Trans Tasman Radiation Oncology Group Inc Melbourne C30 TROG 08 03 RAVES Protocol Amendment 2 Dated 29 May 2014
User Manual: Melbourne C30
Open the PDF directly: View PDF ![]() .
.
Page Count: 114 [warning: Documents this large are best viewed by clicking the View PDF Link!]
TRANS TASMAN RADIATION
ONCOLOGY GROUP LTD
(ACN 132 672 292)
In collaboration with the Urological Society of
Australia and New Zealand (USANZ) and the
Australian and New Zealand Urogenital and Prostate
Cancer Trials Group (ANZUP)
TROG 08.03
RAVES – RADIOTHERAPY ADJUVANT
VERSUS EARLY SALVAGE
A PHASE III MULTI-CENTRE RANDOMISED
TRIAL COMPARING ADJUVANT RADIOTHERAPY
(RT) WITH SURVEILLANCE AND EARLY
SALVAGE RT IN PATIENTS WITH POSITIVE
MARGINS OR EXTRAPROSTATIC DISEASE
FOLLOWING RADICAL PROSTATECTOMY
Final Protocol Version: 7 August 2008
Amendment 1: 8 July 2011
Amendment 2: 29 May 2014
Trial Co-Chairpersons:
Dr Maria Pearse
Department of Radiation Oncology
Auckland City Hospital
P.O. BOX 92-024
Auckland 1142. New Zealand
Tel: +64 9 3074949 ext. 23044
mariap@adhb.govt.nz
A/Prof Andrew Kneebone
Royal North Shore Hospital
Pacific Highway St Leonards NSW 2065
Ph: + 61 2 9926 7483
Fax: + 61 2 9906 6833
AKneebone@nsccahs.health.nsw.gov.au
Trial Coordinator:
Carol Fraser-Browne
Auckland Regional Cancer and Blood
Service
Po Box 92024 Auckland
New Zealand
Tel: +64 9 3074949 ext. 23044
Fax: + 64 9 3599981
Email: carolfb@adhb.govt.nz

TRIAL MANAGEMENT COMMITTEE
Trial Co-Chairpersons
Dr Maria Pearse
Auckland Hospital, New Zealand
A/Prof Andrew Kneebone
Royal North Shore Hospital, Sydney
Executive Committee
Members
Prof. Gillian Duchesne
(Radiation Oncology)
Peter MacCallum Cancer Centre, Melbourne
A/Prof. Richard Fisher
(Trial Statistician)
Centre for Biostatistics and Clinical Trials (BaCT),
Peter MacCallum Cancer Centre, Melbourne
Mr Mark Frydenberg
(Urology)
Monash Medical Centre, Melbourne
Dr Scott Williams
(Radiation Oncology)
Peter MacCallum Cancer Centre, Melbourne
Radiation Oncology
Assoc Prof Chris Atkinson
Christchurch Hospital, New Zealand
Dr Chakiath Jose
Auckland Hospital, New Zealand
Prof David Joseph
Sir Charles Gairdner Hospital, Perth
Dr Liz Kenny
QRI, Royal Brisbane Hospital
Dr John Matthews
Auckland Hospital, New Zealand
A/Prof Jeremy Millar
WBRC, The Alfred Hospital, Melbourne
Prof Nigel Spry
Sir Charles Gairdner Hospital, Perth
Dr Sandra Turner
Westmead Hospital, Sydney
Dr Kirsty Wiltshire
Peter MacCallum Cancer Centre, Melbourne
Urology
Mr Tom Shannon
Hollywood Private Hospital, Perth
Mr Manish Patel
Westmead Hospital, Sydney
Dr John Yaxley
Brisbane Private Hospital
Mr Peter Swindle
Mater Private Hospital, Brisbane
Mr Michael Rice
Auckland Hospital, New Zealand
Mr Henry Woo
Westmead Hospital, Sydney
Trial Genetic Epidemiologist
Dr Julie Marsh
Centre for Genetic Epidemiology, University of
Western Australia, Perth
Trial Physicist
A/Prof Annette Haworth
Peter MacCallum Cancer Centre, Melbourne
Central Pathology Review
Prof Warick Delprado
Douglass Hanly Moir Pathology, Sydney
Dr Ronnie Cohen
Uropath, Perth
Trial Coordinator
Carol Fraser-Browne
Auckland Hospital, New Zealand
Consumer Representatives
David Farley; Ross Gomersall

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 3 of 114
Amendment 2: dated 29 May 2014
Technical Quality Assurance Committee
Chairperson:
A/Prof Annette Haworth
Peter MacCallum Cancer Centre, Melbourne
Committee:
A/Prof Andrew Kneebone
Royal North Shore Hospital, Sydney
Dr Maria Pearse
Auckland Hospital, New Zealand
Dr Mark Sidhom
Liverpool Hospital, Liverpool
Dr Colin Tang
Sir Charles Gairdner Hospital, Perth
Ann Thompson
Peter MacCallum Cancer Centre, Melbourne
Dr Scott Williams
Peter MacCallum Cancer Centre, Melbourne
Dr Kirsty Wiltshire
Peter MacCallum Cancer Centre, Melbourne
Andrea Paneghel
Peter MacCallum Cancer Centre, Melbourne
FOREWORD
This document is intended to describe a Trans Tasman Radiation Oncology Group
(TROG) trial and to provide information about procedures for screening, enrolling and
treating trial participants. It is not intended that the Protocol be used as a guide for
the treatment of patients who are not enrolled on this trial.
TROG will not accept any data for analysis unless each Trial Site has Human
Research Ethics Committee (HREC) approval for patient enrolment and participation
in this trial.
Amendments to the document may be necessary; when approved by TROG, these
will be circulated by the Trial Coordinating Centre, on behalf of TROG, to Trial Sites
participating in the Trial.
The Protocol and all other trial related documentation including the Participant
Information Sheet and Consent Form and Case Report Forms must be written in
English and under no circumstances be translated into another language without
prior written approval from TROG.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 4 of 114
Amendment 2: dated 29 May 2014
CONTENTS
1.0 EXECUTIVE SUMMARY .................................................................................................................. 9
2.0 RAVES SCHEMA ............................................................................................................................ 12
3.0 INTRODUCTION ...................................................................................................................... 13
4.0 OBJECTIVES AND ENDPOINTS ............................................................................................ 17
4.1 PRIMARY OBJECTIVE ..................................................................................................................... 17
4.2 SECONDARY OBJECTIVES .............................................................................................................. 17
4.3 ENDPOINTS ................................................................................................................................... 18
4.3.1 Primary Endpoint ................................................................................................................. 18
4.3.2 Secondary endpoints ........................................................................................................... 18
5.0 TRIAL DESIGN ........................................................................................................................ 18
5.1 TRIAL DESCRIPTION ....................................................................................................................... 18
5.2 STUDY ARMS ................................................................................................................................. 18
5.2.1 Stratification ......................................................................................................................... 18
6.0 PATIENT SELECTION............................................................................................................. 19
6.1 INCLUSION CRITERIA ...................................................................................................................... 19
6.2 EXCLUSION CRITERIA..................................................................................................................... 19
6.3 PATIENT WITHDRAWAL ................................................................................................................... 19
7.0 PATIENT CONSENT AND RANDOMISATION ....................................................................... 20
7.1 SCREENING AND RANDOMISATION .................................................................................................. 20
7.2 CONSENT FOR OPTIONAL BIOLOGICAL SUB-STUDIES ......................................................................... 20
8.0 TECHNICAL GUIDELINES ............................................................................................................. 20
8.1 STATEMENT OF TREATMENT AIM ..................................................................................................... 20
8.2 TREATMENT SCHEDULE ................................................................................................................. 20
8.3 PLANNING SIMULATION .................................................................................................................. 20
8.4 DAILY TREATMENT POSITION ........................................................................................................... 21
8.5 TARGET VOLUME DEFINITIONS/FIELD BORDERS ............................................................................... 21
8.5.1 Clinical Target Volume (adapted from FROGG consensus guidelines45) ........................... 21
8.5.2 Planning target volume (PTV).............................................................................................. 22
8.6 DOSE PRESCRIPTION AND FRACTIONATION ..................................................................................... 23
8.7 TREATMENT PLANNING AND DOSIMETRY ......................................................................................... 24
8.8 DOSE DISTRIBUTION/REPORTING ................................................................................................... 25
8.9 NORMAL TISSUE CONTOURING AND DOSE CONSTRAINTS .................................................................. 27
8.9.1 Dose constraints .................................................................................................................. 28
8.10 TREATMENT EQUIPMENT SPECIFICATIONS/PHYSICAL FACTORS...................................................... 28
8.11 TREATMENT VERIFICATION ........................................................................................................... 28
8.12 QUALITY ASSURANCE PROGRAM .................................................................................................. 29
8.12.1 Dry run ............................................................................................................................... 29
8.12.2 IMRT credentialing program .............................................................................................. 30
8.12.3 Radiation Therapy Technical Review ................................................................................ 30
8.12.4 Data required for timely review .................................................................................... 31
8.12.5 Data required following RT completion ....................................................................... 32

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 5 of 114
Amendment 2: dated 29 May 2014
8.13 SITE VISITS .................................................................................................................................. 32
9.0 TREATMENT OF RELAPSE AFTER RADIOTHERAPY ........................................................ 32
9.1 INDICATIONS FOR ANDROGEN DEPRIVATION TREATMENT .................................................................. 32
9.2 ANDROGEN DEPRIVATION TREATMENT ........................................................................................... 32
9.3 CONTINUOUS OR INTERMITTENT ANDROGEN DEPRIVATION .............................................................. 33
10.0 PATIENT ASSESSMENT ........................................................................................................ 33
10.1PRE-RANDOMISATION ASSESSMENT ............................................................................................... 33
10.1.1 Disease Status, Medical History and Co-Morbidities ........................................................ 33
10.1.2 Imaging .............................................................................................................................. 33
10.1.3 Laboratory Studies ............................................................................................................. 33
10.1.4 Patient-completed questionnaires ..................................................................................... 33
10.2 ASSESSMENT DURING TREATMENT PHASE ..................................................................................... 34
10.3 FOLLOW-UP ASSESSMENTS .......................................................................................................... 34
10.3.1 Clinical Assessment ........................................................................................................... 34
10.3.2 Biochemical assessment ................................................................................................... 34
10.3.3 Imaging .............................................................................................................................. 35
10.4 QUALITY OF LIFE, ANXIETY/DEPRESSION AND ADVERSE EVENT ASSESSMENTS ................................. 35
10.4.1 Quality of Life ..................................................................................................................... 35
10.4.2 Anxiety and Depression ..................................................................................................... 35
10.4.3 Adverse Events .................................................................................................................. 35
10.4.4 Purpose of Quality of life, anxiety/depression and adverse event assessments .............. 35
10.4.5 Health Economics Analysis ......................................................................................... 36
11.0 PATHOLOGY REVIEW............................................................................................................ 36
12.0 BIOLOGICAL SUB-STUDIES ................................................................................................. 38
12.1 RAVES GENETIC STUDIES .......................................................................................................... 38
12.2. RAVES Genetic Study Aims ................................................................................................ 38
12.2.1 RAVES Genetic Study Logistics .................................................................................. 38
12.3. RAVES TISSUE BANKING STUDY AIMS ........................................................................................ 39
12.3.1 RAVES Tissue Banking Study Logistics ............................................................................ 39
12.3.2. Australian Prostate Cancer BioResource ......................................................................... 39
12.3.3. Tissue management policy ............................................................................................... 40
12.3.4 Potential tissue usage ........................................................................................................ 40
13.0 CRITERIA FOR ASSESSING TREATMENT OUTCOMES ..................................................... 40
13.1 BIOCHEMICAL FAILURE (BF) .......................................................................................................... 40
13.2 TIME TO BIOCHEMICAL FAILURE (TTBF) ......................................................................................... 41
13.3 BIOCHEMICAL FAILURE-FREE SURVIVAL (BFFS) ............................................................................. 41
13.4 QUALITY OF LIFE (QOL) AND ANXIETY/DEPRESSION ...................................................................... 41
13.5 ADVERSE EVENTS ........................................................................................................................ 41
13.5.1. NCI Common Terminology Criteria for Adverse Effects (CTCAE) v 3.0 .......................... 41
13.5.2. The Sexual Health Inventory for Men (SHIM) ................................................................... 41
13.6 OVERALL SURVIVAL...................................................................................................................... 41
13.7 DISEASE-SPECIFIC SURVIVAL ........................................................................................................ 42
13.8 TIME TO DISTANT FAILURE ............................................................................................................ 42

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 6 of 114
Amendment 2: dated 29 May 2014
13.9 TIME TO LOCAL FAILURE ............................................................................................................... 42
13.10 TIME TO THE INITIATION OF ANDROGEN DEPRIVATION (AD) .......................................................... 42
13.11 QUALITY ADJUSTED LIFE YEARS AND COST-UTILITY .................................................................... 42
14.0 DATA MANAGEMENT AND DOCUMENTATION .................................................................. 43
14.1 CASE REPORT FORMS (CRFS) .................................................................................................... 43
14.2 RECORDING OF DATA AND RETENTION OF DOCUMENTS .................................................................. 43
14.3 DATABASE MANAGEMENT AND QUALITY CONTROL .......................................................................... 44
15.0 ADVERSE EVENT SCORING AND REPORTING ....................................................................... 44
15.1 DEFINITIONS ........................................................................................................................... 44
15.1.1 Adverse Event (AE) ........................................................................................................... 44
15.1.2 Unexpected Adverse Event (UAE) .................................................................................... 44
15.1.3 SERIOUS ADVERSE EVENT (SAE) ....................................................................................... 45
15.1.4 ATTRIBUTION OF CAUSE OF AN ADVERSE EVENT ................................................................. 45
15.2 REPORTING ................................................................................................................................. 46
15.2.1 Adverse Event Reporting ................................................................................................... 46
15.2.2 Serious Adverse Event Reporting ............................................................................... 46
15.2.3 Other Situations requiring expedited Reporting................................................................. 48
16.0 STATISTICAL CONSIDERATIONS ............................................................................................. 48
16.1 TRIAL DESIGN.............................................................................................................................. 48
16.2 TREATMENT ASSIGNMENT ............................................................................................................ 49
16.3 STATISTICAL METHODS ................................................................................................................ 49
16.3.1 Patient Subsets to be Analysed ......................................................................................... 49
16.3.2 Pre-Randomisation Statistics and General Methods ......................................................... 49
16.3.3 Analysis of the Primary Objective ...................................................................................... 50
16.3.4 Analyses of Secondary Objectives .................................................................................... 51
16.4 SAMPLE SIZE AND POWER ........................................................................................................... 51
16.4.1 Anticipated Accrual Rate ................................................................................................... 51
16.4.2 Sample Size Calculation .................................................................................................... 51
16.5 ANALYSIS PLAN ........................................................................................................................... 53
16.5.1 Interim status and safety reports ....................................................................................... 53
16.5.2 Interim analysis No. 1 ........................................................................................................ 53
16.5.3 Interim analysis No. 2 ........................................................................................................ 53
16.5.4 Interim analysis No. 3 ........................................................................................................ 54
16.5.5 Interim analysis No. 4 ........................................................................................................ 54
16.5.6 Final analysis ..................................................................................................................... 54
16.6 EARLY CLOSURE CRITERIA .......................................................................................................... 54
17.0 RESPONSIBILITIES ..................................................................................................................... 55
17.1 INVESTIGATOR RESPONSIBILITIES ................................................................................................. 55
18.0 INDEPENDENT DATA MONITORING COMMITTEE (IDMC) ...................................................... 55
19.0 ETHICAL CONSIDERATIONS ..................................................................................................... 55
19.1 ETHICAL PRINCIPLES AND REGULATORY COMPLINCE ..................................................................... 55
19.2 ADHERENCE TO THE PROTOCOL ................................................................................................... 56
19.3 INFORMED CONSENT ................................................................................................................... 56

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 7 of 114
Amendment 2: dated 29 May 2014
19.4 CONFIDENTIALITY ........................................................................................................................ 56
20.0 INSURANCE AND COMPENSATION .......................................................................................... 57
21.0 PUBLICATION AND PRESENTATION POLICY ......................................................................... 57
21.1 REPORTING OF RESULTS ........................................................................................................ 57
21.2 TRIAL REGISTRY ..................................................................................................................... 57
REFERENCES ...................................................................................................................................... 58
APPENDIX I. AJCC STAGING SYSTEM (6TH EDITION) ................................................................... 63
APPENDIX II. ECOG PERFORMANCE STATUS ................................................................................ 64
APPENDIX III. CO-MORBIDITY INDEX ............................................................................................... 65
APPENDIX IV. EORTC QLQ-C30 & QLQ-PR25 .................................................................................. 66
APPENDIX V. HOSPITAL ANXIETY AND DEPRESSION SCALE (HADS) ....................................... 70
APPENDIX VI. NCI CTCAE V 3.0 ......................................................................................................... 73
APPENDIX VII. STUDY SCHEDULE FOR PATIENT ASSESSMENTS AND FOLLOW-UP .............. 75
APPENDIX VIII. PICTORIAL CONTOURING GUIDELINES ............................................................... 77
APPENDIX IX: BIOLOGICAL SUB-STUDY PATIENT QUESTIONNAIRE ......................................... 79
APPENDIX IX: CENTRAL PATHOLOGY REVIEW SYNOPTIC REPORT TEMPLATE ..................... 91
APPENDIX XI: SEXUAL HEALTH INVENTORY FOR MEN (SHIM) .................................................. 92
APPENDIX XII: HEALTH RESOURCE USAGE QUESTIONNAIRE ................................................... 93
APPENDIX XIII: RAVES PATIENT INFORMATION AND CONSENT FORM ........................... 103
APPENDIX XIV: RAVES BIOLOGICAL SUB-STUDY PATIENT INFORMATION AND CONSENT
FORM .................................................................................................................................................. 104

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 8 of 114
Amendment 2: dated 29 May 2014
List of Abbreviations
AC
Anal Canal
AD
Androgen Deprivation
AJCC
American Joint Committee on Cancer
ART
Adjuvant Radiotherapy
BaCT
Biostatistics and Clinical Trials, Peter MacCallum Cancer Centre
bF
Biochemical Failure
bFFR
Biochemical Failure-Free Rate
bFFS
Biochemical Failure-Free Survival
CPMP
Committee for Proprietary Medicinal Products
CRF
Case Record/Report Form
CT
Computed Tomography
CTCAE
Common Terminology Criteria for Adverse Effects
CTV
Clinical Target Volume
DICOM
Digital Imaging and Communications in Medicine
DRR
Digitally Reconstructed Radiographs
DVH
Dose Volume Histogram
ECOG
Eastern Cooperative Oncology Group
EORTC
European Organisation for Research and Treatment of Cancer
FROGG
Faculty of Radiation Oncology Genito-Urinary Group
EPE
Extraprostatic Extension
GCP
Good Clinical Practice
HADS
Hospital Anxiety and Depression Scale
HR
Hazard Ratio
HREC
Human Research Ethics Committee
I.V.
Intravenous
ICH
International Conference on Harmonisation
ICRU
International Commission on Radiation Units
IDMC
Independent Data Monitoring Committee
IMRT
Intensity-Modulated Radiation Therapy
LF
Left Femur
MLC
Multileaf Collimators
MRI
Magnetic Resonance Imaging
NCI
National Cancer Institute (USA)
N-IM
Non-Inferiority Margin
PSA
Prostate Specific Antigen
PSADT
Prostate Specific Antigen Doubling Time
PTV
Planning Target Volume
QA
Quality Assurance
QALY
Quality Adjusted Life Year
QoL
Quality of Life
RCT
Randomised Controlled Trial
RP
Radical Prostatectomy
RT
Radiotherapy
RTOG
Radiation Therapy Oncology Group
SAE
Serious Adverse Event
SRT
Salvage Radiotherapy
SVI
Seminal Vesicle Involvement
TMC
Trial Management Committee
TROG
Trans Tasman Radiation Oncology Group

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 9 of 114
Amendment 2: dated 29 May 2014
1.0 Executive Summary
Background
Radical Prostatectomy (RP) is the most frequently employed treatment modality for
clinically localised prostate cancer. Twenty to 50% of patients with clinically localised
disease will have positive margins, extraprostatic (extracapsular) extension or
seminal vesicle involvement with a subsequent high risk of relapse. Three recent
randomised trials have demonstrated a significant benefit of adjuvant post
prostatectomy radiotherapy (RT) in such high-risk patients, and many now regard this
approach as the new standard of care. However, adopting this approach will expose
nearly half of such patients to unnecessary radiotherapy and hence additional
inconvenience and potential treatment morbidity. Salvage radiotherapy, given early,
is recognised to be effective. There is, therefore, a need to compare these two
approaches to determine the utility of adjuvant versus early salvage timing of
therapy.
Aim
Primary Aim
To test the hypothesis that active surveillance with early salvage radiotherapy can be
considered non-inferior to standard treatment with adjuvant (immediate) radiotherapy
with respect to risk of biochemical failure (bF) in patients with extraprostatic (pT3)
disease and/or positive margins following RP.
Secondary Aims
To compare differences in quality of life (QoL), adverse event rates, survival duration
and time to the initiation of androgen deprivation between the two arms. A secondary
aim is to also assess the simultaneous effects of several potential prognostic factors
on each outcome.
Primary endpoint
Biochemical failure (bF) rate, defined as a prostate specific antigen (PSA) level ≥
0.40 ng/mL and rising following radiotherapy.
Secondary endpoints
QoL (EORTC QLQ-C30 and QLQ-PR25), anxiety/depression (Hospital Anxiety and
Depression Scale), adverse events (NCI CTCAE v3.0 and Sexual Health Inventory
for Men), biochemical failure-free survival, overall survival, disease-specific survival,
time to distant failure, time to local failure, cost utility analysis, quality-adjusted life
years and time to androgen deprivation.
Patient Eligibility
Inclusion Criteria
Prior Radical Prostatectomy (RP) for adenocarcinoma of the prostate.
Histological confirmation of adenocarcinoma of the prostate with the Gleason
score reported (Radical Prostatectomy specimen).
Patients must have at least one of the following risk factors:
o Positive margins
o Extraprostatic extension (EPE) with or without seminal vesicle
involvement (pT3a or pT3b) (Appendix I)

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 10 of 114
Amendment 2: dated 29 May 2014
Capable of starting RT within 6 months of RP (a requirement if randomised to
adjuvant RT arm). Note: RT commencement within 4 months of RP is
recommended, but up to 6 months is permitted.
Most recent PSA ≤ 0.10 ng/ml following RP and prior to randomisation
Eastern Cooperative Oncology Group (ECOG) performance status 0 – 1
(Appendix II)
Patient able to adhere to the specified follow-up schedule and complete the
Quality of Life and anxiety/depression self-assessments
Written informed consent obtained prior to randomisation
Completion of all pre-treatment evaluations
18 years and older
Exclusion Criteria
Previous pelvic RT
Androgen deprivation (AD) prior to or following RP
Evidence of nodal or distant metastases
Co-morbidities that would interfere with the completion of treatment and/or 5
years of follow-up
Concurrent cytotoxic medication
Hip prosthesis
Study Schema
This is a phase III multicentre randomised controlled trial. Eligible patients are
randomised to either:
Arm 1: Standard arm
Adjuvant RT (ART) commencing within 4 months of RP is recommended,
but up to 6 months is permitted.
64 Gy in 32 fractions to the prostate bed, or
Arm 2: Experimental arm)
Active surveillance with early salvage RT (SRT)
64 Gy in 32 fractions to the prostate bed
Trigger for SRT is PSA level ≥ 0.20 ng/ml. RT should commence as soon
as possible (no later than 4 months) following the first PSA measurement ≥
0.20 ng/mL.
Stratification
Patients will be stratified by seminal vesicle invasion, Gleason Score, pre-operative
PSA, margin positivity (no/yes), and radiotherapy institution.
Follow-up and evaluation
Patients will be assessed as follows. See study schedule (Appendix VII) for further
detail.
Toxicity of treatment: Clinical assessments of adverse events (urinary, bowel and
sexual function).
Disease status (PSA):
- Surveillance phase (Arm 2): 3 monthly PSA for first 5 years and 6 monthly
thereafter.
- Following radiotherapy (both arms): 6 monthly PSA until the end of the study.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 11 of 114
Amendment 2: dated 29 May 2014
Patient completed questionnaires:
- Hospital Anxiety and Depression Score (HADS), and EORTC QoL
assessment at pre-randomisation, day 1 of radiotherapy (immediate and
salvage arms), at the end of RT, 6 weeks following RT, annually post
randomisation..
- Sexual Health Inventory for Men (SHIM) pre-randomisation and annually post
randomisation.
- Health Resource Usage Questionnaire annually until trial follow-up ceases.
Quality Assurance
A quality assurance programme will provide timely feedback to participating clinicians
and improve quality of post-prostatectomy radiotherapy.
Treatment of biochemical failure following radiotherapy
A patient will be deemed as having a biochemical failure if the PSA is ≥ 0.40 ng/ml
and rising following adjuvant or salvage radiotherapy. It is recommended that the
initiation of androgen deprivation should not occur before 2 years unless one or more
of the following clinical criteria are met or exceeded. It is not mandatory to start
treatment if a criterion is exceeded:
a) A PSA doubling time (PSADT) of less than 12 months with a PSA level of
10.00 ng/ml or more. Investigators are free to delay intervention to higher PSA
levels.
b) A PSADT ≤ 6 months
c) Development of metastases
Statistical considerations
Patients will be randomised with a 1:1 allocation ratio. In order to determine non-
inferiority of SRT with respect to ART with 80% power, using a 10% non-inferiority
margin in the 5-year biochemical failure-free rate (assumed 74% versus 64%), 160
events are required to be observed. It is estimated that this will require a sample size
of 470 patients expected to be accrued over 4.7 years and followed for a further 5
years.
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 12 of 114
Amendment 2: dated 29 May 2014
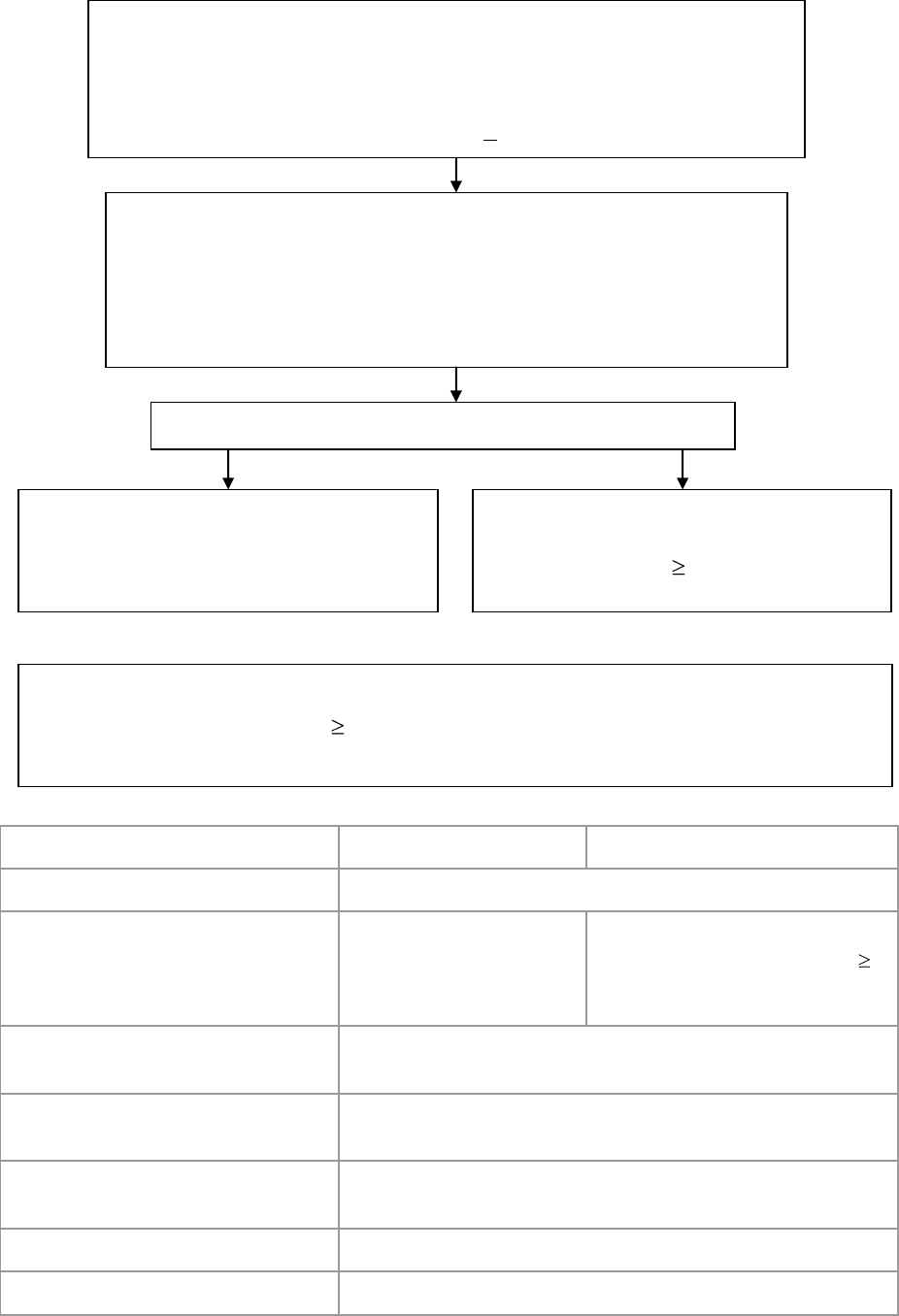
2.0 RAVES Schema
Assessment
ART
SRT
PSA – pre-randomisation
Pre-operative and post-operative
PSA – prior to RT
n/a
3 monthly for 5 years then 6
monthly, until rising PSA (
0.20 ng/mL), then day 1 of RT
PSA – following RT
6 weeks following completion of RT, then
6 monthly from randomisation until end of trial
QoL, HADS, Toxicity, SHIM -
routinely
Pre-randomisation, then annually from randomisation
QoL, HADS, Toxicity – around RT
Day 1 of RT, last day of RT, and 6 weeks after RT
completion
Follow-up visits
6 monthly from randomisation for 5 years; then annually
Health Resource Usage
Annually until trial follow-up ceases
Patient Selection
Radical prostatectomy
Histopathologically confirmed adenocarcinoma
pT3 or positive margins
Post-operative PSA < 0.10 ng/mL
Stratification
Pre-operative PSA
Gleason score
Margin positivity
Seminal vesicle involvement
Radiotherapy institution
RANDOMISATION
Arm 1 - ART
(Immediate) Adjuvant RT
64 Gy in 32 fractions
Arm 2 - SRT
Surveillance with early RT following
rising PSA ( 0.20 ng/mL)
64 Gy in 32 fractions
Primary endpoint: biochemical failure
Defined as 0.40 ng/mL following ART or SRT
(Also includes clinical failure or initiation of androgen deprivation therapy)

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 13 of 114
Amendment 2: dated 29 May 2014
3.0 Introduction
Radical Prostatectomy (RP) is the most frequently employed treatment modality for
clinically localised prostate cancer. In Australasia approximately 3,300 men undergo
RP per annum. While RP is an extremely effective treatment for organ confined
disease, clinical staging of prostate cancer is notoriously inaccurate and frequently
underestimates the extent of disease. In up to 50% of patients with clinically localised
disease prior to RP, the final pathology reveals extracapsular extension, seminal
vesicle involvement or positive margins1-4.
Factors that predict for biochemical recurrence after Radical Prostatectomy
There are a number of factors that predict for biochemical recurrence following RP5-
14. These include
a) Pre-operative PSA level
b) Rapid PSADT prior to RP
c) Gleason score
d) Lymph node involvement
e) Extracapsular extension (pT3a)
f) Seminal vesicle involvement (pT3b)
g) Positive margins
Risk of positive margins
Following RP the positive margin rate ranges between 13% and 40%9,15-23, with
multiple positive margins (≥ 2) in up to 30% of patients20,21,24. The prostate apex is
the most common site of positive margins17,18,20,24. On multivariate analysis, a
positive surgical margin is an adverse prognostic factor with a 5 year biochemical
progression rate between 20 to 40%9,20-26 and a relative risk of recurrence ranging
between 1.5 to 1.99,20,22,23.
Radiotherapy following Radical Prostatectomy
In patients with positive margins and/or pT3 disease, the 5 year biochemical disease
free survival is between 37 to 74% and local control rate 60 to 80%6,7,27,28.
Adjuvant radiotherapy (ART) to the prostate bed is frequently employed in the setting
of pT3 disease or positive margins (node negative and postoperative PSA
undetectable). The aim of ART in these high risk patients is to eradicate local
microscopic disease in the prostate bed.
The indications for salvage RT (SRT) include a persistently elevated PSA following
RP, a rising PSA after an initial nadir, and a palpable or biopsy proven local
recurrence. Sixty to 90% of patients will have a reduction in PSA following SRT,
suggesting the prostate bed, at least in part, is a site of failure in most patients29,30. If
delivered early, (PSA < 1.0 ng/mL), salvage RT is effective in controlling local
disease with up to 68% of patients free from subsequent PSA relapse29,31-33.
However, in many retrospective series salvage RT was not “early,” and the relapse
rate was greater than 80%34.
In the post-prostatectomy setting the optimal timing of radiation is yet to be defined,
although there is increasing evidence supporting the use of ART in patients with pT3
disease and/or positive margins. Three recent randomised controlled trials (RCTs)

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 14 of 114
Amendment 2: dated 29 May 2014
have shown improved biochemical progression free survival and local control in
patients treated with ART compared with observation35-37.
In the landmark EORTC study, 1005 patients were randomised to ART (60Gy within
16 weeks of RP) or wait-and-see. Patients were eligible if they had at least one of the
following risk factors: positive margins, extracapsular extension or seminal vesicle
invasion. At a median follow-up of 5 years, the biochemical progression free survival
was 74.0% in the ART arm and 52% in the wait-and-see arm (p <0.0001, HR 0.48)35.
Collette et al investigated the homogeneity of this benefit across all subgroups. The
benefit of ART was substantial in all subgroups (including both those with seminal
vesicle involvement and extracapsular extension). However, the authors noted that
patients with negative margins may benefit to a lesser extent than the other
subgroups (heterogeneity P = 0.0568)38.
There was no significant difference in grade 3 or 4 late toxicity (4.2% versus 2.6%, p
= 0.07), but continence and potency were not formally assessed. Quality of Life
(QoL) was analysed in a subgroup of 100 patients. Patients completed the EORTC
QLQ-C30 and the prostate specific PR25 questionnaires. There was no difference in
global QoL, although, there was a significant detriment in the genitourinary,
gastrointestinal and sexual quality of life in the patients receiving adjuvant RT39.
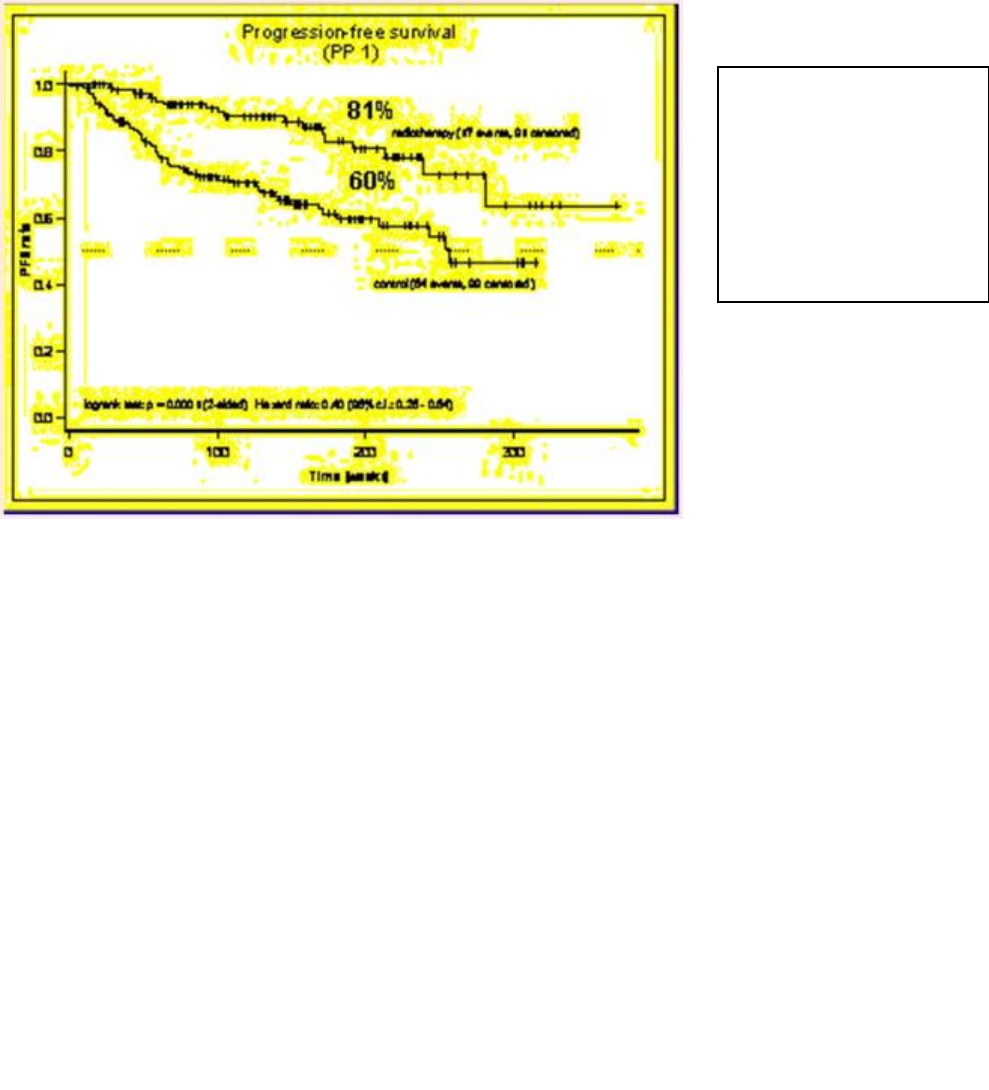
Wiegel et al also randomly assigned patients with positive margins and/or pT3
disease to ART (60Gy) or wait-and-see (ARO 96-02 AP09/95). At a median follow-up
of 40 months there was a 20% improvement in biochemical control at 4 years in the
ART arm (80% versus 60%, p < 0.0001, HR = 0.4). In this study 32 patients (21%)
randomised to ART were told by their Urologist not to have RT and did not receive
RT. An intention to treat analysis was used37.
In a third trial (SWOG 8794), 473 patients with pT3 disease and/or positive margins
were randomised to ART (60 to 64 Gy) or observation. At a median follow-up of 9.7
years, the 10 year biochemical disease free survival (bDFS) was 45% for the ART
arm and 20% for the observation arm. In addition, patients in the ART arm had a
reduced need for androgen deprivation (30% versus 40%) and a delayed time to
androgen deprivation (12.5 versus 10 years). There was a trend to both a reduction
in the rate of distant metastases and an improvement in overall survival, but these
were not significant. At 2 years there was no difference in QoL. Key results from
these studies are shown in figures 1-3 and table 136.
Figure 1:
Biochemical disease free
survival with adjuvant
radiotherapy (2005)
Bolla et al35

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 15 of 114
Amendment 2: dated 29 May 2014
Adjuvant RT Obs HR p
PSA relapse 53% 77% 0.51 <0.001
(<0.4ng/ml)
PFS 67% 48% 0.59 0.001
Mets -free 71% 61% 0.8 0.17
O/S 74% 63% 0.76 0.11
Table 1:
Endpoints of 2005
SWOG study.
Swanson et al36
Figure 2:
Planned subgroup analysis
of the Bolla study35 (2005)
demonstrates that patients
with seminal vesicle
involvement benefit from
adjuvant radiotherapy.
L. Collette et al38
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 16 of 114
Amendment 2: dated 29 May 2014
A major criticism of the three randomised trials is that there was no standard
management for patients on observation that developed relapse. Relapsing patients
were managed with observation, SRT, or androgen deprivation. Only 35% to 55% of
patients in the observation group that relapsed received SRT. In addition, the pre-RT
PSA and PSA doubling time prior to SRT were not reported. Salvage RT was often
delivered too late as a number of patients had documented locoregional recurrence
at the time of SRT35-37.
Although these three RCTs show that compared with observation, ART improves the
5 year biochemical control by approximately 20%, active surveillance and delaying
RT until the salvage setting has a number of advantages. Between 45% and 60% of
the patients in the observation arm do not develop a biochemical relapse. This
suggests that approximately 50% of patients treated with ART receive unnecessary
treatment and are exposed to the potential toxicities of RT. Therefore, delaying RT
until the salvage setting allows a better selection of patients that are more likely to
benefit from RT. Furthermore, if SRT is required, there is a longer interval between
RP and RT, allowing more time for potential recovery of urinary function and potency.
Radiotherapy is most effective when the tumour burden is at its lowest. With the
availability of serum PSA tests, a reliable tumour marker following RP, delaying RT
until early biochemical recurrence is probably as effective as ART. However, there
are a number of series in the literature reporting the inefficacy of SRT. In these series
many patients received SRT for gross disease (a palpable or biopsy proven local
recurrence) or a high PSA. As the SRT was delivered too late, the results were
suboptimal. There is increasing evidence supporting SRT, provided the RT is
delivered at early biochemical recurrence. A number of authors have reported
improved biochemical control if SRT is delivered when the PSA ≤ 1.0 ng/mL
compared with a PSA > 1.0 ng/mL14,40,41. Furthermore, Stephenson et al noted better
results in patients with a pre-RT PSA of ≤ 0.6 ng/mL, compared with those with a
PSA between 0.61 and 2.0 ng/mL. Based on this evidence, it is critical SRT is
delivered early, preferably when the PSA is ≤ 0.6 ng/mL42.
Figure 3:
Biochemical
progression free
survival with 2005
ARO 96-02 study.
T. Wiegel et al37

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 17 of 114
Amendment 2: dated 29 May 2014
Quality of Life
Quality of life (QoL), the assessment of the subjective perception of social,
psychological and somatic well-being and function, becomes a very important
outcome measure when evaluating different treatment approaches that may have
similar survival outcomes. Understanding the implications of toxicities can be central
to the utility analysis that follows a finding of lack of inferiority in the salvage program.
There are a number of series comparing the QoL in patients following RP, external
beam radiotherapy (EBRT) and brachytherapy, showing that urinary, erectile and
bowel dysfunction do impact negatively on QoL. Pearce et al examined the effect of
salvage RT plus 2 years of androgen suppression on QoL. The authors reported
bowel and bladder function deteriorated markedly at the end of RT but recovered
after, and showed only a minor persistent disturbance in the 2 year post RT period
which was of minimal clinical significance43.
Although it appears salvage RT has minimal impact on QoL, to date a direct
comparison between the QoL in men treated with adjuvant RT and those with
salvage RT has not been evaluated in a randomised trial.
Co-morbidity index
A significant covariate potentially affecting survival are co-morbidities. An index of
risk of death from non-cancer causes has been devised by Post et al44, using a
modification of the Charlson co-morbidity index (Appendix IV). Using a simple
cumulative scoring of major disease categories, a highly significant impact was found
in men aged less than 70 years with prostate cancer.
Summary
In the setting of extraprostatic (pT3) disease and/or positive margins following RP,
there is retrospective evidence and three RCTs showing an improvement in
biochemical and local control in patients treated with ART compared with
observation. What has not been addressed in a RCT is whether surveillance with
early SRT is as effective as ART.
4.0 Objectives and Endpoints
4.1 Primary Objective
The principal objective of the trial is to test the hypothesis that active surveillance
with early salvage radiotherapy can be considered non-inferior to standard treatment
with adjuvant (immediate) radiotherapy with respect to risk of biochemical failure (bF)
in patients with pT3 disease and/or positive margins following RP.
4.2 Secondary Objectives
Secondary objectives will include a comparison of the two treatment arms with
respect to each of the secondary endpoints outlined in section 4.3.2.
In addition, a prognostic factors analysis will be performed for each time-to-event
endpoint (sections 4.3.1 and 4.3.2).

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 18 of 114
Amendment 2: dated 29 May 2014
4.3 Endpoints
4.3.1 Primary Endpoint
Biochemical failure (bF): PSA ≥ 0.40 ng/ml and rising following RT
4.3.2 Secondary endpoints
Quality of life (QoL)
Adverse events
Anxiety/depression
Biochemical failure-free survival
Overall survival
Disease-specific survival
Time to distant failure
Time to local failure
Time to the initiation of androgen deprivation
Quality-adjusted life years
Cost-utility
5.0 Trial Design
5.1 Trial Description
The research design is a two-arm, randomised, phase III, multicentre, non-inferiority
trial.
5.2 Study Arms
Eligible patients will be randomised to either:
Arm 1 (standard arm): Adjuvant RT commenced within 6 months of RP. Note:
RT commencement within 4 months is recommended, but up to 6 months is
permitted.
Arm 2 (experimental arm): Active surveillance with early salvage RT following a
rising PSA (PSA level ≥ 0.20 ng/mL prior to radiotherapy).
For both arms, RT consists of 64 Gy in 32 fractions delivered over 6.5 weeks to the
prostate bed.
5.2.1 Stratification
Randomised patients will be stratified (using the minimisation technique) according to
the following criteria:
Preoperative PSA (as a continuous variable)
Gleason Score (from RP specimen; as a continuous variable)
Surgical margins (positive/negative)
Seminal vesicle involvement (pT3b) (Yes/No)
Radiotherapy institution
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 19 of 114
Amendment 2: dated 29 May 2014
6.0 Patient Selection
6.1 Inclusion Criteria
All of the following must apply:
Prior Radical Prostatectomy (RP) for adenocarcinoma of the prostate.
Histological confirmation of adenocarcinoma of the prostate with the Gleason
score reported (Radical Prostatectomy specimen).
Patients must have at least one of the following risk factors:
o Positive margins
o Extraprostatic extension (EPE) with or without seminal vesicle
involvement (pT3a or pT3b) (Appendix I)
Capable of starting RT within 6 months of RP (a requirement if randomised to
adjuvant RT arm). Note: RT commencement within 4 months is
recommended, but up to 6 months is permitted.
Most recent PSA ≤ 0.10 ng/ml following RP and prior to randomisation
Eastern Cooperative Oncology Group (ECOG) performance status 0 – 1
(Appendix II)
Patient able to adhere to the specified follow-up schedule and complete the
Quality of Life and anxiety/depression self-assessments
Written informed consent obtained prior to randomisation
Completion of all pre-treatment evaluations
18 years or older
6.2 Exclusion Criteria
None of the following must apply:
Previous pelvic RT
Androgen deprivation (AD) prior to or following RP
Evidence of nodal or distant metastases
Co-morbidities that would interfere with the completion of treatment and/or 5
years of follow-up
Concurrent cytotoxic medication
Hip prosthesis
6.3 Patient Withdrawal
A patient may withdraw, or be withdrawn, from trial treatment for the following
reasons:
Deterioration whilst on therapy
Unacceptable toxicity
Intercurrent illness which prevents further treatment
Withdrawal of consent by patient
Any alterations in the patient’s condition which justifies the discontinuation of
treatment in the investigator’s opinion
If the patient discontinues the study treatment due to an adverse event potentially
related to study treatment, they must be followed weekly for 4 weeks or until
resolution or stabilisation of the event, whichever occurs first. Patients who
discontinue study treatment but have neither died nor withdrawn consent should be
encouraged to complete the scheduled evaluations and continue to be followed up
according to the protocol.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 20 of 114
Amendment 2: dated 29 May 2014
7.0 Patient Consent and Randomisation
7.1 Screening and Randomisation
Patients w ill be randomised via an internet-based randomisation system. The
Trial Centre will provide each participating site w ith user account s to access the
web-based system. Sites will be notified of treatment arm allocation at the time of
randomisation.
To randomise a patient, sites must complete electronic case report forms (CRFs)
to document eligibility and stratification factors. Prior to patient randomisation,
the investigator should ensure that all of the follow ing requirements are met:
Informed consent has been obtained prior to performing any study specific
procedures.
The patient meets all inclusion criteria and none of the exclusion criteria
must apply.
All pre-randomisation assessments and investigations have been
performed.
The eligibility checklist has been completed, signed and dated.
Patients must not start treatment before the site has received confirmation of
randomisation results. Patients will not be randomised if treatment has started.
7.2 Consent for optional biological sub-studies
Patients will be approached about the optional biological sub-studies six months after
randomisation. If patients wish to participate, they must sign a separate consent form
for the sub-studies. If for any reason consent to the sub-studies is delayed, patients
may choose to join the sub-studies at any point up until 6 months after recruitment
has closed.
8.0 Technical Guidelines
8.1 Statement of treatment aim
In both treatment arms, radiotherapy will consist of a conventionally fractionated
radical course of treatment that aims to deliver 64 Gy in 32 fractions over 6.5 weeks
to the prostate bed (adapted from FROGG consensus guidelines45).
8.2 Treatment Schedule
Arm 1: Standard arm
Adjuvant RT commenced within 6 months of RP. Note: RT commencement within 4
months of RP is recommended, but up to 6 months is permitted.
Arm 2: Experimental arm
Active surveillance with early salvage RT following a rising PSA (PSA level ≥ 0.20
ng/ml prior to RT). SRT should be delivered as soon as possible (no later than 4
months) following the first PSA measurement ≥ 0.20 ng/mL.
8.3 Planning Simulation
A planning CT scan is required to define the clinical target volume (CTV), planning
target volume (PTV) and the organs at risk (OAR). Intravenous contrast with delayed

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 21 of 114
Amendment 2: dated 29 May 2014
scanning (at least 10 minutes) and MRI may be employed to aid delineation of the
anastomosis/penile bulb. If the bladder has been filled with contrast then a pixel-by-
pixel density correction is inappropriate and a “bulk” correction, using typical values
for normal tissue, should be applied.
Additional planning guidelines are as follows:
1. Contiguous axial slices are taken from the bottom of the SI joints to 2 cm
below the ischial tuberosities.
2. The maximal CT slice thickness recommended is 2.5 - 3.0 mm and will be no
more than 5.0 mm.
3. The planning CT is acquired with the patient in the supine position. The patient
is simulated and treated with a full bladder. It is recommended that patients be
encouraged to maintain an empty rectum at simulation and during treatment46.
As a guide, it is expected that the rectal diameter shall be < 5cm. Centres shall
provide details of their rectal and bladder filling protocols on the Facility
Questionnaire, available from http://www.trog.com.au/TROG-0803-trial-
documents.
4. Immobilisation is as per the treating centres policy. Immobilisation and
positioning will be consistent between the planning CT scan and throughout
the treatment.
8.4 Daily treatment position
Daily treatment position shall be consistent between CT planning and throughout
treatment. Similarly, bladder and rectal filling and use of any immobilisation devices
shall be consistent between simulation and treatment.
8.5 Target volume definitions/Field Borders
Pictorial representation of target volumes may be found in Appendix VIII.
8.5.1 Clinical Target Volume (adapted from FROGG consensus guidelines45)
In addition to the planning CT scan and modalities outlined in section 8.3, the
following information may also be utilised to assist in CTV delineation:
Preoperative imaging
Operation report and discussion with the operating surgeon
Histopathology report
Technique:
The CTV structure will be named “CTV” at the planning computer. Delineation of the
CTV surgical bed (based on CT slice thickness of 2.5-3.0 mm) shall be as defined
below:
1. Inferior border: The inferior border of the CTV will be 5-6 mm inferior to the
vesicourethral anastomosis (depending on CT slice thickness), but should be
extended inferiorly if necessary to include all tumour bed clips (i.e. non-vascular).
i. The anastomosis can be identified on axial, coronal and sagittal
reconstructions as the slice inferior to the last slice where urine is visible. To
assist with the treatment plan review process, the CT slice containing the
anastomosis shall contain a contour or some other identifier indicating the

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 22 of 114
Amendment 2: dated 29 May 2014
position of the anastomosis. This contour (or appropriate identifier) shall be
labelled “anastomosis”.
ii. When the anastomosis is not clearly defined, the inferior border will be the first
slice superior to the penile bulb.
2. Anterior border:
i. From the inferior border of the CTV to 3cm superior, the anterior border of the
CTV is the posterior aspect of the symphysis pubis. In certain circumstances
(eg positive margins at bladder neck) it may be necessary to extend this
height up to the level of the superior pubic symphysis.
ii. More superiorly, the anterior border of the CTV should encompass at least the
posterior 1.5cm of the bladder.
3. Posterior border: The space delineated by the levator ani and anterior rectal wall
is at risk for recurrence and should be encompassed in the CTV if rectal dose
constraints allow. As a minimum, the lateral posterior border must approximate
the anterior rectal wall in the inferior portion. Ensure a minimum 2 cm margin
from the posterior extent of the CTV to the posterior rectal wall to prevent the
entire circumference of rectum receiving the full radiation dose. In creating the
CTV posterior border, take into consideration that the PTV expansion and the
95% isodose must not encompass the full circumference of the rectal wall.
More superiorly, the posterior border of the CTV is the anterior mesorectal
fascia. This is often delineated by the posterior border of the residual seminal
vesicles/seminal vesicle bed.
4. Lateral border: The medial border of the levator ani muscle or obturator internus
muscle (pelvic side wall) superiorly.
5. Superior border:
i. If the seminal vesicles are not involved, the superior border should encompass
all of the seminal vesicle bed as defined by post surgical changes and non
vascular clips. The tips of the residual seminal vesicles do not need to be
included.
ii. If the seminal vesicles are pathologically involved by tumour, ensure any
residual seminal vesicles are also included in CTV.
8.5.2 Planning target volume (PTV)
The PTV is created by adding a 10 mm margin in all directions to the CTV, in order to
account for day-to-day variation in patient positioning/set-up and patient and organ
motion.
Planning target volume delineation:
a) Defined as a uniform margin of 10mm from CTV to PTV for the entire dose,
the PTV shall be named “PTV” at the treatment planning computer.
Ensure the PTV expansion and the 95% isodose do not encompass the full
circumference of the rectal wall.
b) If the V40 rectal DVH constraints (section 8.9) cannot be met (i.e. more
than 60% of the rectum is receiving > 40Gy), then we recommend reducing
the posterior margin to a minimum of 0.5 cm keeping other margins to 1 cm
using the auto expansion tool in the planning software. If the V40 still
cannot be met, please submit case to the reviewing team who will then be
in contact to discuss case.
c) If the V60Gy constraint cannot be met despite reducing the posterior
margin to 0.5cm, in some cases it may be necessary to consider a two
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 23 of 114
Amendment 2: dated 29 May 2014
phase technique. Any case where a two phase technique is being
considered should first be discussed with a member of the RAVES QA
committee.
Note that for IMRT treatments a 10 mm uniform expansion is mandatory due to the
increased potential for geographic miss. In some clinical scenarios, it may be
appropriate to manually alter the posterior margin, if sound clinical justification is
provided and daily imaging is done. For advice or additional information, contact the
RAVES QA team via the web site: http://www.trog.com.au/TROG-0803-trial-
documents.
Centres should consult the QA Technical Advisory Committee if the rectal DVH
constraints cannot be met.
8.6 Dose Prescription and Fractionation
The radiation dose for both arms is 64 Gy in 32 fractions over 6.5 weeks to the ICRU
50 reference point which is the centre of the planning target volume, but may be the
intersection of the beam axes if this is close to the centre of the volume47 When using
an IMRT technique (permitted following RAVES IMRT site credentialing), the dose is
typically prescribed to a volume. For IMRT plans, the dose should be prescribed as
follows: the D98 (dose covering 98% of the PTV) shall be at least 95% of the total
dose. The mean and median doses will be within -1% and +2% of 64 Gy (63.4 – 65.3
Gy). The maximum dose (D2, dose to 2% of the PTV) shall be no more than 107% of
the total dose.
Fractional dose will be 2 Gy delivered once a day, 5 days per week or 9 days per
fortnight according to departmental policies.
For 3DCRT techniques the prescribed dose shall be reported and normalised to the
reference point and labelled as a separate point called “ICRU reference point”. The
reference point must adhere to ICRU50 criteria for reference points. The 100%
isodose or equivalent absolute dose in Gray (Gy) should intersect this point.
The ICRU Reference point shall be selected according to the following general
criteria:
The dose at the point should be clinically relevant and representative of the
dose throughout the Planning Target Volume (PTV).
The point should be easy to define in a clear and unambiguous way.
The point should be selected where the dose can be accurately determined
(physical accuracy).
The point should be selected in a region where there is no steep dose
gradient.
These recommendations will be fulfilled if the ICRU Reference point is located:
Always at the centre, or in the central parts, of the Planning Target Volume,
and
When possible on or near the intersection of the beam axes.
When using an IMRT technique (permitted following RAVES IMRT site credentialing),
for the purpose of plan review using the SWAN software, it will be necessary to report

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 24 of 114
Amendment 2: dated 29 May 2014
the dose to a point that is representative of the traditional ICRU 50 point. For IMRT,
this point may be selected after the plan has been optimised, and can be anywhere
in the centre of the volume as long as it is clinically significant. The dose to the ICRU
50 point will be 64 Gy total dose.
8.7 Treatment Planning and Dosimetry
Treatment planning will be carried out with a 3D planning system which shall have,
as a minimum, the following capabilities:
Able to handle at least 40 axial CT slices at 256 x 256 pixel resolution.
Allows definition of multiple structures in 3D from CT data.
Provides a 3D dose calculation algorithm (e.g. convolution / superposition
algorithm) capable of performing calculations which account for variations in
scatter in the presence of 3D-(CT) defined heterogeneities.
Can provide permanent record of each treatment plan, both in electronic form
(data backup) and hard copy.
Can provide hardcopy of superimposed isodose distributions on axial CT
images (sagittal and coronal planes desirable).
Can provide digitally reconstructed radiographs (DRRs) with superimposed
target volume, critical structure contours and treatment aperture.
Provide planning data in DICOM RT or RTOG format that can be uploaded to
CQMS. See Section 8.8, number 4 for further details.
Sites that have completed the RAVES IMRT credentialing program (see Section
8.12.2) may use an inverse planned IMRT technique. All IMRT plans must be
independently verified by the local physics department. The treatment planning
computer must therefore be capable of exporting the treatment plan to the local dose
measurement/verification software for direct measurement/verification by the
physicist.
Treatment planning guidelines include:
1. During treatment planning, if the bladder has been filled with contrast then a
pixel-by-pixel density correction is inappropriate and a “bulk” correction,
using typical values for normal tissue, should be applied.
2. An isocentric technique will be used.
3. All fields are delivered each day.
4. Treatment is delivered with a linear accelerator with ≥ 6 megavoltage
photons.
5. A minimum of 3 fields shall be used.
6. Shielding using blocks or multileaf collimators (MLC) is required to conform
the high dose region to the PTV using at least 5 half-value-layers of
attenuating material thereby minimising dose to normal structures. Real or
virtual wedges may be employed if necessary to achieve the required target
volume homogeneity.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 25 of 114
Amendment 2: dated 29 May 2014
7. IMRT techniques are permitted in this trial ONLY when the site has
completed the RAVES-specific IMRT credentialing process. Centres wishing
to use IMRT techniques should contact the Technical Advisory Committee
for details of the credentialing process at http://www.trog.com.au/TROG-
0803-trial-documents. The IMRT credentialing process is described in
Section 8.12.2 and treatment planning guidelines in Section 8.8.
8.8 Dose Distribution/Reporting
The following guidelines shall be followed for dose distribution and reporting:
1. The absorbed dose at the ICRU reference point shall be reported (see
Section 8.6 for details).
2. The mean and median doses will be within -1% and +2% of 64 Gy (63.4 –
65.3 Gy).
3. PTV homogeneity shall be constrained as follows:
a. The maximum dose (D2, dose to 2% of the PTV) shall be no more
than 107% of the total dose.
b. Minimum isodose covering the PTV (D98, dose covering 98% of the
PTV) shall be at least 95% of the total dose.
4. For treatment plans created with an inverse IMRT technique (RAVES IMRT
credentialed sites only):
a. The maximum dose should be contained within the CTV, and must be
contained within the volume bounded by the PTV.
b. The minimum dose (defined as the D98) of the CTV shall be 64 Gy
c. The dose outside the PTV will be minimised.
d. It is recognised that treatment plans created with an IMRT technique
demand extra precision in treatment delivery due to the presence of
high dose gradients. Therefore planning techniques shall be robust in
the presence of inter (and intra-) fraction organ motion.
5. Centres should provide the below data for all participants for QA review.
This data should be DE-IDENTIFIED (and submitted for review via the
Central Quality Management System (CQMS) as zipped files). Information
on CQMS user accounts and training are available on the TROG website
(www.trog.com.au).
The radiotherapy treatment plan export will be automatically de-identified at
the point of upload to CQMS. The participant name and ID will be replaced
with the trial identifier and the participant reference, eg 13.02 ABC123.
Digital Export of Radiotherapy Treatment Plan

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 26 of 114
Amendment 2: dated 29 May 2014
An electronic export of the radiotherapy treatment planning data file from the
treatment planning system is required. The preferred file format is DICOM-
RT; however RTOG can be accepted if no other format is available. The use
of RTOG is discouraged as this format is not widely accepted by plan review
software.
This electronic export must be in absolute dose (Gy) and include the:
CT dataset or other imaging dataset used for plan calculation,
ensuring all files are included.
Planning files. Please ensure the plan is calculated to the
correct specifications and approved. There will be one plan file,
generally prefixed RP.
Structure files, Please ensure all structures including CTV/PTVs
are exported and named according to protocol specifications.
There will only be one file, generally prefixed RS. The exported
plan should contain only those structures required for the QA
review. All other structures (e.g. those created for the purpose of
IMRT optimisation) shall be removed prior to export.
Dose files. Please ensure the dose matrix is inclusive of all
structures. There will be one dose, generally prefixed RD.
DVH data. The sampling resolution for DVH data shall be 0.1 cm
for contoured structures, 0.2 cm for all other tissue. The bin width
shall be 0.010 Gy. Data shall be presented in absolute dose.
All these exported files need to be uploaded as a single zipped file into
CQMS.
Screen Captures of ICRU reference point and isodose distribution
In addition to the electronic data file, a screen capture from the planning
system (JPEG image) is required. It is essential the screen capture follows
the format below; as it will be used to verify the accuracy of the electronic
plan imported into the plan review software.
Format
The screen capture should be taken at the intersection of the
ICRU reference point, and demonstrate the absolute dose (Gy)
at this point.
The image should display the location of ICRU reference point
(x, y, z co-ordinates) in three (3) viewing planes (sagittal,
transverse and coronal).
Relevant isodose lines should be displayed in absolute dose
(Gy), colour and clearly labelled. As a minimum the max, 100%,
98%, 95%, 90%, 70%, 50%, 20%, 2% isodose lines should be
included.
CT imaging needs to be clearly visible.
Dose Volume Histogram

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 27 of 114
Amendment 2: dated 29 May 2014
Screen Capture (JPEG image) of the DVH, clearly showing the CTV, PTV,
(seminal vesicles if delineated as a separate structure), rectum, left femur
and bladder. This image will be used to verify accuracy of DVH display in
the plan review software and must be in colour.
7. Each treatment plan shall be computed with the following specifications:
Dose matrix maximum grid spacing will be no greater than 2.5mm x
2.5mm x 2.5mm.
Data shall be presented in “absolute dose” as export in relative dose
mode is not fully supported by some commercial systems.
All exported data shall be contained in a single directory for each patient.
The sampling resolution for the dose volume histogram data shall be 0.1
cm for contoured structures, 0.2 cm for all other tissue. The bin width shall
be 0.1 Gy or 10 cGy.
Exported data shall include the DRR for each field.
The target and organs at risk will be named as defined in section 8.5.1:
CTV, PTV, Rectum, LF (for the left femur), Bladder, Anastomosis and AC
(for the anal canal if contoured).
Contouring shall be included on all relevant CT slices for all structures.
The interpolation algorithm on the treatment planning computer may be
used if it is not normal clinical practice to contour on all slices.
Size restrictions for files uploaded to CQMS: Exported data files should
not exceed 50 Mb.
8. DRRs may be provided in jpeg image format
Prior to submission for QA case reviews, all RT material must be DE-IDENTIFIED in
terms of patient names, medical record numbers and other personal identifying
information, and re-labelled according to the registration numbers allocated to the
patient for the trial.
8.9 Normal tissue contouring and dose constraints
Rectum: The external surface of the rectum shall be named “Rectum” at the
treatment planning computer and should be contoured as a solid organ superiorly
from the recto-sigmoid junction (where the rectum turns horizontally into the sigmoid,
usually at the inferior border of the sacro-iliac joint) to 15mm inferior to the inferior
border of the CTV. The rectal contours should extend at least 15mm superior and
inferior to the CTV. It is recommended that patients be encouraged to maintain an
empty rectum at simulation and during treatment. As a guide, it is expected that the
rectal diameter shall be <5cm.
Left femur: Shall be named “LF” at the treatment planning computer and will be
contoured from the acetabulum to the inferior edge of the treatment field.
Bladder: The whole external wall of the bladder shall be named “Bladder” at the
treatment planning computer and should be contoured to the slice superior to the

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 28 of 114
Amendment 2: dated 29 May 2014
anastomosis. Note: During treatment planning, if the bladder has been filled with
contrast then a pixel-by-pixel density correction is inappropriate and a “bulk”
correction, using typical values for normal tissue, should be applied.
Anal canal: Whilst it is not a protocol requirement to delineate the anal canal, if it is
delineated, it shall be named “AC.”
8.9.1 Dose constraints
Rectum: The rectal dose shall be constrained as follows:49-51
volume of rectum receiving 60Gy shall be < 40%
volume of rectum receiving 40Gy shall be < 60%
Femoral heads: The tolerance doses for femoral heads (FH) are poorly defined but
the recommended volume irradiated should not exceed these constraints:
volume of left femur (LF) receiving 35Gy shall be < 100%
volume of LF receiving 45Gy shall be < 60%
volume of LF receiving 60Gy shall be < 30%
8.10 Treatment Equipment Specifications/Physical Factors
Patients will be treated on a megavoltage linear accelerator with the following
facilities:
Capable of delivering at least 6 MV photons
The minimum source-to-axis distance is 100cm
Beam modification (i.e. real or virtual wedges; blocks and/or MLC);
A treatment couch with vertical movement < 3 mm for patients up to 150 kg;
Facilities for taking routine images, with electronic portal imaging devices
(EPID), radiographic film, kV imaging or cone beam CT (CBCT) which can be
used to verify orientation and position of the radiation fields relative to
anatomical structures to within 1 mm.
All monitor unit calculations will be independently verified (i.e. independent of the
normal planning system).
8.11 Treatment Verification
To verify field size and shielding, each portal shall be visually checked on at least
one occasion during the first week of treatment.
To verify patient position, at least two port films or images (e.g. AP and one lateral)
will be acquired in the first week of treatment and then weekly. Films or images will
be compared with DRRs to detect systematic differences between the position of the
radiation field and the intended (planned) field.
It is recognised that treatment plans created with an IMRT technique demand extra
precision in treatment delivery due to the presence of high dose gradients. Without
the use of soft tissue imaging, it may be difficult to verify accurately the target
position. Soft tissue imaging is not mandatory in this trial. However, centres using
IMRT techniques are expected to demonstrate that an imaging policy is in place,
which is appropriate for the margins specified in this protocol. Sites will be required to
describe their imaging protocol in detail on the Facility Questionnaire.
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 29 of 114
Amendment 2: dated 29 May 2014
8.12 Quality Assurance Program
RT technical reviews involving audit of the planning and treatment data will be
conducted. Reviewed parameters and protocol deviations will be in accordance with
the TROG Policy and Procedures Manual (Quality Assurance Statement of Minimum
Requirements for Clinical Trials). Results will be reported to the TMC at least 6
monthly, and at the TROG meetings bi-annually.
The target volume specifications in this protocol are based on the recommendations
of the Faculty of Radiation Oncology Genito-Urinary Group (FROGG) Consensus
Guidelines on post-prostatectomy radiation therapy45. To ensure consistency in
contouring treatment volumes and adherence to the technical requirements of the
trial protocol, centres will participate in a “dry run” and submit electronic treatment
planning data in either RTOG Data Exchange or DICOM-RT format for all trial
patients for timely review. Each centre shall successfully complete the dry run before
they commence registering patients into the trial.
Centres that wish to use inverse IMRT planning techniques must complete the
RAVES IMRT credentialing process summarised in Section 8.12.2.
Contact details for the RT QA team are listed on the RT QA website:
http://www.trog.com.au/TROG-0803-trial-documents
8.12.1 Dry run
8.12.1.1 Each treating clinician will be required to contour a CT dataset. A minimum
of one treatment planning exercise must be completed by each department prior to
site activation, but a plan must be submitted for each clinician who plans to
participate.
8.12.1.2 Contouring and treatment planning will be in accordance with the trial
protocol.
8.12.1.3 The CT dataset will be provided in DICOM format and participating centres
will transfer this dataset to their treatment planning computer to complete the
contouring and planning exercise.
8.12.1.4 The completed exercise will be saved in electronic format (either RTOG
Data Exchange or DICOM-RT format) and submitted electronically along with the Dry
Run Data Submission Form, available from http://www.trog.com.au/TROG-0803-trial-
documents. Data should be uploaded to the TROG QA Centre via CQMS.
8.12.1.5 Specific instructions for saving the completed exercise in electronic format
and uploading data via CQMS may be found on the TROG web site:
www.trog.com.au or requested by emailing qa@trog.com.au .
8.12.1.6 Centres wishing to use IMRT techniques including VMAT should refer to
section 8.12.2 and to the document TROG 08.03: Quality assurance and
credentialing requirements for sites using inverse planned IMRT Techniques for more
detailed instructions on completing the dry run. This document may be found on the
website: http://www.trog.com.au/TROG-0803-trial-documents.
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 30 of 114
Amendment 2: dated 29 May 2014
The RT QA team will review the completed exercise and provide timely feedback to
the participating centre. The completed exercise will contain all data specified in the
instructions provided with the datasets (and may be reviewed on the RT QA website).
Any missing or incorrect data will delay the review process. Successful completion of
the dry run is required (i.e. no major deviations identified by the reviewer) by each
investigator prior to registering a patient on the trial. Should a major deviation be
identified, it may be necessary to complete a second dry run prior to registering
patients in this trial.
8.12.2 IMRT credentialing program
The QA procedures for sites using IMRT techniques are summarised below:
Facility Information: All sites must first complete the Facility Questionnaire,
including the section specifically related to IMRT techniques.
External Audit: Sites must satisfactorily complete an external dosimetry audit
(phantom study). Sites that have not yet completed a TROG approved
external IMRT dosimetry audit should contact the Coordinating Trial Centre to
discuss options for a site visit.
Submission of RAVES-specific case: All clinicians must complete the
contouring and planning benchmarking exercise (Dry Run, see Section 8.12.1)
prior to trial recruitment. For a site to be credentialed to use IMRT in the
RAVES trial, at least one site benchmarking case must use an IMRT
technique AND must be verified by direct measurement using the approved in-
house physics IMRT dosimetry QA protocol. The physics QA dosimetry report
will be submitted with the treatment plan for the RAVES-specific case for
review using CQMS.
More detailed instructions about the IMRT credentialing programs, including VMAT,
may be obtained from the document TROG 08.03: Quality assurance and
credentialing requirements for sites using inverse planned IMRT Techniques,
available from the RT QA website: http://www.trog.com.au/TROG-0803-trial-
documents.
8.12.3 Radiation Therapy Technical Review
Radiation therapy technical reviews will be conducted in two stages. Planning data,
outlined in Section 8.12.4, is due at least one week prior to the patient’s RT start
date. Treatment data, outlined in section 8.12.5 is due within four weeks of the RT
end date.
8.12.3.1 The treatment plans for all registered patients from each treating centre will
undergo timely review by the designated RT QA team. The treatment plan shall be
submitted at least 1 week prior to the commencement of radiotherapy. The treatment
plan will be reviewed by the RT QA team and feedback provided in a timely manner.
Should a major deviation be identified during the review process, the RT QA team
will contact the treating centre to discuss timely modification of the treatment plan.
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 31 of 114
Amendment 2: dated 29 May 2014
8.12.3.2 Should it be necessary to re-plan the patient part way through treatment, the
revised plan shall be submitted for review within 1 week of the revised plan being
created.
8.12.3.3 The review will be conducted using SWAN software.
8.12.3.4 Review parameters and treatment violations will be in accordance with the
TROG Policy and Procedures Manual (Quality Assurance Statement of Minimum
Requirements for Clinical Trials). Results will be reported to the TMC at least 6
monthly and to the TROG Trials Review Meeting biannually.
8.12.3.5 All data submitted to the RT QA team must be de-identified. The unique
patient identification number assigned to each patient at registration must be used on
all data and documents submitted. Details for removing patient identifiers from the
digitally exported treatment planning data may be requested from the TROG QA
office by email: qa@trog.com.au .
8.12.3.6 All clinical documentation and RT data for registered patients should be
stored and remain available for auditors for at least 15 years after completion of the
trial.
8.12.4 Data required for timely review
Form QA2 Checklist RT Planning Data, available from
http://www.trog.com.au/TROG-0803-trial-documents, will be completed for each
patient at least one week prior to the RT start date, and uploaded to CQMS with the
electronic dataset. Data to be included with the forms include:
Trial ID assigned at registration and patient initials
Clinical data as appropriate (e.g. site of positive margins)
Pathology report
Prescription data
o Total dose
o Number of fractions
o Dose per fraction
o Total treatment time
o Prescription point relative to the DICOM origin of the CT slices used
Beam arrangement
o Field sizes
o Radiation energy for each beam
o Gantry angle
o Blocks/MLC margin
Supplemental treatment plan data
o Treatment planning computer and version of software used
Screen dump(s)/ screen shots as described at section 8.8 number 5.
The treatment plan for all trial patients planned using an IMRT technique
(credentialed sites only) must be verified using the in-house dosimetry QA
protocol. The physics dosimetry QA report must be submitted with the plan for
review. This report shall include details of absolute dose verification and the
relative dose fluence check. The in-house QA protocol forms part of the
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 32 of 114
Amendment 2: dated 29 May 2014
credentialing requirements, and therefore the RAVES QA committee must be
alerted to any changes to the in-house QA processes prior to submitting
patient reports. Refer to http://www.trog.com.au/TROG-0803-trial-documents
for contact details.
8.12.5 Data required following RT completion
Form QA3 Checklist RT Treatment Data, available from
http://www.trog.com.au/TROG-0803-trial-documents will be completed for each
patient within four weeks after the RT end date. The following data are to be
submitted:
Documentation of any changes made to the treatment plan since submission
of planning data for timely review. If any changes have been made, the QA
team should be contacted to determine if Form QA2 Checklist RT planning
must be resubmitted.
Radiation therapy summary
Portal images log
8.13 Site Visits
No QA dosimetry site visits are scheduled for this trial (unless an external dosimetry
audit is requested by the site for IMRT credentialing). However, the RT QA team may
undertake a site visit if requested or if appropriate support could not be given
remotely.
At the discretion of the TMC, site visits may be undertaken as part of the data
monitoring activities of this study. The identification of variables requiring verification
from the source data, and the percentage of patients to be audited, will be
determined by the Independent Data Monitoring Committee (IDMC).
9.0 Treatment of relapse after radiotherapy
9.1 Indications for androgen deprivation treatment
The initiation of androgen deprivation (AD) is independent of the nature of the failure
(biochemical, local or distant). It is recommended that PSA doubling time (PSADT)
be based on at least 3 consecutive measurements, at least 2 months apart between
each pair. It is also recommended that the initiation of AD should not occur before 2
years from the diagnosis of biochemical failure unless one or more of the following
minimum clinical criteria are met. However, it is not mandatory to start treatment if a
criterion is met:
a) A PSA doubling time (PSADT) of less than 12 months with a PSA of 10.00
ng/ml or more. Investigators are free to delay intervention to higher PSA
levels.
b) A PSADT ≤ 6 months
c) Development of metastases
9.2 Androgen Deprivation Treatment
AD treatment may include either a luteinising hormone releasing hormone (LHRH)
agonist or bilateral orchidectomy, with or without antiandrogen treatment, or
monotherapy with an antiandrogen.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 33 of 114
Amendment 2: dated 29 May 2014
9.3 Continuous or Intermittent Androgen Deprivation
Androgen deprivation (AD) treatment may be continuous or intermittent. If
intermittent, AD is recommended to be for a 9 month period, and to employ
monotherapy or combined therapy at the clinician’s discretion. If the PSA is < 4.00
ng/ml after 9 months of AD, the patient may stop AD and be monitored with 3
monthly PSAs. When the PSA is > 5 ng/ml, or at a level greater than that at initiation
of AD (whichever is lower), the patient is to be recommenced on AD.
10.0 Patient Assessment
Refer to Appendix VII for the table of assessment and follow-up visits.
10.1Pre-randomisation assessment
10.1.1 Disease Status, Medical History and Co-Morbidities
Medical History and Adverse events assessed using CTCAE v 3.0.
Eastern Cooperative Oncology Group (ECOG) performance status (Appendix
II)
Co-morbidity Index (Appendix III), including the number of prescription
medications, number of co-morbid conditions, and smoking history
10.1.2 Imaging
The most recent PSA at entry into the study is ≤ 0.10 ng/ml and therefore no pre-
randomisation imaging is mandated. Bone scan, CT scan and MRI scan are
performed only if clinically indicated and at the discretion of the investigator.
10.1.3 Laboratory Studies
a) Histopathological confirmation of adenocarcinoma of the prostate with at least
one of the following factors: positive margins, extraprostatic extension (pT3)
with (pT3b) or without (pT3a) seminal vesicle involvement.
b) Pre-operative PSA level.
c) Confirmation of the most recent post-operative serum total PSA ≤ 0.10 ng/ml
following RP and prior to randomisation. An acceptable serum total PSA assay
is required with a lower limit of detection (LLD) ≤ 0.10 ng/ml.
10.1.4 Patient-completed questionnaires
a) Quality of Life: Pre-randomisation Quality of Life (QoL) questionnaires:
EORTC global (QLQ-C30) and EORTC QLQ-PR25 (Appendix IV)
b) Anxiety: Pre-randomisation anxiety and depression: Hospital Anxiety and
Depression Scale (HADS)52 (Appendix V).
c) Adverse Events: Sexual Health Inventory for Men (SHIM)53, (Appendix XI)

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 34 of 114
Amendment 2: dated 29 May 2014
10.2 Assessment during treatment phase
For both ART and SRT, clinical assessments, quality of life, anxiety/depression and
adverse event assessments (excluding the SHIM) are required on day 1 of RT, the
last day of RT and 6 weeks following completion of RT (see Appendix VII and section
10.4 for details).
10.3 Follow-up assessments
10.3.1 Clinical Assessment
For both arms, follow-up will be 6 monthly from randomisation for the first 5 years,
then annually until the end of the trial. Clinical follow-up will be conducted as per the
study schedule (Appendix VII), but can be more frequent if clinically indicated. For
patients randomised to Arm 2 (SRT), it is recommended that patients be followed 6
monthly from randomisation for 2 years following salvage RT. However, data will be
reported only for those time points specified in the trial follow-up schedule. Rectal
examination is recommended if a rising PSA is detected.
10.3.2 Biochemical assessment
Arm 1 (ART): Following adjuvant radiotherapy, serum PSA is to be measured 6
weeks after RT, and then 6 monthly relative to randomisation until the end of the trial.
Arm 2 (SRT): In the surveillance phase, serum PSA evaluations are to be performed
every 3 months from randomisation during the first 5 years, then 6 monthly thereafter
until the occurrence of a rising PSA (≥ 0.20 ng/mL prior to radiotherapy) or the end of
the trial. For patients proceeding to SRT, serum PSA is measured on day 1 of RT. If
a PSA result that triggers SRT falls between two scheduled assessments, the
relevant PSA result must be reported as an unscheduled PSA assessment.
Arm 2 (SRT): Following salvage radiotherapy, serum PSA is measured 6 weeks after
RT, and then 6 monthly relative to randomisation until the end of the trial.
If the first PSA level following SRT is ≥ 0.40 ng/mL, then:
(a) bF is diagnosed if the PSA is rising when compared to the PSA from day 1 of
RT.
(b) If the PSA is falling relative to the level measured on day 1 of RT, the patient is
followed up as usual (this does not constitute bF).
Thereafter, PSA will continue to be measured 6 monthly (relative to randomisation)
until the end of the trial. If a patient’s 6 monthly PSA and clinic visit are within 28 days
of the 6 week post-RT assessment, then the 6 monthly PSA and 6 month clinic visit
can be omitted.
Both Arms: If biochemical failure occurs (PSA level ≥ 0.40 ng/mL following
radiotherapy), a confirmatory PSA may be performed if clinically indicated. The date
of diagnosis of the bF will be that of the first PSA ≥ 0.40 ng/mL. Follow-up PSA
evaluations can be performed as clinically indicated for patients on either arm, but
only PSA results from the time points specified in the study schedule (Appendix VII)

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 35 of 114
Amendment 2: dated 29 May 2014
will be reported. Any other laboratory investigations are to be conducted as clinically
indicated. If a PSA result that meets the criteria for bF falls between two scheduled
assessments, the relevant PSA result must be reported as an unscheduled PSA
assessment.
10.3.3 Imaging
When clinically indicated, appropriate imaging will be arranged at the discretion of the
investigator.
10.4 Quality of life, anxiety/depression and adverse event assessments
All questionnaires are to be completed prior to the patient’s receiving the serum PSA
result.
10.4.1 Quality of Life
QoL patient self-assessment questionnaires are assessed prior to randomisation,
day 1 of radiotherapy (ART and SRT), at the end of RT, and 6 weeks following RT.
QoL assessments are then completed annually from randomisation.
10.4.2 Anxiety and Depression
Hospital Anxiety and Depression Scale (HADS) data will be collected at the same
time points as QoL data (Section 10.4.1), with the aim of comparing the prevalence
of anxiety and depression in both treatment arms.
10.4.3 Adverse Events
Clinician assessed adverse events are assessed prior to randomisation, day 1 of
radiotherapy (ART and SRT), at the end of RT, and 6 weeks following RT, and then
annually until the end of the trial. Patient assessed adverse events (erectile function)
are assess prior to randomisation and then annually from randomisation.
10.4.4 Purpose of Quality of life, anxiety/depression and adverse event
assessments
The purposes of these assessments are:
1. Prior to randomisation: To provide a cross-sectional description of early
postoperative QoL, anxiety/depression, and adverse events.
2. Day 1 of RT: To record QoL anxiety/depression, and clinician assessed
adverse events as a reference level for the effect of RT.
3. End of RT: To record impact of peak RT-related adverse events
4. Six weeks following RT: To record QoL anxiety/depression, and clinician
assessed adverse events if the acute effects of RT are settling.
5. Annual assessments:

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 36 of 114
Amendment 2: dated 29 May 2014
a. Year 1: To assess recovery from surgery (and recovery from RT, if
applicable)
b. All years: To determine chronic and late developing toxicities and
compare the two RT approaches with respect to QoL, anxiety/depression
and adverse events. To additionally characterise toxicities arising from
salvage radiotherapy. To identify if the interval from RP to RT influences
the pattern and intensity of toxicities.
10.4.5 Health Economics Analysis
A within trial and an extrapolated health economic analysis will be conducted to
evaluate differences in costs and Quality Adjusted Life Years (QALYs) between the
two arms. Economic data collected during the trial will include resource use data on
the use of radiotherapy and experience of inpatient admissions. Patients will self
report hospitalisations annually for the duration of the trial, using the Health Resource
Usage Questionnaire (Appendix 12) .
11.0 Pathology Review
It is well established there is significant variability in the reporting of prostate
pathology specimens. The purpose of a central pathology review is to standardise
the evaluation of the Gleason grade and score, positive margins, extraprostatic
extent and seminal vesicle involvement. This also provides the opportunity to
evaluate inter-observer variation between the local and central pathologists.
Central pathology review will be conducted by Dr Warick Delprado MBBS FRCPA
FIAC and Dr Ronnie Cohen MBBS FRCPA.
For all patients entered into RAVES in Western Australia, all slides from the
Prostatectomy specimen will be sent from the patient’s local pathology laboratory
with the local pathology report to the following address:
Dr Ronnie Cohen
PO Box 1337
West Leederville
Western Australia 6901
For patients from the remainder of Australia and New Zealand, slides will be sent
from the patient’s local pathology laboratory with the local pathology report to the
following address:
Dr Warick Delprado
Director – Histopathology,
Douglass Hanly Moir Pathology,
14 Giffnock Avenue
Macquarie Park NSW 2113
Australia.
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 37 of 114
Amendment 2: dated 29 May 2014
Slides must be de-identified and labelled with the patient initials and trial registration
number. Once reviewed, the slides will be returned to the patient’s local pathology
laboratory with a copy of the review performed. Dr Cohen or Dr Delprado will then
send the data and report to the Trial Coordinating Centre. The report template is
included as Appendix X.
For the purposes of the Pathology Review the following definitions will be used53:
Seminal Vesicle Invasion (SVI)
Tumour infiltrating the muscular coat of the seminal vesicle. Cases with invasion of
the ‘‘intraprostatic portion’’ of the seminal vesicle should be regarded as invasion of
the ejaculatory duct.
Extra-prostatic Extension
Tumour that has extended out of the prostate into periprostatic soft tissue (because
the prostate lacks a discrete capsule, the term ‘‘extraprostatic extension’’ (EPE) will
be used instead of ‘‘extracapsular extension’’ to describe tumour that has extended
out of the prostate into periprostatic soft tissue).
Focal extraprostatic extension: 1 field identified with EPE replacing a total
volume <1 High powered field (HPF) (0.5mm) or 2 fields with EPE totalling
<1HPF (0.5mm)
Established: 1 field identified with EPE replacing >1 HPF (0.5mm) or 2 or more
fields with EPE totalling >1HPF (0.5mm).
The location of EPE will be recorded.
Positive Margins
Tumour extending to the inked surface of the prostatectomy specimen which the
surgeon has cut across. At many sites the radical prostatectomy specimen is
surrounded by < 1 mm of periprostatic soft tissue. As tumour is not actually cut
across at the ink, close margins (< 0.1 mm) should not be designated as positive
margins, as they are not associated with tumour that would be left in the patient or
with an increased risk of postoperative progression.
Focal Involved margin will be defined as involvement of the surgical resection
margin in one or two foci, each less than or equal to 1 mm.
Extensive Involved margin will be defined as involvement of the surgical
resection margin by greater than 1 mm in one or more regions.
The location and Gleason score at margin will be recorded.
Gleason Score
The sum of the predominant Gleason grade (primary pattern) and second
commonest Gleason grade (secondary pattern) forms the Gleason Score (ISUP 2005
Modification). This will be based at the index tumour site. The tertiary pattern should
be recorded in a comment if higher than the primary and secondary patterns. The
percent grade 4/5 will be recorded.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 38 of 114
Amendment 2: dated 29 May 2014
12.0 Biological Sub-studies
12.1 RAVES Genetic Studies
Rapid advances in genotyping technology, concomitant with a more detailed
understanding of human sequence variation, have recently made high-density
genome-wide association (GWA) studies in adequately-powered sample sets a real
prospect and technically feasible for many conditions.54,55
These developments enable translational genetic research to be an important
component of this study. There are two optional biological sub-studies in which
patients who have consented to the trial may participate:
1. RAVES Genetic Study: Saliva or blood collection for DNA extraction
2. RAVES Tissue Banking Study: Tissue banking of tumour blocks
Patients will be approached at their 6 month post-randomisation follow-up visit about
consenting to these optional biological sub-studies. If patients are unable to consent
at 6 months post-randomisation, they can join the biological sub-studies at any time
up until 6 months after recruitment closes.
12.2. RAVES Genetic Study Aims
The power of modern genetic epidemiology arises from not assuming a priori
mechanisms and the ability to define novel pathogenic causes. Recent GWA studies
have provided proof of principle that dense screening across the human genome in
well-designed studies can discover novel pathogenic mechanisms.56,57 For instance,
the Wellcome Trust Case Control Consortium has demonstrated that GWA studies
represent a powerful approach to the identification of 27 new genes causally involved
across 8 different common human diseases.58 Results from this Consortium detected
association between common diseases and genetic variants outside genetic regions
previously thought to be involved in disease progression. In addition Zheng et al
demonstrated that genetic variants in five chromosomal regions plus a family history
of prostate cancer have a cumulative association with prostate cancer diagnosis.59
It remains to be seen if a similar cumulative effect is evident in disease progression.
There is, however, consensus that even the current large GWA studies are
underpowered to detect small effects attributable to individual genetic variants.60 The
RAVES Genetic Study will provide linked clinical and genetic data that can be
combined with similar international studies for final genetic analysis.
12.2.1 RAVES Genetic Study Logistics
For the RAVES Genetic Study, DNA will be extracted from saliva or blood samples
and stored for future analyses to improve understanding of how biological processes
are altered in prostate cancer. Molecular, protein and genetic factors will be
evaluated. This research aims to discover how to predict which patients are more
likely to relapse and require radiotherapy, and also which patients are more likely to
respond to radiotherapy.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 39 of 114
Amendment 2: dated 29 May 2014
Sites will be provided with collection materials and instructions for collecting saliva
specimens. In addition to providing the saliva sample, patients will complete one
questionnaire (Appendix IX). Details regarding saliva or blood collection, sample
collection protocols, processing, storage and submission are available from the
Centre for Genetic Epidemiology and Biostatistics (CGEB) at the University of
Western Australia. Contact details are below:
Tegan McNab
Manager Western Australian DNA Bank
Tel: (08) 6488 6738
Fax: (08) 6488 6750
Email: tegan.mcnab@uwa.edu.au
www.wadb.org.au
12.3. RAVES Tissue Banking Study Aims
The aim of this sub-study is to develop a comprehensive annotated tissue
bioresource to enable exemplary future molecular research into prostate cancer. By
matching tissue to a prospective clinical outcomes dataset, molecular research
capabilities will be optimised.
12.3.1 RAVES Tissue Banking Study Logistics
The tissue banking sub-study will not require any additional testing or specimen
collection for the patient. All tissue will be gathered from formalin-fixed paraffin
embedded (FFPE) blocks previously used for histopathological diagnosis of prostate
cancer. During Central Pathology Review (Section 11) index cancer blocks will be
identified. The local site will request the index cancer tissue blocks from the relevant
pathology laboratory for each patient who consents to the sub-study. The blocks plus
a de-identified copy of the pathology report will be sent to the Australian Prostate
Cancer BioResource (APCBR) national manager. Standard operating procedures of
the APCBR will be used to determine the whether each potential sample is suitable
for tissue banking. If unsuitable, the tissue block will be returned to the local site. If
the tissue is banked, it will be assigned a unique sample identifier, which will be
relayed to the RAVES study coordinator to enable future linkage with clinical
outcome information.
12.3.2. Australian Prostate Cancer BioResource
The Australian Prostate Cancer BioResource (APCBR) is a multi-state “virtual tissue
bank” comprising four physically separate tissue repositories, each with its own
database for collection of tissue-associated clinical and pathological data. Each
database is linked by a web-based central database, containing minimum clinical and
pathological datasets downloaded by the individual repositories and which can be
queried to assemble specific research cohorts. A BioResource Management
Committee to oversee operation is drawn from key stakeholders and experts in tissue
bank collections and clinical databases across Australia.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 40 of 114
Amendment 2: dated 29 May 2014
12.3.3. Tissue management policy
All tissues will be subject to the APCBR’s standard Materials Transfer Agreements.
Tissue gathered in connection with the RAVES study will be quarantined within the
APCBR and cannot be accessed without meeting the following criteria:
a. RAVES trial management committee approval of the suitability of the proposed
tissue research.
b. A Human Research Ethics Committee approved study protocol.
c. APCBR Tissue Management Committee approval.
If permission is granted to access tissue, it can be released along with de-identified
(coded) clinical data. For all applications, cost recovery fees may be applicable.
12.3.4 Potential tissue usage
It is likely that the tissue extracted from FFPE specimens will be predominately
limited to tissue microarray (TMA) production and DNA extraction, and in some
cases, RNA extraction. Future requests to use the tissue are, therefore, likely to be
for studies including but not limited to biomarker validation, DNA
sequencing/mutation analysis, microarray analysis and DNA methylation detection.
13.0 Criteria for Assessing Treatment Outcomes
All time-to-event outcomes are measured from randomisation unless otherwise
stated.
13.1 Biochemical failure (bF)
For both arms, biochemical failure (bF) will be diagnosed on the first occasion
following radiotherapy that the serum PSA is > 0.40 ng/mL and rising (from the
previous value). A confirmatory PSA test may be performed if clinically indicated. The
date of bF will be the date of the first PSA level > 0.40 ng/mL. For Arm 2 (SRT), a
PSA result that is > 0.40 ng/mL but less than the PSA result from day 1 of RT does
not constitute biochemical failure. See section 13.2 for additional information on time
to biochemical failure.
For patients randomised to ART who, for one reason or another, do not receive ART,
bF will be diagnosed if the serum PSA is > 0.40 ng/mL on an occasion beyond one
year from randomisation. The date of bF will be the date of the first PSA level (> 0.40
ng/mL beyond one year from randomisation).
For patients randomised to SRT who, for one reason or another, do not receive SRT
following a rising PSA level (> 0.20 ng/mL), bF will be diagnosed if the serum PSA is
≥ 0.40 ng/mL on any occasion beyond one year from the date the PSA was ≥ 0.20
ng/mL. The date of bF will be the date of the first PSA level (≥ 0.40 ng/mL beyond
one year from the date of PSA ≥ 0.20 ng/mL).
For the purposes of this trial, the definition of ‘biochemical failure’ will also include
either or both of the following:
(i) Clinically diagnosed local, regional or distant failure;
(ii) Commencement of Androgen Deprivation (AD) for failure.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 41 of 114
Amendment 2: dated 29 May 2014
The date of such failure will be the date of diagnosis of the clinical failure or
commencement of AD.
13.2 Time to biochemical failure (TTbF)
Time to biochemical failure will be measured from the date of randomisation to the
date of bF. It will be censored by death (without previous bF), the closeout date and
loss to follow-up. Analyses of TTbF, using logrank, Cox regression and Kaplan-Meier
methods, will address the primary objective of assessing the risk of bF between the
two arms.
13.3 Biochemical failure-free survival (bFFS)
Biochemical failure-free survival will be measured from the date of randomisation to
the date of bF or death from any cause. bFFS will be censored by the closeout date
and loss to follow-up.
13.4 Quality of Life (QoL) and Anxiety/Depression
QoL and anxiety/depression will be evaluated pre-randomisation and at regular
intervals, and at the start, end, and following RT. QoL and anxiety/depression will be
analysed using the difference in scores from pre-randomisation. Average scores,
adjusted for pre-randomisation levels, will be plotted against time. If the data permit,
a repeated measures analysis of QoL and anxiety/depression will be performed as a
global comparison of the arms, but this will be supplemented by comparisons of arms
at fixed time points, especially since the pattern of QoL over time is likely to be quite
different between the two arms, and by comparisons around RT.
13.5 Adverse events
13.5.1. NCI Common Terminology Criteria for Adverse Effects (CTCAE) v 3.0
NCI Common Terminology Criteria for Adverse Effects (CTCAE) v 3.0 is the tool
used by clinicians to assess toxicities (Appendix VI). Time to late toxicity (of a given
maximum grade and type) will be used to summarise and compare late toxicities.
Time to late toxicity is defined as the time from treatment start to the given late
toxicity and is censored by death, loss to follow-up and the closeout date for the trial.
The frequency and grade of adverse events (genitourinary, gastrointestinal and
sexual) will be assessed and compared between the two arms. (Appendix XI)
13.5.2. The Sexual Health Inventory for Men (SHIM)
The Sexual Health Inventory for Men (SHIM) is a 5-item patient questionnaire
developed to diagnose the presence and severity of erectile dysfunction (Appendix
XI). This questionnaire is psychometrically sound, and has been linguistically
validated in 8 languages. It is readily self-administered in research or clinical settings.
The SHIM demonstrates sensitivity and specificity for detecting treatment-related
changes in patients with erectile dysfunction. Sexual function scores will be
compared between the two arms.
13.6 Overall survival
Overall survival will be measured from the date of randomisation to the date of death
from any cause.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 42 of 114
Amendment 2: dated 29 May 2014
13.7 Disease-specific survival
Disease-specific survival is defined as the time from the date of randomisation to the
date of death due to prostate cancer. Death ‘due to prostate cancer’ will include any
death following hormone-resistant prostate cancer and any treatment-related death.
Disease-specific survival will be censored by death due to other or unknown causes,
loss to follow-up, and the closeout date of the trial.
13.8 Time to distant failure
Time to distant failure is defined as the time from the date of randomisation to the
date of documented regional, nodal or distant failure. Time to distant failure will be
censored by death without prior distant failure, loss to follow-up and the closeout date
of the trial. In particular, time to distant failure will not be censored by local failure or
bF without evidence of distant failure.
a) Nodal failure
The diagnosis of nodal failure must be confirmed by CT scan or MRI scan
of the abdomen and pelvis.
The date of nodal failure will be the date of the CT scan or MRI scan
confirming the first nodal failure.
b) Bone Metastasis
The diagnosis of bone metastasis must be confirmed on imaging: plain X-
ray, bone scan, CT scan or MRI scan.
The date of bone metastasis will be the date of the first radiological
investigation that documented bone metastasis.
13.9 Time to local failure
Time to local failure is defined as the time from the date of randomisation to the date
of documented palpable or biopsy-proven local failure.
13.10 Time to the initiation of Androgen Deprivation (AD)
Time to the initiation of androgen deprivation will be measured from the date of
randomisation to the date of initiation of androgen deprivation.
13.11 Quality Adjusted Life Years and Cost-utility
Patients will self-report hospital admissions using the Health Resource Usage
Questionnaire. To estimate QALY gains, utility weights will be estimated using a
mapping algorithm that has been estimated for the EORTC QLQ-C30, which is being
used to assess quality of life in all patients 12 monthly from randomisation. To
estimate the longer term costs and benefits, a health economic decision model will
be built and populated. The model will use trial data to describe the pathway of
patients between prostate cancer-specific health states (e.g. disease-free, local
recurrence and distant recurrence). External data will be sought to predict
subsequent pathways between these states (and death) so that costs and QALYs
gained over the lifetime of the trial population can be estimated.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 43 of 114
Amendment 2: dated 29 May 2014
14.0 Data Management and Documentation
14.1 Case Report Forms (CRFs)
The study site will retain a copy of the completed CRFs, with the original CRFs sent
as soon as practicable to:
RAVES Trial Coordinating Centre
Auckland Regional Cancer and Blood Service
Private Bag 92024
Auckland 1142
New Zealand
All corrections to CRFs must be made so that the original entry remains legible.
Corrections should be crossed out with a single line using a black pen, and must be
initialled and dated.
Data clarification queries will be sent to participating institutions as necessary. Sites
must return their response to data clarification queries to the Trial Coordinating
Centre, with a copy retained at site, and amend their copy of the CRF as necessary.
14.2 Recording of data and retention of documents
The investigator or suitably trained staff at each site must complete the CRFs,
transmit the data as instructed, and retain a copy of the CRFs for each patient at their
site in a secure place. Data on subjects collected on CRFs will be documented in an
anonymous fashion, and the subject will only be identified by the subject number,
and by his initials. If, as an exception, it is necessary for safety or regulatory reasons
to identify the subject, TROG and the investigator are bound to keep this information
confidential.
The investigator must maintain source documents for each patient in the study,
consisting of all demographic and medical information reported on the CRFs,
including laboratory data, etc. Sites must keep a copy of the signed informed consent
form and completed CRFs for each patient. All information on CRFs must be
traceable to these source documents kept in the patient's file.
Essential documents, as listed below, must be retained by the investigator to comply
with national and international regulations. Essential documents include:
1. Human Research Ethics Committee (HREC) approvals for the study protocol
and all amendments
2. All source documents and laboratory records
3. CRF copies
4. Patients' informed consent forms (with study number and title of trial)
5. Any other pertinent study documents

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 44 of 114
Amendment 2: dated 29 May 2014
14.3 Database management and quality control
Data from the CRFs are to be checked by Trial Centre staff and entered into the
study database. Possible errors or omissions will be alerted to the investigational site
for resolution.
15.0 Adverse Event Scoring and Reporting
15.1 Definitions
15.1.1 Adverse Event (AE)
An Adverse Event (AE) is any untoward medical occurrence in a patient or clinical
investigation subject administered a pharmaceutical product (or any other protocol
specified intervention including Radiation Therapy, surgery or use of a device) and
which does not necessarily have to have a causal relationship with this treatment.
An AE can therefore be any unfavourable and unintended sign, symptom or disease
temporally associated with the use of a medicinal product, whether or not related to
the medicinal product (or associated with the use of any other protocol specified
intervention including radiation therapy, surgery or use of a device).
AEs include: ‘Adverse Drug Reactions’, i.e.
A reaction, in contrast to an event, is characterised by the fact that a causal
relationship between the drug and the occurrence is suspected.
For unapproved medicines: all noxious and unintended responses to a
medicinal product related to any dose should be considered adverse drug
reactions. The phrase ‘responses to a medicinal product’ means that a
causal relationship between a medicinal product and an adverse event is at
least reasonably possible, i.e. the relationship cannot be ruled out.
Regarding marketed medical products: a response to a drug which is
noxious and unintended and which occurs at doses normally used in man
for prophylaxis, diagnosis, or therapy of diseases or for modification of
physiological function.
15.1.2 Unexpected Adverse Event (UAE)
An Unexpected Adverse Event is an AE for which the nature or severity of the event
is not consistent with the information in the relevant source documents (or with the
applicable side effect risk profile for radiation therapy, surgery or use of a device).
UAEs include: ‘Unexpected Adverse Drug Reactions’, i.e.
The nature and severity of the ADR is not consistent with the information in
the Investigators Brochure for an unapproved investigational product, or the
product information/package insert/summary of product characteristics for
an approved product.
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 45 of 114
Amendment 2: dated 29 May 2014
15.1.3 Serious Adverse Event (SAE)
Adverse events and adverse drug reactions are considered ‘serious’ if they threaten
life or function.
SAEs include: ‘Serious Adverse Drug Reactions’, i.e.
During clinical investigations, adverse events may occur, which if suspected
to be medicinal product related (‘adverse drug reactions’) might be
significant enough to lead to important changes in the way the medicinal
product is developed (e.g. change in dose, population, monitoring, consent).
This is particularly true for reactions, which in their most severe form
threaten life or function.
Due to the significant information they provide, Serious Adverse Events (including
Serious Adverse Drug Reactions) require expedited reporting. SAEs are defined as
any adverse event or adverse drug reaction which:
Results in death (i.e. fatal/grade 5 CTC AE)
Is life-threatening (i.e. grade 4 CTC AE)
Requires inpatient hospitalisation or prolongation of existing
hospitalisation*
Results in persistent or significant disability/incapacity; or
Is a congenital anomaly/birth defect
Note: The term ‘life-threatening’ in the definition of ‘serious’ refers to an event in
which the participant was immediately at risk of death at the time of event; it does not
refer to an event which hypothetically might have caused death if it were more
severe.
*An event that results in hospitalisation or prolongs an existing hospitalisation will not
be considered a serious adverse event if the only reason for the hospitalisation or
prolongation was:
administration of chemotherapy
administration of trial procedures
placement of a permanent intravenous catheter
hospice placement for terminal care
pre-trial scheduled elective surgery
out-patient hospitalisation for procedures such as:
- Elective day surgery
- Convenience purposes (eg. transportation difficulties)
- Planned admission as part of supportive care for insertion of PEG
tube or naso-gastric tube for commencement of enteral feeding
(ie. did not occur following urgent admission as a result of weight
loss or other patient medical events)
15.1.4 Attribution of Cause of an Adverse Event
Attribution of cause requires at least a reasonable possibility of a causal relationship
between the event and the use of a pharmaceutical product (or any other protocol
specified intervention including radiation therapy, surgery or use of a device), i.e. the
relationship cannot be ruled out. One or more of the following categories should be
attributed as the cause of an event:
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 46 of 114
Amendment 2: dated 29 May 2014
Pharmaceutical product (an SAE which is drug related is considered a
Serious Adverse Drug Reaction)
Radiation Therapy
Medical Device
Surgery
Unrelated to trial treatment (i.e. progressive disease, concurrent
medication, concurrent disorder or other
All protocol specified interventions (including pharmaceutical products, radiation
therapy, surgery or use of a device) administered prior to the date of the event must
be attributed a degree of causality from one of the following codes:
Unrelated
Unlikely to be related
Possibly related
Probably related
Definitely related
15.2 Reporting
15.2.1 Adverse Event Reporting
15.2.1.1 Trial Sites/Investigators
All relevant adverse events (including those that are non-serious or expected)
which occur whilst the participant is enrolled on the trial must be reported in
the patients’ medical records and recorded on the relevant CRF. The Common
Terminology Criteria for Adverse Events (CTCAE version 3.0 – see appendices) must
be used to grade the severity of an event.
15.2.1.2 Trial Coordinating Centre (TCC)
Data from the CRF will be entered onto the trial database at the Trial Coordinating
Centre and accessed when required by TROG. Regular analyses of cumulative AE
data should occur at the TCC. If significant safety issues are identified from analyses
the TCC must inform the TSC, other investigators and responsible HRECs.
15.2.2 Serious Adverse Event Reporting
15.2.2.1 Trial Sites/Investigators
Serious Adverse Events must be reported to the Trial Coordinating Centre
(TCC) and the TROG Central Operations Office (TCOO) whether or not
considered related to the treatment under investigation. For Arm A (ART), all
SAEs that occur from the time a participant starts radiation therapy to within 30
days of the final protocol specified treatment are required to be reported. For
Arm B (SRT), all SAEs that occur from commencement of radiotherapy to

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 47 of 114
Amendment 2: dated 29 May 2014
within 30 days of the final protocol-specified treatment are required to be
reported.
The Principal Investigator (PI) must:
Determine whether an AE is ‘Serious’ (refer to criteria at 10.1.3)
For SAEs, the PI must then ascertain the suspected cause
The attribution to the SAE must be recorded in the patients’ medical
records and reported on the SAE form.
The PI must then determine whether the SAE (or Serious Adverse Drug
Reaction) is expected or unexpected.
Both expected and unexpected Serious Adverse Events and Serious
Adverse Drug Reactions must be recorded in the patients’ medical
records and reported to the TCC and TCOO
No pharmaceutical products that meet CTN criteria are used in this trial and therefore
TGA reporting is not required.
SAEs must be reported by completing the TROG SAE form and FAXING it to
the following:
Fax To:
Fax Number:
Trial Coordinating Centre (TCC)
+ 64 9 359 9981
TROG Central Operations Office (TCOO)
+61 (0)2 401 43902
SAE forms are required at the following points:
Initial
Report
Within one working day/24 hours of discovery or notification of the
event. If the reporting of an SAE is delayed by more than 24 hours,
an explanation must be provided in the comments section of the
SAE form.
Incomplete
Reports*
If all details are not available at the time of the initial report a
completed report must be sent within the next 10 days.
Updated
Report
If the event is not resolved (or is ‘on-going’) at the time of the initial
report, the ‘UPDATE: Outcome of Event’ section’ of the SAE Form
must be completed and the form submitted to the TCC and the
TCOO as soon as the event is resolved (with or without sequelae) or
if death has occurred.
*The Investigator is ultimately responsible for reporting the SAE and must sign the
SAE report(s). Should this Investigator not be available to sign the initial SAE form
within the 24 hour period, a comment to this effect must be written on the form and
the form faxed without signature to the TCC and TCOO. The investigator must sign
the SAE form as soon as possible and re-fax to the TCC and the TCOO.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 48 of 114
Amendment 2: dated 29 May 2014
The Investigator at the Trial Site is responsible for determining the local SAE
reporting requirements of the responsible HREC and subsequently notifying the
HREC of SAEs as required.
All Serious ADRs (expected and unexpected) are required to be reported to the
TCC and the TCOO according to section 15.2.2 above.
15.2.2.2 Trial Coordinating Centre (TCC)
The TCC will ensure all SAEs are forwarded for immediate clinical review by a
qualified independent reviewer to ensure any safety issues and subsequent actions
are identified as soon as possible.
The Investigator at the Trial Site is responsible for reporting Serious ADRs to the
responsible HREC according to local requirements.
No pharmaceutical products that meet CTN criteria are used in this trial and therefore
TGA reporting is not required.
15.2.3 Other Situations requiring expedited Reporting
15.2.3.1 Overdoses
Overdoses (drug or radiation) must be reported to the TCC and TCOO (see section
10.4.2) if the event(s) associated with the overdose meet the SAE definitions in
section 10.1.4. If no serious adverse events are experienced the overdose must be
reported in the patients medical record and transcribed onto the relevant trial CRF.
15.2.3.2 New Cancers
The development of new cancers at any time during the trial must be reported in the
patients’ medical record and transcribed onto the relevant trial CRF. If any events
associated with the new cancer meet the SAE definitions listed at section 15.1.4
above, then they should also be reported to the TCC and TCOO (see section 15.4.2).
16.0 Statistical Considerations
16.1 Trial Design
This is a two-arm, randomised, non-inferiority trial whose main aim is to determine, in
patients with clinically localised prostate cancer with positive margins and/or
extraprostatic (pT3) disease following radical prostatectomy, whether active
surveillance with early salvage (delayed) radiotherapy (SRT) can be considered non-
inferior to standard treatment with adjuvant (immediate) radiotherapy (ART) with
respect to risk of biochemical failure (bF).
Secondary aims are to compare arms with respect to quality of life, treatment-related
adverse events, overall survival, biochemical failure-free survival (bFFS), disease-
specific survival, time to distant failure, time to local failure, and time to the initiation
of androgen deprivation. A secondary aim also is to assess the simultaneous effects
of several potential prognostic factors on each outcome. Simultaneous assessment
of efficacy and quality of life will be undertaken using Quality Adjusted Life Years
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 49 of 114
Amendment 2: dated 29 May 2014
(QALYs) analyses. A cost-utility analysis will be undertaken to determine and
compare the cost implications of the two strategies being evaluated.
16.2 Treatment Assignment
Patients will be randomised in the ratio of 1:1 between the two arms: ART and SRT.
Allocation to treatment arms will be balanced by radiotherapy institution, Gleason
score (continuous), preoperative PSA level (continuous), surgical margin status
(positive/negative) and seminal vesicle involvement (pT3b: yes/no), using the
minimisation technique.
16.3 Statistical Methods
16.3.1 Patient Subsets to be Analysed
The following defined patient subsets will be used for the different types of analyses
to be used.
As-randomised subset: All patients who are randomised on the trial and who have
a diagnosis of prostate cancer. Analysis is performed according to the intention-
to-treat (ITT) principle.
Per-protocol subset: All patients who comply with entry criteria receive adequate
treatment and do not otherwise deviate from the protocol in an important way.
Analysis will be according to the treatment actually received.
As-treated subset: All patients who begin treatment. Analysis will be according to
treatment actually received. The as-treated population will be used for safety
analyses.
The analysis of the primary objective will be conducted according to both ITT and
per-protocol methods. The ITT principle says that patients will be analysed according
to the arms to which they were randomised, regardless of compliance with entry
criteria or the treatment they actually receive or whether they subsequently withdraw
from treatment or deviate from the protocol. A definitive conclusion regarding non-
inferiority will follow if these analyses are consistent. (See the comments on this at
the end of the section, Analysis of the Primary Objective.) Secondary objectives will
be conducted similarly, except that safety analyses will be performed using the as-
treated subset of patients.
16.3.2 Pre-Randomisation Statistics and General Methods
Pre-randomisation characteristics by treatment arm will be summarised in frequency
tables and by the use of descriptive statistics for quantitative variables. Summary
tables will be prepared giving numbers of patients by treatment arm and by
randomisation irregularities, treatment compliance, eligibility infringements, and
losses to follow-up (as per CONSORT guidelines).
All times to events will be measured from the date of randomisation unless otherwise
stated. A close-out date will be determined at the time of final analysis as the earliest
date of last contact of all patients alive and not lost to follow-up. All follow-up beyond
this date will be ignored for the purposes of analysis in order to minimise bias arising
from the differential reporting of follow-up for patients who do or do not experience an
event.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 50 of 114
Amendment 2: dated 29 May 2014
Ninety-five percent confidence intervals (95% CI) for differences between arms of all
important endpoints will be calculated, and p-values will be two-sided. An exception
is the analysis of the primary objective, as described below.
16.3.3 Analysis of the Primary Objective
The primary endpoint is bF. (Note: For the purposes of the trial, bF will include any
clinically diagnosed failure and initiation of AD for failure.
Non-inferiority of SRT with respect to ART will be assessed by first nominating a non-
inferiority margin (N-IM), defined as the maximum ‘difference’ between the arms
consistent with the assertion that SRT is effectively non-inferior to ART. The N-IM will
be specified as a hazard ratio (SRT:ART) corresponding to specified 5-year bF-free
rates (bFFR). The N-IM will, therefore, be represented by a hazard ratio greater than
one. (See section 16.4.2 below for rates and ratio used to define the N-IM for this
trial.)
A one-sided 95% confidence interval (CI) for the hazard ratio (SRT:ART) of the form
(0 to upper-limit), (or, equivalently, a two-sided 90% CI for the hazard ratio) will be
calculated and if this CI lies below the N-IM it will be concluded that SRT is,
effectively, non-inferior to ART.
The primary analysis will be performed using a Cox regression model to calculate a
2-sided 90% CI for the hazard ratio which adjusts for the stratification variables
(excluding institution), viz Gleason score, preoperative PSA, seminal vesicle
involvement and surgical margin status. Secondary analyses will include an
unadjusted analysis. The Kaplan-Meier method will be used to estimate curves for
time to bF (TTbF).
The equivalent analysis in terms of hypothesis testing is to test the null hypothesis:
H0: HR(SRT:ART) > N-IM (inferiority),
versus the one-sided alternative hypothesis:
H1: HR(SRT:ART) N-IM (non-inferiority),
where HR(SRT:ART) is the hazard ratio of SRT relative to ART. HR = 1 means exact
equivalence and HR > 1 means SRT has a higher bF rate than ART.
It will be important in this trial to ensure that data quality is very high. Any noise in the
data would have the effect of narrowing any difference between the groups being
compared, thereby increasing the likelihood of demonstrating equivalence when it
does not exist, i.e. increasing the chance of a false positive result (of non-inferiority).
Hence, analysis of the primary objective will include both: (i) an ITT analysis; and (ii)
analysis using the per-protocol patient subset. A conclusion of non-inferiority will be
made if these analyses are consistent in indicating non-inferiority.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 51 of 114
Amendment 2: dated 29 May 2014
16.3.4 Analyses of Secondary Objectives
Frequencies of early toxicity rates will be tabulated and compared between arms
using exact contingency table methods. Late toxicity rates will be estimated using
Kaplan-Meier curves and compared using logrank and Cox regression methods.
The Kaplan-Meier method will be used to estimate curves for bFFS, overall survival,
disease-specific survival, and times to distant failure, local failure, and androgen
deprivation. Cumulative rates of local failure and distant failure will also be estimated
from cumulative incidence curves obtained from competing risks analyses (see Time-
to-event Definitions for details). Cox regression will be used to compare arms for the
various time-to-event endpoints, including those adjusting for prognostic factors and
for prognostic factor analyses.
16.4 Sample Size and Power
16.4.1 Anticipated Accrual Rate
As a result of surveying likely participating sites, the accrual rate to the trial is
expected to be 100 evaluable patients per year.
16.4.2 Sample Size Calculation
It is assumed that the 5-year bFFR of the standard arm (ART) is 74% (35). SRT will
be considered to be non-inferior to ART if its 5-year bFFR is at most 10% lower than
the 5-year bFFR for ART. That is, given 74% bFFR for ART, the experimental arm
(SRT) will be considered non-inferior if its 5-year bFFR is 64% or higher. Assuming
proportional hazards, 74% versus 64% corresponds to a hazard ratio (HR) for
SRT:ART of 1.482 (1/0.675). (The N-IM is 1.482.)
A difference of 10% in 5-year bFFR was chosen because it is felt that if there is a
less than 10% difference in biochemical control between arms the likelihood of any
meaningful difference in clinical endpoints such as distant failure or overall survival
will be very small. Also, it is expected that many patients would accept an increase in
risk of bF of up to 10% at 5 years in order to obtain a better expected QoL (from the
likely avoidance of the need for radiotherapy).
The sample size is based on the primary outcome variable of time to biochemical
failure (time from randomisation to bF).
The following assumptions are made in the calculation of the required sample size:
Biochemical failure-free curves follow exponential distributions;
The biochemical failure-free rate for ART at 5 years is 74%;
The N-IM is that corresponding to a biochemical failure-free rate for SRT at 5
years of 64%, viz a hazard ratio (SRT:ART) of 1.482;
There are no competing risks;
The required power of the trial to declare non-inferiority when the two arms are
identical (74% bFFR at 5 years for both arms) is 80%;
The significance level is 5% (for a one-sided test with type I error, alpha = 0.05);

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 52 of 114
Amendment 2: dated 29 May 2014
Patients will be allocated to treatment arms in the ratio of 1:1.
The accrual rate for the trial will be 100 patients per year;
Patients will be followed up for a further 5 years following close of accrual.
The above assumptions require that 160 biochemical failures be observed by the
time of the main analysis. This number of biochemical failures can be expected if 454
patients are accrued. To allow for losses to follow-up and patients who are not fully
evaluable the trial will aim to accrue 470 patients, which is expected to take 4.7
years. So, in summary:
In order to determine non-inferiority of SRT with respect to ART
with 80% power, using a 10% non-inferiority margin in the 5-year
biochemical failure-free rate (assumed 74% versus 64%: HR =
1.482) 160 events are required to be observed. It is estimated that
this will require a sample size of 470 patients expected to be
accrued over 4.7 years and followed for a further 5 years.
Sample sizes that would be required for other N-IMs and for 90% power, given the
bFFR for ART = 74%, are:
Power = 80%
Power = 90%
Difference in 5-
year bFFR
N-IM (HR)
Events required
Target N
Target N
5%
1.232
570
1300
1600
6%
1.281
410
960
1250
7%
1.330
310
780
1000
8%
1.380
240
650
850
9%
1.431
200
550
700
10%
1.482
160
470
600
About one year prior to the end of accrual a confidential assessment by the IDMC,
with the assistance of the trial statistician, of the assumptions underlying the
estimation of sample size will be made. This assessment will examine:
the assumptions made about the time to bF: viz exponential curve for ART, and
proportional hazards;
an assessment (based on the bF rate for ART) of whether the required number
of events will be observed following 5 years of follow-up after end of accrual;
the likely number of patients who would not be fully evaluable for a per-protocol
subset analysis.
If it is judged to be necessary, an adjustment to the required number of patients to be
accrued will be made to ensure the required number of bFs will be observed. No
analysis of difference or non-inferiority will be made at this time.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 53 of 114
Amendment 2: dated 29 May 2014
16.5 Analysis Plan
It is planned to do interim analyses of adverse events after approximately 2, 3 and 4
years of accrual, a futility analysis after about 3 years, an interim analysis of efficacy
2 years following close of accrual, and a final (main) analysis after 160 events have
been observed. The schedule for analyses may vary if the accrual rate is significantly
different from that expected.
Table: Summary of main events and analyses.
Legend: SOA = start of accrual; EOA = end of accrual; EOF = end of follow-up; IAn = nth interim
analysis; FA = final analysis.
Event
Purpose
Trigger
Expected
Date
Years from
SOA
Expected
accrual
Expected no.
of events *
SOA
Aug 2008
0
0
0
IA1
Toxicity
Earlier of 200
patients and 2
years after SOA
Aug 2010
2
200
11
IA2
Toxicity, futility
Earlier of 300
patients and 3
years after SOA
Aug 2011
3
300
26
IA3
Toxicity, sample
size assumptions
Earlier of 400
patients and 4
years after SOA
Aug 2012
4
400
45
EOA
470 patients
May 2013
4.7
470
64
IA4
Efficacy
2 years after EOA
May 2015
6.7
470
102
EOF
160 events
May 2018
9.7
160
FA
Assess all
objectives
Aug 2018
10
470
160
* given 74% vs 64% bFFR at 5 years in ART and SRT arms, respectively.
16.5.1 Interim status and safety reports
Regular progress reports will be done six-monthly for the TMC and TROG meetings.
These will comprise a) reports of accrual to TROG and b) accrual and data timeliness
and quality to the TMC. The IDMC will receive safety reports by arm. An analysis will
be conducted on the first 100 patients randomised to ART who received the protocol
treatment and had at least one year of follow-up. The aim of this analysis is to
document the late toxicities experienced for the purposes of a publication designed to
improve accrual to the trial. The IDMC will review the analysis and plans for
publication.
16.5.2 Interim analysis No. 1
This will be an analysis of toxicity two years after start of accrual or after 200 patients
have accrued, whichever occurs first. An analysis of toxicity will be submitted to the
IDMC for consideration. The analysis will include rates of grade 2 or worse late
urinary and bowel toxicity. It is not expected that the trial will be stopped for reasons
of toxicity but the IDMC may recommend modification of the treatments.
16.5.3 Interim analysis No. 2
This will be an analysis of toxicity and one for futility three years after start of accrual
or after 300 patients have accrued, whichever occurs first. The futility analysis will

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 54 of 114
Amendment 2: dated 29 May 2014
test at an early stage whether there is any evidence that the risk of bF for SRT is
significantly inferior to that of ART at the 5% one-sided significance level.
16.5.4 Interim analysis No. 3
This will be an analysis of toxicity and to assess the sample size assumptions, four
years after start of accrual or after 400 patients have accrued, whichever occurs first.
The sample size reassessment will examine (i) the shapes of the bFF curves to
assess whether the proportional hazards assumption is reasonable and that the
curves are approximately exponential, and (ii) whether the bFF rate in the ART arm is
consistent with 74% at five years. The analysis will be strictly confidential and the
IDMC will advise on whether the sample size should be modified.
16.5.5 Interim analysis No. 4
This will be an analysis of efficacy two years after the end of accrual for the purposes
of presentation of interim results at an international forum. The results will not
influence the decision to continue to full follow-up until 160 events (bFs) are observed
(expected to be five years after accrual ends) and so will not affect the alpha level
used at final analysis. The final analysis will be carried out if 160 events have not
occurred by seven years following end of accrual.
16.5.6 Final analysis
This will occur after 160 events have been observed, expected to be five years after
the end of accrual. If, five years after the end of accrual, 160 events have not
occurred, or won’t occur soon, a confidential futility analysis will be performed
involving calculation of the conditional power (probability that a statistically significant
outcome will be achieved by the earlier of 160 events or 7 years from end of accrual,
given the alternative hypothesis of no difference and the data to hand). If this
conditional power is sufficiently low (say < 0.20), the IDMC will recommend that the
trial be analysed, otherwise continue to the earlier of 160 events or 7 years accrual.
16.6 Early Closure Criteria
Early stopping of the trial will be considered in the event of:
Inadequate recruitment: once a consistent accrual rate has been established. If
this is less than 50 patients per year the TMC will review the viability of the trial.
Unacceptable toxicity. However, as the experimental arm is the administration of
less intensive treatment, then it is not foreseen that there will be a need to
terminate accrual based on toxicity. If the rates of toxicity are high, then treatment
guidelines will be reviewed.
Evidence becoming available, during the accrual phase of the trial, which clearly
demonstrates that it is unethical to randomise patients to one or both of the trial
arms.
Evidence of inferiority of the SRT arm.
The IDMC will review confidential reports of relevant analyses of trial data and
recommend whether the trial should be stopped or undergo modification or continue
without change.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 55 of 114
Amendment 2: dated 29 May 2014
17.0 Responsibilities
The study will be performed in accordance with the CPMP/ICH Note for Guidance on
Good Clinical Practice (CPMP/ICH/135/95) in Australia and the Interim Good Clinical
Research Practice Guidelines in New Zealand, issued by Medsafe.
17.1 Investigator responsibilities
The investigator is responsible for ensuring that written informed consent by the
patient is obtained before study entry. The investigator is responsible for informing
the ethics committee of any serious adverse events and/or major amendments to the
protocol as per local requirements.
The investigator is responsible for ensuring that all regulatory requirements are
followed.
The investigator is required to ensure compliance with respect to all aspects of the
protocol. It is the responsibility of the investigator to maintain adequate and accurate
case report forms (CRFs) as per sections 14.1 and 14.2.
18.0 Independent Data Monitoring Committee (IDMC)
An Independent Data Monitoring Committee has been established. It comprises three
members, a radiation oncologist, a urologist and a statistician. The committee will
meet biannually.
The role of the IDMC in general is to monitor patient safety and the scientific integrity
of the trial and, more specifically, to:
a) Provide confidential and expert review of the interim analyses. The IDMC can
recommend early closure of the trial.
b) Monitor AE and SAE data
c) Assess adequacy of accrual
d) Review protocol violations
e) Review the frequency of, and reasons for, patient withdrawal (includes both
withdrawal of consent and loss to follow-up)
Following each IDMC meeting, the IDMC Chair will provide a progress report,
including any recommendations, to the Trial Chairs.
19.0 Ethical Considerations
19.1 Ethical principles and Regulatory Complince
The trial will be conducted according to the following regulations and guidelines:
Note for Guidance on Good Clinical Practice (CPMP/ICH/135/95) annotated with
TGA comments (Australia, July 2000)
The Australian Code for Responsible Conduct of Research (August 2007)
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 56 of 114
Amendment 2: dated 29 May 2014
Declaration of Helsinki: Ethical Principles for Medical Research Involving Human
Subjects (last amended by the World Medical Association, 2008)
Interim Good Clinical Research Practice Guidelines (New Zealand, August 1998),
National Statement on Ethical Conduct in Human Research, (Australia, 2007)
Guidelines on Ethics in Health Research (NZ, 2005), and
Current TROG Policy Statements
This Protocol, including the Participant information Sheet and Consent Form (PIC)
must be approved by the responsible HREC before enrolment of trial participants.
No pharmaceutical products that meet CTN criteria are used in this trial.
19.2 Adherence to the protocol
Except for an emergency situation in which proper care for the protection, safety and
well being of the trial participant requires that an alternative treatment be used, the
trial shall be conducted exactly as described in the approved protocol. Any deviation
from the protocol must be recorded and explained.
19.3 Informed Consent
The Principal Investigator is responsible for ensuring that written Informed Consent is
obtained from trial participants before trial entry.
A template PIC for this trial is provided in the protocol appendices. The Principal
Investigator must insert site specific information into the template and have this
approved by the TCC prior to submitting the PIC to the responsible HREC for final
approval.
19.4 Confidentiality
The trial will be conducted in accordance with applicable Privacy Acts and
Regulations. All information regarding trial participants must be treated in strict
confidence. Data, which identify any trial participant, must not be revealed to anyone
not directly involved in the trial or the clinical care of that participant. An exception is
where the trial participant has provided written consent for his/her records to be
included in source document verification. In this instance, the records may be
inspected by (a) a representative of TROG for the purposes of source document
verification or quality audit as stipulated in the ICH GCP Guidelines, or (b) a
representative of a government regulatory authority for the purposes of official
inspection. Records must be made available for inspection on the understanding that
all information relating to trial participants will be treated in strict professional
confidence.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 57 of 114
Amendment 2: dated 29 May 2014
20.0 Insurance and Compensation
TROG endorses the principles of the Medicines Australia Guidelines for
Compensation for Injury Resulting from Participation in a Company Sponsored Trial
and the Research Medicines Industry equivalent in New Zealand.
To provide protection for trial participants involved in TROG Clinical Trials, TROG
maintains a clinical trials insurance policy.
21.0 Publication and Presentation Policy
21.1 Reporting of Results
The Trial Management Committee will be responsible for decisions regarding
presentations and publications arising from this trial according to the TROG
Authorship, Publication and Spokesmanship Guidelines.
Access to data during the trial will be limited to the TCC, the TCOO as required for
QA reviews, the TMC, the DMC and appropriate regulatory bodies. The primary
analysis of trial results for publication will be performed by the TMC statistician. The
primary trial results will be published by TROG.
Acknowledgement of TROG is required in all publications, abstracts and
presentations. Publications and abstracts must be presented to the TMC for review
and approved prior to submission. In addition, publications must be reviewed by the
TROG Publications Committee prior to submission.
21.2 Trial Registry
The TCOO is responsible for registering all TROG trials with an appropriate clinical
trials registry prior to the accrual of the first participant. All TROG trials are registered
at www.clinicaltrials.gov and Australian and New Zealand Clinical Trials Registry
(ANZCTR) www.anzctr.org.au.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 58 of 114
Amendment 2: dated 29 May 2014
REFERENCES
1. Catalona WJ, Smith DS, Ratliff TL, Basler JW. Detection of organ-confined
prostate cancer is increased through prostate-specific antigen-based screening.
JAMA 1993; 270(8):948-954.
2. Partin AW, Pound CR, Clemens JQ, Epstein JI, Walsh PC. Serum PSA after
anatomic radical prostatectomy. The Johns Hopkins experience after 10 years.
Urol Clin North Am 1993; 20(4):713-725.
3. Pound CR, Partin AW, Epstein JI, Walsh PC. Prostate-specific antigen after
anatomic radical retropubic prostatectomy. Patterns of recurrence and cancer
control. Urol Clin North Am 1997; 24(2):395-406.
4. Partin AW, Kattan MW, Subong EN, Walsh PC, Wojno KJ, Oesterling JE et al.
Combination of prostate-specific antigen, clinical stage, and Gleason score to
predict pathological stage of localized prostate cancer. A multi-institutional
update. JAMA 1997; 277(18):1445-1451.
5. Paulson DF, Moul JW, Walther PJ. Radical prostatectomy for clinical stageT1-
T2, N0, M0 prostatic adenocarcinoma: long-term results. J Urol 1990; 144:1180-
1184.
6. Zietman AL, Edelstein RA, Coen JJ, Babayan RK, Kren JR. Radical
prostatectomy for adenocarcinoma of the prostate: the influence of preoperative
pathologic findings on biochemical disease-free outcome. Urology 1994;
43:828-833.
7. Frazier HA, Robertson JE, Humphrey PA, Paulson DF. Is prostate specific
antigen of clinical importance in evaluating outcome after radical
prostatectomy? J Urol 1993; 149:516-518.
8. Crawford ED, Eisenberger MA, McLeod DG, Spaulding JT, Benson R, Door FA,
Blumenstein BA, Davis MA,Goodman PJ. A controlled trial of leuprolide with
and without flutamide in prostatic carcinoma. NEJM 1989; 321:419-424.
9. Swindle P, Eastham JA, Ohori M, Kattan MW, Wheeler T, Maru N, Slawin K,
Scardino PT. Do margins matter? The prognostic significance of positive
surgical margins in radical prostatectomy specimens. J Urol 2005;
Sep;174(3):903-7.
10. Epstein JI, Partin AW, Sauvageot J, Walsh PC. Prediction of progression
following radical prostatectomy. A multivariate analysis of 721 men with long-
term follow-up. Am J Surg Pathol 1996 Mar;20(3):286-92.
11. MacDonald OK, Schild SE, Vora SA, et al: Radiotherapy for men with isolated
increase in serum prostate specific antigen after radical prostatectomy. J Urol
2003; 170: 1833 – 1837.
12. Pisansky TM, Kozelsky TF, Myers RP, Hillman DW, Blute ML, Buskirk SJ et al.
Radiotherapy for isolated serum prostate specific antigen elevation after
prostatectomy for prostate cancer. J Urol 2000; 163(3):845-850.
13. Stevenson AJ, Shariat SF, Zelefsky MJ, et al. Salvage radiotherapy for
recurrent prostate cancer after radical prostatectomy. JAMA 2004, 291: 1325 –
1332.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 59 of 114
Amendment 2: dated 29 May 2014
14. Katz MS, Zelefsky MJ, Venkatraman ES, et al. Predictors of biochemical
outcome with salvage conformal radiotherapy after radical prostatectomy for
prostate cancer. J Clin Oncol 2003; 21: 483 – 489.
15. Anscher, M. S. and L. R Prosnitz. Multivariate analysis of factors predicting local
relapse after radical prostatectomy – possible indications for post-operative
radiotherapy. Int J Radiat Oncol Biol Phys 1991; 21:941-947.
16. van den Ouden, D., F. M. Bentvelsen, E. R. Boeve, and F. H. Schroder. 1993.
Positive margins after radical prostatectomy: correlation with local recurrence
and distant progression. British Journal of Urology 72:489-494.
17. Ackerman, D.A., J.M. Barry, R.A. Wicklund, N. Olson, and B. A. Lowe. Analysis
of risk factors associated with prostate cancer extension to the surgical margin
and pelvic node metastasis at radical prostatectomy. J Urology 1993; 150:1845-
1850.
18. Watson, R. B., F. Civantos, and M. S. Soloway. 1996. Positive surgical margins
with radical prostatectomy: detailed pathological analysis and prognosis.
Urology 48:80-90.
19. D'Amico, A. V., R. Whittington, B. Malkowicz, K. R. Loughlin, D. Schultz, M.
Schnall, C. M. Tempany, J. E. Tomaszewski, A. Renshaw, and A. Wein. An
analysis of the time course of postoperative prostate-specific antigen failure in
patients with positive surgical margins: implications on the use of adjuvant
therapy. Urology 1996; 47:538-547.
20. Kausik SJ, Blute ML, Sebo TJ, Leibovich BC, Bergstralh EJ, Slezak J, Zincke H.
Prognostic significance of positive surgical margins in patients with
extraprostatic carcinoma after radical prostatectomy. Cancer 2002; Sep
15;95(6):1215-9.
21. Obek C, Sadek S Lai S, Civantos F, Rubinowicz D, Soloway MS. Positive
surgical margins with radical retropubic prostatectomy: anatomic site-specific
pathologic analysis and impact on prognosis. Urology 1999; Oct;54(4):682-8.
22. Cheung L, Darson MF, Bergstralh EJ, Slezak J, Myers RP, Bostwick DG.
Correlation of margin status and extraprostatic extension with progression of
prostate carcinoma. Cancer 1999; 86(9):1775-82.
23. Blute ML, Bostwick DG, Bergstralh EJ, Slezak JM, Martin SK, Amling CL,
Zincke H. Anatomic site-specific positive margins in organ-confined prostate
cancer and its impact on outcome after radical prostatectomy. Urology 1997;
Nov;50(5):733-9.
24. Leibovich BC, Engen DE, Patterson DE, Pisansky TM, Alexander EE,Blute ML,
Bergstralh EJ, Zincke H. Benefit of adjuvant radiation therapy for localized
prostate cancer with a positive surgical margin. J Urol. 2000 Apr;163(4):1178-
82.
25. Han M, Pound CR Potter SR, Partin AW, Epstein JI, Walsh PC. Isolated local
recurrence is rare after radical prostatectomy in men with Gleason 7 prostate
cancer and positive surgical margins: therapeutic implications. J Urol. 2001
Mar;165(3):864-6.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 60 of 114
Amendment 2: dated 29 May 2014
26. Ohori, M., T. M. Wheeler, M. W. Kattan, Y. Goto, and P. T. Scardino. 1995.
Prognostic significance of positive surgical margins in radical prostatectomy
specimens. J Urol 154:1818-1824.
27. Stein A, deKernion J B, Smith R B, Dorey F, and Patel H. Prostate-specific
antigen levels after radical prostatectomy in patients with organ-confined and
locally extensive prostate cancer. J Urol 1992;147: 942-946.
28. Partin A W, Borland R N, Epstein J I and Brendler C B. Influence of wide
excision of the neurovascular bundles(s) on the prognosis in men with clinically
localized prostate cancer with established capsular penetration. J Urol 1993,
150: 142-148.
29. Lange PH, Lightner DJ, Medini E, Reddy PK, Vessella RL. The effect of
radiation therapy after radical prostatectomy in patients with elevated prostate
specific antigen levels. J Urol 1990; 144(4):927-932.
30. Schild SE, Wong WW, Grado GL, Buskirk SJ, Robinow JS, Frick LM et al.
Radiotherapy for isolated increases in serum prostate-specific antigen levels
after radical prostatectomy. Mayo Clin Proc 1994; 69(7):613-619.
31. Zietman AL, Coen JJ, Shipley WU, Althausen AF. Adjuvant radiation following
radical prostatectomy: analysis freedom from PSA failure. Urology 1993;
42:292-298.
32. Link P, Freiha FS, Stamey TA. Adjuvant radiation therapy in patients with
detectable prostate specific antigen following radical prostatectomy. J Urol
1991;145:532-534.
33. McCarthy JF, Catalona WJ, Hudson MA. Effect of radiation therapy on
detectable serum PSA levels following radical prostatectomy: Early vs. delayed
treatment. J Urol 1994;151:1575-1578.
34. Schild SE, Sem in Radonc 1998. Radiation therapy after prostatectomy: now or
later? Semin Radiat Oncol. 1998; Apr;8(2):132-9. Review.
35. Bolla M, van Poppel M, Collette L, et al. Postoperative radiotherapy after radical
prostatectomy: a randomised controlled trial (EORTC trial 22911). Lancet 2005;
366:572 – 578.
36. Swanson G, Thompson I, Tangen C, et al. Phase III randomised study of
adjuvant radiotherapy versus observation in patients with pathological T3
prostate cancer (SWOG 8794). Proc ASTRO 2005: 1 (abstr).
37. Wiegel T, Botte D, Willich N, et al. Phase III results of adjuvant radiotherapy
(RT) versus “wait and see” (WS) in patients with pT3 prostate cancer following
radical prostatectomy (RP) (ARO 96-02/AUO AP 09/05). Proc ASCO 2005:
4513 (abstr).
38. Collette L, van Poppel H, Bolla M, van Cangh P, Vekemans K, Da Pozzo L, de
Reijke TM, Verbaeys A, Bosset JF, Pierart M; European Organisation for
Research and Treatment of Cancer (EORTC) Radiotherapy and Genito-urinary
Group. Patients at high risk of progression after radical prostatectomy: Do they
all benefit from immediate post-operative irradiation? (EORTC trial 22911)
European Journal of Cancer. 2005; 41:2662–2672.
39. B.Tombal et al, ASCO Prostate 2006 (abstr).

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 61 of 114
Amendment 2: dated 29 May 2014
40. Freedland SJ, Sutter ME, Dorey F, et al: Defining the ideal cutpoint for
determining PSA recurrence after radical prostatectomy: Prostate-specific
antigen. Urology 2003; 61:365-369.
41. Zelefsky MJ, Aschkenasy E, Kelsen S, Leibel SA. Tolerance and early outcome
results of postprostatectomy three-dimensional conformal radiotherapy. Int J
Radiat Oncol Biol Phys. 1997; 39(2):327-333.
42. Stephenson AJ, Shariat SF, Zelefsky MJ, et al. Salvage radiotherapy for
recurrent prostate cancer after radical prostatectomy. JAMA 2004; 291: 1325–
1332.
43. Pearce A, Choo R, Danjoux C, Morton G, Loblaw A, Szumacher E, Cheung P,
Deboer G, Chander S. Effect of combined treatment with salvage radiotherapy
plus androgen suppression on quality of life in patients with recurrent prostate
cancer after radical prostatectomy. Int J Radiat Oncol Biol Phys. 2006; 65(1):
78-83.
44. Post PN, Hansen BE, Kil PJM, Janssen-Heijnen MLG, Coebergh JWW. The
independent prognostic value of comorbidity among men aged < 75 years with
localized prostate cancer: a population-based study. BJU International. 2001;
87(9): 821–826.
45. Sidhom MA, Kneebone AB, Lehman M, Wiltshire KL, Millar JL, Mukherjee RK,
Shakespeare TP, Tai KH. Post-prostatectomy radiation therapy: Consensus
guidelines of the Australian and New Zealand Radiation Oncology Genito-
Urinary Group. Radiother Oncol 2008; 88(1): 10-19.
46. de Crevoisier R. Tucker SL. Dong L. Mohan R. Cheung R. Cox JD. Kuban DA.
Increased risk of biochemical and local failure in patients with distended rectum
on the planning CT for prostate cancer radiotherapy. Int J Radiat Oncol Biol
Phys. 2005; 62(4):965-73.
47. ICRU Report 62, Prescribing, Recording and Reporting Photon Beam Therapy
(Supplement to ICRU Report 50). International Commission on Radiation Units
and Measurement 1999
48. ICRU Report 50, Prescribing, Recording, and Reporting Photon Beam Therapy
International Commission on Radiation Units and Measurement 1993.
49. Peeters S, Heemsbergen W, van Putten W, Slot A, Tabak H, Mens J, Lebesque
J, Koper P. Acute and late complications after radiotherapy for prostate cancer:
Results of a multicenter randomized trial comparing 68 Gy to 78 Gy. Int J Radiat
Oncol Biol Phys. 2005; 61(4): 1019-34.
50. Cozzarini C, Fiorino C, Ceresoli G, Cattaneo G, Bolognesi A, Calandrino R Villa
E. Significant correlation between rectal DVH and late bleeding in patients
treated after radical prostatectomy with conformal or conventional radiotherapy
(66.6–70.2 Gy). Int J Radiat Oncol Biol Phys. 2004; 55(3): 688-94.
51. Huang E, Pollack A, Levy L, Starkschall G, Dong L, Rosen I, Kuban D Late
rectal toxicity: dose-volume effects of conformal radiotherapy for prostate
cancer Int J Radiat Oncol Biol Phys. 2002; 54(4): 1314-1321.
52. Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta
Psychiatr Scand. 1983; Jun;67(6):361-70.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 62 of 114
Amendment 2: dated 29 May 2014
53. Rosen RC, Riley A, Wagner G, Osterloh IH, Kirkpatrick J, Mishra A.
The international index of erectile function (IIEF) a multidimensional scale for
assessment of erectile dysfunction. Urology. 1997 Jun; 49(6):822-30.
54. Epstein JI, Amin M, Boccon-Gibod L, et al. Prognostic factors and reporting of
prostate carcinoma in radical prostatectomy and pelvic lymphadenectomy
specimens. Scand J Urol Nephrol Suppl. 2005 May;(216):34-63. Review.
55. Altmuller J, Palmer LJ, Fischer G, et al (2001). Genomewide scans of complex
human diseases: true linkage is hard to find. Am J Hum Genet 69, 936-50.
56. Barrett JC and Cardon LR (2006). Evaluating coverage of genome-wide
association studies. Nature Genetics 38(6):659-62.
57. Neale BM and Sham PC (2004). The future of association studies: gene-based
analysis and replication. Am J Hum Genet 75:353-62.
58. Carlson CS, Eberie MA, Kruglyak L, Nickerson DA (2004). Mapping complex
disease loci in whole-genome association studies. Nature 429:446-452
59. The Wellcome Trust Case Control Consortium. Genome-wide association study
of 14,000 cases of seven common diseases and 3,000 shared controls. Nature
447: 661-683 (2007).
60. SL Zheng, J Sun, F Wiklund, S Smith, P Stattin, G Li, H Adami, F Hsu, Y Zhu, K
Bälter, AK Kader, AR Turner, W Liu, ER Bleecker, DA Meyers, D Duggan, et al.
Cumulative association of five genetic variants with prostate cancer. NEJM
10.1056/NEJMoao75819, 16 January 2008 [Epub ahead of print]
61. J Marchini, B Howie, S Myers, G McVean and P Donnelly. A new multipoint
method for genome-wide association studies by imputation of genotypes.
Nature Genetics 39: 906-913 (2007).

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 63 of 114
Amendment 2: dated 29 May 2014
APPENDIX I. AJCC Staging System (6th Edition)
Primary Tumor, Pathologic (pT)
Stage
Definition
pT2*
Organ confined (Note: There is no pathologic T1 classification)
pT2a Unilateral
Unilateral
pT2b Bilateral
Bilateral
pT3
Extraprostatic extension
pT3a
Extraprostatic extension (i.e. without seminal vesicle invasion)
pT3b
Seminal vesicle invasion
pT4
Invasion of bladder, rectum
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 64 of 114
Amendment 2: dated 29 May 2014
APPENDIX II. ECOG Performance Status
Eastern Cooperative Oncology Group (ECOG) performance status
ECOG PERFORMANCE STATUS*
Grade
ECOG
0
Fully active, able to carry on all pre-disease performance without restriction
1
Restricted in physically strenuous activity but ambulatory and able to carry
out work of a light or sedentary nature, e.g., light house work, office work
2
Ambulatory and capable of all self care but unable to carry out any work
activities. Up and about more than 50% of waking hours
3
Capable of only limited self care, confined to bed or chair more than 50% of
waking hours
4
Completely disabled. Cannot carry on any self care. Totally confined to bed
or chair
5
Dead
* Oken, M.M., Creech, R.H., Tormey, D.C., Horton, J., Davis, T.E., McFadden, E.T., Carbone, P.P.:
Toxicity And Response Criteria Of The Eastern Cooperative Oncology Group. Am J Clin Oncol 5:649-
655, 1982.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 65 of 114
Amendment 2: dated 29 May 2014
APPENDIX III. Co-morbidity Index
Does the patient have a history of, or presently have:
Tick if
present
Myocardial infarction
Heart failure
Angina, intermittent claudication, or abdominal aortic aneurysm
Other malignancies (except basal skin carcinoma)
Peptic ulcer disease (medically or surgically treated, but not reflux)
Cerebrovascular accident (stroke) or Transient Ischaemic Attacks
Diabetes mellitus (medically treated)
Chronic obstructive pulmonary diseases
Dementia
Connective tissue / autoimmune diseases: eg. Sarcoid, SLE, Wegener's granulomatosis,
Rheumatoid
Liver disease (any LFT > 1.5 times normal)
Kidney diseases (renal function >1.5 times normal)
Bowel diseases: Crohn's disease, ulcerative colitis
Hypertension (or on antihypertensive therapy)
Hypercholesterolaemia (or using cholesterol lowering agents)
Number of other medical diagnoses
Write Number
Number of regular prescription medications taken (including aspirin)
Write Number
Validated co-morbidity index of Post et al.44

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 66 of 114
Amendment 2: dated 29 May 2014
APPENDIX IV. EORTC QLQ-C30 & QLQ-PR25
EORTC QLQ-C30 (version 3)
We are interested in some things about you and your health. Please answer all of the
questions yourself by circling the number that best applies to you. There are no
"right" or "wrong" answers. The information that you provide will remain strictly
confidential.
Please fill in your initials: l l l l l
Your Birth Date (Day, Month, Year): l l l l l l l l l
Today's date (Day, Month, Year): l l l l l l l l l
Not at
All
A
Little
Quite
a Bit
Very
Much
1.
Do you have any trouble doing strenuous activities,
like carrying a heavy shopping bag or a suitcase?
1
2
3
4
2.
Do you have any trouble taking a long walk?
1
2
3
4
3.
Do you have any trouble taking a short walk outside
of the house?
1
2
3
4
4.
Do you need to stay in bed or a chair during the day?
1
2
3
4
5.
Do you need help with eating, dressing, washing
yourself or using the toilet?
1
2
3
4
During the past week:
Not at
All
A
Little
Quite
a Bit
Very
Much
6.
Were you limited in doing either your work or other
daily activities?
1
2
3
4
7.
Were you limited in pursuing your hobbies or other
leisure time activities?
1
2
3
4
8.
Were you short of breath?
1
2
3
4
9.
Have you had pain?
1
2
3
4
10.
Did you need rest?
1
2
3
4`
11.
Have you had trouble sleeping?
1
2
3
4
12.
Have you felt weak?
1
2
3
4
13.
Have you lacked appetite?
1
2
3
4
14.
Have you felt nauseated?
1
2
3
4
15.
Have you vomited?
1
2
3
4
16.
Have you been constipated?
1
2
3
4

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 67 of 114
Amendment 2: dated 29 May 2014
During the past week:
Not at
All
A
Little
Quite
a Bit
Very
Much
17.
Have you had diarrhoea?
1
2
3
4
18.
Were you tired?
1
2
3
4
19.
Did pain interfere with your daily activities?
1
2
3
4
20.
Have you had difficulty in concentrating on things, like
reading a newspaper or watching television?
1
2
3
4
21.
Did you feel tense?
1
2
3
4
22.
Did you worry?
1
2
3
4
23.
Did you feel irritable?
1
2
3
4
24.
Did you feel depressed?
1
2
3
4
25.
Have you had difficulty remembering things?
1
2
3
4
26.
Has your physical condition or medical treatment
interfered with your family life?
1
2
3
4
27.
Has your physical condition or medical treatment
interfered with your social activities?
1
2
3
4
28.
Has your physical condition or medical treatment
caused you financial difficulties?
1
2
3
4
For the following questions please circle the number between 1 and 7 that best
applies to you.
29.
How would you rate your overall health during the past week?
1 2 3 4 5 6 7
Very poor Excellent
30.
How would you rate your overall quality of life during the past week?
1 2 3 4 5 6 7
Very poor Excellent
© Copyright 1995 EORTC Quality of Life Group. All rights reserved. Version 3.0

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 68 of 114
Amendment 2: dated 29 May 2014
EORTC QLQ – PR25
Patients sometimes report that they have the following symptoms or problems. Please
indicate the extent to which you have experienced these symptoms or problems
during the past week. Please answer by circling the number that best applies to you.
During the past week:
Not
at all
A
little
Quite
a bit
Very
much
31.
Have you had to urinate frequently during the day
1
2
3
4
32.
Have you had to urinate frequently at night?
1
2
3
4
33.
When you felt the urge to pass urine, did you have to hurry to
get to the toilet?
1
2
3
4
34.
Was it difficult for you to get enough sleep, because you
needed to get up frequently at night to urinate?
1
2
3
4
35.
Have you had difficulty going out of the house because you
needed to be close to a toilet?
1
2
3
4
36.
Have you had any unintentional release (leakage) of urine?
1
2
3
4
37.
Did you have pain when you urinated?
1
2
3
4
38.
Answer this question only if you wear incontinence aid.
Has wearing an incontinence aid been a problem for you?
1
2
3
4
39.
Have your daily activities been limited by your urinary
problems?
1
2
3
4
40.
Have your daily activities been limited by your bowel problems?
1
2
3
4
41.
Have you had any unintentional release (leakage) of stools?
1
2
3
4
42.
Have you had blood in your stools?
1
2
3
4
43.
Did you have a bloated feeling in your abdomen?
1
2
3
4
44.
Did you have hot flushes?
1
2
3
4
45.
Have you had sore or enlarged nipples or breasts?
1
2
3
4
46.
Have you had swelling in your legs or ankles?
1
2
3
4

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 69 of 114
Amendment 2: dated 29 May 2014
During the last 4 weeks:
Not
at all
A
little
Quite
a bit
Very
much
47.
Has weight loss been a problem for you?
1
2
3
4
48.
Has weight gain been a problem for you?
1
2
3
4
49.
Have you felt less masculine as a result of your illness or
treatment?
1
2
3
4
50.
To what extent were you interested in sex?
1
2
3
4
51.
To what extent were you sexually active
(with or without intercourse)?
1
2
3
4
PLEASE ANSWER THE NEXT FOUR QUESTIONS ONLY IF YOU HAVE BEEN
SEXUALLY ACTIVE OVER THE LAST 4 WEEKS.
52.
To what extent was sex enjoyable for you?
1
2
3
4
53.
Did you have difficulty getting or maintaining an erection?
1
2
3
4
54.
Did you have ejaculation problems (eg dry ejaculation)?
1
2
3
4
55.
Have you felt uncomfortable about being sexually intimate?
1
2
3
4
© Copyright 1999 EORTC Quality of Life Group. All rights reserved. (phase III module)

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 70 of 114
Amendment 2: dated 29 May 2014
APPENDIX V. Hospital Anxiety and Depression Scale (HADS)
Patients are asked to choose one response from the four given for each interview.
They should give an immediate response and be dissuaded from thinking too long
abut their answers. The questions relating to anxiety are marked “A”, and to
depression “D”. The score for each answer is given in the right column. Instruct the
patient to answer how it currently describes their feelings.
A
I feel tense or ‘wound up’:
Most of the time
3
A lot of the time
2
From time to time, occasionally
1
Not at all
0
D
I still enjoy the things I used to enjoy:
Definitely as much
0
Not quite so much
1
Only a little
2
Hardly at all
3
A
I get a sort of frightened feeling as if something awful is about to
happen:
Very definitely and quite badly
3
Yes, but not too badly
2
A little, but it doesn’t worry me
1
Not at all
0
D
I can laugh and see the funny side of things:
As much as I always could
0
Not quite so much now
1
Definitely not so much now
2
Not at all
3
A
Worrying thoughts go through my mind:
A great deal of the time
3
A lot of the time
2
From time to time, but not too often
1
Only occasionally
0
D
I feel cheerful:
Not at all
3
Not often
2
Sometimes
1
Most of the time
0

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 71 of 114
Amendment 2: dated 29 May 2014
A
I can sit at ease and feel relaxed:
Definitely
0
Usually
1
Not often
2
Not at all
3
D
I feel as if I am slowed down:
Nearly all the time
3
Very often
2
Sometimes
1
Not at all
0
A
I get a sort of frightened feeling like ‘butterflies’ in the stomach:
Not at all
0
Occasionally
1
Quite often
2
Very often
3
D
I have lost interest in my appearance:
Definitely
3
I don’t take as much care as I should
2
I may not take quite as much care
1
I take just as much care as ever
0
A
I feel restless as I have to be on the move:
Very much indeed
3
Quite a lot
2
Not very much
1
Not at all
0
D
I look forward with enjoyment to things:
As much as I ever did
0
Rather less than I used to
1
Definitely less than I used to
2
Hardly at all
3
A
I get sudden feelings of panic:
Very often indeed
3
Quite often
2
Not very often
1
Not at all
0

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 72 of 114
Amendment 2: dated 29 May 2014
D
I can enjoy a good book or radio or TV program:
Often
0
Sometimes
1
Not often
2
Very seldom
3
Scoring (add the As = Anxiety. Add the Ds = Depression). The norms below will give
you an idea of the level of Anxiety and Depression.
0 – 7 = Normal
8 – 10 = Borderline abnormal
11 – 21 = Abnormal
Reference: Zigmond and Snaith (1983)52
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 73 of 114
Amendment 2: dated 29 May 2014
APPENDIX VI. NCI CTCAE v 3.0
NCI Common Terminology Criteria for Adverse Effects (CTCAE) v 3.0 (August 9th 2006)
Genitourinary
GRADE
1
2
3
4
5
Cystitis
Asymptomatic
Frequency with dysuria;
macroscopic haematuria
Transfusion; IV pain
medications; bladder irrigation
indicated
Catastrophic bleeding; major
non-elective intervention
indicated
Death
Urinary Incontinence
Occasional (e.g., with
coughing, sneezing, etc.),
pads not indicated
Spontaneous, pads indicated
Interfering with ADL; intervention
indicated (e.g., clamp, collagen
injections)
Operative intervention indicated
(e.g.,
cystectomy or permanent
urinary diversion)
N/A
Urethral
Stricture/Stenosis
Asymptomatic, radiographic
or endoscopic findings only
Symptomatic but no
hydronephrosis, sepsis or
renal dysfunction; dilation or
endoscopic repair or stent
placement indicated
Symptomatic and altered organ
function (eg, sepsis,
hydronephrosis, or renal
dysfunction); operative
intervention indicated
Life-threatening consequences;
organ failure or operative
intervention requiring organ
resection indicated
Death
Urinary
Frequency/Urgency
Increase in frequency or
nocturia up to 2 x normal;
enuresis
Increase >2 x normal but <
hourly
> 1 x/hr; urgency or catheter
indicated
N/A
N/A
Urinary Retention
Hesitancy or dribbling, no
significant residual urine;
retention occurring during the
immediate postoperative
period
Hesitancy requiring
medication; or operative
bladder atony requiring
indwelling catheter beyond
immediate postoperative
period but for <6 weeks
More than daily catheterization
indicated; urological intervention
indicated (e.g., TURP,
suprapubic tube, urethrotomy)
Life-threatening consequences;
organ failure (e.g., bladder
rupture); operative intervention
requiring organ resection
indicated
N/A
Haemorrhage, GU
Minimal or microscopic
bleeding intervention not
indicated
Gross bleeding, medical
intervention or urinary tract
irrigation indicated
Transfusion, interventional
radiology, endoscopic, or
operative intervention indicated;
radiation therapy (ie
haemostasis of bleeding site)
Life threatening consequences;
major urgent intervention
indicated
Death
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 74 of 114
Amendment 2: dated 29 May 2014
Gastrointestinal
GRADE
1
2
3
4
5
Diarrhoea1
Increase of <4 stools per day
over baseline; mild increase
in ostomy output compared
to baseline
Increase of 4 – 6 stools per day
over baseline; IV fluids
indicated <24hrs; moderate
increase in ostomy output
compared to baseline; not
interfering with ADL
Increase of ≥7 stools per day
over baseline; incontinence; IV
fluids ≥24 hrs; hospitalization;
severe increase in ostomy output
compared to baseline; interfering
with ADL
Life-threatening consequences
(e.g., hemodynamic collapse)
Death
Proctitis
Rectal discomfort
intervention not indicated
Symptoms not interfering with
ADL; medical intervention
indicated
Stool incontinence or other
symptoms interfering with ADL;
operative intervention indicated
Life-threatening consequences
(e.g., perforation)
Death
Haemorrhage, GI
(Rectal)
Mild, intervention (other than
iron supplements) not
indicated
Symptomatic and medical
intervention or minor
cauterization indicated
Transfusion, interventional
radiology, endoscopic, or
operative intervention indicated;
radiation therapy (i.e.,
haemostasis of bleeding site)
Life-threatening
consequences; major urgent
intervention indicated
Death
Incontinence (Anal) 2
Occasional use of pads
Daily use of pads
Interfering with ADL; operative
intervention indicated
Permanent bowel diversion
indicated
Death
1 Diarrhoea includes diarrhoea of small bowel or colonic origin, and/or ostomy diarrhoea.
2 Incontinence, anal is to be used for loss of sphincter control as sequelae of operative or therapeutic intervention
Sexual Function
GRADE
1
2
3
4
5
Erectile Dysfunction
Decrease in erectile function
(frequency/rigidity of
erections) but erectile aids
not indicated
Decrease in erectile function
(frequency/rigidity of erections),
erectile aids indicated
Decrease in erectile function
(frequency/rigidity of erections)
but erectile aids not helpful;
penile prosthesis indicated
N/A
N/A
Others
GRADE
1
2
3
4
5
Refer to CTCAE v3.0
(http://ctep.cancer.gov/r
eporting/ctc.html) and
use exact term or
specify other
Mild Adverse Event
Moderate Adverse Event
Severe and undesirable Adverse
Event
Life-threatening or disabling
Adverse Event
Death
related to
Adverse
Event

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 75 of 114
Amendment 2: dated 29 May 2014
APPENDIX VII. Study Schedule for patient assessments and follow-up
Arm 1 (Adjuvant Radiotherapy – ART)
1 Not required prior to randomisation.
2 Quality of Life (QoL) questionnaires, SHIM and hospital anxiety and depression scale (HADS) - to be done prior to the PSA result being revealed.
3 Includes rectal examination if rising PSA till documented local failure
4 If the first 6 monthly assessment coincides within 28 days of the 6 week post-RT assessment, the 6 monthly assessment can be omitted.
5 If it is not feasible to approach the patient about the optional biological sub-studies at 6 months post-randomisation, they can choose to participate at any
time up until 6 months after recruitment closes.
6PSA tests should be +/- 28 days from target date(s) calculated relative to randomisation. If a PSA result that triggers SRT or meets the criteria for bF falls
between two scheduled assessments, the relevant PSA result must be reported as an unscheduled PSA assessment.
Assessment
(relative to randomisation)
Pre-
randomisation
Day 1 RT
End RT
6 wks
post-RT
Follow up relative to randomisation
0-5 yr
5 yrs to trial end
Randomisation data
(eligibility criteria, stratification factors)
X
Medical history/ Physical exam
X
ECOG performance status
X
Co-morbidity Index
X
Clinical assessment 3
X
X
X4
6 monthly4
12 monthly
Adverse events (CTCAE V 3.0)
X
X
X
X
12 monthly
12 monthly
Central pathology review 1
X
PSA
Pre-operative
Post-operative
X
6 monthly4, 6
6 monthly6
Patient Self-Assessments
Adverse Events
SHIM2
X
12 monthly
12 monthly
Quality of Life 2
EORTC QLQ-C30
EORTC QLQ-PR25
X
X
X
X
12 monthly
12 monthly
Anxiety/depression 2
HADS
X
X
X
X
12 monthly
12 monthly
Hospitalisation questionnaire
12 monthly
12 monthly
Optional biological sub-study
questionnaire
6 months post-
randomisation5
Optional Biological Substudies:
Sputum collection
Tumour blocks
6 months post-
randomisation5
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 76 of 114
Amendment 2: dated 29 May 2014
Arm 2 (Salvage radiotherapy – SRT)
1 Not required prior to randomisation.
2 Quality of Life (QoL) questionnaires, SHIM and hospital anxiety and depression scale (HADS) - to be done prior to the PSA result being revealed.
3 Includes rectal examination if rising PSA till documented local failure
4 If a 6 monthly assessment coincides within 28 days of the 6 week post-RT assessment, the 6 monthly assessment can be omitted.
5 If it is not feasible to approach the patient about the optional biological sub-studies at 6 months post-randomisation, they can choose to participate at any time up until 6 months after
recruitment closes.
6PSA tests should be +/- 28 days from target date(s) calculated relative to randomisation. f a PSA result that triggers SRT or meets the criteria for bF falls between two scheduled
assessments, the relevant PSA result must be reported as an unscheduled PSA assessment.
PSA < 0.2 ng/mL
During and following RT
Assessment
(relative to randomisation)
Pre-
randomisation
Surveillance, relative to randomisation
Day 1 RT
End RT
6 wks
post-RT
Follow up, relative to randomisation
0-5 yrs
5 yrs to trial end
0-5 yrs
5 yrs to trial end
Randomisation data (eligibility
criteria, stratification factors)
X
Medical history/ Physical exam
X
ECOG performance status
X
Co-morbidity Index
X
Clinical assessment 3
6 monthly
12 monthly
X
X
X
6 monthly4
12 monthly
Adverse events (CTCAE V 3.0)
X
12 monthly
12 monthly
X
X
X
12 monthly
12 monthly
Central pathology review 1
X
PSA6
Pre-operative
Post-operative
3 monthly6
6 monthly6
X
X
6 monthly4
6 monthly
Patient Self-Assessments
Adverse events
SHIM2
X
12 monthly
12 monthly
12 monthly
Quality of Life 2
EORTC QLQ-C30
EORTC QLQ-PR25
X
12 monthly
12 monthly
X
X
X
12 monthly
12 monthly
Anxiety/depression 2
HADS
X
12 monthly
12 monthly
X
X
X
12 monthly
12 monthly
Hospitalisation
questionnaire
12 monthly
12 monthly
12 monthly
12 monthly
Optional Biological sub-
study questionnaire
At 6 months
post-
randomisation
At 6 months
post-
randomisation
Optional Biological Substudies:
Sputum collection
Tumour blocks
At 6 months
post-
randomisation
At 6 months
post-
randomisation
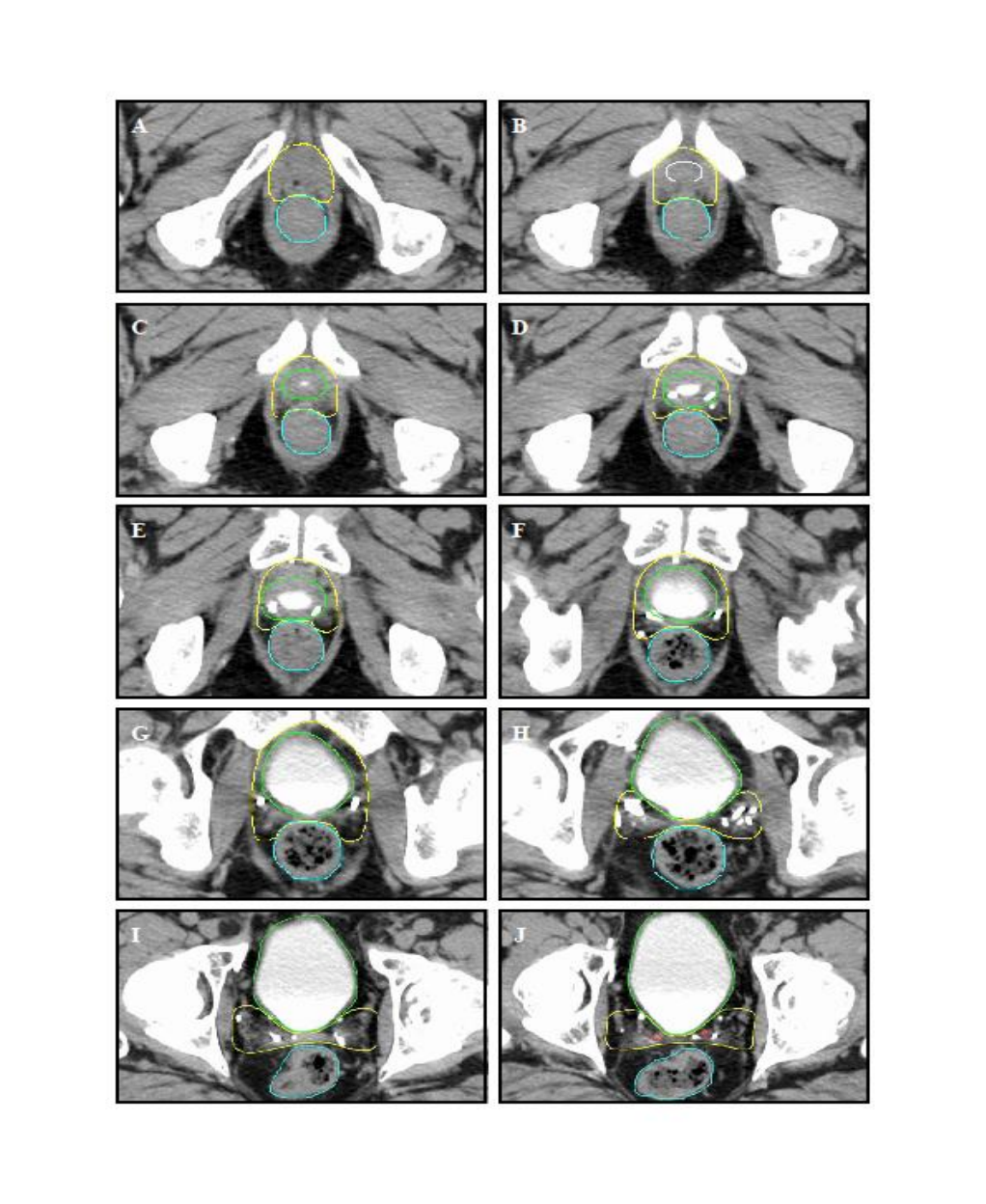
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 77 of 114
Amendment 2: dated 29 May 2014
Appendix VIII. Pictorial Contouring Guidelines
Figure 1
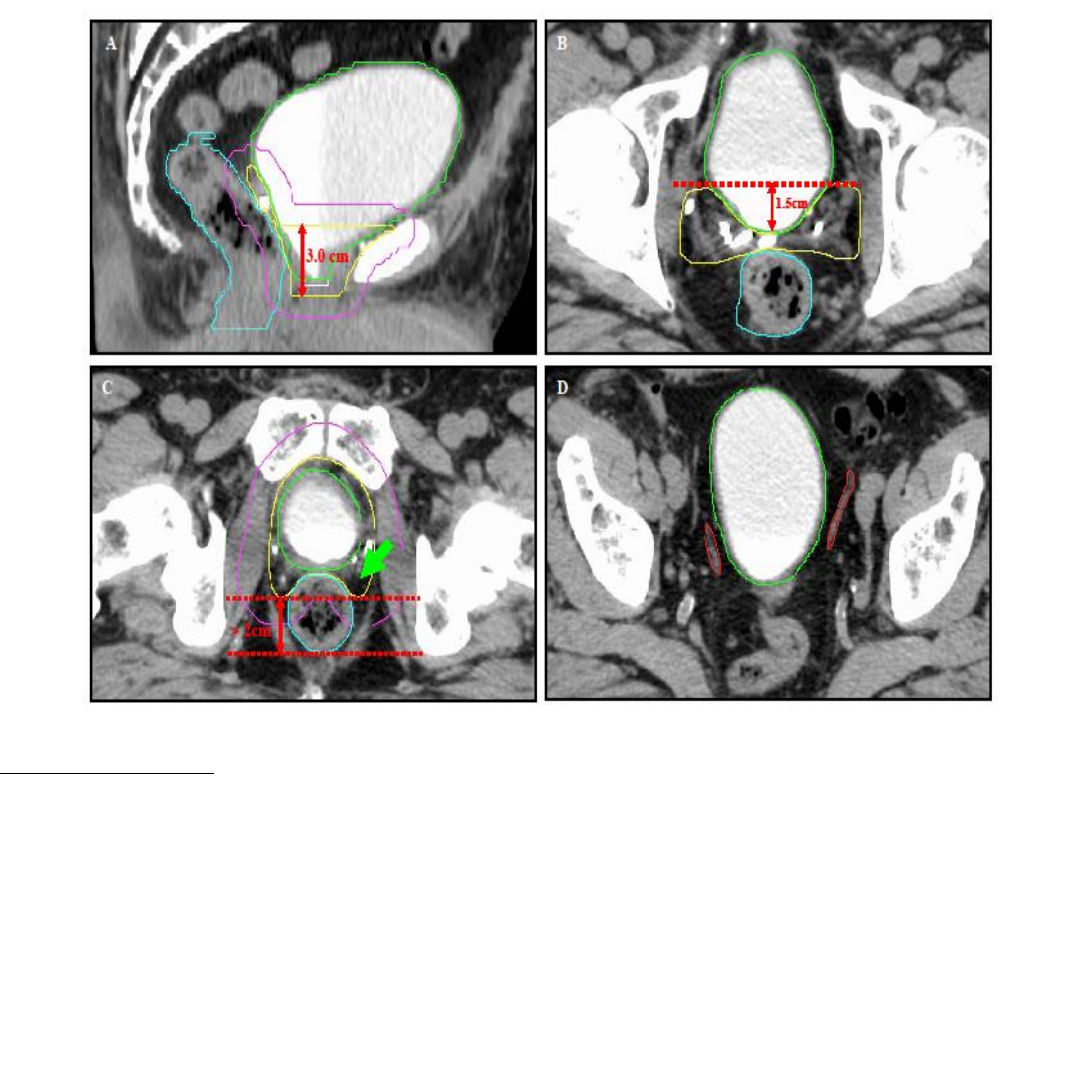
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 78 of 114
Amendment 2: dated 29 May 2014
Figure 2
FIGURE LEGENDS
Figure 1: [CTV – yellow, vesicourethral anastomosis – white, rectum – blue, bladder – green, vas
deferens – red]. Serial CT slices of a sample post-prostatectomy patient demonstrating the contouring
guidelines. Delayed scanning following intravenous contrast allowed contrast accumulation in the
bladder so as to ascertain the most inferior slice where urine is last visible (C). The anastomosis is
located one slice below this (B), and the most inferior CTV slice a further 5mm lower (A). Images D to G
illustrate that the anterior border of the inferior 3cm of the CTV lies behind the symphysissymphisis
pubis. More superiorly (H – J) the anterior border encompasses the posterior 1.5cm of the bladder, and
posteriorly extends to the mesorectal fascia. The most superior slice of the CTV (J) encompasses the
last slice where the vas deferens is visible and all non-vascular surgical clips.
Figure 2: [PTV – pink, other structures as per Figure 1]
A. Sagittal reconstruction illustrating the anterior margin of the CTV extending to the border of the pubic
symphysis for the inferior 3cm of the CTV. The PTV is a 10mm uniform expansion upon the CTV
contours.
B. More superiorly, the anterior border of the CTV should encompass the posterior 1.5cm of the bladder
wall.
C. The space bounded by the levator ani and the rectum anteriorly (arrow) is a significant site of
recurrence and should be encompassed if the volume of rectum being treated is not prohibitive. Ideally,
the distance between the posterior margin of the CTV and the posterior rectal wall should be no less
than 2cm to avoid treating the entire circumference of the rectum.
D. The vas deferens are usually identified superiorly as thin horizontal cylindrical structures which
become small round structures more inferiorly.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 79 of 114
Amendment 2: dated 29 May 2014
Appendix IX: Biological Sub-study Patient Questionnaire
1Not required prior to randomisation.
2PSA - to be evaluated 6 monthly post-RT. Upon suspected biochemical failure, a follow-up evaluation is required 4-6 weeks later.
For ARM 2 patients only – during surveillance phase, PSA to be evaluated 3 monthly for first 5 years and 6 monthly thereafter.
3Toxicity assessment according to CTCAE v3.0
4Quality of Life (QoL) questionnaires - to be done prior to the PSA result
5QoL, anxiety and toxicity to be assessed on the annual anniversary of radical prostatectomy up through 5 years
6Hospital anxiety and depression scale (HADS) - to be done prior to the PSA result.
7 Includes rectal examination if rising PSA till documented local recurrence.
For ARM 2 patients only – during surveillance phase, clinical assessment every 6 months.
1Not required prior to randomisation.
2PSA - to be evaluated 6 monthly post-RT. Upon suspected biochemical failure, a follow-up evaluation is required 4-6 weeks later.
For ARM 2 patients only – during surveillance phase, PSA to be evaluated 3 monthly for first 5 years and 6 monthly thereafter.
3Toxicity assessment according to CTCAE v3.0
4Quality of Life (QoL) questionnaires - to be done prior to the PSA result
5QoL, anxiety and toxicity to be assessed on the annual anniversary of radical prostatectomy up through 5 years
6Hospital anxiety and depression scale (HADS) - to be done prior to the PSA result.
7 Includes rectal examination if rising PSA till documented local recurrence.
For ARM 2 patients only – during surveillance phase, clinical assessment every 6 months.
1Not required prior to randomisation.
2PSA - to be evaluated 6 monthly post-RT. Upon suspected biochemical failure, a follow-up evaluation is required 4-6 weeks later.
For ARM 2 patients only – during surveillance phase, PSA to be evaluated 3 monthly for first 5 years and 6 monthly thereafter.
3Toxicity assessment according to CTCAE v3.0
4Quality of Life (QoL) questionnaires - to be done prior to the PSA result
5QoL, anxiety and toxicity to be assessed on the annual anniversary of radical prostatectomy up through 5 years
6Hospital anxiety and depression scale (HADS) - to be done prior to the PSA result.
7 Includes rectal examination if rising PSA till documented local recurrence.
For ARM 2 patients only – during surveillance phase, clinical assessment every 6 months.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 80 of 114
Amendment 2: dated 29 May 2014

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 81 of 114
Amendment 2: dated 29 May 2014

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 82 of 114
Amendment 2: dated 29 May 2014

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 83 of 114
Amendment 2: dated 29 May 2014

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 84 of 114
Amendment 2: dated 29 May 2014
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 85 of 114
Amendment 2: dated 29 May 2014
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 86 of 114
Amendment 2: dated 29 May 2014

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 87 of 114
Amendment 2: dated 29 May 2014

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 88 of 114
Amendment 2: dated 29 May 2014

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 89 of 114
Amendment 2: dated 29 May 2014

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 90 of 114
Amendment 2: dated 29 May 2014

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 91 of 114
Amendment 2: dated 29 May 2014
APPENDIX IX: Central Pathology Review Synoptic Report Template
Central Pathology Review Synoptic Report
Submitting laboratory:
Name:____________________________________
Collection Date: ____________________________
Laboratory Ref. No: _________________________
Review Component
Review Parameters
Prostate Gland
Measurement: ___by ____ mm
Weight: _________gms
Tumour
Type: NOS/ or subtype
Tumour Location
Regions: Anterior, Posterior
Zones: Peripheral, transitional, central
Index Cancer
Block:
Index Cancer
Gleason Score
Primary Grade
Secondary Grade
Tertiary
% High Grade (4/5)
Intraduct Carcinoma
Present or Absent
Extra Prostatic Extension
Negative
Focal - 1 or 2 fields with EPE extending ≤1 HPF (0.5mm)
Established - 1 or more fields EPE extending >1 HPF (0.5mm)
Location: Bladder Neck, Anterior, Posterolateral
Margin Involvement
Negative
Focal - 1 or 2 regions involved by ≤ 1 mm
Extensive - 1 or more regions involved by > 1 mm
Location: Bladder Neck, Apex, Posterolateral, Anterolateral
Grade at margin: Gleason Grade
Seminal Vesicle
Right side Not Involved / Involved
Left side Not Involved / Involved
Lymph Nodes
Involved / Not involved
Number of total involved: ... / ... (eg: 1/3 or 0/5 etc)
Stage (pT)
2 confined to prostate
3A 3A extra prostatic extension
3B seminal vesicle involvement
Variation from submitting pathologist
Yes/No
If yes, brief comment

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 92 of 114
Amendment 2: dated 29 May 2014
APPENDIX XI: Sexual Health Inventory for Men (SHIM)

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 93 of 114
Amendment 2: dated 29 May 2014
APPENDIX XII: Health Resource Usage Questionnaire
Have you been admitted to hospital in the last 12 months? Yes No
If yes:
How many times were admitted to hospital?
Please describe the reason you were admitted to hospital, and the number of nights
you spent in hospital (please complete a separate line for each admission if more
than 1):
1. Reason for admission:_____________________________________
Number of nights in hospitals:
2. Reason for admission:_____________________________________
Number of nights in hospitals:
3. Reason for admission:_____________________________________
Number of nights in hospitals:
Questionnaire developed by:
Jon Karnon
Professor in Health Economics
School of Population Health and Clinical Practice
University of Adelaide
Do not complete
remainder of form

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 94 of 114
Amendment 2: dated 29 May 2014
APPENDIX XIII: RAVES Patient Information and Consent Form
PATIENT INFORMATION AND CONSENT FORM
Amendment 1: 8 July 2011
Site: [Add your institution’s name]
Full Project Title: A phase III multi-centre randomised trial comparing
adjuvant radiotherapy (RT) with surveillance and early
salvage RT in patients with positive margin or
extraprostatic disease following radical
prostatectomy
Short Title: RAVES: Radiotherapy – Adjuvant Versus Early Salvage
Study Number: TROG 08.03
Principal Researcher: [Add name]
Please make sure you have all [Add number] pages of this document.
Introduction
You are invited to participate in a clinical trial (a type of research study). This Patient
Information Sheet explains the research project and the procedures involved. Its purpose is
to help you decide if you want to take part in this research. Please take your time to make
your decision. You may wish to discuss it with your friends and family.
Participation in this research is voluntary and you may refuse to participate or withdraw from
the trial at any time, without penalty or loss of benefits to which you are entitled. If you decide
you want to take part in the research project, then you will be asked to sign the Consent
Form. By signing it you are telling us that you:
Understand what you have read in the information sheet
Consent to taking part in the research project
Consent to the use of your personal and health information as described in the
information sheet.
You will be given a copy of this Patient Information Sheet and Consent Form to keep as a
record.
Background and Purpose of Study
You have been invited to take part in this research study because:
You have already had surgery to remove your prostate cancer (called a “radical
prostatectomy”)
The features of your cancer (as determined by a pathologist after the surgery)
indicate the risk of the PSA (prostate specific antigen) rising again may be higher
than other patients. This rise in PSA can indicate the cancer is coming back although
this can take months to years to occur. In some patients the cancer may never cause
symptoms even though the PSA rises.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 95 of 114
Amendment 2: dated 29 May 2014
The features the pathologist describes which determine whether you are at higher risk of the
PSA rising again include finding prostate cancer in one of the following areas:
At the edges of the prostate gland and surrounding tissue that was surgically
removed.
Outside the capsule that encloses the prostate.
In the seminal vesicles (the glands that sit above the prostate).
If you have one or more of these risk factors, you have nearly a 50% chance the PSA can
rise again at some stage in the future. There are currently two ways of managing men who
have these high risk features after a radical prostatectomy.
1. The first option is to give radiotherapy immediately after surgery to everyone. We
know from several large studies that giving radiotherapy to everyone immediately
after surgery will halve the risk of the PSA rising again. This means that only half of
the men who have radiotherapy benefit from it. Unfortunately we don’t know which
men benefit from radiotherapy and which don’t. It would be simple to give everyone
radiotherapy if there were no side effects but many men experience unpleasant side
effects (these are described in section 8).
2. The second alternative is to watch men very closely (“active surveillance”) with
regular clinic visits and regular PSA blood tests. The PSA blood test can detect
activity of the cancer months or years before other tests and usually long before any
symptoms appear. Radiotherapy is only given if the PSA starts to rise. In this situation
nearly half the patients would be spared the possible side effects of radiotherapy.
It is not known whether these 2 ways of managing men with high risk features are the same
in controlling the PSA in the long term. In one group of men radiotherapy is given to
everybody with high risk features after surgery where as in the other group radiotherapy is
only given if the PSA starts to rise. The aim of the study is to therefore see whether patients
treated with active surveillance (and giving radiotherapy as soon as the PSA rises) is as
good as immediate radiotherapy. This study will also compare the side effects occurring in
each treatment group and what affect the treatments have on quality of life. It will also
compare whether the 2 ways of managing men with high risk features results in the same
percentage of men who are alive and without prostate cancer.
It is important to note that even if you have radiotherapy (either immediately after the
operation or when the PSA rises after active surveillance) that the PSA may continue to
climb afterwards indicating that the cancer is still active. Reasons why the radiotherapy may
not be of benefit include that there are prostate cancer cells in some other part of the body or
the radiotherapy did not get rid of the cancer remaining in the area where the prostate was.
What Is Involved In The Study?
Participation:
In order to clearly determine the most effective treatment, following surgery, in men that have
been diagnosed with the particular type of prostate cancer that you have, approximately 470
men from New Zealand and Australia are required for this study.
Randomisation (placement in a group):
If you decide to participate you will be "randomised" into one of the study groups described
below. Randomisation means that you are put into a group by chance. Neither you nor your
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 96 of 114
Amendment 2: dated 29 May 2014
doctor can choose what group you will be in. You will have an equal chance of being placed
in either group. You will be told which treatment you are to get.
1. Immediate radiotherapy (radiotherapy will start within 6 months of previous prostate
surgery)
or
2. To be watched very closely with regular clinic visits and PSA blood tests (“active
surveillance”) and to receive radiotherapy only if your PSA level begins to rise.
Treatment:
Group 1: Immediate radiotherapy
If you are allocated to the immediate (within 6 months of surgery) radiotherapy group, you
will receive a course of radiotherapy to the area where the prostate gland used to be (the
prostate bed). Before starting the radiotherapy you will have a CT scan of this area, this is
used to plan the radiotherapy. The radiotherapy is given over about 6 and half weeks. You
will need to come in for treatment 5 times a week and you will have a total of 32 treatments.
Each radiotherapy treatment will take approximately 15 minutes each day. Following the
radiotherapy, review appointments and investigations will be carried out every 6 months
including PSA blood testing. We are also interested in the after effects of radiotherapy and
your doctor will ask you about any changes to your urinary, bowel or sexual function which
may be side-effects of the radiotherapy.
Group 2:”Active surveillance” and delayed radiotherapy if necessary
If you are allocated to the “active surveillance” group, you will be seen in clinic at regular
intervals by your doctor for routine check-ups and blood tests. The PSA blood test is done
every 3 months in the first 5 years then every 6 months thereafter. It is expected that only
half of the patients in the “active surveillance” group will have a rise in the PSA. If the PSA
rises above a low set level you will receive radiotherapy in exactly the same way as Group 1
patients (described above). Following radiotherapy you will be clinically monitored every 6
months as described for Group 1.
Procedures and Medical Tests:
The following tests will be done at the hospital or clinic to make sure that you are eligible for
this study. None of these tests are experimental. They are routine and would be done before
starting your cancer treatment, even if you were not taking part in this study.
Medical history
PSA blood tests (2 teaspoons of blood required per test)
The needles used to take blood or inject substances for body scans might be uncomfortable.
You might get a bruise, or rarely, an infection at the site of the needle puncture.
Many of these tests will also be repeated during the study. Some of these tests may be done
more frequently than if you were not taking part in this research study.
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 97 of 114
Amendment 2: dated 29 May 2014
Questionnaires:
Four (4) questionnaires are included in this study that are designed to help us understand
how your treatment and illness affect your quality of life and feelings (mood).
You will be asked to complete most of these questionnaires at the following times:
Before starting the study
On the first day you receive radiation therapy,
On the last day of radiation therapy,
6 weeks following the completion of radiotherapy
Annually after enrolling on this study
The questionnaires ask about how you are feeling and take about 10 minutes to complete.
Some of the questions are personal; you can refuse to answer these if you wish. One
additional questionnaire will ask for information about some types of medical care you may
have received between clinic visits. This questionnaire will be given to you annually
throughout your participation in the study.
The information you provide is for research purposes only and will remain strictly confidential.
The individuals (e.g. doctors, nurses, etc.) directly involved in your care will not usually see
your responses to these questions—if you wish them to know this information please bring it
to their attention.
Other Treatments Whilst on Study
It is important to tell your doctor and the research staff about any treatments or medications
you may be taking, including non-prescription medications, vitamins or herbal remedies and
any changes to these during your participation in the study. You should check with your
doctor before taking any new medications while on the study, unless such medications are
prescribed by another doctor in an emergency situation, in which case, you should notify the
doctor who has enrolled you on this study of these medications as soon as possible.
Collection and Use of Tissue Samples
By consenting to take part in this study, you are also consenting to the collection and use of
tissue samples for diagnostic testing as outlined below.
To confirm your cancer diagnosis
To make sure you have the type of prostate cancer that is being studied in this research
study, a small sample of your tumour will be sent to a central laboratory in Australia where it
will be examined to confirm your diagnosis. This is called “Central Pathology Review” and is
a necessary part of this study. The tumour sample needed for this review will be from your
cancer that has already been removed by surgery. The samples will be returned to your
hospital’s pathology department after completion of the review.
How Long Will I Be in the Study?
After enrolment into the study you will be followed for 10 years (minimum).
The researchers can take you off the study treatment early however for reasons such as:
The treatment does not work for you
You are unable to tolerate the study treatment

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 98 of 114
Amendment 2: dated 29 May 2014
New information shows that the study treatment is no longer in your best interest
Your doctor no longer feels this is the best treatment for you
Decisions made by the sponsor (TROG) or local regulatory/health authorities
Voluntary Participation and Withdrawal
Participation in this research is voluntary and you may refuse to participate or withdraw from
the trial at any time, without penalty or affecting your routine treatment, your relationship with
those treating you or your relationship with the institution where you are receiving your
treatment.
If you join the research study and then decide to leave, please notify a member of the
research team beforehand. This notice will allow a member of the research team to inform
you of any health risks or special requirements linked to withdrawing or stopping your study
treatment. If you do decide to leave the research study it will not be possible to withdraw the
information that has been collected before your decision to withdraw.
What are the Risks of the Study?
Most men will experience some side effects from radiotherapy. Your doctor will watch you
closely to see if you have side effects. Should you suffer any side-effects please notify the
research team (your doctor or research nurse) as soon as possible. When possible other
drugs will be given to you to make side effects less serious and uncomfortable. Many side
effects go away shortly after therapy is stopped but in some cases side effects can be
serious, long-lasting or permanent.
Radiotherapy Side Effects:
Short-term:
Tiredness/fatigue during radiotherapy.
You may pass urine more often, experience possible discomfort and slowing of the
stream when passing urine.
In patients with urinary incontinence (leakage of urine) after surgery the incontinence
may become worse following radiotherapy. Radiotherapy may also cause urinary
incontinence in patients who have no leakage after surgery
You may notice more frequent bowel motions and there may be mucus or spotting of
blood with the bowel motion. It may be uncomfortable to pass a bowel motion.
Change in colouration of the skin (reddening or tanning) and hair loss in the treatment
area.
Long-term:
Some men notice that their bowel habit changes permanently (often more frequent). It
is quite common to have the occasional minor spotting (bleeding) with the bowel
motion.
A very few people have more persistent bleeding or bowel irritation, and this very
occasionally (1-2%) needs surgery to deal with it.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 99 of 114
Amendment 2: dated 29 May 2014
Radiotherapy is also likely to have a negative effect on the recovery of erections or
sexual functioning. This is more likely to happen if there were difficulties prior to
treatment.
Serious injury to the bladder, bowel, urethra or other tissues in the pelvis is rare.
As with any medical treatment, there is also the chance of experiencing an unknown or
unforeseen side-effect.
In the group of patients receiving radiotherapy straight after surgery there are the risks of the
side effects of radiotherapy (as described above). However, if you start radiotherapy later, it
is possible that the radiotherapy may be less effective. We do not know which is best, which
is why we are conducting the study.
Are There Benefits to Taking Part in the Study?
If you agree to take part in this study, there may or may not be any benefit to you. We hope
that the information from this study will benefit other patients in the future.
New Information Arising During the Study
During the research study, new information about the risks and benefits of the study may
become known to the researchers. If this occurs, you will be told about this new information
and your doctor will discuss whether this new information affects you.
Alternatives to Joining this Research Study
If you decide not to take part in this study, your doctor will discuss other treatment options
with you. These may include other experimental therapy or no therapy at this time. Please
talk to your doctor about the known benefits and risks of these other treatment options.
Privacy, Confidentiality and Disclosure of Information
Any information obtained in connection with this research study that can identify you will
remain confidential and will only be used for the purpose of this research study. It will only be
disclosed with your permission, except as may be required by law.
Your medical records and any information obtained during the trial are strictly confidential
and will not be made publicly available. Only authorised individuals from the following
organisations will have access to your information:
The Trans-Tasman Radiation Oncology Group (TROG) who co-ordinate the study in
Australia and New Zealand
Institution where you are receiving your treatment.
Central review laboratories (because they examine tissue for confirmation of
diagnosis)
Regulatory Authorities
Your de-identified, trial-related information will be securely stored by Auckland Hospital for a
period of 15-years. When the results of the trial are presented at scientific meetings or
published in a medical journal no individual participant will be recognisable from the data
presented. In any publication, information will be provided in such a way so you cannot be
identified. By signing the attached Consent Form, you authorise release of, or access to, this
confidential information to the relevant study personnel and regulatory authorities.
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 100 of 114
Amendment 2: dated 29 May 2014
Your family doctor will be advised of your decision to participate in this study after you have
signed the Consent Form. By signing this form you give permission for doctors, other health
professionals, hospitals or laboratories outside this hospital to release information to [name
of institution].concerning your disease and treatment that is relevant for this study. This
information will remain confidential as described above. By signing this form, you authorise
the release of, or access to the information collected about you as described in this section,
even in the event of your death.
In accordance with the Freedom of Information Act [Year, State], you have the right to
access and to request correction of information held about you by [name of institution].
Results of the Project
It may be a number of years before the results of this research are available. The results are
published in medical journals that are available to the public. Your study doctor will be
informed of the results of the study once they are known. Please ask your doctor if you want
to know more about this.
What are the Costs?
You will not be paid for taking part in this study.
Participating in this study will not incur additional costs to you compared to not participating
and receiving standard treatment.
Insurance and Compensation
This cancer research trial is sponsored and coordinated by the Trans Tasman Radiation
Oncology Group (TROG), a not-for-profit research group involving many cancer researchers
in Australia and New Zealand, as well as internationally.
If you suffer any injuries or complications that may be as a result of your participation in this
cancer research trial, you should immediately contact your doctor, who will assist you in
arranging appropriate medical treatment, at no cost to you.
In the unlikely event of an injury caused by your participation in this cancer research trial,
compensation may be payable to you. TROG maintains a clinical trials insurance policy to
protect you in these circumstances.
The insurance policy has principles about when compensation is payable and the amount to
which you may be entitled, which include:
Compensation will depend on the nature, severity and ongoing nature of the injury.
Compensation will only be paid for injury of a serious and enduring character and not
for temporary pain or discomfort or less serious or readily curable complaints.
Compensation will only be paid when, on the balance of probabilities, the injury was
caused by the administration or use of any drug, product or treatment involved in the
trial or was directly caused by your participation in the trial.
Compensation will not be paid for the failure of a drug, product or treatment under
trial to perform its intended purpose.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 101 of 114
Amendment 2: dated 29 May 2014
The amount of any compensation should be agreed between you and the sponsor of
the cancer research trial (TROG), or by an independent lawyer if agreement cannot
be reached.
Funding/Conflict of Interest
Apart from their standard salary, the researchers will not be paid for doing this research.
The institution where you are receiving your treatment is receiving funds from a number of
funding bodies to help offset the costs of conducting this research.
Approval by a Research Ethics Committee
This project will be carried out according to the National Statement on Ethical Conduct in
Research Involving Humans (2007) produced by the National Health and Medical Research
Council of Australia. This statement has been developed to protect the interests of people
who agree to take part in research.
The ethical aspects of this research project have been approved by [add the name of the
HREC that approved this study].
Further Information or Any Problems
If you have questions concerning this study or if you suffer a study-related medical problem
(for example, any side effects) you can contact the principal researcher [insert name and
ph. number] or any of the following people: [list names and contact phone numbers,
including after hours number(s)].
Name: Telephone:
If you have a complaint about this project or would like any advice regarding your rights as a
patient, you can talk to someone who is not involved in the study at all. That person is:
Name: [e.g. Executive Officer of the HREC]
Position:
Telephone:
You will need to tell [insert name] the name of one of the researchers given above.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 102 of 114
Amendment 2: dated 29 May 2014
CONSENT FORM
Amendment 1: 8 July 2011
Site: [Add your institution’s name]
Full Study title: A phase III multi-centre randomised trial comparing adjuvant radiotherapy
(RT) with surveillance and early salvage RT in patients with positive margin or extraprostatic
disease following radical prostatectomy.
Short Title: RAVES – Radiotherapy Adjuvant versus Salvage
Study Number: TROG 08.03
Principle Researcher: [Add name]
My signature on this consent form means the following:
The study has been fully explained to me and all of my questions have been
answered,
I understand the requirements and the risks of the study,
I agree to be selected, at random, into one of the study groups as described in
section 3 of this Patient Information and Consent Form,
I authorize access to my medical records as explained in this consent form,
I authorise the collection and testing of my tissue samples for the mandatory
components of this study, and
I agree to take part in this study.
Participant’s Name (printed) …………………………………………………………………
Signature Date
Name of Impartial Witness as required (printed) ………………………………………….
Signature Date
Declaration by researcher*: I have given a verbal explanation of the research project, its
procedures and risks and I believe that the participant has understood that explanation.
Researcher’s Name (printed) ………………………………………………………………..
Signature Date
* A senior member of the research team must provide the explanation and provision of
information concerning the research project.
Note: All parties signing the Consent Form must date their own signature.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 103 of 114
Amendment 2: dated 29 May 2014
REVOCATION OF CONSENT FORM
Site: [Add your institution’s name]
Full Study title: A phase III multi-centre randomised trial comparing adjuvant radiotherapy
(RT) with surveillance and early salvage RT in patients with positive margin or extraprostatic
disease following radical prostatectomy.
Short Title: RAVES – Radiotherapy Adjuvant versus Salvage
Study Number: TROG 08.03
Principle Researcher: [Add name]
I hereby wish to WITHDRAW my consent to participate in the research proposal
named above and understand that such withdrawal WILL NOT jeopardise any
treatment or my relationship with the institution where I am receiving treatment.
Participant’s Name (printed) …………………………………………………………………
Signature Date

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 104 of 114
Amendment 2: dated 29 May 2014
APPENDIX XIV: RAVES Biological Sub-study Patient Information and Consent
Form
I
PARTICIPANT INFORMATION SHEET
Optional Genetic Studies
TROG 08.03 RAVES: Radiotherapy – Adjuvant Versus Early Salvage
A phase III multi-centre randomized trial comparing adjuvant radiotherapy with surveillance and early
salvage RT in patients with positive margin or extra prostatic disease following radical prostatectomy
OPTIONAL TISSUE STUDIES
A. RAVES Genetic Study: Genetic basis of response to radiotherapy in
prostate cancer patients
B. RAVES Tissue Banking Study
PRINCIPAL INVESTIGATOR: Add local details here
If you require an interpreter, please let us know as one can be provided.
INTRODUCTION
You are invited to take part in two separate projects because you have consented to
participate in the Trans-Tasman Radiation Oncology Group 08.03 (RAVES) study, which is
looking at treatment for prostate cancer.
Participation is voluntary. You may choose to participate in both, one or neither of the
projects. You will receive the best possible care whether you take part or not.
Please read this participant information sheet carefully. Feel free to ask questions about any
information contained in this sheet. Before deciding whether or not to take part, you may
wish to discuss the project with your family, friends, or your doctor.
If you decide to take part in any of the projects, you will be asked to sign the consent form(s).
By signing it you are telling us that you:
Understand what you have read;
Consent to take part in the research project;
The University of Western Australia
(M409)
35 Stirling Hwy
Crawley WA 6009
Tel: (08) 6488 6730
Local Logo

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 105 of 114
Amendment 2: dated 29 May 2014
Consent to the use of your personal and health information as described.
You will be given a copy of this Participant Information and Consent Form to keep for your
records.
WHO CAN PARTICIPATE IN THE RESEARCH?
All patients who have been diagnosed with prostate cancer and are enrolled in the Trans-
Tasman Radiation Oncology Group (TROG) 08.03 RAVES trial will be asked whether they
would like to participate in the RAVES Genetic Study and/or the RAVES Tissue Banking
Study.
WHAT DOES THE RESEARCH INVOLVE?
Both studies involve asking your permission for researchers to collect and store samples for
future research projects:
Option A: RAVES Genetic Study
Donation of a saliva or blood sample and completion of one questionnaire
You will be given the option of providing either a saliva or blood sample. Your saliva
or blood will provide a sample of your DNA, which is the chemical compound that
genes are made of. Genes are inherited from both parents. The mixture of genes a
person has, or alterations in these genes, may lead to a medical problem or vary the
response to the treatment of a medical problem.
If you consent to the RAVES Genetic Study and wish to donate saliva, you will be
asked to collect into a specially prepared container. Alternatively, if you would rather
donate blood, the sample will be taken by a suitably qualified person at your local
hospital. DNA will be extracted from your saliva or blood and stored in a small plastic
tube in a secure freezer.
Option B: RAVES Tissue Banking Study
Donation of the prostate tissue sample already collected at the time of your surgery
for prostate cancer and potential completion of questionnaires
When your prostate was removed it was sent to a pathology laboratory for
examination. The first step in this process was to put pieces of it into paraffin (wax)
blocks. These blocks are cut into very thin slices so that a pathologist can examine
them under a microscope and send a report to your doctor. After the diagnosis has
been made, there is usually tissue left over in these wax blocks, which are generally
stored or “archived” indefinitely in the laboratory. These blocks are normally not used
again following your diagnosis, although on rare occasions they are retrieved and
looked at again. We are asking for your permission to obtain this stored material so
that it may be used for research in the future. A collection of many of these blocks for
research purposes is termed a tissue bank.
If you consent to Option B, there is no guarantee that any of your tissue will be able to
be put into the tissue bank. The pathologist at your local laboratory will decide if there
is enough tissue available to donate. We are asking your permission to contact the
laboratory where your biopsy specimen is held, and ask them to give us a small piece
of that specimen that they no longer needed. Retrieving and processing the tissue
does not require anything extra from you.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 106 of 114
Amendment 2: dated 29 May 2014
For both Option A and Option B:
Questionnaires: For both studies, you will be asked to complete questionnaires about
your personal background, ethnicity, family cancer history, current medical conditions
and medications. This will help researchers understand how a person’s genetic
makeup might affect response to treatment for prostate cancer.
Access to your medical records: We will ask your permission to access medical
information collected from you for the RAVES trial and during your routine clinic visits.
Your medical information from the trial is necessary to provide background
information about you, as well as information about your diagnosis, treatment and
response to treatment for prostate cancer. In addition, we would like to follow your
progress after your treatment by collecting information from various health agencies
(such as medical practitioners, pathology laboratories or hospitals), your medical
records or cancer registries and attaching it to you specimen in an anonymous
manner. This information is very important in understanding findings related to the
specimens you have donated. No personal information will be collected without your
permission. By signing this form you authorise the release of, or access to the
information collected about you as described in this section, even in the event of your
death.
WHAT IS A TISSUE BANK?
A Tissue Bank collects and stores tissue, blood and other samples for future research
purposes. These samples are labelled with a unique code number so as to protect the
identity of the donor. When researchers wish to study certain diseases, such as prostate
cancer, they can apply to the Tissue Bank to obtain coded samples. These samples can only
be released to future researchers if the Tissue Bank has written evidence of Human
Research Ethics Committee approval for their research projects. Relevant medical history
and clinical information of people who have donated tissue and blood samples are also
collected by the Tissue Bank. This information is stored in a secure database in a separate
location from the tissue.
The purpose of the tissue collection and “banking” is to enable future research to be done on
cancer. In your case, researchers are interested in understanding how to make the treatment
of prostate cancer better by examining the cancer (and sometimes normal tissue) in great
detail in the laboratory.
The first step in doing this is to obtain sample(s) from many men with prostate cancer, along
with their treatment records. This research depends on people like you to donate samples for
research as a “gift.”
The Trans Tasman Radiation Oncology Group is collaborating with two established tissue
banks to bank and process the specimens:
RAVES Genetic Study (Option A): DNA will be extracted from your saliva or blood
and stored at the Western Australian DNA Bank, which is led by the Centre for
Genetic Epidemiology and Biostatistics (CGEB) at the University of Western
Australia. The WA DNA Bank has received formal recognition from a number of
Human Research Ethics Committees. It does not recruit donors but provides the
infrastructure and personnel to handle and process the bio-specimens for medical
researchers using this facility. For safety the DNA collections are housed in two
secure cryo-facilities in the basement of E-Block at Sir Charles Gardner Hospital in
Perth and Royal Perth Hospital.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 107 of 114
Amendment 2: dated 29 May 2014
RAVES Tissue Banking Study (Option B): Prostate specimens from your surgery
will be banked at the Australian Prostate Cancer BioResource. The tissue bank
maintains tissue storage facilities in several capital cities in Australia, which are linked
by a comprehensive database. Each facility has received approval from its
Institutional Ethics Committee to collect and store blood and tissue from patients for
future research purposes.
WHAT WILL HAPPEN TO MY DONATED SPECIMEN(S)?
Your saliva, blood or tissue will be packaged in a secure manner and shipped to whichever
facility will store it. Specimens will be stored indefinitely, until they are used up or until you
contact RAVES trial staff in the event that you change your mind about participating. In the
case of your surgery specimen, your local laboratory may request that it be returned, if
needed.
Any DNA donated for research will be stored in accordance with the National Health &
Medical Research Council’s Guidelines for Genetic Registers and Associated Genetic
Material. Storage will be conducted under a coded system, to ensure that your confidentiality
is maintained.
WHAT KINDS OF RESEARCH WILL MY SALIVA, BLOOD OR TISSUE BE USED FOR?
Because new techniques to look at genes in relation to cancer treatment are regularly
developed, it is not possible for researchers to specify now how your tissue or DNA will be
studied. However, having access to a collection of donated specimens allows researchers to
make discoveries as new techniques become available. The most important discoveries will
be directed to improvements in the diagnosis, monitoring and treatment of prostate cancer.
In addition to providing information that will help develop future treatments, your blood or
saliva sample will be used to investigate whether genetic make-up has an effect on
radiation therapy adverse experiences or “side effects”. This information may identify new
ways that DNA is linked with radiotherapy side effects, which in turn would require further
investigation. Your DNA will not be used for research that involves reproductive cloning.
No researchers will be permitted to derive a genetic profile of you as an individual.
Your sample may be provided to approved researchers from hospitals, universities, medical
research institutes, government affiliated institutions and sometimes commercial
organisations, such as pharmaceutical companies. Data provided for other research studies
will be identified only by a study identification number and not with your name. Researchers
will be required to have approval from a Human Research Ethics Committee, who will ensure
access to the data abides by the Australian guidelines and statutes that govern the Tissue
Banks. Some research studies in cancer may involve sending your DNA interstate and
overseas. If this is the case, these researchers are required to demonstrate that they meet
the appropriate Australian standards of ethics and privacy as detailed below.
WHAT ABOUT CONFIDENTIALITY?
The RAVES study managers are committed to making sure that information about you is
kept safe and in strict confidence. Personal information that can identify you, such as your
name or date of birth is removed and replaced with your RAVES identifying code before it is
sent to the tissue bank. Only your local hospital staff will be able to link your name with your
trial code. The Tissue Bank will assign a further unique number code to your specimen(s),
and anyone who studies your specimens will only be supplied with your coded sample(s) and
data. Only authorised Tissue Bank staff will be able to break this code. This ensures that
nothing that can identify you or your family will ever appear on any public or published
reports. Access to your DNA donation for future research will be managed by an Advisory
Committee and only released where the research project that wishes to use it has been
approved by a Human Research Ethics Committee. International research collaborations

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 108 of 114
Amendment 2: dated 29 May 2014
using your information and/or tissue or DNA sample will only take place where researchers
abide by equal or more stringent regulations of privacy and ethics as those in Australia, as
assessed by a Human Research Ethics Committee.
Personal information, such as health information will remain privileged and confidential,
except as required by law. By signing the attached Consent Form, you authorise release of,
or access to, this confidential information to the relevant study personnel and regulatory
authorities.
To ensure that these policies are followed, Human Research Ethics Committees monitor all
research projects they approve. This is usually done annually, by requesting a report from
the Chief Investigator for the research project. This report covers matters such as the
maintenance and security of records and compliance with the project as originally approved.
WILL I FIND OUT THE RESULTS OF THE GENETIC STUDY OR TISSUE BANK
RESEARCH?
You have the legal right to access your permanent medical records at any time, but specific
research results will not be part of this record. Information from the sample(s) you may
donate is not intended to be used in your diagnosis or treatment. Feedback will not normally
be given to participants regarding personal information. This is because prostate cancer is
thought not to be a single-gene disease (such as cystic fibrosis), but a complex disease with
many genes and other factors contributing to its development and progression. We do not
yet know exactly how these elements all interact, so releasing personal information would not
be clinically meaningful or useful to you or your family.
WILL MY FAMILY BE ABLE TO REQUEST STUDY RESULTS IN THE FUTURE?
You should consider whether or not any significant health information obtained as a result
of the genetic studies should be disclosed to your family members or descendents in the
future. We will ask whether you consent to the disclosure of your information to your family
members or descendents on the attached consent form. Family members would have to
supply a written request to the Chief Investigator, and a Human Research Ethics
Committee would review and consider the request to disclose information. If you do decide
to participate, please consider advising your family members of the purpose for which you
have provided your samples.
WILL MY SAMPLES EVER BE SOLD?
Your samples will NOT be sold. The Tissue Banks may charge researchers a fee to recover
some of the costs of storing and administering its collection of tissue. Although knowledge
acquired through medical research may lead to discoveries that are of commercial value to
the researcher and their institution, there will be no financial benefit to yourself, your family or
the Tissue Banks.
WHAT ARE THE BENEFITS OF TAKING PART?
It is unlikely that there will be a direct benefit to you or your family from participating in these
studies. Your samples may not be used for many years until a new and as yet undiscovered
approach to study cancer is developed. These studies may ultimately benefit other patients
with prostate cancer, through new understandings about treatment and responses to
treatment. The aim of these sub-studies is to improve the health of patients with prostate
cancer. However, sometimes research on your tissue may lead to findings that result in the
development of a commercial test or treatment that may be overseen by other groups,
including private companies. There will be no financial reward or payment to you in such an
event and by participating, you will waive all claims to patents, commercial returns, property
or any material or products which may arise from these studies.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 109 of 114
Amendment 2: dated 29 May 2014
WHAT ARE THE RISKS OF TAKING PART?
There are no documented risks or adverse experiences from saliva sample collection.
If you opt to donate a blood sample, it may hurt for a short time and you may get a bruise.
The amount of blood to be taken is small and you should not notice its loss. Infection at the
site where blood is taken is very unlikely, as the skin is cleaned first with an antiseptic and
sterile equipment is used. This procedure can also occasionally cause light headedness or
fainting. These reactions are usually mild, of short duration and limited to a feeling of
weakness, accompanied by sweating, slowing of heart beat, and a decrease in blood
pressure.
For the patient surveys, it is possible that answering the questions could be upsetting. For
instance, the surveys may arrive at a time when you are not feeling well or you are feeling
anxious. Of course, you may decline to participate in a survey or to answer a specific
question in the survey.
There is a risk that your information may be released without your permission, and
measures have been taken to minimize this risk, as described in the section on
Confidentiality above. It is important to note that there may be circumstances where
disclosure of your health information kept by the study teams could be obtained, for
example, if requested by law as a result of a court order.
Genetic research risks:
Genetic research involves the study of genetic material (DNA), which is shared with your
blood relatives. Genetic research raises many important issues. It is unlikely that these
issues will arise, but you should think about them carefully. Genetic research is undertaken
to discover more accurate ways of predicting diseases within a group of people or in people
where there is a strong family history or predisposition of disease. To perform such studies, a
researcher must demonstrate to a Human Research Ethics Committee that procedures are in
place to deal with certain issues which may arise.
The vast majority of research projects for which your tissue might be used are unlikely to
reveal anything of medical importance specifically for you or your family. However, if that
should happen and a discovery is made that predicts a possible medical condition, the
researcher is required to inform the appropriate Human Research Ethics Committee. If the
Committee considers that this knowledge may be of medical significance to you and/or your
family, a member of the research team would contact you through your doctor and give you
the option of learning more about the findings. If we are unable to contact your doctor, we will
make a reasonable attempt to contact you at your address provided or any updated address
that you might care to provide.
Learning the results from genetic research might create uncertainty or be upsetting; if for
instance, the risk of developing a disease is identified which has no known prevention,
treatment or cure. Some people may learn disturbing information about inherited diseases or
a disorder involving their children, brothers or sisters. This could interfere with family
relationships. You may be faced with the question of “Should I tell the family?” Other family
members may or may not wish to know this information. It is important to understand that
results from genetic research usually will not indicate that you have a disease or disorder, or
whether you will develop it. The results can only show that you have an increased risk of
developing a disease or disorder, but there would still be no guarantee that you will develop
the condition. Any research results that could be of significance to you or your family will
need to be repeated and verified. This may involve having a blood sample taken and having
it retested in an official testing laboratory. This is standard practice for all patients receiving
the results of genetic testing and would be provided free of charge to you. Counselling may
also be provided free of charge if it is appropriate. Before a test is repeated to verify a

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 110 of 114
Amendment 2: dated 29 May 2014
research finding you will be informed about the possible risks involved. This is especially
important for individuals who are found to have a gene mutation, which increases their risk of
disease or cancer. Knowing such information could lead to job discrimination and difficulties
in obtaining some forms of insurance.
On the Consent Form you will be given the option of having your sample excluded from
Study B (RAVES Tissue Bank) research that identify genes or diseases that run in families,
for example, diseases that can be passed on (through DNA) to blood relatives. Whatever
choice you make, your privacy will be protected and your decision will be respected.
We ask you to keep your local hospital up-to-date with your contact details even after
the RAVES study has finished.
WHAT ARE THE COSTS TO ME?
You would not be charged for any tests or samples taken as part of the study. You will not
be paid for taking part in this study. There will be no financial reward or remuneration to
you.
Participating in this study will not incur additional costs to you compared to not participating.
WHAT HAPPENS TO MY SAMPLE(S) AFTER MY LIFETIME?
In the event of your death, the study teams will continue to store your samples and make
them available for use by researchers. Your gift will continue to be used in cancer and other
bio-medical research subject to the same legal and ethical standards discussed in this
document. On the Consent Form you may nominate a representative to be contacted about
any matters that we would have contacted you about. This person should be a blood
relative or spouse/partner and you should discuss the implications of your donation with
whomever you nominate. If you decide to participate, please consider advising your family
members or the Executor of your Will of the existence of your sample(s); perhaps even
provide them with a copy of this Information Sheet and your signed Consent Form.
WHAT DO I NEED TO DO TO PARTICIPATE?
If you have read this information sheet and would like to participate, then please:
Discuss any questions you may have with the clinic staff at your hospital.
Once your questions have been answered, if you still wish to participate, please
carefully read the consent form and sign it together with your doctor.
Indicate which study option(s) you have selected.
Clinic staff will then provide instructions about the study:
WHAT IF I DO NOT WISH TO PARTICIPATE IN EITHER STUDY?
If you do not wish to participate, you do not need to do anything further. Your decision will in
no way affect your level of healthcare, your relationship with your doctor, or your continuing
participation in the TROG 08.03 RAVES trial.
WHAT IF I CHANGE MY MIND?
You are free to withdraw from either study at any point without giving a reason. If you decide
to withdraw from the study, you can choose to let the tissue banks keep and use the DNA
sample or tumour block for future cancer research or request that the sample(s) be
destroyed or returned. If you would like to withdraw from either study, please contact study
staff in writing:
[Insert details of local Principal Investigator or Study Coordinator]
XXXXX
XXXXX
XXXXX
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 111 of 114
Amendment 2: dated 29 May 2014
STATEMENT OF APPROVAL:
The [INSERT LOCAL ETHICS COMMITTEE NAME] has reviewed this study and has given
its approval for the conduct of this research trial. In doing so this study conforms to the
principles set out by the National Statement on Ethical Conduct in Human Research and
according to the Good Clinical Practice Guidelines.
All current and future studies will only be conducted after approval of an Institutional Review
Board (IRB)/Human Research Ethics Committee (HREC). No tissue or health information
will be released to a third party unless it is to carry out research that has been approved by a
Human Research Ethics Committee.
CAN I CONTACT ANYONE WHO IS INDEPENDENT OF THE STUDIES?
If you have a complaint about this project or would like any advice regarding your rights as a
patient, you can talk to someone who is not involved in the study at all.
Name:
Position:
Telephone:
You will need to tell [INSERT LOCAL CONTACT] the name of one of the researchers listed
at the beginning of this document.
FURTHER INFORMATION
Please feel free to ask your doctor any questions at your next clinic appointment. If you need
further information, please contact:
[Insert local study coordinator details here]
Thank you for considering this invitation.
You will be given a copy of this Participant Information
Sheet to keep for your records.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 112 of 114
Amendment 2: dated 29 May 2014
PARTICIPANT CONSENT FORM
TROG 08.03 OPTIONAL TISSUE STUDIES
Study A. RAVES Genetic Study: Genetic basis of response to radiotherapy
in prostate cancer patients
Study B. RAVES Tissue Banking Study
TROG 08.03 RAVES: Radiotherapy – Adjuvant Versus Early Salvage
A phase III multi-centre randomized trial comparing adjuvant radiotherapy with surveillance and early
salvage RT in patients with positive margin or extra prostatic disease following radical prostatectomy
PRINCIPAL INVESTIGATOR: Local Investigator Details
Please read this consent form carefully.
My signature on this consent form confirms the following:
I have read, or have had read to me in a language I understand, the Patient
Information Sheet and Consent Form (Version 2, dated 10/05/11).
I have had an opportunity to ask questions, and I am satisfied with the answers I
received. The research objectives have been fully explained to me. I have understood
the Patient Information Sheet, which describes the purpose of the studies and what
my participation involves. I have been given the opportunity to have a member of my
family or a friend present while the study was explained to me.
I understand that my involvement in the study is voluntary and it will not affect my
relationship with my medical advisers in their management of my health. I also
understand that I am free to withdraw from the study at any stage without my future
treatment being affected. I may do this by contacting RAVES trial staff at my local
hospital
I understand that the results of the research may be of interest to my immediate
family, including my descendants, and I may decide whether or not the information
may be disclosed to my family in accordance with ‘Options for disclosure to family
members’ section detailed below.
The Chief Investigator of this project and his associates involved with this project will
not be liable for any loss or damage to the saliva, blood or tissue taken or used in
accordance with this form.
I give permission for the release of information concerning my disease and treatment
as outlined in the Patient Information Sheet
The University of Western Australia
(M409)
35 Stirling Hwy
Crawley WA 6009
Tel: (08) 6488 6730
LOCAL LOGO
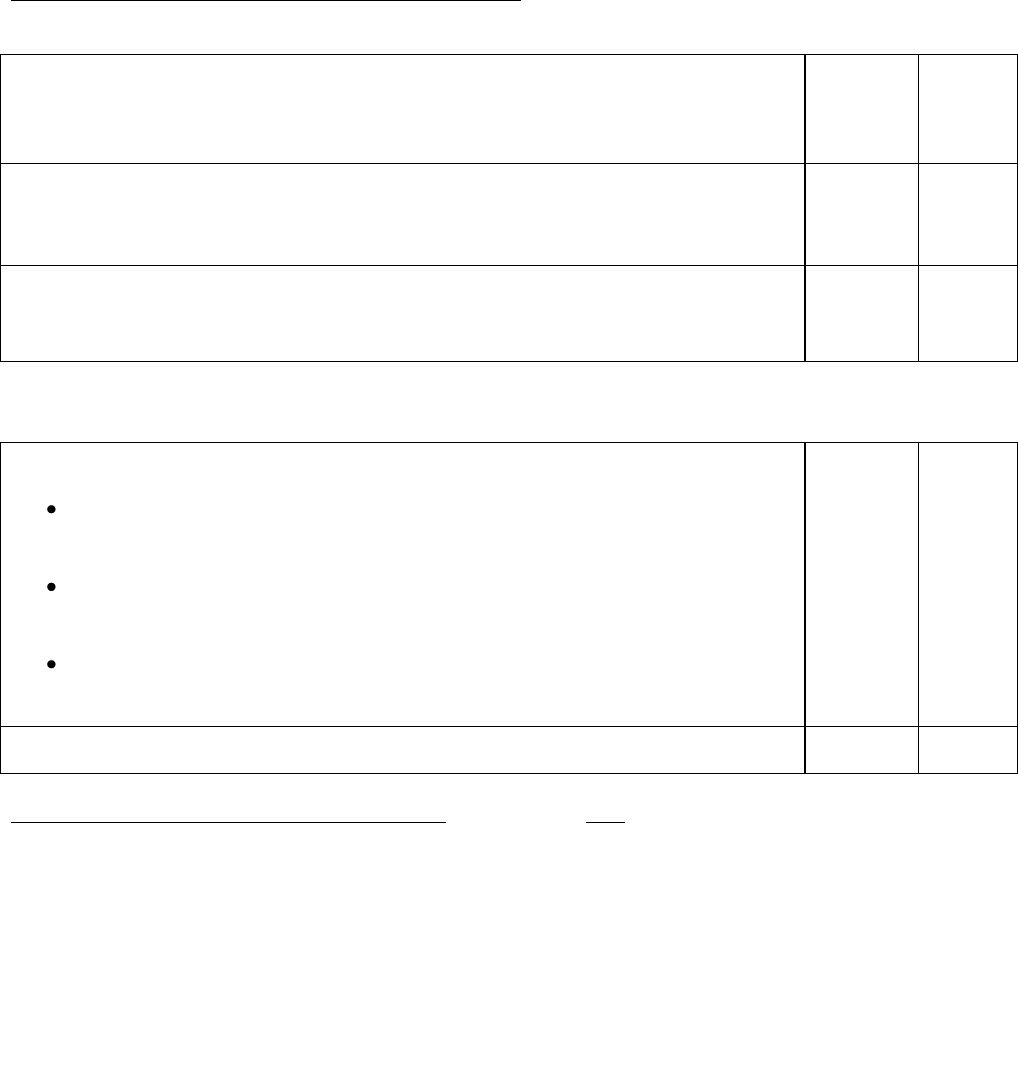
TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 113 of 114
Amendment 2: dated 29 May 2014
Consent to participate in Study A and/or Study B:
Study A:
I consent to donate a saliva or blood sample to the RAVES Genetic Study /
Centre for Genetic Epidemiology for DNA extraction, storage, testing and
research. I have been advised what sample is being collected, what the purpose
of this is and what will be done with the sample.
Yes
No
I give my consent for the RAVES Genetic Study, the Centre for Genetic
Epidemiology and Biostatistics at the University of Western Australia to use my
information and samples, including my health information collected in this study,
for future medical research.
Yes
No
I consent to my information and tissue sample to be available to researchers
working with the Western Australia DNA Bank, some of which may be in
different countries.
Yes
No
Study B:
I agree to allow my archived tissue surplus to diagnostic requirements to be
obtained from the place of its storage and provided as a “gift”.
I understand that the tissue samples (including their constituents and
anything derived from them) will be stored indefinitely in the Australian
Prostate Cancer BioResource Tissue Bank
I understand that my tissue may be used for future studies of prostate
cancer. These may be biochemical or genetic studies provided I have
not stipulated otherwise below.
I give permission for my tissue to be used in any way that The Australian
Prostate Cancer BioResource and the RAVES study managers deem
beneficial.
Yes
No
I consent for genetic research to be performed on my tissue samples
Yes
No
Options for disclosure to family members: Please tick one box only
I understand that future DNA (genetic) testing may result in new information about diseases
or potential diseases that may have health implications for my family, including my
descendants. I understand that extensive testing and validation would be required before the
information can be determined to be useful and that information can only be provided after
careful consideration by a Human Research Ethics committee, through approved medical
channels, and when there is clear evidence of the medical importance to my family.
I DO consent to genetic information obtained from my DNA as a result of this study
being revealed to my family members, upon their written request to the Chief
Investigator, in the event of my death.
I DO nominate a representative to be contacted about any matters that study staff
would need to contact me about, in case I am unable to be reached:
Name:______________________________
Address:_____________________________
I DO NOT consent to genetic information obtained from my DNA, as a result of this
study, being revealed to my family members, upon their written request to the Chief
Investigator, in the event of my death.

TROG 08.03 RAVES Final Protocol Version: 7 August, 2008 Page 114 of 114
Amendment 2: dated 29 May 2014
If you are unclear about anything you have read in the Participant Information Sheet or
this Consent Form, please speak to your doctor before signing this Consent Form.
PATIENT STATEMENT & SIGNATURE
Statement by Participant: I hereby consent to take part in this study and I will be given a
copy of this signed and dated consent form.
________________________________ (printed full name)
________________________________ (signature) _____ / _____ / _____ (personally
dated)
Witness (optional)
In my opinion consent was given freely
________________________________ (printed full name)
________________________________ (signature) _____ / _____ / _____ (personally
dated)
Interpreter (if applicable)
In my opinion consent was given freely
________________________________ (printed full name)
________________________________ (signature) _____ / _____ / _____ (personally
dated)
Statement of Investigator
In my opinion consent was given freely
________________________________ (printed full name)
________________________________ (signature) _____ / _____ / _____ (personally
dated)
