Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 13

LTR_retriever User Manual
Shujun Ou and Ning Jiang
oushujun@msu.edu & jiangn@msu.edu
Department of Horticulture, Michigan State University, East Lansing, MI, 48824, USA
In any research documents using LTR_retriever please cite the following paper:
Shujun Ou and Ning Jiang (2017) LTR_retriever: a highly accurate and sensitive program for
identification of LTR retrotransposons (in preparation)
LTR_retriever is licensed under GNU GPLv3.
Questions and Issues Please See: https://github.com/oushujun/LTR_retriever
Sept. 21, 2017
1 Introduction
LTR_retriever is a command line program (in Perl) for accurate identification of LTR
retrotransposons (LTR-RTs) from outputs of LTRharvest (1), LTR_FINDER (2), and MGEScan-
LTR (3, 4) and generation of a non-redundant LTR-RT library for genome annotations.
As one of the most prevalent transposable elements (TEs), LTR-RT comprises the largest portion
of most plant genomes (5). Due to the sequence diversity of LTR-RTs, identification of such elements
based on sequence homology is inefficient. Instead, LTR-RTs are conserved in terms of element
structure across different species. Several programs have been developed to search for LTR-RTs using
relevant structural characteristics. These programs are very sensitive; however, they are not very
accurate and specific for LTR-RT identifications. LTR_retriever was developed to address the
accuracy and specificity needs, with several new functions to facilitate genome annotation and other
downstream studies.
LTR_retriever aims to identify high-quality LTR-RT exemplars (Figure 1A) that are intact
and non-redundant from a variety of LTR-RT candidates. To retain sensitivity, sequences of nested
LTRs and truncated LTRs (Figure 1CD) that are not represented by intact LTR-RTs will also be
included in the exemplar. This package excludes the vast majority of the non-LTR false positives. The
most common false positives were introduced by two adjacent non-LTR repeats which are found as
SINEs, LINEs, DNA TEs, or solo-LTRs that are derived from different elements (Figure 3). In
addition, LTR_retriever excludes non-LTR open reading frames derived from LINEs, DNA TEs,
or plant coding sequences to reduce misannotations of non-LTR coding sequences as LTR elements.
LTR_retriever identifies and removes LTR-RT nested insertions in the identified intact LTR-RTs,
which also reduces library redundancy. This program can also accurately identify rare non-canonical
LTR-RTs that have terminal motifs different from the canonical 5'-TG..CA-3' motif. The program was
built with a variety of Perl scripts that can be utilized for downstream analyses.
1.1 Main features of LTR_retriever
- A command line Perl program;
- Supports multi-threading;
- Identifies intact LTR-RTs with accurate boundaries;
- Identifies rare LTR-RTs with non-canonical (non-'TGCA') motifs;
- Supports multiple inputs: LTRharvest, LTR_FINDER, and/or MGEScan_LTR;
- Sequence input: FASTA format (contigs, scaffolds, genomes, corrected PacBio reads, and
etc.);
- Output: a non-redundant LTR-RT library (FASTA), GFF3 for all intact LTR-RTs, whole-
genome LTR-RT annotation (GFF), and a comprehensive table.
2 The Structure and Characteristics of LTR-RTs
The structure of an LTR retrotransposon (LTR-RT) is characterized by long terminal repeat
ranging from 75 bp to 5000 bp (Figure 1A). The region between the 5' LTR and 3' LTR is termed the
internal region, which encodes proteins for transposition. At the very termini of the LTRs are the bi-
nucleic motifs, which is 5'-TG..CA-3' in most cases. However, various other motifs have been
detected in the sacred lotus (Nelumbo nucifera) genome and in the rice (Oryza sativa) genome during
our manual annotation, and also found in other studies (e.g., Tos17 (6) ; AtRE1 (7); and TARE1 (8)).
Flanking the terminal motifs is the target site duplication (TSD), which is generated by staggered cuts
from integrase activity (Figure 2) during LTR-RT insertion. TSDs are typically 5 bp in plants but
could vary between 3-6 bp, and the 5' and 3' TSD should be identical because of the mechanism of
their formation (Figure 2). The recently inserted LTR-RT has a highly similar LTR region that is
recognizable by sequence alignment, which is the primary searching scheme for LTR search programs
(1, 2, 4, 9). However, if two highly similar repetitive elements other than LTR (e.g., DNA, LINE,
SINE, solo-LTR, tandem repeat, etc.) are located close to each other (Figure 3), searching tools may
falsely choose them and report them as LTR-RT candidates. These are the most frequent false
positives that occur in de novo searches for LTR-RTs. Given that LTR-RTs are following the "copy-
and-paste" duplication scheme, the regions flanking the newly inserted LTR-RT are unlikely to be
identical to the termini of the internal region. For example, in an intact LTR-RT (Figure 1A), region
“a” is not identical to region “c”, and region “b” is not identical to region “d”. Thus, by aligning the
flanking regions of the two LTR fragments (Figure 3), LTR_retriever can obtain the boundary
information for the candidate.
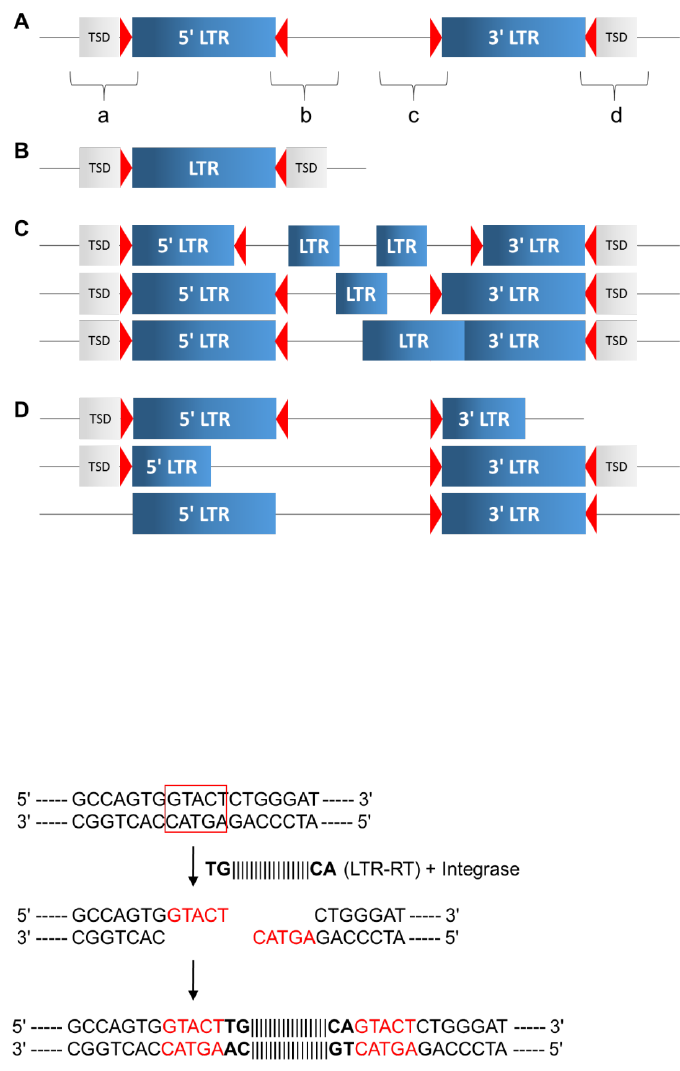
Figure 1. The structures of LTR retrotransposons (LTR-RT). Gray boxes: target site
duplications (TSD); red triangles: LTR motifs; blue boxes: long terminal repeat (LTR);
sequence between 5' LTR and 3' LTR denotes the internal region. (A) The structure of an
intact LTR-RT. Regions a, b, c, and d are main targets analyzed by LTR_retriever. (B)
The structure of a solo-LTR. (C) The structures of nest-inserted LTR-RTs. (D) The structures
of truncated LTR-RTs. Drawing is not on scale.
Figure 2. Formation of target site duplications (TSD). Integrases coded by LTR-RTs
generate staggered cuts (in this case 5'-GTACT-3') on the sequence before new LTR-RT
insertions. By gap filling and sequence ligation, a pair of TSD is formed flanking the newly
inserted LTR.
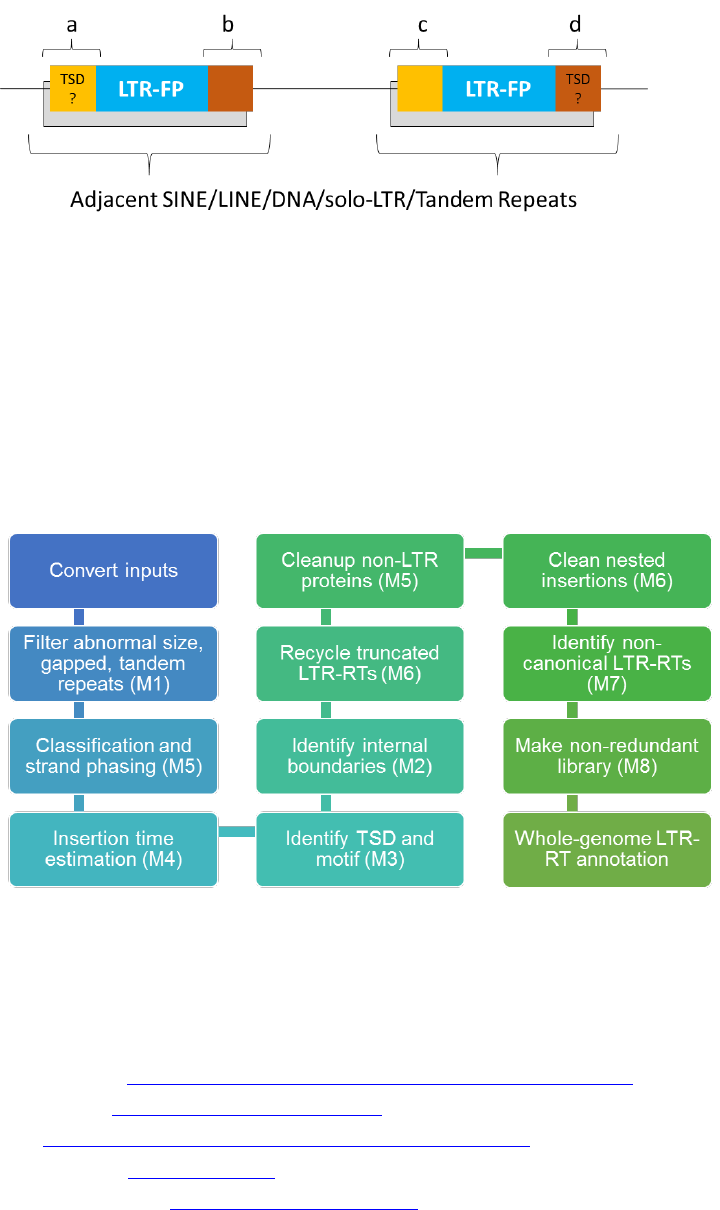
Figure 3. The most common false positives in de novo searches for LTR-RTs. Gray boxes:
two closely positioned SINE/LINE/DNA elements/solo-LTRs/tandem repeats. Light blue boxes:
false LTR regions reported by de novo searches. The false positive also has TSD-like structure
but commonly has extended sequence identity on one or both termini (orange and brown
boxes).
3 Workflow of LTR_retriever
In LTR_retriever, there are eight modules developed to screen and filter out false positives
and construct non-redundant LTR exemplars (Figure 4). More details can be found in our article.
Figure 4. Workflow of LTR_retriever. Modules 1-8 are indicated in parentheses.
4 Installation
LTR_retriever is a command line Perl program that incorporates several programs for
analysis and runs in UNIX-like systems. These programs include:
BLAST+ (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/),
CDHIT (http://weizhongli-lab.org/cd-hit/) OR BLAST
(ftp://ftp.ncbi.nlm.nih.gov/blast/executables/legacy/2.2.25/),
HMMER (http://hmmer.org/), and
RepeatMasker (http://www.repeatmasker.org/).
To run LTR_retriever, you need to provide the paths to the following dependent programs:

makeblastdb, blastn, and blastx in the BLAST+ package,
cd-hit-est in the CDHIT package OR blastclust in the BLAST package,
hmmsearch in the HMMER package, and
RepeatMasker
If the above programs are all accessible through ENV in the UNIX-like system (i.e. paths exported
to .bashrc), no installation is needed. Otherwise, users need to modify the paths file under the
your_path_to/LTR_retriever/ directory. If specifying a path, the required program(s) must
be directly contained in that path but not in any subdirectories.
For example:
Edit the paths file using vi/vim (you may use other text editors such as emacs)
vi /your_path_to/LTR_retriever/paths
Modify the following lines
BLAST+=/your_path_to/BLAST+2.2.30/bin/
RepeatMasker=/your_path_to/RepeatMasker4.0.0/
HMMER=/your_path_to/HMMER3.1b2/bin/
CDHIT=/your_path_to/CDHIT4.6.1/
BLAST=/your_path_to/BLAST2.2.26/bin/ #not required if CDHIT provided
Save changes to paths and exit. The installation is done.
5 Inputs
Two types of inputs are needed for LTR_retriever:
1. Genomic sequence
2. LTR-RT candidates
5.1 Genomic sequence
The sequence must be in FASTA format. Users should backup the original input FASTA file because
LTR_retriever will modify sequence names that are longer than 20 characters to fit the naming
space of RepeatMasker. Thus, FASTA sequence names are recommended to be less than 20
characters without spaces and special punctuation marks other than dots (.) and underscores
(_). For long sequencing reads (e.g., PacBio), self-corrected reads are needed unless the sequence
error rate is lower than 10%.
5.2 LTR-RT candidates
LTR_retriever takes multiple LTR-RT candidate inputs including the standard output of
LTRharvest, the standard output of LTR_FINDER, and the candidate output of MGEScan-LTR.
Users need to obtain the input file(s) from the aforementioned programs before running
LTR_retriever. Either a single input source or a combination of multiple inputs is acceptable. The
following command lines provide examples and suggestions to obtain inputs.
Input from LTRharvest (a program of GenomeTools (10)): (“\” indicates this line and next

line belong to the same command line. Please delete “\” if you run into errors.)
gt suffixerator \
-db genome.fa \
-indexname genome.fa \
-tis -suf -lcp -des -ssp -sds -dna
gt ltrharvest \
-index genome.fa \
-similar 90 -vic 10 -seed 20 -seqids yes \
-minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 \
-motif TGCA -motifmis 1 > genome.harvest.scn
Input from LTR_FINDER:
ltr_finder -D 15000 -d 1000 -L 7000 -l 100 \
-p 20 -C -M 0.9 genome.fa > genome.finder.scn
Input from MGEScan_LTR (a modified version obtained from DAWGPAWS (11)):
perl find_ltr_DAWGPAWS.pl \
-seq=genome.fa \
-min-ltr=100 -max-ltr=7000 -min_iden=90
The –nonTGCA input can be obtained without specifying -motif TGCA -motifmis 1 in
LTRharvest:
gt ltrharvest \
-index genome.fa \
-similar 90 -vic 10 -seed 20 -seqids yes \
-minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 \
> genome.harvest.nonTGCA.scn
6 Outputs
The output of LTR_retriever includes:
A summary table for the identified intact LTR-RTs with coordinate and structural information

(*.pass.list)
A non-redundant LTR-RT library (exemplar) in the FASTA format (*.LTRlib.fa)
A GFF3 format file for all intact LTR-RTs (*.pass.list.gff3)
A GFF format file for the whole-genome LTR-RT annotation (*.gff)
Example of an intact LTR-RT list:
#LTR_loc Category Motif TSD 5'_TSD 3'_TSD Internal Similarity Strand Family
Superfamily Insertion_Time
Chr10:10211053..10223177 pass motif:TGCC TSD:GGTGG 10211048..10211052
10223178..10223182 IN:10211491..10222739 0.9794 - Gypsy LTR 1030000
Chr10:11328988..11335399 pass motif:TGCA TSD:CAGTC 11328983..11328987
11335400..11335404 IN:11329472..11334915 0.9587 - Copia LTR 2065000
Chr2:16844554..16849613 pass motif:TGCA TSD:GCATG 16844549..16844553
16849614..16849618 IN:16845219..16848945 0.9416 ? unknown NA 2920000
Chr2:17280296..17282788 pass motif:TGCA TSD:TATAC 17280291..17280295
17282789..17282793 IN:17280474..17282616 0.9497 + unknown LTR 2515000
Chr2:17891339..17904598 pass motif:TGCA TSD:CCCTC 17891334..17891338
17904599..17904603 IN:17892615..17903320 0.9867 ? Gypsy LTR 665000
Example of an LTR library:
>Chr10:1057194..1057414_LTR#LTR/Copia
TGTTGGCGAACGGCTTCGTCAGACTCTCGCGGCGCGCTCCACGCGCACGACGCGCACCC
CGCGCACGACGCGCAGCGCTCCTCGCTCCGCTCGCACCGCTGCACGTCCGTTAGACCAG
GGGATTAGTTAGGCCCAGGCAACTCCCAAGCCTTGTTGTACATGTATAAATGTAAGCTC
CATTGATCAATGAAAGTTACGGTTGATCCAAATCTCCTTCTACA
>Chr10:12110230..12110468_LTR#LTR/unknown
TGTCATGGGCTTTGGGCCGGGAGTCCTAGGCCCATGAGATAGAATTAGGGTTTGTTAGG
ATTAGATAAGGTTTGTTAGGATTAGATTAAGTAGCCCTCCATCTATATAAGGAGGGATC
CTATCCCAGGTCAGTTAGGCATTAGATCAATATTTATCTTAGTGCCCATCGGCCTGCCT
TCTCAGTGCGACGGAGAGCGTCGCGCCGTTTAGGTTCAGGACCGTATTCCTTGTTCGTG
ACA
>Chr10:10063621..10068275_INT#LTR/Gypsy
AATCCACCCCCCTTACAAGAATTTCGTCCCCGAGATTCGAGGAGGCTAGCATGAAGATA
…
CTACATGCCGCTAGCGGATCCTGCAGTCTTCCGGAGCTTCGGCAAACAATTGGCGACAT
CTTCTTCTTCGCAAAGCTGACCATCTAGTACTAGTTGAAAATCCGAAGGAGGAAGAAGA
CAATAAACATTTTGCAAAT

7 Usage
7.1 LTR_retriever is called as follows:
LTR_retriever -genome genomefile -inharvest LTRharvest_input [options]
where -genome specifies the genome sequence and is also used as the root file name of outputs;
the -inharvest parameter specifies the LTR-RT candidate file obtained from LTRharvest.
Multiple candidate sources can be used (see Table 1).
For example,
User provides only one candidate source:
e.g. 1
LTR_retriever -genome genome.fa -infinder genome.finder.scn
e.g. 2
LTR_retriever -genome genome.fa -inharvest genome.harvest.scn
e.g. 3
LTR_retriever -genome genome.fa -inmgescan genome.MGEScan.scn
User provides multiple candidate sources:
e.g. 4
LTR_retriever \
-genome genome.fa \
-inharvest genome.harvest.scn \
-infinder genome.finder.scn
e.g. 5
LTR_retriever \
-genome genome.fa \
-inharvest genome.harvest.scn \
-infinder genome.finder.scn \
-inmgescan genome.MGEScan.scn

To recover non-canonical LTR-RTs, you may use the –nonTGCA option to provide extra
candidates along with other input(s) (either one source or multiple sources).
e.g. 6
LTR_retriever \
-genome genome.fa \
-inharvest genome.harvest.scn \
–nonTGCA genome.harvest.nmtf.scn
e.g. 7
LTR_retriever \
-genome genome.fa \
-infinder genome.finder.scn \
-inharvest genome.harvest.scn \
-inmgescan genome.MGEScan.scn \
–nonTGCA genome.harvest.nonTGCA.scn
7.2 WARNINGS
LTR_retriever will alter sequence names longer than 20 characters to fit the naming requirement
of RepeatMasker. Please backup your original genome file before using LTR_retriever.
LTR_retriever can take multiple sources as inputs for one single run, but running multiple
instances of LTR_retriever in the same folder at the same time may cause errors.
7.3 An overview of all parameters
Table 1. All parameters for LTR_retriever.
Input options
-genome
[FASTA File]
specify the genome sequence file (in FASTA format)
-inharvest
[File]
LTR-RT candidates obtained from the screen output of LTRharvest
with -motif TGCA parameters
-infinder
[File]
LTR-RT candidates obtained from the screen output of LTR_FINDER
-inmgescan
[File]
LTR-RT candidates obtained from the output of MGEScan_LTR
(the .ltrloc file)
-nonTGCA [File]
Non-canonical LTR-RT candidates obtained from the screen output of
LTRharvest with default parameters
Output options
-verbose
retain intermediate outputs (developer mode)

-noanno
disable whole genome LTR-RT annotation (no GFF output)
Filter options
-misschar [CHR]
specify the character for ambiguous sequences in the genome (default N)
-Nscreen
disable filtering ambiguous sequence in LTR-RT candidates (default
enable (by not specifying this flag))
-missmax [INT]
specify the maximum number of ambiguous bp allowed in an LTR-RT
candidate (default 10)
-missrate [0-1]
specify the maximum percentage of ambiguous length bp allowed in an
LTR-RT candidate (default 0.8)
-minlen [INT]
specify the minimum length (bp) of the LTR region (default 100)
-max_ratio
[FLOAT]
specify the maximum length ratio of the internal region length over the
LTR region length (default 50)
-minscore [INT]
specify the minimum alignment length (INT/2) to identify and filter out
tandem repeats in an LTR-RT candidate (default 1000)
-flankmiss [1-
60]
specify the maximum gap length (bp) allowed in 60bp-flanking
sequences (default 25), smaller number indicates higher stringency
-flanksim [0-
100]
specify the minimum percentage of identity for flanking sequence
alignment (default 60)
-flankaln [0-1]
specify the maximum alignment portion allowed for 60bp-flanking
sequences (default 0.6)
-motif
[[STRING]]
specify a list of (known) motifs in square brackets as the prior knowledge
to search for non-canonical LTR-RTs (default -motif [TCCA TGCT
TACA TACT TGGA TATA TGTA TGCA])
-notrunc
Discard sequence information from truncated LTR-RTs and nested LTR-
RTs (will dampen sensitivity) (default retain (not specifying this flag))
-procovTE [0-1]
specify the maximum portion of an LTR-RT candidate allowed for
cumulated alignments to the DNA TE database and the LINE database
(default 0.7)
-procovPL [0-1]
specify the maximum portion of an LTR-RT candidate allowed for
cumulated alignments to the plant protein database (default 0.7)
-prolensig
[INT]
specify the minimum alignment length (bp) to be counted for LINE/DNA
transposase/plant protein alignment (default 90)
Library options
-blastclust
[[STRING]]
specify blastclust parameters in square brackets (default -
blastclust [-L .9 -b T -S 80]). By triggering this tag
without specifying any parameters (-blastclust), blastclust
will be turned on (default off) with default parameters. blastclust
settings refer to
http://www.ncbi.nlm.nih.gov/Web/Newsltr/Spring04/blastlab.html
-cdhit
specify cd-hit-est parameters in square brackets (default -cdhit
[[STRING]]
[-c 0.8 -G 0.8 -s 0.9 -T 20 -aL 0.9 -aS 0.9]). By
triggering this tag without specifying any parameters, cd-hit-est will
be turned on (default on) with default parameters. cd-hit-est
settings refer to http://weizhongli-lab.org/cd-hit/wiki/doku.php?id=cd-
hit_user_guide
-linelib
[FASTA File]
specify a custom LINE transposase database for LINE TE exclusion
(default LTR_retriever/database/Tpases020812LINE)
-dnalib
[FASTA File]
specify a custom DNA TE transposase database for DNA TE exclusion
(default LTR_retriever/database/Tpases020812DNA)
-plantprolib
[FASTA File]
specify a custom plant protein database for protein coding sequence
exclusion (default
LTR_retriever/database/alluniRefprexp082813)
-TEhmm
[Pfam File]
specify a custom Pfam database for TE identification (default
LTR_retriever/database/TEfam.hmm)
Miscellaneous
-u [FLOAT]
specify the neutral mutation rate of the target species (per bp per year)
(default 1.3e-8 (from rice (12)))
-threads [INT]
specify the number of threads (≤ total available threads, default 4)
--help (-h)
display the help information
8 Benchmarks
Run time of LTR_retriever is roughly proportional to total candidate number (input) as shown in
Table 2. The size and the LTR-RT fraction of a genome together determine the number of LTR-RT
candidates identified by prediction programs.
Table 2. Benchmark of LTR_retriever in model genomes.
Arabidopsis
Drosophila
Rice (MSU v7)
Sacred lotus
Maize (B73 v4)
Genome size (Mb)
120
144
374
708
2,134
Raw candidates
2335
2642
5436
12011
114048
Intact LTR-RT
232
517
2129
918
43227
Fraction masked
7.4%
12.4%
25.3%
29.6%
70.1%
Library entry
233
359
1529
1467
12360
Run time (-threads 20)*
10 min
10 min
42 min
2.1 hr
94.9 hr
*not including the time of whole-genome LTR-RT annotation.
9 Reusable Scripts
LTR_retriever was built on several flexible Perl scripts which are useful for other research
purposes. This section describes some of the most useful ones based on the developer’s experience.
These reusable scripts include many more others are located in LTR_retriever/bin/
Script: annotate_gff.pl
Description: Annotate the GFF file generated by RepeatMasker using the LTR library generated by
LTR_retriever.
Usage: perl annotate_gff.pl genome_LTRlib.fa genome.gff >
genome.anno.gff
Options: None.
Script: call_seq_by_list.pl
Description: Extract sequence from the user provided genome (FASTA format) using a file containing
a list of coordinates (one line each) in the MSU locus format (e.g., target1 Chr01:10000..11000). The
script can output sequence in its minus direction. If the locus coordinate is written backward (e.g.,
Chr1:2000..1000), it would be treated as a negative strand request.
Usage: perl call_seq_by_list.pl MSU_format_list -C genome.fa [options]
Options:
itself (default) extract the sequence specified by the coordinate
up_[INT] extract the sequence [INT] bp upstream of the specified coordinate
down_[INT] extract the sequence [INT] bp downstream of the specified coordinate
-rmvoid if this is triggered (default off), skip printing any void sequence (e.g., no such
sequence in the genome)
-ex if this is triggered (default off), execute the exclude function instead of the
extract function
-purge [0/1] use with -ex, if this is triggered (1, default 0), sequence specified by the
list file will be excluded from the provided FASTA file
-cov [0-1] use with -ex, sequence with removal length longer than the specified portion
(default 0.7) will be entirely excluded
Script: purger.pl
Description: Purge the provided FASTA file with BLAST alignment output.
Usage: perl purger.pl -blast blast_outfmt6 -seq FASTA [options]
Options:
-eval [FLOAT] for BLAST hits, e-values (e.g. 1e-10) lower than this cutoff (default 0.001)
is considered a real alignment
-len [INT] length of alignment hits (bp, default 90) to be considered as a real alignment
-cov [0-1] if the excluded portion of a sequence exceeds the specified value (default 1),
discard the entire sequence
-purge [0/1] if this is triggered (1, default 1), sequence regions identified by BLAST will
be excluded from the provided FASTA file. If this is not triggered, the entire
sequence will be excluded if it achieves the cutoff defined in “-cov”. Otherwise
the matched portion will be retained.
Script: PacBio_processor.pl
Description: Convert fastq files (e.g., PacBio reads) into FASTA files with simple filtering options.
Usage: perl PacBio_processor.pl PacBio.fastq > PacBio.fasta
Options (modified in the script):
minLength minimal read length (bp) (default 500)
maxLength maximal read length (bp) (default 50000)
minRQ minimal read quality (default 0.8)
Bibliography
1. Ellinghaus D, Kurtz S, Willhoeft U. LTRharvest, an efficient and flexible software for de novo detection
of LTR retrotransposons. BMC bioinformatics. 2008;9(1):18.
2. Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons.
Nucleic acids research. 2007;35(Web Server issue):W265-8.
3. Lee H, Lee M, Mohammed Ismail W, Rho M, Fox GC, Oh S, et al. MGEScan: a Galaxy-based system for
identifying retrotransposons in genomes. Bioinformatics. 2016.
4. Rho M, Choi J-H, Kim S, Lynch M, Tang H. De novo identification of LTR retrotransposons in eukaryotic
genomes. BMC genomics. 2007;8(1):90.
5. Jiang N. Plant Transposable Elements. eLS: John Wiley & Sons, Ltd; 2016.
6. Hirochika H, Sugimoto K, Otsuki Y, Tsugawa H, Kanda M. Retrotransposons of rice involved in
mutations induced by tissue culture. Proceedings of the National Academy of Sciences of the United
States of America. 1996;93(15):7783-8.
7. Kuwahara A, Kato A, Komeda Y. Isolation and characterization of copia-type retrotransposons in
Arabidopsis thaliana. Gene. 2000;244(1-2):127-36.
8. Yin H, Liu J, Xu Y, Liu X, Zhang S, Ma J, et al. TARE1, a mutated Copia-like LTR retrotransposon followed
by recent massive amplification in tomato. PloS one. 2013;8(7):e68587.
9. McCarthy EM, McDonald JF. LTR_STRUC: a novel search and identification program for LTR
retrotransposons. Bioinformatics. 2003;19(3):362-7.
10. Gremme G, Steinbiss S, Kurtz S. GenomeTools: A Comprehensive Software Library for Efficient
Processing of Structured Genome Annotations. IEEE/ACM transactions on computational biology and
bioinformatics / IEEE, ACM. 2013;10(3):645-56.
11. Estill JC, Bennetzen JL. The DAWGPAWS pipeline for the annotation of genes and transposable
elements in plant genomes. Plant Methods. 2009;5(1):1-11.
12. Ma J, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proceedings of the
National Academy of Sciences of the United States of America. 2004;101(34):12404-10.
