Phyloscanner Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 27
- I Inferring within- and between-host phylogenies in windows along the genome, using mapped reads
- II Analysing within- and between-host phylogenies
phyloscanner
available from github.com/BDI-pathogens/phyloscanner
Coding lead by Chris Wymant and Matthew Hall,
with contributions from Oliver Ratmann and Christophe Fraser
This manual last updated October 22, 2018
next-gen
sequencing
gives reads
mapping
+ ... + ...
phyloscanner
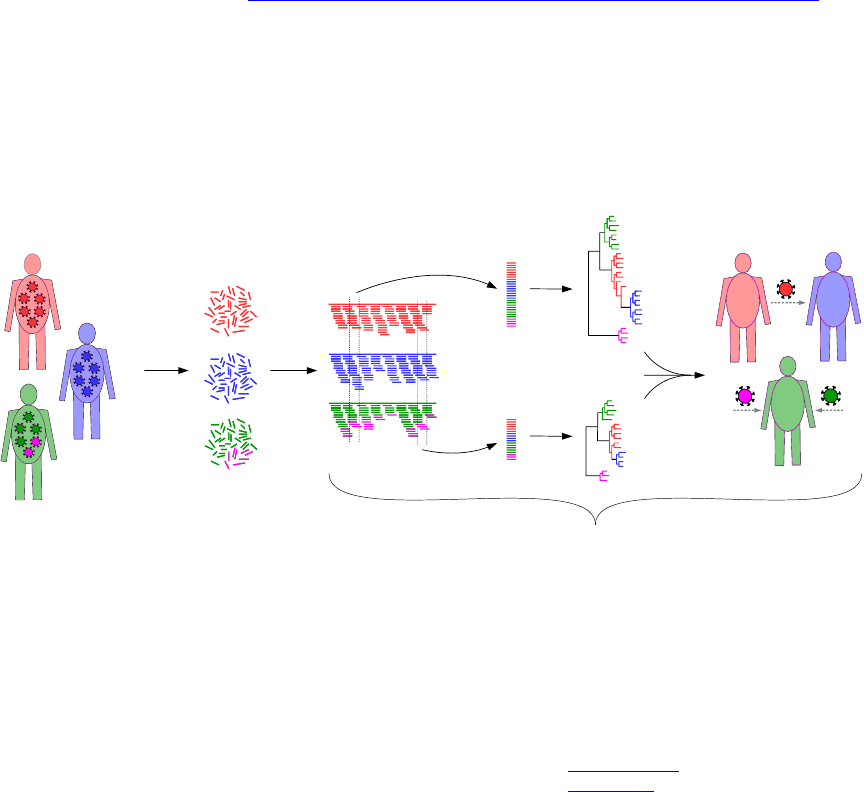
phyloscanner analyses pathogen genetic diversity and relationships between and within hosts at
once, in windows along the genome using mapped reads (fragments of DNA produced by next-generation
sequencing), or using phylogenies with multiple sequences per host previously generated by the user via
any method. An example showing its action on simulated data (which is included with the code) can be
seen at the GitHub repository linked to above.
If you have a query about phyloscanner, ask it publicly at the google group. If you have a problem
getting the code to run or think you’ve found a bug, please create a New issue on the GitHub repository.
The usual bug-reporting etiquette applies. For non-trivial bugs, to allow us to reproduce the problem it’s
helpful to have input that causes the problem. This needn’t be all your data. For example if one read in
a bam file is causing a problem, you could make a new bam file containing only that read, allowing us to
see the problem very quickly. Isolate the problem as much as you can first – help us to help you! If you
get an error when you run phyloscanner as part of a larger piece of code you’ve written yourself (e.g.
a job submission script to a cluster), please don’t make us debug that – the error might be in your code
rather than ours. Isolate the problem to the relevant phyloscanner command.
If you use phyloscanner, please cite it [1] and its dependencies: SAMtools [2], MAFFT [3], RAxML [4] and
ggtree [5]. Details of these are found in the file CitationDetails.bib in phyloscanner’s InfoAndInputs
subdirectory.
Throughout the manual we’ll use phylogeny and tree interchangeably. Forgive us.
1
Contents
I Inferring within- and between-host phylogenies in windows along the
genome, using mapped reads 5
1 The basic command 5
1.1 ThePythonversionyouuse................................... 6
2 What do the window coordinates mean exactly? 6
3 What windows should I choose? 6
3.1 Start&end............................................ 6
3.2 Windowwidth .......................................... 7
3.3 Windowoverlap.......................................... 8
4 What output files are produced? 8
5 Optional arguments 10
5.1 Windowoptions ......................................... 10
5.2 Recommendedoptions...................................... 11
5.3 Readqualityoptions....................................... 12
5.4 Otherassortedoptions...................................... 12
5.5 Options for bioinformatic interrogation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
5.6 Partialprocessingoptions .................................... 14
5.7 Deprecatedoptions........................................ 14
II Analysing within- and between-host phylogenies 16
6 The basic command 16
7 Name format for input files 17
8 Output 17
9 Preparing the trees 17
10 Tip label and file suffix regular expressions 18
11 Branch length normalisation 18
11.1 Parallelising tools/CalculateTreeSizeInGenomeWindows.py ................ 19
12 Blacklisting 20
12.1Duplicateblacklisting ...................................... 20
12.2 Parsimony-based blacklisting for contaminants . . . . . . . . . . . . . . . . . . . . . . . . 20
12.3Dualinfectionblacklisting.................................... 21
12.4Userblacklisting ......................................... 21
12.5Downsampling .......................................... 21
12.6Pruningtheblacklist....................................... 21
13 Parsimony reconstruction 22
14 Summary statistics 23
15 Classification of pairwise relationships 24
16 Pairwise relationship summary 25
2

A tool in two parts
Conceptually, phyloscanner can be neatly split into two steps. Firstly, inferring phylogenies that contain
both within- and between-host diversity, in windows along the genome, using mapped reads as input.
Secondly, analysing those phylogenies or other similar ones (i.e. containing within- and between-host
diversity, but created however the user likes) in order to infer interesting things like transmission and
multiply infected individuals, and give summaries of diversity.
This conceptual split is reflected in the code: phyloscanner make trees.py runs the first step and
phyloscanner analyse trees.R runs the second step. Why didn’t we merge the two steps into one?
•For some uses, step one may not be needed. For example if you don’t have mapped reads, but have
sequence data in another format. We presented an example of this in the phyloscanner paper: a
dataset consisting of multiple whole genomes of Streptococcus pneumoniae for each person carrying
the bacterium, obtained by separately sequencing multiple colonies.
•For some uses, step two may not be needed. Having extracted the within- and between-host diversity
information in each window (first as a multiple sequence alignment and then as a phylogeny), you
might want to do your own totally different analysis of it.
•Each step is controlled by parameters and options to tailor its usage to different kinds of data, and
we don’t (yet) have a way to automatically optimise these for each data set, so you will need to
have a little play to explore what’s appropriate in your case. If you are running both steps, it’s
best to think first about getting step one right first, and then getting step two right. Step one is
primarily about bioinformatics, step two about phylogenetic analysis.
Note that to run either phyloscanner make trees.py or phyloscanner analyse trees.R, like run-
ning any executable file, you must either specify the full path to the executable (e.g.
/path/to/my/phyloscanner/code/phyloscanner make trees.py) or add the directory that contains
the executable to the $PATH environment variable in your terminal (google this if needed). In the manual
we’ll refer to scripts named like tools/SomeCode; such scripts live in the tools subdirectory inside the
main phyloscanner code directory.
Fun fact: step one was written by Chris, step two mostly by Matthew. We sat next to each other
while coding, and we hope that’s reflected in how easy it is to make the two steps work together. If it
isn’t, let us know.
4

Part I
Inferring within- and between-host
phylogenies in windows along the genome,
using mapped reads
1 The basic command
As input to phyloscanner make trees.py, you need to have generated files of mapped reads in bam
format – bam files henceforth. The bam format does not include the sequence to which the reads were
mapped – the reference – which we also need. With the initial $traditionally indicating that what follows
is a command to be run on the command line / in a terminal (i.e. don’t type that initial $), the basic
phyloscanner make trees.py command looks like
$ phyloscanner_make_trees.py ListOfMyInputFiles.csv --windows 1,300,301,600,...
where
•ListOfMyInputFiles.csv is a plain-text, comma-separated-variable (csv) format file in which the
first column contains the bam files, the second column contains the corresponding reference files.
An optional third column, if present, contains aliases – things to rename each bam file to in
phyloscanner make trees.py output; if not present the base name of the bam, i.e. the file name
not including the directory path, will be used. e.g. This input file list might look like
PatientA.bam,PatientA ref.fasta,A
PatientB.bam,PatientB ref.fasta,B
Bam file base names, and aliases if present, must be unique and free of whitespace. Quotation marks
are interpreted as wrapping/protecting fields, allowing them to contain commas that are not field
separators. You can generate this input file list however you like, including manually. The following
block of code illustrates how you might generate it automatically from the command line. It also
checks that your files exist, which phyloscanner make trees.py does anyway, but it’s always best
to catch your errors soonest.
$ for ID in PatientA PatientB PatientC; do
$ bam=MyBamsDir/"$ID".bam
$ ref=MyRefsDir/"$ID".fasta
$ if [[ ! -f "$bam" ]]; then
$ echo "$bam does not exist" >&2
$ break
$ elif [[ ! -f "$ref" ]]; then
$ echo "$ref does not exist" >&2
$ break
$ fi
$ echo "$bam","$ref","$ID"
$ done > MyPhyloscannerInputFile.csv
If those IDs were stored in a plain-text file MyIDs.txt separated by whitespace, you could replace
the first line of that loop with
for ID in $(cat MyIDs.txt); do
•The --windows (or -W) option is used to specify an even number of comma-separated positive
integers: these are the coordinates of the windows to analyse, interpreted pairwise, i.e. the first two
are the left and right edges of the first window, the third and fourth are the left and right edges of
the second window, . . . i.e. in the above example we have windows 1-300, 301-600, . . .
You shouldn’t run multiple phyloscanner make trees.py commands simultaneously in the same direc-
tory, as it writes to and reads from files with fixed names, so the commands could clash. If you’re running
5

multiple jobs in parallel on a computing cluster that starts jobs in the same place, ensure that each job
runs in a different directory (e.g. using the mkdir and cd commands to make and change into a directory
specific to that job).
1.1 The Python version you use
Note that phyloscanner make trees.py can either be run like this:
phyloscanner make trees.py [input and options]
or like this:
python phyloscanner make trees.py [input and options]
In the former case, the version of Python used to execute phyloscanner make trees.py is whatever
version gets called when you simply run the command python in the terminal. In the latter case, you
can explicitly replace python by a specific version of Python on your machine, for example python2.7 or
/usr/bin/python. The version of Python that’s used must (a) be Python 2, not Python 3, and (b) contain
the Python dependencies described in phyloscanner/InfoAndInputs/InstallationNotesForMakingTrees.sh.
The version of Python used can be specified the same way for all of the Python tools in the tools sub-
directory.
2 What do the window coordinates mean exactly?
By default the references used for mapping, together with any extra references if included using the
--alignment-of-other-refs option, are all aligned together and window coordinates are interpreted
with respect to the alignment (i.e. position nrefers to the nth column of that alignment, which could
be a gap for some of the sequences). This alignment can be found in the file RefsAln.fasta after
running phyloscanner make trees.py, should you want to inspect it and possibly run again. You
can manually specify window coordinates with respect to this alignment, using the --windows option,
or have windows automatically chosen using --auto-window-params, which attempts to minimise the
affect of insertions and deletions in the references on your window width and overlap preferences. Alter-
natively, if you are using the --alignment-of-other-refs option to include extra references, you can
use --pairwise-align-to to name one of these references to be a kind of reference reference: instead
of aligning of all the bam file references to each other, they will be sequentially and separately pairwise-
aligned to your named reference, and window coordinates are interpreted with respect to that named
reference (i.e. position nrefers to the position of the nth base of the named reference, not counting
any gaps inside the reference). Using the --pairwise-align-to option is expected to more stable than
--windows or --auto-window-params if your bam file references are many and diverse, since pairwise
alignment is easier than multiple sequence alignment. It also has the advantage that when running
phyloscanner make trees.py more than once with different bam files, the coordinates mean the same
thing each time.
3 What windows should I choose?
I’m glad you asked. It’s important.
3.1 Start & end
You might as well fully cover the genomic region you’re interested in. That requires choosing where to
start and where to end. If you’re interested in the whole genome, the start is 1 and the end is the genome
length, or more precisely the length of the alignment of all references together (or the length of your
named reference if you used the --pairwise-align-to option). You may know that your reads don’t
start right at the beginning of the genome. If this is the case, a good place to start your first window
would be the genome position at which you start having reads.
If your reads were generated by amplifying the sample using primers, the primers should ideally have
been trimmed from the reads as part of whatever bioinformatic pipeline produced your input bam files.
(This can be done for example using fastaq, which is called as part of the shiver pipeline.) Then a
sensible choice for the start of the first window would be the first position after the first primer, and a
sensible choice for the end of the last window would be the last position before the last primer.
6

You might as well fully cover the genomic region you’re interested in. Let’s revisit that. In what
part of the genome would you like to do phylogenetics? Perhaps all of it. Then again, remember that
phylogenetics assumes neutral evolution at every included site. Though phyloscanner make trees.py
allows individual specified sites to be excised after alignment of all the reads from all the bam files,
there may be regions of the genome where selected sites are so dense that it’s simpler to skip the region
altogether. There may also be regions of the genome where there are so many insertions and deletions
(indels) that you’re sceptical that the reads will align correctly in the first place. You can choose to
specify no windows covering such regions.
3.2 Window width
As well as choosing where your first window should start and where your last one should end, you need
to choose how wide each window is. On the one hand, increasing the width of a window increases
its phylogenetic resolution. To see this effect using just some existing reference sequences (forgetting
momentarily about any read data you have), the script tools/CalculateTreeSizeInGenomeWindows.py
could be useful: it calculates trees from references in sliding windows along the genome. (It then calculates
patristic distances, for a purpose discussed later in section 11.) You may be able to see from visual
inspection, or from some automated analysis of your own, that one’s ability to construct well-resolved
phylogenies increases with window width. On the other hand, a read must fully span a window to be
included in it, so the wider you make a window the fewer reads it will contain. To quantify this effect,
you can run
$ tools/EstimateReadCountPerWindow.py ListOfMyInputFiles.csv
(where ListOfMyInputFiles.csv is the same file as we discussed at the top of section 1). This command
uses the information in each bam about the number of reads and how long1they are; it then estimates
the number of reads expected to span a window of width Wby assuming reads are distributed ran-
domly over the whole genome (ignoring the actual read location information in the bam files). If reads
are found to be paired, the calculation is also done in the context of phyloscanner make trees.py’s
--merge-paired-reads option, which merges each pair of reads into a single longer read if they over-
lap with each other (this will give exactly the same result as considering reads separately if reads in
a pair never overlap). The calculation is done as a function of W, showing how the number of reads
decreases monotonically2. A good choice of window width, roughly, would be as large as you can get
before the number of reads per window gets too low to make useful within-host phylogenies. Skip ahead
to section 3.3 if that’s good enough for you and you don’t want to think about it any more.
The --explore-window-widths option of phyloscanner make trees.py was written with the fol-
lowing balancing act in mind. If a window is too wide, too few reads overlap it, as discussed. However
if it is too small, there aren’t enough positions at which a variant can be observed and so few unique
reads will be observed in the window. Somewhere between these two extremes therefore maximises the
number of unique reads. The --explore-window-widths option reports, for each in a list of window
widths to try, a two-dimensional matrix of count values – the number of unique reads found in each
bam and in each successive window along the genome. One way of visually summarising this matrix
of counts as a function of window widths is to give the file containing this data as the argument to
tools/PlotExplorationOfWindowWidths.py, which calculates and plots various percentiles of the ma-
trix of counts. e.g. for a given window width, the 50th percentile of the counts means that half of all the
windows from all the bams had more unique sequences than this, and half had fewer. Anecdotally, for
our HIV data, I did not find that maximising the number of unique sequences per window gave the best
results: manually examining phylogenies revealed that a larger window width produced more revealing
1In this particular instance we define read length as the length of the mapping reference covered by the read, from
its start to its end. This can differ from the actual read length due to insertions or deletions in the read relative to the
reference. Defining the read length this way is appropriate since we’re interested in reads spanning windows, and windows
get defined relative to the reference. This length will also differ from the nominal read length of your sequencing run (which
is typically one fixed value) if the reads have been trimmed as part of bioinformatic processing, e.g. removing primers,
adapters, low-quality bases, or bits of the read that could not be mapped.
2The number of positions at which we could place a window of width Wequals the length of the reference −W+ 1.
The number of positions at which we could place a window of width Wsuch that it is wholly inside a read is the length of
the read −W+ 1. The probability of a randomly placed read overlapping a window of width Wis therefore (read length
−W+ 1) / (reference length −W+ 1). The number of reads and their different lengths are fixed in the data; increasing W
will decrease this probability (except for reads that span the entire reference).
7

trees. But this option can still be helpful for quantifying the decline in how many unique sequences you
get as window width is increased out towards the largest possible values.
The regular --explore-window-widths option calculates the number of unique reads after all read
processing has been done, including aligning the reads from all bams using mafft, and other options
you may have specified that affect how many unique reads survive, for example --excision-coords,
--merging-threshold,--min-read-count,--quality-trim-ends,--min-internal-quality, and
--discard-improper-pairs options. The first two of these can result in two or more unique reads
being merged into one; the rest can simply discard some reads. You could choose values for the
associated parameters immediately and then use --explore-window-widths, or else come back to
--explore-window-widths later on once you’ve got the hang of phyloscanner make trees.py and
investigated the effect of those other options in your data.)
There is also the --explore-window-widths-speedy option which is almost identical: the difference
is that this counts the number of unique reads in the window straight away after they are extracted from
the bam, before any processing of them has been done (including aligning them all with mafft). If you
don’t care about read processing steps that change the number of unique reads, use this because it’s
faster.
Power users might want to optimise their own measure of phylogenetic information as a function of
window width; one of the first metrics to pop into your head might be the mean bootstrap of all nodes in
the tree. That’s not advised because within a sample there may be many very similar sequences, and the
set of nodes connecting these may have poor bootstrap support, but this is not something that ought to
be penalised. Also in theory you might be able to increase the window width until only a single read is
found spanning the window in each patient; your bootstraps might then be great, because between-host
diversity is greater than that within-host, but you’ve thrown out all the within-host information.
3.3 Window overlap
How much should neighbouring windows overlap?
•A good, simple answer to this is zero, i.e. each window starts right after the previous one ends. e.g.
1-99, 100-199, 200-299, . . .
•Negative overlap means unused space in between windows, e.g. 1-99, 200-299, 400-499, . . . The only
reason for this is if you want to exclude certain parts of the genome. (Though it’s hard to imagine
that the regions you want to exclude are perfectly regularly spaced.)
•Positive overlap means the next window begins before the current one ends, e.g. 1-99, 50-149,
100-199, . . . There’s a redundancy in doing this, because you’re analysing the same information
multiple times. However there’s a little bit of stochasticity involved when performing algorithmic
multiple sequence alignment then phylogenetic inference, so even if two windows overlap heavily
some minor differences in the results could arise by chance. Positive overlap may therefore smooth
out some of this stochasticity, at the expense of additional compute time. (You’ll also need to take
into account the non-independency of overlapping windows if you interpret results from multiple
windows in a probabilistic framework.)
4 What output files are produced?
By default, those files that are produced for each window are grouped into subdirectories by file type.
•RefsAln.fasta. This contains the mapping reference from each bam file, together with any
extra references included with --alignment-of-other-refs. This alignment is not created if
--pairwise-align-to is used.
•AlignedReads/AlignedReadsInWindow X to Y.fasta (where Xand Yare window coordinates).
Contains the reads from all bam files in the window X-Y, after processing and alignment, but before
excision of any positions specified with --excision-coords. Reads are automatically assigned a
name of the form A read M count N, where Aidentifies the bam from which the read came, Nis
the number of times that read was found (either identical copies of the same sequence, or merged
similar sequences if --merging-threshold is used), and Mis the rank of the read when they are
ordered by count (i.e. M= 1 is the most common read, M= 2 the second most common etc.)
8

•AlignedReads/AlignedReads PositionsExcised InWindow X to Y.fasta. Only produced if you
specified some positions to excise with --excision-coords, and at least one of those positions was
inside3the window X-Y. If this file exists, the tree in window X-Yis based on this alignment
(not AlignedReadsInWindow X to Y.fasta). Note that excising positions may result in reads that
were previously distinct becoming identical – if they differed only at the excised positions. This is
checked after positions are excised, and reads from the same bam that have become identical are
merged with an updated count. Similarly, if the merging of similar reads (in addition to identical
reads) has been specified, the merging is rerun after excision of positions. If the excision of positions
results in reads becoming merged, clearly there will not be a one-to-one correspondence between
reads before and after excision: after merging there may be fewer unique reads, though with the
same total count.
•Consensuses/Consensuses InWindow X to Y.fasta. In this alignment, the set of unique reads re-
tained from each bam file is collapsed into a single sequence: their consensus (taking the most com-
mon base/gap at each position, weighting each unique read by its count). Note that in general this
consensus could differ from the corresponding part of a consensus called using all reads mapped along
the full length of the reference, because the only reads retained by phyloscanner make trees.py
in a window are those fully spanning it. This process can result in some columns containing only
gaps; these columns are kept, so that the coordinates of this file match those of
AlignedReadsInWindow X to Y.fasta.
•Consensuses/Consensuses PositionsExcised InWindow X to Y.fasta. The consensus as previ-
ously, but with any specified positions to excise excised. Only produced if there are such positions.
•DiscardedReads/DiscardedReads A.bam, where A identifies one of the input bams. Only produced
if --inspect-disagreeing-overlaps is used; see that option’s description in 5.5. These files will
be accompanied by DiscardedReads A ref.fasta (the reference for the bam file).
•DuplicationData/DuplicateReadCountsRaw InWindow X to Y.csv. Each row in this csv-format
file corresponds to one instance of exactly the same unique read being found in two different bams,
before any processing of the reads (merging, excising positions etc.). The first two columns show
the two bams involved, and the third and fourth columns show the count of the read in the two
bams. If a read is exactly duplicated in Ndifferent bams, then there will be N(N−1)/2 associated
lines in this file – one for each different pair of bams. See also the information in the next point.
These files will not be produced if --dont-check-duplicates is specified.
•DuplicationData/DuplicateReadCountsProcessed InWindow X to Y.csv. Each row in this csv-
format file corresponds to one instance of exactly the same unique read being found in at least
two different bams, after all processing of the reads (merging, excising positions etc.). If the same
read is found in Ndifferent bams, there will be one row for this, containing Nfields separated by
commas (each row in general therefore has a different number of columns, though 2 is the most
common). Each field is the name of the read, assigned based on the bam in which it was found.
These files will not be produced if --dont-check-duplicates is specified.
Why is information about the duplication of reads checked and recorded twice, in these two different
files? Duplication information after processing is easiest to work with, because it is the processed
reads that are used to infer phylogenies. However instances of exact duplication before any process-
ing of the reads is a better indicator of cross-sample contamination. Firstly because the options for
similarity-based merging of reads within each bam, and for a minimum read count for inclusion,
result in fewer unique reads to compare between bams. Secondly because excising positions makes
reads shorter and increases the possibility for genuine cross-sample duplication (i.e. not contami-
nation). Anecdotally, we found that being trigger-happy with deleting all codons associated with
HLA escape in a conserved part of the HIV genome resulted in considerable duplication in the reads
after processing that was not present before. Note that during both similarity-based merging of
reads and excision of positions (which is followed by merging newly identical reads), we lose track of
the original identity of reads, i.e. we don’t know what the correspondence is between reads before
processing and after processing.
3Note that if --pairwise-align-to is used, the window coordinates Xand Ymean the same thing as the excision coor-
dinates. If --pairwise-align-to is not used, the window coordinates Xand Ymean positions Xand Yin RefsAln.fasta
as discussed in section 2; therefore a position to excise, Z, being inside the window X-Yis not the same as X≤Z≤Y.
9

•DuplicationData/DuplicateReads contaminants InWindow X to Y.fasta. Only produced if
--contaminant-count-ratio is used; see that option’s description in section 5.7.
•ReadNames/ReadNames1 InWindow X to Y InBam A.txt. Only produced if --read-names-1 is used;
see that option’s description in section 5.5.
•ReadNames/ReadNames2 InWindow X to Y InBam A.csv. Only produced if --read-names-2 is used;
see that option’s description in section 5.5.
•RecombinationData/RecombinantReads InWindow X to Y.fasta. Only produced if
--check-recombination is used; see that option’s description in section 5.2.
Some temporary files are also written to the working directory; by default these are deleted. (They are
kept with --keep-temp-files.)
5 Optional arguments
Information about all optional arguments and what they do can also be seen by running
phyloscanner make trees.py with the --help option. That will give information guaranteed to be
synchronised to your current version of the code, as well as showing all option shorthands (e.g. --help =
-h); however we go into slightly more detail here. We particularly encourage you to familiarise yourself
with the Window options and Recommended options below, which are particularly important for running
phyloscanner make trees.py well.
5.1 Window options
You must choose exactly one of these: --windows,--auto-window-params,--explore-window-widths.
•--windows: used to specify a comma-separated series of paired coordinates defining the boundaries
of the windows. e.g. specifying 1,300,301,600,601,900 would define windows 1-300, 301-600,
601-900.
•--auto-window-params: used to specify 2, 3 or 4 comma-separated integers controlling the auto-
matic creation of regular windows.
–The first integer is the width you want windows to be. If the --pairwise-align-to option
is not being used, we create an alignment containing all references, as mentioned already. In
this case, we weight each column in this alignment by its non-gap fraction, so that windows in
which many references have gaps become correspondingly wider. If the --pairwise-align-to
option is being used, width is simply interpreted with respect to the named reference (i.e. a
width of Wmeans each window contains Wbases from that reference).
–The second is the overlap between the end of one window and the start of the next. This can
be negative, implying unused space in between windows; the recommended value of 0 means
each window starts right after the previous one ends.
–The third integer, if specified, is the start position for the first window. If not specified, this
is 1.
–The fourth integer, if specified, is the end position for the last window. If not specified,
windows will continue up to the end of the reference or references.
•--explore-window-widths: use this option to explore how the number of unique reads found in
each bam file in each window, all along the genome, depends on the window width. After this option
specify a comma-separated list of integers. The first integer is the starting position for stepping
along the genome, in case you’re not interested in the very beginning. Subsequent integers are
window widths to try. For example, if you specified 1000,100,150,200 we would count the number
of unique reads in windows 1000-1099, 1100-1199, 1200-1299, ... and in 1000-1149, 1150-1299, 1300-
1449 ... and in 1000-1199, 1200-1399, 1400-1599, ... where the dots denote continuation to the end
of the genome. Output is written to the file specified with the --explore-window-width-file
option.
10
•--explore-window-width-file: used to specify an output file for window width data, when the
--explore-window-widths option is used. Output is in csv format.
5.2 Recommended options
•--alignment-of-other-refs: used to specify an alignment of reference sequences for inclusion
with the reads, for comparison. These references need not be those used to produce the bam
files. This option is required if phyloscanner is to analyse the trees it produces (i.e if you are
going to run phyloscanner analyse trees.R after phyloscanner make trees.py). The full set
of processed, unaligned reads extracted from each window are aligned to the corresponding section
of the reference alignment using mafft --add.
•--pairwise-align-to: by default, phyloscanner make trees.py figures out where correspond-
ing windows are in different bam files by creating a multiple sequence alignment containing all
of the mapping references used to create the bam files (plus any extra references included with
--alignment-of-other-refs), and window coordinates are interpreted with respect to this align-
ment. However using this option, the mapping references used to create the bam files are each sep-
arately pairwise aligned to one of the extra references included with --alignment-of-other-refs,
and window coordinates are interpreted with respect to this reference. The reference to use
should be specified after this option. Using this option is necessary if you want to run the
tools/CalculateTreeSizeInGenomeWindows.py script and feed its output into phyloscanner analyse trees.R
via its --normRefFileName option (see )
•--x-raxml: use this option to tell phyloscanner make trees.py how to run RAxML, including both
the executable (with the path to it if needed), and the options. If you do not specify anything, we
will try to find the fastest RAxML exectuable available (first raxmlHPC-AVX, then raxmlHPC-SSE3,
then raxmlHPC) and use the options -m GTRCAT -p 1 --no-seq-check. You will need to specify
something if the path to your RAxML executable is not contained in your PATH environment variable
(i.e. if you need to specify the path to the executable in order to run it), or if you want to use different
RAxML options. -m tells RAxML which evolutionary model to use, and -p specifies a random number
seed for the parsimony inferences; both are compulsory. You may include any other RAxML options in
this command (including multi-threading, provided you use one of the multi-threaded executables).
The set of things you specify with --x-raxml need to be surrounded with one pair of quotation
marks (so that they’re kept together as one option for phyloscanner make trees.py and only split
up for RAxML). If you include a path to your RAxML executable, it may not include whitespace, since
whitespace is interpreted as separating RAxML options. Do not include options relating to bootstraps:
use phyloscanner make trees.py’s --num-bootstraps and --bootstrap-seed options instead.
Do not include options relating to the naming of files.
•--merge-paired-reads: this is only relevant for paired-read data for which the mates in a pair
(sometimes) overlap with each other, but is very useful for such data. With this option, over-
lapping mates in a pair are merged into a single (longer) read. This allows wider windows to be
used, enhancing the phylogenetic resolution in a single window. Pairs will only be merged if they
agree on the bases in the overlap and where they have been mapped to; if the reads disagree, that
pair is discarded. Discarded pairs can be inspected by using --inspect-disagreeing-overlaps.
If reads are paired but never overlap, trying to merge them will do nothing except increase
phyloscanner make trees.py’s run time and memory usage; if you’re not sure about read pair
overlap (i.e. you don’t know the insert size distribution for your data), run
tools/EstimateReadCountPerWindow.py as discussed at the start of section 3.2.
•--check-recombination: calculate a metric of recombination for each sample’s set of reads in
each window. (Recommended only if you’re interested, of course.) How the metric is calculated:
for each possible set of three sequences, one is considered the putative recombinant and the other
two the parents. For each possible crossover point (the point at which recombination occurred), we
calculate dLas the difference between the Hamming distance from the recombinant to one parent
and the Hamming distance from the recombinant to the other parent, looking to the left of the
crossover point only; similarly we calculate dRlooking to the right of the crossover point only. dL
and dRare signed integers, such that their differing in sign indicates that the left and right sides of
11

the recombinant look like different parents. We maximise the difference between dLand dR(over
all possible sets of three sequences and all possible crossover points), take the smaller of the two
absolute values, and normalise it by half the length of the alignment of sequences. The resulting
metric is constrained to be between 0 and 1, inclusive. The maximum possible score of 1 is obtained
if and only if the two parents disagree at every site, the crossover point is exactly in the middle,
and either side of the crossover point the recombinant agrees perfectly with one of the parents e.g.
AAAAAAA
AAAACCC
CCCCCCC
Calculation time scales cubically with the number of unique sequences each sample has per window,
and so the option is turned off by default. You can save time by only turning it on only after you’ve
settled on the values of other parameters that affect the number of unique sequences per window
(notably window width, a merging threshold and a minimum read count).
5.3 Read quality options
•--discard-improper-pairs: discard all reads that are were flagged, at the time of mapping, as
improperly paired: in the wrong orientation, or one mate unmapped, or too far apart. For paired-
read data.
•--quality-trim-ends: used to specify a quality threshold for trimming the ends of reads. We trim
each read inwards until a base of this quality is met.
•--min-internal-quality: used to specify an internal quality threshold for reads. Reads are
allowed at most one base below this quality; any read with two or more bases below this quality
are discarded. (If used in conjunction with the --quality-trim-ends option, the trimming of
the ends is done first.)
•--min-read-count: used to specify a minimum count for each unique read. Reads with a count
(i.e. the number of times that sequence was observed, after merging if merging is being done) less
than this value are discarded. The default value of 1 means all reads are kept. You might want to
discard rare reads to protect against sequencing error. Retaining fewer reads will also speed up all
subsequent processing and analysis of the reads.
5.4 Other assorted options
•--excision-coords: used to specify a comma-separated set of integer coordinates that will be
excised from the aligned reads before phylogenies are made. Useful for sites of non-neutral evolution,
which distort phylogenies. Requires the --excision-ref flag.
•--excision-ref: used to specify the name of a reference (which must be present in the file you
specify with --alignment-of-other-refs) with respect to which the coordinates specified with
--excision-coords are interpreted. If you are also using the --pairwise-align-to option, you
must specify the same reference there and here.
•--merging-threshold-a: when multiple reads in the same bam file have exactly the same sequence,
phyloscanner make trees.py always collapses these to a single read with an associated count (i.e.
the number of times that exact sequence was found in distinct reads in the bam file). With this
option, similar reads are merged as well as identical reads. Use this option to specify a positive
integer as the threshold for merging. If read X differs from read Y by this threshold or less, and X
has the smaller count, we keep only read Y and update its count to be the sum of the two counts.
Note that with --merging-threshold-a, if X is similar enough to Y for merging, but Y has already
been merged into Z, X will only be merged into Z too if X and Z are similar enough (contrast with
--merging-threshold-b below). What this algorithm does is to start with the most common read,
see if any less common reads are similar enough to into it, and if so remove those reads and use
their count to bolster the more common read. The procedure is repeated starting with the second
most common read, skipping any reads that have already been ‘used up’ in merging; then the third
most common read and so on.
12

•--merging-threshold-b: similar to --merging-threshold-a above, except that if X is similar
enough to Y for merging, but Y has already been merged into Z, X will automatically be merged
into Z. This algorithm effectively partions the set of reads into groups such that each member of a
group is similar enough to at least one other group member (and each group can be split no further,
i.e. no read is similar enough to any read outside its own group), and uses only the read with the
highest count to represent each group. For the same value of the threshold, merging bwill merge
as much or more than merging a(i.e. bwill tend to result in fewer unique reads).
•--num-bootstraps: used to specify the number of bootstraps to be calculated for RAxML trees (by
default, none, i.e. only the maximum-likelihood tree is calculated).
•--bootstrap-seed: used to specify the random-number seed for running RAxML with bootstraps.
The default is 1.
•--output-dir: used to specify the name of a directory into which output files will be moved. If it
does not exist, it will be created; however we cannot create a new directory inside a directory that
does not yet exist. Temporary and output files are always created in the working directory, i.e. the
directory in which the phyloscanner make trees.py command was run, but with this option the
output files are copied to the specified directory at the end.
•--time: print the times taken by different steps.
•--x-mafft: used to specify the command you need in order to run mafft. The default is simply
mafft; if your mafft executable is not in the $PATH environment variable for your terminal (google
this if you don’t know what it means) you will need to include the directory where this executable
lives, e.g. /path/to/where/I/installed/mafft/mafft.
•--x-samtools: used to specify the command you need in order to run samtools. The default is
simply samtools. See the points raised for the --x-mafft option above.
•--keep-output-together: by default, subdirectories are made for different kinds of output. With
this option, all output files will be in the same directory (either the working directory, or whatever
you specify with --output-dir).
•--keep-temp-files: keep temporary files we create on the way (these are deleted by default).
5.5 Options for bioinformatic interrogation
Options for detailed bioinformatic interrogation of the input bam files, not intended for normal usage.
•--inspect-disagreeing-overlaps: with –merge-paired-reads, those pairs that overlap but dis-
agree are discarded. With this option, these discarded pairs are written to a bam file (one per
patient, with their reference file copied to the working directory) for your inspection.
•--read-names-1: produce a file for each window and each bam, listing the names (as they appear
in the input bam file) of the reads that phyloscanner make trees.py used. If you like this you
may also like tools/ExtractNamedReadsFromBam.py, which is run separately from the command
line (run it initially with --help for more information).
•--read-names-2: as --read-names-1, except the files will show the correspondence between read
names and which unique sequence they correspond to. This option cannot be used with either of the
--merging-threshold or --excision-coords options (because they change the correspondence
initially established between unique sequences and reads).
•--exact-window-start: normally phyloscanner make trees.py retrieves all reads that fully over-
lap a given window, i.e. starting at or anywhere before the window start, and ending at or anywhere
after the window end. If this option is used without --exact-window-end, the reads that are re-
trieved are those that start at exactly the start of the window, and end anywhere (ignoring all
the window end coordinates specified). If this option is used with --exact-window-end, for a
read to be kept it must start at exactly the window start AND end at exactly the window end.
If --merge-paired-reads is also used, this explanation applies to inserts (read pairs) instead of
individual reads.
13
•--exact-window-end: with this option, the reads that are retrieved are those that end at exactly
the end of the window. Read the --exact-window-start help.
•--recover-clipped-ends: the default behaviour of phyloscanner make trees.py is to keep only
reads that fully span the window in question. A read which is long enough in principle to reach
the edge of the window but is not mapped at its end, i.e. the end is clipped, will therefore not be
included. With this option, clipped ends are recovered by considering any bases at the ends of the
read that are unmapped to be mapped instead to 1 more than the base to their left (at the right
end) or 1 less than the base to their right (at the left end), iterating out from the centre. e.g. a
9bp read mapped to positions None,None,10,11,13,14,None,None,None (i.e. clipped on the left by
2bp, and on the right by 3bp, with a 1bp deletion in the middle), is taken to be mapped instead
to positions 8,9,10,11,13,14,15,16,17. In this example, if the window left edge is 8 or 9 and the
right edge is 15, 16 or 17, the read with its clipped ends recovered spans the window but the read
without clipped ends does not. WARNING: mapping software clips the ends of reads for a reason,
namely that that stretch of sequence does not look anything like the reference at that point. The
clipped sequence could be just junk, or genuine sample from a distant part of the genome (i.e. the
read is chimeric); in this case the clipped sequence should be discarded and not recovered. As such,
this option should not be used as part of normal phyloscanner make trees.py usage. Its intended
usage is specifically the following: you have identified a window in a bam file in which reads are
clipped, but you believe the reads to be correct, i.e. the clipping is an artefact of the mapper
being unable to find the correct local alignment. You should combine this option with --no-trees
because the inclusion of clipped sequence, which by definition is very different, increases the chance
of misalignment. You should inspect the aligned reads manually before doing anything else (and
hopefully get some insight into how the reference in this window should be changed in order to have
subsequent remapping get the local alignment right, in particular by contrasting the reference with
the consensus of the aligned reads).
5.6 Partial processing options
Options to only partially run phyloscanner make trees.py, stopping early or skipping steps.
•--align-refs-only: align the mapping references used to create the bam files (plus any extra
reference sequences specified with --alignment-of-other-refs), then quit without doing anything
else. The point of this is to allow inspection of that alignment, whose coordinates are used to
interpret window coordinates (unless --pairwise-align-to is used).
•--read-names-only: to be combined with --read-names-1 or --read-names-2: quit after writing
the read names to a file (which means the reads are not aligned).
•--no-trees: process and align the reads from each window, then quit without making trees.
•--dont-check-duplicates: don’t compare reads between samples to find duplicates – a possible
indication of contamination. (By default this check is done.)
5.7 Deprecated options
Left in phyloscanner make trees.py for backward compatibility or interest.
•--contaminant-count-ratio: used to specify a numerical value which is interpreted in the follow-
ing way: if a sequence is found exactly duplicated between any two bam files, and is more common
in one than the other by a factor at least equal to this value, the rarer sequence is deleted and goes
instead into a separate contaminant read fasta file. This is considered deprecated because including
the exact duplicates in the tree and dealing with them during tree analysis is more convenient –
phyloscanner make trees.py does not need to be rerun if you change your mind about count
thresholds for removing duplicates – it also allows the offending duplicates to be inspected in the
tree.
•--flag-contaminants-only: for each window, just flag contaminant reads then move on (without
aligning reads or making a tree). Only makes sense with the --contaminant-count-ratio flag.
14
•--forbid-read-repeats: using this option, if a read with the same name is found to span each of
a series of consecutive, overlapping windows, it is only used in the first window. Consecutive means
next to each other in the order you specified. For example, if you specified windows 10-20, 15-25,
20-30 and 31-40, and there was a read that spanned all four windows (i.e. it started at or before
position 10 and ended at or after position 40), it would be used in window 10-20, not used in 15-25
because it spanned the last window, not used in 20-30 because it spanned the last window (even
though it was skipped there), and used in 31-40 because this window does not overlap with the last
one. NB with paired read data, mates in a pair have the same name; using this option without the
--merge-paired-reads option will mean at most one of the two mates will be used (in a given
window and consecutive overlapping windows), and with the --merge-paired-reads option mates
will be merged into a single read, which is used only the first time it is encountered in consecutive
overlapping windows.
•--keep-overhangs: keep the part of the read that overhangs the edge of the window. (By default
this is trimmed, i.e. only the part of the read inside the window is kept.) Keeping overhangs means
that, within each bam file, reads that are identical inside the window but have different overhangs
will not be merged into a single sequence (with a count greater than 1). Differences in overhangs
may be SNPs, or simply because the overhangs start or end at different points; this option is
therefore a bit weird, because it’s nice to merge all reads that are identical inside the window of
interest.
•--ref-for-coords: if the --pairwise-align-to option is not used, then a multiple sequence
alignment is created with all the mapping references (used to create the bam files) plus any ex-
tra references included with --alignment-of-other-refs. By default, window coordinates are
interpreted with respect to this alignment, i.e. they are in the alignment coordinates. With this
option (–ref-for-coords), the multiple sequence alignment is still created but window coordinates
are interpreted with respect to a named reference, which must be one of those included with
--alignment-of-other-refs. Use this option to specify the name of the reference. This option
is deprecated because the --pairwise-align-to option also interprets window coordinates with
respect to a named reference, but without needing to construct a multiple sequence alignment –
using just pairwise alignment.
•--recombination-gap-aware: by default, when calculating Hamming distances for the recombi-
nation metric, positions with gaps are ignored. This means that e.g. the following three sequences
would have a metric of zero:
A-AAAAA
A-AAA-A
AAAAA-A
With this option, the gap character counts as a fifth base and so (dis)agreement in gaps con-
tributes to Hamming distance. This increases sensitivity of the metric to cases where indels are
genuine signals of recombination, but decreases specificity, since misalignment may falsely suggest
recombination.
15

Part II
Analysing within- and between-host
phylogenies
The tree analysis procedure takes as input one or more phylogenies, and analyses them to reconstruct
transmission, and identify multiply infected individuals and contaminants. In order, the following steps
are performed:
1. Identify and exclude tree tips corresponding to reads that should not be used to reconstruct trans-
mission (blacklisting)
2. Perform an ancestral state reconstruction on each tree in turn, and use this to identify host sub-
graphs (contiguous regions of the phylogeny that have been assigned to the same host)
3. Calculate per-host summary statistics, and graph them across the genome if multiple trees are given
as input
4. In each tree, determine the relationship between each pair of hosts determined by the relative
positions of their subgraphs
5. Summarise these pairwise relationships over all trees (if more than one tree was given).
The code forms an Rpackage entitled phyloscannerR which appears as a subfolder in the phyloscanner
directory. The package manual can also be found in this directory and users who prefer to perform the
analysis within Ritself may wish to consult that. This package and its dependencies must be installed
prior to use, whether within Ror at the UNIX command line; all dependencies are available in CRAN
except for ggtree [5], which is part of Bioconductor.
To install phyloscannerR (and ggtree), use the command line to navigate to the “phyloscannerR”
subdirectory of the “phyloscanner” directory, then start Rand enter the following commands:
> install.packages("devtools")
> library(devtools)
> source("https://bioconductor.org/biocLite.R")
> biocLite("ggtree")
> install("../phyloscannerR", dependencies = T)
(Obviously if you have already installed ggtree then the second and third lines can be skipped.)
A single command line script, phyloscanner analyse trees.R, can be used to perform a full analysis.
Each step identified above also has its own standalone script, which can be used to, for example, analyse
multiple trees in parallel on a cluster. We do not document all the command line options for these
additional scripts here, and the user is directed to their --help options.
6 The basic command
The basic command for phyloscanner analyse trees.R is
$ phyloscanner_analyse_trees.R TreeInput OutputString ReconstructionModeArguments
OutputString is simply a string identifying all file output. The tree input can be either a single tree file or
a single string that begins all files (see below). For full instructions regarding
ReconstructionModeArguments, see section 13; for a quick start, s,0 gives a parsimony reconstruc-
tion with no within-host diversity penalty. However, be aware that this version of the algorithm will not
be aware of potential contamination.
16

7 Name format for input files
Tree files should be in Newick or Nexus format. These can be the product of
phyloscanner make trees.py, but need not be; the script will work on any set of trees if the tip labels
can be interpreted properly (see below). Each tree should reside in a single file.
The tree files, when the analysis is to be performed on multiple trees, are expected to all be located in
the same directory and have names that all begin with the same nonempty string. The remainders of the
file names, excluding the file extensions, are referred to as file suffixes. If the genome window approach
is used (i.e. each tree represents a window of the genome), the suffixes will be expected to contain
the coordinates of the genome window for each tree. (Output from phyloscanner make trees.py will
automatically be formatted in this way.) A regular expression described below (see section 10) is used
to identify these coordinates; if it fails to identify them, then the script will assume that the windowed
approach was not used.
Any accompanying per-window files specified in optional arguments are assumed to follow the same
pattern: a string beginning every file, the same set of suffixes, and a file extension. This will automatically
be the case for files produced by phyloscanner make trees.py: the values of the recombination metric
(see section 14), and the list of reads that are identical amongst different hosts (see section 12.1). Any
user-specified blacklists (see section 12.4) must also have file names in this format.
Tree files are by default assumed to have .tree as a file extension and csv files .csv, but this can be
overridden with the --treeFileExtension and -csvFileExtension options.
If the analysis is instead to be performed on a single tree, the file name of that tree should be given
as the TreeInput argument instead. The same goes for any other files to be used as optional arguments.
8 Output
The default output of phyloscanner analyse trees.R varies depending on whether it is run on one or
multiple trees.
If the input is a single tree file, the default output is an annotated pdf version of that tree, a csv file
of summary statistics for the hosts present in that tree, and a csv file outlining the relationship between
each pair of hosts in that tree.
If the input is multiple trees, then the default output is an annotated pdf version of all trees, a csv file
of summary statistics for every host across all the trees, a pdf graphing those statistics across all trees,
and a csv file summarising which pairs of hosts meet the criteria for being closely related.
9 Preparing the trees
Two options determine processing of the phylogeny before any further operations are performed. A
named outgroup can be specified with --outgroupName; this is recommended unless the input trees are
known to be correctly rooted. It is always assumed that the lineage represented by the root of the
entire phylogeny was not present in any sampled host, even if no outgroup is given. Trees output by
phyloscanner make trees.py will need to be re-rooted
The output of many phylogeny reconstruction packages, including RAxML, is a binary phylogeny. While
multifurcations may seem to appear when the trees are viewed by eye, these will have a hidden binary
branching order. The branches connecting the nodes within these “multifurcations” may all be of an
extremely short but nonzero minimum length (in RAxML output) or actually zero length (in, e.g., PhyML
output). Since this branching order is largely arbitrary, it is recommended that these regions of the tree be
collapsed into genuine multifurcating nodes for phyloscanner analyse trees.R. A length threshold to
determine pairs of nodes that should be so collapsed can be specified with --multifurcationThreshold.
This is normally numerical, but if gis given it will be guessed from the trees itself. If the trees have
interpretable genome window coordinates, the smallest branch length will be compared to the branch
length corresponding to one SNP. If the former is smaller than 0.25 times the latter, the former will
be used as the threshold; if not, the tree is assumed to contain no multifurcations and no collapsing is
done. If no genome window coordinates are available, the shortest branch length is simply used as the
threshold. Before specifying gthe user should ideally ensure that the trees genuinely do contain apparent
multifurcations.
17
10 Tip label and file suffix regular expressions
The tip labels in the input phylogenies are expected to follow a regular expression that identifies host
IDs, and optionally also read identifiers and read counts. The exact format is specified using a regular
expression with three capture groups: for host ID, read ID, and count. The count is expected to be
an integer. If a third group is not found then it assumed that every tip represents only a single read.
The default regular expression is "^(.*)_read_([0-9]+)_count_([0-9]+)$", but the user can specify an
alternative with the --tipRegex option. In plainer language, this is of the form “NAME read X count Y”
where NAME is the host name, X the read identifier and Y the read count. The tip names of the
outgroup (if it exists), and of any other reference sequences that are included, should not match this
regular expression.
The default regex is appropriate for analysing trees output by phyloscanner make trees.py, when
each bam contains reads from a different host. A different regular expression may be required if
the input is not from phyloscanner make trees.py. It may also be required for trees output by
phyloscanner make trees.py if multiple bam files contain sequences derived from the same host, and
part of the bam file name identifies the host. For example if you had three bam files named PatientA-1.bam,
PatientA-2.bam and PatientB-1.bam, where tips from the first two are from the same host. Changing
the tip regex to "^(.*)-[0-9+]\\.bam_read_([0-9]+)_count_([0-9]+)$" means that the first group
in the regex will match “PatientA”, “PatientA” and “PatientB” for the three bams respectively; all tips
from the first two bams are then associated to the same host, PatientA.
Similarly, the set of file suffixes (see above) may contain genome window coordinates, and a regular
expression is used to identify these. Two capture groups are expected for the start and end of the window.
The default is "^\\D*([0-9]+)_to_([0-9]+)\\D*$". In plainer language this is any number of non-digit
characters, followed by “X to Y” where X is the window start and Y the end, followed by any number of
non-digit characters. An alternative can be specified with the --fileNameRegex option.
11 Branch length normalisation
Two hosts may be classified as closely linked by phyloscanner analyse trees.R on the grounds that
they have host subtrees separated by a patristic distance that lies below a given threshold. However
because we are often interested in a genomic window approach, a fixed distance threshold for all trees
may not be appropriate because of variations in nucleotide diversity in different areas of the genome.
This is because a distance threshold suggestive of a close epidemiological relationship in one window will
not be in others. To remedy this, phyloscanner analyse trees.R can be asked to normalise branch
lengths across windows.
One approach to generating appropriate normalisation values over the genome is implemented by the
script tools/CalculateTreeSizeInGenomeWindows.py. Run from the command line (see it’s --help
option for details on how) taking a whole-genome alignment of existing reference sequences as input,
it infers phylogenies in sliding windows, and characterises the size of the tree by taking the median of
the set of patristic distances between all pairs of references. One such value is obtained per window;
one value is then assigned to each integer position in the genome by taking the mean of the values
from those windows spanning this position (with windows chosen to overlap, so that multiple values
for each position should smooth some stochasticity in phylogenetic inference). Note that you must
specify one sequence in the alignment whose coordinates will be used to define genomic position; this
sequence must match the one you specified with --pairwise-align-to in phyloscanner make trees.py
if we are to use this information for branch length normalisation. This ensures that the coordinates in
the output of tools/CalculateTreeSizeInGenomeWindows.py and the coordinates in the output of
phyloscanner make trees.py (e.g. in the per-window file names) mean the same thing.
Q: what alignment of existing reference sequences should you use for this? A: ideally, one which is in
some sense representative of between-host diversity. We find all pairwise patristic distances, so if your
set of sequences is biased to be overly representative of a particular subset, these patristic distances will
become biased towards zero. That said, all we’re interested in here is how patristic distances change
across the genome, not their absolute values.
The output from tools/CalculateTreeSizeInGenomeWindows.py, or alternatively any other csv file
containing genomic positions with associated normalisation constants, can be used with the
--normRefFileName option. This will scale all normalisation constants by the same factor such that
18

their mean is 1. A normalisation constant is then determined for each window by taking the mean of
the values for all positions in the window. This constant for the window is used to scale all branch
lengths in the tree for subsequent processing by phyloscanner analyse trees.R. The similar option
--normStandardiseGagPol, specific to HIV genomes, instead scales normalisation constants so that the
mean of the constants on the gag and pol genes is 1; distances anywhere in the genome are then inter-
pretable as standard distances in gag/pol.
The --normalisationConstants option is used to directly specify the normalisation to be used for
each tree file. If the argument is numerical, it is used as a normalising constant for every tree. If it is
instead the path to a .csv file, then this file is expected to consist of a first column listing all input tree file
names, and a second of normalising constants. The presence of --normalisationConstants overrides
any other normalisation options, which will be disregarded.
The standalone script normalisation lookup writer.R can be used to separately write a .csv file
for use with the --normalisationConstants option using the procedure described in this section. This
is not normally necessary for use of phyloscanner analyse trees.R, but can be used as an argument
for other standalone scripts.
The normalisation is used for the parsimony reconstruction (see section 13) and for the identification
of closely-related hosts (see sections 15 and 16). However, output trees have the same branch lengths as
input trees, and summary statistics (see section 14) are calculated using raw branch lengths.
11.1 Parallelising tools/CalculateTreeSizeInGenomeWindows.py
The --threads option of tools/CalculateTreeSizeInGenomeWindows.py can be used to specify multi-
ple threads, allows multiple windows to be analysed in parallel using multiple cores on the same machine.
Alternatively, power users may want to massively parallelise, e.g. over multiple different machines on a
computing cluster. Read on if interested; skip ahead to section 12 if not. First choose your desired start,
end, window width and increment parameters. Then generate a set of
tools/CalculateTreeSizeInGenomeWindows.py commands, with each command running only a single
window, namely the window after the one of the previous command. For example say you wanted to start
at position 1000, end at 9000, have a window width of 300 and an increment of 10. Your first window is
1000 −1299, your second is 1010 −1309, . . . and these can be run as separate commands thus:
$ tools/CalculateTreeSizeInGenomeWindows.py MyAln.fasta MyChosenSeqName \
1000 300 output_1000-1299 -E 1299
$ tools/CalculateTreeSizeInGenomeWindows.py MyAln.fasta MyChosenSeqName \
1010 300 output_1010-1309 -E 1309
...
Generating these commands is easily done as part of a loop, e.g.
$ for start in $(seq 1000 10 8700); do
$ end=$((start + 299))
$ command="tools/CalculateTreeSizeInGenomeWindows.py MyAln.fasta MyChosenSeqName"\
" $start 300 output_${start}-${end} -E $end"
$ done
and each command could be put inside a separate file to be run as a separate job. In this example, output
files will be produced named output 1000-1299 ByWindow.csv,output 1000-1299 ByWindow.csv,. . .;
these can be combined with
$ cat output_*_ByWindow.csv | sort -n | uniq > output_all_ByWindow.csv
(the sort and uniq commands preventing the csv header repeating). Finally, running
$ tools/FromPerWindowStatsToPerPositionStats.py output_all_ByWindow.csv \
> output_all_ByPosition.csv
converts the per-window tree sizes to per-position tree sizes, as required as input for the
--normRefFileName option of phyloscanner analyse trees.R.
(We used this approach for parallelisation, as we covered the HIV genome with windows of width 301
bp and an increment of 1 bp, necessitating 9179 windows. Running these as 9179 separate commands in
parallel on a cluster sped things up considerably.)
19
12 Blacklisting
Blacklisting refers to the exclusion of some tips in the phylogeny from the full analysis. This may be
done for a variety of reasons:
•Tips from suspected contaminant reads (i.e. for which we suspect the true host is not the recorded
host) should not be used to reconstruct transmission
•Possible multiple infections may complicate an analysis and the user may prefer to deal only with
the largest collection of reads from such a host
•Uneven tip or read counts between different hosts have the potential to bias the inference of trans-
mission. The user may wish to remove some tips in order to mitigate this.
phyloscanner includes a number of utilities to perform blacklisting, all of which can be run as part of
phyloscanner analyse trees.R. The script does all blacklisting internally, but other scripts in the tools
directory of the phyloscanner code can be used to generate lists of suspect tips in an input phylogeny.
In addition, a script entitled phyloscanner clean alignment.R may be used to perform blacklisting and
remove blacklisted sequences from the input alignments without performing a full phyloscanner run; see
the --help for this for more details. The various types of blacklisting are listed below. All, except user
blacklisting, assume that the tree tips are annotated with read counts.
A CSV file listing every tip that was blacklisted in every tree, and why, will be output if the
--blacklistReport option is given to phyloscanner analyse trees.R.
12.1 Duplicate blacklisting
This procedure blacklists reads from one host which are identical to reads from another, but are present
in sufficiently small numbers that it would be suspected that they are merely contaminants. It is enabled
with the --duplicateBlacklist option to phyloscanner analyse trees.R, which takes a single argu-
ment identifying the duplicate output files from phyloscanner make trees.py. Tips are blacklisted if
the corresponding sequence is exactly identical to the sequence for a tip from another host, and either
the raw read count from the former tip, or the ratio of the read count from the former tip to the read
count from the latter tip, is less than a specified threshold. The raw read count is specified with the -rwt
option and the ratio threshold with the -rtt option. It is acceptable to specify both. As the default
values for both are 0, nothing will be blacklisted unless at least one is given a positive value.
phyloscanner analyse trees.R will append “ X DUPLICATE” to the tip names of tips black-
listed by this procedure, which will appear in annotated tree output. The standalone R script is
duplicate blacklister.R.
12.2 Parsimony-based blacklisting for contaminants
While in many cases contaminant reads will be identical to those from another host in the dataset, the
user may not always be so fortunate; the contaminant may be from another dataset entirely, and hence
have no exact matches amongst the reads that are used to reconstruct the phylogeny. Similarly, a variant
may be sequenced from the dataset that is never assigned the correct host. The --parsimonyBlacklistK
option to phyloscanner analyse trees.R can be used to identify such tips, using the same parsimony
procedure that is employed for ancestral state reconstruction (see section 13, and the phyloscanner
paper). The single additional numerical argument to --parsimonyBlacklistK is the value of kused to
calculate a within-host diversity penalty.
We use this procedure because excessive amounts of within-host diversity may be just as suggestive of
contamination as of a genuine multiple infection. For each host in turn, the tree is pruned so that only the
tips from that host, and the outgroup, remain. (If no such outgroup is given,
phyloscanner analyse trees.R will attempt to find one.) The reconstruction is them performed on
this pruned tree, using the kparameter specified with --parsimonyBlacklistK, and the result used to
see whether the tips of the tree were grouped into one subgraph or more than one subgraph. If the for-
mer, then the phylogeny does not suggest sufficient diversity within this host to suggest either a multiple
infection or contamination. If the latter, then one or the other is likely to be true. We suggest that a
subgraph is more likely to be contamination when it contains very few reads. The arguments used in
20
duplicate blacklisting are reused here. If the total number of reads associated with the tips in a sub-
graph is smaller than the raw threshold (--rawBlacklistThreshold), then all tips in the subgraph are
blacklisted. If the proportion of reads from the host that belong to a subgraph is smaller than the ratio
threshold (--ratioBlacklistThreshold), then all tips in that subgraph are blacklisted. The former
threshold is applied even if there is only one subgraph, in which case it is assumed that so few reads exist
from the host in question that all of them could well be contaminants, with genuine sequencing failing in
this part of the genome.
phyloscanner analyse trees.R will append “ X CONTAMINANT” to the tip names of tips black-
listed by this procedure, which will appear in annotated tree output. The standalone R script is
parsimony based blacklister.R.
12.3 Dual infection blacklisting
If the parsimony-based blacklisting procedure identifies a host with suspicious amounts of within-host di-
versity where the smaller subgraphs have too many reads to be flagged as likely contaminants, a dual infec-
tion (or larger multiple infection) may be suspected. The current version of
phyloscanner analyse trees.R is only able to summarise the relationships of such a host with the
neighbours of all its subgraphs in the transmission tree; it cannot separate the neighbours of each sub-
graph. This may result in spurious inferences being drawn, where a close neighbour to one subgraph and
a close neighbour to another are identified as closely related to each other. If this behaviour is regarded
as particularly undesirable, a suggested stopgap remedy is to assume that the subgraph with the largest
read count represents the same infection across the entire genome, and blacklist the rest.
The --dualBlacklist flag, which can be used only if --parsimonyBlacklistK is also specified, will
do this blacklisting. phyloscanner analyse trees.R will append “ X DUAL” to the tip names of tips
blacklisted by this procedure, which will appear in annotated tree output. The standalone R script is
dual host blacklister.R.
12.4 User blacklisting
The user may wish to specify his or her own blacklist before phyloscanner analyse trees.R is run. A
separate plain text file for each window should be prepared, with a blacklisted tip on each line, ending
with the same file suffix as the corresponding tree file. The path to these files and the string beginning
each should be specified with the --userBlacklist option. These tips will be blacklisted from the start,
and phyloscanner analyse trees.R will append “ X USER” to tip labels.
12.5 Downsampling
The intention of the downsampling procedure is to ensure that the same numbers of reads are in-
cluded in the analysis from each host in the dataset. This is done by taking a random sample of
reads, without replacement, and blacklisting tips where the corresponding reads was never selected
at all. The --maxReadsPerHost option is used to specify a number of reads to downsample to. If
--blacklistUnderrepresented is specified then hosts without enough reads in totality are blacklisted,
but if it is absent then all tips from those hosts are retained.
phyloscanner analyse trees.R rewrites tip labels to reflect the number of times a tip was randomly
selected. “ X DOWNSAMPLED” is appended to tips that are blacklisted because the corresponding
reads were never selected, in which case the read count in the tip label will have been rewritten to 0.
“ X UNDERREPRESENTED” is appended to those that are blacklisted because they are from a host
without enough reads in total (if -dsb is given). The standalone R script is downsample reads.R.
12.6 Pruning the blacklist
Default behaviour is to leave blacklisted tips in the phylogeny; they are not annotated with any host and
are instead given the unsampled state in the parsimony reconstruction, but they remain so as to be exam-
ined in the annotated tree output. The --pruneBlacklist option to phyloscanner analyse trees.R
can be used to prune these tips instead, in which case they will not appear in output at all.
21
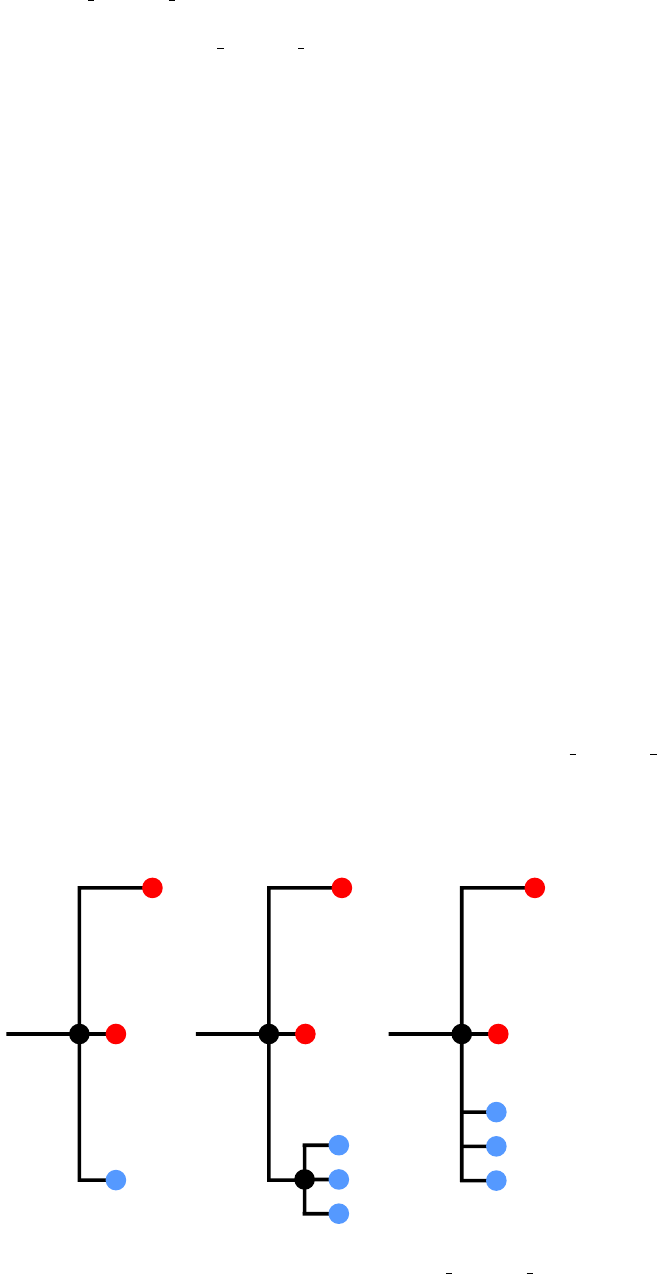
13 Parsimony reconstruction
The core of the phyloscanner analyse trees.R analysis is an ancestral state reconstruction of hosts to
internal nodes of each phylogeny. We offer several algorithms for this; the exact one is specified with the
final compulsory argument to phyloscanner analyse trees.R. For full details, see the supplementary
information of the phyloscanner paper.
•A fast, non-rigorous (for more than two hosts) adaptation of the procedure outlined in Romero-
Severson et al. (PNAS 2016). This is specified by r. It will not reliably reconstruct multiple
infections.
•A parsimony reconstruction using the Sankoff algorithm that allows for an additional “unassigned”
state and can be configured to penalise the reconstruction of single infections to hosts which imply
unreasonable amounts of diversity. This is specified by two or three arguments, separated by
commas. The first is s. The second is the value of the kparameter that penalises within-host
diversity; if this is set to 0 then no penalty is applied and the procedure is equivalent to standard
parsimony with the unassigned state. It is recommended that users explore their data to pick
suitable values of k, as it is likely to be pathogen- and sequencing method-dependent. As a rule
of thumb, it should be the reciprocal of a patristic distance so large that it would be surprising to
encounter a host with an infection so diverse that a phylogeny built from pathogen sequences from
just that host had a branch that long or longer, if the host was infected only once.
The latter parsimony reconstruction algorithm uses a very large matrix for large trees and occasionally R
will run out of memory; the argument --useff will cause the script to use the ff library instead, which
solves many memory issues at the cost of speed.
Of concern in either of the two proper parsimony reconstruction algorithms is used is how to handle
terminal branches of zero or negligible length. In particular, if a tip with a read count of nis connected
to its parent node by such a branch, should that tip be considered as one tip or ntips for the parsimony
reconstruction? As an illustrated example, see below. Phylogenetic software output, with minimum-
length branches collapsed to zero (see above) might appear as in diagram a) in Fig. 13 below, where
colours represent hosts and the numbers in the coloured tips read counts, while numbers on branches are
branch lengths (with zero-length branches displayed with nonzero lengths for clarity). The question is
whether a parsimony reconstruction should consider this clade as b), where all the reads represented by
the blue tip in a) form separate tips but their MRCA mis not the MRCA nof the entire clade, or b),
where the MRCAs are the same. Under simple parsimony, the cost of reconstructing nas red is 1 in b) and
3 in c), but the cost of reconstructing it as blue is 2 in both. By default, phyloscanner analyse trees.R
treats this situation as b), which is the same as ignoring read counts in the parsimony reconstruction
- as it will always be most parsimonious to reconstruct mas blue to avoid turning one transition into
three, the three blue tips can be ignored and the colour of mfixed, so we are back to diagram a) - but
the --readCountsMatterOnZeroLengthBranches option, if specified, will treat it as c) instead.
0
1
1
1
1
1
1
0
0
0
0
0
1
1
1
1
1
1
0
0
0
0
0
1
1
1
3
a) b) c)
nn n
m
Annotated versions of all input trees will be output by phyloscanner analyse trees.R, in pdf format
by default. The --outputNexusTree option can be used to make it provide these trees in Nexus format
22

instead.
The standalone R script to perform parsimony reconstruction is split hosts to subtrees.R.
14 Summary statistics
By default, phyloscanner analyse trees.R produces a csv file of summary statistics with the following
columns:
•id: the host ID for the statistics in this row.
•file suffix: the file suffix of the associated tree file.
•xcoord: used to draw the graphs (see below); this is the midpoint of the genome window if the
windowed approach is used, and the position of the file suffix in an alphabetical ordering if not.
•Tips: the number of tips from this host in this tree.
•Reads: the number of reads from this host in this tree (if read counts were not detected then this
column will be identical to the tip counts, as each tip is assumed to represent one read).
•Subgraphs: the number of subgraphs that the parsimony reconstruction identified for this host in
this tree.
•clades: the number of separate clades monophyletic clades from this host in this tree.
•overall.rtt: the mean patristic distance from tips from this host to the MRCA of the tips from this
host, weighted by read counts if these are given
•largest.rtt: the mean patristic distance from tips from the largest (by read count) subgraph from
the host to the MRCA of that subgraph, weighted by read counts if these are given.
•max.branch.length: the length of the longest branch in the tree obtained by pruning all tips from
the tree except the ones from this host.
•max.patristic.distance: the largest patristic distance between any pair of tips from this host.
•global.mean.pat.distance: the mean patristic distance between tips from this host.
•subgraph.mean.pat.distance: the mean patristic distance between tips in the largest subgraph.
•recombination.metric: this column only appears if the recombination metric files output by
phyloscanner make trees.py have been given to phyloscanner analyse trees.R using the
--recombinationFiles argument. It gives the value of the recombination metric for this host in
this tree.
•prop.gp.1 to prop.gp.X: A variable number of columns recording what proportion of the reads from
this host occur in each subgraph, ordered by size. The total number of columns is the largest
number of subgraphs present for any host in any tree (and so many columns will be 0 for most
hosts).
Also generated, if more than one phylogeny is given as input, is a pdf file graphing these statistics
across all trees for every host. Each host occupies a separate page in this file, with five or six graphs per
host:
1. Number of reads and number of tips
2. Number of subgraphs and number of clades
3. Mean root-to-tip distance in the largest subgraph and in the whole tree
4. Mean pairwise patristic distance in the largest subgraph and in the whole tree
5. The proportion of reads appearing in each subgraph
23
6. (If recombination metric files are specified with -R) The value of the recombination metric across
the genome
If the script could detect genome window coordinates from the file suffixes, and the starts and ends
of those windows occur regularly across the genome, these graphs will be annotated with light grey
rectangles for windows where a host is missing from a single window, and dark grey rectangles where a
window appears to be entirely missing (i.e. it had no tree file).
The standalone R script to produce summary statistics is summary statistics.R.
15 Classification of pairwise relationships
The parsimony reconstruction has annotated all internal nodes in each phylogeny with either a host or
the unassigned state, and from this information, host subgraphs have been identified. The next step in
the script is to document the relationships between each pair of hosts in every tree. By default (when
the script is run on multiple tree files) this is done silently, but two types of output file can be generated.
The --collapsedTrees option produces the collapsed tree (see the phyloscanner paper), in csv
format. This file has four columns:
•The collapsed tree node ID, which is the host ID or “unassigned region” followed by -S and an
identifying number
•The collapsed tree node ID of that node’s unique host, or “root” if it has none
•The ID for the host, or “unassigned region”, associated with this node
•The ID for the host, or “unassigned region”, associated with the parent node
If the script is run on a single tree file only, the classification csv file is produced by default, but the
collapsed tree is not.
The --allClassifications option produces per-tree csv files describing all pairwise relationships
between hosts. The columns of this file are as follows:
•host.1, host.2: the pair of hosts. The order does not signify anything on its own but is relevant to
interpretation of the path.classification column.
•adjacent: This is TRUE if there exist two nodes, one each from subgraphs from the two hosts, such
that the path between the two traverses only nodes that are either also from these host subgraphs,
or unassigned. (In other words, it is possible to move through the phylogeny from one host to the
other without moving through the subgraph of another sampled patient.)
•contiguous: This is TRUE if for any two nodes, one each from subgraphs from the two hosts, the
path between the two intersects no nodes that are not either also from these host subgraphs, or
unassigned. This is similar to adjacent but a little stronger.
•paths12, paths21: paths12 is the number of nodes from host.2 in the collapsed tree which have an
ancestor that is from host.1, and paths21 is the opposite.
•nodes1, nodes2: these are the number of collapsed tree nodes for host.1 and host.2 respectively.
•path.classification: this describes the topological relationship between the two hosts in the cablol-
lapsed tree:
–“none” indicates that no nodes from host.1 are ancestral to nodes from host.2 or vice versa
–“anc” indicates that there is only one node from host.2 and it is a descendant of a node from
host.1
–“desc” indicates that there is only one node from host.1 and it is a descendant of a node from
host.2
–“multiAnc” indicates that there is more than one node from host.2 and all are descended from
a node from host.1
24

–“multiDesc” indicates that there is more than one node from host.1 and all are descended from
a node from host.2
–“complex” indicates that none of the above are true (which is suggestive of an ancestral
relationship and often direct transmission, but with unknown direction)
•min.distance.between.subgraphs: this is the smallest patristic distance between a node in a host.1
subgraph and a host.2 subgraph
•normalised.min.distance.between.subgraphs: If the tree branch lengths were normalised, this is the
above distance but divided by the normalisation constant for this tree
The standalone script to classify pairwise host relationships is classify relationships.R.
16 Pairwise relationship summary
The next stage in the script is to gather the pairwise relationships between hosts in each window and
summarise this across the whole run. Pairs are inferred to be related in a tree if they are both adjacent
and (optionally) within the patristic distance threshold specified by the --distanceThreshold option.
If --distanceThreshold is not given then it is assumed to be infinite and adjacency is the only criterion
that will be used to identify related pairs. If tree branch lengths were normalised then the distance
threshold is applied to normalised distances.
By default, two outputs are produced. The first is a .csv file whose first three columns are a pair of
hosts and a classification of the topological relationship between the pair. Rows for pairs which are never
related at all or never show a particular topological relationship are omitted.
The --windowThreshold option is used to specify the minimum proportion of trees in which a pair
of hosts were found to be related to make rows for that pair appear in the output file. The default is 0.5,
meaning that only pairs that are related in at least half of the trees appear. If this is instead set to 0, all
relationships are reported if they ever occur at all.
The .csv file has the following columns:
•host.1, host.2: the two hosts.
•ancestry: the type of ancestry (see below).
•ancestry.tree.count: the number of trees in which this pair of hosts are related (by the definition
given above) and the relationship is of the type given under “ancestry”.
•both.exist: the number of trees in which both hosts are present.
•fraction: the number of windows showing this relationship over the number of windows where both
occur (“count” over “both.exist”), as text.
•any.relationship.tree.count: the number of trees in which this pair of hosts are related (by the
definition given above).
The ancestry relationships are as follows:
•“none”: no node in the collapsed tree from host.1 is descended from a node from host.2 or vice
versa.
•“trans”: there is just one collapsed tree node from host.2 and it is descended from a node from
host.1. Since they must be adjacent, this makes host.1 its immediate ancestor.
•“multi trans”: there is more than one collapsed tree node from host.2 and all are descended from
a node from host.1. This classification only appears if the --allowMultiTrans option is given; if
not these relationships count as “complex”. This is because this relationship is less indicative of
the direction of transmission than “trans”; it may simply indicate that the tips from the two hosts
are very intermingled, and it is prone to bias introduced by differences in tip counts. By default,
the script is conservative and does not take this relationship as a signal of directionality. However,
as with “complex”, it is quite strongly indicative of direct transmission (Romero-Severson et al.
PNAS 2016).
25

•“complex”: where none of the above are true; if --allowMultiTrans is not specified then
“multi trans” relationships are also allocated “complex”.
The standalone script to summarise these relationships is transmission summary.R.
Unless the --skipSummaryGraph option is specified, a PDF file displaying a simplified summary of
between-patient relationships will also be produced. The dimensions of this plot (in inches) can be
controlled with the --summaryPlotDimensions options; the default here is 25 ×25 inches. If the output
plot is too small and cluttered, try using a larger number. The output here will simplify the between-
host relationships as in figure 4B of the phyloscanner paper, such that there is only one link between
any pair, and that link will appear as an arrow if the proportion of windows showing transmission in
that direction is above a threshold, but simply as a line otherwise. Links with arrows are labelled with
two numbers separated by a forward slash: the first is the proportion of trees showing the directional
relationship implied by the arrow, while the second is the proportion of trees showing any relationship at
all. Links without arrows are just labelled with a the proportion showing any relationship. The default
threshold for the display of arrows is 0.33, but this can be overridden with the --directionThreshold
option. Links will only appear at all if a relationship occurs in more that the --windowThreshold option,
and it is required that --directionThreshold be less than or equal to --windowThreshold.
Graph visualisation is a complicated affair, and we cannot ensure that this PDF file will always
display a satisfactory arrangement of nodes and labels. Users wishing to make their own graphs may
use an analysis package such as Cytoscape with the summary .csv file as input. A standalone script,
simplify graph.R, can be used to simplify the information in this file in the manner described above.
See the command line help for that script for more information.
17 Miscellaneous options
Remaining command line options are as follows:
•--verbose: Specifies the level of verbose output. The default is “1”, which gives minimal output
to the command line. “0” gives no output and “2” full output.
•--noProgressBars: if present and --verbose is given, progress bars will not appear (useful when
the output stream is written to file).
•--outputDir: the directory to write output files into; if not specified this is the current working
directory.
•--pdfWidth: the width of the pdf tree file output
•--pdfRelHeight: the height, per tip, of the pdf tree file output
•--pdfScaleBarWidth: The width of the scale bar in the pdf tree file output (in the same units as
the branch lengths in the tree)
•--outputRDA: If present, an R list containing the objects used in the calculations is written as a
workspace image at the end of the script
•--seed: Two parts of the process may use randomisation: the downsampling, and occasional tie-
breaking in parsimony reconstruction.
•--overwrite: This option tells phyloscanner analyse trees.R to overwrite any existing output
file with the same names. This option sets a random number seed. The script will try to identify
this automatically, but this may not work on all systems.
26
References
[1] C. Wymant, M. Hall, O. Ratmann, D. Bonsall, T. Golubchik, M. de Cesare, A. Gall, M. Cornelissen,
C. Fraser, T. M. P. C. STOP-HCV Consortium, and T. B. Collaboration, “Phyloscanner: Infer-
ring transmission from within- and between-host pathogen genetic diversity,” Molecular Biology and
Evolution, vol. 35, no. 3, pp. 719–733, 2018.
[2] H. Li, B. Handsaker, A. Wysoker, T. Fennell, J. Ruan, N. Homer, G. Marth, G. Abecasis, R. Durbin,
and 1000 Genome Project Data Processing Subgroup, “The sequence alignment/map (sam) format
and samtools,” Bioinformatics, 2009.
[3] K. Katoh, K. Misawa, K. Kuma, and T. Miyata, “Mafft: a novel method for rapid multiple sequence
alignment based on fast fourier transform,” Nucleic Acids Research, vol. 30, no. 14, pp. 3059–3066,
2002.
[4] A. Stamatakis, “Raxml version 8: a tool for phylogenetic analysis and post-analysis of large phyloge-
nies,” Bioinformatics, vol. 30, no. 9, pp. 1312–1313, 2014.
[5] G. Yu, D. K. Smith, H. Zhu, Y. Guan, and T. T.-Y. Lam, “ggtree: an r package for visualization and
annotation of phylogenetic trees with their covariates and other associated data,” Methods in Ecology
and Evolution, vol. 8, no. 1, pp. 28–36, 2017.
27
