (in number of steps) of writes into the partial energies file.
(default value = 0)
nbrhood_cutoff: Denotes the near neighbors cutoff (in Angstroms) regarding the bonded interactions. (default
value = 5.0)
hbond_cutoff: Denotes the cutoff distance (in Angstroms) for hydrogen bond interactions.(default value = 7.5.
A value of 0.0 turns off hydrogen bonds)
bond_graph_cutoff: is the threshold used in determining what is a physical bond, what is not. Bonds and
angles reported in the trajectory file rely on this cutoff. (default value = 0.3)
thb_cutoff: cutoff value for the strength of bonds to be considered in three body interactions. (default value =
0.001)
thb_cutoff_sq: cutoff value for the strength of bond order products to be considered in three body interactions.
(default value = 0.00001)
write_freq: Frequency of writes into the trajectory file. (default value = 0)
traj_title: Title of the trajectory - not the name of the trajectory file.
atom_info: 1 means print only atomic positions + charge (default = 0)
atom_forces: 1 adds net forces to atom lines in the trajectory file (default = 0)
atom_velocities: 1 adds atomic velocities to atoms line (default = 0)
bond_info: 1 prints bonds in the trajectory file (default = 0)
LAMMPS Users Manual
1624

angle_info: 1 prints angles in the trajectory file (default = 0)
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify mix, shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This pair style is part of the USER-REAXC package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
The ReaxFF potential files provided with LAMMPS in the potentials directory are parameterized for real
units. You can use the ReaxFF potential with any LAMMPS units, but you would need to create your own
potential file with coefficients listed in the appropriate units if your simulation doesn't use "real" units.
Related commands:
pair_coeff, fix qeq/reax, fix reax/c/bonds, fix reax/c/species, pair_style reax
Default:
The keyword defaults are checkqeq = yes, lgvdw = no, safezone = 1.2, mincap = 50.
(Chenoweth_2008) Chenoweth, van Duin and Goddard, Journal of Physical Chemistry A, 112, 1040-1053
(2008).
(Aktulga) Aktulga, Fogarty, Pandit, Grama, Parallel Computing, 38, 245-259 (2012).
LAMMPS Users Manual
1625
(Liu) L. Liu, Y. Liu, S. V. Zybin, H. Sun and W. A. Goddard, Journal of Physical Chemistry A, 115,
11016-11022 (2011).
LAMMPS Users Manual
1626

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style resquared command
pair_style resquared/gpu command
pair_style resquared/omp command
Syntax:
pair_style resquared cutoff
cutoff = global cutoff for interactions (distance units)•
Examples:
pair_style resquared 10.0
pair_coeff * * 1.0 1.0 1.7 3.4 3.4 1.0 1.0 1.0
Description:
Style resquared computes the RE-squared anisotropic interaction (Everaers), (Babadi) between pairs of
ellipsoidal and/or spherical Lennard-Jones particles. For ellipsoidal interactions, the potential considers the
ellipsoid as being comprised of small spheres of size sigma. LJ particles are a single sphere of size sigma. The
distinction is made to allow the pair style to make efficient calculations of ellipsoid/solvent interactions.
Details for the equations used are given in the references below and in this supplementary document.
Use of this pair style requires the NVE, NVT, or NPT fixes with the asphere extension (e.g. fix nve/asphere)
in order to integrate particle rotation. Additionally, atom_style ellipsoid should be used since it defines the
rotational state and the size and shape of each ellipsoidal particle.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands:
A12 = Energy Prefactor/Hamaker constant (energy units)• sigma = atomic interaction diameter (distance units)• epsilon_i_a = relative well depth of type I for side-to-side interactions• epsilon_i_b = relative well depth of type I for face-to-face interactions• epsilon_i_c = relative well depth of type I for end-to-end interactions• epsilon_j_a = relative well depth of type J for side-to-side interactions• epsilon_j_b = relative well depth of type J for face-to-face interactions• epsilon_j_c = relative well depth of type J for end-to-end interactions• cutoff (distance units)•
The parameters used depend on the type of the interacting particles, i.e. ellipsoids or LJ spheres. The type of a
particle is determined by the diameters specified for its 3 shape parameters. If all 3 shape parameters = 0.0,
then the particle is treated as an LJ sphere. The epsilon_i_* or epsilon_j_* parameters are ignored for LJ
spheres. If the 3 shape parameters are > 0.0, then the particle is treated as an ellipsoid (even if the 3
parameters are equal to each other).
LAMMPS Users Manual
1627
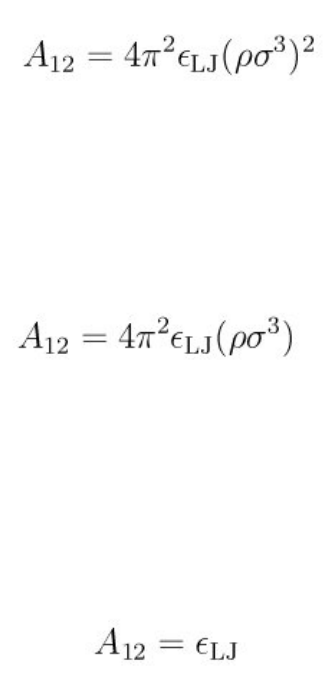
A12 specifies the energy prefactor which depends on the types of the two interacting particles.
For ellipsoid/ellipsoid interactions, the interaction is computed by the formulas in the supplementary
document referenced above. A12 is the Hamaker constant as described in (Everaers). In LJ units:
where rho gives the number density of the spherical particles composing the ellipsoids and epsilon_LJ
determines the interaction strength of the spherical particles.
For ellipsoid/LJ sphere interactions, the interaction is also computed by the formulas in the supplementary
document referenced above. A12 has a modified form (see here for details):
For ellipsoid/LJ sphere interactions, a correction to the distance- of-closest approach equation has been
implemented to reduce the error from two particles of disparate sizes; see this supplementary document.
For LJ sphere/LJ sphere interactions, the interaction is computed using the standard Lennard-Jones formula,
which is much cheaper to compute than the ellipsoidal formulas. A12 is used as epsilon in the standard LJ
formula:
and the specified sigma is used as the sigma in the standard LJ formula.
When one of both of the interacting particles are ellipsoids, then sigma specifies the diameter of the
continuous distribution of constituent particles within each ellipsoid used to model the RE-squared potential.
Note that this is a different meaning for sigma than the pair_style gayberne potential uses.
The epsilon_i and epsilon_j coefficients are defined for atom types, not for pairs of atom types. Thus, in a
series of pair_coeff commands, they only need to be specified once for each atom type.
Specifically, if any of epsilon_i_a, epsilon_i_b, epsilon_i_c are non-zero, the three values are assigned to
atom type I. If all the epsilon_i values are zero, they are ignored. If any of epsilon_j_a, epsilon_j_b,
epsilon_j_c are non-zero, the three values are assigned to atom type J. If all three epsilon_i values are zero,
they are ignored. Thus the typical way to define the epsilon_i and epsilon_j coefficients is to list their values
in "pair_coeff I J" commands when I = J, but set them to 0.0 when I != J. If you do list them when I != J, you
should insure they are consistent with their values in other pair_coeff commands.
Note that if this potential is being used as a sub-style of pair_style hybrid, and there is no "pair_coeff I I"
setting made for RE-squared for a particular type I (because I-I interactions are computed by another hybrid
LAMMPS Users Manual
1628
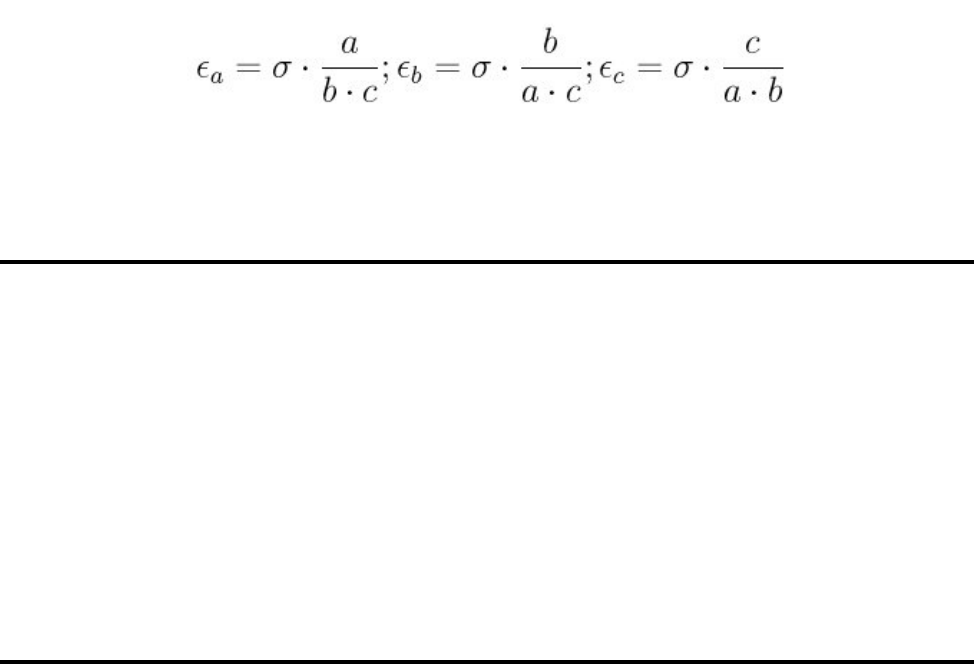
pair potential), then you still need to insure the epsilon a,b,c coefficients are assigned to that type in a
"pair_coeff I J" command.
For large uniform molecules it has been shown that the epsilon_*_* energy parameters are approximately
representable in terms of local contact curvatures (Everaers):
where a, b, and c give the particle diameters.
The last coefficient is optional. If not specified, the global cutoff specified in the pair_style command is used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance can be mixed, but
only for sphere pairs. The default mix value is geometric. See the "pair_modify" command for details. Other
type pairs cannot be mixed, due to the different meanings of the energy prefactors used to calculate the
interactions and the implicit dependence of the ellipsoid-sphere interaction on the equation for the Hamaker
constant presented here. Mixing of sigma and epsilon followed by calculation of the energy prefactors using
the equations above is recommended.
This pair styles supports the pair_modify shift option for the energy of the Lennard-Jones portion of the pair
interaction, but only for sphere-sphere interactions. There is no shifting performed for ellipsoidal interactions
due to the anisotropic dependence of the interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
LAMMPS Users Manual
1629

This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords of the run_style command.
Restrictions:
This style is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
This pair style requires that atoms be ellipsoids as defined by the atom_style ellipsoid command.
Particles acted on by the potential can be finite-size aspherical or spherical particles, or point particles.
Spherical particles have all 3 of their shape parameters equal to each other. Point particles have all 3 of their
shape parameters equal to 0.0.
The distance-of-closest-approach approximation used by LAMMPS becomes less accurate when high-aspect
ratio ellipsoids are used.
Related commands:
pair_coeff, fix nve/asphere, compute temp/asphere, pair_style gayberne
Default: none
(Everaers) Everaers and Ejtehadi, Phys Rev E, 67, 041710 (2003).
(Berardi) Babadi, Ejtehadi, Everaers, J Comp Phys, 219, 770-779 (2006).
LAMMPS Users Manual
1630

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style lj/sdk command
pair_style lj/sdk/gpu command
pair_style lj/sdk/kk command
pair_style lj/sdk/omp command
pair_style lj/sdk/coul/long command
pair_style lj/sdk/coul/long/gpu command
pair_style lj/sdk/coul/long/omp command
Syntax:
pair_style style args
style = lj/sdk or lj/sdk/coul/long• args = list of arguments for a particular style•
lj/sdk args = cutoff
cutoff = global cutoff for Lennard Jones interactions (distance units)
lj/sdk/coul/long args = cutoff (cutoff2)
cutoff = global cutoff for LJ (and Coulombic if only 1 arg) (distance units)
cutoff2 = global cutoff for Coulombic (optional) (distance units)
Examples:
pair_style lj/sdk 2.5
pair_coeff 1 1 lj12_6 1 1.1 2.8
pair_style lj/sdk/coul/long 10.0
pair_style lj/sdk/coul/long 10.0 12.0
pair_coeff 1 1 lj9_6 100.0 3.5 12.0
Description:
The lj/sdk styles compute a 9/6, 12/4, or 12/6 Lennard-Jones potential, given by
LAMMPS Users Manual
1631
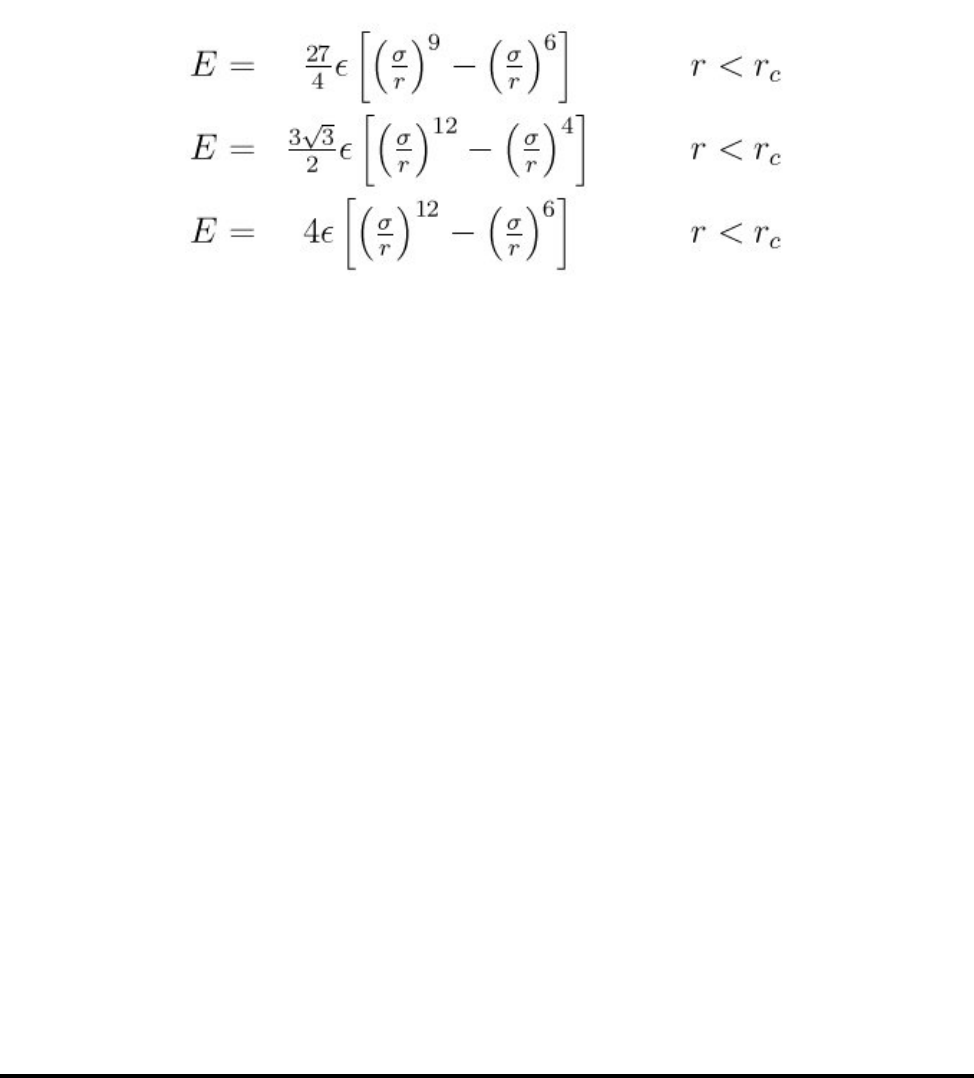
as required for the SDK Coarse-grained MD parametrization discussed in (Shinoda) and (DeVane). Rc is the
cutoff.
Style lj/sdk/coul/long computes the adds Coulombic interactions with an additional damping factor applied so
it can be used in conjunction with the kspace_style command and its ewald or pppm or pppm/cg option. The
Coulombic cutoff specified for this style means that pairwise interactions within this distance are computed
directly; interactions outside that distance are computed in reciprocal space.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
cg_type (lj9_6, lj12_4, or lj12_6)• epsilon (energy units)• sigma (distance units)• cutoff1 (distance units)•
Note that sigma is defined in the LJ formula as the zero-crossing distance for the potential, not as the energy
minimum. The prefactors are chosen so that the potential minimum is at -epsilon.
The latter 2 coefficients are optional. If not specified, the global LJ and Coulombic cutoffs specified in the
pair_style command are used. If only one cutoff is specified, it is used as the cutoff for both LJ and
Coulombic interactions for this type pair. If both coefficients are specified, they are used as the LJ and
Coulombic cutoffs for this type pair.
For lj/sdk/coul/long only the LJ cutoff can be specified since a Coulombic cutoff cannot be specified for an
individual I,J type pair. All type pairs use the same global Coulombic cutoff specified in the pair_style
command.
Styles with a gpu, intel, kk, omp or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP, and OPT packages
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
LAMMPS Users Manual
1632

You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, and rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of the lj/sdk
pair styles cannot be mixed, since different pairs may have different exponents. So all parameters for all pairs
have to be specified explicitly through the "pair_coeff" command. Defining then in a data file is also not
supported, due to limitations of that file format.
All of the lj/sdk pair styles support the pair_modify shift option for the energy of the Lennard-Jones portion of
the pair interaction.
The lj/sdk/coul/long pair styles support the pair_modify table option since they can tabulate the short-range
portion of the long-range Coulombic interaction.
All of the lj/sdk pair styles write their information to binary restart files, so pair_style and pair_coeff
commands do not need to be specified in an input script that reads a restart file.
The lj/sdk and lj/cut/coul/long pair styles do not support the use of the inner, middle, and outer keywords of
the run_style respa command.
Restrictions:
All of the lj/sdk pair styles are part of the USER-CG-CMM package. The lj/sdk/coul/long style also requires
the KSPACE package to be built (which is enabled by default). They are only enabled if LAMMPS was built
with that package. See the Making LAMMPS section for more info.
Related commands:
pair_coeff, angle_style sdk
Default: none
(Shinoda) Shinoda, DeVane, Klein, Mol Sim, 33, 27 (2007).
(DeVane) Shinoda, DeVane, Klein, Soft Matter, 4, 2453-2462 (2008).
LAMMPS Users Manual
1633

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style smd/hertz command
Syntax:
pair_style smd/hertz scale_factor
Examples:
pair_style smd/hertz 1.0 pair_coeff 1 1
Description:
The smd/hertz style calculates contact forces between SPH particles belonging to different physical bodies.
The contact forces are calculated using a Hertz potential, which evaluates the overlap between two particles
(whose spatial extents are defined via its contact radius). The effect is that a particles cannot penetrate into
each other. The parameter has units of pressure and should equal roughly one half of the Young's modulus (or
bulk modulus in the case of fluids) of the material model associated with the SPH particles.
The parameter scale_factor can be used to scale the particles' contact radii. This can be useful to control how
close particles can approach each other. Usually, scale_factor=1.0.
Mixing, shift, table, tail correction, restart, rRESPA info:
No mixing is performed automatically. Currently, no part of USER-SMD supports restarting nor
minimization. rRESPA does not apply to this pair style.
Restrictions:
This fix is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1634

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style smd/tlsph command
Syntax:
pair_style smd/tlsph args
Examples:
pair_style smd/tlsph
Description:
The smd/tlsph style computes particle interactions according to continuum mechanics constitutive laws and a
Total-Lagrangian Smooth-Particle Hydrodynamics algorithm.
This pair style is invoked with the following command:
pair_style smd/tlsph
pair_coeff i j *COMMON rho0 E nu Q1 Q2 hg Cp &
*END
Here, i and j denote the LAMMPS particle types for which this pair style is defined. Note that i and j must be
equal, i.e., no tlsph cross interactions between different particle types are allowed. In contrast to the usual
LAMMPS pair coeff definitions, which are given solely a number of floats and integers, the tlsph pair coeff
definition is organised using keywords. These keywords mark the beginning of different sets of parameters for
particle properties, material constitutive models, and damage models. The pair coeff line must be terminated
with the *END keyword. The use the line continuation operator & is recommended. A typical invocation of
the tlsph for a solid body would consist of an equation of state for computing the pressure (the diagonal
components of the stress tensor), and a material model to compute shear stresses (the off-diagonal components
of the stress tensor). Damage and failure models can also be added.
Please see the SMD user guide for a complete listing of the possible keywords and material models.
Mixing, shift, table, tail correction, restart, rRESPA info:
No mixing is performed automatically. Currently, no part of USER-SMD supports restarting nor
minimization. rRESPA does not apply to this pair style.
Restrictions:
This fix is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1635

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style smd/tri_surface command
Syntax:
pair_style smd/tri_surface scale_factor
Examples:
pair_style smd/tri_surface 1.0 pair_coeff 1 1
Description:
The smd/tri_surface style calculates contact forces between SPH particles and a rigid wall boundary defined
via the smd/wall_surface fix.
The contact forces are calculated using a Hertz potential, which evaluates the overlap between a particle
(whose spatial extents are defined via its contact radius) and the triangle. The effect is that a particle cannot
penetrate into the triangular surface. The parameter has units of pressure and should equal roughly one half of
the Young's modulus (or bulk modulus in the case of fluids) of the material model associated with the SPH
particle
The parameter scale_factor can be used to scale the particles' contact radii. This can be useful to control how
close particles can approach the triangulated surface. Usually, scale_factor=1.0.
Mixing, shift, table, tail correction, restart, rRESPA info:
No mixing is performed automatically. Currently, no part of USER-SMD supports restarting nor
minimization. rRESPA does not apply to this pair style.
Restrictions:
This fix is part of the USER-SMD package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1636

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style smd/ulsph command
Syntax:
pair_style smd/ulsph args
these keywords must be given
keyword = *DENSITY_SUMMATION or *DENSITY_CONTINUITY and *VELOCITY_GRADIENT or
*NO_VELOCITY_GRADIENT and *GRADIENT_CORRECTION or *NO_GRADIENT_CORRECTION
Examples:
pair_style smd/ulsph *DENSITY_CONTINUITY *VELOCITY_GRADIENT
*NO_GRADIENT_CORRECTION
Description:
The smd/ulsph style computes particle interactions according to continuum mechanics constitutive laws and
an updated Lagrangian Smooth-Particle Hydrodynamics algorithm.
This pair style is invoked similar to the following command:
pair_style smd/ulsph *DENSITY_CONTINUITY *VELOCITY_GRADIENT *NO_GRADIENT_CORRECTION
pair_coeff i j *COMMON rho0 c0 Q1 Cp hg &
*END
Here, i and j denote the LAMMPS particle types for which this pair style is defined. Note that i and j can be
different, i.e., ulsph cross interactions between different particle types are allowed. However, i--i
respectively j--j pair_coeff lines have to precede a cross interaction. In contrast to the usual LAMMPS pair
coeff definitions, which are given solely a number of floats and integers, the ulsph pair coeff definition is
organised using keywords. These keywords mark the beginning of different sets of parameters for particle
properties, material constitutive models, and damage models. The pair coeff line must be terminated with
the *END keyword. The use the line continuation operator & is recommended. A typical invocation of the
ulsph for a solid body would consist of an equation of state for computing the pressure (the diagonal
components of the stress tensor), and a material model to compute shear stresses (the off-diagonal
components of the stress tensor).
Note that the use of *GRADIENT_CORRECTION can lead to severe numerical instabilities. For a general
fluid simulation, *NO_GRADIENT_CORRECTION is recommended.
Please see the SMD user guide for a complete listing of the possible keywords and material models.
Mixing, shift, table, tail correction, restart, rRESPA info:
No mixing is performed automatically. Currently, no part of USER-SMD supports restarting nor
minimization. rRESPA does not apply to this pair style.
Restrictions:
•
LAMMPS Users Manual
1637

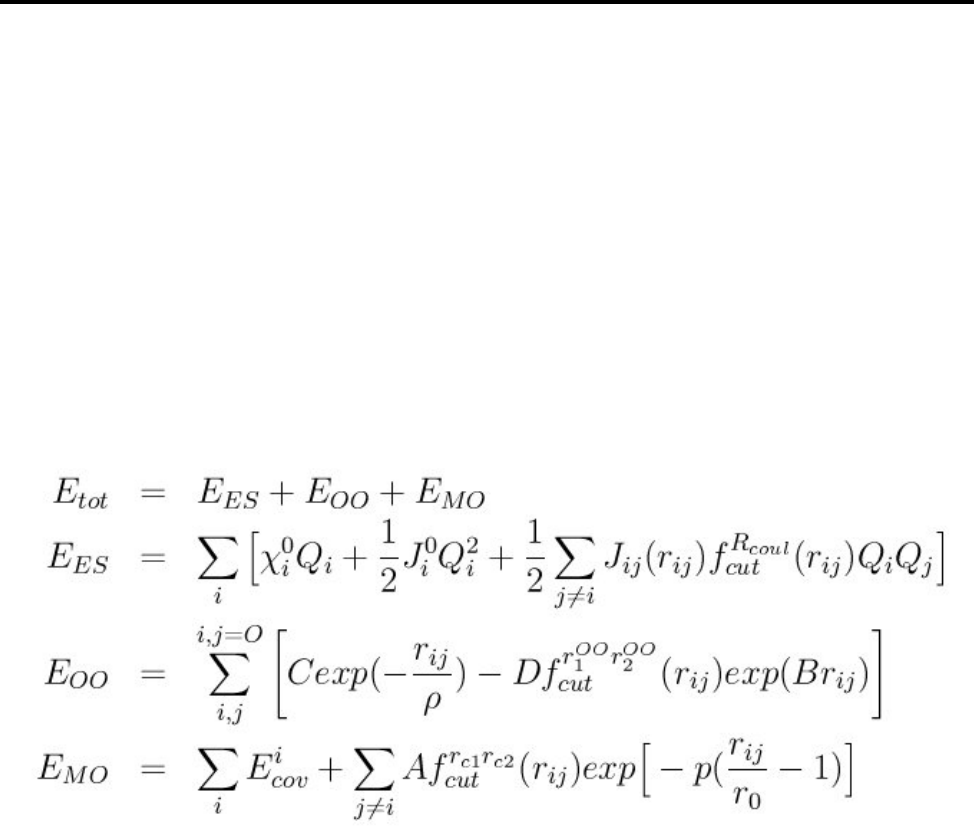
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style smtbq command
Syntax:
pair_style smtbq
Examples:
pair_style smtbq
pair_coeff * * ffield.smtbq.Al2O3 O Al
Description:
This pair stylecomputes a variable charge SMTB-Q (Second-Moment tight-Binding QEq) potential as
described in SMTB-Q_1 and SMTB-Q_2. Briefly, the energy of metallic-oxygen systems is given by three
contributions:
where Etot is the total potential energy of the system, EES is the electrostatic part of the total energy, EOO is the
interaction between oxygens and EMO is a short-range interaction between metal and oxygen atoms. This
interactions depend on interatomic distance rij and/or the charge Qi of atoms i. Cut-off function enables
smooth convergence to zero interaction.
The parameters appearing in the upper expressions are set in the ffield.SMTBQ.Syst file where Syst
corresponds to the selected system (e.g. field.SMTBQ.Al2O3). Examples for TiO2, Al2O3 are provided. A
single pair_coeff command is used with the SMTBQ styles which provides the path to the potential file with
parameters for needed elements. These are mapped to LAMMPS atom types by specifying additional
arguments after the potential filename in the pair_coeff command. Note that atom type 1 must always
correspond to oxygen atoms. As an example, to simulate a TiO2 system, atom type 1 has to be oxygen and
atom type 2 Ti. The following pair_coeff command should then be used:
pair_coeff * * PathToLammps/potentials/ffield.smtbq.TiO2 O Ti
The electrostatic part of the energy consists of two components
LAMMPS Users Manual
1639
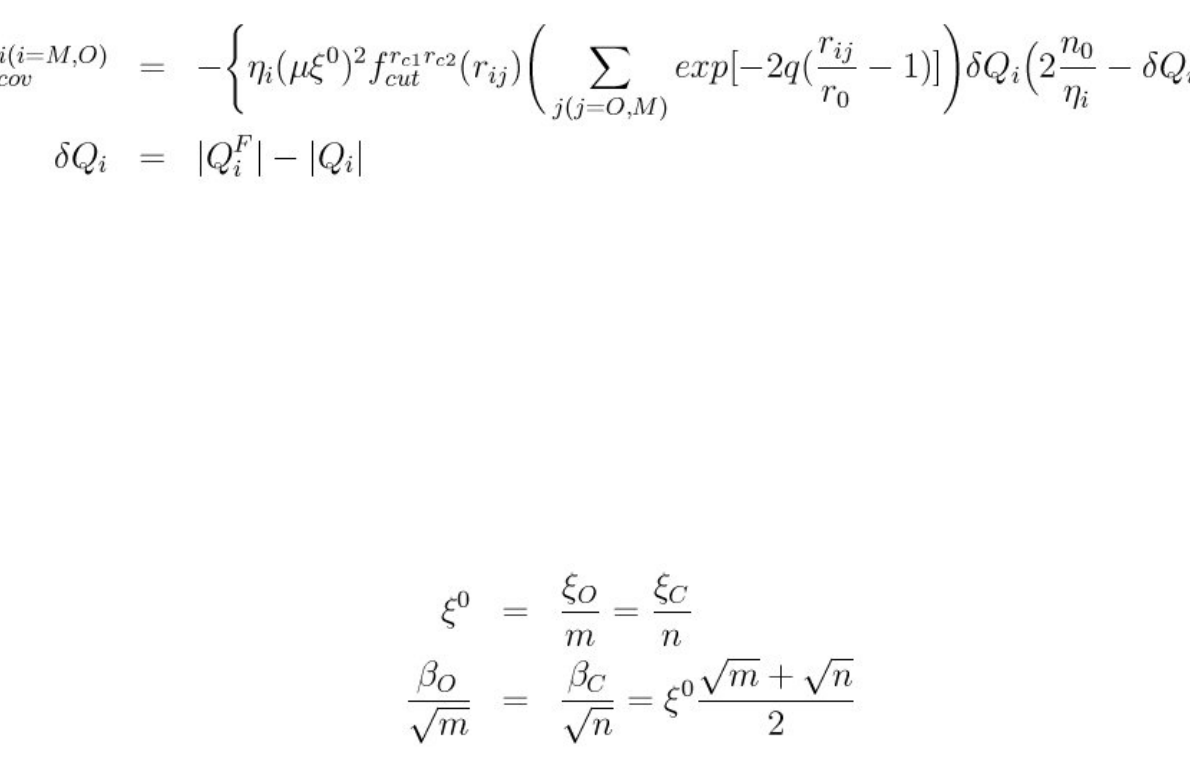
self-energy of atom i in the form of a second order charge dependent polynomial and a long-range Coulombic
electrostatic interaction. The latter uses the wolf summation method described in Wolf, spherically truncated
at a longer cutoff, Rcoul. The charge of each ion is modeled by an orbital Slater which depends on the principal
quantum number (n) of the outer orbital shared by the ion.
Interaction between oxygen, EOO, consists of two parts, an attractive and a repulsive part. The attractive part is
effective only at short range (< r2OO). The attractive contribution was optimized to study surfaces
reconstruction (e.g. SMTB-Q_2 in TiO2) and is not necessary for oxide bulk modeling. The repulsive part is
the Pauli interaction between the electron clouds of oxygen. The Pauli repulsion and the coulombic
electrostatic interaction have same cut off value. In the ffield.SMTBQ.Syst, the keyword 'buck' allows to
consider only the repulsive O-O interactions. The keyword 'buckPlusAttr' allows to consider the repulsive and
the attractive O-O interactions.
The short-range interaction between metal-oxygen, EMO is based on the second moment approximation of the
density of states with a N-body potential for the band energy term, Eicov, and a Born-Mayer type repulsive
terms as indicated by the keyword 'second_moment' in the ffield.SMTBQ.Syst. The energy band term is given
by:
where ηi is the stoichiometry of atom i, δQi is the charge delocalization of atom i, compared to its
formal charge QFi. n0, the number of hybridized orbitals, is calculated with to the atomic orbitals shared di and
the stoichiometry ηi. rc1 and rc2 are the two cutoff radius around the fourth neighbors in the cutoff
function.
In the formalism used here, ╬ is the energy parameter. ╬ is in tight-binding approximation the
hopping integral between the hybridized orbitals of the cation and the anion. In the literature we find many
ways to write the hopping integral depending on whether one takes the point of view of the anion or cation.
These are equivalent vision. The correspondence between the two visions is explained in appendix A of the
article in the SrTiO3SMTB-Q_3 (parameter β shown in this article is in fact the βO). To
summarize the relationship between the hopping integral ╬ and the others, we have in an oxide CnOm
the following relationship:
Thus parameter μ, indicated above, is given by : μ = (√n + √m) ⁄ 2
LAMMPS Users Manual
1640
The potential offers the possibility to consider the polarizability of the electron clouds of oxygen by changing
the slater radius of the charge density around the oxygens through the parameters rBB, rB and rS in the
ffield.SMTBQ.Syst. This change in radius is performed according to the method developed by E. Maras
SMTB-Q_2. This method needs to determine the number of nearest neighbors around the oxygen. This
calculation is based on first (r1n) and second (r2n) distances neighbors.
The SMTB-Q potential is a variable charge potential. The equilibrium charge on each atom is calculated by
the electronegativity equalization (QEq) method. See Rick for further detail. One can adjust the frequency, the
maximum number of iterative loop and the convergence of the equilibrium charge calculation. To obtain the
energy conservation in NVE thermodynamic ensemble, we recommend to use a convergence parameter in the
interval 10-5 - 10-6 eV.
The ffield.SMTBQ.Syst files are provided for few systems. They consist of nine parts and the lines beginning
with '#' are comments (note that the number of comment lines matter). The first sections are on the potential
parameters and others are on the simulation options and might be modified. Keywords are character type and
must be enclosed in quotation marks ('').
1) Number of different element in the oxide:
Nelem= 2 or 3• Divided line•
2) Atomic parameters
For the anion (oxygen)
Name of element (char) and stoichiometry in oxide• Formal charge and mass of element• Principal quantic number of outer orbital (n), electronegativity (◆i) and hardness (J0i)• Ionic radius parameters : max coordination number (coordBB = 6 by default), bulk coordination
number (coordB), surface coordination number (coordS) and rBB, rB and rS the slater radius for each
coordination number. (note : If you don't want to change the slater radius, use three identical
radius values)
•
Number of orbital shared by the element in the oxide (di)• Divided line•
For each cations (metal):
Name of element (char) and stoichiometry in oxide• Formal charge and mass of element• Number of electron in outer orbital (ne), electronegativity (◆i), hardness (J0i) and rSalter the
slater radius for the cation.
•
Number of orbitals shared by the elements in the oxide (di)• Divided line•
3) Potential parameters:
Keyword for element1, element2 and interaction potential ('second_moment' or 'buck' or
'buckPlusAttr') between element 1 and 2. If the potential is 'second_moment', specify 'oxide' or 'metal'
for metal-oxygen or metal-metal interactions respectively.
•
Potential parameter:•
LAMMPS Users Manual
1641
If type of potential is 'second_moment' : A (eV), p, ╬ (eV) and q
rc1 (Å), rc2 (Å) and r0 (Å)
If type of potential is 'buck' : C (eV) and ρ (Å)
If type of potential is 'buckPlusAttr' : C (eV) and ρ (Å)
D (eV), B (Å-1), r1OO (Å) and r2OO (Å)
Divided line•
4) Tables parameters:
Cutoff radius for the Coulomb interaction (Rcoul)• Starting radius (rmin = 1,18845 Å) and increments (dr = 0,001 Å) for creating the potential
table.
•
Divided line•
5) Rick model parameter:
Nevery : parameter to set the frequency (1/Nevery) of the charge resolution. The charges are evaluated
each Nevery time steps.
•
Max number of iterative loop (loopmax) and precision criterion (prec) in eV of the charge resolution• Divided line•
6) Coordination parameter:
First (r1n) and second (r2n) neighbor distances in Å• Divided line•
7) Charge initialization mode:
Keyword (QInitMode) and initial oxygen charge (Qinit). If keyword = 'true', all oxygen charges are
initially set equal to Qinit. The charges on the cations are initially set in order to respect the neutrality
of the box. If keyword = 'false', all atom charges are initially set equal to 0 if you use
"create_atom"#create_atom command or the charge specified in the file structure using read_data
command.
•
Divided line•
8) Mode for the electronegativity equalization (Qeq)
Keyword mode:
QEqAll (one QEq group) | no parameters
QEqAllParallel (several QEq groups) | no parameters
Surface | zlim (QEq only for z>zlim)
•
Parameter if necessary• Divided line•
9) Verbose
If you want the code to work in verbose mode or not : 'true' or 'false'• If you want to print or not in file 'Energy_component.txt' the three main contributions to the energy of
the system according to the description presented above : 'true' or 'false' and NEnergy. This option
•
LAMMPS Users Manual
1642
writes in file every NEnergy time step. If the value is 'false' then NEnergy = 0. The file take into account
the possibility to have several QEq group g then it writes: time step, number of atoms in group g,
electrostatic part of energy, EES, the interaction between oxygen, EOO, and short range metal-oxygen
interaction, EMO.
If you want to print in file 'Electroneg_component.txt' the electronegativity component (∂Etot
⁄∂Qi) or not: 'true' or 'false' and NElectroneg.This option writes in file every NElectroneg time
step. If the value is 'false' then NElectroneg = 0. The file consist in atom number i, atom type (1 for
oxygen and # higher than 1 for metal), atom position: x, y and z, atomic charge of atom i, electrostatic
part of atom i electronegativity, covalent part of atom i electronegativity, the hopping integral of atom
i (Z⓶)i and box electronegativity.
•
NOTE: This last option slows down the calculation dramatically. Use only with a single processor simulation.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify mix, shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you needs to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restriction:
This pair style is part of the USER-SMTBQ package and is only enabled if LAMMPS is built with that
package. See the Making LAMMPS section for more info.
This potential requires using atom type 1 for oxygen and atom type higher than 1 for metal atoms.
This pair style requires the newton setting to be "on" for pair interactions.
The SMTB-Q potential files provided with LAMMPS (see the potentials directory) are parameterized for
metal units.
Citing this work:
Please cite related publication: N. Salles, O. Politano, E. Amzallag and R. Tetot, Comput. Mater. Sci. 111
(2016) 181-189
(SMTB-Q_1) N. Salles, O. Politano, E. Amzallag, R. Tetot, Comput. Mater. Sci. 111 (2016) 181-189
(SMTB-Q_2) E. Maras, N. Salles, R. Tetot, T. Ala-Nissila, H. Jonsson, J. Phys. Chem. C 2015, 119,
10391-10399
(SMTB-Q_3) R. Tetot, N. Salles, S. Landron, E. Amzallag, Surface Science 616, 19-8722 28 (2013)
LAMMPS Users Manual
1643
(Wolf) D. Wolf, P. Keblinski, S. R. Phillpot, J. Eggebrecht, J Chem Phys, 110, 8254 (1999).
(Rick) S. W. Rick, S. J. Stuart, B. J. Berne, J Chem Phys 101, 6141 (1994).
LAMMPS Users Manual
1644
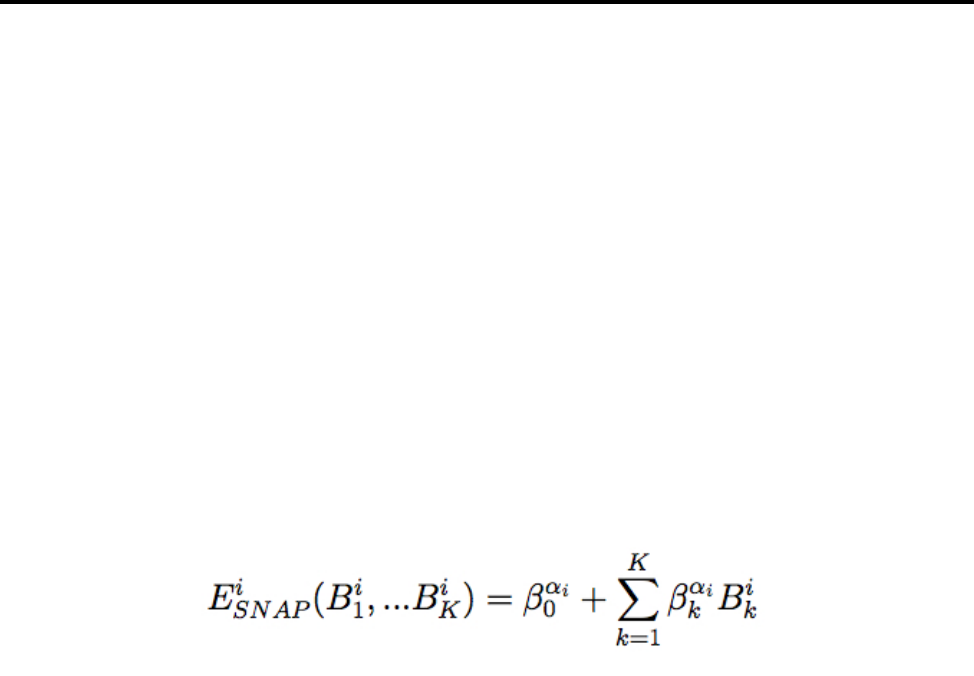
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style snap command
Syntax:
pair_style snap
Examples:
pair_style snap
pair_coeff * * InP.snapcoeff In P InP.snapparam In In P P
Description:
Style snap computes interactions using the spectral neighbor analysis potential (SNAP) (Thompson). Like the
GAP framework of Bartok et al. (Bartok2010), (Bartok2013) it uses bispectrum components to characterize
the local neighborhood of each atom in a very general way. The mathematical definition of the bispectrum
calculation used by SNAP is identical to that used by compute sna/atom. In SNAP, the total energy is
decomposed into a sum over atom energies. The energy of atom i is expressed as a weighted sum over
bispectrum components.
where B_k^i is the k-th bispectrum component of atom i, and beta_k^alpha_i is the corresponding linear
coefficient that depends on alpha_i, the SNAP element of atom i. The number of bispectrum components used
and their definitions depend on the values of twojmax and diagonalstyle defined in the SNAP parameter file
described below. The bispectrum calculation is described in more detail in compute sna/atom.
Note that unlike for other potentials, cutoffs for SNAP potentials are not set in the pair_style or pair_coeff
command; they are specified in the SNAP potential files themselves.
Only a single pair_coeff command is used with the snap style which specifies two SNAP files and the list
SNAP element(s) to be extracted. The SNAP elements are mapped to LAMMPS atom types by specifying N
additional arguments after the 2nd filename in the pair_coeff command, where N is the number of LAMMPS
atom types:
SNAP element file• Elem1, Elem2, ...• SNAP parameter file• N element names = mapping of SNAP elements to atom types•
As an example, if a LAMMPS indium phosphide simulation has 4 atoms types, with the first two being
indium and the 3rd and 4th being phophorous, the pair_coeff command would look like this:
pair_coeff * * snap InP.snapcoeff In P InP.snapparam In In P P
LAMMPS Users Manual
1645
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The two filenames are for the
element and parameter files, respectively. The 'In' and 'P' arguments (between the file names) are the two
elements which will be extracted from the element file. The two trailing 'In' arguments map LAMMPS atom
types 1 and 2 to the SNAP 'In' element. The two trailing 'P' arguments map LAMMPS atom types 3 and 4 to
the SNAP 'P' element.
If a SNAP mapping value is specified as NULL, the mapping is not performed. This can be used when a snap
potential is used as part of the hybrid pair style. The NULL values are placeholders for atom types that will be
used with other potentials.
The name of the SNAP element file usually ends in the ".snapcoeff" extension. It may contain coefficients for
many SNAP elements. Only those elements listed in the pair_coeff command are extracted. The name of the
SNAP parameter file usually ends in the ".snapparam" extension. It contains a small number of parameters
that define the overall form of the SNAP potential. See the pair_coeff doc page for alternate ways to specify
the path for these files.
Quite commonly, SNAP potentials are combined with one or more other LAMMPS pair styles using the
hybrid/overlay pair style. As an example, the SNAP tantalum potential provided in the LAMMPS potentials
directory combines the snap and zbl pair styles. It is invoked by the following commands:
variable zblcutinner equal 4
variable zblcutouter equal 4.8
variable zblz equal 73
pair_style hybrid/overlay &
zbl ${zblcutinner} ${zblcutouter} snap
pair_coeff * * zbl 0.0
pair_coeff 1 1 zbl ${zblz}
pair_coeff * * snap ../potentials/Ta06A.snapcoeff Ta &
../potentials/Ta06A.snapparam Ta
It is convenient to keep these commands in a separate file that can be inserted in any LAMMPS input script
using the include command.
The top of the SNAP element file can contain any number of blank and comment lines (start with #), but
follows a strict format after that. The first non-blank non-comment line must contain two integers:
nelem = Number of elements• ncoeff = Number of coefficients•
This is followed by one block for each of the nelem elements. The first line of each block contains three
entries:
Element symbol (text string)• R = Element radius (distance units)• w = Element weight (dimensionless)•
This line is followed by ncoeff coefficients, one per line.
The SNAP parameter file can contain blank and comment lines (start with #) anywhere. Each non-blank
non-comment line must contain one keyword/value pair. The required keywords are rcutfac and twojmax.
Optional keywords are rfac0, rmin0, diagonalstyle, switchflag, and bzeroflag.
LAMMPS Users Manual
1646
The default values for these keywords are
rfac0 = 0.99363• rmin0 = 0.0• diagonalstyle = 3• switchflag = 0• bzeroflag = 1•
Detailed definitions of these keywords are given on the compute sna/atom doc page.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, where types I and J correspond to two different element types, mixing is
performed by LAMMPS with user-specifiable parameters as described above. You never need to specify a
pair_coeff command with I != J arguments for this style.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This style is part of the SNAP package. It is only enabled if LAMMPS was built with that package. See the
Making LAMMPS section for more info.
Related commands:
compute sna/atom, compute snad/atom, compute snav/atom
Default: none
(Thompson) Thompson, Swiler, Trott, Foiles, Tucker, J Comp Phys, 285, 316 (2015).
(Bartok2010) Bartok, Payne, Risi, Csanyi, Phys Rev Lett, 104, 136403 (2010).
(Bartok2013) Bartok, Gillan, Manby, Csanyi, Phys Rev B 87, 184115 (2013).
LAMMPS Users Manual
1647

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style soft command
pair_style soft/gpu command
pair_style soft/omp command
Syntax:
pair_style soft cutoff
cutoff = global cutoff for soft interactions (distance units)•
Examples:
pair_style soft 1.0
pair_coeff * * 10.0
pair_coeff 1 1 10.0 3.0
pair_style soft 1.0
pair_coeff * * 0.0
variable prefactor equal ramp(0,30)
fix 1 all adapt 1 pair soft a * * v_prefactor
Description:
Style soft computes pairwise interactions with the formula
It is useful for pushing apart overlapping atoms, since it does not blow up as r goes to 0. A is a pre-factor that
can be made to vary in time from the start to the end of the run (see discussion below), e.g. to start with a very
soft potential and slowly harden the interactions over time. Rc is the cutoff. See the fix nve/limit command for
another way to push apart overlapping atoms.
The following coefficients must be defined for each pair of atom types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
A (energy units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global soft cutoff is used.
NOTE: The syntax for pair_coeff with a single A coeff is different in the current version of LAMMPS than in
older versions which took two values, Astart and Astop, to ramp between them. This functionality is now
available in a more general form through the fix adapt command, as explained below. Note that if you use an
LAMMPS Users Manual
1648

old input script and specify Astart and Astop without a cutoff, then LAMMPS will interpret that as A and a
cutoff, which is probably not what you want.
The fix adapt command can be used to vary A for one or more pair types over the course of a simulation, in
which case pair_coeff settings for A must still be specified, but will be overridden. For example these
commands will vary the prefactor A for all pairwise interactions from 0.0 at the beginning to 30.0 at the end
of a run:
variable prefactor equal ramp(0,30)
fix 1 all adapt 1 pair soft a * * v_prefactor
Note that a formula defined by an equal-style variable can use the current timestep, elapsed time in the current
run, elapsed time since the beginning of a series of runs, as well as access other variables.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the A coefficient and cutoff distance for this pair style can be mixed. A is
always mixed via a geometric rule. The cutoff is mixed according to the pair_modify mix value. The default
mix value is geometric. See the "pair_modify" command for details.
This pair style does not support the pair_modify shift option, since the pair interaction goes to 0.0 at the
cutoff.
The pair_modify table and tail options are not relevant for this pair style.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_coeff, fix nve/limit, fix adapt
LAMMPS Users Manual
1649
Default: none
LAMMPS Users Manual
1650
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style sph/heatconduction command
Syntax:
pair_style sph/heatconduction
Examples:
pair_style sph/heatconduction
pair_coeff * * 1.0 2.4
Description:
The sph/heatconduction style computes heat transport between SPH particles. The transport model is the
diffusion equation for the internal energy.
See this PDF guide to using SPH in LAMMPS.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above.
D diffusion coefficient (length^2/time units)• h kernel function cutoff (distance units)•
Mixing, shift, table, tail correction, restart, rRESPA info:
This style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
This style does not support the pair_modify shift, table, and tail options.
This style does not write information to binary restart files. Thus, you need to re-specify the pair_style and
pair_coeff commands in an input script that reads a restart file.
This style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the USER-SPH package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
pair_coeff, pair_sph/rhosum
Default: none
LAMMPS Users Manual
1651
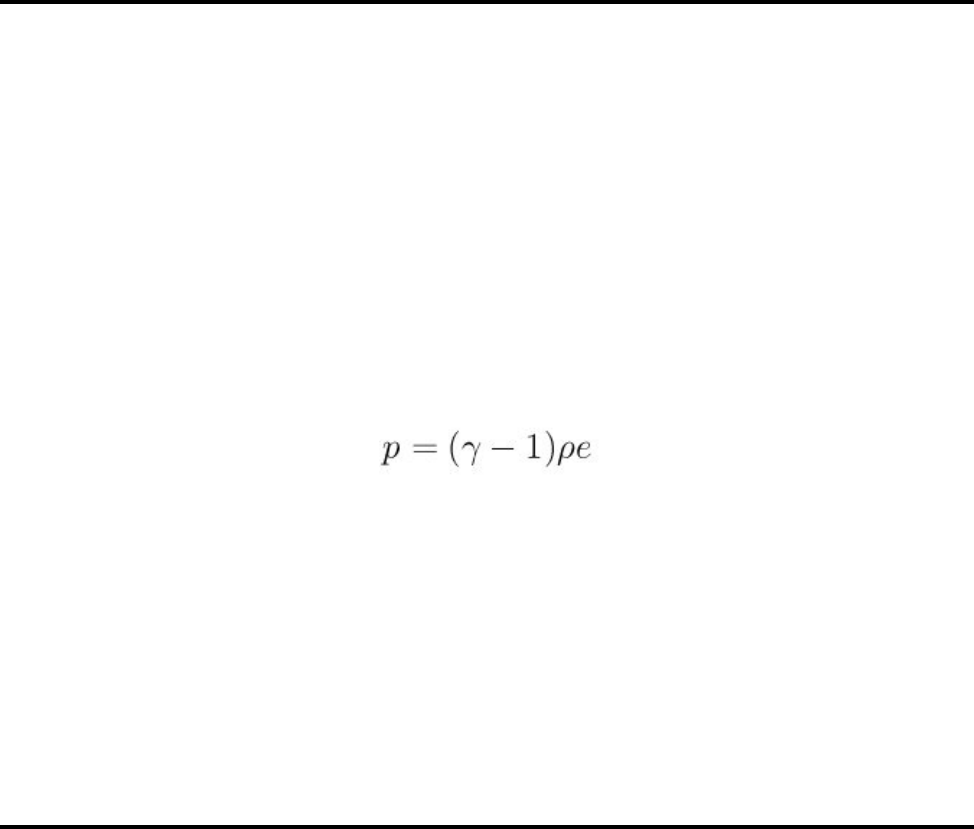
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style sph/idealgas command
Syntax:
pair_style sph/idealgas
Examples:
pair_style sph/idealgas
pair_coeff * * 1.0 2.4
Description:
The sph/idealgas style computes pressure forces between particles according to the ideal gas equation of state:
where gamma = 1.4 is the heat capacity ratio, rho is the local density, and e is the internal energy per unit
mass. This pair style also computes Monaghan's artificial viscosity to prevent particles from interpenetrating
(Monaghan).
See this PDF guide to using SPH in LAMMPS.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above.
nu artificial viscosity (no units)• h kernel function cutoff (distance units)•
Mixing, shift, table, tail correction, restart, rRESPA info:
This style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
This style does not support the pair_modify shift, table, and tail options.
This style does not write information to binary restart files. Thus, you need to re-specify the pair_style and
pair_coeff commands in an input script that reads a restart file.
This style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the USER-SPH package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
LAMMPS Users Manual
1652

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style sph/lj command
Syntax:
pair_style sph/lj
Examples:
pair_style sph/lj
pair_coeff * * 1.0 2.4
Description:
The sph/lj style computes pressure forces between particles according to the Lennard-Jones equation of state,
which is computed according to Ree's 1980 polynomial fit (Ree). The Lennard-Jones parameters epsilon and
sigma are set to unity. This pair style also computes Monaghan's artificial viscosity to prevent particles from
interpenetrating (Monaghan).
See this PDF guide to using SPH in LAMMPS.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above.
nu artificial viscosity (no units)• h kernel function cutoff (distance units)•
Mixing, shift, table, tail correction, restart, rRESPA info:
This style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
This style does not support the pair_modify shift, table, and tail options.
This style does not write information to binary restart files. Thus, you need to re-specify the pair_style and
pair_coeff commands in an input script that reads a restart file.
This style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
As noted above, the Lennard-Jones parameters epsilon and sigma are set to unity.
This pair style is part of the USER-SPH package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
pair_coeff, pair_sph/rhosum
LAMMPS Users Manual
1654

Default: none
(Ree) Ree, Journal of Chemical Physics, 73, 5401 (1980).
(Monaghan) Monaghan and Gingold, Journal of Computational Physics, 52, 374-389 (1983).
LAMMPS Users Manual
1655
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style sph/rhosum command
Syntax:
pair_style sph/rhosum Nstep
Nstep = timestep interval•
Examples:
pair_style sph/rhosum 10
pair_coeff * * 2.4
Description:
The sph/rhosum style computes the local particle mass density rho for SPH particles by kernel function
interpolation, every Nstep timesteps.
See this PDF guide to using SPH in LAMMPS.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above.
h (distance units)•
Mixing, shift, table, tail correction, restart, rRESPA info:
This style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
This style does not support the pair_modify shift, table, and tail options.
This style does not write information to binary restart files. Thus, you need to re-specify the pair_style and
pair_coeff commands in an input script that reads a restart file.
This style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the USER-SPH package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
pair_coeff, pair_sph/taitwater
Default: none
LAMMPS Users Manual
1656
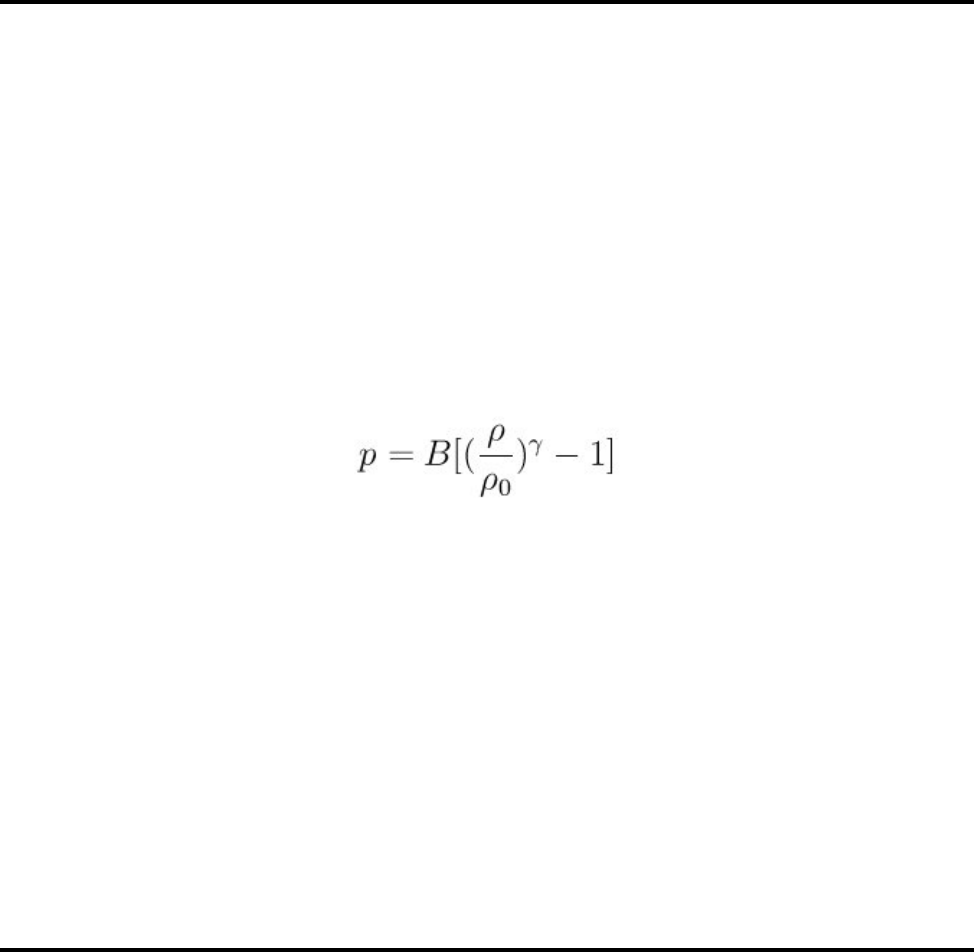
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style sph/taitwater command
Syntax:
pair_style sph/taitwater
Examples:
pair_style sph/taitwater
pair_coeff * * 1000.0 1430.0 1.0 2.4
Description:
The sph/taitwater style computes pressure forces between SPH particles according to Tait's equation of state:
where gamma = 7 and B = c_0^2 rho_0 / gamma, with rho_0 being the reference density and c_0 the
reference speed of sound.
This pair style also computes Monaghan's artificial viscosity to prevent particles from interpenetrating
(Monaghan).
See this PDF guide to using SPH in LAMMPS.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above.
rho0 reference density (mass/volume units)• c0 reference soundspeed (distance/time units)• nu artificial viscosity (no units)• h kernel function cutoff (distance units)•
Mixing, shift, table, tail correction, restart, rRESPA info:
This style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
This style does not support the pair_modify shift, table, and tail options.
This style does not write information to binary restart files. Thus, you need to re-specify the pair_style and
pair_coeff commands in an input script that reads a restart file.
This style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
LAMMPS Users Manual
1657

This pair style is part of the USER-SPH package. It is only enabled if LAMMPS was built with that package.
See the Making LAMMPS section for more info.
Related commands:
pair_coeff, pair_sph/rhosum
Default: none
(Monaghan) Monaghan and Gingold, Journal of Computational Physics, 52, 374-389 (1983).
LAMMPS Users Manual
1658
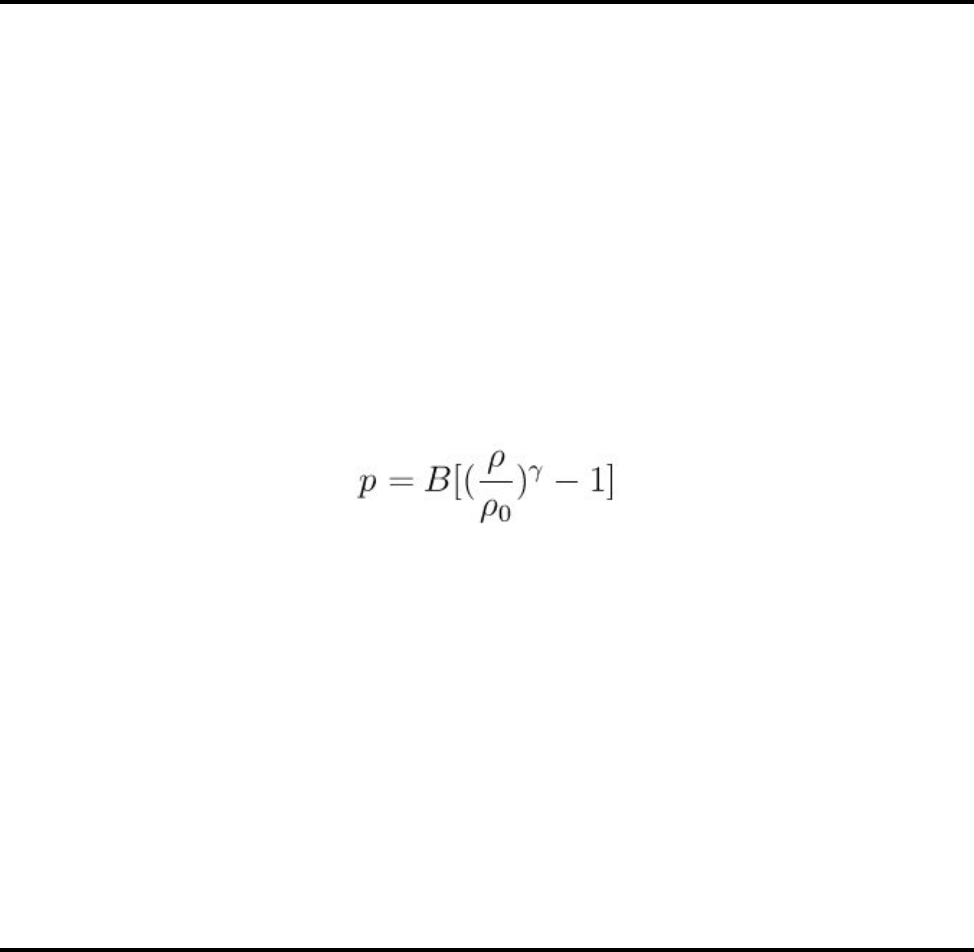
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style sph/taitwater/morris command
Syntax:
pair_style sph/taitwater/morris
Examples:
pair_style sph/taitwater/morris
pair_coeff * * 1000.0 1430.0 1.0 2.4
Description:
The sph/taitwater/morris style computes pressure forces between SPH particles according to Tait's equation of
state:
where gamma = 7 and B = c_0^2 rho_0 / gamma, with rho_0 being the reference density and c_0 the
reference speed of sound.
This pair style also computes laminar viscosity (Morris).
See this PDF guide to using SPH in LAMMPS.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above.
rho0 reference density (mass/volume units)• c0 reference soundspeed (distance/time units)• nu dynamic viscosity (mass*distance/time units)• h kernel function cutoff (distance units)•
Mixing, shift, table, tail correction, restart, rRESPA info:
This style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
This style does not support the pair_modify shift, table, and tail options.
This style does not write information to binary restart files. Thus, you need to re-specify the pair_style and
pair_coeff commands in an input script that reads a restart file.
This style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
LAMMPS Users Manual
1659


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style srp command
Syntax:
pair_style srp cutoff btype dist keyword value ...
cutoff = global cutoff for SRP interactions (distance units)• btype = bond type to apply SRP interactions to (can be wildcard, see below)• distance = min or mid• zero or more keyword/value pairs may be appended• keyword = exclude
bptype value = atom type for bond particles
exclude value = yes or no
•
Examples:
pair_style hybrid dpd 1.0 1.0 12345 srp 0.8 1 mid exclude yes
pair_coeff 1 1 dpd 60.0 4.5 1.0
pair_coeff 1 2 none
pair_coeff 2 2 srp 100.0 0.8
pair_style hybrid dpd 1.0 1.0 12345 srp 0.8 * min exclude yes
pair_coeff 1 1 dpd 60.0 50 1.0
pair_coeff 1 2 none
pair_coeff 2 2 srp 40.0
pair_style hybrid srp 0.8 2 mid
pair_coeff 1 1 none
pair_coeff 1 2 none
pair_coeff 2 2 srp 100.0 0.8
Description:
Style srp computes a soft segmental repulsive potential (SRP) that acts between pairs of bonds. This potential
is useful for preventing bonds from passing through one another when a soft non-bonded potential acts
between beads in, for example, DPD polymer chains. An example input script that uses this command is
provided in examples/USER/srp.
Bonds of specified type btype interact with one another through a bond-pairwise potential, such that the force
on bond i due to bond j is as follows
where r and rij are the distance and unit vector between the two bonds. Note that btype can be specified as an
asterisk "*", which case the interaction is applied to all bond types. The mid option computes r and rij from
the midpoint distance between bonds. The min option computes r and rij from the minimum distance between
bonds. The force acting on a bond is mapped onto the two bond atoms according to the lever rule,
LAMMPS Users Manual
1661

where L is the normalized distance from the atom to the point of closest approach of bond i and j. The mid
option takes L as 0.5 for each interaction as described in (Sirk).
The following coefficients must be defined via the pair_coeff command as in the examples above, or in the
data file or restart file read by the read_data or read_restart commands:
C (force units)• rc (distance units)•
The last coefficient is optional. If not specified, the global cutoff is used.
NOTE: Pair style srp considers each bond of type btype to be a fictitious "particle" of type bptype, where
bptype is either the largest atom type in the system, or the type set by the bptype flag. Any actual existing
particles with this atom type will be deleted at the beginning of a run. This means you must specify the
number of types in your system accordingly; usually to be one larger than what would normally be the case,
e.g. via the create_box or by changing the header in your data file. The fictitious "bond particles" are inserted
at the beginning of the run, and serve as placeholders that define the position of the bonds. This allows
neighbor lists to be constructed and pairwise interactions to be computed in almost the same way as is done
for actual particles. Because bonds interact only with other bonds, pair_style hybrid should be used to turn off
interactions between atom type bptype and all other types of atoms. An error will be flagged if pair_style
hybrid is not used.
The optional exclude keyword determines if forces are computed between first neighbor (directly connected)
bonds. For a setting of no, first neighbor forces are computed; for yes they are not computed. A setting of no
cannot be used with the min option for distance calculation because the minimum distance between directly
connected bonds is zero.
Pair style srp turns off normalization of thermodynamic properties by particle number, as if the command
thermo_modify norm no had been issued.
The pairwise energy associated with style srp is shifted to be zero at the cutoff distance rc.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair styles does not support mixing.
This pair style does not support the pair_modify shift option for the energy of the pair interaction. Note that as
discussed above, the energy term is already shifted to be 0.0 at the cutoff distance rc.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
LAMMPS Users Manual
1662

This pair style writes global and per-atom information to binary restart files. Pair srp should be used with
pair_style hybrid, thus the pair_coeff commands need to be specified in the input script when reading a restart
file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the USER-MISC package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
This pair style must be used with pair_style hybrid.
This pair style requires the newton command to be on for non-bonded interactions.
Related commands:
pair_style hybrid, pair_coeff, pair dpd
Default:
The default keyword value is exclude = yes.
(Sirk) Sirk TW, Sliozberg YR, Brennan JK, Lisal M, Andzelm JW, J Chem Phys, 136 (13) 134903, 2012.
LAMMPS Users Manual
1663

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style sw command
pair_style sw/gpu command
pair_style sw/intel command
pair_style sw/kk command
pair_style sw/omp command
Syntax:
pair_style sw
Examples:
pair_style sw
pair_coeff * * si.sw Si
pair_coeff * * GaN.sw Ga N Ga
Description:
The sw style computes a 3-body Stillinger-Weber potential for the energy E of a system of atoms as
where phi2 is a two-body term and phi3 is a three-body term. The summations in the formula are over all
neighbors J and K of atom I within a cutoff distance = a*sigma.
Only a single pair_coeff command is used with the sw style which specifies a Stillinger-Weber potential file
with parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N
additional arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom
types:
filename• N element names = mapping of SW elements to atom types•
LAMMPS Users Manual
1664
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, imagine a file SiC.sw has Stillinger-Weber values for Si and C. If your LAMMPS simulation
has 4 atoms types and you want the 1st 3 to be Si, and the 4th to be C, you would use the following pair_coeff
command:
pair_coeff * * SiC.sw Si Si Si C
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Si arguments map
LAMMPS atom types 1,2,3 to the Si element in the SW file. The final C argument maps LAMMPS atom type
4 to the C element in the SW file. If a mapping value is specified as NULL, the mapping is not performed.
This can be used when a sw potential is used as part of the hybrid pair style. The NULL values are
placeholders for atom types that will be used with other potentials.
Stillinger-Weber files in the potentials directory of the LAMMPS distribution have a ".sw" suffix. Lines that
are not blank or comments (starting with #) define parameters for a triplet of elements. The parameters in a
single entry correspond to the two-body and three-body coefficients in the formula above:
element 1 (the center atom in a 3-body interaction)• element 2• element 3• epsilon (energy units)• sigma (distance units)• a• lambda• gamma• costheta0• A• B• p• q• tol•
The A, B, p, and q parameters are used only for two-body interactions. The lambda and costheta0 parameters
are used only for three-body interactions. The epsilon, sigma and a parameters are used for both two-body and
three-body interactions. gamma is used only in the three-body interactions, but is defined for pairs of atoms.
The non-annotated parameters are unitless.
LAMMPS introduces an additional performance-optimization parameter tol that is used for both two-body
and three-body interactions. In the Stillinger-Weber potential, the interaction energies become negligibly
small at atomic separations substantially less than the theoretical cutoff distances. LAMMPS therefore defines
a virtual cutoff distance based on a user defined tolerance tol. The use of the virtual cutoff distance in
constructing atom neighbor lists can significantly reduce the neighbor list sizes and therefore the
computational cost. LAMMPS provides a tol value for each of the three-body entries so that they can be
separately controlled. If tol = 0.0, then the standard Stillinger-Weber cutoff is used.
The Stillinger-Weber potential file must contain entries for all the elements listed in the pair_coeff command.
It can also contain entries for additional elements not being used in a particular simulation; LAMMPS ignores
those entries.
LAMMPS Users Manual
1665

For a single-element simulation, only a single entry is required (e.g. SiSiSi). For a two-element simulation, the
file must contain 8 entries (for SiSiSi, SiSiC, SiCSi, SiCC, CSiSi, CSiC, CCSi, CCC), that specify SW
parameters for all permutations of the two elements interacting in three-body configurations. Thus for 3
elements, 27 entries would be required, etc.
As annotated above, the first element in the entry is the center atom in a three-body interaction. Thus an entry
for SiCC means a Si atom with 2 C atoms as neighbors. The parameter values used for the two-body
interaction come from the entry where the 2nd and 3rd elements are the same. Thus the two-body parameters
for Si interacting with C, comes from the SiCC entry. The three-body parameters can in principle be specific
to the three elements of the configuration. In the literature, however, the three-body parameters are usually
defined by simple formulas involving two sets of pair-wise parameters, corresponding to the ij and ik pairs,
where i is the center atom. The user must ensure that the correct combining rule is used to calculate the values
of the threebody parameters for alloys. Note also that the function phi3 contains two exponential screening
factors with parameter values from the ij pair and ik pairs. So phi3 for a C atom bonded to a Si atom and a
second C atom will depend on the three-body parameters for the CSiC entry, and also on the two-body
parameters for the CCC and CSiSi entries. Since the order of the two neighbors is arbitrary, the threebody
parameters for entries CSiC and CCSi should be the same. Similarly, the two-body parameters for entries
SiCC and CSiSi should also be the same. The parameters used only for two-body interactions (A, B, p, and q)
in entries whose 2nd and 3rd element are different (e.g. SiCSi) are not used for anything and can be set to 0.0
if desired. This is also true for the parameters in phi3 that are taken from the ij and ik pairs (sigma, a, gamma)
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
When using the USER-INTEL package with this style, there is an additional 5 to 10 percent performance
improvement when the Stillinger-Weber parameters p and q are set to 4 and 0 respectively. These parameters
are common for modeling silicon and water.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, where types I and J correspond to two different element types, mixing is
performed by LAMMPS as described above from values in the potential file.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
LAMMPS Users Manual
1666

This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the MANYBODY package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
This pair style requires the newton setting to be "on" for pair interactions.
The Stillinger-Weber potential files provided with LAMMPS (see the potentials directory) are parameterized
for metal units. You can use the SW potential with any LAMMPS units, but you would need to create your
own SW potential file with coefficients listed in the appropriate units if your simulation doesn't use "metal"
units.
Related commands:
pair_coeff
Default: none
(Stillinger) Stillinger and Weber, Phys Rev B, 31, 5262 (1985).
LAMMPS Users Manual
1667

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style table command
pair_style table/gpu command
pair_style table/kk command
pair_style table/omp command
Syntax:
pair_style table style N keyword ...
style = lookup or linear or spline or bitmap = method of interpolation• N = use N values in lookup, linear, spline tables• N = use 2^N values in bitmap tables• zero or more keywords may be appended• keyword = ewald or pppm or msm or dispersion or tip4p•
Examples:
pair_style table linear 1000
pair_style table linear 1000 pppm
pair_style table bitmap 12
pair_coeff * 3 morse.table ENTRY1
pair_coeff * 3 morse.table ENTRY1 7.0
Description:
Style table creates interpolation tables from potential energy and force values listed in a file(s) as a function of
distance. When performing dynamics or minimization, the interpolation tables are used to evaluate energy and
forces for pairwise interactions between particles, similar to how analytic formulas are used for other pair
styles.
The interpolation tables are created as a pre-computation by fitting cubic splines to the file values and
interpolating energy and force values at each of N distances. During a simulation, the tables are used to
interpolate energy and force values as needed for each pair of particles separated by a distance R. The
interpolation is done in one of 4 styles: lookup, linear, spline, or bitmap.
For the lookup style, the distance R is used to find the nearest table entry, which is the energy or force.
For the linear style, the distance R is used to find the 2 surrounding table values from which an energy or
force is computed by linear interpolation.
For the spline style, a cubic spline coefficients are computed and stored for each of the N values in the table,
one set of splines for energy, another for force. Note that these splines are different than the ones used to
pre-compute the N values. Those splines were fit to the Nfile values in the tabulated file, where often Nfile <
N. The distance R is used to find the appropriate set of spline coefficients which are used to evaluate a cubic
polynomial which computes the energy or force.
LAMMPS Users Manual
1668

For the bitmap style, the specified N is used to create interpolation tables that are 2^N in length. The distance
R is used to index into the table via a fast bit-mapping technique due to (Wolff), and a linear interpolation is
performed between adjacent table values.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above.
filename• keyword• cutoff (distance units)•
The filename specifies a file containing tabulated energy and force values. The keyword specifies a section of
the file. The cutoff is an optional coefficient. If not specified, the outer cutoff in the table itself (see below)
will be used to build an interpolation table that extend to the largest tabulated distance. If specified, only file
values up to the cutoff are used to create the interpolation table. The format of this file is described below.
If your tabulated potential(s) are designed to be used as the short-range part of one of the long-range solvers
specified by the kspace_style command, then you must use one or more of the optional keywords listed above
for the pair_style command. These are ewald or pppm or msm or dispersion or tip4p. This is so LAMMPS can
insure the short-range potential and long-range solver are compatible with each other, as it does for other
short-range pair styles, such as pair_style lj/cut/coul/long. Note that it is up to you to insure the tabulated
values for each pair of atom types has the correct functional form to be compatible with the matching
long-range solver.
Here are some guidelines for using the pair_style table command to best effect:
Vary the number of table points; you may need to use more than you think to get good resolution.• Always use the pair_write command to produce a plot of what the final interpolated potential looks
like. This can show up interpolation "features" you may not like.
•
Start with the linear style; it's the style least likely to have problems.• Use N in the pair_style command equal to the "N" in the tabulation file, and use the "RSQ" or
"BITMAP" parameter, so additional interpolation is not needed. See discussion below.
•
Make sure that your tabulated forces and tabulated energies are consistent (dE/dr = -F) over the entire
range of r values. LAMMPS will warn if this is not the case.
•
Use as large an inner cutoff as possible. This avoids fitting splines to very steep parts of the potential.•
The format of a tabulated file is a series of one or more sections, defined as follows (without the parenthesized
comments):
# Morse potential for Fe (one or more comment or blank lines)
MORSE_FE (keyword is first text on line)
N 500 R 1.0 10.0 (N, R, RSQ, BITMAP, FPRIME parameters)
(blank)
1 1.0 25.5 102.34 (index, r, energy, force)
2 1.02 23.4 98.5
...
500 10.0 0.001 0.003
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections. The first line begins with a keyword which identifies the section.
The line can contain additional text, but the initial text must match the argument specified in the pair_coeff
LAMMPS Users Manual
1669

command. The next line lists (in any order) one or more parameters for the table. Each parameter is a keyword
followed by one or more numeric values.
The parameter "N" is required and its value is the number of table entries that follow. Note that this may be
different than the N specified in the pair_style table command. Let Ntable = N in the pair_style command, and
Nfile = "N" in the tabulated file. What LAMMPS does is a preliminary interpolation by creating splines using
the Nfile tabulated values as nodal points. It uses these to interpolate energy and force values at Ntable
different points. The resulting tables of length Ntable are then used as described above, when computing
energy and force for individual pair distances. This means that if you want the interpolation tables of length
Ntable to match exactly what is in the tabulated file (with effectively no preliminary interpolation), you
should set Ntable = Nfile, and use the "RSQ" or "BITMAP" parameter. This is because the internal table
abscissa is always RSQ (separation distance squared), for efficient lookup.
All other parameters are optional. If "R" or "RSQ" or "BITMAP" does not appear, then the distances in each
line of the table are used as-is to perform spline interpolation. In this case, the table values can be spaced in r
uniformly or however you wish to position table values in regions of large gradients.
If used, the parameters "R" or "RSQ" are followed by 2 values rlo and rhi. If specified, the distance associated
with each energy and force value is computed from these 2 values (at high accuracy), rather than using the
(low-accuracy) value listed in each line of the table. The distance values in the table file are ignored in this
case. For "R", distances uniformly spaced between rlo and rhi are computed; for "RSQ", squared distances
uniformly spaced between rlo*rlo and rhi*rhi are computed.
NOTE: If you use "R" or "RSQ", the tabulated distance values in the file are effectively ignored, and replaced
by new values as described in the previous paragraph. If the distance value in the table is not very close to the
new value (i.e. round-off difference), then you will be assigning energy/force values to a different distance,
which is probably not what you want. LAMMPS will warn if this is occurring.
If used, the parameter "BITMAP" is also followed by 2 values rlo and rhi. These values, along with the "N"
value determine the ordering of the N lines that follow and what distance is associated with each. This
ordering is complex, so it is not documented here, since this file is typically produced by the pair_write
command with its bitmap option. When the table is in BITMAP format, the "N" parameter in the file must be
equal to 2^M where M is the value specified in the pair_style command. Also, a cutoff parameter cannot be
used as an optional 3rd argument in the pair_coeff command; the entire table extent as specified in the file
must be used.
If used, the parameter "FPRIME" is followed by 2 values fplo and fphi which are the derivative of the force at
the innermost and outermost distances listed in the table. These values are needed by the spline construction
routines. If not specified by the "FPRIME" parameter, they are estimated (less accurately) by the first 2 and
last 2 force values in the table. This parameter is not used by BITMAP tables.
Following a blank line, the next N lines list the tabulated values. On each line, the 1st value is the index from
1 to N, the 2nd value is r (in distance units), the 3rd value is the energy (in energy units), and the 4th is the
force (in force units). The r values must increase from one line to the next (unless the BITMAP parameter is
specified).
Note that one file can contain many sections, each with a tabulated potential. LAMMPS reads the file section
by section until it finds one that matches the specified keyword.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
LAMMPS Users Manual
1670

5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
The pair_modify shift, table, and tail options are not relevant for this pair style.
This pair style writes the settings for the "pair_style table" command to binary restart files, so a pair_style
command does not need to specified in an input script that reads a restart file. However, the coefficient
information is not stored in the restart file, since it is tabulated in the potential files. Thus, pair_coeff
commands do need to be specified in the restart input script.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_coeff, pair_write
Default: none
(Wolff) Wolff and Rudd, Comp Phys Comm, 120, 200-32 (1999).
LAMMPS Users Manual
1671

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style table/rx command
Syntax:
pair_style table style N ...
style = lookup or linear or spline or bitmap = method of interpolation• N = use N values in lookup, linear, spline tables• weighting = fractional or molecular (optional)•
Examples:
pair_style table/rx linear 1000
pair_style table/rx linear 1000 fractional
pair_style table/rx linear 1000 molecular
pair_coeff * * rxn.table ENTRY1 h2o h2o 10.0
pair_coeff * * rxn.table ENTRY1 1fluid 1fluid 10.0
pair_coeff * 3 rxn.table ENTRY1 h2o no2 10.0
Description:
Style table/rx is used in reaction DPD simulations,where the coarse-grained (CG) particles are composed of m
species whose reaction rate kinetics are determined from a set of n reaction rate equations through the fix rx
command. The species of one CG particle can interact with a species in a neighboring CG particle through a
site-site interaction potential model. Style table/rx creates interpolation tables of length N from pair potential
and force values listed in a file(s) as a function of distance. The files are read by the pair_coeff command.
The interpolation tables are created by fitting cubic splines to the file values and interpolating energy and
force values at each of N distances. During a simulation, these tables are used to interpolate energy and force
values as needed. The interpolation is done in one of 4 styles: lookup, linear, spline, or bitmap.
For the lookup style, the distance between 2 atoms is used to find the nearest table entry, which is the energy
or force.
For the linear style, the pair distance is used to find 2 surrounding table values from which an energy or force
is computed by linear interpolation.
For the spline style, a cubic spline coefficients are computed and stored at each of the N values in the table.
The pair distance is used to find the appropriate set of coefficients which are used to evaluate a cubic
polynomial which computes the energy or force.
For the bitmap style, the N means to create interpolation tables that are 2^N in length. The pair distance is
used to index into the table via a fast bit-mapping technique (Wolff) and a linear interpolation is performed
between adjacent table values.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above.
filename• keyword•
LAMMPS Users Manual
1672

species1• species2• cutoff (distance units)•
The filename specifies a file containing tabulated energy and force values. The keyword specifies a section of
the file. The cutoff is an optional coefficient. If not specified, the outer cutoff in the table itself (see below)
will be used to build an interpolation table that extend to the largest tabulated distance. If specified, only file
values up to the cutoff are used to create the interpolation table. The format of this file is described below.
The species tags define the site-site interaction potential between two species contained within two different
particles. The species tags must either correspond to the species defined in the reaction kinetics files specified
with the fix rx command or they must correspond to the tag "1fluid", signifying interaction with a product
species mixture determined through a one-fluid approximation. The interaction potential is weighted by the
geometric average of either the mole fraction concentrations or the number of molecules associated with the
interacting coarse-grained particles (see the fractional or molecular weighting pair style options). The
coarse-grained potential is stored before and after the reaction kinetics solver is applied, where the difference
is defined to be the internal chemical energy (uChem).
Here are some guidelines for using the pair_style table/rx command to best effect:
Vary the number of table points; you may need to use more than you think to get good resolution.• Always use the pair_write command to produce a plot of what the final interpolated potential looks
like. This can show up interpolation "features" you may not like.
•
Start with the linear style; it's the style least likely to have problems.• Use N in the pair_style command equal to the "N" in the tabulation file, and use the "RSQ" or
"BITMAP" parameter, so additional interpolation is not needed. See discussion below.
•
Make sure that your tabulated forces and tabulated energies are consistent (dE/dr = -F) along the
entire range of r values.
•
Use as large an inner cutoff as possible. This avoids fitting splines to very steep parts of the potential.•
The format of a tabulated file is a series of one or more sections, defined as follows (without the parenthesized
comments):
# Morse potential for Fe (one or more comment or blank lines)
MORSE_FE (keyword is first text on line)
N 500 R 1.0 10.0 (N, R, RSQ, BITMAP, FPRIME parameters)
(blank)
1 1.0 25.5 102.34 (index, r, energy, force)
2 1.02 23.4 98.5
...
500 10.0 0.001 0.003
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections. The first line begins with a keyword which identifies the section.
The line can contain additional text, but the initial text must match the argument specified in the pair_coeff
command. The next line lists (in any order) one or more parameters for the table. Each parameter is a keyword
followed by one or more numeric values.
The parameter "N" is required and its value is the number of table entries that follow. Note that this may be
different than the N specified in the pair_style table/rx command. Let Ntable = N in the pair_style command,
and Nfile = "N" in the tabulated file. What LAMMPS does is a preliminary interpolation by creating splines
LAMMPS Users Manual
1673

using the Nfile tabulated values as nodal points. It uses these to interpolate as needed to generate energy and
force values at Ntable different points. The resulting tables of length Ntable are then used as described above,
when computing energy and force for individual pair distances. This means that if you want the interpolation
tables of length Ntable to match exactly what is in the tabulated file (with effectively no preliminary
interpolation), you should set Ntable = Nfile, and use the "RSQ" or "BITMAP" parameter. The internal table
abscissa is RSQ (separation distance squared).
All other parameters are optional. If "R" or "RSQ" or "BITMAP" does not appear, then the distances in each
line of the table are used as-is to perform spline interpolation. In this case, the table values can be spaced in r
uniformly or however you wish to position table values in regions of large gradients.
If used, the parameters "R" or "RSQ" are followed by 2 values rlo and rhi. If specified, the distance associated
with each energy and force value is computed from these 2 values (at high accuracy), rather than using the
(low-accuracy) value listed in each line of the table. The distance values in the table file are ignored in this
case. For "R", distances uniformly spaced between rlo and rhi are computed; for "RSQ", squared distances
uniformly spaced between rlo*rlo and rhi*rhi are computed.
If used, the parameter "BITMAP" is also followed by 2 values rlo and rhi. These values, along with the "N"
value determine the ordering of the N lines that follow and what distance is associated with each. This
ordering is complex, so it is not documented here, since this file is typically produced by the pair_write
command with its bitmap option. When the table is in BITMAP format, the "N" parameter in the file must be
equal to 2^M where M is the value specified in the pair_style command. Also, a cutoff parameter cannot be
used as an optional 3rd argument in the pair_coeff command; the entire table extent as specified in the file
must be used.
If used, the parameter "FPRIME" is followed by 2 values fplo and fphi which are the derivative of the force at
the innermost and outermost distances listed in the table. These values are needed by the spline construction
routines. If not specified by the "FPRIME" parameter, they are estimated (less accurately) by the first 2 and
last 2 force values in the table. This parameter is not used by BITMAP tables.
Following a blank line, the next N lines list the tabulated values. On each line, the 1st value is the index from
1 to N, the 2nd value is r (in distance units), the 3rd value is the energy (in energy units), and the 4th is the
force (in force units). The r values must increase from one line to the next (unless the BITMAP parameter is
specified).
Note that one file can contain many sections, each with a tabulated potential. LAMMPS reads the file section
by section until it finds one that matches the specified keyword.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
The pair_modify shift, table, and tail options are not relevant for this pair style.
This pair style writes the settings for the "pair_style table/rx" command to binary restart files, so a pair_style
command does not need to specified in an input script that reads a restart file. However, the coefficient
information is not stored in the restart file, since it is tabulated in the potential files. Thus, pair_coeff
commands do need to be specified in the restart input script.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
LAMMPS Users Manual
1674


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style tersoff command
pair_style tersoff/table command
pair_style tersoff/gpu
pair_style tersoff/intel
pair_style tersoff/kk
pair_style tersoff/omp
pair_style tersoff/table/omp command
Syntax:
pair_style style
style = tersoff or tersoff/table or tersoff/gpu or tersoff/omp or tersoff/table/omp
Examples:
pair_style tersoff
pair_coeff * * Si.tersoff Si
pair_coeff * * SiC.tersoff Si C Si
pair_style tersoff/table
pair_coeff * * SiCGe.tersoff Si(D)
Description:
The tersoff style computes a 3-body Tersoff potential (Tersoff_1) for the energy E of a system of atoms as
LAMMPS Users Manual
1676
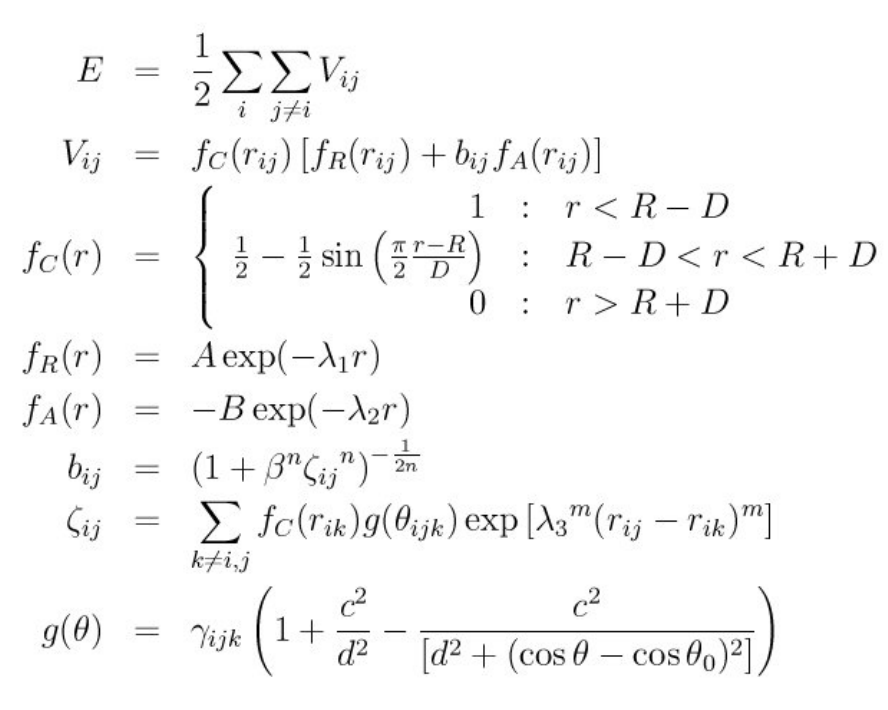
where f_R is a two-body term and f_A includes three-body interactions. The summations in the formula are
over all neighbors J and K of atom I within a cutoff distance = R + D.
The tersoff/table style uses tabulated forms for the two-body, environment and angular functions. Linear
interpolation is performed between adjacent table entries. The table length is chosen to be accurate within
10^-6 with respect to the tersoff style energy. The tersoff/table should give better performance in terms of
speed.
Only a single pair_coeff command is used with the tersoff style which specifies a Tersoff potential file with
parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional
arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N element names = mapping of Tersoff elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, imagine the SiC.tersoff file has Tersoff values for Si and C. If your LAMMPS simulation has
4 atoms types and you want the 1st 3 to be Si, and the 4th to be C, you would use the following pair_coeff
command:
pair_coeff * * SiC.tersoff Si Si Si C
LAMMPS Users Manual
1677
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Si arguments map
LAMMPS atom types 1,2,3 to the Si element in the Tersoff file. The final C argument maps LAMMPS atom
type 4 to the C element in the Tersoff file. If a mapping value is specified as NULL, the mapping is not
performed. This can be used when a tersoff potential is used as part of the hybrid pair style. The NULL values
are placeholders for atom types that will be used with other potentials.
Tersoff files in the potentials directory of the LAMMPS distribution have a ".tersoff" suffix. Lines that are not
blank or comments (starting with #) define parameters for a triplet of elements. The parameters in a single
entry correspond to coefficients in the formula above:
element 1 (the center atom in a 3-body interaction)• element 2 (the atom bonded to the center atom)• element 3 (the atom influencing the 1-2 bond in a bond-order sense)• m• gamma• lambda3 (1/distance units)• c• d• costheta0 (can be a value < -1 or > 1)• n• beta• lambda2 (1/distance units)• B (energy units)• R (distance units)• D (distance units)• lambda1 (1/distance units)• A (energy units)•
The n, beta, lambda2, B, lambda1, and A parameters are only used for two-body interactions. The m, gamma,
lambda3, c, d, and costheta0 parameters are only used for three-body interactions. The R and D parameters are
used for both two-body and three-body interactions. The non-annotated parameters are unitless. The value of
m must be 3 or 1.
The Tersoff potential file must contain entries for all the elements listed in the pair_coeff command. It can
also contain entries for additional elements not being used in a particular simulation; LAMMPS ignores those
entries.
For a single-element simulation, only a single entry is required (e.g. SiSiSi). For a two-element simulation, the
file must contain 8 entries (for SiSiSi, SiSiC, SiCSi, SiCC, CSiSi, CSiC, CCSi, CCC), that specify Tersoff
parameters for all permutations of the two elements interacting in three-body configurations. Thus for 3
elements, 27 entries would be required, etc.
As annotated above, the first element in the entry is the center atom in a three-body interaction and it is
bonded to the 2nd atom and the bond is influenced by the 3rd atom. Thus an entry for SiCC means Si bonded
to a C with another C atom influencing the bond. Thus three-body parameters for SiCSi and SiSiC entries will
not, in general, be the same. The parameters used for the two-body interaction come from the entry where the
2nd element is repeated. Thus the two-body parameters for Si interacting with C, comes from the SiCC entry.
The parameters used for a particular three-body interaction come from the entry with the corresponding three
elements. The parameters used only for two-body interactions (n, beta, lambda2, B, lambda1, and A) in entries
whose 2nd and 3rd element are different (e.g. SiCSi) are not used for anything and can be set to 0.0 if desired.
LAMMPS Users Manual
1678
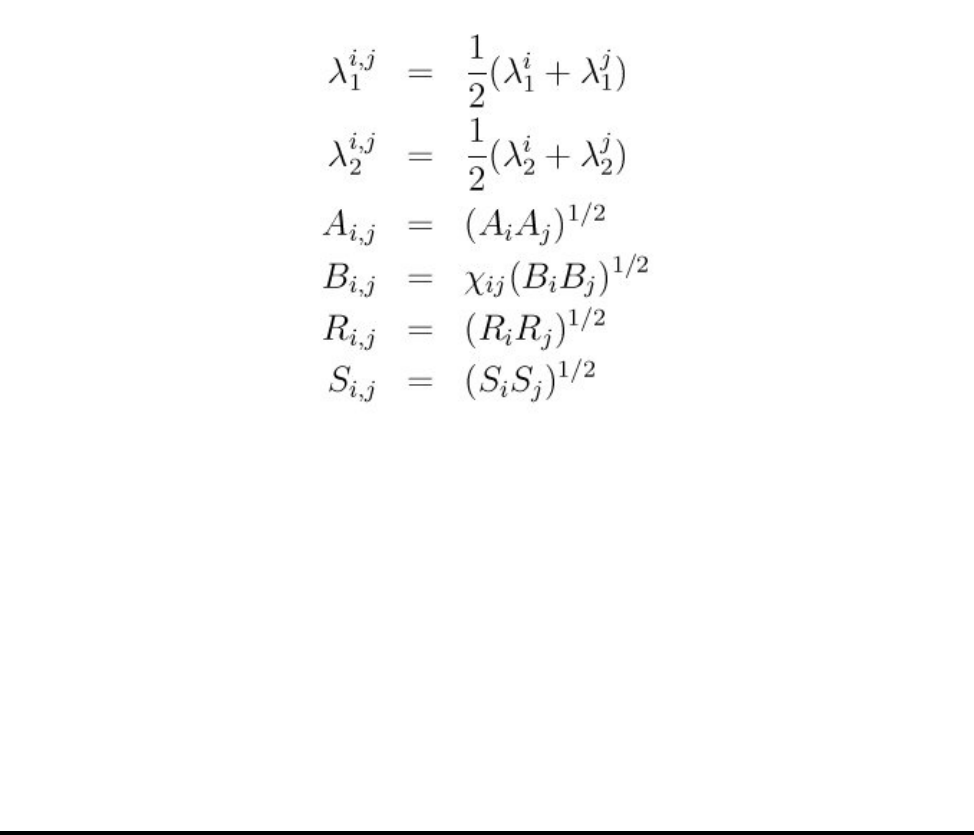
Note that the twobody parameters in entries such as SiCC and CSiSi are often the same, due to the common
use of symmetric mixing rules, but this is not always the case. For example, the beta and n parameters in
Tersoff_2 (Tersoff_2) are not symmetric.
We chose the above form so as to enable users to define all commonly used variants of the Tersoff potential.
In particular, our form reduces to the original Tersoff form when m = 3 and gamma = 1, while it reduces to
the form of Albe et al. when beta = 1 and m = 1. Note that in the current Tersoff implementation in
LAMMPS, m must be specified as either 3 or 1. Tersoff used a slightly different but equivalent form for
alloys, which we will refer to as Tersoff_2 potential (Tersoff_2). The tersoff/table style implements Tersoff_2
parameterization only.
LAMMPS parameter values for Tersoff_2 can be obtained as follows: gamma_ijk = omega_ik, lambda3 = 0
and the value of m has no effect. The parameters for species i and j can be calculated using the Tersoff_2
mixing rules:
Tersoff_2 parameters R and S must be converted to the LAMMPS parameters R and D (R is different in both
forms), using the following relations: R=(R'+S')/2 and D=(S'-R')/2, where the primes indicate the Tersoff_2
parameters.
In the potentials directory, the file SiCGe.tersoff provides the LAMMPS parameters for Tersoff's various
versions of Si, as well as his alloy parameters for Si, C, and Ge. This file can be used for pure Si, (three
different versions), pure C, pure Ge, binary SiC, and binary SiGe. LAMMPS will generate an error if this file
is used with any combination involving C and Ge, since there are no entries for the GeC interactions (Tersoff
did not publish parameters for this cross-interaction.) Tersoff files are also provided for the SiC alloy
(SiC.tersoff) and the GaN (GaN.tersoff) alloys.
Many thanks to Rutuparna Narulkar, David Farrell, and Xiaowang Zhou for helping clarify how Tersoff
parameters for alloys have been defined in various papers.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
LAMMPS Users Manual
1679

These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, where types I and J correspond to two different element types, mixing is
performed by LAMMPS as described above from values in the potential file.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the MANYBODY package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
This pair style requires the newton setting to be "on" for pair interactions.
The Tersoff potential files provided with LAMMPS (see the potentials directory) are parameterized for metal
units. You can use the Tersoff potential with any LAMMPS units, but you would need to create your own
Tersoff potential file with coefficients listed in the appropriate units if your simulation doesn't use "metal"
units.
Related commands:
pair_coeff
Default: none
(Tersoff_1) J. Tersoff, Phys Rev B, 37, 6991 (1988).
(Albe) J. Nord, K. Albe, P. Erhart, and K. Nordlund, J. Phys.: Condens. Matter, 15, 5649(2003).
(Tersoff_2) J. Tersoff, Phys Rev B, 39, 5566 (1989); errata (PRB 41, 3248)
LAMMPS Users Manual
1680

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style tersoff/mod command
pair_style tersoff/mod/c command
pair_style tersoff/mod/gpu command
pair_style tersoff/mod/kk command
pair_style tersoff/mod/omp command
pair_style tersoff/mod/c/omp command
Syntax:
pair_style tersoff/mod
pair_style tersoff/mod/c
Examples:
pair_style tersoff/mod
pair_coeff * * Si.tersoff.mod Si Si
pair_style tersoff/mod/c
pair_coeff * * Si.tersoff.modc Si Si
Description:
The tersoff/mod and tersoff/mod/c styles computes a bond-order type interatomic potential (Kumagai) based
on a 3-body Tersoff potential (Tersoff_1), (Tersoff_2) with modified cutoff function and angular-dependent
term, giving the energy E of a system of atoms as
LAMMPS Users Manual
1681
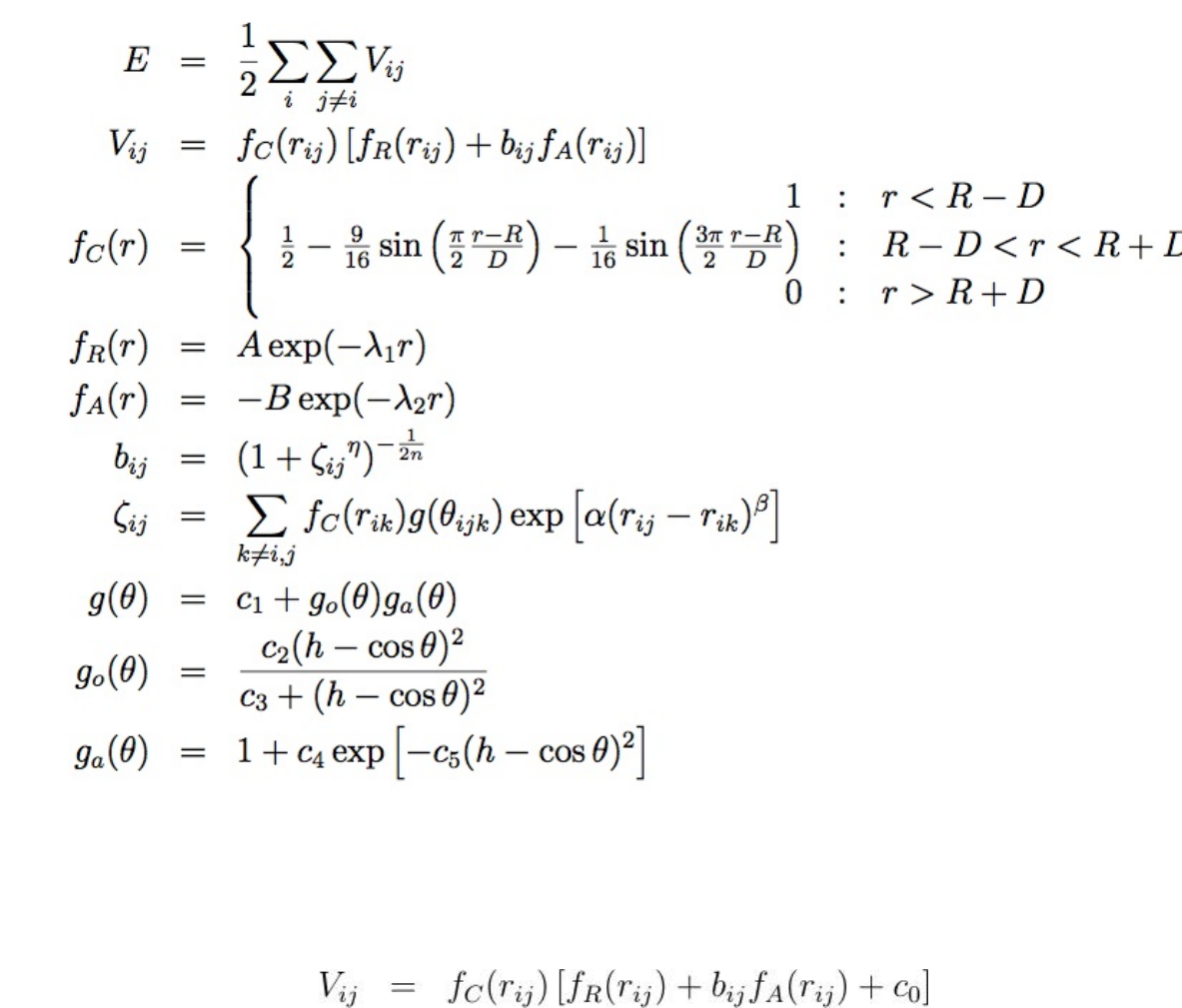
where f_R is a two-body term and f_A includes three-body interactions. The summations in the formula are
over all neighbors J and K of atom I within a cutoff distance = R + D. The tersoff/mod/c style differs from
tersoff/mod only in the formulation of the V_ij term, where it contains an additional c0 term.
The modified cutoff function f_C proposed by (Murty) and having a continuous second-order differential is
employed. The angular-dependent term g(theta) was modified to increase the flexibility of the potential.
The tersoff/mod potential is fitted to both the elastic constants and melting point by employing the modified
Tersoff potential function form in which the angular-dependent term is improved. The model performs
extremely well in describing the crystalline, liquid, and amorphous phases (Schelling).
Only a single pair_coeff command is used with the tersoff/mod style which specifies a Tersoff/MOD potential
file with parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N
additional arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom
types:
LAMMPS Users Manual
1682

filename• N element names = mapping of Tersoff/MOD elements to atom types•
As an example, imagine the Si.tersoff_mod file has Tersoff values for Si. If your LAMMPS simulation has 3
Si atoms types, you would use the following pair_coeff command:
pair_coeff * * Si.tersoff_mod Si Si Si
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The three Si arguments map
LAMMPS atom types 1,2,3 to the Si element in the Tersoff/MOD file. If a mapping value is specified as
NULL, the mapping is not performed. This can be used when a tersoff/mod potential is used as part of the
hybrid pair style. The NULL values are placeholders for atom types that will be used with other potentials.
Tersoff/MOD file in the potentials directory of the LAMMPS distribution have a ".tersoff.mod" suffix.
Potential files for the tersoff/mod/c style have the suffix ".tersoff.modc". Lines that are not blank or comments
(starting with #) define parameters for a triplet of elements. The parameters in a single entry correspond to
coefficients in the formulae above:
element 1 (the center atom in a 3-body interaction) element 2 (the atom bonded to the center atom) element 3
(the atom influencing the 1-2 bond in a bond-order sense) beta alpha h eta beta_ters = 1 (dummy parameter)
lambda2 (1/distance units) B (energy units) R (distance units) D (distance units) lambda1 (1/distance units) A
(energy units) n c1 c2 c3 c4 c5 c0 (energy units, tersoff/mod/c only):ul
The n, eta, lambda2, B, lambda1, and A parameters are only used for two-body interactions. The beta, alpha,
c1, c2, c3, c4, c5, h parameters are only used for three-body interactions. The R and D parameters are used for
both two-body and three-body interactions. The c0 term applies to tersoff/mod/c only. The non-annotated
parameters are unitless.
The Tersoff/MOD potential file must contain entries for all the elements listed in the pair_coeff command. It
can also contain entries for additional elements not being used in a particular simulation; LAMMPS ignores
those entries.
For a single-element simulation, only a single entry is required (e.g. SiSiSi). As annotated above, the first
element in the entry is the center atom in a three-body interaction and it is bonded to the 2nd atom and the
bond is influenced by the 3rd atom. Thus an entry for SiSiSi means Si bonded to a Si with another Si atom
influencing the bond.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
LAMMPS Users Manual
1683

Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the MANYBODY package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
This pair style requires the newton setting to be "on" for pair interactions.
The Tersoff/MOD potential files provided with LAMMPS (see the potentials directory) are parameterized for
metal units. You can use the Tersoff/MOD potential with any LAMMPS units, but you would need to create
your own Tersoff/MOD potential file with coefficients listed in the appropriate units if your simulation doesn't
use "metal" units.
Related commands:
pair_coeff
Default: none
(Kumagai) T. Kumagai, S. Izumi, S. Hara, S. Sakai, Comp. Mat. Science, 39, 457 (2007).
(Tersoff_1) J. Tersoff, Phys Rev B, 37, 6991 (1988).
(Tersoff_2) J. Tersoff, Phys Rev B, 38, 9902 (1988).
(Murty) M.V.R. Murty, H.A. Atwater, Phys Rev B, 51, 4889 (1995).
(Schelling) Patrick K. Schelling, Comp. Mat. Science, 44, 274 (2008).
LAMMPS Users Manual
1684

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style tersoff/zbl command
pair_style tersoff/zbl/gpu command
pair_style tersoff/zbl/kk command
pair_style tersoff/zbl/omp command
Syntax:
pair_style tersoff/zbl
Examples:
pair_style tersoff/zbl
pair_coeff * * SiC.tersoff.zbl Si C Si
Description:
The tersoff/zbl style computes a 3-body Tersoff potential (Tersoff_1) with a close-separation pairwise
modification based on a Coulomb potential and the Ziegler-Biersack-Littmark universal screening function
(ZBL), giving the energy E of a system of atoms as
LAMMPS Users Manual
1685
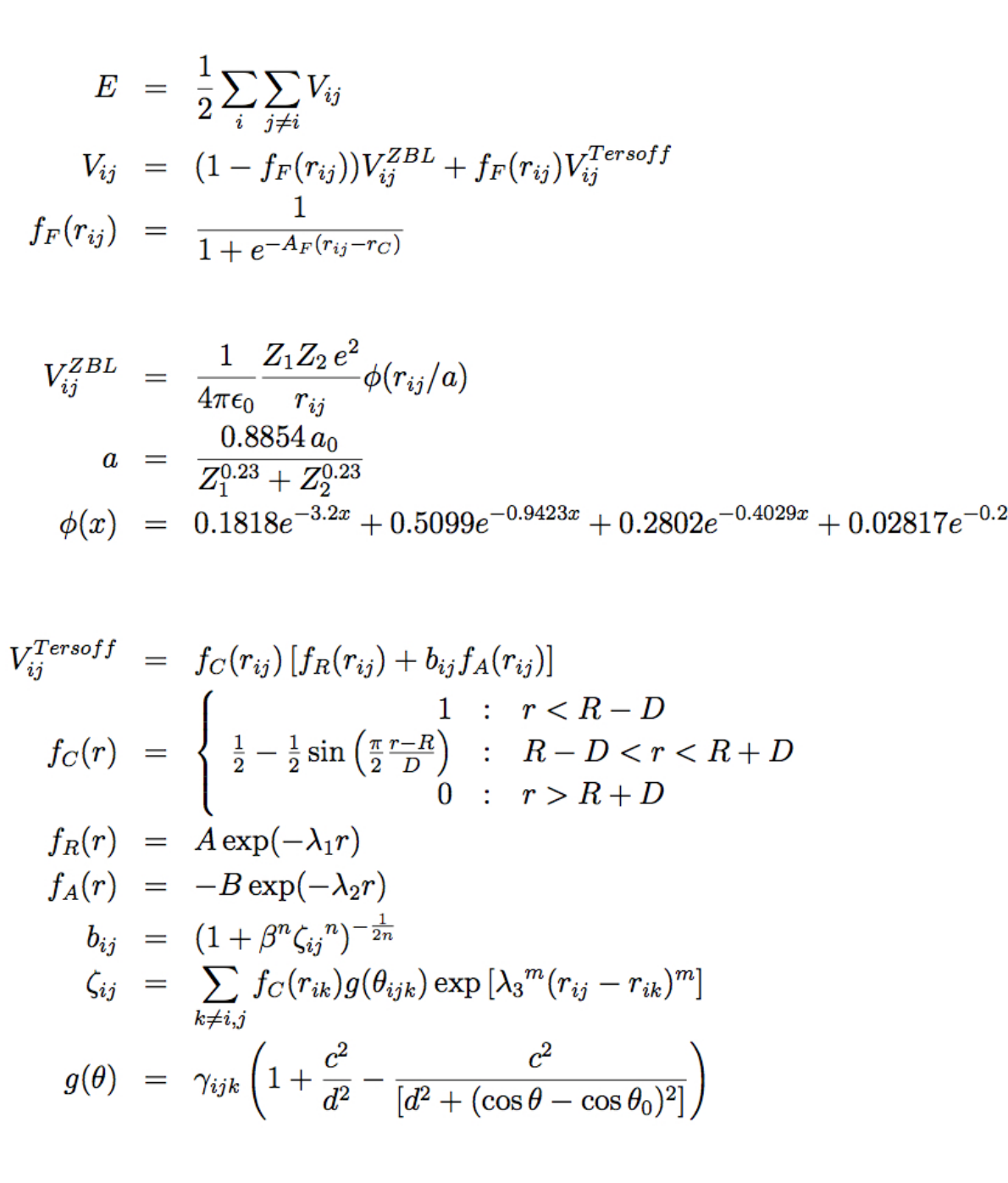
LAMMPS Users Manual
1686
The f_F term is a fermi-like function used to smoothly connect the ZBL repulsive potential with the Tersoff
potential. There are 2 parameters used to adjust it: A_F and r_C. A_F controls how "sharp" the transition is
between the two, and r_C is essentially the cutoff for the ZBL potential.
For the ZBL portion, there are two terms. The first is the Coulomb repulsive term, with Z1, Z2 as the number
of protons in each nucleus, e as the electron charge (1 for metal and real units) and epsilon0 as the permittivity
of vacuum. The second part is the ZBL universal screening function, with a0 being the Bohr radius (typically
0.529 Angstroms), and the remainder of the coefficients provided by the original paper. This screening
function should be applicable to most systems. However, it is only accurate for small separations (i.e. less
than 1 Angstrom).
For the Tersoff portion, f_R is a two-body term and f_A includes three-body interactions. The summations in
the formula are over all neighbors J and K of atom I within a cutoff distance = R + D.
Only a single pair_coeff command is used with the tersoff/zbl style which specifies a Tersoff/ZBL potential
file with parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N
additional arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom
types:
filename• N element names = mapping of Tersoff/ZBL elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, imagine the SiC.tersoff.zbl file has Tersoff/ZBL values for Si and C. If your LAMMPS
simulation has 4 atoms types and you want the 1st 3 to be Si, and the 4th to be C, you would use the following
pair_coeff command:
pair_coeff * * SiC.tersoff Si Si Si C
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Si arguments map
LAMMPS atom types 1,2,3 to the Si element in the Tersoff/ZBL file. The final C argument maps LAMMPS
atom type 4 to the C element in the Tersoff/ZBL file. If a mapping value is specified as NULL, the mapping is
not performed. This can be used when a tersoff/zbl potential is used as part of the hybrid pair style. The NULL
values are placeholders for atom types that will be used with other potentials.
Tersoff/ZBL files in the potentials directory of the LAMMPS distribution have a ".tersoff.zbl" suffix. Lines
that are not blank or comments (starting with #) define parameters for a triplet of elements. The parameters in
a single entry correspond to coefficients in the formula above:
element 1 (the center atom in a 3-body interaction)• element 2 (the atom bonded to the center atom)• element 3 (the atom influencing the 1-2 bond in a bond-order sense)• m• gamma• lambda3 (1/distance units)• c• d• costheta0 (can be a value < -1 or > 1)• n• beta•
LAMMPS Users Manual
1687
lambda2 (1/distance units)• B (energy units)• R (distance units)• D (distance units)• lambda1 (1/distance units)• A (energy units)• Z_i• Z_j• ZBLcut (distance units)• ZBLexpscale (1/distance units)•
The n, beta, lambda2, B, lambda1, and A parameters are only used for two-body interactions. The m, gamma,
lambda3, c, d, and costheta0 parameters are only used for three-body interactions. The R and D parameters are
used for both two-body and three-body interactions. The Z_i,Z_j, ZBLcut, ZBLexpscale parameters are used
in the ZBL repulsive portion of the potential and in the Fermi-like function. The non-annotated parameters are
unitless. The value of m must be 3 or 1.
The Tersoff/ZBL potential file must contain entries for all the elements listed in the pair_coeff command. It
can also contain entries for additional elements not being used in a particular simulation; LAMMPS ignores
those entries.
For a single-element simulation, only a single entry is required (e.g. SiSiSi). For a two-element simulation, the
file must contain 8 entries (for SiSiSi, SiSiC, SiCSi, SiCC, CSiSi, CSiC, CCSi, CCC), that specify Tersoff
parameters for all permutations of the two elements interacting in three-body configurations. Thus for 3
elements, 27 entries would be required, etc.
As annotated above, the first element in the entry is the center atom in a three-body interaction and it is
bonded to the 2nd atom and the bond is influenced by the 3rd atom. Thus an entry for SiCC means Si bonded
to a C with another C atom influencing the bond. Thus three-body parameters for SiCSi and SiSiC entries will
not, in general, be the same. The parameters used for the two-body interaction come from the entry where the
2nd element is repeated. Thus the two-body parameters for Si interacting with C, comes from the SiCC entry.
The parameters used for a particular three-body interaction come from the entry with the corresponding three
elements. The parameters used only for two-body interactions (n, beta, lambda2, B, lambda1, and A) in entries
whose 2nd and 3rd element are different (e.g. SiCSi) are not used for anything and can be set to 0.0 if desired.
Note that the twobody parameters in entries such as SiCC and CSiSi are often the same, due to the common
use of symmetric mixing rules, but this is not always the case. For example, the beta and n parameters in
Tersoff_2 (Tersoff_2) are not symmetric.
We chose the above form so as to enable users to define all commonly used variants of the Tersoff portion of
the potential. In particular, our form reduces to the original Tersoff form when m = 3 and gamma = 1, while it
reduces to the form of Albe et al. when beta = 1 and m = 1. Note that in the current Tersoff implementation in
LAMMPS, m must be specified as either 3 or 1. Tersoff used a slightly different but equivalent form for
alloys, which we will refer to as Tersoff_2 potential (Tersoff_2).
LAMMPS parameter values for Tersoff_2 can be obtained as follows: gamma = omega_ijk, lambda3 = 0 and
the value of m has no effect. The parameters for species i and j can be calculated using the Tersoff_2 mixing
rules:
LAMMPS Users Manual
1688

Tersoff_2 parameters R and S must be converted to the LAMMPS parameters R and D (R is different in both
forms), using the following relations: R=(R'+S')/2 and D=(S'-R')/2, where the primes indicate the Tersoff_2
parameters.
In the potentials directory, the file SiCGe.tersoff provides the LAMMPS parameters for Tersoff's various
versions of Si, as well as his alloy parameters for Si, C, and Ge. This file can be used for pure Si, (three
different versions), pure C, pure Ge, binary SiC, and binary SiGe. LAMMPS will generate an error if this file
is used with any combination involving C and Ge, since there are no entries for the GeC interactions (Tersoff
did not publish parameters for this cross-interaction.) Tersoff files are also provided for the SiC alloy
(SiC.tersoff) and the GaN (GaN.tersoff) alloys.
Many thanks to Rutuparna Narulkar, David Farrell, and Xiaowang Zhou for helping clarify how Tersoff
parameters for alloys have been defined in various papers. Also thanks to Ram Devanathan for providing the
base ZBL implementation.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
LAMMPS Users Manual
1689

For atom type pairs I,J and I != J, where types I and J correspond to two different element types, mixing is
performed by LAMMPS as described above from values in the potential file.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This pair style is part of the MANYBODY package. It is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
This pair style requires the newton setting to be "on" for pair interactions.
The Tersoff/ZBL potential files provided with LAMMPS (see the potentials directory) are parameterized for
metal units. You can use the Tersoff potential with any LAMMPS units, but you would need to create your
own Tersoff potential file with coefficients listed in the appropriate units if your simulation doesn't use
"metal" units.
Related commands:
pair_coeff
Default: none
(Tersoff_1) J. Tersoff, Phys Rev B, 37, 6991 (1988).
(ZBL) J.F. Ziegler, J.P. Biersack, U. Littmark, 'Stopping and Ranges of Ions in Matter' Vol 1, 1985,
Pergamon Press.
(Albe) J. Nord, K. Albe, P. Erhart and K. Nordlund, J. Phys.: Condens. Matter, 15, 5649(2003).
(Tersoff_2) J. Tersoff, Phys Rev B, 39, 5566 (1989); errata (PRB 41, 3248)
LAMMPS Users Manual
1690

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style thole command
pair_style lj/cut/thole/long command
pair_style lj/cut/thole/long/omp command
Syntax:
pair_style style args
style = thole or lj/cut/thole/long or lj/cut/thole/long/omp• args = list of arguments for a particular style•
thole args = damp cutoff
damp = global damping parameter
cutoff = global cutoff (distance units)
lj/cut/thole/long or lj/cut/thole/long/omp args = damp cutoff (cutoff2)
damp = global damping parameter
cutoff = global cutoff for LJ (and Thole if only 1 arg) (distance units)
cutoff2 = global cutoff for Thole (optional) (distance units)
Examples:
pair_style hybrid/overlay ... thole 2.6 12.0
pair_coeff 1 1 thole 1.0
pair_coeff 1 2 thole 1.0 2.6 10.0
pair_coeff * 2 thole 1.0 2.6
pair_style lj/cut/thole/long 2.6 12.0
Description:
The thole pair styles are meant to be used with force fields that include explicit polarization through Drude
dipoles. This link describes how to use the thermalized Drude oscillator model in LAMMPS and polarizable
models in LAMMPS are discussed in this Section.
The thole pair style should be used as a sub-style within in the pair_hybrid/overlay command, in conjunction
with a main pair style including Coulomb interactions, i.e. any pair style containing coul/cut or coul/long in its
style name.
The lj/cut/thole/long pair style is equivalent to, but more convenient that the frequent combination
hybrid/overlay lj/cut/coul/long cutoff thole damp cutoff2. It is not only a shorthand for this pair_style
combination, but it also allows for mixing pair coefficients instead of listing them all. The lj/cut/thole/long
pair style is also a bit faster because it avoids an overlay and can benefit from OMP acceleration. Moreover, it
uses a more precise approximation of the direct Coulomb interaction at short range similar to coul/long/cs,
which stabilizes the temperature of Drude particles.
The thole pair styles compute the Coulomb interaction damped at short distances by a function
LAMMPS Users Manual
1691

\begin{equation} T_{ij}(r_{ij}) = 1 - \left( 1 + \frac{s_{ij} r_{ij} }{2} \right) \exp \left( - s_{ij} r_{ij} \right)
\end{equation}
This function results from an adaptation to point charges (Noskov) of the dipole screening scheme originally
proposed by Thole. The scaling coefficient \(s_{ij} \) is determined by the polarizability of the atoms, \(
\alpha_i \), and by a Thole damping parameter \( a \). This Thole damping parameter usually takes a value of
2.6, but in certain force fields the value can depend upon the atom types. The mixing rule for Thole damping
parameters is the arithmetic average, and for polarizabilities the geometric average between the atom-specific
values.
\begin{equation} s_{ij} = \frac{ a_{ij} }{ (\alpha_{ij})^{1/3} } = \frac{ (a_i + a_j)/2 }{
[(\alpha_i\alpha_j)^{1/2}]^{1/3} } \end{equation}
The damping function is only applied to the interactions between the point charges representing the induced
dipoles on polarizable sites, that is, charges on Drude particles, \( q_{D,i} \), and opposite charges, \( -q_{D,i}
\), located on the respective core particles (to which each Drude particle is bonded). Therefore, Thole
screening is not applied to the full charge of the core particle \( q_i \), but only to the \( -q_{D,i} \) part of it.
The interactions between core charges are subject to the weighting factors set by the special_bonds command.
The interactions between Drude particles and core charges or non-polarizable atoms are also subject to these
weighting factors. The Drude particles inherit the 1-2, 1-3 and 1-4 neighbor relations from their respective
cores.
For pair_style thole, the following coefficients must be defined for each pair of atoms types via the pair_coeff
command as in the example above.
alpha (distance units^3)• damp• cutoff (distance units)•
The last two coefficients are optional. If not specified the global Thole damping parameter or global cutoff
specified in the pair_style command are used. In order to specify a cutoff (third argument) a damp parameter
(second argument) must also be specified.
For pair style lj/cut/thole/long, the following coefficients must be defined for each pair of atoms types via the
pair_coeff command.
epsilon (energy units)• sigma (length units)• alpha (distance units^3)• damps• LJ cutoff (distance units)•
The last two coefficients are optional and default to the global values from the pair_style command line.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
LAMMPS Users Manual
1692

These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing:
The thole pair style does not support mixing. Thus, coefficients for all I,J pairs must be specified explicitly.
The lj/cut/thole/long pair style does support mixing. Mixed coefficients are defined using
\begin{equation} \alpha_{ij} = \sqrt{\alpha_i\alpha_j}\end{equation} \begin{equation} a_{ij} = \frac 1 2 (a_i
+ a_j)\end{equation}
Restrictions:
These pair styles are part of the USER-DRUDE package. They are only enabled if LAMMPS was built with
that package. See the Making LAMMPS section for more info.
This pair_style should currently not be used with the charmm dihedral style if the latter has non-zero 1-4
weighting factors. This is because the thole pair style does not know which pairs are 1-4 partners of which
dihedrals.
The lj/cut/thole/long pair style should be used with a Kspace solver like PPPM or Ewald, which is only
enabled if LAMMPS was built with the kspace package.
Related commands:
fix drude, fix langevin/drude, fix drude/transform, compute temp/drude pair_style lj/cut/coul/long
Default: none
(Noskov) Noskov, Lamoureux and Roux, J Phys Chem B, 109, 6705 (2005).
(Thole) Chem Phys, 59, 341 (1981).
LAMMPS Users Manual
1693

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style tri/lj command
Syntax:
pair_style tri/lj cutoff
cutoff = global cutoff for interactions (distance units)
Examples:
pair_style tri/lj 3.0
pair_coeff * * 1.0 1.0
pair_coeff 1 1 1.0 1.5 2.5
Description:
Style tri/lj treats particles which are triangles as a set of small spherical particles that tile the triangle surface
as explained below. Interactions between two triangles, each with N1 and N2 spherical particles, are
calculated as the pairwise sum of N1*N2 Lennard-Jones interactions. Interactions between a triangle with N
spherical particles and a point particle are treated as the pairwise sum of N Lennard-Jones interactions. See the
pair_style lj/cut doc page for the definition of Lennard-Jones interactions.
The cutoff distance for an interaction between 2 triangles, or between a triangle and a point particle, is
calculated from the position of the triangle (its centroid), not between pairs of individual spheres comprising
the triangle. Thus an interaction is either calculated in its entirety or not at all.
The set of non-overlapping spherical particles that represent a triangle, for purposes of this pair style, are
generated in the following manner. Assume the triangle is of type I, and sigma_II has been specified. We want
a set of spheres with centers in the plane of the triangle, none of them larger in diameter than sigma_II, which
completely cover the triangle's area, but with minimal overlap and a minimal total number of spheres. This is
done in a recursive manner. Place a sphere at the centroid of the original triangle. Calculate what diameter it
must have to just cover all 3 corner points of the triangle. If that diameter is equal to or smaller than sigma_II,
then include a sphere of the calculated diameter in the set of covering spheres. It the diameter is larger than
sigma_II, then split the triangle into 2 triangles by bisecting its longest side. Repeat the process on each
sub-triangle, recursing as far as needed to generate a set of covering spheres. When finished, the original
criteria are met, and the set of covering spheres shoule be near minimal in number and overlap, at least for
input triangles with a reasonable aspect-ratio.
The LJ interaction between 2 spheres on different triangles of types I,J is computed with an arithmetic mixing
of the sigma values of the 2 spheres and using the specified epsilon value for I,J atom types. Note that because
the sigma values for triangles spheres is computed using only sigma_II values, specific to the triangles's type,
this means that any specified sigma_IJ values (for I != J) are effectively ignored.
For style tri/lj, the following coefficients must be defined for each pair of atoms types via the pair_coeff
command as in the examples above, or in the data file or restart files read by the read_data or read_restart
commands:
epsilon (energy units)• sigma (distance units)•
LAMMPS Users Manual
1694

cutoff (distance units)•
The last coefficient is optional. If not specified, the global cutoff is used.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance for all of this pair
style can be mixed. The default mix value is geometric. See the "pair_modify" command for details.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
This style is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
Defining particles to be triangles so they participate in tri/tri or tri/particle interactions requires the use the
atom_style tri command.
Related commands:
pair_coeff, pair_style line/lj
Default: none
LAMMPS Users Manual
1695

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style vashishta command
pair_style vashishta/omp command
pair_style vashishta/kk command
pair_style vashishta/table command
pair_style vashishta/table/omp command
Syntax:
pair_style style args
style = vashishta or vashishta/table or vashishta/omp or vashishta/table/omp• args = list of arguments for a particular style•
vashishta or vashishta/omp args = none
vashishta/table or vashishta/table/omp args = Ntable cutinner
Ntable = # of tabulation points
cutinner = tablulate from cutinner to cutoff
Examples:
pair_style vashishta
pair_coeff * * SiC.vashishta Si C
pair_style vashishta/table 100000 0.2
pair_coeff * * SiC.vashishta Si C
Description:
The vashishta and vashishta/table styles compute the combined 2-body and 3-body family of potentials
developed in the group of Priya Vashishta and collaborators. By combining repulsive, screened Coulombic,
screened charge-dipole, and dispersion interactions with a bond-angle energy based on the Stillinger-Weber
potential, this potential has been used to describe a variety of inorganic compounds, including SiO2
Vashishta1990, SiC Vashishta2007, and InP Branicio2009.
The potential for the energy U of a system of atoms is
LAMMPS Users Manual
1696
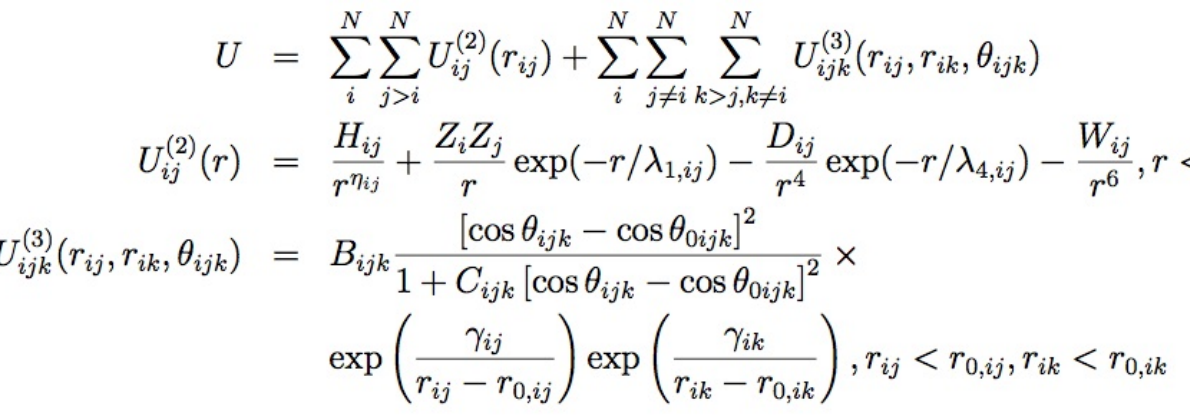
where we follow the notation used in Branicio2009. U2 is a two-body term and U3 is a three-body term. The
summation over two-body terms is over all neighbors J within a cutoff distance = rc. The twobody terms are
shifted and tilted by a linear function so that the energy and force are both zero at rc. The summation over
three-body terms is over all neighbors J and K within a cut-off distance = r0, where the exponential screening
function becomes zero.
The vashishta style computes these formulas analytically. The vashishta/table style tabulates the analytic
values for Ntable points from cutinner to the cutoff of the potential. The points are equally spaced in R^2
space from cutinner^2 to cutoff^2. For the two-body term in the above equation, a linear interpolation for each
pairwise distance between adjacent points in the table. In practice the tabulated version can run 3-5x faster
than the analytic version with with moderate to little loss of accuracy for Ntable values between 10000 and
1000000. It is not recommended to use less than 5000 tabulation points.
Only a single pair_coeff command is used with either style which specifies a Vashishta potential file with
parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional
arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
filename• N element names = mapping of Vashishta elements to atom types•
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, imagine a file SiC.vashishta has parameters for Si and C. If your LAMMPS simulation has 4
atoms types and you want the 1st 3 to be Si, and the 4th to be C, you would use the following pair_coeff
command:
pair_coeff * * SiC.vashishta Si Si Si C
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Si arguments map
LAMMPS atom types 1,2,3 to the Si element in the file. The final C argument maps LAMMPS atom type 4 to
the C element in the file. If a mapping value is specified as NULL, the mapping is not performed. This can be
used when a vashishta potential is used as part of the hybrid pair style. The NULL values are placeholders for
atom types that will be used with other potentials.
LAMMPS Users Manual
1697
Vashishta files in the potentials directory of the LAMMPS distribution have a ".vashishta" suffix. Lines that
are not blank or comments (starting with #) define parameters for a triplet of elements. The parameters in a
single entry correspond to the two-body and three-body coefficients in the formulae above:
element 1 (the center atom in a 3-body interaction)• element 2• element 3• H (energy units)• eta• Zi (electron charge units)• Zj (electron charge units)• lambda1 (distance units)• D (energy units)• lambda4 (distance units)• W (energy units)• rc (distance units)• B (energy units)• gamma• r0 (distance units)• C• costheta0•
The non-annotated parameters are unitless. The Vashishta potential file must contain entries for all the
elements listed in the pair_coeff command. It can also contain entries for additional elements not being used
in a particular simulation; LAMMPS ignores those entries. For a single-element simulation, only a single
entry is required (e.g. SiSiSi). For a two-element simulation, the file must contain 8 entries (for SiSiSi, SiSiC,
SiCSi, SiCC, CSiSi, CSiC, CCSi, CCC), that specify parameters for all permutations of the two elements
interacting in three-body configurations. Thus for 3 elements, 27 entries would be required, etc.
Depending on the particular version of the Vashishta potential, the values of these parameters may be keyed to
the identities of zero, one, two, or three elements. In order to make the input file format unambiguous, general,
and simple to code, LAMMPS uses a slightly confusing method for specifying parameters. All parameters are
divided into two classes: two-body and three-body. Two-body and three-body parameters are handled
differently, as described below. The two-body parameters are H, eta, lambda1, D, lambda4, W, rc, gamma,
and r0. They appear in the above formulae with two subscripts. The parameters Zi and Zj are also classified as
two-body parameters, even though they only have 1 subscript. The three-body parameters are B, C, costheta0.
They appear in the above formulae with three subscripts. Two-body and three-body parameters are handled
differently, as described below.
The first element in each entry is the center atom in a three-body interaction, while the second and third
elements are two neighbor atoms. Three-body parameters for a central atom I and two neighbors J and K are
taken from the IJK entry. Note that even though three-body parameters do not depend on the order of J and K,
LAMMPS stores three-body parameters for both IJK and IKJ. The user must ensure that these values are
equal. Two-body parameters for an atom I interacting with atom J are taken from the IJJ entry, where the 2nd
and 3rd elements are the same. Thus the two-body parameters for Si interacting with C come from the SiCC
entry. Note that even though two-body parameters (except possibly gamma and r0 in U3) do not depend on
the order of the two elements, LAMMPS will get the Si-C value from the SiCC entry and the C-Si value from
the CSiSi entry. The user must ensure that these values are equal. Two-body parameters appearing in entries
where the 2nd and 3rd elements are different are stored but never used. It is good practice to enter zero for
these values. Note that the three-body function U3 above contains the two-body parameters gamma and r0. So
U3 for a central C atom bonded to an Si atom and a second C atom will take three-body parameters from the
LAMMPS Users Manual
1698
CSiC entry, but two-body parameters from the CCC and CSiSi entries.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, where types I and J correspond to two different element types, mixing is
performed by LAMMPS as described above from values in the potential file.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus,
you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions:
These pair style are part of the MANYBODY package. They is only enabled if LAMMPS was built with that
package. See the Making LAMMPS section for more info.
These pair styles requires the newton setting to be "on" for pair interactions.
The Vashishta potential files provided with LAMMPS (see the potentials directory) are parameterized for
metal units. You can use the Vashishta potential with any LAMMPS units, but you would need to create your
own potential file with coefficients listed in the appropriate units if your simulation doesn't use "metal" units.
Related commands:
pair_coeff
Default: none
(Vashishta1990) P. Vashishta, R. K. Kalia, J. P. Rino, Phys. Rev. B 41, 12197 (1990).
LAMMPS Users Manual
1699
(Vashishta2007) P. Vashishta, R. K. Kalia, A. Nakano, J. P. Rino. J. Appl. Phys. 101, 103515 (2007).
(Branicio2009) Branicio, Rino, Gan and Tsuzuki, J. Phys Condensed Matter 21 (2009) 095002
LAMMPS Users Manual
1700

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style yukawa command
pair_style yukawa/gpu command
pair_style yukawa/omp command
Syntax:
pair_style yukawa kappa cutoff
kappa = screening length (inverse distance units)• cutoff = global cutoff for Yukawa interactions (distance units)•
Examples:
pair_style yukawa 2.0 2.5
pair_coeff 1 1 100.0 2.3
pair_coeff * * 100.0
Description:
Style yukawa computes pairwise interactions with the formula
Rc is the cutoff.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
A (energy*distance units)• cutoff (distance units)•
The last coefficient is optional. If not specified, the global yukawa cutoff is used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
LAMMPS Users Manual
1701

You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the A coefficient and cutoff distance for this pair style can be mixed. A is an
energy value mixed like a LJ epsilon. The default mix value is geometric. See the "pair_modify" command for
details.
This pair style supports the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_coeff
Default: none
LAMMPS Users Manual
1702

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style yukawa/colloid command
pair_style yukawa/colloid/gpu command
pair_style yukawa/colloid/omp command
Syntax:
pair_style yukawa/colloid kappa cutoff
kappa = screening length (inverse distance units)• cutoff = global cutoff for colloidal Yukawa interactions (distance units)•
Examples:
pair_style yukawa/colloid 2.0 2.5
pair_coeff 1 1 100.0 2.3
pair_coeff * * 100.0
Description:
Style yukawa/colloid computes pairwise interactions with the formula
where Ri and Rj are the radii of the two particles and Rc is the cutoff.
In contrast to pair_style yukawa, this functional form arises from the Coulombic interaction between two
colloid particles, screened due to the presence of an electrolyte, see the book by Safran for a derivation in the
context of DVLO theory. Pair_style yukawa is a screened Coulombic potential between two point-charges and
uses no such approximation.
This potential applies to nearby particle pairs for which the Derjagin approximation holds, meaning h << Ri +
Rj, where h is the surface-to-surface separation of the two particles.
When used in combination with pair_style colloid, the two terms become the so-called DLVO potential,
which combines electrostatic repulsion and van der Waals attraction.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the
examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
A (energy/distance units)• cutoff (distance units)•
LAMMPS Users Manual
1703

The prefactor A is determined from the relationship between surface charge and surface potential due to the
presence of electrolyte. Note that the A for this potential style has different units than the A used in pair_style
yukawa. For low surface potentials, i.e. less than about 25 mV, A can be written as:
A = 2 * PI * R*eps*eps0 * kappa * psi^2
where
R = colloid radius (distance units)• eps0 = permittivity of free space (charge^2/energy/distance units)• eps = relative permittivity of fluid medium (dimensionless)• kappa = inverse screening length (1/distance units)• psi = surface potential (energy/charge units)•
The last coefficient is optional. If not specified, the global yukawa/colloid cutoff is used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the A coefficient and cutoff distance for this pair style can be mixed. A is an
energy value mixed like a LJ epsilon. The default mix value is geometric. See the "pair_modify" command for
details.
This pair style supports the pair_modify shift option for the energy of the pair interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need
to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
LAMMPS Users Manual
1704

Restrictions:
This style is part of the COLLOID package. It is only enabled if LAMMPS was built with that package. See
the Making LAMMPS section for more info.
This pair style requires that atoms be finite-size spheres with a diameter, as defined by the atom_style sphere
command.
Per-particle polydispersity is not yet supported by this pair style; per-type polydispersity is allowed. This
means all particles of the same type must have the same diameter. Each type can have a different diameter.
Related commands:
pair_coeff
Default: none
(Safran) Safran, Statistical Thermodynamics of Surfaces, Interfaces, And Membranes, Westview Press,
ISBN: 978-0813340791 (2003).
LAMMPS Users Manual
1705

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style zbl command
pair_style zbl/gpu command
pair_style zbl/omp command
Syntax:
pair_style zbl inner outer
inner = distance where switching function begins• outer = global cutoff for ZBL interaction•
Examples:
pair_style zbl 3.0 4.0
pair_coeff * * 73.0 73.0
pair_coeff 1 1 14.0 14.0
Description:
Style zbl computes the Ziegler-Biersack-Littmark (ZBL) screened nuclear repulsion for describing
high-energy collisions between atoms. (Ziegler). It includes an additional switching function that ramps the
energy, force, and curvature smoothly to zero between an inner and outer cutoff. The potential energy due to a
pair of atoms at a distance r_ij is given by:
where e is the electron charge, epsilon_0 is the electrical permittivity of vacuum, and Z_i and Z_j are the
nuclear charges of the two atoms. The switching function S(r) is identical to that used by pair_style
lj/gromacs. Here, the inner and outer cutoff are the same for all pairs of atom types.
The following coefficients must be defined for each pair of atom types via the pair_coeff command as in the
examples above, or in the LAMMPS data file.
Z_i (atomic number for first atom type, e.g. 13.0 for aluminum)•
Z_j (ditto for second atom type)•
LAMMPS Users Manual
1706

The values of Z_i and Z_j are normally equal to the atomic numbers of the two atom types. Thus, the user
may optionally specify only the coefficients for each I==I pair, and rely on the obvious mixing rule for cross
interactions (see below). Note that when I==I it is required that Z_i == Z_j. When used with hybrid/overlay
and pairs are assigned to more than one sub-style, the mixing rule is not used and each pair of types
interacting with the ZBL sub-style must be included in a pair_coeff command.
NOTE: The numerical values of the exponential decay constants in the screening function depend on the unit
of distance. In the above equation they are given for units of angstroms. LAMMPS will automatically convert
these values to the distance unit of the specified LAMMPS units setting. The values of Z should always be
given as multiples of a proton's charge, e.g. 29.0 for copper.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the Z_i and Z_j coefficients can be mixed by taking Z_i and Z_j from the
values specified for I == I and J == J cases. When used with hybrid/overlay and pairs are assigned to more
than one sub-style, the mixing rule is not used and each pair of types interacting with the ZBL sub-style must
be included in a pair_coeff command. The pair_modify mix option has no effect on the mixing behavior
The ZBL pair style does not support the pair_modify shift option, since the ZBL interaction is already
smoothed to 0.0 at the cutoff.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy
and pressure, since there are no corrections for a potential that goes to 0.0 at the cutoff.
This pair style does not write information to binary restart files, so pair_style and pair_coeff commands must
be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the
inner, middle, outer keywords.
Restrictions: none
Related commands:
LAMMPS Users Manual
1707


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
pair_style zero command
Syntax:
pair_style zero cutoff nocoeff
zero = style name of this pair style cutoff = global cutoff (distance units) nocoeff = ignore all pair_coeff
parameters (optional)
Examples:
pair_style zero 10.0
pair_style zero 5.0 nocoeff
pair_coeff * *
pair_coeff 1 2*4 3.0
Description:
Define a global or per-type cutoff length for the purpose of building a neighbor list and acquiring ghost
atoms, but do not compute any pairwise forces or energies.
This can be useful for fixes or computes which require a neighbor list to enumerate pairs of atoms within
some cutoff distance, but when pairwise forces are not otherwise needed. Examples are the fix bond/create,
compute rdf, compute voronoi/atom commands.
Note that the comm_modify cutoff command can be used to insure communication of ghost atoms even
when a pair style is not defined, but it will not trigger neighbor list generation.
The optional nocoeff flag allows to read data files with a PairCoeff section for any pair style. Similarly, any
pair_coeff commands will only be checked for the atom type numbers and the rest ignored. In this case,
only the global cutoff will be used.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in
the examples above, or in the data file or restart files read by the read_data or read_restart commands, or by
mixing as described below:
cutoff (distance units)•
This coefficient is optional. If not specified, the global cutoff specified in the pair_style command is used.
If the pair_style has been specified with the optional nocoeff flag, then a cutoff pair coefficient is ignored.
Mixing, shift, table, tail correction, restart, rRESPA info:
The cutoff distance for this pair style can be mixed. The default mix value is geometric. See the
"pair_modify" command for details.
This pair style does not support the pair_modify shift, table, and tail options.
•
LAMMPS Users Manual
1709

This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not
need to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support
the inner, middle, outer keywords.
Restrictions: none
Related commands:
pair_style none
Default: none
LAMMPS Users Manual
1710
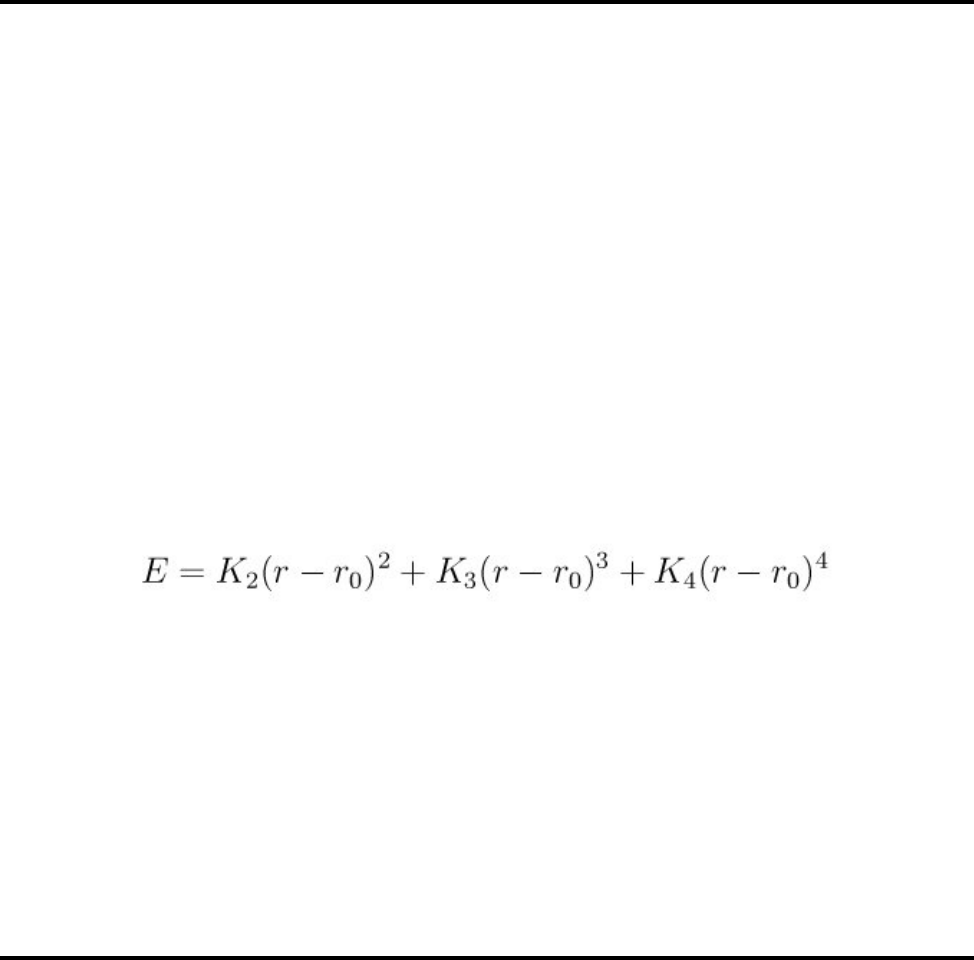
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style class2 command
bond_style class2/omp command
bond_style class2/kk command
Syntax:
bond_style class2
Examples:
bond_style class2
bond_coeff 1 1.0 100.0 80.0 80.0
Description:
The class2 bond style uses the potential
where r0 is the equilibrium bond distance.
See (Sun) for a description of the COMPASS class2 force field.
The following coefficients must be defined for each bond type via the bond_coeff command as in the example
above, or in the data file or restart files read by the read_data or read_restart commands:
R0 (distance)• K2 (energy/distance^2)• K3 (energy/distance^3)• K4 (energy/distance^4)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
LAMMPS Users Manual
1711

See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This bond style can only be used if LAMMPS was built with the CLASS2 package. See the Making
LAMMPS section for more info on packages.
Related commands:
bond_coeff, delete_bonds
Default: none
(Sun) Sun, J Phys Chem B 102, 7338-7364 (1998).
LAMMPS Users Manual
1712
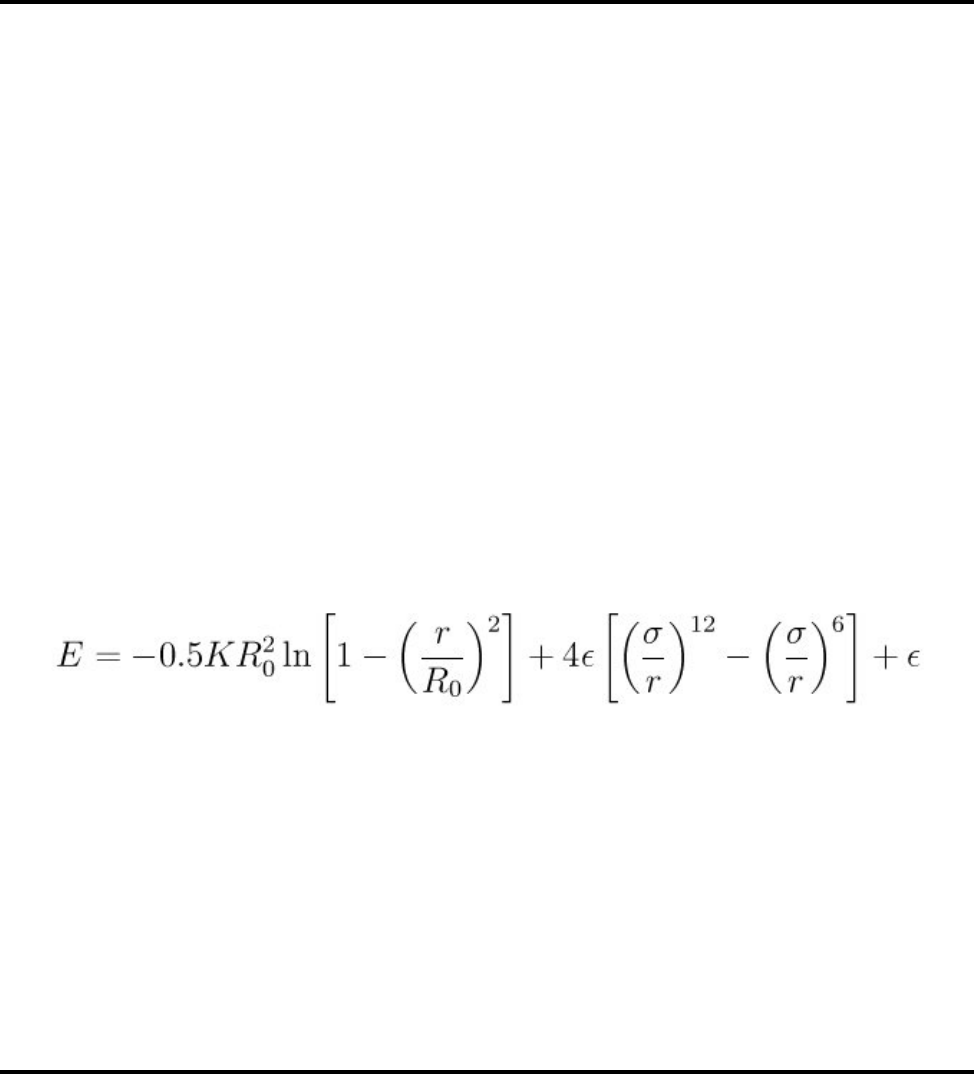
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style fene command
bond_style fene/intel command
bond_style fene/kk command
bond_style fene/omp command
Syntax:
bond_style fene
Examples:
bond_style fene
bond_coeff 1 30.0 1.5 1.0 1.0
Description:
The fene bond style uses the potential
to define a finite extensible nonlinear elastic (FENE) potential (Kremer), used for bead-spring polymer
models. The first term is attractive, the 2nd Lennard-Jones term is repulsive. The first term extends to R0, the
maximum extent of the bond. The 2nd term is cutoff at 2^(1/6) sigma, the minimum of the LJ potential.
The following coefficients must be defined for each bond type via the bond_coeff command as in the example
above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy/distance^2)• R0 (distance)• epsilon (energy)• sigma (distance)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
LAMMPS Users Manual
1713

You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This bond style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
You typically should specify special_bonds fene or special_bonds lj/coul 0 1 1 to use this bond style.
LAMMPS will issue a warning it that's not the case.
Related commands:
bond_coeff, delete_bonds
Default: none
(Kremer) Kremer, Grest, J Chem Phys, 92, 5057 (1990).
LAMMPS Users Manual
1714
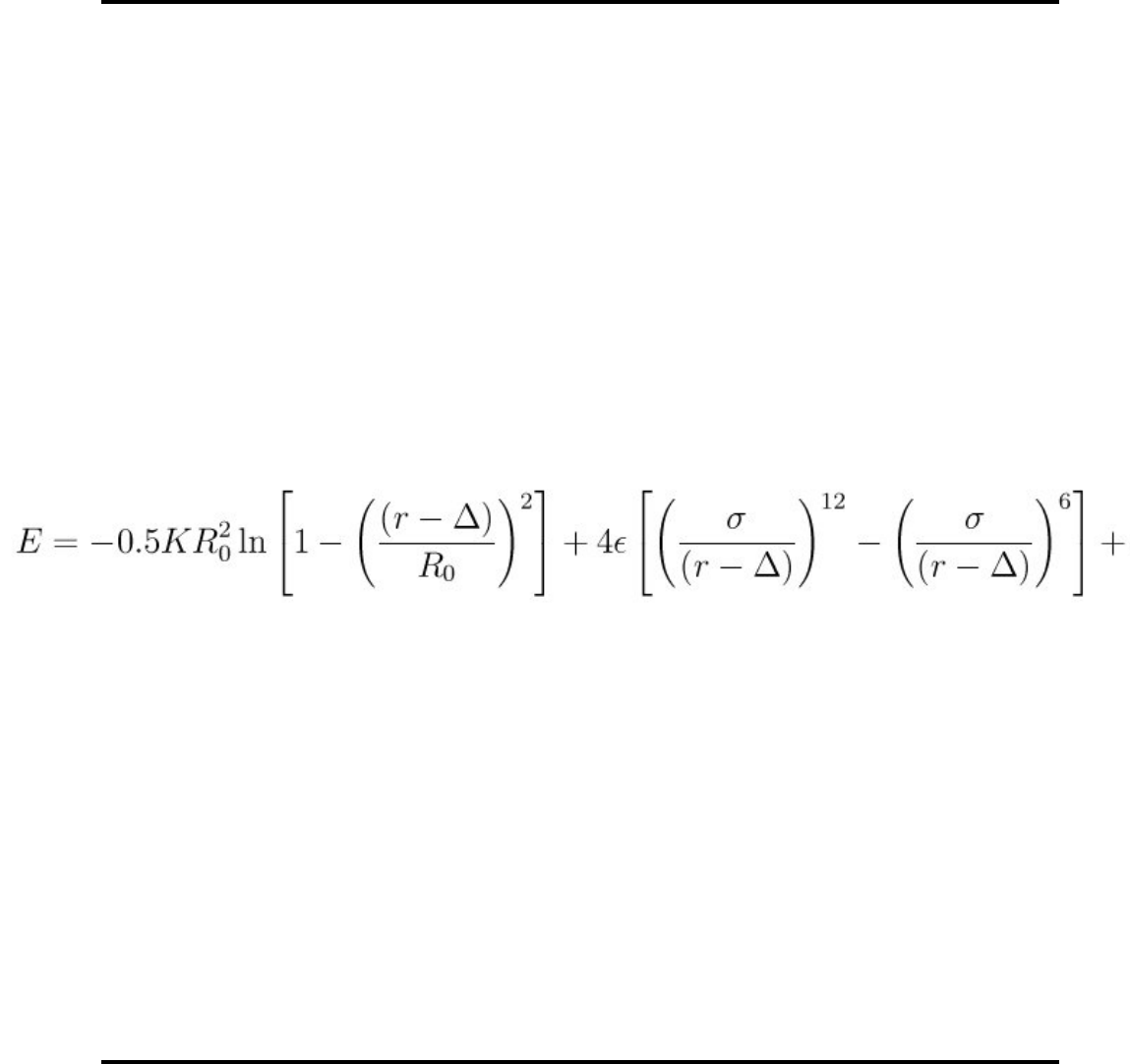
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style fene/expand command
bond_style fene/expand/omp command
Syntax:
bond_style fene/expand
Examples:
bond_style fene/expand
bond_coeff 1 30.0 1.5 1.0 1.0 0.5
Description:
The fene/expand bond style uses the potential
to define a finite extensible nonlinear elastic (FENE) potential (Kremer), used for bead-spring polymer
models. The first term is attractive, the 2nd Lennard-Jones term is repulsive.
The fene/expand bond style is similar to fene except that an extra shift factor of delta (positive or negative) is
added to r to effectively change the bead size of the bonded atoms. The first term now extends to R0 + delta
and the 2nd term is cutoff at 2^(1/6) sigma + delta.
The following coefficients must be defined for each bond type via the bond_coeff command as in the example
above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy/distance^2)• R0 (distance)• epsilon (energy)• sigma (distance)• delta (distance)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
LAMMPS Users Manual
1715

You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This bond style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
You typically should specify special_bonds fene or special_bonds lj/coul 0 1 1 to use this bond style.
LAMMPS will issue a warning it that's not the case.
Related commands:
bond_coeff, delete_bonds
Default: none
(Kremer) Kremer, Grest, J Chem Phys, 92, 5057 (1990).
LAMMPS Users Manual
1716

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style oxdna/fene command
bond_style oxdna2/fene command
Syntax:
bond_style oxdna/fene
bond_style oxdna2/fene
Examples:
bond_style oxdna/fene
bond_coeff * 2.0 0.25 0.7525
bond_style oxdna2/fene
bond_coeff * 2.0 0.25 0.7564
Description:
The oxdna/fene and oxdna2/fene bond styles use the potential
to define a modified finite extensible nonlinear elastic (FENE) potential (Ouldridge) to model the connectivity
of the phosphate backbone in the oxDNA force field for coarse-grained modelling of DNA.
The following coefficients must be defined for the bond type via the bond_coeff command as given in the
above example, or in the data file or restart files read by the read_data or read_restart commands:
epsilon (energy)• Delta (distance)• r0 (distance)•
NOTE: The oxDNA bond style has to be used together with the corresponding oxDNA pair styles for
excluded volume interaction oxdna/excv, stacking oxdna/stk, cross-stacking oxdna/xstk and coaxial stacking
interaction oxdna/coaxstk as well as hydrogen-bonding interaction oxdna/hbond (see also documentation of
pair_style oxdna/excv). For the oxDNA2 (Snodin) bond style the analogous pair styles and an additional
Debye-Hueckel pair style oxdna2/dh have to be defined.
The coefficients in the above example have to be kept fixed and cannot be changed without reparametrizing
the entire model.
Example input and data files for DNA duplexes can be found in examples/USER/cgdna/examples/oxDNA/
and /oxDNA2/. A simple python setup tool which creates single straight or helical DNA strands, DNA
duplexes or arrays of DNA duplexes can be found in examples/USER/cgdna/util/. A technical report with
more information on the model, the structure of the input file, the setup tool and the performance of the
LAMMPS Users Manual
1717

LAMMPS-implementation of oxDNA can be found here.
Restrictions:
This bond style can only be used if LAMMPS was built with the USER-CGDNA package and the
MOLECULE and ASPHERE package. See the Making LAMMPS section for more info on packages.
Related commands:
pair_style oxdna/excv, pair_style oxdna2/excv, fix nve/dotc/langevin, bond_coeff
Default: none
(Ouldridge) T.E. Ouldridge, A.A. Louis, J.P.K. Doye, J. Chem. Phys. 134, 085101 (2011).
(Snodin) B.E. Snodin, F. Randisi, M. Mosayebi, et al., J. Chem. Phys. 142, 234901 (2015).
LAMMPS Users Manual
1718
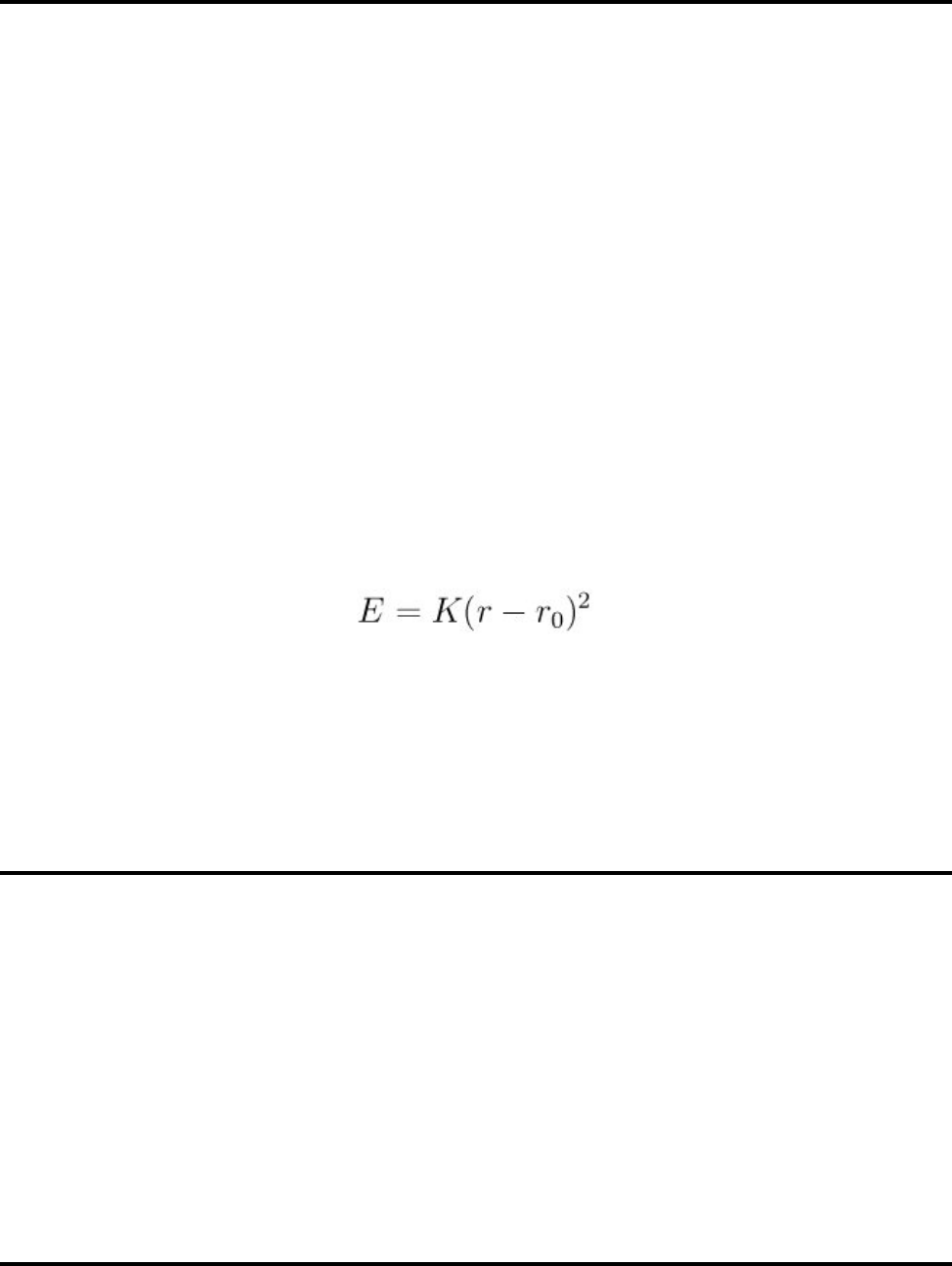
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style harmonic command
bond_style harmonic/intel command
bond_style harmonic/kk command
bond_style harmonic/omp command
Syntax:
bond_style harmonic
Examples:
bond_style harmonic
bond_coeff 5 80.0 1.2
Description:
The harmonic bond style uses the potential
where r0 is the equilibrium bond distance. Note that the usual 1/2 factor is included in K.
The following coefficients must be defined for each bond type via the bond_coeff command as in the example
above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy/distance^2)• r0 (distance)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
LAMMPS Users Manual
1719
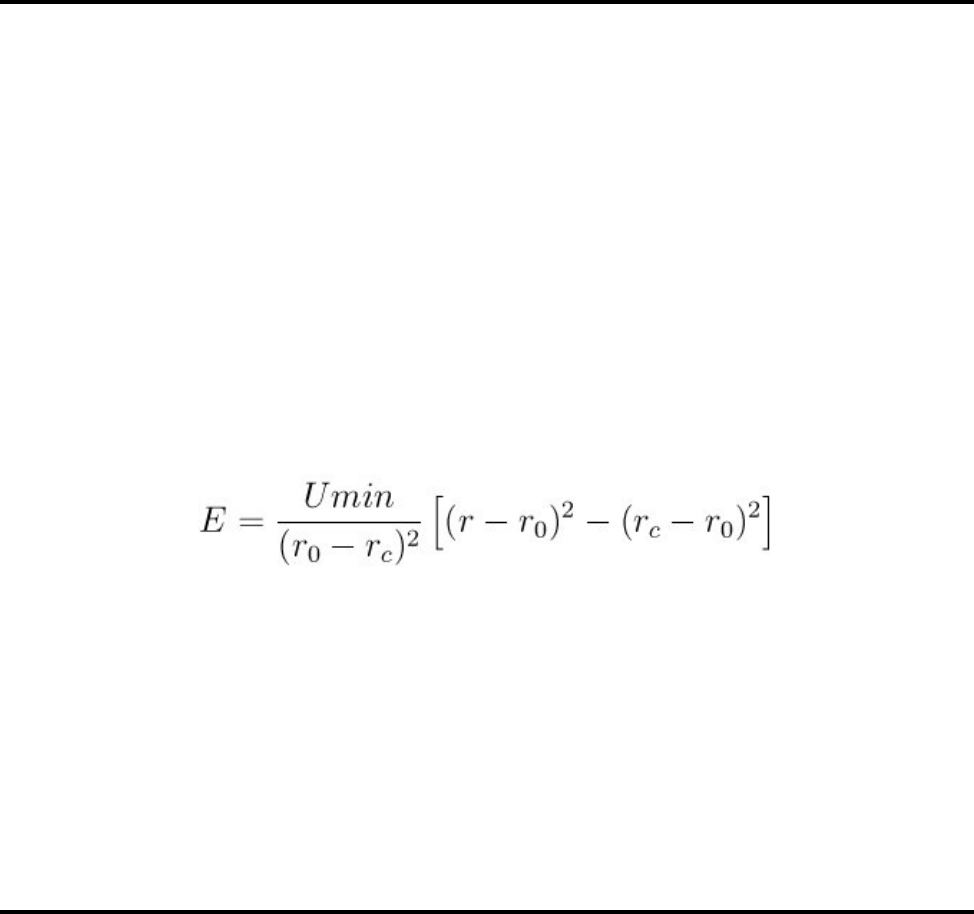
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style harmonic/shift command
bond_style harmonic/shift/omp command
Syntax:
bond_style harmonic/shift
Examples:
bond_style harmonic/shift
bond_coeff 5 10.0 0.5 1.0
Description:
The harmonic/shift bond style is a shifted harmonic bond that uses the potential
where r0 is the equilibrium bond distance, and rc the critical distance. The potential is -Umin at r0 and zero at
rc. The spring constant is k = Umin / [ 2 (r0-rc)^2].
The following coefficients must be defined for each bond type via the bond_coeff command as in the example
above, or in the data file or restart files read by the read_data or read_restart commands:
Umin (energy)•
r0 (distance)•
rc (distance)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
LAMMPS Users Manual
1721


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style harmonic/shift/cut command
bond_style harmonic/shift/cut/omp command
Syntax:
bond_style harmonic/shift/cut
Examples:
bond_style harmonic/shift/cut
bond_coeff 5 10.0 0.5 1.0
Description:
The harmonic/shift/cut bond style is a shifted harmonic bond that uses the potential
where r0 is the equilibrium bond distance, and rc the critical distance. The bond potential is zero for distances
r > rc. The potential is -Umin at r0 and zero at rc. The spring constant is k = Umin / [ 2 (r0-rc)^2].
The following coefficients must be defined for each bond type via the bond_coeff command as in the example
above, or in the data file or restart files read by the read_data or read_restart commands:
Umin (energy)• r0 (distance)• rc (distance)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
LAMMPS Users Manual
1723

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style hybrid command
Syntax:
bond_style hybrid style1 style2 ...
style1,style2 = list of one or more bond styles•
Examples:
bond_style hybrid harmonic fene
bond_coeff 1 harmonic 80.0 1.2
bond_coeff 2* fene 30.0 1.5 1.0 1.0
Description:
The hybrid style enables the use of multiple bond styles in one simulation. A bond style is assigned to each
bond type. For example, bonds in a polymer flow (of bond type 1) could be computed with a fene potential
and bonds in the wall boundary (of bond type 2) could be computed with a harmonic potential. The
assignment of bond type to style is made via the bond_coeff command or in the data file.
In the bond_coeff commands, the name of a bond style must be added after the bond type, with the remaining
coefficients being those appropriate to that style. In the example above, the 2 bond_coeff commands set bonds
of bond type 1 to be computed with a harmonic potential with coefficients 80.0, 1.2 for K, r0. All other bond
types (2-N) are computed with a fene potential with coefficients 30.0, 1.5, 1.0, 1.0 for K, R0, epsilon, sigma.
If bond coefficients are specified in the data file read via the read_data command, then the same rule applies.
E.g. "harmonic" or "fene" must be added after the bond type, for each line in the "Bond Coeffs" section, e.g.
Bond Coeffs
1 harmonic 80.0 1.2
2 fene 30.0 1.5 1.0 1.0
...
A bond style of none with no additional coefficients can be used in place of a bond style, either in a input
script bond_coeff command or in the data file, if you desire to turn off interactions for specific bond types.
Restrictions:
This bond style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Unlike other bond styles, the hybrid bond style does not store bond coefficient info for individual sub-styles in
a binary restart files. Thus when restarting a simulation from a restart file, you need to re-specify bond_coeff
commands.
Related commands:
bond_coeff, delete_bonds
LAMMPS Users Manual
1725
Default: none
LAMMPS Users Manual
1726

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style morse command
bond_style morse/omp command
Syntax:
bond_style morse
Examples:
bond_style morse
bond_coeff 5 1.0 2.0 1.2
Description:
The morse bond style uses the potential
where r0 is the equilibrium bond distance, alpha is a stiffness parameter, and D determines the depth of the
potential well.
The following coefficients must be defined for each bond type via the bond_coeff command as in the example
above, or in the data file or restart files read by the read_data or read_restart commands:
D (energy)• alpha (inverse distance)• r0 (distance)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
1727

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style none command
Syntax:
bond_style none
Examples:
bond_style none
Description:
Using a bond style of none means bond forces and energies are not computed, even if pairs of bonded atoms
were listed in the data file read by the read_data command.
See the bond_style zero command for a way to calculate bond statistics, but compute no bond interactions.
Restrictions: none
Related commands: none
bond_style zero
Default: none
LAMMPS Users Manual
1729

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style nonlinear command
bond_style nonlinear/omp command
Syntax:
bond_style nonlinear
Examples:
bond_style nonlinear
bond_coeff 2 100.0 1.1 1.4
Description:
The nonlinear bond style uses the potential
to define an anharmonic spring (Rector) of equilibrium length r0 and maximum extension lamda.
The following coefficients must be defined for each bond type via the bond_coeff command as in the example
above, or in the data file or restart files read by the read_data or read_restart commands:
epsilon (energy)• r0 (distance)• lamda (distance)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
1730

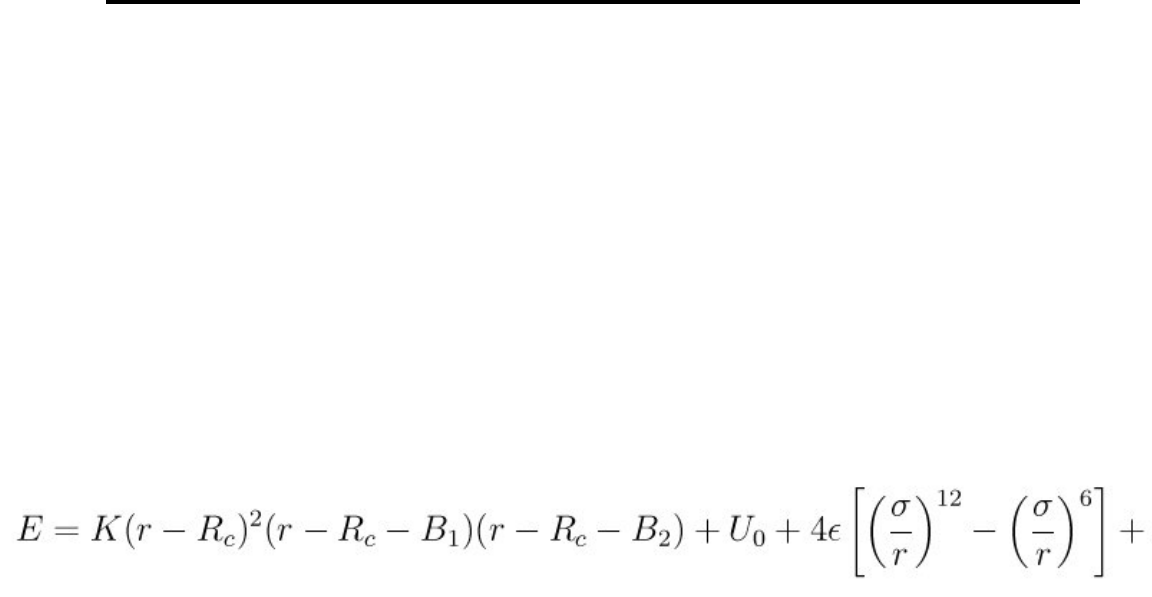
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style quartic command
bond_style quartic/omp command
Syntax:
bond_style quartic
Examples:
bond_style quartic
bond_coeff 2 1200 -0.55 0.25 1.3 34.6878
Description:
The quartic bond style uses the potential
to define a bond that can be broken as the simulation proceeds (e.g. due to a polymer being stretched). The
sigma and epsilon used in the LJ portion of the formula are both set equal to 1.0 by LAMMPS.
The following coefficients must be defined for each bond type via the bond_coeff command as in the example
above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy/distance^4)• B1 (distance)• B2 (distance)• Rc (distance)• U0 (energy)•
This potential was constructed to mimic the FENE bond potential for coarse-grained polymer chains. When
monomers with sigma = epsilon = 1.0 are used, the following choice of parameters gives a quartic potential
that looks nearly like the FENE potential: K = 1200, B1 = -0.55, B2 = 0.25, Rc = 1.3, and U0 = 34.6878.
Different parameters can be specified using the bond_coeff command, but you will need to choose them
carefully so they form a suitable bond potential.
Rc is the cutoff length at which the bond potential goes smoothly to a local maximum. If a bond length ever
becomes > Rc, LAMMPS "breaks" the bond, which means two things. First, the bond potential is turned off
by setting its type to 0, and is no longer computed. Second, a pairwise interaction between the two atoms is
turned on, since they are no longer bonded.
LAMMPS does the second task via a computational sleight-of-hand. It subtracts the pairwise interaction as
part of the bond computation. When the bond breaks, the subtraction stops. For this to work, the pairwise
interaction must always be computed by the pair_style command, whether the bond is broken or not. This
LAMMPS Users Manual
1732

means that special_bonds must be set to 1,1,1, as indicated as a restriction below.
Note that when bonds are dumped to a file via the dump local command, bonds with type 0 are not included.
The delete_bonds command can also be used to query the status of broken bonds or permanently delete them,
e.g.:
delete_bonds all stats
delete_bonds all bond 0 remove
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This bond style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
The quartic style requires that special_bonds parameters be set to 1,1,1. Three- and four-body interactions
(angle, dihedral, etc) cannot be used with quartic bonds.
Related commands:
bond_coeff, delete_bonds
Default: none
LAMMPS Users Manual
1733

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style table command
bond_style table/omp command
Syntax:
bond_style table style N
style = linear or spline = method of interpolation• N = use N values in table•
Examples:
bond_style table linear 1000
bond_coeff 1 file.table ENTRY1
Description:
Style table creates interpolation tables of length N from bond potential and force values listed in a file(s) as a
function of bond length. The files are read by the bond_coeff command.
The interpolation tables are created by fitting cubic splines to the file values and interpolating energy and
force values at each of N distances. During a simulation, these tables are used to interpolate energy and force
values as needed. The interpolation is done in one of 2 styles: linear or spline.
For the linear style, the bond length is used to find 2 surrounding table values from which an energy or force
is computed by linear interpolation.
For the spline style, a cubic spline coefficients are computed and stored at each of the N values in the table.
The bond length is used to find the appropriate set of coefficients which are used to evaluate a cubic
polynomial which computes the energy or force.
The following coefficients must be defined for each bond type via the bond_coeff command as in the example
above.
filename• keyword•
The filename specifies a file containing tabulated energy and force values. The keyword specifies a section of
the file. The format of this file is described below.
The format of a tabulated file is as follows (without the parenthesized comments):
# Bond potential for harmonic (one or more comment or blank lines)
HAM (keyword is the first text on line)
N 101 FP 0 0 EQ 0.5 (N, FP, EQ parameters)
(blank line)
1 0.00 338.0000 1352.0000 (index, bond-length, energy, force)
2 0.01 324.6152 1324.9600
LAMMPS Users Manual
1734

...
101 1.00 338.0000 -1352.0000
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections. The first line begins with a keyword which identifies the section.
The line can contain additional text, but the initial text must match the argument specified in the bond_coeff
command. The next line lists (in any order) one or more parameters for the table. Each parameter is a keyword
followed by one or more numeric values.
The parameter "N" is required and its value is the number of table entries that follow. Note that this may be
different than the N specified in the bond_style table command. Let Ntable = N in the bond_style command,
and Nfile = "N" in the tabulated file. What LAMMPS does is a preliminary interpolation by creating splines
using the Nfile tabulated values as nodal points. It uses these to interpolate as needed to generate energy and
force values at Ntable different points. The resulting tables of length Ntable are then used as described above,
when computing energy and force for individual bond lengths. This means that if you want the interpolation
tables of length Ntable to match exactly what is in the tabulated file (with effectively no preliminary
interpolation), you should set Ntable = Nfile.
The "FP" parameter is optional. If used, it is followed by two values fplo and fphi, which are the derivatives
of the force at the innermost and outermost bond lengths. These values are needed by the spline construction
routines. If not specified by the "FP" parameter, they are estimated (less accurately) by the first two and last
two force values in the table.
The "EQ" parameter is also optional. If used, it is followed by a the equilibrium bond length, which is used,
for example, by the fix shake command. If not used, the equilibrium bond length is to the distance in the table
with the lowest potential energy.
Following a blank line, the next N lines list the tabulated values. On each line, the 1st value is the index from
1 to N, the 2nd value is the bond length r (in distance units), the 3rd value is the energy (in energy units), and
the 4th is the force (in force units). The bond lengths must range from a LO value to a HI value, and increase
from one line to the next. If the actual bond length is ever smaller than the LO value or larger than the HI
value, then the bond energy and force is evaluated as if the bond were the LO or HI length.
Note that one file can contain many sections, each with a tabulated potential. LAMMPS reads the file section
by section until it finds one that matches the specified keyword.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
LAMMPS Users Manual
1735

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
bond_style zero command
Syntax:
bond_style zero nocoeff
Examples:
bond_style zero
bond_style zero nocoeff
bond_coeff *
bond_coeff * 2.14
Description:
Using an bond style of zero means bond forces and energies are not computed, but the geometry of bond pairs
is still accessible to other commands.
As an example, the compute bond/local command can be used to compute distances for the list of pairs of
bond atoms listed in the data file read by the read_data command. If no bond style is defined, this command
cannot be used.
The optional nocoeff flag allows to read data files with a BondCoeff section for any bond style. Similarly, any
bond_coeff commands will only be checked for the bond type number and the rest ignored.
Note that the bond_coeff command must be used for all bond types. If specified, there can be only one value,
which is going to be used to assign an equilibrium distance, e.g. for use with fix shake.
Restrictions: none
Related commands:
bond_style none
Default: none
LAMMPS Users Manual
1737
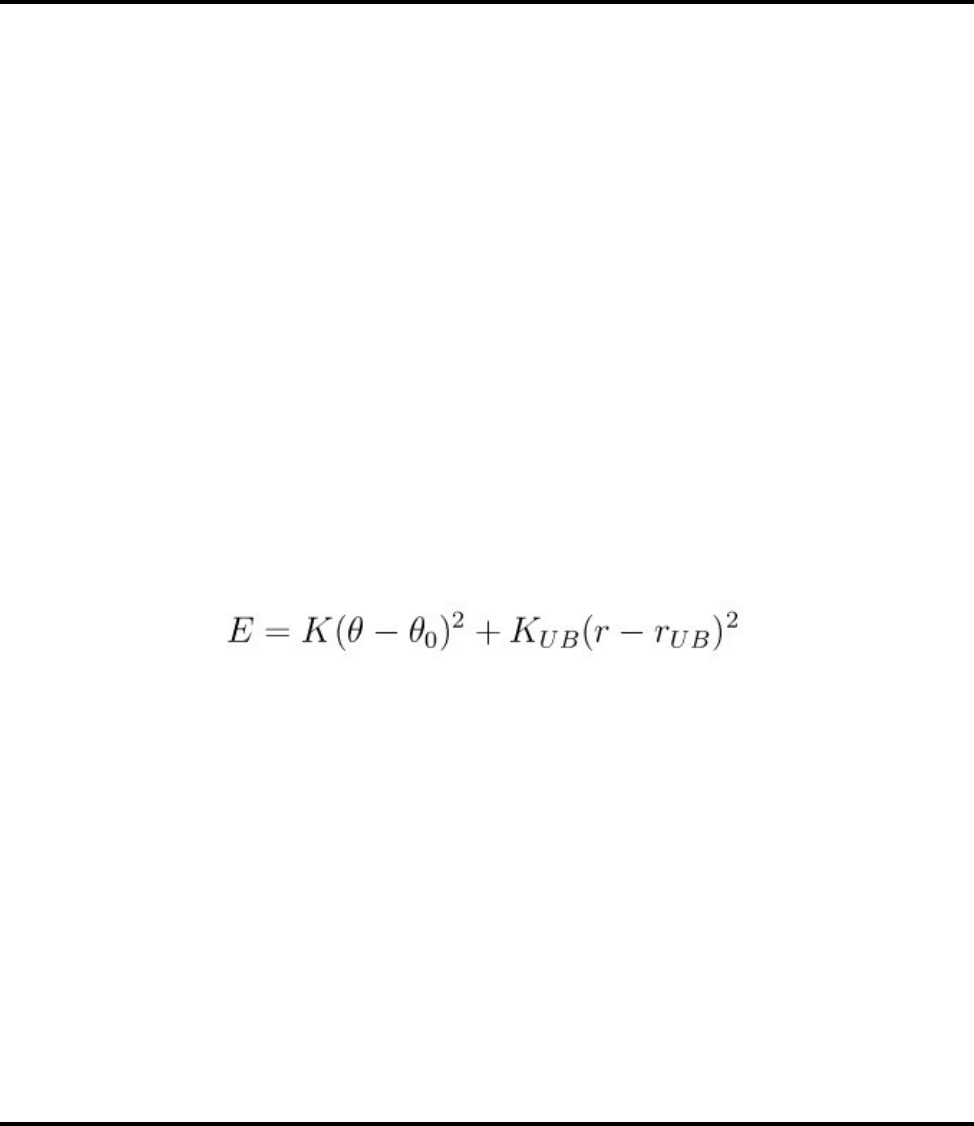
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style charmm command
angle_style charmm/intel command
angle_style charmm/kk command
angle_style charmm/omp command
Syntax:
angle_style charmm
Examples:
angle_style charmm
angle_coeff 1 300.0 107.0 50.0 3.0
Description:
The charmm angle style uses the potential
with an additional Urey_Bradley term based on the distance r between the 1st and 3rd atoms in the angle. K,
theta0, Kub, and Rub are coefficients defined for each angle type.
See (MacKerell) for a description of the CHARMM force field.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy/radian^2)• theta0 (degrees)• K_ub (energy/distance^2)• r_ub (distance)•
Theta0 is specified in degrees, but LAMMPS converts it to radians internally; hence the units of K are in
energy/radian^2.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
LAMMPS Users Manual
1738

These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Related commands:
angle_coeff
Default: none
(MacKerell) MacKerell, Bashford, Bellott, Dunbrack, Evanseck, Field, Fischer, Gao, Guo, Ha, et al, J Phys
Chem, 102, 3586 (1998).
LAMMPS Users Manual
1739

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style class2 command
angle_style class2/omp command
angle_style class2/kk command
Syntax:
angle_style class2
Examples:
angle_style class2
angle_coeff * 75.0
angle_coeff 1 bb 10.5872 1.0119 1.5228
angle_coeff * ba 3.6551 24.895 1.0119 1.5228
Description:
The class2 angle style uses the potential
where Ea is the angle term, Ebb is a bond-bond term, and Eba is a bond-angle term. Theta0 is the equilibrium
angle and r1 and r2 are the equilibrium bond lengths.
See (Sun) for a description of the COMPASS class2 force field.
Coefficients for the Ea, Ebb, and Eba formulas must be defined for each angle type via the angle_coeff
command as in the example above, or in the data file or restart files read by the read_data or read_restart
commands.
These are the 4 coefficients for the Ea formula:
theta0 (degrees)• K2 (energy/radian^2)• K3 (energy/radian^3)• K4 (energy/radian^4)•
LAMMPS Users Manual
1740

Theta0 is specified in degrees, but LAMMPS converts it to radians internally; hence the units of the various K
are in per-radian.
For the Ebb formula, each line in a angle_coeff command in the input script lists 4 coefficients, the first of
which is "bb" to indicate they are BondBond coefficients. In a data file, these coefficients should be listed
under a "BondBond Coeffs" heading and you must leave out the "bb", i.e. only list 3 coefficients after the
angle type.
bb• M (energy/distance^2)• r1 (distance)• r2 (distance)•
For the Eba formula, each line in a angle_coeff command in the input script lists 5 coefficients, the first of
which is "ba" to indicate they are BondAngle coefficients. In a data file, these coefficients should be listed
under a "BondAngle Coeffs" heading and you must leave out the "ba", i.e. only list 4 coefficients after the
angle type.
ba• N1 (energy/distance^2)• N2 (energy/distance^2)• r1 (distance)• r2 (distance)•
The theta0 value in the Eba formula is not specified, since it is the same value from the Ea formula.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the CLASS2 package. See the Making
LAMMPS section for more info on packages.
Related commands:
angle_coeff
LAMMPS Users Manual
1741

Default: none
(Sun) Sun, J Phys Chem B 102, 7338-7364 (1998).
LAMMPS Users Manual
1742

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style cosine command
angle_style cosine/omp command
Syntax:
angle_style cosine
Examples:
angle_style cosine
angle_coeff * 75.0
Description:
The cosine angle style uses the potential
where K is defined for each angle type.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
LAMMPS Users Manual
1743

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style cosine/delta command
angle_style cosine/delta/omp command
Syntax:
angle_style cosine/delta
Examples:
angle_style cosine/delta
angle_coeff 2*4 75.0 100.0
Description:
The cosine/delta angle style uses the potential
where theta0 is the equilibrium value of the angle, and K is a prefactor. Note that the usual 1/2 factor is
included in K.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy)• theta0 (degrees)•
Theta0 is specified in degrees, but LAMMPS converts it to radians internally.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
1745
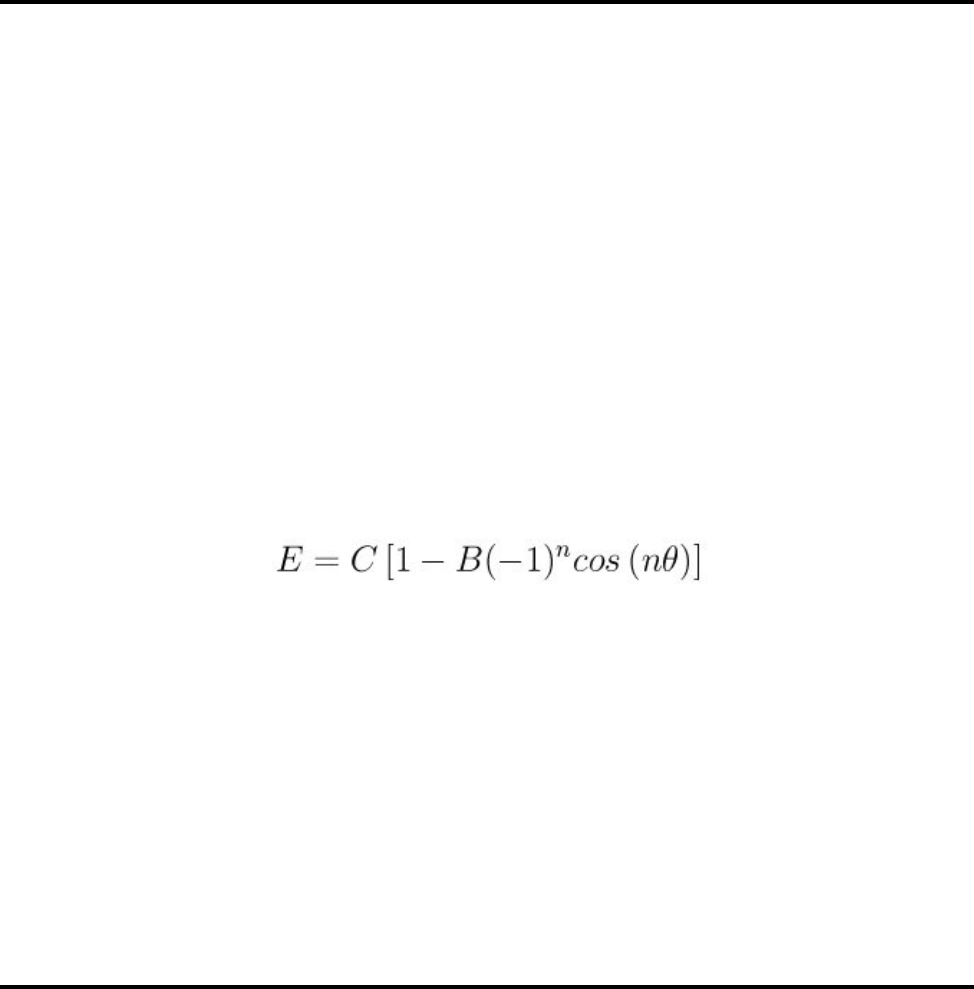
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style cosine/periodic command
angle_style cosine/periodic/omp command
Syntax:
angle_style cosine/periodic
Examples:
angle_style cosine/periodic
angle_coeff * 75.0 1 6
Description:
The cosine/periodic angle style uses the following potential, which is commonly used in the DREIDING force
field, particularly for organometallic systems where n = 4 might be used for an octahedral complex and n = 3
might be used for a trigonal center:
where C, B and n are coefficients defined for each angle type.
See (Mayo) for a description of the DREIDING force field
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
C (energy)• B = 1 or -1• n = 1, 2, 3, 4, 5 or 6 for periodicity•
Note that the prefactor C is specified and not the overall force constant K = C / n^2. When B = 1, it leads to a
minimum for the linear geometry. When B = -1, it leads to a maximum for the linear geometry.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
LAMMPS Users Manual
1747

input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Related commands:
angle_coeff
Default: none
(Mayo) Mayo, Olfason, Goddard III, J Phys Chem, 94, 8897-8909 (1990).
LAMMPS Users Manual
1748

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style cosine/shift command
angle_style cosine/shift/omp command
Syntax:
angle_style cosine/shift
Examples:
angle_style cosine/shift
angle_coeff * 10.0 45.0
Description:
The cosine/shift angle style uses the potential
where theta0 is the equilibrium angle. The potential is bounded between -Umin and zero. In the neighborhood
of the minimum E=- Umin + Umin/4(theta-theta0)^2 hence the spring constant is umin/2.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
umin (energy)• theta (angle)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
1749
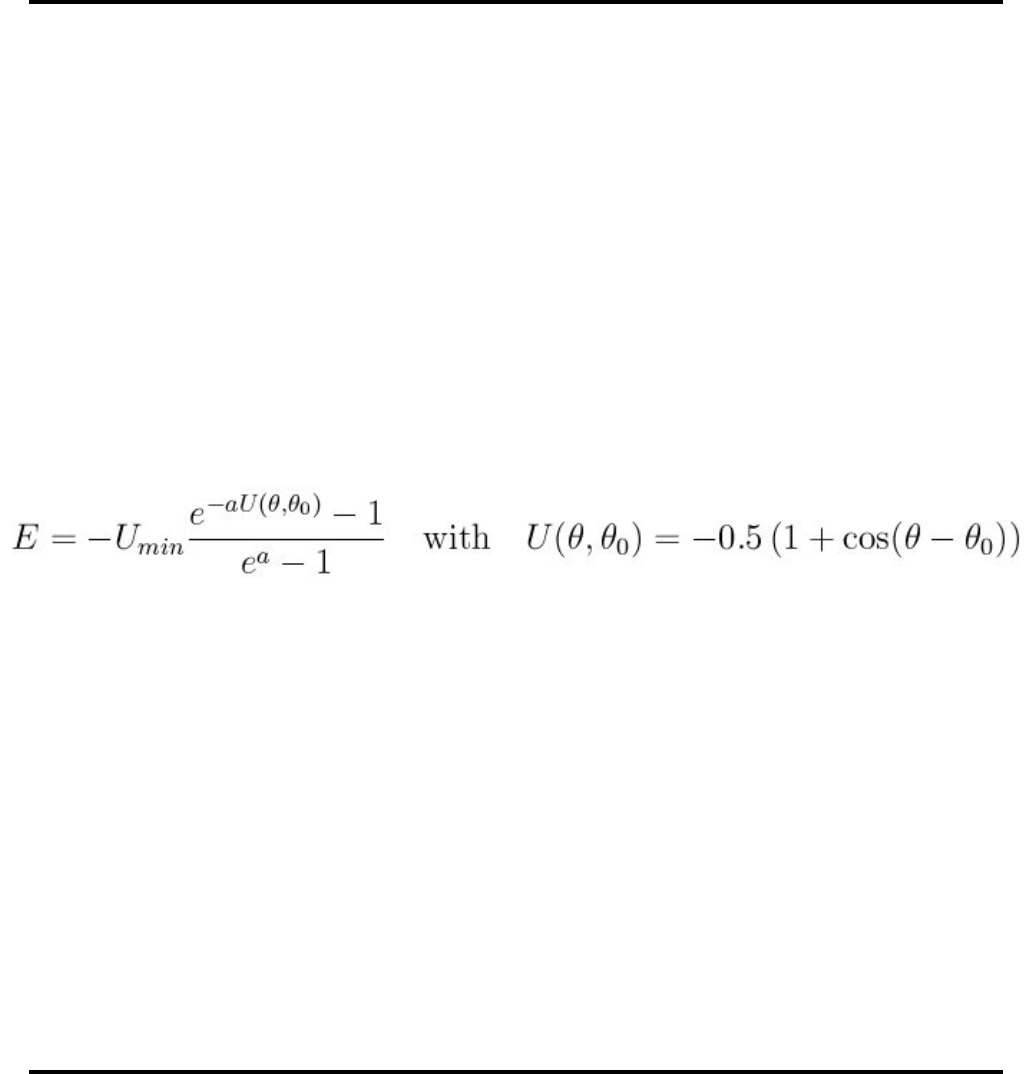
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style cosine/shift/exp command
angle_style cosine/shift/exp/omp command
Syntax:
angle_style cosine/shift/exp
Examples:
angle_style cosine/shift/exp
angle_coeff * 10.0 45.0 2.0
Description:
The cosine/shift/exp angle style uses the potential
where Umin, theta, and a are defined for each angle type.
The potential is bounded between [-Umin:0] and the minimum is located at the angle theta0. The a parameter
can be both positive or negative and is used to control the spring constant at the equilibrium.
The spring constant is given by k = A exp(A) Umin / [2 (Exp(a)-1)]. For a > 3, k/Umin = a/2 to better than 5%
relative error. For negative values of the a parameter, the spring constant is essentially zero, and anharmonic
terms takes over. The potential is furthermore well behaved in the limit a -> 0, where it has been implemented
to linear order in a for a < 0.001. In this limit the potential reduces to the cosineshifted potential.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
umin (energy)• theta (angle)• A (real number)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
LAMMPS Users Manual
1751

You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info on packages.
Related commands:
angle_coeff, angle_cosine_shift, dihedral_cosine_shift_exp
Default: none
LAMMPS Users Manual
1752
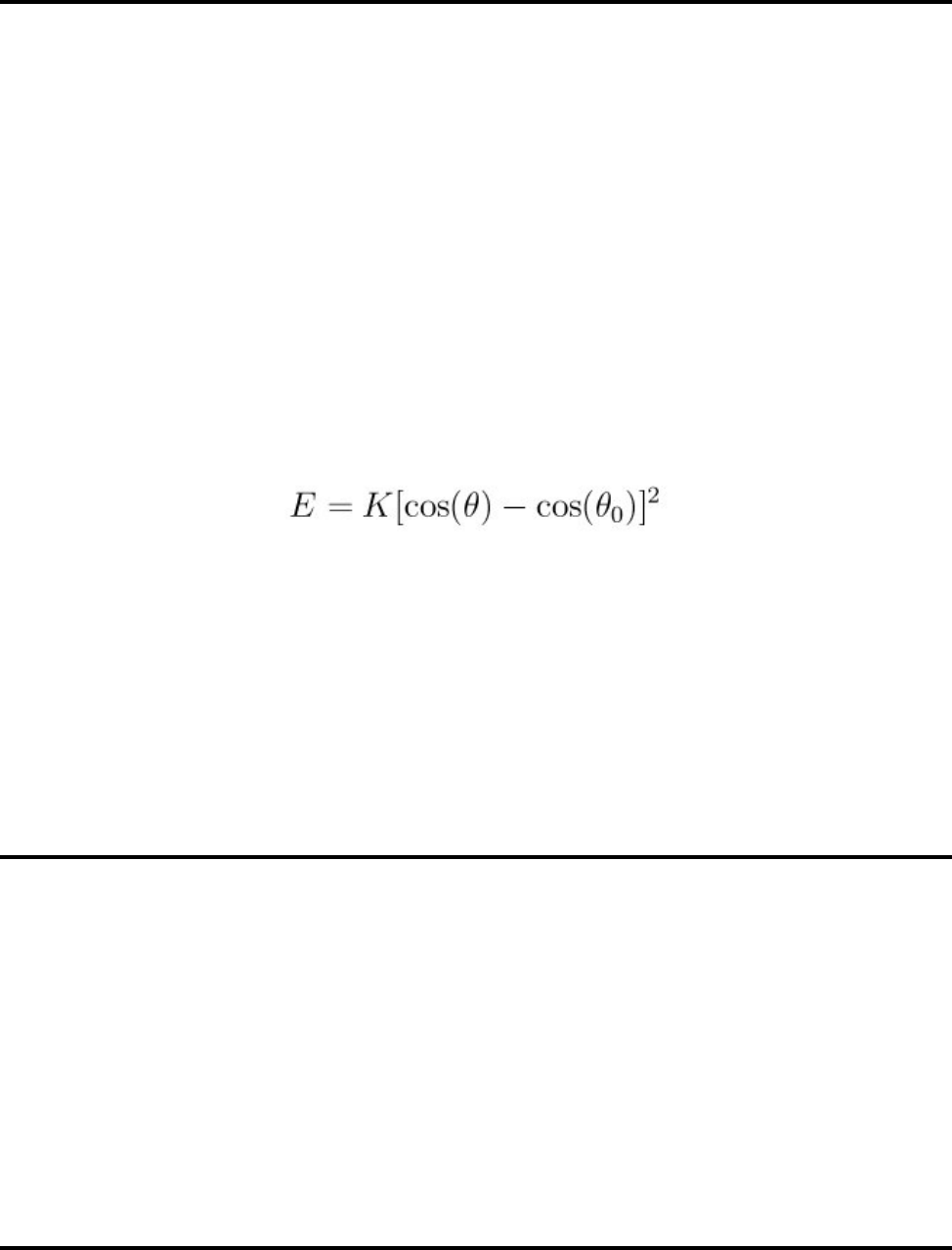
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style cosine/squared command
angle_style cosine/squared/omp command
Syntax:
angle_style cosine/squared
Examples:
angle_style cosine/squared
angle_coeff 2*4 75.0 100.0
Description:
The cosine/squared angle style uses the potential
where theta0 is the equilibrium value of the angle, and K is a prefactor. Note that the usual 1/2 factor is
included in K.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy)• theta0 (degrees)•
Theta0 is specified in degrees, but LAMMPS converts it to radians internally.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
LAMMPS Users Manual
1753
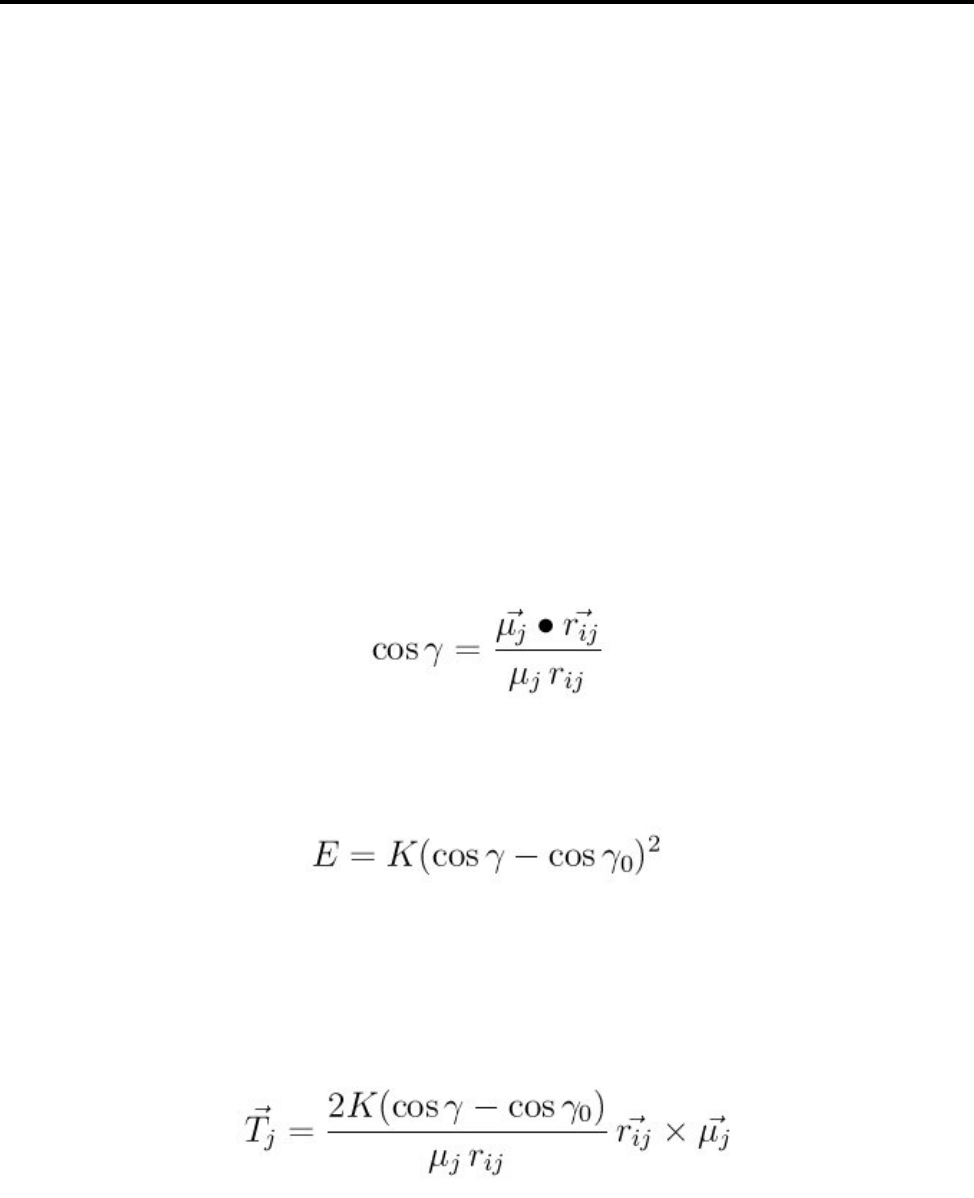
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style dipole command
angle_style dipole/omp command
Syntax:
angle_style dipole
Examples:
angle_style dipole
angle_coeff 6 2.1 180.0
Description:
The dipole angle style is used to control the orientation of a dipolar atom within a molecule (Orsi).
Specifically, the dipole angle style restrains the orientation of a point dipole mu_j (embedded in atom 'j') with
respect to a reference (bond) vector r_ij = r_i - r_j, where 'i' is another atom of the same molecule (typically, 'i'
and 'j' are also covalently bonded).
It is convenient to define an angle gamma between the 'free' vector mu_j and the reference (bond) vector r_ij:
The dipole angle style uses the potential:
where K is a rigidity constant and gamma0 is an equilibrium (reference) angle.
The torque on the dipole can be obtained by differentiating the potential using the 'chain rule' as in appendix
C.3 of (Allen):
Example: if gamma0 is set to 0 degrees, the torque generated by the potential will tend to align the dipole
along the reference direction defined by the (bond) vector r_ij (in other words, mu_j is restrained to point
towards atom 'i').
LAMMPS Users Manual
1755
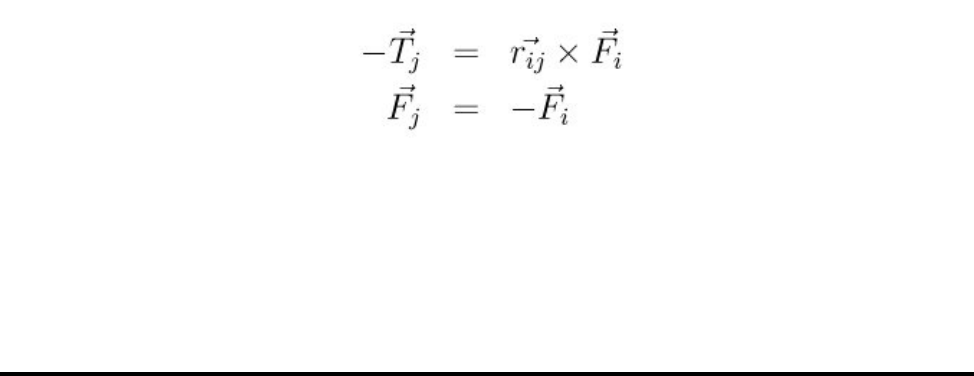
The dipolar torque T_j must be counterbalanced in order to conserve the local angular momentum. This is
achieved via an additional force couple generating a torque equivalent to the opposite of T_j:
where F_i and F_j are applied on atoms i and j, respectively.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy)• gamma0 (degrees)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info on packages.
NOTE: In the "Angles" section of the data file, the atom ID 'j' corresponding to the dipole to restrain must
come before the atom ID of the reference atom 'i'. A third atom ID 'k' must also be provided, although 'k' is
just a 'dummy' atom which can be any atom; it may be useful to choose a convention (e.g., 'k'='i') and adhere
to it. For example, if ID=1 for the dipolar atom to restrain, and ID=2 for the reference atom, the corresponding
line in the "Angles" section of the data file would read: X X 1 2 2
The "newton" command for intramolecular interactions must be "on" (which is the default).
This angle style should not be used with SHAKE.
Related commands:
angle_coeff, angle_hybrid
LAMMPS Users Manual
1756

Default: none
(Orsi) Orsi & Essex, The ELBA force field for coarse-grain modeling of lipid membranes, PloS ONE 6(12):
e28637, 2011.
(Allen) Allen & Tildesley, Computer Simulation of Liquids, Clarendon Press, Oxford, 1987.
LAMMPS Users Manual
1757
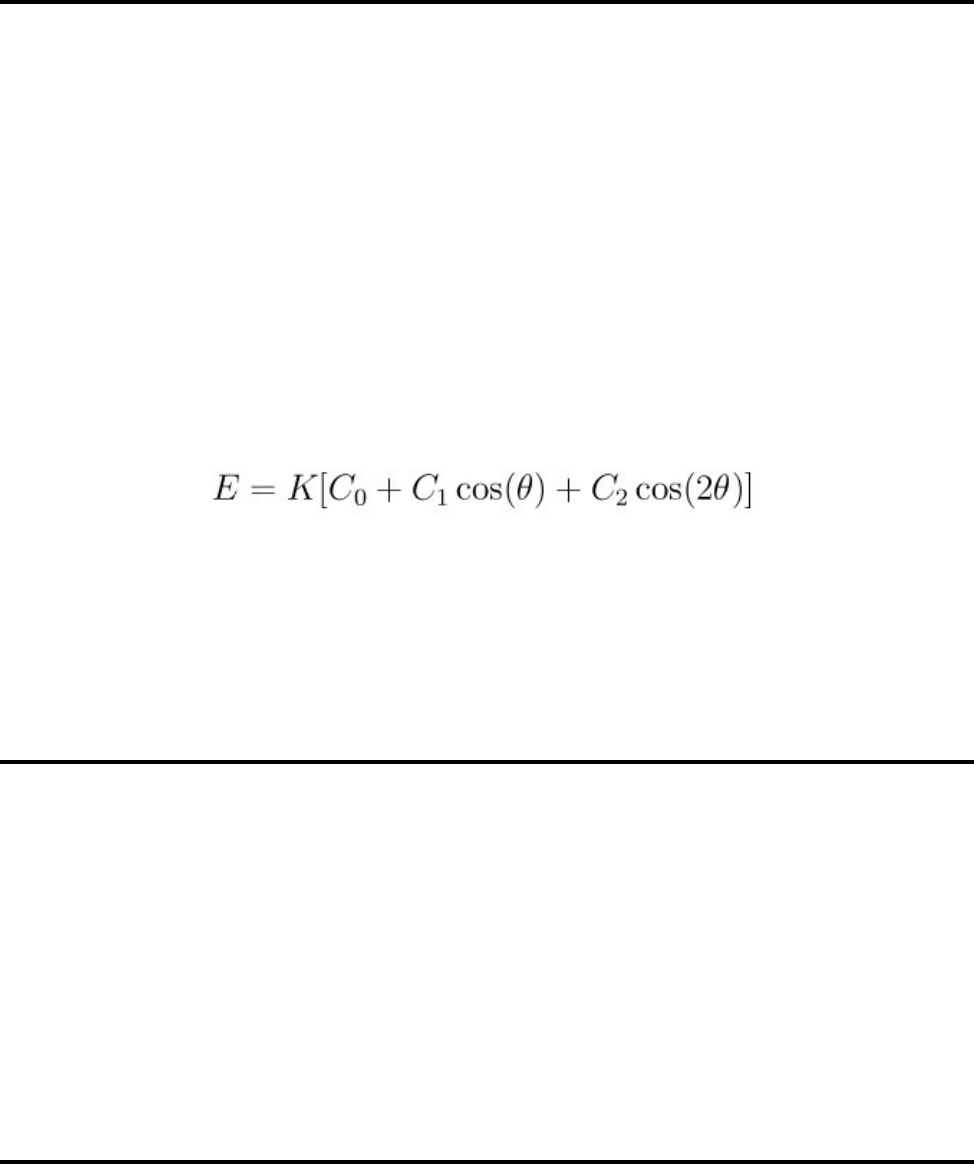
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style fourier command
angle_style fourier/omp command
Syntax:
angle_style fourier
Examples:
angle_style fourier angle_coeff 75.0 1.0 1.0 1.0
Description:
The fourier angle style uses the potential
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy)• C0 (real)• C1 (real)• C2 (real)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the USER_MISC package. See the Making
LAMMPS section for more info on packages.
LAMMPS Users Manual
1758

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style fourier/simple command
angle_style fourier/simple/omp command
Syntax:
angle_style fourier/simple
Examples:
angle_style fourier/simple angle_coeff 100.0 -1.0 1.0
Description:
The fourier/simple angle style uses the potential
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy)• c (real)• n (real)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the USER_MISC package. See the Making
LAMMPS section for more info on packages.
LAMMPS Users Manual
1760
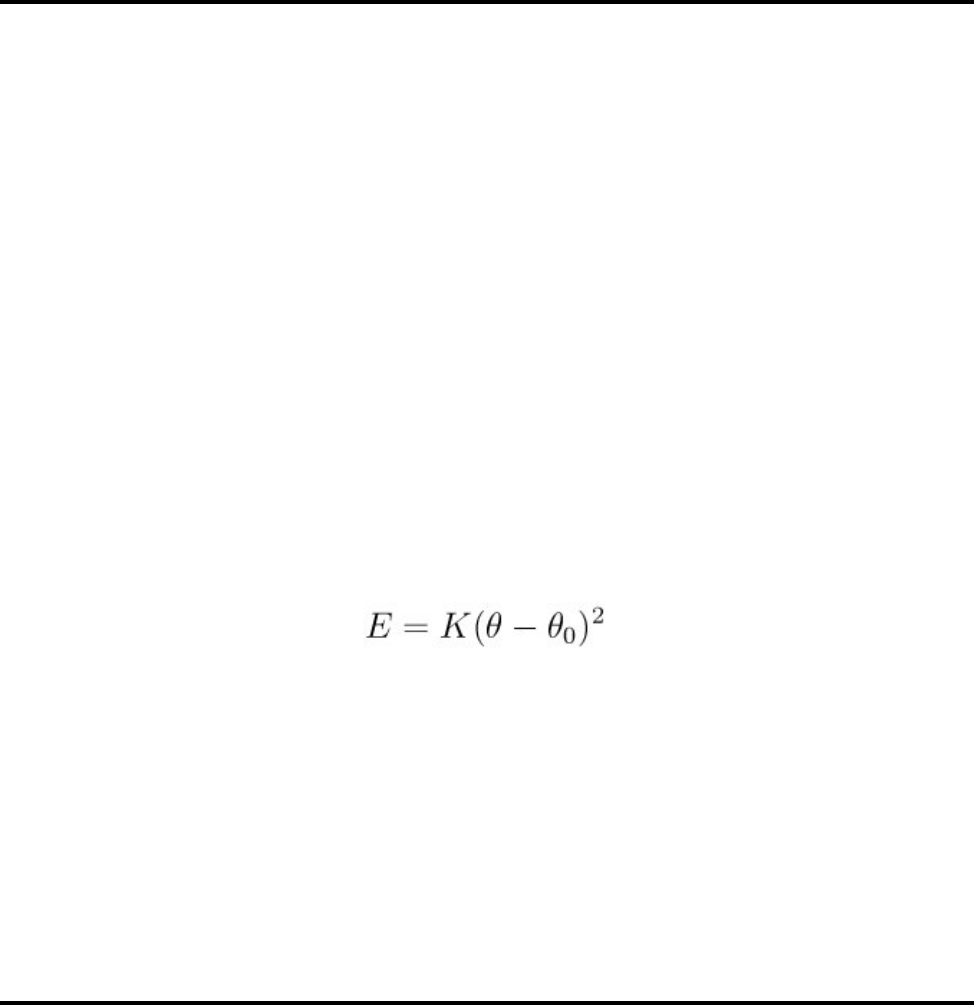
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style harmonic command
angle_style harmonic/intel command
angle_style harmonic/kk command
angle_style harmonic/omp command
Syntax:
angle_style harmonic
Examples:
angle_style harmonic
angle_coeff 1 300.0 107.0
Description:
The harmonic angle style uses the potential
where theta0 is the equilibrium value of the angle, and K is a prefactor. Note that the usual 1/2 factor is
included in K.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy/radian^2)• theta0 (degrees)•
Theta0 is specified in degrees, but LAMMPS converts it to radians internally; hence the units of K are in
energy/radian^2.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
LAMMPS Users Manual
1762

input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Related commands:
angle_coeff
Default: none
LAMMPS Users Manual
1763

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style hybrid command
Syntax:
angle_style hybrid style1 style2 ...
style1,style2 = list of one or more angle styles•
Examples:
angle_style hybrid harmonic cosine
angle_coeff 1 harmonic 80.0 30.0
angle_coeff 2* cosine 50.0
Description:
The hybrid style enables the use of multiple angle styles in one simulation. An angle style is assigned to each
angle type. For example, angles in a polymer flow (of angle type 1) could be computed with a harmonic
potential and angles in the wall boundary (of angle type 2) could be computed with a cosine potential. The
assignment of angle type to style is made via the angle_coeff command or in the data file.
In the angle_coeff commands, the name of an angle style must be added after the angle type, with the
remaining coefficients being those appropriate to that style. In the example above, the 2 angle_coeff
commands set angles of angle type 1 to be computed with a harmonic potential with coefficients 80.0, 30.0
for K, theta0. All other angle types (2-N) are computed with a cosine potential with coefficient 50.0 for K.
If angle coefficients are specified in the data file read via the read_data command, then the same rule applies.
E.g. "harmonic" or "cosine", must be added after the angle type, for each line in the "Angle Coeffs" section,
e.g.
Angle Coeffs
1 harmonic 80.0 30.0
2 cosine 50.0
...
If class2 is one of the angle hybrid styles, the same rule holds for specifying additional BondBond (and
BondAngle) coefficients either via the input script or in the data file. I.e. class2 must be added to each line
after the angle type. For lines in the BondBond (or BondAngle) section of the data file for angle types that are
not class2, you must use an angle style of skip as a placeholder, e.g.
BondBond Coeffs
1 skip
2 class2 3.6512 1.0119 1.0119
...
Note that it is not necessary to use the angle style skip in the input script, since BondBond (or BondAngle)
coefficients need not be specified at all for angle types that are not class2.
LAMMPS Users Manual
1764

An angle style of none with no additional coefficients can be used in place of an angle style, either in a input
script angle_coeff command or in the data file, if you desire to turn off interactions for specific angle types.
Restrictions:
This angle style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Unlike other angle styles, the hybrid angle style does not store angle coefficient info for individual sub-styles
in a binary restart files. Thus when restarting a simulation from a restart file, you need to re-specify
angle_coeff commands.
Related commands:
angle_coeff
Default: none
LAMMPS Users Manual
1765

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style none command
Syntax:
angle_style none
Examples:
angle_style none
Description:
Using an angle style of none means angle forces and energies are not computed, even if triplets of angle atoms
were listed in the data file read by the read_data command.
See the angle_style zero command for a way to calculate angle statistics, but compute no angle interactions.
Restrictions: none
Related commands:
angle_style zero
Default: none
LAMMPS Users Manual
1766
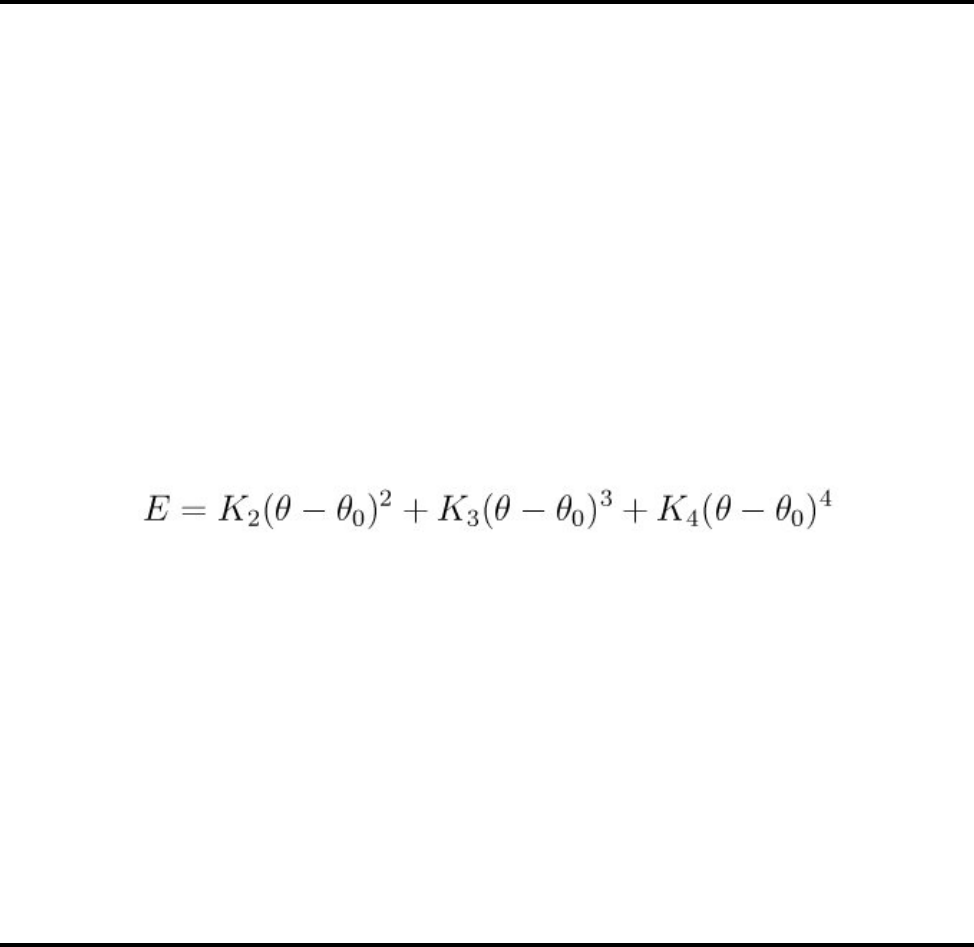
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style quartic command
angle_style quartic/omp command
Syntax:
angle_style quartic
Examples:
angle_style quartic
angle_coeff 1 129.1948 56.8726 -25.9442 -14.2221
Description:
The quartic angle style uses the potential
where theta0 is the equilibrium value of the angle, and K is a prefactor. Note that the usual 1/2 factor is
included in K.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
theta0 (degrees)• K2 (energy/radian^2)• K3 (energy/radian^3)• K4 (energy/radian^4)•
Theta0 is specified in degrees, but LAMMPS converts it to radians internally; hence the units of K are in
energy/radian^2.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
LAMMPS Users Manual
1767

See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the USER_MISC package. See the Making
LAMMPS section for more info on packages.
Related commands:
angle_coeff
Default: none
LAMMPS Users Manual
1768
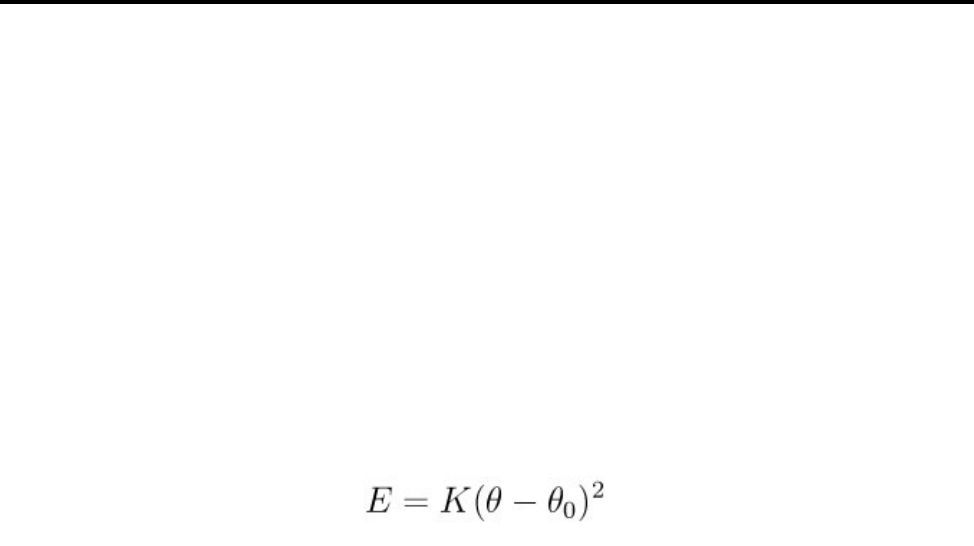
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style sdk command
Syntax:
angle_style sdk
angle_style sdk/omp
Examples:
angle_style sdk
angle_coeff 1 300.0 107.0
Description:
The sdk angle style is a combination of the harmonic angle potential,
where theta0 is the equilibrium value of the angle and K a prefactor, with the repulsive part of the non-bonded
lj/sdk pair style between the atoms 1 and 3. This angle potential is intended for coarse grained MD simulations
with the CMM parametrization using the pair_style lj/sdk. Relative to the pair_style lj/sdk, however, the
energy is shifted by epsilon, to avoid sudden jumps. Note that the usual 1/2 factor is included in K.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above:
K (energy/radian^2)• theta0 (degrees)•
Theta0 is specified in degrees, but LAMMPS converts it to radians internally; hence the units of K are in
energy/radian^2. The also required lj/sdk parameters will be extracted automatically from the pair_style.
Restrictions:
This angle style can only be used if LAMMPS was built with the USER-CG-CMM package. See the Making
LAMMPS section for more info on packages.
Related commands:
angle_coeff, angle_style harmonic, pair_style lj/sdk, pair_style lj/sdk/coul/long
Default: none
LAMMPS Users Manual
1769

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style table command
angle_style table/omp command
Syntax:
angle_style table style N
style = linear or spline = method of interpolation• N = use N values in table•
Examples:
angle_style table linear 1000
angle_coeff 3 file.table ENTRY1
Description:
Style table creates interpolation tables of length N from angle potential and derivative values listed in a file(s)
as a function of angle The files are read by the angle_coeff command.
The interpolation tables are created by fitting cubic splines to the file values and interpolating energy and
derivative values at each of N angles. During a simulation, these tables are used to interpolate energy and
force values on individual atoms as needed. The interpolation is done in one of 2 styles: linear or spline.
For the linear style, the angle is used to find 2 surrounding table values from which an energy or its derivative
is computed by linear interpolation.
For the spline style, a cubic spline coefficients are computed and stored at each of the N values in the table.
The angle is used to find the appropriate set of coefficients which are used to evaluate a cubic polynomial
which computes the energy or derivative.
The following coefficients must be defined for each angle type via the angle_coeff command as in the
example above.
filename• keyword•
The filename specifies a file containing tabulated energy and derivative values. The keyword specifies a
section of the file. The format of this file is described below.
The format of a tabulated file is as follows (without the parenthesized comments):
# Angle potential for harmonic (one or more comment or blank lines)
HAM (keyword is the first text on line)
N 181 FP 0 0 EQ 90.0 (N, FP, EQ parameters)
(blank line)
N 181 FP 0 0 (N, FP parameters)
1 0.0 200.5 2.5 (index, angle, energy, derivative)
LAMMPS Users Manual
1770

2 1.0 198.0 2.5
...
181 180.0 0.0 0.0
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections. The first line begins with a keyword which identifies the section.
The line can contain additional text, but the initial text must match the argument specified in the angle_coeff
command. The next line lists (in any order) one or more parameters for the table. Each parameter is a keyword
followed by one or more numeric values.
The parameter "N" is required and its value is the number of table entries that follow. Note that this may be
different than the N specified in the angle_style table command. Let Ntable = N in the angle_style command,
and Nfile = "N" in the tabulated file. What LAMMPS does is a preliminary interpolation by creating splines
using the Nfile tabulated values as nodal points. It uses these to interpolate as needed to generate energy and
derivative values at Ntable different points. The resulting tables of length Ntable are then used as described
above, when computing energy and force for individual angles and their atoms. This means that if you want
the interpolation tables of length Ntable to match exactly what is in the tabulated file (with effectively no
preliminary interpolation), you should set Ntable = Nfile.
The "FP" parameter is optional. If used, it is followed by two values fplo and fphi, which are the 2nd
derivatives at the innermost and outermost angle settings. These values are needed by the spline construction
routines. If not specified by the "FP" parameter, they are estimated (less accurately) by the first two and last
two derivative values in the table.
The "EQ" parameter is also optional. If used, it is followed by a the equilibrium angle value, which is used,
for example, by the fix shake command. If not used, the equilibrium angle is set to 180.0.
Following a blank line, the next N lines list the tabulated values. On each line, the 1st value is the index from
1 to N, the 2nd value is the angle value (in degrees), the 3rd value is the energy (in energy units), and the 4th
is -dE/d(theta) (also in energy units). The 3rd term is the energy of the 3-atom configuration for the specified
angle. The last term is the derivative of the energy with respect to the angle (in degrees, not radians). Thus the
units of the last term are still energy, not force. The angle values must increase from one line to the next. The
angle values must also begin with 0.0 and end with 180.0, i.e. span the full range of possible angles.
Note that one file can contain many sections, each with a tabulated potential. LAMMPS reads the file section
by section until it finds one that matches the specified keyword.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
LAMMPS Users Manual
1771


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
angle_style zero command
Syntax:
angle_style zero nocoeff
Examples:
angle_style zero
angle_style zero nocoeff
angle_coeff *
angle_coeff * 120.0
Description:
Using an angle style of zero means angle forces and energies are not computed, but the geometry of angle
triplets is still accessible to other commands.
As an example, the compute angle/local command can be used to compute the theta values for the list of
triplets of angle atoms listed in the data file read by the read_data command. If no angle style is defined, this
command cannot be used.
The optional nocoeff flag allows to read data files with AngleCoeff section for any angle style. Similarly, any
angle_coeff commands will only be checked for the angle type number and the rest ignored.
Note that the angle_coeff command must be used for all angle types. If specified, there can be only one value,
which is going to be used to assign an equilibrium angle, e.g. for use with fix shake.
Restrictions: none
Related commands:
angle_style none
Default: none
LAMMPS Users Manual
1773

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style charmm command
dihedral_style charmm/intel command
dihedral_style charmm/kk command
dihedral_style charmm/omp command
dihedral_style charmmfsh command
Syntax:
dihedral_style style
style = charmm or charmmfsh•
Examples:
dihedral_style charmm
dihedral_style charmmfsh
dihedral_coeff 1 0.2 1 180 1.0
dihedral_coeff 2 1.8 1 0 1.0
dihedral_coeff 1 3.1 2 180 0.5
Description:
The charmm and charmmfsh dihedral styles use the potential
See (MacKerell) for a description of the CHARMM force field. This dihedral style can also be used for the
AMBER force field (see comment on weighting factors below). See (Cornell) for a description of the
AMBER force field.
NOTE: The newer charmmfsh style was released in March 2017. We recommend it be used instead of the
older charmm style when running a simulation with the CHARMM force field and Coulomb cutoffs, via the
pair_style lj/charmmfsw/coul/charmmfsh command. Otherwise the older charmm style is fine to use. See the
discussion below and more details on the pair_style charmm doc page.
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy)• n (integer >= 0)• d (integer value of degrees)• weighting factor (1.0, 0.5, or 0.0)•
LAMMPS Users Manual
1774

The weighting factor is required to correct for double counting pairwise non-bonded Lennard-Jones
interactions in cyclic systems or when using the CHARMM dihedral style with non-CHARMM force fields.
With the CHARMM dihedral style, interactions between the 1st and 4th atoms in a dihedral are skipped
during the normal non-bonded force computation and instead evaluated as part of the dihedral using special
epsilon and sigma values specified with the pair_coeff command of pair styles that contain "lj/charmm" (e.g.
pair_style lj/charmm/coul/long) In 6-membered rings, the same 1-4 interaction would be computed twice
(once for the clockwise 1-4 pair in dihedral 1-2-3-4 and once in the counterclockwise dihedral 1-6-5-4) and
thus the weighting factor has to be 0.5 in this case. In 4-membered or 5-membered rings, the 1-4 dihedral also
is counted as a 1-2 or 1-3 interaction when going around the ring in the opposite direction and thus the
weighting factor is 0.0, as the 1-2 and 1-3 exclusions take precedence.
Note that this dihedral weighting factor is unrelated to the scaling factor specified by the special bonds
command which applies to all 1-4 interactions in the system. For CHARMM force fields, the special_bonds
1-4 interaction scaling factor should be set to 0.0. Since the corresponding 1-4 non-bonded interactions are
computed with the dihedral. This means that if any of the weighting factors defined as dihedral coefficients
(4th coeff above) are non-zero, then you must use a pair style with "lj/charmm" and set the special_bonds 1-4
scaling factor to 0.0 (which is the default). Otherwise 1-4 non-bonded interactions in dihedrals will be
computed twice.
For simulations using the CHARMM force field with a Coulomb cutoff, the difference between the charmm
and charmmfsh styles is in the computation of the 1-4 non-bond interactions, though only if the distance
between the two atoms is within the switching region of the pairwise potential defined by the corresponding
CHARMM pair style, i.e. within the outer cutoff specified for the pair style. The charmmfsh style should only
be used when using the pair_style lj/charmmfsw/coul/charmmfsh to make the Coulombic pairwise
calculations consistent. Use the charmm style with long-range Coulombics or the older pair_style
lj/charmm/coul/charmm command. See the discussion on the CHARMM pair_style doc page for details.
Note that for AMBER force fields, which use pair styles with "lj/cut", the special_bonds 1-4 scaling factor
should be set to the AMBER defaults (1/2 and 5/6) and all the dihedral weighting factors (4th coeff above)
must be set to 0.0. In this case, you can use any pair style you wish, since the dihedral does not need any
Lennard-Jones parameter information and will not compute any 1-4 non-bonded interactions. Likewise the
charmm or charmmfsh styles are identical in this case since no 1-4 non-bonded interactions are computed.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
1775

This dihedral style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Related commands:
dihedral_coeff
Default: none
(Cornell) Cornell, Cieplak, Bayly, Gould, Merz, Ferguson, Spellmeyer, Fox, Caldwell, Kollman, JACS 117,
5179-5197 (1995).
(MacKerell) MacKerell, Bashford, Bellott, Dunbrack, Evanseck, Field, Fischer, Gao, Guo, Ha, et al, J Phys
Chem B, 102, 3586 (1998).
LAMMPS Users Manual
1776
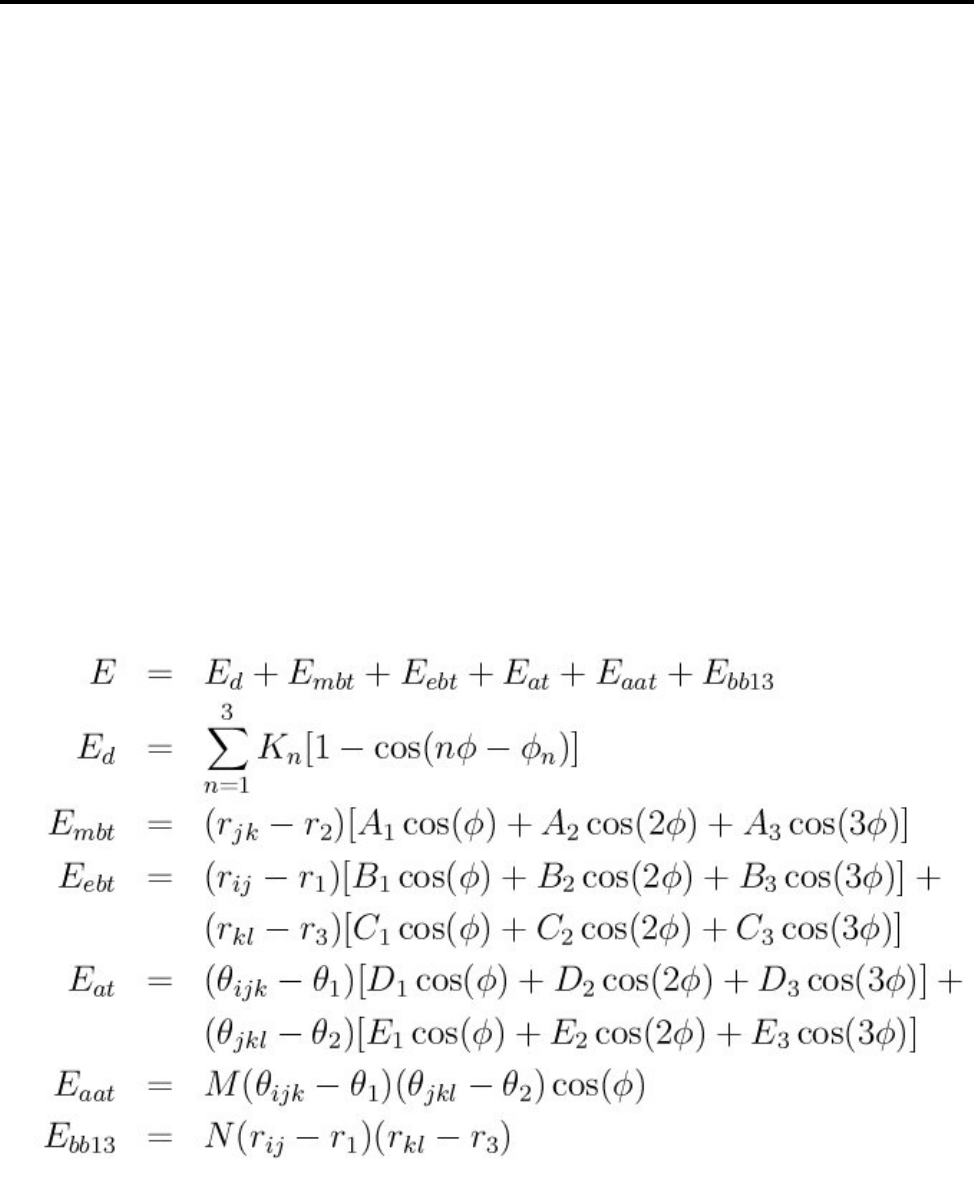
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style class2 command
dihedral_style class2/omp command
dihedral_style class2/kk command
Syntax:
dihedral_style class2
Examples:
dihedral_style class2
dihedral_coeff 1 100 75 100 70 80 60
dihedral_coeff * mbt 3.5945 0.1704 -0.5490 1.5228
dihedral_coeff * ebt 0.3417 0.3264 -0.9036 0.1368 0.0 -0.8080 1.0119 1.1010
dihedral_coeff 2 at 0.0 -0.1850 -0.7963 -2.0220 0.0 -0.3991 110.2453 105.1270
dihedral_coeff * aat -13.5271 110.2453 105.1270
dihedral_coeff * bb13 0.0 1.0119 1.1010
Description:
The class2 dihedral style uses the potential
where Ed is the dihedral term, Embt is a middle-bond-torsion term, Eebt is an end-bond-torsion term, Eat is an
angle-torsion term, Eaat is an angle-angle-torsion term, and Ebb13 is a bond-bond-13 term.
Theta1 and theta2 are equilibrium angles and r1 r2 r3 are equilibrium bond lengths.
LAMMPS Users Manual
1777
See (Sun) for a description of the COMPASS class2 force field.
Coefficients for the Ed, Embt, Eebt, Eat, Eaat, and Ebb13 formulas must be defined for each dihedral type via
the dihedral_coeff command as in the example above, or in the data file or restart files read by the read_data
or read_restart commands.
These are the 6 coefficients for the Ed formula:
K1 (energy)• phi1 (degrees)• K2 (energy)• phi2 (degrees)• K3 (energy)• phi3 (degrees)•
For the Embt formula, each line in a dihedral_coeff command in the input script lists 5 coefficients, the first of
which is "mbt" to indicate they are MiddleBondTorsion coefficients. In a data file, these coefficients should
be listed under a "MiddleBondTorsion Coeffs" heading and you must leave out the "mbt", i.e. only list 4
coefficients after the dihedral type.
mbt• A1 (energy/distance)• A2 (energy/distance)• A3 (energy/distance)• r2 (distance)•
For the Eebt formula, each line in a dihedral_coeff command in the input script lists 9 coefficients, the first of
which is "ebt" to indicate they are EndBondTorsion coefficients. In a data file, these coefficients should be
listed under a "EndBondTorsion Coeffs" heading and you must leave out the "ebt", i.e. only list 8 coefficients
after the dihedral type.
ebt• B1 (energy/distance)• B2 (energy/distance)• B3 (energy/distance)• C1 (energy/distance)• C2 (energy/distance)• C3 (energy/distance)• r1 (distance)• r3 (distance)•
For the Eat formula, each line in a dihedral_coeff command in the input script lists 9 coefficients, the first of
which is "at" to indicate they are AngleTorsion coefficients. In a data file, these coefficients should be listed
under a "AngleTorsion Coeffs" heading and you must leave out the "at", i.e. only list 8 coefficients after the
dihedral type.
at• D1 (energy/radian)• D2 (energy/radian)• D3 (energy/radian)• E1 (energy/radian)•
LAMMPS Users Manual
1778

E2 (energy/radian)• E3 (energy/radian)• theta1 (degrees)• theta2 (degrees)•
Theta1 and theta2 are specified in degrees, but LAMMPS converts them to radians internally; hence the units
of D and E are in energy/radian.
For the Eaat formula, each line in a dihedral_coeff command in the input script lists 4 coefficients, the first of
which is "aat" to indicate they are AngleAngleTorsion coefficients. In a data file, these coefficients should be
listed under a "AngleAngleTorsion Coeffs" heading and you must leave out the "aat", i.e. only list 3
coefficients after the dihedral type.
aat• M (energy/radian^2)• theta1 (degrees)• theta2 (degrees)•
Theta1 and theta2 are specified in degrees, but LAMMPS converts them to radians internally; hence the units
of M are in energy/radian^2.
For the Ebb13 formula, each line in a dihedral_coeff command in the input script lists 4 coefficients, the first
of which is "bb13" to indicate they are BondBond13 coefficients. In a data file, these coefficients should be
listed under a "BondBond13 Coeffs" heading and you must leave out the "bb13", i.e. only list 3 coefficients
after the dihedral type.
bb13• N (energy/distance^2)• r1 (distance)• r3 (distance)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This dihedral style can only be used if LAMMPS was built with the CLASS2 package. See the Making
LAMMPS section for more info on packages.
LAMMPS Users Manual
1779

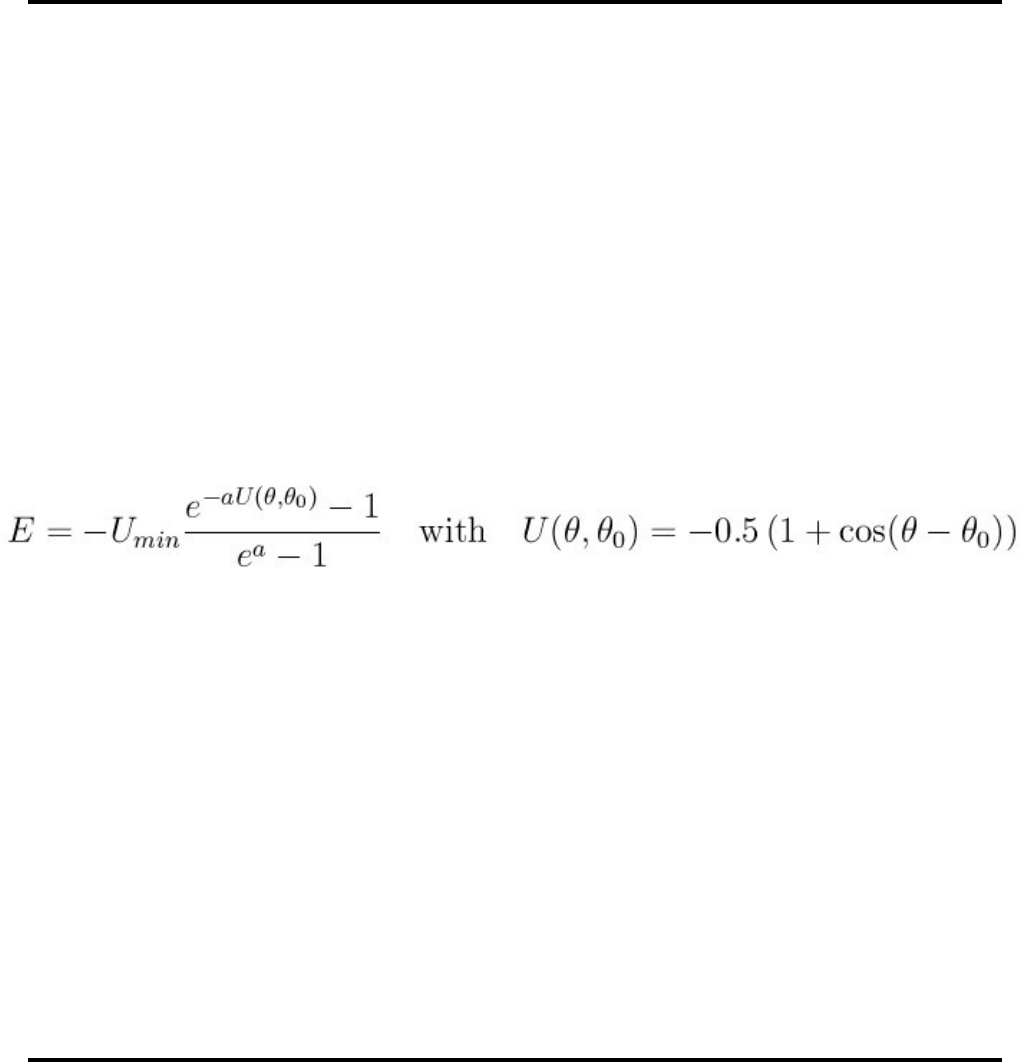
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style cosine/shift/exp command
dihedral_style cosine/shift/exp/omp command
Syntax:
dihedral_style cosine/shift/exp
Examples:
dihedral_style cosine/shift/exp
dihedral_coeff 1 10.0 45.0 2.0
Description:
The cosine/shift/exp dihedral style uses the potential
where Umin, theta, and a are defined for each dihedral type.
The potential is bounded between [-Umin:0] and the minimum is located at the angle theta0. The a parameter
can be both positive or negative and is used to control the spring constant at the equilibrium.
The spring constant is given by k=a exp(a) Umin/ [2 (Exp(a)-1)]. For a>3 k/Umin = a/2 to better than 5%
relative error. For negative values of the a parameter, the spring constant is essentially zero, and anharmonic
terms takes over. The potential is furthermore well behaved in the limit a->0, where it has been implemented
to linear order in a for a < 0.001.
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
umin (energy)• theta (angle)• A (real number)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
LAMMPS Users Manual
1781

You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This dihedral style can only be used if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info on packages.
Related commands:
dihedral_coeff, angle_cosine_shift_exp
Default: none
LAMMPS Users Manual
1782
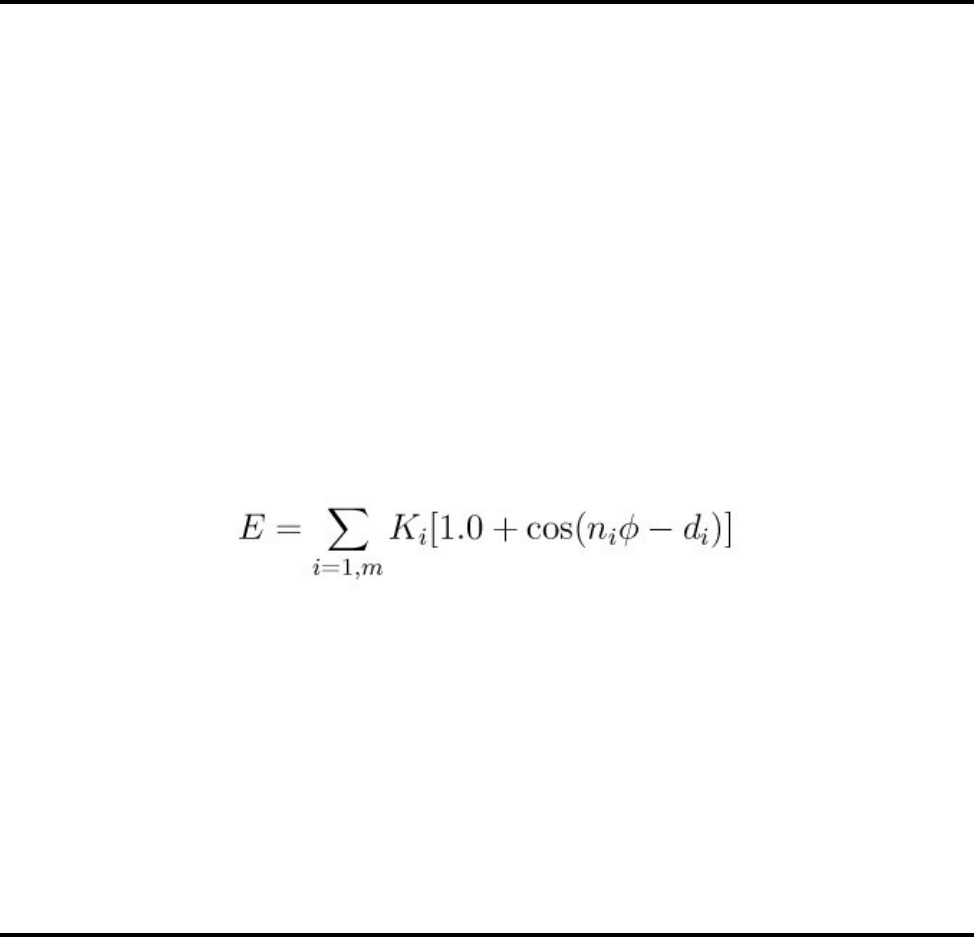
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style fourier command
dihedral_style fourier/omp command
Syntax:
dihedral_style fourier
Examples:
dihedral_style fourier
dihedral_coeff 1 3 -0.846200 3 0.0 7.578800 1 0 0.138000 2 -180.0
Description:
The fourier dihedral style uses the potential:
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
m (integer >=1)• K1 (energy)• n1 (integer >= 0)• d1 (degrees)• [...]• Km (energy)• nm (integer >= 0)• dm (degrees)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
LAMMPS Users Manual
1783

See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the USER_MISC package. See the Making
LAMMPS section for more info on packages.
Related commands:
dihedral_coeff
Default: none
LAMMPS Users Manual
1784

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style harmonic command
dihedral_style harmonic/intel command
dihedral_style harmonic/omp command
Syntax:
dihedral_style harmonic
Examples:
dihedral_style harmonic
dihedral_coeff 1 80.0 1 2
Description:
The harmonic dihedral style uses the potential
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy)• d (+1 or -1)• n (integer >= 0)•
NOTE: Here are important points to take note of when defining LAMMPS dihedral coefficients for the
harmonic style, so that they are compatible with how harmonic dihedrals are defined by other force fields:
The LAMMPS convention is that the trans position = 180 degrees, while in some force fields trans =
0 degrees.
•
Some force fields reverse the sign convention on d.• Some force fields let n be positive or negative which corresponds to d = 1 or -1 for the harmonic style.•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
LAMMPS Users Manual
1785

You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This dihedral style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Related commands:
dihedral_coeff
Default: none
LAMMPS Users Manual
1786
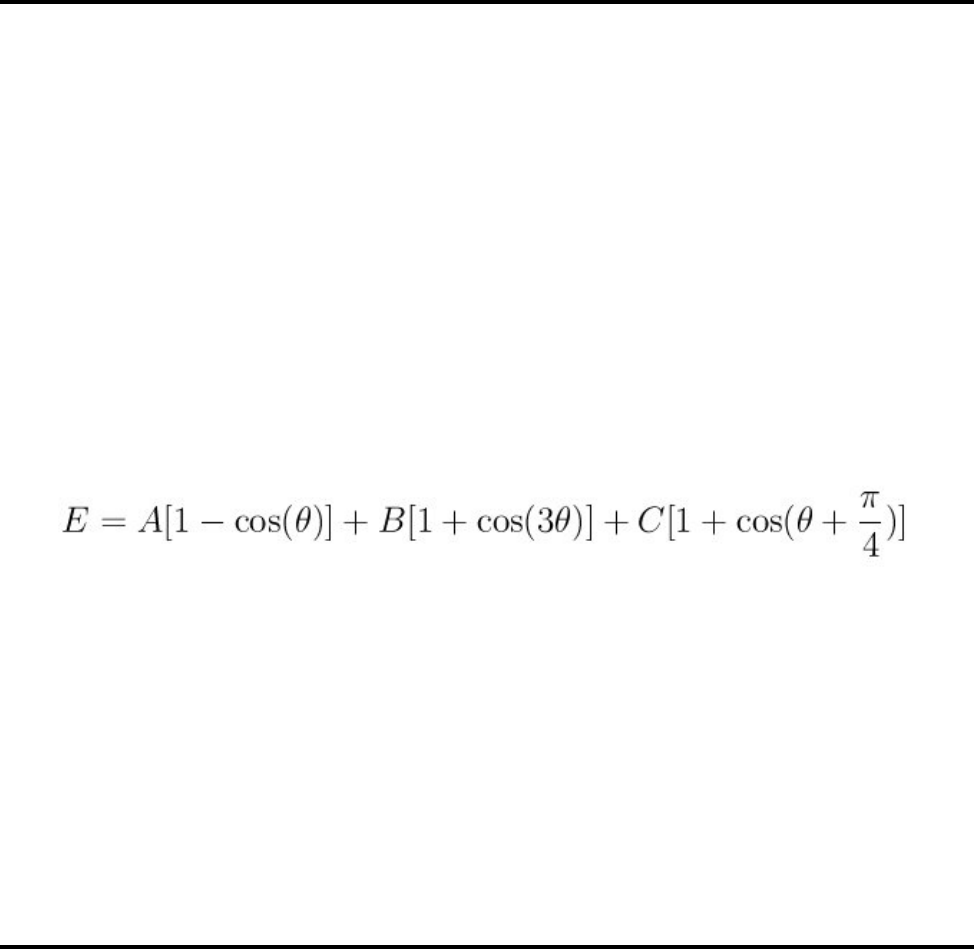
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style helix command
dihedral_style helix/omp command
Syntax:
dihedral_style helix
Examples:
dihedral_style helix
dihedral_coeff 1 80.0 100.0 40.0
Description:
The helix dihedral style uses the potential
This coarse-grain dihedral potential is described in (Guo). For dihedral angles in the helical region, the energy
function is represented by a standard potential consisting of three minima, one corresponding to the trans (t)
state and the other to gauche states (g+ and g-). The paper describes how the A,B,C parameters are chosen so
as to balance secondary (largely driven by local interactions) and tertiary structure (driven by long-range
interactions).
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
A (energy)• B (energy)• C (energy)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
LAMMPS Users Manual
1787

See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This dihedral style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Related commands:
dihedral_coeff
Default: none
(Guo) Guo and Thirumalai, Journal of Molecular Biology, 263, 323-43 (1996).
LAMMPS Users Manual
1788

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style hybrid command
Syntax:
dihedral_style hybrid style1 style2 ...
style1,style2 = list of one or more dihedral styles•
Examples:
dihedral_style hybrid harmonic helix
dihedral_coeff 1 harmonic 6.0 1 3
dihedral_coeff 2* helix 10 10 10
Description:
The hybrid style enables the use of multiple dihedral styles in one simulation. An dihedral style is assigned to
each dihedral type. For example, dihedrals in a polymer flow (of dihedral type 1) could be computed with a
harmonic potential and dihedrals in the wall boundary (of dihedral type 2) could be computed with a helix
potential. The assignment of dihedral type to style is made via the dihedral_coeff command or in the data file.
In the dihedral_coeff commands, the name of a dihedral style must be added after the dihedral type, with the
remaining coefficients being those appropriate to that style. In the example above, the 2 dihedral_coeff
commands set dihedrals of dihedral type 1 to be computed with a harmonic potential with coefficients 6.0, 1,
3 for K, d, n. All other dihedral types (2-N) are computed with a helix potential with coefficients 10, 10, 10 for
A, B, C.
If dihedral coefficients are specified in the data file read via the read_data command, then the same rule
applies. E.g. "harmonic" or "helix", must be added after the dihedral type, for each line in the "Dihedral
Coeffs" section, e.g.
Dihedral Coeffs
1 harmonic 6.0 1 3
2 helix 10 10 10
...
If class2 is one of the dihedral hybrid styles, the same rule holds for specifying additional AngleTorsion (and
EndBondTorsion, etc) coefficients either via the input script or in the data file. I.e. class2 must be added to
each line after the dihedral type. For lines in the AngleTorsion (or EndBondTorsion, etc) section of the data
file for dihedral types that are not class2, you must use an dihedral style of skip as a placeholder, e.g.
AngleTorsion Coeffs
1 skip
2 class2 1.0 1.0 1.0 3.0 3.0 3.0 30.0 50.0
...
Note that it is not necessary to use the dihedral style skip in the input script, since AngleTorsion (or
EndBondTorsion, etc) coefficients need not be specified at all for dihedral types that are not class2.
LAMMPS Users Manual
1789

A dihedral style of none with no additional coefficients can be used in place of a dihedral style, either in a
input script dihedral_coeff command or in the data file, if you desire to turn off interactions for specific
dihedral types.
Restrictions:
This dihedral style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Unlike other dihedral styles, the hybrid dihedral style does not store dihedral coefficient info for individual
sub-styles in a binary restart files. Thus when restarting a simulation from a restart file, you need to re-specify
dihedral_coeff commands.
Related commands:
dihedral_coeff
Default: none
LAMMPS Users Manual
1790

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style multi/harmonic command
dihedral_style multi/harmonic/omp command
Syntax:
dihedral_style multi/harmonic
Examples:
dihedral_style multi/harmonic
dihedral_coeff 1 20 20 20 20 20
Description:
The multi/harmonic dihedral style uses the potential
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
A1 (energy)• A2 (energy)• A3 (energy)• A4 (energy)• A5 (energy)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
1791

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style nharmonic command
dihedral_style nharmonic/omp command
Syntax:
dihedral_style nharmonic
Examples:
dihedral_style nharmonic
dihedral_coeff 3 10.0 20.0 30.0
Description:
The nharmonic dihedral style uses the potential:
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
n (integer >=1)• A1 (energy)• A2 (energy)• ...• An (energy)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
1793

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style none command
Syntax:
dihedral_style none
Examples:
dihedral_style none
Description:
Using a dihedral style of none means dihedral forces and energies are not computed, even if quadruplets of
dihedral atoms were listed in the data file read by the read_data command.
See the dihedral_style zero command for a way to calculate dihedral statistics, but compute no dihedral
interactions.
Restrictions: none
Related commands:
dihedral_style zero
Default: none
LAMMPS Users Manual
1795
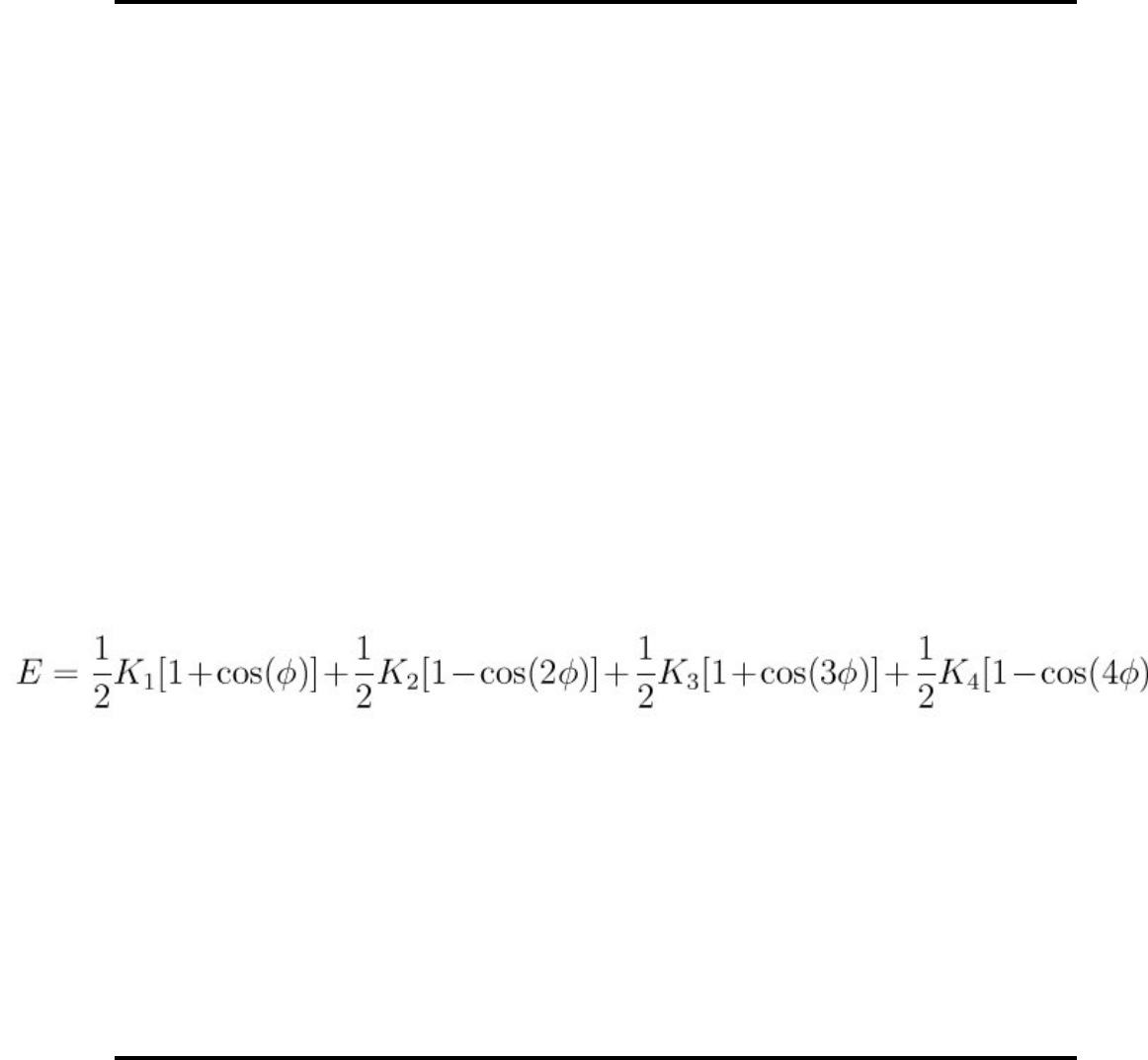
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style opls command
dihedral_style opls/intel command
dihedral_style opls/kk command
dihedral_style opls/omp command
Syntax:
dihedral_style opls
Examples:
dihedral_style opls
dihedral_coeff 1 1.740 -0.157 0.279 0.00 # CT-CT-CT-CT
dihedral_coeff 2 0.000 0.000 0.366 0.000 # CT-CT-CT-HC
dihedral_coeff 3 0.000 0.000 0.318 0.000 # HC-CT-CT-HC
Description:
The opls dihedral style uses the potential
Note that the usual 1/2 factor is not included in the K values.
This dihedral potential is used in the OPLS force field and is described in (Watkins).
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K1 (energy)• K2 (energy)• K3 (energy)• K4 (energy)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
LAMMPS Users Manual
1796

You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This dihedral style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Related commands:
dihedral_coeff
Default: none
(Watkins) Watkins and Jorgensen, J Phys Chem A, 105, 4118-4125 (2001).
LAMMPS Users Manual
1797

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style quadratic command
dihedral_style quadratic/omp command
Syntax:
dihedral_style quadratic
Examples:
dihedral_style quadratic
dihedral_coeff 100.0 80.0
Description:
The quadratic dihedral style uses the potential:
This dihedral potential can be used to keep a dihedral in a predefined value (cis=zero, right-hand convention
is used).
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy/radian^2)• phi0 (degrees)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
LAMMPS Users Manual
1798
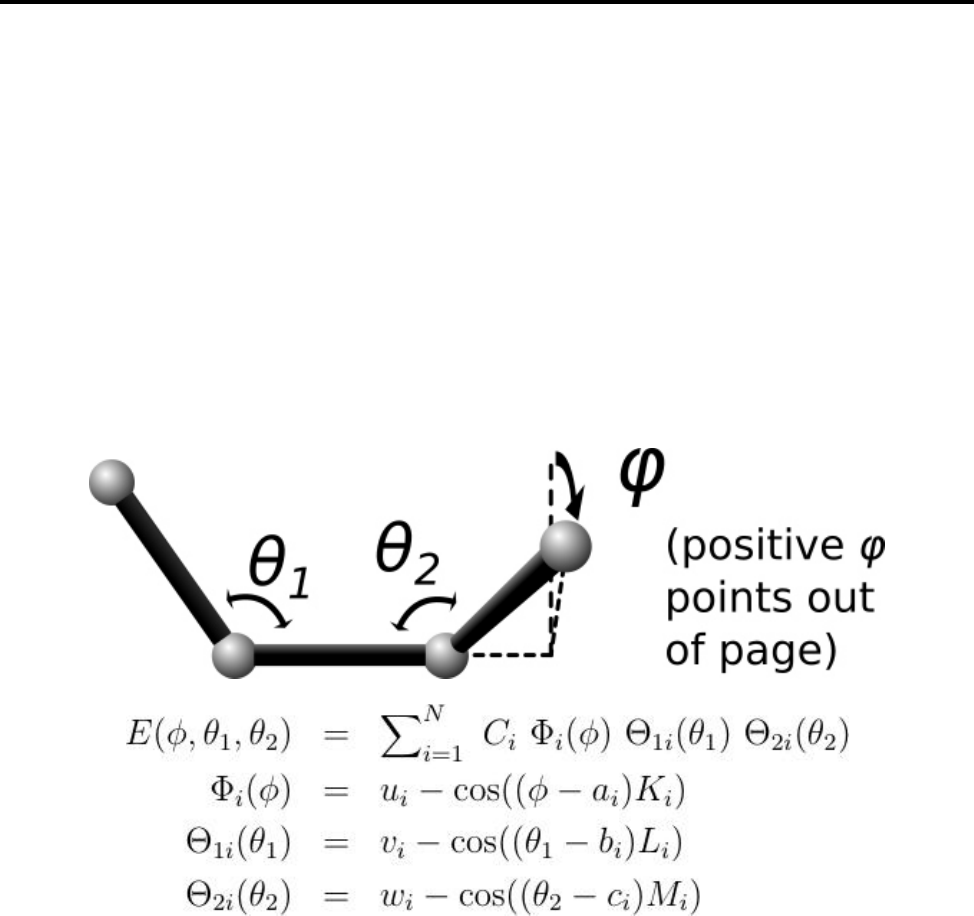
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style spherical command
Syntax:
dihedral_style spherical
Examples:
dihedral_coeff 1 1 286.1 1 124 1 1 90.0 0 1 90.0 0
dihedral_coeff 1 3 286.1 1 114 1 1 90 0 1 90.0 0 &
17.3 0 0.0 0 1 158 1 0 0.0 0 &
15.1 0 0.0 0 0 0.0 0 1 167.3 1
Description:
The spherical dihedral style uses the potential:
For this dihedral style, the energy can be any function that combines the 4-body dihedral-angle (phi) and the
two 3-body bond-angles (theta1, theta2). For this reason, there is usually no need to define 3-body "angle"
forces separately for the atoms participating in these interactions. It is probably more efficient to incorporate
3-body angle forces into the dihedral interaction even if it requires adding additional terms to the expansion
(as was done in the second example). A careful choice of parameters can prevent singularities that occur with
traditional force-fields whenever theta1 or theta2 approach 0 or 180 degrees. The last example above
corresponds to an interaction with a single energy minima located at phi=114, theta1=158, theta2=167.3
degrees, and it remains numerically stable at all angles (phi, theta1, theta2). In this example, the coefficients
17.3, and 15.1 can be physically interpreted as the harmonic spring constants for theta1 and theta2 around
their minima. The coefficient 286.1 is the harmonic spring constant for phi after division by
sin(158)*sin(167.3) (the minima positions for theta1 and theta2).
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above, or in the Dihedral Coeffs section of a data file file read by the read_data command:
LAMMPS Users Manual
1800

n (integer >= 1)• C1 (energy)• K1 (typically an integer)• a1 (degrees)• u1 (typically 0.0 or 1.0)• L1 (typically an integer)• b1 (degrees, typically 0.0 or 90.0)• v1 (typically 0.0 or 1.0)• M1 (typically an integer)• c1 (degrees, typically 0.0 or 90.0)• w1 (typically 0.0 or 1.0)• [...]• Cn (energy)• Kn (typically an integer)• an (degrees)• un (typically 0.0 or 1.0)• Ln (typically an integer)• bn (degrees, typically 0.0 or 90.0)• vn (typically 0.0 or 1.0)• Mn (typically an integer)• cn (degrees, typically 0.0 or 90.0)• wn (typically 0.0 or 1.0)•
Restrictions:
This dihedral style can only be used if LAMMPS was built with the USER_MISC package. See the Making
LAMMPS section for more info on packages.
Related commands:
dihedral_coeff
Default: none
LAMMPS Users Manual
1801

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style table command
dihedral_style table/omp command
Syntax:
dihedral_style table style Ntable
style = linear or spline = method of interpolation• Ntable = size of the internal lookup table•
Examples:
dihedral_style table spline 400
dihedral_style table linear 1000
dihedral_coeff 1 file.table DIH_TABLE1
dihedral_coeff 2 file.table DIH_TABLE2
Description:
The table dihedral style creates interpolation tables of length Ntable from dihedral potential and derivative
values listed in a file(s) as a function of the dihedral angle "phi". The files are read by the dihedral_coeff
command.
The interpolation tables are created by fitting cubic splines to the file values and interpolating energy and
derivative values at each of Ntable dihedral angles. During a simulation, these tables are used to interpolate
energy and force values on individual atoms as needed. The interpolation is done in one of 2 styles: linear or
spline.
For the linear style, the dihedral angle (phi) is used to find 2 surrounding table values from which an energy
or its derivative is computed by linear interpolation.
For the spline style, cubic spline coefficients are computed and stored at each of the Ntable evenly-spaced
values in the interpolated table. For a given dihedral angle (phi), the appropriate coefficients are chosen from
this list, and a cubic polynomial is used to compute the energy and the derivative at this angle.
The following coefficients must be defined for each dihedral type via the dihedral_coeff command as in the
example above.
filename• keyword•
The filename specifies a file containing tabulated energy and derivative values. The keyword specifies a
section of the file. The format of this file is described below.
The format of a tabulated file is as follows (without the parenthesized comments). It can begin with one or
more comment or blank lines.
# Table of the potential and its negative derivative
LAMMPS Users Manual
1802
DIH_TABLE1 (keyword is the first text on line)
N 30 DEGREES (N, NOF, DEGREES, RADIANS, CHECKU/F)
(blank line)
1 -168.0 -1.40351172223 0.0423346818422
2 -156.0 -1.70447981034 0.00811786522531
3 -144.0 -1.62956100432 -0.0184129719987
...
30 180.0 -0.707106781187 0.0719306095245
# Example 2: table of the potential. Forces omitted
DIH_TABLE2
N 30 NOF CHECKU testU.dat CHECKF testF.dat
1 -168.0 -1.40351172223
2 -156.0 -1.70447981034
3 -144.0 -1.62956100432
...
30 180.0 -0.707106781187
A section begins with a non-blank line whose 1st character is not a "#"; blank lines or lines starting with "#"
can be used as comments between sections. The first line begins with a keyword which identifies the section.
The line can contain additional text, but the initial text must match the argument specified in the
dihedral_coeff command. The next line lists (in any order) one or more parameters for the table. Each
parameter is a keyword followed by one or more numeric values.
Following a blank line, the next N lines list the tabulated values. On each line, the 1st value is the index from
1 to N, the 2nd value is the angle value, the 3rd value is the energy (in energy units), and the 4th is -dE/d(phi)
also in energy units). The 3rd term is the energy of the 4-atom configuration for the specified angle. The 4th
term (when present) is the negative derivative of the energy with respect to the angle (in degrees, or radians
depending on whether the user selected DEGREES or RADIANS). Thus the units of the last term are still
energy, not force. The dihedral angle values must increase from one line to the next.
Dihedral table splines are cyclic. There is no discontinuity at 180 degrees (or at any other angle). Although in
the examples above, the angles range from -180 to 180 degrees, in general, the first angle in the list can have
any value (positive, zero, or negative). However the range of angles represented in the table must be strictly
less than 360 degrees (2pi radians) to avoid angle overlap. (You may not supply entries in the table for both
180 and -180, for example.) If the user's table covers only a narrow range of dihedral angles, strange
numerical behavior can occur in the large remaining gap.
Parameters:
The parameter "N" is required and its value is the number of table entries that follow. Note that this may be
different than the N specified in the dihedral_style table command. Let Ntable is the number of table entries
requested dihedral_style command, and let Nfile be the parameter following "N" in the tabulated file ("30" in
the sparse example above). What LAMMPS does is a preliminary interpolation by creating splines using the
Nfile tabulated values as nodal points. It uses these to interpolate as needed to generate energy and derivative
values at Ntable different points (which are evenly spaced over a 360 degree range, even if the angles in the
file are not). The resulting tables of length Ntable are then used as described above, when computing energy
and force for individual dihedral angles and their atoms. This means that if you want the interpolation tables
of length Ntable to match exactly what is in the tabulated file (with effectively nopreliminary interpolation),
you should set Ntable = Nfile. To insure the nodal points in the user's file are aligned with the interpolated
table entries, the angles in the table should be integer multiples of 360/Ntable degrees, or 2*PI/Ntable radians
LAMMPS Users Manual
1803

(depending on your choice of angle units).
The optional "NOF" keyword allows the user to omit the forces (negative energy derivatives) from the table
file (normally located in the 4th column). In their place, forces will be calculated automatically by
differentiating the potential energy function indicated by the 3rd column of the table (using either linear or
spline interpolation).
The optional "DEGREES" keyword allows the user to specify angles in degrees instead of radians (default).
The optional "RADIANS" keyword allows the user to specify angles in radians instead of degrees. (Note:
This changes the way the forces are scaled in the 4th column of the data file.)
The optional "CHECKU" keyword is followed by a filename. This allows the user to save all of the Ntable
different entries in the interpolated energy table to a file to make sure that the interpolated function agrees
with the user's expectations. (Note: You can temporarily increase the Ntable parameter to a high value for this
purpose. "Ntable" is explained above.)
The optional "CHECKF" keyword is analogous to the "CHECKU" keyword. It is followed by a filename, and
it allows the user to check the interpolated force table. This option is available even if the user selected the
"NOF" option.
Note that one file can contain many sections, each with a tabulated potential. LAMMPS reads the file section
by section until it finds one that matches the specified keyword.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This dihedral style can only be used if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info on packages.
Related commands:
dihedral_coeff
Default: none
LAMMPS Users Manual
1804

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
dihedral_style zero command
Syntax:
dihedral_style zero nocoeff
Examples:
dihedral_style zero
dihedral_style zero nocoeff
dihedral_coeff *
Description:
Using a dihedral style of zero means dihedral forces and energies are not computed, but the geometry of
dihedral quadruplets is still accessible to other commands.
As an example, the compute dihedral/local command can be used to compute the theta values for the list of
quadruplets of dihedral atoms listed in the data file read by the read_data command. If no dihedral style is
defined, this command cannot be used.
The optional nocoeff flag allows to read data files with a DihedralCoeff section for any dihedral style.
Similarly, any dihedral_coeff commands will only be checked for the dihedral type number and the rest
ignored.
Note that the dihedral_coeff command must be used for all dihedral types, though no additional values are
specified.
Restrictions: none
Related commands: none
dihedral_style none
Default: none
LAMMPS Users Manual
1805
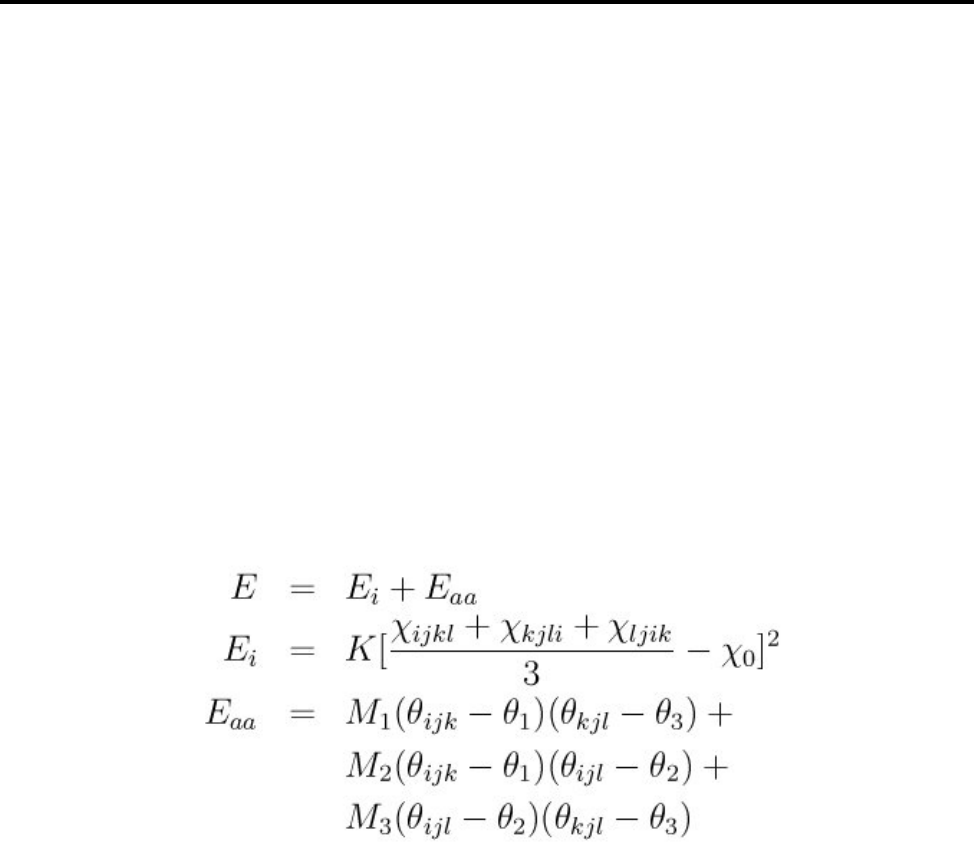
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style class2 command
improper_style class2/omp command
improper_style class2/kk command
Syntax:
improper_style class2
Examples:
improper_style class2
improper_coeff 1 100.0 0
improper_coeff * aa 0.0 0.0 0.0 115.06 130.01 115.06
Description:
The class2 improper style uses the potential
where Ei is the improper term and Eaa is an angle-angle term. The 3 X terms in Ei are an average over 3
out-of-plane angles.
The 4 atoms in an improper quadruplet (listed in the data file read by the read_data command) are ordered
I,J,K,L. X_IJKL refers to the angle between the plane of I,J,K and the plane of J,K,L, and the bond JK lies in
both planes. Similarly for X_KJLI and X_LJIK. Note that atom J appears in the common bonds (JI, JK, JL) of
all 3 X terms. Thus J (the 2nd atom in the quadruplet) is the atom of symmetry in the 3 X angles.
The subscripts on the various theta's refer to different combinations of 3 atoms (I,J,K,L) used to form a
particular angle. E.g. Theta_IJL is the angle formed by atoms I,J,L with J in the middle. Theta1, theta2, theta3
are the equilibrium positions of those angles. Again, atom J (the 2nd atom in the quadruplet) is the atom of
symmetry in the theta angles, since it is always the center atom.
Since atom J is the atom of symmetry, normally the bonds J-I, J-K, J-L would exist for an improper to be
defined between the 4 atoms, but this is not required.
LAMMPS Users Manual
1806

See (Sun) for a description of the COMPASS class2 force field.
Coefficients for the Ei and Eaa formulas must be defined for each improper type via the improper_coeff
command as in the example above, or in the data file or restart files read by the read_data or read_restart
commands.
These are the 2 coefficients for the Ei formula:
K (energy/radian^2)• X0 (degrees)•
X0 is specified in degrees, but LAMMPS converts it to radians internally; hence the units of K are in
energy/radian^2.
For the Eaa formula, each line in a improper_coeff command in the input script lists 7 coefficients, the first of
which is "aa" to indicate they are AngleAngle coefficients. In a data file, these coefficients should be listed
under a "AngleAngle Coeffs" heading and you must leave out the "aa", i.e. only list 6 coefficients after the
improper type.
aa• M1 (energy/distance)• M2 (energy/distance)• M3 (energy/distance)• theta1 (degrees)• theta2 (degrees)• theta3 (degrees)•
The theta values are specified in degrees, but LAMMPS converts them to radians internally; hence the units of
M are in energy/radian^2.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This improper style can only be used if LAMMPS was built with the CLASS2 package. See the Making
LAMMPS section for more info on packages.
LAMMPS Users Manual
1807

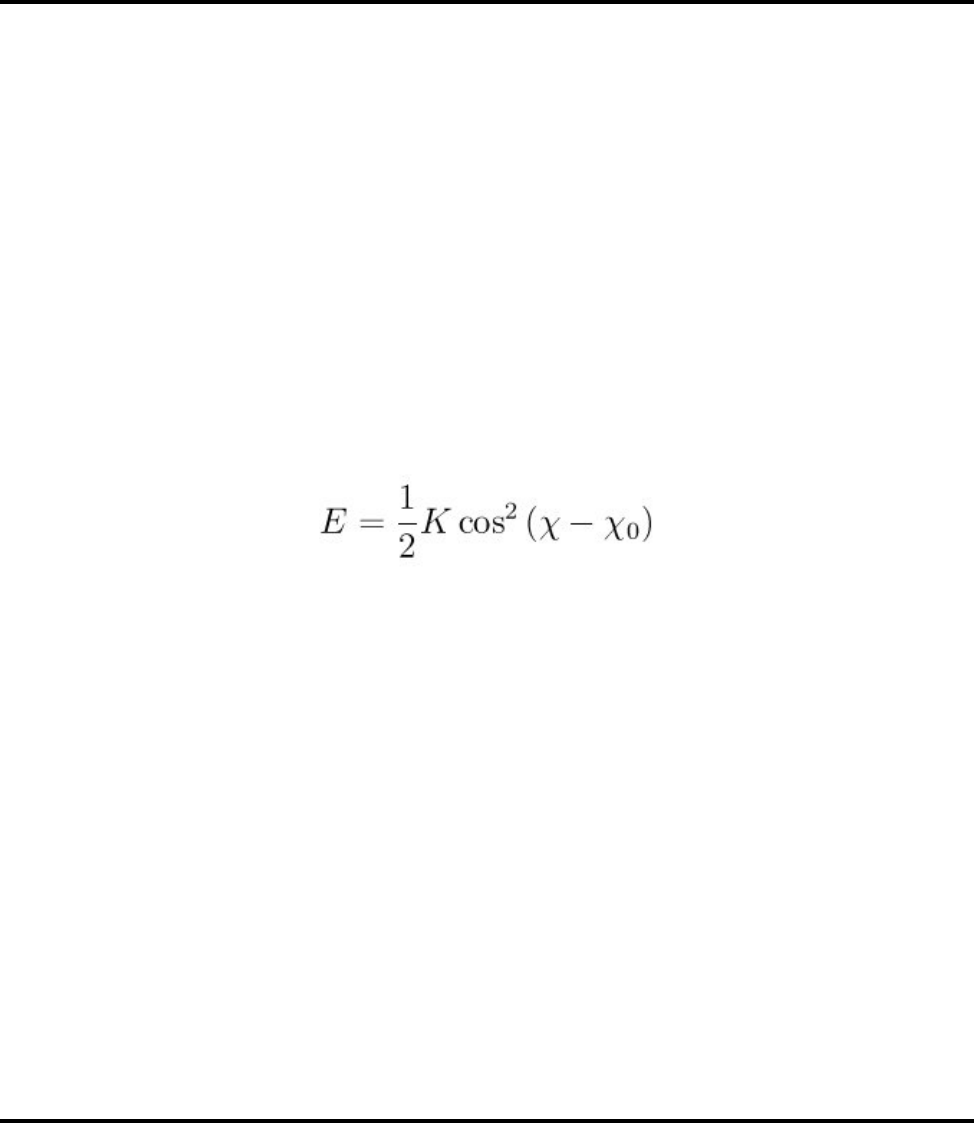
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style cossq command
improper_style cossq/omp command
Syntax:
improper_style cossq
Examples:
improper_style cossq
improper_coeff 1 4.0 0.0
Description:
The cossq improper style uses the potential
where x is the improper angle, x0 is its equilibrium value, and K is a prefactor.
If the 4 atoms in an improper quadruplet (listed in the data file read by the read_data command) are ordered
I,J,K,L then X is the angle between the plane of I,J,K and the plane of J,K,L. Alternatively, you can think of
atoms J,K,L as being in a plane, and atom I above the plane, and X as a measure of how far out-of-plane I is
with respect to the other 3 atoms.
Note that defining 4 atoms to interact in this way, does not mean that bonds necessarily exist between I-J, J-K,
or K-L, as they would in a linear dihedral. Normally, the bonds I-J, I-K, I-L would exist for an improper to be
defined between the 4 atoms.
The following coefficients must be defined for each improper type via the improper_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy/radian^2)• X0 (degrees)•
X0 is specified in degrees, but LAMMPS converts it to radians internally; hence the units of K are in
energy/radian^2.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
LAMMPS Users Manual
1809

These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This improper style can only be used if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info on packages.
Related commands:
improper_coeff
Default: none
LAMMPS Users Manual
1810

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style cvff command
improper_style cvff/intel command
improper_style cvff/omp command
Syntax:
improper_style cvff
Examples:
improper_style cvff
improper_coeff 1 80.0 -1 4
Description:
The cvff improper style uses the potential
where phi is the improper dihedral angle.
If the 4 atoms in an improper quadruplet (listed in the data file read by the read_data command) are ordered
I,J,K,L then the improper dihedral angle is between the plane of I,J,K and the plane of J,K,L. Note that
because this is effectively a dihedral angle, the formula for this improper style is the same as for
dihedral_style harmonic.
Note that defining 4 atoms to interact in this way, does not mean that bonds necessarily exist between I-J, J-K,
or K-L, as they would in a linear dihedral. Normally, the bonds I-J, I-K, I-L would exist for an improper to be
defined between the 4 atoms.
The following coefficients must be defined for each improper type via the improper_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy)• d (+1 or -1)• n (0,1,2,3,4,6)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
LAMMPS Users Manual
1811

section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This improper style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Related commands:
improper_coeff
Default: none
LAMMPS Users Manual
1812
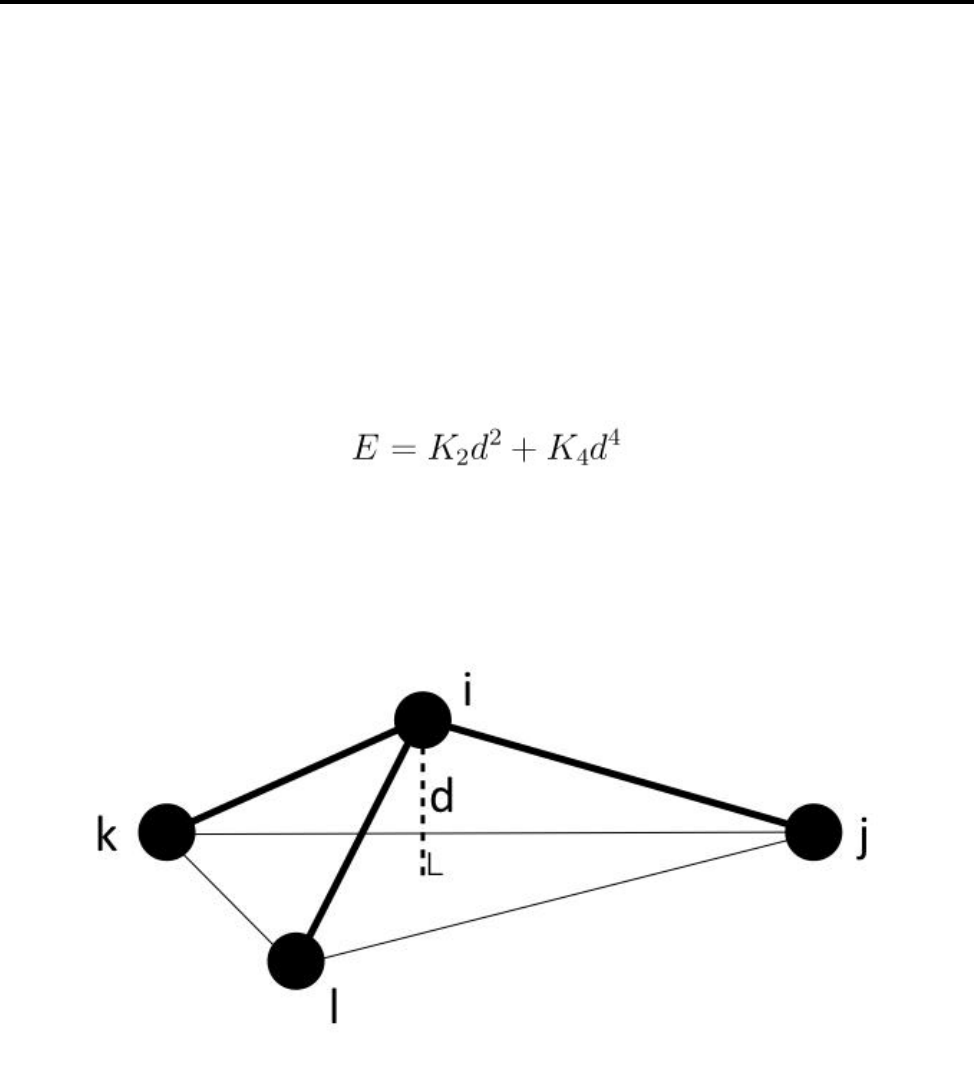
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style distance command
Syntax:
improper_style distance
Examples:
improper_style distance
improper_coeff 1 80.0 100.0
Description:
The distance improper style uses the potential
where d is the distance between the central atom and the plane formed by the other three atoms. If the 4 atoms
in an improper quadruplet (listed in the data file read by the read_data command) are ordered I,J,K,L then the
I-atom is assumed to be the central atom.
Note that defining 4 atoms to interact in this way, does not mean that bonds necessarily exist between I-J, J-K,
or K-L, as they would in a linear dihedral. Normally, the bonds I-J, I-K, I-L would exist for an improper to be
defined between the 4 atoms.
The following coefficients must be defined for each improper type via the improper_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K_2 (energy/distance^2)•
LAMMPS Users Manual
1813


LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style fourier command
improper_style fourier/omp command
Syntax:
improper_style fourier
Examples:
improper_style fourier
improper_coeff 1 100.0 180.0
Description:
The fourier improper style uses the following potential:
where K is the force constant and omega is the angle between the IL axis and the IJK plane:
If all parameter (see bellow) is not zero, the all the three possible angles will taken in account.
The following coefficients must be defined for each improper type via the improper_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy)• C0 (real)• C1 (real)• C2 (real)• all (integer >= 0)•
LAMMPS Users Manual
1815

Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This angle style can only be used if LAMMPS was built with the USER_MISC package. See the Making
LAMMPS section for more info on packages.
Related commands:
improper_coeff
Default: none
LAMMPS Users Manual
1816
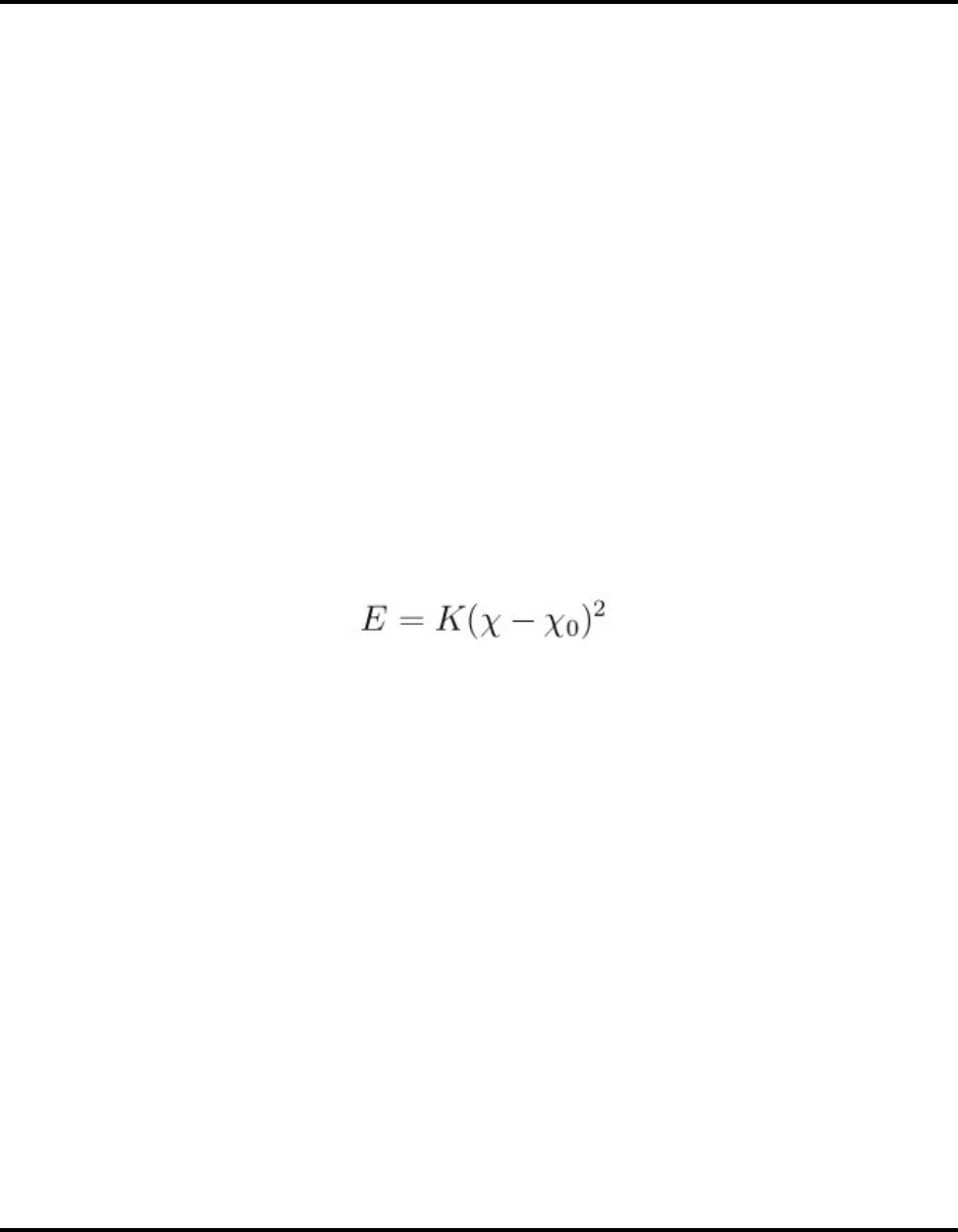
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style harmonic command
improper_style harmonic/intel command
improper_style harmonic/kk command
improper_style harmonic/omp command
Syntax:
improper_style harmonic
Examples:
improper_style harmonic
improper_coeff 1 100.0 0
Description:
The harmonic improper style uses the potential
where X is the improper angle, X0 is its equilibrium value, and K is a prefactor. Note that the usual 1/2 factor
is included in K.
If the 4 atoms in an improper quadruplet (listed in the data file read by the read_data command) are ordered
I,J,K,L then X is the angle between the plane of I,J,K and the plane of J,K,L. Alternatively, you can think of
atoms J,K,L as being in a plane, and atom I above the plane, and X as a measure of how far out-of-plane I is
with respect to the other 3 atoms.
Note that defining 4 atoms to interact in this way, does not mean that bonds necessarily exist between I-J, J-K,
or K-L, as they would in a linear dihedral. Normally, the bonds I-J, I-K, I-L would exist for an improper to be
defined between the 4 atoms.
The following coefficients must be defined for each improper type via the improper_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy/radian^2)• X0 (degrees)•
X0 is specified in degrees, but LAMMPS converts it to radians internally; hence the units of K are in
energy/radian^2.
LAMMPS Users Manual
1817

Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This improper style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Related commands:
improper_coeff
Default: none
LAMMPS Users Manual
1818
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style hybrid command
Syntax:
improper_style hybrid style1 style2 ...
style1,style2 = list of one or more improper styles•
Examples:
improper_style hybrid harmonic helix
improper_coeff 1 harmonic 120.0 30
improper_coeff 2 cvff 20.0 -1 2
Description:
The hybrid style enables the use of multiple improper styles in one simulation. An improper style is assigned
to each improper type. For example, impropers in a polymer flow (of improper type 1) could be computed
with a harmonic potential and impropers in the wall boundary (of improper type 2) could be computed with a
cvff potential. The assignment of improper type to style is made via the improper_coeff command or in the
data file.
In the improper_coeff command, the first coefficient sets the improper style and the remaining coefficients are
those appropriate to that style. In the example above, the 2 improper_coeff commands would set impropers of
improper type 1 to be computed with a harmonic potential with coefficients 120.0, 30 for K, X0. Improper
type 2 would be computed with a cvff potential with coefficients 20.0, -1, 2 for K, d, n.
If the improper class2 potential is one of the hybrid styles, it requires additional AngleAngle coefficients be
specified in the data file. These lines must also have an additional "class2" argument added after the improper
type. For improper types which are assigned to other hybrid styles, use the style name (e.g. "harmonic")
appropriate to that style. The AngleAngle coeffs for that improper type will then be ignored.
An improper style of none can be specified as the 2nd argument to the improper_coeff command, if you desire
to turn off certain improper types.
Restrictions:
This improper style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Unlike other improper styles, the hybrid improper style does not store improper coefficient info for individual
sub-styles in a binary restart files. Thus when restarting a simulation from a restart file, you need to re-specify
improper_coeff commands.
Related commands:
improper_coeff
Default: none
LAMMPS Users Manual
1819

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style none command
Syntax:
improper_style none
Examples:
improper_style none
Description:
Using an improper style of none means imroper forces and energies are not computed, even if quadruplets of
improper atoms were listed in the data file read by the read_data command.
See the improper_style zero command for a way to calculate improper statistics, but compute no improper
interactions.
Restrictions: none
Related commands:
improper_style zero
Default: none
LAMMPS Users Manual
1820
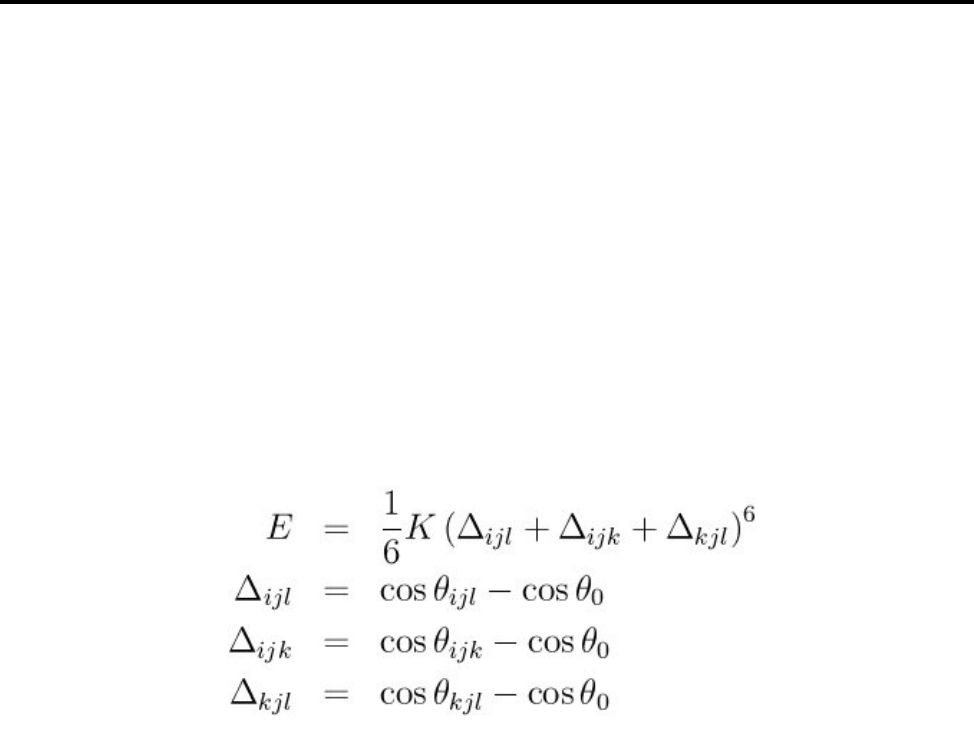
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style ring command
improper_style ring/omp command
Syntax:
improper_style ring
Examples:
improper_style ring
improper_coeff 1 8000 70.5
Description:
The ring improper style uses the potential
where K is a prefactor, theta is the angle formed by the atoms specified by (i,j,k,l) indices and theta0 its
equilibrium value.
If the 4 atoms in an improper quadruplet (listed in the data file read by the read_data command) are ordered
i,j,k,l then theta_ijl is the angle between atoms i,j and l, theta_ijk is the angle between atoms i,j and k,
theta_kjl is the angle between atoms j,k, and l.
The "ring" improper style implements the improper potential introduced by Destree et al., in Equation (9) of
(Destree). This potential does not affect small amplitude vibrations but is used in an ad-hoc way to prevent the
onset of accidentally large amplitude fluctuations leading to the occurrence of a planar conformation of the
three bonds i-j, j-k and j-l, an intermediate conformation toward the chiral inversion of a methine carbon. In
the "Impropers" section of data file four atoms: i, j, k and l are specified with i,j and l lying on the backbone of
the chain and k specifying the chirality of j.
The following coefficients must be defined for each improper type via the improper_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
K (energy/radian^2)• theta0 (degrees)•
LAMMPS Users Manual
1821

theta0 is specified in degrees, but LAMMPS converts it to radians internally; hence the units of K are in
energy/radian^2.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This improper style can only be used if LAMMPS was built with the USER-MISC package. See the Making
LAMMPS section for more info on packages.
Related commands:
improper_coeff
(Destree) M. Destree, F. Laupretre, A. Lyulin, and J.-P. Ryckaert, J Chem Phys, 112, 9632 (2000).
LAMMPS Users Manual
1822
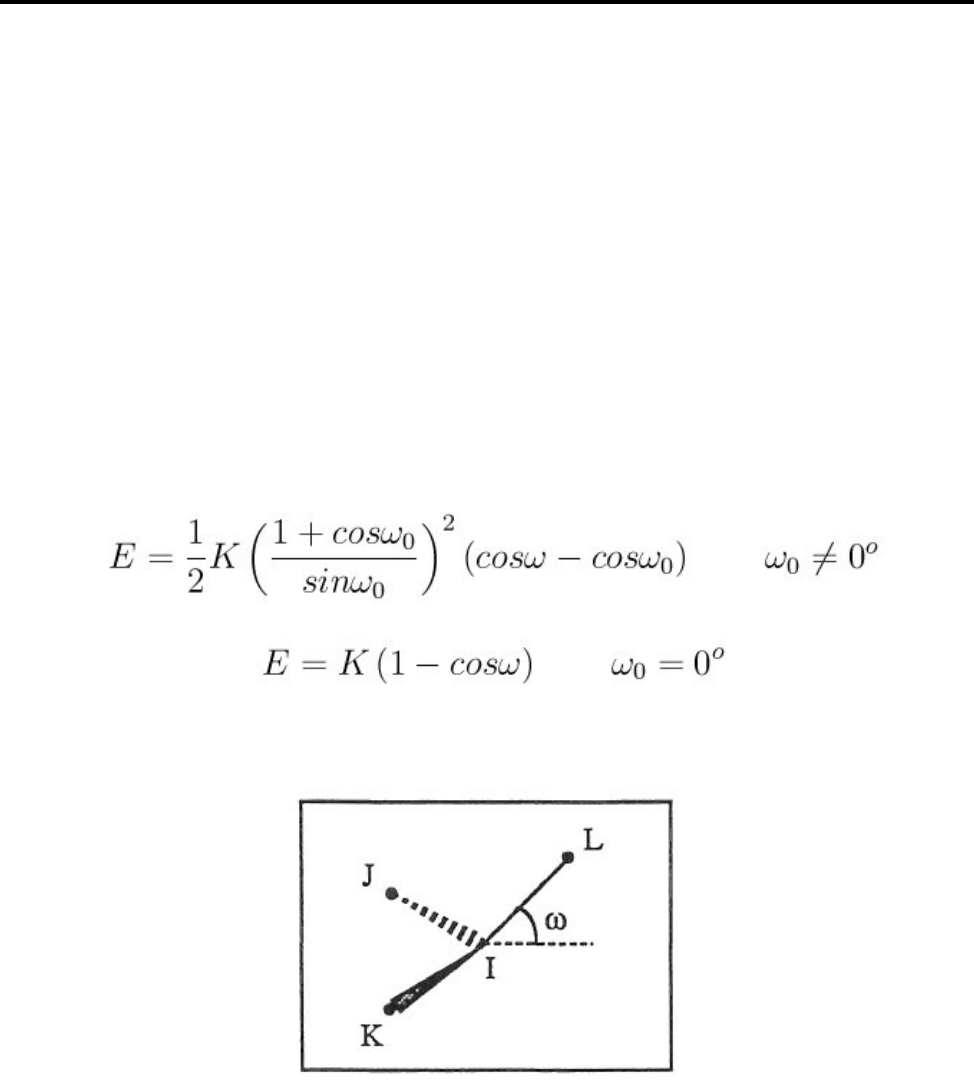
LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style umbrella command
improper_style umbrella/omp command
Syntax:
improper_style umbrella
Examples:
improper_style umbrella
improper_coeff 1 100.0 180.0
Description:
The umbrella improper style uses the following potential, which is commonly referred to as a classic
inversion and used in the DREIDING force field:
where K is the force constant and omega is the angle between the IL axis and the IJK plane:
If omega0 = 0 the potential term has a minimum for the planar structure. Otherwise it has two minima at +/-
omega0, with a barrier in between.
See (Mayo) for a description of the DREIDING force field.
The following coefficients must be defined for each improper type via the improper_coeff command as in the
example above, or in the data file or restart files read by the read_data or read_restart commands:
LAMMPS Users Manual
1823

K (energy)• omega0 (degrees)•
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the
suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section
5 of the manual. The accelerated styles take the same arguments and should produce the same results, except
for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages,
respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS
section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use
the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your
input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Restrictions:
This improper style can only be used if LAMMPS was built with the MOLECULE package. See the Making
LAMMPS section for more info on packages.
Related commands:
improper_coeff
Default: none
(Mayo) Mayo, Olfason, Goddard III, J Phys Chem, 94, 8897-8909 (1990),
LAMMPS Users Manual
1824

LAMMPS WWW Site - LAMMPS Documentation - LAMMPS Commands
improper_style zero command
Syntax:
improper_style zero nocoeff
Examples:
improper_style zero
improper_style zero nocoeff
improper_coeff *
Description:
Using an improper style of zero means improper forces and energies are not computed, but the geometry of
improper quadruplets is still accessible to other commands.
As an example, the compute improper/local command can be used to compute the chi values for the list of
quadruplets of improper atoms listed in the data file read by the read_data command. If no improper style is
defined, this command cannot be used.
The optional nocoeff flag allows to read data files with a ImproperCoeff section for any improper style.
Similarly, any improper_coeff commands will only be checked for the improper type number and the rest
ignored.
Note that the improper_coeff command must be used for all improper types, though no additional values are
specified.
Restrictions: none
Related commands: none
improper_style none
Default: none
LAMMPS Users Manual
1825
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
LAMMPS Users Manual
1826

fix_modify AtC add_molecule
syntax
fix_modify_AtC add_molecule <small|large> <TAG> <GROUP_NAME>
small|large = can be small if molecule size < cutoff radius, must be large otherwise• <TAG> = tag for tracking a species• <GROUP_NAME> = name of group that tracking will be applied to•
examples
group WATERGROUP type 1 2
fix_modify AtC add_molecule small water WATERGROUP
description
Associates a tag with all molecules corresponding to a specified group.
restrictions
related
default
No defaults for this command.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1827

fix_modify AtC add_species
syntax
fix_modify_AtC add_species <TAG> <group|type> <ID>
<TAG> = tag for tracking a species• group|type = LAMMPS defined group or type of atoms• <ID> = name of group or type number•
examples
fix_modify AtC add_species gold type 1
group GOLDGROUP type 1
fix_modify AtC add_species gold group GOLDGROUP
description
Associates a tag with all atoms of a specified type or within a specified group.
restrictions
related
default
No defaults for this command.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1828

fix_modify AtC atom_element_map
syntax
fix_modify AtC atom_element_map <eulerian|lagrangian> <frequency>
frequency (int) : frequency of updating atom-to-continuum maps based on the current configuration -
only for eulerian
•
examples
fix_modify atc atom_element_map eulerian 100
description
Changes frame of reference from eulerian to lagrangian and sets the frequency for which the map from atoms
to elements is reformed and all the attendant data is recalculated.
restrictions
Cannot change map type after initialization.
related
default
lagrangian
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1829

fix_modify AtC atom_weight
syntax
fix_modify AtC atom_weight <method> <arguments>
<method> =
value: atoms in specified group assigned constant value given
lattice: volume per atom for specified lattice type (e.g. fcc) and parameter
element: element volume divided among atoms within element
region: volume per atom determined based on the atom count in the MD regions and their volumes.
Note: meaningful only if atoms completely fill all the regions.
group: volume per atom determined based on the atom count in a group and its volume
read_in: list of values for atoms are read-in from specified file
•
examples
fix_modify atc atom_weight constant myatoms 11.8
fix_modify atc atom_weight lattice
fix_modify atc atom_weight read-in atm_wt_file.txt
description
Command for assigning the value of atomic weights used for atomic integration in atom-continuum coupled
simulations.
restrictions
Use of lattice option requires a lattice type and parameter is already specified.
related
default
lattice
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1830

fix_modify AtC atomic_charge
syntax
fix_modify AtC <include | omit> atomic_charge
<include | omit> = switch to activiate/deactiviate inclusion of intrinsic atomic charge in ATC•
examples
fix_modify atc compute include atomic_charge
description
Determines whether AtC tracks the total charge as a finite element field
restrictions
Required for: electrostatics
related
default
if the atom charge is defined, default is on, otherwise default is off
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1831

fix_modify AtC boundary
syntax
fix_modify AtC boundary type <atom-type-id>
<atom-type-id> = type id for atoms that represent a ficticious boundary internal to the FE mesh•
examples
fix_modify AtC boundary type ghost_atoms
description
Command to define the atoms that represent the ficticious boundary internal to the FE mesh. For fully
overlapped MD/FE domains with periodic boundary conditions no boundary atoms should be defined.
restrictions
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1832

fix_modify AtC boundary_dynamics
syntax
fix_modify AtC boundary_dynamics < on | damped_harmonic | prescribed | coupled | none > [args]
description
Sets different schemes for controlling boundary atoms. On will integrate the boundary atoms using the
velocity-verlet algorithm. Damped harmonic uses a mass/spring/dashpot for the boundary atoms with added
arguments of the damping and spring constants followed by the ratio of the boundary type mass to the desired
mass. Prescribed forces the boundary atoms to follow the finite element displacement. Coupled does the same.
restrictions
Boundary atoms must be specified. When using swaps between internal and boundary atoms, the initial
configuration must have already correctly partitioned the two.
related
default
prescribed on
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1833

fix_modify AtC boundary_faceset
syntax
fix_modify AtC boundary_faceset <is | add> [args]
examples
fix_modify AtC boundary_faceset is obndy
description
This command species the faceset name when using a faceset to compute the MD/FE boundary fluxes. The
faceset must already exist.
restrictions
This is only valid when fe_md_boundary is set to faceset.
related
default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1834

fix_modify AtC output boundary_integral
syntax
fix_modify AtC output boundary_integral [field] faceset [name]
field (string) : name of hardy field• name (string) : name of faceset•
examples
fix_modify AtC output boundary_integral stress faceset loop1
description
Calculates a surface integral of the given field dotted with the outward normal of the faces and puts output in
the "GLOBALS" file
restrictions
Must be used with the hardy/field type of AtC fix ( see fix atc command )
related
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1835

fix_modify AtC consistent_fe_initialization
syntax
fix_modify AtC consistent_fe_initialization <on | off>
<on|off> = switch to activiate/deactiviate the initial setting of FE intrinsic field to match the projected
MD field
•
examples
fix_modify atc consistent_fe_initialization on
description
Determines whether AtC initializes FE intrinsic fields (e.g., temperature) to match the projected MD values.
This is particularly useful for fully overlapping simulations.
restrictions
Can be used with: thermal, two_temperature. Cannot be used with time filtering on. Does not include
boundary nodes.
related
default
Default is off
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1836

fix_modify AtC output contour_integral
syntax
fix_modify AtC output contour_integral [field] faceset [name] <axis [x | y | z ]>
field (string) : name of hardy field• name (string) : name of faceset• axis (string) : x or y or z•
examples
fix_modify AtC output contour_integral stress faceset loop1
description
Calculates a surface integral of the given field dotted with the outward normal of the faces and puts output in
the "GLOBALS" file
restrictions
Must be used with the hardy/field type of AtC fix ( see fix atc command )
related
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1837

fix_modify AtC control
syntax
fix_modify AtC control <physics_type> <solution_parameter>
physics_type (string) = thermal | momentum• solution_parameter (string) = max_iterations | tolerance•
fix_modify AtC transfer <physics_type> control max_iterations <max_iterations>
max_iterations (int) = maximum number of iterations that will be used by iterative matrix solvers•
fix_modify AtC transfer <physics_type> control tolerance <tolerance>
tolerance (float) = relative tolerance to which matrix equations will be solved•
examples
fix_modify AtC control thermal max_iterations 10
fix_modify AtC control momentum tolerance 1.e-5
description
Sets the numerical parameters for the matrix solvers used in the specified control algorithm. Many solution
approaches require iterative solvers, and these methods enable users to provide the maximum number of
iterations and the relative tolerance.
restrictions
only for be used with specific controllers : thermal, momentum
They are ignored if a lumped solution is requested
related
default
max_iterations is the number of rows in the matrix
tolerance is 1.e-10
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes•
1838
Files• Directories•
LAMMPS Users Manual
1839

fix_modify AtC control momentum
syntax
fix_modify AtC control momentum none
fix_modify AtC control momentum rescale <frequency>
frequency (int) = time step frequency for applying displacement and velocity rescaling•
fix_modify AtC control momentum glc_displacement
fix_modify AtC control momentum glc_velocity
fix_modify AtC control momentum hoover
fix_modify AtC control momentum flux [faceset face_set_id, interpolate]
face_set_id (string) = id of boundary face set, if not specified (or not possible when the atomic
domain does not line up with mesh boundaries) defaults to an atomic-quadrature approximate
evaulation
•
examples
fix_modify AtC control momentum glc_velocity
fix_modify AtC control momentum flux faceset bndy_faces
description
restrictions
only to be used with specific transfers : elastic
rescale not valid with time filtering activated
related
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1840
fix_modify AtC control thermal
syntax
fix_modify AtC control thermal <control_type> <optional_args>
control_type (string) = none | rescale | hoover | flux•
fix_modify AtC control thermal rescale <frequency>
frequency (int) = time step frequency for applying velocity rescaling•
fix_modify AtC control thermal hoover
fix_modify AtC control thermal flux <boundary_integration_type(optional)> <face_set_id(optional)>
boundary_integration_type (string) = faceset | interpolate• face_set_id (string), optional = id of boundary face set, if not specified (or not possible when the
atomic domain does not line up with mesh boundaries) defaults to an atomic-quadrature approximate
evaulation, does not work with interpolate
•
examples
fix_modify AtC control thermal none
fix_modify AtC control thermal rescale 10
fix_modify AtC control thermal hoover
fix_modify AtC control thermal flux
fix_modify AtC control thermal flux faceset bndy_faces
description
Sets the energy exchange mechansim from the finite elements to the atoms, managed through a control
algorithm. Rescale computes a scale factor for each atom to match the finite element temperature. Hoover is a
Gaussian least-constraint isokinetic thermostat enforces that the nodal restricted atomic temperature matches
the finite element temperature. Flux is a similar mode, but rather adds energy to the atoms based on
conservation of energy. Hoover and flux allows the prescription of sources or fixed temperatures on the
atoms.
restrictions
only for be used with specific transfers : thermal (rescale, hoover, flux), two_temperature (flux)
rescale not valid with time filtering activated
related
1841


fix_modify AtC control thermal
correction_max_iterations
syntax
fix_modify AtC control thermal correction_max_iterations <max_iterations>
max_iterations (int) = maximum number of iterations that will be used by iterative matrix solvers•
examples
fix_modify AtC control thermal correction_max_iterations 10
description
Sets the maximum number of iterations to compute the 2nd order in time correction term for lambda with the
fractional step method. The method uses the same tolerance as the controller's matrix solver.
restrictions
only for use with thermal physics using the fractional step method.
related
default
correction_max_iterations is 20
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1843

fix_modify AtC decomposition
syntax
fix_modify AtC decomposition <type>
<type> =
replicated_memory: nodal information replicated on each processor
distributed_memory: only owned nodal information on processor
•
examples
fix_modify atc decomposition distributed_memory
description
Command for assigning the distribution of work and memory for parallel runs.
restrictions
replicated_memory is appropriate for simulations were the number of nodes << number of atoms
related
default
replicated_memory
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1844

fix_modify AtC extrinsic electron_integration
syntax
fix_modify AtC extrinsic electron_integration <integration_type> <num_subcyle_steps(optional)>
integration_type (string) = explicit | implicit | steady• num_subcycle_steps (int), optional = number of subcycle steps for the electron time integration•
examples
fix_modify AtC extrinsic electron_integration implicit
fix_modify AtC extrinsic electron_integration explicit 100
description
Switches between integration scheme for the electron temperature. The number of subcyling steps used to
integrate the electron temperature 1 LAMMPS timestep can be manually adjusted to capture fast electron
dynamics.
restrictions
For use only with two_temperature type of AtC fix ( see fix atc command )
default
implicit
subcycle_steps = 1
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1845

fix_modify AtC equilibrium_start
syntax
fix_modify AtC equilibrium_start <on|off>
examples
fix_modify atc equilibrium_start on
description
Starts filtered calculations assuming they start in equilibrium, i.e. perfect finite element force balance.
restrictions
only needed before filtering is begun
related
see fix_modify AtC filter
default
on
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1846

fix_modify AtC extrinsic exchange
syntax
fix_modify AtC extrinsic exchange <on|off>
examples
fix_modify AtC extrinsic exchange on
description
Switches energy exchange between the MD system and electron system on and off
restrictions
Only valid for use with two_temperature type of AtC fix.
related
see fix atc command
default
on
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1847

fix_modify AtC fe_md_boundary
syntax
fix_modify AtC fe_md_boundary <faceset | interpolate | no_boundary> [args]
examples
fix_modify atc fe_md_boundary interpolate
description
Specifies different methods for computing fluxes between between the MD and FE integration regions.
Faceset defines a faceset separating the MD and FE regions and uses finite element face quadrature to
compute the flux. Interpolate uses a reconstruction scheme to approximate the flux, which is more robust but
less accurate if the MD/FE boundary does correspond to a faceset. No boundary results in no fluxes between
the systems being computed.
restrictions
If faceset is used, all the AtC non-boundary atoms must lie within and completely fill the domain enclosed by
the faceset.
related
see for how to specify the faceset name.
default
Interpolate.
Generated on 21 Aug 2013 for ATC by 1.6.1
1848

fix_modify AtC fem create mesh
syntax
fix_modify AtC fem create mesh <nx> <ny> <nz> <region-id> <f|p> <f|p> <f|p>
nx ny nz = number of elements in x, y, z• region-id = id of region that is to be meshed• f p p = perioidicity flags for x, y, z•
examples
fix_modify AtC fem create mesh 10 1 1 feRegion p p p
description
Creates a uniform mesh in a rectangular region
restrictions
creates only uniform rectangular grids in a rectangular region
related
default
none
Generated on Mon Aug 17 09:35:16 2009 for ATC by 1.3.9.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1849

fix_modify AtC filter scale
syntax
fix_modify AtC filter scale <scale>
scale (real) = characteristic time scale of the filter•
examples
fix_modify AtC filter scale 10.0
description
Filters the MD dynamics to construct a more appropriate continuous field. Equilibrating first filters the time
derivatives without changing the dynamics to provide a better initial condition to the filtered dynamics
restrictions
only for be used with specific transfers: thermal, two_temperature
related
fix_modify AtC filter fix_modify AtC filter type
default
0.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1850

fix_modify AtC filter type
syntax
fix_modify AtC filter type <exponential | step | no_filter>
examples
fix_modify AtC filter type exponential
description
Specifies the type of time filter used.
restrictions
only for be used with specific transfers: thermal, two_temperature
related
fix_modify AtC filter fix_modify AtC filter scale
default
No default.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1851
fix atc command
syntax
fix <fixID> <group> atc <type> <parameter_file>
fixID = name of fix• group = name of group fix is to be applied• type
= thermal : thermal coupling with fields: temperature
= two_temperature : electron-phonon coupling with field: temperature and electron_temperature
= hardy : on-the-fly post-processing using kernel localization functions (see "related" section for
possible fields)
= field : on-the-fly post-processing using mesh-based localization functions (see "related" section for
possible fields)
•
parameter_file = name of the file with material parameters.
note: Neither hardy nor field requires a parameter file
•
examples
fix AtC internal atc thermal Ar_thermal.dat
fix AtC internal atc two_temperature Ar_ttm.mat
fix AtC internal atc hardy
fix AtC internal atc field
description
This fix is the beginning to creating a coupled FE/MD simulation and/or an on-the-fly estimation of
continuum fields. The coupled versions of this fix do Verlet integration and the /post-processing does not.
After instantiating this fix, several other fix_modify commands will be needed to set up the problem, e.g.
define the finite element mesh and prescribe initial and boundary conditions.
The following coupling example is typical, but non-exhaustive:
# ... commands to create and initialize the MD system
# initial fix to designate coupling type and group to apply it to
# tag group physics material_file
fix AtC internal atc thermal Ar_thermal.mat
# create a uniform 12 x 2 x 2 mesh that covers region contain the group
# nx ny nz region periodicity
fix_modify AtC mesh create 12 2 2 mdRegion f p p
# specify the control method for the type of coupling
# physics control_type
fix_modify AtC thermal control flux
# specify the initial values for the empirical field "temperature"
1852
# field node_group value
fix_modify AtC initial temperature all 30.
# create an output stream for nodal fields
# filename output_frequency
fix_modify AtC output atc_fe_output 100
run 1000
likewise for this post-processing example:
# ... commands to create and initialize the MD system
# initial fix to designate post-processing and the group to apply it to
# no material file is allowed nor required
fix AtC internal atc hardy
# for hardy fix, specific kernel function (function type and range) to #
be used as a localization function
fix AtC kernel quartic_sphere 10.0
# create a uniform 1 x 1 x 1 mesh that covers region contain the group
# with periodicity this effectively creats a system average
fix_modify AtC mesh create 1 1 1 box p p p
# change from default lagrangian map to eulerian
# refreshed every 100 steps
fix_modify AtC atom_element_map eulerian 100
# start with no field defined
# add mass density, potential energy density, stress and temperature
fix_modify AtC fields add density energy stress temperature
# create an output stream for nodal fields
# filename output_frequency
fix_modify AtC output nvtFE 100 text
run 1000
the mesh's linear interpolation functions can be used as the localization function
by using the field option:
fix AtC internal atc field
fix_modify AtC mesh create 1 1 1 box p p p
...
LAMMPS Users Manual
1853
Note coupling and post-processing can be combined in the same simulations using separate fixes.
For detailed exposition of the theory and algorithms please see:
Wagner, GJ; Jones, RE; Templeton, JA; Parks, MA, An atomistic-to-continuum coupling method for
heat transfer in solids. Special Issue of Computer Methods and Applied Mechanics (2008) 197:3351.
•
Zimmerman, JA; Webb, EB; Hoyt, JJ;. Jones, RE; Klein, PA; Bammann, DJ, Calculation of stress in
atomistic simulation. Special Issue of Modelling and Simulation in Materials Science and Engineering
(2004), 12:S319.
•
Zimmerman, JA; Jones, RE; Templeton, JA, A material frame approach for evaluating continuum
variables in atomistic simulations. Journal of Computational Physics (2010), 229:2364.
•
Templeton, JA; Jones, RE; Wagner, GJ, Application of a field-based method to spatially varying
thermal transport problems in molecular dynamics. Modelling and Simulation in Materials Science
and Engineering (2010), 18:085007.
•
Jones, RE; Templeton, JA; Wagner, GJ; Olmsted, D; Modine, JA, Electron transport enhanced
molecular dynamics for metals and semi-metals. International Journal for Numerical Methods in
Engineering (2010), 83:940.
•
Templeton, JA; Jones, RE; Lee, JW; Zimmerman, JA; Wong, BM, A long-range electric field solver
for molecular dynamics based on atomistic-to-continuum modeling. Journal of Chemical Theory and
Computation (2011), 7:1736.
•
Mandadapu, KK; Templeton, JA; Lee, JW, Polarization as a field variable from molecular dynamics
simulations. Journal of Chemical Physics (2013), 139:054115.
•
Please refer to the standard finite element (FE) texts, e.g. T.J.R Hughes The finite element method , Dover
2003, for the basics of FE simulation.
restrictions
Thermal and two_temperature (coupling) types use a Verlet time-integration algorithm. The hardy type does
not contain its own time-integrator and must be used with a separate fix that does contain one, e.g. nve, nvt,
etc.
Currently,
the coupling is restricted to thermal physics• the FE computations are done in serial on each processor.•
related
fix_modify commands for setup:
fix_modify AtC mesh create• fix_modify AtC mesh quadrature• fix_modify AtC mesh read• fix_modify AtC mesh write• fix_modify AtC mesh create_nodeset• fix_modify AtC mesh add_to_nodeset• fix_modify AtC mesh create_faceset box• fix_modify AtC mesh create_faceset plane• fix_modify AtC mesh create_elementset• fix_modify AtC mesh delete_elements•
LAMMPS Users Manual
1854
fix_modify AtC mesh nodeset_to_elementset• fix_modify AtC boundary• fix_modify AtC internal_quadrature• fix_modify AtC time_integration (thermal)• fix_modify AtC time_integration (momentum)• fix_modify AtC extrinsic electron_integration• fix_modify AtC internal_element_set• fix_modify AtC decomposition•
fix_modify commands for boundary and initial conditions:
fix_modify AtC initial• fix_modify AtC fix• fix_modify AtC unfix• fix_modify AtC fix_flux• fix_modify AtC unfix_flux• fix_modify AtC source• fix_modify AtC remove_source•
fix_modify commands for control and filtering:
fix_modify AtC control• fix_modify AtC control thermal• fix_modify AtC control thermal correction_max_iterations• fix_modify AtC control momentum• fix_modify AtC control localized_lambda• fix_modify AtC control lumped_lambda_solve• fix_modify AtC control mask_direction• fix_modify AtC filter• fix_modify AtC filter scale• fix_modify AtC filter type• fix_modify AtC equilibrium_start• fix_modify AtC extrinsic exchange• fix_modify AtC poisson_solver•
fix_modify commands for output:
fix_modify AtC output• fix_modify AtC output nodeset• fix_modify AtC output elementset• fix_modify AtC output boundary_integral• fix_modify AtC output contour_integral• fix_modify AtC mesh output• fix_modify AtC write_restart• fix_modify AtC read_restart•
fix_modify commands for post-processing:
fix_modify AtC kernel• fix_modify AtC fields• fix_modify AtC gradients•
LAMMPS Users Manual
1855

fix_modify AtC rates• fix_modify AtC computes• fix_modify AtC on_the_fly• fix_modify AtC pair_interactions/bond_interactions• fix_modify AtC sample_frequency• fix_modify AtC set•
miscellaneous fix_modify commands:
fix_modify AtC atom_element_map• fix_modify AtC atom_weight• fix_modify AtC write_atom_weights• fix_modify AtC reset_time• fix_modify AtC reset_atomic_reference_positions• fix_modify AtC fe_md_boundary• fix_modify AtC boundary_faceset• fix_modify AtC consistent_fe_initialization• fix_modify AtC mass_matrix• fix_modify AtC material• fix_modify AtC atomic_charge• fix_modify AtC source_integration• fix_modify AtC temperature_definition• fix_modify AtC track_displacement• fix_modify AtC boundary_dynamics• fix_modify AtC add_species• fix_modify AtC add_molecule• fix_modify AtC remove_species• fix_modify AtC remove_molecule•
Note: a set of example input files with the attendant material files are included with this package
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
LAMMPS Users Manual
1856

fix_modify AtC fix_flux
syntax
fix_modify AtC fix_flux <field> <face_set> <value | function>
<field> = field name valid for type of physics, temperature | electron_temperature• <face_set> = name of set of element faces•
examples
fix_modify atc fix_flux temperature faceSet 10.0
description
Command for fixing normal fluxes e.g. heat_flux. This command only prescribes the normal component of
the physical flux, e.g. heat (energy) flux. The units are in AtC units, i.e. derived from the LAMMPS length,
time, and mass scales.
restrictions
Only normal fluxes (Neumann data) can be prescribed.
related
see fix_modify AtC unfix_flux
default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1857

fix_modify AtC fix
syntax
fix_modify AtC fix <field> <nodeset> <constant | function>
<field> = field name valid for type of physics• <nodeset> = name of set of nodes to apply boundary condition• <constant | function> = value or name of function followed by its parameters•
examples
fix_modify AtC fix temperature groupNAME 10.
fix_modify AtC fix temperature groupNAME 0 0 0 10.0 0 0 1.0
description
Creates a constraint on the values of the specified field at specified nodes.
restrictions
keyword 'all' reserved in nodeset name
related
see fix_modify AtC unfix
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1858

fix_modify AtC computes
syntax
fix_modify AtC computes <add | delete> [per-atom compute id] <volume | number>
add | delete (keyword) = add or delete the calculation of an equivalent continuum field for the
specified per-atom compute as volume or number density quantity
•
per-atom compute id = name/id for per-atom compute, fields can be calculated for all per-atom
computes available from LAMMPS
•
volume | number (keyword) = field created is a per-unit-volume quantity or a per-atom quantity as
weighted by kernel functions
•
examples
compute virial all stress/atom
fix_modify AtC computes add virial volume
fix_modify AtC computes delete virial
compute centrosymmetry all centro/atom
fix_modify AtC computes add centrosymmetry number
description
Calculates continuum fields corresponding to specified per-atom computes created by LAMMPS
restrictions
Must be used with the hardy/field type of AtC fix ( see fix atc command )
Per-atom compute must be specified before corresponding continuum field can be requested
related
See manual page for compute
default
No defaults exist for this command
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1859
fix_modify AtC fields
syntax
fix_modify AtC fields <all | none>
fix_modify AtC fields <add | delete> <list_of_fields>
all | none (keyword) = output all or no fields• add | delete (keyword) = add or delete the listed output fields• fields (keyword) =
density : mass per unit volume
displacement : displacement vector
momentum : momentum per unit volume
velocity : defined by momentum divided by density
projected_velocity : simple kernel estimation of atomic velocities
temperature : temperature derived from the relative atomic kinetic energy (as done by )
kinetic_temperature : temperature derived from the full kinetic energy
number_density : simple kernel estimation of number of atoms per unit volume
stress : Cauchy stress tensor for eulerian analysis (atom_element_map), or 1st Piola-Kirchhoff stress
tensor for lagrangian analysis
transformed_stress : 1st Piola-Kirchhoff stress tensor for eulerian analysis (atom_element_map), or
Cauchy stress tensor for lagrangian analysis
heat_flux : spatial heat flux vector for eulerian, or referential heat flux vector for lagrangian
potential_energy : potential energy per unit volume
kinetic_energy : kinetic energy per unit volume
thermal_energy : thermal energy (kinetic energy - continuum kinetic energy) per unit volume
internal_energy : total internal energy (potential + thermal) per unit volume
energy : total energy (potential + kinetic) per unit volume
number_density : number of atoms per unit volume
eshelby_stress: configurational stress (energy-momentum) tensor defined by Eshelby [References:
Philos. Trans. Royal Soc. London A, Math. Phys. Sci., Vol. 244, No. 877 (1951) pp. 87-112; J.
Elasticity, Vol. 5, Nos. 3-4 (1975) pp. 321-335]
vacancy_concentration: volume fraction of vacancy content
type_concentration: volume fraction of a specific atom type
•
examples
fix_modify AtC fields add velocity temperature
description
Allows modification of the fields calculated and output by the transfer class. The commands are cumulative,
e.g.
fix_modify AtC fields none
followed by
fix_modify AtC fields add velocity temperature
will only output the velocity and temperature fields.
1860

restrictions
Must be used with the hardy/field type of AtC fix, see fix atc command. Currently, the stress and heat flux
formulas are only correct for central force potentials, e.g. Lennard-Jones and EAM but not Stillinger-Weber.
related
See fix_modify AtC gradients , fix_modify AtC rates and fix_modify AtC computes
default
By default, no fields are output
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
LAMMPS Users Manual
1861

fix_modify AtC gradients
syntax
fix_modify AtC gradients <add | delete> <list_of_fields>
add | delete (keyword) = add or delete the calculation of gradients for the listed output fields• fields (keyword) =
gradients can be calculated for all fields listed in fix_modify AtC fields
•
examples
fix_modify AtC gradients add temperature velocity stress
fix_modify AtC gradients delete velocity
description
Requests calculation and ouput of gradients of the fields from the transfer class. These gradients will be with
regard to spatial or material coordinate for eulerian or lagrangian analysis, respectively, as specified by
atom_element_map (see fix_modify AtC atom_element_map )
restrictions
Must be used with the hardy/field type of AtC fix ( see fix atc command )
related
default
No gradients are calculated by default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1862

fix_modify AtC kernel
syntax
fix_modify AtC kernel <type> <parameters>
type (keyword) = step, cell, cubic_bar, cubic_cylinder, cubic_sphere, quartic_bar, quartic_cylinder,
quartic_sphere
•
parameters :
step = radius (double)
cell = hx, hy, hz (double) or h (double)
cubic_bar = half-width (double)
cubic_cylinder = radius (double)
cubic_sphere = radius (double)
quartic_bar = half-width (double)
quartic_cylinder = radius (double)
quartic_sphere = radius (double)
•
examples
fix_modify AtC kernel cell 1.0 1.0 1.0
fix_modify AtC kernel quartic_sphere 10.0
description
restrictions
Must be used with the hardy AtC fix
For bar kernel types, half-width oriented along x-direction
For cylinder kernel types, cylindrical axis is assumed to be in z-direction
( see fix atc command )
related
default
No default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1863

fix_modify AtC on_the_fly
syntax
fix_modify AtC on_the_fly <bond | kernel> <optional on | off>
- bond | kernel (keyword) = specifies on-the-fly calculation of bond or kernel matrix elements
on | off (keyword) = activate or discontinue on-the-fly mode•
examples
fix_modify AtC on_the_fly bond on
fix_modify AtC on_the_fly kernel
fix_modify AtC on_the_fly kernel off
description
Overrides normal mode of pre-calculating and storing bond pair-to-node a nd kernel atom-to-node matrices. If
activated, will calculate elements of t hese matrices during repeated calls of field computations (i.e.
"on-the-fly") and not store them for future use.
on flag is optional - if omitted, on_the_fly will be activated for the s pecified matrix. Can be deactivated using
off flag.
restrictions
Must be used with the hardy/field type of AtC fix ( see fix atc command )
related
default
By default, on-the-fly calculation is not active (i.e. off). However, code does a memory allocation check to
determine if it can store all needed bond and kernel matrix ele ments. If this allocation fails, on-the-fly is
activated.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1864

fix_modify AtC rates
syntax
fix_modify AtC rates <add | delete> <list_of_fields>
add | delete (keyword) = add or delete the calculation of rates (time derivatives) for the listed output
fields
•
fields (keyword) =
rates can be calculated for all fields listed in fix_modify AtC fields
•
examples
fix_modify AtC rates add temperature velocity stress
fix_modify AtC rates delete stress
description
Requests calculation and ouput of rates (time derivatives) of the fields from the transfer class. For eulerian
analysis (see fix_modify AtC atom_element_map ), these rates are the partial time derivatives of the nodal
fields, not the full (material) time derivatives.
restrictions
Must be used with the hardy/field type of AtC fix ( see fix atc command )
related
default
No rates are calculated by default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1865

fix_modify AtC initial
syntax
fix_modify AtC initial <field> <nodeset> <constant | function>
<field> = field name valid for type of physics, temperature | electron_temperature• <nodeset> = name of set of nodes to apply initial condition• <constant | function> = value or name of function followed by its parameters•
examples
fix_modify atc initial temperature groupNAME 10.
description
Sets the initial values for the specified field at the specified nodes.
restrictions
keyword 'all' reserved in nodeset name
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1866

fix_modify AtC internal_atom_integrate
syntax
fix_modify AtC internal_atom_integrate <on | off> fix_modify AtC internal_atom_integrate
on
description
Has AtC perform time integration for the atoms in the group on which it operates. This does not include
boundary atoms.
default
on for coupling methods, off for post-processors off
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1867

fix_modify AtC internal_element_set
syntax
fix_modify AtC internal_element_set <element-set-name>
<element-set-name> = name of element set defining internal region, or off•
examples
fix_modify AtC internal_element_set myElementSet fix_modify AtC
internal_element_set off
description
Enables AtC to base the region for internal atoms to be an element set. If no ghost atoms are used, all the AtC
atoms must be constrained to remain in this element set by the user, e.g., with walls. If boundary atoms are
used in conjunction with Eulerian atom maps AtC will partition all atoms of a boundary or internal type to be
of type internal if they are in the internal region or to be of type boundary otherwise.
restrictions
If boundary atoms are used in conjunction with Eulerian atom maps, the Eulerian reset frequency must be an
integer multiple of the Lammps reneighbor frequency
related
see atom_element_map_type and boundary
default
off
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1868

fix_modify AtC internal_quadrature
syntax
fix_modify atc internal_quadrature <on | off> [region]
examples
fix_modify atc internal_quadrature off
description
Command to use or not use atomic quadrature on internal elements fully filled with atoms. By turning the
internal quadrature off these elements do not contribute to the governing PDE and the fields at the internal
nodes follow the weighted averages of the atomic data.
optional
Optional region tag specifies which finite element nodes will be treated as being within the MD region. This
option is only valid with internal_quadrature off.
restrictions
related
default
on
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1869

fix_modify AtC kernel
syntax
fix_modify AtC kernel <type> <parameters>
type (keyword) = step, cell, cubic_bar, cubic_cylinder, cubic_sphere, quartic_bar, quartic_cylinder,
quartic_sphere
•
parameters :
step = radius (double)
cell = hx, hy, hz (double) or h (double)
cubic_bar = half-width (double)
cubic_cylinder = radius (double)
cubic_sphere = radius (double)
quartic_bar = half-width (double)
quartic_cylinder = radius (double)
quartic_sphere = radius (double)
•
examples
fix_modify AtC kernel cell 1.0 1.0 1.0 fix_modify AtC kernel quartic_sphere 10.0
description
restrictions
Must be used with the hardy AtC fix
For bar kernel types, half-width oriented along x-direction
For cylinder kernel types, cylindrical axis is assumed to be in z-direction
( see fix atc command )
related
default
No default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1870

fix_modify AtC control localized_lambda
syntax
fix_modify AtC control localized_lambda <on|off>
examples
fix_modify atc control localized_lambda on
description
Turns on localization algorithms for control algorithms to restrict the influence of FE coupling or boundary
conditions to a region near the boundary of the MD region. Control algorithms will not affect atoms in
elements not possessing faces on the boundary of the region. Flux-based control is localized via row-sum
lumping while quantity control is done by solving a truncated matrix equation.
restrictions
related
default
Default is off.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1871

fix_modify AtC control lumped_lambda_solve
syntax
fix_modify AtC control lumped_lambda_solve <on|off>
examples
fix_modify atc control lumped_lambda_solve on
description
Command to use or not use lumped matrix for lambda solve
restrictions
related
default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1872

fix_modify AtC control mask_direction
syntax
fix_modify AtC control mask_direction <direction> <on|off>
examples
fix_modify atc control mask_direction 0 on
description
Command to mask out certain dimensions from the atomic regulator
restrictions
related
default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1873

fix_modify AtC mass_matrix
syntax
fix_modify AtC mass_matrix <fe | md_fe>
<fe | md_fe> = activiate/deactiviate using the FE mass matrix in the MD region•
examples
fix_modify atc mass_matrix fe
description
Determines whether AtC uses the FE mass matrix based on Gaussian quadrature or based on atomic
quadrature in the MD region. This is useful for fully overlapping simulations to improve efficiency.
restrictions
Should not be used unless the FE region is contained within the MD region, otherwise the method will be
unstable and inaccurate
related
default
Default is off
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1874

fix_modify AtC material
syntax
fix_modify AtC material [elementset_name] [material_id]
examples
fix_modify AtC material gap_region 2
description
Sets the material model in elementset_name to be of type material_id.
restrictions
The element set must already be created and the material must be specified in the material file given the the
atc fix on construction
related
default
All elements default to the first material in the material file.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1875

fix_modify AtC mesh add_to_nodeset
syntax
fix_modify AtC mesh add_to_nodeset <id> <xmin> <xmax> <ymin> <ymax> <zmin> <zmax>
<id> = id of FE nodeset to be added to• <xmin> <xmax> <ymin> <ymax> <zmin> <zmax> = coordinates of the bounding box that contains
the desired nodes to be added
•
examples
fix_modify AtC mesh add_to_nodeset lbc -11.9 -11 -12 12 -12 12
description
Command to add nodes to an already existing FE nodeset.
restrictions
related
default
Coordinates are assumed to be in lattice units.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1876

fix_modify AtC mesh create
syntax
fix_modify AtC mesh create <nx> <ny> <nz> <region-id> <f|p> <f|p> <f|p>
nx ny nz = number of elements in x, y, z• region-id = id of region that is to be meshed• f p p = periodicity flags for x, y, z•
examples
fix_modify AtC mesh create 10 1 1 feRegion p p p
description
Creates a uniform mesh in a rectangular region
restrictions
Creates only uniform rectangular grids in a rectangular region
related
fix_modify AtC mesh quadrature
default
When created, mesh defaults to gauss2 (2-point Gaussian) quadrature. Use "mesh quadrature" command to
change quadrature style.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1877

fix_modify AtC mesh create_elementset
syntax
fix_modify AtC create_elementset <id> <xmin> <xmax> <ymin> <ymax> <zmin> <zmax>
<id> = id to assign to the collection of FE element• <xmin> <xmax> <ymin> <ymax> <zmin> <zmax> = coordinates of the bounding box that contains
only the desired elements
•
examples
fix_modify AtC mesh create_elementset middle -4.1 4.1 -100 100 -100 1100
description
Command to assign an id to a set of FE elements to be used subsequently in defining material and mesh-based
operations.
restrictions
Only viable for rectangular grids.
related
default
Coordinates are assumed to be in lattice units.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1878

fix_modify AtC mesh create_faceset box
syntax
fix_modify AtC mesh create_faceset <id> box <xmin> <xmax> <ymin> <ymax> <zmin> <zmax> <in|out>
[units]
<id> = id to assign to the collection of FE faces• <xmin> <xmax> <ymin> <ymax> <zmin> <zmax> = coordinates of the bounding box that is
coincident with the desired FE faces
•
<in|out> = "in" gives inner faces to the box, "out" gives the outer faces to the box• units = option to specify real as opposed to lattice units•
examples
fix_modify AtC mesh create_faceset obndy box -4.0 4.0 -12 12 -12 12 out
description
Command to assign an id to a set of FE faces.
restrictions
Only viable for rectangular grids.
related
default
The default options are units = lattice and the use of outer faces
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1879

fix_modify AtC mesh create_faceset plane
syntax
fix_modify AtC mesh create_faceset <id> plane <x|y|z> <val1> <x|y|z> <lval2> <uval2> [units]
<id> = id to assign to the collection of FE faces• <x|y|z> = coordinate directions that define plane on which faceset lies• <val1>,<lval2>,<uval2> = plane is specified as the x|y|z=val1 plane bounded by the segments x|y|z =
[lval2,uval2]
•
units = option to specify real as opposed to lattice units•
examples
fix_modify AtC mesh create_faceset xyplane plane y 0 x -4 0
description
Command to assign an id to a set of FE faces.
restrictions
Only viable for rectangular grids.
related
default
The default option is units = lattice.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1880

fix_modify AtC mesh create_nodeset
syntax
fix_modify AtC mesh create_nodeset <id> <xmin> <xmax> <ymin> <ymax> <zmin> <zmax>
<id> = id to assign to the collection of FE nodes• <xmin> <xmax> <ymin> <ymax> <zmin> <zmax> = coordinates of the bounding box that contains
only the desired nodes
•
examples
fix_modify AtC mesh create_nodeset lbc -12.1 -11.9 -12 12 -12 12
description
Command to assign an id to a set of FE nodes to be used subsequently in defining boundary conditions.
restrictions
related
default
Coordinates are assumed to be in lattice units.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1881

fix_modify AtC mesh delete_elements
syntax
fix_modify AtC mesh delete_elements <element_set>
<element_set> = name of an element set•
examples
fix_modify AtC delete_elements gap
description
Deletes a group of elements from the mesh.
restrictions
related
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1882

fix_modify AtC mesh nodeset_to_elementset
syntax
fix_modify AtC nodeset_to_elementset <nodeset_id> <elementset_id> <max/min>
<nodeset_id> = id of desired nodeset from which to create elementset• <elementset_id> = id to assign to the collection of FE element• <max/min> = flag to choose either the maximal or minimal elementset•
examples
fix_modify AtC mesh nodeset_to_elementset myNodeset myElementset min
description
Command to create an elementset from an existing nodeset. Either the minimal element set of elements with
all nodes in the set, or maximal element set with all elements with at least one node in the set, can be created
restrictions
None.
related
default
Unless specified, the maximal element set is created
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1883

fix_modify AtC mesh output
syntax
fix_modify AtC mesh output <file_prefix>
examples
fix_modify AtC mesh output meshData
description
Command to output mesh and associated data: nodesets, facesets, and elementsets. This data is only output
once upon initialization since currently the mesh is static. Creates (binary, "gold" format) Ensight output of
mesh data.
restrictions
none
related
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1884

fix_modify AtC mesh quadrature
syntax
fix_modify AtC mesh quadrature <quad>
quad = one of <nodal|gauss1|gauss2|gauss3|face> --- when a mesh is created it defaults to gauss2, use
this call to change it after the fact
•
examples
fix_modify AtC mesh quadrature face
description
(Re-)assigns the quadrature style for the existing mesh.
restrictions
related
fix_modify AtC mesh create
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1885

fix_modify AtC mesh read
syntax
fix_modify AtC mesh read <filename> <f|p> <f|p> <f|p>
filename = name of file containing mesh to be read• f p p = periodicity flags for x, y, z•
examples
fix_modify AtC mesh read myComponent.mesh p p p
fix_modify AtC mesh read myOtherComponent.exo
description
Reads a mesh from a text or exodus file, and assigns periodic boundary conditions if needed.
restrictions
related
default
periodicity flags are false by default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1886

fix_modify AtC mesh write
syntax
fix_modify AtC mesh write <filename>
filename = name of file to write mesh•
examples
fix_modify AtC mesh write myMesh.mesh
description
Writes a mesh to a text file.
restrictions
related
default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1887

fix_modify AtC time_integration (momentum)
syntax
fix_modify AtC time_integration <descriptor>
descriptor (string) = time integration type•
various time integration methods for the finite elements
description
verlet - atomic velocity update with 2nd order Verlet, nodal temperature update with 2nd order Verlet,
kinetostats based on controlling force
fractional_step - atomic velocity update with 2nd order Verlet, mixed nodal momentum update, 2nd order
Verlet for continuum and exact 2nd order Verlet for atomic contributions, kinetostats based on controlling
discrete momentum changes
gear - atomic velocity update with 2nd order Verlet, nodal temperature update with 3rd or 4th order Gear,
kinetostats based on controlling power
examples
fix_modify atc time_integration verlet
fix_modify atc time_integration fractional_step
description
related
see fix atc command
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1888

fix_modify AtC output
syntax
fix_modify AtC output <filename_prefix> <frequency> [text | full_text | binary | vector_components |
tensor_components ] fix_modify AtC output index [step | time ]
filename_prefix (string) = prefix for data files• frequency (integer) = frequency of output in time-steps• options (keyword/s):
text = creates text output of index, step and nodal variable values for unique nodes
full_text = creates text output index, nodal id, step, nodal coordinates and nodal variable values for
unique and image nodes
binary = creates binary Ensight output
vector_components = outputs vectors as scalar components
tensor_components = outputs tensor as scalar components (use this for Paraview)
•
examples
fix_modify AtC output heatFE 100
fix_modify AtC output hardyFE 1 text tensor_components
fix_modify AtC output hardyFE 10 text binary tensor_components
fix_modify AtC output index step
description
Creates text and/or binary (Ensight, "gold" format) output of nodal/mesh data which is transfer/physics
specific. Output indexed by step or time is possible.
restrictions
related
see fix atc command
default
no default format output indexed by time
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1889

fix_modify AtC output elementset
syntax
fix_modify AtC output volume_integral <eset_name> <field> {`
set_name (string) = name of elementset to be integrated over• fieldname (string) = name of field to integrate csum = creates nodal sum over nodes in specified
nodeset
•
examples
fix_modify AtC output eset1 mass_density
description
Performs volume integration of specified field over elementset and outputs resulting variable values to
GLOBALS file.
restrictions
related
see fix atc command
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1890

fix_modify AtC output nodeset
syntax
fix_modify AtC output nodeset <nodeset_name> <operation>
nodeset_name (string) = name of nodeset to be operated on• operation (keyword/s):
sum = creates nodal sum over nodes in specified nodeset
•
examples
fix_modify AtC output nodeset nset1 sum
description
Performs operation over the nodes belonging to specified nodeset and outputs resulting variable values to
GLOBALS file.
restrictions
related
see fix atc command
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1891

fix_modify AtC pair_interactions/bond_interactions
syntax
fix_modify AtC pair_interactions <on|off>
fix_modify AtC bond_interactions <on|off>
examples
fix_modify AtC bond_interactions on
description
include bonds and/or pairs in the stress and heat flux computations
restrictions
related
default
pair interactions: on, bond interactions: off
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1892

fix_modify AtC poisson_solver
syntax
fix_modify AtC poisson_solver mesh create <nx> <ny> <nz> <region-id> <f|p> <f|p> <f|p>
nx ny nz = number of elements in x, y, z• region-id = id of region that is to be meshed• f p p = perioidicity flags for x, y, z•
examples
fix_modify AtC poisson_solver mesh create 10 1 1 feRegion p p p
description
Creates a uniform mesh in a rectangular region
restrictions
creates only uniform rectangular grids in a rectangular region
related
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1893

fix_modify AtC read_restart
syntax
fix_modify AtC read_restart [file_name]
examples
fix_modify AtC read_restart ATC_state
description
Reads the current state of the fields from a named text-based restart file.
restrictions
The restart file only contains fields and their time derivatives. The reference positions of the atoms and the
commands that initialize the fix are not saved e.g. an identical mesh containing the same atoms will have to be
recreated.
related
see write_restart fix_modify AtC write_restart
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1894

fix_modify AtC remove_molecule
syntax
fix_modify_AtC remove_molecule <TAG>
<TAG> = tag for tracking a molecule type•
examples
fix_modify AtC remove_molecule water
description
Removes tag designated for tracking a specified set of molecules.
restrictions
related
default
No defaults for this command.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1895

fix_modify AtC remove_source
syntax
fix_modify AtC remove_source <field> <element_set>
<field> = field name valid for type of physics• <element_set> = name of set of elements•
examples
fix_modify atc remove_source temperature groupNAME
description
Remove a domain source.
restrictions
keyword 'all' reserved in element_set name
related
see fix_modify AtC source
default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1896

fix_modify AtC remove_species
syntax
fix_modify_AtC delete_species <TAG>
<TAG> = tag for tracking a species•
examples
fix_modify AtC remove_species gold
description
Removes tag designated for tracking a specified species.
restrictions
related
default
No defaults for this command.
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1897

fix_modify AtC reset_atomic_reference_positions
syntax
fix_modify AtC reset_atomic_reference_positions
examples
fix_modify atc reset_atomic_reference_positions
description
Resets the atomic positions ATC uses to perform point to field
operations. In can be used to use perfect lattice sites in ATC but a
thermalized or deformed lattice in LAMMPS.
restrictions
related
default
Default is off
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1898

fix_modify AtC reset_time
syntax
fix_modify AtC reset_time
examples
fix_modify atc reset_time 0.0
description
Resets the simulation time counter.
restrictions
related
default
syntax
fix_modify AtC kernel_bandwidth
examples
fix_modify atc reset_time 8
description
Sets a maximum parallel bandwidth for the kernel functions during parallel communication. If the command
is not issued, the default will be to assume the bandwidth of the kernel matrix corresponds to the number of
sampling locations.
restrictions
Only is used if kernel functions are being used.
related
default
Number of sample locations.
Generated on 21 Aug 2013 for ATC by 1.6.1
1899
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
LAMMPS Users Manual
1900

fix_modify AtC sample_frequency
syntax
fix_modify AtC sample_frequency [freq]
freq (int) : frequency to sample field in number of steps•
examples
fix_modify AtC sample_frequency 10
description
Specifies a frequency at which fields are computed for the case where
time filters are being applied.
restrictions
Must be used with the hardy/field AtC fix ( see fix atc command ) and is
only relevant when time filters are being used.
related
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1901

fix_modify AtC set
syntax
fix_modify AtC set reference_potential_energy <value_or_filename(optional)>
value (double) : optional user specified zero point for PE in native LAMMPS energy units• filename (string) : optional user specified string for file of nodal PE values to be read-in•
examples
fix_modify AtC set reference_potential_energy
fix_modify AtC set reference_potential_energy -0.05
fix_modify AtC set reference_potential_energy myPEvalues
description
Used to set various quantities for the post-processing algorithms. It sets the zero point for the potential energy
density using the value provided for all nodes, or from the current configuration of the lattice if no value is
provided, or values provided within the specified filename.
restrictions
Must be used with the hardy/field type of AtC fix ( see fix atc command )
related
default
Defaults to lammps zero point i.e. isolated atoms
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1902

fix_modify AtC source
syntax
fix_modify AtC source <field> <element_set> <value | function>
<field> = field name valid for type of physics• <element_set> = name of set of elements•
examples
fix_modify atc source temperature middle temporal_ramp 10. 0.
description
Add domain sources to the mesh. The units are consistent with LAMMPS's units for mass, length and time
and are defined by the PDE being solved, e.g. for thermal transfer the balance equation is for energy and
source is energy per time.
restrictions
keyword 'all' reserved in element_set name
related
see fix_modify AtC remove_source
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1903


fix_modify AtC temperature_definition
syntax
fix_modify AtC temperature_definition <kinetic|total>
examples
fix_modify atc temperature_definition kinetic
description
Change the definition for the atomic temperature used to create the finite element temperature. The kinetic
option is based only on the kinetic energy of the atoms while the total option uses the total energy (kinetic +
potential) of an atom.
restrictions
This command is only valid when using thermal coupling. Also, while not a formal restriction, the user should
ensure that associating a potential energy with each atom makes physical sense for the total option to be
meaningful.
default
kinetic
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1905

fix_modify AtC time_integration (thermal)
syntax
fix_modify AtC time_integration <descriptor>
descriptor (string) = time integration type•
various time integration methods for the finite elements
description
gear - atomic velocity update with 2nd order Verlet, nodal temperature update with 3rd or 4th order Gear,
thermostats based on controlling power
fractional_step - atomic velocity update with 2nd order Verlet, mixed nodal temperature update, 3/4 Gear for
continuum and 2 Verlet for atomic contributions, thermostats based on controlling discrete energy changes
examples
fix_modify atc time_integration gear
fix_modify atc time_integration fractional_step
description
related
see fix atc command
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1906

fix_modify AtC filter
syntax
fix_modify AtC filter <on | off | equilibrate>
on | off (keyword) = turns filter on or off• equilibrate = runs dynamics without filtering but initializes filtered quantities•
examples
fix_modify atc transfer filter on
description
Filters the MD dynamics to construct a more appropriate continuous field. Equilibrating first filters the time
derivatives without changing the dynamics to provide a better initial condition to the filtered dynamics
restrictions
only for be used with specific transfers: thermal, two_temperature
related
fix_modify AtC filter scale
fix_modify AtC equilibrium_start
default
off
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1907

fix_modify AtC track_displacement
syntax
fix_modify AtC track_displacement <on/off>
examples
fix_modify atc track_displacement on
description
Determines whether displacement is tracked or not. For solids problems this is a useful quantity, but for fluids
it is not relevant.
restrictions
Some constitutive models require the displacement field
default
on
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1908

fix_modify AtC unfix_flux
syntax
fix_modify AtC fix_flux <field> <face_set> <value | function>
<field> = field name valid for type of physics, temperature | electron_temperature• <face_set> = name of set of element faces•
examples
fix_modify atc unfix_flux temperature faceSet
description
Command for removing prescribed normal fluxes e.g. heat_flux, stress.
restrictions
related
see fix_modify AtC unfix_flux
default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1909

fix_modify AtC unfix
syntax
fix_modify AtC unfix <field> <nodeset>
<field> = field name valid for type of physics• <nodeset> = name of set of nodes•
examples
fix_modify AtC unfix temperature groupNAME
description
Removes constraint on field values for specified nodes.
restrictions
keyword 'all' reserved in nodeset name
related
see fix_modify AtC fix
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1910

fix_modify AtC write_atom_weights
syntax
fix_modify AtC write_atom_weights <filename> <frequency>
<filename> = name of file that atomic weights are written to• <frequency> = how often writes will occur•
examples
fix_modify atc write_atom_weights atm_wt_file.txt 10
description
Command for writing the values of atomic weights to a specified file.
restrictions
related
default
Generated on 21 Aug 2013 for ATC by 1.6.1
Main Page• Related Pages• Namespaces• Classes• Files• Directories•
1911

fix_modify AtC write_restart
syntax
fix_modify AtC write_restart [file_name]
examples
fix_modify AtC write_restart restart.mydata
description
Dumps the current state of the fields to a named text-based restart file. This done when the command is
invoked and not repeated, unlike the similar lammps command.
restrictions
The restart file only contains fields and their time derivatives. The reference positions of the atoms and the
commands that initialize the fix are not saved e.g. an identical mesh containing the same atoms will have to be
recreated.
related
see read_restart fix_modify AtC read_restart
default
none
Generated on 21 Aug 2013 for ATC by 1.6.1
1912...............................................................................................................................................................................1
