NCPA User Guide
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 28
ANeCA: User Guide
Author: Mahesh Panchal
This software was
Developed during my PhD
Supervisor: Dr. M. A. Beaumont
Funding: BBSRC, 10/02-10/05
July 22, 2008
Contents
1 Software Documentation 3
1.0.1 Updates from v1.0 . . . . . . . . . . . . . . . . . . . . 3
1.0.2 Updates from v1.1 . . . . . . . . . . . . . . . . . . . . 3
1.1 Basic’sofNCPA ......................... 4
1.1.1 Summary of the Method. . . . . . . . . . . . . . . . . . 4
1.1.2 Contraversy of NCPA . . . . . . . . . . . . . . . . . . . 5
1.2 Programcitation ......................... 5
1.3 TheInputFiles .......................... 6
1.3.1 The Nexus or Phylip DNA sequence file . . . . . . . . 6
1.3.2 Geographic information file . . . . . . . . . . . . . . . 7
1.4 TheOutputFiles......................... 7
1.4.1 TheGraphFile...................... 8
1.4.2 The Geodis Input file . . . . . . . . . . . . . . . . . . . 8
1.4.3 TheGMLfile ....................... 8
1.4.4 TheNestfile ....................... 8
1.4.5 The NANOVA file . . . . . . . . . . . . . . . . . . . . 8
1.4.6 The GeoDis Output file . . . . . . . . . . . . . . . . . 9
1.4.7 The GeoDis Summary file . . . . . . . . . . . . . . . . 9
1.4.8 The Inference File . . . . . . . . . . . . . . . . . . . . 9
1.5 Using the software for NCPA . . . . . . . . . . . . . . . . . . 9
1.5.1 Using the Graphical Interface . . . . . . . . . . . . . . 9
1.5.2 Using the Command Line . . . . . . . . . . . . . . . . 14
1.5.3 Drawing the nested design . . . . . . . . . . . . . . . . 14
1.6 AWorkedexample ........................ 15
1.6.1 Creating the Geographic information file . . . . . . . . 15
1.6.2 Creating the Nexus file . . . . . . . . . . . . . . . . . . 16
1.6.3 Create the haplotype network . . . . . . . . . . . . . . 16
1.6.4 Creating the Nested Design . . . . . . . . . . . . . . . 17
1.6.5 Drawing the Nested Design . . . . . . . . . . . . . . . 18
1.6.6 Performing the Permutation Analysis . . . . . . . . . . 18
1
1.6.7 Applying the Automated Inference Key. . . . . . . . . 18
1.7 Using the software for genotype-phenotype studies . . . . . . . 19
1.7.1 File simplifications . . . . . . . . . . . . . . . . . . . . 19
1.7.2 Using the Graphical Interface . . . . . . . . . . . . . . 20
1.7.3 Using the Command line . . . . . . . . . . . . . . . . . 20
1.8 Limitations of Software . . . . . . . . . . . . . . . . . . . . . . 20
1.9 Common Exceptions thrown . . . . . . . . . . . . . . . . . . . 21
1.10KnownIssues ........................... 21
1.11 Frequently Asked Questions . . . . . . . . . . . . . . . . . . . 22
1.12Disclaimer............................. 24
1.13 Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . 25
2
Chapter 1
Software Documentation
This software is a basic fully automated implementation of Nested Clade
Phylogeographic Analysis (NCPA). NCPA is a method of phylogeographic
inference developed by Alan Templeton and colleagues.
Please become familiar with this manual. It will solve a lot of
the common problems encountered using this software. Also please
check your results. This software is not intended to be a black box,
and is still in early stages of development.
1.0.1 Updates from v1.0
1. The inference key implemented has been updated from 14th July 2004
to the inference key dated 11th November 2005.
Unfortunately I have not had the time to work on including updates of TCS
or GeoDis.
1.0.2 Updates from v1.1
1. There has been a change in implementation of Question 19 and 20
on the inference key. These questions will no longer regard popula-
tions (sampled or unsampled) within the convex hull of all the clades
as within the clade boundary if they are within the convex hull of a
subclade.
2. ANeCA now uses TCS v1.21.
3. ANeCA now uses GeoDis v2.5.
3

1.1 Basic’s of NCPA
This is a summary of the method only and my opinion of the state of NCPA.
Please familiarise yourself with the literature surrounding the method includ-
ing criticisms and their replies. If you are unfamiliar with NCPA please use
the references within as a guide to familiarise yourself with the method.
1.1.1 Summary of the Method.
Many researchers use NCPA in various ways augmenting the method with
various results and techniques. The basic methodology is to generate a hap-
lotype network from your data, nest the haplotype network, calculate statis-
tics, and use an inference key. This is normally applied to a single locus, for
example, the Cytochrome b region in mtDNA.
Note that NCPA is a method that has evolved over the years. As such
there is some discrepancy as to how NCPA should be applied. The cur-
rent method of applying NCPA suggested by Templeton et al. (2005) is the
following:
1. Generate a haplotype network.
2. Use the nesting algorithm provided in the original literature (Templeton
et al. 1987; Templeton and Sing 1993).
3. Calculate the Dnand Dcstatistics using GeoDis (Posada et al. 2000).
4. Apply the key either by hand, using AUTOINFER (Zhang et al. 2006)1,
or this software (Check the differences).
5. Validate these inferences using other loci (Templeton et al. 2005).
However in practice many of the articles published by Templeton use the
following steps:
1. Generate a haplotype network.
2. Use the criteria in Crandall and Templeton (1993) to find the best
resolution of the network.
3. If there are still loops, enumerate all possible trees within the network.
4. Nest all trees (This program is adequate for that purpose).
1This software has recently been withdrawn, but may become available in the near
future.
4
5. Calculate the Dnand Dcstatistics using GeoDis (Posada et al. 2000).
6. Apply the key either by hand, using AUTOINFER (Zhang et al. 2006),
or this software (Check the differences).
7. Check that all the trees provide inferences that are concordant. Discard
inferences that do not match.
8. Validate these inferences using other loci (Templeton et al. 2005).
1.1.2 Contraversy of NCPA
The field of phylogeography is currently split as to whether the method works
or not. Much of the reasoning behind NCPA is as yet not explained in the lit-
erature. Simulations by Knowles and Maddison (2002) indicated that NCPA
did not work when they simulated fragmentation. These simulations were
criticised by Templeton (2004), however they still provide valuable insights
into the performance of NCPA and its usage. Moreover, Templeton (2004)
is often considered proof that NCPA is not prone to false positives, and is
actually conservative, however it is not the entire story. Much of NCPA is
in fact untested. What has been tested is the performance of NCPA under
fragmentation, and range expansion, using real data sets with strong a priori
expectations on a single loci, using steps 2. and 3. above. NCPA has also
been tested using simulations under the scenario of panmixia without using
steps 2. and 3. on single loci. These simulations shows that NCPA is prone
to false positives on a single locus when there is no signal, but remember that
this is not the full story. Multi-locus NCPA may still provide the answer to
the false positives from multiple testing, but it still needs to be tested.
There is still the question of what effect the criteria of Crandall and
Templeton (1993) have on the performance of NCPA. The features used in
the criteria are also used later in the calculation of Dcand Dn. How exactly
(what is the algorithm) does one say that inferences are concordant? When
enumerating all trees in a haplotype network, can a missing intermediate
be a tip? If a rooted and unrooted haplotype network both give different
inferences in the same clade, is the inference from the unrooted haplotype
network false?
1.2 Program citation
This is the citation for this software.
M. Panchal. 2007. The automation of Nested Clade Phylogeographic Anal-
5
ysis. Bioinformatics,23:509-510.
Please include the citations for TCS and GeoDis as well, as this software
relies heavily upon it.
M. Clement, D. Posada, and K. A. Crandall. 2000. TCS: A computer pro-
gram to estimate gene genealogies. Molecular Ecology,9(10):1657-1659.
D. Posada, K. A. Crandall, A. R. Templeton. 2000. GeoDis: A program
for the cladistic nested analysis of the geographical distribution of genetic
haplotypes. Molecular Ecology,9(4):487-488.
1.3 The Input Files
This section gives a description of the input files required to generate the
output files. Each file is a plain text file (do not save the files as RTF or
DOC files; they are not plain text).
1.3.1 The Nexus or Phylip DNA sequence file
This software uses a modified version of the Nexus file. Labels for each
DNA sequence must start with a letter and only contain letters or digits
(no whitespace). Each label should also have a period followed by a number
indicating which location (see 1.3.2) the sequence came from. Here is an
example of a Nexus file.
#NEXUS
begin data;
dimensions ntax=11 nchar=38;
format datatype=dna missing=? gap=-;
matrix
mxh1.1 CGTAAAGTTATACCCGAAAGGGAGAGAGTGAGAGTGTG
mxh2.1 CGTAAAGTTTTACCCGAAAGGGAGAGAGTGAGAGTGTG
mxs1.1 CGTAAAGTTATACCCGAAAGGGAGAGAGTGAGAGTGTG
mxs2.1 CGTAAAGTTATAGGCGAAAGGGAGAGAGTGAGAGTGTG
mxs3.2 CGTAAAGTTATAGGGGAAAGGGAGAGAGTGAGAGTGTG
mxs4.2 CGTAAAGTAATAGCCGAAAGGGAGAGAGTGAGAGTGTG
mxs5.2 CGTAAAGTAATAGCCGAAAGGGAGAGAGTGAGAGTGTG
txl1.3 CGTAATGTTATACCCGAAAGGCAGAGAGTGAGAGTGTG
txl2.3 CGTAATGTTATACCCGAAAGGCTCAGAGTGAGAGTGTG
txl3.4 CGTAATGTTATACCCGAAAGGCTCAGAGTGAGAGTGTG
txl4.4 CGTAAAGTTATAGCCGAAAGGCTGAGAGTGAGAGTGTG
6
;
end;
Note: Although not in the specification of the NEXUS file format, each label
and its corresponding DNA sequence should be on a single line (NEXUS
sequential). Also the nexus file should not contain any other information
other than that which can be seen in the example.
1.3.2 Geographic information file
The first line of the file should contain an identifier for the data set. The
second line is a number nthat indicates the number of locations sampled
(this does not include the unsampled locations specified). The next 2nlines
contain the entries for each sampled location. Each geographic location spec-
ified is written on two lines. The first line of the entry contains the number
(id) of the geographic location, followed by the name of the location (letter
followed by letters or digits and no whitespace). The second line specifies
the sample size, the location in decimal degrees, and the radius (spread) of
the sample location (in Km). Further habitable unsampled locations (where
prior knowledge indicates the species is present, or there is no prior knowledge
about the area) should also be specified after the sampled locations (This is
IMPORTANT information that is used to detect sampling inadequacies).
This is an example of a geographic information file.
mydataset // Name of the data set
2 // Number of sampled geographic locations
1 pop1 // Population number and name
7 10.50 -0.23 3 // Sample size, latitude, longitude, radius
2 pop2
5 -5.60 4.50 5.0
3 pop3 // This is an unsampled habitable location
0 -4.50 -3.50 5.4
Note: The number for each geographic location should be unique, sequential
and start from 1. Furthermore if a geographic region is not included, the
assumption made is that the area was sampled and the species was absent
in that area, or that the area is uninhabitable for that species.
1.4 The Output Files
This section describes the files that are given as output to the analysis.
7
1.4.1 The Graph File
TCS automatically writes out a *.graph file when it constructs a haplotype
network from the nexus file. The graph file is written in the GML format, and
specifies the structure of the haplotype network. It also contains haplotype
frequency data and outgroup weights for each sampled haplotype.
1.4.2 The Geodis Input file
This is produced from the *.graph file and the geographic information file.
The geodis input file (*.gdin) can be written in two formats. The first format
specifies the geographic information as decimal degrees, and is automatically
written out. The second format writes the geographic information as a dis-
tance matrix where the distances are the great circle distances between each
geographic location. The clade information written for both formats are
exactly the same.
1.4.3 The GML file
A *.gml file is also written. This contains the structure of the nested clado-
gram. The file contains a graph in the GML format for each level of the
nested cladogram. This file was not intended to be used to construct the
nested cladogram design manually. It was intended to be used by the soft-
ware only. To see what the nested cladogram design looks like please use the
*.nest file.
1.4.4 The Nest file
An optional file (*.nest) that can be written so the user can manually recon-
struct the nested cladogram design on a diagram of the haplotype network
(which can be obtained through TCS). This contains the id of each clade in
the nested design. For the 0-step clades, the frequency data is included as
well as the label for the clade, and which clades it is connected to. For n-step
clades (n≥1), the information available is the label, the subclades within
the clade and the clades that are connected to it. The subclades are speci-
fied in the order of tip clades, interior clades, followed by the symmetrically
stranded clades.
1.4.5 The NANOVA file
An optional file (*.nanova) can also be written which will provide nested in-
formation for each individual in the analysis. The data is provided as partial
8
data (as Comma Separated Values) that can be combined with individuals’
phenotype data for analysis with a Nested Analysis of Variance. The file is
primarily produced for users that wish to perform genotype-phenotype stud-
ies rather than geographic association studies of a species. See 1.7 for more
information.
1.4.6 The GeoDis Output file
This file (*.gdout) is written by GeoDis and contains the Dcand Dnproba-
bilities for each clade.
1.4.7 The GeoDis Summary file
This file can be produced by the automated inference key. The input files
required for this and the inference file are the geographic information file,
the Geodis Input (decimal degrees format), the GeoDis Output file, and the
GML file. The GeoDis input file needs to be in the decimal degrees format
even if the distance matrix format was analysed by GeoDis. The file contains
a summary of the information contained in the GeoDis Output file and marks
statistically significant values with a star (and s or l if it is statistically small
or statistically large). It also contains geographic locations and their spread.
The geographic distribution is also given for each clade.
1.4.8 The Inference File
This file contains the inference made for each clade and the chain of inference
showing which questions were answered to reach that conclusion.
1.5 Using the software for NCPA
This is a basic guide to using the software for geographic association studies
of a species.
1.5.1 Using the Graphical Interface
To run the software either select NCPA.jar in your file explorer or type
java -jar NCPA.jar
from the command line.
To create the haplotype network using TCS v1.18, select
9
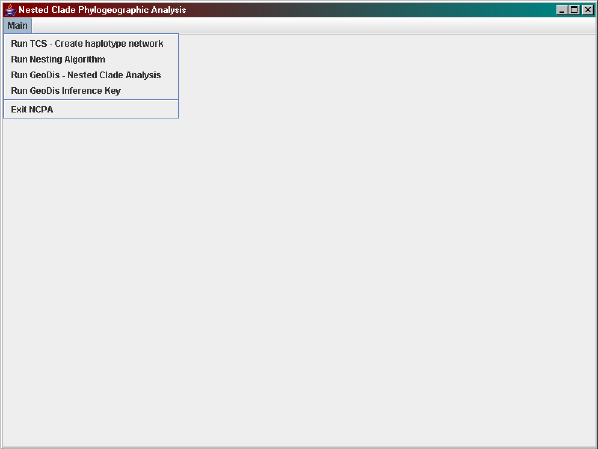
Figure 1.1: How to start TCS.
Main > Run TCS - Create haplotype network
from the menu.
Then select your nexus file from the menu using the following command,
File > Select NEXUS/PHYLIP Sequence file
and then click RUN, which will automatically save a *.graph file. Once the
haplotype network is created it is a good idea to save a picture of the haplo-
type network as well, although this can also be done at a later stage. Click
File > Save network as postscript
to save the image as a postscript file. Please refer to the TCS documentation
for further information about its functionality.
The next step is to nest the haplotype network and is done by either
selecting
Main > Run Nesting Algorithm
from the menu or by clicking the Run Nesting Algorithm button.
The following interface is displayed, and if TCS was used before, the
textfields should be automatically completed providing the files can be found.
The user must provide the locations of the TCS graph file and the file con-
10
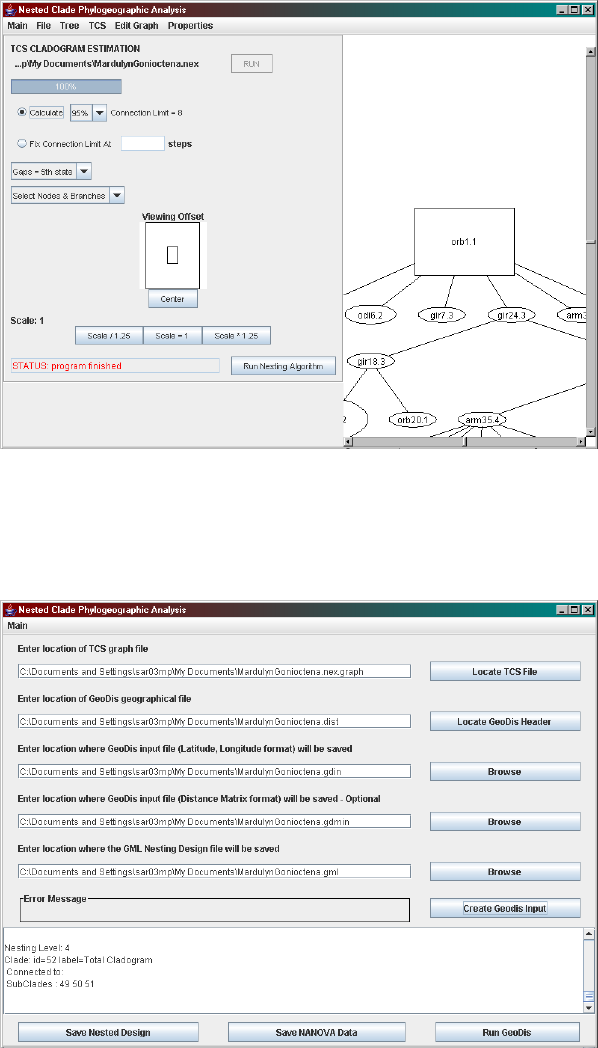
Figure 1.2: A screen shot of TCS.
Figure 1.3: A screen shot of the nesting software.
11
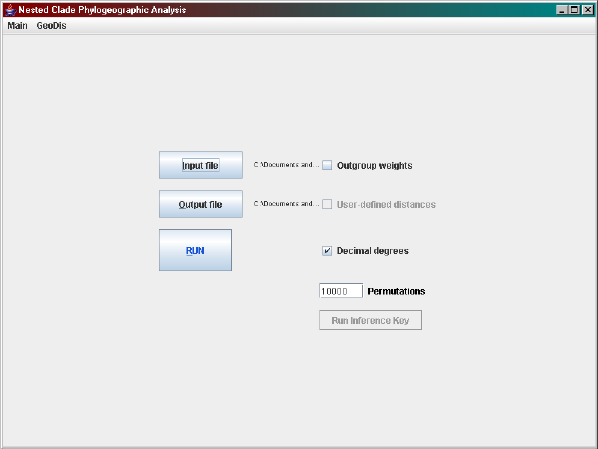
Figure 1.4: A screen shot of GeoDis.
taining the geographic information. The names of the GeoDis input files and
the name of the GML nested design file also need to be specified as they
will be required later on. Although the Distance Matrix format form of the
GeoDis file is optional it might be a good idea to compare the analyses from
both forms of input.
By clicking the Create GeoDis Input button, a summary of the nested
design will be shown in the text area that can be used to reconstruct it on the
haplotype network. This can also be saved by clicking Save Nested Design.
To move on to GeoDis select,
Main > Run GeoDis - Nested Clade Analysis
from the menu or click on the button Run GeoDis, which displays the GeoDis
interface.
If the nesting algorithm has just been run, the default input and output
files specified will be for the latitude-longitude format. Select the Decimal
degrees checkbox and then click RUN. If you want to analyse the distance ma-
trix format, then the input and output files need to be reselected. Following
that select the user-defined distances checkbox and then click RUN.
The automated inference key is then run by selecting
Main > Run GeoDis Inference Key
12
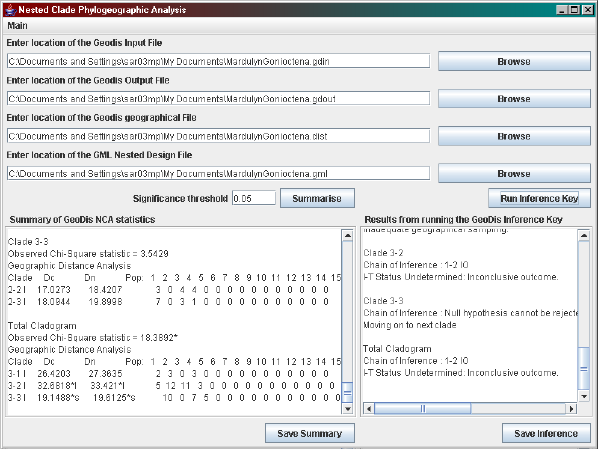
Figure 1.5: A screen shot of the automated inference key.
from the menu or clicking on the Run Inference Key button. Again the
textfields are automatically filled in if GeoDis was used before. The files
required are the GeoDis input file in latitude-longitude format (even if you
are analysing the distance matrix format output from GeoDis), the GeoDis
output (in either latitude-longitude or distance matrix format; the default is
latitude-longitude format), the geographic information and the GML nested
design. By clicking Summarise, a summary of the geodis statistics are cre-
ated. The summary contains the location of the sample sites and the clades
analysed. For each clade analysed the summary marks which distances are
significant at the 5% level with a *s or a *l (significantly small or large re-
spectively), and also the sample location distribution. Click Run inference
key to run the automated inference key. This will analyse each clade and
give the chain of inference and the final outcome, but nothing more. To find
out which locations were involved in which inferences, the automated infer-
ence key must be traced manually using the summary file and the chain of
inference.
Note: GeoDis and the automated inference key may take a long time to run
for large data sets. Please be patient (To be sure an error has not occured it
may be better to run the graphical interface from the command line).
13
1.5.2 Using the Command Line
By using the command line all the files described above are written to disk
with the same name stem as the DNA sequence file (i.e., the NEXUS or
PHYLIP file), including the analyses with the distance matrix form (these
are tagged with an m before the file ending e.g., filename.minfer or file-
name.mgdin). To execute the software, type on the command line,
java -jar NCPA.jar [dnaSeqFile] [geographicfile]
e.g.,
java -jar NCPA.jar mydataset.nex mydataset.dist
Currently there is no provision to run individual components of the au-
tomated software, and so if you want to conduct a portion of the analysis, it
must be done from the graphical interface (or you can modify the code).
Additional options are available that must be specified in the given order.
These are the number of permutations that GeoDis uses (default is 10,000),
whether to use the distance matrix format (0 - false, 1 - true), and the signif-
icance threshold to use (default is 0.05). Again this is run on the command
line as follows.
java -jar NCPA.jar [dnaSeqFile] [geographicfile]
[numberOfPermutations] 0 [significanceThreshold]
All five parameters must be included even if you only wish to change one of
them.
1.5.3 Drawing the nested design
When the nesting software has finished writing the GeoDis Input file, you
have the option of saving a summary of the nested design that can be used
to draw it out. The first step is to print out a picture of the haplotype tree
from TCS. If you have disjoint networks present TCS will only draw out one
of them, but by using the options from the menu the other networks can be
drawn out separately as well.
The summary of the nesting design contains all the levels of nesting start-
ing from the haplotype network. Each clade, including missing intermediates,
have a corresponding id with which to identify them. By using the labels of
the clades, haplotypes on the picture of the network can be assigned their ids.
Each clade also indicates which clade it was connected to allowing missing
intermediates to be assigned their ids. 1-step and higher level clades also
14
specify the ids of their subclades, and so the nesting design can be gradually
built up. The subclade ids are specified in the order of tip clades, interior
clades, and symmetrically stranded clades, which is helpful to determine how
the network was nested.
1.6 A Worked example
The files in this example are provided in the directory WorkedExample. The
data set provided is a real data set from (Mardulyn 2001). This example uses
the decimal degrees notation, however it is still possible to analyse distances
in GeoDis using the distance matrix format. I explain how this is done at
the appropriate point in the example.
1.6.1 Creating the Geographic information file
Here is a step by step guide to creating the Geographic information file.
1. Type the name of the dataset on the first line of the file.
2. On the second line of the file type the number of populations sampled.
3. On the following lines we then type the information for each sampled
area. The information for each sampled area takes 2 lines. On the
first is the population identification number (starting at 1) followed by
the name of the area (at least 3 characters long, consisting of letters
and numbers and no white space). The second line of the information
contains the sample size, latitude (in decimal degrees), longitude (in
decimal degrees), and the radius (in Km) of the sample size.
4. After the sampled areas are the unsampled habitable areas. These are
locations where the species is known to be but was unsampled (e.g.
inaccessable areas), or where the area is habitable but the presence
of the species is unknown. This information takes 2 lines for each
area. The first line is the population identification number (continuing
numbering from the last sampled population), followed by the location
name. The second line is the sample size (which is 0), the latitude
(decimal degrees), the longitude (decimal degrees), and the area radius
(Km).
5. Save the file as a plain text file.
See GonioctenaPallida.dist for the example file.
15
1.6.2 Creating the Nexus file
This is a step by step guide to creating the Nexus file in sequential NEXUS
format.
1. On the first line of the file type #NEXUS and then leave a line.
2. Type begin data;.
3. Following this type dimensions ntax= followed by the number of in-
dividuals in your data set. On the same line type nchar= followed by
the length (number of bases) of the DNA sequences and a semicolon.
(including insertions and missing characters represented by a -and ?
respectively).
4. On the next line type format datatype=dna missing=? gap=-;.
5. Then matrix on the next line.
6. On the next ntax lines enter a label for each of your dna sequences.
Ensure the labels start with a letter and contain only letters or digits
(no whitespace or special characters).
7. At the end of each label type a period followed by the identification
number of the population/deme it came from.
8. Follow each label with a space or tab followed by the DNA sequence.
9. Once all the DNA sequences are written, on a new line type ;.
10. On the new line type end;.
11. Then save the file as a plain text file with the file ending .nex.
See GonioctenaPallida.nex for the example file.
1.6.3 Create the haplotype network
Open ANeCA, and select Main > Run TCS (or the latest version of TCS).
Open the Nexus file and click Run. At this stage it is your choice how you wish
to resolve loops, if they are present. In this example we follow the steps of
Mardulyn (2001) and resolve loops using two criteria suggested by Crandall
and Templeton (1993): (i) rare haplotypes are more likely to be found at
the tip, and more common haplotypes at interior nodes, of a cladogram;
and (ii) a singleton is more likely to be connected to haplotypes from the
16
same population than to haplotypes from different populations (However,
note warning above about resolving loops). Once we have identified which
edges/branches and missing intermediate haplotypes need to be removed,
we need to identify the identification numbers of the missing intermediate
haplotypes and the haplotypes which the branches connect to in order to
modify the *.graph file. To do this, visually locate the nodes and edges which
need to be removed. By double-clicking on a node that needs to be removed
or is connected to edge that needs to be removed, a dialog box will popup. In
the title bar will be the words Node x, where xis the identification number of
the node. For each node that needs to be removed, record this number. For
each edge that needs to be removed, record both nodes the edge is connected
to. Open the graph file (in this case GonioctenaPallida.nex.graph) written
by TCS in a plain text editor. Using the search function, search for the string
id xwhere xis the identification number of the node to be removed. This
string (id x) will be nested in a node [ ...] block. Delete this block to
remove the node. For each edge to be removed, search for the string source
x. This will identify edge [ ...] blocks where one of the haplotypes is
connected to the edge to be removed. Check that the target ycorresponds
to the other haplotype the edge is connected to. If this is true, then remove
that edge [ ...] block. Repeat this, searching for target xand checking
the source y, to ensure the edges that need to be removed have been. Save
this file as plain text and then continue with the nesting. Note that although
it is possible to modify visually the haplotype network within TCS, this
cannot be saved as a graph file. However please take the opportunity to save
a picture of the haplotype network.
1.6.4 Creating the Nested Design
Click Run Nesting Algorithm from TCS or from the menu. Select the
graph file and the geographic information file to be used in nesting. Select
the location to save the GeoDis input file (GonioctenaPallida.gdin) and the
GML nested design (GonioctenaPallida.gml), and then click Create GeoDis
Input. In order to draw and check the nested design you will need to save
the summary of the nested design (GonoictenaPallida.nest), by clicking Save
Nested Design. If you wish to write out the GeoDis input with geographic
information as a distance matrix then specify the name of that file too (Go-
nioctenaPallida.gdmin).
17
1.6.5 Drawing the Nested Design
Print out a picture of the haplotype network, and open the nest file (Go-
nioctenaPallida.nest) in a plain text editor. At the start of the file will be
a count of the number of edges in the graph and a count of how many are
part of loops. Next is the clade information at Nesting Level: 0, the
haplotype network level. Each Clade: gives the identification number of
the haplotype/clade and the label it has on the diagram of the haplotype
network. Clades with label=" " indicate the clade is a missing interme-
diate haplotype. On the diagram of the haplotype network label each node
with the id of the clade, including missing intermediate haplotypes. Labeling
missing intermediate clades may be difficult, however each Clade: shows the
id’s of the clades it is connected to. Using this information it is possible to
deduce which id’s belong to which missing intermediate clades. When all the
haplotypes on the diagram have been labeled, move onto Nesting Level:
1. For each Clade: in this level use SubClades : identify which group of
haplotypes/clades have been nested, and draw the clade onto the diagram.
Mark the clade id next to each clade drawn onto the diagram. Continue
this for each nesting level, checking that clades are connected to the correct
clades.
1.6.6 Performing the Permutation Analysis
Click Run GeoDis, and select the GeoDis input file to be analysed (either Go-
nioctenaPallida.gdin or GonioctenaPallida.gdmin), and select either Decimal
degrees or User-defined distances depending on the input file chosen.
Both input files have been analysed here (GonioctenaPallida.gdout corre-
sponds to the input file GonioctenaPallida.gdin, and GonioctenaPallida.gdmout
corresponds to the input file GonioctenaPallida.gdmin).
1.6.7 Applying the Automated Inference Key.
Click Run Inference Key to open the automated inference key. Select the
GeoDis input file in decimal degree format (GonioctenaPallida.gdin), the
GeoDis output file from either format (GonioctenaPallida.gdout or Goniocte-
naPallida.gdmout) depending on the statistics you wish to analyse, the ge-
ographical information file (GonioctenaPallida.dist), and the GML file (Go-
nioctenaPallida.gml). Then click Summarise and Run Inference Key. This
will summarise the information required to apply the key, and present the
inferences made for each clade using the automated key. To check each in-
ference (GonioctenaPallida.infer for the analysis using decimal degrees, and
18
GonioctenaPallida.minfer for the analysis using the user-defined distances)
follow the inference key using the summary file (GonioctenaPallida.gdsum
for the analysis using decimal degrees format, GonioctenaPallida.gdmsum
for the analysis using the user-defined distances). For each clade the sum-
mary file contains the Dcand Dnvalues and their significance, and also
the number of individuals at each location. This allows the user to mark
out a clades geographic boundaries on a map and see which individuals and
locations are involved in an inference.
1.7 Using the software for genotype-phenotype
studies
Nested Clade Analysis was originally designed for genotype-phenotype asso-
ciation studies (although this software was initially designed only for NCPA).
A short guide is provided for using the software for genotype-phenotype as-
sociation studies.
Note: NCA for genotype-phenotype association studies has been superceded
by TreeScan (Posada et al. 2006), in particular to address the multiple testing
problem.
1.7.1 File simplifications
Although some users may also want to look at geographic associations, the
data specified for NCPA is more than is required for genotype-phenotype
association studies. If the user is not concerned about geographic location
then the distance information file need only contain the following few lines.
data_set_name
1
1 pop_1
1 0.0 0.0 0
The DNA sequence file normally would include the geographic location
in the label of each individual, however this should be replaced by the index
1 (corresponding to the dummy location in the geographic information file).
#NEXUS
begin data;
dimensions ntax=11 nchar=38;
format datatype=dna missing=? gap=-;
19
matrix
mxh1.1 CGTAAAGTTATACCCGAAAGGGAGAGAGTGAGAGTGTG
mxh2.1 CGTAAAGTTTTACCCGAAAGGGAGAGAGTGAGAGTGTG
mxs1.1 CGTAAAGTTATACCCGAAAGGGAGACAGTGAGTGTGTG
mxs2.1 CGTAAAGTTATAGGCGAAAGGGAGACAGTGAGAGTGTG
mxs3.1 CGTAAAGTTTTAGGGGAAAGGGAGACAGTGAGAGTGTG
mxs4.1 CGTAAAGTAATAGCCGAAAGGGAGAGAGTGAGAGTGTG
mxs5.1 CGTAAAGTAATAGCCGAAAGGGAGAGAGTGAGAGTGTG
1.7.2 Using the Graphical Interface
TCS and the automated nesting algorithm is used in the same way as for
NCPA. The only difference is that an extra file needs to be saved. Once the
nesting is complete, Click on Save NANOVA Data. This saves the data for
each individual, row by row specifying which clade the individual is in at
each step of the nested design. Note that TCS does not necessarily produce
a haplotype tree, and therefore the graph file will need to be modified before
nesting is performed. Often techniques from Coalescent Theory are used to
resolve loops (see Crandall and Templeton (1993) and Pfenninger and Posada
(2002)), or all possible trees are explored.
1.7.3 Using the Command line
To run NCA from the command line type:
java -jar NCPA.jar [dnaSeqFile] [geographicfile] -NANOVA
This causes the software to omit the GeoDis and automated inference key
stages of a normal NCPA run.
1.8 Limitations of Software
Careful consideration needs to be given to regions crossing the +180/-180 line
of longitude. For smaller areas of coverage all the geographic co-ordinates
can be offset such that the area does not lie over the line. This assumes the
species cannot migrate in both directions around the globe to reach locations
either side of the +180/-180 line of longitude.
The automated inference key uses spherical geometry to some extent and
great circle distances between locations, and so is unsuitable for use in situ-
ations in which the distance matrix format in GeoDis would traditionally be
used (e.g., for riparian species).
20
Lastly, if NCPA appears not to respond then it is likely that an ex-
ception has been thrown. Please run the application from the command
line using java -jar NCPA.jar. Then duplicate the problem and note the
full output from the command line (redirect the output to a file is you
area able to using java -jar NCPA.jar > output.txt). Then send this
to m.panchal@rdg.ac.uk explaining how the bug occurred, how to replicate
it, the input files, and the output from the command line.
Note: If the Exception thrown is an OutOfMemoryException you can try
the following solution. Type the following on the command line.
java -Xms256m -Xmx256m -jar NCPA.jar ([additional arguments])
Lastly please consult the developers documentation for further informa-
tion regarding the software, such as the design and implementation details.
1.9 Common Exceptions thrown
We have attempted to make this software as easy to use as possible however
you may encounter certain exceptions while running ANeCA. Most excep-
tions generally note the problem and not the cause. We try to explain the
cause here and how they can be corrected.
OutOfMemoryException This exception indicates that the Java Virtual
Machine (JVM) has used up the memory allocated to this process. To
correct this the amount of memory the JVM uses must be increased,
using the flags -Xms and -Xmx. Type the following on the command
line.
java -Xms256m -Xmx256m -jar NCPA.jar ([additional arguments])
This exception most often occurs in large datasets.
NumberFormatException:”unable to parse number” This indicates that
the geographic information file has been incorrectly written. Please
check this against the examples given.
1.10 Known Issues
Random exceptions thrown for Mac users Various exceptions have been
thrown running this software on Mac Operating Systems. Often the
exception cannot be reliably replicated and therefore the source of these
seemingly random exceptions is unknown.
21
Rerunning GeoDis After GeoDis has been run once, under certain cir-
cumstances it does not run again unless the software is restarted. The
circumstances are as yet unknown.
1.11 Frequently Asked Questions
How do I specify the radius of a sample location?
The radius of a sample location in effect describes the approximate movement
range of individuals in that location, and also how far the sampled individuals
were spread out. For example, plants may be sampled from a large area, or
insects may be caught using a single trap but will have traveled to it within a
general radius. This parameter is slightly subjective, however it means that
the sample location covers a certain area rather than a point location.
How do I specify the unsampled locations?
This is done in the same way as for the sampled locations. These locations
are important because you either don’t know if something is there or you
know something is there but haven’t been able to sample it. Here the radius
can be used to describe the approximate size of the unknown area. It is up
to the user to determine how many of these location need to be specified and
to what granularity.
Why should I include unsampled locations?
If no unsampled locations are included, the software assumes that the species
is absent in that area. This also means the safeguards introduced in the in-
ference key to guard against sampling inadequacies are bypassed.
My study area is too small/large to specify the radius in Km.
What do I do?
NCPA is insensitive to scale, and so geographic co-ordinates can be safely
scaled, translated, and rotated to make radius distances in Km more sensible.
I want to resolve loops. How can I do this?
This software currently offers no option of resolving loops. It is still possi-
ble to resolve them though and continue to use the software. First run the
nesting software on the reticulate network and save the *.nest file. Print a
picture of the haplotype network, and then use the *.nest file to assign the
ids to the haplotypes. Use your favoured method of breaking/resolving loops
to determine which edges should be removed. Open up a plain text editor
and open up the *.graph file automatically written by TCS (should be in the
22
same directory as your nexus file). In front of you now will be what looks
nonsensical information. Scroll down until you reach lines that looks like
this.
edge [
source 3
target 5
]
This is a representation of an edge. The numbers after source and target
are the ids of the haplotypes. Find the edges that you want to remove and
delete those four lines corresponding to each edge. For example if there is an
edge that links haplotype with id 4 to a haplotype with id 7 then the code
will look either like this,
edge [
source 4
target 7
]
or like this.
edge [
source 7
target 4
]
By removing these lines you have removed the edge connecting haplotype 4
to haplotype 7. Make sure you save this file as a plain text file. Open up the
nesting software and specify the file you just saved as the TCS graph file.
You can then continue using the software and nest the modified haplotype
network.
I have external information that makes some of the clades inte-
rior clades rather than tip clades. How do I change this in the
program?
Changing the tip/interior status of a clade is not currently possible within
the program. Once the GeoDis input file has been written, it can be changed
manually in a plain text editor. See the GeoDis documentation for further
information regarding the GeoDis input specification. Once editing of the
files is complete, it can be read into GeoDis.
23
I want to use another haplotype network estimation method in-
stead of Statistical Parsimony. How do I use it with this software?
The software currently nests haplotype networks that have been written in
the GML format that is written as standard by the TCS software. To use a
haplotype network that is produced by another haplotype network estima-
tion method, it must first be converted/written in the GML format. Unfor-
tunately there is no software that currently does this, and so the graph file
must be written by hand.
I have found a bug in the software. Who do I contact?
Please check if it is a bug, and there is no solution mentioned in the manuals
(either this or the TCS or GeoDis manuals). If it is a problem regarding the
correctness of TCS output or GeoDis output, please use the latest version of
the program available on David Posada’s website. If the problem persists,
please contact David Posada. If the problem is to do with the graphical
interface not functioning correctly (in TCS or GeoDis as well), or regarding
the nesting software or the automated inference key, please contact me.
A newer version of TCS/GeoDis is available. Why is it not in-
cluded?
I haven’t had the time to incorporate the new versions yet.
The software has stopped responding. What happened?
If it is a large data set please be patient with the software. If you have made
modifications to the input files then it is possible that there may be an error
reading the files. If you think a problem has occured, close the software, and
run it again from the command line using java -jar NCPA.jar from where
the NCPA.jar file is located.
1.12 Disclaimer
This software is provided free of charge. This program is distributed in the
hope that it will be useful, but without any warranty; without even the
implied warranty of merchantability or fitness for a particular purpose. The
utmost effort has been made to ensure the program is correct and free from
bugs, however problems may arise.
24
1.13 Acknowledgements
I would like to thank Mario Pulqu´erio, Oscar Moya, Jesus G´omez-Zurita,
Samantha O’Loughlin, and Gernot Segelbacher, for testing and providing
feedback regarding the software. I’m also grateful to the Biotechnology and
Biological Sciences Research Council for funding my PhD during which this
software was developed. I would also like to thank Keith Crandall, David
Posada, and an anonymous reviewer for comments and suggestions on the
software and its documentation.
25
Bibliography
K. A. Crandall. Multiple interspecies transmissions of human and simian
t-cell leukemia/lymphoma virus type i sequences. Molecular Biology and
Evolution, 13(1):115–131, 1996.
K. A. Crandall and A. R. Templeton. Empirical tests of some predictions
from coalescent theory with applications to intraspecific phylogeny recon-
struction. Genetics, 134(3):959–969, 1993.
L. L. Knowles and W. P. Maddison. Statistical phylogeography. Molecular
Ecology, 11(12):2623–2635, 2002.
P. Mardulyn. Phylogeography of the vosges mountains populations of go-
nioctena pallida (coleoptera : Chrysomelidae): a nested clade analysis of
mitochondrial dna haplotypes. Molecular Ecology, 10(7):1751–1763, 2001.
M. Pfenninger and D. Posada. Phylogeographic history of the land snail can-
didula unifasciata (helicellinae, stylommatophora): Fragmentation, corri-
dor migration, and secondary contact. Evolution, 56(9):1776–1788, 2002.
D. Posada, K. A. Crandall, and A. R. Templeton. Geodis: a program for
the cladistic nested analysis of the geographical distribution of genetic
haplotypes. Molecular Ecology, 9(4):487–488, 2000.
D. Posada, K. A. Crandall, and A. R. Templeton. Nested clade analysis
statistics. Molecular Ecology Notes, 0(0):Published before print., 2006.
A. R. Templeton. Statistical phylogeography: methods of evaluating and
minimizing inference errors. Molecular Ecology, 13(4):789–809, 2004.
A. R. Templeton and C. F. Sing. A cladistic analysis of phenotypic associ-
ations with haplotypes inferred from restriction endonuclease mapping .4.
nested analyses with cladogram uncertainty and recombination. Genetics,
134(2):659–669, 1993.
26
A. R. Templeton, E. Boerwinkle, and C. F. Sing. A cladistic analysis of
phenotypic associations with haplotypes inferred from restriction endonu-
clease mapping .1. basic theory and an analysis of alcohol dehydrogenase
activity in drosophila. Genetics, 117(2):343–351, 1987.
A. R. Templeton, E. Routman, and C. A. Phillips. Separating population
structure from population history : a cladistic analysis of the geographi-
cal distribution of mitochondrial dna haplotypes in the tiger salamander,
ambystoma tigrinum. Genetics, 140(2):767–782, 1995.
A. R. Templeton, T. Maxwell, D. Posada, J. H. Stengard, E. Boerwinkle,
and C. F. Sing. Tree scanning: A method for using haplotype trees in
phenotype/genotype association studies. Genetics, 169:441–453, 2005.
A.-B. Zhang, S. Tan, and T. Sota. Autoinfer 1.0: a computer program to
infer biogeographical events automatically. Molecular Ecology Notes, 6:
597–599, 2006.
27
