Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 41
User Manual
LRSDAY: Long-read Sequencing Data Analysis for Yeasts
Release v1.1.0
(2018-07-11)
Author Contact:
Jia-Xing Yue (岳家兴)
Email: yuejiaxing@gmail.com
GitHub: yjx1217
Twitter: iAmphioxus
Website: http://www.iamphioxus.org
Table of Contents
INTRODUCTION .......................................................................................................................... 1!
Background+......................................................................................................................................................................................+1!
Overview+of+the+LRSDAY+workflow+.......................................................................................................................................+2!
Comparison+with+other+methods+............................................................................................................................................+3!
Experimental+Design+....................................................................................................................................................................+4!
Limitations+and+potential+adaptation+.................................................................................................................................+8!
Expected+improvements+.............................................................................................................................................................+8!
CITATIONS .................................................................................................................................. 9!
MATERIALS ................................................................................................................................. 9!
Hardware,+operating+system+and+network........................................................................................................................+9!
Software+or+library+requirements+..........................................................................................................................................+9!
Input+data+......................................................................................................................................................................................+10!
Example+data+...............................................................................................................................................................................+10!
PROCEDURE .............................................................................................................................. 12!
Download,+install+and+configure+LRSDAY+++●+TIMING&<1&h+...................................................................................+12!
Run+LRSDAY+with+the+testing+example+●+TIMING&50&h+...........................................................................................+14!
? TROUBLESHOOTING .............................................................................................................. 25!
●
TIMING ................................................................................................................................ 28!
ANTICIPATED RESULTS ............................................................................................................. 29!
REFERENCES ............................................................................................................................. 33!
APPENDIX ................................................................................................................................. 36!
Appendix+1:+Pre-shipped+supporting+data+for+LRSDAY+.............................................................................................+36!
Appendix+2:+Tips+for+adapting+LRSDAY+to+other+eukaryotic+organisms+...........................................................+38!
1
INTRODUCTION
Background
Twenty years ago, the genome sequence of the budding yeast Saccharomyces cerevisiae was
published1. As the first complete eukaryotic genome ever sequenced, this marked a major
scientific milestone in biology. Since then, the genomes of many model and non-model organisms
have been sequenced, with this process accelerating after the emergence of next-generation
sequencing (NGS) technologies. Despite the notably improved throughputs, NGS technologies
suffer from the limitation of short reads and usually result in highly fragmented genome
assemblies containing numerous gaps and local mis-assemblies. The recently developed long-
read sequencing technologies represented by Pacific Biosciences (PacBio) and Oxford Nanopore
offer compelling alternatives to overcome such hurdles, producing high-quality genome
assemblies with substantially improved continuity and accuracy. Although initially tested in
microbial genome sequencing, their recent applications in complex mammalian and plant
genomes also achieved high-quality results2–6. With such new sequencing technologies,
challenging genomic regions with enriched repetitive elements, strongly biased GC-content, or
complex structural variants can often be correctly resolved. It is therefore anticipated that genome
sequencing projects will routinely adopt long-read-based sequencing technologies in the coming
years to gain insight in these complex genomic regions.
Yeast is a leading model organism with great importance in both basic biomedical research and
biotechnological applications. Its small genome size makes it particularly suitable for long-read-
based high-coverage genome sequencing. The resulting complete genome assembly with fully-
resolved subtelomere structure can in turn illuminate the genetic basis of many complex
phenotypic traits with unprecedented resolution. Recently, we used the long-read sequencing
technologies to generate the first panel of population-level end-to-end reference genomes of 12
yeast strains representing major subpopulations of the partially domesticated S. cerevisiae and
its sister species Saccharomyces paradoxus7. In addition, there have been a number of other
studies carrying out long-read sequencing for many S. cerevisiae strains8–11. Given the vast
genomic and phenotypic diversity of S. cerevisiae, we expect the incoming collection of long-read-
based high-quality genome assemblies of strains from widespread geographic locations and
ecological niches will substantially deepen our understanding in the S. cerevisiae natural genetic
variation and its associated biotechnological values.
2
Overview of the LRSDAY workflow
Here we present a highly organized and modular computational framework named Long-Read
Sequencing Data Analysis for Yeasts (LRSDAY), which enables automated high-quality yeast
genome assembly and annotation production from raw long-read sequencing data. The prototype
of LRSDAY has been developed to generate the yeast population reference panel (YPRP) in our
previous study7. Under the hood, LRSDAY contains a series of task-specific modules handling
long-read-based de novo genome assembly, short-read-based assembly polishing, reference-
guided assembly scaffolding, as well as comprehensive genomic feature annotations. These
tasks can be run individually, selectively or coordinately depending on users’ needs. LRSDAY
supports both leading long-read sequencing technologies: PacBio and Oxford Nanopore.
Running the full LRSDAY workflow, the final output is a chromosome-level genome assembly with
high-quality annotations of centromeres, protein-coding genes, tRNAs, transposable elements
(TEs; Ty1-Ty5 for S. cerevisiae and S. paradoxus), and telomere associated core X and Y’
elements. LRSDAY is shipped with various auxiliary scripts, configuration files and supporting
data that enable its semi-automatic installation, configuration, and execution with minimal manual
intervention (Appendix 1). This design concept greatly alleviates the technical barrier for bench
biologists with limited bioinformatics experiences. In addition, a real case example and its final
outputs are also provided for users’ test and comparison. All these task-specific modules, auxiliary
files, installed tools, sample outputs, together with the user-created project directories for the
testing example and users’ own data are hosted under the same home directory
($LRSDAY_HOME) in a self-contained fashion (Fig. 1). This design makes LRSDAY well-isolated
from the rest of the system and therefore greatly improves its portability. To sum up, LRSDAY is
a highly transparent, automated and powerful computational framework that handles both
genome assembly and annotation, which suits the needs of the yeast community in performing
long-read-based genome sequencing projects. In the PROCEDURE section of this article, we
provide a step-by-step walkthrough on how to install, configure, and run LRSDAY with our
prepared testing example.
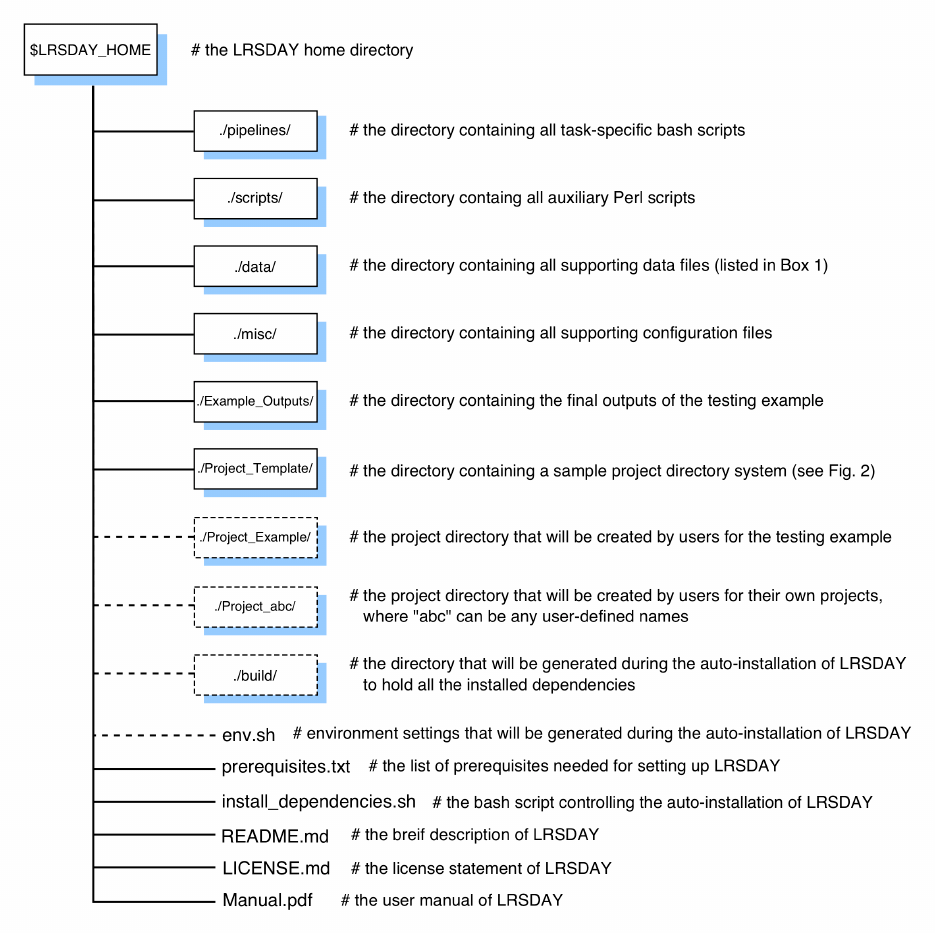
3
Figure 1. Overview of the LRSDAY directory system. All the top-level directories (boxes, solid
lines) and individual files of LRSDAY are listed and briefly described. Additional directories and
files will be generated during the installation or execution of LRSDAY (boxes, dashed lines).
Comparison with other methods
Genome assembly is a rapidly moving field, co-evolving with the fast-paced development of
sequencing technologies. In recent years, both hybrid (i.e. using both long and short reads) and
4
native (i.e. using long reads only) assemblers that support long-read sequencing data have been
developed and tested on many different organisms8,12–17. As for gene annotation, there is also a
wide range of choices that perform gene model prediction in an ab initio fashion or based on
additional evidences (e.g. mRNA transcripts, protein-sequence alignment)18–20. Specifically for
yeasts, a web-based gene annotation tool has been developed that combines both approaches21.
However, there is currently no integrated solution that handles both genome assembly and
annotation in a seamless way. To fill this gap, we revamped our original workflow for deriving the
yeast population-level reference genomes7 into a self-contained package to considerably
streamline this process with modular design and automated implementation. Moreover, rather
than simply combining the existing tools for genome assembly and gene annotation, LRSDAY
assembled a well-integrated workflow with many other functionalities (e.g. reference-guided
scaffolding, gene orthology identification, and additional genomic feature annotation) built in,
which makes it a unique one-stop solution for high-quality genome assembly and annotation
production from long-read sequencing data.
Experimental Design
Genome assembly and annotation are complex computational processes with many intermediate
steps and inputs/outputs involved. With LRSDAY, we designed a highly structured project
directory system to help users to run the whole workflow in an organized and modular way (Fig.
2). Within such project directory system, the three subdirectories holding the pre-shipped
reference genome as well as the user-supplied long (PacBio or Oxford Nanopore) and short
(Illumina) reads are numbered as “00” and the task-specific subdirectories are numbered
sequentially from “01” to “13” according to their execution orders. For each subdirectory, a self-
explained name is attached after the number index to help users to navigate through the workflow.
To run each task, users only need to edit (i.e. to specify the input and output file paths and certain
task-specific parameters) and execute the task-specific pipeline scripts pre-placed in these
subdirectories. These pipeline scripts will automatically set environment variables, process the
data, and formulate the results. All computationally intensive tasks can be processed using
multiple threads to substantially save computation time. Although LRSDAY is mainly designed for
yeasts, most of these tasks can be further adapted for the analysis on any eukaryotic organisms
(See Appendix 2 for details). Below we briefly describe the computational processes executed by
each task-specific module in LRSDAY with the corresponding PROCEDURE Step labeled in
parentheses.
5
01. Long-read-based_Genome_Assembly (Step 13): Long reads generated from PacBio or
Oxford Nanopore technologies are used to perform de novo genome assembly using an
overlap-layout-consensus (OLC) algorithm.
02. Illumina-read-based_Assembly_Polishing (Step 15; optional): Illumina reads are first
clipped and trimmed to remove potential sequencing adapters and low-quality regions.
The cleaned reads are subsequently mapped to the raw long-read-based genome
assembly. The resulting BAM file is further processed for alignment sorting, mate
information and read group fixing, duplicates removal as well as local realignment. Finally,
the processed bam file is used for correcting base-level errors of the long-read-based
assembly.
03. Reference-guided_Assembly_Scaffolding (Step 16): The contigs from the polished
genome assembly are first aligned to the reference genome to identify their shared
sequence homology, based on which reference-guided assembly scaffolding is
subsequently performed. The chromosomal identity of each scaffold is labeled
accordingly. Structural rearrangements captured in the contigs will remain untouched
during the scaffolding.
04. Centromere_Identity_Profiling (Step 18): The pre-shipped S. cerevisiae centromere
sequences are searched against the scaffolded assembly for chromosome-specific
centromere identity profiling.
05. Mitochondrial_Genome_Assembly_Improvement (Step 19): The polished contigs
corresponding to the mitochondrial genome are re-collected from the scaffolded assembly.
The mitochondrial contigs spanning over the designated starting point (the ATP6 gene by
default) are broken into subsegments to prevent assembly problems caused by the
circular organization of the mitochondrial genome. The resulting contigs are then re-
assembled into a single linear sequence, which is further circularized by the designated
ATP6 starting point. The nuclear scaffolds and the circularized mitochondrial sequence
together form the improved genome assembly.
06. Supervised_Final_Assembly (Steps 20-22): A modification list containing the ordering,
orientation and naming information of each sequence from the improved genome
assembly is generated for users to review and to make manual adjustment when needed.
The final genome assembly is further generated based on the user-edited modification
list.
07. Centromere_Annotation (Step 23): The pre-shipped S. cerevisiae centromere
sequences are searched against the final genome assembly for centromere annotation.

6
08. Gene_Annotation (Step 25): De novo protein-coding and tRNA gene annotations are
performed for the final genome assembly, which are further leveraged by the mRNA
transcripts and protein sequences alignments.
09. TE_Annotation (Step 28): The pre-shipped curated TE library (containing the long
terminal repeats (LTRs) and internal sequences of the S. cerevisiae and S. paradoxus
Ty1-Ty5 by default) is searched against the final genome assembly to identify TEs. The
identified TEs are further curated and classified into the full-length, truncated, and solo-
LTRs of Ty1-Ty5.
10. Core_X_Element_Annotation (Step 30): The pre-shipped curated hidden Markov model
(HMM) of the S. cerevisiae core X elements is searched against the final genome
assembly to annotate core X elements.
11. Y_Prime_Element_Annotation (Step 32): The pre-shipped representative S. cerevisiae
Y’ element sequence is searched against the final genome assembly to annotate Y’
elements. Note that Y’ elements can have long, short or degenerated forms22, and we
used a representative long-form Y’ element as the query to maximize detection power.
12. Gene_Orthology_Identification (Step 34): The annotated protein-coding genes are
compared with the reference protein-coding genes based on both sequence homology
and gene order conservation to identify gene orthology relationship between these two
sets. Based on such gene orthology relationships, the Saccharomyces Genome Database
(SGD; http://www.yeastgenome.org/) systematic names are assigned to the annotated
protein-coding genes.
13. Annotation_Integration (Step 35): The annotations of centromeres, TEs, protein-coding
genes, tRNAs, as well as core X and Y’ elements are combined and sorted to form a final
integrated multi-feature annotation.
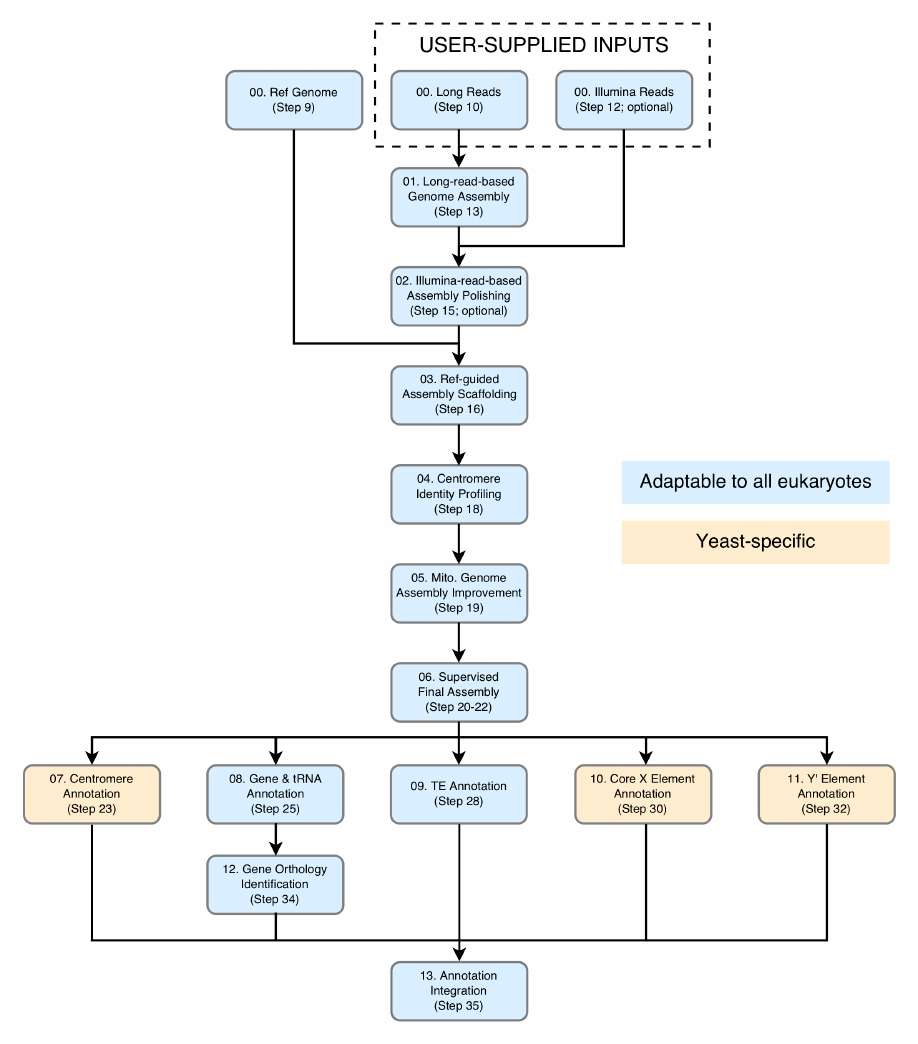
7
Figure 2. The workflow of LRSDAY. Each box represents an individual module. These modules
are numbered according to the tasks described in Experimental Design, with the corresponding
protocol step numbers also indicated. Modules that can be adapted for other eukaryotes are
colored in light blue while those yeast-specific are colored in orange.

8
Limitations and potential adaptation
In its distributed form, LRSDAY is tailored for the model budding yeast S. cerevisiae and its closely
related sister species S. paradoxus with a number of pre-shipped auxiliary data files configured
accordingly. However, given its modular design, the backbone of LRSDAY can be adapted for
virtually any eukaryotic organisms to perform genome assembly, sequence polishing, reference-
guided scaffolding, protein-coding genes and tRNA annotations, gene orthology identification,
and annotation integration. In Appendix 2, we provide some tips with regard to such adaptation.
Moreover, those assembly polishing, scaffolding and various annotation modules (See
Experimental Design) can also be used to analyze existing genome assemblies derived from any
or any combination of sequencing technologies. Such flexibility makes LRSDAY very useful for
expanded use cases and therefore suits the needs of a broader audience.
Expected improvements
As thousands of yeast strains have been or are currently under sequencing23–26, our knowledge
of the overall genome content diversity27 of this important model organism is expanding rapidly,
revealing a whole new picture of the pan-genome diversity of S. cerevisiae. For example, our lab
is currently working on characterizing the pan-genome of >1,000 S. cerevisiae isolates across the
globe (The 1002 Yeast Genomes Project26; http://1002genomes.u-strasbg.fr/). Future
developments of LRSDAY will incorporate such pan-genome dataset to provide additional
annotation information for those non-reference genes, especially with regard to their evolutionary
origin, population prevalence, and putative functions. Such information will greatly help users to
dissect and interpret complex genotype-phenotype interactions in diverse ecological and
biotechnological settings. An additional potential future direction of our research is the direct
integration with the downstream synteny analysis tools (e.g. CHROnicle28, MCScanX29, etc) to
perform automatic large-scale structural variants discovery, which exploits one of the major
benefits of having a high-quality genome assembly derived from long reads. Finally, we envision
developing a dedicated web-based tool to implement such database and tool integration at a
larger scale towards fully automated genomics analysis for the yeast community in the long run.

9
CITATIONS
Jia-Xing Yue & Gianni Liti. (2018) Long-read sequencing data analysis for yeasts. Nature
Protocols, 13:1213–1231.
Jia-Xing Yue, Jing Li, Louise Aigrain, Johan Hallin, Karl Persson, Karen Oliver, Anders
Bergström, Paul Coupland, Jonas Warringer, Marco Cosentino Lagomarsino, Gilles Fischer,
Richard Durbin, Gianni Liti. (2017) Contrasting evolutionary genome dynamics between
domesticated and wild yeasts. Nature Genetics, 49:913-924.
MATERIALS
Hardware, operating system and network
This protocol is designed for a desktop or computing server running an x86-64-bit Linux operating
system. Multi-threaded processors are preferred to speed up the process since many steps can
be configured to use multiple threads in parallel. For assembling and analyzing the budding yeast
genomes (genome size = ~12 Mb), at least 16 Gb of RAM and 100 Gb of free disk space are
recommended. When adapted for other eukaryotic organisms with larger genome sizes, the RAM
and disk space consumption will scale up, majorly during de novo genome assembly (performed
by Canu17). Please refer to Canu’s manual (http://canu.readthedocs.io/en/latest/) for suggested
RAM and disk space consumption for assembling large genomes. Stable Internet connection is
required for the installation and configuration of LRSDAY as well as for retrieving the testing data.
Software or library requirements
● Bash (https://www.gnu.org/software/bash/)
● Bzip2 (http://www.bzip.org/)
● Cmake (https://cmake.org/)
● GCC and G++ v4.7 or newer (https://gcc.gnu.org/)
● Ghostscript (https://www.ghostscript.com)
● Git (https://git-scm.com/)
● GNU make (https://www.gnu.org/software/make/)
● Gzip (https://www.gnu.org/software/gzip/)
● Java runtime environment (JRE) v1.8.0 or newer (https://www.java.com)

10
● Perl v5.12 or newer (https://www.perl.org/)
● Python v2.7.9 or newer (https://www.python.org/)
● Python v3.6 or newer (https://www.python.org/)
● Tar (https://www.gnu.org/software/tar/)
● Unzip (http://infozip.sourceforge.net/UnZip.html)
● Virtualenv v15.1.0 or newer (https://virtualenv.pypa.io)
● Wget (https://www.gnu.org/software/wget/)
● Zlib (https://zlib.net/)
Input data
● Long reads: A single FASTQ file containing PacBio or Oxford Nanopore reads is needed,
which will be used for long-read based de novo genome assembly (Task 01).
● Short reads: Short reads are optional for LRSDAY but LRSDAY could take advantage of
short reads when such data is available to perform additional assembly polishing (Task
02). If paired-end Illumina sequencing is performed, two FASTQ files containing the
forward and reverse Illumina reads respectively are needed. If only single-end Illumina
sequencing data is available, one FASTQ file containing the single-end reads is needed.
● Reference genome: For the budding yeast S. cerevisiae, we pre-shipped two reference
genome files (one original assembly and one with hard-masked subtelomeres and
chromosome-ends based on our previous study7). The masked version is used for
chromosomal scaffolding to minimize the confounding effect due to interchromosomal
subtelomeric rearrangemnets. When working with organisms of which the subtelomeric
regions are undefined, users can just use a single raw reference genome instead. The
reference genome file(s) will be used for reference-guided scaffolding, mitochondrial
genome assembly improvement and supervised final genome assembly (Task 03, 05 and
06 respectively).
● A number of S. cerevisiae-specific auxiliary data have been pre-shipped with LRSDAY for
genomic feature annotation and gene orthology identification (Task 07-12).
Example data
● The S. cerevisiae reference genome pre-shipped with LRSDAY is taken from our
previous study7 with the Genbank accession number GCA_002057635.1
(https://www.ncbi.nlm.nih.gov/assembly/GCA_002057635.1/). The sequencing reads

11
used for the testing example come from the same study, which consists of both PacBio
and Illumina reads produced from the S. cerevisiae strain SK1. The PacBio reads can be
retrieved with the ENA analysis accession number ERZ448251
(https://www.ebi.ac.uk/ena/data/view/ERZ448251). The Illumina reads can be retrieved
with the SRA sequencing run accession number SRR4074258
(https://www.ncbi.nlm.nih.gov/sra/?term=SRR4074258). In LRSDAY, we have provided
bash scripts to automatically download and setup these data for the testing example.

12
PROCEDURE
Download, install and configure LRSDAY ● TIMING <1 h
1) Download the latest LRSDAY release (current version: v1.1.0) by entering the following
commands in a terminal window:
$ wget https://github.com/yjx1217/LRSDAY/releases/download/v1.1.0/LRSDAY-v1.1.0.tar.gz
$ tar xvzf LRSDAY-v1.1.0.tar.gz
$ cd LRSDAY-v1.1.0
$ bash install_dependencies.sh
▲CRITICAL STEP Make sure you have a fast and stable Internet connection when
running this step since many tools will be downloaded here. Check that all the
prerequisites (see Software or library requirements in EQUIPMENT or the prerequisite.txt
file in the downloaded LRSDAY directory) have been installed on your system.
▲CRITICAL STEP Upon the successful completion of executing the bash script, you
should see a confirmation message prompted out: “LRSDAY message: This bash script
has been successfully processed! :)”. Otherwise, it means an error has occurred during
the execution of the bash script, which interrupted the automatic installation process. This
also applies to all the bash scripts used in Steps 8-35. Whenever an error is encountered,
please check the error message and refer to the troubleshooting section when available.
When the cause of the error is fixed, re-run this step to initiate a new installation. The
installer will prompt for the confirmation of deleting the build directory generated by the old
run, always answer “yes” to authorize such action so that the new installation can start.
▲CRITICAL STEP Pay attention to the final message prompted by the installation script
for a number of notes with regard to the additional manual setup steps as well as the
licensing information for commercial users.
? TROUBLESHOOTING
2) Load the environment settings for LRSDAY by entering:
$ source env.sh
After loading the pre-configured environment settings, the current directory should be
assigned to the environment variable $LRSDAY_HOME. You can check to see if the full
path to your current directory is displayed after entering:
$ echo $LRSDAY_HOME

13
▲CRITICAL STEP Although most required tools have been automatically installed and
configured, manual downloading and/or configuration are needed for GATK30 and
RepeatMasker31 due to license restriction of these tools or of their dependent libraries.
Make sure to run this command to load the pre-configured environment settings before
such manual setup (described in Steps 3-7). If you exited your terminal session before or
in the middle of your manual setup, you need to re-load the environment settings before
proceeding. These environment settings will be automatically loaded each time the task-
specific bash pipelines of LRSDAY are executed.
? TROUBLESHOOTING
3) Download and set up GATK3. Go to the official website of GATK
(https://software.broadinstitute.org/gatk/download/archive) and download GATK v3.8 (the
recently released GATK4 will not work). Registration might be needed for unregistered
GATK users. Place the downloaded GATK package (file name: GenomeAnalysisTK-3.8-
1- gf15c1c3ef.tar.bz2) in the GATK installation directory and set up GATK by entering the
following command in terminal:
$ mv GenomeAnalysisTK-3.8-1- gf15c1c3ef.tar.bz2 $LRSDAY_HOME/build/GATK3
$ cd $LRSDAY_HOME/build/GATK3
$ tar xjf GenomeAnalysisTK-3.8-1.tar.bz2
$ mv ./GenomeAnalysisTK-3.8-1*/GenomeAnalysisTK.jar .
$ chmod 755 GenomeAnalysisTK.jar
4) Set up TRF for RepeatMasker. Go to the TRF website
(http://tandem.bu.edu/trf/trf.download.html) to download the TRF v4.09 executable built
for 64-bit Linux. Place the downloaded file (file name: trf409.linux64) in the RepeatMasker
installation directory and set up TRF by entering:
$ mv trf409.linux64 $LRSDAY_HOME/build/RepeatMasker/
$ cd $LRSDAY_HOME/build/RepeatMasker/
$ chmod 755 trf409.linux64
$ ln -s trf409.linux64 trf
5) Set up the RepBase library for RepeatMasker. Go to the RepBase website
(http://www.girinst.org/repbase/) and register a user account (using a non-profit
institutional email address, e.g. the “.com” extension will not work) to download the
RepeatMasker edition of the RepBase library (version: 20170127). Place the downloaded
file (file name: RepBaseRepeatMaskerEdition-20170127.tar.gz) in the RepeatMasker
installation directory and uncompress it by entering:
$ mv RepBaseRepeatMaskerEdition-20170127.tar.gz $LRSDAY_HOME/build/RepeatMasker/
14
$ cd $LRSDAY_HOME/build/RepeatMasker/
$ tar xzf RepBaseRepeatMaskerEdition-20170127.tar.gz
? TROUBLESHOOTING
6) Get the installation path of TRF and rmblastn/makeblastdb by entering:
$ echo $trf_dir
$ echo $rmblast_dir
Remember these two paths since they will be used for the RepeatMasker configuration in
Step 7.
? TROUBLESHOOTING
7) Run the configuration script for RepeatMasker by entering:
$ perl ./configure
This configuration script will prompt for several questions. Please do the following to
answer these questions. Enter “env” for the question about the installation path of Perl.
Just press enter for the question about the installation path of RepeatMasker. Enter the
first path that you obtained in Step 6 for the question about the installation path of TRF.
Enter “2” for the question about selecting a search engine. Then enter the second path
that you obtained in Step 6 for the question about the installation path of rmblastn and
makeblastdb. Just press enter for the question about the default search engine. And finally
enter “5” to complete the configuration.
Run LRSDAY with the testing example ● TIMING 50 h
8) Create the project directory. When running LRSDAY with your own data, it is
recommended to make a copy of our Project_Template directory to create your own
project directory such as Project_abc, where “abc” can be any string containing letters,
numbers, or underscores. For this testing example, we make a copy of the
Project_Template directory and name it as Project_Example by entering:
$ cd $LRSDAY_HOME
$ cp -r Project_Template Project_Example
▲CRITICAL STEP Before proceeding to your own project, it is advised to first run our
prepared testing example to check if LRSDAY is working properly as well as to get
acquainted with the logic and workflow of LRSDAY.
9) Prepare the reference genome files. When running LRSDAY with your own data, you can
directly put the reference genome (in FASTA format without compression) in the
00.Ref_Genome subdirectory of your project directory (e.g. Project_abc). If your
15
sequenced organism is S. cerevisiae or S. paradoxus, you can use the reference genome
pre-shipped with LRSDAY. Here we prepare the pre-shipped reference genome for the
testing example by entering:
$ cd ./Project_Example/00.Ref_Genome
$ bash LRSDAY.00.Prepare_Sc_Ref_Genome.sh
10) Prepare the long reads. When running LRSDAY with your own data, you can directly put
the long reads in the 00.Long_Reads subdirectory of your project directory (e.g.
Project_abc). The reads file should be in FASTQ or FASTA format. Compressed files with
file extensions of “.gz”, “.bz2”, and “.xz” are also supported. For this testing example, you
can download the sample long reads by entering:
$ cd ./../00.Long_Reads
$ bash LRSDAY.00.Retrieve_Sample_PacBio_Reads.sh
11) (Optional) If your long reads are generated from the PacBio Sequel platform, your reads
are likely to be in BAM format. In this case, convert it to our supported format (FASTA or
FASTQ) using the following commands:
$ source ./../../env.sh
$ $bedtools_dir/bedtools bamtofastq -i long_reads.bam -fq long_reads.fastq
$ gzip long_reads.fastq
12) (Optional) Prepare the Illumina reads. Like for the long reads, directly put your Illumina
reads in the 00.Illumina_Reads subdirectory of your project directory (e.g. Project_abc)
when running LRSDAY for your own data. The reads file should be in FASTQ format with
“gzip” comprehension (identified by the “.gz” extension). For this testing example, you can
download the Illumina reads by entering:
$ cd ./../00.Illumina_Reads
$ bash LRSDAY.00.Retrieve_Sample_Illumina_Reads.sh
? TROUBLESHOOTING
13) Perform long-read-based de novo genome assembly by running the following commands:
$ cd ./../01. Long-read-based_Genome_Assembly
$ bash LRSDAY.01.Long-read-based_Genome_Assembly.sh
Upon the completion of this step, a summary file (SK1.canu.stats.txt for this testing
example) will be generated to report some basic summary statistics (e.g. total assembly
size, N50 (i.e. the contig length such that 50% of the total assembly size is contained in
contigs of at least this size), L50 (i.e. the number of longest contigs such that 50% of the
total assembly size is contained), GC-content, etc) to assist gauging the genome
assembly quality (Table 1). Two VCF files (SK1.canu.filter.mummer2vcf.SNP.vcf and

16
SK1.canu.filter.mummer2vcf.INDEL.vcf for the testing example) will also be generated to
report base-level differences between the raw genome assembly and the reference
genome for their uniquely alignable regions, which could also help for assessing the
genome assembly quality.
▲CRITICAL STEP This step can be run with multiple threads to speed up the process.
Depending on the CPU configuration of your Linux server/desktop, you can edit the
“threads=” option in the bash script LRSDAY.01.Long-read-based_Genome_Assembly.sh
to enable multi-threading. You can do the same for all the following tasks whenever the
“threads=” option is provided in the corresponding task-specific bash script. Simple text
editors such as emacs, vim, gedit or pico are highly recommended for such editing. Rich
text editors might not work
▲CRITICAL STEP Note this step will take long to finish (see TIMING), so we recommend
running this step and all the other time-consuming steps with “nohup”
(https://en.wikipedia.org/wiki/Nohup), which allows the process to continue running after
you exit the terminal or logout from the server. As an example, you can run the bash script
using nohup as follows:
$ nohup bash LRSDAY.01.Long-read-based_Genome_Assembly.sh >run_log.txt 2>&1 &
▲CRITICAL STEP Starting from v1.1.0, LRSDAY added the
"customized_canu_parameters” option to support customized parameter settings for the
Canu assembler. In addition, two additional assemblers, Flye and smartdenovo, were
further supported. Based on our test, Flye32 (https://github.com/fenderglass/Flye) and
smartdenovo (https://github.com/ruanjue/smartdenovo) run much faster than Canu but
also came with understandable tradeoff in assembly precision, as reflected by the higher
base-level error rate of their resulting assemblies. Therefore, when assembling with Flye
or smartdenovo, post-assembly polishing is strongly recommended.
▲CRITICAL STEP When running LRSDAY with your own data, modify the bash script to
specify the input reads and reference genome, the input reads type (e.g. “pacbio_raw”,
“pacbio_corrected”, “nanopore_raw” or “nanopore_corrected”), the estimated genome
size for the assembled genome, as well as the prefix for the output data. Remember to do
similar project-specific adjustment for all the following steps.
? TROUBLESHOOTING
14) (Optional) Polishing genome assembly with long-reads. When running LRSDAY for your
own data, if you performed PacBio sequencing and also have access to a locally installed
PacBio SMRT Analysis software package (http://www.pacb.com/products-and-

17
services/analytical-software/smrt-analysis/), we recommend running the first-pass
polishing for the assembly generated in Step 13 based on raw PacBio reads by using
PacBio’s own Quiver/Arrow pipeline13. If you performed Oxford Nanopore sequencing, we
recommend using nanopolish (https://github.com/jts/nanopolish) or equivalent tools to
perform the assembly polishing based on raw Nanopore reads in this step. We expect to
directly pack nanopolish into LRSDAY in our next release. We will also try to the same for
PacBio’s Quiver/Arrow pipeline but it seems quite challenging. Finally, we also tested
Racon (https://github.com/isovic/racon) for polishing but its performance was not
satisfying.
15) (Optional) Polishing genome assembly with Illumina reads. When Illumina reads are
available, we recommend running this additional polishing step either for the raw assembly
generated in Step 13 (when Step 14 is skipped as in our testing example) or for the long-
read-polished assembly generated in Step 14. Use the following commands to perform
Illumina-read-based assembly polishing. This step can be run with multiple threads.
$ cd ./../02.Illumina-read-based_Assembly_Polishing
$ bash LRSDAY.02.Illumina-read-based_Assembly_Polishing.sh
TABLE 1 | Assembly statistics for the genome of S. cerevisiae strain SK1 assembled in the
testing example.
Assembly statistics
Raw
assembly
Scaffolded
assembly
Final
assembly
Total sequence count
35
34
33
Total sequence length (bp)
12505999
12511058
12489291
Min sequence length (bp)
1248
1248
1248
Max sequence length (bp)
1480196
1480203
1480203
Mean sequence length (bp)
357314.26
367972.29
378463.36
Median sequence length (bp)
103703.00
74951.00
84648.00
N50 (bp)
818058
923633
923633
L50
6
6
6
N90 (bp)
341459
341463
341463
L90
15
14
14
GC%
38.27
38.25
38.29
18
N%
0.00
0.08
0.04
Note
N50: the contig length such that 50% of the total assembly size is contained in contigs of at
least this size.
L50: the number of longest contigs such that 50% of the total assembly size is contained.
N90: the contig length such that 90% of the total assembly size is contained in contigs of at
least this size.
L90: the number of longest contigs such that 90% of the total assembly size is contained.
GC%: the percentage of guanine (G) and cytosine (C) bases in the nucleotide sequences.
N%: the percentage of the N bases in the nucleotide sequence. In genome assembly, the N
bases are usually used to represent scaffolding gaps.
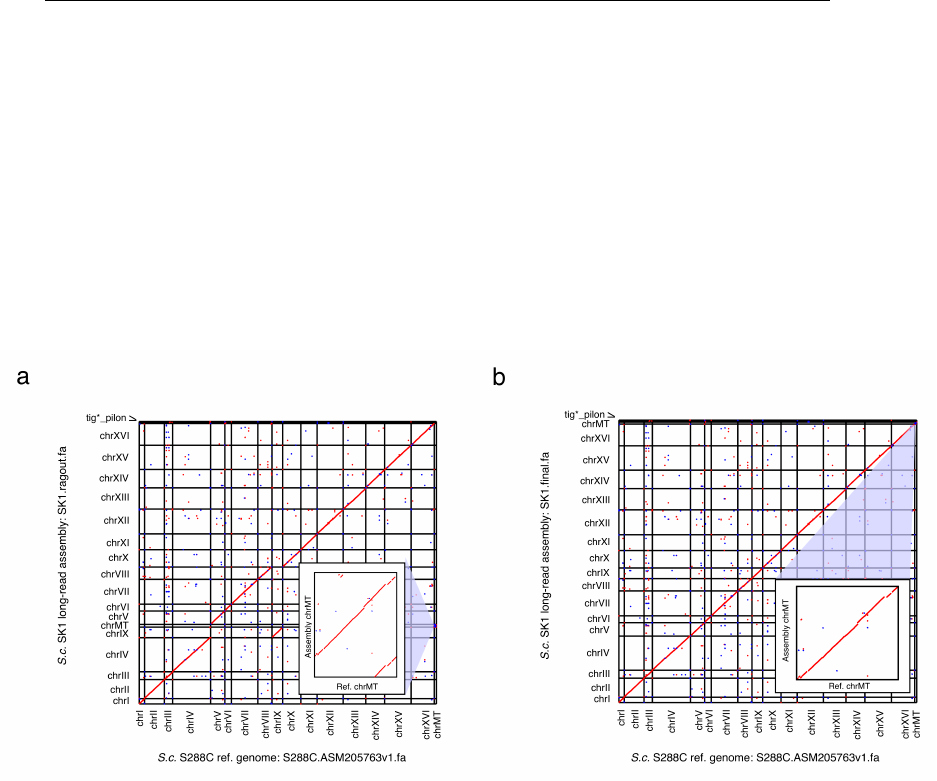
Figure 3. Genome-wide dotplots of the S. cerevisiae SK1 genome assembly generated in
the LRSDAY testing example. Both the raw scaffolded assembly (panel a; generated in Step
16) and the final assembly (panel b, generated in Step 22) are analyzed. The forward and reverse
sequence matches are depicted in red and blue respectively, while the zoomed-in views of the
mitochondrial genome (chrMT) comparison are shown in insets. In addition to the sixteen nuclear
chromosomes and the mitochondrial genome, the scaffolded and final assemblies also contain
some short contigs (named as tig*_pilon) that are derived from highly repetitive regions.
16) Perform chromosome-level scaffolding for the long-read-based assembly. Run the
following commands:
$ cd ./../03.Reference-guided_Assembly_Scaffolding
$ bash LRSDAY.03.Reference-guided_Assembly_Scaffolding.sh
19
This step can be run with multiple threads. Upon completion, a list of summary statistics
(SK1.ragout.stats.txt for this testing example) will be generated for the scaffolded
assembly (Table 1).
▲CRITICAL STEP Please check the generated genome-wide dotplot
(SK1.ragout.filter.pdf for the testing example) (Fig. 3a) to verify the correctness of
chromosomal identity assignment performed by Ragout33 and apply manual adjustment in
Step 21 when necessary. When running LRSDAY with your own data, you might see a
single scaffold corresponds to more than one reference chromosomes, which could be
due to shared sequence homology between duplicated regions or interchromosomal
rearrangements. Both types of events can be correctly interpreted based on the genome-
wide dotplot generated in this Step. In either case, LRSDAY can correctly assign
chromosomal identity of the corresponding scaffold based on its encompassed
centromere identity as annotated in Step 18. Check the generated AGP file
(SK1.ragout.agp for the testing example) for the details of reference-based scaffolding.
▲CRITICAL STEP Due to the high AT and repeat contents and the circular conformation
of the mitochondrial genome, multiple contigs corresponding to the mitochondrial genome
are often obtained from the raw genome assembly, as shown in the generated
mitochondrial genome dotplot (SK1.ragout.chrMT.filter.pdf for the testing example) (Fig.
3a, inset). A list of such mitochondrial contigs will also be generated (SK1.mt_contig.list
for the testing example), which will be used in Step 19 for improving mitochondrial genome
assembly.
17) (Optional) When running LRSDAY for your own data, if you have strong evidence for mis-
scaffolding based on prior knowledge or other experimental data (e.g. mate-pair libraries
or chromosomal contact data), break the corresponding ragout scaffolds back to contigs
and re-joined them with corrected order using the pre-shipped Perl scripts
break_scaffolds_by_N.pl, join_contigs_by_N.pl and extract_region_from_genome.pl in
the $LRSDAY_HOME/scripts directory by running the following commands:
perl $LRSDAY_HOME/scripts/break_scaffolds_by_N.pl -i <the input FASTA file containing the
scaffold sequence(s) to break> -o < the output FASTA file containing the scaffolds after the
breaking> -g <the minimal length of runs of Ns in the input scaffold(s) for breaking, e.g. 100>
perl $LRSDAY_HOME/scripts/join_contigs_by_N.pl -i <the input FASTA file containing contigs for
joining in a sequential order> -o <the output FASTA file containing scaffold sequence after the
contig joining> –g <gap size, i.e. the number of Ns to be inserted between two joined contigs> -t
<sequence name for the newly joined scaffold>
20
perl $LRSDAY_HOME/scripts/extract_region_from_genome.pl -i <the input FASTA file containing
the genome assembly> -o <the output FASTA file containing the extracted query sequence> -q
<specially formatted query string (sequence:start-end:strand) containing the genomic coordinates
for the region to be extracted, e.g. using the query string chrI:1000-4000:+ for extracting the
sequence in the region 1000-4000 bp on the + strand of chrI in the input genome assembly> -f <the
length of flanking sequences to be extracted as well, e.g. 100 for 100-bp flanking region>
A scenario for such use case is when the breakpoints of structural rearrangements are
also the breakpoints of the genome assembly. In this case, the reference-based
scaffolding will arrange contigs according to the reference genome configuration and
therefore un-do the genome rearrangement.
18) Perform centromere profiling for the scaffolded genome assembly by running the following
commands:
$ cd ./../04.Centromere_Identity_Profiling
$ bash LRSDAY.04.Centromere_Identity_Profiling.sh
▲CRITICAL STEP The chromosome-specific centromere identities profiled here will be
used as another layer of information for the final chromosomal identity assignment in Step
21. The profiled centromere identities usually agree well with the chromosomal identities
labeled in Step 16, so that chrI will have the CEN1 centromere and chrII will have the
CEN2 centromere, etc. Exception can occur when interchromosomal rearrangements are
involved in your sequenced genome. In such case, we recommend naming those
rearranged chromosomes according to their encompassed centromeres in Step 21.
19) Perform mitochondrial genome assembly improvement by running the following
commands:
$ cd ./../05.Mitochondrial_Genome_Assembly_Improvement
$ bash LRSDAY.05.Mitochondrial_Genome_Assembly_Improvement.sh
▲CRITICAL STEP Check the generated final mitochondrial genome dotplot
(SK1.mt_improved.chrMT.filter.pdf for the testing example) and compare it with the
mitochondrial genome dotplot generated in Step 16 to see how the mitochondrial genome
assembly has been improved when aligning with the reference mitochondrial genome (Fig.
3b, inset). When running this step for your own data, the degree of such improvement may
vary because it depends on both the complexity of the assembled mitochondrial genome
and the quality of library preparation and sequencing experiments.
? TROUBLESHOOTING
20) Generate the assembly modification list file for performing the final chromosome
assignment by running the following commands:
21
$ cd ./../06.Supervised_Final_Assembly
$ bash LRSDAY.06.Supervised_Final_Assembly.1.sh
21) Edit the generated assembly modification list file (SK1.modification.list for the testing
example) based on the genome-wide dotplot generated in Step 16 and the centromere
profiles generated in Step 18. The modification list file consists of three comma-separated
columns, which correspond to the original sequence name, sequence orientation, and new
sequence name respectively. With this file, you can do three types of editing:
If you need to change the current sequence order, you can move the corresponding
rows upward or downward to reflect the correct order.
If you need to invert the orientation of a given sequence, you can change its
orientation from “+” to “-” in column 2.
If you need to rename a given sequence, you can specify the new name in the third
column.
For this testing example here, we need to move the row “chrIX,+,chrIX” downward to place
it after the row “chrVIII,+,chrVIII”, so that chrIX will be placed after chrVIII in the final
assembly. Also, we need to change the row “chrMT_Contig1,+,chrMT_Contig1” to
“chrMT_Contig1,+,chrMT” for renaming the assembled sequence corresponding to the
mitochondrial genome.
22) Once all the modifications have been specified, run the following bash script to generate
the final genome assembly as well as the associated genome-wide dotplot (Fig. 3b),
assembly statistics (Table 1), and VCF files:
$ bash LRSDAY.06.Supervised_Final_Assembly.2.sh
23) Re-run centromere annotation for the final genome assembly using the following
commands:
$ cd ./../07.Centromere_Annotation
$ bash LRSDAY.07.Centromere_Annotation.sh
24) (Optional) Customize the configuration file for gene annotation. When running LRSDAY
with your own data, edit the configuration file
$LRSDAY_HOME/misc/maker_opts.customized.ctl if your sequenced organisms is
neither S. cerevisiae nor S. paradoxus (See Appendix 2 for the details). If your sequenced
organism is S. cerevisiae or S. paradoxus, no customization is needed unless you have
native transcriptome or expressed sequence tag (EST) data for the strain that you
sequenced. In this case, you can edit the line 16 of this file to provide the full path of the
native transcriptome or EST assembly for your sequenced strain.

22
25) Annotate protein-coding genes and tRNAs for the final genome assembly, using the
following commands. This step can be run with multiple threads.
$ cd ./../08.Gene_Annotation
$ bash LRSDAY.08.Gene_Annotation.sh
26) (Optional) A manual checklist file (SK1.EVM.manual_check.list for the testing example)
containing a list of genes with suspicious annotations will be generated in Step 25. As
labeled in this file, these annotated gene models could be fragmented, frameshifted or
containing internal stop codons. Potentially, these genes could be good candidates for
pseudogenes. Manually inspect the annotated gene models of these genes by loading the
annotation result (SK1.EVM.gff3 for the testing example) together with the protein/EST-
alignment evidence files generated during the annotation in Step 25
(SK1.protein_evidence.gff3 and SK1.est_evidence.gff3 for the testing example) into IGV34
to check how well these suspicious gene models are supported by the corresponding
protein/EST-alignment evidence and to tag or remove those truly problematic ones in your
downstream analysis.
27) (Optional) Perform dedicated mitochondrial protein-coding and RNA annotation. If you are
interested in studying mitochondrial genomes, we highly recommend running dedicated
mitochondrial feature annotation with specialized software such as MFannot
(http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl). Although
MFannot does not support local installation so far, you can run MFannot conveniently via
its web portal. Be sure to select the correct genetic code table (e.g. “3 Yeast Mitochondrial”
for annotating yeast mitochondrial genomes) for your analysis.
28) Annotate transposable elements (TEs) for the final genome assembly. This step can be
run with multiple threads using the following commands:
$ cd ./../09.TE_Annotation
$ bash LRSDAY.09.TE_Annotation.sh
29) (Optional) TE activity can be highly dynamic in the genome with many complex cases
such as fragmentation and nested insertion. In LRSDAY, we used REannotate35 to
automatically resolve these complex cases, which works well for most cases but it could
occasionally misjoin two adjacent TEs when they are closely spaced. Further inspect and
curate the LRSDAY TE annotation output (SK1.TE.gff3 for the testing example) by
visualizing it in IGV34 together with the raw REannotate annotation (SK1.REannotate.gff
for the testing example). For each TE found in the LRSDAY TE annotation output, examine
its corresponding LTR and internal region structure based on the raw REannotate
23
annotation to check for misjoinings. If needed, you can manually edit the corresponding
TE annotation output file to decouple the misjoinings.
30) Annotate yeast telomere-associated core X elements for the final genome assembly, using
the following commands:
$ cd ./../10.Core_X_Element_Annotation
$ bash LRSDAY.10.Core_X_Element_Annotation.sh
31) (Optional) Inspect the alignment file for annotated core X element. In LRSDAY, we label
the identified core X elements to be “partial” if they are shorter than 300 bp. This should
work for most cases but we recommend inspecting the generated alignment file
(SK1.X_element.aln.fa for the testing example) for further curation. Upon the curation,
manually adjust the “partial” labeling in the annotation file (SK1.X_element.gff3 for the
testing example) when needed.
32) Annotate yeast telomere-associated Y’ elements for the final genome assembly by using
the following commands:
$ cd ./../11.Y_Prime_Element_Annotation
$ bash LRSDAY.11.Y_Prime_Element_Annotation.sh
33) (Optional) Inspect the alignment file for annotated Y’ element. Like for the core X element
annotation, we used a hard length cutoff (3500 bp) to label if the identified Y’ elements are
“partial”. We recommend manually inspecting the generated alignment file
(SK1.Y_prime_element.aln.fa for the testing example) to check if the “partial” labeling is
needed and edit the annotation file (SK1.Y_prime_element.gff3 for the testing example)
accordingly.
34) Perform orthology identification for protein coding genes by using the following
commands:
$ cd ./../12.Gene_Orthology_Identification
$ bash LRSDAY12.Gene_Orthology_Identification.sh
In this step, a gene orthology relationship list is created between the annotated proteome
and the SGD S. cerevisiae reference proteome based on both sequence similarity and
synteny conservation. Based on this list, we further attach SGD systematic names to our
gene annotation as shown in the “Name=” field of the generated GFF3 file
(SK1.updated.gff3 for the testing example). For a given annotated gene, when more than
one orthologous gene can be found in the SGD reference proteome, we will label all of its
co-orthologs in the “Name=” filed with “/” between the alternative SGD systematic names
(e.g. “YAR071W/YHR215W”), whereas when no orthologous gene can be found, we will
label its gene name as “Name=NA”. This step can be run with multiple threads.
24
35) Integrate the annotation of different genomic features into a unified GFF3 file by using the
following commands:
$ cd ./../13.Annotation_Integration
$ bash LRSDAY.13.Annotation_Integration.sh

25
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
TABLE 2 | Troubleshooting table.
Step
Problem
Possible reason
Solution
1
Downloading errors or
unresponsive remote
servers
Unstable internet
connection or temporary
problems of the remote
servers where the tools for
downloading are hosted.
Stabilizing the internet connection
and give another try.
1
Compilation or
installation error for
specific tools.
Prerequisites not satisfied
or corner cases due to
your specific system
settings.
If missing prerequisites are found
(see software and library
requirements in EQUIPMENT),
install the missing prerequisites
and give another try as described
above. Otherwise, record the error
message and email it to the
developers of the problematic tools
or the authors of this protocol for
further problem diagnosis.
2
Cannot find the file
“env.sh”.
Step 1 has failed.
Check the error message that you
got when running Step 1 and refer
to the troubleshooting for Step 1.
5
Rejected by RepBase
for the license
registration.
A personal or commercial
email address (e.g. with a
.com extension) was used
for the registration.
Use a non-commercial institutional
email address for the registration
or purchase the commercial
license if you work in a commercial
entity. Also, the installation of
RepBase can be potentially
skipped although not
recommended by RepeatMasker.
Users can still follow the remaining
steps of this protocol even if they
skip the RepBase installation here.
26
6
“echo $rmblast_dir”
returns nothing.
Step 2 has failed or your
terminal session got
interrupted before this
step.
Re-load environment settings in
Step 2 and then re-try this step.
12
Downloading
warnings/errors
encountered.
Temporary SRA server
problems.
Directly download the sample
Illumina reads for the testing
example by running the following
commands:
$ wget
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SR
R407/008/SRR4074258/SRR4074
258_1.fastq.gz
$ ln -s SRR4074258_1.fastq.gz
SRR4074258_pass_1.fastq.gz
$ wget
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SR
R407/008/SRR4074258/SRR4074
258_2.fastq.gz
$ ln -s SRR4074258_2.fastq.gz
SRR4074258_pass_2.fastq.gz
13
The total assembly
size is much smaller
than the expected
value or the assembly
is too fragmented.
Insufficient sequencing
depth of coverage.
Obtain more reads. We
recommend a minimal sequencing
depth of 20X.
13
Assembly runs too
slow.
This likely will happen for
Canu with high-depth
nanopore reads.
Three options are available:
1) Use the pre-shipped perl
script
subsampling_seqeunces.p
l in the
$LRSDAY_HOME/scripts
directory to downsample
(either randomly or
preferably sampling the

27
longer ones) the reads.
Example:
perl
subsampling_sequences.p
l -i input.fq(.gz) -f fastq -s
0.1 -m random -p output #
randomly sampling the
10% sequences
or
perl
subsampling_sequences.p
l -i input.fq(.gz) -f fastq -s
0.1 -m longest -p output #
sampling the 10% longest
sequences
2) Use the customized
parameters as suggested
in the bash script
LRSDAY.01.Long-read-
based_Genome_Assembly
.sh
3) Use alternative
assemblers such as Flye
or smartdenovo for this
step.
19
Only partial assembly
is obtained for the
mitochondrial genome.
High AT and repeat
content of the
mitochondrial genome is
challenging for de novo
genome assembly.
If the sequencing is done with
PacBio, obtaining more reads
should solve the problem. If
sequencing is done with Oxford
Nanopore data, currently there is
no satisfying solution. Future
improvement of the Oxford
Nanopore sequencing technology
will likely solve this problem given
the rapid development of this
technology.
28
● TIMING
The following timing information was measured on a Linux computing server with an Intel Xeon
CPU E5-2630L v3 (1.80GHz) using 4 threads. Enabling multithreading can substantially decrease
the processing time. All optional steps except Steps 12 and 15 were not processed when
measuring the computation time.
Steps 1-7, set up LRSDAY: < 1 h.
Step 8, prepare the project directory for the testing example: 5 s.
Step 9, prepare the reference genomes for the testing example: 1 s.
Step 10, prepare the long reads for the testing example: 22 min.
Step 12 (optional), prepare the Illumina reads for the testing example: 20 min.
Step 13, de novo assembly using long-reads: 26 h.
Step 15 (optional), assembly polishing using Illumina reads: 1h.
Step 16, reference-guided scaffolding for the raw assembly: 12 min.
Step 18, centromere identity profiling for the scaffolded assembly: 1 s.
Step 19, mitochondrial genome assembly improvement: 4 min.
Step 20, generate the assembly modification list file: 1s.
Step 21, manual editing the assembly modification list file: 30 s.
Step 22, finalize genome assembly based on the assembly modification list file: 20 s.
Step 23, centromere annotation for the final assembly: 1s.
Step 25, protein-coding gene and tRNA annotation for the final assembly: 19 h.
Step 28, TE annotation for the final assembly: 5 min.
Step 30, core X element annotation for the final assembly: 2 min.
Step 32, Y’ element annotation for the final assembly: 2.5 min.
Step 34, gene orthology identification for the annotated protein-coding genes: 20 min.
Step 35, final annotation integration: 1 min.

29
ANTICIPATED RESULTS
Upon the completion of the LRSDAY workflow described above, users can expect to obtain a
chromosome-level genome assembly with comprehensive genomic feature annotation, which will
lay a solid foundation for all kinds of downstream genomic and functional analyses. As
demonstrated in Fig. 3b, the final genome assembly is highly continuous, with each chromosome
assembled in an end-to-end fashion. Both genome-wide dotplots (like those shown in Fig. 3) and
summary statistics (like those listed in Table 1) will be generated to help users to evaluate the
genome assembly quality both graphically and quantitatively. As for the annotation, LRSDAY
profiles a full spectrum of genomic features for the assembled yeast genome, which include
centromeres, protein-coding genes, tRNAs, Ty1-Ty5 transposable elements, as well as the
telomere-associated core X and Y’ elements. The availability of such rich information can be very
valuable for users working on diverse biological questions. In Table 3, we further summarized the
major outputs of the testing example. The final genome assembly and annotation outputs
generated with this testing example are also provided in the directory Example_Outputs for users
to make direct comparison with their own results.
TABLE 3 | Major LRSDAY outputs from the testing example.
Task
Step
Output file or directory
Explanation for the output
01
13
SK1.canu.fa
The long-read-based de novo genome
assembly containing all the contigs
assembled by Canu17.
SK1.canu.stats.txt
The summary table reporting basic
assembly statistics, such as the number
of the assembled sequences, the total
length of the assembled sequences, the
minimal, maximal, mean and median
lengths of the assembled sequences, the
N50, L50, N90, and L90 of the assembled
sequences, as well as the base
composition (A%, T%, G%, C%, AT%,
GC% and N%) of the assembled
sequences. Users can compare this file
with the similar file generated in Step 16
and Step 22. We also summarized such
comparison in Table 1.
SK1.canu.filter.pdf
The genome-wide dotplot for the
comparison between the raw genome
assembly and the reference genome.
SK1.canu.filter.mummer2vcf.SNP
.vcf
The VCF file showing SNP differences
between the raw genome assembly and
the reference genome. This file can be
used for assessing the raw assembly
quality.

30
SK1.canu.filter.mummer2vcf.IND
EL.vcf
The VCF file showing INDEL differences
between the raw genome assembly and
the reference genome. This file can be
used for assessing the raw assembly
quality.
SK1_canu_out
the directory containing all the output files
of Canu
02
15
SK1.pilon.fa
The polished genome assembly
generated by Pilon36.
SK1.pilon.vcf
The VCF file reporting the variants
identified by Pilon based on short reads
mapping against the input genome
assembly.
SK1.pilon.changes
The space-delimited record of all the
changes that Pilon made during the
assembly polishing. The four columns
are: the original sequence coordinate, the
new sequence coordinate after the
correction, the original base, the new
base after the correction.
SK1.realn.bam
The BAM file of short reads mapping
against the input genome assembly.
03
16
SK1.ragout.fa
The scaffolded genome assembly based
on the reference genome.
SK1.ragout.stats.txt
The summary table reporting basic
assembly statistics of the scaffolded
genome assembly. Users can compare
this file with the similar file generated in
Step 13 and Step 22. We also
summarized such comparison in Table 1.
SK1.ragout.agp
the AGP file reporting the order and
orientation of each input contig used
during scaffolding.
SK1_ragout_out
The directory containing all the output
files of Ragout33.
SK1.ragout.filter.pdf
The genome-wide dotplot for the
comparison between the scaffolded
assembly and the reference genome.
SK1.mt_contig.list
The list of assembled contigs
corresponding to the mitochondrial
genome. This file will be used for Step 19.
SK1.mt_contig.fa
The assembled contig sequences
corresponding to the mitochondrial
genome.
SK1.ragout.chrMT.filter.pdf
The dotplot for the comparison between
the scaffolded mitochondrial genome
assembly and the reference mitochondrial
genome.
04
18
SK1.centromere.gff3
The profiled centromere identities for the
scaffolded genome assembly.
05
19
SK1.mt_improved.fa
The improved genome assembly with
better processing (re-assembling and
circularization) of the mitochondrial
genome.
SK1.mt_improved.chrMT.filter.pdf
The dotplot for the comparison between
the improved mitochondrial genome

31
assembly and the reference mitochondrial
genome. You should see improved
collinearity in this plot than the similar plot
generated in Step 16.
06
20-22
SK1.modification.list
The assembly modification list file for
manual editing to guide the final genome
assembly.
SK1.final.fa
The final genome assembly generated by
LRSDAY.
SK1.final.filter.pdf
The genome-wide dotplot for the
comparison between the final genome
assembly and the reference genome.
SK1.final.stats.txt
The summary table reporting basic
assembly statistics of the final genome
assembly. Users can compare this file
with the similar file generated in Step 13
and Step 16. We also summarized such
comparison in Table 1.
SK1.final.filter.mummer2vcf.SNP.
vcf
The VCF file showing SNP differences
between the final genome assembly and
the reference genome. This file can be
used for assessing the final assembly
quality.
SK1.final.filter.mummer2vcf.INDE
L.vcf
The VCF file showing INDEL differences
between the final genome assembly and
the reference genome. This file can be
used for assessing the final assembly
quality.
07
23
SK1.centromere.gff3
The centromere annotation for the final
genome assembly.
08
25
SK1.maker.raw.gff3
The raw MAKER37 annotation for protein-
coding genes and tRNA genes.
SK1.EVM.gff3
The final EVM38 annotation for protein-
coding genes and tRNA genes with
systematically assigned gene IDs.
SK1.protein_evidence.gff3
The protein-to-genome alignment
evidences generated by MAKER. This file
can be used for manual curation of those
suspicious annotations.
SK1.est_evidence.gff3
The EST-to-genome alignment evidences
generated by MAKER. This file can be
used for manual curation of those
suspicious annotations.
SK1.EVM.trimmed_cds.fa
The CDSs of the final EVM protein-coding
annotation with the out-of-frame parts
trimmed.
SK1.EVM.trimmed_cds.log
The log file of the CDS trimming for the
final EVM protein-coding gene annotation.
SK1.EVM.pep.fa
The translated protein sequences of the
trimmed CDSs derived from the final EVM
protein-coding gene annotation.
SK1.EVM.manual_check.list
The list of suspicious gene annotations for
manual curation.
SK1.EVM.PoFF.gff
The gene synteny information derived
from SK1.EVM.gff3, which will be used for
Task 12 (Step 34).

32
SK1.EVM.PoFF.ffn
Same as SK1.EVM.trimmed_cds.fa but
with simpler sequence IDs, which could
be used for Task 12 (Step 34).
SK1.EVM.PoFF.ffa
Same as SK1.EVM.pep.fa but with
simpler sequence IDs, which will be used
for Task 12 (Step 34).
09
28
SK1.REannotate.gff
The raw TE annotation from REannotate.
This file can be used for further curating
TE annotation.
SK1.TE.gff3
The final TE annotation from LRSDAY.
10
30
SK1.X_element.gff3
The final core X element annotation from
LRSDAY.
SK1.X_element.fa
The sequences of all the annotated core
X elements.
SK1.X_element.aln.fa
The sequence alignment of all the
annotated core X elements for further
checking whether the annotated feature is
complete or partial as well as whether this
is consistent with the labeling in the
annotation file SK1.X_element.gff3.
11
32
SK1.Y_prime_element.gff3
The final Y’ element annotation from
LRSDAY.
SK1.Y_prime_element.fa
The sequences of all the annotated Y’
elements.
SK1.Y_prime_element.aln.fa
The sequence alignment of all the
annotated Y’ elements for further
checking whether the annotated feature is
complete or partial as well as whether this
is consistent with the labeling in the
annotation file
SK1.Y_prime_element.gff3.
12
34
SK1.proteinortho
The gene orthology mapping between the
annotated genes and the reference gene
sets based only on sequence similarity.
SK1.poff
The gene orthology mapping between the
annotated genes and the reference gene
sets based on both sequence similarity
and synteny conservation.
SK1.updated.gff3
The updated gene annotation with
reference-based gene name labeling.
13
35
SK1.final.gff3
The final integrated annotation from
LRSDAY.
SK1.final.trimmed_cds.fa
The CDSs of the final protein-coding gene
annotation with the out-of-frame parts
trimmed.
SK1.final.trimmed_cds.log
SK1.final.trimmed_cds.log: the log file of
the CDS trimming for the final protein-
coding gene annotation.
SK1.final.pep.fa
The translated protein sequences of the
trimmed CDSs derived from the final
protein-coding gene annotation.
SK1.final.manual_check.list
The list of suspicious gene annotations for
manual curation.
SK1.final.fa
A copy of the final genome assembly
generated in Task 06 (Step 22).
33
REFERENCES
1. Goffeau, A. et al. Life with 6000 Genes. Science 274, 546–567 (1996).
2. Gordon, D. et al. Long-read sequence assembly of the gorilla genome. Science 352,
aae0344 (2016).
3. VanBuren, R. et al. Single-molecule sequencing of the desiccation-tolerant grass
Oropetium thomaeum. Nature 527, 508–11 (2015).
4. Bickhart, D. M. et al. Single-molecule sequencing and chromatin conformation capture
enable de novo reference assembly of the domestic goat genome. Nat. Genet. 49, 643–
650 (2017).
5. Badouin, H. et al. The sunflower genome provides insights into oil metabolism, flowering
and Asterid evolution. Nature 546, 148–152 (2017).
6. Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long
reads. Nat. Biotechnol. 36, 338–345 (2018).
7. Yue, J.-X. et al. Contrasting evolutionary genome dynamics between domesticated and
wild yeasts. Nat. Genet. 49, 913–924 (2017).
8. Goodwin, S. et al. Oxford Nanopore sequencing, hybrid error correction, and de novo
assembly of a eukaryotic genome. Genome Res. 25, 1750–1756 (2015).
9. McIlwain, S. J. et al. Genome sequence and analysis of a stress-tolerant, wild-derived
strain of Saccharomyces cerevisiae used in biofuels research. G3 (Bethesda). 6, 1757–
66 (2016).
10. Istace, B. et al. de novo assembly and population genomic survey of natural yeast
isolates with the Oxford Nanopore MinION sequencer. Gigascience 6, 1–13 (2017).
11. Giordano, F. et al. De novo yeast genome assemblies from MinION, PacBio and MiSeq
platforms. Sci. Rep. 7, 3935 (2017).
12. Koren, S. et al. Hybrid error correction and de novo assembly of single-molecule
sequencing reads. Nat. Biotechnol. 30, 693–700 (2012).
13. Chin, C.-S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT
sequencing data. Nat. Methods 10, 563–9 (2013).
14. Berlin, K. et al. Assembling large genomes with single-molecule sequencing and locality-
sensitive hashing. Nat. Biotechnol. 33, 623–630 (2015).
15. Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time
sequencing. Nat. Methods 13, 1050–1054 (2016).
16. Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long
34
sequences. Bioinformatics 32, 2103–2110 (2016).
17. Koren, S. et al. Canu: Scalable and accurate long-read assembly via adaptive κ-mer
weighting and repeat separation. Genome Res. 27, 722–736 (2017).
18. Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA.
Genome Res. 10, 516–522 (2000).
19. Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: A web server for
gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
20. Besemer, J. & Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes,
eukaryotes and viruses. Nucleic Acids Res. 33, (2005).
21. Proux-Wéra, E., Armisén, D., Byrne, K. P. & Wolfe, K. H. A pipeline for automated
annotation of yeast genome sequences by a conserved-synteny approach. BMC
Bioinformatics 13, 237 (2012).
22. Louis, E. J. & Haber, J. E. The structure and evolution of subtelomeric Y’ repeats in
Saccharomyces cerevisiae. Genetics 131, 559–574 (1992).
23. Strope, P. K. et al. The 100-genomes strains, an S. cerevisiae resource that illuminates
its natural phenotypic and genotypic variation and emergence as an opportunistic
pathogen. Genome Res. 25, 762–774 (2015).
24. Almeida, P. et al. A population genomics insight into the Mediterranean origins of wine
yeast domestication. Mol. Ecol. 24, 5412–5427 (2015).
25. Gallone, B. et al. Domestication and divergence of Saccharomyces cerevisiae beer
yeasts. Cell 166, 1397–1410.e16 (2016).
26. Peter, J. et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates.
Nature 556, 339–344 (2018).
27. Bergström, A. et al. A high-definition view of functional genetic variation from natural
yeast genomes. Mol. Biol. Evol. 31, 872–888 (2014).
28. Drillon, G., Carbone, A. & Fischer, G. SynChro: A fast and easy tool to reconstruct and
visualize synteny blocks along eukaryotic chromosomes. PLoS One 9, (2014).
29. Wang, Y. et al. MCScanX: A toolkit for detection and evolutionary analysis of gene
synteny and collinearity. Nucleic Acids Res. 40, (2012).
30. McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing
next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
31. Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0. 2013-2015 .
http://www.repeatmasker.org (2013).
32. Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. Assembly of Long Error-Prone Reads
35
Using Repeat Graphs. bioRxiv 247148 (2018). doi:10.1101/247148
33. Kolmogorov, M., Raney, B., Paten, B. & Pham, S. Ragout - A reference-assisted
assembly tool for bacterial genomes. Bioinformatics 30, (2014).
34. Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
35. Pereira, V. Automated paleontology of repetitive DNA with REANNOTATE. BMC
Genomics 9, 614 (2008).
36. Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection
and genome assembly improvement. PLoS One 9, (2014).
37. Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database
management tool for second-generation genome projects. BMC Bioinformatics 12, 491
(2011).
38. Haas, B. J. et al. Automated eukaryotic gene structure annotation using
EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7
(2008).
39. Lowe, T. M. & Eddy, S. R. A computational screen for methylation guide snoRNAs in
yeast. Science 283, 1168–71 (1999).
40. Minkin, I., Patel, A., Kolmogorov, M., Vyahhi, N. & Pham, S. Sibelia: A scalable and
comprehensive synteny block generation tool for closely related microbial genomes. in
Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial
Intelligence and Lecture Notes in Bioinformatics) 8126 LNBI, 215–229 (2013).
36
APPENDIX
Appendix 1: Pre-shipped supporting data for LRSDAY
With LRSDAY, we pre-ship the following supporting datasets for the automatic execution of
LRSDAY. Unless labeled otherwise, most of these pre-shipped datasets were either described or
generated in our previous study7.
1) ATP6.cds.fa # The coding sequence (CDS) of the S. cerevisiae S288C ATP6 gene.
2) fuzzy_defragmentation.txt # the fuzzy defragmentation file for REannotate35.
3) Proteome_DB_for_annotation.CDhit_I95.fa # our curated proteome dataset for S.
cerevisiae and other closely related yeast sensu stricto species.
4) query.Y_prime_element.long.fa # the sequence of a representative S. cerevisiae Y’
element.
5) S288C.ASM205763v1.fa.gz # the S. cerevisiae S288C genome assembly.
6) S288C.ASM205763v1.noncore_masked.fa.gz # the S. cerevisiae S288C genome
assembly with subtelomeres and chromosome-ends hard-masked.
7) S288C.centromere.fa # the centromere sequence of S. cerevisiae S288C.
8) S288C.gene.hmm # the hidden Markov model (HMM) for de novo gene annotation based
on S. cerevisiae S288C.
9) S288C.X_element.hmm # the hidden Markov model (HMM) for the core X element
annotation based on S. cerevisiae S288C.
10) Sc-meth.sites # the S. cerevisiae methylation sites (shipped with snoScan39).
11) Sc-rRNA.fa # the S. cerevisiae rRNA sequences (shipped with snoScan39).
12) SGDref.PoFF.faa # the proteinortho proteome file generated for the SGD reference
genome.
13) SGDref.PoFF.ffn # the proteinortho CDS file generated for the SGD reference genome.
14) SGDref.PoFF.gff # the proteinortho gene order gff file generated for the SGD reference
genome.
15) te_proteins.fasta # protein sequences for genes encoded within TEs (shipped with
MAKER37).
16) TY2_specific_region.fa # the sequence of a Ty2 specific regions for differentiating Ty1
and Ty2.
17) TY_lib.Yue_et_al_2017_NG.fa # a custom RepeatMasker library for Ty annotation in S.
cerevisiae and S. paradoxus.
37
18) TY_lib.Yue_et_al_2017_NG.LTRonly.fa # representative Ty LTR sequences of S.
cerevisiae and S. paradoxus.

38
Appendix 2: Tips for adapting LRSDAY to other eukaryotic organisms
The backbone modules of LRSDAY can be easily adapted for other eukaryotic organisms. Here
are some tips with regard to this:
1) For Task 01 (long-read-based genome assembly; Step 13), be sure to adjust the genome
size parameter in line 13 of the bash script LRSDAY.01.Long-read-
based_Genome_Assembly.sh based on the estimated genome size of the organism that
you sequenced.
2) For Task 03 (reference-guided assembly scaffolding; Step 16), be sure to modify the bash
script LRSDAY.03.Reference-guided_Assembly_Scaffolding.sh to provide the reference
genome file of your sequenced organisms to guide the scaffolding and chromosome
assignment. It is very likely that the chromosomal cores and subtelomeres of your
reference genome have not been clearly defined. In such case, you can provide the same
reference genome file for both the “ref_genome_raw=” and
“ref_genome_noncore_masked=” parameters. The “chrMT_tag=” and “gap_size=”
parameters should also be adjusted for your own project.
3) By default, Task 03 (reference-guided assembly scaffolding; Step 16) performs reference-
guided scaffolding using the whole genome alignment constructed by Sibelia40. Sibelia is
designed for processing small genomes (<100 Mb). For organisms with large genomes,
you should install Progressive Cactus (https://github.com/glennhickey/progressiveCactus)
and use it to build the whole genome alignment in HAL format and feed it directly to
Ragout33. Please refer to Ragout’s user manual
(http://fenderglass.github.io/Ragout/usage.html) for this advanced usage. We have pre-
shipped the HAL tools (https://github.com/ComparativeGenomicsToolkit/hal) to enable
Ragout to process the HAL file generated by Progressive Cactus.
4) For Task 05 (mitochondrial genome assembly improvement; Step 19), be sure to modify
the “gene_start=”, “ref_genome_raw=”, and “chrMT_tag=” parameters in the bash script
LRSDAY.05.Mitochondrial_Genome_Assembly_Improvement.sh based on your own
project.
5) While many of the genomic feature annotation tasks are S. cerevisiae and S. paradoxus
specific, Task 08 (protein-coding genes and tRNA annotation; Step 25) can be adapted
for any eukaryotic organism. In general, you will need to edit the lines 16, 22, 34, 36, 44,
45, and 68-71 in the $LRSDAY_HOME/misc/maker_opts.customized.ctl file to feed
organism-specific parameters into MAKER. Also, please refer to MAKER’s own Wiki page
(http://weatherby.genetics.utah.edu/MAKER/wiki/index.php/Main_Page) and protocols

39
(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4286374/) for technical details and
advanced usage. Similarly, you can learn more about EVM from its website
(http://evidencemodeler.github.io/).
6) While Task 09 (TE annotation; Step 28) is heavily tuned for yeasts, the same tools that
we used here (RepeatMasker31 and REannotate35) can be used for any eukaryotic
organism. We recommend reading their respective manuals (distributed with the
corresponding software) for adapting these tools in your own study.
7) For Task 12 (gene orthology identification; Step 34), you need to edit the “ref_PoFF_faa=”
and “ref_PoFF_gff=” parameters based on the reference gene annotation that you used.
Please check ProteinOrtho’s manual (https://www.bioinf.uni-
leipzig.de/Software/proteinortho/manual.html) for more details on required file formats.
The pre-shipped Perl script prepare_PoFFfaa_simple.pl and prepare_PoFFgff_simple.pl
in the $LRSDAY_HOME/scripts directory should help for this. You can run these two
scripts as follows:
$ source ./../../env.sh
$ perl $LRSDAY_HOME/scripts/prepare_PoFFffn_simple.pl -i prefix.pep.fa -o
prefix.PoFF.faa
$ perl $LRSDAY_HOME/scripts/prepare_PoFFgff_simple.pl -i prefix.raw.gff -o
prefix.PoFF.gff
