SPOT Instructions V1
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 21

sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
1
1. Rationale
2. Installing SPOT
3. Running SPOT – Quick Start
4. Running SPOT
5. Data input formats
6. Data output formats
7. Setting up an AWS account
8. Setting up personal AWS interface on your laptop
9. Starting an AWS instance
10. Logging into you AWS instance
11. References
Cite: A.M. King, C.K. Vanderpool, and P.H. Degnan. sRNA-target Prediction Organizing
Tool (SPOT) integrates computational and experimental data to facilitate functional
characterization of bacterial small RNAs
1. Rationale
Computational approaches for sRNA target prediction have limitations but are relied
upon to generate testable hypotheses for sRNA function. Some algorithms are available
online or downloadable (e.g., TargetRNA2, IntaRNA), however these tools frequently
yield distinct results, have different data output formats and default search parameters.
Therefore, manually compiling results from these disparate tools and integrating the
predictions with existing experimental data is not trivial. We have generated an
innovative approach to streamline use of multiple existing sRNA target prediction
algorithms and integrate predictions with experimental data to generate a unified set of
target predictions. To this end, we have developed SPOT a flexible software pipeline
that searches for sRNA-mRNA binding sites in parallel using separate search tools,
collates the predictions, and integrates experimental data using customizable results
filters.
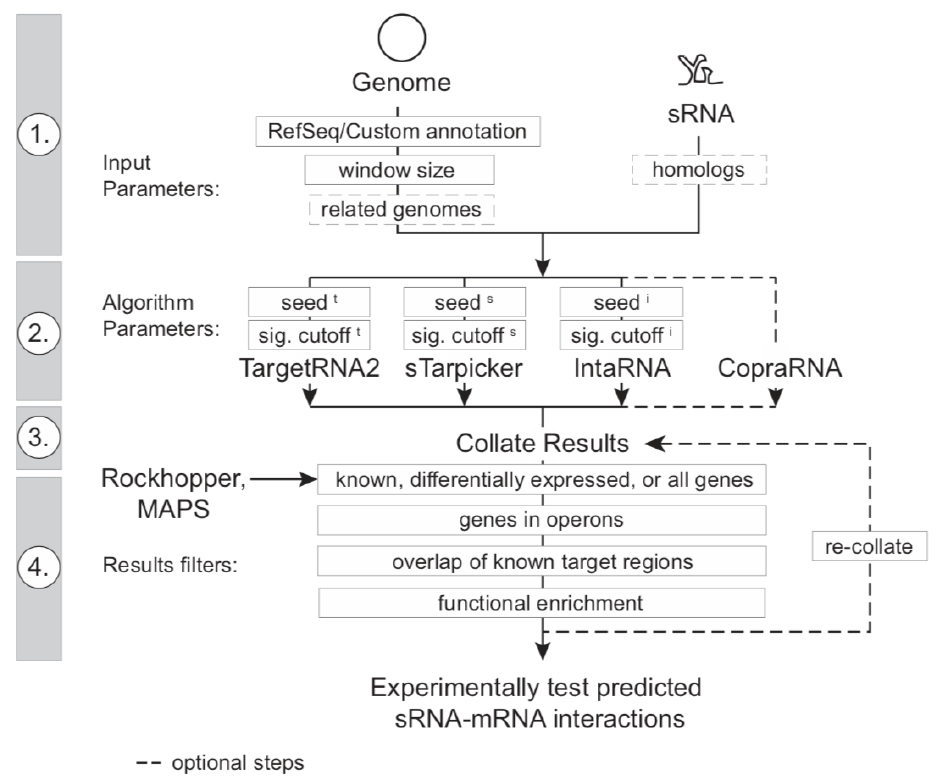
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
2
Figure 1. Schematic of SPOT pipeline analysis (King et al.)
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
3
2. Installing SPOT
SPOT is a PERL program that runs TargetRNA2, IntaRNA, StarPicker and CopraRNA
in parallel, and collates the results to find consensus sRNA-mRNA targets (Figure 1).
Furthermore, additional data types can be utilized to filter the results including
expression differences, known binding sites, operon predictions and window size of
possible binding sites.
As written the program can run on any Unix/Linux based system, however it has a
number of dependencies. To facilitate its use we have set up an Amazon Web Service
(AWS) cloud Amazon Machine Image (AMI) with all of the required software installed.
Skip to sections 4-7 for setting up your own SPOT AMI. However, using the code
available here you can set up and run SPOT on a local server.
First, download and install the following software tools and all of their dependencies
according the authors’ instructions:
• TargetRNA v2
• StarPicker
• IntaRNA v1.0.4
• CopraRNA v1.2.9
Several modifications were made to the StarPicker and IntaRNA code to accommodate
demands of the pipeline.
Replace the following programs with those provided in the GitHub link. Modifications in
the code are marked with ## comments and/or initials (PHD). Descriptions of edits
made are listed briefly below.
StarPicker:
sTarPicker_global2.pl : changes made to input of command line arguments
IntaRNA v1.0.4:
add_GI_genename_annotation.pl : distinguish GeneIDs vs GI Nos
get_refseq_from_ftp.pl : Replacement code for get_refseq_from_ftp.sh
IntaRNA_wrapper.pl : Option added to use local GenBank files, use
get_refseq_from_ftp.pl
rerun_enrichment.pl : code snippet re-running enrichment analysis from
IntaRNA_Wrapper.pl
termClusterReport.pl : code modified to handle GeneIDs vs GI Nos
CopraRNA v1.2.9:
get_refseq_from_ftp.pl : Replacement code for get_refseq_from_ftp.sh
termClusterReport.pl : code modified to handle GeneIDs vs GI Nos
get_CDS_from_gbk.pl : code modified to skip and flag GenBank files not present
in kegg2refseqnew.csv list
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
4
Note: D3 Javascript libraries may or may not be accessible using existing framework
to generate functional enrichment heatmaps (http://d3js.org/d3.v3.min.js). If problems
are encountered, it is possible to edit the master html files in IntaRNA and CopraRNA
to use a local version of d3.v3.min.js .
Be sure all programs are added to the user path and all path references in
StarPicker, IntaRNA, and CopraRNA match your system installation.
The statistics program R is installed as a requirement for IntaRNA and CopraRNA. As
such add the following two packages:
• RColorBrewer
• gplots
$ sudo R
> install.packages(c('RColorBrewer', 'gplots'))
Some of the output from the SPOT program will be written in an xlsx format using the
Excel Writer PERL module:
• Excel-Writer-XLSX-0.98
$ sudo cpan Excel::Writer::XLSX
Most existing Unix/Linux installations should have sendmail installed. If not, install the
appropriate package
• sendmail
$ sudo apt install sendmail-bin
SPOT can work with local copies of genomes and annotations. However, to access
genomes from NCBI install the efetch program from the Entrez Direct (edirect)
toolkit.
• edirect
Retrieve and decompress the SPOT directory from GitHub containing core pipeline
script and its additional required support PERL scripts.
• SPOT
Make sure SPOT and all of the programs are in your user path. Modify the core pipeline
script with the absolute path locations for TargetRNA2, IntaRNA, StarPicker and
CopraRNA, and other support PERL scripts.
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
5
3. Running SPOT - Quick Start
SPOT is a pipeline script that when run without arguments will print all of the possible
program options:
$ spot.pl
Usage ./spot.pl
Input parameters:
-r Fasta file of sRNA query
-a RefSeq Accession number (assumes any local files have RefSeq
number as their prefix)
...
The minimum data required for a SPOT search are:
1. A fasta file of the small RNA sequence
2. A RefSeq genome accession number
$ spot.pl -r sgrS.fasta -a NC_000913
This will initiate a job using the SgrS as the sRNA query and the E. coli str. K12
(NC_000913) as the reference genome. Progress of the search will be printed to the
screen. Run time will depend on the number of processors available as each search
tool is distributed to a separate sub process. By default CopraRNA is not run unless
specifically requested.

sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
6
4. Running SPOT
SPOT has an array of actions that control the input, algorithm parameters, and results
filtering.
$ spot.pl
Usage ./spot.pl
Input parameters:
-r Fasta file of sRNA query
-a RefSeq Accession number (assumes any local files have RefSeq
number as their prefix)
-o output file prefix (default = TEST)
-g Use local GenBank or PTT&FNA files for all Programs? (default = N
use latest from GenBank, CopraRNA cannot use local files)
-n Other genome RefSeq ids for CopraRNA listed in quotes '' ,
current max is 5 genomes (default ='')
-m Multisequence sRNA file for each genome in CopraRNA list
(default ='')
-x Email address for job completion notification (default ='')
Algorithm parameters:
-u Number of nt upstream of start site to search (default = 60)
-d Number of nt downstream of start site to search (default = 60)
-s seed sizes for I, T, S e.g., '6 7 6' (defaults TargetRNA = 7,
IntaRNA & Starpicker = 6)
-c P/Threshold value Cutoff for T, S, I e.g., '0.5 .001 un'
(defaults Target = 0.05, Starpicker = 0.5, IntaRNA = top)
Results Filters:
-b Number of nt upstream of start site to filter results
(default = -20)
-e Number of nt downstream of start site to filter results
(default = 20)
Note: -b and -e ignored if using a list (-l) or Rockhopper
results (-t)
Note: Set -b and -e to -u and -d to get all possible matches in
results
-l List of up and/or down regulated genes, include binding coord if
known e.g.,
b1101\tdown\n
b3826\tup\tsRNA_start\tsRNA_stop\tmRNA_start\tmRNA_stop\n
OR
-t transcriptome expression file from Rockhopper *_transcripts.txt
-f Rockhopper fold change cutoff (default = 1.5)
-q Rockhopper q value cutoff (default = 0.01)
-k Rockhopper Expression cutoff value (default = 100)
-p Operon file from DOOR-2 (http://csbl.bmb.uga.edu/DOOR/index.php)
(optional)
-w Report all genes even if List or Rockhopper provided?
(default = No)
-y Exclude target predictions by only 1 method? (default = Yes)
Note: Does not apply to genes on List or significantly expressed
from Rockhopper
-z Skip sRNA-mRNA detection steps, and just re-analyze data [Yy]es
(default = No) (Run in the same directory & requires original
results files from each program)

sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
7
Given the time SPOT runs can take it is recommended to use a queueing tool on large
distributed servers (qsub, slurm). Alternatively, on the AWS server, laptop or other
smaller computers it is recommended to use screen to ensure that jobs are not
prematurely aborted if the user account is logged out of.
$ screen -L spot.pl -r sgrS.fasta -a NC_000913
Four test datasets and precomputed output files are included in the folder
example_files. The following examples correspond to the four provided test
datasets.
test01 - Examine entire E. coli str. K12 genome for SgrS sRNA target mRNAs. This
folder only has the sRNA sequence in a fasta file, uses the individual program default
SEED size and significance settings and retrieves the genome sequence for E. coli from
GenBank. The final option is to have an email sent to the user after the job has
completed.
$ cd test01
$ ls
sgrS.fasta
$ spot.pl -r sgrS.fasta -o stringent -a NC_000913 -x username@email.edu
=========Prepping RefSeq Files====================
[Thu Aug 23 23:14:27 UTC 2018]
...
test02 - Examine E. coli str. K12 genome for SgrS sRNA target mRNA matches among
a set of defined differentially expressed genes (sgrS_diff.txt). In this case the user
has a fasta file and a traditional GenBank protein translation file (PTT). The user also
indicates a larger window size 150 nt upstream of the CDS start position and 100 nt
downstream to search for binding sites.
$ cd test02
$ ls
sgrS.fasta
sgrS_diff.txt
NC_000913.fna
NC_000913.ptt
$ spot.pl -r sgrS.fasta -l sgrS_diff.txt -u 150 -d 100 -c '0.5 0.001 un' -o
relaxed -a NC_000913 -g Y
Note: PTT files can be easily generated in Excel. Allowing for customization of gene
annotations and subsequent analyses. A script included with SPOT is fnaptt2gbk.pl
which can be used to generate GenBank files using the genome PTT and fasta files
as inputs. However, always make sure that MAC or DOS line breaks are converted into
UNIX line breaks.

sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
8
test03 - SPOT was designed to allow re-analysis of existing results. This example code
block is rung in a folder containing the results of test02’s search. In this case even
though the upstream/downstream region searched was 150nt and 100nt, the reanalysis
eliminates any binding sites found outside of 50nt upstream and 30nt downstream. This
search also does not use the list of differentially expressed genes.
$ cd test03
$ ls
sgrS.fasta
sgrS_diff.txt
NC_000913.fna
NC_000913.ptt
...
$ spot.pl -r sgrS.fasta -u 150 -d 100 -c '0.5 0.001 un' -o changed_50_30 -a
NC_000913 -g Y -b -50 -e 30 -z Y
test04 – SPOT can also be run using a *transcript.txt file generated by the
RNAseq analysis program Rockhopper directly (instead of list as in example test02). In
this example default expression cutoffs are used, however these can be specified by the
user. In addition, when provided a set of sRNA homologs and target genomes
CopraRNA can be run. In these instances only genomes in RefSeq can be used.
Custom genome annotations cannot be utilized.
$ cd test04
$ ls
NC_000913_SgrS_transcripts.txt
sgrS.fasta
sgrS_homologs.fasta
$ spot.pl -r sgrS.fasta -t NC_000913_SgrS_transcripts.txt -o express -a
NC_000913 -m sgrS_homologs.fasta -n 'NC_002695 NC_011740' -u 150 -d 100
When the jobs have completed compare your results to the files in the corresponding
_results folder.
5. Data input formats
sRNA fasta file – DNA sequence of sRNA in a standard fasta file. File extension does
not matter (.fasta, .fa, .fna, .frn, .ffn)
RefSeq ID – Standard RefSeq IDs can be used and GenBank files (.gbff) will be
retrieved using efetch. Program will retrieve additional replicons (e.g., plasmids) or
scaffolds associated with the provided RefSeq IDs, however, the search will only be
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
9
carried out on the file with a name corresponding to the input RefSeq ID. By default the
.gbff is renamed to a .gb file, and.fna and .ptt files are generated.
Local Files – Different combinations of local files can be used. They all must have the
same prefix and end in the following suffixes:
.fna
Genome fasta sequence
.ptt
Protein translation table – gene annotation
.gb or .gbk
Genbank file
Files without these suffixes will be ignored. All must have Unix linebreaks and the .ptt
file must be tab separated. Allowed input combinations include:
.fna
.ptt
.gb or
.gbk
Status
Action
1.
Ö
Ö
Ö
okay
Start run
2.
Ö
Ö
okay
Make .gb file, start run
3.
Ö
Ö
okay
Make .ptt file, start run
4.
Ö
Ö
okay
Make .fna file, start run
5.
Ö
okay
Make .fna and .ptt file, start run
6.
Ö
bad
Abort run
7.
Ö
bad
Abort run
.ptt Files – This is a legacy GenBank annotation format. However, the StarPicker
algorithm used here requires this format. This format is very easy to generate in Excel
and can allow users of SPOT to customize their annotations. See example:
Escherichia coli str. K-12 substr. MG1655, complete genome - 1..4641652
4141 proteins
Location
Strand
Length
PID
Gene
Synonym
Code
COG
Product
190..255
+
21
thrL
b0001
-
-
thr operon leader peptide
337..2799
+
820
thrA
b0002
-
-
Bifunctional aspartokinase
2801..3733
+
310
thrB
b0003
-
-
homoserine kinase
3734..5020
+
428
thrC
b0004
-
-
L-threonine synthase
Note: As indicated above, customization of PTT files allows users to correct or change
annotations based on new data. Furthermore, by modifying PTT files RNAs can be
included in the annotation. First, this allows for sRNA – RNA interactions to be
identified. Second, this approach was used in the manuscript to perform a ‘reverse’
search. For a ‘reverse’ search the PTT file is edited to ONLY include the known sRNAs.
Then, the user supplies the UTR or putative sRNA binding region to SPOT as a fasta
file if it were the sRNA. ‘Reverse’ searches cannot use CopraRNA and as sRNAs do not
have GI numbers and may not have GeneIDs - no functional enrichment plots will be
produced. This may result in several warnings when the SPOT is run, however it should
not influence the final composite predictions.

sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
10
Differentially expressed genes – Lists of differential genes can be formatted as tab
separated files one of two ways. Do not include a header line
Simple:
Locus
Expression
b1101
down
b3826
up
With known binding sites:
Locus
Expression
sRNA_start
sRNA_stop
mRNA_start
mRNA_stop
b1101
down
168
187
-30
-9
b3826
up
168
187
-96
-76
Rockhopper *transcript.txt files – SPOT can read default output files of
Rockhopper from simple pairwise RNAseq experiments. Files generated with the
verbose output option in Rockhopper cannot be read. Files should have 12 columns
including the normalized expression values for the treatment and control, the q Values
and the estimated fold-change.
sRNA Multisequence Fasta Files – If running CopraRNA, sRNA files must conform to
expectations of the CopraRNA program:
1. RNA sequence must have Us instead of Ts
2. The sequence names must correspond to the individual genome RefSeq IDs
3. Must include the focal genome sRNA sequence as well
6. Data output formats
Data from each individual algorithm is preserved in the output folder for manual
investigation.
TargetRNA2_*txt = TargetRNA2 Primary report
*.output = Starpicker Primary report
intarna_websrv_table_truncated.csv = IntaRNA Primary report
*_ hIntaRNA.csv = CopraRNA Primary report
SPOT generates several output files for further analysis:
XLSX file – Primary file containing consensus table of sRNA-mRNA predictions from the
3 or 4 tools used in the run. File name prefix corresponds to run output prefix that was
assigned (-o , default= TEST).
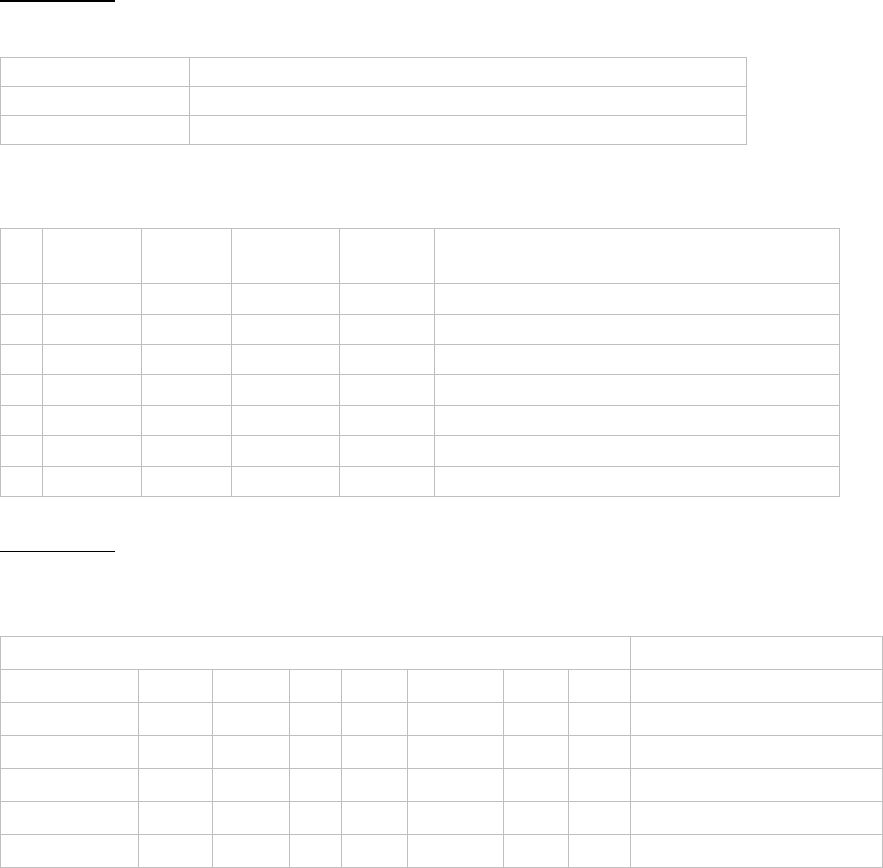
• Sheet 1 (complete.txt) shows the aligned predictions, p values, and coordinates
for the predicted interaction for each gene.
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
11
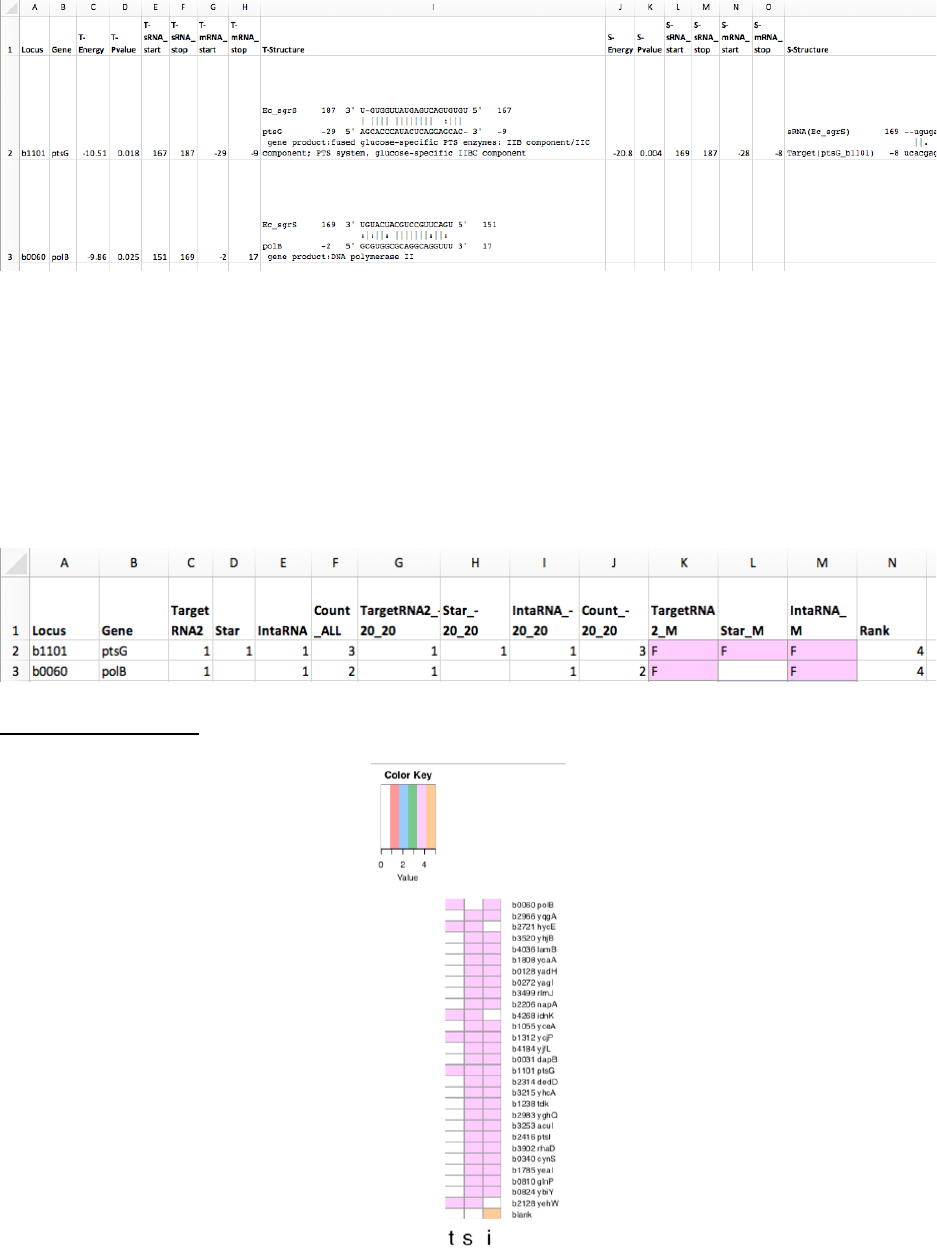
• Sheet 2 (summary.txt) has the counts predicted by each gene, and a summary
letter and ranking based location and on the number of algorithms that found the
same prediction.
A Prediction overlaps a known binding site
B
à
E Predictions that are not coincident with a known binding site when one was
provided for that gene. Shared letters overlap the same site.
F
à
I Predictions when no known binding site was provided. Shared letters overlap the
same site.
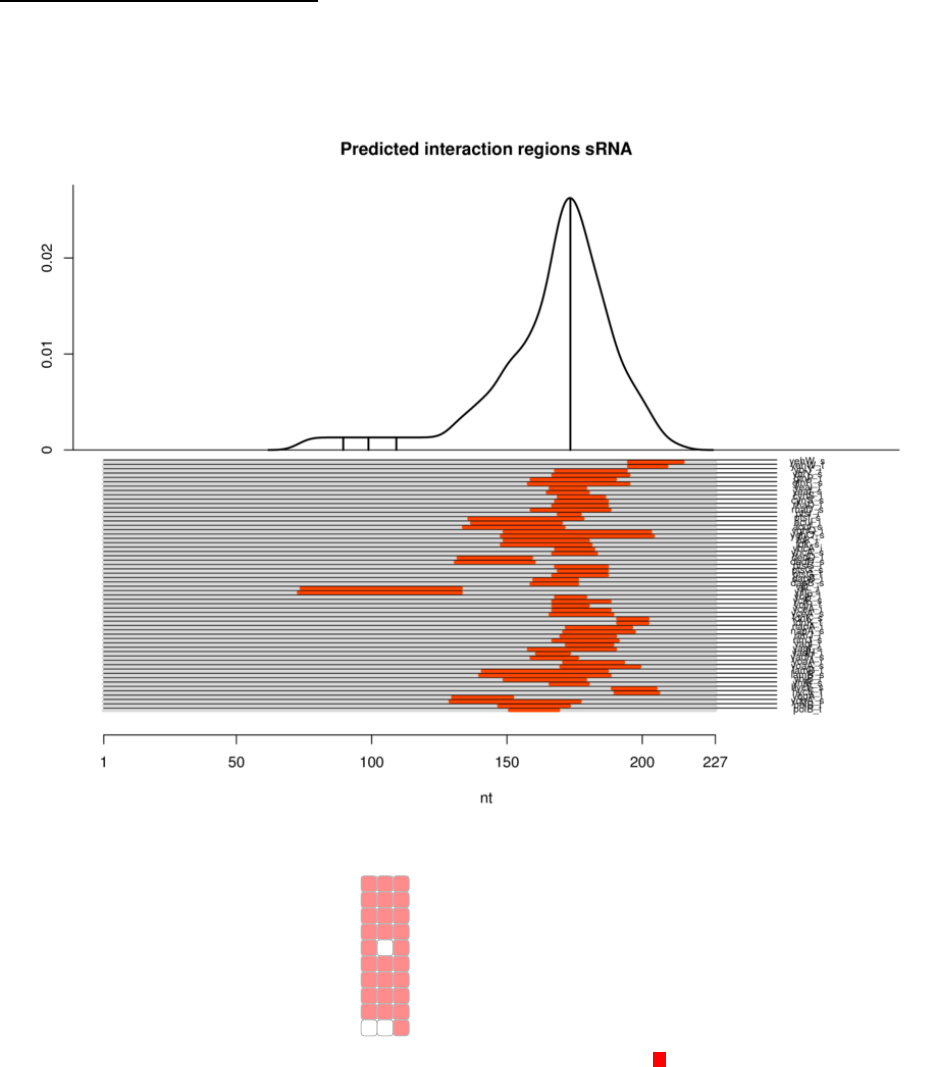
*_summary.pdf file – This file has a R generated plot that corresponds to Sheet 2
(summary.txt) which can be imported to Illustrator.
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
12
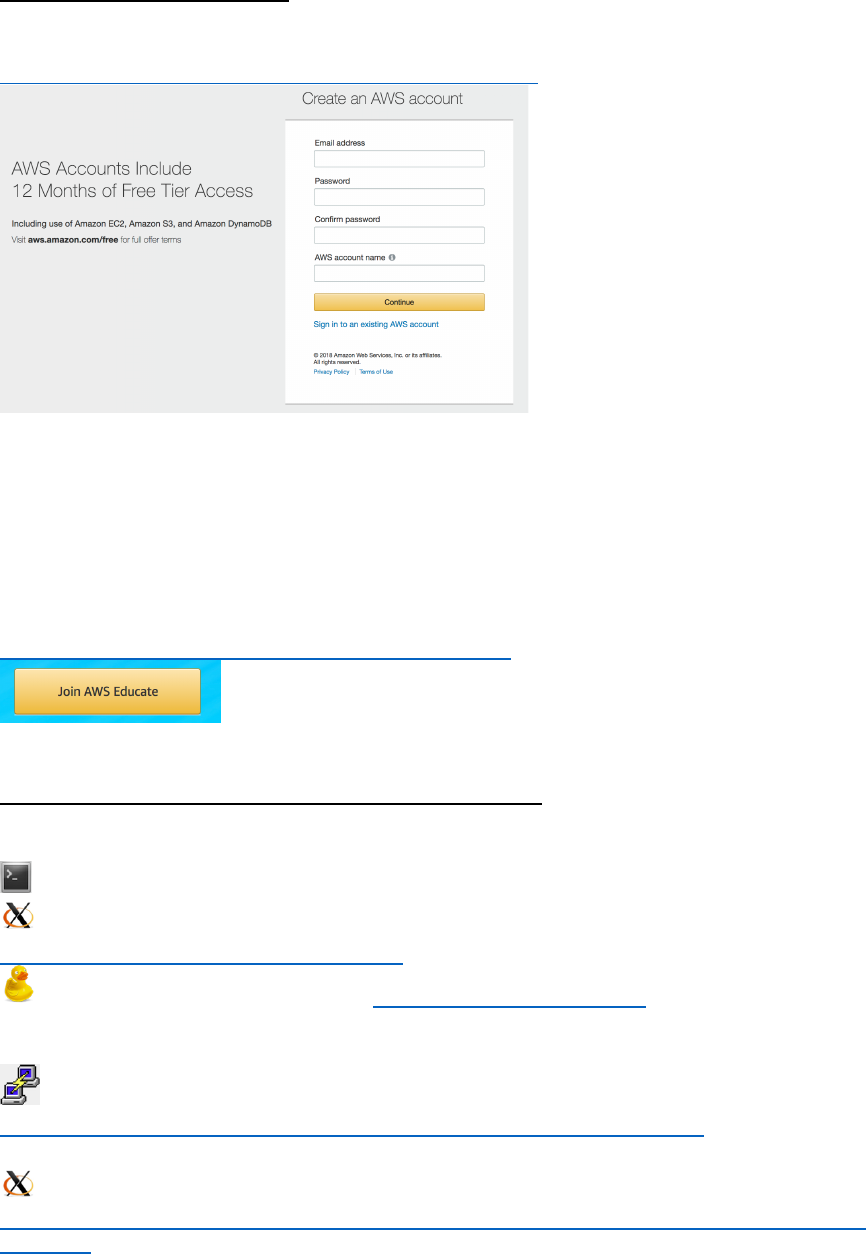
COLLATED_RESULTS folder – This folder contains plots generated based on IntaRNA
tools showing the localization of binding sites of the mRNA and sRNA as *pdf, *png and
*ps files. In addition, a functional enrichment heatmap is included as a *pdf file similar to
those individually provided by IntaRNA and CopraRNA - however it represents the
collated results.
Example result files for the sRNA SgrS and corresponding test datasets are available
with the SPOT software distribution.
b4184-yjfL
b1312-ycjP
b1785-yeaI
b0128-yadH
b4036-lamB
b0810-glnP
b2128-yehW
b2983-yghQ
b1101-ptsG
b2966-yqgA
123
1:2.34topologicaldomain:Periplasmic
2:2.08topologicaldomain:Cytoplasmic
3:1.7transmembraneregion
group1:1.35
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
13
7. Setup an AWS account
Navigate to the new account setup page:
https://portal.aws.amazon.com/billing/signup#/start
For now, set up your home region as “U.S. East (W. Virginia)” later you can switch this
as necessary.
Unfortunately, when setting up an account you will need a credit card number
Input Education credit - Depending on your application it may be possible to apply for
education credits to defray the cost of the AWS server time:
https://aws.amazon.com/education/awseducate/
8. Setup personal AWS interface on your laptop
People with MACs:
Terminal will already be installed /Applications/Utilities
Download & Install XQuartz if not already installed
http://xquartz.macosforge.org/landing/
Download & Install Cyberduck https://cyberduck.io/?l=en
People with PCs:
Download & Install PuTTY
http://www.chiark.greenend.org.uk/~sgtatham/putty/download.html
Download & Install xMing
http://sourceforge.net/project/downloading.php?group_id=156984&filename=Xming-6-9-0-31-
setup.exe
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
14
How to setup xMing : http://www.geo.mtu.edu/geoschem/docs/putty_install.html
Download & Install WinSCP http://winscp.net/eng/download.php
or
Download & Install Cyberduck https://cyberduck.io/?l=en
9. Starting an AWS instance
For in-depth instructions regarding starting an AWS instance please see:
https://docs.aws.amazon.com/AWSEC2/latest/UserGuide/launching-instance.html
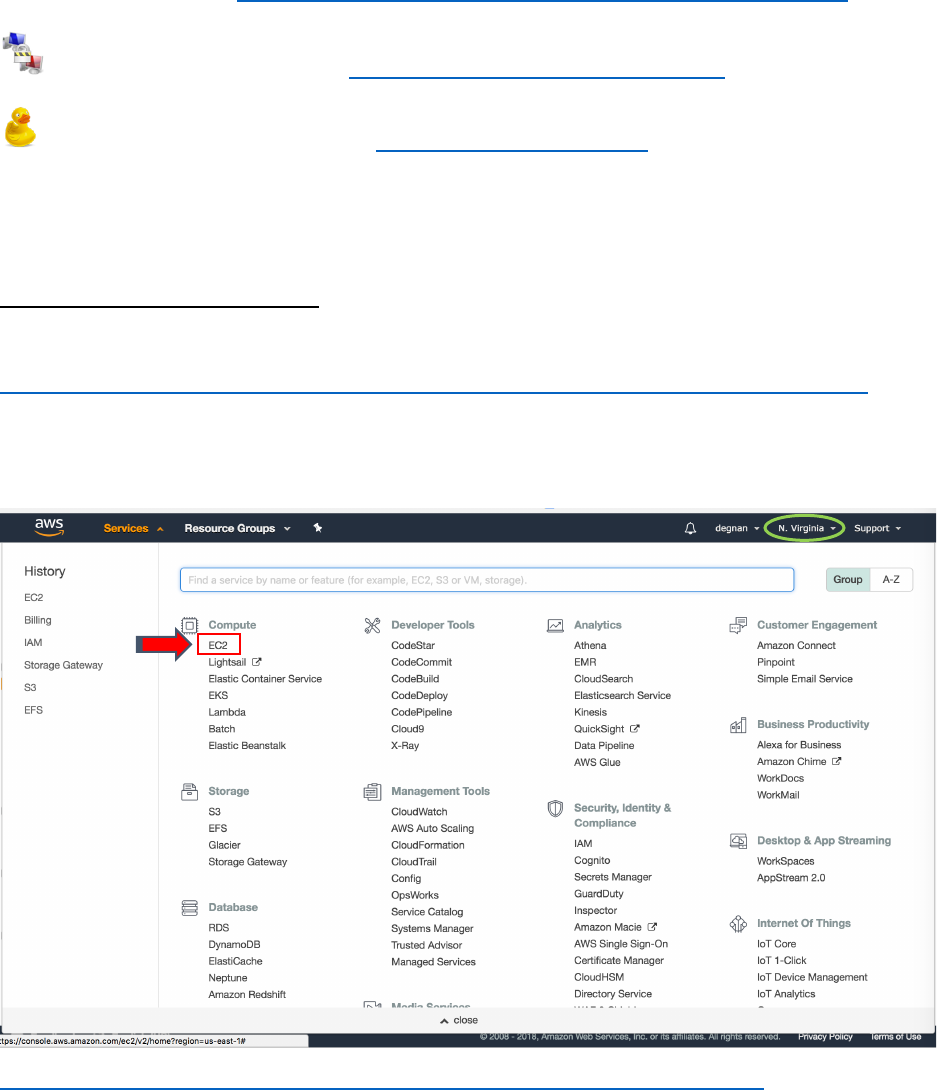
1. After making and logging into your AWS account find your way to the EC2 (Elastic
Computing Cloud) page. You can find it under “Services” menu on the upper left-hand
corner of the page:
https://console.aws.amazon.com/ec2/v2/home?region=us-east-1#Home:
2. Make sure your home region as “U.S. East (W. Virginia)”. Your region is indicated in
the upper right-hand corner of the page (circled above)
1
2
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
15
3. On the right-hand side bar under “IMAGES” select “AMIs”
4. In the search bar switch from “Owned by me” to “Public images” and search for
“SPOTv1”
5. Select the blue “Launch” button
3
4
5
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
16
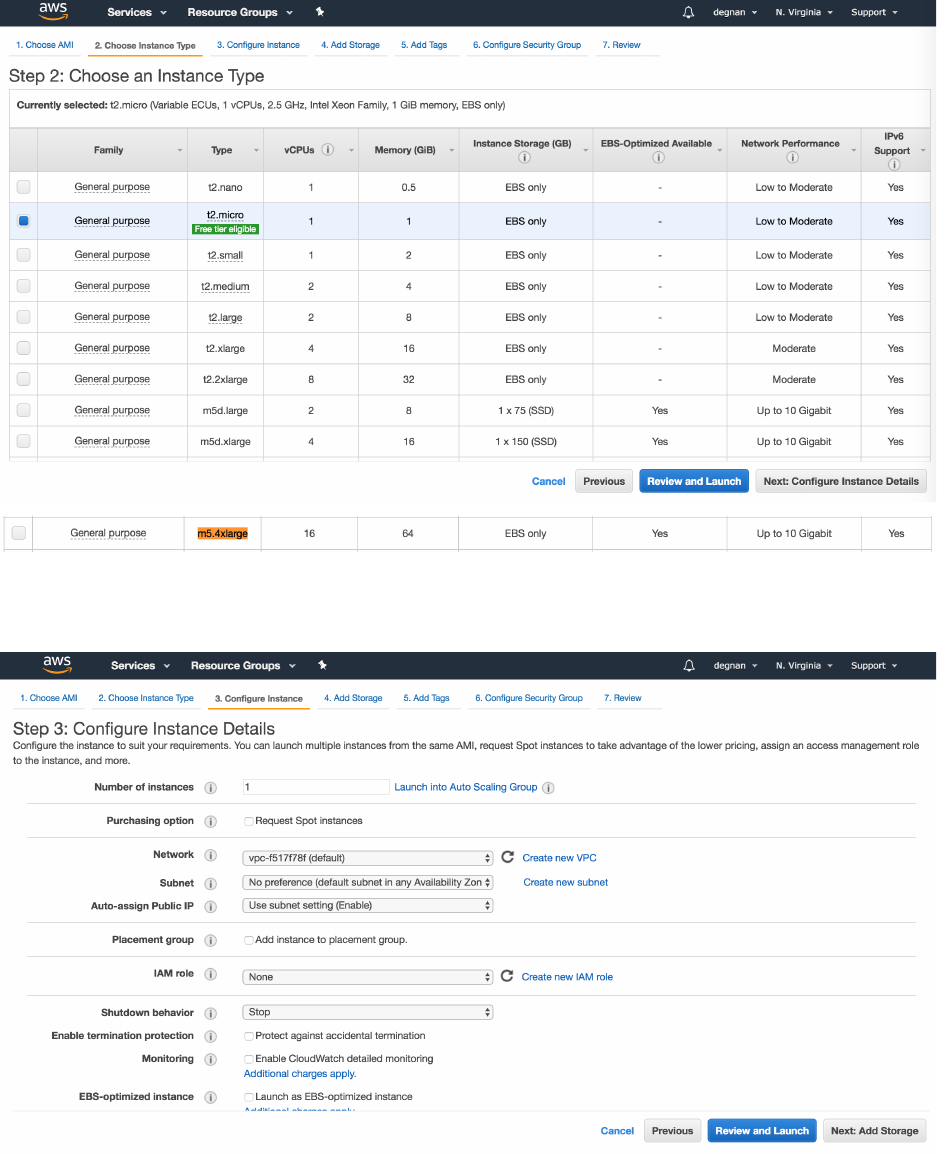
6. Now you are on AWS “Step 2: Choose and Instance Type” – Select your computer:
t2.micro is the only free option, however it is maxed out at 1GiB of RAM, 1
processor and 30GiB of storage. Very slow
m5.2xlarge 8 virtual processors, 64 GiB of RAM
7. Select “Next: Configure Instance Details” button on bottom-right
8. On “Step 3: Configure Instance Details” page – leave defaults as-is
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
17
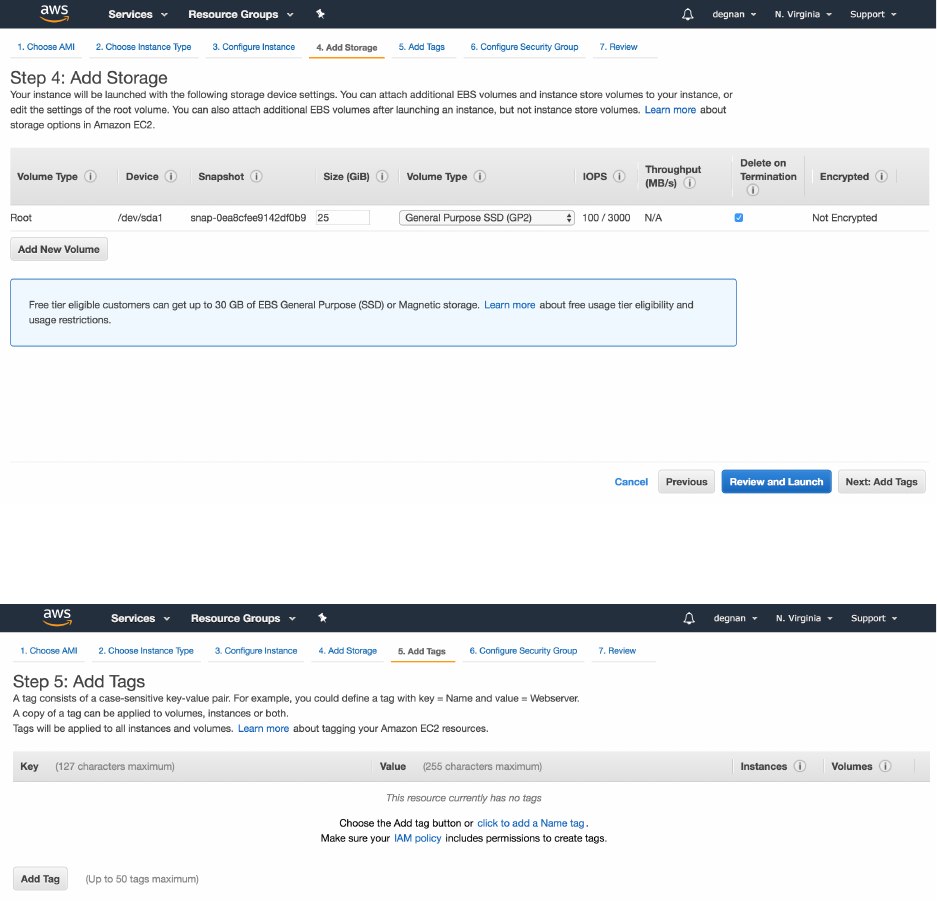
9. Select the “Next: Add Storage” button on bottom-right, to move to the next step
10. On the “Step 4: Add Storage” adjust local disk size to 30 GiB
11. Select the “Next: Add Tags” button on bottom-right, to move to the next step
12. On the “Step 5: Add Tags” optionally hit the “Add Tag” button OR skip to step 14
13. For example Add a key = “Name” and value = “my-SPOT” or “SPOT-server”
14. Select the “Next: Configure Security Group” button on bottom-right, to move to the
next step
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
18
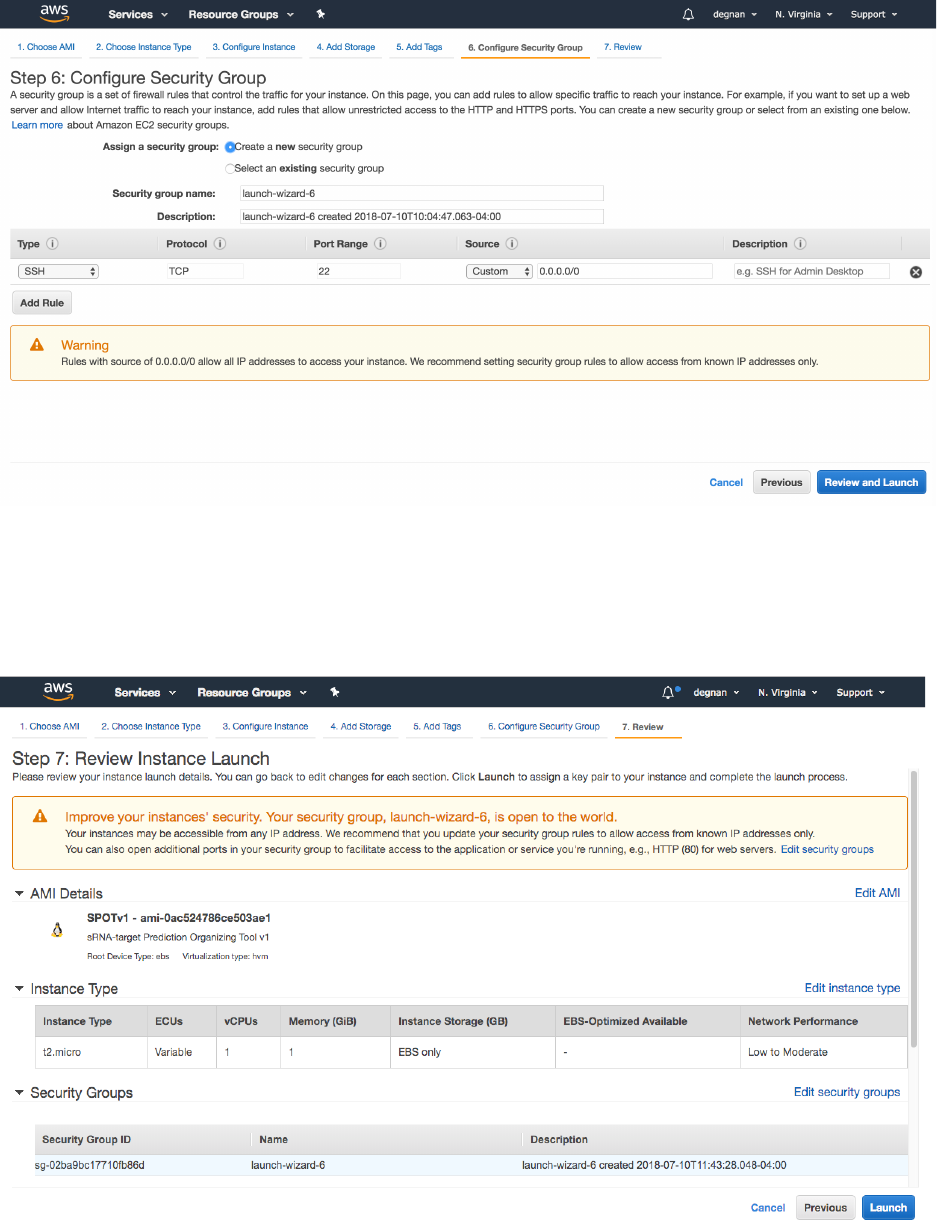
15. On “Step 6: Configure Security Group” page – leave defaults as-is
**Note: For now, we will ignore the Warning. In the future consider making your
instances harder to access by non-users in your lab/group**
16. Select the “Review and Launch” button on bottom-right, to move to the next step
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
19
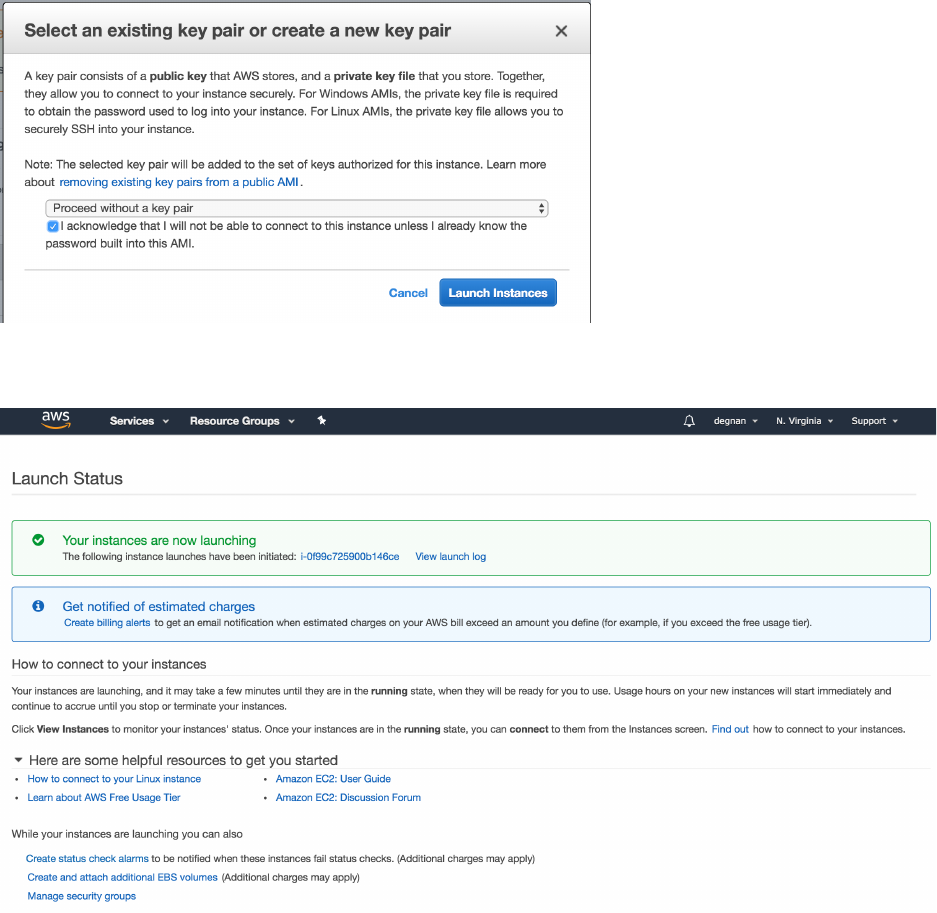
17. You can inspect the settings before hitting the “Launch” button. As before ignore
warnings.
18. Now it asks you to select or create a key pair.
19. You will need to download the key and save it to a private location on your computer
(e.g., the folder ~/.ssh/) .
20. From here you can navigate using the left-hand side bar to your “Instances”
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
20
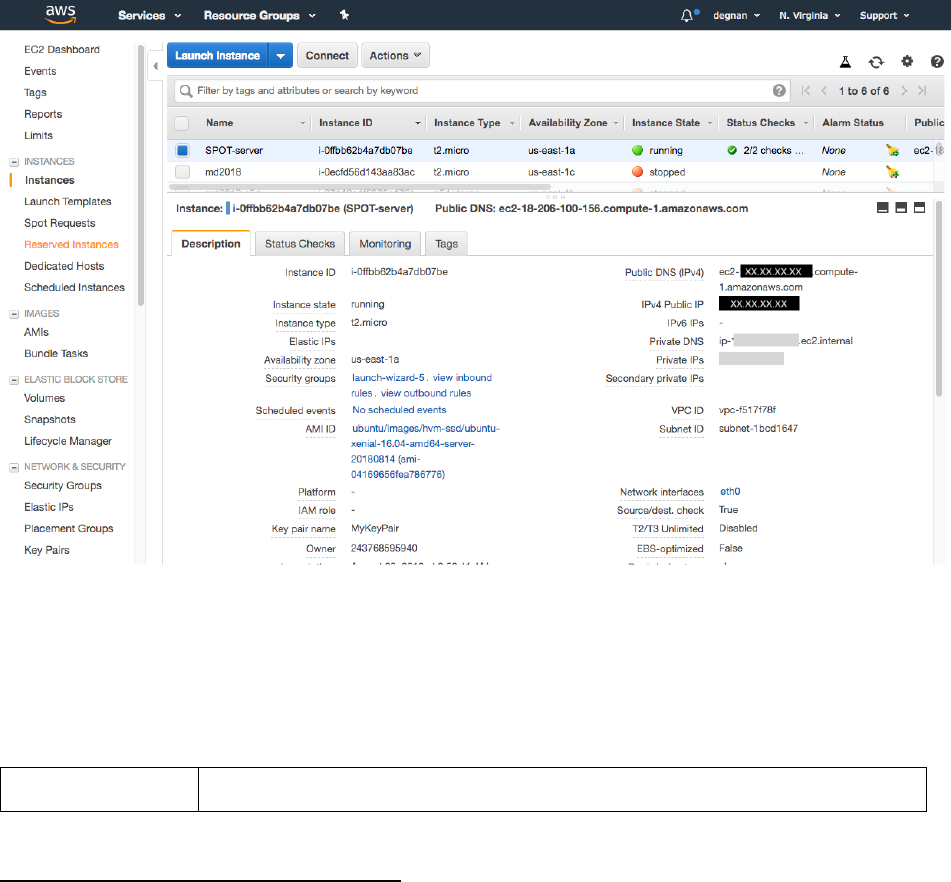
21. Instance state will be “Initializing” until the computer has “booted” up.
22. Once the Instance state switches to “running” and you select the instance, details of
the instance will be shown below.
23. Find and copy the “IPv4 Public IP” address for your instance. You will use this to
login to your server.
IPv4 Public IP:
10. Logging into you AWS instance
To log into the server you will need your:
1. Private ssh key yourid_key.pem
2. username = first name and last initial as one word (e.g., Jane Doe = janed)
3. XX-XX-XX-XX = Your specific IPv4 Public IP from above
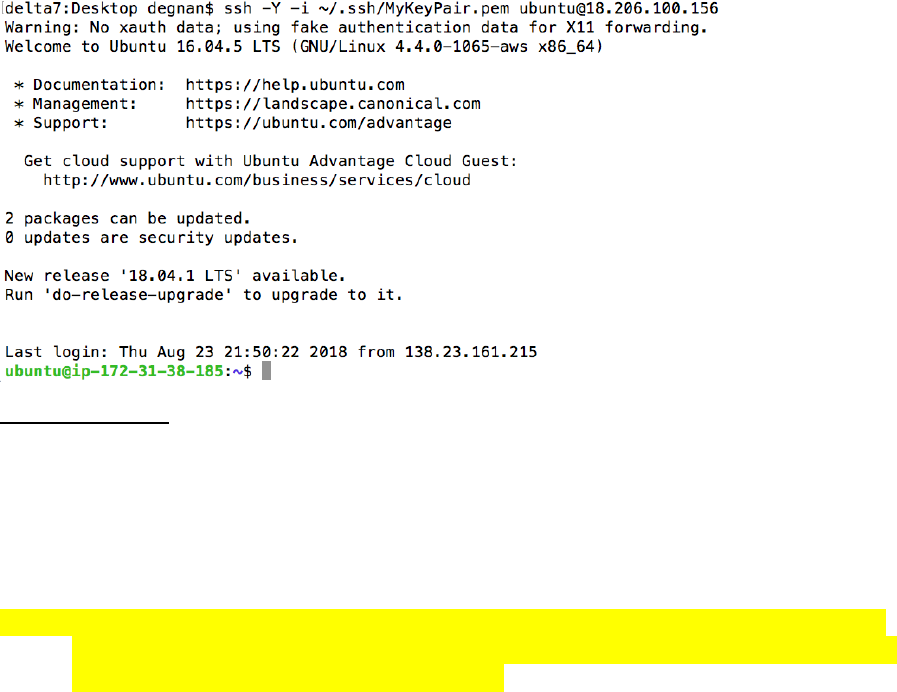
Login using Terminal on a MAC or UNIX.
$ ssh -Y -i ~/.ssh/yourid_key.pem username@XX-XX-XX-XX
Login from Windows using PuTTY
a. Open PuTTY
b. Under Category, click on SSH > Auth
c. Click browse
sRNA-target Prediction Organizing Tool v1 – Manual 10 Sept-18
21
d. Find your private key (yourid_key.pem) and select it
e. Under Category, click Session and input address of your EC2 instance (XX-XX-XX-
XX) in the "host name" box
f. Type "SPOT" in the box under saved sessions and click save.
g. Double-click on the "SPOT" that appears under saved sessions.
h. Log in with your username. Your key should be used automatically.
i. For future logins, just double-click the "SPOT" saved session.
Once entered you will find yourself on the command line interface:
11. References
Busch A, Richter AS, Backofen R. 2008. IntaRNA: efficient prediction of bacterial sRNA
targets incorporating target site accessibility and seed regions. Bioinformatics
24:2849-2856.
Kery MB, Feldman M, Livny J, Tjaden B. 2014. TargetRNA2: identifying targets of small
regulatory RNAs in Bacteria. Nucleic Acids Research 42:W124-129.
King AM, Vanderpool CK, and Degnan PH. sRNA-target Prediction Organizing Tool
(SPOT) integrates computational and experimental data to facilitate functional
characterization of bacterial small RNAs.
McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA,
Vanderpool CK, Tjaden B. 2013. Computational analysis of bacterial RNA-Seq
data. Nucleic Acids Research 41:e140.
Wright PR, Georg J, Martin M, Sorescu DA, Richter, AS, Lott S, Kleinkauf R, Hess WR,
Backofen R. 2014. CopraRNA and IntaRNA: predicting small RNA targets,
networks and interaction domains. Nucleic Acids Research 42:W119-W123.
Ying X, Cao Y, Wu J, Liu Q, Cha L, Li W. 2011. sTarPicker: a method for efficient
prediction of bacterial sRNA targets based on a two-step model for hybridization.
PloS One 6:e22705.
