R User Guide
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 26

AC-PCA: simultaneous dimension reduction and
adjustment for confounding variation
User’s Guide
Zhixiang Lin
October 31, 2016
If you use AC-PCA in published research, please cite:
Z. Lin, C. Yang, Y. Zhu, J. C. Duchi, Y. Fu, Y. Wang, B. Jiang, M. Zamanighomi,
X. Xu, M. Li, N. Sestan, H. Zhao, W. H. Wong:
AC-PCA: simultaneous dimension reduction and adjustment for confounding variation
bioRxiv, http://dx.doi.org/10.1101/040485
Contents
1 Introduction 2
1.1 Scope ................................. 2
1.2 How to get help . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.3 Input for AC-PCA . . . . . . . . . . . . . . . . . . . . . . . . . . 2
2 Theory behind AC-PCA 2
2.1 AC-PCA in a general form . . . . . . . . . . . . . . . . . . . . . . 2
2.2 AC-PCA with sparse loading . . . . . . . . . . . . . . . . . . . . 3
3 Specific experimental designs and case studies 3
3.1 Categorical confounding factors . . . . . . . . . . . . . . . . . . . 3
3.2 Continuous confounding factors . . . . . . . . . . . . . . . . . . . 7
3.3 Experiments with replicates . . . . . . . . . . . . . . . . . . . . . 10
3.4 Application to a human brain exon array dataset . . . . . . . . . 12
3.5 Application to a model organism ENCODE (modENCODE) RNA-
Seqdataset .............................. 16
3.6 AC-PCA with sparsity . . . . . . . . . . . . . . . . . . . . . . . . 23
1
1 Introduction
1.1 Scope
Dimension reduction methods are commonly applied to visualize datapoints in a
lower dimensional space and identify dominant patterns in the data. Confound-
ing variation, technically and biologically originated, may affect the performance
of these methods, and hence the visualization and interpretation of the results.
AC-PCA simultaneously performs dimension reduction and adjustment for con-
founding variation. The intuition of AC-PCA is that it captures the variations
that are invariant to the confounding factors. One major application area for
AC-PCA is the transcriptome data, and it can be implemented to other data
types as well.
1.2 How to get help
This user’s guide addresses many scenarios for confounding factors. Additional
questions about AC-PCA can be sent to linzx06@gmail.com. Comments on
possible improvements are very welcome.
1.3 Input for AC-PCA
AC-PCA requires two inputs: the data matrix Xand the confounder matrix Y.
Xis of dimension n×p, where nis the number of samples and pis the number
of variables (genes). Yis of dimension n×q, where nis the number of samples
and qis the number of confounding factors. Missing data is allowed in both X
and Y. The columns in Xand Yare assumed to be centered.
Confounding factors depend on the experimental design and can also depend
on the scientific question: confounding factors can be different for the same
experiment. As an example, consider a transcriptome experiment where gene
expression levels were measured in multiple tissues from multiple species. If
one wants to capture the variation across tissues that is shared among species,
then the species labels are the confounding factors. In constrast, if the variation
across species but shared among tissues are desirable, the tissue labels are the
confounding factors. AC-PCA is suitable for experiments with complex designs.
In the following sections, we implement AC-PCA and provide details on how
to design Yfor various types of confounders and various experimental designs.
2 Theory behind AC-PCA
2.1 AC-PCA in a general form
Let Xbe the N×pdata matrix and Ybe the N×lmatrix for lconfounding
factors. Let K=Y Y T. We propose the following objective function to adjust
2
for confounding variation:
maximize
v∈RpvTXTXv −λvTXTKXv
subject to ||v||2
2≤1,
(1)
We can choose Yso that vTXTKXv represents the confounding variation in
the projected data. Formula 1 can be generalized to other kernels on Y. In the
R package, we provide options for linear kernel(i.e. Y Y T) and Gaussian kernel.
2.2 AC-PCA with sparse loading
A sparse solution for vcan be achieved by adding `1constraint:
maximize
v∈RpvTXTXv subject to vTXTKXv ≤c1,||v||1≤c2,||v||2
2≤1.(2)
3 Specific experimental designs and case studies
3.1 Categorical confounding factors
Categorical confounding factors are commonly observed in biological data, it
can be technical (different experimental batches, etc.) and biological (donor
labels, different races, species, etc.). we treat samples with the same confounder
label as a group. Here are the group labels for simulated example 1:
> library(acPCA)
> data(data_example1)
> data_example1$group ### the group labels
[1]111111111122222222223333333333
> X <- data_example1$X ### the data matrix
we perform regular PCA and compare the result with the true simulated
pattern:
> pca <- prcomp(X, center=T) ###regular PCA
> par(mfrow=c(1,2), pin=c(2.5,2.5), mar=c(4.1, 3.9, 3.2, 1.1))
> plot(pca$x[,1], pca$x[,2], xlab="PC 1", ylab="PC 2", col="black", type="n", main="PCA")
> text(pca$x[,1], pca$x[,2], labels = data_example1$lab, col=data_example1$group+1)
> plot(data_example1$true_pattern[1,], data_example1$true_pattern[2,],
+ xlab="Factor 1", ylab="Factor 2", col="black", type="n", main="True pattern",
+ xlim=c(min(data_example1$true_pattern[1,])-0.3,
+ max(data_example1$true_pattern[1,])+0.3) )
> text(data_example1$true_pattern[1,], data_example1$true_pattern[2,], labels = 1:10)
3
−20 0 20 40
−20 −10 0 10 20 30 40
PCA
PC 1
PC 2
1
2
3
4
5
6
7
8
9
10 1
2
3
4
5
6
7
8
9
10
1
2
3
4
5
6
7
8
9
10
−1.5 −0.5 0.5 1.5
0.0 0.5 1.0 1.5
True pattern
Factor 1
Factor 2
1
2
3
45
6
7
8
9
10
In the PCA plot, each color represents a group of samples with the same
confounder label. The confounding variation dominates variation of the true
pattern.
To adjust for the confounding variation, the Ymatrix is desiged such that
the penalty term equals the between groups sum of squares:
> Y <- data_example1$Y ### the confounder matrix
> Y
[,1] [,2] [,3]
[1,] 0.6666667 -0.3333333 -0.3333333
[2,] 0.6666667 -0.3333333 -0.3333333
[3,] 0.6666667 -0.3333333 -0.3333333
[4,] 0.6666667 -0.3333333 -0.3333333
[5,] 0.6666667 -0.3333333 -0.3333333
[6,] 0.6666667 -0.3333333 -0.3333333
[7,] 0.6666667 -0.3333333 -0.3333333
[8,] 0.6666667 -0.3333333 -0.3333333
[9,] 0.6666667 -0.3333333 -0.3333333
[10,] 0.6666667 -0.3333333 -0.3333333
[11,] -0.3333333 0.6666667 -0.3333333
[12,] -0.3333333 0.6666667 -0.3333333
4
[13,] -0.3333333 0.6666667 -0.3333333
[14,] -0.3333333 0.6666667 -0.3333333
[15,] -0.3333333 0.6666667 -0.3333333
[16,] -0.3333333 0.6666667 -0.3333333
[17,] -0.3333333 0.6666667 -0.3333333
[18,] -0.3333333 0.6666667 -0.3333333
[19,] -0.3333333 0.6666667 -0.3333333
[20,] -0.3333333 0.6666667 -0.3333333
[21,] -0.3333333 -0.3333333 0.6666667
[22,] -0.3333333 -0.3333333 0.6666667
[23,] -0.3333333 -0.3333333 0.6666667
[24,] -0.3333333 -0.3333333 0.6666667
[25,] -0.3333333 -0.3333333 0.6666667
[26,] -0.3333333 -0.3333333 0.6666667
[27,] -0.3333333 -0.3333333 0.6666667
[28,] -0.3333333 -0.3333333 0.6666667
[29,] -0.3333333 -0.3333333 0.6666667
[30,] -0.3333333 -0.3333333 0.6666667
Here are the results when we implement AC-PCA:
> par(mfrow=c(1,1))
> ### first tune lambda
> result_tune <- acPCAtuneLambda(X=X, Y=Y, nPC=2, lambdas=seq(0, 10, 0.05),
+ anov=T, kernel = "linear", quiet=T)
> ###run with the best lambda
> result <- acPCA(X=X, Y=Y, lambda=result_tune$best_lambda,
+ kernel="linear", nPC=2)
> ###the signs of the PCs are meaningless
> plot(result$Xv[,1], -result$Xv[,2], xlab="PC 1", ylab="PC 2",
+ type="n", main="AC-PCA")
> text(result$Xv[,1], -result$Xv[,2], labels = data_example1$lab,
+ col=data_example1$group+1)
5
−30 −20 −10 0 10 20 30
−10 −5 0 5
AC−PCA
PC 1
PC 2
1
2
3
456
7
8
9
10
1
2
3
4
56
7
8
9
10
1
2
3
4
56
7
8
9
10
We set anov =Tas the penalty term has the ANOVA interpretation. AC-
PCA is able to recover the true latent structure. We can also implement AC-
PCA with Gaussian kernel (bandwidth h= 0.5):
> h <- 0.5
> result_tune <- acPCAtuneLambda(X=X, Y=Y, nPC=2, lambdas=seq(0, 10, 0.05),
+ anov=F, kernel = "gaussian",
+ bandwidth=h, quiet=T)
> result <- acPCA(X=X, Y=Y, lambda=result_tune$best_lambda,
+ kernel="gaussian", bandwidth=h, nPC=2)
> ###the signs of the PCs are meaningless
> plot(result$Xv[,1], -result$Xv[,2], xlab="PC 1", ylab="PC 2",
+ col="black", type="n", main="Gaussian kernel, h=0.5")
> text(result$Xv[,1], -result$Xv[,2], labels = data_example1$lab,
+ col=data_example1$group+1)
6
−30 −20 −10 0 10 20 30
−10 −5 0 5
Gaussian kernel, h=0.5
PC 1
PC 2
1
2
3
456
7
8
9
10
1
2
3
4
56
7
8
9
10
1
2
3
4
56
7
8
9
10
We set anov =Fas the penalty term does not have the ANOVA interpre-
tation. Gaussian kernel also works well. For detailed discussion on bandwidth
selection, please refer to Chapter 3 in [4].
3.2 Continuous confounding factors
Continuous confounding factors may be present in biological data, for example,
age.
Simulated example 2: the confounder is assumed to be continuous and it con-
tributes a global trend to the data.
We first perform regular PCA:
> data(data_example2)
> X <- data_example2$X ###the data matrix
> Y <- data_example2$Y ###the confounder matrix
> Y
[,1]
[1,] 2.5
[2,] -0.5
[3,] -3.5
[4,] 4.5
[5,] 0.5
7
[6,] -4.5
[7,] -1.5
[8,] 3.5
[9,] 1.5
[10,] -2.5
> pca <- prcomp(X, center=T) ###regular PCA
> par(mfrow=c(2,2), pin=c(2.5,2.5), mar=c(4.1, 3.9, 3.2, 1.1))
> plot(pca$x[,1], pca$x[,2], xlab="PC 1", ylab="PC 2", col="black", type="n", main="PCA")
> text(pca$x[,1], pca$x[,2], labels = data_example2$lab)
> plot(data_example2$true_pattern[1,], data_example2$true_pattern[2,],
+ xlab="Factor 1", ylab="Factor 2", col="black", type="n", main="True pattern",
+ xlim=c(min(data_example2$true_pattern[1,])-0.3,
+ max(data_example2$true_pattern[1,])+0.3) )
> text(data_example2$true_pattern[1,], data_example2$true_pattern[2,],
+ labels = data_example2$lab)
> plot(data_example2$confound_pattern[1,], data_example2$confound_pattern[2,],
+ xlab="", ylab="", col="black", type="n", main="Confounding pattern",
+ xlim=c(min(data_example2$confound_pattern[1,])-0.3,
+ max(data_example2$confound_pattern[1,])+0.3) )
> text(data_example2$confound_pattern[1,], data_example2$confound_pattern[2,], labels =
+ paste(data_example2$lab, '(',data_example2$Y, ')', sep=""), cex=0.5 )
−40 −20 0 20 40
−20 0 10 20 30
PCA
PC 1
PC 2
abc
def
g
hij
−1.5 −0.5 0.5 1.5
0.0 0.5 1.0 1.5
True pattern
Factor 1
Factor 2
a
b
c
def
g
h
i
j
−1.5 −0.5 0.5 1.5
0.2 0.4 0.6 0.8
Confounding pattern
a(2.5)
b(−0.5)
c(−3.5)
d(4.5)
e(0.5)
f(−4.5)
g(−1.5)
h(3.5)
i(1.5)
j(−2.5)
8
Next, we implement AC-PCA with linear and Gaussian kernels:
> ### linear kernel
> result_tune <- acPCAtuneLambda(X=X, Y=Y, nPC=2, lambdas=seq(0, 10, 0.05),
+ anov=F, kernel = "linear", quiet=T)
> result <- acPCA(X=X, Y=Y, lambda=result_tune$best_lambda,
+ kernel="linear", nPC=2)
> par(mfrow=c(1,2), pin=c(2.5,2.5), mar=c(4.1, 3.9, 3.2, 1.1))
> plot(result$Xv[,1], result$Xv[,2], xlab="PC 1", ylab="PC 2",
+ col="black", type="n", main="Linear kernel")
> text(result$Xv[,1], result$Xv[,2], labels = data_example2$lab)
> ### Gaussian kernel
> result_tune <- acPCAtuneLambda(X=X, Y=Y, nPC=2, lambdas=seq(0, 10, 0.05),
+ anov=F, kernel = "gaussian",
+ bandwidth=mean(abs(Y)), quiet=T)
> result <- acPCA(X=X, Y=Y, lambda=result_tune$best_lambda,
+ kernel = "gaussian", bandwidth=mean(abs(Y)), nPC=2)
> plot(result$Xv[,1], result$Xv[,2], xlab="PC 1", ylab="PC 2",
+ col="black", type="n", main=paste("Gaussian kernel, h=", mean(abs(Y)), sep="") )
> text(result$Xv[,1], result$Xv[,2], labels = data_example2$lab)
−20 0 10 20 30
−10 −5 0 5
Linear kernel
PC 1
PC 2
a
b
c
d
ef
g
h
i
j
−30 −10 0 10 20
−5 0 5 10
Gaussian kernel, h=2.5
PC 1
PC 2
a
b
c
d
e
f
g
h
i
j
The results of linear and Gaussian kernels are comparable.
9

3.3 Experiments with replicates
Consider an experiment where the gene expression levels are measured under
multiple biological conditions with several replicates. It may be desirable to
capture the variation across biological conditions but shared among replicates.
Simulated example 3: there are 10 biological conditions, each with n= 3 repli-
cates. The variation is shared among replicates for half of the genes and not
shared for the other genes. Suppose we want to capture the variation shared
among replicates, the confounder matrix can be chosen such that samples within
the same biological condition are “pushed” together:
> data(data_example3)
> X <- data_example3$X ### the data matrix
> data_example3$lab ### the biological conditions
[1]1112223334445556667778889
[26] 9 9 10 10 10
> Y <- data_example3$Y ### the confounder matrix
> dim(Y)
[1] 30 30
> Y[,1]
[1] 0.5773503 -0.5773503 0.0000000 0.0000000 0.0000000 0.0000000
[7] 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
[13] 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
[19] 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
[25] 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
In each column of Y, there is one 1/√nand one −1/√ncorresponding to a
pair of samples from the same biological condition. So the number of columns
in Yis 10 ×3×2/2. For a biological condition, treating the 3 replicates as
3 groups, it can be shown that the penalty term vTXTY Y TXv equals the
summation of the between groups sum of squares over the biological conditions.
When the number of replicates across the biological conditions is not the same, to
maintain the ANOVA interpretation, we can change the corresponding entries in
Yto 1/√niand 1/√ni, where niis the number of replicates in the ith biological
condition. We first implement PCA and compare it with the true pattern:
> pca <- prcomp(X, center=T) ###regular PCA
> par(mfrow=c(1,2), pin=c(2.5,2.5), mar=c(4.1, 3.9, 3.2, 1.1))
> plot(pca$x[,1], pca$x[,2], xlab="PC 1", ylab="PC 2", col="black", type="n", main="PCA")
> text(pca$x[,1], pca$x[,2], labels = data_example3$lab)
> plot(data_example3$true_pattern[1,], data_example3$true_pattern[2,],
+ xlab="Factor 1", ylab="Factor 2", col="black", type="n", main="True pattern",
+ xlim=c(min(data_example3$true_pattern[1,])-0.3,
10
+ max(data_example3$true_pattern[1,])+0.3) )
> text(data_example3$true_pattern[1,], data_example3$true_pattern[2,],
+ labels = 1:10)
−40 0 20 40 60
−40 −30 −20 −10 0 10 20 30
PCA
PC 1
PC 2
1
1
1
2
2
2
3
3
3
4
4
4
5
5
5
66
6
7
7
7
8
8
89
9
9
10
10
10
−1.5 −0.5 0.5 1.5
0.0 0.5 1.0 1.5
True pattern
Factor 1
Factor 2
1
2
3
45
6
7
8
9
10
Next we implement AC-PCA:
> result_tune <- acPCAtuneLambda(X=X, Y=Y, nPC=2, lambdas=seq(0, 10, 0.05),
+ anov=T, kernel = "linear", quiet=T)
> result <- acPCA(X=X, Y=Y, lambda=result_tune$best_lambda,
+ kernel="linear", nPC=2)
> par(mfrow=c(1,1))
> plot(-result$Xv[,1], result$Xv[,2], xlab="PC 1", ylab="PC 2",
+ col="black", type="n", main="Linear kernel")
> text(-result$Xv[,1], result$Xv[,2], labels = data_example3$lab)
11
−20 −10 0 10 20
−10 −5 0 5
Linear kernel
PC 1
PC 2
1
1
1
2
2
2
3
3
3
4
4
4
5
5
56
6
6
7
7
7
8
8
8
9
9
9
10
10
10
3.4 Application to a human brain exon array dataset
We analyze the human brain exon array dataset [3]. The dataset was down-
loaded from the Gene Expression Omnibus (GEO) database under the acces-
sion number GSE25219. The dataset was generated from 1,340 tissue samples
collected from 57 developing and adult post-mortem brains. In the dataset,
gene expression levels were measured in different brain regions in multiple time
windows. We use a subset of 1,000 genes for demonstration purpose. For time
window 5, we first perform PCA to visualize the brain regions:
> data(data_brain_w5)
> X <- data_brain_w5$X;
> table(data_brain_w5$hemispheres) ###1: left hemisphere, 3: right hemisphere
1 3
45 29
> pca <- prcomp(X, center=T) ###regular PCA
> plot(pca$x[,1], pca$x[,2], xlab="PC 1", ylab="PC 2",
+ col="black", type="n", main="PCA, window 5, brain data")
> text(pca$x[,1], pca$x[,2], labels = data_brain_w5$regions,
+ col=data_brain_w5$donor_labs, font=data_brain_w5$hemispheres)
12
−20 −10 0 10
−10 −5 0 5 10
PCA, window 5, brain data
PC 1
PC 2
MFC
VFC
DFC
STC
ITC
A1C
IPC
S1C
M1C MFC
OFC
VFC
DFC STC
ITC
A1C
IPC
S1C
M1C
MFC
OFC VFC
DFC
STC
ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC
ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC
ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC
ITC
IPC S1C
MFC
OFC
VFC
DFC
STC
ITC
A1C
IPC
S1C M1C
VFC
STC
ITC
A1C
IPC
S1CM1C
In window 5, samples from the same donor tend to form a cluster. To adjust
for the donor’s effect, we selected Yso that the penalty term vTXTY Y TXv
equals the donor to donor variation in the projected data.
> Y <- data_brain_w5$Y
We first tune λ:
> result_tune <- acPCAtuneLambda(X=X, Y=Y, nPC=2, lambdas=seq(0, 20, 0.1),
+ anov=T, kernel = "linear", quiet=T)
> result_tune$best_lambda
[1] 2.9
We set anov =Tas the penalty term has the ANOVA interpretation. Next
we use the tuned λ:
> result1 <- acPCA(X=X, Y=Y, lambda=result_tune$best_lambda,
+ kernel="linear", nPC=2)
> Xv1 <- result1$Xv
> plot(Xv1[,1], Xv1[,2], xlab="PC 1", ylab="PC 2",
+ col="black", type="n", main=paste("AC-PCA, window 5, lambda=",
+ result_tune$best_lambda, sep="") )
> text(Xv1[,1], Xv1[,2], labels = data_brain_w5$regions,
+ col=data_brain_w5$donor_labs, font=data_brain_w5$hemispheres)
13
−4 −2 0 2 4
−3 −2 −1 0 1 2 3 4
AC−PCA, window 5, lambda=2.9
PC 1
PC 2
MFC
VFC
DFC
STC ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC
ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC DFC
STC ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC ITC
IPC
S1C
MFC
OFC
VFC
DFC
STC ITC
A1C
IPC
S1C
M1C
VFC
STC ITC
A1C
IPC
S1C
M1C
In the manuscript, we used λ= 5:
> result2 <- acPCA(X=X, Y=Y, lambda=5, kernel="linear", nPC=2)
> Xv2 <- result2$Xv
> plot(-Xv2[,1], Xv2[,2], xlab="PC 1", ylab="PC 2",
+ col="black", type="n", main="AC-PCA, window 5, lambda=5")
> text(-Xv2[,1], Xv2[,2], labels = data_brain_w5$regions,
+ col=data_brain_w5$donor_labs, font=data_brain_w5$hemispheres)
14
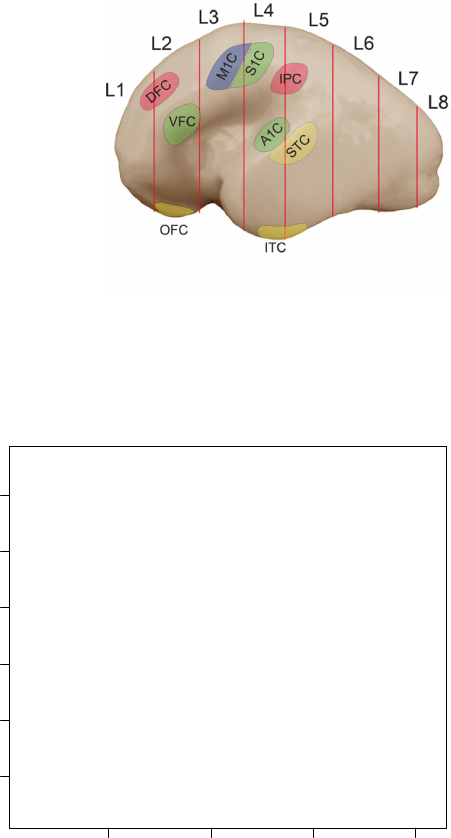
Figure 1: Representative fetal human brain, lateral surface of hemisphere [3]
−2 0 2 4
−2 −1 0 1 2 3
AC−PCA, window 5, lambda=5
PC 1
PC 2
MFC
VFC
DFC
STC ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC DFC
STC ITC
A1C
IPC
S1C
M1C
MFC
OFC
VFC
DFC
STC ITC
IPC
S1C
MFC
OFC
VFC
DFC
STC ITC
A1C
IPC
S1C
M1C VFC
STC ITC
A1C
IPC
S1C
M1C
The overall patterns for the two λs are similar. The pattern is in consistent
with the anatomical structure of neocortex (Figure 1).
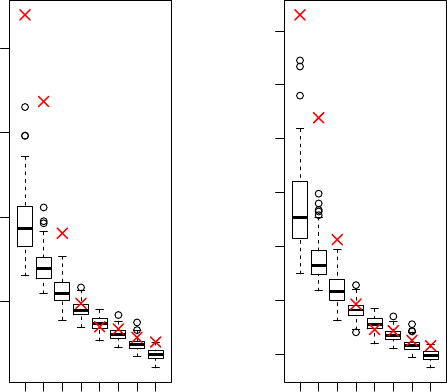
We can use permutation to evaluate the significance of the first 8 PCs:
> result <- acPCA(X=X, Y=Y, lambda=5, kernel="linear", nPC=8, eval=T)
> result$sigPC
[1] 3
15
1 3 5 7
100 150 200 250
PC
Eigenvalue
1 3 5 7
1.0 1.5 2.0 2.5 3.0 3.5 4.0
PC
Variance
Data: red cross; Permutation: boxplot
The first 3 PCs are significant.
3.5 Application to a model organism ENCODE (modEN-
CODE) RNA-Seq dataset
The modENCODE project generates the transcriptional landscapes for model
organisms during development[1, 2]. In the analysis, we used the time-course
RNA-Seq data for fly and worm embryonic development. The modENCODE
RNA-Seq dataset was downloaded from https://www.encodeproject.org/comparative/transcriptome/.
For the orthologs in fly that map to multiple orthologs in worm, we took median
to get a one to one match, resulting in 4831 ortholog paris. We first perform
some exploratory analysis:
> library(acPCA)
> data(data_fly_worm)
> data_fly <- data_fly_worm$data_fly ###the data matrix for fly
> data_worm <- data_fly_worm$data_worm ###the data matrix for fly
> dim(data_fly)
[1] 12 4831
> dim(data_worm)
[1] 24 4831
16
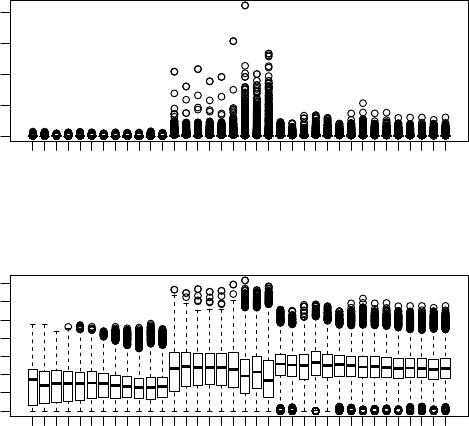
> par(mfrow=c(2,1), pin=c(2.5,2.5), mar=c(4.1, 3.9, 3.2, 1.1))
> boxplot(t(rbind(data_fly, data_worm)),las = 2, names =
+ c(rep('fly', 12), rep('worm', 24)), ylab="expression level")
> boxplot(log2(t(rbind(data_fly, data_worm))+1),las = 2,
+ names = c(rep('fly', 12), rep('worm', 24)), ylab="log2(expression+1)")
fly
fly
fly
fly
fly
fly
fly
fly
fly
fly
fly
fly
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
0
5000
10000
15000
20000
expression level
fly
fly
fly
fly
fly
fly
fly
fly
fly
fly
fly
fly
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
worm
0
2
4
6
8
10
12
14
log2(expression+1)
We can clearly see that the expression levels in worm samples tend to be
higher. Next we perform PCA on fly and worm separately on the log2 scale:
> pca_fly <- prcomp(log2(data_fly+1), center=T)
> pca_worm <- prcomp(log2(data_worm+1), center=T)
> par(mfrow=c(1,1), pin=c(3, 3))
> plot(pca_fly$x[,1], -pca_fly$x[,2], xlab="PC 1", ylab="PC 2",
+ col="red", type="n", main="fly PCA",
+ xlim=c(-(max(abs(pca_fly$x[,1]))+13), max(abs(pca_fly$x[,1]))+13),
+ ylim=c(-(max(abs(pca_fly$x[,2]))+13), max(abs(pca_fly$x[,2]))+13))
> text(pca_fly$x[,1], -pca_fly$x[,2], labels =
+ data_fly_worm$fly_time, col="red")
17

−60 −20 0 20 40 60
−60 −40 −20 0 20 40 60
fly PCA
PC 1
PC 2
0−2
2−4
4−6
6−8
8−10
10−12
12−14
14−16
16−18
18−20
20−22
22−24
> par(mfrow=c(1,1), pin=c(3, 3))
> plot(pca_worm$x[,1], pca_worm$x[,2], xlab="PC 1", ylab="PC 2",
+ col="red", type="n", main="worm PCA",
+ xlim=c(-(max(abs(pca_worm$x[,1]))+13), max(abs(pca_worm$x[,1]))+13),
+ ylim=c(-(max(abs(pca_worm$x[,2]))+13), max(abs(pca_worm$x[,2]))+13))
> text(pca_worm$x[,1], pca_worm$x[,2], labels =
+ data_fly_worm$worm_time, col="blue")
18

−100 −50 0 50 100
−50 0 50
worm PCA
PC 1
PC 2
0
0.5
11.5
2
2.5
33.5
4
55.5
66.5
77.5
8
8.5
9
9.5
10
10.5
11
11.5
12
In the PCA plots, each data point represents a time window (fly) or a time
point (worm), in the unit of hours(h). For fly, there is a smooth temporal
pattern. For worm, there seems to be three clusters, which may originate from
the dominated effect of a small subset of highly expressed genes. To adjust for
the highly expressed genes, we used the rank across samples within the same
species. The rank matrix was then scaled to have unit variance. Another benefit
of using rank is that we simultaneously make adjustment for the observation that
worm genes tend to have higher expression levels. For ties in the expression
level, we used ’average’ option in the R’s rank function. Here is PCA on the
rank matrix:
> X <- data_fly_worm$X ###this is the scaled rank matrix. Fly and worm are combined.
> pca_fly_rank <- prcomp(X[1:12,], center=T)
> pca_worm_rank <- prcomp(X[13:36,], center=T)
> par(mfrow=c(1,1), pin=c(3, 3))
> plot(pca_fly_rank$x[,1], -pca_fly_rank$x[,2], xlab="PC 1", ylab="PC 2",
+ col="red", type="n", main="fly PCA, rank",
+ xlim=c(-(max(abs(pca_fly_rank$x[,1]))+13), max(abs(pca_fly_rank$x[,1]))+13),
+ ylim=c(-(max(abs(pca_fly_rank$x[,2]))+13), max(abs(pca_fly_rank$x[,2]))+13))
> text(pca_fly_rank$x[,1], -pca_fly_rank$x[,2], labels =
+ data_fly_worm$fly_time, col="red")
19
−50 0 50
−40 −20 0 20 40
fly PCA, rank
PC 1
PC 2
0−2
2−4
4−6
6−8
8−10
10−12
12−14
14−16
16−18
18−20
20−22
22−24
> par(mfrow=c(1,1), pin=c(3, 3))
> plot(pca_worm_rank$x[,1], pca_worm_rank$x[,2], xlab="PC 1", ylab="PC 2",
+ col="red", type="n", main="worm PCA, rank",
+ xlim=c(-(max(abs(pca_worm_rank$x[,1]))+13), max(abs(pca_worm_rank$x[,1]))+13),
+ ylim=c(-(max(abs(pca_worm_rank$x[,2]))+13), max(abs(pca_worm_rank$x[,2]))+13))
> text(pca_worm_rank$x[,1], pca_worm_rank$x[,2], labels =
+ data_fly_worm$worm_time, col="blue")
20
−50 0 50
−50 0 50
worm PCA, rank
PC 1
PC 2
0
0.5
11.5
22.5
3
3.5
4
55.5
6
6.5
77.5
8
8.5
99.5
10
10.5
11
11.5
12
Using rank gives a better visualization, as the data points are more spread
out. Next we implement PCA on fly and worm jointly, using the rank matrix:
> pca_rank <- prcomp(X, center=T)
> par(mfrow=c(1,1), pin=c(3, 3))
> plot(pca_rank$x[,1], pca_rank$x[,2], xlab="PC 1", ylab="PC 2",
+ col="red", type="n", main="fly+worm, PCA, rank",
+ xlim=c(-(max(abs(pca_rank$x[,1]))+13), max(abs(pca_rank$x[,1]))+13),
+ ylim=c(-(max(abs(pca_rank$x[,2]))+13), max(abs(pca_rank$x[,2]))+13))
> text(pca_rank$x[,1], pca_rank$x[,2],
+ labels=data_fly_worm$X_time,
+ col=c(rep("red", 12), rep("blue", 24)) )
> legend("topright", legend = c("fly", "worm"), lty=1,
+ col=c("red","blue"),lwd=1.5,seg.len=1)
21
−50 0 50
−60 −20 0 20 40 60
fly+worm, PCA, rank
PC 1
PC 2
0−2
2−4
4−6
6−8
8−10
10−12
12−14
14−16
16−18
18−20
20−22
22−24
0
0.5
11.5
22.5
3
3.5
4
55.5
6
6.5
77.5
8
8.5
99.5
10
10.5
11
11.5
12
fly
worm
The variation of species confounds the PCA result, as we observed that
samples from the same species tend to be close.
Next we implement AC-PCA. Without prior knowledge on the alignment
of the developmental stages in fly and worm, we shrink the data points in fly
towards the mean of the data points in worm: for example, time window 0-2h
in fly is shrunk to the mean of time points 0h, 0.5h and 1h in worm, as the
embryonic stage for fly is 24h, and for worm it is 12h. Time window 8-10h in
fly is shrunk to the mean of 4h and 5h in worm, as the worm sample 4.5h is
missing.
> Y <- data_fly_worm$Y
> Y[,1]
[1] 1.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
[7] 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
[13] -0.3333333 -0.3333333 -0.3333333 0.0000000 0.0000000 0.0000000
[19] 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
[25] 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
[31] 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000
> result_tune <- acPCAtuneLambda(X=X, Y=Y, nPC=2, lambdas=seq(0, 10, 0.05),
+ anov=F, kernel = "linear", quiet=T)
> result <- acPCA(X=X, Y=Y, lambda=result_tune$best_lambda,
22
+ kernel="linear", nPC=2)
> par(mfrow=c(1,1), pin=c(3, 3))
> plot(-result$Xv[,1], result$Xv[,2], xlab="PC 1", ylab="PC 2",
+ col="red", type="n", main="fly+worm, AC-PCA, rank",
+ xlim=c(-(max(abs(result$Xv[,1]))+13), max(abs(result$Xv[,1]))+13),
+ ylim=c(-(max(abs(result$Xv[,2]))+13), max(abs(result$Xv[,2]))+13))
> text(-result$Xv[,1], result$Xv[,2],
+ labels=data_fly_worm$X_time,
+ col=c(rep("red", 12), rep("blue", 24)) )
> legend("topright", legend = c("fly", "worm"),
+ lty=1, col=c("red","blue"),lwd=1.5,seg.len=1)
−60 −20 0 20 40 60
−60 −40 −20 0 20 40 60
fly+worm, AC−PCA, rank
PC 1
PC 2
0−2
2−4
4−6
6−8
8−10
10−12
12−14
14−16
16−18
18−20
20−22
22−24
0
0.5
11.5
2
2.5
3
3.5
4
5
5.5
6
6.5
77.5
8
8.5
99.5
10
10.5
11
11.5
12
fly
worm
We set anov =Fas the penalty term does not have the ANOVA inter-
pretation. The variation of different species is adjusted. The genes with top
loadings in AC-PCA tend to have consistent temporal pattern in fly and worm
(see manuscript).
3.6 AC-PCA with sparsity
For AC-PCA with sparsity constrains, there are two tuning parameters, c1and
c2:c1controls the strength of the penalty, and c2controls the sparsity of v. The
parameters are tuned sequentially: c1is tuned without the sparsity constrain,
23
and then c2is tuned with c1fixed. We implement the procedure on the brain
exon array data, time window 2:
> set.seed(10)
> data(data_brain_w2)
> X <- data_brain_w2$X ###the data matrix
> Y <- data_brain_w2$Y
> result1cv <- acPCAtuneLambda(X=X, Y=Y, nPC=2, lambdas=seq(0, 20, 0.1),
+ anov=T, kernel = "linear", quiet=T)
> result1 <- acPCA(X=X, Y=Y, lambda=result1cv$best_lambda, kernel="linear", nPC=2)
> v_ini <- as.matrix(result1$v[,1])
> par(mfrow=c(1,2), pin=c(2.5,2.5), mar=c(4.1, 3.9, 3.2, 1.1))
> c2s <- seq(1, 0, -0.1)*sum(abs(v_ini))
> resultcv_spc1_coarse <- acSPCcv( X=X, Y=Y, c2s=c2s, v_ini=v_ini,
+ kernel="linear", quiet=T, fold=10)
> c2s <- seq(0.9, 0.7, -0.02)*sum(abs(v_ini))
> resultcv_spc1_fine <- acSPCcv( X=X, Y=Y, c2s=c2s, v_ini=v_ini,
+ kernel="linear", quiet=T, fold=10)
> result_spc1 <- acSPC( X=X, Y=Y, c2=resultcv_spc1_fine$best_c2,
+ v_ini=v_ini, kernel="linear")
> v1 <- result_spc1$v
> sum(v1!=0)
[1] 736
24
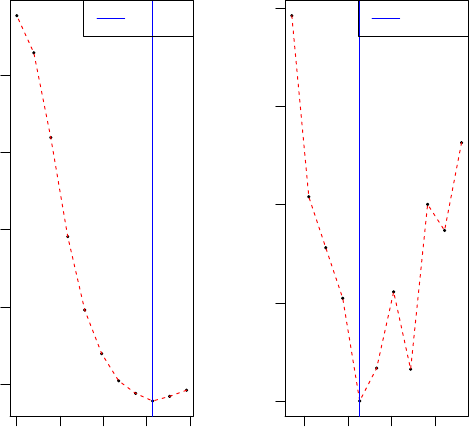
0 5 10 15 20
2650 2700 2750 2800 2850
Cross−validation
Sparsity parameter c2
mean squared error(MSE)
Best c2
14 15 16 17
2641 2642 2643 2644 2645
Cross−validation
Sparsity parameter c2
mean squared error(MSE)
Best c2
To speed-up the computational time, a coarse search for c2was performed
first, followed by a finer grid.
Multiple sparse principal components can be obtained by substracting the
first several principal components, and then implement the algorithm:
> v_substract <- as.matrix(result1$v[,1])
> v_ini <- as.matrix(result1$v[,2])
> par(mfrow=c(1,2), pin=c(2.5,2.5), mar=c(4.1, 3.9, 3.2, 1.1))
> c2s <- seq(1, 0, -0.1)*sum(abs(v_ini))
> resultcv_spc2_coarse <- acSPCcv(X=X, Y=Y, c2s=c2s, v_ini=v_ini,
+ v_substract=v_substract, kernel="linear",
+ quiet=T, fold=10)
> c2s <- seq(0.5, 0.2, -0.02)*sum(abs(v_ini))
> resultcv_spc2_fine <- acSPCcv(X=X, Y=Y, c2s=c2s, v_ini=v_ini,
+ v_substract=v_substract, kernel="linear",
+ quiet=T, fold=10)
> result_spc2 <- acSPC(X=X, Y=Y, c2=resultcv_spc2_fine$best_c2,
+ v_ini=v_ini, v_substract=v_substract, kernel="linear")
> v2 <- result_spc2$v
> sum(v2!=0)
[1] 68
25
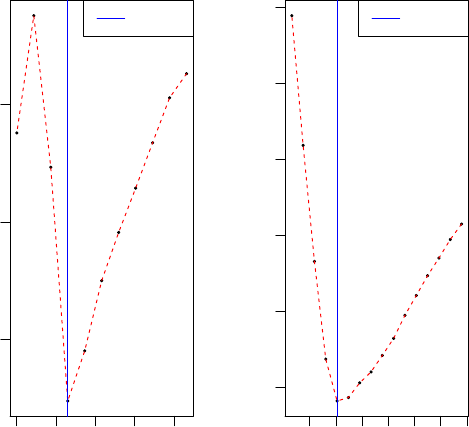
0 5 10 15 20
2880 2885 2890
Cross−validation
Sparsity parameter c2
mean squared error(MSE)
Best c2
5 6 7 8 9 10
2874 2876 2878 2880 2882 2884
Cross−validation
Sparsity parameter c2
mean squared error(MSE)
Best c2
In the example, the non-sparse first PC was substracted. We can also sub-
stract the sparse PC:
> v_substract <- v1
References
[1] Susan E Celniker, Laura AL Dillon, Mark B Gerstein, Kristin C Gunsalus,
Steven Henikoff, Gary H Karpen, Manolis Kellis, Eric C Lai, Jason D Lieb,
David M MacAlpine, et al. Unlocking the secrets of the genome. Nature,
459(7249):927–930, 2009.
[2] Mark B Gerstein, Joel Rozowsky, Koon-Kiu Yan, Daifeng Wang, Chao
Cheng, James B Brown, Carrie A Davis, LaDeana Hillier, Cristina Sisu,
Jingyi Jessica Li, et al. Comparative analysis of the transcriptome across
distant species. Nature, 512(7515):445–448, 2014.
[3] Hyo Jung Kang, Yuka Imamura Kawasawa, Feng Cheng, Ying Zhu, Xuming
Xu, Mingfeng Li, Andr´e MM Sousa, Mihovil Pletikos, Kyle A Meyer, Goran
Sedmak, et al. Spatio-temporal transcriptome of the human brain. Nature,
478(7370):483–489, 2011.
[4] Matt P Wand and M Chris Jones. Kernel smoothing. Crc Press, 1994.
26
