SAT Mix Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 7

SAT_mix manual
SAT_mix = SNPhylo + Admixture + Treemix
Original script of SAT_mix is SNPhylo’s which was customized, and modified for PAPGI study by
JaeJin Choi, KOBIC 2014
Purpose: Integrate three different methods and provide “Big picture”
SNPhylo + Admixture + Treemix
Requirements/Pre-installation
1. Interpreter(compiler): R, Python, Perl
2. External program: MUSCLE, DNAML, Admixture, Treemix, Plink, (SNPhylo)
Run ./setup.sh for configuration
Input file formats: VCF, Hapmap, PED, GDS, simple SNP file; Contain AGCT, not integer
Primary parameters:
Linkage Disequilibrium(LD)
Minor Allele Frequency(MAF)
MISS, PNSS – recommend to set = 0
Function specific parameters
1. SNPhylo
Prefixed; Support 3 options based on the length of SNP sequence
2. Admixture
Prefixed; ancestor k = 2 ~ 7
3. Treemix
-t group index
-R number of migration
-r root (is in group index)
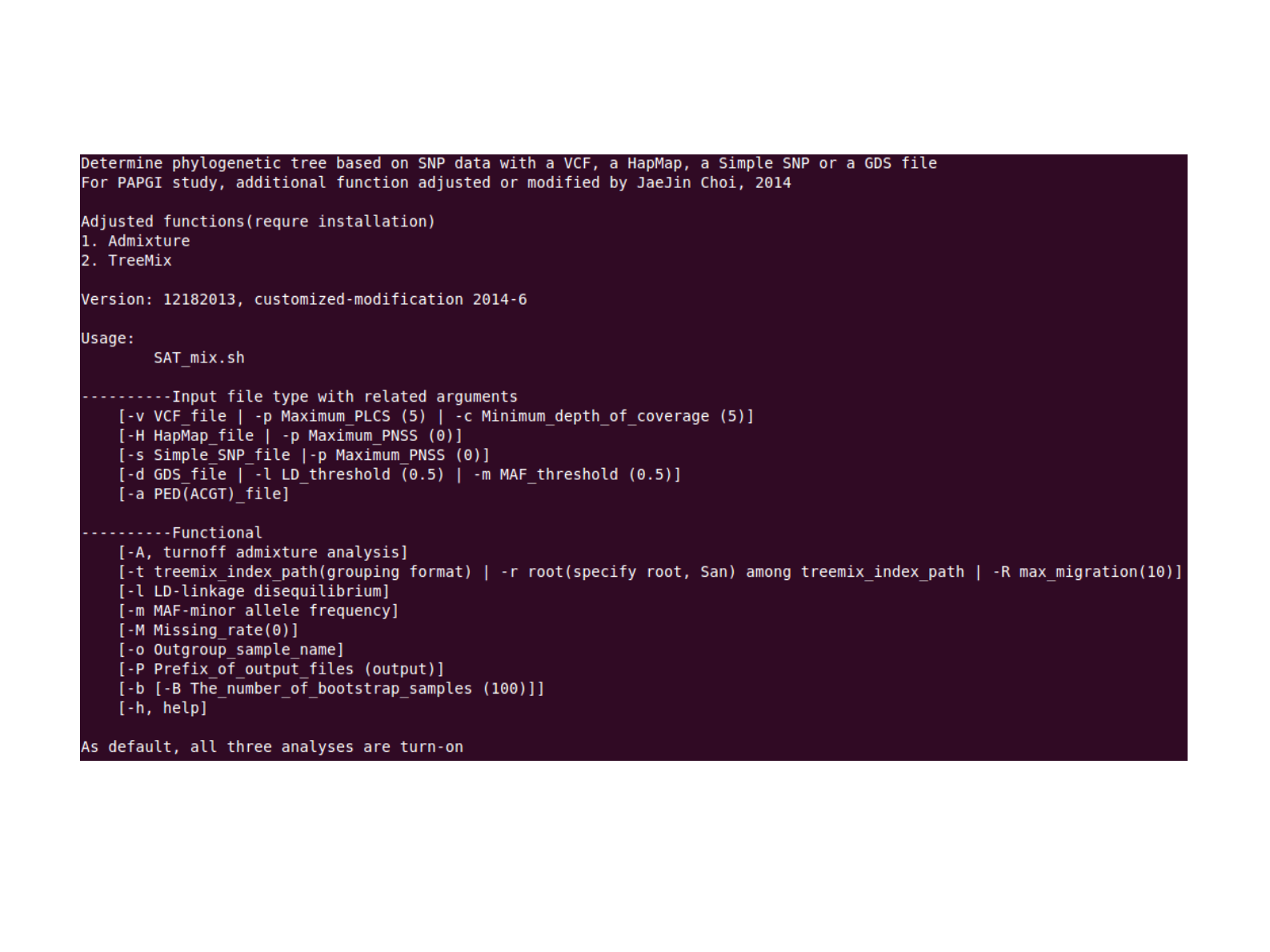
SAT_mix manual
For more detail; -h for help
Original script is “SNPhylo”
Any file path should be direct in absolute path(full length path)
Example; sh [root of]/SAT_mix.sh -l 0.05 -m 0.01 -p 0 -M 0 -P [root of]/out -b -H [root of]/any.hapmap -t [root of]/group_index -R 10 -r San
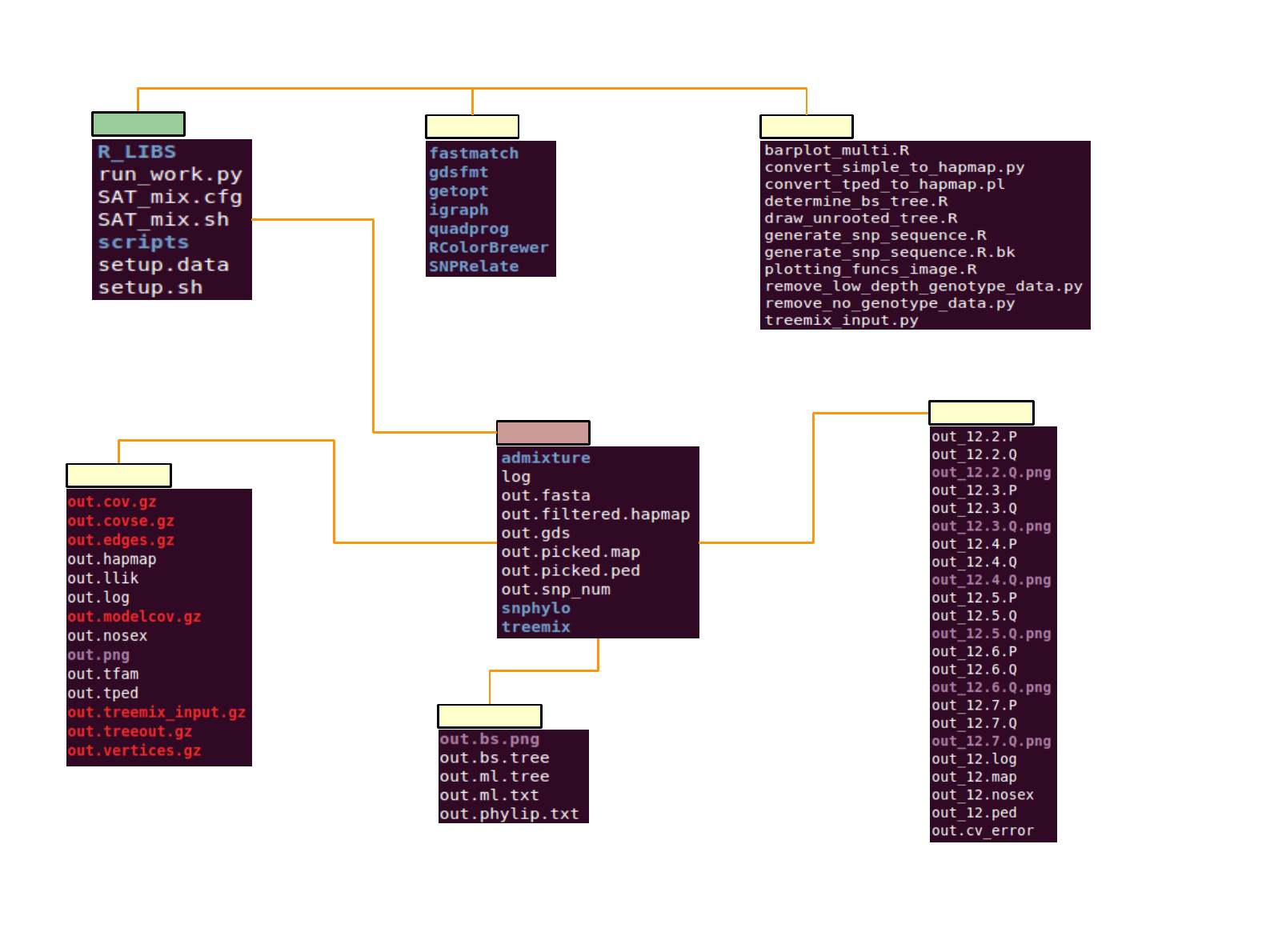
SAT_mix file structure
All scripts acquired for process, include visualization
If necessary,
any R library will be stored here
Recommend to use
independent output folder
scriptsR_LIBS
output
admixture
snphylo
treemix
Treemix acquire gzipped(*.gz) input file
out.png is image file of result
Take [output]/out.fasta as a input
1. out.bs.tree in newick format
2. out.bs.png is image file of out.bs.tree
Run k = 2 ~7
Determine optimum ‘k’ from out.cv_error, which
have smallest CV error rate
Main
SAT_mix file; how script run
output
Assume run;
sh [root of]/SAT_mix.sh -l 0.05 -m 0.05 -p 0 -M 0 -P [root of]/out -b -H [root pf]/any.hapmap -t [root of]/group_index -R 10 -r San > log
LD | -l = 0.05
MAF | -m = 0.05
MISS | -M , and PNSS | -p = 0
-t [group_index]
Root | -R = ‘San’
Maximum migration event | -r = 10
157 Individuals
Start to remove low quality data.
23669 low quality lines were removed
Start HapMap2GDS ...
Scanning ...
file: [root of]/l0.05-m0.05/out.filtered.hapmap
content: 135018 rows x 168 columns
Wed Jun 25 23:08:02 2014 store sample id, snp id, position, and chromosome.
start writing: 157 samples, 135017 SNPs ...
file: [root of]/l0.05-m0.05/out.filtered.hapmap
Wed Jun 25 23:16:08 2014 Done.
Finally picked; 5348 SNPs
--admixture start
Prepare Admixture...
Obtain; [root pof/l0.05-m0.05/admixture/out_12.ped(map), --recode12
Admixture analysis proceed...
(k = 2 ~ 7)
--admixture done
TreeMix analysis proceed...
/San
--treemix start
(obtain treemix input file by several conversion)
--treemix done
--snphylo start
MSA proceed using 5348 SNPs
BS tree draw proceed
Adding species:
1. M_39
2. M_40
3. M_69
.
.
.
157. M_15
Output written to file "outfile"
Tree also written onto file "outtree"
Done.
--snphylo done
!End without notable errors
Remove no genotype SNPs
(low quality, and missing)
After LD, MAF, and MISS filtration,
we obtain 5348 SNPs
Treemix
Output; out.png → ML tree image with n
migration events in arrow
Admixture
‘K’ = 2 ~ 7, prefixed
Output; out_12.’K’.Q.png.
SNPhylo
Output;
1. out.bs.tree → ML tree with bootstrap
support in newick format
2. out.bs.png → image file of out.bs.tree
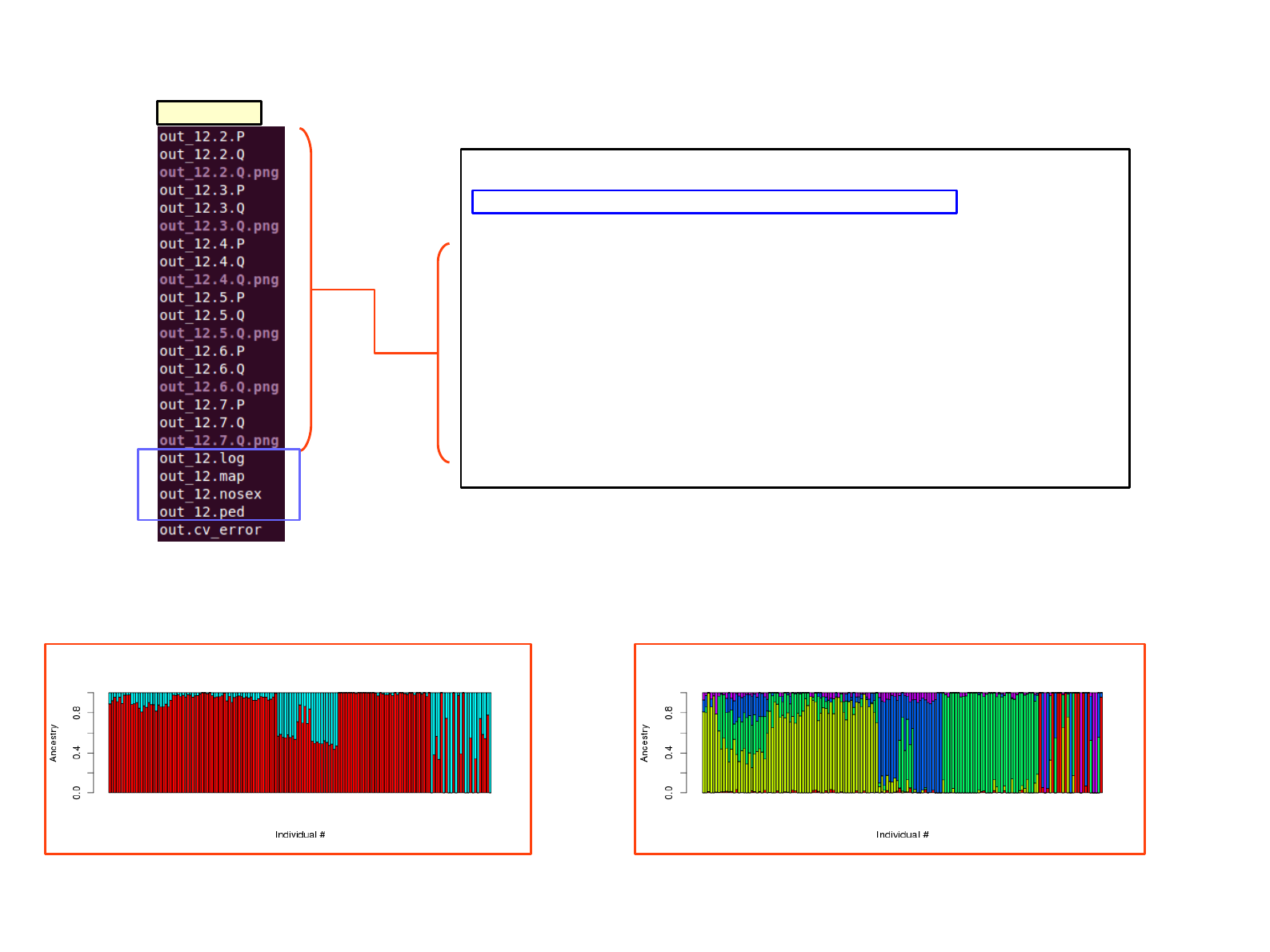
SAT_mix output; admixture
admixture
--admixture start
Prepare Admixture...
Obtain;[root of]/l0.05-m0.05/admixture/out_12.ped(map), --recode12
Admixture analysis proceed...
1- tree k=2
2- obtain figure [root of]/l0.05-m0.05/admixture/out_12.2.Q.png
1- tree k=3
2- obtain figure [root of]/l0.05-m0.05/admixture/out_12.3.Q.png
1- tree k=4
2- obtain figure [root of]/l0.05-m0.05/admixture/out_12.4.Q.png
1- tree k=5
2- obtain figure [root pf]/l0.05-m0.05/admixture/out_12.5.Q.png
1- tree k=6
2- obtain figure [root of]/l0.05-m0.05/admixture/out_12.6.Q.png
1- tree k=7
2- obtain figure [root pf]/l0.05-m0.05/admixture/out_12.7.Q.png
--admixture done
out_12.2.Q.png out_12.5.Q.png
…... ….K=7
out.cv_error; K with smallest CV is optimal suggested from ‘admixture’
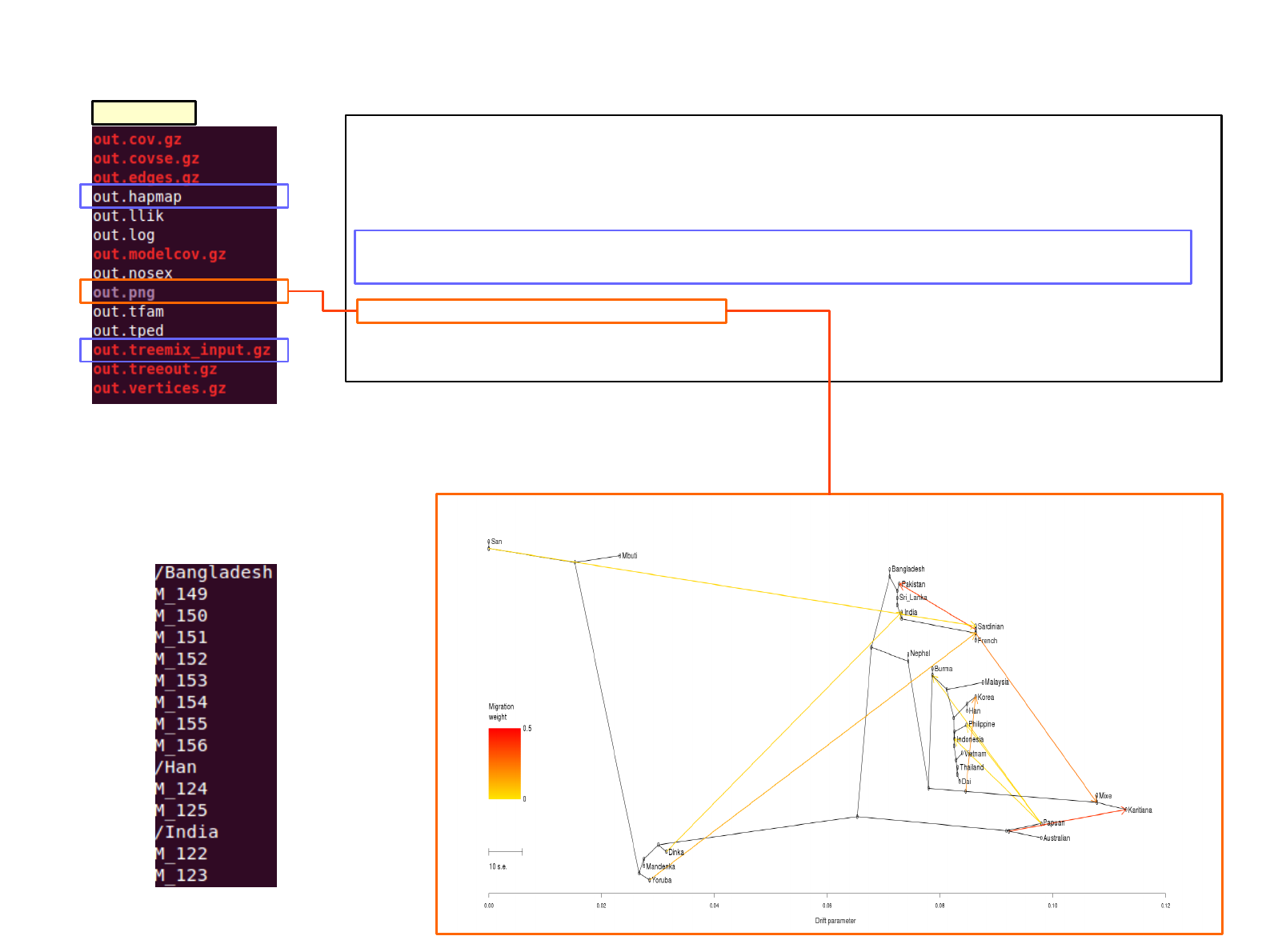
SAT_mix output; treemix
treemix
group_index in file use with argument ‘-t’
In this case, grouping is based on individual’s nationality
/[name of group] #’/’ at the front!
..
… [name of individual]
TreeMix analysis proceed...
/San
--treemix start
Prepare TreeMix...
Convert [root of]/l0.05-m0.05/out.picked.ped(map) -> [root of]/l0.05-m0.05/treemix/out.hapmap
Obtain;[root of]/l0.05-m0.05/treemix/out.hapmap
1- convert hapmap -> treemix input format
2- gzip compress [root of]/l0.05-m0.05/treemix/out.treemix_input -> [root of]/l0.05-m0.05/treemix/out.treemix_input.gz
3- run treemix, -m 10 -root San
4- obtain figure[root of]/l0.05-m0.05/treemix/out.png
--treemix done

SAT_mix output; snphylo
--snphylo start
MSA proceed using 5348 SNPs
BS tree draw proceed
(spaces)
Nucleic acid sequence Maximum Likelihood method, version 3.695
Settings for this run:
U Search for best tree? Yes
T Transition/transversion ratio: 2.0000
F Use empirical base frequencies? Yes
C One category of sites? Yes
R Rate variation among sites? constant rate
W Sites weighted? No
S Speedier but rougher analysis? Yes
G Global rearrangements? No
J Randomize input order of sequences? No. Use input order
O Outgroup root? No, use as outgroup species 1
M Analyze multiple data sets? No
I Input sequences interleaved? Yes
0 Terminal type (IBM PC, ANSI, none)? ANSI
1 Print out the data at start of run No
2 Print indications of progress of run Yes
3 Print out tree Yes
4 Write out trees onto tree file? Yes
5 Reconstruct hypothetical sequences? No
Y to accept these or type the letter for one to change
Adding species:
1. M_39
.
.
.
157. M_15
Output written to file "outfile"
Tree also written onto file "outtree"
Done.
--snphylo done
!End without notable errors
snphylo
out.bs.tree; newick tree with bootstrap score
out.bs.png; image file of out.bs.tree
out.ml.tree; newick tree
MUSCLE options; multiple sequence alignment
1. SNP sequence <= 50000
Muscle -phyi -in [input].fasta -out [output]
2. 50000 <= SNP sequence < 100000
Muscle -phyi -in [input].fasta -out [output] -maxiters 2
3. SNP sequence >= 100000
Muscle -phyi -in [input].fasta -out [output] -maxiters 1 -diags -sv
As sequence get longer, alignment accuracy decrease
